






























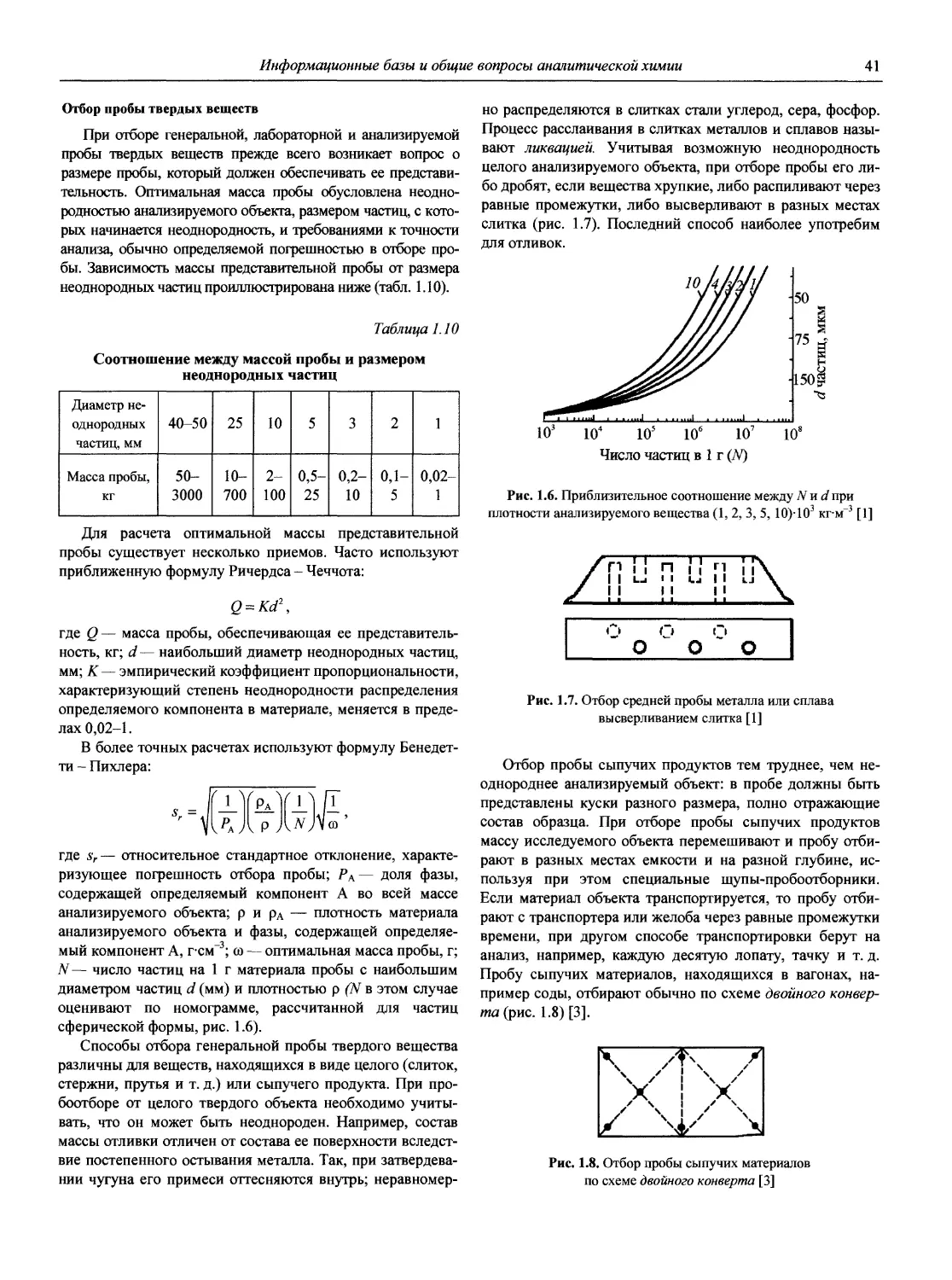













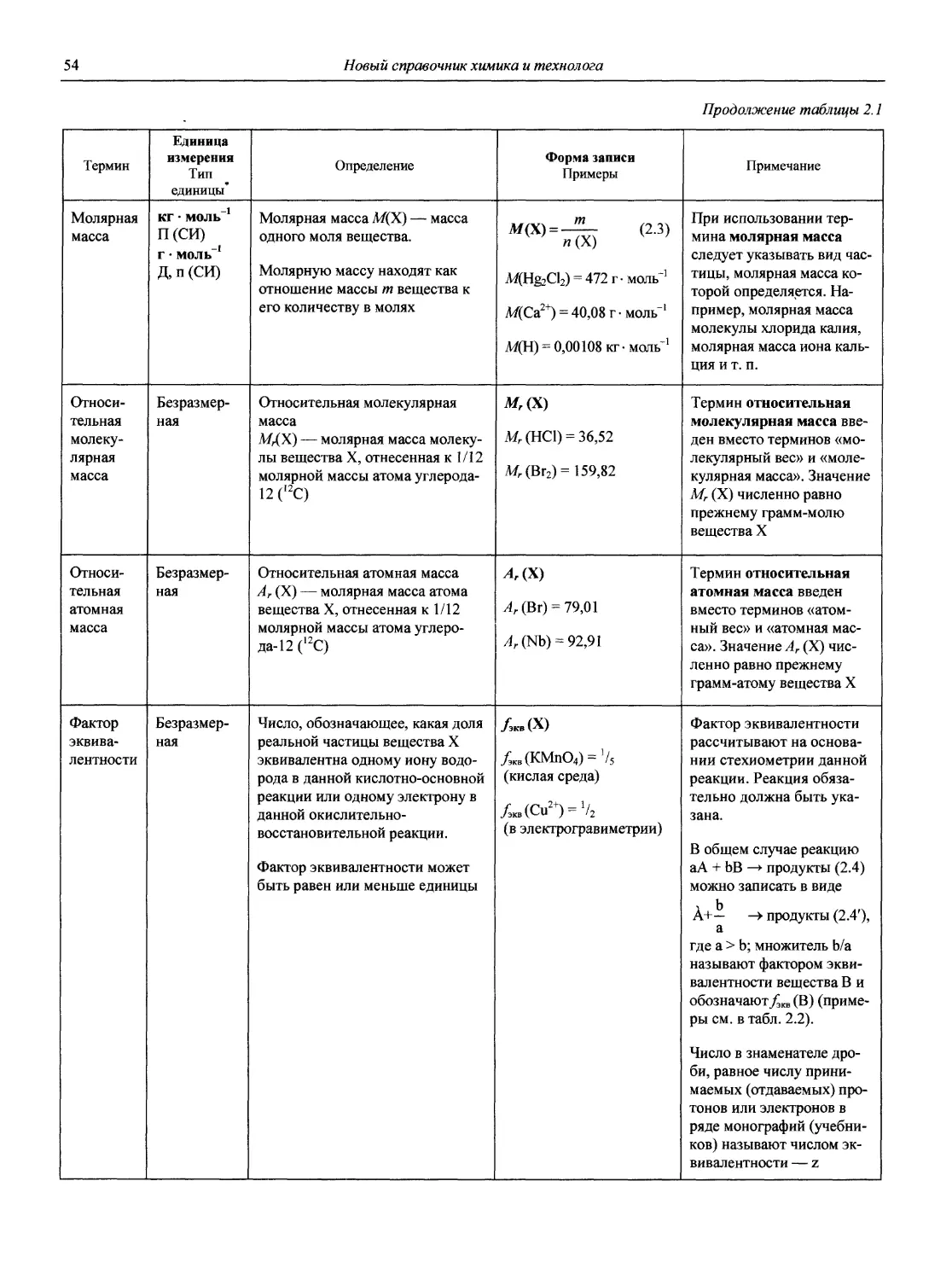
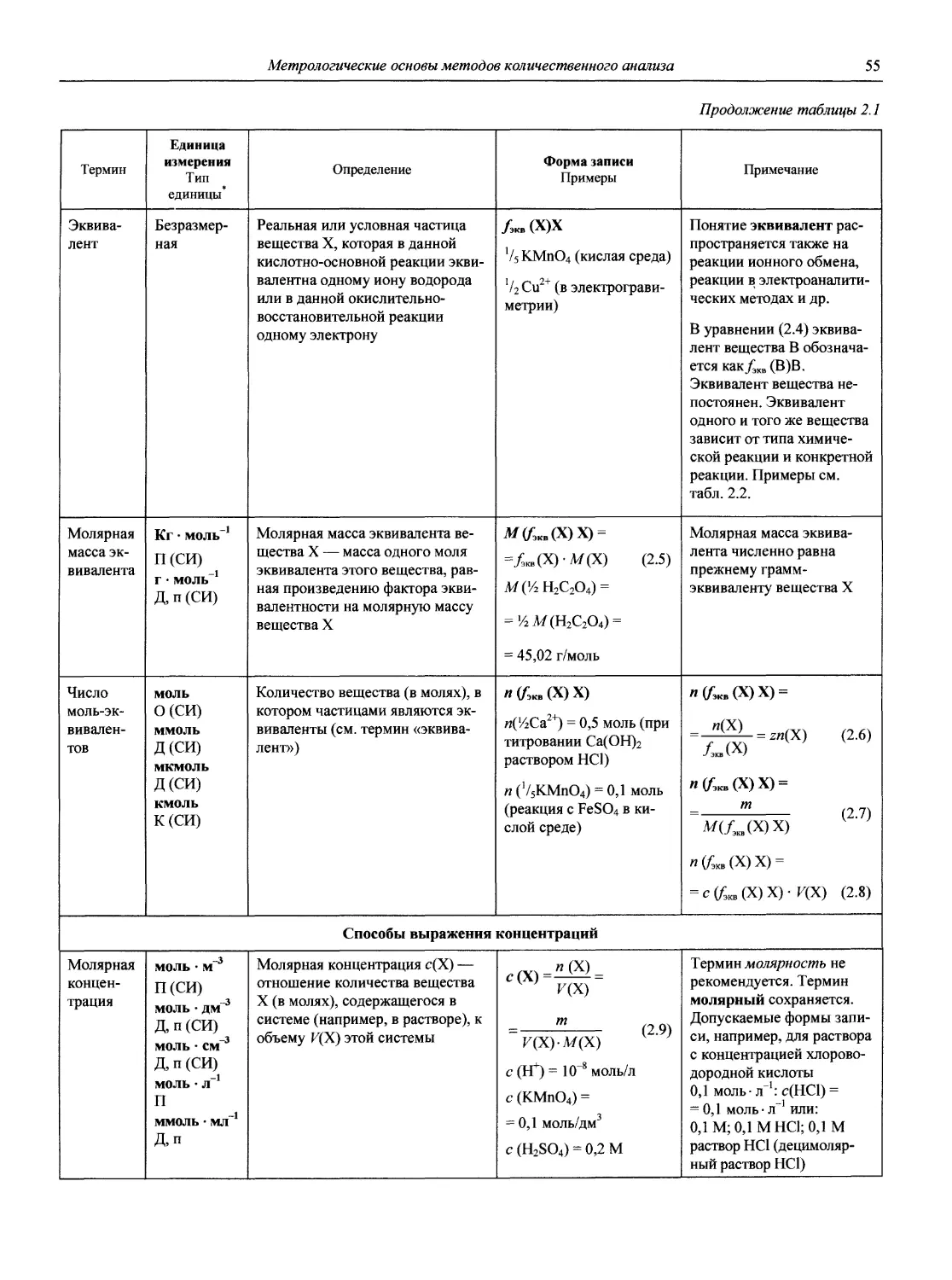
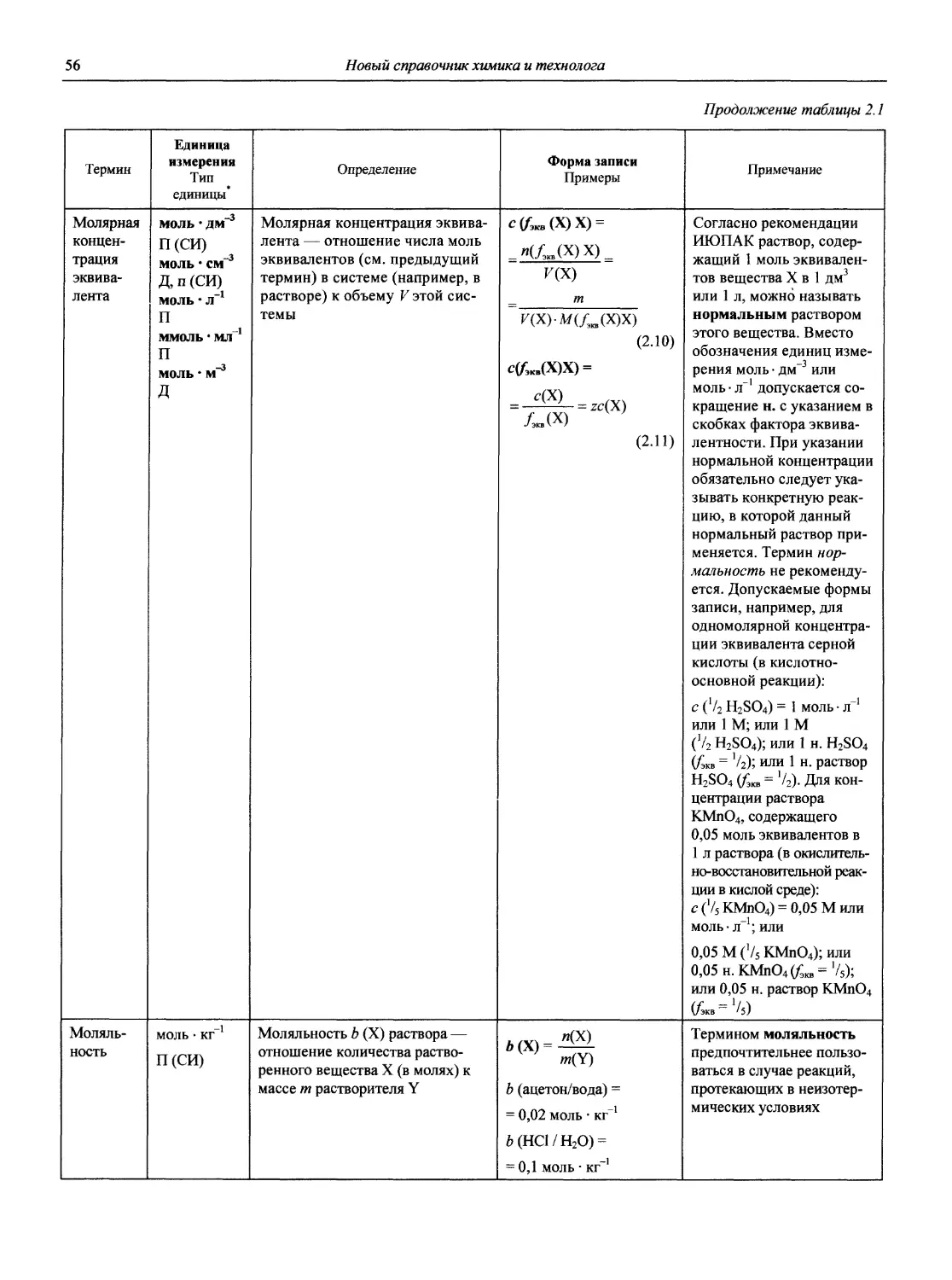










































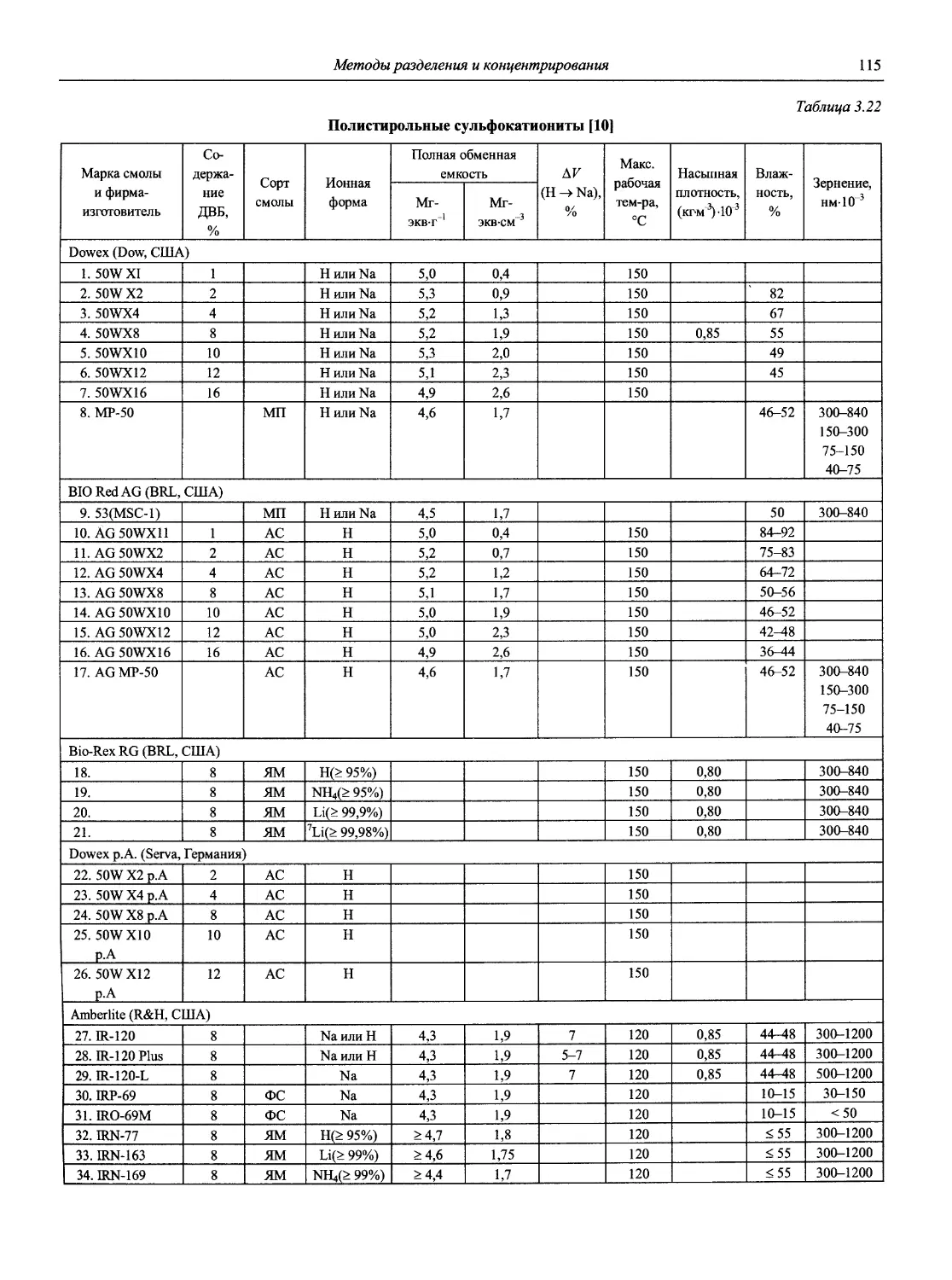



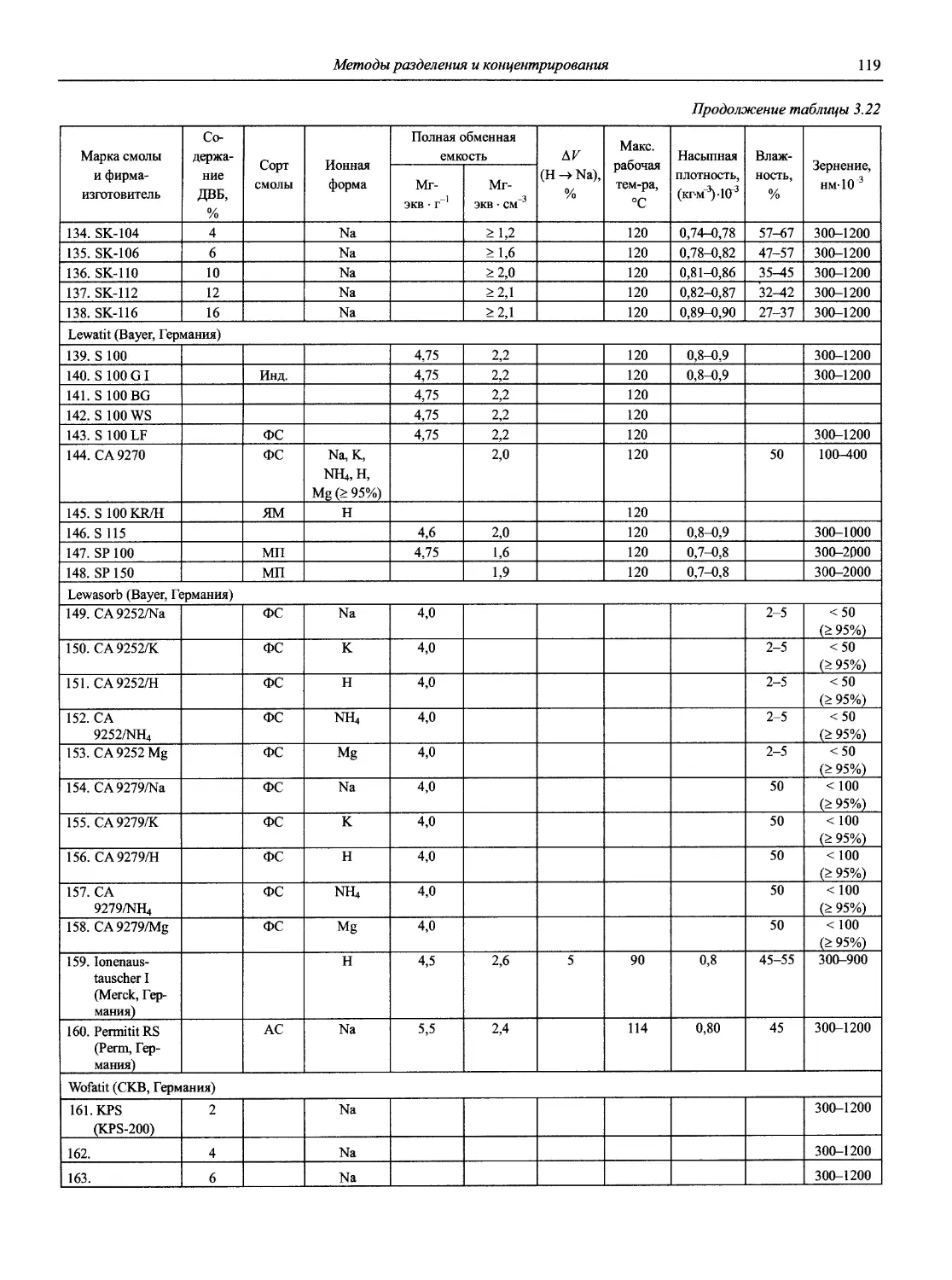
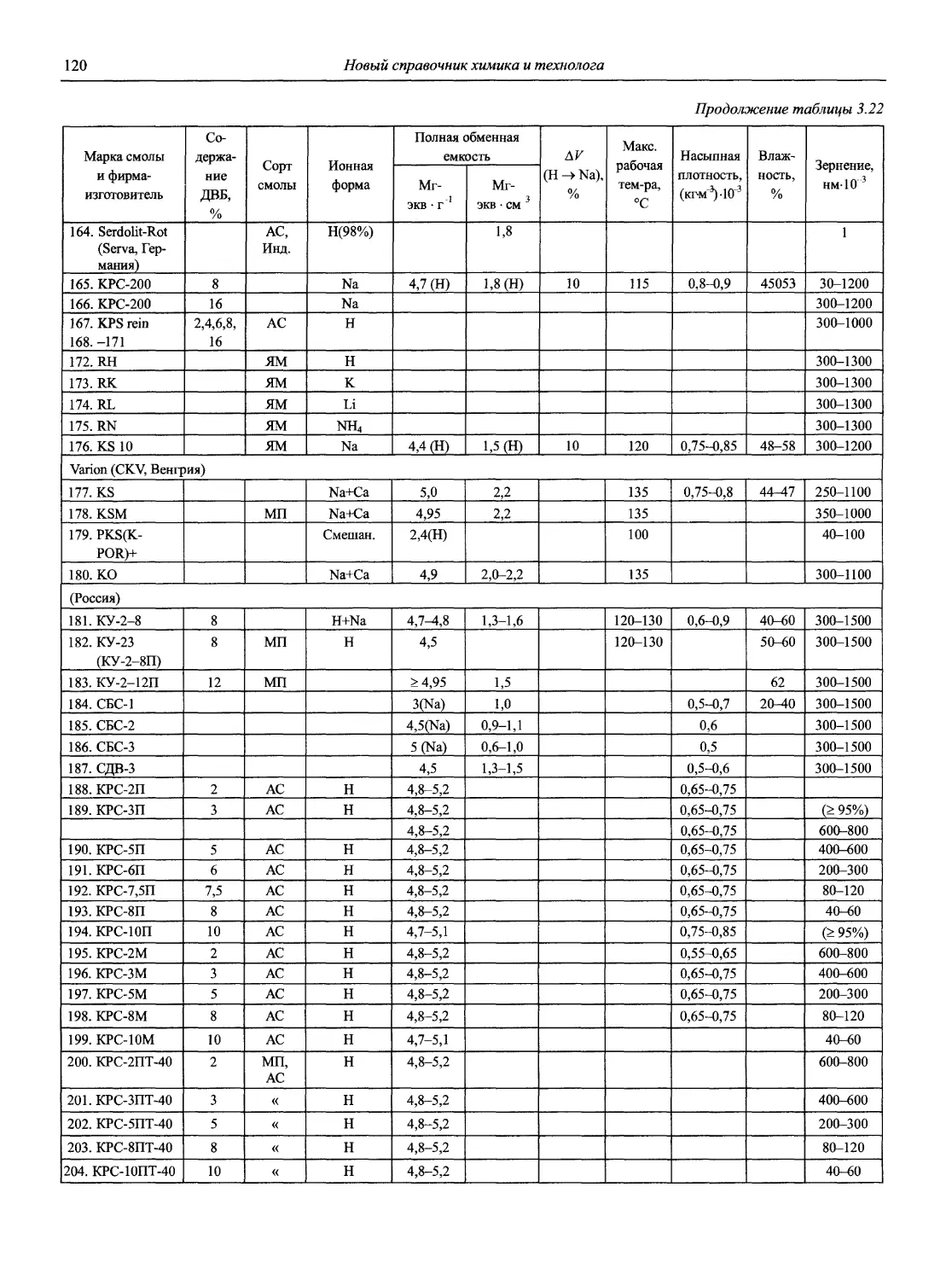
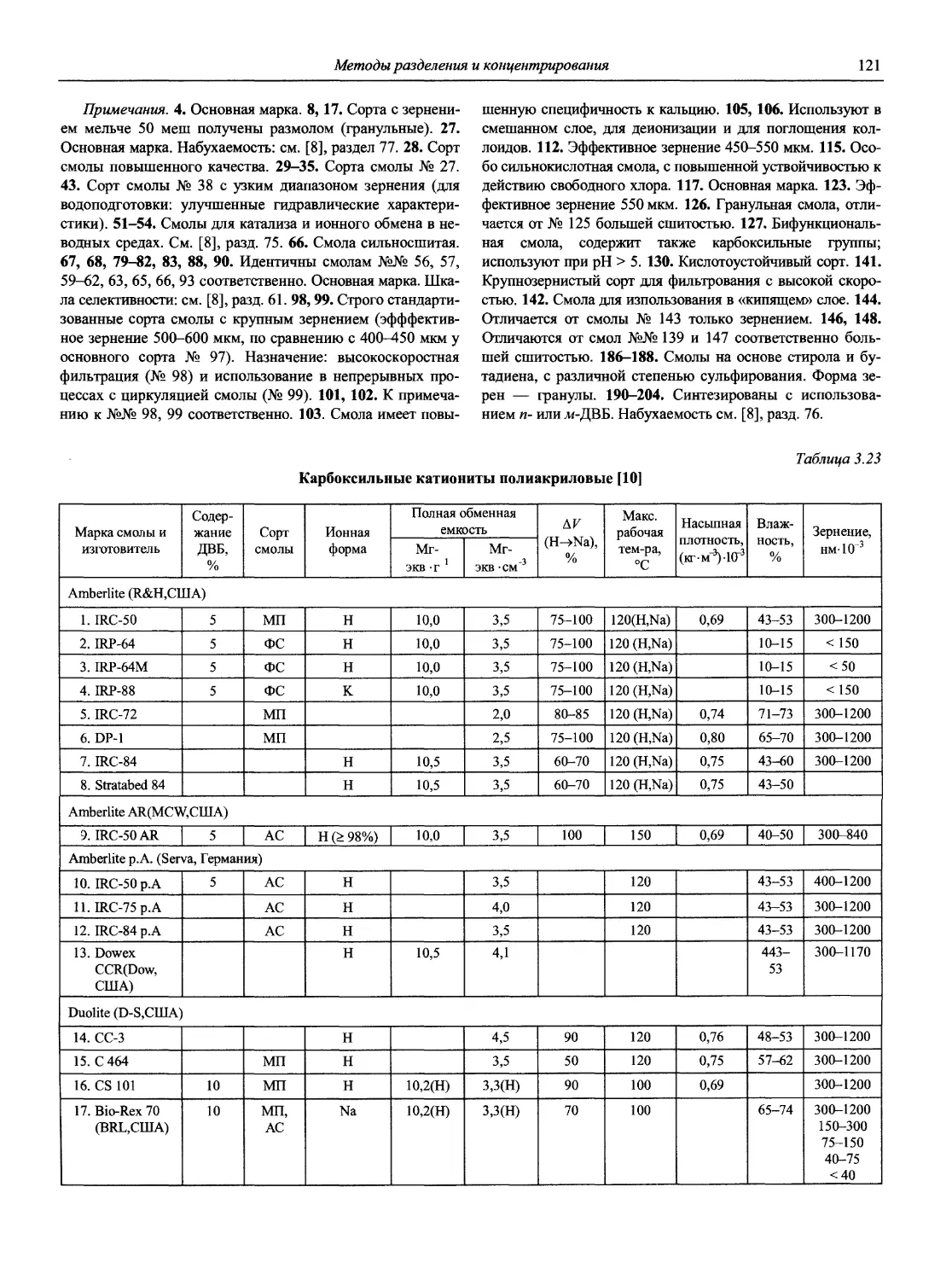









































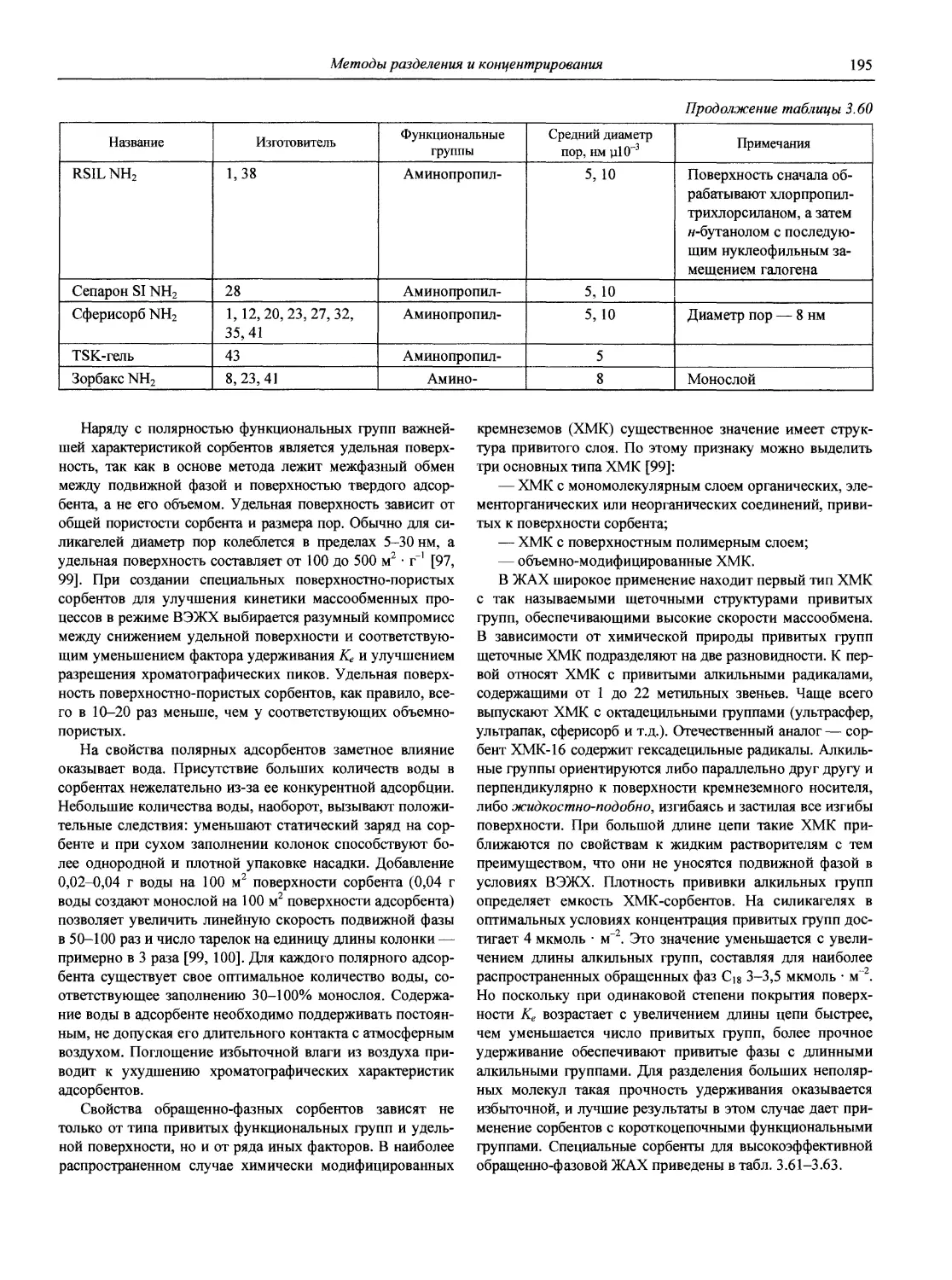
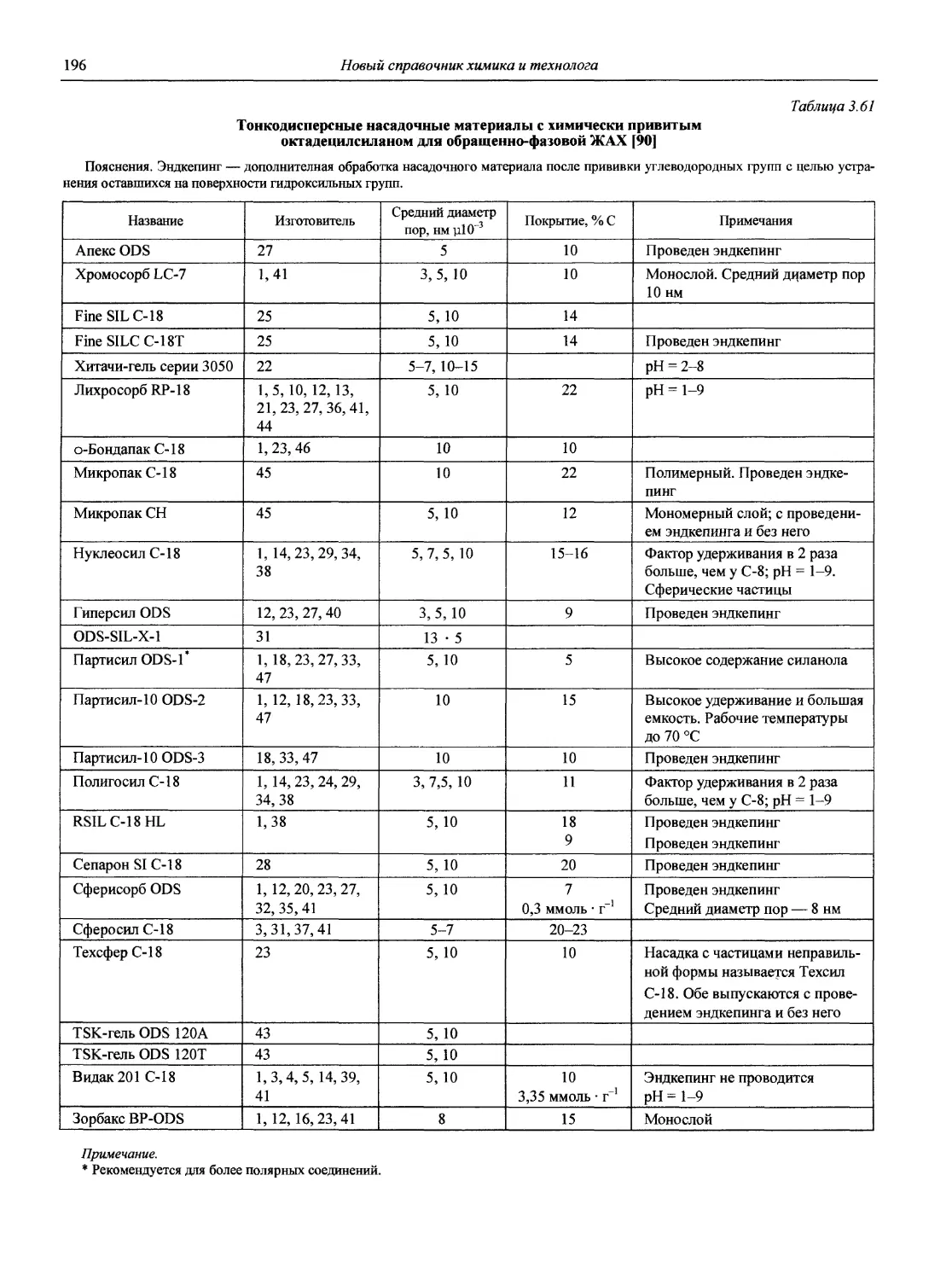
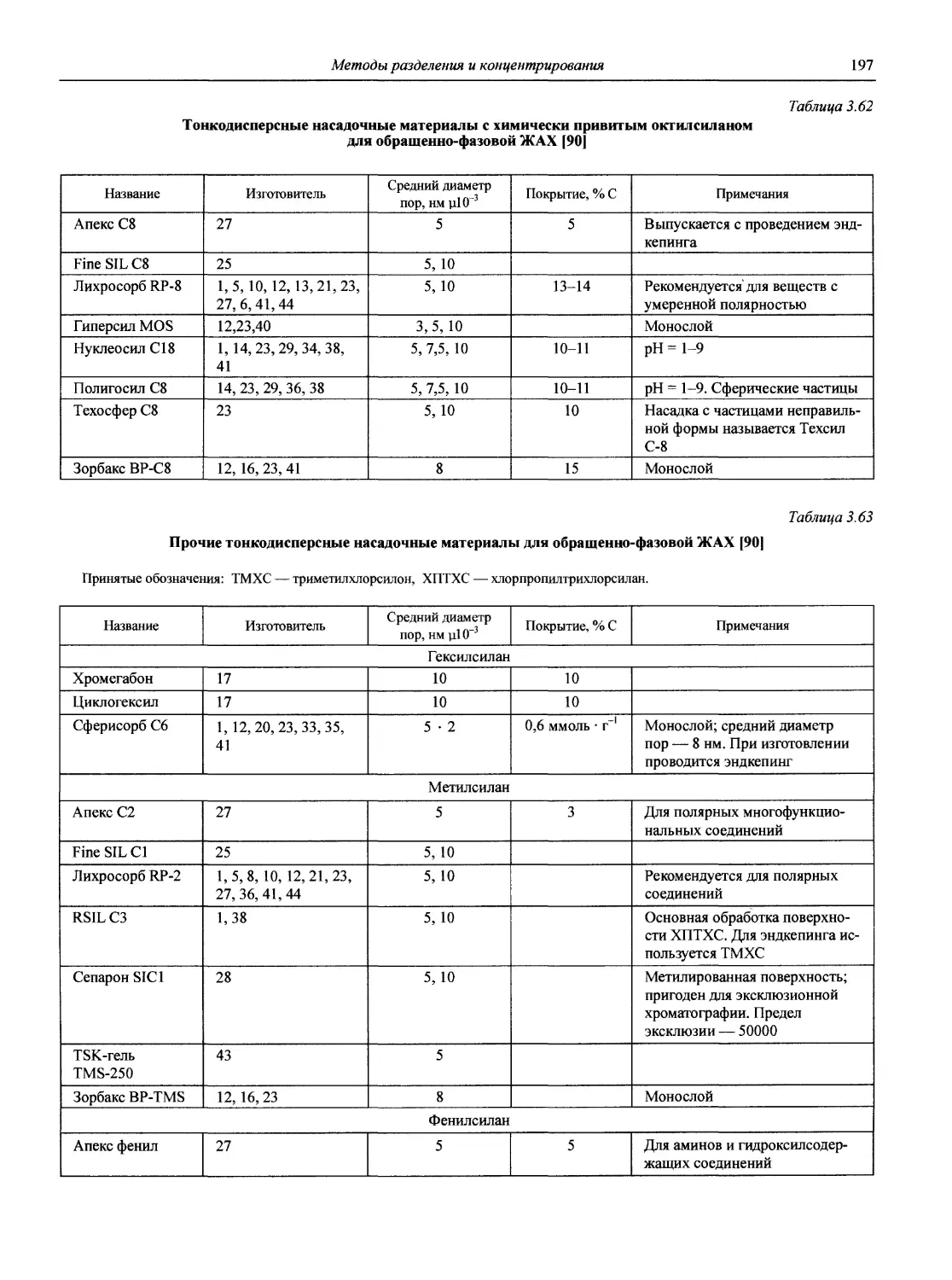
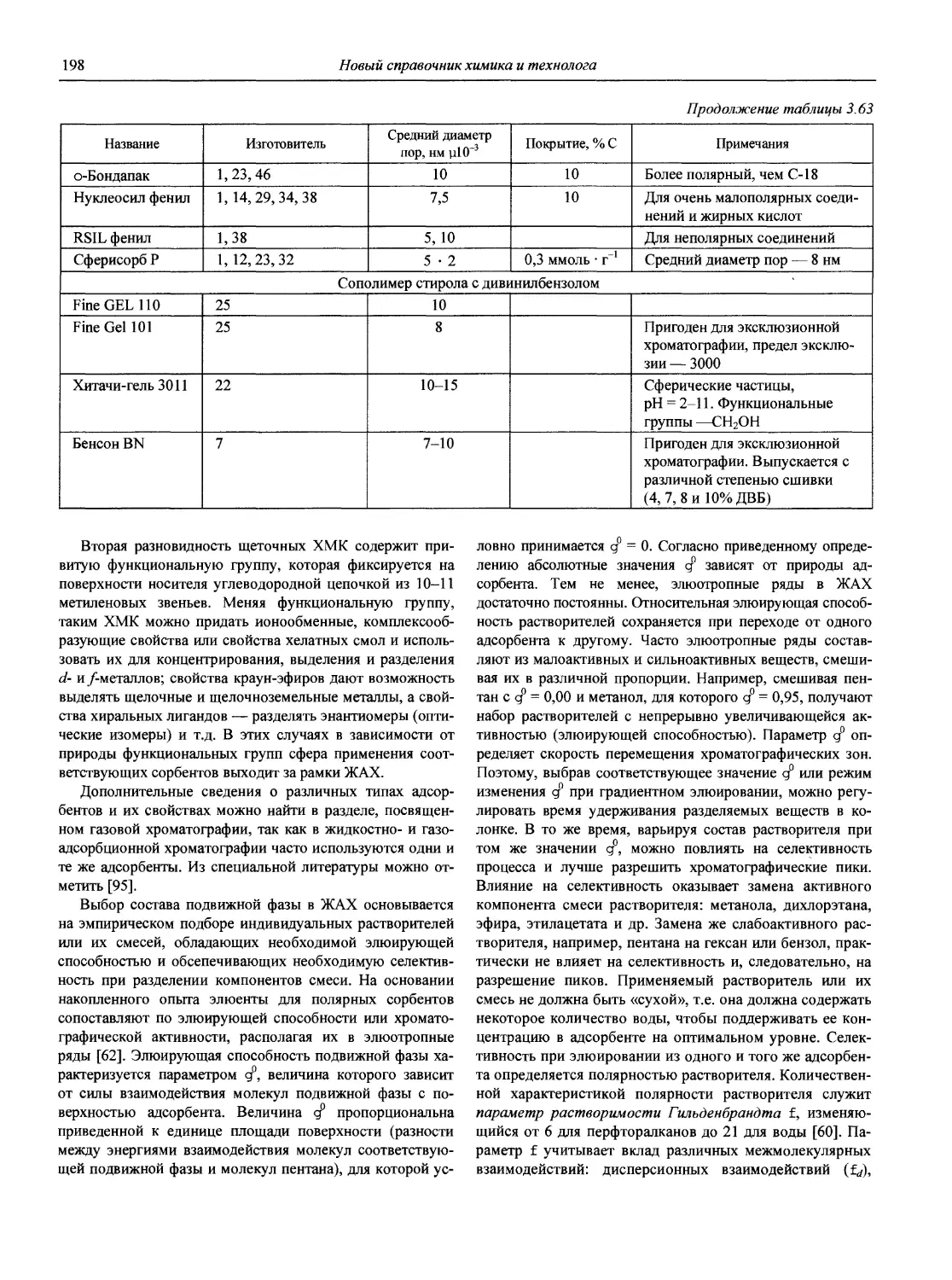







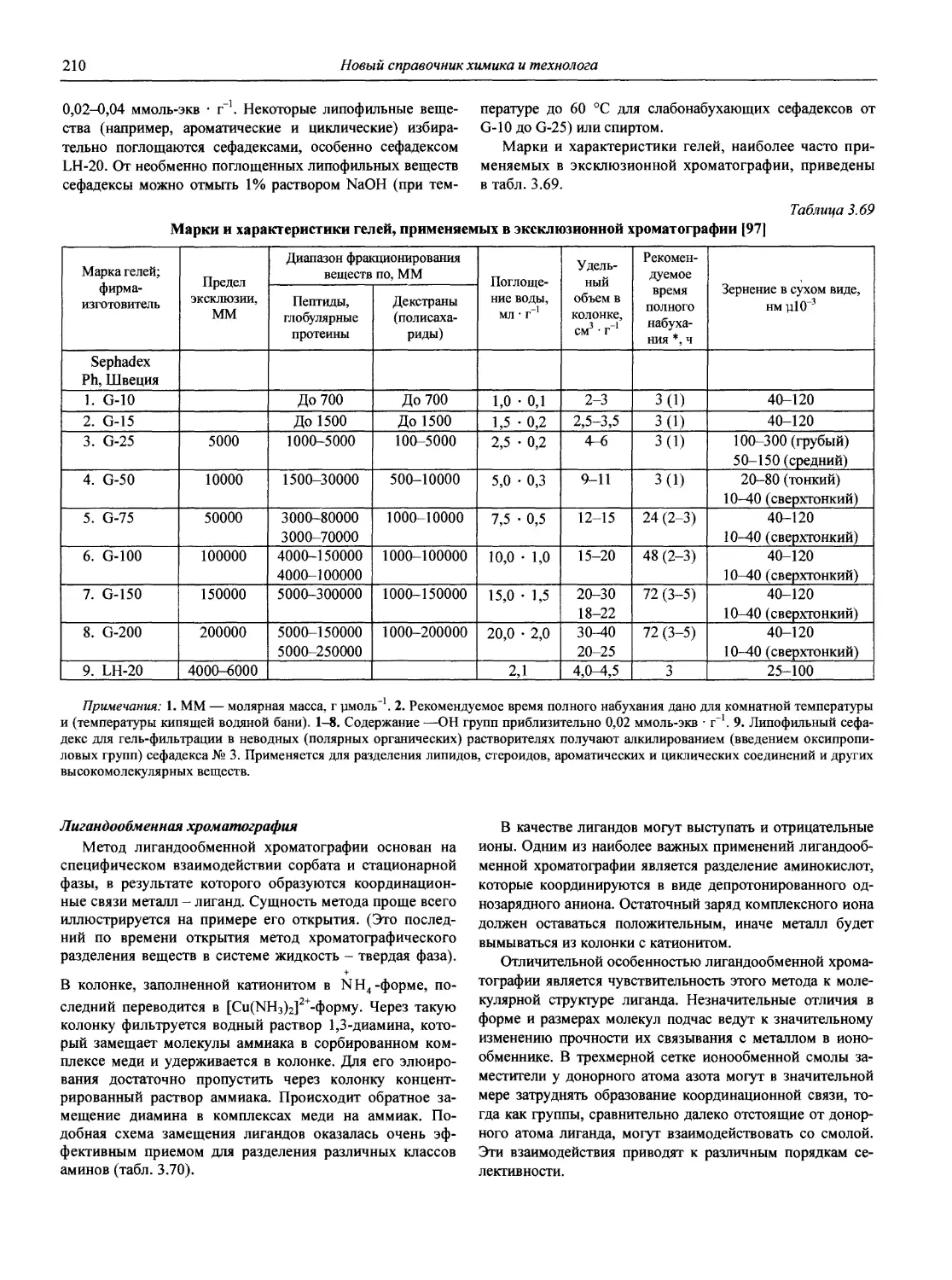
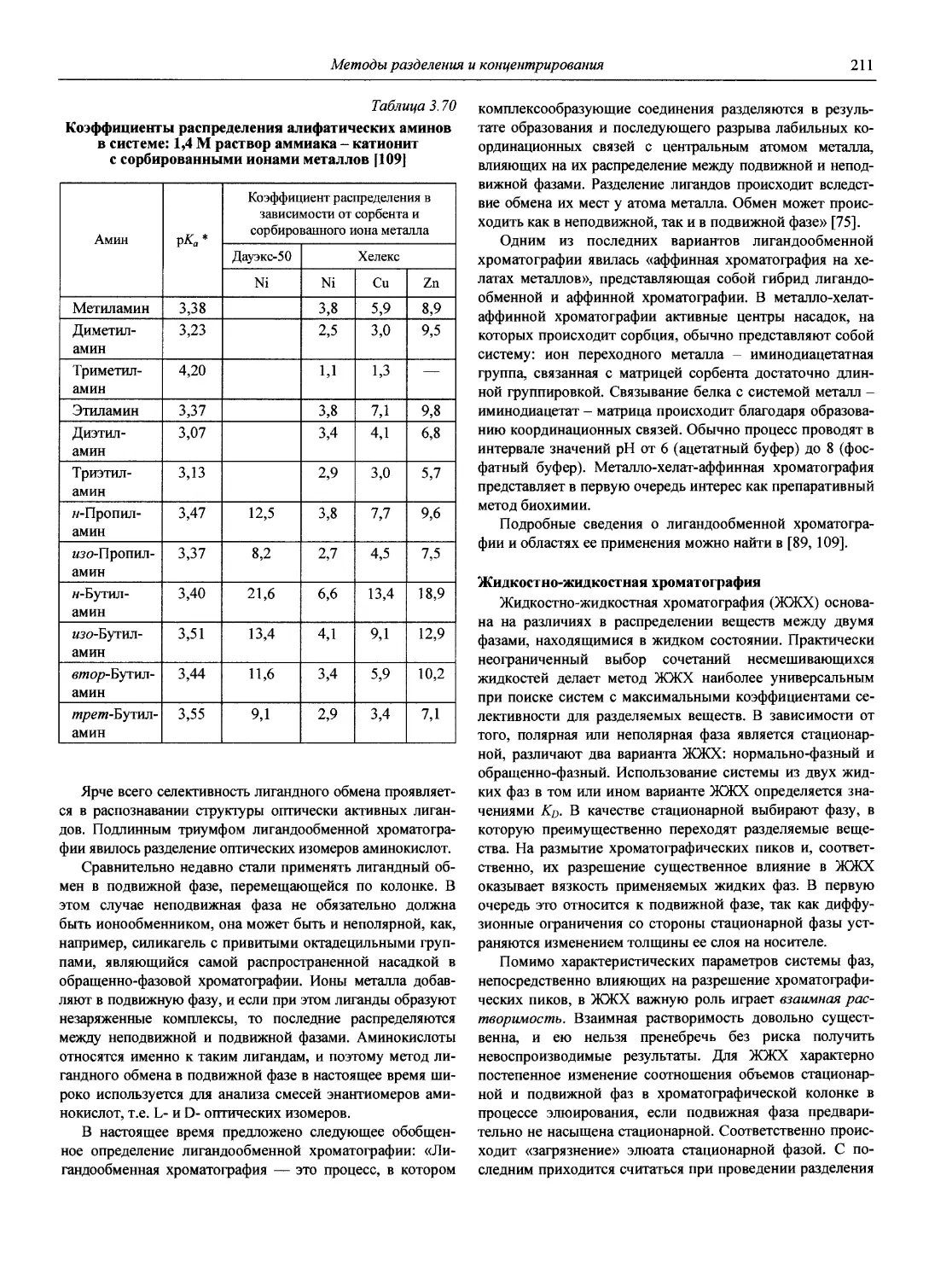



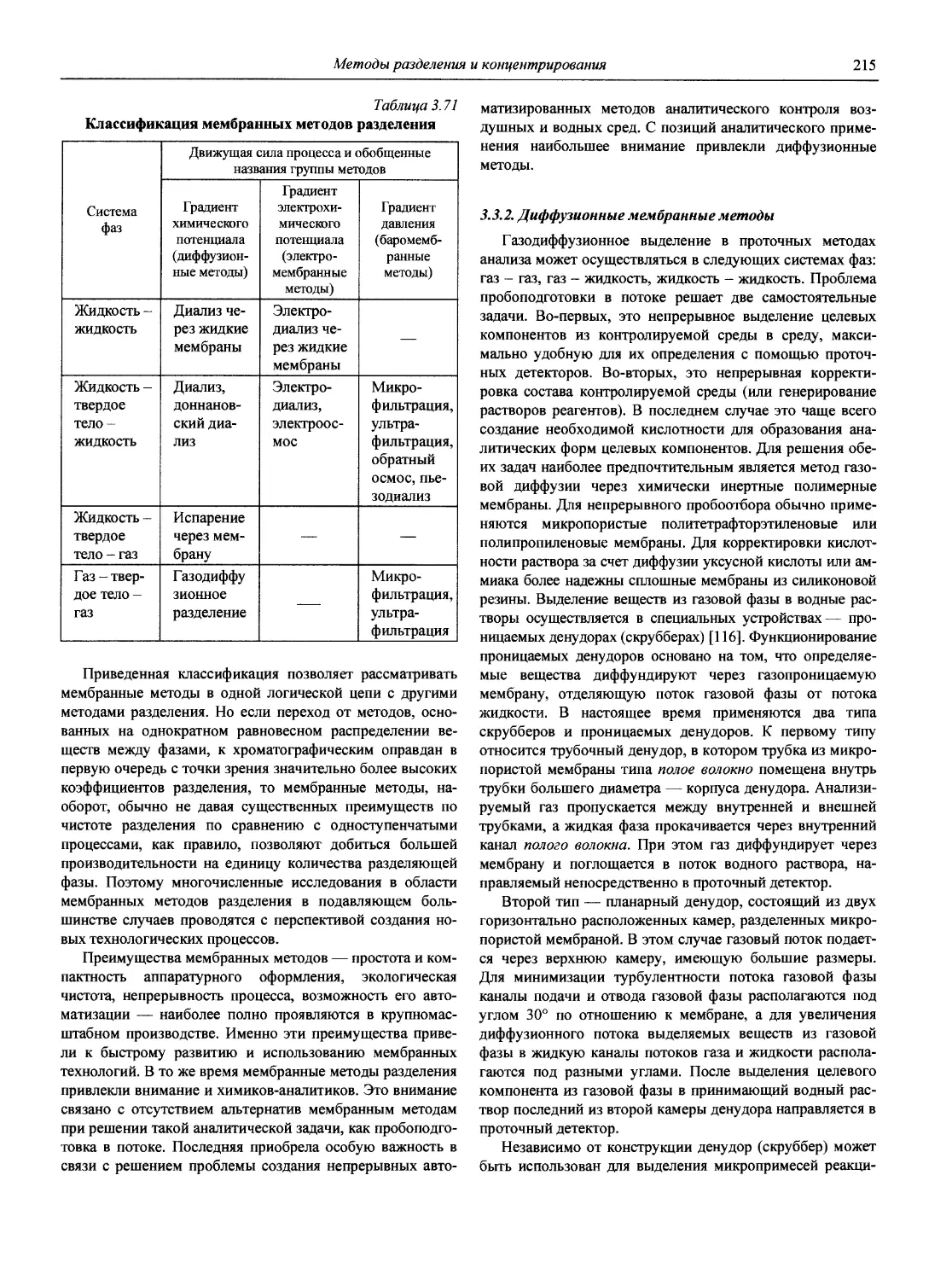
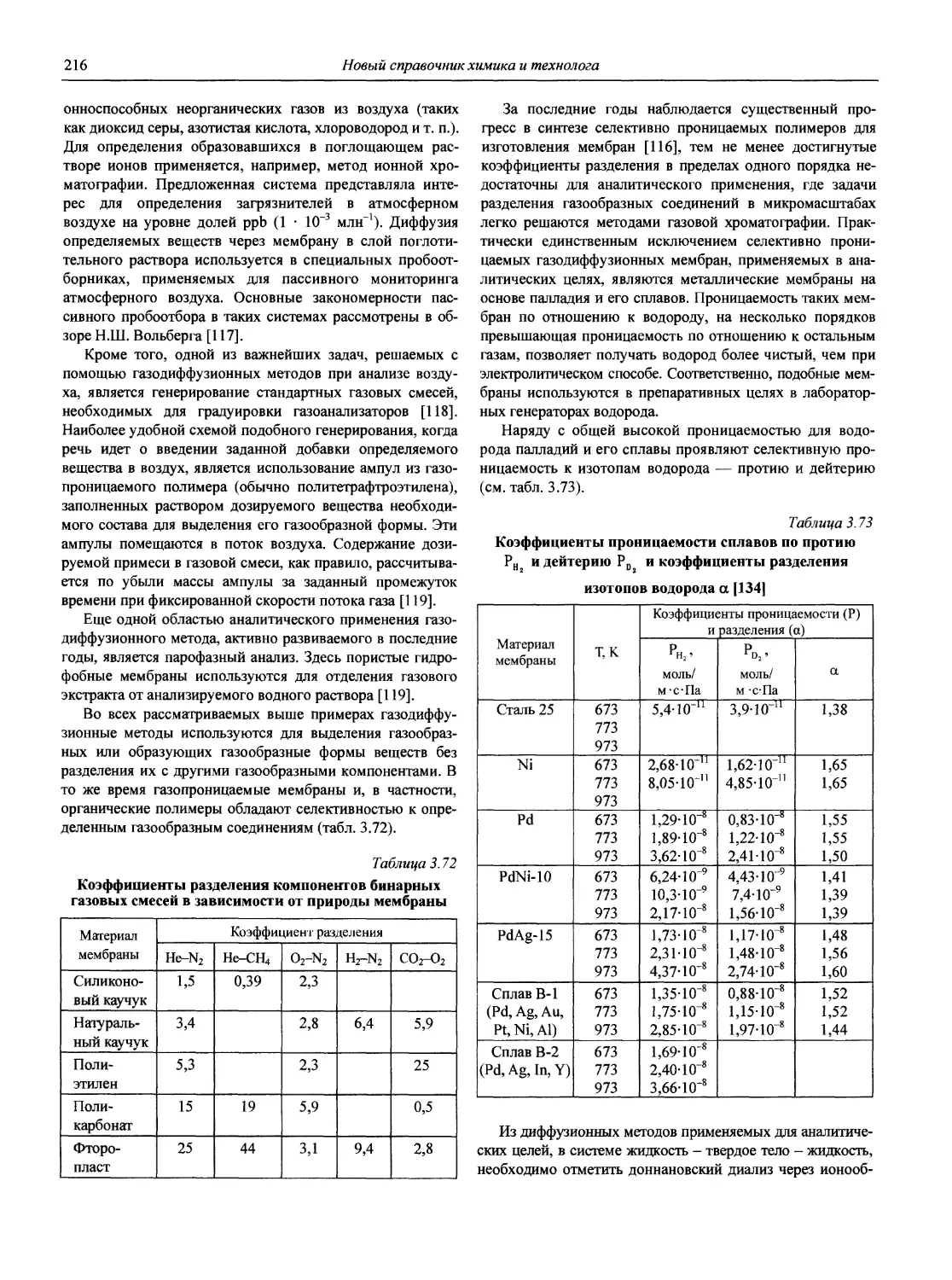








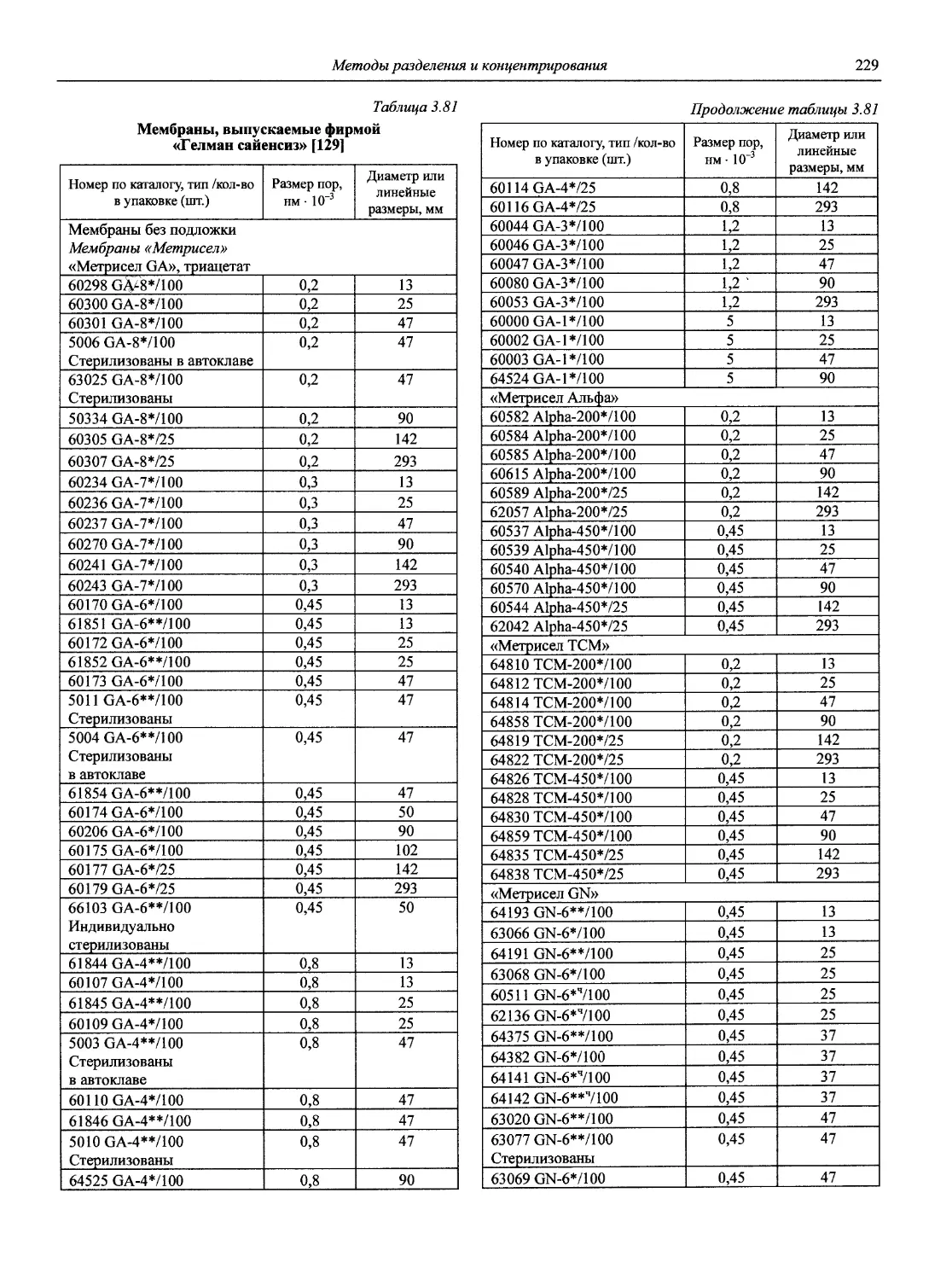
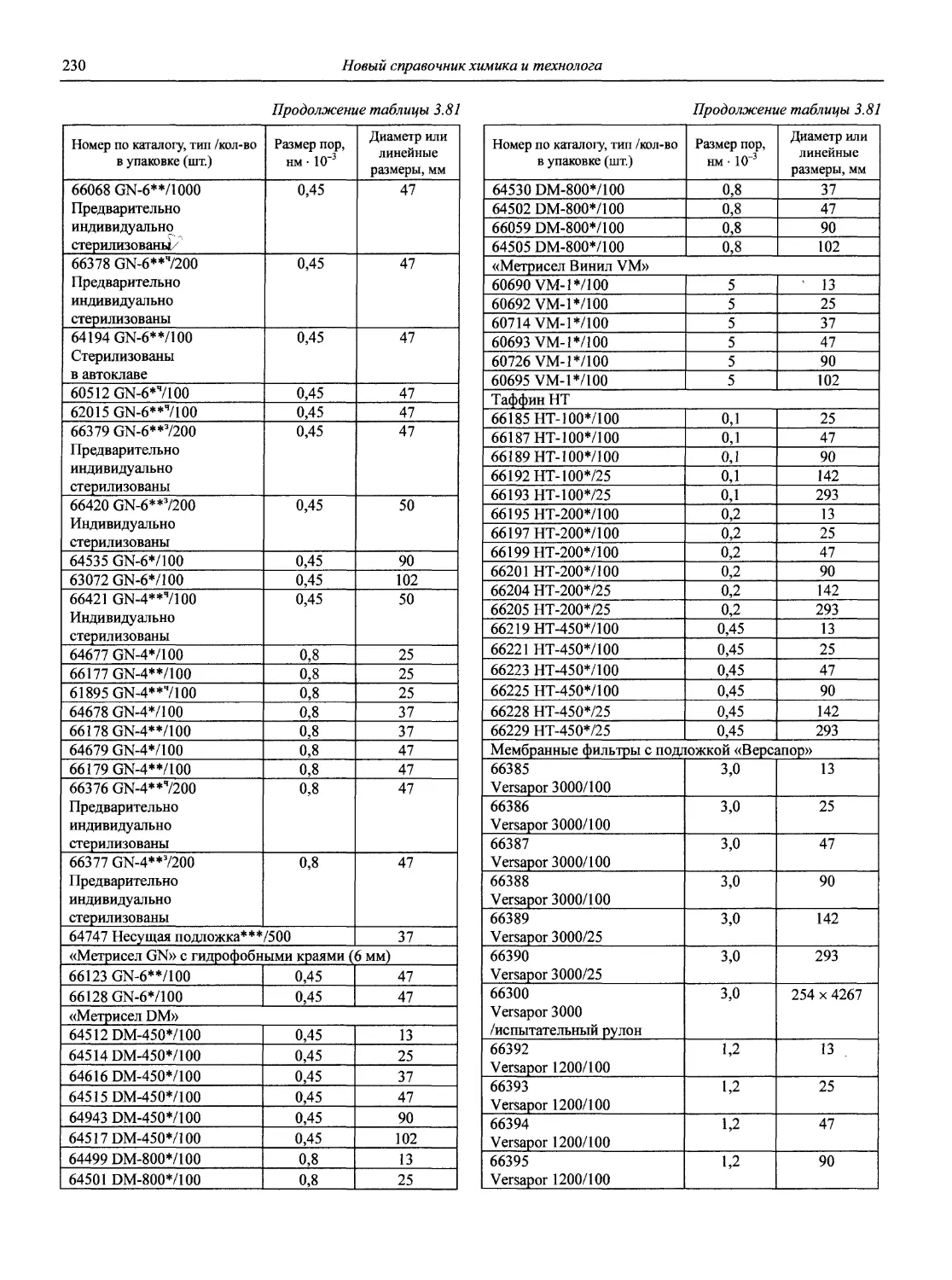
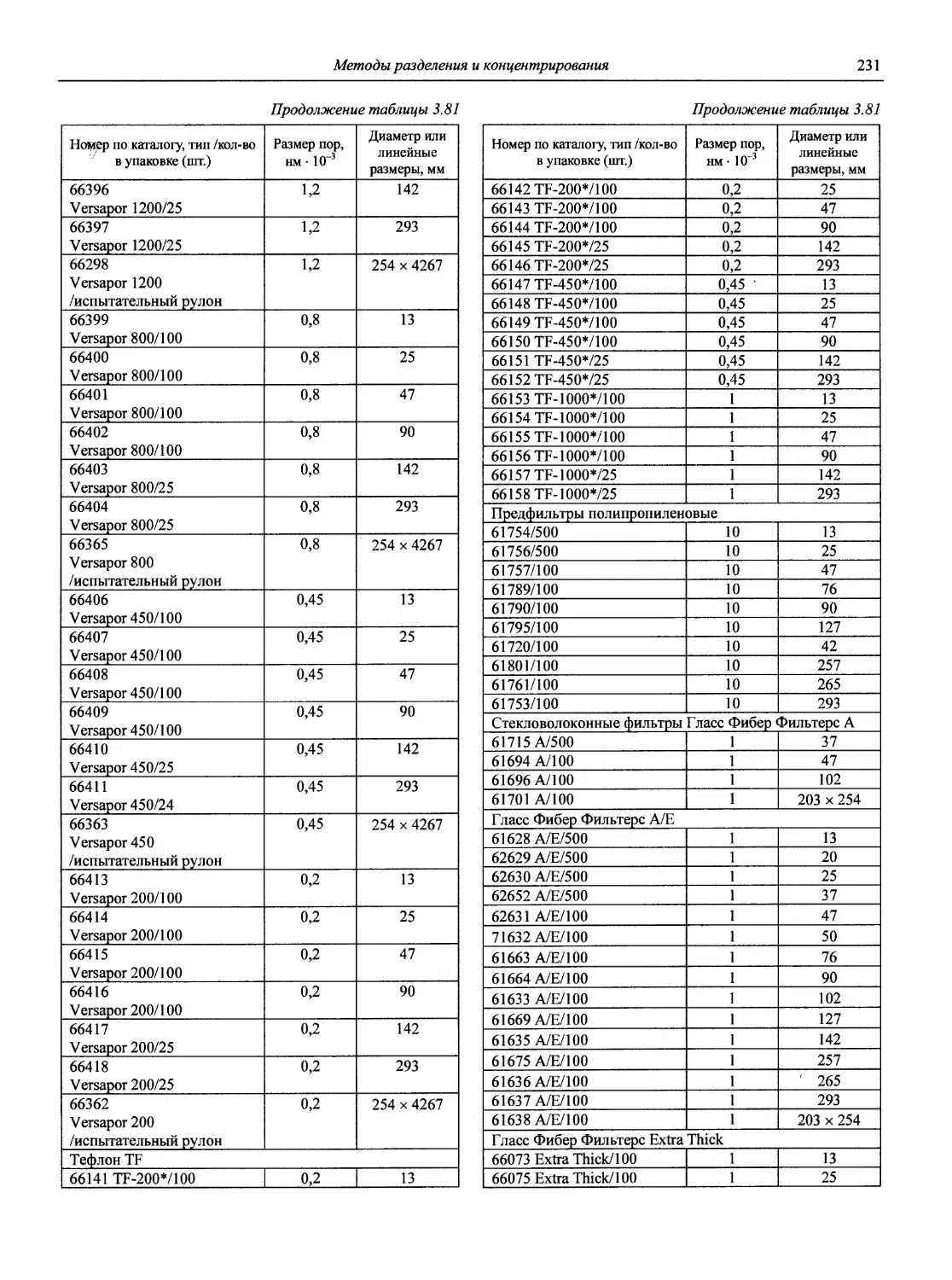

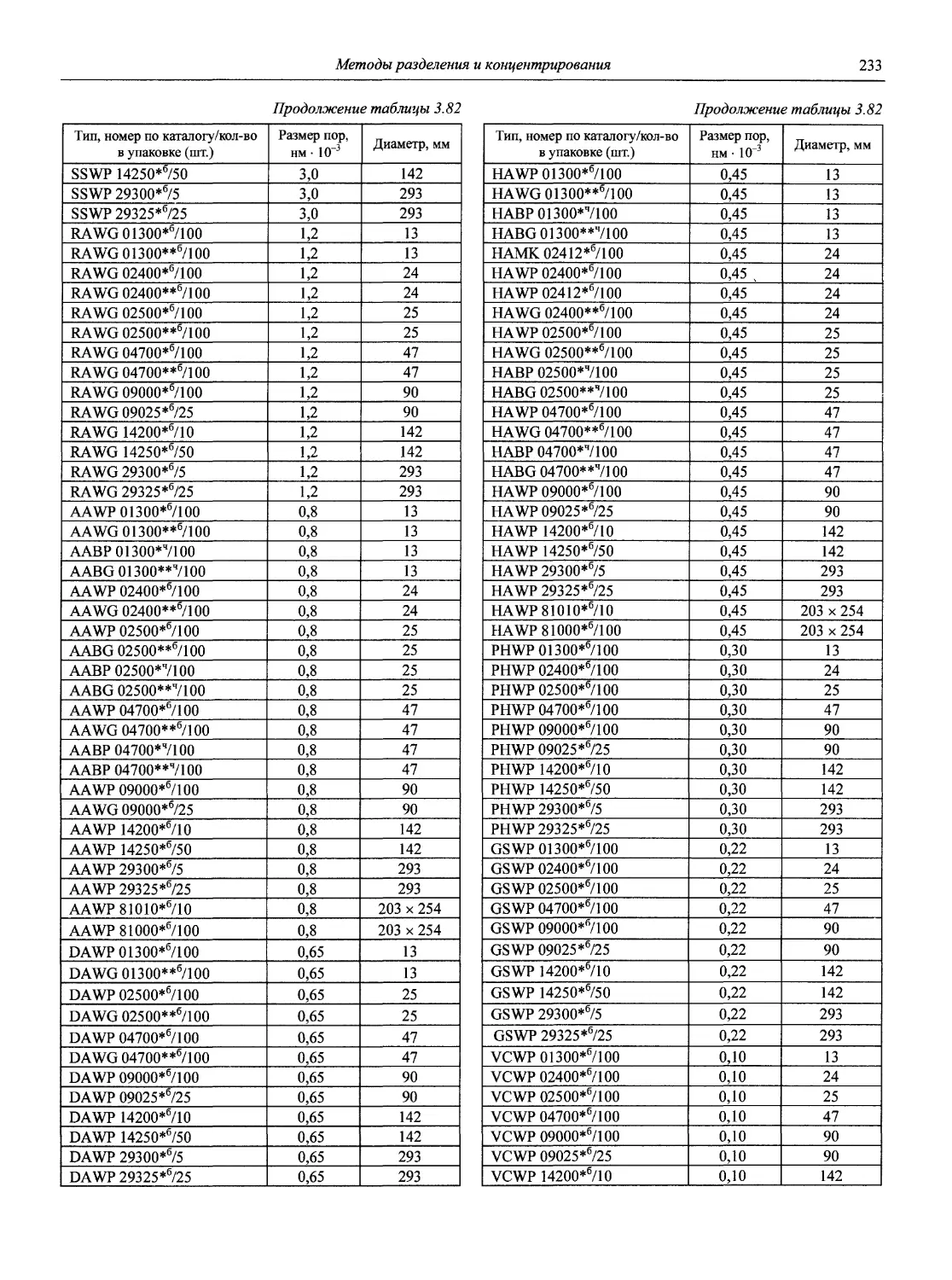






































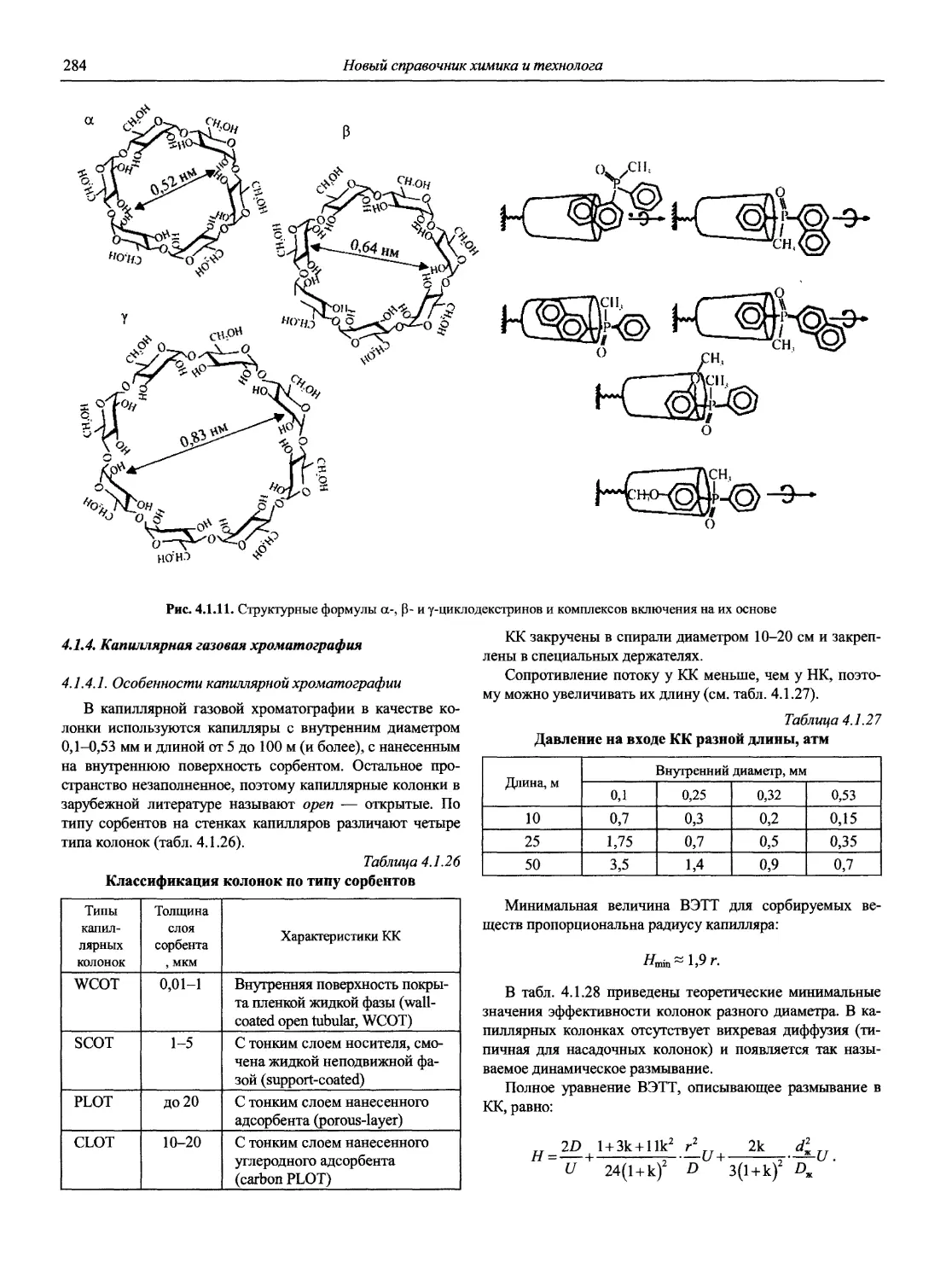


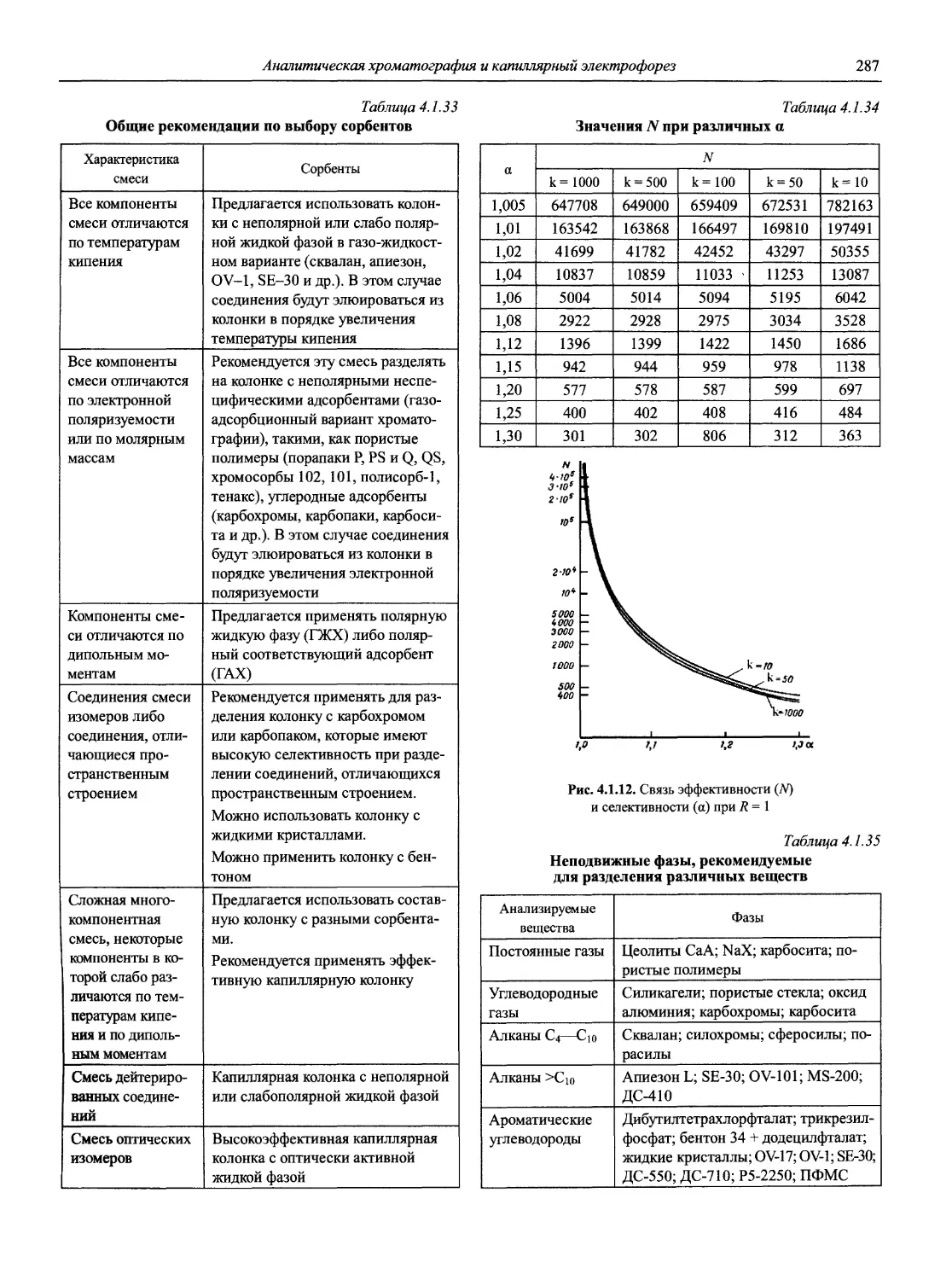

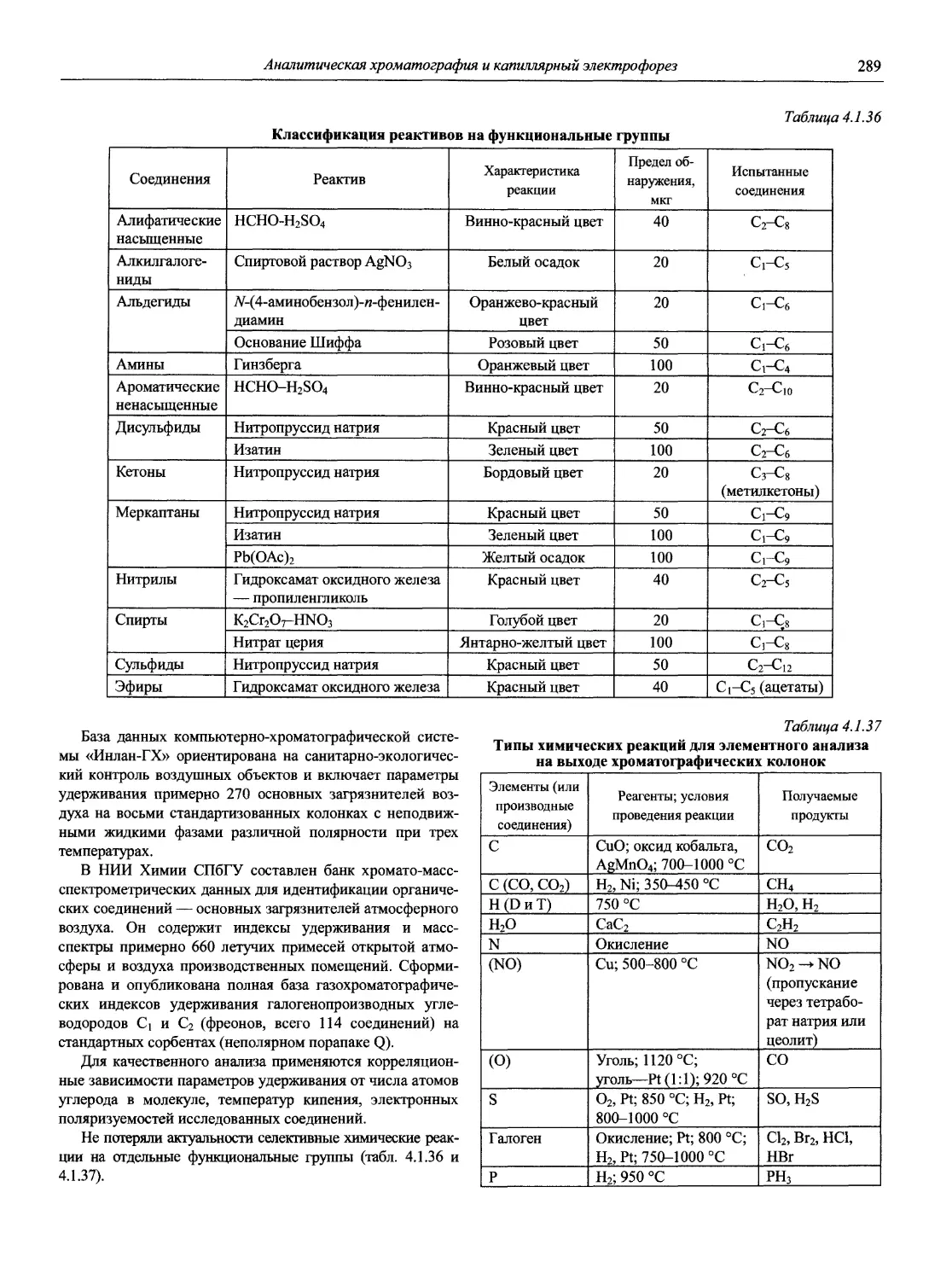



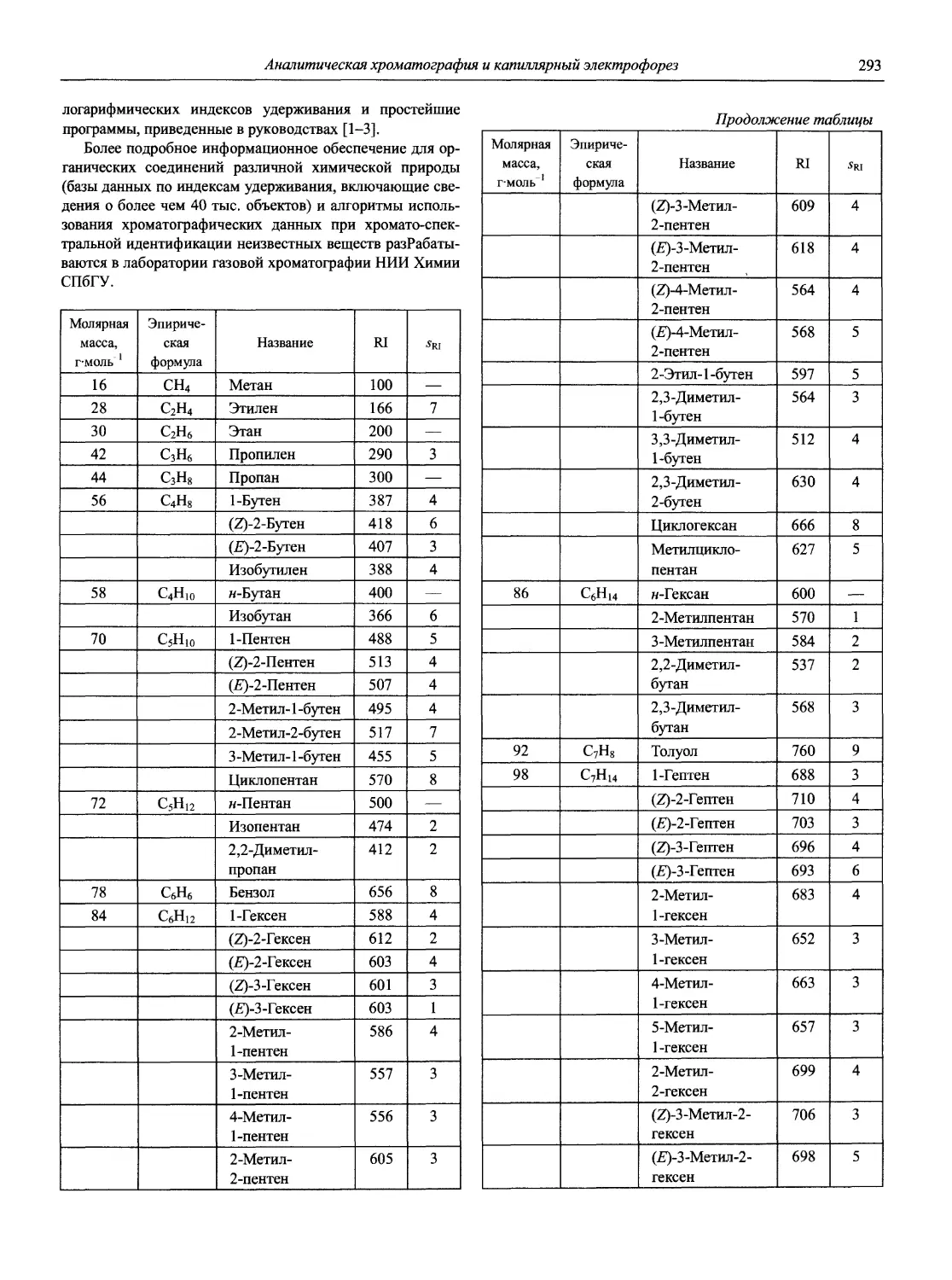















































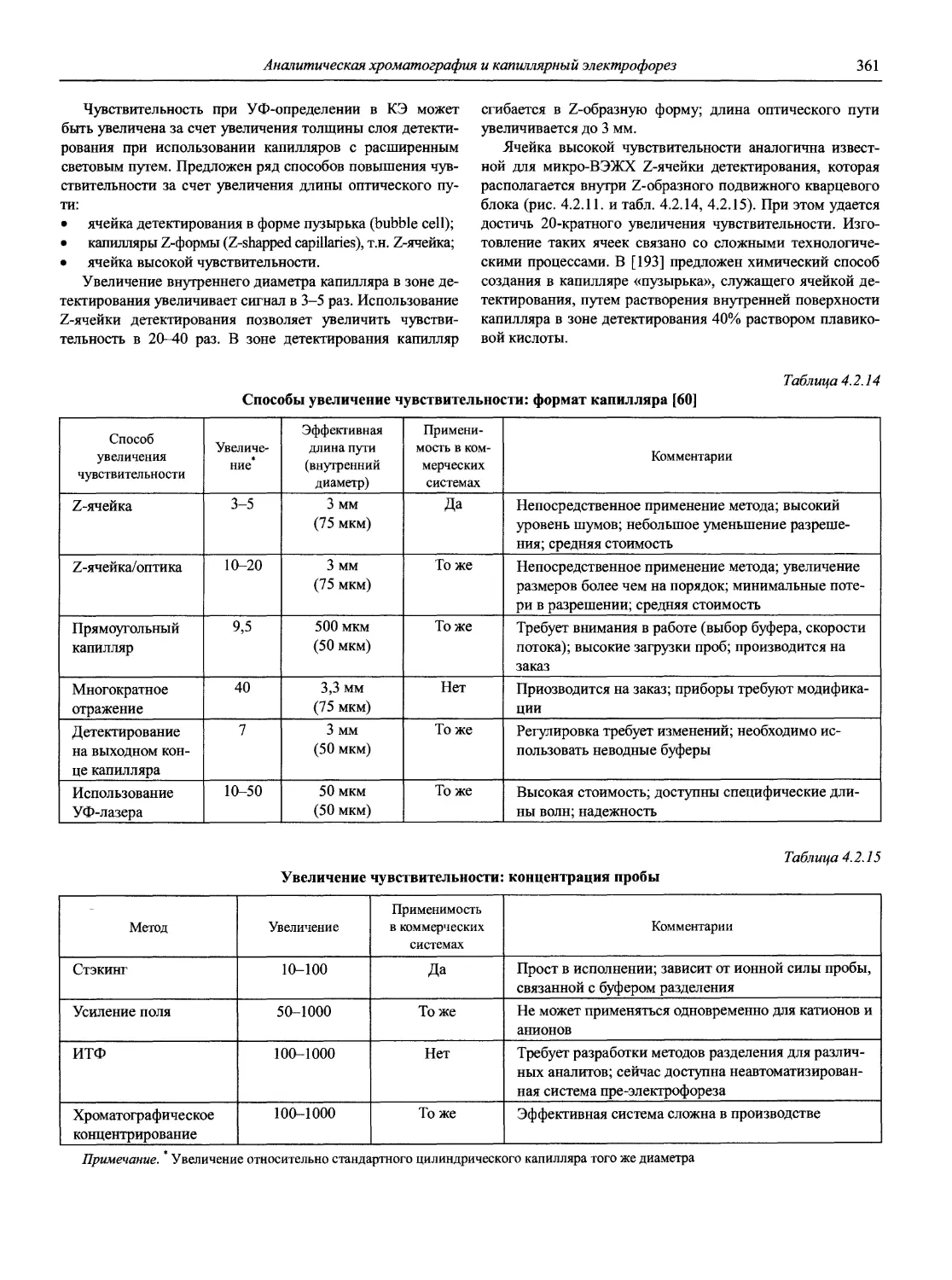





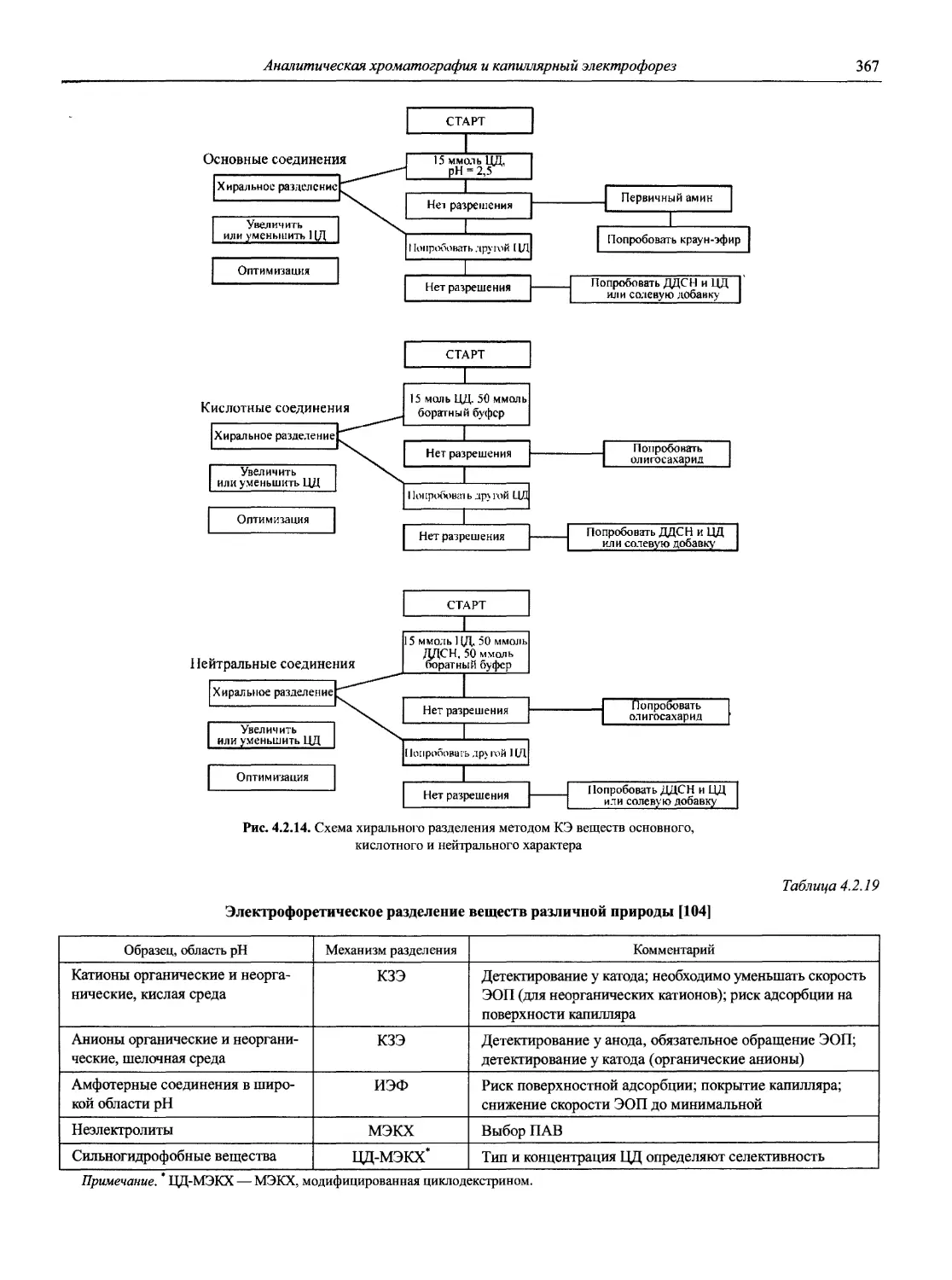

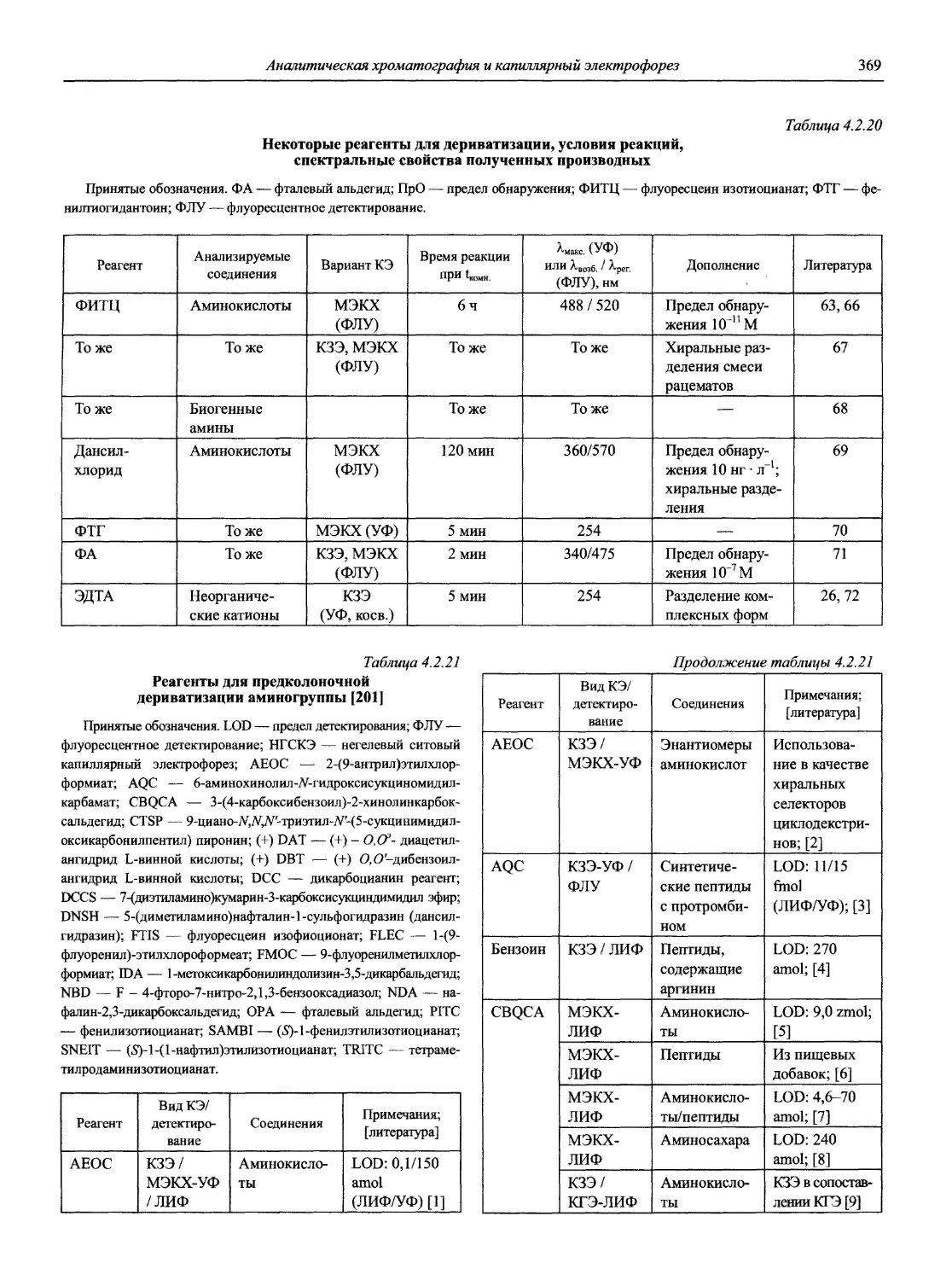



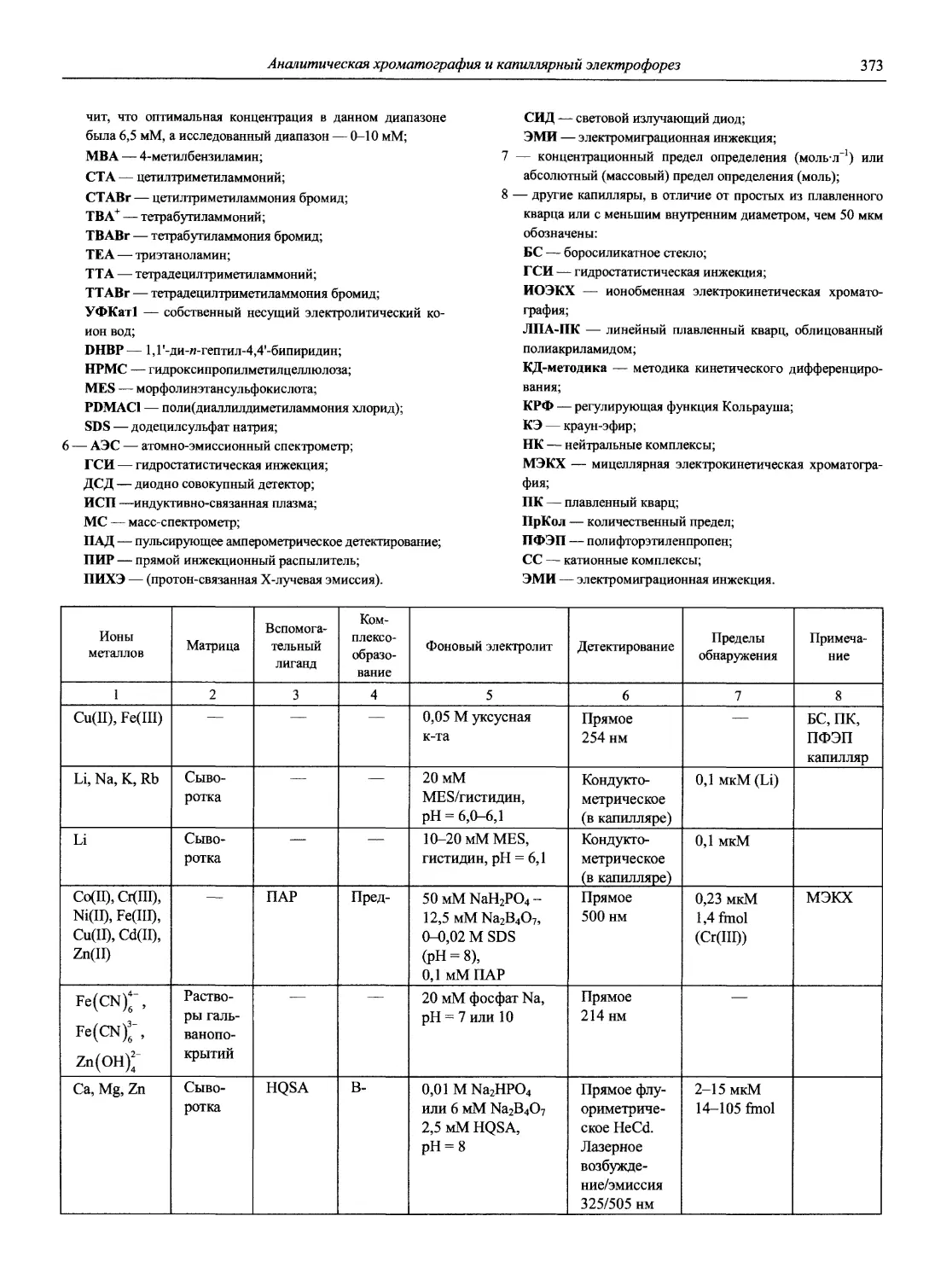

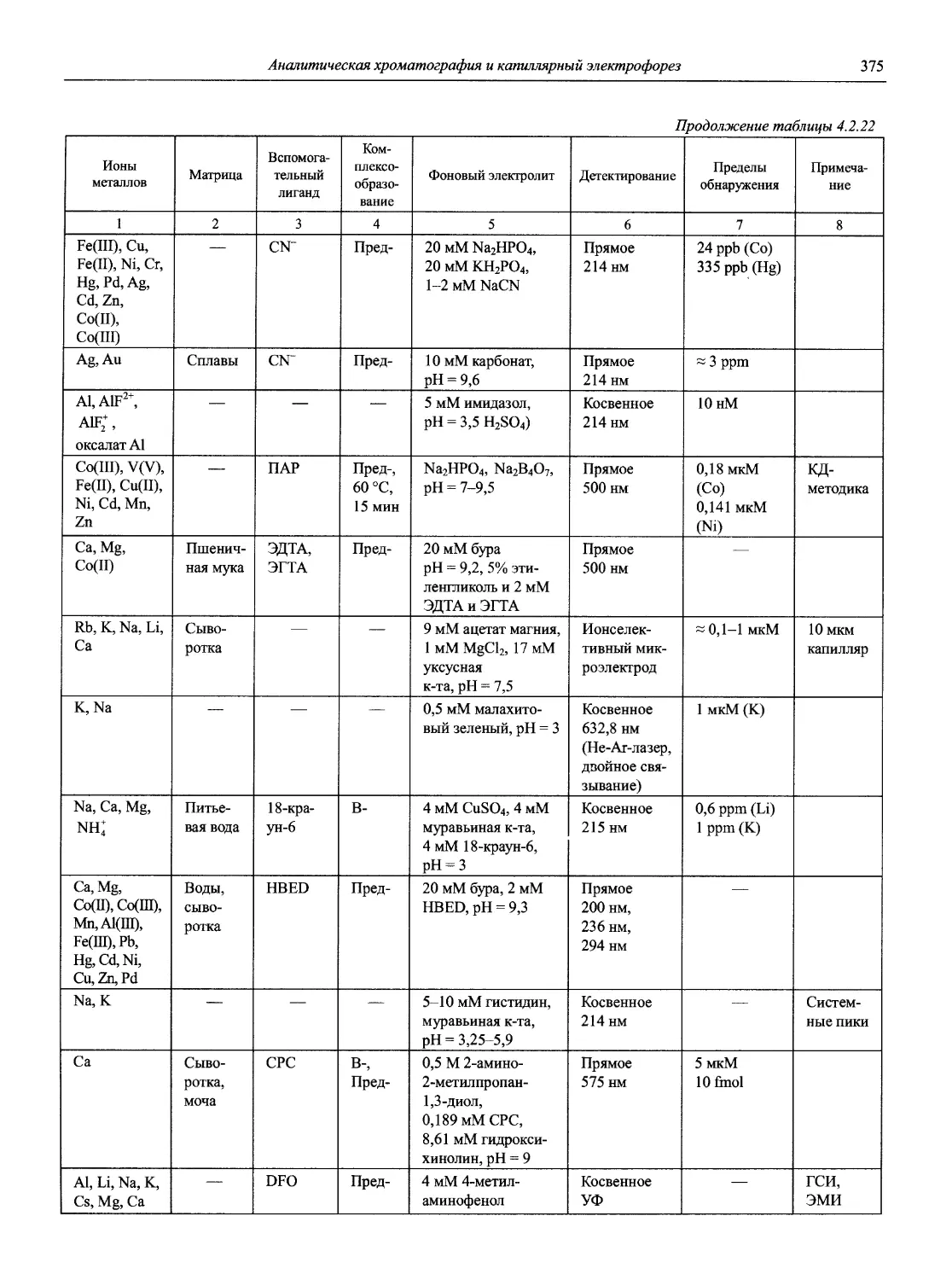
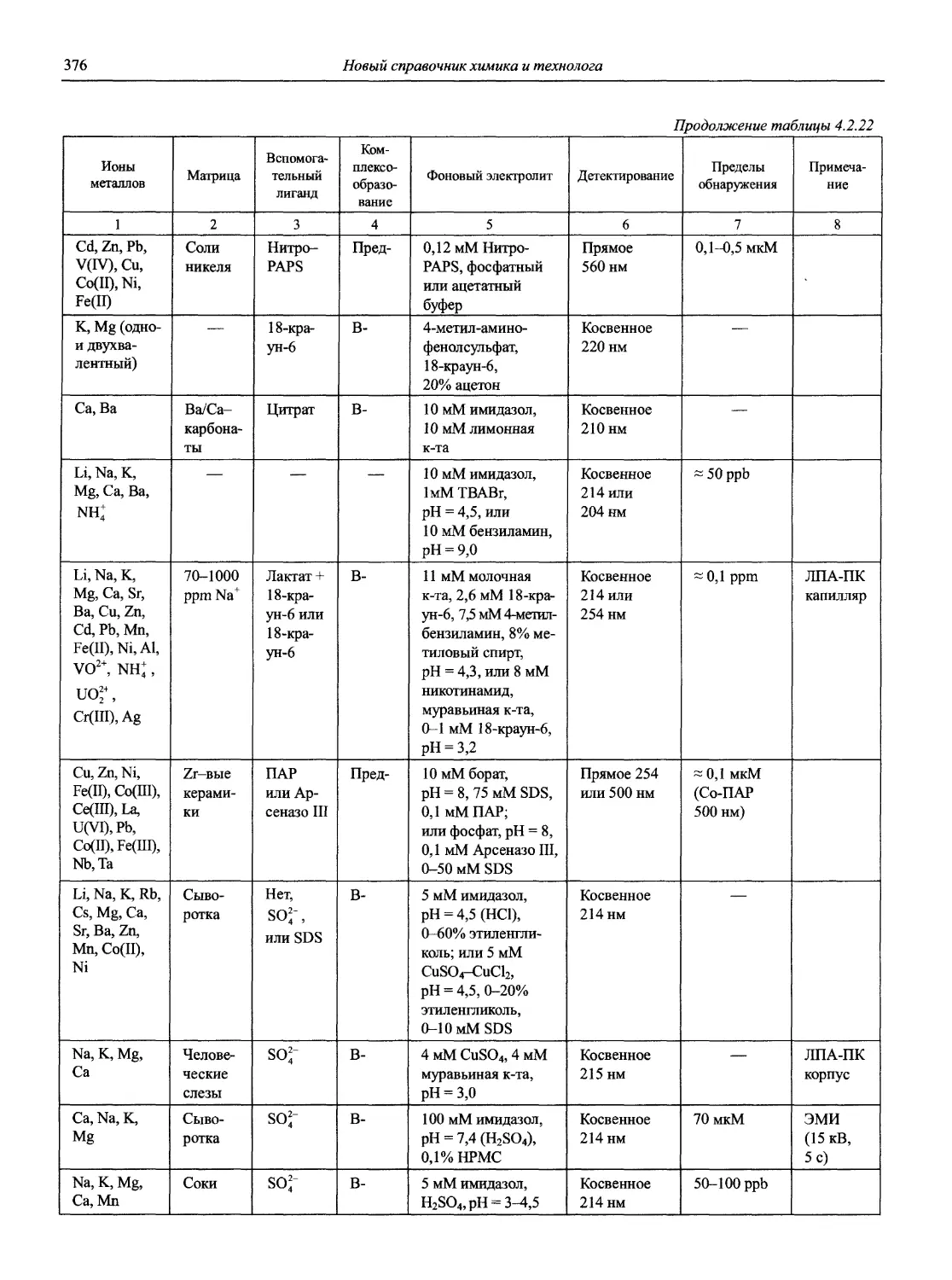



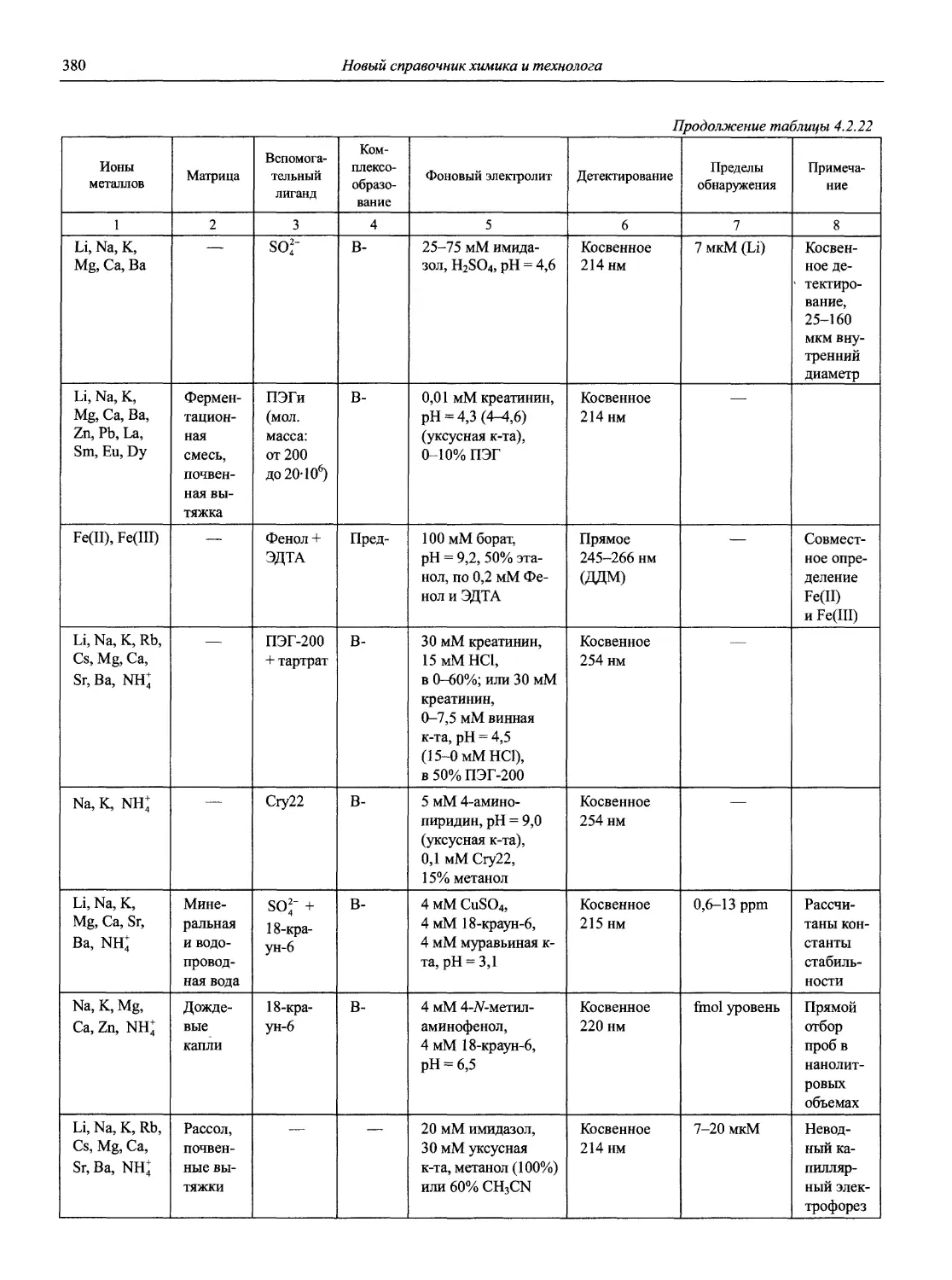
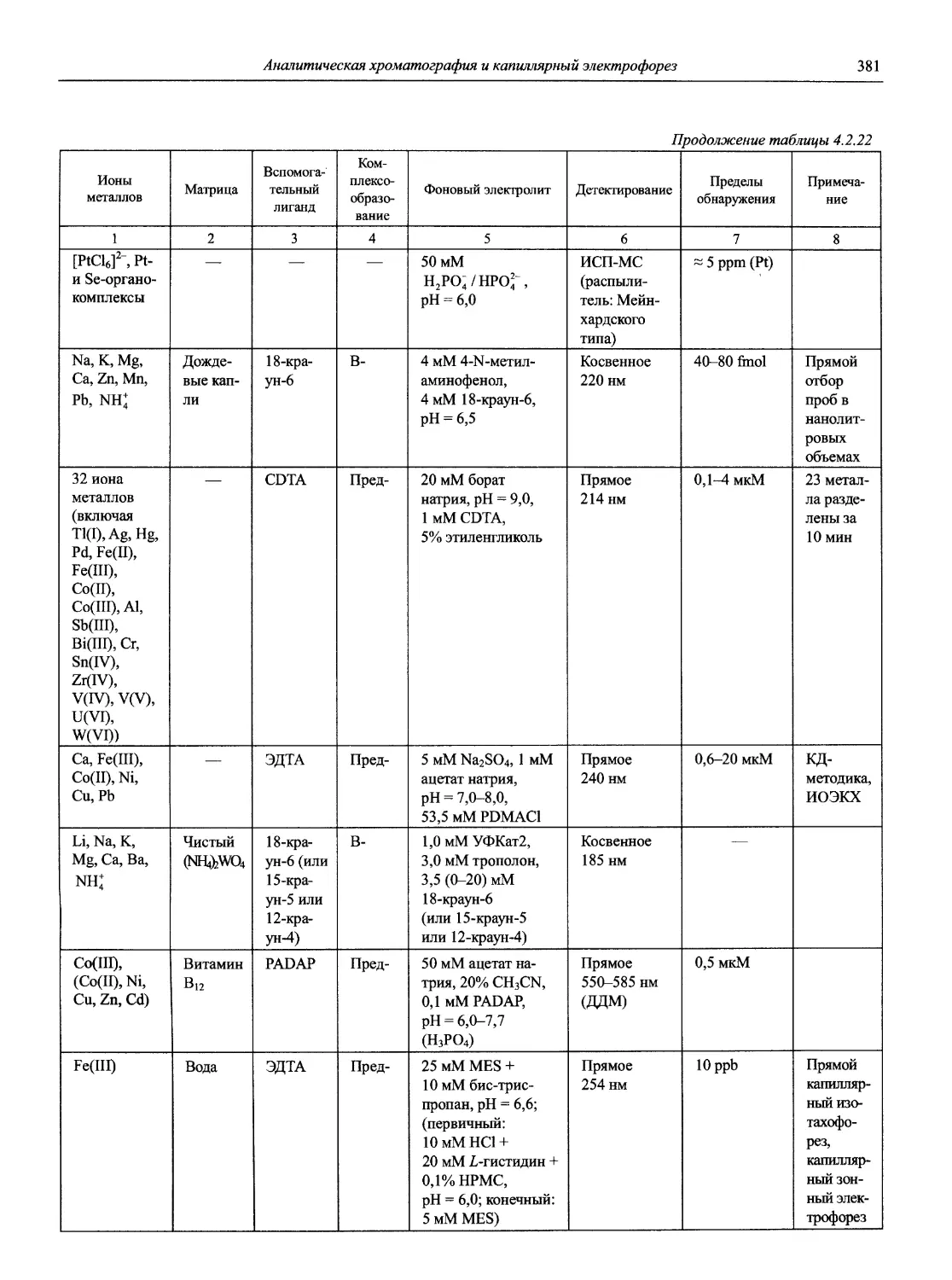












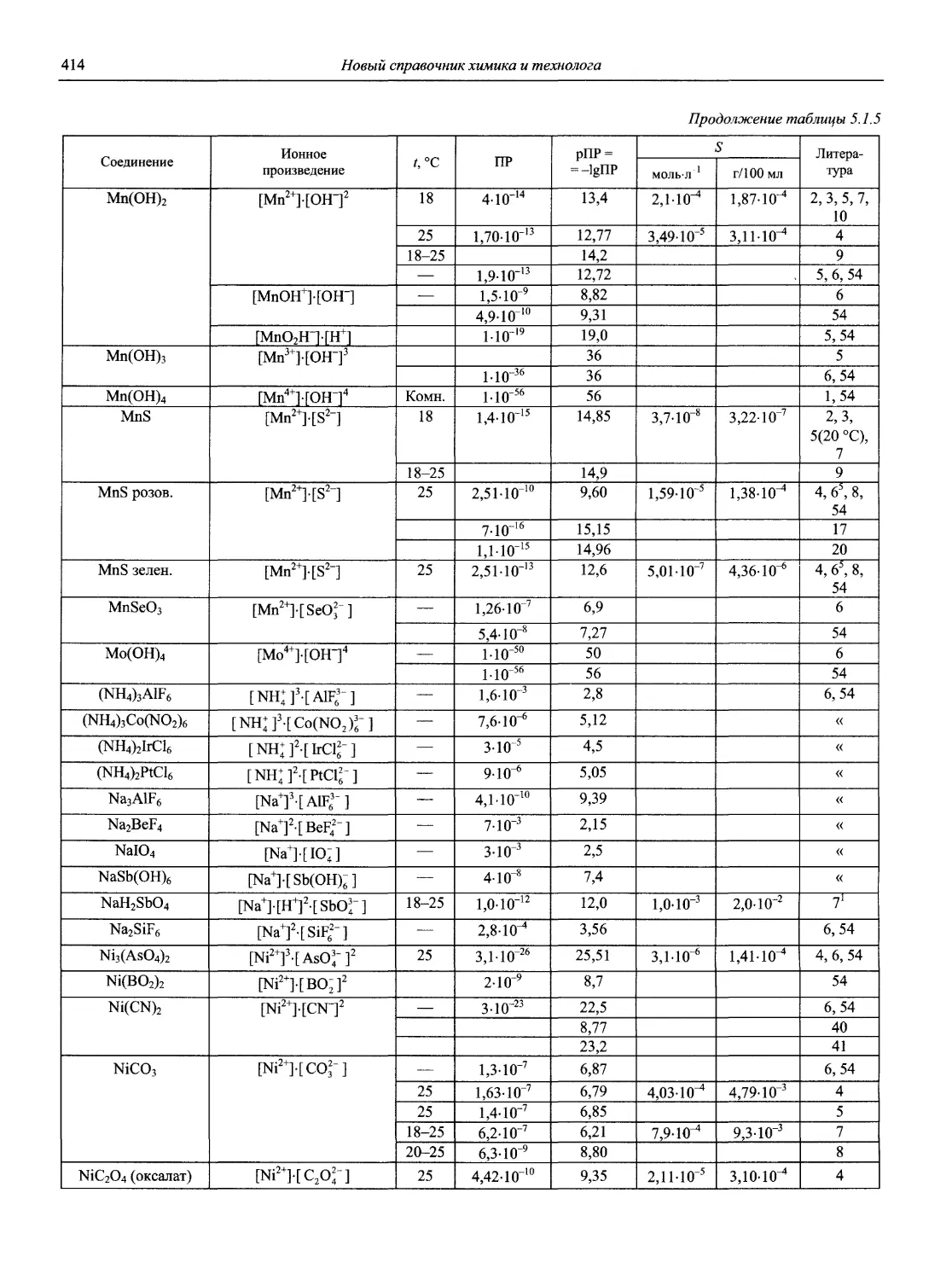
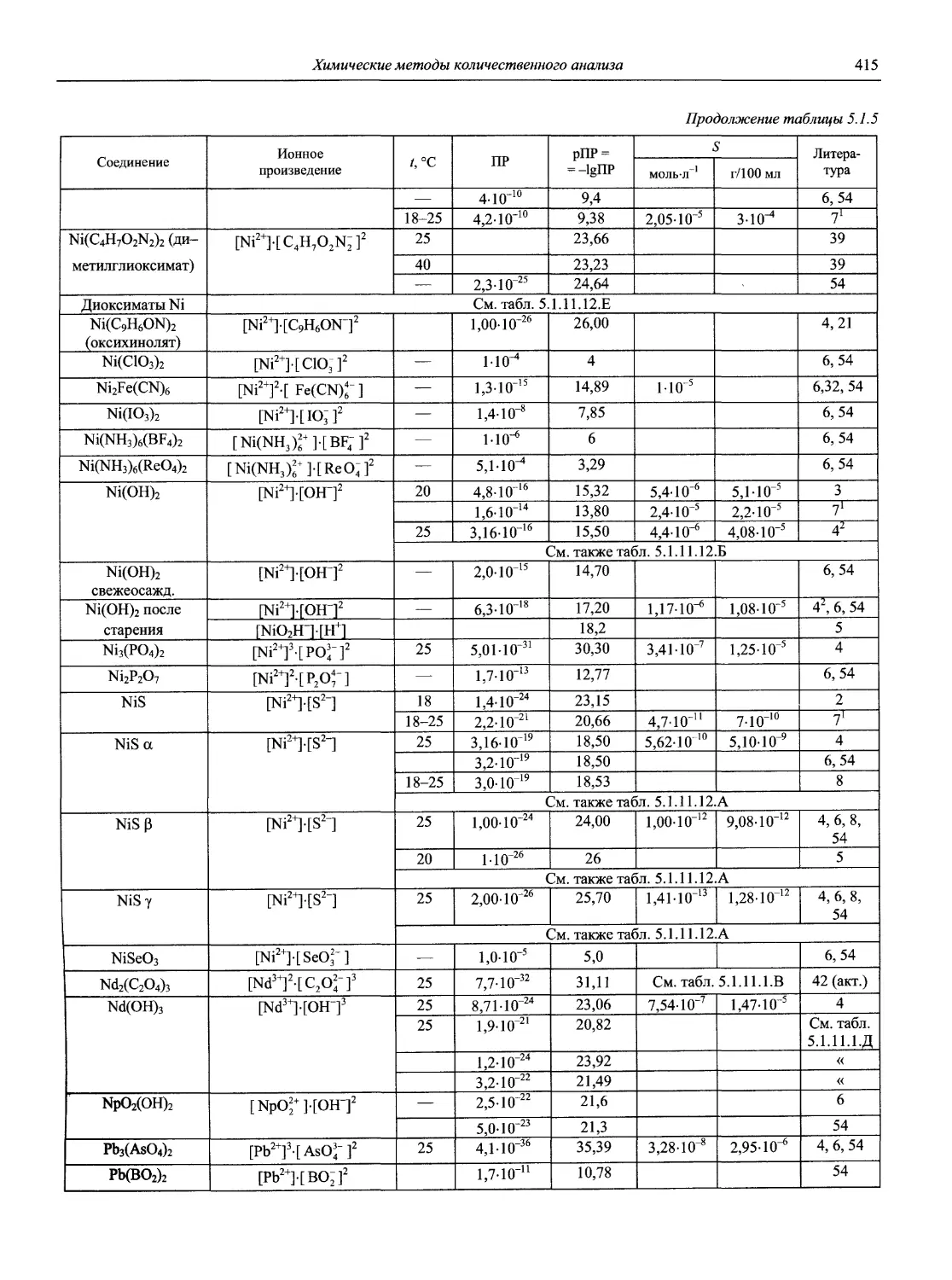
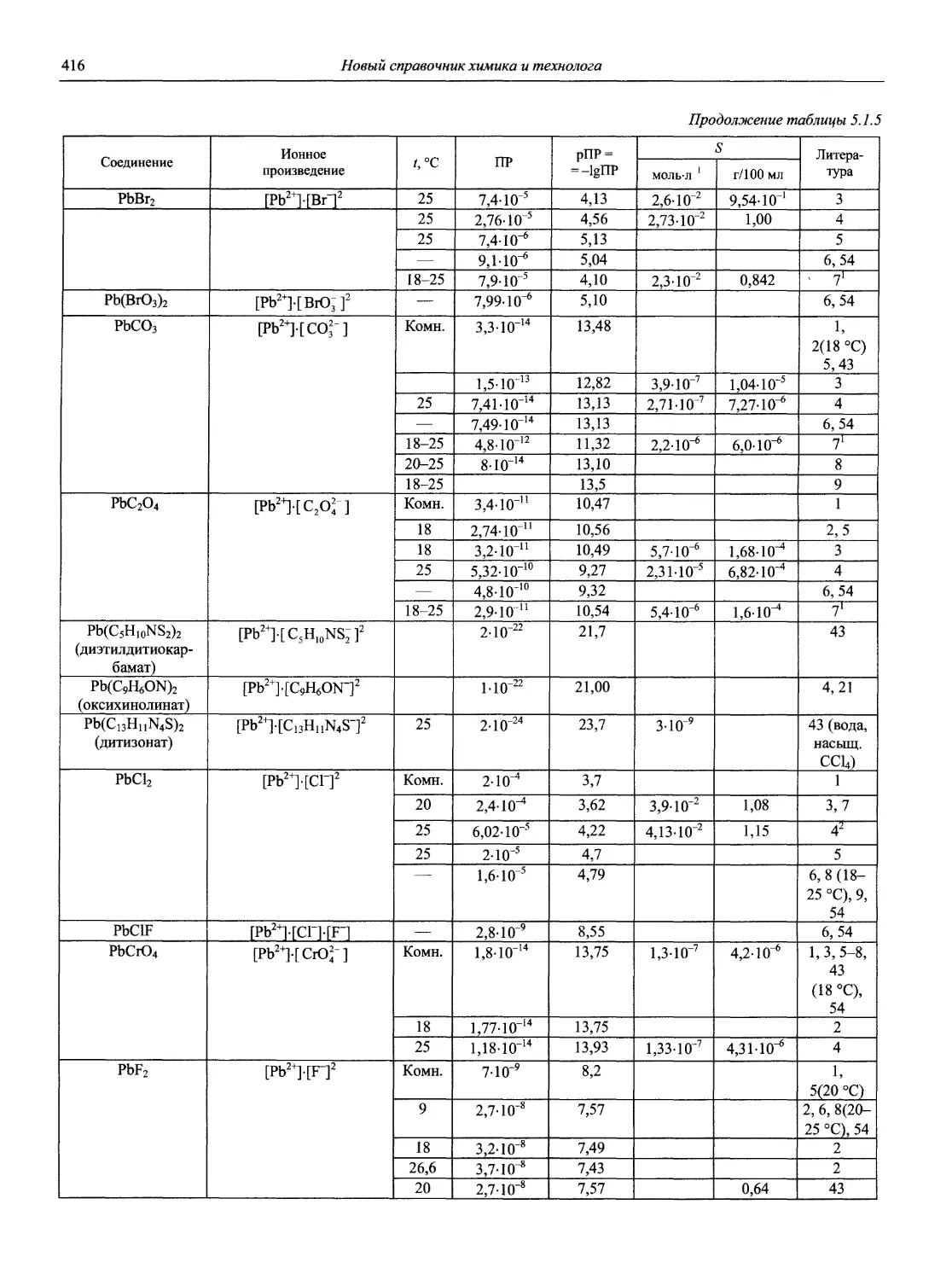


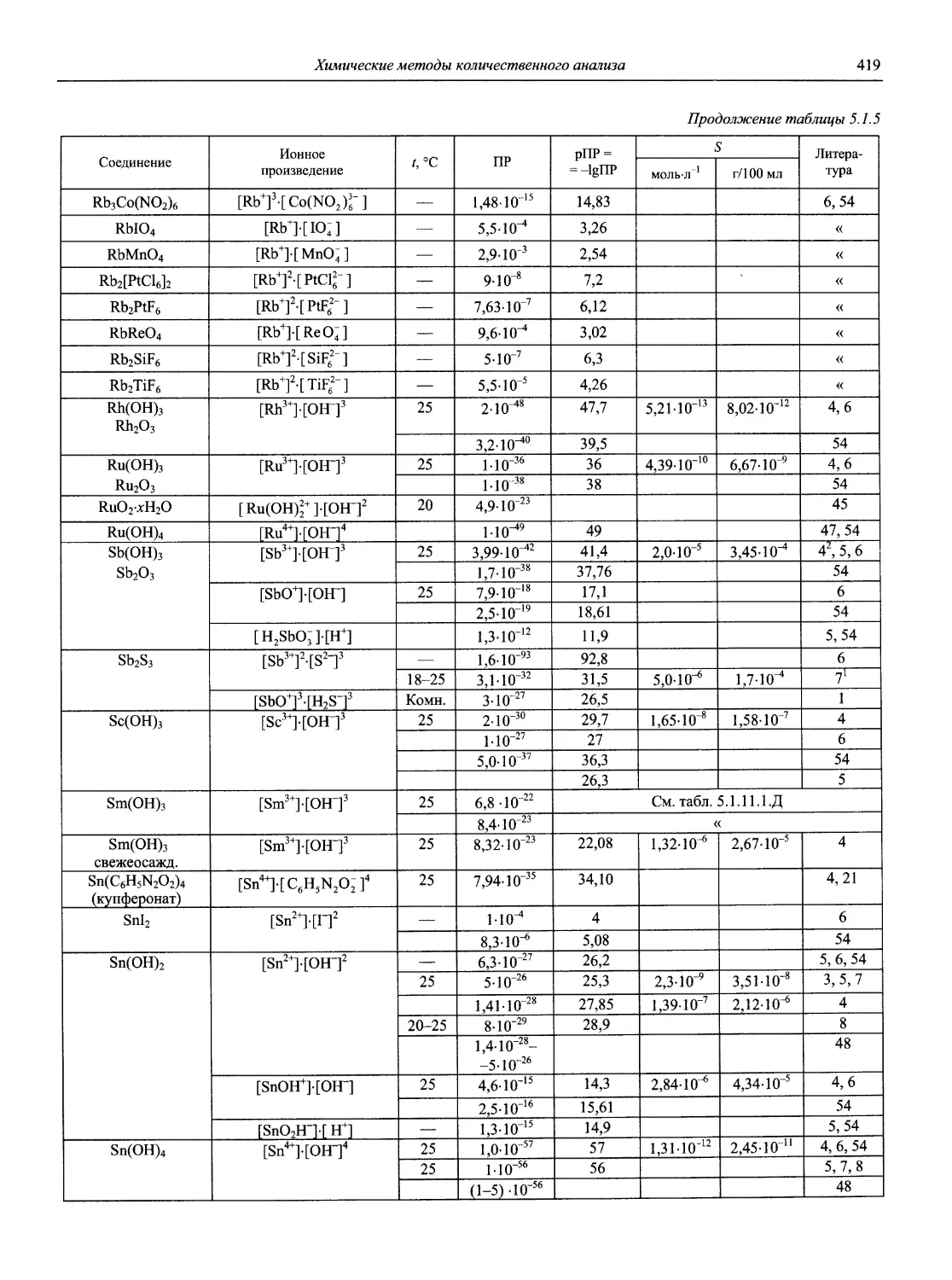














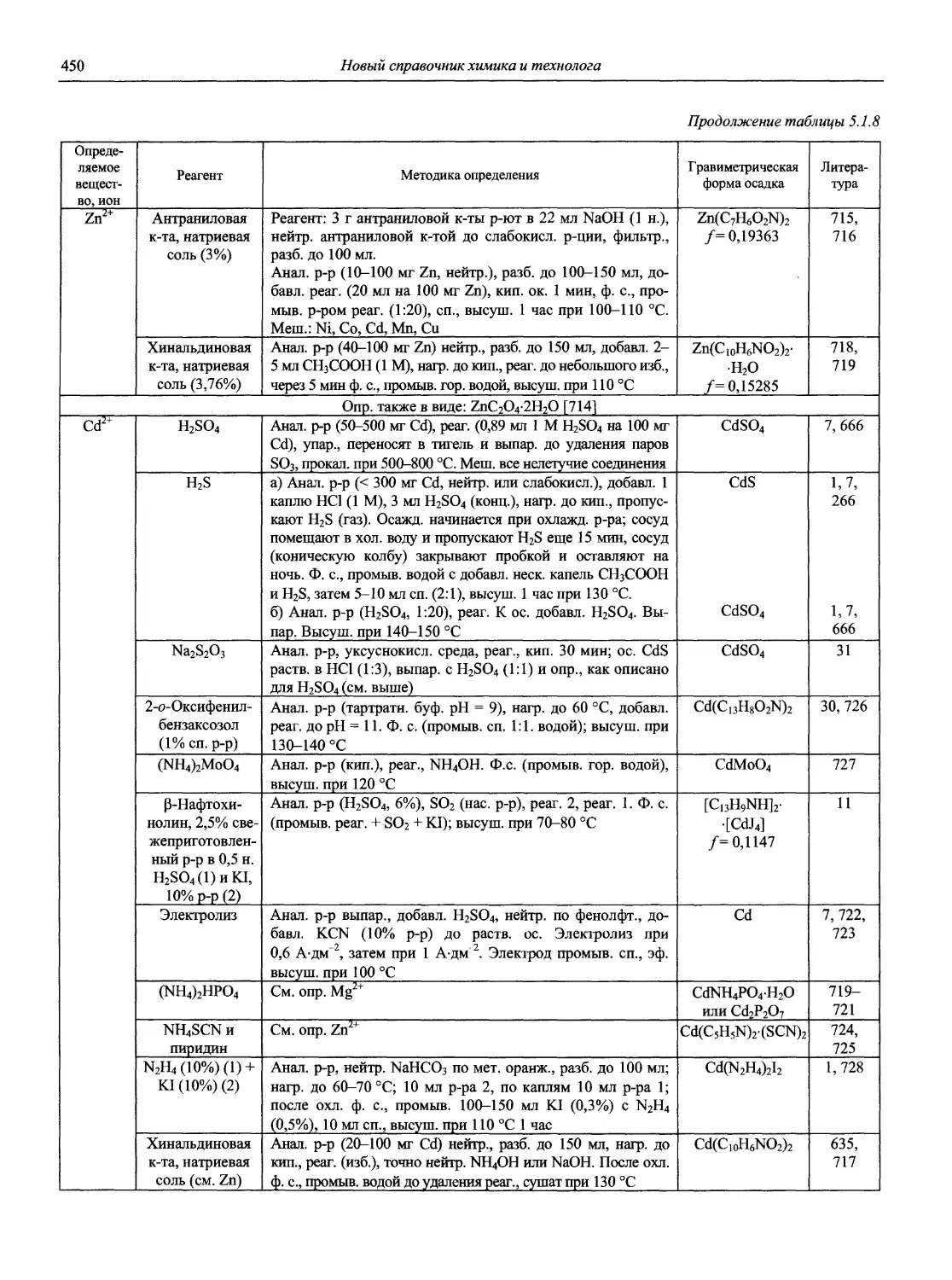
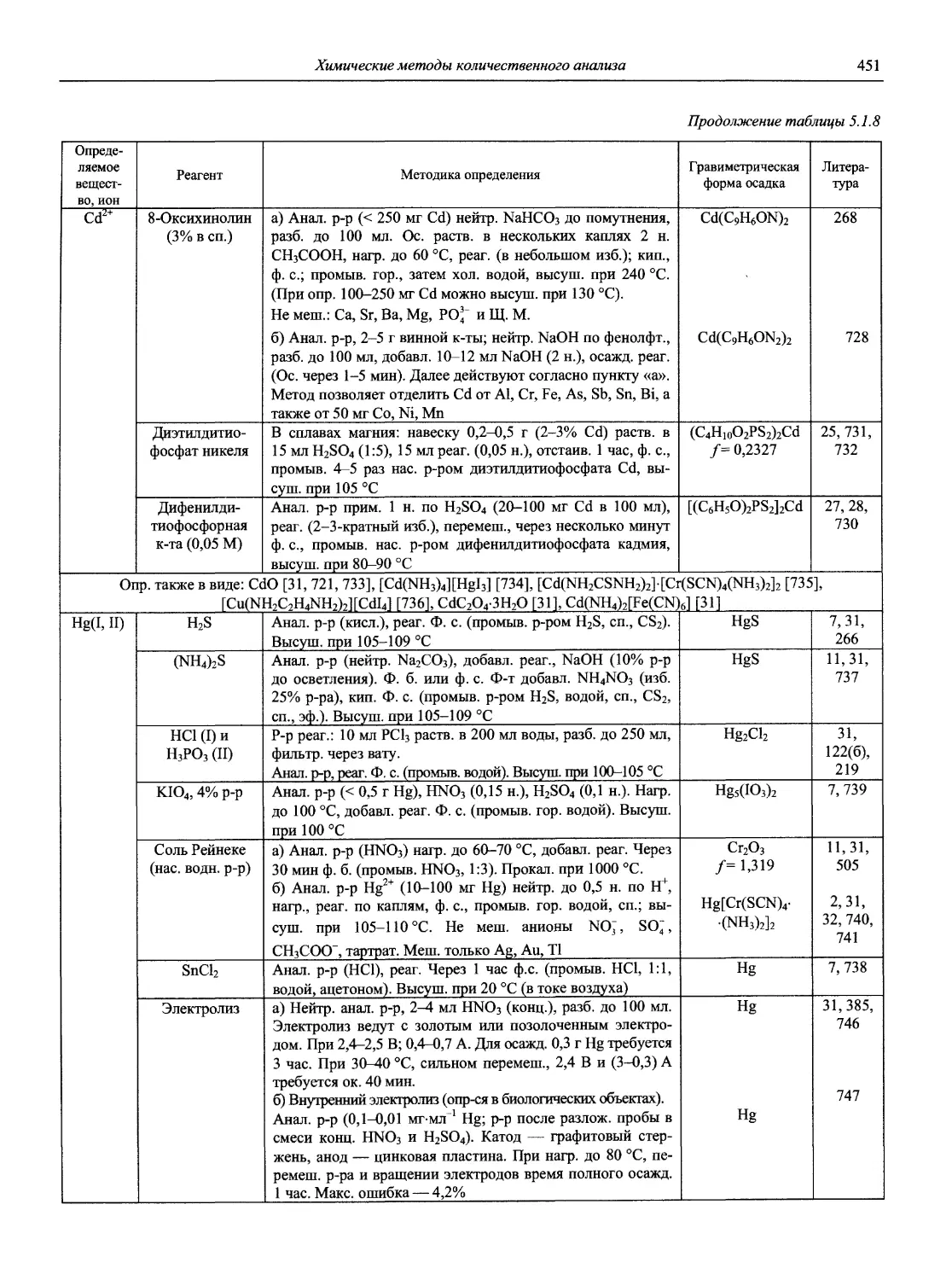

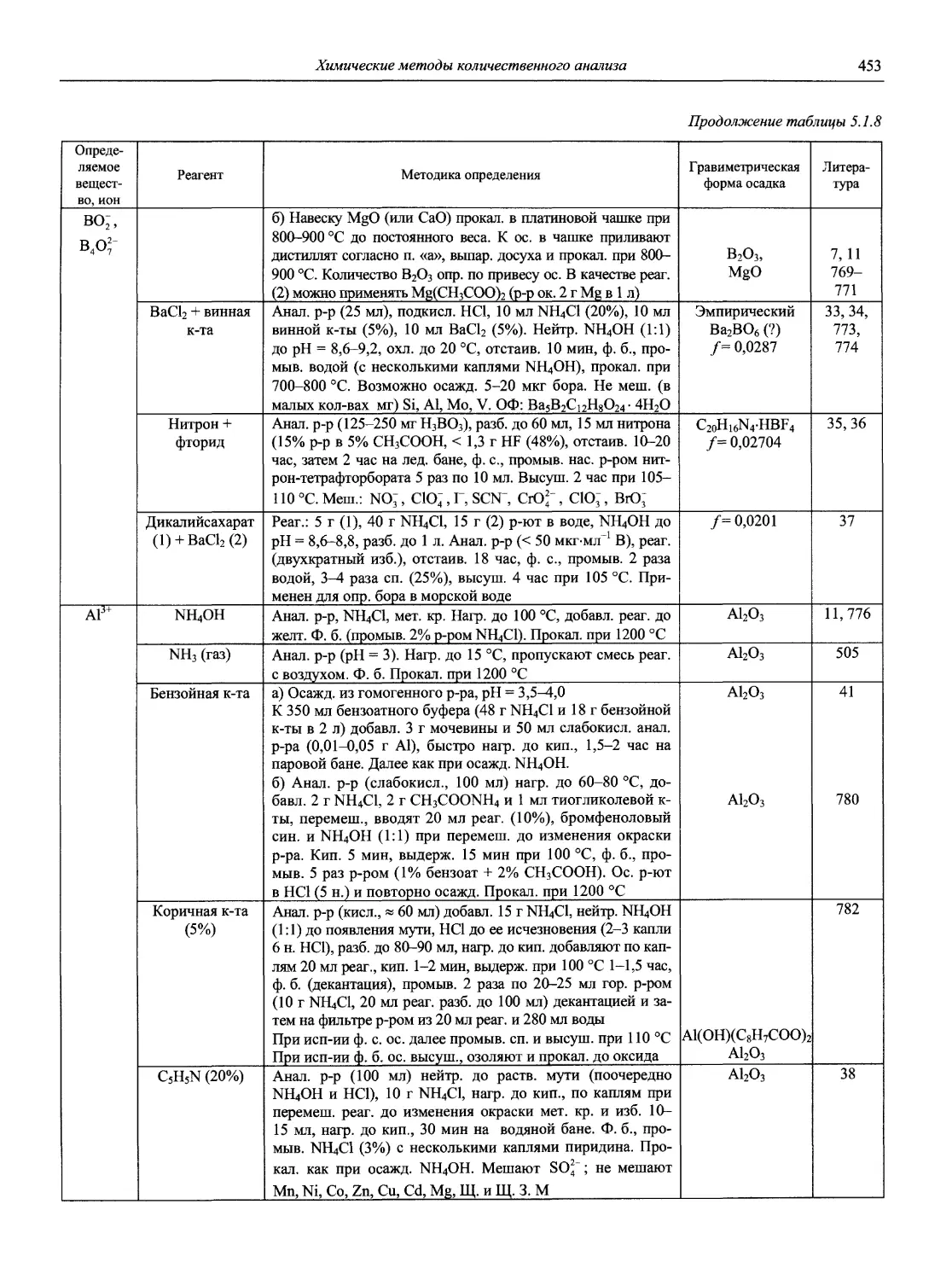
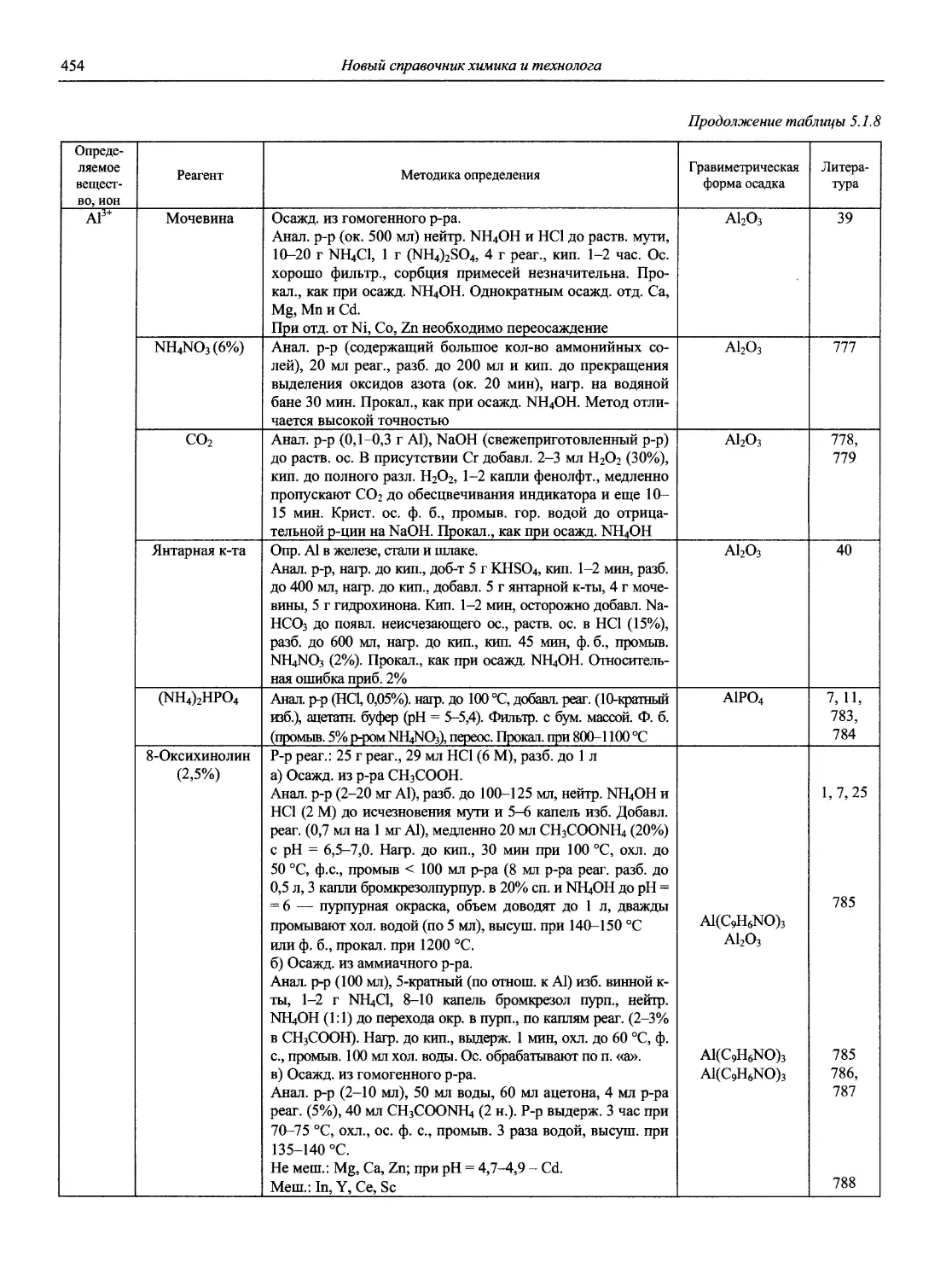







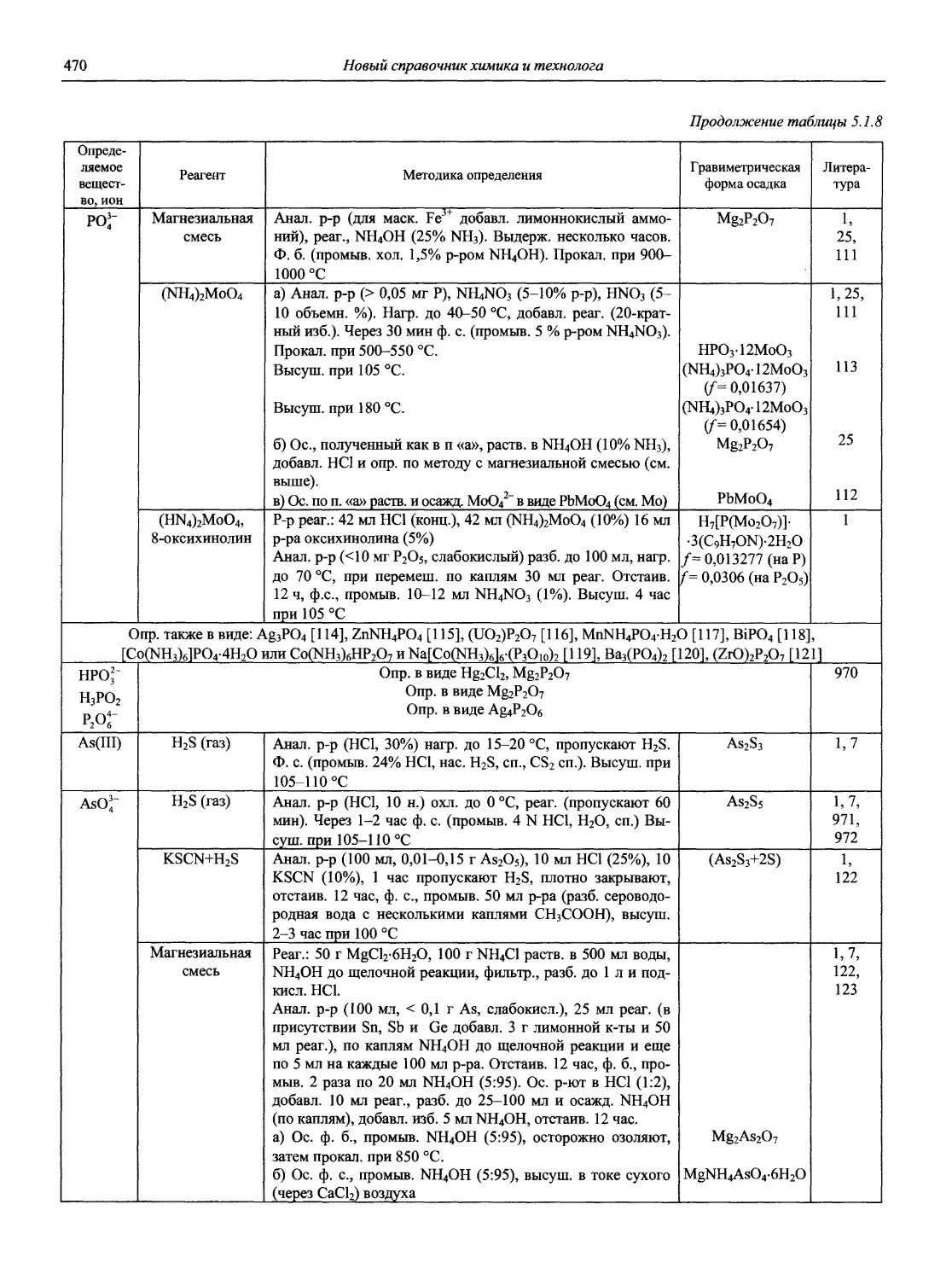
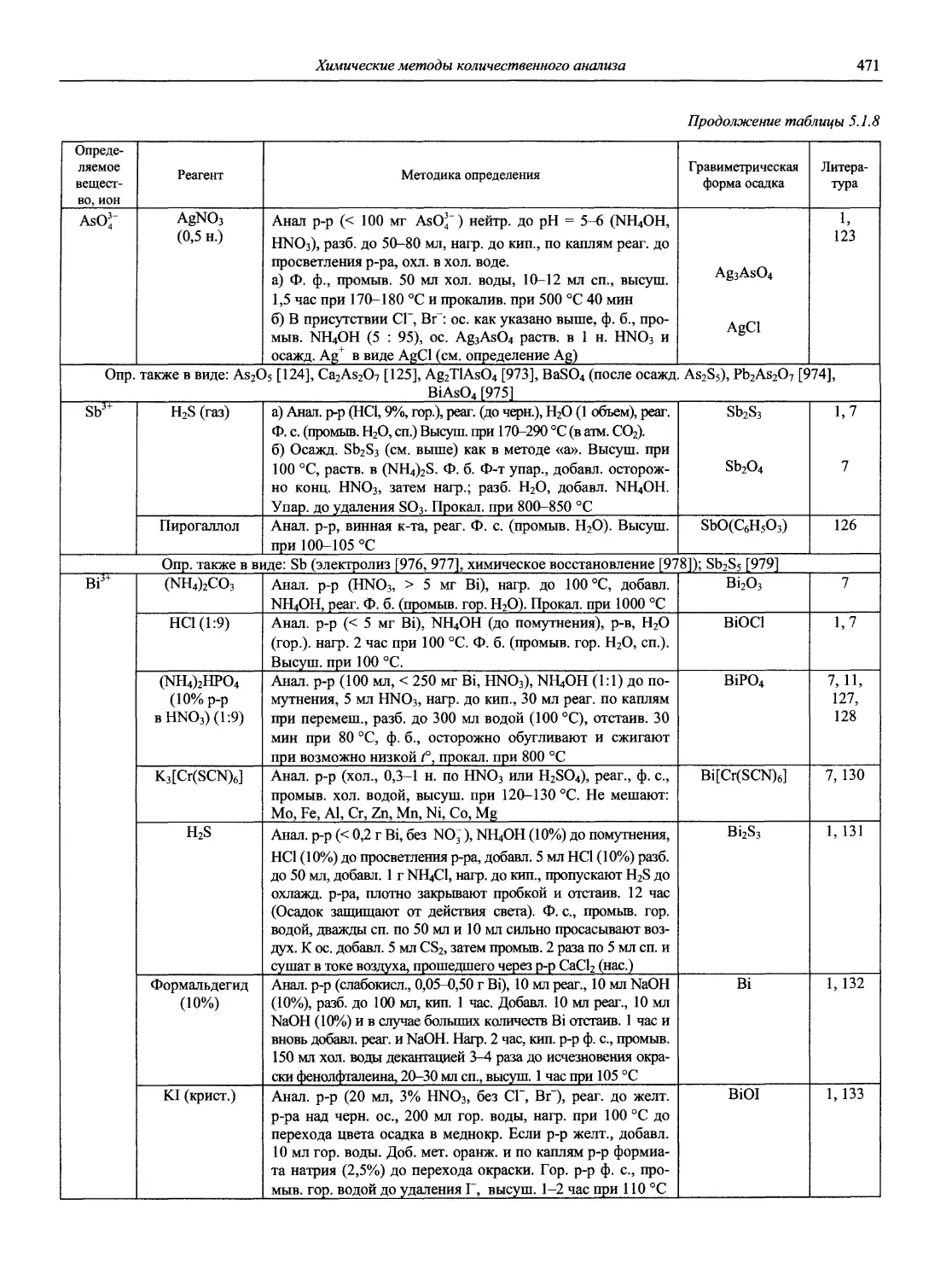
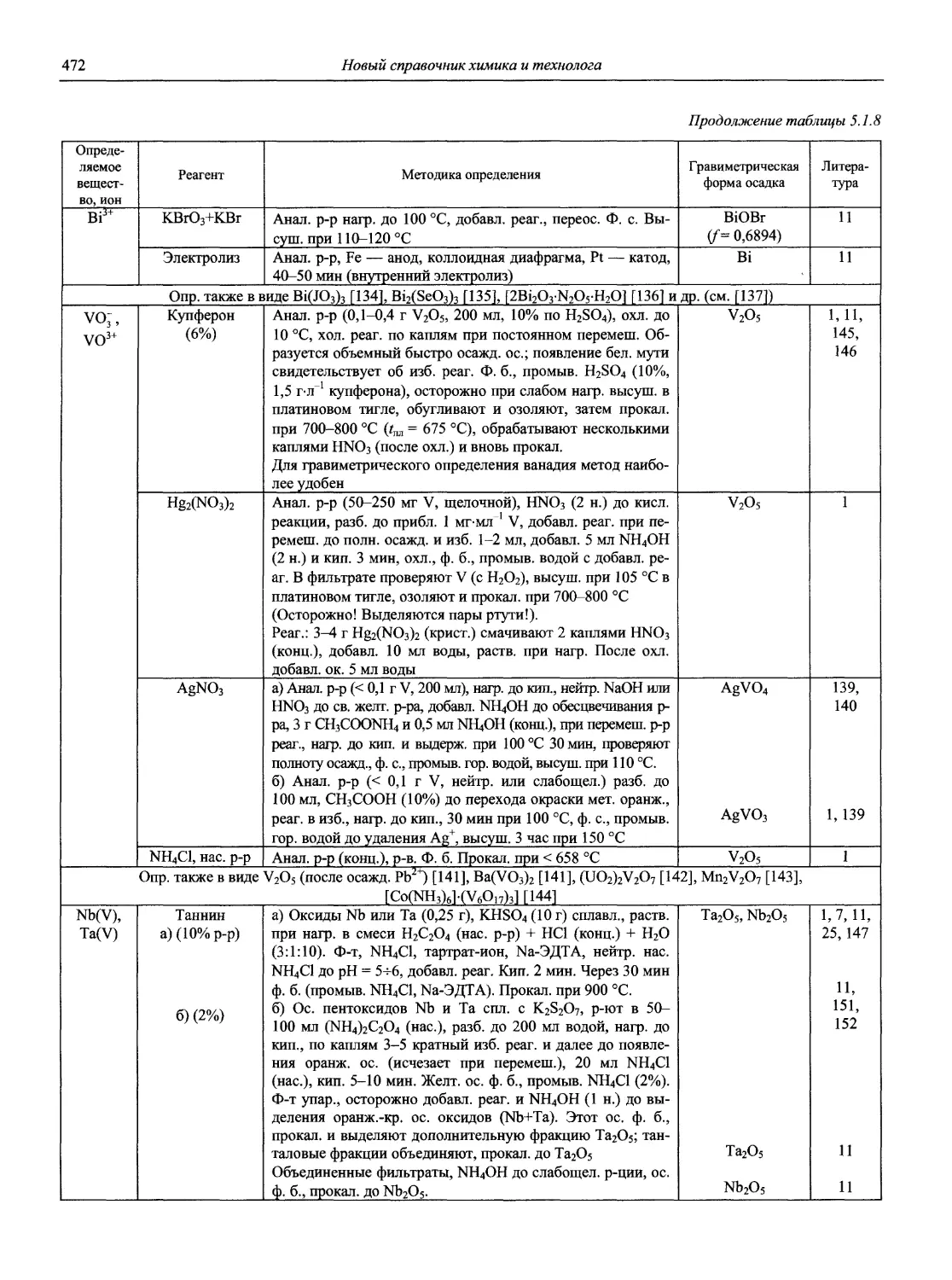

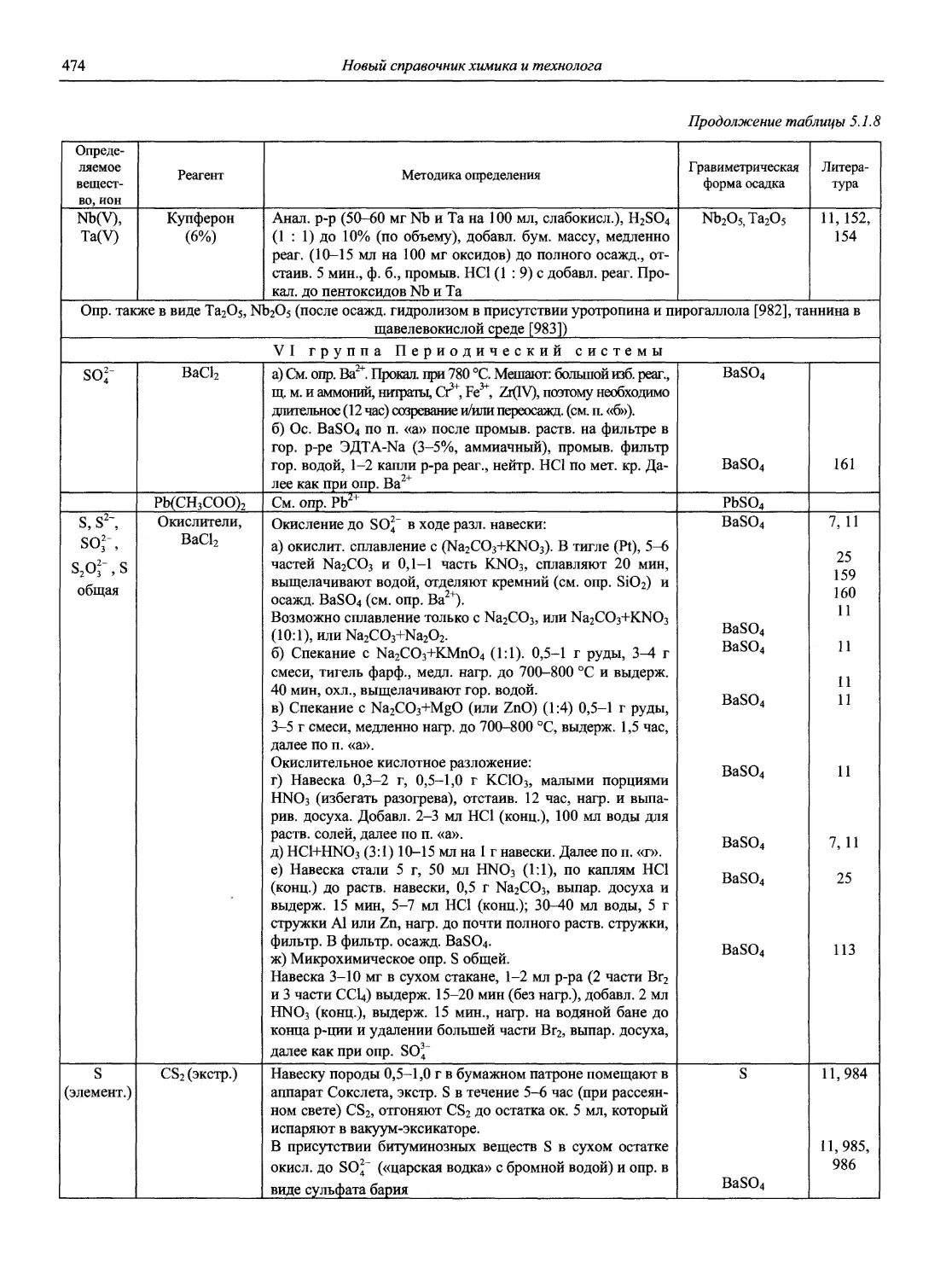




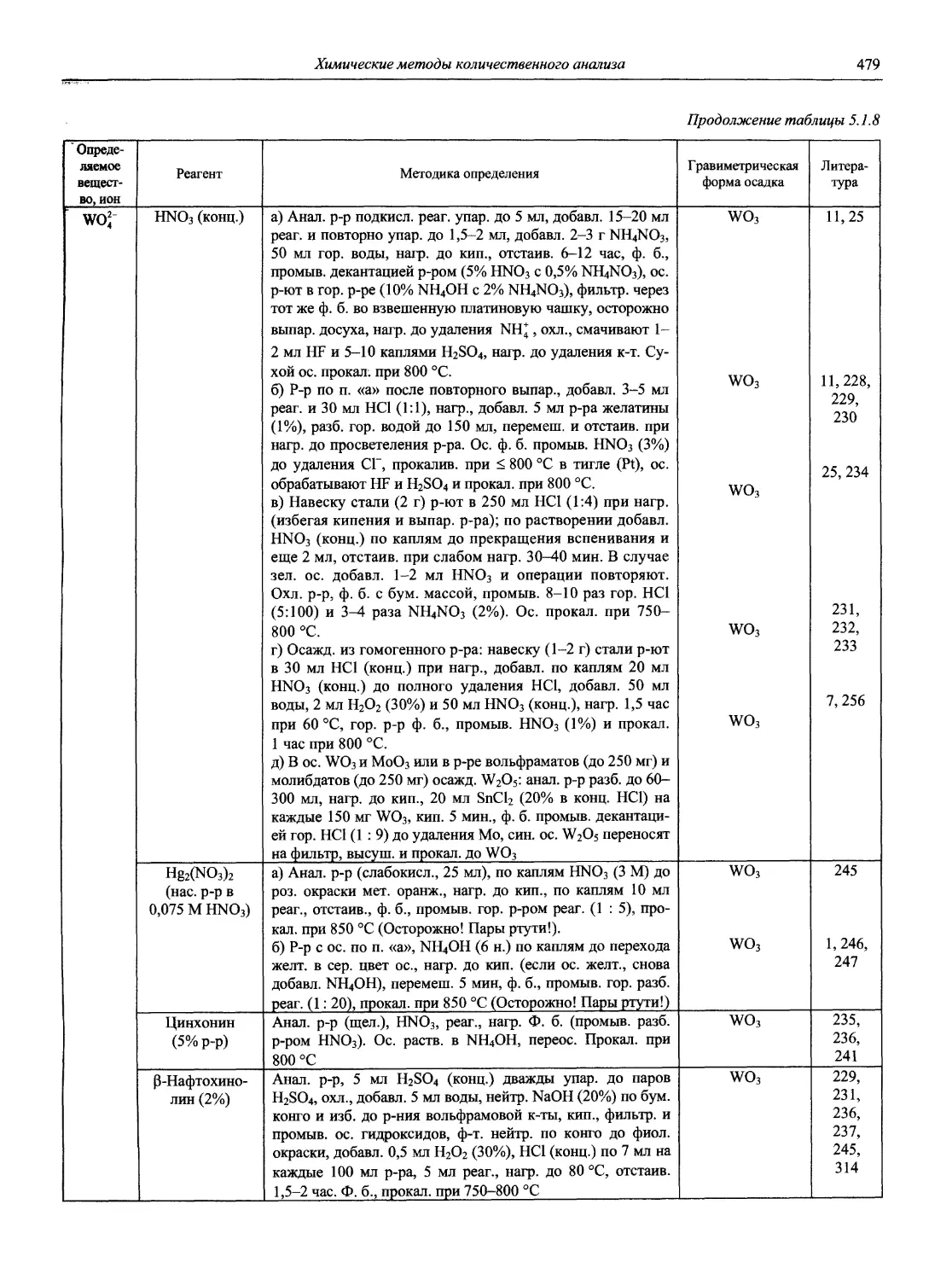

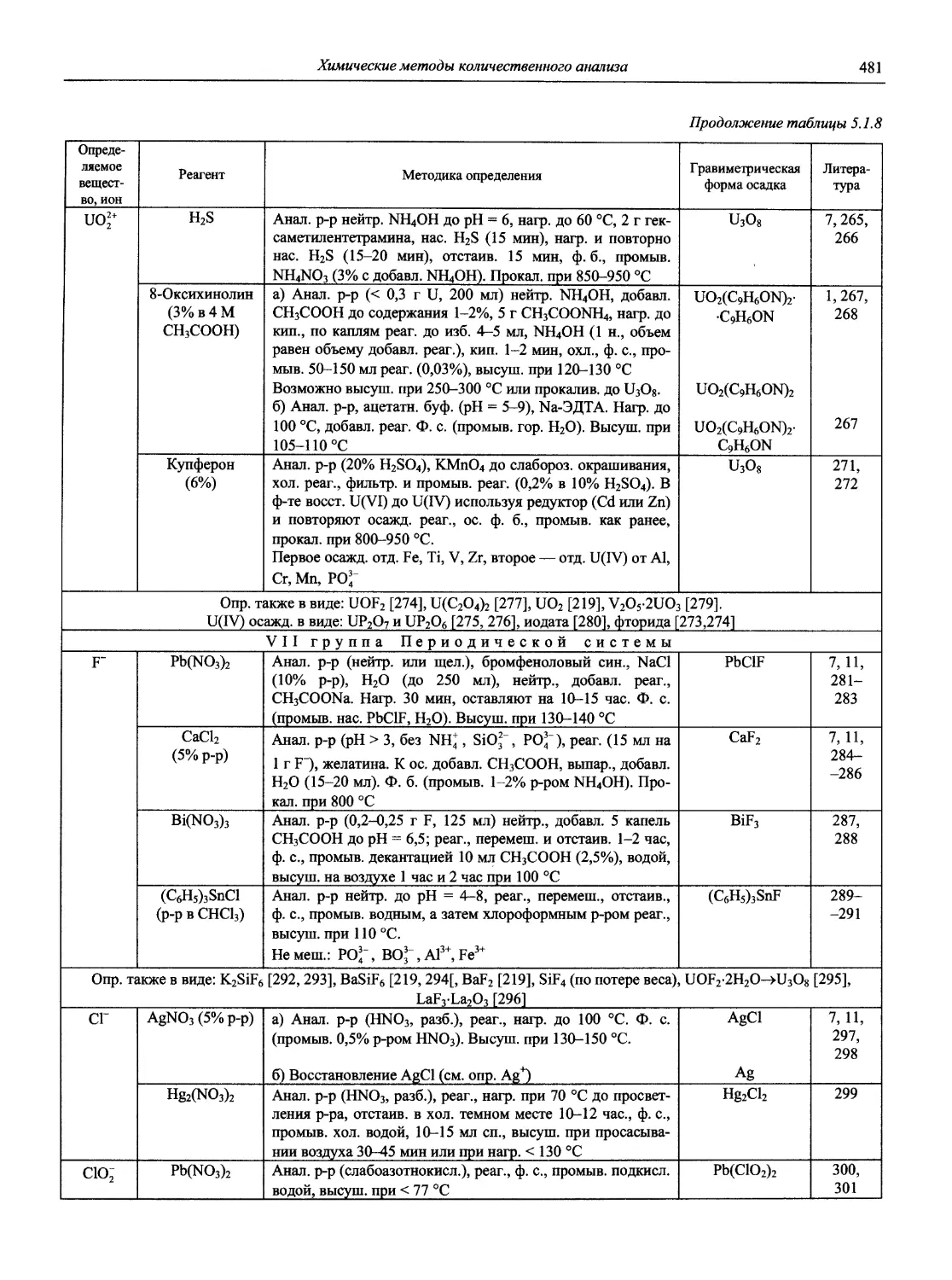
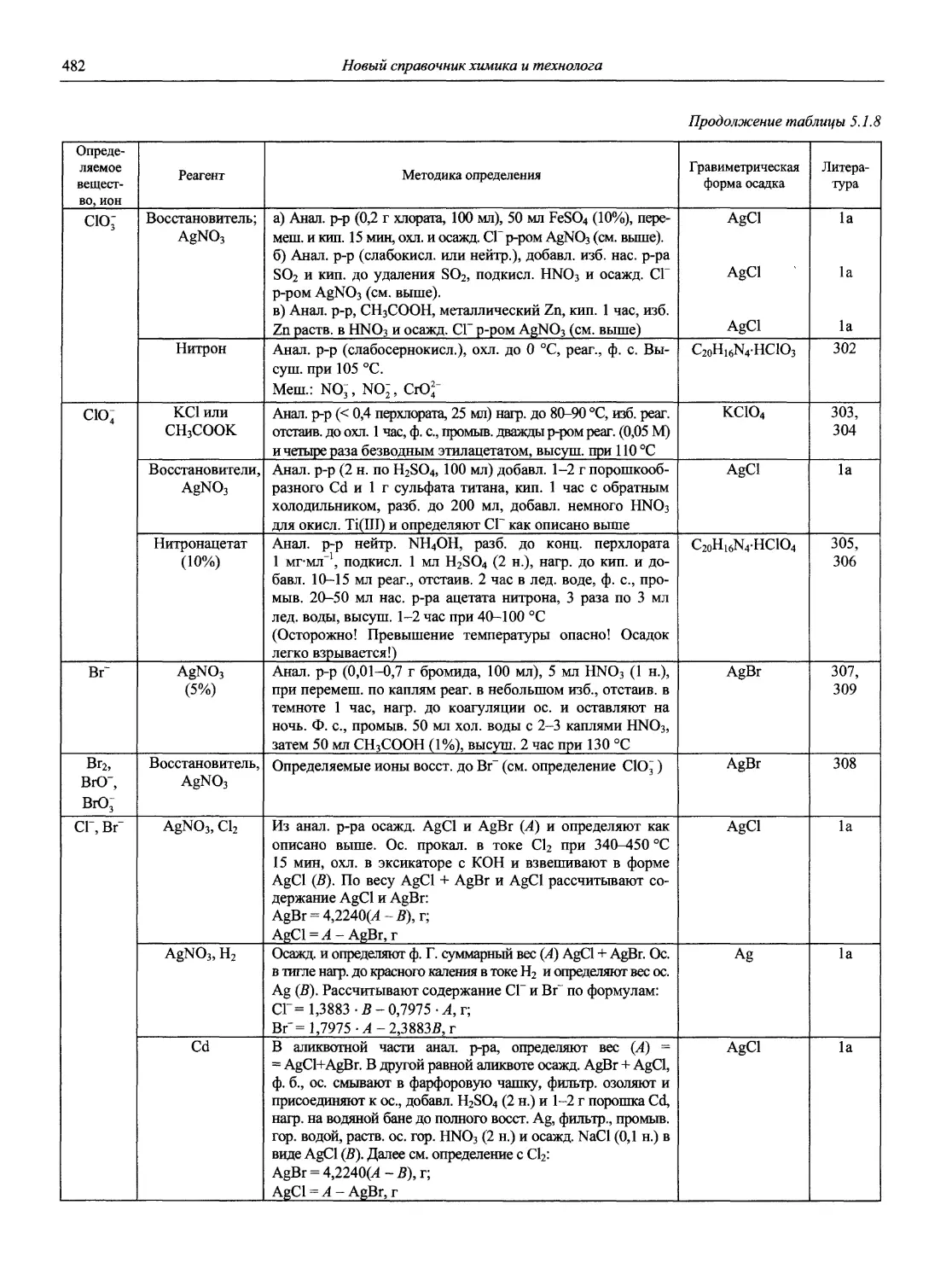
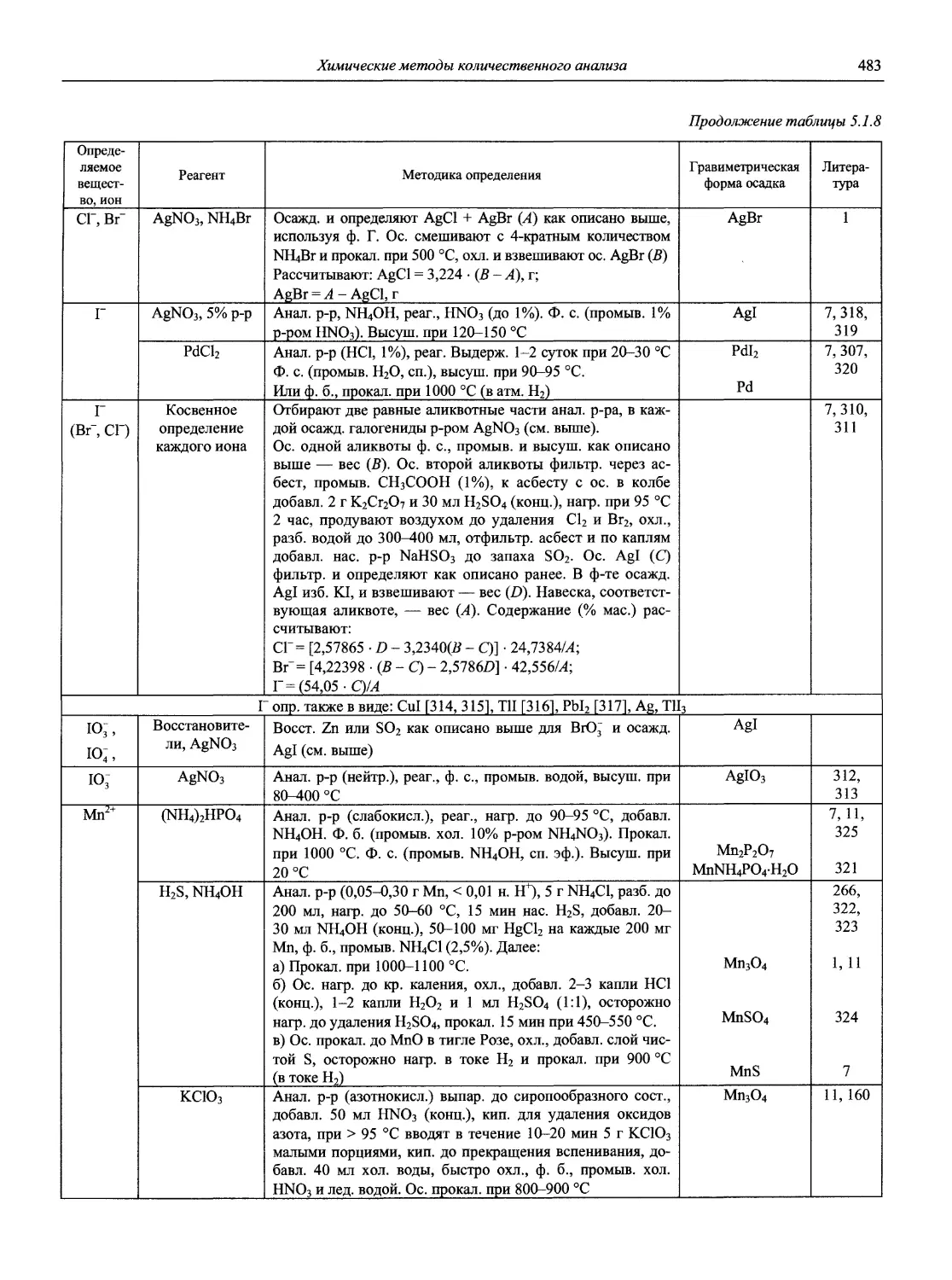





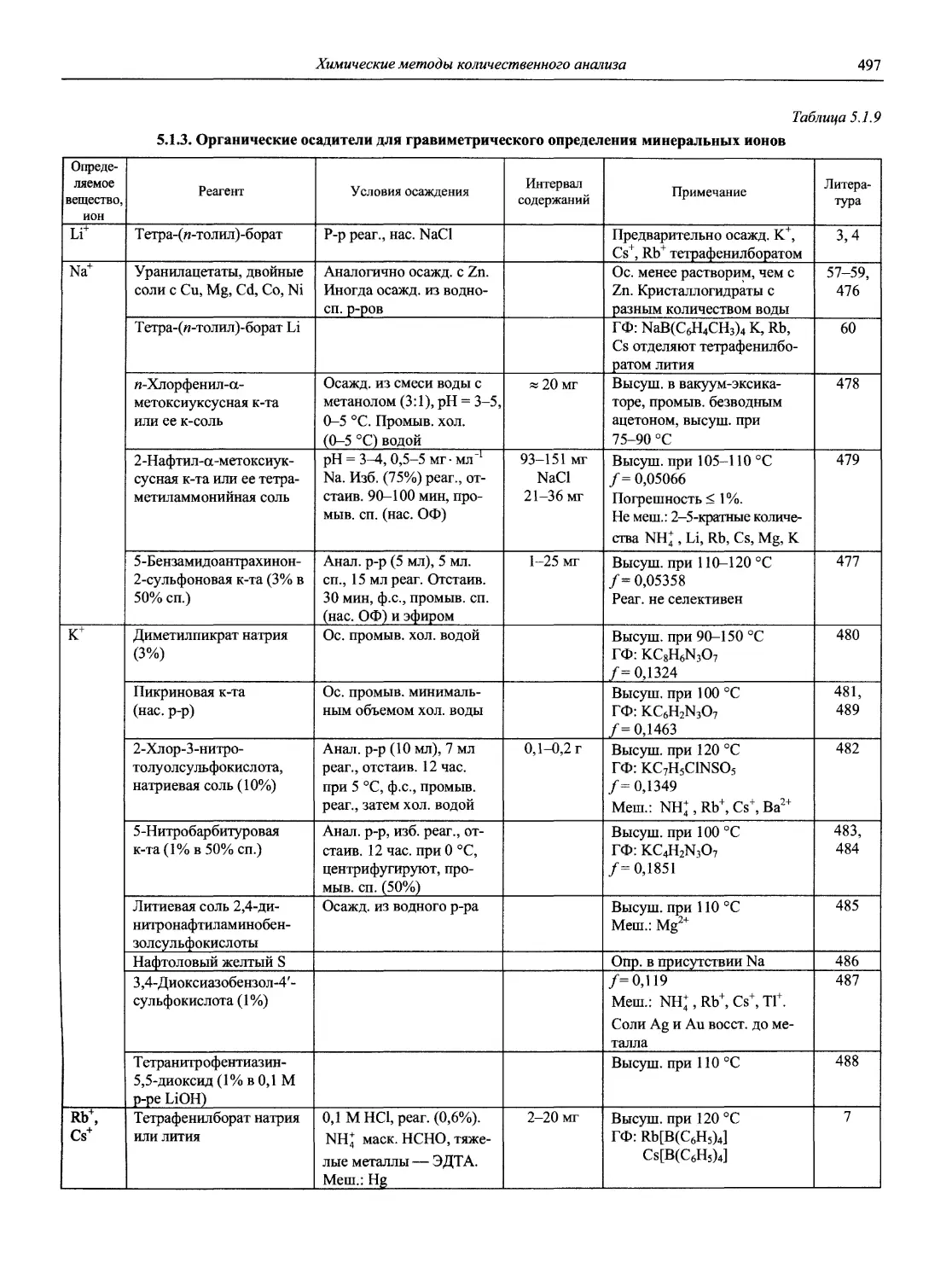
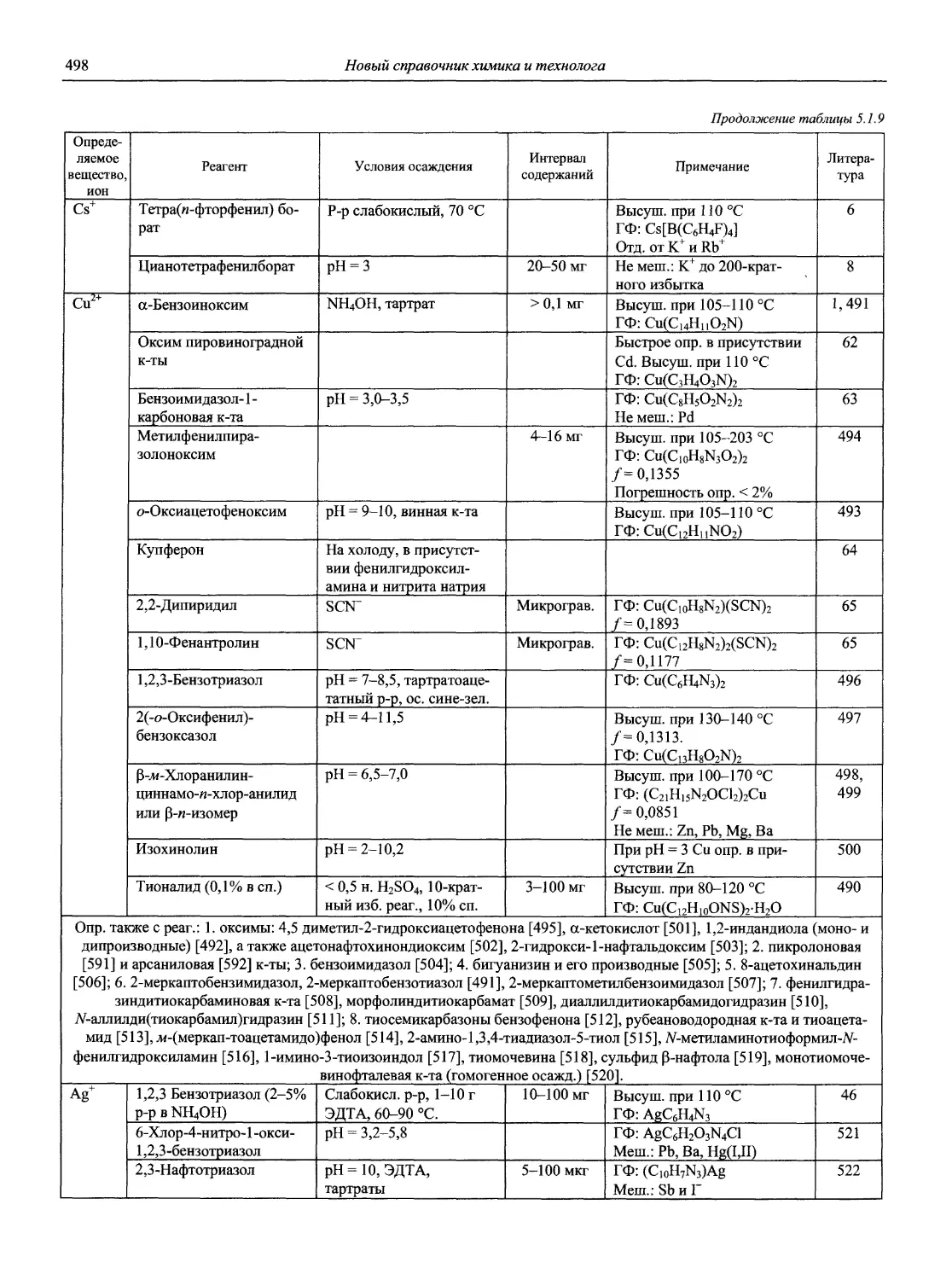


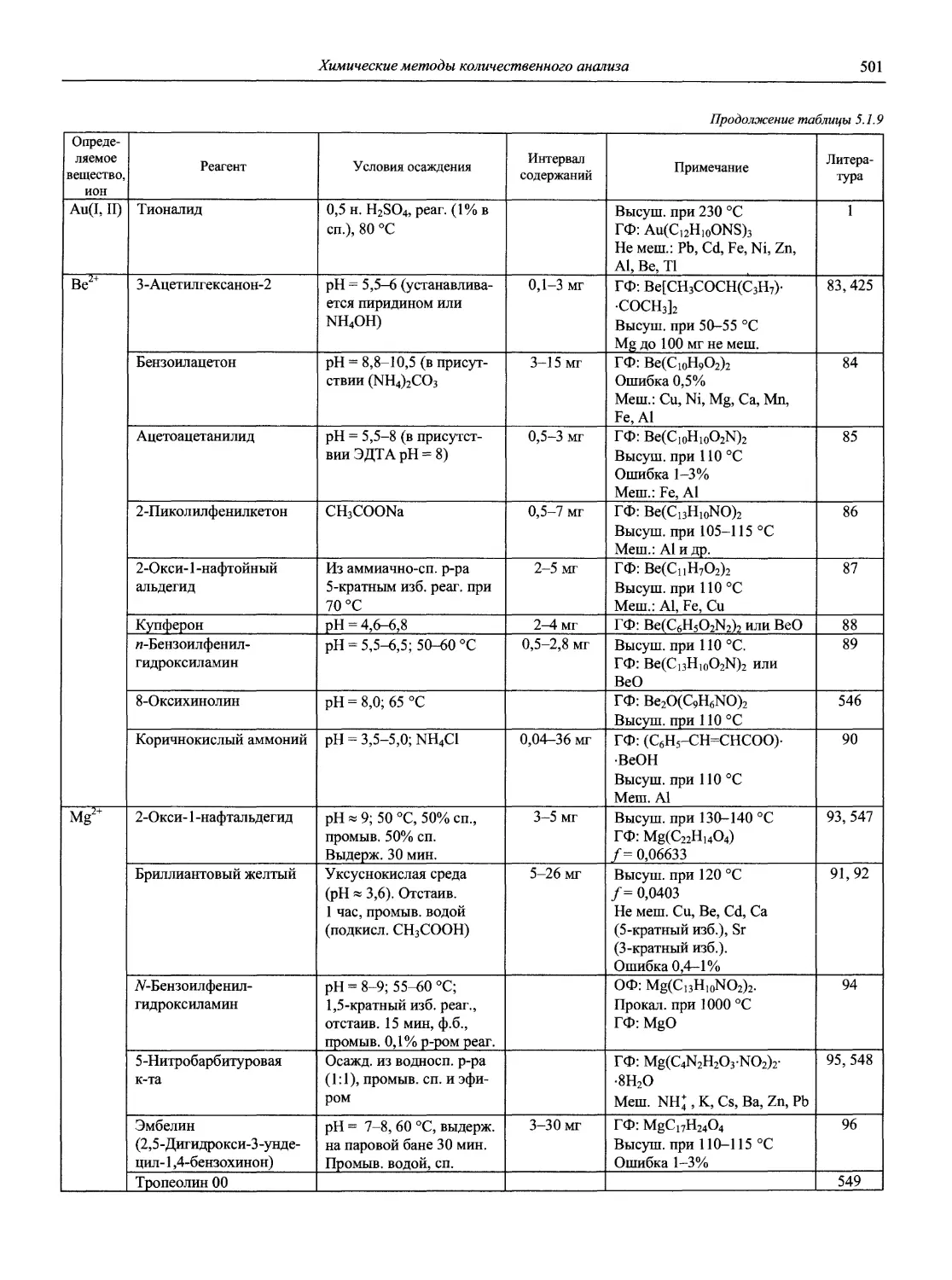
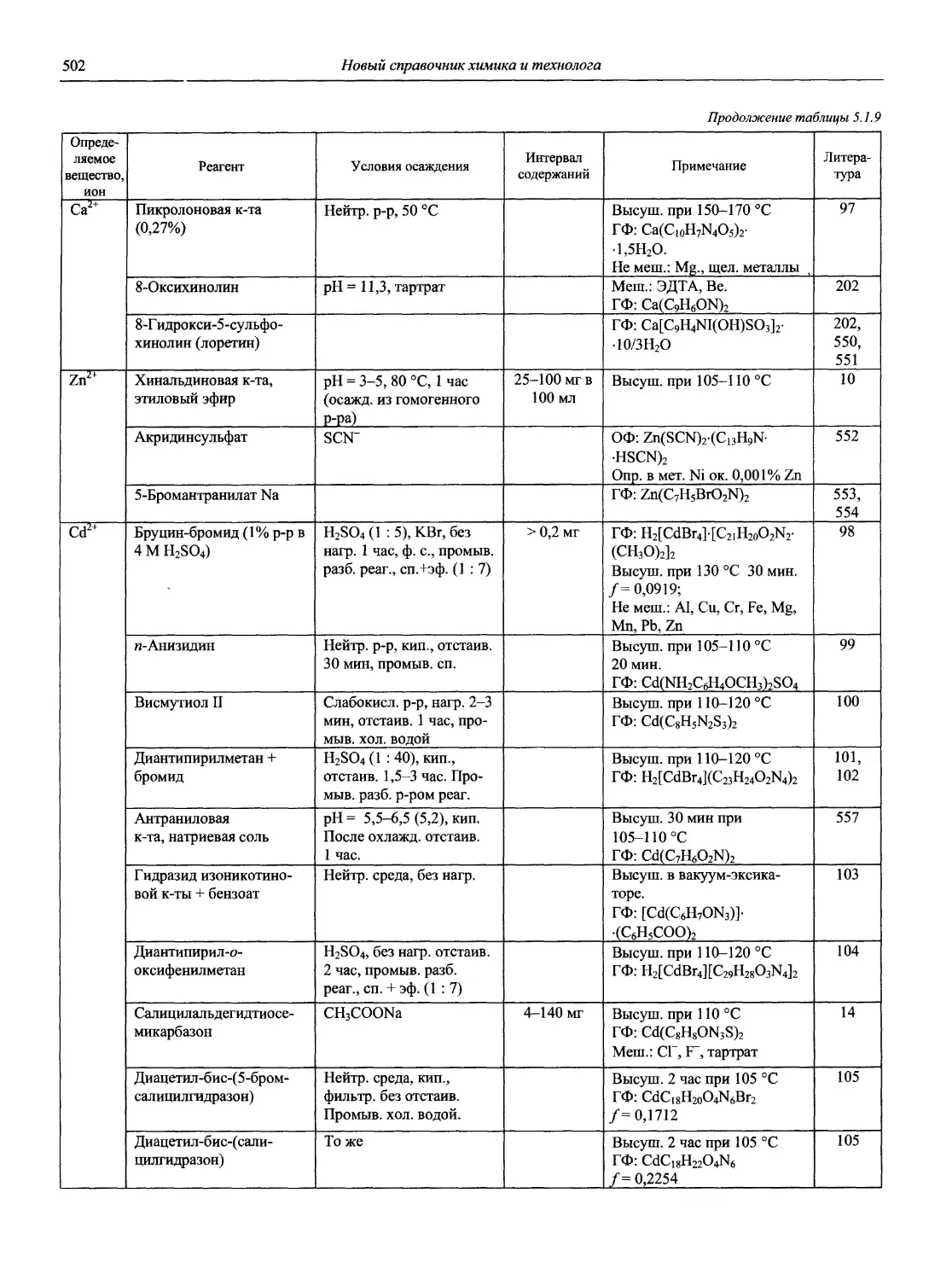




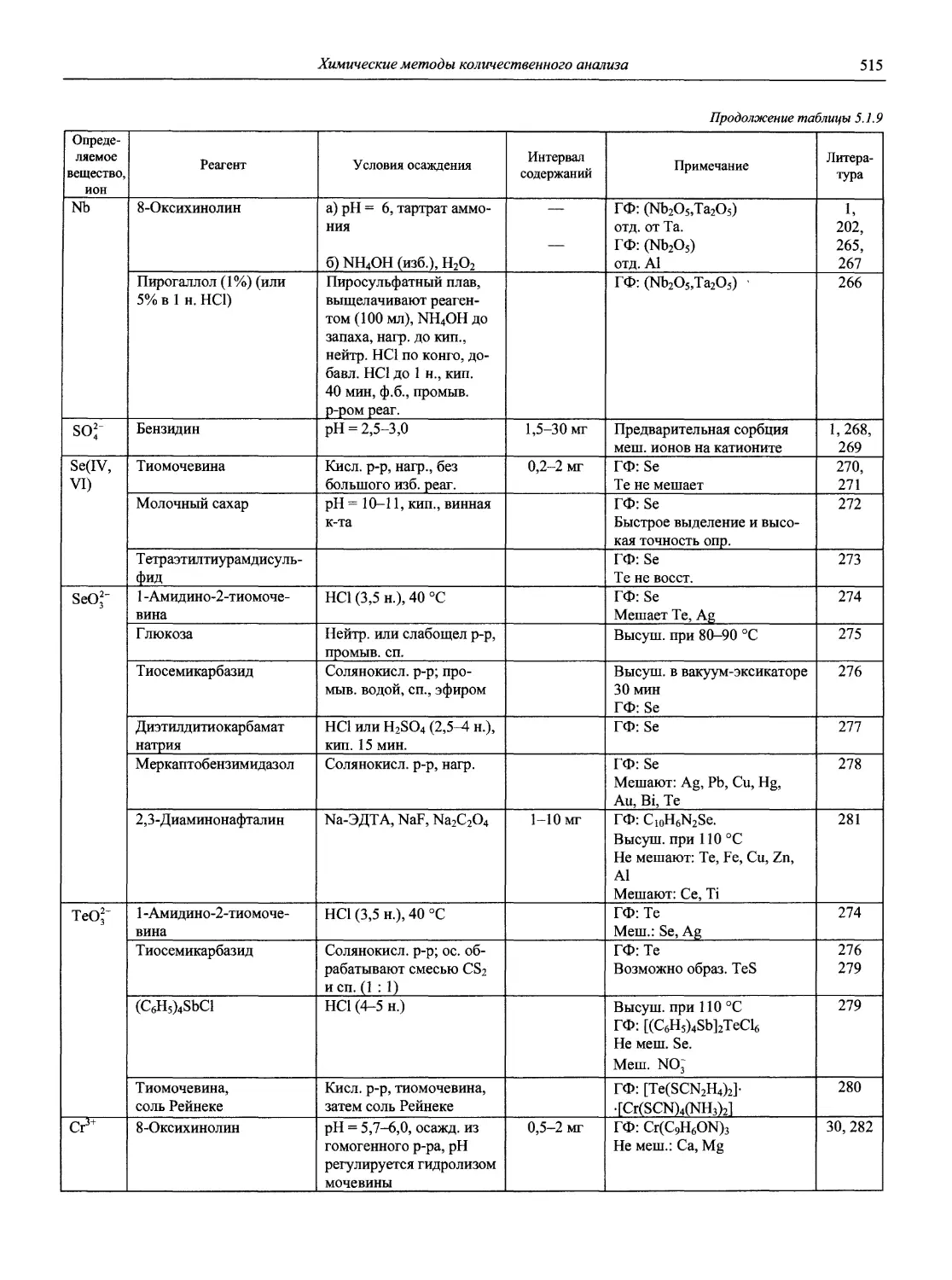


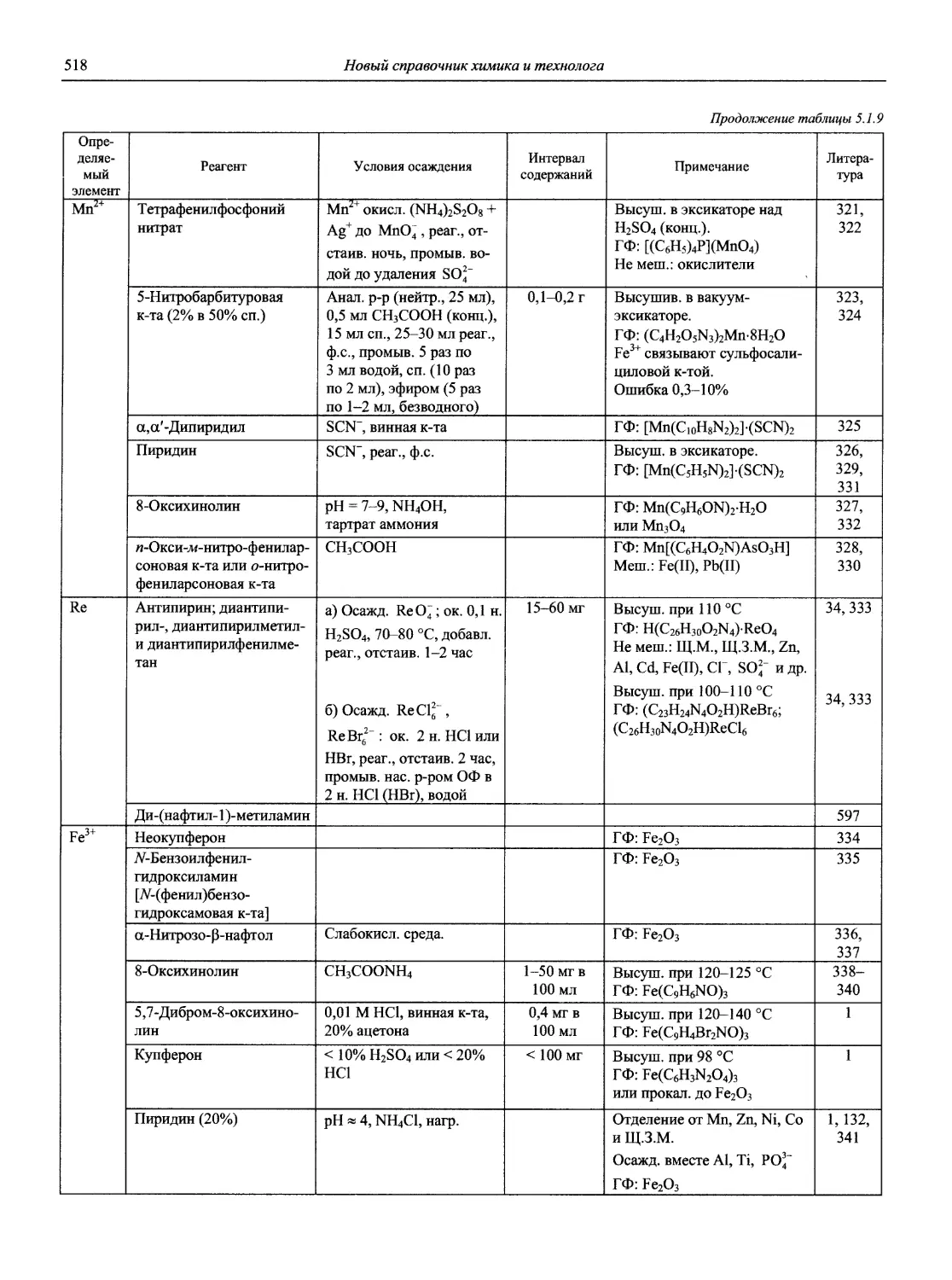



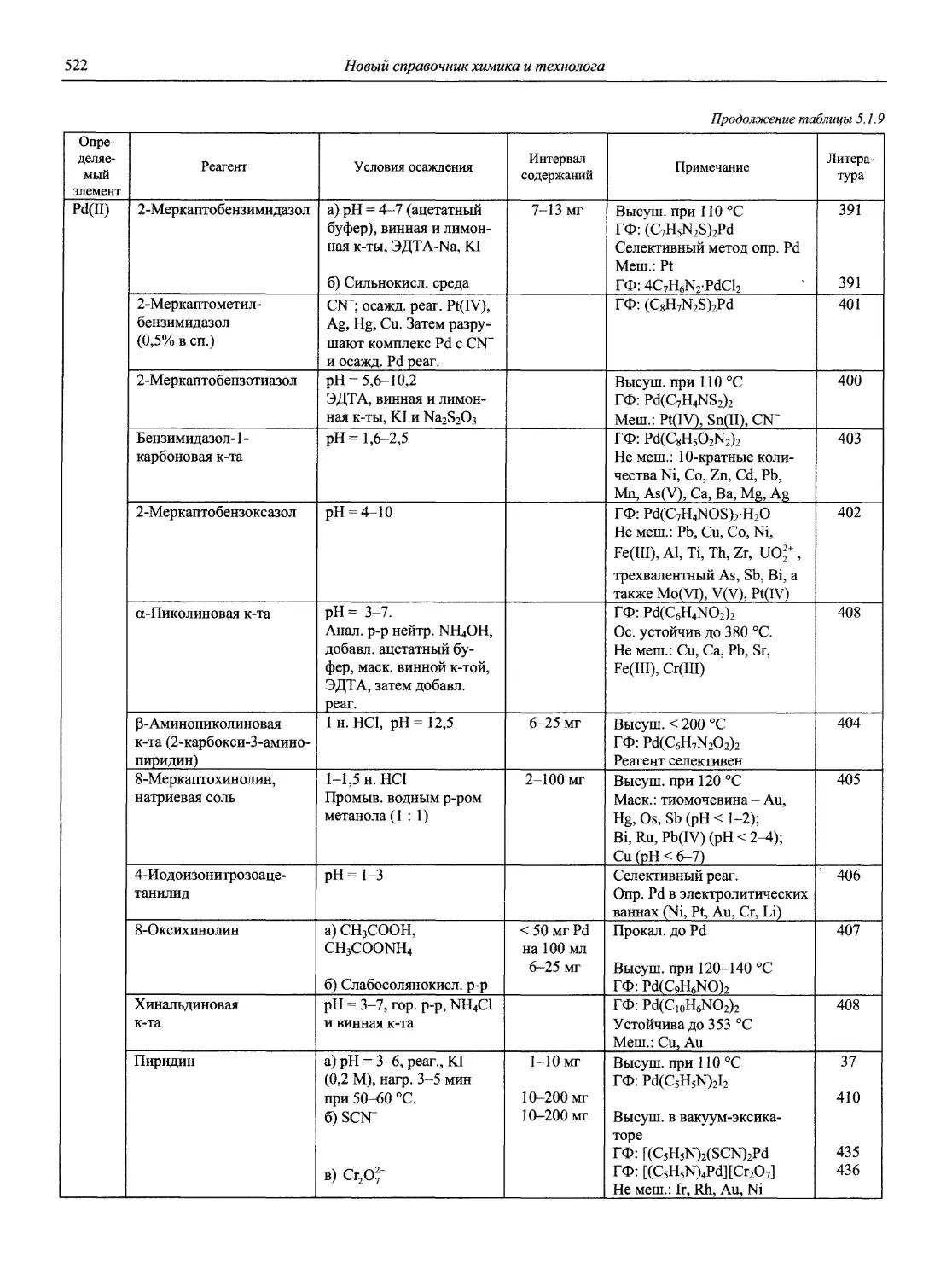


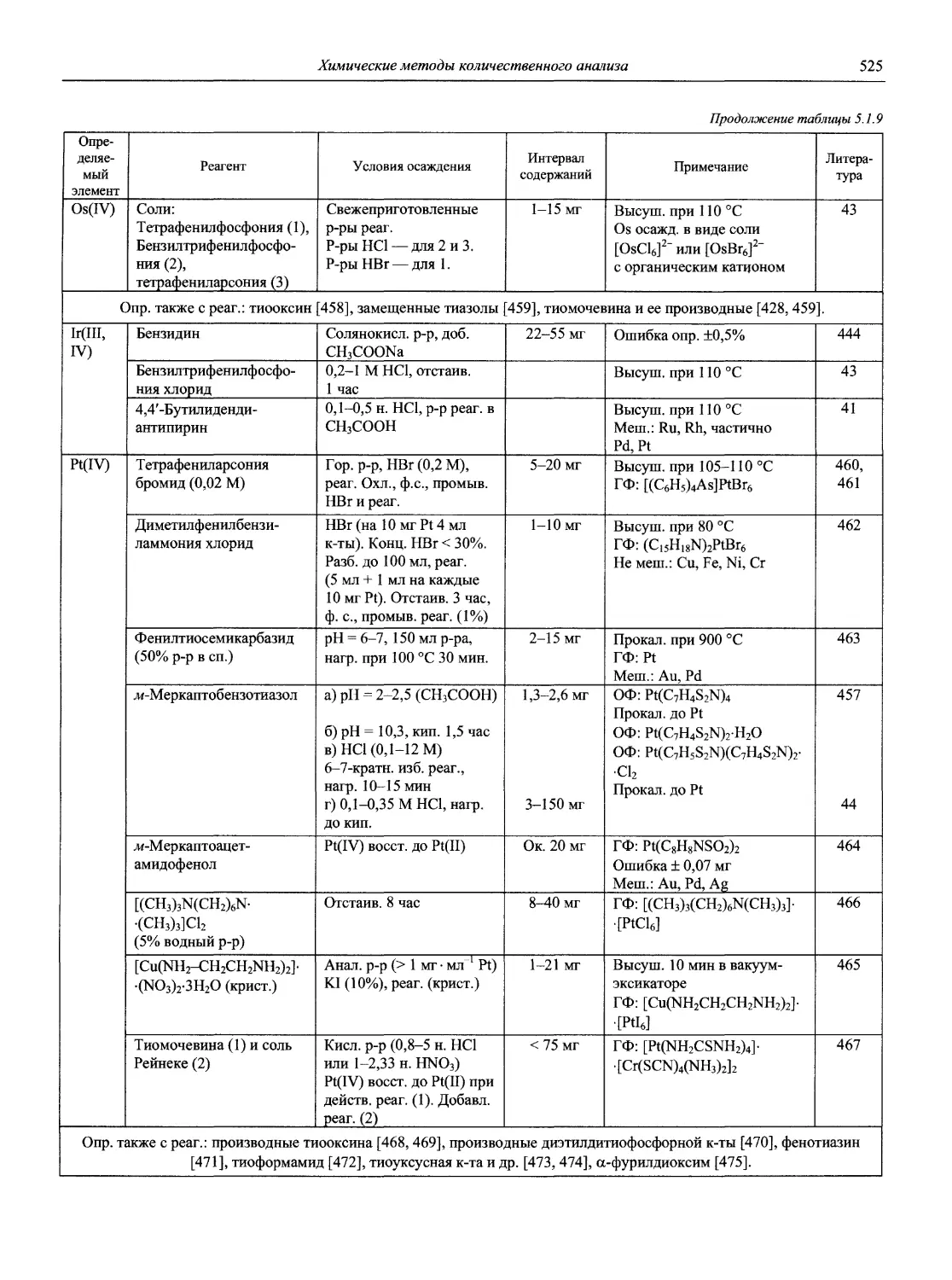

















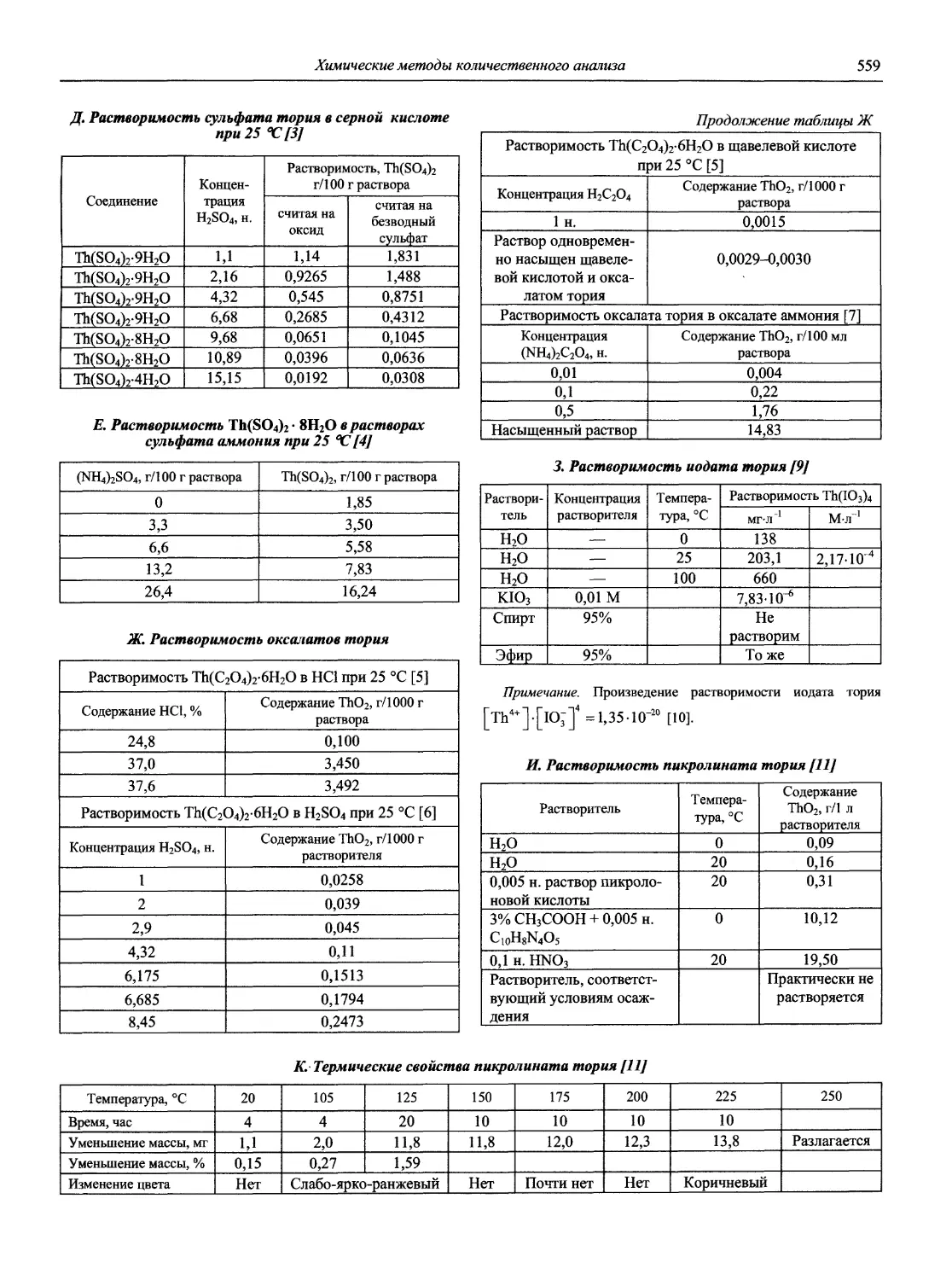


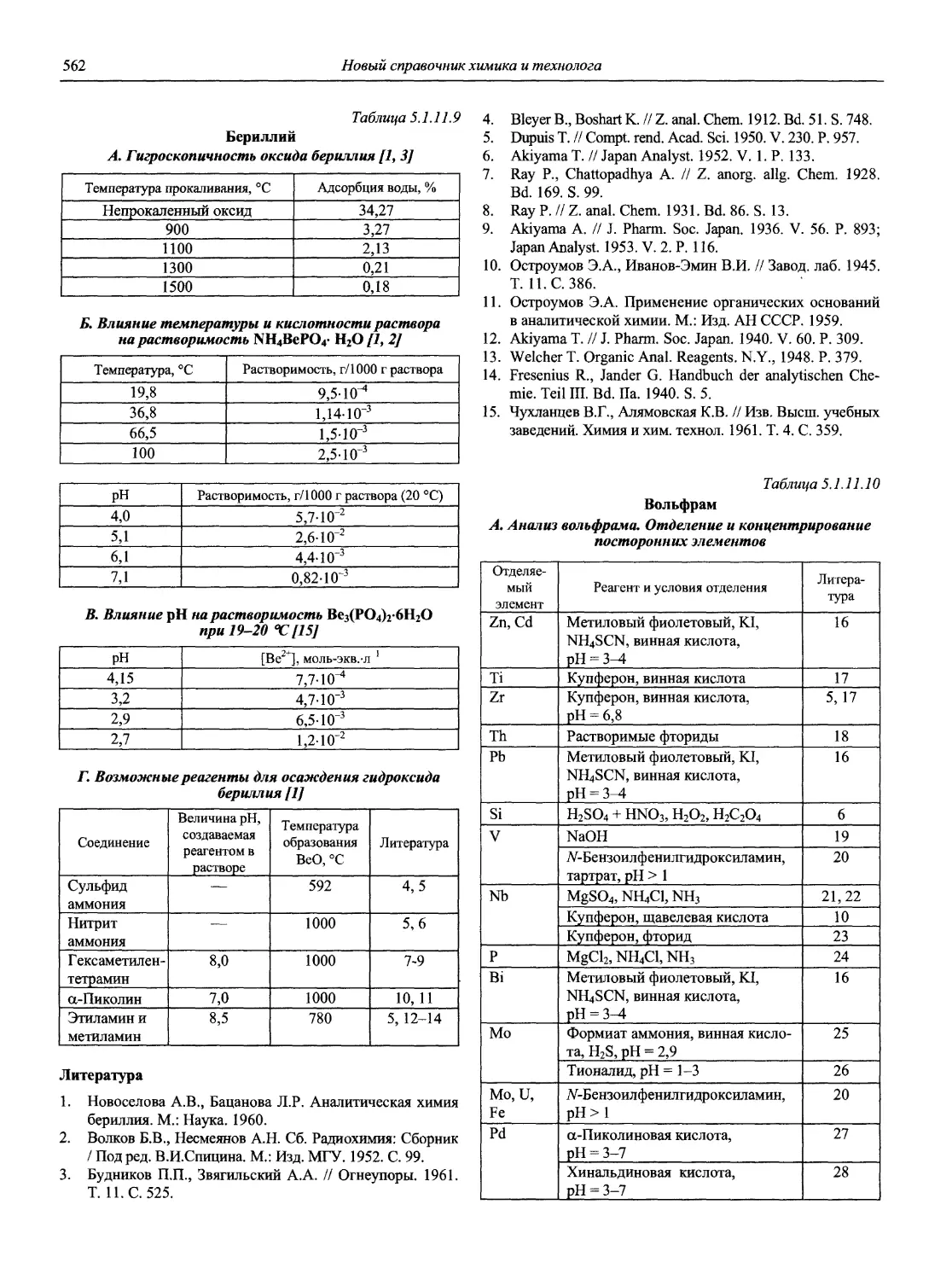
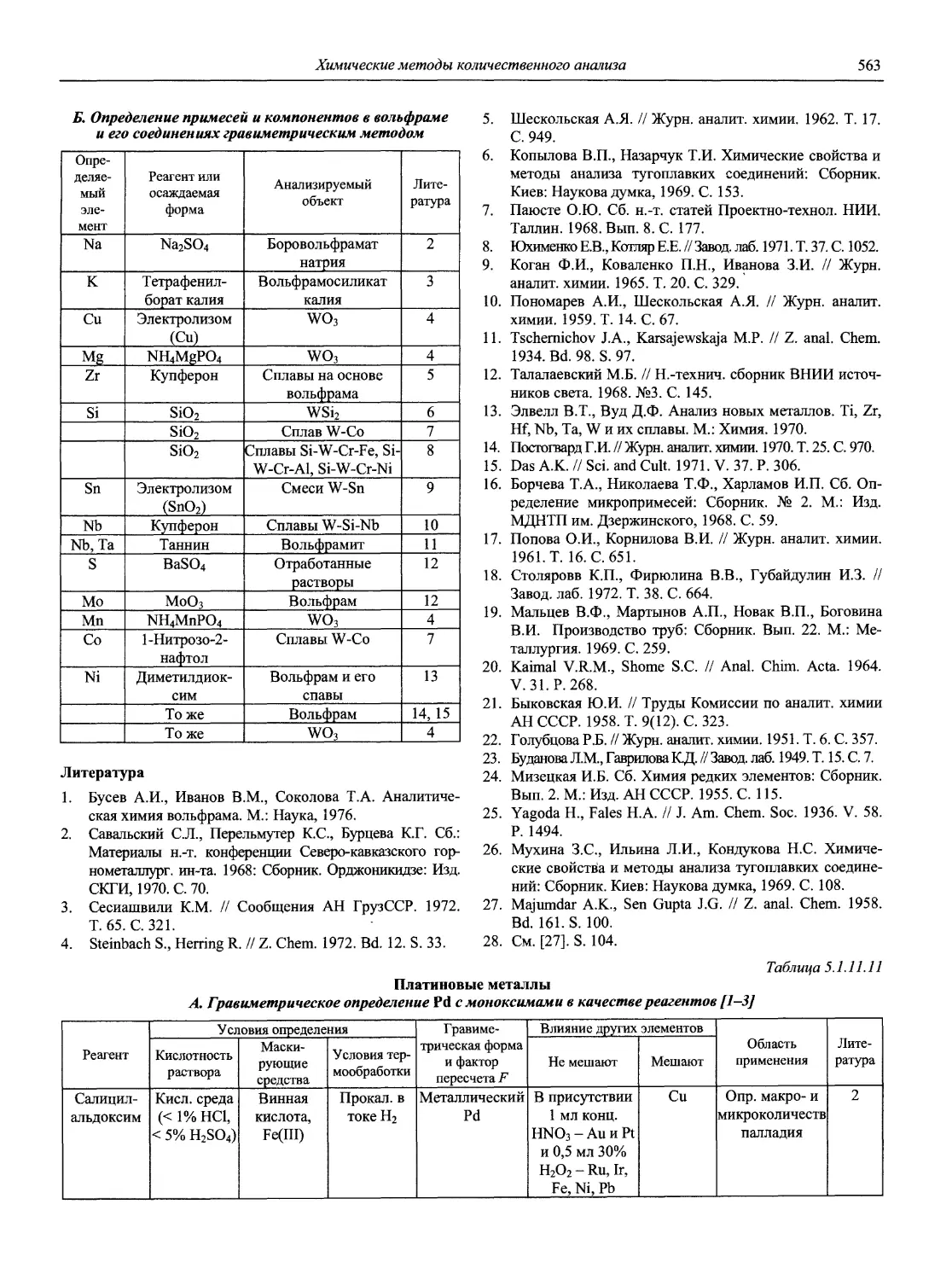
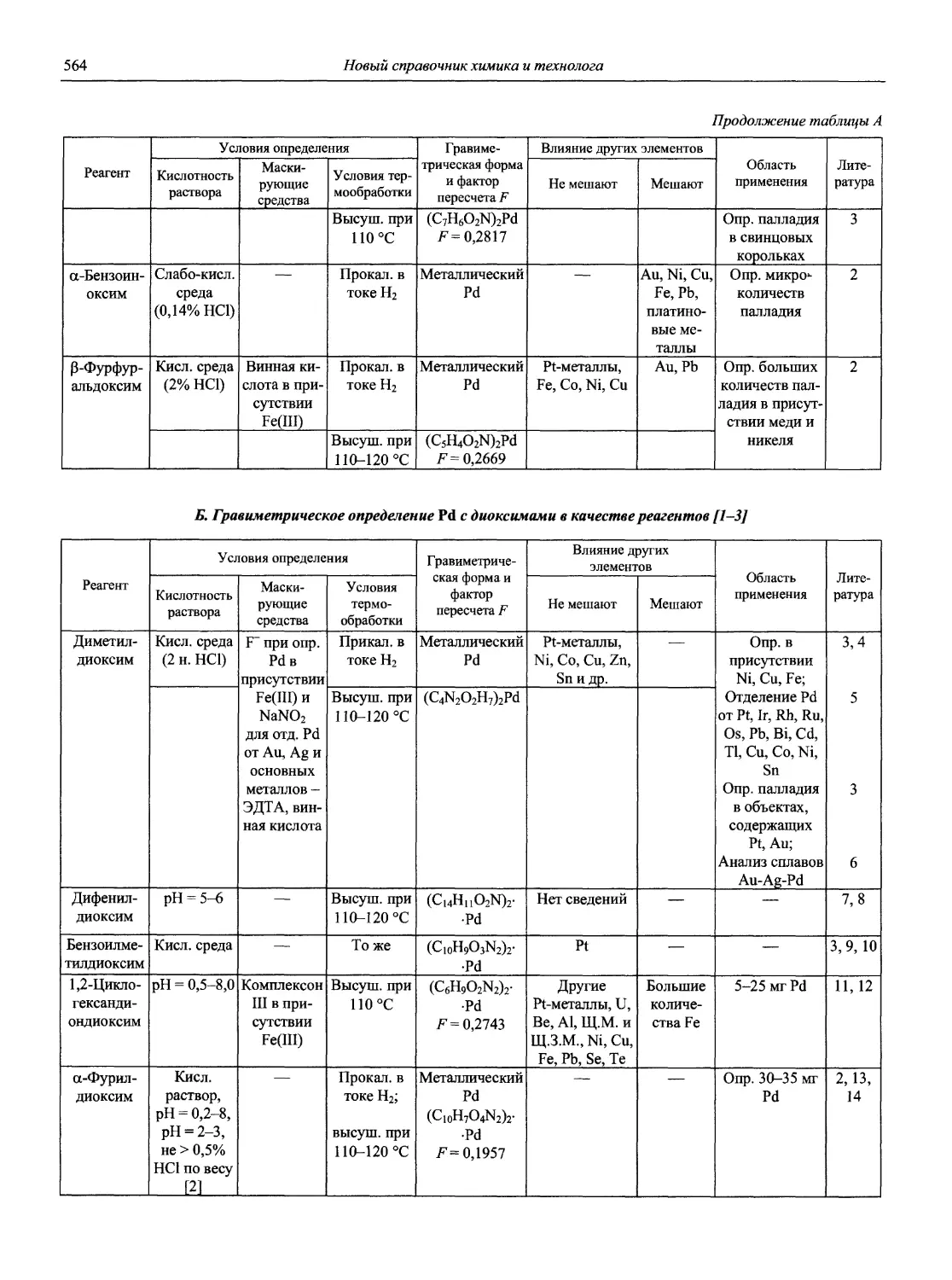
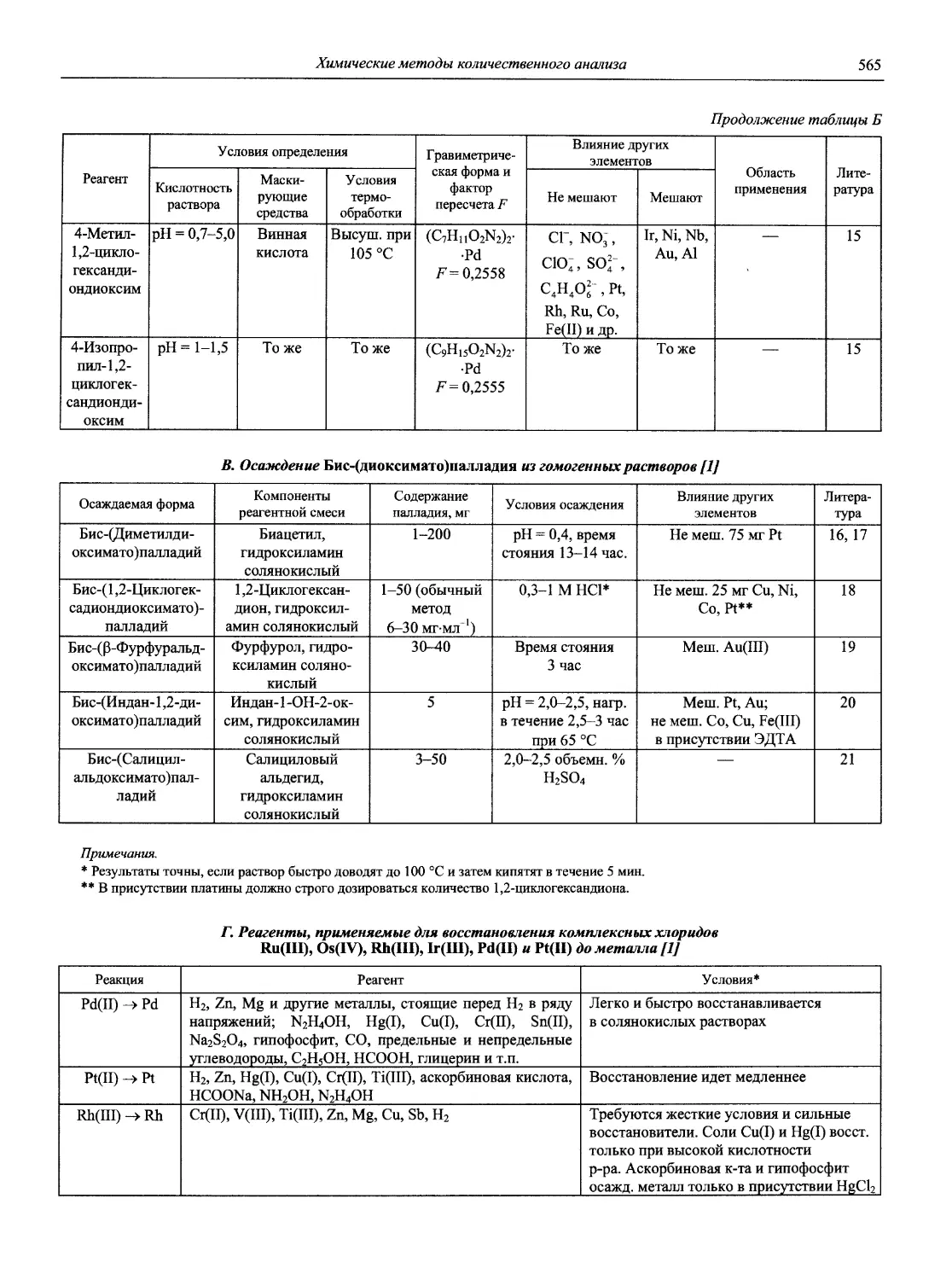

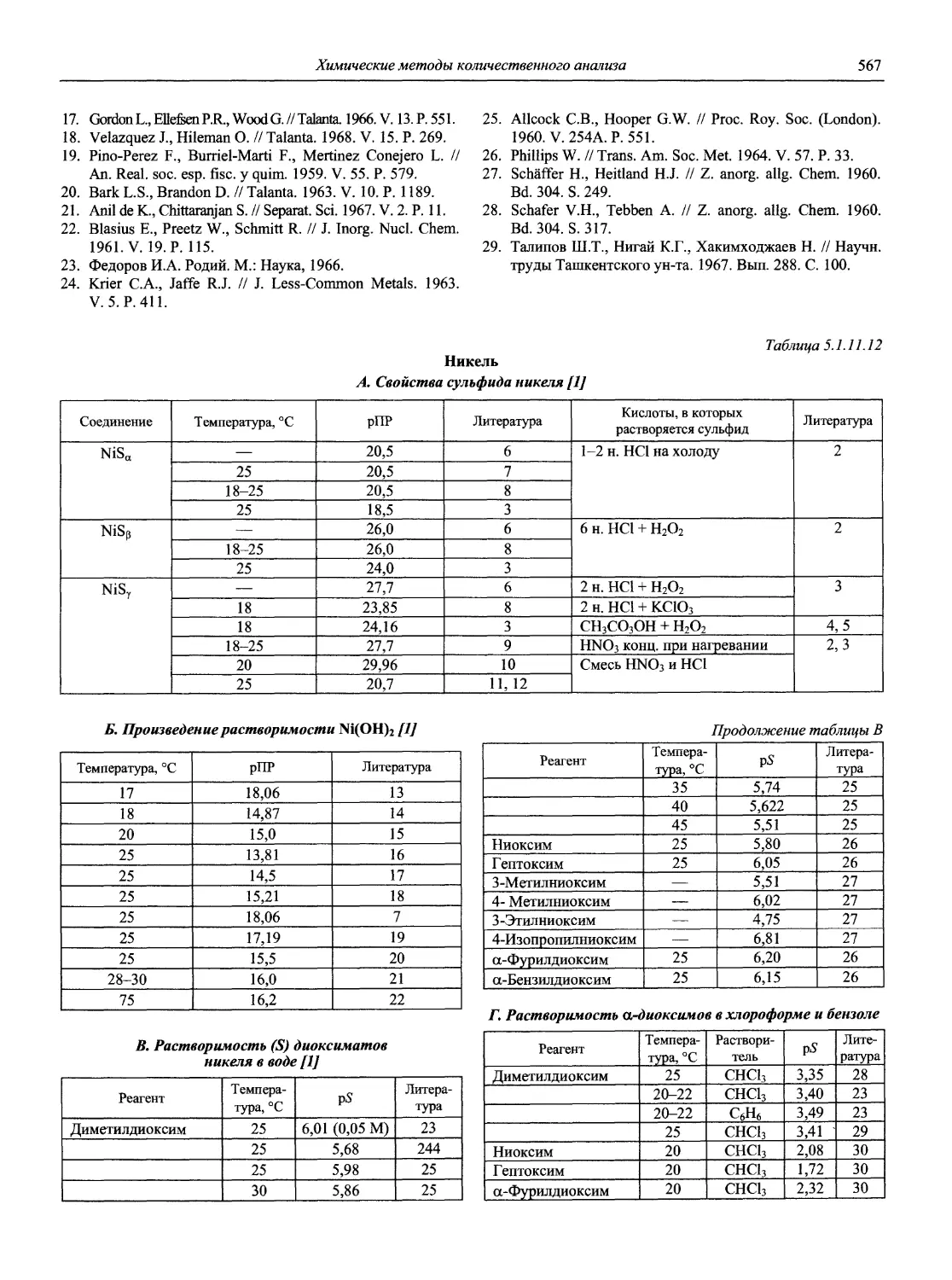

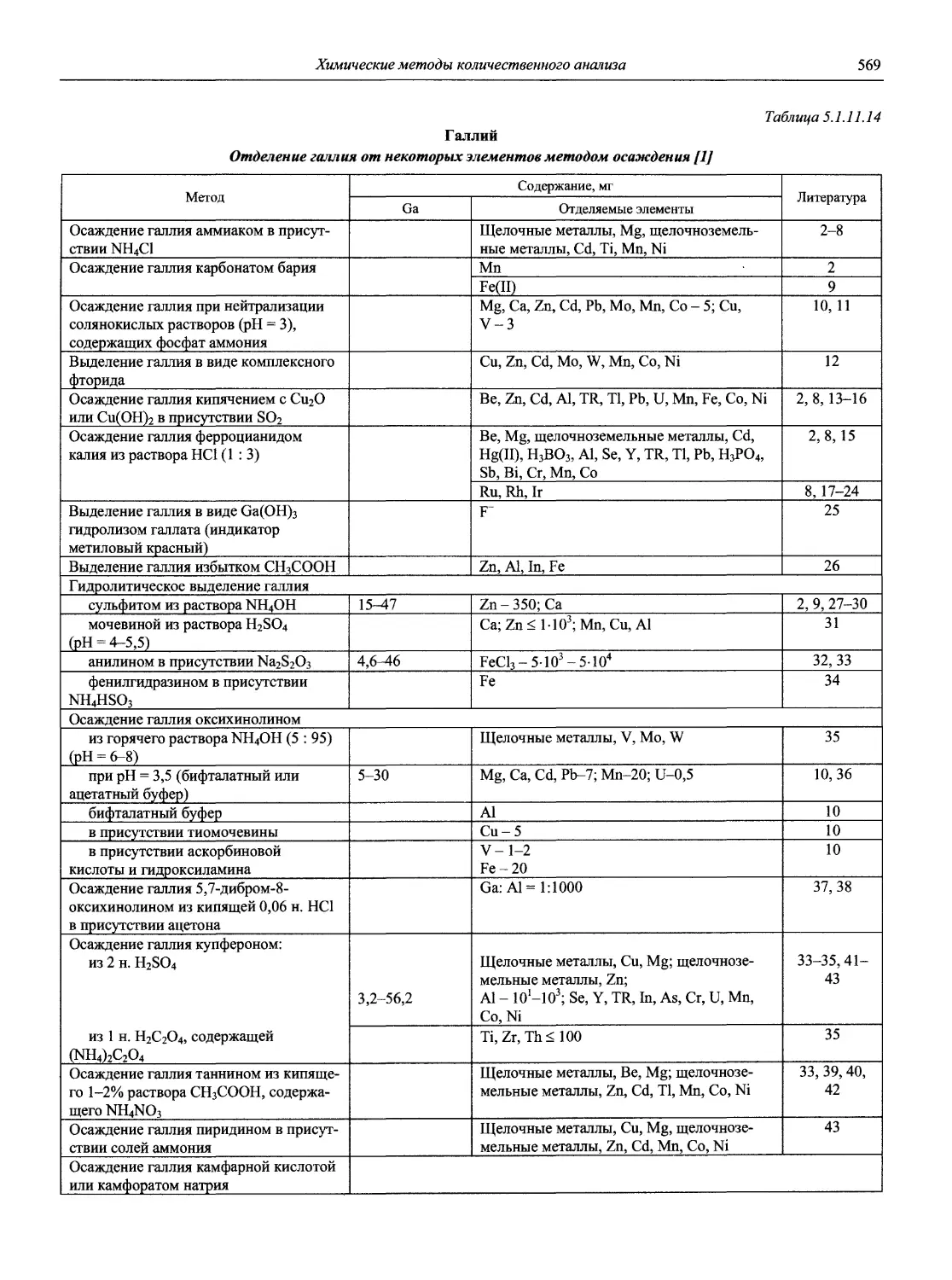
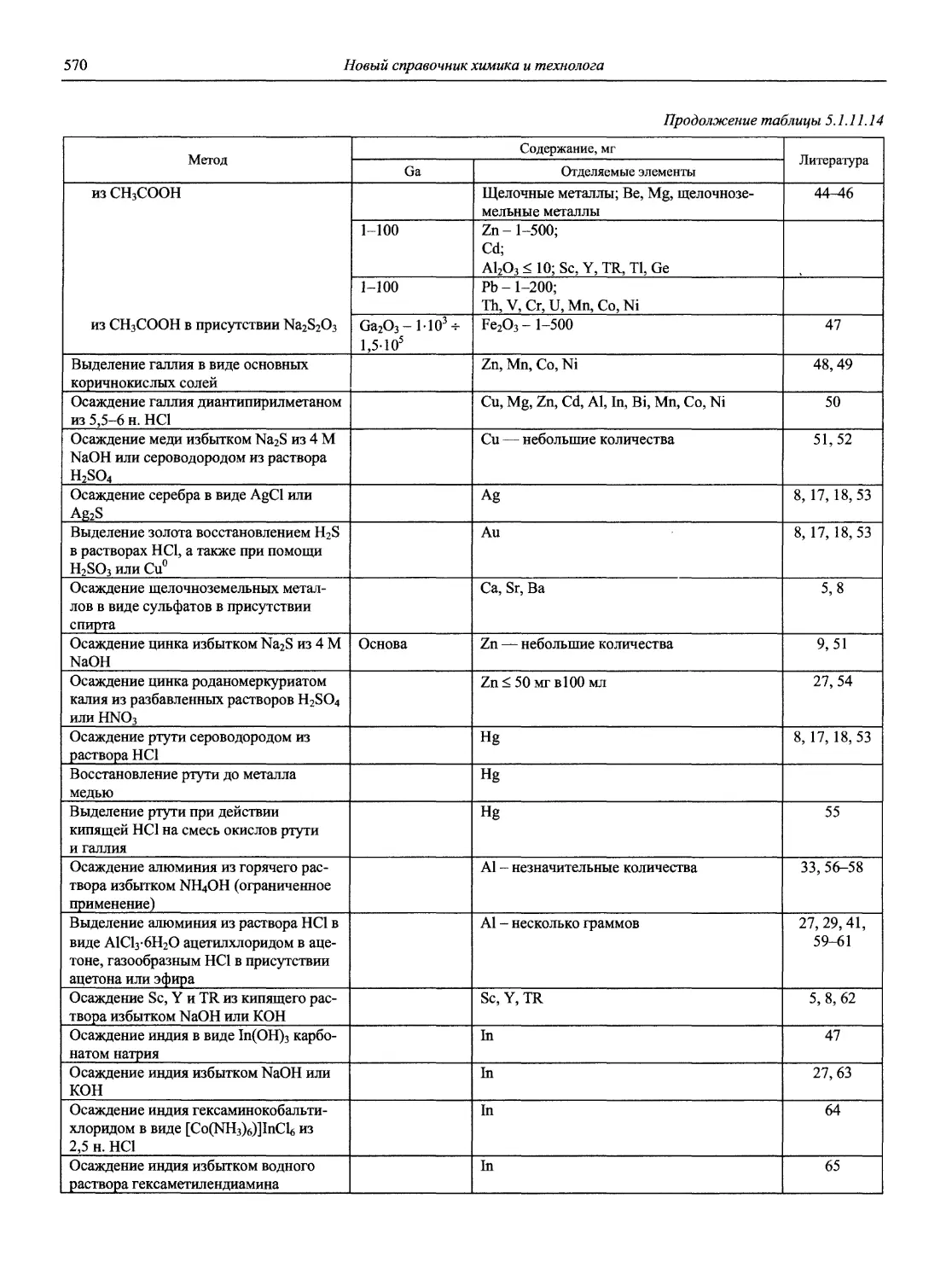








































































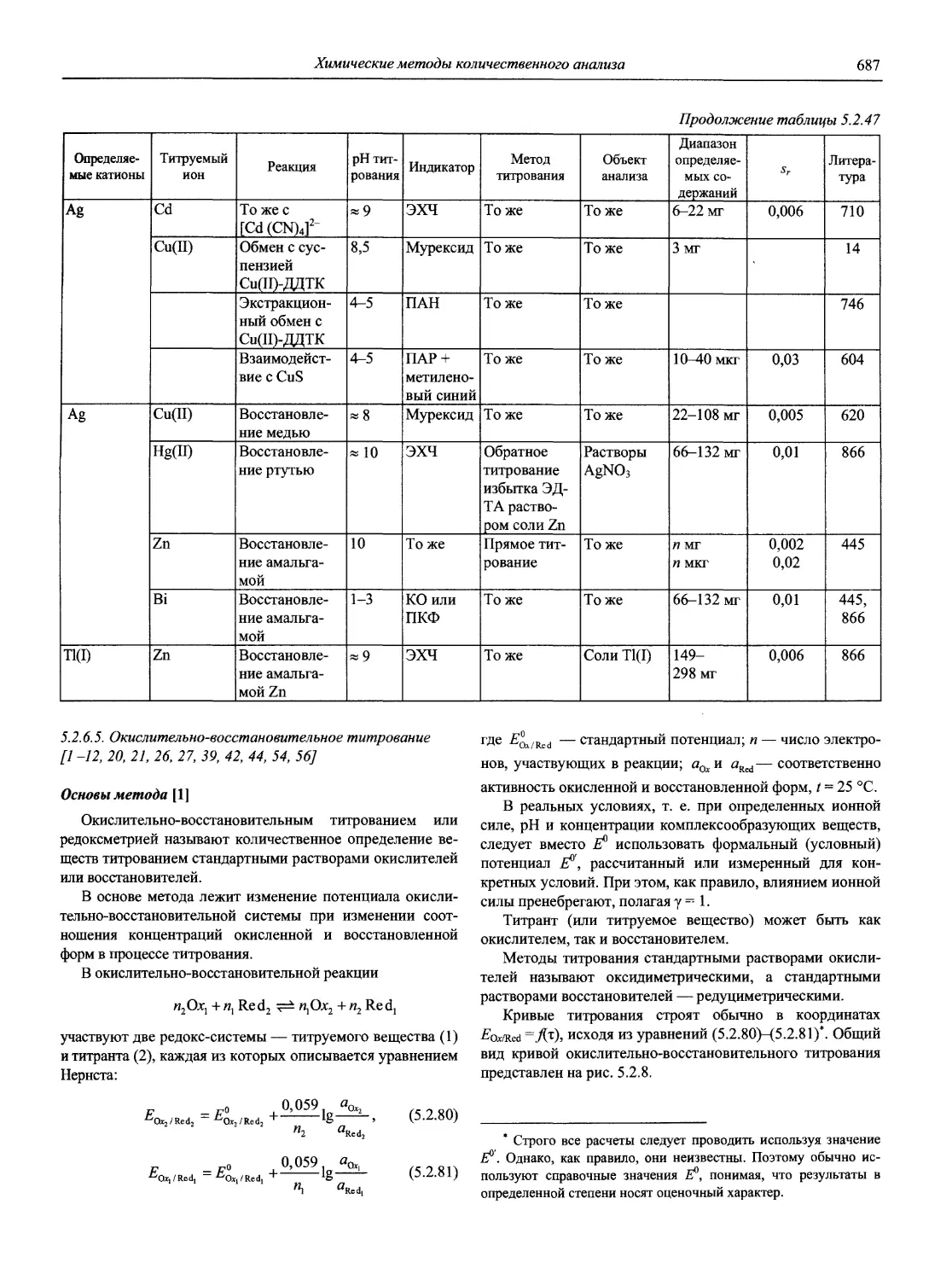




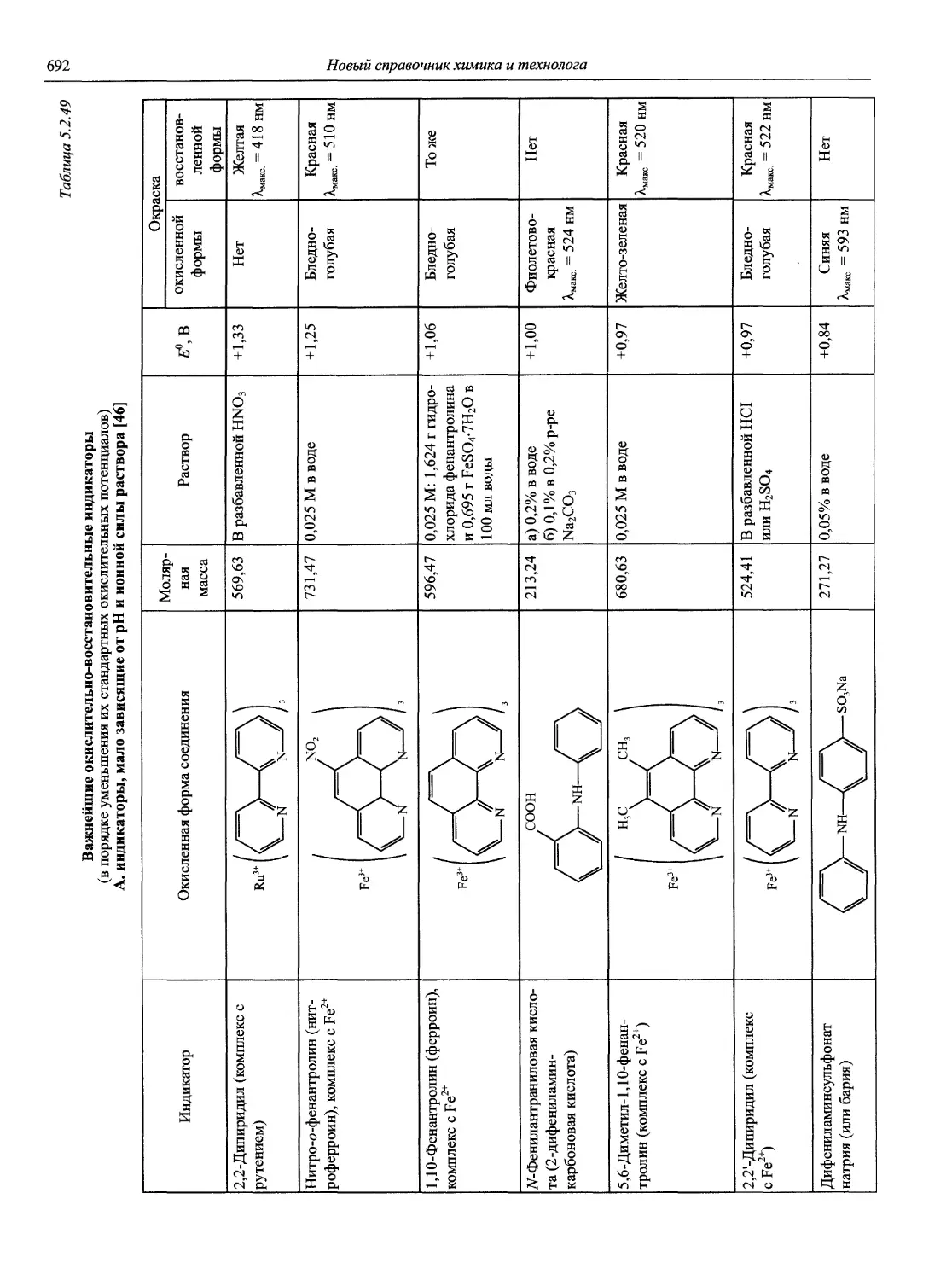
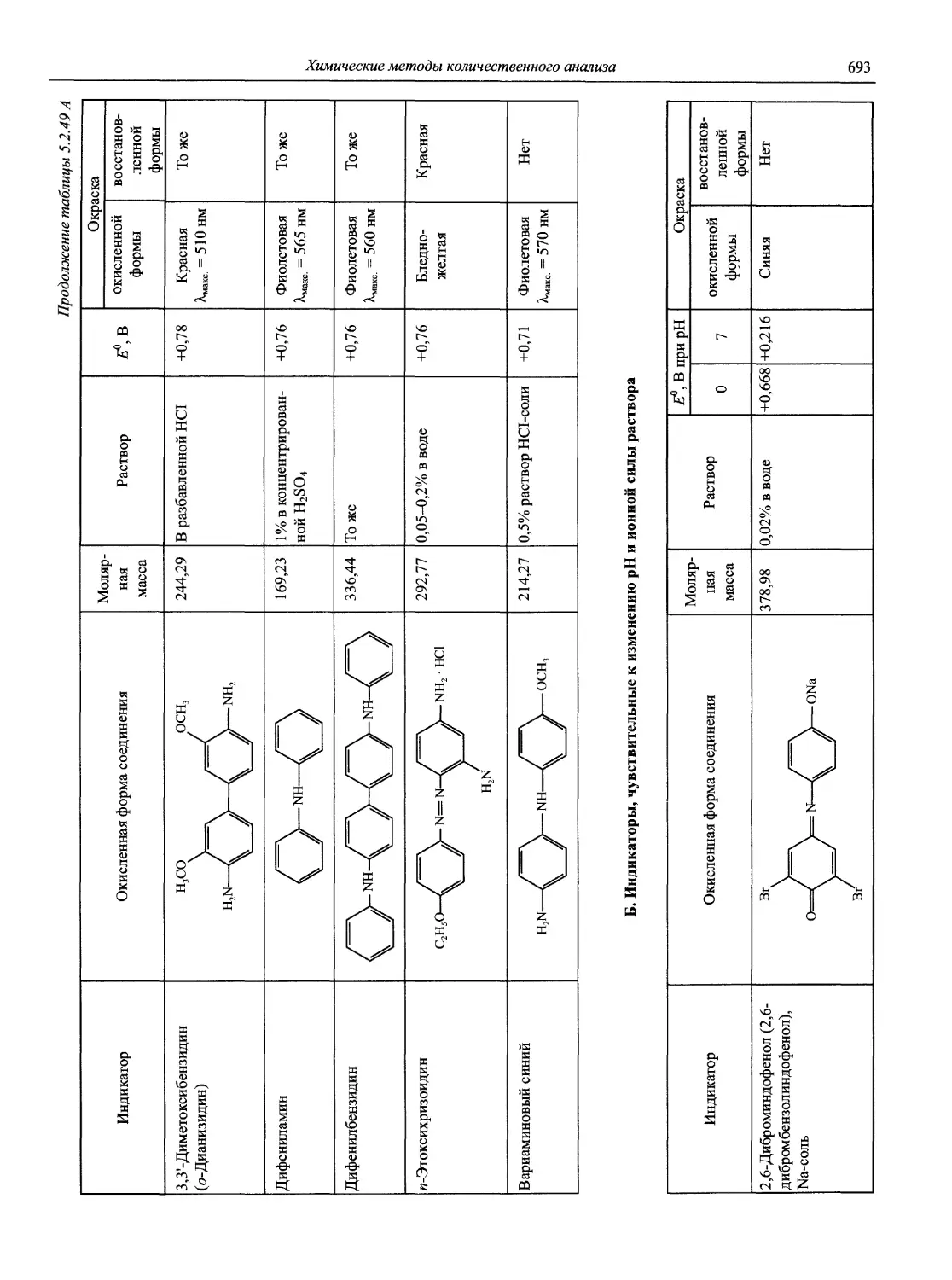


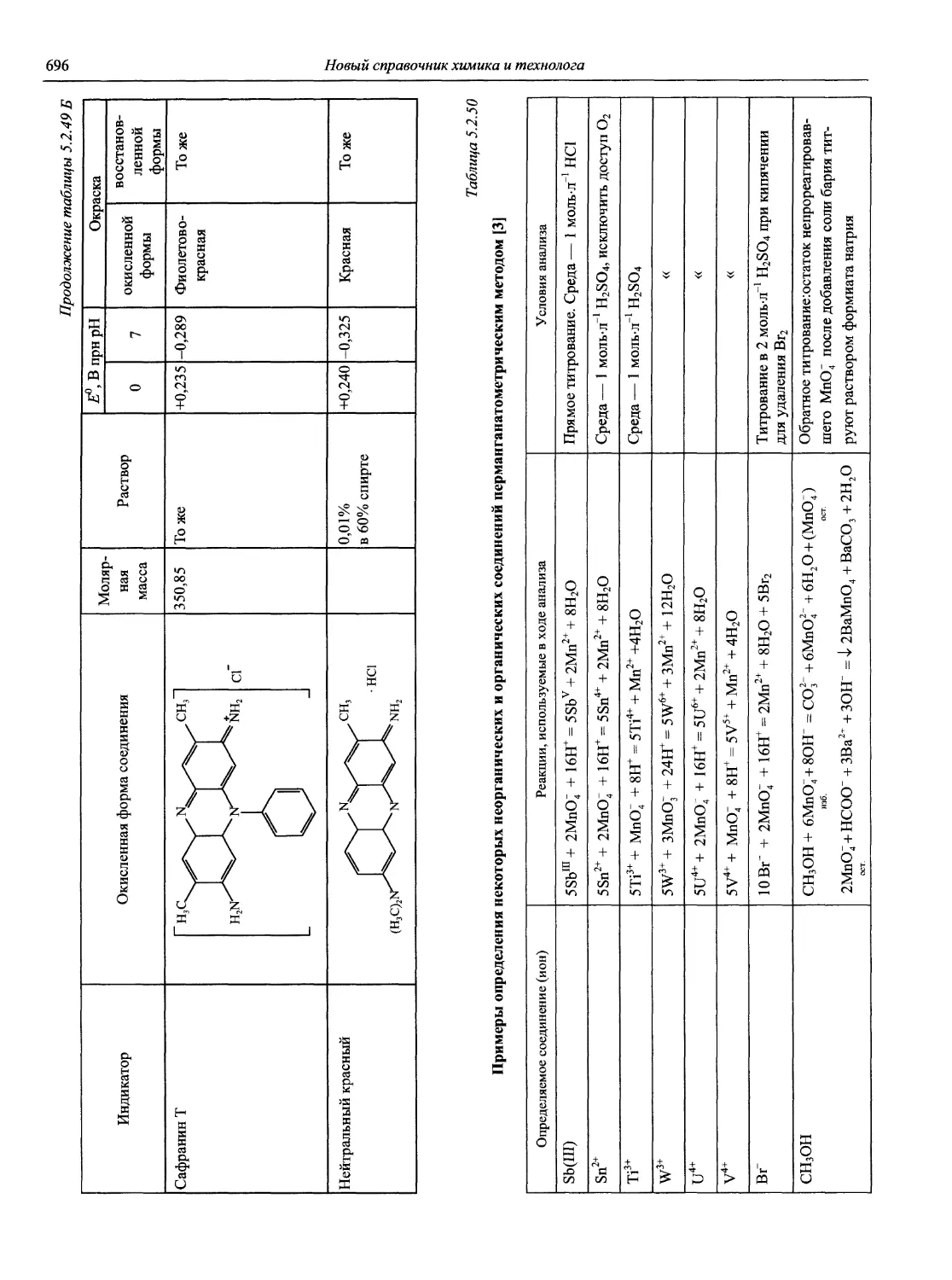


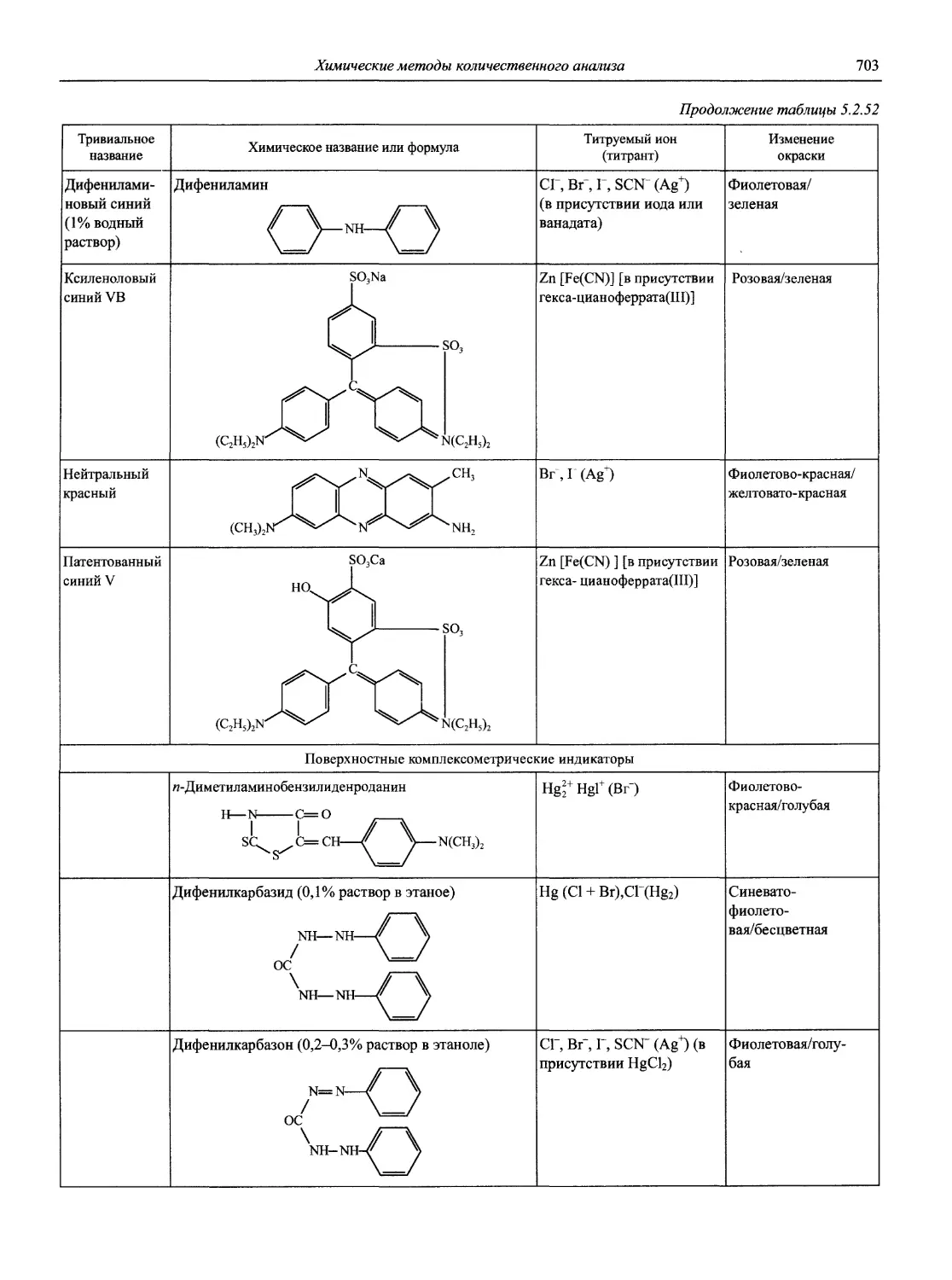
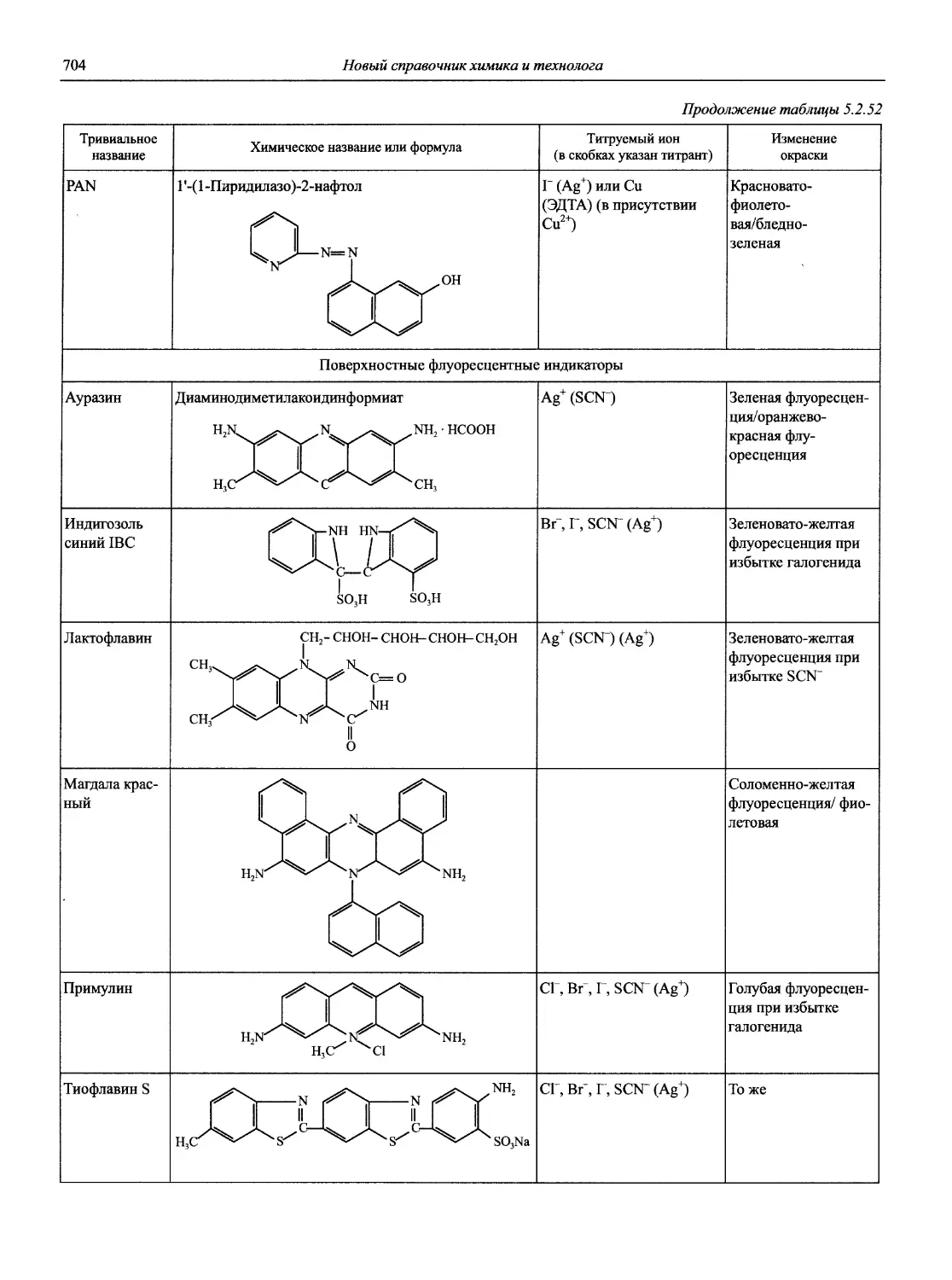
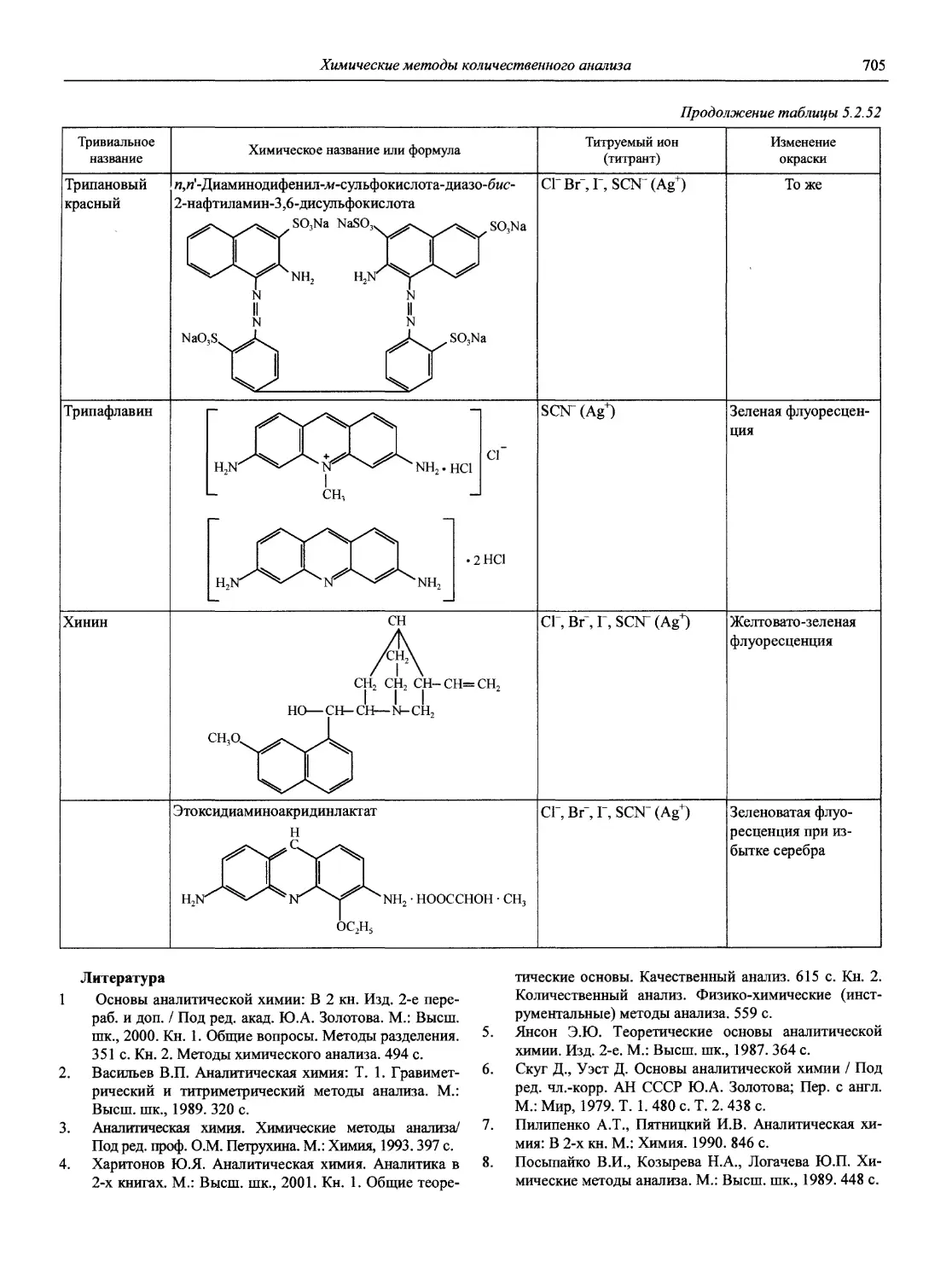








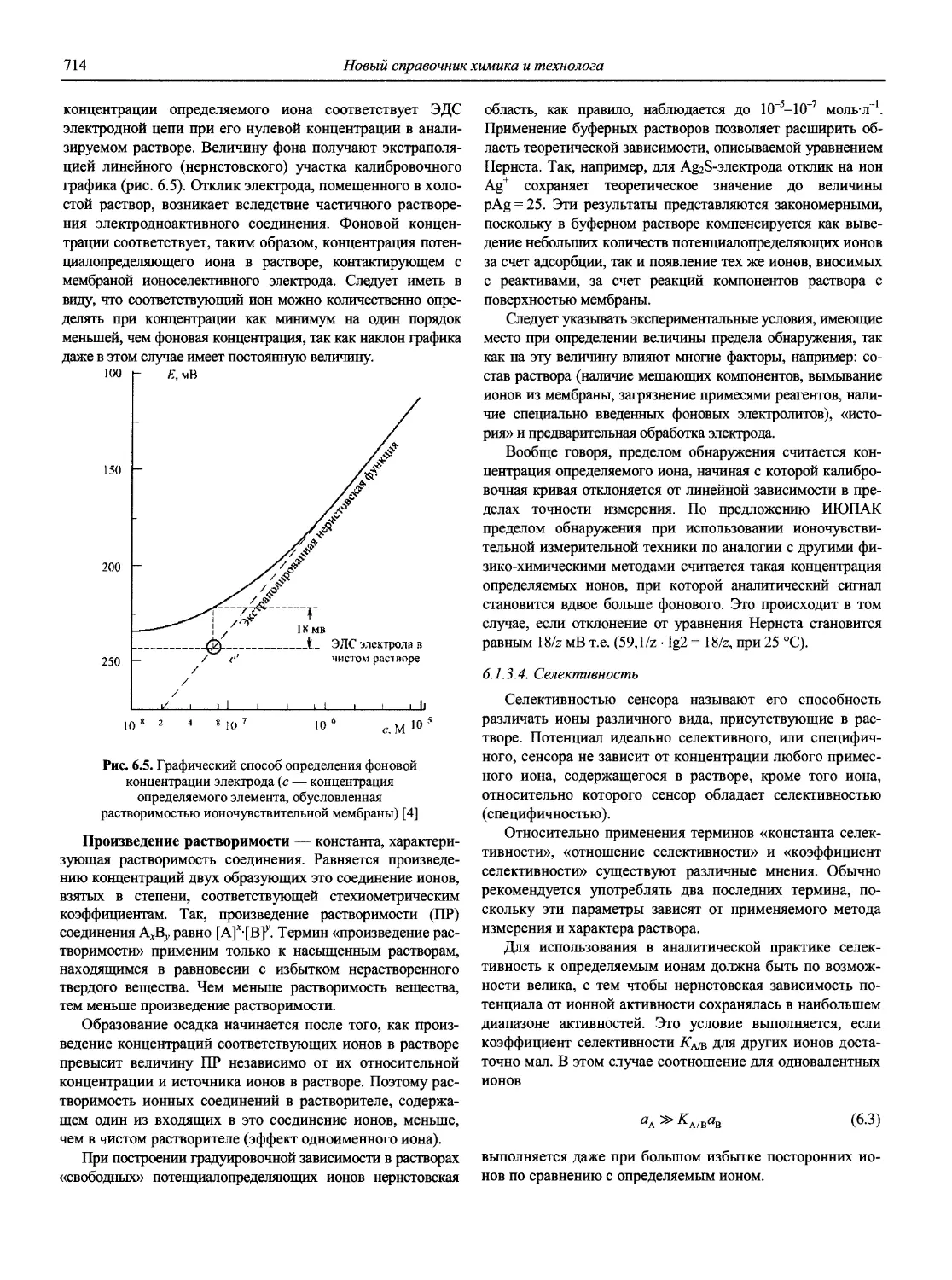
























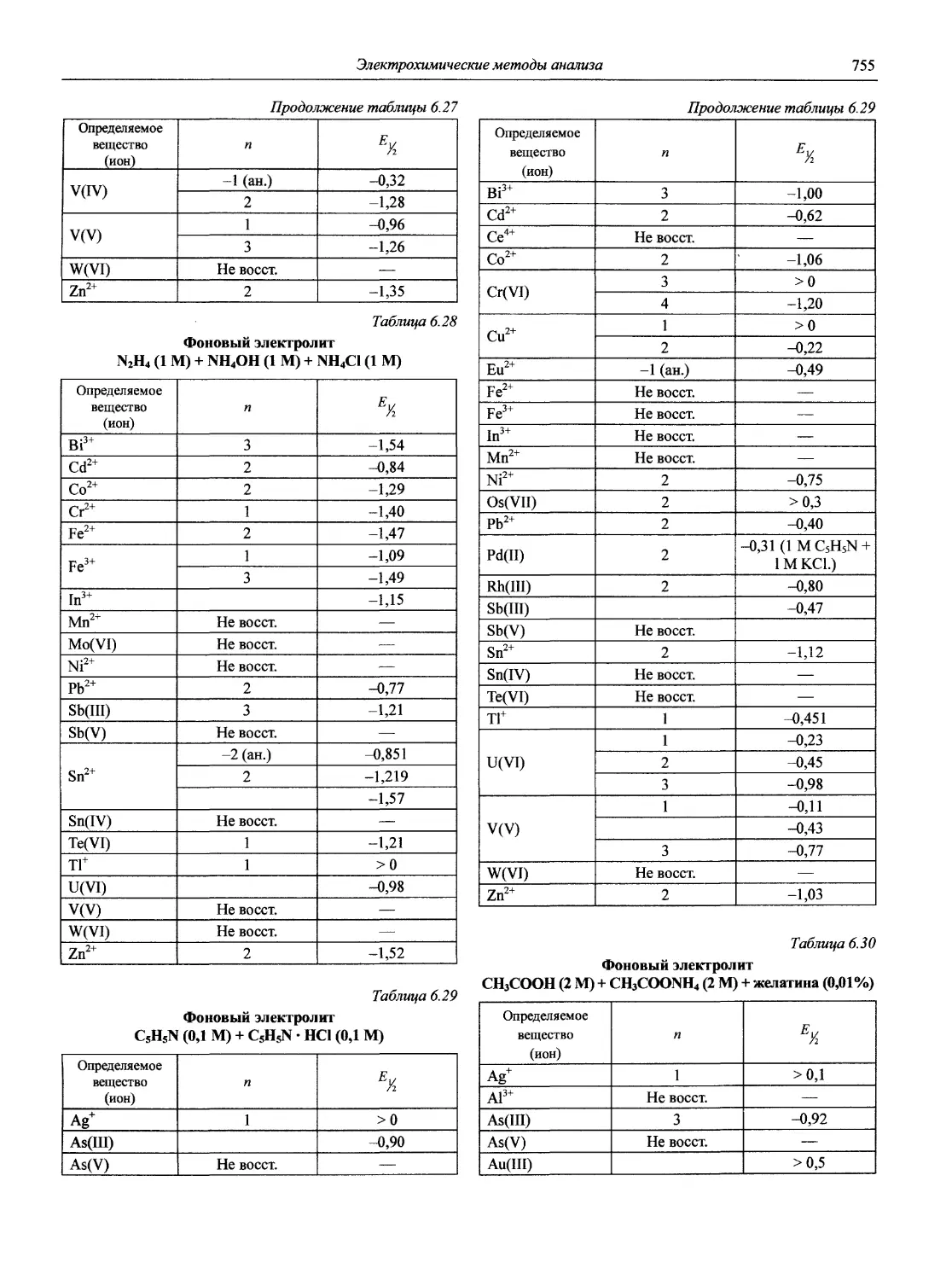
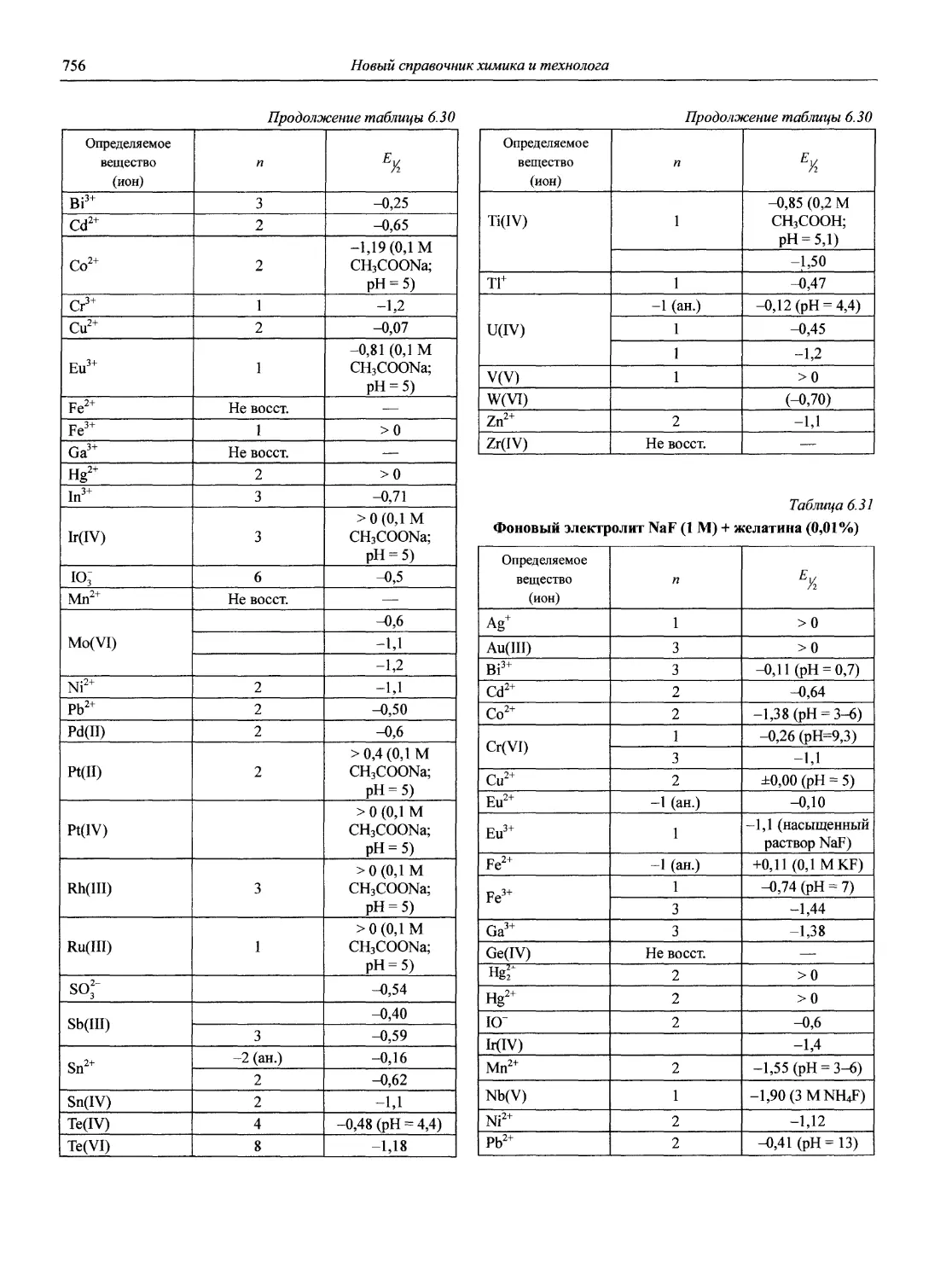
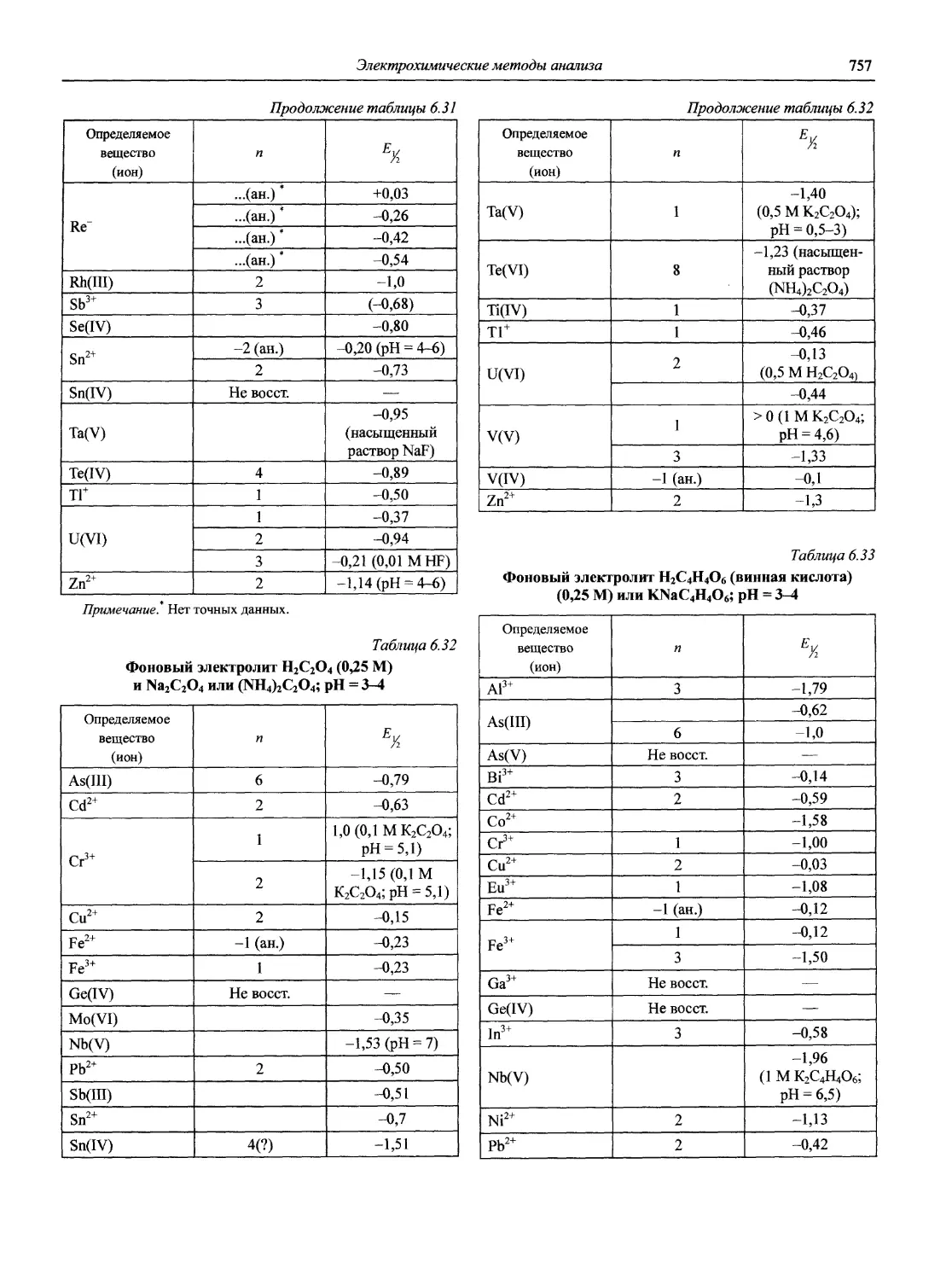
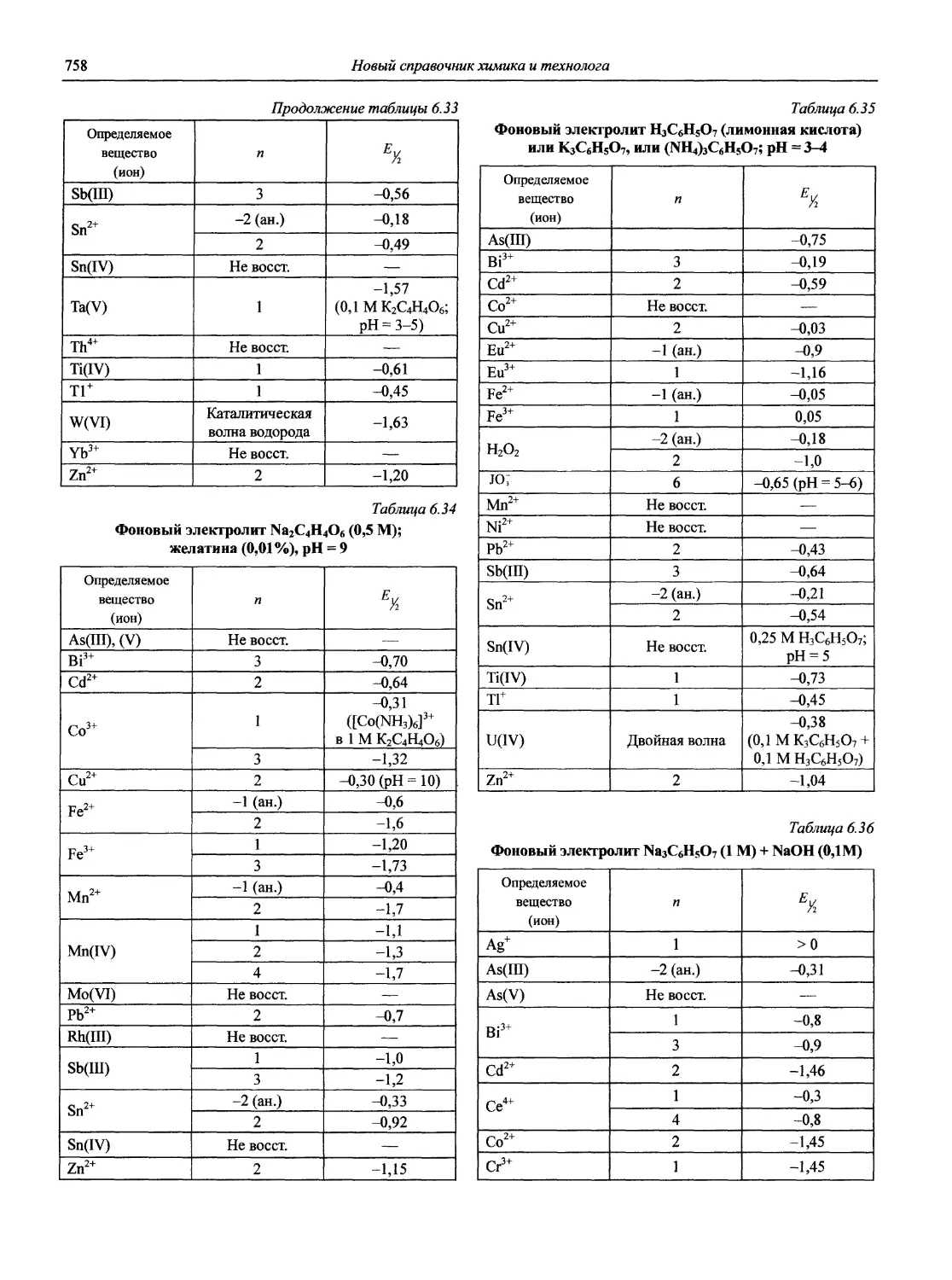




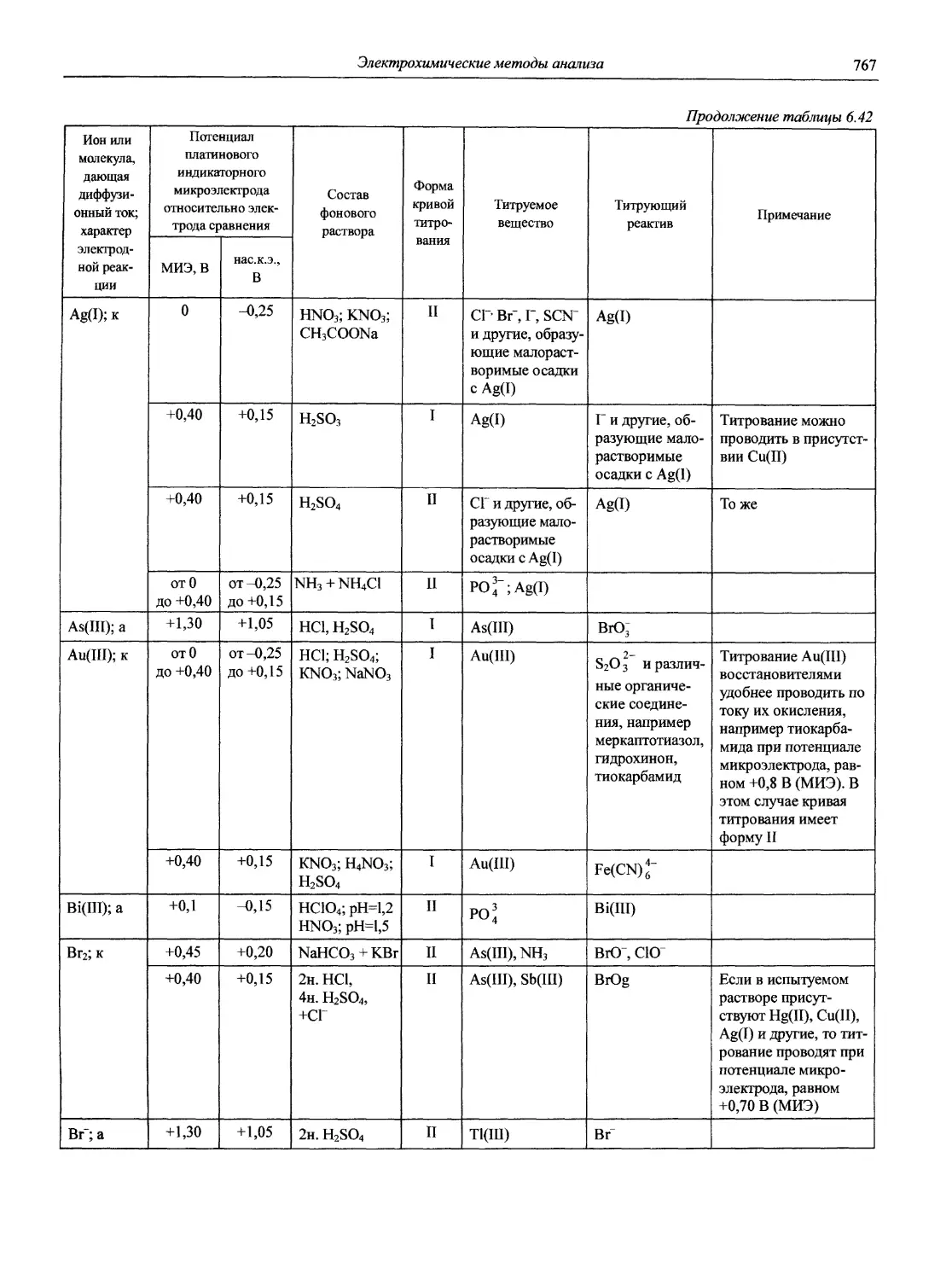
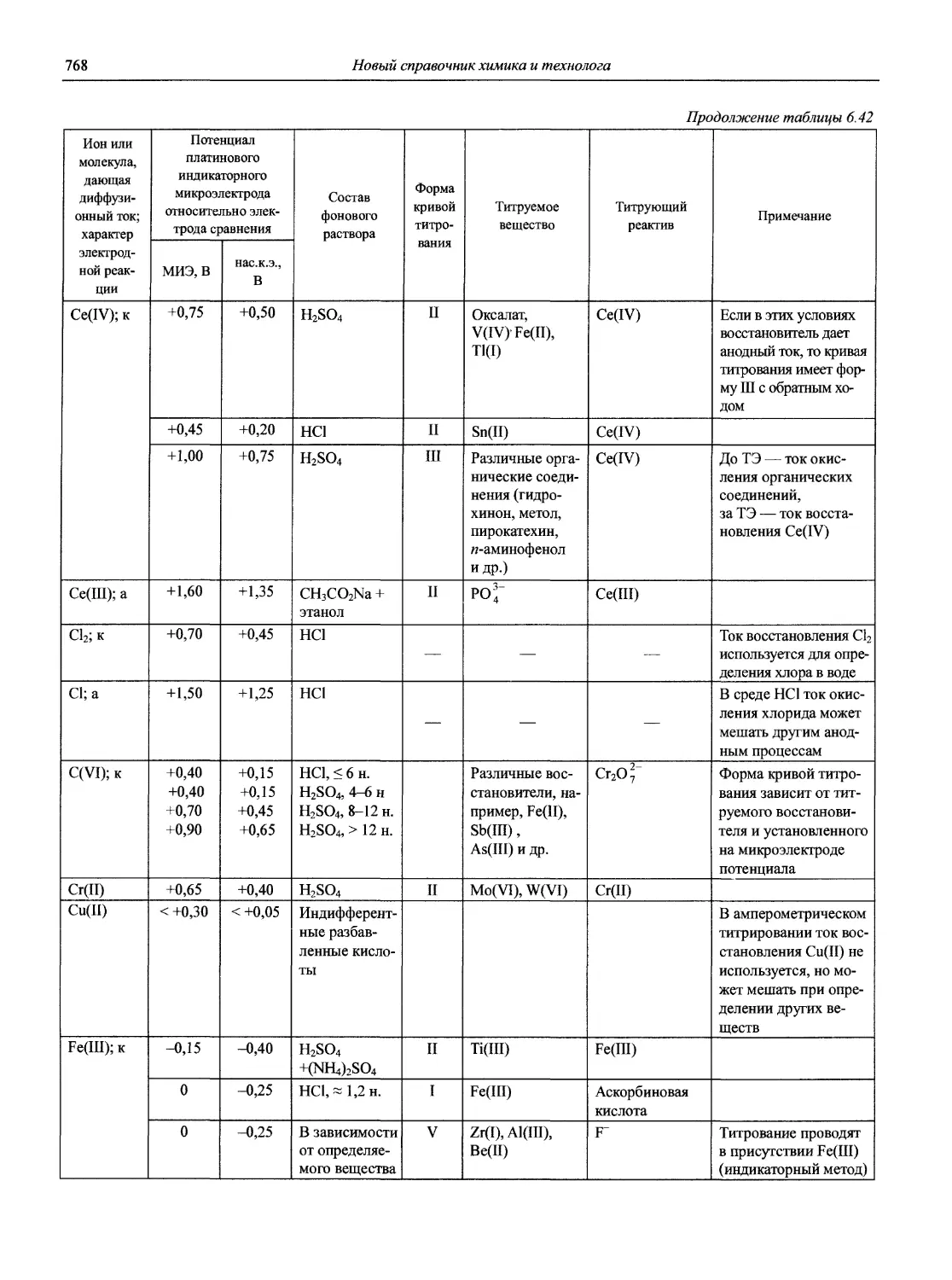



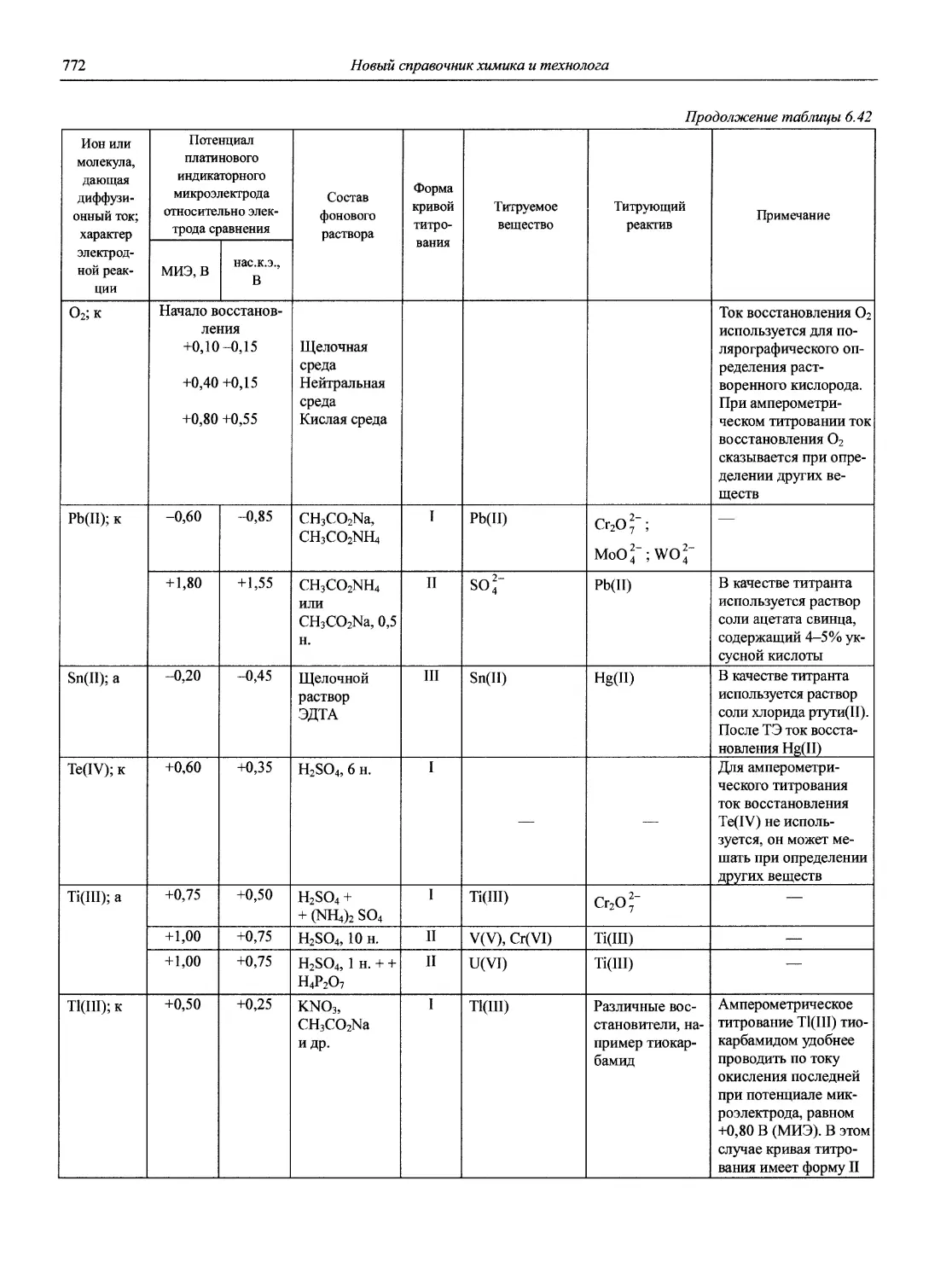
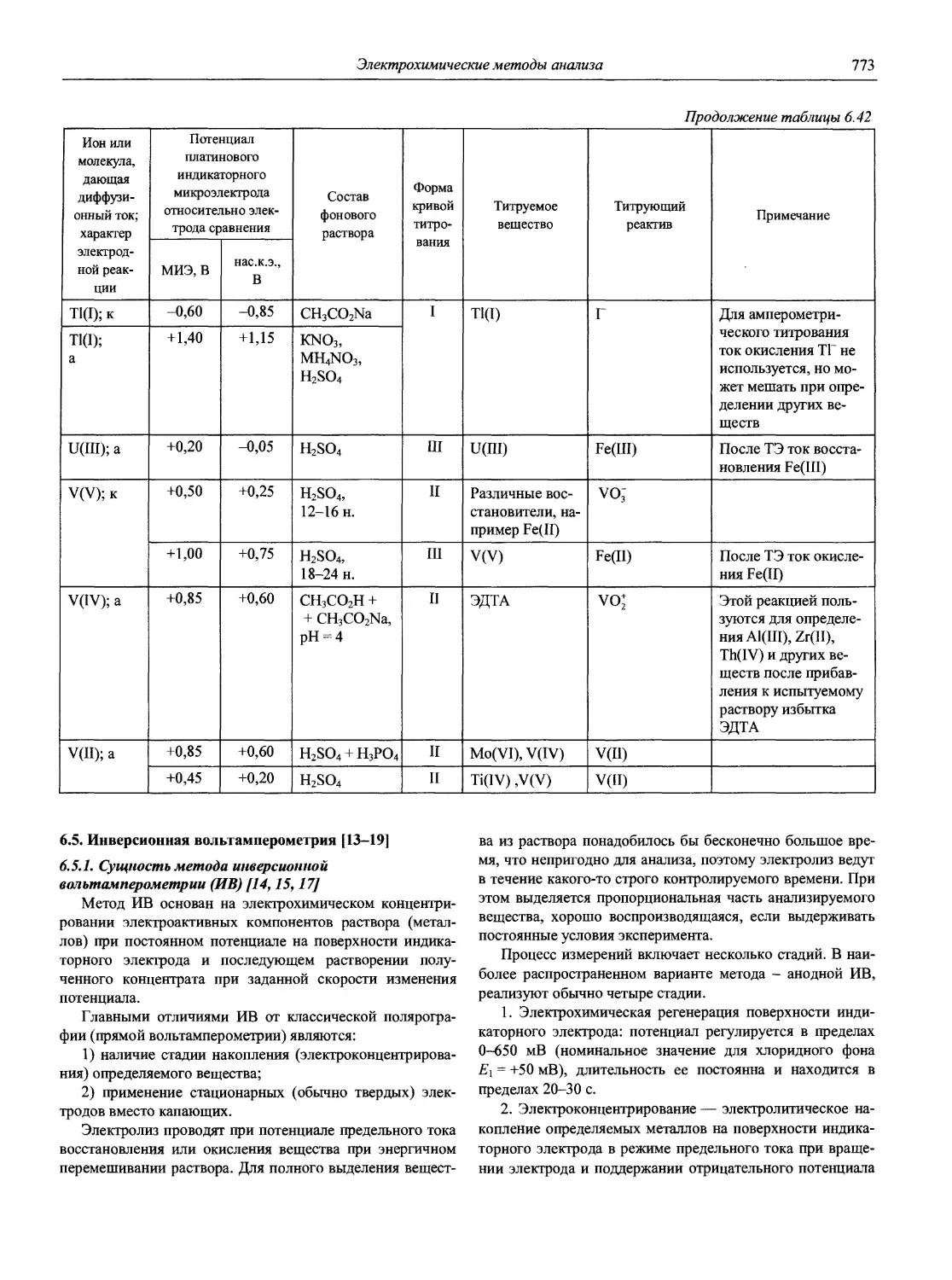


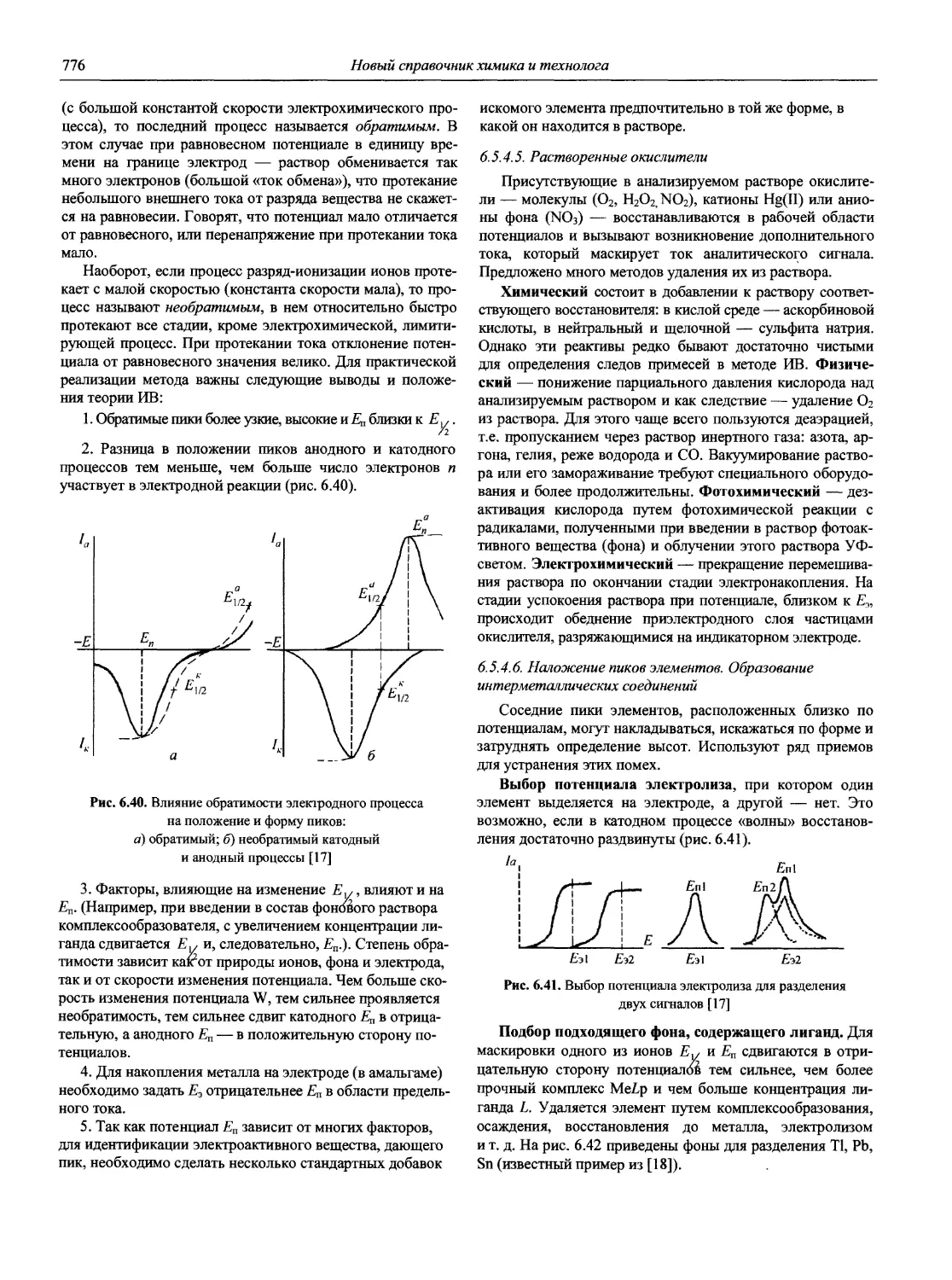

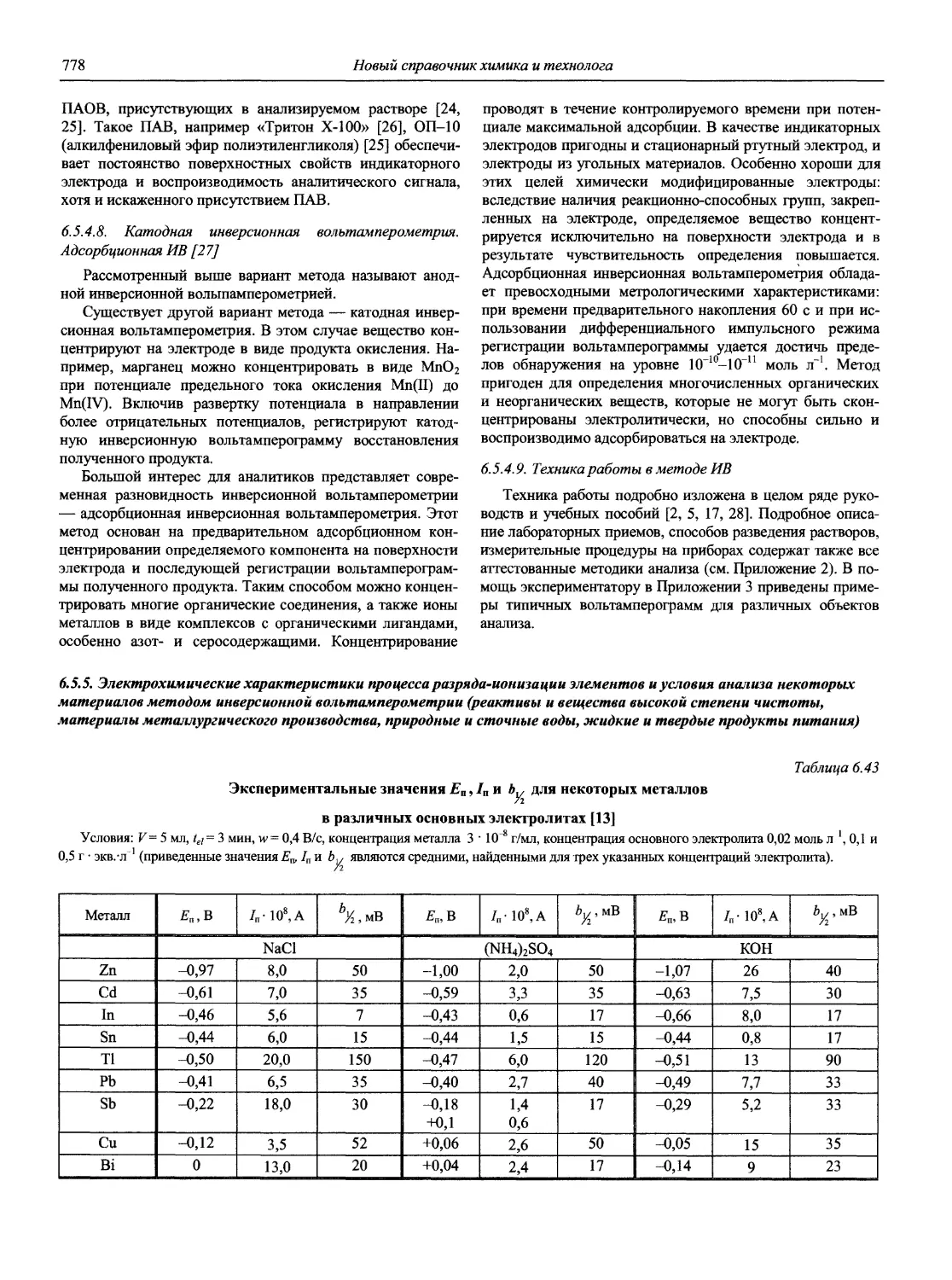

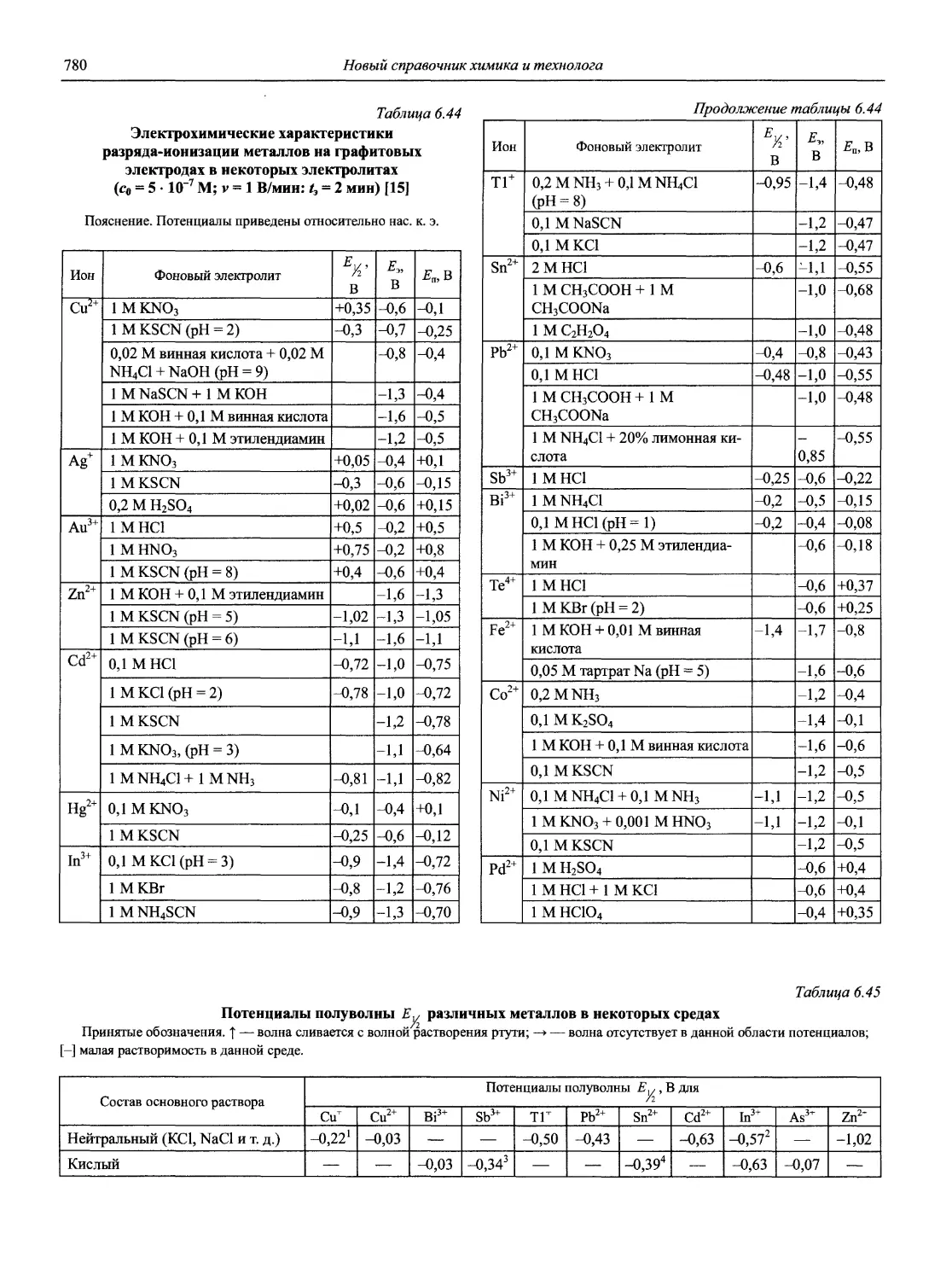
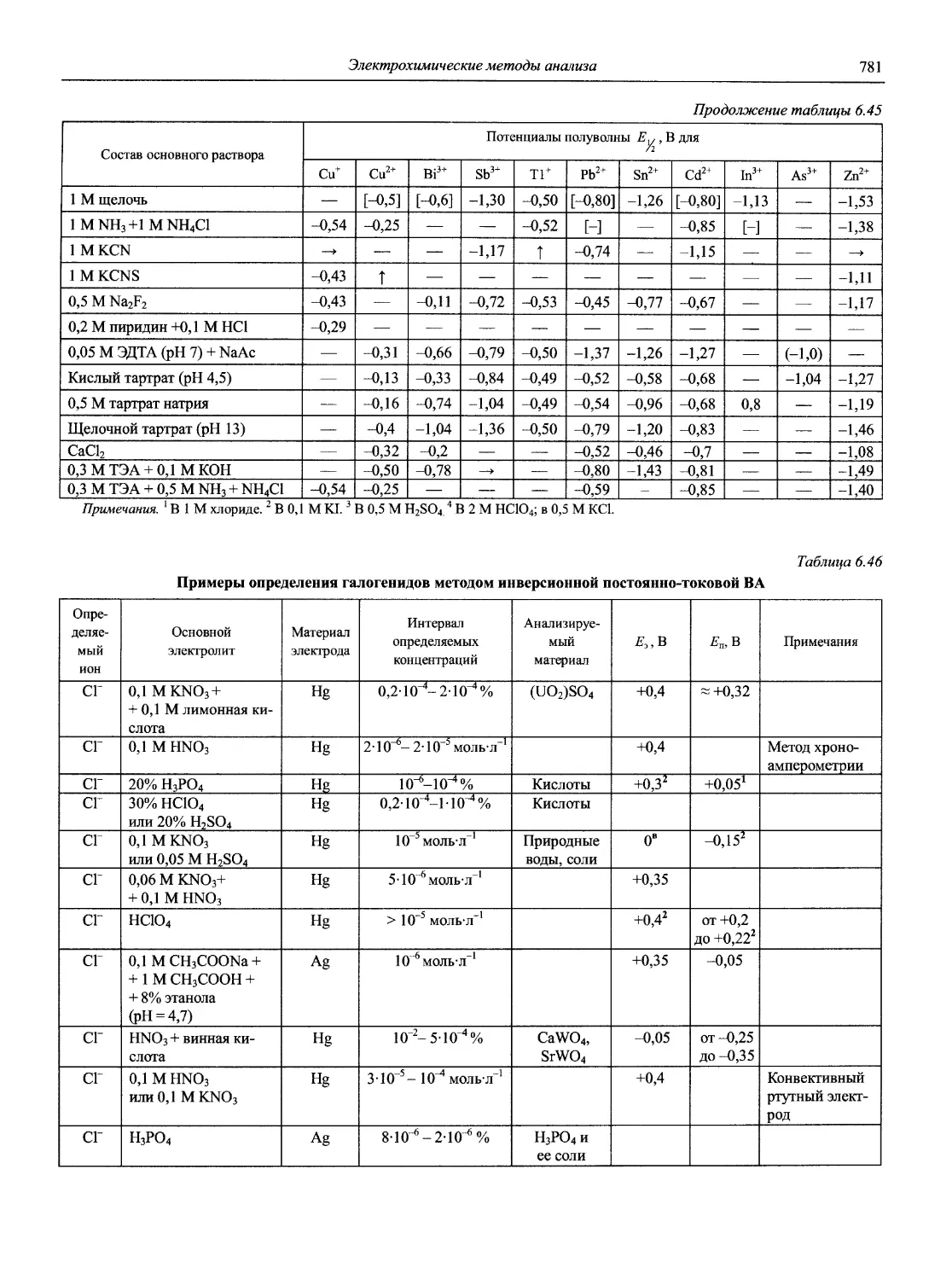




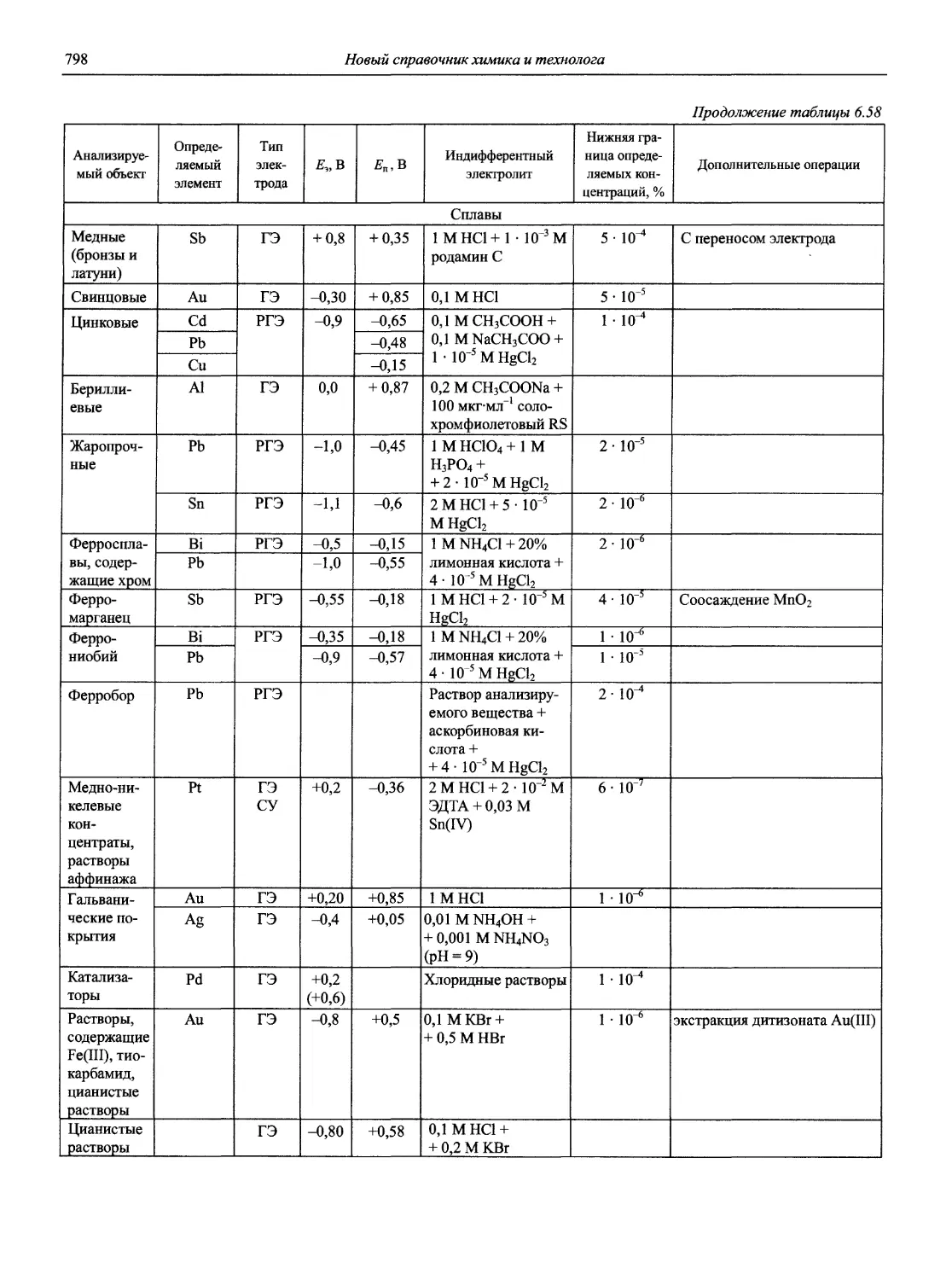

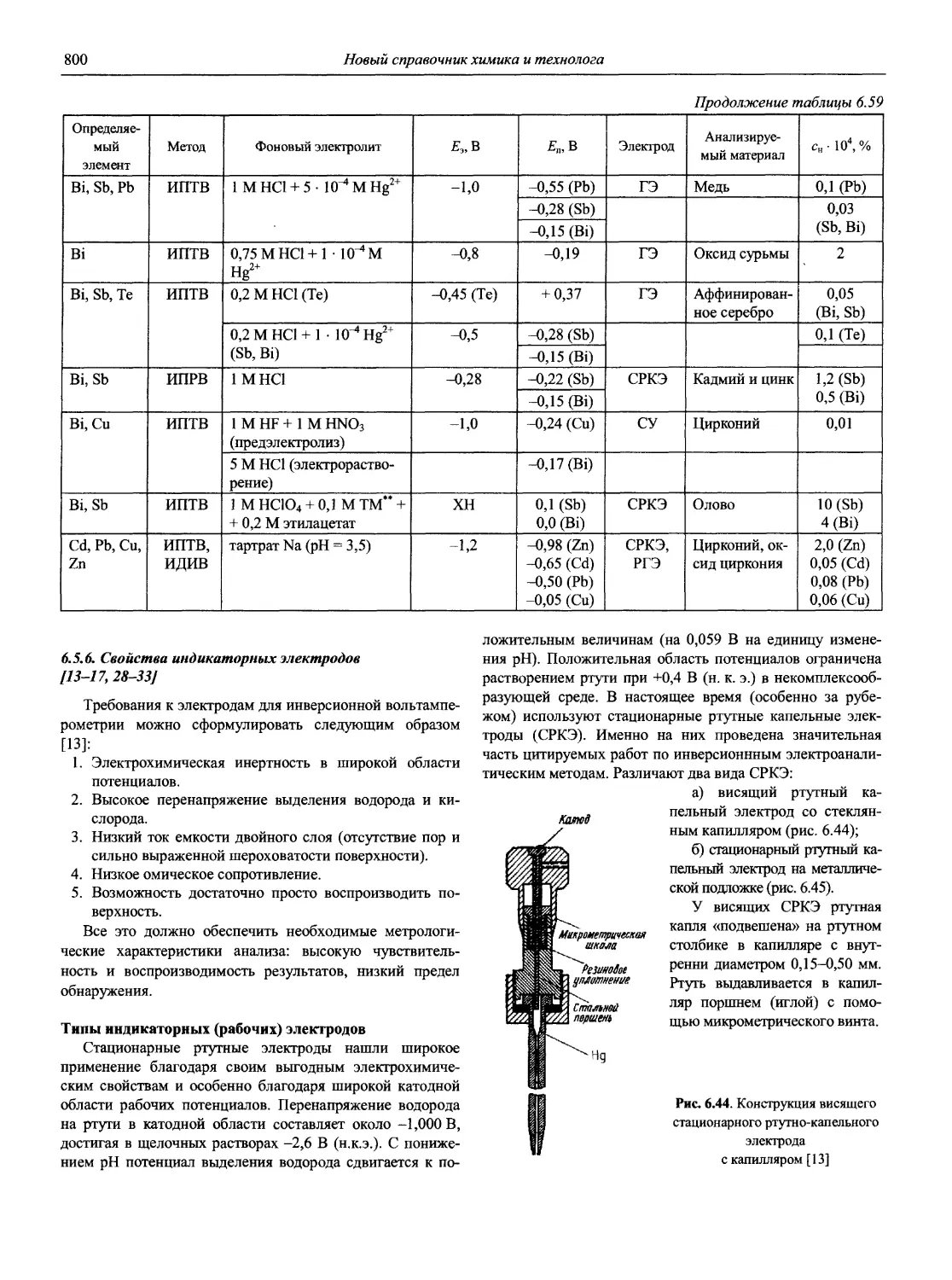


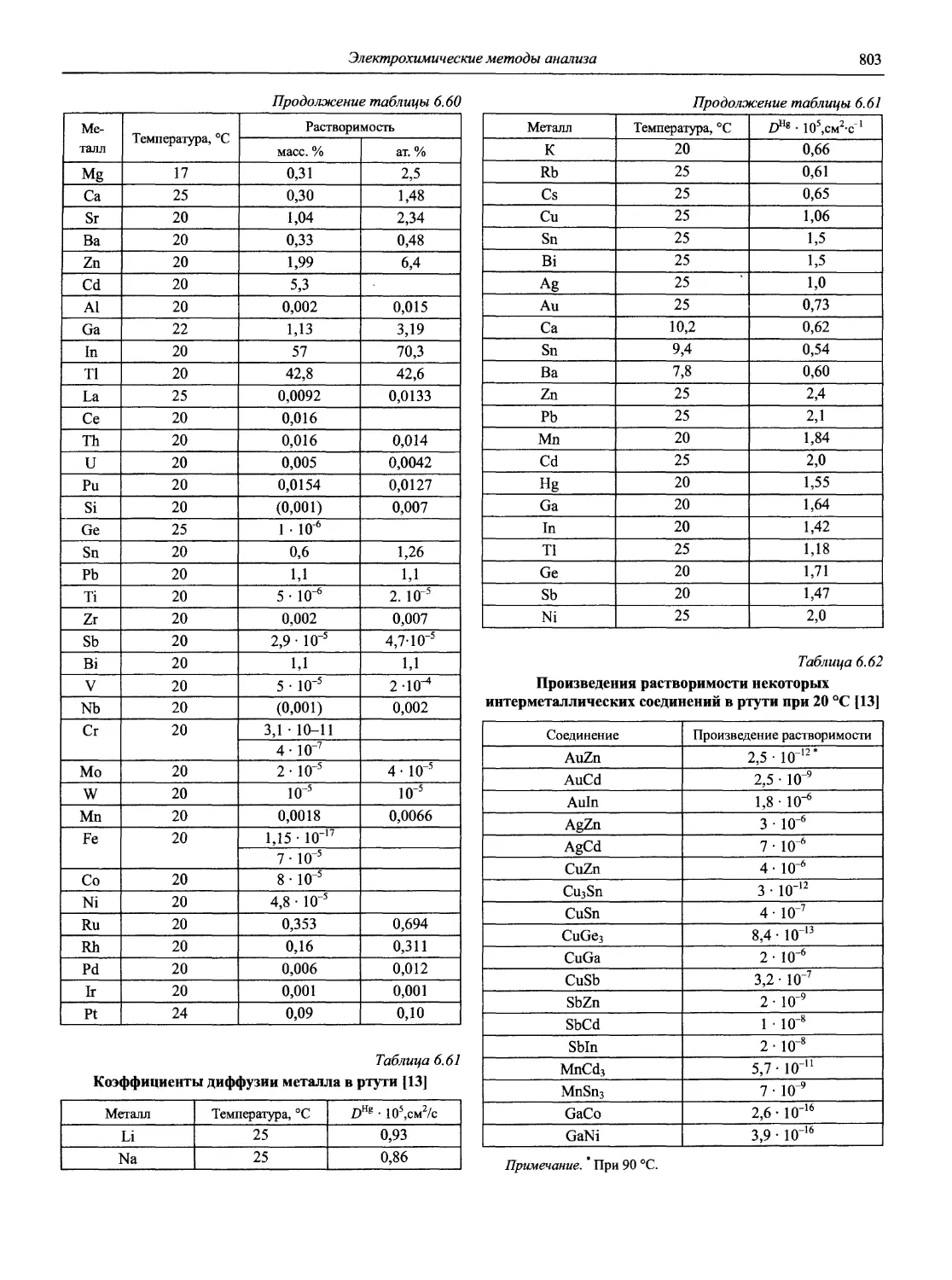


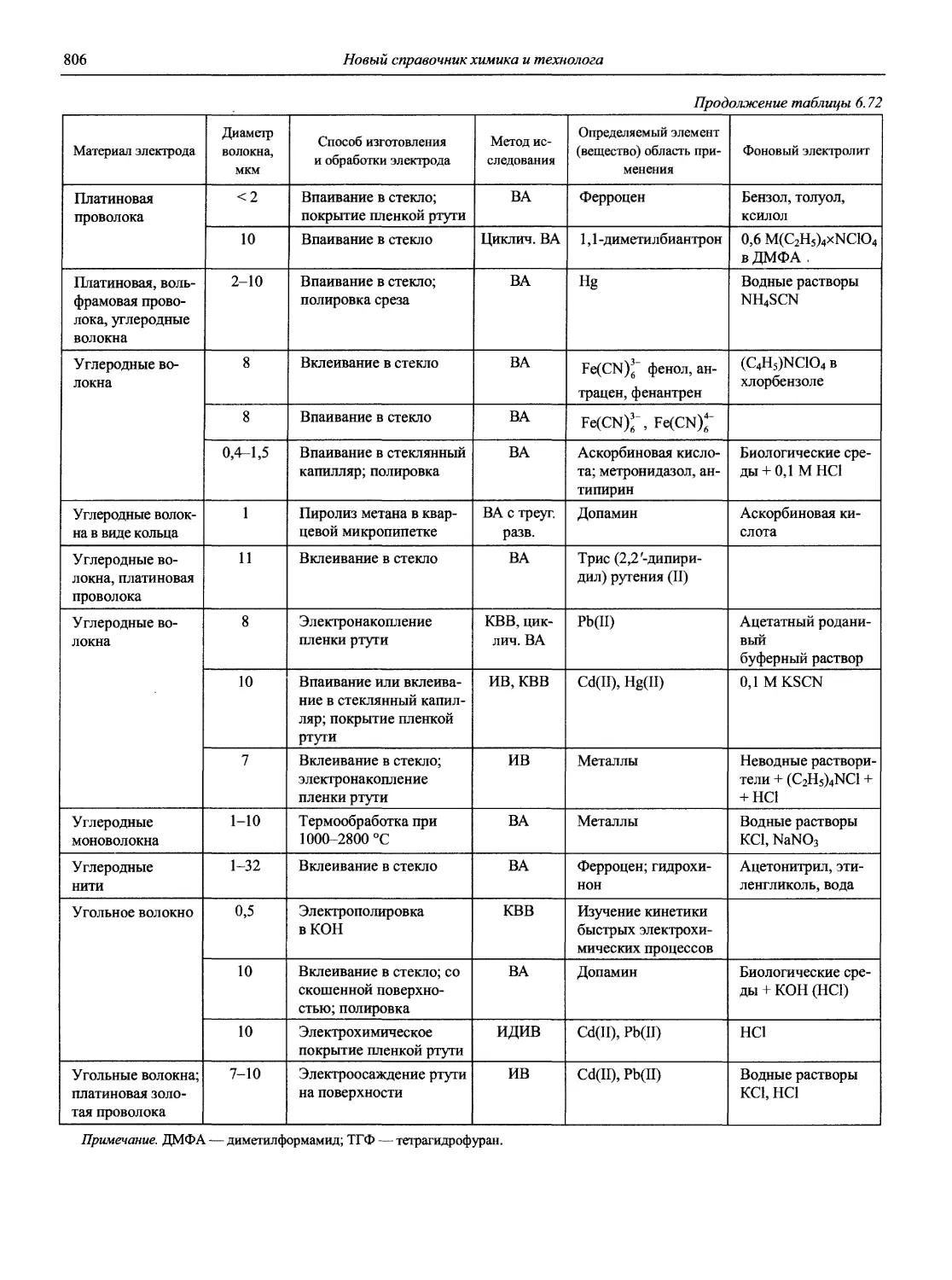



































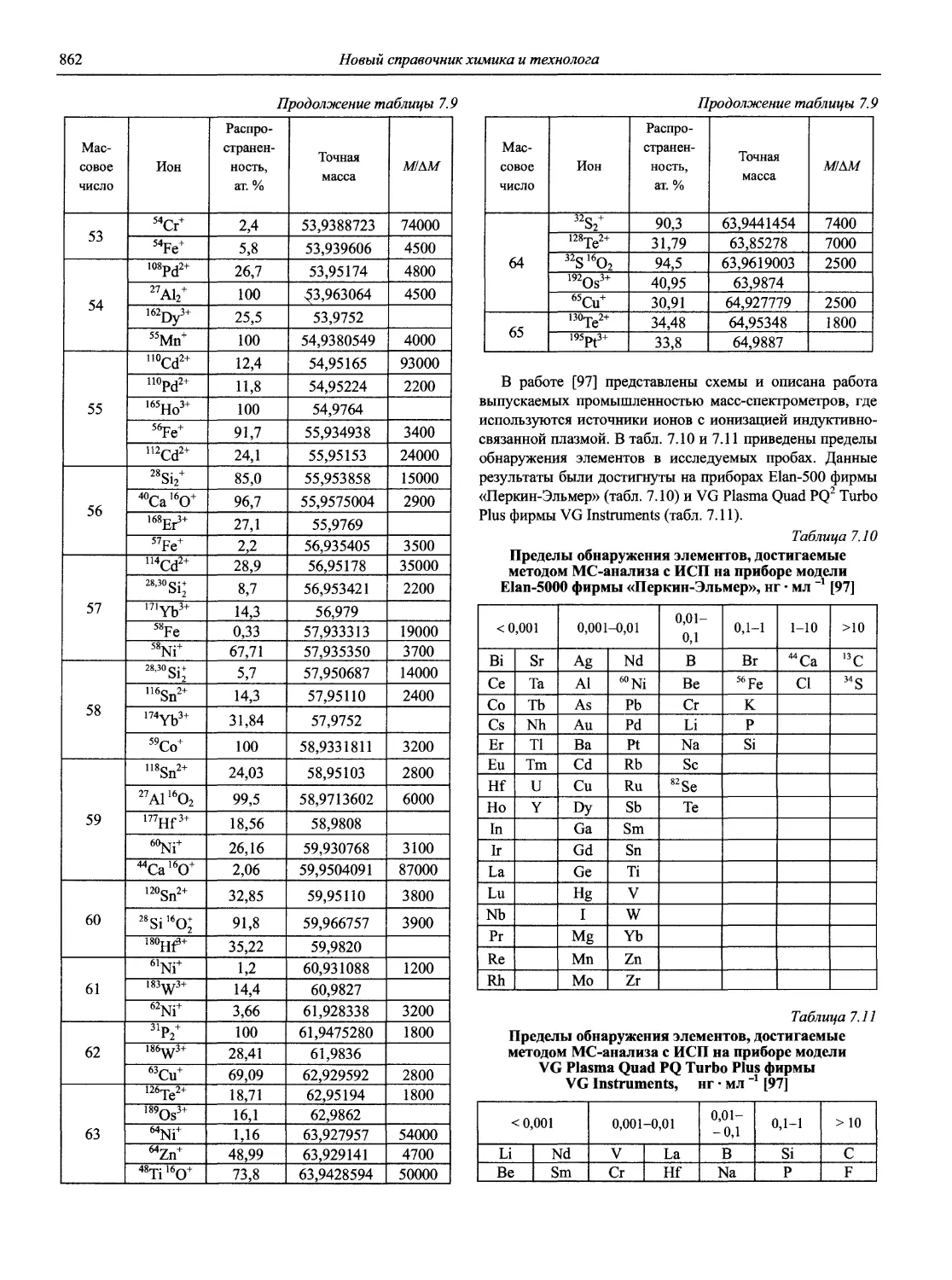




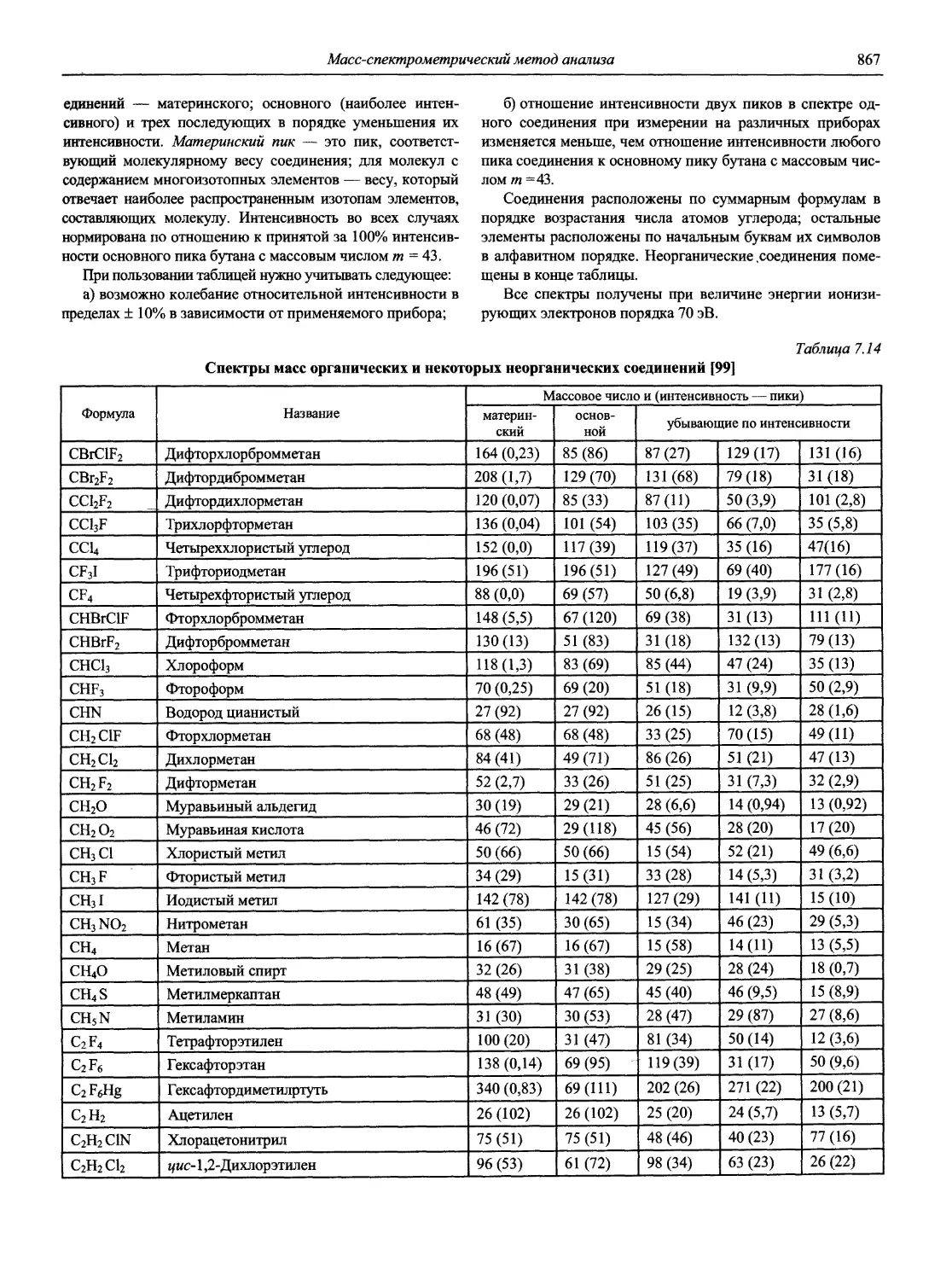
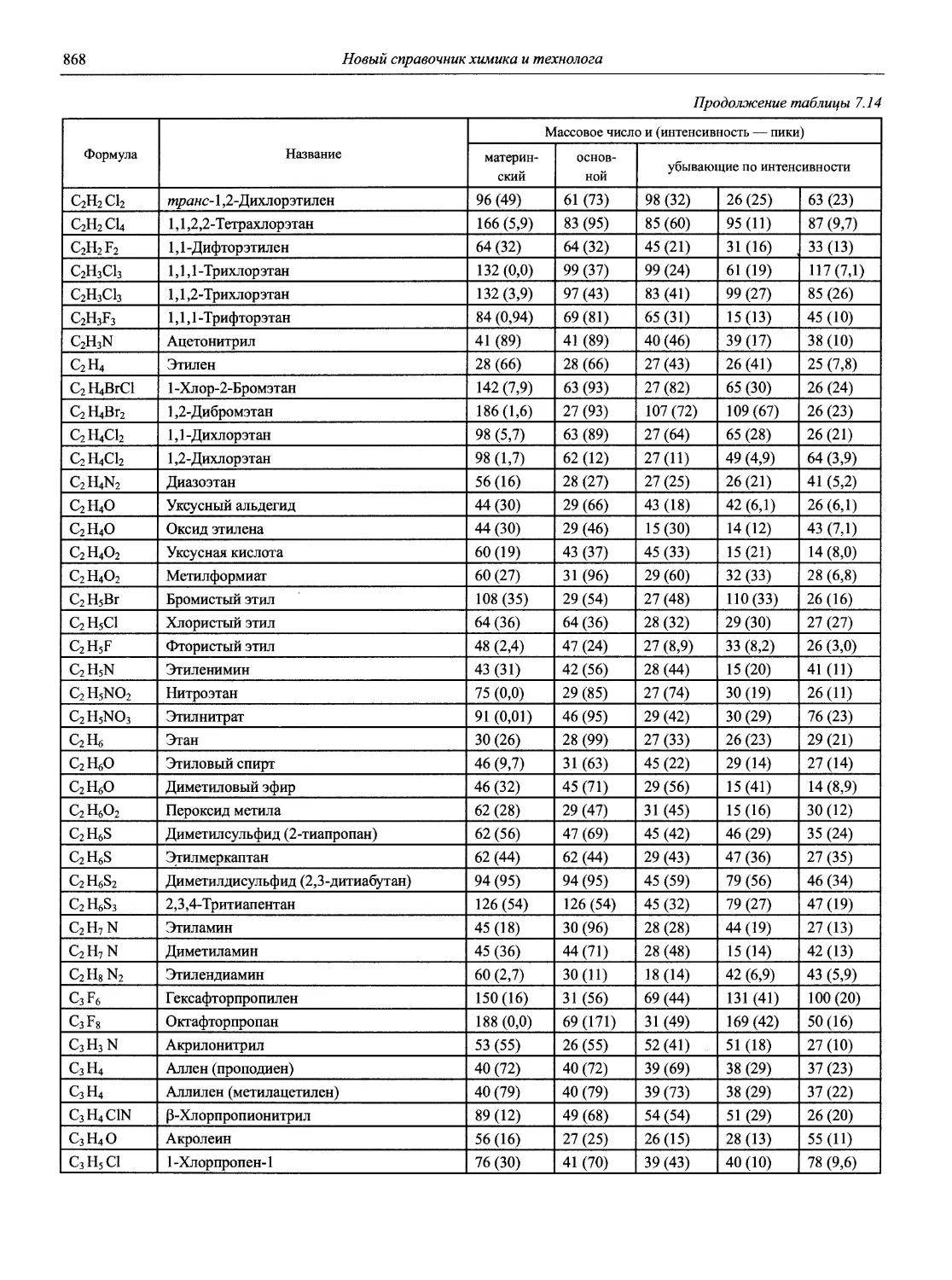
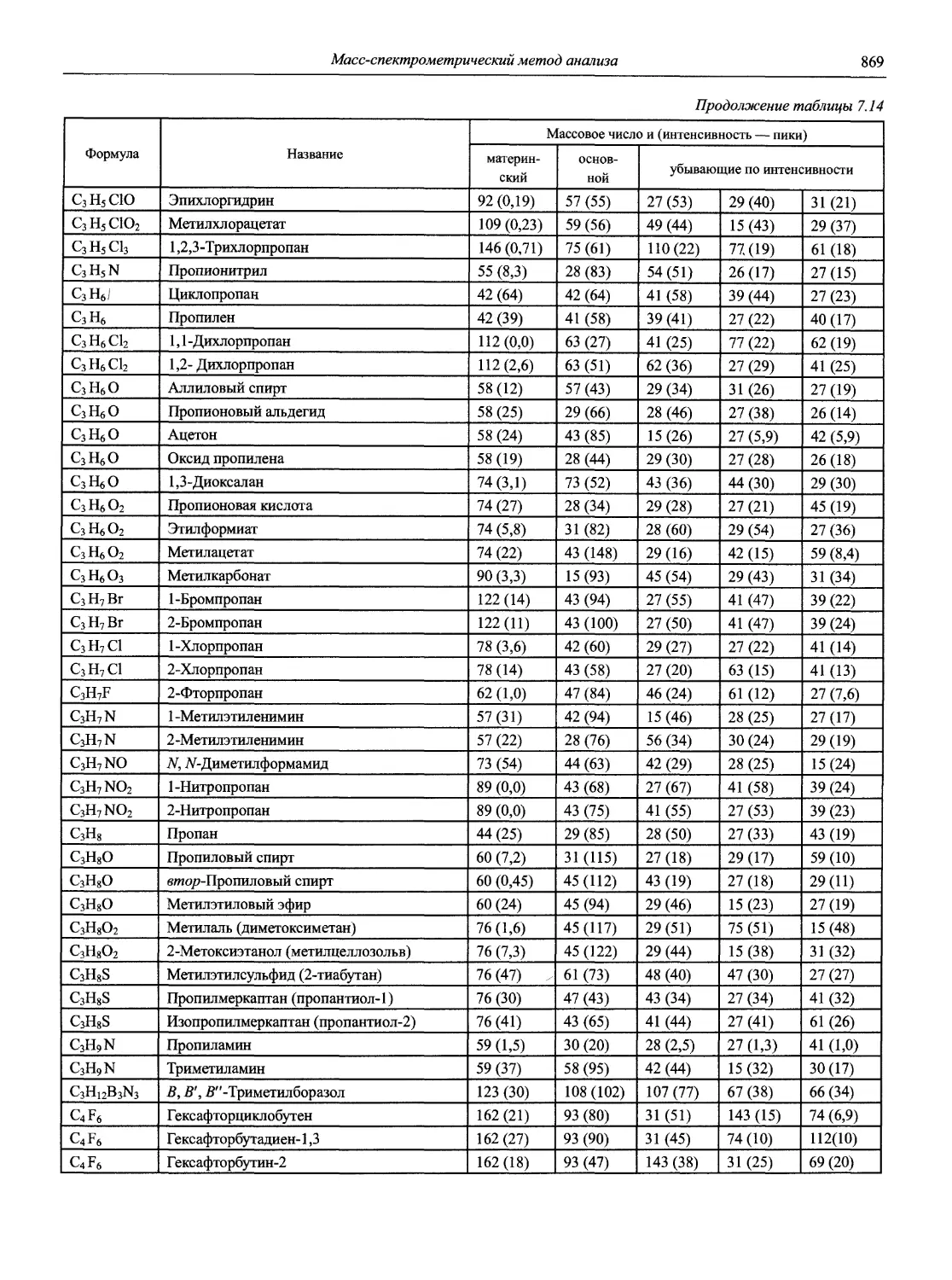






































































Автор: Карцова А.А. Калинкин И.П. Булатов М.И. Мосичев В.И.
Теги: аналитическая химия химия химическая промышленность справочник коллоидная химия химическая кинетика химическая технология химические реакции справочник химика технолога серия профессионал электродные процессы химическая диффузия
ISBN: 5-94365-046-6
Год: 2002




‘ЯО(ВЫ(Й~
СПРАВОЧНИК ХИМИКА и ТЕХНОЛОГА
Аналитическая химия
ББК 24.4
24.46
Н72
Авторы:
доц., к.х.н. Ю.А. Барбалат
проф., акад. РАЕН, д.х.н. Ю.Г. Власов к.х.н. В.А. Демин
проф., д.х.н. Ю.Е. Ермоленко
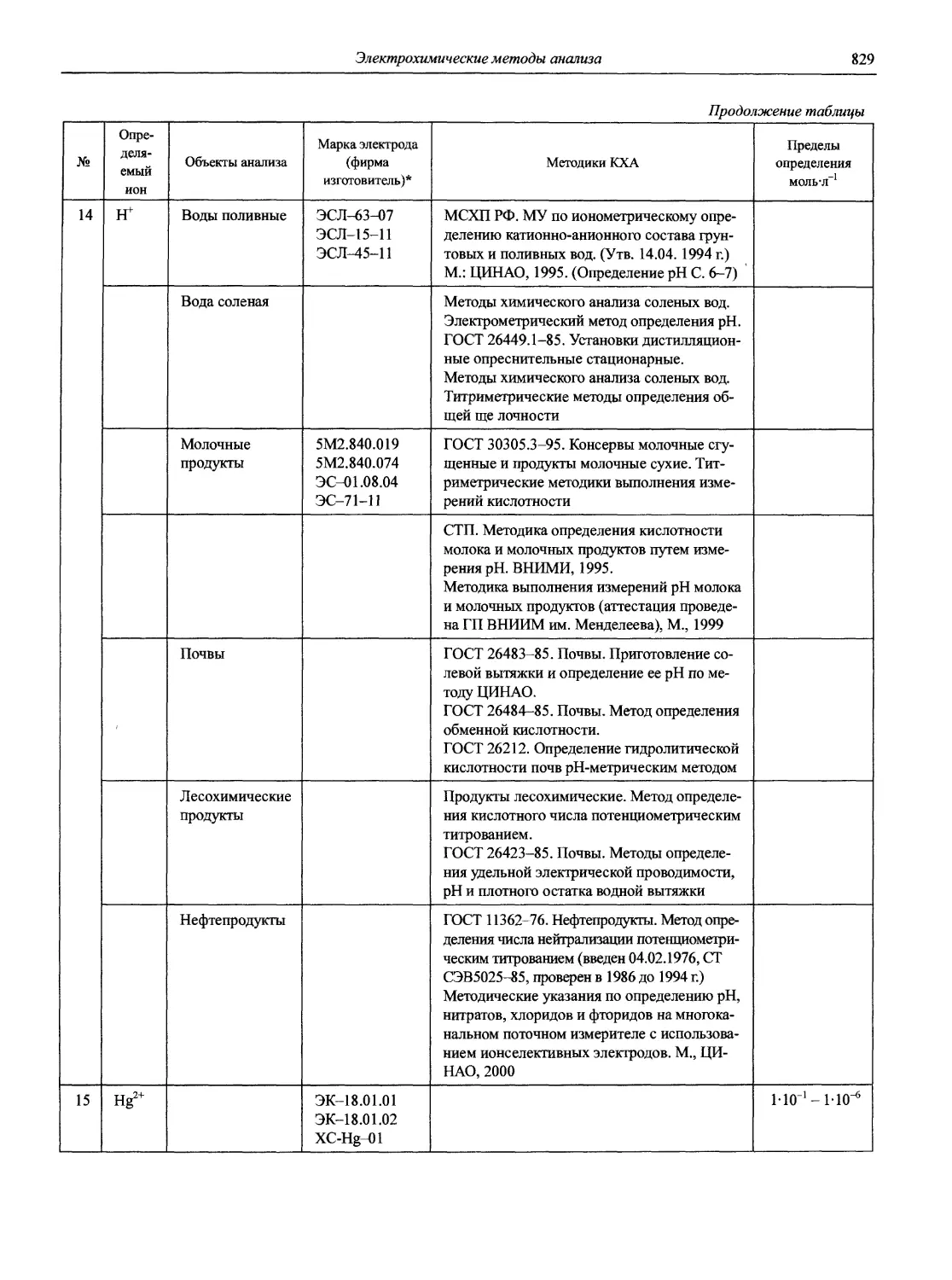
акад. РАН, проф., д.х.н. Ю.А. Золотов проф., д.х.н. В.М. Иванов
проф., д.х.н. И.П. Калинкин
проф. д.х.н. А.А. Карцова н.с. В.В. Колодников
к.х.н. В.И. Мосичев
проф., акад. РАЕН, д.х.н., засл. деят. науки Л.Н. Москвин
|Мир и Семья, Профессионал, СПб
проф., д.т.н. В.М. Немец
к.х.н. Т.Г. Никитина
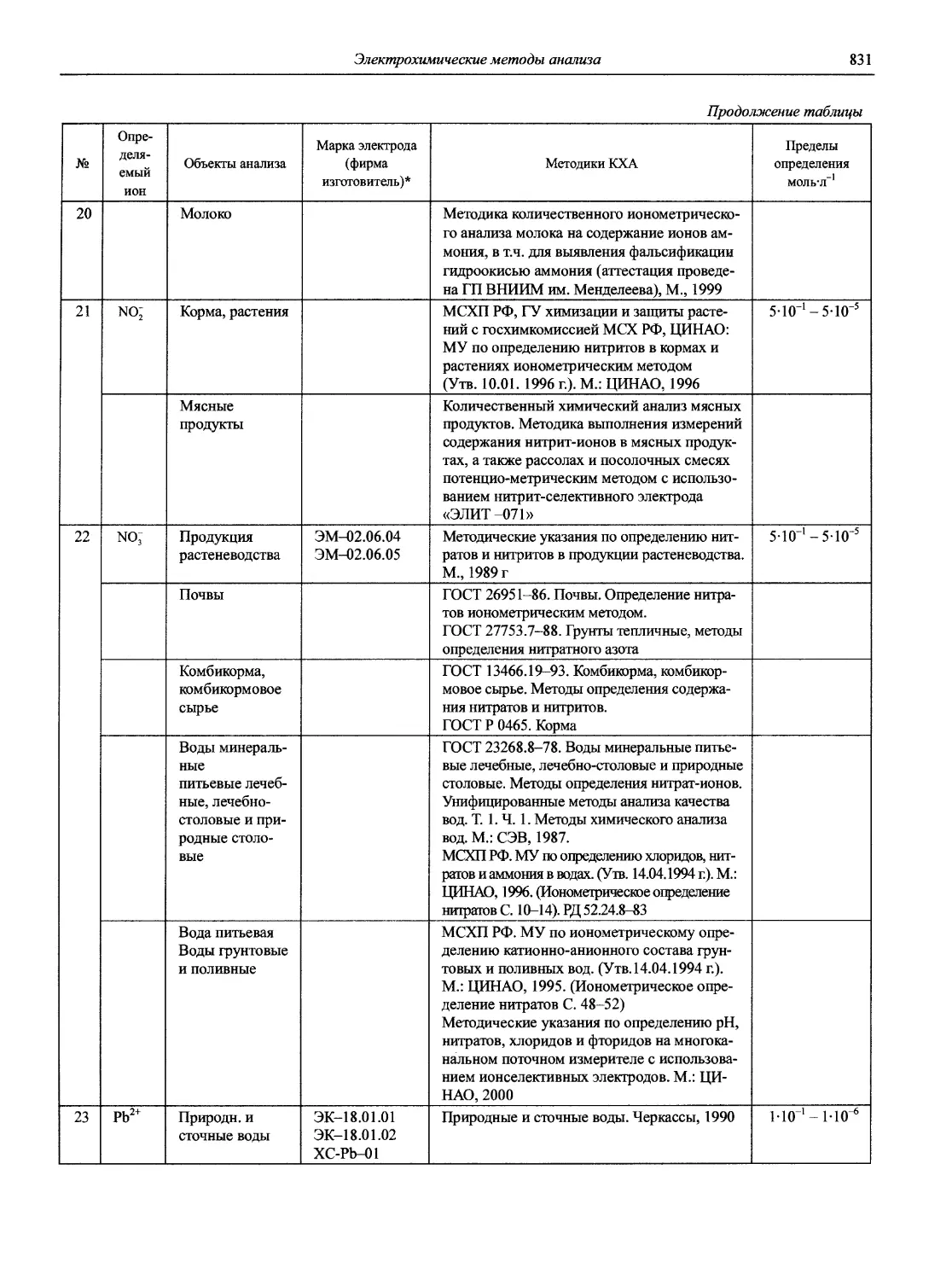
доц., к.х.н. О.В. Родинков
проф., д.х.н. Г.В. Саидов
доц., к.ф-м.н. О.В. Свердлова
с.н.с., к.т.н. А.А. Соловьев
доц., к.х.н. В.Ф. Теплых
доц., к.х.н. А. С. Халонин
доц., к.х.н. Н.М. Якимова
к.х.н. А.Я. Яшин
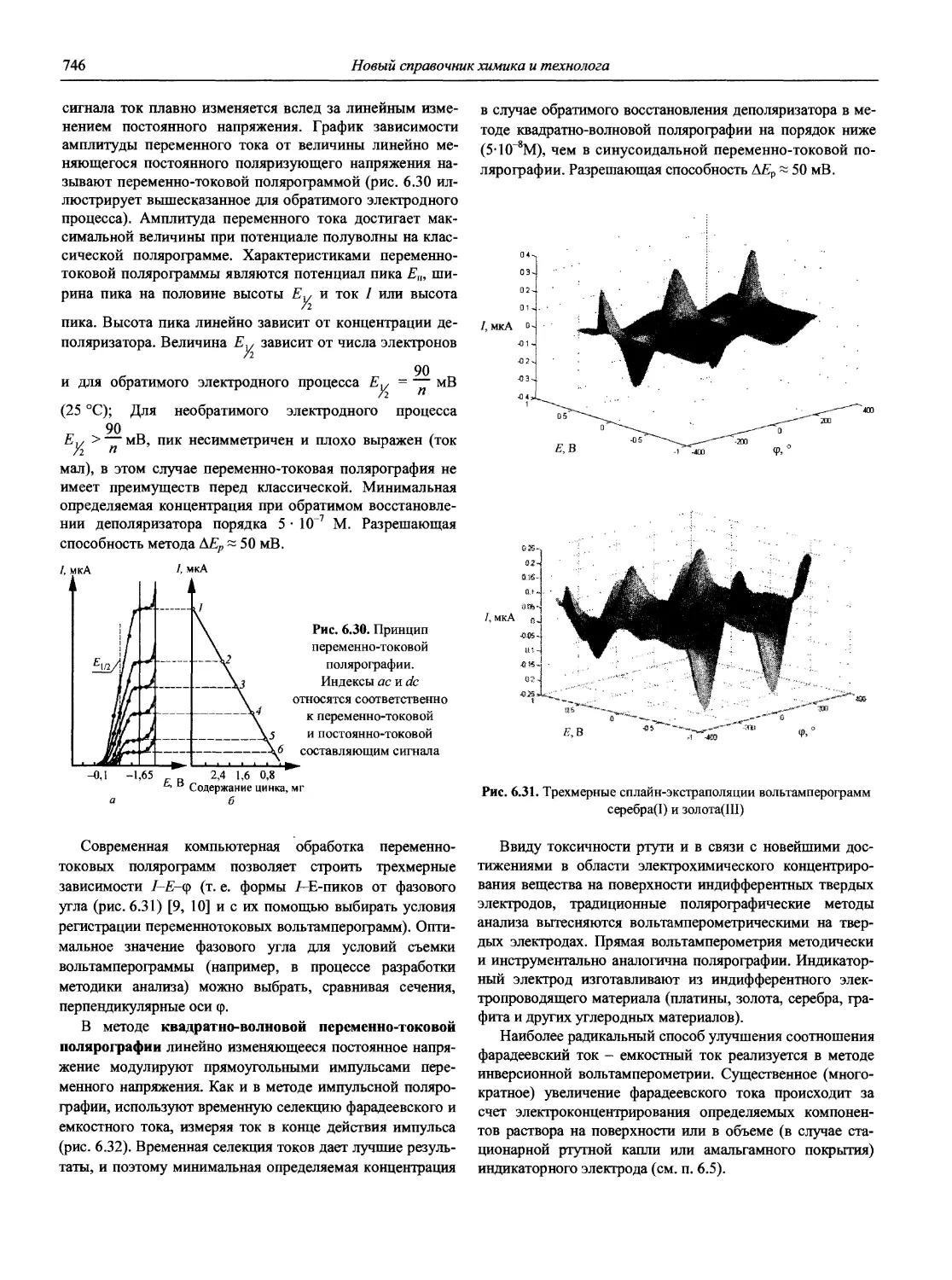
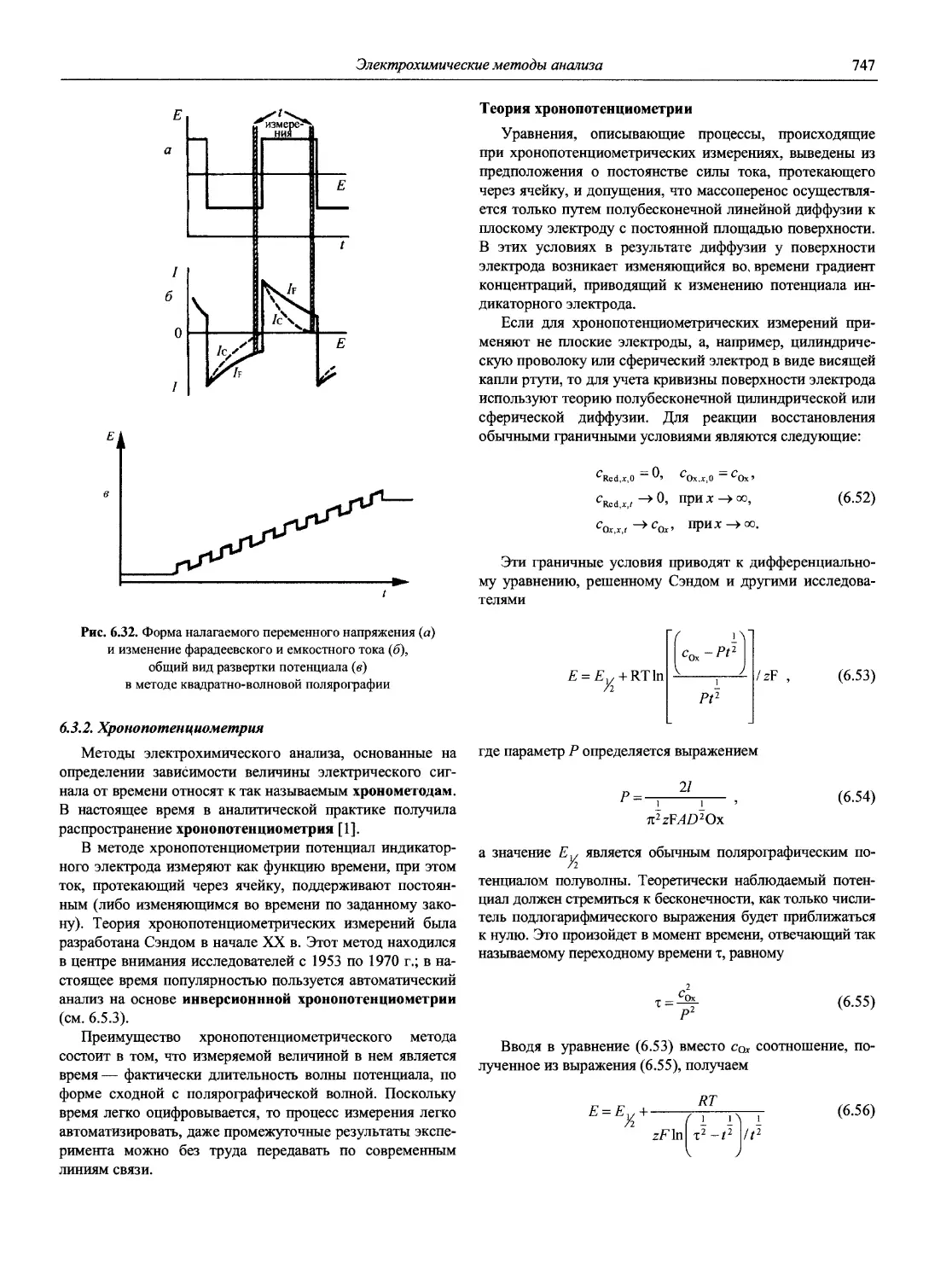
проф., д.х.н.,
лауреат Гос. Премии СССР и РФ Я.И. Яшин
Редакторы тома:
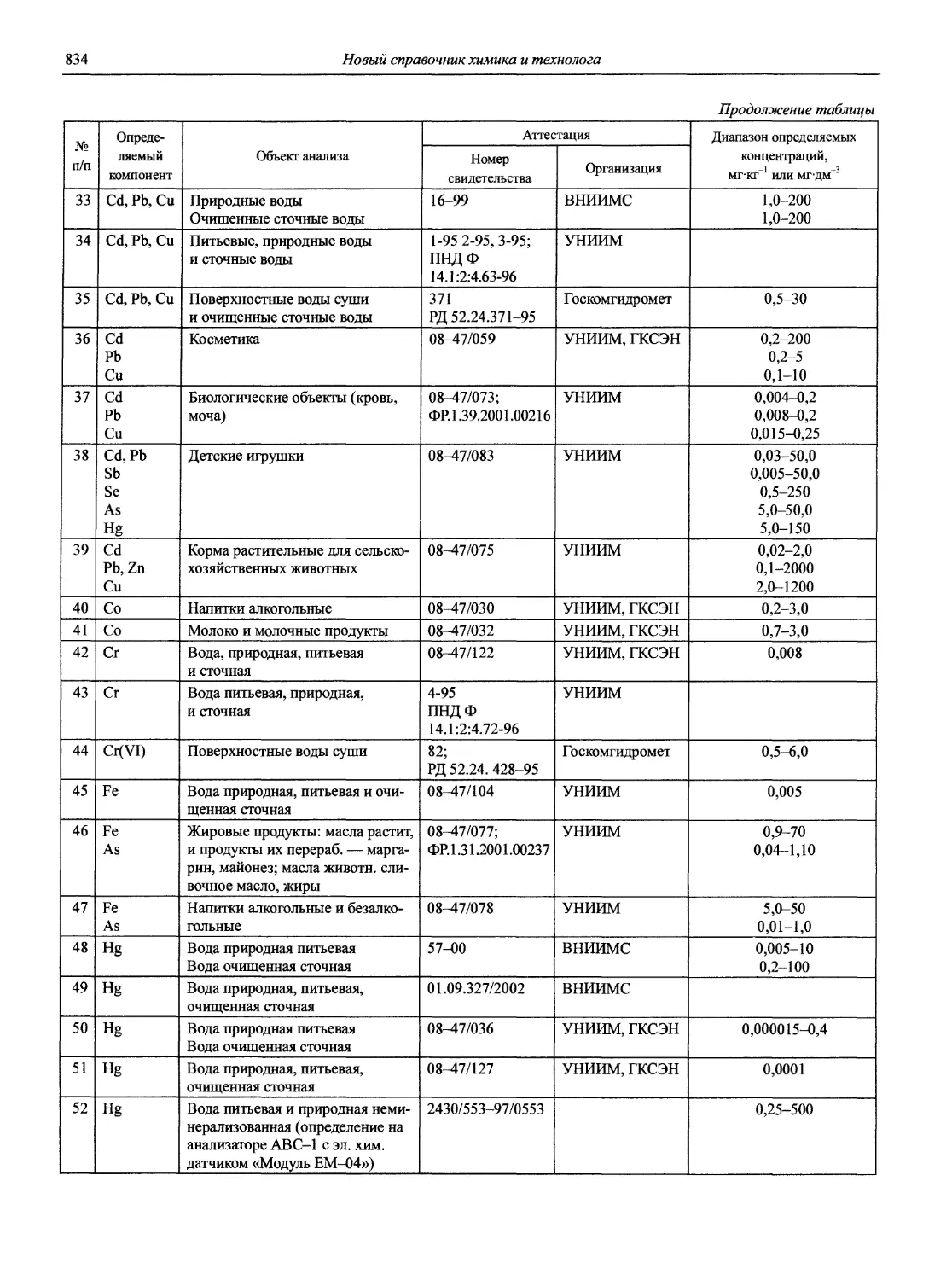
проф., д.х.н. И.П. Калинкин (общая редакция)
к.х.н. В.И. Мосичев (общая редакция)
проф. д.х.н. А.А. Карцова
доц., к.х.н. М.И Булатов
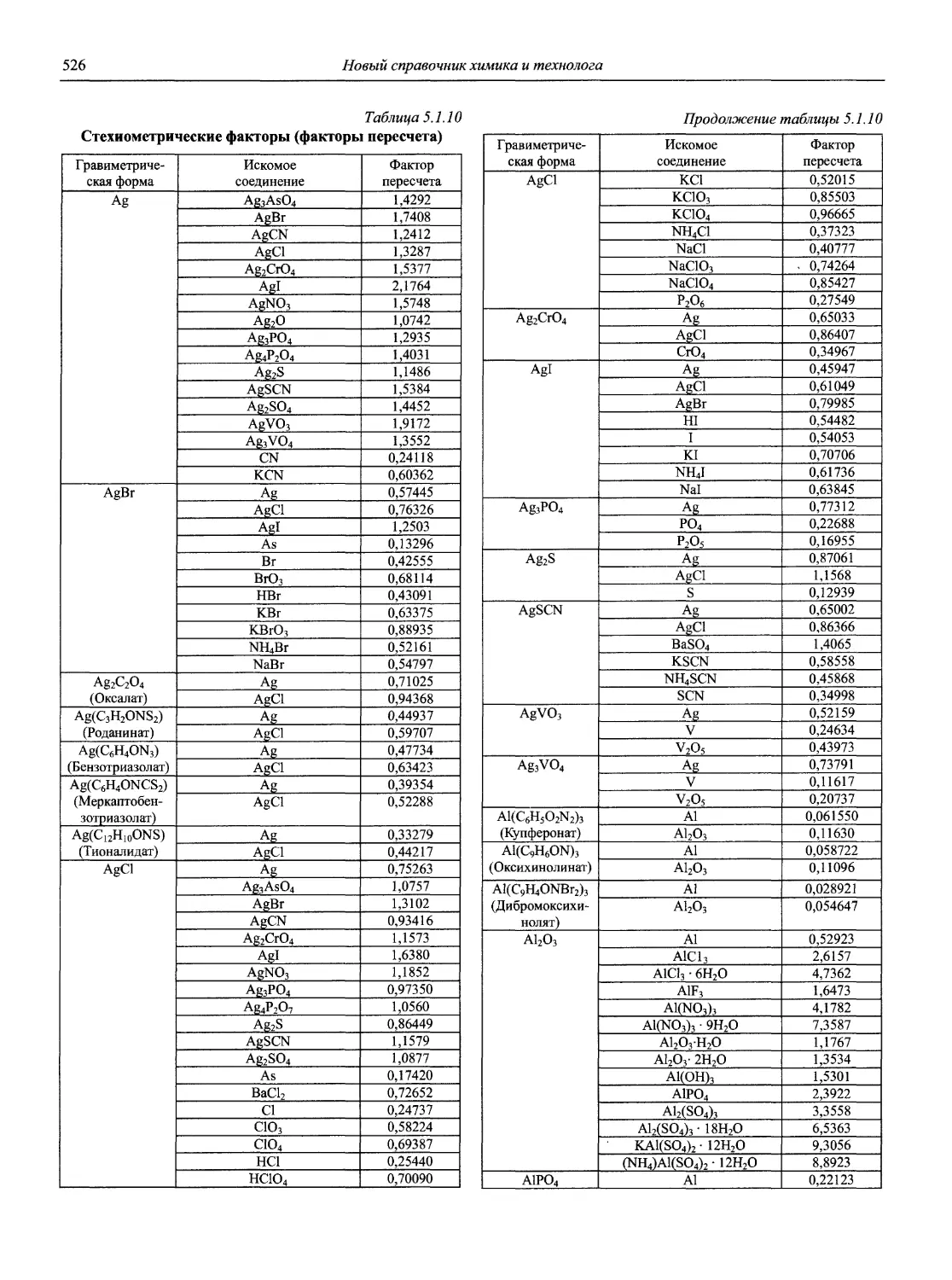
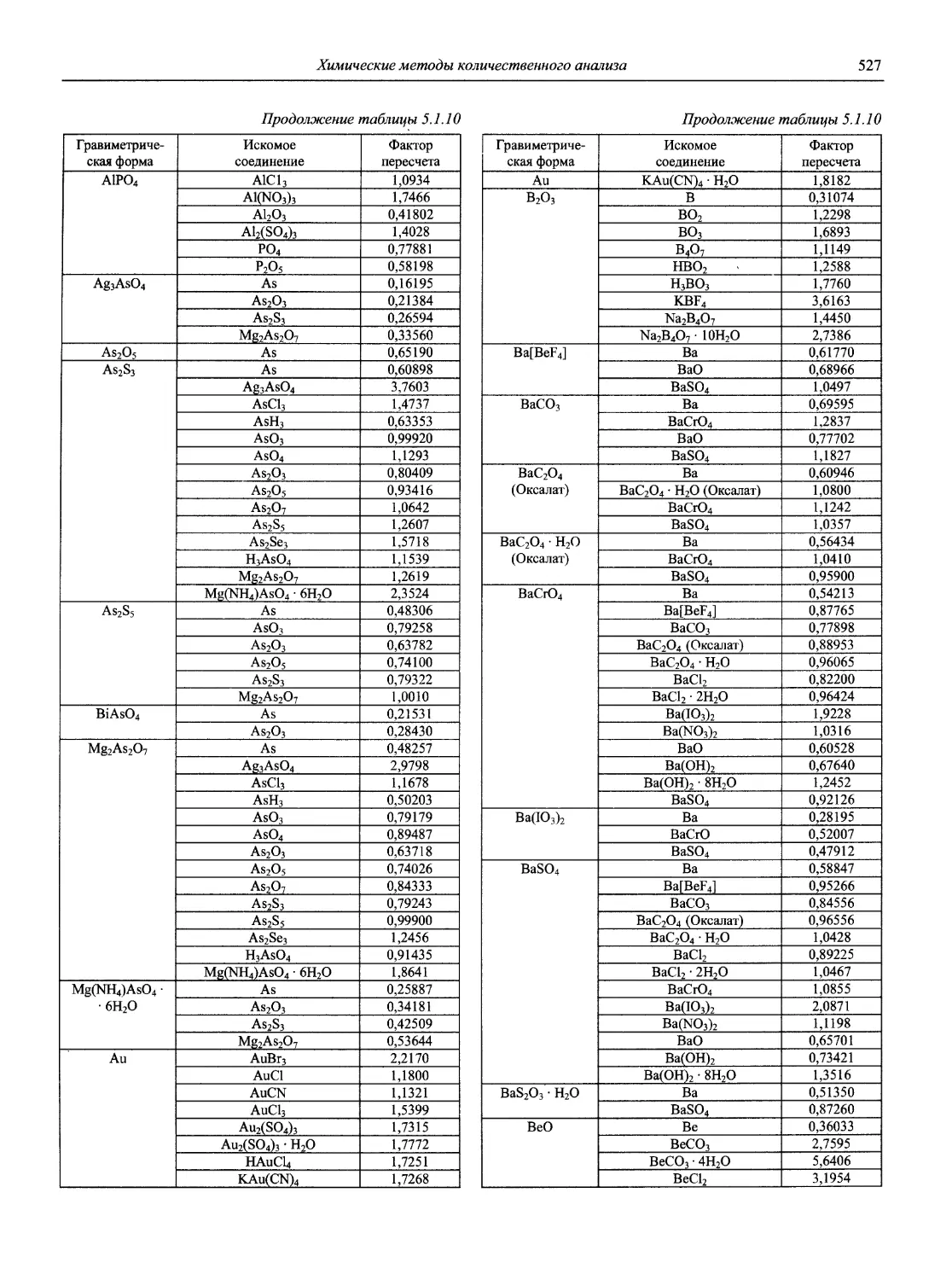
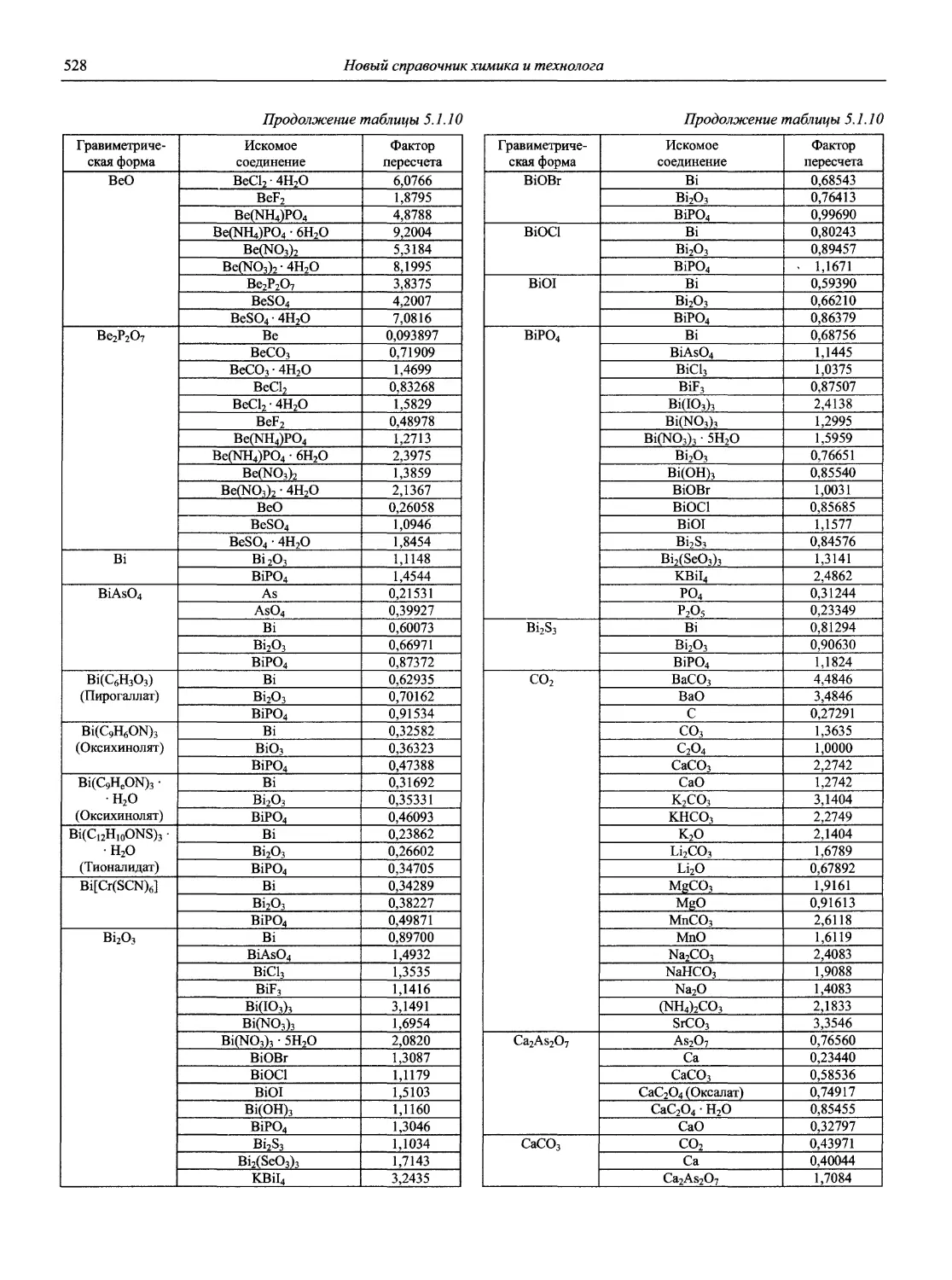
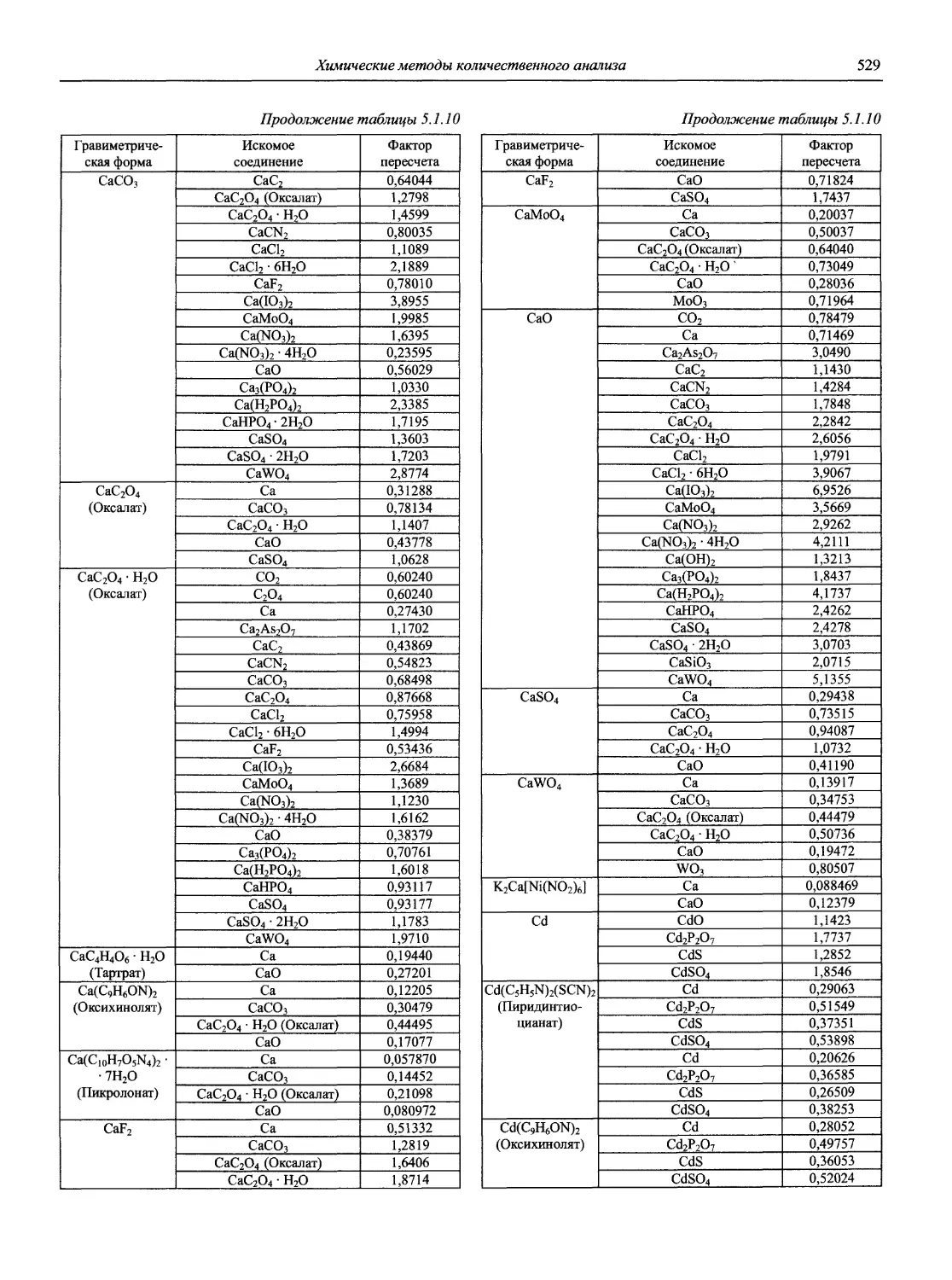
Н 72 Новый справочник химика и технолога. Аналитическая химия. Ч. I. — С.-Пб.: АНО НПО «Мир и Семья», 2002. — 964 с.
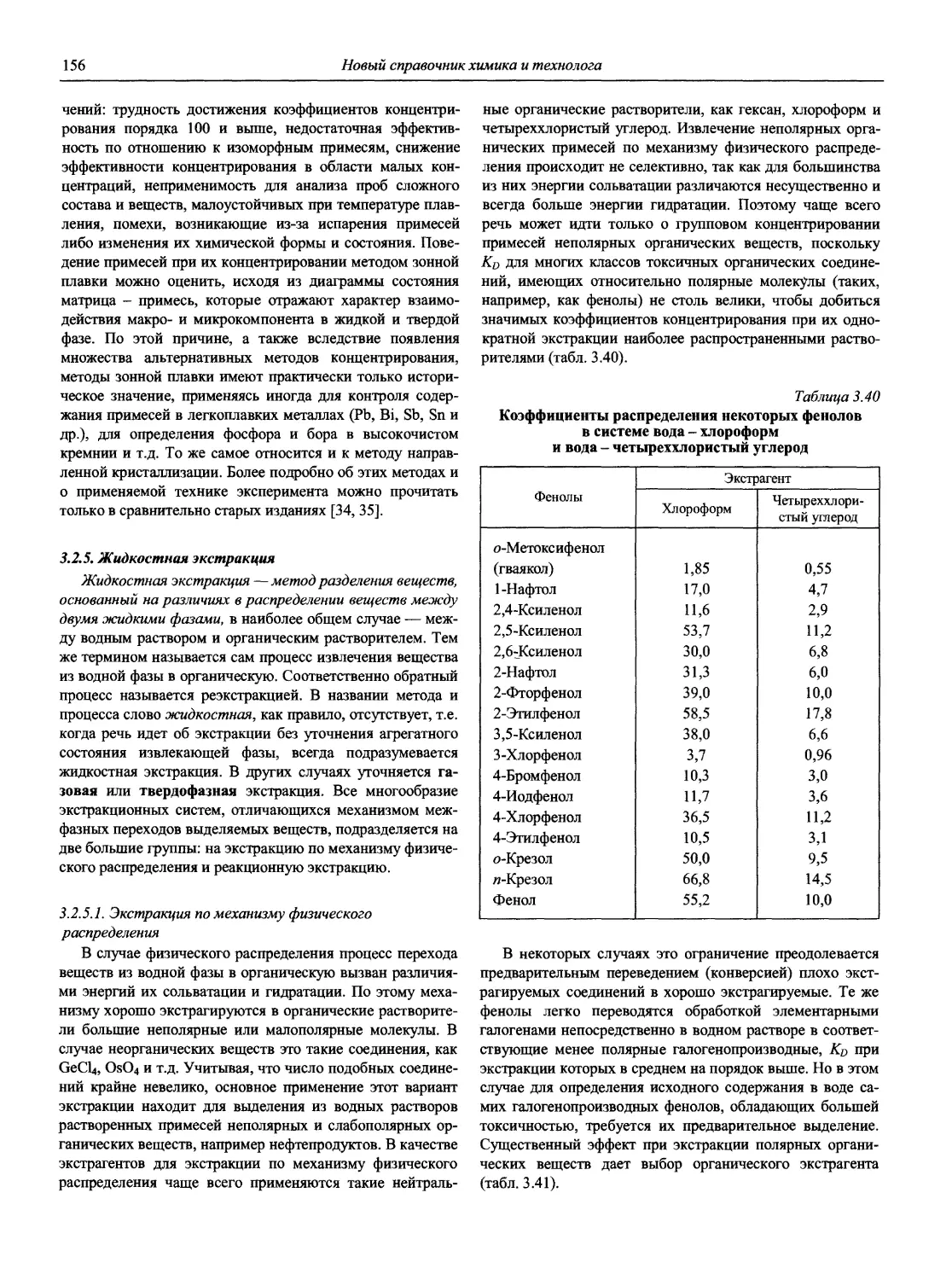
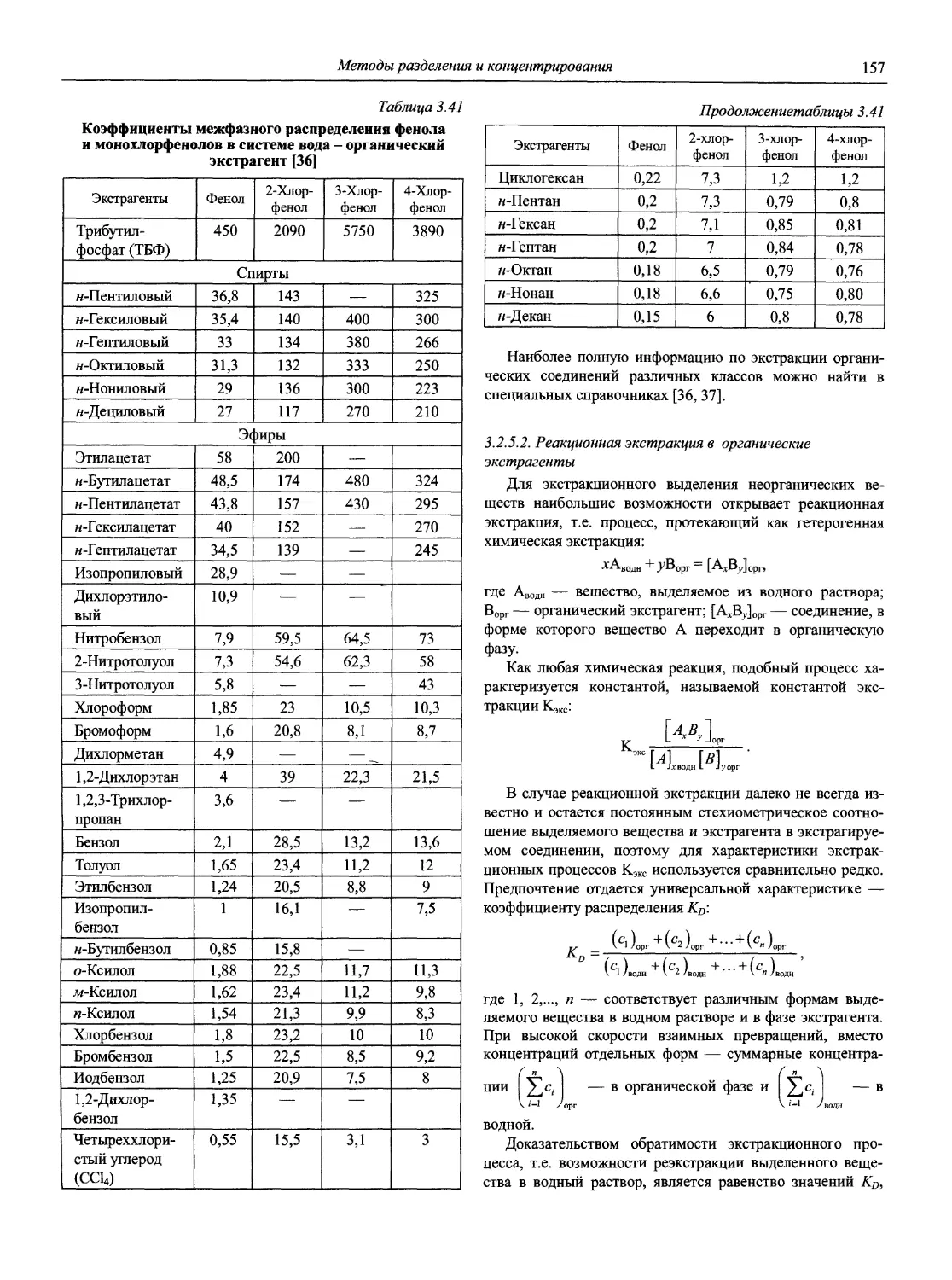
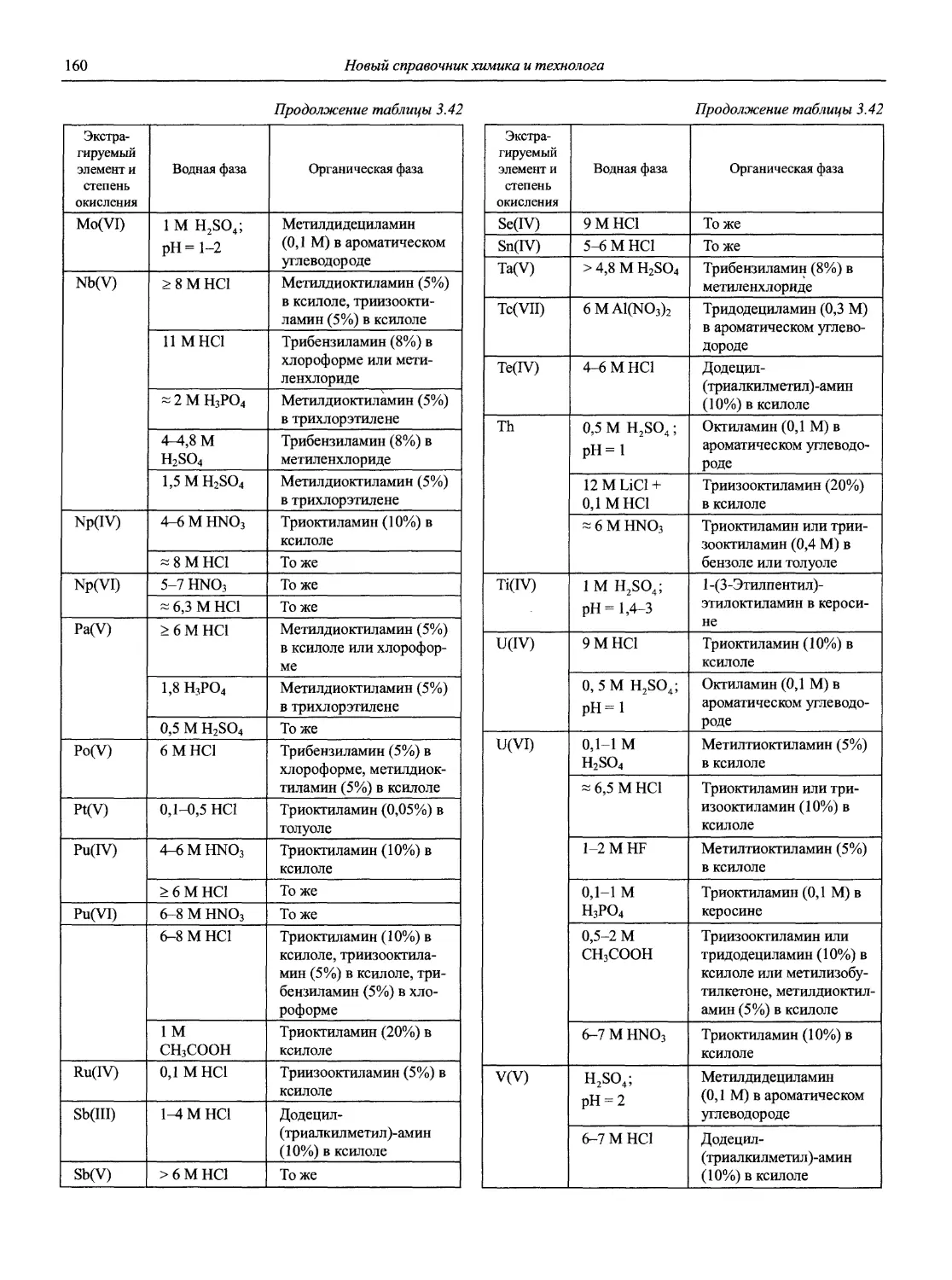
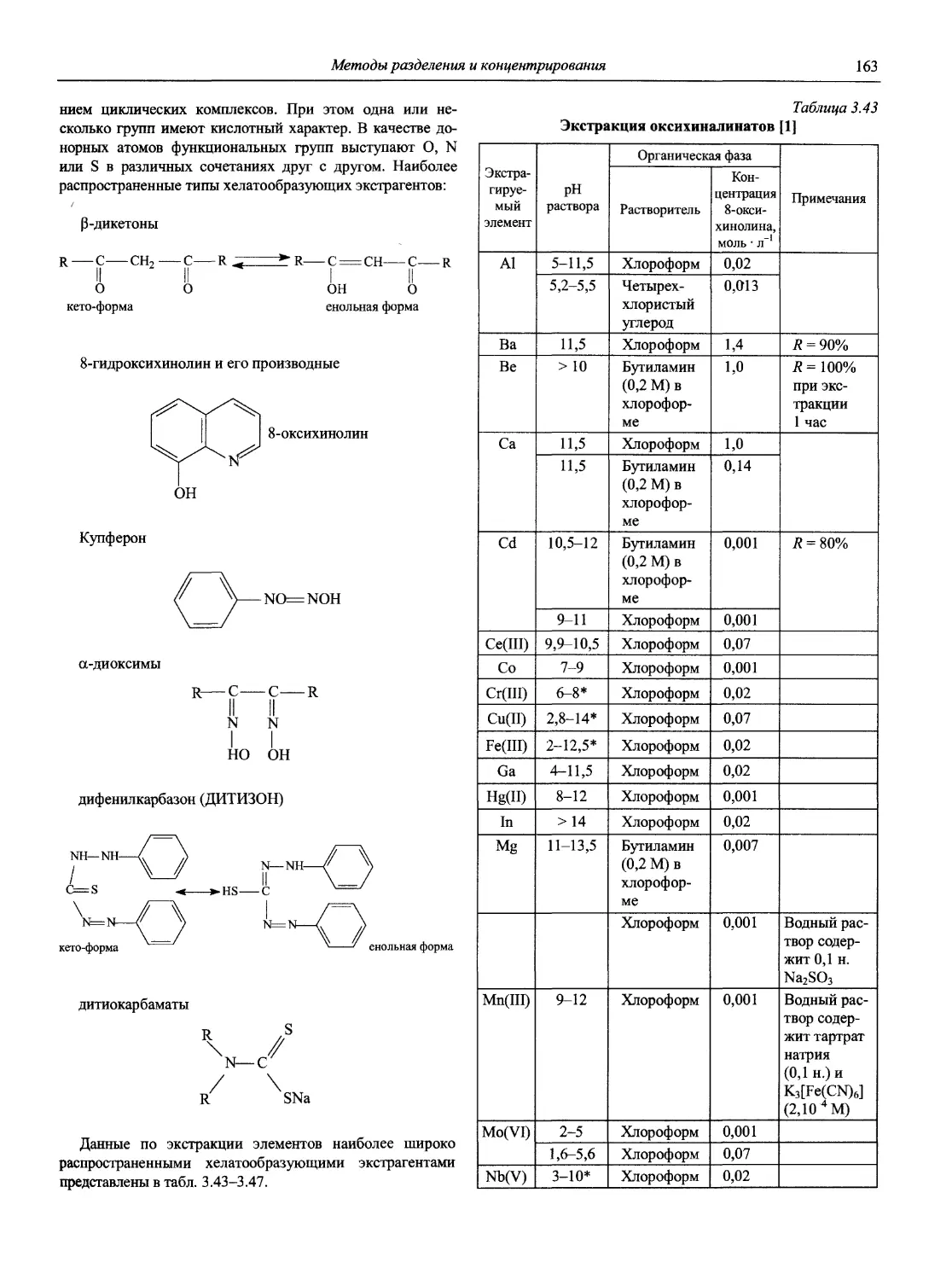
Справочник в 2-х частях отражает современное состояние, возможности и многообразие методов аналитической химии. В сжатом виде изложены теоретические основы всех ведущих методов анализа, аппаратура и обширнейший обобщенный справочный материал. Рассмотрены области применения, диапазоны определяемых содержаний, пределы обнаружения методов. Значительное внимание уделено анализу объектов окружающей среды.
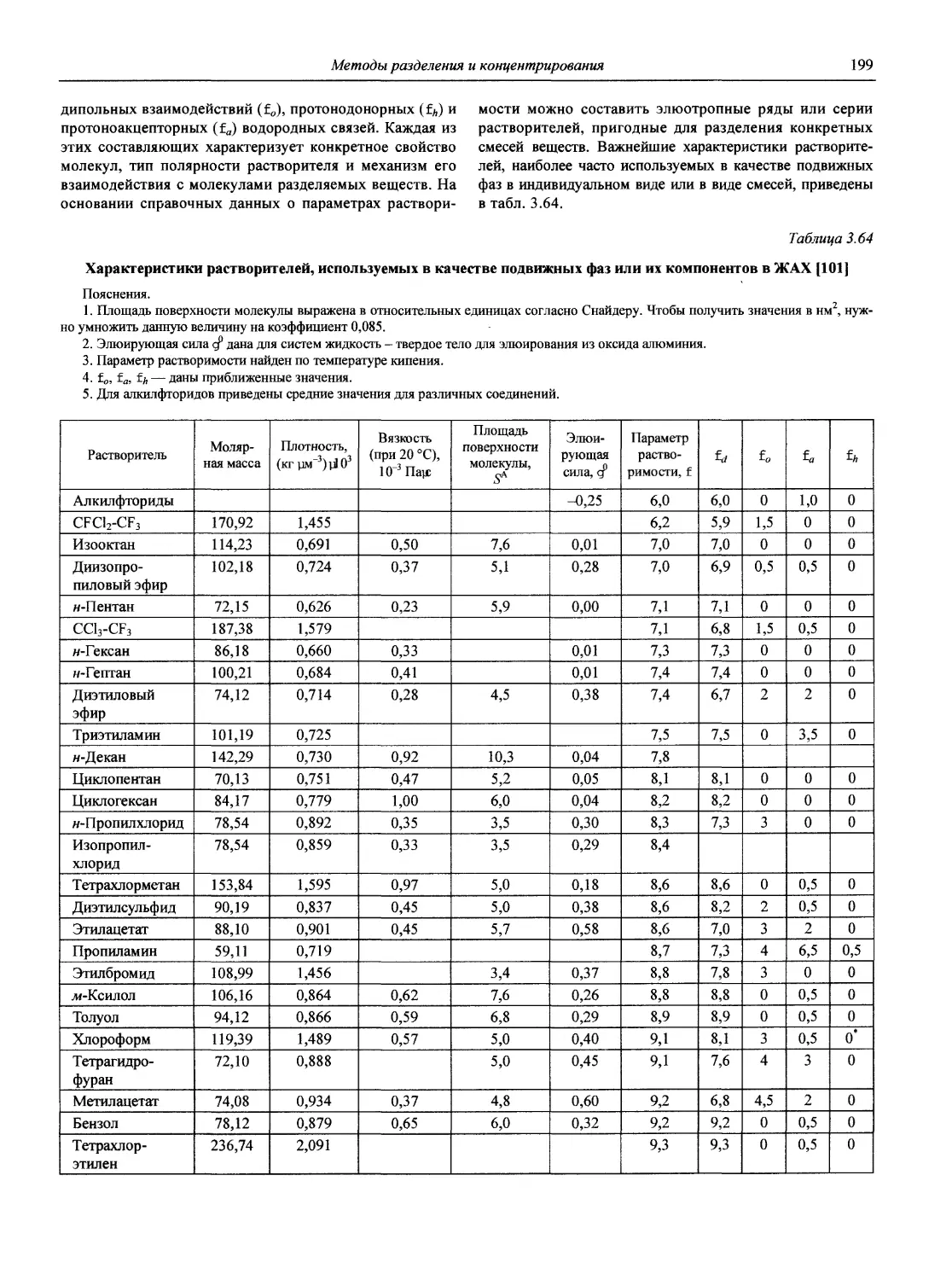
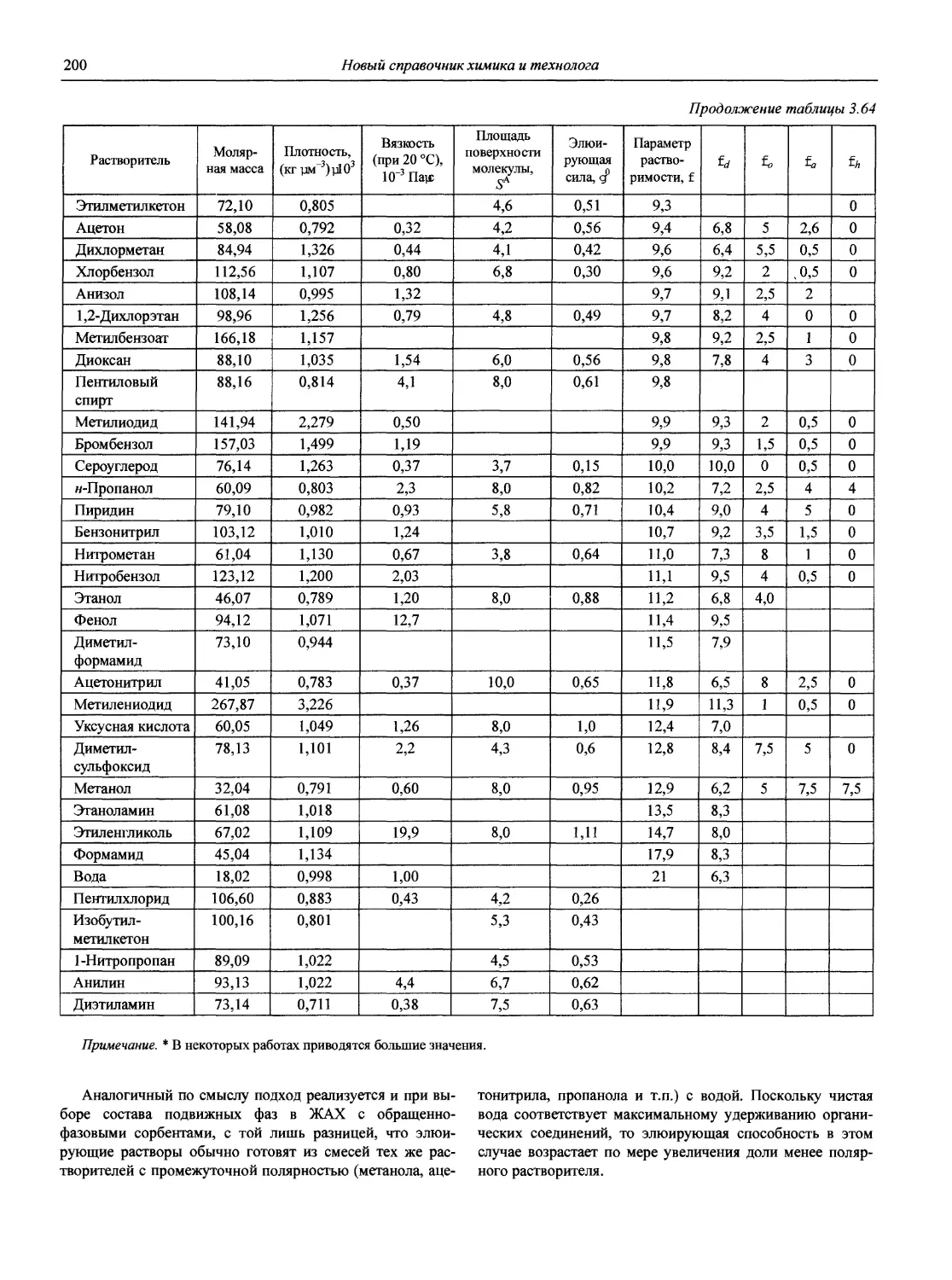
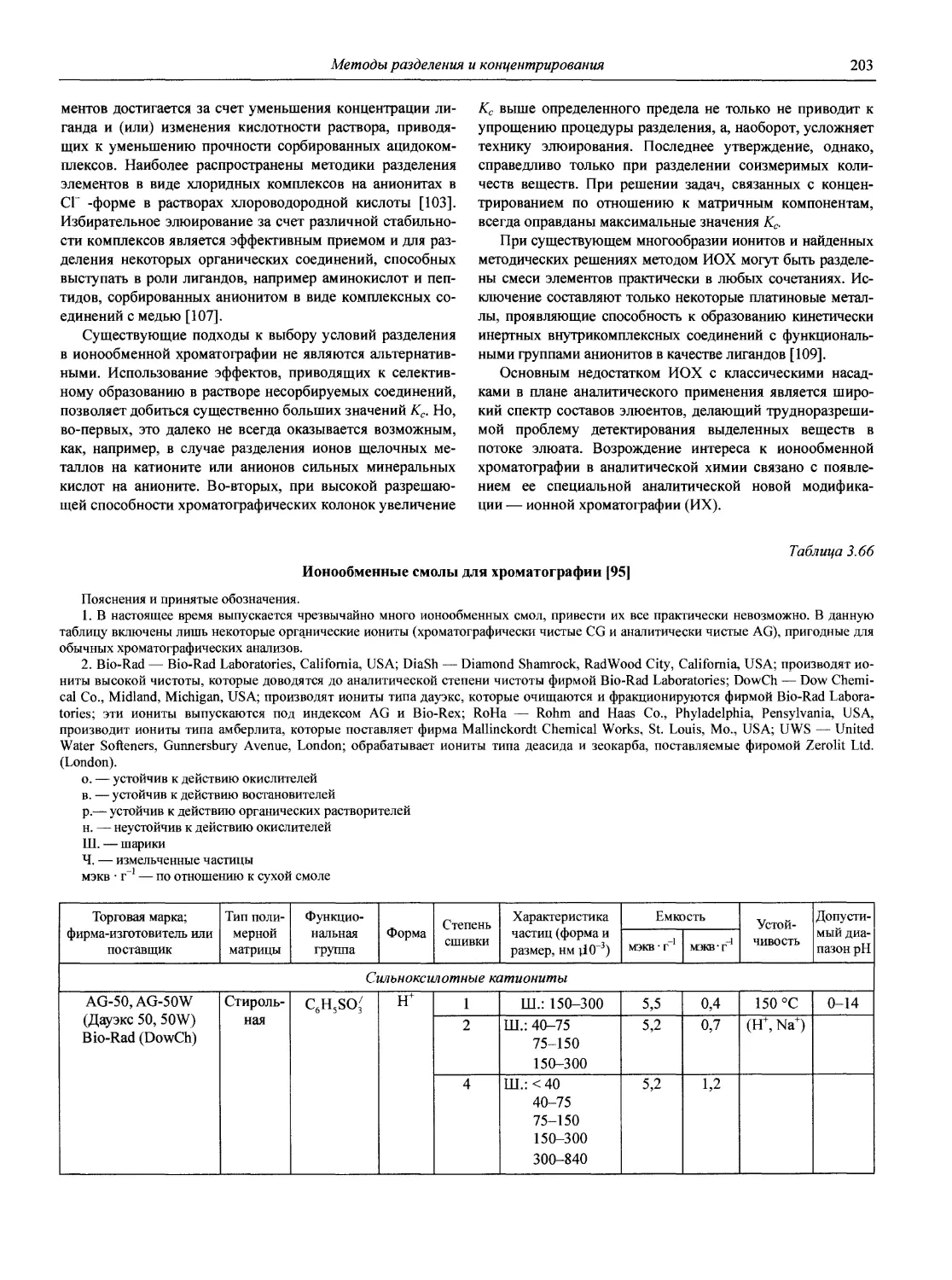
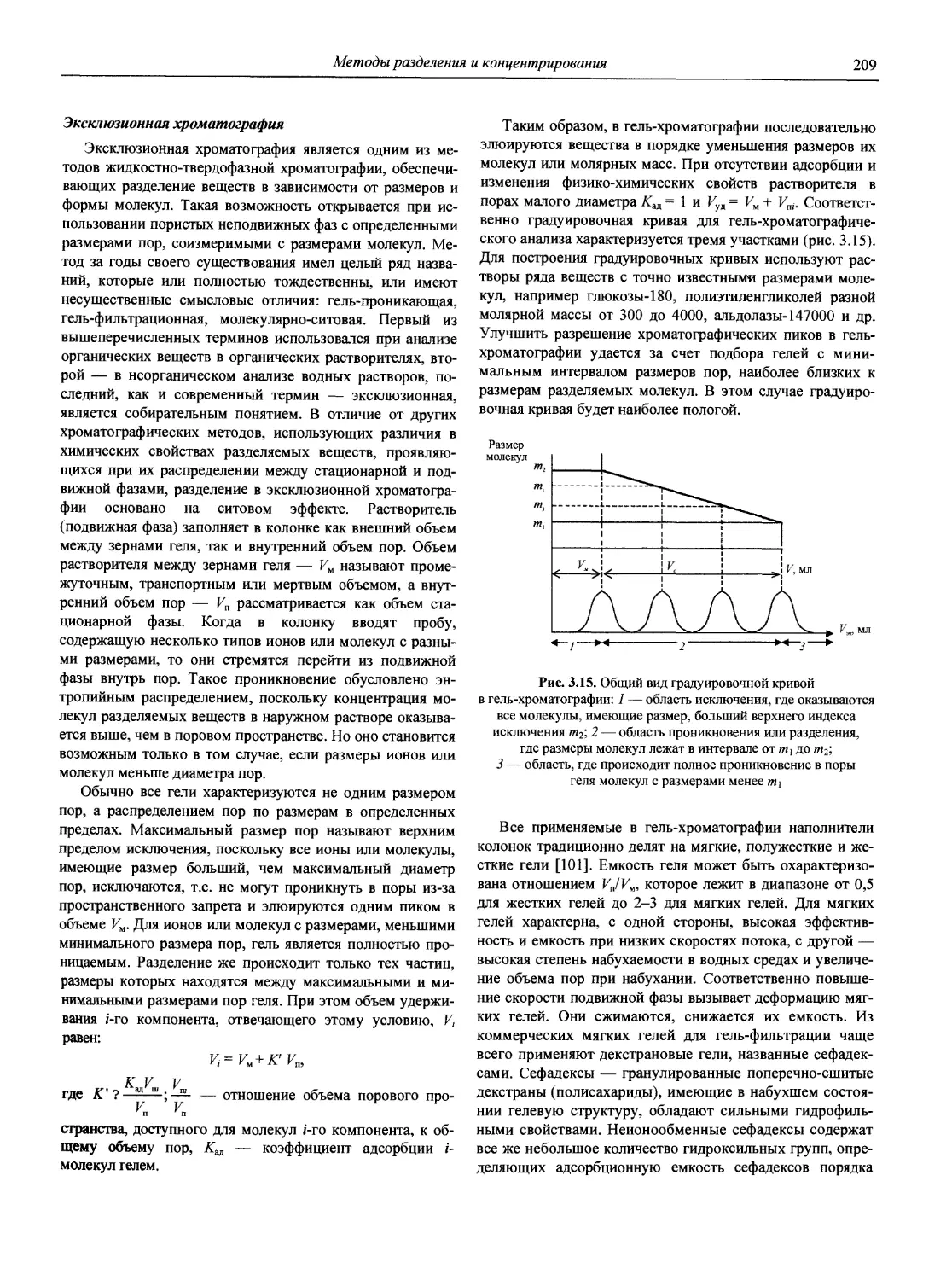
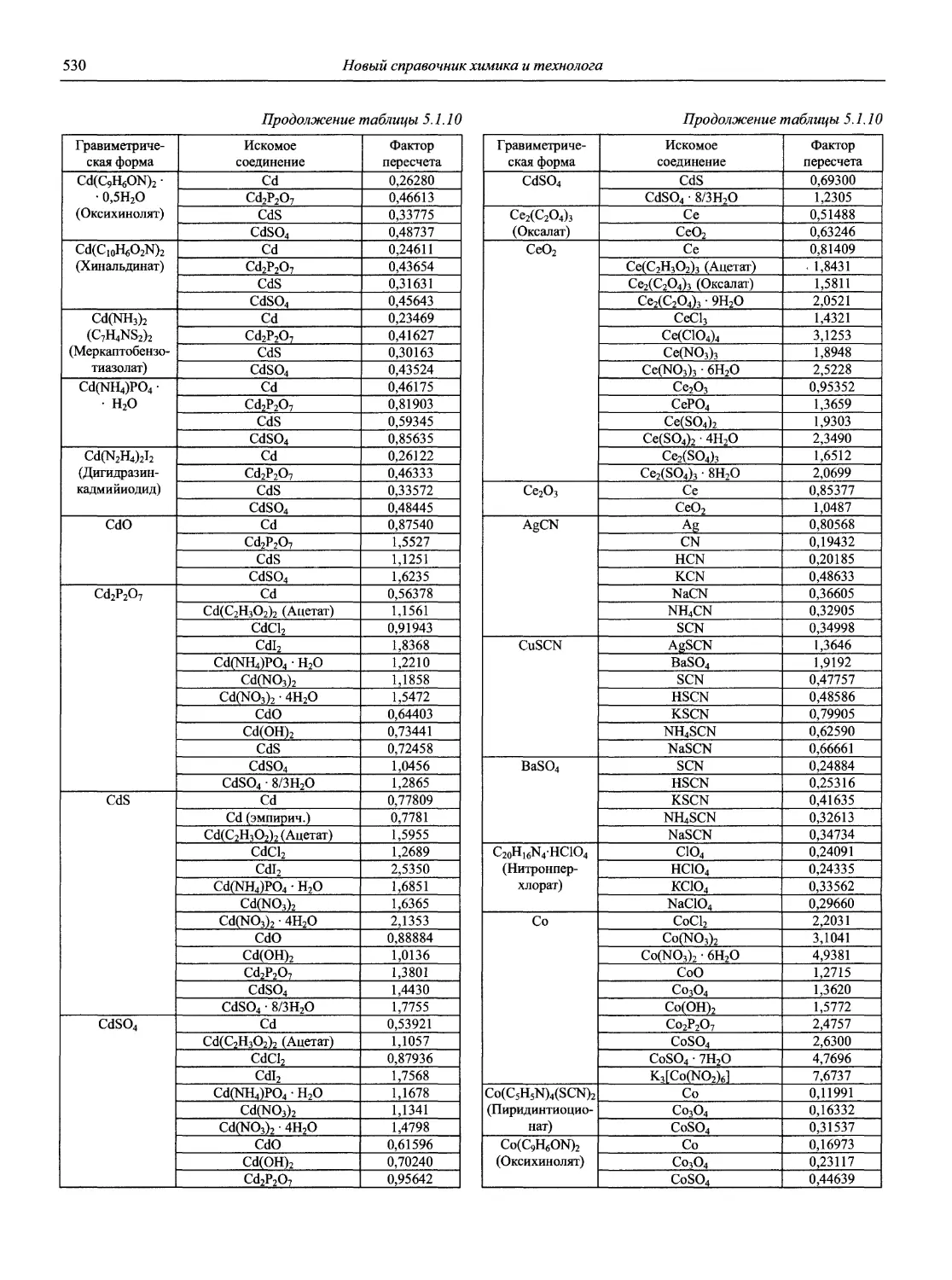
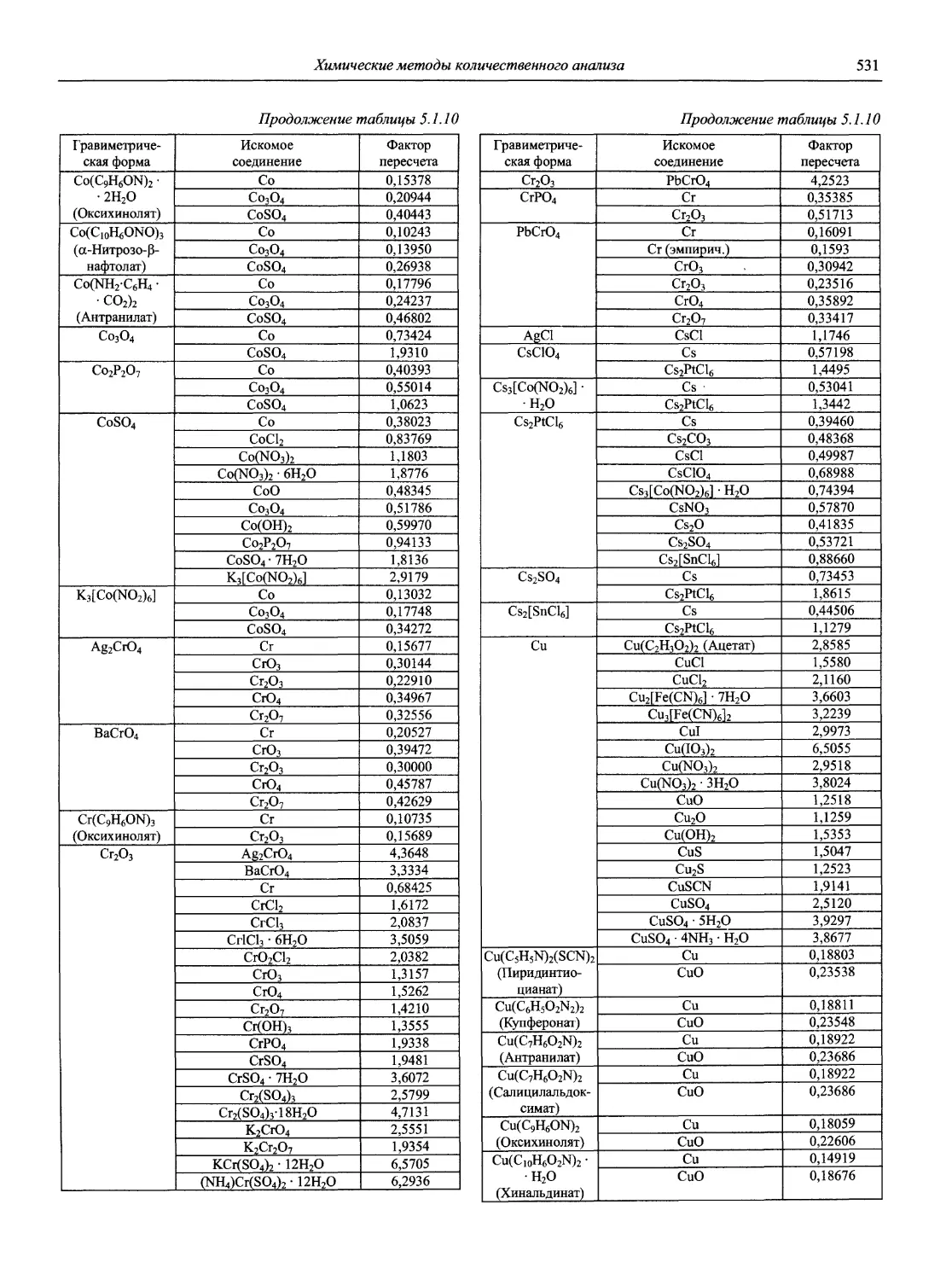
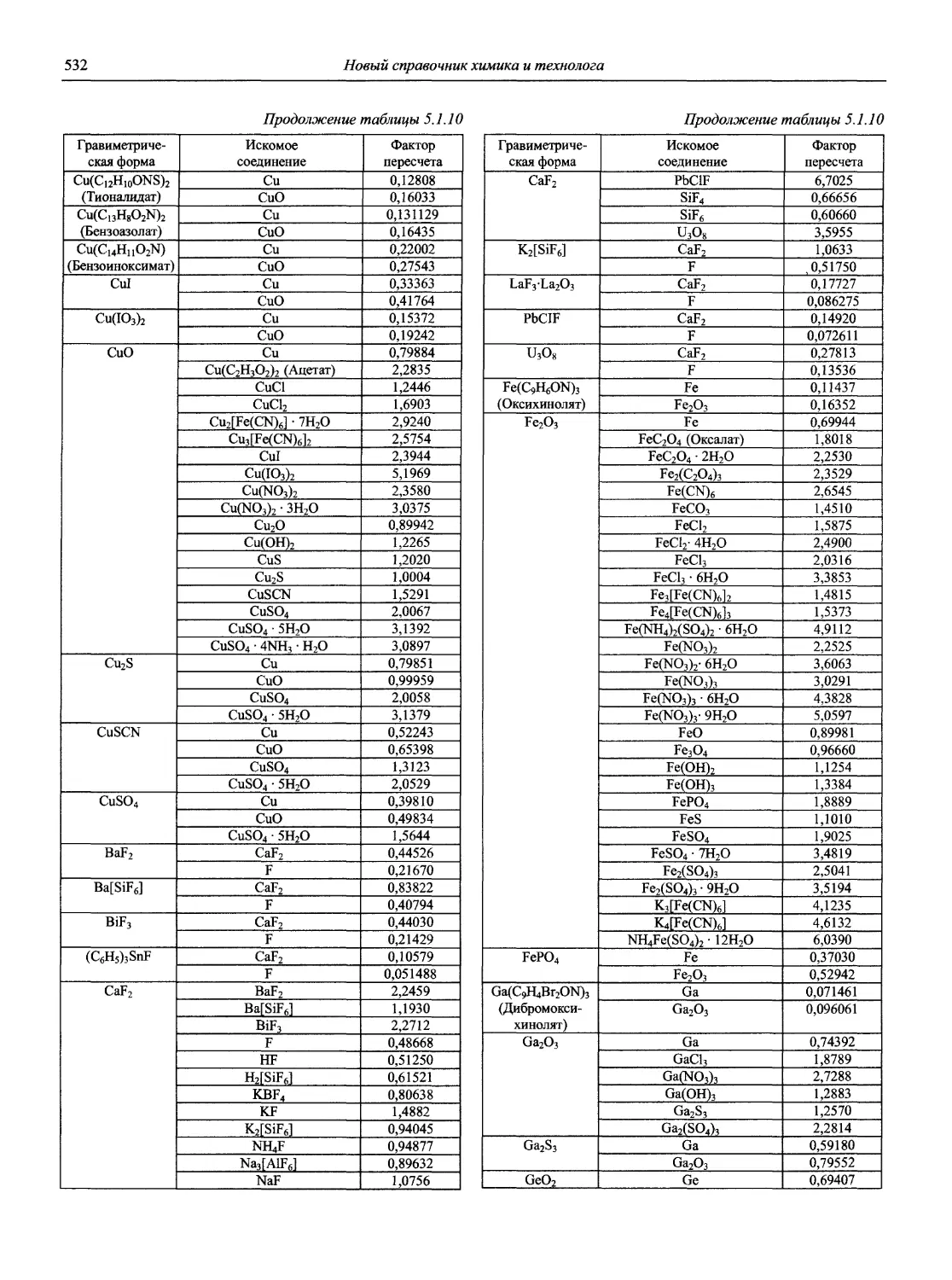
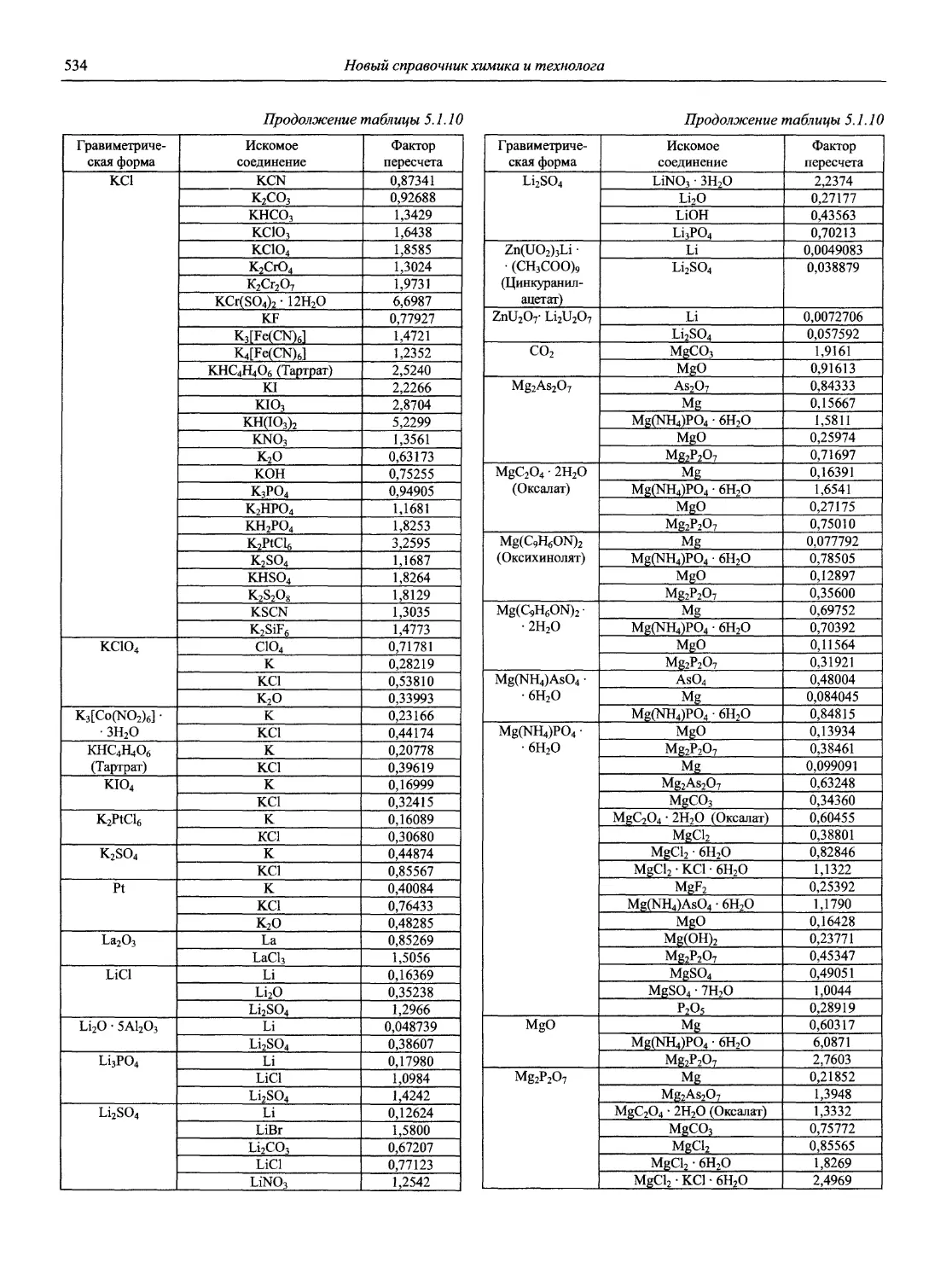
В первой части тома представлены информационные базы и общие вопросы аналитической химии, метрологические основы методов количественного анализа, методы разделения и концентрирования, хроматографические методы и капиллярный электрофорез, гравиметрические, титриметрические и электрохимические методы анализа, масс-спектрометрический метод и газовый анализ.
Предназначен для научного и инженерно-технического персонала аналитических лабораторий, инженеров химиков-технологов, преподавателей и студентов университетов, химико-технологических, химико-фармацевтических вузов, а также для специалистов смежных профессий, применяющих методы количественного анализа.
Новая, наиболее важная для специалистов информация по темам, вошедшим в данный том, будет в дальнейшем размещаться на сайте издательства www.npomis.com
Все права защищены и принадлежат издателю.
Любое использование материала данной книги, полностью или частично, без разрешения АНО НПО «Мир и Семья» запрещено и будет преследоваться по закону.
ISBN 5-94365-046-6
© АНО НПО «Мир и Семья», 2002
ОТ ИЗДАТЕЛЬСТВА
Петербургское издательство «Мир и семья» предлагает специалистам-химикам и всем заинтересованным читателям 7-томный «Новый справочник химика и технолога» (Справочник). («Справочник химика» в 7 томах, под общей редакцией чл.-корр. Б.П. Никольского, последний раз переиздан в 1962-1966 годах.)
Название Справочника отражает основную задачу издателей и авторов: помимо базовых, «академических», публиковавшихся в научной и справочной литературе (например, в «Справочнике химика») сведений, представить в максимально возможном объеме новую информацию, данные, полученные за последние 40 лет в области не только химической науки, но и химических технологий— и таким образом сделать Справочник интересным, нужным, необходимым широкой «химической» аудитории — как ученых и студентов, так и производственников-практиков.
В создании Справочника участвуют крупные ученые и ведущие специалисты химических отраслей из Москвы, Санкт-Петербурга, других городов страны (всего около 150 авторов); материалы являются оригинальными авторскими разработками либо подготовлены на основе современной литературы. Справочник обобщает опыт работы российских и зарубежных ученых и технологов 2-ой половины XX века и показывает перспективу развития химической и смежных областей науки и производства.
В процессе работы неоднократно возникала необходимость привлечения дополнительных материалов, разработ
ки новых тем, более расширенного и углубленного изложения уже включенных в Справочник разделов. В связи с этим значительно увеличился, по сравнению с первоначально запланированным, объем (некоторые тома выйдут в двух книгах), сроки выпуска данного и следующих томов отодвинулись. Однако авторско-издательский коллектив считает эти производственные потери и трудности оправданными необходимостью оптимального решения заявленной глобальной задачи.
Продолжает издание том «Аналитическая химия» в двух частях. Настоящий том дает представление о современном состоянии, достижениях, возможностях одной из самых значительных для человечества XXI века, динамично развивающихся областей химической науки.
Основные темы других томов:
♦ общие сведения о строении и физических свойствах веществ;
♦ свойства растворов, электродные процессы, коррозия и противокоррозионные конструкционные материалы;
♦ сырье и продукты промышленности;
♦ процессы и аппараты химических технологий;
♦ вредные химические вещества.
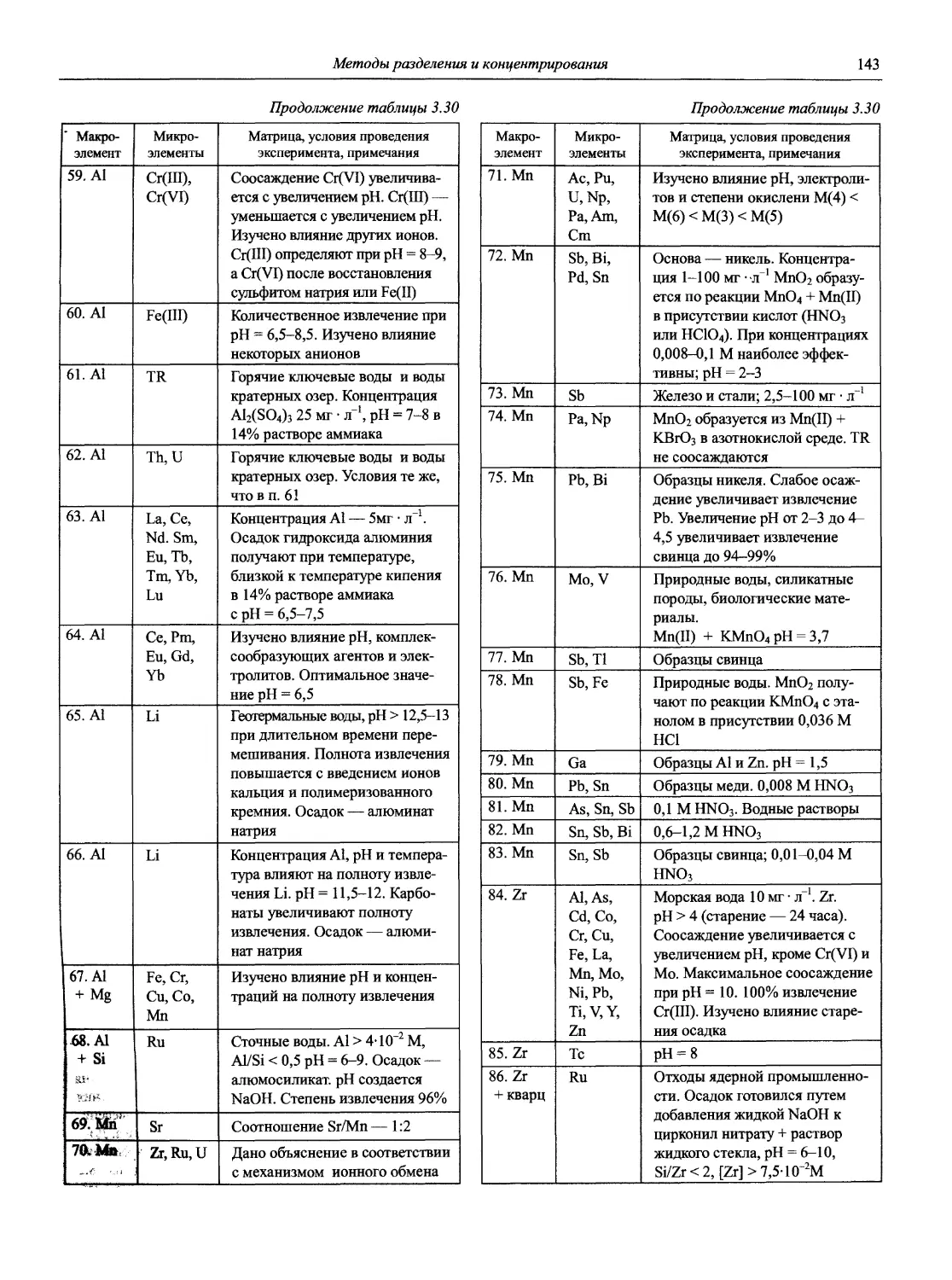
Руководители издательства выражают благодарность всем тем, без чьей самоотверженной работы выпуск сего основополагающего труда не состоялся бы.
ОТ АВТОРОВ ТОМА
Аналитическая химия (АХ) — наука об определении химического состава веществ и отчасти — их химического строения; ее целями являются разработка теоретических основ различных методов анализа, установление границ их применимости, оценка метрологических характеристик аналитических методов, создание конкретных методик анализа различных объектов [1].
Единого определения АХ нет. На VIII Европейской конференции по аналитической химии («Евроанализ-VIII», Эдинбург, 5-11 сентября 1993 г.) специально созданной рабочей группой была выработана и одобрена следующая формулировка:
«Analytical chemistry is a scientific discipline which develops and applies methods, instruments and strategies to obtain information on the composition and nature of matter in space and time» («Аналитическая химия — это научная дисциплина, которая развивает и применяет методы, средства и общую методологию получения информации о составе и природе вещества в пространстве и во времени». — Перевод акад. Ю.А. Золотова).
Аналитическая химия — не только важнейшая составляющая химической науки, но и одна из самых актуальных прикладных областей человеческих знаний.
Уже с 1980-х годов каждый пятый американский химик считает себя аналитиком. С 90-х годов XX века в промышленно развитых странах 10% стоимости валового национального продукта приходится на сертификацию качества продукции, в том числе на аналитический контроль химического состава. В Великобритании на сегодня ежегодная стоимость аналитических определений превышает 7 миллиардов фунтов стерлингов.
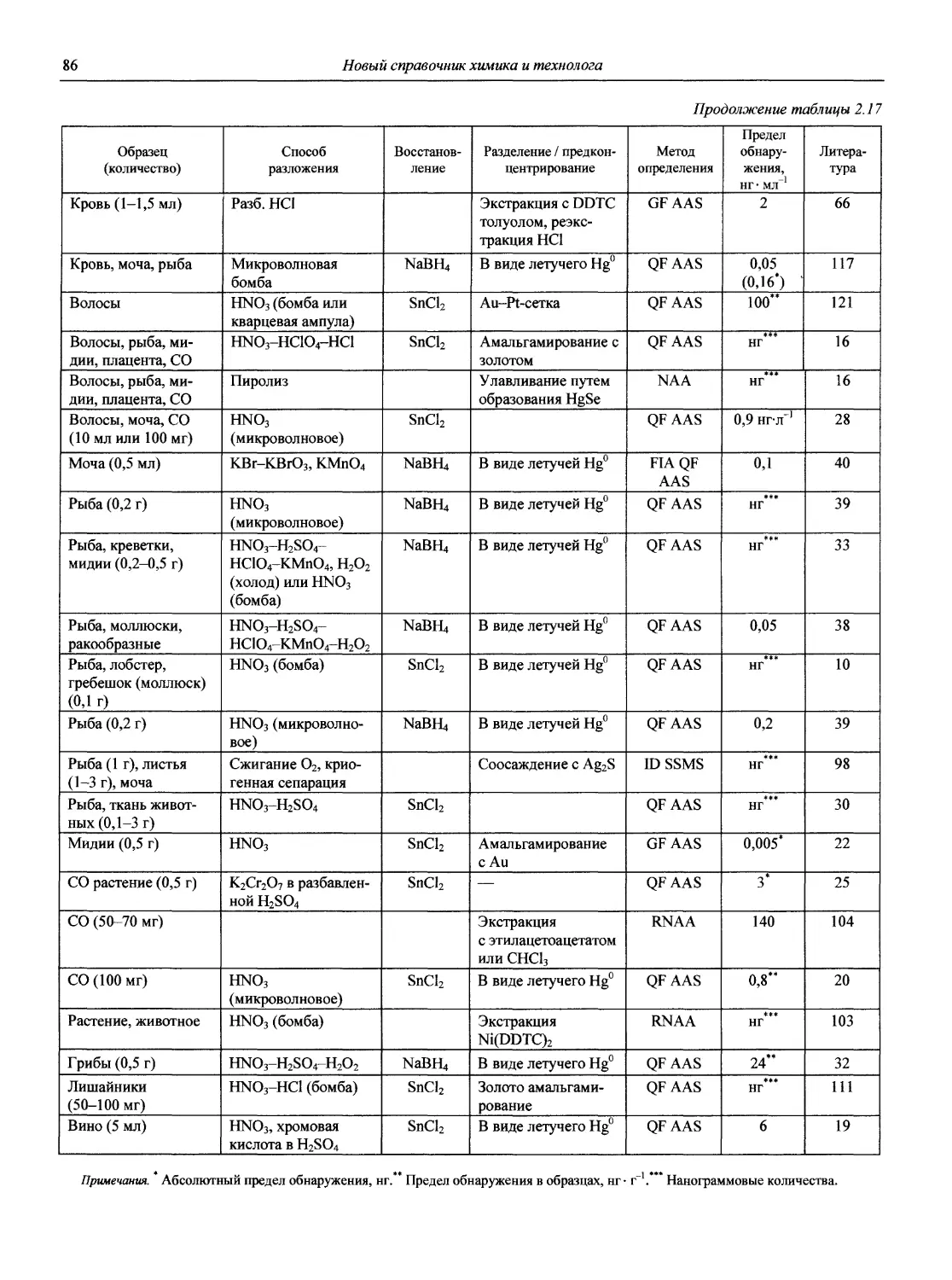
Почти 40 лет прошло после выхода в нашей стране тома «Справочника химика», посвященного АХ [2]. За это время неузнаваемо изменился мир и столь же радикально, качественно изменилась АХ, что наглядно представлено в работе [3].
В аналитическом контроле (важной области практического применения АХ) основную роль стали играть физические, главным образом спектрометрические методы, хотя классические химические методы1 (гравиметрия, титриметрия) не потеряли своего значения в обеспечении метрологического единства аналитических измерений в целом.
1 Под этим термином в ряде случаев объединяют химические и физико-химические методы.
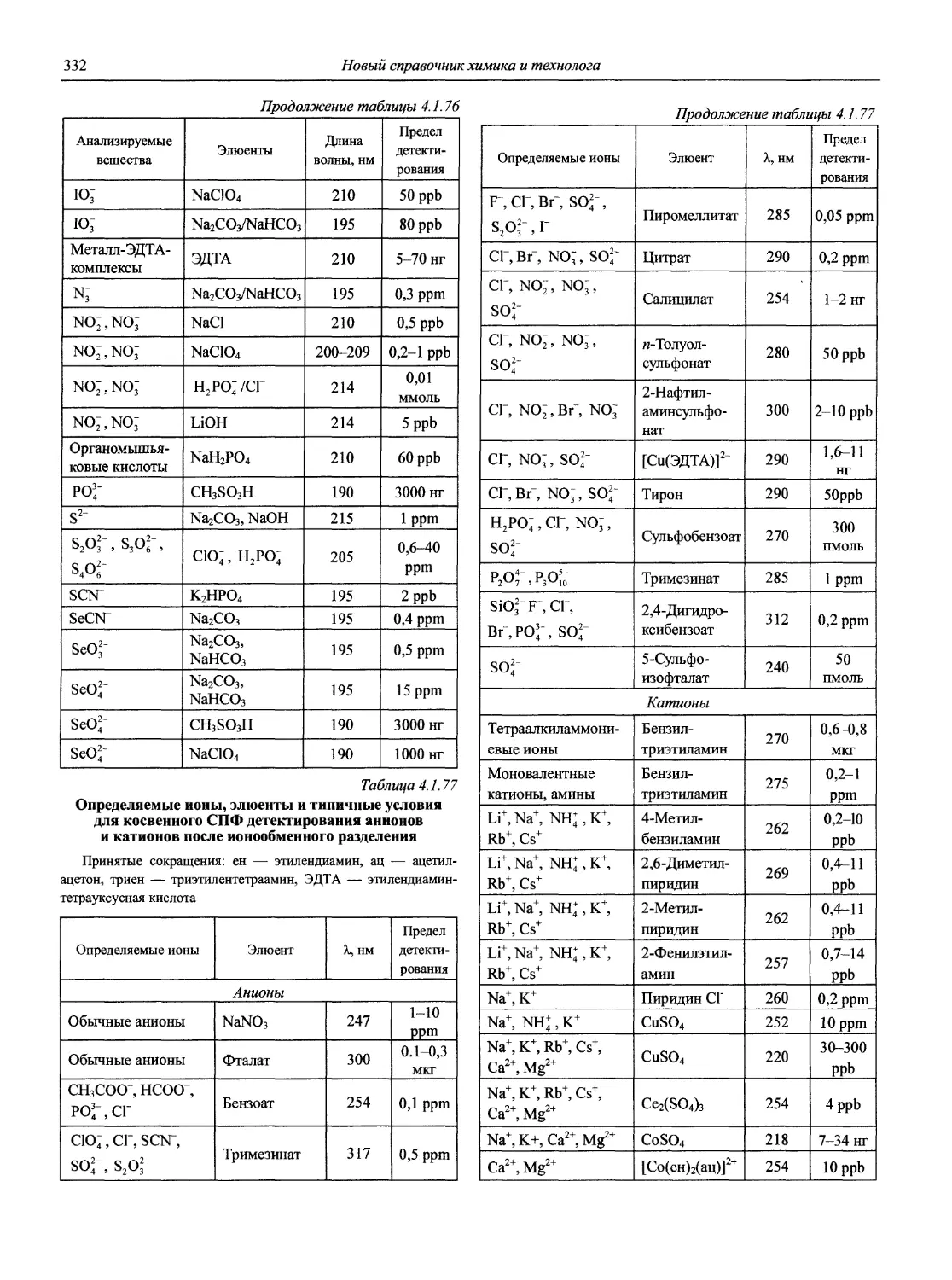
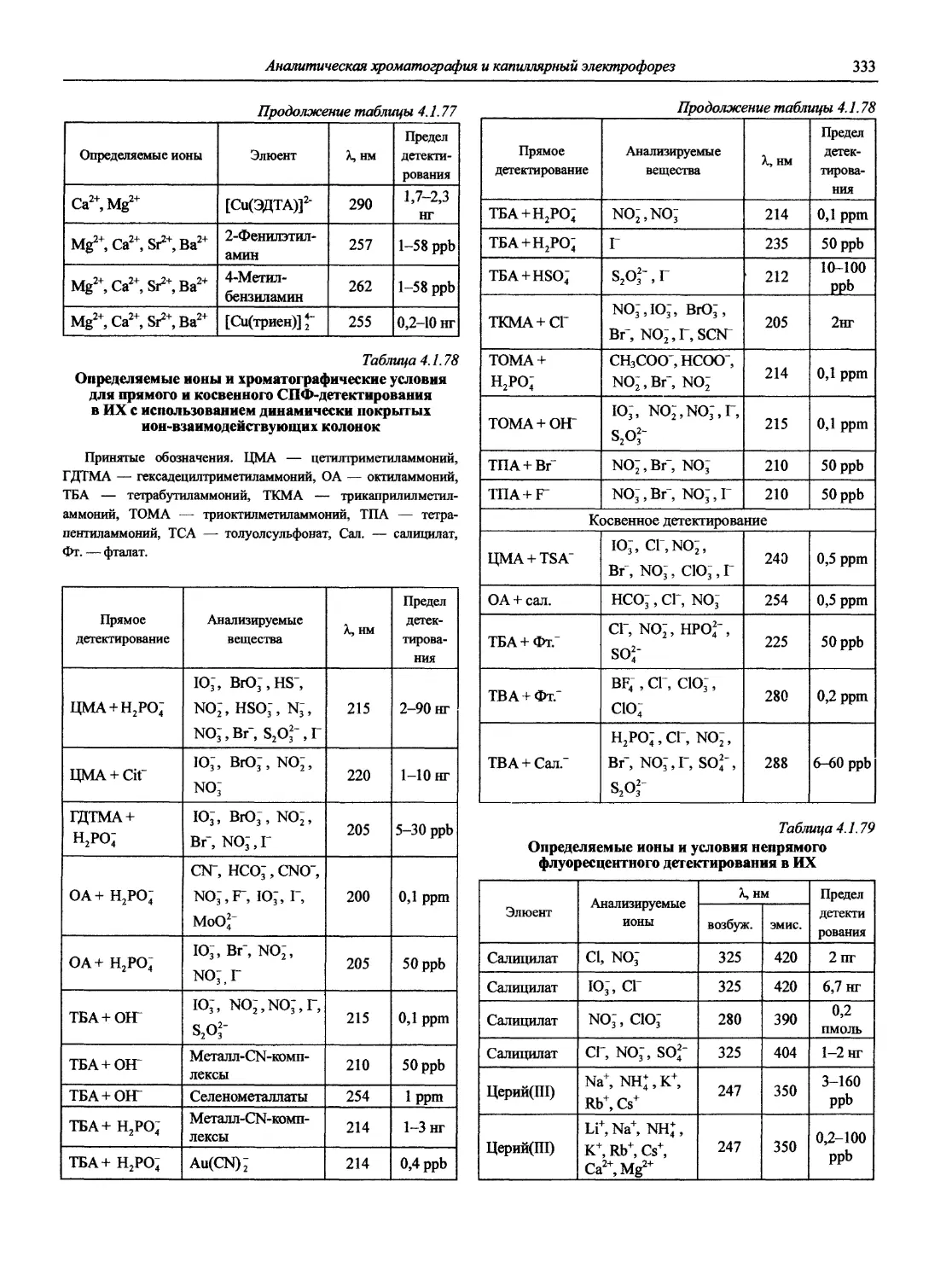
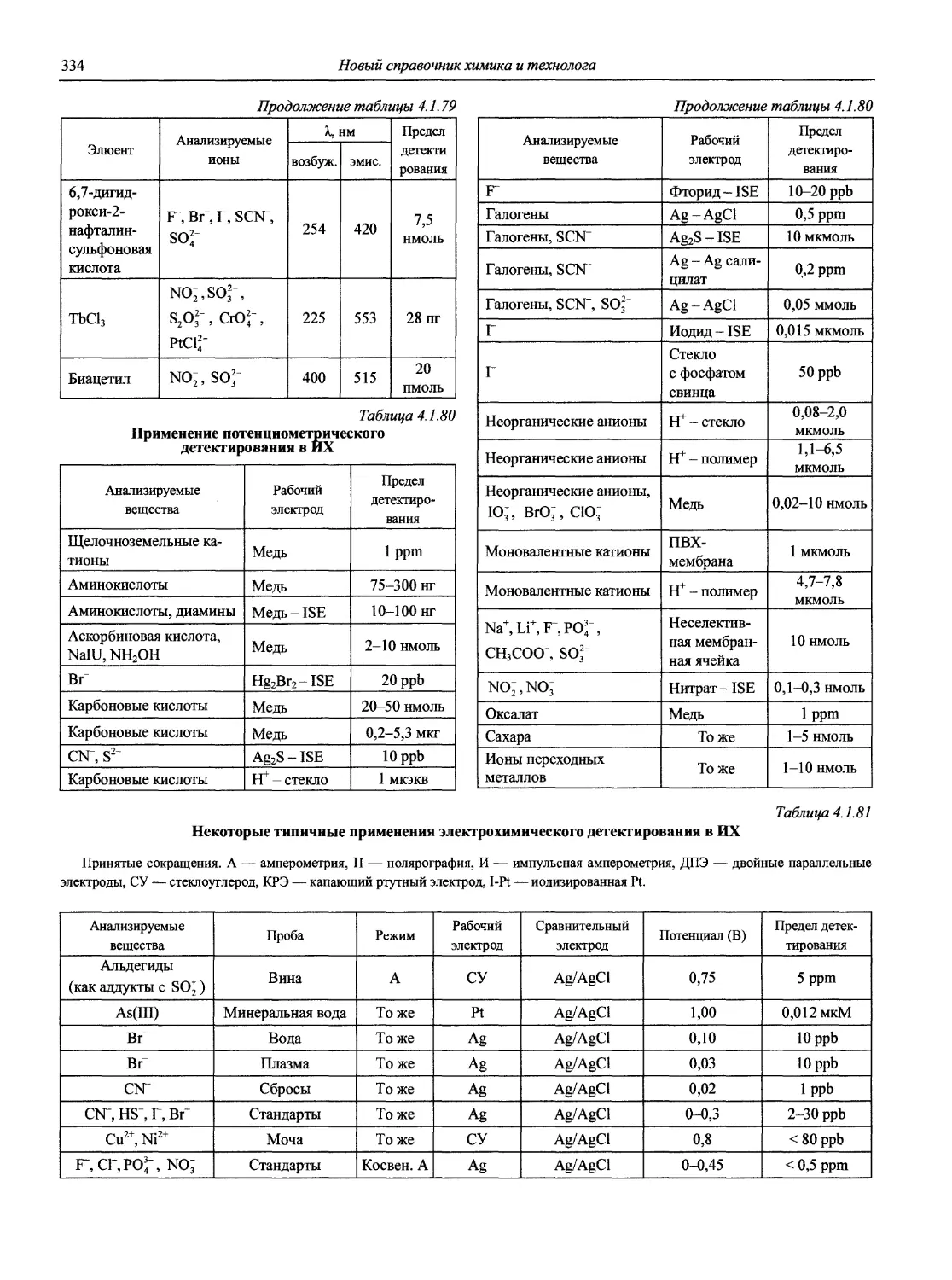
Именно в эти годы получили основное развитие и широкое применение совершенно новые методы анализа, такие как атомно-абсорбционная и атомно-флуоресцентная спектрометрия, рентгенофлуоресцентная спектрометрия, рентге-носпекгральный микроанализ, хромато-масс-спектрометрия, высокоэффективная жидкостная хроматография, парофазный анализ, проточно-инжекционный анализ; в электрохимических методах новое интенсивно развиваемое направление — электрохимические сенсоры, тест-методы и т.д. Поразительного прогресса достигли хроматографические методы.
Возникло новое направление — наноаналититическая химия, оперирующая с пико- и нанолитровыми объемами анализируемых веществ и реагентов. Методы наноанали-тической химии играют решающую роль в установлении полного сиквенса генома человека, в выяснении причин многих генетических заболеваний и создании генно-инженерных лекарств.
Стали доступными тонкие методы исследования состава и структуры поверхности твердых тел (Оже-электронная спектроскопия, рентгеновская и ультрафиолетовая фотоэлектронная спектроскопия, спектроскопия дальней тонкой структуры рентгеновских спектров поглощения, масс-спектрометрия вторичных ионов и др.), в немалой степени обусловившие бурное развитие микроэлектроники в последние десятилетия.
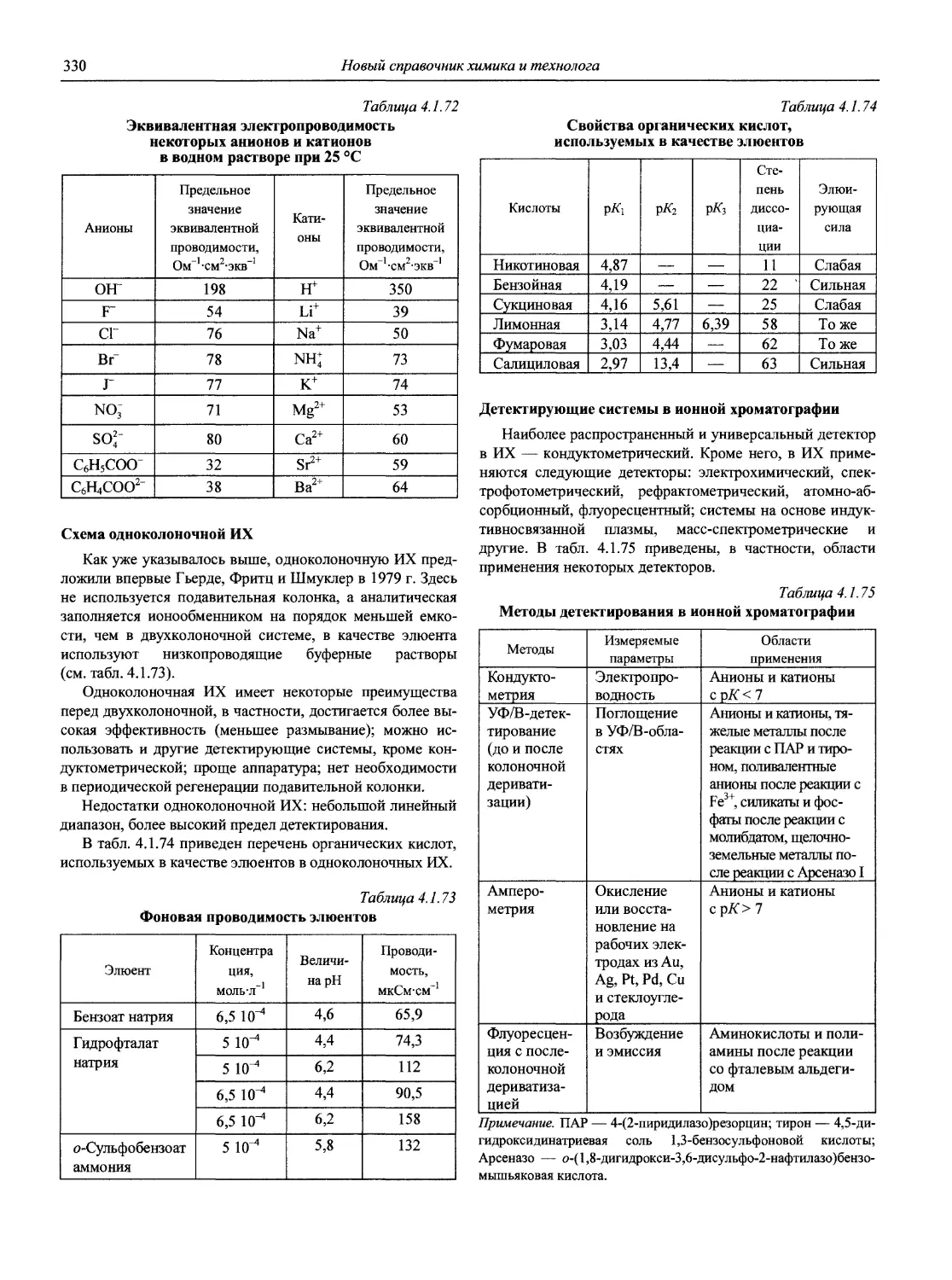
Созданы новые поколения атомно-эмиссионных и атомно-абсорбционных спектрометров, сканирующих и многоканальных рентгенофлуоресцентных спектрометров, масс-спектрометров, переносных и мобильных анализаторов различного типа и т.д. Программное обеспечение современных аналитических приборов позволяет не только управлять процедурой анализа, но и автоматизировать сам процесс разработки конкретных методик анализа, выполнять статистическую обработку получаемых результатов (с построением диаграмм контроля качества результатов анализа), обеспечивает практически неограниченный объем хранения данных, возможность использования нескольких языков, передачу информации на периферийные устройства и т.д. Столь совершенные приборы позволяют решать задачи многоэлементного анализа сложных по составу материалов с привлечением многофакторных градуировочных моделей, а высокая селективность и чувствительность новых методов анализа обеспечивает снижение пределов обнаружения многих элементов на несколько порядков по сравнению с методами АХ 60-х годов XX века.
Значительно изменилось соотношение основных анализируемых объектов: уменьшилась доля органических и неорганических объектов. Во много раз возросло внимание к исследованию экологических и биологических объектов [3].
Современная АХ позволяет выполнять определения в огромном диапазоне содержаний компонентов — от 102 до 10 10 масс % (для отдельных элементов). Немалая заслуга в этом принадлежит советской и российской аналитической науке и практике, особенно в создании и развитии целого ряда научных направлений АХ, теоретических основ многих современных методов анализа, методов пред
варительного концентрирования следов неорганических и органических микрокомпонентов, в синтезе большого количества органических реагентов, селективных сорбентов и т.д., что частично отражено в справочнике [4]. В монографиях Ю.А. Золотова [5-7] в широком плане рассматриваются и анализируются достижения отечественной и мировой АХ за последние 40-50 лет: развитие теоретических и методологических основ, перспективные методы анализа, методы разделения и концентрирования, новые аналитические задачи, аналитическое приборостроение, необходимость совершенствования программ преподавания, подготовки кадров; акцент сделан на одном из приоритетных направлений современной АХ — экоаналитике (анализе объектов окружающей среды).
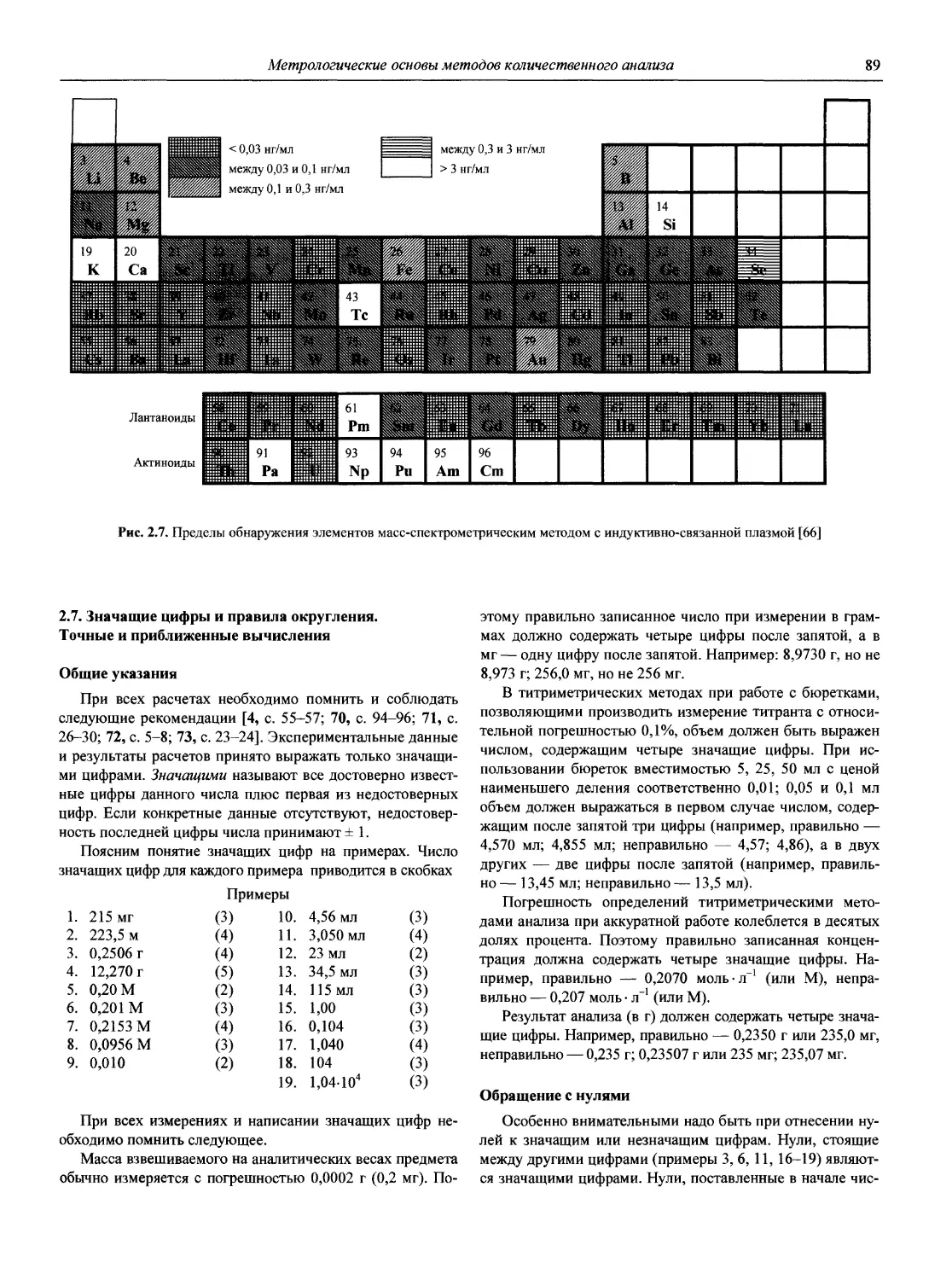
Наиболее полное представление о современном состоянии методов АХ дает недавно (2002 г.) вышедшая в США Энциклопедия по аналитической химии (Encyclopedia of Analytical Chemistry. Applications, Theory and Instrumentation. Edited by R.A. Meyers, John Wiley and Sons Ltd.), состоящая из 15 томов общим объемом 14000 страниц (плюс 6500 иллюстраций).
К сожалению, в связи с общим упадком экономики и соответствующим снижением финансирования науки, в последнее десятилетие XX в. в России значительно снизилась интенсивность фундаментальных и прикладных исследований в области АХ.
Ведущие российские библиотеки не могли приобретать даже крайне необходимую научную литературу, тогда как за рубежом в это время наблюдался настоящий бум по изданию монографий, учебников и справочников, посвященных новейшим исследованиям в области АХ. В итоге подавляющая часть наших химиков-аналитиков оказалась изолированной от последних достижений мировой АХ.
Именно это критическое положение современной отечественной научной и справочной литературы по АХ побудило издательство «Мир и семья» — издателей Справочника — и авторский коллектив этого тома поставить перед собой следующие нестандартные для справочника цели и задачи:
— продемонстрировать современное состояние и возможности аналитической химии, многообразие ее методов, позволяющих решать самые сложные задачи качественного и количественного определения элементов и веществ;
— кратко изложить теоретические основы методов АХ с тем, чтобы Справочник одновременно мог служить своего рода практическим руководством по АХ;
— организовать материал разделов Справочника по принципу исчерпания темы, т.е. по каждому направлению дать необходимый объем теоретических, методологических и справочных сведений, достаточных для практического применения конкретного метода АХ;
— предоставить возможность химикам-практикам ориентироваться во всем многообразии методов АХ, а химикам-исследователям — получить полное представление об информационных базах современной АХ, монографической литературе, библиографии журнальных статей, имея в виду возможность ознакомления с ними через межбиблио
течные абонементы ведущих российских библиотек, связанных с научными библиотеками Европы;
Перечисленные положения демонстрируют принципиальные различия между предыдущим справочником (1965 г.), который отражал состояние АХ первой половины XX века, и настоящим Справочником, информационной базой которого являются в основном теоретические и экспериментальные данные, свидетельствующие о прогрессе методов АХ во второй половине XX века.
При подготовке Справочника широко использовались электронные каталоги библиотек США, ФРГ, Англии, материалы отечественных и зарубежных учебников (прежде всего [1]), многочисленных монографий, приведенных в списках рекомендуемой литературы, а также обзорные статьи в таких изданиях, как «Журнал аналитической химии», «Заводская лаборатория» (Россия), «Analytical Chemistry» (США) и др. за последние 10 лет. Обширнейшая справочная информация по пределам обнаружения и совместному определению следов элементов спектрохимическими методами имеется в монографии [8], а по органическим реагентам — в справочнике [9].
В Справочник включены русско-английский и англорусский словари терминов по аналитической химии, рекомендации ИЮПАК и Научного совета по аналитической химии РАН по терминологии, единицам измерений, которыми руководствовались авторы при изложении материала.
Авторы выражают благодарность:
— заместителю директора издательства «Мир и семья» А. А. Полуде за идею проекта и всем сотрудникам издательства за поистине титаническую работу, проделанную при подготовке Справочника к изданию;
— главным редакторам издательства Н.Н. Атаманенко и Л.В. Белкановой за создание нашего творческого коллектива и плодотворную совместную работу над Справочником;
— ведущему библиографу справочного отдела Библиотеки РАН Н.А. Волковой за помощь в библиографических по
исках в Интернете при формировании информационной базы Справочника.
Авторы будут признательны читателям за конструктивную критику и пожелания.
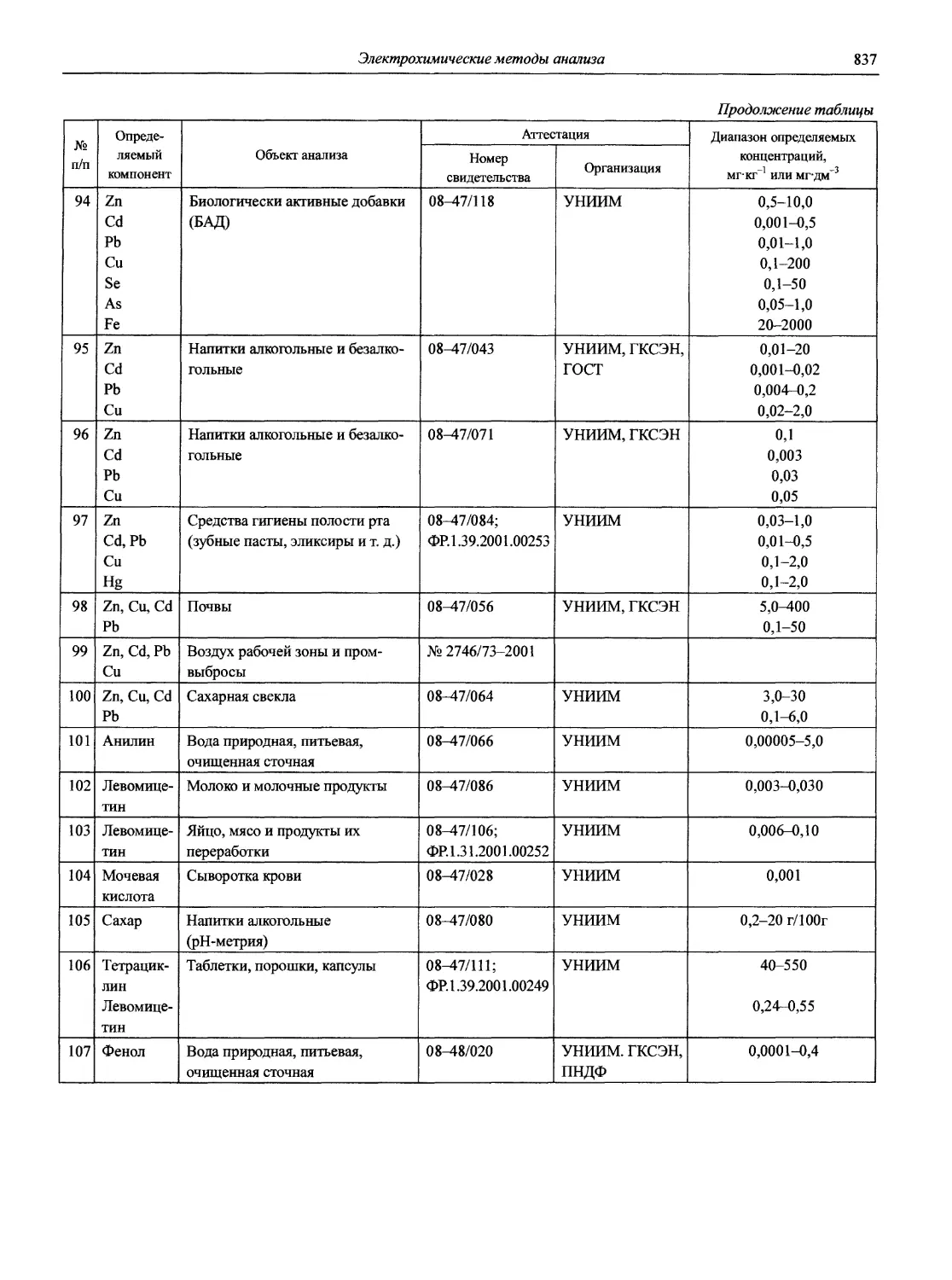
Литература:
1. Основы аналитической химии: В 2 кн. 2-е изд., пере-раб. и доп. / Под ред. акад. РАН Золотова Ю.А. М.: Высшая школа, 2000. Кн. 1. Общие вопросы. Методы разделения. 351 с.; Кн. 2. Методы химического анализа. 494 с.
2. Справочник химика: В 7 т. Т. 4. Аналитическая химия. Спектральный анализ. Показатели преломления / Под ред. чл.-корр. АН СССР Никольского Б.П. М,-Л.: Химия, 1965. 920 с.
3. Архипов Д.Б., Березкин В.Г. Развитие аналитической химии во второй половине XX столетия (наукометрический анализ) // Журн. аналит. химии. 2002. Т. 57, № 7. С. 699-703 (см. компакт-диск).
4. Саввин С.Б. Кто есть кто в российской аналитической химии. М.: Наука, 2000.234 с.
5. Золотов Ю.А. Аналитическая химия: проблемы и достижения. М.: Наука, 1992.225 с.
6. Золотов Ю.А. Аналитическая химия: фрагменты картины / ОНТИ ГЕОХИ РАН. М„ 1999.144 с.
7. Золотов Ю.А. Перемена столетий. М: ГЕОХИ, 2001. 212 с.
8. Lobinski R., Marchenko Z. Spectrochemical Trace Analysis of Metals and Metalloids // Wilson and Wilson’s. Comprehensive Analytical Chemistry. Vol. 30. Amsterdam etc: Elsevier, 1997. 808 p.
9. Dictionary of Analytical Reagents / Editorial board. A. Townshend (et al.). Series: Chapman and Hall Chemical database: London: Chapman a. Hall, 1993.1370 p.
Профессор, доктор химических наук И.П. Калинкин, кандидат химических наук В.И. Мосичев
К ЧИТАТЕЛЯМ
Издательство с благодарностью примет и учтет при последующих изданиях все ваши замечания, предложения и пожелания
Раздел 1
ИНФОРМАЦИОННЫЕ БАЗЫ И ОБЩИЕ ВОПРОСЫ АНАЛИТИЧЕСКОЙ ХИМИИ
Авторы-составители: проф., д. х. н. И.П. Калинкин,
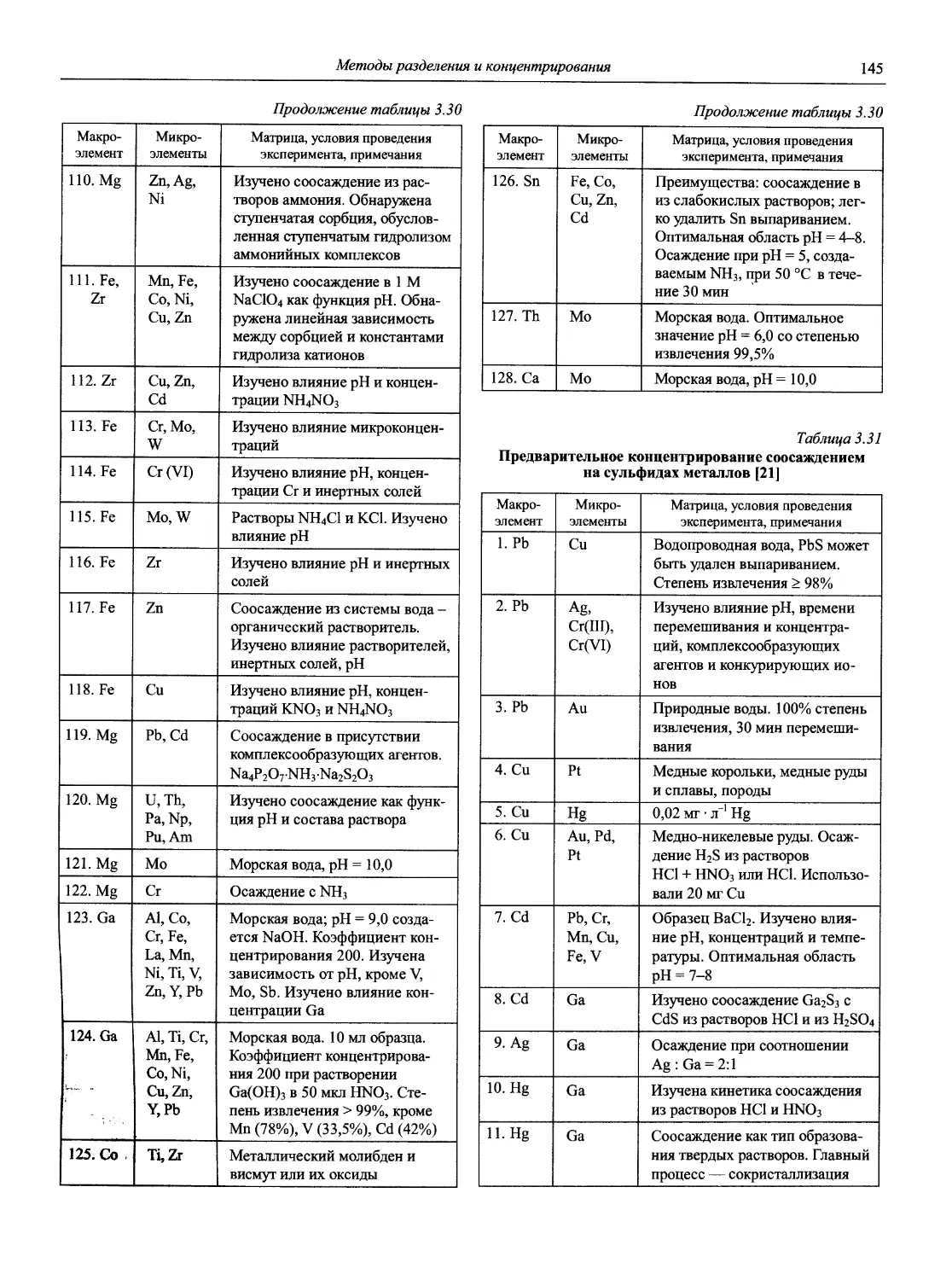
к. х. н. В.А. Демин
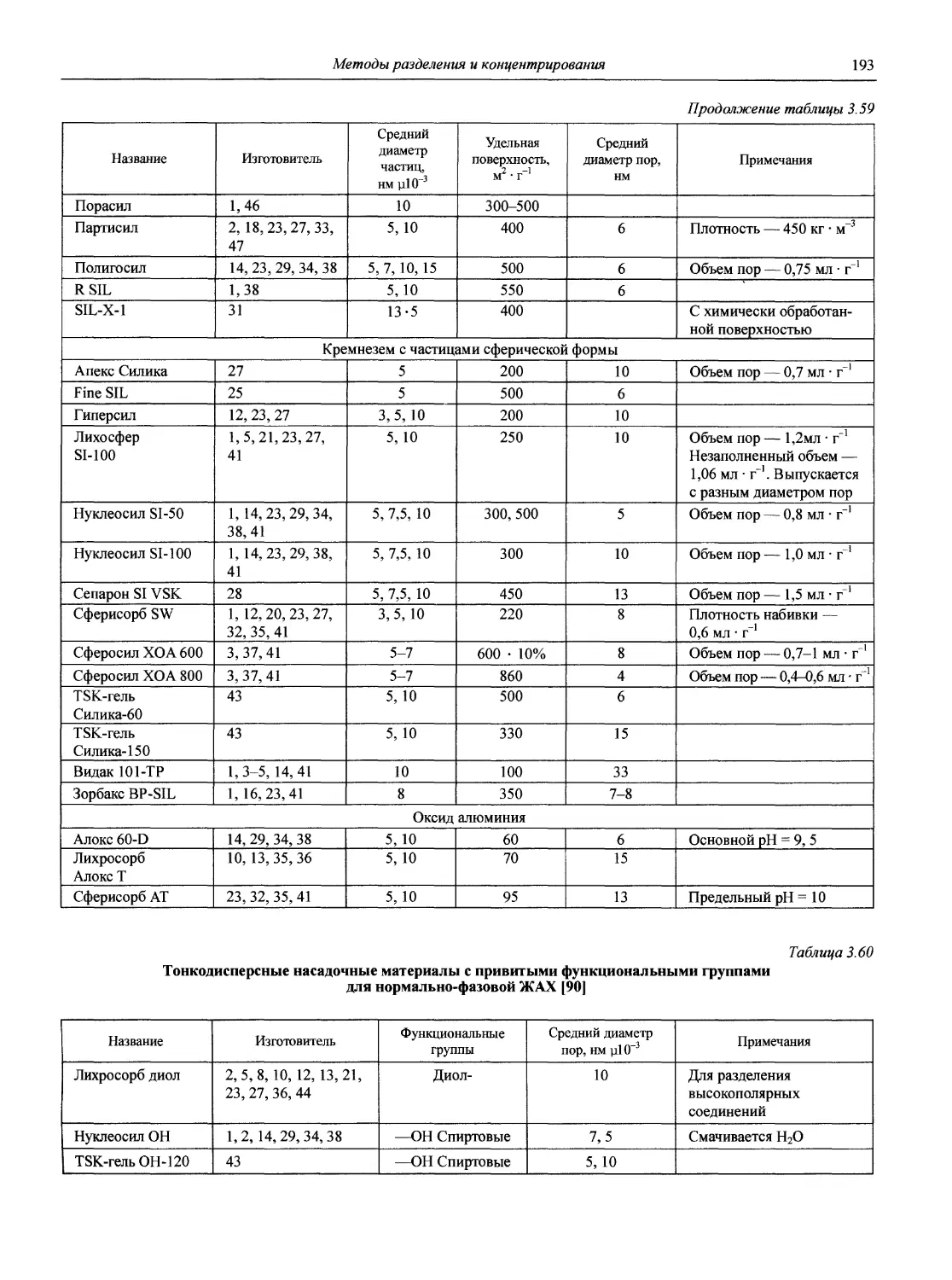
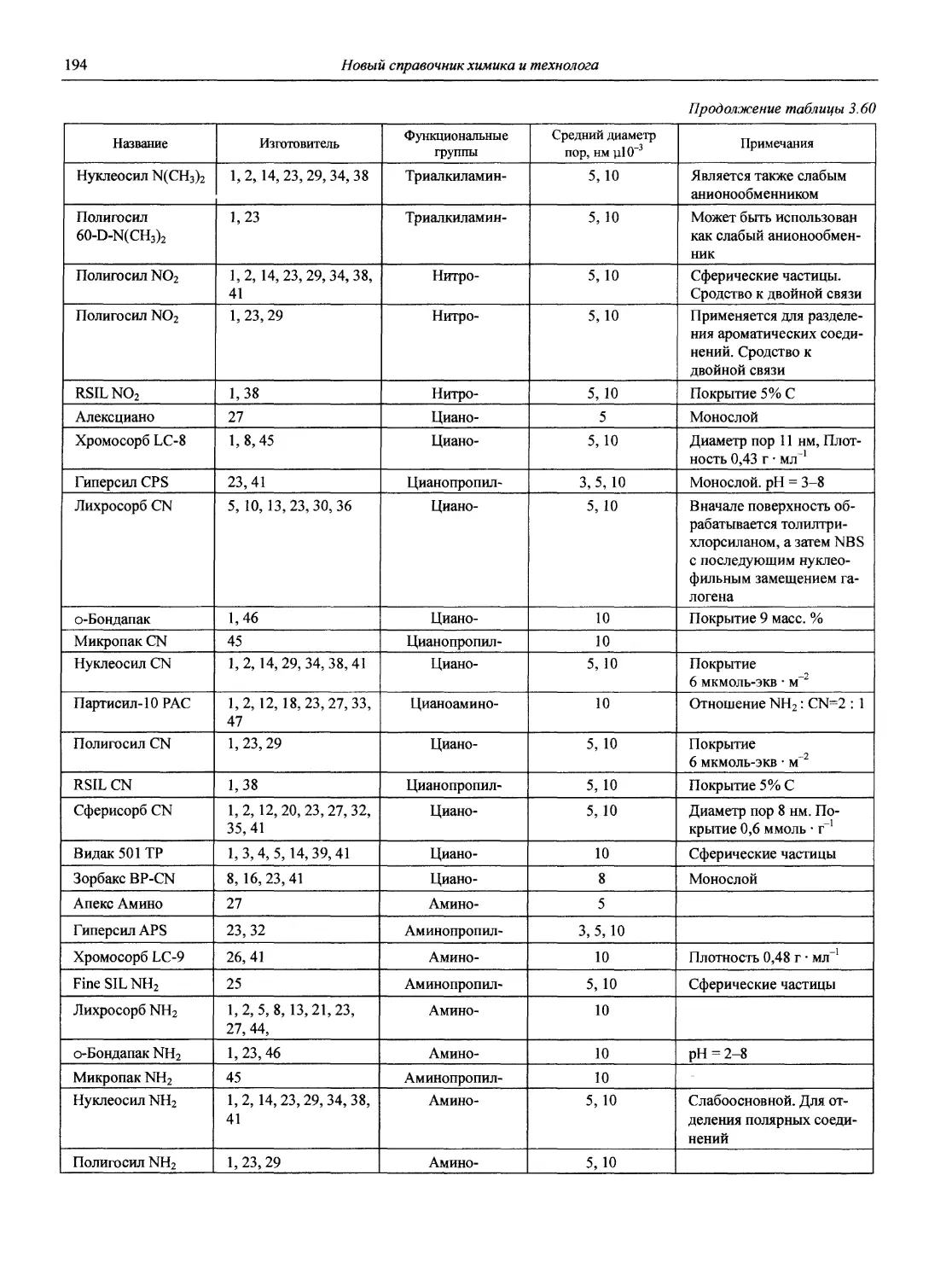
1.1. Основные печатные и электронные справочные источники по аналитической химии
1.1.1. Рекомендации Международного союза теоретической и прикладной химии (ИЮПАК) по вопросам номенклатуры аналитической химии и их перевод в России
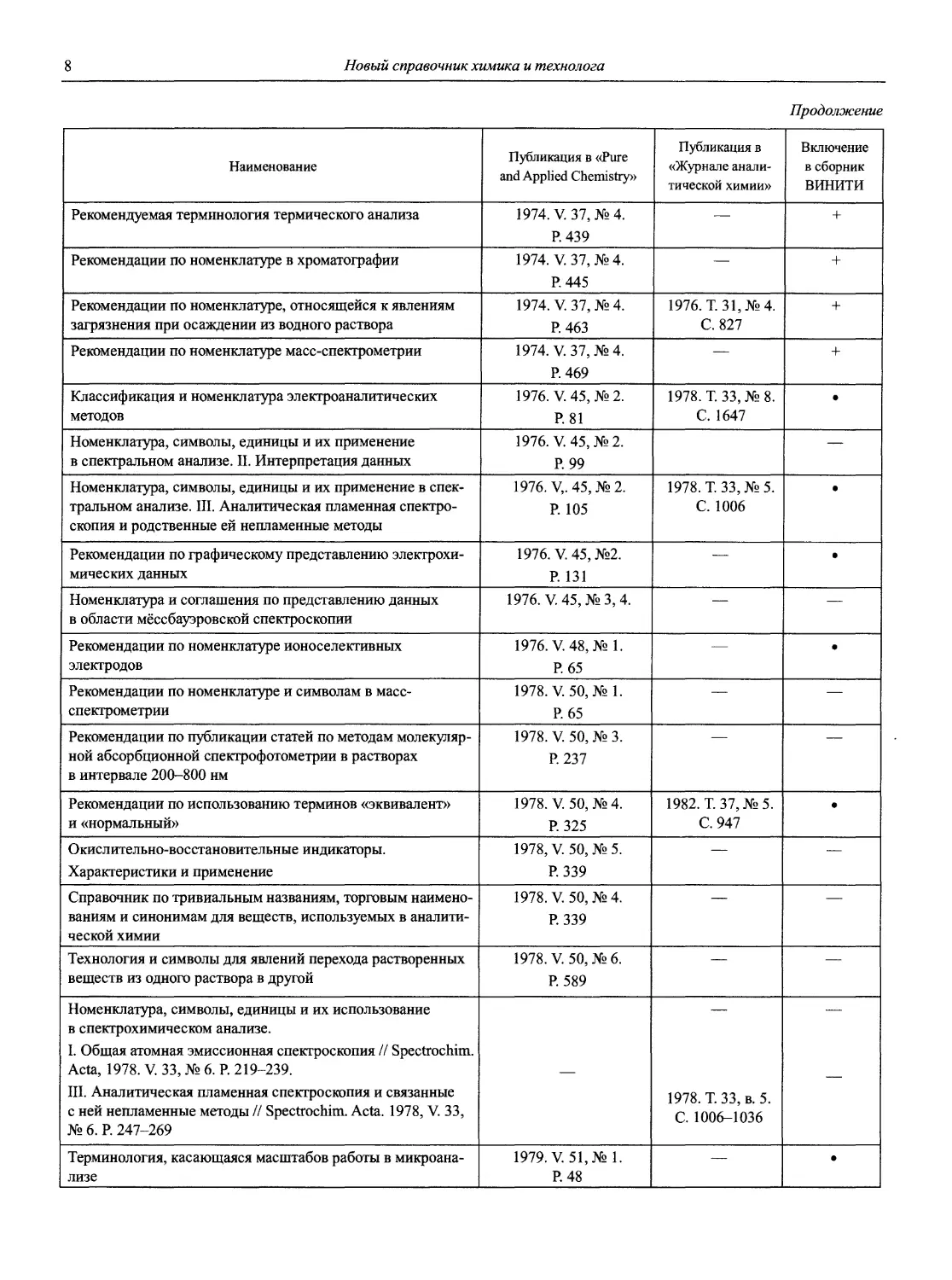
Наименование Публикация в «Риге and Applied Chemistry» Публикация в «Журнале аналитической химии» Включение в сборник ВИНИТИ
Рекомендации по терминологии, относящейся к использованию прецизионных весов 1960. V. l.P. 171 — —
Рекомендации по представлению материалов и номенклатуре в газовой хроматографии 1964. V. 8. P. 553 — +
Практическое измерение pH в амфипротонных и смешанных растворителях 1969. V. 18, №3. P.421 — •
Рекомендуемая номенклатура для титриметрического анализа 1969. V. 18, №3. P.427 — +
Рекомендации по представлению результатов химического анализа 1969. V. 18, №3. P.437 1971. Т. 26, №4. С.1021 +
Рекомендуемые символы для характеристики равновесий в растворах 1969. V. 18, №3. P.459 — +
Рекомендуемая номенклатура для жидкость - жидкостного распределения 1970. V. 21, № 1. P. 109 1971. Т. 26, №4. С.1019 +
Рекомендуемая номенклатура для автоматического анализа 1970. V. 21, №4. P. 527 — +
Рекомендации по номенклатуре ионного обмена 1972. V. 29, № 4. P. 617 1975. Т. 29, № 6. С. 1448 +
Номенклатура, символы, единицы и их использование в спектральном анализе. I. Обычная атомно-эмиссионная спектроскопия 1972. V. 30, №3,4. P. 651 — +
8
Новый справочник химика и технолога
Продолжение
Наименование Публикация в «Риге and Applied Chemistry» Публикация в «Журнале аналитической химии» Включение в сборник ВИНИТИ
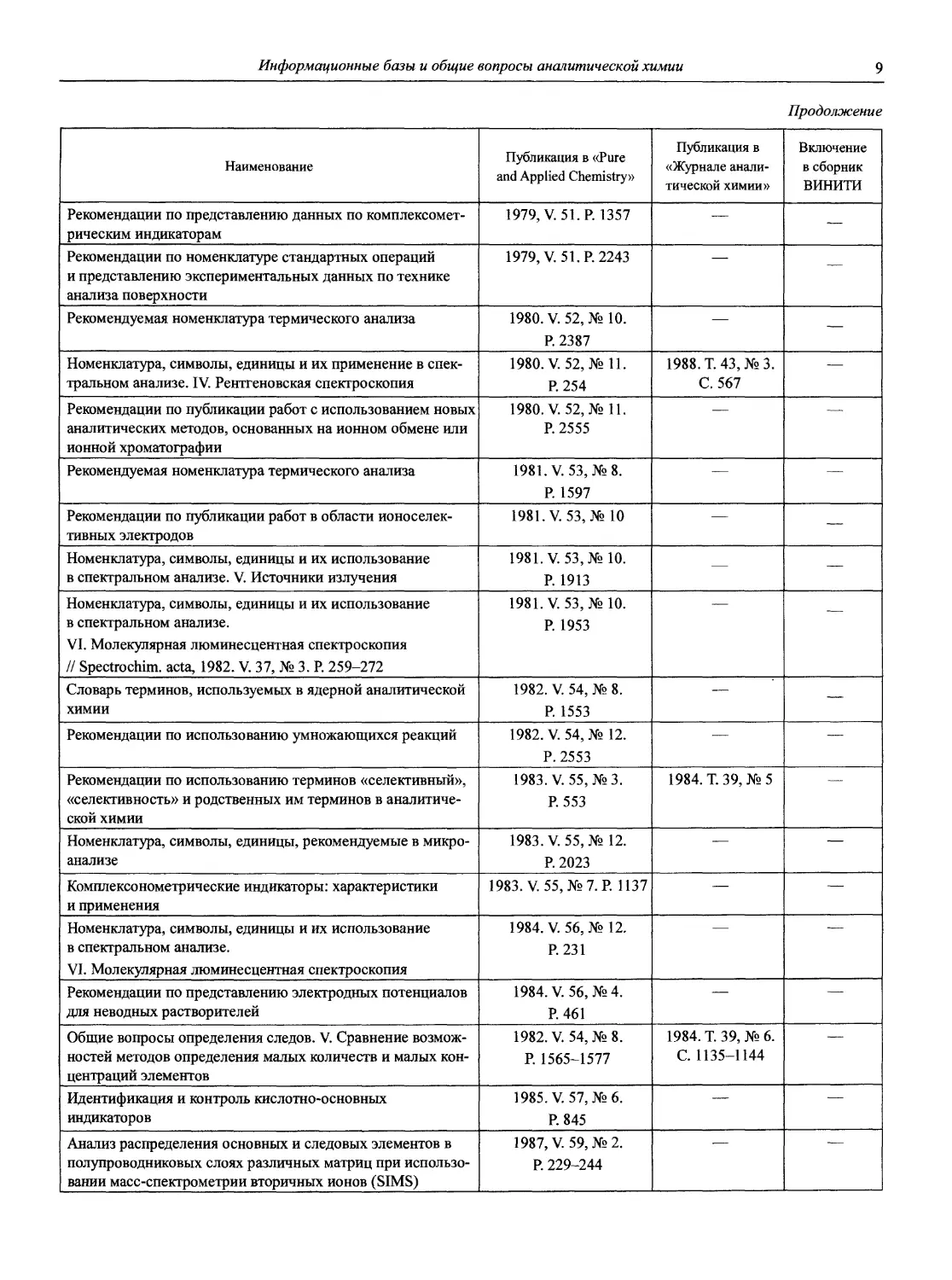
Рекомендуемая терминология термического анализа 1974. V. 37, №4. Р.439 — +
Рекомендации по номенклатуре в хроматографии 1974. V. 37, №4. Р445 — +
Рекомендации по номенклатуре, относящейся к явлениям загрязнения при осаждении из водного раствора 1974. V. 37, № 4. Р.463 1976. Т. 31, №4. С. 827 +
Рекомендации по номенклатуре масс-спектрометрии 1974. V. 37, № 4. Р.469 — +
Классификация и номенклатура электроаналитических методов 1976. V. 45, №2. Р. 81 1978. Т. 33, № 8. С.1647 •
Номенклатура, символы, единицы и их применение в спектральном анализе. II. Интерпретация данных 1976. V. 45, №2. Р. 99 —
Номенклатура, символы, единицы и их применение в спектральном анализе. III. Аналитическая пламенная спектроскопия и родственные ей непламенные методы 1976. V,. 45, №2. Р. 105 1978. Т. 33, № 5. С.1006 •
Рекомендации по графическому представлению электрохимических данных 1976. V. 45, №2. Р. 131 — •
Номенклатура и соглашения по представлению данных в области мёссбауэровской спектроскопии 1976. V. 45, №3,4. — —
Рекомендации по номенклатуре ионоселективных электродов 1976. V. 48, № 1. Р. 65 — •
Рекомендации по номенклатуре и символам в масс-спектрометрии 1978. V. 50, № 1. Р. 65 — —
Рекомендации по публикации статей по методам молекулярной абсорбционной спектрофотометрии в растворах в интервале 200-800 нм 1978. V. 50, № 3. Р. 237 — —
Рекомендации по использованию терминов «эквивалент» и «нормальный» 1978. V. 50, №4. Р. 325 1982. Т. 37, № 5. С. 947 •
Окислительно-восстановительные индикаторы. Характеристики и применение 1978, V. 50, № 5. Р. 339 — —
Справочник по тривиальным названиям, торговым наименованиям и синонимам для веществ, используемых в аналитической химии 1978. V. 50, №4. Р. 339 — —
Технология и символы для явлений перехода растворенных веществ из одного раствора в другой 1978. V. 50, № 6. Р. 589 — —
Номенклатура, символы, единицы и их использование в спектрохимическом анализе. I. Общая атомная эмиссионная спектроскопия // Spectrochim. Acta, 1978. V. 33, № 6. Р. 219-239. III. Аналитическая пламенная спектроскопия и связанные с ней непламенные методы // Spectrochim. Acta. 1978, V. 33, № 6. Р. 247-269 — 1978. Т. 33, в. 5. С. 1006-1036 —
Терминология, касающаяся масштабов работы в микроанализе 1979. V. 51, № 1. Р.48 — •
Информационные базы и общие вопросы аналитической химии
9
Продолжение
Наименование Публикация в «Риге and Applied Chemistry» Публикация в «Журнале аналитической химии» Включение в сборник ВИНИТИ
Рекомендации по представлению данных по комплексометрическим индикаторам 1979, V. 51. Р. 1357 — —
Рекомендации по номенклатуре стандартных операций и представлению экспериментальных данных по технике анализа поверхности 1979, V. 51. Р. 2243 — —
Рекомендуемая номенклатура термического анализа 1980. V. 52, № 10. Р. 2387 — —
Номенклатура, символы, единицы и их применение в спектральном анализе. IV. Рентгеновская спектроскопия 1980. V. 52, № 11. Р.254 1988. Т. 43, №3. С. 567 —
Рекомендации по публикации работ с использованием новых аналитических методов, основанных на ионном обмене или ионной хроматографии 1980. V. 52, № 11. Р. 2555 — —
Рекомендуемая номенклатура термического анализа 1981. V. 53, №8. Р. 1597 — —
Рекомендации по публикации работ в области ионоселективных электродов 1981. V. 53, № 10 — —
Номенклатура, символы, единицы и их использование в спектральном анализе. V. Источники излучения 1981. V. 53, № 10. Р. 1913 — —
Номенклатура, символы, единицы и их использование в спектральном анализе. VI. Молекулярная люминесцентная спектроскопия И Spectrochim. acta, 1982. V. 37, № 3. Р. 259-272 1981. V. 53, № 10. Р. 1953 — —
Словарь терминов, используемых в ядерной аналитической химии 1982. V. 54, № 8. Р. 1553 — —
Рекомендации по использованию умножающихся реакций 1982. V. 54, № 12. Р.2553 — —
Рекомендации по использованию терминов «селективный», «селективность» и родственных им терминов в аналитической химии 1983. V. 55, №3. Р. 553 1984. Т. 39, № 5 —
Номенклатура, символы, единицы, рекомендуемые в микроанализе 1983. V. 55, № 12. Р. 2023 — —
Комплексонометрические индикаторы: характеристики и применения 1983. V. 55, № 7. Р. 1137 — —
Номенклатура, символы, единицы и их использование в спектральном анализе. VI. Молекулярная люминесцентная спектроскопия 1984. V. 56, № 12. Р.231 — —
Рекомендации по представлению электродных потенциалов для неводных растворителей 1984. V. 56, № 4. Р. 461 — —
Общие вопросы определения следов. V. Сравнение возможностей методов определения малых количеств и малых концентраций элементов 1982. V. 54, №8. Р. 1565-1577 1984. Т. 39, № 6. С.1135-1144 —
Идентификация и контроль кислотно-основных индикаторов 1985. V. 57, №6. Р. 845 — —
Анализ распределения основных и следовых элементов в полупроводниковых слоях различных матриц при использовании масс-спектрометрии вторичных ионов (SIMS) 1987, V. 59, № 2. Р. 229-244 — —
10
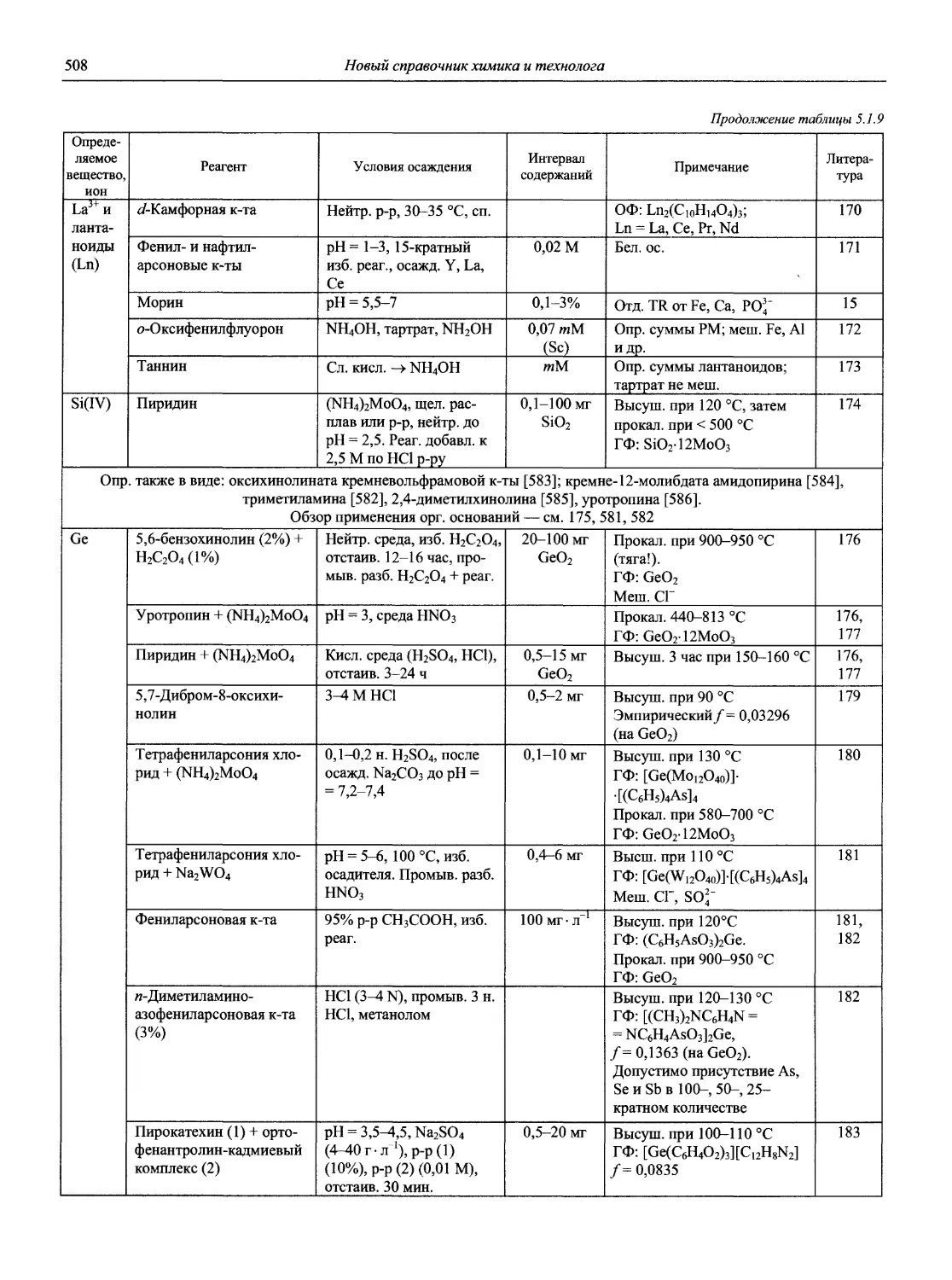
Новый справочник химика и технолога
Продолжение
Наименование Публикация в «Риге and Applied Chemistry» Публикация в «Журнале аналитической химии» Включение в сборник ВИНИТИ
Описательная классификация электронной спектроскопии 1987. V. 59, № 10. Р. 1343-1406 — —
Критическая оценка градуировочных операций при анализе распределения легирующих элементов в кремнии и арсениде галлия 1988. V. 60, № 3. Р. 437^444 — —
Номенклатура, символы, единицы и их использование в спектрохимических анализах. VII. Молекулярная абсорбционная спектроскопия: ультрафиолетовая и видимая (UV/VIS) 1988. V. 60, №9. Р. 1450-1460 — —
Определение и классификация мешающих примесей при аналитических операциях 1989. V. 61. Р. 91-95 — —
Номенклатура по пробоотбору в аналитической химии (Рекомендации 1990) 1990. V. 62, № 6. Р. 1193-1208 — —
Обзор экспериментальной техники в исследовании поверхности 1990. V. 62, № 12. Р. 2297-2322 — —
Номенклатурная система по рентгентовской спектроскопии (X-Ray Spectroscopy) 1991. V. 63. Р. 735-746 — —
Английская аббревиатура экспериментальной техники исследования поверхности и химической спектроскопии 1991. V. 63, №6. Р. 887-893 — —
Представление результатов химического анализа 1994. V. 66. Р. 595-608 1998. Т. 53, №9. Р. 999-1008 —
Методы разделения 1994. V. 66. Р. 2501-2512 — —
Номенклатура по масс-спектрометрии 1994. V. 66. Р. 305-334 — —
Термоаналитические и энтальпиметрические методы 1994. V. 66. Р. 2487-2492 — —
Автоматический анализ 1994. V. 66. Р. 2493-2500 — —
Электрохимический анализ 1995. V. 67. Р. 507-518 — —
Магнитные методы анализа 1995. V. 67. Р. 593-596 — —
Номенклатура кинетических методов анализа 1993. V. 65, № 10. Р. 2291-2298 1995. V. 67. Р. 601-613 1998. Т. 53, № 1. С. 110-116 —
Номенклатура по радиоаналитической химии 1995. V. 67. Р. 1943-1949 1998. Т. 53, № 10. С.1112-1120 —
Характеристики качества анализа 1995. V. 67. Р. 331-343; 649-669 — —
Применение 1995. V. 67. Р. 615-648; 1377-1406; 1533-1548; 1563-1608 — —
Номенклатура при оценке определения аналитических включений и возможности их количественного определения 1995. V. 67. Р. 1699-1723 — —
Информационные базы и общие вопросы аналитической химии
11
Продолжение
Наименование Публикация в «Риге and Applied Chemistry» Публикация в «Журнале аналитической химии» Включение в сборник ВИНИТИ
Каталитические методы анализа: характеристика, классификация и методология 1995. V. 67, № 4. Р. 601-613 — —
Селективность в аналитической химии (ИЮПАК Рекомендации 2001) 2001. V. 73, №8. Р. 1381-1386 — —
Примечание. Таблица составлена по данным журнала Pure and Applied Chemistry и публикаций [1-7]. Переводы, отмеченные +, опубликованы в [1]; • — в [2].
Литература
1. Номенклатурные правила ИЮПАК по химии. Том 1, полутом 2. Неорганическая химия, физическая химия, аналитическая химия. М.: ВИНИТИ, 1979. 660 с.
2. Номенклатурные правила ИЮПАК. Т. 4. Аналитическая химия. М.: ВИНИТИ, 1985. 177 с.
3. В Научном совете АН СССР по аналитической химии. Рекомендации ИЮПАК по использованию терминов «эквивалент» и «нормальный» // Журнал аналитической химии. 1982. Т. 37, вып. 5. С. 947-961.
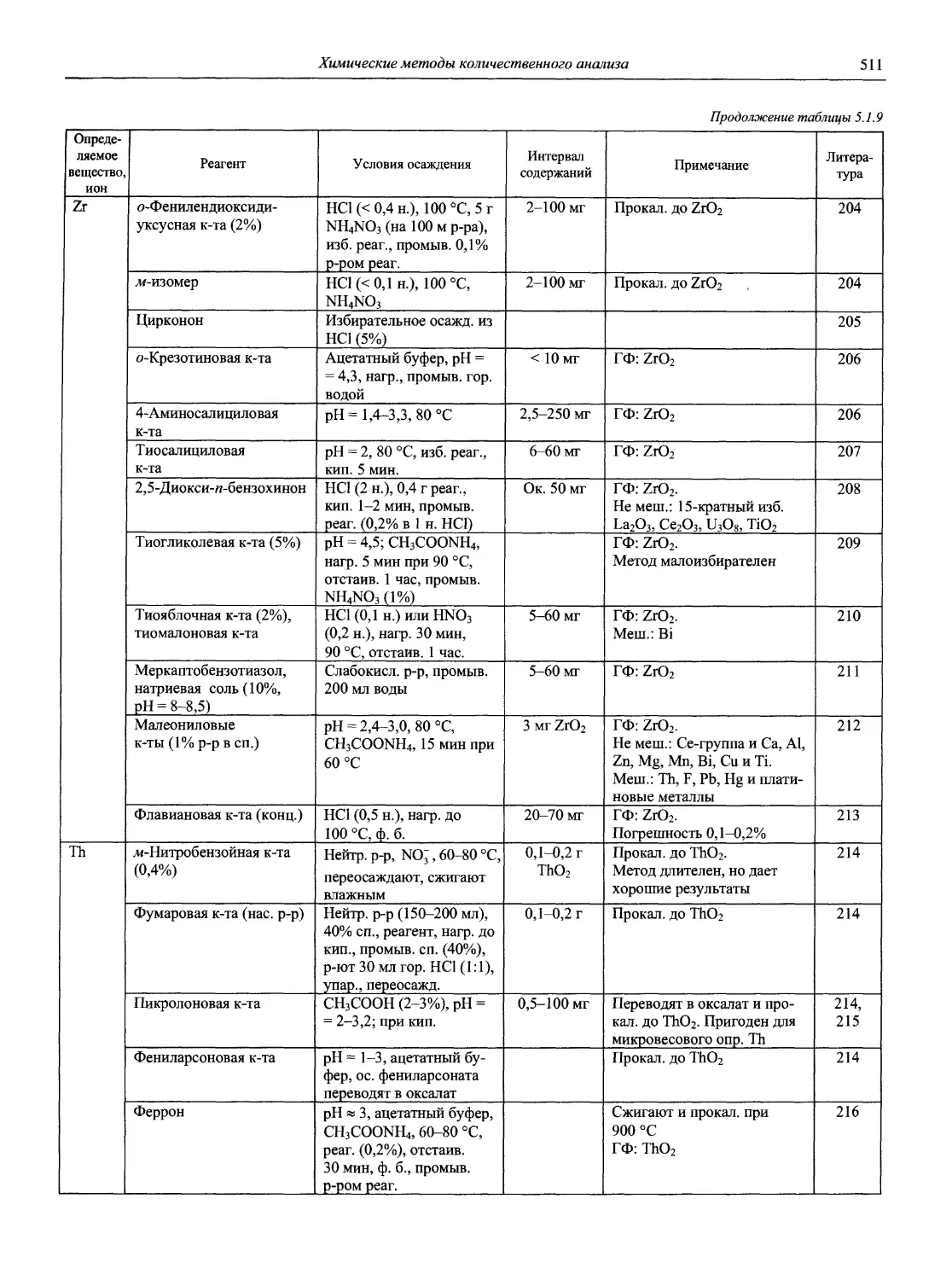
4. Нейман Е. Я. // Журнал аналитической химии. 1991. Т. 46. С. 393-405.
5. Берлин А. А. // Журнал аналитической химии. 1989. Т. 44, вып. 4. С. 762-764.
6. Иванов В.М. // Журнал аналитической химии. 1999. Т. 54, вып. 3. С. 335-336.
7. Compendium of Analytical Nomenclature: Definitive Rules 1997 / Prepared for publications by J. Inczedy, T. Lenguel, A. M. Ure. 3rd ed. Oxford: Blackwell Science, 1998. ISBN 0865426255m. (Подробное содержание см. компакт-диск).
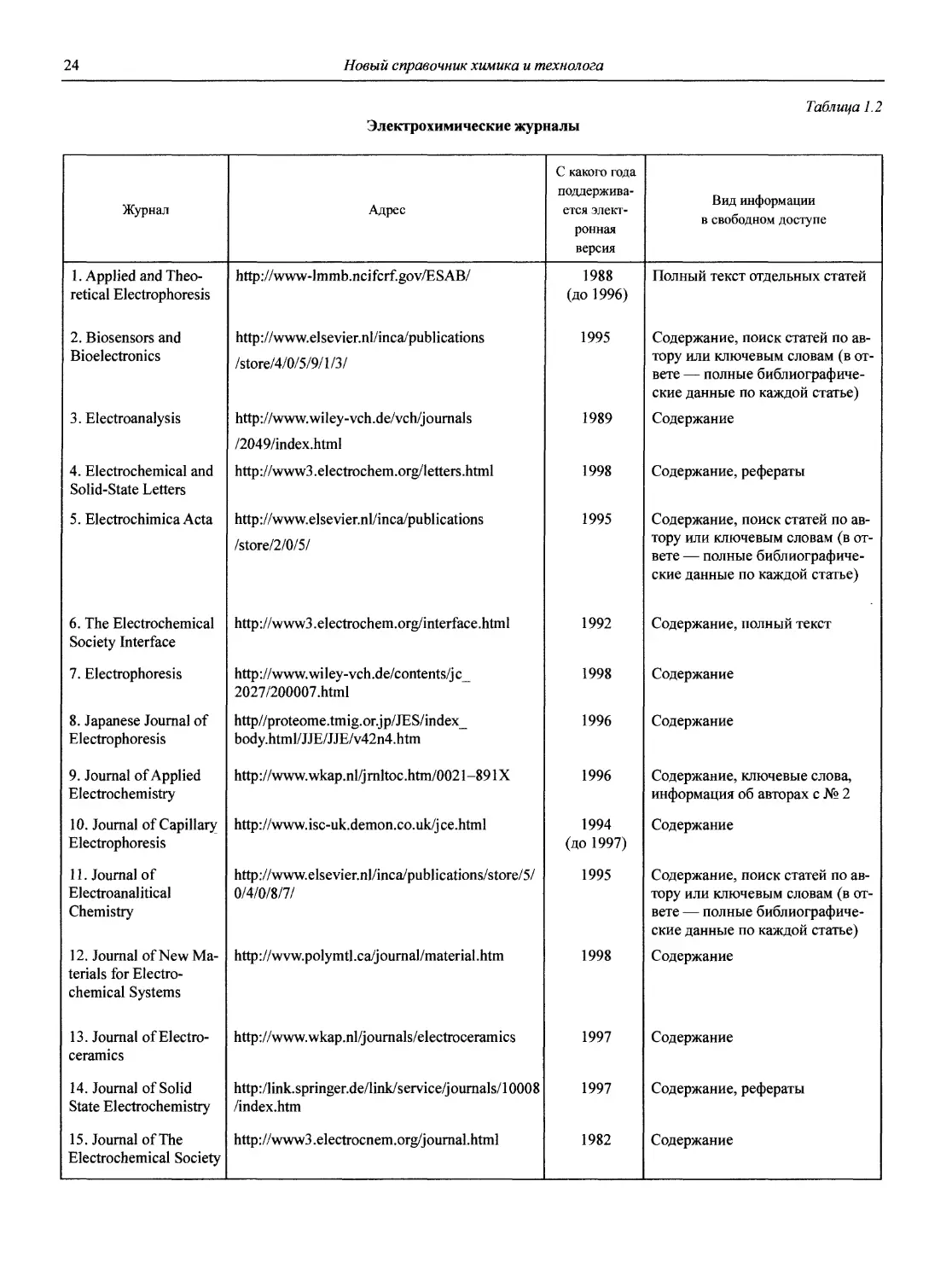
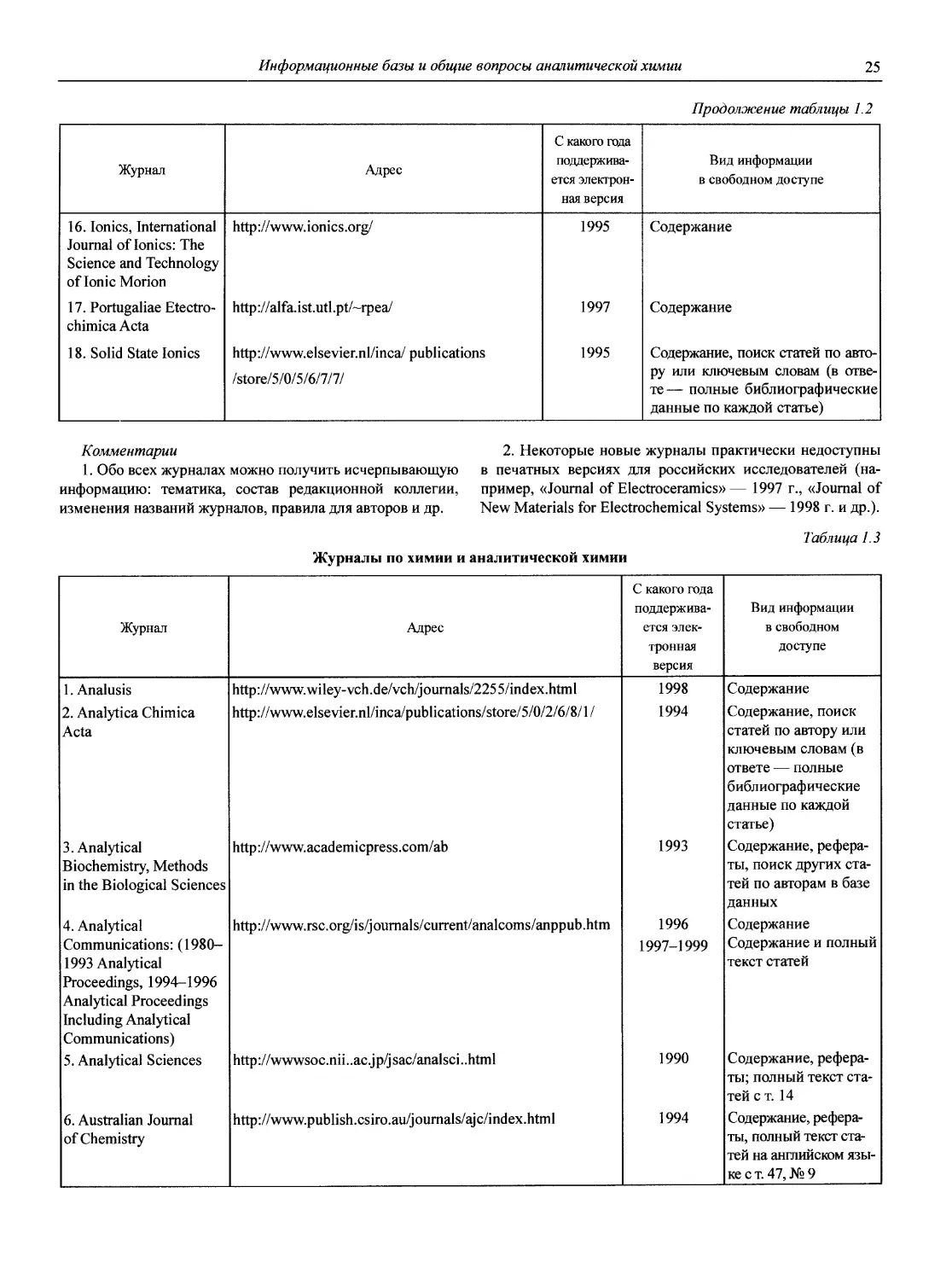
1.1.2. Журналы по аналитической химии
1. American Laboratory (Shelton, СТ: International
Scientific Communicatoin, Inc. USA).
2. Analusis (Rueil-Malmaison: Societe de production documentaires. France).
3. Analyst (London: Royal Society of Chemistry. Great
Britain).
4. Analytica Chimica Acta (Amsterdam: Elsevier Pub. Co. Netherlands).
5. Analytical Abstracts (London: Royal Society of Chemistry. Great Britain).
6. Analytical Biochemistry (New York: Academic Press. USA).
7. Analytical Chemistry (Washington, DC. USA).
8. Analytical Communications (до 1999 г. Analytical Proceedings) (Cambridge. Great Britain).
9. Analytical and Bioanalytical Chemistry (до 2001 г. Fresenius’ Journal of Analytical Chemistry) (Berlin; Heidelberg. Germany).
10. Analytical Letters (New York: M. Dekker. USA).
11. Analytical Methods and Instrumentation (Chichester: Wiley. England).
12. Analytical Sciences (Tokyo. Japan).
13. Applied Spectroscopy (Baltimore, Md: Society for Applied Spectroscopy. USA).
14. Atomic Spectroscopy (Norwalk, Conn.: Perkin/Elmer Corp. USA).
15. Chemia Analityczna (Warszawa: Pamstwowe Wydawn. Naukowe. Poland).
16. Chromatographia (Long Island City, N.Y. USA).
17. Chromatographic Reviews (Amsterdam: Elsevier. Netherlands).
18. Clinica Chimica Acta (Amsterdam: Elsevier Scientific Pub. Co. Netherlands).
19. Clinical Chemistry (Baltimore, Md: P.B. Hoeber. USA).
20. CP Information Newsletter (inductively coupled plasma discharges for spectrochemical analysis) (Amherst, Mass. USA).
21. Critical Reviews in Analytical Chemistry (Boca Raton, Fla.: CRC Press. USA).
22. Electrophoresis.
23. Environmental Science and Technology (Washington, DC. USA).
24. International Journal of Environmental Analytical Chemistry (New York: Gordon and Breach. USA).
25. International Journal of Mass Spectrometry and Ion Processes (Amsterdam: Elsevier. Netherlands).
26. International Labmate (St. Albans, Herts. Great Britain).
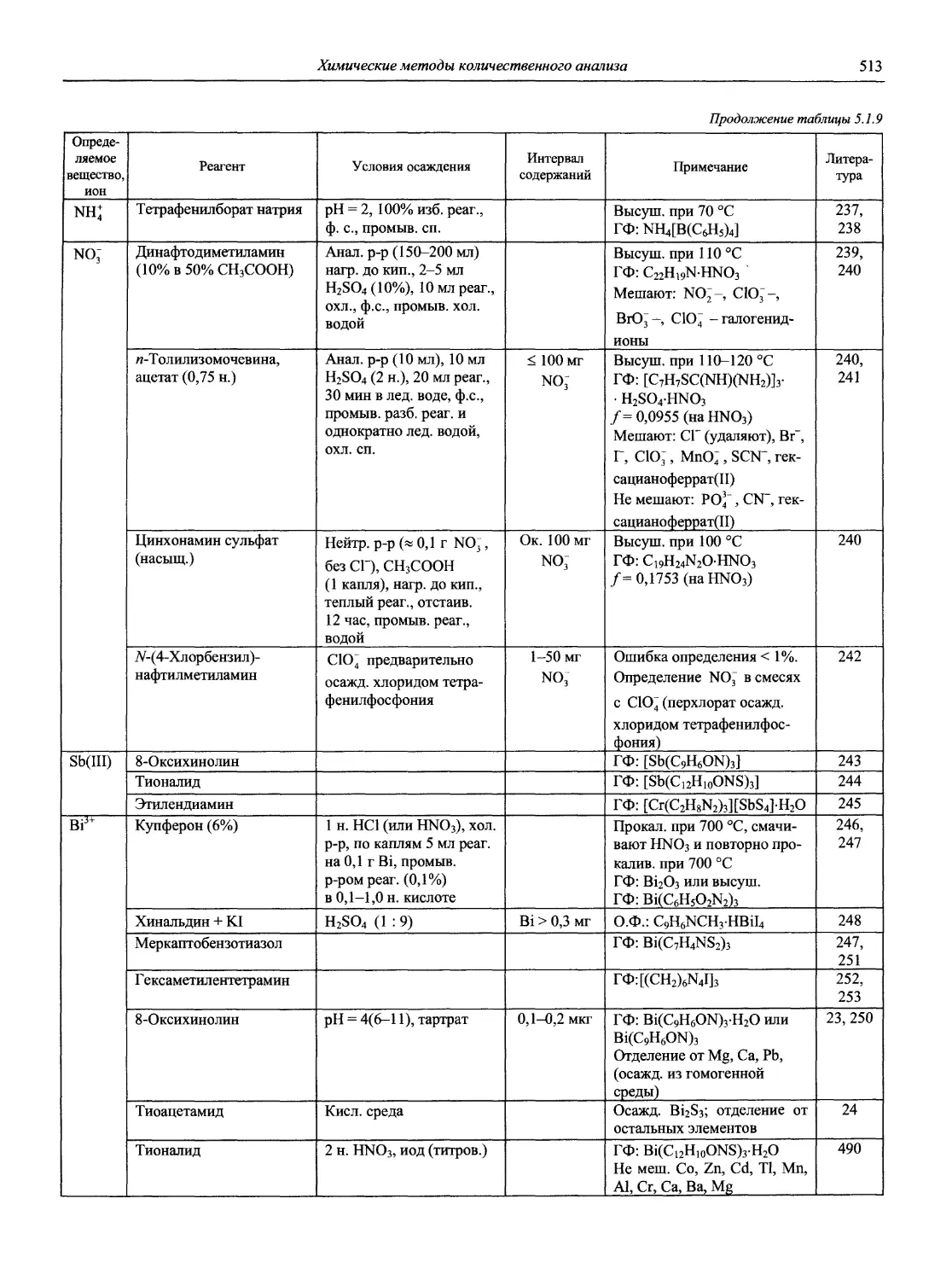
27. International Laboratory (Greens Farms, Conn. USA).
28. Journal of Analytical Atomic Spectrometry (London: Royal Society of Chemistry, Great Britain).
29. Journal of Analitical Toxicology.
30. Journal of Association of Official Analytical Chemists (Washington, DC. USA).
31. Journal of Chromatographic Science (Niles, Ill.: Preston Publications, USA).
32. Journal of Chromatography (Amsterdam. Neitherlands).
33. Journal of Chromatography Biomedical Application (Netherland).
34. Journal of Electroanalytical Chemistry (Lausanne: Elsevier. Switzerland).
3 5. Journal of High Resolution Chromatography and Chromatography Communications.
36. Journal of Pharmaceutical and Biomedical Analysis (New York. USA).
37. Journal of Radioanalytical and Nuclear Chemistry (Articles and Letters) (Lausanne: Elsevier Sequoia. Switzerland).
38. Journal of Separation Science (Weinheim: Wiley-VCH. Germany).
12
Новый справочник химика и технолога
39. Laboratory Practice (London: United Trade Pr. Great Britain).
40. Microchemical Journal (New York: Academic Press. USA).
41. Mikrochimica Acta (New York; Vienna).
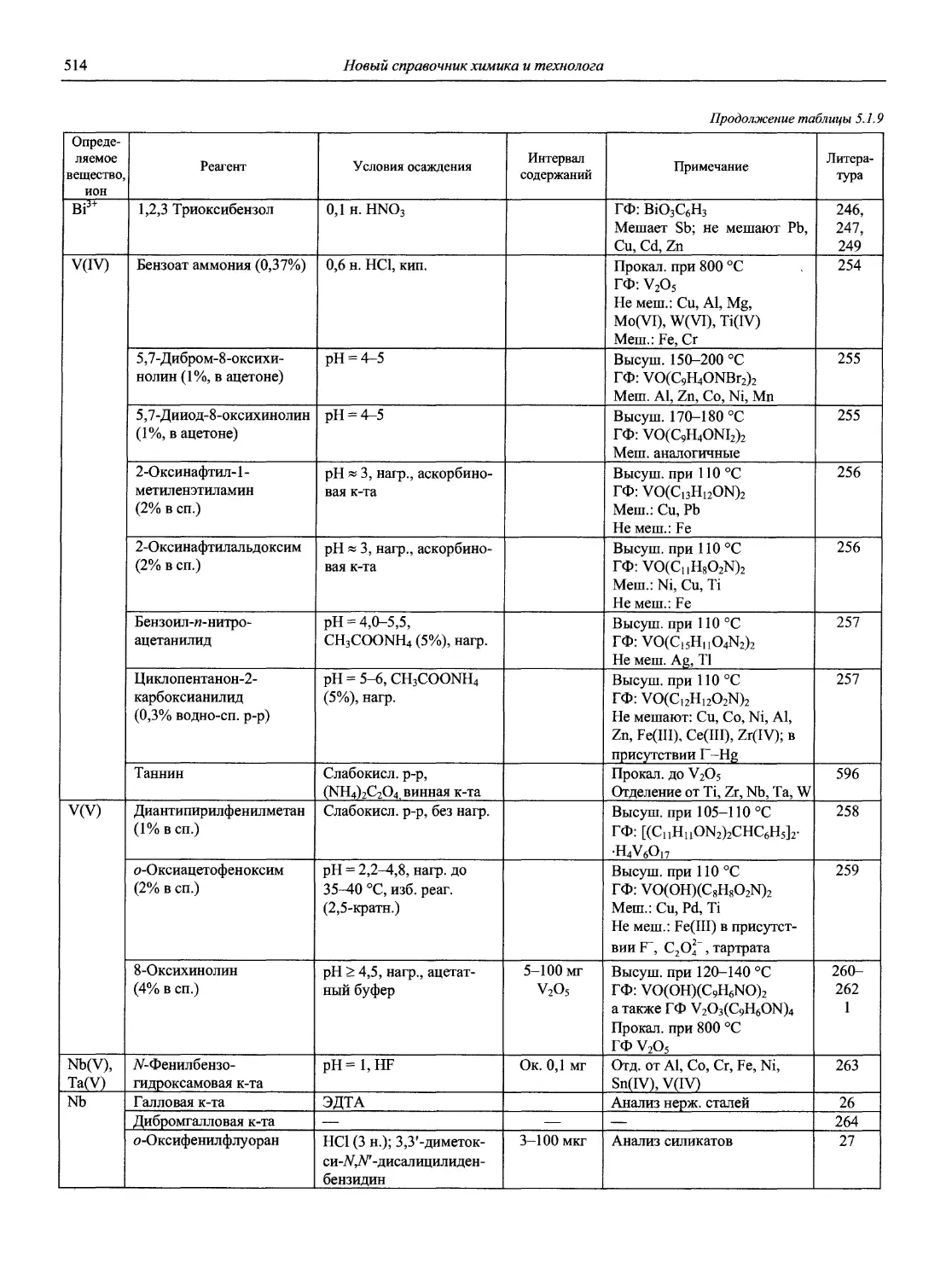
42. Sensors and Actuators В Chemical.
43. Separation Science and Technology (New York: M. Dekker. USA).
44. Solvent Extraction and Ion Exchange.
45. Spectrochimica Acta. Part A: Molecular Spectroscopy (Oxford; New York: Pergamon Press).
46. Spectrochimica Acta. Part B: Atomic Spectroscopy (Amsterdam; New York: Elsevier).
47. Surface and Interface Analysis (London; Philadelphia: PA: Heyden & Son).
48. Taianta (Oxford; New York: Pergamon Press).
49. Thermochimica Acta (Amsterdam: Elsevier. Netherlands).
50. Trends in Analytical Chemistry (Amsterdam: Elsevier Scientific Pud. Co. Neitherlands).
51. X-Ray Spectrometry (London: Heyden. Great Britain).
52. Бунсэки кагаку (Япония).
53. Высокочистые вещества (журнал прекратил существование в 1999 г.) (Россия).
54. Датчики и системы (Россия).
55. Журнал аналитической химии (РАН. Россия).
56. Журнал прикладной спектроскопии (Минск).
57. Заводская лаборатория (Россия).
58. Феньси хуасюэ (Китай).
59. Сенсоры (с 2001 г.) (Россия).
Комментарий
Кроме перечисленных, существует много журналов более широкого профиля, в которых публикуются труды по аналитической химии. Например, из изданий, выходящих на русском языке, можно назвать «Доклады Российской академии наук», «Успехи химии», «Журнал физической химии», «Электрохимию», «Известия вузов. Химия и химическая технология» и др.
1.1.3. Справочники и руководства общего характера
1. Артеменко А.И., Тикунова И.В., Малеванный В.А. Справочное руководство по химии. 2-е изд. М.: Высшая школа, 2002.
2. Бишоп Э. Индикаторы: Справочное рук-во / Пер. с англ.; Под ред. д. х. н. Марова И.Н. М.: Мир, 1976. Т. 1,495 с.;Т. 2,446 с.
3. Бурдун Г.Д. Справочник по международной системе единиц. М.: Изд-во стандартов, 1971.
4. Гиллебранд В.Ф., Лендль Г.Э., Брайт Г.А., Гофман Д.И. Практическое руководство по неорганическому анализу. 3-е изд. / Пер. с англ.; Под. ред. проф. Лурье Ю.Ю. М.: Химия, 1957. 1016 с.
5. Коренман И.М. Органические реагенты в неорганическом анализе. М.: Химия, 1980. 448 с.
6. Коростелев П.П. Реактивы для технического анализа: Справочник. М.: Металлургия, 1988. 383 с.
7. Коростелев П.П. Титриметрический и гравиметрический анализ в металлургии: Справочник. М.: Металлургия, 1986. 320 с.
8. Коростелев П.П. Фотометрический и комплексонометрический анализ в металлургии: Справочник: М.: Металлургия, 1984. 272 с.
9. Лазарев А.И. Органические реактивы в анализе металлов: Справочник. М.: Металлургия, 1980. 232 с.
10. Лурье Ю.Ю. Справочник по аналитической химии. 6-е изд., перераб. и доп. М.: Химия, 1989. 448 с.
11. Номенклатурные правила ИЮПАК (IUPAC) по химии. Полутом 1. 1993. 406 с.; 2. 1993. 447 с. М.: ВИНИТИ.
12. Руководство по аналитической химии / Пер. с нем.; Под ред. проф. Клячко Ю.А. М.: Мир, 1975. 462 с.
13. Русско-английский и англо-русский словари терминов по аналитической химии: Рекомендации ИЮПАК (IUPAC) // Журнал аналитической химии. 2001. Т. 56, № 8. С. 883-892; № 9. С. 992-1000; № 11. С. 1217-1227; № 12. С. 1310-1319; 2002. Т. 57, № 1. С. 101-110; №2. С. 206-215; № 4. С. 434-443; № 5. С. 550-558 (см. компакт-диск).
14. Указатель основных терминов по аналитической химии на русском и английском языках / Под. ред. Золотова Ю.А. и Неймана Е.Я. М.: ВИНИТИ, 1988. 108 с.
15. Химическая энциклопедия: В 5 т. М.: Большая российская энциклопедия, 1988-1998.
16. Bard A.J., Parsons R. and Jordan J. Standard Potential in Aqueous Solutions, IUPAC Publication. New York: M. Dekker Inc., 1985.
17. Chend K.L., Ueno K., Imamura T. Handbook of Organic Analytical Reagents. Boca Raton: CRC Press, 1982.
18. Compendium of Chemical Terminology. 2nd. ed., comp. / McNaught A.D. and Wilkinson A. Oxford: Blackwell Science, 1997.
19. CRC Handbook of Basic Tables for Chemical Analysis / Bruno T.J., Svoronos P.D.N. Boca Raton, Fla.: CRC Press, 1989. 517 p. ISBN 0849339359.
20. Dictionary of Analytical Reagents I Townshend A., Bums D.T., Guilbault G.G., Lobinski R.T., Marchenko Z., Newman E.J. and Onishi (editors). London: Chapman and Hall, 1993. 1370 p. ISBN 0412351501 (информация о 5000 аналитических реагентов на прилагаемом компакт-диске).
21. Encyclopedia of Analytical Chemistry: Applications, Theory, and Instrumentation / Editor-in-chief Meyers R.A. New York: Willey, 2000. ISBN 0471976709.
22. Encyclopedia of Analytical Science / Editor-in-chief Alan Townshend. London; San Diego: Academic Press, 1995. 10 vs. (6059 p.). ISBN 0122267001: 012226701X (V. 1), 0122267028 (V. 2). Contents: v. 1. A - Che; v. 2. Chi-Fla; v. 3. Flow - Gas; v. 4. Gast - Lip; v. 5. Lig-Micros; v. 6. Microw - Pha; v. 7. Pha - Rut; v. 8. Sam - Sur; v. 9. Sew - Z; v. 10. Index, directories, and appendices.
23. Handbook on Metals in Clinical and Analytical Chemistry / Ed. by Seiler H.G. et al. New York etc.: Dekker, 1994. 751 p. ISBN 0824790944.
Информационные базы и общие вопросы аналитической химии
13
24. James S. Using Literature / Ed. Norman В. Chapman. Chichester etc.: Wiley, 1987. 598 p. ISBN 0471912204.
25. Multilingual Dictionary of Analytical Terms. English, French, German, Spanish, Russian, Chinese and Japanese. / Ed. Chalmers R.A., Oxford etc: Blackwell S. Publ., 1994. 275 p.
26. Official and Standardized Methods of Analysis. 3rd ed. / Ed. by C. Watson. Cambridge: The Royal Society of Chemistry, 1994. 778 p. ISBN 0851864414.
27. Quantities, Units and Symbols in Physical Chemistry. Oxford: Blackwell Sci. Publ, 1992.
28. Rauscher K. u. a. Chemische Tabellen und Rechentafeln fur dir Analytischem. Praxis. 8 stark uberarbeitete aufl. Leipzig: Deutscher Verlag fur Grundstoffindustrie, 1986. 320 s. ISBN 3342001070.
29. Reagent Chemico Chemicals: American Chemical Society Specifications. 8th ed. Anon. Publ., American Chemical Soc. Washington, D C, USA, 1993. 803 p.
30. Stability Constants Database (1993) (IUPAC, Academic Software, Otley, UK).
31. Stability Constants of metal -ion complexes: Part A. Inorganic Ligands (1982); Part B. Organic Ligands (1980) Oxford: Pergamon Press.
1.1.4. Серии монографий и обзорная информация по аналитической химии
1. Аналитическая химия элементов. М.: Наука, 1960— 1990. — Монографии с обширной библиографией, издано более 40 томов.
2. Проблемы аналитической химии. М.: Наука (Ранее: Труды Комиссии по аналитической химии. АН СССР)— Сборники статей по анализу отдельных объектов или коллективные монографии, также посвященные объектам анализа.
3. Бырько В.М. Дитиокарбаматы. М.: Наука, 1984.
4. Виноградов А.В., Елинсон С.В. Оксихинолин. М.: Наука, 1978. 342 с.
5. Иванов В.М. Гетероциклические азотсодержащие азосоединения. М.: Наука, 1982.
6. Мясоедова Г.В., Саввин С.Б. Хелатообразующие сорбенты. М.: Наука, 1984. 173 с.
7. Назаренко В.А., Антанович В.П. Триоксифлуораны. М.: Наука, 1973.
8. Обзорная информация. Серия «Реактивы и особо чистые вещества». Ассортимент реактивов на конкретные неорганические ионы. М.: НИИЭХИМ и ИРЕА.
9. Пешкова В.М, Мельникова Н.В. |3-Ди кетоны. М.: Наука, 1980. 200 с.
10. Пешкова В.М., Савостина В.М., Иванова Е.К. Оксимы. М.: Наука, М.: Наука. 1977. 236 с.
11. Пилипенко А.Т., Зульфигарова О.С. Гидроксамовые кислоты. М.: Наука, 1989.
12. Пилипенко А.Т., Шевченко Л.Л., Зульфигарова О.С. Купферон. М.: Наука. 1988.
13. Пятницкий И.В., Сухан В.В. Маскирование и демаскирование в аналитической химии. М.: Наука, 1990.220 с.
14. Саввин С.Б., Чернова Р.К., Штыков С.Н. Поверхностно-активные вещества. М.: Наука, 1991.
15. Юрист И.М., Талмуд М.М. Селективное комплексометрическое титрование. М.: Наука, 1993. 232 с.
16. Advances in Analytical Chemistry and Instrumentation / Ed. by Reilley Ch. N.. New York; London, Interscience publ.
17. Analytical Spectroscopy Library Amsterdam: Elsevier; Chichester: Publ. On behalf of ACOL by J. Wiley.
18. Analytical Chemistry by Open. Learning. J. Wiley a. Sons. Ltd.
Analytical Chemistry by Open Learning
(Project Director Brian R. Currell Thames Polytechnic)
Titles in Series:
♦Samples and Standards
♦Sample Pretreatment
♦Classical Methods
♦Measurement, Statistics and Computation
♦Using Literature
♦Instrumentation
♦Chromatographic Separations
♦Electrophoresis
♦Thin Layer Chromatography
♦Fluorescence and Phosphorescence Spectroscopy
♦Infra Red Spectroscopy
♦Atomic Absorption and Emission Spectroscopy
♦Nuclear Magnetic Resonance Spectroscopy
♦X-Ray Methods
♦Scanning Electron Microscopy and Microanalysis
♦Principles of Electroanalytical Methods
♦Potentiometry and Ion Selective Electrodes
♦Polarography and Other Voltammetric Methods
♦Radiochemical Methods
♦Clinical Specimens
♦Diagnostic Enzymology
♦Quantitative Bioassay
♦Assessment and Control of Biochemical Methods
♦Thermal Methods
♦Microprocessor Applications
Lindsay S. High Performance Liquid Chromatography / author Sandie Lindsay; editor John Barnes. Edition Information: 2nd ed. Chichester, England; New York: Published on behalf of Thames Polytechnic, London, by Wiley, 1992. ISBN 0471931802 (cloth), 0471931152 (paper).
Reeve R.N. Environmental Analysis / author Roger N. Reeve; editor John D. Barnes. Edition information: Chichester; New York: Published on behalf of ACOL (University of Greenwich) by J. Wiley, 1994. ISBN 0471938335 (paper) 047195134X (cloth).
Fowlis Ian A. Gas Chromatography: Analytical Chemistry by Open Learning Edition Information: 2nd ed. Chichester; New York: Published on behalf of ACOL (University of Greenwich) by Wiley, 1995. ISBN 0471954675 (cloth),0471954683 (pbk.). Book (Print, Microform, Electronic, etc.).
14
Новый справочник химика и технолога
Stuart B. Modem Infrared Spectroscopy / author Barbara Stuart; editor David J. Ando. Edition information: New York: Published on behalf of ACOL (University of Greenwich) by Wiley, 1996. 180 p. ISBN 0471959162 (cloth), 0471959170 (paper). Book (Print, Microform, Electronic, etc.). Vol. 4.
Thomas M. J. K.Ultraviolet and Visible Spectroscopy / author Michael J.K. Thomas; editor David J. Ando. Edition Informa- Vol. 5.
tion: 2nd ed. Chichester; New York: Published on behalf of ACOL (University of Greenwich) by J. Wiley, 1996. 229 p. ISBN 0471967424 (cloth) 0471967432 (pbk.). Vol. 6.
Stuart B. Biological Applications of Infrared Spectroscopy / Vol. 7.
author Barbara Stuart; editor David J. Ando. Edition information: Chichester; New York: Published on behalf of ACOL Vol. 8.
(University of Greenwich) by John Wiley, 1997. 191p.
ISBN 0471974137 (cloth) 0471974145 (pbk.). Vol. 9.
Dean J.R. Atomic Absorption and Plasma Spectroscopy /author John R. Dean; editor David J. Ando. Edition Information: 2nd Vol. 10.
ed. Chichester; New York: Published on behalf of ACOL (University of Greenwich) by J. Wiley, 1997. Vol. 11.
ISBN 0471972541 (cloth). 047197255X (pbk.). Book (Print, Microform, Electronic, etc.). Vol. 12.
Barker J. Mass Spectrometry. Edition Information: 2nd ed. / autho, James Barkers; editor David J. Ando. Edition informa- Vol. 13.
tion: New York: John Wiley & Sons, 1999. ISBN 0471967645 (cloth), 0471967629 (paper). Vol. 14.
19. Chemical Analysis. A Series of Monographs on Analytical Vol. 15.
Chemistry and its Applications. J. D. Winefordner, Series Editor (New York etc): Wiley (A. Wiley— Interscience Vol. 16.
publ.) (перечень томов см. ниже). 20. Treatise on Analytical Chemistry / Ed. by Ph. Kolthoff and Ph.J. Elwing (первые 4 тома); / Ed. by J. Elwing et al. (последующие тома). 2d ed., Pt.l. Vol. 1. 1978 - Vol. 14, Vol. 17.
1986. New York etc: Wiley. 21. Treatise on Analytical Chemistry / Ed. by Kolthoff and Vol. 18.
Ph.J. Elwing. Part 2. Analytical Chemistry of inorganic and organic compounds. Vol. 1 (1961) - Vol. 17 (1980). New York etc: Wiley. Vol. 19.
Chemical Analysis. A Series of Monographs on Analytical Chemistry and Its Applications Vol. 20. Vol. 21. Vol. 22.
(J. D. Winefordner, Series Editor. A. Willey - Interscience Publication. Willey a. Sons. New York etc.) Vol. 23.
Vol. 1. The Analytical Chemistry of Industrial Poisons, Hazards, and Solvents. Second Edition . By the late Vol. 24.
Morris B. Jacobs. Vol. 25.
Vol. 2. Chromatographic Adsorption Analysis. By Harold H. Strain (out of print). Vol. 26.
Vol. 3. Photometric Determination of Traces of Metals. Fourth Edition Part I: General Aspects. By E. B. Sandell and Hiroshi Onishi Part ПА: Individual Vol. 27.
Metals, Aluminum to Lithium. By Hiroshi Onishi Part IIB: Individual Metals, Magnesium to Zirconium. By Hiroshi Onishi.
Organic Reagents Used in Gravimetric and Volumetric Analysis. By John F. Flagg (out of print).
Aquametry: A Treatise on Methods for the Determination of Water. Second Edition (in three parts). By John Mitchell, Jr. and Donald Milton Smith.
Analysis of Insecticides and Acaricides. By Francis A. Gunther and Roger C. Blinn (out of print).
Chemical Analysis of Industrial Solvents. By the late Morris B. Jacobs and Leopold Schetlan.
Colorimetric Determination of Nonmetals. Second Edition. Edited by the late David F. Boltz and James A. Howell.
Analytical Chemistry of Titanium Metals and Compounds. By Maurice Codell.
The Chemical Analysis of Air Pollutants. By the late Morris B. Jacobs.
X-Ray Spectrochemical Analysis. Second Edition. By L. S. Birks.
Systematic Analysis of Surface-Active Agents. Second Edition. By Milton J. Rosen and Henry A. Goldsmith.
Alternating Current Polarography and Tensam-metry. By B. Breyer and H. H. Bauer.
Flame Photometry. By R. Herrmann and J. Alke-made.
The Titration of Organic Compounds (in two parts).
By M.R.F. Ashworth.
Complexation in Analytical Chemistry: A Guide for the Critical Selection of Analytical Methods Based on Complexation Reactions. By the late Anders Ringbom.
Electron Probe Microanalysis. Second Edition. By L.S. Birks.
Organic Complexing Reagents: Structure, Behavior, and Application to Inorganic Analysis. By D. D. Perrin.
Thermal Analysis. Third Edition. By Wesley Wm. Wendlandt.
Amperometric Titrations. By John T. Stock.
Reflectance Spectroscopy. By Wesley Wm. Wendlandt and Harry G. Hecht.
The Analytical Toxicology of Industrial Inorganic Poisons. By the late Morris B. Jacobs.
The Formation and Properties of Precipitates. By Alan G. Walton.
Kinetics in Analytical Chemistry. By Harry B.
Mark, Jr. and Garry A. Rechnitz.
Atomic Absorption Spectroscopy. Second Edition. By Morris Slavin, 1978.
Characterization of Organometallic Compounds (in two parts). Edited by Minoru Tsutsui.
Rock and Mineral Analysis. Second Edition. By Wesley M. Johnson and John A. Maxwell.
Информационные базы и общие вопросы аналитической химии
15
Vol. 28. The Analytical Chemistry of Nitrogen and Its Compounds (in two parts). Edited by C. A. Streuli and Philip R. Averell.
Vol. 29. The Analytical Chemistry of Sulfur and Its Compounds (in three parts). By J. H. Karchmer.
Vol. 30. Ultramicro Elemental Analysis. By Gunther Tolg.
Vol. 31. Photometric Organic Analysis (in two parts). By Eugene Sawicki.
Vol. 32. Determination of Organic Compounds: Methods and Procedures. By Frederick T. Weiss.
Vol. 33. Masking and Demasking of Chemical Reactions. By D.D. Perrin.
Vol. 34. Neutron Activation Analysis. By D. De Soete, R. Gijbels, and J. Hoste.
Vol. 35. Laser Raman Spectroscopy. By Marvin C. Tobin.
Vol. 36. Emission Spectrochemical Analysis. By Morris Slavin.
Vol. 37. Analytical Chemistry of Phosphorus Compounds. Edited by M. Halmann.
Vol. 38. Luminescence Spectrometry in Analytical Chemistry. By J. D. Winefordner, S. G. Schulman and T.C. O'Haver, 1972.
Vol. 39. Activation Analysis with Neutron Generators. By Sam S. Nargolwalla and Edwin P. Przybylowicz.
Vol. 40. Determination of Gaseous Elements in Metals. Edited by Lynn L. Lewis, Laben M. Melnick and Ben D. Holt.
Vol. 41. Analysis of Silicones. Edited by A. Lee Smith.
Vol. 42. Foundations of Ultracentrifugal Analysis. By H. Fujita.
Vol. 43. Chemical Infrared Fourier Transform Spectroscopy. By Peter R. Griffiths.
Vol. 44. Microscale Manipulations in Chemistry. By T.S. Ma and V. Horak.
Vol. 45. Thermometric Titrations. By J. Barthel.
Vol. 46. Trace Analysis: Spectroscopic Methods for Elements. Edited by J. D. Winefordner.
Vol. 47. Contamination Control in Trace Element Analysis. By Morris Zief and James W. Mitchell.
Vol. 48. Analytical Applications of NMR. By D. E. Leyden and R. H. Cox.
Vol. 49. Measurement of Dissolved Oxygen. By Michael L. Hitchman.
Vol. 50. Analytical Laser Spectroscopy. Edited by Nicolo Omenetto.
Vol. 51. Trace Element Analysis of Geological Materials. By Roger D. Reeves and Robert R. Brooks, 1978.
Vol. 52. Chemical Analysis by Microwave Rotational Spectrscopy. By Ravi Varma and Lawrence W. Hrubesh.
VA 53. Information Theory As Applied to Chemical Analysis. By Karel Eckschlager and Vladimir Stepanek.
Vol. 54. Applied Infrared Spectroscopy: Fundamentals, Techniques and Analytical Problem solving. By A. Lee Smith, 1979.
Vol. 55. Archaeological Chemistry. By Zvi Goffer.
Vol. 56.
Vol. 57.
Vol. 58.
Vol. 59.
Vol. 60.
Vol. 61.
Vol. 62.
Vol. 63.
Vol. 64.
Vol. 65.
Vol. 66.
Vol. 67.
Vol. 68.
Vol. 69.
Vol. 70.
Vol. 71.
Vol. 72.
Vol. 73.
Vol. 74.
Vol. 75.
Vol. 76.
Vol. 77.
Vol. 78.
Vol. 79.
Vol. 80.
Vol. 81.
Immobilized Enzymes in Analytical and Clinical Chemistry. By P. W. Carr and L. D. Bowers.
Photoacoustics and Photoacoustic Spectroscopy. By Allan Rosencwaig.
Analysis of Pesticide Residues. Edited by H. Anson Moye, 1981.
Affinity Chromatography. By William H. Scouten.
Quality Control in Analytical Chemistry. Second Edition. By G. Kateman and L. Buy dens, 1982; 2nd ed. —1993.
Direct Characterization of Fineparticles. By Brian H. Kaye.
Flow Injection Analysis. By J. Ruzicka and E.H. Hansen, 1981.
Applied Electron Spectroscopy for Chemical Analysis. Edited by Hassan Windawi and Floyd Ho. 1982. Analytical Aspects of Environmental Chemistry. Edited by David F. S. Natusch and Philip К. Норке. 1983.
The Interpretation of Analytical Chemical Data by the Use of Cluster Analysis. By D. Luc Massart and Leonard Kaufman.
Solid Phase Biochemistry: Analytical and Synthetic Aspects. Edited by William H. Scouten.
An Introduction to Photoelectron Spectroscopy. By Pradip K. Ghosh, 1983.
Room Temperature Phosphorimetry for Chemical Analysis. By Tuan Vo-Dinh.
Potentiometry and Potentiometric Titrations. By E. P. Seijeant, 1984.
Design and Application of Process Analyzer Systems. By Paul E. Mix.
Analysis of Organic and Biological Surfaces. Edited by Patrick Echlin, 1984.
Small Bore Liquid Chromatography Columns: Their Properties and Uses. Edited by Raymond P.W. Scott. Modem Methods of Particle Size Analysis. Edited by Howard G. Barth.
Auger Electron Spectroscopy. By Michael Thompson, M. D. Baker, Alee Christie and J. F. Tyson, 1985.
Spot Test Analysis: Clinical, Environmental, Forensic and Geochemical Applications. By Ervin Jun-greis, 1985.
Receptor Modeling in Environmental Chemistry. By Philip К. Норке.
Molecular Luminescence Spectroscopy: Methods and Applications (in three parts). Edited by Stephen G. Schulman. Pt. 1 1985; Pt. 2. 1988; Pt. 3 1993.
Inorganic Chromatographic Analysis. Edited by John C. MacDonald, 1985.
Analytical Solution Calorimetry. Edited by J.K. Grime.
Selected Methods of Trace Metal Analysis: Biological and Environmental Samples. By J. C. VanLoon, 1985.
The Analysis of Extraterrestrial Materials. By Isidore Adler.
16
Новый справочник химика и технолога
Vol. 82.
Vol. 83.
Vol. 84.
Vol. 85.
Vol. 86.
Vol. 87.
Vol. 88.
Vol. 89.
Vol. 90.
Vol. 91.
Vol. 92.
Vol. 93.
Vol. 94.
Vol. 95.
Vol. 96.
Vol. 97.
Vol. 98.
Vol. 99.
Vol. 100.
Vol. 101.
Vol. 102.
Vol. 103.
Vol. 104.
Vol. 105.
Chemometrics. By Muhammad A. Sharaf, Deborah L. Ulman, and Bruce R. Kowalski.
Fourier Transform Infrared Spectrometry. By Peter R. Griffiths and James A. de Haseth, 1986.
Trace Analysis: Spectroscopic Methods for Molecules. Edited by Gary Christian and James B. Callis, 1986.
Ultratrace Analysis of Pharmaceuticals and Other Compounds of Interest. Edited by S. Ahuja.
Secondary Ion Mass Spectrometry: Basic Concepts, Instrumental Aspects, Applications and Trends. By A. Benninghoven, F.G. Rudenauer and H. W. Werner, 1987, 1227 p. ISBN 0471010561.
Analytical Applications of Lasers. Edited by Edward H. Piepmeier, 1986.
Applied Geochemical Analysis. By С. O. Ingamells and F. F. Pitard, 1986.
Detectors for Liquid Chromatography. Edited by Edward S. Yeung.
Inductively Coupled Plasma Emission Spectroscopy: Part I: Methodology, Instrumentation and Performance, 1987. 583 p. ISBN 0471096865(V. 1); Part П: Applications and Fundamentals. Edited by J.M. Boumans. 1987. 486 p. ISBN 047185378X (V. 2).
Applications of New Mass Spectrometry Techniques in Pesticide Chemistry. Edited by Joseph Rosen, 1987.
X-Ray Absorption: Principles, Applications, Techniques of EXAFS, SEXAFS and XANES. Edited by D. C. Konnigsberger.
Quantitative Structure-Chromatographic Retention Relationships. By Roman Kaliszan.
Laser Remote Chemical Analysis. Edited by Raymond M. Measures.
Inorganic Mass Spectrometry. Edited by F. Adams, R. Gijbels and R. Van Grieken, 1988. 404 p.
Kinetic Aspects of Analytical Chemistry. By Horacio A. Mottola, 1988.
Two-Dimensional NMR Spectroscopy. By Jan Schrami and Jon M. Bellama.
High Performance Liquid Chromatography. Edited by Phyllis R. Brown and Richard A. Hartwick, 1989.
X-Ray Fluorescence Spectrometry. By Ron Jenkins. 1988, 175 p. ISBN 0471836753.
Analytical Aspects of Drug Testing. Edited by Dale G. Deutsch, 1989.
Chemical Analysis of Polycyclic Aromatic Compounds. Edited by Tuan Vo-Dinh, 1989.
Quadrupole Storage Mass Spectrometry. By Raymond E. March and Richard J. Hughes, 1989.
Determination of Molecular Weight. Edited by Anthony R. Cooper.
Selectivity and Detectability Optimizations in HPLC. By Satinder Ahuja, 1989.
Laser Microanalysis. By Lieselotte Moenke-Blankenburg, 1989.
Vol. 106. Clinical Chemistry. Edited by E. Howard Taylor, 1989.
Vol. 107. Multielement Detection Systems for Spectrochemi-cal Analysis. By Kenneth W. Busch and Marianna A. Busch, 1990.
Vol. 108. Planar Chromatography in the Life Sciences. Edited by Joseph C. Touchstone, 1990.
Vol. 109. Fluorometric Analysis in Biomedical Chemistry: Trends and Techniques Including HPLC Applications. By Norio Ichinose, George Schwedt, Frank Michael Schnepel and Kyoko Adochi, 1991.
Vol. 110. An Introduction to Laboratory Automation. By Victor Cerda and Guillermo Ramis.
Vol 111. Gas Chromatography: Biochemical, Biomedical, and Clinical Applications. Edited by Ray E. Clement, 1991.
Vol. 112. The Analytical Chemistry of Silicones. Edited by A. Lee Smith, 1991.
Vol. 113. Modem Methods of Polymer Characterization. Edited by Howard G. Barth and Jimmy W. Mays, 1991.
Vol. 114. Analytical Raman Spectroscopy. Edited by Jeannette Graselli and Bernard J. Bulkin, 1991.
Vol. 115. Trace and Ultratrace Analysis by HPLC. By Satinder Ahuja, 1992.
Vol. 116. Radiochemistry and Nuclear Methods of Analysis. By William D. Ehmann and Diane E. Vance, 1991. 531 p. ISBN 0471600768(c).
Vol. 117. Applications of Fluorescence in Immunoassays. By Ilkka Hemmila, 1991.
Vol. 118. Principles and Practice of Spectroscopic Calibration. By Howard Mark, 1991.
Vol. 119. Activation Spectrometry in Chemical Analysis. By S. J. Parry, 1991.
Vol. 120. Remote Sensing by Fourier Transform Spectrometry. By Reinhard Beer.
Vol. 121. Detectors for Capillary Chromatography. Edited by Herbert H. Hill and Dennis McMinn, 1991.
Vol. 122. Photochemical Vapor Deposition. By J. G. Eden.
Vol. 123. Statistical Methods in Analytical Chemistry. By Peter C. Meier and Richard Zund, 1993.
Vol. 124. Laser Ionization Mass Analysis. Edited by Akos Vertes, Renaat Gijbels and Fred Adams, 1993. 559 p.
Vol. 125. Physics, Chemistry, and Technology of Solid State Gas Sensor Devices. By Andreas Mandelis and Constantinos Christofides, 1993.
Vol. 126. Electroanalytical Stripping Methods. By Kh. Brain-ina and E. Neyman, 1993. 198 p.
Vol. 127. Air Monitoring by Spectroscopic Techniques / Ed. by Markus W. Sigris, 1994. —XXV, 531 p. ISBN 0471558753.
Vol. 128. Information Theory in Analytical Chemistry 1 Karel Eckschlager; Klaus Danzer, 1994. — XV, 275 p. ISBN 0471595071.
Vol. 129. Flame Chemiluminescence Analysis by Molecular Emission Cavity Detection / Ed. by David A. Stiles, 1994. — VII, 205 p. ISBN 0471943401.
Информационные базы и общие вопросы аналитической химии
17
Vol. 130. Hybride Generation Atomic Absorption Spectrometry / Jiri Dedina; Dimiter L. Tsalev, 1995. — XVIII, 526 p. ISBN 0471953644.
Vol. 131. Selective Detectors: Environmental, Industrial, and Biomedical Applications / Ed. by Robert E. Sievers, 1995. —XXI, 261 p. ISBN 0471013439.
Vol. 132. High-speed Countercurrent Chromatography / Ed. by Yoichiro Ito, 1996. — ХХШ, 454 p. ISBN 0471637491.
Vol. 133. Particle-induced X-Ray Emission Spectrometry (PIXE) / Ed. by Sven A. E. Johansson, 1995-ХХШ, 451 p.ISBN 0471589446.
Vol. 134. Photothermal Spectroscopy Methods for Chemical Analysis I Stephen E. Bialkowski, 1996. — XXIX, 584 p. ISBN 0471574678.
Vol. 135. Element Speciation in Bioinorganic Chemistry I Ed. by Sergio Caroli, 1996. — XXVII, 474 p.
ISBN 0471576417.
Vol. 136. Laser Enhanced Ionization Spectrometry I Ed. by John C. Travis, 1996. — XXI, 334 p.
ISBN 0471576840.
Vol. 137. Fluorescence Imaging Spectroscopy and Microscopy / Ed. by Xue Feng Wang, 1996. — XXIX, 483 p. ISBN 047101527X.
Vol. 138. Introduction to X-Ray Powder Diffractometry I Ron Jenkins; Robert L. Snyder, 1996. — ХХШ, 403 p. ISBN 0471513393.
Vol. 139. Modem Techniques in Electroanalysis / ed. by Petr Vanysek, 1996. — XVII, 369 p. ISBN 0471555142.
Vol. 140. Total-reflection X-Ray Fluorescence Analysis I Reinhold Klockenkamper 1997. 245 p.
ISBN/ISSN 0471305243.
Vol. 141. Spot Test Analysis: Clinical, Environmental, Forensic, and Geochemical Applications /Ervin Jungreis, 2nd ed. 1997. 377 p. ISBN 0471124125.
Vol. 142. The Impact of Stereochemistry on Drug Development and Use I Hassan Y. Aboul-Enein, 1997.— XXVII, 695 p. ISBN 0471596442.
Vol. 143. Macrocyclic Compounds in Analytical Chemistry / Ed. by Yu. A. Zolotov, 1997.— XXIV, 424 p. ISBN 0471172626.
Vol. 144. Surface Launched Acoustic Wave Sensors: Chemical Sensing and Thin-film Characterization I Michael Thompson and David C. Stone, 1997.— XVHI, 196 p. ISBN 0471127949.
Vol. 145. Modem Isotope Ratio Mass Spectrometry / 1. T. Platzner, 1997. — XVI, 514 p. ISBN 0471974161.
Vol. 146. High Performance Capillary Electrophoresis: Theory, Techniques and Applications I d. by Morteza G. Khaledi, 1998. — ХХХП, 1047 p. ISBN 0471148512.
Vol. 147. Solid Phase Extraction: Principles and Practice / E. M. Thurman; M. S. Mills, 1998. — XXVI, 344 p. ISBN 04716I422X.
VoL 148. Commercial Biosensors: Applications to Clinical, Bioprocess, and Environmental Samples I Ed. by Graham Ramsay, 1998. — XVI, 304 p.
ISBN 047158505X.
Vol. 149. A Practical Guide to Graphite Furnace Atomic Absorption Spectrometry I David J. Butcher, Joseph Sneddon, 1998. 250 p. ISBN 0471125539.
Vol. 150. Principles of Chemical and Biological Sensors I Ed. by Dermot Diamond, 1998. — XXVII, 334 p. ISBN 0471546194.
Vol. 151. Pesticide Residues in Foods: Methods, Techniques, and Regulation I W. George Fong et al. 1999. — XIV, 358 p. ISBN 0471574007.
Vol. 152. X-Ray Fluorescence Spectrometry / Ron Jenkins — 2nd ed. 1999. — XVIII, 207 p. ISBN 0471299421.
Vol. 153. Statistical Methods in Analytical Chemistry I Peter C. Meier and Richard E. Zund. — 2nd ed. 2000. — XXVI, 424 p. ISBN 0471293636.
Vol. 154. Modem Analytical Methodologies in Fat- and Water-soluble Vitamins I Edited by Won O. Song, Gary R. Beecher, Ronald R. Eitenmiller, 2000. 471 p. ISBN 0471179426.
Vol. 155. Modem Analytical Methods in Art and Archaeology / Enrico Ciliberto, 2000. — XXIV, 755 p.
ISBN 04712936IX.
Vol. 156. Shpol'skii Spectroscopy and Other Site Selection Methods: Applications in Environmental Analysis, Bioanalytical Chemistry and Chemical Physics I Ed. by Cees Gooijer et al., 2000. — XXI, 552 p. ISBN 0471245089.
Vol. 157. Raman Spectroscopy for Chemical Analysis I Richard L. McCreery, 2000. — XXIV, 420 p.
ISBN 0471252875.
Vol. 158. Large (C > 24) Polycyclic Aromatic Hydrocarbons: Chemistry and Analysis I John C. Fetzer, 2001.— XVI, 288 p. ISBN 0471363545.
Vol. 159. Handbook of Petroleum Analysis I James G. Speight, 2001. — XVIII, 489 p. ISBN 0471361674.
Примечание. Многие из томов, изданных по 1997 год (включительно), т. е. до т. 145, имеются в Библиотеке РАН (Санкт-Петербург).
22. Chromatographic Science: a series monographs. New York; Dekker NT: Chromatographic Science, (перечень томов см. ниже).
Chromatographic Science (с 1990 г.)
Vol. 47
Packings and Stationary Phases in Chromatographic Techniques I Edited by Klaus K. Unger. New York: M. Dekker, 1990. 936 p. ISBN 0824779401.
Vol. 48
Detection-oriented Derivatization Techniques in Liquid Chromatography / Edited by Henk Lingeman, Willy J.M. Under-berg. New York: M. Dekker, 1990. 389 p. ISBN 0824782879.
18
Новый справочник химика и технолога
Vol. 49
Chromatographic Analysis of Pharmaceuticals / Edited by John A. Adamovics. New York: M. Dekker, 1990. 661 p.
ISBN 0824779533.________________________________________
Vol. 50
Multidimensional Chromatography: Techniques and Applications I Edited by Hernan J. Cortes. New York: M. Dekker, 1990. 378 p. ISBN 0824781368.___________________________
Vol. 51
HPLC of Biological Macromolecules: Methods and Applications I Edited by Karen M. Gooding, Fred E. Regnier.
New York: M. Dekker, 1990. 676 p. ISBN: 0824778790.
Vol. 52
Modem Thin-layer Chromatography I Edited by Nelu Grinberg. New York: M. Dekker, 1990. 490 p. ISBN 0824781384,_____________________________________________
Vol. 53
Popl Milan et al. Chromatographic Analysis of Alkaloids. New
York: M, Dekker, 1990. 667 p, ISBN 0824781406.__________
Vol. 54
Papadoyannis LN. HPLC in Clinical Chemistry New York: M, Dekker, 1990. 488 p. ISBN 0824781392.
Vol. 55
Handbook of Thin-layer Chromatography I Edited by Joseph Sherma, Bernard Fried. New York: M. Dekker, 1991. 1047 p.
ISBN 0824783352.________________________________________
Vol. 56
Berezkin V.G. Gas-liquid-solid Chromatography. New York: M. Dekker, 1991. 231 p. ISBN 0824784251 (acid-free paper).
Vol. 57
Complexation Chromatography I Edited by Cagniant D. New
York: M, Dekker, 1992. 294 p. ISBN 0824785770.__________
Vol. 58
Niessen W.M.A., Greef J. van der Jan. Liquid Chromatography - Mass Spectrometry: Principles and Applications. New York:
M. Dekker, 1992. ISBN 0824786351._______________________
Vol. 59
KrejcH Milos. Trace Analysis with Microcolumn Liquid Chromatography. New York: M. Dekker, 1992. 206 p.
ISBN 0824786416.________________________________________
Vol. 60
Modem Chromatographic Analysis of Vitamins I Edited by
Andrew P. De Leenheer, Willy E. Lambert, Hans J. Nelis. 2nd ed. New York, N.Y.: Dekker, 1992. 575 p. ISBN 0824786262.
Vol. 61
Preparative and Production Scale Chromatography I Edited by G. Ganetsos, P.E. Barker. New York: M. Dekker, 1993. 786 p.
ISBN 0824787382.________________________________________
Vol. 63
Handbook of Affinity Chromatography / Edited by Toni Kline.
New Yoik: Dekker, 1993.332 p, ISBN 0824789393.__________
Vol. 64
Capillary Electrophoresis Technology I Edited by Norberto A. Guzman. New York: Dekker, 1993. 857 p.
ISBN 0824790421.
Vol. 65
Lipid Chromatographic Analysis I Edited by Takayuki Shi-bamoto. New York: M. Dekker, 1994. 412 p.
ISBN 0824789415.
Vol. 66
Thin-layer Chromatography: Techniques and Applications I
Bernard Fried, Joseph Sherma. 3rd ed., rev. and expanded.
New York: M. Dekker, 1994.451 p.
ISBN 0824791711.________________________________________
Vol. 67
Scott Raymond P.W. Liquid Chromatography for the Analyst.
New York: M. Dekker, 1994, 328 p. ISBN 0824791843.
Vol. 69
Handbook of Size Exclusion Chromatography I Edited by Chisan Wu. New York: M. Dekker, 1995. 453 p.
ISBN 0824792882.________________________________________
Vol. 70
Scott Raymond P. W. Techniques and Practice of Chromatography. New York: M. Dekker, 1995. 395 p.
ISBN 0824794605.________________________________________
Vol. 71
Handbook of Thin-layer Chromatography I Edited by Joseph
Sherma, Bernard Fried. 2nd ed., rev. and expanded. New York:
M. Dekker, 1996. 1104 p. ISBN 0824794540._______________
Vol. 73
Scott Raymond P.W. Chromatographic Detectors: Design, Function, and Operation. New York: M. Dekker, 1996. 514 p. ISBN 0824797795.
Vol. 74
Chromatographic Analysis of Pharmaceuticals I Edited by John
A. Adamovics. 2nd ed., rev. and expanded. New York: M. Dek-ker, 1997. 527 p. ISBN 0824797760.
Vol. 75
Supercritical Fluid Chromatography with Packed Columns: Techniques and Applications I Edited by Klaus Anton, Claire
Berger. New York: M. Dekker, 1998. 483 p.
ISBN 0824700139.
Vol. 76
Scott R. P.W. Introduction to Analytical Gas Chromatography. 2nd ed. New York: M. Dekker, 1998. 397 p.
ISBN 0824700163.
Vol. 77
Chromatographic Analysis of Environmental and Food Toxicants I Edited by Takayuki Shibamoto. New York: M.
Dekker, 1998. 331 p.
ISBN 0824701453.________________________________________
Vol. 78
Handbook of HPLC I Edited by Elena Katz, et al. New York:
M. Dekker, 1998. 989 p. ISBN 0824794443.________________
Vol. 79
Niessen W.M.A. Liquid Chromatography - Mass Spectrometry. 2nd ed., rev. and expanded. New York: M. Dekker, 1999. 634 p. ISBN 0824719360.
Информационные базы и общие вопросы аналитической химии
19
Vol. 80
Wehr Tim et al. Capillary Electrophoresis of Proteins. New York: M. Dekker, 1999. 286 p.
ISBN: 0824702050._________________________________________
Vol. 81
Fried Bernard, Sherma Joseph. Thin-layer Chromatography. 4th ed., rev. and expanded. New York: M. Dekker, 1999. 499 p. ISBN 0824702220.___________________________________
Vol. 83
Berthod G.-A.-C. Micellar. Liquid Chromatography. New York:
M. Dekker, 2000.603 p. ISBN 0824799933.___________________
Vol. 84
Modem Chromatographic Analysis of Vitamins. 3rd ed., rev. and expanded / Edited by A.P. Leenheer, W.E. Lambert, J.F.
Van Bocxlaer. New York: M. Dekker, 2000. 616 p.
ISBN: 0824703162.
Vol. 85
Beesley T.E., Buglio B., Scott R.P.W. Quantitative Chromatographic Analysis. New York: M. Dekker, 2001. 378 p.
ISBN 0824705033.
Vol. 86
Current Practice of Gas Chromatography - Mass Spectrometry I Edited by W.M.A. Niessen. New York: M. Dekker, 2001. 507 p. ISBN 0824704738.
Vol. 88
Cazes J., Scott R.P.W. Chromatography theory. New York:
Dekker, 2002. ISBN 0824707788.
23. Ellis Horwood Series in Analytical Chemistry. Chichester: Horwood.
24. Modem Analytical Chemistry. Chemical Analysis. New York: Publisher; London: Plenum.
25. Studies in Analytical Chemistry. Amsterdam etc.: Elsevier Scientific Publ. Co.
26. Practical Spectroscopy. A Series / Ed. by Edward G. Brame. The CECON Group Wilmington, Delaware, Dekker, Inc.; New York etc. (перечень томов см. ниже).
Series: Practical Spectroscopy
1. Infrared and Raman Spectroscopy (in three parts). Edited by Edward G. Brame Jr. and Jeanette G. Grasselli.
2. X-Ray Spectrometry. Edited by H. K. Herglotz and L.S. Birks.
3. Mass Spectrometry (in two parts). Edited by Charles Merritt and Charles N. McEwen.
4. Infrared and Raman Spectroscopy of Polymers. Edited by H. W. Siesler and K. Holland-Moritz.
5. NMR Spectroscopy Techniques. Edited by Cecil Dy-bowski and Robert L. Lichter.
6. Infrared Microspectroscopy: Theory and Applications. Edited by Robert G. Messerschmidt and Matthew A. Harthcock.
7. Flow Injection Atomic Spectroscopy. Edited by Jose Luis Burguera.
8. Mass Spectrometry of Biological Materials. Edited by Charles N. McEwen and Barbara S. Larsen.
9. Field Desorption. Mass Spectrometry. Ed. by Leszld Prdkai.
10. Chromatography/Fourier Transform Infrared Spectroscopy and Its Applacations. Ed. By Robert White.
11. Modem NMR Techniques and Their Application in Chemistry. Edited by Alexander I. Popov and Klaas Hallenga.
12. Luminescence Techniques in Chemical and Biochemical Analysis Edited by Willy R. G. Baeyens, Denis De Keu-keleire and Katherine Korkidis.
13. Handbook of Near-Infrared Analysis. Edited by Donald A. Bums and Em. W. Ciurczak.
14. Handbook of X-ray Spectrometry: Methods and Techniques. Edited by Rene E. Van Grieken and Andrzej A. Markowicz.
15. Internal Reflection Spectroscopy: Theory and Applications. Edited by Francis M. Mirabella.
16. Microscopic and Spectroscopic Imaging of the Chemical State. Edited by Michael D. Morris.
17. Mathematical Analysis of Spectral Orthogonality. Ed. by John H. Kalivas and Patrick M. Lang.
18. Laser Spectroscopy: Techniques and Applications. Ed. by E. Roland Menzel.
19. Practical Guide to Infrared Microspectroscopy. Edited by Howard J. Humecki.
20. Quantitative X-ray Spectrometry: Second Edition. Ed. by Ron Jenkins, R. W. Gould and Dale Gedcke.
21. NMR Spectroscopy Techniques. Second Edition, rev. and exp.. Edited by Martha D. Bruch.
22. Spectrophotometric Reactions. Ed. by Ludmila Cermak-ova and Jiri Gasparic.
Vol. 24
Infrared and Raman Spectroscopy of Biological Materials / Edited by Hans-Ulrich Gr., Bing Y. New York: M. Dekker, 2001. 581 p. ISBN 0824704096.
Vol. 25
Near-infrared Applications in Biotechnology / Edited by Ramesh Raghavachari. New York: M. Dekker, 2001.382 p.
ISBN 0824700090.
Vol. 27
Handbook of Near-infrared Analysis / Edited by Donald A.
Bums, Emil W. Ciurczak. 2nd ed., rev. and expanded. New York: M. Dekker, 2001. 814 p.
ISBN 0824705343.__________________________________________
Vol. 28
Handbook of Raman Spectroscopy: from the Research Laboratory to the Process Line / Edited by Ian R. Lewis, Howell G.M. Edwards. New York: M. Dekker, 2001. 1054 p.
ISBN 0824705572.
Vol. 29
Handbook of X-ray Spectometry / Edited by R.E. Van Grieken, Andrzej A. Markowicz. 2nd ed., rev. and expanded. New York: M. Dekker, 2002. Projected Pub. Date: 0111.
ISBN: 0824706005.
20
Новый справочник химика и технолога
Vol. 31 Ciurczak Emil W. and Drennen J.K. Pharmaceutical and Me- Vol. IX
dicinal Applications of Near-infrared Spectroscopy. New York: M. Dekker, 2002. Projected Pub. Date: 0203 ISBN 0824794532. Vol. X
Vol. 32 Vol. XI
Applied Electrospray Mass Spectrometry / Edited by Birendra N. Pramanik, A. K. Ganguly, Michael L. Gross. New York: M. Dekker, 2002. Projected Pub. Date: 0202 ISBN 0824706188 Vol. XII
27. Pergamon Series in Analytical Chemistry. Oxford etc.: Pergamon Press.
28. Techniques and Instrumentation in Analytical Chemistry. Amsterdam: Elsevier.
29. Wilson and Wilson’s. Comprehensive Analytical Chemistry I Ed. By G. Svehla. Amsterdam, etc.: Elsevier (перечень томов см. ниже).
Comprehensive analytical chemistry
Vol. IA Analytical Processes ♦Gas Analysis ♦Inorganic Qualitative Analysis ♦Organic Qualitative Analysis ♦Inorganic Gravimetric Analysis.
Vol. IB Inorganic Titrimetric Analysis ♦Organic. Quantitative Analysis.
Vol. IC Analytical Chemistry of the Elements.
Vol. IIA Electrochemical Analysis ♦Electrodeposition
♦Potentiometric Titrations Conductometric Titrations ♦High-Frequency Titrations.
Vol. IIB Liquid Chromatography in Columns ♦Gas Chromatography ♦Ion Exchangers ♦Distillation.
Vol. IIC Paper and Thin Layer Chromatography ♦Radiochemical Methods ♦Nuclear Magnetic Resonance and Electron Spin Resonance
Methods ♦X-Ray Spectrometry.
Vol. IID Coulometric Analysis.
Vol. Ill Elemental Analysis with Minute Samples
♦Standards and Standardization ♦Separations by Liquid Amalgams ♦Vacuum Fusion Analysis of Gases in Metals ♦Electroanalysis in Molten Salts.
Vol. IV Instrumentation for Spectroscopy ♦Atomic Absorption and Fluorescence Spectroscopy ♦Diffuse Reflectance Spectroscopy.
Vol. V Emission Spectroscopy ♦Analytical Micro wave Spectroscopy ♦Analytical Applications of Electron Microscopy.
Vol. VI Analytical Infrared Spectroscopy.
Vol. VII Thermal Methods in Analytical Chemistry ♦Substoichiometric Analytical Methods.
Vol. VIII Enzyme Electrodes in Analytical Chemistry ♦Molecular Fluorescence Spectroscopy ♦Photometric Titrations ♦Analytical Applications of Interferometry.
Vol. XIII
Vol. XIV Vol. XV Vol. XVI
Vol. XVII Vol. XVIII
Vol. XIX
Vol. XX
Vol. XXI
Vol. XXII Vol. XXIII
Vol. xxrv Vol. XXV
Vol. XXVI
Vol. XXVII Vol. XXVIII
Vol. XXIX
Vol. XXX
Vol. XXXI
Ultraviolet Photoelectron and Photoion Spectroscopy ♦Auger Electron Spectroscopy ♦Plasma Excitation in Spectrochemical Analysis.
Organic Spot Tests Analysis, 1980 ♦The History of Analytical Chemistry.
The Application of Mathematical Statistics in Analytical Chemistry ♦Mass Spectrometry ♦Ion Selective Electrodes, 1981.
Thermal Analysis: Part A. Simultaneous Ther-moanalytical Examination by Means of the Deri vatograph, 1981 ♦Part B. Biochemical and Clinical Applications of Thermometric and Thermal Analysis, 1982 ♦Part C. Emanation Thermal Analysis and other Radiometric Emanation Methods, 1984 ♦PartD. Thermophysical Properties of Solids, 1984 ♦Part E. Pulse Method of Measuring Basic Thermophysical Parameters, 1990.
Analysis of Complex Hydrocarbons, 1981: Part A. Separation Methods ♦Part B. Group Analysis and Detailed Analysis.
Ion-Exchangers in Analytical Chemistry, 1982. Methods of Organic Analysis.
Chemical Microscopy, 1982.
♦Thermomicroscopy of Organic Compounds. Gas and Liquid Analysers, 1982.
Kinetic Methods in Chemical Analysis, 1983. ♦Application of Computers in Analytical Chemistry.
Analytical Visible and Ultraviolet Spectrometry, 1986.
Photometric Methods in Inorganic Trace Analysis.
New Developments in Conductimetric and Oscillometric Analysis, 1988.
Titrimetric Analysis in Organic Solvents, 1986. Analytical and Biomedical Applications of Ion-Selective Field-Effect Transistors, 1988.
Energy Dispersive X-Ray Fluorescence Analysis. Preconcentration of Trace Elements, 1990 I By Yu. A. Zolotov, N. M. Kuzmin.
Radionuclide X-Ray Fluorescence Analysis with Environmental Applications.
Analytical Voltammetry, 1992.
Analysis of Substances in the Gaseous Phase I By E. Smolkova-Keulemansova and L. Feltl, 1991. 479 p. ISBN: 0444891226.
Chemiluminescence Immunoassay / By lan Weeks. Amsterdam, 1992. — XVI, 293 p.
ISBN 0444890351.
Spectrochemical Trace Analysis for Metals and Metalloids I By R. Lobinski and Z. Marczenko. 1997. 808 p. ISBN 044482368.
Surfactants in Analytical Chemistry: Applications of Organized Amphiphilic Media I By E. Pramauro and E. Pelezetti, 1996. 521 p.
ISBN 0444890335.
Информационные базы и общие вопросы аналитической химии
21
Vol. XXXII Environmental Analytical Chemistry / By D. Perez-Bendito and S. Rubio. 1st ed. 1999. 842 p. ISBN 0444822054.
Vol. XXXIII Elemental Speciation: New Approaches for Trace Element Analysis / Edited by Joseph A. Caruso, Karen L. Sutton, Kathryn L. Ackley. 1st ed. 2000. 581 p. ISBN 0444500472.
Vol. XXXIV Discrete Sample Introduction Techniques for Inductively Coupled Plasma Mass Spectrometry / Diane Beauchemin 1st ed. 2000. 575 p. ISBN 0444899510.
Vol. XXXV Modem Fourier Transform Infrared Spectroscopy / Alfred A. Christy, Yukihiro Ozaki, Vasilis G. Gregoriou, 2001. 356 p.
ISBN 0444500448.
Vol. XXXVI Chemical Test Methods of Analysis / Yu. A. Zolotov, V.M. Ivanov, V.G. Amelin. 2002. ISBN 0444502610.
30. Chapman and Hall Chemical Database. London: Chapman and Hall.
31. Fundamental Reviews: Analytical Chemistry: 1994, v. 66, № 12, p. 1R-684R; 1996, v. 68, № 12, p. 1R-652R; 1998, v. 70, № 12, p. 1R-645R; 2000, v. 72, № 12, p. 1R-211R.
32. Treatise on Analytical Chemistry / Ed. I. M. Kolthoff and Ph. J. Elving. New York, etc.: J Willey a. Sons. 1959— 1989. — Многотомное издание: часть I посвящена теоретическим вопросам, часть II — методам анализа органических и неорганических соединений, часть III — методам анализа технических материалов.
33. American Society for Testing and Materials: Annual Book of Standards. Philadelphia: American Society for Testing and Materials.
34. Day R.A. Quntitative Analysis / R.A. Day Jr., A.L.Underwood. 6th ed. Englewood Cliffs; London: Prentice-Hall, 1991. 685p. ISBN 0137473613(intemational.).
35. Fifield F.W. Principles and Practice of Analytical hemistry / F.W. Fifield and D. Kealey. Malden, MA: Blackwell Science, 2000. ISBN 0632053844.
36. Harvei David. Modem Analitical Chemistry. Boston: McGraw-Hill, 2000. 798p. ISBN 0072375477, ISBN 0071169539 (international ed.).
37. Robinson James W. Undergraduate Instrumental Analysis/ 5th ed. New York: M. Dekker, 1995. 858p.
1.1.5. Основная учебная литература
no аналитической химии
1. Аналитическая химия / Под ред. Кельнера Р., Мерме Ж.-М., Ono М., Видмер М; Пер. с англ. М.: Мир, 2002.
2. Аналитическая химия. Химические методы анализа / Под ред. Петрухина О.М. М.: Химия, 1993. 397 с.
3. Булатов М.И. Расчеты равновесий в аналитической химии. Л.: Химия, 1984. 184 с.
4. Васильев В.П. Аналитическая химия: В 2 ч. М.: Высшая школа, 1989.
5. Дёрффель К. Статистика в аналитической химии / Пер. с нем.; Под ред. Адлера Ю.П. М.: Мир, 1994. 268 с. ISBN 5030027998.
6. Дорохова Е.Н., Прохорова Г.В. Задачи и вопросы по аналитической химии. М.: Мир, 2001. 267 с.
7. Кунце У., Шведт Г. Основы качественного и количественного анализа / Пер. с нем. Гармаша А.В.. М.: Мир, 1997. 424 с.
8. Основы аналитической химии: Практическое руководство / Под ред. акад. Золотова Ю.А. М.: Высшая школа, 2001. 463 с.
9. Основы аналитической химии: В 2 кн. 2-е изд., пере-раб. и доп. / Под ред. акад. Золотова Ю.А. М.: Высшая школа, 2000. Кн. 1. Общие вопросы. Методы разделения. 351 с.
10. Основы аналитической химии: В 2 кн. 2-е изд., пере-раб. и доп. / Под ред. акад. Золотова Ю.А. М.: Высшая школа, 2000. Кн. 2. Методы химического анализа. 494 с.
И. Петерс Д., Хайес Дж., Хифтье Г. Химическое разделение и измерение: теория и практика аналитической химии. В 2 кн. М.: Химия, 1978.
12. Пиккеринг У.Ф. Современная аналитическая химия. М.: Химия, 1977. 559 с.
13. Пилипенко А.Т., Пятницкий И.В. Аналитическая химия. Т.1, 2. М.: Химия, 1990.
14. Скуг Д., Уэст Д. Основы аналитической химии: В 2 т. 3-е изд. / Пер. с англ.; Под ред. чл.-корр. АН СССР Золотова Ю.А. М.: Мир, 1979. Т. 1. 480 с.; Т. 2. 438 с.
15. Фритц Дж., Шенк Г. Количественный анализ. М.: Мир, 1978.557 с.
16. Харитонов Ю.Я. Аналитическая химия (аналитика): В 2 кн. М.: Высшая школа, 2001. Кн. 1. Общие теоретические основы. Качественный анализ. 615 с.; Кн. 2. Физико-химические (инструментальные) методы анализа. 559 с.
17. Чарыков А.К. Математическая обработка результатов химического анализа. Л.: Химия, 1984. 168 с.
18. Янсон Э.Ю. Теоретические основы аналитической химии. 2-е изд. М.: Высшая школа, 1987. 364 с.
19. Analitical Chemistry / Ed. by Keller R., Mermet J.-M., Otto M., Widmer H.M. Weinheim: Wiley- VCH, 1998.916 p.
20. Crystian G. D. Analytical Chemistry. 5th ed. New York: Wiley a. Sons, 1994. 812 p.
21. Day R.A., Underwood A.L. Quantitative Analysis. 6th ed. Englewood Cliffs. London: Prentice-Hall, 1991. 685 p. ISBN / ISSN 0137473613(Intemational ed.); 0137473613 (Intemationaled); 0137471556.
22. De Levier R. Principles of Quantitative. New York: McCraw-Hill, 1997. 737 p. ISBN / ISSN 0071142886, 0070163626.
23. Dean J.A. Analytical Chemistry Handbook. New York: McGraw-Hill, 1995. ISBN 0070161976.
24. Filield F.W. and Kealey D. Principles and Practice of Analytical Chemistry. Malden, MA: Blackwell Science, 2000. ISBN 0632053844.
25. Freiser H. Concepts and Calculations in Analytical Chemistry. A. Spreadsheet Approach. CRC Press. London ; Tokyo: Boca Raton An Arbor, 1992. 315 p.
22
Новый справочник химика и технолога
26. Fritz Y.S., Schenk G.A. Quantitative Analytical Chemistry. 5th ed. Boston; London: Allyn and Bacon, 1987. 690 p. ISBN I ISSN 0205105548; 0205104800.
27. Handbook of Analytical Techniques / Ed by H. Gunzler and A. Williams / Weinheim; New York: Wiley - VCH, 2001.
28. Harris D.S. Quantitative Chemical Analysis Fifth Edition. W.H. Freeman and Company. New York, 1999. 898 p. + Appendix + CD. ISBN 0716728818. (оглавление на компакт-диске).
29. Harvey D. Modem Analytical Chemistry. Boston: McCraw-Hill, 2000. 798 p.
ISBN 0072375477; 0071169539(Intemational ed.).
ISBN 0471981370(cloth); 0471972614(pbk).
ISBN 0849347173.
30. Robinson J. W. Undergraduate Instrumental Analysis. 5th ed. New York: M. Dekker, 1995. 858 p.
31. Rouessac Fr. Chemical Analysis: Modem Instrumental Methods and Techniques / Fr. Rouessac, An. Rouessac; Translated by M. Bertrand and K. Waldron. Chichester; New York: Wiley, 2000. 445 p.
32. Skoog D.A. et al. Analytical Chemistry: An Introduction. Series: Saunders Golden Sunburst Series. 7th ed. Fort Worth. London: Saunders College, 2000. 773 p. ISBN/ISSN 0030202930.
33. Skoog D.A., West D.M. Holler F.J. Fundamentals of Analytical Chemistry. 7th ed. Philadelphia: Saunders College Publishing, 1996. 870 p. ISBN 0030059380.
34. Skoog D.A., West D.M., Holler F.J. Analytical Chemistry. An Introduction. 6th edition. Saunders Golden Sunburst Series. Sounders College Publishing Harcourt Bace Col
lege Publishers. Philadelphia etc., 1994. 612 p. + Appendix A1-A45 + Index II - I 18. ISBN 003097285X (оглавление на компакт-диске).
35. Vogel A.I. Vogel’s Textbook of Quantitative Chemical Analysis. 6th ed. /Ed. by J. Mendham et al.; Harlow: Prentice Hall, 2000. 806 p. ISBN /ISSN 0582226287.
36. Пиккеринг У.Ф. Современная аналитическая химия.
М.: Химия, 1977.
1.1.6. Интернет: обзор основных сайтов по аналитической химии1
В Интернете сейчас можно найти тысячи файлов на «химические» темы. Если библиотеки в традиционном виде постепенно начинают терять свои позиции [3], то электронная информация, доступная через Интернет, последние 5-6 лет ощутимо растет не только количественно, но и качественно [2]. Так, пять лет назад через Интернет были доступны около 300 научных журналов, а сейчас их — более 5000. В публикациях уже присутствуют полноправные ссылки на электронные источники данных.
Из огромного числа сайтов по аналитической химии отобраны и представлены в данном издании (табл. 1.1-1.8) только наиболее «устойчивые» и информативные. К сайтам, представляющим наибольший интерес, даны комментарии.
1 Из публикации: В.П. Колотов, В.И. Широкова. Информационные ресурсы Интернета в области аналитической химии // Журнал аналитической химии. 2001. Т. 56, № 7. С. 678-689. http://www.geokhi.ru/rasanalytchem/Resources/default.htm.
Таблица 1.1
Электрохимические общества, организации, группы
Название Адрес
1. Academic centers and national laboratories 2. Deutsche Electrophorese Gesellschaft 3. Electroanalytical Chemistry Related Sites 4. Electrochemical Science and Technology Information Resource (ESTIR) 5. Electrochemiluminescence 6. Electrochemical Science and Technology Information Resource (ESTIR) 7. Ernest B. Yeager Center for Electrochemical Sciences(YCES) 8. The Electrochemical Society (ECS) 9. International Society of Electrochemistry (ISE) http://electrochem.cwru.edu/estir/inet.htm#acad http ://www. weihenstephan.de/blm/deg/ deg.htm 1 http://seac.tufts.edu/related.html http://electrochem.cwru.edu/yeager/default.htm http://www.liv.ac.uk/electrochem.htmlhttp://www.electrochem.cwru.edu/estir/ http://electrochem.cwru.edu/veager/default.htm http.7/www. electrochem.org/. http://www.access.ch/ise/
Информационные базы и общие вопросы аналитической химии
23
Продолжение таблицы 1.1
Название Адрес
10. International Union of Pure and Applied Chemistry: Commission on Eltctrochemistry 11. Japan Association of Chemical Sensors, The Electrochemical Society of Japan 12. Japanese Electrophoresis Society (JES)J. 13. Heyrovsky Institute of Physical Chemistry 14. International Union of Pure and Applied Chemistry: Commission on Electrochemistry 15. Royal Society of Chemistry, Analytical Division, Electroana-lytical Group 16. Royal Society of Chemistry, Faraday Division, Electrochemistry Group 17. Society for Electroanalytical Chemistry (SEAC) 18. Societe Francaise d'Electrophorese 19. Swiss Electrophoresis Society 20. The British Electrophoresis Society 21. The Electrochemical Society 22. The Electroforesis Society 23. The Electrophoresis Society (USA) http://www.iupac.Org/divisions/I/I.3/index.html http://chemsens.mase.nagasaki-u.ac.jp/eng/eng.htm http://proteome.tmig. or.jp/JESindex_body.html/ http:www.jh-inst.cas.cz/science/dept3-t.html http://www.iupac.Org/divisions/I/I.3/index.html http ://www.rsc. org/lap/rsccom/dab/ana006. htm http://www.rsc.org/lap/rsccom/dab/fara005.htm http://seac.tufts.edu http://www-ics.u-strasbg.fr/sfe http ://dcwww. epfl .ch ./icp/chel_de .html http://www.harefield.nthames.nhs.uk/nhli/bes/ http://www.electrochem.org/ http://www.aesociety.org/ http://www-lmmb.ncifcrf.gov/ESAB/indexEP.html
Комментарии
1. ESTIR [4]: ♦ огромное количество гипердокументов, например исчерпывающий список книг (более 800 наименований) и обзоров (более 2000 наименований), изданных за рубежом с 1950 г. по настоящее время; ♦ список трудов по материалам конференций (более 600 наименований); ♦ информация более чем о 400 высших учебных заведениях разных стран; ♦ гиперссылки на тематически близкие источники и базы данных, на специализированные журналы, словари, справочники; ♦ номенклатура и стандарты; ♦ данные о конференциях на ближайшие годы; ♦ сведения о научно-технических обществах, академических центрах и национальных лабораториях.
2. Сайт Международного электрохимического общества (одного из старейших научных обществ, основанного в 1902 г.)— «The Electrochemical Society (ECS)» [5]: ♦ информация о преподавании химии твердого тела, электрохимии и технологии; ♦ гиперссылки на источники по электроосаждению, коррозии, физической электрохимии, сенсорам, органической и биологической электрохимии, промышленному электролизу и электрохимической технологии.
3. Сайт Международного научного общества по электрохимии (общество создано в 1949 г. ведущими европейскими и американскими учеными) — «International Society of Electrochemistry» (ISE) [6]: ♦ информация более чем о 30 национальных и региональных секциях общества, о еже
годных международных научно-технологических конгрессах по новым материалам, сенсорной технологии и др.
4. Сайт ИЮПАК [7]: ♦ информация для широкого круга специалистов, в том числе и материалы Комиссии ИЮПАК по электрохимии.
5. Сайт научного Общества по электроаналитической химии (общество создано в 1984 г.)— «Society for Electroana-lytical Chemistiy» (SEAC) [8]: ♦ список конференций; ♦ гиперссылки на коммерческие сайты, в том числе на сайты фирм-производителей электрохимического оборудования; ♦ гиперссылки на 10 специализированных журналов по электроаналитической химии в режиме on-line и ряд других журналов и изданий; ♦ электронные версии выпусков «SEACcommu-nications» [9].
6. В таблице представлены адреса организаций и научных обществ по электрофорезу, люминесценции и др.
Сайты химических журналов
Электронные версии журналов публикуются со значительным опережением по сравнению с печатными.
Следует обратить внимание на то, что создатели и держатели массивов информации обеспечивают различные возможности свободного (бесплатного) доступа к базам данных: в большинстве случаев это только содержание выпусков, иногда — рефераты статей, значительно реже — полный текст (как правило, в html- или pdf-форматах).
24
Новый справочник химика и технолога
Таблица 1.2
Электрохимические журналы
Журнал Адрес С какого года поддерживается электронная версия Вид информации в свободном доступе
1. Applied and Theoretical Electrophoresis http://www-lmmb.ncifcrf.gov/ESAB/ 1988 (до 1996) Полный текст отдельных статей
2. Biosensors and Bioelectronics http ://www. el se vier.nl/inca/publ ications /store/4/0/5/9/1/З/ 1995 Содержание, поиск статей по автору или ключевым словам (в ответе — полные библиографические данные по каждой статье)
3. Electroanalysis http://www.wiley-vch.de/vch/joumals /2049/index.html 1989 Содержание
4. Electrochemical and Solid-State Letters http ://www3. electrochem.org/letters.html 1998 Содержание, рефераты
5. Electrochimica Acta http://www.elsevier.nl/inca/publications /store/2/0/5/ 1995 Содержание, поиск статей по автору или ключевым словам (в ответе — полные библиографические данные по каждой статье)
6. The Electrochemical Society Interface http://www3.electrochem.org/interface.html 1992 Содержание, полный текст
7. Electrophoresis http://www.wiley-vch.de/contents/jc 2027/200007.html 1998 Содержание
8. Japanese Journal of Electrophoresis http//proteome .tm ig. or.j p/JE S/in dex_ body.html/JJE/JJE/v42n4.htm 1996 Содержание
9. Journal of Applied Electrochemistry http ://www. wkap.nl/j mltoc .htm/0021-891X 1996 Содержание, ключевые слова, информация об авторах с № 2
10. Journal of Capillary Electrophoresis http://www.isc-uk.demon.co.uk/jce.html 1994 (до 1997) Содержание
11. Journal of Electroanalitical Chemistry http://www.elsevier.n1/inca/publications/store/5/ 0/4/0/8/7/ 1995 Содержание, поиск статей по автору или ключевым словам (в ответе — полные библиографические данные по каждой статье)
12. Journal of New Materials for Electrochemical Systems http ://wvw.polymtl. ca/j oumal/material. htm 1998 Содержание
13. Journal of Electroceramics http://www.wkap.nl/joumals/electroceramics 1997 Содержание
14. Journal of Solid State Electrochemistry http:/link.springer.de/link/service/joumals/10008 /index.htm 1997 Содержание, рефераты
15. Journal of The Electrochemical Society http://www3.electrocnem.org/joumal.html 1982 Содержание
Информационные базы и общие вопросы аналитической химии
25
Продолжение таблицы 1.2
Журнал Адрес С какого года поддерживается электронная версия Вид информации в свободном доступе
16. Ionics, International Journal of Ionics: The Science and Technology of Ionic Morion http://www.ionics.org/ 1995 Содержание
17. Portugaliae Etectro-chimica Acta http://alfa.ist.utl.pt/~rpea/ 1997 Содержание
18. Solid State Ionics http://www.elsevier.nl/inca/ publications /store/5/0/5/6/7/7/ 1995 Содержание, поиск статей по автору или ключевым словам (в ответе— полные библиографические данные по каждой статье)
Комментарии
1. Обо всех журналах можно получить исчерпывающую информацию: тематика, состав редакционной коллегии, изменения названий журналов, правила для авторов и др.
2. Некоторые новые журналы практически недоступны в печатных версиях для российских исследователей (например, «Journal of Electroceramics»— 1997 г., «Journal of New Materials for Electrochemical Systems» — 1998 г. и др.).
Таблица 1.3
Журналы по химии и аналитической химии
Журнал Адрес С какого года поддерживается электронная версия Вид информации в свободном доступе
1. Analusis http://www.wiley-vch.de/vch/joumals/2255/index.html 1998 Содержание
2. Analytica Chimica Acta http://www.elsevier.n1/inca/publications/store/5/0/2/6/8/l/ 1994 Содержание, поиск статей по автору или ключевым словам (в ответе — полные библиографические данные по каждой статье)
3. Analytical Biochemistry, Methods in the Biological Sciences http://www.academicpress.com/ab 1993 Содержание, рефераты, поиск других статей по авторам в базе данных
4. Analytical http://www.rsc.org/is/joumals/current/analcoms/anppub.htm 1996 Содержание
Communications: (1980-1993 Analytical Proceedings, 1994-1996 Analytical Proceedings Including Analytical Communications) 1997-1999 Содержание и полный текст статей
5. Analytical Sciences http ://wwwsoc.nii. .ac.j p/j sac/analsci. .html 1990 Содержание, рефераты; полный текст статей с т. 14
6. Australian Journal of Chemistry http://www.publish.csiro.au/joumals/ajc/index.html 1994 Содержание, рефераты, полный текст статей на английском языке с т. 47, № 9
26
Новый справочник химика и технолога
Продолжение таблицы 1.3
Журнал Адрес С какого года поддерживается электронная версия Вид информации в свободном доступе
7. Biochemistry (Moscow) http://www.protein.bio.msu.su/biokhimiya/contents.htm 1996 Содержание, рефераты
8. Biophysical Chemistry http://www.elsevier.n1/inca/publications/store/5/2/2/4/9/9/ 1994 Содержание, поиск статей по автору или ключевым словам (в ответе — полные библиографические данные по каждой статье)
9. Bulletin of the Chemical Society of Japan http://www.chemistry.or.jp/joumals/bcsj/index-e.html 1996 Содержание, рисунки и подписи к ним на англ, языке
10. Chemical Sensors http://chemsens.mase.nagasaki-u.ac.jp/joumal /eng/eng j.htm 1990 Содержание
11. Chemistry International http://www.iupac.org/publications/ci/ index.html 1997 Содержание, полный текст публикаций
12. Chemistry Letters http://www.chemistry.or.jp/joumals/chem-lett/index-e.html 1997 Содержание, рефераты и рисунки с подписями на английском языке
13. Computers & Chemistry http://www.elsevier.nl/inca/ publications/store/3/7/9/ 1995 Содержание, поиск статей по автору или ключевым словам (в ответе — полные библиографические данные по каждой статье)
14. Critical Reviews in Analytical Chemistry (CRAC) http://www.crcpress.com/jour/online/crac/ Не указан в свободном доступе Избранные статьи
15. Field Analytical Chemistry & Technology http://www3. interscience .wiley. com/cgibin/j toc/lD=3 8876 1996 Содержание, рефераты
16. Helvetica Chimica Acta http://www.interscience.wiley.com/jpages/0018-019X/ info.html 1998 Содержание, рефераты
17. Hung. J. Industrial. Chem. http://www.vein.hu/HJIC 1973 Содержание, рефераты
18. Journal of Analytical Chemistry (Zhumal Analiticheskoi Khimii) http://www.maik.rssi.ru/jounals/anchem.htm 1996 Содержание, рефераты на английском языке
19. Journal of Analytical Toxicology http://www.jatox.com/ 1996 Содержание, рефераты
20. Journal of Biological Chemistry http://www-jbc.stanford.edu/jbc/ 1980 - май 1995 с июня 1995 Содержание и рефераты Содержание, рефераты и полный текст статей
Информационные базы и общие вопросы аналитической химии
27
Продолжение таблицы 1.3
Журнал Адрес С какого года поддерживается электронная версия Вид информации в свободном доступе
21. Journal of Chemical Physics http ://oj ps.aip.org/j cpo/ 1975-1996 1997 Содержание и рефераты Содержание, рефераты и полный текст статей
22. Journal of Chemometrics http://www3.interscience.wiley.com/cgi-bin/jtoc/ID=4425 1996 Содержание, рефераты
23. Journal of Chromatographic Science (JCS) http ://www.j-chrom-sci.com/ 1995 Содержание, рефераты
24. Journal of Colloid and Interface Science http://www.academicpress.com/jcis 1993 Содержание, рефераты, поиск других статей авторов в базе данных
25. Journal of Computational Chemistry http://www3.interscience.wiley.com/cgibin/jtoc/ID=33822 1996 Содержание, рефераты
26. Journal of Environmental Monitoring (JEM) http://www.rsc.org/is/joumals/current/jem/jempub.htm 1999 Содержание
27. Journal of High Resolution Chromatography http ://www3. interscience .wiley. com/cgibin /jtoc/ID=5008460 1996 Содержание, рефераты
28. Journal of Solid State Chemistry http://www.academicpress.eom/j ssc 1993 Содержание, рефераты, поиск других статей авторов в базе данных
29. Kagaku to Kogyo (Chemistry and Chemical Industry) (Japanese) http ://www. chemistry, or.j p/j oumals/kakou/index-e .html 1998 Содержание, рефераты и рисунки с подписями на английском языке
30. Kagaku to Kyoiku (Chemical Education) (Japanese) http://www.chemistry.or.jp/joumals/chem.-edu/index-e.html 1998 Содержание, рефераты и рисунки с подписями на английском языке
31. Mendeleev Communications http://Mendeleev Communications 1997 Содержание
32. Microchemical Journal http://www.academicpress.com/microchem 1993 Содержание, рефераты, поиск других статей авторов в базе данных
33. Nippon Kagaku Kaishi (Journal of The Chemical Society of Japan, Chemistry and Industrial Chemistry) http://www.chemistry.or.jp/joumals/nikka/index-e/html 1997 Содержание
34. Platinum Metals Review http://tisbe.catchword.com/vl=20969122/cl=l 4/nw=l/rpsv /catchword/matthey/00321400/contpl.htm 1998 С № 2 — содержание, полный текст статей
35. Pure and Applied Chemistry http://www. iupac.org/publications/рас/index, html 1997 Полный текст отдельных статей
28
Новый справочник химика и технолога
Продолжение таблицы 1.3
Журнал Адрес С какого года поддерживается электронная версия Вид информации в свободном доступе
36. Research Journal of Chemistry and Environment http://www.chemenviron.com/ 1999 Оглавление, рефераты
37. Reviews in Computational Chemistry http://chem.iupui.edu/~boyd/rccontents.html 1990 Содержание
38. Russian Journal of Inorganic Chemistry http://www.maik.rssi.ru/eng/online/index.htm 1996 Содержание, рефераты на английском языке
39. Russian Journal of Physical Chemistry http://www.maik.rssi.ru/eng/online/index.htm 1996 Содержание, рефераты на английском языке
40. Russian Journal of Physical Chemistry (Zhumal Fizicheskoi Khimii) http://www.maik.rssi.ru/jouinals/physchem.htm 1996 Содержание, рефераты на английском языке
41. Sensors and Actuators B: Chemical http://www.elsevier.n1/inca/publications/store/5/0/4/l/0/4/ 1995 Содержание, поиск статей по автору или ключевым словам (в ответе — полные библиографические данные по каждой статье)
42. Surface and Interface Analysis http ://www3. inter sci ence .wiley. com/cgi bin/j toc/ID=2009 1996 Содержание, рефераты, ключевые слова
43. Taianta http://www.elsevier.nl/inca/publictions/ store/5/2/5/4/3/8/ 1995 Содержание, поиск статей по автору или ключевым словам (в ответе — полные библиографические данные по каждой статье)
44. The Analyst http://www.rsc.org/is/joumals/current/analyst/anlpub.htm 1998 Содержание
45. The Journal of Biochemistry http://jb.bcasj.or.jp/ 1996 Содержание, полный текст статей
46. The Journal of Chemical Education http://jchemed.chem.wisc.edu/Joumal/Issues/Current/index. html 2000 Содержание, рефераты с т. 77, № 8
47. TrAC - Trends in Analytical Chemistry http://www.elsevier.n1/inca/publications/store/5/0/2/6/9/5/ 1995 Содержание, поиск статей по автору или ключевым словам (в ответе — полные библиографические данные по каждой статье)
48. Zcitschrift fur anorganische und allgemeine Chemie http ://www3. interscience, wiley. com/cgibin /jtoc/ID-10005159 1998 Содержание, рефераты
Информационные базы и общие вопросы аналитической химии
29
Продолжение таблицы 1.3
Журнал Адрес С какого года поддерживается электронная версия Вид информации в свободном доступе
49. Вестник Москов- http://www.chem. msu.su:8081/rus/vmgu/001/ 1995 Содержание
ского Университета. Химия 1996 с 1998 Содержание, рефераты, с т. 36 Содержание, рефераты, полные тексты статей с т. 39, № 4
50. Журнал аналитической химии http://www.geokhi.ru/~zhakh 1999 Содержание, страница главного редактора, информация о семинарах по аналит. химии (ГЕОХИ и МГУ)
51. Журнал Российского химического общества им. Д.И. Менделеева http://www.chem.msu.su: 8081/rus/ 1999 Содержание с № 2
52. Успехи химии http ://rcr. ioc. ас .ru/ukh .html 1994 Содержание, полный текст статей
Комментарии
1. Данные о журналах, не имеющих электронного варианта (или не предоставляющих свободного доступа на сайт), можно найти на сайтах соответствующих издательств (см. табл. 1.4).
2. Информацию о журналах на японском языке можно найти на сайте Японского химического общества (табл. 1.1, № 12) [11].
3. «Журнал аналитической химии» (один из немногих представленных в Интернете журналов на русском языке): ♦ содержание выпусков; ♦ страница главного редактора; ♦ правила для авторов; ♦ рекомендации ИЮПАК; ♦ информация о деятельности Научного совета РАН.
4. Российские журналы на английском языке — сайт International Academic Publishing Company (IAPC) «Nauka» [13].
Таблица 1.4
Издательства
Издательство Адрес
1. Academic Press http ://www. academicpress.com/www/j oumal/
2. Blackwell Science Ltd http://www.blacksci.co.uk/uk/joumals.htm
3. CRC Press http ://www. crcpress.com/
4. Elsevier http://www.elsevier.nl/
5. International Academic Publishing Company http://www.maik.rssi.ru/
6. Kluwer Academic Publishers http://www.wkap.nl/
7. Springer http://www.springer.de/product/joumals/subjects.html
8. The Gordon and Breach Publishing Group http ://www. gbhap-us. com/j omals .htm
10. Wiley Interscience http ://www3. intersc ience .wiley. com/j omalfinder.html
11. Wiley-VCH http://www.wiley-vch.de/
Комментарии
1. Elsevier (один из лидеров электронной информации в области химии и химической технологии) [14]: ♦ информация по 15 направлениям( химия и химическая технология, математика, физика, астрономия); ♦ электронные версии около 120 журналов (по общей, аналитической, неорганиче-
ской, органической, физической и теоретической химии; по электрохимии, спектроскопии, химической технологии); ♦ в открытом доступе возможность поиска публикаций по автору или ключевым словам; ♦ еженедельные новости.
2. Wiley-VCH [15]: ♦ электронные версии новых (начали выходить в 2001, 2002 гг.) журналов по химии, а также анонс новых изданий.
30
Новый справочник химика и технолога
Таблица 1.5
Электронные библиотеки и каталоги
Электронная библиотека Адрес
1. Научная электронная библиотека РФФИ http://www.elibrary.ru/menu_info.asp
2. Academic Press-Journal in Chemistry http://www.academicpress.com/www/joumal/chem.htm
3. ACS Free Search http ://www.acs .org/j oumals/aoc/aocsearch .html
4. ADFA Library (Search the ADFA Library catalogue) http:/library.adfa. edu.au/
5. Cambridge University Chemical Laboratory (Chemistry Journals) http://www.ch.cam.ac.uk/ChemJoumals.html
6. Chemistry Journals (Cambridge University Chemical Laboratorv) http://seac.tufts.edu/related.html
7. Electroanalytical Chemistry Related Journals on the WWW http :///seac. tufts. edu/i oumals .html
8. Electronic Access to Kluwer Journals http://www.wkap.nl/kaphtml.htm/kluweronline
9. Elsevier Science: search of jomal tables of contents http://www.elsevier.nl/inca/search
10. German Virtual Library - Chemie http://www.rz.uni-karlsmhe.de/Outerspace/VirtualLibrary/54.html
11. Journals under subject "Chemistry" - RS&C http://www3.intercience.wiley.com/cgi-bin/browsepj isu/subi ect=Chemistry
12. Journals (Monthly) published by The Chemical Society of Japan http ://w ww. ch emistry.or.jp/j oumal s/index-e.html
13. Library-New Mexico State University http://lib.nmsu.edu/
14. Society of Chemical Industry: Publications http://sci.mond.org/pubs.html
15. The Library and Information Centre (RS&C) http://www.rsc. org/lic/collections.htm
16. The University of Liverpool, Chemical Literature & Publishing http://www.liv.ac.uk/Chemistry/Links/links.html
17. WWW Virtual Library - Chemistry http://www.chem.ucla.edu/chempointers.html
Комментарий
Первой и пока единственной практически реализованной российской электронной библиотекой стала «Научная электронная библиотека РФФИ», созданная в конце 1998 г. [19]. Подробную информацию о ней можно
найти в статье [3]. Ожидается, что в будущем число электронных журналов на библиотечном сервере РФФИ превысит 1000 (по всем областям знаний). При финансовой поддержке РФФИ организован доступ и на зарубежные серверы некоторых издательств: Elsevier, Springer, Academic Press и некоторых других.
Таблица 1.6
Химические общества
Научное общество Адрес
1. American Chemical Society (ACS) http://www.acs.org/
2. Asian Coordinating Group for Chemistry (ACGC) http://ozchemnet.adfa.oz.au/FACS/ACGC/default.html
3. The Chemical Society of Japan (CSJ) http ://www.chemistry.or. j p/index-e.html
4. Federation of Asian Chemical Societies (FACS) http://ozchemnet.adfa.oz.au/FACS/
5. Learned Societies (Chemical) http://www.ch.cam.ac.uk/ChemSitesOther.html
6. International Union of Pure and Applied Chemistry http://www.iupac.org/index.html
7. Royal Society of Chemistry (RS&C) http://www.rsc.org/
8. Society of Chemical Industry (SCI) http://sci.mond.org/ groups.html
9. The Chemical Society of Japan (CSJ) http://www.chemistry.or.ip/index-e.html
10. University of Waterloo Scholarly Societies Project http ://www. lib. u Waterloo. ca/society/
Комментарий. Наиболее обширный и детальный список web-сайтов научных и технических обществ — на сайте Университета Ватерлоо [20].
Информационные базы и общие вопросы аналитической химии
31
Таблица 1.7
Организации
Организации Адрес
1. Australian Defence Force Academy (ADFA) http://www.ch.adfa.edu.au/ozchemnet/
2. Institute of Analytical Chemistry at Vienna University of Technology http://www.iac.tuwien.ac.at/
3. New Mexico State University http ://www.nmsu. edu/
4. The University of Liverpool, Department of Chemistry http://www.liv.ac.uk/Chemistry.html
5. Внешние информационные ресурсы РФФИ: российские научные организации http://193.233.79. 157/science_rus.htm
Комментарий. Сайт Ливерпульского университета: примерно 5800 источников информации по химии в Интернете [21].
Таблица 1.8
Полезная химическая и электрохимическая информация
Название Адрес
1. Analytical Chemistry at Chemical Analysis, com http://vvww.chemicalanalysis.com/
2. Analytical Chemistry Network (ACN) http ://www.chemsoc.org/net works/can/index .htm
3. Analytical Methods, Products and Services http://www.che.ufl.edu/www-che/topics/analvtical.html
4. Analytical Reference Materials Group http://www.j-chrom-sci.com/
5. Bioanalytical Systems, Inc. http ://www.bioanalytical .com/
6. Center for Ion Conducting Membranes http://www.cicm.ic.ac.uk/home.html
7. Center for Process Analytical Chemistry (CPAC) http://www.cpac.washington.edu/
8. ChemConnect http://www.chemconnect.com/news/ioumals.html
9. Chemical Data http://www.ch.cam.ac.uk/chemdata.html
10. Chemical Engineering http://www.che.ufl.edu/www-che/
11. Chemistry Indexes http://qsprO3 .tuwien.ac.at/lo/
12. Chemistry on the Internet http://argon.ch.ic.ac.uk/infobahn/boc.html
13. Chemistry Software List http://qsprO3 .tuwien.ac.at/lo/
14. Chemistry Resources on the Internet - Sheffield http ://www.shef.ac .uk/~chem/
15. Sheffield’s Chemdex™ http://www.shef.ac.uk/chemistry/chemdex/
16. Analytical Chemistry http://www.chemdex.org/chemdex/analvtical.html/analvtical
17. Chemical Journals http://www.chemdex.org/chemdex/ioumals.html
18. Chemometrics Sites http://qsprO3 .tuwien.ac.at/lo/
19. Chemometrics World http ://www. wiley.co.uk/wileychi/ chemometrics/
20. Chromatography http://www.rohan.sdsu.edu/stafl/driacknVchemistrv/chemlink/analvtic/
analyt5.html
21. Global Instructional Chemistry http://http://www.ch.ic.ac.uk/GIC/
22. International Chromatography Guide http://www.iatox.com/chrom guide/index.htm
23. Galaxy-Chemistry-Analytical http://galaxy.einet.net/galaxy/Science/Chemistry/Analytical.html
24. Gravimetric Methods of Analysis http://ull.chemistry.uakron.edu/analytical/gravimetry/
25. Guide to Chemistry Resources on the Internet - RPI http://www.mi.edU/dept/chem./cheminfo/chemres.html
26. List of Russian WWW-servers http://www.ras.ru/map list.html
27. Mass Spectrometry on the Internet http://base-peak.wiley.com/
28. On-line Chemistry Clubs http ://www.ch.cam.ac .uk/ChemS itesOther.html
29. PS&C — for information about the Society's Journals, Books, Databases & Databanks and related services http://www.rsc.org/is/producthome.htm
32
Новый справочник химика и технолога
Продолжение таблицы 1.8
Название Адрес
3 0. Chemical Abstracts Service http ://info,cas .org/welcome .html
31. Science Citation Index http://wos.isitrial.com/ClW.cgi
32. The Best World Collections of Pointers http://argon.ch.ic.ac.uk/infobahn/boc.html
33. The Chemistry Hypermedia Project On-line Resources for Students, Educators and Scientists. http://www.chem.vt.edu/chem-ed/vt-chem-ed.html
3 4. The Yahoo Collection of Chemical Pointers http://dir.yahoo.com/science/chemistry/
3 5. Two Thousand of the Best Chemistry Sites http ://www.ch.cam.ac .uk/ChemSitesIndex.html
36. World Fuel Cell Council http://members.aol.com/fuelcells/index.html
37. Yahoo guide to WWW Resources in Chemistry http://dir.yahoo.com/science/chemistry/
3 8. Princeton Applied Research Products http://www.par-online.com/applications/appindex.html
39. Внешние информационные ресурсы РФФИ: научные фонды http://l 93.233.79.157/foundations.htm
Комментарии
1. Сайт РФФИ [3] представляет интерес не только как один из немногих российских сайтов, но и тем, что периодически предоставляет возможность бесплатного доступа к зарубежным базам данных.
2. Наибольший интерес представляют базы данных Кембриджского и Шеффилдского университетов [17, 22].
3. ISI Web of Science (Institute for Scientific Information, Филадельфия) [24] и «Science Citation Index Expanded» [25]: база данных научного цитирования.
Заключение. Пользуясь интернет-источниками, следует помнить:
4 изменение web-сайтов, включая их исчезновение, происходит без предупреждения;
♦ ссылка на сайт при его использовании обязательна.
Литература
1. Крол Э. Все об Интернете / Пер. с англ. К0 «Торговоиздательское бюро BHV», 1995. 592 с. ISBN 5874190015.
2. Anderson J.L., Conry L.A., Leddy Y. Anal. 1998. V. 70, № 12.P. 519R.
3. Алфимов Л.В., Новиков В. Научная электронная библиотека РФФИ сегодня // Поиск. 2000. 26.05; сайт РФФИ:
http://193.233.79.157/pub/poisk/poisk.asp.article=content. htm.
4. Electrochemical Science and Technology Inform Resource (ESTIR): http://electrochem.cwru.edu/yeager/default.htm.
5. The Electrochemical Society: http://www.electrochem.org/.
6. International Society of Electrochemistry (ISE): http://www.access.ch./ise.c/.
7. International Union of Pure and Applied Chemistry: http://www.iupac.org/index.html.
8. Society for Electroanalytical Chemistry (SEAC): http://seac.tufts.edu.
9. SEACcommunications: htlp://seac.tufts.edu;communica-tions.html.
10. American Chemical Society (ACS): http://www.acs.org/.
11. The Chemical Society of Japan (CSJ): http://www. chem-istry.or.jp/index-e.html.
12. Журнал аналитической химии: http://www.geokhi. ru/~zhakh.
13. International Academic Publishing Company (IAPC) «Nauka»: http ://www. maik. rssi.ru/.
14. Elsevier WWW Publications: http://www.elsevier.nl/.
15. Wiley-VCH: http://www.wiley-vch.de/berlin/joumals/.
16. Wiley InterScience: http://www3.interscience.wiley.
Com/jounalfinder.html.
17. http://www.ch.cam.ac.uk/.
18. The Library and Information Centre (RS&C): http://www.rsc.org/lic/collections.htm.
19. Научная электронная библиотека РФФИ: http://www.elibrary.ru/menu_info.asp.
20. University of Waterloo, Scholarly Societies Project: http://www.hb.uwaterloo.ca/society/.
21. The University of Liverpool, Department of Chemistry: http ://www. li v. ac.uk/Chemistry .html.
22. Chemistry Resources on the Internet. Sheffield: http://www.shef.ac.uk;~chem/.
23. Chemometrics World: http://www.wiley.co.uk/wileychi/ chemometrics/.
24. http://wos.isitrial.com/CIW.cgi.
25. «Индексы цитирования — от первоисточника»: http://194.85.223,6/rfbr/pub/poi sk/isi_01 .htm.
26. http://www.geokhi.ru/~rusanalytchem/Resources/default. htm.
1.1.7. Compendium of Analytical Nomenclature (см. компакт-диск)
1.1.8. Русско-английский и англо-руский словари терминов по аналитической химии (рекомендации ИЮПАК) (см. компакт-диск)
Информационные базы и общие вопросы аналитической химии
33
1.2. Структура и методы аналитической химии (АХ) [1-3]
Можно выделить три функции АХ как области знания: 1) решение общих вопросов анализа (например, развитие его метрологии); 2) разработка аналитических методов; 3) решение конкретных задач анализа.
Классификация видов анализа. Можно выделить качественный и количественный анализы. Первый определяет, какие компоненты включает анализируемый объект, второй дает сведения о количественном содержании всех или отдельных компонентов. При определении микропримесей грань между этими видами анализа подчас стирается.
Классификация может базироваться на масштабе работы, объеме или массе пробы: макро-, полумикро-, микро-, ультрамикро- и субмикроанализы.
Под масштабами работы [2] в АХ, прежде всего, подразумевают величину взятой навески пробы (анализируемой части пробы, аналитической навески). В тех пределах, в которых возможен выбор, величина навески пробы S определяется применяемым методом анализа (в определенных случаях методикой анализа), относительным со-держанием определяемого компонента С и другими факторами, например требуемой точностью.
Для описания и классификаций масштабов работы методов (методик) применяется двойное обозначение: S - С, т.е. величина (масса) — содержание компонента (в % или млн-1).
На этой основе методы классифицируются четко, и их области применения могут быть представлены в прямоугольных координатах, в которых массы навесок проб откладываются по абсциссе (5), а относительные содержания (Q — по ординате. Принятые единицы для S — грамм, а для С — % или млн-1. Из-за широких областей S и С график построен в билографмических координатах (рис. 1.1). Наклонные прямые (диагонали) на рис. 1.1 представляют абсолютные количества Q конкретного компонента.
Для более точного обозначения и классификации методов области S и С описывают соответствующими терминами. * **
«Комиссия по терминологии Научного совета АН СССР по аналитической химии отмечает, что рекомендуемая ниже терминология в значительной степени нова для английского и русского научно-технического языка. К тому же в русском языке вместо термина примесный (следовый) анализ (объекта) обычно используется термин определение примесей (следов) в объекте. Новая классификация методов и методик анализа по относительным содержаниям определяемых компонентов и величинам анализируемых навесок проб четко определяет основные термины в данной области, которые ранее использовались неоднозначно».
** См. также раздел 2, п. 2.1. Авторы [1а, б] предлагают также более общее определение: «Содержание Со— массовая доля, объемная доля, концентрация, масса в некотором количестве вещества, объем в некотором количестве вещества или другая характеристика абсолютного или относительного количества вещества». В случае возможных недоразумений оговаривают конкретное понимание термина с указанием единиц измерения.
Классификация по массе навески пробы (5). Величины навесок проб можно классифицировать как граммовые (1-Юг), дециграммовые (0,1-1 г), сантиграммовые (0,01-0,1 г), миллиграммовые (0,001-0,01 г), микрограм-мовые (Ю^-Ю^г), нанограммовые (10-9—10-6 г), пикограммовые (10-12-10-9 г), фемтограммовые (10-15-10-12 г) и т.п. (см. А на рис. 1.1).
Макро-, полумикро- и микро- являются терминами, использовавшимися долгие годы для обозначения величин навесок проб и соответственно масштабов аналитических операций. Эти термины стоит сохранить.
Общепринято, что макронавеску пробы следует рассматривать как навеску, имеющую массу свыше 0,1 г. Верхняя граница для такой навески не оговаривается, но большинство методов, рассматриваемых как макрометоды, используют навески проб в области 0,1-1 г.
Термин полумикроанализ целесообразно заменить на мезомикроанализ. Мезонавеской пробы (полумикронавеской) можно считать навеску пробы в интервале 0,1-0,01 г.
Навески пробы в области 10-2-10-3могут быть названы микронавесками; в области 10 3 101 г— субмикронавесками, а в области ниже 10~*г— ультрамикронавесками без каких-либо ограничений нижней границы для последнего вида (см. В на рис. 1.1).
В соответствии с величиной навески пробы различают макро-, мезо- (по традиции полумикро-), микро-, субмикро-, ультрамикро- анализ (метод).
Классификация по относительному содержанию компонента (Q. Термины основной, неосновной и примесный (следовый) используют в общей классификации на основе их относительных содержаний следующим образом:
Основной компонент....................100-1%
Неосновной компонент..................1-0,01%
Примесный (следовый) компонент........< 0,01%
< 100%)
Имеются веские основания (исторические и практические) верхней границей относительных содержаний примесей (следов) считать величину 100 млн-1 и так же, как и до настоящего времени, не устанавливать никакого нижнего предела. При этом любое содержание компонента ниже 100 млн-1 следует рассматривать как примесное (следовое). Однако развитие техники аналитических работ в настоящее время требует, чтобы область примесных следовых содержаний была далее подразделена следующим образом:
Примеси (следы)................10 2—10^* млн-1
Микропримеси (микроследы)......10^-10-7 млн-1
Нанопримеси (наноследы)........10-7—1О-10 млн-1
Пикопримеси (пикоследы)........10-10—10-13 млн-1
В микроанализе обычно используют S-классификацию, т.е. аналитик в основном обращает внимание на малую величину навески своей пробы и много меньше связан с относительным содержанием в ней определяемого компонента. Часто на практике в микроанализе и субмикро- или ультрамикроанализе определяемый компонент является основным, т.е. С> 1 %.
34
Новый справочник химика и технолога
g ч s g -1 е. 3 млн § °
К Л /7 11 S 10 -10
довые) содержание 1 Микропримеси 1 (микроследы) ООО ? ? '* ООО Jo Д Д о
о t О 2 к о S 10-Ю4 ИГ-104
1
При . Примеси (следы) о о 1 1 н-* 5 °, 1 я -е*
§ S
▼ ’ lo'-io-3 10-10 4 и
Основной Неосновной компонент компонент о о о о LXt N. LU J- J- 1 1 Ом О~ Оо О,
ч\ ю4
-2ю4
"10 < X
1 Дециграмм о4 1 Сантиграмм У2 1 Миллиграмм о-3 ю4 ю’5 10 Микрограмм
в * — Макро — Мезо- Микро- Субмикро - - Ультрамшфо ►
использующего микронавески проб, но возможны случаи, когда возникает потребность дать более точное определение вида анализа, подобно приведенным ниже:
— улыпрапримесный (ультраследовый) анализ, т.е. 5 < 10“4 г, С < 100 млн1;
— субмикропримесный (субмикроследовый) анализ, т.е. S = 10'-10 4 г, С< 100 млн4;
— микропримесный анализ, т.е. 5 = 10~2-10~3 г, С < 100 млн-1;
— мезопримесный (мезоследовый) анализ, т.е. 5= 104-10“2; С< 100 млн4 (0,01%).
Классификация видов анализа может быть основана на природе обнаруживаемых или определяемых частиц; в этом случае говорят об анализе изотопном, элементном (атомноионном), структурно-групповом (функциональном), молекулярном, вещественном, фазовом.
Когда говорят «сделайте химический анализ», часто имеют в виду определение элементного состава образца: из каких элементов состоит данный объект, каковы их концентрации или количество. Этот вид анализа называют элементным.
Изотопным анализом профессиональные аналитики занимаются мало; такие анализы чаще проводят физики, геологи, биологи.
В вещественном анализе определяют, в каких формах присутствует интересующий нас компонент в анализируемом объекте и каково содержание этих форм. Например, медь в минерале может быть в виде оксида, сульфида или смеси этих соединений. Вещественный анализ имеет много общего с молекулярным и фазовым.
Молекулярный анализ — это обнаружение и
определение химических соединений. Типичным примером является анализ смеси газов. Среди методов молекулярного анализа ныне главенствующее место занимают хроматографические.
Для химиков-органиков существен еще один вид анализа, промежуточный между элементным и молекулярным,— структурно-групповой анализ. Это прежде всего определение функциональных групп, т. е. отдельных групп органических соединений — карбоксильной, гидроксильной, аминной и др.
Фазовый анализ — анализ включений в неоднородном объекте, например минералах. Так, сульфид и оксид меди не распределены в минерале гомогенно, а образуют отдельные фазы.
В основе другой классификации могут лежать природа и особенности анализируемого объекта. Так, различают АХ неорганических, органических веществ, высокочистых веществ, металлов и сплавов, объектов окружающей среды ит.д.
Можно предложить другие классификации видов анализа: валовый - локальный, деструктивный - недеструк
S -классификация
Рис. 1.1. Классификация аналитических методов и методик по основным величинам навески пробы и относительному содержанию компонента [2]
В примесном (следовом) анализе наоборот: значение С имеет определяющее значение, а величина навески пробы 5 играет не основную роль. Соответственно С может быть 10“5 млн-1 при S в области от 1 до 100 г.
Однако существуют случаи, когда содержание С может находиться на уровне млн-1 или ниже (типичная задача определения примесей), но при этом величина навески 5 составляет только 100 мкг, т.е. возникает проблема, типичная для микроанализа. В таком случае ощущается, что приведенные выше S- и С- классификации уже недостаточны и становится необходимой смешанная S—("-классификация.
По указанной причине предлагается термин ультра-примесный или ультраследовый, т.е. ультрамикро-примесный или ультрамикроследовый зарезервировать для подобных случаев аналитической практики.
В общем случае этот термин может быть использован для описания целой области примесного (следового) анализа,
Информационные базы и общие вопросы аналитической химии
35
тивный, контактный - дистанционный, дискретный - непрерывный.
Методы аналитической химии
Все существующие методы АХ можно разделить на методы пробоотбора, разложения проб, разделения компонентов, обнаружения (идентификации) и определения. Существуют гибридные методы, сочетающие разделение и определение. Методы обнаружения и определения имеют много общего.
Наибольшее значение имеют методы определения. В АХ методы определения основаны на разных принципах. Принципы разные, но практически все методы основаны на зависимости между составом вещества и его свойствами. Обычно измеряют свойство (например, интенсивность окраски, радиоактивность, интенсивность излучения или поглощения света) и по полученному сигналу судят о составе вещества, точнее о содержании интересующего компонента.
Можно классифицировать методы определения по характеру измеряемого свойства или по способу регистрации соответствующего сигнала. Методы определения делятся на химические, физические и биологические. Химические методы базируются на химических (в том числе электрохимических) реакциях. Сюда можно отнести и методы, называемые физико-химическими. Физические методы основаны на физических явлениях и процессах (взаимодействие вещества с потоком энергии), биологические* — на явлениях живой природы. Эта классификация условна. Так, например, фотометрические методы могут быть и химическими (в большинстве случаев), и чисто физическими.
Можно классифицировать методы определения по видам анализа, для которых они предназначены. Можно говорить о методах изотопного, элементного, молекулярного анализа и т. д.
Литература
1. Основы аналитической химии: В 2 кн. / Под ред. акад. Золотова Ю.А. Кн. 1. Общие вопросы. Методы разделения. 2-е изд., перераб. и доп. М: Высшая школа, 2000. С. 6-8.
2. Номенклатурные правила ИЮПАК. Т. 4. Аналитическая химия. М.: ВИНИТИ, 1985. С. 9-12.
3. Туманов А.А., Крестьянинов П.А. Обзор «Биологический метод анализа: состояние, перспектива» // Журн. аналиг. химии. 2002. Т. 57, № 5. С. 45А470.
1.3. Общая схема количественного анализа
Типичный количественный анализ включает последовательные стадии, представленные на рис. 1.2. В отдельных случаях могут быть исключены одна или несколько промежуточных операций.
* Полный обзор см. [3] (к п. 1.2).
Рис. 1.2. Стадии количественного анализа
* Представительные аналитические навески или аликвоты — это части (доли) материала (раствора) примерно одинаковой массы (или одинакового объема), которые проводятся через все аналитические операции одновременно и параллельно.
* * Мешающее вещество (interference substance) в методике анализа— это вещество, которое вызывает предопределенную систематическую погрешность результатов анализа. Вывод о том, мешает данное вещество определению или нет, зависит от количества мешающего вещества и определяемого компонента в анализируемом образце [3, 4].
Литература
1. Основы аналитической химии: В 2 кн. / Под ред. акад. Золотова Ю.А. Кн. 1. Общие вопросы. Методы разделения. 2-е изд., перераб. и дополн. М.: Высшая школа, 2000. С. 59-78.
36
Новый справочник химика и технолога
2. Skoog D.A., West D.M., Holler F.J. Analytical Chemistry. An Introduction. Sixth Edition. Saunders Golden Sunburst. Series. Saunders Gollege Publishing Harcourt Bace College Publishers. Philadelphia etc., 1994. 612 p.
3. Журнал аналит. химии. 1984. T. 39, № 5. С. 945-947.
4. Номенклатурные правила ИЮПАК. Т 4. Аналитическая химия. М: ВИНИТИ, 1985. С. 169-173.
1.4. Выбор метода анализа [1]
Выбирая метод анализа, необходимо четко знать цель анализа, задачи, которые при этом нужно решить, оценить достоинства и недостатки доступных методов анализа [1,2].
Прежде чем рассматривать факторы, которые необходимо учитывать при выборе того или иного метода анализа, следует обсудить понятия метод и методика. Метод — это совокупность принципов, положенных в основу анализа безотносительно к конкретному объекту и определяемому веществу; методика — подробное описание всех условий и операций проведения анализа определенного объекта. При определении компонента в конкретном объекте в методику вводят также описание операций отбора пробы и подготовки ее к анализу (например, растворение образца в подходящем растворителе и устранение влияния веществ, мешающих определению).
Ниже рассмотрены основные факторы, которые нужно принимать во внимание, выбирая метод и методику анализа.
Содержание компонента. При выборе метода анализа необходимо учитывать ожидаемое содержание обнаруживаемого или определяемого компонента. При этом важно не только оценить процентное содержание компонента в образце, его концентрацию в анализируемом растворе, но и количество вещества, которое может быть взято на анализ. Таким образом, выбор метода анализа обусловливается абсолютным содержанием компонента.
Концентрация определяемого компонента и количество образца, предоставляемого на анализ, могут меняться в широких пределах
Количество образца, получаемое на анализ, в одних случаях может быть не лимитировано, а в других (определение вкраплений в минералах, анализ крови, биомасс, космических объектов и т.д.) очень мало (миллиграммы или даже доли миллиграмма).
Чувствительность метода или методики определяется тем минимальным количеством вещества, которое можно обнаруживать или определять данным методом, по данной методике (более строгое определение этого понятия и его количественное выражение см. раздел 2, п. 2.2). На рис. 1.3 приведена относительная характеристика чувствительности некоторых методов. Нижняя граница определяемого содержания демонстрирует возможности метода и наилучший результат, достигаемый при определении ряда веществ (см. раздел 2, п. 2.6). Так, при анализах в цветной металлургии нижняя граница определяемых содержаний многих элементов методом искровой масс-спектрометрии составляет КГ5-10~7%, методом нейтронно-активационного анализа —10 7—1014г, химикоспектральным анализом — 10“5—10 7%; электрохимические, фотометрические и другие методы с предварительным
концентрированием позволяют определять ряд примесей на уровне 10 5—10 7 % [2].
Сопоставляя чувствительность различных методов и оценивая примерное содержание компонента в образце, химик выбирает тот или иной метод анализа.
Природа (особенности) анализируемого вещества и из-бирателъност метода. При проведении анализа имеют дело с самыми разнообразными объектами — продуктами промышленного и сельскохозяйственного производства, объектами окружающей среды, космическими объектами, произведениями искусства и т. д. Естественно, что выбор метода и методики анализа при этом определяется не только задачей анализа, но также свойствами и особенностями образца. Необходимо учитывать физические свойства анализируемого объекта: его агрегатное состояние, летучесть, гигроскопичность, механическую прочность и т. д. Определяющими при выборе метода анализа являются химические свойства образца. При этом важно знать и принимать во внимание химические свойства основы образца, часто называемой матрицей анализируемого объекта; качественный химический состав образца; химические свойства определяемого компонента и сопутствующих ему примесей.
Рис. 1.3. Нижние границы определяемых содержаний компонентов (-1g Q, г) [1]
Зная химические свойства основы и ожидаемых компонентов анализируемого объекта, оценив возможные помехи, выбирают как можно более избирательный метод, т. е. метод, с помощью которого в данных условиях можно обнаружить или определить нужные компоненты без помех со стороны других присутствующих компонентов. В химической литературе, наряду с термином избирательность, используют термин селективность. Метод, методику или реакцию, позволяющие обнаружить или
Информационные базы и общие вопросы аналитической химии
37
определить только немногие (малое число) компоненты (аналиты) в смеси или в матрице без помех со стороны других компонентов подобного поведения, называют селективными. Метод, методику или реакцию, позволяющие обнаружить или определить только один компонент (ана-лит) в смеси или в матрице без помех со стороны других компонентов, называют специфичным. Таким образом, специфичность следует рассматривать как предельную селективность. Подробнее см. [3,4].
Можно говорить об избирательности метода, методики и отдельной реакции, положенных в основу обнаружения или определения компонента. Так, высокой избирательностью характеризуются такие методы, как ионометрия, атомноабсорбционный и ферментативный методы. Многие реакции, лежащие в основе методик, также высоко избирательны.
Методику химического анализа можно сделать более избирательной (селективной), изменив условия проведения анализа (pH среды, концентрацию реагентов, растворитель и т. д.); устранив влияние мешающих компонентов переведением их в нереакционноспособную форму (маскирование) или отделением (осаждение, экстракция, хроматография) их от основного компонента (см. раздел 3).
Рассматривая методы и методики, следует сказать об их универсальности — возможности обнаруживать или определять многие компоненты. Особенно ценно иметь возможность обнаруживать или определять многие компоненты одновременно из одной пробы, т. е. проводить многокомпонентный анализ. Высокая избирательность метода и его универсальность не противоречат друг другу: многие универсальные методы анализа отличаются высокой избирательностью определения отдельных компонентов, например такие методы, как хроматография, некоторые виды вольтамперометрии, атомно-эмиссионная спектроскопия. Методами атомно-эмиссионной спектроскопии с применением индуктивно-связанной плазмы и квантометров можно определять из одной пробы (без разделения) 25-30 различных элементов.
Точность анализа — это собирательная характеристика метода или методики, оценивающая их правильность и воспроизводимость. Когда говорят о высокой точности, предполагают, что результаты правильные и разброс данных анализа незначителен. Точность часто характеризуют относительной погрешностью (ошибкой) определения в процентах.
Требования к точности анализа обычно определяются целью и задачами анализа, природой объекта. Необязательно всегда стремиться к высокой точности. Например, при текущем контроле многих металлургических и химических производств определение компонентов можно проводить с погрешностью в 10-15%. В том случае, когда важно более точно знать как содержание основного компонента, так и содержание вредных примесей (например, в фармацевтической и пищевой промышленности), погрешность не должна быть выше 0,1-1%. Для полупроводников же погрешность определения основных компонентов должна быть ниже 0,1%, а по возможности и 0,01%, так как физические свойства этих соединений в значительной
степени зависят от постоянства их стехиометрического состава.
Достаточно точны гравиметрические и титриметрические методы, погрешность которых обычно составляет соответственно 0,05-0,2 и 0,1-0,5%. Из современных методов наиболее точен кулонометрический, позволяющий проводить определение компонентов с погрешностью 0,001-0,01%.
Как правило, требования к точности химического анализа диктуют технологи, геологи, медики, физики и т. д. Но у химиков-аналитиков всегда должно быть собственное понимание необходимости достижения той или иной точности при проведении анализа. Неоправданное требование высокой точности определения обычно удлиняет и удорожает химический анализ. Так, при увеличении точности определения ряда компонентов с 2 до 0,2% время анализа увеличивается более чем в 20 раз. Завышение требований к точности часто приводит к необходимости использовать сложную и дорогостоящую аппаратуру. Таким образом, у исследователя должен быть разумный подход к выбору более или менее точного метода, особенно при проведении массовых химических анализов.
Экспрессностъ метода. Требование к экспрессности, т.е. быстроте проведения анализа часто выдвигается как одно из основных требований при выборе метода или методики анализа. Задачи анализа иногда диктуют необходимость выбора экспресс-метода.
Есть методы, которые позволяют проводить анализ очень быстро. Так, методы атомно-эмиссионной спектроскопии с применением квантометров дают возможность определять 15-20 элементов за несколько секунд; в методе ионометрии используют ион-селективные, в том числе ферментные электроды, время отклика которых на содержание компонента составляет 0,5-1 мин.
Следует отметить, что в большинстве методик измерение сигнала, связанного с содержанием, как правило, довольно быстрая стадия. Основное время при проведении химического анализа затрачивается на подготовку пробы. Поэтому для уменьшения времени анализа при прочих равных условиях следует выбирать наиболее избирательные, не требующие специальной подготовки проб, методики.
Стоимость анализа. При выборе метода анализа нередко большую роль, особенно при проведении серийных и массовых анализов, играет стоимость химического анализа, куда входит стоимость используемой аппаратуры, реактивов, рабочего времени аналитика и иногда самой анализируемой пробы.
Методы различны по стоимости аппаратурного оформления. Наиболее дешевые — титриметрические, гравиметрические, потенциометрические методы. Аппаратура большей стоимости используется, например, в вольтамперометрии, спектрофотометрии, люминесценции, атомной абсорбции. Наиболее высока стоимость аппаратуры, используемой в нейтронно-активационном методе анализа, масс-спектрометрии, ЯМР- и ЭПР-спектроскопии (ядерно-магнитно-резонансная и электронно-парамагнитно-резонансная), в атомно-эмиссионной спектроскопии с индуктивно связанной плазмой.
38
Новый справочник химика и технолога
Таблица 1.9
Оценочные характеристики методов количественного анализа [2]
Метод Экспрессность метода Относительная стоимость анализа Диапазон f определяемых концентраций (ре) Точность анализа
Гравиметрия ОН Н 1-2 В
Титриметрия У Н 1-4 В
Кулонометрия ОН-У Н-У 1-4 В
Вольтамперометрия У У 3-10 У
Потенциометрия У-В Н-У 1-7 У
Спектрофотометрия У-В Н-У 3-6 У
Атомная спектрометрия в У-В 3-9 У
Эмиссионная спектроскопия с индуктивно-связанной плазмой в в 5-9 У
Хроматография в У-В 3-9 У
Нейтронно-активационный анализ он в (а) У
Рентгенофлуоресцентный анализ в в (б) в
Примечания. 1. Принятые сокращения: В — высокая; У — умеренная; Н — низкая; ОН — очень низкая.
2./ указан примерно: (а) — 10 5-10 12гл4, (б) — Ю'-Ю^гл’1. 3. pc = 1g—, где с— концентрация в моль-л1.
Оценивая стоимость анализа, учитывают также стоимость и доступность реактивов; время, затрачиваемое на обнаружение или определение одного компонента; массу анализируемой пробы, особенно в тех случаях, когда дорогостоящим является сам материал анализируемого объекта (сплавы и слитки платиновых металлов, золота и т. п.). При прочих равных условиях для решения поставленной задачи следует выбирать наиболее дешевые метод и методику проведения анализа. Некоторая информация, относящаяся к выбору подходящего метода анализа, представлена в сжатом виде в табл. 1.9: «классические» методы, избранные инструментальные методы и недеструктивные методы.
Автоматизация анализа. При проведении массовых однородных анализов следует выбирать метод, допускающий автоматизацию анализа, которая позволяет облегчить труд аналитика, заменив многие «ручные» трудоемкие операции автоматическими, снизить погрешности отдельных операций, увеличить скорость проведения анализа, снизить его стоимость, проводить анализ на расстоянии и т.д.
Другие требования к методам анализа. Помимо приведенных выше факторов, которые принимают во внимание при выборе метода и методики, к методу, в зависимости от задачи анализа, могут предъявляться и другие специфические требования. Например, проведение анализа без разрушения образца (недеструктивный анализ) необходимо при анализе произведений искусства, археологических объектов, предметов судебной экспертизы и т. п. В этом случае анализ часто проводят с применением рентгено-флуоресцентного и ядерно-физи-ческих методов.
При химическом анализе вкраплений, микрофаз металлических слитков, геологических и археологических образцов; при послойном анализе пленок; при выяснении состава пятен, штрихов в рукописях, в объектах судебной экспертизы и т. д. требуется проводить локальный анализ. При таком анализе вводят новую характеристику метода— пространственное разрешение, т. е. способность различать близко расположенные участки образца. Пространственное разрешение определяется диаметром и глубиной области, разрушаемой при анализе. Наиболее высокое пространственное разрешение, достигаемое современными методами локального анализа, — 1 мкм по поверхности и до 1 нм (т. е. несколько моноатомных слоев) по глубине. В локальном анализе используют рентгеноспектральные методы (электронно-зондовый микроанализатор), атомно-эмиссионные спектральные методы с лазерным возбуждением, масс-спектрометрию.
Одна из важных задач современной аналитической химии — проведение химического анализа на расстоянии (дистанционный анализ). Такая проблема возникает при анализе космических объектов, исследовании дна Мирового океана, при анализе радиоактивных или других вредных для здоровья человека веществ. Проблему анализа на расстоянии часто решают с применением ядерно-физических, масс-спектрометрических и других методов.
Таким образом, большое число факторов, которые необходимо оценить и учесть при выборе метода анализа и оптимальной методики обнаружения или определения компонентов, делает этот этап химического анализа достаточно сложным. Определенную помощь аналитик может получить, решая эту задачу с применением моделирования на ЭВМ.
Информационные базы и общие вопросы аналитической химии
39
Литература
1. Основы аналитической химии: В 2 кн. 2-е изд., перераб. и доп. / Под ред. акад. Золотова Ю.А. Кн. 1. Общие вопросы. Методы разделения. М.: Высшая школа, 2000. С. 20-30.
2 а. Каплан Б.Я., Майоров И.А., Филимонов Л.Н. Методы аналитического контроля в цветной металлургии: Руководство. Т. 9. Часть 1. Метрология и стандартизация методик аналитического контроля производства в цветной металлургии. М.: Гиредмет, 1984. С. 216-225.
2 б. Каплан Б.Я, Филимонов Л.Н., Майоров И.А. Метрология аналитического контроля производства в цветной металлургии. М.: Металлургия, 1989. 200 с.
3. Рекомендации по использованию терминов «селективный», «селективность» и родственных им терминов в аналитической химии // Pure a. Appl. Chem. V. 55, № 3. Р. 553 / Публикация подготовлена Г. ден Буфом и А. Галуцким; Пер. с англ. В.И. Игнатова и ЕЯ. Неймана, а) Журн. аналит. химии, 1984. Т. 39, № 5. С. 945-947;
б) Номенклатурные правила ИЮПАК. Т 4. // Аналитическая химия. М.: ВИНИТИ, 1985. С. 169-173.
4. Селективность в аналитической химии (IUPAC Recommendations 2001) // Pure a. Appl. Chem., 2001. V. 73, № 8. P. 1381-1386.
5. Vogel's Textbook of Quantitative Chemical Analysis. Fifth Edition. Longman Scientific and Technical / Copublished in the United States with Wiley a. Sons Inc., New York, 1989. 877 p.
1.5. Отбор и подготовка
проб к анализу (способы вскрытия)1 [1-23] (использованы материалы И.Ф. Долмановой [1])
Проведение химического анализа начинают с отбора и подготовки пробы к анализу. Следует отметить, что все стадии анализа связаны между собой. Так, тщательно измеренный аналитический сигнал не дает правильной информации о содержании определяемого компонента, если неправильно проведен отбор или подготовка пробы к анализу. Погрешность при отборе пробы часто определяет общую точность определения компонента и делает бессмысленным использование высокоточных методов. В свою очередь отбор и подготовка пробы зависят не только от природы анализируемого объекта, но и от способа измерения аналитического сигнала. Приемы и порядок отбора пробы и ее подготовки настолько важны при проведении химического анализа, что обычно регламентируются Государственным стандартом (ГОСТ) [1].
1.5.1. Отбор пробы
Для проведения анализа, как правило, берут так называемую среднюю (представительную) пробу. Это небольшая часть анализируемого объекта, средний состав и свойства которой должны быть идентичны во всех отношениях
1 См. также раздел 7 и [18].
среднему составу и свойствам исследуемого объекта. Различают генеральную, лабораторную и анализируемую пробы.
Генеральная (называемая иногда первичной, большой или грубой) проба отбирается непосредственно из анализируемого объекта. Она достаточно большая — обычно 1-60 кг, для некоторых объектов (например, руды) составляет иногда 0,6-5 т. Из генеральной пробы путем ее сокращения отбирают лабораторную пробу (обычно от 1 до 25 кг). Одну ее часть используют для предварительных исследований, другую — для арбитражных анализов, третью— непосредственно для анализа (анализируемая проба). В случае необходимости пробу измельчают и усредняют. Для анализируемой пробы проводят несколько определений компонента: из отдельных навесок 10-1000 мг (если анализируемый объект— твердое вещество) или аликвот (если анализируемый объект — жидкость или газ).
Содержание определяемого компонента в анализируемой пробе должно отражать среднее содержание этого компонента во всем исследуемом объекте, т. е. анализируемая проба должна быть представительной. Чем больше материала отобрано для пробы, тем она представительнее. Однако с очень большой пробой трудно работать, это увеличивает время анализа и расходы на него. Таким образом, отбирать пробу нужно так, чтобы она была представительной и не очень большой.
При отборе пробы необходимо учитывать:
1) агрегатное состояние анализируемого объекта (способы отбора пробы различны для газов, жидкостей и твердых веществ);
2) неоднородность анализируемого материала (чем однороднее вещество, тем проще отобрать пробу);
3) размер частиц, с которых начинается неоднородность;
4) требуемую точность оценки содержания компонента во всей массе анализируемого объекта в зависимости от задачи анализа и природы исследуемого объекта.
Один из факторов, который нужно учитывать при выборе способа отбора пробы,— возможность изменения состава объекта и содержания определяемого компонента во времени. Например, переменный состав воды в реке, колебания состава дымовых газов промышленного предприятия, изменение концентрации компонентов в пищевых продуктах и т. д.
Отбор пробы газов
Степень однородности газов велика (неоднородность наблюдается на молекулярном уровне). Смеси газов гомогенны, поэтому генеральная проба может быть относительно небольшой и отбор пробы обычно не представляет трудностей. Пробу газа отбирают, измеряя его объем при помощи вакуумной мерной колбы или бюретки с соответствующей запорной жидкостью, часто конденсируют газ в ловушках разного типа при низких температурах. По-разному отбирают пробу газа из замкнутой емкости и из потока. В замкнутой
Информационные базы и общие вопросы аналитической химии
41
Отбор пробы твердых веществ
При отборе генеральной, лабораторной и анализируемой пробы твердых веществ прежде всего возникает вопрос о размере пробы, который должен обеспечивать ее представительность. Оптимальная масса пробы обусловлена неоднородностью анализируемого объекта, размером частиц, с которых начинается неоднородность, и требованиями к точности анализа, обычно определяемой погрешностью в отборе пробы. Зависимость массы представительной пробы от размера неоднородных частиц проиллюстрирована ниже (табл. 1.10).
Таблица 1.10
Соотношение между массой пробы и размером неоднородных частиц
Диаметр неоднородных частиц, мм 40-50 25 10 5 3 2 1
Масса пробы, кг 50- 3000 10- 700 2- 100 0,5- 25 0,2- 10 0,1-5 0,02-1
Для расчета оптимальной массы представительной пробы существует несколько приемов. Часто используют приближенную формулу Ричердса - Чеччота:
Q = Kd\
где Q— масса пробы, обеспечивающая ее представительность, кг; d — наибольший диаметр неоднородных частиц, мм; К— эмпирический коэффициент пропорциональности, характеризующий степень неоднородности распределения определяемого компонента в материале, меняется в пределах 0,02-1.
В более точных расчетах используют формулу Бенедет-ти - Пихлера:
5 =
г у1лЛ р Jwv® ’
где sr— относительное стандартное отклонение, характеризующее погрешность отбора пробы; РА— доля фазы, содержащей определяемый компонент А во всей массе анализируемого объекта; р и рд — плотность материала анализируемого объекта и фазы, содержащей определяемый компонент А, гем-3; о — оптимальная масса пробы, г; N— число частиц на 1 г материала пробы с наибольшим диаметром частиц d (мм) и плотностью р (N в этом случае оценивают по номограмме, рассчитанной для частиц сферической формы, рис. 1.6).
Способы отбора генеральной пробы твердого вещества различны для веществ, находящихся в виде целого (слиток, стержни, прутья и т. д.) или сыпучего продукта. При про-боотборе от целого твердого объекта необходимо учитывать, что он может быть неоднороден. Например, состав массы отливки отличен от состава ее поверхности вследствие постепенного остывания металла. Так, при затвердевании чугуна его примеси оттесняются внутрь; неравномер
но распределяются в слитках стали углерод, сера, фосфор. Процесс расслаивания в слитках металлов и сплавов называют ликвацией. Учитывая возможную неоднородность целого анализируемого объекта, при отборе пробы его либо дробят, если вещества хрупкие, либо распиливают через равные промежутки, либо высверливают в разных местах слитка (рис. 1.7). Последний способ наиболее употребим для отливок.
Рис. 1.6. Приблизительное соотношение между Nwd при плотности анализируемого вещества (1, 2, 3, 5, 10)-103 кг-м~3 [1]
Рис. 1.7. Отбор средней пробы металла или сплава высверливанием слитка [1]
Отбор пробы сыпучих продуктов тем труднее, чем неоднороднее анализируемый объект: в пробе должны быть представлены куски разного размера, полно отражающие состав образца. При отборе пробы сыпучих продуктов массу исследуемого объекта перемешивают и пробу отбирают в разных местах емкости и на разной глубине, используя при этом специальные щупы-пробоотборники. Если материал объекта транспортируется, то пробу отбирают с транспортера или желоба через равные промежутки времени, при другом способе транспортировки берут на анализ, например, каждую десятую лопату, тачку и т. д. Пробу сыпучих материалов, находящихся в вагонах, например соды, отбирают обычно по схеме двойного конверта (рис. 1.8) [3].
Рис. 1.8. Отбор пробы сыпучих материалов по схеме двойного конверта [3]
42
Новый справочник химика и технолога
Для получения усредненной пробы, характеризующей большие количества вещества (руда, минеральные удобрения), находящегося в железнодорожном составе, используют довольно сложную процедуру. Лучше всего отбирать пробы при погрузке или выгрузке материалов. Отбор проб из вагонов, если они только что загружены и материал не подвергся сегрегации, производят по определенной схеме. Например, из материала (руда или др.), загруженного в 60-70 железнодорожных вагонов, нужно отобрать 60-70 проб, по одной из каждого вагона. Поверхность материала в вагоне разравнивают и намечают на ней точки отбора проб, как показано на рис. 1.9. Из первого вагона берут пробу в точке № 1, из второго — в точке № 2, из пятнадцатого — в точке № 15 — и так до 20-го вагона. В 21-м вагоне пробы берут снова в точке № 1, повторяют всю схему отбора и т. д. Если число вагонов оказывается меньше числа точек отбора, предусмотренных схемой, то из оставшихся вагонов (менее 15) берут пробы через одну точку, т. е. из 61-го вагона — в точке № 1, из 62-го вагона — в точке № 3 и т. д. до конца. Пробы отбирают не с поверхности, а с некоторой глубины. Глубина расположения точки отбора от поверхности материала указана на рис. 1.9.
Рис. 1.9. Разметка железнодорожного вагона по площади и по глубине отбора для получения генеральной пробы [2]
Перемешивание проводят механически в емкостях (ящики, коробы и т. д.), перекатыванием из угла в угол на различных плоскостях (брезентовые полотнища, листы бумаги и т. д.), перемешиванием методом конуса и кольца [1,3].
Способ кольца и конуса заключается в том, что исходную пробу рассыпают попеременно в конусообразную кучу и окружающее ее кольцо (рис. 1.10). Затем конусообразную кучу рассыпают на кольцо и полученное кольцо большого объема снова ссыпают в конус. В вершину последнего вставляют доску и «развертывают» конус. Снова раскидывают материал в кольцо и повторяют все сначала. Этот способ применим для проб массой 200-2500 кг и размеров кусков не более 50-60 мм. Малые по объему пробы хорошо перемешиваются при растирании в шаровых мельницах.
Рис. 1.10. Перемешивание пробы по способу кольца и конуса [3]
После отбора генеральной (или лабораторной) пробы твердого вещества осуществляют процесс гомогенизации, включающий операции измельчения (дробления) и просеивания. Пробы, содержащие крупные куски, разбивают в дробильных машинах и мельницах разного типа, меньшие измельчают в шаровых мельницах и специальных ступках (из закаленной инструментальной стали), состоящих из плиты— основания, закрепляющего кольца и пестика (ступки Абиха или Платтнера). Для тонкого измельчения используют фарфоровые, агатовые, яшмовые и кварцевые ступки с пестиками из такого же материала.
Так как в процессе дробления куски разного размера растирают по-разному (мягкие материалы измельчаются гораздо быстрее, чем твердые), то возможны потери в виде пыли, приводящие к изменению состава пробы. Чтобы избежать этого, в процессе измельчения периодически делят крупные и мелкие частицы просеиванием, и крупные частицы растирают отдельно. Операции измельчения и просеивания чередуют до тех пор, пока не получат достаточно растертую однородную пробу.
Следующий этап отбора пробы— усреднение, включающее операции перемешивания и сокращения пробы.
Сокращение пробы проводят различными способами (рис. 1.11).
Рис. 1.11. Приспособления для сокращения пробы: а — квартование; б — шахматный способ отбора; в — механический делитель [1]
а
б
4'1/2 от 1
Информационные базы и общие вопросы аналитической химии
43
Действие приспособлений а) и б) понятны из рисунка. Процесс в), как правило, многостадийный, включающий повторное перемешивание и деление. Способы квартования и шахматного отбора проиллюстрированы также на рис. 1.12 а, б.
Рис. 1.12. Подробная иллюстрация способов квартования и шахматного отбора [2]
Степень сокращения может быть определена заранее на основании расчета величины генеральной и анализируемой проб, которые получают в результате последовательного уменьшения объема анализируемого объекта.
1.5.2. Потери и загрязнения при отборе пробы.
Хранение пробы
В процессе отбора и хранения пробы возможны потери определяемого компонента, внесение загрязнений, изменение химического состава. Все это приводит к увеличению общей погрешности анализа.
Потери в виде пыли при измельчении твердых образцов горных пород могут составлять до 3% массы объекта. Если состав пылевой фракции отличается от состава образца, то это приводит к неправильному определению содержания компонентов. Потери в виде пыли можно в заметной степени уменьшить просеиванием пробы при измельчении.
Другой возможный источник ошибок при отборе и хранении пробы — потеря летучих продуктов вследствие изменения температурного режима при хранении или разогрева
при измельчении твердых образцов. Так, при измельчении горных пород, руд и минералов наблюдаются заметные потери таких летучих компонентов, как вода, ртуть, сера, таллий. При изменении температуры особенно велики потери летучих органических соединений, определяемых в различных природ ных и промышленных объектах.
Могут быть также большие потери вследствие адсорбции определяемого компонента на поверхностях емкостей для отбора и хранения пробы.
Состав анализируемого объекта может меняться за счет проходящих в нем химических реакций (за счет разложения компонентов, окисления их при взаимодействии с атмосферным кислородом). Например, концентрация пестицидов в растениях, почве и т.п. со временем значительно понижается, что обусловлено, прежде всего, химическими превращениями пестицидов. При анализе геологических образцов в процессе отбора пробы наблюдаются заметные потери определяемых компонентов вследствие окисления: сера, рений, железо(П) — или восстановления (ртуть). Потери ртути в пробе, если не принять особых мер предосторожности, могут достигать 60% [1].
Погрешности, обусловленные внешними загрязнениями, особенно велики при определении примесей компонентов, их следовых количеств. Вот почему при растирании образцов используют ступки из особо твердых материалов (агат или кварц) и хранят пробы в посуде из особых сортов стекла или полиэтилена. Например, пробы воды для определения кремния отбирают только в полиэтиленовые бутыли, а не в стеклянную посуду; при определении органических соединений, наоборот, предпочтительнее посуда из стекла.
Исходя из сказанного, методики отбора пробы строго регламентируются нормативными документами (ГОСТы, аттестованные МВИ): число и последовательность операций измельчения и просеивания, время и интенсивность растирания, способы отделения представительной пробы.
Особо рассматривают вопрос о хранении и консервации пробы. Допустимый промежуток времени между отбором и анализом зависит от состава пробы, природы определяемых компонентов и условий хранения пробы. Чем больше вероятность изменения содержания определяемых компонентов, тем скорее должен быть проведен анализ; если невозможно провести анализ сразу после отбора, то пробу консервируют. Некоторые определяемые вещества устойчивы длительное время и не требуют особых условий консервации (резкое охлаждение, изменение pH среды, добавление стабилизирующих веществ). В отдельных случаях для сохранения определяемого компонента его экстрагируют органическими растворителями или сорбируют на различных твердых веществах. Для получения достоверных результатов пробу природной воды, например, анализируют обычно в течение 1-2 ч после отбора. Пробы можно стабилизировать на несколько часов охлаждением до 0 °C и на несколько месяцев — резким охлаждением до - 20 °C. Для консервирования определяемых компонентов добавляют разные консерванты, чаще всего это кислоты и вещества, образующие комплексные соединения.
44
Новый справочник химика и технолога
Хранят пробы в условиях, гарантирующих постоянство их состава в отношении тех компонентов, которые предполагается определять, при этом учитывают комплекс условий (температура, освещенность, материал посуды и т. д.); однако в ряде случаев до сих пор не найдено удовлетворительного способа хранения пробы.
1.5.3. Подготовка пробы к анализу
Подготовка пробы — важный этап проведения химических анализов. При подготовке пробы к анализу можно выделить три основные стадии: 1) высушивание; 2) разложение (чаще с переведением пробы в раствор); 3) устранение влияния мешающих компонентов.
Устранение влияния мешающих компонентов обычно производят в процессе измерения либо непосредственно перед ним, исходя из требований конкретной методики анализа и возможностей аналитической аппаратуры. Эта стадия подготовки рассматривается в описаниях конкретных аналитических методов и методик.
Вода в пробах. Высушивание образцов
Анализируемый образец содержит, как правило, переменное количество воды. Это может быть химически несвязанная вода, например: адсорбированная на поверхности пробы твердого вещества; сорбированная щелями и капиллярами аморфных веществ (цеолит, крахмал, белок); окклюдированная полостями минералов, руд, горных пород. Такая вода присутствует в пробе как загрязнение из атмосферы или раствора, в котором формировалось анализируемое вещество. Количество воды может меняться в зависимости от температуры и влажности окружающей среды, способа отбора и хранения пробы, приемов и степени измельчения твердого вещества, времени и способа его хранения и т. п. Например, при измельчении и хранении базальта содержание в нем адсорбированной воды может увеличиваться от 0,2 до 2%. Количество же сорбированной воды в таких аморфных веществах, как силикагель, уголь растительного и животного происхождения, может составлять 20% от массы образца.
Анализируемый объект может также содержать химически связанную воду (иногда называемую стехиометрической водой), т. е. являющуюся неотъемлемой частью молекулярной или кристаллической структуры твердого вещества. Это может быть кристаллизационная (например, в соединениях ВаСЬ’ 2Н2О, CaSO4- 2Н2О, Na2B4O7 • ЮН2О) или конституционная вода, выделяющаяся в результате разложения вещества при нагревании (Са(ОН)2 —> СаО + Н2О; 2KHSO4 K2S2O7 + Н2О). Часть химически связанной воды может теряться в процессе отбора и хранения пробы. Например, при измельчении CaSO4 • 2Н2О вследствие разогрева пробы при растирании содержание воды может уменьшиться от 20 до 5%.
Для правильного установления состава объекта и получения воспроизводимых результатов необходимо удалить влагу из образца, высушив его до постоянной массы, или определить содержание воды, так как результат анализа следует пересчитать на постоянную массу. Чаще всего анализируемый
образец высушивают на воздухе или в сушильных шкафах при относительно высокой температуре (105-120 °C). Получить воздушно-сухую массу образца можно лишь для таких негигроскопичных веществ, как металлы, сплавы, некоторые виды стекол и минералов. В отдельных случаях пробы высушивают в эксикаторах над влагопоглощающими веществами (прокаленный хлорид кальция, фосфорный ангидрид, перхлорат магния, драйерит — 0,5 CaSO4- Н2О). Длительность и температуру высушивания образца, зависящие от его природы, устанавливают заранее экспериментально (например, методом термогравиметрии). Если какие-либо особые указания на этот счет в методике отсутствуют, образцы сушат в сушильных шкафах при 110 °C в течение 1-2 ч. Содержание определяемого компонента рассчитывают, исходя из навески высушенного при определенных условиях образца. Если нужно установить состав первоначально отобранного материала, то следует определить массу, потерянную при высушивании. Если полностью удалить воду из пробы или высушить ее до постоянной массы не удается, то воду определяют в нескольких пробах, отобранных для анализа на другие компоненты.
Известно большое число методов определения воды. Так, воду определяют гравиметрически косвенным или прямым методом. В косвенном методе о содержании воды судят по потере массы анализируемой пробы при ее высушивании или прокаливании. Этот метод часто не дает правильных результатов, что связано с трудностью определения температуры, необходимой для полного выделения воды, и потерей с водой летучих компонентов образца. Прямой гравиметрический метод основан на поглощении выделившейся из образца воды подходящим поглотителем, чаще всего безводным перхлоратом магния. О содержании воды судят по увеличению массы предварительно взвешенного поглотителя.
Часто для определения воды применяют титриметриче-ский метод с использованием реагента Фишера. Для определения воды часто используют и такие методы, как газожидкостная хроматография и ИК-спектроскопия.
Разложение образцов. Переведение пробы в раствор
Существуют методы анализа (например, некоторые спектроскопические или ядерно-физические), в которых для измерения аналитического сигнала используют анализируемые пробы, в том числе и твердые объекты (без предварительного разрушения) в виде гомогенных образцов, порошков, таблеток, полученных прессованием, и т. п. В большинстве же методов анализа требуется предварительное переведение определяемого компонента в раствор.
Выбор способа разложения пробы и переведения ее компонентов в раствор зависят от нескольких факторов, которые необходимо учитывать при обосновании схемы химического анализа. Прежде всего обращают внимание на неорганическую или органическую природу основы (матрицы) объекта, химический состав образца, химические свойства определяемого компонента. Так, при определении одного и того же элемента (например, кобальта, цинка, железа) в крови, пищевых продуктах или сплавах и минералах способ разложения образцов определяется со
Информационные базы и общие вопросы аналитической химии
45
ответственно органической или неорганической природой объекта. Разложение и перевод в раствор проб силикатов проводят в зависимости от определяющего их состав соотношения МеО : SiO2. Если в составе силиката преобладают оксиды металлов, то пробу растворяют в кислотах, если оксид кремния, то проводят сплавление или спекание.
При определении содержания железа, титана, алюминия в силикате пробу сплавляют со щелочными плавнями; при определении суммы щелочных металлов — спекают с СаО и СаСО3. Способ разложения пробы и переведения ее в раствор определяется также целью анализа и во многом зависит от выбранного аналитического метода. Например, различаются подготовки проб при определении органических соединений в биологических объектах хроматографическими или спектрофотометрическими методами.
Выбрав способ разложения пробы, необходимо оценить источники всех возможных погрешностей на этой стадии анализа. Наиболее типичные ошибки обусловлены потерей летучих компонентов при использовании высоких температур; загрязнением из материалов посуды и приспособлений для разложения проб; наличием мешающих проведению анализа примесей в реактивах и растворителях, используемых при разложении образцов.
Способы разложения делят на сухие и мокрые, к первым относят термическое разложение, сплавление и спекание с различными веществами (солями, оксидами, щелочами и их смесями) [4]; ко вторым — растворение анализируемой пробы в различных растворителях, преимущественно в кислотах и их смесях.
Растворение
В идеальном случае растворитель должен растворять пробу быстро, в достаточно мягких условиях и не мешать на последующих стадиях анализа.
Лучший растворитель— вода. Многие неорганические соли (особенно соли щелочных металлов, аммония и магния) и некоторые органические соединения (низшие и многоатомные спирты, аминокислоты, гидрохлориды аминов, соли щелочных металлов органических кислот, мочевина и т. д.) легко растворяются в воде. Иногда в воду добавляют немного кислоты для предотвращения гидролиза и частичного осаждения некоторых катионов металлов. В отдельных случаях для растворения органических веществ используют смесь воды и смешивающегося с ней органического растворителя (например, смесь воды и этанола).
Для растворения органических соединений применяют органические растворители. Как правило, это спирты, хлорированные углеводороды, кетоны. Так, для растворения полимерных материалов разного типа используют диме-тилформамид, диметилацетамид, метилизобутилкетон, циклогексанон, метанол.
При мокром способе разложения пробы часто применяют различные кислоты и их смеси при нагревании. При этом в пробу не вводят посторонние катионы, а сами кислоты сравнительно легко удаляются из сферы реакции в процессе нагревания; желательно, чтобы кислота образовывала с ионом определяемого элемента растворимую соль. Лучшим растворителем для руд и многих металлов является соляная кислота [2].
Кислоты в зависимости от их природы и концентрации могут проявлять окислительные (конц. HNO3 и H2SO4) или комплексообразующие свойства (HF и Н3РО4). Иногда к кислотам добавляют пероксид водорода Н2О2), органические оксикислоты и т.п. Наиболее часто используемые для растворения кислоты и их смеси представлены в табл. 1.11 [1].
Таблица 1.11
Кислоты и их смеси, используемые при растворении
Кислота Растворяемый объект Примечание
НС1 Металлы, оксиды металлов, железные руды, карбонаты, органические амины Кислота улетучивается при нагревании
HF Силикатные горные породы и минералы, стекла, керамика 1. Кислота удаляется при нагревании с H2SO4. 2. Устраняется кремний в виде SiF4. 3. Избыток кислоты мешает, так как образуются комплексные соединения с некоторыми металлами
HNO3 (конц.) Металлы (кроме Au, Pt, Cr, Al),сплавы, сульфиды, арсениды, органические соединения Окислитель
H2SO4 (конц.) Металлы (сурьма, олово), оксиды металлов, арсениды, ферротитан, органические соединения Окислитель, возможно разрушение стекла посуды
НС1О4 (конц.) Сплавы железа, нержавеющая сталь Сильный окислитель, взрывоопасна!
HNO3 + H2SO4 Большинство неорганических веществ, органические соединения Часто используемая смесь HNO3 и H2SO4 переменного соотношения
HF + HNO3 Сплавы вольфрама, молибдена, тантала, циркония, силикаты, ферромолибден Образуются фторидные комплексные соединения
HF + Н3ВО3 Сплавы многих редких металлов, природные ные фосфаты, керамические материалы 1. Смесь кислот часто используют в автоклавах. 2. В присутствии Н3ВО3 заметно ускоряются многие процессы растворения
40
Новый справочник химика и технолога
емкости (например, цех предприятия, рабочая комната и т. д.) пробу газа отбирают в разных точках (в зависимости от задачи), объемы газа смешивают — или анализируют отдельно каждую пробу. При отборе пробы из потока газа обычно используют метод продольных струй и метод поперечных сечений. Метод продольных струй применяют, когда состав газа вдоль потока не меняется.
В этом случае поток делят на ряд струй вдоль потока и пробы газа отбирают в струях через одну (рис. 1.4а). Если состав газа вдоль потока меняется, то пробы берут на определенных расстояниях (часто через специальные отверстия в трубах) вдоль потока (рис. 1.46).
Рис. 1.4. Отбор проб газа в потоке [2]
а): метод продольных струй; б): метод поперечных сечений; в): чертеж устройства для отбора газа из выбранной точки потока
Так как состав анализируемых газов часто меняется во времени (например, в зависимости от графика работы предприятий, состояния атмосферы, температуры в помещениях и т. д.), то в зависимости от требуемой информации пробы усредняют или анализируют отдельно объемы газов, отобранные в разное время.
Отбор пробы жидкостей
Способы отбора гомогенных и гетерогенных жидкостей различны.
Гомогенные жидкости отличаются высокой степенью однородности, поэтому, как и для газов, способы отбора пробы относительно просты. Пробу гомогенной жидкости отбирают при помощи соответствующих пипеток, бюреток и мерных колб. Отбор пробы из общей емкости проводят после тщательного перемешивания. Если по какой-либо причине (например, из-за большого объема) жидкость нельзя хорошо перемешать, то отбор пробы проводят на разной глубине и в разных местах емкости.
Отбор гомогенной жидкости из потока проводят через определенные интервалы времени и в разных местах (рис. 1.5а). Для отбора проб на разной глубине используют специальные пробоотборные устройства — батометры различной конструкции. Основная часть батометра— цилиндрический сосуд вместимостью 1-3 л, закрывающийся сверху и снизу крышками. После погружения цилиндра в
жидкость на заданную глубину крышки закрывают, и сосуд с пробой поднимают на поверхность. Место и время отбора жидкости выбирают в зависимости от решаемой задачи. Например, при оценке загрязнения водоема, обусловленного сточными водами, резонно анализировать не только воду самих стоков, но и воду водоема ниже и выше впадения в него стока, учитывая время максимального сброса сточных вод.
Пробы гетерогенных жидкостей отбирают не только по объему, но и по массе. Чтобы отобрать пробу, поступают по-разному: в одних случаях жидкость гомогенизируют, в других, наоборот, добиваются полного ее расслоения. Гомогенизацию проводят, изменяя температуру, перемешивая жидкость или подвергая ее вибрации. Если гомогенизировать жидкость невозможно, то ее расслаивают и отбирают пробу каждой фазы, используя при этом специальные пробоотборники, с большим числом забирающих камер (рис. 1.56). Так отбирают на анализ различные фракции продуктов и полупродуктов нефтеперерабатывающей промышленности. Обычно пробу берут после отстаивания смеси жидкостей в чанах или цистернах.
Размер генеральной пробы жидкости хотя и меняется в известных пределах, но все же обычно невелик и не превышает нескольких литров или килограммов.
Места отбора пробы
-z—r-'TtomoK
——
в)
Рис. 1.5. Отбор проб жидкости [2]:
а) из потока;
б) гетерогенной, разделяющейся на слои;
в) чертеж крана для отбора жидкости из трубопровода
46
Новый справочник химика и технолога
Продолжение таблицы 1.11
Кислота Растворяемый объект Примечание
Царская водка HC1 + HNO3 (3:1) Металлы (Au, Pt, Pd), сплавы, сульфидные руды, органические соединения Сильные окислительные свойства
H2SO4 + HC1O4 + + Н3РО4 Ферросплавы, железные руды Связывание Fe3+ в фосфатные комплексы
НС1 + H2SO4+ Br2 Металлы (In, Sb), некоторые органические соединения
Источниками загрязнений при растворении в кислотах могут быть примеси в используемых реагентах или частичное растворение материала посуды. Поэтому для растворения применяют кислоты высокой степени очистки и подбирают сосуды из соответствующего материала. Например, растворение в HF ведут в чашках и стаканчиках из платины, фторопласта (политетрафторэтилена), стеклоуглерода. При растворении в кислотах возможны также механические потери при вскипании и разбрызгивании, потери летучих (H2S, SO2, СО2, GeCLj, SbCl3, SnCl4 и т. п.) или труднорастворимых веществ (CaF2, SrF2, BaF2, MgF2 и т. п.).
Автоклавное вскрытие
Растворение проб часто проводят в автоклавах (рис. 1.13.) [1], что имеет некоторые преимущества: обеспечивается разложение веществ, не взаимодействующих с реагентами при обычных температуре и давлении; уменьшается количество расходуемых реагентов; увеличивается скорость разложения; удается избежать потерь летучих продуктов реакции, за исключением газов после охлаждения автоклава и его разгерметизации.
Рис. 1.13. Автоклав для разложения веществ кислотами (поперечное сечение):
/ — корпус; 2 — фторопластовый стакан; 3 — пружина;
4 - кольцо; 5 — винт для регулирования давления;
6 — крышка
Для ускорения разложения кислотами иногда используют катализаторы. Так, окисление органических материалов концентрированной серной кислотой ускоряется в присутствии CuSO4, HgSO4 и H2SeO3.
При разложении проб растворы гидроксидов, карбонатов щелочных металлов или аммиака применяют значительно реже, чем кислоты. Эти соединения используют в основном для перевода в раствор анионов; при этом многие неорганические катионы и органические соединения, входящие в состав образца, остаются в осадке. Гидроксид натрия (или калия) растворяет некоторые металлы (алюминий) и оксиды кислотного характера (WO3, МоО3, GeO3, V2O5).
В отдельных случаях для растворения используют ферменты. Один из способов растворения высокомолекулярных соединений, например белков, — гидролиз в присутствии трипсина, папаина и других протеаз.
Выбор сухого способа разложения (сплавление, спекание и термическое разложение) определяется задачей анализа, природой разлагаемого вещества, выбранным методом определения компонентов, наличием необходимой аппаратуры.
Сухое озоление
(термическое разложение)
Этот способ наиболее распространен при вскрытии проб органического происхождения в токсикологическом анализе следовых содержаний примесей металлов (в лабораториях Санэпиднадзора, Госкомэкологии, на станциях защиты растений, в ветеринарных лабораториях ит. п.) [1, 5].
Сухое сожжение органических веществ (ОВ) проводят под действием кислорода воздуха или кислорода из баллона [6, 7, 8]. Большинство пищевых продуктов сгорает при температуре 550-600 °C (табл. 1.12). Преимуществом этого способа является простота аппаратуры (термопечи и тигли), минимум внимания оператора, отсутствие загрязнений от реактивов; недостатком— возможность потерь легколетучих элементов (Hg, As, Se, Те), взаимодействие с материалом тигля и длительность процесса. Получило широкое распространение сухое сожжение с озоляющими добавками. Это могут быть окислители, разбавители, плавни, вещества, препятствующие улетучиванию элементов. Например, при определении мышьяка при прокаливании добавляют соли магния. После прокаливания остаток растворяют в подходящем водном растворе кислоты.
Информационные базы и общие вопросы аналитической химии
47
Таблица 1.12
Температура озоления некоторых материалов (определение общей зольности) [7]
Анализируемый материал Навеска, г t,°C
Злаки 3-5 600
Мука, мучные продукты 3-5 550
Крахмал 3-5 800
Варенье, фруктовый сок 25 525
Кофе, чай 5-10 525
Какао 2-5 600
Сахар 5-10 525
Мед 5-10 600
Орехи 5-10 525
Пряности 2 550-600
Молоко, сливки 5 500
Сыр 1 550
Желатин 5 550
Мясо 3-7 550
Ниже приводятся свойства кислот (и пероксида водорода), используемых для разложения органических веществ (ОВ) по ГОСТ 26929-94 [6].
НС1 — хлороводородная кислота:
не является окислителем и не разрушает большинство ОВ; растворяет ОВ основного характера — амины, алкалоиды;
разлагает металлоорганические соединения Си, Fe, Zn, Sn, As; эти ионы выделяют из биологических жидкостей, пищевых продуктов, минеральных масел экстракцией НС1;
гидролизует многие природные ОВ (протеины, аминокислоты, полисахариды;
вызывает улетучивание хлоридов As(III), Sb(III), Ge(IV), Se(IV), Sn(IV); менее летучи хлориды Hg(II), Re(VII), 1п(Ш), добавки окислителей предотвращают потери As и Sb.
H2SO4 — серная кислота:
взаимодействие H2SO4 с ОВ может вызвать окисление, сульфирование, этерификацию, гидролиз эфиров, дегидратацию или полимеризацию;
почти все ОВ разлагаются методом Кьельдаля с применением закрытых сосудов, а также в открытых сосудах с добавками:
• катализаторов (CuSO4, SeO2, HgSO4),
• солей, повышающих (сульфаты ка-
лия, натрия),
• окислителей (НС1О4, КМпО4, Н2О2), не-разложившийся остаток окисляют HNO3 или Н2О2;
серная кислота усиливает действие других окислителей в смеси за счет повышения Ткип;
• могут быть потери нерастворимых сульфатов (РЬ) при анализе веществ, богатых щелочно-земельными металлами (молоко, кости);
• могут быть потери легколетучих галогенидов, As и Se;
• избыток H2SO4 трудно удаляется из-за высокой Ткип.
HNO3 — азотная кислота:
• окисление ОВ одной HNO3 протекает с трудом. Возможны реакции этерификации и 'нитрования с ароматическими и алифатическими ОВ;
• многие нитросоединения являются более устойчивыми к деструкции, чем исходные вещества, тогда разложение начинают с разбавленной HNO3 или сначала разрушают ОВ серной кислотой;
• разложение ОВ ускоряется в смесях HNO3 с H2SO4, НС1О4 и HNO3, а также в закрытых сосудах из-за повышения давления и Ткип кислоты;
• избыток HNO3 легко удаляется при нагревании более 120 °C;
• в присутствии HNO3 отгоняются летучие НО, НВг, Н3ВО3, S.
НСЮ4 — хлорная кислота:
• окислительные свойства проявляет при Т-160 °C, вызывая полное разрушение ОВ;
• из-за взрывоопасности редко применяется одна кислота, используют предварительное окисление ОВ азотной или серной кислотой, особенно если соединения содержат спиртовые группы;
• требует тщательного соблюдения правил безопасности и специально защищенных вытяжных шкафов.
Н2О2 — пероксид водорода:
• является слабой кислотой, но сильным окислителем, особенно в кислой среде, например в H2SO4;
• в щелочной среде окислительно-восстановительный потенциал ниже, но реакции протекают быстрее;
• для разложения различных ОВ широко применяют смеси с H2SO4h HNO3;
• преимущества обработки пероксидом: высокая чистота, продукт окисления— вода, избыток пероксида легко удаляется кипячением;
• разложение Н2О2 катализируется ионами переходных металлов (Fe, Си, Мп, Сг), причем образуются промежуточные частицы — радикалы-окислители;
• под действием УФ-облучения генерируются сильные окислители — ОН-радикалы. В присутствии солей Fe, оксикислот и УФ-облучения процесс окисления ОВ приобретает циклический характер.
Проводя сухую минерализацию, приходится учитывать неизбежные потери, связанные с летучестью ряда неорганических соединений определяемых элементов (табл. 1.13).
48
Новый справочник химика и технолога
Таблица 1.13
Неорганические соединения, температура кипения которых не превышает 200 °C
Тип соединения Элементы
Хлориды As, В, Ge, Р, Ti, Sn, V, Si
Фториды As, В, Ge, Si, P, W, Mo, U, Re, Ir, Os
Оксиды Os, Ru, S, C
Гидриды As, B, Sn, Pb, Si, Те, Ge, S, P, Se
Сухой способ используют тогда, когда мокрый способ не дает удовлетворительных результатов. Сухой способ менее предпочтителен, чем растворение в кислотах, поскольку возрастает вероятность и величина погрешностей, особенно при сплавлении. Это связано, во-первых, с высокой температурой обработки образца и с большими потерями летучих веществ, а также с разрушением материала посуды и, следовательно, загрязнениями пробы. Во-вторых, источником ошибок может служить большой (по сравнению с массой пробы) избыток плавня и разлагающих агентов. При этом происходит загрязнение анализируемого материала, а растворы после обработки сплава или спека содержат много солей, которые могут мешать определению компонентов на последующих стадиях анализа.
Сплавление
Сплавление как метод разложения пробы сухим способом чаще используют при анализе неорганических веществ.
При сплавлении тонко измельченный образец перемешивают с 8-10-кратным избытком реагента (плавня) и нагревают (300-1000 °C) до получения прозрачного плава. Время плавления определяют опытным путем. В зависимости от природы разлагаемой пробы оно может варьироваться от нескольких минут до нескольких часов. Сплавление считается законченным, когда масса в тигле становится совершенно однородной, прозрачной и легкоподвижной. После охлаждения застывшую массу растворяют в воде или кислотах. При сплавлении используют щелочные, кислые и окислительные плавни.
В качестве щелочных плавней часто применяют карбонаты, гидроксиды, бораты щелочных металлов и их смеси. При обработке щелочным плавнем металлические компоненты превращаются в растворимые в кислотах оксиды и соли, неметаллические— в растворимые соли щелочных металлов.
Из плавней, обладающих кислыми свойствами, чаще используют пиросульфат калия, гидросульфат калия и ВгО3. При этом в плаве образуются сульфаты и бораты соответствующих металлов.
В качестве окислительных плавней используют щелочные плавни (Na2CO3, Na2B4O7, NaOH и др.) с добавкой окисляющих веществ (KNO3, NaNO3, КС1О3 и др.). Наиболее активный окислительный плавень — пероксид натрия Na2O2 , сплавление с которым проводят только тогда, когда
сплавление с другими плавнями не дает результатов. При этом для предотвращения разрушения тиглей сплавление часто проводят в присутствии гидроксидов или карбонатов щелочных металлов.
Наиболее употребимые плавни и их смеси [2]
Натрия карбонат (безводный). Na2CO3 х. ч. (химически чистый), Tra= 850 °C. Щелочной плавень. Применяют при анализе силикатов, нерастворимых (кислых) шлаков, огнеупоров, глин, нерастворимых в кислотах остатков, при разложении трудноразложимых сульфатов. Сплавление проводят с 6-8-кратным количеством плавня в платиновых, железных и никелевых тиглях.
Натрия гидрокарбонат. NaHCO3 х. ч. При 300 °C разлагается, образуя нейтральный карбонат. Щелочной плавень. Сплавление проводят с 12-14-кратным количеством плавня в платиновых, железных и никелевых тиглях.
Натрия пероксид. Na2O2, Тм = 460 °C. Щелочно-окислительный плавень. Применяют при определении серы, хрома, ванадия, марганца, кремния, фосфора в рудах и ферросплавах, молибдена в молибденовом блеске и др. Сплавление проводят с 6-8-кратным количеством плавня в железных, никелевых и серебряных тиглях.
Натрия (калия) гидрокид (едкий натр, едкое кали). NaOH, Тпл = 328 °C. (КОН, Тпл = 360 °C.) Щелочной плавень. Применяют при определении олова в оловянном камне, при отделении титана от алюминия в присутствии железа и т. д. Сплавление проводят с 8-10-кратным количеством плавня в железных, никелевых и серебряных тиглях.
Калия (натрия) гидросульфат. KHSO4, Тпл = 214 °C. (NaHSO4, Тпл = 185 °C.). Кислый плавень. Применяют при разложении силикатов, вольфрамовых руд, для отделения вольфрамовой и кремниевой кислот, при сплавлении оксидов титана, алюминия, железа, меди и др. Сплавление проводят с 12-14-кратным количеством плавня в платиновых, фарфоровых и кварцевых тиглях.
Калия (натрия) дисульфат. K2S2O7, Тпл = 414 °C. (Na2S2O7, Тпл = 400 °C). Кислый плавень. Применяют в тех же случаях, что и бисульфат калия. Сплавление проводят с 8-12-кратным количеством плавня в платиновых, фарфоровых и кварцевых тиглях.
Дибора(Ш) триоксид (плавленый, мелкоизмельчен-ный). В2О3, Тпл = 577 °C. Получают обезвоживанием борной кислоты при 1000 °C в платиновой чашке. Применяют при разложении силикатов. Сплавление проводят с 5-6-кратным количеством плавня в платиновых тиглях.
Натрия тетраборат (бура). Na2B4O7 (Na2B4O7 • ЮН2О), Тпл = 878 °C. Технический препарат перекристаллизовывают и обезвоживают нагреванием сначала на водяной бане, а затем в муфельной печи при 800-900 °C. Плавень применяют для разложения циркониевых, танталовых, ниобиевых и титановых руд. Сплавление проводят с 8-10-кратным количеством плавня в платиновых тиглях.
Калия гидрофторид. KHF2, Тпл = 239 °C. Кислый плавень; применяют для разложения ниобатов, танталатов и
Информационные базы и общие вопросы аналитической химии
49
циркона. Сплавление проводят с 8-10-кратным количеством плавня в платиновых тиглях.
Натрия тиосульфат, NasS2O3, высушенный при 212 °C. Разлагается при 225 °C, образуя пентасульфид и сульфат натрия. Щелочно-сульфирующий плавень. Сплавление проводят с 8-10-кратным количеством плавня в фарфоровых и кварцевых тиглях.
Смесь 1 части натрия карбоната (безводного) и 1 части калия карбоната (безводного). Na2CO3+ К2СО3 х.ч. Тпл = 500 °C. Щелочной плавень. Сплавление проводят с 6—8-кратным количеством плавня в платиновых, железных и никелевых тиглях.
Смесь 6 частей карбоната натрия (безводного) и 0,5 части калия нитрата.
Щелочно-окислительный плавень. Применяют при определении серы, мышьяка, хрома, ванадия, фосфора в рудах, для отделения титана от ванадия, хрома и др. Сплавление проводят с 8-10-кратным количеством плавня в платиновых, железных и никелевых тиглях.
Смесь Дитмара. Смесь 3 частей калия-натрия карбоната или натрия карбоната (безводного) с 2 частями тетрабората натрия (плавленого, измельченного до порошка). Щелочно-окислительный плавень. Применяют для разложения хромистого железняка, ильменита и др. Сплавление проводят с 10-12-кратным количеством плавня в платиновых, фарфоровых и кварцевых тиглях.
Смесь Ротэ. Смесь 2 частей натрия карбоната (безводного) с 1 частью оксида магния. Щелочно-окислительный плавень (пек). Применяют для разложения ферросплавов и хромистого железняка, при определении хрома, марганца и др. Сплавление проводят с 10-14-кратным количеством плавня в платиновых, железных, фарфоровых и кварцевых тиглях.
Смесь Эшка. Смесь 1 части натрия карбоната (безводного) с 2 частями оксида магния. Щелочно-окислительный плавень (пек). Применяют при определении серы в углях и для разложения ферросплавов. Сплавление проводят с 4-10-кратным количеством плавня в платиновых, железных, никелевых, фарфоровых и кварцевых тиглях.
Смесь 2 частей натрия карбоната (безводного) с 1 частью оксида магния. Щелочно-окислительный плавень (пек). Применяют при определении серы в рудах, преимущественно сульфидных. Сплавление проводят с 8-10-кратным количеством плавня в фарфоровых и кварцевых тиглях.
Смесь 4 частей калия-натрия карбоната с 1 частью калия тартрата. Щелочно-восстановительный плавень. Сплавление проводят с 8-10-кратным количеством плавня в платиновых и кварцевых тиглях.
Смесь фрейбергская. Смесь 3 частей натрия карбоната (безводного) с 4 частями кристаллической мелкоизмель-ченной серы (или 5 частей К2СО3 с 3 частями серы). Ще-лочно-сульфидирующий плавень. Применяют при отделении молибдена, сурьмы, мышьяка и олова от свинца, меди, серебра и др.; для разложения продуктов обжига руд цветных металлов (штейнов, шпейзов и др.); при разделении
титана и ванадия. Сплавление проводят с 8-10-кратным количеством плавня в фарфоровых и кварцевых тиглях.
Смесь Лоу. Смесь 5 частей натрия пероксида с 1 частью натрия карбоната. Щелочно-окислительный плавень. Сплавление проводят с 6-8-кратным количеством плавня в железных, никелевых и серебряных тиглях.
Смесь 6 частей натрия (калия) гидроксида и 0,5 части натрия (калия) нитрата. Щелочно-окислительный плавень; применяют вместо пероксида натрия. Сплавление проводят с 4-6-кратным количеством плавня в железных, никелевых и серебряных тиглях.
Смесь 2 частей натрия карбоната (безводного) с 1 частью буры. Плавень для корунда. Сплавление проводят с 8-10-кратным количеством плавня в платиновых тиглях.
Смесь Смита. Смесь карбоната кальция СаСО3 х.ч., свободного от щелочей, и хлорида аммония NH4CI х.ч. (8:1). Применяют при определении щелочных металлов по методу Смита. Спекание проводят с 8-10-кратным количеством смеси в специальном пальцевидном платиновом тигле.
Спекание
Спекание— это взаимодействие веществ при повышенной температуре в твердой фазе. Прокаливание пробы с подходящим твердым реагентом не всегда сопровождается образованием расплава; в отдельных случаях смесь не расплавляется, а только спекается. Спекание — сложный, полностью не изученный процесс. Предполагается, что спекание основано на высоком химическом сродстве компонентов пробы к введенным реагентам, на диффузии и реакциях обмена.
В отдельных случаях спекание позволяет провести разложение пробы быстрее и проще, способствует уменьшению загрязнений, поскольку при этом часто используют меньший (двух- или четырехкратный) избыток реагентов и менее высокие температуры.
Спекание проводят обычно со смесью карбонатов щелочных металлов и оксидов магния, кальция или цинка. Рекомендуется использовать спекание при разложении проб силикатов, сульфидов, оксидов металлов.
Так, при разложении силикатов с целью определения в них щелочных металлов проводят спекание пробы со смесью СаСО3 и NH4CI в соотношении 8:1 (метод Лоуренса -Смита). Сначала при слабом нагревании хлорид аммония сублимируется и диссоциирует с образованием аммиака и хлорида водорода, последний взаимодействует с СаСО3 с образованием мелких частиц СаС12.
При повышении температуры до 1000-1100 °C медленно выделяется СО2, а смесь СаО и СаС12 взаимодействует с пробой, образуя силикат кальция и хлориды щелочных металлов. Спек обрабатывают горячей водой.
Пиролиз
Пиролизом называется процесс термического разложения в отсутствие веществ, реагирующих с разлагаемым соединением.
50
Новый справочник химика и технолога
При пиролизе органических веществ характеристические фрагменты органических соединений появляются главным образом в интервале 300-700 °C. При более высоких температурах увеличивается степень образования простых веществ, таких как СИ», СО, СО2, Н2О. Неорганические вещества разлагаются, как правило, при температурах 1000-1500 °C.
Скорость нагрева должна быть большой: при медленном повышении температуры образовавшиеся продукты разложения могут вступать в реакции. Особенно часто это наблюдается в присутствии кислорода, поэтому пиролиз желательно проводить в атмосфере инертного газа (азот, гелий) или в вакууме.
Пиролиз проводят различными способами: прокаливают пробу в тигле или небольшой лодочке в печи; наносят образец на металлическую проволоку или спираль и нагревают их до нужной температуры; помещают вещество в вакуумированную или заполненную инертным газом стеклянную или кварцевую трубку и также нагревают ее до необходимой температуры. Помимо указанных (часто используемых) способов применяют термическое разложение при облучении лазером, потоком электронов высокой энергии, нагревание смеси пробы с ферромагнитным материалом (например, с порошком железа) в высокочастотном электрическом поле и т. д.
Пиролиз чаще используют при анализе органических веществ, особенно полимеров. Впервые термическое разложение этого типа было применено при анализе каучуков. Для разложения неорганических соединений пиролиз используют значительно реже, например при разложении сульфатов с выделением О2 и SO2 (1350 °C); стекла (1650 °C); оксидов алюминия и редкоземельных элементов (1000 °С)ит. д.
Газообразные продукты пиролиза поглощают твердыми сорбентами или соответствующими химическими реагентами и затем определяют различными аналитическими методами. Продукты пиролиза чаще анализируют методами газовой хроматографии, УФ- и ИК-спектроскопии, масс-спектрометрии.
Низкотемпературное озоление кислородной плазмой
Высокоэффективным способом окислительной минерализации является разложение образцов с помощью возбужденного кислорода (низкотемпературной кислородной плазмы), который получают, пропуская газообразный кислород под давлением 133-665 Па через высокочастотное электрическое поле [7].
Навеску пробы (0,1-0,2 г) помещают в кварцевую чашечку и при температуре 150-180 °C обрабатывают кислородной плазмой, полученной с помощью высокочастотного генератора (13,5 МГц, 1 кВ, 300 W), направленной перпендикулярно к пробе. При такой минерализации органическая часть и вода отгоняются (их можно анализировать отдельно), а многие элементы (Ag, As, Со, Сг и др.) образуют оксиды, которые растворяют в кислотах и определяют различными методами. Некоторые примеры низкотемпературного озоления различ
ных материалов приведены в [7]. В работе [9] этот способ успешно использовался для определения Zn, Cd, Pb и Си методом дифференциальной ИВ (инверсионной вольтамперометрии) в стандартном образце «листьев оливы» наряду с методом мокрого озоления в смеси хлорной и азотной кислот. Для активации кислорода можно использовать микроволновое электрическое поле. Достоинствами этого метода являются отсутствие опасности загрязнения пробы материалом сосуда или реагентами (единственным реагентом является кислород), а также селективность (отделение органической части от неорганической), что важно при анализе почв, медико-биологических образцов, объектов животного и растительного происхождения.
Микроволновое разложение
Этому способу подготовки пробы посвящена книга Кингстона и Джесси [10], обзор [11]. Здесь источником тепла для мокрой минерализации веществ является энергия микроволнового (МВ) излучения. МВ — это вид электромагнитного излучения (в диапазоне частот 300-30000 МГц), вызывающий движение ионов и вращение диполей и приводящий к быстрому разогреву всего объема образца, поглощающего MB-энергию. Например, при частоте 2450 МГц ориентация молекул и их возвращение в беспорядочное состояние происходит 5109 раз в секунду. В отличие от традиционного нагрева на плитке или печи, где тепло передается путем молекулярных столкновений через поверхность нагреваемого сосуда, MB-излучение нагревает весь объем жидкости, не нагревая сосуда, проницаемого для МВ (фторопласт, кварц). В результате вместо 1-2 ч для полного разложения проб кислотой требуется 10-15 мин, а Ткип достигается за 2 мин.
Современные способы измерения температуры и давления непосредственно в MB-печи позволили сделать интересные выводы об условиях разложения основных компонентов пищевых продуктов азотной кислотой под давлением. Температуры разложения основных компонентов: углеводы — 140 °C, белки — 150 °C, жиры — 160 °C. Достаточно 10 мин для полного разложения азотной кислотой всех компонентов пищевых продуктов, кроме ароматических нитросоединений. Однако эти соединения могут влиять на определение других компонентов (например, тяжелых металлов вольтамперометрическим методом) и требуют дальнейшего разложения [5].
Использование MB-печей позволяет автоматизировать процесс подготовки пробы и значительно ускорить весь анализ. При разложении различных проб в микроволновом поле в большинстве случаев используют смесь (HNO3 + Н2О2). Некоторые исследователи используют МВ-энергию для ускорения извлечения тяжелых металлов из илов и торфов.
Использование ультразвука в подготовке пробы
Звуковые колебания с частотой, превышающей частоту, воспринимаемую человеческим ухом (т. е. выше 16 кГц), называются ультразвуком (УЗ). Ультразвук можно получить в пьезокристалле за счет превращения электрической
Информационные базы и общие вопросы аналитической химии
51
энергии в колебательную. Различают высокочастотный УЗ (2-10 кГц), не влияющий на химические реакции, и низкочастотный УЗ (20-100 кГц), вызывающий эффект кавитации — образование и схлопывание пузырьков в жидкости. Энергия, выделяемая при этом, приводит к ускорению химических реакций. Эти явления изучает сонохимия [12]. При распространении волны разрежения низкочастотного УЗ в жидкой среде (20 тыс. колебаний в с), развивается отрицательное давление, достаточное для преодоления межмолекулярных сил сцепления. Молекулы отделяются друг от друга, образуя в среде крошечные пустоты (микропузырьки). В цикле сжатия микропузырьки схлопываются с выделением большого количества энергии. При 25 °C под давлением мощного УЗ схлопывание микропузырьков может привести к повышению температуры (до 5000 К) и давления (до 1000 атм).
Влияние УЗ на химические реакции проявляется через повышение температуры, концентрации реагентов, увеличение давления. Кроме этого под влиянием УЗ в кавитационном пузырьке могут образовываться радикалы, изменяться сольватация, разрываться водородные связи и полимерные цепи. При УЗ-обработке гетерогенной системы (твердое тело - жидкость, жидкость - жидкость) происходит дробление частиц, увеличение поверхности перемешивания, образование эмульсий с большой поверхностью контакта. УЗ в подготовке проб пищевых продуктов и объектов окружающей среды применяется для перемешивания и измельчения материалов, получения вытяжек из почв, аэрозольных фильтров, генерации реакционноспособных радикалов, очистке поверхностей посуды и электродов. В электрохимических системах применение УЗ облегчает транспорт ионов (подобно перемешиванию), удаляет пузырьки газа с поверхности, активирует электрод, улучшает качество металлических покрытий, влияет на скорость электрохимических реакций.
В [5] применение УЗ в подготовке пробы продемонстрировано на примере определения микроэлементов в комбикормах. Навеску образца (1-2 г) смачивали небольшим количеством концентрированных неорганических кислот, добавляли дистиллированную воду, а затем пробу подвергали УЗ-воздействию в течение 2 мин с погружением излучателя в раствор. После фильтрации обработанную пробу и вытяжку можно анализировать любым аналитическим методом на содержание Си, Zn, Мп, Fe, Со.
Фотохимическая подготовка пробы
Ультрафиолетовое облучение широко используется при определении органических веществ, углерода, азота и фосфора, присутствующих в водах [13, 14]. При облучении растворов, содержащих органические вещества, светом ртутной лампы с длинами волн короче 400 нм протекают первичные или вторичные фотореакции. Как известно, энергия кванта света обратно пропорциональна длине волны и при 200-300 нм составляет 600-400 кДж-моль1, в то время как энергия химических связей лежит в пределах 250-500 кДж-моль'1. Количество распавшихся под дейст
вием УФ молекул пропорционально квантовому выходу, интенсивности и продолжительности облучения [13].
К первичным фотореакциям относятся реакции фотодиссоциации, фотоактивации, внутримолекулярного фотоокисления и восстановления и другие приводящие к образованию высокоактивных частиц с неспаренным электроном — радикалов. В присутствии в водах кислорода, примесей металлов, взвесей и т. д. главную роль начинают играть вторичные процессы, например фотоокисление органических веществ.
Вторичные процессы существенно зависят от температуры, перемешивания и концентрации катализатора или окислителя. В качестве эффективных окислителей выступают озон, кислород, пероксид водорода, персульфаты и др., в присутствии которых подавляющее количество органических веществ может быть разложено до углекислого газа и воды. Современные исследования показали, что в реакциях фотоокисления с участием кислорода, перокида водорода и озона образуются высокоактивные ОН-радикалы, окислительно-восстановительный потенциал которых близок к паре F2-F“ и которые с большой скоростью (порядка 107 - IО10 Л'моль1-с ') взаимодействуют с органическими молекулами, приводя к их распаду. Согласно стандарту DIN 38406 Е16 (Германия), при определении Zn, Cd, Pb, Си, Ni, Со в водах используется окислительный фотолиз.
За последние годы увеличилось применение УФ в подготовке проб биологических и пищевых продуктов. Особое место занимает УФ-минерализация органических веществ в катодной адсорбционной вольтамперометрии, где ОВ могут препятствовать адсорбции металлокомплексов на поверхности электрода или вступать в конкурирующие реакции. При анализе летучих соединений, таких как ртуть, УФ-подго-товка пробы занимает исключительное положение.
Особенности подготовки пробы в некоторых видах анализа
В ряде случаев сам ход аналитического определения может включать элементы подготовки пробы. Например, в ходе инверсионно-вольтамперометрического определения тяжелых металлов для сокращения общей длительности анализа используют электрохимическую обработку проб [5].
Электрохимический метод подготовки пробы основан на окислении органических веществ в присутствии обычно хлор-ионов путем прямого анодного окисления органических веществ или косвенного — через реакции с частицами генерированных окислителей (вероятно, радикального характера). При определении тяжелых металлов в присутствии ПАВов пробу помещают в стеклоуглеродный тигель, являющийся анодом. Наилучший эффект разрушения ОВ получают при потенциале анода от + 1,4 до + 1,5 В, концентрации хлор-ионов 0,75 - 1,0 М и времени 10-20 мин. Избыток окислителя (С1) восстанавливают при меняющемся потенциале от -1,4 до +0,2 В с периодичностью 15 с. В ИВ-анализаторе ИВА-ЗА осуществляется совместная электрохимическая подготовка пробы и ИВ-определение металлов, например определение свинца и меди в крови.
52
Новый справочник химика и технолога
Известно применение мембранной обработки с наложением переменного тока для очистки вод от ПАВов и определения ряда элементов. Преимуществом этого метода является минимальное загрязнение проб из-за отсутствия окисляющих реактивов и возможность совмещения подготовки пробы с определением тяжелых металлов.
Данный метод эффективен при обработке проб, содержащих органические вещества в малых количествах, например в природных водах [15].
Особенности разложения проб при определении платиновых металлов в углеродистых породах рассмотрены в работе [16]. При этом авторы учитывают опыт подготовки проб методами кислотного разложения, автоклавного разложения, пробирной плавки, предлагая способ разложения смесью H2SO4 и СгО3.
Радиохимический анализ природных и синтетических объектов в цепочке основных химических операций (помимо типичного мокрого озоления) включает концентрирование радионуклидов соосаждением на коллекторах, сорбцией на ионитах и комплексообразующих сорбентах или выпариванием [17].
Литература
1. Основы аналитической химии: В 2 кн. 2-е изд., пере-работ. и доп. / Под ред. акад. Золотова Ю.А. М.: Высш, шк., 2000. Кн. 1. Общие вопросы. Методы разделения. 351 с.
2. Коростелев П.П. Лабораторная техника химического анализа / Под ред. д.х.н. Бусева А.И. М.: Химия, 1981. 312 с.: илл.
3. Краткая химическая энциклопедия. Том 3 (М-П). М.: Советская энциклопедия, 1964. С. 748-757 (опробование материалов).
4. Ушакова Н.Н., Николаева Е.Р., Моросанова С.А. Пособие по аналитической химии. М.: МГУ, 1978. 224 с.
5. Современные методы пробоподготовки пищевых продуктов. / Сост. Захарова Э.А. Томск: ТПУ, ВНПФ ЮМХ, 1997. 42 с.
6. ГОСТ 26929-94. Сырье и продукты пищевые. Подготовка проб. Минерализация для определения токсичных элементов.
7. Бок Р. Методы разложения в аналитической химии. М.: Химия, 1984. 316 с.
8. Gorsuch Т.Т. The Destruction of Organic Matter. Perga-mon Press, 1970.
9. Rosali bin Othman, Hill J.O., Magee R.J. // Fr. Z. Anal.
Chem. 1987. V. 326. P. 350.
10. Пробоподготовка в микроволновых печах / Ред. Кингстон Г.М., Джесси Л.Б. М.: Мир, 1991.
11. Кузьмин Н.М., Кубракова И.В. Микроволновая пробоподготовка // Журн. аналит. химии. 1996. Т. 51, № 1.С. 44-49.
12. Маргулис М.А. Основы звукохимии. М.: Высшая школа, 1984.
13. Немодрук А.А., Безрогова Е.В. Фотохимические реакции в аналитической химии. М.: Химия, 1972.
14. Шелковников В.В. Интенсификация пробоподготовки природных объектов под воздействием СВЧ-, УФ- и УЗ-полей с целью определения тяжелых металлов методом ИВА. Автореф. дисс. к. х. н. Томск, 1994.
15. Свинцова Л.Д., Чернышева Н.Н. Электрохимическая пробоподготовка при инверсионно-вольтамперометрическом определении токсичных металлов в природных водах. Влияние активной среды на выделение кадмия, свинца и меди из комплексных соединений с гуминовыми и фульвокислотами // Журн. аналит. химии. 1993. Т. 48, № 9. С. 1450-1457.
16. Бельский Н.К., Небольсина Л.А., Оксеноид К.Г. Разложение проб при определении платиновых металлов в углеродистых породах // Журн. аналит. химии. 1997. Т. 52, №2. С. 150-153.
17. Павлоцкая Ф.И. Основные принципы радиохимического анализа объектов природной среды и методы определения радионуклидов стронция и трансурановых элементов // Журн. аналит. химии. 1997. Т. 52, №2. С. 126-143.
18. Nomenclature for Sampling in Analitical Chemistry (IUPAC Recommendation 1990) // Pure and Applied Chemistry. 1990. Vol. 62, № 6. P. 1193-1208.
19. Smith R. and James G.V. The Sampling of Bulk Materials. RSC, London, 1981.
20. Gy P. M. Sampling of Heterogeneous and Dynamic Materials Systems. Theeries of Heterogeneity, Sampling and Homogenizing. Elsevier, Amsterdam. 1992.
21. Sulcek Z. and Povondra P. Methods of Decomposition of Inorganic Analysis. CRC Press, Boca Raton, FL, 1989.
22. Kingston H.M. and Jassie L.B. (Eds.). Introduction to Microwave Sample Preparation. ACS, Washington, DC, 1988.
23. Wilson a. Wilson's Comprehensive Analytical Chemistry. Vol. 30. Spectrochemical Trace Analysis for metals and metalloids. By L. Lobinski and Z. Marczenko. Elsevier., Amsterdam etc. 1997. 808 p.
Раздел 2
МЕТРОЛОГИЧЕСКИЕ ОСНОВЫ МЕТОДОВ
КОЛИЧЕСТВЕННОГО АНАЛИЗА
Автор-составитель: проф., д. х. н. И.П. Калинкин
2.1. Количество вещества, эквивалентность, способы выражения концентраций
Химику-аналитику практически повседневно приходится иметь дело с расчетами в химических методах анализа. В связи с введением в действие системы СИ, в которой основной единицей массы является килограмм, а основной единицей количества вещества — моль, использование таких единиц массы, как грамм-атом, грамм-моль, грамм-эквивалент, грамм-ион и соответствующих им единиц концентрации (например, грамм-эквивалент/литр) не допускается.
Научный совет АН СССР по аналитической химии уже в 1982 г. [1] счел необходимым (подробно см. компакт-диск)
дать разъяснения по вопросу дальнейшего использования терминов эквивалент и нормальность. Затем этим вопросам были посвящены публикации [2, 3]. Изменения в основных терминах, определениях, единицах измерения в связи с введением международной системы СИ отражены практически во всей отечественной учебной литературе [4-11] и в ведущих зарубежных учебниках, например [12-14].
В основу нижеприведенной справочной таблицы 2.1 положены термины, определения и единицы измерения, представленные в [16]. Она дополнена расчетными формулами, которые необходимы для конкретных количественных расчетов.
Таблица 2.1
Основные термины, определения, единицы измерения и расчетные формулы, связанные с количеством вещества, эквивалентностью и выражением концентраций
Пояснения.* Принятые обозначения: О (СИ) — основная (СИ); Д (СИ) — дольная (СИ); К (СИ) — кратная (СИ); П (СИ) — производная (СИ); Д п (СИ) — дольная, производная (СИ); П — производная (внесистемная); Д — дольная, производная (внесистемная).
Миллионные, миллиардные, триллионные доли [4, 12]. Для очень разбавленных растворов и для очень низких содержаний элементов удобно выражать долю компонента от общего количества вещества в частях на миллион (млн4; ppm).
Термин Единица измерения Тип единицы Определение Форма записи Примеры Примечание
Количество вещества моль (моль) О (СИ) миллимоль (ммоль) Д(СИ) микромоль (мкмоль) Д(СИ) киломоль (кмоль) К (СИ) Моль — количество вещества X, содержащее столько реальных или условных частиц, сколько атомов содержится в 0,012 кг углерода-12 (12С). При использовании моля как единицы количества вещества следует четко указать, какие именно реальные или условные частицы имеются в виду «(X) n (НС1) = 2 моль п (Са2+) = 3 ммоль n (F) = 6 мкмоль n (H2SO4) = 0,5 моль л(Х)=^— (2.1) А/(Х) n (X) - с (X) V (2.2) Под реальными частицами следует понимать атомы, ионы, молекулы, радикалы, электроны и т. п., а под условными частицами — такие, как, например, 1/5 молекулы КМпО4 в случае окислительно-восстановительной реакции в кислой среде (см. термин «эквивалент»). Слово моль после числа и в заголовках таблиц не склоняют
54
Новый справочник химика и технолога
Продолжение таблицы 2.1
Термин Единица измерения Тип единицы* Определение Форма записи Примеры Примечание
Молярная масса кг • моль-1 П(СИ) г • моль-1 Д,п(СИ) Молярная масса Л/(Х) — масса одного моля вещества. Молярную массу находят как отношение массы т вещества к его количеству в молях /И M(X)%(X) <2'3) МН&С12) = 472 г- моль-1 Л/(Са2+) = 40,08 г моль-1 Л/(Н)--0.00108 кг-моль1 При использовании термина молярная масса следует указывать вид частицы, молярная масса которой определяется. Например, молярная масса молекулы хлорида калия, молярная масса иона кальция и т. п.
Относительная молекулярная масса Безразмерная Относительная молекулярная масса Л/ДХ) — молярная масса молекулы вещества X, отнесенная к 1/12 молярной массы атома углерода-12 (12С) М,(Х) Mr (НС1) = 36,52 Мг (Вг2) = 159,82 Термин относительная молекулярная масса введен вместо терминов «молекулярный вес» и «молекулярная масса». Значение Mr (X) численно равно прежнему грамм-молю вещества X
Относительная атомная масса Безразмерная Относительная атомная масса Лг (X) — молярная масса атома вещества X, отнесенная к 1/12 молярной массы атома углеро-да-12 (,2С) Л,(Х) Л (Вг)-79,01 Л(ИЬ) = 92,91 Термин относительная атомная масса введен вместо терминов «атомный вес» и «атомная масса». Значение Ar (X) численно равно прежнему грамм-атому вещества X
Фактор эквивалентности Безразмерная Число, обозначающее, какая доля реальной частицы вещества X эквивалентна одному иону водорода в данной кислотно-основной реакции или одному электрону в данной окислительно-восстановительной реакции. Фактор эквивалентности может быть равен или меньше единицы /ЭКВ(Х) 7эКВ(КМпО4) =’/5 (кислая среда) /э„(Си2*) = ‘/2 (в электрогравиметрии) Фактор эквивалентности рассчитывают на основании стехиометрии данной реакции. Реакция обязательно должна быть указана. В общем случае реакцию аА + ЬВ —> продукты (2.4) можно записать в виде ь А+— -> продукты (2.4'), а где а > Ь; множитель Ь/а называют фактором эквивалентности вещества В и обозначают/^ (В) (примеры см. в табл. 2.2). Число в знаменателе дроби, равное числу принимаемых (отдаваемых) протонов или электронов в ряде монографий (учебников) называют числом эквивалентности — Z
Метрологические основы методов количественного анализа
55
Продолжение таблицы 2.1
Термин Единица измерения Тип единицы Определение Форма записи Примеры Примечание
Эквивалент Безразмерная Реальная или условная частица вещества X, которая в данной кислотно-основной реакции эквивалентна одному иону водорода или в данной окислительно-восстановительной реакции одному электрону /экв(Х)Х '/5 КМпО4 (кислая среда) V2 Cu2+ (в электрогравиметрии) Понятие эквивалент распространяется также на реакции ионного обмена, реакции в электроаналити-ческих методах и др. В уравнении (2.4) эквивалент вещества В обозначается как^экв (В)В. Эквивалент вещества непостоянен. Эквивалент одного и того же вещества зависит от типа химической реакции и конкретной реакции. Примеры см. табл. 2.2.
Молярная масса эквивалента Кг • моль-1 П(СИ) г • моль-1 Д,п(СИ) Молярная масса эквивалента вещества X — масса одного моля эквивалента этого вещества, равная произведению фактора эквивалентности на молярную массу вещества X М(/;кв(Х)Х) = =/экв(Х)-М(Х) (2.5) A/C/2H2C2O4) = = /2 Л/(Н2С2О4) = = 45,02 г/моль Молярная масса эквивалента численно равна прежнему грамм-эквиваленту вещества X
Число моль-эк-вивален-тов моль О (СИ) ммоль Д(СИ) мкмоль Д(СИ) кмоль К (СИ) Количество вещества (в молях), в котором частицами являются эквиваленты (см. термин «эквивалент») «(АЭКВ(Х)Х) н('/2Са2+) = 0,5 моль (при титровании Са(ОН)2 раствором НС1) п (1/5КМпО4) = 0,1 моль (реакция с FeSO4 в кислой среде) и(Акв(Х)Х) = = _н(Х)_ = AJX) «(АЭКВ(Х)Х) = = (2-7) W,KB(X)X) и«экв(Х)Х) = = с«экв(Х)Х)-Г(Х) (2.8)
Способы выражения концентраций
Молярная концентрация моль • м-3 П(СИ) моль • дм-3 Д, п (СИ) -3 моль • см Д» п (СИ) моль • л-1 П ммоль • мл1 Д,п Молярная концентрация с(Х) — отношение количества вещества X (в молях), содержащегося в системе (например, в растворе), к объему 1'(Х) этой системы с(Х)=-^-^ = Г(Х) = (2.9) К(Х)-М(Х) с (Н ) = Ю“8 моль/л с (КМпО4) = = 0,1 моль/дм3 с (H2SO4) = 0,2 М Термин молярность не рекомендуется. Термин молярный сохраняется. Допускаемые формы записи, например, для раствора с концентрацией хлороводородной кислоты 0,1 моль - л-1: с(НС1) = = 0,1 моль - л-1 или: 0,1 М; 0,1МНС1; 0,1 М раствор НС1 (децимоляр-ный раствор НС1)
56
Новый справочник химика и технолога
Продолжение таблицы 2.1
Термин Единица измерения Тип единицы Определение Форма записи Примеры Примечание
Молярная концентрация эквивалента моль • дм-3 П(СИ) моль • см-3 Д, п (СИ) моль • л 1 П ммоль • мл1 п моль • м-3 д Молярная концентрация эквивалента — отношение числа моль эквивалентов (см. предыдущий термин) в системе (например, в растворе) к объему V этой системы с(Аэкв(Х)Х) = _»(/эи(Х)Х)_ Г(Х) Г(Х).Л/(/эга(Х)Х) (2.10) <Э1ев(Х)Х) = = =zc(X) /ЭКВ(Х) (2.П) Согласно рекомендации ИЮПАК раствор, содержащий 1 моль эквивалентов вещества X в 1 дм3 или 1 л, можно называть нормальным раствором этого вещества. Вместо обозначения единиц измерения моль • дм-3 или моль • л’1 допускается сокращение н. с указанием в скобках фактора эквивалентности. При указании нормальной концентрации обязательно следует указывать конкретную реакцию, в которой данный нормальный раствор применяется. Термин нормальность не рекомендуется. Допускаемые формы записи, например, для одномолярной концентрации эквивалента серной кислоты (в кислотноосновной реакции): с C/2 H2SO4) = 1 моль • л-1 или 1 М; или 1 М (72 H2SO4); или 1 н. H2SO4 (4кв = ’/2); или 1 н. раствор H2SO4 (f3KB = ’/2). Для концентрации раствора КМпО4, содержащего 0,05 моль эквивалентов в 1 л раствора (в окислительно-восстановительной реакции в кислой среде): с (% КМпО4) = 0,05 М или моль • л-1; или 0,05 М (’/5 КМпО4); или 0,05 H.KMnO4(f9KB = 75); или 0,05 н. раствор КМпО4 (4. = '/,)
Моляльность моль • кг-1 П(СИ) Моляльность b (X) раствора — отношение количества растворенного вещества X (в молях) к массе т растворителя Y ь (X) = -^29- zn(Y) b (ацетон/вода) = = 0,02 моль • кг-1 6(НС1/Н2О) = = 0,1 моль • кг-1 Термином моляльность предпочтительнее пользоваться в случае реакций, протекающих в неизотермических условиях
Метрологические основы методов количественного анализа
57
Продолжение таблицы 2.1
Термин Единица измерения Тип единицы Определение Форма записи Примеры Примечание
Массовая доля Относительная Отношение массы данного компонента, содержащегося в системе, к общей массе этой системы ®(Х)=-^2- (2.12) ^навески (в гравиметрии, титри-метрии) Допускается выражать массовую долю в долях единицы, процентах (%), промилле (тысячная часть %) и в миллионных долях (млн1)**
Молярная доля Относительная Отношение количества вещества (в молях) компонента, содержащегося в данной системе, к общему количеству вещества системы (в молях) П, а = —— Хп См. примечание к термину массовая доля
Объемная доля Относительная Отношение объема компонента, содержащегося в системе (растворе), к общему объему системы V. ф = —— V IF То же
Массовая концентрация кг • м3 П(СИ) кг • дм3 Д,п(СИ) кг • см 3 Тоже г • дм3 То же г • см 3 То же г • л-1 П г • мл-1 д Отношение массы компонента, содержащегося в системе (растворе), к объему этой системы (раствора) Для растворов р(Х)=^2 (2.13) 7 F(X) C(X)=-P2L (2.14) Л/(Х) </,и(Х)Х)= Р(Х) (2.15) W3KB(X)X) Выражать концентрацию (молярную и массовую) в процентах не рекомендуется
Плотность раствора г • см3 Д,п(СИ) кг • м3 Д, п (СИ) Отношение массы раствора к его объему Р =р (2.16) Связь между плотностью раствора, массовой долей и молярной концентрацией выражается формулами: c(X) = ffl(X)‘p'10 , (2.17) Л/(Х) ✓ , -у'У'- со(Х)-р10 С(/экв ( ) ) (Y1Y1 W3Kb(X)X) (2.18)
58
Новый справочник химика и технолога
Продолжение таблицы 2.1
Термин Единица измерения Тип единицы Определение Форма записи Примеры Примечание
Титр раствора кг • см-3 Д, п (СИ) г* см-3 Дп(СИ) г • мл 1 д Масса вещества X, содержащегося в одном кубическом сантиметре или одном миллилитре раствора Г(Х)=^> (2.19) Г(Х) 7 на практике обычно выражается в г • мл1 Связь между титром раствора, молярной концентрацией и молярной концентрацией эквивалента выражается формулами: Т(Х)-103 с(Х) (2.20) Л/(Х) с(/„(Х)Х)= (221) ЖЭКВ(Х)Х)’ } где 7(Х) выражен в г • мл-1
Титр раствора по определяемому компоненту (веществу) кг • см-3 Д,п(СИ) г • см3 Дп(СИ) г • мл 1 д Масса определяемого компонента X, эквивалентная массе титранта Y, содержащегося в 1 см3 или в (1 мл р-ра) или отношение массы определяемого компонента (вещества) т(Х) к эквивалентному объему l'(Y) стандартного раствора TpQ = w(X) (2.22) C(Y) на практике выражается в г мл-1 <Y^ Т\ — показывает, какая <х; масса определяемого компонента (вещества) X реагирует с 1 мл стандартного раствора реагента Y. Связь между титром по определяемому компоненту (веществу) и молярной концентрацией стандартного раствора реагента выражается формулами: (YA а Т — МО3 C(Y)--=_._k2£2— . а М(Х) (2.23) где а и г — стехиометрические коэффициенты веществ X и Y в уравнении реакции: аХ + rY-+ продукты; т(—}103 - — (2.24) W3KB(X)X)
_, _к масса растворенного компонента , л6
С (млн )=--------------------------------10
масса раствора
масса компонента 6 масса образца
(2.25)
Плотность разбавленных растворов близка к 1,00 г • мл1. В этом случае: 1 млн-1 (1 ppm) = 1 мкг мл-1 (Ipg/ml) = = 1 мг • л-1; 1 млн1 (1 ppm) = 1 • 10“4% (масс.)
Для еще более разбавленных растворов или более низких содержаний компонента результаты чаще представляют числом частей на миллиард:
Метрологические основы методов количественного анализа
59
масса растворенного компонента 1л9
С (рро)--------------------------------10 =
масса раствора
_ масса компонента -
масса образца
Тогда: 1 ppb = 1 нг • мл-1 (lng/ml) = 1 мкг • л-1 (1 p.g/1);
1 ppb = 1 • Ю7%.
При еще меньших содержаниях компонента оперируют триллионными долями: 1 ppt (одна часть на триллион частей) = 1 • 1012%.
Когда говорят о газах, то долю, например млн-1, относят не к массе, а к объему. Так 12 млн1 (12 ppm) СО2 в воздухе означают 12 микролитров (12 ц1) СО2 на 1 л воздуха.
Таблица 2.2
Типичные титриметрические реакции и соответствующие значения факторов эквивалентности и эквивалентов*
Аналитическая реакция Фактор эквивалентности для первого вещества: /экв (X) Эквивалент, /экв (X) X
1. НС1 + NaOH = NaCl + Н2О /ЭКВ(НС1)=1 НС1; NaOH**
2. Na2CO3 + НС1 = NaHCO3 + NaCl /кв (Na2CO3)=l Na2CO3; НС1
3. Na2CO3 + 2HC1 = 2NaCl + H2CO3 /кв (Na2CO3)=l/2 1/2 Na2CO3; HC1
4. H2SO4 + 2NaOH = Na2SO4 + 2H2O /кв (H2SO4) = 1/2 1/2 H2SO4; NaOH
5. H3PO4 + KOH = KH2PO4 + H2O /ЭКВ(Н3РО4)=1 H3PO4; KOH
6. H3PO4 + 2KOH = K2HPO4 + 2H2O /экв (Н3РО4) = 1/2 1/2 H3PO4; KOH
7. H3PO4 + 3KOH = K3PO4 + 3H2O /кв (Н3РО4) = 1/3 1/3 H3PO4; KOH
8. H2C2O4 + 2KOH = K2C2O4 + 2H2O /кв (Н2С2О4) = 1/2 1/2 H2C2O4; KOH
9. Ba(OH)2 + 2HC1 = BaCl2 + 2H2O /кв (Ва(ОН)2) = 1/2 1/2 Ba(OH)2; HC1
10. FeCl3 + Na2H2Y*** NaFeY + 2HC1 + NaCl /KB(FeCl3)=l FeCl3; Na2H2Y
11. 2KMnO4 + 5H2C2O4 + 3H2SO4 = K2SO4+ 2MnSO4 + 10CO2 + 8H2O /экв(КМпО4)=1/5 1/5 KMnO4; 1/2 H2C2O4
12. 2KMnO4 + 10FeSO4 + 8H2SO4 = K2SO4 + 2MnSO4 + 5Fe2(SO4)3 + 8H2O /кв (КМпО4) = 1/5 1/5 KMnO4; FeSO4
13. 2KMnO4 + 5H2O2 + 3H2SO4 = K2SO4 + 2MnSO4 + 8H2O + SO2 /кв (КМпО4)=1/5 1/5 KMnO4; 1/2 H2O2
14. K2Cr2O7 + 6KI + 14HC1 = 2CrCl3 + 3I2 + 8KC1 + 7H2O /кв (К2Сг2О7) = 1/6 1/6 K2Cr2O7; У212
15. I2 + 2Na2S2O3 = Na2S4O6 + 2NaI /kb(I2) = 1/2 1/212; Na2S2O3
16. K2Cr2O7 + 6FeCl2 + 14HC1 = 2CrCl3 + FeCl3 + 2KC1 + 7H2O /кв (К2Сг2О7) = 1/6 1/6 K2Cr2O7; FeCl3
17. SnCl2 + Fe2(SO4)3 + 2HC1 = SnCl4 + 2FeSO4 + H2SO4 /кв (SnCl2) = 1/2 1/2 SnCl2; 1/2 Fe2(SO4)3
Примечания. * На основе табл. [1а].** Когда коэффициент равен 1, то записывают только формулу соединения. *** Комплексон III (ЭДТА) Y — анион ЭДТА.
2.2. Аналитический сигнал. Методы измерения [4]
После отбора и подготовки пробы наступает стадия химического анализа, на которой и проводят обнаружение компонента или определение его количества. С этой целью измеряют аналитический сигнал. В отдельных случаях возможно непосредственное определение содержания. Так, например, в гравиметрическом методе иногда прямо измеряют массу определяемого компонента, например элементарной серы или углерода. В большинстве же методов аналитическим сигналом служит среднее из измерений физической величины, функционально связанной с содержанием определяемого компонента. Это может быть сила тока, ЭДС системы, оптическая плотность, интенсивность излучения и т. д.
В случае необходимости обнаружения какого-либо компонента обычно фиксируют появление аналитического сигнала — появление осадка, окраски, линии в спектре и т. д. Появление аналитического сигнала должно быть надежно зафиксировано. При определении количества компонента измеряется величина аналитического сигнала: масса осадка, сила тока, интенсивность линии спектра и т. д. Затем рассчитывают содержание компонента с использованием функциональной зависимости аналитический сигнал-содержание: у = //), которая устанавливается расчетным или опытным путем и может быть представлена в виде формулы, таблицы или графика. Содержание при этом может быть выражено абсолютным количеством определяемого компонента в
60
Новый справочник химика и технолога
молях, в единицах массы или через соответствующие концентрации.
При измерении аналитического сигнала учитывают наличие полезного аналитического сигнала, являющегося функцией содержания определяемого компонента, и аналитического сигнала фона, обусловленного примесями определяемого компонента и мешающими компонентами в растворах, растворителях и матрице образца, а также «шумами», возникающими в измерительных приборах, усилителях и другой аппаратуре. Эти шумы не имеют отношения к определяемому компоненту, но накладываются на его собственный аналитический сигнал. Задача аналитика состоит в том, чтобы максимально снизить величину аналитического сигнала фона и, главное, сделать минимальными его колебания. Обычно аналитический сигнал фона учитывают при проведении контрольного (холостого) опыта, когда через все стадии химического анализа проводится проба, не содержащая определяемого компонента. Полезным сигналом при этом будет аналитический сигнал, равный разности измеренного аналитического сигнала и аналитического сигнала фона.
На основании существующей зависимости между аналитическим сигналом и содержанием находят концентрацию определяемого компонента. Обычно при этом используют методы градуировочного графика, стандартов или добавок. Описанные в литературе другие способы определения содержания компонента, как правило, являются модификацией этих трех основных методов .
Наиболее распространен метод градуировочного графика. При этом в координатах аналитический сигнал-содержание компонента строят график с использованием образцов сравнения с различным и точно известным содержанием определяемого компонента. Затем, измерив величину аналитического сигнала в анализируемой пробе, находят содержание определяемого компонента по градуировочному графику (рис. 2.1).
Рис. 2.1. Метод градуировочного графика
В химическом анализе чаще всего используют прямолинейные градуировочные графики, построенные для определенного диапазона определяемых содержаний, т. е. в области значений, предусмотренных данной методикой.
* ГТ
Для учета возможных потерь определяемого компонента при проведении анализа, изменения тех или иных условий в процессе регистрации аналитического сигнала в атомно-эмиссионной спектроскопии, в хроматографии, вольтамперометрии используют метод внутреннего стандарта.
Расчет метрологических характеристик линейного графика у = а + Ьх или у = Ь'х и результата анализа проводят методами регрессионного анализа (методом наименьших квадратов — МНК) и статистической обработки результатов определений (см. п. 2.4) [4, 15, 22, 23, 25].
В методе стандартов измеряют аналитический сигнал в образце сравнения (эталонном образце) с известным содержанием компонента и в анализируемой пробе: уэт = = &эт и ух = Scx, где S — коэффициент пропорциональности. Если определенное в идентичных условиях значение 5 заранее известно, то можно провести расчет по формуле у у с
сх= —. Обычно же применяют соотношение —— = —,
S Л сх
откуда
с, (2.26)
-Уэт
Иногда используют два эталонных образца, в которых содержание компонента отличается от предполагаемого в анализируемой пробе в одном случае в меньшую, в другом — в большую сторону. Этот вариант метода стандартов называют иногда методом ограничивающих растворов. Содержание определяемого компонента рассчитывают по формуле
сх
Уэт,2 Уэт,1
(2.27)
В тех случаях, когда при определении малых количеств компонента нужно учесть влияние матрицы образца на величину аналитического сигнала, часто используют метод добавок — расчетный и графический.
При определении содержания расчетным методом берут две аликвоты раствора анализируемой пробы и в одну из них вводят добавку определяемого компонента известного содержания. В обеих пробах измеряют аналитический сигнал — ух и ух + доб. Неизвестную концентрацию определяемого компонента рассчитывают по формуле
_________У х^доб^доб______ Л + доб^доб+СУх + доб-ЛЖ’
(2.28)
где ЕдОб и сдоб — объем и концентрация добавленного раствора определяемого компонента; И — аликвота анализируемой пробы.
При определении содержания компонента графическим методом берут п аликвот анализируемой пробы: 1, 2, 3, ..., п. В аликвоты 2, 3, ..., п вводят известные, возрастающие количества определяемого компонента. Во всех аликвотах измеряют аналитический сигнал и строят график в координатах: аналитический сигнал - содержание определяемого компонента, приняв за условный нуль содержание определяемого компонента в аликвоте без добавки (аликвота 1). Экстраполяция полученной прямой до пересечения с осью абсцисс дает отрезок, расположенный влево от условного нуля координат, величина которого в выбранном масштабе и единицах измерения соответствует искомому содержанию (сх) определяемого компонента (рис. 2.2).
Метрологические основы методов количественного анализа
61
Метод стандартов и метод добавок применимы для линейной градуировочной функции. Метод градуировочного графика допускает использование как линейной, так и нелинейной функций: аналитический сигнал-содержание. В последнем случае требуется большее число экспериментальных данных и результат определения содержания компонента бывает, как правило, менее точным.
Во всех методах определения неизвестного содержания компонента используют функциональную зависимость у = = Sx. Коэффициент чувствительности S (иногда его называют просто чувствительность) характеризует отклик аналитического сигнала на содержание компонента. Коэффициент чувствительности — это значение первой производной градуировочной функции при данном определенном содержании. Для прямолинейных градуировочных графиков — это тангенс угла наклона прямой (см. рис. 2.1):
(229)
Ах с2 - с, с3 - с,
Чем больше коэффициент чувствительности S, тем меньшие количества компонента можно обнаруживать и определять, получая один и тот же аналитический сигнал. Чем выше S, тем точнее можно определить одно и то же количество вещества. Вот почему при разработке нового метода или методики химического анализа исследователь, стремясь увеличить коэффициент чувствительности, использует различные приемы: концентрирование, усовершенствование аппаратуры, создание новых реагентов и т. п.
Во всех рассмотренных способах используют образцы сравнения (эталоны), т. е. образцы, пробы, растворы с точно установленным содержанием компонента. Методы анализа, использующие образцы сравнения — это так называемые относительные методы химического анализа. Абсолютных (безэталонных) методов в аналитической химии немного— например, методы гравиметрии, прямой кулонометрии, некоторые варианты радиохимических методов.
Образцы сравнения для относительных методов анализа могут быть приготовлены из химически чистых, устойчивых веществ известного состава (стандартные вещества). В этом случае содержание определяемого компонента вычисляют по химической формуле стандартного вещества. Возможно приготовление образцов сравнения в отдельной лаборатории, учреждении, отрасли, когда содержание компонента устанавливают разными методами на разных приборах многие аналитики. Наиболее надежные результа
ты получают, когда в качестве образцов сравнения используют стандартные образцы (СО) — специально приготовленные материалы, состав и свойства которых достоверно установлены и официально аттестованы специальными государственными метрологическими учреждениями.
При проведении химического анализа обычно не ограничиваются единичным определением, а проводят несколько параллельных определений (как правило, 3-5) для одной и той же пробы в одинаковых условиях. Средний результат параллельных определений называют результатом анализа и обозначают через с или х . Отклонение результата анализа от истинного содержания определяемого компонента (ц) называют погрешностью (или ошибкой) определения. Наряду с обнаружением или определением содержания компонента важна оценка достоверности полученных результатов, погрешностей измерения (см. п. 2.3)
2.3. Погрешности количественного химического анализа.
Представление результатов химического анализа
Результат количественного химического анализа*, как и результат любых измерений, сопровождается погрешностью (ошибкой), которая может быть снижена лишь до определенного уровня. Определение величины этой погрешности нередко является сложной задачей, которую требуется решать химику, поскольку результат анализа, выполненного с неизвестной степенью надежности, не представляет научной и практической ценности.
При анализе пробы, как правило, проводят несколько параллельных определений. Отдельные результаты этих определений должны лежать как можно ближе друг к другу и соответствовать истинному содержанию пробы.
В связи с этим существуют две основные метрологические характеристики, по которым химик судит о результатах анализа:
1) воспроизводимость результатов определений;
2) правильность, т.е. соответствие полученного результата содержанию определяемого компонента в пробе.
Согласно методическим указаниям «Метрологическое обеспечение количественного химического анализа. Основные положения. РД 50-674-88» (М.: Изд. стандартов, 1989. 8 с.): Количественный химический анализ пробы вещества (КХА) — экспериментальное определение содержания (массовой и объемной доли, молярной концентрации и т.д.) одного или ряда компонентов вещества в пробе физическими, физико-химическими, химическими и другими методами. КХА проводят согласно методике анализа, узаконенной в установленном порядке, посредством косвенных измерений либо путем прямых измерений с использованием приборов специального назначения. Процедура КХА, как правило, включает операции по преобразованию пробы в форму, обеспечивающую надежное и точное определение компонента данным методом. Результат КХА — установленное содержание компонента вещества в пробе — следует выражать в единицах физических величин, допущенных к использованию в стране, с указанием характеристик его погрешности или их статистических оценок.
62
Новый справочник химика и технолога
Метрологическое обеспечение количественного химического анализа основывается на методах математической статистики [1-7,12-30].
В настоящем разделе кратко рассмотрены типы погрешностей, возникающих при проведении анализа, способы их оценки и рекомендации ИЮПАК [15-21] по представлению результатов химического анализа, которыми руководствуются обычно при публикациях в научной литературе.
В заводских аналитических лабораториях, в литературе по метрологии действуют нормативные документы — государственные стандарты (ГОСТы), руководства и методические инструкции (МИ), которые необходимо выполнять при обработке результатов наблюдений и представлении результатов измерений (анализа).
Термины, символы, определения, форма представления результатов измерений (анализа) в рекомендациях ИЮПАК и ГОСТах или МИ по ряду моментов существенно различаются, например, ГОСТ Р ИСО 5725-1-2000 (см. компакт-диск). Вопросы метрологии аналитического контроля производств представлены в [31-44].
В настоящем разделе рассматривается статистическая обработка результатов прямых равноточных наблюдений (определений).
КХА является косвенным измерением, при котором сначала проводят прямые измерения физических величин (хь х2, х3, ..., xt, ..., х„), а затем, по определенным формулам, связывающим эти величины с измеряемой величиной у, вычисляют ее значение. Поэтому расчеты метрологических характеристик результатов анализа в конкретных случаях рассматриваются в соответствующих разделах справочника с учетом уравнений, представленных в табл. 2.4.
2.3.1. Общие термины [/, 15-21, 32, 33, 35]
Измеренное значение {measured value). Наблюдаемое значение массы или объема, показание прибора или другая величина, найденная при анализе образца.
Результат {result). Окончательное значение для измеренного или рассчитанного значения, найденное по окончании измерения, включая все вспомогательные процедуры и численные оценки.
Это определение, по нашему мнению, требует уточнения. В соответствии с классификацией, принятой в метрологии, различают:
результат наблюдения — значение величины, получаемое при отдельном наблюдении;
результат измерения — значение величины, найденное путем ее измерения; за результат измерения принимают среднее арифметическое результатов наблюдений, в которые предварительно введены поправки для исключения систематических погрешностей;
результат определения — значение содержания определяемого компонента в пробе, найденное при единичном определении;
результат анализа — среднее значение регламентированного числа результатов параллельных определений компонента в одной пробе.
Переменная {variable; х), (случайная величина). Измеренная или рассчитанная численная величина или характеристика. Соответствующая численная величина может быть использована для статистической обработки. Переменная величина, например, может быть измеренной величиной или результатом.
Серия {series), (выборка*). Ряд измеренных величин (хь х2,..., х„ ..., хп), которые эквивалентны друг другу с точки зрения статистического исследования, т. е. результаты повторяющихся анализов, использующих только один аналитический метод для вещества, которое считается гомогенным.
Истинная величина {true value; ц, т). Величина, которая характеризует некий параметр, однозначно определенный в условиях, существующих в то время, когда данный параметр рассматривается. Это — идеальная величина, которую можно достичь только в случае, когда устранены все источники погрешностей измерения и выбрана вся генеральная совокупность.
Правильность {accuracy). Степень близости между полученным результатом и истинным значением. Правильность является качественной характеристикой и включает комбинацию компонентов случайных погрешностей и обычную систематическую погрешность. Это качество измерений, отражающее близость к нулю систематических погрешностей. Отсутствие в химическом анализе систематических погрешностей обеспечивает его правильность (рис. 2.3). Количественной оценкой правильности результата анализа (оценкой систематической погрешности) служит разность между средним (средним арифметическим результатов наблюдений) и истинным значением определяемой величины.
Случайная погрешность
Случайная погрешность
мала.
Случайная погрешность
Случайная погрешность
велика. мала. велика.
Системати-
Система-
Систематиче- Систематиче-
ческая тическая ская
екая
погрешность погрешность
погрешность погрешность
отсутствует
отсутствует положительна отрицательна
Рис. 2.3. Систематические и случайные погрешности, результаты наблюдений (определений):
а — хорошо воспроизводимы и правильные;
б — плохо воспроизводимы, но правильные; в — хорошо воспроизводимы, но неправильны; г — плохо воспроизводимы и неправильны [8]
Выборка — совокупность значений случайной величины. Рассматривается как случайная выборка из генеральной совокупности значений случайной величины. Генеральная совокупность — гипотетическая совокупность всех значений одной и той же случайной величины [32].
Метрологические основы методов количественного анализа
63
Воспроизводимость" (precision). Степень близости ме-лщунсэависимыми результатами измерений, полученными при использовании экспериментальной методики при оговоренных условиях (рис. 2.3). Чем меньше случайная погрешность эксперимента, влияющая на результат, тем точнее данная методика. Мерой воспроизводимости (или невоспроизводимости) служит абсолютное (5) или относительное (sr) стандартное отклонение, вычисляемое из результатов нескольких параллельных наблюдений.
Комментарий. Как следует из Международного словаря основных и общих метрологических терминов (International Vocabulary of Basic and General Terms in Metrology. ISO, 1993), термин «precision» часто используют в смысле «правильность». Чтобы избежать путаницы в употреблении терминов, следует четко представлять, что воспроизводимость (precision) относится только к дисперсии, но не к отклонению от истинного (в традиционном понимании) значения.
Сходимость* (repeatability). Степень согласованности независимых результатов, порученных при помощи одного и того же метода или идентичного анализируемого материала в одинаковых условиях (один и тот же исполнитель, тот же прибор, та же лаборатория и незначительные интервалы между измерениями). Мерой сходимости является стандартное отклонение (standard deviation), употребляемое с уточняющим термином, т. е. стандартное отклонение сходимости (repeatability standard deviation).
2.3.2. Классификация погрешностей
Существует несколько различных, частично перекрывающих друг друга подходов к классификации погрешностей. Наиболее распространенные варианты классификации погрешностей с указанием главного принципа, положенного в их основу, представлены в [24, с. 22-23].
По способу выражения (вычисления) погрешности подразделяют на абсолютные и относительные.
Погрешность результата (измерения, определения и т.п.) (error of result', е). Отклонение результата (измерения, определения и т.п.) от истинного значения (р) * измеряемой величины:
е=х-ц. (2.30)***
Если необходимо, то рассчитывают погрешности единичных определений:
е( = xt - ц .
Погрешности могут быть положительными или отрицательными — в зависимости от того, завышает или занижает погрешность результат анализа.
* Иные формулировки [35, 45-46].
Истинное значение величины неизвестно, его применяют только в теоретических исследованиях. На практике используют действительное значение величины хя, в результате чего погрешность измерения Д хизм определяют по формуле [35]:
А хизм = Хщм - х» гДе хизм — измеренное значение величины.
В отечественной литературе погрешность измерения, определения и т.д. обозначают разными индексами: D, Дх, Ахизм и др.
(2-31)
Погрешность измерения, выраженная в единицах измеряемой величины, называется абсолютной погрешностью измерения.
Комментарий. Если результат, например концентрация определяемого вещества, выражен в процентах, данный параметр также, естественно, будет выражен в процентах. При этих обстоятельствах во избежание путаницы с термином относительная погрешность, выраженная в процентах данную величину можно указывать как абсолютную погрешность, выраженную в процентах (percent absolute error).
Относительная погрешность (relative error, ег). Погрешность, деленная на истинное значение:
е |х-ег = —= ---
ц ц
Относительная погрешность в процентах (percentage relative error, er, (%)). Получают умножением величины относительной погрешности на 100:
ег(о/о) = к2±1.юо. (2.32)
Ц
Комментарий. Относительная погрешность обычно знака не имеет. Во избежание путаницы ее нельзя называть просто погрешностью или погрешностью, выраженной в процентах.
По характеру причин, вызывающих погрешности, различают систематические, случайные погрешности и промахи.
Случайная погрешность. Составляющая погрешности результата измерения, определения и т.п., изменяющаяся случайным образом (по знаку и значению) при повторных измерениях, определениях и т. п., проведенных с одинаковой тщательностью [32].
Причины появления случайных погрешностей неизвестны. Случайные погрешности определяют воспроизводимость метода (методики) анализа; делают неточным результат анализа (рис. 2.3).
Систематическая погрешность. Составляющая погрешности результата измерения (определения), остающаяся постоянной или закономерно изменяющаяся при повторных измерениях, определениях и т. п. одной и той же величины [32].
Систематическая погрешность вызывается постоянно действующей причиной. Наличие и величина систематических погрешностей характеризуют правильность методики анализа и ее результата. Систематические погрешности делают неверным сам анализ (рис. 2.3).
Грубыми погрешностями (промахами) называют погрешности измерения, определения, которые существенно превышают ожидаемые при данных условиях. Они, как правило, обусловлены небрежностью или некомпетентностью экспериментатора.
64
Новый справочник химика и технолога
2.3.3. Систематические погрешности.
Способы их выявления [7, 4, 22-24, 32,36, 38, 47, 48]
Систематические погрешности — погрешности, величину которых можно измерить и учесть, хотя это порой бывает далеко не просто. По характеру влияния на конечный результат систематические погрешности подразделяют на аддитивные (постоянные), не зависящие от содержания определяемого компонента, и мультипликативные (пропорциональные), зависящие от содержания определяемого компонента. Аддитивные погрешности возникают, например, при неучете холостого сигнала в инструментальных методах анализа; мультипликативные— в титриметрии при неправильной установке титра (концентрации) титранта. Мультипликативные погрешности весьма эффективно обнаруживаются и устанавливаются методом стандартных добавок. Перечислить все источники систематических погрешностей невозможно. Типичными составляющими погрешности измерений (определений) являются: методические составляющие, инструментальные составляющие и погрешности, вносимые оператором — субъективные (индивидуальные) погрешности. Источники и причины указанных составляющих погрешностей измерений, определений подробно рассмотрены в ГОСТ Р 8.563-96 [38].
Индивидуальные (субъективные) погрешности возникают в результате незнания, небрежности, предвзятости или физических недостатков экспериментатора. Например, они могут появляться при неправильном перенесении пробы, в частности, во время отбора и перенесения аликвотного объема мерной пипеткой при выдувании из нее раствора для «ускорения» анализа.
Типичные индивидуальные погрешности в титриметрии обусловлены различной степенью дальтонизма, когда определяют конечную точку титрования по переходу окраски индикатора, с оценкой положения мениска раствора между двумя ближайшими делениями бюретки и т. д.
Инструментальные погрешности. Эти погрешности обусловлены несовершенством приборов, с которыми работает химик, или влиянием на них внешних факторов, прежде всего температуры окружающего воздуха.
Часто объем мерной посуды (бюреток, пипеток и колб) несколько отличается от объема, установленного при ее калибровке. Систематические погрешности этого вида можно устранить, калибруя мерную посуду при соответствующей температуре [47, 48].
Периодическая поверка аналитических приборов (весов, спектрофотометров, полярографов, хроматографов и т. д.) сводит к минимуму систематическую составляющую инструментальных погрешностей.
Методические погрешности. Основной вклад в общую погрешность вносят методические погрешности, которые обусловлены методикой определения. Методические погрешности могут быть обусловлены погрешностями отбора пробы, переведения пробы в удобную для анализа форму, операциями концентрирования и разделения компонентов.
Особо следует выделить методические погрешности, связанные с отклонением поведения реагентов или реакций, на которых основано определение, от идеального. Причина
ми таких погрешностей могут быть малая скорость реакции титрования, неполнота протекания реакций, протекание побочных реакций, мешающих определению и т. д.
Также следует обращать особое внимание на чистоту (марку) и содержание основного вещества в реактивах, которые используются в определениях или для стандартизации титрантов и которые должны отвечать требованиям ГОСТов на них.
В первом случае в реактивах может оказаться в качестве примеси определяемый компонент, а во втором — посторонние примеси, содержание которых может быть недопустимо высоким для первичного стандарта. Результатом является реактивная ошибка.
Типичной и наиболее широко распространенной методической погрешностью титриметрических методов анализа является индикаторная ошибка . Она возникает при фиксировании конечной точки титрования.
Обычно конечная точка титрования и точка эквивалентности в большей или меньшей степени не совпадают, т. е. объем титранта, израсходованного на титрование, не отвечает строго объему Г(К)стех. Обусловлено это тем, что даже тщательно подобранный индикатор меняет цвет несколько раньше или позже точки эквивалентности. Индикаторные систематические ошибки могут вносить существенный вклад в общую погрешность анализа. Методики их расчета и оценки их значимости представлены, например, в [4, 6, 7, 49, 50].
При обработке результатов химического анализа систематические погрешности должны быть выявлены и устранены или, по возможности, оценены.
Для обнаружения систематической погрешности одним из нижеприведенных способов проверяют значимость различия [4, 22, 24, 32,51,52]:
— между результатом анализа стандартного образца (СО) состава (SRM — standard reference materials) или аттестованной смеси (АС), который получают с помощью разработанной или используемой методики, и аттестованным содержанием (паспортными данными) определяемого компонента в СО или в АС. Это самый надежный способ выявления систематической погрешности и аттестации на
Общий термин индикаторная ошибка, применяемый в учебной литературе, объединяет, по существу, три вида ошибок: химическую, визуальную (дискриминационную) и собственно индикаторную ошибки. Химическая ошибка обусловлена несовпадением pH (при кислотно-основном титровании), рМ (-lg[M] — при комплексонометрическом титровании), pl (—lg[CT], —lg[Br ]. -lg[r] — при аргентометрическом титровании) в конечной точке титрования и в точке эквивалентности. Визуальная ошибка обусловлена ограниченной способностью глаза оценивать интенсивность и цветность окраски. Индикаторная ошибка обусловлена взаимодействием индикатора с титрантом или титруемым веществом.
Типы СО см. ГОСТ 8.315-97. Стандартные образцы. Основные положения. В зависимости от установленного порядка утверждения стандартные образцы подразделяются на следующие категории: • государственные стандартные образцы (ГСО); • отраслевые стандартные образцы (ОСО); • стандартные образцы предприятий (СОП).
Метрологические основы методов количественного анализа
65
.правильность метода или методики анализа. Необходимое условие применения СО и АС в химическом анализе — максимальная близость состава и свойства стандартного образца или аттестованной смеси и анализируемой пробы;
— между результатами анализа, полученными с помощью данной и альтернативной (арбитражной) методик анализа;
— между результатами анализа, полученными в данной и альтернативной (арбитражной) лабораториях;
— между результатами анализа, полученными с использованием двух разных навесок анализируемого вещества (метод варьирования, варьирования величины пробы). Удваивая (способ удвоения) или увеличивая размер пробы в кратное число раз, можно обнаружить по изменению найденного содержания определяемого компонента постоянную (аддитивную) систематическую погрешность;
— между результатом анализа данного вещества и содержанием определяемого компонента в этом веществе, рассчитанном из результата анализа вещества с известной добавкой определяемого компонента (метод добавок).
Во всех указанных случаях значимость различия между сравниваемыми результатами а{ и а2 устанавливают при помощи /-критерия (см. табл. 2.3): систематическую погрешность считают значимой, если:
h-«2|
5 V«1 «2
>t(P,f = пх+п2-2),
(2.33)
_ Is^n, -l) + sl(n2-l)
где S=J~——~—-—-—,
у w, + n2 - 2
sf и s2— дисперсии, характеризующие случайное рассеяние результатов для а{ и а2 соответственно; п< и п2 — число опытов при получении результатов at и а2, Р — доверительная вероятность; f— число степеней свободы.
После выявления систематической погрешности она должна быть оценена и устранена путем изменения методики определения или замены средств измерения, или исключением из получаемых результатов путем исправления их на величину этой погрешности.
Алгоритмы и типичные способы оценивания характеристик погрешности результатов количественного химического анализа описаны также в ГОСТе Р 8.563-96 [38] ив МИ 2336-95 [41].
При оценке систематических ошибок можно условно выделить погрешности трех типов [4].
К первому типу относят погрешности известной природы, которые могут быть рассчитаны a priori (предварительно) до определения компонента и учтены введением соответствующей поправки. Примеры таких погрешностей — индикаторные ошибки и ошибки измерения объемов в титриметрии, ошибки взвешивания в гравиметрическом методе анализа.
Таблица 2.3
Значения /-критерия Стьюдента в зависимости от доверительной вероятности Р (двусторонняя постановка задачи) и Р (односторонняя постановка задачи) и степени свободы f [22]*
f р
0,75 0,90 0,95 0,98 0,99
Р
0,875 0,95 0,975 0,99 0,995
1 2,41 6,31 12,7 ' 31,82 63,7
2 1,60 2,92 4,30 6,97 9,92
3 1,42 2,35 3,18 4,54 5,84
4 1,34 2,13 2,78 3,75 4,60
5 1,30 2,01 2,57 3,37 4,03
6 1,27 1,94 2,45 3,14 3,71
7 1,25 1,89 2,36 3,00 3,50
8 1,24 1,86 2,31 2,90 3,36
9 1,23 1,83 2,26 2,82 3,25
10 1,22 1,81 2,23 2,76 3,17
11 1,21 1,80 2,20 2,72 3,11
12 1,21 1,78 2,18 2,68 3,05
13 1,20 1,77 2,16 2,65 3,01
14 1,20 1,76 2,14 2,62 2,98
15 1,20 1,75 2,13 2,60 2,95
16 1,19 1,75 2,12 2,58 2,92
17 1,19 1,74 2,И 2,57 2,90
18 1,19 1,73 2,10 2,55 2,88
19 1,19 1,73 2,09 2,54 2,86
20 1,18 1,73 2,09 2,53 2,85
21 1,18 1,72 2,08 2,52 2,83
22 1,18 1,72 2,07 2,51 2,82
23 1,18 1,71 2,07 2,50 2,81
24 1,18 1,71 2,06 2,49 2,80
25 1,18 1,71 2,06 2,49 2,79
26 1,18 1,71 2,06 2,48 2,78
28 1,17 1,70 2,05 2,47 2,76
30 1,17 1,70 2,04 2,46 2,75
00 1,15 1,64 1,96 2,33 2,58
Примечание. * Доверительная вероятность — доля случаев, в которых среднее (арифметическое) при данном числе определений будет лежать в определенных пределах. Доверительная вероятность Р связана с двусторонней — верхней и нижней — границей разброса среднего значения выборки. В аналитической химии, как правило, пользуются доверительной вероятностью Р — 0,95 и значительно реже Р = 0,90 или Р = 0,99. Часто устанавливают одностороннее требование, например, чтобы значение х не превышало некоторой верхней границы. В этом случае говорят об односторонней границе с соответствующей вероятностью Р .
— — р
Между Р и Р существует соотношение р = о 5 + — • Подробнее ’ 2
см. [22].
Ко второму типу можно отнести погрешности известной природы, значения которых могут быть оценены в ходе химического анализа или при постановке специального эксперимента. К ним относятся инструментальные, реактивные ошибки, ошибки отдельных стадий химического анализа — методические погрешности. Если исследова-
66
Новый справочник химика и технолога
тель может оценить ошибки отдельных стадий и операций, то, по закону сложения погрешностей, он может вычислить общую погрешность результата анализа. В табл. 2.4 приведены расчеты абсолютных и относительных погрешностей некоторых функций.
При расчете систематических погрешностей следует различать два важных случая.
Если известны и величины, и знаки погрешностей отдельных составляющих, то расчет суммарной погрешности производится по формулам, приведенным в столбце а табл. 2.4. Величина суммарной погрешности при этом получается с определенным знаком.
Если известны лишь максимально возможные погрешности отдельных стадий (это равносильно тому, что известны лишь абсолютные величины, но не знаки этих погрешностей), то расчет производится по формулам, указанным в столбце б табл. 2.4. При этом результат расчета также является абсолютной величиной суммарной погрешности.
Аналитика часто интересуют не только выявление и оценка систематической погрешности, а в большей мере — способы ее уменьшения и устранения. Один из таких способов — релятивизация (от англ, relative — относительный), когда в идентичных условиях проводят отдельные аналитические операции таким образом, что происходит нивелирование систематических погрешностей. Так, в тит-риметрии отбирают аликвоты стандартного и анализируемого растворов одними и теми же пипетками, в гравиметрии взвешивают пустой тигель и тигель с осадком на
одних и тех же весах, с одними и теми же разновесами и т.д. Один из приемов релятивизации погрешностей — проведение контрольного опыта. При этом происходит нивелирование погрешностей, обусловленных загрязнениями из реактивов, воды, используемой посуды; погрешностей стадии пробоподготовки и т. д.
К третьему типу относят погрешности невыясненной природы, значения которых неизвестны. Эти погрешности трудно выявить и исключить. Их можно обнаружить лишь после устранения прочих систематических погрешностей и последующего тщательного исследования всех стадий, операций и условий проведения анализа. Обычно в таких случаях используют прием рандомизации (от англ, random — случайно) — переведение систематических погрешностей в разряд случайных. Возможность рандомизации основана на том, что систематическая погрешность единичного явления (метода, прибора, исполнителя анализа) при рассмотрении ее в более широком классе однотипных явлений (группа методов, серия приборов, коллектив аналитиков) становится величиной переменной, т. е. приобретает черты случайной погрешности и оценивается с применением методов математической статистики.
2.3.4. Статистическая обработка результатов прямых равноточных наблюдений {определений)
Перед расчетом основных метрологических характеристик результата химического анализа методами математической статистики систематические погрешности должны
Таблица 2.4
Суммирование погрешностей*
Функция Систематические погрешности Общие (суммарные) случайные погрешности
а Б
у = Х1 + х2 Ду = Axi + Ах2 |Ду| = |ДХ1| + |Дх2| (2-34>
У = Х1 —х2 Ду = Axt - Дх2 |Ду| = |Дх1| + |Дх2| sy = (2.35)
У = Х1 -х2 Ду _ Дх, Дхг у X] х2 Ду _ Дх, Дх, у х, х2 z \2 z \2 1 \ 1 1 1 = р_ + (2.36) у Км 1м
Xj у=— х2 Ду Дх, Дх2 у X, х2 Ау _ Дх, Дх2 У хг х2 z \2 z \2 •S’ 1 1 ( «г 1 -М pL + -S- (2.37) у Км 1м
У=хР Ду Дх — = Р— Ду Дх — = Р— sy Sx — = р—
У х У х У X
у = 1пх ж Дх 1,1 &х s
Ду = — Ду & г sy =
X X у X
у = lgx Дх Ду = 0,434— |Ду| = 0,434 — S = 0,434 —
X X у X
Примечание* При необходимости из относительной погрешности рассчитывают абсолютную и наоборот.
Метрологические основы методов количественного анализа
67
быть выявлены и устранены или переведены в разряд случайных.
В химическом анализе содержание вещества в пробе устанавливают, как правило, по небольшому числу параллельных определений (п = 3-7). Для расчета погрешностей в этом случае пользуются методами современной математической статистики, разработанной для малого числа определений. Полученные результаты рассматривают как случайную (малую) выборку из некоторой гипотетической генеральной совокупности, состоящей из бесконечного числа выполненных в данных условиях наблюдений. Соответственно различают выборочные параметры (параметры малой выборки} случайной величины, которые зависят от числа наблюдений, и параметры генеральной совокупности, не зависящие от числа наблюдений.
Все измерения в метрологии делятся на прямые и косвенные.
При прямых непосредственных измерениях числовое значение измеряемой величины х получают непосредственным сравнением этой величины с эталоном (например, массы предмета при взвешивании на чашечных весах — с массой разновесок, объема раствора — с проградуированной шкалой бюретки и т. п.). Обычно результаты таких измерений сразу получают из показаний измерительного прибора.
Результат каждого прямого измерения включает случайную погрешность, которая зависит от большого числа случайных факторов.
Если отклонения, вызванные случайными факторами, сравнимы по абсолютному значению с чувствительностью прибора, то они обнаруживаются приборами, и при п измерениях одной и той же величины получаются результаты хь х2, ..., Xj, ..., хп, которые могут отличаться друг от друга в пределах чувствительности данных измерений.
Статистическую обработку результатов наблюдений (определений) малой выборки (и) , оценку их воспроизводимости и правильности проводят по следующей схеме (см. п. 2.3.4.1).
2.3.4.1. Термины, относящиеся к параллельным измерениям. Оценка воспроизводимости
В описание результатов, полученных при параллельных (replicate) измерениях (определениях), следует включать характеристики: число измерений, среднее арифметическое, стандартное отклонение (или размах, см. комментарии к обоим терминам), границы доверительного интервала и, если известно, истинное значение, а также оценку границ систематической погрешности.
Число измерений (number of observations; п). Общее число полученных данных (измеренных значений, определений) в серии, объем выборки (sample size).
Комментарий. Это число необходимо указывать всегда.
В случае, если рассматривается генеральная совокупность, используют обозначение N.
* Предполагается, что выборка имеет приближенно нормальный закон распределения [22, 24].
Число степеней свободы (degrees of freedom; f). Статистическая величина, показывающая число переменных, которые могут быть присвоены произвольно при характеристике выборки. В наиболее простом случае, когда имеют п измерений (определений) и один исследуемый параметр (среднее значение), /= п - 1.
Уровень доверительной вероятности (confidence level; Р = 1 - а), или доверительная вероятность. Вероятность того, что ожидаемая величина исследуемого параметра лежит внутри некоторого интервала. Доверительная вероятность Р — доля случаев, в которых среднее (х) при данном числе определений будет лежать в определенных пределах. Доверительная вероятность связана с двусторонней — верхней и нижней — границей разброса среднего значения выборки.
С точки зрения математической статистики надежность полученного в лаборатории результата анализа тем выше, чем больше доверительная вероятность.
В аналитической химии, как правило, пользуются доверительной вероятностью Р = 0,95 или двухсигмовым (2о) критерием, но в особо важных случаях принимают Р = = 0,99 — критерием Зо. Доверительная вероятность может быть выражена и в процентах. Комплементарная величина а известна как уровень значимости (significance level).
Уровень значимости а = (1 - Р) — максимальная вероятность того, что погрешность превзойдет некое предельное (критическое) значение ± А хкр, т. е. такое значение, что появление этой погрешности можно рассматривать как следствие значимой (неслучайной) причины [22, 24].
В разных литературных источниках уровень значимости обозначается по-разному: а, 0, р.
Среднее арифметическое, средняя величина (arithmetic mean, average; х ). Сумма всех значений серии (выборки) наблюдений, деленная на число наблюдений:
(2.38)
х ==-п
Комментарий. Во всех процессах определения суммы (здесь и далее, если это не оговорено особо) пределы суммирования от 1 до п.
Отклонение (deviation; d). Разность между случайной величиной и арифметическим средним выборки, к которой она принадлежит:
dt =Х'-х. (2.39)
Размах (выборки) (range; R). Разность между наибольшей и наименьшей из наблюдаемых величин в выборке:
R -^макс -^мин- (2.40)
Комментарий. Этот параметр особенно удобен для малых выборок (п < 10) как альтернативная мера дисперсии.
Стандартное отклонение (standard deviation). Оценивается как положительный квадратный корень величины, получаемой при делении суммы квадратов разностей всех элементов выборки и среднего этой выборки на число степеней свободы (в простейшем случае — число измерений минус единица). Обозначения: 5 — выборочное стандартное отклонение (estimated standard deviation)'.
68
Новый справочник химика и технолога
s= Х<х,-х)2
V и-1
(2.41/
Относительное стандартное отклонение {relative standard deviation; 57). Стандартное отклонение, деленное на среднее выборки:
(2.42)
Относительное стандартное отклонение, выраженное в процентах (percentage standard deviation; sr, (%). Получают умножением величины относительного стандартного отклонения на 100.
Комментарий. Рекомендуется при описании результатов использовать относительное стандартное отклонение, не выраженное в процентах, во избежание путаницы в том случае, когда результаты также выражены в процентах. Термин коэффициент вариации (coefficient of variation) вместо термина относительное стандартное отклонение использовать не рекомендуется.
В общем случае метод (методика) анализа оптимален в той области содержаний (концентраций), в которой и абсолютное (а) и относительное (sr) стандартные отклонения имеют минимальные значения.
Дисперсия (variance; V). Квадрат стандартного отклонения.
5V У(х,-х)2
V- = . (2.43)
и-1 и-1
Дисперсия среднего (выборки), стандартное отклонение среднего (выборки).
При оценке воспроизводимости полученных результатов вычисляют также дисперсию среднего:
,Л_. £(х'~*)2
и(и-1)
и стандартное отклонение среднего
5
% =~Г УП
£(х,-х)2 и(и-1)
(2.45)
Для обозначения стандартного отклонения среднего в англоязычной литературе использует термин population standard deviation, или стандартная погрешность (standard error) с символом о.
2.3.4.2. Оценка правильности результата измерения (анализа)
После того как осуществлена проверка грубых погрешностей (в случае отдельных подозрительных измеренных значений) и их исключение, производят оценку границ доверительного интервала (С), доверительного интервала х±С и при необходимости — оценку правильности результата.
Границы доверительного интервала (confidence levels about the mean). Симметричные границы доверительного интервала (±Q для оценки среднего, в который с доверительной вероятностью Р попадает математическое ожидание (среднее генеральной совокупности). Численное значение С рассчитывают по уравнению
t р * “ s ,
С = ^=- (2.46)
у/п
или
C = tPJ-s„ (2.46')
где tPf — табличное значение /-критерия Стьюдента (табл. 2.3).
Обычно для расчета границ доверительного интервала пользуются значением Р = 0,95, но при ответственных измерениях требуется более высокая надежность (Р = 0,99).
Необходимо отметить, что если при отработке методики выполняют п параллельных измерений, а методика анализа в дальнейшем предусматривает выдачу результатов из т параллельных измерений (обычно п > 10, т = 2 - 3), то границы доверительного интервала для рядовых анализов следует рассчитывать по формуле [53]
а не по формулам (2.46), (2.46'), где 5 — стандартное отклонение для выборки из п опытов. В противном случае значение С рядового анализа окажется слишком заниженным.
Доверительный интервал (confidence interval) описывается как х ± С.
Если воспроизводимость измеренных значений (результатов наблюдений, определений) характеризуют стандартным отклонением, то результат (измерения, анализа) характеризуют доверительным интервалом, который описывается как х ± С.
Доверительный интервал ограничивает область, внутри которой, при отсутствии систематических погрешностей, находится истинное значение результата (измерения, анализа) с заранее заданной доверительной вероятностью Р:
х±С; (2.47)
При подсчете значений s всегда используют неокругленные результаты измерения (анализа) с ненадежным знаком после запятой [22].
* В отечественной литературе выражения (2.46), (2.46') иногда именуются доверительным интервалом, а вместо индекса С используется индекс Ах.
Метрологические основы методов количественного анализа
69
х-С <ц<х+С;
х -tp f • s- < ц < х + tp f • 5-;
(2.48) Для полной характеристики правильности методики необходима оценка систематических погрешностей вблизи (2 49) нижней и верхней границ интервала концентраций сн - св или содержаний определяемого элемента тя - т&.
_ s _
X-tp f —j= < Ц < X +tp f y/n
(2.50)
Из уравнений (2.48-2.50) следует, что значение доверительного интервала зависит от объема выборки, т. е. от числа проведенных опытов: с уменьшением числа измерений увеличивается доверительный интервал (при той же доверительной вероятности) или при заданном доверительном интервале уменьшается надежность измерений.
Обработанные данные можно представить в виде табл. 2.5.
Таблица 2.5
Представление экспериментальных данных (образец)
х,- п X V (s или sy) tp f s с=-^Г-yjn — _L ' S X + -—J-2-yjn
2.3.5. Оценка грубых погрешностей (промахов)
В литературе приводятся различные методы оценки и исключения грубых погрешностей:
— исключение грубых погрешностей методом вычисления максимального относительного отклонения; метод рекомендован для метрологических служб [24, 31, 54-56];
— проверка годности результатов измерений по правилу За [57];
— определение грубых погрешностей по (2-критерию [4, 22].
В аналитической химии, как правило, речь идет о сериях с малым числом измерений. В этом случае определение промахов лучше оценивать при помощи размаха варьирования. Для этого п результатов упорядочивают по величине; значение, которое может рассматриваться как грубая погрешность, обозначают Х]. Затем вычисляют:
Значимость систематической погрешности. Значение систематической погрешности характеризует меру правильности результатов определений (анализа). О значимости систематической погрешности, т.е. правильности результата анализа, судят в зависимости от того, попадает ли истинное значение определяемой величины в установленный доверительный интервал или находится вне его. Если | х - ц | > С , то можно говорить о значимой систематической погрешности Дхс, интервальное значение которой заключено в пределах:
для
и = 3-7 Q =
R
(2.52а)
для a II oo 1 о to II Xj -x2 , (2.526)
х-ц-С< Дхс <х-ц + С. (2.51)
В этом случае необходимо выяснить причину появления систематической погрешности. Следует отметить, что задача освобождения результатов измерений от систематических погрешностей требует глубокого анализа всей совокупности данных измерений.
Наиболее вероятные источники систематических погрешностей и способы проверки правильности результатов количественных определений рассмотрены в п. 2.3.3.
Для обнаружения и исключения систематических погрешностей широко применяют также регрессионный и корреляционный анализы (см., например, [22,24, 34]).
где R ~ х, - хп — размах варьирования (разница между наибольшим и наименьшим значениями ряда измерений).
Вычисленное значение Q сравнивают с критическим значением (2крит при доверительной вероятности Р = 0,90 (табл. 2.6). Если Q > QK?m, то результат х} является промахом и его отбрасывают. Если Q < то исключать результат нельзя — он принадлежит выборочной совокупности.
Таблица 2.6
Значения (2-критерия (доверительная вероятность Р = 0,90) [4]
n Окрит n Окрит
3 0,94 7 0,51
4 0,76 8 0,47
5 0,64 9 0,44
6 0,56 10 0,41
* При оформлении окончательного результата придерживаются следующего правила: погрешность должна иметь одну-две значащие цифры, а число, выражающее среднее значение результата (измерения), должно оканчиваться разрядом, которым начинается погрешность, т. е. значения среднего результата и границ доверительного интервала должны быть выражены числами с одинаковым числом знаков после запятой; их положено округлять в одинаковой степени. Правило округления чисел см. п. 2.7.
Согласно авторам [4] Q критерий неприменим к малым выборкам (п < 5); в этом случае необходимо набрать большее число данных или использовать другие способы выявления промаха. После исключения промаха данные выборочной совокупности можно обрабатывать с применением методов математической статистики.
70
Новый справочник химика и технолога
2.3.6. Сравнение двух средних [4,22]
В аналитической практике нередко возникает необходимость сравнения двух дисперсий (Pi и К2) и двух средних (Х] их,), которые получены из двух независимых друг от друга серий (выборок) с гц и п2 измерениями. Например, в случае несколько различающихся результатов анализа одного и того же объекта, полученных двумя разными методами, в двух разных лабораториях, разными химиками и т.д.
Необходимо установить, обусловлено ли это несовпадение случайной погрешностью или разница результатов статистически значима. С этой целью сначала выясняется, нет ли значимой разницы между дисперсиями обеих серий. Сравнение ведется при помощи F-распределения (F-критерия, критерия Фишера).
Расчитывают F3KCn:
V s2
= <2-53)
Г2 s2
где V\ > V2 или s2 > si.
Полученное значение F3KCn: сравнивают с табличными FTa6n (табл. 2.7) при выбранной доверительной вероятности и числе степеней свободы f\ = пх- 1 nf2= п2 - 1. В таблицах число степеней свободы большей дисперсии приводится в горизонтальном ряду, меньшей — в вертикальном ряду. Если F3scn >F(P,f1,f2) при выбранной доверительной вероятности (обычно Р = 0,95 или Р = 0,99), то расхождение между дисперсиями значимо и рассматриваемые выборочные совокупности отличаются по воспроизводимости. Если F3Kcn <F(P,/1,/2), то различие в воспроизводимости имеет случайный характер и обе дисперсии Ki и Т2 являются приближенными оценками одной и той же общей для обеих выборок дисперсии ст2 генеральной совокупности.
Таблица 2.7
Значения F-распределения в зависимости от числа степеней свободы /1 и f2 для Р ~ 0,95 [22 б]*
fl /1
1 2 3 4 6 8 10 12 20
1 161 200 216 225 234 239 242 244 248
2 18,51 19,00 19,16 19,25 19,33 19,37 19,39 19,41 19,44
3 10,13 9,55 9,28 9,12 8,94 8,84 8,78 8,74 8,66
4 7,71 6,94 6,59 6,39 6,16 6,04 5,96 5,91 5,80
5 6,61 5,79 5,41 5,19 4,95 4,82 4,74 4,68 4,56
6 5,99 5,14 4,76 4,53 4,28 4,15 4,06 4,00 3,87
7 5,59 4,74 4,35 4,12 3,87 3,73 3,63 3,57 3,44
8 5,32 4,46 4,07 3,84 3,58 3,44 3,34 3,28 3,15
9 5,12 4,26 3,86 3,63 3,37 3,23 3,13 3,07 2,93
10 4,96 4,10 3,71 3,48 3,22 3,07 2,97 2,91 2,77
11 4,84 3,98 3,59 3,36 3,09 2,95 2,86 2,79 2,65
12 4,75 3,88 3,49 3,26 3,00 2,85 2,76 2,69 2,54
13 4,67 3,80 3,41 3,18 2,92 2,77 2,67 2,60 2,46
14 4,60 3,74 3,34 3,11 2,85 2,70 2,60 2,53 2,39
15 4,54 3,68 3,29 3,06 2,79 2,64 2,55 2,48 2,33
16 4,49 3,63 3,24 3,01 2,74 2,59 2,49 2,42 2,28
17 4,45 3,59 3,20 2,96 2,70 2,55 2,45 2,38 2,23
18 4,41 3,55 3,16 2,93 2,66 2,51 2,41 2,34 2,19
19 4,40 3,52 3,13 2,90 2,63 2,48 2,38 2,31 2,15
20 4,35 3,49 3,10 2,87 2,60 2,45 2,35 2,28 2,12
Примечание. * При f н/2 > 20 и для промежуточных значений/ и/2 значения F можно найти в приложении монографии [22 б]. Там же приведена таблица значений F для Р =0,99.
Метрологические основы методов количественного анализа
71
Если расхождение между дисперсиями незначимо, можно сравнить среднее х, и х2 двух серий (выборок), т.е. выяснить, есть ли статистически значимая разница в результатах анализа, полученных двумя различными методами, по двум разным методикам, на двух разных приборах, разными аналитиками и т.д. Для решения этой задачи используется /-распределение.
Рассчитывают среднее взвешенное двух дисперсий
?2 (и, - l)Ft + (и2 - 1)К2 «и + п2 - 2
и
t _5~*2 I П1П2
(2.54)
(2-55)
Сравнивают /эксп с /табл (см. табл. 2.3) при числе степеней свободы f = «1 + и2 - 2 и доверительной вероятности Р = 0,99. Если при этом /эксп > /табл, то расхождение между xj и х2 значимо, выборки не принадлежат одной генеральной совокупности и цх цл,;. Если /эксп < /табл, то цл. - = 0 и можно все данные рассматривать как еди-
ную выборочную совокупность в («1 + и2) результатов.
Пример [4]
При анализе золы растений на содержание меди получено (мкг):
спектрофотометрическим 0,75 0,72 0,73 0,74 0,72
методом
полярографическим 0,74 0,76 0,75 0,73
методом
Рассчитывают jq = 0,73, х2 = 0,74 и дисперсии V\ = = 0,000170 и Г2 = 0,000125, F3Kcn = 1,36; = 9,12 (табл. 2.7)
при/ = 4,/ = 3; Р = 0,95; F3Kcn < Fra6jl. Следовательно, воспроизводимость обоих методов одинакова.
Для сравнения средних рассчитывают У2 = 0,00014 и /эксп = 1,86. Поскольку /эксп < /табл (1,86 < 3,50, табл. 2.3) при числе степеней свободы /= 7 и Р = 0,99, то расхождение между средними незначимо и обе выборочные совокупности принадлежат одной генеральной совокупности. Результаты спектрофотометрического и полярографического определений меди можно рассматривать как результаты одной выборки.
2.4. Расчет уравнения линейного градуировочного графика, его метрологических характеристик и метрологических характеристик результата анализа ]4,22,23,25,56]
Вычисление метрологических характеристик линейного графика
Вычисление параметров а и Ъ. В общем случае линейная зависимость выражается уравнением:
у = а + Ъх. (2.56)
Уравнение (2.56) называют линейной регрессией. Параметр а градуировочной функции характеризует отрезок,
отсекаемый на оси ординат (intercept), который соответствует неизбежному значению холостого опыта, а коэффициент регрессии Ь, характеризующий наклон градуировочной зависимости (slope), представляет чувствительность метода (методики) анализа.
Значения параметров а и Ъ вычисляются методом регрессионного анализа.
Если имеется п взаимосвязанных пар значений (х„ у,), то можно записать:
У! = a + bxi,
у2 = а + Ьх2,
(2.57)
yt =а + bXj,
yn=a + bxn.
Здесь п — число измерений; х( — известное содержание (концентрация) определяемого компонента в z-м стандартном растворе; у1 — результат прямых измерений аналитического сигнала z-го стандартного раствора.
Непременным при построении градуировочного графика является условие, чтобы погрешности определения значения х были существенно (намного) меньше погрешностей измерения у*.
В левой части системы уравнений (2.57) находятся измеренные значения у,, а в правой — вычисленные значения Yt = а + Ьхг. Разность между обеими величинами дает погрешность. Аналитически задача метода наименьших квадратов может быть выражена в следующей форме:
т т 2
se=Z(y,-i;)2 <2-58)
»=1 /=1
Если у, - (а + Ьх,) = 5,, тогда
т
SQ = ^, i =
(2.59)* **
где т — общее число данных (измеренных значений), использованных при построении градуировочного графика.
Следовательно, задача линейного регрессионного анализа (метода наименьших квадратов) состоит в том, чтобы сумма квадратов отклонений SQ экспериментальных точек (х(, у;) вдоль ординаты от проведенной прямой была минимальной.
При определении компонентов в растворах в первую очередь необходимо выбирать соответствующие аналитические навески и мерную, хорошо отградуированную посуду, обеспечивающую малые погрешности отбираемых для анализа аликвотных объемов.
т
** В дальнейшем с целью упрощения символ суммы бу-дем обозначать просто / .
72
Новый справочник химика и технолога
Для того, чтобы найти параметры а и Ь, удовлетворяющие минимуму SQ, берут частные производные выражения (2.58) относительно а, затем относительно Ь, полученные выражения приравнивают нулю, и, решая уравнения, находят:
(2.60)
или
V-1 2 —2 ’
fx, ~тх
(2.60')
где
- 5>, _ £у,
X = —— и у = —— т т
V у - Й V X *
= ^=dl---^-^ = y-bx (2.61)
т
или в несколько ином варианте:
(2.6Г)
Константы а и b являются случайными величинами, ввиду чего необходима оценка их доверительных интервалов.
Дисперсии констант а и Ь. Дисперсии констант а и b
вычисляются по уравнениям:
_ ms2y
2(х,-х)2=„2:^-(2х,)2;
(2.62)
Сумму квадратов в уравнении (2.64) удобнее определять, пользуясь следующим выражением:
SQ^y.-rf =^(«-2) =
=Т.у2-аТ.У'^ьТ.х.у- <2.65)
При проведении вычислений по формуле (2.65) все расчеты необходимо выполнять при достаточно большом числе знаков после запятой, так как искомую сумму квадратов часто ищут для весьма близких значений. Поэтому на данном этапе даже незначительные погрешности в вычислении могут привести к большим погрешностям.
Анализ уравнения (2.62) показывает, что для константы b дисперсия si тем меньше, чем дальше значение xt лежит от его среднего значения х , т. е. чем шире интервал содержаний (концентраций), выбранный для построения градуировочной кривой.
Стандартное отклонение точек от найденной зависимости (standard deviation of points about the fitted line; s или sy). Оценка воспроизводимости измерений (зависимой переменной). Этот параметр также носит название остаточной суммы отклонений (residual standard deviation). Данный параметр рассчитывают исходя из уравнения (2.66):
L о. J ’ <2-66)
V т-2
где (т - 2) есть число степеней свободы.
Стандартное отклонение рассчитанного значения наклона градуировочной зависимости (standard deviation of the slope; sh). Параметр, характеризующий воспроизводимость расчета наклона найденной регрессионной зависимости, находят по уравнению
Ж
s2 = 2=-----------------г = ^5>,2 (2.63)
mS(x,-x)2 m^-Q»2 т
ms2y
№*2'
(2.67)
со степенями свободыf = т-2.
Дисперсию sy, характеризующую рассеяние результатов относительно прямой, вычисляют по формуле
' (т-2) (т-2) ( '
со степенями свободы f = т-2.
Однако, если для каждой из т проб проводят по к, параллельных измерений, так что имеется mkt = т' результатов измерений, то с уравнением (2.64) будет связано не f = = т-2, & f=m'-2, которое и будет в знаменателе.
* Если а мало отличается от 0, то уравнение может иметь вид у = Ь'х. См. проверку значимости константы а.
Стандартное отклонение рассчитанного значения отрезка, отсекаемого на оси ординат (standard deviation of the intercept; sa). Параметр, характеризующий воспроизводимость расчета отрезка, отсекаемого найденной регрессионной зависимостью на оси ординат, находят по уравнению
(2.68)
Границы доверительного интервала рассчитанного значения наклона регрессионной зависимости (confidence limits about the slope; ± AZ>). Границы доверительного интервала, соответствующего доверительной вероятности Р, рассчитывают по уравнению
Метрологические основы методов количественного анализа
73
M) = tPf-sb (2.69)*
при/= т - 2, или /= т' - 2.
Границы доверительного интервала для рассчитанного значения отрезка, отсекаемого на оси ординат (confidence limits about the intercept; : Да). Границы доверительного интервала, соответствующего доверительной вероятности Р, рассчитывают по уравнению
Да - tP f • sa (2.70)
приf= т-2, или /= т' -2.
Зная Да и Л/?, определяют число необходимых знаков после запятой для констант а и Ь.
Вычисление метрологических характеристик результата анализа:
xw, 51ан, Дх^.
После того как определена функция зависимости у = а + + Ьх и рассчитаны значения а, Ь, Ла, ЛЬ по данным измерения аналитического сигнала у анализируемых проб, рассчитывают метрологические характеристики результата анализа.
Среднее значение результата анализа (хан). Проба определяемого компонента с неизвестным содержанием — оно должно быть в интервале содержаний, для которых вычислена функциональная зависимость у = fix) — анализируется п раз (обычно п > 3). При этом получают п значений аналитического сигнала у^у^, У;ш В случае подозрительно выделяющегося результата ут осуществляют его проверку по ^-критерию (п. 2.3.5). Рассчитывают среднее ут, а по уравнению
ь
находят среднее значение содержания компонента в анализируемой пробе.
Стандартное отклонение результата анализа. В случае градуировочного графика вида у = а + Ьх погрешность метода анализа состоит из трех частных погрешностей, обусловленных погрешностями констант а и Ь, и значения Уан- Эти три погрешности суммируются по закону распределения погрешностей. Если каждый из т (стандартных растворов) анализируется без повторений, а проба с неизвестным содержанием — и7 раз и для этих и7 проб среднее
_ 2л„, _ 2^-
значение у =-------, а для эталонов у = ——, то
и, m
1 , 1 । fa. "У)* =
", т
* В данном тексте используются индексы границ доверительного интервала Ла, ЛЬ, Лх^, АУ как более традиционные по сравнению с рекомендованными ИЮПАК [15] -Са, Св, Сх, Су.
Значения tP,f берут из табл. 2.3 при Р = 0,95.
(2.72)
или:
Анализ уравнения (2.73) показывает, что стандартное отклонение результата анализа тем меньше:
а) чем круче градуировочный график (больше коэффициент регрессии);
б) чем больше число измерений т при построении градуировочного графика. В общем случае т должно быть не менее 5. При фотометрических определениях погрешность измерения у (оптической плотности А) зависит еще и от абсолютного значения оптической плотности. Ввиду этого в области высоких значений погрешности оптических плотностей следует предусмотреть дополнительные стандартные растворы;
в) меньше разность значений ут - у. Она будет наименьшей при уш = у . При больших различиях уш и у значения s- возрастают. Поэтому градуировочный график можно использовать только в той области содержаний (концентраций), где лежат эталонные пробы. Интервал содержаний (концентраций), для которого строится градуировочный график, не должен быть слишком растянут.
Границы доверительного интервала результата анализа Дх^ (границы доверительного интервала для рассчитанного значения независимой переменной — confidence limits about the fitted value of the independent variable; ±CX [15])
tew=tPJ-sXm (f=m-2). (2.74)
Доверительный интервал результата анализа находится по формуле
*ан±АХан- (2-75)
Границы доверительного интервала для рассчитанного значения зависимой переменной (confidence limits about the fitted value of dependent variable; ±Cy). Полученную функцию у = a + bx можно использовать для расчета Yk и ЛУк для одного значения хк. Границы доверительного интервала для вычисления значения Yk рассчитывают по формуле
(2.76)
* Если проводят параллельные измерения для каждого из т стандартных растворов и число параллельных измерений равно кр то в уравнении вместо т берется т' - mkj Hf=m’-2.
74
Новый справочник химика и технолога
Из уравнения (2.76) следует, что границы доверительного интервала зависят от разницы (хк -х) и будут тем больше, чем дальше х* лежит от среднего значения х . Поэтому экстраполяция даже при наличии линейной связи может сопровождаться большими погрешностями.
Из градуировочного графика можно непосредственного определить предел обнаружения [22].
Пример расчета [22 б]
Для построения градуировочного графика при фотометрическом определении бензола в УФ-области измерены оптические плотности семи стандартных растворов. Полученные результаты приведены в табл. 2.8. Далее проанализированы две серии двух растворов неизвестной концентрации бензола в спирте (по три параллельные пробы). По экспериментальным данным рассчитаны средние значения Уан(Лн) = 1^2 (для первой серии) и ^ан(лан) = 0,93 (для второй серии). Рассчитать значения xw и Дх^.
Таблица 2.8
Вычисление коэффициентов регрессии
Расчет характеристик линейного графика
Вычисление коэффициентов регрессии удобно проводить в табличной форме, где приводятся результаты расчетов сумм х, у, и средних значе-
ний у и х :
х=^- = 1,53; у = о,951; (Ух,У =114,49. п п ' 7
Проверку правильности вычислений осуществляют по уравнению
£(*,)2 л+£л2- (2-77)
Значения сумм из табл. 2.8 подставляют в уравнение (2.77). При условии правильности вычислений значения сумм левой и правой частей уравнения будут равны и можно будет приступить к расчету констант а и b по уравнениям (2.60) и (2.61) или (2.60') и (2.61'), подставляя в них найденные значения для сумм из табл. 2.8.
№ п/п Концентрация Х(С,), г/л Оптическая плотность yi(At) х2 у2 х,у, х. + У, (*+*)’
1 0,2 0,20 0,04 0,0400 0,040 0,40 0,1600
2 0,5 0,37 0,25 0,1369 0,185 0,87 0,7569
3 1,0 0,64 1,00 0,4096 0,640 1,64 2,6896
4 1,5 0,93 2,25 0,8649 1,395 2,43 5,9049
5 2,0 1,22 4,00 1,4884 2,440 3,22 10,3684
6 2,5 1,50 6,25 2,2500 3,750 4,00 16,0000
7 3,0 1,80 9,00 3,2400 5,400 4,80 23,0400
1=’ 10,7 х=1,53 6,66 у = 0,951 22,79 8,4298 13,850 17,36 58,9198
Примечание. Согласно уравнению (2.77) и табличным данным: 22,79 + 2 • 13,850 + 8,4298 = 58,9198;
58,9198 = 58,9198, т.е. вычисления в табл. 2.8 выполнены правильно.
Из уравнений (2.60), (2.61) вычисляют:
7-13,850-10,7-6,66 7-22,79-114,49
= 0,570337;
6,66-0,570337-10,7
а =------------------
7
= 0,079628.
По уравнению (2.65) рассчитывают сумму квадратов для дисперсии, характеризующей отклонение измеренных значений^/ относительно вычисленных У,:
^(у, - Y,)2 = 8,429800-0,079628 • 6,66 -
-0,570337 • 13,850 = 0,000311;
х = °’00031 = о,0000622; sv = 0,007887.
у 5
Из уравнений (2.62) и (2.63) находят дисперсии для констант b и а:
2 = 7-0,0000622 = 0 00000967 = 0 003! j (Су*= 5);
ь 7-22,79-114,49 7
2 = 0,0000622 22,79 = 0 00003147. s = 0 00561 (с /= 5).
а (7-22,79-114,49) V J ’
При Р = 0,95 tp.f = 2,57 (см. табл. 2.3). Отсюда AZ> =tp,f-Sb = = 0,00799 и &а = tp/ • sa = 0,014. Следовательно, при Р = = 0,95 b = 0,570 ± 0,008 (= чувствительность); а = 0,079 ± ± 0,014 (= фон).
Метрологические основы методов количественного анализа
75
Расчет характеристик результатов анализа
Для серии растворов ут (Аж ) = 1,53, согласно уравнениям (2.73)-(2.75), вычисляют среднее значение результата анализа:
1,533-0,070 0,570
= 2,55 г/л.
При Р - 0,95 и /= 5 границы доверительного интервала для Хан равны
0,0000622
А- _ 2’57
“ 0,570Х
7|"1,533-—^
1 1 у 7 J
3 7 0,5702 (7-22,79-114,49)
= 0,028 г/л.
Следовательно, доверительный интервал при Р = 0,95 равен 2,55 ± 0,03 г/л.
Аналогичные расчеты для второй серии растворов Ян(^ан) = 0,93 дают значение х^ = 1,39 г/л. Так как в этом случае ® у , то третий член под корнем уравнения (2.72) мал. При Р - 0,95 абсолютная погрешность анализа становится меньше (Д хт = 0,02 г/л), и доверительный интервал равен 1,39 ± 0,02 г/л.
Проверка значимости константы а
В ряде случаев значение а настолько мало, что этим членом уравнения прямой можно пренебречь. При зависимости v = Ьх вычисления значительно упрощаются. Тогда:
(2.76)
(5.)! = Хи Д (с/= «-!); (2.77)
' 7 т-1
Se' = Z(?, -Г, / =(</(т-1) = £к-Ь^у;, (2.78)
(2.79)
(2.80)
В ситуации, когда значение а уравнения^ = а + Ьх мало отличается от нуля и возникает предположение, что уравнение может иметь вид у = Ь'х, осуществляют проверку константы а по следующей схеме. Вычисляют сумму квадратов SQ по (2.65) и SQ по (2.78) соответственно при
/= т- 2 uf = т- 1 степенями свободы. При этом всегда SQ > SQ. Затем находят соотношение:
F=^SQ. (2.81)
Sy
Здесь sy — дисперсия, вычисленная по уравнению (2.64).
Сравнивают значения F и F(p,f} =1, /2 = т-2^ (табл. 2.7). Если F < то имеются основания
для перехода к уравнению у = Ь'х.
Пример
На основании данных фотометрических определений рассчитано уравнение градуировочного графика^ = а + Ьх. Требуется проверить: возможно ли более простое уравнение вида v = Ь'х.
Имеются следующие данные:
у = 0,101 + 115,567%; ^= 119,625%;
SQ = s2y (т-2) = 0,4107; SQ' = (s'y)' (т-1) = 0,4682; т = 19.
Результаты представляют по схеме:
SQ f 4
У = Ь'х 0,4682 18
у = а + Ьх 0,4107 17 0,0242
Разность 0,0575 1
F = д’Q242 = 2'^ • Из Данныхтабл. 2.7 получаютF(P = = 0,95,/ = 1,/ = 17) = 4,45. Следовательно, между обеими дисперсиями: sy - 0,0242, вычисленной по уравнению (2.64) и!sy) = 0,0260 — по уравнению (2.77) — нет значимой разницы, поэтому переход к уравнению^ = Ь'х допустим.
Проверку гипотезы линейности градуировочного графика можно найти в [22, 23, 56].
2.5. Термины по представлению результатов химического анализа (в алфавитном порядке) [15]
воспроизводимость выборочное стандартное отклонение precision estimated standard deviation
выброс границы доверительного интервала границы доверительного интервала для рассчитанного значения зависимой переменной outlier confidence levels about the mean confidence limits about the fitted value of the dependent variable
границы доверительного интервала для рассчитанного значения наклона регрессионной зависимости confidence limits about the slope
76
Новый справочник химика и технолога
границы доверительного интервала для рассчитанного значения независимой переменной
границы доверительного интервала для рассчитанного значения отрезка, отсекаемого на оси ординат дисперсия
доверительный интервал зависимая переменная измеренное значение истинная величина коэффициент вариации коэффициент корреляции математическое ожидание медиана
минимально обнаруживаемое содержание
минимальный значимый сигнал
мода
наклон градуировочного графика независимая переменная
объем выборки остаточная сумма отклонений
отклонение
относительная погрешность относительная погрешность в процентах
относительное стандартное отклонение
относительное стандартное отклонение в процентах отрезок, отсекаемый на оси ординат
параллельные измерения
переменная
погрешность результата полная систематическая погрешность
пороговое значение критерия правильность
предел обнаружения повторяемость
размах выборки рассчитанное значение независимой переменной рассчитанное значение зависимой переменной Результат серия
среднее арифметическое
среднее взвешенное
среднее гармоническое
confidence limits about the fitted value of the independent variable confidence limits about the intercept
variance
confidence interval dependent variable measured value true value
coefficent of variation correlation coefficient population mean median
minimum detectable quantity
minimum significant signal
mode
slope
independent variable
sample size
residual standard de-
viation
deviation
relative error percentage relative error
relative standard deviation
percentage standard deviation
intercept
replicate measurements
variable
error of result bias
decision threshold
accuracy
detection limit
repeatability
range
estimated value of the independent variable estimated value of the dependent variable result
series
arithmetic mean weighed mean harmonic mean
среднее геометрическое geometric mean,
(логарифмическое) logorithmic mean
среднее квадратичное quadratic mean
среднеквадратичное отклонение root mean square deviation
среднее значение average
стандартная погрешность standard error
стандартное отклонение standard deviation
стандартное отклонение standard deviation
для групп данных from grouped data
стандартное отклонение для пар standard deviation
данных from paired data
стандартное отклонение повто- reproducibility
ряемости standard deviation
стандартное отклонение рассчи- standard deviation
танного значения наклона градуировочной кривой of the slope
стандартное отклонение рассчи- standard deviation
танного значения отрезка, отсекаемого на оси ординат of the intercept
стандартное отклонение repeatability standard
сходимости deviation
стандартное отклонение точек standard deviation
от найденной зависимости of points about the fitted line
суммарное стандартное pooled standard
отклонение deviation
сходимость repeatability
уравнение функциональной equation for calibra-
(градуировочной) зависимости tion relation
уровень доверительной вероятности confidence level
уровень значимости significance level
устойчивая статистика robust statistics
число измерений number of observations
число степеней свободы degrees of freedom
2.6. Предел обнаружения.
Диапазон определяемых содержаний.
Нижняя граница определяемых содержаний
Возможности методов анализа по обнаружению малых количеств и малых концентраций лучше всего выражаются через статистический термин предел обнаружения (detection limit-DL) [4, 22, 23, 58 69]. Предел обнаружения (сыт Р или смин. fc) — наименьшее содержание, при котором по данной методике можно обнаружить присутствие определяемого компонента с заданной доверительной вероятностью Р или при коэффициенте достоверности к, численное значение которого выбирается в соответствии с уровнем доверительной вероятности [58].
Существуют и другие варианты определения предела обнаружения.
Понятие предела обнаружения, как следует из определения, относится к области качественного анализа и определяет минимальное количество щмии (или концентрацию
Метрологические основы методов количественного анализа
77
смин) компонента, которое может быть обнаружено с высокой заданной вероятностью.
Анализ литературных данных, посвященных проблеме оценки предела обнаружения элементов различными методами, показывает, что расчетные значения этого параметра для одного элемента даже при работе одним методом могут колебаться в пределах порядка и более. Обусловлено это рядом причин: степенью корректности постановки холостого опыта; степенью учета влияния примесей в реактивах; недостаточным вниманием к многочисленным погрешностям, источники и значения которых также изменяются с изменением материала пробы и условиями анализа; отсутствием единого мнения о значении доверительной вероятности, которое должно приниматься при расчетах, о методике учета сигнала фона (холостого опыта) — у(|)0Н (ухол) и методике расчета стандартного отклонения фона, которые входят в расчетные уравнения, принятые ИЮПАК:
Лин^фон+^Д (2.81)
Умкя-У^=^ (2-82)
^мин, к (ИЛИ Смин />) f (у'фон).
(2.83)
Здесь умин — минимальный аналитический сигнал, который еще может быть измерен; уфон— среднее значение сигнала холостого (контрольного) опыта, рассчитываемое из числа определений ЛфОН(Пхол) > 20; s — стандартное отклонение сигнала холостого (контрольного) опыта:
S
1
где J мин _ 37фон — минимальный разностный аналитический сигнал.
В литературе применяются различные варианты обозначения величин, входящих в уравнение (2.81) и (2.83). ИЮПАК [65] рекомендует использовать обозначения, предложенные Кайзером [59 а]:
х = + к|sK0IITp | при с = f (х), (2.84)
которые в смысловом значении полностью идентичны символам уравнений (2.81), (2.83).
Использование значения к = 3 обеспечивает уровень доверительной вероятности Р = 99,86%; в этом случае учин - (Уфон + 3^ ) при условии, что случайные погрешности определения сигнала y^H подчиняются закону нормального распределения. Однако распределение значений у/фм вблизи предела обнаружения может не следовать закону нормального распределения. В этом случае, согласно
неравенству Чебышева, вероятность того, что Л™ (Уфон + 3 V ) * **> Окажется равной 89 %.
Следовательно, для расчетов предела обнаружения нельзя использовать к < 3.
Количественно предел обнаружения можно определить, пользуясь выражением
X
=-?=-. (2.85)
Л
где sy^ — стандартное отклонение аналитического сигнала фона (холостого опыта); S — коэффициент чувствительности (см. п. 2.2).
Существуют и другие способы расчета предела обнаружения, но уравнение (2.85) используют чаще всего. Методики расчета см., например, в [22, 23 (с. 61-68, 317— 319)]. Многочисленные сводные данные по пределам обнаружения следов металлов и неметаллов спектроскопическими методами приведены в монографии [66].
В работе [69] рассмотрены вопросы, связанные с пределом обнаружения в вольтамперометрических методах.
В количественном химическом анализе обычно приводят диапазон определяемых содержаний — область значений определяемых содержаний, предусмотренная данной методикой и ограниченная нижней и верхней границами определяемых содержаний. Верхняя граница (тв, св) — наибольшее значение количества или концентрации компонента, определяемое по данной методике. Оно ограничено, как правило, изученным интервалом либо возможностью измерения аналитического сигнала с достаточной точностью.
Область применимости любого метода при определении следов элементов определяется нижней границей определения содержаний (тн, сн) — наименьшим содержанием компонента, определяемого по данной методике. В литературе приводится много способов расчета сн например, согласно [65] аналитический сигнал рассчитывается из уравнения (2.84), но при к = 10.
Обычно за нижнюю границу определяемых содержаний принимают то минимальное количество или концентрацию, которые можно определить с относительным стандартным отклонением sr < 0,33.
В табл. 2.9-2.17 приведены пределы обнаружения ряда экологически опасных элементов спектрохимическими
* Разностный аналитический сигнал Ay = у„ т — у. = 3s,t •s г МИН фин лхол
(или х — Хконтр = 3sKomp) принимается за минимально обнаруживаемый. Если Ay < 3 s — элемент не обнаруживается, если 10s > Ay > 3s„ — элемент возможно обнаружить только
.Уфой ./фон
качественно, а при Ay > lOs^, возможно количественное определение элемента.
** В англоязычной литературе синонимом можно считать предел определения. Сводные данные по пределам определения многих элементов рядом физических методов приведены на компакт-диске.
78
Новый справочник химика и технолога
методами в воде, клинических и биологических материа- FAAS пламенная атомно-абсорбционная
лах [66]. Таблицы по пределам обнаружения элементов в воде методом атомно-эмиссионной спектрометрии с ин- FAES спектрометрия пламенная атомно-эмиссионная
дуктивно-связанной плазмой — с пневматическим распылением, ультразвуковым распылением и электротер- FI спектрометрия проточная инжекция
мическим испарением — представлены в [74]. На рис. FIA проточно-инжекционный анализ
2.4.-2.7 приведены пределы определения элементов раз- GF графитовая печь
личными спектроскопическими методами [66]. HDEHP диэтилгексилфосфорная к-та
Таблицы пределов обнаружения ICP ID INAA индуктивно связанная плазма изотопное разбавление инструментальный нейтронно-
некоторых экологически опасных элементов спектрохимическими методами MIBK активационный анализ метилизобутилкетон
Обозначения методов MIP MS микроволновая индукционная плазма масс-спектрометрия
AAS атомно-абсорбционная спектрометрия NAA нейтронно-активационный анализ
AES атомно-эмиссионная спектрометрия ND недисперсионная
AFS атомно-флюоресцентная спектрометрия QF кварцевая печь
APDT пирролидиндитиокарбамат аммония RNAA радиохимический нейтронный
CF непрерывный поток CRM сертифицированный образец (материал) SSMS активационный анализ масс-спектрометрия вторичных ионов
сравнения TBP трибутилфосфат
CV холодный пар TI термическая ионизация
DBDTC дибензилдитиокарбамат TPhP трифенилфосфат
DCP прямой поток плазмы VIS спектрофотометрия (фотометрия)
DDTC диэтилдитиокарбамат ЕТА электротермическая атомизация XRF в видимой области рентгенофлуоресцентный анализ
Таблица 2.9
Аналитические методы определения общей ртути в воде [66]
Образец (количество) Разделение и/или предконцентрирование Метод определения Предел обнаружения, нг • л-1 Литература
Озерная, речная, минеральная вода (60 мл) Электроосаждение на Pt-электроде GFAAS 40 79
Озерная, дождевая вода (50 мл) Восстановление SnCl2, амальгамирование с Ап GF AAS 0,1 22
Озерная вода (50 мл) Восстановление SnCl2, амальгамирование с Au MIP AES 0,01 11
Вода (200 мл) Восстановление SnCl2, амальгамирование с Au ID ICP MS 0,2 23
Морская, речная вода (100 мл) Восстановление SnCl2, амальгамирование с Au Кольцевой разряд AFS 0,5 12
Морская вода, вода из дельты реки, дождевая вода (2 л) Восстановление SnCl2, амальгамирование с Au GF CV AAS 0,04 13
Морская и сточная воды (50 мл) Восстановление NaBH4, улавливание в графитовой печи (GF), покрытой Pt GF CV AAS 0,6 60
Болотная вода (100мл) Выпаривание с HNO3-H2SO4-KMnO4-K2S2O8; восстан. SnCl2 в H2SO4 или NaBH4 в NaOH, улавливание в GF, покрытой Pt GF CV AAS * нг 23
Метрологические основы методов количественного анализа
79
Продолжение таблицы 2.9
Образец (количество) Разделение и/или предконцентрирование Метод определения Предел обнаружения, нг л-1 Литература
Озерная, речная вода Восстановление NaBH4 ID ICP MS 8** 34
Сточная вода Восстановление NaBH4 после окисления ртутьорганического соединения с S?Os? в присутствии Cd2+ CV AAS 150 36
Вода Восстановление SnCl2 FI CV AFS 180 26
Сточная вода Восстановление SnCl2 в NaOH в присутствии Cu(II) и K2S2O8 CV AAS 100
Примечания* Нанограммовые количества (1 нг = 10 9 г).** Абсолютный предел обнаружения, пг (1 пг = 10 12 г).
Литература к табл. 2.9
[79] Хи В. et al. // Taianta. 1985. V. 32. Р. 1016.
[22] Lee S.H., Jung К. and Lee D.S. // Taianta. 1989. V. 36. P. 999.
[ll]Nogiri Y., Otsuki A. and Fuwa К. И Anal. Chem. 1986. V. 58. P. 544.
[29] Smith R.G. // Anal. Chem. 1993. V. 65. P. 2485.
[12]Wrembel H.Z. И Spectrochim. Acta. 1986. V. 41B. P. 247.
[13] Gill G.A. and Fitzgerald W.F. // Mar. Chem. 1987. V. 20.
P. 227.
[60] Yan X., Ni Z. and Guo Q. // Anal. Chim. Acta. 1993. V. 272. P. 105.
[23] Baxter D.C. and Freeh W. И Anal. Chim. Acta. 1990. V. 236. P. 377.
[34] Haraldsson C., Westerlund S. and Ohman P. И Anal. Chim. Acta. 1989. V. 221. P. 77.
[36] Munaf E., Takeuchi T., Ishii D. and Haraguchi H. // Anal. Sci. 1991. V. 6. P. 605.
[26] Morita H., Sugimoto M. and Shimomura S. // Anal. Sci. 1990. V. 60. P. 91.
[17] Goto M., Munaf E. and Ishii D., Fresenius' Z. И Anal. Chem. 1988. V. 332. P. 745.
Таблица 2.10
Методы определения алюминия в воде и жидкостях для диализа [66]
Образец (количество) Способ подготовки образца Метод определения Предел обнаружения, мкг • мл-1 Литература
Сточная вода Экстракция HDEHP (расплавом жирных КИСЛОТ С]7~•С20) XRF 0,5 81
Водопроводная вода, жидкость для диализа Обмен F', Г анионов, элюирование 1 М NaOH ICP AES 0,02 11
Жидкость для диализа (spiked) Анионный обмен, элюирование 1 М NaOH или FI Chelex-100, элюирование 2,5 М NaOH ICP AES 0,003 5
Поверхностные морские слои, речная вода (250-500 мл) Сорбция на смоле, иммобилизированной 8-оксихинолином, элюирование HC1-HNO3 GF AAS 0,08** 16
Речная и морская вода Сорбция на 8-оксихинолине, на CPG*, элюирование HC1-HNO3 FAAS 0,003 13 15
Речная вода, жидкость для ' диализа Анионный обмен pH = 6,5, элюирование 0,1 М НО DCP AES нг 6
Водопроводная вода Сорбция на Хромотропе 2В, нанесенном на анионообменник, pH = 6, элюирование 0,01 М НО VIS *** нг 8
80
Новый справочник химика и технолога
Продолжение таблицы 2.10
Образец (количество) Способ подготовки образца Метод определения Предел обнаружения, мкг • мл”1 Литература
Питьевая вода Анионный обмен А1- Пирокатехинового фиолетового комплекса, pH = 7, элюирование 0,1 М НС1 DCP AES *** нг 7
Примечания* CPG — стекло с контролируемыми порами. ** пг • мл \ *** Нанограммовые количества, нг (1 нг = 10 9 г).
Литература к таблице 2.10
[81] Zebreva A.I. et al. И Anal. Chim. Acta. 1987. V. 195. P. 357.
[11] Pereiro Garcia M.R. et al. H J. Anal. At. Spectrom. 1987. V. 2. P. 699.
[5] Pereiro Garcia M.R. et al. 11 J. Anal. At. Spectrom. 1990. V. 5.P. 5.
[16] Shiller A.M. 11 Geochim. Cosmochim. Acta. 1988. V. 52. P. 1879.
[13] Mohammad B., Ure A.M. and Littlejohn. // J. Anal. At. Spectrom. 1992. V. 7. P. 695.
[15] Mohammad B., Ure A.M. and Littlejohn. И J. Anal. At. Spectrom. 1993. V. 8. P. 325.
[6] Sarzanini C. et al. И Anal. Chem. 1985. V. 57. P. 1960.
[8] Pesavento M., Profumo A., Riolo C., Soldi T. // Analyst. 1989. V. 114. P. 623.
[7] Sarzanini C. et al. // Anal. Chem. 1987. V. 59. P. 484.
Таблица 2.11
Методы определения урана в природной воде [66]
Вода (количество) Разделение и/или предконцентрирование Метод определения Предел обнаружения, нг • мл”1 Литература
Природная (1л) Сорбция на ТВР, нанесенном на полиуретановую пену VIS 0,05 20,52
Природная (нг) Экстракция А-и-метоксифенил-2-фурилакрилгидроксамовой кислотой (СНС13 и МИБК-метилизобутил кетон) VIS, FAAS 5-6 7
Морская (0,2 л), водопроводная (0,5 л) Соосаждается с ПАН [1-(2-пиридилазо)-2-нафтил] INAA 0,004 48
Морская (1 мл) Анионный обмен TIMS 2 24
Морская (3 мл) Анионный обмен, соосаждение с Fe(OH)3, анионный обмен TIMS 24
Вода из скважины, морская, речная вода (0,25 мл) 20-кратное разбавление ID ICP MS 0,04 32
Сточная вода (200 мл) Сорбция на хелатном волокне; десорбция HNO3 ICP AES 5 51
Вода из скважины (1 мл) Анионный обмен, соосаждение с Fe(OH)3, обмен анионов, на электроосаждение a-spec 0,0003 16
Вода из источника (4 мл), почвенная (0,1 г) SFE с ТТА*-ТВР RNAA Hr 14
Сточная вода (200 мл) Сорбция на хелатном волокне; десорбция HNO3 ICP AEF 51
Примечание.* [-2-тиеноалтрифторацетон].
Метрологические основы методов количественного анализа
81
Литература к таблице 2.11
[20] Aziz М., Beheir S.G. and Shakir К. // J. Radional. Nucl. Chem. 1993. V. 172. P. 319.
[52] Shakir K., Aziz M. and Beheir S.G. // J. Radional. Nucl. Chem. 1992. V. 162. P. 227.
[7] Kamata E., Nakashima R. and Furukawa M. // J. Anal. At. Spectrom. 1987. V. 2. P. 321.
[48] Beam H. and Ryan D.E. // Anal. Chim. Acta. 1984. V. 158. P. 119.
[24] Chen J.H., Edwards R. L. and Wasserburg G.J. // Earth Plan. Sci. Lett. 1986. V. 80. P. 241.
[32] Toole J., McKay K. and Baxter M. // Anal. Chim. Acta. 1991. V. 245. P. 83.
[17] Barbous A.A. // Geochim. Cosmochim. Acta. 1994. V. 58.P. 4591.
[16] Cochran J.K. et al. // Geochim. Cosmochim. Acta. 1986. V. 50. P. 663.
[14] Lin Y. et al. // Environ. Sci. Technol. 1994. V. 28. P. 1190.
[51] Chang X. et al. // Mikrochim. Acta. 1990. V. 1. P. 101.
Таблица 2.12
Аналитические методы определения мышьяка в воде [66]
Вода (количество) Разделение и/или предконцентрирование Метод определения Предел обнаружения, нг • мл-1 Литература
СО В виде летучего AsH3, отделение на мембране ICPMS 0,5 13
СО (10 мл) В виде летучего AsH3 ICPMS 1,5 93
СО поверхность (100 мл) Осаждение с DBDTC NAA 0,02 48
СО морская (10 мл) В виде летучего AsH3, улавливание в графитовой печи (GF) MIPAES 0,012 7
СО морская (0,5 мл) В виде летучего AsH3, улавливание в GF GF AAS 0,060 6
Морская (200-500 мл) Осаждение с Se XRF 1 55
Морская (300 мл) Экстракция APDC (СНС13), разложение с HNO3-HC1O4, AsH3 QF AAS 0,03 44
Морская, речная, из источника (100 мл) Экстракция APDC (СНС13), реэкстракция в HNO3 NAA 0,01 45
Поверхностная, прибрежная (100 мл) Соосаждение Аз(У)-молибдата с TPhP • Cl NAA нг* 50
Поверхностная, прибрежная (100 мл) Соосаждение As(III) с DBDTC NAA нг* 50
Речная В виде летучего AsH3 QF AAS 0,6 26
Речная вода (0,25-0,5 л) В виде летучего AsH3 QF AAS 5 107
Питьевая (5 мл) В виде летучего AsH3 DCP AES 0,6 21,23
Питьевая, минеральная (10 мл) В виде летучего AsH3 FI FAAS нг* 20
Минеральная (50 мл) В виде летучего AsH3, улавливание ратвором Ce(IV)-KI GF AAS 0,06 9
Минеральная (5-25 мл) В виде летучего AsH3 AAS 20 11
Ключевая (100 мл) Осаждение As( У)-молибдата с ТРеР. Вг ICP AES 0,3 53
Грунтовая (1 л) Соосождение As(III и V) на Th(OH)4, в виде летучего AsH3 VIS нг* 108
Сточная Сорбция As(V) на носителе, модифицированном Os2SnCl2, элюирование 2 М НС1 FAAS GF AAS 10 109
Примечание* Нанограммовые количества.
82
Новый справочник химика и технолога
Литература к таблице 2.12
[13] Branch S. et al. // J. Anal. At. Spectrom. 1991. V. 6. P. 155.
[93] Haraldsson C., Pollak M. and Ohman P. // J. Anal. At. Spectrom. 1992. V. 7. P. 1183.
[48] van Elteren J.T. et al. // Anal. Chim. Acta. 1989. V. 222. P. 159.
[7] Matusiewicz H. and Sturgeon R.E. // Spectrochim. Acta. 1990. V. 45B. P. 209.
[6] Sturgeon R.E., Willie S.N. and Berman S.S. I I J. Anal. At. Spectrom. 1986. V. l.P. 115.
[44] Amankwah S.A. and Fasching J.L. // Taianta. 1985. V. 32. P. 111.
[45] Mok W.M., Shah N.K. and Wai C.M. H Anal. Chem. 1986. V. 58. P. 110.
[50] van Elteren J.T. et al. H Anal. Chim. Acta. 1991. V. 252. P. 89.
[26] Narasaki H. 11 J. Anal. At. Spectrom 1988. V. 3. P. 517.
[107] Narasaki H., Kato Y. and Kimura H. H Anal. Sci. 1992. V. 8. P. 893.
[21] Brindle I.D. et al. H Analyst. 1992. V. 117. P. 407.
[23] Chen H., Brindle I.D. and Le X.C. H Anal. Chem. 1992. V. 64. P. 667.
[9] Tsalev D.L., Mandjukov P. B. and Statis J.A. H J. Anal. At. Spectrom. 1987. V. 2. P. 135.
[11] Veber M., Cujes K. and Gomiscek. 11 J. Anal. At. Spectrom. 1994. V. 9. P. 285.
[53] HataN. et al. //Analyst. 1989. V. 114. P. 1255.
[108] Tamari Y. et al. H Anal. Sci. 1989. V. 5. P. 481.
[109] Russeva E., Havezov I. and Detcheva A., Fresenius' Z // J. Anal. Chem. 1993. V. 347. P. 320.
Таблица 2.13
Определение следов кадмия в воде [66]
Вода (количество) Предконцентрирование и/или разделение Метод определения Предел обнаружения, нг • л-1 Литература
Морская (0,35 л) Экстракция DDCT-APDC (фреоном TF), реэкстракция (HNO3) GF AAS 3,5 8
Морская (0,2-2 мл) On-line сорбция APDC комплекса на С! 8-колонке GF AAS 1,3 20
Морская Сорбция на Chelex-100, pH = 6,5, элюирование 2М HNO3 GF AAS * нг 15
Морская (0,5 л) Соосаждение Cd-DBDTC-комплекса фенолфталеином INAA * нг 77
Морская, речная (0,2-0,5 л) Соосаждение с ПАН GF AAS 1,5 18
Речная (0,05 л) Катодное осаждение на W-проволоке ETA AAS 10 25
Речная (0,1 л) Сорбция на модифицированной пиро-катехинфиолетовом или на смоле с ксиленоловым оранжевым FAAS * нг 16
Озерная (2 л) Анионный обмен в виде хлоридного комплекса, элюирование 2 М HNO3 FAAS 5 21
Вода (0,1 л) Соосаждение с 1п(ОН)3 GF AAS 2 91
Вода (0,03 л) Катодное электроосаждение ID TIMS 1 24
Водопроводная (1л) Сорбция на полидитиокарбаматной смоле, элюирование HNO3 FAAS 30 14
Сточная вода Сорбция на иминодиацетатной смоле, элюирование 1 М НС1 ICP AES 50 12
Примечание. Нанограммовые количества.
Литература к таблице 2.13
[8] Statham Р. J. // Anal. Chim. Acta. 1985. V. 169. Р. 149.
[20] Liw Z. and Huang S. // Anal. Chim. Acta. 1993. V. 281.
P. 185.
[15] Pai S. and Chen H. // Mar. Chem. 1994. V. 47. P. 81.
[77] Saleh A.I., Remail S.W. and Milard F.M. // J. Radianal.
Nucl. Chem. 1993. V. 168. P. 23.
[18] Taguchi S. et al. //Analyst. 1988. V. ИЗ. P. 1695.
Метрологические основы методов количественного анализа
83
[23] Majidi V. and Halcombe J.А. // J. Anal. At. Spectrom. 1989. V. 4. P. 439.
[25] Zhang G. et al. // Taianta. 1993. V. 40. P. 409.
[16] Sing A.K. and Dhingra S.K. // Analyst. 1992. V. 117. P. 889.
[21] Tao S. et. al // Analyst. 1994. V. 119. P. 1455.
[91] Hiraide M., Usami T. and Kawaguchi H. // Anal. Sci. 1992. V. 8.P. 31.
Определение <
[24] Trettenbach J. and Heumann K.G. // Fresenius' Z. Anal. Chem. 1985. V. 322. P. 306.
[14] Yebra-Biurrum M.C. et al. // Analyst. 1991. V. 116. P. 1033.
[12] Kumamura T. et al. // Anal. Chim. Acta. 1986. V. 181. P. 271.
Таблица 2.14
ена в воде |66]
Вода (количество) Приготовление образцов Метод определения Предел обнаружения, нг • л Литература
CO морская Выпаривание с H2SO4-HC1O4, в виде летучего H2Se ICP AES 400 105
CO морская (100 мл) В виде летучего H2Se ICP MS 2,5 38
CO (10 мл) В виде летучего H2Se ICP MS 6 1
CO морская (300 мл) Сорбция на Cis в виде APDC комплекса, элюирование МеОН GF AAS 7 134
СО морская (20-50 мл) В виде летучего H2Se, улавливание в графитной печи (GF) GF AAS 1,5 37
СО морская В виде летучего H2Se ND AFS 27а* 16
СО морская, (5 мл) В виде летучего H2Se ND AFS ♦ нг 17
Морская (0,25-0,5 л) Соосаждение с La(OH)3, в виде летучего H2Se QF AAS 8 46
Свежая (100 мл) Анионный обмен после разложения КМпО4 при pH = 2, в виде летучего H2Se FI AAS 5 135
Свежая Соосаждение с DBDTC и фенолфталеином IN A A 48
Свежая (250 мл) Осаждение Se° или в виде летучего H2Se IDTI MS 10 11
Речная (50-500 мл) Выпаривание с HNO3- H2SO4-HC1O4, в виде летучего H2Se QF AAS нг* 29
Речная В виде летучего H2Se QF AAS 1000 34
Озерная, сточная В виде летучего H2Se, улавливание в GF GF AAS нг 3
Питьевая, минеральная, речная, дождевая В виде летучего H2Se, улавливаниие на Cromosorb W QF AAS 6** 31
Водопроводная вода, вода из скважины (6,7 мл) On-line соосаждение с La(OH)3, в виде летучего H2Se FI AAS 1 47
Почвенная вода (200 мл) Анионный обмен Se(IV) и Se(VI) ID TIMS 40 112
Минеральная (5-25 мл) В виде летучего H2Se QF AAS ♦ нг 136
Примечания. * Нанограммовые количества. ** Абсолютный предел обнаружения — пг.
Литература к таблице 2.14
[38] Tao Н., Lam J.W.H. and McLaren J.W. // J. Anal. At. Spectrom. 1993. V. 8. P. 1067.
[105] Tracy M.L. and Moller G. // J. Assoc. Anal. Chem. [1] Haraldsson C., Pollak M. and Ohman P. // J. Anal. At.
1990. V. 73. P. 404. Spectrom. 1992. V. 7. P. 1183.
84
Новый справочник химика и технолога
[134] Sturgeon R.E., Willie S.N. and Berman S.S. H Anal. Chem. 1985. V. 57. P. 6.
[37] Willie S.N., Sturgeon R.E. and Berman S.S. H Anal. Chem. 1986. V. 58. P. 1140.
[16] D'ulivo A., Lampugnani L. and Zamboni R. // J. Anal. At. Spectrom. 1990. V. 5. P. 225.
[17] D'ulivo A. // J. Anal. At. Spectrom. 1989. V. 4. P. 67.
[46] Saisho H. and Fujimura Y. // Anal. Sci. 1990. V. 6. P. 619.
[135] Ornemark U and Olin A. // Taianta. 1994. V. 41. P. 67.
[48] Saleh A.I. et al. // J. Radianal. Nucl. Chem. 1990. V. 140. P. 357.
[Ill] Tanzer D. and Heuman K.G. // Anal. Chem. 1991. V. 63. P. 1984.
[29] Narasaki H., // Fresenius' Z. Anal. Chem. 1985. V. 321. P. 464.
[34] -Narasaki H. 11 J. Anal. At. Spectrom. 1988. V. 3. P. 517.
[31] Piwonka J., Kaiser G. and Tolg G. 11 Fresenius' Z. Anal. Chem. 1985. V. 321. P. 225.
[47] Tao G. and Hansen E.H. // Analyst. 1993. V. 119. P.333.
[112] Heumann K.G. and Grosser R. // Fresenius' Z Anal. Chem. 1988. V. 332. P. 880.
[136] Veber M., Cuyes K. and Gomiscek S. // J. Anal. At. Spectrom. 1993. V. 9. P. 285.
Таблица 2.15
Комбинированные методы определения селена в клинических материалах [66]
Образец (количество) Способ разложения Разделение и/или предконцентрирование Метод определения Предел обнаружения, мкг • г-1 Литература
Физиологическая жидкость (0,1-1 мл) HNO3-H3PO4, Н2О2 В виде летучего H2Se AAS нг* 63
Физиологическая жидкость (0,2-1 мл), моча (1г) HNO3-HC1O4 Экстракция 2,3-нафталиндиамином циклогексаном нг* 63
Сыворотка (2 мл) hno3-h2so4- НС1О4 В виде летучего H2Se FI QF AAS 1,2" 35
Сыворотка, кровь, моча HNO3-H2O2 или hno3-hcio4 В виде летучего H2Se ICP MS * нг 24
Сыворотка, кровь, ногти (0,1-1 мл) HNO3-HC1O4 или hno3-h2o2 (кипячение с использованием микроволнового нагрева) Экстракция 4-нитро-о-фенилендиамином или СНС13 IDGC MS 2** 41
Сыворотка, волосы, ногти (0,3-5 мг) HNO3-HC1O4, (в бомбе), НС1 В виде летучего H2Se QF AAS _.*** 6 31
Кровь (0,5 г), моча (1 г) Mg(NO3)2- HNO3-HC1 или HNO3-HC1O4-H2SO4 В виде летучего H2Se QF AAS нг* 149
Кровь HNO3-HC1O4, MgCl2 Экстракция о-фенилендиамином или (толуолом) RNAA 500 52
Ткань человека (0,1-0,2 г) HNO3-H2SO4 В виде летучего H2Se QF AAS нг* 150
Ткань грудной железы HC1O4 — царская водка Экстракция о-фенилендиамином или бензолом RNAA 53
Волосы (0,5 г) HNO3 Экстракция в виде Sel4 толуолом GF AAS нг* 50
Волосы, 6hoCRM, моча (0,5-1,0 г) HNO3 В виде летучего H2Se DCPAES 0,5** 24
CRM моча (20 мкл) HNO3-HC1O3 (в бомбе) В виде летучего H2Se, улавливание в GF GF AAS нг* 3
Примечания. * Нанограммовые количества. ** В растворе, нг/мл.
Абсолютный предел обнаружения, пг.
Метрологические основы методов количественного анализа
85
Литература к таблице 2.15
[63] Macpherson А.К., Sampson В. and Diplock А.Т. // Analyst. 1988. V. 113. Р. 281.
[35] McLaughlin К. et al. // Analyst. 1990. V. 115. P. 275.
[24] Ek P. and Hulden S.G. // Taianta. 1987. V. 34. P. 495.
[41] Ducros V. et al. // Analyst. 1993. V. 119. P. 1715
[34] Piwonska J., Kaiser G. and Tolg G. // Fresenius' Z. Anal. Chem. 1985. V. 321. P. 225.
[149] Hansson L., Pettersson J. and Olin A. // Taianta. 1987.
V. 34. P. 829.
[52] Kalonskova J. et al. // J. Radianal. Nucl. Chem. 1989. V. 129. P. 59.
[150] Fairhurst J., Lloyd B. and Delves H.T. // Anal. Chim. Acta. 1987. V. 197. P. 97.
[53] Gard A.N. et al. // Biol. Trace. Elem. Res. 1990. V. 26-27. P. 485.
[50] Ejaz M. and Qureshi M.A. // Taianta. 1987. V. 34. P. 337.
[24] Ek P. and Hulden S.G. // Taianta. 1987. V. 34. P. 495.
[3] Li Z., Ni Z. and Shan X. // Spectrochim. Acta. 1989.
V. 44B. P. 339.
Таблица 2.16
Определение кадмия в клинических образцах [66]
Образец (количество) Разложение (предварительная обработка) Разделение и/или предконцентрирование Метод определения Предел обнаружения, мкг • л-1 Литература
Кровь Депротеинизация СС13СООН Экстракция DDTC (MIBK) FAAS 0,6 6
Кровь Депротеинизация HNO3 — FAES 0,2 66
Кровь, сыворотка, коагулят (1 г) HNO3, Н2О2 — GF AAS 0,007’ 92
Кровь, плазма, моча Анионный обмен GF AAS 0,1 102
Моча (20 мл) HNO3 или HC1O4-HNO3 Электроосаждение на W-проволоке ETA AAS 0,01 25
Моча HNO3 Сорбция на А12О3, элюирование HNO3 FAAS 0,4 38
Моча (25 мл) СН3СООН Экстракция в виде Cdl^ три-и-октиламином или и-бутилацетатом FAAS 1 1
Моча (1 мл) — Электроосаждение ICP MS 0,5 28
Печень человека (1г) HNO3 — FAAS 0,1" 103
Примечания. Абсолютный предел обнаружения, нг. ”В образцах мкг• г *.
Литература к таблице 2.16
[6] Karakaya A., Silzen S. and Yiicesay В. // J. Anal. Texicology. 1992. V. 16. P. 403.
[66] FalkH. et al. // Analyst. 1986. V. 111. P. 285.
[92] Gastillo J.R., Concha I. and Martinez C. // At. Spec-trosc. 1987. V. 8. P. 172.
[102] Peterson D.P. et al. // Anal. Biochem. 1991. V. 192. P. 434.
[25] Zhang G. et al. // Taianta. 1993. V. 40. P. 409.
[38] Karakaya A. and Taylor A. // J. Anal. At. Spectrom. 1989. V. 4. P. 261.
[1] Saito I. et al. // J. Assoc. Off. Anal. Chem. 1988. V. 71. P. 829.
[28] Pretty J.R. et al. // J. Anal. At. Spectrom. 1990. V. 5. P. 425.
[103] Burguera M., Burguera J.L. and Alarcn O.M. // Anal. Chim. Acta. 1988. V. 244. P. 421.
Таблица 2.17
Определение общей ртути в биологических материалах [66]
Образец (количество) Способ разложения Восстановление Разделение / предконцентрирование Метод определения Предел обнаружения, нг • мл’1 Литература
Сыворотка, кровь HNO3-H2SO4-HC1O4; окисление (NH4)2S2O8 — Экстракция с Ni-DDTC или СНС13 RNAA нг 101
Сыворотка (0,1 г) СО HNO3 (бомба) SnCl2 Амальгамирование с Au QF AAS 0,1’ 18
86
Новый справочник химика и технолога
Продолжение таблицы 2.17
Образец (количество) Способ разложения Восстановление Разделение / предкон-центрирование Метод определения Предел обнаружения, нг • мл-1 Литература
Кровь (1-1,5 мл) Разб. HCI Экстракция с DDTC толуолом, реэкстракция НС1 GFAAS 2 66
Кровь, моча, рыба Микроволновая бомба NaBH4 В виде летучего Hg° QF AAS 0,05 (0,16*) ' 117
Волосы HNO3 (бомба или кварцевая ампула) SnCl2 Au-Pt-сетка QF AAS 100** 121
Волосы, рыба, мидии, плацента, СО HNO3-HCIO4-HCI SnCl2 Амальгамирование с золотом QF AAS ♦** нг 16
Волосы, рыба, мидии, плацента, СО Пиролиз Улавливание путем образования HgSe NAA ♦ ♦♦ нг 16
Волосы, моча, СО (10 мл или 100 мг) HNO3 (микроволновое) SnCl2 QF AAS 0,9 нгл~1 28
Моча (0,5 мл) КВг-КВгОз, КМпО4 NaBPU В виде летучей Hg° FIAQF AAS 0,1 40
Рыба (0,2 г) HNO3 (микроволновое) NaBHU В виде летучей Hg° QF AAS нг 39
Рыба, креветки, мидии (0,2-0,5 г) HNO3-H2SO4-НС1О4-КМпО4, Н2О2 (холод) ИЛИ HNO3 (бомба) NaBFU В виде летучей Hg° QF AAS *** нг 33
Рыба, моллюски, ракообразные HNO3-H2SO4- НС1О4-КМпО4-Н2О2 NaBPU В виде летучей Hg° QF AAS 0,05 38
Рыба, лобстер, гребешок (моллюск) (0,1 г) HNO3 (бомба) SnCl2 В виде летучей Hg° QF AAS нг 10
Рыба (0,2 г) HNO3 (микроволновое) NaBPU В виде летучей Hg° QF AAS 0,2 39
Рыба (1 г), листья (1-3 г), моча Сжигание О2, криогенная сепарация Соосаждение с Ag2S 1DSSMS *♦* нг 98
Рыба, ткань животных (0,1-3 г) HNO3-H2SO4 SnCl2 QF AAS нг 30
Мидии (0,5 г) HNO3 SnCl2 Амальгамирование с Au GF AAS 0,005* 22
СО растение (0,5 г) К2Сг2О7 в разбавленной H2SO4 SnCl2 — QF AAS 3* 25
СО (50-70 мг) Экстракция с этилацетоацетатом или СНС13 RNAA 140 104
СО (100 мг) HNO3 (микроволновое) SnCl2 В виде летучего Hg° QF AAS 0,8** 20
Растение, животное HNO3 (бомба) Экстракция Ni(DDTC)2 RNAA нг 103
Грибы (0,5 г) HNO3-H2SO4-H2O2 NaBH4 В виде летучего Hg° QF AAS 24** 32
Лишайники (50-100 мг) HNO3-HCI (бомба) SnCl2 Золото амальгамирование QF AAS ♦ ♦♦ нг 111
Вино (5 мл) HNO3, хромовая кислота в H2SO4 SnCl2 В виде летучего Hg° QF AAS 6 19
Примечания. Абсолютный предел обнаружения, нг.** Предел обнаружения в образцах, нг - г \*** Нанограммовые количества.
Метрологические основы методов количественного анализа 87
Литература к таблице 2.17
[101] Versieck J. et al. // Biol. Trace. Elem. Res. 1990. V. 26-27. P. 683.
[18] Bloom N.S. and Watras C.J. // Sci. Total Environ. 1989. V. 87. P. 199.
[66] Emteborg H. et al. // J. Anal. At. Spectrom. 1992. V. 7. P. 405.
[117] Welz B., Tsalev D. and Sperling M. // Anal. Chim. Acta. 1992. V. 261. P. 91.
[121] Bruhn C.G. et al. // J. Anal. At. Spectrom. 1994. V. 9. P. 535.
[16] Horvat M. et al. // Appl. Organomet. Chem. 1988. V. 2. P. 515.
[28] Vermeier G. et al. // Anal. Chim. Acta. 1991. V. 242. P. 203.
[40] Guo T. and Baasne J. Baasner. // Anal. Chim. Acta. 1993. V. 278. P. 189.
[39] Navarro M. et al. // Anal. Chim. Acta. 1992. V. 257. P. 155.
[33] Colina de Vargas M. and Romero R.A. // At. Spectrosc.
1989. V. 10. P. 160.
[38] Colina de Vargas M. and Romero R.A. // Analyst. 1992. V. 117. P. 645.
[10] Welz B. and Melcher M. // Anal. Chem. 1985. V. 57. P. 427.
[98] Moody J.R. and Paulsen P.J. // Analyst. 1988. V. 113. P. 923.
[30] Adeloju S.B. et al. // Anal. Chim. Acta. 1994. V. 285. P. 359.
[25] Landi S. et al. // Analyst. 1990. V. 115. P. 173.
[104] Hkan S.Z. and Turel Z.R. // J. Radianal. Nucl. Chem. 1992. V. 156. P. 103.
[20] Vermeir G., Vandecasteele and Dams R. // Anal. Chim. Acta. 1989. V. 220. P. 257.
[103] Kucera J. and Soukal L. // J. Radianal. Nucl. Chem. 1993. V. 168. P. 185.
[32] Rincon F-Leon and Zurera-Cosano G. // At. Spectrosc. 1986. V. 7. P. 82.
[111] Lupsina V. et al. // Analyst. 1992. V. 117. P. 673.
[19] Cacho J. and Castells J.E. // At. Spectrosc. 1989. V. 10 P. 85.
Лантаноиды
Актиноиды
58 Се 59 Рг 61 Pm
90 Th 91 Ра 92 и 93 Np 94 Pu 95 Am 96 Cm
Рис. 2.4. Пределы обнаружения элементов
методом атомно-абсорбционной спектрометрии (AAS) с пламенным атомизатором [66]
88
Новый справочник химика и технолога
Лантаноиды
Актиноиды
Рис. 2.5. Пределы обнаружения элементов методом атомно-абсорбционной спектрометрии (AAS) с электротермическим (графитовым) атомизатором [66]
Лантаноиды
Актиноиды
Рис. 2.6. Пределы обнаружения элементов методом атомно-эмиссионной спектроскопии (AES) с индуктивно-связанной плазмой [66]
Метрологические основы методов количественного анализа
89
Лантаноиды
Актиноиды
Рис. 2.7. Пределы обнаружения элементов масс-спектрометрическим методом с индуктивно-связанной плазмой [66]
2.7. Значащие цифры и правила округления. Точные и приближенные вычисления
Общие указания
При всех расчетах необходимо помнить и соблюдать следующие рекомендации [4, с. 55-57; 70, с. 94-96; 71, с. 26-30; 72, с. 5-8; 73, с. 23-24]. Экспериментальные данные и результаты расчетов принято выражать только значащими цифрами. Значащими называют все достоверно известные цифры данного числа плюс первая из недостоверных цифр. Если конкретные данные отсутствуют, недостоверность последней цифры числа принимают ± 1.
Поясним понятие значащих цифр на примерах. Число значащих цифр для каждого примера приводится в скобках
Примеры
1. 215 мг (3) 10. 4,56 мл (3)
2. 223,5 м (4) И. 3,050 мл (4)
3. 0,2506 г (4) 12. 23 мл (2)
4. 12,270 г (5) 13. 34,5 мл (3)
5. ОДОМ (2) 14. 115 мл (3)
6. 0,201 М (3) 15. 1,00 (3)
7. 0,2153 М (4) 16. 0,104 (3)
8. 0,0956 М (3) 17. 1,040 (4)
9. 0,010 (2) 18. 104 (3)
19. 1,04-104 (3)
При всех измерениях и написании значащих цифр необходимо помнить следующее.
Масса взвешиваемого на аналитических весах предмета обычно измеряется с погрешностью 0,0002 г (0,2 мг). По
этому правильно записанное число при измерении в граммах должно содержать четыре цифры после запятой, а в мг — одну цифру после запятой. Например: 8,9730 г, но не 8,973 г; 256,0 мг, но не 256 мг.
В титриметрических методах при работе с бюретками, позволяющими производить измерение титранта с относительной погрешностью 0,1%, объем должен быть выражен числом, содержащим четыре значащие цифры. При использовании бюреток вместимостью 5, 25, 50 мл с ценой наименьшего деления соответственно 0,01; 0,05 и 0,1 мл объем должен выражаться в первом случае числом, содержащим после запятой три цифры (например, правильно — 4,570 мл; 4,855 мл; неправильно — 4,57; 4,86), а в двух других — две цифры после запятой (например, правильно — 13,45 мл; неправильно — 13,5 мл).
Погрешность определений титриметрическими методами анализа при аккуратной работе колеблется в десятых долях процента. Поэтому правильно записанная концентрация должна содержать четыре значащие цифры. Например, правильно — 0,2070 моль • л1 (или М), неправильно — 0,207 моль • л’1 (или М).
Результат анализа (в г) должен содержать четыре значащие цифры. Например, правильно — 0,2350 г или 235,0 мг, неправильно — 0,235 г; 0,23507 г или 235 мг; 235,07 мг.
Обращение с нулями
Особенно внимательными надо быть при отнесении нулей к значащим или незначащим цифрам. Нули, стоящие между другими цифрами (примеры 3, 6, 11, 16-19) являются значащими цифрами. Нули, поставленные в начале чис
90
Новый справочник химика и технолога
ла (примеры 3, 5-9, 16) не являются значащими, т.к. они показывают только место запятой в десятичной дроби. Например, массу образца можно выразить в граммах или в миллиграммах (0,2205 г или 220,5 мг), при этом недостоверность массы и количество значащих цифр остаются неизменными. Недостоверность последней значащей цифры в выражении массы по-прежнему составляет ±1(0,0002 г или 0,2 мг).
Нули в конце числа (примеры 4, 5, 9, 11, 15) обычно считаются значащими. Если такой нуль не является значащим и используется только для указания места запятой в десятичной дроби, его следует исключить и записать число, применяя умножение на 10”.
Например, в числе 11 000, записанном в такой форме, значащими считаются все пять цифр. Если же считать, что число значащих цифр только три, то число следует записать в виде 1,10- 104
Абсолютная недостоверность числа в первом случае считается равной ±1, а во втором случае она оставляет ±0,01 Ю4 или ±100.
Округление чисел
При проведении любых расчетов необходимо грамотно провести округление результата, т.е. определить число значащих цифр в числе, полученном в результате арифметических действий с числами, полученными экспериментально, расчетным путем или взятыми из таблицы.
Более строгий подход основан на сравнении относительных недостоверностей сомножителей и произведения (или частного). Относительная недостоверность равна отношению абсолютной недостоверности числа к самому числу. Относительная недостоверность произведения (или частного) равна сумме недостоверностей сомножителей. Например, нужно найти частное 98 : 87,25. Относительные недостоверности составляют (приближенно):
1 : 98 = 1 • 1(Г2 и 0,01 : 87,25 = 1 • 10-4. Следовательно, относительная недостоверность частного 0,01 ± 0,0001 = = 1 • 10“2. При делении чисел с помощью калькулятора получаем число 1,1232... Поскольку недостоверна вторая цифра после запятой, частное следует округлить до 1,12.
Возведение в степень
При возведении числа в степень относительная недостоверность результата увеличивается в число раз, равное степени. Например, при возведении в квадрат она удваивается.
Извлечение квадратного корня
Относительная недостоверность результата извлечения корня вдвое меньше относительной недостоверности подкоренного числа, поэтому в некоторых случаях после извлечения корня число значащих чисел увеличивается. Например, ^/1,00 -1,000 , так как относительная недостоверность числа 1,00 равна 1 • 10'2, а результат извлечения корня 0,005, т.е. неопределенность заключена в третьем знаке после запятой.
Логарифмирование
При логарифмировании число значащих чисел в мантиссе равно числу цифр, которое содержал нестепенной член числа. Характеристика логарифма не входит в число значащих цифр, так как они указывают лишь на порядок логарифмируемого числа. Например, lg0,l • 10 2 = -3,0; lg0,10- Ю 2 = -3,00; lg0,l = -1,0. Абсолютная недостоверность логарифма приблизительно в 2,5 раза меньше относительной недостоверности числа под логарифмом. Например, если логарифм известен с точностью 1 • 10“3, относительная погрешность логарифмируемой величины не меньше чем 2,5- Ю3 При вычислении антилогарифма число значащих цифр уменьшается. Например, antlgl0,23 = 1,7- 1О10.
Точные и приближенные вычисления
Все вычисления в количественном анализе делятся на точные и приближенные. При точных расчетах для получения окончательного результата анализа следует помнить, что точность результата определяется точностью метода, точностью мерной посуды, тщательностью измерений. Точность окончательного результата не может быть повышена путем арифметических действий над результатами измерений. Например, погрешность аккуратных определений титриметрическими методами анализа обычно колеблется в пределах 0,1 - п- 0,1% (отн.) Поэтому при точных экспериментальных измерениях масс на аналитических весах и объемов по бюреткам результаты прямых измерений должны содержать по четыре значащие цифры. Следовательно, все используемые в последующих расчетах величины и конечный результат также должны содержать четыре значащие цифры. Например, правильно записанная концентрация должна содержать четыре значащие цифры: правильно 0,2021 моль -л1, но не 0,202 и не 0,20214; правильно записанный результат вычислений массы: т = = 0,1545 г, но не 0,154 и не 0,15454 г.
При расчетах начальных навесок или объемов жидких проб на стадии приготовления раствора титранта или титрируемого вещества примерно заданной концентрации не требуется большая точность вычислений. В результате таких приближенных вычислений ограничиваются двумя-тремя значащими цифрами.
Литература
1. В Научном совете АН СССР по аналитической химии И Журнал аналитической химии. 1982. Т. 37, № 5.
а) Об использовании понятий и терминов «эквивалент» и «нормальный». С. 946-956.
б) Изменения в основных терминах и единицах измерения в связи с введением Международной системы единиц (СИ). С. 957-961.
2. Номенклатурные правила ИЮПАК. М.: ВИНИТИ, 1985. Том 4. Аналитическая химия. С. 139-168.
3. Берлин А.А. И Журнал аналитической химии. 1989. Т. 44, вып. 4. С. 762-764.
4. Основы аналитической химии: В 2 кн. 2-е изд., перераб. и доп. / Под ред. акад. Золотова Ю.А. М.: Высшая
Метрологические основы методов количественного анализа
91
шк., 2000. Кн. 1. Общие вопросы. Методы разделения. 351 с.
5. Основы аналитической химии: В. 2 кн. 2-е изд., пере-раб. и доп. / Под. ред. акад. Золотова Ю.А. М.: Высшая шк., 2000. Кн. 2. Методы химического анализа. 494 с.
6. Аналитическая химия. Химические методы анализа / Под. ред. Петрухина О.М. М.: Химия, 1993. 397 с.
7. Васильев В.П. Аналитическая химия. М.: Высшая шк., 1989. Том 1. Гравиметрический и титриметрический методы анализа. 320 с.
8. Кунце У., Швед Г. Основы качественного и количественного анализа / Пер. с нем. Гармаша А.В. М.: Мир, 1997. 424 с.
9. Посыпайко В.И., Козырева Н.А., Логачева Ю.П. Химические методы анализа. М.: Высшая шк., 1989. 448 с.
10. Дорохова Е.Н., Прохорова Г.В. Задачи и вопросы по аналитической химии. М.: Мир, 2001. 267 с.
11. Толстоусов В.Н., Эфрос С.М. Задачи по количественному анализу. Л: Химия, 1986. 160 с.
12 .Skoog D.A., West D.M., Holler F.J. Analytical Chemistry. An Introduction. Sixth Edition. Saunders Golden Sunburst Series. Saunders College Publishing, 1994. 612 p. + Appendix A 1-A 45 + Index 11-118. ISBN 003097258X.
13. Harris D.C. Qantitative Chemical Analysis. Fifth Edition. New York: W.H. Freeman and Company, 1999. 899 p. + Appendix + CD ROM. ISBN 0716728818.
14. Vogel's Textbook of Quantitative Chemical Analysis. Fifth Edition / Ed. Jeffery G.H. et al. Longman Scientific and Technical Copublished in the United States with J. Wiley, a. Sons Inc. New York, 1989. 877 p.
15. В Научном совете РАН по аналитической химии. Представление результатов химического анализа. Рекомендации ИЮПАК 1994 г. // Журнал аналитической химии. 1998. Т. 53, № 9. С. 999-1008. (Pure a. Applied Chemistry, 1994. V. 66. Р. 595).
16. Номенклатурные правила ИЮПАК по химии. М.: ВИНИТИ, 1979. Том 1, полутом 2. С. 553-561.
17. ASTM: Standard Terminology for Statistical Methods E456-83a, 1984 Annual Book of American Society for Testing and Materials (Philadelphia, PA USA).
18. ISO Standard ISO 3534-1993 on Statistics - Vocabulary and Symbols. International Organization for Standardization (Geneva, Switzerland).
19. ISO (1993). International Vocabulary of Basic and General Terms in Metrology. 2nd Ed. International Organization for Standardization (Geneva, Switzerland).
20. IUPAC, Ed., H. Freiser, G.H. Nancollas. Compendium of Analytical Nomenclature. 2nd Ed. Oxford: Blackwell Scientific Publ., 1987.
21. Page C.H., Vigoureux P. The International System of Units (SI). NBS Special Publication. 1974. P. 330.
22. ДёрффельК.
а) Статистика в аналитической химии. 5-е изд. М.: Мир, 1994. 268 с.
б) Статистика в аналитической химии / Пер. с нем.; Под ред. В.В. Налимова. М.: Мир, 1969. 247 с.
23. Булатов М.И., Калинкин И.П. Практическое руководство по фотометрическим методам анализа. 5-е изд., перераб. Л.: Химия, 1986. 432 с.
24. Чарыков А.К. Математическая обработка результатов химического анализа. М.: Химия, 1984. 168 с.
25. Налимов В.В. Применение математической статистики при анализе вещества. М.: Физматгиз, 1960. 431 с.
26. Meier Р. С., Zund R.E. Statistical Methods in Analytical Chemistry. Chemical Analysis. A Series of Monographs on Analytical Chemistry and its Applications. V. 153. New York: Wiley, 2000. 424 P. ISBN 0471293636.
27. Anderson R.L. Practical Statistics for Analytical Chemistry. New York: Van Nostrand-Reinhold, 1987.
28. McCormick D. Measurement, Statistics and Computation. Chichester etc.: Wiley, 1987. 760 p. (Analytical Chemistry by Open Learning). ISBN 0471913669.
29. Otto M. Chemometrics: Statistics and Computer Application in Analytical Chemistry. Papeback. Wiley, 1999. 330 p. ISBN 352729628X.
30. Billo E.J. Microsoft Excel for Chemists. New York: Wiley, 1997.
31. Артемьев Б.Г., Голубев С.М. Справочное пособие для работников метрологических служб: В 2-х кн. 3-е изд., перераб. и доп. М.: Изд. стандартов, 1990. Кн. 1. С. 1-528; кн.2 С. 531-960.
32. Каплан Б.Я., Филимонов Л.Н., Майоров И.А. Метрология аналитического контроля производства в цветной металлургии. М.: Металлургия, 1989. 200 с.
33. Каплан Б.Я., Майоров И.А., Филимонов Л.Н. Руководство. Методы аналитического контроля в цветной металлургии. Т. 9. Ч. 1, 2. Метрология и стандартизация методик аналитического контроля производства в цветной металлургии. М.: Гиредмет, 1984.
34. Методы обработки результатов наблюдений при измерениях // Труды метрологических институтов СССР / Под ред. К.П. Широкова. М.-Л.: Изд. стандартов, 1972. Вып. 134(194). 117 с.
35. Рекомендации по межгосударственной стандартизации. Метрология. Основные термины и определения / Межгосударственный совет по стандартизации, метрологии и сертификации. Минск: ИПК Изд. стандартов, 2000. 32 с.
36. МИ 2336-95. Характеристики погрешности результатов количественного химического анализа. Алгоритмы оценивания (рекомендации) Екатеринбург: УНИИМ, 1995. 44 с.
37. РД 50-674-88. Методологические указания. Метрологическое обеспечение количественного химического анализа. Основные положения. М.: Изд. стандартов, 1989. 8 с.
38. ГОСТ Р8. 563-96. Методики выполнения измерений. М.: Госстандарт России, 1996. 11 с.
39. ГОСТ 8. 207-76. Прямые измерения с многократными наблюдениями. Методы обработки результатов наблюдений. Основные положения. М.: Изд. стандартов, 1979.8 с.
40. МИ 2083-90 ГСИ. Измерения косвенные (рекомендации). М.: Изд. стандартов, 1991. 8 с.
41. МИ 2334-95. Смеси аттестованные. Общие требование к разработке (рекомендации). Екатеринбург: УНИИМ, 1995. 17 с.
92
Новый справочник химика и технолога
42. ГОСТ 8.010-90. Методы выполнения измерений. М.: Изд. стандартов, 1991. 16 с.
43. ОСТ 48-189-88. Система метрологического обеспечения цветной металлургии. Порядок определения нормативов точности норм содержания компонентов и примесей в материалах. М.: Минцветмет СССР, 1988. 42 с.
44. Снесарев К.А., Зараковская А.И., Воробьева Г.А. Метрологические основы аналитического контроля химических производств. М.; Л.: Гослесбумиздат, 1960. 206 с.
45. Плинер Ю.Л., Свечникова Е.А., Огурцов В.П. Управление качеством химического анализа в металлургии. М.: Металлургия, 1979.103 с.
46. Разумов В.А. // Журнал аналитической химии. 1983. Т. 38, №7. С. 1157.
47. Алексеев В.Н. Количественный анализ. М.: Химия, 1972. 504 с.
48. Сусленникова В.М., Киселева Е.К. Руководство по приготовлению растворов. Л.: Химия, 1973. 145 с.
49. Булатов М.И. Расчеты равновесий в аналитической химии. Л.: Химия, 1984. 184 с.
50. Янсон Э.Ю. Теоретические основы аналитической химии. 2-е изд. М.: Высшая шк., 1987. 364 с.
51. Бланк А.Б. Метрологические аспекты аналитического контроля состава материалов // Журнал аналитической химии, 1997. Т. 52, № 8. С. 800-807.
52. Смагунова А.Н. Способы оценки правильности результатов анализа // Журнал аналитической химии. 1997. Т. 52, № 8. С. 1022-1029.
53. Бланк А.Б., Бугаевский А.А. // Журнал аналитической химии. 1969. Т. 24, № 9. С. 1445; 1970. Т. 25, № 1. С. 11-17.
54. Пустыльник Е.И. Статистические методы анализа и обработки наблюдений. М.: Наука, 1968.
55. Зайдель А.Н. Элементарные оценки ошибок измерений. 2-е изд.. М.: Наука, 1974. 108 с.
56. Львовский Е.Н. Статистические методы построения эмпирических формул. М.: Высшая шк., 1982. 224 с.
57. Алексеев Р.И., Коровин Ю.И. Руководство по вычислению и обработке результатов анализа. М.: Атомиз-дат, 1972. 72 с.
58. Журнал аналитической химии. 1975. Т. 30. № 10. С. 2058-2063.
59. Kaiser Н.
а) // Anal. Chem., 1970. V. 42, № 2. Р. 24А; № 4. Р. 26А.
б) // Spectrochem. Acta, 1978. V. В 33, № 9. Р. 551-576.
60. Koch O.G. // Anal. Chim. Acta, 1979. V. 107, № 5. P. 373-376.
61. Koch O.G., La Eleur P. D., Marrison G.H. // Pure a. Appl. Chem., 1982. V. 54, № 8. P. 1565-1577; Пер.: Журн. аналитической химии. 1984. Т. 39, № 6. С. 1135-1145.
62. Вайнфорднер Дж. И В кн.: Спектроскопические методы определения следов элементов / Пер. с англ.; Под. ред. Петрухина О.М., Недлера В.В. М: Мир, 1979. С. 9-19, 428-477.
63. Зильберштейн Х.И. // Изв. СО АН СССР. Сер. хим. наук. 1978. № 9/4. С. 30-48.
64. Бланк А.Б. // Журнал аналитической химии. 1979. Т. 34, №1. С. 5-9.
65. Журнал аналитической химии. 1984. Т. 39, № 6. С. 1135-1144. В Научном совете АН СССР по аналитической химии. Общие вопросы определения следов. V. Сравнение возможности методов определения малых количеств или малых концентраций элементов. Пер. из журн. Pure a. Appl. Chem., 1982. V. 52, № 8. Р.1565-1577.
66. Lobinski R. and Marczenko Z. Spectochemical Trace Analysis for Metals and Metalloids. Wilson a. Wilson's Comprehensive Analytical Chemistry. V. 30, Amsterdam etc., Elsevier, 1997. 807 P. ISBN 044482896 (Российская Г-5 национальная библиотека шифр XOl^j. Оглавление см. компакт-диск).
67. Prudnicov E.D., Elgersma J.W. and Smidt H.C. // J. Anal. At. Spectrom., 1994. V. 9. P. 619.
68. Winefordner J.D., Petrucei G.A., Stevenson C.L. and Smith B.W. // J. Anal. At. Spectrum., 1994. V. 9, P. 131.
69. Вясилев M.P. О пределе обнаружения вольтамперометрических методов анализа // Завод, лаб. 1993. Т. 59. № 1. С. 13-15 (см. компакт-диск).
70. Скуг Д., Уэст Д. Основы аналитической химии / Пер. с англ.; Под ред. чл.-корр. АН СССР Золотова Ю.А. М.: Мир, 1979. Том 1. С. 94-96.
71. Фритц Дж., Шенк Г. Количественный анализ / Пер. с англ.; Под ред. чл.-корр. АН СССР Золотова Ю.А. М.: Мир, 1978. С. 26-30.
72. Меркушева С.А. Методика решения задач по аналитической химии. Минск.: Высшая шк., 1985. С.5-8.
73. Петерс Д., Хайес Дж., Хифтье Г. Химическое разделение и измерения. Теория и практика аналитической химии / Пер. с англ.; Под ред. Зорина Н.Б., Агасяна П.К. М.: Химия, 1978. Том 1. С. 23-24.
74. The Determination of Trace Metals in Natural Waters // Blackwell Scientific Publications I Ed. by T. S. West, H. W. Numberg. Oxford etc., 1988.362 p. ISBN 0632020210.
Раздел 3
МЕТОДЫ РАЗДЕЛЕНИЯ И КОНЦЕНТРИРОВАНИЯ
Авторы-составители:
акад. РАЕН, проф., д. х. н. Л.Н. Москвин, доц., к. х. н. О.В. Родинков, доц., к. х. н. Н.М. Якимова
— изохорный потенциал, Дж • моль 1 AG0 — энергия Гиббса, Дж • моль 1
v — скорость подвижной фазы, дм3 • мин 1 ЕН— теплота испарения, Дж • моль 1 d — диаметр зерна сорбента dn—дайна диффузионного пробега в неподвижной фазе
Da — коэффициент продольной диффузии F — число Фарадея, 96500 Кл • моль 1
тМ
i — электродиализный поток вещества
через мембрану
к — константа скорости реакции
Кс — коэффициент селективности
KD — коэффициент распределения
L — длина хроматографической колонки, м I — расстояние, на которое перемещается
компонент
п — число ступеней (тарелок)
N— число теоретических тарелок
Аэфф — число эффективных теоретических тарелок
q — количество вещества, г
R—универсальная газовая постоянная, 8,314 Дж • моль4 • К 1
R, — степень выделения (извлечения)
ПРИНЯТЫЕ ОБОЗНАЧЕНИЯ
7?s — разрешение хроматографических пиков
tR — время удерживания, мин
и, — подвижность ионов в фазе мембраны мэ.ф.— электрофоретическая подвижность V— объем, дм3
VR — объем удерживания, дм3
W— объемные скорости, мл • мин4
УА — мольная доля компонента А в газовой фазе
г, — заряд иона
Z, — ширина хроматографической полосы
Е — разность потенциалов, В
Ке — фактор удерживания (фактор емкости) Кконц — коэффициент концентрирования Км — степень разделения веществ по молярной массе
Кр — коэффициент разделения
Кэкс — константа экстракции
М — молярная масса
Н— высота, эквивалентная теоретической тарелке или ВЭТТ
ПР° — термодинамическое произведение растворимости
р — общее давление над смесью жидкостей, Па
Ра нрв — парциальные давления компонентов А и В, Па
о о
рк и рв — давление насыщенного пара компонентов А и В при данной температуре, Па
pH — отрицательный десятичный логарифм концентрации ионов протония
с — концентрация растворенного вещества, моль • дм 3
Ф — флегмовое число
Х\ и Хв — мольные доли компонентов А и В в растворе
а — коэффициент относительной летучести жидкостей
5 — параметр растворимости Гильден-брандта, (кал • см 3)'4
е° — параметр, характеризующий элюирующую способность подвижной фазы
у,-— коэффициент активности
X, — коэффициент, учитывающий плотность заполнения колонки
Цо,5 — полуширина пика элюирования т — продолжительность миграции
ААС — атомно-абсорбционная спектрометрия
АПДК — аммония пирролидиндитиокарбамат, C5H9NS2 • NH3
АЭС — атомно-эмиссионная спектрометрия
БГКП — бактерии группы кишечных палочек
БХ — бумажная хроматография
ВА — вольтамперометрия
ПРИНЯТЫЕ СОКРАЩЕНИЯ
ВЭЖХ — высокоэффективная жидкостная хроматография
ГАК — газо-адсорбционное концентрирование
ГАХ — газо-адсорбционная хроматография
ГЖТХ — газо-жидкостно-твердофазная хроматография
ГЖХ — газо-жидкостная хроматография
ГХ — газовая хроматография
ДБДТКК — дибензилдитиокарбаминовая кислота, (C6H5CH2)NCS2H
ДДТКК — диэтилдитиокарбаминовая
кислота, (C2H5)2NCS2H
ДМГ — диметилглиоксим, HON=C(CH3)-C(CH3)=NOH
ДОЕ — динамическая обменная емкость
ЖАХ — жидкостно-адсорбционная хроматография
94
Новый справочник химика и технолога
ЖГАХ — жидкостно-газо-адсорбционная хроматография
ЖГХ — жидкостно-газовая хроматография
ЖЖХ — жидкостно-жидкостная хроматография
ЖТХ — жидкостно-твердофазная хроматография
ИВА — инверсионная вольтамперометрия
ИОХ — ионообменная хроматография
ИПХ — ион-парная хроматография
ИС — ионообменные смолы
ИСП-АЭС — атомно-эмиссионная спектрометрия с индуктивно-связанной плазмой
ИХ — ионная хроматография
МСВИ — масс-спектрометрия вторичных ионов
НАА — нейтронно-активационный анализ
НОММ — номинально отсекаемая молярная масса
П — потенциометрия
ПАВ — поверхностно-актвиные вещества
ПАН — 1-(2-пиридилазо)-2-нафтол, Ci5HnN3O
ПДТКК — пирролидиндитиокарбамино-вая кислота (C4H9N)CS2H
ПИД—пламенно-ионизационный детектор
ПОЕ — полная обменная емкость 111И1Ф — поточное проточное фракционирование в поперечном поле
ППФ — проточное фракционирование в поперечном поле
ПР — произведение растворимости
СЕ — сорбционная емкость
СКФЭ — сверхкритическая флюидная экстракция
СППФ — седиментационное проточное фракционирование в поперечном поле
СФХ — сверхкритическая флюидная хроматография
Т — титриметрический анализ
Тв — турбидиметрия
Т1И1Ф — термическое проточное фракционирование в поперечном поле
ТСХ — тонкослойная хроматография ТФЭ — твердофазная экстракция Ф — фотометрия
ФКП — фаги кишечных палочек
Фл — флуориметрия
ХМА — хроматомембранная абсорбция
ХМГЭ — хроматомембранная газовая
экстракция
ХМЖЭ — хроматомембранная жидкостная экстракция
ХМК — химически модифицированные кремнеземы
ЧАО — четвертичные аммониевые основания
ЭДТА — этилендиаминтетрауксусная кислота
Э1111Ф — электрическое проточное фракционирование в поперечном поле
ЭХ — экстракционная хроматография PDC — пирролидиндитиокарбамат, c5h8ns2
TR — редкоземельные элементы
Основные термины и количественные характеристики методов разделения
Разделение — процесс или операция, когда в результате из исходной смеси веществ получается несколько фракций ее компонентов в индивидуальном виде или в виде смесей с новым качественным и количественным составом.
Концентрирование (в технологических процессах синоним — обогащение) — процесс или операция повышения содержания целевых, т.е. выделяемых веществ по отношению к матрице.
Матрица — вещество, в среде которого находятся выделяемые компоненты, как правило вода или водные растворы.
Концентрирование — частный случай разделения. Выделение в самостоятельное направление объясняется спецификой его целей. В аналитической химии цель разделения — обеспечение возможности определения веществ неселективными методами или их получение в препаративных целях; цель концентрирования — снижение нижней границы диапазона определяемых концентраций и упрощение необходимых методов определения. Общая цель— экономическая: удешевление оборудования, необходимого для конечного определения целевых компонентов, и снижение требований к квалификации персонала, необходимого для его обслуживания.
Общие характеристики методов разделения и концентрирования выражаются через соответствующие коэффициенты:
G/l )кон
Хр = Х„ = ^=-100%.
(^)исх
Здесь Кр — коэффициент разделения; Кконц — коэффициент концентрирования; q — количество вещества: i — выделяемого, j — отделяемого, в исходной смеси - «исх» и в конечном препарате - «кон».
Степень выделения:
R_ = Ш^.100о/о (^)исх
Классификация методов разделения
Все методы разделения в зависимости от уровня однородности состава исходной смеси веществ подразделяются на методы разделения гетерогенных (макроскопически неоднородных) и гомогенных (макроскопически однородных) смесей.
Разделение гетерогенных смесей веществ производится в зависимости от агрегатного состояния, фазового или дисперсного состава образующих их компонентов. Эти методы, как правило, основаны на различиях в физических свойствах веществ. Для сплошных сред эти свойства — плотность и вязкость, для дисперсных — масса, размеры и форма частиц. К этой группе методов разделения относятся: фильтрация, седиментация, центрифугирование, флотация и т. п. Целью разделения гетерогенных смесей является фракционирование частиц по агрегатному состоянию, фазовому составу и степени дисперсности. Методы разделения гетерогенных смесей веществ, как правило, обеспечивают высокую эффективность разделения на фракции, отличающиеся по агрегатному состоянию. Разделение по фазам твердотельных смесей или разделение частиц различной дисперсности и плотности сводится к получению обогащенных фракций. Наибольший интерес к этой группе методов проявляется в промышленном производстве при переработке полезных ископаемых, при очистке водных сбросов и газоаэрозольных выбросов промышленных предприятий и т.п.
Принципиальные ограничения по селективности сужают область применения методов разделения гетерогенных
Методы разделения и концентрирования
95
раций на стадиях предварительной подготовки проб и выделения конечных продуктов. В качестве важнейших из них можно упомянуть отделение осадка от раствора, фракционирование по размерам частиц при определении форм существования токсичных примесей в природных водах. Оставаясь важнейшими элементами аналитических процедур, эти методы выполняют вспомогательную роль, не решая проблем разделения смесей веществ на индивидуальные компоненты. Из перечисленных выше методов разделения гетерогенных смесей фильтрация, седиментация и центрифугирование хорошо известны как с точки зрения физических принципов, на которых они основаны, так и с точки зрения областей применения в анализе. Особое место среди этой группы методов занимает флотация, имеющая свои области применения в аналитической химии.
Флотация
Флотация — это метод разделения смесей твердых частиц веществ, основанный на различии в их смачивании. Она широко распространена в гидрометаллургии как высокоэффективный способ обогащения полезных ископаемых — руд, минералов после их предварительного измельчения.
Для этого водную суспензию измельченного сырья обрабатывают реагентами-собирателями (анионо- и катионоактивными, а также неионогенными поверхностно-активными веществами), которые адсорбируются на поверхности частиц извлекаемого компонента и понижают их смачиваемость. Затем через суспензию пропускают воздух в виде мелких пузырьков и флотируют извлекаемые микрокомпоненты. В присутствии реагентов-вспенивателей (соединений, способных адсорбироваться на границе вода -воздух и умеренно стабилизирующих пену) над поверхностью суспензии образуется довольно устойчивый слой пены, обогащенной извлекаемыми микрокомпонентами. В суспензию могут быть также введены реагенты-подавители, или депрессоры, предотвращающие прилипание пузырьков воздуха к балластным веществам и вынос их в концентрат, активаторы для увеличения адсорбции собирателей и их гидрофобизирующего действия, регуляторы кислотности среды.
Сущность элементарного акта флотации заключается в следующем. При сближении пузырька газа и гидрофобной твердой поверхности разделяющая их прослойка воды при достижении некоторой критической толщины становится неустойчивой и самопроизвольно разрушается; происходит прилипание пузырька к поверхности частицы и вынос ее в пену над поверхностью суспензии.
Флотацию микрокомпонентов осуществляют в специальных ячейках, которые обеспечивают введение и диспергирование воздуха во взвеси, перемешивание пульпы с воздухом и поддержание флотируемых частиц во взвешенном состоянии, разделение пульпы и пены, удаление и транспортировку полученных продуктов обогащения. Возможности флотационного концентрирования рассматриваются в специальных обзорах, относящихся к 80-м годам XX века, ссылки на которые можно найти в обобщенной монографии [2].
Разделение гомогенных смесей веществ производится на молекулярном, атомарном или изотопном уровне. Все методы разделения гомогенных смесей можно объединить в четыре группы по общности принципов, на которых они основаны:
— основанные на образовании выделяемым веществом новой фазы;
— основанные на различиях в распределении веществ между фазами;
— основанные на различиях, проявляемых веществами при индуцированном переносе из одной фазы в другую через разделяющую их третью фазу (мембранные методы);
— основанные на различиях в скорости пространственного перемещения веществ в пределах одной фазы.
В рамках каждой группы существует своя классификация по характерным признакам.
3.1. Методы разделения, основанные на образовании выделяемым веществом новой фазы, в зависимости от агрегатного состояния исходной смеси и выделяемых веществ
Таблица 3.1
Внутригрупповая классификация методов разделения, основанных на образовании выделяемым веществом новой фазы
Агрегатное состояние исходной смеси веществ Твердое тело Газ Жидкость
Жидкость Осаждение Отгонка, упаривание, дистилляция, ректификация —
Газ Вымораживание — Вымораживание
Твердое тело — Возгонка, отгонка в парах реагента Селективное растворение
Примечание. Осаждение, отгонка и селективное растворение основаны на химических превращениях выделяемых веществ; упаривание, дистилляция, ректификация, вымораживание — на физических фазовых переходах в результате изменения температуры.
ЗАЛ. Осаждение
Разделение происходит за счет образования малорастворимых соединений одним или несколькими компонентами раствора при их взаимодействии с вводимыми в раствор или генерируемыми в нем веществами-реагентами и последующего отделения осадка от маточного раствора одним из методов разделения гетерогенных смесей: фильтрацией, цент
96
Новый справочник химика и технолога
рифугированием или седиментацией. Существует несколько классов малорастворимых химических соединений, реакции образования которых используются для разделения веществ методом осаждения из водных растворов:
— гидроксиды металлов и слабые минеральные кислоты;
— соли некоторых минеральных кислот и ацидоком-плексов металлов (фториды, хлориды, сульфаты, сульфиды, цианоферраты, хлорплатинаты и т.п.);
— соединения с органическими реагентами (хелаты и ионные ассоциаты);
— вещества в элементарном состоянии (селен, теллур, благородные металлы).
Коэффициент разделения и степень извлечения целевых компонентов определяются растворимостью образующихся соединений. Важнейшая характеристика — произведение растворимости (ПР). Для реакции
т\п+ + пЪт~ о КтВп ПР = [А”Т[В'"Т
ПР важнейших малорастворимых веществ приведены в табл. 3.2. Наиболее распространенной формой осадков являются гидроксиды, образование которых зависит от pH растворов. В табл. 3.3 приведены ориентировочные значения pH начала осаждения различных ионов в виде гидроксидов из кислых растворов; в табл. 3.4 — аналогичные данные для щелочных растворов; в табл. 3.5 приведены значения pH полного осаждения гидроксидов; в табл. 3.6 — перечень реагентов для осаждения гидроксидов, образующихся при указанных в таблице или меньших значениях pH.
Таблица 3.2
Произведения растворимости важнейших малорастворимых соединений [7]
Формула соединения ПР°
Ас2(С2О4)3 21024
Ас(ОН)3 (свежеосажденный) 2,1-IO-19
Ас(ОН)з (после старения) l,3-10"21
Ag3AsO3 1-10’17
Ag3AsO4 l-10~22
AgBO2 4-103
AgBr 5,3-IO’13
AgBrO3 5,5-IO-5
AgC2H3O2 4-10~3
AgCN 1,4-Ю-16
Ag2CO3 1,2-10"12
Ag2C2O4 3,5-10-14
AgCl 1,78-IO-10
AgC102 2-10"*
AgC103 5,0-10"2
Ag2CrO4 1,1-10"12
Ag2Cr2O7 1-Ю'10
Ag3Co(CN)6 3,9-10 26
Продолжение таблицы 3.2
Формула соединения TTP°
Ag3Fe(CN)6 1-10-22
Ag4Fe(CN)6 8,5-IO"*5
Ag2HVO4 2-10’14
Agl 8,3-IO-17
AgIO3 3,0-10"8
AgMnO4 1,6-10~3
Ag2MnO4 2,8-10"12
AgN3 2,9-10~9
AgNO2 6,0-10"*
Ag2O (Ag,OH) 1,95-10"8
AgOCN 2,3-IO"7
Ag2PO3F (2Ag+, PO3F2") 8,9-10"*
Ag3PO4 1,3-IO”20
AgReO4 7,95-IO"5
Ag2S 6,3-IO"50
AgSCN 1,1-10 42
Ag2SO3 1,5-IO"14
AgSO3NH2 1-10’1
Ag2SO4 1,6-105
AgSeCN 4,0-10”16
Ag2SeO3 9,8-10"16
Ag2SeO4 5,6-10~8
AgVO3 5-10"7
Ag2WO4 5,5-1 O’12
A1AsO4 1,6-10"16
A1(OH)3:
(АГ, ЗОН ) 3,2-1034
(АЮН2+, 2OH ) 3,2-IO"25
(H+,A1O~) 1,6-10"13
A1PO4 5,75-10"19
Am(OH)3 5,0-10-24
Am(OH)4 1-10"56
AuBr 5,0-IO-17
AuBr3 4,0-10~36
AuCl 2,0-10"13
AuCl3 3,2-10"5
AuOH 7,9-IO-20
Au(OH)3 3,2-10"*3
Aul l,6-10"23
Aul3 1-10"*6
Ba3(AsO4)2 7,8-10"51
Ba(BrO3)2 5,5-10-6
BaCO3 4,0-10-10
BaC2O4 l,l-10"7
Методы разделения и концентрирования
91
Продолжение таблицы 3.2
Формула соединения ПР0
ВаСгО4 1,2-10^°
BaF2 1,1-Ю-6
Ba3FefCN)6 3108
Ва(Юз)2 1,5-10-9
ВаМпО4 2,5-Ю10
ВаМоО4 410 s
Ва(ОН)2 5,0-10'
Ba3PO4F 4-107
Ва3(РО4)2 6-1039
Ва2Р2О7 3-10"11
BaPt(CN)4 410 3
Ba(ReO4)2 5,25-102
BaSO3 8107
BaSO4 1,1-Ю-10
BaS2O3 1,64 o5
BaSeO4 5108
Be(OH)2:
(Be2+, 2011) 4,9-1 O’22
(BeOH+, OH) 1,95-10 13
BiAsO4 2,8-Ю-10
Bi2(C2O4)3 4-Ю-35
Bil3 8,1-IO-19
BiOCI:
(BiO+, СГ) 7-109
(BiOCl+H2O = Bi3++2OH + СГ) 1,8-1 O’31
BiOOH 4-Ю-10
BiPO4 1,3- Ю23
Bi2S3 1 io-97
Ca3(AsO4)2 6,81019
CaC4H4O4 7,7-107
CaCO3 3,8-10-9
CaC2O4 2,3-1 O’9
CaCrO4 7,1-IO"1
CaF2 4,0-10-11
CaHPO4 2,7-107
Ca(H2PO4)2 1-103
Ca(IO3)2 7,0-107
Ca(NH4)2Fe(CN)6 4-108
Ca(OH)2:
(Ca2+, 2 OH-) 6,5-10 6
(CaOH+, OH) 9,l-10-5
Ca3(PO4)2 2,0-1029
CaPO3F 4-103
Ca5(PO4)3OH 1,6-1 O’58
Продолжение таблицы 3.2
Формула соединения ПР°
CaSO3 3,2-10’7
CaSO4 2,5-IO"5
CaSeO3 4,7-106
CaSiF6 8,1-10^
CaWO4 Cd3(AsO4)3 9,0-109 2,2-1 O’33
Cd(BO2)2 2,3 -10~9
Cd(CN)2 1,0-1 O’8
CdCO3 l,0 10“12
CdC2O4 1,5-10 8
Cd2Fe(CN)6 4,2-1018
Cd(NH3)6(BF4)2 2-106
Cd(OH)2 (Cd2+, 2OH ) (свежеосажденный) 2,2-10“14
Cd(OH)2 (Cd2+, 20НЭ (после старения) 5,9-1045
Cd(OH)2 (H+,HCdO2) 2-1019
CdS 1,6-1 O’28
CdSeO3 5,0-10~9
CdWO4 2-IO-6
Ce2(C2O4) 2,5-10 29
Ce(IO3)3 3,2-1 O’10
Ce(IO3)4 5 10 17
CeO2 (CeO2+, 2OH“) 11O20
CeO2 (CeO4+, 4OH ) 1,6-ю"18
Ce2(SeO3)2 3,7-1025
Co3(AsO4)2 7,6-1029
Co(BO2)2 3,2-10 9
CoCO3 1,05-1 O'10
CoC2O4 6,3-10 s
Co2[Fe(CN)6] 4,8-1 O’38
Co[Hg(SCN)4] 1,5-Ю”6
Co(IO3)2 1,0-Ю"1
Co(NH3)6(BF4)2 410-6
Co(NH3)6(ReO4)2 1,7-Ю-12
Co(OH)2 (голубой) 6,3-10-15
Co(OH)2 (розовый, свежеосажденный) l,6-10-15
Co(OH)2 (розовый после старения) 2,0-10-16
Со(ОН)3 4-Ю"15
a-CoS 4,0-10-21
р-CoS 2,0-10-25
CoSeO3 1,6-10-7
CrAsO4 7,8-10-21
98
Новый справочник химика и технолога
Продолжение таблицы 3.2
Формула соединения TTP0
Cr(NH3)6(BF6)3 6,2-10-5
Cr(NH3)6(MnO4)3 4,0-10-8
Cr(NH3)6(ReO4)3 7,7-10-12
Cr(NH3)6(SO3F)3 4,3-10^
Cr(OH)2 l,0-10-17
Cr(OH)3(Cr3+, ЗОН-) 6,3-10-31
(CrOH2+, 2OH-) 7,9-10-21
(H+, H2CrO-) 4,0-10-15
CrPO4 (фиолетовый) l,010-17
CrPO4 (зеленый) 2,4-10-23
Cs[AuC14] (Cs+, AuC14) 110-3
CsBF4 (Cs+, bf; ) 210-5
CsBH4 (Cs+, BH4) 2,5-Ю-7
CsBrO3 2 10-2
CsC103 410-2
CsC104 410-3
Cs3[Co(NO2)e] (3Cs’,[co(no2)’;]) 5,8-10-16
Cs[HgCl3](Cs+,[HgCl3" ]) 210-3
CsIO3 l,010-2
CsIO4 4,4-10-3
CsMnO4 9,1 10-5
Cs2PtCl6 3-10-8
Cs2PtF6 2,4-Ю-6
CsReO4 4,0-104
Cs2SiF6 l,310-5
Cs2SnCl6 3,6-10-8
Cu3(AsO4)2 7,6-10-36
CuBr 5,25-10-9
CuCN 3,2-IO-20
CuCO3 2,5-10-1 °
CuC2O4 310-9
CuCI 1,2-Ю-6
CuCrO4 3,6-Ю-6
Cu2[Fe(CN)6] 1,3-10-16
Cui l,110-12
Cu(IO3)2 7,4-10-8
CuN3 5,0-10-9
Cu2O 110-14
Cu(OH)2:
(Cu2+, 2OH) 8,3-IO-20
(CuOH ‘, OH-) 8,3-IO-12
Продолжение таблицы 3.2
Формула соединения ПР0
(H+, HCuO- ) 1-1019
Cu2(OH)2CO3 (малахит) 1,7-10-34
Cu3(OH)2(CO3)2 (азурит) 1,1-1(H6
Cu2P 2O2 8,3 IO-16
CuS 6,3-IO-36
Cu2S 2,5-IO"*8
CuSCN 4,8-10-15
CuSe l-10^9
CuSeO3 1,7-10-8
CuWO4 110 5
FeAsO4 5,8-10-21
FeCO3 3,5-10-11
FeC2O4 2-10-7
Fe4[Fe(CN)6]3 3,0-10 41
Fe(OH)2:
(Fe2+, 2OH ) 7,1 IO-16
(FeOH+, OH-) 2,2-10-11
(H+, HFeO-) 8-Ю-20
Fe(OH)3 (Fe3+, 3 OH ) (свежеосажденный) 6,3 10 38
Fe(OH)3 (Fe3+, ЗОН ) (после старения) 3,2-10 40
Fe(OH)3:
( Fe(OH)2, ОН-) 6,8-10-18
(Fe(OH)2+, 2OH ) 2-Ю-28
FePO4 1,3-10-22
FeS 5-Ю-18
FeS2 (Fe2+, S2-) 6,3-10-31
FeSe 1Ю-26
Fe2(SeO3)3 2Ю-31
Ga4[Fe(CN)6] 1,5-10-34
Ga(OH)3:
(Ga3+, ЗОН-) l,610-37
(H+, H2GaO-) 2,5-10-11
GeO2 (Ge4+, 4OH-) 1Ю-57
GeS 3-Ю-35
HfO(OH)2 (HfO2+, 2OH-) 410-26
Hg2Br2 ( Hg2+, 2Br-) 5,8-10-23
Hg2CO3(Hg2+, CO2-) 8,9-10-17
Hg2C2O4(Hg2+, C2O2-) 1Ю-13
Hg2Cl2 ( Hg2+, 2СГ) 1,3-10-18
Hg2CrO4( Hg2+, CrO2-) 5,0-10-9
Методы разделения и концентрирования
99
Продолжение таблицы 3.2
Формула соединения ПР0
Hg2I2( Hg2+, 2Г) 4,5-IO’29
Hg2(IO3)2(Hg2+, 2IO-) 2,45-IO-14
Hg2HPO4( Hg2+, HPO4”) 4,0-10-13
Hg2O ( Hg2+, 2OH ) l,6-10-23
HgO (Hg2+, 2OH) 3,0-10-26
HgS (черный) 1,6-IO"52
HgS (красный) 4,0-10’53
Hg2s(Hg;*,s2-) 1-Ю"*7
Hg2(SCN)2 ( Hg2+, 2SCN-) 3,0-IO’20
Hg2SO3(Hg2*, SO‘ J i-io-27
IIg,SO4(Hg;-, SOJ ) 6,8- IO7
HgSe 1-1059
Hg2SeO3 (Hg2+, SeO2-) 6,3-10 15
Hg2WO4(Hg2+, WO2') 1,1-IO-17
In4[Fe(CN)6] 1,9-Ю"14
In(IO3)2 3-10-3
In(OH)3:
(In3+, ЗОН ) 1,2-10-37
(In(OH)2+, 2OH ) 1,2-IO’27
(H+, H2InO3) 1-Ю-16
In2S3 5,75-IO’74
IrO2 (Ir4+, 4OH ) l,6-10“72
Ir2O3 (2Ir3+, 6OH) 2-Ю"*8
IrS2 l-10“75
K3[A1F6] l,610-9
K[BF4] 2-Ю-3
K[BH4] 1,3- IO’3
кцсжив] 2,25-IO’8
KC1O4 l,110-2
K3[Co(NO2)6] 4,3-IO-10
K2Cu2[Fe(CN)6] 2,2-10-27
K2[GeF6] 3,0-10-5
K2[HfF6] 2-Ю-3
K2[IrCl6] 6,8-10-5
KIO4 8,3-10"*
K2Na[Co(NO2)6] 2,2-10-11
K2[PdC14] 1,6-10-5
K2[PdCl6] 6,0-10-6
K2[PtC14] 8-Ю-3
K2[PtCld 1,1-Ю-5
K2[PtF6] 2,9-10-5
Продолжение таблицы 3.2
Формула соединения ПР°
KReO4 1,9-10-3
K2[SiF6] 8,7-10-7
K2[TiF6] 5-10"*
K2[ZrF6] 5-10"*
La(BrO3) ЗЮ-3
La2(CO3)3 . 4-Ю-34
La2(C2O4)2 1-Ю-25
La(103)3 6,2-10-12
La2(MoO4)3 2,2-10-21
La(OH)3 (свежеосажденный) 6,5-10-20
La(OH)3 (после старения) 2,0-10-22
La2S3 2,0-10-13
La2(SO4)3 ЗЮ-5
Li2CO3 4,0-10-3
LiF 1,7-10-3
LiOH 4-Ю-2
Li3PO4 3,2-10-9
Mg(AsO4)3 2,1-Ю-20
MgCO3 2,1-Ю-5
MgF2 6,5-10-9
Mg(IO3)2 З-Ю-3
MgK2[Fe(CN)6] 5-Ю-9
Mg(NH4)2[Fe(CN)6] 4-Ю-8
MgNH4PO4 2,5-10-13
Mg(OH)2 (свежеосажденный) 6,0-10-10
Mg(OH)2 (после старения):
(Mg2+, 2OH ) 7,1-10-12
(MgOH+, OH ) 2,6-10-9
Mg3(PO4)2 1-Ю-13
MgSO3 З-Ю-3
MgSeO3 4,4-10-6
Mn(AsO4)3 1,9-10-29
MnCO3 1,8-Ю-11
MnC2O4 5-Ю-6
Mn2[Fe(CN)6] 7,9-10-13
MnNH4PO4 1-Ю-12
Mn(OH)2:
(Mn2+, 2OH ) 1,9-10-13
(MnOH+, OH ) 4,9-10-10
(H+, HMnO-) 1-Ю-19
Mn(OH)3 1-Ю-36
Mn(OH)4 1-Ю-56
MnS (телесного цвета) 2,5-10-10
MnS (зеленый) 2,5-10-13
100
Новый справочник химика и технолога
Продолжение таблицы 3.2
Формула соединения ПР0
MnSeO3 5,4-1 O’8
Мо(ОН) 1-1056
(NH4)3A1F6 1-10’3
(NH4)3[Co(NO2)6] 7,6-IO6
(NH4)2IrCl6 3105
(NH4)2PtCl6 9-105
Na3AlF6 4,1-IO’10
Na2BeF6 7-103
NaIO4 3-103
NaSb(OH)6 4-10 s
Na2SiF6 2,8-IO-4
Ni3AsO4 3,11026
Ni(BO3)2 2-10"9
Ni(C4H7O2N2)2 (диметилглиоксимат) 2,3-IO’25
Ni(CN)2 3-1 O’23
NiCO3 l,3-10’7
NiC2O4 4-1 O’10
Ni(C103)2 110-4
Ni2[Fe(CN)6] 1,3-1045
Ni(IO3)2 1,4-1 O’8
Ni(NH3)6(BF6)2 1-10 6
Ni(NH3)6(ReO4)2 5Д-10-4
Ni(OH)2 (свежеосажденный) 2,0-1015
Ni(OH)2 (после старения) 6,3-10-18
Ni2P2O7 1,7-10’13
a-NiS 3,2-10-19
p-NiS 1-Ю’24
y-NiS 2,0-10-26
NiSeO3 1,0-10-5
NpO2(OH)2:
(NpO2,2OJ~T) 5,0-10-23
Pb3(AsO4)2 4,1 10-36
Pb(BO2)2 1,7-10-11
PbBr2 9,1-10 6
Pb(BrO3)2 8,0-10-6
PbCO3 7,5-10-14
PbC2O4 4,8-10-10
PbCl2 1,6-10-5
PbCIF 2,8-10-9
PbCrO4 1,8-10-14
PbF2 2,7-10-8
Pb2[Fe(CN)6] 9,55-10-19
Pbl2 1,1-Ю-9
Продолжение таблицы 3.2
Формула соединения ПР°
РЬ(Ю3)2 2,6-10-13
РЬМоО4 4,0-10-6
Pb(N3)2 2,6-10’9
PbO2 (Pb4+, 40Н ) 3,0- IO4*
РЬ3О4(2РЬ2+, 4РЬО3 ) 5,3-IO’51
Pb(OH)2 (Pb2+, 20Н ) (желтый) 7,9-f О-16
РЬ(ОН)2 (РЬ2+, 2011) (красный) 5-10-16
(РЬОН+, ОН") 1,0-10-9
(Н+, НРЬО’ ) 3,2-10-16
РЬОНВг 2-10-15
РЬ2(ОН)2СО3 3,5-10^6
PbOHCl 2-10’14
РЬ3(РО4)2 7,9-1 О’43
РЬ5(РО4)3С1 7,5-10’80
PbPO3F 1 10“7
PbS 2,5-10-27
Pb(SCN)2 2,0-10-5
PbSO4 1,6-10’8
PbS2O3 4,0-10’7
PbSe 1-Ю’38
PbSeO3 3-10-12
PbSeO4 1,45-10’7
PbWO4 4,5-107
Pd(OH)2 1-Ю’31
Pd(OH)4 6,5-10’71
Po(OH)4 6,3-10-52
PoS 5-10’29
Po(SO4)2 2,6-107
PtBr4 З-Ю^1
PtCl4 8,0-10-29
PtO2 (Pt4+, 40H ) 1,6-10-72
Pt(OH)2 1-Ю"35
PtS 8-Ю’73
Pu(IO3)4 5-10-13
PuO2CO3 1,7-10-13
Pu(OH)3 2,0-10’20
Pu(OH)4 3,2-10-50
PuO2OI l (PuO2, Oi l ) 5-Ю-10
PuO2(OH)2 (PuO2+, 20H ) 2,3-10-20
Ra(IO3)2 8,8-10-10
Ra(NO3)2 6,2-10’3
RaSO4 4,3-10-11
RbBF4 1-Ю"3
Методы разделения и концентрирования
101
Продолжение таблицы 3.2
Формула соединения ПР0
RbBH4 2,5-10^
RbBrO3 2-10 2
RbC104 2,5 -IO3
Rb3[Co(NO2)6] 1,48-IO"15
RbIO4 5,5 104
RbMnO4 2,9-10’
Rb2[PrCI6] 9-108
Rb2[PtF6] 7,6-10-7
RbReO4 9,6-10 4
RbSiF6 5-10"7
Rb2TiF6 5,5-105
Rh2O3 (Rh3+, ЗОН ) 3,2-10^°
Ru2O3 (Ru3+, ЗОН ) 1-1038
Ru(OH)4 110 49
Sb203:
(Sb3+, ЗОН ) l,71038
(SbO+, OH) 2,5-10-19
(3H+, H2SbO3“) 1,3 -io12
Sc(OH)3 5,0-Ю37
Snl2 8,3-Ю-6
Sn(OH)2
(Sn2+, 2OH ) 6,31027
(SnOH+, OH) 2,5 IO-16
(H+, HSnO" ) 1,3 IO-15
Sn(OH)4 11057
SnS 2,5-IO27
Sr3(AsO4)2 1,3 IO-18
SrCO3 1,1-1O“10
SrC2O4 1,6-IO"7
SrCrO4 3,6-10-5
SrF2 2,5-109
Sr(IO3)2 3,3-1 O’7
SrMnO4 2-10“7
Sr(OH)2 3,2-1 O’4
Sr3(PO4)2 110-31
SrPO3F ЗЮ-3
SrSO3 4-10"8
SrSO4 3,2-IO"7
SrSeO3 4,4-106
SrSiF6 l,510-2
SrWO4 2,2- IO-10
Te(OH)4 210-58
Th(C2O4)2 l,110-25
Продолжение таблицы 3.2
Формула соединения ПР°
Th(IO3)4 2,5-10-15
Th(OH)4 (Th4+, 4OH) 2,0-10-50
Th3(PO4)4 2,6-IO"79
Th(SO4)2 4-10-3
Ti(OH)4 (Ti4+, 4OH) 6,3-IO"52
TIBr - 3,9-10-6
TlBrO3 1,7-10 4
T12CO3 4-10-3
T12C2O4 2-104
T1C1 1,7-10^
T1C1O4 4-10-2
Tl3[Co(NO2)6] 1,0-1016
Tl2CrO4 9,8-10-13
TUtFeCCN)^ 5-10'10
TH 5,75-1О-8
T1IO3 3,1106
T1N3 2,2 104
T1(OH)3 1,3-10 46
T13PO4 6,7-10-8
Tl2[PtCl6] 4-10 12
TlReO4 1,2-10’5
T12S 5,0-10-21
T1SCN 1,7-10^
T12SO3 6,3-10^
T12SO4 4-10 3
T12S2O3 2,0-107
T1VO3 5,5-109
T14V2O7 2,6 10 19
UO2CO3 1,9-1012
UO2C2O4 2-104
(UO2)[Fe(CN)6] 7,1 10-14
UO2HAsO4 3,2-10-11
UO2HPO4 2,14-10-11
UO2(IO3)2 ЗЮ-8
UO2KAsO4 2,5-Ю23
UO2KPO4 7,8-10-24
UO2NH4AsO4 1,7-10-24
UO2NH4PO4 4,4-10-27
UO2NaAsO4 1,3-10-22
U(OH)3 1-Ю19
U(OH)4 6,3-10-55
UO2(OH)2 4,0-10-23
VO(OH)2 1,910"24
V2O5 (V02+, 201Г) 1,610-15
(V0)3(P04)2 8-10-25
W(OH)4 1-Ю"50
102
Новый справочник химика и технолога
Продолжение таблицы 3.2
Формула соединения ПР0
Zn3(AsO4)2 1,3-IO"28
Zn(CN)2 2,6- IO13
ZnCO3 1,45-10-11
ZnC2O4 2,75-108
Zn2[Fe(CN)6] 2,l-1016
Zn[Hg(SCN)4] 2,2-10~7
Zn(IO3)2 2,0-108
Zn(OH)2:
(Zn2+, 2OH ) 1,4'1 O’17
(ZnOH+, OIT) 1,4-KT11
Zn3(PO4)2 9,I1033
ZnS (сфалерит) 1,6-1 O'24
ZnS (вюрцит) 2,5-10“22
ZnSe 1-1031
ZnSeO3 1,9-1 O’8
Zr(OH)4:
(Zr4+, 4OH“) 7,9-1 O’55
(Zr(OH)|+, 2OH“) 2,01025
Zr3(PO4)4 i-io132
Таблица 3.3
Последовательный ряд осаждения гидроксидов из кислых растворов (с концентрацией металлов 1 моль • дм-3) [1]
pH начала осаждения Осаждаемые ионы (точные значения pH)
-о Sb(III), Sb(V), Sn(IV), Mo(VI), W(VI), Ge(IV), Ti(IV)
~ 1 Nb(V), Ta(V)(0,6), Ce(IV)(l,2), Tl(III)
«2 Os(IV), Zr(IV), Hf(IV), Sn(II), Fe(III)(2,3), Hg(II), Bi(III) (из хлоридов)
~3 Hg(I), Ga(II), In(III), Th(IV) (3,5)
~4 Al(III), U(IV), Ir(VI), Ti(III)
~5 Cr(III), Mn(IV), Bi(III), U(VI)
~6 Cu(II)(5,5), Be(II), Sc(III)(5,9), Zn(II), Ru(III), Rh(III) и Pd(II) (из Cl-комплексов; Pt при этом не осаждается) Lu(III)(6,l); Yb(III)(6,3); Tu(III)(6,4); Er(III)(6,5)
~7 Y(III), Sm(III), Eu(III), Gd(III)(6,8); Nd(III)(7,3); Pr(III)(7,4); Ce(III)(7,6); La(III)(7,8); Fe(II), Ni(II), Co(II), Pb(II)(7,8)
= 8 Ag(I)(8,0); Cd(II), La(III)(7,8)
~9 Mn(II)(8,7)
«10 Mg(II)
> 12 Ca(II)(12); Sr(II)(14); Ba(II)(14) (при большой концентрации бария)
Таблица 3.4
Последовательный ряд осаждения гидроксидов из щелочных растворов [1]
pH начала осаждения Осаждаемые ионы (точные значения pH)
> 12 Nb(V), Ta(V)(12,6); Pb(II)
~ 12 Zn(II), Be(II)(12,0);
« 11 Al(III), Sb(III)(l 1), Sn(II) и Sn(IV), Cr(IlI)(l 1,5)
« 10 Ga(III)(9,7)
«9 Mo(VI), W(VI)(9,0)
Таблица 3.5
pH количественного осаждения гидроксидов [1]
Ион pH Ион pH
Ag(I) 11-13 Hg(H) 5-12
Al(III) 5-7,5 In(III) 6-10
Be(II) 7-10 La(III) 14
Bi(III) 7-8 Mg(II) > 11
Ca(II) > 14 Mn(II) 10-14
Ce(IV) 3-(> 10) Ni(II) 9-(> 10)
Ce(III) 8-(> 10) Pb(II) 9-10
Cd(II) >9 Sn(II) 1-13
Co(II) 9-14 Sn(IV) 4-10
Cr(III) 6-11 Ta(V) 6-(> 10)
Cu(II) 8-(> 10) Th(IV) 4-10
Fe(III) 4—14 Ti(IV) >2
Fe(II) 9-14 U(VI) ~8
Ga(III) 4—10 Zn(II) 8-10
Hf(IV) 4-(> 10) Zr(IV) 4-<> 10)
Таблица 3.6
Реагенты для осаждения гидроксидов [1]
Реагент Методика осаждения pH
NH4OH (или NH3-H2O) + NH4C1 Анализир. р-р (солянокислый) нагреть, добавлять NH3 Н2О до изменения окраски метилового оранж. или метилового красн. 7-9
CH3COONa + CH3COOH К анализир. р-ру (слабокисл.), добавить CH3COONa, нагр. до 70 °C 4-6
NaHCO3 К анализир. р-ру (слабокисл.) добавить NaHCO3 до изменения окраски бромфенолового синего, кипятить 4
Методы разделения и концентрирования
103
Продолжение таблицы 3.6
Реагент Методика осаждения рн
Пиридин c6h5n Анализир. р-р (нейтрализовать) + NH4C1 нагреть до кипения, добавлять пиридин до изменения окраски метилового красн., + пиридин (в избытке) нагреть 5-6,5
Уротропин (CH2)6N4 Анализир. р-р (pH = 2-4) натр, до 30 °C, добавлять NI14С1, уротропин (в избытке) 5-5,8
Фенилгидразин C6H5NH-NH2 К анализир. р-ру (слабокисл.) нагреть, добавляют фенилгидразин ~5
Бензоат аммония C6H5C(O)O-N1I4 Анализир. р-р нейтрализовать по метиловому оранж., добавлять СН3СООН, бензоат аммония, нагреть и кипятить ~ 6
ВаСО3 К анализир. р-ру добавить суспензию свежеприготовленного ВаСО3, взболтать на холоде или при нагревании 7,3
CdCO3 Осаждать как с ВаСОз 6,5
СаСОз То же 7,5
РЬСОз То же 6,2
ZnO Взболтать с суспензией ZnO 5,5
MgO Взболтать с суспензией MgO 10,5
HgO Взболтать с суспензией HgO 7,4
Na2S2O3 К анализир. р-ру (слабокисл.) добавить Na2S2O3; нагреть ~ 6
КВгОз + HBr Анализир. р-р + реагент (КВгОз + НВг) кипятить до удаления брома 2,7
КВгОз + HC1 То же 1,3
Таблица 3.7
Устранение мешающего влияния матричных элементов их осаждением [3]
Мат- рица Методы определения Осаждаемая форма Определяемые микроэлементы
РЬ ААС, АЭС Pb(NO3)2 (упаривание) Ag,Al, Bi, Cd, Со, Си, Fe, Ga, In, К, Mg, Mn, Na, Ni, Pd, Tl, Zn
РЬ ААС, Ф РЬС12 Ag, Al, Au, Bi, Cd, Co, Си, Fe, Ga, In, K, Mg, Mn, Na, Ni, Sb, Tl
РЬ ААС PbSO4 Al, Cd, Co, Си, Ga, In, Mn, Ni, Pd, Zn
РЬ Ф PbS2O3 Zn
Т1 АЭС, Ф TH Bi, Cd, Co, Си, Fe, In, Ni, Pb
Hg ВА, АЭС, Ф Hg Bi, Cd, Co, Си, Fe, Mg, Mn, Ni, Pb, Tl, Zn
Ag Ф Ag-амальгама As, Cd, Си, Fe, Ga, In, Mn, Ni, Pb, Tl, Zn
Ni Ф [Ni(NH3)6](CIO4)2 Перхлорат гексамминоникеля Co
Те Ф,П TeO2 Си, Pb
Si Ge Na4SiO4 (силикат натрия) или Na2GeO3 (германат натрия) В
Си CuSCN CuS Fe, Pb Cd, Co, Fe, In, Mn, Ni, Pb, Zn
Метод осаждения имеет две области применения. Во-первых, это первая стадия гравиметрического анализа, во-вторых, это традиционный способ пробоподготовки для устранения мешающего влияния матричных компонентов анализируемых смесей, применяемый в сочетании с различными методами конечного определения микроэлементов (табл. 3.7).
Обозначения методов определения: ААС — атомно-абсорбционная спектрометрия; АЭС — атомно-эмиссионная спектрометрия; ВА — вольтамперометрия; ИВА — инверсионная вольтамперометрия; МСВИ — масс-спектрометрия вторичных ионов; П — потенциометрия; Т — тит-риметрический анализ; Тб — турбинометрия; Ф — фотометрия; Фл — флуориметрия.
3.1.2. Отгонка из раствора
Разделение происходит за счет образования в растворах летучих соединений в результате взаимодействия выделяемых веществ с соответствующими реагентами. Выделение образовавшихся летучих соединений в газовую фазу инициируется кипячением раствора (унос с паром) или пропусканием через раствор газа-носителя, не взаимодействующего с выделяемыми веществами.
Отгонка из растворов находит применение как способ пробоподготовки, обеспечивающий устранение мешающего влияния матричных компонентов выделением из летучих соединений (табл. 3.8) или выделением летучих соединений определяемых микроэлементов (табл. 3.9).
104
Новый справочник химика и технолога
Таблица 3.8
Таблица 3.9
Пробоподготовка, основанная на отгонке из растворов матричных элементов [3]
Матрица Методы определения Отгоняемое соединение Реагент Определяемые микроэлементы
Si, SiO2 П, Ф, Фл SiF4 HF + HNO3 HF + HNO3 + HC1O4 HF + H2SO4 Al, Bi, Cd, Fe, In Ni, P, Pb, Tl, Zn
В АЭС BF3 HF + HNO3 Al, Ba, Be, Ca, Co, Cr, Cu, Fe, Mg, Mn, Ni, Ti, V,W
Ge, GeO2 АЭС, Ф GeCl4 HC1 + HNO3 + HC1O4 Ag, Al, Be, Bi, Cd, Cu, Fe, Ga, In, Mg, Mn, Ni, P, Pb, Sb
Сг АЭС CrO2Cl2 HC1 + HC1O4 Al, Cu, Fe, Mg, Mn, Ni, Ti, V
Sn АЭС SnC14 ССЦ + Cl2 Ag, Au, Bi, Co, Cu, Ga, Ni, Pb
As, GaAs АЭС AsBr3, AsCl3 HCl + Br2 + CC14 Al, Be, Bi, Cd, Co, Cr, Mg, Mn, Ni, Si, V, Zr
Sn Ф,П SnBr4, SnCU HCl + HBr + Br2; HBr + Br2 Cu, Fe, Pb
Bi Ф BiBr3 HBr Pb
Se, SeO2 п, Тб, АЭС SeBr4 HBr Cd, Cu, Fe, Ga, Pb, S, Те, Tl, щелочные и щелочноземельные элементы
Se, SeO2 П,Ф SeO2 HNO3 + H2SO4 HNO3, H2SO4 Al, As, Ba, Bi, Ca, Cd, Co, Cu, Ga, In, Ni, Pb, Те, Tl
В АЭС ВСОСНзЬ CH3OH Al, As, Cu, Fe, Mg, Mo, Na, P, Pb, Si
А1 АЭС A1(CH2-CH2Br)3 ЭТИЛ-бромид алюминия C2H5Br Ag, Co, Cr, Cu, Fe, Mn, Ni, Pb
Пробоподготовка, основанная на отгонке из растворов микроэлементов [3]
Матрица Методы определения Микроэлемент Отгоняемая форма Реагенты
Природные воды ААС, АЭС Hg Hg Восстановитель (SnCl2, NaBH4)
Фториды и оксиды урана Ф Br Br2 H2Cr2O7 -H2SO4
Черные и цветные металлы и сплавы Ф,Т N NH3 Щелочной раствор
Ni, Ti Ф, Т, катодная ИВА, МСВИ S H2S HC1, HC1 + HI + H3PO2
Горные породы Т Cl HC1 H3PO4, конц.
А1, морская вода и сточные воды АЭС As, Sb AsH3; SbH3 HC1 + Zn
Природные воды, горные породы, РЬ, Ni-сплавы ИСП-АЭС, ААС As, Те, Bi, Ge, Sb, Se Гидриды Кислотный раствор + NaBH4
Al, Nb, Zr, циркалой АЭС В BF3 Смесь кислот, содержащая HF
Pu, U Ф Si SiF4 НС1О4 + HF + HNO3
Те Ф Sb SbF3 H2SO4 + HBr + HC1
Те Ф As, Sn AsBr3, SnBr4 HC1O4 + HBr + HC1
Морские осадки Ф Se SeBr4 H3PO4 конц. + NH4Br + KIO3
Be, Ni, Th, U, Zr, циркалой Ф В B(OCH3)3 CH3OH
3.1.3. Отгонка в парах реагента (ОПР)
ОПР — метод выделения веществ, способных образовывать газообразные соединения непосредственно из твердофазных смесей за счет гетерогенных реакций с реагентами, находящимися в газовой фазе.
Учитывая, что скорость гетерогенной реакции пропорциональна площади межфазного контакта, метод примени-
Методы разделения и концентрирования
105
ется при анализе тонких пленок или порошков (исходная смесь переводится в мелкодисперсное состояние). Наиболее типичным (аналитическим) применением ОПР является определение углерода и серы в сталях (с использовани
ем отгонки соответствующих диоксидов в токе водорода) Реальный круг элементов, которые могут выделяться отгонкой непосредственно из твердофазных образцов, существенно шире (табл. 3.10).
Таблица 3.10
Примеры отделения микроэлементов путем их отгонки из твердых образцов [5]
Матрица Элемент Температура, °C Степень извлечения, % Отделяемые содержания, ед. изм. Реагент и газ-носитель
ZnO Cd 300/750 100 2-3000, нг h2/n2
Соединения Zn 1000/1200 100 мкг-мг Н2
Al, Ga, In Zn 1000/1150 91,0 20-1000, нг Н2
Бокситы Be 1000 100 5-100, нг О2
Бокситы Zn 1150 100 30, мкг Н2
Твердые образцы Tl 1000 100 2-1000, нг Н2
Си Se 1000 96,7 2, мкг
Си Se 1150 100 1, нг 02
А1 Cd, Zn 600-700 100 20, пг Н2/Аг
Аэрозоли Cd 600-700 100 0,15-0,25, мкг h2/n2
Горные породы Bi, Cd, Tl 1000-1200 95-100 10-10 пг h2/n2
Растительные материалы Pb 1000 100 нг-мкг Н2
Металлы, Si, SiO2 В 190 100 нг-мкг HF и Н2О
При этом, как и в случае отгонки из растворов, возмож- Продолжение таблицы 3.11
ны варианты отгонки матричных элементов и микропримесей. Отгонка в парах реагента возможна в форме раз-личных химических соединений (табл. 3.11) Таблица 3.11 Неорганические летучие соединения, подходящие для концентрирования микроэлементов методом ОПР Тип соединения Элементы, образующие летучие соединения (25-1000 °C)
Фториды Ti, Zr, Hf, V, Nb, Ta, Mo, W, Tc, Re, Ru, Os, Rh, Ir, Hg, Si, Ge, Sn, P, As, Sb, Bi, S, Se, Те
Оксиды S, Se, Те, Po, (As), Tc, Re, Ru, Os, (Mo, W), Zn, Cd, Hg, (Ir)
в температурном диапазоне 25-1000 °C [5] А1С13-комплексы Лантаноиды, актиноиды, Ca, Sr, Ba, Ra, Fe, Co, Ni, Cu, Pd, Pa, Mo
Тип соединения Элементы, образующие летучие соединения (25-1000 °C)
Элементы Инертные газы, галогены, 0, S, Se, Те, Po, N, P, As, Sb, Bi, Sn, Pb, Tl, Zn, Cd, Hg (Li, Na, K, Rb, Cs, Fr)
Хлориды Ti, Zr, Hf, V, Nb, Ta, (Cr), Mo, W, Tc, Re, Mn, Fe, Ru, Os, Au, Zn, Cd, Hg, Al, Ga, In, Tl, Si, Ge, Sn, Pb, P, As, Sb, Bi, S, Se, Те, Po, Ce
Гидриды As, Se, Sn, Pb, Те, Sb, Bi
Расширяя диапазон температур, можно существенно увеличить число элементов, отгоняемых в форме оксидов (табл. 3.12).
Таблица 3.12
Элементы, которые отгоняются в форме оксидов в потоках инертного газа или кислорода при повышенных температурах [5]
Элемент Температура плавления, °C Температура кипения, °C Оксид Температура плавления оксида, °C Температура кипения оксида, °C
Li 179 1317 Li2O 1700 Возгон 1000
Na 98 892 Na2O Возгон 1275
Na2O2 Разл. 460
К 64 774 K2O Разл. 350
K2O2 490 Разл.
Cs 29 690 Cs2O Разл. 400
106
Новый справочник химика и технолога
Продолжение таблицы 3.12
Элемент Температура плавления, °C Температура кипения, °C Оксид Температура плавления оксида, °C Температура кипения оксида, °C
Rb 39 688 Rb2O Разл. 400
Rb2O2 570 Разл. 1011
В 2070±25 Возгон. 2550 Н3ВО3 169 300
Тс 2140±20 4700 Тс2О7 119,5 Разл. 260
Re 3180 5627 Re2O7 300 Возгон. 360
Os 3000+10 5000 OsO4 39 130
Ru 2250 3900 RuO4 26 Разл. 108
Ir 2443 4180 Ir2O3; IrO2 Перенос в потоке
Pt 1772 3800 PtO2 о2
Zn 419 907 ZnO 1975
Cd 320 765 CdO Разл. 900 Возгон. 1559
Hg -39 356 Hg2O Разл. 100
Ga 30 2403 Ga2O 650 Возгон. 500
In 156 2000 In2O3 1910 Возгон, в вакууме 565-700
In2O Возгон, в вакууме 656-700
Tl 303 1457 T12O3 717
Sn 231 2260 SnO Разл. 1080 Возгон. 1800-1900
SnO2 1127 2500
Pb 327 1744 PbO 888 1535
Mg 651 1107 MgO 2800 3600
P 44 280 P2O5 580 Возгон. 300
As Разл. 358 As2O3 Возгон. 193 457
Sb 650 1380 Sb2O3 Возгон. 1550
Bi 271 1560 Bi2O3 860 1900
S 112 444 so2 72 10
Se 217 685 SeO2 340-350 Вак. Возгон. 340
Те 452 1390 TeO2 733 1245
Po 254 962 PoO2 Разл. 500 Возгон. 885
Отгонка в парах реагента в варианте термохроматографии [5] открывает возможность разделения элементов. Разделение осуществляется в колонке с создаваемым в ней определенным градиентом температур (табл. 3.13).
Таблица 3.13
Термохроматографическое отделение элементов и соединений [5]
Отгоняемые элементы или соединения Поверхность колонки Градиент температур, °C
ZrCl4, RuC14, NbCU, МоСЦ ит.д. Кварц, покрытый различными хлоридами 900-25
Cf, Es, Fm, Md Титан 1700-800
I, At, Hg Кварц, покрытый серебром 1000-100
Продолжение таблицы 3.13
Отгоняемые элементы или соединения Поверхность колонки Градиент температур, °C
I, At, Hg, Po, Cd, In, Sb, Tc, Tl, Pb, Bi, Sn (в потоке H2) Кварц 1000-25
Ir, Re, Os в потоке O2 « 1000-25
YBr3, ZnBr2, Ga2Br6, NbBr5, TeBr5, ZrBr4, HfBr4, AsBr3, GeBr4 « 1000-25
СаС12, СеС13, СеСЦ, ZrCl4, TeCl4, TcCl4 « 1000-25
Ir, Os, Re, Ru, Тс в потоке o2 « 1000-25
Методы разделения и концентрирования
107
Продолжение таблицы 3.13
Отгоняемые элементы или соединения Поверхность колонки Градиент температур, °C
ZrCl4, HfCU NbCl5, ТеС15 Кварц, ВеС12 1000-25
ТеС14, ReCl3, RuC14, OsCl4 NaCl, КС1 1000-25
CdCl2, InCi, SnCl2, SbCl3, SbCl5, BiCl3, TeCl4 СаС12 1000-25
ZrBr4, NbBr5, TcBr5, CdBr2 Кварц, NaBr 1000-25
InBr, SnBr2, SnBr4, SbBr3 BiBr3, TeBr4 KBr, CsBr 1000-25
OsCLj, ReCl6, AuCl3, PtCLj Кварц 1000-25
3.1.4. Упаривание, дистилляция, ректификация
Упаривание, дистилляция, ректификация — это методы, основанные на фазовых переходах вещества из жидкой фазы в газообразную без химических превращений при температурах и давлениях фазовых переходов.
Возможность разделения веществ дистилляционными методами следует из различий парциальных давлений веществ при одной и той же температуре:
РА=Рл^А,а^А Рв Рв-^в Л’
где р,\ и /?в — парциальные давления компонентов А и В над раствором, Ад и Хъ— мольные доли компонентов А и В в растворе, р\ и — давления насыщенного пара при данной температуре, а — коэффициент относительной летучести жидкостей.
Давление насыщенного пара над жидкостью р" и рйг при различных температурах и Т2 связаны соотношением: rf АН(Т,-Тг) р° R т,т2 ’
где ЛЯ — теплота испарения.
Это соотношение позволяет, зная АЯ и Гкип при атмосферном давлении, оценить р° при другой температуре.
Коэффициент а, как правило, недостаточно велик, чтобы добиться разделения двух жидкостей за однократный акт испарения - конденсации. Каждый из дистилляционных методов имеет свои методические особенности, определяющиеся его назначением. Упаривание — процесс ис
парения матрицы. Обычно матрицей является вода, часто — нелетучие примеси из растворов. Метод, при кажущейся простоте, имеет свои методические проблемы. Главная — необходимость избегать капельного уноса влаги. Поэтому упаривание, как правило, проводят при температурах ниже температуры кипения и добиваются равномерного объемного нагрева испаряемой жидкости (песчаная баня) или поверхностного нагрева (инфракрасная лампа). Для интенсификации процесса вместо интенсивного нагрева прибегают к сдвигу термодинамического равновесия за счет отвода из системы паровой фазы.
Относительная летучесть двух компонентов а может быть найдена из соотношения:
, А^В-^А
In а =--2----
R7'
где АЯВ и АЯа — молярные энтальпии испарения компонентов А и В.
Энтальпию испарения можно оценить из правила Тру-тона:
~ 97 Дж (моль • К)'1, ^кип
где Гмп — температура кипения, — и затем по формуле рассчитать значение а.
Так, для компонентов, температура кипения которых отличается на 10 К, а = 1,3-1,4.
Из этой группы методов важнейшие — дистилляция и ректификация.
Дистилляция — однократный процесс испарения-конденсации.
Ректификация — многократное повторение актов испарения - конденсации, позволяющее улучшить разделение. Возможности метода определяются числом ступеней (теоретических тарелок), на которых реализуется однократный акт испарения - конденсации, что следует из характеризующей метод ректификации зависимости:
Л *а
—— = а -------—,
1-УА 1А-2ГА
где УА — мольная доля компонента А в газовой фазе; п — число ступеней (тарелок).
В таблице 3.14 представлены расчетные значения а" для идеальных смесей в зависимости от величины ₽ = (УкипА - Липв)/(Т’КипА + Гсипв) Для различного числа тарелок.
Таблица 3.14
Расчетные значения а" в зависимости от 0 для идеальных смесей жидкостей
P a"
n = 1 и = 2 n = 3 и = 4 и = 5 и = 6 и = 7 n = 8 n = 9 n - 10
0,001 1,024 1,048 1,073 1,098 1,124 1,151 1,178 1,206 1,234 1,264
0,002 1,050 1,098 1,151 1,206 1,264 1,324 1,388 1,45 1,52 1,60
0,003 1,073 1,151 1,234 1,324 1,42 1,52 1,63 1,75 1,88 2,02
0,004 1,098 1,206 1,324 1,45 1,60 1,75 1,93 2,11 2,32 2,55
108
Новый справочник химика и технолога
Продолжение таблицы 3.14
р а”
п =1 п = 2 п = 3 л = 4 п = 5 п = 6 и = 7 п = 8 п = 9 п = 10
0,005 1,124 1,263 1,42 1,60 1,80 2,02 2,27 2,55 2,87 3,22
0,006 1,151 1,324 1,52 1,75 2,02 2,32 2,67 3,07 3,54 4,07
0,008 1,205 1,45 1,75 2,11 2,55 3,07 3,71 4,47 5,39 6,50
0,01 1,26 1,60 2,02 2,55 3,22 4,07 5,14 6,5 8,22 10,4
0,02 1,60 2,55 4,07 6,50 10,4 16,6 26,5 42,3 67,5 108
0,03 2,02 4,07 8,21 16,6 33,4 67,5 136 275 554 1120
0,04 2,55 6,50 16,6 42,3 108 275 701 1790 4560 1,16-104
0,05 3,22 10,4 33,4 108 347 1120 3560 1,16-104 3,74-104 1,21-105
0,06 4,07 16,6 67,5 275 1120 4560 1,85104 7,55-Ю4 3,07-105 1,25-106
0,08 6,50 42,2 275 1790 1,16-104 7,55 104 4,91-105 3,19-Ю6 2,07-107 1,34-108
0,10 10,4 108 1120 1,16-104 1,21105 1,25-106 1,30-107 1,35-108 1,40-109 1,45-1О10
Уравнение справедливо для идеальных смесей жидкостей, когда р = ^p*Xt (р — общее давление над смесью жидкостей). При отклонениях от идеального состояния, вызванных образованием азеотропов, ректификация не обеспечивает разделения компонентов.
Важное значение имеет конструкция ректификационных колонок, определяющая достаточное число п при разделении.
При сравнении эффективности ректификационных колонок необходимо принимать во внимание, что для одной и той же колонки п — число переменное, определяемое флегмовым числом Ф, равным отношению объема конденсата (флегмы), возвращаемого в перегонную колбу (Квозвр), к объему отбираемого продукта (Иотбир ):
ф _ возвр
V отбир
Чем меньше флегмовое число, тем меньше п и Кр — коэффициент разделения. Они максимальны при полном возврате флегмы. В подобных условиях производится сравнение эффективности различных ректификационных колонок.
3.2. Методы разделения, основанные на различиях в распределении веществ между фазами
Внутригрупповая классификация методов разделения, основанных на различиях в межфазном распределении веществ по агрегатному состоянию фаз и способу осуществления процесса межфазного распределения, представлена в табл. 3.15.
Таблица 3.15
Агрегатное состояние фаз, между которыми происходит распределение Методы разделения в зависимости от способа осуществления межфазного распределения
Однократное равновесное распределение Многократное перераспределение Хроматографический способ 1
Жидкость - твердое тело Статическая сорбция. Соосаждение. Твердофазная экстракция Динамическая сорбция. Зонная плавка и направленная кристаллизация Жидкостно-твердофазная хроматография (ЖТХ)2
Жидкость - жидкость Жидкостная экстракция Многоступенчатая экстракция Жидкостно-жидкостная хроматография (ЖЖХ)3
Газ - твердое тело Статическая адсорбция Динамическая адсорбция Газо-адсорбционная хроматография (ГАХ)
Жидкость - газ Газовая экстракция абсорбция Динамическая абсорбция Газожидкостная хроматография (ГЖХ). Жидкостно-газовая хроматография (ЖГХ)
Методы разделения и концентрирования
109
Продолжение таблицы 3.15
Агрегатное состояние фаз, между которыми происходит распределение Методы разделения в зависимости от способа осуществления межфазного распределения
Однократное равновесное распределение Многократное перераспределение Хроматографический способ 1
Твердое тело (жидкость) — вещество в сверхкритическом состоянии Сверхкритическая флюидная экстракция (СКФЭ) — Сверхкритическая флюидная хроматография (СФХ)
Примечания.
1 Сущность хроматографического способа осуществления процессов межфазного распределения раскрывается в п. 3.2.9.
2 Варианты ЖТХ в зависимости от механизма удерживания разделяемых веществ твердой фазой: жидкостно-адсорбционная хроматография (ЖАХ), ионообменная хроматография (ИОХ), аффинная хроматография, лигандообменная хроматография (ЛОХ), эксклюзионная, подразделяемая на гель-проникающую и гель-фильтрационную.
3 Варианты ЖЖХ в зависимости от полярности фаз, участвующих в хроматографическом процессе: ЖЖХ с неподвижной полярной фазой, ЖЖХ с обращенными фазами. Синонимом последней применительно к разделению неорганических веществ является экстракционная хроматография.
3.2.1. Общие характеристики методов, основанных на различиях в межфазном распределении
Основной характеристикой является коэффициент распределения KD = с/7с/ , где с{" и с,' — концентрации одного и того же z-ro вещества в двух фазах: извлекающей и отдающей соответственно при установлении межфазного равновесия.
Другие характеристики — коэффициент селективности Кс = KD ]KD (где индексы i и j соответствуют двум различным веществам, находящимся в одной и той же системе фаз) и изотерма, характеризующая зависимость коэффициента распределения от собственной концентрации распределяемого вещества. Известны два варианта представления изотерм межфазного распределения. В первом и наиболее часто встречающемся случае на оси абсцисс откладывается равновесная концентрация вещества в отдающей фазе (с,)ЭТд, а на оси ординат — концентрация в
Рис. 3.1. Изотермы межфазного распределения (1-й вариант представления)
Постоянство величины KD (независимо от изменений концентрации веществ в системе) соответствует случаю
линейной изотермы (кривые 1 на рис. 3.1 и 3.2.). Как правило, изотерма является линейной только в области низких концентраций вещества. С ростом концентрации обычно наблюдается снижение величины KD (выпуклая изотерма, кривые 2 на рис. 3.1 и 3.2), реже с увеличением концентрации KD увеличивается (вогнутая изотерма, кривые 3 на рис. 3.1 и 3.2).
Второй вариант представления изотерм — зависимость KDi от суммарной концентрации вещества в системе — [(с;)отд + (о)изв] (рис. 3.2).
Рис. 3.2. Изотермы межфазного распределения (2-й вариант представления)
Специфической особенностью некоторых процессов межфазного распределения является необратимость перехода вещества из одной фазы в другую, типичная для экстракции хелатов и сорбции ионов металлов хелатообразующими сорбентами. В этих случаях KD теряет смысл как характеристика равновесного распределения вещества и в качестве адекватной характеристики процесса вводится степень извлечения 7?г:
110
Новый справочник химика и технолога
R = 100%,
kJ +(?,) X /изв X Jl / отд
где ((/J™ и (<7/)»гд — количества z-того вещества в извлекающей и отдающей фазе после разделения соответственно.
3.2.2. Сорбционные методы
Сорбция — собирательное название процессов или операций выделения веществ из жидкой или газовой фазы в твердую. Употребляется в тех случаях, когда нет необходимости акцентировать внимание на механизме выделения веществ в твердую фазу, или когда этот механизм неизвестен, или когда имеет место несколько механизмов связывания выделяемых веществ (сорбатов) в фазе выделяющего вещества (сорбента). В отличие от абсорбции — объемного поглощения веществ в жидкую или твердую фазу — выделение веществ из жидкой или газовой фазы поверхностью сорбента называется адсорбцией. В зависимости от механизма удерживания сорбата сорбентом выделяются:
молекулярная адсорбция, вызванная действием ван-дер-ваальсовых сил между молекулами сорбата и атомами на поверхности сорбента;
ионный обмен, являющийся гетерогенной химической реакцией обратимого стехиометрического обмена ионами между контактирующими жидкой и твердой фазами;
комплексообразующая сорбция, при которой функциональные группы сорбента выступают в роли лигандов, координируемых сорбируемыми ионами металлов.
Важнейшей характеристикой всех адсорбентов первого типа является удельная поверхность, а для микропористых адсорбентов дополнительными характеристиками являются удельный объем микропор и средний радиус пор. При прочих равных условиях адсорбция одного и того же вещества возрастает при увеличении удельной поверхности адсорбента и уменьшении радиуса пор, если только размеры самой сорбируемой молекулы не являются препятствием для ее проникновения в поры адсорбента (ситовый эффект). Важной эксплуатационной характеристикой адсорбента в случае выделения веществ из газовой фазы является верхняя температурная граница диапазона его применения, характеризующая условия эффективной термодесорбции адсорбированных примесей.
3.2.2.1. Молекулярная адсорбция
Закономерности молекулярной адсорбции определяются химической природой поверхности адсорбента. Согласно предложенной А.В. Киселевым [6] классификации все адсорбенты в зависимости от природы их поверхности можно разделить на три группы: 1) не содержащие локализованных зарядов; 2) содержащие положительные локализованные заряды и 3) содержащие отрицательные локализованные заряды.
К первой группе адсорбентов, которые называют неполярными, относятся углеродные сорбенты (табл. 3.16), не
полярные полимерные сорбенты (табл. 3.17) и модифицированные неполярными группами кремнеземы (табл. 3.18) [8-10]. Неполярные сорбенты используются для выделения и концентрирования из газовой и жидкой фазы неполярных и слабополярных веществ, молекулы которых не содержат полярных функциональных групп с повышенной электронной плотностью, а также для разделения последних. Межмолекулярное взаимодействие на неполярных адсорбентах обусловлено исключительно дисперсионными силами. Прочность адсорбции увеличивается по мере увеличения числа атомов в молекуле сорбата и соответствующего увеличения их молярной массы. В гомологических рядах прочность адсорбции закономерно возрастает с увеличением числа атомов углерода. Другая особенность проявляется в том, что менее разветвленные и транс-изомеры органических веществ адсорбируются прочнее более разветвленных и z/uc-изомеров, за счет меньших сте-рических препятствий.
При адсорбции из жидкой фазы коэффициенты адсорбции возрастают по мере уменьшения растворимости сорбата в растворителе. При одном и том же числе атомов углерода в молекулах органических соединений адсорбируемость на неполярных сорбентах из водной фазы уменьшается в ряду алкан > алкен > алкин > арен > простой эфир > сложный эфир > кетон > альдегид > спирт > амин.
Ко второй группе адсорбентов относятся силикагели, цеолиты (табл. 3.19) и оксид алюминия, а к третьей группе — полимерные сорбенты с привитыми полярными группами [9, 10] (табл. 3.20). Адсорбенты второй и третьей групп называют полярными. Подобные адсорбенты используются для разделения и концентрирования полярных соединений. Межмолекулярное взаимодействие молекул сорбата с поверхностью этих сорбентов обусловлено (наряду с универсальными, дисперсионными) индукционными и ориентационными силами. Применение этих сорбентов для концентрирования веществ из влажного газа и водных растворов ограничено в силу их высокого сродства к воде, которая адсорбируется гораздо сильнее, чем многие даже относительно высокомолекулярные соединения. Поэтому эти сорбенты используются для концентрирования полярных соединений из неполярных органических и газообразных сред. При этом сорбенты второй группы являются высокоэффективными осушителями последних.
Адсорбируемость на полярных сорбентах возрастает с увеличением полярности сорбатов и для органических веществ с одним и тем же числом атомов углерода уменьшается в ряду амин > спирт > альдегид > кетон > сложный эфир > простой эфир > арен > алкин > алкен > алкан. В случае адсорбции из газовой фазы увеличение молярной массы в гомологических рядах приводит к увеличению адсорбируемости веществ. Однако для полярных адсорбентов этот эффект выражен гораздо слабее, чем для неполярных, которые оказываются более предпочтительными для разделения гомологов.
Методы разделения и концентрирования
111
В случае адсорбции из жидких сред увеличение молярной массы в гомологических рядах для наиболее полярных соединений (амины, спирты, альдегиды) сопровождается уменьшением прочности адсорбции этих соединений на полярных адсорбентах. Это связано с тем, что увеличение числа метиленовых групп в гомологических рядах полярных соединений приводит к значительному уменьшению их полярности, в то время как дисперсионная составляющая межмолекулярного взаимодействия между адсорбентом и сорбатом при этом существенно не изменяется.
Таблица 3.16
Углеродные сорбенты [8,9]
Тип адсорбента Наименование Удельная поверхность, м2т' Диаметр пор, нм Область применения
Графитированная термическая сажа
Карбопак Разделение газов и легких углеводородов и их изо-меров, концен-трирование этих веществ из воздуха
А 15 —
С 12 —
В 100 1,3
В-НТ 100-110 1,3
В-НТ-100 100-110 1,3
Карбосфер 1200 1,3
Карбохром
1 7-10 —
2 80-100 —
Карбограф Концентрирова-ние органических соединений из воды
1 100 —
4 210 —
5 560 1,0-1,2
Угольное молекулярное сито
Карбосив 1000 — Разделение газов и их концентрирование из воздуха
Активированные угли
СКТ 1000- 1500 < 1 —
АГ 800-1000 < 1 Разделение га-зов, концентрирование газов из воздуха и орга-нических ве-ществ из водных растворов
Карбоксин 1000 1200 —
1002 1100 —
Гиперкарб PGC 120-240 —
Таблица 3.17
Пористые неполярные полимерные сорбенты [10]
Сорбент Тип Насыпная плотность, (кг-м 3)-10“3 Удельная поверхность, м2-г 1 Средний радиус пор, нм Температурный предел, °C
Порапак Р Ст-ДВБ 0,39 225- 350 250
Порапак PS ЭВБ-ДВБ 0,35 500- 700 4 250
Порапак Q Сила-низ. Р
Порапак QS Сила-низ. Q 4-5 250
Амберлиты XAD
XAD-1 ЭВБ-ДВБ 0,65 100 10 150
XAD-2 ЭВБ-ДВБ 0,65 330 4,5 150
XAD-4 ЭВБ-ДВБ 0,65 750 2,5 150
PRP-1 ВС-Ст-ДВБ — 415 4 —
Амбер-хром CG-161 ВС-Ст-ДВБ — 900 7-8 —
Лихролют EN ВС-ЭВБ-ДВБ — 1200 1,5 —
Изолют EVN ВС-Ст-ДВБ — 1100 40-45 —
Енви-хром Р ВС-Ст-ДВБ — 900 — —
Стиросорб MN-200 СС- ПСт — 1500 0,5 —
Стиросорб 2 СС- ПСт — 910 — —
Пояснения. АО — аминооксид; ВПир — винилпирролидон; ВС — высокосшитый; ДВБ — дивинилбензол; ПА — полиамид; ПС — полисорб; ПСт — полистирол; СО — сульфоксид; СС — сверхсшитый; Ст — стирол; ЭВБ — этилвинилбензол; ПДВБ — полидивинилбензол.
112
Новый справочник химика и технолога
Таблица 3.18
Объемно-пористые кремнеземы с привитыми неполярными группами [10]
Марка сорбента Основа сорбента Привитая группа Удельная поверхность, м^г1 Средний диаметр пор, нм
я-Октан/Пора-сил С Порасил С я-Октан 50-100 20^40
С18/Порасил В Порасил В С18 125-200 10-20
Фенил/Пора-сил В Порасил В Дифенил-силан 125- 200 10-20
М-Бондапак С18 М-Порасил С18
GC Бондапак С18 Порасил С С18 50-100 20^40
Лихросорб RP-8 SI 60 С8 400- 500 6
Микропак-СН SI60 С18 400-500 6
Видак ТР
201 обращенная фаза Видак 101 ТР С18 100 33
Зорбакс-ODS Зорбакс -SIL С18 250- 350 4
Партисил-10
ODS Партисил 10 С18 >400 4—6
Продолжение таблицы 3.18
Марка сорбента Основа сорбента Привитая группа Удельная поверхность, mV Средний диаметр пор, нм
Сферисорб-ODS Сфери-сорб С18 200
Сферосил
SIA075 Сферосил ХОВО75 Гидрофобная 10'0 30
SIB 075 То же То же То же То же
SIC 075 То же То же То же То же
Нуклеосил
С8 Нуклеосил 100 С8 300 10
С18 С18
Таблица 3.19
Цеолиты — молекулярные сита [8]
Тип адсорбента Марка молекулярного сита Диаметр пор, нм • 103
Калия алюмоси- ЗА 0,3
КА 0,3
Натрия алюмоси-ликат 4А 0,4
NaA 0,4
13Х 1,0
NaX 0,9-1,0
Кальция алюмо-силикат 5А 0,5
СаА 0,5
10Х 0,8
СаХ 0,8
Таблица 3.20
Пористые полимерные полярные сорбенты [10]
Сорбент Тип Насыпная плотность (кг • м 3)- 10 3 Удельная поверхность м2 • г 1 Средний радиус пор, нм Темепера-турный предел, °C
Порапак N Впир 0,39 225-350 4 200
Порапак R Впир 0,33 450-600 4 250
Порапак S Впир 0,35 300-450 5 250
Порапак Т ЭГДА 0,44 250-300 10 200
Амберлиты XAD
XAD-7 АЭ — 450 4 —
XAD-8 АЭ — 140 12,5 —
XAD-9 СО — 70 18 —
XAD-11 ПА — 70 17,5 —
XAD-12 АО — 25 65 —
Хромосорб 104 АКН-ДВБ 0,32 100-200 30-40 250
Хромосорб 105 АЭ 0,34 600-700 20-30 250
Хромосорб 107 АЭ — 100-200 0,25 250
Полисорбонитрил АКН-ДВБ 0,34 80-100 60-70 200
Полисорбатазол П-ВМП-ЭГДМА 0,32 50 60-70 250
Нигрополисорб-1 ПС-1 cNO2 0,30 100-250 40-50 200
Аминополисорб-1 ПС-1 с NH2 0,30 100-250 50-60 250
Методы разделения и концентрирования
113
Продолжение таблицы 3.20
Сорбент Тип Насыпная плотность (кг-м 3)- 10 3 Удельная поверхность м^г"1 Средний радиус пор, нм Темепера-турный предел, °C
Нитрополисорб-2 ПС-2 c NO2 0,32 40-50 180-200 180
Аминополисорб-2 ПС-2 c NH2 0,32 30-50 200-300 250
Тенакс GC ПДФФО 0,37 19 — 380
Хромосорб 108 АЭ — 100-200 0,25 250
Полиамидные порошки
MN-polyamid 0,25 — —- —
SC-6 Поли-£-капроамид 0,25 — — —
SC 6,6 Полигексаметилендиами-надипинат 0,25 — — —
DC 6 Поли-£-капроамид 0,25 — — —
DC 6 UV254 Поли-£-капроамид 0,25 — — —
DC 11 Поли-о-аминоундекамид 0,25 — — —
DC 11 UV254 Поли-о-аминоундекамид 0,25 — — —
DC 6,6 Полигексаметилен-диаминадипинат 0,25 — — —
DC 6,6 UV254 Полигексаметилен-диаминадипинат 0,25 — — —
DC 6 AC Поли-£-капроамид 0,25 — — —
DC 11 AC Поли-о-аминоундекамид 0,25 — — —
DC 6,6 AC Полигекс аметил ен-диаминадипинат 0,25 — — —
Пояснения. АКН — акрилонитрил; АО аминооксид; АЭ — акриловый эфир; ВПир — винилпирролидон; ДВЕ — дивинилбензол; ПА полиамид; ПВМП — н-винил-3(5)-метилпиразол; ПДФФО — р-2,6-дифенилфениленоксид; ПС — полисорб; ПСт — полистирол; СО — сульфоксид; Ст — стирол; ЭГДМА — этиленгликоль диметакрилат.
3.2.2.2. Ионный обмен
Сорбенты, проявляющие способность к ионному обмену (иониты или ионообменники), представляют собой полимерные вещества, которые содержат функциональные группы, способные при контакте с растворами электролитов к обмену ионов. В зависимости от химической природы полимерной матрицы иониты делятся на два больших класса: неорганические и органические. Первые в свою очередь делятся на природные и синтетические.
Неорганические иониты
С природных неорганических ионитов начиналась история ионного обмена и как явления, и как технологического процесса [10]. Сведения о синтетических неорганических ионитах можно найти в [12,13]. Ионообменные свойства проявляют многие малорастворимые неорганические соединения природного и синтетического происхождения: оксиды, гидроксиды, соли минеральных кислот, кислородсодержащие кислоты (молибденовая, вольфрамовая, сурьмяная), гетерополикислоты и их соли, алюмосиликаты, сульфиды, цианоферраты. Выделяют четыре основных типа неорганических ионообменников: окси-гидратные, сульфидные, цианоферратные и сорбенты на основе гетерополярных солей [14]. К этому перечню можно добавить металлосорбенты [15].
Подавляющее большинство минеральных и синтетических ионообменников относят к первому типу. Оксигид-ратные сорбенты соответствуют общей формуле
(НО)хМр2Ор.х • пН 2О,
где М и Э —- элементы 3-6 групп Периодической системы. При значениях z, начиная с нуля, этой формуле соответствуют простые и смешанные оксиды, гидроксиды, кислородсодержащие кислоты и гетерополикислоты и их соли. Для оксигидратных сорбентов возможны различные механизмы сорбции, включая разные виды межмолекулярных взаимодействий молекул сорбата с поверхностными группами сорбента. Как ионообменники они чаще всего проявляют способность к обмену ионов водорода на ионы металла. Менее характерна для них ионообменная функция с замещением ОН" -ионов на анионы минеральных кислот или ацидокомплексы. Из ионообменников этого типа с точки зрения практической значимости в химическом анализе можно выделить фосфоромолибдат аммония, сурьмяную и сурьмяно-кремниевые кислоты, гидратированные диоксиды циркония, титана и олова. Для фосфоромолибда-та аммония характерна высокая селективность к ионам тяжелых щелочных металлов — рубидия и цезия — по отношению к подавляющему большинству других элементов Периодической системы. При этом обеспечивается
114
Новый справочник химика и технолога
обратимость сорбции. Сурьмянокислотные сорбенты в зависимости от условий синтеза могут проявлять селективность к катионам с различной величиной заряда. Гидратированные диоксиды циркония, титана и олова интересны своими анионообменными свойствами, позволившими проводить выделение ацидокомплексов платиновых металлов [16].
Сульфидные сорбенты, представляющие собой индивидуальные или смешанные сульфиды, отвечают общей формуле
MV3.SZ.
Экспериментально доказано, что на поверхности сульфидов наблюдается повышенная концентрация H2S и ионов HS”. Сульфидные сорбенты могут проявлять обменные свойства по отношению к ионам металлов аналогично ок-сигидратным сорбентам, но более вероятной является сорбция с замещением М или Э на ионы других халько-фильных элементов. Сульфидные сорбенты проявляют избирательность к ^-элементам, способным образовывать малорастворимые сульфиды. Практически они могут применяться только как коллекторы при концентрировании.
Цианоферратные сорбенты
M3?[Fe(CN)6]z,
где, как правило, М — однозарядный, а Э — двухзарядный ион соответственно. Эти сорбенты проявляют обменные свойства за счет способности к замещению однозарядного иона М. Наиболее широкое применение из сорбентов данного класса находит смешанный цианоферрат калия и кобальта, который проявляет исключительную селективность к ионам цезия, позволяющую проводить его концентрирование из морской воды [17]. Возможности аналитического применения цианоферратных сорбентов ограничены из-за необратимости сорбции.
Сорбенты на основе гетерополярных солей BaSO4, LaF3 способны к обмену ионов металлов и удерживанию ионов в дефектах структуры кристаллов. Например, BaSO4 после специальной обработки используют для селективного выделения микроколичеств стронция.
В рассмотренных выше случаях неорганические ионо-обменники проявляют селективность при сорбции катионных форм элементов. В качестве примера селективных неорганических анионообменников можно рассматривать сорбенты на основе металлического серебра и висмута, проявляющие высокую селективность к галогенид-ионам. Сорбция происходит по механизму анионного обмена с замещением адсорбированных на поверхности металла ОН- -ионов на галогенид-ионы.
Органические иониты
Основным классом ионообменных материалов являются органические иониты — органические полимерные материалы с привитыми к матрице функциональными груп
пами, способными к обмену ионов при контакте с растворами. Органические иониты различаются по составу и структуре полимерной матрицы и природе ионогенных групп. К органическим ионитам относятся химически модифицированные активированные угли и целлюлоза. Наибольшее же распространение получили ионообменные смолы (ИС) — ионообменники со специальной сетчатой полимерной матрицей. По структуре матрицы ИС подразделяются на гелевые с микропорами молекулярных размеров и макропористые с размерами пор до десятков нм. Матрицей ИС с гелевой структурой чаще всего является сополимер стирола и дивинилбензола с различным процентным содержанием последнего, определяющим жесткость сшивки полимера и, как следствие, такие важнейшие характеристики ИС, как набухаемость и кинетика сорбции (табл. 3.21).
Таблица 3.21
Набухаемость сульфированных сетчатых стирол-дивинилбензольных катионитов [18]
Пояснение. Набухаемость (W, R) характеризуется количеством воды в граммах, связанной 1 г совершенно сухой смолы при ее максимальном набухании.
Процентное содержание дивинилбензола Набухаемость Процентное содержание дивинилбензола Набухаемость
2 3,45 10 0,83
4 1,92 15 0,59
6 1,36 20 0,48
8 1,04 25 0,38
По химической природе фиксированных в полимерном каркасе функциональных групп ИС подразделяются на катиониты и аниониты, представляющие собой поликислоты и полиоснования соответственно. По степени диссоциациии в контактирующих с ИС водных растворах катиониты подразделяются на слабо-, средне- и сильнокислотные; аниониты — на слабо-, средне- и сильноосновные.
Подробную информацию об ионном обмене, ионитах разных классов и областях их практического применения можно найти в [19, 20].
Сведения о промышленно-выпускаемых отечественных и зарубежных ионитах приведены в табл. 3.22-3.26.
В табл. 3.22-3.26 приняты следующие обозначения: АС — аналитический сорт; Инд. — сорт смолы с индикатором на истощение; ИП — изопористые смолы; МП — макропористые сорбенты; ФС — фармацевтический сорт (или сорт для использования в пищевой промышленности; ХС — хроматографический сорт; ЯМ — ядерная марка (сорт для использования в ядерных реакторах).
Методы разделения и концентрирования
115
Таблица 3.22
Полистирольные сульфокатиониты [10]
Марка смолы и фирма-изготовитель Содержание ДВБ, % Сорт смолы Ионная форма Полная обменная емкость АГ (Н Na), % Макс, рабочая тем-ра, °C Насыпная плотность, (кг-м3)-103 Влажность, % Зернение, нм-10 3
Мг-экв-г1 Мг-эквсм-3
Dowex (Dow, США)
1. 50WXI 1 Н или Na 5,0 0,4 150
2. 50W Х2 2 Н или Na 5,3 0,9 150 82
3. 50WX4 4 Н или Na 5,2 1,3 150 67
4. 50WX8 8 Нили Na 5,2 1,9 150 0,85 55
5. 50WX10 10 Н или Na 5,3 2,0 150 49
6. 50WX12 12 Н или Na 5,1 2,3 150 45
7. 50WX16 16 Н или Na 4,9 2,6 150
8. МР-50 МП Н или Na 4,6 1,7 46-52 300-840 150-300 75-150 40-75
BIO Red AG (BRL, США)
9. 53(MSC-1) МП Н или Na 4,5 1,7 50 300-840
10. AG50WX11 1 АС Н 5,0 0,4 150 84-92
11. AG 50WX2 2 АС Н 5,2 0,7 150 75-83
12. AG 50WX4 4 АС Н 5,2 1,2 150 64-72
13. AG 50WX8 8 АС Н 5,1 1,7 150 50-56
14. AG 50WX10 10 АС Н 5,0 1,9 150 46-52
15. AG50WX12 12 АС Н 5,0 2,3 150 42^48
16. AG 50WX16 16 АС Н 4,9 2,6 150 36-44
17. AG MP-50 АС Н 4,6 1,7 150 46-52 300-840 150-300 75-150 40-75
Bio-Rex RG (BRL, США)
18. 8 ЯМ Н(> 95%) 150 0,80 300-840
19. 8 ЯМ NH4(> 95%) 150 0,80 300-840
20. 8 ЯМ Li(> 99,9%) 150 0,80 300-840
21. 8 ЯМ 7Li(> 99,98%) 150 0,80 300-840
Dowex p.A. (Serva, Германия)
22. 50W X2 p.A 2 АС Н 150
23. 50W X4 p.A 4 АС Н 150
24. 50WX8 p.A 8 АС н 150
25. 50WX10 p.A 10 АС н 150
26. 50WX12 p.A 12 АС н 150
Amberlite (R&H, США)
27. IR-120 8 Na или Н 4,3 1,9 7 120 0,85 44-48 300-1200
28. IR-120 Plus 8 Na или Н 4,3 1,9 5-7 120 0,85 44-48 300-1200
29. IR-120-L 8 Na 4,3 1,9 7 120 0,85 44-48 500-1200
30. IRP-69 8 ФС Na 4,3 1,9 120 10-15 30-150
31. IRO-69M 8 ФС Na 4,3 1,9 120 10-15 <50
32. IRN-77 8 ЯМ Н(> 95%) >4,7 1,8 120 <55 300-1200
33. IRN-163 8 ЯМ Li(> 99%) >4,6 1,75 120 <55 300-1200
34. IRN-169 8 ЯМ NH4(> 99%) >4,4 1,7 120 <55 300-1200
116
Новый справочник химика и технолога
Продолжение таблицы 3.22
Марка смолы и фирма-изготовитель Содержание ДВБ, % Сорт смолы Ионная форма Полная обменная емкость АГ (H->Na), % Макс, рабочая тем-ра, °C Насыпная плотность, (кгм^КГ3 Влажность, % Зернение, нм-103
Мг-экв • г1 Мг-экв • см 3
35. IRN-218 8 ЯМ 7Li(> 99%) >4,6 1,75 120 <55 300-1200
36. IR-116 1 Na 0,3 120 0,80 85-95 300-1200
37. IR-118(h) 4,5 Н 4,3 1,5 5-10 120 0,74 56-60 300-1200
38.IR-122 10 Na 4,3 2,1 5-7 120 0,86 39-43 300-1200
39. IR-124 12 Na 4,3 2,2 5-7 120 0,86 37-41 300-1200
40. 200 >20 МП Na 4,3 1,75 3-5 150 0,77-0,83 46-51 300-1200
41. 200С >20 МП Na 4,3 1,75 3-5 150 0,77-0,83 46-51
42. 252 МП Na 1,80 3-5 150 0,77-0,83 45-50 300-1100
43. Stratabed 122 10 Na 2,1 5-7 120 0,86 40-44
Amberlite р.А. (Serva, Германия)
44. IR-120 8 АС H 1,9 120 44-48 400-1200
45. IR-120-HP 1 АС H 0,3 120 85-95 400-1100
46. IR-112 4 АС H 120 56-60 400-1100
47. IR-122 10 АС H 2,1 120 40-44 300-1200
48. IR-124 12 АС H 2,1 120 40-44 400-1100
49. 200 >20 АС H 1,75 140 47-52 400-1000
50. 252 1,75 140 45-50 400-1000
Amberlyst (R&H, США)
51. 15 МП H 4,6 1,8 120 0,34-0,38 <3 300-1200
52. XN-1005 МП H 3,4 1,5 120 <3 300-1200
53. XN-1010 МП H 3,3 1,0 120 <3 300-1200
Amberlyst p.A. (Serva, Германия)
54. 15 p.A. АС H 2,9 120 45-50 400-1200
55. Amberlite IR-120 AR (MCW, США) 8 АС H(> 98%) 5,0 1,75 150 0,77 49-55 300-840
Permitit (Perm, США)
56. Q-100 10 Na 4,6 1,9 120 0,80-0,88 45-55 300-1200
57. Q-101 10 H 4,6 1,9 120 0,77-0,85 45-55 300-1200
58. QHPF 10 АС H 4,6 1,9 120 0,77-0,85 45-55 150-600
59. Q-106 10 ЯМ NH4 4,6 1,8 120 0,77-0,85 40-50 300-1200
60. Q-107 10 ЯМ Li 4,6 1,8 120 0,77-0,85 45-55 300-1200
61. Q-108 10 ЯМ 7Li 4,6 1,8 120 0,77-0,85 45-55 300-1200
62. NQH 10 ЯМ H 4,6 1,8 120 0,77-0,85 45-55 300-1200
63. Q-110 12,5 Na 5,0 2,1 120 0,88-0,93 45-55 300-1200
64. Q-lll 12,5 H 4,6 1,9 120 0,77-0,83 45-55 300-1200
65. Q-130 15 Na 5,0 2,1 120 0,88-0,96 45-55 300-1200
66. Q-220 Na 3,9 1,75 120 0,80-0,88 45-55 300-1200
lonac (ICC, США)
67. C-240 10 Na 4,6 1,9 130(H), 140 (Na) 0,80-0,88 45-55 300-1200
68. C-242 10 H 4,6 1,9 130 (Н), 140 (Na) 0,77-0,85 45-55 300-1200
69. ANGC-242 10 АС H >4,0 130(H), 140 (Na) 0,77-0,85 45-55 300-1200
70. C-252 10 H >4,0 130(H), 140 (Na) 0,77-0,85 45-55 300-1200
Методы разделения и концентрирования
117
Продолжение таблицы 3.22
Марка смолы и фирма-изготовитель Содержание ДВБ, % Сорт смолы Ионная форма Полная обменная емкость ДР (H -> Na), % Макс, рабочая тем-pa, °C Насыпная плотность, (ком-3)-КГ3 Влаж- ность, % Зернение, нм10“3
Мг-экв • г 1 Мг-экв см 3
71. С-244 10 Н >4,0 130 (H), 140 (Na) 0,77-0,85 45-55 200-600
72. ANGC-244 10 АС Н >4,0 130(H), 140 (Na) 0,77-0,85 40-50 200-600
73. С-249 10 ФС Na >4,0 130 (H), 140 (Na) 0,80-0,88 45-55 300-1200
74. С-267 10 ФС Н >4,0 130(H), 140 (Na) 0,80-0,88 45-55 300-1200
75. С-253 10 ФС Na >4,0 130(H), 140 (Na) 0,80-0,88 45-55 600-1200
76. С-257 10 ФС Н >4,0 130 (H), 140 (Na) 0,80-0,88 45-55 600-1200
77. CI-294 10 Инд Na >4,0 130(H), 140 (Na) 0,80-0,88 45-55 300-1200
78. CI-295 10 Инд Н >4,0 130 (H), 140 (Na) 0,80-0,88 45-55 300-1200
79. NC-10 10 ЯМ Н(> 95%) 4,6 1,8 130(H), 140 (Na) 0,77-0,85 45-55 300-1200
80. NC-15 10 ЯМ NH4(> 95%) 4,6 1,8 130 (H), 140 (Na) 0,77-0,85 40-50 300-1200
81.NC-20 10 ЯМ Li(> 95%) 4,6 1,8 130(H), 140 (Na) 0,77-0,85 45-55 300-1200
82. NC-21 10 ЯМ 7Li(> 95%) 4,6 1,8 130(H), 140 (Na) 0,77-0,85 45-55 300-1200
83. С-250 12,5 Na >5,0 >2,3 130(H), 140 (Na) 0,88-0,93 35-45 300-1200
84. С-251 12,5 H >5,0 >2,3 130 (H), 140 (Na) 0,88-0,93 35^45 300-1200
85. С-258 12,5 ФС Na >5,0 >2,3 130(H), 140 (Na) 0,88-0,93 35^45 300-1200
86. С-259 12,5 ФС H >5,0 >2,3 130(H), 140 (Na) 0,88-0,93 35^45 300-1200
87. ANGC-243 12,5 АС H >5,0 >2,3 130(H), 140 (Na) 40-50 300-1200
88. С-255 15 Na 5,0 2,1 130 (H), 140 (Na) 0,88-0,96 35^45 300-1200
89. С-256 15 Н 5,0 2,1 130(H), 140 (Na) 0,88-0,96 35-45 300-1200
90. С-280 Na 3,9 1,7 130(H), 140 (Na) 0,80-0,88 35^45 300-1200
91. С-281 Н 3,9 1,7 130(H), 140 (Na) 0,80-0,88 35^45 300-1200
92. С-350 20 МП Na >4,6 >1,4 120 55-65 300-1200
DuoUte (D-S, США)
93. С 20 8 Na 5,1(H) 2,05 (Na) 7 120 0,86 43^46 300-1200
94.С 20 L 8 Na 5,1(H) 2,05 (Na) 7 120 0,86 43^46 600-1000
95. С 20 А 8 ФС Na 5,1(H) 2,05 (Na) 7 120 0,86 43-46 400-1200
96. С 25 D 5,5 Na 5,1(H) 1,6 (Na) 4 110 0,82 55-60 300-1200
97. С 26 8 МП Na 1,85 7 140 0,85 45-50 300-1200
118
Новый справочник химика и технолога
Продолжение таблицы 3.22
Марка смолы и фирма-изготовитель Содержание ДВБ, % Сорт смолы Ионная форма Полная обменная емкость AE (H -> Na), % Макс, рабочая тем-pa, °C Насыпная плотность, (кгм-3)-103 Влаж- ность, % Зернение, нм-10 3
Мг-экв • г”1 Мг-экв • см 3
98. С 26 С, 99. С 26 CI 8 МП Na 1,85 7 140 400-1200
100. С 261 5,5 МП Na 1,5 10 120 0,78 55-60 300-1200
101. С 261, 102. С 26 CI 5,5 МП Na 1,5 10 120 400-1200
103.С 264 >12 МП Na 2,5 6 140 0,90 40-43 300-1200
104. С 20-Nuclear 8 ЯМ H(> 99%) 5,0(H) 2,1 (Na) 120 0,80 40-50 300-1200
105. Micro-Ionex С-Н ЯМ H(> 99,5%) 5,0(H) 120 40-50 20-130
106. Micro-Ionex c-nh4 ЯМ NH4(> 98%) 4,7 (NH4) 120 40-50 20-130
107. С 27 МП Na 5,0(H) 2,1 (Na) 7 150 0,82 300-1200
108. С20-Х10 10 Na 2,3 (Na) 150 42 300-1200
109. С20-Х12 12 Na 2,5 (Na) 150 38 300-1200
110.С 202 А Na 5,1(H) 1,0 (Na) 14 120 300-1200
111. С204 Na 5,1(H) 2,6 (Na) 6 > 120 0,90 300-1200
112.С 208 Na 1,5 10 120 59 300-1200
Zerolit ZK (Zer, Англия), Zeo-Karb (Perm, Англия)
113. 225 8 Na 4,5-5,0 2,0-2,1 4-9 120 (H), 140 (Na) 0,85 46-52 300-1200 150-300
114. 325 10 H 4,8 2,0 4-9 120 (H), 140 (Na) 0,85 300-1200
115.425 Na 1,9 4-9 120 (H), 140 (Na) 0,85 300-1200
116. Resex Р (Cros, Англия) Na 4,8 2,0 120 0,90 50 300-800
Imac (Imac, Нидерланды)
117. С-12 8-10 Na 5,1 2,0 5 120 0,88 51 300-1200
118. С-14 14 Na 5,1 2,1 120 0,88 49 300-1200
119. С-8-Р 8 МП Na 5,1 1,7 5 120 0,86 57 300-1200
120. С-16-Р 16 МП Na 5,1 2,1 5 120 0,92 48 300-1200
Kastel (ME, Италия)
121. С-300 8 Na 4,25 5 100 0,85 45 300-1200
122. С-300 AGR 12 Na 4,5—4,7 3 120 0,89 45 300-1200
123. С-300Р 8 МП Na 1,7 120 0,84 53 300-1200
Relite (Rdl, Италия)
124. CF МП Na 5,0 2,0 5 120 0,85-0,90 43—45 300-1200
125. CFS МП Na 5,0 1,9 5 120 0,75-0,80 46-47 300-1200
126. CFZ Na 120 300-1200
127. СМ-2 Na+H 0,84 47
128. RedexCF МП
Diaion (M-I, Япония)
129. SK-1A 8 Na > 1,9 120 0,80-0,84 43-50 300-1200
130. SK-1B 8 Na > 1,9 120 0,80-0,84 43-50 300-1200
131. SK-1 AG 8 АС H 2,0-2,2 120 0,86-0,88 50-55 70-150
132. SK-102 2 Na >0,6 120 0,69-0,73 72-82 300-1200
133. SK-103 3 Na >0,9 120 0,71-0,75 64-74 300-1200
Методы разделения и концентрирования
119
Продолжение таблицы 3.22
Марка смолы и фирма-изготовитель Содержание ДВБ, % Сорт смолы Ионная форма Полная обменная емкость АГ (Н -> Na), % Макс, рабочая тем-ра, °C Насыпная плотность, (кгм-УКГ3 Влаж- ность, % Зернение, нм-10 3
Мг-экв • г"1 Мг-экв • см-3
134. SK-104 4 Na > 1,2 120 0,74-0,78 57-67 300-1200
135. SK-106 6 Na >1,6 120 0,78-0,82 47-57 300-1200
136. SK-110 10 Na >2,0 120 0,81-0,86 35-45 300-1200
137. SK-112 12 Na >2,1 120 0,82-0,87 32-42 300-1200
138. SK-116 16 Na >2,1 120 0,89-0,90 27-37 300-1200
Lewatit (Bayer, Германия)
139. S 100 4,75 2,2 120 0,8-0,9 300-1200
140. S 100GI Инд. 4,75 2,2 120 0,8-0,9 300-1200
141. S 100 BG 4,75 2,2 120
142. S 100WS 4,75 2,2 120
143. S 100 LF ФС 4,75 2,2 120 300-1200
144. СА9270 ФС Na, К, nh4, h, Mg (> 95%) 2,0 120 50 100-400
145. S 100KR/H ЯМ H 120
146. S 115 4,6 2,0 120 0,8-0,9 300-1000
147. SP 100 МП 4,75 1,6 120 0,7-0,8 300-2000
148. SP150 МП 1,9 120 0,7-0,8 300-2000
Lewasorb (Bayer, Германия)
149. CA9252/Na ФС Na 4,0 2-5 <50 (> 95%)
150. СА9252/К ФС К 4,0 2-5 <50 (> 95%)
151. СА9252/Н ФС H 4,0 2-5 <50 (> 95%)
152. СА 9252/NH4 ФС nh4 4,0 2-5 <50 (> 95%)
153. СА 9252 Mg ФС Mg 4,0 2-5 <50 (> 95%)
154. CA9279/Na ФС Na 4,0 50 < 100 (> 95%)
155. СА9279/К ФС К 4,0 50 < 100 (> 95%)
156. СА9279/Н ФС H 4,0 50 < 100 (> 95%)
157. СА 9279/NH4 ФС nh4 4,0 50 < 100 (> 95%)
158. CA9279/Mg ФС Mg 4,0 50 < 100 (> 95%)
159.lonenaus-tauscher I (Merck, Германия) H 4,5 2,6 5 90 0,8 45-55 300-900
160. PermititRS (Perm, Германия) АС Na 5,5 2,4 114 0,80 45 300-1200
Wofatit (СКВ, Германия)
161. KPS (KPS-200) 2 Na 300-1200
162. 4 Na 300-1200
163. 6 Na 300-1200
120
Новый справочник химика и технолога
Продолжение таблицы 3.22
Марка смолы и фирма-изготовитель Содержание ДВБ, % Сорт смолы Ионная форма Полная обменная емкость АГ (Н -> Na), % Макс, рабочая тем-ра, °C Насыпная плотность, (кг-м^-Ю3 Влажность, % Зернение, нм-10 3
Мг-экв • г 1 Мг-экв • см 3
164. Serdolit-Rot (Serva, Германия) АС, Инд. Н(98%) 1,8 1
165. КРС-200 8 Na 4,7 (Н) 1,8 (Н) 10 115 0,8-0,9 45053 30-1200
166. КРС-200 16 Na 300-1200
167. KPSrein 168. -171 2,4,6,8, 16 АС Н 300-1000
172. RH ЯМ Н 300-1300
173. RK ЯМ К 300-1300
174. RL ЯМ Li 300-1300
175. RN ЯМ nh4 300-1300
176. KS 10 ЯМ Na 4,4 (И) 1,5 (Н) 10 120 0,75-0,85 48-58 300-1200
Varion (CKV, Венгрия)
177. KS Na+Ca 5,0 2,2 135 0,75-0,8 44—47 250-1100
178. KSM МП Na+Ca 4,95 2,2 135 350-1000
179. PKS(K-POR)+ Смешан. 2,4(H) 100 40-100
180. KO Na+Ca 4,9 2,0-2,2 135 300-1100
(Россия)
181.КУ-2-8 8 H+Na 4,7—4,8 1,3-1,6 120-130 0,6-0,9 40-60 300-1500
182. КУ-23 (КУ-2-8П) 8 МП H 4,5 120-130 50-60 300-1500
183. КУ-2-12П 12 МП >4,95 1,5 62 300-1500
184. СБС-1 3(Na) 1,0 0,5-0,7 20-40 300-1500
185. СБС-2 4,5(Na) 0,9-1,1 0,6 300-1500
186. СБС-3 5 (Na) 0,6-1,0 0,5 300-1500
187. СДВ-3 4,5 1,3-1,5 0,5-0,6 300-1500
188. КРС-2П 2 АС H 4,8-5,2 0,65-0,75
189. КРС-ЗП 3 АС H 4,8-5,2 0,65-0,75 (> 95%)
4,8-5,2 0,65-0,75 600-800
190. КРС-5П 5 АС H 4,8-5,2 0,65-0,75 400-600
191. КРС-6П 6 АС H 4,8-5,2 0,65-0,75 200-300
192. КРС-7,5П 7,5 АС H 4,8-5,2 0,65-0,75 80-120
193. КРС-8П 8 АС H 4,8-5,2 0,65-0,75 40-60
194. КРС-10П 10 АС H 4,7-5,1 0,75-0,85 (> 95%)
195. KPC-2M 2 АС H 4,8-5,2 0,55-0,65 600-800
196. КРС-ЗМ 3 АС H 4,8-5,2 0,65-0,75 400-600
197. КРС-5М 5 АС H 4,8-5,2 0,65-0,75 200-300
198. КРС-8М 8 АС H 4,8-5,2 0,65-0,75 80-120
199. КРС-10М 10 АС H 4,7-5,1 40-60
200. КРС-2ПТ-40 2 МП, АС H 4,8-5,2 600-800
201. КРС-ЗПТ-40 3 « H 4,8-5,2 400-600
202. КРС-5ПТ-40 5 « H 4,8-5,2 200-300
203. КРС-8ПТ-40 8 « H 4,8-5,2 80-120
204. КРС-10ПТ-40 10 « H 4,8-5,2 40-60
Методы разделения и концентрирования
121
Примечания. 4. Основная марка. 8,17. Сорта с зернением мельче 50 меш получены размолом (гранульные). 27. Основная марка. Набухаемость: см. [8], раздел 77. 28. Сорт смолы повышенного качества. 29-35. Сорта смолы № 27. 43. Сорт смолы № 38 с узким диапазоном зернения (для водоподготовки: улучшенные гидравлические характеристики). 51-54. Смолы для катализа и ионного обмена в неводных средах. См. [8], разд. 75. 66. Смола сильносшитая. 67, 68, 79-82, 83, 88, 90. Идентичны смолам №№ 56, 57, 59-62, 63, 65, 66, 93 соответственно. Основная марка. Шкала селективности: см. [8], разд. 61. 98,99. Строго стандартизованные сорта смолы с крупным зернением (эфффектив-ное зернение 500-600 мкм, по сравнению с 400-450 мкм у основного сорта № 97). Назначение: высокоскоростная фильтрация (№ 98) и использование в непрерывных процессах с циркуляцией смолы (№ 99). 101, 102. К примечанию к №№ 98, 99 соответственно. 103. Смола имеет повы-
шенную специфичность к кальцию. 105, 106. Используют в смешанном слое, для деионизации и для поглощения коллоидов. 112. Эффективное зернение 450-550 мкм. 115. Особо сильнокислотная смола, с повышенной устойчивостью к действию свободного хлора. 117. Основная марка. 123. Эффективное зернение 550 мкм. 126. Гранульная смола, отличается от № 125 большей сшитостью. 127. Бифункциональная смола, содержит также карбоксильные группы; используют при pH > 5. 130. Кислотоустойчивый сорт. 141. Крупнозернистый сорт для фильтрования с высокой скоростью. 142. Смола для изпользования в «кипящем» слое. 144. Отличается от смолы № 143 только зернением. 146, 148. Отличаются от смол №№ 139 и 147 соответственно большей сшитостью. 186-188. Смолы на основе стирола и бутадиена, с различной степенью сульфирования. Форма зерен — гранулы. 190-204. Синтезированы с использованием п- или jw-ДВБ. Набухаемость см. [8], разд. 76.
Таблица 3.23
Карбоксильные катиониты полиакриловые [10]
Марка смолы и изготовитель Содержание ДВБ, % Сорт смолы Ионная форма Полная обменная емкость АГ (H->Na), % Макс, рабочая тем-ра, °C Насыпная плотность, (кгм-3)10“3 Влажность, % Зернение, нм-10 3
Мг-экв -г 1 Мг-экв - см"3
Amberlite (R&H,CUIA)
1. IRC-50 5 МП Н 10,0 3,5 75-100 120(H,Na) 0,69 43-53 300-1200
2. IRP-64 5 ФС Н 10,0 3,5 75-100 120 (H,Na) 10-15 < 150
3. IRP-64M 5 ФС Н 10,0 3,5 75-100 120 (H,Na) 10-15 <50
4. IRP-88 5 ФС К 10,0 3,5 75-100 120 (H,Na) 10-15 < 150
5. IRC-72 МП 2,0 80-85 120 (H,Na) 0,74 71-73 300-1200
6. DP-1 МП 2,5 75-100 120 (H,Na) 0,80 65-70 300-1200
7. IRC-84 н 10,5 3,5 60-70 120 (H,Na) 0,75 43-60 300-1200
8. Stratabed 84 н 10,5 3,5 60-70 120 (H,Na) 0,75 43-50
Amberlite AR(MCW,CUIA)
9. IRC-50 AR 5 АС Н (> 98%) 10,0 3,5 100 150 0,69 40-50 300-840
Amberlite p.A. (Serva, Германия)
10. IRC-50 p.A 5 АС н 3,5 120 43-53 400-1200
11. IRC-75 p.A АС н 4,0 120 43-53 300-1200
12. IRC-84 p.A АС н 3,5 120 43-53 300-1200
13. Dowex CCR(Dow, США) н 10,5 4,1 443- 53 300-1170
Duolite (D-S,CUIA)
14. CC-3 н 4,5 90 120 0,76 48-53 300-1200
15. C 464 МП н 3,5 50 120 0,75 57-62 300-1200
16. CS 101 10 МП н 10,2(H) 3,3(Н) 90 100 0,69 300-1200
17. Bio-Rex 70 (BRL,США) 10 МП, АС Na 10,2(H) 3,3(Н) 70 100 65-74 300-1200 150-300 75-150 40-75 <40
122
Новый справочник химика и технолога
Продолжение таблицы 3.23
Марка смолы и изготовитель Содержание ДВБ, % Сорт смолы Ионная форма Полная обменная емкость АГ (H-»Na), % Макс, рабочая тем-ра, °C Насыпная плотность, (кгмУЮ’3 Влажность, % Зернение, нм-10”3
Мг-экв г 1 Мг-экв см 3
18. Permitit Q-210 [Perm, США] Н 6,5 3,5 120 0,75 40-50 300-1200
19. lonac С-270 (ICC,США) 2,5 7,9-8,2 3,5-3,7 95 300-2000
Zerolit ZK (Zer, Англия); Zeo-Karb (Perm, Англия)
20. 226 2,5 Н 9-10 3,5—4,5 100 0,75 41-50 300-1200
21. 236 Н 5-16 100 0,77 300-1200
22. 227 Na 4,0 100 0,87 300-1200
23. Imac Z-5 (Imac, Нидерланды) МП Н 3,0 60 100 0,85 48 300-1200
Relite CC [RDI, Италия] 10 Н 10,0 3,5 18-25 (Н-Са) 120 0,75-0,80 46—48 300-1000
24. Kastel C-100 (ME, Италия) 10 н >6,0 50 110 0,87 50 300-1200
25. Lewatit CNP (Bayer, Германия) МП 3,8 80 0,7-0,8 300-1500
26. Lewasorb CA 9257 (Bayer, Германия) ФС Na, К, Н, Mg, NH4(> 95%) 9 2-5 <50 (> 95%)
'll. lone-naustauscher IV (Merck, Германия) 10 6,5 55 90 0,7 45-55 300-900
28. Permutit (Perm, Германия) 9-10 4,0 100 300-1200
Карбоксильные катиониты (Россия)
29. КБ-4 10 8,5 4,2 35 0,55-0,6 <50 300-2000
30. КБ-4П-2 2,5 Na >9,5 3,5 100 0,68-0,82 <75 250-1000
31. КБ-41 МП Н 6,0-7,5 110 300-2000
Таблица 3.24
Аниониты сильноосновные [10]
Пояснение. В качестве исходных веществ для синтеза сильноосновных анионитов, если это специально не оговорено, используют стирол, ДВБ и триметиламин или диметилэтаноламин. В первом случае образуются иониты с активными группами -N(CH3) («тип 1»), во втором — с группами -N+(CH3)2C2H4OH («тип 2»).
Марка смолы и фирма-изготовитель Тип активных групп Содержание ДВБ, % Сорт смолы Ионная форма Полная обменная емкость АГ (H->Na), % Максимальная рабочая температура, °C Насыпная плотность, (кг-м”3)-10”3 Влажность, % Зернение, нм-10 3
Мг-экв • г 1 Мг-экв • см 3
Dowex (Dow, США)
1. 1-Х1 1 1 С1 3,6 0,4 150(d), 50(ОН) 84
2. 1-Х2 1 2 С1 3,6 0,9 150(С1), 50(ОН) 75
3. 1-Х4 1 4 С1 3,5 1,2
Методы разделения и концентрирования
123
Продолжение таблицы 3.24
Марка смолы и фирма-изготовитель Тип активных групп Содержание ДВЕ, % Сорт смолы Ионная форма Полная обменная емкость АГ (Н—>Na), % Максимальная рабочая температура, °C Насыпная плотность, (кгм3)-103 Влажность, % Зернение, нм-10'3
Мг-экв • г-1 Мг-экв • см3
4. 1-Х8 1 8 С1 3,5 1,4 150(d), 50(ОН) 43
5. 1-Х10 1 10 С1 3,0 1,5 17 150(d), 50(ОН) 0,72
6. 1-Х16 1 16 С1 2,0 1,2 150(d), 50(ОН)
7. 11 1 4-5 С1 4,0 1,24 150(d), 50(ОН) 55 840- 1170 300-840
8. 21К 1 С1 4,5 1,2 17 150(d), 50(ОН) 0,72 57 840- 1170 300-840 300-500
9. МР-1 1 МП С1 4,2 1,2 300-840 150-300 80-150 40-80
10. 31(MSA-1) 1 МП С1 4,2 1,0 64 400-1200
11. 2-Х1 2 1 С1 150(d), ЗО(ОН)
12. 2-Х2 2 2 С1 150(d), ЗО(ОН)
13. 2-Х4 2 4 С1 3,7 1,2 150(d), ЗО(ОН) 53
14. 2-Х8 2 8 С1 3,6 1,4 12 150(d), ЗО(ОН) 0,72 38
15. 2-Х10 2 10 С1 150(d), ЗО(ОН)
16. 2-Х16 2 16 С1 150(d), ЗО(ОН)
Bio-Rad AG (BRL, США)
17. AG 1-X1 1 1 АС С1 3,2 0,4 150(d), 50(ОН) 80-90
18. AG 1-X2 1 2 АС С1 3,5 0,8 150(d), 50(ОН) 70-78
19. AG 1 X4 1 4 АС С1 3,5 1,2 150(d), 50(ОН) 59-65
20. AG 1-X8 1 8 АС С1 3,2 1,4 150(d), 50(ОН) 39-45
21. AG1-X10 1 10 АС С1 3,0 1,5 150(d), 50(ОН) 34-42
22. AG21K 1 АС С1 4,5 1,3 150(d), 50(ОН) 52-62
23. AGMP-1 1 АС С1 4,2 1,2 56-64
24. AG 2-X4 2 4 АС С1 3,2 1,2 150(d), ЗО(ОН) 54-60
25. AG 2-X8 2 8 АС С1 3,2 1,4 150(d), ЗО(ОН) 34-40
26. AG2-X10 2 10 АС С1 3,0 1,5 150(d), ЗО(ОН) 28-36
124
Новый справочник химика и технолога
Продолжение таблицы 3.24
Марка смолы и фирма-изготовитель Тип активных групп Содержание ДВБ, % Сорт смолы Ионная форма Полная обменная емкость дг (H->Na), % Максимальная рабочая температура, °C Насыпная плотность, (кг-м 3)-10~3 Влажность, % Зернение, нм-10 3
Мг-экв • г 1 Мг-экв • см 3
Bio-Rex RG (BRL, США)
27. RG 1-Х8 1 8 ЯМ ОН (> 90%) 0,67 300-840
28. RG2-X8 2 8 ЯМ ОН (> 90%) 300-840
Dowex р. A. (Serva, Германия)
29. IXlp.A. 1 1 АС С1 300-840
30. 1 Х2 р. А. 1 2 АС С1 150-300
31. 1 Х4р. А. 1 4 АС С1 80-150
32. 1 Х8 р. А. 1 8 АС С1 40-80
33. 21 Кр.А. 1 АС С1 840- 1170
34. 2X4 р. А. 2 4 АС С1 300-840
35. 2X8 р. А. 2 8 АС С1 150-300
36. 2X10 р. А. 2 10 АС С1 < 150
Amberlite (R&H, США)
37. IRA-400 1 8 С1 1,4 18-22 75(G), 60(ОН) 0,71 42-48 300- 1200
38. IRA- 400С 1 8 С1 1,4 18-22 75(G), 60(ОН) 0,71 42-48
39. IRN-78 1 8 ЯМ он (> 80%) ЗБ5 0,8 18-22 75(G), 60(ОН) 60 300- 1200
40. IRP-67 1 8 ФС С1 1,2 18-22 75(G), 60(ОН) 10-15 30-150
41. IRP-67M 1 8 ФС С1 18-22 75(G), 60(ОН) 10-15 <50
42. IRA- 4018 1 8 С1 0,8 5-10 75(G), 60(ОН) 0.69 59-65 300- 1200
43. IRA-402 1 8 С1 1,25 20-25 75(G), 60(ОН) 0,69 50-57 300- 1500
44. IRA-425 1 8 С1 1,35 18-22 75(G), 60(ОН) 0,69 49-55 840- 1400
45. IRA-430 1 8 С1 1,1 18-22 75(G), 60(ОН) 0,69 50-57 840- 1400
46. IRA-410 2 8 С1 1,35 10-15 75(G), 40(ОН) 0,71 38-44 300- 1200
47. IRA-458 С1 1,25 10-15 75(G), 40(ОН) 0,72 59-62
48. XE-268 С1 0,76 25-30 75(G), 60(ОН) 0,69 70-80
49. IRA-900 1 8 МП С1 1,0 15-20 75(G), 60(ОН) 0,67 58-64 300- 1200
50. IRA- 900C 1 8 МП С1 1,0 15-20 75(G), 60(ОН) 0,67 58-64
51. IRA-904 1 МП С1 0,7 3-5 75(G), 60(ОН) 0,67 56-62 300- 1000
52. IRA-910 2 4 МП С1 1,0 12-17 75(G), 40(ОН) 0,67 55-60 300- 1200
53. IRA-938 МП С1 0,53 8-10 75(G), 40(ОН) 0,56 69-77 300- 1200
Методы разделения и концентрирования
125
Продолжение таблицы 3.24
Марка смолы и фирма-изготовитель Тип активных групп Содержание ДВБ, % Сорт смолы Ионная форма Полная обменная емкость дк (H->Na), % Максимальная рабочая температура, °C Насыпная плотность, (кг-м”3)-10”3 Влажность, % Зернение, нм-10”3
Мг-экв г1 Мг-экв • см”3
54. Stratea-bed-402 1 6 С1 1,25 20-25 75(С1), 60(ОН) 0,69 50-57
Amberlyst (R&H, США)
55. A26 1 МП С1 1,1 75(С1), 60(ОН) 0,62-0,69, 61 ЗОО-КОО
A27 1 МП С1 0,7 75(С1), 60(ОН) 0,64-0,67 45 ЗОО-КОО
Amberlite p.A. (Serva, Германия)
56. IRA-400 p.A. 1 8 АС С1 3,7 1,2 16 75(С1), 60(ОН) 0,71 45-50 400- 1000
57. IRA-401 p.A. 1 4 АС С1 3,2 0,8 21 75(С1), 60(ОН) 0,69 60-65 400- 1000
58. IRA-401 Sp.A. 1 4 АС С1 3,4 0,8 21 75(С1), 60(ОН) 0,69 60-65 400- 1000
59. IRA-402 p.A. 1 6 АС С1 4,2 1,2 18 75(0), 60(ОН) 0,72 53-60 400- 1000
60. IRA-404 p.A. 1 8 АС С1 4,2 1,6 17 75(0), 60(ОН) 0,72 45—49 400- 1000
61. IRA-425 p.A. 2 8 АС С1 4,2 1,3 17 75(0), 60(ОН) 0,69 50-53 400- 1000
62. IRA-410 p.A. 1 8 АС С1 3,3 1,3 11 75(0), 35(ОН) 0,70 45-50 400- 1000
63. IRA-900 p.A. 2 8 МП, АС С1 4,4 0,7 17 75(0), 60(ОН) 0,67 56-62 400- 1000
64. IRA-904 p.A. 2 8 « С1 2,6 0,8 5 75(0), 60(ОН) 0,69 56-62 400- 1000
65. IRA-911 p.A. 2 8 « С1 2,4 1,1 3 75(0), 35(ОН) 0,69 45-50 400- 1000
Amberlyst p.A. (Serva, Германия)
66. A-26 p.A. 1 « С1 4,1-4,4 1,1 75(d), 60(ОН) 61-65 400- 1000
67. A-27 p.A. 1 « С1 2,5-2,7 0,7 75(0), 60(ОН) 56-62 400- 1000
68. A-29 p.A. 2 « VI 2,6-2,8 1,1 75(0), 35(ОН) 42-48 400- 1000
69. Triiodide-Resin 1 8 МП, ФС h 300-840
Amberlite AR (MCW, США)
70. IRA-400 AR 1 8 АС Cl 3,3 1,2 100(0), 60(ОН) 0,65 42—48 300- 840
71. IRA-401 AR 1 4 АС Cl 3,4 0,8 100(0), 60(ОН) 0,67 59-65 300- 840
72. IRA- 4018 AR 1 4 АС Cl 3,5 0,8
Permitit (Perm, США)
73. S-100 1 8 Солевая 3,6 1,0 10-15 100(0), 60(ОН) 0,69-0,74 50-60 ЗОО-КОО
74. S-101 1 8 ОН 3,5 1,0 10-15 100(0), 60(ОН) 0,64-0,72 50-60 300- 1200
126
Новый справочник химика и технолога
Продолжение таблицы 3.24
Марка смолы и фирма-изготовитель Тип активных групп Содержание ДВБ, % Сорт смолы Ионная форма Полная обменная емкость AF (H->Na), % Максимальная рабочая температура, °C Насыпная плотность, (кг-м“3)-103 Влажность, % Зернение, нм-10 3
Мг-экв • г 1 Мг-экв см 3
75. S-105 1 8 ОН 3,5 1,0 10-15 юо(с1), 60(ОН) 0,64-0,72 50-60 300- 1200
76. S-200 2 8 Солевая 3,5 1,3 10-15 100(С1) 0,69-0,74 45-55 300- 1200
77. S-201 2 8 ОН 3,7 1,3 10-15 60(ОН) 0,64-0,72 45-55 300- 1200
lonac (ICC, США)
78. А-540 1 9 Солевая 3,5-3,6 1,0 10-15 100(С1), 60(ОН) 0,69-0,74 50-60 300- 1200
79. А-542 1 9 ОН 3,5-3,6 1,0 10-15 100(С1), 60(ОН) 0,64-0,72 50-60 300- 1200
80. ANGA- 542 1 9 АС ОН 3,5-3,6 1,0 10-15 юо(с1), 60(ОН) 0,64-0,72 60-70 300- 1200
81.NA-30 1 9 ЯМ ОН 3,5-3,6 1,0 10-15 100(d), 60(ОН) 0,64-0,72 50-60 300- 1200
82. А-544 1 9 ФС Солевая 3,5-3,6 1,0 10-15 100(d), 60(ОН) 0,69-0,74 55-60 300- 1200
83. А-548 1 9 « 3,5-3,6 1,0 10-15 100(С1), 60(ОН) 0,69-0,74 55-60 400- 1200
84. А-546 1 >9 « 3,5 1,3 10-15 100(С1), 70(ОН) 0,77-0,83 50-55 300- 1200
85. А-935 1 Солевая 3,5 1,0 Ю0(С1,ОН) 0,64-0,80 55-60 300- 1200
86. А-936 1 ОН 3,5 1,0 Ю0(С1,ОН) 300- 1200
87. A-IP 1 1,8 Солевая >4,0 Ю0(С1,ОН) 50-60 300- 1200
88. А-641 1 16 МП « >3,7 70 60-70 300- 1200
89. А-550 2 9 « >3,3 1,3 10-15 100(С1), 40(ОН) 0,69-0,74 45-55 300- 1200
90. А-552 2 9 ОН >3,3 1,3 10-15 юо(С1), 40(ОН) 0,64-0,72 45-55 300- 1200
91. А-558 2 9 Солевая >3,3 1,3 10-15 100(С1), 40(ОН) 0,69-0,74 45-55 400- 1200
92. А-2 2 4 ОН 100(d), 40(ОН) 300- 1200
Duolitr (D-S, США)
93. А 101 D 1 ИП С1 4,0 1,3 12 100(С1), 60(ОН) 0,74 50-55 300- 1200
94. A 101D-Nuclear 1 ИП ОН 1,3(С1) 12 юо(с1), 60(ОН) 0,70 300- 1200
95. А 121 1 ИП С1 4,0 1,3 12 100(С1), 60(ОН) 0,74 54-57 300- 1200
96. А 140 1 ИП С1 1,0 15 100(С1), 60(ОН) 0,73 60-65 300- 1200
97. А 161 1 МП С1 3,4(С1) 1,0(С1) 17 100(d), 60(ОН) 0,73 53-58 300- 1200
98. А 161 С 1 ИП С1 3,4(С1) 1,0(С1) 17 юо(С1), 60(ОН)
Методы разделения и концентрирования
\21
Продолжение таблицы 3.24
Марка смолы и фирма-изготовитель Тип активных групп Содержание ДВБ, % Сорт смолы Ионная форма Полная обменная емкость АГ (H-»Na), % Максимальная рабочая температура, °C Насыпная плотность, (кг-м'3)103 Влажность, % Зернение, нм-10'3
Мг-экв • г1 Мг-экв • см-3
99. А 161 С1 МП С1
100. А 171 Р 1 ИН С1 0,8 23 100(С1), 60(ОН) 63-68 300- 1200
101. ES-109 1 МП С1 300- 1200
102. ES-104 1,2 С1 300- 1200
103. А 102D 2 ИП С1 3,8-4,2 1,4 6 75(С1), 35(ОН) 0,76 45-50 300- 1200
104. А 162 2 ИП С1 3,35(С1) 1,0 10 75(С1), 35(ОН) 0,745 50-55 300- 1200
105. А 161 С 2 МП С1 3,35(С1) 1,0 10 75(С1), 35(ОН)
106. А 162 С1 ИП С1
107. Micro-Ionex А-ОН 1 ям он 4,2(ОН) 100 «70 5-90
Zerolit DA (Zer, Англия); De-Acidite (Perm, Англия)
108. FF-IP 1 (3-5) ИП С1 4,0 1,2 6-14 60(С1), 40(ОН) 0,69 52-58 300- 1200
109. FX-IP 1 (3-5) ИП С1 4,0 1,2 6-14 60(С1), 40(ОН) 0,69 300- 1200
110. FF-DVB 1 С1 60(С1), 40(ОН) 300- 1200
111. TR 1 МП С1
112. K-MP 1 (3-5) МП С1 2,8 0,8 60(С1), 40(ОН) 0,74 52-58 300- 1200
113. P-IP 1,2 (3-5) ИП С1 3,5-4,0 1,15 40(С1) 0,72 52-57 300- 1200
114. N-IP 2 (3-5) ИП С1 3,5^1,0 1,1 4-8 40(С1) 0,72 50-57 300- 1200
115. NX 2 С1 1,1 40(С1) 0,69 300- 1200
116. Rezanex HBT (Cros, Англия) 1 С1 3,5 1,5 60 0,69 50 300- 1000
Imac (Imac, Нидерланды)
117. S 5^10 1 МП С1 3,8 1,0-1,2 20 100(С1), 50(ОН) 0,73 58 400-800
118. S5-50 1 МП С1 3,2 1,2 15 юо(с1), 50(ОН) 0,73 53 400- 800
119. S 5-52 2 МП С1 3,0 1,2 15 80(С1), 40(ОН) 0,73 43 400-800
Asmit (Imac, Нидерланды)
120. 259 N 1 МП С1 0,5 100(С1), 60(ОН) 0,71 72 600- 1200
121. 261 1 МП С1 4,0 0,8 100(С1), 50(ОН) 65 400- 800
128
Новый справочник химика и технолога
Продолжение таблицы 3.24
Марка смолы и фирма-изготовитель Тип активных групп Содержание ДВБ, % Сорт смолы Ионная форма Полная обменная емкость (H->Na), % Максимальная рабочая температура, °C Насыпная плотность, (кг-м“3)-10 3 Влажность, % Зернение, нм-103
Мг-экв • г 1 Мг-экв • см3
Kastel (ME, Италия)
122. А-500 1 7 С1 3,0 20 60(ОН) 0,70 50 300- 1200
123. А-500 Р 1 5 С1 3,0 20 60(ОН) 0,64 55 ' 300- 1200
124. А-501 D 1 4 МП С1 0,64 300- 1200
125. А-510 1 4,5 МП С1 0,66 300- 1200
126. А-300 2 7 С1 3,2 13 40(ОН) 0,73 45 300- 1200
127. А-ЗОО Р 2 5 С1 3,о 13 40(ОН) 0,70 53 300- 1200
Relite (RDI, Италия)
128. ЗА 1 С1 3,2 1,8 18 80(С1), 60(ОН) 0,66-0,71 43^15 300- 1000
129. 3 AS 1 МП С1 3,2 1,6 18 80(С1), 60(ОН) 0,65-0,70 45^18 300- 1000
130. 3AZ 1 МП С1 20 80(С1), 60(ОН) 0,65 300- 1000
131. 2А 2 С1 3,3 2,0 18 80(С1), 60(ОН) 0,66-0,71 43 300- 1000
132. 2 AS 2 МП С1 3,3 1,8 18 80(С1), 60(ОН) 0,65-0,70 45 300- 1000
Diaion (М-I, Япония)
133. SA-10-A 1 8 С1 > 1,2 0,63-0,67 43^17 300- 1200
134. SA-10-B 1 8 С1 3,5 1,2 0,63-0,67 54 300- 840
135. SA-11-A 1 4 С1 >0,9 0,62-0,66 55-65 300- 1200
136. SA-ll-B 1 4 С1 >0,85 0,60-0,64 55-65 300- 1200
137. SA-20-A 2 8 С1 > 1,3 0,65-0,69 40-45 300- 1200
138. SA-20-B 2 8 С1 3,5 1,3 0,65-0,69 40-45 300- 840
139. SA-21-A 2 4 С1 0,8 0,60-0,65 55-65 300- 1200
140. SA-21-B 2 4 С1 0,8 0,60-0,65 55-65 300- 1200
Lewatit (Bayer, Германия)
141. M 500 1 4,0 1,6-1,7 70 0,70-0,78 300- 1200
142. M 500 G 2G3 1 Инд. 4,0 1,6-1,7 70 0,70-0,78 300- 1200
143. M 500 BG 1 4,0 1,6-1,7 70
Методы разделения и концентрирования
129
Продолжение таблицы 3.24
Марка смолы и фирма-изготовитель Тип активных групп Содержание ДВБ, % Сорт смолы Ионная форма Полная обменная емкость АГ (H>Na), % Максимальная рабочая температура, °C Насыпная плотность, (кг-м 3)-103 Влажность, % Зернение, нм-10 3
Мг-экв г-1 Мг-экв • см3
144. МР 500 1 МП 4,0 1,2-1,3 70 0,68-0,75 300- 1500
145. МР 500 KR/OH 1 ЯМ ОН 4,0 1,2-1,3 70
146. МР 500 А 1 МП С1 1,0 0,62-0,68 62-66 300- 1500
147. М 600 2 3,7 1,6
148. М 600 WS 2 3,7 1,6 75(С1), 40(ОН)
149. МР 600 2 МП С1 3,7 1,2 75(С1), 40(ОН) 0,70-0,78 48-52 300- 1500
150. МР 600 G2 2 Инд. 3,7 1,2 9 75(С1), 40(ОН) 0,70-0,78 300- 1500
151. Serdilit-Blau р.А. (Serva, Германия) 1 АС СО3 1,2 0,72 45 « 1000
152.lone-naustausc her III (Merck, Германия) 1 ОН 5 2,7 5 40(ОН) 0,8 50-60 300- 900
Permitit (Perm, Германия)
153. ESB 1 АС С1 3,3 1,4 100(С1), 70(ОН) 0,6-0,8 50-60 300- 1200
154. ES 2 АС С1 3,2 1,3 100(С1), 40(ОН) 0,7 50 300- 1200
(Россия)
155. AB-17-8 1 8 С1 3,8-4,5 50 0,66-0,74 40-60 400- 1200
156. AB-17-8 4c 1 8 АС С1 3,8-4,5 50 0,66-0,74 40-60 400- 1200
157., AB-17-6 1 6 С1 >4,3 50 0,66-0,74 40-60 400- 1200
158. AB-17-2 1 2 С1 50 400-1200
159. AB-171 1 8 МП С1 3,5-4,0 300-1500
160. APA-2n 1 2 АС С1 3,8-4,2 600-800
161. АРА-Зп 1 3 АС С1 3,8-4,2 400-600
162. APA-5n 1 5 АС С1 3,7-3,9 200-300
163. APA-8n 1 8 АС С1 3,4-3,6 80-120
164. APA-lOn 1 10 АС С1 3,4-3,6 40-60
165. APA-2m 1 2 АС С1 3,8-4,2 600-800
166. АРА-Зм 1 3 АС С1 3,8-4,2 400-800
167. APA-5M 1 5 АС С1 3,7-3,9 200-300
168. APA-8M 1 8 АС С1 3,3-3,7 80-120
169. АРА-Юм 1 10 АС С1 3,3-3,7 40-60
130
Новый справочник химика и технолога
Аниониты сильноосновные пиридиновые [10]
Таблица 3.25
Марка смолы и фирма-изготовитель Тип активных групп Содержание ДВБ, % Сорт смолы Ионная форма Полная обменная емкость дк (H->Na), % Макс, рабочая тем-ра, °C Насыпная плотность, (кг-м“3)-10 3 Влажность, % Зернение, нм-10 3
Мг-экв • г 1 Мг-экв • см 3
1. Permitit S-180 (Perm, США) 4,3 1,3 100(С1) 0,77-0,83 45-55 300-1200
lonac (ICC, США)
2. А-580 Солевая 4,3 1,3 100(С1) 0,77-0,83 55-65 300-1200
3. А-590 То же 4,3 1,3 100(С1) 850-2000
4. Bio-Rex 9 (BRL, США) АС С1 3,7 1,3 38 46-54 300-1200 150-300 75-150 40-75
Таблица 3.26
Аниониты слабоосновные полимеризационные [10]
Пояснение. Большинство смол этого типа синтезируют на полистирольной, реже — на полиакриловой или поливиниловой основе. Обычная форма зерна — бисер. Активные группы:
—NH2(1), >NH(2), (3), и частично—N+R3 (4).
Марка смолы и фирма-изготовитель Тип активных групп Содержание ДВБ, % Сорт смолы Ионная форма Полная обменная емкость дк (H->Na), % Макс, рабочая тем-ра, °C Насыпная плотность, (кг-м 3)-10 3 Влажность, % Зернение, нм-10 3
Мг-экв • г 1 Мг-экв • см 3
Dowex (Dow, США)
1. 3 3(1,2) 4 ОН 5,5-6,0 2,5-3,6 25 65 0,72 30 300-840
2.4 ОН 7,5 2,5 58 400-1200
3. 70 (MWA-1) 3 МП 4,2 1,7 60 300-840
Bio-Rad AG (BRL, США)
4. AG 3-X4 3(1,2) 4 АС С1 5,5 2,2 25 65 0,72 25-35
5. AG 3-X4A 4 АС С1 2,8 1,9 40-45
Amberlite (R&H, США)
6. IR-45 1,2,3 он 1,9 ЮО(ОН) 0,67 40-45 300-1200
7. IRA-93 3 МП он 1,25 8-12 ЮО(ОН) 0,61 46-54 300-1200
8. IRA-94 3 МП он 1,2 23-28 ЮО(ОН) 0,62 54-58 300-1200
9. IRA-68 3 он 1,6 15-20 60(ОН) 0,72 57-63 300-1200
10. XE-236 он 3,1 15-20 60(ОН) 0,69-0,75 46-52 300-1200
11. Sratabed 93 3 МП он 1,25 15 ЮО(ОН) 0,61 46-54
12. Amberlyst A21 3 МП он 4,8 1,3 23-28 ЮО(ОН) 0,61-0,67 45-53 300-1000
Amberlite AR (MCW, США)
13. IR-45 CP 1,2,3 ХС он 5,0 2,0 10 120(0), ЮО(ОН) 0,66 37-45 300-840
14. IR-45 AR 1,2,3 АС он 5,0 2,0 10 120(0), ЮО(ОН) 0,67 37-45 300-840
Amberlite p.A. (Serva, Германия)
15. IR-45 p.A. 1,2,3 АС он 2,0 40-48 400-1100
Методы разделения и концентрирования
131
Продолжение таблицы 3.26
Марка смолы и фирма-изготовитель Тип активных групп Содержание ДВБ, % Сорт смолы Ионная форма Полная обменная емкость АК (H->Na), % Макс, рабочая тем-ра, °C Насыпная плотность, (кг-м 3)-10”3 Влажность, % Зернение, нм-103
Мг-экв • г4 Мг-экв • см~3
16. IRA-93 р.А. 3 АС ОН 1,8 50-58 400-1100
17. IRA-68 р.А. 3 АС ОН 1,6 57-63 400-1100
18. Amberlyst А-21 р.А. 3 АС ОН 4,5-5,0 1,4-1,7 ЮО(ОН) 45-53 700-1000
lonac (ICC, США)
19. А-315 2 С1 5,7 2,0 95(С1), 40(ОН) 300-2000
20. А-316 9 95(С1), 40(ОН) 35—45
21. ANGA-316 9 АС ОН >5,0 95(С1), 40(ОН) 300-1200
22. CGA-315 7 ХС НС1 >3,5 95(С1), 40(ОН) 75-150
23. CGA-316 7 ХС НС1 >3,5 95(С1), 40(ОН) 40-75
24. CGA-300 3 7 ХС НС1 >3,5 95(С1), 40(ОН) 75-150
25. CGA-301 3 7 ХС НС1 >3,5 95(С1), 40(ОН) 40-75
Duolite (D-S, США)
26. А 303 3,4 ОН 3,7(С1) 1,3 15-20 100(С1), 60(ОН) 0,74 50-60 300-1200
27. А 305 он 3,6(С1) 1,9(ОН) 35 100(С1), 60(ОН) 300-1200
28. А 368 3,4 МП он 1,6 45-50 100(С1), 40(ОН) 0,70 47-53 300-1200
29. А 368 PR 3,4 МП он 1,4 25 100(С1), 40(ОН) 0,70 47-53 300-1200
Zerolit DA (Zer, Англия); De-Acidite (Perm, Англия)
H-IP 3,4 3-5 7-9 ИП ИП С1 С1 3,8 1,3 70 70 0,75 52-58 300-1200 300-1200
30. HX-IP 3,4 7-9 ИП С1 1,3 5-10 70 0,74 300-1200
31. G-IP 3 7-9 ИП С1 4,0 1,5 100 0,68 300-1200
32. M-IP 1,2,3 3-5 ИП С1 5,8 1,9 60 0,74 50-58 300-1200
33. J 1,2,3 7-9 ИП С1 5,5 2,4 60 0,74 300-1200
34. SRA121, 122,123, 124 3,4 2-3 ХС С1 70
35. SRA125, 126, 127, 128 3,4 3-5 ХС С1 3,8 1,3 70
36. SRA129, 130, 131, 132 3,4 7-9 ХС С1 70
34-36. SRA121, 125,129 300-1200
132
Новый справочник химика и технолога
Продолжение таблицы 3.26
Марка смолы и фирма-изготовитель Тип активных групп Содержание ДВБ, % Сорт смолы Ионная форма Полная обменная емкость ЛИ (H->Na), % Макс, рабочая тем-ра, °C Насыпная плотность, (кг-м 3)10”3 Влажность, % Зернение, нм-10 ’3
Мг-экв • г 1 Мг-экв см 3
SRA 122, 126, 130 150-300
SRA 123,127, 131 80-150
SRA 124, 128, 132 <80
37. SRA91, 92, 93, 94 3 2-3 ХС С1 100
38. SRA95, 96, 97, 98 3 305 хс С1
39. SRA 99, 100, 101, 102 3 7-9 ХС С1 4,0 1,5 100
SRA91,95, 99 300-1200
SRA 92, 96, 100 150-300
SRA 93, 97, 101 80-150
SRA 94, 98, 102 <80
40. SRA 151 1,2,3 3-5 хс С1 5,8 1,9 60 300-1200
41. SRA 152 1,2,3 3-5 хс С1 5,8 1,9 60 150-300
42. SRA 153 1,2,3 3-5 хс С1 5,8 1,9 60 80-150
43. SRA 154 1,2,3 3-5 хс С1 5,8 1,9 60 <80
Imac (Imac, Нидерланды)
44. A-20 2,3 МП он 2,1 25 100 0,73 55 300-840
45. A-21 3 МП он 2,1 20 100 0,73 50 300-840
46. A-21D 3 МП он 1,5 20 0,8 55 300-1300
Relite (Rdl, Италия)
47. 4MS 3 20 100 0,78 45
48. MS-170 1,2 МП он 6,2 2,2 10 100 0,7-0,8 45 300-1000
Lewatit (Bayer, Германия)
49. MP 60 2,3 МП 6,3 2,4 100 0,60-0,68 300-1500
50. MP 62 МП 1,9 100 0,60-0,70 300-1500
51. MP 62 LF МП 1,9 100
52. MP 64 МП 1,5 70 0,65-0,75 300-1200
53. MP 7080 GR МП он >4,0 51-57 100-200
54. lone-nauscher II (Merck, Германия) 1,2 он 5 3,3 15 100 0,5 35-45 300-900
Serdolit SA 44 (Serva, Германия, Россия)
55. AB-18 3,4 С1 3,0 1,9 130 0,6-0,8 400-1600
56. AB-20 3,4 С1 >3,5 1,0 130 0,7 250-1000
57. AB-23 3,4 С1 >3,5 1,1 0,7 250-1090
58. AH-18 3 С1 3,5 1,4-1,6 100 0,6-0,7 40-60 400-1200
59. AH-22 1,2 >4,5 400-1200
60. AH-23 3 >5,0 2,3 0,8 >30 250-1000
61. AH-25 3 >5,0 3,1 0,8 30-50 250-1000
Методы разделения и концентрирования
133
Основные характеристики ионообменных смол
Полная обменная емкость (ПОЕ) — число молей веществ в ионной форме, поглощаемое 1 кг или 1 г сорбента. ПОЕ соответствует выраженному в молях общему количеству ионогенных групп на единицу массы сорбента. ПОЕ рассчитывается по следующему уравнению:
ПОЕ = КС^нач~^'^он^ж бт
где (сг)нач и (с,)кон — концентрации поглощаемого ИС вещества в объеме раствора Р'ж, контактирующего с сорбентом в статических условиях, в начальный и конечный момент времени после установления равновесия в системе соответственно; <2т — масса ИС, контактирующей с объемом раствора Иж. Методика определения ПОЕ регламентируется ГОСТ 20255.1-89.
Динамическая обменная емкость (ДОЕ) является характеристикой емкости ионитов при сорбции в динамических условиях (сорбция при фильтрации раствора через слой сорбента). ДОЕ характеризует обменную емкость ИС до начала проскока в фильтрат сорбируемых ионитом ионов (в фиксированных условиях проведения эксперимента) по количеству ИС в слое сорбента, его геометрическим размерам, скорости фильтрации, концентрации сорбируемых ионов в исходном растворе и по концентрации в фильтрате, принимаемой за начало проскока.
(с) V ттгур _ \ * /нач пр
QT ’
где Ипр — объем раствора с концентрацией (сг)нач, пропущенный через слой ИС с массовым количеством последней <2т до достижения в фильтрате концентрации z-ионов (с,)Пр, условно принимаемой за начало проскока. Условия определения ДОЕ стандартизованы ГОСТ 20255.2-89.
ДОЕ является не только емкостной, но и кинетической характеристикой ИС. Чем меньше разница между ПОЕ и ДОЕ, тем эффективнее ИС для динамических сорбционных процессов.
В зависимости от природы противоионов, компенсирующих заряд функциональных групп в ИС, различают формы ИС. Для катионитов может быть кислая или Н+-форма и солевая, чаще всего Na’-форма. В случае анионитов понятие форма распространяется только на сильноосновные аниониты. Может быть основная (011-форма) и солевая, обычно СГ-форма.
Содержание дивинилбензола в полимерной матрице указывается в товарных названиях ионообменных смол и отечественного, и зарубежного производства символами Хи, где п — численная величина его процентного содержания в исходной смеси мономеров. Пример — КУ-2X8 или Dowex 1X4.
Механизм взаимодействия органических ионитов с разделяемыми веществами только в простейших случаях является реакцией ионного обмена. По механизму ионного обмена разделяют, например, катионы щелочных металлов и анионы сильных минеральных кислот. Прочность сорб
ции в этих случаях определяется размерами ионных радиусов гидратированных ионов. Чем меньше радиус гидратированного иона, тем прочнее сорбция. Для сильнокислотных катионитов ряд прочности сорбции ионов щелочных металлов Cs+ > Rb+ > К+ > Na+ > Li+.
Для сильноосновных анионитов ряд прочности сорбции анионов в зависимости от ионных радиусов
сю; > no; > Вг > сг > F-.
На величину KD сорбируемых ИС ионных форм элементов одновременно влияют конкуренция присутствующих в растворе одноименных ионов и комплексообразование с ионами-лигандами, приводящее к образованию комплексных ионов с меньшей величиной заряда (вплоть до нейтральных молекул) или ионов с противоположными знаками заряда. Поэтому для выбора условий разделения элементов на ИС, наиболее информативными являются зависимости KD от состава водного раствора, в котором они находятся. К настоящему времени систематизированы данные практически для всех элементов периодической системы Д.И. Менделеева об их сорбции ИС из растворов наиболее широко используемых в аналитической и технологической практике кислот: НС1, HNO3, HF, H2SO4, СН3СООН. Соответствующие данные можно найти в [1].
3.2.2.3. Комплексообразующая сорбция
В этом случае удерживание веществ сорбентами является следствием донорно-акцепторного взаимодействия сорбатов с функциональными группами сорбента. Комплексообразующие сорбенты иногда называются комплексообразующими ионитами, или комплекситами. Специальным классом комплекситов, нашедших наибольшее распространение в аналитической химии, явились хелатообразующие сорбенты (ХС) — полимерные аналоги хелатообразующих реагентов (табл. 3.27).
В качестве полимерных матриц ХС обычно используются те же сетчатые сополимеры, что и в ионообменных смолах. Чаще всего — это сополимеры стирола и дивинилбензола, метилметакрилата и акрилонитрила, а также целлюлоза. В отличие от ИС, выпускаемых только в гранулированном виде, одной из разновидностей ХС являются волокнистые сорбенты с полимерной матрицей в виде ваты, ткани, нитей и жгутов, обладающие лучшими кинетическими характеристиками по сравнению с гранулированными аналогами.
Для характеристики сорбционных свойств ХС используют ПОЕ, которая, как и в случае ИС, соответствует общему содержанию функциональных групп на единицу массы сорбента. Для определения содержания функциональных групп в ХС применяются методы анализа на характерные для этих групп элементы: азот, серу, фосфор и т.п. — или методы функционального анализа для непосредственного определения этих групп. Величина ПОЕ для ХС является малоинформативным параметром, так как в отличие от ионообменной сорбции в данном случае не выдерживаются стехиометрические соотношения количества
134
Новый справочник химика и технолога
сорбированных ионов в зависимости от величины их знака заряда или координационного числа для мономерных аналогов. Важнейшей характеристикой оказывается сорбционная емкость (СЕ) по каждому сорбируемому металлу, зависящая от числа координируемых его ионами функциональных групп в фазе ХС, которое в свою очередь, помимо химической природы металла зависит от степени сшитости и гибкости полимерной матрицы и пространственного расположения функциональных групп в полимере [9,11]. Так,
например, для сравнительной характеристики сорбентов с иминодиацетатными, оксихинолиновыми и другими группами, входящими во внутреннюю сферу комплексов с ионами Си2+, пользуются величиной СЕ (сорбционной емкостью) этих сорбентов по иону Си2+. Для характеристики сорбентов с гидразинными группами приводят СЕ по цинку. Сорбционная емкость ХС по ионам различных металлов может изменяться в широких пределах табл. 3.28. Подробнее о свойствах ХС см. [11].
Таблица 3.27
Хелатообразующие функциональные группы полимерных сорбентов и мономерные аналоги [11]
Структура хелатообразующих групп сорбентов Мономерные аналоги
^СН2СООН хсн2соон Иминодиуксусная кислота
СН2СН2СООН хсн2сн2соон Иминодипропионовая кислота
N— СН2СН2СООН (СН2)2 1 N— СН2СН2СООН Этилендиамин-Л'.уУ'-дипропионовая кислота
он 1 СН2СООН 1 1| хсн2соон о-Оксифенилиминодиуксусная кислота
CH2N(CH2COOH)2 — сн 1 CH2N(CH2COOH)2 2-Окси-1,3-диаминопропан-Л', N, N', N-тетрауксусная кислота
—— ОСН2СООН Ц. J—ОСН2СООН Пирокатехин-О, О'-диуксусная кислота
/ \ ОСН2СООН Z \ ОСН2СООН 1,8-диоксинафталин-О, О -диуксусная кислота
соон — с / х соон 2,6-Пиридиндикарбоновая кислота
Методы разделения и концентрирования
135
Продолжение таблицы 3.27
Структура хелатообразующих групп сорбентов Мономерные аналоги
— С /7—°Н / N 8-Оксихинолин
ОН ——он Пирокатехин
он он /^^^он Пирогаллол
соон Салициловая кислота
он он so,и Хромотроповая кислота
он JL он HOjS^^^^44 SO3H Пирокатехин-3,5-дисульф окислота
но ——он NO Нитрозорезорцин
/ \ NH / N 8-Аминохинолин
СИ—СИ XN=C— СН3 3 (5)-Метилпиразол
136
Новый справочник химика и технолога
Продолжение таблицы 3.27
Структура хелатообразующих групп сорбентов Мономерные аналоги
N СН \nh— сн Имидазол
N+H— NH— СД15 СО NH— NH— С6Н5 Дифенилкарбазид
NH NH— СХ/ xnh2 Гуанидин
но /—N—\ Н(/ N= N у 4-(2-Пиридилазо)резорцин
ОН ОН n=n-Q 1 -(2-Оксифенилазо)-2-нафтол
— С /7—SH / n 8-Меркаптохинолин
О=С NH СН С= S Роданин
R? уИ-ЭД R2 /\ 1 /\ HN < д NH—N=C—N=N 6 Л NH Ri r/ Формазаны
AsO3H2 N= N OH о-Оксифениларсоновая кислота
OH OH As°3H2 N= N N~ N / HO3Sx^5^''/4x^^^ SO3H Азосоединения группы арсеназо III
Методы разделения и концентрирования
137
Продолжение таблицы 3.27
Структура хелатообразующих групп сорбентов Мономерные аналоги
nh2 он so3h Z \ N=N Z \ / no2 Нитроксаминазо
—с=о 1 NH— ОН Гидроксамовая кислота
C-C-jfX- II ||\/ N N ОН ОН а-Фурилдиоксим
N 1 1 11 /С— sh 2-Меркаптобензотиазол
(C2H5)2N— cz % Диэтилдитиокарбаминат натрия
Т /° N—CH2—Р—OR 1 ОН Аминоэтилфосфоновая кислота
< > С—СН2-С—CF3 II II О О Теноилтрифторацетон
сгЧХ 1 1 Дибензо-18-краун-6 (DB-18-C-6)
Таблица 3.28
Сорбционная емксоть хелатообразующих сорбентов [11]
Хелатообразующая группа Полимерная матрица Сорбционная емкость по ионам металлов
^СН2СООН ^СН2СООН Сополимер стирола с дивинил-бензолом Cu(II) 2,9-3,2 мг-экв г1
Сополимер глицидилметакрилата с этилендиметакрилатом Cu(II) 1,15 ммоль-г-1
Целлюлоза Cu(II) 1,1 мг-эквг-1
138
Новый справочник химика и технолога
Продолжение таблицы 3.28
Хелатообразующая группа Полимерная матрица Сорбционная емкость по ионам металлов
AsO3H2 ОН ОН \ N= N jL jL N= N V \ ТТУ 4=7 1 SO3H Аминополистирол Cu(II) 2 ммоль-г’1
La(III) 1,0 ммоль-г 1
Сополимер стирола с дивинил-бензолом La(III) 8,4 мг-г’1
Целлюлоза Th(IV) 0,15 мг-г1
O=C NH N=N— CH C=S Аминополистирол Pd(II) 220 мг-г’1
Сополимер стирола с дивинил-бензолом (8%) Pd(II) 46,2 мг-г’1
nh2 ho so3h / —N=N—/ / NO, Аминополистирол Pd(II) 94 мг-г’1
Сополимер стирола с дивинил-бензолом Cu(II) 32 мг-г’1
C=NH 1 SH C—SH II s Целлюлозное волокно Pd(II) 93 мг-г’1
ПВС-волокно Pd(II) 1,5 мг-экв-г’1
CH= CH XN=1—CH3 Сополимер стирола с дивинил-бензолом Ag(I) - 263 мг-г’1; Au(III) - 660 мг-г’1
Поливиниленовое волокно Ag(I) - 80 мг-г’1; Au(III) - 219 мг-г’1
§ i 1 Сополимер стирола с дивинил-бензолом Pd(II) - 58 мг-мл”1; Au(III) - 150 мг-мл’1
3.2.2.4. Твердофазная экстракция
Твердофазная экстракция (ТФЭ) — метод выделения веществ, по схеме осуществления процесса аналогичный сорбции из растворов. Специфика термина связана с историей его появления. Среди неполярных сорбентов уже отмечались гидрофобизированные силикагели. В тех случаях, когда последние применяются для выделения и разделения органических веществ, они обычно рассматриваются как одна из разновидностей неполярных сорбентов. Параллельно развивалось другое направление применения алкилированных силикагелей в химическом анализе — выделение из водных растворов ионов металлов в форме хелатов. При этом было доказано, что закономерности сорбции хелатов являются общими с закономерностями их жидкостной экстракции. Соответствующее направление вошло в литературу по методам разделения как ТФЭ. Помимо хелатов для выделения металлов могут быть использованы ионные ассоциаты их заряженных комплексов с гидрофобными органическими противо
ионами. В настоящее время об исторических корнях термина часто забывают, и он используется как синоним сорбции обращеннофазными сорбентами. Более подробную информацию о ТФЭ можно найти в [23-28], а о современной ее модификации — твердофазной микроэкстракции — в [28].
3.2.3. Динамическая сорбция
Сорбционное разделение и концентрирование веществ в аналитической химии, как правило, проводят в динамических условиях, пропуская анализируемую пробу через колонку с сорбентом [29, 30]. Установление взаимосвязи между концентрациями сорбатов на входе в сорбционную колонку и выходе из нее является одной их основных задач динамики сорбции. Решение этой задачи позволяет выбрать оптимальные условия сорбционного разделения: размеры сорбционной колонки, размер частиц сорбента, объем и скорость пропускания пробы через сорбционную колонку.
Методы разделения и концентрирования
139
Коэффициент концентрирования Аконц при условии количественного выделения сорбируемого компонента равен:
К =\ (3.1)
КОНЦ ТГ ’ 7
Г Л
где Vo — объем пробы, пропущенной через сорбционную колонку; Vs — объем сорбента в колонке.
Зависимость концентрации компонента с на выходе из сорбционной колонки от объема V пропущенной через нее пробы с концентрацией выделяемого компонента с0 отражает выходная кривая, имеющая 5-образную форму (см. рис. 3.3). На выходной кривой можно выделить три области. Первая область (до точки А) — область полного извлечения сорбата, вторая область от точки А до точки D — область частичного извлечения сорбата, где происходит проскок сорбата и его концентрацией на выходе из сорбционной колонке уже пренебрегать нельзя, и, наконец, третья область (за точкой D) — область равновесного насыщения сорбента, где концентрация сорбата на выходе из колонки практически равна его концентрации на входе.
Рис. 3.3. Типичная выходная кривая при проведении сорбционного концентрирования
На практике с целью повышения коэффициента концентрирования через сорбционную колонку пропускают объем, превосходящий объем КА, пренебрегая некоторой потерей сорбата. Так, если пропустить через сорбционную колонку объем пробы, равный VB (см. рис. 3.3), степень извлечения сорбата /?, будет равна
я, • с0Кв - s№lt = с/в - ]с(И) Д', (3.2)
О
где со Кв — количество сорбата в объеме пробы Кв, а 5двГв — площадь заштрихованной фигуры АВС, которая соответствует количеству сорбата, проскочившего через сорбционную колонку при пропускании данного объема пробы. Величина Кв, соответствующая объему пробы, который может быть пропущен через сорбционную колонку при незначительной величине проскока через нее сорбата, называется объемом до проскока.
Вид функции с(К) зависит от типа изотермы сорбции, а сама функция содержит ряд параметров, определение которых требует проведения трудоемких исследований [29, 30]. В случае линейной изотермы сорбции, которая имеет
место при концентрировании микропримесей из предельно разбавленных растворов, зависимость с от К может быть представлена в виде следующей функции [31]:
СТр[(К/^Кд-1)2Улг]
°1 + ехр[(К/ад-1)2^] ( )
где N — число эквивалентных теоретических тарелок, соответствующих заданной высоте сорбционной колонки. Как следует из общей теории динамики сорбции и уравнения (3.3), объем пробы, пропущенный через сорбционную колонку, для которого с = со /2, не зависит от числа N и, следовательно, от скорости пропускания пробы и размера частиц сорбента.
N увеличивается пропорционально увеличению длины сорбционной колонки и уменьшению размера частиц сорбента. Зависимость N от скорости потока пробы имеет максимум (подробно этот вопрос рассмотрен в разделе, посвященном теории хроматографии). Увеличение N при прочих равных условиях приводит к увеличению объема пробы Кв:
KB«K,(l-lVw). (3.4)
В пределе, при N, стремящемся к бесконечности, Кв стремится к VR.
N может быть рассчитано на основании экспериментально полученной выходной кривой по формуле [32]:
V2
N=------—(3.5)
где К0; 159 — объем пробы, пропущенной через сорбционную колонку, при котором с = О,159со (см. рис. 3.3).
На практике применение сорбционных колонок с большим числом N, которое может быть достигнуто за счет увеличения ее длины либо за счет применения мелкозернистого сорбента, нерационально. Оптимальными для 95-99%-го извлечения [33] можно считать сорбционные колонки с А =5-10. При сорбционном концентрировании органических веществ из водных растворов подобные значения N достигаются при использовании сорбентов с размерами частиц 100-200 мкм в колонках длиной З-б см и линейной скорости пропускания водной пробы 0,2-0,4 см • с1.
3.2.4. Кристаллизационные методы концентрирования
3.2.4.1. Соосаждение
По технике эксперимента метод соосаждения имеет много общего с обычным осаждением. Обычно в раствор вводят реагент, способный к образованию малорастворимого соединения с одним из макрокомпонентов разделяемой смеси или со специально введенным в раствор веществом и выделяемыми микропримесями. Не исключается и другая схема соосаждения, при которой в раствор вносят готовый осадок коллектора. В этом случае имеет место полная аналогия с процессами сорбционного выделения на неорганических сорбентах. При кажущемся сходстве обе
140
Новый справочник химика и технолога
схемы соосаждения далеко не полностью тождественны Друг Другу. При образовании осадка в растворе в присутствии микрокомпонентов происходит их объемное, объемноповерхностное и поверхностное поглощение твердой фазой. При внесении готового осадка, если специально не создаются условия для его перекристаллизации, происходит только поверхностная сорбция. Но независимо от схемы осуществления процесса соосаждение, в отличие от осаждения, подчиняется закономерностям межфазного распределения.
Соосаждение микрокомпонента по типу объемного распределения между раствором и осадком происходит при изоморфной и изодиморфной сокристаллизации, при образовании аномально смешанных и гриммовских кристаллов. Равновесное распределение микрокомпонента в процессе сокристаллизации описывается уравнением:
—^— = D -У-. а-х 41 Ь-у
Здесь х и у — количества макро- и микрокомпонентов, перешедших в кристаллическую твердую фазу, соответственно; а и Ь — первоначальные количества макро- и микрокомпонентов в системе; — коэффициент кристаллизации.
Коэффициент кристаллизации DKV связан с коэффициентом распределения KD выражением:
D = ^L,
” d.
где с\ — концентрация макрокомпонента в равновесном растворе; б/0 — плотность кристаллической фазы.
Учитывая, что равновесное объемное распределение на практике трудно достижимо, известно уравнение для расчета неравновесного распределения:
In а = Х1п ——, а-х Ь-у где 1 — постоянная кристаллизации, являющаяся экспериментально определяемой характеристикой конкретных условий осаждения.
В случае образования осадка коллектора в мелкокристаллической форме с сильно развитой поверхностью может наблюдаться соосаждение микрокомпонентов не только в результате объемного распределения, т.е. различного рода сокристаллизации, но и благодаря поверхностнообъемному и поверхностному распределению, включающему первичную, вторичную и внутренюю адсорбцию, а также механический захват при высокой скорости формирования осадка. В [21, 22] систематизированы многочисленные конкретные (в основном по решению радиохимических задач) примеры распределения микрокомпонента между твердой и жидкой фазами, обусловленного отдельными видами адсорбции. В радиохимии основное внимание уделяется селективному соосаждению по механизму объемного распределения. В аналитической химии предпочтение отдается групповому концентрированию на осадках-коллекторах. Наиболее типичные химические формы осадков, используемые при групповом соосажде-нии приведены в табл. 3.29. В табл. 3.30-3.39 приведены данные по предварительному концентрированию соосаж-дением на гидроксидах, сульфидах, сульфатах, фосфатах, фторидах, оксалатах, веществах в элементарном виде, органических веществах и металлических носителях, на чистых органических веществах с указанием соосаждаемых микроэлементов, объектов и условий проведения эксперимента [21].
Таблица 3.29
Соосаждение микроэлементов в виде: О — гидроксидов или кислот, □ — сульфидов, • — сульфатов, — фосфатов, Д — фторидов, П — оксалатов, к — элементов, * — оксихинолинатов
зЫ 4Ве О 5B
nNa izMg О* 13 Al ОИ ☆ 14S1 0
19К б • * Я О < 21SC О* 22Ti О* 23V о* 24СГ оп а 25МП оп и 2&Fe on ☆ 27C0 on 2sNi on 29Cu on зо^п О ce згСе О 33AS 0 34Se Ok
7Rb 38Sr ои ☆ 39Y о* 4oZr О* 4iNb О* 42Мо О* 4зТс О 44Ru on 4sRh oa k 4бРб on k 47Ag on Ik 48Cd on H 49In on ☆ 50Sn 0 51Sb on 52Te Ok
55CS 5бВа 57Ьа О* а 72Hf он ☆ 7зТа О* 74W О* 75Re Ok 7бО$ on k 77Ir on k 78Pt on k 79Au on 8oHg on k 81T1 on ☆ 82₽Ь ОИ □ И 83Bi on
87Fr 88^а • 89Ас оП
5вСе
ОД П
9oTh ДИ
С]
94Рп од нП
92U од ниП
Методы разделения и концентрирования
141
Таблица 3.30
Предварительное концентрирование соосаждением на гидроксидах металлов [21]
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
l.Fe Cd, Co, Eu Морская вода, изучена зависимость от pH, концентраций и старения. Предлагается pH = 9 и 35 мг • л-1 Fe для соосаждения
2. Fe Ga Водные растворы, изучена зависимость от pH, концентрации сорбента и продолжительности контакта
3. Fe Tc Растворимые ядерные топлива (fuels)
4. Fe Sr Соосаждение изучено как функция от pH, от соосаждающего агента, температуры и концентрации
5. Fe Sr Изучено влияние разных концентраций различных однозарядных катионов
6. Fe Mo Морская вода, изучена зависимость от pH. Соосаждение происходит только в области pH = 4-9
7. Fe Zn, Cd, Mn, Sr, Cr La, Ce, Eu, Gd, Er Yb, Ag, Cs, Rb Низкая степень растворения. Изучено влияние pH. Несильная абсорбция Cs+ и Rb+. Зависит от pH, степень соосаждения низкая (< 20%). Изучена степень соосаждения в зависимости от pH, извлечение 100%
8. Fe Pb, Cd, Zn, Tl Изучено влияние pH и концентрации
9. Fe Cr(VI) Изучено влияние других анионов и pH. Cr(VI) соосаждается только при pH = 4-7
10. Fe Eu Изучено влияние pH и комплексообразующих агентов
11. Fe Ga Изучено влияние pH и комплексообразующих агентов
12. Fe Ru Изучено влияние pH и восстанавливающих агентов
13. Fe Ru,Mn, Eu Изучено влияние pH и концентрации
14. Fe Ru Изучено влияние pH и примесей
15. Fe СДП1) Моча. Сг (П1) соосаждается при pH = 10, в то время как Cr(VI) остается в растворе
Продолжение таблицы 3.30
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
16. Fe V Речная вода. Оптимальная область pH = 6-9. Лучшие результаты получены с Fe(OH)3 + Ti(OH)4
17. Fe Se, As, W, Ge, Те, Cr, P Оптимальная область pH = 5-7 для Se и Sb, 5-8 для As и W, 5-10 для Ge, Те и Сг и 6-7 для Р. Степень извлечения 50% для всех, кроме Se — 5%
18. Fe Cd, Ni, Pb Гуминовые вещества увеличивают абсорбцию/соосаждение Cd и Ni
19. Fe Ni, Cu, Zn, Pb, Mn Природные воды. Содержание Fe 15 мг • л’1, pH = 9 (NaOH). Предел обнаружен. 1 мкг • л1
20. Fe Zn, Cu, Pb,Mg Основа — фосфаты, маскирующий агент — Са. Соосаждение при pH = 8,5
21. Fe Cd, Cu, Zr, Cr Сточные воды. pH = 10 для Cd, Cu, Zn. pH = 8 для Сг(Ш). Изучено влияние pH, концентрации, времени и мешающих ионов
22. Fe As, W, V, Nb, Mo Для коагуляции (1л образца) пробу встряхивали в течение 15 мин и нагревали до 70-80 °C. pH и степень извлечения: 7,8-9 (100%) для V; 7,8-9 (75-90%) для As, Nb; 7 (80%) для Мо; 7-9 (40-50%) для W
23. Fe(II) Tc Радиоактивные отработанные растворы. Fe используется од-но при pH = 6-12; вместе с Zr4+ при pH = 4,2-12 или с Si4+ при pH = 8-12
24. Fe Cr(III) Морская вода. Извлечение 99%. pH = 7,0-9,0 для 0,4 мкг • л1. Предел обнаруж. — 0,02 мкг • л1
25. Fe Zn, Pb Природные воды. 10-100 мкг • л1. Оптимальная область pH = 9
26. Fe Be pH = 6-7. Матрица — стандартные руды
27. Fe V Морские и природные воды, биологические материалы, силикаты pH от 3,2 до 8,0
28. Fe Co, Zn, Cd, Ni, Cu, Ag Изучено влияние pH и других параметров
29. Fe Ge Фосфориты после сплавления с NaOH
142
Новый справочник химика и технолога
Продолжение таблицы 3.30
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
30. Fe Po Изучено влияние pH и комплексующих агентов
31. Fe Лантаноиды, Sc Раствор Na2CO3. Количественное соосаждение при pH = 9-11 для 0,1 М Na2CO3. Повышение концентрации Na2CO3 увеличивает соосажадение
32. Fe Cu Изучено влияние pH, концентраций и времени перемешивания
33. Fe U Количественное соосаждение при pH < 12. Изучено влияние концентрации и времени перемешивания
34. Fe Co, Zn Кривая зависимости степени извлечения от pH имеет максимум при pH = 8. Изучено вымывание из осадка
35. Fe As, P Осаждение в присутствии NH4OH при pH = 8
36. Fe Sr Изучена зависимость от pH и концентраций
37. Fe Ce Изучено влияние pH, NH4+ и soj-
38. Fe P Sr, Ce, Cs Изучено влияние pH и концентраций. Фосфат-ионы повышают соосаждение Sr2+, Се3+ и Cs+
39. Fe Se Морская вода, силикаты и морские образцы. pH = 4-6. Для морской воды предел обнаруж. 0,4 мкг • л-1
40. Fe Mg, Zn, Cd, Mn, Co, Ni, Hg, Cu Обсуждается механизм сооса-жаденияс окислительно-восстановительными реакциями
41. Fe Pd, Pt, Rh, Ru, Ir, Os Изучено влияние pH, температуры, концентрации и времени перемешивания
42. Fe Ac, Pu, U, Np, Pa, Am, Cm Изучено влияние pH и концентрации электролитов. Полнота соосаждения зависит от состояния ионов в растворе: мо+>м3+>мо2+>м4+
43. Fe Am, Pu, Np Изучено влияние pH и концентрации электролитов. Ат (pH > 3), Pu (pH > 4), Np (pH = 9-10)
Продолжение таблицы 3.30
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
44. Fe Cr(III), Cr(VI), Mo(VI), W(VI) Изучено влияние pH и солевых эффектов. Сг(Ш) осаждается при pH = 5-9, Cr(VI) ) осаждается при pH = 5-6, Мо при pH = 4-5, W при pH = 6-8
45. Fe Co, Ni, Ca, Zn, Cd Соосаждение в растворе аммиака
46. Fe Cu, Ni Изучено влияние концентраций и инертных солей
47. Fe Cu, Ni, Zn, Mn Увеличение концентрации аммиака увеличивает соосаждение
48. Fe U и другие элементы Изучена десорбция горячим раствором соды и использована для отделения 30 микроэлементов из матрицы U
49. Fe Ru Полное извлечение при pH = 10
50. Fe Au, Ag, Cu Изучено влияние pH от концентраций. Ag, Си при pH = 9,5-11; Au при pH = 5-8
51. Fe(II) + Fe(III) + Fe (порошок) Tc Fe(II) и Fe (порошок) используются для получения ТсО2 при pH = 1. ТсО2 осаждается Fe(OH)+ ’ Fe(OH)3 при pH > 8 в присутствии NaOH. Изучено влияние pH, концентраций и времени взаимодействия
52. Fe + Ti V Речная вода; pH = 4-8
53. Mg + Fe Be, Cu, Zn, Pb, Bi, Co, Ni, Cd Даны условия для количественного соосаждения каждого металла
54. Fe + Ti V Морская вода; pH = 6,0
55. Al Au Природные воды. Степень извлечения более 99%
56. Al Cr(III), Cr(VI), Mo, W Количественное осаждение Сг(П1) (pH = 5-9), Мо (pH = 4-5), W (pH = 6-8). Максимальное, но не количественное извлечение Cr(VI) при pH = 5-6
57. Al Ac, Pu, U, Np, Pa, Am, Cu Полнота извлечения актиноидов зависит от степени окисления (5 > 3 > 6 > 4) и pH
58. Al + аминокислоты Co Изучено влияние pH в области 4-9. Присутствие NaCl увеличивает область соосаждения
Методы разделения и концентрирования
143
Продолжение таблицы 3.30
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
59. А1 Сг(Ш), Cr(VI) Соосаждение Cr(VI) увеличивается с увеличением pH. Сг(1П) — уменьшается с увеличением pH. Изучено влияние других ионов. Сг(Ш) определяют при pH = 8-9, a Cr(VI) после восстановления сульфитом натрия или Fe(II)
60. А1 Fe(III) Количественное извлечение при pH = 6,5-8,5. Изучено влияние некоторых анионов
61. А1 TR Горячие ключевые воды и воды кратерных озер. Концентрация A12(SO4)3 25 мг • л1, pH = 7-8 в 14% растворе аммиака
62. А1 Th, U Горячие ключевые воды и воды кратерных озер. Условия те же, что в п. 61
63. А1 La, Ce, Nd. Sm, Eu, Tb, Tm, Yb, Lu Концентрация А1 — 5мг • л1. Осадок гидроксида алюминия получают при температуре, близкой к температуре кипения в 14% растворе аммиака с pH = 6,5-7,5
64. А1 Ce, Pm, Eu, Gd, Yb Изучено влияние pH, комплексообразующих агентов и электролитов. Оптимальное значение pH = 6,5
65. А1 Li Геотермальные воды, pH > 12,5-13 при длительном времени перемешивания. Полнота извлечения повышается с введением ионов кальция и полимеризованного кремния. Осадок — алюминат натрия
66. А1 Li Концентрация А1, pH и температура влияют на полноту извлечения Li. pH = 11,5-12. Карбонаты увеличивают полноту извлечения. Осадок — алюминат натрия
67. А1 + Mg Fe, Cr, Cu, Co, Mn Изучено влияние pH и концентраций на полноту извлечения
68. Al + Si gl* Ru Сточные воды. А1 > 4- НГ2 М, Al/Si < 0,5 pH = 6-9. Осадок — алюмосиликат. pH создается NaOH. Степень извлечения 96%
69. Sr Соотношение Sr/Mn —1:2
7O.Mn Zr, Ru, U Дано объяснение в соответствии с механизмом ионного обмена
Продолжение таблицы 3.30
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
71. Мп Ас, Ри, U,Np, Ра, Ат, Ст Изучено влияние pH, электролитов и степени окислени М(4) < М(6) < М(3) < М(5)
72. Мп Sb, Bi, Pd, Sn Основа — никель. Концентрация 1-100 мг - л1 МпО2 образуется по реакции МпО4 + Mn(II) в присутствии кислот (HNO3 или НС1О4). При концентрациях 0,008-0,1 М наиболее эффективны; pH = 2-3
73. Мп Sb Железо и стали; 2,5-100 мг • л~1
74. Мп Pa, Np МпО2 образуется из Мп(П) + KB1O3 в азотнокислой среде. TR не соосаждаются
75. Мп Pb, Bi Образцы никеля. Слабое осаждение увеличивает извлечение РЬ. Увеличение pH от 2-3 до 4— 4,5 увеличивает извлечение свинца до 94-99%
76. Мп Mo, V Природные воды, силикатные породы, биологические материалы. Мп(П) + КМпО4рН = 3,7
77. Мп Sb, Tl Образцы свинца
78. Мп Sb, Fe Природные воды. МпО2 получают по реакции КМпО4 с этанолом в присутствии 0,036 М НС1
79. Мп Ga Образцы А1 и Zn. pH = 1,5
80. Мп Pb, Sn Образцы меди. 0,008 М HNO3
81. Мп As, Sn, Sb 0,1 М HNO3. Водные растворы
82. Мп Sn, Sb, Bi 0,6-l,2MHNO3
83. Мп Sn, Sb Образцы свинца; 0,01-0,04 М HNO3
84. Zr Al, As, Cd, Co, Cr, Cu, Fe, La, Mn, Mo, Ni, Pb, Ti, V, Y, Zn Морская вода 10 мг • л-1. Zr. pH > 4 (старение — 24 часа). Соосаждение увеличивается с увеличением pH, кроме Cr(VI) и Мо. Максимальное соосаждение при pH = 10. 100% извлечение Сг(Ш). Изучено влияние старения осадка
85. Zr Tc pH = 8
86. Zr + кварц Ru Отходы ядерной промышленности. Осадок готовился путем добавления жидкой NaOH к цирконил нитрату + раствор жидкого стекла, pH = 6-10, Si/Zr <2, [Zr] > 7,5-10~2М
144
Новый справочник химика и технолога
Продолжение таблицы 3.30
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
87. Zr Ca, Fe, Ti, Si, V, Zn, Mn А12О3 и другие соединения А1. Соосаждение с Zr(NO3)4 при pH >9
88. Zr Pb, Fe, Mn, Cd, Cu, Zn, Ni, Co Природные воды, соосаждение при pH = 10. NaOH подходит для всех, NH4OH — для первых трех
89. Zr Fe, Zn, Mn, Cu Стандарты объектов окружающей среды
90. Zr Fe, Zn, Mn, Cu, Cd, Pb Стандарты на растительные материалы
91. Zr Fe, Si, Ca, Mg, Cr А12О3
92. Zr Cd, Co, Cu, Mn, Fe, Pb, Zn Природные воды; pH = 9,9-10,1. Встряхивать 10 минут и оставлять на ночь
93. Zr Fe, Zn, Mn, Cu, Cd, Co, Pb, Ni Стандарты объектов окружающей среды
94. Zr Fe, Zn, Mn, Cu, Cd, Co Стандарты объектов окружающей среды
95. Zr Sb, Sn, Bi, Pb, As, Fe Образцы меди; pH = 9,4-9,5 с 6 м nh4oh
96. Zr Sb Природные воды, водоросли, силикаты; pH = 5,0
97. La As, Bi, Sb, Se, Те Молибден; pH = 9,0-11,0. Изучено соосаждение элементов в высших степенях окисления. Se извлекается не очень хорошо. Использовано двойное соосаждение. Концентрация La 120 мг • лч. Нужный pH создается NaOH
98. La Co, Cr, Cu, Fe, Mn, Ni, Ti, V, Zn, Zr Молибден и МоО3. Все элементы извлекаются количественно. Изучено влияние pH и концентрации La. Использовано двойное соосаждение с La(OH)3. Оптимальный pH = 11,0
99. La Au, Cu, Pb, Bi, Cd, Zn, Ni, Co, Fe, Al Серебро. Осаждение с NH4OH. Количественное извлечение
Продолжение таблицы 3.30
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
100. La Bi Образцы свинца. Осаждение из растворов HNO3 с 1 М NH4OH
101. La Mo, Fe, Ni, Co, Zn Морская вода, 5 мг • л1 La , pH = 9,8 создается 1 М Na2CO3. Осаждение при 80 °C в течение 30 мин и дольше, старение — несколько часов
102. La Se, Те, As, Sb, Fe, Pb, Sn, Bi Медь. pH = 9-10. Полное извлечение. При pH ниже 8,5 неполное осаждение La(OH)3. Фильтрование при pH > 9
103. La Fe, Ni, Cr, Mn, Co, Ag, As, Cd, Ga, Mn, P, Rh, Si, V, W, Y, Zn Na. Осаждение с NaOH до покраснения .и-крезола
104. La Mo, Fe, Ni, Cu, Zn Морская вода. Определение ААС
105. La Pb, Mn Водные растворы. Большая схема разделения. РЬ и Мо отделяются от металлов, образующих растворимые аммиачные комплексы (Cd, Zn, Со, Ni) соосаж-дением с La(OH)3 в присутствии избытка NH4OH
106. La Неспецифичное Высокочистые материалы. Используется для предконцентри-рования Со, Ni, Zn, которые не соосаждаются с La(OH)3 в избытке аммиака
107. La Ti Морская вода. Концентрация La 10 мг • л-1. pH = 9,8 создается 1 М Na2CO3. Выдерживают 30 мин при 80 °C
108. Ti Hg, Cu, Zn, Ni, Co, Cd Раствор NH4CI. Изучена зависимость от pH
109. Ti(OH)4 Fe, Cr, Co, Cu, Ni Стандарты. 2 мкг Ti позволили решить проблемы фильтрования и увеличить степень извлечения. pH = 3,8-4,5 создавали введением газообразного аммиака. Добавляли диэтилдитиокарбаминат натрия и доводили pH до 8,0-8,2
Методы разделения и концентрирования
145
Продолжение таблицы 3.30
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
ПО. Mg Zn, Ag, Ni Изучено соосаждение из растворов аммония. Обнаружена ступенчатая сорбция, обусловленная ступенчатым гидролизом аммонийных комплексов
111. Fe, Zr Mn, Fe, Co, Ni, Cu, Zn Изучено соосаждение в 1 М NaC104 как функция pH. Обнаружена линейная зависимость между сорбцией и константами гидролиза катионов
112. Zr Cu, Zn, Cd Изучено влияние pH и концентрации NH4NO3
113. Fe Cr, Mo, W Изучено влияние микроконцентраций
114. Fe Cr (VI) Изучено влияние pH, концентрации Сг и инертных солей
115. Fe Mo, W Растворы NH4CI и КС1. Изучено влияние pH
116. Fe Zr Изучено влияние pH и инертных солей
117. Fe Zn Соосаждение из системы вода -органический растворитель. Изучено влияние растворителей, инертных солей, pH
118. Fe Cu Изучено влияние pH, концентраций KNO3 И NH4NO3
119. Mg Pb, Cd Соосаждение в присутствии комплексообразующих агентов. Na4P2O7NH3Na2S2O3
120. Mg U, Th, Pa, Np, Pu, Am Изучено соосаждение как функция pH и состава раствора
121. Mg Mo Морская вода, pH = 10,0
122. Mg Cr Осаждение с NH3
123. Ga Al, Co, Cr, Fe, La, Mn, Ni,Ti,V, Zn, Y, Pb Морская вода; pH = 9,0 создается NaOH. Коэффициент концентрирования 200. Изучена зависимость от pH, кроме V, Мо, Sb. Изучено влияние концентрации Ga
124. Ga Al, Ti, Cr, Mn, Fe, Co, Ni, Cu, Zn, Y, Pb Морская вода. 10 мл образца. Коэффициент концентрирования 200 при растворении Ga(OH)3 в 50 мкл HNO3. Степень извлечения > 99%, кроме Мп (78%), V (33,5%), Cd (42%)
125. Co Ti,Zr Металлический молибден и висмут или их оксиды
Продолжение таблицы 3.30
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
126. Sn Fe, Со, Cu, Zn, Cd Преимущества: соосаждение в из слабокислых растворов; легко удалить Sn выпариванием. Оптимальная область pH = 4-8. Осаждение при pH = 5, создаваемым NH3, при 50 °C в течение 30 мин
127. Th Мо Морская вода. Оптимальное значение pH = 6,0 со степенью извлечения 99,5%
128. Са Мо Морская вода, pH = 10,0
Таблица 3.31
Предварительное концентрирование соосаждением на сульфидах металлов [21]
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
1. РЬ Си Водопроводная вода, PbS может быть удален выпариванием. Степень извлечения > 98%
2. РЬ Ag, Cr(III), Cr(VI) Изучено влияние pH, времени перемешивания и концентраций, комплексообразующих агентов и конкурирующих ионов
3. РЬ Au Природные воды. 100% степень извлечения, 30 мин перемешивания
4. Си Pt Медные корольки, медные руды и сплавы, породы
5. Си Hg 0,02 мг • л’1 Hg
6. Си Au, Pd, Pt Медно-никелевые руды. Осаждение H2S из растворов НС1 + HNO3 или НС1. Использовали 20 мг Си
7. Cd Pb, Cr, Mn, Cu, Fe, V Образец ВаС12. Изучено влияние pH, концентраций и температуры. Оптимальная область pH = 7-8
8. Cd Ga Изучено соосаждение Ga2S3 с CdS из растворов НС1 и из H2SO4
9. Ag Ga Осаждение при соотношении Ag:Ga = 2:l
Ю. Hg Ga Изучена кинетика соосаждения из растворов НС1 и HNO3
H. Hg Ga Соосаждение как тип образования твердых растворов. Главный процесс — сокристаллизация
146
Новый справочник химика и технолога
Продолжение таблицы 3.31
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
12. As Ga Соосаждение за счет образования аморфных твердых растворов. Изучены концентрационные диаграммы
13. Zn Ga Изучено влияние температуры, времени, концентрации NaOH, Zn и Cd. Соосаждение — изоаморфно
14. Mo Ag, As, Bi, Cd, Cu, Ge, In, Pb, Sb, Sn, Те, Tl Различные металлы. Кислые растворы (pH = 1,2; значение pH важно). Осаждение путем насыщения H2S, добавление H2SO4 до начала осаждения H2S приводит к быстрому и полному осаждению
15. Bi Tl Образцы мочи. Сначала разрушается органическая матрица. Соосаждение с восстановлением FeSO4 + Bi(NO3)3+H2S
16. В As, Sb, Sc Образцы вод. Раствор 1,2 М НС1 Минимальное количество As(III) - 10 нг, Sb(III) - 50 нг, Se - 20 нг
17. Sb Cu,Pb Образцы Zn. pH = 1-2. Осаждение тиоацетамидом
Таблица 3.32
Предварительное концентрирование соосаждением на сульфатах металлов [21]
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
1. РЬ Sc(IV) Слабокислые и нейтральные растворы. Для фотометрического определения PbSO4 растворяют в тартрате аммония
2. РЬ Zn Степень извлечения Zn — 100%
3. РЬ Cr Водопроводные и морские воды. Cr(VI) количественно соосаждается при pH > 3,5. Сг(Ш) начинает соосаждаться при pH = 3,5. При pH > 6 обе формы количественно соосаждаются из морской воды со степенью извлечения 85-87%
4. Ва Np, Pu, Am Изучено соосаждение для различных кислот и концентраций, различных концентраций К, Na, Ва
Продолжение таблицы 3.32
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
Ва Оказалось, что степень соосаждения возрастает при добавлении ионов натрия и калия сверх определенного значения. Кислотность должна быть до 0,7 М. Использование окислителей и восстановителей позволяет разделить соосаждаемые элементы
5. Ва U, Th Изучено влияние кислот, кислотности и содержания ионов калия и натрия
6. Ва U, Pu, Am Ядерные воды
7. Ва Fe(III) Изучено влияние концентрации
8. Ва Po, Pb При перекристаллизации Ba(Ra)SO4 не более 0,03% Ро и РЬ
9. Ва Th К+ способствует соосаждению, в то время как многовалентные ионы стараются предотвратить ее
10. Ва Th, U, Pu, Am Образцы почвы. Соосаждение из растворов НС1
И. Ва, Са, Sr La Обсуждается влияние состава
12. Sr, К TR Изучено влияние состава растворов
13. Th + Co(NH3)6 Eu, U, Np, Am, Cm Th(IV) — сульфатный комплекс [Co(NH3)6]2[Th(SO4)5] • иН2О соосаждает трехвалентные лантаноиды и актиноиды, а также шестивалентные ионы актиноидов при образовании смешанных кристаллов
14. Li, К Ca, Sr, Ba Растворы KNO3. Для понижения растворимости сульфатов лития или калия добавляют этиловый спирт
Таблица 3.33
Предварительное концентрирование соосаждением на фосфатах [21]
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
1. РЬ Сг(Ш), Cr(VI) Морская вода. При pH > 6 соосаждаются количественно обе формы. Отношение концентраций РЬ к фосфату должно быть больше 3
Методы разделения и концентрирования
147
Продолжение таблицы 3.33
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
2. РЬ Ag, Cd, U, Сг(П1), Cu, Mn(II), Th, Zr Кислотность растворов морской воды создается раствором аммония по индикатору метиловому красному. Степень извлечения больше 90%
3. А1, Sr Cr, Zn, Fe, Mn, Ru,U Изучено влияние pH. Оптимальная область pH = 4-5
4. Al U Природные и морские воды
5. Ca TR Водные растворы
6. Ca U, Th Пищевые продукты и стандарты. Степень извлечения 95-99%
7. Bi Fe, Cu, Zn, Mn, Pb pH = 5,2
8. Bi Fe, Cu Показано, что поверхностная адсорбция является основной причиной соосаждения
9. Bi Np, Pu, Am, Cm Изучено влияние типа кислот, кислотности, содержания висмута и других элементов. Для соосаждения используется 0,2 М фосфорная кислота и 1 мг Bi
Таблица 3.34
Предварительное концентрирование соосаждением на фторидах [21]
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
1. Са Fe, Си Показано, что основной причиной соосаждения является поверхностная адсорбция. Си(П) осаждается как противоион фторидных комплексов, тогда как Fe(III) соосаждается в виде комплексных ионов [FeF6]3
2. Са Fe, Со, Ni, Си, Zn, Мп Растворы NH4F и HF. Осадок растворяется в НС1 (1:3) для последующего атомно-абсорбционного определения. Полнота извлечения (> 99%) лантанидов достигается после осаждения 30% кальция. Неполное осаждение кальция мешает сосаж-дению многих элементов
З.Са La, Рг, Nd, Sm, Ей, Tb, Dy, Но, Ег, Тш Водные растворы
Продолжение таблицы 3.34
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
4. Са Th, Y, La, Ce, Gd, Lu Природные грунтовые воды
5. Са U Водные растворы
6. Са U Водные растворы
7. Са Sc, Zr, Yb Водные раствЬры
8. Са + Mg Eu, Dy, Gd, Sm Be, U, Zr, Ti — металлы и сплавы. Иттрий добавляется в качестве носителя для увеличения извлечения
9. Th TR Для осаждения используется NH4F. Осаждение на NH4ThF5-H2O в качестве носителя обеспечивает количественное соосаждение TR
10. La Th Урановые руды. Лантан в качестве носителя и HF для осаждения LaF3. Степень извлечения 98 ± 3%
И. Nd U, Pu, Am Образцы почв и вод
Таблица 3.35
Предварительное концентрирование соосаждением на оксалатах [21]
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
1. Са Ра Стандарты и пищевые продукты после предварительного соосаждения на Са3(РО4)2. Отделение Ра от урана и продуктов распада
2. Са РЬ 100 мл раствора (2-15 мкг РЬ) + + 5 мл 1:1 HNO3 + 10 мл 1% СаС12 + 10 мл 5% раствора аммиака до пожелтения индикатора метилового красного. Оксалаты прокаливаются при температуре 850-900 °C в течение 10 мин для образования оксидов
3. Са Bi Та же процедура, что и для РЬ, но использовали 4 мл СаС12 и раствор оксалата аммония. Прокаливание оксалатов при той же температуре в течение 15 мин
4. Са Pb Использовали 5 мл 5% раствора СаС12 и 1 мл 5% оксалата аммония
148
Новый справочник химика и технолога
Продолжение таблицы 3.35
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
5. Li, К Са Для уменьшения растворимости оксалатов лития и калия добавляли этанол
6. Са Sr, Y, Pm, Се Моча и водные растворы. Степень извлечения Sr (100%), Y (65%), Pm (90%), Се (87%)
7. Са TR, Re(III) Обнаружено, что соосаждение протекает по ионообменному механизму трех ионов Са(П) на два иона Re(III). Определены константы равновесия для семи TR
8. Са Sc, La, Ce, Pr, Nd, Sm, Eu, Tb, Dy, Ho, Yb, Lu Биологические материалы
9. Са Sc, La, Sm, Eu, Dy Природные термальные и морские воды
10. Са TR Водные растворы
11. Са TR Геологические образцы
Таблица 3.36
Предварительное концентрирование соосаждением на различных солях [21]
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
1. СаСО3+ Mg(OH)2 V, Cu, Cd Природные и морские воды. Проведено многократное соосаждение для исключения мешающего влияния
2. МпСОз Ag, Bi, Cd, Си, Fe, Co, Ni, Pb, Tl, Zn Водные растворы солей марганца. Осаждается около 7,5% Мп. Изучено влияние времени взаимодействия, концентраций носителя, микрокомпонентов и анионов марганца. Осаждение в присутствии карбоната аммония
3. К2СО3 Sr Растворы KNO3. Для понижения растворимости К2СОз к водному раствору добавляют этиловый спирт
4. Арсенат циркония Ti Природные воды
5. Соли серебра Hg, Cu, Pd Чистые металлы
Таблица 3.37
Предварительное концентрирование соосаждением на веществах в элементарной форме [21]
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
1. Те Ag Неорганические материалы (в основном металлы). Осадок теллура образуется при восстановлении 1 мг соединения теллура с хлоридом олова в 2М НС1. Увеличение концентрации хлорид-ионов приводит к неполному осаждению. Мешают большие количества меди. В этом случае проводят пе-реосаждение
2. S Au Чистое серебро. Серебро в виде Ag2S осаждается с золотом. Удаляют серебро кипячением в горячей концентрированной азотной кислоте
3. As Se, Те Геологические и биологические образцы. После разложения образцов смесью (HNO3 + HCIO4) добавляют арсенит натрия (—1,5 мг As). As(III) восстанавливают до элементарного мышьяка гипофосфорной кислотой при 80 °C в течение 15 мин. Старение при комнатной температуре 8 часов
4. As Те Медь и медные сплавы. Образцы растворяют в концентрированной азотной кислоте. Добавляют арсенат натрия (0,5 мг As) и элементарный мышьяк осаждают фосфиновой кислотой. Определяют нанограммовые содержания теллура
5. As Те Термостойкие сплавы на Ni-основе. После растворения добавляются арсенит натрия. НС1 и фосфиновую кислоту нагревают при 90-100 °C в течение 20 мин, затем оставляют на 5 часов при комнатной температуре. Изучено влияние концентрации компонентов и реагентов
Методы разделения и концентрирования
149
Продолжение таблицы 3.37
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
6. As Se Термостойкие сплавы на Ni-основе. После растворения добавляются арсенит натрия, НС1 и гипофосфор-ную кислоту. Изучено влияние концентраций всех реагентов. Растворы нагревали при 80-100 °C для полного восстановления As и извлечения Se. Время старения осадка не менее 2 часов
7. Se + Те Pd, Pt, Au, Ag, Ru Геологические образцы. Se и Те восстанавливали SnCl?
8. Pb Ag, Au, Bi, Cu, Pd Образцы свинца. Некоторые микроэлементы можно концентрировать из различных материалов путем осаждения с небольшим количеством матрицы NaBH4. Осадок металла служит в качестве коллектора всех микроэлементов, электрохимически менее активных, чем матрица
9. Fe + Pd Au, Cd, Co, Cr, Cu, Hg, Mn, Mo, Ni, Pb, Pt, Sn, Tl, V, Zn Образцы вод. Концентрации Fe и Pd — 5 мг • л1 100 мл 5 М раствора NaBH4 использовали для восстановления. Раствор оставляли в течение 20 часов. Степень извлечения — от 90 до 95%
10. Fe + Pd Ag, As(III), As(V), Bi, Cd, Co, Cr(lII), Cr(VI), Cu, Mn, Ni, Pb, Sb(III), Sb(V), Sc(IV), Sn, Те, Tl, Zn Океанские воды. К 900 мл добавляли 5 мг • л-1 Fe и Pd + 5 мл 6% NaBH4. Оставляли на 15 часов
11. Hg Bi, Co, Cd, Pb, Ni, Sn, Tl, In, Ag, Au, Pd Различные металлические образцы. Металлы покрывали тонким слоем ртути. Растворяли металл в НС1 или разбавленной HNO3, оставляя небольшой осадок ртути, обогащенной микроэлементами
Продолжение таблицы 3.37
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
12. As(Se) Se As Сталь. После растворения Se и As восстанавливали солями олова (II). Se оказался подходящим носителем для As и наоборот. Те и Sb не соосаждаются
Таблица 3.38
Предварительное концентрирование соосаждением на органических соединениях и металлических носителях [21]
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
1. Pb(PDC)2 или Bi(PDC)3 Cd, Co, Mo, Sc, Ti, V Природные воды. 200-500 мл образца. pH = 4,0±0,1; 0,7 мг ам-монийпиролидиндитио-карбамата (АПДК), 3 мг нитратов свинца или висмута
2. Pb(PDC)2 или Bi(PDC)3, или Ni (PDC)2 Cd, Co, Cr, Fe, Mo, Ni, Sc, Ti, V, Zn Биологические флюиды: сыворотка крови, молоко
3. Pb(PDC)2 или Bi(PDC)3 Sc Сыворотка крови человека
4. Pb(PDC)2 As, Se, Sb, Co, Zn, Hg, Cd, Cu, Fe, Mo, Ni, Sc, U Морская вода. Изучено влияние pH
5. Pb(PDC)2 Cr(IIl), Cr(VI) Природные воды. Cr(VI) соосаждается при pH = = 4,0. Сг(Ш) — при pH = = 9,0
6. Pb(PDC)2 Co, Cu, Hg Поверхностные воды Мертового моря. 1 л образца + 2 мг РЬ, pH = 3,6 + 20 мг АПДК
7. Fe(PDC)3 V, Zn, As, Hg, Pb, Se 500 мл образца + 200 мкг Ре3+ + АПДК. Изучено влияние кон-г» 3+ центрации ге
8. Тетраметилендитио-карбамат железа (III) Cd, Co, Cu, Ni, Pb Минеральные воды. 200-250 мл образца воды + 500 мкг Fe3+ при pH = 2-3 + 2,5 мг тетраметилендитиокарбамата аммония. Железо выбрано благодаря его высокой концентрации в минеральной воде. Степень извлечения > 95%
150
Новый справочник химика и технолога
Продолжение таблицы 3.38
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
9. Дибензилдитиокарбамат железа U Природные воды. Степень извлечения >95%
10. Диэтилдитиокарбамат железа Mn(II), Cr(III), Cr(VI), Co, Ni, Cu, Zn, Pb Природные воды. 100 мл образца воды, при pH = 7. Количественное извлечение
ll.Fe(PDC)3 Cd, Cu, Pb, Ni, Ag, Zn Речные раковины
12. Co(PDC)3 Cu, Ni, Cd Морская вода. 100 мл образца воды + 50 мкг Со(СоС12) + 10 мг АПДК. В течение 5 мин старение осадка. pH = 2
13. Co(PDC)3 Pb, Ni, Cr(VI), Cd Питьевые воды. 1 л образца pH = 4 + 500 мкг Со + 100 мг АПДК. Осадок отстаивается 30 мин. Достаточно высокая степень извлечения
14. Co(PDC)3 или Co(PDC)2 V, Cr, Fe, Co, Ni, Cu, Zn, As(III), Sc(IV), Hg, Pb, Cd, Zr, Mo, Pd, Ag, Sn Природные воды. Для повышения соосаждения необходима предварительная подготовка проб для исключения влияния органических веществ. Органические вещества разрушаются кипячением с персульфатом калия в течение 45 мин. 0,25-0,90 мл образца воды + 250 мкг Со; pH = 4 + 250 мг АПДК
15. Y(PDC)3 Fe, Ni, Cu, Zn, Pb Биологические материалы. 60 мл, pH = 3-4 + 100 мкг Y + 80 мг АПДК. Осадок отстаивается в течение 1 часа
16. ТХДДТК) + Ti(OH)4 Fe, Cr, Co, Cu, Ni, Mn 2 мкг Ti, pH = 3,8-4,5. 2 мг натриевой соли ДДТК. Далее pH меняется до 8,0-8,2. Осаждение 5 мин
17. Mo(PDC)3 Fe, Co, Ni, Cu, Zn, Cd, Pb Природные воды, содержания на уровне мкг • л’1
18. Mo(PDC)3 Большинство переходных металлов Водные образцы. Осаждение с добавлением АПДК
Продолжение таблицы 3.38
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
19. Индия ок-синат + тан-нин + тионалид Ag, Al, Bi, Cd, Co, Cr, Cu, Fe, Ga, Ge, Mo, Ni, Pb, Sn, Ti, V, Zn 100 мл образца биоматериала + 5 мг In + 5 мг охина pH = 5,2 + 0,2 г танниновой кислоты + 20 мг тионалида. Танниновая кислота добавляется для увеличения степени извлечения Сг и V. Тионалид — для количественного извлечения РЬ и Sn
20. Индия ок-синат + тан-нин + тионалид Al, Co, Ge, Mn, Ni, Ti, V, Bi, Pb, Mo, Cd, Zn, Водные образцы. 100 мл образца + 30 мг In + 0,5 г оксина pH 5,2 + 0,2 г танниновой кислоты + 20 мг тионалида. Старение в течение 12 часов
21. Алюминия оксинат или индия оксинат Cu, Co, Mo Образцы с/х культур. Оксинат алюминия количественно извлекает Со и Мо. Оксинат индия частично соосаждает все. Добавление танниновой кислоты и тионалида количественно извлекает все указанные компоненты. 500 мл образца + 15 мг А1 + 500 мг оксина + 0,2 г танниновой кислоты + 20 мг тионалида
22. Алюминия оксинат + тионалид Cu, Fe, Pb, Mn, Ni, Sn, Zn Некоторые неорганические соли (KBr, КО, KI, KNO3, NaCl, NaBr, NaNO3) pH = 9. Нагревать при температуре SO-85 °C в течение 1 часа
23. Индия оксинат + тан-нин + тионалид Co, Zr, Ti, V, Mo, Cr, W, Ni, Pb Образцы КО, pH = 5,2. Оставлять на ночь
24. Индия оксинат + тан-нин + тионалид Sn, V, Ti, Mo, Cu, Mn, Pb, Zn, Fe Загрязнители воздуха, собранные на стекловолоконный фильтр и обработанные 50 мл горячей 6 М НС1. 10 мг In + 1 г оксина + 100 мг тионалида + 400 мг танниновой кислоты; pH = 5,2. Оставлять на ночь
Методы разделения и концентрирования
151
Продолжение таблицы 3.38
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
25. Алюминия оксинат + таннин + тионалид Ag, Au, Cu, Pb Почва и сточные сбросы. Коэффициент концентрирования — 400. Степень извлечения 80%
26. Оксинат магния Al, Cd, Co, Cu, Mn, Ni, Pb, Zn Водные растворы. 100 мл раствора + 20 мг Mg2+ + 100-200 мг оксина, pH = 9. Старение — 1 час при 70 °C. Степень извлечения —100% для всех, кроме CR(83%), Мо(64%), V(70%). Сг — 98% при pH = 10,5
27. Оксинат магния или алюминия Co Природные воды. 100-500 мл образца воды + 20 мг Mg2+ + 40-100 мг оксина, pH = 9,5. Старение 1 час при 70 °C. Изучено влияние pH на соосаждение Со с оксинатом магния, алюминия, никеля. Органические вещества не влияют на соосаждение с оксинатом магния
28. Оксинат магния Cd, Pb, Zn Такая же процедура, как в двух предыдущих примерах. Оптимальное значение pH = 8,0-10,0
29. Оксинат магния Cu, Mn Та же процедура. Старение при 80 °C. Полное извлечение Си при pH = 6,7-8,7; Мп при pH = 6,5-9,7
30. Оксин + тионалид + Bi Se, As, Sb, Hg, La, Cd, Co Природные воды. Изучено влияние pH. Оптимальное значение pH = 7 для большинства микроэлементов. Для La — 8-9
31. Ni + ДМГ Pd Водные растворы. pH ~ 6,5. Pt(IV) тоже со-осаждается
32. Ni + ДМГ + ПАН Cd, Cu, Mn, Pb, Zn Озерные и морские воды. 10-350 мл образца воды + 4 мг никеля + 17,5 мг ДМГ + 2 мг ПАН при pH = 10. Старение — 1 час при 60 °C. ПАН добавляется в связи с тем, что Ni-ДМГ не обсепечивает полноты извлечения
Продолжение таблицы 3.38
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
32. Ni + ДМГ + ПАН Степень извлечения Си, РЬ (100%), Cd(86%), Мп (10%). Оптимальные pH: для Си — 5-10,5; для Мп — 9-10,5; для РЬ — 10-10,5
33. Fe + Купферон Zr Несколько типов сталей. Fe + купферон ведет себя также как гидроксид железа (III). Содержание Fe в осадке — 70-75 мг
34. Al-салицилат Sn Zn-Al сплавы. Отделение Sn от Си, РЬ
35. R-Роди-зонат Ba, Sr, Са, Ra, Pu, Ce, Zr Водные растворы, морская вода, моча. Осаждение родизоната калия происходит при добавлении или хлорида аммония или этилового спирта. Осаждение в присутствии этилового спирта (pH = 5) приводит к большему извлечению микроэлементов, чем в присутсвии хлорида аммония
36. Си или Си + Fe ДДТК Ni, Co, Hg, Tc, Os, Pd, Sn, Sb, Bi, In, Fe, Ti, Nb, Tl, Pb, Zn, Cd, Mn Некоторые элементы могут осаждаться даже при pH = 1, другие — при pH = 4, Мп — при pH = 9,5. 5 мкг Си или Си + Fe (pH = 9). Старение — 5 мин
Таблица 3.39
Предварительное концентрирование соосаэедением на чистых органических носителях [21]
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
1. ДДТКК Ni, Со, Си, Hg, Тс, Os, Pd, Sn, Sb, Bi, In, Fe Осаждение осуществляется при введении в водные растворы Na-ДДТКК и дальнейшем подкислении раствора. Соосаждаются комплексные ионы металлов с ДДТКК
152
Новый справочник химика и технолога
Продолжение таблицы 3.39
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
2. ДДТКК Fe, Mn, Zn, Cu, Cd, As, Pb, Sc, Mn Образцы вод. Ni и Си полностью извлекаются при pH = 1-11. Zn, Cd, Pb, Fe(III) начинают осаждаться при pH = 1-2, но полное осаждение при pH = 4. Используют pH = 5. Мп количественно не извлекается без металлического носителя. As — при pH = 5,0-5,5
3. ДДТКК + Дибензили-ден-D-сорбитол Pb, Fe, Cu, Zn, Mn, Cd, Cr, Sb, As Промышленные сточные воды. Диапазон определяемых концентраций 1-50 мкг. pH = 5,0-5,5 (ацетатный буферный раствор). 100 мг Na-ДДТКК + 17 мг дибензилидена D-сорбитола
4. ДДТКК Cu, Zn, Hg, Fe Соленые воды. Обогащенные NaCl водные растворы. 250 мл образца воды + 400 мг Na- ДДТК, pH = 4
5. АПДК + зефирамин Cr, Mn, Fe, Co, Ni, Cu, Zn, Pb Биологически материалы. Осаждение при pH = 6-8
6. ДДТКК Zn, Co, Mn, Eu, Образцы природных вод. Добавление Na-ДДТК (400 мг), pH = 5
7. Диэти-ламмоний N,N'~ дитиокарбамат Cr, Ni, Fe, Cu, Zn, Cd, Pb, As, Hg, Sc, Pd Питьевые и сточные воды. Биологические материалы. Степень извлечения > 80%
8. ДДТКК Fe, Co, Ni, Cu, Zn, Sb, Hg, Pb, Ag, Cd 100 мл образца воды. pH = 4; 5 мг Na-ДДТК. Старение —15 мин.
9. ПДТКК Fe, Co, Ni, Cu, Zn, As, Se, Ag, Cd, Sb, Hg,Pb 100 мл образца воды. pH = 4. 10 мг аммониевой соли ПДТКК. Старение — 20 мин.
10. ДБДТКК Mn, Fe, Co, Ni, Cu, Zn, As, Se, Ag, Cd, Sb, Hg, Tl,Pb 100 мл образца воды. pH = 4. 10 мг Na-соли ДБДТКК в метаноле. Старение — 30 мин.
11. ДДТКК + силикагель Cu, Pb, Mn Растворы сахаров. pH = 5,3-5,4
Продолжение таблицы 3.39
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
12. 3,5-Дифенил пи-разолинди-тиокарбами-новая кислота Си, РЬ, Мо Водные вытяжки почв. pH = 1-2
13. ДБДТКК As(III), As(V), Cd, Fe, Zn Водные растворы. pH ~ 2. 100 мл образца воды + 10 мг натриевой соли ДБДТКК в метаноле. Соосаждается As(III). As(V) нужно восстанавливать до As(III). Степень извлечения —100% при pH =1-3. Высокие степени извлечения и для других элементов
14. ДБДТКК + фенолфталеин Sc(IV) Пресные и морские воды, 500 мл образца воды, pH = 2. 10 мг натриевой соли ДБДТКК в метаноле. 10 мг фенофталенина в метаноле. Без фенолфталеина степень извлечения 97%, но уменьшается со старением осадка. В присутствии фенолфталеина старение не уменьшает степень извлечения
15. Купферон Sn, Sb, Nb, Ta, Zr, Hf, Fe, Ti, V, Ga, Bi, U 1% H2SO4 (водные растворы). 50 мг купферона
16. Фенил-флуорон Ge, Mo, W, Th, In, Mn, Zn, Pb, V, Ti, Cu, Fe, Co, Ni 1% H2SO4 (водные растворы)
17. ПАН Mn, Zn, Pb, V, TI, Cu, Fe, Co, Ni 30-100 мл образца воды. Изучено влияние pH и маскирующих агентов на полноту извлечения
18. ПАН Mn, Fe, Co, Ni, Cu, Zn 50 мл раствора воды, pH = 10. ПАН в спиртовом растворе, 5 мг ПАН
19. ПАН Mn, Fe, Co, Ni, Cu, Zn Высокочистые Мо и W и их оксиды. 1 г образца. Предел обнаруж. — 0,2-1 мг • л-1
Методы разделения и концентрирования
153
Продолжение таблицы 3.39
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
20. ПАН Fe, Мп, Си, Zn, Cd, Pb Водные растворы; pH =10. Для формиро-вания осадка добавляют ПАВ. Изучено влияние маскирующих агентов
21. ПАН Мп, Со, Zn, Ей 250 мл образца воды + 20 мг ПАН в этаноле. Нагревание до 80 °C, разрешается охлаждение
22. ПАН Сг(Ш), Мп, Ni, Си, Zn, Hg, Ей 0,5-4 л образца воды + 20 мг ПАН в этаноле. Нагревание при 70-80 °C в течение 10 мин. Изучено влияние pH. Ni, Zn, Си извлекаются при pH = 6,5-10, Cr(III) и Мп — при pH = 10
23. ПАН Сг(Ш), Мп, Ni, Си, Zn, РЗЭ Геотермальные воды. 0,5-2,5 л; pH = 9, 20 мг ПАН в этаноле. 70-80 °C в течение 10 мин
24. ПАН и Морская и водопроводная вода. Биологические материалы. pH = 4,5-6,5. Степень извлечения — 85-94%
25. Хром-пиразол (II) фторид Bi, Pb, Sb, Sn Водные образцы
26. Хром-пиразол (И) иодид Pd, Rh,Au, Ag Ni электролитические ванны. 100 мл, концентрации 5-200 мг • л-1
27. Хром-пиразол (II) фторид + SCN” U(VI) Кислые водные растворы. Указанная система была изучена для соосаждения U(VI)
28. Хром-пиразол (II) фторид + галогениды Cd, Bi, In, TI Несколько красителей было исследовано в качестве соосадителей для галогенидов металлов
29. a-Ni-Бен-зилдиоксим Ni Водные растворы. 100 мл образца воды. 2-200 мкг • л-1 Ni; pH ~ 10. Извлечение при pH = 8-10
30. а-Бен-зилдиоксим Ni Морская вода. 500 мл образца воды + 1 мг реагента. Время старения снижается при pH = 9,5
31. а-Бен-зилдиоксим W, Mo Образцы воды. Соосаждение неполное
Продолжение таблицы 3.39
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
32. Ниоксим Ni Водные растворы 100 мл). pH = 4,5. 32 мг 1,2 циклогексано-дионодиоксима (ниоксим). Маскирующие агенты — тартраты и ЭДТА
33. а-Нафтон-оксим Мо Морские воды. 100 мл образца воды, pH = 2. 100-200 мг реагента требуется для количественного соосаждения 10 мкг Мо. Старение — 15 мин, включая встряхивание
34. Оксин (8-хинолинол или 8-гидроксихинолин) Fe, Се, Рг, Pu Железо сокристалли-зуется с 8-хинолинолом (оксин). Старение — 20 мин
35. Оксин Al Водные растворы. 100 мл. 10 мг • л"1 А1, pH = 6,8-6,9. 160 г оксина. Старение — 1 час
36. Поли-5-винил-8-гидроксихи-нолин Al, Cu, MN, Fe, V, Ni, Pb, Zn, Co Указанный реагент придает оксину хелатные свойства. Растворяется в 75% уксусной кислоте (35 мг • л”1). Изучено влияние pH и концентрации реагента на соосаждение. pH = 7-9 полного извлечения элементов, кроме V, для V — 4-5.
37. Оксин Cd, Cu, Mn, Pb, Zn Морская вода; pH = 7-8. 100-150 мл образца воды. 100 мг оксина. Выдерживать в течение 1 часа при 70 °C. При комнатной температуре — в течение нескольких часов. Степень извлечения > 98%
38. 8,8'-Дихинолилдисульфид + 8-меркаптохинолин (тиоксин) Mn, Co, Cu, Zn, Sb, Hg, Ag, Cd, Fe, Ni, Au, Sc, Rb, Cs 95-98% степень извлечения после 1 часа старения. Na, R, Sm, Мо не соосаждаются. Оптимальный pH = 2-12. Температура 35-85 °C
39. Тионалид (тиогликоль-Р-амино-нафталид) Au, Os, Ta, In, Hg, Ag, W, Zn, Sn, TI, Co, Ir, Ru, Mn, Cr, Hf Водные растворы. Морская вода. Степень извлечения изучена для pH = 0,3; 5; 7; 10
154
Новый справочник химика и технолога
Продолжение таблицы 3.39
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
40. То же As(lli) Морская вода, биологически материалы. Соосажда-ется As(III). As(V) — нет
41. Тионалид (2-меркапто-А-2-нафтил-ацетамид) Оксин As(III), Cu, Sb(III), Sb(V) Морская вода. 0,01-0,5 н. H2SO4. Количественное соосаждение из 0,4 н. H2SO4. Sb, Си соосажда-ются при pH = 6-9
42. Тионалид + поливинил пирролидин Cu, Fe, Zn, Sc, Cd, Sn, Tc, Hg, Pb Образцы вод. 0,25-1 л образца воды + 15 мл 0,1% тионалида в 50% уксусной кислоте + 15 мл 1% водного раствора поливинилпиролидина; pH = 4. Встряхивание 15 мин. Старение — 5 мин. Степень извлечения: Cd(70%), Zn(80%), Fe, Cu(90%), Sc(IV), Sn(IV), Tc(IV), Hg, Pb— 95%
43. Тионалид Ag, Cd, Co, Cr(III), Cr(VI), Cu, Fe(II), Fe(III), Hg, Zn, Sc, Sb(III), Sb(V) Образцы воды 500 мл + 50 г тионалида. 1 час старения при встряхивании при температуре 40-50 °C. Старение при комнатной температуре — 20 час; pH = 9. Степени извлечения: Со (67%), Cr(VI) (70%), Fe(III), Cr(III) (87%), Ag (88%), Cd(89%), Fe(II) (91%), для остальных > 97%
44. Антипу-риновые красители + тио-цинат или таннат Co, Mn, Cu, Zn Водные растворы. Изучено несколько антипу-риновых красителей
45. Дианти-пирилметан TR Образцы воды. pH = 3-5. Изучено влияние анионов
46. Дитизон Ag, Bi, Cd, Cu, Hg, Pb, Pd, Zn Солянокислые и азотнокислые растворы. Степень извлечения зависит от концентрации кислоты. Метод использован для анализа образцов алюминия, сульфата алюминия и кальция
Продолжение таблицы 3.39
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
47. Дитизон + фенолфталеин Si, Со Поверхностные воды. Фенолфталеин не влияет на извлечение, но помогает сформировать осадок. 7 мг • л“1 дитизона. 0,8-4 л воды. При pH = 1 соосаждается только Ag. Со извлекается при pH = 6,5-8. Дитизон растворен в уксусной кислоте, фенолфталеин — в этиловом спирте. Старение — 50 час
48. Оксин + фенолфталеин U, Со, Ni, Zn Образцы вод; 1-4 л. 1,25 г • л1 оксина (в 20% уксусной кислоте). Фенолфталеин — 1 г. pH = 6-7. Старение — 50 час
49. а-Нитрозо-0-нафтол Со, Fe, U, Се, Сг(Ш), Zr Морская вода. 30-50% кристаллизация реагента достаточна для 100% со-кристаллизации микроэлементов. Значения pH: Со (4-9); Fe (2-9); Zn(3-7); U (6,6-8); Сг(Ш) (6,5-7,5); Zr (5-6)
50. Тионалид + фенолфталеин Ag, Cd 200 мл водного раствора; pH = 5,5. 1 г фенолфталеина + 2 мг тионалида
51. Оксин + нафталин Fe(III) А1- сплавы, А1, речная вода. pH = 4,8 при 30-40 °C. Изучено влияние pH и концентраций реагентов
52. Оксин + нафталин Cd Водные растворы. Нафталин в растворе ацетона. Изучено влияние pH, концентраций реагентов и мешающих ионов
53. Морфо-лин-4-карбо-дитиат + нафталин Zn, Cd, Pb Сплавы, образцы воды. Оптимальные значения pH: Zn (3,7-9,4); Cd (3,3-11,0); Pb (5,4-10,2)
54. 2-Мер-каптобензи-мидазол Au, Sn, Hg, Ag, Ta Морская вода. Изучено влияние pH = 11 мл образца воды + 15 мг реагента. 48 час старения при 5 °C. Высокая степень извлечения при pH: Au(l); Sn(5); Hg(l-5); Ag (1-5); Та (1-3)
Методы разделения и концентрирования
155
Продолжение таблицы 3.39
Макроэлемент Микроэлементы Матрица, условия проведения эксперимента, примечания
55. Динаф-толметан Be Водные растворы. Биологические материалы. 100 мл образца. pH = 7,2 + 3 мл 0,4 М раствора реагента в диметилсульфоксиде. Старение — 30 мин
56. 1,10-Фе-нантролин + тетрафенил-боран Mn, Fe, Ni, Cu, Со, Zn, Cd, Sn, Hg, Pb Образцы вод. 50 мл - 1 л образца воды. pH = 5. Для 1 л пробы 1 мл 0,2 М тетрафенилборана натрия и 0,25 М 1,10 фенантролина. Степень извлечения для всех элементов 99-100% при pH = 5-6. Для свинца — 80%. Старение — 30 с
57. Тетрафе-ниларсениум перхлорат 96TcO4 Водные перхлоратные растворы. Степень извлечения не зависит от pH = 2-12. Зависит от концентрации реагента. Соосаждается только пе-ретехнат. Процент соосаждения Tc(IV) очень низкий
3.2.4.2. Зонная плавка и направленная кристаллизация
Классическая схема разделения веществ методом соосаждения ориентирована на определение микропримесей в растворе. В других схемах осуществления подобных процессов, которые объединяются общим термином кристаллизационное концентрирование, метод позволяет концентрировать микропримеси, находящиеся в твердой матрице. В основе кристаллизационного концентрирования лежит процесс перераспределения матричных и примесных компонентов анализируемого образца между жидкой фазой расплава или солевого раствора эвтектического состава и твердой кристаллической фазой при условии управляемого перемещения межфазной границы. В результате управляемой кристаллизации микропримеси, оттесняясь фронтом кристаллизации, концентрируются в тонкой пленке, образующейся на поверхности кристаллического образца.
В отличие от соосаждения метод кристаллизационного концентрирования является безреагентным. Поскольку после концентрирования микропримесей и отделения концентрата матрица может быть возвращена в производство, этот метод позволяет экономить анализируемый материал. Это становится важным положительным свойством метода в характерных для него областях применения: при анализе высокочистых материалов, предназначенных для изготовления полупроводников, сцинтилляторов, лазерных кри
сталлов, изделий для волоконной оптики и т.д. Другим отличием метода управляемой кристаллизации от соосаждения является возможность только группового концентрирования микропримесей без разделения.
Управляемая кристаллизация имеет две схемы практической реализации процесса концентрирования: зонную плавку и метод направленной кристаллизации, отличающиеся исходной формой анализируемого образца и техникой проведения анализа. Если в зонной плавке исходное состояние анализируемого вещества твердое, то в направленной кристаллизации образец предварительно переводят в жидкое состояние в виде расплава или водно-солевого раствора эвтектического состава (рис. 3.4).
Рис. 3.4. Схема реализации методов управляемой кристаллизации:
а — зонная плавка: 1 — до начала процесса; 2 — с одной зоной расплава, перемещающейся вдоль слитка; 3 — с несколькими одновременно перемещающимися зонами расплава;
б — направленная кристаллизация: 4 — до начала процесса;
5 — при проведении процесса
1—1 — твердая фаза; I_I — расплавленный раствор
эвтектического состава в случае направленной кристаллизации
При направленной кристаллизации между жидкой и образующейся твердой фазой устанавливается граница раздела — фронт кристаллизации или затвердевания, который благодаря заданному градиенту температуры может перемещаться с определенной скоростью. В зонной плавке концентрирование микропримесей и одновременно очистка твердого образца происходят при направленном перемещении вдоль него одной или нескольких узких зон расплава. При медленном перемещении зоны расплава формируются два фронта или две межфазные границы: фронт расплавленного вещества — фронт кристаллизации.
Концентрирование примесей в твердых веществах зонной плавкой осуществляют однократным или многократным проведением узкой зоны расплава через горизонтально или вертикально расположенный образец. При зонной перекристаллизации участки твердой фазы, образующиеся в первую очередь, обогащаются компонентами с коэффициентами распределения, превышающими единицу; чем выше коэффициент распределения, тем быстрее это вещество кристаллизуется. Конечные участки слитка обогащаются компонентами с коэффициентом распределения меньше единицы. У метода есть ряд объективных ограни
156
Новый справочник химика и технолога
чений: трудность достижения коэффициентов концентрирования порядка 100 и выше, недостаточная эффективность по отношению к изоморфным примесям, снижение эффективности концентрирования в области малых концентраций, неприменимость для анализа проб сложного состава и веществ, малоустойчивых при температуре плавления, помехи, возникающие из-за испарения примесей либо изменения их химической формы и состояния. Поведение примесей при их концентрировании методом зонной плавки можно оценить, исходя из диаграммы состояния матрица - примесь, которые отражают характер взаимодействия макро- и микрокомпонента в жидкой и твердой фазе. По этой причине, а также вследствие появления множества альтернативных методов концентрирования, методы зонной плавки имеют практически только историческое значение, применяясь иногда для контроля содержания примесей в легкоплавких металлах (Pb, Bi, Sb, Sn и др.), для определения фосфора и бора в высокочистом кремнии и т.д. То же самое относится и к методу направленной кристаллизации. Более подробно об этих методах и о применяемой технике эксперимента можно прочитать только в сравнительно старых изданиях [34, 35].
3.2.5. Жидкостная экстракция
Жидкостная экстракция — метод разделения веществ, основанный на различиях в распределении веществ между двумя жидкими фазами, в наиболее общем случае — между водным раствором и органическим растворителем. Тем же термином называется сам процесс извлечения вещества из водной фазы в органическую. Соответственно обратный процесс называется реэкстракцией. В названии метода и процесса слово жидкостная, как правило, отсутствует, т.е. когда речь идет об экстракции без уточнения агрегатного состояния извлекающей фазы, всегда подразумевается жидкостная экстракция. В других случаях уточняется газовая или твердофазная экстракция. Все многообразие экстракционных систем, отличающихся механизмом межфазных переходов выделяемых веществ, подразделяется на две большие группы: на экстракцию по механизму физического распределения и реакционную экстракцию.
3.2.5.1. Экстракция по механизму физического распределения
В случае физического распределения процесс перехода веществ из водной фазы в органическую вызван различиями энергий их сольватации и гидратации. По этому механизму хорошо экстрагируются в органические растворители большие неполярные или малополярные молекулы. В случае неорганических веществ это такие соединения, как GeCl4, OsO4 и т.д. Учитывая, что число подобных соединений крайне невелико, основное применение этот вариант экстракции находит для выделения из водных растворов растворенных примесей неполярных и слабополярных органических веществ, например нефтепродуктов. В качестве экстрагентов для экстракции по механизму физического распределения чаще всего применяются такие нейтраль
ные органические растворители, как гексан, хлороформ и четыреххлористый углерод. Извлечение неполярных органических примесей по механизму физического распределения происходит не селективно, так как для большинства из них энергии сольватации различаются несущественно и всегда больше энергии гидратации. Поэтому чаще всего речь может идти только о групповом концентрировании примесей неполярных органических веществ, поскольку KD для многих классов токсичных органических соединений, имеющих относительно полярные молекулы (таких, например, как фенолы) не столь велики, чтобы добиться значимых коэффициентов концентрирования при их однократной экстракции наиболее распространенными растворителями (табл. 3.40).
Таблица 3.40 Коэффициенты распределения некоторых фенолов в системе вода - хлороформ и вода - четыреххлористый углерод
Фенолы Экстрагент
Хлороформ Четыреххлористый углерод
о-Метоксифенол
(гваякол) 1,85 0,55
1-Нафтол 17,0 4,7
2,4-Ксиленол 11,6 2,9
2,5-Ксиленол 53,7 11,2
2,6-Ксиленол 30,0 6,8
2-Нафтол 31,3 6,0
2-Фторфенол 39,0 10,0
2-Этилфенол 58,5 17,8
3,5-Ксиленол 38,0 6,6
3-Хлорфенол 3,7 0,96
4-Бромфенол 10,3 3,0
4-Иодфенол 11,7 3,6
4-Хлорфенол 36,5 11,2
4-Этилфенол 10,5 3,1
о-Крезол 50,0 9,5
л-Крезол 66,8 14,5
Фенол 55,2 10,0
В некоторых случаях это ограничение преодолевается предварительным переведением (конверсией) плохо экстрагируемых соединений в хорошо экстрагируемые. Те же фенолы легко переводятся обработкой элементарными галогенами непосредственно в водном растворе в соответствующие менее полярные галогенопроизводные, KD при экстракции которых в среднем на порядок выше. Но в этом случае для определения исходного содержания в воде самих галогенопроизводных фенолов, обладающих большей токсичностью, требуется их предварительное выделение. Существенный эффект при экстракции полярных органических веществ дает выбор органического экстрагента (табл. 3.41).
Методы разделения и концентрирования
157
Таблица 3.41
Коэффициенты межфазного распределения фенола и монохлорфенолов в системе вода - органический экстрагент [36]
Экстрагенты Фенол 2-Хлор-фенол 3-Хлорфенол 4-Хлор-фенол
Трибутилфосфат (ТБФ) 450 2090 5750 3890
Спирты
w-Пентиловый 36,8 143 — 325
«-Гексиловый 35,4 140 400 300
«-Гептиловый 33 134 380 266
«-Октиловый 31,3 132 333 250
«-Нониловый 29 136 300 223
«-Дециловый 27 117 270 210
Эс )ИрЫ
Этилацетат 58 200 —
н-Бутилацетат 48,5 174 480 324
н-Пентилацетат 43,8 157 430 295
н-Гексилацетат 40 152 — 270
н-Гептилацетат 34,5 139 — 245
Изопропиловый 28,9 — —
Дихлорэтиловый 10,9 — —
Нитробензол 7,9 59,5 64,5 73
2-Нитротолуол 7,3 54,6 62,3 58
3-Нитротолуол 5,8 — — 43
Хлороформ 1,85 23 10,5 10,3
Бромоформ 1,6 20,8 8,1 8,7
Дихлорметан 4,9 — —
1,2-Дихлорэтан 4 39 22,3 21,5
1,2,3-Трихлор-пропан 3,6 — —
Бензол 2,1 28,5 13,2 13,6
Толуол 1,65 23,4 И,2 12
Этилбензол 1,24 20,5 8,8 9
Изопропилбензол 1 16,1 — 7,5
«-Бутил бензол 0,85 15,8 —
о-Ксилол 1,88 22,5 И,7 11,3
.м-Ксилол 1,62 23,4 11,2 9,8
«-Ксилол 1,54 21,3 9,9 8,3
Хлорбензол 1,8 23,2 10 10
Бромбензол 1,5 22,5 8,5 9,2
Иодбензол 1,25 20,9 7,5 8
1,2-Дихлор-бензол 1,35 — —
Четыреххлористый углерод (CCI4) 0,55 15,5 3,1 3
Продолжениетаблицы 3.41
Экстрагенты Фенол 2-хлор-фенол 3-хлор-фенол 4-хлор-фенол
Циклогексан 0,22 7,3 1,2 1,2
«-Пентан 0,2 7,3 0,79 0,8
«-Гексан 0,2 7,1 0,85 0,81
«-Гептан 0,2 7 0,84 0,78
«-Октан 0,18 6,5 0,79 0,76
«-Нонан 0,18 6,6 0,75 0,80
«-Декан 0,15 6 0,8 0,78
Наиболее полную информацию по экстракции органических соединений различных классов можно найти в специальных справочниках [36, 37].
3.2.5.2. Реакционная экстракция в органические экстрагенты
Для экстракционного выделения неорганических веществ наибольшие возможности открывает реакционная экстракция, т.е. процесс, протекающий как гетерогенная химическая экстракция:
•*-АВодц + уВорг — [АхВДорг,
где Аводн — вещество, выделяемое из водного раствора; Ворг — органический экстрагент; [АхВДорг — соединение, в форме которого вещество А переходит в органическую фазу.
Как любая химическая реакция, подобный процесс характеризуется константой, называемой константой экстракции Кэкс:
к
В случае реакционной экстракции далеко не всегда известно и остается постоянным стехиометрическое соотношение выделяемого вещества и экстрагента в экстрагируемом соединении, поэтому для характеристики экстракционных процессов Кэкс используется сравнительно редко. Предпочтение отдается универсальной характеристике — коэффициенту распределения KD:
(с.) + (с2) + ... + (с„)
ГЛ __ х 1/орг \ z/орг х И /орг
где 1, 2,..., п — соответствует различным формам выделяемого вещества в водном растворе и в фазе экстрагента. При высокой скорости взаимных превращений, вместо концентраций отдельных форм — суммарные концентра-
[п Л / п Л
| — в органической фазе и ^с. — в
‘=1 /орг V '=1 /водн
ВОДНОЙ.
Доказательством обратимости экстракционного процесса, т.е. возможности реэкстракции выделенного вещества в водный раствор, является равенство значений KD,
158
Новый справочник химика и технолога
полученных для прямого процесса экстракции и обратного процесса реэкстракции при одинаковом составе водной и органической фаз:
-^Т>экс -^Ореэкс •
Чаще всего за основу классификации экстрагентов, применяемых для реакционной экстракции принимается тип экстрагента: нейтральный, кислотный, основный. Но подобная классификация слишком условна, т.к. многие экстрагенты меняют свои свойства в зависимости от состава водного раствора. Выпадают из этой классификации хелатообразующие экстрагенты, имеющие одновременно кислотные и основные группы. Поэтому более рациональной является классификация экстрагентов по природе донорных атомов, ответственных за образование химической связи с экстрагируемым веществом, и по структурному подобию молекул экстрагентов. По этим признакам можно выделить пять основных классов экстрагентов: кислород-, азот- и серосодержащие, хелатообразующие и макроциклические.
Кислородсодержащие экстрагенты
Наиболее распространенные типы кислородсодержащих экстрагентов приведены ниже.
Нейтральные экстрагенты Кислотные экстрагенты
Простые эфиры Ri — О — R2 Карбоновые кислоты R—С = О ОН
Спирты R-OH
Кетоны R.|-C=O r2 Кислые эфиры фосфорной кислоты
Полные эфиры фосфорной кислоты Ri“Ox r2- О-Р = О R3-Oz О О и и о. о. / 1 \ / 1 \ ООО 000 III III df £ I i I
Сульфоксиды Rl"> = О
В зависимости от характера химической связи кислорода в молекуле экстрагента и свойств экстрагируемого вещества возможны три механизма образования экстраги
руемых соединений с кислородсодержащими экстрагентами. Во-первых, все рассматриваемые нейтральные экстрагенты при контакте с кислыми водными растворами способны к образованию ионных ассоциатов с анионными формами экстрагируемых соединений: анионами сильных минеральных кислот и ацидокомплексами металлов. Во-вторых экстрагенты, имеющие кислотные группы (карбоновые и фосфорорганические кислоты), способны экстрагировать катионные формы по механизму ионообменного замещения в молекуле экстрагента протона на ион металла. Наконец, кислородсодержащие экстрагенты, являющиеся жесткими основаниями (по Пирсону), способны входить во внутреннюю сферу «жесткой» кислоты. В последнем случае имеет место экстракция по механизму образования координационных соединений.
Экстрагируемость анионов по механизму образования ионных ассоциатов увеличивается в ряду экстрагентов от простых эфиров к сульфоксидам (см. табл. 3.42). Этот ряд можно рассматривать как ряд основности кислородсодержащих экстрагентов, понимая под последней способность к сольватации протона. Экстракция по подобному механизму не исключена и для кислотных экстрагентов, но она проявляется только как эффект, сопутствующий экстракции по ионообменному механизму. Существующие представления о структуре катионной части ионного ассоциата не однозначны. Гидратное число протона в водных растворах равно четырем, т.е. каждый ион гидроксония Н3О+ гидратирован тремя молекулами воды. При взаимодействии гидратированного протона с молекулами кислородсодержащих экстрагентов последние координируются вокруг него с частичным замещением молекул воды на молекулы экстрагента или просто сольватируя протон вместе с гидратной оболочкой. Соответствующие катионные образования имеют название гидрато-сольватов, а механизм экстракции анионных форм элементов за счет образования ионных ассоциатов с гидрато-сольватами протонов получил название гидратно-сольватного. Гидратное число 4 не всегда выдерживается в катионной части гидрато-сольватов и может варьироваться в пределах от 1 до 12, т.е. гидратные и сольватные числа далеко не постоянны и могут изменяться в зависимости от состава фаз. Таким образом, с точки зрения образования экстрагируемой катионной части ионного ассоциата экстракционная способность кислородсодержащих экстрагентов определяется конкуренцией процессов гидратации и сольватации ионов гидроксония, а селективность экстракции — процессами образования экстрагируемых анионных форм элементов в водной фазе.
Схематично процесс экстракции ацидокомплексов металлов кислородсодержащими экстрагентами можно представить следующим образом. При контакте условного экстрагента R=O с кислым раствором происходит образование в органической фазе катионных гидрато-сольватов, заряд которых скомпенсирован анионами соответствующей кислоты:
Н+ + X* + nR = О + wH2O ->[H(H2O)OT(R=O)„] 'X .
Методы разделения и концентрирования
159
Переход экстрагируемого соединения [МХ7] (</ <г) в органическую фазу можно рассматривать как реакцию ионного обмена:
{[H(H2O)4R=O)J*X-}0pr + о
о {[H(H2O)„(R=O),]‘ [МХ,Г<’-2)}ч,г + X-.
При экстракции по гидратно-сольватному механизму существенную роль играет форма существования экстрагируемого вещества в водном растворе. Экстрагируемость ацидокомплексов определяется их прочностью и различиями энергий их гидратации и гидратации лиганда или других анионов, присутствующих в водном растворе. Экстрагируемость будет тем выше, чем меньше заряд и чем больше размеры ацидокомплекса, т.е. чем меньше он гидратирован. Наиболее характерные формы хорошо экстрагируемых соединений [МХд]- и [МХ6]~, где X — галогенид-ион. Среди наиболее широко известных соединений — [FeCl4] , [AuC14] , [SbCl6] . Различия в экстрагируемое™ однотипных комплексов, например [РеС14] и [АиС14]“, наряду с размерами определяются их устойчивостью в водных растворах. Относительно непрочные тетрахлоридные комплексы железа образуются в водном растворе только при значительном избытке хлорид-ионов. Поэтому при экстракции наиболее основными кислородсодержащими экстрагентами, не требующими высокой концентрации протонов для образования ионных ассоциатов, например, трибутил фосфатом (ТБФ), зависимости KD от концентрации НО для Fe(III) и Au(III) резко различаются [32]. При экстракции слабоосновным экстрагентом эти различия в значительной степени стираются, так как высокая кислотность водного раствора, необходимая для образования катионных гидрато-сольватов, одновременно обеспечивает условия для образования тетрахлоридного комплекса железа (Ш). Отсюда для экстракционного разделения элементов, образующих однотипные комплексы, более эффективны последние экстрагенты в ряду основности (табл. 3.42). Для их отделения от элементов, не образующих хорошо экстрагируемых форм, — первые.
Данные по экстракции элементов алифатическими аминами приведены в табл. 3.42, где указаны экстракционные системы, обеспечивающие степень извлечения R > 80 %.
Таблица 3.42
Экстракция элементов растворами алифатических аминов [1]
Экстрагируемый элемент и степень окисления Водная фаза Органическая фаза
Ag 1 М LiCl + на Метилдиоктил амин (10%) в трихлорэтилене
Am(III) 12MLiCl + 0,1 м на Триизоктиламин (20%) в ксилоле
As(III) 8-п м на Додецил-(триалкилме-тил)-амин (10%) в ксилоле
Продолжение таблицы 3.42
Экстрагируемый элемент и степень окисления Водная фаза Органическая фаза
Be 0,1 MH2C2O4; pH = 3-4 Триизооктиламин (0,1 М) в хлороформе
Bi 0,5-2 м на Октиламин (0,1 М) в хлороформе
Cd 5-6 м на Трибензиламин (0,1 М) в бензоле
Со 6-ю м на Метилдиоктиламин (8%) в трихлорэтилене
> 6 м на Триизооктиламин (0,1 М) в керосине
Cr(VI) Слабощелочная Метилдиоктиламин (5%) в хлороформе
Раствор Н2Сг2О7 в 6 М на Трибензиламин (5%) в хлороформе, триизооктиламин (5%) в ксилоле
Cu(Il) > 6 м на Метилдиоктиламин (8%) в трихлорэтилене
6,5 м на Триоктиламин (0,1 М) в ксилоле
Fe(III) > 2 м на Метилдиоктиламин (8%) в трихлорэтилене, триизооктиламин (10%) в ксилоле
5МНС1 Трибензиламин (0,2 М) в хлороформе
Ga 7МНС1 Дециламин в хлороформе
Ge(IV) 8-п м на Додецил-(триалкилметил)-амин (10%) в ксилоле
Hf > 8 м на Метилдиоктиламин (5%) в ксилоле, триизооктиламин (5%) в ксилоле
8МНС1 Триизооктиламин (0,2 М) в циклогексане
0,1-1,ом H2SO4 Метилдиоктиламин (5%) в ксилоле, триоктиламин или триизооктиламин (0,1 М) в смеси керосина (97%) и тридеканола (3%)
Hg(H) 1-8 м на Додецил-(триалкилметил)-амин (10%) в ксилоле
In 6-7 м на Додецил- (триалкилметил)-амин (10%) в ксилоле
Mn(II) 8МНС1 Триоктиламин (0,1 М) в ксилоле
160
Новый справочник химика и технолога
Продолжение таблицы 3.42
Экстрагируемый элемент и степень окисления Водная фаза Органическая фаза
Mo(VI) 1 M H2SO4; pH = 1-2 Метилдидециламин (0,1 M) в ароматическом углеводороде
Nb(V) >8MHC1 Метилдиоктиламин (5%) в ксилоле, триизоокти-ламин (5%) в ксилоле
11 MHC1 Трибензиламин (8%) в хлороформе или мети-ленхлориде
~ 2 M H3PO4 Метилдиоктиламин (5%) в трихлорэтилене
4-4,8 M H2SO4 Трибензиламин (8%) в метиленхлориде
1,5MH2SO4 Метилдиоктиламин (5%) в трихлорэтилене
Np(IV) 4-6 M HNO3 Триоктиламин (10%) в ксилоле
« 8 M HC1 То же
Np(VI) 5-7 HNO3 Тоже
~ 6,3 M HC1 То же
Pa(V) > 6 M HC1 Метилдиоктиламин (5%) в ксилоле или хлороформе
1,8 H3PO4 Метилдиоктиламин (5%) в трихлорэтилене
0,5 M H2SO4 То же
Po(V) 6MHC1 Трибензиламин (5%) в хлороформе, метилдиоктиламин (5%) в ксилоле
Pt(V) 0,1-0,5 HC1 Триоктиламин (0,05%) в толуоле
Pu(IV) 4-6MHNO3 Триоктиламин (10%) в ксилоле
> 6 M HC1 То же
Pu(VI) 6-8 M HNO3 То же
6-8 M HC1 Триоктиламин (10%) в ксилоле, триизооктила-мин (5%) в ксилоле, трибензиламин (5%) в хлороформе
1 M CH3COOH Триоктиламин (20%) в ксилоле
Ru(IV) 0,1 MHC1 Триизооктиламин (5%) в ксилоле
Sb(III) 1-4 M HC1 Додецил- (триалкилметил)-амин (10%) в ксилоле
Sb(V) > 6 M HC1 То же
Продолжение таблицы 3.42
Экстрагируемый элемент и степень окисления Водная фаза Органическая фаза
Se(IV) 9MHC1 To же
Sn(IV) 5-6 M HC1 To же
Ta(V) > 4,8 M H2SO4 Трибензиламин (8%) в метиленхлориде
Tc(VII) 6MA1(NO3)2 Тридодециламин (0,3 М) в ароматическом углеводороде
Te(IV) 4-6MHC1 Додецил-(триалкилметил)-амин (10%) в ксилоле
Th 0,5 M H2SO4; pH - 1 Октиламин (0,1 М) в ароматическом углеводороде
12 M LiCl + 0,1 MHC1 Триизооктиламин (20%) в ксилоле
~ 6 M HNO3 Триоктиламин или триизооктиламин (0,4 М) в бензоле или толуоле
Ti(IV) 1 M H2SO4; pH = 1,4-3 1 -(3 -Этил пентил )-этилоктиламин в керосине
U(IV) 9MHC1 Триоктиламин (10%) в ксилоле
0, 5M H2SO4; pH-1 Октиламин (0,1 М) в ароматическом углеводороде
U(VI) 0,1-1 M H2SO4 Метилтиоктиламин (5%) в ксилоле
~ 6,5 M HC1 Триоктиламин или триизооктиламин (10%) в ксилоле
1-2 M HF Метилтиоктиламин (5%) в ксилоле
0,1-1 M H3PO4 Триоктиламин (0,1 М) в керосине
0,5-2 M CH3COOH Триизооктиламин или тридодециламин (10%) в ксилоле или метилизобу-тилкетоне, метилдиоктиламин (5%) в ксилоле
6-7 M HNO3 Триоктиламин (10%) в ксилоле
V(V) H2SO4; pH = 2 Метилдидециламин (0,1 М) в ароматическом углеводороде
6-7 M HC1 Додецил-(триалкилметил)-амин (10%) в ксилоле
Методы разделения и концентрирования
161
Продолжение таблицы 3.42
Экстрагируемый элемент и степень окисления Водная фаза Органическая фаза
Zn > 2 М НС1 Метилтиоктиламин (8%) в трихлорэтилене или ксилоле, трибензиламин (5%) в хлороформе, триизооктиламин (5%) в метилизобутилкетоне
Zr 0,1-1,ОМ H2SO4 Метилтиоктиламин (5%) в ксилоле, триоктиламин или триизооктиламин (0,1 М) в смеси керосина (97%) и тридеканола (3%)
Экстрагируемость при переходе к многозарядным анионным комплексам резко падает. Заметный эффект в данном случае достигается только при использовании экстрагентов последних трех типов, начиная с эфиров фосфорной кислоты. Об экстракции двухзарядных комплексов можно говорить чисто условно, так как наиболее вероятный механизм экстракции в этом случае связан с переходом в органическую фазу соединений типа H[MXq]~. Из класса основных кислородсодержащих экстрагентов наибольший интерес представляют эфиры фосфорной кислоты и в первую очередь ТБФ [1, 39].
Кислородсодержащие экстрагенты, имеющие кислотные группы, часто называют жидкими катионообменни-ками. Из широко распространенных кислотных экстрагентов наибольшей селективностью при экстракции катионных форм элементов обладают фосфорорганические кислоты. Существенные различия в экстрагируемо-сти в данном случае проявляются как для катионов с различной величиной заряда, так и для катионов, отличающихся только размерами ионных радиусов. Например, типичный экстрагент этого класса ди-2-этилгексилортофософорная кислота (Д2ЭГФК) обеспечивает возможность разделения таких близких по химическим свойствам элементов, как лантаноиды и актиноиды. Среднее значение Кс для соседней пары этих элементов превышает 2. Селективность экстракции карбоновыми кислотами значительно ниже, поэтому в общем случае их применение более оправдано для суммарного концентрирования катионных форм элементов, чем для их разделения. Подробные сведения о кислотных экстрагентах и их свойствах можно найти в работе [39]. Данные по экстракции элементов из солянокислых растворов Д2ЭГФК приведены в [1].
Координационный механизм реализуется при экстракции кислородсодержащими экстрагентами из нейтральных и слабокислых растворов. Экстракционная способность кислородсодержащих экстрагентов, извлекающих металлы по координационному механизму, увеличивается в ряду основности кислородсодержащих экстрагентов. Она опре
деляется в первую очередь электронной плотностью на электронодонорном атоме молекулы экстрагента. В случае одинаковых функциональных групп последняя зависит от электроотрицательности других групп, входящих в состав экстрагента: чем она выше, тем меньше электронная плотность на электронодонорном атоме молекулы экстрагента, ответственном за образование химической связи с ионами металла. Так, при переходе в ряду: фосфаты, фосфонаты, фосфинаты, фосфиноксиды — коэффициенты распределения возрастают из-за снижения суммарной электроотрицательности при замене групп R — О — на R —.На практике с координационным механизмом экстракции кислородсодержащими экстрагентами чаще всего приходится сталкиваться при выделении урана, тория, редкоземельных и трансурановых элементов из растворов нитратов. Процессы внутрисферного замещения лигандов часто замедлены во времени, но для кислородсодержщих экстрагентов в общем случае кинетические ограничения не характерны. Поэтому даже в случае координационного механизма они с успехом применяются в многоступенчатых экстракционных процессах, включая экстракционную хроматографию.
Данные по экстракции элементов фосфиноксидом, для которого характерен координационный механизм экстракции, а также наиболее полную информацию об экстракции другими кислородсодержащими экстрагентами можно найти в [1, 38, 39].
Азотсодержащие экстрагенты
К этому классу экстрагентов относят первичные (RNH2), вторичные (R2NH), третичные (R3N) амины и соли четвертичных аммониевых оснований (R4NX), где R — алкильный или ароматический радикал, X — анион любой кислоты. Экстракционная способность аминов определяется наличием у атома азота неподеленной пары электронов, благодаря чему они способны присоединять протон в кислых растворах или образовывать координационные связи с центральными атомами, образующими комплексные соединения. Отсюда — три механизма экстракции азотсодержащими экстрагентами: по реакции присоединения, ионообменный и координационный механизмы.
По реакции присоединения аминами экстрагируются кислоты:
(Н+)ВОдн + (Х-)вода + (R3N)opr (R3NHX)opr.
В данном случае амины выступают в роли оснований в гетерогенной реакции кислотно-основного взаимодействия с образованием соли амина в органической фазе. Способность кислот экстрагироваться по реакции кислотноосновного взаимодействия повышается с увеличением силы кислоты и возрастанием радиуса аниона. Рост радиуса аниона приводит к снижению энергии его гидратации, что облегчает переход кислоты из водной фазы в органическую (рис. 3.5) [44].
162
Новый справочник химика и технолога
Г no; Bf СГ
сю;
Рис. 3.5. Зависимость константы экстракции кислоты от энергии гидратации аниона (экстрагент — триоктиламин в циклогексане)
Изменение константы в зависимости от энергии гидратации аниона наиболее значительно в случае экстракции третичными аминами. Для вторичных и особенно первичных аминов характер зависимости становится более пологим. Влияние строения амина на экстракцию кислот связано с двумя факторами: изменением подвижности электронной пары атома азота (индукционный эффект заместителя) и проявлением стерических эффектов. При этом чем значительнее проявляется отрицательный индукционный эффект, тем меньше экстрагируемость кислоты. Так, константа экстракции в ряду вторичных аминов заметно снижается (на 3-5 порядков) при замене алкильной группы у атома азота на фенильную. Замена радикала у атома азота приводит не только к изменению подвижности электронной пары, но и к изменению стерических факторов, которые сильнее влияют на константу экстракции, чем электронные. Энергия связи в солях аминов между катионом и анионом в первом приближении определяется электростатическим притяжением. Рост числа углеводородных цепочек, их длины и степени разветвления способствует увеличению расстояния между катионом и анионом, т.е. снижению энергии связи и, следовательно, уменьшению константы экстракции. Поэтому, например, устойчивость солей аминов, образующихся по реакции кислотноосновного взаимодействия, изменяется в последовательности: первичные > вторичные > третичные.
Соли аминов и четвертичных аммониевых оснований (ЧАО) проявляют способность к анионному обмену.
(w-z)(R4NX)opr + [МХ„]^г)вода о {(R4N)UMX„]}opr +
+ (п—г))(Х")вода-
Аналогичная реакция может протекать с солью амина, образовавшейся в результате реакции присоединения.
Отличия в случае экстракции ЧАО проявляются только в том, что для протекания реакции анионного обмена не требуется предварительная «протонизация» молекул экстрагента. Поэтому ЧАО проявляют экстракционные свойства в нейтральных и даже слабощелочных растворах. При экстракции аминами из кислых растворов реакции присоединения и ионного обмена выделяются как последователь
ные стадии чисто условно. В присутствии нескольких анионных форм в водном растворе процесс экстракции может рассматриваться как протекание нескольких параллельных реакций присоединения. В конкуренции за образование ионного ассоциата с экстрагентом преимуществом будут обладать наименее гидратированные анионы.
Экстракция аминами и ЧАО по механизму ионного обмена представляет наибольший практический интерес при извлечении из растворов ацидокомплексов металлов. Закономерности экстракционных процессов при этом близки к наблюдаемым при экстракции наиболее основными кислородсодержащими экстрагентами. Различия проявляются только в лучшей экстрагируемое™ многозарядных анионных форм элементов за счет более ярко выраженных основных свойств азотсодержащих экстрагентов.
Соответствующие данные по экстракции аминами можно найти в [1, 44].
Серосодержащие экстрагенты
К этой группе относят аналоги многих кислородсодержащих экстрагентов, в которых атом кислорода замещен на атом серы. Из серосодержащих экстрагентов наибольшее распространение нашли тиоэфиры. В отличие от своих кислородсодержащих аналогов они практически не проявляют способности к «протонированию» и, соответственно, для них не характерен гидрато-сольватный механизм экстракции. Экстракционные свойства серосодержащих экстрагентов определяются их склонностью к участию в гетерогенных реакциях внутрисферного замещения лигандов в комплексных соединениях, образованных халькофильны-ми элементами. При этом наиболее вероятны два варианта образования экстрагируемого соединения:
[МХ^^вода + (и-гХВДорт о [MXXRzS^opr + (я-г)(Х’)водн, [МХ„]4н-г)водн + «(R2S)opr о ([M(R2S)„]Xz)opr + (и-г)(Х-)Водн.
Как «мягкие» основания, серосодержащие экстрагенты образуют наиболее прочные связи с легкополяризуемыми ионами (меди, серебра, ртути, золота и платиновых металлов), относящимися к классу «мягких» кислот по классификации Пирсона. Длина и строение алкильного радикала оказывают существенное влияние на экстракционную способность серосодержащих экстрагентов. Удлинение и разветвление алкильных радикалов обычно приводит к уменьшению KD. Аналогичный, но еще более сильный эффект вызывает замена алкильных радикалов на фенильные. Образующиеся в органической фазе комплексы, как правило, кинетически инертны, и процесс реэкстракции затруднен. Соответственно сфера применения серосодержащих экстрагентов ограничена групповым концентрированием или групповым выделением халькофильных элементов [45].
Хелатообразующие экстрагенты
Характерной структурной особенностью молекул хелатообразующих экстрагентов является наличие, по крайней мере, двух функциональных групп, способных взаимодействовать с ионами экстрагируемых элементов с образова-
Методы разделения и концентрирования
163
нием циклических комплексов. При этом одна или несколько групп имеют кислотный характер. В качестве донорных атомов функциональных групп выступают О, N или S в различных сочетаниях друг с другом. Наиболее распространенные типы хелатообразующих экстрагентов:
/
Р-дикетоны
Таблица 3.43
Экстракция оксихиналинатов [1]
*R---С—СН-----С---R
ОН О
енольная форма
R — С—СН2—С—
О О
кето-форма
Купферон
NO=NOH
а-диоксимы
R—С----С R
II II
N N
НО ОН
дифенилкарбазон (ДИТИЗОН)
дитиокарбаматы
SNa
Данные по экстракции элементов наиболее широко распространенными хелатообразующими экстрагентами представлены в табл. 3.43-3.47.
Экстрагируемый элемент pH раствора Органическая фаза Примечания
Растворитель Концентрация 8-окси-хинолина, моль л-1
А1 5-11,5 Хлороформ 0,02
5,2-5,5 Четыреххлористый углерод 0,013
Ва 11,5 Хлороформ 1,4 R = 90%
Be > 10 Бутиламин (0,2 М) в хлороформе 1,0 R = 100% при экстракции 1 час
Са 11,5 Хлороформ 1,0
11,5 Бутиламин (0,2 М) в хлороформе 0,14
Cd 10,5-12 Бутиламин (0,2 М) в хлороформе 0,001 R = 80%
9-11 Хлороформ 0,001
Се(Ш) 9,9-10,5 Хлороформ 0,07
Со 7-9 Хлороформ 0,001
Сг(Ш) 6-8* Хлороформ 0,02
Cu(II) 2,8-14* Хлороформ 0,07
Fe(III) 2-12,5* Хлороформ 0,02
Ga 4-11,5 Хлороформ 0,02
Hg(II) 8-12 Хлороформ 0,001
In > 14 Хлороформ 0,02
Mg 11-13,5 Бутиламин (0,2 М) в хлороформе 0,007
Хлороформ 0,001 Водный раствор содержит 0,1 н. Na2SO3
Mn(III) 9-12 Хлороформ 0,001 Водный раствор содержит тартрат натрия (0,1 н.) и K3[Fe(CN)6] (2,10 4 М)
Mo(VI) 2-5 Хлороформ 0,001
1,6-5,6 Хлороформ 0,07
Nb(V) 3-10* Хлороформ 0,02
164
Новый справочник химика и технолога
Продолжение таблицы 3.43
Экстрагируемый элемент pH раствора Органическая фаза Примечания
Растворитель Концентрация 8-окси-хин олина, моль•л 1
Ni 8,7 Хлороформ 0,001
8,7-12,5 Бутиламин (0,2 М) в хлороформе 0,001
Pb 8,5-12,5 Хлороформ 0,01
Pd(II) 3,5 Хлороформ 0,001 R ~ 75%
Sc 9,5-10,5 Бензол 0,002
Sn(II) 7,5-9,5 Хлороформ 0,001
Sn(IV) 2,5-5,5 Хлороформ 0,07
Sr 11,3 Бутиламин (0,2 М) в хлороформе 0,35
11,4 Хлороформ 1,0
Ti(IV) 4-9 * Хлороформ 0,02
Tl(III) 4-9 * Хлороформ 0,02
Th 7-10 * Хлороформ 0,02
U(VI) 7-9,9 * Хлороформ 0,02
6,5-9,0 Хлороформ 0,002
V(V) 3,4-4 Хлороформ 0,001
W(VI) 2 Хлороформ 0,01
4,5 * Хлороформ 0,02
Y 7-8 Хлороформ 0,2
Zn 8-12,5 Бутиламин (0,2 М) в хлороформе 0,02
4,6-13,4 Хлороформ 0,027
Примечания.
* Соотношение объемов водной и органической фаз равно 5:1.
Во всех случаях, кроме специально оговоренных, R = 100%.
Таблица 3.44
Экстракция купферонатов [1]
Экстрагируемый элемент Водная фаза Органическая фаза
А1 pH = 2-5 Хлороформ
Bi НС1 или H2SO4 Толуол, метилэтилкетон
Cd Нейтральный раствор Диэтиловый эфир (при т-ре кипения)
Ce(IV) pH = 2 Бутил ацетат
Со Разбавленная уксусная кислота Этилацетат, диэтиловый эфир
Cu(II) НС1 (1:10)* Хлороформ
Продолжение таблицы 3.44
Экстрагируемый элемент Водная фаза Органическая фаза
Fe(III) H2SO4(l:10)* Хлороформ, диэтилацетат, иэтило-вый эфир
Hg(H) Нейтральный раствор Бензол, хлороформ
In Разбавленная кислота Бензол, хлороформ
Mn(II) Нейтральный раствор Диэтиловый эфир
Mo(VI) НС1 (2:11)* Этилацетат, хлороформ
Nb(V) Кислый раствор Хлороформ
Pa(V) 0,1—4 н. кислота Бензол, хлороформ, диэтиловый эфир
Sb(III) H2SO4(l:10)* Хлороформ
Sn(II) 1,5 н. на Бензол, хлороформ
Sn(IV) НС1 (1:10)* Этилацетат
Ti(II) на (1:10)* Хлороформ, этилацетат, диэтиловый эфир
Th на (1:10)* Этилацетат, бутилацетат
U(VI) H2SO4(l:10)* Диэтиловый эфир
V(IV) 0,5 н. на Этилацетат
V(V) на или h2so4 (1:10)* Диэтиловый эфир, этилацетат
W(VI)** на (1:10)* Этилацетат
Zn** Нейтральный раствор Диэтиловый эфир
Zr H2SO4(l:10)* Этилацетат
Примечания.
* Объемное соотношение кислота : вода.
** Извлечение неполное.
Таблица 3.45
Экстракция диэтилдитиокарбаматов [1]
Экстрагируемый элемент pH водного раствора Органическая фаза
Ag 3 Этилацетат
4-11 Четыреххлористый углерод
As(III) 4-5,8 Четыреххлор истый углерод
Au(III) 4-11 Четыреххлористый углерод
Методы разделения и концентрирования
165
Продолжение таблицы 3.45
Экстрагируемый элемент pH водного раствора Органическая фаза
Bi 4-11 Четыреххлористый углерод
1-10 Хлороформ, диэтиловый эфир
Cd 4-11 Четыреххлористый углерод
3 Этилацетат
Со 4-11 Четыреххлористый углерод
Cr(VI) 0-6 Хлороформ
Cu(II) 1-3,5 Хлороформ
4-11 Четыреххлористый углерод
Fe(II) 4-11 Четыреххлористый углерод
Fe(III) 4-9,8 Четыреххлористый углерод
0-10 Хлороформ
Ga 5,5 Четыреххлористый углерод
3 Этилацетат
Hg(H) 3 Этилацетат
4-11 Четыреххлористый углерод
In 4-10 Четыреххлористый углерод
3 Этилацетат
Mn(II) 6-9 Четыреххлористый углерод
6,5 Этил ацетат
Mo(VI) 3 Этилацетат
Nb(V) 4-5,5 Четыреххлористый углерод
Ni 0-10 Хлороформ
4-11 Четыреххлористый углерод
Os 7-9 Четыреххлористый углерод
Pb 4-11 Четыреххлористый углерод
~ 0,2 Этилацетат
Pd(II) 4-11 Четыреххлористый углерод
Pt(IV) 4-11 Четыреххлористый углерод
Pu(VI) 3 Пентилацетат, пентиловый спирт
Re Конц. HC1 Этилацетат
Продолжение таблицы 3.45
Экстрагируемый элемент pH водного раствора Органическая фаза
Sb(III) 4-9,5 Четыреххлористый углерод
Se(IV) 4-6,2 Четыреххлористый углерод
3 Этилацетат
Sn(IV) 5-6 Четыреххлористый углерод
Te(IV) 4-8,8 Четыреххлористый углерод
0,001-5,0 сильная кислота Хлороформ, бензол
T1(I) 4-11 Четыреххлористый углерод
Tl(III) 4-11 Четыреххлористый углерод
U(VI) 1-5 Бензол
4—8 Четыреххлористый углерод
6,5-8,5 Хлороформ, пентилацетат, диэтиловый эфир
V(V) 4-5,9 Четыреххлористый углерод
w 1-1,5 Этилацетат
Zn 3 Этилацетат
4-11 Четыреххлористый углерод
Таблица 3.46
Экстракция ацетилацетонатов [1]
Экстрагируемый элемент pH водного раствора Органическая фаза R, %
А1 4-6 Ацетилацетон (50%) в хлороформе 90
1-4,4 Ацетилацетон 10-90
Be 1,0 Ацетилацетон 100
Bi 1,0 Ацетилацетон 100
Ce(III) >4 Ацетилацетон 100
Co(III) 1 Ацетилацетон (50%) в хлороформе 100
0,0-2,0 Ацетилацетон 95-99,5
Cr(III) 0,0-2,0 Ацетилацетон 100
Cu(II) 2,5-6,0 Ацетилацетон 85
Dy 5,3 Ацетилацетон 45,8
Er 5,5 Ацетилацетон 65,5
166
Новый справочник химика и технолога
Продолжение таблицы 3.46
Экстрагируемый элемент PH водного раствора Органическая фаза R, %
Fe(III) 1,5 Ацетилацетон 100
1,0 Ацетилацетон (50%) в хлороформе 96
Ga 3,0 Ацетилацетон 83
3,0 Ацетилацетон (50%) в хлороформе 95
Gd 5,8 Ацетилацетон 39,3
Hf 2,3 Ацетилацетон >95
Hg(H) 3,0 Ацетилацетон 48
Ho 5,75 Ацетилацетон 57,5
In 3,0 Ацетилацетон 94
Mn(II) 4-6,5 Ацетилацетон 100
Mo(VI) 0,01-6,0 н. H2SO4 Ацетилацетон (50%) в хлороформе 96-98
Nb(V) 2-5 Ацетилацетон (20%) в хлороформе 90
Nd 5,6 Ацетилацетон 30,0
Pb 5-7 Ацетилацетон 80-98,5
Pu(IV) 2-10 Ацетилацетон 0,1 М в бензоле 100
Ru(III) 4-6 Ацетилацетон (33%) в хлороформе 90
Sn 5,5 Ацетилацетон 32,0
Tb 5,7 Ацетилацетон 48,0
Th >4 Ацетилацетон 100
Ti(IV) 0,0-1,6 Ацетилацетон (50%) в хлороформе 10-76
U(IV) 2,5 Ацетилацетон 73
U(VI) 4-6 Ацетилацетон 100
V(IH) 2,0 Ацетилацетон (50%) в хлороформе 93
V(IV) >2,3 Ацетилацетон (50%) в хлороформе 80
V(V) 2,15 Ацетилацетон (50%) в хлороформе 68
Y 5,7 Ацетилацетон 52,9
Yb 5,8 Ацетилацетон 84,6
Zn 5,5-7 Ацетилацетон 70
Zr 2-3 Ацетилацетон 73
3-8 Ацетилацетон (20%) в хлороформе 98
Таблица 3.47
Экстракция тиофенкарбонилтрифторацетонатов [1]
Экстрагируемый элемент pH водного раствора Органическая фаза
Растворитель Концентрация тиофен-карбонил-трифтораце-тона, моль-л 1
Ас >5,5 Бензол 0,25
А1 5,5 Бензол 0,02
5,5-6,0 Метилизобу-тилкетон 0,1
Am(III) >3,5 Бензол 0,2
Be 6-7 Бензол 0,02
Bi 2,0 Бензол 0,25
Bk >2,5 Бензол 0,2
Са 8,2 Бензол 0,5
Ce(III) * 1 н. H2SO4 Ксилол 0,5
Ce(III) ** 2,9 Трибутилфосфат (0,5 М) в бензоле 0,2
Cf 3,0 Бензол 0,2
Cm 3,5 Бензол 0,2
Co(II) 5,1-6,8 Ацетон + бензол (3:1) 0,15
Cr(III) 5,75 Бензол 0,15
Cs 8,7 Нитрометан, нитробензол 0,5
Cu(II) 2,4 6,0 Бензол 0,15
Es ~3,0 Бензол 0,2
Eu >3,4 Толуол 0,2
Fe(III) 2-3 Бензол 0,02
Fm ~3,0 Бензол 0,2
Hf 2 н HclO4 Бензол 0,1
In 2,5-4 Бензол 0,5
La >4,5 Бензол 0,2
Mn(II) 6,7-8,0 Ацетон + бензол (3:1) 0,15
Na *** 8,7 Нитрометан, нитробензол 0,5
Ni 6 Ацетон + бензол (3:1) 0,15
Np(IV) l,0MHCl или HNO3 Ксилол 0,5
Pa 4 h. HNO3 Бензол 0,4
Pa **** 2-6 н. HC1 Бензол 0,5
Pb 4 Бензол 0,25
Po(IV) 1,5-2,0 Бензол 0,25
Pu(IV) 0,5 M HC1 или HNO3 Бензол 0,15
Sc 2,5-6 Ксилол 0,2
Sr > 10 Бензол 0,02
Методы разделения и концентрирования
167
Продолжение таблицы 3.47
Экстрагируемый элемент pH водного раствора Органическая фаза
Растворитель Концентрация тиофен-карбонил-трифтораце-тона, моль-л-1
Th 0,8 Бензол 0,25
2,0 Четыреххлористый углерод 0,5
Т1(1) >6,5 Бензол 0,25
Т1(Ш) >3,5 Бензол 0,25
U(VI) >3 Бензол 0,2
V(V) 2,5-4,1 Бутиловый спирт 0,3
Y >6 Бензол 0,02
4,2-10 Метил изобу-тилкетон 0,1
Yb >3,4 Толуол 0,2
Zr 6MHC1 Ксилол 0,5
2-10 M HNO3 Ксилол 0,5
Примечания.
* Я = 80%;** Я = 91%; ***Я = 45%; **** R = 90%.
Макроциклические экстрагенты
Открытие Педерсеном в 60-х гг. XX века гетероцепных соединений, так называемых краун-эфиров [38], положило
начало новому, интенсивно развивающемуся направлению в области экстракционного разделения элементов. Первые синтезированные соединения этого класса представляли собой макроциклы из нескольких чередующихся эфирных фрагментов, пространственное строение которых напоминало корону, отсюда и появилось общее название этого класса соединений:
Конформационная гибкость полиэфирной молекулы позволяет атомам кислорода эфирных групп ориентироваться внутрь цикла, создавая тем самым полость определенного размера с высокой электронодонорной активностью. В водных растворах, вследствие близости сольватационных свойств атомов кислорода, краун-эфира и молекул воды, константы устойчивости комплексов катионов металлов, как правило, невелики (< 102), а их различия
в ряду металлов одной группы незначительны. Но благодаря липофильности молекул краун-эфиров, регулируемой за счет введения различных заместителей при углеродных атомах кольца, появляется возможность увеличить эти различия при экстракции катионов металлов в органическую фазу.
В общем случае экстракция катионов металлов с помощью нерастворимого в воде краун-эфира осуществляется в виде ионной пары:
(М+)
ВОДИ + (А)води +(L)oPr (ЕМА)орГ,
где L — молекула краун-эфира.
При этом в экстрагент селективно извлекаются ионы тех металлов, радиусы которых в наибольшей степени соответствуют размеру полости макроцикла. Например, 18-краун-6 с размером полости 0,28-0,32 нм особенно селективен к катиону калия (ионный диаметр — 0,266 нм). Введение боковых заместителей в полиэфир влияет как на основность атомов кислорода в цикле, так и на его конформационную гибкость, что также изменяет значение константы экстракции металла. Краун-соединения с пространственно жесткой структурой более предпочтительно проявляют так называемый пик селективности, т.е. особенно избирательны к одному из ряда близких по свойствам ионов. Природа аниона, в зависимости от энергии его гидратации, влияет в основном на KD. Однако помимо общей тенденции изменения коэффициента распределения может наблюдаться и изменение селективности экстракционного процесса, связанное как со специфическим взаимодействием аниона с макроциклическим лигандом, так и с частичной электролитической диссоциацией ионных пар.
Помимо геометрии полости наиболее существенный вклад в селективность комплексообразования вносит природа активных центров. Наличие в молекулах макроциклов разнообразных электронодонорных атомов существенно расширяет их комплексообразующие и экстракционные свойства и создает дополнительные возможности для варьирования селективности. Замещение атомов кислорода в кольце на «мягкие» атомы серы благоприятствует комплексообразованию с «мягкими» ионами переходных металлов и, напротив, снижает устойчивость комплексов с «жесткими» ионами. Атом азота в качестве донорного центра играет промежуточную роль. Предполагается, что макроциклические соединения, содержащие в качестве гетероатомов одновременно серу, азот и кислород особенно перспективны как избирательные экстрагенты для переходных металлов [48].
Еще большую катионную избирательность по сравнению с моноциклическими аналогами проявляют бис-краун-эфиры, в которых два полиэфирных цикла находятся на концах относительно короткой соединительной цепочки. Их более высокая селективность объясняется структурной подготовленностью к образованию внутримолекулярных сэндвичевых комплексов [50]. Следует отметить, что комплексообразующие и ионоселективные свойства бис-краун-эфиров определяются не только размером полиэфирных циклов, но и природой связывающей их цепочки. Введение в моноциклический краун-эфир дополнительной
168
Новый справочник химика и технолога
олигоэфирной цепи в качестве моста приводит к образованию нового лиганда бициклического типа, так называемого криптанда. Ниже показан криптанд [2,2,2].
Криптанды в большей степени, чем краун-эфиры, обладают способностью проявлять пики селективности (например, криптанд [2,2,2] особенно селективен к катиону калия), поскольку они имеют хорошо выраженную полость. В то же время процессы образования и распада комплексов металлов с этими бициклическими соединениями сильно заторможены, так как должны сопровождаться существенной конформационной перестройкой молекулы лиганда. В связи с этим при осуществлении экстракционного процесса в неравновесных условиях (например, при переносе через экстракционные мембраны) зачастую происходит резкое снижение селективности экстрагента [51, 52].
Экстракция металлов краун-эфирами и криптандами благодаря их высокой селективности находит применение, как в аналитической химии, так и при разработке новых экстракционных технологий [50-52]. В частности, предложены методы экстракционного выделения калия, таллия, свинца, стронция и многих других металлов. Ведутся поиски способов разделения таких традиционно трудноразделяемых смесей веществ, как ионы лантаноидов, изотопы щелочных и щелочноземельных элементов [50, 53]. Гетероциклические лиганды применяют в качестве активных компонентов мембран ионоселективных электродов и селективно-проницаемых жидких мембран [54, 55]. Есть основания полагать, что аналитические и технологические возможности макроциклических экстрагентов еще далеко не исчерпаны. Более подробную информацию о макроциклических соединениях и возможностях их применения в качестве экстрагентов можно найти в [56, 57].
Выбор экстрагентов и условий экстракционного выделения
На основании анализа накопленного опыта в области применения экстракционных методов в химическом анализе для экстракционного разделения следов элементов предлагается использовать экстрагенты, показанные на рис. 3.7.
3Li V 4Ве 5B
iiNa V 12Mg V 13A1 B* i4Si
19К V 2оСа V 21SC 22Ti 23V OB 24Cl OB 25M11 OB 2бГе OB BO 27C0 OB 2gNi OB 29Cu OB BO зо^п OB 0 a q t» згОе О 33AS 00 34Se OO
7Rb V 38$Г V 39y 40Zr ★ 4iNb В 42Mo OB 43TC 0 44Ru О 45Rh 46Pd BO 47^g OB 0 48Cd OB BO 49In OB BO 50$П 00 51Sb 00 5гТе OO
55CS V 5бВа V 57 La 72Hf ★ 7зТа 74W 75&e 76OS BO 77k 0 7»Pt BO 79Au В soHg BO 81T1 OB 0 82?b OB 83Bi OB 0
9oTh
94Pu ★
Рис. 3.6. Предпочтительные экстрагенты для выделения микроэлементов: О — дитиокарбоматы, • — дитизонаты, О—галогениды, — 8-гидроксихинолинаты, * —р-дикетонаты, V — соединения с краун-эфирами [55]
3.2.5.3. Реакционная экстракция в расплаве металлов (пробирная плавка)
Метод пробирной плавки является специфическим методом концентрирования благородных металлов, основанном на их выделении в расплавы металлов, чаще всего в свинец или серебро. Пробирая плавка является комплексным методом пробоподготовки, объединяющим стадии разложения пробы и концентрирования микроэлементов.
Общую схему пробирной плавки в классическом варианте на примере определения в материалах содержания золота можно представить следующим образом: шихтование (смешение пробы с необходимыми количествами восстановителя или окислителя, флюса и коллектора), тигельная или шерберная плавка.
В зависимости от состава объекта анализа и определяемых элементов коллектором для извлечения благородных
Методы разделения и концентрирования
169
металлов могут являться соединения свинца: оксид свинца (глет), иногда сурик, ацетат свинца, металлический свинец; применяют также сплав свинца с серебром, металлические серебро, медь, никель, олово, некоторые виды штейнов (сплавы сульфидов), медно-никелевый коллектор и т.п. Медь и медно-никелевые сплавы образуют с платиноидами непрерывные ряды твердых растворов. В качестве флюсов, переводящих тугоплавкие соединения в форму легкоплавких шлаков, используют буру, соду, поташ; в качестве восстановителей — порошкообразный древесный уголь, пшеничную или ржаную муку, винный камень (тартрат К), крахмал, серу, железные опилки, бумагу; в роли окислителей (помимо кислорода воздуха) — селитру, глет, сурик.
Тигельная плавка — это восстановительно-экстракционный процесс: глет или иной коллектор восстанавливается до металла углем, крахмалом или иным углеродсодержащим веществом и растворяет благородные металлы, образуя с ними сплавы, собирающиеся в виде отдельной фазы на дне тигля. Температура плавки зависит от используемого коллектора. Так, плавку на свинец ведут при 900-950 °C, на медный королек — при 1150-1200 °C. Полученный королек и представляет собой концентрат благородных металлов. Тигельную плавку используют для анализа руд и продуктов переработки с содержанием благородных металлов выше 1 г на 1 т исходного вещества. Для анализа
богатых руд (сотни и более граммов благородных металлов на 1 т) используют шерберную плавку.
Шерберная плавка — окислительно-восстановительный процесс, протекающий в присутствии пробирного зерненого свинца, буры и кварца. Примеси, окисляясь, переходят в шлак, в то время как благородные металлы собираются металлическим свинцом. Окислительная атмосфера создается поступающим в печь воздухом и глетом, образующимся из свинца. В этом случае шихту расплавляют при 900-950 °C при закрытых дверцах муфеля и затем окисляют расплавленный свинец при доступе воздуха; свинец частично улетучивается в виде оксида, но большая часть остается. Сплав выливают в изложницу, и после охлаждения свинцовый сплав освобождают от шлака. Подробнее с методом пробирной плавки можно познакомиться в [2].
3.2.6. Сверхкритическая флюидная экстракция (СКФЭ)
Как следует из самого названия метода, в нем в качестве экстрагента, т.е. извлекающей фазы, используются вещества в состоянии сверхкритического флюида (СФ), т.е. в агрегатном состоянии, соответствующем условиям, когда температура и давление превышают критические значения для данного вещества. В табл. 3.48 приведены характеристики веществ, чаще всего используемых в качестве сверхкритических флюидов.
Таблица 3.48
Характеристики химических соединений, наиболее часто используемых в качестве сверхкритических флюидов [59]
Соединение Характеристики флюидов
Критическая температура Тс, °C Критическое давление рс, атм Критическая плотность рс, г мл"1 Плотность при 400 атм, Р400» г ' мл Плотность в жидком состоянии Pl, г • мл 1
со2 31,3 72,9 0,47 0,96 0,93 (63,4 атм; 25 °C)
n2o 36,5 72,5 0,45 0,94 0,91 (нас.; 0 °C) 0,64 (59 атм; 25 °C)
NH3 132,5 112,5 0,24 0,40 0,68 (нас.;-33,7 °C) 0,60 (10,5 атм; 25 °C)
н-С5 (алканы, алкены, алкины) 196,6 33,3 0,23 0,51 0,75 (1 атм; 25 °C)
H-C4 (алканы, алкены, алкины) 152,0 37,5 0,23 0,50 0,58 (нас.; 20 °C) 0,57 (2,6 атм; 25 °C)
SF6 45,5 37,1 0,74 1,61 1,91 (нас.;-50 °C) 3,08 (нас.; 111,75 °C)
Хе 16,6 58,4 1,10 2,30 1,53 (нас.Н5,6 °C)
CC12F2 111,8 40,7 0,56 U2 1,30 (6,7 атм; 25 °C)
CHF3 25,9 46,9 0,52 — 1,51 (нас.;-100 °C)
Из приведенных в таблице соединений в СКФЭ наиболее предпочтительным по своим свойствам и доступности является СФ СО2.
Выбор СФ в качестве экстрагентов вызван тем, что вещества в сверхкритическом состоянии по плотности и растворяющей способности приближаются к жидкостям и в то же время выделенные в СФ вещества легко освобождаются от экстрагента при изменении температуры и давления,
что делает их привлекательными для решения препаративных задач. Извлекаемыми веществами чаще всего являются высокомолекулярные органические соединения. Основная область применения процессов межфазного распределения с использованием СФ — сверхкритическая флюидная хроматография (СФХ), в которой СФ играют роль подвижной фазы (см. п. 3.2.9) [57-59].
170
Новый справочник химика и технолога
3.2.7. Газовая экстракция
Газовая экстракция —метод разделения, основанный на распределении веществ между конденсированной (жидкой или твердой) и газовой фазами. При осуществлении газовой экстракции выделяемые компоненты переходят из конденсированной фазы в газовую. В качестве коэффициента распределения KD в газовой экстракции принято использовать отношение концентрации компонента в жидкой (твердой) фазе cL(S} (моль • дм 3) к его концентрации в газовой фазе сG (моль • дм 3) при равновесии:
Ад —
CG
Для идеальных и предельно разбавленных растворов KD является константой и не зависит от концентрации компонента. Коэффициент распределения зависит от изменения стандартного изохорного потенциала ЛГ° при растворении компонента, находящегося в газовой фазе, в жидкости и температуры Т:
*£,=етрГгГ Г
I KI J
Природа газовой фазы практически не влияет на величину KD. Повышение температуры на 1 °C (при комнатных температурах) приводит к уменьшению KD на 3-8%. Для газообразных веществ, не способных к диссоциации в жидкости, величина KD равна их растворимости в данной жидкости. Справочные данные по коэффициентам распределения различных веществ приведены в табл. 3.49-3.51.
Таблица 3.49
Значения коэффициентов распределения KD газообразных и летучих компонентов в системе вода - газ [60]
Компонент KD °C
Гелий 0,00995 20
Неон 0,0112 20
Аргон 0,0377 20
Криптон 0,063 20
Ксенон 0,109 20
Водород 0,0196 20
Азот 0,014 20
Кислород 0,033 20
Озон 0,355 20
Сероводород 2,86 20
Сероуглерод 1,263 20
Диоксид серы 43,3 20
Аммиак 762,6 20
Оксид азота (I) 0,4844 36
Оксид азота (II) 0,0402 40
Гидрид фосфора 0,2765 17
Гидрид мышьяка 0,2 15
Гидрид сурьмы 0,2 15
Оксид углерода 0,0249 20
Диоксид углерода 0,878 20
Тетраэтилсвинец 0,0152 20
Метан 0,0355 20
Продолжение таблицы 3.49
Компонент KD °C
Этан 0,0334 40
Гексан 0,021 20
Гептан 0,019 20
Этилен 0,125 20
Пропилен 0,146 20
Бутилен-1 0,186 20
Пропадиен 0,326 20
Бутадиен-1,3 0,376 20
Ацетилен 1,118 20
Циклопропан 0,315 20
Циклогексан 0,37 22
Бензол 3,73 22
Толуол 2,5 37
Этилбензол 1,8 16
Пропилбензол 0,986 16
Хлорметан 2,36 30
Дихлорметан 11,4 20
Хлороформ 8,91 20
Тетрахлорметан 1,04 20
Хлорэтан 2,30 20
1,2-Дихлорэтан 26,3 20
1,1-Дихлорэтан 6,6 20
1-Хлорпропан 1,59 30
Трихлорэтилен 3,0 20
Бромметан 2,95 23
Бромэтан 3,9 20
1-Бромпропан 3,3 20
Иодэтан 4,3 20
Хлорбензол 9,04 20
Этанол 1412 40
Пентанол-1 900 37
Пентанол-2 760 37
Пентанол-3 714 37
2-Метилбутанол-4 848 37
2-Метил бутанол-1 756 37
2-Метилбутанол-З 587 20
2-Метилбутанол-2 900 20
Диметиловый эфир 37 18
Диэтиловый эфир 40 20
Метилформиат 149 20
Этилформиат НО 20
Пропилформиат 90,4 20
Метилацетат 351 20
Этилацетат 226 20
Пропилацетат 164 20
Изопропилацетат 88 20
Бутилацетат 102 20
Изопентилацетат 80 20
Метилакрилат 196 20
Метилметакрилат 75 20
Метиламин 511 60
Диметиламин 230 60
Этиламин 321 60
Диэтиламин 83 60
Анилин 8040 40
Методы разделения и концентрирования
171
Таблица 3.50
Коэффициенты распределения органических веществ в системах жидкость - воздух (или азот) при температурах от 10 до 30 °C. В скобках указаны экстраполированные данные [61]
Экстрагируемое вещество Жидкая фаза Интервал концентраций, масс. % Коэффициенты распределения
10 °C 15 °C 20 °C 25 °C 30 °C
Углеводороды
Бензол Вода Ю^-Ю 5 7,6 6,2 4,8 4,0 3,4
65% СНзСООН Ю^-ОД 276 (0 °C) 146 (124) 106 72*
80% СНзСООН 10^-0,5 684 (0 °C) 385 313 270 219
СНзСООН 10 4-0,5 — — 895 780 620
ПЭГ-300 0,02-1 — — 641 601 456
ПЭГ-400 10 4-0,7 — — (741) 586 375*
Сквалан 10 2-1,5 — — (598) (500) 420
Толуол Вода 104-105 8Д 6,0 4,6 3,6 2,9
65% СНзСООН ю^-од 602 (0 °C) 294 (236) 194 125*
80% СНзСООН ю^-од 1715 (0 °C) 854 665 518 415
СНзСООН 10 4-10 5 — — 2480 2135 1760
ПЭГ-300 0,01-1,0 — — (1457) 1154 1046
м-Ксилол Вода 1 О’4-! О5 9,7 7,4 5,9 4,0 3,9
65% СНзСООН ю^-од 1012(0 °C) 499 (406) 320 229*
80% СНзСООН 104-0,5 3680 (0 °C) 1 880 1580 1300 1110
СНзСООН 10 410 5 6370 5570 4570
Этилбензол 65% СНзСООН 1 О’4-! О5 849 (0 °C) 398 (322) 257 172*
80% СНзСООН 104-0,5 2276 (0 °C) 1284 (1062) 983 581*
Кислородосодержащие соединения
Этиловый спирт Вода 0,01-1,0 (10200) 7970 7020 5260 (4440)*
Пропиловый спирт » 0,01-1,0 (9280) 6950 5480 4090 (3210)*
Бутиловый спирт » 0,01-1,0 (8610) 6410 4660 3600 (2710)*
Ацетон » 0,1-1,0 1418 1171 752 551 484*
Метилэтилкетон » 0,01-1,0 1150 810 600 380 (283)*
Диоксан » 0,01-1,0 14840 10340 8000 5750 (4330)*
Этилацетат » 0,04—1,0 431 299 210 150 (108)*
Бутилацетат » 0,01-0,7 296 189 126 87 (59)*
Сераорганические соединения
Метилмеркаптан Бензол — 178 150 124 111 91
Этилмеркаптан » — 592 486 406 336 323
Диметилсульф ид » — 728 563 488 438 419
Примечание. * Данные при 35 °C.
172
Новый справочник химика и технолога
Таблица 3.51
Коэффициенты распределения KD компонентов в системе жидкость - газ при 25 °C [61]
Жидкая фаза Компоненты
м-Октан Толуол Этиловый спирт Метилэтилкетон Диоксан Нитрометан
Сероуглерод 14300 8900 143 661 2654 264
Циклогексан 13500 3900 80 355 1137 126
Триэтиламин 13300 4450 690 677 1692 458
Диэтиловый эфир 13200 6760 2114 1003 2616 1170
Этилбромид 11100 11800 — 5777 8907 2115
н-Гексан 9900 3520 82 355 974 135
Изооктан 9800 2680 61 310 886 118
Тетрагидрофуран 8870 10800 2168 2136 6141 5379
Диизопропиловый эфир 8800 4020 719 786 1725 719
Толуол 8705 — 273 1339 3956 1452
и-Ксилол 7620 5120 268 1260 3632 1191
Бензол 8310 7930 320 1880 6597 1994
Хлороформ 7150 10500 779 9632 35646 2686
Четыреххлористый углерод 7030 5620 149 997 4342 384
Дибутиловый эфир 6920 3750 368 592 1586 490
Метилендихлорид 6302 11100 632 — 3368 4994
Декан 6302 2571 62 265 820 114
Хлорбензол 5972 6261 265 1901 5237 1468
Бромбензол 4808 5762 248 1600 4812 1327
Фторбензол 4567 7245 336 2339 6361 2460
2,6-Лутидин 3805 4875 3681 1516 3870 2739
Сквалан 3512 1775 42 184 640 84
Гексафторбензол 3512 8181 — — 4342 1408
Фенетол 3043 4609 337 1371 3708 1812
2-Пиколин 2945 5174 4705 2035 4685 3778
Этилендихлорид 2852 7459 559 4442 — 4369
Этилацетат 2852 5020 1279 — 4946 5179
Иодбензол 2852 4567 216 1224 4140 1031
Метилэтилкетон 2535 5121 1878 — 4812 6994
Диэтиловый эфир диэтиленгликоля 2466 4260 1384 1174 3235 4369
Анизол 2466 4224 431 1783 5087 2736
Октиловый спирт 2339 1895 2168 639 1518 411
Циклогексанон 2225 5020 1690 1747 4239 5379
/ирети-Бутиловый спирт 2172 1561 6051 — 2869 705
Т етраметилгуанидин 2027 4738 19715 1600 4046 7362
Изопентиловый спирт 1982 1631 3527 907 1953 603
Пиридин 1982 5452 5294 2404 5936 5594
1,4-Диоксан 1900 5070 1878 1729 — 5594
Бутиловый спирт 1823 2016 4758 1039 — 759
Пропиловый спирт 1657 1902 4705 1085 2403 903
Изопропиловый спирт 1688 1566 — 1318 2195 858
Дифениловый эфир 1628 3620 230 980 3490 1 180
Методы разделения и концентрирования
173
Продолжение таблицы 3.51
Жидкая фаза Компоненты
л/-Октан Толуол Этиловый спирт Метил-этилкетон Диоксан Нитрометан
Ацетон 1599 4121 — 2838 5237 8800
Бензонитрил 1519 4260 926 2307 5936 4994
Тетраметилмочевина 1401 5282 7636 1840 4140 9994
Дибензиловый эфир 1301 3089 356 1009 3225 1861
Ацетофенон 1282 3899 1054 1646 4946' 4112
Гексаметилфосфортриамид 1264 4447 38539 1747 3632 15724
Этиловый спирт 1264 2221 — 1490 2695 1499
Хинолин 1247 3424 2918 1416 3708 2911
Нитробензол 1152 3782 424 1729 5396 3883
jw-Крезол 891 2270 12284 24091 99028 2973
Диметилацетамид 776 4155 9211 2035 4685 13202
Уксусная кислота 750 2075 7061 3331 17820 3778
Нитроэтан 714 3782 834 2935 7422 8377
Метиловый спирт 608 1487 — 2247 3870 2851
Бензиловый спирт 581 1881 3527 2247 8907 2084
Диметилформамид 577 3840 6778 2163 4946 13994
Трикрезилфосфат 566 1487 463 603 1241 1452
Метоксиэтанол 526 1961 2821 1371 3956 5179
Нонилфенилоксиэтилат 481 1067 651 380 1049 1394
А-Метил-2-пирролидон 459 3960 7368 1662 4342 12607
Ацетонотрил 418 2666 1761 2935 8097 13327
Анилин 356 2240 1837 3685 13707 5594
Метилформамид 285 1528 5647 1516 3632 5594
А-(Цианоэтил)формалин 235 1532 936 853 2280 3494
Бутиролактон 208 2799 2286 1901 5087 9994
Нитрометан 205 2041 2125 2886 9898 —
Додекафторгептанол 187 837 6051 26282 162000 3778
Формилморфолин 170 1874 2644 1121 3235 7362
Пропиленкарбонат 168 1769 1205 1543 4451 8745
Диметилсульфоксид 121 1745 7061 1270 3870 13994
Тетрафторпропанол 119 880 8560 28437 71298 8229
Сульфолан 61,0 1248 1407 997 3870 7362
Три(цианоэтокси)пропан 53,0 775 662 649 1953 4236
Оксидипропионитрил 49,8 1010 794 1039 3490 6358
Диэтиленгликоль 38,9 240 1407 221 699 927
Триэтиленгликоль 24,6 263 1140 201 699 869
Этиленгликоль — 105 2644 270 1081 743
Формамид — 138 3527 857 3295 2739
Вода — — 5647 469 5396 1105
Для идеальных и предельно разбавленных растворов
Коэффициент селективности а в газовой экстракции определяется отношениями давлений насыщенного пара разделяемых компонентов р° и коэффициентов активности
в растворе у:
P2I2
Основная область применения газовой экстракции в аналитической химии — парофазный газохроматографиче-
174
Новый справочник химика и технолога
ский анализ водных растворов, биологических жидкостей (кровь, моча и т.п.), пищевых продуктов и полимерных материалов.
Парофазный анализ — метод получения информации о природе, составе или состоянии жидких и твердых тел путем анализа контактирующей с ними газовой фазы.
Взаимосвязь между концентрациями компонента в газе-экстрагенте cG и анализируемой пробе cL0 зависит от способа осуществления процесса газовой экстракции. По способу осуществления все варианты газовой экстракции подразделяются на статические, динамические и проточные.
В статических вариантах известный объем анализируемой жидкости VL приводится в равновесие с определенным объемом газа-экстрагента VG в замкнутой системе. После установления равновесия
= c°L Cg kd + vg/vl-
В динамических вариантах газовой экстракции поток газа-экстрагента проходит через объем или над объемом неподвижной пробы. В этом случае при условии установления равновесия между фазами:
где VG — объем пропущенного газа-экстрагента; VG — объем газа-экстрагента в сосуде над пробой.
В проточных вариантах газовой экстракции поток газа-экстрагента пропускают через поток или над потоком анализируемой жидкости. При условии установления равновесия:
G KD + WGIWL’
где WG и W[ — объемные скорости потока газа-экстрагента и анализируемой жидкости, соответственно, мл • мин"1.
3.2.8. Жидкостная абсорбция
Жидкостная абсорбция основана на распределении веществ между газовой и жидкой фазами. При осуществлении жидкостной абсорбции абсорбируемые компоненты (абсорбаты) переходят из газовой фазы в объем жидкой фазы. Для характеристики абсорбционных процессов используют тот же коэффициент распределения в системе жидкость - газ, что и в газовой экстракции. Значения коэффициентов распределения газообразных веществ в системе жидкость - газ приведены в табл. 3.52.
Различают физическую и химическую абсорбцию (хемосорбцию). При физической абсорбции (растворении) энергия межмолекулярного взаимодействия молекул аб-сорбата и абсорбента невелика (менее 20 кДж • моль"1). В случае хемосорбции происходит химическое взаимодействие растворенного абсорбата с компонентами абсорбента, например ионизация молекул кислот и оснований с образованием соответствующих ионов. Энергия хемосорбции-онного взаимодействия более 25 кДж • моль"1.
Основная область применения жидкостной абсорбции в аналитической химии — выделение и концентрирование летучих микропримесей при анализе воздуха. Как правило, используют динамический вариант абсорбционного выделения путем пропускания потока анализируемого воздуха через сосуд, заполненный жидким абсорбентом. Наибольшее распространение находят абсорберы с пластинкой из пористого стекла, в которых обеспечивается равномерное диспергирование микропузырьков воздуха в объеме жидкого абсорбента.
Взаимосвязь между концентрациями абсорбата в жидкой фазе cL и анализируемом воздухе cG при условии его количественного выделения следующая:
где VG — объем анализируемого воздуха, который пропущен через абсорбент объема VL.
При осуществлении динамического абсорбционного выделения объем до проскока определяемых веществ может быть найден на основании взаимосвязи между концентрациями абсорбата в газовой фазе на выходе из абсорбера cG и входе в него cG В случае физической абсорбции:
cg = с°| 1-ехРт^“1’ (2)
\ & D*L )
в случае хемосорбции
, ( vc v.ykA
1-ехр-----?---—?-
„ Ч KDVL Wr+1
cG = c°G--------—--------, (3)
° VLkKDl(WG+V)
где k — константа скорости реакции сорбата с хемосорбентом; WG — объемная скорость потока анализируемого воздуха через абсорбер.
Довольно часто в практике химического анализа с целью повышения степени извлечения определяемых веществ анализируемый воздух пропускают через ряд последовательно соединенных абсорберов. В этом случае уравнения (2) и (3) переходят соответственно в уравнения (4) и (5):
vg-n
_____ KD-vL, KdVl h
f vg-n vl^kd !
\KD-VL nwg J
_______5!______
V^D , !
Wc
(5)
где N — число абсорберов; 5 — счетчик суммирования; ! — символ факториала.
Методы разделения и концентрирования
175
Таблица 3.52
Коэффициенты распределения (KD) газообразных веществ в системе жидкость - газ при 20 °C [62]
Абсорбируемый компонент Kd
Тетрахлорид углерода Этанол Гексан Бензол Метилацетат Метанол Ацетон Вода
Гелий — 0,0281 0,0480 0,0180 — 0,0313 0,0309 0,0159
Неон — 0,402 0,0680 0,271 — 0,043 ; 0,0104
Водород 0,0686 0,071 — 0,58 0,077 0,085 0,083 0,0178
Азот 0,157 0,12 0,206 0,116 0,175 0,135 0,135 0,0150
Оксид углерода 0,21 — — 0,164 0,236 0,170 0,235 0,0230
Аргон 0,305 0,240 0,412 0,221 — 0,250 0,273 0,0340
Кислород 0,278 0,220 — 0,220 0,27 0,237 0,254 0,0310
Оксид азота(П) 0,321 0,266 -— 0,272 — — — 0,0479
Метан 0,72 0,505 0,56 0,532 0,56 0,494 0,584 0,0339
Криптон 1,12 0,473 1,00 0,680 — — 0,91 0,0580
Диоксид углерода 2,34 2,670 — 2,3 6,5 3,32 7,2 0,878
Этан 5,72 2,15 3,1 4,05 2,92 1,87 3,15 0,0500
Этилен 3,92 0,252 2,81 3,33 3,44 — 3,67 0,1220
Ацетилен 3,1 5,8 — 4,85 22,05 — 22,8 1,0500
Оксид азота(1) 4,26 2,78 — 3,38 5,74 3,09 5,45 0,6200
Диоксид серы 18,45 112,5 — 64,3 167 201,0 198 33,2
Сероводород 9,3 7,42 5,43 13,1 — — — 2,42
При анализе воздуха хемосорбция используется гораздо чаще физической абсорбции, поскольку позволяет достигать существенно больших коэффициентов концентрирования. В табл. 3.53 приведены составы абсорбирующих растворов для выделения ряда неорганических и органических веществ при анализе воздуха.
Таблица 3.53
Компоненты абсорбирующих растворов при анализе воздуха [61-63]
Абсорбируемое вещество Состав абсорбирующего раствора
Диоксид азота 8% KI в воде
Триэтаноламин
50% С6Н6 + 50% Н2 SO4 + Н2О
Аммиак 0,02 н. H2SO4
Озон 1% KI в воде
Хлор 1% KI в воде
0,15% 2,6 Диметилфенола + 0,4% H2SO4
Бром 1 МКОН
Хлороводород Дистиллированная вода
Фтороводород Дистиллированная вода
Бромоводород 1 МКОН
Арсин 0,025 н. КМпО4 + 0,05 н. H2SO4
Фосфин 0,025 н. КМпО4 + 0,05 н. H2SO4
Продолжение таблицы 3.53
Абсорбируемое вещество Состав абсорбирующего раствора
Цианистый водород 0,1 н. раствор NaOH
Дистиллированная вода
Сероводород 0,5% (NH4)2CO3 + 0,2% Na3AsO3
Диоксид серы 0,04 М Na2(HgCl4)
3% КС1О3
0,05 М Моноэтаноламин
Гидразин 0,1 МНС1
Ацетон
Сероуглерод 1,5% диэтиламина в этаноле
Оксид углерода 30% K3Fe(CN)6
Анилин 0,01 н. H2SO4
Ацетон Дистиллированная вода
Ацетофенон Этанол
Ароматич. углеводороды 10% KNO3 в H2SO4
Гексаметилендиамин Дистиллированная вода
Гексен,гептен 30% H2SO4
Дикетен 16MH2SO4
Диметилбензин-амин 0,01 МНС1
Диметиламин 0,01 МНС1
176
Новый справочник химика и технолога
Продолжение таблицы 3.53
Абсорбируемое вещество Состав абсорбирующего раствора
Диметилфенол 0,8%Na2CO3
Дициклопентадиен 16MH2SO4
Диэтиламин Этанол
Диэтилкетон Дистиллированная вода
А, А-Диметилацетамид Дистиллированная вода
Диметилэтанол-амин 6% NaHCO3
Ксилидин 2% H2SO4 в этаноле
Масляный альдегид 1% гидроксиламин в этаноле
Метанол Дистиллированная вода
Метакрилат 2,5% NaOH
Метиламин 0,1 МНС1
Метилэтилкетон Дистиллированная вода
Метил пропилкетон Дистиллированная вода
Муравьиная кислота 0,1 М NaOH
Нитрофенол 0,1 MNaOH
Нитроанизол 10%KNO3bH2SO4
Нитроциклогексан Этанол
Оксид этилена 8 М H2SO4
Пиридин 0,1 МНС1
Пропаргиловый спирт Дистиллированная вода
Скипидар Уксусная кислота
Сложные эфиры органич. кислот Этанол
Толуидин 0,1МНС1
Фенол 0,8% Na2CO3
10% NaOH + 30% глицерина
Формальдегид Дистиллированная вода
50% изопропанола
0,2% ацетилацетона + 0,3% СН3СООН + 15% ацетата аммония
2,4-Динитрофенилгидразин в 0,05 М H2SO4
Фосген Дистиллированная вода
Фенилгидразин 2% этанол
Фурфурол Этанол
Хлорметан Уксусная кислота
Хлорбензил Хинолин
Четыреххлористый углерод Пиридин
Эпихлоргидрин Пиридин
Этилендиамин Дистиллированная вода
Метафос, тиофос, фосфамид Диметилформамид
3.2.9. Хроматография и хроматографические методы разделения веществ (см. также раздел 4)
Хроматография и хроматографические методы разделения веществ находят широчайшее применение в аналитической, препаративной химии и химической промышленности [1-144]. Об их значимости свидетельствуют также целая серия специальных международных журналов по хроматографии (см. раздел 1.1.2) и специальная серия монографий (Chomatographic Science — см. раздел 1.1.4).
3.2.9.1. Классификация хроматографических методов разделения веществ. Основные термины
В аналитической химии нет другого столь неоднозначно трактуемого термина, как хроматография. Трудно найти двух специалистов в области хроматографических методов, которые дали бы идентичные определения хроматографии [64, 65]. Чтобы понять, чем вызвана подобная ситуация, необходимо совершить краткий экскурс в историю хроматографии.
Открытием явления расторжения зон при прохождении смеси веществ через слой адсорбента и появлением самого термина хроматография мир обязан русскому ботанику М.С. Цвету [66]. На заседании общества естествоиспытателей 14 марта 1903 г. он выступил с сообщением об открытии нового метода разделения сложных смесей, названного им хроматографией, которую он рассматривал как метод адсорбционного анализа. Подробно познакомится с историей открытия М.С. Цвета можно в [72]. Не умаляя значения открытия М.С. Цвета, необходимо с современных позиций уточнить, что он осуществил хроматографический процесс только в одной из возможных двухфазных систем: в системе жидкость - твердое тело, и, соответственно, предложил только один из многочисленных хроматографических методов — жидкостноадсорбционную хроматографию. Но вся дальнейшая история хроматографии явилась подтверждением плодотворности идеи М.С. Цвета и универсальности предложенного им принципа разделения веществ в двухфазных системах.
После того как в 1941 году А. Мартин и Р. Синдж предложили метод «распределительной хроматографии», осуществив хроматографический процесс в системе двух жидких фаз, стало очевидным, что хроматография — это нечто большее, чем адсорбционный анализ. Можно спорить, было ли оправданным называть предложенный ими вариант метода распределительной хроматографией: хроматография М.С. Цвета также была распределительной, только в другой системе фаз. Важно другое. После работ А. Мартина и Р. Синджа начинает вырисовываться первый основной классификационный признак хроматографических методов разделения веществ: система фаз, в которой осуществляется хроматографический процесс. Однако потребовалось еще несколько десятилетий, чтобы формирование основных направлений хроматографии, характеризуемых агрегатным состоянием контактирующих фаз и их ролью в хроматографическом процессе, подошло к логическому завершению (табл. 3.54) [73].
Методы разделения и концентрирования
177
Таблица 3.54
Классификация хроматографических методов разделения веществ по агрегатному состоянию фаз и механизму удерживания разделяемых веществ стационарной фазой*
Фазы, участвующие в хроматографическом процессе, и их роль Хроматографические методы и их вариации в зависимости от механизма удерживания разделяемых веществ стационарной фазой Начало развития метода (год первых публикаций)
Стационарная (неподвижная) фаза или фазы Подвижная фаза
Твердое тело Жидкость Жидкостно-твердофазная (ЖТХ)
Жидкостно-адсорбционная (ЖАХ) 1903
Ионообменная (ИОХ) 1938
Аффинная 1951
Лигандообменная 1961
Эксклюзионная 1959
Жидкостно-абсорбционная хроматография с обращенными фазами 1969
Полярная жидкость Неполярная жидкость Нормально фазовая жидкостно-жидкостная хроматография (ЖЖХ) 1941
Неполярная жидкость Полярная жидкость ЖЖХ с обращенными фазами 1953
Органический экстрагент Водный раствор Экстракционная хроматография (ЭХ) 1959
Твердое тело Газ Газо-адсорбционная (ГАХ) 1941
Жидкость Газ Газо-жидкостная (ГЖХ) 1952
Газ Жидкость Жидкостно-газовая (ЖГХ) 1981
Твердое тело Вещество в сверхкритическом состоянии. Сверхкритический флюид Сверхкритическая (флюидная) (СФХ) 1962
Твердое тело и жидкость Газ Газо-жидкостно-твердофазная (ГЖТХ) 1966
Твердое тело и газ Жидкость Жидкостно-газо-адсорбционная (ЖГАХ) 1989
Примечание.
«По цели хроматографирования выделяют аналитическую хроматографию (качественный и количественный анализ); препаративную хроматографию (для получения веществ в чистом виде, для концентрирования и выделения веществ); промышленную (производственную) хроматографию для автоматического управления процессом (при этом целевой продукт поступает в датчик)» [Основы аналитический химии. Кн. 1. Общие вопросы. Методы разделения. 2-е изд., перераб. и доп. / Под ред. акад. Ю.А. Золотова М.: Высшая школа, 2000. 351 с.]
В любом хроматографическом методе, независимо от агрегатного состояния фаз, имеет место многократное перераспределение веществ между двумя фазами — подвижной и стационарной (неподвижной). В потоке подвижной фазы компоненты исходной смеси веществ перемещаются относительно неподвижной, помещенной в ограниченное пространство, имеющей форму протяженного цилиндрического или плоского слоя. При этом стационарная фаза находится в мелкодисперсном состоянии или в виде тонкой пленки на поверхности стенок, ограничивающих это пространство. Разделительное пространство чаще всего имеет форму протяженного цилиндра, называемого колонкой. Если колонка заполнена стационарной фазой, находящейся в мелкодисперсном состоянии, то слой стационарной фазы
в колонке называется насадкой, а сами колонки насадочными в отличие от капиллярных колонок, в которых стационарная фаза фиксирована на внутренней поверхности стенок, ограничивающих разделительное пространство. Если стационарными являются жидкие или газовые фазы, не имеющие своей формы и размеров, их диспергирование в пределах разделительного пространства достигается за счет пористых твердотельных носителей стационарной фазы. Как правило, в качестве носителей используются вещества, инертные по отношению к компонентам разделяемых смесей и обеим фазам, участвующим в хроматографическом процессе, и, следовательно, не влияющие на процесс межфазного распределения между удерживаемой на носителе стационарной фазой и подвижной фазой, пе
178
Новый справочник химика и технолога
ремещающейся относительно нее. Если же твердофазный носитель участвует в процессе межфазного распределения, реализуются хроматографические методы, в названии которых упоминаются все три фазы, участвующие в межфазном распределении, например газо-жидкостно-твердофазная хроматография.
Влияние адсорбции на твердом носителе проявляется в той или иной степени в любом хроматографическом процессе со стационарными жидкой и особенно газовой фазами. В гель-проникающей хроматографии можно рассматривать процесс распределения веществ в системе — твердое тело-жидкость в пространстве — поры-жидкость в межчастичном пространстве, т.е. находящемся между частицами геля. При этом процессы адсорбции на поверхности гелей вносят существенный вклад в удерживание веществ в хроматографической колонке. Наконец, в ионообменной хроматографии при использовании в качестве подвижных фаз водно-органических растворов происходит обогащение фазы ионообменника водой по сравнению с подвижной фазой. Более того, известны примеры использования в качестве подвижных фаз органических экстрагентов, не смешивающихся с водой. При этом в хроматографической колонке вещества распределяются в трехфазной системе — ионооб-менник - водный раствор - органический экстрагент. Но во всех случаях речь идет о сопутствующих явлениях и процессах. Появление каких-либо специальных методов жидкостно-жидкостно-твердофазной хроматографии остается проблематичным.
Тем не менее можно считать, что любому из возможных сочетаний фаз в двухфазных и трехфазных системах соответствуют определенные хроматографические методы. При этом возможны самые различные механизмы взаимодействия разделяемых веществ со стационарной твердой фазой, которые являются дополнительными классификационными признаками хроматографических методов в системе — жидкая - твердая фаза: адсорбционная, аффинная, гелъ-проникающая, ионообменная, лигандообменная и другие разновидности жидкостно-твердофазной хроматографии.
Таким образом, хроматография может рассматриваться как универсальный способ осуществления процессов межфазного распределения веществ, заключающийся в относительном перемещении контактирующих фаз в ограниченном пространстве в условиях, когда одна из фаз постоянно находится в диспергированном состоянии или в виде пленки на поверхности стенок, ограничивающих это пространство. Подобное перемещение может происходить в цилиндрической колонке или в плоском слое. В первом случае говорят о колоночной, во втором — о планарной или плоскостной хроматографии. При этом независимо от геометрической формы ограниченного пространства, в котором осуществляется хроматографический процесс, одна из фаз может быть неподвижной или обе будут находиться в движении относительно друг друга. В результате перераспределения веществ между фазами проявляются различия в скоростях движения фронтов или дискретных зон отдельных веществ в разделительном пространстве в потоке подвижной фазы
(в зависимости от величин их коэффициентов распределения в соответствующей системе фаз).
При одновременном движении обеих фаз в различных направлениях в пределах разделительного пространства проявляются и различия в направлениях движения зон, и соответствующие разновидности хроматографических методов разделения. При встречном движении потоков двух фаз это противоточная хроматография, при взаимно перпендикулярном — двухмерная. Для характеристики обычного хроматографического процесса, когда одна из фаз является неподвижной, а вторая перемещается относительно нее, иногда используется термин прямоточная хроматография.
Элюент
Разделяемые вещества
Рис. 3.7. Схема формирования зон разделения веществ (н) и кривая изменения концентрации разделяемых веществ в элюате на выходе из колонки (б) при элюентной схеме хроматографического разделения
Для наиболее общего случая прямоточной (обычной) хроматографии возможно несколько схем достижения конечного эффекта разделения вещества за счет многократного повторения актов их межфазного распределения. Первая схема, традиционно называемая проявительной и иногда элюентной [74], (последнее название более точное) подразумевает следующую последовательность операций
Методы разделения и концентрирования
179
при хроматографическом разделении. В разделительное пространство вводится небольшая аликвота исходной смеси веществ в том агрегатном состоянии, в котором находится подвижная фаза. После этого в колонку или в плоский слой направляется поток подвижной фазы — элюента. В этом случае в качестве элюентов используют вещества или растворы веществ, сродство которых к стационарной фазе меньше, чем у компонентов разделяемой смеси. Иногда в качестве элюентов используют растворы веществ, образующих с компонентами менее прочно сорбируемые или несорбируемые формы. Подбирая составы
элюентов для создания условий получения необходимых значений Кс и Кр, достигают эффекта разделения этих компонентов (рис. 3.7).
Второй из возможных схем хроматографического разделения является фронтальная, при которой через слой стационарной фазы пропускается непосредственно исходная смесь разделяемых веществ. Схема формирования зон разделяемых веществ в стационарной фазе и кривая изменения состава подвижной фазы на выходе из колонки при фронтальном хроматографическом процессе представлены на рис. 3.8.
А+В+Е
а)
А+В+Е
I
p g — жидкая или газообразная среда Е, в которой находится исходная смесь веществ А и В
Рис. 3.8. Схема формирования зон определяемых веществ А и В (а) и кривая изменения состава подвижной фазы на выходе из колонки (б) при фронтальном хроматографическом процессе
Первым на выходе из колонки (Рис. 3.86) появляется наименее сорбируемый компонент А в той среде Е, в которой он подается в колонку. В последующих порциях элюата будет присутствовать трехкомпонентная смесь: продолжает элюироваться компонент А и появляется компонент В. Составы элюата и исходной смеси веществ выравниваются. В случае многокомпонентной разделяемой смеси на выходе из колонки после компонента А будут последовательно появляться более прочно удерживаемые подвижной фазой компоненты до того момента, пока из колонки не начнет выходить исходная смесь веществ.
Последняя третья схема хроматографического разделения — вытеснительная. В данном случае, как и при элю-ентной схеме сначала в колонку вводят некоторое количество разделяемой смеси веществ (например, компоненты А, В и С, растворяют в Е), а далее подают в Е раствор вещества Д, обладающего наибольшим сродством к стационарной фазе. Это вещество и является вытесняющим компонентом, который вытесняет из последней фазы все ранее содержащиеся в ней вещества А, В и С в порядке изменения их сродства к стационарной фазе. Первым вытесняется наименее прочно удерживаемое вещество А, далее В и С. При этом зоны вытесняемых веществ в колонке и на выхо
де из нее взаимно перекрываются, а зона последнего из выделяемых компонентов С перекрывается с зоной вытеснителя Д (рис. 3.9).
Рис. 3.9. Изменение состава элюата и концентрации компонентов разделяемой смеси на выходе из хроматографической колонки при вытеснительной схеме разделения: интегральная кривая---------,
кривые изменения концентрации индивидуальных компонентов------------------
Предпочтительной в подавляющем большинстве случаев и потому общепринятой является элюентная схема осуществления хроматографического процесса. Собственно методом разделения является любая из возможных схем
180
Новый справочник химика и технолога
осуществления хроматографического процесса в приложении к конкретной системе фаз, а методом анализа — сочетание одного из хроматографических методов разделения с определенным методом определения концентрации разделенных веществ в элюате.
Существуют две основные принципиально различные схемы хроматографического анализа. Первая, которой в наибольшей степени соответствует термин элюентная, соответствует случаю, когда после хроматографического разделения по элюентной схеме последующее определение разделенных веществ осуществляется в потоке элюата, выходящего из колонки. Чтобы не вносить дополнительной терминологической путаницы, эта схема хроматографического анализа в дальнейшем будет рассматриваться как традиционная. Вторая схема — хроматографическое разделение с определением разделенных веществ непосредственно в хроматографической колонке или в плоском слое. Наибольшее распространение нашла первая схема, причем на начальном этапе развития хроматографии стадии разделения и последующего определения веществ были разнесены во времени и в пространстве. Для определения каждого из выделенных компонентов мог применяться свой метод определения в отдельных фракциях элюата, но при этом хроматографический анализ был лишен своих основных достоинств — универсальности и экспрессное™. Качественным скачком в развитии аналитической хроматографии явилось создание газового хроматографа, в котором были совмещены принципы хроматографического разделения и неселективного детектирования разделенных веществ непосредственно в потоке подвижной газовой фазы, называемой газом-носителем. Подобно тому, как создание газового хроматографа привело к появлению первого важнейшего раздела в науке о хроматографических методах анализа — газовой хроматографии, решение проблемы непрерывного детектирования веществ в потоках жидких фаз способствовало появлению и развитию второго аналитического направления — жидкостной хроматографии.
Необходимо отметить, что любые варианты схем хроматографического анализа, основанных на детектировании выделенных веществ в элюате, имеют один общий недостаток. Концентрация веществ в элюате в несколько раз меньше, чем в стационарной фазе, а именно тем меньше, чем больше коэффициент распределения, соответствующий условиям элюирования вещества из колонки. Вынужденное разбавление при элюировании приводит к увеличению значений пределов обнаружения. Большие возможности для снижения пределов обнаружения открывает схема хроматографического анализа с непосредственным определением веществ в хроматографической колонке или в плоском слое. Эта схема пока применяется сравнительно редко, что объясняется ограниченными техническими возможностями детектирования веществ непосредственно в стационарной фазе. Во всех видах плоскостной хроматографии, где такую схему детектирования реализовать значительно проще, чем в колоночной, ей отдается предпочтение. В последнее время наметилась тенденция распространения сферы применения этой схемы и на коло
ночную хроматографию с использованием люминесцентных методов детектирования. На новом этапе развития хроматографии фактически происходит возврат к «цветов-скому варианту» наблюдения хроматограммы непосредственно на колонке. Однако вместо визуального наблюдения на качественном уровне или извлечения и анализа ее отдельных фрагментов производят сканирование колонки или плоского слоя по всей длине и определяют содержание веществ в разделенных зонах с выдачей количественных результатов. В соответствии с логикой М.С. Цвета и смыслом содержания термина именно эта схема хроматографического анализа является проявительной хроматографией.
Предложенная выше трактовка термина хроматография не является традиционной. Она помогает понять сущность хроматографического процесса и объединяющее начало многочисленных хроматографических методов. В то же время представление о хроматографии как о способе осуществления процесса межфазного распределения веществ не является альтернативой традиционным представлениям о ней как о методе разделения или методе анализа, а точнее о совокупности методов разделения и анализа. Скорее наоборот, триединое понимание смыслового содержания термина хроматография позволяет прийти к логически завершенной схеме формирования терминологии для характеристики хроматографических методов в отличие от рекомендованного ИЮПАК определения: «Хроматография — это физический метод разделения, в котором разделяемые компоненты распределяются между двумя фазами, одна из которых неподвижна (стационарная фаза), тогда как другая (подвижная фаза) движется в определенном направлении» [74]. Общая схема формирования терминологии для характеристики хроматографических методов разделения и анализа веществ приведена в табл. 3.55.
Таблица 3.55
Общая терминологическая схема для характеристики хроматографических методов в зависимости от содержания, придаваемого термину хроматография [75]
Смысловое содержание термина хроматография Характеристические признаки хроматографических методов Соответствующие смысловому содержанию хроматографические методы: хроматография
Способ осуществления процесса межфазного распределе-НИЯ Геометрия пространства, в пределах которого осуществляется хроматографический процесс Колоночная: насадочная, капиллярная; плоскостная: бумажная, тонкослойная
Степень дисперсности или толщина пленки стационарной фазы Обычная, высокоэффективная
Направление относительного перемещения фаз Обычная (прямоточная), противоточная, двухмерная
Методы разделения и концентрирования
181
Продолжение таблицы 3.55
Смысловое содержание термина хроматография Характеристические признаки хроматографических методов Соответствующие смысловому содержанию хроматографические методы: хроматография
Совокупность методов разделения Агрегатное состояние фаз и их роль в хроматографическом процессе Жидкостнотвердофазная, жидкостножидкостная, газожидкостная и т. п. (см. табл. 3.15)
Механизм взаимодействия веществ со стационарной фазой Жидкостноадсорбционная, ионообменная, газоадсорбционная и т. п.
Схема процесса разделения веществ Элюентная, фронтальная, вытеснительная
Назначение Аналитическая, препаративная
Совокупность методов анализа Агрегатное состояние подвижной фазы или химическая форма определяемых веществ Газовая, жидкостная, сверхкритическая флюидная, ионная, хиральная
Условия элюирования Изотермическая с программированием температуры, градиентная, изократическая и т.п.
Схема определения разделенных веществ Традиционная (элюентная), проявительная
Приведенная схема позволяет видеть, что для исчерпывающей характеристики хроматографических методов разделения необходимо включение признаков хроматографии как способа осуществления процесса межфазного распределения. Соответственно для характеристики методов анализа — включение признаков хроматографии как способа осуществления процесса межфазного распределения и совокупности методов разделения. Очевидно, что в практической деятельности необходимость в таком обилии определений к слову хроматография отсутствует. Обычно достаточно одного, раскрывающего смысл решаемой аналитической или препаративной задачи. Однако для понимания сущности используемого метода необходимо иметь в виду все характеристические признаки методов.
Помимо объяснявшихся выше терминов, в табл. 3.55 используются термины, нуждающиеся в дополнительном пояснении на основании [74].
Изотермическая хроматография — процесс, в котором элюирование разделяемых веществ осуществляется при постоянной температуре.
Хроматография с программированием температуры — процесс, в котором температура колонки изменяется по заданной программе в течение процесса разделения.
Градиентное элюирование — процедура элюирования, в которой состав подвижной фазы изменяется непрерывно или ступенчато в течение процесса разделения.
Изократическое элюирование — процедура элюирования при неизменном составе подвижной фазы.
Наконец, в специальных комментариях не нуждается выделение в хроматографических методах двух направлений по назначению методов — аналитического и препаративного. С последним приходится сталкиваться существенно реже. Наиболее подробную информацию о препаративной хроматографии можно найти в [76].
Для того чтобы раскрыть содержание отдельных терминов, используемых для характеристики хроматографических методов, необходимо кратко остановиться на основных положениях теории хроматографии и их практических следствиях.
Более подробно с хроматографическими методами анализа и техникой аналитических хроматографических методов можно ознакомиться в разделе 4.
3.2.9.2. Теоретические основы хроматографии
Основные понятия
Основными характеристиками хроматографических пиков разделяемых веществ при элюентной схеме хроматографического процесса является объем VRi или время tRi удерживания вещества в хроматографической колонке, ширина хроматографической полосы или пика элюирования Z,. Эти характеристики наглядно представлены на рис. 3.10, на котором схематически приведена хроматограмма трехкомпонентной смеси.
Рис. 3.10. Хроматограмма и ее основные параметры
Согласно определению, принятому ИЮПАК [74], под хроматограммой понимается «графическое или другое представление отклика детектора, концентрации аналита в вытекающем потоке или другой величины, используемой в качестве меры концентрации в вытекающем потоке, как функции объема вытекающего потока или времени». Ширина пика Z определяется как расстояние между точками
182
Новый справочник химика и технолога
пересечения касательных в точках перегиба пика элюирования с осью абсцисс и выражается в единицах времени или объема, в зависимости от того, чем характеризуется положение максимума пика: временем удерживания tR или объемом удерживания VR. Иногда в качестве характеристики размытия зон хроматографируемых веществ используют [3-ширину пика на высоте смакс/е, где смакс — концентрация вещества в максимуме пика; е — основание натурального логарифма. Величина |3 связана с величиной Z соотношением: |3 = ^-Z. Дополнительно на рис. 3.10 указано время выхода несорбирующегося вещества — мертвое время /0 и мертвый или свободный объем колонки Ко, равный объему подвижной фазы, находящейся в колонке:
V0=Wt0,
где W— объемная скорость подвижной фазы.
Соответственно выражение для объема элюента, который необходимо пропустить через колонку до выхода из нее зоны z-го вещества VRi с максимальной концентрацией, имеет вид:
VRi=V0 + KDiVc^.,
где Кс.ф — объем, занимаемый в колонке стационарной фазой.
Для характеристики межфазного распределения в хроматографической колонке ранее вводилось понятие коэффициента или фактора емкости Ке', являющегося отношением приведенного времени или объема удерживания к мертвому времени или свободному объему:
ю- ' _ (tRi — )
• —'t----------V—’
10 к0
Содержание понятий приведенное время или объем следует из выражений в числителях.
В настоящее время вместо фактора емкости говорят о факторе удерживания. Фактор удерживания является более адекватным понятием, чем коэффициент или фактор емкости, так как он, как и коэффициент распределения, характеризует систему фаз, в которой осуществляется хроматографический процесс, при концентрациях распределяемых веществ, соответствующих области линейности изотермы межфазного распределения, и функционально никак не связан с сорбционной или экстракционной емкостью стационарной фазы.
Разделительные возможности той или иной системы фаз в хроматографии, как и в других методах, основанных на различиях в распределении веществ между фазами, характеризуются коэффициентом селективности:
где индексы i и j соответствуют двум разделяемым компонентам. По физическому смыслу селективность является мерой термодинамического различия в распределении компонентов между фазами:
с Rr
где Д(Д6°) — разность энергии Гиббса межфазного перехода двух разделяемых компонентов.
В жидкостной хроматографии селективность зависит как от состава стационарной фазы, так и состава подвижной фазы. Поэтому необходимо говорить о селективности системы фаз в целом, а не только о селективности стационарной фазы, как в газовой хроматографии, где, подвижная фаза представляет собой практически инертный по отношению к разделяемым веществам газ-носитель. Тем не менее существует точка зрения, что межмолекулярными взаимодействиями в газовой фазе также нельзя полностью пренебрегать и что они могут влиять на селективность разделения веществ в газовой хроматографии [64].
Степень разделения двух веществ характеризуется разрешением хроматографических пиков Rs:
Rs=2
^R2 6?1) . Z] + Z2 _
Разрешение пиков тем лучше, чем больше различия времен или объемов удерживания и меньше размытость пиков.
Взаимосвязь между отдельными характеристиками хроматографического процесса устанавливает «тарелочная» теория хроматографии.
Теория тарелок
Впервые общую теорию хроматографии, основанную на концепции теоретических тарелок, создали в начале 40-х годов А. Дж. Мартин и Р. Л. Синдж [77]. Они предложили модель, описывающую хроматографический процесс движения зон разделяемых веществ в колонке и их размывания в процессе перемещения по ней аналогично процессам, происходящим при ректификации или противоточной экстракции. Впоследствии теория тарелок была дополнена и развита Глюкауфом, Ван-Деемтером, Гиддингсом.
Теория тарелок основана на ряде допущений и упрощений:
— хроматографическая колонка представляется условно разделенной на ряд идентичных слоев, названных теоретическими тарелками;
— каждый слой, т.е. теоретическая тарелка, содержит определенный объем стационарной и подвижной фаз;
— объемы стационарной и подвижной фазы одинаковы во всех слоях;
— в каждом слое в процессе хроматографирования устанавливается состояние равновесия между фазами и, следовательно, в пределах каждой тарелки отношение концентраций вещества в стационарной и подвижной фазах остается постоянным, равным коэффициенту распределения
I ^ci I I ^ci I I ^ci I
a D : — = — = — = KDi, где cci и cni— концентра-
( c . I ( C - / [ c . I
\ ш/ \ ш/2 \ Ш Jn
ции z-вещества в стационарной и подвижной фазах в каждом из условных слоев, обозначенных индексами от 1 до п;
Методы разделения и концентрирования
183
— загрузка хроматографической колонки не превышает определенного предела, гарантирующего сохранение линейности изотермы межфазного распределения и соответственно независимость KD от концентрации.
Теория тарелок устанавливает взаимосвязь между параметрами хроматографических пиков и числом теоретических тарелок N, характеризующих хроматографическую
колонку:
N = 1б1^|
I Z J
Рис. 3.11. Изменение числа эффективных теоретических тарелок Дэфф, требуемого для разделения двух компонентов, в зависимости от коэффициента селективности
ИЛИ
[\2 z \2 t А (у А
— =4 —
PJ IP )
Кроме того, исходя из экспериментальных удобств, на
практике для расчета числа тарелок в случае неполного разделения используют полуширину (ширину на половине высоты) пика Цо.5 (см. рис. 3.10):
А2 С t I V
-М =5,55 2k.
М-0,5 ) \ Но,5
N = 5,55
Помимо общего числа теоретических тарелок N, разделительная способность колонки харктеризуется числом эффективных теоретических тарелок А'>фф, определяемым с учетом фактора удерживания:
/ \2
А = 16 kzM
I Z J
Число Л',фф более адекватно характеризует разделяющую способность колонки, чем N; если N велико, а коэффициент Ке незначителен, даже может оказаться недостаточным для разделения.
Для сравнения эффективности хроматографических колонок используют высоту, эквивалентную теоретической тарелке (ВЭТТ или //):
Н = L/N, где L — длина колонки.
В рамках теории тарелок доказано, что разрешение хроматографических пиков Rs описывается выражением:
Отсюда видно, что разрешение пиков зависит от двух параметров: коэффициента селективности Кс и числа Афф. При этом число тУэфф , требуемое для разделения двух веществ, резко увеличивается с уменьшением (рис. 3.11 и табл. 3.56).
Таблица 3.56
Число Аэфф для разделения с заданным разрешением, как функция Кс
1 l?s=l,5
Кс Л^эфф кс А^фф
0,01 160000 1,01 360000
1,10 940 1,10 4360
1,15 940 1,15 2110
1,2 575 1,2 810
Теория тарелок дает удобный математический аппарат для формализованной оценки эффективности хроматографического процесса, но не обладает предсказательной способностью с точки зрения оптимизации условий хроматографического разделения, так как не учитывает реальные физические процессы, происходящие в колонке. Принятые допущения далеко не всегда адекватны условиям хроматографического разделения в различных системах фаз.
Кинетическая теория
Попытка учесть неидеальность процесса, а именно: ограниченность скорости переноса вещества между фазами (т.е. кинетику межфазного перехода) скорости продольной и поперечной диффузии, влияние размеров частиц стационарной фазы — предпринята Ван-Деемтером в теории, получившей название кинетической теории хроматографии [78].
По Ван-Деемтеру, ВЭТТ складывается из трех независимых составляющих, обусловленных неравномерностью потока подвижной фазы, продольной диффузией вещества и замедленностью установления равновесия между фазами:
Н=НХ +Н2 + Нз.
Он же предложил выражения для каждого члена этого уравнения:
Н} =А + [1 j + Cv , k v )
где А, В, С — постоянные для конкретного хроматографического процесса — учитывают вклад неравномерности потока подвижной фазы, продольной диффузии и отклонения от равновесного распределения в применяемой системе фаз соответственно; v — скорость подвижной фазы.
Слагаемое А зависит от структуры, плотности и равномерности заполнения колонки, от размера зерна носителя и приблизительно равно 2Хс/, где X — коэффициент, учитывающий плотность заполнения колонки, d — диаметр зерна. Если X = 0,5, то А определяет нижнее предельное значение Н\, равное d. В этом случае будет достигнут максимум эффективности колонки. В высокоэффективной газовой хроматографии /7МИН составляет (3—5)б/.
184
Новый справочник химика и технолога
Вклад продольной диффузии в подвижной фазе (£>п) в ВЭТТ описывается уравнением:
т.е. В = 2£>п- По мере увеличения скорости v значение Я2 уменьшается. Диффузия в стационарной фазе также вносит свой вклад в Н2, связанный с v обратно пропорциональной зависимостью. Благодаря существенной разнице в величинах коэффициентов диффузии в подвижной и стационарной фазах, в случае газовой хроматографии диффузионным размыванием в стационарной фазе можно пренебречь.
Слагаемое Н2 = С v учитывает конечную скорость процессов распределения хроматографируемого вещества между фазами при перемещении зоны этого вещества по колонке. Сопротивление мгновенному массопереносу оказывают диффузионный подвод вещества к межфазной границе в подвижной и стационарной фазах и замедленная кинетика перехода через межфазную границу. Если допустить, что лимитирующей стадией процесса является поперечная диффузия в стационарной фазе, характеризующаяся коэффициентом диффузии Dc, для газохроматографического процесса справедливо
Н -Г к* хл. Ч э ?
L(l + ^e) J
где dn — длина диффузионного пробега вещества в неподвижной фазе.
Из этого уравнения следует, что ВЭТТ растет с повышением линейной скорости подвижной фазы, удлинением диффузионного пробега, т.е. с увеличением толщины слоя стационарной фазы, и уменьшается с ростом и Dc.
Отправной точкой при создании Ван-Деемтером кинетической теории явилась экспериментально полученная для газовой хроматографии зависимость Н от v (рис. 3.12, кривая /). Это предопределяет адекватность кинетической теории в первую очередь реальным газохроматографическим процессам.
Если слагаемое Н\ дает постоянный вклад в ВЭТТ, то влияние второго и третьего члена суммы изменяется в зависимости от v. При малых скоростях превалирует вклад продольной диффузии в подвижной фазе, при больших — замедленность в установлении межфазного распределения.
Рис. 3.12. Зависимости высоты, эквивалентной теоретической тарелке (ВЭТТ), от скорости подвижной фазы для газовой (7) и жидкостной (2) хроматографии
Зависимость Н от v в жидкостной хроматографии имеет несколько иной вид (рис. 3.12, кривая 2). На ней при практически реализуемых скоростях подвижной фазы закономерно отсутствует восходящая начальная ветвь, определяемая продольной диффузией в подвижной фазе, скорость которой в жидких средах относительно мала.
3.2.9.3. Практические следствия теоретических представлений о хроматографическом процессе
Эффективность любого метода разделения и анализа определяется скоростью получения результата, отвечающего заданным техническим или метрологическим требованиям. Соответственно критерием эффективности хрома-
тографических методов является отношение —-, где — Г.
время, необходимое для получения результата, a Rs — разрешение, требуемое для получения препарата требуемой чистоты или для определения содержания компонента с заданной точностью. Чем больше это отношение, тем более эффективен метод. Из теории тарелок следует, что разрешение пиков определяется величинами Кс и Н и в незначительной степени зависит от Кс. Фактор удерживания оказывает заметное влияние на разрешение, когда его значение относительно невелико. И это влияние проявляется тем сильнее, чем меньше Кс. С другой стороны, учитывая, что увеличение Кй приводит к соответствующему росту времени разделения, оказывается неоправданным элюирование веществ из хроматографической колонки при больших значения Кс. Оптимальное значение Ке обычно лежит в интервале от 1 до 10.
Важнейшей характеристикой системы фаз, используемой для хроматографического разделения, является Кс. Даже незначительное увеличение Кс приводит к резкому улучшению разрешения. При прочих равных условиях можно воспользоваться колонкой меньшей длины и получить соответствующий выигрыш во времени разделения. С позиций многовариантности выбора условий достижения максимальных Кс жидкостная хроматография существенно превосходит газовую. В ГХ, помимо того, что селективность определяется только различиями во взаимодействии молекул разделяемых веществ с неподвижной фазой, сами эти взаимодействия имеют физическую природу. Селективность же, как правило, в наибольшей степени проявляется при образовании химической связи.
Выбор системы фаз, обеспечивающей проявление максимальных значений Кс, практически безоговорочно оправдан при решении препаративных задач и задач, связанных с предварительным концентрированием или выделением веществ в качестве отдельной самостоятельной стадии анализа. Не столь однозначен подобный подход в хроматографическом анализе.
Создание условий разделения при максимальных Кй для каждой пары компонентов требует изменения условий разделения, включающих изменение составов подвижной и неподвижной фаз. При изменении составов элюентов требуется перестройка всей системы детектирования, а при
Методы разделения и концентрирования
185
изменении состава неподвижной требуется замена насадки. То и другое существенно усложняет схему анализа и оказывается оправданным только в тех случаях, когда в пределах одной системы фаз не удается достигнуть минимально необходимого разрешения хроматографических пиков. Поэтому для хроматографических методов анализа важнейшее значение имеют другие факторы, которые наряду с Кс влияют на разрешение пиков.
Общим способом повышения эффективности хроматографических методов является уменьшение значения Н. Оценить относительный вклад в Н различных параметров хроматографического процесса позволяет теория Ван-Деемтера. Эти параметры можно подразделить на две группы.
К первой группе относятся физико-химические параметры, характеризующие систему фаз:
— коэффициенты диффузии в подвижной и стационарной фазах;
— константы скоростей реакции межфазного обмена.
Вторую группу составляют параметры, характеризующие технику эксперимента в хроматографическом способе осуществления процесса межфазного распределения:
— диаметр и форма частиц насадок, ширина диапазона их разброса по размерам, кривизна и соответственно длина каналов в твердой фазе (для пористых сорбентов) или толщина пленки стационарной жидкой фазы;
— диаметр, длина и равномерность набивки колонки;
— линейная скорость подвижной фазы.
Характер влияния на Н коэффициентов диффузии в подвижной и стационарной фазах следует из ранее приведенных уравнений для Н2 и Н3. Среди параметров, характеризующих технику эксперимента при хроматографическом разделении веществ, главным является размер и форма частиц насадок. Диаметр частиц или толщина пленки неподвижной фазы определяют длину диффузионного пробега вещества к границе раздела фаз. Очевидно, что чем меньше размеры частиц, тем меньше диффузионные ограничения, но всегда существует нижняя граница размеров частиц, определяемая проницаемостью слоя насадки в хроматографической колонке для подвижной фазы. В свою очередь проницаемость колонки для одной и той же подвижной фазы зависит не только от диаметра частиц, но и от высоты колонки. Получается замкнутый круг. Чем меньше Кс, тем больше требуется N^. Для получения необходимого числа Мфф следует или уменьшить Н до соответствующего значения при сохранении длины колонки, или увеличить ее длину при сохранении Н. Оба требования выполнимы только до определенных пределов, ниже которых колонки оказываются непроницаемыми для подвижной фазы при допустимом давлении. Одновременным решением проблем снижения диффузионных ограничений со стороны стационарной фазы и обеспечения необходимой проницаемости колонок для подвижных фаз, явилось создание пленочных и поверхностно-пористых сорбентов, позволяющих без существенного уменьшения размеров частиц и соответственно без принципиального увеличения сопротивления колонки потоку подвижной фазы в произ
вольных пределах регулировать толщину слоя стационарной фазы. Но и в этом случае размер частиц ограничен определенным верхним пределом, так как увеличение их размеров приводит к соответствующему возрастанию межчастичных расстояний в насадке. Лимитирующей стадией становится диффузия в подвижной фазе. Аналогичный результат снижения диффузионных ограничений достигается в ГЖХ и ЖЖХ при использовании поверхностнопористых носителей стационарной жидкой фазы. В ЖЖХ при соизмеримых скоростях диффузий в стационарной и подвижной фазах применение поверхностно-пористых носителей дает существенное уменьшение ВЭТТ. Эффект проявляется сильнее при использовании стационарных фаз, коэффициенты диффузии в которых существенно меньше, чем в подвижных. Суммарный объем стационарной фазы при использовании поверхностно-пористых сорбентов и носителей изменяется в широких пределах, определяемых тем, что важнее при решении конкретной задачи: сорбционная емкость или разрешение пиков. Диапазон изменений объемов стационарной фазы и, соответственно, динамической сорбционной емкости составляет от долей до десятков процентов объема стационарной фазы и емкости, соответствующих обычным сорбентам и объемно-пористым носителям.
Создание поверхностно-пористых и пленочных сорбентов дало качественный скачок в повышении эффективно-
сти хроматографического процесса. Отношение — увели-
чилось в 102—103 и более раз. Появился и соответствующий термин: высокоэффективная хроматография, в отличие от обычной, так называемой классической хроматографии. Но поскольку, как уже отмечено выше, применение специальных сорбентов и соответствующее техническое оформление — это не самостоятельный метод разделения, а общий методический прием, применяемый для всех сочетаний фаз, под высокоэффективной хроматографией подразумевается высокоэффективный режим проведения хроматографического процесса.
Из-за многообразия хроматографических процессов, применяемых стационарных фаз, размеров хроматографических колонок и решаемых задач практически невозможно говорить в общем случае об оптимальном размере частиц насадки хроматографических колонок. К тому же выбор размеров часто определяется не оптимальным решением, а наличием готовых сорбентов определенной дисперсности. Обычно в классической жидкостной и газовой хроматографии используют сорбенты размером от 100 до 200 мкм, в высокоэффективной — 3—10 мкм. Однако известны примеры применения и в классическом варианте сорбентов размером 15-20 мкм [79], а в высокоэффективной — до 75 мкм [80]. Поэтому можно говорить только об общей тенденции уменьшения размеров частиц сорбентов и носителей до определенных пределов, определяемых диаметром хроматографических колонок.
Важное значение имеет разброс размеров и форма частиц насадок, используемых для набивки хроматографических колонок. Широкий разброс размеров и неправильная
186
Новый справочник химика и технолога
форма приводят к неоднородности слоя сорбента в хроматографической колонке, локальным различиям в проницаемости, появлению застойных зон. Следствием является неравномерность потока подвижной фазы, появление существенных различий в длине диффузионного пробега разделяемых веществ к границе раздела фаз. В результате происходит дополнительное размытие зон разделяемых веществ, хроматографические пики становятся асимметричными. Отрицательный эффект проявляется в увеличении ВЭТТ в результате роста величины Разрешение хроматографических пиков будет всегда лучше на колонке, заполненной частицами сорбента, размеры которых лежат в более узком интервале, например, интервал 30-50 мкм лучше, чем 10-50 мкм.
При традиционной схеме осуществления хроматографического процесса на гранулированных сорбентах эффекты, связанные с неоднородной проницаемостью насадок для подвижной фазы, проявляются в случае применения колонок больших диаметров для решения препаративных задач и задач, связанных с концентрированием по отношению к матрице или матричным компонентам. По мере увеличения диаметров колонок всегда необходимо укрупнять сорбент, чтобы избежать неравномерности потока подвижной фазы. Это приводит к вынужденному увеличению ВЭТТ при переходе к большим колонкам. Уплотнение порошкообразных сорбентов в процессе работы колонок нарушает соотношение объемов стационарной и подвижной фаз, приводит к ухудшению их проницаемости.
Альтернативным решением при заполнении насадками колонок больших диаметров является применение насадок в виде пористых блоков [81]. Пористые блочные насадки, пригодные для использования в хроматографии, имеют два типа пор, различающихся по размерам: поры, характерные для материала сорбента и носителя, и поры, размеры которых соизмеримы с межчастичными расстояниями в обычных хроматографических колонках с гранулированными насадками. Насадки этого типа созданы для газоадсорбционной и ионообменной (на неорганических ионообменни-ках) хроматографии [83, 84]. Аналогично решается задача улучшения хроматографических характеристик колонок большого диаметра в экстракционной хроматографии [85, 86]. В качестве полимерной основы таких сорбентов и носителей стационарной жидкой фазы используется пористый политетрафторэтилен. Наиболее широкое практическое применение подобные сорбенты находят для концентрирования радионуклидов в радиохимическом анализе [87]. Постоянство геометрической формы и размеров сорбентов позволяет в данном случае отказаться от стадии элюирования выделенных радионуклидов и использовать их непосредственно в качестве источников радиоактивного излучения. Аналогичная схема группового хроматографического выделения может быть использована в рентгенофлуоресцентном анализе.
Длина колонки влияет на эффективность хроматографического анализа в двух прямо противоположных направлениях. Увеличение длины приводит к улучшению разрешения пиков, но соответственно возрастает и время
удерживания. Поэтому при выборе длины колонки критерием оказывается Уэфф, необходимое для получения требуемого разрешения самых трудноразделяемых компонентов анализируемой смеси. Но опять-таки приготовление колонки под отдельную частную задачу слишком трудоемко, чтобы на практике часто менять колонки. Более того, развитие хроматографического анализа идет по пути стандартизации хроматографических колонок и их промышленного выпуска. Поэтому требуемое УЭфф определяет только нижнюю границу высоты колонки при> определенном диаметре и зернении сорбента.
Обычная длина колонок в классической жидкостной хроматографии 10-25 см, в газовой — 1-3 м. В высокоэффективной жидкостной хроматографии длина может увеличиваться до 50-100 см и более. Для увеличения числа теоретических тарелок в газовой хроматографии иногда изготавливают колонки большей длины, равной десяткам и даже сотням метров. Длинные колонки для газовой хроматографии сворачивают в спираль, в жидкостной обычно их делают секционными, используя соединительные устройства с минимальным мертвым объемом. Различие приемов объясняется тем, что эффективность свернутых в спираль колонок в жидкостной хроматографии оказывается ниже, чем прямых секционных колонок той же длины. Эффект неравномерности потока подвижной фазы при изменении угла наклона колонки относительно оси земного притяжения проявляется тем сильнее, чем больше ее диаметр.
Выбор диаметра колонки лимитируется двумя главными факторами: увеличением ВЭТТ за счет поперечной диффузии с ростом диаметра и снижением сорбционной емкости колонки при его уменьшении. Очевидно, что в тех случаях, когда процесс разделения не лимитируется количеством макрокомпонентов в пробе, оправдано уменьшение диаметра. Так, в капиллярной хроматографии используют микроколонки диаметром вплоть до 20 мкм. Применение подобных колонок вводит существенные ограничения по количеству разделяемых веществ. При анализе смесей с большим разбросом по содержанию отдельных компонентов в режиме высокоэффективной хроматографии оправдано увеличение диаметра колонок до 2-3 мм. Это позволяет увеличивать количество пробы без нарушения линейности изотермы межфазного распределения для компонентов, определяющих загрузку колонки. В обычной жидкостной и газовой хроматографии диаметр колонки составляет 5-10 мм (в жидкостной) и 2-4 мм (в газовой) хроматографии. Но и здесь в соответствии с решаемой задачей возможны существенные отклонения в обе стороны: минимально до 2-3 мм, максимально до 50-60 мм и более. Причем верхний предел определяется решением не только препаративных задач, но и чисто аналитических, например в эксклюзионной хроматографии.
При определении оптимальной скорости подвижной фазы, как и большинства уже рассмотренных параметров хроматографического процесса, выбор оптимума строится на компромиссе. На кривой, отражающей зависимость Н
Методы разделения и концентрирования
187
от v , в случае газовой хроматографии есть минимум (рис. 3.12, кривая 7), а в жидкостной практически, чем меньше v, тем меньше Н. Но при минимизации скорости потока увеличивается время удерживания:
ts=N(l + Ke)^.
Поэтому, несмотря на проигрыш в величине Н, разделение обычно производится при скоростях подвижной фазы, соответствующих восходящим ветвям кривых. Согласно приведенному уравнению, время, затрачиваемое на разделение,
Н „
пропорционально величине —. Исходя из характера зави-v
77 м
симости между — от v, представленной на рис. 3.13, по-V
еле определенного предела дальнейшее увеличение v заметно не сказывается на продолжительности разделения. Иначе говоря, существует оптимальная скорость подвижной фазы. При превышении этой скорости усложняется техника эксперимента (повышение давления) без ощутимого выигрыша во времени получения результата. Оптимальная скорость в обычной жидкостной хроматографии составляет 0,001-0,01 см с-1, а в высокоэффективной — 1-3 см • с 1 для колонок диаметром 2-3 мм, в газовой — 2-10 см • с \ Чтобы обеспечить эту скорость в газовой хроматографии работают при избыточных давлениях 0,12— 0,25 МПа, а в высокоэффективной жидкостной хроматографии интервал рабочих давлений простирается от нескольких единиц до десятков МПа. Однако иногда используемый в качестве синонима высокоэффективной жидкостной хроматографии термин хроматография высоких давлений смысла не имеет.
v
3.2.9.4. Хроматографические методы, являющиеся вариациями хроматографического способа осуществления процесса межфазного распределения
Колоночная и планарная (плоскостная) хроматографии
Наиболее распространенным способом осуществления хромоатографического процесса является колоночный. Хроматографическая колонка в наиболее общем случае представляет собой трубку определенных размеров, заполненную насадкой, являющейся стационарной фазой. Подобные колонки называются насадочными. Материа
лами для изготовления колонок служат стекло, кварц, нержавеющая сталь, реже — медь и другие металлы. При одинаковом диаметре и одном и том же способе изготовления наиболее эффективны стеклянные и кварцевые колонки, калиброванные колонки из нержавеющей стали. Существенное влияние на эффективность колонки оказывает равномерность ее заполнения, которая, в свою очередь, зависит от способа заполнения. Общеприняты два способа заполнения колонок: суспензионный и сухой. Суспензионный способ применяют, если размер частиц насадки менее 30-50 мкм. В ионообменной и гель-проникающей хроматографии им пользуются независимо от размеров частиц сорбента. Суспензию в подходящем растворителе, контакт с которым не приводит к изменению сорбционных свойств насадки, готовят обычно с объемным содержанием твердой фазы 10-25%. Колонку заполняют в фильтрационном, а не в седиментационном режиме: суспензию вводят в колонку с достаточно высокой скоростью под давлением, это позволяет предотвратить седиментационное разделение частиц по размерам и плотности и способствует получению равномерной насадки. В случае очень мелких частиц суспензионный способ предохраняет частицы от слипания и образования более крупных агломератов.
Общие принципы выбора высоты и диаметра колонок рассмотрены выше. По мере совершенствования техники хроматографического анализа (создания микродозирующих устройств и высокочувствительных микродетекторов) наметилась общая тенденция уменьшения диаметра колонок. В современной аналитической практике стали обычным явлением микроколонки диаметром 1 мм и менее [88]. По-видимому, колонки с насадкой диаметром 50 мкм являются тем разумным пределом минимизации обычных хроматографических колонок, технические трудности преодоления которого вряд ли оправдают достигнутые результаты.
В качестве альтернативы микроколонкам с насадкой развивается капиллярный вариант колоночной хроматографии, в котором, как следует из названия, хроматографическая колонка представляет собой капиллярную трубку. Наибольшее распространение капиллярные колонки находят в газовой хроматографии, но известны и многочисленные примеры их применения в жидкостной капиллярной хроматографии [88]. В этом случае жидкую стационарную фазу наносят в виде тонкой пленки на стенки колонки. Возможно и изготовление специальных капилляров с привитыми на внутренней поверхности сорбентами. Толщина пленки стационарной фазы обычно равна 1-5 мкм при диаметре капилляра от 20 до 250 мкм.
Основные ограничения для капиллярных колонок связаны с их малой емкостью. Обычно масса разделяемых веществ не превышает микрограммовых количеств, а объем пробы составляет доли микролитра (0,1 нл - 0,1 мкл). Объемная скорость потока подвижной фазы в капиллярных колонках сравнительно невысока, но линейные скорости в них выше, чем в обычных колонках для
188
Новый справочник химика и технолога
высокоэффективной хроматографии. Значит, капиллярные колонки обеспечивают более высокую скорость проведения разделения и, соответственно, большую эффективность. Преимущество капиллярных колонок в разрешающей способности может быть достигнуто за счет их лучшей относительной проницаемости для подвижной фазы и отсутствия технических ограничений при изготовлении протяженных колонок. Длина в несколько десятков метров является нормой для капиллярных колонок. Число теоретических тарелок, обеспечиваемых такими колонками, обычно составляет 104-105, а в рекордных случаях достигает 106. В силу своих особенностей капиллярные колонки наиболее удобны для разделения небольших количеств очень сложных смесей. Они нашли применение в биохимии, медицине, фармацевтике, для клинических анализов физиологических жидкостей, в нефтехимии и других областях. Расширение сферы применения капиллярных колонок непосредственно связано с решением технических проблем изготовления систем дозирования малых объемов пробы в поток газа-носителя и микродетектирующих устройств.
Планарная или плоскостная, хроматография объективно имеет более узкую сферу применения, чем колоночная. Последний вариант осуществления хроматографического процесса является универсальным, применимым при любом сочетании фаз. Плоскостную хроматографию в любом из ее вариантов: бумажной хроматографии (БХ имеет скорее историческое значение) или так называемой тонкослойной хроматографии (ТСХ применяется чаще) — используют только для разделения в условиях, когда подвижная фаза находится в жидком состоянии. В отличие от колоночного хроматографического процесса, как следует из обобщенного названия этого направления в хроматографии, БХ и ТСХ осуществляют на плоскости: на листе специальной хроматографической бумаги или на пластине из тонкого слоя адсорбента, закрепленного на жесткой подложке.
К специфическим достоинствам плоскостного способа осуществления хроматографического процесса относится легкость реализации двухмерной схемы разделения. Пластинку или бумажную ленту можно последовательно обрабатывать двумя проявляющими растворами, подаваемыми со стороны двух взаимно перпендикулярных граней. В этом случае удается в пределах одного эксперимента разделить большее число компонентов.
По окончании разделения возможны самые разнообразные способы идентификации и определения содержания выделенных веществ: от прямого визуального наблюдения (без использования или с использованием предварительной обработки хроматограмм раствором подходящего проявляющего хроматограмму реагента) до точных измерений с помощью сканирующих детектирующих устройств.
В ТСХ используют обычно те же сорбенты и носители, что и в колоночной хроматографии (см. табл. 3.57).
Таблица 3.57
Классификация сорбентов для тонкослойной хроматографии [89, с. 855]
Класс сорбентов Сорбенты
Полярные неорганические (гидрофильные) Силикагель или кремниевая кислота, оксид алюминия, кизельгур, силикат магния (флорисил)
Полярные органические Целлюлоза, хитин, полиамид -6 или -11
Полярные связные фазы Аминопропил-, цианопропил-, ди-ол-модифицированный кварц
Гидрофобные связанные фазы С2 С8 -, Cig - и фенил-модифици-рованный силикагель; триацетат- и трифенилкарбамат целлюлозы
Ионообменники Полистирольные анионо- и катио-но-обменники, анионо- и катионо-обменники на основе целлюлозы
Неорганические Фосфат циркония, висмутат и молибдат, молибдофосфат и висмутофосфат аммония, гидроксиды
Органические Полиметакриловые кислоты; вещество — специфический комплексообразующий лиганд
Гель-фильтрационная среда Поперечносшитые полимерные декстрановые гели (сефадекс)
Хиральные фазы Целлюлоза, триацетат целлюлозы, силанизированные силикагели с привитыми комплексами меди(П)
Как и в колоночной ЖЖХ, в тонкослойной реализуются два варианта — нормально-фазовый и обращенно-фазовый, для каждого из которых, используются соответствующие носители. Предпочтение тому или иному носителю отдается и в зависимости от природы разделяемых веществ (см. табл. 3.58).
Таблица 3.58
Применение нормально-фазовой и обращенно-фазовой ТСХ [89, с. 855]
Сорбент Классы разделяемых веществ
Силикагель Альфатоксины, спирты, анаболические соединения, барбитураты, бензодиазепины, желчь, карбогидраты, эфирные масла, жирные кислоты, флавоноиды, гликозиды, липиды, микотоксины, нитроанилины, нуклеотиды, пептиды, пестициды, стероиды, сульфамиды, поверхностно-активные вещества, сахариды, тетрациклины, витамины
Оксид алюминия Ароматические углеводороды, гербициды, инсектициды, ионы металлов, растворимые в жирах витамины, липиды, липофильные красители, полиаро-матические углеводороды
Методы разделения и концентрирования
189
Продолжение таблицы 3.58
Сорбент Классы разделяемых веществ
Целлюлоза Амины, аминокислоты, антибиотики, искусственные сахара, карбогидраты, катехины, флавоноиды, полиаромати-ческие углеводороды, пептиды
Алкил- и арил-связанные фазы Спирты, амиды, амины, аминокислоты, аминофенолы, анитибиотики, антиоксиданты, барбитураты, лекарства, жирные кислоты, производные индола, нуклеоснования, олигопептиды, оптические изомеры, полиароматические углеводороды, пептиды, фармацевтические препараты, фенолы, фталаты, порфирины, консерванты, стероиды, поверхностно-активные вещества, тетрациклины
Амино-модифициро ванный силикагель Нуклеозиды, нуклеотиды, пестициды, производные пурина, стероиды, витамины
Цианомодифицированный силикагель Анальгетики, антибиотики, бензодиазепины, карбоновые кислоты, каркоте-ноиды, пестициды, фенолы, стероиды
Диол-модифици-рованный силикагель Нуклеозиды, пестициды, фармацевтические препараты, фосфолипиды
Инообмен-ники на основе целлюлозы Анализ ДНК, фрагментов ДНК и РНК, пищевые красители, неорганические ионы, стероиды
Ионообмен-ники на основе полистирола Амины, аминокислоты, неорганические ионы, пептиды, пуриновые и пиримидиновые производные
Висмутофосфат аммония Амины, аминокислоты, производные индола, олигопептиды, полиамины, сульфамиды
Силикагель, обработанный парафином, силиконом и растительными маслами Барбитураты, карбоксильные соединения, производные жирных кислот, нитрофенолы, полихлорированные бифенилы, пептиды, пестициды, фенолы, стероиды, поверхностно-активные вещества, триазины
Силавизированный силикагель, обработанный анионо-и катионоактивными ПАВами Алифатические и ароматические амины, спирты, аминокислоты, аминосахара, карбоновые и сульфокислоты, лекарства, производные индола, нуклеоснования, нуклеозиды, нуклеотиды, пептиды, дипептиды, полипептиды, фенолы, фенотиазиновые основания, стероиды, сульфамиды, растворимые в воде пищевые красители
Продолжение таблицы 3.58
Сорбент Классы разделяемых веществ
Силакагель, обработанный серебром Цис- и юранс-изомеры сложных эфиров, цис- транс-изомеры диеновых эфиров, жирные кислоты, холисти-рольные сложные эфиры, позиционные и геометрические изомеры жирнокислотных метильных эфиров, терпеноиды, простагландины
Для жидкостно-жидкостной ТСХ со стационарной неполярной фазой поверхность диоксида кремния подвергается гидрофобизации обработкой алкилсиланами. Первоначально, как и в классической колоночной хроматографии, при формировании тонкослойных пластинок использовались сорбенты и носители жидких фаз с размерами частиц 200-250 мкм. В настоящее время чаще применяют мелкодисперсные сорбенты с узким диапазоном размеров частиц от 1 до 25 мкм. Появился и соответствующий термин — высокоэффективная тонкослойная хроматография. [90]. Подробно с техникой тонкослойной хроматографии и областями ее применения можно познакомиться в [91-94].
Противоточная и двухмерная хроматография
При традиционной схеме хроматографического процесса, когда одна из фаз неподвижна, а другая перемещается относительно нее, операции разделения производятся последовательно по времени. Возможность хроматографического разделения в непрерывном режиме появляется при противоточном и двухмерном способе относительного перемещения фаз.
При кажущейся простоте схемы противоточного способа осуществления хроматографического процесса его практическая реализация требует сложных технических решений для осуществления взаимного перемещения фаз во встречных направлениях и непрерывного выделения из их потоков целевых компонентов. Целый ряд попыток создания противоточных хроматографических устройств для газовой и ионообменной хроматографии закончился неудачей из-за технических трудностей. К тому же в подавляющем большинстве случаев их преодоление не оправдано достигаемым конечным эффектом разделения исходной смеси веществ только на две фракции. Неожиданным техническим решением проблемы осуществления противоточного хроматографического процесса явилась противоточная центрифужная хроматография Countercurrent centrifugal Chromatography (ССС), которая по смыслу механизма разделения может называться в русскоязычной литературе ЖЖХ в поле центробежных сил. Этот вариант хроматографического процесса был впервые реализован в системе двух жидких фаз. И до сих пор ЖЖХ в поле центробежных сил остается основным направлением развития этого метода. В этом методе, в отличие от традиционных направлений ЖЖХ, не требуется носитель стационарной фазы. Диспергирование стационарной фазы и ее удержи
190
Новый справочник химика и технолога
вание в потоке подвижной фазы осуществляется за счет центробежных сил, возникающих в хроматографической колонке спиралевидной формы при ее вращении. Хроматографический процесс в поле центробежных сил может быть осуществлен в двух принципиально отличных вариантах, характеризуемых как гидростатическая и гидродинамическая равновесные системы. Различие этих двух систем заключается в том, что в первом случае спиральная колонка не вращается вокруг собственной оси, а только вокруг внешней оси центрифуги, а во втором — колонка вращается вокруг двух осей.
Если вращающуюся спиральную колонку заполнить с одного конца стационарной фазой, а через другой конец спирали ввести подвижную фазу, в объеме колонки произойдет их взаимное диспергирование. При этом вытеснению стационарной фазы будет препятствовать действующая в противоположном направлении сила архимедова винта, возникающая вследствие вращения колонки относительно оси центрифуги. Если эта сила, зависящая от скорости вращения центрифуги, будет перемещать частицы стационарной фазы со скоростью, превышающей скорость движения подвижной фазы, реализуются условия для осуществления противоточного хроматографического разделения. Если скорости движения фаз сбалансированы, отпадает необходимость в постоянной подпитке колонки стационарной фазой и появляется возможность хроматографического разделения в обычном режиме пропускания через колонку подвижной фазы. Случай строго сбалансированного по скорости противоположно направленного движения обеих фаз, при котором стационарная фаза оказывается неподвижной относительно стенок колонки, практически трудно реализуем и не является оптимальным для данного процесса. Поэтому метод вошел в литературу как противоточная центрифужная хроматография. В то же время, несмотря на наличие встречного движения фаз, соответствующего формальным признакам противоточного хроматографического процесса, ЖЖХ в поле центробежных сил по конечному эффекту разделения ближе к обычной колоночной хроматографии.
При осуществлении противоточного хроматографического процесса возможны два случая принципиально различные по конечному эффекту разделения. Первый соответствует условиям, когда
v у
(1 -xj >Vc > (1 -Хе;)
где ус — линейная скорость стационарной фазы, индексы i и j соответствуют компонентам с большей и меньшей прочностью удерживания стационарной фазой.
Таким образом, если скорость движения стационарной фазы больше скорости движения одного или нескольких компонентов разделяемой смеси с потоком элюента и меньше скорости движения одного или нескольких слабее удерживаемых компонентов, то создаются условия для непрерывного противоточного разделения исходной смеси веществ на две фракции, выделяемые в потоках обеих фаз, участвующих в хроматографическом процессе. Именно
этот случай и рассматривается в связи с противоточной хроматографией.
Второй случай, обычно реализуемый в ЖЖХ в поле центробежных сил, представляет интерес при разделении наиболее близких по свойствам веществ в дискретном варианте элюирования в потоке одной фазы. Он реализуется при условии:
v Vc>(l-X/)
(индекс п соответствует компоненту, наиболее прочно удерживаемому стационарной фазой), т.е. когда скорость стационарной фазы меньше скорости движения зоны самого прочно удерживаемого компонента. В этом случае встречное движение стационарной фазы создает эффект увеличения длины хроматографической колонки и соответственно приводит к улучшению разрешения хроматографических пиков. Очевидно, что возможности такого способа повышения эффективности хроматографического процесса ограничены разделением смесей веществ с близкими объемами удерживания.
В гидродинамической равновесной системе осуществляется более эффективное диспергирование фаз и, следовательно, достигается более высокая эффективность разделения при более высоких скоростях потока подвижной фазы, чем в гидростатической системе.
Противоточная центрифужная ЖЖХ разрабатывалась и использовалась для препаративных разделений органических и биоорганических веществ. Исследования последних лет показали, что подобная техника может быть применима для аналитических и радиохимических разделений, предконцентрирования, очистки неорганических веществ в растворах в лабораторных масштабах с использованием различных экстракционных систем.
Возможности разделения веществ в ЖЖХ в поле центробежных сил определяются выбором двуфазной системы, характеризующейся определенными соотношениями плотностей, полярностей и коэффициентов распределения разделяемых веществ. При этом в большинстве случаев для разделения могут использоваться те же экстракционные системы, что и в традиционных вариантах ЖЖХ. В последние годы появились новые варианты ЖЖХ в поле центробежных сил: в частности, методы осадочной и пенной ЖЖХ в поле центробежных сил.
Непрерывное двухмерное хроматографическое разделение смеси веществ на произвольное число фракций становится возможным при одновременном равномерном перемещении обеих фаз в направлениях, перпендикулярных друг другу. Сущность такого процесса поясняет рис. 3.14. Слой стационарной фазы ABCD движется с равномерной скоростью в направлении MN относительно неподвижных систем АВ' и СТ)', предназначенных соответственно для подачи потока второй фазы по границе АВ и для сбора элюата по границе CD. Разделяемая смесь непрерывно подается в слой сорбента в точке 0. В направлении АС компоненты смеси движутся по слою стационарной фазы в соответствии с закономерностями хроматографического процесса. В направлении АВ зоны разделяемых веществ смещаются в пространстве вместе со слоем стационарной фазы.
Методы разделения и концентрирования
191
Рис. 3.14. Схема непрерывного двухмерного хроматографического разделения
Траектория движения каждого из компонентов исходной смеси веществ, вводимых в точке 0, будет являться результирующей сложения двух векторов: вектора скорости движения его зоны в слое стационарной фазы в потоке второй фазы и вектора скорости движения самой стационарной фазы. Величина первого вектора определяется законами движения зоны хроматографируемого вещества в стационарной фазе и потому является для него характеристической величиной. Соответственно траектории движения зон отдельных компонентов исходной смеси будут пересекать границу стационарной фазы CD в определенной точке пространства и попадать в соответствующий приемник элюата в системе его сбора С D'.
Если вместо условного непрерывного слоя стационарной фазы перейти к хроматографическому устройству в виде цилиндрического кольца, вращающегося относительно систем подачи разделяемой смеси веществ и элюента по верхнему срезу цилиндра, и системы фракционного сбора элюата по нижнему срезу, открывается возможность непрерывного хроматографического разделения. Аналогичного эффекта можно добиться в плоскостном варианте хроматографии, придав замкнутому пространству, в пределах которого осуществляется хроматографический процесс, форму плоского кольца. В этом случае подача разделяемой смеси и элюентов будет осуществляться по внутреннему периметру вращающегося кольца, сбор фракций элюата — по внешнему или наоборот.
Подробнее с историей метода непрерывной двухмерной хроматографии, его теорией, устройством двухмерных хроматографов и областями применения можно познакомиться в [3, с. 116-126].
3.2.9.5. Варианты хроматографических методов разделения в зависимости от агрегатного состояния фаз и механизма удерживания разделяемых веществ стационарной фазой
Жидкостно-твердофазная хроматография
Независимо от схемы и условий осуществления хроматографического процесса, важнейшими характеристическими признаками методов, образующих совокупность хроматографических методов разделения является агре
гатное состояние фаз, участвующих в хроматографическом процессе, и механизм взаимодействия разделяемых веществ со стационарной фазой.
В соответствии с первым из этих признаков наиболее распространенным вариантом хроматографических методов является жидкостно-твердофазная хроматография. Но подобный обобщенный термин не нашел широкого распространения. Обычно принято выделять отдельные методы в пределах этого направления по второму классификационному признаку — механизму взаимодействия хроматографируемого вещества со стационарной фазой: жидкостноадсорбционную, аффинную, ионообменную, лигандообменную, эксклюзионную и другие. Каждый из этих методов имеет свою специфику и требует отдельного рассмотрения.
Жидкостно-адсорбционная хроматография (ЖАХ)
ЖАХ является родоначальником хроматографических методов. Именно на примере этого варианта хроматографического процесса М.С. Цвет открыл хроматографию.
Стационарной фазой в этом методе служит твердый, как правило, пористый адсорбент, обеспечивающий большую поверхность межфазного контакта, и способный к различным межмолекулярным взаимодействиям с веществами, находящимися в растворенном состоянии в подвижной фазе. Прочность удерживания хроматографируемых веществ в колонке проявляется в зависимости от различий в энергиях адсорбции молекул сорбата и растворителя. Чем больше энергия адсорбции первых превышает энергию адсорбции молекул растворителя, тем прочнее удерживаются вещества в колонке. Интервалы изменения значений энергии адсорбции зависят от вида межмолекулярного взаимодействия, определяемого природой поверхности и адсорбирующихся веществ.
В наиболее общем случае адсорбционные процесы в ЖАХ вызваны слабыми ван-дер-ваальсовыми силами. При сорбции на полярных адсорбентах, поверхность которых покрыта гидроксильными группами, часто происходит образование более прочных водородных связей с молекулой адсорбата. В обоих случаях полностью исключаются кинетические ограничения в установлении сорбционных равновесий в системе — жидкая фаза - адсорбент. Наконец, между молекулой адсорбата и функциональными группами на поверхности адсорбента может протекать химическая реакция с образованием химической связи. Последствием такого взаимодействия может быть прочная, часто необратимая хемосорбция или образование новых химических соединений, отсутствовавших в разделяемой смеси. Как правило, химическое взаимодействие разделяемых веществ с поверхностью адсорбента является крайне нежелательным процессом, вероятность протекания которого учитывается при выборе условий разделения. Механизм хемосорбционного удерживания является неприемлемым для ЖАХ.
В пределах ван-дер-ваальсовых взаимодействий прочность сорбции зависит от относительной полярности молекул адсорбата и функциональных групп на поверхности
192
Новый справочник химика и технолога
сорбента. В зависимости от полярности адсорбента в ЖАХ в наиболее общем случае выделяются два направления: нормально-фазный и обращенно-фазный варианты. Первый относится к случаю применения полярных адсорбентов, второй — неполярных. На начальном этапе развития ЖАХ использовались полярные адсорбенты. Наиболее распространенные типы полярных адсорбентов — силикагели, целлюлоза, оксид алюминия. В настоящее время около 90% всех анализов в режиме ВЭЖХ выполняется на обращенно-фазовых неполярных сорбентах на основе силикагеля. Максимальный эффект обращения полярности достигается при прививке на поверхности адсорбента алкильных групп. В качестве промежуточного варианта используют сорбенты с привитыми диольными, ширильными и другими подобными им функциональными группами. Выпуск специальных сорбентов для ЖАХ, включая ее высокоэффективный вариант, освоен многими фирмами, перечень которых приведен ниже; он необходим для пользования последующими таблицами, в колонке «Изготовитель» числа соответствуют номерам из этого списка.
Список основных фирм-изготовителей сорбентов для жидкостно-адсорбционной хроматографии [90]
1. Altech
2. Altex
3. Analabs
4. Anspec Copmpany
5. Applied Science
6. Beckman
7. Benson Company
8. Biolab Products
9. Bio-Rad Laboratories
10. Brownlee
11. Calbiochem-Benring Corporation
12. Chromanetics
13. Chromatix
14. Chrompak (Holland)
15. Dionex
16. DuPont
17. ES Industries
18. Gow Mac
19. Hamilton
20. Hetp
21. Hewlett Packard
22. Hitachi
23. HPLC Technology Ltd.
24. OCN Igc.
25. Jasco
26. Johns-Manville
27. Jones Chromatography
28. Laboratory Instrument Works (Prague, Chehia)
29. Machery-Nagel (Germany)
30. MCB
31. Perkin-Elmer
32. Phase Sep
33. Pierce
34. Rainen
35. Regis
36. Rheodyne
37. Rhone Poulence (France)
38. RSL
39. Separation Group
40. Shandon
41. Supelco
42. Synchrom
43. Toyo Soda
44. Unimetrics Corporation
45. Varian
46. Waters Associates
47. Whatman
Характеристики наиболее часто используемых сорбентов для высокоэффективной нормально-фазовой ЖАХ приведены в табл. 3.59 и табл. 3.60.
Таблица 3.59
Тонкодисперсные насадочные материалы для нормально-фазовой жидкостно-адсорбционной хроматографии [90]
Название Изготовитель Средний диаметр частиц, нм ц10~3 Удельная поверхность, м2^1 Средний диаметр пор, нм Примечания
Кремнезем с частицами неправильной формы
Хромосорб LC-6 1,41 5, 10 400 12 Плотность — 410 кг • м 3
Хитачи-гель серии 3030 22 5-7 500
Силикагель фирмы ICN 24 3-7, 7-12 500-600
Лихросорб SI-60 2, 5,10, 12,13, 23, 27, 36,41,44 5,10 550 6
Лихросорб SI-100 2,5, 10, 12,13, 23, 27, 36, 41, 44 5, 10 300 10
Методы разделения и концентрирования
193
Продолжение таблицы 3.59
Название Изготовитель Средний диаметр частиц, нм ц10~3 Удельная поверхность, м2 • г-1 Средний диаметр пор, нм Примечания
Порасил 1,46 10 300-500
Партисил 2, 18, 23,27, 33, 47 5, 10 400 6 Плотность — 450 кг • м“3
Полигосил 14, 23, 29, 34, 38 5,7,10, 15 500 6 Объем пор — 0,75 мл • г1
RSIL 1,38 5, 10 550 6
SIL-X-1 31 13-5 400 С химически обработанной поверхностью
Кремнезем с частицами сферической формы
Апекс Силика 27 5 200 10 Объем пор — 0,7 мл г 1
Fine SIL 25 5 500 6
Гиперсил 12, 23, 27 3,5, 10 200 10
Лихосфер SI-100 1,5,21,23,27, 41 5, 10 250 10 Объем пор — 1,2мл г'1 Незаполненный объем — 1,06 мл • г-1. Выпускается с разным диаметром пор
Нуклеосил SI-50 1, 14, 23,29, 34, 38,41 5,7,5, 10 300, 500 5 Объем пор — 0,8 мл г-1
Нуклеосил SI-100 1, 14, 23, 29, 38, 41 5, 7,5, 10 300 10 Объем пор — 1,0 мл • г-1
Сепарон SI VSK 28 5, 7,5, 10 450 13 Объем пор — 1,5 мл • г-1
Сферисорб SW 1, 12, 20, 23,27, 32, 35,41 3,5, 10 220 8 Плотность набивки — 0,6 мл • г-1
Сферосил ХОА 600 3,37,41 5-7 600 • 10% 8 Объем пор — 0,7-1 мл • г-1
Сферосил ХОА 800 3,37,41 5-7 860 4 Объем пор — 0,4-0,6 мл • г1
TSK-гель Силика-60 43 5, 10 500 6
TSK-гель Силика-150 43 5, 10 330 15
Видак 101-ТР 1, 3-5, 14, 41 10 100 33
Зорбакс BP-SIL 1, 16, 23,41 8 350 7-8
Оксид алюминия
Алокс 60-D 14, 29, 34, 38 5, 10 60 6 Основной pH = 9, 5
Лихросорб Алокс Т 10, 13,35,36 5, 10 70 15
Сферисорб АТ 23,32,35,41 5, 10 95 13 Предельный pH = 10
Таблица 3.60
Тонкодисперсные насадочные материалы с привитыми функциональными группами для нормально-фазовой ЖАХ [90]
Название Изготовитель Функциональные группы Средний диаметр пор, нм ц10“3 Примечания
Лихросорб диол 2,5, 8, 10, 12, 13,21, 23, 27, 36, 44 Диол- 10 Для разделения высокополярных соединений
Нуклеосил ОН 1, 2,14, 29, 34, 38 —ОН Спиртовые 7,5 Смачивается Н2О
TSK-гель ОН-120 43 —ОН Спиртовые 5, 10
194
Новый справочник химика и технолога
Продолжение таблицы 3.60
Название Изготовитель Функциональные группы Средний диаметр пор, нм ц10”3 Примечания
Нуклеосил N(CH3)2 1, 2,14,23,29,34, 38 Триалкил амин- 5,10 Является также слабым анионообменником
Полигосил 60-D-N(CH3)2 1,23 Триалкил амин- 5, 10 Может быть использован как слабый анионообмен-ник
Полигосил NO2 1,2, 14, 23, 29, 34,38, 41 Нитро- 5, 10 Сферические частицы. Сродство к двойной связи
Полигосил NO2 1, 23,29 Нитро- 5, 10 Применяется для разделения ароматических соединений. Сродство к двойной связи
RSIL NO2 1,38 Нитро- 5, 10 Покрытие 5% С
Алексциано Т1 Циано- 5 Монослой
Хромосорб LC-8 1,8,45 Циано- 5, 10 Диаметр пор 11 нм, Плотность 0,43 г • мл 1
Гиперсил CPS 23,41 Цианопропил- 3, 5,10 Монослой. pH = 3-8
Лихросорб CN 5, 10, 13, 23, 30, 36 Циано- 5, 10 Вначале поверхность обрабатывается толилтри-хлорсиланом, а затем NBS с последующим нуклеофильным замещением галогена
о-Бондапак 1,46 Циано- 10 Покрытие 9 масс. %
Микропак CN 45 Цианопропил- 10
Нуклеосил CN 1,2, 14, 29, 34,38,41 Циано- 5, 10 Покрытие 6 мкмоль-экв • м 2
Партисил-10 РАС 1,2, 12,18,23,27, 33, 47 Цианоамино- 10 Отношение NH2: CN~-2 : 1
Полигосил CN 1,23,29 Циано- 5, 10 Покрытие 6 мкмоль-экв • м 2
RSIL CN 1,38 Цианопропил- 5, 10 Покрытие 5% С
Сферисорб CN 1, 2,12, 20, 23, 27, 32, 35,41 Циано- 5, 10 Диаметр пор 8 нм. Покрытие 0,6 ммоль • г1
Видак 501 ТР 1,3,4,5,14,39,41 Циано- 10 Сферические частицы
Зорбакс BP-CN 8, 16, 23, 41 Циано- 8 Монослой
Апекс Амино 27 Амино- 5
Гиперсил APS 23,32 Аминопропил- 3, 5,10
Хромосорб LC-9 26,41 Амино- 10 Плотность 0,48 г • мл1
Fine SILNH2 25 Аминопропил- 5, 10 Сферические частицы
Лихросорб NH2 1,2, 5, 8,13,21,23, 27, 44, Амино- 10
о-Бондапак NH2 1, 23, 46 Амино- 10 pH - 2-8
Микропак NH2 45 Аминопропил- 10 -
Нуклеосил NH2 1,2,14,23,29,34,38, 41 Амино- 5, 10 Слабоосновной. Для отделения полярных соединений
Полигосил NH2 1,23,29 Амино- 5,10
Методы разделения и концентрирования
195
Продолжение таблицы 3.60
Название Изготовитель Функциональные группы Средний диаметр пор, нм ц10“3 Примечания
rsilnh2 1,38 Аминопропил- 5, 10 Поверхность сначала обрабатывают хлорпропил-трихлорсиланом, а затем н-бутанолом с последующим нуклеофильным замещением галогена
Сепарон SI NH2 28 Аминопропил- 5, 10
Сферисорб NH2 1, 12, 20, 23, 27, 32, 35,41 Аминопропил- 5, 10 Диаметр пор — 8 нм
TSK-гель 43 Аминопропил- 5
Зорбакс NH2 8,23,41 Амино- 8 Монослой
Наряду с полярностью функциональных групп важнейшей характеристикой сорбентов является удельная поверхность, так как в основе метода лежит межфазный обмен между подвижной фазой и поверхностью твердого адсорбента, а не его объемом. Удельная поверхность зависит от общей пористости сорбента и размера пор. Обычно для силикагелей диаметр пор колеблется в пределах 5-30 нм, а удельная поверхность составляет от 100 до 500 м2 г-1 [97, 99]. При создании специальных поверхностно-пористых сорбентов для улучшения кинетики массообменных процессов в режиме ВЭЖХ выбирается разумный компромисс между снижением удельной поверхности и соответствующим уменьшением фактора удерживания Ке и улучшением разрешения хроматографических пиков. Удельная поверхность поверхностно-пористых сорбентов, как правило, всего в 10-20 раз меньше, чем у соответствующих объемнопористых.
На свойства полярных адсорбентов заметное влияние оказывает вода. Присутствие больших количеств воды в сорбентах нежелательно из-за ее конкурентной адсорбции. Небольшие количества воды, наоборот, вызывают положительные следствия: уменьшают статический заряд на сорбенте и при сухом заполнении колонок способствуют более однородной и плотной упаковке насадки. Добавление 0,02-0,04 г воды на 100 м2 поверхности сорбента (0,04 г воды создают монослой на 100 м2 поверхности адсорбента) позволяет увеличить линейную скорость подвижной фазы в 50-100 раз и число тарелок на единицу длины колонки — примерно в 3 раза [99, 100]. Для каждого полярного адсорбента существует свое оптимальное количество воды, соответствующее заполнению 30-100% монослоя. Содержание воды в адсорбенте необходимо поддерживать постоянным, не допуская его длительного контакта с атмосферным воздухом. Поглощение избыточной влаги из воздуха приводит к ухудшению хроматографических характеристик адсорбентов.
Свойства обращенно-фазных сорбентов зависят не только от типа привитых функциональных групп и удельной поверхности, но и от ряда иных факторов. В наиболее распространенном случае химически модифицированных
кремнеземов (ХМК) существенное значение имеет структура привитого слоя. По этому признаку можно выделить три основных типа ХМК [99]:
— ХМК с мономолекулярным слоем органических, эле-менторганических или неорганических соединений, привитых к поверхности сорбента;
— ХМК с поверхностным полимерным слоем;
— объемно-модифицированные ХМК.
В ЖАХ широкое применение находит первый тип ХМК с так называемыми щеточными структурами привитых групп, обеспечивающими высокие скорости массообмена. В зависимости от химической природы привитых групп щеточные ХМК подразделяют на две разновидности. К первой относят ХМК с привитыми алкильными радикалами, содержащими от 1 до 22 метильных звеньев. Чаще всего выпускают ХМК с октадецильными группами (ультрасфер, ультрапак, сферисорб и т.д.). Отечественный аналог — сорбент ХМК-16 содержит гексадецильные радикалы. Алкильные группы ориентируются либо параллельно друг другу и перпендикулярно к поверхности кремнеземного носителя, либо жидкостно-подобно, изгибаясь и застилая все изгибы поверхности. При большой длине цепи такие ХМК приближаются по свойствам к жидким растворителям с тем преимуществом, что они не уносятся подвижной фазой в условиях ВЭЖХ. Плотность прививки алкильных групп определяет емкость ХМК-сорбентов. На силикагелях в оптимальных условиях концентрация привитых групп достигает 4 мкмоль м 2. Это значение уменьшается с увеличением длины алкильных групп, составляя для наиболее распространенных обращенных фаз С]8 3-3,5 мкмоль • м 2. Но поскольку при одинаковой степени покрытия поверхности Ке возрастает с увеличением длины цепи быстрее, чем уменьшается число привитых групп, более прочное удерживание обеспечивают привитые фазы с длинными алкильными группами. Для разделения больших неполярных молекул такая прочность удерживания оказывается избыточной, и лучшие результаты в этом случае дает применение сорбентов с короткоцепочными функциональными группами. Специальные сорбенты для высокоэффективной обращенно-фазовой ЖАХ приведены в табл. 3.61-3.63.
196
Новый справочник химика и технолога
Таблица 3.61
Тонко дисперсные насадочные материалы с химически привитым октадецилсиланом для обращенно-фазовой ЖАХ [90]
Пояснения. Эндкепинг — дополнителная обработка насадочного материала после прививки углеводородных групп с целью устранения оставшихся на поверхности гидроксильных групп.
Название Изготовитель Средний диаметр пор, нм ц10~3 Покрытие, % С Примечания
Апекс ODS 27 5 10 Проведен эндкепинг
Хромосорб LC-7 1,41 3, 5, 10 10 Монослой. Средний диаметр пор 10 нм
Fine SIL С-18 25 5, 10 14
Fine SILC С-18Т 25 5, 10 14 Проведен эндкепинг
Хитачи-гель серии 3050 22 5-7,10-15 pH = 2-8
Лихросорб RP-18 1,5,10, 12,13, 21, 23, Т1, 36, 41, 44 5, 10 22 pH = 1-9
о-Бондапак С-18 1, 23, 46 10 10
МикропакС-18 45 10 22 Полимерный. Проведен эндкепинг
Микропак СН 45 5, 10 12 Мономерный слой; с проведением эндкепинга и без него
Нуклеосил С-18 1, 14,23,29, 34, 38 5,7,5, 10 15-16 Фактор удерживания в 2 раза больше, чем у С-8; pH = 1-9. Сферические частицы
Гиперсил ODS 12, 23, 27, 40 3,5, 10 9 Проведен эндкепинг
ODS-SIL-X-1 31 13 • 5
Партисил ODS-1* 1, 18,23,27,33, 47 5, 10 5 Высокое содержание силанола
Партисил-10 ODS-2 1, 12, 18,23,33, 47 10 15 Высокое удерживание и большая емкость. Рабочие температуры до 70 °C
Партисил-10 ODS-3 18, 33,47 10 10 Проведен эндкепинг
Полигосил С-18 1, 14, 23, 24, 29, 34,38 3, 7,5, 10 11 Фактор удерживания в 2 раза больше, чем у С-8; pH = 1-9
RSIL C-18HL 1,38 5, 10 18 9 Проведен эндкепинг Проведен эндкепинг
Сепарон SI С-18 28 5, 10 20 Проведен эндкепинг
Сферисорб ODS 1, 12, 20, 23, 27, 32, 35, 41 5, 10 7 0,3 ммоль • г-1 Проведен эндкепинг Средний диаметр пор — 8 нм
Сферосил С-18 3,31,37,41 5-7 20-23
Техсфер С-18 23 5, 10 10 Насадка с частицами неправильной формы называется Техсил С-18. Обе выпускаются с проведением эндкепинга и без него
TSK-гель ODS 120А 43 5, 10
TSK-гель ODS 120Т 43 5, 10
Видак 201 С-18 1,3,4,5, 14, 39, 41 5, 10 10 3,35 ммоль • г-1 Эндкепинг не проводится pH - 1-9
Зорбакс BP-ODS 1,12, 16,23,41 8 15 Монослой
Примечание.
* Рекомендуется для более полярных соединений.
Методы разделения и концентрирования
197
Таблица 3.62
Тонкодисперсные насадочные материалы с химически привитым октилсиланом для обращенно-фазовой ЖАХ [90]
Название Изготовитель Средний диаметр пор, нм ц10 3 Покрытие, % С Примечания
Апекс С8 27 5 5 Выпускается с проведением энд-кепинга
Fine SIL С8 25 5, 10
Лихросорб RP-8 1,5,10, 12,13,21,23, 27, 6,41,44 5, 10 13-14 Рекомендуется для веществ с умеренной полярностью
Гиперсил MOS 12,23,40 3,5, 10 Монослой
Нуклеосил С18 1,14, 23,29, 34,38, 41 5, 7,5, 10 10-11 pH = 1-9
Полигосил С8 14, 23, 29, 36, 38 5, 7,5, 10 10-11 pH = 1-9. Сферические частицы
Техосфер С8 23 5, 10 10 Насадка с частицами неправильной формы называется Техсил С-8
Зорбакс ВР-С8 12, 16, 23,41 8 15 Монослой
Таблица 3.63
Прочие тонкодисперсные насадочные материалы для обращенно-фазовой ЖАХ [90]
Принятые обозначения: ТМХС — триметилхлорсилон, ХПТХС — хлорпропилтрихлорсилан.
Название Изготовитель Средний диаметр пор, нм ц10~3 Покрытие, % С Примечания
Гексилсилан
Хромегабон 17 10 10
Циклогексил 17 10 10
Сферисорб С6 1, 12, 20, 23,33,35, 41 5 • 2 0,6 ммоль • г"1 Монослой; средний диаметр пор — 8 нм. При изготовлении проводится эндкепинг
Метилсилан
Апекс С2 27 5 3 Для полярных многофункциональных соединений
Fine SIL Cl 25 5, 10
Лихросорб RP-2 1,5,8, 10, 12,21,23, 27, 36,41,44 5,10 Рекомендуется для полярных соединений
RSIL СЗ 1,38 5, 10 Основная обработка поверхности ХПТХС. Для эндкепинга используется ТМХС
Сепарон SIC1 28 5, 10 Метилированная поверхность; пригоден для эксклюзионной хроматографии. Предел эксклюзии — 50000
TSK-гель TMS-250 43 5
Зорбакс BP-TMS 12, 16, 23 8 Монослой
Фенилсилан
Апекс фенил 27 5 5 Для аминов и гидроксилсодержащих соединений
198
Новый справочник химика и технолога
Продолжение таблицы 3.63
Название Изготовитель Средний диаметр пор, нм ц10~3 Покрытие, % С Примечания
о-Бондапак 1,23,46 10 10 Более полярный, чем С-18
Нуклеосил фенил 1, 14,29, 34,38 7,5 10 Для очень малополярных соединений и жирных кислот
RSIL фенил 1,38 5, 10 Для неполярных соединений
Сферисорб Р 1, 12,23,32 5 • 2 0,3 ммоль • г-1 Средний диаметр пор — 8 нм
Сополимер стирола с дивинилбензолом
Fine GEL ПО 25 10
Fine Gel 101 25 8 Пригоден для эксклюзионной хроматографии, предел эксклю-зии — 3000
Хитачи-гель ЗОН 22 10-15 Сферические частицы, pH = 2-11. Функциональные группы —СН2ОН
Бенсон BN 7 7-10 Пригоден для эксклюзионной хроматографии. Выпускается с различной степенью сшивки (4, 7, 8 и 10% ДВБ)
Вторая разновидность щеточных ХМК содержит привитую функциональную группу, которая фиксируется на поверхности носителя углеводородной цепочкой из 10-11 метиленовых звеньев. Меняя функциональную группу, таким ХМК можно придать ионообменные, комплексообразующие свойства или свойства хелатных смол и использовать их для концентрирования, выделения и разделения d- и /-металлов; свойства краун-эфиров дают возможность выделять щелочные и щелочноземельные металлы, а свойства хиральных лигандов — разделять энантиомеры (оптические изомеры) и т.д. В этих случаях в зависимости от природы функциональных групп сфера применения соответствующих сорбентов выходит за рамки ЖАХ.
Дополнительные сведения о различных типах адсорбентов и их свойствах можно найти в разделе, посвященном газовой хроматографии, так как в жидкостно- и газоадсорбционной хроматографии часто используются одни и те же адсорбенты. Из специальной литературы можно отметить [95].
Выбор состава подвижной фазы в ЖАХ основывается на эмпирическом подборе индивидуальных растворителей или их смесей, обладающих необходимой элюирующей способностью и обсепечивающих необходимую селективность при разделении компонентов смеси. На основании накопленного опыта элюенты для полярных сорбентов сопоставляют по элюирующей способности или хроматографической активности, располагая их в элюотропные ряды [62]. Элюирующая способность подвижной фазы характеризуется параметром д°, величина которого зависит от силы взаимодействия молекул подвижной фазы с поверхностью адсорбента. Величина g пропорциональна приведенной к единице площади поверхности (разности между энергиями взаимодействия молекул соответствующей подвижной фазы и молекул пентана), для которой ус-
ловно принимается д° = 0. Согласно приведенному определению абсолютные значения д° зависят от природы адсорбента. Тем не менее, элюотропные ряды в ЖАХ достаточно постоянны. Относительная элюирующая способность растворителей сохраняется при переходе от одного адсорбента к другому. Часто элюотропные ряды составляют из малоактивных и сильноактивных веществ, смешивая их в различной пропорции. Например, смешивая пентан с д° = 0,00 и метанол, для которого д° = 0,95, получают набор растворителей с непрерывно увеличивающейся активностью (элюирующей способностью). Параметр д° определяет скорость перемещения хроматографических зон. Поэтому, выбрав соответствующее значение д° или режим изменения д° при градиентном элюировании, можно регулировать время удерживания разделяемых веществ в колонке. В то же время, варьируя состав растворителя при том же значении д°, можно повлиять на селективность процесса и лучше разрешить хроматографические пики. Влияние на селективность оказывает замена активного компонента смеси растворителя: метанола, дихлорэтана, эфира, этилацетата и др. Замена же слабоактивного растворителя, например, пентана на гексан или бензол, практически не влияет на селективность и, следовательно, на разрешение пиков. Применяемый растворитель или их смесь не должна быть «сухой», т.е. она должна содержать некоторое количество воды, чтобы поддерживать ее концентрацию в адсорбенте на оптимальном уровне. Селективность при элюировании из одного и того же адсорбента определяется полярностью растворителя. Количественной характеристикой полярности растворителя служит параметр растворимости Гилъденбрандта f, изменяющийся от 6 для перфторалканов до 21 для воды [60]. Параметр f учитывает вклад различных межмолекулярных взаимодействий: дисперсионных взаимодействий (frf),
Методы разделения и концентрирования
199
дипольных взаимодействий (fo), протонодонорных (Ц) и протоноакцепторных (fa) водородных связей. Каждая из этих составляющих характеризует конкретное свойство молекул, тип полярности растворителя и механизм его взаимодействия с молекулами разделяемых веществ. На основании справочных данных о параметрах раствори-
мости можно составить элюотропные ряды или серии растворителей, пригодные для разделения конкретных смесей веществ. Важнейшие характеристики растворителей, наиболее часто используемых в качестве подвижных фаз в индивидуальном виде или в виде смесей, приведены в табл.3.64.
Таблица 3.64
Характеристики растворителей, используемых в качестве подвижных фаз или их компонентов в ЖАХ [101]
Пояснения.
1. Площадь поверхности молекулы выражена в относительных единицах согласно Снайдеру. Чтобы получить значения в нм2, нужно умножить данную величину на коэффициент 0,085.
2. Элюирующая сила дана для систем жидкость - твердое тело для элюирования из оксида алюминия.
3. Параметр растворимости найден по температуре кипения.
4. fo, fa, Ц — даны приближенные значения.
5. Для алкилфторидов приведены средние значения для различных соединений.
Растворитель Молярная масса Плотность, (кг рм~3)р103 Вязкость (при 20 °C), 10"3 Паус Площадь поверхности молекулы, 5а Элюирующая сила, Параметр растворимости, f £о
Алкилфториды -0,25 6,0 6,0 0 1,0 0
CFC12-CF3 170,92 1,455 6,2 5,9 1,5 0 0
Изооктан 114,23 0,691 0,50 7,6 0,01 7,0 7,0 0 0 0
Диизопропиловый эфир 102,18 0,724 0,37 5,1 0,28 7,0 6,9 0,5 0,5 0
я-Пентан 72,15 0,626 0,23 5,9 0,00 7,1 7,1 0 0 0
CCI3-CF3 187,38 1,579 7,1 6,8 1,5 0,5 0
я-Гексан 86,18 0,660 0,33 0,01 7,3 7,3 0 0 0
н-Гептан 100,21 0,684 0,41 0,01 7,4 7,4 0 0 0
Диэтиловый эфир 74,12 0,714 0,28 4,5 0,38 7,4 6,7 2 2 0
Триэтилам ин 101,19 0,725 7,5 7,5 0 3,5 0
я-Декан 142,29 0,730 0,92 10,3 0,04 7,8
Циклопентан 70,13 0,751 0,47 5,2 0,05 8,1 8,1 0 0 0
Циклогексан 84,17 0,779 1,00 6,0 0,04 8,2 8,2 0 0 0
я-Пропилхлорид 78,54 0,892 0,35 3,5 0,30 8,3 7,3 3 0 0
Изопропил-хлорид 78,54 0,859 0,33 3,5 0,29 8,4
Т етрахлорметан 153,84 1,595 0,97 5,0 0,18 8,6 8,6 0 0,5 0
Диэтилсульф ид 90,19 0,837 0,45 5,0 0,38 8,6 8,2 2 0,5 0
Этил ацетат 88,10 0,901 0,45 5,7 0,58 8,6 7,0 3 2 0
Пропиламин 59,11 0,719 8,7 7,3 4 6,5 0,5
Этилбромид 108,99 1,456 3,4 0,37 8,8 7,8 3 0 0
jw-Ксилол 106,16 0,864 0,62 7,6 0,26 8,8 8,8 0 0,5 0
Толуол 94,12 0,866 0,59 6,8 0,29 8,9 8,9 0 0,5 0
Хлороформ 119,39 1,489 0,57 5,0 0,40 9,1 8,1 3 0,5 0*
Тетрагидрофуран 72,10 0,888 5,0 0,45 9,1 7,6 4 3 0
Метил ацетат 74,08 0,934 0,37 4,8 0,60 9,2 6,8 4,5 2 0
Бензол 78,12 0,879 0,65 6,0 0,32 9,2 9,2 0 0,5 0
Тетрахлорэтилен 236,74 2,091 9,3 9,3 0 0,5 0
200
Новый справочник химика и технолога
Продолжение таблицы 3.64
Растворитель Молярная масса Плотность, (кг рм-3)р103 Вязкость (при 20 °C), 10’3 Паус Площадь поверхности молекулы, S* Элюирующая сила, Параметр растворимости, f fo fa fA
Этилметилкетон 72,10 0,805 4,6 0,51 9,3 0
Ацетон 58,08 0,792 0,32 4,2 0,56 9,4 6,8 5 2,6 0
Дихлорметан 84,94 1,326 0,44 4,1 0,42 9,6 6,4 5,5 0,5 0
Хлорбензол 112,56 1,107 0,80 6,8 0,30 9,6 9,2 2 .0,5 0
Анизол 108,14 0,995 1,32 9,7 9,1 2,5 2
1,2-Дихлорэтан 98,96 1,256 0,79 4,8 0,49 9,7 8,2 4 0 0
Метил бензоат 166,18 1,157 9,8 9,2 2,5 1 0
Диоксан 88,10 1,035 1,54 6,0 0,56 9,8 7,8 4 3 0
Пентиловый спирт 88,16 0,814 4,1 8,0 0,61 9,8
Метилиодид 141,94 2,279 0,50 9,9 9,3 2 0,5 0
Бромбензол 157,03 1,499 1,19 9,9 9,3 1,5 0,5 0
Сероуглерод 76,14 1,263 0,37 3,7 0,15 10,0 10,0 0 0,5 0
н-Пропанол 60,09 0,803 2,3 8,0 0,82 10,2 7,2 2,5 4 4
Пиридин 79,10 0,982 0,93 5,8 0,71 10,4 9,0 4 5 0
Бензонитрил 103,12 1,010 1,24 10,7 9,2 3,5 1,5 0
Нитрометан 61,04 1,130 0,67 3,8 0,64 11,0 7,3 8 1 0
Нитробензол 123,12 1,200 2,03 П,1 9,5 4 0,5 0
Этанол 46,07 0,789 1,20 8,0 0,88 П,2 6,8 4,0
Фенол 94,12 1,071 12,7 11,4 9,5
Диметил-формамид 73,10 0,944 11,5 7,9
Ацетонитрил 41,05 0,783 0,37 10,0 0,65 11,8 6,5 8 2,5 0
Метилениодид 267,87 3,226 П,9 11,3 1 0,5 0
Уксусная кислота 60,05 1,049 1,26 8,0 1,0 12,4 7,0
Диметилсульфоксид 78,13 1,101 2,2 4,3 0,6 12,8 8,4 7,5 5 0
Метанол 32,04 0,791 0,60 8,0 0,95 12,9 6,2 5 7,5 7,5
Этаноламин 61,08 1,018 13,5 8,3
Этиленгликоль 67,02 1,109 19,9 8,0 1,11 14,7 8,0
Формамид 45,04 1,134 17,9 8,3
Вода 18,02 0,998 1,00 21 6,3
Пентилхлорид 106,60 0,883 0,43 4,2 0,26
Изобутил-метилкетон 100,16 0,801 5,3 0,43
1-Нитропропан 89,09 1,022 4,5 0,53
Анилин 93,13 1,022 4,4 6,7 0,62
Диэтил амин 73,14 0,711 0,38 7,5 0,63
Примечание. * В некоторых работах приводятся большие значения.
Аналогичный по смыслу подход реализуется и при выборе состава подвижных фаз в ЖАХ с обращенно-фазовыми сорбентами, с той лишь разницей, что элюирующие растворы обычно готовят из смесей тех же растворителей с промежуточной полярностью (метанола, аце
тонитрила, пропанола и т.п.) с водой. Поскольку чистая вода соответствует максимальному удерживанию органических соединений, то элюирующая способность в этом случае возрастает по мере увеличения доли менее полярного растворителя.
Методы разделения и концентрирования
201
В рассмотренных вариантах ЖАХ используется исключительно как метод разделения органических веществ. В качестве одного из вариантов метода параллельно с обращенно-фазной ЖАХ возникло направление, получившее название ЖАХ хелатов. В этом варианте появилась возможность разделения металлов в форме хелатов. При этом селективность разделения определяется как устойчивостью хелатных комплексов, так и межмолекулярными взаимодействиями хелатов с поверхностью адсорбентов. Широта областей использования метода определяется возможностью осуществления хроматографического процесса как в колоночном, так и в плоскостном вариантах. Учитывая огромный арсенал данных по экстракции хелатов (см. п. 3.2.5), которые могут быть использованы для выбора условий их разделения методом ЖАХ хелатов, к настоящему времени предложено множество методик разделения ионов металлов этим методом, сведения о которых систематизированы в [102].
Аффинная хроматография
Аффиная хроматография является самостоятельной областью жидкостно-адсорбционной хроматографии, выделяемой по специфическому механизму взаимодействия разделяемых веществ с сорбентом. Метод основан на характерной особенности биологически активных веществ селективно и обратимо связывать определенные вещества, называемые аффинными лигандами или аффиантами. Таким образом, ферменты связывают соответствующие ингибиторы, антитела — антигены, гормоны — их рецепторы и т.п. Если по аналогии с обращенными фазами приготовить сорбенты с привитыми аффинными лигандами, появляется возможность селективного хроматографического выделения близких по свойствам биологически активных соединений и их разделения между собой. Наиболее часто применяемые аффинные лиганды приведены в табл. 3.65.
Таблица 3.65
Лиганды, используемые в аффинной хроматографии [89]
Тип лиганда Примеры удерживаемых веществ
Высокоспецифичные лиганды
Антитела Различные агенты (лекарства, гормоны, пептиды, протеины, вирусы и т.д.)
Инигибиторы энзимов и аналогов Энзимы
Нуклеокислоты Нуклеокислоты (фрагменты), ДНК и РНК— связующие протеины
Общие лиганды
Лектины Малые сахара, полисахариды, гликопротеины, гликолипиды
Протеин А и протеин G Неповрежденные антитела
Продолжение таблицы 3.65
Тип лиганда Примеры удерживаемых веществ
Боронаты Катехины и соединения с содержанием остатков сахаров, таких как полисахариды и гликопротеины
Синтетические красители Дегидрогенезис, протеины
Металло-хелаты Аминокислоты, образующие связь с металлами, пептиды или протеины
Элюирование из колонки в аффинной хроматографии может вызываться каким-либо растворимым аффиантом, образующим более прочное соединение с выделяемым веществом, чем привитые аффинные лиганды, изменением ионной силы раствора или температуры. В наиболее общем случае элюирование достигается путем изменения pH раствора, приводящего к подавлению диссоциации функциональных групп, ответственных за образование химической связи между сорбатом и сорбентом. Учитывая селективность сорбции, разделение в аффинной хроматографии часто реализуется по простейшей схеме динамической сорбции: поглощение целевого биологически активного вещества на колонке с удалением в фильтрат несорбируе-мых примесей и его последующее элюирование в виде единственного хроматографического пика. Подобная схема оказывается наиболее удобной при решении препаративных задач.
Высокая селективность аффинных сорбентов может рассматриваться одновременно как достоинство и недостаток метода аффинной хроматографии. Метод часто не имеет аналогов по возможности выделения лабильных биологически активных веществ. Специфика взаимодействия сорбата и сорбента зависит не от химической структуры и молекулярных размеров аффиантов и выделяемых веществ, а от биологической природы. С другой стороны, необходимость использования специальных сорбентов с привитыми аффинными лигандами определенного типа для выделения не только каждой группы веществ, но чаще— каждого индивидуального биологически активного вещества порождает свои специфические трудности, связанные с необходимостью самостоятельного приготовления сорбентов. При этом, учитывая многообразие биологически активных веществ, практически невозможно дать универсальный рецепт приготовления аффианга с носителем. Помимо того, что условия связывания должны учитывать стабильность аффианга, важное значение имеют стери-ческие факторы. Молекулы аффианта должны находиться на таком расстоянии от поверхности носителя, чтобы исключалась возможность их деформации и не возникали стерические препятствия для образования химической связи с выделяемым веществом. Немаловажен и тот факт, что большинство аффиантов сами являются биологически активными веществами и не всегда поддаются длительному хранению. И хотя стабильность целого ряда аффинных
202
Новый справочник химика и технолога
лигандов возрастает в результате их связывания с носителем, приготовленные сорбенты можно хранить ограниченное время при низких температурах и в стерильных условиях [89, с. 20].
Несмотря на объективные недостатки, метод аффинной хроматографии во многих случаях не имеет альтернативы в биохимическом анализе. С методиками приготовления аффинных сорбентов, техникой эксперимента и конкретными примерами практического применения метода можно познакомиться в специальных изданиях [89,96].
Ионообменная хроматография
Развитие метода ионообменной хроматографии связано с созданием органических ионообменных смол (см. п. 3.2.2), обеспечивающих наряду с проявлением способности к ионному обмену в широком интервале составов жидких фаз относительно высокие значения коэффициентов диффузии ионов в фазе ионита. В 50-е годы XX века найдены многочисленные методические решения для разделения самых разнообразных смесей неорганических веществ на катионитах и анионитах, в последующие годы обобщенные в ряде монографий [95, 103, 104]. В случае применения ИОХ для разделения органических веществ важным этапом явилось создание макропористых ионитов и ионитов на целлюлозной и силикагелевой основах, матрица которых оказалась проницаемой для больших органических молекул, таких, например, как аминокислоты.
В качестве стационарной фазы в ИОХ можно использовать любые ионообменные смолы (см. п. 3.2.2). На практике насадками хроматографических колонок чаще всего служат специально синтезированные и предварительно фракционированные по размерам ионообменные смолы для хроматографии (см. табл. 3.22-3.26). Иониты со слабокислотными, слабоосновными и хелатообразующими функциональными группами применяют для решения частных задач, в основном связанных с предварительным избирательным концентрированием отдельных компонентов из большого объема раствора. Неорганические ионо-обменники природного и синтетического происхождения не дали значимого толчка в развитии метода, найдя сравнительно ограниченное применение для селективного выделения отдельных компонентов или группового концентрирования [71, 72]. Основной причиной этого является плохая воспроизводимость результатов и замедленность кинетики ионного обмена. Учитывая, что для насадок хроматографических колонок важное значение имеет не только природа сорбентов, но и степень их дисперсности, налажен выпуск специальных ионообменных смол для хроматографии (табл. 3.66).
Для ионообменной хроматографии характерны два общих подхода при выборе условий разделения. В простейшем случае рассматриваются различия в прочности сорбции ионов, определяемых величиной заряда и их радиусом в соответствии с рядами селективности (см. п. 3.2.2). Для последовательного элюирования по этому механизму в элюентном режиме в качестве элюента выбирается рас
твор, содержащий любой из одноименно заряженных ионов, менее прочно удерживаемый ионитом по сравнению с разделяемыми. Например, для сильнокислотных катионитов универсальными элюентами являются растворы сильных минеральных кислот, так как ионы Н+ являются наименее прочно сорбируемыми катионами. Выбор концентрации Н+ в элюенте основывается на многочисленных справочных данных по коэффициентам распределения в системах иониты — растворы минеральных кислот (см. рис. 3.15, 3.16). При реализации подобной схемы разделения, основанной на ионообменном замещении одноименнозаряженными ионами, присутствующими в элюенте, достигаются минимальные значения Кс, которые может обеспечить система ионит - раствор, так как селективность определяется только реакциями ионного обмена в фазе ионитов. Соответственно увеличение Кс в подобных условиях разделения может быть достигнуто за счет использования ионитов с другими функциональными группами. При одинаковых функциональных группах в случае органических ионитов некоторый эффект дает также изменение степени поперечной сшивки полимерной матрицы. Например, при разделении Cs+ и Na+ на катионите Dowex-50 увеличение доли дивинилбензола в матрице ионита от 4 до 6% приводит к возрастанию Кс от 1,7 до 2.
Значительно большие возможности для повышения Кс в системе ионит - раствор дает использование реакций комплексообразования в жидкой фазе. В этом случае элюирование из ионита достигается не за счет конкурентной сорбции другого иона, а в результате смещения равновесия в растворе в сторону образования (нейтральных или одноименно заряженных с функциональными группами ионита) комплексных соединений. При использовании растворов комплексообразователей в качестве элюентов значения Кс близки к отношению констант устойчивости комплексов разделяемых элементов. Поэтому при отсутствии справочных данных по коэффициентам распределения в растворах того или иного комплексообразователя для ориентировочной оценки значений Кс можно воспользоваться данными по константам устойчивости комплексов разделяемых ионов металлов с лигандами, присутствующими в элюенте.
Об относительных преимуществах комплексообразовательного принципа элюирования в ИОХ можно судить, например, по схеме разделения редкоземельных элементов (TR) на катионитах. В растворах хлорной кислоты, где комплексообразование практически не проявляется, значения Кс для любой пары элементов иттриевой подгруппы близки к единице и значимого разделения даже при высокой эффективности колонки добиться не удается. При элюировании TR-растворами таких комплексообразователей, как с-оксиизобутилат, лактат, ЭДТА, наблюдаются хорошо разрешенные пики всех редкоземельных и актиноидных элементов [103]. Значения Кс для соседней пары элементов изменяются в пределах от 1,3 до 3. Аналогичный подход преимущественно используют и при разделении ионов металлов на анионитах. Анионит предварительно переводят в форму соответствующего лиганда. Последовательное или градиентное элюирование разделяемых эле
Методы разделения и концентрирования
203
ментов достигается за счет уменьшения концентрации лиганда и (или) изменения кислотности раствора, приводящих к уменьшению прочности сорбированных ацидоком-плексов. Наиболее распространены методики разделения элементов в виде хлоридных комплексов на анионитах в СГ -форме в растворах хлороводородной кислоты [103]. Избирательное элюирование за счет различной стабильности комплексов является эффективным приемом и для разделения некоторых органических соединений, способных выступать в роли лигандов, например аминокислот и пептидов, сорбированных анионитом в виде комплексных соединений с медью [107].
Существующие подходы к выбору условий разделения в ионообменной хроматографии не являются альтернативными. Использование эффектов, приводящих к селективному образованию в растворе несорбируемых соединений, позволяет добиться существенно больших значений Кс. Но, во-первых, это далеко не всегда оказывается возможным, как, например, в случае разделения ионов щелочных металлов на катионите или анионов сильных минеральных кислот на анионите. Во-вторых, при высокой разрешающей способности хроматографических колонок увеличение
Кс выше определенного предела не только не приводит к упрощению процедуры разделения, а, наоборот, усложняет технику элюирования. Последнее утверждение, однако, справедливо только при разделении соизмеримых количеств веществ. При решении задач, связанных с концентрированием по отношению к матричным компонентам, всегда оправданы максимальные значения Кс.
При существующем многообразии ионитов и найденных методических решениях методом ИОХ могут быть разделены смеси элементов практически в любых сочетаниях. Исключение составляют только некоторые платиновые металлы, проявляющие способность к образованию кинетически инертных внутрикомплексных соединений с функциональными группами анионитов в качестве лигандов [109].
Основным недостатком ИОХ с классическими насадками в плане аналитического применения является широкий спектр составов элюентов, делающий трудноразрешимой проблему детектирования выделенных веществ в потоке элюата. Возрождение интереса к ионообменной хроматографии в аналитической химии связано с появлением ее специальной аналитической новой модификации — ионной хроматографии (ИХ).
Таблица 3.66
Ионообменные смолы для хроматографии [95]
Пояснения и принятые обозначения.
1. В настоящее время выпускается чрезвычайно много ионообменных смол, привести их все практически невозможно. В данную таблицу включены лишь некоторые органические иониты (хроматографически чистые CG и аналитически чистые AG), пригодные для обычных хроматографических анализов.
2. Bio-Rad — Bio-Rad Laboratories, California, USA; DiaSh — Diamond Shamrock, RadWood City, California, USA; производят иониты высокой чистоты, которые доводятся до аналитической степени чистоты фирмой Bio-Rad Laboratories; DowCh — Dow Chemical Co., Midland, Michigan, USA; производят иониты типа дауэкс, которые очищаются и фракционируются фирмой Bio-Rad Laboratories; эти иониты выпускаются под индексом AG и Bio-Rex; RoHa — Rohm and Haas Co., Philadelphia, Pensylvania, USA, производит иониты типа амберлита, которые поставляет фирма Mallinckordt Chemical Works, St. Louis, Mo., USA; UWS — United Water Softeners, Gunnersbury Avenue, London; обрабатывает иониты типа деасида и зеокарба, поставляемые фиромой Zerolit Ltd. (London).
о. — устойчив к действию окислителей
в. — устойчив к действию востановителей
р.— устойчив к действию органических растворителей
н. — неустойчив к действию окислителей
Ш. — шарики
Ч. — измельченные частицы
мэкв • г-1 — по отношению к сухой смоле
Торговая марка; фирма-изготовитель или поставщик Тип полимерной матрицы Функциональная группа Форма Степень сшивки Характеристика частиц (форма и размер, нм ДО-3) Емкость Устой-чивость Допустимый диапазон pH
мэкв г 1 мэкв-г1
Силъноксилотные катиониты
AG-50, AG-50W (Дауэкс 50, 50W) Bio-Rad (DowCh) Стирольная C6H,SO3' Н+ 1 Ш.: 150-300 5,5 0,4 150 °C 0-14
2 Ш.: 40-75 75-150 150-300 5,2 0,7 (H+,Na+)
4 Ш.:<40 40-75 75-150 150-300 300-840 5,2 1,2
204
Новый справочник химика и технолога
Продолжение таблицы 3.66
Торговая марка; фирма-изготовитель или поставщик Тип полимерной матрицы Функциональная группа Форма Степень сшивки Характеристика частиц (форма и размер, нм ДО'3) Емкость Устой-чивость Допустимый диапазон pH
мэкв-г 1 мэкв-г1
8 Ш.: < 40 40-75 75-150 150-300 300-1200 5,1 1,7 о. в., р.
10 Ш.: 300-1200 5,0 1,9
12 <40 40-75 75-150 150-300 300-840 5,0 2,3
16 Ч.: 300-840
16 Ч.: 40-75 75-150 150-300 4,9 2,6
AG МР-50 Bio-Rad Стирольная c6h,so; H+ Мелкопористый Ч.: 40-75 75-150 150-300 1,6 1,8
Амберлит CG-120 RoHa Стирольная C6H,SO3' 8 Ш.: < 75 75-150 4,3 120 °C 1-14
Биорекс 40 (дуолит С-3) Bio-Rad Фенольная RCH2SOZ H+ Ч.: <40 40-75 75-150 150-300 300-840 2,9 1,2 40 °C; в., р., н.
Зеокарб 225 (зеролит 225) UWS Стирольная C6HsSO3z Na+ 1 Ш.:<75 75-150 150-300 300-1200 4,5 0-14
2 Ш.: < 75 75-150 150-300 300-1200 2,1
4,5 <75
8 Ш.: < 75 75-150 150-300 300-1200
12 Ш.:<75 75-150 150-300 300-1200
20 Ш.:<75 75-150 150-300 300-1200
Среднекислотные катиониты
Биорекс 63 (Дуолит С-63) Bio-Rad (DiaSh) Стирольная C6H5SOZ Na+ Крупнозернистый Ш.: 40-75 75-150 150-300 300-840 6,6 3,1 100 °C; о., в., р. 0-14
Методы разделения и концентрирования
205
Продолжение таблицы 3.66
Торговая марка; фирма-изготовитель или поставщик Тип полимерной матрицы Функциональная группа Форма Степень СШИВКИ Характеристика частиц (форма и размер, нм Ц0-3) Емкость Устой-чивость Допустимый диапазон pH
мэкв•г 1 мэкв-г1
Слабокислотные катиониты
Амберлит CG-50 RoHa Метакриловая RCOO- Н+ 2-3 Ш.:>75 75-150 10 120 4-14
Биорекс 70 (Дуолит CS-101) Bio-Rad Акриловая RCOO Na+ Мелкопористая Ш.: 300-840 10,2 3,3 100 °C о.,в.,р.
Зеокарб 226 (зеролит 226) UWS Акриловая RCOO Н+ 2-5 LLL: > 75 75-150 150-300 300-1200 9-10 3,5 о.,в.,р. 4-14
Силъноосновные аниониты
AG-1 (Дауэкс 1AG) Bio-Rad (DowCh) Стирольная СбН5СН2К+-•(СНз) СГ 1 Ш.: 150-300 3,2-3,6 0,4 50 °C (он-) 0-14
2 Ш.:40 40-75 75-150 150-300 3,5-3,6 0,8-0,9 150 °C (СГ)
4 Ш.: 40-75 300-840 3,5 1,2
8 Ш.: 40-75 300-840 3,2-3,5 1,4
10 LLL: 40-75 3,2 1,5
AG-2 (Дауэкс 2AG) Bio-Rad (DowCh) Стирольная C6H5CH2N+-•(СН3) С2Н5ОН сг 1 Ш.: 300-840 3,2-3,7 1,2 30 °C (ОН) 0-14
8 LLL: 40-75 75-150 150-300 300-840 3,2-3,6 1,4 150 °C (СГ) р.,в.,н.
10 Ш.: 40-75 75-150 150-300 3,0 1,5
AG-2 IK (Дауэкс 21 KAG) Bio-Rad (DowCh) Стирольная адсн^- •(СНз) сг 4 Ш.: 150-300 300-840 840-1000 4,5 1,3 50 °C (ОН) 0-14
AG MP-1 Bio-Rad Стирольная СбН5СН2К+-•(СНз) сг Мелкопористая Ш.: 300-840 Ч.: 40-75 75-150 150-300 1,0
Амберлит CG-400 RoHa Стирольная c6h5ch2n+-•(СНз) сг Ш.:>75 75-100 з,з 1,2 60 °C (ОН) 75 °C (СГ) 0-12
Биорекс 9 Bio-Rad Стирольная Фенилпиридиновая сг Ш.: 300-840 Ч.: 40-75 75-150 150-300 3,7 1,3 38 °C 0
Деасидит FF UWS Стирольная СбНзСНХ-•(СНз) сг 2-3 Ш.:>75 75-150 150-300 300-1200 4,2 1,5 60 °C (ОН’) 0-14
3-5 Ш.: > 75 75-150 150-300 300-1200
206
Новый справочник химика и технолога
Продолжение таблицы 3.66
Торговая марка; фирма-изготовитель или поставщик Тип полимерной матрицы Функциональная группа Форма Степень сшивки Характеристика частиц (форма и размер, нм ДО”3) Емкость Устой-чивость Допустимый диапазон pH
мэкв•г 1 мэкв-г1
7-9 Ш.: > 75 75-150 150-300 300-1200 в.,р.,н.
Среднеосновные аниониты
Биорекс 5 Bio-Rad Полиалкиновая —RN(CH3)2- •С2Н5ОН СГ Ш.: 40-75 75 300-840 8,8 2,8 60 °C о.,в.,р.
(Дуолит АЗО) (DiaSh) Аминовая —RNH-•(СН3)2 3,8
Деасидит Н UWS Стирольная c6h5ch2n+-•(СН3) и (С6Н5СН2)Х-•(СН3)2 СГ 2-3 3-5 7-9 100-300 500-1200 75 75-150 75 75-150 150-300 500-1200
Слабоосновные аниониты
АС-3(дуэкс 3) Bio-Rad (DowCh) Стирольная c6h5n+hr2 СГ 4 Ш.: 300-840 Ч.: 40-75 75-150 2,8 1,9 65 °C 0-7
Амберлит CG-45 RoHa Стирольная Полиамин-ная ОН” Ш.:>75 75-150 5 2 100 °C 0-9
Амберлит CG-4B RoHa Фенольная Аминная ОН” Ч.: 40-75 75-150 10 2,5 40 °C 0-7
Амберлист А-21 RoHa трет-Аминная Мелкопористая Ч.: 50-100 1,7
Деасидит G UWS Стирольная CH2N+H-•(СН2С6Н5) СГ 2-3 Ш.: >75 75-150 150-300 300-1200
3-5 ILL: >75 75-150 150-300 300-1200
7-9 Ш.: > 75 75-150 150-300 300-1200
Деасид М (зеролит М) UWS Аминная СГ 3-5 Ш.:>75 75-150 150-300 300-1200 6
В классическом варианте заполнения колонок ионообменными смолами метод ИОХ наиболее интересен для решения препаративных задач. Переход от классической схемы ИОХ к аналитическому варианту метода — ионной хроматографии потребовал решения целого комплекса проблем. Во-первых, ориентируясь на наиболее универсальный и технически простой способ неселективного определения концентрации разделенных ионов в растворе — измерение
электрической проводимости, было необходимо найти условия ионообменного разделения при минимальной ионной силе растворов, применяемых в качестве элюентов. Последнее при прочих равных условиях может быть достигнуто за счет снижения обменной емкости сорбента в колонке, приводящего к соответствующему уменьшению фактора удерживания. Во-вторых, при снижении обменной емкости одновременно было необходимо повысить эффективность
Методы разделения и концентрирования
207
колонки, т.е. перейти к высокоэффективной хроматографии, чтобы обеспечить разделение нескольких компонентов при одном составе элюента Помимо ионной хроматографии вариант высокоэффективной ИОХ используется для разделе
ния и определения органических веществ. Перечень специальных ионообменных сорбентов для высокоэффективной ИОХ приведен в табл. 3.67 и 3.68, где принятые условные обозначения: S — прочный, W — непрочный.
Таблица 3.67
Тонко дисперсные пористые насадки для анионообменной хроматографии [90]
Название Изготовитель Средний диаметр частиц, нм ц10“3 Прочность Функциональные группы Ионообменная емкость, мэкв • г-1 Примечания
На основе SiO2
Лихросорб 5, 8, 10,13,27, 36 10 S —NR3 0,55 Основа — силикагель с порами размером 10 нм
Микропак АХ 45 5, 10 W Бифункциональный Рекомендуется для разделения компонентов нуклеиновых кислот
Микропак SAX 45 10 S —NR3 Анионит общего назначения. Пригоден для разделения нуклеотидов
Нуклеосил SB 1,2,10, 14, 23, 34,38 5, 10 S —NCH3C1 1 pH = 1-9
Партисил 10 SAX 2, 18, 23, 27,33, 47 10 S —NR3 < 1 (оценка) pH = 1,5-7,5
RSILAN 1,38 5,10 S —NR3 < 1 (оценка)
Синхропак АХ-300 4, 5, 35, 42, 45 10 W Полимерный амин Средний диаметр пор 30 нм, pH = 2-8. Рекомендуется для разделения белков и ферментов. Монослой
Видак 301 ТР 1,3,4, 5, 14,39, 41 10 S —NR3 0,2 pH = 1-9. Силикагель покрыт гидрофобной фазой высокой плотности
Зорбакс SAX 16, 23 7 S —NR3 < 1 (оценка) pH = 2-9
На полимерной основе
Аминокс А-28 А-29 9,45 9 • 2 7 • 10 S —NR3 3,2 Степень сшивки всех насадок этого типа — 8%
Бенсон ВА-Х 1,7, И 7-10 S — NR3C1Z 5 pH = 1-12. Степень сшивки различная (4, 6, 8,10%)
Бенсон BWA 7 7-10 W — NR2HC1“ 5 Выпускаются также насадки со степенью сшивки 10-15%
Хитачи GEL 3011-N 22 10-15 S — NR3 pH = 2-11. Сферические частицы
208
Новый справочник химика и технолога
Таблица 3.68
Тонкодисперсные пористые насадки для катионообменной хроматографии [90]
Название Изготовитель Средний диаметр частиц, нм ц10“3 Прочность Функциональные группы Ионообменная емкость, мэкв • Г1 Примечания
На основе SiO2
Хромеганбонд SCX 6 10 S —SO3H < 1 (оценка) Средний диаметр пор 6 нм
Лихросорб КАТ 1,5,8, Ю, 13, 23, 26 10 S —SO3Z 1,2 Основа — кремнезем с порами диаметром 10 нм
Нуклеосил SA 1,2,14,23,29, 34,38 5, 10 S —SO3Na -1 pH = 1-9
Партисил-10 SCX 1,2, 18,23, 27, 33, 47 10 S —SOZ -1(оценка) pH - 1,5-7,5
RSIL-CAT 1,38 5, 10 S —SO3Z Покрытие — 5%
Видак 401 ТР 1,3,4, 14, 15, 39,41 10 S —SO3Z -1 (оценка)
Зорбакс SCX 16, 23 6-8 S —so3z 5 pH = 2-9
На полимерной основе
Аминекс А А-5 А-7 А-8 А-9 9, 45 13 • 2 7-11 5-8 11,5 • 0,5 S —SO3Z 5 Степень сшивки всех насадок этого типа — 8%
Аминекс НРХ-87 9 9 S —SOZ 5
Векман АА W-1 W-2 W-3 W-4 6 12,5 • 2,5 9,5 • 2,0 8,5 • 2,5 8,5 • 1,0 —SO3Z 5 Степень сшивки всех насадок этого типа — 8%. Применяются преимущественно для разделения аминокислот
Бенсон ВСООН 7 7-10 W —COOH 10
Бенсон ВС-Х 1,7,11 7-10 S —SOzNa 5,2 pH = 1-14. Степень сшивки — от 0,40 до 0,32. Пригоден также для обращенно-фазовой хроматографии
Хромекс катион 15 11 • 1 S —SO3Z 5 Степень сшивки — 8 или 12%. Катионит общего назначения
Гамильтон НС 19 10-15 S —SO3Na 5
Хитачи-гель зоне 22 10-15 W —COOH 5 pH = 2-11. Сферические частицы
Хитачи-гель ЗОН 22 10-15 S —SO3Z
Методы разделения и концентрирования
209
Эксклюзионная хроматография
Эксклюзионная хроматография является одним из методов жидкостно-твердофазной хроматографии, обеспечивающих разделение веществ в зависимости от размеров и формы молекул. Такая возможность открывается при использовании пористых неподвижных фаз с определенными размерами пор, соизмеримыми с размерами молекул. Метод за годы своего существования имел целый ряд названий, которые или полностью тождественны, или имеют несущественные смысловые отличия: гель-проникающая, гель-фильтрационная, молекулярно-ситовая. Первый из вышеперечисленных терминов использовался при анализе органических веществ в органических растворителях, второй — в неорганическом анализе водных растворов, последний, как и современный термин — эксклюзионная, является собирательным понятием. В отличие от других хроматографических методов, использующих различия в химических свойствах разделяемых веществ, проявляющихся при их распределении между стационарной и подвижной фазами, разделение в эксклюзионной хроматографии основано на ситовом эффекте. Растворитель (подвижная фаза) заполняет в колонке как внешний объем между зернами геля, так и внутренний объем пор. Объем растворителя между зернами геля — 1М называют промежуточным, транспортным или мертвым объемом, а внутренний объем пор — Кп рассматривается как объем стационарной фазы. Когда в колонку вводят пробу, содержащую несколько типов ионов или молекул с разными размерами, то они стремятся перейти из подвижной фазы внутрь пор. Такое проникновение обусловлено энтропийным распределением, поскольку концентрация молекул разделяемых веществ в наружном растворе оказывается выше, чем в поровом пространстве. Но оно становится возможным только в том случае, если размеры ионов или молекул меньше диаметра пор.
Обычно все гели характеризуются не одним размером пор, а распределением пор по размерам в определенных пределах. Максимальный размер пор называют верхним пределом исключения, поскольку все ионы или молекулы, имеющие размер больший, чем максимальный диаметр пор, исключаются, т.е. не могут проникнуть в поры из-за пространственного запрета и элюируются одним пиком в объеме Км. Для ионов или молекул с размерами, меньшими минимального размера пор, гель является полностью проницаемым. Разделение же происходит только тех частиц, размеры которых находятся между максимальными и минимальными размерами пор геля. При этом объем удерживания /-го компонента, отвечающего этому условию, V, равен:
К=УМ + К'УП,
К V
где А?' ? ад ;-а- — отношение объема порового про-странства, доступного для молекул /-го компонента, к общему объему пор, Аад — коэффициент адсорбции /-молекул гелем.
Таким образом, в гель-хроматографии последовательно элюируются вещества в порядке уменьшения размеров их молекул или молярных масс. При отсутствии адсорбции и изменения физико-химических свойств растворителя в порах малого диаметра Кяя = 1 и 1\д = Км + Fnj. Соответственно градуировочная кривая для гель-хроматографиче-ского анализа характеризуется тремя участками (рис. 3.15). Для построения градуировочных кривых используют растворы ряда веществ с точно известными размерами молекул, например глюкозы-180, полиэтиленгликолей разной молярной массы от 300 до 4000, альдолазы-147000 и др. Улучшить разрешение хроматографических пиков в гель-хроматографии удается за счет подбора гелей с минимальным интервалом размеров пор, наиболее близких к размерам разделяемых молекул. В этом случае градуировочная кривая будет наиболее пологой.
Рис. 3.15. Общий вид градуировочной кривой
в гель-хроматографии: 1 — область исключения, где оказываются все молекулы, имеющие размер, больший верхнего индекса исключения т2; 2 — область проникновения или разделения, где размеры молекул лежат в интервале от ггц до т2;
3 — область, где происходит полное проникновение в поры геля молекул с размерами менее пц
Все применяемые в гель-хроматографии наполнители колонок традиционно делят на мягкие, полужесткие и жесткие гели [101]. Емкость геля может быть охарактеризована отношением Vn/VM, которое лежит в диапазоне от 0,5 для жестких гелей до 2-3 для мягких гелей. Для мягких гелей характерна, с одной стороны, высокая эффективность и емкость при низких скоростях потока, с другой — высокая степень набухаемости в водных средах и увеличение объема пор при набухании. Соответственно повышение скорости подвижной фазы вызывает деформацию мягких гелей. Они сжимаются, снижается их емкость. Из коммерческих мягких гелей для гель-фильтрации чаще всего применяют декстрановые гели, названные сефадек-сами. Сефадексы — гранулированные поперечно-сшитые декстраны (полисахариды), имеющие в набухшем состоянии гелевую структуру, обладают сильными гидрофильными свойствами. Неионообменные сефадексы содержат все же небольшое количество гидроксильных групп, определяющих адсорбционную емкость сефадексов порядка
210
Новый справочник химика и технолога
0,02-0,04 ммоль-экв • г1. Некоторые липофильные вещества (например, ароматические и циклические) избирательно поглощаются сефадексами, особенно сефадексом LH-20. От необменно поглощенных липофильных веществ сефадексы можно отмыть 1% раствором NaOH (при тем
пературе до 60 °C для слабонабухающих сефадексов от G-10 до G-25) или спиртом.
Марки и характеристики гелей, наиболее часто применяемых в эксклюзионной хроматографии, приведены в табл. 3.69.
Таблица 3.69
Марки и характеристики гелей, применяемых в эксклюзионной хроматографии [97]
Марка гелей; фирма-изготовитель Предел эксклюзии, ММ Диапазон фракционирования веществ по, ММ Поглоще-ние воды, мл • г'1 Удельный объем в колонке, см3 • г-1 Рекомендуемое время полного набухания *, ч Зернение в сухом виде, нм цЮ-3
Пептиды, глобулярные протеины Декстраны (полисахариды)
Sephadex Ph, Швеция
1. G-10 До 700 До 700 1,0 • 0,1 2-3 3(1) 40-120
2. G-15 До 1500 До 1500 1,5 • 0,2 2,5-3,5 3(1) 40-120
3. G-25 5000 1000-5000 100-5000 2,5 • 0,2 4—6 3(1) 100-300 (грубый) 50-150 (средний)
4. G-50 10000 1500-30000 500-10000 5,0 • 0,3 9-11 3(1) 20-80 (тонкий) 10-40 (сверхтонкий)
5. G-75 50000 3000-80000 3000-70000 1000-10000 7,5 • 0,5 12-15 24 (2-3) 40-120 10-40 (сверхтонкий)
6. G-100 100000 4000-150000 4000-100000 1000-100000 10,0 • 1,0 15-20 48 (2-3) 40-120 10 40 (сверхтонкий)
7. G-150 150000 5000-300000 1000-150000 15,0 • 1,5 20-30 18-22 72 (3-5) 40-120 10-40 (сверхтонкий)
8. G-200 200000 5000-150000 5000-250000 1000-200000 20,0 • 2,0 30-40 20-25 72 (3-5) 40-120 10-40 (сверхтонкий)
9. LH-20 4000-6000 2,1 4,0-4,5 3 25-100
Примечания: 1. ММ — молярная масса, г рмоль'1. 2. Рекомендуемое время полного набухания дано для комнатной температуры и (температуры кипящей водяной бани). 1-8. Содержание —ОН групп приблизительно 0,02 ммоль-экв • г”1. 9. Липофильный сефадекс для гель-фильтрации в неводных (полярных органических) растворителях получают алкилированием (введением оксипропиловых групп) сефадекса № 3. Применяется для разделения липидов, стероидов, ароматических и циклических соединений и других высокомолекулярных веществ.
Лигандообменная хроматография
Метод лигандообменной хроматографии основан на специфическом взаимодействии сорбата и стационарной фазы, в результате которого образуются координационные связи металл - лиганд. Сущность метода проще всего иллюстрируется на примере его открытия. (Это последний по времени открытия метод хроматографического разделения веществ в системе жидкость - твердая фаза).
В колонке, заполненной катионитом в NH4-форме, последний переводится в [Си(МН3)2]2+-форму. Через такую колонку фильтруется водный раствор 1,3-диамина, который замещает молекулы аммиака в сорбированном комплексе меди и удерживается в колонке. Для его элюирования достаточно пропустить через колонку концентрированный раствор аммиака. Происходит обратное замещение диамина в комплексах меди на аммиак. Подобная схема замещения лигандов оказалась очень эффективным приемом для разделения различных классов аминов (табл. 3.70).
В качестве лигандов могут выступать и отрицательные ионы. Одним из наиболее важных применений лигандообменной хроматографии является разделение аминокислот, которые координируются в виде депротонированного однозарядного аниона. Остаточный заряд комплексного иона должен оставаться положительным, иначе металл будет вымываться из колонки с катионитом.
Отличительной особенностью лигандообменной хроматографии является чувствительность этого метода к молекулярной структуре лиганда. Незначительные отличия в форме и размерах молекул подчас ведут к значительному изменению прочности их связывания с металлом в ионо-обменнике. В трехмерной сетке ионообменной смолы заместители у донорного атома азота могут в значительной мере затруднять образование координационной связи, тогда как группы, сравнительно далеко отстоящие от донорного атома лиганда, могут взаимодействовать со смолой. Эти взаимодействия приводят к различным порядкам селективности.
Методы разделения и концентрирования
211
Таблица 3.70
Коэффициенты распределения алифатических аминов в системе: 1,4 М раствор аммиака - катионит с сорбированными ионами металлов [109]
Амин рХа* Коэффициент распределения в зависимости от сорбента и сорбированного иона металла
Дауэкс-50 Хелекс
Ni Ni Си Zn
Метиламин 3,38 3,8 5,9 8,9
Диметиламин 3,23 2,5 3,0 9,5
Триметиламин 4,20 1,1 1,3 —
Этиламин 3,37 3,8 7,1 9,8
Диэтил-амин 3,07 3,4 4,1 6,8
Триэтил-амин 3,13 2,9 3,0 5,7
н-Пропил-амин 3,47 12,5 3,8 7,7 9,6
г/уо-Пропил-амин 3,37 8,2 2,7 4,5 7,5
//-Бутиламин 3,40 21,6 6,6 13,4 18,9
изо-Бутил-амин 3,51 13,4 4,1 9,1 12,9
в/иор-Бутил-амин 3,44 11,6 3,4 5,9 10,2
/м/?е»/-Бугил-амин 3,55 9,1 2,9 3,4 7,1
Ярче всего селективность лигандного обмена проявляется в распознавании структуры оптически активных лигандов. Подлинным триумфом лигандообменной хроматографии явилось разделение оптических изомеров аминокислот.
Сравнительно недавно стали применять лигандный обмен в подвижной фазе, перемещающейся по колонке. В этом случае неподвижная фаза не обязательно должна быть ионообменником, она может быть и неполярной, как, например, силикагель с привитыми октадецильными группами, являющийся самой распространенной насадкой в обращенно-фазовой хроматографии. Ионы металла добавляют в подвижную фазу, и если при этом лиганды образуют незаряженные комплексы, то последние распределяются между неподвижной и подвижной фазами. Аминокислоты относятся именно к таким лигандам, и поэтому метод лигандного обмена в подвижной фазе в настоящее время широко используется для анализа смесей энантиомеров аминокислот, т.е. L- и D- оптических изомеров.
В настоящее время предложено следующее обобщенное определение лигандообменной хроматографии: «Лигандообменная хроматография — это процесс, в котором
комплексообразующие соединения разделяются в результате образования и последующего разрыва лабильных координационных связей с центральным атомом металла, влияющих на их распределение между подвижной и неподвижной фазами. Разделение лигандов происходит вследствие обмена их мест у атома металла. Обмен может происходить как в неподвижной, так и в подвижной фазе» [75].
Одним из последних вариантов лигандообменной хроматографии явилась «аффинная хроматография на хелатах металлов», представляющая собой гибрид лигандообменной и аффинной хроматографии. В металло-хелат-аффинной хроматографии активные центры насадок, на которых происходит сорбция, обычно представляют собой систему: ион переходного металла - иминодиацетатная группа, связанная с матрицей сорбента достаточно длинной группировкой. Связывание белка с системой металл -иминодиацетат - матрица происходит благодаря образованию координационных связей. Обычно процесс проводят в интервале значений pH от 6 (ацетатный буфер) до 8 (фосфатный буфер). Металло-хелат-аффинная хроматография представляет в первую очередь интерес как препаративный метод биохимии.
Подробные сведения о лигандообменной хроматографии и областях ее применения можно найти в [89, 109].
Жидкостно-жидкостная хроматография
Жидкостно-жидкостная хроматография (ЖЖХ) основана на различиях в распределении веществ между двумя фазами, находящимися в жидком состоянии. Практически неограниченный выбор сочетаний несмешивающихся жидкостей делает метод ЖЖХ наиболее универсальным при поиске систем с максимальными коэффициентами селективности для разделяемых веществ. В зависимости от того, полярная или неполярная фаза является стационарной, различают два варианта ЖЖХ: нормально-фазный и обращенно-фазный. Использование системы из двух жидких фаз в том или ином варианте ЖЖХ определяется значениями Kd- В качестве стационарной выбирают фазу, в которую преимущественно переходят разделяемые вещества. На размытие хроматографических пиков и, соответственно, их разрешение существенное влияние в ЖЖХ оказывает вязкость применяемых жидких фаз. В первую очередь это относится к подвижной фазе, так как диффузионные ограничения со стороны стационарной фазы устраняются изменением толщины ее слоя на носителе.
Помимо характеристических параметров системы фаз, непосредственно влияющих на разрешение хроматографических пиков, в ЖЖХ важную роль играет взаимная растворимость. Взаимная растворимость довольно существенна, и ею нельзя пренебречь без риска получить невоспроизводимые результаты. Для ЖЖХ характерно постепенное изменение соотношения объемов стационарной и подвижной фаз в хроматографической колонке в процессе элюирования, если подвижная фаза предварительно не насыщена стационарной. Соответственно происходит «загрязнение» элюата стационарной фазой. С последним приходится считаться при проведении разделения
212
Новый справочник химика и технолога
не только в препаративных целях, но и в аналитических, когда примеси стационарной фазы в элюате влияют на значение аналитического сигнала при последующем определении выделенных веществ. Для устранения нежелательных эффектов, связанных с взаимной растворимостью фаз, процедура разделения в ЖЖХ в общем случае включает операции предварительного насыщения элюента стационарной фазой и очистки от нее элюата. Простейшим методическим приемом насыщения элюента является установка дополнительной периодически заменяемой колонки с той же стационарной фазой на линии его подачи в основную разделительную колонку. Для очистки элюата на выходе из основной колонки подключается другая дополнительная колонка, поглощающая селективно по отношению к разделяемым веществам примеси стационарной фазы. Размеры этой колонки и мертвые объемы соединительных устройств должны быть минимальными, чтобы исключить дополнительное размытие хроматографических пиков.
Носители стационарной фазы в ЖЖХ выбирают в зависимости от способности к удерживанию жидкостей различной полярности. Прочность удерживания определяется их индивидуальными химическими свойствами и структурой. Максимальная прочность удерживания характерна для носителей, набухающих в стационарной фазе. Но в этом случае под влиянием сил сольватационного взаимодействия с носителем происходит изменение физико-химических свойств стационарной фазы, проявляющееся в снижении значений коэффициентов распределения и скорости диффузии. Замедленность диффузионных процессов приводит к резкому увеличению ширины хроматографических пиков. Поэтому в качестве носителей обычно применяют пористые вещества, не набухающие в стационарной фазе.
Чем больше общая пористость носителя, тем больший объем стационарной фазы он может удерживать. Последнее утверждение справедливо при условии, что размер пор несоизмеримо меньше размера частиц носителя, т.е. исключается механическое вытеснение стационарной фазы из порового пространства потоком подвижной фазы. У объемно-пористых носителей удельный объем пор составляет 1-1,3 мл • г1, что позволяет довести соотношение объемов стационарной и подвижной фаз до значения, близкого к 1. У поверхностно-пористых носителей суммарный объем пор существенно ниже, и максимальный объем, заполненный стационарной фазой, в колонке не превышает нескольких процентов от объема подвижной фазы. Оптимальный диаметр пор носителя находится в интервале 10-50 нм. При большем диаметре пор неподвижная фаза начинает вымываться, причем тем интенсивнее, чем выше линейная скорость подвижной фазы. Уменьшение диаметра пор ниже 10 нм может приводить к появлению ситового эффекта. При этом вещества с большими размерами молекул будут вымываться вместе с не-удерживаемыми компонентами независимо от значений коэффициентов распределения в используемой системе жидких фаз.
Носитель должен быть инертным по отношению не только к стационарной фазе, но и к подвижной и к самим
разделяемым веществам. Химическая стойкость носителя во многом определяет сферу его применимости. В ЖЖХ часто используют одновременно растворы различного состава. Универсальный носитель должен выдерживать воздействие любых агрессивных сред, применяемых при хроматографическом разделении. Нежелательные побочные эффекты могут вызывать процессы адсорбции разделяемых веществ на поверхности носителя. Обратимая адсорбция влияет на межфазное распределение веществ в хроматографической колонке и может приводить' к дополнительному размытию хроматографических пиков. Необратимая адсорбция вызывает неконтролируемые потери разделяемых веществ. Носители, как и сорбенты, обычно применяют в хроматографии в виде порошков определенной степени дисперсности. Критерии выбора размеров частиц общие для хроматографических методов. Улучшение кинетики межфазного обмена достигается использованием поверхностно-пористых носителей. Для решения аналитических задач, связанных с концентрированием примесей из большого объема раствора, иногда применяют носители в виде пористых блоков.
Нанесение неподвижной фазы на носитель и заполнение колонок в ЖЖХ можно проводить двумя приемами. В первом случае в зависимости от размера частиц носителя и его свойств колонку заполняют сухим или суспензионным способом, а затем через слой носителя пропускают жидкость, которая будет служить стационарной фазой. Жидкость может быть использована в чистом виде, но чаще ее разбавляют легколетучим растворителем. Затем насадку сушат током воздуха или азота для удаления растворителя и промывают подвижной фазой. Содержание стационарной фазы в колонке определяется ее концентрацией в исходном растворе. Максимальный объем стационарной фазы достигается при последовательном пропускании через колонку неразбавленной подвижной фазы и подвижной фазы того же состава, что используется при последующем разделении.
Во втором случае нанесение стационарных жидких фаз на носитель производят вне колонки. Носитель смешивают с раствором стационарной фазы в легколетучем растворителе, смесь осторожно перемешивают током воздуха до полного удаления (испарения) растворителя. Хроматографическую колонку заполняют готовой насадкой, содержащей определенное количество стационарной фазы.
Объем стационарной фазы выбирают в зависимости от характера решаемых задач. Если решают препаративные задачи или выделяют из раствора матричные компоненты при высоких значениях Кс, используют колонки с максимальным объемом стационарной фазы. Когда определяющую роль играет разрешение хроматографических пиков, существенным становится нахождение оптимального объема стационарной фазы для применяемого носителя. Любое отклонение от оптимума ведет к ухудшению разделения. Большая толщина пленки стационарной фазы способствует агрегации частиц носителя, нарушению однородности потока подвижной фазы. Снижение количества стационарной фазы ниже определенного предела приво
Методы разделения и концентрирования
213
дит к уменьшению поверхности межфазного контакта и ухудшению хроматографических характеристик колонок. Обычно для объемно-пористых носителей оптимальный объем стационарной фазы составляет 5-10% от общего объема колонки. В случае поверхностно-пористых носителей— 0,5-1,5%.
ЖЖЧ со стационарной полярной фазой
ЖЖХ со стационарной полярной фазой обеспечивает возможность разделения веществ менее полярных, чем стационарная фаза, и более полярных, чем подвижная. Полярность жидкостей оценивают по эмпирическим шкалам или по параметру растворимости Гильденбрандта [ПО]. Примером шкалы относительной полярности являются так называемые миксотропные серии Мачека и Прохазки. Одна из таких серий в порядке убывания полярности выглядит следующим образом: вода > формамид > ацетонитрил > метанол > уксусная кислота > этанол > изопропанол > ацетон > диоксан > тетрагидрофуран > метилэтилкетон > фенол > бутанол > пентанол > этилацетат, этиловый эфир > бутилацетат > нитрометан > изопропиловый эфир > ди-хлорметан > трихлорметан > 1,2-дихлорэтан > бромбензол > этилбромид > бензол > хлористый пропил > толуол > ксилол > тетрахлорметан > сероуглерод > циклогексан > гексан > гептан > керосин. Исходя из приведенной шкалы можно приблизительно выбрать условия разделения. Если полярности разделяемых компонентов близки к полярности подвижной фазы, то Ке для любого из них окажется близким к нулю и разделения добиться практически невозможно. Если близки полярности разделяемых компонентов и стационарной фазы, то значения Ке окажутся далеко за пределами оптимального интервала 1 < Ке < 10. Поскольку далеко не всегда удается подобрать систему из двух несме-шивающихся растворителей, обеспечивающих оптимальное значение Ке, часто в качестве подвижной фазы используют двухкомпонентную смесь. При этом достигается эффект уменьшения различий в полярности подвижной и стационарной фаз и соответственно возможность направленного изменения значений Ке для разделяемых веществ.
Количественной характеристикой полярности растворителя служит параметр растворимости Гильденбрандта f, о котором подробнее см. в описании ЖАХ.
Носителями стационарных фаз в данном варианте хроматографии обычно служат гидрофильные вещества, поскольку именно воду или водные растворы чаще всего используют в качестве стационарной фазы. Наиболее распространены носители, изготовленные на основе древесной или хлопковой целлюлозы, силикагеля и кремниевой кислоты, оксида алюминия, кизельгура, пористых стекол и т. п.
ЖЖХ с обращенными фазами
ЖЖХ со стационарной неполярной фазой вошла в литературу как ЖЖХ с обращенными фазами. Последнее название является данью истории развфития хроматографических методов. Понятие обращенные фазы подчерки
вает отличие от ранее возникшего направления ЖЖХ со стационарной полярной фазой. ЖЖХ с обращенными фазами имеет еще одно название — экстракционная хроматография, возникшее в период развития этого направления применительно к разделению неорганических веществ, когда отправными данными при выборе условий хроматографического разделения служили данные статической экстракции (п. 3.2.5). Иногда экстракционную хроматографию с позицией разделения органических соединений отождествляют с одним из частных случаев ЖЖХ со стационарной неполярной фазой — ион-парной хроматографией (ИПХ) [111]. В данном случае речь идет о выделении самостоятельного метода на основании механизма образования экстрагируемого соединения в стационарной фазе по аналогии с выделением ионообменной хроматографии в системе жидкость - твердое тело. Этот механизм реализуется, например, если в водном растворе соединение диссоциировано на противоионы, а в органическую фазу переходит в молекулярной форме или если один из противоионов, образующих экстрагируемое соединение, находится в стационарной фазе и удерживает другой ион из подвижной фазы. Но эти случаи далеко не исчерпывают возможные механизмы образования экстрагируемых соединений в системах фаз, используемых в экстракционной хроматографии. Поэтому понятия экстракционная хроматография и ион-парная хроматография далеко не тождественны.
При экстракции неорганических веществ межфазный переход в большинстве случаев связан с их химическим взаимодействием с неполярной фазой. Поэтому критерии относительной полярности стационарной и подвижной фаз и разделяемых веществ в этом случае оказываются, как правило, неприемлемыми. Химические соединения, образуемые разделяемыми веществами в фазе экстрагента, обычно не могут быть выделены в свободном состоянии, и априорная информация об их полярности отсутствует. Выбор экстракционных систем для осуществления экстракционнохроматографического процесса, помимо общих требований минимальной взаимной растворимости фаз и максимальных значений Кс, основывается на двух предпосылках: значения коэффициентов распределения разделяемых веществ при нанесении на колонку должны отвечать условию Ке > 10, скорость химической реакции межфазного перехода не должна быть лимитирующей стадией в установлении экстракционного равновесия в хроматографической колонке. Иными словами, для экстракционно-хроматографического разделения приемлемы системы, обеспечивающие высокие значения Ке и не имеющие кинетических ограничений.
Последнему условию практически без исключения удовлетворяют кислородсодержащие экстрагенты. В случае аминов ограничения появляются при разделении платиновых металлов, когда нельзя исключить вероятность образования в фазе экстрагента внутрикомплексных соединений с замедленной кинетикой обмена лигандами. Для серосодержащих и хелатообразующих экстрагентов, как правило, характерна замедленность стадии реэкстракции. Поэтому их применение в условиях экстракционной хро
214
Новый справочник химика и технолога
матографии ограничивается решением аналитических задач индивидуального и группового концентрирования из растворов без последующего избирательного элюирования.
При выборе условий разделения органических веществ в ЖЖХ с обращенными фазами используют критерии, аналогичные ЖЖХ со стационарной полярной фазой. В отличие от последней разделяемые вещества по полярности должны быть ближе к неполярной фазе, выступающей в данном случае в качестве стационарной. Оба направления ЖЖХ взаимно дополняют друг друга. ЖЖХ со стационарной полярной фазой обеспечивает возможность разделения полярных соединений, ЖЖХ с обращенными фазами — менее полярных.
Носителями стационарных фаз в этом варианте хроматографии служат неполярные химически инертные полимерные материалы, чаще всего фторсодержащие. Кроме того, носители фаз получают путем модифицирования поверхности силикагеля, заключающемся в замещении силанольных групп Si-OH органофильными группами при обработке моно-, ди- и трихлорсиланами. С появлением обращенно-фазовой ЖАХ интерес к ЖЖХ с обращенными фазами снизился. Интенсивно развивающимся направлением в ЖЖХ становится ее противоточный центрифужный вариант.
Хроматография в системе жидкость - газ
Для системы газ - жидкость вариант хроматографического процесса — газожидкостная хроматография (ГЖХ) — осуществляется в условиях подвижной газовой фазы и стационарной жидкой. Соответственно разделяемые вещества при температуре хроматографической колонки находятся в газообразном состоянии. Различия в межфазном распределении разделяемых веществ между жидкой и газовой фазой определяются разницей в энергии их межмолекулярного взаимодействия в жидкой фазе. Поэтому селективность метода целиком зависит от состава стационарной фазы, а прочность удерживания в хроматографической колонке регулируется изменением температуры. Интервал рабочих температур в ГЖХ простирается от -196 до 400 °C, но крайние значения, особенно в области низких температур, используют сравнительно редко. Верхний предел температур определяется термической устойчивостью стационарных жидких фаз и разделяемых органических веществ.
К стационарным фазам, применяемым в газожидкостной хроматографии, предъявляется ряд общих требований:
— высокая термическая стойкость и низкое давление пара при рабочих температурах;
— химическая инертность по отношению к разделяемым веществам;
— максимальное проявление различий в энергии межмолекулярного взаимодействия с разделяемым веществом.
Современная ГЖХ наиболее распространена в капиллярном варианте. В аналитической практике вполне обычным является использование капиллярных колонок до 100 и более метров длиной с эффективностью 105 тарелок. Но достаточно широко представлен и обычный колоночный вариант метода. В качестве носителей стационарной жидкой фазы в ГЖХ используют практически те же материа
лы, что и ЖЖХ. Поскольку ГЖХ наряду с газоадсорбционной хроматографией является основой аналитического метода газовой хроматографии, особенности методов ГЖХ и ГАХ подробно рассмотрены в соответствующем разделе.
Жидкостно-газовая хроматография
Подобно тому, как в ЖЖХ перемена мест стационарной и подвижной фаз привела к появлению нового хроматографического метода, обращение фаз оказалось возможным и для системы газ - жидкость. Жидкостно-газовая хроматография является одним из самых молодых хроматографических методов. Экспериментальное доказательство принципиальной осуществимости ЖГХ-процесса и первые примеры его практического применения относятся к 1982 году [112]. Оно несколько опередило теоретические предсказания принципиальной возможности осуществления подобного процесса и обоснования его перспективности [113]. К настоящему времени можно считать доказанным, что данный метод является эффективным способом пробоподготовки при анализе «постоянных» газов, растворенных в воде и водных растворах.
3.2.9.6. Хроматографические методы анализа
Согласно классификации, приведенной в табл. 3.71, важнейшую группу хроматографических методов составляют методы анализа. Поэтому методы газовой, жидкостной, сверхкритической флюидной, ионной и хиральной хроматографии рассматриваются в специальном разделе, посвященном аналитической хроматографии. При этом учитывается и тот факт, что понимание и трактовки хроматографии специалистами в области методов разделения веществ и в области хроматографических методов анализа далеко не идентичны и приводимые ими сведения взаимно дополняют друг друга и позволяют лучше понять специфику хроматографии [114, 115].
3.3. Мембранные методы разделения веществ
3.3.1. Внутригрупповая классификация методов
Согласно общей групповой классификации методов разделения, основанной на принципах фазовых превращений и межфазных переходов, третья группа включает методы, в которых разделение достигается за счет различных свойств, проявляемых веществами при их индуцированном, т.е. вызванном воздействием каких-либо сил, переносе из одной фазы в другую через разделяющую их третью фазу. Промежуточная фаза является перегородкой между двумя первыми или мембраной {мембрана в переводе с латинского и означает перепонка); соответственно методы разделения, происходящего в этой фазе, называются мембранными. Как и в двух предыдущих случаях, основным критерием внутригрупповой классификации является агрегатное состояние фаз, участвующих в процессе разделения. Специфическим классификационным признаком для этой группы является движущая сила процесса межфазного переноса веществ (таблица 3.71).
Методы разделения и концентрирования
215
Таблица 3.71
Классификация мембранных методов разделения
Система фаз Движущая сила процесса и обобщенные названия группы методов
Градиент химического потенциала (диффузионные методы) Градиент электрохимического потенциала (электро-мембранные методы) Градиент давления (баромембранные методы)
Жидкость -жидкость Диализ через жидкие мембраны Электродиализ через жидкие мембраны —
Жидкость -твердое тело -жидкость Диализ, доннанов-ский диализ Электродиализ, электроосмос Микрофильтрация, ультрафильтрация, обратный осмос, пьезодиализ
Жидкость -твердое тело - газ Испарение через мембрану — —
Газ-твердое тело — газ Газодиффу зионное разделение — Микрофильтрация, ультрафильтрация
Приведенная классификация позволяет рассматривать мембранные методы в одной логической цепи с другими методами разделения. Но если переход от методов, основанных на однократном равновесном распределении веществ между фазами, к хроматографическим оправдан в первую очередь с точки зрения значительно более высоких коэффициентов разделения, то мембранные методы, наоборот, обычно не давая существенных преимуществ по чистоте разделения по сравнению с одноступенчатыми процессами, как правило, позволяют добиться большей производительности на единицу количества разделяющей фазы. Поэтому многочисленные исследования в области мембранных методов разделения в подавляющем большинстве случаев проводятся с перспективой создания новых технологических процессов.
Преимущества мембранных методов — простота и компактность аппаратурного оформления, экологическая чистота, непрерывность процесса, возможность его автоматизации — наиболее полно проявляются в крупномасштабном производстве. Именно эти преимущества привели к быстрому развитию и использованию мембранных технологий. В то же время мембранные методы разделения привлекли внимание и химиков-аналитиков. Это внимание связано с отсутствием альтернатив мембранным методам при решении такой аналитической задачи, как пробоподго-товка в потоке. Последняя приобрела особую важность в связи с решением проблемы создания непрерывных авто
матизированных методов аналитического контроля воздушных и водных сред. С позиций аналитического применения наибольшее внимание привлекли диффузионные методы.
3.3.2. Диффузионные мембранные методы
Газодиффузионное выделение в проточных методах анализа может осуществляться в следующих системах фаз: газ - газ, газ - жидкость, жидкость - жидкость. Проблема пробоподготовки в потоке решает две самостоятельные задачи. Во-первых, это непрерывное выделение целевых компонентов из контролируемой среды в среду, максимально удобную для их определения с помощью проточных детекторов. Во-вторых, это непрерывная корректировка состава контролируемой среды (или генерирование растворов реагентов). В последнем случае это чаще всего создание необходимой кислотности для образования аналитических форм целевых компонентов. Для решения обеих задач наиболее предпочтительным является метод газовой диффузии через химически инертные полимерные мембраны. Для непрерывного пробоотбора обычно применяются микропористые политетрафторэтиленовые или полипропиленовые мембраны. Для корректировки кислотности раствора за счет диффузии уксусной кислоты или аммиака более надежны сплошные мембраны из силиконовой резины. Выделение веществ из газовой фазы в водные растворы осуществляется в специальных устройствах— проницаемых денудорах (скрубберах) [116]. Функционирование проницаемых денудоров основано на том, что определяемые вещества диффундируют через газопроницаемую мембрану, отделяющую поток газовой фазы от потока жидкости. В настоящее время применяются два типа скрубберов и проницаемых денудоров. К первому типу относится трубочный денудор, в котором трубка из микропористой мембраны типа полое волокно помещена внутрь трубки большего диаметра — корпуса денудора. Анализируемый газ пропускается между внутренней и внешней трубками, а жидкая фаза прокачивается через внутренний канал полого волокна. При этом газ диффундирует через мембрану и поглощается в поток водного раствора, направляемый непосредственно в проточный детектор.
Второй тип — планарный денудор, состоящий из двух горизонтально расположенных камер, разделенных микропористой мембраной. В этом случае газовый поток подается через верхнюю камеру, имеющую большие размеры. Для минимизации турбулентности потока газовой фазы каналы подачи и отвода газовой фазы располагаются под углом 30° по отношению к мембране, а для увеличения диффузионного потока выделяемых веществ из газовой фазы в жидкую каналы потоков газа и жидкости располагаются под разными углами. После выделения целевого компонента из газовой фазы в принимающий водный раствор последний из второй камеры денудора направляется в проточный детектор.
Независимо от конструкции денудор (скруббер) может быть использован для выделения микропримесей реакци
216
Новый справочник химика и технолога
онноспособных неорганических газов из воздуха (таких как диоксид серы, азотистая кислота, хлороводород и т. п.). Для определения образовавшихся в поглощающем растворе ионов применяется, например, метод ионной хроматографии. Предложенная система представляла интерес для определения загрязнителей в атмосферном воздухе на уровне долей ppb (1 • ПГ3 млн'1). Диффузия определяемых веществ через мембрану в слой поглотительного раствора используется в специальных пробоотборниках, применяемых для пассивного мониторинга атмосферного воздуха. Основные закономерности пассивного пробоотбора в таких системах рассмотрены в обзоре Н.Ш. Больберга [117].
Кроме того, одной из важнейших задач, решаемых с помощью газодиффузионных методов при анализе воздуха, является генерирование стандартных газовых смесей, необходимых для градуировки газоанализаторов [118]. Наиболее удобной схемой подобного генерирования, когда речь идет о введении заданной добавки определяемого вещества в воздух, является использование ампул из газопроницаемого полимера (обычно политетрафтроэтилена), заполненных раствором дозируемого вещества необходимого состава для выделения его газообразной формы. Эти ампулы помещаются в поток воздуха. Содержание дозируемой примеси в газовой смеси, как правило, рассчитывается по убыли массы ампулы за заданный промежуток времени при фиксированной скорости потока газа [119].
Еще одной областью аналитического применения газодиффузионного метода, активно развиваемого в последние годы, является парофазный анализ. Здесь пористые гидрофобные мембраны используются для отделения газового экстракта от анализируемого водного раствора [119].
Во всех рассматриваемых выше примерах газодиффузионные методы используются для выделения газообразных или образующих газообразные формы веществ без разделения их с другими газообразными компонентами. В то же время газопроницаемые мембраны и, в частности, органические полимеры обладают селективностью к определенным газообразным соединениям (табл. 3.72).
Таблица 3.72
Коэффициенты разделения компонентов бинарных газовых смесей в зависимости от природы мембраны
Материал мембраны Коэффициент разделения
HeN2 Не-СН4 o2-n2 H2-N2 СО2-О2
Силиконовый каучук 1,5 0,39 2,3
Натуральный каучук 3,4 2,8 6,4 5,9
Полиэтилен 5,3 2,3 25
Поликарбонат 15 19 5,9 0,5
Фторопласт 25 44 3,1 9,4 2,8
За последние годы наблюдается существенный прогресс в синтезе селективно проницаемых полимеров для изготовления мембран [116], тем не менее достигнутые коэффициенты разделения в пределах одного порядка недостаточны для аналитического применения, где задачи разделения газообразных соединений в микромасштабах легко решаются методами газовой хроматографии. Практически единственным исключением селективно проницаемых газодиффузионных мембран, применяемых в аналитических целях, являются металлические мембраны на основе палладия и его сплавов. Проницаемость таких мембран по отношению к водороду, на несколько порядков превышающая проницаемость по отношению к остальным газам, позволяет получать водород более чистый, чем при электролитическом способе. Соответственно, подобные мембраны используются в препаративных целях в лабораторных генераторах водорода.
Наряду с общей высокой проницаемостью для водорода палладий и его сплавы проявляют селективную проницаемость к изотопам водорода — протию и дейтерию (см. табл. 3.73).
Таблица 3.73
Коэффициенты проницаемости сплавов по протию РН2 и дейтерию и коэффициенты разделения
изотопов водорода а [134]
Материал мембраны T,K Коэффициенты проницаемости (P) и разделения (a)
Рн2’ моль/ м-с-Па PDP моль/ м -с-Па a
Сталь 25 673 773 973 5,4-10'11 3,9-10'11 1,38
Ni 673 773 973 2,68-10'11 8,05-10'” 1,62-10'11 4,85-10'” 1,65 1,65
Pd 673 773 973 1,29-10’8 1,89-10'8 3,62-10'8 0,83-1О'8 1,22-10’8 2,41-10'8 1,55 1,55 1,50
PdNi-10 673 773 973 6,24-109 10,3-10'9 2,17-10'8 4,43-1О'9 7,4-10'9 1,56-10'8 1,41 1,39 1,39
PdAg-15 673 773 973 1,73-10'8 2,31-Ю'8 4,37-10'8 1,17-10'8 1,48-10'8 2,74-10'8 1,48 1,56 1,60
Сплав В-1 (Pd, Ag, Au, Pt, Ni, Al) 673 773 973 1,35-10'8 1,75-10“8 2,85-1О'8 0,88-10'8 1,15-10'8 1,97-10'8 1,52 1,52 1,44
Сплав B-2 (Pd, Ag, In, Y) 673 773 973 1,69-1 О'8 2,40-1О'8 3,66-10'8
Из диффузионных методов применяемых для аналитических целей, в системе жидкость - твердое тело - жидкость, необходимо отметить доннановский диализ через ионооб
Методы разделения и концентрирования
217
менные мембраны, по своей структуре близкие к ионообменным смолам. Сведения о выпускаемых промышленностью ионообменных мембранах можно найти в [97].
В отличие от обычного диализа, движущей силой которого является концентрационный градиент, в доннанов-ском диализе движущей силой процесса переноса противоионов является разность доннановских потенциалов, возникающих на границе раздела фаз со стороны отдающего и принимающего растворов [121]. Подбирая состав принимающего раствора, так называемого реверсивного электролита, можно создать условия для переноса каких-либо ионов против градиента концентрации, тем самым добиваясь их концентрирования. В состав реверсивного раствора электролита вводят ионы, не мешающие последующему анализу. В табл. 3.74 приведены результаты работ по доннановскому диализу с целью разделения и концентрирования микропримесей из водных растворов.
Таблица 3.74
Применение доннановского диализа в аналитической химии [122]
Решаемая задача Реверсивный электролит Коэффициент концентрирования
Разделение Са, Со, Cu(II) MgSO4 (или LiCl) + ЭДТА —
Разделение Pt, Rh, Pd, Ir в виде хлоридных комплексов 0,5 М NH4C1 —
Концентрирование Cd, Pb, Zn, Cu(II) 0,2 М MgSO4 60-70
Концентрирование Co, Cu(II) 2 М MgSO4 54 ±5
Концентрирование Cu, Pb, Zn, Co из растворов, содержащих глицин и гуминовые кислоты 0,6 М Na2SO4 2,5 ± 0,2
Концентрирование СГ, POj“, no:, SO*’ 0,04 М Na2CO3 + 0,16М NaHCO3 14-16
Концентрирование Cu, Cd, Pb, Zn с отделением от ПАВ, ком-плексообразователей и мешающих электро-активных веществ 0,2 М MgSO4 + 5 • Ю^М A12(SO4)3 50-70
Необходимо отметить, что в последующие годы этот метод не нашел развития. Учитывая, что твердофазные ионообменные мембраны имеют селективность, ограниченную знаками заряда разделяемых ионов, поиски селективно проницаемых мембран для разделения веществ в растворах были продолжены среди класса жидких мембран (ЖМ), которые могут рассматриваться в качестве ближайших аналогов биологических мембран.
Поскольку индивидуальность веществ в наибольшей степени проявляется в химических реакциях, то прежде всего проявление избирательности характерно именно для мембран, проницаемость которых определяется образованием новых соединений в фазе мембраны. Само понятие селективность в описании мембранных процессов было впервые использовано для характеристики биологических мембран, которых можно рассматривать как важнейшую группу мембран реакционного типа. Принципы функционирования биологических мембран рассматриваются в ряде монографий [123-125]. Обратим внимание только на аналитический аспект их применения.
Развивается новое направление в определении общей загрязненности природных вод токсичными веществами по реакции на них живых организмов. Учитывая, что обмен веществ в жизнедеятельности организмов определяется функционированием клеточных мембран, это направление можно рассматривать как один из возможных вариантов использования биологических мембран в химическом анализе. Так, в обзоре [126] рассматриваются варианты осуществления автоматизированного измерения качества воды с использованием рыб (форель, карп, золотая рыбка), высокочувствительных к присутствию в воде токсичных веществ [127].
Диффузионный мембранный метод в системе жидкость- твердое тело - газ получил название испарение через мембрану или первапорация. Метод основан на селективной проницаемости некоторых материалов для различных компонентов жидких смесей. Явление селективной проницаемости впервые обнаружено на каучуковых мембранах для смесей углеводород - спирт. Отличительной особенностью процесса мембранного испарения от других мембранных процессов является переход проникающих через мембрану веществ из жидкого состояния в парообразное, для чего требуется подвод к системе энергии, по меньшей мере равной теплоте испарения пермеата. Из этого следует, что испарение через мембрану может быть использовано практически лишь тогда, когда селективность переноса гораздо выше, чем при простом испарении, в частности, для разделения азеотропных и близко кипящих смесей. Движущей силой процесса мембранного испарения является разность химических потенциалов по обе стороны мембраны. Для поддержания химического потенциала на достаточно высоком уровне необходимо предотвратить конденсацию пермеата на поверхности мембраны со стороны пара. Это достигается непрерывным отводом пара, обдувом инертным газом или вакуумированием.
Коэффициент разделения для данного метода выражается как
где ХА и Хв — концентрации компонентов А и В в исходной смеси, а УА и YB — концентрации тех же компонентов в проникшем через мембрану паре. Об эффективности разделения жидких смесей этим методом в зависимости от природы мембраны можно судить по данным, приведнным в табл. 3.75.
218
Новый справочник химика и технолога
Таблица 3.75
Возможности разделения жидких смесей методом первапорации [128]
Раствор ХА Ya Материал мембраны tx, °C Проницаемость G, л • м~2 • ч“!
Вода-изопро-панол 12 50 81 89 Целлофан 80 80 1,9 4,8
Вода-метанол 18 63 То же 80 4,6
Вода-гидразин 77 30 92 59 То же 40 40 2,4 1,0
Вода-диметил-гидразин 38 90 99,5 99,5 Ацетилцеллюлоза 40 40 6,0 3,3
Бензол -изопропанол 10 90 48 96 Полиэтилен То же 60 60 0,2 3,2
н-Гексан - н-октан 42,2 78 То же 53 1,4
Бензол -н-гексан 57,3 61,9 То же 53 4,9
Природа взаимодействия между исходным раствором и материалом мембраны будет оказывать значительное влияние как на равновесную концентрацию разделяемых веществ в мембранной фазе, так и на скорость транспорта компонентов смеси через мембрану. Необходимо отметить, что выбор полимера для процесса испарения связан с большими ограничениями. Первапорационные мембраны должны обладать не только высокими показателями селективности, производительности и механической прочности, но и выдерживать прямой контакт с органическими растворителями при повышенной температуре. Со стороны пермеата мембрана бывает почти сухой, по крайней мере, при работе по вакуумной схеме, поэтому набухает неравномерно, что влечет за собой дополнительную нагрузку на мембрану. Оптимально удовлетворяют этим требованиям композитные мембраны, в которых механическую, термическую и химическую стойкость обеспечивает практически инертная по отношению к пермеату пористая подложка, а характеристики массопереноса и селективности определяются тонким активным слоем.
Метод первапорации находит практическое применение для решения технологических задач, в частности задач разделения водно-спиртовых смесей, где применение дистилляционных методов ограничено из-за образования азеотропов. Сведения об использовании первапорации в аналитических целях отсутствуют.
3.3.3. Электромембранные методы
Упоминавшийся в предыдущем разделе доннановский диализ является частным случаем применения ионообменных мембран в решении задач разделения веществ. Основной областью их применения является электродиализ.
Электродиализный поток веществ через мембрану определяется выражением:
м..uj&dq
dx
где щ, Zi нс. — соответственно подвижность, заряд и кон-а. х d(p
центрация ионов в фазе мембраны; — — градиент потен-dx
циала на мембране.
Таким образом, индивидуальными характеристическими свойствами вещества, которые определяют величину его электродиализного потока через мембрану, являются нг и z,. Подвижность — характеристика, близкая по физическому смыслу коэффициенту диффузии Д в уравнении для диализного потока и связанная с ним уравнением Нернста-Эйнштейна:
D,zl-и, = ——.
(RT)
Здесь F — число Фарадея.
В общем случае селективность ионообменных мембран ограничена избирательным переносом катионов (катионообменные мембраны) или анионов (анионообменные мембраны). Соответственно основная область их применения в электродиализе — суммарное выделение катионов или анионов из растворов с целью обессоливания морской воды или очистки сточных вод. Применение электродиализа для суммарного концентрирования ионных форм элементов в аналитических целях ограничено, с одной стороны, неполнотой концентрирования и, с другой стороны, протеканием электрохимических реакций на электродах с участием концентрируемых форм, что приводит к усложнению их последующего аналитического определения.
Интерес к методу связан, в первую очередь, с решением технологической задачи опреснения воды, но и этот интерес существенно снизился после появления рассматриваемого в следующем разделе метода обратного осмоса. Практически единственной успешно решаемой с помощью электродиализа через ионообменные мембраны аналитической задачей является выделение и концентрирование микропримесей из нерастворимых в воде соединений.
В качестве аналитического электромембранного метода может рассматриваться электроосмотическая фильтрация. Традиционно электроосмос рассматривается как одно из электрокинетических явлений, проявляющееся в движении жидкости вдоль заряженной поверхности под влиянием внешнего электрического поля. Возникновение электроосмотического потока объясняется теорией двойного электрического слоя как эффект, вызываемый коллективным движением ионов одного знака заряда вдоль границы раздела фаз. Долгое время электроосмос рассматривался исключительно как явление переноса растворителя через капиллярно-пористые тела, и вопрос о возможности концентрационных изменений, происходящих в растворе, не обсуждался. Тот факт, что при электроосмосе из водных растворов солей мембрана оказывается непроницаемой для
Методы разделения и концентрирования
219
ионов, имеющих знак заряда одноименный с зарядом поверхности мембраны, впервые отмечен при изучении элек-трокинетических свойств мембран из пористого политетрафторэтилена. Механизм удерживания ионных примесей при электроосмосе можно объяснить с позиций противоточного электромиграционного процесса [129]. В случае отрицательно заряженных политетрафторэтиленовых мембран направление электроосмотического переноса воды противоположно направлению электромиграции анионов под действием электрического поля. И если линейная скорость электромиграции анионов к аноду превышает по абсолютному значению линейную скорость электроосмотического переноса воды, то наблюдается количественное удерживание анионов в анодной камере. Аналогичные рассуждения справедливы и для условий удерживания катионов в катодной камере при электроосмосе через положительно заряженные мембраны.
Проявление избирательной проницаемости пористых мембран для ионов одного знака заряда при электроосмотическом переносе воды положено в основу методик электроосмотического концентрирования анионных и катионных форм элементов из крайне разбавленных растворов. Возможности метода объективно ограничены суммарным содержанием в растворе ионных примесей на уровне 1 • 10-3 моль • л-1, так как объемная скорость электроосмотического потока резко падает с увеличением общего солесодержания. Поэтому достоинства метода в наибольшей степени проявляются при анализе воды высокой чистоты. Здесь, среди безреагентных методов суммарного концентрирования ионных форм, электроосмос оказывается наиболее привлекательной альтернативой упариванию, существенно превосходя его по экспрессности и полноте выделения.
На параметры электроосмотического концентрирования практически не влияет присутствие в растворе недис-социированных или малодиссоциированных соединений. Поэтому электроосмос применяют для концентрирования заряженных форм примесей, в том числе и коллоидных, из водных растворов полярных органических и малодиссоциированных неорганических соединений (этиленгликоль, глюкоза, пероксид водорода и борная кислота). Метод можно также применять для концентрирования электроза-ряженных органических примесей, например продуктов деструкции ионообменных смол.
Сочетание последовательных операций электроосмотической фильтрации воды через две мембраны с разным знаком заряда поверхности позволяет осуществить процесс деионизации воды, обеспечивающий глубокую очистку воды от электрозаряженных примесей [3, с. 203].
3.3.4. Баромембранные методы (мембранная фильтрация)
В основе баромембранных методов лежит перенос вещества через пористую перегородку вследствие конвективного движения среды под действием градиента давления. В зависимости от диаметра пор мембраны различают процессы обратного осмоса (мембраны с диаметром пор
1 • 10-3—1 • 10“2 мкм), ультрафильтрации (1 • 10-2—0,1 мкм) и микрофильтрации (0,1-10 мкм). Два последних метода одинаково эффективны для выделения примесей, как из жидких, так и из газообразных сред [129]. Движение потока жидкости через пористые среды подчиняется закону Дарси, согласно которому скорость такого потока прямо пропорциональна пористости мембраны, градиенту давления на мембране и обратно пропорциональна вязкости жидкости. На перенос растворенного вещества через мембрану оказывает влияние концентрационная поляризация. Это явление увеличения концентрации растворенного вещества, задерживаемого мембраной у ее поверхности вследствие избирательного перемещения растворителя через мембрану. Например, в случае обратноосмотического процесса растворенное вещество, не имея возможности проникнуть через мембрану, накапливается в тонком пограничном слое у ее поверхности, где его концентрация становится выше, чем в основном объеме раствора. Эта увеличившаяся концентрация приводит к увеличению осмотического давления и, соответственно, величины давления, под которым раствор может фильтроваться через мембрану, так как эта величина определяется концентрацией солей именно в пограничном слое. В некоторых случаях концентрация растворенного вещества в этой тонкой пленке может стать настолько высокой, что последняя делается очень вязкой и желеобразной. Диффузия через слой образовавшегося геля идет более медленно через саму мембрану, даже при увеличении разности давлений по обе стороны мембраны. Поэтому при использовании мембран в процессе фильтрационного разделения важно уметь определять, контролируется ли процесс переноса вещества проникновением через мембрану или же через гелевый слой; результаты контроля дают информацию о необходимости регенерации или замены мембраны. Образование гелевого слоя, если его нельзя предотвратить, может произойти в течение нескольких минут от начала фильтрации. Коль скоро такой слой геля образовался, он становится определяющим фактором, который влияет на скорость потока больше, чем сама мембрана.
Некоторые растворенные вещества образуют гелевый слой быстрее, чем другие. Способность молекулы к гелеобразованию определяется главным образом ее коэффициентом диффузии — величиной, зависящей от формы молекулы и ее молярной массы. Молекулы меньшего размера диффундируют быстрее, чем молекулы большего размера, а сферические — быстрее, чем линейные, имеющие ту же молярную массу. На скорость гелеобразования влияет концентрация вещества, склонного к гелеобразованию. С достижением определенной концентрации молекулы таких веществ начинают быстро образовывать гелеобразный слой у поверхности мембраны, удаляясь при этом из раствора; если же их концентрация достаточно низка, то предел растворимости вещества не будет достигнут и образования такого пограничного слоя не произойдет.
Другой проблемой является эффект Доннана, который возникает, когда одно из растворенных веществ (например, нуклеиновая кислота или белок) находится в ионизован
220
Новый справочник химика и технолога
ном состоянии, но не способно диффундировать через поры мембраны [121]. В результате диффузии растворенных веществ происходит неравномерное распределение ионов по обе стороны мембраны. Поскольку электронейтральность должна сохраняться, на мембране возникает так называемый потенциал Доннана, который препятствует дальнейшей диффузии любого типа ионов с зарядом, противоположным тому, которым обладают неспособные диффундировать молекулы.
С учетом этих особенностей процесса необходимо предпринимать следующие меры для того, чтобы интенсифицировать процесс мембранной фильтрации:
— из тех мембран, которые способны задерживать молекулы необходимого сорта, выбрать для работы мембрану с наибольшим размером пор;
— использовать максимальное давление, которое еще не приводит к гелеобразованию;
— использовать перемешивание, чтобы жидкость вблизи от поверхности мембраны находилась в движении;
— начинать фильтрацию с самой низкой с практической точки зрения концентрации; разбавление может оказаться полезным;
— поддерживать температуру фильтруемых растворов по возможности настолько высокой, насколько указанные растворы это допускают;
— подобрать такое значение pH и ионной силы, при которых растворимое вещество, подлежащее фильтрации, имело бы максимальную растворимость.
При этом механизм выделения частиц из фильтруемой среды только в самом простейшем случае может рассматриваться как механическое удерживание за счет того, что размеры частиц превосходят размеры пор в мембране. Специальные исследования механизма выделения частиц полистирольных латексов из водных сред при фильтрации через положительно заряженные мембраны показали, что основной вклад в удерживание частиц дают электроосмотические взаимодействия. Поэтому мембраной удерживаются частицы значительно меньших размеров, чем размеры пор. В газовой фазе электроосмотические взаимодействия дают еще больший вклад по сравнению с ситовым эффектом. Поэтому в случае микро- и ультрафильтрации нельзя гарантировать четкие пределы исключения частиц по размерам, ориентируясь на диаметр пор.
Соответственно методы мембранной фильтрации находят применение преимущественно для суммарного отделения частиц, превосходящих определенные размеры.
3.3.4. 1. Мембранная фильтрация жидких сред
В пробоподготовке при анализе природных вод на содержание тяжелых металлов общепринята операция микрофильтрации через фильтры типа «миллипор» (амер.) или их отечественные аналоги «владипор» с размерами пор 0,45 мкм. В микробиологическом анализе фильтрация применяется, например, для определения суммарного содержания в воде бактерий группы кишечных палочек (БГКП). Метод мембранной фильтрации является более
точным по сравнению с ранее применявшимися методами, основанными на определении косвенных признаков присутствия БГКП [131]. Мембраны должны соответствовать определенным технологическим условиям (ТУ). При их изготовлении следует учитывать требования к толщине, пористости, показателю преломления и нанесению разметочной сетки. Требования предъявляются и к таким характеристикам как точка пузырька, удельная производительность, задержка микроорганизмов, диффузионная проницаемость и противобактериальноые свойства. Важным является также требование к способности мембраны выдерживать автоклавную обработку. (В США действуют предварительные Государственные ТУ на мембранные фильтры с размерами пор 0,01-8,0 мкм NNN-D-00370 и ТУ на мембраны с размером пор 0,45 мкм «Millitary Specif-cation» MJL-D-37005A, Disk, Filtering, Microporus, в котором оговариваются все требования к мембранам).
Преимущественные области применения мембранных фильтров определяются не только размерами пор, но и материалом, из которого изготовлена мембрана. О самих областях применения и рекомендуемых для них типах мембран можно судить по данным, приведенным в табл. 3.76.
Таблица 3.76
Мембраны, рекомендуемые для лабораторных исследований [129]
Область применения Рекомендуемые размеры пор, нм • 10”3 Материал мембраны
Мембраны, рекомендуемые для лабораторных исследований (Мембраны «Нуклеопор», с размерами пор 0,03-8 нм 10 3, могут использоваться во всех лабораторных иследованиях)
Осветление и извлечение частиц из жидкости 1 Эфир целлюлозы
Фильтрация вязких жидкостей 3-8 То же
Анализ свободных от азота веществ 0,45 Ацетилцеллюлоза
Извлечение или сбор крупных вирусов и фагов 0,1 Эфир целлюлозы
Фильтрация вирусов 0,025 То же
Извлечение микоплазм 0,1 То же
Извлечение и сбор коллоидных осадков 0,1 То же
Сверхтонкая очистка для получения реагентов, используемых в методах светорассеяния 0,1 То же
Сверхтонкая очистка растворителей для ВЭЖХ 0,5 Тефлон, регенерированная целлюлоза
Стерильная фильтрация водных систем 0,2 Эфир целлюлозы, найлон
Методы разделения и концентрирования
221
Продолжение таблицы 3.76
Область применения Рекомендуемые размеры пор, нм • 103 Материал мембраны
Стерильная фильтрация неводных систем 0,2 Тефлон, регенерированная целлюлоза
Стерильная фильтрация спиртовых растворов 0,2 Ацетилцеллюлоза
Извлечение бактерий 0,45 Эфир целлюлозы
Аналитическое отделение осадка 0,65 То же
Разделение водных суспензий Нуклеопор
Гравиметрический анализ промышленных гидравлических жидкостей 5 Эфир целлюлозы
Стерилизация вентиляционного воздуха в помещениях 0,2 Тефлон
Фильтрация органических растворов и растворителей Регенерированная целлюлоза
Стерилизация агрессивных жидкостей 0,2 Тефлон
Осветление агрессивных жидкостей 1 То же
Стерилизация воздуха и газов 0,2 То же
Анализ радиоактивных веществ Эфир целлюлозы
Мембраны для микробиологического анализа воды
Концентрирование вирусов 0,1 Нитроцеллюлоза
Общий подсчет бактерий 0,2 Эфир целлюлозы
Подсчет БГКП 0,45 То же
Подсчет ФКП 0,7 Эфир целлюлозы (анизотропный)
Извлечение фитопланктона для анализа 1-3 Эфир целлюлозы
Общее извлечение частиц из суспензий для анализа 5 То же
Извлечение тонких осадков для анализа 0,45 То же
Анализ питательной котловой воды на содержание в ней частиц оксида железа 0,45 Эфир целюлозы (с нанесенной счетной сеткой)
Продолжение таблицы 3.76
Область применения Рекомендуемые размеры пор, нм • 103 Материал мембраны
Мембраны для контроля качества пищевых продуктов и напитков
Подсчет бактерий 0,2 Эфир целлюло-' зы
Подсчет БГКП 0,45 То же
Определение БГКП в молоке 0,65 То же
Выделение дрожжей и плесенных грибков для подсчета колоний 0,65 То же
Выделение дрожжей и плесенных грибков для микроскопического анализа 0,8 То же
Мембраны для испытаний на стерильность
Фармацевтические и косметические средства, не содержащие бактериостатики 0,2 0,45 Эфир целлюлозы То же
Фармацевтические и косметические средства, содержащие бактериостатики 0,2 0,45 То же То же (с гидрофобным краем)
Мази, пасты, не содержащие бактериостатики 0,3 0,45 То же То же
Мази, пасты, содержащие бактериостатики о,з 0,45 То же То же (с гидрофобным краем)
Мембраны для цитологических исследований и хемотаксиса
Выделение человеческих клеток для послойного цитологического анализа 1-5 Эфир целлюлозы
Камеры для хемотаксиса (изучение миграции клеток под действием химических «приманок») 3-12 То же Нуклеопор
Мембраны для стерилизации культуральных средметодомпоследовательной фильтрации
Грубая или предварительная фильтрация 0,8-1 Эфир целлюлозы
Тонкая фильтрация 0,45 То же
Окончательная фильтрация 0,3 То же
Стерилизующая фильтрация 0,2 Эфир целлюлозы
Извлечение микоплазм 0,1 То же
Предварительная фильтрация сыворотки 1 То же
222
Новый справочник химика и технолога
Продолжение таблицы 3.76
Область применения Рекомендуемые размеры пор, нм • 1 (Г3 Материал мембраны
Мембраны для биомедицинских и клинических химических исследований
Подсчет клеток в воде, идущей на приготовление лекарств 0,8 Эфир целлюлозы
Связывание РНК и ДНК 0,45 Нитроцеллюлоза
Связывание белков 0,45 То же
Отделение несвязанных белков от связанных 0,45 Ацетилцеллюлоза
Радиоиммунологиче-ские исследования (отделение связанных с антителами антигенов от свободных антигенов) 0,45 То же
Выделение белков из воды 0,025 Нитроцеллюлоза
Флуоресцентный анализ антител 0,45 Нитроцеллюлоза (черная мембрана)
Полупроницаемые перегородки в диффузионных камерах 0,6 0,2 и 0,4 Поливинилхлорид Нуклеопор
Мембраны общепромышленного назначения и для производства лекарственных средств
Стабилизация пива 1,2 Эфир целлюлозы
Стабилизация вин и соков 0,8 То же
Окончательная фильтрация напитков 1 Ацетилцеллюлоза (ни в коем случае не нитроцеллюлоза)
Сверхтонкая очистка воды 0,45 1,2 Эфир целлюлозы
Сверхтонкая очистка спиртов 1 Ацетилцеллюлоза
Стерилизующая фильтрация спиртовых растворов 0,2 То же
Осветление фоторезистов 10 Тефлон
Осветление концентрированных кислот и щелочей 10 То же
Осветление прочих реактивов и коррозионноактивных жидкостей 10 То же
Продолжение таблицы 3.76
Область применения Рекомендуемые размеры пор, нм • 10“3 Материал мембраны
Осветление ракетных топлив и криогенных веществ 10 Тоже
Производство полупроводников 0,2 Ацетилцеллюлоза
Осветление вакцин 0,45 Эфир целлюлозы
Стерилизующая фильтрация вакцин 0,2 То же
Производство различных растворов для внутреннего введения 0,2 То же
Фильтрация гидравлических жидкостей 0,8 Ацетилцеллюлоза
Перечень приведенных в табл. 3.76 примеров применения микрофильтрации, относящихся в основном к решению микробиологических задач, можно дополнить примерами применения метода в неорганическом анализе. Помимо отделения взвешенных форм тяжелых металлов при фильтрации проб природных вод через фильтры 0,45 мкм, микрофильтрация через фильтры с узкой дисперсией пор по размерам позволяет определять формы существования микроэлементов в водных средах (табл. 3.77).
Таблица 3.77
Определение физико-химических форм следов элементов в водах с использованием мембранной фильтрации [136]
Проба Определяемый элемент Характеристики мембран Метод определения
Природная вода Cu, Cd, Hg — ААС
То же Li, В, Be, С Ядерные трековые фильтры Ядерно-физиче-ский ана- лиз
То же Hg, Zn, Co Ядерные трековые фильтры, 0,45 мкм НАА
То же 58Co, 60Co, 54Mn, 64Zn Полые волокна «Амикон» Гамма-спектроскопия
То же Al Поликарбо-натные мембранные фильтры, 0,1-0,45 мкм Спектрофотометрия ААС
Методы разделения и концентрирования
223
Продолжение таблицы 3.77
Проба Определяемый элемент Характеристики мембран Метод определения
Природные и рудничные воды Be Пленочные фильтры, 0,45 мкм ИСП-АЭС
Речные воды Fe,Al Полимерные фильтры, 8 мкм, 1 мкм и 0,4 мкм ИСП-АЭС
То же 32 элемента Фильтры (Миллипор 0,05; 0,45 мкм) НАА
Озерная вода Al, Fe, Zn Полые волокна (Амикон) ААС
Морские и другие воды Np, Am Пленочные фильтры, 0,45 мкм Радиометрия
Морские воды Tl,Ag, Cu, Cd, Pb, Ni, Zn Мембраны (Нуклеопор) ИР-МС
То же 16 элементов Мембраны (Нуклеопор) НАА
Расширение возможностей микрофильтрационного разделения частиц возможно при одновременном воздействии градиента давления и электрического поля. Процесс, происходящий при одновременном воздействии на обратноосмотическую мембрану градиентов давления и электрического потенциала, назван электроосмотической фильтрацией [132]. В процессе электроосмофильтрации катионы и анионы, проникающие в результате миграции соответственно в катодную и анодную камеры ячейки, отводятся пермеатом.
Однотипные мембранные фильтры выпускают различные зарубежные фильтры. При выборе мембран для мембранной фильтрации не последнюю роль играют цены на них, установленные различными фирмами. Перечень мембран:
Название или материал мембраны
HAWP
Тиран-М/Е WCN Поликарбонат Смесь эфиров Нитроцеллюлоза Нитроцеллюлоза Смесь эфиров
Ультипор Нитроцеллюлоза
Фирма
«Миллипор» «Пюропор» «Ватман» «Нуклеопор» «Нуклеопор»
«Майкрофилтрейшнстемс» «Шляйхер и Шуль» «Гелман»
«Пол»
«Сарториус»
Целый ряд мембран подобных типов выпускается отечественным НПО «Мембраны» в г. Владимире. При этом цены на них существенно ниже, чем на зарубежные аналоги.
Вторым из баромембранных методов в порядке уменьшения размеров выделяемых частиц является ультрафильтрация. По своему назначению этот метод характеризуется как процесс разделения и фракционирования растворов, при котором макромолекулы отделяются от раствора и низкомолекулярных соединений фильтрацией через мембраны. Как и в случае микрофильтрации, основной областью применения ультрафильтрации является суммарное концентрирование отделяемых на мембранах частиц (в данном случае макромолекул). Реже ультрафильтрация применяется для фракционирования макромолекул по размерам, дополняя тем самым метод эксклюзионной хроматографии. В отличие от последней при ультрафильтрации вместо пределов исключения говорят о номинальной отсекаемой молярной массе (НОММ). Речь идет о способности мембраны задерживать молекулы в зависимости от их размера. Термин номинальный вводится по той причине, что в реальных условиях форма и заряд молекулы влияют на скорость ее прохождения через мембрану. Величина задержки, определяемая НОММ, означает, что 90% сферических заряженных молекул данной молярной массы будут задержаны. Однако молекулы, вытянутые в длину и имеющие большую молярную массу, чем НОММ, могут пройти через соответствующую мембрану, в то время как заряженные молекулы с массой, меньшей НОММ, могут оказаться задержанными. В табл. 3.78 и 3.79 приведены задерживающие способности двух больших серий мембран, предназначенных для ультрафильтрации.
Таблица 3.78 Номинальные величины отсекаемых молярных масс (НОММ) при ультрафильтрации для ультрафильтров фирмы «Нуклеопор» [129]
НОММ Тип мембраны
А С F
500 А 0,5 С 0,5 —
1000 А1 С 1 —
5000 А5 С5 F5
10000 А 10 С 10 F 10
20000 — С 20 F20
50000 А 50 С 50 F50
100000 А 100 С 100 F 100
300000 — — F300
500000 — — F500
1000000 — — F 1000
Примечания. Мембраны типов А, С, F изготовлены из трех различных полимерных материалов, предназначенных для работы с различными растворителями. Мембраны типа А пригодны для работы в диапазоне pH от 2 до 12 и выпускаются в виде полых волокон со значениями НОММ 2000, 10000 и 50000. Мембраны типа С предназначены для работы в диапазоне pH от 2 до 10 и более устойчивы к органическим растворителям, чем мембраны типа А. Мембраны типа F работают в диапазоне pH от 1 до 14, устойчивы к хлору и большинству органических растворителей, могут подвергаться обработке в автоклаве, а также использоваться при температурах до 130 °C.
224
Новый справочник химика и технолога
Таблица 3.79
Номинальные величины отсекаемых молекулярных масс для ультрафильтров фирмы «Амикон» [129]
Дисковые мембраны НОММ Полое волокно НОММ
UMO5 500 ХМЗОО 300000
UM2 1000 Н1Р2 2000
YM5 5000 Н5Р2 2000
РМ10 10000 Н1Р5 5000
YM10 10000 Н10Р5 5000
YM10U 10000 HIP 10 10000
UM10 10000 Н5Р10 10000
UM20 20000 Н10Р10 10000
РМ30 30000 HIX50 50000
YM30 30000 Hl 0X50 50000
ХМ50 50000 HIP100 100000
ХМ100А 100000 Н10Х100 100000
Для баромембранных процессов вообще и для ультрафильтрации в частности существенную роль играют конструкции фильтрационных устройств.
Для ультрафильтрации небольших объемов (1-50 мл) фирма «Миллипор» производит так назывемые погружаемые ультрафильтры «Иммерсибл-СХ», которые состоят из пористой полипропиленовой сердцевины, покрытой ультрафильтрационной мембраной со значениями НОММ 10000 и 30000. Выходное отверстие устройств подсоединяют к вакуумному насосу, а само устройство помещают в сосуд с образцом. Вода, соли или другие низкомолекулярные вещества проходят через мембрану, а выделяемый продукт остается в сосуде сконцентрированным и обессоленным. Для снижения концентрационной поляризации это устройство можно подсоединить к специальной мешалке, которую фирма выпускает отдельно.
Фирма «Байо-Молекуляр дайнэмикс» производит удобное приспособление «Микропродикон» для вакуумного концентрирования образцов различных объемов от больших до исключительно малых (25 мкл). В этом автономном вакуумируемом приборе диализная или ультрафильтрационная мембрана поддерживается в вертикальном положении пластмассовым стержнем, который погружен в рабочую камеру. Образец, содержащийся в открытом резервуаре, заполняет пространство между мембраной и стержнем и, по мере того как вода и соли удаляются, концентрируется и собирается в небольшом сборнике, расположенном на дне. Размерами этого сборника определяется конечный объем сконцентрированного образца. После того как прибор собран и вакуумирован, он становится полностью автономным и может быть помещен в холодную комнату или водяную баню.
Для препаративной ультрафильтрации предназначена кассетная система «Пелликон», предлагаемая фирмой «Миллипор». Сам ультрафильтр состоит из очень тонкой (20-30 нм) полупроницаемой мембраны, прочно связанной с пористой подложкой. Последняя обеспечивает механиче
скую устойчивость и оказывает малое сопротивление потоку, а поскольку сама мембрана очень тонка, достигается высокая скорость фильтрации. Ультрафильтры «Пелликон» выпускаются в виде дисков, пленок, пакетов или кассет, причем последние используются в кассетной системе «Пелликон». Существует пять типов мембран со значениями НОММ от 1000 до 100000. Эти мембраны можно использовать в любого рода устройствах, уже описанных выше, либо в кассетной системе «Пелликон». Кассеты могут использоваться при давлениях до 7 атм (в режиме обычной фильтрации либо в режиме диафильтрации). Общий объем раствора над мембранами для этой системы составляет 200 мл. Кроме того, фирмой выпускается подобная кассетная система из нержавеющей стали, рассчитанная на фильтрацию больших объемов.
Основные области применения ультрафильтрации — промышленные технологии в фармакологии, в микроэлектронике, молочной промышленности и т. п., т. е. всюду, где существует потребность в выделении высокомолекулярных примесей из жидких сред (подробнее см. [129, с. 363]).
Метод обратного осмоса по физико-химической природе процесса, лежащего в его основе, отличается от других баромембранных методов. Для его понимания достаточно вспомнить сущность явления осмоса — движение растворителя через мембрану из раствора с низкой концентрацией солей в концентрированный раствор (рис. 3.16).
Высота подъема раствора
а)
Мембрана
Поток Менее кони,
воды раствор
Более
концентрированный раствор
Рис. 3.16. Различие между осмосом (а) и обратным осмосом (б) [127]
Если ультрафильтрацию, как правило, применяют в тех случаях, когда необходимо задержать нужное вещество, то при обратном осмосе конечным продуктом за редкими исключениями является фильтрат. Мембраны, используемые в обратном осмосе, имеют значительно более низкие значения НОММ и работают при гараздо более высоких давлениях, чем мембраны, применяемые для ультрафильтрации. Первоначально обратный осмос был разработан для обессоливания только морской воды, но в настоящее время
Методы разделения и концентрирования
225
он широко используется также для понижения концентрации солей в умеренно или слабо соленых водах в ряде промышленных отраслей. Так, с его помощью может быть удалено от 80 до 95% солей из питающей воды, используемой для промышленных нужд энергетики.
Литература по обратному осмосу обширна и разнообразна. Основные принципы этого процесса подробно обсуждаются Хвангом и Каммермейером [120].
Движущей силой процесса в данном случае является разность приложенного к концентрированному раствору давления р и осмотического давления л: (р-п). С учетом концентрационной поляризации процесс обратного осмоса проводят при давлениях, значительно превышающих осмотическое при исходной концентрации солей в воде. Так, п для усредненного состава морской воды равно 0,25 МПа, обессоливание морской воды осуществляют при р = 0,7-0,8 МПа, а иногда и выше.
Процесс обратного осмоса приобрел практическую значимость лишь после того, как были разработаны соответствующие мембраны. Мембрана должна обладать необходимой прочностью для работы при высоких давлениях, химической стойкостью и устойчивостью к микробиологической атаке. Вначале большинство мембран для обратного осмоса изготавливались из ацетилцеллюлозы, причем ацетилцеллюлоза для этих мембран несколько отличается от используемой в микрофильтрационных мембранах: она содержит меньше ацетильных групп на остаток глюкозы. Теоретически на один остаток глюкозы могут приходиться три ацетильные группы, но при высокой степени замещения скорость прохождения воды через мембрану оказывается небольшой. С другой стороны, если содержание ацетильных групп низко, скорость прохождения воды велика, однако селективность таких мембран (задержка ими соли) мала. По-виднмому, оптимальная степень замещения должна быть в пределах 2,1-2,5, что обеспечивает задержку соли на 90-95% и расход через единицу поверхности мембраны (100-200) • 10”5 г • см~2 • с-1 [120].
К другим важным характеристикам мембран относятся их стойкость по отношению к высоким и низким pH, устойчивость к хлору, к гидролизу к высушиванию, к действию бактерий и способность выдерживать динамические нагрузки под давлением [133].
Кроме ацетилцеллюлозы, для изготовления мембран был применен ряд других материалов, а именно полиамиды (найлоны), полибензимидазолы, сульфонированный диметилполифенилоксид и полиэтиленимин. Для обратного осмоса был также испытан другой класс мембран, называемых динамическими (или мембранами in situ, т.е. применяемыми непосредственно в объекте анализа). Это жидкие мембраны, которые фактически образуются непосредственно на микропористой подложке, когда на ее поверхность наносится раствор, содержащий поверхностно-активные вещества [132]. Некоторые пористые материалы, не обладающие достаточной селективностью для обратного осмоса, могут быть использованы в этих целях, если они сформированы как динамические мембраны.
Большинство целлюлозных мембран для обратного осмоса выполнено в виде полых волокон с использованием соответствующих способов намотки синтетического волокна. Главное преимущество полых волокон в применении к обратному осмосу заключается в том, что они обеспечивают в высшей степени выгодное отношение поверхности мембран к объему мембранного аппарата. Таким образом, используя относительно компактное оборудование, можно достичь высоких скоростей потока.
Как уже отмечалось выше, интерес К обратному осмосу на первом этапе развития этого метода подогревался в связи с необходимостью решения проблем обессоливания воды. На сегодня область его промышленного применения существенно расширилась. В эту область входят:
— обработка промышленных отходов: регулирование загрязнений на кожевенных заводах, гальванических и текстильных производствах и т. п.;
— пищевая промышленность: регулирование загрязнений или возврат таких полезных продуктов, как сыворотка;
— очистка сточных вод: обратный осмос может быть использован как третья стадия обработки бытовых стоков, чтобы удалить аммоний, органические примеси или соли. Позволяет повторно использовать отработанные воды для бытовых нужд (причем перечень производств, где обратный осмос применяется для очистки сбросов, постоянно растет и в частности начал применяться для очистки радиоактивных сбросов на предприятиях атомной энергетики);
— городское водоснабжение: обратный осмос позволяет удалять токсичные примеси из источников бытового водоснабжения и делать воду пригодной для потребления;
— химическая промышленность: здесь применение обратного осмоса состоит в извлечении соединений, находящихся в разбавленном виде, из больших количеств жидкости. При этом, как правило, конечной целью является извлекаемый продукт, а не фильтрат.
Обратный осмос нередко используют вместо дистилляции для получения высококачественной воды. Одно из главных преимуществ обратного осмоса заключается в значительной экономии энергии, поскольку дистилляция требует большого расхода тепла. Энергетические затраты в случае обратного осмоса составляют лишь 25% тех, что идут на дистилляцию. Однако стоимость энергии составляет лишь часть общих затрат на проведение процесса обратного осмоса. Необходимо учесть также стоимость мембранных модулей, предварительных и последующих фильтров, финансовые затраты на обслуживание, куда входит контроль установки, необходимый для ее нормальной работы. Такой контроль должен быть непрерывным во всех случаях, где от чистоты конечного продукта зависят проблемы безопасности. В частности, потребность непрерывного технологического контроля существует в фармацевтической промышленности, особенно при получении исключительно чистой воды, используемой для приготовления инъекционных растворов, поскольку малейшее повреждение мембраны, не обнаруженное немедленно, может привести к попаданию в воду пирогенных примесей.
226
Новый справочник химика и технолога
Расширению сфер промышленного применения обратного осмоса не привело к его использованию для решения аналитических задач. Эта закономерность объясняется тем, что преодоление сложных технических проблем для его осуществления в микромасштабе при наличии множества альтернатив не компенсируется достигаемым эффектом неполного выделения ионных примесей.
3.3.4. 2. Мембранная фильтрация воздуха
Мембранные фильтры первоначально были разработаны для фильтрации воды, но их вскоре стали использовать также для фильтрации воздуха (табл. 3.80). Вначале это было сделано для анализа загрязнений воздушной среды, но интенсивное развитие отраслей промышленности с высокими технологиями привело к необходимости осуществлять особо тщательный контроль за производственной атмосферой, поэтому фильтры и мембраны стали широко применять для очистки и анализа промышленного воздуха.
Таблица 3.80
Мембраны для анализа загрязненного воздуха [129]
Область применения Рекомендуемые размеры пор, мкм Материал мембраны
Разделение разных по размерам частиц, содержащихся в воздухе 0,4-8 Нуклеопор
Обычный отбор проб из воздуха 0,8 Эфир целлюлозы
Определение содержания вирусов в воздухе 0,2 Нитроцеллюлоза
Выделение содержащихся в воздухе частиц для последующего микроскопического анализа 0,45 Нитроцеллюлоза (зеленая мембрана с нанесенной сеткой)
Выделение слабоокра-шенных частиц для последующего микроскопического анализа 0,45 Нитроцеллюлоза (черная мембрана с нанесенной сеткой)
Контроль на содержание волокон асбеста 0,8 Нитроцеллюлоза
Контроль содержания капель масла в тумане 0,8 То же
Продолжение таблицы 3.80
Область применения Рекомендуемые размеры пор, мкм Материал мембраны
Количественное выделение респираторной пыли 1 То же
Контроль содержания частиц в горячих газах 0,8 Ацетилцеллюлоза
Выделение хлопковой пыли для гравиметрического анализа 0,5 Нитроцеллюлоза
Выделение влажных частиц для последующего анализа 1 Эфир целлюлозы
Контроль содержания аэрозолей 3 Тефлон
Контроль биологически опасных компонентов выхлопных двигателей 0,2 Эфир целлюлозы
В воздухе можно обнаружить великое множество частиц (аэрозолей) как естественного, так и искусственного происхождения; эти частицы могут быть как твердыми, так и жидкими (аэрозоли с твердой и жидкой дисперсной фазой) и иметь самые различные размеры. На рис. 3.17 приведены основные характеристики частиц, которые могут находиться в воздухе.
Эти частицы во многих случаях ведут себя как динамические системы, причем мелкие частицы агрегируют, образуя крупные, а крупные частицы дробятся на более мелкие (рис. 3.18).
Первичной называется такая частица, размер которой может быть уменьшен далее только разрывом химических связей. Эти частицы могут легко ассоциировать под действием таких сил, как ван-дер-ваальсовы силы и электростатическое притяжение. Подобные взаимодействия оказываются существенными для частиц размером 10 мкм и менее, а для частиц больших размеров становятся незначительными по сравнению с другими действующими на них силами, такими как, например, сила тяжести. Средний диаметр самых мелких частиц, которые можно видеть невооруженным глазом, составляет 20 мкм.
Методы разделения и концентрирования
227
Диаметр частиц, мкм
0,001 0,01 0,1
10 100 1000 10000
Типичные частицы < Смолистый аэрозоль » !*• Масляный аэрозоль
* Газовая са 4— А эрозоль Коллоидный кремнезем Аэрозоль хло аммония 1 I Аэро ка—*1* олеу * . . 11Ы I* ин П ылс с\хос <— Щелочные аз
Час Кристаллы Jj морской СОЛИ п", 1К м, цщы сажи и/альвр
" Вирус! ” для —м 1*
т V Типы пвоочищайощего оборудования — — — — — к—
— Выс окоэффективньк — Термическое о - Аниараш для г Иио,н~|; воздушные фил 1 3. ICKC 1рОСТЙ1ИЧСС10
Частицы, диспергир в газе Твердые
Жидкие
Почва —
Атмосфера
4 Лету ЛЬ И ДЫМ рила »[< Цеме | Сериокислотиь 1 * аэрозоль ПОЛЬ _ | Удобрении» чин пепел <ая пыль » » нтная пыль » 4ч ! Распыленный , грунтовый »] известияк В— Морской пссс к->| сади
ума 11 ялеобразные __ нсектициды “* Гр\ нто вы if тал образное .... с молоко зрою.ш 1 Распыленные хинам легких 1 'ЭрПТрО! V ил и* - Фдоташюннь р\ДЫ ьк Ц Споры растений Цветочная пылыта к * я пыль »| —н -*1 I*-Капли лневматич^ <t премией ' Медово *1 1С_*| Капли гидравличс форсунки ЧССКИЙ БОЛОС
—— Центр fic скрубберы (гаю прикладки — |4- — —. —. ВО!Д> ьтры — -4Й4- — 1—к-сою осаждения
тфужныс ccnupurq очистители! -• —4 иные фильтры + -* Ударные н —
1ЛЬ — — — -
t — — а. —
-J-* Ит Мелкий Крчпкый jJ— г.«=.
л* -I- песок f песок
( Облака и пеной _ ।Т>ман,Ииюросъ^
э1г план *1* । *1* *1*
Рис. 3.17. Характеристики и размеры атмосферных частиц [129]
кристаллиты шньу $ 0°
Ч&тИртурпые. ^^ <7 ^ частицы Ч' ' ""
проникновения частиц в человеческий организм при дыхании (рис. 3.19).
Скопление хлопьев Скопление агломерата*
Рис. 3.18. Динамическая природа атмосферных частиц [129]
Вредное воздействие аэрозольных частиц на человека определяется не только токсичностью образующих их веществ, но и их дисперсностью, определяющей глубину
Рис. 3.19. Дыхательная система человека и размеры частиц, проникающих на разную ее глубину [129]:
1 — носовая полость; 2 — ротовая полость; 3 — гортань;
4 — трахея; 5 — первичные бронхи; 6 — вторичные бронхи;
7 — третичные бронхи; 8 — бронхиола; 9 — альвеолярные ходы;
10 — альвеолярные сумки; 11 — альвеолы
228
Новый справочник химика и технолога
Один из наилучших способов оценки распределения аэрозолей по размерам — фракционирование их на калиброванных мембранах «Нуклеопор».
Существует несколько механизмов извлечения частиц мембраной: прямой перехват (эффект касания), инерционное улавливание, диффузия, электростатическое притяжение и гравитационное осаждение (седиментация). При этом механизм прямого перехвата, определяющий удерживание частиц, размеры которых превышают размеры пор, дополняется удерживанием по другим механизмам. Об относительном вкладе различных механизмов удерживания можно судить по экспериментально полученной зависимости эффективности удерживания частиц аэрозолей из воздуха при его фильтрации через мембрану «Нуклеопор» (рис. 3.20). Эффективность выражается как отношение числа уловленных мембраной частиц к количеству частиц, находившихся в исходной пробе воздуха.
Эффективность
захвата частиц
Рис. 3.20. Эффективность захвата частиц различных размеров мембранами «Нуклеопор» с порами 5 мкм
Как видно из приведенного рисунка, эффективность улавливания оказывается наибольшей для очень мелких и очень крупных частиц и наименьшей для частиц размером 0,05-0,10 мкм. Представленная зависимость заставляет критически подходить к данным фракционного анализа размеров аэрозольных частиц по результатам мембранной фильтрации даже через хорошо откалиброванные мембраны, такие как «Нуклеопоры». Важным параметром, влияющим на эффективность, является «лобовая» скорость воздушного потока, т.е. скорость его движения в направлении, перпендикулярном поверхности мембраны. Наибольшая эффективность, улавливания достигается при малых лобовых скоростях на уровне 1 см • с-1 и меньше. В действительности эффективность улавливания сложным образом зависит как от лобовой скорости, так и от размера пор, и существует оптимальная лобовая скорость для сепарации различных частиц по размерам. Таким образом, хотя фракционирование по размерам может быть осуществлено с помощью набора мембран «Нуклеопор», действующих совместно как одно устройство, причем воздух проходит последовательно от одной мембраны к другой, использо
вание отдельных мембран «Нуклеопор» позволяет провести даже более тонкую сепарацию, если подобрать для каждой мембраны с определенным размером пор оптимальную скорость.
Метод мембранной микрофильтрации дополняет диффузионные методы пробоподготовки, используемые при анализе воздуха (см. раздел 3.3.2) и часто используется в сочетании с ними. Кроме того, ультра- и микрофильтрационная пробоподготовка легко сочетается с инструментальными методами анализа [116].
Для мембранной микрофильтрации воздуха могут использоваться те же мембраны, что применяются для фильтрации водных растворов. Но есть и специальные мембраны. В частности, широкое применение находят серебряные мембраны, состоящие из мельчайших частиц серебра, сплавленных вместе с образованием пористой структуры. Никаких связующих агентов не используется. Достоинство серебряных мембран заключается в их высокой термической стойкости, благодаря чему они могут работать при температурах до 400 °C. В этих условиях другие мембраны или фильтры были бы разрушены. Их можно применять и при более низких температурах, при весовом анализе частиц в газовых потоках такого состава, который вызвал бы у обычных фильтрующих материалов большие потери в массе. Однако необходимо заметить, что серебряные мембраны не выдерживают действия на них таких химических агентов, как цианиды и сероводород.
Вторым специфическим типом мембран, применяемых для определения содержания микроорганизмов в воздухе, являются мембраны из желатины. По своей структуре (пористость — 80%, плотность пор — 107 г • см-3 или 1О10 кг м 3) такие мембраны, производимые фирмой «Сарториус», сходны с обычными мембранными фильтрами, но обладают уникальным свойством растворяться в воде, что очень удобно при микробиологическом анализе.
В заключение необходимо отметить, что при анализе аэрозольных частиц в воздухе мембранные фильтры обладают рядом объективных преимуществ по сравнению с фильтрами из волокнистых материалов. Задержанные частицы остаются на поверхности мембраны, где их можно легко анализировать микроскопически или с помощью химических методов. До того как были разработаны мембранные фильтры, микроскопический анализ частиц по размерам был более трудоемким. Особенно хорошо для анализа частиц в воздухе подходят мембраны «Нуклеопор». Они ведут себя как истинные сита и могут быть классифицированы по калибрам в виде отдельных комплектов, каждый из которых предназначен для задержки частиц конкретного размера. Благодаря своей малой массе, а также тому, что они практически не набухают в воде, мембраны «Нуклеопор» могут с успехом применяться в гравиметрическом анализе, что позволяет определять распределение частиц в атмосфере по массе.
Перечни мембран различных типов и назначения, выпускаемых в промышленных масштабах, приведены в табл. 3.81-3.90.
Методы разделения и концентрирования
229
Таблица 3.81
Мембраны, выпускаемые фирмой «Гелман сайенсиз» [129]
Номер по каталогу, тип /кол-во в упаковке (шт.) Размер nop, нм 10“3 Диаметр или линейные размеры, мм
Мембраны без подложки Мембраны «Метрисел» «Метрисел GA», триацетат
60298 GV8*/100 0,2 13
60300 GA-8*/100 0,2 25
60301 GA-8*/100 0,2 47
5006 GA-8*/100 Стерилизованы в автоклаве 0,2 47
63025 GA-8*/100 Стерилизованы 0,2 47
50334 GA-8*/100 0,2 90
60305 GA-8*/25 0,2 142
60307 GA-8*/25 0,2 293
60234 GA-7*/100 0,3 13
60236 GA-7*/100 0,3 25
60237 GA-7*/100 0,3 47
60270 GA-7*/100 0,3 90
60241 GA-7*/100 0,3 142
60243 GA-7*/100 0,3 293
60170 GA-6*/l00 0,45 13
61851 GA-6**/100 0,45 13
60172 GA-6*/l00 0,45 25
61852 GA-6**/100 0,45 25
60173 GA-6*/100 0,45 47
5011 GA-6**/100 Стерилизованы 0,45 47
5004 GA-6**/100 Стерилизованы в автоклаве 0,45 47
61854 GA-6**/100 0,45 47
60174 GA-6*/100 0,45 50
60206 GA-6*/100 0,45 90
60175 GA-6*/100 0,45 102
60177 GA-6*/25 0,45 142
60179 GA-6*/25 0,45 293
66103 GA-6**/100 Индивидуально стерилизованы 0,45 50
61844 GA-4**/100 0,8 13
60107 GA-4 */100 0,8 13
61845 GA-4**/100 0,8 25
60109 GA-4*/100 0,8 25
5003 GA-4**/100 Стерилизованы в автоклаве 0,8 47
60110 GA-4*/100 0,8 47
61846 GA-4**/100 0,8 47
5010 GA-4**/100 Стерилизованы 0,8 47
64525 GA-4*/100 0,8 90
Продолжение таблицы 3.81
Номер по каталогу, тип /кол-во в упаковке (шт.) Размер nop, нм • 10 3 Диаметр или линейные размеры, мм
60114 GA-4*/25 0,8 142
60116 GA-4*/25 0,8 293
60044 GA-3*/100 1,2 13
60046 GA-3*/100 1,2 25
60047 GA-3*/100 1,2 47
60080 GA-3*/100 1,2 ' 90
60053 GA-3*/100 1,2 293
60000 GA-1 */100 5 13
60002 GA-1 */100 5 25
60003 GA-1 */100 5 47
64524 GA-1 */100 5 90
«Метрисел Альфа»
60582 Alpha-200*/100 0,2 13
60584 Alpha-200*/100 0,2 25
60585 Alpha-200*/100 0,2 47
60615 Alpha-200*/100 0,2 90
60589 Alpha-200*/25 0,2 142
62057 Alpha-200*/25 0,2 293
60537 Alpha-450*/100 0,45 13
60539 Alpha-450*/l00 0,45 25
60540 Alpha-450*/100 0,45 47
60570 Alpha-450*/100 0,45 90
60544 Alpha-450*/25 0,45 142
62042 Alpha-450*/25 0,45 293
«Метрисел ТСМ»
64810 TCM-200*/100 0,2 13
64812 TCM-200*/100 0,2 25
64814 TCM-200*/100 0,2 47
64858 TCM-200*/100 0,2 90
64819 TCM-200*/25 0,2 142
64822 TCM-200*/25 0,2 293
64826 TCM-450*/100 0,45 13
64828 TCM-450*/100 0,45 25
64830 TCM-450*/100 0,45 47
64859 TCM-450*/l 00 0,45 90
64835 TCM-450*/25 0,45 142
64838 TCM-450*/25 0,45 293
«Метрисел GN»
64193 GN-6**/100 0,45 13
63066 GN-6*/100 0,45 13
64191 GN-6**/100 0,45 25
63068 GN-6*/100 0,45 25
60511 GN-6*7100 0,45 25
62136 GN-6*7100 0,45 25
64375 GN-6**/100 0,45 37
64382 GN-6*/100 0,45 37
64141 GN-6*7100 0,45 37
64142 GN-6**4/100 0,45 37
63020 GN-6**/100 0,45 47
63077 GN-6**/100 Стерилизованы 0,45 47
63069 GN-6*/100 0,45 47
230
Новый справочник химика и технолога
Продолжение таблицы 3.81
Номер по каталогу, тип /кол-во в упаковке (шт.) Размер nop, нм • 10“3 Диаметр или линейные размеры, мм
66068 GN-6**/1000 Предварительно индивидуально стерилизованы 0,45 47
66378 GN-6**4/200 Предварительно индивидуально стерилизованы 0,45 47
64194 GN-6**/100 Стерилизованы в автоклаве 0,45 47
60512 GN-6*4/100 0,45 47
62015 GN-6**7100 0,45 47
66379 GN-6**7200 Предварительно индивидуально стерилизованы 0,45 47
66420 GN-6**7200 Индивидуально стерилизованы 0,45 50
64535 GN-6*/100 0,45 90
63072 GN-6*/100 0,45 102
66421 GN-4**7100 Индивидуально стерилизованы 0,45 50
64677 GN-4*/100 0,8 25
66177 GN-4**/100 0,8 25
61895 GN-4**7100 0,8 25
64678 GN-4*/100 0,8 37
66178 GN-4**/100 0,8 37
64679 GN-4*/100 0,8 47
66179 GN-4**/100 0,8 47
66376 GN-4* *7200 Предварительно индивидуально стерилизованы 0,8 47
66377 GN-4**7200 Предварительно индивидуально стерилизованы 0,8 47
64747 Несущая подложка* **/500 37
«Метрисел GN» с гидрофобными краями (6 мм)
66123 GN-6**/100 0,45 47
66128 GN-6*/100 0,45 47
«Метрисел DM»
64512 DM-450*/l00 0,45 13
64514 DM-450*/100 0,45 25
64616 DM-450*/100 0,45 37
64515 DM-450*/l00 0,45 47
64943 DM-450*/100 0,45 90
64517 DM-450*/! 00 0,45 102
64499 DM-800*/100 0,8 13
64501 DM-800*/100 0,8 25
Продолжение таблицы 3.81
Номер по каталогу, тип /кол-во в упаковке (шт.) Размер nop, нм • 10"3 Диаметр или линейные размеры, мм
64530 DM-800*/100 0,8 37
64502 DM-800*/100 0,8 47
66059 DM-800*/100 0,8 90
64505 DM-800*/100 0,8 102
«Метрисел Винил VM»
60690 VM-l*/100 5 ' 13
60692 VM-l*/100 5 25
60714 VM-1 */100 5 37
60693 VM-1 */100 5 47
60726 VM-1 */100 5 90
60695 VM-1 */100 5 102
Таффин HT
66185 HT-100*/100 0,1 25
66187 HT-100*/100 0,1 47
66189 HT-100*/100 0,1 90
66192 HT-100*/25 0,1 142
66193 HT-100*/25 0,1 293
66195 HT-200*/100 0,2 13
66197 HT-200*/100 0,2 25
66199 HT-200*/100 0,2 47
66201 HT-200*/100 0,2 90
66204 HT-200*/25 0,2 142
66205 HT-200*/25 0,2 293
66219 HT-450*/l 00 0,45 13
66221 HT-450*/100 0,45 25
66223 HT-450*/100 0,45 47
66225 HT-450*/100 0,45 90
66228 HT-450*/25 0,45 142
66229 HT-450*/25 0,45 293
Мембранные фильтры с подложкой «Версапор»
66385 Versapor 3000/100 3,0 13
66386 Versapor 3000/100 3,0 25
66387 Versapor 3000/100 3,0 47
66388 Versapor 3000/100 3,0 90
66389 Versapor 3000/25 3,0 142
66390 Versapor 3000/25 3,0 293
66300 Versapor 3000 /испытательный рулон 3,0 254 х 4267
66392 Versapor 1200/100 1,2 13
66393 Versapor 1200/100 1,2 25
66394 Versapor 1200/100 1,2 47
66395 Versapor 1200/100 1,2 90
Методы разделения и концентрирования
231
Продолжение таблицы 3.81
Номер по каталогу, тип /кол-во в упаковке (шт.) Размер пор, нм • 10 3 Диаметр или линейные размеры, мм
66396 Versapor 1200/25 1,2 142
66397 Versapor 1200/25 1,2 293
66298 Versapor 1200 /испытательный рулон 1,2 254 х 4267
66399 Versapor 800/100 0,8 13
66400 Versapor 800/100 0,8 25
66401 Versapor 800/100 0,8 47
66402 Versapor 800/100 0,8 90
66403 Versapor 800/25 0,8 142
66404 Versapor 800/25 0,8 293
66365 Versapor 800 /испытательный рулон 0,8 254 х 4267
66406 Versapor 450/100 0,45 13
66407 Versapor 450/100 0,45 25
66408 Versapor 450/100 0,45 47
66409 Versapor 450/100 0,45 90
66410 Versapor 450/25 0,45 142
66411 Versapor 450/24 0,45 293
66363 Versapor 450 /испытательный рулон 0,45 254 х 4267
66413 Versapor 200/100 0,2 13
66414 Versapor 200/100 0,2 25
66415 Versapor 200/100 0,2 47
66416 Versapor 200/100 0,2 90
66417 Versapor 200/25 0,2 142
66418 Versapor 200/25 0,2 293
66362 Versapor 200 /испытательный рулон 0,2 254 х 4267
Тефлон TF
66141 TF-200*/100 0,2 13
Продолжение таблицы 3.81
Номер по каталогу, тип /кол-во в упаковке (шт.) Размер nop, нм • I03 Диаметр или линейные размеры, мм
66142 TF-200*/100 0,2 25
66143 TF-200*/100 0,2 47
66144 TF-200*/100 0,2 90
66145 TF-200*/25 0,2 142
66146 TF-200*/25 0,2 293
66147 TF-450*/100 0,45 ' 13
66148 TF-450*/100 0,45 25
66149 TF-450*/100 0,45 47
66150 TF-450*/100 0,45 90
66151 TF-450*/25 0,45 142
66152 TF-450*/25 0,45 293
66153 TF-1000*/100 1 13
66154 TF-1000*/100 1 25
66155 TF-1000*/100 1 47
66156 TF-1000*/100 1 90
66157 TF-1000*/25 1 142
66158 TF-1000*/25 1 293
Предфильтры полипропиленовые
61754/500 10 13
61756/500 10 25
61757/100 10 47
61789/100 10 76
61790/100 10 90
61795/100 10 127
61720/100 10 42
61801/100 10 257
61761/100 10 265
61753/100 10 293
Стекловолоконные фильтры Гласс Фибер Фильтерс А
61715 А/500 1 37
61694 А/100 1 47
61696 А/100 1 102
61701 А/100 1 203 х 254
Гласс Фибер Фильтерс А/Е
61628 А/Е/500 1 13
62629 А/Е/500 1 20
62630 А/Е/500 1 25
62652 А/Е/500 1 37
62631 А/Е/100 1 47
71632 А/Е/100 1 50
61663 А/Е/100 1 76
61664 А/Е/100 1 90
61633 А/Е/100 1 102
61669 А/Е/100 1 127
61635 А/Е/100 1 142
61675 А/Е/100 1 257
61636 А/Е/100 1 265
61637 А/Е/100 1 293
61638 А/Е/100 1 203 х 254
Гласс Фибер Фильтерс Extra Thick
66073 Extra Thick/100 1 13
66075 Extra Thick/100 1 25
232
Новый справочник химика и технолога
Продолжение таблицы 3.81
Номер по каталогу, тип /кол-во в упаковке (шт.) Размер nop, нм • IO"3 Диаметр или линейные размеры, мм
66077 Extra Thick/1 ООО 1 45
66078 Extra Thick/100 1 47
66080 Extra Thick/100 1 76
66081 Extra Thick/100 1 90
66084 Extra Thick/50 1 127
66085 Extra Thick/50 1 142
66086 Extra Thick/25 1 257
66087 Extra Thick/25 1 265
66088 Extra Thick/25 1 293
Метригард
64794 Metrigard/100 0,7 13
64795 Metrigard/100 0,7 20
64796 Metrigard/100 0,7 25
64798 Metrigard/100 0,7 47
64790 Metrigard/100 0,7 90
64802 Metrigard/100 0,7 127
64803 Metrigard/100 0,7 142
64805 Metrigard/25 0,7 265
64807 Metrigard/25 0,7 293
Микрокварц
66096 Glass Fiber/100 1 25
66097 Glass Fiber/100 1 37
66098 Glass Fiber/100 1 47
66099 Glass Fiber/100 1 102
66100 Glass Fiber/100 1 203 х 254
Спектро Грейд
64876 Glass Fiber/100 N/A 25
64877 Glass Fiber/100 N/A 37
64878 Glass Fiber/100 N/A 47
64879 Glass Fiber/100 N/A 50
64881 Glass Fiber/100 N/A 102
64884 Glass Fiber/100 N/A 142
64948 Glass Fiber/100 N/A 203 х 254
Примечания. *Гладкие. **С сеткой. ***Целлюлозные. 4 Черные. З3еленые.
Таблица 3.82
Мембраны, выпускаемые фирмой «Миллипор корпорейшн» [129]
Тип, номер по каталогу/кол-во в упаковке (шт.) Размер пор, нм • 10~3 Диаметр, мм
Дисковые мембраны «Дюпар» гидрофобные
GVHP 01300*б/100 0,22 13
GVHP 02500*б/100 0,22 25
GVHP 04700*6/100 0,22 47
GVHP 09050*б/50 0,22 90
GVHP14250*6/50 0,22 142
GVHP 29325*ё/25 0,22 293
HVHP01300*6/100 0,45 13
HVHP 02500*б/100 0,45 25
Продолжение таблицы 3.82
Тип, номер по каталогу/кол-во в упаковке (шт.) Размер пор, нм • 1(Г3 Диаметр, мм
HVHP 04700*б/100 0,45 47
HVHP 09050*б/50 0,45 90
HVHP 14250*б/50 0,45 142
HVHP 29350*б/25 0,45 293
Гидрофобные складчатой конфигурации
CVHP 293МР*б/25 0,22 < 293
HVHP 293 МР*б/25 0,45 293
Гидрофильные
GVWP 01300*ж/100 0,22 13
GVWP 02500*ж/100 0,22 25
GVWP 04700*ж/100 0,22 47
GVWP 09050*ж/50 0,22 90
GVWP14250*’K/50 0,22 142
GVWP 29325*ж/25 0,22 293
HVLP 01300*ж/100 0,45 13
HVLP 02500*ж/100 0,45 25
HVLP 04700*ж/100 0,45 47
HVLP 09050*ж/50 0,45 90
HVLP 14250*ж/50 0,45 142
HVLP 29325 *ж/25 0,45 293
Гидрофобные складчатой конфигурации
GVWP 293 МР*ж/25 0,22 293
HVLP 293 МР*ж/25 0,45 293
Мембраны «Миллипор» MF
SCWP 01300*б/100 8,0 13
SCWP 02400*б/100 8,0 24
SCWP 02500*б/100 8,0 25
SCWP 04700*б/100 8,0 47
SCWP 09000*б/100 8,0 90
SCWP 09025*б/25 8,0 90
SCWP 14200*б/10 8,0 142
SCWP 14250*б/50 8,0 142
SCWP 29300*б/5 8,0 293
SCWP 29325*б/25 8,0 293
SMWP 01300*б/100 5,0 13
SMWP 02400*б/100 5,0 24
SMWP 02500*б/100 5,0 25
SMWP 04700*б/100 5,0 47
SMWP 09000*б/100 5,0 90
SMWP 09025*б/25 5,0 90
SMWP 14200*б/10 5,0 142
SMWP 14250*б/50 5,0 142
SMWP 29300*б/5 5,0 293
SMWP 29325*б/25 5,0 293
SSWP 01300*б/100 3,0 13
SSWP 02400*б/100 3,0 24
SSWP 02500*б/100 3,0 25
SSWP 04700*б/100 3,0 47
SSWP 09000*б/100 3,0 90
SSWP 09025*б/25 3,0 90
SSWP 14200*б/10 3,0 142
Методы разделения и концентрирования
233
Продолжение таблицы 3.82
Тип, номер по каталогу/кол-во в упаковке (шт.) Размер nop, нм • IO-3 Диаметр, мм
SSWP 14250*750 3,0 142
SSWP 29300*75 3,0 293
SSWP 29325*725 3,0 293
RAWG 01300*7100 1,2 13
RAWG 01300**7100 1,2 13
RAWG 02400*7100 1,2 24
RAWG 02400**7100 1,2 24
RAWG 02500*б/100 1,2 25
RAWG 02500**б/100 1,2 25
RAWG 04700*б/100 1,2 47
RAWG 04700**7100 1,2 47
RAWG 09000*7100 1,2 90
RAWG 09025 *б/25 1,2 90
RAWG 14200*710 1,2 142
RAWG 14250*750 1,2 142
RAWG 29300*75 1,2 293
RAWG 29325 *б/25 1,2 293
AAWP 01300*б/100 0,8 13
AAWG 01300**7100 0,8 13
ААВР 01300*7100 0,8 13
AABG 01300**7100 0,8 13
AAWP 02400*7100 0,8 24
AAWG 02400**б/100 0,8 24
AAWP 02500*7100 0,8 25
AABG 02500**7100 0,8 25
ААВР 02500*7100 0,8 25
AABG 02500**7100 0,8 25
AAWP 04700*7100 0,8 47
AAWG 04700**7100 0,8 47
ААВР 04700*7100 0,8 47
ААВР 04700**7100 0,8 47
AAWP 09000*7100 0,8 90
AAWG 09000*725 0,8 90
AAWP 14200*710 0,8 142
AAWP 14250*750 0,8 142
AAWP 29300*75 0,8 293
AAWP 29325*725 0,8 293
AAWP 81010*710 0,8 203 x 254
AAWP 81000*7100 0,8 203 x 254
DAWP 01300*7100 0,65 13
DAWG 01300**7100 0,65 13
DAWP 02500*7100 0,65 25
DAWG 02500**7100 0,65 25
DAWP 04700*7100 0,65 47
DAWG 04700**7100 0,65 47
DAWP 09000*7100 0,65 90
DAWP 09025*725 0,65 90
DAWP 14200*710 0,65 142
DAWP 14250*750 0,65 142
DAWP 29300*75 0,65 293
DAWP 29325*725 0,65 293
Продолжение таблицы 3.82
Тип, номер по каталогу/кол-во в упаковке (шт.) Размер nop, нм • 10-3 Диаметр, мм
НАWP 01300*7100 0,45 13
HAWG 01300**7100 0,45 13
НАВР 01300*7100 0,45 13
HABG 01300**7100 0,45 13
НАМК 02412*7100 0,45 24
HAWP 02400*7100 0,45 , 24
HAWP 02412*7100 0,45 24
НА WG 02400**7100 0,45 24
HAWP 02500*7100 0,45 25
HAWG 02500**7100 0,45 25
НАВР 02500*7100 0,45 25
HABG 02500**7100 0,45 25
HAWP 04700*7100 0,45 47
HAWG 04700**7100 0,45 47
НАВР 04700*7100 0,45 47
HABG 04700**7100 0,45 47
HAWP 09000*7100 0,45 90
HAWP 09025*725 0,45 90
HAWP 14200*710 0,45 142
HAWP 14250*750 0,45 142
HAWP 29300*75 0,45 293
HAWP 29325*725 0,45 293
HAWP 81010*710 0,45 203 x 254
HAWP 81000*7100 0,45 203 x 254
phwp 01300*7100 0,30 13
PHWP 02400*7100 0,30 24
PHWP 02500*7100 0,30 25
PHWP 04700*7100 0,30 47
PHWP 09000*7100 0,30 90
PHWP 09025*725 0,30 90
PHWP 14200*710 0,30 142
PHWP 14250*750 0,30 142
PHWP 29300*75 0,30 293
PHWP 29325*725 0,30 293
GSWP 01300*7100 0,22 13
GSWP 02400*7100 0,22 24
GSWP 02500*7100 0,22 25
GSWP 04700*7100 0,22 47
GSWP 09000*7100 0,22 90
GSWP 09025*725 0,22 90
GSWP 14200*710 0,22 142
GSWP 14250*750 0,22 142
GSWP 29300*75 0,22 293
GSWP 29325*725 0,22 293
vcwp 01300*7100 0,10 13
VCWP 02400*7100 0,10 24
VCWP 02500*7100 0,10 25
VCWP 04700*7100 0,10 47
VCWP 09000*7100 0,10 90
VCWP 09025*725 0,10 90
VCWP 14200*710 0,10 142
234
Новый справочник химика и технолога
Продолжение таблицы 3.82
Тип, номер по каталогу/кол-во в упаковке (шт.) Размер пор, нм • 10-3 Диаметр, мм
VCWP 14250*б/50 0,10 142
VCWP 29300*6/5 0,10 293
VCWP 29325*6/25 0,10 293
VMWP 01300*6/100 0,05 13
VMWP 02400*6/100 0,05 24
VMWP 02500*6/100 0,05 25
VMWP 04700*6/100 0,05 47
VMWP 09025*6/100 0,05 90
VMWP 09000*6/100 0,05 90
VMWP 14200*6/10 0,05 142
VMWP 14250*6/50 0,05 142
VMWP 29300*6/5 0,05 293
VMWP 29325*6/25 0,05 293
VSWP 01300*6/100 0,025 13
VSWP 02400*6/100 0,025 24
VSWP 02500*6/100 0,025 25
VSWP 04700*6/100 0,025 47
VSWP 09000*6/100 0,025 90
VSWP 09025*6/25 0,025 90
VSWP 14200*6/10 0,025 142
VSWP 14250*6/50 0,025 142
VSWP 29300*6/5 0,025 293
VSWP 29325*6/25 0,025 293
Складчатой конфигурации
SMMP 29300*6/100 293 5,0
SSMP 29320*6/20 293 3,0
RAMP 29300*6/100 293 1,2
RAMP 29320*6/20 293 1,2
AAMP 29300*6/100 293 0,8
AAMP 29320*6/20 293 0,8
DAMP 29300*6/100 293 0,65
DAMP 29320*6/20 293 0,65
HAMP 29300*6/100 293 0,45
HAMP 29320*6/20 293 0,45
GSMP 29300*6/100 293 0,12
GSMP 29320*6/20 293 0,22
Для заправки мониторов, с тонкими абсорбирующими подложками
SCWP 03700*6/100 37 8,0
SMWP 03700*6/100 37 5,0
SSWP 03700*6/100 37 3,0
RAWP 03700*6/100 37 1,2
AAWP 03700*6/100 37 0,8
AAWG 03700**6/100 37 0,8
AABG 03700**4/100 37 0,8
HAWP 03700*6/100 37 0,45
HAWG 03700**6/100 37 0,45
HABG 03700**4/100 37 0,45
GSWP 03700*6/100 37 0,22
Для заправки мониторов, с толстыми абсорбирующими подложками
Продолжение таблицы 3.82
Тип, номер по каталогу/кол-во в упаковке (шт.) Размер пор, нм • 103 Диаметр, мм
SMWP 037 РО*б/Ю0 37 5,0
AAWP 037 РО*6/ЮО 37 0,8
AAWG 037РО**б/Ю0 37 0,8
HAWG 037РО**б/Ю0 37 0,45
YAWP 037РО*6/ЮО 37 0,45
Для заправки мониторов, взвешенные попарно, с толстыми абсорбирующими подложками
AAWP 037 РМ 37 0,8
Упакованные для автоклавирования, с абсорбирующими подложками
RAWP 047 АО*б/100 47 1,2
AAWP 047 АО*6/ЮО 47 0,8
AAWG 047 АО**6/ЮО 47 0,8
DAWP 047 АО*б/100 47 0,65
DAWG 047 АО**6/ЮО 47 0,65
HAWP 047 АО*б/100 47 0,45
HAWG 047 АО**6/ЮО 47 0,45
GSWP 047 АО*б/100 47 0,22
Стерильно упакованные, с абсорбирующими подложками
AAWP 047 SO*6/100 47 0,8
AAWG 047 SO**6/100 47 0,8
ААВР 047 SO*4/100 47 0,8
AABG 047 SO**’/100 47 0,8
DAWP 047 SO*6/100 47 0,65
DAWG 047 SO**6/100 47 0,65
HAWP 047 SO*6/100 47 0,45
HAWG 047 SO**6/100 47 0,45
HABP 047 SO*4/100 47 0,45
HABG 047 SO**4/100 47 0,45
GSWP 047 SO*6/100 47 0,22
S-pak, стерильные, индивидуально запечатанные
HAWG 047 SI *6/100 47 0,45
HCWG047 SI**6/100 47 0,7
HCWG 047 S3*6/100 47 0,7
S-pak, стерильные, индивидуально запечатанные, с абсорбирующими подложками
HAWG 047 S2**6/1000 47 0,45
HCWG 047 S4* *6/200 47 0,7
Взвешенные попарно
AAWP 047 ОМ*б/50 пар 47 0,8
HAWP 047 ОМ*б/50 пар 47 0,45
Сверхтонкие (толщиной 25 мкм)
ТНМР 01300*6/100 13 0,30
THWP 01400*6/100 14 0,30
THWP 02500*6/100 25 0,30
THWP 04700*6/100 47 0,30
THWP 10200*6/100 102 х 102 0,30
Сверхтонкие, взвешенные попарно
THWP 013 ОМ*б/20 пар 13 0,30
Прямоугольные
SCWP 019GR*6/100 — 8,0
Методы разделения и концентрирования
235
Продолжение таблицы 3.82
Тип, номер по каталогу/кол-во в упаковке (шт.) Размер nop, нм • 10-3 Диаметр, мм
SMWP 0190R*6/100 — 5,0
SSWP 0190R*6/100 — 3,0
С гидрофобными краями (6 мм)
SMEP 0470W*7100 47 5,0
SMEP 0900W*6/100 90 5,0
SSEP 047 OW*6/100 47 3,0
SSEP 090 OW*6/100 90 3,0
RAEP 047 OW*7100 47 1,2
AAEP 047 OW*6/100 47 0,8
HAEP 047 OW*6/100 47 0,45
HAEP 090 OW*6/100 90 0,45
GSEP 047 OW*7100 47 0,22
С гидрофобными краями (6 мм), упакованные для автоклавирования
НАЕР 047 AW*7100 47 0,45
HAEG 047 AW*7100 47 0,45
С гидрофобными краями (6 мм), стерильно упакованные
НАЕР 047 SW*6/100 47 0,45
С гидрофобными краями (3 мм), стерильно упакованные
НАЕР 047 S0*6/100 47 0,45
HAEG 047 S0*6/100 47 0,45
GSEP 047 SO*6/100 47 0,22
Мембраны TF, слабо экстрагируемые водой
RATF 14250*750 142 1,2
RATF 29325 *6/25 293 1,2
AATF 29325*6/25 293 0,8
DATF 29325*725 293 0,65
HATF 01300*7100 13 0,45
HATF 02500*7100 25 0,45
HATF 04700*7100 47 0,45
HATF 14250*750 142 0,45
HATF 29325*725 293 0,45
PHTF 29325 *6/25 293 0,30
GSTF 01300*7100 13 0,22
GSTF 02500*7100 25 0,22
GSTF 04700*6/100 47 0,22
GSTF 14200**710 142 0,22
GSTF 14250**750 142 0,22
GSTF 29325**725 293 0,22
Складчатой конфигурации
RAMT 29300**7100 293 1,2
HAMT 29300**7100 293 0,45
Мембраны «Поливик»
BSWP 01300**7100 13 2,0
BSWP 02500**7100 25 2,0
BSWG 02500***7100 25 2,0
BSWP 04700**7100 47 2,0
BSWG 04700***7100 47 2,0
BSWP 09000**7100 90 2,0
BSWP 09025**725 90 2,0
BSWP 14200**710 142 2,0
Продолжение таблицы 3.82
Тип, номер по каталогу/кол-во в упаковке (шт.) Размер nop, нм • KT3 Диаметр, мм
BSWP 14250**750 142 2,0
BSWP 29300**75 293 2,0
BSWP 29325**725 293 2,0
BDWP 01300**7100 13 0,6
BDWP 02500*7100 25 0,6
BDWP 04700*7100 47 0,6
BDWG 04700**7100 47 0,6
BDWP 09000*7100 90 0,6
BDWP 14200*710 142 0,6
BDWP 14250*750 142 0,6
BDWP 29325*725 293 0,6
Складчатой конфигурации
BDMP 29300*7100 293 0,6
Для заправки мониторов, с тонкими абсорбирующими подложками
BSWP 03700*7100 37 2,0
BDWP 03700*7100 37 0,6
Мембраны для щелочного элюирования
BSAE 02500*7100 25 2,0
Мембраны «Флюоропор»
FSLW 01300*7100 13 3,0
FSLW 02500*7100 25 3,0
FSLW 04700*7100 47 3,0
FSLW 09000*725 90 3,0
FSLW 14200*710 142 3,0
FSLW 29300*75 293 3,0
FSLW 81005*75 203 x 254 3,0
FSLW 81050*750 203 x 254 3,0
FALP 01300*7100 13 1,0
FALP 02500*7100 25 1,0
FALP 04700*7100 47 1,0
FALP 09025*725 90 1,0
FALP 09050*750 90 1,0
FALP 14200*710 142 1,0
FALP 14250*750 142 1,0
FALP 29300*75 293 1,0
FALP 29325*725 293 1,0
FALP 81005*75 203 x 254 1,0
FALP 81050*750 203 x 254 1,0
FHLP 01300*7100 13 0,5
FHLP 02500*7100 25 0,5
FHLP 04700*7100 47 0,5
FHLP 09025*725 90 0,5
FHLP 09050*750 90 0,5
FHLP 14200*710 142 0,5
FHLP 14250*750 142 0,5
FHLP 29300*75 293 0,5
FHLP 29325*725 293 0,5
FGLP 01300*7100 13 0,2
FGLP 02500*7100 25 0,2
FGLP 04700*7100 47 0,2
236
Новый справочник химика и технолога
Продолжение таблицы 3.82
Тип, номер по каталогу/кол-во в упаковке (шт.) Размер nop, нм • 10~3 Диаметр, мм
FGLP 09025*б/25 90 0,2
FGLP 09050*6/10 90 0,2
FGLP 14200*6/25 142 0,2
FGLP 14250*б/100 142 0,2
FGLP 29300*6/120 293 0,2
FGLP 29325 *6/100 293 0,2
Складчатой конфигурации
FSMP 29320*б/20 293 3,0
FAMP 29320*6/20 293 1,0
FHMP 29320*б/20 293 0,5
FGMP 29320*6/20 293 0,2
Для заправки мониторов, с тонкими абсорбирующими подложками
FSLW 03700*6/100 37 3,0
FALP 03700*б/100 37 1,0
FHLP 03700*6/100 37 0,5
FHUP 02500*6/100 25 0,5
FHUPW 04700*6/100 47 0,5
Мембраны «Селотейт»
EAWP 01300*6/100 13 1,0
EAWP 02500*б/100 25 1,0
EAWP 04700*6/100 47 1,0
EAWP 09025 *6/25 90 1,0
EAWP 14200*6/10 142 1,0
EAWP 29300*б/25 293 1,0
EHWP 01300*6/100 13 0,5
EHWP 02412*6/120 24 0,5
EHWP 02500*6/100 25 0,5
EHWP 04700*б/100 47 0,5
EHWP 09025 *6/25 90 0,5
EHWP 14200*6/10 142 0,5
EHWP 29300*6/25 293 0,5
EGWP 01300*6/100 13 0,2
EGWP 02500*6/100 25 0,2
EGWP 04700*6/100 47 0,2
EGWP 09025*6/25 90 0,2
EGWP 14200*6/10 142 0,2
EGWP 29300*6/25 293 0,2
Мембраны «Митекс»
LCWP 01300*6/100 13 10,0
LCWG 01300**6/100 13 10,0
LCWP 02500*6/100 25 10,0
LCWG 02500**6/100 25 10,0
LCWP 04700*6/100 47 10,0
LCWG 04700* *6/l 00 47 10,0
LCWP 09000*6/100 90 10,0
LCWP 09025 *6/25 90 10,0
LCWP 14200*6/10 142 10,0
LCWP 14250*6/50 142 10,0
LCWP 29300*6/5 . 293 10,0
LCWP 29325 *6/25 293 10,0
Продолжение таблицы 3.82
Тип, номер по каталогу/кол-во в упаковке (шт.) Размер пор, нм • 10“3 Диаметр, мм
LSWP 01300*6/100 13 5,0
LSWG 01300**б/100 13 5,0
LSWP 02500*6/100 25 5,0
LSWG 02500**6/100 25 5,0
LSWP 04700*б/100 47 5,0
LSWG 04700* *6/100 47 5,0
LSWP 09000*б/100 90 5,0
LSWP 09025*6/25 90 5,0
LSWP 14200*6/10 142 5,0
LSWP 14250*6/50 142 5,0
LSWP 29300*6/5 293 5,0
LSWP 29325*6/25 293 5,0
Для заправки мониторов, с тонкими абсорбирующими подложками
LSWP 03700*6/100 37 5,0
Мембраны для контороля воздуха PVC Мембраны, не содержащие диоксида кремния
PVC5 03700*б/100 37 5,0
Серебряные мембраны для контороля воздуха
AG80 03700*б/100 37 0,8
AG45 02500*6/100 25 0,45
Мембраны для гибридизации ДНК, РНК Нитроцеллюлозные мембраны для процесса Сазерна
HAHY 08250*6/50 82 0,45
HAHY 08550*6/50 85 0,45
HAHY 13750*6/50 137 0,45
HAHY 00010*6/303 х 3048 305 х 3048 0,45
HAHY 304FO*6/10 305 х 305 0,45
Мембраны для выращивания бактериальных колоний
НАТТ 08250*б/50 82 0,45
HATF 08550*6/50 85 0,45
НАТТ 13750*6/50 137 0,45
Мембраны гидрофобные «Дюрапор» для переноса пятен
GVHP 02500*6/100 25 0,2
GVHP 29325 *6/25 293 0,2
GVHP 00010*6/305 х 3048 305 х 3048 0,2
Примечания. *Гладкие. **С сеткой. 6 Белые. жЖелтые.
Таблица 3.83
Мембраны и сепараторы для кассетной системы «Пелликон» [129]
Тип и номер по каталогу НОММ или размер пор, мкм Материал
Кассеты в комплекте (0,45 м2 на упаковку)
РСАС 00005 1000 Целлюлоза
PTGC 00005 10000 Полисульфон
PLGC 00005 10000 Целлюлоза
РТТК 00005 30000 Полисульфон
РТНК 00005 100000 Полисульфон
Методы разделения и концентрирования
237
Продолжение таблицы 3.83
Тип и номер по каталогу HOMM или размер пор, мкм Материал
РТНК 00005 100000 Полисульфон (жесткий задерживающий)
PSVP 00005 1000000 Целлюлоза
HYLP 00С5 0,5 Дюрапор (жесткий задерживающий)
Пакеты в комплекте (0,05 м2 на упаковку; требуются задерживающие экраны)
PTGC 00001 10000 Полисульфон
РТТК 00001 30000 Полисульфон
РТНК 00001 PSVP 00001 100000 1000000 Полисульфон
Листы (0,002 м2 каждый, 20 шт. в упаковке; требуются экраны для осадка и фильтрата)
GSWP OHV20 0,2 Нитроцеллюлоза
HAWP OHV20 0,45 Нитроцеллюлоза
RAWP OHV20 1,2 Нитроцеллюлоза
EGWP OHV20 0,2 Ацетицеллюлоза
GVWP OHV20 0,2 Дюрапор
HVLP OHV20 0,5 Дюрапор
Таблица 3.84
Предфильтры и подложки для мембран фирмы «Миллипор корпорейшн» [129]
Тип и номер по каталогу Диаметр, ММ Кол-во в упаковке, шт Тип и номер по каталогу Диаметр, мм К-во в упаковке, шт
Дисковые мембраны АР 15
API 50 2200 22 100 АР 150 9000 90 100
API 50 2500 25 100 АР151 2450 124 50
АР150 4200 42 100 АР151 4250 142 50
API 50 4700 47 100 АР152 5725 257 25
API 50 7500 75 100 АР 152 9325 293 25
Дисковые мембраны АР20
АР20 010 00 10 100 АР20 075 00 75 100
АР20 013 00 13 100 АР20 090 00 90 100
АР20 020 00 20 100 АР20 124 50 124 50
АР20 022 00 22 100 АР20 142 50 142 50
АР20 025 00 25 100 АР20 257 25 257 25
АР20 035 00 35 100 APMS 275 00 275 100
АР20 042 00 42 100 АР20 293 25 293 25
Продолжение таблицы 3.84
Тип и номер по каталогу Диаметр, мм Кол-во в упаковке, шт Тип и номер по каталогу Диаметр, мм К-во в упаковке, шт
АР20 047 00 47 100 APMS 293 00 293 100
Дисковые мембраны АР25
АР25 010 00 10 100 АР25 090 00 90 100
АР25 013 00 13 100 АР25 124 50 124 50
АР25 018 00 18 100 АР25 127 50 127 50
АР25 020 00 20 100 АР25 142 50 142 50
АР25 022 00 22 100 АР25 257 25 257 25
АР25 025 00 25 100 APTS 275 20 275 20
АР25 035 00 35 100 АР25 293 25 293 25
АР25 042 00 42 100 АРТР 293 20 293 20
АР25 047 00 47 100 APTS 293 20 293 20
АР25 075 00 75 100
Дисковые стекловолоконные микрофильтры АР40 (только для анализа; нескрепленное волокно)
АР40 025 05 24 500 АР40 047 05 47 500
АР40 037 05 37 500
Таблица 3.85
Подложки для мембран фирмы «Миллипор корпорейшн» [129]
Номер по каталогу Диаметр, мм Кол-во в упаковке, шт Описание
Абсорбирующие подложки АР 10
АР10 013 00 13 100 Подложки
АР10 024 00 24 100 То же
АР10 025 00 25 100 То же
АР10 037 00 37 100 То же
АР 10 047 00 47 100 Стерильные
АР10 047 00 47 200 Стерильные, с распределителем
АР 10 047 00 47 100 Подложки
АР10 142 S0 142 50 То же
Толстые поддерживающие подложки АРЗО
АРЗО 034 Р0 34 100
Тканые сетчатые прокладки АР32
АР32 042 00 42 100
АР32 075 00 75 100
АР32 124 00 124 10
АР32 124 50 124 50
АР32 257 00 257 5
АР32 257 25 257 25
АР32 275 OS 275 100
238
Новый справочник химика и технолога
Таблица 3.86
Ультрафильтры фирмы «Миллипор корпорейшн» (количество штук в упаковке: 10 — для дисковых мембран диаметром 13-76 мм; 5 — для мембран диаметром 90-150 мм) [129]
Материал Тип НОММ Диаметр, мм
13 25 43 47 62 76 90 142 150
Целлюлоза РСАС 1000 01310 02510 04310 04710 06210 07610 09005 14205 15005
То же PLGC 10000 01310 02510 04310 04710 06210 07610 09005 14205 15005
Полисульфон PTGC 10000 01310 02510 04310 04710 06210 07610 09005 14205 15005
То же РТТК 30000 01310 02510 04310 04710 06210 07610 09005 14205 15005
То же РТНК 100000 01310 02510 04310 04710 06210 07610 09005 14205 15005
Целлюлоза PSVP 1000000 01310 02510 04310 04710 06210 07610 09005 14205 15005
Таблица 3.87
Дисковые мембраны фирмы «Нуклепор корпорейшн» (номера по каталогу) [129]
Диаметр, мм Кол-во в упаковке, шт Размер пор, нм • 10 3
12,0 10,0 8,0 5,0 3,0 2,0 0,1 0,8
13 100 110416 110415 110414 110413 110412 110411 110410 110409
25 100 110616 110615 110614 110613 110612 110611 110610 110609
37 100 110816 110815 110814 110813 110812 110811 110810 110809
43 100 111016 111015 111014 111013 111012 111011 111010 111009
47 100 111116 111115 111114 111113 111112 111111 111110 111109
50 100 111216 111215 111214 111213 111212 111211 111210 111209
76 100 111516 111515 111514 111513 111512 111511 111510 111509
90 10 111616 111615 111614 111613 111612 111611 111610 111609
90 25 111716 111715 111714 111713 111712 111711 111710 111709
90 100 111816 111815 111814 111813 111812 111811 111810 111809
142 10 112016 112015 112014 112013 112012 112011 112010 112009
142 25 112116 112115 112114 112113 112112 112111 112110 112109
142 50 112216 112215 112214 112213 112212 112211 112210 112209
13 100 110408 110407 110406 110405 110404 110403 110402 110401
25 100 110608 110607 110606 110605 110604 110603 110602 110601
37 100 110808 110807 110806 110805 110804 110803 110802 110801
43 100 111008 111007 111006 111005 111004 111003 111002 111001
47 100 111108 111107 111106 111105 111104 111103 111102 —
50 100 111208 111207 111206 111205 111204 111203 111202 —
76 100 111508 111507 111506 111505 111504 111503 111502 —
90 10 111608 111607 111606 111605 111604 111603 111602 —
90 25 111708 111707 111706 111705 111704 111703 111702 —
90 100 111808 111807 111806 111805 111804 111803 111802 —
142 10 112008 112007 112006 112005 112004 112003 112002 —
142 25 112108 112107 112106 112105 112104 112103 112102 —
142 50 112208 112207 112206 112205 112204 112203 112202 —
293 10 112708 112707 112706 112705 112704 112703 112702 —
293 25 112808 112807 112806 112805 112804 112803 112802 —
293 100 112908 112907 112906 112905 112904 112903 112902 —
Методы разделения и концентрирования
239
Таблица 3.88
Прямоугольные мембраны фирмы «Нуклепор корпорейшн» [129]
Размер пор, нм • 10-3 Размер мембраны, мм Кол-во в упаковке, шт Номер по каталогу
12,0 19x42 100 113316 113308
12,0 203 х 254 10 113416 113408
12,0 203 х 254 25 113516 113508
12,0 203 х 254 100 113616 113608
10,0 19x42 100 113315 113307
10,0 203 х 254 10 113415 113407
10,0 203 х 254 25 113515 113507
10,0 203 х 254 100 113615 113607
8,0 19x42 100 113314 113306
8,0 203 х 254 10 113414 113406
8,0 203 х 254 25 113514 113506
8,0 203 х 254 100 113614 113606
5,0 19x42 100 113313 113305
5,0 203 х 254 10 113413 113405
5,0 203 х 254 25 113513 113505
5,0 203 х 254 100 113613 113605
3,0 19x42 100 113312 113304
3,0 203 х 254 10 113412 113404
3,0 203 х 254 25 113512 113504
3,0 203 х 254 100 113612 113604
1,0 19x42 100 113310 113303
1,0 203 х 254 10 113410 113403
1,0 203 х 254 25 113510 113503
1,0 203 х 254 100 113610 113603
0,8 10x42 100 113309 113302
0,8 203 х 254 10 113409 113402
0,8 203 х 254 25 113509 113502
0,8 203 х 254 100 113609 113602
Таблица 3.89
Мембранные фильтры фирмы «Сарториус» (белые) [129]
Размер пор, нм • 10-3 Материал мембраны Номер по каталогу Диаметр дисковых мембран и тип упаковки
13 20 24 25 30 37 40
8 Нитроцеллюлоза SM 11301 N N N N N N N
5 Нитроцеллюлоза SM 11342 N N N N N N
5 Тефлон SM 11842 N N N
3 Нитроцеллюлоза SM 11302 N N N N N N
1,2 Нитроцеллюлоза SM11 303 N N N N N N N
1,2 Тефлон SM 11803 N N N
Продолжение таблицы 3.89
Размер пор, нм • 10~3 Материал мембраны Номер по каталогу Диаметр дисковых мембран и тип упаковки
13 20 24 25 30 37 40
1,2 Полиамид SM 11903 N N N
1,2 Ацетилцеллюлоза SM 12303 N N
0,8 Ацетилцеллюлоза SM 11104 N N N N N N N
0,8 Нитроцеллюлоза SM 11304 N N N N N N N
0,8 Регенерированная целлюлоза SM 11604 N N N N N N
0,8 Полиамид SM 11904 N N N N
0,65 Ацетилцеллюлоза SM 11105 N N N N N
0,65 Нитроцеллюлоза SM 11305 N N N N N N
0,65 Регенерированная целлюлоза SM 11605 N N N N
0,65 Полиамид SM 11905 N N N N N
0,45 Ацетилцеллюлоза SM 11106 N N N N N N
0,45 Нитроцеллюлоза SM 11306 N N N N N N N
0,45 Регенерированная целлюлоза SM 11606 N N N N N N
0,45 Полиамид SM 11806 N N N
0,45 Селласарт SM 12006 N N N N N
0,3 Нитроцеллюлоза SM 11347 N N N N N N
0,2 Ацетилцеллюлоза SM 11107 N N N N N N
0,2 Нитроцеллюлоза SM 11307 N N N N N N N
0,2 Регенерированная целлюлоза SM 11607 N N N N N
0,2 Полиамид SM 11807 N N N N
0,2 Селласарт SM 12007 N N N
0,15 Нитроцеллюлоза SM 11378 N N N
0,1 Нитроцеллюлоза SM 11358 N N N N
0,05 Нитроцеллюлоза SM 11328 N N N
0,01 Нитроцеллюлоза SM 11318 N N N N N
Примечание-. N — 100 шт.
240
Новый справочник химика и технолога
Таблица 3.90
Мембранные фильтры фирмы «Сарториус» для микробиологических и аналитических целей [129]
Пояснения: ACN — стерильные, отдельно упакованные, 100 шт.; ACR — стерильные, отдельно упакованные, 1000 шт.; APR — стерильные, отдельно упакованные с абсорбирующими подложками (10 пачек по 100) и двумя распределителями; AHN — без сетки, 100 шт.; ALN — стерильные, упакованные вместе, 100 шт.; HCN — с гидрофобными краями (6 мм), 100 шт.; N — 100 шт.; PEN — свободные от фосфатов, 100 шт.
Номер по каталогу Особенности Материал мембраны Размер пор, мкм Цвет Цвет сетки Диаметр мембран (мм) и тип упаковки
13 24 25 47 50 80
SM 12500 Нитроцеллюлоза 12 Белый N N, ACN N, ACN
SM 13001 Нитроцеллюлоза 8 Серый Белый N N N
SM 13101* Гидрофобные края Нитроцеллюлоза 8 Белый Черный N N,AHN N, AHN
SM 12602* Желатин 3 Белый ALN ALN ALK
SM 11403* Нитроцеллюлоза 1,2 Белый Черный N N, ACN
SM 11404** Нитроцеллюлоза 0,8 Белый Черный N N, ACN N
SM 13004** Нитроцеллюлоза 0,8 Серый Белый N N, ACN N
SM 13704** Стерильные, с абсорбционной подложкой Нитроцеллюлоза 0,8 Белый Черный ALN
SM 11405 Нитроцеллюлоза 0,65 Белый Черный N N, ACN
SM 13005 Нитроцеллюлоза 0,65 Серый Белый N N, ACN
SM 11306*** Свободные от фосфатов Нитроцеллюлоза 0,45 Белый PEN PEN
SM 11336*** Нитроцеллюлоза 0,45 Белый N N N N, ACN N, ACN
SM 11406*** Нитроцеллюлоза 0,45 Белый Черный N N, ACN N, ACN
SM 13006 Нитроцеллюлоза 0,45 Серый Белый N N, ACN N, ACN
SM 13106 Гидрофобные края Нитроцеллюлоза 0,45 Белый Черный N HCN, PEN
SM 13506 Гидрофобные края Ацетилцеллюлоза 0,45 Белый Черный N, ACN, HCN N, ACN
SM 13706 Стерильные, с абсорбционной подложкой Нитроцеллюлоза 0,45 Белый Черный ALN ALN
SM 13806 Нитроцеллюлоза 0,45 Зеленый Зеленый N, ACN, ACR N, ACN, ACR
SM 13906 Нитроцеллюлоза 0,45 Белый Зеленый N, ACN, ACR, APR N, ACN, ACR, APR
SM 11407 Нитроцеллюлоза 0,2 Белый Черный N N, ACN N, ACN
SM 13107 Гидрофобные края Нитроцеллюлоза 0,2 Белый Черный N N, ACN N, ACN
Методы разделения и концентрирования
241
Продолжение таблицы 3.90
Номер по каталогу Особенности Материал мембраны Размер пор, МКМ Цвет Цвет сетки Диаметр мембран (мм) и тип упаковки*
13 24 25 47 50 80
SM 13507 Гидрофобные края Ацетилцеллюлоза 0,2 Белый Черный N, ACN N, ACN
SM 13707 Стерильные, с абсорбционной подложкой Ацетилцеллюлоза 0,2 Белый Черный ALN ALN
Примечание.
* Выпускаются также стерильные упакованные вместе мембраны диаметром 80 мм (по 50 шт. в упаковке).
** Выпускаются также мембраны диаметром 80 мм (по 100 шт. в упаковке).
*** Выпускаются также мембраны диаметром 13 и 24 мм (по 100 шт. в упаковке).
3.4. Методы внутрифазного разделения
Последняя из групп методов разделения объединяет методы, основанные на различиях в свойствах ионов, атомов или молекул, проявляемых в пределах одной гомогенной системы при воздействии электрического, магнитного, гравитационного, теплового полей или центробежных сил. При этом не исключается возможность фазовых превращений при переводе исходной смеси веществ в то агрегатное состояние, в котором происходит разделение, или при выделении фракций ее отдельных компонентов. Эффект разделения достигается за счет различного пространственного перемещения веществ в пределах фазы, в которой происходит их разделение. Различия в скорости пространственного перемещения ионов, атомов или молекул будут проявляться в зависимости от их массы, размеров, заряда, энергии взаимодействия частиц с ионами и молекулами, образующими среду, в которой происходит разделение. Относительная роль тех или иных факторов в достижении конечного эффекта разделения, в свою очередь, зависит от природы действующих на них сил. Наиболее очевидный случай — электрофоретическое или, как его иногда называют, элекгромиграционное разделение ионов в растворах за счет различных скоростей их движения в электрическом поле. Здесь важнейшими факторами оказываются размер и заряд иона. Различия в массе и заряде в наибольшей степени проявляются при воздействии на ионизованные частицы ускоряющего электрического поля и отклоняющего магнитного. Этот способ воздействия на систему лежит в основе масс-сепарационного метода. При разделении под воздействием центробежных сил — ультрацентрифугировании определяющим фактором оказывается масса молекул.
Помимо этих, давно уже ставших традиционными, методов внутрифазного разделения, использующих различия в скоростях пространственного перемещения веществ, в последние два десятилетия появилась целая группа методов, получивших в англоязычной литературе название FFF-методов. Буквальный перевод этой аббревиатуры: поле - поток - фракционирование. Соответствующая русская аббревиатура — ППФ. В русскоязычную литературу эти методы вошли как проточное фракционирование в поперечном поле [136]. Сущность методов заключается в проявлении эффекта разделения веществ при движении однофазного потока через узкий канал перпендикулярно направлению действия силового поля. Поле в зависимости от индивидуальных свойств ионов или молекул вещетва в различной степени смещает их из объема потока к одной из стенок канала. Учитывая параболическую форму профиля скоростей потока в узком канале, вещества, плотнее «прижатые к стенке» действующей силой, медленнее смещаются по каналу с потоком и наоборот. Соответственно вещества выходят из канала с различными временами удерживания, а кривая изменения концентрации в потоке — фрактограмма оказывается аналогичной хроматограмме. Поэтому эту группу методов иногда называют однофазной хроматографией, хотя по сущности процессов, вызывающих эффект разделения, хроматографические и ППФ-методы не имеют ничего общего. Отдельные методы этой группы выделяются в зависимости от природы поперечного поля, действующего на разделяемые вещества.
Следовательно, любой из известных методов внутрифазного разделения может быть охарактеризован агрегатным состоянием фазы, в пределах которой происходит разделение, и видом сил, вызывающих пространственные перемещения ионов, атомов или молекул (табл. 3.91).
Таблица 3.91
Методы внутрифазного разделения
Агрегатное состояние фазы, в которой происходит разделение Природа поперечного поля и сил, вызывающих пространственное перемещение ионов, атомов или молекул
электрическое поле электрическое и магнитное поля центробежная сила или гравитационное поле тепловое поле механическое перемещение
Жидкость Электрофорез (электромиграция); ЭППФ — Ультрацентрифугирование; СППФ ТППФ ПППФ
Газ (вакуум) Электрофорез Масс-сепарация Ультрацентрифугирование — —
242
Новый справочник химика и технолога
Для большинства методов этой группы характерно отсутствие четкой границы в приложении к разделению гомогенных и гетерогенных смесей веществ. Например, электрофорез возник и до сих пор иногда рассматривается только как метод разделения коллоидных частиц. Более того, по сути своей — это метод разделения заряженных частиц за счет их различных подвижностей в электрическом поле. В общем случае размеры частиц не оговариваются, и область применения метода охватывает и простые ионы, и макроионы аминокислот, и заряженные частицы коллоидов и взвесей. Аналогично обстоит дело с ультрацентрифугированием и ППФ-методами. Даже в тех случаях, когда метод имеет достаточно четкие границы применимости по размерам или массам разделяемых частиц, их положение на условной шкале дисперсности частиц различной природы не привязано к принятой границе гомогенности. Существование верхней границы чаще всего определяется принципом целесообразности: если задача легко решается более простым методом, нет необходимости использовать более сложный. Наличие нижней границы может быть связано как с объективными факторами, определяемыми природой явления, используемого для разделения, так и с техническими возможностями практической реализации условий, необходимых для осуществления процесса разделения. Наиболее наглядный пример — ультрацентрифугирование. Очевидно, что с помощью ультрацентрифуги можно выделить взвешенные частицы из раствора, но в этом нет необходимости. А при переходе к разделению частиц на молекулярном уровне в случае жидких фаз возможности метода ограничены фракционированием макромолекул. Добиться фракционирования простых молекул удается только в газовой фазе, но при условии разряжения и чрезвычайно высоких скоростей вращения, реализуемых только при магнитной «подвеске» ротора центрифуги.
Для методов внутрифазного разделения в целом характерны сложные аппаратурные решения, и целесообразность их применения в аналитической химии оправдана пропорционально возможностям, которых не имеют другие методы. Самым простым по техническому оформлению является метод электрофоретического (электромигра-ционного) разделения ионов в растворе, имеющий широкие области применения в аналитической химии. Масс-сепарация как метод разделения интересна прежде всего тем, что является основой одного из вариантов широко распространенных методов химического анализа — масс-спектрометрии. Здесь произошло еще более тесное слияние метода разделения и метода конечного определения, чем в случае хроматографических методов анализа. При описании масс-спектрометрического метода обычно даже не упоминается, что он является одним из гибридных методов анализа. Сложность аппаратурного оформления и высокие энергозатраты в масс-сепарационном методе компенсируются универсальностью и практически неограниченной разделительной способностью. Этим объясняется тот факт, что масс-сепарация является одним из основных препаративных технологических методов разделения изотопов.
ППФ-методы, как правило, также отличающиеся относительной сложностью разделительных устройств, привлекли большое внимание аналитиков и успешно развиваются благодаря существенно расширившимся возможностям разделения веществ по размерам и конфигурации молекул.
Остальные методы внутрифазного разделения или находят только технологическое применение, или их применение в химическом анализе ограничено решением частных задач. Так, ультрацентрифугирование является одним из основных методов обогащения изотопов урана. Аналитическое применение ограничено фракционированием макромолекул органических веществ. Из сферы применения электрофореза в газовой фазе можно вспомнить улавливание взвешенных частиц из газовых потоков, в первую очередь, минеральных составляющих или частиц несгоревшего топлива в выбросах тепловых электростанций, котельных и т.п.
3.4.1. Электрофорез в растворе
Электрофорез, ионофорез, ионография, электрохрома-тофорез, элекгрофореграфия, электромиграция — вот далеко не полный перечень синонимов, возникших за более чем вековую историю метода. Наряду с субъективными факторами — желанием авторов каждого нового названия открыть новый метод — есть и объективные причины такого явления. Электрофорез возник в рамках коллоидной химии, как одно из электрохимических явлений, обратных электроосмосу. Применение его для разделения ионов объясняет появление методов ионофореза и ионографии. Наиболее часто употребляемым обобщенным термином для различных вариаций метода является электрофорез, т.е. имеет место возврат к первоначальному названию.
Разделение в электрофоретическом методе осуществляется за счет различия в подвижностях ионов:
z,qE ui - ~, 6лг,т|
где Ui — подвижность иона; zt — абсолютная величина заряда z-ro иона; е — заряд электрона; Е — разность потенциалов; гг — радиус z-ro иона; т| — коэффициент вязкости.
Соответственно коэффициент селективности для элек-тромиграционного метода можно записать как отношение подвижностей разделяемых ионов:
Сам электромиграционный принцип разделения не обладает высокой селективностью, т. к. диапазон изменения z невелик и, как правило, малы различия в радиусах ионов. Чтобы добиться значимого разделения, необходимы методические приемы для проявления максимальных различий в подвижностях или направленное изменение состава раствора с целью изменения величины или даже знака заряда разделяемых веществ. Последний подход основывается на более широком понимании термина за
Методы разделения и концентрирования
243
ряд иона в уравнении (*). При наличии в растворе мгновенно устанавливающихся ионных равновесий, например между различными комплексными соединениями одного элемента, электромиграционная подвижность будет определяться усредненным эффективным зарядом. Величина эффективного заряда определяется соотношением констант устойчивости отдельных комплексных форм или констант диссоциации при образовании в растворе слабо-диссоциированных соединений. Она может непрерывно изменяться в интервале значений от заряда исходной ионной формы до заряда комплекса с внутренней сферой, полностью заполненной электрозаряженными лигандами. Соответственно становится неограниченным диапазон возможных значений Кс.
В плане этих общих подходов электромиграционный метод близок к хроматографии (те же два основных направления повышения эффективности разделения) — поиск методических приемов лучшего разрешения зон при постоянных Кс и использование химических превращений с целью увеличения Кс. Похожи и основные схемы практического осуществления процесса разделения: на колонке, на бумаге, в тонком слое. Возникший на заре развития электромиграции метод подвижной границы внешне аналогичен фронтальному анализу в хроматографии. В этом случае движение разделяемых ионов в электрическом поле происходит непосредственно из раствора их смеси. В наиболее распространенном случае зонного электрофореза просматривается общность с проявительным режимом элюирования в хроматографии. Узкая полоса исходной смеси веществ в среде определенного электролита разделяется на индивидуальные зоны. Существует внешняя аналогия противоточного и двухмерного электромиграци-онного разделения с соответствующими способами осуществления хроматографического процесса. Поэтому при всем принципиальном различии методов по природе химических процессов, лежащих в их основе, хроматографию и электрофорез иногда даже рассматривают как смежные методы [95].
Сходство дополнительно усиливается близостью общих схем осуществления процесса разделения. Чтобы реализовать минимальные различия в подвижностях ионов, необходимо минимизировать влияние конвективного перемешивания раствора. Поэтому электромиграционное разделение проводят в пористой среде или в капиллярах. Опять-таки по аналогии с хроматографическим процессом раствору электролита, в котором производится электромиграционное разделение, придается форма тонкого слоя или цилиндрической колонки. Основное различие заключается в том, что пористый материал, формирующий тонкий слой или заполняющий колонку, выступает только в роли стабилизатора пространственного расположения раствора электролита. Межфазное распределение между раствором электролита и пористым носителем в общем случае не является фактором, определяющим скорость электромиграции ионов в растворе. Возможен и смешанный вариант, когда электромиграционнный процесс накладывается на хроматографический, т.е. носитель одновременно выступает в
роли стабилизатора потока и сорбента. В этом случае происходит сложение векторов скорости электромиграцион-ного и хроматографического движения зон разделяемых веществ.
Среди возможных схем электромиграционного разделения по распространенности и практической значимости предпочтение отдается зонному электрофорезу, т.е. разделению одной стартовой зоны — на отдельные зоны индивидуальных компонентов; происходит это в неподвижном слое раствора электролита. Для характеристики процесса введено понятие электрофоретической подвижности, определяемой в условиях конкретного эксперимента:
где мэ.ф — электрофоретическая подвижность; I — расстояние, на которое переместился компонент; т — продолжительность миграции.
Расстояние между зонами двух компонентов А/ пропорционально разности подвижностей А мэ.ф, Е и т:
AZ = А мэ.ф, Е т.
На электрофоретическую подвижность оказывают влияние параметры частицы (знак и величина заряда, размеры и форма), параметры раствора (состав, ионная сила, pH, вязкость, температура) и параметры носителя (структура, адсорбционные и электрокинетические свойства). Указанные параметры взаимозависимы. Так, при увеличении концентрации электролита растет сила тока в системе, а градиент потенциала уменьшается. Ионная сила раствора влияет на электроосмотический поток, зависящий от электрокинети-ческих свойств наполнителя или стенок капилляра: знака заряда поверхности и дзета(0-потенциала. Истинное перемещение мигранта 4 складывается из экспериментально зафиксированного расстояния /эксп и расстояния /осм, пройденного вместе с электроосмотическим потоком:
I 4>КСП ± /осм.
Знак в последнем выражении характеризует направление перемещения мигранта в электроосмотическом потоке: «+» — перемещение мигранта «по течению» потока (в одном с ним направлении), «-» — перемещение мигранта «против течения» (в противоположную сторону). Обычно поправку на электроосмос определяют при помощи электроосмотических индикаторов, являющихся в условиях разделения нейтральными и недиссоциированнными веществами.
На перемещение мигранта оказывает влияние, помимо электроосмоса, еще реофорез, саморазогрев раствора и адсорбция. Реофорез — это явление дополнительного смещения зон компонентов, вызванное встречными потоками электролита из электродных пространств. Существенным оказывается и влияние саморазогрева системы с точки зрения зависимости подвижности ионов от температуры. Известно, что повышение температуры только на 1 °C приводит к увеличению подвижности ионов приблизительно на 2%.
244
Новый справочник химика и технолога
Как отмечено выше, на электромиграционное разделение определяющее влияние оказывают процессы комплексообразования в растворе. Для этого случая при отсутствии кинетических ограничений в установлении ионных равновесий для разности подвижностей двух компонентов справедливо выражение [138]:
Awt = и[ - и'^ -
м;+«;р;[А] ^+<p,U]
i+p;[A] 1+ряа] ’
где ис — суммарные подвижности всех индивидуальных форм каждого из компонентов; ик — подвижность катиона; щ — подвижность комплексного иона; р/ — константа устойчивости комплексов; [А] — концентрация лиганда.
Значение Awc достигает максимума при условии:
Jp;/p;-1 7 (uk-ut).
Pz' Pz + J
(Аыс)макс называется фактором разделения. Он может быть выбран на основании справочных данных о константах устойчивости комплексов. Максимальное расстояние между центрами зон разделяемых компонентов рассчитывается по уравнению:
AZMaKC = Е т (uk - uj) (пт - 1) («,/2 + 1),
Из этого уравнения очевидно, что А/ увеличивается с ростом напряженности приложенного электрического поля и продолжительности миграции. При этом чем выше Е, тем быстрее будут перемещаться ионы. Ограничивающими факторами в увеличении Е и, следовательно, в уменьшении т являются саморазогрев системы и скорость установления равновесия в реакциях комплексообразования. При высоких градиентах потенциала даже при незначительной замедленности реакций образования или диссоциации комплексных соединений может произойти электродиф-фузное размытие зоны или ее расщепление на отдельные участки, соответствующие разным ионным формам одного компонента. Этот недостаток при суммарном выделении целевого компонента оборачивается достоинством при изучении форм существования элементов в растворе.
Эффективность электромиграционного разделения определяется подобно хроматографическому не только фактором разделения, но и степенью размытия зоны. Для разделения двух компонентов необходимо, чтобы А/ было больше За, где а — стандартное отклонение распределения концентрации компонента в зоне. Величина а зависит от коэффициента тепловой диффузии Dx, коэффициента электродиффузии D3 и дисперсии длины пробега иона между последовательными столкновениями с частицами носителя а2:
а = ^а2 + 2(£>э +Dx)t.
Отсюда очевидно, что выбор оптимальных условий осуществления электрофоретического процесса представляет достаточно трудную задачу. Стремление сократить
время разделения приводит к разогреву и, соответственно к диффузионному и электродиффузионному размытию зоны. Важное значение имеет выбор однородного носителя раствора электролита, обеспечивающего минимальную дисперсию длин пробегов ионов. В качестве носителей электролита при зонном электромиграционном разделении используют: бумагу, ацетат целлюлозы, кварцевый порошок, гели на основе крахмала, желатину, агар, полиакриламид и т.п.
Но наиболее предпочтительной схемой электрофоретического разделения является стабилизация слоя электролита в капилляре. Капиллярный вариант электрофоретического разделения известен сравнительно давно, но интерес к нему особенно усилился в последние годы по мере совершенствования техники микродетектирования. Объединение электрофоретического разделения в капилляре с проточными детекторами привело к появлению нового гибридного метода анализа — капиллярного электрофореза. Этому методу посвящен специальный раздел настоящего справочника.
3.4.2. Проточное фракционирование в поперечном поле (ППФ-методы)
Как уже отмечалось выше, последняя по времени возникновения группа методов внутрифазного разделения основывается на эффекте фракционирования смеси макромолекул в потоке жидкости, проходящем через узкий канал, который проявляется при наложении силового поля, перпендикулярного направлению потока. Природа сил полей, воздействующих на поток жидкого носителя, может быть различна. К настоящему времени практически реализовано в лабораторном исполнении и даже в серийно выпускаемых приборах по крайней мере четыре варианта ППФ-процессов под действием гравитационных (центробежных), гидродинамических, тепловых и электрических сил. Соответственно выделяют седиментационные (СППФ), поточные (1ШПФ), термические (ТППФ) и электрические (ЭППФ) разновидности метода.
Схематично процесс разделения независимо от природы действующих сил можно представить следующим образом (рис. 3.21). Смесь двух компонентов А и В с различными молекулярными массами (МА > Мв) вводят в поток носителя, движущийся в плоском канале толщиной ®. Величину (о выбирают такой, чтобы обеспечить достаточно крутой параболический профиль скоростей потока жидкости в канале. Например, в первых выпускаемых промышленных приборах-фракционаторах толщина канала не превышает 250 мкм. Под влиянием любого поля, действующего перпендикулярно направлению потока жидкого носителя, молекулы компонентов А и В будут смещаться к одной из стенок канала. Концентрированию компонентов у соответствующей стенки канала, называемой иногда аналитической стенкой, препятствует встречный диффузионный поток. В итоге устанавливается динамическое равновесие, в котором реализуется экспоненциальное распределение молекул по сечению потока от стенки к его
Методы разделения и концентрирования
245
центру. Условная толщина слоев /А и /в, образуемая компонентами, будет зависеть от природы и силы приложенного поля, скорости потока носителя, коэффициента диффузии, молярной массы и структуры макромолекул. Учитывая, что скорость потока жидкого носителя в канале растет по мере удаления от его стенок, компонент В, распределенный в более толстом диффузионном слое /в, будет двигаться быстрее компонента А и первым выйдет из канала.
Поле
Рис. 3.21. Схема разделения смеси компонентов ППФ-методами: ю — толщина канала; А и В — компоненты с разными молярными массами (МА> Мв); /А и /в — толщины диффузионных слоев компонентов А и В
Измеряя концентрацию разделяемых веществ в носителе на выходе из канала, можно получить кривую в координатах время (объем) удерживания - отклик детектора, аналогичную хроматограммам (рис. 3.10).
Максимальная степень разделения компонентов смеси по молярной массе Л?м, выражаемая уравнением
d\nVR
d InM
(где VR — объем удерживания; М — молярная масса), достигается в каждом варианте ППФ при объеме удерживания VR> 100 VK, (VK — свободный объем канала). Различия в достигаемых значениях Км для каждого варианта ППФ в зависимости от молекулярной массы компонентов иллюстрирует рис. 3.22.
В СППФ значения разделяемых молярных масс определяются силой центробежного поля. В плоском канале под действием только гравитационного поля легко осаждаются крупные частицы размером более 1-2 мкм. Разделение более мелких частиц требует помещения канала в поле центробежных сил. Указанного нижнего предела молярной массы, равного ~ 5 • 105 (рис. 3.22, кривая 2), можно достичь при увеличении центробежного ускорения до значения « 100 000 g.
Для преодоления возможного влияния сорбционных эффектов, искажающих результаты разделения, стенки каналов тщательно полируют, металлические стенки покрывают инертным материалом. Тщательность полировки аналитической стенки канала важна для всех методов ППФ. Например, молекулы линейного полистирола при грубообработанных стенках делятся в течение 16 часов; Сравним с результатом, полученным при тщательно полированных стенках канала (рис. 3.23).
Рис. 3.22. Зависимость Км от молярной массы разделяемых компонентов в различных вариантах ППФ:
7 и 2 — СППФ, отличающиеся значением центробежной силы (в случае 7 больше, чем в случае 2); 3 — ТППФ; 4 — ПППФ
Рис. 3.23. Фрактограмма молекул линейного полистирола в Т1ШФ: длина канала — 42 см; разность температур стенок канала — 60 °C; толщина канала — 51 мкм
В термическом варианте ППФ верхнюю стенку канала нагревают, а нижнюю (аналитическую) охлаждают, устанавливая разность температур между стенками в интервале 50-100 °C, чаще 80 °C, что соответствует градиенту температур ~ 1000 °C • см1. Изменение температур в канале может вызвать кипение носителя у верхней стенки, преодолеваемое увеличением давления и соответствующим повышением температуры кипения. Температура нижней стенки отвечает условиям предельной растворимости компонентов смеси в носителе. Именно эти два фактора определяют выбор температур в канале. Как правило, температуру собирающей аналитической стенки поддерживают на уровне « 25 °C. Как видно из рис. 3.22, селективность термического варианта ППФ ниже, чем седиментационного, и составляет 0,33 для структур молекул в виде компактных сфер и 0,65 для молекул линейных полимеров. Однако диапазон разделяемых по массе и размеру молекул у ТППФ существенно шире, чем у СППФ. С помощью ТППФ можно разделять молекулы, начиная от молярных
246
Новый справочник химика и технолога
масс, равных ~ 103, а наилучших результатов в СППФ добиваются в диапазоне молекулярных масс 107-109.
В ПППФ селективность разделения еще ниже, чем в ТППФ, поскольку в этом варианте разделение основано на различиях только одного параметра — коэффициента диффузии. Поле в этом варианте ППФ представляет собой простой поток носителя, движущийся в направлении, перпендикулярном основному потоку носителя в канале. Образование в канале перекрестного потока требует, чтобы по крайней мере одна из стенок (аналитическая) была проницаемой для носителя. Для этой цели используют пористые пленочные материалы. Нижний предел разрешающихся молярных масс и, следовательно, размеров молекул зависит от размеров пор пористого материала, а верхний предел достигает 1 мкм. В 1Ш11Ф применяют как водные, так и органические носители, например этилбензол. Скорость основного продольного потока носителя составляет « 5 мл • ч-1, скорость поперечного потока ~ 20 мл • ч1. Объем канала и его ширина здесь могут быть несколько большими, чем в других вариантах ППФ.
ЭППФ является наиболее технически сложным для практической реализации методом, и пока еще рано оценивать его аналитические возможности.
В целом ППФ оказались удачным дополнением эксклюзионной хроматографии, позволяющим быстро и эффективно разделять смеси высокомолекулярных органических веществ — латексов, полимерных материалов, протеинов, ДНК, полимеров. Поскольку объем канала невелик (от 1 до 5 мл), то и объем пробы, вводимой в канал, должен быть также мал. Обычно объем пробы находится в пределах от 1 до 500 мкл при содержании разделяемых компонентов в пробе ориентировочно равном ~ 1%. Это не ограничивает возможности применения ППФ для анализа биологических объектов.
Сравнение аналитических возможностей эксклюзионной хроматографии и ППФ-методов показало, что диапазон разделяемых веществ по молярным массам в первом случае ограничен величиной 106 атомных единиц массы, а во втором простирается до 1018 [1, 3 с. 231].
3.5. Комбинированные и прочие методы разделения
Приведенная выше общая и внутригрупповая классификация методов разделения, охватывающая подавляющее большинство известных методов, оказалась к тому же столь удачной, что в нее хорошо вписались открытые гораздо позже ППФ-методы. Есть и исключение из этого правила — дополнительная группа методов разделения, которые не уложились в прокрустово ложе общей классификационной схемы.
В эту группу входят методы, в которых осуществляется суммирование принципов разделения, относящихся к рассмотренным ранее основным группам методов или по совокупности признаков не попадающих ни в одну из групп. Используемый термин комбинированные не тождественен термину гибридные методы, основанному на сочетании методов разделения и методов определения в одной анали
тической операции. Типичные примеры последних: хроматографические методы анализа и метод капиллярного электрофореза. Широко известными примерами комбинированных методов являются методы электроосаждения (образование новой фазы вызвано электрохимическими процессами) и методы электрохроматографического разделения, в котором объединены принципы электромиграции и ионобменной хроматографии. Менее известен метод электромембранного разделения изотопов водорода на металлических водопроницаемых мембрана^, в котором суммируются изотопные эффекты, проявляющиеся при электролизе и диффузии через мембрану [139]. Наибольший интерес из не вошедших в общую классификационную схему методов анализа представляют две группы методов — фотохимические и хроматомембранные.
Первые относятся к методам, не соответствующим по своим принципам ни одной из групп, вторые, как следует из названия, сочетают принципы хроматографических и мембранных методов.
3.5.1. Фотохимические методы
Фотохимические методы включают три последовательные стадии: стадию селективного фотовозбуждения определенных молекул с переводом их в реакционноспособное состояние, стадию переведения возбужденных молекул в определенные химические формы и, наконец, собственно разделение образованных возбужденными молекулами химических форм от других компонентов разделяемой смеси с применением любых известных методов разделения. Две первые стадии часто оказываются совмещенными. Селективность фотохимических методов определяется различиями в спектрах поглощения разделяемых молекул и уровнем монохроматизации возбуждающего излучения.
Фотохимические методы развиваются преимущественно с ориентацией на разделение изотопов одного элемента в препаративных и технологических целях. В качестве источника монохроматического электромагнитного излучения обычно используют лазеры. В этом случае относительная сложность процесса разделения компенсируется уникальной селективностью метода, определяемой малой спектральной шириной лазерного излучения. Для эффективного разделения необходимо, чтобы в спектре поглощения выбранного газообразного соединения или паров элемента наблюдался изотопный сдвиг, т.е. различие положений линий в спектрах отдельных изотопов. С максимальной точностью также должна совпадать длина волны лазерного излучения и длина волны, соответствующая энергии перехода из основного в возбужденное состояние одного из изотопов. Дополнительным обязательным условием является необратимое превращение исходного соединения изотопа в новую химическую форму в результате индуцированной фотохимической реакции или достаточное время жизни изотопа, возникшего в результате фотовозбуждения, следствием чего может бытьреализован процесс последующего выделения изотопа под действием электрического поля.
Методы разделения и концентрирования
247
Диапазон длин волн лазерного излучения, пригодного для селективного фотовозбуждения веществ в ионном, атомарном или молекулярном состоянии, охватывает области спектра от ультрафиолетовой до дальней инфракрасной [139]. Кроме того, известны способы разделения изотопов при использовании различия в колебательновращательных спектрах радиочастотной области [140]. Радиочастотный вариант метода основан на известном явлении парамагнитного резонанса — избирательном поглощении электромагнитных волн парамагнитным веществом, находящемся в магнитном поле. Под действием магнитного поля уровни энергии молекул расщепляются на магнитные подуровни (эффект Зеемана). При облучении молекул электромагнитным излучением радиочастотного диапазона с энергией, равной «шагу» магнитного расщепления для молекул с определенным изотопным составом, происходит резонансное поглощение излучения, вызывающее изменение их угловых моментов. При попадании далее смеси веществ в разделяющее магнитное поле наблюдается пространственное разделение молекул, соответствующих различным изотопам. Переход к более длинноволновому диапазону (радиочастотному и микроволновому) позволяет увеличить разрешающую способность благодаря большему различию в спектрах изотопов в этой области по сравнению с видимой или инфракрасной областями.
Радиочастотное возбуждение рассматривается как альтернатива лазерному возбуждению в оптической области спектра. Но пока еще недостаточно опубликованных данных, чтобы делать обоснованные выводы об относительных преимуществах того или иного способа возбуждения. Большинство предложенных схем фотохимического разделения по-прежнему основывается на использовании лазеров [141]. Главное внимание уделяется проблеме разделения изотопов урана. Сложности при практической реализации метода возникают при выборе газообразной химической формы разделяемых изотопов с приемлемыми спектральными характеристиками и соответствующих лазеров. Обсуждаются различные варианты использования летучих молекулярных соединений: гексафторида урана, его р-дикетонатов и атомного пара. Несмотря на большие энергозатраты на испарение металла, пока предпочтение отдается лазерному разделению изотопов в парах металлического урана. При переходе к разделению изотопов других элементов проблема упрощается пропорционально многовариантности выбора летучих соединений и увеличению изотопного сдвига в спектрах поглощения с уменьшением изотопных масс [139].
Из других направлений применения фотохимических реакций в процессах разделения можно отметить фотохимическое восстановление платиновых металлов [142]. В этом случае монохроматичность электромагнитного излучения не является обязательным условием проведения процесса. Луч света с широким диапазоном длин волн направляется на раствор, в котором диспергированы частицы фотохимического катализатора, например диоксида титана. В результате фотовозбуждения в поверхностном слое
катализатора создается пара электрон - дырка. Образующиеся электроны участвуют в гетерогенной реакции восстановления ионов металлов с выделением их на частицах катализатора. В данном случае можно говорить о близкой аналогии с методами электроосаждения, с той лишь разницей, что подвод энергии к системе осуществляется электромагнитным излучением.
3.5.2. Хроматомембранный массообменный процесс и хроматомембранные методы
Хроматомембранный процесс предложен как общий способ осуществления массообменных процессов в системах — жидкость - газ и жидкость - жидкость в условиях относительного перемещения фаз, аналогично хроматографическому процессу. При этом одна из фаз может быть неподвижной, а вторая перемещается относительно нее, с последующей инверсией ролей фаз: поток подвижной фазы останавливается, а неподвижная становится подвижной. При необходимости обе фазы перемещаются в пространстве навстречу или перпендикулярно друг другу по схеме, аналогичной непрерывной противоточной и двухмерной хроматографии соответственно. Подобная схема массо-обмена между потоками двух фаз позволяет решить проблему непрерывного разделения веществ.
Сущность хроматомембранного процесса заключается в следующем. Массообмен между потоками двух несмеши-вающихся жидкостей или жидкости и газа осуществляется в пористой матрице из гидрофобного материала с открытыми порами. Чтобы обеспечить возможность независимого движения потоков двух фаз, пористый материал, из которого изготовлена матрица, имеет два типа пор, существенно отличающихся по размерам.
Размеры макропор выбраны такими, чтобы величина возникающего в них капиллярного давления была пренебрежимо малой и не препятствовала прохождению через них потока полярной фазы, не смачивающей материал матрицы. Поры второго типа, условно называемые в дальнейшем микропорами, наоборот, настолько малы, что возникающее в них капиллярное давление препятствует попаданию в них полярной жидкой фазы. В то же время они обеспечивают достаточную проницаемость для потока газов или неполярных жидкостей, смачивающих поверхность пористой гидрофобной матрицы.
Схема осуществления хроматомембранного процесса представлена на рис. 3.24. Она соответствует прохождению пересекающихся под прямым углом потоков двух фаз: полярная жидкость протекает через бипористую среду, образующую массообменное пространство в соответствии с градиентом давления Др„ =р\ - /?2, где /?1 и/?2 — давление полярной фазы на входе и на выходе из массообменной камеры соответственно, а в перпендикулярном направлении создается поток неполярной жидкости или газа (рз и /?4 — давление неполярной фазы на входе и на выходе соответственно). Взаимное смешение потоков двух фаз в каналах, образованных порами того и другого типа, исключается вследствие разности давлений, под которыми
248
Новый справочник химика и технолога
каждая из фаз находится в матрице. Давление неполярной фазы поддерживается меньшим, чем давление полярной фазы, т.е. давление полярной фазы на выходе из массообменной камеры рг превышает давление на входе в нее неполярной жидкой фазы или газар^‘.
Р1> Рз-
В результате газ или неполярная жидкая фаза не могут перейти из занимаемых ими микропор в макропоры. В свою очередь, их вытеснению из микропор полярной фазой препятствует капиллярное давление, т.к. выполняется условие:
Р\ </>4+ |рК|,
где |рк| — абсолютная величина капиллярного давления, имеющего в данном случае отрицательное значение.
Рис. 3.24. Схема проведения хроматомембранного массообменного процесса:
1 — бипористая гидрофобная матрица; 2 — пористые гидрофобные мембраны; 3,4 — вход и выход неполярной жидкой или газообразной фазы; 5,6 — вход и выход полярной жидкой фазы
Теоретический анализ и экспериментальные исследования показали, что оптимальным материалом для изготовления бипористых матриц является политетрафторэтилен с размерами микропор в интервале от долей до единиц мкм при условии минимальной дисперсии их размеров в пределах одной массообменной матрицы. В то же время размер макропор можно варьировать в пределах от 0,25 до 3 мм, как и в случае микропор с минимальной дисперсией размеров. При этом критерий выбора размеров макропор аналогичен критерию выбора размеров частиц сорбентов и носителей в хроматографии. Этот критерий — компромисс между улучшением проницаемости колонки или матрицы в данном случае для полярной фазы и ухудшением разрешения зон разделяемых веществ. По аналогии с насадками в классической хроматографии таким компромиссом являются макропоры в интервале 0,25-0,5 мм.
Подробную информацию о выборе условий хроматомембранного разделения веществ можно найти в [144].
Подобно хроматографическим методам разделения веществ хроматомембраннные методы подразделяются в зависимости от направленности массообменного процесса или той роли, которую играют обе фазы, участвующие в хроматомембранном процессе. Это следующие методы;
— хроматомембранная абсорбция, ХМА (применяется при выделении веществ из газовой фазы в полярную жидкую);
— хроматомембранная газовая экстракция, ХМГЭ (обратный процесс);
— хроматомембранная жидкостная экстракция, ХМЖЭ (выделение из водных растворов в органические растворители). Все вышеперечисленные методы оказались очень эффективными при решении задач пробоподготовки в сочетании с инструментальными методами анализа, особенно когда речь идет о пробоподготовке в потоке для непрерывного анализа. Здесь хроматомембранные методы превосходят все известные аналоги. Об аналитических возможностях хроматомембранных методов можно судить по данным, приведенным в табл. 3.92.
Таблица 3.92
Аналитические возможности подготовки проб атмосферного воздуха и природных вод хроматомембранными методами [114]
Пояснение. ИХ — ионная хроматография; ГХ — газовая хроматография; ПИД — пламенно-ионизационный детектор; ГАК — газоадсорбционное концентрирование; РГЭ — реакционная газовая экстракция.
Определяемое вещество Метод определения Достигнутое значение нижней границы диапазона определяемых концентраций, мкг • л-1 Метод разделения
Атмосферный воздух
so2 Фото-метрия; ИХ 0,025 ХМА
0,003
Диоксид азота Фотометрия 0,12 ХМА
hno2,nox ИХ 0,005 ХМА
Аммиак pH-электрод; ИХ 1,0 ХМА
0,1 ХМА
HF, НС1 ИХ 0,001 ХМА
0,002 ХМА
Метанол, этанол ГХсПИД 0,05 ХМА
0,001 ХМА РГЭ в форме алкил-нитрита
Методы разделения и концентрирования
249
Продолжение таблицы 3.92
Определяемое вещество Метод определения Достигнутое значение нижней границы диапазона определяемых концентраций, мкг • л-1 Метод разделения
Метанол, этанол ГХсПИД 0,06 ХМА
0,002 ХМА РГЭ в форме ал-килнитрита
Гидразин Фотометрия 0,01 ХМА
1,1-Диме-тилгидра-зин Фотометрия 0,05 ХМА
Формаль- дегид Фотометрия 0,005 ХМА
Природные воды
Медь(И) Фотометрия 1 ХМЖЭ
Бензол; Толуол; хлороформ; СС14 ГХ с ПИД 6 ХМГЭ
0,04 ХМГЭ+ГА К
8 ХМГЭ
0,06 ХМГЭ+ГА К
100 ХМГЭ
0,4 ХМГЭ+ГАК
200 ХМГЭ
0,8 ХМГЭ+ГАК
Метилацетат этилацетат То же 300 ХМГЭ
2 ХМГЭ+ГА К
200 ХМГЭ
1 ХМГЭ+ГАК
Углеводороды С1-С4 То же 0,1-0,3 ХМГЭ
Нитриты Фотометрия 0,2 ХМЖЭ
Фенолы То же 1 ХМЖЭ
Анионные ПАВ 8 ХМЖЭ
50 (в присутствии гуминовых кислот) ХМЖЭ
Нефтепродукты Фотометрия 1 ХМЖЭ
Фенолы То же 1 ХМЖЭ
Сульфид-ионы ионометрия 0,5 ХМЖЭ
Ионы аммония Кондуктометрия 50 ХМЖЭ
Сульфит-ионы Фотометрия 10 ХМЖЭ
Литература к разделу 3
1. Справочник Химика: В 7 т. Т. 4. 2-е изд-е. Л.: Химия, 1967. 920 с.
2. Золотов Ю.А., Кузьмин Н.М. Концентрирование микроэлементов. М.: Химия, 1982. 430 с.
3. Мицуике А. Методы концентрирования микроэлементов в неорганическом анализе. М.: Химия, 1986. 152 с.
4. Москвин Л.Н., Царицына Л.Г. Методы разделения и концентрирования в аналитической химии. Л.: Химия, 1991.256 с.
5. K.Bachmann. // Taianta. 1982. Vol. 29. Р. 1-25.
6. Киселев А.В., Яшин Я.И. Газоадсорбционная хроматография. М.: Наука, 1967. 256 с.
7. Лурье Ю.Ю. Справочник по аналитической химии. М.: Химия, 1989. 450 с.
8. Модифицированные кремнеземы в сорбции, катализе и хроматографии / Под ред. Г.В. Лисичкина. М.: Химия, 1986. 248 с.
9. Филиппов О.А., Тихомирова Т.И., Цизин Г.И. и др. // Журн. аналит. химии. 2003. Т. 58, № 5.
10. Лурье А.А. Хроматографические материалы. М.: Химия, 1978. 440 с.
11. Мясоедова Г.В., Саввин С.Б. Хелатообразующие сорбенты. М.: Наука, 1984. 176 с.
12. Самуэльсон О. Ионообменные разделения в аналитической химии / Пер. с англ. А.Б. Шейнина М.: Химия, 1966.416 с.
13. Амфлет И. Неорганические иониты. М.: Мир, 1966.188 с.
14. Иониты в химической технологии / Под ред. акад. Б.П. Никольского и чл.-корр. АН СССР П.Г. Роман-кова. Л.: Химия, 1982. 416 с.
15. Вольхин В.В., Егоров Ю.В., Белинская Ф.А. и др. Ионный обмен / Под ред. М.М. Синявина. М.: Наука, 1981. С. 25.
16. Мельников В.А., Москвин Л.Н., Четвериков В.В. и др. // Радиохимия. 1986. Т. 28. Вып. 2. С. 208-212.
17. Симанова С.А., Бойчинова Е.С. // Ионный обмен и ионометрия. Л.: Изд-во ЛГУ, 1988. Вып. 6. С. 3-16.
18. PeperK.W. И J. Appl. Chem. 1951. V. 1. Р. 124.
19. Сейфер Г.Б. // Неорганические ионообменные материалы. Л.: Изд-во ЛГУ, 1980. Вып. 2. С. 9-17.
20. Гельферих Ф. Иониты М.: ИЛ. 1962. 460 с.
21 Preconcentration Techniques for Trace Elements / Ed. by Alfassi Z.B. Wai Ch. M. CRC Press Boca Raton Ann Arbor. London. 1992. 461 p.
22. Радиохимия и химия ядерных процессов / Под ред. А.Н. Мурина, В.Д. Нефедова, В.П. Шведова. Л.: Гос-химиздат, 1960. 784 с.
23. Спиваков Б.Я., Петрухин О.М., Малофеева Г.И. и др. // Журн. аналит. химии. 1992. Т. 47, № 9. С. 1601.
24. Малофеева Г.И., Данилова Т.В., Петрухин О.М. и др. // Журн. аналит. химии. 1994. Т. 49, № 6. С. 635.
25. Малофеева Г.И., Петрухин О.М., Рожкова Л.С. и др. И Журн. аналит. химии. 1996. Т. 51, № 3. С. 279.
26. Малофеева Г.И., Чмутова М.К., Рожкова Л.С. и др. // Радиохимия. 1998. Т. 40, № 3. С. 235.
27. Малофеева Г.И., Седых Э.М., Рожкова Л.С. и др. // Журн. аналит. химии. 1999. Т. 54, № 1. С. 167.
28. Pawliszyn J. Solid Phase Microextraction. Theory and Practice / By Willey-VCH. 1997. 276 p.
250
Новый справочник химика и технолога
29. Рачинский В.В. Введение в общую теорию динамики сорбции и хроматографии. М.: Наука, 1964. 135 с.
30. Когановский А.М., Клименко Т.М., Левченко Т.М. Адсорбция органических веществ из воды. Л.: Химия, 1990. 256 с.
31. Родинков О.В., Москвин Л.Н., Синицына Т.В. Хроматомембранная абсорбция микропримесей полярных органических веществ из воздуха водными растворами //Журн. аналит. химии. 1998. Т. 53, № 4. С. 373-378.
32. Немировский А.М. Расчеты во фронтальной хроматографии // Завод, лаб. 1996. № 3. С. 13
33. Родинков О.В., Москвин Л.Н. Выбор оптимальных условий сорбционного концентрирования летучих органических веществ из водных растворов // Журн. аналит. химии. 1999. Т 54, № 2. С. 144-147.
34. Пфанн В. Зонная плавка / Под ред. В.Н. Вигдоровича. Пер. с англ. М.: Мир, 1970. 366 с.
35. Бланк А.Б. Анализ чистых веществ с применением кристаллизационного концентрирования. М.: Химия, 1986. 184 с.
36. Коренман Я.И. Коэффициенты распределения органических соединений: Справочник. Воронеж: Изд-во Воронежского ун-та, 1992.
37. Коренман Я.И. Экстракция фенолов. Горький: Волго-Вятское изд-во, 1973. 216 с.
38. Мартынов Б.В. Экстракция органическими кислотами и их солями: Справочник. М.: Энерготомиздат, 1989.270 с.
39. Николотова З.И., Карташова Н.А. Экстракция нейтральными органическими соединениями. М.: Атом-издат, 1976. 560 с.
40. Ishimori Т., Watanobe К., Nakumura Е. И Bull. Chem. Soc. 1960. V. 33, № 10. Р. 1443.
41. Ishimori Т., Watanobe К., Nakumura Е. // Bull. Chem. Soc. 1960. V. 33, № 5. P. 636.
42. Kimura K. // Bull. Chem Soc. Japan. 1961. V. 33, № 8. P. 1038.
43. Шмидт B.C., Межов Э.А., Рыбаков К.A. // Журн. неорг. хим. 1978. Т. 23. С. 2756.
44. Межов Э.А. Экстракция аминами, солями аминов и четвертичных аммониевых оснований. М.: Атомиздат, 1977. 304 с.
45. Ванифатова Н.Г., Серакова И.В., Золотов Ю.А. Экстракция металлов нейтральными серосодержащими соединениями. М.: Наука, 1980. 104 с.
46. Pedersen C.J. // J. Amer. Chem. Soc. 1967. V. 89. P. 2497.
47. Золотов Ю.А. Журн. Всесоюз. хим. о-ва им. Д.И. Менделеева. 1985. Т. 30, № 5. С. 584.
48. Coordination Chemistry of Macrocyclic Compounds / Ed. by G.A.Melson N.Y.; London: Plenum Press, 1979.
49. Progress in Macrocyclic Chemistry. V. 1. / Ed. by R.M. Izatt, J.J. Crestensen. N.Y.: John Wiley and Sons, 1979. 191 p.
50. Химия комплексов «гость — хозяин». Синтез, структура,применение / Под ред. Ф. Фештле, Э. Вебера. М.: Мир, 1988. 511 с.
51. Jepson В.Е., Dewitt R. // J. Inorg. Chem, 1976. V. 38. P. 1175.
52. Овчинников Ю.А., Иванов B.T., Шкраб А.М. Мембраноактивные комплексоны. М.: Наука, 1974. 463 с.
53. Ammann D., Morf W.E., Anker et al. 11 Ion Selective Electrode Rev. 1983. V. 5. P. 3.
54. Хираока M. Краун-соединения / Пер. с англ. М.: Мир, 1986. 363 с.
55. Lobinski R., Matchenko Z.. // Spectrochemical Trace Analysis for Metals and Metalloids. Amsterdam // Wilson & Wilson’s Comprehensive Analytical Chemistry. Amsterdam ets.: Elsevier, 1997. V. 30. 808 p.
56. TsukuleM. //J. Coord. Chem, Ser. B. 1987. V. 16. P. 101.
57. Сверхкритическая флюидная хроматография I Под ред. Р. Смита. М.: Мир, 1991. 280 с.
58. Вредные вещества в промышленности: Справочник для химиков, инженеров и врачей: В 3 т. 7-е изд-е / Под общ. ред. Н.В. Лазарева. Л.: Химия, 1976.
59. Витенберг А.Г, Иоффе Б.В. Газовая экстракция в хроматографическом анализе. Л.: Химия, 1982. С. 22.
60. Химическая энциклопедия: В 5 т. Т.1 / Под ред. Кнунянц И.Л. и др. М.: Сов. Энцикл, 1988. 623 с.
60а. Перегуд Е.А, Горелик Д.О. Инструментальные методы контроля загрязнения атмосферы. Л.: Химия, 1982. 384 с.
61. Методические указания по определение вредных веществ в воздухе. М.: ЦРИА «Морфлот», 1981. 252 с.
62. Дмитриев М.Т, Казнина Н.И, Пинигина И.А. Санитарно-химический анализ загрязняющих веществ в окружающей среде: Справочник. М.: Химия, 1989. 368 с.
63. Другое Ю.С, Родин А.А. Газохроматографическая идентификация загрязнений воздуха, воды и почвы: Практическое руководство. СПб.: Теза, 1999. 622 с.
64. Березкин В .Г. // Завод, лаб. 2002. Т. 68, № 1. С. 44-49.
65. Москвин Л.Н. И Журн. аналит. химии. 1996. Т. 51, № 8. С. 901-903.
66. Цвет М.С. Труды Варшавского об-ва естествоиспытателей. Отдел биологии. 1903. Т. 14. С. 1.
67. Сеченкова Е.М. М.С.Цвет — создатель хроматографии. Москва: Янус-К, 1997. 440 с.
68. Tswett M.S. // Вег. Dtsch. Botan. Ges. 1906. Bd. 24. S. 316-323, S. 384.
69. Kuhn R, Winterstein A, Lederer E. // Hoppe Seyler’s Z. Physiol. Chem. 1931. Bd. 197. S. 141.
70. Zechmeister L, Chelnoky L. Die Chromatographische Absorption Methode: Grundlegen, Methodik Anwendun-gen. Wien: Springer Verlag, 1937.
71. Hesse G.E. Adsorption Methoden in Chemischen Labora-torium. Berlin, 1943.
72. Цвет М.С. Хроматографический адсорбционный анализ / Под ред. А.А. Рихтера и Т.А. Красносельской. М.: Изд-во АН СССР, 1946.
73. Москвин Л.Н, Катыхин Г.С. Хроматография: метрологический и терминологический аспекты // Журн. аналит. химии. 1992. Т. 47, № 8. С. 1508-1515.
74. Nomenclature for Chromatography (JUPAC Recomendations 1993) Pure and Appl. Chem. 1993. V. 65, № 4. P. 819.
75. Compendium of Analytical Nomenclatur: Definitive Rules 1977 11 Prepared for Publications bu J. Inczedy, T. Len-guel, A.M. Ure. Third Editions. Blackwell Sciens. Oxford, 1988. ISBN 0865426255 m.
76. Препаративная жидкостная хроматография / Сб. под ред. Б. Бидингемейера; Пер. с англ. М.: Мир, 1990. 358 с.
Методы разделения и концентрирования
251
77. Жидкостная колоночная хроматография / Под ред.
3. Дейла, К. Мацека, Я. Янока; Пер. с англ, под ред. В.Г. Березкина. М.: Мир, 1978. Т. 1. 554 с.; Т. 2. 471 с.; Т. 3. 428 с.
78. Van de Deemter J.J., Zuiderwang F.G., Klinkenberg A. // Chem. Eng. Sci. 1956. V. 5. P. 271.
79. Москвин Л.Н., Шматко А.Г., Красноперов B.M. // Электрохимия. 1987. Т. 23, № 1. С. 30-35.
80. Радиохимия и химия ядерных процессов / Под ред. А.Н. Мурина, В.Д. Нефедова, В.П. Шведова. Л.: Гос-химиздат, 1960. 784 с.
81. Король А.Н. Неподвижные фазы в газожидкостной хроматографии. М.: Химия, 1985. 162 с.
82. Преображенский Б.К., Москвин Л.Н., Калямин А.В. и др. //Радиохимия. 1968. Т. 10, № 3. С. 377.
83. Москвин Л.Н., Гумеров М.Ф., Горшков А.И. // ЖПХ. 1974. Т. 4, №7. С. 1973-1976.
84. Москвин Л.Н., Мельников В.А. // Радиохимия. 1974. Т. 16, № 1.С. 53-57.
85. Москвин Л.Н., Царицына Л.Г. // Радиохимия. 1970. Т. 12, № 1.С. 187-189.
86. Москвин Л.Н., Царицына Л.Г. // Радиохимия, 1970. Т. 12, №5. С. 731-743.
87. Москвин Л.Н., Гумеров М.Ф., Ефимов А.А. и др. Методы химического и радиохимического контроля в атомной энергетике / Под ред. Л.Н. Москвина. Л.: Энергоатомиздат, 1988. 216 с.
88. Беленький В.Г., Ганкина Э.С., Макаров В.Г. Капиллярная жидкостная хроматография. Л.: Наука, 1987. 208 с.
89. Encyclopedia of Chromatography / Ed. Jack Cazes. Marcel Dekker, Inc. N.Y.: Basel. 1390 p.
90. Введение в микромасштабную высокоэффективную жидкостную хроматографию / Сб. под ред. Д. Исиа; Пер с англ. М.: Мир, 1991. 240 с.
91. Pittsburgh Conf, on Analytical Chemistry and Applied Spectroscopy. USA: New Orleans, 1985. Abstracts. 1076.
92. Количественный анализ хроматографическими методами / Под ред. Э. Кэц; Пер. с англ. М.: Мир, 1990.
93. Столяров Б.В., Саввинов И.М. и др. Практическая газовая и жидкостная хроматография. СПб.: Изд-во СПбГУ, 1998.
94. Руководство по современной тонкослойной хроматографии / Сб. под ред. проф. О.Г. Ларионова. М.: Научный совет РАН по хроматографии, 1994. 312 с.
95. Лабораторное руководство по хроматографическим и смежным методам / Сб. под ред. О. Микеш; Пер. с англ. М.: Мир, 1982. С. 152.
96. Аффинная хроматография. Методы / Под ред. П. Дина, У. Джонсона, Ф. Мидла; Пер. с англ. М.: Мир, 1988.129 с.
97. Лурье А.А. Сорбенты и хроматографические носители. М.: Химия, 1972.319 с.
98. Snyder L.R.. Principles of Adsorption Chromatography. N. Y.: M. Dekker, 1968.
99. Лисичкин Г.В., Кудрявцев Г.В., Сердон А.А. и др. Модифицированные кремнеземы в сорбции, катализе и хроматографии. М.: Химия, 1986. 248 с.
100. Энгельгардт X. Жидкостная хроматография при высоких давлениях / Пер. с англ. М.: Мир, 1980. 245 с.
101. Современное состояние жидкостной хроматографии / Под ред. Дж. Крикленда; Пер. с англ. М.: Мир, 1974. 325 с.
1О2. Тилербаев А.Р., Петрухин О.М. Жидкостная адсорбционная хроматография хелатов. М.: Наука, 1989.287 с.
103. Самуэльсон О. Ионообменные разделения в аналитической химии / Пер. с англ. А.Б. Шейнина. М.: Химия 1966.416 с.
Ю4.Тремийон Б. Разделение на ионообменных смолах / Пер. с франц. К.В. Чмутова и Н.А. Бремера. М.: Мир, 1967. 431 с.
105. Мархол М. Ионообменники в аналитической химии. М.: Мир, 1985. Т. 2. 362 с.
106. Фринтц Дж., Гьерде Д., Поланд К. Ионная хроматография. М.: Мир, 1984. 224 с.
107. Хроматография. Практическое приложение метода / Под ред. Э. Хефтмана; Пер. с англ. А.В. Родионова под ред. В.Г. Березкина. М.: Мир, 1989. Т.1. 335 с. Т. 2. 422 с.
108. Симанова С.А., Кукушкин Ю.Н. // Изв. высш. учеб, заведений. Серия химия и хим. техн. 1985. Т. 28, вып. 8. С. 3-15.
109. Даванков В.А., Навратил Дж., Уолтон X. Лигандообменная хроматография. М.: Мир, 1989. 295 с.
ПО. Хроматография на бумаге / Под ред. И.М. Хайса и К.М. Мачека. М.: ИЛ, 1962. 851 с.
111. Высокоэффективная жидкостная хроматография в биохимии / Под ред. А. Хеншина и др.; Пер. с англ, под ред. И.В. Березина. М.: Мир, 1988. 688 с.
112. Москвин Л.Н., Горшков А.И., Гумеров М.Ф. // ДАН СССР. 1982. Т. 265, № 2. С. 378.
113. Giddings J.C., Meyers M.N. // J. High Resolution Chromatography. 1983. V. 6. P. 381.
114. Moskvin L.N., Rodinkov O.V. Analytical application of Liquid-gas and Liquid-gas-Solid Chromatography // Critical Reviews in Analytic. Chemistry. 1994. V. 24 (5, 6). P. 317-325.
115. Родинков O.B., Москвин Л.Н. Жидкостно-газовая хроматография и ее аналитические возможности // Журн. аналит. химии. 1996. Т. 51, № 1. С. 102-109.
116. Москвин Л.Н., Никитина Т.Г. // Мембраны. Сер. Критические технологии. 2000. № 8. С. 3-16.
117. Больберг Н.Ш. // Экологическая химия. 1995. Т. 4, № 2. С. 129-140.
118. А.с. 879455 СССР, МПК G 01 N 31/08. Способ градуировки газоаналитических приборов / З.Л. Баскин, В.И. Калмановский, Ю.П. Рыбалченко. (СССР). — Опубл. 07.11.81. Бюл. № 41.
119. Витенберг А.Г., Новикайте Н.В., Бурейко А.С. // Журн. аналит. химии. 1996. Т.51, №8. С. 865-869.
120. Хванг С.Т., Каммербайер К. Мембранные процессы разделения. М.: Химия, 1981. 464 с.
121. Москвин Л.Н., Гурский В.С. // Журн. аналит. хим. Т. 43, №4. С. 581-591.
122. Овчинников Ю.А., Иванов В.Т., Шкроб А.М. Мембранно-активные комплексоны. М.: Наука, 1974. 464 с.
123. Маркин С.В., Чизмаджев Ю.А. Индуцированный ионный транспорт. М.: Наука, 1974. 252 с.
124. Лев А.А. Моделирование ионной избирательности клеточных мембран. Л.: Наука, 1976. 210 с.
252
Новый справочник химика и технолога
125. Rodinger W // Wasser und Abwasser. 1983. В. 27, № 1. S. 135.
126. Ивахно С.Ю., Афанасьев А.В., Ягодин Г.А. Мембранная экстракция неорганических веществ // Итоги науки и техники. Сер. неорг. химия. Т. 13. М.: ВИНИТИ АН СССР, 1985. 127 с.
127. Дытнерский Ю.И. Мембранные процессы разделения жидких смесей. М.: Химия, 1975. 227 с.
128. Москвин Л.Н., Годон Л.А., Турский В.С. и др. // II Всесоюзная конф, по аналитической химии радиоактивных элементов. Тез. докл. М.: Наука, 1986. С. 48.
129. Брок Т. Мембранная фильтрация. М.: Мир, 1987. 462 с.
130. Мамет В.А., Копчинский Г.А., Доленко А.В. и др. // Теплоэнергетика. 1979. № 12. С. 53.
131. Spivakov В.Ya., Geckler К., Bayer Е. // Nature. 1985. V. 1985, №6017. Р. 313.
132. Resting R.E. Asymmetric Cellulose Acetate Membranes. // In: Reverse Osmosis and Synthetic Membranes./ Ed. Sourirajan S. Nat. Res. Council Canada Public. No. 15627. Canada. 1977. P. 89-110
133. Thomas D.J. Dynamics Membranes - Their Technological and Engineering Aspects // In: Reverse Osmosis and Synthetic Membranes. Nat. Res. Council Canada Public. No. 15627. Canada. 1977. P. 295-312.
134. Spivakov B.Ya., Shkinev V.M., Geckeler K.E. // Rure & Appl. Chem. 1994. V. 66, № 3. P. 631-640.
135. Николаев Н.И. Диффузия в мембранах. М.: Химия, 1980. 232 с.
136. Янга Й. Проточное фракционирование в поперечном поле / Пер. с англ. М.: Мир, 1992. 294 с.
137. Степанов А.В., Корчемная Е.К. Электромиграцион-ный метод в неорганическом анализе. М.: Наука, 1979. 328 с.
138. Москвин Л.Н., Турский В.С. // Электрохимия. 1984. Т. 20, № 5. С. 640.
139. Индуцированные лазером химические процессы / Под * ред. Дж. Стейнфелда. М.: Мир, 1984. 312 с.
140. Amirov A., Even U.// Nucl. Eng. Intern. 1979. V. 24, № 282. P. 8.
141. Суглобов Д.Н., Сидоренко Г.В., Легин Е.К. Летучие органические и комплексные соединения f-элементов. М.: Энергоатомиздат, 1987. 208 с.
142. Nishi Т., Uetake N., Kawamura F., Yusa H. / Trans. Amer. Nucl. Soc. 1987. V. 55. P. 242.
143. Москвин Л.Н. Хромато-мембранный метод разделения веществ. Аналитические и технологические возможности // Российский химический журнал. 1996. Т. 40, № 1. С. 67-75.
Специальная литература по методам разделения и концентрирования в аналитической химии
1. Золотов Ю.А. и Кузьмин Н.М. Концентрирование микроэлементов. М.: Химия, 1982.
2. Мицуике А. Методы концентрирования микроэлементов в неорганическом анализе. М.: Химия; 1986.
3. Москвин Л.Н. и Царицына Л.Г. Методы разделения и концентрирования в аналитической химии. Л.: Химия, 1991.
4. Основы аналитической химии: В 2 кн. Книга 1. Общие вопросы. Методы разделения / Под ред. акад. Ю.А. Золотова. М.: Высшая школа. 1998.
5. Valcarcel М., de Castro M.D. Luque. Non-Chromatographic Continuous Seperarion Techniques. The Royal Soc. Of Chem. 1991.
6. Новый справочник химика и технолога: Основные свойства неорганических, органических и элементоорганических соединений. СПб.: Мир и семья, 2002. 1271 с.
Раздел 4
АНАЛИТИЧЕСКАЯ ХРОМАТОГРАФИЯ И КАПИЛЛЯРНЫЙ ЭЛЕКТРОФОРЕЗ
Авторы-составители.*
проф., д.х.н. А.И. Яшин, проф., д.х.н. А.А. Карцова, к.х.н. А.Я. Яшин
НФХ — нормально-фазовая хроматография
ОФХ — обращенно-фазовая хроматография
ДЕТЕКТОРЫ
ПИД — пламенно-ионизационный
ДТП — по теплопроводности (катарометр)
ЭЗД — электронного захвата
ФИД — фотоионизационный
ТИД — термоионный
ПФД — пламенно-фотометрический
АЭД — атомно-эмиссионный
МСД — масс-спектрометрический
КОЛОНКИ
КК — капиллярные
НК — насадочные
Ак — чувствительность концентрационного детектора
Лп — чувствительность потокового детектора
с — концентрация сорбируемого вещества
сн — концентрация сорбата в неподвижной фазе
сп — концентрация сорбата в подвижной фазе
D — коэффициент молекулярной диффузии
ПРИНЯТЫЕ ОБОЗНАЧЕНИЯ
£>ж — коэффициент диффузии в жидкой фазе
£>кин — кинетическое размывание хроматографической зоны
£>м — размывание хроматографической зоны за счет молекулярной диффузии
DCT — размывание хроматографической зоны за счет стеночного эффекта
£>эф — эффективный коэффициент диффузии
Дшхр — вихревая диффузия
<7Ж — толщина пленки жидкой фазы
<7ср — средний диаметр пор
ф — диаметр зерна
Fr — объемная скорость газа-носителя
I— индекс удерживания Ковача gc — масса сорбента
j — поправочный коэффициент на сжимаемость газа-носителя
Н— высота, эквивалентная теоретической тарелке (ВЭТТ)
К— коэффициент распределения
Къ — константа вихревой диффузии
к — фактор удерживания (фактор емкости)
L — длина колонки
N— число теоретических тарелок (эффективность)
Pi — давление на входе в колонку
j>o — давление на выходе из колонки
A(AG) — разность свободных энергий сорбции двух компонентов
Q — теплота сорбции
R — газовая постоянная
г — радиус капиллярной колонки Rs — разрешение
Ак — площадь поверхности адсорбента в колонке
S — удельная поверхность адсорбента 5П — площадь пика
5К — сечение колонки tR — время удерживания /м — время удерживания несорбируемого компонента (мертвое время)
t'R — приведенное время удерживания — удерживаемый объем
VR — приведенный удерживаемый объем
Vd — мертвый (свободный) объем, объем пустот в колонке
VN — чистый удерживаемый объем
V? — удельный удерживаемый объем
Котн — относительный удерживаемый объем
Рпор — объем пор в адсорбенте Рпод — объем подвижной фазы Рнеп — объем неподвижной фазы U — линейная скорость газа-носителя X — доля свободного сечения
Wc— ширина пика при основании
Ио 5 — ширина пика на половине высоты а — фактор разделения (селективность) 8 — уровень шума нулевой линии р] — коэффициент массопередачи внешней диффузии
р2 — коэффициент массопредачи внутренней диффузии
р — фазовое отношение
254
Новый справочник химика и технолога
4.1. Аналитическая хроматография
4.1.1. Газовая хроматография*
4.1.1.1. Параметры удерживания в газовой хроматографии
Время удерживания и удерживаемый объем
Разделение в хроматографии основано на различной сорбируемости анализируемых соединений при движении их по слою сорбента в колонке. Если соединение не сорбируется, то оно не удерживается сорбентом в колонке и бу
дет выходить из колонки со скоростью потока газа-носителя. Если же вещества сорбируются, то они удержатся в колонке, это бу
дет определяться их сорбционной способностью: чем сильнее сорбция соединения, тем дольше оно будет удерживаться в колонке.
Рис. 4.1.1. Параметры удерживания Параметры удерживания, по существу, характеризуют сорбционную способность анализируемых соединений. Различие в сорбируемости в конечном итоге определяется различием межмолекулярных взаимодействий вещество -сорбент.
Время от момента ввода пробы в колонку до выхода максимума пика называется временем удерживания tR (рис. 4.1.1). Оно складывается из двух составляющих: времени нахождения молекул соединения в газовой фазе (zm) и времени
нахождения молекул соединения в сорбируемом состоянии
М:
h ~ + h •
Время нахождения молекул исследуемого соединения в газовой фазе зависит от доли пустот в насадочной или капиллярной колонке. В разных насадочных колонках плотность набивки может изменяться, будет также изменяться и величина ZM поэтому для характеристики истинной удерживающей способности необходимо определять величину t'R — так называемое приведенное время удерживания:
Величину ZM определяют по времени выхода несорби-руемого соединения (иногда называемого мертвым временем). В газовой хроматографии эту величину определяют по времени выхода гелия или водорода в случае применения детектора по теплопроводности и метана при использовании пламенно-ионизационного детектора.
Приведенное время удерживания зависит от скорости газа-носителя: чем больше скорость газа-носителя, тем меньше время удерживания, поэтому на практике удобнее использовать удерживаемый объем yR — произведение времени удерживания на объемную скорость газа-носителя Fr:
Уя = tR-Fr.
Удерживаемый объем — это объем газа-носителя, который необходимо пропустить через хроматографическую колонку, чтобы элюировать данное анализируемое соединение.
Приведенный удерживаемый объем (1^) соответственно равен:
V'R = (tR -tu}Fr = tRF ~tuFr = VR-Vd, К \ л M / г КГ МГ К а ’
где Vd— объем пустот в колонке (мертвый объем).
В хроматографе Vd реально складывается из объемов всех пустот в газовом тракте (дозатора, переходных соединений, колонок, детектора).
Объемную скорость газа-носителя чаще всего измеряют на выходе из колонки. Из-за сжимаемости газа-носителя при повышении давления объемная скорость неодинакова по длине колонки. В начале колонки она меньше, чем на выходе, поэтому для определения средней скорости в колонке вводится специальная поправка j, учитывающая перепад давления:
[х2
А -1
/у
J г. Z -,3 ’
A _i
W
где р[ — входное давление, — давление на выходе колонки.
Приведенный удерживаемый объем с поправкой на среднее давление называется чистым удерживаемым объемом:
VN=Vr-J-
Чистый удерживаемый объем можно считать физико-химической константой, так как он не зависит от скорости газа-носителя при постоянной температуре и доли пустот в колонке.
Чистый удерживаемый объем зависит от количества сорбента в колонке, поэтому для точных физико-химических измерений используют понятие удельного удерживаемого объема У?. Величина У? — это чистый удерживаемый объем, отнесенный к массе сорбента g в колонке или к площади поверхности адсорбента при усредненном давлении в хроматографической колонке и температуре Т колонки:
См. также разделы 3.2.9.2, 3.2.9.3.
уТ _ VRj . уТ _ VrJ 8 g ’ 8 SK
Аналитическая хроматография и капиллярный электрофорез
255
Для особо точных физико-химических измерений вводят поправку на давление пара воды вследствие того, что измерения обычно проводят мыльно-пенным измерителем, а также на разность температур на выходе из колонки и в колонке.
Относительные параметры удерживания
Все рассматриваемые выше параметры удерживания зависят от случайных небольших колебаний параметров опыта, в частности расхода газа-носителя и температуры термостата колонки.
Для исключения этих влияний используют относительные параметры удерживания.
При расчете относительного параметра удерживания (времени или объема) берут отношение чистого объема удерживания исследуемого вещества к чистому объему удерживания стандартного соединения:
v
^ОТН ту 5 *ОГН I
*Nc lRc
В качестве стандартных соединений используют н-алканы, с параметрами удерживания, близкими к параметрам удерживания исследуемого вещества. В этом случае при случайных колебаниях расхода или температуры абсолютные параметры удерживания будут изменяться, а их отношения — практически нет.
В качестве относительного параметра широко используют индекс Ковача:
I = 100
IgT1
-ь 100л?,
где 1 — приведенные времена удерживания н-алканов, с числом атомов углерода в молекуле п и п+1, t'R — приведенное время удерживания исследуемого соединения.
Индекс Ковача — безразмерная величина и может быть подсчитана с большой точностью, например в капиллярных колонках — до сотых долей процента. Индексы Ковача в первую очередь применяют для идентификации неизвестных веществ (проведение качественного анализа).
Изменения индексов Ковача для соединений, отличающихся природой функциональной группы, используют для оценки межмолекулярных взаимодействий. Индексами удерживания определенного набора стандартных веществ характеризуют полярность неподвижных жидких фаз и адсорбентов (см. Приложение).
4.1.1.2. Параметры хроматографического пика
Выходной сигнал анализируемого соединения имеет форму треугольника или пика, обычно это участок нулевой линии, на котором возникает сигнал при выходе анализируемого соединения из хроматографической колонки. Нулевая или базовая линия соответствует участку нулевой концентрации анализируемого соединения. Запись пика исследуемого соединения вместе с участками нулевой ли
нии до и после пика называется хроматограммой. Высота пика — это расстояние от максимума пика до его основания, измеренное параллельно оси отклика детектора. Ширина пика у основания — отрезок основания пика, отсекаемый двумя касательными, проведенными в точках перегибов восходящей и нисходящей ветвей хроматографического пика. Ширина пика на полувысоте — отсекаемый пиком отрезок линии, проведенной параллельно основанию пика на середине его высоты. Площадь пика — это площадь части хроматограммы, заключенной между пиком и его основанием.
Важным параметром пика является коэффициент асимметрии, который применяется для сравнения различных твердых носителей, адсорбентов и всей газовой системы хроматографа в целом. В идеальных условиях пик по форме близок к кривой Гаусса, то есть симметричен. На практике пики по разным причинам в основном несимметричны.
Асимметрия пиков ухудшает разделение и затрудняет количественную обработку.
Асимметричные пики появляются при разделении на неоднородных сорбентах, когда концентрации анализируемых соединений соответствуют нелинейным участкам изотермы сорбции. Кроме того, это может быть вызвано в некоторых случаях кинетикой сорбции (замедленный процесс десорбции), наличием непроду-ваемых полостей. Асимметрию пиков оценивают относительно полуширин пиков на половине высоты (рис. 4.1.2) отношением отрезка БВ от основания отношением
отрезков ДЕ к ГД. Точнее пользоваться отношением площадей половин пика — отношением заштрихованной части пика к незаштрихованной части (см. рис. 4.1.2).
4.1.1.3. Основные виды размывания полос в колонке
Обычно выделяют три вида размывания полос [1-3].
• Размывание, связанное с различной скоростью движения по слою сорбента зон с разной концентрацией (зависит от формы изотермы сорбции).
• Диффузионные размывания (молекулярная диффузия, вихревая диффузия, динамическое размывание, сте-ночный эффект).
• Кинетическое размывание, связанное со скоростью внешнего и внутреннего массообмена.
Первый вид размывания наблюдается в том случае, когда концентрация вещества соответствует нелинейному участку изотермы сорбции. На рис. 4.1.3 показано размывание полос для различных типов изотерм.
В случае нелинейности изотермы сорбции линейная скорость перемещения зоны вещества по слою сорбента (77с) равна:
Рис. 4.1.2. Оценка асимметричности пиков
к АБ либо на 1/10 высоты пика
256
Новый справочник химика и технолога
D = KRUd3 , вихр ,
где U — линейная скорость газа-носителя.
Рис. 4.1.3. Размывание полос для различных типов изотерм: а — линейная изотерма; б — изотерма Лэнгмюра (выпуклая изотерма); в — вогнутая изотерма
При линейной изотерме сорбции (изотерма Генри) отношение а/с постоянно во всем диапазоне концентраций, поэтому искажение формы зон не происходит, следовательно, зарегистрированный пик будет симметричным.
В случае нелинейной изотермы сорбции отношение а к с изменяется с концентрацией. Для выпуклой изотермы (изотермы Лэнгмюра) с повышением концентрации величина а/с уменьшается, а скорость продвижения участков зон с большими концентрациями возрастает. В случае вогнутой изотермы (рис. 4.1.Зв), наоборот, скорость продвижения участков зон с меньшими концентрациями будет больше. Это единственный вид размывания, который можно полностью исключить. Для этого нужно выбрать адсорбент с однородной поверхностью с линейной изотермой для анализируемых концентраций.
Молекулярная диффузия. В хроматографических колонках это размывание рассматривают вдоль колонки, размывание в других направлениях ограничено стенками колонки, поэтому в этом случае часто используют термин «продольная диффузия». Ширина полосы на слое Wc связана с коэффициентом молекулярной диффузии D уравнением:
где К — коэффициент распределения; SK — сечение колонки; X — доля свободного сечения; L — длина колонки.
Вихревая диффузия имеет место только в насадочных колонках и определяется неоднородностью набивки, разным сопротивлением потоку в разных частях сечения колонки. Общий поток газа-носителя при попадании в колонку распадается на отдельные микропотоки между зернами. Если сопротивление по сечению неоднородно, то там, где сопротивление меньше, микропоток будет продвигаться быстрее; наоборот, там, где сопротивление больше, микропоток будет двигаться медленнее. Это приведет к дополнительному размыванию. Уравнение для вихревой диффузии (Дихр) имеет следующий вид:
где Кв — константа вихревой диффузии, зависит от условий конкретной колонки; d3— диаметр зерна сорбента.
Чтобы уменьшить вклад вихревой диффузии, нужно уменьшать размер зерен, использовать более узкий фракционный состав зерен, исключать пыль и заполнять колонку с максимальной плотностью.
Динамическое размывание наблюдается в пустых незаполненных (т.е. капиллярных) колонках, оно связано с профилем скоростей по сечению в пустых трубках. В центре скорость потока больше, чем у стенок, в результате происходит искажение формы полосы и имеет место дополнительное размывание.
Выражение для динамического размывания Dam:
D
ДИН
где г — радиус капилляра; D — коэффициент молекулярной диффузии.
Стеночный эффект. Плотность набивки на единицу объема около стенок всегда меньше, а доля пустот больше, чем в центре колонки, особенно при использовании зерен крупного размера. Это приводит к тому, что скорость газа-носителя около стенок больше, чем в центре колонки. В результате возникает дополнительное размывание, т.н. стеночный эффект, определяемый соотношением:
ст W
где b — постоянная, равная отношению доли пустоты около стенки к доле пустоты в центре; d3— диаметр зерен; U — линейная скорость; D — коэффициент молекулярной диффузии. По данным Гелэя, величина b может колебаться от 3 до 6. Стеночный эффект может вносить большой вклад, когда диаметр колонок значительно больше высоты, эквивалентной теоретической тарелке (ВЭТТ), т.е. в препаративной хроматографии. В аналитических колонках небольшого диаметра (внутренний диаметр 2-4 мм) размывание за счет стеночного эффекта мало и им пренебрегают.
Кинетическое размывание. Задержка массообмена с поверхностью адсорбента вследствие медленности процессов адсорбции и десорбции также приводит к размыванию полосы. Задержка при адсорбции приводит к продвижению компонента в газовой фазе вперед, т.е. к размыванию переднего фронта полос, а задержка при десорбции приводит к размыванию заднего фронта. В том случае, когда скорости адсорбции и десорбции неодинаковы, такое размывание может быть несимметричным. Кинетическое размывание равно:
D ,------_________
- 2(1+JQ (₽.+М
где Pi, р2— коэффициенты массопередачи соответственно для внешней и внутренней диффузии; К — коэффициент распределения.
Аналитическая хроматография и капиллярный электрофорез
257
Для внешнего массообмена
Внутренняя массопередача в жидкой пленке
dx
где Dx — коэффициент диффузии в жидкой фазе; dx — толщина пленки жидкой фазы на носителе.
С учетом этих зависимостей
-У з \
U2K2 d2 d\ .
Общее размывание, оцениваемое эффективным коэффициентом диффузии для аналитической колонки, равно:
Дф Вм + ОВИХр + £)кин,
K2d2U2
D^YD+KBUd3 +
2(\ + K)2D2
4.1.1.4. Основные уравнения в газовой хроматографии
Коэффициент распределения — отношение концентраций исследуемого соединения в неподвижной и подвижной фазах в равновесных условиях:
Фазовое отношение — это отношение объемов подвижной и неподвижной фаз в колонке:
о = под
р V г иеп
Фактор удерживания (фактор емкости) — это отношение приведенных времен удерживания к мертвому времени:
к = —.
tK
Фактор разделения — величина, характеризующая селективность разделительной системы, равная отношению факторов удерживания или приведенных времен удерживания двух соседних пиков на хроматограмме:
Дф связано с величиной Н (ВЭТТ):
1 20
эф 2 и
Общее уравнение зависимости Н от скорости носителя:
газа-
Эффективность колонки — характеристика степени размывания полос в колонке, характеризуется числом теоретических тарелок N или Н.
ы 2D _ , 2уО К2 d2 К d2
Н = —= 2КЛ + ——+------+-----------U •
и Вз и (i+£) D\ (\+k)2dx
В более простом виде
где Ио,5 — ширина пика на половине высоты.
В
U
'2
В первоначальном уравнении Ван-Деемтера:
H = A+—+cU-U
На графике зависимости Н oi U (рис. 4.1.4) можно в общем виде оценить величины А, В и с.
где L — длина колонки; N — безразмерная величина; Н имеет размерность длины обычно в мм.
Разрешение Rs — это отношение расстояния между максимумами исследуемых соседних пиков к сумме их полуширин, выраженных в одних и тех же единицах измерения:
Рис. 4.1.4. График зависимости Н от U (уравнение Ван-Деемтера)
R =2——— s
Связь степени разрешения с фактором разделения а, фактором удерживания к и эффективностью N:
а-1 а
4
258
Новый справочник химика и технолога
Число тарелок, необходимое для полного разделения при7?, = 1:
.. 1 ( a Y(k+1Y
16va-lJ < k )
при k > 10 это уравнение можно упростить:
№1бГ—1 I a-lj
Определение различий свободных энергий сорбции двух соседних компонентов:
A(AG) = -/?sTlna.
Связь удерживаемого объема с температурой колонки
lg^ = В' +
Q 2,3RST
График зависимости lg VR от обратной температуры (1/7) — прямая линия, тангенс угла наклона которой равен:
tga=-f-5 e = 2,3/?stga; g«19,23tga, 2,Зл4.
т.о. из наклона прямой температурной зависимости удерживаемого объема можно определить теплоту сорбции.
4.1.1.5. Влияние экспериментальных параметров
на хроматографическое разделение (на величины a, R и N)
Влияние природы сорбента. Природа сорбента играет решающую роль в разделении компонентов. Разделение происходит за счет различий межмолекулярных взаимодействий разделяемых молекул с сорбентом. Селективность разделения определяется природой сорбента. В большинстве случаев селективность разделения (величина а) уменьшается с повышением температуры.
Длина колонки. Степень разделения пропорциональна квадратному корню от длины колонки. Эффективность прямо пропорциональна длине колонки.
Сечение колонки. С уменьшением сечения колонки (диаметра) возрастает эффективность и степень разрешения колонки.
Размер зерен сорбента. Размывание хроматографических полос, эффективность в значительной степени зависят от размера зерен сорбента. Вихревая диффузия и внешний массообмен сильно зависят от диаметра зерен сорбента.
Толщина жидкой пленки на носителе. С увеличением толщины пленки жидкой фазы (в случае газо-жидкостной хроматографии) увеличиваются времена удерживания, затрудняется внутренний массообмен и уменьшается эффективность
VR=Vd+KV„, где Vd — мертвый объем, К — коэффициент распределения, Кж — объем жидкой фазы.
Природа газа-носителя. В газовой хроматографии при небольших давлениях инертные газы-носители практически не адсорбируются, особенно в газо-жидкостной хроматографии. Поэтому природа газа-носителя практически не влияет на селективность разделения, за исключением некоторых случаев в газо-адсорбционной хроматографии при разделении газов на активных тонкопористых адсорбентах.
Природа газа-носителя влияет на эффективность колонок особенно при высоких скоростях. Сопротивление колонки, перепад давления в ней определяется? вязкостью газа-носителя.
Давление газа-носителя. В большинстве случаев на входе в колонку используют избыточное давление в пределах от 0,1 до 2 атм (в очень редких случаях выше). Изменение давления в этих пределах практически не влияет ни на селективность, ни на эффективность разделения. В ряде работ применялись пониженные давления на выходе из колонки, т.е. вакуумная хроматография для разделения малолетучих высококипящих соединений. Описаны варианты газовой хроматографии при повышенных давлениях. При повышении давления возрастает сорбция газа-носителя и уменьшается сорбция разделяемых соединений, особенно если в качестве подвижной фазы используются пары жидкостей, в частности воды. Парофазная хроматография расширяет аналитические возможности газовой хроматографии.
Размер пробы. Размер введенной пробы анализируемой смеси должен быть таким, чтобы не вызывать перегрузку колонки. При введении пробы больше максимально допустимой изменяются времена удерживания. Особенно важно не перегружать капиллярную колонку, так как ее эффективность сильно падает с перегрузкой.
Способ дозирования. Пробу можно ввести быстро, в виде узкой полосы или медленно, в виде разбавленной полосы. Первый способ введения — «метод поршня» — идеальный, второй — «способ экспоненциального разбавления» может приводить к дополнительному размыванию полосы. Способ дозирования в реальных случаях занимает промежуточное состояние между этими крайними случаями. В общем случае ширина дозируемой пробы должна быть значительно меньше ширины полосы вещества, получаемого на выходе из колонки.
Чувствительность, линейность, инерционность и стабильность детектора. Назначение детектора — регистрация выходных кривых в виде сигналов (пиков) достаточной амплитуды, необходимых для количественных измерений. Для точных количественных измерений необходимо, чтобы
• детектор не искажал истинную форму полосы, образующейся на слое сорбента, другими словами, детектор должен быть малоинерционен, постоянная времени — небольшой;
• показания детектора были пропорциональны концентрации или количеству дозируемых веществ, т.е. детектор должен обладать достаточно широкой областью линейности;
Аналитическая хроматография и капиллярный электрофорез
259
• запись сигнала была устойчивой, не должно быть флуктуации нулевой линии или же монотонного смещения нулевой линии в течение длительного времени (дрейфа нулевой линии).
4.1.1.6. Аппаратура для газовой хроматографии
Функциональная схема газового хроматографа
В аналитических хроматографах используют проявительный вариант хроматографии, в этом случае газ-носитель непрерывно продувается через хроматографическую колонку. Расход газа-носителя создается за счет перепада давления на входе и выходе колонки.
Схема современного газового хроматографа изображена на рис. 4.1.5. Для создания перепада давления через колонку хроматограф подсоединяют к источнику со сжатым газом 1 (баллонная или лабораторная линия со сжатым газом). Через колонку поток газа-носителя должен проходить с постоянной и определенной скоростью, по
этому на входе в колонку на линии газа-носителя устанавливают регулятор и стабилизатор расхода газа-носителя 2 и измеритель расхода газа 3. Если газ-носитель загрязнен нежелательными примесями, то в этом случае устанавливается еще фильтр 4. Таким образом, на входе в колонку подключается ряд устройств, часто объединяемых в один блок (блок подготовки газа), назначение которого — установка, стабилизация, измерение и очистка потока газа-носителя. Перед входом в колонку устанавливается устройство для ввода анализируемой пробы в колонку — дозатор-испаритель 5. Обычно анализируемую пробу вводят микрошприцем 8 через самозатекающее термостойкое резиновое уплотнение в дозаторе, газовые пробы вводят до
зирующим шестиходовым краном.
Анализируемая проба, введенная в дозатор, захватывается потоком газа-носителя (если анализируемая проба — жидкость, то она предварительно переходит в дозаторе-испарителе в парообразное состояние) и направляется в хроматографическую колонку 6. За счет различной сорбируемости компоненты смеси будут с разной скоростью
Рис. 4.1.5. Функциональная схема газового хроматографа
продвигаться по колонке. Вещества, которые сорбируются слабо, будут продвигаться по колонке с большей скоростью и выходить первыми. Сильносорбируемые вещества будут продвигаться по колонке медленнее.
Если выбран достаточно селективный сорбент и подобраны оптимальные условия, то на выходе колонки компоненты смеси будут полностью разделены. Детектор 11 зарегистрирует присутствие разделенных компонентов в газе-носителе. Эти сигналы в случае необходимости усиливаются (усилитель 13) и регистрируются на шкале вторичного самопишущего прибора 14 или дисплея ПЭВМ в виде выходных кривых (или пиков). Для обеспечения стабильного режима работы детектора используется блок питания детектора 12.
Сорбируемость веществ зависит от температуры. Для исключения влияния колебания температуры на результаты разделения, колонку помещают в специальную камеру-термостат, температура которой устанавливается и поддерживается терморегулятором 9. В случае необходимости
температура колонки в процессе разделения может изменяться по определенной программе с помощью блока программирования температуры 10.
Высота или площадь пика пропорциональны количеству или концентрации компонента в смеси. Площадь пика может быть измерена с помощью электронного интегратора 15 или ПЭВМ. Значения площадей пиков могут быть отпечатаны на бумажном носителе.
Таким образом, перед хроматографическим анализом необходимо провести следующие операции на приборе: • открыть вентиль баллона со сжатым газом и установить по манометру или специальному измерителю определенный расход газа-носителя;
включить питание детектора;
установить необходимую температуру в термостате колонок;
включить самопишущий прибор, интегратор или ПЭВМ, после выхода прибора на устойчивый режим (через 30-60 мин.) микрошприцем отобрать и ввести в дозатор-испаритель анализируемую пробу.
Все дальнейшие операции проходят без участия оператора: компоненты пробы разделяются на колонке, регистрируются в детекторе, записываются на диаграммной ленте вторичного прибора, интегратор или ПЭВМ определяет площадь пика, а в случае применения ПЭВМ с принтером можно сразу получить полный протокол — хроматограмму с распечатанной рядом таблицей концентраций разделенных компонентов.
Элементы блока подготовки газов
Как было указано выше, назначение блока подготовки газов (БПГ) или сис
темы подготовки газов — очистка, установка, регулировка стабилизация и измерение газовых потоков: газа-носителя,
260
Новый справочник химика и технолога
воздуха, водорода и других дополнительных газовых потоков. Поддержание стабильного потока газа-носителя важно для получения воспроизводимых значений параметров удерживания и параметров пиков. Колебания расходов газа-носителя влияют на шумы (флуктуации) детектирующих систем.
Основные элементы БПГ: дроссель, регулятор давления и регулятор расхода.
Дроссель изменяет расход газа путем изменения сопротивления канала, по которому проходит газ.
Регулятор давления стабилизирует давление на входе в колонку при возможных внешних колебаниях давления газа. Специальная мембрана в регуляторе давления воспринимает изменение давления газа и передает соответствующее смещение исполнительному механизму.
В режиме программирования температуры термостата сопротивление колонки повышается, а расход падает. В этом случае для сохранения постоянного расхода в колонке используется регулятор расхода. При падении расхода в связи с увеличением сопротивления в колонке регулятор расхода повышает входное давление настолько, чтобы восстановился первоначальный расход газа-носителя. Расход газов измеряют мыльно-пенным измерителем, реометром, ротаметром или специальным электронным измерителем расхода на принципе теплового расходомера. Фильтры для очистки газа-носителя заполняют адсорбентами (активированный уголь, силикагель, цеолит).
В современных хроматографах используются БПГ с электронным заданием и управлением расходов газов.
Дозирующие устройства (дозаторы)
Дозаторы предназначены для ввода в хроматографическую колонку точно выбранного количества анализируемой пробы. Общие требования к дозаторам: воспроизводимость ввода пробы (желательно ниже 1-2%), сохранение состава исходной анализируемой пробы. Кроме того, ввод пробы должен происходить быстро, без сильного размывания исходной смеси. Различают дозаторы для ввода газообразных, жидких и твердых проб. Для быстрого ввода газообразных проб используют микрошприцы, мембранные краны (чаще всего в автоматических промышленных хроматографах), золотниковые, поршневые и вращающиеся поворотные краны. В современных лабораторных хроматографах чаще всего применяются поворотные краны. Такой кран состоит из неподвижного корпуса со штуцерами для подвода газа-носителя и анализируемого газа и сверху движущейся поворотной втулки с каналами, соединяющими линии газа-носителя и анализируемого газа. На корпусе устанавливается трубка-доза для точного ввода пробы. Корпус и вращающаяся втулка сильно прижаты друг к другу, их контактирующие поверхности тщательно отполированы и при повороте должны плавно скользить относительно друг друга. Такие краны могут быть 6, 8, 10 и даже 14-ходовые (или портовые). Чаще всего для дозирования применяются 6-ходовые краны. Схема ввода газовой пробы таким краном показана на рис. 4.1.6. Поворот крана может проводиться вручную или автоматически,
электрическим или пневматическим приводом. При изготовлении крана используются следующие материалы: нержавеющая сталь, хостеллой, тефлон, наполненный тефлон, веспел и др.
Рис. 4.1.6. Ввод
краном: а — заполнение пробоотборной петли крана пробой 5; б — ввод пробы в потоке газа-носителя G
Жидкие пробы вводятся в газовые хроматографы микрошприцами на 1, 5, 10, 50 мкл через термостойкое резиновое уплотнение испарителя. Величина дозируемой пробы легко регулируется в широких диапазонах. Общий вид таких микрошприцев изображен на рис. 4.1.7. Эти шприцы
сравнительно недороги и удобны для очистки.
Для автоматиче
Рис. 4.1.7. Шприц
ского ввода жидких
проб применяют специальные поршневые, вращающиеся и золотниковые дозирующие краны. В поршневом кране движущийся поршень имеет сбоку кольцевую канавку, глубина которой определяет объем введенной пробы. Поршень двигается между полостью, промываемой непрерывным потоком анализируемого вещества, и нагретым испарителем.
Твердые пробы в основном вводят в пиролизных устройствах через специальные шлюзы.
Детекторы для газовой хроматографии
Всего для газовой хроматографии предложено более 60 типов детектирующих систем. По общепринятой классификации детекторы подразделяются на дифференциальные и интегральные по форме зарегистрированного сигнала. Дифференциальные детекторы измеряют мгновенное различие в концентрации вещества в потоке газа-носителя. Хроматограмма, зарегистрированная таким детектором, представляет собой ряд пиков, площадь которых пропорциональна количеству разделенных соединений. Интегральные детекторы измеряют суммарные количества соединений, выходящих из колонки. Хроматограмма в этом случае ступенчатая, высота ступеней пропорциональна количеству соответствующих соединений.
В зависимости от однократной или многократной регистрации молекул анализируемых соединений выделяют концентрационные и потоковые детекторы. В концентрационных детекторах сигнал пропорционален концентрации соединения в подвижной фазе (элюенте). Здесь имеет место многократная регистрация молекул анализируемых соединений. В потоковых (или массовых) детекторах сигнал пропорционален количеству пробы компонента, дости
Аналитическая хроматография и капиллярный электрофорез
261
гаемому ячейки детектора в единицу времени. В этом случае происходит только однократная регистрация.
По селективности детекторы классифицируются на универсальные, селективные и специфические. В универсальных детекторах регистрируются все компоненты смеси, выходящие из колонки, за исключением подвижной фазы. Селективные детекторы регистрируют определенные группы соединений на выходе из колонки. Специфические детекторы регистрируют только один компонент или ограниченное число компонентов с подобными химическими характеристиками.
Основные технические характеристики детекторов:
• чувствительность или предел детектирования;
• линейность (динамический диапазон);
• инерционность (постоянная времени, быстродействие);
• стабильность (уровень шума и дрейфа);
• величина эффективного объема чувствительной ячейки.
Чувствительность концентрационных детекторов Ак определяется следующим выражением:
где S„ — площадь пика, см2; V — шкала самописца, мВ см1; Fr — скорость газа-носителя, мл • с-1; q — масса соединения, мг; F — скорость движения ленты самописца, см • с.
Размерность чувствительности в этом случае мВ • мг мл1.
Чувствительность потоковых детекторов (мВ • мг с1) равна:
Fq '
В последние годы чаще всего определяют предел детектирования. Для оценки минимально обнаруживаемой концентрации необходимо, кроме чувствительности, знать уровень флуктуаций (шума) нулевой линии. Минимальным сигналом, поддающимся измерению, обычно принято считать сигнал, высота которого в несколько раз (2-5) превышает уровень шумов 8:
25 С =---
mm л •
А
Величина cmm — предел детектирования — определяет предельные возможности прибора.
Под линейностью детекторов понимают диапазон концентраций, в пределах которых наблюдается линейность зависимости сигнал - концентрация. Для определения величины линейности строят соответствующий график. Обычно диапазон линейности расположен от предела детектирования до концентраций, в которых уже наблюдается отклонение от линейности на 5-10%.
Под инерционностью (быстродействием, постоянной времени) подразумевается скорость реагирования детектора на быстрое изменение концентрации на выходе из колонки. Детектор должен иметь такое быстродействие, чтобы при регистрации не искажать формы полосы соедине
ния, выходящего из колонки. В современных, особенно ионизационных детекторах постоянная времени — менее 0,1—0,01 с. В некоторых катарометрах, чаще всего устаревших конструкций, постоянная времени может составлять около 1 с и даже выше.
Быстродействие сильно зависит от величины эффективного объема ячейки.
Уровень шума нулевого сигнала детектора определяется кратковременными флуктуациями. Дрейф — это монотонное смещение нулевой линии. Величину смещения оценивают в течение 1 часа. Обычно требования к этим показателям таковы: шум 0,5% рабочей шкалы и дрейф не более 3% в час.
В табл. 4.1.1 приведены технические характеристики детекторов, применяемых в современных газовых хроматографах.
Таблица 4.1.1
Технические характеристики наиболее часто применяемых детекторов для ГХ
Детектор Предел детектирования (S/N= 2) Линейный динамический дапазон Тип Анализируемые соединения
ПИД 5 IO’12 ГС • c J 107 Селек-тив. Регистрирует органические соединения, ионизируемые в пламени водорода
ДТП 4 • 10’1 г • мл-1 106 Универе. Регистрирует все соединения, отличающиеся по теплопроводности от газа-носителя
эзд 1 • 10’14 г с-1 103-104 Селек-тив. Регистрирует в основном галогенорга-нические соединения
ФИД 2 • 10’12 г • с-1 ю7 То же Регистрирует все соединения за счет УФ-излучений с потенциалом ионизации менее 10,7 эВ или 11,7 эВ
тид 4 • 10’13 r(N) с’1 2 • 10~13 г(Р) • с’1 ю4 То же Селективно определяет гетеросоединения, имеющие атомы N и Р в молекуле
262
Новый справочник химика и технолога
Продолжение таблицы 4.1.1
Дете ктор Предел детектирования (S/N=2) Линейный динамический дапазон Тип Анализируемые соединения
ПФД 2 • ПГ11 r(S) • с-1 9 • ПГ13 г(Р) с-1 103 104 Спе-циф. Специфичен к S- и Р- содержащим соединениям
АЭД 1 ПГ13 2 • ПГ11 г • с-1 104 Универе. Регистрирует все соединения, имеющие в своем составе 12 основных элементов (Н, С, S,N, Ридр.)
мед 1 • ПГ11 г • с’1 1 • ПГ9 г • с-1 105 Универе. Регистрирует все соединения и может по масс-спектрам идентифицировать соединение
Механизм работы детекторов
Пламенноионизационный детектор (ПИД) основан на ионизации органических соединений в пламени водорода. Точный механизм ионизации не выяснен. С использованием масс-спектрометрометрии проведено исследование и обнаружено, что механизм новообразования связан с термодеструкцией и последующей хемоионизацией.
В ПИД одним из электродов служит горелка, второй электрод — коллектор — располагается над горелкой. Малые токи (1 • 109- 1О“12А) усиливаются, т.к. шумы самого детектора малы. Из-за высокой чувствительности, большого диапазона линейности ПИД стал наиболее распространенным детектором. В табл. 4.1.2 приведены атомные инкременты для показаний ПИД к соединениям разных классов.
Таблица 4.1.2
Атомные инкременты для показания ПИД
Атом Тип атома Вклад в общий сигнал (эффективное углеродное число)
С Алифатический 1
С Ароматический 1
С Олефиновый 0,95
с Ацетиленовый 1,30
с Карбонильный 0
с Нитрильный 0,30
О Простой эфир -1
О Первичный спирт -0,60
Продолжение таблицы 4.1.2
Атом Тип атома Вклад в общий сигнал (эффективное углеродное число)
О Вторичный спирт -0,75
О Третичный спирт -0,25
С1 У алифатического углерода -0,12
С1 У атома углерода при двойной связи +0,05
Детектор по теплопроводности (ДТП) — катарометр
Чувствительными элементами в ДТП являются нагретые нити (филаменты) из ряда металлов (платина, вольфрам, сплав вольфрам-рений и др.), помещенные в специальные камеры, продуваемые газом-носителем. Филаменты включены в плечи моста Уинстона. Через сравнительную камеру проходит поток чистого газа-носителя, через рабочую камеру — газ-носитель с примесями разделяемых соединений. Сопротивление нитей зависит от температуры. При изменении состава газа в рабочей камере теплопроводность его изменяется, изменяется теплопередача от нити к стенкам камеры, температура нити и, следовательно, сопротивление нити по сравнению с сопротивлением нити в сравнительной камере. Происходит разбаланс моста, возникает сигнал на нулевой линии. В табл. 4.1.3 приведены значения теплопроводности газов-носителей и некоторых органических веществ.
Таблица 4.1.3
Значения теплопроводимостей некоторых газов и паров
Соединение Теплопроводность при 100 °C - 103 Вт • (м • К)”1 Теплопроводность по отношению к гелию, %
Водород 223,6 128
Гелий 174,2 100
Азот 31,4 18,0
Диоксид углерода 22,2 12,7
Аргон 21,8 12,5
Этан 30,6 17,5
Бутан 23,4 13,5
Нонан 18,8 10,8
Бензол 17,2 9,9
Ацетон 16,7 9,6
Этанол 22,2 12,7
Этилацетат 17,2 9,9
Хлороформ 10,5 6,0
Метилиодид 7,9 4,6
Электронно-захватный детектор (ЭЗД)
ЭЗД предназначен для анализа веществ, обладающих электронным сродством, в частности галогенноорганиче
Аналитическая хроматография и капиллярный электрофорез
263
ских соединений. Полезный сигнал детектора — это уменьшение начального тока, однозначно связанного с количеством анализируемого соединения.
В ионизационной камере ЭЗД помещается радиоактивный источник (например, 63Ni). Под воздействием радиации молекулы газа-носителя (азот, аргон, гелий) ионизируются с высвобождением электрона:
N2 -> N2+ + е.
В камере между электродами приложено напряжение, фоновый ток создается в основном электронами, т.к. их подвижность на три порядка выше, чем подвижность ионов. Кроме того, большая часть ионов рекомбинирует, не доходя до электродов. При попадании в ячейку детектора соединений, обладающих сродством по отношению к электрону, происходит захват ими свободных электронов:
М + е —> М ,
что приводит к снижению начального фонового тока.
ЭЗД обладает высокой ионизационной эффективностью. В газе-носителе недопустимо присутствие кислорода, влаги и др. соединений, снижающих количество электронов или их подвижность.
Предел детектирования ЭЗД на два-три порядка ниже ПИД, он сильно зависит от числа и положения атомов галогенов в молекулах. В табл. 4.1.4 приведены данные по относительной чувствительности (относительно хлормета-на) ЭЗД к некоторым соединениям.
Таблица 4.1.4
Относительная чувствительность ЭЗД к некоторым соединениям
Соединения Относительная чувствительность Соединения Относительная чувствительность
Хлорметан 1 Фторбензол 0,3
Дихлорметан 11 Хлорбензол 10
Хлороформ 4 • 105 Бромбензол 10
Четыреххлористый углерод 5 106 Иодбензол 3 104
Термоионный детектор (ТИД)
ТИД селективен к N- и P-со держащим соединениям за счет введения в пламя водорода паров солей щелочных металлов (К, Na, Rb и Cs). Скорость введения паров щелочных металлов должна быть стабилизирована. ТИД чувствителен к стабильности поддержания скорости водорода, воздуха и газа-носителя. Селективность ТИД к N- и Р- органическим соединениям по сравнению с ПИД — порядка 102-103.
Пламенно-фотометрический детектор (ПФД)
ПДФ селективен к S- и P-содержащим соединениям, при сжигании которых в пламени, обогащенном водородом, по сравнению с ПИДом, излучаемый свет от этих
элементов направляется в фотоумножитель через специальные фильтры (394 нм для S и 526 нм для Р).
Особенности детектора:
• чувствительность ПФД к S-и P-содержащим соединениям тем больше, чем выше содержание этих элементов в соединениях;
• сигнал к P-содержащим соединениям пропорционален концентрации этого вещества в газе-носителе;
• сигнал к S-содержащим соединениям пропорционален логарифму потока вещества.
Фотоионизационный детектор (ФИД)
В ФИДе ионизация анализируемых соединений происходит за счет УФ-излучения в специальной камере с двумя электродами. При фотоионизации молекулы анализируемых соединений диссоциируются на ион и электрон:
А + hv -> А+ + е-.
Образуемые ионы собираются электродами. Ионизируются только те соединения, потенциал которых ниже энергии фотонов. В зависимости от лампы энергия фотонов может быть 9,5; 10,2 и 11,7 эВ.
ФИД как и ПИД обладает высокой чувствительностью ко всем органическим соединениям. К ароматическим соединениям ФИД имеет в 10-50 раз большую чувствительность, чем ПИД.
В отличие от ПИД, ФИД может регистрировать H2S, РН3, NH3, AsH3 и др.
В табл. 4.1.5 приведены потенциалы ионизации некоторых веществ, в табл. 4.1.6 — относительная чувствительность ФИД (относительно бензола) к соединениям разных классов.
Масс-спектрометрический детектор (МСД)
В последние годы достигнут прогресс в создании небольших настольных МСД для газовых хроматографов. В настоящее время этот высокочувствительный детектор — самый совершенный прибор для идентификации неизвестных веществ. Имеется библиотека масс для более 250 тыс. соединений. МСД обычно включает вакуумный насос, ионный источник и систему обработки. Для газовых хроматографов используются в основном два вида ионизации: электронный удар и химическая ионизация.
В качестве анализатора ионов могут применяться магнитные, квадрупольные и ионные ловушки, анализаторы ионно-циклотронного резонанса, с двойной фокусировкой (магнитные и электростатические), времяпролетные.
В качестве детектора, регистрирующего пучки ионов, используются электронный и фотоэлектронный умножитель, коллектор Фарадея, плоская электронная матрица.
МСД — ионизационный деструктивный потоковый детектор, универсальный и одновременно селективный, т.к. всегда можно найти массу, типичную только для данного соединения. При исследовании МСД в режиме детектирования отдельных ионов чувствительность его очень высока (в 1000 раз больше, чем в режиме сканирования) около 10“13 г (100 фемтограмм). Международный стандарт ионизации 70 эВ (1,1 • 1017 Дж) общепризнан, на многих современных хромато-масс-спектрометрах предусмотрен только такой фиксированный режим ионизации. Создана библиотека масс с этим источником ионизации.
264
Новый справочник химика и технолога
Таблица 4.1.5
Потенциалы ионизации некоторых соединений
Соединения эВ Соединения эВ Соединения эВ Соединения эВ
Простые молекулы Хлорированные углеводороды Ксилол 8,45 Диметилдисуль-фид 8,46
Азот 15,58 Стирол 8,47
Кислород 12,08 Метилхлорид 11,28 Анилин 7,70 Альдегиды, кетоны, спирты, кислоты
Вода 12,59 Четыреххлористый углерод 11,47 Азотосодержащие соединения
Формальдегид * 10,87
Оксид углерода 14,01
Диоксид углерода 13,79 Хлороформ 11,42 Аммиак 10,15 Ацетальдегид 10,21
Оксид азота 9,25 1,2-Дихлорэтан 11,12 Метиламин 8,97 Акролеин 10,10
Диоксид азота 9,78 Винилиденхлорид 9,83 Ацетонитрил 12,22 Ацетон 9,69
Метанол 10,85
Хлор 11,48 Винилхлорид 10,00 Акрилонитрил 10,91
Иод 9,28 Трихлорэтилен 9,45 Сернистые соединения Этанол 10,48
Предельные и непредельные углеводороды Гетероциклические и ароматические углеводороды Диоксид серы 12,34 Муравьиная кислота 11,05
Сероводород 10,46
Уксусная кислота 10,37
Метан 12,98 Фенол 8,50 Карбонилсульфид 11,18
Дисульфид углерода 10,08
Этилен 10,52 Пиридин 9,32
Ацетилен 11,41
Бензол 9,25 Метилмеркаптан 9,44
1-Бутен 9,58
Толуол 8,82
Гексан 10,17 Диметилсульфид 8,69
Таблица 4.1.6
Относительная чувствительность ФИД к соединениям разных классов (относительно бензола)
Соединения эВ Соединения эВ
н-Алканы Докозан 1,13
Гентан 0,032 Разветвленные алканы
Октан 0,08 2,2-Диметилбутан 0,037
Нонан 0,14 2,3-Диметилбутан 0,032
Декан 0,23
2-Метилпентан 0,011
Ундекан 0,30 Циклогексан 0,18
Додекан 0,37
Метилциклогексан 0,28
Тридекан 0,46
Декагидронафтен 1,04
Тетрадекан 0,53
1-Алкены
Пентадекан 0,59
Гексадекан 0,71 1-Гептен 0,54
Гептадекан 0,72 1-Октен 0,55
Октадекан 0,79 1-Пентадецен 0,92
Нонадекан 0,86 2-Гептен 0,51
Эйкозан 0,93 3-Гептен 0,58
Соединения эВ Соединения эВ
1,9-Декандиен 1,07 Бензол 1,00
2-Октен 2,75 Толуол 1,09
Альдегиды Этилбензол 1,16
Пентаналь 0,36 н-Пропилбензол 1,21
Октаналь 0,49 н-Бутилбензол 1,27
Спирты н-Гексилбензол 1,29
1-Бутанол 0,023 н-Октил бензол 1,52
1-Пентанол 0,053 Децилбензол 1,69
1-Гексанол 0,086 о-Ксилол 1,14
Изобутанол 0,029 jw-Ксилол 1,15
Эфиры п-Ксилол 1,20
Метилпропионат 0,01 Кумол 1,22
Бутилацетат 0,044 р-Кумол 1,27
Ароматические углеводороды Мезитилен 1,27
Колонки для газовых хроматографов
Колонки в газовой хроматографии подразделяются на насадочные (НК): препаративные, аналитические, микро-насадочные и капиллярные (КК). В табл. 4.1.7 приведены характеристики этих колонок.
В насадочных, микронасадочных колонках сорбент находится внутри трубки и имеет форму цилиндра. Набивка должна быть плотной и однородной, без пустот. Чем плот
нее и однороднее набивка, тем меньше размывание полос и больше эффективность колонки.
В КК слой сорбентов наносится на внутреннюю поверхность капилляра в виде слоя жидкой неподвижной фазы или в виде слоя адсорбента.
На рис. 4.1.8 изображены разные типы колонок.
По форме НК бывают прямые, U-образные, W-образ-ные и спиральные с разным радиусом кривизны.
Аналитическая хроматография и капиллярный электрофорез
265
Таблица 4.1.7
Характеристики колонок для газовых хроматографов
Типы колонок Внутренний диаметр колонок, мм Длина колонки, м
Препаративные насадочные Более 4 0,5-2
Аналитические насадочные 2-4 0,2-6
Микронасадочные 0,5-1 0,5-3
Капиллярные 0,2-0,3 5-100
Узкие капиллярные 0,05-0,2 5-100
Капиллярные широкого диаметра 0,3-0,8 10-60
Поликапиллярные 0,04 0,2; 1
Прямые и U-образные НК легко и наиболее плотно заполняются сорбентом без специальных приспособлений. W-образные и спиральные колонки заполняют под давлением на входе, либо с вакуумом на выходе из колонки.
На спиральных колонках при большом радиусе кривизны витков появляется дополнительное размывание, связанное с неоднородностью скоростей по сечению. Сопротивление потоку у ближней (к центру окружности) стенки трубки меньше, чем у дальней, так как пути прохождения газовых потоков у ближней стенки меньше.
Рис. 4.1.8. Типы колонок Колонки изготавливаются из металла (нержавеющая сталь, никель, медь), стекла, тефлона и других материалов. Чаще всего в аналитической практике применяются колонки из нержавеющей стали (для особо агрессивных смесей — колонки из никеля). Для разделения неустойчивых соединений (каталитически разлагающихся при контакте с металлической поверхностью) используют стеклянные и тефлоновые колонки; в частно
сти, стеклянные колонки широко применяются при анали
зе пестицидов.
КК изготавливались из нержавеющей стали, меди и латуни, затем начали использовать стекло (была предложена специальная лабораторная установка доя вытягивания капилляров из толстостенной стеклянной трубки с внешним диаметром 6-10 мм). Позднее (с 1980 г.) начали применять кварцевые КК, которые имеют наиболее инертную поверхность. Кварцевые капилляры для придания гибкости и прочности с внешней поверхности покрываются тонким слоем высокотемпературного полиамидного лака (до 350 °C) или слоем алюминия. Кварцевые КК со слоем лака допускают изгиб до 8-10 мм. В последние годы вновь появился интерес к металлическим КК, но с инертной (пассивированной) внутренней поверхностью.
Основные типы газовых хроматографов
В табл. 4.1.8 приведены основные типы газовых хроматографов, выпускаемых серийно.
Среди хроматографов разных типов отметим газовые хроматографы с масс-спектрометрическим и инфракрасными детекторами, а также газовые анализаторы, под которыми обычно понимаются газовые хроматографы, укомплектованные для решения конкретных аналитических задач «под ключ», т.е. оснащенные специальными колонками, аттестованной методикой и стандартами для градуировки. Иногда приборы такого типа называют газохроматографическими комплексами [4].
Таблица 4.1.8
Основные типы газовых хроматографов
Типы хроматографов Назначение
Лабораторные Работают в стационарном режиме в лабораториях разного профиля
Промышленные (хроматографы на потоке) Применяются для контроля производственных процессов в автоматическом режиме. Чаще всего имеют взрывобезопасное исполнение
Портативные
Малогабаритные Для передвижных и стационарных лабораторий. Экономия средств, энергии, места, расходных материалов при сохранении аналитических характеристик. Вес 10-25 кг
Портативные, транспортируемые, полевые Для анализа на месте расположения обследуемого объекта. Оперативность анализа, автономность по электрическому и газовому питанию. Вес 5-15 кг
Микрохроматографы (кремниевая технология), переносные, персональные, карманные Для оперативных относительно простых аналитических задач. Полная автономность. Ограниченные аналитические возможности. Вес 0,2-3 кг
Специальные микрохроматографы Для космических исследований. Автоматизация анализа, малая масса, устойчивость к ударам и тряске
Препаративные С широкими колонками для выделения веществ в чистом виде
В табл. 4.1.9 приведен перечень лабораторных газовых хроматографов, выпускаемых в нашей стране, характеристики основных лабораторных газовых хроматографов зарубежных фирм рассмотрены в [3].
В табл. 4.1.10 приведен перечень портативных отечественных газовых хроматографов, в табл. 4.1.11 — зарубежных портативных хроматографов.
В табл. 4.1.12 перечислены портативные хромато-масс-спектрометры, а в табл. 4.1.13 — портативные хроматографы для космических исследований.
266
Новый справочник химика и технолога
Таблица 4.1.9
Отечественные лабораторные газовые хроматографы
Название модели Фирма, город Детекторы Термостаты Отличительные особенности
1. Кристалл-2000М ЗАО СКВ «Хроматэк» г. Йошкар-Ола ПИД, ЭЗД, ПФД, ДТП, ФИД, ТИД 40-400 °C, изотермич. + программир. Блочное исполнение детекторов; термостат 6 л для многоколоночных систем недостаточен
2. Кристалл-5000 То же То же То же Термостат увеличен до 9 л
3. Цвет-800 ОАО «Цвет» г. Дзержинск То же 50-400 °C Большой объем термостата — 22 л
4. Кристал-люкс-4000 НПФ «Мета-Хром» г. Йошкар-Ола То же То же Блочное исполнение детекторов
5. ЛХМ-2000 «Милаб» г. Москва ПИД, ДТП, ЭЗД, тид 50-400 °C Разработан на базе старой модели 3700
6. Кристалл 2000 ФГУП «Купол» г. Ижевск ПИД, ДТП, ПФД, ЭЗД, ТИД, ФИД 40-400 °C Устаревшая конструкция по сравнению с Кристалл-2000М
7. Хромое ГХ-1000 ЗАО «Химаналит-сервис» г. Дзержинск ПИД, ЭЗД, тид, ДТП 50-450 °C На базе Цвет 500, снятого с производства. Все узлы и блоки заимствованы
8. ГАЛС-311 НПФ АП «Люмэкс» г. Санкт-Петербург ПИД, ЭЗД, ДТП, ПФД, тид 50-399 °C Прибор на базе модели 4890 (Hewlett Packard, КНР)
9. Цвет Яуза Т НПО «Химавтома-тика» г. Москва ПИД, ДТП, ФИД 50-400 °C изотермич. Надежный специализированный хроматограф для технологического контроля
10. Цвет Яуза, модель 100 НПО «Химавтома-тика» г. Москва ПИД, ДТП, ДЭЗ, ТИД, ПФД, ФИД 50-400 °C изотермич. + программир. Универсальный хроматограф
Таблица 4.1.10
Технические характеристики отечественных портативных хроматографов
Название-модели Фирма Технические характеристики
Детекторы Дозаторы Термостат Колонки Мощность, Вт Вес, кг Питание, В
ХПМ-1 ОАО «Цвет» ПИД Насос Изотермич. НК 1,5-40 10 12
ХПМ-2 ОАО «Цвет» ПИД Кран, шприц Изотермич. 50-200 °C НК 1-3 м 1,5-40 16 220-пер., 12 - пост.
ХПМ-4 То же ПИД, ДТП То же 50-200 °C НК 0,5-3 м 20 11 То же
Цвет П-182 То же ФИД То же 50-200 °C КК 40 12 То же
Цвет-МХ То же ПИД, ФИД, ТИД, ДТП То же 50-200 °C КК 6 3 12
ЭХО-М ООО «Си-бер-тех» ЭЗД Шприц, пред. КОНЦ. 40-175 °C Полика-пиллярные 40 11 12 пост.
ЭХО-EW То же ФИД, ЭЗД Шприц-кран 50-180 °C То же 25,60 12,9 То же
ПГМХ Хромдет ФИД То же 50-100 °C 10 То же
ФГХ-1 НПП «Хим-рибор» ФИД То же КК, 20-25 м 10 То же
Аналитическая хроматография и капиллярный электрофорез
267
Продолжение таблицы 4.1.10
Название-модели Фирма Технические характеристики
Детекторы Дозаторы Термостат Колонки Мощность, Вт Вес, кг Питание, В
ФГХ-2 НПП «Хим-прибор» ФИД, ДТП, ТИД 50-250 °C КК, 20-25м 10 12 пост.
АХГ-002 Эконокс ДТП Кран — — 70 16,5 То же
МХК НИИ ХРОМ ДТП Кран 50-150 °C НК 1,2,4, 8 м 60,400 6-8 220
мхп Тоже ПИД Шприц То же
МХФ Тоже ФИД То же
Таблица 4.1.11
Технические характеристики зарубежных портативных хроматографов
Название модели Фирма Технические характеристики
Детекторы Дозаторы Термостат Колонки Питание, В Вес, кг
311DGC HNU Systems ПИД, ЭЗД, ФИД, ПФД, ДТП Кран 10-ходовый 50-200 °C, программир. КК или НК ПО, 115,230 25
Model 4100 Electronic Sensor Technology Поверхн. аку-стич. резонатор Криофокусировка 50-200 °C, программир. КК 120,12 15,9
MSI-301 Microsensor Systems То же Дозирование насосом 5 40 °C КК или НК 120,12 5,4
Р200 Р200Н MTI Analytical Instruments ДТП Инжектор на кремниевой технологии 50-180 °C — 12 10,4
Fm-2000 O.I. Analytical ПИД, ФИД, ПФД, ППФД, ПИД/ФИД Кран, предконц. — КК 110 8,1
Snapshot PE Photovac ФИД Кран 45 °C КК широкого диаметра 12 4,4
Voyager PE Photovac ФИД, ЭЗД Шприц, кран 55-80 °C КК широкого диаметра, микронасос 12 6,8
Scento-screen Sentex Systems ЭЗД, ФИД, ДТП, микроар-гонный, ионизационный Шприц, кран, предконц. До 180 °C программир. КК 30 м, насад. 3,6 м 110/220 14
Scento-graph Plus II Sentex Systems ЭЗД, ФИД, ДТП, микроар-гонный, ионизационный Шприц, кран, предконц. До 180 °C программир. КК до 105 м 12 21,8
Micro GC CP-2002 Chrompack Varian ДТП Кран 30-180 °C изотермич. НК 3,6 м, две колонки НКиКК 110,12 6,7
268
Новый справочник химика и технолога
Таблица 4.1.12
Технические характеристики портативных газовых хромато-масс-спектрометров
Фирма Bruker Instr. Inficon Viking Instr.
Модель ЕМ640, EM640S Hapsite Spectra Trak 572
Термостат 50-300 °C изотерм, и три скорости программир. — 50-325 °C изотерм, и программ.
Колонки 30 м х 0,2 мм 30 м х 0,32 мм 60 м х 0,32 мм
Газ-носитель N2, Не n2 Не, N2, Н2
Дозатор Капил, ввод с делением и без деления потока — Капил, ввод с делением и без деления потока
Диапазон масс 1-640 1-300 1,6-700
Максим, скорость сканирования 2000 а.е.м. • с ! 1000 а.е.м. • сек1 1800 а.е.м. • сек1
Тип ионизации Электронный удар Электронный удар, 70 эВ Электронный удар, 70 эВ
Вакуумная система: насос Геттерный Геттерный Турбомолекулярный диафрагменный
Чувствительность 1 нг • с1 (при S/N 10:1) 100 нг для гексахлорбензола —
Динамический диапазон 109 — 106
Вес, кг 60 16 34
Размеры: высота х ширина х глубина, см 55 х 45 х 35 18x43 х 46 46 х 61 х 29
Таблица 4.1.13
Технические характеристики портативных газовых хроматографов для космических кораблей
Назначение Программа полета Детекторы Колонки Термостат Газ-носитель Дозатор
Анализ атмосферы и грунта Марса «Viking» 1975-1976 г.г. (США) ДТП, МС 7,6 м х 1,1 мм с по-рапаком Q; 2 м х 0,75 мм с тенаксом 24 °C Не, 13,5 см3мин-1 Краны
Анализ атмосферы Венеры «Венера 11-14» ГХ «Сигма» 1978-1982 г.г. Неоновый ионизационный Три колонки с поли-сорбом цеолитом и оксидом магния 2 м; 2,5 м 70 °C Ne То же
Анализ атмосферы Венеры и кометы Галлея «Вега» 1984-1985 г.г. ГХ «Сигма-3» Гелиевый ионизационный ДТП, два ДЭЗ Три колонки с пора-паками Q, S, N и Е 70 °C Не, N2 То же
Анализ атмосферы Венеры «Pioneer-Venus» 1978 г. ДТП 15,85 м х 1,1мм с по-рапаком N; 2,1 м х 1,1 мм с poly-DVB 18 °C Не, 35 см3-мин 1 То же
Анализ атмосферы Титана (спутник Сатурна) «Cassini-Huygens» 1997-2004 г.г. МС Три парал. колонки: рога PLOTQ PLOT А12О3, цеолит Программир. — —
Дополнительные устройства для газовой хроматографии
Криогенное устройство — система термостатирования колонок (диапазон температур: от комнатных до -100 °C) для разделения трудно разделяемых газовых смесей. Для этих целей используется жидкий азот из сосуда Дьюара.
Система обратной продувки: шестиходовый кран-дозатор и четырехходовый кран, — включает обратную продувку колонки для быстрого элюирования суммы тяжелых компонентов, в частности, при определении природного газа Ci - С5 и ЕС6.
Обогатительные устройства для концентрирования тяжелых примесей из газовых потоков с последующей десорбцией и дозирования в аналитическую колонку. Кон
Аналитическая хроматография и капиллярный электрофорез
269
центрирование примесей происходит в охлаждаемой небольшой обогатительной колонке. После обогащения десорбция производится специальной разогретой печкой.
Криофокусирующее устройство позволяет концентрировать примеси в начале охлажденной капиллярной колонки. Сильносорбируемые высококипящие соединения удерживаются на начальном участке колонки, а газ-носитель и легкие соединения проходят через колонку, не сорбируясь. После окончания процесса концентрирования происходит быстрый нагрев (тепловой удар) для того, чтобы при десорбции проба не размывалась, но вводилась в колонку в виде узкой полосы с десорбированными сконцентрированными компонентами.
Устройство для концентрирования методом выдувания и накопления (purge and trap) предназначено для выдувания из загрязненных вод летучих и малолетучих примесей и накопления их на специальной адсорбционной ловушке с последующей тепловой десорбцией и переводом в хроматографическую колонку.
Устройство парофазного концентрирования (headspace) позволяет повысить чувствительность определения легкокипящих соединений, растворенных в воде, имеющих коэффициенты распределения менее 10. Эти устройства позволяют также извлекать и дозировать легкие анализируемые соединения из биологических проб, из твердых материалов (пород, почв, полимерных материалов и др.).
Устройство пиролизное. Пиролизная газовая хроматография применяется для анализа нелетучих высокотемпературных соединений (полимеров, каучуков, смол, олигомеров, биополимеров и др.) по продуктам их разложения в инертной среде (пиролиз).
Пиролиз проводят с помощью обычного термического нагрева, высокочастотного нагрева (до точки Кюри), лазерного разогрева и разряда. Устройство для пиролиза изготавливается в виде приставки к стандартным газовым хроматографам, которые включают вместо узла ввода пробы или параллельно ему. Пиролизные устройства бывают трех типов: филаментного, печного и высокочастотного с ферромагнитными держателями. Различают мягкий пиролиз до 500 °C, в основном для биологических объектов (бактерий, белков, крахмала и др); средний пиролиз при 500-800 °C для исследования полимеров; жесткий пиролиз при 800-1100 °C, полимеры разрушаются на небольшие фрагменты, образуется много продуктов разложения.
Автоматические дозирующие устройства (автосам-плеры). Автосамплер включает от 8 до 120 стеклянных пробирок с пробами, которые подаются к месту отбора и дозирования по специальной программе. Кроме пробирок с пробами, имеются пробирки с промывочными растворителями для промывки дозирующего узла после ввода анализируемой пробы. Последовательность операций задается и контролируется микропроцессорным блоком управления и может быть откорректирована под конкретные задачи.
Метанатор. В некоторых аналитических задачах чувствительность детектора по теплопроводности недостаточна для определения СО и СО2. В этих случаях проводят конверсию СО и СО2 до метана в специальной трубке
с Ni-катализатором в потоке водорода после разделения СО и СО2 на хроматографической колонке. Получаемые пики метана регистрируют ионизационно-пламенным детектором на уровне < 10 4%.
Прочие устройства. В составе газовых хроматографов применяют иногда измерители потоков, разные интерфейсы (ГХ-МС, ГХ-ИКС, ЖХ-ГХ и др.), генераторы водорода, азота, сверхчистого воздуха, фильтры-очистители газов.
Технические требования к программному обеспечению верхнего уровня газовых хроматографов
1. Программное обеспечение (ПО) должно быть рассчитано на обслуживание до четырех (в редких случаях до восьми) хроматографов одновременно.
2. В программе должно быть обеспечено выполнение следующих функций:
• задание режимов работы хроматографа;
• индикация текущих режимов;
• отслеживание аварийных ситуаций;
• прием и фильтрация хроматографических сигналов;
• обработка хроматограмм;
• идентификация компонентов смеси по абсолютному времени удерживания;
• создание паспорта хроматограммы;
• создание методики проведения анализа;
• расчет параметров хроматографических колонок;
• расчет статистических параметров;
• распечатка хроматограммы и протокола анализа;
• осуществление тестирования устройств хроматографа;
• осуществление тестирования самого ПО;
• наличие оперативной подсказки.
3. ПО должно иметь возможность подстройки под уровень сложности задач потребителя (исследовательский вариант, рутинный, специальной задачи).
4. ПО должно работать в операционной среде Windows 95, 98 и выше.
5. Размер хроматограмм — неограниченный.
6. Число обрабатываемых пиков — до 1000.
7. Должна быть предусмотрена ручная коррекция — раз-метка хроматограмм, начало и конец счета, введены функции оптимизации, настройки системы.
8. Расчет индексов удерживания (линейных, логарифмических).
9. Должна быть предусмотрена возможность работы одной и той же копии программы на разных компьютерах.
10. Должна быть предусмотрена возможность расчета концентрации компонентов по группам пиков.
11. Должна быть предусмотрена возможность оценки погрешности расчетов высоты и площади пиков.
12. Должна быть предусмотрена возможность проверки с помощью внешнего устройства «Генератор хроматограмм» и возможность внутреннего тестирования.
13. Результаты обработки должны быть распечатаны в виде стандартного протокола: хроматограмма (с расшифровкой) и таблица с результатами анализа.
14. В памяти ПЭВМ результаты должны храниться в течение года (в частности, для анализов при сертификации пищевых продуктов).
270
Новый справочник химика и технолога
15. Программа должна проводить калибровку как по одной точке, так и многоточечную калибровку с использованием как линейных, так и нелинейных калибровочных зависимостей.
Литература
1. Столяров Б.В. и др. И Практическая газовая и жидкостная хроматография. СПб.: СПбГУ, 1998. С. 81.
2. Verker P.J. // Chromatogr. 1984. V. 300. Р. 249.
3. Erickson В. // Anal. Chem. News and Features. 1999.
Apr. P. 271-276.
4.1.2. Газо-жидкостная хроматография
4.1.2.1. Преимущества и недостатки газо-жидкостной хроматографии
В газо-жидкостной хроматографии разделение происходит за счет различной растворимости компонентов смеси в пленке жидкой фазы (ЖФ), нанесенной на поверхность макропористого твердого тела, так называемого твердого носителя. В настоящее время для многих аналитических задач газо-жидкостная хроматография — наиболее эффективный метод.
Применение ЖФ дает ряд преимуществ:
1. На ЖФ изотерма абсорбции линейна при обычных рабочих условиях в широком диапазоне концентраций, а это означает, что пики получаются симметричными.
2. Не вызывает затруднений выбор достаточно селективной ЖФ ввиду их большого многообразия.
3. Количество ЖФ в колонке можно легко менять, изменяя удерживаемые объемы и селективность разделения. Используя одну и ту же ЖФ, можно приготовить как пре
паративные, так и высокоэффективные аналитические колонки (насадочные или капиллярные).
4. ЖФ доступны, имеют достаточно высокую степень чистоты и стабильные свойства, таким образом, параметры удерживания на них воспроизводимы.
Однако применение ЖФ встречает и ряд серьезных трудностей. Основные недостатки газо-жидкостной хроматографии связаны с летучестью и нестабильностью ЖФ, что затрудняет применение этого метода для разделения и анализа ряда важных смесей.
Основные требования к неподвижным ЖФ:
• малая летучесть при рабочих температурах (это связано с временем работы колонки);
• химическая инертность и термическая стабильность;
• низкая вязкость при рабочих температурах;
• растворимость в наиболее доступных растворителях.
В табл. 4.1.14 приведена классификация ЖФ по полярности, а в табл. 4.1.15 перечислены основные типы неподвижных ЖФ.
Таблица 4.1.14
Классификация жидких фаз по полярности
Тип ЖФ Примеры
Неполярные Сквалан, апиезоны, полиметилси-локсаны: OV-1, SE-30 и др.
Средней полярности Полиэфиры, сложные жиры: фталаты, фосфаты, себацинаты, эфиры полиэтиленгликоля и др.
Полярные Полиэтиленгликоли, глицерин, нит-рильные фазы и др.
Таблица 4.1.15
Основные типы неподвижных жидких фаз
Неподвижная ЖФ Молярная масса, г • моль'1 Относительная плотность при 293,2 К Диэлектрин, постоянная Полярность по Рор-шнайдеру Максимальная рабочая температура колонки, °C Рекомендуемые растворители Применение
Неполярные
Апиезоны 15,000 1,086 2,65 8 тип L-320, типы М, N, W 270-300 Метиленхлорид, ксилол, гексан, диэтиловый эфир, толуол Для разделения высоко-кипящих веществ
Вазелиновое масло — — — 0 100 Этанол, диэтиловый эфир Углеводороды, метиловые эфиры жирных кислот
Гексадекан 226,45 0,7734 2,07 -2 60 Гексан, диэтиловый эфир, петролейный эфир Легкие углеводороды
Сквалан 422,83 0,80785 2,09 0 130 Ацетон, дихлорметан Нормальные и терпеновые углеводороды, спирты, кетоны, простые и сложные эфиры
Сквален 410,73 0,8562 2,3:3 12 100 То же То же
Аналитическая хроматография и капиллярный электрофорез
271
Продолжение табл. 4.1.15
Неподвижная ЖФ Молярная масса, г моль4 Относительная плотность при 293,2 К Диэлектрин, постоянная Полярность по Рор-шнайдеру Максимальная рабочая т-ра колонки, °C Рекомендуемые растворители Применение
ПМС-100 5000 0,968 9 250 Диэтиловый эфир Универсальное
Е-301 —- — 9 250 Дихлорметан, хлороформ Тоже
SE-30 — 9 300 При нагревании То же
Среднеполярные
Дибутил-фталат 278,35 1,0489 6,10 38 100 Дихлорметан Насыщенные и ненасыщенные углеводороды, кислородсодержащие вещества
Динонилфталат 418,62 0,9668 4,75 26 130 То же Универсальное
Дидецилфталат 446,68 0,960 — 25 150 То же То же
Бензилдифенил 244,34 1,191 — 40 100 То же Ароматические углеводороды, их изомеры, амины, жирные кислоты
Диоктил-себацинат 426,69 0,913 4,01 28-4,05 150 Дихлорметан, ацетон, хлороформ Насыщенные и ароматические углеводороды, низшие жирные кислоты, эфиры, кетоны, спирты
ПФМС-4 1500 1,10 — 24 350 Диэтиловый эфир Универсальное
ПФМС-6 20000 1,15 3,2 33 400 То же То же
НПС-50 1200 1,0493 — 37 200 Ацетон То же
НПС-100 1290 1,09 — 62 180 То же То же
СКТФТ-50 — — — 21 350 Ацетон То же
СКТФТ-100 — — — 32 350 То же То же
QF-1 — — — — 220 Хлороформ Стероиды, пестициды, сахара (при программировании температуры колонки)
Полифениловый эфир (5Ф4Э) 446 — — 44 200 То же Полициклические углеводороды
Триэтил ен-гликоль-бутират 290,34 1,037 50 100 Ацетон Нормальные и изомерные низшие углеводороды
Трикрезилфосфат 368,4 1,16 6,7 48 130 Ацетон, этанол Ароматические и алифатические углеводороды, кетоны, сложные эфиры
Полярные
Адипонитрил 108,15 0,962 — 83 50 Хлороформ, метанол, диэтиловый эфир, этанол Легкие углеводороды
Глицерин 92,09 1,2613 42,4 80 70 — Кислородсодержащие соединения, (для разделения смеси аммиака с метиламинами)
Диглицерин 166,18 1,30 — 79 130 — Спирты, кетоны в присутствии воды. Алифатические амины
272
Новый справочник химика и технолога
Продолжение табл. 4.1.15
Неподвижная ЖФ Молярная масса, г • моль 1 Относительная плотность при 293,2 К Диэлектрин, постоянная Полярность по Рор-шнайдеру Максимальная рабочая т-ра колонки, °C Рекомендуемые растворители Применение
Полиэтилен гликоли
ПЭГ-300 300 1,125 16 83 90 Дихлорметан Универсальцое
ПЭГ-400 400 1,125 13,7 75 100 Ацетон
ПЭГ-1000 1000 — 11,0 71 150 Хлороформ
ПЭГ-20М 20,000 — 68 200
Полиэтилен-гликольади-пинат (ПЭГА) — 1,0577 95 78 200 Хлороформ Высшие жирные кислоты, другие кислородосодержащие соединения
Реоплекс-400 16000 323,2 — — — Дихлорметан
-Тиодипропионитрил 140,22 1,111 — 82-90 90 Ацетон Универсальное
(3,(3 -Оксидипропио-нитрил 124,15 1,05 100 70 Углеводороды, спирты, для отделения ароматических углеводородов от нафтеновых и парафиновых
(3,(3 - Аминодипропио-нитрил 123,11 — — — 80 Хлороформ, метанол Легкие углеводороды и ароматические углеводороды
Триэтаноламин 149,18 1,124 29,36 87 75-100 Дихлорметан, хлороформ Легкие углеводороды, гетероциклические соединения
Формамид 45,04 1,1334 — 109 40 Метанол Легкие углеводороды (доСб)
ASL, США Apolar (silar) (цианопро-пилсилико-ны) 7СР, 9СР, 10СР 250 Хлороформ, ацетон Метиловые эфиры цис-и транс- изомеров высших жирных кислот
SP Sup. США SP-216-PS (полиэфир, модифицированный Н3РО4) 200 Хлороформ Высшие жирные кислоты
SP-4OO (хлорфенил-метилсилико-новое масло) — — — — 350 Ацетон, хлороформ Универсальное
SP-1000 20,000 — — — 275 Хлороформ Свободные жирные кислоты (С6 и выше)
SP-2100 (ме-тилсилконо-вое масло) — — — — 350 То же Универсальное
SP-2340 (цианопро-пилсиликон) — — — — 275 Хлороформ, ацетон Изомеры положения, гщс-щраис-изомеры
Аналитическая хроматография и капиллярный электрофорез
273
Для оценки полярности ЖФ используется система Рорш-найдера [1]. Эта система основана на определении разности индексов удерживания Ковача на полярных и неполярных фазах для таких тестовых соединений как бензол, этанол, метилэтилкетон, нитрометан и пиридин. Фазой с нулевой полярностью считают сквалан, а в качестве фазы со 100% полярностью — РД'-оксидипропионитрил. В табл. 4.1.16 при-
ведены значения разностей индексов Ковача для ряда наиболее распространенных ЖФ (индексы Ковача получены при 100 °C, ЖФ в количестве 20% нанесены на хромосорб W). В настоящее время чаще всего неподвижные ЖФ прививают к поверхности носителя и сшивают в объеме пленки за счет различных пероксидов (дибензоила, дикумила и др.). В табл. 4.1.17 приведены наиболее распространенные ЖФ.
Таблица 4.1.16
Приращения индексов Ковача для наиболее распространенных жидких фаз
ЖФ Константа Роршнайдера
бензол этанол метилэтилкетон нитрометан пиридин
Апиезон L 0,32 0,39 0,25 0,48 0,55
Бентон-34 2,41 5,13 4,30 - -
Диизодецилфталат 0,83 1,65 1,43 2,53 1,54
Динонилфталат 0,84 1,76 1,48 2,70 1,53
Диэтиленгликольсукцинат 4,93 7,58 6,14 9,50 8,37
Неопентилгликольизофталат 2,07 3,56 3,15 4,99 3,77
Р,Р -Оксидипропионитрил 5,88 8,48 8,14 12,58 9,19
Полипропиленгликоль-себацинат 1,93 3,38 2,58 4,36 3,27
Полифениловый эфир (5 колец) 1,75 2,27 2,34 3,26 2,84
Полиэтиленгликоль-4000 3,22 5,46 3,86 7,15 5,17
Силиконовое масло DC-560 0,31 1,47 0,75 1,33 0,75
Силиконовое масло QF-1 1,09 0,86 3,00 3,94 2,41
OV-1 0,16 0,20 0,50 0,85 0,48
OV-3 0,42 0,81 0,85 1,52 0,89
OV-7 0,70 1,12 1,19 1,98 1,34
OV-11 1,13 1,57 1,69 2,66 1,95
OV-17 1,30 1,66 1,79 2,83 2,47
OV-22 1,58 1,80 2,04 3,27 2,59
OV-25 1,76 2,00 2,15 3,34 2,81
OV-101 0,16 0,20 0,50 0,85 0,48
OV-210 1,41 2,13 3,55 4,73 3,04
OV-225 2,17 3,20 3,33 5,16 3,69
Твин 80 2,14 4,20 2,78 5,20 3,65
Трикрезилфосфат 1,74 3,22 2,58 4,14 2,95
Этиленгликольадипинат 3,43 5,46 4,52 7,11 6,00
Этиленгликольизофталат 3,00 4,76 4,10 6,51 5,17
Этиленгликольсукцинат 4,51 7,06 5,67 9,04 7,69
Индексы Ковача на сквалане 649 384 531 457 695
TH
Новый справочник химика и технолога
Таблица 4.1.17
Неподвижные жидкие фазы
Тип жидкой фазы Краткое обозначение Структурная формула
Полимеры
Полидиметилсилоксан (100%) SE-30, OV-1 и др. CH3 1 O—Si— CH3 100%
Полибисцианопропилсило-ксан (100%) SP-2340 0- ch2ch2ch2cn Si
ch2ch2cch2cn 100%
Полиэтиленгликоль (и) Supelcowax 10 -OCH2-CH^- -OH n
Полиэтиленгликоль (я), модифицированный нитроте-рефталевой кислотой Nukol О HO2C C“ no2 -OCH2-CH2- 0 -0—c—C^~ co2H n no2
Сополимеры
Диметилсилоксана (95%) с дифенилсилоксаном (5%) SPB-5 1 1 ' /-Х ? г i c 1 -0—s 1 c Из
)_ 5% - H3 95%
Диметилсилоксана (80%) с дифенилсилоксаном (20%) SPB-20 1 2! Cy—c? Су о 1 1 1 20% CH3 — O—Si CH3 80%
Диметилсилоксана (65%) с дифенилсилоксаном (35%) SPB-35 ’ 91 — 0—Si 35% CH3 — O— Si CH3 65%
Дифенилсилоксана (50%) с диметилсилбксаном (50%) SPB-50 1 1 1 <0—i,_Q, 1 1 50% CH3 1 — O—Si 1 CH3 50%
Бис-цианопропилсилоксана (80%) с цианопропилфенил-силоксаном (20%) SP-2330 c 0— s c h2ch2ch2cn - 0- ch2ch2ch2cn
h2ch2ch2cn 80% - 6 J 20%
Аналитическая хроматография и капиллярный электрофорез
275
Тип жидкой фазы
Краткое обозначение
Продолжение таблицы 4.1.17
Структурная формула
Бис-цианопропилсилоксана (90%) с цианопропилфенил-силоксаном (10%)
ch2ch2ch2cn
О—Si----------
ch2ch2ch2cn
ch2ch2ch2cn
10%
Диметилсилоксана (86%) с бис-цианопропилфенил-силоксаном (14%)
SPB-1701
Октилсилоксана (50%) с метил сил оксаном (50%)
SPB-Octyl
СН3
I
Si-----------------------
ch2ch2ch2ch2ch2ch2ch2cn
n
Этиленгликоля (и) с пропи-ленгликолем (w)
PAG
СН3
но—сн2—сн2—о
СН2-СН-О----н
п
т
4.1.2.2. Твердые носители
Возможности газо-жидкостной хроматографии могут быть реализованы лишь при наличии подходящего твердого носителя. Идеальный носитель химически неактивен, с однородными макропорами, на поверхности которых равномерно распределена пленка неподвижной жидкой фазы. Он, как правило, должен обладать высокой термической стабильностью, механической прочностью и хорошей смачиваемостью.
Чаще всего в качестве твердых носителей используют материалы на основе природных диатомитов. Реже применяются синтетические кремнеземные носители — макропористые силикагели, макропористые стекла, аэросилоге-ли. Используются также носители на основе туфов и перлитов. В отдельных случаях применяют в качестве твердых носителей стеклянные шарики, хлорид натрия, металлические спирали, пористый тефлон, обожженную керамику и графитированную сажу [2-6].
Носители на основе диатомитов. Носители с высокой механической прочностью гранул получают прокаливанием природного диатомита при 900 °C и выше. Такие носители, имеющие розовый цвет, относятся к I типу. Их удельная поверхность может иметь величину 5-8 м2-г-1; размер пор небольшой, в основном 0,4-2 мкм; pH 5% водной суспензии составляет 6-7; термостойкость достигает 1000 °C. Носители этого типа обеспечивают высокую эффективность разделения неполярных и слабополярных веществ, но проявляют большую адсорбционную способ
ность по отношению к сильно полярным соединениям (например, спиртам, аминам, кислотам).
Носители II типа (кальцинированные или белые) приготавливаются спеканием природного диатомита с щелочными флюсами и последующим прокаливанием при 900 °C. Адсорбционная активность белых носителей меньше, чем розовых, поэтому их применяют для хроматографирования полярных соединений. Носители II типа имеют удельную поверхность 1-3 м2-г-1, pH 5% водной суспензии 8-10, средний размер пор 8-9 мкм. Термостойкость до 1000 °C.
В нашей стране выпускаются носители обоих типов; на основе Инзенских диатомитов — Сферохромы 1, 2, 3. Производство носителя включает образование водной суспензии, просушку ее при 450 °C, последующее прокаливание в течение 24-36 часов и рассеивание. В процессе производства сферохрома 1 в суспензию добавляют карбонаты натрия и калия и прокаливают при 920-940 °C. Перед нанесением ЖФ носитель следует отсеять от пыли и просушить при температуре 300 °C. Дополнительное прокаливание при высоких температурах (950 °C) приводит к снижению адсорбционной активности, но эффективность разделения снижается. Установлено, что наилучшая эффективность на сферохроме 1 наблюдается при нанесении 5-10% (по массе) ЖФ.
Сферохромы 1, 2, 3 имеют различные физические и химические свойства. Сферохром 1 используют главным образом для анализа полярных соединений основного и нейтрального характера, Сферохром 2 является более универ
276
Новый справочник химика и технолога
сальным носителем, его поверхность нейтральна, и он рекомендуется для разделения органических соединений различного строения. Сферохром 3 имеет большую удельную поверхность (10-15 м2т '), заметную адсорбционную активность и рекомендуется для особых случаев разделения. На основе Инзенских диатомитов выпускаются также твердые носители — динохромы Н и П [5]. Производство включает в себя обработку дробленого кирпича марки К-600 кипящей соляной кислотой с последующим прокаливанием при 900 °C. В результате прокаливания отмытой кислотой массы с добавкой гидрооксида натрия получают динохром П, а в результате прокаливания без введения щелочного реагента — носитель динохром Н. Последний рекомендуется применять для аналитической и препаративной газо-жидкостной хроматографии неполярных и слабополярных веществ, преимущественно в сочетании с неполярными и слабополярными неподвижными фазами. На носитель можно наносить до 35% неподвижной ЖФ (по массе). Оптимальное количество жидкости для аналитических колонок —12-15%.
Носитель динохром П рекомендуется использовать для разделения полярных веществ в сочетании со слабополярными и полярными неподвижными ЖФ. На носитель можно наносить до 30% жидкости; оптимум для аналитических колонок— 10-15%.
На основе Джрадзорского диатомита, тщательно отмытого кислотой и водой, путем сплавления с 5-7% соды получают твердые носители — порохромы 1, 2, 3. По данным авторов [7,8] порохромы отличаются от большинства других носителей однородной макропористой структурой, высокой геометрической однородностью пор. Кроме того, они адсорбционно и каталитически инертны. Порохромы можно применять для аналитической и препаративной газо-жидкостной хроматографии в сочетании как с неполярными, так и с полярными неподвижными ЖФ. Из диатомита Джрадзорского месторождения в последнее время изготавливаются также твердые носители — цветхромы [9].
Фирма «Лахема» производит твердые носители для газо-жидкостной хроматографии — Хезасорб и Хроматон N, Хроматон N-cynep, Хроматон М-супер, инертон и инертон-супер. Указанные носители по своим основным параметрам (пористая структура, химический состав, эффективность) близки к американским Хромосорбам W и Р [2, 10], Хроматон N обладает узким распределением пор и не содержит микропор, снижающих эффективность разделения. Низкое содержание каталитически активных оксидов типа R2O3, прежде всего Fe2O3, позволяет работать при высоких температурах и низкой степени смачивания, не опасаясь каталитического разложения разделяемых веществ. Высокая химическая чистота и малая удельная поверхность обуславливают адсорбционную инертность носителя, которая очень важна, особенно при разделении сильнополярных веществ на носителе с низкой степенью смачивания.
Хезасорб по сравнению с Хроматоном N содержит больше Fe2O3 и соответственно обладает большей катали
тической активностью. Носитель обладает высокой химической и термической стабильностью до 1000 °C, механически прочен. Основная область применения хезасорба — высокоэффективное разделение неполярных и слабополярных соединений. Оценке свойств носителей типа Хроматон N и Хезасорб посвящено большое количество работ [2]. Основные характеристики вышеперечисленных носителей приведены в табл. 4.1.18.
Хроматон N-cynep представляет собой отмытый кислотой и щелочью силализированный Хроматон N, инертный по отношению к веществам, чувствительным к каталитическому разложению.
Хроматон М-супер применяется для разделения сложных веществ методом газовой хроматографии, прежде всего при высоких температурах (напр. пестицидов).
Инертон — это носитель, в котором удачно совмещаются адсорбционная инертность белых диатомитовых носителей (напр. Хроматон N) с механической прочностью розовых (напр. Хезасорб). Для инертона характерно узкое распределение пор, а также отсутствие микропор. Эти свойства в сочетании с низкой каталитической активностью гарантируют хорошую разделяющую способность носителя.
Инертон-супер представляет собой отмытый кислотой и щелочью сил авизированный инертон. Исследованию хроматографических свойств новых твердых носителей фирмы «Лахема» посвящена работа [10].
Полимерные твердые носители. Наиболее широкое применение нашли носители на основе политетрафторэтилена, обладающие высокой химической стойкостью и инертностью поверхности.
Из полимерных твердых носителей наиболее часто применяется Полихром-1 при анализе водных растворов спиртов и особенно жирных кислот. К недостаткам этого носителя следует отнести низкую механическую прочность, высокую электризуемость, трудность нанесения неподвижной фазы, а также слабую смачиваемость полярными неподвижными фазами, что вызывает неравномерность их распределения на поверхности носителя. Лучше всего поверхность Полихрома-1 смачивается метилсиликоновыми и фторированными жидкостями. Специальная обработка Полихрома-1 и фторопласта-4 улучшает смачиваемость поверхности носителя и повышает эффективность колонки [11].
Полихром-1 легко комкуется, поэтому для равномерного заполнения колонки его необходимо охлаждать до 0°С. При охлаждении готового сорбента в жидком азоте колонка заполняется значительно равномернее, и ее эффективность заметно повышается, однако при этом на сорбенте конденсируется влага из воздуха, и колонку необходимо дополнительно кондиционировать. Выше 180 °C использовать Полихром-1 не рекомендуется. При работе с детектором электронного захвата верхний температурный предел использования составляет 140-150 °C [11].
Некоторые характеристики полимерных твердых носителей приведены в табл. 4.1.19.
Аналитическая хроматография и капиллярный электрофорез
277
Таблица 4.1.18
Характеристики твердых носителей на основе диатомитов
Марка носителя Тип Насыпная плотность (кг-м 3)-10 3 Удельная поверхность, м2т-1 Механическая прочность, % истирания в кипящем слое pH Адсорбционная активность, мл Общий объем пор, (м’-кг^ПГ3 Эффективный радиус пор, мкм
Отечественные
Сферохром-1 (ТНД-ТС-М)* II 0,43 0,8-2,1 1,75 8,8 45 0,65 1
Сферохром-2 I 0,54 4 2,5 7,5 — 0,6 1
Сферохром-3 I 0,6 10-15 — — — — —
Динохром Н I 0,45 5-8 1,5 7,5-8 — 0,7 0,42
Динохром П II 0,5-0,6 1-1,5 1 8-9 — 0,6 0,7
Порохром 1 II 0,2-0,4 0,1-1,5 — 8 — 1,5-1,8 3,5
Порохром 2 II 0,3-0,4 0,5-2 — 8 — 1,5-1,8 3,5
Порохром 3 II 0,23-0,3 2-4 — — — 1,5-1,8 —
Цветхром 1 I 0,4-0,6 1,5-3,5 — 6-7 — 0,6 —
Цветхром 2 II 0,4-0,6 0,5-0,9 — 6,5-8 — 0,5 —
Зарубежные
Хезасорб III 0,6 1,9 — 6,2-7,3 — 0,6-Ю,7 0,4-0,7
Хроматон N II 0,24 1,0 — 9-10 — 1,3-1,4 6
Инертон II 0,55 0,4-0,65 —- 7,0-8,5 — 0,4-0,6 —
Хромосорб W II 0,33 1,25 19,8 8,4 47 1,80 2,0
Хромосорб Р I 0,45 4,0 9,8 6,5 110 0,9 0,4
Хромосорб G II 0,58 0,5 8,5 — 0,36-Ю,45 —
Хромосорб R-6470-1W («Джон Мэн-вилл» США) I — 5-6 — — — — 1-4
Anakrom Ana II 0,28-0,31 1,0-1,4 8-9,5 — — —
Supelcoport Sup. II — — — — — — —
Примечание. Каталитическая активность образования циклогексена — 5,2%
Таблица 4.1.19
Характеристики полимерных носителей
Марки носителей Насыпн. ПЛОТНОСТЬ, (кг-м“3)-1О“3 Удельная поверхность, м2т 1 Молярная масса, г-моль 1 Размер частиц, мм Общий объем пор, (м3-кгч)1О3 Максим, рабочая температура, °C Максим, кол-во фазы, масс. %
Хромосорб-Т J-М (США) 0,42 7-8 0,25-0,42
Фторопласт-4Д 0,32 8-9 100000-140000 0,25-0,5 0,30 320 До 30
Полихром-1 0,68 5,6 То же 0,25-0,5 0,25-0,30 280 20
Полихром-2 0,69 6,6 Тоже 0,25-0,5 0,20-0,25 200 20
4.1.2.3. Процесс приготовления сорбента
В хроматографической практике используют различные варианты нанесения неподвижной ЖФ на твердый носитель; пленка должна быть по возможности равномерно нанесена. Хорошее смачивание зависит от сил притяжения
между жидкостью и поверхностью твердого носителя, а также от сил взаимодействия между молекулами самой жидкости. Чем больше силы притяжения жидкость - твердое тело, тем лучше смачивание. Положительный эффект достигается подбором пары: твердый носитель - жидкая фаза: например, гидрофильные поверхности плохо смачи
278
Новый справочник химика и технолога
ваются углеводородами, поэтому для неподвижных фаз этого состава предпочтительнее твердые носители, содержащие алкилсилильные группы. Разумеется, на распределение неподвижной ЖФ влияет не только смачивание, но и
геометрическая структура твердого носителя, т. е. удельная поверхность, объем и размер пор.
Основной принцип нанесения неподвижной ЖФ на твердый носитель заключается в том, что ее растворяют в
подходящем растворителе, из которого она затем выделя
ется в виде пленки на твердом носителе по одному из ни
жеописанных методов.
От природы неподвижной ЖФ зависит, какой растворитель следует применять. Растворитель должен кипеть
при низких температурах и очень хорошо растворять неподвижную ЖФ. Чаще всего для этого применяют диэтиловый эфир, петролейный эфир, хлористый метилен, ацетон, хлороформ, метанол, четыреххлористый углерод, этанол, бензол или смеси этих растворителей.
Степень пропитки (содержание неподвижной ЖФ) выражают или в граммах неподвижной ЖФ на 100 г твердого носителя (т. е. в виде массовых процентов неподвижной фазы по отношению к твердому носителю) или в граммах неподвижной ЖФ на 100 г сорбента (т. е. в виде массовых процентов неподвижной фазы по отношению к твердому носителю, пропитанному жидкой фазой).
Существуют различные варианты нанесения неподвижной ЖФ на твердый носитель с применением летучих растворителей [2, 12-15] (рис. 4.1.9).
газ-носитсль
Рис. 4.1.9. Варианты нанесения ЖФ на твердые носители: а, б— метод испарения, в — фильтрационный метод, г — метод нанесения в «кипящем» слое
Метод испарения. Необходимое для нанесения количество неподвижной ЖФ растворяют в подходящем растворителе. Объем растворителя должен быть такой, чтобы в него полностью погружался обрабатываемый твердый носитель. Равномерно перемешивая образовавшуюся суспензию, раствори
тель испаряют вначале при невысокой температуре (на водяной бане), а затем сорбент досушивают при более высокой температуре (на паровой бане). Полученный сорбент прогревают еще
несколько часов в сушильном шкафу.
Часто носитель насыщают раствором неподвижной ЖФ и выпаривают растворитель под вакуумом. В этом случае недостатки обычного метода испарения — неравномерность распределения неподвижной жидкой фазы на носителе, механическое дробление носителя — в определенной
мере уменьшаются.
Фильтрационный метод. Твердый носитель смешивают с раствором неподвижной ЖФ, затем суспензию наносят на фильтр, растворитель частично отсасывают, а полученный «мокрый» сорбент затем подсушивают.
Используют и несколько иной вариант фильтрационного метода, когда раствор неподвижной ЖФ наносят на пористый фильтр, добавляют твердый носитель, массу перемешивают в течение нескольких минут, затем фильтруют под вакуумом и высушивают.
Фильтрационный метод целесообразно применять для приготовления сорбентов с низкими концентрациями неподвижной фазы (1-3%). Количество неподвижной ЖФ, оставшейся на твердом носителе, определяется по линейному уравнению
сн=«к-ср’
где сн и ср — концентрация неподвижной ЖФ соответственно на носителе и в растворителе, %; ак — коэффициент корреляции.
Метод является достаточно быстрым, однако его существенный недостаток состоит в том, что для определения фактического содержания НЖФ на твердом носителе требуется проведение дополнительных экспериментов. При этом фильтрационный метод используется для приготовления сорбента с различной концентрацией неподвижной жидкой фазы (напр. от 5 до 30%). Затем строится график зависимости концентрации неподвижной ЖФ на твердом носителе от ее концентрации в растворе. Зная последнюю, находят концентрацию неподвижной ЖФ на носителе или наоборот.
Для некоторых носителей существуют таблицы, по которым можно рассчитать требуемую концентрацию раствора, необходимую для нанесения данного процента ЖФ на твердый носитель.
Фронтальный метод основан на пропускании раствора неподвижной ЖФ через колонку, заполненную твердым носителем. Для его осуществления раствор ЖФ медленно пропускают через хроматографическую колонку или специальную емкость с твердым носителем до установления равновесия между сорбированным количеством ЖФ на твердом носителе и подаваемым раствором. После установления равновесия растворитель удаляют нагреванием колонки под вакуумом в потоке инертного газа. Для равномерной пропитки твердого носителя по всей длине колонки рекомендуется пропускать приблизительно два объема раствора на объем носителя.
Концентрация неподвижной ЖФ на твердом носителе в этом методе может быть определена при взвешивании колонки после удаления растворителя.
С хорошим приближением можно рассчитать количество неподвижной ЖФ на твердом носителе, если известна исходная концентрация раствора, масса твердого носителя и объем пропущенного раствора неподвижной ЖФ.
Особенно целесообразно применение этого метода при использовании носителей с непористой внешней поверх
Аналитическая хроматография и капиллярный электрофорез
279
ностью (стеклянные и металлические шарики, стальные порошки и др).
Способ, предложенный в работе [15], отличается от предыдущего тем, что после непродолжительной продувки колонки инертным газом для удаления избытка раствора сорбент выгружается из колонки и подвергается окончательной досушке. Существует большое количество модификаций этих трех основных способов нанесения неподвижной ЖФ на носитель [16-18].
Метод приготовления сорбента в «кипящем» слое лишен некоторых недостатков, которые присущи методам нанесения неподвижной ЖФ на носитель, изложенным выше. Он заключается в том, что неподвижную ЖФ наносят на внешнюю поверхность частиц носителя в виде мелких капель путем распыления раствора инертным газом [19, 20]. Для его оценки сравнивали критерии разделения и эффективности колонок, заполненных сорбентом, приготовленным двумя различными методами: в «кипящем» слое и обычном методе испарения. Полученные результаты показывают, что колонки с сорбентом, приготовленным в «кипящем» слое, имеют большую эффективность [19].
В заключение необходимо отметить особенности нанесения ЖФ на фторополимерные сорбенты. Необходимость особого внимания при нанесении на тефлон определяется его специфическими свойствами, а именно: возникновением статического заряда при перемешивании или просеивании и слипанием частиц носителя.
Рекомендуется наносить ЖФ на тефлон-6 из раствора летучих растворителей, медленно испаряя растворитель без нагревания и осторожно перемешивая сорбент шпателем. Для испарения растворителя рекомендуется пропускать над чашкой с сорбентом поток сухого азота.
Если частицы тефлона агрегируются, его помещают в охлажденную до 0 °C емкость и периодически встряхивают.
Перспективным направлением в приготовлении сорбентов на основе политетрафторэтилена можно считать вариант предварительной модификации носителя химическими реагентами. Показано, что обработка тефлона натрий-нафталиновым комплексом повышает эффективность колонки в два раза.
Литература
1. Rohrschneider L.J. // Chromatogr. Sci. 1963. V. 32. Р. 2.
2. Березкин В.Г., Пахомов В.П., Сакодынский К.И.
Твердые носители в газовой хроматографии. М.: Химия, 1975.
3. Гаврилова Т.Б., Киселев А.В. Газовая хроматография. Дзержинск: Изд. Дзержинского филиала ОКБА, 1966.
4. Киселев А.В., Яшин Я.И. Газоадсорбционная хроматография. М.: Наука, 1967.
5. Лурье А.А. Хроматографические материалы. М.: Химия, 1978.
6. Головитиков Ю.Н. // Завод, лаб. 1972. № 38. С. 532.
7. Твердые носители для газовой хроматографии. Проспект ВНИЦ монокристаллов, М. 1969.
8. Брызгалова В.И., Гаврилова Т.Б., Киселев А.В. Газовая хроматография. Вып. 15. М.: НИИТЭХИМ, 1971.
9. А.с. 269574 СССР / А.В. Киселев, Т.Б. Гаврилова, Н.И. Брызгалова. (СССР). БИ № 15, 1970.
10. Лефтер В.Е., Дуброва П.Л., Ефименкова П.М., Прокопенко Н.А. Методы повышения эффективности хроматографических колонн. М.: НИИТЭХИМ, 1977.
11. Баландина В.А. и др. Анализ полимеризационных пластмасс. Л.: Химия, 1967.
12. Guvemator G., Gager F., Robb E. et al. // J. Gas Chroma-tog. V. 6. 1965.
13. Ногаре С.Д., Джувет P.C. Газо-жидкостная хроматография. Л.: Недра, 1966.
14. Столяров Б.В., Савинов И.М., Витенберг А.Г. Руководство к практическим работам по газовой хроматографии. Л.: ЛГУ, 1973. 223 с.
15. Супина В.Р. Насадочные колонки в газовой хроматографии. М.: Мир, 1977.148 с.
16. Smith E.D. // Anal. Chem. 1960. V. 32. Р. 1049.
17. А. А. Жуховицкий и др. // Журн. аналитич. химии. 1972. №97. С. 748.
18. Сато К. Газовая хроматография. Вып. 7. М.: НИИТЭХИМ, 1967.
19. Шкуратов В.И., Литяева З.А., Валько Г.М.. Газовая хроматография. Вып. 11. М.: НИИТЭХИМ, 1969.
20. Березкин В.Г., Пахомов В.П. // Химия и технология топлив и масел. 1965. № 9. С. 58.
21. Бардышев И.И. и др. // Завод, лаб. 1971. № 37. С. 1438.
4.1.3. Газоадсорбционная хроматография
4.1.3.1. Особенности газоадсорбционной хроматографии
В газоадсорбционной хроматографии (ГАХ) разделение соединений происходит за счет различной адсорбируемости на поверхности адсорбента. ГАХ — один из основных методов газовой хроматографии наряду с газо-жидкостной хроматографией. ГАХ широко используется для разделения газов и паров легкокипящих соединений, структурных изомеров, а также для разделения высококипящих соединений. Адсорбция на плоских поверхностях более чувствительна к геометрической структуре молекул по сравнению с растворением, т.к. в первом случае молекула испытывает одностороннее межмолекулярное взаимодействие с адсорбентом, а во втором она окружена молекулами растворителя со всех сторон. Для ГАХ разработаны однородные неорганические, полимерные и углеродные адсорбенты. Возможности ГАХ значительно расширила разработка различных методов геометрического, адсорбционного, ионообменного и химического модифицирования. Колонки с неорганическими и углеродными адсорбентами не имеют собственного фона, в отличие от колонки с сорбентами на основе жидких фаз. Это обстоятельство позволяет работать на таких колонках и при более высоких температурах в режиме программирования, используя более чувствительные шкалы.
280
Новый справочник химика и технолога
4.1.3.2. Классификация адсорбентов по химической природе поверхности
В газовой хроматографии различные межмолекулярные взаимодействия подразделяют на неспецифические и специфические. Неспецифические, в основном ориентационные, вызываются особенностями локального распределения электронной плотности во взаимодействующих молекулах. Эти особенности связаны с локальным концентрированием отрицательного и положительного зарядов на отдельных связях или звеньях специфически взаимодействующих молекул. Водородная связь представляет собой частный случай таких специфических, но еще межмолекулярных взаимодействий. Такое подразделение взаимодействий в известной степени условно, однако оно помогает систематизации разрозненных фактов и позволяет создать удобную качественную классификацию. В табл. 4.1.20 приведена классификация адсорбентов и молекул по характеру межмолекулярных взаимодействий.
Таблица 4.1.20
Классификация молекул и адсорбентов по их способности к неспецифическим и специфическим молекулярным взаимодействиям
[Киселев А.В., Ковалева Н.В., Торбина Л.И. / В сб. Газовая хроматография. Вып. 15. М.: НИИТЭХИМ, 1971. С. 5]
4.1.3.3. Классификация адсорбентов по геометрической структуре
Геометрическая структура адсорбентов (см. табл. 4.1.21) характеризуется удельной поверхностью S, средним диаметром пор фр, объемом пор Ипор. Удельная поверхность одного грамма адсорбента выражается в м2 гч. Удельная поверхность адсорбентов для газовой хроматографии колеблется в пределах 10—1000 м2 г-1. Средний диаметр пор выражается в ангстремах, А (10 8 см) или в нанометрах (10-9 нм) и колеблется от 0,4 до 100 нм. Средний объем пор — это объем всех пор одного грамма (килограмма) адсорбента (размерность — см3т' и м3-кг 1 соответственно) при допущении, что все поры цилиндрической формы. Величины S, dcp, Ипор для адсорбентов одной природы в простейшем случае связаны между собой:
3V d = — ср S
Группы молекул Типы адсорбентов
I | II | III
А — со сферически симметричными оболочками или о-связями (благородные газы, насыщенные углеводороды) Неспецифические взаимодействия, определяемые в основном дисперсии-онными силами
В — с электронной плотностью, локально сосредоточенной на периферии отдельных связей или звеньев; л-связи (ненасыщенные и ароматические углеводороды) и свободные электронные пары (эфиры, кетоны, третичные амины, нитрилы и т. п.) Неспецифические взаимодействия Неспецифические + специфические взаимодействия
С — с положительным зарядом, локально сконцентрированным на периферии звеньев (например, некоторые металлоорганические соединения)
D — с функциональными группами, в которых на периферии соседних звеньев локально сконцентрированы как электронная плотность, так и положительный заряд (молекулы с группами —ОН и >NH)
Примечания.
I. Графитированные сажи (обработанные в среде водорода) не несущие ионов или активных групп .
II. Несущие локально сконцентрированные положительные заряды (кислые гидроксилы, обменные катионы малого радиуса).
Ш. Несущие локально сконцентрированные отрицательные заряды (эфирные, нитрильные, карбонильные и другие группы).
В ряду таких адсорбентов при близких величинах Ипор величина dcp обратно пропорциональна S, т.е. тонкопористые адсорбенты имеют большую удельную поверхность и, наоборот, макропористые адсорбенты — небольшую.
Адсорбенты можно разделить на непористые, объемнопористые и поверхностно-пористые. В непористых адсорбентах используются зерна без внутренней пористости, обычно это тонкодисперсные адсорбенты.
Таблица 4.1.21
Классификация адсорбентов по геометрической структуре [3]
Тип адсорбентов S, м2- г 1 <4р, нм V * пор’ (м3-кгч)-103
Непористые 5-200 — —
Однородномакропористые 5-100 >50 до 1,5
Однороднотонкопористые 500-1000 0,4—10 0,5-0,8
Неоднороднопористые 250-800 1-20 0,3-1,2
Рис. 4.1.10. Схематическое изображение зерен адсорбентов: а — объемно-пористые; б — поверхностно-пористые
У объемно-пористых адсорбентов (рис. 4.1.10а) поры имеются во всем объеме зерна, а в поверхностно-пористых адсорбентах (рис. 4.1.106) центральная часть зерна непористая. Внутренний массообмен на таких адсорбентах происходит быстрее, чем в объемно-пористых адсорбентах.
Аналитическая хроматография и капиллярный электрофорез
281
4.1.3.4. Методы модифицирования адсорбентов в газовой хроматографии
Методы модифицирования чаще всего применяются для улучшения химической и геометрической однородности заведомо неоднородных адсорбентов.
Геометрическое модифицирование — это расширение пор тонкопористых адсорбентов, превращение неоднородно тонкопористых адсорбентов в макропористые однородные адсорбенты. В частности, гидротермальная обработка силикагелей в автоклаве (воздействие на силикагель перегретого пара, давления и температуры до 700-800 °C) сильно сокращает удельную поверхность (с 300 до 30 м2- г !) и расширяет средний диаметр пор (с 7 до 50 нм).
Адсорбционное модифицирование — блокирование активных центров поверхности адсорбента за счет адсорбции молекул сильнополярных высокомолекулярных соединений, которые при температуре разделения не десорбируются из колонки.
Механическое или физическое модифицирование — отложение или накопление на основной поверхности слоя или пленки другого твердого нелетучего вещества, менее активного в адсорбционном отношении.
Полимерные адсорбенты. Для газовой хроматографии разработаны пористые полимерные адсорбенты с разной
химической природой поверхности и разной геометрической структурой (см. табл. 4.1.22).
Углеродные адсорбенты. В табл. 4.1.23 приведены характеристики углеродных адсорбентов как однородных тонкопористых, так и макропористых.
Химическое модифицирование — это проведение поверхностных химических реакций. Наиболее типичная реакция химического модифицирования — силирование гидроксилированной поверхности кремнеземов, в частности силикагелей:
^Si—ОН + ClSi(CH3)3 ^Si—О—Si(CH3)3 + НС1.
В табл. 4.1.22 приведены характеристики состава модифицирующих слоев на силикагеле.
4.1.3.5. Основные типы адсорбентов
В газо-адсорбционной хроматографии применяют кремнеземные адсорбенты (табл. 4.1.22), цеолиты (молекулярные сита) (табл. 4.1.23), пористые полимерные адсорбенты (табл. 4.1.24) и углеродные тонкопористые и макропористые адсорбенты (табл. 4.1.25).
В последние годы для разделения изомеров применяются а-, [3- и у-циклодекстрины (рис. 4.1.11).
Таблица 4.1.22
Основные адсорбенты для газовой хроматографии.
Характеристики промышленных макропористых кремнеземных адсорбентов
Наименование S, м2- г 1 Диаметр пор, нм Кпор, (м3-кг *)• 103 Размер зерен, нм-10 3 Форма зерен
1. Макропористые силикагели
Силикагель
МСА-1 15-25 80-120
МСА-2 60-90 40-70 0,7-1 100-800 Сферическая
мен 2-6 150 То же
Порасил
В 125-250 10-20
С 50-100 2040
д 25-45 4089 0,6-1 37-75
Е 10-20 80-150 75-125 Сферическая
Сферосил
ХОА 200 125-250 10-20
ХОВ 075 50-100 20-40 0,6-1 40-60 Сферическая
ХОВ 030 25-45 40-80 60-80
ХОВ 015 10-20 80-150
ХОС 005 2-6 15-20 100-150
Меркосорб и меркогель
Si-150 150-200 150 37-75
Si-500 50-79 50 0,7-1 75-125 Несферическая
Si-1000 20-30 100 То же
282
Новый справочник химика и технолога
Продолжение таблицы 4.1.22
Наименование S, м2- г 1 Диаметр пор, нм Упор, (м3кг ‘)-103 Размер зерен, нм-10 3 Форма зерен
2. Макропористые аэросил огелй (силохромы)
Силохром
СХ-1 25-35 220-250
СХ-2 45-60 130-100 1,5-1,9 100-800 Сферическая
СХ-3 70-90 70-100 То же
С-80 70-90 40-50 1,2-1,4 80-160, 160-315, 315-500 Несферическая
С-120 110-130
Таблица 4.1.23
Цеолиты (молекулярные сита)
Размеры молекул возрастают
Не, Ne, Аг, СО н2,о2 n2,nh3 Н2О Граница размеров цеолита Са-мор-денита (0,38 нм) Кг, Хе, СН4 С2Нб СН3ОН CH3CN CH3NH2 СН3С1 СН3Вг СО2 С2н2 cs2 С3Нб «-СдНю н-СуНю «-с14н30 С2Н5С1 С2Н5Вг С2Н5ОН c2h5nh2 СН2С12 СН2Вг2 CHF2C1 CHF3 (СН3)2 NH CH3I В2Нб cf4 c2f6 cf2ci2 CF3C1 CHFC12 SF6 (CH3)3N C6H6 Нафталин изо-С4Ню (C2H5)3N C5H5CH3 Хинолин U3O-C5H12 C(CH3)4 C6H4(CH3)2 6-децил- CHC13 C(CH3)3C1 Циклогексан 1,2,3,4- CHBr3 C(CH3)3Br Циклогексен тетра- CHI3 C(CH3)3OH Тиофен гидро- (CH3)CHOH СС1| Фуран нафта- (СН3)2СНС1 СВг4 Пиридин лин- w-C3Fs C2F2CI4 Диоксан 2-бугил- н-СдНю ВюН14 1-гекси- н-С7Н1б линдан В5Н9 C6FHCF3 1,3,5-Три-этил-бен-зол (н-С^)^
Тип 5
Тип 4 Граница размеров цеолита NaA ~ 0,4 нм)
Граница размеров Тип 3 цеолита СаА (~ 0,49 нм)
Тип 2 Граница размеров цеолита СаХ (» 0,8 нм)
Граница размеров
Тип 1 цеолита NaX (~ 1 нм)
Таблица 4.1.24
Характеристики полимерных адсорбентов
Сорбент Состав полимера S, м2- г 1 <4р, НМ Насыпная масса, (кг-м 3)-10 3 Температурный предел, °C
Порапак Р Стирол, дивинилбензол 100-200 0,28 250
Порапак Q Этилстирол, дивинилбензол 600-650 7,5 0,35 250
Порапак QS Силанизированный порапак Q 600-800 — 0,35 250
Порапак PS Силанизированный порапак Р — — — —
Аналитическая хроматография и капиллярный электрофорез
283
Продолжение табл. 4.1.24
Сорбент Состав полимера S, м2- г 1 ^ср? НМ Насыпная масса, (кг-м 3)10“3 Температурный предел, °C
Порапак N Стирол, дивинилбензол, винилпирролидон 500-550 7,5 0,39 200
Порапак R То же 500-550 7,6 0,33 250
Порапак S 500-550 7,5 0,35 200
Порапак Т Стирол, этиленгликольдиметакрилат 300 9,0 0,44 200
Хромосорб 101 Стирол, дивинилбензол 30-40 350,0 0,30 300
Хромосорб 102 То же 300-400 8,5 0,29 250
Хромосорб 103 Сшитый полистирол 15-25 350,0 0,22 275
Хромосорб 104 Акрилонитрил, дивинилбензол 100-200 60,0-80,0 0,32 250
Хромосорб 105 Полиароматический сорбент 600-700 40,0-60,0 0,34 250
Хромосорб 106 Сшитый полистирол 700-800 — — 250
Хромосорб 107 Сшитый акриловый эфир 400-509 — — 250
Хромосорб 108 То же — — —
Синахром Стирол, дивинилбензол 500-600 4,5 250
Полисорб-1 То ж е 200-250 13,0 0,29 250
Полисорб-10 Поли-и-дивинилбензол 300-350 — 0,23 250
ПАР-1 Стирол, дивинилбензол 100 20,0 0,69 250
ПАР-2 То же 300 9,0 0,67 250
Фазепак Р Сшитый полистирол 400 7,5 0,50 250
Фазепак PQ Сшитый полистирол 300 15,0 0,50 250
Тенакс Поли-и-2,6-дифенилфенилен-оксид 18,8 140,0 400
Таблица 4.1.25
Углеродные адсорбенты для газовой хроматографии
Название Структурные характеристики: S, м2- г-1, <7ср Способ получения
Графитированная термическая сажа (ГТС) 7 Получено нагреванием сажи до 3000 °C в инертной или восстановительной среде [1]
Карбохром-8 6-8 Получено из графитированной термической сажи [1]
Карбохром-80 80-90, 53 нм Получено из ацетиленовой сажи [1]
Карбохром А и В 6 Из ГТС и не ГТС [2]
Карбохром С 60 Из канальной сажи путем отложения пирографита в зернах сажи [3]
Карбопак А и С 10-15 Изготовляется фирмой Supelco (США)
Карбопак В 90-100 То же
Карбосито В 1000, 1,0-1,2 нм То же
Карбоцвет-БГ* 1000, 1,0-1,2 нм Изготовляется по методике Базаевой-Гвоздович
По характеристикам не уступает американскому карбоситу.
284
Новый справочник химика и технолога
Рис. 4.1.11. Структурные формулы а-, р- и у-циклодекстринов и комплексов включения на их основе
4.1.4. Капиллярная газовая хроматография
4.1.4.1. Особенности капиллярной хроматографии
В капиллярной газовой хроматографии в качестве колонки используются капилляры с внутренним диаметром 0,1-0,53 мм и длиной от 5 до 100 м (и более), с нанесенным на внутреннюю поверхность сорбентом. Остальное пространство незаполненное, поэтому капиллярные колонки в зарубежной литературе называют open — открытые. По типу сорбентов на стенках капилляров различают четыре типа колонок (табл. 4.1.26).
Таблица 4.1.26
Классификация колонок по типу сорбентов
Типы капиллярных колонок Толщина слоя сорбента , мкм Характеристики КК
WCOT 0,01-1 Внутренняя поверхность покрыта пленкой жидкой фазы (wall-coated open tubular, WCOT)
SCOT 1-5 С тонким слоем носителя, смочена жидкой неподвижной фазой (support-coated)
PLOT до 20 С тонким слоем нанесенного адсорбента (porous-layer)
CLOT 10-20 С тонким слоем нанесенного углеродного адсорбента (carbon PLOT)
КК закручены в спирали диаметром 10-20 см и закреплены в специальных держателях.
Сопротивление потоку у КК меньше, чем у НК, поэтому можно увеличивать их длину (см. табл. 4.1.27).
Таблица 4.1.27
Давление на входе КК разной длины, атм
Длина, м Внутренний диаметр, мм
0,1 0,25 0,32 0,53
10 0,7 0,3 0,2 0,15
25 1,75 0,7 0,5 0,35
50 3,5 1,4 0,9 0,7
Минимальная величина ВЭТТ для сорбируемых веществ пропорциональна радиусу капилляра:
1,9 г.
В табл. 4.1.28 приведены теоретические минимальные значения эффективности колонок разного диаметра. В капиллярных колонках отсутствует вихревая диффузия (типичная для насадочных колонок) и появляется так называемое динамическое размывание.
Полное уравнение ВЭТТ, описывающее размывание в КК, равно:
ТТ 2D l + 3k + llk2 г2 2k d2
Н = +------------5---U +-------7 ——U .
и 24(1 +k)2 D 3(1 +к)2 пж
Аналитическая хроматография и капиллярный электрофорез
285
Таблица 4.1.28
Минимальные значения эффективности колонок
Характери-стики колонок Внутренний диаметр, мм
0,1 0,25 0,32 0,53
N макс/м длины 11000 4400 3500 2200
ВЭТТ, мм 0,09 0,22 0,29 0,45
Декан Дициклогексиламин
Ундекан Метилундеканоат
Нонаналь Метилдодеканоат
2,3-Бутандиол 2,6-Диметиланилин
1-Октанол 2,6-Диметилфенол
Метилдеканоат 2-Этилгексановая кислота
Оптимальные объемы и линейные скорости в КК приведены в табл. 4.1.29.
Таблица 4.1.29
Оптимальные объемы и линейные скорости в КК
Скорость потока Внутренний диаметр, мм
0,1 0,25 0,32 0,53
Линейная, см-с-1 40-50 25-35 20-35 18-27
Объемная, 3 -1 см мин 0,2-0,3 0,7-1,0 1,0-1,7 2,4-3,5
Наилучшими газами-носителями для КК считаются гелий и водород. Очень важна чистота газа-носителя (отсутствие кислорода), т.к. тонкая пленка жидкой фазы при повышенных температурах может сильно окисляться.
Для оценки качества КК предложена тестовая смесь К. Гроба.
Спирты и диолы из этой смеси используются для определения активных силанольных групп на кварцевой поверхности КК.
Емкость КК мала по сравнению с насадочными (табл. 4.1.30). Дозируемые пробы должны быть не более 10-6—10-9 г, чтобы не перегружать колонки. Для ввода таких небольших проб в КК используются специальные методы ввода проб: с делением потока, без деления потока, наколоночный ввод, ввод твердых проб, ввод с отдувкой растворителя и ввод проб с программированием температуры. Разработан специальный метод концентрирования на начальном охлажденном участке КК — метод криофокусировки, с последующим быстрым нагревом — «тепловым ударом».
Наиболее распространенные жидкие фазы, используемые в капиллярных колонках, приведены в табл. 4.1.31. Весьма популярными стали адсорбционные капиллярные колонки типа PLOT (табл. 4.1.32).
Таблица 4.1.30
Емкость капиллярных колонок [г]
Диаметр колонки, мм Толщина пленки, мкм
0,1 0,25 0,5 1,0 1,2 2,5 5,0
0,1 1-Ю’8 2,5-10’8 5-10’8 1-107 — — —
0,25 2,5-10’8 1-Ю’8 1-Ю’8 1-Ю"8 1-Ю’8 1-10~8 1-Ю’8
0,32 3,2-10’8 8-10’8 1,6-10’7 3,2-10’7 3,8-10’8 8-Ю7 1,6-10"6
0,53 5,3-108 1,3-10’7 2,6-10 7 5,3-107 6,4-10’7 1,3-10‘6 2,6-10"6
Таблица 4.1.31
Наиболее распространенные жидкие фазы, используемые в WCOT-колонках
Состав Полярность Определяемые вещества Фазы с близкими константами Мак-Рейнольдса Диапазон рабочих температур, °C
100% диметилполисилоксан (каучук) Неполярная Фенолы, углеводороды, амины, серосодержащие соединения, пестициды, полихлорированные бензолы OV-1, SE-30 -60-325
100% диметилполисилоксан (вязкая жидкость) То же Производные аминокислот, эфирные масла OV-101, SP-2100 0-280
5% дифенил, 95% диметилполисилоксан То же Жирные кислоты, метиловые сложные эфиры, алкалоиды, лекарственные препараты, галогенсодержащие соединения SE-52, OV-23, SE-54 60-325
14% цианопропилфенил-полисилоксан Средняя Лекарственные средства, стероиды, пестициды OV-1701 -20-280
50% фенил, 50% метилполисилоксан То же Лекарственные средства, стероиды, пестициды, гликоли OV-17 60-240
286
Новый справочник химика и технолога
Продолжение таблицы 4.1.31
Состав Полярность Определяемые вещества Фазы с близкими константами Мак-Рейнольдса Диапазон рабочих температур, °C
50% цианопропилметил, 50% фенил То же Жирные кислоты, метиловые сложные эфиры, ацетаты альдитола OV-225 60-240
50% трифторпропил-полисилоксан То же Галогенсодержащие соединения + ароматические соединения OV-210 45-240
ПЭГ, модифицированный терефталевой кислотой Полярная Кислоты, спирты, альдегиды, акрилаты, нитрилы, кетоны OV-101, SP-2100 60-240
ПЭГ То же Свободные кислоты, спирты, простые эфиры, эфирные масла, гликоли, растворители Карбовакс 20М 60-220
Таблица 4.1.32
Адсорбционные капиллярные колонки типа PLOT
Тип Области применения, комментарии
Chrompack
5А PLOT He, Ne, H2, Ar, Kr, Xe, HD, D2, ht,dt,t2, o2,n2, CH4,C2H4, cd4, c2d6, CO, NO. N=2000
ai2o3 PLOT Ci-C5
А12О3/КС1 PLOT Ci-Сю
Рога PLOT
Q Спирты, вода, полярные летучие нитрилы, углеводороды, газы
S Кетоны, эфиры, галогенуглеводороды, углеводороды
и Все летучие полярные нитрилы нитросоединения, спирты альдегиды, этан, этилен, сернистые газы, СО2 в воздухе, ppm воды в газах
Carbo PLOT N2, О2, СО, СО2, Cj-C2
Supelco
Carboxen-1006 PLOT Ci - С3, формальдегид-вода-метанол, примеси в этилене, пористые мол. сита S = 750 м2т-1, програмир. До 250 °C
Mol Sieve 5A PLOT N2,02, СО, СН4 (за 5 мин), Аг, О2
Alumina PLOT С]-Сю все Ci-С4, бензол, толуол и ксилолы
Supel-Q PLOT ДВБ, Ci-C4, сернистые газы, спирты, кетоны, альдегиды и полярные соединения
Alumina KC1 PLOT До 200 °C деакгивир. КС1
Carboxen-1010 PLOT Н2, N2, СО, СН4, СО2, С2- и С3-углевод., О2 отдельно от N2
Carbopack X PLOT Порист. ГТС
Supel G45 PLOT До 200 °C
Supel -N PLOT До 200 °C
Supel -R PLOT До 200 °C
CLOT Со слоем Карбопака В
CP-Carbo Bond Для разделения по ASTM D2505 (до 350 °C)
4.1.5. Основные рекомендации по выбору сорбентов и условий хроматографического разделения смесей конкретных веществ
В практике хроматографического анализа смеси компонентов могут иметь известный или неизвестный состав. Во втором случае предварительно нужно установить состав смеси, а потом выбирать условия хроматографического разделения либо брать самую эффективную КК, провести на ней разделение и идентифицировать компоненты хроматографическим способом.
Очень часто, особенно в производственных смесях, состав заранее полностью известен. В этом случае выбору оптимальных условий хроматографического разделения помогут сведения о следующих свойствах компонентов смеси: температура кипения, дипольный момент, электронная поляризуемость и молярная масса.
В зависимости от различий свойств компонентов смеси можно воспользоваться рекомендациями, изложенными в табл. 4.1.33.
В табл. 4.1.35 приводятся рекомендации по применению неподвижных фаз для разделения определенных классов соединений.
Если для компонентов исследуемой смеси есть опубликованные справочные данные по удерживанию, то в этом случае можно не только выбрать оптимальный сорбент, но и рассчитать минимальную длину колонки. Для этого нужно из этих данных определить величину а (отношение исправленных времен удерживания двух соседних пиков) для самой трудно разделяемой пары соединений и по графику (рис. 4.1.12) и табл. 4.1.34 определить минимально необходимую эффективность колонки для полного разделения этих соединений. Затем, принимая среднюю эффективность равной 1500-2000 теоретических тарелок на 1 метр длины колонки, можно легко определить необходимую длину колонки. Например, при а = 1,04 для полного разделения необходимо взять колонку эффективностью 10 тыс. теоретических тарелок, т. е. при указанной выше средней эффективности необходима колонка длиной от 5 до 6 м.*
* См.также раздел 3.2.9.3.
Аналитическая хроматография и капиллярный электрофорез
287
Таблица 4.1.33
Общие рекомендации по выбору сорбентов
Характеристика смеси Сорбенты
Все компоненты смеси отличаются по температурам кипения Предлагается использовать колонки с неполярной или слабо полярной жидкой фазой в газо-жидкостном варианте (сквалан, апиезон, OV-1, SE-ЗО и др.). В этом случае соединения будут элюироваться из колонки в порядке увеличения температуры кипения
Все компоненты смеси отличаются по электронной поляризуемости или по молярным массам Рекомендуется эту смесь разделять на колонке с неполярными неспецифическими адсорбентами (газоадсорбционный вариант хроматографии), такими, как пористые полимеры (порапаки Р, PS и Q, QS, хромосорбы 102, 101, полисорб-1, тенакс), углеродные адсорбенты (карбохромы, карбопаки, карбосита и др.). В этом случае соединения будут элюироваться из колонки в порядке увеличения электронной поляризуемости
Компоненты смеси отличаются по дипольным моментам Предлагается применять полярную жидкую фазу (ГЖХ) либо полярный соответствующий адсорбент (ГАХ)
Соединения смеси изомеров либо соединения,отличающиеся пространственным строением Рекомендуется применять для разделения колонку с карбохромом или карбопаком, которые имеют высокую селективность при разделении соединений, отличающихся пространственным строением. Можно использовать колонку с жидкими кристаллами. Можно применить колонку с бен-тоном
Сложная многокомпонентная смесь, некоторые компоненты в которой слабо различаются по температурам кипения и по дипольным моментам Предлагается использовать составную колонку с разными сорбентами. Рекомендуется применять эффективную капиллярную колонку
Смесь дейтерированных соединений Капиллярная колонка с неполярной или слабополярной жидкой фазой
Смесь оптических изомеров Высокоэффективная капиллярная колонка с оптически активной жидкой фазой
Таблица 4.1.34
Значения N при различных а
а N
к = 1000 к = 500 к = 100 к = 50 к= 10
1,005 647708 649000 659409 672531 782163
1,01 163542 163868 166497 169810 197491
1,02 41699 41782 42452 43297 50355
1,04 10837 10859 11033 ' 11253 13087
1,06 5004 5014 5094 5195 6042
1,08 2922 2928 2975 3034 3528
1,12 1396 1399 1422 1450 1686
1,15 942 944 959 978 1138
1,20 577 578 587 599 697
1,25 400 402 408 416 484
1,30 301 302 806 312 363
Рис. 4.1.12. Связь эффективности (N) и селективности (а) при R = 1
Таблица 4.1.35
Неподвижные фазы, рекомендуемые для разделения различных веществ
Анализируемые вещества Фазы
Постоянные газы Цеолиты СаА; NaX; карбосита; пористые полимеры
Углеводородные газы Силикагели; пористые стекла; оксид алюминия; карбохромы; карбосита
Алканы С4—Сю Сквалан; силохромы; сферосилы; по-расилы
Алканы >С10 Апиезон L; SE-ЗО; OV-101; MS-200; ДС-410
Ароматические углеводороды Дибутилтетрахлорфталат; трикрезилфосфат; бентон 34 + додецилфталат; жидкие кристаллы; OV-17; OV-1; SE-30; ДС-550; ДС-710; Р5-2250; ПФМС
288
Новый справочник химика и технолога
Продолжение табл. 4.1.35
Анализируемые вещества Фазы
Галогеносодержащие углеводороды ДС-200; ДС-500; апиезон; додецилфталат; сквалан; трикрезилфосфат; OV-101; SP-2100
Терпеновые углеводороды ДС-550; этиленгликольсукцинат; эти-ленгликольадипинат; трикрезилфосфат; диэтиленгликольсукцинат
Альдегиды Порапак Q; ДС-550; Ucon 280Х; ПЭГ-20М; додецилфталат; карбохро-мы; карбопаки; полипропилен-гликольадипинат; ПЭГ-4000
Эфиры (простые и сложные) Порапак Q; SE-30; динонилфталат; ПЭГ-20М; этиленгликольсукцинат; ПФМС-4, СКТФТ-50; неопентил-гликольсукцинат
Амины ПЭГ-20М; трикрезилфосфат; порапак Q; ДС-550; карбопак В + 40% ПЭГ-20М + 0,8% КОН; Хромосорб + 10% ПЭГ-20М + 2% КОН; SP-96 + 15% квадрол + 5 КОН, Tenax-OC, SE-52
Амиды Versamid 900; апиезон L
Спирты Порапаки, ПЭГ-20М, карбопак С + 0,5% ПЭГ-1500; карбопак С + 0,1% SP-1000; карбохромы
Фенолы 0,1% SP-1000 на карбопаке С; OV-17; ХЕ-60.
Кислоты С2—С5 Карбопак В + 3% ПЭГ-20М + 0,5% Н3РО4; 10% SP-1200 + 1% Н3РО4 на хромосорбе W; порапаки
Кислоты ^С6 FFAP, SP-1000; ПЭГ-20М, обработанный терефталевой кислотой; DEGS-PS8 на Supelcoport; апиезон L; SE-52
Гликоли Порапак Q; тенакс
Кетоны Порапак Q; ПЭГ-20М; ДС-550; додецилфталат; трикрезилфосфат
Нитрилы ПЭГ-400; ХЕ-1150; THEED; ДС-710
Олефины Сквалан; AgNO3 + этиленгликоль; трикрезилфосфат
Стероиды QF-1; SE 30; ХЕ-60; OV-1; OV-17; SP-2100; SP-2250; SP-2300; OV-225
Сахара (производные) SE-52; QF-1; 3% OV-225 на Supelcoport; 10% SP-2401 на Supelcoport; ДЭГС; ПЭГ-6000; 15% ЭГА на хромосорбе W-AW
Аминокислоты 0,65% ЭГА на хромосорбе W, 2% OV-17; 10% OV-11 на Supelcoport (для ТМС-производных); апиезон М на хромосорбе W HP (или газ-хроме Q) или 3% SE-30 на газ-хроме Q (для триметил силильных производи.) 1,7% OV-17 на хромосорбе GHP
Продолжение табл. 4.1.35
Анализируемые вещества Фазы
Жирные кислоты (метиловые эфиры) 3% SP-2100 на Supelcoport; 15% OV-275 на хромосорбе Р, диэтиленгликольсукцинат; 10% SP-2330, EGSS-X; ПЭГ-20М
Желчные кислоты (метиловые эфиры) 3% SP-2250 на Supelcoport
Алкалоиды 3% SP-2250 на Supelcoport; SE-30; OV-17
Барбитураты 3% SP-2250 на Supelcoport; OV-1; OV-17
Пестициды 1,5% SP-2250 + 2% SP-2410 на Supelcoport; QF-1; SE-30; OV-17; OV-1
4.1.6. Качественный анализ в газовой хроматографии
Аналитические задачи в газовой хроматографии можно разделить на два типа:
• анализ смесей, качественный состав которых заранее известен; это в основном технологические задачи в химической, нефтехимической промышленности и др. областях;
• анализ смесей, качественный состав которых не известен; такие задачи встречаются в сфере контроля загрязнений окружающей среды, в биохимии, медицине, санитарной и судебной химии и др. областях.
Во втором случае перед проведением количественного анализа необходимо провести идентификацию неизвестных компонентов смеси.
Методы идентификации основываются на
• применении параметров удерживания;
• использовании химических реакций до хроматографических колонок;
• использовании качественных химических реакций на выходе из колонки;
• использовании специальных детектирующих систем;
• сборе разделенных компонентов в чистом виде и применении других физико-химических методов;
• селективном удалении (поглощении) некоторых разделенных компонентов смеси;
• сочетании параметров удерживания и масс-спектрометрии;
• корреляционных зависимостях параметров удерживания.
Для идентификации используют, как правило, относительные параметры удерживания, индексы Ковача. С целью повысить достоверность сопоставляют индексы Ковача на колонках с полярными и неполярными фазами.
Автоматизированная база данных унифицированной системы хроматографического анализа (УСХА) «Свето-хром-IEC» содержит около 200 тыс. опубликованных за тридцатилетний период (1960-1992 гг.) сведений по индексам удерживания, относительному удерживанию и удельным объемам удерживания 15 тыс. органических веществ на 350 неподвижных фазах в изометрических режимах.
Аналитическая хроматография и капиллярный электрофорез
289
Таблица 4.1.36
Классификация реактивов на функциональные группы
Соединения Реактив Характеристика реакции Предел обнаружения, мкг Испытанные соединения
Алифатические насыщенные HCHO-H2SO4 Винно-красный цвет 40 C2-Cg
Алкилгалоге-ниды Спиртовой раствор AgNO3 Белый осадок 20 Ci-C5
Альдегиды У-(4-аминобензол)-«-фенилен-диамин Оранжево-красный цвет 20 С1-С6
Основание Шиффа Розовый цвет 50 С1-С6
Амины Гинзберга Оранжевый цвет 100 Сг-С4
Ароматические ненасыщенные HCHO-H2SO4 Винно-красный цвет 20 С2-Сю
Дисульфиды Нитропруссид натрия Красный цвет 50 с2-с6
Изатин Зеленый цвет 100 С2-Сб
Кетоны Нитропруссид натрия Бордовый цвет 20 Сз-Cg (метилкетоны)
Меркаптаны Нитропруссид натрия Красный цвет 50 С4-С9
Изатин Зеленый цвет 100 С1-С9
РЬ(ОАс)2 Желтый осадок 100 С1-С9
Нитрилы Гидроксамат оксидного железа — пропиленгликоль Красный цвет 40 С2-С5
Спирты К2Сг2О7—HNO3 Голубой цвет 20 Ci-Cs
Нитрат церия Янтарно-желтый цвет 100 Cj-Cg
Сульфиды Нитропруссид натрия Красный цвет 50 C2-Ci2
Эфиры Гидроксамат оксидного железа Красный цвет 40 Ci-C5 (ацетаты)
База данных компьютерно-хроматографической системы «Инлан-ГХ» ориентирована на санитарно-экологический контроль воздушных объектов и включает параметры удерживания примерно 270 основных загрязнителей воздуха на восьми стандартизованных колонках с неподвижными жидкими фазами различной полярности при трех температурах.
В НИИ Химии СПбГУ составлен банк хромато-масс-спектрометрических данных для идентификации органических соединений — основных загрязнителей атмосферного воздуха. Он содержит индексы удерживания и масс-спектры примерно 660 летучих примесей открытой атмосферы и воздуха производственных помещений. Сформирована и опубликована полная база газохроматографических индексов удерживания галогенопроизводных углеводородов Ci и С2 (фреонов, всего 114 соединений) на стандартных сорбентах (неполярном порапаке Q).
Для качественного анализа применяются корреляционные зависимости параметров удерживания от числа атомов углерода в молекуле, температур кипения, электронных поляризуемостей исследованных соединений.
Не потеряли актуальности селективные химические реакции на отдельные функциональные группы (табл. 4.1.36 и 4.1.37).
Таблица 4.1.37
Типы химических реакций для элементного анализа на выходе хроматографических колонок
Элементы (или производные соединения) Реагенты; условия проведения реакции Получаемые продукты
С СиО; оксид кобальта, AgMnO4; 700-1000 °C со2
С (СО, СО2) Н2, Ni; 350-450 °C сн4
Н (D и Т) 750 °C н2о, н2
Н2О СаС2 С2н2
N Окисление NO
(NO) Си; 500-800 °C NO2 -> NO (пропускание через тетраборат натрия или цеолит)
(О) Уголь; 1120 °C; уголь—Pt (1:1); 920 °C СО
S О2, Pt; 850 °C; Н2, Pt; 800-1000 °C SO, H2S
Галоген Окисление; Pt; 800 °C; Н2, Pt; 750-1000 °C Cl2, Br2, НС1, НВг
Р Н2; 950 °C РН3
290
Новый справочник химика и технолога
4.1.7. Основные методы количественного анализа в газовой хроматографии
Хроматографический метод — относительный метод анализа. Измеряемая концентрация на выходе сильно отличается от концентрации в анализируемой пробе. При вводе пробы в колонку она сразу же разбавляется в детекторе газом-носителем, при прохождении пробы через колонку происходит дополнительное размывание, в результате на выходе концентрация компонентов совсем другая. Для проведения количественного анализа необходима градуировка — установление связи между первоначальными концентрациями в пробе и сигналами, полученными на выходе из колонки.
Метод абсолютной градуировки заключается в построении графика зависимости площади или высоты пика от содержания соединения в пробе. При выполнении количественного анализа по методу абсолютной градуировки предъявляются высокие требования к точности и воспроизводимости дозирования пробы. Кроме того, необходимо строго соблюдать тождественность условий при градуировке прибора и при реальных анализах. Наиболее воспроизводимое дозирование выполняется газовым краном-дозатором или специальными дозаторами жидкости.
Метод внутреннего стандарта: к известному количеству анализируемого образца добавляется известное количество не содержащегося в нем эталонного соединения — внутреннего стандарта. Содержание (концентрация) соединения в анализируемой смеси определяется по следующей формуле:
100
где St, SCT — площади пиков анализируемого соединения и стандарта; qCT и qn — количества стандартного соединения и анализируемой пробы. Преимущество метода внутреннего стандарта заключается в том, что случайные изменения температуры колонки, расхода газа-носителя и др. должны одинаково влиять на площади (или высоты) пиков как анализируемого соединения, так и стандартного вещества, следовательно, их отношения будут изменяться слабо. В этом случае нет необходимости точно дозировать анализируемую пробу.
Недостатки метода внутреннего стандарта:
• трудности в некоторых случаях при выборе внутреннего стандарта;
• необходимость подготовки пробы — добавка стандарта и возможное введение дополнительной погрешности.
Метод внутренней нормализации. Здесь процентное отношение анализируемого соединения в пробе определяют отношением его площади к сумме площадей всех компонентов на хроматограмме. Необходимо измерять количественные параметры всех пиков и приводить с помощью поправочных коэффициентов к единой шкале чувствительности детектирования. Содержание соединения в анализируемой пробе определяют по формуле:
где S, — площадь пика, f — градуировочный коэффициент.
Литература
1. 75 years of Chromatography — a Historical Dialogue / Ed. by Ettre L.S. and Zlatkis A. Amsterdam: Elsevier, 1979.502 р.
2. Сенченкова E.M. Рождение идеи и метода адсорбционной хроматографии. М.: Наука, 1991. 227 с.
3. Сенченкова Е.М. М.С. Цвет — создатель хроматографии. М.: Янус-К, 1997. 439 с.
4. Хроматография газовая. Термины и определения. ГОСТ 17567-72, введен 1.07.73 г.
5. Recommendations on Nomenclature for Chromatography. Rules approved 1973 // Pure and Appl. Chem. 1974. V. 7, № 4. P. 445-462.
6. Ettre L., Nomenclature for Chromatography // Pure and Appl. Chem. 1993. V. 65, № 4. P. 819-872.
7. Хроматография. Основные понятия. Терминология / Под ред. В.А. Даванкова М.: РАН, 1997. 48 с.
8. Гольберт К.А., Вигдергауз М.С. Курс газовой хроматографии. М.: Химия, 1974.
9. Ногаре С.Д., Джувет Р.С. Газо-жидкостная хроматография. Теория и практика / Пер. с англ. Л.:Недра, 1966.
10. McNair Н.М., Miller J.M. Basic Gas Chromatography. New York: Wiley, 1998. 200 p.
11. Smolkova-Keulemansova E., Feltl L. Analysis of Substances in the Gaseous Phase. Amsterdam: Elsevier, 1991.
12. Poole C.F., Poole S.K. Chromatography Today. Amsterdam: Elsevier, 1991. 1026 р.
13. Giddings J.C. Unified Separation Science. Chichester: Wiley, 1991.320 р.
14. Blum W., Aichholz R., Hochtemperatur Gas-Chromato-graphie. Heidelberg: Huthing, 1991. 166 S.
15. Theoretical Advancement in Chromatography and Related Separation Techniques / Ed. F. Dandi, G. Guiochon NATO Asi Ser V. 383. Dordrecht; Boston; London: Kluwer, 1992. 641 p.
16. Карабанов H.T. Физико-химические основы хроматографического процесса, Н. Новгород: НГУ им. Н.И. Лобачевского, 1994. 123 с.
17. Miller J.M. Chromatography: Concepts and Contrasts. New York: John Wiley and Sons, 1999.
18. Trenchant J. Manuel Practique de Chromatographie en Phase Gazeuse. Paris; Milan; Barcelone: Masson, 1995. 700 p.
19. Jennings W, Mittlefehldt E., Stremple P. Analytical Gas Chromatography. 2nd Ed. San Diego: Academic, 1997. 389 p.
20. Scott R.P.W. Introduction to Analytical Gas Chromatography. New York: M. Dekker, 1997. 416 p.
21. Березкин В.Г., Малюкова И.В. Влияние природы газа-носителя на удерживание и величину ВЭТТ в ГАХ // Успехи химии, 1998. Т. 67. С. 839-860.
Аналитическая хроматография и капиллярный электрофорез
291
22. Киселев А.В., Яшин Я.И. Адсорбционная газовая и жидкостная хроматография. М.: Химия, 1979. 288 с.
23. Kiselev A.V., Yashin Ya. I. Gas und Flussigkeits. Heidelberg: Verlag, 1985. 391 p.
24. Киселев A.B., Пошкус Д.П., Яшин Я.И. Молекулярные основы адсорбционной хроматографии. М.: Химия, 1986. 272 с.
25. Kiselev A.V., Yashin Ya. I. Adsorpcni plynova a kapali-nova chromatographie. Praha: SNTL-Nakladatelstvi Technicke Literatury, 1988. 400 p.
26. Alan Jones A. An Introduction to Gas-Liquid Chromatography. London; N.Y.: Academic Press, 1970. 202 p.
27. Gas-Liquid Chromatography by D.W. Grant. London: Van Nostrand Reinhold Co, 1971. 215 p.
28. Microbore Column Chromatography: Unified Approach to Chromatography / Ed. F. Yang New York: M. Dekker, 1989. 424 p.
29. Rood D. A Practical Guide to the Care. Maintenance and Troubleshooting of Capillary Gas Chromatographic Systems. Heidelberg: Huthig, 1991. 191 p.
30. Capillary Chromatography. The Applications / Ed. W.G. Jennings, J.G. Nikelly Heidelberg: Huthig, 1991. 153 p.
31. Grob K. On-Column Injection in Capillary Gas Chromatography. Heidelberg: Huthig, 1991. 591 p.
32. Van Es. A. High Speed Narrow Bore Capillary Gas Chromatography. Heidelberg: Huthig, 1992. 143 p.
33. Grob K. Split and Splitless Injection in Capillary GC. 3rd rev. ed. Oxford: Huthig Publishing, 1993. 574 p.
34. Berezkin V.G. and De Zeeuw J. Capillary Gas Adsorption Chromatography. Heidelberg: Huthig, 1996. 320 p.
35. Высокоэффективная газовая хроматография / Под ред. К. М. Хайвер. Мир, 1993. 288 с.
36. К. Тесаржик, К. Комарек. Капиллярные колонки в газовой хроматографии / Пер. с чеш. М.: Мир, 1987. 221с.
37. Яшин Я.И. Физико-химические основы хроматографического разделения. М.: Химия, 1976. 216 с.
38. Березкин В.Г. и др. Газовая хроматография в нефтехимии. М.: Наука, 1975.. 267 с.
39. Березкин В.Г. Газо-жидко-твердофазная хроматография. М.: Химия, 1986. 112 с.
40. Сакодынский К.И., Волков С.А. Препаративная газовая хроматография. М.: Химия, 1972. 208 с.
41. Препаративная газовая хроматография / Пер. с англ.; под ред. В.Г. Березкина и К.И. Сакодынского М.: Мир, 1974. 408 с.
42. Ma T.S., Lados A.S. Organic Functional Group Analysis by Gas Chromatography. London; N. Y.: Academic Press, 1976. 186 p.
43. Cagniant D. Complexation Chromatography // Chroma-togr. Science. Ser. № 57. New York: M. Dekker, 1991.
44. Киселев А.В. и др. Физико-химическое применение газовой хроматографии / А. В. Киселев, К.И. Сакодынский, А. В. Йогансен и др. М.: Химия, 1973. 255 с.
45. Методы-спутники в газовой хроматографии / Пер. с англ.; Под ред. В. Г. Березкина. М.: Мир, 1972. 398 с.
46. Me Fadden W.H. Techniques of Combined Gas Chromatography-mass-spectrometry. Application in Organic Analysis. N. Y.: Wiley-Intersci, 1973. 458 p.
47. Коган Л.А. Количественная газовая хроматография. М.: Химия, 1975. 184 с.
48. Novak J. Quantitative Analysis by Gas Chromatography. N. Y.: M. Dekker, 1975. 218 p.
49. Сакодынский К.И., Бражников B.B., Волков С.А. и др. Аналитическая хроматография. М.: Химия, 1993.
50. Гиошон Ж., Гийемен К.. Количественная газовая хроматография для лабораторных анализов и промышленного контроля: 4.1, 2. М.: Мир, 1991. 380 с.
51. Березкин В.Г., Лощилова В.Д., Панков А.Г., Ягодов-ский В.Д. Хроматораспределительный метод. М.: Наука, 1976. 112 с.
52. Авгуль Н.Н., Киселев А.В., Пошкус Д.П. Адсорбция газов и паров на однородных поверхностях. М.: Химия, 1975. 384 с.
53. Baiulescu G.E., Hie V.A. Stationary Phases in Gas Chromatography. Oxford: Pergamon Press, 1975. 370 p.
54. Bonded Stationary Phases in Chromatography / Ed. E. Gryshka Michigan: Ann Arbor Sci Publ, 1975. 273 p.
55. Березкин В.Г., Пахомов В.П., Сакодынский К. И. Твердые носители в газовой хроматографии. М.: Химия, 1975. 200 с.
56. Unger К. Packing and Stationary Phases in Chromatographic Techniques. New York: M. Dekker, 1990.
57. Hetem M. J.J. Chemically Modified Silica Surfaces in Chromatography. Heidelberg: Huthig, 1992. 235 p.
58. Слижков Ю.С., Гавриленко M.A. Применение внут-рикомплексных соединений в газовой хроматографии. Томск: ТГУ, 2000. 137 с.
59. Лурье А.А. Хроматографические материалы: Справочник. М.: Химия, 1978. 440 с.
60. Король А.Н. Неподвижные фазы в газо-жидкостной хроматографии: Справочник. М: Химия, 1985. 220 с.
61. Senate Commission for Clinical Toxicological Analysis. Gas Chromatographic Retention Indices of Toxicologically Relevant Substances on SE-30, OV-1. Weinheim: Verlag Chemie, 1991. 200 p.
62. Pacakova V., Feltl L. Chromatographic Retention Indices — an Aid to the Identification of Organic Compounds. London: Ellis Horwood, 1992. 285 p.
63. Pfleger K., Maurer H.H., Weber A. Mass Spectral and GC Data of Drugs, Poisons, Pesticides, Pollutants and Their Metabolites: Part 1-3. 2nd ed. Weinheim; New York: VCH, 1992. 3030 p.
64. De Zeeum RA. et al. Gas Chromatographic Retention Indices of Solvents and Other Volatile Substances for Use in Toxicological Analysis. Weinheim: VCH; Verlags, 1992. 130 p.
65. De Zeeum R.A. et al. Gas Chromatographic Retention Indices of Toxicologically — Relevant Substances on Packed or Capillary Columns with Dimethylsilicone Stas-tionary Phases. Weinheim: VCH; Verlags, 1992. 403 p.
66. Retention Standartization and Phase Comparisions I Ed. R.M. Smith // J. of Chrom. Libr. 1995. V. 57. 480 p.
67. Пецев H., Коцев H. Справочник по газовой хроматографии / Пер. с болг. М.: Мир, 1987. 260 с.
68. Гвоздович Т.Н., Худяков В.Л., Яшин Я.И. Атлас-справочник хроматографических разделений. М.: МИХМ, 1978.
292
Новый справочник химика и технолога
69. Гуревич А.Л., Коломийцев Л.А., Русинов Л.А. Автоматизация обработки хроматографической информации. М.: Энергия, 1973. 112 с.
70. Ротин В.А. Радиоионизационное детектирование в газовой хроматографии. М.: Атомиздат, 1974. 189 с.
71. David D.J. Gas Chromatographic Detectors. N. Y.: Wiley-Intersci, 1974. 296 p.
72. Sevcik J. Detectors in Gas Chromatography. Elsevier Scientific Publishing Company, 1976. 192 p.
73. Jentzsch D., Otte E., Detectoren in der Gas-Chromato-graphie. Fr. am Main: Akademische-Verlags gesellschaft, 1970. 454 s.
74. Бражников Б.В. Детекторы для хроматографии. М.: Машиностроение, 1992. 318с.
75. Hill Н.Н., McMinn D. Detectors for Capillary Chromatography. London: Wiley, 1992.
76. Walker J.G., Maynard J.B. Chromatographic Systems: Maintenance and Troubleshooting, N. Y.; London: Academic Press, 1972. 289 p.
77. Ворона Э.Н., Лошак В.И., Юдович В.Б. Основные параметры и метрологические характеристики отечественных хроматографов. М.: ВНИИКИ, 1975. 64 с.
78. Кеилеманс А. Хроматография газов / Пер. с англ. М.: ИЛ, 1975.
79. Экспериментальные методы в адсорбции и хроматографии / Под ред. А.В. Киселева М.: МГУ, 1972.
80. Вяхирев Д.А., Шушунова А.Ф. Руководство по газовой хроматографии. М.: Высшая школа, 1975. 301 с.
81. Modem Practice of Gas Chromatography / Ed. R.L. Grob N.Y.; London: Wiley-Interscience, 1977. 654 p.
82. Прикладная хроматография / Под. ред. К.И. Сако-дынского. М.: Наука, 1984. 302 с.
83. Столяров Б.В. Савинов И.М., Витенберг А.Г. и др. Практическая газовая и жидкостная хроматография: Учеб, пособие. СПб.: СПбГУ, 1998. 610 с.
84. Руководство по газовой хроматографии: В 2х ч. / Пер. с нем.; под ред. Э. Лейбница, Х.Г. Штруппе М.: Мир, 1988. 478 с.
85. Sofer G.K., Nystroem L.E. Process Chromatography. A Practical Guide. San Francisco: Academic, 1989. 160 p.
86. Frank H. Chromatographic Separation of Optical Isomers. Heidelberg: Huthig, 1993.
87. Konig W.A. Gas Chromatography Enantiomer Separation with Modified Cyclodextrins. Heidelberg: Huthig, 1992.168 c.
88. Beesley T.E., Scott R.P.W. Chiral Chromatography. New York: John Wiley and Sons, 1999.
89. Bertsch W. // JHRC, 1999. V. 22. P. 647-665.
90. Multidimensional Chromatography. Techniques and Applications / Ed. H.J. Cortes New York: M. Dekker, 1989.
91. Карасек Ф., Клемент P. Введение в хромато-масс-спектрометрию / Пер. с англ. М.: Мир, 1993. 237с.
92. McMaster М. and McMaster С. GC/Ms: A Practical User’s Guide. New York: Wiley-VCH, 1998. 167 p.
93. Grob K. On-line Coupled LC-GC. Heidelberg: Huthig, 1991.462 р.
94. Supercritical Fluid Extraction and Its Use in Chromatographic Sample Preparation / Ed. S.A. Westwood London: Chapman and Hall, 1992. 184 p.
95. Supercritical Fluid Science and Technology / Ed.: K.P. Johnson, J.M.L. Penninger Washington: American Chemical Society, 1989. 550 p.
96. Hyphenated Techniques in Supercritical Fluid Chromatography and Extraction / Ed. K. Jinno Togohashi: Toy-chaskii university of technology, 1992. 334 p.
97. Westwood S.A. Supercritical Fluid Extraction and Its Use in Chromatographic Sample Preparation. London: Blackle academic and professional, 1993. 170 p.
98. Belgacem M.H., Gandini A. // Sci. Ser. 1999. V. 80. P. 41-124.
99. Voelkel A. // Crit. Rev. Anal. Chem., 1999. V. 22. P. 411-439.
100. Inverse Gas Chromatography. Characterization of Polymers and Other Materials, ACS Symposium Series / D.R. Lloyd, Th.C. Word, P. Schreiber Washigton: American Chemical Society, 1989. 331 p.
Приложение
Газохроматографические индексы удерживания нефтяных углеводородов на стандартных неполярных полидиметилсилоксановых неподвижных фазах
Приложение включает статистически обработанные (рандомизованные) значения газохроматографических индексов удерживания (RI) основных классов нефтяных углеводородов (алканы, циклоалканы, алкены и арены с молярными массами не более 200 Да) на всех известных стандартных неполярных полидиметилсилоксановых неподвижных фазах [-Si(CH3)2-O-]n, в том числе SE-30, OV-101, HP-1, DB-1, RTX-1, SP-2100, Е-301 (устаревшая), SF-96 (устаревшая), ПМС-100 и др. В соответствии с концепцией рандомизации межлабораторных данных [1], температурная зависимость индексов удерживания [3 = <Ж1/б7Т (обычно [3 > 0) не принимается во внимание и обусловленный ею разброс характеризуется стандартными отклонениями 5М. Критерием газохроматографической идентификации является попадание экспериментальных значений индексов удерживания в диапазоны (RI) ± .vR1. Для всех перечисленных ниже соединений приведены молярные массы (М), молекулярные формулы, названия, средние значения индексов удерживания (RI) и соответствующие стандартные отклонения (a'ri). Общее число усредняемых данных (N), заимствованных из всех доступных литературных источников начиная с середины 1970-х г.г., а также экспериментально определенных авторами, не указано. Для простейших представителей перечисленных гомологических рядов приведены индексы удерживания всех возможных изомеров, тогда как по мере увеличения молярных масс информация ограничивается только изомерами с наименее разветвленным углеродным скелетом.
Реперными компонентами для вычисления газохроматографических индексов удерживания являются н-алканы, которым присвоены значения RI = Ю0лс, где пс —- число атомов углерода в молекуле. Для расчета RI в чаще всего используемых режимах линейного программирования температуры рекомендуется применять систему линейно
Аналитическая хроматография и капиллярный электрофорез
293
логарифмических индексов удерживания и простейшие программы, приведенные в руководствах [1-3].
Более подробное информационное обеспечение для органических соединений различной химической природы (базы данных по индексам удерживания, включающие сведения о более чем 40 тыс. объектов) и алгоритмы использования хроматографических данных при хромато-спектральной идентификации неизвестных веществ разрабатываются в лаборатории газовой хроматографии НИИ Химии СПбГУ.
Молярная масса, г-моль 1 Эпириче-ская формула Название RI Sri
16 сн4 Метан 100 —
28 С2н4 Этилен 166 7
30 с2н6 Этан 200 —
42 С3н6 Пропилен 290 3
44 С3н8 Пропан 300 —
56 С4н8 1-Бутен 387 4
(7)-2-Бутен 418 6
(Е)-2-Бутен 407 3
Изобутилен 388 4
58 с4н10 н-Бутан 400 —
Изобутан 366 6
70 С5Н1О 1-Пентен 488 5
(2)-2-Пентен 513 4
(£)-2-Пентен 507 4
2-Метил-1-бутен 495 4
2-Метил-2-бутен 517 7
3 -Метил-1 -бутен 455 5
Циклопентан 570 8
72 С5н12 н-Пентан 500 —
Изопентан 474 2
2,2-Диметил-пропан 412 2
78 СбНб Бензол 656 8
84 СбН12 1-Гексен 588 4
(7)-2-Гексен 612 2
(£)-2-Гексен 603 4
(Z)-3-Гексен 601 3
(£)-3-Гексен 603 1
2-Метил-1-пентен 586 4
З-Метил-1-пентен 557 3
4-Метил-1-пентен 556 3
2-Метил- 2-пентен 605 3
Продолжение таблицы
Молярная масса, г-моль 1 □лирическая формула Название RI Sri
(Z)-3-Метил -2-пентен 609 4
(£)-3 -Метил -2-пентен 618 4
(И)-4-Метил-2-пентен 564 4
(£)-4-Метил-2-пентен 568 5
2-Этил-1-бутен 597 5
2,3-Диметил-1-бутен 564 3
3,3-Диметил-1-бутен 512 4
2,3-Диметил-2-бутен 630 4
Циклогексан 666 8
Метилциклопентан 627 5
86 С6Н14 н-Гексан 600 —
2-Метилпентан 570 1
З-Метилпентан 584 2
2,2-Диметил-бутан 537 2
2,3-Диметил-бутан 568 3
92 С7н8 Толуол 760 9
98 С7н14 1-Гептен 688 3
(2)-2-Гептен 710 4
(£)-2-Гептен 703 3
(7)-3-Гептен 696 4
(£)-3-Гептен 693 6
2-Метил-1-гексен 683 4
З-Метил-1-гексен 652 3
4-Метил-1-гексен 663 3
5-Метил-1-гексен 657 3
2-Метил-2-гексен 699 4
(Z)-3-Метил-2 -гексен 706 3
(£)-3-Метил-2-гексен 698 5
294
Новый справочник химика и технолога
Продолжение таблицы
Молярная масса, г-моль 1 □лирическая формула Название RI Sri
98 с7н14 (7)-4-Метил-2-гексен 663 4
(Е)-4-Метил-2-гексен 663 4
(У)-5-Мети.[-2-гексен 678 4
(£)-5-Метил- 2-гексен 668 3
(7)-2-Метил-3-гексен 661 2
(£)-2-Метил- 3-гексен 653 7
(7)-3-Метил-3-гексен 699 4
(£)-3-Метил-3-гексен 691 3
2-Этил-1-пентен 690 2
2,3-Диметил-1-пентен 654 3
2,4-Диметил- 1 -пентен 643 4
3,3-Диметил-1-пентен 629 5
3,4-Диметил-1-пентен 642 2
4,4-Диметил-1-пентен 610 3
3-Этил-1-пентен 656 3
3 -Этил-2-пентен 702 4
2,3-Диметил-2-пентен 708 4
2,4-Диметил-2-пентен 649 6
(7)-3,4-Диметил- 2-пентен 677 2
(£)-3,4-Диметил-2-пентен 682 4
(7)-4,4-Диметил- 2-пентен 642 3
(£)-4,4-Диметил- 2-пентен 622 8
З-Метил-2-этил-1-бутен 665 2
2,3,3-Триметил-1-бутен 636 4
Циклогептан 803 10
Метилциклогексан 725 8
Продолжение таблицы
Молярная масса, г-моль1 □лирическая формула Название RI Sri
98 с7н14 Этилциклопентан 734 5
1,1-Диметилциклопентан 673 И
z/wc-l ,2-Диметил-циклопентан 724 6
транс-\,2-Диметилцикло-пентан 689 6
цис-1,3-Диметил-циклопентан 684 5
транс-\,3-Диметилцикло-пентан 687 5
106 с8н10 Этилбензол 854 9
о-Ксилол 885 9
jw-Ксилол 865 8
и-Ксилол 863 9
112 С8н16 1-Октен 788 3
(7)-2-Октен 811 2
(£)-2-Октен 802 3
З-Октены 798 2
4-Октены 794 2
2-Метил-1-гептен 779 5
З-Метил-1-гептен 744 6
4-Метил-1-гептен 748 4
5-Метил-1-гептен 752 4
6-Метил-1-гептен 752 3
2-Метил- 2-гептен 796 5
З-Метил- 2-гептены 794 3
4-Метил- 2-гептены 755 3
5-Метил- 2-гептены 776 2
6-Метил- 2-гептены 770 1
(7)-2-Метил- 3-гептен 746 5
(Е)-2-Метил-3-гептен 749 4
Аналитическая хроматография и капиллярный электрофорез
295
Продолжение таблицы
Молярная масса, г-моль-1 Эпириче-ская формула Название RI •Sri
112 С«Н16 З-Метил-3-гептены 791 7
4-Метил- 3-гептены 794 3
5-Метил- 3-гептены 752 11
6-Метил- 3-гептены 758 3
2-Этил-1-гексен 784 2
3-Этил-1-гексен 738 6
4-Этил-1 -гексен 750 4
Пропилциклопентан 835 8
Изопропилциклопентан 813 7
1 -Метил-1 -этилциклопентан 797 14
г/wc-l-Метил-2 -этилциклопентан 823 7
транс-1 -Метил-2-этилцикло-пентан 790 6
z/wc-1-Метил-3-этилцикло пентан 789 6
транс-1 -Метил-3-этил цикло пентан 787 6
1,1,2-Триметил-циклопентан 763 9
1,1,3-Триметилциклопентан 723 11
цис-цис-1,2,3 -Триметилциклопентан 796 7
цис-транс-1,2,3-Триметилцикло-пентан 773 7
транс-цис-1,2,3 -Триметилциклопентан 745 7
цис-цис-1,2,4-Триметилцикло-пентан 771 6
цис-транс-1,2,4-Триметилцикло-пентан 769 7
Продолжение таблицы
Молярная масса, г-моль-1 Эпириче-ская формула Название RI ‘Sri
транс-транс-1,2,4-Триметил-циклопентан 740 3
Этилциклогексан 838 8
1,1-Диметилциклогексан 783 6
транс-1,2- Диметилциклогексан 800 7
цис-1,2-Диметил-циклогексан 828 6
транс-1,3-Диметилцикло-гексан 805 9
цис-1,3-Диметил-циклогексан 781 6
транс-1,4-Диметилцикло-гексан 780 6
цис-1,4-Диметилциклогексан 797 13
114 //-Октан 800 —
2-Метилгептан 766 2
З-Метилгептан 773 2
4-Метилгептан 676 2
2,2-Диметил-гексан 721 3
2,3-Диметил-гексан 761 3
2,4-Диметил-гексан 733 2
2.5-Диметил-гексан 730 2
3,3-Диметил -гексан 744 4
3,4-Диметил-гексан 771 3
З-Этилгексан 774 1
2,2.3-Триметил-пентан 738 4
2,2,4-Триметил-пентан 691 3
2,3,3-Триметил-пентан 762 5
2,3,4-Триметил-пентан 753 4
296
Новый справочник химика и технолога
Продолжение таблицы
Молярная масса, г-моль1 □лирическая формула Название RI 5ri
114 с8н18 2-Метил-З-этил-пентан 762 2
3-Метил-3-эти л-пентан 775 5
2,2,3,3-Тетра-метилбутан 725 3
116 с9н8 1Я-Инден 1035 14
118 ОДо Индан 1018 14
120 С9Н12 Пропилбензол 945 9
Изопропилбензол 914 6
1 -Метил-2-этил-бензол 967 4
1-Метил-3-этилбензол 950 4
1-Метил-4-этил-бензол 951 4
1,2,3-Триметил-бензол 1011 4
1,2,4-Триметил-бензол 984 10
1,3,5-Триметил-бензол 963 13
126 С9Н18 1-Нонен 887 2
(7)-2-Нонен 912 2
(£)-2-Нонен 902 2
(Z)-3-Нонен 898 2
(£)-3-Нонен 895 2
(7)-4-Нонен 897 2
(£)-4-Нонен 893 2
2-Метил-1-октен 875 7
7-Метил-1 -октен 852 3
2-Метил-2-октен 893 2
3-Метил-2-октены 902 8
4-Метил- 2-октены 873 12
4-Этил- 2-гептены 902 13
Бутилциклопентан 930 5
Изобутилциклопентан 891 3
(1-Метил-пропил)цикло-пентан 922 7
Продолжение таблицы
Молярная масса, г-моль 1 □лирическая формула Название RI Sri
(1,1-Диметил-этил)цикло-пентан 881 6
1-Метил- 1 -пропилцикло-пентан 887 3
z/wc-1-Метил-2-пропилцикло-пентан 914 6
транс-1 -Метил-2-пропилцикло-пентан 887 3
1-Метил-3-пропилциклопентаны 891 4
1 -Метил- 1 -изопропилциклопентан 893 5
z/wc-1-Метил-2-изопропил-циклопентан 893 5
транс-1 -Метил-2-изопропил-циклопентан 866 6
1-Метил-З-изо-пропилцикло-пентаны 882 16
1,1-Диэтилциклопентан 902 8
цис-1,2-Диэтилциклопентан 914 8
транс-1,2-Диэтилцикло-пентан 891 8
1,3-Диэтил-циклопентаны 900 10
Пропилциклогексан 933 9
Изопропилцикло гексан 924 7
1,1,2-Триметил-циклогексан 884 8
1,1,3-Триметил-циклогексан 840 10
1,1,4-Триметил-циклогексан 839 10
1-Метил-1-этил-циклогексан 904 4
Аналитическая хроматография и капиллярный электрофорез
297
Продолжение таблицы
Молярная масса, г-моль1 □лирическая формула Название RI -Sri
126 с9н18 г/мс-1-Метил-2-этилциклогексан 922 3
транс-1 -Метил-2-этилцикло-гексан 906 7
цис-1 -Метил-3 -этилциклогексан 888 6
транс-1 -Мети л-3-этилциклогексан 908 5
г/мс-1-Метил-4-этилциклогексан 907 5
транс-1 -Метил-4-этилцикло-гексан 897 10
128 с10н8 Нафталин 1163 13
с9н10 н-Нонан 900 —
2-Метилоктан 864 2
3-Мети л октан 871 2
4-Метилоктан 863 2
2,2-Диметил-октан 817 4
2,3-Диметил-октан 854 4
2,4-Диметил-октан 822 2
2,5-Диметил-октан 833 2
2,6-Диметил-октан 827 2
3,3-Диметил-октан 838 2
3,4-Диметил-октан 859 3
3,5-Диметил-октан 834 2
4,4-Диметил-октан 830 2
3-Этилоктан 868 2
4-Этилоктан 858 2
2,2,3-Триметил-гексан 822 6
2,2,4-Триметил-гексан 791 3
2,2,5-Триметил-гексан 779 4
Продолжение таблицы
Молярная масса, г-моль1 □лирическая формула Название RI Sri
2,3,3-Триметил-гексан 841 6
2,3,4-Триметил-гексан 852 3
2,3,5-Триметил-гексан 814 2
2,4,4-Триметил-гексан 810 3
3,3,4-Триметил-гексан 858 5
2-Мети л-3 -эти л-гексан 846 2
З-Метил-З-этил-гексан 858 4
3 -Мети л-4-этил-гексан 858 4
2-Метил-4-этил-гексан 826 2
2,2,3,3-Тетра-метилпентан 858 6
2,2,3,4-Тетра-метилпентан 823 4
2,2,4,4-Тетра-метилпентан 775 4
2,3,3,4-Тетра-метилпентан 862 5
2,2-Диметил-3 -этилпентан 825 4
2,3-Диметил-3 -этилпентан 879 6
2,4-Диметил-3 -этилпентан 839 3
3,3-Диэтил-пентан 881 5
130 CioH10 1-Метил- 1//-инден 1104 13
2-Метил- 1//-инден 1140 13
З-Метил-1//-инден 1128 12
4-Метил- 1//-инден 1146 11
5-Мети л- 1//-инден 1137 И
6-Метил-1//-инден 1137 11
298
Новый справочник химика и технолога
Продолжение таблицы
Молярная масса, г-моль1 Эпириче-ская формула Название RI -Sri
130 СюН10 7-Метил- 1//-инден 1146 11
132 с10н12 1-Метилиндан 1073 9
2-Метил ин дан 1070 11
4-Метилиндан 1134 9
5-Метилиндан 1125 9
134 с10н14 Бутилбензол 1046 5
Изобутилбензол 997 6
1 -Метилпропил-бензол 1003 7
1,1-Диметил-этилбензол 984 7
1-Метил-2-пропил бензол 1050 6
1-Метил-3-пропилбензол 1036 5
1-Метил-4-пропилбензол 1042 5
1-Метил-2-изо-пропил бензол 1024 8
1-Метил-З-изо-пропил бензол 1006 7
1-Метил-4-изо-пропилбензол 1013 5
1,2-Диэтил-бензол 1040 7
1,3-Диэтил-бензол 1035 5
1,4-Диэтил-бензол 1040 8
1,2-Диметил-3-этилбензол 1091 6
1,2-Диметил-4-этил бензол 1074 5
1,3-Диметил-2-этил бензол 1076 6
1,3-Диметил- 4-этилбензол 1067 4
1,4-Диметил-2-этил бензол 1064 4
1,3-Диметил-5-этилбензол 1045 3
1,2,4,5-Тетра-метил бензол 1103 4
1,2,3,5-Тетра-метилбензол 1107 6
1,2,3,4-Тетра-метилбензол 1137 6
136 С1он1б Адамантан 1124 13
Продолжение таблицы
Молярная масса, г-моль1 Эпириче-ская формула Название RI -Sri
138 с10н18 транс- Декалин 1059 11
г/ис-Декалин 1113 13
140 С10Н20 1-Децен 998 2
(2)-2-Децен 1010 2
(£)-2-Децен 1004 4
3-Децены 997 3
4-Децены 992 2
5-Децены 989 4
2-Мети л-1-нонены 982 4
5-Метил-1-нонены 952 4
7-Мети л-1-нонены 961 4
8-Метил-1-нонены 955 5
Бутилциклогексан 1031 4
Изобутилциклогексан 986 6
1-Метилпропил-циклогексан 1024 6
1,1-Диметил-этилциклогексан 990 5
Пентилциклопентан 1034 3
Изопентилцикло пентан 1000 4
1-Метилбутил-циклопентан 1016 4
1 -Метил-2-пропилциклогексаны 994 6
1-Метил-2-пропилциклогексаны 1001 7
1-Метил-3-изопропилциклогексаны 970 4
транс-1 -Мети л-4-изопропил-циклогексан 976 5
z/мо 1-Метил-4-изопропил-циклогексан 991 6
142 СнНю 1-Метил-нафталин 1292 14
Аналитическая хроматография и капиллярный электрофорез
299
Продолжение таблицы
Молярная масса, г-моль-1 □лирическая формула Название RI Sri
142 СцН1о 2-Метил-нафталин 1285 20
С10Н22 н-Декан 1000 —
2-Метилдекан 964 2
З-Метилдекан 970 2
4-Метилдекан 959 2
5-Метилдекан 957 2
148 СцН1б Пентилбензол 1143 9
Изопентилбензол 1112 7
1-Метилбутил-бензол 1081 10
2-Метилбутил-бензол 1105 7
1-Этилпропил-бензол 1076 12
1,2-Диметил-пропилбензол 1068 4
2,2-Диметил-ропилбензол 1048 5
1,1-Диметил-пропил бензол 1083 12
1 -Бутил-2-метил-бензол 1151 4
1 -Бутил-3-метил-бензол 1138 4
1 -Бутил-4-метил-бензол 1138 6
Пентаметил-бензол 1259 4
152 С12Н8 Аценафтилен 1424 15
Бифенилен 1371 12
154 С12Н10 Бифенил 1356 12
Аценафтен 1461 15
С11Н22 1-Ундецен 1089 4
(2)-2-Ундецен 1113 4
(Е)-2-Ундецен 1102 2
З-Ундецены 1094 2
4-Ундецены 1092 2
5-Ундецены 1090 2
Гексилциклопентан 1135 4
Пентилциклогексан 1135 5
156 С12Н12 1,2-Диметил-нафталин 1436 6
Продолжение таблицы
Молярная масса, г-моль“1 □лирическая формула Название RI Sri
1,3-Диметил-нафталин 1401 14
1,4-Диметил-нафталин 1416 14
1,5-Диметил-нафталин 1418 И
1,6-Диметил-нафталин 1405 14
1,7-Диметил-нафталин 1404 10
1,8-Диметил-нафталин 1456 9
2,3-Диметил-нафталин 1418 11
2,6-Диметил-нафталин 1388 11
2,7-Диметил-нафталин 1386 6
1 -Эти лнафталин 1375 11
2-Этилнафталин 1373 15
С11Н24 н-Ундекан 1100 —
2-Метилдекан 1063 3
3-Мети л декан 1071 2
4-Метилдекан 1062 3
5-Метилдекан 1058 2
162 с12н18 Гексилбензол 1240 9
1-Метил- 2-пентилбензол 1249 9
1-Метил- 3-пентилбензол 1235 4
1-Метил- 4-пентил бензол 1244 9
1-Бутил-2-этил-бензол 1217 11
1-Бутил-З-этил-бензол 1221 9
1-Бутил-4-этил-бензол 1232 7
1,3-Дипропил-бензол 1204 9
1,4-Дипропил-бензол 1219 6
Гексаметил-бензол 1417 14
166 С13Н10 Флуорен 1559 17
168 С13Н12 Дифенилметан 1403 19
300
Новый справочник химика и технолога
Продолжение таблицы
Молярная масса, г-моль”1 Эпириче-ская формула Название RI Ум
С12Н24 1-Додецен 1189 3
(2)-2-Додецен 1215 6
(£)-2-Додецен 1202 2
З-Додецены 1194 2
4-Додецены 1191 2
5-Додецены 1188 2
6-Додецены 1186 2
Гексил циклогексан 1238 6
Гептилциклопентан 1234 7
170 С13Н14 1-Пропил-нафталин 1453 15
2-Пропил-нафталин 1459 15
С12Н26 «-Додекан 1200 —
2-Метилундекан 1163 2
3 -Метил ундекан 1172 2
4-Метил ундекан 1160 2
5 -Мети лунд екан 1154 2
6-Метилундекан 1152 4
176 С13Н20 Гептил бензол 1339 15
178 С14Н10 Антрацен 1742 15
Фенантрен 1744 22
182 с13н26 1-Тридецен 1287 3
(2)-2-Тридецен 1310 2
(£)-2-Тридецен 1300 2
3-Тридецены 1290 5
4-Тридецены 1284 6
5-Тридецены 1281 6
6-Тридецены 1278 8
Гептилциклогексан 1330 7
184 С14Н16 1 -Бутилнафталин 1563 13
2-Бутилнафталин 1566 12
С13Н28 «-Тридекан 1300 —
2-Метилдодекан 1264 2
3 -Метилдод екан 1271 3
4-Метилдодекан 1258 2
5 -Метилдодекан 1252 2
6-Метилдодекан 1250 3
190 С14н22 Октилбензол 1451 4
196 С14Н28 1-Тетрадецен 1389 2
198 С14Н30 н-Тетрадекан 1400 —
Продолжение таблицы
Молярная масса, г-моль1 Эпириче-ская формула Название RI •Ум
2-Метилтри-декан 1362 2
3-Метилтридекан 1371 2
4-Метилтридекан 1359 3
5-Метилтри-декан 1350 3
6-Метилтридекан 1344 2
7-Метилтри-декан 1346 6
Литература
1. Зенкевич И.Г. // Журн. аналит. химии. 1996. Т. 51, № 11. С. 1140-1148.
2. Зенкевич И.Г. // Журн. аналит. химии. 1984. Т. 39, № 7. С. 1297-1307.
3. Zenkevich I.G., Ioffe B.V. // J. Chromatogr. 1988. V. 439. P. 185-194.
4.1.8. Высокоэффективная жидкостная хроматография (ВЭЖХ)
4.1.8.1. Основные особенности метода и аппаратуры
Хроматографическое разделение было открыто М. С. Цветом в 1903 г. в виде жидкостно-адсорбционного варианта. Однако жидкостная хроматография (ЖХ) в течение долгого времени не получала столь широкого распространения в аналитической практике, как газовая хроматография (ГХ), хотя последняя была предложена значительно позднее и охватывала более узкий круг веществ. Одной из причин медленного развития колоночной жидкостной хроматографии было отсутствие соответствующей аппаратуры, в частности высокочувствительных и достаточно универсальных детектирующих устройств и малопроизводительных беспульсационных насосов высокого давления.
До недавнего времени разделение методом ЖХ в колонках было очень медленным, так как элюирование подвижной фазы осуществлялось лишь под действием силы тяжести, а регистрацию разделенных полос из-за отсутствия подходящих детекторов проводили по отдельным собранным фракциям.
ЖХ стала усиленно развиваться, начиная с 1960 г., благодаря разработке новых методов детектирования и в связи с тем, что предельные возможности анализа малолетучих и термически неустойчивых веществ методом ГХ к этому времени уже были использованы (реакционная и пиролизная ГХ), а также выявились ее серьезные ограничения. Этому также способствовало все большее возрастание требований к анализу жидких растворов высокомолекулярных
Аналитическая хроматография и капиллярный электрофорез
301
органических веществ как синтетических, так и природных (биополимеры).
Среди других хроматографических методов (бумажная, тонкослойная, газовая) ЖХ в колонках является наилучшим методом, позволяющим проводить разделение не только в наиболее мягких условиях (температуры колонки — комнатная или близкая к ней), но и благодаря возможности изменения природы подвижной фазы. В отличие от бумажной и тонкослойной ЖХ в хроматографии на колонках сорбент и разделяемые вещества не подвержены влиянию окружающей атмосферы.
Вследствие всех этих преимуществ ЖХ в последние годы все более широко внедряется в аналитическую практику, интенсивно разрабатывается хроматографическая аппаратура, исследуются физико-химические основы селективности и размывания. Большим достижением, способствовавшим значительному сокращению времени разделения в ЖХ, явилось повышение давления на входе в колонку до десятков и сотен атмосфер. При использовании более мелких фракций зерен сорбента и поверхностнопористых сорбентов это позволило увеличить скорость массообмена и соответственно повысить эффективность колонок. По времени разделения высококипящих соединений ВЭЖХ на современной аппаратуре с высокими входными давлениями успешно конкурирует с ГХ, а по эффективности значительно превосходит ее (уже достигнута эффективность порядка 200 тыс. теоретических тарелок на метр).
Ниже рассмотрены только основные особенности теории и аппаратуры для ЖХ. Для более детального ознакомления можно обратиться к специальным руководствам и обзорам (см. список литературы)*.
4.1.8.2. Классификация хроматографических систем: сорбент — элюент - сорбат по типам межмолекулярных взаимодействий
В ЖХ имеются исключительно большие возможности управления селективностью разделения. В ГХ с практически неадсорбирующимся газом-носителем вещества разделяются за счет различий только неспецифических (в основном дисперсионных) межмолекулярных взаимодействий или суммы специфических и неспецифических межмолекулярных взаимодействий сорбат - сорбент. В ЖХ за счет влияния подвижной фазы удерживание веществ и селективность разделения может определяться значительно большим разнообразием межмолекулярных взаимодействий. Здесь можно реализовать случай, когда удерживание определяется преимущественно специфическим взаимодействием с сорбентом при применении неполярного или слабополярного элюента (так называемый нормальнофазовый вариант ЖХ) или преимущественно неспецифическим взаимодействием с сорбентом при применении полярного элюента (так называемый обращенно-фазовый вариант ЖХ), а также их различными комбинациями. В молекулярной адсорбционной ЖХ удерживание веществ и
* См. также раздел 3.2.9.4.
селективность их разделения, как упоминалось выше, даже в простейших случаях определяется тремя видами межмолекулярных взаимодействий: вещество - сорбент, элюент -сорбент и вещество - элюент. При применении смешанных элюентов число возможных видов взаимодействии возрастает. Однако в некоторых случаях те или иные взаимодействия преобладают. В общем случае можно сказать, что удерживание в адсорбционной ЖХ определяется специфическими или неспецифическими межмолекулярными взаимодействиями вещества как с сорбентом и с элюентом у поверхности адсорбента, так и с элюентом в объеме жидкой подвижной фазы.
Селективность к разным классам соединений оценивается по разностям изменений свободной энергии адсорбции Д(Дб) из бесконечно разбавленных растворов:
-Д(ДС) = КПп-^-,
^ст
где t'R — приведенное время удерживания исследуемого вещества; /'ст — время удерживания стандартного вещества, в качестве которого в случае силикагеля удобно использовать бензол.
Величины Д(ДС7) определяются по отношению исправленных времен удерживания соединений, полученных при одинаковых условиях, часто из одной хроматограммы; следовательно, значения Д(ДС) могут быть рассчитаны с большой точностью. По изменениям Д(ДС7) производных бензола (относительно самого бензола) можно оценить селективность той или иной хроматографической системы к данным классам соединений. Как уже было отмечено, поскольку в ЖХ элюент выполняет не только транспортную функцию, необходимо говорить о селективности системы в целом, а не о селективности сорбента.
Таблица 4.1.38
Классификация систем: сорбент - элюент - разделяемые вещества по полярности и по видам преобладающих межмолекулярных неспецифических (нс) и специфических (с) взаимодействий
Сорбент Элюент Разделяемые вещества Виды взаимодействий
Вещество - сорбент Вещество - элюент
Неполярный Неполярный Неполярные нс нс
То же Полярный То же То же То же
То же То же Полярные То же с
То же Полярный (+ спец, вещества) То же с + нс То же
Полярный Полярный То же с с
То же Неполярный (+ спец, вещества) То же То же нс
302
Новый справочник химика и технолога
Многообразие видов взаимодействий, влияющих на удерживание в ВЭЖХ, с одной стороны, затрудняет теоретический расчет вкладов тех или иных взаимодействий в общее удерживание, но, с другой стороны, когда механизм этих взаимодействий понятен, можно направленно регулировать селективность разделения. В некоторых простых случаях всегда можно выделить преобладающие взаимодействия.
В табл. 4.1.38 приведена классификация систем: сорбент - элюент - разделяемые вещества (сорбаты).
Прежде чем перейти к рассмотрению этих систем, необходимо напомнить, что следует принимать во внимание строение молекул адсорбируемых веществ, а именно: плоская или неплоская молекула анализируемого вещества, экранированы или доступны полярные функциональные группы, имеют место или нет внутримолекулярные (в частности, водородные) связи в о-положении. Все это, в конечном счете, будет определять характер ориентации молекул относительно поверхности сорбента и удерживание веществ.
Механизмы удерживания в обращенно-фазовой хроматографии
В качестве неполярных адсорбентов в жидкостной хроматографии используют в основном силикагели с привитыми углеводородными группами. В этом случае применяют полярные элюенты — в основном, водно-спиртовые. Это объясняется простотой, универсальностью применения и высокой стабильностью работы колонки и воспроизводимостью получаемых характеристик удерживания.
Предложены различные механизмы удерживания на таких колонках.
1. Предполагается, что удерживание определяется растворением, поскольку принималось, что привитые к поверхности силикагеля длинные алкильные цепи образуют обычную жидкость. Однако это представление мало правдоподобно, так как расстояние между углеводородными цепями на «щеточном сорбенте» значительно больше, чем расстояние между молекулами в жидкости. Кроме того, само представление о «щетках» неверно, так как в случае полярного элюента конформационная подвижность цепей способствует их выталкиванию и переплетению у поверхности.
2. Предполагается, что органический компонент подвижной фазы (элюента) преимущественно адсорбируется на неполярном адсорбенте, при этом на его поверхности создается новая жидкая неподвижная фаза. Тогда удерживание определялось бы распределением между подвижной фазой и новой жидкой стационарной фазой. Однако известно, что адсорбция хорошо растворимого в воде органического вещества из водного элюента ограничивается монослоем.
3. Свойства углеводородных цепей в «щеточных» сорбентах сравнивают со свойствами жидких кристаллов. Предполагается, что удерживание определяется закономерностями растворения в жидких кристаллах. Однако конформационная подвижность этих цепей при примене
нии полярных элюентов приводит к неправильному их переплетению и изменению толщины слоя.
4. Предполагается, что удерживание определяется размещением в слое алкильных радикалов, иммобилизованных к поверхности силикагеля, т. е. неспецифическим межмолекулярным взаимодействием — вещество - адсорбент. Вместе с тем в данном методе активную роль играет межмолекулярное взаимодействие — вещество - элюент, особенно в тех случаях, когда вещество полярное, так как это взаимодействие будет специфическим. В настоящее время число сторонников этих адсорбционных представлений увеличивается, тем более, что они единственно правильны в том случае, когда модифицирующий слой образован жесткими группами, например фенильными, способными совершать лишь заторможенные вибрационные колебания вокруг связей с атомами кремния поверхности силикагеля.
5. Предполагается, что механизм удерживания сложный и включает разные процессы. Вероятно, в той или иной степени это возможно в случае прививки к поверхности силикагеля конформационно-подвижных цепей. В случае же прививки жестких групп единственно возможным является предположение 4.
Представления о механизме удерживания на неспецифических адсорбентах описаны в работах К. Хорвата и др. Это так называемый сольвофобный или гидрофобный механизм удерживания. Рассматривается обратимое взаимодействие между адсорбируемым веществом и привитым углеводородным модификатором поверхности. Коэффициент емкости колонки связывается со свойствами углеводородных цепей и составом элюента. Сольвофобная теория разработана для низких коэффициентов распределения веществ в растворителях. Процесс растворения в этом случае — двухступенчатый. Первая ступень — образование полости в растворителе (элюенте) по форме и размерам входящей молекулы растворенного вещества. Изменение свободной энергии A(AG), требуемой для этого процесса, есть произведение площади поверхности полости и поверхностного натяжения растворителя (элюента). Вторая ступень — размещение молекул в полости. Энергия этого процесса определяется ван-дер-ваальсовым и электростатическим взаимодействиями. Окончательное выражение принимает в расчет различие свободного объема между конденсированной и газовой фазами.
Были сделаны попытки использования теории регулярных растворов Гильденбрандта для объяснения механизма удерживания. Установлено, что наблюдается линейная зависимость удерживания сорбатов от логарифма молярной концентрации ацетонитрила в подвижной фазе:
Igk = b + plgc,
где Ь, р — константы; с — молярная концентрация ацетонитрила в подвижной фазе. Коэффициенты корреляции таких зависимостей — порядка 0,99.
Однако кроме взаимодействия — вещество - элюент необходимо учитывать взаимодействие вещества и элюен
Аналитическая хроматография и капиллярный электрофорез
303
та с адсорбентом. Удерживание на неполярных адсорбентах из водных элюентов повышается с повышением мольного объема адсорбируемого вещества, с увеличением различий в полярности между веществом и элюентом (водой) и с уменьшением различий в полярности между адсорбируемым веществом и адсорбентом.
Неполярный сорбент - неполярный элюент - неполярные разделяемые вещества
В этой системе удерживание определяется только неспецифическими взаимодействиями, и они близки для членов гомологических рядов. Разделения на многих сорбентах практически нет. На углеродных адсорбентах, которые сильно адсорбируют углеводороды, можно достичь различия в удерживании алканов за счет различий в упаковке адсорбатов на поверхности угля. Установлено незначительное различие в удерживании алканов на углеродном адсорбенте, значение A(AG') между гомологами в этом случае мало (80 Дж-моль”1).
Полярный абсорбент - неполярный элюент - полярные разделяемые вещества
В жидкостной хроматографии на полярном адсорбенте (например, на силикагеле с гидроксилированной поверхностью) из неполярного углеводородного или слабополярного элюента удерживание полярных молекул, не имеющих длинных углеводородных цепей, преимущественно определяется специфическими межмолекулярными взаимодействиями — сорбат - сорбент. Неспецифическое взаимодействие с сорбентом метильных и метиленовых групп сорбата в этом случае не играет существенной роли, так как эти группы имеются и в углеводородном элюенте. Вклад неспецифического взаимодействия — вещество -элюент в этом случае также мал по сравнению с вкладом специфического взаимодействия — адсорбент - вещество.
Поскольку специфическое взаимодействие в этом случае включает образование направленных водородных связей или является электростатическим ориентационным, удерживание, кроме протонодонорной способности или значения дипольного и квадрупольного момента молекулы, в сильной степени определяется доступностью полярных групп поверхности для полярных групп вещества и возможностью нужной для специфического взаимодействия ориентации его молекул относительно полярных групп на поверхности адсорбента. В этом варианте наблюдается высокая селективность разделения изомеров и других соединений, отличающихся как пространственным строением, так и распределением электронной плотности в молекуле. Например, жидкостно-адсорбционная хроматография на полярном адсорбенте, в частности на силикагеле с гидроксилированной поверхностью из неполярного или слабополярного элюента позволяет хорошо разделять о-, м- и «-изомеры ароматических соединений, содержащих полярные группы в этих положениях.
Селективность к таким изомерам в ЖХ значительно выше селективности в ГХ на том же силикагеле. Это связано с тем, что в ЖХ на полярном адсорбенте разделение
происходит преимущественно за счет различий в специфических взаимодействиях между полярными группами разделяемых веществ и адсорбента, так как неспецифические взаимодействия разделяемых молекул и элюента с адсорбентом не сильно различаются. В этом случае большее влияние оказывает ориентация разделяемых молекул, а также экранирование полярных функциональных групп его молекул неполярными группами, в результате чего удерживание рр/ио-замещенных производных обычно значительно меньше.
Большую роль играет возможность образования в молекулах разделяемых веществ внутримолекулярной водородной связи. Если две полярные функциональные группы в o/w/о-положении способны образовывать внутримолекулярную водородную связь, то удерживание на полярном (специфическом) адсорбенте уменьшается, так как ослабляется специфическое взаимодействие — вещество - адсорбент. Изменение A(AG) для производных фенола на силикагеле с гидроксилированной поверхностью при элюировании смесью гексан - хлороформ - изопропанол составляет для пирокатехина — 530, пирогаллола — 640, резорцина — 3800, гидрохинона — 4940 и флороглюцина — 8660 Дж-моль”1 соответственно. Влияние внутримолекулярной водородной связи на уменьшение A(AG') можно оценить по разности этих величин для гидрохинона и пирокатехина, флороглюцина и пирогаллола.
Из колонки с силикагелем бензол и его производные элюируют в следующем порядке: бензол, толуол, хлорбензол, анизол, нитробензол, метиловый эфир бензойной кислоты, бензонитрил, бензальдегид, бензилбензоат, ацетофенон, бензиловый спирт, анилин, фенол.
Этот вариант жидкостной хроматографии характеризуется следующими особенностями:
а) высокой селективностью разделения соединений, отличающихся природой и числом полярных функциональных групп; при разделении соединений, сильно различающихся по полярности, часто необходимо градиентное элюирование;
б) высокой селективностью разделения молекул, различающихся геометрическим строением, в том числе и изомеров;
в) гомологи в этом варианте обычно не разделяются, однако этот недостаток может стать достоинством, так как позволяет проводить групповое разделение насыщенных алканов, различных ароматических углеводородов и полярных соединений в нефтяных фракциях;
г) в этом варианте адсорбционные характеристики полярных адсорбентов могут изменяться во времени за счет адсорбции воды или других сильнополярных веществ, присутствующих в элюенте.
При постоянной температуре и влажности состав элюента остается постоянным, равновесие между элюентом и адсорбентом сохраняется, и колонка работает стабильно. Однако при изменении температуры и состава элюента характеристики удерживания изменяются. В случае разделения о-, м- и «-изомеров (производных бензола) возможный порядок их выхода определяется изменением распре
304
Новый справочник химика и технолога
деления электронной плотности в бензольном кольце в зависимости от природы заместителей (заместители, отдающие или оттягивающие электронную плотность), изменением поляризуемости, ориентацией молекул изомеров относительно поверхности адсорбента, экранированием полярных функциональных групп неполярными группами и возможностью образования внутримолекулярной водородной связи между соседними полярными функциональными группами.
Общие закономерности удерживания на неспецифических (неполярных) сорбентах
При адсорбции веществ на неполярных адсорбентах из сильнополярных элюентов, чаще всего — водно-спирто-вых смесей (так называемый обращенно-фазовый вариант хроматографии), сильнее адсорбируются молекулы веществ, содержащих алкильные неполярные циклы или группы. В основном эти вещества удерживаются на неполярной (гидрофобной) поверхности за счет адсорбции неполярных частей их молекул, т. е. за счет неспецифического взаимодействия. Полярные группы молекул сорбата только уменьшают удерживание, так как они взаимодействуют с полярными группами молекул элюента, а это взаимодействие ослабляет межмолекулярное взаимодействие разделяемых молекул с сорбентом и облегчает их возвращение в объем полярного элюента. Таким образом, в этом случае удерживание в основном определяется, во-первых, неспецифическим взаимодействием — вещество - сорбент и, во-вторых, специфическим взаимодействием — вещество - элюент, причем последнее уменьшает удерживание. Этот молекулярный механизм удерживания надо иметь в виду, так как распространенный термин «обращенно-фазовая хроматография» не передает сущности явления, тем более, что органические вещества удерживаются и на гидроксилированной поверхности силикагеля из водных растворов.
ЖХ на неполярных сорбентах из полярных элюентов имеет следующие особенности.
1. Хорошо разделяются гомологи, так как их молекулы удерживаются за счет неспецифических межмолекулярных взаимодействий с неполярными группами поверхности неспецифического сорбента в тем большей степени, чем больше их углеводородная часть.
2. Слабо удерживаются сильнополярные вещества, они элюируют раньше неполярных; если на полярном сорбенте сильнополярные вещества удерживаются сильно и для их десорбции необходимы ступенчатое или градиентное элюирование с увеличением полярности элюента, то на неполярном сорбенте подобной трудности нет, сильнополярные вещества выходят достаточно быстро.
3. Сильно проявляется межмолекулярное взаимодействие — вещество - элюент. Изменяя природу элюента, можно иногда в большей степени изменить удерживаемые объемы и селективность разделения, чем при изменении природы сорбента; в некоторых случаях взаимодействие вещество — элюент усиливают за счет добавки в элюент специальных веществ (в частности, растворов солей сереб
ра) для увеличения селективности разделения ароматических и непредельных соединений за счет образования комплексов с ионом серебра; в этом случае увеличивается селективность при общем уменьшении удерживания.
4. Колонки с неполярным адсорбентом стабильны, поскольку в них не накапливается адсорбируемая вода. При правильной эксплуатации они могут работать сколь угодно долго без регенерации.
5. Колонки с неполярным адсорбентом имеют, тем не менее, меньшую эффективность по сравнению’ с аналогичными по размерам колонками с полярным адсорбентом с такими же размерами зерен, удельной поверхностью и пористостью, так как вязкость полярных элюентов (в частности, водно-спиртовых смесей) значительно выше, чем у таких углеводородов, как гексан.
Колонки с неполярными адсорбентами представляют интерес для анализа многих биологических систем, так как процессы адсорбции, происходящие на таких адсорбентах, особенно на полученных направленным химическим модифицированием поверхности, близки к процессам, происходящим в клетках живых организмов: в обоих случаях происходит адсорбция из водных сред. На полярных адсорбентах можно реализовать положительную адсорбцию и удерживание из водных растворов даже смешивающихся с водой веществ.
Неполярный адсорбент - полярный элюент — неполярные или слабополярные разделяемые вещества
Примеров разделения гомологов на неполярных адсорбентах из полярных элюентов описано много. Представляет интерес удерживание гомологов и изомеров полиметил- и моноалкилбензолов. В табл. 4.1.39 приведены измеренные для полиметил- и моноалкилбензолов на колонке с силани-зированным силикагелем с элюентом изопропанол - вода времена удерживания и факторы емкости к (равные объемным отношениям). Исправленные времена удерживания tR, факторы емкости колонки k = tR/tcKWi и относительные (к бензолу) изменения свободной энергии при адсорбции Д(Дб) получены на колонке размером 15 х 0,6 см, заполненной силанизированным триметилхлорсиланом силикагелем КСС-4, элюент изопропанол - вода (2:3), скорость потока 2,4 см3 мин1 исправленного времени удерживания изучаемых веществ к времени удерживания практически неадсор-бируемого вещества.
В отличие от адсорбции на гидроксилированной поверхности силикагеля, удерживание в этом случае с ростом числа атомов углерода в молекуле возрастает как в ряду поли-метилбензолов, так и в ряду моно-н-алкилбензолов. Поскольку при использовании силанизированного силикагеля и смеси изопропилового спирта и воды в качестве элюента удерживание преимущественно определяется неспецифическим межмолекулярным взаимодействием вещество - адсорбент и специфическим межмолекулярным взаимодействием вещество - элюент, н-алкилбензолы удерживаются сильнее соответствующих полиметилбензо-лов с тем же числом атомов углерода в молекуле.
Аналитическая хроматография и капиллярный электрофорез
305
Группа —СН3 является донорным заместителем, поэтому электронная плотность на бензольном кольце в по-лиметилбензолах увеличивается. В случае .м-алкилбен-золов метиленовые ^СН2 и метильные — СН3 группы в углеводородной цепи очень слабо влияют на распределение электронной плотности в бензольном кольце, поэтому электронная плотность в бензольном кольце изменяется слабо. В связи с этим специфическое межмолекулярное взаимодействие — адсорбат - элюент для полиметилбен-золов больше, чем для соответствующих moho-jw-алкилбензолов. Неспецифическое же межмолекулярное взаимодействие — сорбат - адсорбент на поверхности си-ланизированного силикагеля увеличивается с числом метильных и метиленовых групп в молекуле адсорбата, следовательно, для изомерных аналогов полиметилбензолов и н-алкилбензолов оно не должно сильно различаться, несмотря на зависимость от ориентации молекулы относительно поверхности. Поскольку специфическое межмолекулярное взаимодействие — вещество - элюент уменьшает общее удерживание адсорбатов в колонне с неполярным адсорбентом из полярного элюента, полиметилбензолы удерживаются слабее моно-.и-ал кил бензолов.
Таблица 4.1.39
Параметры удерживания ароматических углеводородов
Ароматические углеводороды с к -Д(ДС) Дж • моль 1
Бензол 204 2,72 —
Толуол 254 3,39 545
Этилбензол 329 4,38 1180
Изопропилбензол 382 5,10 1555
н-Пропилбензол 415 5,53 1758
тр ет-Бут илбензол 493 6,57 2185
втор-Бутил бензол 507 6,76 2255
н-Бутилбензол 535 7,14 2390
Пентилбензол 681 9,08 2985
о-Ксилол 303 4,04 980
jw-Ксилол 317 4,29 ИЗО
и-Ксилол 328 4,37 1175
1,2,3-Триметил-бензол 355 4,74 1375
1,3,5-Триметил-бензол 390 5,21 1610
1,2,3,4-Тетраметил-бензол 400 5,33 1665
1,2,3,5-Тетраметил-бензол 440 5,86 1900
1,2,4,5-Тетраметил- 4 бензол 474 6,32 2090
' т Пентаметилбензол 521 6,96 2330
Гексаметилбензол 598 7,98 2665
Зависимость Igk от числа атомов углерода п в молекуле этих классов соединений линейная. Аналогичная зависимость получена на силикагеле с гидроксилированной по
верхностью с элюентом гексаном. Зависимость Igk от и для моно-к-алкилбензолов на неполярном сорбенте также близка к линейной. Это показывает, что соответствующие значения A(AG) аддитивны по группам ^СН2. Вклад в A(AG’) одной группы (—СНз) составляет около 600 Дж-моль1. Эта величина характеризует неспецифическое межмолекулярное взаимодействие группы ^СН2 молекулы м-алкилбензола с этим адсорбентом.
Соответствующая зависимость для .некоторых изомеров диметилбензолов отклоняется от линейной. К линейной зависимости A(AG) от п ближе изомеры, в которых заместители (—СНз) находятся в орто-положении. Вклад группы ^СН2 в A(AG) для полиметилбензолов в этом случае составляет около 440 Дж-моль1.
Порядок удерживания изомеров полиметилбензолов, а также изопропилбензола, .м-пропилбензола и соответственно трет- втор- и н-бутилбензолов противоположен порядку удерживания этих соединений на силикагеле с гидроксилированной поверхностью из элюента - гексана. Это связано с тем, что в случае гидроксилированной поверхности силикагеля, как было отмечено выше, удер-ивание определяется специфическим межмолекулярным взаимодействием — адсорбат - адсорбент и неспецифическим межмолекулярным взаимодействием — адсорбат -элюент, а в случае силанизированной поверхности силикагеля — неспецифическим межмолекулярным взаимодействием адсорбат - адсорбент и преимущественно специфическим межмолекулярным взаимодействием — адсорбат -элюент.
Установление вкладов, вносимых в термодинамические характеристики удерживания отдельными звеньями и функциональными группами молекул и олигомеров, весьма важно, поскольку они, как уже отмечалось выше, должны составить экспериментальную основу развития полу-эмпирической теории удерживания в жидкостной хроматографии. Отметим, что определение таких вкладов в случае белковых аминокислот позволило предсказывать времена удерживания (при определенных pH) ряда пептидов.
Неполярный адсорбент - полярный элюент -полярные разделяемые вещества
Удерживание веществ за счет неспецифического взаимодействия — вещество - адсорбент и специфического взаимодействия — вещество - элюент возможно в системе: неполярный адсорбент - полярные разделяемые вещества - сильнополярный элюент.
При хроматографии на неполярном адсорбенте с использованием полярного элюента соединений (адсорбатов), имеющих полярные функциональные группы, значительную роль играет межмолекулярное взаимодействие этих соединений с полярным элюентом. Но межмолекулярное взаимодействие является специфическим и часто определяется возникновением водородных связей между полярными функциональными группами молекул полярных соединений и полярными группами элюента. Доля специфического межмолекулярного взаимодействия полярного соединения с элюентом в общем межмолекуляр
306
Новый справочник химика и технолога
ном взаимодействии данного соединения с адсорбентом и элюентом может быть значительной.
Влияние межмолекулярного взаимодействия производных ароматических углеводородов с полярным элюентом на удерживаемые объемы и селективность разделения на силанизированном силикагеле легче всего проследить на примере производных, отличающихся природой полярных функциональных групп. Влияние межмолекулярного взаимодействия с полярным элюентом можно оценить по бензолу или толуолу и их производным с полярными функциональными группами в иара-положении.
На удерживание анализируемых веществ в ВЭЖХ влияют также структура и состав молекул элюента.
4.1.8.3. Общие требования к элюентам в ВЭЖХ
Большое влияние на удерживание и селективность разделения в ВЭЖХ оказывает природа элюента. Для каждой разделяемой смеси помимо адсорбента надо выбрать определенный по природе и составу элюент, чтобы достигнуть оптимальной степени разделения: полного разделения за возможно короткое время.
Для выбранного сорбента (по геометрии зерен, пор и химии поверхности) от природы и состава элюента зависят все основные величины, определяющие степень разделения R: селективность системы адсорбент - элюент а, факторы емкости анализируемых веществ к, эффективность колонки. Природа элюента влияет на работу детектирующих систем.
К элюентам для ВЭЖХ предъявляются следующие требования. Элюент должен:
• обеспечить достаточно высокую селективность за приемлемое время; для малокомпонентных смесей значения к должны лежать в пределах 2 < к < 5, а для многокомпонентных — в пределах 0,5 < к < 20;
• быть маловязким (не более 0,3-0,5 сантипуаза), чтобы обеспечить небольшое сопротивление потоку и высокую эффективность;
• растворять анализируемые вещества;
• быть дешевым, доступным и безопасным в работе.
Элюент не должен:
• быть химически активным (не должен химически реагировать ни с анализируемыми веществами, ни с адсорбентом);
• содержать сильносорбирующихся примесей, в частности воду и другие полярные вещества при разделении на полярных адсорбентах;
• регистрироваться детектором.
Способы очистки элюентов приведены в табл. 4.1.40. Некоторые элюенты применяют только в свежеперегнан-ном виде, пока не накопились продукты их окисления.
Основные элюенты и их классификация
Влияние элюента на удерживание и селективность анализируемых веществ проявляется двумя путями. Молекулы анализируемых веществ в большинстве случаев вынуждены вытеснять молекулы элюента с поверхности адсорбента, затрачивая на это часть своей энергии. Чем
сильнее молекулы элюента адсорбируются на поверхности, тем слабее адсорбируются молекулы анализируемых веществ. Взаимодействие — вещество - элюент направлено в объем элюента, оно отрывает молекулы анализируемых веществ от поверхности адсорбента. Таким образом, это взаимодействие также уменьшает удерживание анализируемых веществ.
Таблица 4.1.40
Очистка растворителей-элюентов, для жидкостной хроматографии
Типы очистки элюентов Способ очистки
Удаление влаги Пропускание через слой AI2O3 или цеолита
Удаление полярных примесей Пропускание через колонку с силикагелем или А12О3
Удаление продуктов окисления, стабилизирующих добавок и т. д. Перегонка либо фронтальная хроматография
Удаление олефинов из алканов Пропускание через колонку с силикагелем, пропитанным AgNO3
Удаление кислорода Длительное барботирование через элюент потока чистого гелия
Удаление НС1 и НВг и др. из галогензамещенных элюентов Пропускание через колонку с А12О3 в щелочной форме
Удаление микрочастиц суспензии Пропускание через метало-керамические или металлические фильтры с диаметром отверстия 0,5 мкм
Элюирующая способность различных растворителей определяется суммой этих взаимодействий. По элюирующей способности все элюенты располагают в элюотропные ряды, которые для полярных и неполярных элюентов различаются. Выделяют следующие типы взаимодействия молекул элюента с молекулами анализируемых веществ: дисперсионные, диполь-дипольные, диполь-ионные, водородные связи.
Под диполь-ионными взаимодействиями понимают взаимодействие ионов анализируемых веществ с элюентами, характеризующимися высокой диэлектрической постоянной (вода, спирты и т. п.). В результате этих взаимодействий возникают электростатические притяжения ионов и молекул элюента.
Для оценки полярности и элюирующей способности элюентов используют параметр полярности Р, параметр адсорбционной силы растворителя, параметр растворимости Гильденбрандта. В зависимости от типов взаимодействий элюенты классифицируют также по группам, в частности особо выделяются элюенты — акцепторы протонов (эфиры), доноры протонов (хлороформ), доноры-акцепторы (спирты).
Аналитическая хроматография и капиллярный электрофорез
307
Для НФХ в основном применяют в качестве элюентов н-алканы (пентан, гексан, гептан) как в чистом виде, так и с добавками различных полярных веществ (метанол, этанол, изопропанол, простые и сложные эфиры, хлороформ и другие полярные вещества).
Для ОФХ применяют следующие растворители (около растворителей указана относительная элюирующая способность растворителей): вода — 0; метанол — 3,0; ацетонитрил — 3,1; ацетон — 3,4; диоксан — 3,5; этанол — 3,6; изопропанол — 4,2; тетрагидрофуран — 4,4. В табл. 4.1.41 приведены физические характеристики этих элюентов. Чаще всего в ОФХ в качестве элюентов используют водоспиртовые смеси, в некоторых случаях применяют и неводные смеси на основе ацетонитрила и тетрагидрофурана.
В табл. 4.1.42 указаны основные свойства элюентов для ВЭЖХ. Элюирующая способность растворителей меняется от природы адсорбента (см. табл. 4.1.43). По селективности элюенты разделяются на группы, приведенные в табл. 4.1.44).
В последнее время в качестве элюента использовали воду при разделении на гидроксилированном силикагеле. Вода как элюент представляет интерес с экономической точки зрения (доступна, дешева), а также с точки зрения безопасности (нетоксична). При использовании воды в качестве элюента появляется возможность более эффективно применить пламенный ионизационный и другие типы детекторов. В качестве элюентов в ВЭЖХ с ИКС-детек-тором с Фурье-преобразованием применяли дейтерииро-ванные соединения (CD3CN, D2O).
Таблица 4.1.41
Свойства элюентов для ОФХ
Принятые обозначения. nRi — коэффициент рефракции при 25 °C; УФ — нижний предел применения в УФ-области; р — плотность при 20 °C; ц — вязкость при 20 °C; £ — диэлектрическая проницаемость; ц -— дипольный момент; v — поверхностное натяжение.
Название УФ, нм nRi р, (кг-м 3)-10 3 т), сП £ Р, Д v, Дина-см1
Ацетонитрил 190 1,342 0,787 0,358 38,8 3,37 29
Ацетон 330 1,357 0,791 0,322 20,7 2,72 23
Вода 170 1,333 0,998 1,00 78,5 1,84 73
Этанол 205 1,359 0,789 1,19 24,5 1,68 22
Метанол 205 1,326 0,792 0,584 32,7 1,66 22
Изопропанол 205 1,375 0,785 2,39 19,9 1,68 21
Диоксан 215 1,420 1,034 1,26 2,21 0,45 33
Тетрагидрофуран 210 1,404 0,889 0,51 7,58 1,70 27,6
Примечание. Коэффициент перевода в СИ 1 сП = 10 3 Па с; 1 Д = 3,33564-10 30 Кл-м; 1 Дина-см 1 = 10 3 Н-м *.
Таблица 4.1.42
Свойства растворителей для ВЭЖХ
Растворитель Температура кипения, °C Плотность, (кг-м“3)-10 3 Вязкость (25 °C), сП Коэффициент преломления nw Предел прозрачности для УФ-свет, нм Элюирующая сила, е°
на оксиде алюминия на силикагеле
Ацетон 56 0,79 0,30 1,356 330 0,56
Ацетонитрил 82 0,78 0,34 1,341 190 0,65 0,50
Бензол 80 0,88 0,60 1,498 280 0,32
Вода 100 1,00 0,89 1,333
Гексан 69 0,66 0,30 1,372 190 0,01 0,01
Гептан 98 0,68 0,40 1,385 195 0,01 0,01
Дибутиловый эфир 142 0,77 0,64 1,397 220
Диметилформамид 153 0,94 0,80 1,428 268
Диоксан 101 1,03 1,2 1,420 215 0,56
Дихлорэтан 83 1,25 0,78 1,442 228 0,44
Диэтиловый эфир 35 0,71 0,24 1,350 218 0,38
Изооктан 99 0,69 0,47 1,389 197 0,01
Изопропиловый эфир 68 0,73 0,38 1,365 220 0,25 0,01
308
Новый справочник химика и технолога
Продолжение таблицы 4.1.42
Растворитель Температура кипения, °C Плотность, (кг-м3)-103 Вязкость (25 °C), сП Коэффициент преломления и20 "D Предел прозрачности для УФ-свет, нм Элюирующая сила, е°
на оксиде алюминия на силикагеле
Метанол 65 0,79 0,54 1,326 205 0,95 0,34
Метиленхлорид 40 1,33 0,41 1,421 233 0,42 0,7
Метилэтилкетон 80 0,86 0,38 1,376 329 0,51 0,32
Метилцеллозольв 125 0,97 1,60 1,400 210
Пентан 36 0,63 0,22 1,355 205
1-Пропанол 97 0,80 1,9 1,385 205 0,82
2-Пропанол 82 0,79 1,9 1,384 205 0,82
Тетрагидрофуран 66 0,89 0,46 1,405 212 0,57 0,44
Толуол НО 0,87 0,55 1,494 285 0,54
Триэтиламин 89 0,73 0,36 1,398
Уксусная кислота 118 1,05 1,1 1,370 230 0,40 0,26
Хлороформ 61 1,49 0,53 1,443 245 0,04
Циклогексан 81 0,78 0,90 1,423 200
Четыреххлористый углерод 77 1,60 0,90 1,457 265 0,18
Этанол 78 0,79 1,08 1,359 210 0,88
Этилацетат 77 0,90 0,43 1,370 256 0,58 0,38
Этиленгликоль 182 1,1 6,5 1,431 210 1,Н
Примечание. В ряде случаев в таблице отсутствуют значения элюирующей силы для некоторых растворителей. Однако, получаемая из построенных элюотропных рядов качественная информация полезна: каждый последующий член ряда обладает большей элюирующей силой, чем предыдущий.
Таблица 4.1.43
Элюирующая сила растворителей
Растворитель Сорбент
силикагель оксид алюминия сажа
Гексан 0,01 0,01 0,10
Бензол 0,32 0,20
Хлористый бутил 0,20 0,26 0,13
Хлороформ 0,26 0,40 0,18
Метиленхлорид 0,32 0,42 0,13
Изопропиловый эфир 0,34 0,28
Этил ацетат 0,38 0,58 0,13
Тетрагидрофуран 0,44 0,57 0,14
Ацетонитрил 0,50 0,65 0,04
Метанол 0,70 0,95 0,00
Таблица 4.1.44
Характеристика различных групп селективности
Группа Класс веществ
I Алифатические простые эфиры, амины
II Алифатические спирты
III Пиридин, тетрагидрофуран, амиды (кроме формамида), эфиры гликолей, сульфоксиды
IV Гликоли, уксусная кислота, формамид
Продолжение таблицы 4.1.44
Группа Класс веществ
V Метиленхлорид, этиленхлорид
VI Сульфоны, нитрилы
Via Алифатические кетоны и сложные эфиры, диоксан
VII Ароматические углеводороды, нитросоединения
VIII Фторированные спирты, вода, хлороформ
4.1.8.4. Адсорбенты для ВЭЖХ
Общие требования к адсорбентам
Требования к адсорбентам для ВЭЖХ отличаются от требований к адсорбентам для ГАХ, это связано со многими причинами. Во-первых, в ВЭЖХ подвижная фаза -элюент всегда адсорбируется, поэтому требования к однородности поверхности снижаются по сравнению с требованиями в ГАХ. Во-вторых, массообмен в жидкости происходит значительно медленнее, чем в газовой фазе, поэтому тонкопористые адсорбенты мало пригодны к высокоэффективной ВЭЖХ из-за медленности массообмена. В-третьих, для работы при высоких давлениях необходимо, чтобы зерна адсорбентов для ВЭЖХ обладали значительно более высокой механической прочностью, а форма зерен по возможности должна быть сферической.
Аналитическая хроматография и капиллярный электрофорез
309
Влияние геометрической структуры адсорбентов на удерживание в ВЭЖХ
В ВЭЖХ применяют адсорбенты разной геометрической структуры (с разной удельной поверхностью, разным диаметром пор, объемом пор и разным распределением пор по эффективным диаметрам). Однако влияние геометрической структуры на удерживаемые объемы, селективность разделения и эффективность колонок изучено недостаточно. Была сделана попытка оптимизации геометрической структуры для достижения наилучшей степени разделения и показано, что лучшее разделение достигается на силикагеле с S = 400 м2т“1 и <Уср = 10 нм.
Для жидкостной адсорбционной хроматографии важна общая поверхность адсорбента в колонке А = Sm (S— удельная поверхность адсорбента в колонке, т — масса адсорбента в колонке). Исправленное время удерживания пропорционально длине колонки L и произведению Sm. В одной и той же по природе системе время удерживания можно менять, изменяя либо L, либо S. Величина S влияет на селективность разделения: на колонке длиной 10 см, заполненной силикагелем с S = 470 м2т-1, фенантрен и антрацен не разделяются, при 5 = 590 mV1 (средний размер пор dcp = 6 нм) разделяются частично, а при S = 800 mV1 (с^ = 4 нм) разделяются полностью. Однако как в ГХ, так и в ЖХ селективность при одной и той же удельной поверхности может зависеть от размеров пор адсорбента.
Были исследованы зависимости удерживаемых объемов от удельной поверхности в интервале 80-700 м2т-1 и от среднего диаметра пор силикагелей в интервале 2-100 нм.
Удерживаемые объемы производных бензола на колонках с силикагелями с близкими поверхностями (около 540 м2тч), но с разным средним диаметром пор (различие в 2,5 раза) различаются, причем для более тонкопористого силикагеля удерживаемые объемы меньше. Эффективность колонок для тонкопористого силикагеля также меньше. Данные эффекты сохранились и при повышенных температурах как в нормально-, так и в обращенно-фазовом вариантах хроматографии. Эти явления можно связать с медленностью массообме-на в тонких порах, поэтому силикагели с порами размером менее 4 нм применять в ВЭЖХ нецелесообразно.
В настоящее время коммерческие силикагели выпускаются со средними размерами пор от 4 до 200 нм. Для аналитических разделений в ВЭЖХ используются силикагели с размерами пор 6-12 нм. Для разделений больших молекул предпочтительнее силикагели с размерами пор 20-30 нм.
Роль химической природы поверхности адсорбента
Для ВЭЖХ разработан и выпускается достаточно широкий ассортимент адсорбентов, более 50 фирм производят около 200 наименований. Однако многие адсорбенты разных фирм близки по химической природе поверхности и отличаются только названиями. Перечень адсорбентов, выпускаемых для различных вариантов жидкостной хроматографии, приведен в табл. 4.1.45. Чаще всего применяют в ВЭЖХ чистые силикагели и силикагели с привитыми неполярными и полярными функциональными группами (табл. 4.1.46).
В ВЭЖХ в качестве адсорбентов чаще всего используются следующие материалы: силикагели — 75%, полимеры (полиметакрилаты, полистиролдивинилбензолы, полиэтиленгликоли, целлюлозы и др.) — 20%, пористые углеродные адсорбенты на основе графитированной сажи, оксид циркония, гидроапатиты — 4%, оксид алюминия (нейтральная, кислая) — 1%.
Таблица 4.1.45
Относительная доля применения разных методов ВЭЖХ на разных сорбентах
Методы хроматографии Процент пользователей
Обращенно-фазовый 50,4
С18 24
с8 15,9
Фенил 7,1
с4 2,3
Ci-Q 1,1
Нормально-фазовый 24,1
8,9
Силикагель 8,5
nh2- 4,7
Диол- 2
Ионообменная и ионная 14
Анионы 7,4
Катионы 6,6
Эксклюзионная 6,7
Водная 3,5
Неводная 3,2
Хиральная 2,8
Гидрофобная 1,1
Прочие 1,1
Таблица 4.1.46
Наиболее распространенные сорбенты для ВЭЖХ и ИХ на основе силикагеля
Неподвижные фазы Формула
Октадецил С18 < Г3 \ О— Si— (СН2)17СН3 ' СН3
Октил С8 < Г3 У О— Si— (СН2)7СН3 \ сн3
Гексил С6 / сн3 J> о— Si— (СН2)5СН3 ' сн3
Метил Ci / сн3 О—Si—СН3 \ СН3
310
Новый справочник химика и технолога
Продолжение таблицы 4.1.46
Неподвижные фазы Формула
Фенил- / СН3 , \ СН3
Нитрил- / СИз У О— Si— (CH2)3CN ' СН3
Амино- У (У^ Si— (CH2)3NH2 <-сг^
Анионообменник У <У^ Si— СН2СН2СН2Н+(СН3)3СГ
Катионообменник У (У^ Si— CH2CH2CH2SO3H
Силикагели
Адсорбционная активность силикагелей сильно зависит от термической обработки (300-900 °C), вызывающей уменьшение поверхностной концентрации гидроксильных групп.
Предельно гидроксилированная поверхность силикагеля содержит 8,7 ± 0,2 ммольм2 гидроксильных групп или четыре гидроксильные группы на площадке в 1 нм. Поверхность чистого гидроксилированного силикагеля должна быть слабокислой, pH водной вытяжки частиц силикагеля ~ 5. Однако pH водных вытяжек выпускаемых силикагелей сильно различается: от 3,9 до 9,9. Значение pH поверхности силикагелей сильно влияет на удерживание полярных веществ, особенно веществ основного или кислого характера. Для оптимизации селективности разделения необходимо регулировать pH поверхности силикагеля.
Среди неполярных адсорбентов наиболее популярны силикагели с химически привитыми углеводородными группами — н-алкильными (адсорбенты типа молекулярных «щеток») и фенильными. Исследовано влияние длины углеводородной цепи, модифицирующей поверхность адсорбента, на селективность щеточных адсорбентов, с целью выбора оптимальной длины углеродной цепи. Было показано, что существенного различия между селективностью разделения на адсорбентах с короткими и длинными цепями нет, однако с увеличением длины цепи увеличивается фактор емкости. При изучении влияния длины цепи с числом атомов углерода от Q до С18 на удерживание было показано, что фактор емкости возрастает до определенной длины цепи, выше которой остается постоянным. Для данного адсорбируемого вещества имеется «критическая» длина цепи, не зависящая от состава элюента. Она возрастает с ростом размеров молекул адсорбируемого вещества от минимальной величины (для С6) до максимальной (С14).
На удерживание веществ влияет содержание углерода в привитых цепях. Максимальное содержание углерода для С22 = 19,6%, для Cis = 17,2% и для С8 = 9,6% от массы насадки. Для сравнения рекомендуется стандартизация различных связанных фаз по количеству углерода на единицу объема колонки. С увеличением длины цепи от Ci до С22 увеличивается относительное удерживание и емкость колонки. Это всегда справедливо для неполярных веществ, для полярных веществ может быть наоборот. Необходимо отметить, что высокая емкость к необходима для многокомпонентных систем. Для простой смеси предпочтительны низкие значения к, чтобы увеличить скорость анализа. Силикагели с длинными цепями (С8-С[8) рекомендуется применять для разделения неполярных веществ, а силикагели с короткими цепями (С1-С4) — для разделения полярных веществ.
Проведено сравнение удерживания на адсорбентах с привитыми к поверхности линейными и разветвленными цепями при применении разных элюентов. Было найдено, что удерживание веществ на адсорбентах с разветвленными цепями значительно меньше, чем на адсорбентах с не-разветвленными цепями.
Для разделения сердечных гликозидов из водноспиртового элюента оптимальным как в отношении селективности, так и эффективности колонки является модифицирование силикагеля дифенилхлорсиланом. Концентрация привитых модифицирующих групп разной природы и конформационной подвижности количественно изучалось химическими и спектроскопическими методами.
В результате спектроскопических, химических и адсорбционных исследований химического модифицирования поверхности кремнезема было показано, что, в зависимости от концентрации силанольных групп на поверхности до модифицирования, от условий модификаций и природы модификатора, под слоем привитых алкилсилильных групп остаются в том или ином количестве поверхностные силанольные группы, из-за стерических препятствий не вошедшие в реакцию с модификатором. Наличие этих силанольных групп вызывает повышенную адсорбцию полярных молекул малых размеров, прежде всего молекул воды и метанола, в то время как теплота адсорбции крупных органических молекул остается меньше теплоты конденсации.
Подробно изучено влияние длины углеводородной цепи неспецифического адсорбента на удерживание и селективность разделения н-алканов, полиядерных ароматических соединений (ПАС) и алициклических соединений. Для изучения были взяты адсорбенты, химически модифицированные привитыми углеводородными цепями Сь С8 и С18. Удерживание ПАС и алканов с водно-метанольным элюентом значительно больше на адсорбенте с Ci8, чем на адсорбентах с Q и С8. Это различие в удерживании меньше при работе с индивидуальным органическим элюентом, содержащим ацетонитрил. Однако изучение влияния длины углеводородной цепи, привитой к поверхности силикагеля, нельзя рассматривать, как указывалось выше, отдельно от изучения влияния поверхностной концентрации привитых групп.
Аналитическая хроматография и капиллярный электрофорез
311
Изменение доли органического компонента в элюенте сильно изменяет селективность разделения ПАС и насыщенных углеводородов. Повышение концентрации органического компонента в элюенте дает большее различие в удерживании жестких плоских молекул ПАС относительно неплоских на модифицированном адсорбенте с Ci8 по сравнению с адсорбентами с Ci и С8. Было обнаружено, что адсорбенты с С- и С¥ близки по характеристикам удерживания. Селективность разделения на адсорбенте с Ci8 для некоторых смесей несомненно выше. Однако селективность в целом определяется не только длиной цепей, привитых к поверхности адсорбента, но и природой элюента и, как отмечено выше, поверхностной концентрацией привитых групп.
При изучении адсорбции органических веществ из водных растворов было обнаружено, что углеводородные цепи в «щеточных» адсорбентах взаимодействуют друг с другом за счет дисперсионных сил, что уменьшает адсорбцию и удерживание органических веществ. При добавлении в элюент метанола или других органических соединений органические компоненты раздвигают цепи модификатора, дисперсионные взаимодействия между этими цепями уменьшаются. В этом случае на «щеточных» адсорбентах достигаются постоянные характеристики удерживания. Конформационная подвижность алкильных цепей приводит к тому, что из полярных элюентов они выталкиваются на поверхность силикагеля, образуя слои непостоянной толщины и структуры. На адсорбентах «нещеточных», например модифицированных прививкой жестких органических циклов или жесткими полимерными пленками, таких эффектов не наблюдается.
В связи с этим лучшие результаты, как уже отмечалось, должны достигаться в случае прививки жестких углеводородных групп, в частности фенильных, циклогексильных и т. п.
Особая проблема, активно обсуждаемая в литературе, — зависимость удерживания разных веществ от состава подвижной фазы на силикагелях с привитыми алкильными цепями. Показано, что квадратическая связь между логарифмом фактора емкости и объемной долей органического модификатора имеется только при содержании воды в элюенте до 90%, при большем содержании воды эта связь не выполняется.
Силикагели с (нитрильными и аминными группами на поверхности) широко применяют как в НФХ, так и в ОФХ. Силикагели с аминными группами на поверхности обладают хорошей селективностью при разделении сахаров. Силикагели с аминными и нитрильными группами, наряду с чистым силикагелем, применяют для группового разделения нефтепродуктов. В литературе предложены и описаны силикагели с другими привитыми функциональными группами: силикагели с ароматическими группами на поверхности сильнее удерживают ароматические соединения, особенно 2-(3-пиренил)этилдиметилсилоксан. Силикагели с сульфидными группами оказались селективными к галогенсодержащим веществам, в частности к галогенсодержащим пестицидам, а с привитой органортутной цепью селективны к гетероциклическим соединениям. Силикаге
ли с перфторированными группами Ci8 более селективны к фторированным соединениям; к соединениям других классов они более гидрофобны, чем силикагели с алкильной цепью С]8.
Разработаны методы прививки к поверхности 1,3-диа-зол ов (имидазола, бензимидазола, гистамина), 1,3,4-триа-золов (аминотриазол), а-аминокислот (гистидина, триптофана, цистеина), серосодержащих соединений (аминотиазола, тиокарбамида, тиосемикарбазида, сульфаниловой кислоты). Силикагели с привитым рибофлавином применяли для разделения гелиценов.
Силикагели с привитыми цепями неустойчивы в щелочных средах, в связи с этим разработаны методы прививки гидролитически стабильных химически связанных фаз.
Углеродные адсорбенты
Углеродные адсорбенты наиболее перспективны для ОФХ из-за стабильности и селективности. Впервые были применены силохромы и макропористые силикагели со слоем пирографита на поверхности. Углеродные адсорбенты на основе ГТС механически непрочны, и их трудно применять при высоких давлениях.
В 1982 г. Дж. Ноксом и М. Гильбертом была предложена технология получения пористых углеродных адсорбентов на основе графито-термической сажи. Эти адсорбенты в настоящее время используются для разделения разных смесей: ароматических соединений, структурных изомеров, катионов и анионов, фенольных соединений, всевозможных фармацевтических препаратов.
Углеродные сорбенты марки «НурегсагЬ» — универсальные адсорбенты для разделения полярных и неполярных соединений — также обладают высокой селективностью при разделении изомеров. В ВЭЖХ НурегсагЬ интересен своей стабильностью во всем диапазоне pH (0-14), он не растворяется и не гидролизуется, не набухает в любом растворителе, выдерживает давление до 400 атм.; колонки, наполненные НурегсагЬ, обладают высокой эффективностью до 60 тыс. теоретических тарелок на метр. В табл. 4.1.47 приведена селективность углеродных адсорбентов к гомологам.
Таблица 4.1.47
Селективность к гомологам углеродных адсорбентов в ВЭЖХ
Элюенты Гомологи а -A(AG), кДж • моль 1
Метанол н-Алканы 2,09 1,84
Ацетонитрил То же 1,74 1,38
Дихлорметан То же 1,58 1,15
Диэтиловый эфир То же 1,35 0,75
Пентан То же 1,38 0,8
Гептан То же 1,29 0,68
Ацетонитрил Алкилбензолы 1,74 1,38
То же Бром-н-алканы 1,74 1,38
То же Хлор-н-алканы 1,74 1,38
То же н-спирты 1,74 1,38
312
Новый справочник химика и технолога
Структурные характеристики Hypercarb: удельная поверхность 120 м2т-1, средний диаметр — 2,5 нм, пористость — 75%, размер частиц для ВЭЖХ — 5 или 7 мкм.
Пористые полимеры
В ВЭЖХ применяют пористые полимеры разной природы, особенно в последние годы (около 20%). На их основе имеются сорбенты разной пористости и разной механической прочности. В ВЭЖХ в качестве сорбентов применяются следующие полимерные материалы: сополимеры стирола с дивинилбензолом, полибутадиены, целлюлоза, полиамиды, этилвинилбензолы, производные поливинилового спирта, полисахариды, полиэтиленгликоли и др. Чаще всего используются сополимеры стирола с дивинилбензолом. Пористые полимеры в отличие от силикагелей стабильны во всем диапазоне pH (0-14).
Полимерные материалы предпочтительнее в ионообменной и ионной хроматографии.
Полимерные сорбенты применяются для разделения и определения сахаров в пище и напитках.
Для аналитических и препаративных разделений пептидов и белков применяются такие гидрофильные полимерные материалы, как полидекстрины. В эксклюзионной хроматографии используют пористые полимеры с широкими порами. Эксклюзионная хроматография с органическими элюентами называется гель-проникающей хроматографией и применяется для разделения полимеров. Эксклюзионная хроматография с водными элюентами — гель-фильтрационная хроматография — используется для разделения биомолекул.
Другие адсорбенты
В ВЭЖХ в качестве адсорбентов применяют ионообменники, бентоны, полиуретан, адсорбенты со слоем жидких кристаллов, циклодекстрины, силикагели с химически привитыми к поверхности акцепторными группами (три-нитрофлюорены) и др.
Как уже упоминалось выше, наибольшее применение в ВЭЖХ находят сорбенты с привитыми алкильными группами. Фирмы выпускают сотни типов таких сорбентов. Для правильного выбора сорбентов для обращенно-фазовой хроматографии предложены разные системы оценок, по одной из них проводят измерения эффективности, факторов удерживания, гидрофобности, стерической селективности, оценки водородной связи, оценки ионообменной емкости при разных значениях pH элюента. В табл. 4.1.48 приведено описание этих параметров, а в табл. 4.1.49 по этим параметрам сопоставлены наиболее известные коммерческие сорбенты Cig.
Таблица 4.1.48
Основные технические характеристики сорбентов
Обозначение Параметр Метод оценки
N Эффективность Число теоретических тарелок на метр длины колонки
к Фактор удерживания Время выхода несорбиру-емого компонента определяется по метанолу, оценивается плотность прививки к поверхности силикагеля
асн2 Гидрофобность или гидрофобная селективность Отношение факторов удерживания пентилбензола и бутил-бензола агн = к™. /ксс . Это Ltij 1 1Ь 1 dd мера плотности покрытия поверхности фазой и его лигандной емкости
Ит/О Стерическая селективность Отношение факторов удерживания между трифениленом и о-терфенилом ат/о = kT/ko. Этот показатель оценивает селективность к молекулам разной геометрической формы, а также связи с типом силилирующего реагента
Ик/Ф Емкость взаимодействия по типу водородной связи Отношение факторов удерживания между кофеином и фенолом ак/ф = кк/кф. Это оценивает число свободных гидроксильных групп и степень эндкеппинга (экранирования)
ЙБ/Ф Ионообменная емкость при pH = 7,6 Отношение факторов удерживания бензиламина и фенола «б/ф= кБ/кф. Эта величина определяет общую силанольную активность
аБ/Ф Ионообменная емкость при pH = 2,7 Отношение факторов удерживания бензиламина и фенола, но в кислой среде. Этот параметр определяет общую кислотную активность силанольных групп
Таблица 4.1.49
Характеристика коммерческих сорбентов Cis для обращено-фазовой хроматографии
Тип фазы Фирма k aCH2 aT/O «К/Ф «Б/Ф pH = 7,6 «Б/Ф pH = 2,7 N на 1 м
Develosil ODS Phenomenex 6,7 1,49 1,24 0,51 0,1 0,07 63000
Discovery С18 Supelco 3,32 1,48 1,51 0,39 0,28 0,1 80000
Genesis Cu Jones Chrom 6,25 1,5 1,41 0,44 0,29 0,1 73000
Аналитическая хроматография и капиллярный электрофорез
313
Продолжение таблицы 4.1.49
Тип фазы Фирма k асн. CCt/O аК/Ф аБ/Ф pH =7,6 аБ/Ф pH = 2,7 N на 1 м
Hypersil 100 С is TCS 7,66 1,53 1,4 0,42 1,01 0,25 79000
Hypersil Elite Cu TCS 3,2 1,47 1,6 0,37 0,29 0,1 79000
Intersil ODS Hichrom 6,31 1,47 1,57 0,36 0,53 0,01 44000
Jupiter Cu 300A Phenomenex 2,26 1,48 1,65 0,37 0,47 0,27 25000
Kromasil Cu Hichrom 7,01 1,48 1,53 0,4 0,31 , 0,11 85000
Lichrosphere RP18 Merck 7,92 1,48 1,73 0,54 1,39 0,19 46000
Luna C18 (16,5%C) Phenomenex 5,97 1,47 1,17 0,4 0,24 0,68 90000
Luna C18 (19%C) Phenomenex 6,34 1,47 1,23 0,41 0,26 0,06 81000
Novapak Cu Waters 4,49 1,49 1,44 0,48 0,27 0,14 70000
Nucleosil Cu Hichrom 4,8 1,44 1,68 0,7 2,18 0,13 50000
Optimal-ODS H Capital HPLC 6,15 1,48 1,38 0,44 0,24 0,09 83000
Prodigy ODS 3 Phenomenex 7,27 1,49 1,26 0,42 0,27 0,09 73000
PurospherRP18 Merck 4,78 1,44 1,93 0,72 1,29 0,07 28000
Purospher RP18c Merck 6,51 1,48 1,75 0,46 0,34 0,08 66000
Selectosil Cu Phenomenex 4,94 1,45 1,69 0,68 1,98 0,14 61000
Spherisorb ODS1 Waters 1,78 1,47 1,64 1,57 2,84 2,55 86000
Spherisorb ODSB Waters 5,09 1,46 1,78 0,8 3,56 0,06 51000
Summit ODS (W) Crawford 5,45 1,47 1,29 0,56 0,4 o,l 88000
Supercosil LCu Supelco 4,82 1,47 1,42 0,46 1,93 0,89 61000
Superspher RP18c Merck 5,47 1,47 1,64 0,44 0,42 0,11 50000
Symmetry Cu Waters 6,51 1,46 1,49 0,41 0,68 0,01 56000
Symmetry Shield RP 18 Waters 4,66 1,41 1,22 0,27 0,2 0,04 83000
Xterra MC Cu Waters 3,52 1,42 1,26 0,42 0,35 0,1 42000
XterraRP18 Waters 2,38 1,29 1,83 0,33 0,2 0,07 41000
YMC Pro C18 YMC 7,42 1,53 1,29 0,46 0,26 0,08 80000
Zorbax Rx Cu Agilent 5,68 1,57 1,61 0,54 0,55 0,11 88000
Zorbax SB- Cu Agilent 6 1,49 1,2 0,65 1,46 0,13 77000
Zorbax Extend Cu Agilent 6,66 1,5 1,49 0,38 0,55 0,11 88000
Среднее 5,4 1,47 1,52 0,52 0,81 0,21 65000
Новые сорбенты для ВЭЖХ, предложенные в 2001 г
В табл. 4.1.50, 4.1.51 приведены списки специализированных колонок для ВЭЖХ. В табл. 4.1.52 указаны сорбенты, приготовленные по новым технологиям.
Таблица 4.1.51
Специализированные колонки в ВЭЖХ
Таблица 4.1.50
Специальные колонки
Типы Число
Хиральные 54
Аффинные 38
Для разделения карбогидратов 29
Гидрофобные 24
Для контроля загрязнений окружающей среды 15
Для разделения аминокислот 9
Для органических кислот 6
Для лекарств 4
Для разделения катехоламинов 3
Название сорбентов Назначение колонок
Hypersil Green Env Для анализа загрязнений окружающей среды: фенолов, фталатов, пестицидов и др.
Hypersil Green PAH Для анализа полиаромати-ческих соединений, включая и бензопирен
Hypersil Green Carbamate Для анализа карбаматных пестицидов (по методу ЕРА)
Hypersil PeP Для анализа и выделения синтетических и природных пептидов и белков (размер пор 10 и 30 нм)
314
Новый справочник химика и технолога
Продолжение таблицы 4.1.51
Название сорбентов Назначение колонок
Hypersil Duet (Cig/SAX, C18/SCX) Колонки co смешанными фазами, для анализа одновременно гидрофобных и ионных соединений, для анализа метаболизма лекарственных препаратов
HyperREZ XP Carbo-Hydrate Для анализа, спиртов, сахаров (высокая стабильность при низких значениях pH)
Таблица 4.1.52
Сорбенты для ОФХ в ВЭЖХ, приготовленные по нетрадиционной технологии либо не на силикагелевой основе
Обозначение сорбента Фирма-изготовитель Характеристики
Diamond Band Ci8 ZirChrom Separation Ci8 привита на оксид циркония, для работы при высоких pH и температурах
Hydrocell RP 10D BioChrom Labs. Inc На основе полистиролди-винилбензола, размер пор 100 нм, сферические частицы 10 мкм, рекомендуется для разделения белков
Poroshell 300SB-Ci8 Agilent Technologies Cis на поверхностно-пористом сферическом силикагеле 5 мкм колонка 75 х 2,1 мм для быстрого разделения белков
Presto FT-Ci6 Imtant Corp Cis на непористых частицах сферического кремнезема размером 2 мкм, колонки 30 х 4,6 мм для сверхбыстрого разделения
Aquasil Ci8 Thermo Hypersil Наличие гидрофильных и гидрофобных групп, для анализа полярных соединений (в частности, водорастворимые витамины), устойчив в водных элюентах (до 100%)
Fluophase PFP To же Перфторированные фенильные группы
Fluophase RP To же Перфторированные алкильные цепи
Fluophase WP To же Перфторированные алкильные цепи на силикагеле с размером пор 30 нм, все перфторированные сорбенты рекомендуются для разделения фосфолипидов, галогенсодержащих соединений, фенолов, изомеров
Продолжение таблицы 4.1.52
Обозначение сорбента Фирма-изготовитель Характеристики
Aster Polymer Cis Astec На основе поливинилового спирта
Spherisorb ASY Waters На основе оксида алюминия
Supelcogel ODS (W) Supelco На основе сополимера стирола с дивинилбензол ом
ZirChrom PBD ZirChrom Separation На основе оксида циркония
Hy Purity THKSo Сферические частицы силикагеля 3 и 5 мкм (диаметр пор — 18 нм) С18, С8 и CN для приготовления капиллярных колонок
Pico Frit New Objective Inc Для капиллярных колонок 75 х 10,50 мкм или 100 мм
4.1.8.5. Динамическое модифицирование адсорбентов
При динамическом модифицировании в элюент постоянно добавляют специальные вещества в небольшом количестве (в концентрациях порядка 0,001 М). Данные вещества преимущественно адсорбируются на поверхности адсорбента и изменяют химическую природу поверхности. Анализируемые вещества чаще всего сильно и специфически взаимодействуют с этими специальными веществами на поверхности адсорбента, при этом удерживание будет определяться кроме межмолекулярных ван-дер-ваальсовых взаимодействий и другими видами взаимодействия (вещество — адсорбент), в частности ионными, донорно-акцепторными, комплексообразующими.
Динамическому модифицированию подвергают как полярные, так и неполярные адсорбенты. При использовании элюентов со специальными добавками на одной колонке с неполярным адсорбентом можно реализовать различные варианты хроматографии: ион-парную, ионную, лигандную, донорно-акцепторную.
Метод динамического модифицирования имеет и свои ограничения: во-первых, специальные добавки в элюент не должны нарушать стабильность работы детекторов, во-вторых, нужно тщательно поддерживать параметры режима работы постоянными, чтобы не было нарушения равновесия в колонке.
В табл. 4.1.53 приведены некоторые примеры применения динамического модифицирования адсорбентов. Чаще всего, как видно из таблицы, динамическое модифицирование адсорбентов применяют при анализе биологически активных соединений.
Аналитическая хроматография и капиллярный электрофорез
315
Таблица 4.1.53
Жидкостная хроматография с динамическим модифицированием адсорбентов
Вещества, добавляемые в элюент Анализируемые вещества
Органические амины Трициклические антидепрессанты
Амины Лекарства
Хлорид тетрабутиламмония Ароматические кислоты
Цетримид Ароматические кислоты
Краун-эфиры Антибиотики
Хелаты металлов Дансилпроизводные аминокислот
Хиральные добавки а-Изомеры аминокислот
Цвиттер-ионы Нуклеотиды
Азотсодержащие ароматические производные Сахара
Акцепторы (или доноры) Смесь доноров (или акцепторов)
AgNO3 Изомеры феромонов
Карбоновые кислоты Различные смеси полярных веществ
4.1.8.6. Общая схема жидкостного хроматографа
Современный жидкостный хроматограф включает в себя следующие системы и устройства: систему подготовки и подачи элюента, в которую входит резервуар для элюента и насос, система ввода пробы, хроматографические колонки, термостаты колонок и детекторов, детекторы и сборники фракций. Кроме того, к нему может быть присоединен интегратор или ЭВМ.
Дополнительно жидкостные хроматографы комплектуются автодозатором, дегазатором, вторым насосом для градиентного элюирования.
Конструкции современных жидкостных хроматографов обычно пердставлены различными вариантами:
• блочно-модульная компоновка,
• компоновка в одном моноблоке,
• промежуточное исполнение: модульное в едином блоке.
Выбор конфигурации (модели хроматографа) из отдельных моделей определяется аналитической задачей. Модульная система позволяет легко и быстро сконструировать конкретную систему с небольшими затратами. Гибкая блочно-модульная система позволяет создавать различные по количеству блоков приборы, что позволяет решать как простые рутинные производственные задачи, так и проводить сложные научно-исследовательские измерения.
Интегрированная моноблочная система в некоторых случаях выгодна для специализированных конкретных задач. Преимущества этих крайних систем определяет интегрированная система с заменяемыми блоками.
Типы выпускаемых жидкостных хроматографов рассмотрены в табл. 4.1.54. В табл. 4.1.55 приведены технические характеристики отечественных жидкостных хроматографов.
Таблица 4.1.54 Типы жидкостных хроматографов
Типы Характеристика
Простые Один детектор, без термостата
Универсальные Несколько детекторов, термостат, автоматическое дозирование
Автоматизированные Микропроцессорное управление заданием режима, обработкой результатов и диагностикой неисправностей
Препаративные Большие колонки
Специальные: для молекулярноситовой хроматографии, для ионной хроматографии, микроколоночные и капиллярные, хроматографы на ЧИПах Прецизионный насос Коррозионностойкое исполнение Насос на малые расходы Наноколонки, микрорасходы элюента
Таблица 4.1.55
Технико-экономические показатели отечественных жидкостных хроматографов
Название модели Разработчик Детекторы, диапазоны длин волн Наличие термостати-рования колонок, детекторов Давление насоса, атм Прочие характеристики
Стайер-УФ/Вид «Аквилон», г. Москва СПФ (спектрофотометрический) Нет 200
Стайер-Fluor «Аквилон», г. Москва Флуоресцентный То же 200
Миллихром А-02 ЗАО «Эко Нова», г. Новосибирск УФ (190-360 нм) 35-90 °C 70 Автомат, дозатор вес - 17 кг, колонки только 75 х 2 мм
316
Новый справочник химика и технолога
Продолжение таблицы 4.1.55
Название модели Разработчик Детекторы Наличие термостати-рования колонок, детекторов Давление насоса, атм Прочие характеристики
Миллихром-5 ЗАО «Научприбор», г. Орел СПФ (190-360 нм) Нет 70 Вес 17 кг; длина колонки в пределах 60-120 мм
Минихром ГПЭЦ «Аналитэк», г. Москва УФ (254 нм, 280 нм) Тоже 250 Малогабаритный вес - 9,5 кг
ВЭЖХ-3 «Люмекс», г. С.-Перербург СПФ (210-730 нм); Флуор. (200-650 нм) То же 250 Вес - 10 кг; хроматографическая приставка
ВЭЖХ-4 «Люмекс», г. С.-Перербург То же 250 Градиентное элюирование
Цвет-4000 ОАО «Цвет», г. Дзержинск Нижегородской обл. СПФ (210-700 нм) 40-100 °C 200 В стадии разработки
Цвет Яуза НПО «Химавтоматика», г. Москва ФЛУ (250-650); хемилюм. на S и N, УФ (254, 280 нм) 30-80 °C 250; 400 Разработка с 2002 г., колонки от 50 до 250 мм
Системы подготовки и подачи элюента
Весьма важный узел жидкостного хроматографа представляет собой система подготовки и подачи элюента. В нее входят резервуар с элюентом, где должна быть предусмотрена возможность дегазации растворителя (в вакууме, при нагревании или ультразвуком), а также соответствующие фильтры для удаления воды и взвешенных твердых частиц из органических растворителей. Система подачи элюента должна обеспечить, во-первых, беспульсацион-ный поток жидкости через колонку и детектор. Пульсация или монотонное изменение скорости потока элюента приводит к невоспроизводимости удерживаемых объемов, что, в свою очередь, затрудняет качественный и количественный анализ. Кроме того, пульсация потока элюента искажает работу детектирующих систем, это также отражается на погрешности измерений и чувствительности анализа. Система подачи элюента должна обеспечить, во-вторых, возможность изменения в сравнительно широком диапазоне скоростей потока (в пределах двух порядков); в-третьих, возможность работы при высоких давлениях, необходимых для сокращения времени разделения, и, в-четвертых, возможность работы в режиме градиентного элюирования.
Применяемые в жидкостных хроматографах насосы можно разделить на следующие типы.
1. Насосы плунжерного типа, у которых элюент периодически подается в хроматографическую колонку. Эти насосы позволяют создавать входное давление до 50,0 МПа. Чтобы сгладить пульсации, неизбежные при таком способе подачи элюента, применяются буферные системы. Однако полностью исключить возникающие толчки очень трудно, поэтому в настоящее время такие
насосы используют систему обратной связи, что может уменьшить пульсацию, таким образом, скорость потока поддерживается постоянной с точностью менее 1%.
2. Насосы с гидропневматическим усилением, где давление на жидкость создается за счет давления газа. В этом случае газ из баллона давит на поршень большого сечения, который передает давление на поршень меньшего сечения. В результате происходит «усиление» давления, пропорциональное отношению сечения площадей этих поршней. Максимальное давление при этом может быть развито до 100 МПа и выше. Здесь пульсация не наблюдается. Единственный недостаток системы — изменение скорости потока при изменении сопротивления колонки. Для повторного забора жидкостей периодически проводят рециклы, т.е. быстрое заполнение всей емкости жидкостью. В последнее время такие насосы применяются для заполнения колонок.
3. Насосы так называемого шприцевого типа по конструкции близки к насосам второго типа. В них давление на поршень осуществляется штоком специального шагового электродвигателя, что позволяет исключить недостаток всех известных насосов: скорость потока в колонке будет постоянной даже при изменении сопротивления, так как подача элюента происходит в этом случае с постоянной, заданной, неизменяющейся скоростью. Максимальное давление составляет ® 50 МПа. Подача элюента этим насосом осуществляется без пульсации. Для получения смеси растворителей можно использовать два параллельных насоса.
При использовании градиентного элюирования смесь растворителей создается в специальных устройствах, где концентрация одного из компонентов (обычно сильнее адсорбирующегося) нарастает во времени. Основные технические характеристики насосов приведены в табл. 4.1.56.
Аналитическая хроматография и капиллярный электрофорез
317
Таблица 4.1.56
Технические характеристики насосов для ВЭЖХ
Тип насоса * L-7110 G1354A Альянс LC-2010 200LC PU-1580
1. Точность поддержания скорости потока, % <0,2 0,3 0,075 0,25 о,з 0,1
2. Диапазон скоростей потоков, см • мин-1 0,001-9,999 0,001-10,0 0,1-10,0 0,1-10,0 0,01-10,0 0,001-10,0
3. Максимальное давле- ** ние, атм 400 400 >400 >400 420 ' 500 (350)
Примечание. Насосы могут быть изготовлены из нержавеющей стали, керамики, тефлона, сапфира, рубина.
** Коэффициент перевода в СИ: 1 атм. = 1,01325-101 МПа.
Системы ввода пробы
Пробы вводятся без остановки либо автосамплерами. Характеристики автосамплеров приведены в табл. 4.1.57. Автоматический ввод последовательных проб необходим для увеличения производительности анализов.
Таблица 4.1.57
Характеристики автосамплеров для жидкостных хроматографов
Модель L-7200 G1313A Альянс LC2010 200
Воспроизводимость, % <0,3 <0,5 <0,5 <0,3 <0,5
Число пробирок (объемом 1,5 мл) 80(120) 100 120 210 99
Диапазон дозируемых объемов, мкл 0,1^55 0,1-5000 0,1-1500 0,1-2000 0,1-2500
Колонки
В жидкостной хроматографии чаще всего применяются колонки небольшой длины — от 3 до 25 см. Внутренний диаметр колонок колеблется от 1 до 4,6 мм.
Очень важной деталью, особенно при применении высоких давлений, являются пробки, закрепляющие сорбент, они обычно прочно запрессовываются на концах колонки. Лучшие пробки изготовляют из пористого металлокерамического материала. Применение стеклянной ваты и даже металлических сеток, особенно у выхода из колонки, недопустимо, так как мелкие зерна сорбента диаметром около 0,005-0,01 мм могут при этом попадать в ячейку детектора, нарушая его работу.
Наиболее распространенные колонки имеют следующие размеры, мм: 100 х 4,6, 150 х 4,6, 250 х 4,6 и 100 х 4, 150x4, 100x 3, 150x 3.
Микронасадочные колонки имеют внутренний диаметр 1-2,1 мм. В последнее время появились колонки с внутренним диаметром 0,32 мм и капиллярные с внутренним диаметром 0,05 и 0,01 мм. Преимущества микронасадоч-ных и капиллярных колонок:
• повышение чувствительности за счет уменьшения размывания;
• достижение оперативности разделения;
• возможность прямого подсоединения с МС;
• использование детекторов для ГХ: ПИД, ЭЗД, ТИД и др.;
• программирование температуры колонки (сокращение времени разделения и повышения чувствительности);
• микроколонки более экономичны (снижение затрат в работе за счет снижения расхода элюента и сорбента).
Монолитные колонки
До создания монолитных колонок трубки обычно заполняли по специальной технологии под давлением зернами силикагеля нерегулярной формы 5 или 10 мкм (в начале 1970 г.), затем в 1975 г. было налажено производство сферических частиц силикагеля, что облегчило более полное заполнение колонок. Были созданы сферические пористые частицы размером 3 мкм и непористые частицы размером 1,3 мкм.
Монолитные колонки представляют собой обычные трубки (100 х 4,6 мм или 150 х 4,6 мм) внутри которых создается сплошная пористая среда силикагеля, состоящего из пор двух типов: макропор диаметром 2 мкм (2-103 нм) и мезопор 13 нм. Сквозные макропоры обеспечивают хорошую проницаемость таких колонок для потока элюента и быстрый массообмен молекул анализируемых веществ. Общая пористость монолитной силикагелевой матрицы около 80%, удельная поверхность 300 м^г'1. На монолитных колонках можно проводить разделения в 2-4 раза быстрее обычных колонок, заполненных зернами сорбента. Время разделения можно дополнительно уменьшить при использовании программирования расхода элюента в процессе разделения.
Фирма «Мегк» освоила выпуск таких колонок «Chromo-lith» совместно с Х.Танака, К.Наканиши. Зависимость величины высоты, эквивалентной теоретической тарелки (Н) от скорости потока самая пологая.
318
Новый справочник химика и технолога
Термостаты
Разделения с помощью жидкостной хроматографии в большинстве случаев проводят при комнатной температуре, реже (например, при хроматографии синтетических полимеров) используют нагревание, причем обычно не выше 100 °C. Для термостатирования часто применяют водяные термостаты. В современных жидкостных хроматографах используют обычно воздушное термостатирова-ние с открытым нагревателем и быстрым принудительным перемешиванием. Чтобы исключить при этом опасность взрыва в случае негерметичности жидкостных систем хроматографа, через термостат иногда продувают поток инертного газа (азота или аргона). Программирование температуры колонки в жидкостной хроматографии так же эффективно, как и в газовой.
В последние годы выпускаются термостаты с контактным нагревом без принудительного перемешивания воздуха. Колонки в термостатах размещаются как в вертикальном, так и горизонтальном положении. Диапазон температур обычно 40-80 °C. Для разделения биологических неустойчивых соединений используют температуры от 0 или 5 °C до 80-90 °C, для этих целей применяют элементы Пельтье (нагрев и охлаждение).
Детектирующие системы для ВЭЖХ
В ВЭЖХ применяются десятки типов детектирующих систем. Основные технические требования к детекторам:
— низкий предел детектирования, определяемый отношением уровня шума к чувствительности;
— широкий линейный диапазон;
— стабильность показаний (низкий уровень шума и малый дрейф во времени);
— малая инерционность (постоянная во времени).
Детекторы подразделяются на универсальные и селективные. Для некоторых целей выгодно применять универсальные детекторы (определение всех компонентов смеси) или специфические (селективные) детекторы, в частности при анализе микропримесей из сложных смесей или из сложных матриц: загрязнения окружающей среды, пищевые продукты, клинические анализы и др.
Основные типы применяемых детекторов в ВЭЖХ можно классифицировать следующим образом (см. табл. 4.1.58).
В аналитической практике наибольшее распространение имеют следующие детекторы: фотометрический, спектрофотометрический, флуоресцентный, рефрактометрический, электрохимический, масс-спектрометрический. Сравнение разных детектирующих систем приведено в табл. 4.1.59.
Таблица 4.1.58
Типы детекторов в ВЭЖХ
Группы Тип
Элементный анализ Атомно-абсорбционные Индуктивно связанная плазма Микроволновая индуктивносвязанная плазма
Оптические Фотометрические Спектрофотометрические с переменной длиной волны Спектрофотометрические на диодной матрице Инфракрасные Раман-спектроскопические Рефрактометрические Светорассеивающие
Люминесцентные Флуоресцентные Лазерные флуоресцентные Хемилюминесцентные
Электрохимические Кондуктометрические Амперометрические Импульсные амперометрические Кулонометрические
Масс-спектрометрические МС с электроспреем МС с термоспреем Времяпролетные МС
ЯМР ЯМР
Газохроматографические детекторы Ионизационнопламенные Термоионные Пламенно-фотометрический
Радиоактивность Радиоактивные
Сенсоры Биосенсоры в ВЭЖХ (энзимамперометрическая)
Импульсная Иммунохимическая
Акустика Акустические
Чаще всего фотометрические детекторы применяют с фиксированными длинами волн: 245 нм и 280 нм. В нижеприведенных таблицах указаны технические характеристики лучших детектирующих систем: программируемых спектрофотометрических (табл. 4.1.60), спектрофотометрических на диодной матрице (табл. 4.1.61), флуоресцентных (табл. 4.1.62), рефрактометрических (табл. 4.1.63).
Таблица 4.1.59
Сравнение характеристик разных детекторов для ВЭЖХ
Детектор Предел детектирования, моль-л”1 Селективность Преимущества и недостатки детекторов
Кулонометрический 10’11-10-12 Высокая Полное окисление или восстановление анализируемого вещества. Возможно трехмерное измерение сигнала
Амперометрический 10-10-10”n То же Низкая стоимость. Селективность для электроактивных соединений
Аналитическая хроматография и капиллярный электрофорез
319
Продолжение таблицы 4.1.59
Детектор Предел детектирования, моль-л1 Селективность Преимущества и недостатки детекторов
Масс-спектрометрический ПГ10 Очень высокая Высокая стоимость. Позволяет идентифицировать неизвестные вещества
Флуоресцентный 1О7-1О10 Высокая Высокая стоимость. Необходимость дериватизации проб
Хемилюминисцентный 1О'7-1О*10 Очень высокая Высокая стоимость. Ограниченность применения, только для N- и S- содержащих соединений
Спектрофотометрический на диодной матрице 10^-10’7 Высокая Высокая стоимость. Возможность трехмерной регистрации сигналов
Спектрофотометрический 105-107 Низкая Низкая стоимость. Универсальная применимость
Детектор на основе светорассеивания 10^-10’5 То же Универсальная применимость
Рефрактометрический пг5 То же Универсальный детектор, весьма чувствительный к изменению температуры и скорости элюента
Таблица 4.1.60
Сравнение технических характеристик программируемых спектрофотометрических детекторов
Модель 785А 486 490 HP 1100 APD-10 L-7400
Тип оптики Двухлучевая Двухлучевая Двухлучевая Однолучевая сравнительный диод Двухлучевая —
Диапазон длин волн, нм 190-700 190-600 190-600 190-800 190-600 (900) 195-600
Ширина щели, нм 5 8 4 8,5 8 10
Источник света — тип ламп Дейтериевая (вольфрамовая) Дейтериевая Ксеноновая пульсирующая Дейтериевая Дейтериевая (вольфрамовая) —
Уровень шума, е.о.п. (диапазон длин волн, условие, постоянное время сигнала) 1-Ю’5 (210-280 нм, 1 с) I105 (254 нм, сухая ячейка) 5-Ю5 (сухая ячейка) 7,5-10’6 (254 нм, сухая ячейка, 2 с) 5-Ю5 (250 нм, сухая ячейка) 4-Ю-6 (254 нм)
Пределы поглощения, е.о.п. 0,0005-3,0 0,001-2,0 0,001-3,0 0,001-3,0 0,001-2,5 —
Дрейф, е.о.п.- ч1 < 1-КГ4 2,5-10^ 2,5-1 О'4 < 1-104 3-104 2-104
Объем ячейки, мкл 12 8 8 14 8 —
Длина пути, мм 8 10 10 10 10 —
Специальные функции Сканирование 0,2-1,0 нм-с1, диагностика определение утечек Автокалибровка длин волн, одноканальный Сканирование, оценка чистоты пиков по отношениям на разных длинах до 12 длин волн Сканирование, проверка утечек Сканирование, автокорректировка и мониторинг за длинами волн
Программируемые функции Время, диапазон длин волн 6 программ Время, диапазон длин волн Программируемая длина волны Время, диапазон длин волн —
Примечание: е.о.п. — единица оптической плотности.
320
Новый справочник химика и технолога
Таблица 4.1.61
Сравнение технических характеристик спектрофотометрических детекторов на диодной матрице
Модель LC-235C 996 HP 1100 SPD-M L-7455
Тип оптики Двухлучевая Однолучевая Однолучевая Однолучевая —
Диапазон длин волн, нм 195-365 190-800 190-950 200-600(800) 190-800
Разрешение диодов 5 нм (35 диодов) 1,2 нм 1 нм (1024 диодов) 1,2 нм (512 диодов) —
Точность установки ± 1 нм ± 2 нм ± 1 нм ± 1 нм —
Источник света — тип ламп Дейтериевая Дейтериевая Дейтериевая (вольфрамовая) Дейтериевая (вольфрамовая) —
Уровень шума, е.о.п. < 1,5-105 <2,5-10 5 < 1-Ю’5 <2,5-10’5 7,5-10"6
Дрейф, е.о.п.-ч -1 < 1104 <2-10"3 <210’3 < 1-Ю"3 1-10~3
Контроль Пульт, программа «Турбохром» Программа «Миллениум 2010» Программа «Хемстейшн» Мембранный пульт, программ-ма от ПЭВМ —
Объем ячейки, мкл 8 8 13 8 —
Длина оптического пути, мм 10 10 10 10 —
Специальные функции Диагностика Самокалибровка — программа 1-16 нм Связь световодом —
Таблица 4.1.62
Сравнение технических характеристик флуоресцентных детекторов
Модель LC-240 474 HP 1046А RF-551 L-7485
Тип оптической системы Двухлучевая, голографическая с полным отражением Двухлучевая Однолучевая Двухлучевая, голографическая решетка
Источник света — тип ламп Ксеноновая, пульсирующая, 8,3 Вт Ксеноновая, 150 Вт Ксеноновая, пульсирующая, 5 Вт Ксеноновая, 5 Вт Ксеноновая, 150 Вт
Возбуждение- Монохроматор 230-650 нм полоса 10 нм 200-700 нм 18 или 30 нм 200-800 нм 25 нм (10 нм) 220-750 нм 15 нм 200-850 нм 15 нм
Эмиссия монохроматор 250-750 нм 10 нм 200-700 нм 18 или 30 нм 200-750 нм 25 нм, 10 нм 220-650 нм 15 нм 250-900 нм 15 нм, 30 нм
Объем ячейки 7 мкл, 4 мкл 16 мкл 5 мкл 12 мкл 12 мкл
Предел детектирования 4-Ю14 по антрацену 4-Ю’14 по антрацену 8-10’14 по антрацену 4-Ю’14 по антрацену
Таблица 4.1.63
Сравнение технических характеристик рефрактометрических детекторов
Модель 200 RI 410 HP 1047А RID-6A RI2000T RiAS 100 475
Тип Отражающий, контроль микропроцессором Отражающий, контроль микропроцессором Отражающий Отражающий Отражающий Дифферен циальный рефрактометр Дифференциальный рефрактометр
Источник света Вольфрамовая лампа Пульсирующий LED Вольфрамовая лампа Вольфрамовая лампа — Отражающий диод 940 нм
Диапазон, ед. Rf 1,00-1,75 1,00-1,75 1,00-1,75 1,00-1,75 1,00-1,75 — —
Уровень шума, ед. Rf 2,5-10’9 2-10’8 2,5-10'9 2,5-10-9 5-1O10 1-Ю’8 1-Ю’8
Объем ячейки, мкл 8 10 7 10 9 0,25 8
Диапазон термоста-тирования 35 и 55 °C — 30, 35, 40, 45, 50 °C — 35 и 55 °C — -—
Аналитическая хроматография и капиллярный электрофорез
321
Электрохимические детекторы
К электрохимическим детекторам относят: амперометрические, потенциометрические, амперометрические в импульсном режиме, кондуктометрические, полярографические и кулонометрические. Наибольшее применение находят амперометрические детекторы (АД) для анализа соединений, в молекулах которых есть функциональные группы, способные окисляться или восстанавливаться. Для расширения областей применения АД используют, во-первых, до- и послеколоночную дериватизацию анализируемых соединений; во-вторых, рабочие электроды разной природы (см. табл. 4.1.64). Три типа чувствительных ячеек: струя-стенка (flow-at), тонкослойные с параллельным потоком (flow-by), с потоком, проходящим через пористый рабочий электрод (flow-throuth).
АД имеет большие преимущества:
— низкий предел детектирования ® 1-10”15 г;
— селективность;
— широкий линейный диапазон « 106;
— малый объем ячеек ® неск. нл;
— дешевизна;
— универсальность;
— надежность.
АД в режиме полного окисления или восстановления переходит в кулонометрический режим, позволяющий определение в трех измерениях. Для повышения чувствительности или для расширения областей применения детекторов применяют химическую дериватизацию проб. Преимущества и недостатки до- и после колоночных реакций приведены в табл. 4.1.65.
Таблица 4.1.64
Перечень типов рабочих электродов и соединений, определяемых на них
Материал рабочего электрода Определяемые соединения
Стеклоуглерод Универсальный, но наиболее пригоден для анализа катехоламинов и их метаболитов, фенолов, хлорфенолов, нафтолов, катехолов, ароматических аминов, нитроароматических соединений, хинонов, полиенов, тиолов, дисульфидов и др.
Золото Алифатические спирты, моносахара, дисахара, олигосахара, алифатические амины, аминоспирты, аминосахара, нитроароматические соединения, аминокислоты, серасодержащие пестициды, этилентиомочевина
Платина Спирты, гликоли, альдегиды, гипохлориты, арсениты, гидразины, ацетилхолин, пероксиды
Серебро Цианиды, сульфиды, сульфиты, тио-сульфиты, тиоцианаты, бромиды, йодиды, гидросульфиды
Продолжение таблицы 4.1.64
Материал рабочего электрода Определяемые соединения
Ртуть Тиолы, дисульфиды, нитрозомины, восстанавливаемые металлы
Медь Сахара, аминокислоты, пептиды, полипептиды, белки
Никель Сахара, спирты, аминокислоты
Палладий Ароматические углеводороды
Прочие электроды: — электроды, модифицированные энзимами; — электроды, модифицированные токопроводящими полимерами; — электроды, модифицированные фталоцианинами, ферроценом и др. Смеси и сплавы: Ni-Ti, Ni - стеклоуглерод, Ni-Cr, Ni-Cu, Ni-Cr-Fe, Pt-стеклоуглерод, Au-Cr, Co и др.
Таблица 4.1.65
Оценка предколоночных и послеколоночных методов получения производных
Метод получения производных Преимущества Недостатки
Предколо-ночный Нет ограничений по растворителям; на условия реакций нет ограничений; возможен широкий выбор реактивов Возможно образование производных других веществ; необходим внутренний стандарт; воспроизводимость низкая, т.к. реакция может не дойти до конца
Послеко-лоночный Подготовка пробы минимальная; получение производных автоматическое и точность высокая; образование побочных продуктов менее вероятно Ограничения по хроматографическому элюенту из-за нерастворимости или химической реакции; реакция должна быть быстрой (от нескольких секунд до 2—3 минут); разделение может ухудшаться из-за послеколоночного перемешивания
322
Новый справочник химика и технолога
Литература
1. Перри С., Атос Р., Брюер П. Практическое руководство по жидкостной хроматографии. М.: Мир, 1974.260 с.
2. Лабораторное руководство по хроматографическим и смежным методам / Под ред. О. Микеш. М.: Мир, 1982. 395 с.
3. Melander W.R., Horvath С. High Performance Liquid Chromatography. Advances and Perspectives. I Ed. C. Horvath. New York, 1983.
4. Poole C.F., Schuettte S.A. Contemporary Practice of Chromatography. Amsterdam: Elsevier, 1984.
5. Андерсон А.А. Жидкостная хроматография аминосоединений. Рига: Зинатне, 1984. 295 с.
6. Parris N.A. Instrumental Liquid Chromatography: A Practical Manual of High Performance Liquid Chromatographic Methods. 2nd compl. rev. ed. Amsterdam: Elsevier, 1984. 432 c.
7. Киселев A.B., Пошкус Д.П., Яшин Я.И. Молекулярные основы адсорбционной хроматографии. М.: Химия, 1986.
8. Gilbert М.Т. High Performance Liquid Chromatography. Bristol: Wright, 1987. 288 p.
9. Meger V. Practical High Performance Liquid Chromatography. New York, 1988. 280 p.
10. Snyder L.R., Glajch J.L., Kirkland J.J. Practical HPLC Method Development. New York: John Wiley and Sons, 1988.260 p.
11. Smith R.M. Gas and Liquid Chromatography in Analytical Chemistry. Chichester: Wiley, 1988.402 p.
12. Eppert G. Introduction in Liquid Chromatography High Pressure. 2nd ed. Berlin: Akad.Verlag, 1988. 211 p.
13. Lim C.K. HPLC of Small Molecule: Practical. Oxford: IRL. Press, 1986. 333 p.
14. Sewell P.A. Clarke B. Chromatographic Separation: Analytical Chemistry. Chichester et al.: Wiley, 1987. 335 p.
15. High Performance Liquid Chromatography / Ed. P.R Brown, R.A. Hartwirk. 11 Chemical Analysis. V. 18. New York: Wiley, 1989. 688 p.
16. HPLC Advances and Perspectives. Vol. 5 I Ed. C. Horvath San Diego: Academic Press, 1988. 331 p.
17. Ravindranath B. Principles and Practice of Chromatography. Chichester: Horwood, 1989. 502 p.
18. Ahuia S. Trace and Ultratrace Analysis by High Performance Liquid Chromatography. New York: Wiley, 1992. 419 p.
19. Poppe H. Column Liquid Chromatography. Elsevier: Amsterdam, 1992. 135 p.
20. Стыскин Е.Л., Ициксон Л.Б., Брауде E.B. Практическая высокоэффективная жидкостная хроматография. М.: Химия, 1986. 288 с.
21. Scott R.P.W. Liquid Chromatography Column Theory. Chichester: Wiley, 1992. 279 p.
22. Intelligible Approach to HPLC I Ed. Miyazaki M. Tokyo: Hirokawa Pablishing Co, 1991. 208 p.
23. Chromatography. Fundamentals and Applications of Chromatography and Related Differential Migration Methods. Part A: Fundamentals and Techniques I Ed. E. Heftmann. Elsevier, 1992. 630 p.
24. Snyder L.R., Glajch J.L., Kirkland J.J. Practical HPLC Method Development. Tokyo: Kagaku; Dojin, 1992.272 p.
25. Haddad P.R., Jackson P.E. Ion Chromatography: Principles and Application. Amsterdam: Elsevier, 1990. 798 p.
26. Japan Society for Analytical Chemistry Ion Chromatography Data Book. Tokyo: Kagaku Shinbunsha, 1991. 548 p.
27. Walton I. Ion-Exchange Chromatography. Part A. Amsterdam: Elsevier, 1992. P. 227-265.
28. Touchstone J.C. Techniques and Applications of Thin Layer Chromatography. New York, 1985. 395 S.
29. Kaiser R., Reider R. Planar Chromatography I Heidelberg: Huthig, 1986. V. 1.205 p.
30. Kaiser R. Einfiihrung in die HPPLC. Heidelberg: Huthig, 1987.256 S.
31. Introduction to High-Pressure Thin Layer Chromatography I Ed. R. Kaiser. Bad Duerkheim: Inst. Chromatogr. 1976. 235 p.
32. Березкин В.Г., Бочков H.C. Количественная тонкослойная хроматография. Инструментальные методы. М.: Наука, 1980. 183 с.
33. Geiss F. Fundamentals of Thin Layer Chromatography (Planar Chromatography). Heidelberg: Huthig, 1987.482 p.
34. Hamilton R.J., Hamilton S. Thin Layer Chromatography / Ed. D. Kealey. Chichester: Wiley, 1987.129 p.
35. Thin Layer Chromatography: Reagents and Detection Methods. V.la: Physical and Chemical Detection Methods: Fundamentals, Reagents 1. / Ed. H. York et al. Weinheim: VCH, 1990. 464 p.
36. Planar Chromatography in the Life Sciences / Ed. Touchstone J.C. New York: Wiley, 1990. 199 p.
37. Touchstone J.C. Practice of Thin Layer Chromatography. 3d ed. New York: Wiley and Sons, 1992. 377 p.
38. Fried B., Sherma J. Thin-Layer Chromatography. Techniques and Applications: 3d ed. New York: M. Dekker, 1994. 45Ip.
39. Руководство по современной тонкослойной хроматографии I Под ред. О.Г. Ларионова. М., 1994. 311с.
40. Thin Layer Chromatography: Reagents and Detection Methods. V. lb: Physical and Chemical Detection Methods: Activation Reactions, Reagents Sequences, Reagents II. IH. York, W. Funk, W. Fisher et al. Weinheim: VCH, 1994. 496 p.
41. Fried B., Sherma J. Practical Thin Layer Chromatography. A Multidisciplinary Approach. Boca Raton CRC Press Inc. 1996. 336 p.
42. Dubin P.L. Aqueous Size-Exclusion Chromatography. Amsterdam: Elsevier, 1988. 454 p.
43. High-Performance Liquid Chromatography of Biopolymers and Biooligomers. Part B: Separation of Individual Compound Classes I Ed. O. Mikes. Amsterdam; Oxford: Elsevier, 1988. 772 p.
44. Hagel L. Janson J.C. Size-Exlusion Chromatography. Part A. Amsterdam: Elsevier, 1992. 160 p.
45. HPLC of Macromolecules I Ed. R.W.A. Oliver. Tokyo: Hirokawa, 1992. 256 p.
Аналитическая хроматография и капиллярный электрофорез
323
46. Chromatography of Polymers: Characterization by SES and FFF, AC5 Symp. Ser. ACS / Ed. T. Provder. Washington, 1993.337 p.
47. Handbook of Size Exclusion Chromatography / Ed. C.-S. Wa. New York: M. Dekker, 1995. 463 p.
48. Самсонов Г.В. Препаративная селективная хроматография биологически активных соединений // Ж. прикл. хим. 1992. Т. 65. С. 961-975.
49. Prepative and Production Scale Chromatography / Ed. G. Ganetsos, P.E. Barker. New York: M. Dekker, 1993.798 p.
50. Preparative Liquid Column Chromatography / Ed K.K. Unger. Darmstadt: GIT-Verlag, 1994. 280 p.
51. Hostettman K., Hostettman M., Morston A. Preparative Chromatography Techniques: Application in Natural Product Isolation. Tokyo: Kagaku Dojin, 1990. 204 p.
52. Самсонов Г.В. Препаративная хроматография как система биоразделений // Химич, промышленность. 1993. №7. С. 292-295.
53. Process Scale Liquid Chromatography / Ed. G. Subramanian Weinheim: VCH, 1995. 225 p.
54. Беленький Б.Г., Ганкина Э.С., Мальцев В.Г. Капиллярная жидкостная хроматография. Л.: Наука, 1987. 208 с.
55. Microbore Column Chromatography. Unified Approach to Chromatography I Ed. F. Yang, New York: M. Dekker, 1989. 424 p.
56. Введение в микромасштабную высокоэффективную жидкостную хроматографию / Под ред. Д. Иона. Пер. с англ.; Под ред. В.Г. Березкина. М.: Мир. 1991. 240 с.
57. Туркова Я. Аффинная хроматография. М.: Мир, 1980. 472 с.
58. Fassina G., Chaiken I.M. // Adv. Chromatogr. 1987. V. 27. P. 247-297.
59. Аффинная хроматография. Методы / Под ред. П. Дин, У. Джонсон, Ф. Мида / Пер. с англ. М.: Мир, 1988.278 с.
60. Phillips Т.М. Affinion Chromatography: Part A. Amsterdam: Elsevier, 1991. P. 309-338.
61. Kasai K., Matsumoto L, Beppu M. Affinity Chromatography. Tokyo: Kagaku. Dojin, 1991.284 p.
62. Handbook of Affinity Chromatography in: Chromatogr. Sci. / Ed. T. Kline. New York: M. Dekker, 1993. 332 p.
63. Даванков В.А., Навратил Дж., Уолтон X. Лигандообменная хроматография / Под ред. В.А. Даванкова. Пер. с англ.; М.: Мир, 1989.295 с.
64. Computerized Multiple Input Chromatography / Ed. M. Kaljurand and E. Kullik. New York: Ellis Horwood, Chichester, Halsted (Wiley), 1989. 226 p.
65. Computer-Assisted Method Development for High Performance Liquid Chromatography / Ed. J.L. Glaich, L.R. Snyder. Amsterdam: Elsevier, 1990.676 p.
66. Countercurrent Chromatography - Theory and Practice / Ed. N.B. Mandova, Y. Yto. New York: M. Dekker, 1998. 841 p.
67. Multidimensional Chromatography. Techniques and Applications / Ed. H.J. Cortes. New York: M. Dekker, 1990. 400 p.
68. Sover G.K., Nustroem L.S. Process Chromatography: A Practical Guide. San Francisko: Academic, 1989. 160 p.
69. Sofer G. and Hagel L. Handbook of Process Chromatography: A Guide to Optimization Scale up and Validation. San Diego: Academic, 1997. 450 p.
70. Cagniant D. Charge-Transfer Chromatography // Chromatogr. Sci. 1992. V. 57. P. 97-142.
71. Compexation Chromatography I Ed. D. Cagniant. New York: M. Dekker, 1992. 294 p.
72. Алесковская B.H. Титриметрическая хроматография. Л.: ЛГУ, 1991. 175 с.
73. Остерман Л.А. Хроматография белков и нуклеиновых кислот. М.: Наука, 1985. 536 с.
74. Fallon A., Booth R.F.G., Bell L.D. Applications of HPLC in Biochemistry. Amsterdam: Elsevier, 1987. 338 p.
75. Rapid Identification of Bacteria by Chromatography. Application of New Chromatography Technology / Ed. Y. Ikuya. Tokyo: Saikon Publishing Co, 1988. 131 p.
76. Chromatography and Isolation of Insect Hormones and Pheromones / Ed. A.R. McCaffery, I.D. Wilson. New York: Plenum, 1990. 376 p.
77. Performance Liquid Chromatography in Biotechnology / Ed. W.S. Hancock. New York: Wiley, 1990. 564 p.
78. HPLC of Proteins, Peptides and Polynucleotides: Contemporary Topics and Application / Ed. T.W. Hearn. New York: VCH, 1991. 776 p.
79. Practical Protein Chromatography Methods Mol. Biol. / Ed. A. Kenney and S. Fowell. Totowa: Humana Press, 1992. 327 p.
80. Therapeutic Drug Monitoring and Toxicology by Liquid Chromatogr. / Ed. H.Y. Steven, H. Wond. New York: M. Dekker, 1985.
81. Chromatography and Modification of Nucleosides / Ed. C.W. Gehrke, K.C.T. Kuo. Amsterdam: Elsevier, 1990.400 p.
82. Liquid Chromatography in Biomedical Analysis / Ed. T. Hunai. Amsterdam: Elsevier, 1991. 196 p.
83. Investigation of Metabolic Disorders by Chromatography / Ed. H. Liebich. Heidelberg: Huehtig, 1996. 220 p.
84. Chromatographic Analysis of Pharmaceuticals / Ed. J.A. Adamovics. New York: M. Dekker, 1989.680 p.
85. HPLC in Pharmacy and Biochemistry / Ed. M. Sarsunova, О. Напе, В. Kakac. Heidelberg: Huethig, 1990. 325 p.
86. Chromatography of Drugs and Other Toxic Compounds / Ed. Z. Deyl, К Macek. Amsterdam: Elsevier, 1990. 559 p.
87. HPLC in the Pharmaceutical Industry / Ed. G.M. Fong, S.K. Lam. New York: M. Dekker, 1991. 328 p.
88. Lunn G. and Schmuff N.R. HPLC Methods for Pharmaceutical Analysis. New York: Wiley, 1997. 1609 p.
89. Liquid Chromatographic Separation of Carbohydrates. A Collection of Invited Papers // Carbohydr. Res. 1991. V. 215, №1.P. 1-227.
90. Chromatographia of Micotoxins: Techniques and Applications / Ed. Y. Betina. Amsterdam: Elsevier, 1993. 440 p.
91. Carbohydrate Analysis - High Performance Liquid Chromatography and Capillary Electrophoresis / Ed. El Z. Rossi. Amsterdam: Elsevier, 1995. 672 p.
324
Новый справочник химика и технолога
92. Chromatographic Enantioseparation - Methods and Application / Ed. S.G. Alienmark. New York: Ellis Hor-wood, Chichester Halsted (Wiley), 1988. 224 p.
93. Chiral Liquid Chromatography / Ed. W.J. Lough. Glasgow: Blackie, 1989. 276 p.
94. Chiral Separation by HPLC / Ed. A.V. Krstulovic. New York: Ellis Horwood, Chichester, 1989. 548 p.
95. Шведт Г. Хроматографические методы в неорганическом анализе / Пер. с англ.; Под ред. В.Г. Березкина. М.: Мир, 1984. 254 с.
96. Inorganic Chromatographic Analysis / Ed. J.C. Macdonald. New York: Wiley, 1985. 450 p.
97. Methods in Chromatography. Porphyrins / Ed. C.K. Lim. Singapore: World Sci., 1997. V. 2. 250 p.
98. Тимербаев A.P., Петрухин O.M. Жидкостная адсорбционная хроматография хелатов. М.: Наука, 1989. 284 с.
99. Modem Methods in Plant Analysis, New Series. High Performance Liquid chromatography / Ed. H.F. Linskens, J.F. Jackson. Berlin: Springar-Verlag, 1987. 248 p.
100. Гугля Е.Б. Определение изоцианатов в воздухе // Журн. аналит. химии. 2000. Т. 55. С. 508.
101. Sazzanini С. In: Giangazza A. u.a. Chem. Processes Mar. Environ. Berlin: Springer-Verlag, 2000. S. 339-356.
102. Kataoka H. // J. Chromatogr. 1997. V. 774. P. 121.
103. Snyder L.R. Theory of Chromatography: Part A. Amsterdam: Elsevier, 1992. 142 p.
104. Pefferkom E. Interfacial Phenomena in Chromatography. New York: M. Dekker, 1999. 448 p.
105. Тимербаев A.P., Петрухин O.M. Механизм удерживания анионных хелатов в ионпарной хроматографии // Журн. аналит. хим. 1991. Т. 46, № 2. С. 213-224.
106. Electric Field Applications in Chromatography, Industrial and Chemical Processes / Ed. T. Tsuda. Weinheim: VCH, 1995.311 p.
107. The HPLC Solvent Guide / Ed. P.C. Sadek. Chichester: Wiley, 1996.352 p.
108. Retention and Selectivity in Liquid Chromatography I Ed. R.M. Smith. Amsterdam: Elsevier, 1995. 402 p.
109. Structure and Retention in Chromatography: A Chemometric Approach / Ed. R. Kaliszan. Amsterdam: Harwood, 1997. 224 p.
110. Jinno K. Chromatographic Separation Based on Molecular Recognition. Weinheim: VCH, 1996. 250 p.
111. Kaliszan R. Quantitative Structure Chromatographic Retention Relationships // Chem. Anal. (U.K.) 1987. V. 93. P.303
112. Detectors for Liquid Chromatography / Ed. E.S. Yeung. New York: Wiley, 1986. 366 p.
113. Selective Sample Handling and Detection in High-Performance Liguid Chromatography: Part A / Ed R.W. Frei. andK. Zech. Amsterdam: Elsevier, 1988. 458 p.
114. Detection-Oriented Derivatization Techniques in Liquid Chromatography I Ed. H. Lingeman, W.J.M. Underberg. New York: M. Dekker, 1990. 389 p.
115. Practical Spectroscopy. Fourier Transform Insfrared Spectroscopy and its Application / Ed. R. White. New York: M. Dekker, 1990. 328 p.
116. Selective Detectors: Environmental, Industrial and Biomedical Applications / Ed. R.S. Sievers. New York: Wiley. 1995. 261 p.
117. Scott R.P.W. Chromatography Detectors. New York: M. Dekker, 1997, 536 p.
118. HPLC Detection, Newer Methods / Ed. G. Pananay. New York: VCH, 1992. 236 p.
Приложение
Индексы удерживания (ИУ) некоторых органических соединений в обращенно-фазовой ВЭЖХ
Использование системы индексов удерживания для представления параметров удерживания в обращенно-фазовой высокоэффективной жидкостной хроматографии (ВЭЖХ) хорошо известно [6], но все же не получило столь же широкого признания и распространения, как в газовой хроматографии. Чаще всего параметры удерживания выражают в виде принципиально не обладающих межлабораторной воспроизводимостью факторов емкости k = (U / to - 1) или их логарифмов. Это связано с преобладающим использованием такого аналитического метода как ВЭЖХ для подтверждающих анализов (определение априорно известных веществ при наличии стандартных образцов), а не для разведочных (идентификация неизвестных соединений). Другой объективной причиной меньшей «популярности» индексов удерживания в ВЭЖХ является их больший межлабораторный разброс, обусловленный как их зависимостью от состава элюента (особенно от pH), так и от такого трудно контролируемого параметра как степень «старения» хроматографических колонок. Если стандартные отклонения рандомизованных значений RI (ГЖХ) для стандартных неполярных полидиметилсилоксановых неподвижных фаз для большинства веществ составляют 2-10 ед. инд., то для RI (ВЭЖХ) — 10-30 ед. инд. при меньшем рабочем диапазоне хроматографических колонок в шкале индексов удерживания. Как правило, наибольшей воспроизводимостью ИУ характеризуются органические соединения, не имеющие активных атомов водорода.
Приведенные ниже значения RI (ВЭЖХ) некоторых наиболее подробно охарактеризованных органических соединений пока еще не могут рассматриваться как достаточно полная база данных, скорее иллюстрируя возможную форму представления справочной информации в этом аналитическом методе. При усреднении межлабораторных данных учитывали параметры удерживания, определенные при использовании наиболее распространенных элюентов вода-ацетонитрил и вода-метанол с pH в диапазоне от 2-6.
Реперными компонентами для вычисления И1(ВЭЖХ) являются н-алкилфенилкетоны QHsCOQH^i, которым, как и в ГЖХ, присваивают значения RI = 100 пс, где Пс — число атомов углерода в молекуле. Расчет линейно-логарифмических индексов удерживания, применимых как в изократи-ческих условиях, так и в градиентных режимах элюирования, проводится по тем же формулам, что и RI (ГЖХ) [2,3], и с использованием тех же программ [4,5].
Аналитическая хроматография и капиллярный электрофорез
325
Молярная масса, г-моль"1 Эмпирическая формула Название RI Sri
78 с6н6 Бензол 932 17
92 С7Н8 Толуол 1016 19
93 GJI7N Анилин 686 10
94 СбН6О Фенол 688 16
96 С5Н4О2 Фурфурол 593 9
103 c7h5n Бензонитрил 807 8
104 с8н8 Стирол 1058 16
106 С7Н6О Бензальдегид 777 8
с8н10 Этилбензол 1100 10
107 c9h9n Л’-Мстиланилин 821 15
Бензиламин 768 8
108 с7н8о Анизол 907 12
Бензиловый спирт 643 13
Крезол 760 23
ПО С6Н6О2 Гидрохинон 593 32
112 С6Н5С1 Хлорбензол 1039 17
117 c8h7n Индол 838 5
СбН51Чз 1Я-Бензотриазол 665 23
C7H5NO Фенилизоцианат 938 5
120 С8Н8О Ацетофенон 800 —
122 С7Н6О2 Салициловый альдегид 813 36
С()Н6№20 Никотинамид 509 21
123 c(,h5no2 Нитробензол 854 18
126 С7Н7С1 Бензилхлорид 989 11
128 СюН8 Нафталин 1124 16
С6Н5ОС1 2-Хлорфенол 779 18
3-Хлорфенол 806 26
4-Хлорфенол 804 29
134 С9НюО Пропиофенон 900 —
135 C8H9NO Ацетанилид 689 10
136 С8Н8О2 Метил бензоат 888 13
137 C7H7NO2 4-Нитротолуол 954 5
139 c6h5no3 2-Нитрофенол 792 21
3-Нитрофенол 732 30
4-Нитрофенол 755 43
144 СюНбО 1-Нафтол 925 31
146 С9НбО2 Кумарин 791 13
148 С10н12о Анетол 1142 10
150 с10н14о Тимол 1027 10
151 C8H7NO2 Парацетамол 601 26
152 С8Н8О3 Метилсали-цилат 934 9
Продолжение таблицы
Малярная масса, г-моль"1 Эмпирическая формула Название RI Sri
152 С8Н8О3 Ванилин 702 22
154 С12Ню Дифенил 1192 14
157 C6H4NO2C1 1-Нитро- 2-хлорбензол 976 20
160 С9Н4О3 Нингидрин 574 17
164 СюН12О2 Эвгенол 910 14
165 C9HuNO2 Анестезин 823 11
167 c12h9n Карбазол 1133 23
169 C12HnN Дифениламин 1134 13
169 C8HnNO3 Пиридоксин (витамин Вб) 513 22
170 C7H7Br 4-Бромтолуол 1155 11
172 СбН5ОВг 4-Бромфенол 824 28
C6HeN2O2S Сульфамид 473 14
178 C14H10 Антрацен 1298 8
Фенантрен 1290 24
179 c13h9n Акридин 1102 25
182 c12h10n2 Азобензол 1259 28
c13H10o Бензофенон 1058 16
184 C8H]2N2O3 Барбитал 614 28
CuHeS Дибензотиофен 1299 26
191 c12h17no Диэтиламид лг-толуиловой кислоты 905 18
194 c8h10n4o2 Кофеин 633 27
CioHio04 Диметилфталат 852 24
202 c16H10 Пирен 1361 15
Флуорантен 1345 15
208 Ci4H8O2 1,4-Антрахинон 1110 24
212 C14H12O2 Бензоин 889 11
226 CnH16N2O3 Пентобарбитал 871 22
232 Ci2Hi2N2O3 Фенобарбитал 729 48
240 Ci4H8O4 Ализарин 940 12
284 Ci6Hi3N2O3 Диазепам 996 27
285 c17h19no3 Морфин 609 27
299 Ci6H14N3OC1 Хлордиазеп-оксид 948 29
354 C2iH26N2O3 Йохимбин 848 23
Литература
1. Зенкевич И.Г. // Журн. аналит. химии. 1996. Т. 51, № 11. С. 1140-1148.
2. Зенкевич И.Г. // Журн. аналит. химии. 1984. Т. 39, № 7. С. 1297-1307.
3. Zenkevich LG., Ioffe B.V. П J. Chromatogr. 1988. V. 439. P.185-194.
326
Новый справочник химика и технолога
4. Мариничев А.Н., Турбович МЛ., Зенкевич И.Г. Физико-химические расчеты на микро-ЭВМ: Справочник. Л.: Химия, 1990. 254 с.
5. Столяров Б.В., Савинов И.М., Витенберг А.Г. и др. Практическая газовая и жидкостная хроматография: Учебное пособие. СПб.: СПбГУ, 1998. 611 с.
6. Retention and Selectivity in Liquid Chromatography / Ed. R.M. Smith. Amsterdam: Elsevier, 1995.462 p.
4.1.9. Ионная хроматография
Отличие ионной хроматографии от классической ионообменной хроматографии
Ионная хроматография (ИХ) — аналитический вариант классической ионообменной хроматографии, ее отличает оперативность, большая эффективность и более высокая чувствительность анализа. Это достигается применением ионообменников меньшей емкости (0,1 мэкв-г"'), меньшим размером диаметров зерен (для увеличения скорости мас-собмена). Для снижения предела детектирования Смолл, Стивенс и Боумен [1] в 1975 г. предложили применение двух последовательно соединенных колонок с кондуктометрическим детектированием. Первая колонка — разделительная, вторая — подавительная (компенсационная), значительно снижающая фоновый ток элюента и понижающая предел обнаружения, т.к. реальный предел детектирования определяется отношением уровня шума к полезному сигналу. В 1979 г. был предложен одноколоночный вариант ИХ (без подавительной колонки) за счет применения ионообменников еще меньшей емкости (около 0,01 мэкв-г"1) и элюентов со слабой проводимостью [2].
Основные методы ИХ
В классической ионообменной хроматографии разделение происходит за счет ионного обмена. ИХ применяется для разделения как неорганических, так и органических анионов и катионов (см. табл. 4.1.66). Разделение анионов в основном проводят на анионообменниках полимерной основы с четвертичными аммонийными группами. Катионы разделяются на катионообменниках с сульфогруппами.
Ионообменники могут быть выполнены как на основе силикагеля, так и на полимерной основе. Механизм разделения в ион-эксклюзионной хроматографии определяется эксклюзией по Доннану, стерической эксклюзией и сорбционными процессами [3]. В этом методе в качестве ионо-обменника используется сульфированный полистирольный материал высокой емкости. Ион-эксклюзионная хроматография применяется для разделения слабых неорганических и органических кислот. Сильные кислоты не удерживаются и элюируют неразделенными, как несорбируемые компоненты. В комбинации с подходящими методами детектирования этот метод может применяться для разделения и определения аминокислот, спиртов, альдегидов и сахаров.
В ион-парной хроматографии разделение происходит за счет адсорбции. В качестве адсорбентов применяются силикагели с химически привитыми (октил- или октадецил-) алкильными группами. В элюент, кроме органических модификаторов, добавляются ион-парные реагенты (табл. 4.1.67). Ион-парная хроматография применяется для разделения поверхностно-активных анионов и катионов, комплексов переходных металлов.
Иногда для разделения ионов также применяется обращенно-фазовая жидкостная хроматография с привитыми аминопропильными фазами.
Таблица 4.1.66
Основные ионохроматографические методы разделения
Методы Функционалы! ые группы Анализируемые смеси Элюенты
Ионная хроматография анионов -(NR3)+ F", СГ, SCN , CN", РО/, Н2РО", НРО32~, НРО2", Р2О/, Р3О]0", NO", NO", S2", SO32", SO2", S2O|", S2O2", S2O2 , ОСГ, CIO", CIO", C1O4", SeO3”, SeO2", HAsO32", WO2", MoO2", CrO2" Na2CO3/NaHCO3
Ионная хроматография катионов -SO" Li+, Na+, NH/, K+, Rb+, Cs+, Mg2+, Ca2+, Sr2\ Ba2+ НС1, 2,3-диаминопропионовая кислота
Ион-эксклюзионная -SO2" Алифатические кислоты, спирты, альдегиды, бораты, силикаты, карбонаты НС1, октилсульфоновая кислота
Ион-парная хроматография анионов Неполярная фаза — С18 Ароматические кислоты, анионные ПАВ (моющие средства), металлоциановые комплексы, многие неорганические вышеперечисленные анионы NH4OH, гидроксид тетрабутил аммония
Ион-парная хроматография катионов Неполярная фаза — С18 Аммониевые основания, алкиламины, алканоламины, катионные ПАВ (моющие средства), сульфоновые соединения и др. НС1, октил сульфоновая кислота
Аналитическая хроматография и капиллярный электрофорез
327
Таблица 4.1.67
Реагенты для ион-парной хроматографии в порядке повышения гидрофобности
Анализ анионов Анализ катионов Анализ анионов Анализ катионов
Гидроксид аммония Гидрохлорная кислота, перхлорат Гидроксид тетрапропил-аммония Гептасульфоновая кислота
Гидроксид тетраэтил-аммония Гексасульфоновая кислота Гидроксид бутиламмония Октасульфоновая кислота
Преимущества ИХ
Анализ неорганических и органических ионов в растворах — сложная аналитическая задача. Внедрение ИХ для этих целей в последнее десятилетие явилось большим достижением во многих актуальных областях. Если для анализа катионов существуют альтернативные экспрессные и чувствительные методы (например, атомно-абсорбционная спектрометрия, спектроскопия на основе индуктивносвязанной плазмы), то для анализа анионов они отсутствуют. Такой чувствительный и оперативный метод, как ИХ, позволяет определять все типы анионов (табл. 4.1.68). Методы гигрометрии, фотометрии, колориметрии сильно уступают ИХ.
Таблица 4.1.68
Перечень анионов, анализируемых ИХ
Класс Анионы
Галогенид-ионы F’, СГ, Вг , Г
Кислородсодержащие галогенид-ионы осг, сю;, сю;, сю4, вю;, ю;
Кислородфосфор-содержащие РО32’, РО3“, РО3“, Р2О<’, p3os0-, р4о*;, po3f2-
С ерасодержащие S2“, SOf’, SO4“, S2O3", SCN“
Азотсодержащие CN-, ocn", no;, no;, n;
Кремнесодержащие SiO3”, SiF62’
Борсодержащие b4o2~, bf;
Оксиметаллические Aso;, AsO3’, SeO32’, SeO2 MoO2’, WO2’, CrO2’
Одно из главных преимуществ ИХ — быстрое одновременное определение многокомпонентных смесей катионов или анионов (до 10 и более) в течение 2-15 мин. Основные анионы (фторид, хлорид, нитрат, сульфат, фосфат) можно разделить на хороших ионообменниках за 2-5 мин., за 15-20 мин. можно разделить все катионы группы лантанидов. ИХ способна разделить и определить т- 2+ катионы в разных валентных состояниях, например, Fe и Fe3+, Сг3+ и Сг6+, чего не может сделать атомно-адсорбционная спектрометрия.
Следующее преимущество — низкий предел обнаружения (110 3 мкг-мл1) без предварительного концентрирования за счет подавления фонового тока. Применение предва
рительной концентрирующей колонки позволяет снизить предел обнаружения еще на 2-3 порядка, т.е. до величины 110 s- КГ6 мкг-мл’1. В последние годы в особочистой воде анионы определяют на уровне ppt, т.е. Ю10%.
Линейность кондуктометрического детектора в ИХ — в пределах 105—106 порядков (в пределах 0,01-100 мг-мл1). что позволяет определять как микро, так и макрокомпоненты анализируемой смеси. Следует отметить, что величина анализируемой пробы обычно небольшая (10-50 мкл).
При анализе водных проб в большинстве случаев про-боподготовка проста или же ее вообще нет, поэтому можно использовать разные детекторы или их комбинации, создать полностью автоматический режим анализа. В настоящее время цена одного анализа анионов в питьевой воде методом ИХ самая низкая, недаром этот метод сертифицирован в США, Германии и России.
На удерживание ионов, в частности анионов в ИХ влияют пять параметров (см. табл. 4.1.69). Изменяя элюирующую способность подвижной фазы, можно в широких пределах изменять параметры удерживания (рис. 4.1.13).
Таблица 4.1.69
Влияние разных параметров элюента на удерживание анионов
Параметр Результат
Температура Влияет на селективность и эффективность разделения. С повышением температуры, с одной стороны, уменьшается удерживание и селективность разделения, с другой — повышается эффективность за счет увеличения скорости массобмена и уменьшения вязкости элюента
Скорость потока С увеличением скорости удерживание уменьшается. Наибольшая эффективность наблюдается при оптимальной скорости элюента в соответствии с зависимостью ВЭТТ от скорости
pH Времена удерживания анионов слабых кислот увеличиваются с повышением pH
Ионная сила Элюирующая способность повышается с повышением ионной силы
Природа буферного раствора Элюирующая способность и селективность разделения анионов определяются природой элюента. Изменение природы буфера будет также изменять pH элюента
328
Новый справочник химика и технолога
Типы ионообменных колонок
Основой современных ионообменников для ИХ являются силикагели и органические пористые полимеры. Последние в настоящее время доминируют, т.к. более стабильны в щелочных средах при pH > 8. Силикагели устойчивы в пределах pH = 2-8.
Ионообменники характеризуются ионообменной емкостью — числом ионообменных центров на грамм ионооб-
менника. Обычно она выражается в единицах миллиэквивалента на грамм полимерной основы (или в миллимоль-г-1 для моновалентных групп).
В ИХ с кондуктометрическим детектором следует использовать ионообменники с низкой емкостью (менее 0,1 мэкв-г1). Ионообменники с емкостью около 1 мэкв-г-1 применяются только с амперометрическим и фотометрическим методами детектирования.
сильнее
слабее------->
Тримезат Фталат Цитрат л-гидрокси- Тартрат- Фосфатный Глюконат- л-толуол кон 1 мМ pH = 11 Бензоат Тартрат
натрия калия амония бензоат борат буфер борат сульфаты калия натрия
0,5 мМ pH = 8,6 1 мМ pH = 6,5 0,5 мМ pH = 6,5 1,8 мМ pH = 8,7 1 мМ pH = 4,0 5 мМ pH = 6,5 1,3 мМ pH = 8,6 3,5 мМ pH = 6,0 1 мМ pH = 6,0 2 мМ pH = 3,2
и ^,н2ро; - сГ F~_ —,HS - сГ "F" ✓ F" Z/НСОз 0
5- __ SU4 -\NOj -Л. Cit — С1 _ — N02 — Вг~ - NOj 2- 1 m 1 О 'l. G se L IJ 1 IN. In 1-0 ko uz « z к (6 1 гм I 1 к , О и о 1 Z м Z 1 1 L - СГ - N02 - Вт” —С1 _ _/№2 Вг" —NO” IN 1 -О о □ z z U 1 H3SiO4 /,оз Z>F" H2AsO3 ^BrO3 - F" — Ac — Form - BrO3 ^H2PO4 -h2asoj r-ioj -5
10- — SO4 - SeOj” -CIO4 - SO4" - HPO2 " N<>3 - НРО4” 4 .Cl - HS“ J - Cl" ^H2PO2 -10
- СгО42" — SCN - SeO4 “ soj" - CN -^no2 — no; ^BrOJ xcr
- so4 — SO4 2- “^CNO" 2
15- — I - cro; — SO4 ^Cl°3 -15
Q Л2” ” CN” — Оха! — SO* Br “ Br - C10J
5- S2O3 -NO3 - CIO^
—> МоО. 4 - no; - Br"
20- L20
Рис. 4.1.13. Сравнение удерживания анионов с элюентами разной элюирующей способностью на колонке 50 х 4,6 мм с анионообменником Waters IC Рак А или TSK IC PW (емкость 30 мкэкв г-1):
Ас — ацетат, Cit — цитрат, Form — формиат, Оха! — оксалат
В настоящее время наиболее распространенной основой для анионообменников являются сополимеры стирола с дивинилбензолом, т.к. они стабильны во всем диапазоне pH (0-14), имеют высокую степень сшивки (для увеличения механической прочности). При синтезе этих материалов легко получить сферические частицы. Удельная поверхность сополимеров стирола с дивинилбензолом может варьироваться в пределах 20-700 м2т-1, а средний диаметр пор колебаться от 2 до 50 нм.
Для получения ионообменника в матрицу полимера вводят ионогенные группы, которые фиксируются за счет ковалентной связи. Обычно их называют функциональными группами.
В соответствии с природой функциональных групп ионообменники подразделяются на
• катионообменники с фиксированными анионами: -
-SO-, -COO-, -РО2- и др.;
• анионообменники с фиксированными катионами: -NR,, -NHR;, -NH2R+ и др.
Ионообменники классифицируются (см. табл. 4.1.70) в зависимости от силы сопряженной кислоты (или основания).
Таблица 4.1.70
Классификация ионообменников в зависимости от силы сопряженной кислоты (или основания)
Ионообменник Тип Функциональные группы
Анионообменники Сильноосновной -(NR3)+
Среднеосновной -(NR3)+
Слабоосновной 4NH2R)
Катионообменники Сильнокислотный -SO-
Среднекислотный -PO32-;-AsO32-
Слабокислотный -COOH
Схема двухколоночной ИХ
Основная особенность двухколоночной ИХ состоит в наличии подавительной колонки и кондуктометрического детектирования, что позволяет реализовать наименьший
Аналитическая хроматография и капиллярный электрофорез
329
предел детектирования за счет снижения фонового тока (см. рис. 4.1.14).
Рис. 4.1.14. Схема двухколоночной ИХ
Элюент из емкости 1 насосом 2 с постоянной скоростью прокачивается через дозирующий кран 3, защитную колонку 4, разделительную колонку 5, подавительную колонку 6, ячейку кондуктометрического детектора 7 и сливную емкость 8. Аналоговый сигнал от детектора преобразуется в цифровой и подается на ПЭВМ 9 для регистрации и обработки. Результаты разделения и анализа можно распечатать на принтере 10 в виде протокола, состоящего из хроматограммы с разделенными компонентами и таблицы с количественными результатами.
Работу подавительной колонки можно рассмотреть на примере разделения катионов.
Аналитическая колонка заполнена катионообменником, в ней проходит следующий обмен:
-R-H + Na+(lC) + NO’ ё* -R-Na(K) + NO; + Н.
Подавляющая колонка заполнена ионообменником в -ОН-форме, в этой колонке анион NO3 замещает ОН, соединяясь с Н , переходит в Н2О:
-r-oh+no; + н -r-no;+н2о.
Сильнопроводящий анион остается на анионообменни-ке подавительной колонки.
В отличие от малой емкости разделительной колонки (0,01-0,1 мэквт’1), подавительная колонка имеет большую емкость — от 1 до 5 мэквт’1.
В табл. 4.1.71 приведен перечень элюентов, используемых при кондуктометрическом детектировании с химическим подавителем фоновой проводимости при разделении анионов и катионов. Эквивалентная проводимость некоторых анионов и катионов приведена в табл. 4.1.72.
Таблица 4.1.71
Элюенты, используемые в двухколоночной ионной хроматографии
Элюент Элюирующий ион Элюирующая способность Реакция подавления
При разделении анионов
NaOH ОН’ Низкая R-H + Na+ + ОН’ R-Na + Н2О
Na2B4O7 во; То же R-H + Na++ ВО: R-Na + НВО2
NaHCO3/Na2CO3 нсо;, со32’ Средняя 3R-H + 3Na++ НСО32” <=* 3R-Na + Н2СО3 + Н2О
Na2CO3 со32 То же 2R-H + 2Na+ + СО2’ <=* 2R-Na + H2CO3
NaHCO3/NaOH СО32’, ОН' Высокая 3R-H + 3Na+ + СО32’ +ОНГ ё* 3R-Na + Н2СО3 + Н2О
NaNO3/NaOH no;, он Средняя 2R-H + 2Na+ + NO; + ОН’ <=* 2R-Na + Н + NO; + Н2О
NaOC6H5 СбН5О’ То же R-H + Na++ СбН5О’ ё* R-Na + СбН5ОН
NH2CHRCOOH H2NCHRCOO“ Зависит от свойств кислоты 2R-H + Na++ H2NCHRCOO’ ё* ё* R-Na++ R-(NH3CHR)+COOH
При разделении катионов
HNO3 н+ Низкая r-oh + h++ no; r-no3 + h2o
AgNO3 Ag* Средняя R-Cl+Ag++ no; ё* R-NO3+AgC14<
C6H4(NH2)2 -2HC1 C6H4(NH2)H;+ Высокая 2R-OH+C6H4 (NH2)2 H22+ +2СГё± 2R-d+C6H4 (NH2)2 +2HO
BaCl2 Ва2+ То же 2R-SO4 + Ba2++2С1“ё^ 2R-C1 + BaSOj
Pb(NO3)2 Pb2+ То же 2R-IO3 + Pb2+ + 2 NO; 2R-NO3 + Pb(103)4
Zn(NO3)2/HNO3 Zn2+, Н* То же 5R-OH + Zn2+ + H + 3 NO; 3R-NO3 + R2-ZnO2 + 3H2O
KC1/HC1 К, Н Средняя 2R-OH + K+ + H" + 2СГ 2R-C1 + K+ + OH + H2O
NH2CHRCOOH + HC1 (H3NCHR)+COOH Зависит от свойств кислоты 2R-OH + (H3NCHR)+COOH - CM ё* R-Cl + 2H2O + ROOCRNFT
330
Новый справочник химика и технолога
Таблица 4.1.72
Эквивалентная электропроводимость некоторых анионов и катионов в водном растворе при 25 °C
Анионы Предельное значение эквивалентной проводимости, 0м-1-см2-экв4 Катионы Предельное значение эквивалентной проводимости, Ом-1-см2-экв-1
ОН 198 ЬГ 350
F" 54 Li+ 39
СГ 76 Na+ 50
Вг“ 78 nh; 73
Г 77 к+ 74
NO' 71 Mg2+ 53
so2 80 Ca2+ 60
свесит 32 Sr2* 59
С6Н4СОО2 38 Ba2+ 64
Схема одноколоночной ИХ
Как уже указывалось выше, одноколоночную ИХ предложили впервые Гьерде, Фритц и Шмуклер в 1979 г. Здесь не используется подавительная колонка, а аналитическая заполняется ионообменником на порядок меньшей емкости, чем в двухколоночной системе, в качестве элюента используют низкопроводящие буферные растворы (см. табл. 4.1.73).
Одноколоночная ИХ имеет некоторые преимущества перед двухколоночной, в частности, достигается более высокая эффективность (меньшее размывание); можно использовать и другие детектирующие системы, кроме кондуктометрической; проще аппаратура; нет необходимости в периодической регенерации подавительной колонки.
Недостатки одно колоночной ИХ: небольшой линейный диапазон, более высокий предел детектирования.
В табл. 4.1.74 приведен перечень органических кислот, используемых в качестве элюентов в одноколоночных ИХ.
Таблица 4.1.73
Фоновая проводимость элюентов
Элюент Концентра ЦИЯ, моль-л4 Величина pH Проводимость, мкСм-см4
Бензоат натрия 6,5 104 4,6 65,9
Гидрофталат натрия 5 104 4,4 74,3
5 104 6,2 112
6,5 104 4,4 90,5
6,5 104 6,2 158
о-Сульфобензоат аммония 5 104 5,8 132
Таблица 4.1.74
Свойства органических кислот, используемых в качестве элюентов
Кислоты P#i рА’г Р^З Степень диссоциации Элюирующая сила
Никотиновая 4,87 — — И Слабая
Бензойная 4,19 — — 22 ' Сильная
Сукциновая 4,16 5,61 — 25 Слабая
Лимонная 3,14 4,77 6,39 58 То же
Фумаровая 3,03 4,44 — 62 То же
Салициловая 2,97 13,4 — 63 Сильная
Детектирующие системы в ионной хроматографии
Наиболее распространенный и универсальный детектор в ИХ — кондуктометрический. Кроме него, в ИХ применяются следующие детекторы: электрохимический, спектрофотометрический, рефрактометрический, атомно-абсорбционный, флуоресцентный; системы на основе индуктивносвязанной плазмы, масс-спектрометрические и другие. В табл. 4.1.75 приведены, в частности, области применения некоторых детекторов.
Таблица 4.1.75
Методы детектирования в ионной хроматографии
Методы Измеряемые параметры Области применения
Кондуктометрия Электропроводность Анионы и катионы срА’<7
УФ/В-детек-тирование (до и после колоночной деривати-зации) Поглощение в УФ/В-обла- стях Анионы и катионы, тяжелые металлы после реакции с ПАР и тиро-ном, поливалентные анионы после реакции с Fe3+, силикаты и фосфаты после реакции с молибдатом, щелочноземельные металлы после реакции с Арсеназо I
Амперометрия Окисление или восстановление на рабочих электродах из Au, Ag, Pt, Pd, Cu и стеклоуглерода Анионы и катионы ср#>7
Флуоресценция с после-колоночной дериватиза-цией Возбуждение и эмиссия Аминокислоты и полиамины после реакции со фталевым альдегидом
Примечание. ПАР — 4-(2-пиридилазо)резорцин; тирон — 4,5-ди-гидроксидинатриевая соль 1,3-бензосульфоновой кислоты; Арсеназо — о-(1,8-дигидрокси-3,6-дисульфо-2-нафтилазо)бензо-мышьяковая кислота.
Аналитическая хроматография и капиллярный электрофорез
331
Основные требования к детекторам для ИХ: низкий предел детектирования (определяется уровнем шума и чувствительностью), линейность сигнала детектора в широком диапазоне концентраций, стабильность детектора при колебаниях скорости потока элюента и температуры, малый объем ячейки и др.
Принципы работы основных детекторов рассмотрены в главе, посвященной ВЭЖХ. Что касается кондуктометрического детектора, то принцип его работы основан на свойствах раствора электролита, который будет проводить электрический ток, если в него поместить два электрода и между ними приложить напряжение. Чем выше ток, проходящий через раствор, тем выше его электропроводность. По закону Ома:
U = IR,
(где U — приложенное напряжение, В; I — ток, A; R — сопротивление, Ом) сопротивление раствора будет определяться концентрацией и природой ионных смесей в растворе, а также температурой.
Электропроводность раствора G — величина, обратная его сопротивлению:
Удельная электропроводность К (Ом • см ) определяется следующим соотношением:
где L — расстояние между электродами, см; А — площадь электродов, см2.
К — постоянная кондуктометрической ячейки, определяется уравнением:
К = А [см-1].
Таким образом, удельная электропроводность:
X=GK.
Эквивалентная электропроводность X определяется концентрацией анализируемого вещества в растворе:
_ 1000Х Г см2 Л —------ ------ ,
с L0M3KB
где с — концентрация анализируемого вещества в эквивалентах на 1000 см3.
Измеряемая электропроводность связана с эквивалентной электропроводностью:
1000К ’
Детекторы в зависимости от областей применения подразделяются на универсальные и селективные. Кондуктометрический детектор — универсальный, все остальные можно отнести к селективным.
Некоторые ионы поглощают в УФ-области и их можно определять прямым УФ-детектированием (табл. 4.1.76). В табл. 4.1.77 приведены анализируемые ионы, элюенты области длин волн для косвенного (непрямого) спектрофотометрического детектирования (СПФ).
Перечень анализируемых ионов и хроматографические условия для прямого и непрямого СПФ-детектирования с использованием динамически покрытых ион-взаимодей-ствующих колонок приведен в табл. 4.1.78. Для снижения предела детектирования применяют непрямое флуоримет-рическое детектирование (табл. 4.1.79).
В последние годы широкое применение находят потенциометрические и амперометрические методы детектирования. Некоторые виды типичного применения потенциометрического детектирования приведены в табл. 4.1.80.
В табл. 4.1.81, 4.1.82 даны примеры использования и пределы детектирования амперометрического, импульсного амперометрического, кулонометрического, полярографического и других методов электрохимического детектирования.
Таблица 4.1.76 Перечень анализируемых ионов и некоторых рабочих
условий для прямого СПФ-детектирования после ионообменного разделения
Анализируемые вещества Элюенты Длина волны, нм Предел детектирования
АзОд”, AsO^’ НС1 200 1,5 ppm
Вг“ К2НРО4 195 0,6 нг
Вг“ NaCl 214 50 ppb
ВгО. CH3SO3H 205 50 ppb
ВгО; NaC104 210 50 ppb
ВгО; Na2CO3/NaHCO3 195 160 ppb
с2о2- Трис/ SO4" 205 50 нг
Цитрат Трис/БОд’ 205 300 нг
СГ CH3SO3H 190 30 ppb
СГ NaC104 190 3 ppb
СЮ’ Na2CO3/NaHCO3 195 0,2 ppm
С1О3’ Na2CO3/ NaHCO3 195 4 ppm
СгО2’ HNO3, NaOH 365 <1 ppb
Сг(1П), Сг(У1)-ЭДТА ЭДТА 350,220 2 ppm
Cr-, Pt-, Au- Cl-комплексы HC1 225 0,4 ppm
HCOO’ CH3SO3H 190 20 нг
332
Новый справочник химика и технолога
Продолжение таблицы 4.1.76
Анализируемые вещества Элюенты Длина волны, нм Предел детектирования
Юз NaCJO4 210 50 ppb
Ю3“ Na2CO3/NaHCO3 195 80 ppb
Металл-ЭДТА-комплексы ЭДТА 210 5-70 нг
N3" Na2CO3/NaHCO3 195 0,3 ppm
NO;, NO; NaCl 210 0,5 ppb
NO’, NO" NaC104 200-209 0,2-1 ppb
NO’, NO’ h2po; /СГ 214 0,01 ммоль
no;, no; LiOH 214 5 ppb
Органомышья-ковые кислоты NaH2PO4 210 60 ppb
PO34’ CH3SO3H 190 3000 нг
S2’ Na2CO3, NaOH 215 1 ppm
S2O2’, S3O2’, S4O2' CIO;, H2PO4 205 0,6-40 ppm
SCN K2HPO4 195 2 ppb
SeCNT Na2CO3 195 0,4 ppm
SeO2’ Na2CO3, NaHCO3 195 0,5 ppm
SeO2' Na2CO3, NaHCO3 195 15 ppm
SeO2' CH3SO3H 190 3000 нг
SeO2' NaClO4 190 1000 нг
Таблица 4.1.77
Определяемые ионы, элюенты и типичные условия для косвенного СПФ детектирования анионов и катионов после ионообменного разделения
Принятые сокращения: ен — этилендиамин, ац — ацетил-ацетон, триен — триэтилентетраамин, ЭДТА — этилендиамин-тетрауксусная кислота
Определяемые ионы Элюент X, нм Предел детектирования
Анионы
Обычные анионы NaNO3 247 1-10 РРт
Обычные анионы Фталат 300 0.1-0,3 мкг
СН3СОО", НСОО’, РО3’,СГ Бензоат 254 0,1 ppm
сю;, СГ, SCN“, SO2’, S2O2’ Тримезинат 317 0,5 ppm
Продолжение таблицы 4.1.77
Определяемые ионы Элюент X, нм Предел детектирования
F’, СГ, Br’, SO2’, S2O32’, Г Пиромеллитат 285 0,05 ppm
сг,Br“, no;, so2’ Цитрат 290 0,2 ppm
сг, no;, no;, SO2’ Салицилат 254 1-2 нг
cr, no;, no;, SO2’ и-Толуол-сульфонат 280 50 ppb
cr, no;,bt’, no; 2-Нафтил-аминсульфо-нат 300 2-10 ppb
cr, no;, so42’ [Си(ЭДТА)]2- 290 1,6-11 нг
cr,Br’, no;, so2' Тирон 290 50ppb
h2po; , cr, no; , SO2’ Сульфобензоат 270 300 пмоль
p2or,p3o;0 Тримезинат 285 1 ppm
SiO32’F’, СГ, Br’,PO3’, SO2’ 2,4-Дигидро-ксибензоат 312 0,2 ppm
SO2’ 5-Сульфо-изофталат 240 50 пмоль
Катионы
T етраалкил аммониевые ионы Бензил-триэтиламин 270 0,6-0,8 мкг
Моновалентные катионы, амины Бензил- триэтил амин 275 0,2-1 PPm
Li+, Na+, NH4+, К+, Rb+, Cs+ 4-Метил-бензиламин 262 0,2-10 ppb
Li+, Na+, NH; , K+, Rb+, Cs+ 2,6-Диметил-пиридин 269 0,4-11 ppb
Li+, Na+, NH4+, K+, Rb+, Cs+ 2-Метил-пиридин 262 0,4-11 ppb
Li+, Na+, NH; , K+, Rb+, Cs+ 2-Фенилэтил-амин 257 0,7-14 ppb
Na+, K+ Пиридин СГ 260 0,2 ppm
Na+, NH;, K+ CuSO4 252 10 ppm
Na+, K+, Rb+, Cs+, Ca2+, Mg2+ CuSO4 220 30-300 PPb
Na+, K+, Rb+, Cs+, Ca2+, Mg2+ Ce2(SO4)3 254 4 ppb
Na+, K+, Ca2+, Mg2+ CoSO4 218 7-34 нг
Ca2+, Mg2+ [Со(ен)2(ац)]г+ 254 10 ppb
Аналитическая хроматография и капиллярный электрофорез
333
Продолжение таблицы 4.1.77
Определяемые ионы Элюент X, нм Предел детектирования
Са2*, Mg2+ РЖЭДТА)]2- 290 1,7-2,3 нг
Mg2*, Са2*, Sr2*, Ва2* 2-Фенилэтил-амин 257 1-58 ppb
Mg2+, Са2*, Sr2*, Ва2* 4-Метил-бензиламин 262 1-58 ppb
Mg2*, Са2*, Sr2*, Ва2* [Си(триен)] *" 255 0,2-10 нг
Таблица 4.1.78
Определяемые ионы и хроматографические условия для прямого и косвенного СПФ-детектирования в ИХ с использованием динамически покрытых ион-взаимодействующих колонок
Принятые обозначения. ЦМА — цетилтриметиламмоний, ГДТМА — гексадецилтриметиламмоний, ОА — октил аммоний, ТБА — тетрабутиламмоний, ТКМА — трикаприлилметил-аммоний, ТОМА — триоктилметиламмоний, ТПА — тетра-пентиламмоний, ТСА — толуолсульфонат, Сал. — салицилат, Фт. — фталат.
Прямое детектирование Анализируемые вещества X, нм Предел детектирования
цма+н2ро; IO", BrO”,HS", NO", HSO3", N", no; , Br", s2o2", г 215 2-90 нг
ЦМА + Cit" io;, Bro;, no;, no; 220 1-10 нг
ГДТМА + н2ро; io;, Bro;, no;, Br", no; , г 205 5-30 ppb
оа+ н2ро; CN", HCO;, CNO", no;,f", io;, r, MoO2" 200 0,1 ppm
оа+ н2ро; io;, вг, no;, no;, г 205 50 ppb
ТБА + ОН" io;, no;,no;,f, S2O2" 215 0,1 ppm
ТБА + ОН Металл-CN-комплексы 210 50 ppb
ТБА + ОНГ Селенометаллаты 254 1 ppm
ТБА+ н2ро; Металл-CN-KOMn-лексы 214 1-3 нг
ТБА+ н2ро4 Au(CN); 214 0,4 ppb
Продолжение таблицы 4.1.78
Прямое детектирование Анализируемые вещества X, нм Предел детектирования
тба+н2ро; no;, no; 214 0,1 ppm
тба+н2ро4 г 235 50 ppb
тба+hso; S2O3", г 212 10-100 ррь
ТКМА+СГ no;,io;, Bro;, вг, no;,г, scn" 205 2нг
ТОМА + н2ро; СНзСОО", HCOO", no; , Br", no; 214 0,1 ppm
ТОМА + ОН" io;, no;,no;,f, S2O2" 215 0,1 ppm
ТПА + Вг" NO;,Br", no; 210 50 ppb
ТПА+F" No;,Br~, no;,г 210 50 ppb
Косвенное детектирование
ЦМА + TSA" io;, cf,no;, вг, no;, сю;,г 240 0,5 ppm
ОА + сал. hco; , cr, no; 254 0,5 ppm
ТБА + Фт." cr, no;, hpo2’, SO2" 225 50 ppb
ТВ А + Фт." bf4 ,cr, cio;, C1O4" 280 0,2 ppm
ТВ А + Сал." h2po;,cf, no;, вг, no;,г, so2", s2o32 288 6-60 ppb
Таблица 4.1.79
Определяемые ионы и условия непрямого флуоресцентного детектирования в ИХ
Элюент Анализируемые ионы X, нм Предел детекти рования
возбуж. ЭМИС.
Салицилат ci, no; 325 420 2 пг
Салицилат ю;, сг 325 420 6,7 нг
Салицилат no; , сю; 280 390 0,2 пмоль
Салицилат cr, no;, so2" 325 404 1-2 нг
Церий(П1) Na*, NH;, К* Rb* Cs* 247 350 3-160 ррь
Церий(Ш) Li* Na*, NH;, K*, Rb*, Cs*, Ca2*, Mg2* 247 350 0,2-100 ppb
334
Новый справочник химика и технолога
Продолжение таблицы 4.1.79 Продолжение таблицы 4.1.80
Элюент Анализируемые ионы X, нм Предел детекти рования Анализируемые вещества Рабочий электрод Предел детектирования
возбуж. ЭМИС.
6,7-дигид-рокси-2-нафталин-сульфоновая кислота F, Вг, Г, SCN , SO*~ 254 420 7,5 нмоль F Фторид - ISE 10-20 ppb
Галогены Ag-AgCl 0,5 ppm
Галогены, SCN- Ag2S-ISE 10 мкмоль
Галогены, SCN- Ag - Ag салицилат 0,2 ppm
ТЬС13 NO;, SO*-, S2O*-, CrOf, PtClf 225 553 28 пг
Галогены, SCN, SO," Ag-AgCl 0,05 ммоль
Г Иодид - ISE 0,015 мкмоль
Г Стекло с фосфатом свинца 50 ppb
Биацетил NO;, SO*- 400 515 20 пмоль
Таблица 4.1.80 Применение потенциометрического детектирования в ИХ Неорганические анионы Н - стекло 0,08-2,0 мкмоль
Неорганические анионы ГТ - полимер 1,1-6,5 мкмоль
Анализируемые вещества Рабочий электрод Предел детектирования
Неорганические анионы, IO;, ВгО;, CIO3 Медь 0,02-10 нмоль
Щелочноземельные катионы Медь 1 ppm Моновалентные катионы ПВХ-мембрана 1 мкмоль
Аминокислоты Медь 75-300 нг Моновалентные катионы Н+ - полимер 4,7-7,8 мкмоль
Аминокислоты, диамины Медь-ISE 10-100 нг
Na+, Li+,F, РО*-, СН3СОО-, so*- Неселективная мембранная ячейка 10 нмоль
Аскорбиновая кислота, NalU, NH2OH Медь 2-10 нмоль
ВГ Hg2Br2-ISE 20 ppb NO;, NO; Нитрат - ISE 0,1-0,3 нмоль
Карбоновые кислоты Медь 20-50 нмоль
Оксалат Медь 1 ppm
Карбоновые кислоты Медь 0,2-5,3 мкг Сахара Тоже 1-5 нмоль
СЕТ, S2 Ag2S-ISE 10 ppb Ионы переходных металлов То же 1-10 нмоль
Карбоновые кислоты FT - стекло 1 мкэкв
Таблица 4.1.81
Некоторые типичные применения электрохимического детектирования в ИХ
Принятые сокращения. А — амперометрия, П — полярография, И — импульсная амперометрия, ДПЭ — двойные параллельные электроды, СУ — стеклоуглерод, КРЭ — капающий ртутный электрод, I-Pt — иодизированная Pt.
Анализируемые вещества Проба Режим Рабочий электрод Сравнительный электрод Потенциал (В) Предел детектирования
Альдегиды (как аддукты с SO") Вина А СУ Ag/AgCl 0,75 5 ppm
As(III) Минеральная вода То же Pt Ag/AgCl 1,00 0,012 мкМ
ВС Вода То же Ag Ag/AgCl 0,10 10 ppb
Вг- Плазма То же Ag Ag/AgCl 0,03 10 ppb
СЬГ Сбросы То же Ag Ag/AgCl 0,02 Ippb
CN“, HS , Г, вг Стандарты То же Ag Ag/AgCl 0-0,3 2-30 ppb
Cu2+, Ni2+ Моча То же СУ Ag/AgCl 0,8 < 80 ppb
F-, сг,ро*-, no; Стандарты Косвен. А Ag Ag/AgCl 0-0,45 < 0,5 ppm
Аналитическая хроматография и капиллярный электрофорез
335
Продолжение таблицы 4.1.81
Анализируемые вещества Проба Режим Рабочий электрод Сравнительный электрод Потенциал (В) ПД
HS , CN Питьевая вода П КРЭ Ag/AgCl 0,00 1-2,5 ppm
Г, S2O2 Фармацевтика А, ДПЭ СУ Ag/AgCl 1,05; 1,15 10-50 ppb
Г Йодированная соль А I-Pt Ag/AgCl 0,8 10 ppb
Г Морская вода Тоже СУ Ag/AgCl 1,0 5 ppb
Лактоза Чипсы И Au Ag/AgCl Пульсир. ' —
Мальтоза Кукурузные чипсы То же Au Ag/AgCl Пульсир. 50 ppb
NO’, NO’ Копченое мясо, рыба А, ДПЭ СУ Ag/AgCl 1,10,1,00 50
Органические кислоты Моча А СУ — 1,10 —
SO32’ Пища То же Pt Ag/AgCl 0,4 0,1 ppm
S2O32’ Моча, плазма А, ДПЭ СУ Ag/AgCl 0,7,0,9 10 пмоль
S2’ Туман А Ag Ag/AgCl 0,00 19 ppb
S2’, SO2’, S2O32’ Уголь Косвен. П КРЭ — 0,08 —
S2’, S2O32’ Ликер А Ag — — —
S’,Br’,CN’ Кислые воды То же Ag Ag/AgCl 1,0 —
Серийно выпускаемые ионные хроматографы
В нашей стране специализированные ионные хроматографы с кондуктометрическими и другими детекторами производят три организации (табл. 4.1.82).
Таблица 4.1.82
Ионные хроматографы, выпускаемые в России
Название модели Организация-производитель Детекторы Технические характеристики
Цвет-Яуза модель 2 Цвет-Яуза модель 3 ОАО НПО «Хим-автоматика» (г. Москва) Кондукт. Кондукт.н- ампер. Колонки и детектор термостатируются, насос до 250 атм, дозирующий кран Rheodyne
Стайер-CD Стайер-А НПКФ «Аквилон» (г. Москва) Кондукт. То же Насос до 250 атм., кран-дозатор Rheodyne. Предназначен для анализа особочистых вод
Цвет-4000 ОАО «Цвет» (г. Дзержинск, Нижегородской обл.) То же Насос до 200 атм., кран-дозатор собственного производства
Впервые в нашей стране ионные хроматографы были произведены ОАО «Цвет» в 1982 г. Длительное время серийно выпускались лабораторные ионные хроматографы «Цвет-3006» и портативные (перевозимые) ионные хроматографы ХПИ-1.
За рубежом лидирующее положение в области ионохроматографического детектирования занимает фирма Dionex (США), выпускающая портативные ионные хроматографы (модели DX-80 и DX-100), лабораторные ионные хроматографы (модели DX-120, DX-500), а также автоматические промышленные ионные хроматографы. Фирма Dionex производит все необходимые компоненты для ионной хроматографии: экстрактор, автосамплер, генератор элюента EG40, химические подавители (в т.ч. саморегени-рирующиеся подавители), выпускает также аминокислот
ный анализатор на основе анионообменной хроматографии и импульсного амперометрического детектирования. Этот анализатор осуществляет прямой анализ аминокислот (без химической дериватизации), он в 50 раз более чувствителен, чем классические аминокислотные анализаторы с применением нингидриновой реакции. Dionex выпускает весь ряд аналитических колонок для ионной хроматографии.
Фирма Metrohm (Швейцария) производит компактный ионный хроматограф (модель 761) с двухплунжерным насосом, дозирующим краном, подавителем и набором колонок. Кондуктометрический детектор термостатируется с точностью до 0,01 °C.
Ионные хроматографические системы на базе жидкостных хроматографов выпускает фирма «А1тех Associated» с
336
Новый справочник химика и технолога
введением автоподавителя фонового тока типа ERIS, а также фирмы «Jand М Analytische Mess - and Regeltechnik & Gmbh» (Германия), «LabAlliance» (США), «Lachat Instruments» (Zellweger Analytics), «Prior Separation Technology», «SeQuant АВ».
Ниже (см. табл. 4.1.83) рассмотрены некоторые типичные неисправности в ИХ системах.
Таблица 4.1.83
Основные неисправности в ИХ системах
Нарушения Возможные причины Действия по устранению
Высокая фоновая проводимость, но небольшой шум Элюент загрязнен анионами сильных кислот Проверить электропроводность деионизированной воды, используемой для приготовления элюента
Высокий фон и большой шум Подавитель не работает. Высокое давление в детекторе Проверить работу подавителя. Промыть ячейку
Высокий (периодический) шум с нормальным уровнем фона Возможные пузырьки в насосе. Неисправность насоса Удалить воздух из насоса и дегазировать элюент. Исправить ячейку
Чувствительность для анионов слабых кислот уменьшается Неполное подавление; pH после подавителя низкий Проверить и отрегулировать работу подавителя. Проверить pH элюента после подавителя
Высота пика сульфата уменьшается и ширина увеличивается Ионы металла в подавителе Промыть мембрану щелочным раствором соли магния ЭДТА
Дрейф нулевой линии Система еще не вышла на режим. Утечки жидкости в системе, колебания температуры, грязные фильтры Подождать выхода на режим. Проверить стабильность параметров системы
Отрицательные пики Высокий фон Отрегулировать подавитель
Высокое обратное давление Входной колоночный фильтр забит. Неисправен инжектор, он может занять положение между указателями «анализ» и «ввод» Отсоединить колонку. Сравнить с нормальным давлением, если давление остается высоким, то проверить кран
Области применения ИХ
Ниже перечислены основные области применения ИХ: контроль пищевых продуктов и лекарств; анализ биологических жидкостей в медицине, кислотности почв; контроль в производстве полупроводников и в энергетических отраслях; анализ детергентов в сточных водах; анализ растворов гальванических ванн и проявляющих растворов; определение серы, анионов и катионов в нефтепродуктах; контроль выбросов в целлюлозно-бумажной, химической, металлургической промышленности.
В табл. 4.1.84 дан перечень аттестованных методик, выполняемых методом ИХ.
В табл. 4.1.85 приведены примеры промышленного применения ИХ, опубликованные в [3-5]. В табл. 4.1.86 перечислены анионы, определяемые в пищевых продуктах, в табл. 4.1.87 приведены примеры применения ИХ в медицине, в табл. 4.1.88 и 4.1.89 — примеры анализов газов, аэрозолей и состава микрочастиц в атмосфере.
Таблица 4.1.84
Перечень аттестованных и включенных в Госреестр методик
Название методики Номер регистрации
Метод выполнения измерений (МВИ) массовых концентраций диоксида азота и азотной кислоты (суммарно), оксида азота, триоксида серы и серной кислоты (суммарно), диоксида серы, хлороводорода, фтороводорода, ортофосфорной кислоты и аммиака в пробах промышленных выбросов, атмосферного воздуха и воздуха рабочей зоны ПНДФ 13.1:2:3.19-98
МВИ массовых концентраций ионов натрия, калия, магния, кальция, бария и аммония в пробах природной, питьевой и сточной воды ПНДФ 14.1:2:4.131-98
МВИ массовых концентраций ионов, NO”, NO', СГ, F", SO^’, РО3’ в пробах природной, питьевой и сточной воды ПНДФ 14.1:2:4.132-98
МВИ массовых концентраций ионов в no; , no; , СГ, F-, SO,, РО34 пробах почв (в водорастворимой форме) ПНДФ 16.1.8-98
МВИ массовой концентрации 1,1-диметилгидразина в образцах природных вод методом ИХ с амперометрическим детектором Свидетельство № 1-99 о метрологической аттестации МВИ
МВИ массовой концентрации фенола в питьевых, хозяйственно-быто-вых и поверхностных водах ПНДФ 14.1:2:4.8-95
Аналитическая хроматография и капиллярный электрофорез
337
Таблица 4.1.85
Промышленные применения ионной хроматографии [3—5J
Таблица 4.1.87
Применение ИХ в медицине (на основании 23 публикаций)
Области применения Анализируемые ионы
Анализ промышленных сточных вод, сбросов и выбросов Фторид, хлорид, нитрат, сульфат, сульфит, бисульфид, карбонат, фосфат, сульфид, цианид, тиосульфат, тиоцианат, диэтилфосфат, диэтилфосфотиоат, бутират, дибутилфосфат, монобутилфосфат, тетрафторборат. Фенол, моноэтиламин, диэтиламин, триэтиламин, гидроксиламин, катионы лития, натрия, аммония, калия, магния, кальция, Fe2+, Fe3+, меди, свинца, цинка, никеля, кобальта, кадмия, Сг2 *, Сг3'
Сбросы целлюлозно-бумажной промышленности Сульфид, сульфит, сульфат, оксалат, тиосульфат, ацетат, хлорид, бикарбонат, гипохлорид, хлорит
Анализ детергентов (синтетических моющих средств) Фосфат, диполифосфат, сульфат, триполифосфат, ортофосфат, октилсуль-фатнатрия, децилсульфат натрия, додецилсульфат натрия, тетраэтилсульфат натрия, алкилбензо сульфаты Ci - С]6
Таблица 4.1.86
Анионы, определяемые в пищевых продуктах
Анион Объекты анализа
СГ NO; sof ро3 Фруктовый сок, газированные напитки, листья чая, молоко, вино, овощи, морковный сок, картофельные чипсы, свиное мясо, детское питание, шпинат, пиво, компоты, фрукты, попкорн
NO" Апельсиновый и фруктовые соки, шпинат, зерно, кофе, компоты, мясо, пищевые экстракты, консервы, пиво
SO2’ Напитки, красное вино, лимонный и фруктовые соки, трибы, овощи, кока-кола, крахмал, пиво, виноград
н2ро; Чай, апельсиновый сок, печень, шпинат
г Молочные продукты, соевые детские продукты, салаты, рыба
Вг’ Попкорн, хлеб, мясо, овощи, томатный сок
СЮ’ Овощи
ВгО; Бакалейные продукты, хлеб
Ю3’ Молоко, попкорн, столовая соль
СгО2’ Апельсиновый сок, картофельные чипсы
SeO2”, SeO2’ То же
сьг Абрикосы, зерна фруктов, консервы
Анализиру емые объекты Анализируемые ионы
Кровь, сыворотка, плазма Хлорид, фосфат, бромид, нитрат, сульфат, тиосульфат, азид, нитрит, цианид, тиоцианат, сукцинат, иодид, лактат, 3-гидрокси-бутират, креатинин, мочевая кислота, катионы натрия, аммония, калия, магния, кальция, меди, цинка, железа
Моча Фторид, лактат, хлорид, бромид, нитрат, сульфат, оксалат, фосфат, иодид, нитрит, тиоцианат, тиосульфат, цитрат. Кислоты: оксалиновая, оксиуксусная, аскорбиновая, ванилилминдалевая, кетоглутамаровая, лимонная, изолимонная, сукциновая, катионы натрия, калия, аммония, кальция, магния, цинка
Слюна Хлорид, фосфат, сульфат, лактат, нитрит, тиоцианат, катионы магния, кальция
Почечные камни Оксалаты
Спиномозговая жидкость Кислоты: мочевая, аскорбиновая, гомованилиновая
Экстракт тканей Катионы натрия, калия, аммония
Экстракты кала Нитрат
Таблица 4.1.88
Фильтры для сбора газов, аэрозолей и частиц
Проба Среда фильтров Экстрагенты Смеси, количественно определяемые ИХ
Аэрозоли Кварцевая вата 0,05 мМ НС1О4 БОд", NO“,Na+
То же Мембрана 0,05мМНСЮ4, 0°С SO2’, SO32’, no;
То же ПТФЭ-мембрана 1мМ фталат cr, no;, so2’
Аэрозоли, краски ПВХ-мембраны 2%NaOH, 3%Na2CO3 CrO2’
Частицы ПТФЭ-мембраны Деионизированная вода cr, no;, no;, SO2’
Кислые газы Бумага, пропитанная 1МК0Н Деионизированная вода cr, no;, no;, SO2’
Диоксид серы Бумага, пропитанная Na2CO3 Н2О2 SO2’
Аммиак Мембрана, пропитанная 10%Н3РО4 Деионизированная вода nh;
338
Новый справочник химика и технолога
Таблица 4.1.89
Свойства и применение некоторых адсорбционных трубок для улавливания из газовой фазы соединений с последующим определением ИХ
Принятые обозначения. МЭА — моноэтаноламин, ДЭА — диэтиламин, ТЭА — триэтаноламин.
Проба Материалы в адсорбционной трубке Смеси, определяемые ИХ
Аммиак Уголь, пропитанный H2SO4 nh;
Аммиак Силикагель, пропитанный H2SO4 NH^, амины
Хлорацетил-хлорид Силикагель Хлорацетат, СГ
Этаноламины Оксид алюминия МЭА, ДЭА, ТЭА
Формальдегид Уголь, пропитанный окислителем НСОО~
Муравьиная кислота Хромосорб 103 НСОО
HF Силикагель F
НС1, Н3РО4, НВг HNO3, H2SO4 Силикагель СГ, РО^Вг" no;, so2-
H2S, SO2 Анионообменники в ОН-форме so$-
Неорганические кислоты Силикагель СГ, Br, NO3, so*
Иод Уголь с NaOH г
no2 Трубка с ТЭА no;
no2 Sep-Pak Cj8 с ТЭА no;
SO2 Активированный уголь SO*-
SO2, no2 Sep-Pak Ci 8 с ТЭА-КОН no;, no;, so32,so2-
SO2, no2 Цеолиты с TEA no;, so2-
Литература
1. Weiss J. Ion Chromatography. 2nd ed. Weinheim: VCH, 1994. 467 p.
2. Шпигун O.A., Золотов Ю.А. Ионная хроматография. М.: МГУ, 1990. 198 с.
3. Haddad P.R., Jackson Р.Е. Ion Chromatography Principles and Application. Amsterdam: Elsevier, 1990. 748 p.
4. Gjerde D.T. and Fritz J.S. Ion Chromatography. 2nd. ed. N.Y.: Huthig; Mamaroneck, 1987. 278 p.
5. Smitt R. Ion Chromatography Applications, CRC Press, Boca Raton FL. 1988.
6. Weiss J. Handbook of Ion Chromatography. CA: Dionex Sunnyvale, 1986.
7. Smith F.C., Chang R.C. The Practice of Ion Chromatography. N.Y.: John Wiley, 1983. 218 p.
8. Haddad P.R., Jackson P.E. Ion Chromatography Principles and Applications // Chromat. Libr. Amsterdam: Elsevier, 1990. V. 146. 748 p.
9. Macdonald J.C. Inorganic Chromatographic Analysis. N.Y.: J. Willey, 1985.450 p.
10. Japan Society for Analytical Chemistry Ion Chromatography Data Book, Tokyo: Kagaku Shinbunska, 1991. 548 p.
11. Нестеренко П.Н., Тарасенко Д.А., Шпигун О.А. Одновременное определение анионов и катионов ИХ И Журн. аналит. химии. 1994. Т. 49. С. 244-256.
12. Шпигун О.А., Обрезков О.Н. Современная ИХ И Российский хим. журнал. 1994. Т. 38. С. 16-20.
4.1.10. Метод тонкослойной хроматографии (ТСХ)*
Тонкослойная (или планарная) хроматография (ТСХ) — простой, дешевый, оперативный метод хроматографии для анализа всех классов соединений. В рассматриваемом методе для разделения используются стеклянные, металлические, пористые керамические и пластмассовые пластины размером 100 х 100 мм и другие, с однородным слоем адсорбента толщиной 50-300 мкм. Фракция зерен адсорбента может быть в пределах 3-15 мкм. Применяют стеклянные пластинки, на поверхности которых спекают слои адсорбента (такие пластины можно использовать повторно), а также пластины на основе стекловолокна — Instant thin-layer chromatography.
Основные адсорбенты для пластин в ТСХ: силикагель, оксид алюминия, микрокристаллическая целлюлоза, триацетат целлюлозы, циклодекстрины и их производные, флорисил (силикат магния), модифицированные силикагели, силикагели, пропитанные 8-оксихинолином для разделения комплексов металлов, неорганические ионообмен-ники, хитозан и др. В табл. 4.1.90 приведены типы используемых пластин.
Таблица 4.1.90
Пластины для ТСХ
Тип ТСХ Диаметр зерен адсорбента, мкм Толщина адсорбционного слоя, мкм
Классическая ТСХ 10-15 300
Высокоэффективная ТСХ (ВЭТСХ) 5-7 <200
Микро-ТСХ 2-5 50-70
Препаративная ТСХ 500-2000
Для препаративной хроматографии используют пластины с изменяющейся толщиной адсорбционного слоя.
Классификация пластин по адсорбционной активности: • полярные (на основе силикагелей и оксидов алюминия); • неполярные (на основе силанизированных силикагелей);
См. также раздел 3.2.9.4.
Аналитическая хроматография и капиллярный электрофорез
339
• неполярные (обращеннофазовые) на основе модифицированных силикагелей с алкильными цепями Cz-C8, Ci8;
• средней полярности на основе привитых амино-, циано- и диольных групп.
В качестве связующего материала для закрепления адсорбционного слоя применяют гипс, крахмал, полиакриламид, силикагель. Связующие материалы добавляются в пределах 2-15%.
К некоторым пластинам для повышения чувствительности детектирования добавляются флуоресцирующие вещества (флуоресцеин, пирен и др.).
Кроме пластин, в ТСХ для разделения используются стержни (150 х 0,9 мм) из стали, меди или кварца. После разделения смеси и высушивания стержни с постоянной скоростью пропускаются через пламя ионизационнопламенного детектора. Эти стержни можно использовать многократно (до 100 раз).
Основные преимущества ТСХ:
• одновременное разделение нескольких проб (до 20 и более);
• возможность двумерного разделения;
• можно наблюдать весь процесс разделения;
• высокая производительность;
• полный анализ (на старте можно проверить остатки пробы);
• быстрота разделения;
• низкая стоимость;
• небольшое количество элюента;
• возможность широкого выбора цветных реакций;
• непосредственный контакт при детектировании радиоактивных соединений;
• возможность применения селективных и специифиче-ских детекторов.
Недостатки ТСХ:
• пластины в ТСХ — открытые системы, поэтому некоторые неустойчивые соединения (чувствительные к кислороду, влаге и т.п.) могут разлагаться;
• в простейших вариантах ТСХ затруднен количественный анализ; в этих случаях можно говорить о по-луколичественном методе;
• по эффективности разделения метод ТСХ сильно уступает высокоэффективной жидкостной хроматографии.
Исторически в ТСХ количественное измерение проводили посредством визуальных наблюдений. В последние годы созданы совершенные, но дорогие детектирующие системы: денситометрия, масс-спектрометрия для ТСХ, радиосканирование изотопов, ТСХ-ИКС и др., лазерное сканирование для флуоресцентных измерений. Сканирование пятен с помощью УФ-сканеров позволяет детектировать на уровне нанограмм. Флуоресцентные сканеры позволяют достичь предела детектирования в наиболее благоприятных случаях пикограммового уровня.
В нашей стране ТСХ применяют около 10 тысяч лабораторий.
Основные организации и лаборатории, использующие ТСХ:
— центры Госсанэпидемнадзора;
— центры стандартизации и метрологии;
— экологические центры;
— аналитические лаборатории по контролю качества пищевых продуктов;
— лаборатории станций защиты растений;
— ветеринарные лаборатории;
— лаборатории агрохимслужбы;
— лаборатории судмедэкспертизы;
— наркологические диспансеры; 5
— медико-генетические лаборатории;
— экспертно-криминалистические лаборатории МВД.
В нашей стране пластины для ТСХ выпускает ЗАО «Сор-полимер» на полимерной (лавсановой) подложке ПТСХ-П и на алюминиевой подложке ПТСХ-АФ (см. табл. 4.1.91). Полимерные лавсановые подложки для пластин устойчивы до 130 °C и практически устойчивы ко всем растворителям.
Пластины выпускаются трех типов: 10 х 10 см, 10 х 15 см и 10 х 20 см.
Таблица 4.1.91
Пластины «Сорбофил»
Типы Обозначения
Аналитические без люминофора ПТСХ-П-А
Аналитические с люминофором ПТСХ-П-А-УФ, ПТСХ-АФ-А-УФ
Высокоэффективные без люминофора ПТСХ-П-В, ПТСХ-АФ-В
Высокоэффективные с люминофором ПТСХ-П-В-УФ, ПТСХ-АФ-В-УФ
ЗАО «Сорбполимер» поставляет комплектные наборы:
— универсальные наборы;
— наборы с лампой на 254 нм;
— наборы с лампой на 313 и 365 нм;
— генетические наборы;
— наборы для анализа микотоксинов.
В состав набора входят: хроматографические камеры двух типов, распылитель с камерой для распыления реагента, термостолик (компактный нагрев для подсушки пластин), специальные микрошприцы с укороченной иглой, УФ-осветители с длинами волн 254, 313, 365 нм, трафареты, камеры опрыскивания с подставкой.
Для сканирования пятен выпускается видеоденситометр «Сорбитол». Измерение пятен в денситометрии основано на определении прошедшего через пятно или отраженного от пятна света по отношению к интенсивности света, прошедшего через чистую пластину или отраженного от чистой поверхности.
Дальнейший прогресс в ТСХ связывают с применением: • автоматических приспособлений для дозирования;
• центрифугирования;
• средств автоматизации;
• электрического поля для ускорения миграции веществ (тонкослойная электрохроматография);
340
Новый справочник химика и технолога
а также с
• разработкой новых методов детектирования веществ на хроматограммах;
• проведением процессов в ТСХ под давлением или непрерывном потоке элюента (процесс, приближающийся к ВЭЖХ);
• антикруговым вариантом ТСХ.
Для повышения эффективности метода используют:
— слой сорбента с градиентом pH;
— градиент состава подвижной фазы;
— многократное элюирование;
— проведение процесса под давлением;
— насыщение слоя сорбента парами подвижной фазы.
Современная ТСХ становится инструментальной и количественной, это несомненный прогресс. Однако в этом случае оборудование дорожает и такие преимущества ТСХ, как дешевизна и простота, теряются.
За рубежом оборудование для ТСХ выпускают следующие фирмы: Camag, Analtech, Camag Sci., Fisher Sci., Hellamco SA., Interchim, Laborgeratere, Labor Exchange и др.
Пластины для ТСХ, кроме вышеуказанных, выпускают фирмы: Merck, Supelco, Whatman, Tokyo Kasei, Macherey-Nagel, ICN Bioanalitical, Bioscan, Stratlab, Capital и др. [32].
Выходит специализированный международный журнал «Journal of Planar Chromatography» и периодически проводятся Международные симпозиумы по ТСХ.
Литература
1. Zlatkis A., Kaiser R. HPTLC: High Performance Thin-Layer Chromatography. Amsterdam: Elsevier, 1977.
2. Волынец М.П. Тонкослойная хроматография в неорганическом анализе. М.: Наука, 1974.
3. Kaiser R. Einfuhrung in die HPPLC. Heidelberg: Huthig, 1987. 256 s.
4. Jork H., Funk W., Fischer W., and Wimmer H. Thin layer chromatography — Reagents and Detectionsmethods VCH-Verlags-gesellschaft. Weinheim, 1990.
5. Maslowska J., Mlodzikowski X. Chitosan a Ew Adsorbent For Thin-Layer Chromatography. Separation of Metal Ions Countuined in Polfamins Mixtures. // Chem. Anal. 1986. V. 31, №2.P. 193-199.
6. Методы определения микроколичеств пестицидов в продуктах питания, кормах и внешней среде: Справочник. М.: Колос, 1992.
4.1.11. Особенности хроматографии с подвижной фазой в сверхкритическом состоянии
Газовая хроматография при невысоких давлениях с использованием слабо адсорбирующихся (практически не адсорбирующихся) газов-носителей применяется для разделения и анализа достаточно летучих веществ (с давлением пара не менее 0,13 Па). Однако многие вещества, хроматографический анализ которых имеет важное практическое значение, во-первых, недостаточно летучи и, во-вторых, очень сильно адсорбируются даже при высоких температурах колонки или претерпевают термо деструкцию. Речь идет о соединениях с большой молекулярной массой
(полимеры), соединениях со средней молекулярной массой (олигомеры), сильно полярных соединениях, в частности аминокислотах, лекарственных веществах, белках, а также очень сильно адсорбирующихся даже на слабо специфических адсорбентах труднолетучих неорганических соединениях.
Чтобы увеличить летучесть некоторых из таких соединений и понизить коэффициенты их распределения (константы Генри), не прибегая к сильному повышению температуры колонки, можно вместо практически не Адсорбирующегося газа-носителя в качестве подвижной фазы использовать довольно сильно адсорбирующиеся вещества с большой концентрацией (в подвижной фазе). Эти вещества применяют в сверхкритическом состоянии, когда достаточно высокая их концентрация достигается за счет высокого давления.
Хроматография с подвижной фазой в сверхкритическом состоянии (СФХ) занимает промежуточное положение между газовой и высокоэффективной жидкостной хроматографией по возможностям разделения высококи-пящих соединений.
В свою очередь, сверхкритическое состояние вещества занимает промежуточное положение по характеристикам между газом и жидкостью (табл. 4.1.92). Физические параметры некоторых сверхкритических флюидов приведены в табл. 4.1.93.
Таблица 4.1.92
Сравнение хроматографических методов
Подвиж ная фаза Плотность, г-мл-1 Вязкость, сП Диффузия, см2-с-1 Тип колонок
Газ ~10’3 (0,5-3.5) •ю-4 0,01-1,0 КК
Сверхкритический флюид 0,2-0,9 (0,2-1,0) •10"* 3,3-0.1 (-10"4) ККиНК
Жидкость 0,8-1,0 (0,3-24) •10'* 0,5-2,0 (10'5 НК
Примечание. Коэффициент перевода в сист. СИ: I сП = 10 3 Па-с.
Для регулирования величин удерживаемых объемов и селективности разделения в методе флюидо-адсорбционной хроматографии по сравнению с обычной газоадсорбционной хроматографией можно использовать две дополнительные возможности: выбор природы подвижной фазы и выбор давления. Если вместо практически не адсорбирующегося газа-носителя использовать заметно адсорбирующееся вещество в сверхкритическом состоянии, то адсорбционное равновесие между разделяемыми компонентами и адсорбентом изменится в сторону уменьшения константы Генри этих компонентов. Во-первых, это вызвано тем, что взаимодействие в подвижной фазе между молекулами разделяемых веществ и молекулами флюида (подвижного вещества-носителя) будет способствовать ослаблению взаимодействия разделяемых веществ с адсорбентом, т. е. приведет к «повышению летучести» разделяемых веществ на данном адсорбенте. Этот эффект может
Аналитическая хроматография и капиллярный электрофорез
341
быть весьма значительным (возможно повышение «летучести» на четыре порядка). Во-вторых, молекулы самого флюида в этих условиях (высокие давления, т. е. высокая концентрация молекул флюида) довольно сильно адсорбируются, вследствие чего адсорбция молекул разделяемых веществ, концентрация которых во флюиде мала, уменьшится. Адсорбируясь на наиболее адсорбционно-активных местах поверхности, молекулы флюида по существу играют роль «модификаторов», во многих случаях повышая однородность поверхности адсорбентов, что позволяет получать значительно более симметричные пики. В этом случае такое модифицирование поверхности происходит непосредственно в самой колонке в процессе разделения. Степень этого модифицирования можно варьировать, изменяя давления флюида вплоть до появления на поверхно
сти адсорбента плотно адсорбированного слоя из молекул флюида. При использовании в качестве флюидов наиболее доступных растворителей сверхкритическое состояние может достигаться при давлении от 4 до 20 МПа и при температурах не выше 300 °C.
Давление флюида может влиять не только на удерживаемые объемы, но в некоторых случаях и на порядок элюирования. Из табл. 4.1.94, 4.1.95 видно, что порядок элюирования флюидом (изопропанолом) 9,10-дифенилантрацена и пицена из колонки с оксидом алюминия при 245 °C при повышении давления от 4,5 до 5,0 МПа будет меняться. Изменение порядка выхода из колонки с оксидом алюминия при 213 °C при повышении давления флюида (н-пентана) происходит и при разделении 1,1 -динафтила и фенантрена [6].
Таблица 4.1.93
Свойства некоторых веществ, используемых в качестве флюидов
Вещество Ткип, °C Давление пара* при 25 °C, атм. Критическое состояние
Температура, °C Давление, атм. Плотность, (кг • м)-10”3
Диоксид углерода -78,5 63,4 31,3 72,9 0,448
Аммиак -33,4 3,8 132,3 111,3 0,24
Хлортрифторметан -81,4 35,7 28,8 39,0 0,580
Оксид азота(1) -89 56,0 36,5 71,4 0,457
Этан -88 41,0 32,4 48,3 0,203
н-Пропан -44,5 9,4 96,8 42,0 0,220
н-Бутан -0,5 2,4 152,0 37,5 0,228
Дихлордифторметан -29,8 6,2 111,7 39,4 0,558
н-Пентан 36,3 0,74 196,6 33,3 0,232
н-Гексан 69,0 0,33 234,2 29,6 0,234
н-Гептан 98,4 0,06 267,0 27,0 0,235
2,3 -Диметилбутан 58,0 0,31 226,8 31,0 0,241
Бензол 80,1 0,13 288,9 48,3 0,302
Метанол 64,7 0,16 240,5 78,9 0,272
Этанол 78,4 0,07 243,4 63,0 0,276
Изопропанол 82,5 0,05 235,3 47,0 0,273
Дихлорфторметан 8,9 1,7 178,5 51,0 0,522
Трихлорфторметан 23,7 1,05 196,6 41,7 0,554
Дихлортетрафторэтан 3,5 2,3 146,1 35,5 0,682
Вода 100 0,03 374,4 226,8 0,344
Примечание. * Коэффициент перевода в сист. СИ: 1 атм. = 1,01325-10 1 МПа.
Таблица 4.1.94
Значения константы Генри Кг для некоторых углеводородов во флюидно-адсорбционной хроматографии на оксиде алюминия при различных давлениях
Углеводороды Флюид Температура колонки,°C Кг при давлениях
4,1 МПа 4,5 МПа 5 МПа
9,10-Дифенилантрацен Изопропанол 245 — 6,9 1,57
Пинен То же 245 — 7,9 1,39
1,Г-Динафтил и-Пентан 213 4,38 — 1,89
Фенантрен То же 213 3,61 —- 2,19
342
Новый справочник химика и технолога
Флюидо-адсорбционная хроматография обеспечивает высокую селективность при разделении изомеров (табл. 4.1.92) — в частности, смеси фенантрена (Т,^ = 336,8 °C) и антрацена (Тиш = 339,9 °C); смеси 1,2-бензопирена (Т^ = 493 °C) и 3,4-бензопирена (Тищ = 495 °C). Такие смеси плохо разделяются меодом капиллярной газожидкостной хроматографии. Вместе с тем разделение 1,2- и 3,4-бензопиренов представляет большой практический интерес, так как они сильно различаются по своим канцерогенным свойствам.
Флюидо-адсорбционную хроматографию проводили также на пористых полимерах. При этом была показана возможность полного разделения кислородсодержащих многоядерных ароматических соединений, а также олигомеров с молекулярными массами свыше 1000.
Таблица 4.1.95
Константа Генри Кг для антрацена и фенантрена на оксиде алюминия при разных температурах (давление флюида — и-пентана-4 МПа)
T, °C Kr Кг аятр/Кг фен
Фенантрен Антрацен
200 2,68 4,74 1,77
215 6,28 10,3 1,64
225 13,8 21 1,52
250 17,2 24,9 1,45
275 13,9 19,2 1,38
300 12,8 15,2 1,19
Функциональная схема СФ хроматографа приведена на рис. 4.1.15. Подвижная фаза в СФХ подается насосом как жидкость, затем сжатый флюид нагревается до температуры выше критической и подается в колонку через кран. Ограничитель давления после колонки сохраняет сверхкритические условия во всей системе. Для повышения элюирующей способности флюидов применяются следующие органические модификаторы: ацетонитрил, 1,4-диоксан, диэтиловый эфир, пентан, гексан, метанол, изопропанол, тетрагидрофуран.
Рис. 4.1.15. Общая схема сверхкритического флюидного хроматографа
Детекторы, используемые в СФХ: УФ-детекгоры, пламенно-ионизационный, МС, ИКС, флуоресцентный, пламенно-фотометрический, термоионный. Ниже приведен список разделяемых соединений методом СФХ.
Анализируемые смеси
Олигомеры полистирола
Полиядерные ароматические соединения
Высококипящие углеводороды
Нефтепродукты (дизельное топливо, керосин и др.), продукты переработки угля
Органические кислоты
Липиды, глицериды
Эфиры карбогидратов
Витамины
Гербициды
Лекарства
Микотоксины
Эритромицин
Простогландины
Хелаты металлов
Неподвижные жидкие фазы
для газовой хроматографии
Жидкие кристаллы
Смолы
Пищевые продукты: растительные и животные масла, компоненты сыров, кофе и др.
Литература
1. Supercritical Fluid Chromatography and Extraction. Settle F.A. Ed. Prentice Hall Upper. NJ: Saddle River, 1997.
2. Supercritical Fluid Extraction and Chromatography / Ed. M.S. Verrail. Chichester: Wiley, 1996.
3. Supercritical Fluid in Separation Sciences / Ed. O. Kaiser. Dusseldorf: Incom, 1997.
4. Berger T.A. Packed Column SFC. Cambridge: The Royal Society of Chemistry, 1995.
5. Anton K., Berger C. Supercritical Fluid Chromatography with Packed Columns —- Techniques and Applications. N.Y.: M. Dekker, 1998.
6. Saito M. et al. Preparative Supercritical Fluid Chromatography and Its Application to Chiral Separation. Singapore: World Scientific, 1995.
7. Basic Principles and the Role of Supercritical Fluid Chromatography in Lipid Analysis / Eds. King J.W., List G.R. Champaign: Aocs. Press, 1996.
8. Supercritical Fluid Chromatography of Trace Components in Oils and Fats / Eds. King J.W., List G.R. Champaign: Aocs Press, 1996.
9. Supercritical Fluid Chromatography for the Analysis of Oleochemicals / Eds. King J.W., List G.R. Champaign: Aocs Press, 1996.
10. Use of Semipreparative Supercritical Fluid Chromatography for the Separation and Isolation of Flavor and Food Constituents / Ed. Rizvi S.S.H. Glasgow: Blackie, 1994.
11. Verillion F. et al. H Intern. Lab. 1992. Jul/Aug. P. 29.
12. Supercritical fluid Chromatography / Ed. Smith R.M., RSC Chromatography monographs. London, 1988. 238 p.
Аналитическая хроматография и капиллярный электрофорез
343
4.2. Капиллярный электрофорез
Принятые сокращения и обозначения
АПАВ — анионное поверхностно-активное вещество ВЭЖХ — высокоэффективная жидкостная хроматография ДДСН — додецилсульфат натрия (англ. SDS)
ДЭС — двойной электрический слой
ИТФ — изотахофорез
ИЭР — ионизация электрораспылением ИЭФ — изоэлектрическая фокусировка КАЭ — капиллярный аффинный электрофорез КГЭ — капиллярный гель-электрофорез КЗЭ — капиллярный зонный электрофорез ККМ — критическая концентрация мицеллообразования КЛАВ — катионное поверхностно-активное вещество КЭ — капиллярный электрофорез
ЛИФ — лазерно-индуцируемая флуоресценция
МЦ — макроцикл
МЭКХ — мицеллярная электрокинетическая хроматогра-
фия (англ. МЕКС)
ПАВ — поверхностно-активное вещество
ПЭГ — полиэтиленгликоль
ТФЭ — твердофазная экстракция ЦД — циклодекстрины
ЦТАБ — цетилтриметиламмоний бромид
ЭОП — электроосмотический поток ЭХ — электрохроматография
D — коэффициент диффузии вещества
Е — напряженность электрического поля к — фактор емкости
£общ. — общая длина капилляра £Эф„— эффективная длина капилляра N — эффективность (число теоретических тарелок) р/ — изоэлектрическая точка
R — разрешение соседних пиков
TRIS — 3-(гидроксиметил)-аминометан
U — разность потенциалов а — селективность е — диэлектрическая константа С, — дзета-потенциал
Цобщ. — общая (кажущаяся) электрическая подвижность частицы
Рэоп — подвижность электроосмотического потока
Г| — вязкость
Наряду с хроматографическими методами разделения, в аналитическую практику, начиная с 1990 г., активно внедряется метод капиллярного электрофореза (КЭ). Уже к 1999 г. число публикаций на эту тему превысило 700 [1]. Далеко не полный перечень книг, монографий, обзоров, посвященных теоретическим основам и практическим приложениям метода КЭ, представлен ссылками [3-58]. Капиллярный электрофорез становится альтернативой и дополнением к методу высокоэффективной жидкостной хроматографии, в первую очередь, для ионогенных и полярных соединений, воплотив в себе достоинства капиллярной газовой хроматографии, ВЭЖХ и традиционного электрофореза.
В начале 80-х гг. XX в. Дж. Йоргенсон и К. Лукас продемонстрировали сепарационные возможности кварцевых капилляров с внутренним диаметром 75 мкм [50]. Предпосылкой для его развития стала возможность использования кварцевого капилляра с высокой прозрачностью в УФ-области и с равномерным внутренним диаметром от 25 до 100 мкм.
Метод КЭ основан на разделении сложных смесей компонентов, находящихся в электролите, заполняющем кварцевый капилляр, при приложении к нему разности потенциалов и прямого УФ-детектирования в потоке жидкости. Эффект разделения достигается за счет различия в электрофоретической подвижности ионов в присутствии электроосмотического потока. Использование кварцевых капилляров позволяет реализовать процессы разделения с градиентом потенциала до 2,5 кВ-см"1, эффективностью до 1,5 107 теоретических тарелок (т. т.) [59].
Главные достоинства метода КЭ: выигрыш в эффективности (миллионы т. т.), малый объем дозы, более простая процедура пробоподготовки, простое и дешевое аппаратурное оформление, возможность on-line детектирования и автоматизации, включая процедуру пробоподготовки [60]. Использование узких капилляров способствует интенсивному «рассеиванию» джоулева тепла, позволяя применять высокие напряжения [52]. В классическом электрофорезе максимальная величина прилагаемого напряжения составляет ~ 300 В. В методе КЭ значение этой величины лежит в диапазоне 5000-30 000 В.
Высокая чувствительность лазер-индуцированного флуоресцентного (ЛИФ) детектирования в сочетании с различными приемами концентрирования пробы (т.н. стэкинг) позволяет использовать КЭ для анализа предельно малых количеств вещества (1016 10 21 М).
Дальнейший прогресс микросепарационной техники связан с обращением к микроэлектромеханическим системам. Это, в первую очередь, чип-приборы КЭ: одноканальные чип-сенсоры и многоканальные чип-линейки. КЭ в чип-формате и многокапиллярным системам посвящена целая серия работ [1, 63-68].
4.2.1. Основы метода КЭ
Общая схема прибора
Система, реализующая принципы электрофоретического разделения включает: кварцевый капилляр, источник высокого напряжения, устройство ввода пробы, детектор и систему вывода информации (рис. 4.2.1).
344
Новый справочник химика и технолога
Как правило, используют:
• кварцевые капилляры с внешним полиимидным защитным покрытием (внутренний диаметр 50-75 мкм, внешний — 365 мкм, общая длина — 30-100 см);
• положительные и отрицательные напряжения до 30 кВ (сменные высоковольтные блоки или режим с переключением полярности);
• для ввода пробы применяют избыточное давление (гидродинамический способ) или высокое напряжение (электрокинетический). Объем вводимой в капилляр пробы составляет несколько нанолитров;
• для регистрации сигналов чаще всего используют УФ-детектирование непосредственно в капилляре, в прямом и косвенном вариантах.
Дополнительные устройства позволяют автоматизировать подачу образцов (автосамплер), осуществлять отвод тепла от капилляра (системы охлаждения капилляра), управлять прибором, собирать и обрабатывать полученные данные.
Кварцевый капилляр в электрическом поле
Находящиеся на поверхности плавленого кварца силоксановые группы > Si = О при контакте с водой подвергаются гидролизу с образованием силанольных гидроксилов ОН
>sZ
ОН
, которые затем гидратируются.
Константа диссоциации поверхностных силанольных групп Ki = 4 • 10~3; при pH > 2,5 они в значительной степени диссоциированы, обеспечивая поверхности капилляра отрицательный заряд. При pH < 2 диссоциация силанольных групп практически полностью подавлена, и поверхность становится нейтральной.
На границе раздела кварц - водный раствор электролита возникает т. н. двойной электрический слой (ДЭС). Его первую обкладку составляют отрицательно заряженные гидратированные силанольные группы. В приповерхностном слое электролита к отрицательно заряженной поверхности кварца примыкают гидратированные катионы, образуя вторую обкладку ДЭС. Из-за электростатического взаимодействия слой катионов, непосредственно контактирующий с поверхностью капилляра, становится мало подвижным. Остальная часть катионов распространяется в толщу раствора, составляя диффузную часть второй обкладки ДЭС. Распределение катионов между неподвижным и диффузным слоями, а, следовательно, и толщина двойного слоя, зависит от общей концентрации электролита в растворе: чем она больше, тем большая часть положительного заряда диффузного слоя перемещается в неподвижный слой и тем меньше толщина диффузного слоя [69]. При концентрации бинарного однозарядного электролита Ю^-КГ4 М толщина двойного электрического слоя составляет в среднем 30-50 мкм. При диаметре внутреннего канала кварцевого капилляра 50-100 мкм практически вся жидкость, заполняющая капилляр, представляет собой диффузную часть ДЭС. Полиимидный слой в области детектирования удаляется механически или выжиганием.
Микрообъем анализируемого раствора (~ 2 нл) вводится в кварцевый капилляр, предварительно заполненный электролитом, обладающим буферными свойствами (ведущий электролит). После подачи высокого напряжения (до 30 кВ) к концам капилляра компоненты смеси начинают двигаться с разной скоростью, зависящей от их заряда и массы, достигая в разное время зоны детектирования.
Для разделения предпочтительно использование узких капилляров (табл. 4.2.1), однако при этом уменьшается толщина слоя при УФ-детектировании в режимё реального времени, что ведет к снижению чувствительности детектирования.
Таблица 4.2.1
Характеристики капилляров [29]
Внутренний диаметр, мкм Вводимый объем (нл) при длине 1 мм Площадь поверхности капилляра (мм2) на метр Относительное сопротивление, %
250 49,4 785 625
160 20,1 502 256
100 7,9 314 100
75 4,4 236 56
50 2,0 157 25
25 0,5 79 6
Электроосмотический поток
При наложении продольного электрического поля в капилляре возникает движение носителей электрических зарядов во взаимно противоположных направлениях — электрофорез [61]. Избыточная концентрация катионов в диффузной части двойного электрического слоя увлекает за собой всю массу жидкости в капилляре. Возникает течение жидкости в капилляре под действием электрического поля, т. н. электроосмотический поток (ЭОП), который осуществляет пассивный перенос раствора внутри капилляра.
ЭОП характеризуется плоским профилем потока (в отличие от параболического в ВЭЖХ), который при движении зон компонентов внутри капилляра практически не вызывает их уширения (рис. 4.2.2). Этим и определяется очень высокая эффективность КЭ.
Рис. 4.2.2. Влияние профиля потока на ширину зоны вещества: а — электроосмотический поток; б — ламинарный поток
Величина ЭОП пропорциональна диэлектрической проницаемости е, напряженности приложенного поля Е и количеству зарядов на стенке капилляра, или возникаю
Аналитическая хроматография и капиллярный электрофорез
345
щему при этом ^-потенциалу и обратно пропорциональна вязкости электролита т|. Она может быть описана с помощью уравнения Гельмгольца
4 л т|
Скорость ЭОП зависит от pH раствора (в сильнокислых растворах ЭОП отсутствует, в слабокислых — незначительна, а при переходе в нейтральную и щелочную область pH скорость ЭОП возрастает до максимально возможной) и и от концентрации электролита в ведущем буфере — чем она больше, тем меньшая часть катионов ДЭС остается в диффузионном слое и, соответственно, уменьшается скорость ЭОП. ЭОП также уменьшается при добавлении к буферу органических растворителей.
Если в капилляр со стороны анода ввести небольшой объем раствора пробы, то ЭОП перенесет эту зону к катоду (в область детектирования). Заряженные компоненты пробы переместятся в соответствии с их электрическими подвижностями. Катионы, двигаясь к катоду, будут обгонять ЭОП (рис. 4.2.3).
Рис. 4.2.3. Электрофоретическая миграция ионов в присутствии ЭОП
Скорость их движения складывается из скорости ЭОП и скорости электромиграции, поэтому на выходе капилляра катионы появляются первыми и тем раньше, чем больше их электрическая подвижность.
Нейтральные компоненты пробы способны перемещаться только под действием электроосмотического потока, тогда как анионные будут перемещаться к аноду со скоростями меньшими, чем скорость ЭОП. Медленно мигрирующие анионы появятся на выходе после ЭОП, а те, чья скорость электромиграции по абсолютной величине превышает скорость ЭОП, будут выходить из капилляра в прианодное пространство.
В традиционном варианте КЭ электроосмотический поток движется против миграции анионов.
Если добавить к ведущему электролиту ПАВ, то на по верхности капилляра адсорбируется положительный заряд. ЭОП меняет направление и переносит разделительный буфер в направлении анода.
4.2.2. Способы ввода пробы. Зона пробы в капилляре
В КЭ различают три способа ввода пробы: гидродинамический (пневматический), гидростатический (перепадом давления), электрокинетический.
Пневматический способ наиболее часто используют в коммерчески доступных приборах. В герметичном узле ввода создается небольшое избыточное давление воздуха, которое вдавливает пробу в капилляр. Величина давления и время ввода подбираются экспериментально. Объем вводимой пробы составляет несколько сотых долей мкл. Среднее квадратичное отклонение не превышает 3%; используя внутренний стандарт, можно снизить эту величину до 1%.
Объем вводимой пробы в единицу времени определяется следующим образом:
у __
1 128г|£ ’
где Др — давление, D — внутренний диаметр капилляра, L — длина капилляра.
В гидростатическом методе проба вводится в капилляр за счет перепада уровней растворов, находящихся на входном и выходном концах капилляра. Ошибка ввода может достигать 3%. Гидростатический эффект может вносить определенные погрешности как при вводе пробы, так и при анализе. Необходимо при выполнении измерений следить, чтобы уровни растворов во входных и выходных пробирках были одинаковы.
а Rb
Рис. 4.2.4. Способы ввода пробы: а — гидростатический ввод — 10 см, 10 с;
б — электрокинетический ввод — 10 кВ, 1 с
При электрокинетическом способе на капилляр, опущенный в раствор пробы на входном конце, и в раствор рабочего буфера на выходном подают высокое напряжение. «Втягивание» образца в капилляр происходит за счет возникающего ЭОП и электрофореза. Особенность этого варианта заключается в том, что под воздействием высокого напряжения раствор пробы подвергается некоторой дифференциации еще до попадания пробы в капилляр. Компоненты с различной электрофоретической подвижностью будут вводиться по-разному (обладающие большей электрической подвижностью ионы рубидия вводятся с
346
Новый справочник химика и технолога
большей скоростью, чем ионы лития и серебра) и, соответственно, по-разному детектироваться (рис. 4.2.4) [70]. Ошибка может достигать 4-5%. Тем не менее, данный способ получает все большее распространение для концентрирования с помощью т. н. стэкинга (проба концентрируется в несколько сотен раз [51]).
Ведущий электролит
Ведущий электролит всегда содержит буфер, поскольку pH — важный параметр в электрофорезе. Величина pH ведущего электролита определяет как скорость течения жидкости в капилляре (ЭОП), так и форму нахождения компонента в растворе. Чувствительность ЭОП к изменению pH раствора регламентирует использование ведущих электролитов с высокой буферной емкостью, при этом диапазон pH, как правило, имеет значения pk ± 1 [71]. Благодаря высокой стабильности кварцевого капилляра при электрофоретическом разделении можно использовать буферные системы от 2 до 12 ед. pH. При анализе кислот наиболее часто используют боратный буфер (pH = 9,3), оснований — фосфатный (pH = 2,5). Эти буферные системы имеют высокую буферную емкость и низкое поглощение в УФ-излучении. Если основания не растворяются в фосфатном буфере, используется ацетатный (pH = 4,8). В табл. 4.2.2 представлены наиболее распространенные буферные системы, используемые в КЭ.
Таблица 4.2.2
Наиболее распространенные буферные системы в КЭ
Буфер рка
Фосфатный 2,14 (pKai); 7,21 (рКЛ); 13,3 (рКа3)
Цитратный 3,12 (рК,1); 4,76 (рКо2); 6,40 (рад
Формиатный 3,75
Лактатный 3,85
Ацетатный 4,75
Имидазольный 7,00
TRIS 8,30
Боратный 9,24
CHES (2-(А-циклогексил-амино)этансульфоновая кислота) 9,50
В качестве буферов в КЭ используются также цвиттерионные и двойные буферные системы [72].
Концентрация ионов буфера должна быть больше, чем концентрация ионов в растворе пробы. При низкой концентрации буфера достигается более короткое время анализа.
Типичные концентрации находятся в диапазоне 20-150 мМ. Использование более концентрированных буферов снижает сгеночные эффекты, приводит к улучшенной форме пика, по
вышает стэкинг (концентрирование) образца. Буферы низкой концентрации (менее 20 мМ) обеспечивают максимально быстрое разделение, поскольку подвижность аналитов и ЭОП обратно пропорциональны корню квадратному от концентрации буфера. Проводимость разбавленных буферов низка, возможно использование высоких электрических полей, при этом отмечается усиление стеночных эффектов и ослабление стэкинга.
Важная роль отводится ионной силе. Ее повышение приводит к снижению ^-потенциала и, соответственно, снижению ЭОП. Наоборот, низкая ионная сила усиливает ЭОП, снижает время анализа за счет увеличения ^-потенциала.
Идеальный буфер для капиллярного электрофореза должен обладать следующими свойствами:
• достаточной буферной емкостью в выбранном диапазоне pH;
• малым поглощением на длине волны детектирования;
• низкой подвижностью ведущего иона (т. е. в составе электролита должны быть ионы большого радиуса и минимального заряда).
Добавки к ведущему электролиту
Среди используемых в КЭ добавок к ведущему электролиту, реализующих т.н. вторичные равновесия, наиболее популярны поверхностно-активные вещества — мицеллы и циклодекстрины [18, 38, 49]. Их введение в состав буферных растворов позволяет в разной степени влиять на селективность. Определяющими факторами являются тип ПАВ и его концентрация [56]. Наиболее используемая система: додецил сульфат натрия (20-150 мМ) в 20 мМ боратного (pH = 9,3) или фосфатного (pH = 7,0) буфера.
В КЭ могут быть использованы как ионогенные (катионные (КЛАВ), анионные (АПАВ) и цвиттер-ионные [72, 92]), так и нейтральные поверхностно-активные вещества (см. табл. 4.2.3).
Таблица 4.2.3
ПАВ, используемые в капиллярном элекгрофорезе[18]
ТипПАВ Название Формула ккм, мМ
Анионные Додецилсульфат натрия (SDS) [С12Н25 О SO’ ] Na+ 8,2
Катионные Цетилтриметиламмония бромид (СТАВ) [СиВДЛСНэЫВг- 1,3
Неионные Полиоксиэтилен (23), додеканол (BRD35) С12Н25(ОСН2СН2)23ОН 0,1
Цвиттер-ионные н-Додецил-сульфаин СвН^СНз^СВДвО^ 1,2
Аналитическая хроматография и капиллярный электрофорез
347
При концентрации ниже критической концентрации мицеллообразования (ККМ) мономерные формы ионогенных ПАВ могут выступать как ион-парные добавки (АПАВ, КЛАВ), а также влиять на растворимость гидрофобных компонентов смеси и модифицировать стенки капилляра (ЦТАБ).
Добавки ПАВ в ведущий электролит влияют не только на поведение зоны пробы в капилляре, но и на стенки самого капилляра, модифицируя ЭОП (уменьшая, увеличивая или обращая) [42]. При концентрации ПАВ в растворе электролита больше ККМ реализуется вариант мицеллярной элекгрокинетической хроматографии (см. далее) и принцип разделения на основе распределения компонентов пробы между гидрофильной (водной) и гидрофобной (мицеллярной) фазами.
В качестве модификаторов ЭОП широко распространены алкиламмониевые основания и соли как модификаторы ЭОП [73-76].
Использование макроциклических реагентов (краун-эфиров, макроциклических антибиотиков (рис. 4.2.5), каликсаренов, циклодекстринов и т.д.) как добавок в ведущий электролит (табл. 4.2.4-4.2.6) позволяет управлять селективностью разделения сложных смесей органических и неорганических соединений, в первую очередь, за счет уникальной способности макроциклов образовывать комплексы включения с анализируемыми веществами по типу «гость» - «хозяин» [93-98]. В образующихся комплексах «хозяином» выступает макроциклический реагент, а «гостем» — аналит. В зависимости от своего строения макроциклы могут связывать молекулярные, катионные или анионные субстраты. Многие из них используются в качестве хиральных селекторов. Типичные концентрации добавляемых в ведущий электролит циклодекстринов (ЦД), используемых для хиральных и ахиральных разделений, — от 1 до 20 мМ в 20-50 мМ боратном (pH = 9,3) или фосфатном (pH = 2,5) буферах [99].
Таблица 4.2.4
Различные добавки к ведущему электролиту [99]
Цель Реагент Механизм
Модификация подвижности Боратный буфер Комплексы с углеводами, диолами
Каликсарены Комплексы включения
Продолжение таблицы 4.2.4
Цель Реагент Механизм
Модификация подвижности Хелатирующие агенты Комплексы с металлами
Краун-эфиры Комплексы включения
Циклодекстрины Комплексы , включения
Дендримеры Комплексы включения
Макроциклические антибиотики Комплексы включения
Модификация подвижности Органические растворители Сольватация
Сульфоновые кислоты Образование ионных пар
Детергенты Взаимодействие с мицеллами
Ионы переходных металлов Образование комплекса
Четвертичные аммониевые соединения Образование ионных пар
Модификация ЭОП Катионные детергенты Динамическое покрытие, обращение ЭОП
Линейные полимеры Динамическое покрытие
Органические растворители Изменение вязкости
Снижение сорбции на стенках капилляра Катионные детергенты Динамическое покрытие, обращение ЭОП
Линейные полимеры Динамическое покрытие
Полиамины Взаимодействие с силанольными гидроксилами
Улучшение растворимости Органические растворители Гидрофобные взаимодействия
348
Новый справочник химика и технолога
Гликопептиды
Амнногликозвды
Ванкомицин
Тейкопланин
Фрадиомицин В Ri=H r2oh2nh2
Фрадиомицин С Ri=CH2NH2 r2=h
Ансамицины
Римафицин В R=OCH2COOH
Римафицин SV ROH
Римафицин
Стрептомицин
Рис. 4.2.5. Структурные формулы макроциклических антибиотиков, используемых в качестве хиральных селекторов
Аналитическая хроматография и капиллярный электрофорез
349
Таблица 4.2.5
Природа и функция добавок в КЭ [42]
Таблица 4.2.6
Влияние буферных добавок на ЭОП
Добавка Пример Влияние Литература
ПАВ (катионные, анионные и нейтральные) ДДСН, ЦТАБ Модификация ЭОП, растворение гидрофобных компонентов, ион-парные взаимодействия, мицеллообразова-ние при концентрации > ККМ 77-83
Органические модификаторы Метанол, ацетонитрил, трифторуксусная кислота Изменение скорости ЭОП (чаще снижение), изменение селективности в хиральном анализе и в МЭКХ 84
Макроциклические агенты Циклодекстрины, краун-эфиры, макроциклические антибиотики Образование комплексов включения, хиральное распознавание, растворимость гидрофобных компонентов 87-90
Амины, четвертичные аммониевые соединения Диаминопропан Образование ионных пар, обращение ЭОП 42
Линейные гидрофильные полимеры Полиакриламид, метилцел-люлоза Снижение скорости ЭОП, уменьшение адсорбции компонентов пробы на стенках кварцевого капилляра (при концентрациях добавки приблизительно сотые доли %), гель-электрофорез (при концентрации полимера 1-1%) 85, 86
Цвиттерионные компоненты TRIS Возрастание ионной силы раствора без увеличения электропроводности среды, влияние на селективность при разделении белков 42
Реагенты, способные к образованию водородных связей Мочевина Изменение селективности в МЭКХ, растворение белков 91
Буферные добавки Используемые концентрации, мМ Влияние Проблемы
ЦТАБ 0,2-1,0 Обращение ЭОП Мицелл ообра-зование, растворимость до 50 мМ
ДДТАБ 0,2-2,0 То же Мицелл ©образование
ГДПХ 0,2-1,5 Тоже Мицелл ©образование, поглощение в УФ-области
SB 14 1-10 Очень малый ЭОП
Спермин 2-10 То же Образование ионных пар
Диэтилентриамин 1-5 То же
Полиэти-ленамин 0,5-2,0 Обращение ЭОП
Покрытие SAX — То же Низкая стабильность
Наряду с перечисленными добавками используют введение хелатирующих агентов при определении переходных металлов, ион-парных агентов при анализе кислот и оснований, ионов серебра при определении алкенов, ионов магния — для нуклеозидов [99].
4.2.4. Основные варианты капиллярного электрофореза
Метод капиллярного электрофореза может быть представлен в нескольких вариантах (табл. 4.2.7). Для аналитических целей используются следующие разновидности КЭ (табл. 4.2.7).
Таблица 4.2.7
Основные варианты метода капиллярного электрофореза
ВидКЭ Основа разделения
Зонный капиллярный электрофорез (КЗЭ) Подвижность компонентов пробы в растворе электролита
Мицеллярная электро-кинетическая хроматография (МЭКХ) Гидрофобные / ионные взаимодействия с мицеллами
Капиллярный гель-электрофорез (КГЭ) Размер и заряд частиц
Изоэлектрическая фокусировка (ИЭФ) Изоэлектрические точки компонентов
350
Новый справочник химика и технолога
КЗЭ, используемый в основном для разделения биополимеров (белков, полипептидов, нуклеотидов и т.д.), реализуется в капилляре, заполненном гелем (агарозой, целлюлозой, акриламидом и т.д.). На электрофоретическую подвижность биополимеров оказывает влияние характер пористости матрицы геля. Разделение молекул осуществляется по их размерам (т.н. ситовый эффект). В случае изотахофореза (ИТФ) проба движется между двумя электролитами с различными подвижностями ионов. Как вариант вытеснительной хроматографии ИТФ может быть использован на стадии предварительного концентрирования перед разделением методом КЭ. В последние годы достаточное число работ опубликовано по капиллярному аффинному электрофорезу (КАЭ) и электрохроматографии (ЭХ). В последнем варианте воплощается техника высокоэффективного капиллярного электрофореза и высокооэф-фективной жидкостной хроматографии: разделение осуществляется в кварцевых капиллярах с внутренним диаметром 50-100 мкм, упакованных ультрасорбентом (диаметр частиц сорбента < 3 мкм). Течение элюента и перенос пробы происходит за счет электроосмотического потока.
Наиболее распространенными вариантами КЭ являются зонный и мицеллярный [18].
4.2.4.1. Капиллярный зонный электрофорез (КЗЭ)
В приборах КЗЭ чаще всего полярность входного конца положительная (анод), и ЭОП переносит зону пробы к катоду, вблизи которого расположен детектор. Компоненты смеси движутся в среде электролита с разными скоростями, образуя дискретные зоны. Катионные компоненты пробы, перемещаясь к катоду, обгоняют ЭОП и первыми регистрируются на электрофореграмме. Затем детектора достигает зона исходного раствора, в которой перемещаются нейтральные компоненты. Нейтральные соединения мигрируют с одинаковой скоростью, равной скорости ЭОП, и не могут быть разделены.
В зависимости от их способности поглощать в УФ-излучении на электрофореграмме регистрируется пик, называемый системным. Для его идентификации в пробу часто добавляют специальные вещества — маркеры (ацетон, бензиловый спирт, оксид мезитила).
Поведение анионных компонентов пробы зависит от соотношения скоростей миграции ЭОП и анионов. Если скорость электромиграции аниона меньше скорости ЭОП, он может быть зарегистрирован на той же электрофореграмме после системного пика. В противном случае анион выйдет из капилляра в прианодное пространство. Описанный вариант носит название капиллярного зонного электрофореза, в режиме которого могут определяться катионы и медленно мигрирующие анионы [100-105].
Для определения анионных компонентов методом КЗЭ надо изменить полярность прикладываемого напряжения (входной конец отрицательный), при этом изменится и направление ЭОП. Чтобы предотвратить это, поверхность кварцевого капилляра модифицируют введением в состав электролита катионного поверхностно-активного вещества (например, ЦТАБ). Катион динамически сорбируется от
рицательно заряженной поверхностью кварцевого капилляра, которая приобретает гидрофобный характер за счет неполярных цетильных групп (-Ci6H33). Дальнейшая промывка рабочим буферным раствором, содержащим ЦТАБ, сорбирует следующий слой детергента, и поверхность капилляра приобретает положительный заряд, а диффузная часть двойного электрического слоя — отрицательный. Таким образом, ЭОП снова движется от входного конца к детектору.
Рассмотренные выше обстоятельства приходится учитывать в процессе разделения белков и пептидов [106, 107]. При КЗЭ белков с немодифицированным капилляром рекомендуется после каждого проведенного разделения при вводе пробы из биологических матриц тщательно промывать капилляр раствором едкого натра. При этом молекулы, адсорбированные на стенках капилляра, удаляются. Если значения pH выше изоэлектрической точки (р7), то белки находятся в анионной форме, т.е. имеют тот же заряд, что и стенки кварцевого капилляра. Предпочтительный pH буфера составляет 9-11. При pH < 2 адсорбция белков уменьшается вследствие протонирования силанольных групп. Возникают проблемы иного рода: очень малый ЭОП и возможная денатурация белков. Для предотвращения сорбции белков стенками капилляра к буферу добавляют соли щелочных металлов, низших полиаминов, цвиттер-ионов, обладающих большой буферной емкостью. Перспективно использование неионных ПАВ в качестве динамических покрытий.
В табл. 8а [107] (см. компакт-диск) представлены условия разделения белков методом КЭ. Отличительная особенность КЗЭ состоит в том, что он пригоден для разделения только ионогенных компонентов пробы, тогда как нейтральные соединения, не обладающие собственной электрофоретической подвижностью, движутся со скоростью ЭОП.
4.2.4.2. Мицеллярная электрокинетическая хроматография (МЭКХ)
МЭКХ (англ. МЕКС) или мицеллярная электрокинетическая капиллярная хроматография (англ. МЕСС) объединяет принципы электрофореза и хроматографии. Введенная в 1984 г. японским ученым Терабе, МЭКХ получила наиболее широкое распространение среди других вариантов капиллярного электрофореза, в первую очередь, за счет способности разделять как ионогенные, так и незаряженные компоненты проб [78, 79]. Разделение нейтральных соединений стало возможным благодаря введению в состав ведущего электролита ПАВ - мицеллообразователей. Чаще всего используют анионные ПАВ (например, ДДСН) в концентрациях, превышающих ККМ, которая для ДДСН в водном растворе составляет 8 мМ [77, 78, 90, 108]. В этом случае в растворе электролита находятся преимущественно мицеллы и небольшая доля мономерной формы ПАВ. Мономеры состоят из гидрофильнго хвоста и отрицательно заряженной головы (в случае анионного ПАВ). При формировании мицелл мономерные фрагменты агрегируются неполярными концами внутрь, а внешняя сфери
Аналитическая хроматография и капиллярный электрофорез
351
ческая часть становится полярной и отрицательно заряженной. В полярных средах молекулы дифильных веществ (типа ПАВ) объединяются неполярными фрагментами, образуя прямые мицеллы или везикулы. В неполярных средах возникают обратные мицеллы (рис. 4.2.6.) [113].
Рис. 4.2.6. Примеры структур детергентов в растворе а — мицелла; б — обратные мицеллы;
в — двуслойная визикула; г — двойной слой
Ни мицеллярная, ни мономерная форма АПАВ не взаимодействуют со стенкой кварцевого капилляра, но при подаче на капилляр высокого напряжения обе формы движутся по направлению к аноду, в то время как ЭОП направлен к катоду [78]. Если в капилляр на анодной стороне ввести пробу, содержащую нейтральные и заряженные компоненты, то ЭОП будет переносить их к катоду, а навстречу будет двигаться поток отрицательно заряженных мицелл АПАВ. Скорость ЭОП превышает электрофоретическую миграцию мицелл, и поэтому мицеллы так же, как и ЭОП, в конечном итоге, мигрируют к катоду, но с меньшей скоростью. Компоненты пробы распределяются между фазой раствора и псевдостационарной фазой — мицеллой, причем константа этого распределения специфична для каждого сорта молекул пробы [79]. Менее полярные аналиты мигрируют к детектору с меньшей скоростью, чем более полярные, и все разделяемые соединения элюируются в «окне удерживания» между временем ЭОП (самое короткое) и временем миграции мицеллы (самое длительное). На электрофореграмме регистрируются нейтральные компоненты, а также медленно мигрирующие анионы пробы. В большинстве работ используются анионные детергенты в качестве мицелл. Катионные детергенты обращают ЭОП
вследствие изменения заряда стенок капилляра. ЭОП движется к аноду, а мицеллы — к катоду. При использовании катионных детергентов ЭОП не подавляется даже в кислых условиях из-за сильной адсорбции катионных частиц. Типы различных мицелл, используемых в МЭКХ, представлены в табл. 86 (см. компакт-диск). Модифицируя либо мицеллу [119-121], либо ведущий электролит, можно менять селективность и эффективность разделения [53]. Модификация ведущего электролита может быть осуществлена путем добавок органических растворителей: метанола, ацетонитрила, тетрагидрофурана [54, 117]; комплексообразующих агентов [96,118].
4.2.5. Детектирование в КЭ
Детектирование в системах КЭ может осуществляться различными способами:
• непосредственно в капилляре в части, близкой к выходному концу, в режиме реального времени (оп-capillary, on-columri) [81, 121-125]. Этот способ характерен для большинства коммерческих систем капиллярного электрофореза. В зоне детектирования с внешней стенки капилляра снимают защитное полиимидное покрытие непосредственно на выходном конце капилляра (end-capillary, end-column) [122];
• вне системы КЭ (off-capillary, off-column). При этом, как правило, детектор представляет собой отдельный самостоятельный прибор (например, масс-спектрометр), соединенный с системой КЭ интерфейсом [129].
Используются те же принципы детектирования, что и в ВЭЖХ. Важным преимуществом КЭ перед ВЭЖХ (помимо плоского профиля ЭОП) является отсутствие соединительных гидравлических линий между узлами инжектор -капилляр и капилляр - детектор, которые приводят к уширению зоны вещества за счет внеколоночного размывания.
Некоторые характеристики детекторов, применяемых в КЭ, представлены в табл. 4.2.9.
• УФ/В (прямое и косвенное) [81, 83, 89, 90, 94, 100, 121, 127, 137, 145];
• флуоресцентное (прямое и косвенное), включая индуцированную лазером флуоресценцию [122, 123, 130, 133-136, 138];
• масс-спектрометрическое [127, 136, 139];
• кондуктометрическое [128];
• амперометрическое (прямое и косвенное) [38, 39, 131,
137, 138];
• радиометрическое [124];
• рефрактометрическое [123, 132].
Таблица 4.2.9
Характеристики детекторов, применяемых в КЭ [58]
Способ детектирования Селективность Универсальность Детектирование в капилляре Примерный предел детектирования, моль-л1 (определяется объемом вводимой пробы) Процент использования, % [литература]
УФ/В поглощение + — + 10 5-10 7 55 [38]
Косвенное поглощение — + + Ю^-Ю'6 5 [121]
352
Новый справочник химика и технолога
Продолжительность таблицы 4.2.9
Способ детектирования Селективность Универсальность Детектирование в капилляре Примерный предел детектирования, моль/л (определяется объемом вводимой пробы) Процент использования, % [литература]
Флуоресценция + + 10 7-1(Г9 15 [104]
Косвенная флуоресценция — + + 10^-10 8 2[104]
Индуцированная лазером флуоресценция + — + ю 13-10 16 5-7 [58]
Масс-спектрометрия + + — 10 8 - ю10 10 [58]
Амперометрия + — — 1071010 2 [104]
Косвенная амперометрия — + — 10 6-10 8 < 1 [58]
Кондуктометрия — + — 10 7-10 9 < 1 [58]
Рефрактометрия + + 10-6- 10“8 < 1 [58]
Радиометрия + — + 10 10 - 10 12 < 1 [58]
Наиболее распространенным вариантом детектирования является УФ/В, основанное на поглощении веществом УФ- или видимого света. Внутри этого спектрального диапазона чаще всегоиспользуют длину волны 254 нм, т.к. более половины всех анализируемых компонентов поглощают УФ-излучение в этой области спектра (ароматические соединения; вещества, молекулы которых содержат подвижные л- и р-электроны, функциональные группы ^-С-—О, ^C=S, —C=N, —N=N—, а также системы сопряженных связей).
При УФ-детектировании в КЭ концентрационная чувствительность в 30-100 раз ниже, чем в ВЭЖХ.
Фотометрические детекторы, используемые в КЭ, подразделяют на
а) детекторы с фиксированной длиной волны — источники света: ртутная (254 нм), кадмиевая (229 нм) и цинковая (214 нм) лампы. Это наиболее простые и недорогие системы;
б) детекторы с изменяемой длиной волны — источники света: дейтериевые и вольфрамовые лампы (рабочий диапазон 190-350 нм и 340-850 нм соответственно);
в) детекторы на диодной матрице ДМД [44]. Регистрация и запись сигналов обеспечиваются в режиме сканирования. Данные, полученные одновременно на различных длинах волн, обрабатываются с помощью компьютеров, выделяющих сигнал на оптимальной длине волны и вычитающих фон.
Для соединений, анализируемых с помощью КЭ и не поглощающих в УФ-диапазоне, существует возможность регистрации методом косвенного УФ-детектирования [58]. В состав ведущего электролита вводят небольшое количество хромофора — вещества, поглощающего на требуемой длине волны. Для определения анионов поглощающий ион тоже должен быть анионом, например, хромат-, фталат-ионы, анионы пиромеллитовой кислоты [145] (в концентрации 2-10 мМ), а при определении катионов чаще используют катионы ароматических аминов, в частности, ион протонированного бензимидазола [73] или ионы Си2+
[145]. Непрямое детектирование используется при определении хлорид- и сульфат-ионов, ионов калия и натрия, а также алифатических аминов, карбоновых кислот, некоторых сахаров. Так как ионная сила ведущего электролита в процессе разделения остается постоянной, в зоне, где находится непоглощающий ион, уменьшается концентрация поглощающего иона. Обмен происходит строго эквивалентно, на электрофореграмме наблюдаются обратные (отрицательные) пики, площади которых пропорциональны концентрациям определяемых ионов. Косвенное УФ-детектирование является универсальным вариантом детектирования, позволяя регистрировать все присутствующие в анализируемом растворе компоненты.
Флуоресцентное детектирование используют для регистрации веществ, обладающих собственной флуоресценцией или тех, для которых возможно получение флуоресцирующих производных [122, 130, 133, 135, 136].
Среди известных люминофоров — фталевый альдегид, 1-диметиламино-5 нафталинсульфохлорид (дансилхлорид), фенилизотиоцианат. Метод отличается высокой селективностью, а его чувствительность на 2-3 порядка превышает возможности УФ-детектирования. Часть капилляра облучается УФ-светом соответствующей длины волны, а испускаемый свет регистрируется перпендикулярно направлению входящего излучения. Простые модификации приборов обеспечивают фиксированные длины волн возбуждения и регистрации флуоресценции, те, что сложнее, позволяют варьировать длину волны возбуждения [122]. Более дорогие приборы с этим видом детектирования имеют монохроматоры как для возбуждающего, так и для флуоресцентного света, благодаря чему детектирование становится высоко специфичным. Необходимо обращать внимание на явление тушения флуоресценции и предотвращать его возникновение (если детектирование не проводят по косвенному варианту, в основе которого лежит тушение люминесценции ведущего электролита) [130]. Флуоресцентное детектирование требует применения импульсных источников света, в качестве которых использу
Аналитическая хроматография и капиллярный электрофорез
353
ются ксеноновые лампы, а также более дорогие лазерные системы: аргон-ионные, гелий-кадмиевые, гелий-неоновые и другие (табл. 4.2.10) [58, 138]. Лазеры обеспечивают большой выход флуоресцентного сигнала, что позволяет относить этот вид детектирования к самым чувствительным (10“13-10~16 моль-л '). Основными характеристиками лазерных источников являются: рабочая длина волны (ар-гон-ионный — 488 нм, гелий-кадмиевый — 325 нм), мощность излучения (чем больше мощность лазера, тем выше чувствительность детектирования) и возможность фокусирования лучей в узкую зону, соизмеримую с внутренним диаметром капилляра.
Таблица 4.2.10
Источники лазерного излучения для ЛИФ-детектирования [138]
Лазер Доступные длины волн, нм
Ar-ионный (воздушное охлаждение) 457,472,476,488,496, 501,514
Аг-ионный 275, 300, 305, 333,351, 364, 385,457, 472, 476, 488,496, 501,514
Аг-ионный, (с удвоением частоты) 229, 238, 244, 248, 257
АгКг 350-360,457, 472, 476, 488, 496, 501,514, 521, 514, 521,531,568, 647, 752
HeNe 543, 594, 604, 612, 633
Эксимерные
ХеС1 (импульсный) 308
KrF (импульсный) 248
Азотный (импульсный) 337
Азотный с перестраиваемой длиной волны в зависимости от давления 360-950
Твердотельные
YAG (с удвоением частоты) 532
YAG (учетверенная частота) 266
Диодные лазеры
Удвоенной частоты (LiNbO3) 415
Удвоенной частоты (КТР) 424
Утроенной частоты (YLiF) 349
Широкие возможности для использования в качестве детектора в КЭ имеет масс-спектрометр [58, 139]. Он в достаточной мере чувствителен и универсален: регистрируются все вещества, имеющие молярные массы в рабочем диапазоне масс этого детектора. Встроенная и легко дополняемая библиотека спектров позволяет идентифицировать анализируемые компоненты в сложных смесях. Наиболее часто используют ионизацию электрораспылением (ИЭР, electrospray ionization (ESI)) [91], в таких системах
выход капилляра располагают непосредственно в интерфейсе ИЭР.
К электрохимическим методам детектирования в КЭ относят амперометрический (прямое и косвенное определение), кондуктометрический и потенциометрический. Амперометрическое детектирование для КЭ впервые было предложено в 1987 г. для анализа катехоламинов [140] и может быть использовано для обнаружения электрохимически активных веществ. В основе метода лежит измерение тока, протекающего в электрохимической ячейке при происходящих на рабочем электроде реакциях окисления или восстановления; величина тока прямо пропорциональна концентрации анализируемого соединения. Обычно в электрохимической ячейке находятся три электрода: рабочий (из стеклоуглерода, угольной пасты или амальгамированного золота), вспомогательный и электрод сравнения; типичные потенциалы детектирования 0,4-1,2 В. Подавляющее большинство амперометрических исследований в КЭ проводят по окислению (анализ ароматических гидроксисоединений, ароматических аминов, индолов, меркаптанов и т.д.) [58]. Детектирование по восстановлению практически не используют из-за мешающего влияния растворенного кислорода. Недостаток амперометрического детектирования — отравление рабочего электрода ввиду сильной сорбции промежуточных продуктов окислительно-восстановительных реакций поверхностью электрода, следствием является снижение его активности [44]. Замена угольного электрода медным позволяет увеличить срок службы рабочего электрода в неимпульсной схеме амперометрического детектирования [49].
Кондуктометрическое детектирование традиционно используют в анализе ионов, ввиду отсутствия у последних собственного поглощения и люминесценции. При появлении в зоне детектирования анализируемого иона электропроводность раствора меняется. Одна из проблем этого вида детектирования связана с возникновением помимо фоновой электропроводности электролита некоторой электропроводности в зоне вещества, которая может быть решена использованием подавительной схемы детектирования [141]. В результате ионного обмена буферный противоион образует слабо диссоциирующие кислоту (анализ анионов) или основание (анализ катионов). Снижается общая проводимость буферного раствора, но возрастает разница между электропроводностью пробы и буфера. По-давительная техника практически не используется при анализе катионов из-за необходимости работы с низкими значениями pH электролитов, а, следовательно, малыми скоростями ЭОП и затруднениями в транспортировке пробы в зону детектирования. Эта схема приводит также к экстраколоночному размыванию зон компонентов, что ограничивает эффективность разделения.
4.2.5.1. Зарубежные коммерческие системы капиллярного электрофореза
Первые коммерческие приборы для КЭ появились в 1988 г. в США [142]. Разработкой и выпуском аппаратуры занимаются такие всемирно известные компании, как
354
Новый справочник химика и технолога
Agilent Technologies (Hewlett Packard), Beckman Coulter, BioRad Laboratories, Waters, Perkin-Elmer, Prince Technologies (табл. 4.2.11). Различия в аппаратуре, как правило, связаны с числом и видом используемых детекторов [60, 42]. Наиболее востребованная зарубежными специалистами система капиллярного электрофореза Agilent 3D СЕ (фирма Agilent Technologies) характеризуется [47]:
• наличием диодно-матричного детектора, обеспечивающего получение спектрограмм в широком диапазоне длин волн в реальном масштабе времени;
• автоматической идентификацией веществ благодаря библиотечному поиску;
• возможностью подключения к масс-спектрометру;
• программируемым переключением полярности в ходе анализа;
• быстрой и простой сменой капилляра;
• возможностью применения различных вариантов метода КЭ;
• наличием профессиональных программных продуктов для сбора и обработки данных, а также для управления прибором.
Таблица 4.2.11
Зарубежные коммерческие системы КЭ [47]
Модель Система капиллярного электрофореза Р/АСЕТМ MDQ Methods Development system PrinCE MCE 2000 Ultra-Plus II СЕ
Компания Agilent Technologies 2850 Centerville Rd. Wilmington, DE, 19808 800-227-9770 www. agilent. corn/c hem Beckman Coulter, Inc. 4300 N. Harbor Blvd. P.O. Box 3100 Fullerton, CA 92834-3100 714-871-4848 www.beckmancoulter. com/ Prince Tehnologies P.O. Box 2194 7801 CD Emmen The Netherlands 30(0)591 629184 www.princetechno-logies.com CombiStep 1915 Scholl Road Applied Science Building II Ames, Iowa 50011 515-294-2837 www. combistep. com Micro-Tech Scientific 140 South Wolfe Rd. Sunnyvale, CA 94086 408-730-8324 www.microlc.com
Тип ввода пробы Под давлением, вакуумный или электрокинетиче -ский Под давлением, вакуумный или электрокинетиче -ский Электрокинетиче-СКИЙ Вакуумный и электрокинетиче ский Под высоким давлением, время ввода контролируется специальным клапаном; гидродинамический и элек-трокинетический
Капилляры
Минимальная длина 8,5 см (от точки ввода до детектора); 33 см общая; 25-, 50-, 75 или 100 мкм внутренний диаметр 7 см (от ввода до детектора); общая -27 см; максимальная длина переменна Различная длина и диаметр Общая длина 40 см; эффективная - 20 см; внутренний диаметр 75 мкм 5-50 см
Совместимость 25-, 50-, 75-мкм расширенный оптический путь, высокочувствительная ячейка (75 мкм) Все размеры капилляров Внутренний диаметр 50, 75, 100, 250 и 300 мкм; открытый или заполненный капилляр
Напряжение, кВ ±30 ±30 ±30 ±20 ±60
Ток, мА 0-300 0-300 от -200 до 200 0-4,0 (общий ток) 1,2
Мощность, Вт 0-6 0-9 0-6 Программируемый контроль напряжения
Аналитическая хроматография и капиллярный электрофорез
355
Продолжение таблицы 4.2.11
Модель Система капиллярного электрофореза Р/АСЕТМ MDQ Methods Development system PrinCE MCE 2000 Ultra-Plus 11 СЕ
Автоввод пробы Термостатируемая карусель на 48 мест; флаконы от 100 мкл до 2 мл Термостатируемый автосэмплер; 2 мл и ПЦР-флаконы; 0,5 мл и 100 мкл трубки; 96-местная площадка 4-х позиционный или 38/40-позици-онный автосэмплер; различные размеры флаконов ПЦР-тарелка на 96 проб; объем пробы 0,2-1,0 мл Тарелки на 96 и 384 места; флаконы на 2 мл
Детекторы УФ/В диодная матрица Модульная УФ/В диодная матрица, УФ/В с выбором длины волны Позволяет использовать любые совместимые с КЭ детекторы, включая МС, флуоресцентный, ЛИФ, кондуктометриче -ский, УФ/В, фотодиодную матрицу Стандартный УФ с длиной волны 214 нм; также доступны 254,280 и 305 нм УФ/В, МС
Дополнительные особенности Высокочувствительные оптические ячейки; самонастраивающиеся капиллярные картриджи: высокое давление на концах для капиллярной хроматографии; программа ChemStation; автоматический коллектор фракций; интегрированный КЭ/МС (квадрупольный и ионная ловушка масс-спектрометры) Автоматический коллектор фракций; высокое давление на концах для капиллярной хроматографии; температурный контроль циркулирующей жидкости; независимый температурный контроль буфера и пробы; графики подвижности; алгоритм для расчетов; 32 К программа (совмещенная платформа для ЖХ/КЭ); большой набор-химического оборудования и реактивов Полностью модульный; высокое давление на входе, выходе или на обоих концах для КЭХ*; легкое подключение к масс-спектрометру; гибкость; различные модели; специально разработанные программы расчета подвижностей; контроль окончания по току 96 одновременных разделений; программа обсчитывает 96 проб менее чем за 15 мин Охлаждение поддона для проб (элементы Пельтье); интегрированный капиллярный градиент по увеличению pH; 8 нм х 20 нл потоковая ячейка; программируемое напряжение и градиент растворителя
Примечание Прямая связь с масс-спектрометром Agilent 1100 серии; подключается к Picometrics’ ЛИФ; подключается к настраиваемому флуоре сцентному детектору Leap Technology Одно- или двухканальный ЛИФ; адаптер для внешнего детектора; ионная ловушка МС/МС; интегрированный КЭ/МС/МС и МС” одноточечная регулировка и анализ Одиночный подъемник с дополнительным выбором температурного контроля или высокого давления; двойной подъемник с дополнительным контролем температуры; двойной подъемник со стандартным давлением; интегрированный КЭ и капиллярная хроматография Тестовый набор для пептидов, рН/буферные тесты; предварительное концентрирование; log Р; хираль-ный скрининг; определение Р^а Может быть сконфигурирован под многомерную ЖХ, КЭ и КЭХ*; работает под Windows 98, NT 4,0; программа для масс-спектроскопии Finnigan X’Calibur; способен быть полностью автоматическим
Примечание. * КЭХ — капиллярная электрохроматография
356
Новый справочник химика и технолога
Фирмой Agilent Technologies созданы капилляры с внутренним пузырьком, позволяющие увеличить длину оптического пути и тем самым повысить чувствительность определения при использовании УФ-детектора [20, 145].
4.2.5.2. Отечественные системы капиллярного электрофореза
В России (С.-Петербург) занимаются разработкой и выпуском систем КЭ научно-производственная фирма аналитического приборостроения «Люмэкс» (системы КЭ «Капель») и Институт аналитического приборостроения (приборы высокоэффективного капиллярного электрофореза «Нанофор»: приборы комплекса «Нанофор-1» с СФ-детектором и «Нанофор-2» с лазер-индуцируемым флуоресцентным детектором лвдаб. =473 нм). Приборы комплекса находят применение в том числе и в медицинской диагностике. «Нанофор-1» — при анализе гликогемоглобина и белков сыворотки крови, а также определении ионного состава питьевой воды. «Нанофор-2» — сверхчувствительный автоматический прибор иммуноанализа и генетической диагностики (реализованы методики фракционирования биополимеров: пептидов, белков, разделение олиго- и полинуклеотидов); возможен анализ ароматических полинитросоединений, редкоземельных элементов, солей тяжелых металлов.
Фирма «Люмэкс» первой в России начала выпуск серии приборов «Капель» («Капель-103Р», «Капель-104РТ» и «Капель-105»), внесенных в Госреестр средств измерений [143], которые могут применяться для анализа катионов металлов, неорганических и органических анионов, аминокислот, витаминов, наркотических средств, пигментов и красителей, белков, пептидов, анализа фармпрепаратов и пищевых продуктов, для контроля качества вод и напитков, технологического контроля производств, входного контроля сырья, в криминалистике, медицине и биохимии [144], в том числе для расшифровки генетического кода живых организмов и т.д.
Некоторые характеристики указанных моделей приведены в табл. 4.2.12.
Сравнение технических параметров отечественных и зарубежных приборов с одинаковым принципом детектирования (приборы Quanta 4000-Е (фирма Waters) и «Капель-103» (фирма «Люмэкс»)) показало равные возможности систем капиллярного электрофореза при анализе ионного состава водных объектов [55].
В «Капели» предусмотрено два способа ввода пробы.
• Пневматический (с помощью давления): в герметичном узле ввода создается избыточное давление, которое вдавливает пробу в капилляр; величина давления и время ввода подбираются экспериментально. Объем вводимой пробы составляет несколько сотых долей мкл. Необходимо следить, чтобы раствор пробы, смачивающий наружную поверхность капилляра и электрода, не попадал в раствор ведущего электролита.
• Электрокинетический: проба вводится в капилляр ЭОП. Для этого подается высокое напряжение на капилляр на входном конце, опущенном в раствор пробы, и на выходном — в раствор рабочего буфера. Возникающий ЭОП втягивает пробу в капилляр.
Таблица 4.2.12
Сравнительные характеристики модификаций системы «Капель»
Характеристики Модификация
«Капель-103Р» «Капель-104» «Капель-105»
Детектор Фотометрический, 254 нм Фотометрический,, 254 нм Фотометрический, 190-400 нм
Ввод пробы Электрокинетический, пневматический
Охлаждение капилляра Воздушное, принудительное, без контроля температуры Жидкостное, с контролем температуры
Смена образцов Ручная Ручная или автоматическая
Число пробирок на входе капилляра 4 10
Число пробирок на выходе капилляра 2 10
Управление Функциональная клавиатура, интерфейс RS 232
Индикация ЖК-дисплей 4 строки по 20 символов ЖК-дисплей 30 строк по 16 символов
Обработка данных По «МультиХром» для Windows-95/98 NT
Источник питания 220 В ± 15%, 50/60 Гц
Потребляемая мощность, Вт 60 80 150
Размер, мм 420 х 330 х 360 420 х 360 х 440
Масса, кг 16 22 24
Опытная модификация «Капель-103РЕ» — электро-инжекционный анализатор со специальной кассетой, на которой окно детектора расположено в середине капилляра, благодаря чему эффективная длина капилляра одинакова как для анионных, так и катионных компонентов проб. Компоненты вводятся в капилляр с двух концов. Двигаясь навстречу друг другу, они могут взаимодействовать, образуя новые соединения, обладающие иной электрофоретической подвижностью, чем исходные, регистрируясь при прохождении зоны детектирования.
4.2.6. Характеристики удерживания
Важнейшей характеристикой массопереноса вещества при электрофорезе является электрофоретическая подвижность частиц — скорость перемещения, отнесенная к единице напряженности электрического поля. Время
Аналитическая хроматография и капиллярный электрофорез
357
единице напряженности электрического поля. Время миграции заряженных веществ в КЭ определяется двумя параметрами: собственной электрофоретической подвижностью частицы Цэф и подвижностью ЭОП Цэоп — течением буфера под действием электрического поля, определяющим время выхода незаряженных молекул.
Общая (наблюдаемая, кажущаяся) подвижность иона Робщ определяется:
Цобщ. Рэф. + Рэоп, (4.2.1)
где Цэф — электрофоретическая подвижность иона;
Рэоп — подвижность ЭОП.
Рэф может быть найдена:
(М.= -3—, (4.2.2)
блцг
где q — заряд иона; ц — вязкость раствора; г — радиус иона.
Подвижность ЭОП находят из
Р»„=—, (4.2.3)
п
где е — диэлектрическая константа; £ — потенциал (характеризует заряд поверхности капилляра).
На практике ц(Кзш рассчитывают, исходя из экспериментальных данных:
М«ж= (4-2.4)
U Сигр.
где £Общ — общая длина капилляра (от входного до выходного конца), см; £эф — эффективная длина капилляра (от входного конца до зоны детектирования), см; U — величина рабочего напряжения, В; /мигр — время миграции компонента, с.
Аналогично (4.2.4) рассчитывают |Х,ОП, подставляя время миграции нейтрального компонента — маркера ЭОП (например, ацетона, бензилового спирта).
Параметры электромиграции специфичны для каждого сорта заряженных частиц. Такие факторы как диффузионные, сорбционные, конвекционные, гравитационные и т.д. в капилляре существенно ослаблены, благодаря чему и достигаются рекордные эффективности разделений.
Для точной оценки времени миграции при электрофоретическом разделении важен контроль ЭОП. Факторы, влияющие на его величину: вязкость электролита (ц), диэлектрическая проницаемость (е), электрокинетический потенциал (^-потенциал). Величина последнего определяется pH, ионной силой, температурой фонового электролита, типом составляющих его катионов. Незначительные колебания ЭОП резко снижают воспроизводимость времен миграции. Характер этих колебаний определяется изменением поверхности капилляра (^-потенциала). Этот эффект особенно ощутим при анализе реальных образцов (матричный эффект). Следовательно, при идентификации компонентов реального объекта необходима процедура пред
варительной стандартизации/нормализации, результатом которой явились бы стабилизация ЭОП и улучшенная воспроизводимость времен миграции. В ряде работ (например, [70]) рекомендовано временную шкалу заменить на шкалу эффективной подвижности (т.е. собственной электрофоретической подвижности Цэф = Цобщ - ЦэОП), используя ЭОП-маркеры или внутренние стандарты с известной или рассчитанной подвижностью. Использование электрофоретической подвижности вместо времен миграции позволяет корректно сопоставлять результаты разделения сложных смесей, полученные в разных условиях (рис. 4.2.7) [70].
а
.11।
30 35
Рис. 4.2.7. МЭКХ разделение триазинов: а — время миграции;
б — электрофоретическая подвижность
Скорость миграции аналитов в МЭКХ зависит от концентрации ПАВ в мицеллосодержащем рабочем электролите сПАВ, который контролирует концентрацию мицелл в ведущем электролите смиц (смиц = спав - ККМ).
Между логарифмом фактора удерживания аналита и логарифмом концентрации мицелл в ведущем электролите — линейное соотношение:
log k “ а + т log смиц.. (4.2.5)
Механизм разделения для нейтральных соединений в МЭКС основан на распределении аналитов между двумя движущимися фазами: управляемой ЭОП водной и псевдо-стационарной — мицеллой
/ Ивод ,
К —[^миц
(4.2.6)
где [5]миц — концентрация аналита в мицелле; [5]вод — концентрация аналита в ведущем электролите.
358
Новый справочник химика и технолога
Удерживание в МЭКХ описывается фактором удерживания к [141]:
к = Zr - /эоп/ ^эоп(1 ~ ZrZ/миц.) , (4.2.7)
где Zr, Zgon и ?миц. — времена миграции аналита, нейтрального полярного соединения, не удерживаемого «псевдо-стационарной фазой» и мигрирующего с ЭОП (маркер электроосмотического потока) и соединения, полностью удерживаемого мицеллярной псевдостационарной фазой (маркер мицеллы), соответственно.
Фактор удерживания зависит от концентрации мицелл в ведущем электролите:
к К Рмиц. / Кводц К const смиц / Кводн., (4.2.8) где Емнц — объем мицеллярной «псевдостационарной фазы»; Кводн — объем водной фазы в разделительном капилляре.
Используя логарифмическую форму уравнения (4.2.7), получим уравнение (5) при т = 1:
log k = а + log Смиц, = а + log (спав,- КМК) (4.2.9)
В условиях, когда ЭОП подавлен [146]
ZR=(1 + l/k)ZMHU. (4.2.10)
Хорошо зарекомендовавшие себя в газовой и высокоэффективной жидкостной хроматографии индексы удерживания были использованы и в МЭКХ для характеристики нейтральных ароматических соединений и способности удерживания «псевдостационарных фаз». Индексы удерживания лучше воспроизводятся, чем факторы удерживания, и не зависят от отношения фаз, т.е. от концентрации детергента [147].
4.2.7. Количественный и качественный анализ в КЭ
Для качественного и количественного анализа (как и в хроматографии) в КЭ используются два важнейших параметра: площадь (или высота) пика на электрофореграмме и время миграции. Относительное стандартное отклонение для времени миграции и площади пика с использованием двух внутренних стандартов достигает 1%. Использование большего количества стандартов (до четырех маркеров с известной электрофоретической подвижностью) позволяет добиться величины стандартного отклонения 0,01-0,03%. [198].
На точность определения площади (высоты) пика особенно влияют два фактора: вводимый объем и стеночные эффекты. Точность улучшается с повышением концентрации образца, особенно когда используется площадь, а не высота [195, 199].
Температура также является критическим фактором, от которого зависит вопроизводимость анализа в КЭ [199].
4.2.8. Эффективность, разрешение и селективность метода КЭ
Метод КЭ характеризуется высокой эффективностью. Эффективность, выраженная числом теоретических тарелок N, может быть определена по уравнению (4.2.11) или
непосредственно из электрофореграммы (при условии гауссовой формы пика) по уравнению (4.2.12):
У __
2^щ.
(4.2.11)
где Цэф. — электрофоретическая подвижность компонента; 7-эф. — эффективная длина капилляра; £общ. — общая длина капилляра; U — величина рабочего напряжения; D — коэффициент диффузии аналита.
(4.2.12)
где ZM — время миграции компонента; Wo>5 — ширина пика на половине высоты.
N возрастает с увеличением напряжения и уменьшением коэффициента диффузии. При напряжении от 100 до 35 000 В, а также эффективном заряде от 1 до 10 достигается величина 107 т. т. • м ’, т.е. КЭ по эффективности существенно превосходит ВЭЖХ. На практике это было продемонстрировано для молекул ДНК в капилляре, заполненном полиакриламидным гелем (т.е. когда отсутствуют взаимодействия аналита со стенками капилляра).
Как правило, теоретическое значение N (уравнение 4.2.11) превышает величину эффективности, полученную в реальном эксперименте (уравнение 4.2.12), поскольку в (4.2.11) учитывается только продольная диффузия: компоненты с низкими коэффициентами диффузии образуют более узкие пики. Если эффективность лимитирована диффузией, более высокое напряжение приведет к ее увеличению [148].
Ниже перечислены основные факторы, влияющие на уширение зон компонентов и, следовательно, величину N в капиллярном электрофорезе [149].
Продольная диффузия. Поскольку профиль потока в КЭ формируется с помощью ЭОП, его вкладом в уширение полос можно пренебречь. В идеальном случае во внимание принимается исключительно параметр продольной диффузии (вихревая и молекулярная диффузия, как в случае ВЭЖХ, — отсутствуют).
Длина вводимой зоны пробы, определяемая длительностью ее ввода: в идеале должна быть как можно меньше, однако достижение низких пределов обнаружения требует увеличения объема пробы или ее концентрирования.
На рис. 4.2.8 представлена зависимость эффективности от длины зоны вводимого образца.
Длина зоны ввода {см)
Рис. 4.2.8.
Зависимость числа теоретических тарелок для небольших молеул от длины вводимой зоны образца
Аналитическая хроматография и капиллярный электрофорез
359
Температурный градиент: при наложении электрического поля в капилляре, заполненном буфером, протекает электрический ток, величина которого зависит от удельной проводимости буфера и диаметра капилляра. Отвод тепла происходит через стенки капилляра, что приводит к возникновению в буфере радиального температурного градиента. Разница в температуре между серединой и стенками капилляра возрастает пропорционально квадрату диаметра капилляра. Температура в центре капилляра может быть на 10 °C выше, чем на внутренней стенке. Возникающий вследствие этого градиент вязкости приводит к тому, что вещество у стенки перемещается медленнее, чем в центре, это влечет за собой уширение полос и снижение эффективности [150].
На рис. 4.2.9 представлена зависимость фактора удерживания от температуры.
Рис. 4.2.9. Зависимость фактора удерживания к от температуры
Адсорбция на стенках капилляра: взаимодействие веществ со стенками капилляра ведет к искажению формы пиков (появление «хвостов»).
Различия в электропроводности пробы и ведущего электролита. Уширение пика, обусловленное электрофоретическими эффектами, пропорционально проводимости (и ионной силе) раствора образца относительно буфера. В случае высокой концентрации образца сила электрического поля (и, следовательно, линейные скорости) в зоне образца много ниже, чем в ведущем электролите. Благодаря этому происходит разбавление образца (дес-тэкинг) — уширение. Обратная ситуация в соотношении проводимостей (стэкинг), наоборот, приводит к узким пикам на электрофореграмме [175].
Наличие гидродинамического потока из-за различия в уровне жидкостей. Незначительная разность уровней между сосудами с электролитами на входном и выходном концах капилляра приводит к возникновению гидродинамического потока с параболическим профилем; чем больше диаметр капилляра, тем значительней это сказывается на эффективности разделения.
Всеми этими параметрами можно управлять, создавая оптимальные схемы разделения [71, 130, 151, 152].
4.2.9. Чувствительность метода КЭ
Среди возможных причин недостаточной чувствительности метода КЭ выделяют две: детектирование происходит в зоне в слое малой толщины (что обусловлено внутренним диаметром капилляра); очень малый объем вводимой пробы.
Несмотря на успехи лазерно-индуцированной флуоресценции и электрохимического детектирования для улучшения чувствительности метода КЭ по-црежнему основным способом детектирования остается спектрофотометрический. При этом УФ-детекторы не обладают достаточной концентрационной чувствительностью из-за небольших вводимых объемов и малой длины оптического пути в сравнении с хроматографическими методами [195]. Содержание большинства лекарственных препаратов при терапевтическом мониторинге — 1-30 мг л ', некоторых — даже ниже 1 мг-л-1. Чтобы обеспечить определение таких уровней концентраций в присутствии высоких концентраций белков (гл4), необходимо концентрирование вне или внутри капилляра. Особую трудность представляют реальные объекты: в сыворотке много белков (60 000 мг-л ') и солей (содержание хлорида натрия « 140 ммоль-л-1), что приводит к уширению полос; белки, связываясь со стенками капилляра, вызывают вторичные взваимодействия, что приводит к ухудшению воспроизводимости. Проблемы, связанные с воспроизводимостью ввода пробы при КЭ, обусловлены небольшой разницей в давлении и малым временем ввода пробы. Большие объемы при нормальных условиях быстро уменьшают эффективность разделения из-за перегрузки. Поэтому, вводя большие объемы, необходимо концентрировать зоны перед разделением. В табл. 4.2.13 представлены основные методы концентрирования, используемые в КЭ. Эффекты концентрирования достигаются, если работать с негомогенными буферными системами. Простейший случай: проба вводится из чистого водного раствора. Указанные эффекты особенно возрастают, когда увеличивается размер образца.
Таблица 4.2.13
Методы концентрирования (стэкинг-методы) в КЭ
Метод Снижение порога обнаружения Применимость Примечание
Электро-стэкинг 10-100 Прямая Быстро развивающийся метод. Результат зависит от электропроводности раствора и подвижности ионов пробы
Обращение поля 50-1000 Прямая Развитие методов для катионов и анионов различно
360
Новый справочник химика и технолога
Продолжение таблицы 4.2.13
Метод Снижение порога обнаружения Применимость Примечание
ИЭФ 10-100 Прямая Хорошо применимо для слабых кислот и оснований
ИТФ 100-1000 Условная Необходима адаптация метода для каждой новой молекулы пробы
Хроматографическое обогащение 100-500 Только со специальным капилляром Необходим дорогой и трудно устанавливаемый капилляр
Подходы к увеличению чувствительности можно подразделить на три категории: стратегия концентрирования образца; увеличение длины оптического пути; использование высокочувствительных селективных детекторов [60].
Стэкинг (stacking) — один из наиболее общих подходов к увеличению концентрационной чувствительности в КЭ. Стэкинг образца происходит, когда ионы аналитов пересекают границу, которая отделяет зону высокой проводимости раствора образца и низкой — ведущего электролита.
Когда матрица образца имеет значительно более низкую проводимость (обычно за счет разбавления буфером или другим растворителем), чем ведущий электролит, в зоне образца возникает относительно высокое электрическое поле. Аналиты внутри зоны образца движутся с более высокой локальной скоростью и, замедляясь на границе с зоной ведущего электролита, концентрируются. Стэкинг образца применителен к заряженным и нейтральным ана-литам [109, ПО]. В [111] сообщается о подобной технике фокусирования для мицеллярного КЭ. Матрица образца, наоборот, в этом случае имеет более высокое содержание солей (большую проводимость), чем ведущий электролит. Использование стэкинга позволяет приблизить чувствительность метода КЭ к ВЭЖХ.
Свипинг (sweeping) — новая техника концентрирования заряженных или нейтральных частиц в МЭКХ, ее суть заключается в том, что аналиты концентрируются псевдо-стационарной фазой (мицеллой), которая проникает в зону образца без псевдостационарной фазы [114]. При этом (в отличие от стэкинга) проводимость раствора образца близка проводимости ведущего электролита. Свипинг некоторых анионных аналитов дает высокие факторы концентрирования (в 1000 раз) без какой-либо стадии предварительного концентрирования (рис. 4.2.10).
На рис. 4.2.10 иллюстрируется пример разделения анионов [114], где объединена техника стэкинга и свипинга: капилляр покрыт полиакриламидом, заполнен высоко-
проводящим буфером без мицелл; с входного конца капилляра — водяная пробка (гидродинамический ввод) (А); далее электрокинетическим путем вводятся анионы аналитов; на границе вода - зона буфера высокой проводимости имеет место стэкинг (ионы аналитов мигрируют через водную пробку с высокой скоростью; у границы с высоко-проводящим буфером замедляются и фокусируются) (Б); аналиты направлены к аноду, ЭОП — к катоду. Далее, с двух концов капилляра вводятся положительно заряженные мицеллы и приложено напряжение (В). Мицеллы взаимодействуют с аналитами. ЭОП и аналиты движутся в одном направлении.
Рис. 4.2.10. Разделение анионов в режиме МЭКХ [114] с использованием техники стэкинга-свипинга
Другая возможность использовать негомогенность буфера для концентрирования компонентов пробы состоит в выборе буфера. Подвижность ионов при прохождении скачка pH [155] на приграничной поверхности между буфером и раствором пробы изменяется, и происходит кон
центрирование.
Рис. 4.2.11. Схема изменения оптической ячейки для увеличения длины оптического пути [60]: а — стандартное детектирование («on-column»);
б — оптимизированная 2-я ячейка; в — прямоугольный капилляр; г — ячейка многократного отражения;
д — детектирование на конце капилляра («end-column»)
Аналитическая хроматография и капиллярный электрофорез
361
Чувствительность при УФ-определении в КЭ может быть увеличена за счет увеличения толщины слоя детектирования при использовании капилляров с расширенным световым путем. Предложен ряд способов повышения чувствительности за счет увеличения длины оптического пути:
• ячейка детектирования в форме пузырька (bubble cell);
• капилляры Z-формы (Z-shapped capillaries), т.н. Z-ячейка;
• ячейка высокой чувствительности.
Увеличение внутреннего диаметра капилляра в зоне детектирования увеличивает сигнал в 3-5 раз. Использование Z-ячейки детектирования позволяет увеличить чувствительность в 20-40 раз. В зоне детектирования капилляр
сгибается в Z-образную форму; длина оптического пути увеличивается до 3 мм.
Ячейка высокой чувствительности аналогична известной для микро-ВЭЖХ Z-ячейки детектирования, которая располагается внутри Z-образного подвижного кварцевого блока (рис. 4.2.11. и табл. 4.2.14, 4.2.15). При этом удается достичь 20-кратного увеличения чувствительности. Изготовление таких ячеек связано со сложными технологическими процессами. В [193] предложен химический способ создания в капилляре «пузырька», служащего ячейкой детектирования, путем растворения внутренней поверхности капилляра в зоне детектирования 40% раствором плавиковой кислоты.
Таблица 4.2.14
Способы увеличение чувствительности: формат капилляра [60]
Способ увеличения чувствительности Увеличение Эффективная длина пути (внутренний диаметр) Применимость в коммерческих системах Комментарии
Z-ячейка 3-5 3 мм (75 мкм) Да Непосредственное применение метода; высокий уровень шумов; небольшое уменьшение разрешения; средняя стоимость
Z-ячейка/оптика 10-20 3 мм (75 мкм) То же Непосредственное применение метода; увеличение размеров более чем на порядок; минимальные потери в разрешении; средняя стоимость
Прямоугольный капилляр 9,5 500 мкм (50 мкм) Тоже Требует внимания в работе (выбор буфера, скорости потока); высокие загрузки проб; производится на заказ
Многократное отражение 40 3,3 мм (75 мкм) Нет Приозводится на заказ; приборы требуют модификации
Детектирование на выходном конце капилляра 7 3 мм (50 мкм) То же Регулировка требует изменений; необходимо использовать неводные буферы
Использование УФ-лазера 10-50 50 мкм (50 мкм) То же Высокая стоимость; доступны специфические длины волн; надежность
Таблица 4.2.15
Увеличение чувствительности: концентрация пробы
Метод Увеличение Применимость в коммерческих системах Комментарии
Стэкинг 10-100 Да Прост в исполнении; зависит от ионной силы пробы, связанной с буфером разделения
Усиление поля 50-1000 Тоже Не может применяться одновременно для катионов и анионов
ИТФ 100-1000 Нет Требует разработки методов разделения для различных аналитов; сейчас доступна неавтоматизированная система пре-электрофореза
Хроматографическое концентрирование 100-1000 То же Эффективная система сложна в производстве
Примечание. * Увеличение относительно стандартного цилиндрического капилляра того же диаметра
362
Новый справочник химика и технолога
4.2.9.1. Селективность
Обычно в КЭ разрешение двух компонентов определяют так же, как в ВЭЖХ:
где /1 и ?2 — времена миграции первого и второго компонента, мин; Wi и W2 — ширина пиков 1 и 2 при основании, мин.
Разделение в КЭ, в первую очередь, управляется эффективностью, а не селективностью. В этом заключается еще одно важное отличие КЭ от ВЭЖХ: благодаря узким зонам компонентов даже очень малые различия в электрической подвижности веществ (иногда < 0,05%) обеспечивают полное разделение [44].
/г= —V2V-I — I (4.2.14)
4 I Н )
ц + ц, где Др = Ц2 - Ць - •
Селективность а разделения двух компонентов в зонном варианте КЭ определяется выражением:
а=—, (4.2.15)
Pi
где Ц! — электрофоретическая подвижность компонента 1; ц2— электрофоретическая подвижность компонента 2.
При концентрации ПАВ в растворе электролита больше ККМ реализуется вариант мицеллярной электрокинетиче-ской хроматографии и принцип разделения на основе распределения компонентов пробы между гидрофильной (водной) и гидрофобной (мицеллярной) фазами, селективность которого будет определяться отношением факторов к'
емкости двух компонентов: а = . В свою очередь, к' в
режиме МЭКХ можно найти:
= (4.2.16)
где za — время миграции анализируемого вещества; t0 — время миграции неудерживаемого компонента; /миц. — время миграции мицеллы.
Для нахождения ?0 и /миц. в пробу вводят маркер ЭОП (ацетон) и метку мицелл (судан 3 или судан 4).
Основным достоинством метода КЭ является высокая эффективность (сотни тысяч теоретических тарелок), при этом селективность в КЗЭ недостаточна [157]. Расширение диапазона селективности может быть достигнуто за счет изменения pH ведущего электролита, введения в состав буфера различных добавок: ПАВ [77-80, 89, 90], макро
циклических агентов [88, 90], смешанных мицелл [161], органических растворителей [84, 188] и т.д. На селективность разделения в КЭ влияют тип мицелл, хиральных селекторов [156], природа органических растворителей; в [96, 118] исследуется влияние ПЭГ различной массы на селективность разделения различных замещенных бензойных кислот в КЭ. Модифицируя либо мицеллу, либо ведущий электролит, можно менять селективность и эффективность разделения [100].
В [87] в режиме мицеллярной электрокинетической хроматографии, применив короткие капилляры, сильное электрическое поле, щелочной буфер (pH = 9,5), позволивший генерировать сильный ЭОП и гидродинамическое давление, удалось разделить незаряженные производные изотиазолонов.
Имеется ряд работ по неводному капиллярному электрофорезу [137, 158].
Неводные растворители в КЭ перспективны для соединений, плохо растворимых в воде, а также имеющих близкие электрофоретические подвижности. Органические растворители (метанол, ацетонитрил, изопропанол), которые вводят в буферный раствор в концентрации от 5 до 20% (объемн.), могут уменьшать гидрофобные взаимодействия между анализируемым компонентом и мицеллой, а также влиять на подвижность ЭОП и собственную электрофоретическую подвижность аналита [47], тем самым изменяя селективность.
В [159] обсуждаются свойства формамида в качестве ведущего электролита, имеющего более высокую диэлектрическую константу, чем вода. В [160, 162] рассматриваются вопросы хирального разделения в неводных растворителях. В [153] исследуется влияние органических растворителей (метанола, этанола, 1-пропанола и 2-пропа-нола, ацетонитрила) в качестве модификаторов в КЭ, улучшающих форму пиков и способствующих разделению позиционных изомеров фенола.
При добавлении веществ, образующих ионные пары, меняется электрофоретическая подвижность. В [76] обсуждается использование т.н. «ионных жидкостей» (типа тэтраэтиламмония тетрафторбората) в качестве ведущего электролита, взаимодействующего с аналитами, и рассчитываются константы такого взаимодействия в режиме КЭ [85].
Влияя на ЭОП модификацией поверхности капилляра, осуществляют разделение ионов с сильно различающимися подвижностями.
Капилляры с покрытием [1, 29] обладают следующими особенностями: позволяют управлять электроосмотическим потоком (направлением и его величиной); способствуют уменьшению адсорбции на стенках аналитов. Однако имеются проблемы с приготовлением и стабильностью подготовленных покрытых капилляров.
Различают методы динамического и статического покрытия. Для разделения белков в КЭ используют покрытия на основе диолов, ПЭГ, мальтозы. Внутренние стенки капилляров можно покрыть не только углеводородами, но и белками, например, альфа-лактальбумином. Широкое
Аналитическая хроматография и капиллярный электрофорез
363
применение для разделения биомолекул и белков, фрагментов ДНК нашли покрытия на основе линейных полиакриламидов, на основе поливинилпирролидона, поли-этиленимина. Используются цвиттер-ионы для модификА-ции поверхности капилляра [92], капилляры с полиакриламидным покрытием [114].
До настоящего времени коммерчески доступны капилляры только с очень ограниченным числом типов покрытий: • Applied Biosystems (Foster City, California) выпускают катионные реагенты, обеспечивающие положительный заряд поверхности капилляра и способствующие обращению ЭОП;
• BioRad (Hercules, California) выпускают капилляры, модифицированные гидрофильными покрытиями;
• Supelco (Pennsylvania) готовят нейтральные гидрофильные, слабо гидрофобные Сц гидрофобные С8 и С18;
• Isco, Inc. (Lincoln, Nebraska) предлагают покрытия следующих типов: ковалентно-связанные Ci8, глицериновое, сульф окис л отные [29].
Селективносью можно управлять и с помощью стэкинга — концентрирования образца в капилляре (см. выше). В самом простом варианте ввод пробы осуществляется из чистого водного раствора. Образец подготавливается в том же самом разделительном буфере, но с более низкой ионной силой (« в 10 раз меньшей). Степень концентрирования достигает »100. Указанный прием недостаточно эффективен при концентрировании ионов с малой подвижностью и в случае, когда ЭОП и ионы ориентированы в одном направлении.
Большее концентрирование пробы происходит, если на короткое время приложить напряжение с полярностью, противоположной той, при которой будет осуществляться разделение. При этом определяемые ионы должны перемещаться в направлении, обратном ЭОП. Можно достичь тысячекратного концентрирования. Ограничение состоит в том, что могут анализироваться либо анионы, либо катионы.
Другой вариант использования негомогенности буферных систем для концентрирования пробы — различия в pH. Концентрирование ионов пробы происходит при прохождении скачка pH на границе раздела между буфером и пробой, что, например, используется при разделении белков. Белки вводятся из раствора с высоким значением pH, а разделяются в буфере с низким pH.
В табл. 4.2.16 суммированы основные параметры, влияющие на селективность в КЭ, и достигаемые результаты.
Таблица 4.2.16
Изменяемые параметры и достигаемые эффекты
Переменная Диапазон Эффект от увеличения переменной
Тип и размер ЦД Большое влияние на хи-ральную селективность
Концентрация ЦД 1-100 мМ Повышение вязкости, понижение ЭОП, повышение миграции раствора в случае комплексообразования
Продолжение таблицы 4.2.16
Переменная Диапазон Эффект от увеличения переменной
Напряжение 5-30 кВ Уменьшение времени анализа; небольшое ухудшение разрешения
Ток 5-250 мкА Уменьшение времени анализа; небольшое ухудшение разрешения
Длина капилляра 20-100 см Увеличение времени анализа; улучшение разрешения
Внутренний диаметр капилляра 25-100 мкм Увеличение силы тока; небольшое ухудшение разрешения
pH 1,5-11,5 Увеличение ЭОП; повышение ионизации кислот, понижение ионизации оснований
Органические растворители 1-30% объемн. Улучшение или ухудшение разрешения
Мочевина 1-7 М Повышение ионизации гидрофобных растворов (и ЦД)
Ион-парный реагент 1-20 мМ Может происходить ухудшение или улучшение разрешения
Аминные модификаторы 1-50 мМ Уменьшение заряда поверхности, уменьшение «хвостов» пиков
Вязкость Различная Уменьшение ЭОП, увеличение времен миграции
Концентрация электролита 5-200 мМ Улучшение разрешения, повышение силы тока, уменьшение ЭОП, ионизация раствора, уменьшение «хвостов»
Катионный детергент 1-20 мМ Обращение ЭОП, увеличение растворимости, увеличение времен миграции
Время ввода 1-20 с Улучшение сигнала, небольшое ухудшение разрешения
Производные целлюлозы или полимеров 0,1-0,5% Может улучшиться разрешение
Тип солевой добавки 10-50 мМ Выбор сильно влияет на хиральную селективность
364
Новый справочник химика и технолога
Примеры увеличения чувствительности детектирования с использованием инструментальных решений представлены на рис. 4.2.12 и 4.2.13.
Рис. 4.2.12. Многокапиллярная система, использующая sheath-flow технику: 1 — капилляр;
2— выходное отверстие капилляра; 3 — входное отверстие капилляра;
4 — емкости с буфером; 5 — микротитровочный столик; 6 — кварцевая камера; 7 — лазер;
8 — лазерный луч;
9 — линзы;
10 — флюидный поток
Рис. 4.2.13. Сочетание КЭ с ЯМР
4.2.10. Объекты для анализа методом капиллярного электрофореза
Первые аналитические приложения КЭ были связаны с разделением полярных заряженных образцов: наиболее «подходящими» оказались неорганические катионы и анионы, а также низкомолекулярные карбоновые кислоты [72, 100, 101, 137, 163]. В это же время происходит частичная замена традиционного электрофореза его капиллярным исполнением, таким образом, КЭ начинают использовать в биотехнологии для анализа нуклеиновых кислот, аминокислот, макромолекул — белков и пептидов [106,149, 176, 177].
Относительная электрофоретическая подвижность пептидов связана с молекулярной массой следующим соотношением:
Цотн.
Z
Vm7’
где Z — заряд биополимера; М — его молекулярная масса. При этом одной из серьезных проблем является сорбция аналитов на стенках кварцевого капилляра. Для предотвращения данного явления осуществляется предварительная химическая модификация силанольных гидроксилов путем силанизации. Используют и динамическую модификацию силанольных групп. Так, поверхностью капилляра сильно сорбируются полимерные полиамины (полибрен — гексадиметрин бромид, полидиметилдиаллиламмоний хлорид, полиэтиленимин и т.д.) Большинство аминокислот и корготкоцепочечных пептидов не поглощают в УФ-свете [101]. Детектирование этих аналитов требует дериватиза-ции с введением хромофорных или флуорофорных групп, тогда достигается возможность детектировать аттомоляр-ные (10 21) количества [164].
Важной областью применения КЭ (см. табл. 4.2.17) стала фармацевтика [87, 142, 149, 154,168, 178, 179].
Таблица 4.2.17
Использование различных вариантов КЭ для характеристики объектов биофармацевтики [169]
ВидКЭ Применения
Зонный капиллярный электрофорез Белки, пептиды, гликопротеины, моноклональные антитела, олигосахариды
Изоэлектрическая фокусировка Белки, пептиды, гликопротеины, моноклональные антитела; определение изоэлектрической точки
Капиллярный гель-электрофорез Олигонуклеотиды, ДНК, полисахариды, определение молекулярных масс белков
Мицеллярная электро-кинетическая хроматография Пептиды, углеводы
В табл. 4.2.18 представлен список лекарственных средств, анализируемых в сыворотке или моче методом КЭ.
Таблица 4.2.18
Список лекарственных средств, анализируемых в сыворотке или моче методом КЭ
Класс/соединение Метод Буфер / условия
Антиэпилептики
Барбитураты МЭКХ Фосфатный буфер 25 мМ; pH = 8,0; 50 мМ ДДСН; 210 нм
То же То же Борат-фосфатный буфер; pH - 7,8; 50 мМ ДДСН; сканирование
Аналитическая хроматография и капиллярный электрофорез
365
Продолжение таблицы 4.2.18
Класс/соединение Метод Буфер / условия
Felbamate МЭКХ Боратный буфер 100 мМ; pH = 8,4; 55мМ ДДСН; 214 нм
Фенобарбитал То же Боратный буфер 100 мМ; pH = 8,4; 55мМ ДДСН; 214 нм
Фенобарбитал КЗЭ Боратный буфер 300 мМ; pH = 8,5; 254 нм
Lamotrigine* То же Ацетатный буфер 130 мМ; pH = 4,8; 214 нм
Gabapentin То же Борат-фосфатный буфер; реакция с флуоре-скамином
Несколько МЭКХ Боратный буфер; pH = 9,3; с экстракцией
Антиаритмики
Амиодарон КЗЭ Фосфатный буфер, стэкинг
Desethyl-amiodarone* То же То же
Прокаинамид То же Фосфатный буфер 50 мМ; 200 нм
Л’-Ацетил-прокаинамид То же То же
Несколько МЭКХ Экстракция пробы
Сердечно-сосудистые
Несколько МЭКХ | Твердофазная экстракция
Анальгетики
Парацитамол МЭКХ Боратный буфер; pH = 10 с ДДСН; 200 нм
Салициловая кислота То же То же
Тоже КЗЭ Боратный буфер 175 мМ; pH = 9,4
Ибупрофен То же Боратный буфер 200 мМ; 214 нм
Кетопрофен То же Боратный буфер 250 мМ; 254 нм
Антиастматические
Теофиллин МЭКХ Боратный буфер 6 мМ; фосфатный буфер 10 мМ; pH ~ 9,0; 75 мМ ДДСН; сканирование
То же КЗЭ Боратный буфер 300 мМ; рН = 8,5; 254 нм
То же МЭКХ Фосфатный буфер 20 мМ; pH = 11; ДДСН
То же КЗЭ/ МЭКХ Борат-фосфатный буфер; pH = 9; детектирование при 280 нм
Продолжение таблицы 4.2.18
Класс/соединение Метод Буфер / условия
Кофеин МЭКХ Боратный буфер 6 мМ; фосфатный буфер pH =10; pH ~ 9,0; 75 мМ ДДСН; сканирование
Контрастные вещества, применяемые при исследовании почек
lohexol* КЗЭ Боратный буфер 220 мМ; pH = 8,8; 254 нм
lopamidol То же Боратный буфер 175 мМ; pH = 9,4; 254 нм
lothalamic acid* То же Боратный буфер 175 мМ; pH = 9,4; 254 нм
Иммуносупрессоры
Циклоспорин МЭКХ Фосфат-боратный буфер; ДДСН; ацетонитрил; 200 нм
Противоопухолевые
Suramin КЗЭ Capso буфер 63 мМ; pH = 9,7; 25 нм
То же То же TRIS-борат; pH = 8,6
Метотрексат То же TRIS-MES буфер; pH = 6,7; экстракция; ЛИФ-детектирование
Таксол МЭКХ TRIS-борат; pH = 8,5; 100 мМ ДДСН; экстракция; 230 нм
Цитозин-P-D-арабинозид КЗЭ Цитратный буфер 40 мМ; pH = 2,5; экстракция
Томоксифен То же Ацетатный буфер; ацетонитрил; метанол; экстракция
Антибиотики
Цефиксим КЗЭ Фосфатный буфер 50 мМ; pH = 6,8
Различные МЭКХ Фосфат-боратный буфер с ДДСН
Амикацин То же Фосфат-боратный буфер; pH = 7,0; флуоресценция
Остальные препараты
Никотиновая кислота КЗЭ Боратный буфер 10 мМ; pH = 9,3 — фосфатный буфер 10 мМ; pH = 2,3; 254 нм
ОН-Кумарин То же Фосфатный буфер pH = 7,5; экстракция
Фосфомицин То же Боратный буфер; 254 нм
366
Новый справочник химика и технолога
Продолжение таблицы 4.2.18
Класс/соединение Метод Буфер / условия
Заменители гепарина МЭКХ Фосфатный буфер сДДСН
P-Блокаторы, различные Тоже ДЦСН 50 мМ в 100 мМ боратного буфера; pH =8,1
P-Блокаторы, различные КЗЭ Фосфатный буфер; рН = 3,1
Р-Блокаторы, различные МЭКХ Фосфатный буфер; pH = 7,0; с бромидом N-ацетил триметил-аммония
Глипивид То же Концентрирование на капилляре
Glyburide* То же Концентрирование на капилляре
Фторцитозин То же Фосфат-боратный буфер; pH = 9,2; 210 нм
Различные препараты То же Экстракция
Флуконазол То же Прямой ввод или экстракция; 190 нм
Примечание. Отсутствует отечественный аналог.
Оценка чистоты лекарственных препаратов и хираль-нЫе разделения [93, 95, 154] на 90% выполняются различными вариантами КЭ.
Несмотря на то, что изомеры имеют часто близкие химические структуры, их биологический эффект резко различен. Хиральное разделение имеет особое значение для КЭ из-за его экспрессное™, высокого разрешения, эффективности, простоты и низкой стоимости анализа в сравнении с хроматографическими вариантами разделения энантиомеров (ТСХ, ВЭЖХ, газовой хроматографией).
Разделение двух энантиомеров в КЭ может быть достигнуто с использованием хиральных селекторов, введенных в состав ведущего электролита, взаимодействие которого с рацемической смесью приводит к образованию диастереомеров, обладающих различными электрофоретическими подвижностями.
Большинство хиральных разделений выполняется в чистых растворах, однако, имеется серия публикаций, посвященная разделению энантиомеров лекарственных препаратов с помощью мицеллярной электрокинетаческой хроматографии, включая использование хиральных мицелл и белков [170] при терапевтическом мониторинге в моче и в сыворотке; решаются вопросы идентификации хиральных лекарственных препаратов методом КЭ, устанавливаются параметры, влияющие на стереоселективность. [170-175]. В качестве хиральных селекторов используются циклодекстрины [75, 95, 184, 192] и их
производные, хиральные краун-эфиры [95], протеины, линейные олигосахариды, гепарин [97, 176], хиральные производные аминокислот с ацилкаликс-4-ареном [98], гликопептидные антибиотики (ванкомицин, канамицин, стрептомицин и др.) [93]. Энантиоселективность контролируется модификацией нескольких параметров: тип антибиотика и его концентрация, pH, ионная сила и концентрация ведущего электролита, природа органического модификатора и т.д. Высокая энантиоселективность, проявляемая этими агентами, характерна для природных соединений. Так, ванкомицин содержит 18 асимметричных центров и несколько функциональных групп (карбоксильные, гидроксильные, амидные, аминогруппы, ароматические кольца). Они хорошо растворимы в воде и в полярных апротонных растворителях. Благодаря высокой стереоселективности, эти хиральные селекторы используют при низких концентрациях (0,5-5 мМ) с прямым и косвенным УФ-детектированием. Во избежание адсорбции антибиотиков на стенках капилляра его промывают длительное время водой, затем раствором ведущего электролита, не содержащего добавок антибиотиков.
Возможная схема, иллюстрирующая оптимизацию процедуры энантиомерного разделения [158], представлена на рис. 4.2.14.
Развитие новых возможностей метода привело к расширению круга соединений и областей, доступных для анализа с использованием КЭ: криминалистика (анализ наркотиков [138, 179, 180]), биомедицинские исследования [49, 136, 181, 182, 187, 194], экологический мониторинг (анализ пестицидов, фунгицидов) [41, 165, 183, 185, 186, 189], анализ продовольственного сырья, пищевых продуктов, напитков (определение кофеина в растворимом кофе, ароматических альдегидов в винах) [190]; исследование и определение отдельных комплексных форм d-элементов в растворе [191].
Повышение чувствительности определения как за счет появления современных способов детектирования (ЛИФ и масс-спектрометрия) [82, 91], так и вследствие разработки оперативных вариантов концентрирования пробы ([109-111, 165, 166]) обеспечили доступ капиллярному электрофорезу к следовому анализу [138]. Многие лекарственные препараты находятся на уровне ниже пределов обнаружения для КЭ и ВЭЖХ. Описаны методы повышения чувствительности при одновременном детектировании нескольких лекарств с использованием комбинированного варианта им-муноферментного, лазерно-индуцированного детектирования и КЭ [182].
Разработка принципов неводного КЭ [167] позволила снять ограничение по растворимости анализируемых образцов в водных системах. Так, соединения, не растворимые (или крайне мало растворимые) в водных или водноорганических (содержание органического компонента < 50 %) растворах, прежде не могли быть проанализированы с помощью капиллярного электрофореза. В табл. 4.2.19 приведены примеры оптамального использования различных видов КЭ для разделения ионов, амфотерных соединений и неэлектролитов.
Аналитическая хроматография и капиллярный электрофорез
367
Рис. 4.2.14. Схема хирального разделения методом КЭ веществ основного, кислотного и нейтрального характера
Таблица 4.2.19
Электрофоретическое разделение веществ различной природы [104]
Образец, область pH Механизм разделения Комментарий
Катионы органические и неорганические, кислая среда КЗЭ Детектирование у катода; необходимо уменьшать скорость ЭОП (для неорганических катионов); риск адсорбции на поверхности капилляра
Анионы органические и неорганические, щелочная среда КЗЭ Детектирование у анода, обязательное обращение ЭОП; детектирование у катода (органические анионы)
Амфотерные соединения в широкой области pH ИЭФ Риск поверхностной адсорбции; покрытие капилляра; снижение скорости ЭОП до минимальной
Неэлектролиты МЭКХ Выбор ПАВ
Сильногидрофобные вещества ЦД-МЭКХ* Тип и концентрация ЦД определяют селективность
Примечание. ЦД-МЭКХ — МЭКХ, модифицированная циклодекстрином.
368
Новый справочник химика и технолога
Определение ионной природы вещества
нейтральный
катионный
анионный
Проведение экспериментов
с использованием следующих буферов
25 мМ фосфатный (pH = 2,5)
25 мМ боратный (pH = 9,3)
25 мМ боратный
+ 25 мМ додецилсульфат натрия (pH = 9,3)
Первый этап:
25 мМ фосфатный буфер (pH = 2,5)
Первый этап:
25 мМ боратный буфер
(pH = 9,3)
Есть пики?
Проверка концентрации пробы
(НЕТ)
(ДА)
Оптимизация разделения
Если проба слишком разбавлена: выбирают более чувствительный детектор, концентрируют пробу, используют электрокинетический ввод пробы.
2.
3.
Если проба имеет высокий солевой фон : проводят обессоливание пробы или увеличивают концентрацию буфера
Рис. 4.2.15. Схема пробного КЭФ-анализа образца неизвестного состава
Для образцов неизвестного состава можно применить следующую схему пробного анализа методом КЭ (рис. 15).
4.2.11. Подготовка пробы к анализу методом капиллярного электрофореза
Первым этапом любого анализа, в том числе с использованием метода КЭ, является отбор и подготовка пробы. Отобранная проба должна быть представительной (т.е. не отличаться по качественному и количественному составу от анализируемого материала), а процедура отбора пробы— легко воспроизводимой [24]. При необходимости проводят консервацию пробы [78]. Время хранения отобранных проб перед анализом должно быть минимальным, а условия и способы хранения должны исключать неконтролируемые изменения состава пробы за счет испарения, окисления и фотодеградации [32]. На этапе подготовки пробы к анализу проводят удаление мешающих веществ, выделение и концентрирование определяемых соединений, их превращение в более удобные аналитические формы [24]. Необходимость выполнения всех или только некоторых из перечисленных процедур обусловливается природой анализируемого объекта и свойствами содержащегося в нем доминирующего вещества. Так, например, зонный вариант капиллярного электрофореза с УФ-детекти-
Первый этап: 25 мМ боратный + 25 мМ додецилсульфата натрия (pH = 9,3)
рованием позволяет анализировать неорганические анионы в водных объектах на уровне десятых долей мг л1 при прямом определении. В этом случае подготовка образца воды (питьевой, природной и сточной) к КЭ-анализу будет заключаться в фильтровании пробы через мембранный фильтр с диаметром пор 0,2 мкм и дегазации фильтрата путем его центрифугирования (4000 об. мин"1, 2 мин) [22]. Однако с целью повышения чувствительности и селективности анализа методом КЭ, прибегают к следующим способам отделения пробы от ее матрицы: адсорбции, твердофазной и жидкофазной экстракции, СВЧ-минерализации и др. [11]. Часто используют комбинацию отдельных названных методов и их разновидностей, включая обработку порции анализируемого материала специфическими химическими реагентами, т.е. проводят дериватизацию [24].
Дериватизация широко используется в КЭ для повышения чувствительности детектирования, особенно в комбинации с селективными детекторами (например, флуоресцентный на лазерах): в анализируемое соединение «вводят» хромофорные или флуоро-форные группы [11], изменяя при этом летучесть, заряд, массу, гидрофобность, оптические свойства и т.д. [56].
Всю информацию по дериватизации принято классифицировать по трем признакам [65]:
• схема получения производных (предкапиллярная, посткапиллярная, on-line);
• реагенты для дериватизации;
• природа функциональной группы, по которой идет реакция.
Среди известных схем получения производных чаще всего выбирают предкапиллярный вариант [65-69], что определяется отсутствием ограничений по реагентам (можно использовать даже те реагенты, для которых скорость реакции невысока или требуется значительное повышение температуры) и возможностью удаления избытка реагента и концентрирования полученных производных перед анализом. В продаже имеются большое количество коммерчески доступных готовых наборов для предкапиллярной дериватизации [65].
В табл. 4.2.20 представлены наиболее часто упоминаемые в литературе последних лет реагенты для дериватизации, а также приведены условия реакций и спектральные свойства полученных производных. В табл. 4.2.21 также представлены реагенты для предколоночной дериватизации аминогруппы.
Аналитическая хроматография и капиллярный электрофорез
369
Таблица 4.2.20
Некоторые реагенты для дериватизации, условия реакций, спектральные свойства полученных производных
Принятые обозначения. ФА — фталевый альдегид; ПрО — предел обнаружения; ФИТЦ — флуоресцеин изотиоцианат; ФТГ — фенилтиогидантоин; ФЛУ — флуоресцентное детектирование.
Реагент Анализируемые соединения Вариант КЭ Время реакции при tK0MH 7-макс. (УФ) или ХВ03б / Хрег (ФЛУ), нм Дополнение Литература
ФИТЦ Аминокислоты МЭКХ (ФЛУ) 6 ч 488 / 520 Предел обнаружения 1011 М 63, 66
То же То же КЗЭ, МЭКХ (ФЛУ) То же То же Хиральные разделения смеси рацематов 67
То же Биогенные амины То же То же — 68
Дансил-хлорид Аминокислоты МЭКХ (ФЛУ) 120 мин 360/570 Предел обнаружения 10 нг • л“1; хиральные разделения 69
ФТГ То же МЭКХ (УФ) 5 мин 254 — 70
ФА То же КЗЭ, МЭКХ (ФЛУ) 2 мин 340/475 Предел обнаружения 10-7М 71
ЭДТА Неорганические катионы КЗЭ (УФ, косв.) 5 мин 254 Разделение комплексных форм 26, 72
Таблица 4.2.21 Реагенты для предколоночной дериватизации аминогруппы [201]
Принятые обозначения. LOD — предел детектирования; ФЛУ — флуоресцентное детектирование; НГСКЭ — негелевый ситовый капиллярный электрофорез; АЕОС — 2-(9-антрил)этилхлор-формиат; AQC — 6-аминохинолил-У-гидроксисукциномидил-карбамат; CBQCA — 3-(4-карбоксибензоил)-2-хинолинкарбок-сальдегид; CTSP — 9-циано-У,У,У-триэтил-У-(5-сукцинимидил-оксикарбонилпентил) пиронин; (+) DAT — (+) - 0,0?- диацетил-ангидрид L-винной кислоты; (+) DBT — (+) <9,<9'-дибензоил-ангидрид L-винной кислоты; DCC — дикарбоцианин реагент; DCCS — 7-(диэтиламино)кумарин-3-карбоксисукциндимидил эфир; DNSH — 5-(диметиламино)нафталин-1-сульфогидразин (дансил-гидразин); FTIS — флуоресцеин изофиоционат; FLEC — 1-(9-флуоренил)-этилхлороформеат; FMOC — 9-флуоренилметилхлор-формиат; IDA— 1 -метоксикарбонилиндолизин-3,5-дикарбальдегид; NBD — F - 4-фторо-7-нитро-2,1,3-бензооксадиазол; NDA — на-фалин-2,3-дикарбоксальдегид; ОРА — фталевый альдегид; PITC — фенилизотиоцианат; SAMBI — (5)-1-фенилэтилизотиоцианат; SNEIT — (5)-1-(1-нафтил)этилизотиоцианат; TRITC — тетраме-тилродаминизотиоцианат.
Реагент Вид КЭ/ детектирование Соединения Примечания; [литература]
АЕОС КЗЭ/ МЭКХ-УФ /ЛИФ Аминокислоты LOD: 0,1/150 amol (ЛИФ/УФ) [1]
Продолжение таблицы 4.2.21
Реагент Вид КЭ/ детектирование Соединения Примечания; [литература]
АЕОС КЗЭ/ МЭКХ-УФ Энантиомеры аминокислот Использование в качестве хиральных селекторов циклодекстринов; [2]
AQC КЗЭ-УФ / ФЛУ Синтетические пептиды с протромбином LOD: 11/15 fmol (ЛИФ/УФ); [3]
Бензоин КЗЭ / ЛИФ Пептиды, содержащие аргинин LOD: 270 amol; [4]
CBQCA мэкх-ЛИФ Аминокислоты LOD: 9,0 zmol; [5]
МЭКХ-ЛИФ Пептиды Из пищевых добавок;[6]
мэкх-ЛИФ Аминокисло-ты/пептиды LOD: 4,6-70 amol; [7]
мэкх-ЛИФ Аминосахара LOD: 240 amol; [8]
КЗЭ/ КГЭ-ЛИФ Аминокислоты КЗЭ в сопоставлении КГЭ [9]
370
Новый справочник химика и технолога
Продолжение таблицы 4.2.21
Реагент Вид КЭ/ детектирование Соединения Примечания; [литература]
CBQCA МЭКХ-ЛИФ Аминосодержащие соединения LOD: 2,6-885 amol; [10]
МЭКХ-ЛИФ Аминосахара LOD: 75 zmol; [И]
CTSP КЗЭ-ЛИФ Аминокислоты LOD: 1,2-6,8 amol; [12]
(+) DAT КЗЭ-УФ D-/L-Триптофан С полимерными буферными добавками; [13]
КЗЭ-УФ Энантиомеры а-амино-кислот С полимерными буферными добавками; [14]
(+) DBT КЗЭ-УФ То же С полимерными буферными добавками; [15]
КЗЭ-УФ То же Эффект растворителя и pH; [16]
DCC мэкх-ЛИФ Аминокисл о-ты/низкомо-лекулярные пептиды LOD: 0,1 amol; [17]
DCCS КЗЭ / ЛИФ Аминокислоты LOD: 100 amol; [18]
DNSH КЗЭ / ЛИФ Высокополярные токсины Из природных источников [19]
FITC КЗЭ/ЛИФ Аминокислоты LOD: 9-150 zmol; [20]
КЗЭ / ЛИФ То же LOD: 2 fmol; [18]
КЗЭ / ЛИФ То же LOD: 1,1 amol; [21]
КЗЭ / ЛИФ То же LOD: 1,7-6 zmol; [22]
КЗЭ/ЛИФ То же LOD: 3,9 fmol; [23]
КЗЭ/ЛИФ То же LOD: уровень amol; [24]
КЗЭ / ЛИФ Пептиды Производные по одной аминогруппе; [25]
FLEC МЭКХ-УФ Энантиомеры аминокислот Хир альное разделение с косвенным детектированием; [26]
Продолжение таблицы 4.2.21
Реагент Вид КЭ/ детектирование Соединения Примечания; [литература]
Флуо-ресци- МЭКХ-ЛИФ Пептиды Из пищевых добавок; [7]
КЗЭ-ЛИФ Высокополярные токсины Из природных источников; [19]'
КЗЭ- ЛИФ Аминокислоты LOD: 23,7 fmol (LIF); [23]
МЭКХ/Ф Полиамины В тканях крыс, LOD: 200 fmol; [27]
КЗЭ-ЛИФ Белки, пептиды, аминокислоты В смеси белковых лекарств; [28]
КЗЭ / ЛИФ То же Конкретные соединения; [29]
МЭКХ- УФ / ЛИФ Белки В фармацевтических препаратах; [30]
FMOC КЗЭ/ мэкх-Ф/ЛИФ Аминокислоты LOD: 0,8 fmol; [23]
МЭКХ- УФ Энантиомеры аминокислот Прямое хи-ральное разделение; [26]
мэкх-ЛИФ Аминокислоты LOD: 0,8 amol; [31]
мэкх-ЛИФ Аминокислоты со вторичной аминогруппой В сыворотке и моче(после гидролиза); [32]
IDA МЭКХ-УФ Аминокислоты LOD: 2 fmol; [33]
Реагент Мэрфи КЗЭ/ МЭКХ- УФ Энантиомеры аминокислот Непрямое хи-ральное разделение; [34]
КЗЭ/ МЭКХ- УФ Изомеры пептидов Непрямое хи-ральное разделение; [34]
NBD-F КЗЭ-ЛИФ Энантиомеры аминокислот LOD: 0,014% L- в D-; [35]
мэкх-ЛИФ Дипептиды Фенилаланин, LOD: 70 amol; [36]
NDA КЗЭ-ЛИФ Аминокислоты LOD: 12 amol; [21]
нгскэ- УФ - ЛИФ Белки LOD: 9,5-59 amol; [30]
Аналитическая хроматография и капиллярный электрофорез
371
Продолжение таблицы 4.2.21
Реагент ВидКЭ/ детектирование Соединения Примечания; [литература]
NDA кзэ- ЛИФ Аминосодержащие полимеры Разделение 30 олигомеров; [37]
КЗЭ-эд Аспарагиновая, глутаминовая кислоты, аланин В коре головного мозга крыс LOD: 39-88 fmol; [38]
КЗЭ/ МЭКХ- ЛИФ Аминокислоты (энантиомеры) LOD: 1,1 /1,2 amol; [39]
КЗЭ-УФ Нейротрансмиттерные аминокислоты КЗЭ-МЭКХ; [40]
КЗЭ-УФ а-Дифтор-метилорнитин В тканях мозга крыс. LOD: 36-60 fmol; В плазме LOD: 7,4 fmol; [41]
OPA МЭКХ- УФ Пептиды Из пищевых добавок; [6]
кзэ- ЛИФ Высокополярные токсины Из природных источников LOD:0,1 amol; [19]
кзэ- ЛИФ Аминокислоты LOD: 80 amol; [23]
нгскэ- УФ/ЛИФ Белки LOD: 4,2 fmol; [30]
КЗЭ/ МЭКХ-ФЛУ Аминокислоты LOD: 3,0-100 amol; [42]
МЭКХ- УФ D- в избытке L-валина LOD: 300 fmol; [43]
МЭКХ- УФ/ФЛУ Энантиомеры аминокислот LOD: 0,3% D-в L-валине; [44]
МЭКХ- УФ/ЛИФ Энантиомеры аспарагиновой и глутаминовой кислот LOD: 4 fmol / 360 fmol (УФ / ЛИФ); [45]
ОРА/ FMOC МЭКХ-ЛИФ Аминокислоты Селективное детектирование аминокислот co вторичной аминогруппой; [32]
Продолжение таблицы 4.2.21
Реагент Вид КЭ/ детектирование Соединения Примечания; [литература]
PITC КЗЭ - УФ Аминосодержащие органические соединения Продукты реакции Малараде (Maillard); [46]
SAMBI КЗЭ-УФ Энантиомеры аминокислот Использование для дериватизации хи-рального реагента; [47]
SNEIT КЗЭ - УФ Энантиомеры аминокислот Использование для дериватизации хи-рального реагента; [45]
TRITC МЭКХ-ЛИФ Аминокислоты LOD: 1 zmol; [48]
Литература к таблице
1. Engstrom A., Andersson Р. Е., Josefsson В. // Anal. Chem. 1995. V. 67. Р. 3018-3022.
2. Wan. Н., Engstrom A., Blomberg L.G. // J. Chrom. A. 1996. V. 731. P. 283-292.
3. De Antonis K.M., Brown P.R., Cheng Y., Cohen S.A. // J. Chromatogr. A. 1994. V. 661. P. 279-285.
4. Cobb K.A., Novotny M.V. // Anal. Biochem. 1992. V. 200. P. 149-155.
5. Arriaga E.A., Zhang Y„ Dovichi N.J. // Anal. Chim. Acta. 1995. V. 299. P. 319-326.
6. Liu J., Cobb K.A., Novotny M. // J. Chromatogr. 1990. V. 519. P. 189-197.
7. Liu J., Hsieh Y., Wiesler D., Novotny M. // Anal. Chem. 1991. V. 63. P. 408-412.
8. Liu J., Shirota O., Novotny M. // Anal. Chem. 1991. V. 63. P. 413^17.
9. Liu J., Dolnik V., Hsieh Y., Novotny M. // Anal. Chem. 1992. V. 64. P. 1328-1336.
10. Bergquest J., Gilman S.D., Ewing A.G., Ekman R. // Anal. Chem. 1994. V. 66. P. 3512-3518.
11. Zhang Y., Arriaga E., Diedrich P., Hindsgaul O., Dovichi N. J. // J. Chromatogr. A. 1995. V. 716. P. 221-229.
12. Fuchigami T., Imasaka T. // Anal. Chim. Acta. 1993. V. 282. P. 209-213.
13. Schutzner W., Fanali S., Rizzi A., Kenndler E. // J. Chromatogr. 1993. V. 639. P. 375-378.
14. Schutzner W., Caponecchi G., Fanali S., Rizzi A., Kenndler E. // Electrophoresis. 1994. V. 15. P. 769-773.
15. Schutzner W., Fanali S., Rizzi A., Kenndler E. 11 Anal. Chem. 1995. V. 67. P. 3866-3870.
16. Schutzner W., Fanali S., Rizzi A., Kenndler E. // J. Chromatogr. A. 1996. V. 719. P. 411^20.
372
Новый справочник химика и технолога
17. Mank A.J., Yeung E.S. И J. Chromatogr. A. 1995. V. 708. P. 309-321.
18. Higashijima T., Fuchigami T., Imasaka T., Ishibashi N. I I Anal. Chem. 1992. V. 64. P. 711-714.
19. Wright B.W., Ross G.A., Smith R.D. J. // Microcol. Sep. 1989. V. l.P. 85-89.
20. Cheng Y.F., Dovichi N.J. // Science. 1988. V. 242. P. 562-564.
21. Nickerson B., Jorgenson J.W. П J. High Resolut. Chromatogr. Com. 1988. V. 11. P. 878-881.
22. Wu S, Dovichi N.J. H J. Chromatogr. 1989. V. 480. P. 141-155.
23. Albin M., Weinberger R., Sapp E., Moring S. // Anal. Chem. 1991. V. 63. P. 417-422.
24. Mattush J., Dittrich K. // J. Chromatogr. A. 1994. V. 680. P. 279-285.
25. Zhao J.Y., Waldron K.C., Miller J., Zhang J.Z., Harke H., Dovichi N. I I J. Chromatogr. A. 1992. V. 608. P. 239-242.
26. Wan H., Andersson P. E., Engstrom A., Blomberg L. G. // J. Chromatogr. A. 1995. V. 704. P. 179 -193.
27. Tsuda T., Kobayashi Y., Hori A., Matsumoto T., Suzuki О. П J. Microcol. Sep. 1990. V. 2. P. 21-25.
28. Guzman N.A., Moschera J., Bailey C.A., Iqbal K., Ma-lick A.W. // J. Chromatogr. A. 1992. V. 598. P. 123-131.
29. Guzman N.A., Moschera J., Iqbal K., Malick A.W. // J. Chromatogr. A. 1992. V. 608. P. 197-204.
30. Gump E.L., Monnig C.A. // J. Chromatogr. A. 1995. V. 715. P. 177.
31. Chan K.C., Janini G.M., Muschik G.M., Issaq H.J. 11 J. Chromatogr. A. 1993. V. 653. P. 93-97.
32. Chan K.C., Janini G.M., Muschik G.M., Issaq H.J. // J. Chromatogr. A. 1993. V. 622. P. 269-273.
33. Oguri S., Uchida C., Miyake Y., Miki Y., Kakehi K. Analyst. 1995. V. 120. P. 63-68.
34. Tran A.D., Blane T., Leopold E.J. I I J. Chromatogr. 1990. V. 516. P.241-249.
35. Ruyters H., van der Wai S. // J. Liq. Chromatogr. 1994. V. 17. P. 1883-1897.
36. Beijersten I., Westerlund D. // J. Chromatogr. A. 1995. V. 716. P. 389-399.
37. Amankwa L.N., Scholl J., Kuhr W.G. // Anal. Chem. 1990. V. 62. P. 2189-2193.
38. O’Shea T.J., Weber Р.1., Bammel B.P., Lunte C.E., Lunte S.M. // J. Chromatogr. 1992. V. 608. P. 189-195.
39. Ueda Y., Mitchell R., Kitamura F., Metcalf T., Kuwana T., Nakamoto A. I I J. Chromatogr. 1992. V. 593 P. 265-274.
40. Weber P.L., O’Shea T. J, Lunte S.M. // J. Pharm. Biomed. Anal. 1994. V. 12. P. 319-324.
41. Hu T., Zuo H., Riley C.M., Stobaugh J.F., Lunte S.M. // J. Chromatogr. A. 1988. V. 716. P. 381-388.
42. Liu J., Cobb K.A., Novotny M. // J. Chromatogr. A. 1988. V. 468 P. 56-65.
43. Houben R.J.H., Gielen H., van der Wai Sj. 11 J. Chromatogr. 1993. V. 634. P. 317-322.
44. Tivesten A., Folestad S. // J. Chromatogr. A. 1995. V. 708. P. 323-337.
45. Tivesten A. / Thesis Goteborg University, 1997.
46. Deyl Z., Miksik I., Struzinsky R. // J. Chromatogr. 1990. V. 516. P. 287-298.
47. Bonfichi R., Dallanoce C., Lociuro S., Spada A. // J. Chromatogr. A. 1995. V. 707. P. 355-365.
48. Zhao J., Chen D., Dovichi N. // J. Chromatogr. 1992. V. 608. P.117-120.
В табл. 4.2.22 представлены различные варианты определения катионов металлов методами КЭ.
Определение ионов металлов с помощью
Пояснения и принятые обозначения.
1 — для простоты, заряды опущены там, где они ясны (наиболее общие степени окисления в водных растворах). Анали-ты или другие важные катионы неметаллов (например, NH^ ) показаны в пояснениях.
3 — вспомогательный лиганд, добавленный к образцу или фоновому электролиту для образования комплексов с ионами металлов, чтобы облегчить их разделение и/или определение:
Арсеназо III — (2,2 -(1,8-дигидрокси-3,6-дисульфонафти-лен-2,7 бис-азо)-бисбензоларсониевая кислота;
ПАР — 4-(2-пиридилазо)резорцин;
ТСРР — Р,Р,Р,Р-тетракис(4-карбоксифенил)порфирин.
ЭГТА — этиленгликоль-бис-(а-аминоэтиловый эфир)-/У'.А'-тетрауксусная кислота;
Br-PAPS — 2-(5-бромо-2-пиридилазо)-5-(А-11ропил-Л'-суль-фопропиламино)фенол;
С — краун-эфир;
CDTA — транс-1,2-диаминоциклогексан-А, N,N', N -тетра-уксусная кислота;
о-СРС — о-крезолфталеиновый комплексон;
Таблица 4.2.22
капиллярного электрофореза [201]
Сгу22 — полиэфирный криптанд-22;
DFO — десферриоксимин;
DHABDS — 2,2'-дигидроксиазобензол-5,5’-дисульфонат;
HBED — N.N'-ди-(2-гидроксибензил)этилендиамин-А,А-диуксусная кислота;
HEDTC — бис(2-гидроксиэтил )дитиокарбамат;
HQSA — 8-гидроксикуинол ин-5 -сульфокислота;
LG — люмогаллион [4-хлоро-3-(2,4-дигидроксифенилазо)-2-гидроксибензен-1 -сульфокислота];
Нитро-PAPS — 2-(5-нитро-2-пиридилазо)-5-(.\'-11ропил-Л'-сульфопропиламино)-фенол;
Нитрозо-NSA — 2-нитрозо-1-нафтол-4-сульфокислота;
PAD АР—2-(5-бромо-2-пиридилазо)-5-(диэтиламино)фенод;
TRIS — 3-(гвдроксимегил)-аминомстав.
4 — только если используется вспомогательный лиганд;
Пред-----предкапиллярное образование комплексов (тем-
пература и время для реакции даны, если применимы);
В----образование комплексов в капилляре.
5 — величины, показанные в пояснениях, — это область исследованных концентраций, например 6,5 (0-10) мМ, зна-
Аналитическая хроматография и капиллярный электрофорез
373
чит, что оптимальная концентрация в данном диапазоне была 6,5 мМ, а исследованный диапазон — 0-10 мМ;
MBA — 4-метилбензиламин;
СТА — цетилтриметиламмоний;
СТАВг — цетилтриметиламмония бромид;
ТВА+ — тетрабугиламмоний;
ТВАВг — тетрабутиламмония бромид;
TEA — триэтаноламин;
ТТА — тетрадецилтриметиламмоний;
ТТАВг — тетрадецилтриметиламмония бромид;
УФКат1 — собственный несущий электролитический коион вод;
DHBP—1,1' -ди-«-гептил-4,4’-бипиридин;
НРМС — гидроксипропилметилцеллюлоза;
MES — морфолинэтансульфокислота;
PDMAC1 — поли(диаллилдиметиламмония хлорид);
SDS — додецилсульфат натрия;
6 — АЭС — атомно-эмиссионный спектрометр;
ГСИ — гидростатистическая инжекция;
ДСД — диодно совокупный детектор;
ИСП —индуктивно-связанная плазма;
МС — масс-спектрометр;
ПАД — пульсирующее амперометрическое детектирование;
ПИР — прямой инжекционный распылитель;
ПИХЭ — (протон-связанная Х-лучевая эмиссия).
сид — световой излучающий диод;
ЭМИ — электромиграционная инжекция;
7 — концентрационный предел определения (моль-л1) или абсолютный (массовый) предел определения (моль);
8 — другие капилляры, в отличие от простых из плавленного кварца или с меньшим внутренним диаметром, чем 50 мкм обозначены:
БС — боросиликатное стекло;
ГСИ — гидростатистическая инжекция;
ИОЭКХ — ионобменная электрокинетическая хроматография;
ЛПА-ПК — линейный плавленный кварц, облицованный полиакриламидом;
КД-методика — методика кинетического дифференцирования;
КРФ — регулирующая функция Кольрауша;
КЭ — краун-эфир;
НК — нейтральные комплексы;
МЭКХ — мицеллярная электрокинетическая хроматография;
ПК — плавленный кварц;
ПрКол — количественный предел;
ПФЭП — полифторэтиленпропен;
СС — катионные комплексы;
ЭМИ — электромиграционная инжекция.
Ионы металлов Матрица Вспомогательный лиганд Ком-плексо-образо-вание Фоновый электролит Детектирование Пределы обнаружения Примечание
1 2 3 4 5 6 7 8
Cu(II), Fe(III) — — — 0,05 М уксусная к-та Прямое 254 нм — БС, ПК, ПФЭП капилляр
Li, Na, К, Rb Сыворотка — — 20 мМ MES/гистидин, pH = 6,0-6,1 Кондуктометрическое (в капилляре) 0,1 MKM(Li)
Li Сыворотка — — 10-20 мМ MES, гистидин, pH = 6,1 Кондуктометрическое (в капилляре) 0,1 мкМ
Со(П), Сг(П1), Ni(II), Fe(III), Cu(II), Cd(II), Zn(II) ПАР Пред- 50мМ№Н2РО4-12,5 мМ Na2B4O7, 0-0,02 М SDS (pH = 8), 0,1 мМПАР Прямое 500 нм 0,23 мкМ 1,4 fmol (Cr(III)) МЭКХ
Fe(C<, Fe(CNt, Zn(OHf Растворы гальванопокрытий 20 мМ фосфат Na, pH = 7 или 10 Прямое 214 нм
Ca, Mg, Zn Сыворотка HQSA В- 0,01 MNa2HPO4 или 6 мМ Na2B4O7 2,5 мМ HQSA, pH = 8 Прямое флу-ориметриче-ское HeCd. Лазерное возбужде-ние/эмиссия 325/505 нм 2-15 мкМ 14-105 fmol
374
Новый справочник химика и технолога
Продолжение таблицы 4.2.22
Ионы металлов Матрица Вспомогательный лиганд Ком-плексо-образо-вание Фоновый электролит Детектирование Пределы обнаружения Примечание
1 2 3 4 5 6 7 8
Щелочные, щелочноземельные — — — 20 мМ ацетат Mg, pH = 7,5 Ионселек-тивный микроэлектрод ~ 10-100 нМ
Pt(II), Сг(Ш), Со(Ш), Rh(III), Pd(II) — Дикетоны Пред- 0-100 мМ SDS, 20 мМ Na2B4O7 pH = 9,2 Прямое 230 нм — НК, МЭКХ
Co(II), Zn(II), Mn(II), Cu(II) ТСРР Пред-, 20 мин 0,02 М SDS, 0,05 М имидазол, 0,05 М NaH2PO4 и 0,0125 М Na2B4O7, pH = 7,0 Прямое 422 нм 80 нМ 0,5 fmol (Zn) НК, МЭКХ
Li, Na, K, Mg, Ca, Sr, Ba, Mn, Fe(II), Со(П), Ni, Cu, Zn, Cd УФКат1 Косвенное 214 нм ~ 0,1 ppm
Li, Na, K, Mg, Ca, Ni, Cd — — — 5 мМ MES, гистидин, pH = 6,0 Кондуктометрическое (в конце капилляра) ~ lO-lO^6 мкМ
K, Na, Li, Ba, Ca, Mg Минеральная вода, уксус — — 3-5 мМ имидазол, pH = 4,5 Косвенное 214 нм 0,05-0,1 ppm
Li, Na, K, Mg, Ca 0,5 мМ сульфат церия (III), 0,5 мМ 2,5-дигидрокси-бензойная к-та, pH = 3,4 Косвенное флуоримет-рическое возбужде-ние/эмиссия 251/346 нм Анионы + катионы совместно
Mg, Ca, Sr, Ba, Zn, Cd, Hg, Cu, Ni, Co(II), Mn, Pb, Pd, Fe(IH) Речная вода, сыворотка ЭДТА в- 2 мМ Na2B4O7, 2 мМ ЭДТА Прямое 200 нм ~ 10 мкМ
Cs, K, NH4, Ca, Mg, Na, Sr, Ba, Li Дождевая вода 18-кра-ун-6, sot в- 500 мкМ СеС13, 2 мМ 18-краун-6 350 мкМ СеС13, 150 мкМ Ce2(SO4)3, 2 мМ 18-краун-6 500 мкМ Ce2(SO4)3, 2 мМ 18-краун-6 Косвенное флуоримет-рическое возбуждение / эмиссия 251/346 нм 1-3 мкМ или 0,1-0,3 мкМ (ЭМИ)
Li, Na, K, NH:, Rb, Mg, Ca, Ba, Be Дождевые капли 18-кра-ун-6, sot в- 350 мкМ Ce2(SO4)3, 250 мкМ Ce2(SO4)3, 1 мМ 18-краун-6 Косвенное флуоримет-рическое возбуждение / эмиссия 251/346 нм 0,2-0,8 pg Также используют мк-ВЭЖХ
Аналитическая хроматография и капиллярный электрофорез
375
Продолжение таблицы 4.2.22
Ионы металлов Матрица Вспомогательный лиганд Ком-плексо-образо-вание Фоновый электролит Детектирование Пределы обнаружения Примечание
1 2 3 4 5 6 7 8
Fe(III), Си, Fe(II), Ni, Cr, Hg, Pd, Ag, Cd, Zn, Co(II), Co(III) CN" Пред- 20 мМ Na2HPO4, 20 мМ КН2РО4, 1-2 мМ NaCN Прямое 214 нм 24 ppb (Со) 335 ppb (Hg)
Ag, Au Сплавы CN Пред- 10 мМ карбонат, pH = 9,6 Прямое 214 нм ~ 3 ppm
Al, A1F2+, aif2+, оксалат Al — — — 5 мМ имидазол, pH = 3,5 H2SO4) Косвенное 214 нм 10 нМ
Co(III), V(V), Fe(II), Cu(II), Ni, Cd, Mn, Zn — ПАР Пред-, 60 °C, 15 мин Na2HPO4, Na2B4O7, pH = 7-9,5 Прямое 500 нм 0,18 мкМ (Со) 0,141 мкМ (Ni) КД-методика
Ca, Mg, Co(II) Пшеничная мука ЭДТА, ЭГТА Пред- 20 мМ бура pH = 9,2, 5% этиленгликоль и 2 мМ ЭДТА и ЭГТА Прямое 500 нм —
Rb, K, Na, Li, Ca Сыворотка — — 9 мМ ацетат магния, 1 мМ MgCl2, 17 мМ уксусная к-та, pH = 7,5 Ионе елек-тивный микроэлектрод ~ 0,1-1 мкМ 10 мкм капилляр
K, Na 0,5 мМ малахитовый зеленый, pH = 3 Косвенное 632,8 нм (Не-Аг-лазер, двойное связывание) 1 мкМ (К)
Na, Ca, Mg, nh; Питьевая вода 18-кра-ун-6 в- 4 мМ CuSO4, 4 мМ муравьиная к-та, 4 мМ 18-краун-6, рН = 3 Косвенное 215 нм 0,6 ppm (Li) 1 PPm (К)
Ca, Mg, Со(П), Co(IH), Mn,Al(IH), Fe(IH),Pb, Hg, Cd,Ni, Cu, Zn,Pd Воды, сыворотка HBED Пред- 20 мМ бура, 2 мМ HBED, pH = 9,3 Прямое 200 нм, 236 нм, 294 нм
Na, К — — — 5-10 мМ гистидин, муравьиная к-та, pH = 3,25-5,9 Косвенное 214 нм — Системные пики
Ca Сыворотка, моча СРС В-, Пред- 0,5 М 2-амино-2-метил пропан-1,3-диол, 0,189 мМ СРС, 8,61 мМ гидроксихинолин, pH = 9 Прямое 575 нм 5 мкМ 10 fmol
Al, Li, Na, K, Cs, Mg, Ca — DFO Пред- 4 мМ 4-метил-аминофенол Косвенное УФ — ГСИ, ЭМИ
376
Новый справочник химика и технолога
Продолжение таблицы 4.2.22
Ионы металлов Матрица Вспомогательный лиганд Комплекс о-образо-вание Фоновый электролит Детектирование Пределы обнаружения Примечание
1 2 3 4 5 6 7 8
Cd, Zn, Pb, V(IV), Cu, Co(II), Ni, Fe(II) Соли никеля Нитро-PAPS Пред- 0,12 мМ Нитро-PAPS, фосфатный или ацетатный буфер Прямое 560 нм 0,1-0,5 мкМ
K, Mg (одно-и двухвалентный) — 18-кра-ун-6 В- 4-метил-амино-фенолсульфат, 18-краун-6, 20% ацетон Косвенное 220 нм —
Ca, Ba Ba/Ca-карбона-ты Цитрат В- 10 мМ имидазол, 10 мМ лимонная к-та Косвенное 210 нм —
Li, Na, K, Mg, Ca, Ba, NH4 10 мМ имидазол, 1мМТВАВг, pH = 4,5, или 10 мМ бензиламин, pH = 9,0 Косвенное 214 или 204 нм ~ 50 ppb
Li, Na, K, Mg, Ca, Sr, Ba, Cu, Zn, Cd, Pb, Mn, Fe(II), Ni, Al, VO2+, nh: , uot+, Cr(III), Ag 70-1000 ppm Na+ Лактат + 18-кра-ун-6 или 18-кра-ун-6 В- 11 мМ молочная к-та, 2,6 мМ 18-кра-ун-6, 7,5 мМ 4-меотл-бензиламин, 8% метиловый спирт, pH = 4,3, или 8 мМ никотинамид, муравьиная к-та, 0-1 мМ 18-краун-6, pH = 3,2 Косвенное 214 или 254 нм « 0,1 ppm ЛПА-ПК капилляр
Cu, Zn, Ni, Fe(II), Со(Ш), Ce(III), La, U(VI),Pb, Со(П), Fe(III), Nb,Ta Zr-вые керамики ПАР или Арсеназо III Пред- 10 мМ борат, pH = 8, 75 мМ SDS, 0,1 мМПАР; или фосфат, pH = 8, 0,1 мМ Арсеназо III, 0-50 мМ SDS Прямое 254 или 500 нм ~ 0,1 мкМ (Со-ПАР 500 нм)
Li, Na, K, Rb, Cs, Mg, Ca, Sr, Ba, Zn, Mn, Co(II), Ni Сыворотка Нет, so2-, или SDS В- 5 мМ имидазол, pH = 4,5 (НС1), 0-60% этиленгликоль; или 5 мМ CuSO4-CuCl2, pH = 4,5, 0-20% этиленгликоль, 0-IOmMSDS Косвенное 214 нм
Na, K, Mg, Ca Человеческие слезы so2- В- 4 мМ CuSO4,4 мМ муравьиная к-та, pH = 3,0 Косвенное 215 нм — ЛПА-ПК корпус
Ca, Na, K, Mg Сыворотка so2- В- 100 мМ имидазол, pH = 7,4 (H2SO4), 0,1% НРМС Косвенное 214 нм 70 мкМ ЭМИ (15 кВ, 5 с)
Na, K, Mg, Ca, Mn Соки so2- В- 5 мМ имидазол, H2SO4, pH = 3-4,5 Косвенное 214 нм 50-100 ppb
Аналитическая хроматография и капиллярный электрофорез
377
Продолжение таблицы 4.2.22
Ионы металлов Матрица Вспомогательный лиганд Ком-плексо-образо-вание Фоновый электролит Детектирование Пределы обнаружения Примечание
1 2 3 4 5 6 7 8
Cu, Cd, Zn, Pb, Co(II), Ni, Fe(II), V(IV) NiCl2 5-Вг- PAPS Пред- 0,12 мМ 5-Br-PAPS, 24 мМ ацетат, pH 4,9 Прямое 550 нм 50 нМ-1 мкМ
Na, К — — — 1-10 мМ гистидин + 1-10 мМ К+, уксусная к-та, pH = 5 Косвенное 214 нм — Системные пики
Fe(III)-ругин, квар-цетин Вытяжка растений — — 50 мМ борат/НС1, pH = 8,5 Прямое 200-360 нм —
Na, К, Mg, Ca Сточные воды Тропо-лон В- 1,2мМУФКат 2/3 мМ трополон Косвенное 185 нм —
Na, K, Mg, Ca Офтальмолог, оптические стекла so*- В- 20 мМ имидазол, до pH = 7,2 (H2SO4) и затем до pH = 6 (НС1), 0,1% НРМС Косвенное 214 нм ЭМИ (15 кВ, 10 сек)
Ca, Li, Na, K, Mg, Ba, NH*, креа- тинин Моча Тартрат + 18-краун-6 В- 5 мМ пиридин, 3,6 мМ винная к-та, 2 мМ 18-краун-6, pH = 4,05 Косвенное 255 нм ~25 мкМ
[Pd(CN)4]2~, [Pt(CN)4]2-, [PdCy2-, [Ptcy2- Анодные покрытия — — 30 мМ NaH2PO4/ Na2HPO4, pH = 7 15 масс. % ацетонитрил Прямое 214 нм 0,18-0,63 мкМ 7-25 amol
Li, Na, K, Mg, Ca, Ba, nh: Минеральная вода, водопроводная вода 10 мМ имидазол, 1мМ ТВАВг, pH = 4,5, или 10 мМ бензиламин, pH = 9,0 Косвенное 214 или 204 нм ~ 50 ppb
Стабильные комплексы Gd(III), Pt(II), Fe(III) Сыворотка — — 20-25 мМ фосфат, pH = 6,7-6,8, 10-30% CH3CN; 0-90 мМ SDS Прямое 200 нм или 214 нм —
Li, Na, K, Cs, Mg, Ca Питьевая вода — — 0,1 мМ малахитовый зеленый, 1 мМ Tris, уксусная к-та, pH = 3,9-6,5 Косвенное 630 нм ~ 5 пМ 0,8 fmol
K, Li, Ca, Ba, Cu, Zn, Mn, Fe(III) ЭДТА в- 2,5 мМ Na2B4O7, pH = 9, 2, 10 мкМ флуоресцеин натрия, или 5 мМ ЭДТА, pH = 7 Косвенное флуоримет-рическое возбужде-ние/эмиссия 488/520 нм 15-390 ppb или 0,7^44 ppb ЭМИ (10 кВ, 30 с); определение базируется на КРФ
Sc, Y, лантаноиды — CDTA Пред- 20 мМ Na2B4O7, рН= 11, 1 мМ CDTA Прямое 214 нм 2-10 мкМ
378
Новый справочник химика и технолога
Продолжение таблицы 4.2.22
Ионы металлов Матрица Вспомогательный лиганд Ком-плексо-образо-вание Фоновый электролит Детектирование Пределы обнаружения Примечание
1 2 3 4 5 6 7 8
Sr, Си, Fe(III), Fe(II), Сг, As, Sn(II), Sn(IV) 0,04 М ацетат натрия, pH = 8,2, (Fe-, Сг-породы) или 0,06 М СаС12, pH = 6,7, Sn(IV), Sn(II), Sr, As(III), As(V), Cr(III), Cr(VI) ИСП-ЭАС,-МС (распылитель Мейнхардов-ского типа) 0,1-100 ppb (ЭАС), 0,06-2 ppb (МС)
Li, К, Li, Ca, Ba, Cu, Zn, Mn, Fe(III) ЭДТА В- 10 мкМ флуоресцеин натрия в 2,5 мМ Na2B4O7, pH = 9,2, или 5 мМ ЭДТА, pH = 7 Косвенное флуоримет-рическое возбужде-ние/эмиссия 488/520 нм 15-390 ppb или 0,7-44 ppb ЭМИ (10 кВ, 30 с); определение базируется на КРФ
К Лекарства K+-co-лей — — 6 мМ имидазол в 4 мМ муравьиной к-те Косвенное 214 нм 0,5 ppm
Na Цефало-тин Na-солей — — 6 мМ имидазол в 4 мМ муравьиной к-те Косвенное 214 нм —
Cu, Pb, Cr, Fe(III) — ЭДТА Пред- 50 мМ ацетат натрия, pH = 5,5, 0-0,5 мМ ТТАВг Прямое 225 нм 6,4-27 мкМ
PbCl2, СдИцРЬХСОьНЬ — — — Нет данных исп-мс (стеклянный сплавленный распылитель) —
Li, Na, K, Mg, Ca, nh: — 18-кра-ун-6 в- 10 (1-15) мМ имидазол, 2,5 (0-300) мМ 18-краун-6, pH = 4,5 (3,5-6) Косвенное 214 нм —
Mg, Li, Na, K, Ca, Sr, Ba Речная вода, моча ЭДТА в- 10 мМ пиридин, 0,8 (0,6-1) мМ ЭДТА, pH = 5 (4,5-5,1) (НС1О4) Косвенное 254 нм —
Na, K, Mg, Ca Воздушные поверхностные флюиды 10 мМ имидазол, pH = 3,5 (НС1), 8% изопропанол Косвенное 214 нм 17-82 мкМ
Li, Na, K, Cs, Mg, Ca, Sr, Ba, Mn, Fe(II), Co(II), Ni, Cu, Zn, Cd, Pb, NH4 Лактат в- 10 (1-15) мМ имидазол, 5 (1-12) мМ молочная к-та, 0,5 (0-1,5) мМ 18-краун-6, pH = 4,5 Косвенное 214 нм
Fe(III), Ni, Cu, Pb 20 мМ NaCl 1)Тартрат 2) ЭДТА в- 10 мМ ацетат натрия, 2 мМ винная к-та, 0,2 мМ ТТАВг, pH = 4,8 Прямое 242 нм 0,5-3 мкМ Двухшаговое комплек-сообразов ание в капилляре
Аналитическая хроматография и капиллярный электрофорез
379
Продолжение таблицы 4.2.22
Ионы металлов Матрица Вспомогательный лиганд Ком-плексо-образо-вание Фоновый электролит Детектирование Пределы обнаружения Примечание
1 2 3 4 5 6 7 8
U(VI) Вода Арсеназо III Пред- 1 мМ фосфат натрия, 0,6 мМ борат натрия, ЮмМНСЮд, 50-75 мМ NaCl, 0,01 мМ Арсеназо III Прямое 650 нм 10 ppb Стэкинг
[АиС14Г, ([PdCI4]2, [PtCU]2) — — — юоммна, 400 мМ NaCl Прямое 220 нм —
Co(II), Ni, Cu, Zn, Cd — PAD АР Пред- 50 мМ ацетат натрия, 20% CH3CN, 0,1 mMPADAP, pH = 6,0-7,7 (Н3РО4) Прямое 550-585 нм 30-550 ppb
Li, Na, K, Mg, Ca, Ba, Mn, Cr, Zn, Cd — — — 15 (3-20) мМ 2-амидопирин, pH = 5 (4-7) (уксусная к-та, ~ 25 мМ) Косвенное 214 нм 0,04-0,6 ppm
[PdCL,]2", [PtClJ2- Катализатор 100-200 мМ ацетат, 400-500 мМ NaCl, pH = 2,8; или 100 мМ ацетат, pH = 5,5, 0,1 мМТТАВг Прямое 270 нм 0,9-1,4 ppm
Fe (Fe(II)+ + Fe(III)) Воды Фенол Пред- 50 мМ ацетат аммония/ уксусная к-та, pH = 5,0 Прямое 270 нм 5 нМ
Cr(III), Mn, Fe(III), Cu, Zn, Cr(VI) Растворы гальванопокрытий CDTA Пред-(70 °C 5 мин) 10 мМ формиат, pH = 3 (совместно Сг(Ш) с СгО^ ); или 10 мМ борат, 1 мМ CDTA, pH = 9,0 Прямое 214 нм 50 и 10 ppb Cr(III) и Cr(VI) Совместно с СгО<’
Li, Na, K, Cs, Mg, Ca, Ba, (NH^, органические основания) — 18-кра-ун-6 В- 30 мМ гистидин, 30 мМ MES, 1 мМ 18-краун-6, pH = 6 Кондуктометрическое (в конце капилляра)
Cr(III), Cr(VI) Растворы гальванопокрытий CDTA Пред-(70 °C 5 мин) 10 мМ формиат, рН = 3 Прямое 214 нм 50 и 10 ppb для Cr(III) и Cr(VI) Совместно с СгО^
[PtCI4]2 , [PtCls]2-, Pt- и Se- органо-комплексы — — — 50 мМ н2ро;/нро$_, pH = 6,0 Прямое 195-320 нм (развертка изображения)
380
Новый справочник химика и технолога
Продолжение таблицы 4.2.22
Ионы металлов Матрица Вспомогательный лиганд Ком-плексо-образо-вание Фоновый электролит Детектирование Пределы обнаружения Примечание
1 2 3 4 5 6 7 8
Li, Na, К, Mg, Са, Ва sot В- 25-75 мМ имидазол, H2SO4, pH = 4,6 Косвенное 214 нм 7 мкМ (Li) Косвенное де-' тестирование, 25-160 мкм внутренний диаметр
Li, Na, К, Mg, Са, Ва, Zn, Pb, La, Sm, Eu, Dy Ферментационная смесь, почвенная вытяжка ПЭГи (мол. масса: от 200 до 20-106) В- 0,01 мМ креатинин, pH = 4,3 (4-4,6) (уксусная к-та), 0-10% ПЭГ Косвенное 214 нм
Fe(II), Fe(III) Фенол + ЭДТА Пред- 100 мМ борат, pH = 9,2, 50% этанол, по 0,2 мМ Фенол и ЭДТА Прямое 245-266 нм (ДДМ) Совместное определение Fe(II) и Fe(III)
Li, Na, K, Rb, Cs, Mg, Ca, Sr, Ba, NH* ПЭГ-200 + тартрат В- 30 мМ креатинин, 15мМНС1, в 0-60%; или 30 мМ креатинин, 0-7,5 мМ винная к-та, pH = 4,5 (15-0мМНС1), в 50% ПЭГ-200 Косвенное 254 нм
Na, K, NH* Сгу22 В- 5 мМ 4-амино-пиридин, pH = 9,0 (уксусная к-та), 0,1 мМ Сгу22, 15% метанол Косвенное 254 нм —
Li, Na, K, Mg, Ca, Sr, Ba, NH* Минеральная и водопроводная вода sot + 18-кра-ун-6 В- 4 мМ CuSO4, 4 мМ 18-краун-6, 4 мМ муравьиная к-та, pH = 3,1 Косвенное 215 нм 0,6-13 ppm Рассчитаны константы стабильности
Na, K, Mg, Ca, Zn, NH* Дождевые капли 18-кра-ун-6 В- 4 мМ 4-А-метил-аминофенол, 4 мМ 18-краун-6, pH = 6,5 Косвенное 220 нм fmol уровень Прямой отбор проб в нанолит- ровых объемах
Li, Na, K, Rb, Cs, Mg, Ca, Sr, Ba, NH4 Рассол, почвенные вытяжки 20 мМ имидазол, 30 мМ уксусная к-та, метанол (100%) или 60% CH3CN Косвенное 214 нм 7-20 мкМ Неводный капиллярный электрофорез
Аналитическая хроматография и капиллярный электрофорез
381
Продолжение таблицы 4.2.22
Ионы металлов Матрица Вспомогательный лиганд Ком-плексо-образо-вание Фоновый электролит Детектирование Пределы обнаружения Примечание
1 2 3 4 5 6 7 8
[PtCl6]2’, Pt-и Se-органо-комплексы 50 мМ н2ро4/нро;, pH = 6,0 исп-мс (распылитель: Мейн-хардского типа) ~ 5 ppm (Pt)
Na, К, Mg, Са, Zn, Мп, Pb, nh; Дождевые капли 18-кра-ун-6 В- 4 мМ 4-№-метил-аминофенол, 4 мМ 18-краун-6, pH - 6,5 Косвенное 220 нм 40-80 fmol Прямой отбор проб в нанолит-ровых объемах
32 иона металлов (включая Tl(I),Ag,Hg, Pd, Fe(II), Fe(III), Co(II), Co(III), Al, Sb(III), Bi(III), Cr, Sn(IV), Zr(IV), V(IV),V(V), U(VI), W(VI)) CDTA Пред- 20 мМ борат натрия, pH = 9,0, 1 мМ CDTA, 5% этиленгликоль Прямое 214 нм 0,1-4 мкМ 23 металла разделены за 10 мин
Ca, Fe(III), Co(II), Ni, Cu, Pb — ЭДТА Пред- 5 мМ Na2SO4, 1 мМ ацетат натрия, pH = 7,0-8,0, 53,5 мМ PDMAC1 Прямое 240 нм 0,6-20 мкМ КД-методика, ИОЭКХ
Li, Na, K, Mg, Ca, Ba, nh; Чистый (NH4hWO4 18-кра-ун-6 (или 15-кра-ун-5 или 12-кра-УН-4) В- 1,0 мМ УФКат2, 3,0 мМ трополон, 3,5 (0-20) мМ 18-краун-6 (или 15-краун-5 или 12-краун-4) Косвенное 185 нм
Co(III), (Co(II), Ni, Cu, Zn, Cd) Витамин В12 PAD АР Пред- 50 мМ ацетат натрия, 20% CH3CN, 0,1 mMPADAP, pH = 6,0-7,7 (Н3РО4) Прямое 550-585 нм (ДДМ) 0,5 мкМ
Fe(IH) Вода ЭДТА Пред- 25 мМ MES + 10 мМ бис-трис-пропан, pH = 6,6; (первичный: 10 мМ НС1 + 20 мМ Т-гистидин + 0,1%НРМС, pH = 6,0; конечный: 5 мМ MES) Прямое 254 нм 10 ppb Прямой капиллярный изо-тахофо-рез, капиллярный зонный электрофорез
382
Новый справочник химика и технолога
Продолжение таблицы 4.2.22
Ионы металлов Матрица Вспомогательный лиганд Ком-плексо-образо-вание Фоновый электролит Детектирование Пределы обнаружения Примечание
1 2 3 4 5 6 7 8
Co PADAP Пред- 50 мМ ацетат натрия, 0,1 мкМ PADAP, pH = 8, 2 мМ СТАВг; для стэкинга в 10 раз развести водой Прямое видимое (быстро сканирующий детектор) 50 нМ Обратное • наряжение и стэкинг
Cr, Co(II), Ni, Pt, Cu, Cd, Hg,Pb, Bi HEDTC Пред- 100 мМ борат натрия, pH = 9,2 (8,2-10,2), IOmMSDS, 0,1 мМ HEDTC, 0 или 4% метанол Прямое 254 нм 0,1-1 мкМ (22-133 ppb) МЭКХ, НК
Sr, Ba, (Mg, Ca) Минеральная вода Сульфоназо III В- 20 мМ MES, 10 мМ Tris, pH -6,2, 0,3 мМ Сульфоназо III Прямое 654 нм (СИД) 0,35 мкМ (50 ppb, Ва) Комплексообразование в капилляре и стэкинг
Mg, Ca, Sr, Ba — Арсеназо I В- 17 мМ муравьиная к-та, pH = 9,5, (диэтаноламин), 1,0 мМ Арсеназо I Прямое 568 нм (СИД) ~ 1-11 мкМ Исследование чистоты лиганда
La, U(VI) Арсеназо III В- 15 мМ лимонная к-та, 20 мМ Tris, pH = 4,5, 0,1 мМ Арсеназо III Прямое 654 нм (СИД) Взаимодействие раствор-стенка капил.
14 лантаноидов, U(VI) Арсеназо III В- 15 мМ лимонная к-та, 20 мМ Tris, pH = 4,3, 0,025 мМ Арсеназо Ш Прямое 654 нм (СИД) ~ 0,6 мкМ (La) или 0,06 мкМ (La, стэкинг) Комплексообразование в капилляре и стэкинг
Литература
1. Altria K.D. Overview of Capillary Electrophoresis and Capillary Electrochromatography (Review) // J. Chromatogr. 1999. V. 856. P. 443-463.
2. Vindevogel J., Sandra P. Introduction to MEKC in: Chromatographic Methods. Heidelberg: Hutig Verlag, 1992.
3. Capillary Electrophoresis Technology. In Chromatographic Science Series. I Ed. Guzman N.A. New York; Basel; Hong Kong: M. Dekker Inc., 1993. V. 64. 857 p.
4. S.F.Y. Li. Capillary Electrophoresis // J. Chrom. Library. Amsterdam: Elsevier. 1992. V. 52. 608 p.
5. P. Jandik, G. Bonn. Capillary Electrophoresis of Small Molecules and Ions. Weinheim: VCN-Verlag, 1993. 298 p.
6. Kuhn R, Hofstetter-Kuhn S. Capillary Electrophoresis, Principle and Practice. Berlin; Heidelberg: Springer; Verlag, 1993. 375 p.
7. Lehmann R., Voelter W., Liebich H.V. Capillary Electrophoresis in Clinical Chemistry// J. Chrom. B. 1997. V. 697. P. 3-35.
8. Capillary Electrophoresis of Small Molecules and Ions / P. Jandik and G. Bonn. New York: VCH, 1993. 298 p.
9. Capillary Zone Electrophoresis / Ed. F. Foret; L. Krivan-kova; P. Bocek. Weinheim: VCH, 1993. 346 p.
10. WeinbergerR. Practical Capillary Electrophoresis. Boston: Acad. Press, 1993. 312 p.
11. Capillary Electrophoresis Technology I Ed. by Norberto A. Guzman. New York: M. Dekker, 1993. 857 p.
12. Capillary Electrophoresis: Theory and Practice / Ed. P. Camilleri Boca Raton: CRC Press, 1993. 495 p.
13. Scholl J.B., De Zwaan J. J. Micellar Electrokinetic Chromatography as a Generalized Alternative to High-performance Liquid Chromatography for Purity Determi
Аналитическая хроматография и капиллярный электрофорез
383
nation of a Class of Investigational Antibacterial Drugs // Chromatogr. B. 1997. V. 695. P. 147-156.
14. Jenkins M.A., Guerrin M.D. Capillary Electrophoresis as a Clinical Tool // J. Chrom. B. 1996. V. 682. P. 23-34.
15. Capillary Electrophoresis / Ed. P. Camilleri Boca Raton: CRC. Press, 1993. 512 p.
16. Klein G. L., Jolif C. R. Capillary Electrophoresis for the Routine Clinical Laboratory. Handbook of Capillary Electrophoresis. Boca Raton: CRC Press, 1994. 420 p.
17. Engelhardt H., Beck W., Schmitt T. Capillarelektropho-rese, Metoden und Moglichkeiten. Wiesbaden: Vieveg Verlag, 1994. 329 s.
18. Handbook of Capillary Electrophoresis / Ed. P. James Landers. Boca Raton et al.: CRC Press, 1994. 649 p.
19. Directory of Capillary Electrophoresis. Amsterdam et al.: Elsevier, 1994. 150 p.
20. Capillary Zone Electrophoresis and Related Techniques / Ed. J. Haleem. New York: Dekker, 1994. P. 3831-4059.
21. Baker D. R. Capillary Electrophoresis. New York et a.: Wiley, 1995. 244 p.
22. Baker D. R. Capillary Electrophoresis (Techniques in Analytical Chemistry). New York: Wiley, 1995. 244 p.
23. Engelhardt H., Beck W., Schmitt Th. Capillary Electrophoresis Methods and Proteins. Wiesbaden: Vieweg Verlag, 1996.215 р.
24. Heiger D.N. High Performance Capillary Electrophoresis. Hewlett-packard GmbH, Waldbronn Analytical Division.
25. Engelhardt H. et al. Capillary Electrophoresis: Methods and Potentials / Heinz Engelhardt; Wolfgang Beck; Thomas Schmitt. Transl. from the German by George Gut-nikov. Braunschweig; Wiesbaden: Vieweg, 1996. 215 p.
26. Capillary Electrophoresis Guidebook: Principles, Operation, and Applications I Ed. D. Kevin Altria; Totowa N.J.: Humana Press, 1996. 349 p.
27. Pharmaceutical and Biomedical Applications of Capillary Electrophoresis / Ed. S.M. Lunte. Oxford: Pergamon, 1996.511 p.
28. Capillary Electrophoresis in Analytical Biotechnology / Ed. P.G. Righetti. Boca Raton: CRC Press, 1996. 551 p.
29. Руководство по капиллярному электрофорезу / Под ред. A.M. Волощука. М., 1996. 231 с.
30. Handbook of Capillary Electrophoresis Applications / Ed. H. Shintani. London: Blackie Acad. & Professional, 1997. 737 p.
31. Analysis of Nucleic Acids by Capillary Electrophoresis / Ed. C. Heller. Braunschweig: Vieweg, 1997. 313 p.
32. Capillary Electrophoresis in the Life Sciences / Ed. A.M. Krstulovic. Amsterdam: Elsevier, 1997. 293 p.
33. Shihabi Z.K. Handbook of Electrophoresis. 2nd ed. Boca Raton: CRC Press, 1997. P. 457-477.
34. Shintani H., Polonsky J. Handbook Capillary Electrophoresis Applications. London: Blackie, 1997. 768 p.
35. Shihabi Z.K. Handbook of Capillary Electrophoresis Applications / Eds. H. Shiutani, J. Polonski. London: Chapman and Hall, 1997. P. 386-408.
36. Oda R.P., Roche M.E., Landers J.P., Shihabi Z.K. Handbook of Capillary Electrophoresis. 2nd ed. Boca Raton: CRC Press, 1997. P. 639-674.
37. Kevin D. A. Analysis of Pharmaceuticals by Capillary Electrophoresis. Braunschweig: Vieweg, 1998. 285 p.
38. Capillary Electrophoresis: Theory and Practice / Ed. P. Camilleri. 2nd ed. Boca Raton: CRC Press, 1998. 552 p.
39. Wehr T., Rodriguez-Diaz R., Zhu M. Capillary Electrophoresis of Proteins. New York: M. Dekker, 1999. 286 p.
40. Sorensen H. Chromatography and Capillary Electrophoresis in Food Analysis. Cambridge, 1999. 470 p.
41. Fukushi K., Takeda S., Chayama K., Wakida S. Application of Capillary Electrophoresis to the Analysis of Inorganic Ions in Environmental Samples (Review) // J. Chromatogr. A. 1999. V. 834. P. 349-362.
42. Heiger D. High Performance Capillary Electrophoresis. / Agilent Technologies. 2000. № 5968-9963E. 135 p.
43. Capillary Electrophoresis and Related Separation Technologies / Ed. J.I. Haleem. New York: M. Dekker, 2000. 183 p.
44. Fritz J.S. Ion Chromatography: Now Including Ion Analysis by Capillary Electrophoresis 3d completely rev. and enl. ed. Weinheim: Wiley-VCH, 2000. 254 p.
45. Capillary Electrophoresis / Ed. C. Horvath. Amsterdam: Elsevier, 2000. 513 p.
46. Frazier R. A., Jennifer M. A. and Harry E. N. Capillary Electrophoresis for Food Analysis: Method Development. Cambridge: Royal Soc. ofChemisty, 2000. 127 p.
47. Capillary Electrophoresis and Electrochromatography Reviews: Theory and Methodology / Ed. Ziad El Rassi. Weinheim: Wiley-VCH, 2000. P. 3873 4192.
48. Capillary Electrophoresis of Nucleic Acids / Ed. Mitchel-son R. K. Totowa; NJ: Humana Press (Methods in Molecular Biology) Band I. Introduction to the Capillary Electrophoresis of Nucleic Acids. 2001. 484 p. Band 2. Practical Applications of Capillary Electrophoresis. 2001. 408 p.
49. Hempel G. Strategies to Improve the Sensitivity in Capillary Electrophoresis for the Analysis of Drugs in Biological Fluids (review) // Electrophoresis. 2000. V. 21. P.691-698.
50. Jorgenson J.W., Lukacs K.D. High-resolution Separations Based on Electrophoresis and Electroosmosis // J. Chromatogr. 1981. V. 218. P. 209-214.
51. Tomlinson A.J., Bensen L.M., Guzmahn N. R., Naylor S. Preconcentration and Microreaction Technology on-line with Capillary Electrophoresis (review) // J. Chromatogr. A. 1996. V. 744. P. 3.
52. Lucy C.A., Yeung К. K.-C., Fu S., Li D., Henselwood T. L., Underhill R.S. 1998 W.A.E. McBride Medal Lecture Searching for the Holy Grail in Analytical Separations (review) // Canad. Joum. Chem. 1999. V. 77, № 3. P. 281-290.
53. Katzuta S., Saitoh K. J. Control of Separation Selectivity in Micellar Electrokineticchromatography by Modification of the Micellar Phase with Solubilized Organic Compounds (review) // J. Chromatogr. A. 1997. V. 780. P. 165-178.
54. Sarmini K., Kenndler E. Influence of Organic Solvents on the Separation Selectivity in Capillary Electrophoresis (review) // J. Chromatogr. A. 1997. V. 792. P. 3-11.
384
Новый справочник химика и технолога
55. Veraart J.R., Lingeman Н., Brinkman V.A. Th. Coupling of Biological Sample Handling and Capillary Electrophoresis (review) // J. Chromatogr. 1999. V. 856. P. 483-514.
56. Polesello S., Valsecchi S. M. Electrochemical Detection in the Capillary Electrophoresis Analysis of Inorganic Compounds (review) // J. Chromatogr. A. 1999. V. 834. P. 103-116.
57. Muijselaar P.G., Otsuka K., Terabe S. Micelles as Pseudo-Stationary Phases in Micellas-Electrokinetic Chromatography (review) // J. Chrom. A. 1997. V. 780. P. 41-61.
58. Swinney K., Bomhop D.J. Detection in Capillary Electrophoresis (review) // Electrophoresis. 2000. V. 21. P. 1239-1250.
59. Беленький Б.Г. Капиллярные электросепарационные методы — новая эра в анализе многокомпонентных проб И Научное приборостроение. 1992. № 1. С. 3-36.
60. Albin М., Grossmann P.D., Moring S.E. Sensitivity Enhancement for Capillary Electrophoresis // Anal. Chem. 1993. V. 65. 489A—497A.
61. Vesterberg O. History of Electrophoretic Methods 11 J. Chrom. 1989. V. 480. P. 3-19.
62. Woolley A.T., Mathies R.A. Ultra-High-Speed DNA Sequencing Using Capillary Electrophoresis Chips H Anal. Chem. 1995. V. 67. P. 3676-3680.
63. Deng Y., Henion J. Chip-Based Capillary Electrophoresis- mass Spectrometry Determination of Carnitines in Human Urine // Anal. Chem. 2001. V. 73. P. 639-646.
64. Waters L.C., et al. Microchip Device for Cell Lysis, Multiplex PCR Amplification and Electrophoretic Sizing // Anal. Chem. 1998. V. 70. P. 158-162.
65. Gfrorer P., Schewitz J., Pusecker K., Bayer E. In-line Coupling of Capillary Separation Techniques with HNMR//Anal. Chem. 1999. V. 71. P. 315A-321A.
66. Jacobson S.C., Ramsey J.M. Electrokinetic Focusing in Microfabricated Channel Structures 11 Anal. Chem. 1997. V. 69. P. 3212-3217.
67. Schultz G.A., et al. Fully Integrated Monolithic Microchip Electrospray Device far Mass Spectrometry // Anal. Chem. 2000. V. 72. P. 4058-4063.
68. Culberston C.T., Ramsey R.S., Ramsey J.M. Electroosmotically Induced Hydraulic Pumping on Microchips Edifferential Ion Transport // Anal. Chem. 2000. V. 72. P.2285-2291.
69. Каменцев Я.С., Ягов Г.В. Капиллярный электрофорез как аналитический метод / Материалы семинара: Применение метода капиллярного электрофореза для анализа ионного состава воды. СПб., 2000. С. 3-17.
70. Schmitt-Kopplin Н., et al. Improving the Use of CE in a Chro-matographer’s World // LC GC. 2001. V. 7. P. 384-388.
71. Bocek P., Vespalec R. Selectivity in Capillary Electrophoresis //Anal. Chem. 2000. V. 1. P. 586-595.
72. Mori M., Hu Wezhi, Haddad P.R., Fritz J.S., Tanaka K., Tsue H., Tanaka S. Capillary Electrophoresis Using High Ionic Strength Background Electrolytes Containing Zwitterionic and Non-Ionic Surfactants and its Application to Direct Determination of Bromide and Nitrate in Seawater П Analyt. and Bioanalyt. Chem. 2002. V. 372. №1. P.181-186.
73. Yanes G., Gratz S. R., Baldwin J., Robinson S.E., Stalcup A.M. Capillary Electrophoretic Application of l-Alkyl-3-methylimidazolium-Based Ionic Liquids // Anal. Chem. 2001. V. 73. P. 3838-3844.
74. Harrold M.P., Wojtusik M. J., Riviello J., Henson P.J. Parameters Influencing Separation and Detection of Anions by Capillary Electrophoresis // J. Chrom. 1993. V. 640. P. 463-471.
75. Quang C., Khaledi M. G. Improved Chiral Separation of Basic Compounds in Capillary Electrophoresis Using p-Cyclodextrin and Tetraalkylammonium Reagents // Anal. Chem. 1993. V. 65. P. 3354-3358.
76. Yanes G., Gratz S. R., Stalcup A.M. Tetraethylammonium Tetrafluorborate: a Novel Electrolyte with a Unique Role in the Capillary Electrophoretic Separation of Polyphenols Found in Grape Seed Extracts // Analyst. 2000. V. 125. P.1919-1923.
77. Swedberg S.A. Use of Non-Ionic and Zwitterionic Sur-factance to Enhance Selectivity in High Performance Capillary Electrophoresis. An Apparent Micellar Electrokinetic Capillary Chromatography Mechanism // J. Chrom. 1990. V. 503. P. 449-452.
78. Terabe. S., Otsuka K., Ichikawa K., Tsuchiya A., Ando T. Electrokinetic Separations with Micellar Solutions and Open-Tubular Capillaries H Anal. Chem. 1984. V. 56. P. 111-113.
79. Terabe. S., Otsuka K., Ando T. Band Broadening in Electrokinetic Chromatography with Micellar Solutions in Open-Tubular Capillaries // Anal. Chem. 1989. V. 61. P. 251-260.
80. Terabe. S. Electrokinetic Chromatography: an Interface between Electrophoresis and Chromatography // Trends Anal. Chem. 1989. V. 8. P. 129-134.
81. Shakulashvili N., Faller T., Engelhardt H. Simultaneous Determination of Alkali, Alkaline Earth and Transition Metal Ions by Capillary Electrophoresis with Indirect UV Detection H J. Chrom. 2000. V. 895. P. 205-212.
82. Craston D.H., Saeed M. Analysis of Carboxylic and Related Acids in the Environment by Capillary Electrophoretic Techniques // J. Chrom. 1998. V. 827. P. 1-12.
83. Tivesten A., Folestad S. Separation of Precolumn-labelled D- and L-Aminoacids by MEKC with UV and Fluorescence Detection // J. Chrom. 1995. V. 708. P.323-337.
84. Sarmini K., Kenndler E. Influence of Organic Solvents on the Separation Selectivity in Capillary Electrophoresis I I J. Chrom. 1997. V. 792. P. 3-11.
85. Guttmann A., Cohen A.S., Heiger D.N., Karger B.L. Analytical and Micropreparative Ultrahigh Resolution of Oligonucleotides by Polyacrylamide Gel High-Performance Capillary Electrophoresis H Anal. Chem. 1990. V. 62. P. 137-141.
86. Guttman A., Arai A., Magyar K. Influence of pH on the Migration Properties of Oligonucleotides in Capillary Gel Electrophoresis // J. Chrom. 1992. V. 608. P. 175-179.
87. Gubitz G., Schmid M.G. Recent Progress in Chiral Separation Principles in CE // Electrophoresis. 2000. V. 21. P. 4112-4135.
Аналитическая хроматография и капиллярный электрофорез
385
88. Kuhn R., Stoecklin F., Emi F. Chiral Separations by Host Guest Complexation with Cyclodextrin and Crown Ether in Capillary Zone Electrophoresis // Chromatographia. 1992. V. 33. P. 32-36.
89. Garrison A.W., Schmitt P., Kettrup A. Separation of Phenoxy Acid Herbicides and Their Enantiomers by HPCE // J. Chromatogr. 1994. V. 688. P. 317-327.
90. Mechref Y., Rassi Z. CE of Herbicides: 1. Precolumn De-rivatization of Chiral and Achiral Phenoxy Acid Herbicides with a Fluorescent Tag for Electrophoretic Separation in the Presence of Cyclodextrins and Micellar Phases //Anal. Chem. 1996. V. 68. P. 1771-1777.
91. Geiser L., Cherkaoui S., Veuthey J.-L. Simultaneous Analysis of Some Amphetamine Derivatives in Urine by Nonaqueous Capillary Electrophoresis Coupled to Electrospray Ionization Mass Spectrometry // J.Chrom. 2000. V. 895. P. 111-121.
92. Baryla N.E., Lucy C.A. Simultaneous Separation of Cationic and Anionic Proteins Using Zwitterionic Surfactants in Capillary Electrophoresis // Anal. Chem. 2000. V. 72. P.2280-2284.
93. Desiderio C., Fanali S. Chiral Analysis by Capillary Electrophoresis Using Antibiotics by Chiral Selector // J. Chromatogr. A. 1998. V. 807. P. 37-56.
94. Manege L.C., Takayanagi T., Oshima M., Motomizu S. Ion Association Reactions in Aqueous Solution between Non-UV-Absorbing Crown-ether-alkali Metal Complexes with Picrate Ion by Capillary Zone Electrophoresis //Analyst. 2000. V. 125, № 11. P. 1928-1932.
95. Koide T., Ueno K. Mechanistic Study of Enantiomeric Recognition of Primary Amino Compounds Using an Achiral Crown Ether with Cyclodextrin by Capillary Electrophoresis and Nuclear Magnetic' Resonance // J. Chrom. 2001. V. 752. P. 241-248.
96. Esaka Y., Yamagachi Y., Kano K., Goto M. Separation of Hydrogen-Bonding Donors in Capillary Electrophoresis Using Polyethers as Matrix // Anal. Chem. 1994. V. 66. P. 2441-2445.
97. Abushoffa A.M., Clark B. J. Resolution of the Enantiomers of Oxamniquine by Capillary Electrophoresis and High-Performance Liquid Chromatography with Cyclodextrins and Heparin as Chiral Selectors // J. Chrom. A. 1995. V. 700. P. 51-58.
98. Pena M.S., Zhang Y., Warner J.M. EnantiomericSepara-tions by Use of Calixarene Electrokinetic Chromatography// Anal. Chem. 1997. V. 69. P. 3239-3242.
99. Weinberger R. Buffer Systems for Capillary Electrophoresis. Encyclopedia of Chromatography / Ed. J. Cazes. Florida: Florida Atlantic University Boca Raton, 2001. P. 94-97.
100. Carou M.I.T., Mahia P.L., Lorenzo S.M., Fernandez E.F., Rodriguez D. P. Capillary Zone Electrophoresis for the Determination of Light-Absorbing Anions in Environmental Samples // J. Chromatogr. A. 2001. V. 918. P. 411-421.
101. Zemann A. J. Sub-minute Separations of Organic and Inorganic Anions with Coelectroosmotic Capillary Electrophoresis // J. Chrom. 1997. V. 787. P. 243-251.
102. Warner M. Migration toward Capillary Electrophoresis // Anal. Chem. 1994. V. 22. P. 1137-1141.
103. Timerbaev A.R., Shpigun O.A. Recent Progress in Capillary Electrophoresis of Metal Ions // Electrophoresis. 2000. V. 21. P. 4179-4191.
104. Андреева M.B., Сметанин Г.Н. Применение метода капиллярного электрофореза при контроле состава питьевых, природных и сточных вод // Материалы семинара: Применение метода капиллярного электрофореза для анализа ионного состава воды. Практические приложения метода для анализа питьевых и сточных вод и сертификации бутылированных вод и напитков. СПб., 2000. С. 35-48.
105. Van den Hoop M.A.G.T., et al. Analysis of Fluoride in Rain Water. Comparison of CE with Ion Chromatography and Ion-Selective Electrode Potentiometry // J. Chrom. 1996. V. 739. P. 241-248.
106. Corradini D. Amino Acids, Peptides and Proteins: Analysis by CE. Encyclopedia of Chromatography / Ed. by J. Cazes. Florida: Florida Atlantic University Boca Raton, 2001. P. 34-38.
107. Krylov S.N., Dovichi N. J. Capillary Electrophoresis for the Analysis of Biopolymers // Anal. Chem. 2000. V. 72. P. 111R-128 R.
108. Penmetsa K.V., Leidy R.B., Shea D. Herbicide Analysis by MEKC // J. Chromatogr. 1996. V. 745. P. 201-208.
109. Quirino J.P., Terabe S. On-line Concentration of Neutral Analytes for Micellar Electrokinetic Chromatography. 3. Stacking with Reverse Migrating Micelles // Anal. Chem. 1998. V. 70. P. 149-157.
110. Quirino J.P., Terabe S. On-line Concentration of Neutral Analytes for Micellar Electrokinetic Chromatography. 5. Field-Enhanced Sample Injection with Reverse Migrating Micelles // Anal. Chem. 1998. V. 70. P. 1893-1901.
111. Palmer J., Munro N., Landers J. P. Universal Concept for Stacking Neutral Analytes in Micellar Capillary Electrophoresis //Anal. Chem. 1999. V. 71. P. 1679-1687.
112. Boyel M.C. Simultaneouws Determination of Antioxidants, Preservatives and Sweeteness Permitted as Additives in Food by Mixed Micellar Electrokinetic Chromatography// J. Chrom. 1999. V. 847. P. 369-375.
ИЗ. Русанов А.И. Удивительный мир наноструктур // Журн. общей химии. 2002. Т. 72, вып. 4. С. 532-549.
114. Kim J.-B., Otsuka К., Terabe S. Anion Selective Exhaustive Injection-Sweep-Micellar Electrokinetic Chromatography// J. of Chrom. 2001. V. 932 (1-2). P. 129-137.
115. Lin C.-E., Chen M.-J. Separation and Selectivity of Benzophenones in Micellar Electrokinetic Chromatography Using Sodium Dodecyl Sulfate Micellar or Sodium Cholate Modified Mixed Micelles // J. Chromatogr. A. 2001. V. 923. P. 241-248.
116. Jandera P., Fischer J., Effenberger H. Characterisation of Retention in Micellar High-Performance Liquid Chromatography and in Micellar Electrokinetic Chromatography Using Lipophilicity and Polarity Indices // J. Chromatogr. A. 1998. V. 807. P. 57-70.
117. Takeda S., Wakilda S., Yamana M., Kamahara A., Higashi K. Migration Behavior of Phtalate Esters in Micellar Electrokinetic Chromatography with or without Added Methanol//Anal. Chem. 1993. V. 65. P. 2489-2492.
386
Новый справочник химика и технолога
118. Esaka Y., Goto М., Haraguchi Н., Ikeda Т., Капо К. Hydrogen-bonding Interaction in Capillary Electrophoresis Using Polyether Matrices // J. Chrom. A. 1995. V. 711. P. 305-311.
119. Bartak P., Bednar P., Friedecky D., Haviger A., Sevcik J. Fast Analysis of Antibacterial Isothiazolones by Capillary Electrophoresis // J. Chromatogr. B. 2001. V. 758. P. 323-325.
120. Wiedmer S.K., Riekkola M.-L. Mixed Micelles of Sodium Dodecyl Sulfate and Sodium Cholate: Micellar Electrokinetic Capillary Chromatography and Nuclear Magnetic Resonance Spectroscopy 11 Anal. Chem. 1997. V. 69. P. 1577-1584.
121. Yand S., Khaledi M. G. Chemical Selectivity in Micellar Electrokinetic Chromatography: Characterization of Solute-Micelle Interactions for Classification of Surfactants // Anal. Chem. 1995. V. 67. P. 499-510.
122. Asthana A., Bose D., Durgbanshi A., Sanghi S.K., Kok W.Th. Determination of Aromatic Amines in Water Samples by Capillary Electrophoresis with Electrochemical And Fluorescence Detection // J. Chrom. 2000. V. 895. P. 197-203.
123. Gamer T. W., Yeung E. S. Indirect Fluorescence Detection of Sugars Separated by Capillary Zone Electrophoresis with Visible Laser Excitation // J. Chrom. 1990. V. 515.P. 639-644.
124. Ivanov A.R., Nazimov I.V., Lobazov A.P., Popkovich G.B. Direct Determination of Amino Acids and Carbohydrates by HPCE With Refractometric Detection // J. Chrom. A. 2000. V. 894. P. 253-257.
125. Pentoney Jr. S. L., Zare R.N., Quint J.F. Semiconductor Radioisotope Detector for Capillary Electrophoresis H J. Chrom. 1989. V. 480. P. 259-270.
126. Lu W., Shamsi S.A., McCarley T.D., Warner I.M. Online Capillary Electrophoresis-Electrospray Ionization Mass Spectrometry Using a Polymerized Anionic Surfactant// Electrophoresis. 1998. V. 19. P. 2193-2199.
127. Столяров Б.В., Савинов И.М., Витенберг А.Г и др. Практическая газовая и жидкостная хроматография. СПб.: СПГУ, 1998. 612 с.
128. Song X., Budde W.L. Determination of Chlorinated Acid Herbicides and Related Compounds in Water by Capillary Electrophoresis-Electrospray Negative Ion Mass Spectrometry // J. Chrom. 1998. V. 829. P. 327-340.
129. Huang X., Gordon M.J., Zare R.N. Bias in Quantitative Capillary Zone Electrophoresis Caused by Electrokinetic Sample Injection 11 Anal. Chem. 1988. V. 60. P. 375-377.
130. Hsieh M.-M., Chang H.-T. Dynamic Control for the Separation of Organic Acids in Capillqary Electrophoresis И J. Chrom. A. 1998. V. 793. P. 145-152.
131. Kuhr W.G., Yeung E.S. Optimization of Sensitivity and Separation in Capillary Zone Electrophoresis with Indirect Fluorescence Detection // Anal. Chem. 1988. V. 60. P. 2642-2646.
132. Lu W., Cassidy R.M. Pulsed Amperometric Detection of Carbohydrates Separated by Capillary Electrophoresis // Anal. Chem. 1993. V. 65. P. 2878-2881.
133. Tarigan H.J., Neill.P. Kenmore C.K., Bomhop D.J. Capillary-scale Refractive Index Detection by Interferometric Backscatter//Anal. Chem. 1996. V. 15. P. 1763-1770.
134. Male K.B., Luong J. Derivatization, Stabilization and Detection of Biogenic Amines by Cyclodextrin-Modified Capillary Electrophoresis-Laser-Induced Fluorescence Detection // J. Chrom. 2001. A. V. 926 (2). P. 309-317.
135. Kaneta T., Imasaka T. Indirect Detection of Aromatic Hydrocarbons by Semiconductor Laser Fluorometry in Micellar Electrokinetic Chromatography П Anal. Chem. 1995. V.67. P. 829-834.
136. Schneede J., Ueland P.M. Application of Capillary Electrophoresis with Laser-Induced Fluorescence Detection for Routine Determination of Methylmalonic Acid in Human Serum // Anal. Chem. 1995. V. 67. P. 812-819.
137. Schneede J., Ueland P.M. Application of Capillary Electrophoresis with Laser-Induced Fluorescence Detection for Routine Determination of Methylmalonic Acid in Human Serum // Anal. Chem. 1995. V. 67. P. 812-819.
138. Salimi-Moosavi H., Cassidy R.M. Capillary Electrophoresis of Inorganic Anions in Nonaqueous Media with Electrochemical and Indirect UV-Detection // Anal. Chem. 1995. V. 67, № 6. P. 1067-1073.
139. Lurie I. S., Anex D.S., Fintschenko Y., Choi W.-Y. Profiling of Impurities in Heroin by Capillary Electrochromatography and Laser-induced Fluorescene Detection // J. Chrom. 2001. V. 924 (1-2). P. 421-427.
140. Lazar M.I., Ramsey R.S., Sundberg S., Ramsey J.M. Subattomole-Sensitivity Microchip Nanoelectrospray Source with Time-of-Flight Mass Spectrometry Detection //Anal. Chem. 1999. V. 71. P. 3627-3631.
141. Wallingford R.A., Ewing A.G. Amperometric Detection of Catechols in Capillary Zone Electrophoresis with Normal and Micellar Solutions // Anal. Chem. 1988. V. 60. P. 258-263.
142. Kar S., Dasgupta P.K., Liu H., Hwang H. Computer-interfaced Bipolar Pulse Conductivity Detection for Capillary Systems H Anal. Chem. 1994. V. 66. P. 2537-2543.
143. Issaq H.J. A Decade of Capillary Electrophoresis // Electrophoresis. 2000. V. 21. P. 1921-1939.
144. Система капиллярного электрофореза «Капель», № 17727-01 в Государственном реестре средств измерений. Сертификат об утверждении типа средств измерений № 10431.
145. Система капиллярного электрофореза. Основы метода. Аппаратура. Примеры использования систем капиллярного электрофореза «Капель-103, -104, -105» /Под ред. Н. В. Комаровой. СПб: Петрополис, 2001. 65 с.
146. Weinberger R. Absorbance Detection in Capillary Electrophoresis. Encyclopedia of Chromatography / Ed. J. Cazes. Florida: Florida Atlantic University Boca Raton, 2001. P. 1-3.
147. Otsuka K., Terabe S. Retention Factor: Effect on MEKC Separation Encyclopedia of Chromatography I Ed. J. Cazes. Florida: Florida Atlantic University Boca Raton, 2001. P. 715-717.
Аналитическая хроматография и капиллярный электрофорез
387
148. Muijselaar P.G., Claessens Н.А., Cramers С.А. Application of the Retention Index Concept in Micellar Electrokinetic Capillary Chromatography 11 Anal. Chem. 1994. V. 66. P. 635-644.
149. Reijenga J.C. Applied Voltage Effect on Mobility, Selectivity and Resolution in Capillary Electrophoresis. Encyclopedia of Chromatography / Ed. J. Cazes. Florida: Florida Atlantic University Boca Raton, 2001. P. 60.
150. Krull I.S., Kazmi S. Application of Capillary Electrochromatography to Biopolymers and Pharmaceuticals. Encyclopedia of Chromatography I Ed. J. Cazes. Florida: Florida Atlantic University Boca Raton, 2001. P. 49-58.
151. Королева E.M. Повышение чувствительности метода капиллярного электрофореза / В сб. Применение метода капиллярного электрофореза для анализа ионного состава воды. СПб. 2000. С. 20-34.
152. Zerbinati О., Trotta F., Giovannoli С. Optimization of the Cyclodextrin-Assisted Capillary Electrophoresis Separation of The Enantiomers of Phenoxyacid Herbicides 11 J. Chrom. 2000. V. 875. P. 423-430.
153. Комарова H.B., Карцева Л.А. Оптимизация условий разделения гербицидов класса хлорфеноксикарбоновых кислот в природных и питьевых водах методом капиллярного зонного электрофореза // Журн. аналит. химии. 2002. Т. 57. С. 766-772.
154. Masseter М., Zemann A.J. Influence of Organic Solvents in Coelectroosmotic Capillary Electrophoresis of Phenols //Anal. Chem. 1995. V. 67, № 6. P. 1047-1053.
155. Penn S. G., Liu G., Begrton F.T., Goodall D.M., Loran J.S. Systematic Approach to Treatment of Enantiomeric Separations in Capillary Electrophoresis and Liquid Chromatography. 1. Initial Evalution Using Propranolol and Dancylated Amino Acids // J. Chrom. A. 1994. V. 680. P. 147-155.
156. Britz-McKibbin P., Chen D.D.Y. Selective Focusing of Catecholamines and Weakly Acidic Compounds by Capillary Electrophoresis Using a Dynamic pH Junction // Anal. Chem. 2000. V. 72. P. 1242-1252.
157. Riekkola M-L., Wiedmer S.K., Valko I. E., Siren H. Selectivity in Capillary Electrophoresis in The Presence of Micelles, Chiral Selectors and Non-Aqueous Media // J. Chrom. A. 1997. V. 792. P. 13-35.
158. Huang X., Coleman W.F., Zare R.N. Analysis of Factors Causing Peak Broadening in Capillary Zone Electrophoresis 11 J. Chrom. 1989. V. 480. P. 95-110.
159. Okada T. Nonaqueous Ion-Exchange Chromatography and Electrophoresis. Approaches to Nonaqueous Solution Chemistry and Design of Novel Separation // J. Chrom. A. 1998. V. 804. P. 17-28.
160. Sahata R.S., Khaledi M.G. Nonaqueous Capillary Electrophoresis // Anal. Chem. 1994. V. 66. P. 1141-1146.
161. Wang F., Khaledi G. Chiral Separation by Nonaqueous Capillary Electrophoresis 11 Anal. Chem. 1996. V. 68. P. 3460-3464.
162. Ahuja E.S., Preston B.P., Foley J.P. Anionic-Zwitterionic Mixed Micelles in Micellar Electrokinetic Chromatography: sodium dodecyl sulfate-V-dodecyl-V,V-dimethyl-
ammonium-3-propane-l-sulfonic acid // J. Chrom. B. 1994. V. 657. P. 271 -284.
163. Wright P.B., Lister A.S., Dorsey J.G. Behaviour and Use of Nonaqueous Media without Supporting Electrolyte in Capillary Electrophoresis and Capillary Electrochromatography// Anal. Chem. 1997. V. 69. P. 3251-3259.
164. Hennion M.C. Solid-phase Extraction: Method Development, Sorbents and Coupling with Liquid Chromatography// J. Chrom. 1999. V. 856. P. 3-54.
165. Chapman J., Hobbs J. Putting Capillary Electrophoresis to Work // LC-GC. 1999. V. 17. P. 266-279.
166. Sabik H., Jeannot R., Rondeau B. Multiresidue Methods Using Solid-Phase Extraction Techniques for Monitoring Priority Pesticides, Including Triazines and Degradation Products in Ground and Surface Water // J. Chrom. 2000. V. 885. P. 217-236.
167. Riekkola. M.-L., Wiedmer S., Valko I.E., Siren H. Selectivity in Capillary Electrophoresis in the Presence of Micelles, Chiral Selectors and Non-Aqueous Media // J. Chrom. 1997. V. 792. P. 13- 35.
168. Riekkola M.-L., Jussila M., Porras. S.P., Valko I.E. Nonaqueous Capillary Electrophoresis // J. Chrom. 2000. V. 892. P. 155- 170.
169. Thorman W., Theurillat R., Wind M., Kuldvee R. Therapeutic Drug Monitoring of Antiepileptics by Capillary Electrophoresis. Characterization of Assays Via Analysis of Quality Control Sera Containing 14 Analytes // J. Chrom. 2001. V. 924 (1-2). P. 429-437.
170. Girard M. Biopharmaceutical by Capillary Electrophoresis. Encyclopedia of Chromatography I Ed. J. Cazes. Florida: Florida Atlantic University Boca Raton, 2001. P. 87-90.
171. Nishi H. Enantiomer Separation of Drugs by Electrokinetic Chromatography (review) // J. Chrom. A. 1996. V. 735. P. 57-76.
172. Fanali S. Identificaqtion of Chiral Drug Isomers by Capillary Electrophoresis (review) 11 J. Chrom. A. 1996. V. 735. P. 77-121
173. Nishi H., Terabe S. Micellar Electrokinetic Chromatography Perspectives in Drug Analysis // J. Chrom. A. 1996. V. 735. P. 3-27.
174. Brunner L.J., Dipiro J.t., Capillary Electrophoresis for Therapeutic Drug Monitoring // Electrophoresis. 1998. V. 19, № 16-17. P. 2848-2855.
175. Penn S.G., Liu G., Begrton F.T., Goodall D.M., Loran J.S. Systematic Approach to Treatment of Enantiomers Separations in Capillary Electrophoresis and Liquid Chromatography. 1. Initial Evaluation Using Propranolol and Dancylated Amino Acids // J. Chrom. A. 1994. V. 680. P. 147-155.
176. Deyl Z., Tagliaro F., Miksik I. J. Biomedical Applications of Capillary Electrophoresis // J. Chrom. B. 1994. V. 656. P. 3-27.
177. Loukas Y.L., Sabbah S., Scriba K.E. Method Development and Vqalidation for the Chiral Separation of Peptides in The Presence of Cyclodextrins Using Capillary Electrophoresis and Experimental Design 11 J. Chrom. A. 2001. V. 931. P. 141-152.
388
Новый справочник химика и технолога
178. Riekkola M-L., Wiedmer S.K., Valko I. E., Siren H. Selectivity in Capillary Electrophoresis in The Presence of Micelles, Chiral Selectors and Non-Aqueous Media // J. Chrom. A. 1997. V. 792. P. 13-35.
179. Shihabi Z.K. Therapeutic Drug Monitiring by Capillary Electrophoresis // J. Chromatogr. A. 1998. V. 807. P. 27-36.
180. Guttmann A., Paulus A., Cohen A.S., Grinberg N., Karger B.L. Use of Complexing Agents for Selective Separation in High-Performance Capillary Electrophoresis; Chiral Resolution Via Cyclodextrins in Incorporated Within Polyacrylamide Gel Columns // J. Chrom. A. 1988. V. 448. P. 41-53.
181. Cikalo M.G., Bartie K.D., Robson M., Meyers P., Euerby M.R. Capillary Electrochromatography // Analyst. 1998. V. 123. P. 87R-102R.
182. Chen S., Lillard J. Continuous Cell Introduction for the Analysis of Individual Cells by Capillary Electrophoresis //Anal. Chem. 2001. V. 73. P. 111-116.
183. Schmalzing D., Nashabeh W., Yao X., Mhatre R. Capillary Electrophoresis-Based Immunoassay for Cortisol in Serum//Anal. Chem. 1995. V. 67. P. 606-612.
184. Jackson P.E., Haddad P.R. Optimization of Injection Technique in Capillary Ion Electrophoresis for the Determination of Trace Level Anions in Environmental Samples // J. Chrom. 1993. V. 640. P. 481 -487.
185. Maruscak W., Trojanowicz M., Margasinska M., Engelhardt H. Application of Carboxymethyl-p-cyclodextrin as a Chiral Selector in Capillary Electrophoresis for Enantiomer Separation of Selected Neurotransmitters // J. of Chrom. A. 2001. V. 926. P. 327-336.
186. Л.А. Карцова, H.B. Комарова. Применение твердофазной экстракции при анализе гербицидов группы хлорфеноксикарбоновых кислот в природных и питьевых водах методом капиллярного зонного электрофореза // Тезисы докл. VIII Всероссийского симпозиума по молекулярной жидкостной хроматографии и капиллярному электрофорезу. 15-19 октября 2001. С. 73.
187. Л.А.Карцова, Н.В. Комарова. Разделение симметричных триазинов методом мицеллярной электрокинети-ческой капиллярной хроматографии с использованием анионного ПАВ // Тезисы докл. VID Всероссийского симпозиума по молекулярной жидкостной хроматографии и капиллярному электрофорезу. 15-19 октября 2001. С. 74.
188. Unger М. Capillary Zone Electrophoresis of Alkaloids Influence of Structure on Electrophoretic Mobility // J. Chrom. 1998. V. 807. P. 81-87.
189. Lopez-Crio S., Garcia-Alvarez-Coque M.C., Hinze W.L., Quina F.H., Berthod A. Effect of a Variety of Organic Additives on Retention and Efficiency in Micellar Liquid Chromatography// J. Chrom. A. 2001. V. 932. P. 4826-4845.
190. Rodriguez R., Pico Y., Font G., Manes J. Analysis of Postharvest Fungicides by Micellar Electrokinetic Chromatography // J. Chrom. 2001. V. 924 (1-2). P. 387-396.
191. Panossian A., Mamikonyan G., Torosyan M., Gabrielyan E., Mkhitaryan S. Analysis of Aromatic Aldehydes in Brandy and Wine by High-Performance Capillary Electrophoresis // Anal. Chem. 2001. V. 73. P. 4379^4-383.
192. Штыкова C.C., Гуменюк А.П., Муштакова С.П. Изучение состояния родия в растворе хлороводородной кислоты методом капиллярного электрофореза // В сб.: Всероссийский симпозиум по теории и практике хроматографии и электрофореза, посвященнный 95-летию открытия хроматографии М.С. Цветом. Самара, 1999. С. 143-150.
193. Boden J., Bachmann J.B.K. Investigation of Matrix Effects in Capillary Zone Electrophoresis // J. Chrom. A. 1996. V. 734. P. 319-324.
194. Мосулишвили Л.М., Барнов В.А., Цибахашвили Н.Я., Энгельгардт X., Бек В. Способ повышения чувствительности детектирования в капиллярном электрофорезе // Журн. аналит. химии. 2001. № 6. С. 579.
195. Tomlinson A.J., Bensen L.M., Landers J.P., Scanlan G.F. Investigation of the Metabolism of the Neuroleptic Drug Haloperidol by Capillary Electrophoresis // J. Chromatogr. B. 1993. V. 652. P. 417.
196. Watzig H. J. Appropriate Calibration Functions for Capillary Electrophoresis. I. Precision and Sensitivity Using Peak Areas and Heights // J. Chrom. A. 1995. V. 700. P. 1-7.
197. Chen S., Lee M.L. Automated Instrumentation for Comprehensive Isotachophoresis-Capillary Zone Electrophoresis // Anal. Chem. 2000. V. 72, № 4. P. 816-820.
198. Chen S., Lee M.L. Counterflow Isotachophoresis-Capillary Zone Electrophoresis on Directly Coupled Columns of Different Diameters // Analyt. Chem. 1998. V. 70. P. 3777-3780.
199. Jumppunen J.H., Reikkola M.-L. Marker Techniques for High-Accuracy Identification in CZE // Anal. Chem. 1995. V. 67. P. 1060-1066.
200. Watzig H. J. Appropriate Calibration Functions for Capillary Electrophoresis. II. Heteroscedasticity and Its Consequences // J. Chromatogr. A. 1995. V. 700. P. 9-20.
201. Маска M., Haddad R. Determination of Metal Ions by Capillary Electrophoresis // Electrophoresis. 1997. V. 18. P. 2482-2501.
202. Bardelmeijer H.A., Waterval J.C.M., Lingeman H. et al. Pre-, On- and Post-column Derivatization in Capillary Electro-phoresis (review) // Electrophoresis. 1997. V. 18. P.2214-2227.
Раздел 5
ХИМИЧЕСКИЕ МЕТОДЫ КОЛИЧЕСТВЕННОГО АНАЛИЗА
Автор-составитель: проф., д.т.н. ИП. Калинкин
доц., к.х.н. А.С. Халонин
К химическим (классическим) методам, применяемым в АХ, обычно относят гравиметрические и титриметрические методы, которые в настоящее время существенно уступили место инструментальным методам. Области применения классических методов — точное определение высоких и средних содержаний (или концентраций) веществ (> 1% масс). Они являются стандартными при оценке правильности и непревзойденными по точности: относительная погрешность определения обычно не превышает 0,1— 0,2%, в то время как погрешность многих инструментальных методов — 2-5%.
5.1. Гравиметрические методы
Теоретические основы и практические примеры гравиметрических методов детально рассмотрены в учебниках и монографиях [1-15], но в них не приводится единое определение метода. Так согласно [3]: «Гравиметрическим анализом называют метод количественного химического анализа, основанный на точном измерении массы определяемого вещества или его составных частей, выделяемых в виде соединений точно известного постоянного состава. Гравиметрические определения можно разделить на три группы: методы отгонки, выделения и осаждения». Первые две группы немногочисленны и применяются для определения ограниченного круга компонентов, например, кристаллизационной воды, СО2 и т.д.
Метод осаждения
В методах осаждения определяемый компонент выделяют в осадок в виде малорастворимого соединения (осаждаемой формы — ОФ); отделяют осадок от надосадочной жидкости (маточного раствора) фильтрованием; промывают осадок для удаления остатков маточного раствора и примесей, адсорбированных на его поверхности; высушивают осадок при низкой или высокой температуре (при необходимости прокаливают) для приведения в форму, удобную для взвешивания (гравиметрическую форму —
ГФ) и взвешивают полученный осадок. Отсюда следует, что одним из существенных недостатков гравиметрических методов является длительность определения.
Ниже перечислены требования к ОФ [1]. Осадок должен:
— быть практически нерастворимым. Определяемый компонент должен выделяться количественно, при этом его концентрация в растворе после осаждения не должна превышать IO 6 М. Осаждение считается количественным, когда остаточное количество осаждаемого вещества в растворе лежит за пределами точности взвешивания на аналитических весах (0,0002 г);
— выделяться в форме, удобной для его отделения от раствора и промывания, и по возможности быть крупнокристаллическим, если он кристаллический, или хорошо скоагулированным, если он аморфен. Важно, чтобы он был однородным по дисперсности;
— быть чистым, т.е. не содержать посторонних примесей;
— количественно переходить в процессе термообработки в гравиметрическую форму.
Что касается требований к гравиметрической форме (ГФ), то она должна:
— быть соединением определенного и постоянного состава;
— быть достаточно устойчивой на воздухе (малогигроскопичной, не взаимодействовать активно с кислородом и СО2);
— обладать по возможности большой молярной массой, что уменьшает относительную ошибку взвешивания.
Необходимая для гравиметрического определения масса ГФ определяется, с одной стороны, погрешностью весов, с другой — оптимальной массой осаждаемой формы. Погрешность обычных аналитических весов одни авторы принимают равной 1 I О 4 г, другие — 2 • 10 4 г. Поскольку относительная погрешность определения обычно не превышает 0,1%, погрешность взвешивания должна составлять не больше 0,1% от минимальной массы ГФ. Следовательно:
Л-2Ы0"4
m>V J , 100; т.е. т > (0,1-0,2) г.
140'1
390
Новый справочник химика и технолога
Обычно рекомендуемая масса ГФ для кристаллических осадков не превышает ® 0,5 г, а для аморфных ® 0,1 г.
Оптимальная масса ОФ зависит от размеров воронки для фильтрования и тигля для высушивания или прокаливания осадка [2].
В зависимости от структуры осадка масса ГФ может колебаться в следующих интервалах (в г):
аморфный (Fe2O3 • иН2О и т.п.) 0,07-0,1
кристаллический, легкий (СаСО3 и т.п.) 0,1-0,15
кристаллический, тяжелый (BaSO4 и т.п.) 0,2-0,4
кристаллический, очень тяжелый (PbSO4) до 0,5
Эти примерные величины служат основанием для оценки массы аналитической навески анализируемого вещества (пробы).
Для получения хорошо фильтрующихся осадков необходимо соблюдать определенные правила и приемы осаждения и последующего созревания осадков различной
Условия осаждения кристаллических осадков
Для уменьшения вероятности образования сразу большого числа зародышевых центров кристаллизации и, следовательно, получения крупнокристаллических осадков с небольшой поверхностью, в начале осаждения стремятся увеличить растворимость осадка и избежать локальных пересыщений. С этой целью:
— осаждение ведут, как правило, из горячих разбавленных растворов;
— раствор осадителя добавляют медленно, при интенсивном непрерывном перемешивании;
— перед осаждением в раствор добавляют вещества, несколько повышающие растворимость осадка.
Эффективное медленное поступление ионов-осадителей может быть достигнуто осаждением из гомогенного раствора (методом возникающих реагентов). Оно осуществляется несколькими способами: регулированием pH, испарением растворителя, генерированием аниона осадителя, синтезом реагента осадителя (см. табл. 5.1.1).
структуры.
Таблица 5.1.1
Методы гомогенного генерирования осадителя [13]
Осадители Реагент Реакция генерирования Осаждаемые элементы
ОН Мочевина 85-100 °C . (NH2)2CO + ЗН2О<==> СО2 + 2NH? + 2ОН“ Al, Ga, Th, Bi, Fe, Sn
poJ- Триметилфосфат Триэтилфосфат (СН3О)3РО + ЗН2О <=± ЗСН3ОН + Н3РО4 (С2Н5О)3РО + ЗН2О <=± ЗС2Н5ОН + Н3РО4 Zr, Hf
c2oj- Этилоксалат (С2Н5)2С2О4+ 2Н2О <=± 2С2Н5ОН + Н2С2О4 Mg, Zn, Ca
so; Диметилсульфат Диэтилсульфат (CH3O)2SO4 + 4Н2О < > 2СН3ОН + so; + 2Н3О+ (C2H5)2SO4 + 4Н2О <=± 2С2Н5ОН + SO2 + 2Н3О+ Ba, Ca, Sr, Pb
СО?' Т рихлоруксусная кислота С13ССООН + 2ОН <==± СНС13 + СО2 + Н2О La, Ba, Ra
H2S* Тиоацетамид CH3CSNH2 + Н2О <=± CH3CONH2 + H2S Sb, Mo, Cu, Cd
DMG** Диацетил + гидроксиламин СН3СОСОСН3 + 2H2NOH DMG + 2Н2О Ni
HOQ*** 8-Ацетокси-хинолин**** CH3COOQ + Н2О СНзСООН + HOQ Al, U, Mg, Zn
Примечания. * **Диметилглиоксим ***8-Оксихинолин **** s OH II он он 1 Y || ^1 сн3 — с—NH2 I | 1 N Ч N N Vх" 11 11 1 II 1 сн3—с—с—сн3 сн3—0-0
Осаждение аморфных осадков
При осаждении аморфных осадков особое внимание обращают на предотвращение образования коллоидов и получение более плотных осадков. С этой целью
— осаждение ведут, как правило, из концентрированных растворов добавлением концентрированных растворов
осадителей, что способствует переходу неустойчивой студенистой формы в более плотную;
— - осаждение ведут из горячих растворов;
— раствор осадителя добавляют быстро, порциями, при перемешивании. Это способствует получению осадка с малой поверхностью и уменьшает адсорбцию примесей;
Химические методы количественного анализа
391
— перед осаждением добавляют достаточное количество электролитов-коагуляторов для предотвращения коллоидообразования;
— по окончании осаждения к раствору с осадком приливают около 100 мл горячей воды, в результате чего часть посторонних адсорбированных ионов переходит в раствор, и осадок становится более чистым.
В отдельных случаях для более глубокого обезвоживания аморфного осадка, его полной коагуляции, уменьшения растворимости и склонности переходить в коллоидное состояние раствор с осадком выпаривают; осадок солей и ОФ высушивают при повышенной температуре (130-200 °C).
Созревание осадков
Существенное улучшение кристаллических осадков с точки зрения упрощения процесса фильтрования и получения более чистого осадка достигают, выдерживая его под маточным раствором (при нагревании или на холоде). Происходит созревание осадка, сопровождающееся укрупнением кристаллов, уменьшением их поверхности и уменьшением концентрации соосаждающихся примесей.
Аморфный осадок фильтруют сразу после разбавления раствора, т.к. при стоянии загрязненность осадка адсорбированными примесями растет.
Фильтрование осадков
Фильтрование позволяет выполнить две важные для аналитика процедуры: отделить осадок от раствора, отмыть его от остатков маточного раствора и сорбированных примесей. Выбор материала фильтра зависит от свойств и структуры осадка, размера частиц, температуры дальнейшей обработки осадка.
Стеклянные фильтры (воронки или тигли) с фильтрующей пористой стеклянной пластинкой выдерживают нагревание до 150 °C. Фарфоровые фильтрующие тигли и тигли Гуча* выдерживают прокаливание выше 500 °C. Прокаливание при 1000-1200 °C проводят в кварцевых или платиновых тиглях.
Фильтрование осадков, требующих для перевода в ГФ высокотемпературной обработки, ведут, как правило, через бумажные беззольные фильтры. Содержание золы в них уменьшено специальной обработкой фильтрованной бумаги, и масса образующейся при сжигании золы известна для каждого фильтра. Характеристики фильтров приведены в табл. 5.1.2.
Таблица 5.1.2
Фильтры для гравиметрического определения
А. Средний диаметр пор фильтров
Тип фильтра Средний диаметр пор, мкм
Стеклянный № 1 100-120
№2 40-50
№3 20-25
№4 10
Тигли Гуча готовят путем фильтрования измельченного и взмученного в воде тонковолокнистого асбеста через фарфоровые тигли с решетчатым дном до получения слоя асбеста 1-2 мм.
Продолжение таблицы 5.1.2
Тип фильтра Средний диаметр пор, мкм
Фильтровальная бумага обыкновенная 3,5-10
уплотненная 1-2,2
Керамиковые фильтры 0,1-0,4
Мембранные фильтры 0,005-0,5
У льтрафильтры 0,001-0,1
Б. Бумажные фильтры
Классификация фильтров Скорость фильтр., см3 • мин”1 (не менее) Область применения
Обеззоленные: медленно-фильтрующие (синяя лента) 10 В гравиметрии для фильтрации тонкодисперсных осадков типа BaSO4
среднефильт-рующие (белая лента) 20 В гравиметрии для фильтрации осадков типа ZnCOs
быстрофильт-рующие (красная лента) 40 В гравиметрии для фильтрации осадков типа Fe(OH)3
Обезжиренные (желтая лента) 20 При количественном анализе жиров и восков
Промывание осадков
Промывание ведут для удаления маточного раствора из пор осадка и сорбированных примесей с поверхности осадка и фильтра. Ниже перечислены требования к промывной жидкости.
— Растворимость осадка в промывной жидкости не должна быть выше его растворимости в маточном растворе. Для уменьшения растворимости в нее добавляют ион-осадитель. Если осадитель в процессе термообработки не удаляется из осадка, после основного промывания осадок промывают минимальным объемом промывной жидкости без осадителя или воды или органических растворителей (ацетон, спирт, эфир).
— При промывании аморфных осадков не должна происходить их пептизация. В случае неорганической природы ОФ необходимо добавлять электролиты-коагуляторы.
— Все компоненты промывной жидкости должны полностью удалятся из осадка в процессе термообработки.
Погрешности в гравиметрическом анализе
Основными источниками погрешностей в гравиметрии являются потери части аналитической формы за счет растворимости ОФ на стадии осаждения и промывания осадка. Величина этих потерь может быть оценена на основе значений произведения растворимости К$ (ПР) . ** ***
** Скорость фильтрации указана для фильтра диаметром 9 см для дистиллированной воды при температуре 17-20 °C и давлении 500 мм вод. ст.
*** В литературе используется как обозначение Ks, так и ПР.
392
Новый справочник химика и технолога
Для реакции осаждения - растворения: /иА + «В АтВ„,
где А — анион, В — катион:
К ° —
S ~ аАаВ>
где К° (ПР°) — термодинамическая константа, аА и ав — активности катиона и аниона соответственно.
Растворимость 5 малорастворимого электролита, выраженная в молях вещества в 1 л раствора, связана с произведением растворимости [1-7, 13-15]:
о /к„(А В„) V ттпп
Следует помнить, что если:
1) ионную силу можно принять равной нулю и протеканием конкурирующих реакций протонирования и/или комплексообразования можно пренебречь, то растворимость осадка вычисляют по величине К°;
2) влиянием ионной силы пренебречь нельзя (имеем дело с заметно растворимым соединением в присутствии посторонних электролитов), но конкурирующие реакции отсутствуют, растворимость вычисляют по величине реального произведения растворимости;
3) конкурирующими реакциями пренебречь нельзя, растворимость вычисляют по величине условного произведения растворимости К3.
Растворимость ОФ не должна быть выше «1 - КГ6 моль-л-1 (см. выше). Для достижения S < 1 • 10-6 моль-л-1 осаждение обычно ведут при 1,5-2 кратном избытке иона-осадителя (по отношению к стехиометрии). Большее количество осадителя может приводить к дополнительному растворению части осадка. Расчеты показывают [2, 4], что в случае бинарных малорастворимых электролитов при 1,5-2 кратном избытке осадителя количественное осаждение достигается лишь при значениях К°(ПР°) < 1 • 10-8. Поэтому в гравиметрии бинарные малорастворимые соединения, для которых К° (ПР°) > 10-8, как правило, не используются.
Осадитель добавляют также к промывной жидкости и/или ведут промывание растворителями, не растворяющими осадок. Количественно влияние избытка осадителя, посторонних электролитов, побочных (сопряженных) равновесий протонирования и комплексообразования на растворимость малорастворимых электролитов (осадков) подробно рассмотрено в [1-7]. Необходимые для расчетов справочные данные значений активности ионов и К" приведены в табл. 5.1.3-5.1.5.
Таблица 5.1.3
Коэффициенты активности различных ионов [11]
Ионы Значения коэффициента активности при ионной силе*
0,0005 0,001 0,0025 0,005 0,01 0,025 0,05 0,1
Неорганических соединений
Н+ 0,975 0,967 0,950 0,933 0,914 0,88 0,86 0,83
Li+ 0,975 0,965 0,948 0,929 0,907 0,87 0,835 0,80
Rb+, Cs+, ЫН} , Ag+, Tl+ 0,975 0,964 0,945 0,924 0,898 0,85 0,80 0,75
K+, СГ, Br-, Г, CN-, NO-, no; 0,975 0,964 0,945 0,925 0,899 0,85 0,805 0,755
oh-, f-, hs-, cio4-, BrO3, io;, Mno;, cio;, ocn-, scn- 0,975 0,964 0,946 0,926 0,900 0,855 0,81 0,76
Na+,cdci+, cio;, io;, hco;, h2po4, hso;, h2Aso; 0,975 0,964 0,947 0,928 0,902 0,86 0,82 0,775
Hg^,sot,s2o;,s4o^-,s2o^, seo;-,cro;-,HPo;- 0,903 0,867 0,803 0,740 0,660 0,545 0,445 0,355
Pb2+,CO32-,MoO;-,SO^- 0,903 0,868 0,805 0,742 0,665 0,55 0,455 0,37
Sr2+, Ba2+, Ra2+, Cd2+, Hg2+, S2-, S2O; , wo;- 0,903 0,868 0,805 0,744 0,67 0,555 0,465 0,38
Ca2+, Cu2+, Zn2+, Sn2+, Mn2+, Fe2+, Ni2+, Co2+ 0,905 0,870 0,809 0,749 0,675 0,57 0,485 0,405
Mg2+, Be2+ 0,906 0,872 0,813 0,755 0,69 0,595 0,52 0,45
PO;,[Fe(CN)6]3' 0,796 0,725 0,612 0,505 0,395 0,25 0,16 0,095
АГ, Fe3+, Crj+, Sc3+, Y3+, La3+, In3+, Ce3+, Pr3 ‘, Nd3+, Sm3+ 0,802 0,738 0,632 0,54 0,445 0,325 0,245 0,18
[Fe(CN)6]4“ 0,668 0,57 0,425 0,31 0,20 0,10 0,048 0,021
Th4+, Zr4+, Ce4+, Sn4+ 0,678 0,588 0,455 0,35 0,255 0,155 0,10 0,065
Химические методы количественного анализа
393
Продолжение таблицы 5.1.3
Ионы Значения коэффициента активности при ионной силе*
0,0005 0,001 0,0025 0,005 0,0] 0,025 0,05 0,1
Органических соединений
нсоо’, ch,nh;, н,с,н,о;, (ch,)2nh; 0,975 0,964 0,946 0,926 0,900 0,855 0,81 0,76
~OOCCH2NH^, (CH3)3NH+, c2h5nh3+ 0,975 0,964 0,947 0,927 0,901 0,855 0,815 0,77
CH3COO , (CH3)4N+, CH2C1COO, NH2CH2COO 0,975 0,964 0,947 0,928 0,902 0,86 0,82 0,775
СбН5соос6п4онсоо, c6h4cicoo C6H5CH2COO, H2C=CHCH2COO(C2H5)4N+, (CH3)2CH CHCOO , (C3H7)2NH^ 0,975 0,965 0,948 0,929 0,907 0,87 0,835 0,80
[OC6H2(NO2)3],(C3H7)3NH+ 0,975 0,965 0,948 0,929 0,909 0,875 0,845 0,81
(coo),', nctn,o; 0,903 0,867 0,804 0,741 0,662 0,55 0,45 0,36
H2C(COOf, (CH2COOf, (CHCOHCOO)2~ 0,903 0,868 0,805 0,744 0,67 0,555 0,465 0,38
C6H4(OOO)*~, H2C(CH2COO)*’, CH2CH2(CW)2 0,905 0,870 0,809 0,749 0,675 0,57 0,485 0,405
c6H5o2-f> Э / 0,796 0,728 0,616 0,51 0,405 0,27 0,18 0,115
ГГ C1Z1 +C2Z2 +C3Z3 +--c„z„
Примечание. * Ионная сила ц = , где сь с2, с3.. .с„ — концентрации всех присутствующих в растворе ионов, a zb
z2,z3 ...zn — соответствующие заряды этих ионов.
Таблица 5.1.4
Коэффициенты активности различных ионов при высоких значениях ионной силы раствора (приближенные значения)
Пояснение. Таблица составлена Л. Мейтесом [29] по следующей формуле Дэвиса: - = —--0,2ц , где ц — ионная сила
z,2 1 + 1,57Й
раствора; / — коэффициент активности иона; z, — заряд иона (от 1 до 6). За эффективный ионный радиус взято некоторое среднее его значение для всех ионов.
Ц IgZ Z,2 Значения / при z,
1 2 3 4 5 6
0,05 0,076 0,84 0,50 0,21 0,062 0,013 0,0019
0,1 0,090 0,81 0,44 0,16 0,037 0,0058 0,00060
0,2 0,097 0,80 0,41 0,14 0,028 0,0038 0,00033
0,3 0,094 0,81 0,42 0,14 0,032 0,0046 0,00043
0,4 0,086 0,82 0,45 0,17 0,042 0,0072 0,00082
0,5 0,075 0,84 0,50 0,21 0,062 0,013 0,0020
0,6 0,063 0,87 0,56 0,27 0,098 0,027 0,0054
0,7 0,050 0,89 0,63 0,36 0,16 0,058 0,016
0,8 0,035 0,92 0,72 0,48 0,27 0,13 0,054
0,9 0,020 0,96 0,83 0,66 0,48 0,31 0,19
1,0 0,0044 0,99 0,96 0,91 0,85 0,78 0,69
394
Новый справочник химика и технолога
Таблица 5.1.5
Произведение растворимости (ПР) и растворимость (5) некоторых малорастворимых соединений
Принятые обозначения и пояснения.
Комн. — комнатная;
(акт.) — приведено значение произведения активностей;
1S дана в г/100 г воды;
2 S дана с учетом наличия в растворе недиссоциированных молекул и ионных пар;
3 Автор не указывает на наличие в растворе молекулярных форм и не приводит значений растворимости соединений;
4 Следует иметь в виду, что при переходе солей Hg(II) в раствор образуется очень мало свободных ионов Hg2+, но относительно много недиссоциированных молекул. Поэтому низкие значения ПР могут создать неверные представления о соответственно малых растворимостях этих солей. О степени диссоциации солей Hg(II) следует судить по константам, помещенным в соответствующих разделах справочника;
5 Значение ПР при допущении, что ПР = ПР° дано по [38]. Оно существенно отличается от найденных ранее [17, 20].
Соединение Ионное произведение t,°C ПР рПР = = -lgIIP S Литера-тура
моль-л 1 г/100 мл
Ас2(С2О4)з [Ac3+]2-[C2O2-]3 — 210“24 23,7 54
Ас(ОН)3 свежеосажд. [Ac3+]-[OH~]3 — 2,1-IO-19 18,68 «
Ас(ОН)3 после старения [AC3+][OHT — 1,3 -1 O’21 20,89 «
Ag[Ag(CN)2] [Ag+]-[ Ag(CN)- ] Комн. 2-10“12 11,70 1
20 2,2-1 O’12 11,66 2
18-25 — ИЛ 9
Ag3AsO4 [Ag+]3-[AsO3’] Комн. 1-10 22 22,0 1,6, 11,54
18-25 l,15-10~19 18,94 1,84-1 O’5 8,5-Ю*4 71
20 l,15-10~19 18,94 1,84-1 O’5 8,5-Ю*4 3
20 1,12-1O20 19,51 3,59-10^ 1,66-Ю*4 4
Ag3AsO3 [Ag+]3-[ AsO3- ] 11017 17,00 6, 54
25 1,22-1 O’17 16,91 2,60-1 O’5 5,99-10*4 4
4,5-IO’19 18,35 1,1-Ю"5 4,91-Ю*4 3
AgBO2 [Ag+]-[BO-] — 4-10-3 2,4 54
AgBr [Ag+]-[Br-] Комн. 4,1-IO43 12,39 1
4,9-10~13 12,31 7,12-1 O’7 1,34-10’5 42
5,3-10’13 12,28 6, 54
18 4,1-IO’13 12,39 2
25 7,7-1013 12,11 8,8-10’7 1,65-ю5 2,3,7, 10
25 7-10’13 12,15 5
20-25 5,25-10“13 12,28 8
18-25 — 12,4 9
5,3-10“13 12,28 11
AgBrO3 [Ag+]-[BrO3-] Комн. 5-1 O’5 4,30 1
Комн. 5,77-10’5 4,24 6,3-10’3 10
20 3,97-10~5 4,40 2
25 5,77-10~5 4,24 2
25 5,8-1 O’5 4,24 5
5,5-10’5 4,26 6
AgCN [Ag+]-[CN-] — 1,4-10’16 15,84 54
Ag2CC>3 [Ag+]2-[CO3~ ] Комн. 6-1 O’12 11,22 1
25 6,15-10~12 11,21 1,15-Ю*4 3,17-10’3 2,3
25 6,2-1 O’12 11,21 5
20-25 1-10’12 12,00 8
18-25 — 11,3 9
Химические методы количественного анализа
395
Продолжение таблицы 5.1.5
Соединение Ионное произведение t,°C ПР рПР = =—ignp S Литера-тура
моль-л 1 г/100 мл
8,2-10-12 11,09 6, 54
25 9,49-Ю'12 11,02 1,33-Ю-4 3,68-Ю“3 4
Ag2C2O4 [Ag*]2-[C2OM Комн. 5-1012 11,30 1
3,5-Ю'11 10,46 6, 54
1,1-Ю'11 10,96 1,4-1 О'4 4,25-Ю“3 3
25 4,1-Ю’11 10,39 2,27-104 6,89-Ю“3 4
3,5-Ю'11 10,46 6
18-25 1,4-Ю'12 11,85 1,12-Ю-4 3,4-10'3 71
Ag(CH3COO) [Ag+]-[CH3COO'] 18-25 — — 6,2-Ю'2 1,04 71
— 4-10'3 2,4 54
Ag2C4H40e (тартрат) [Ag+HC^OJ- 18-25 — — 5,5-Ю'3 0,2 71
AgC5H9O2 (валерианат) [Ag+KC5H9O- ] Комн. 8-10 5 4,1 1
AgC7H5O2 (бензоат) [Ag+]-[c7H5o;] Комн. 9,3-1О5 4,03 «
AgC7H5O3 (салицилат) [Ag+].[C7H5O- ] Комн. 1,4-10’5 4,85 «
AgCl [Ag+]-[CF] Комн. 1,1-Ю'10 9,96 «
1,78-10'10 9,75 6, 54
25 1,78-Ю'10 9,75 1,35-10”5 1,93-Ю-4 42
1,56-10'10 9,81 1,25-Ю'5 1,8-Ю-4 10
4,7 0,21-Ю'10 10,68 2
9,7 0,37-Ю'10 10,43 «
25 1,56-Ю'10 9,81 «
50 13,2-Ю'10 8,88 «
100 215-Ю'10 7,67 «
AgC102 [Ag+]-[C10'] — 2-Ю4 3,7 54
AgC103 [Ag+]-[cio;] — 5-Ю'2 1,3 54
Ag2CrO4 [Ag+]2-[CrO2~] Комн. 2-10'12 11,70 1, 5(20 °C), 9
14,8 1,2-Ю'12 11,92 2
25 9-10'12 11,05 2, 10
20 9-10'12 11,05 1,3-Ю-4 4,31-Ю'3 3, 71
25 1,44-10'12 11,84 7,12-10'5 2,36-10'3 4
— 1,1-Ю'12 11,95 6, 54
20-25 2,45-10'12 11,61 8
Ag2Cr2O7 [Ag+]2-[Cr2O7'] Комн. 2-10'7 6,40 1,8
25 2-Ю'7 6,70 2,5
— 1-Ю'10 10,00 6
Ag4[Fe(CN)6] [Ag+]4-[Fe(CN)£" ] Комн. 1,6-ю-41 40,80 1
Комн. 1,5-Ю-41 40,82 2,2-10'9 1,4-Ю'7 6, 10
18-25 — — « 1,5-10'8 « Ю'7 71
— 8,5-1045 44,07 54
Ag3[Fe(CN)6] [Ag+]3-[ Fe(CN)^ ] 1-Ю'22 22,00 6, 54
Комн. 9,8-Ю'26 25,01 2,4-10'7 1,3-Ю'5 10
18-25 2,1-Ю'24 23,68 1,2-Ю”6 6,6-10'5 71
396
Новый справочник химика и технолога
Продолжение таблицы 5.1.5
Соединение Ионное произведение t,°C ПР рПР = = -lgIIP S Литера-тура
моль-л 1 г/100 мл
Ag2HVO4 [Ag']2'[HVO;-] — 2-Ю'14 13,7 54
Agl [Ag+Hn Комн. 1-Ю’16 16,00 1,5,9
13 0,32-10'16 16,49 2
25 1,5-1016 15,82 1,2-10’8 2,82-10’7 2,3
20-25 8,З-Ю’17 16,08 * 6, 8, 54
18-25 1,5-10’16 15,82 1,2-10’8 2,8-10’7 71, 10
25 9,98-10’17 16,00 1,03-10’8 2,41-10’7 42
AgIO3 [Ag+]-[io;] Комн. 2-Ю’8 7,70 1
9,4 0,92-10’8 8,04 9,6-10’5 2,72-10’3 2,3
25 3,22-10"8 7,49 1,79-1 О’4 5,07-10’3 42
25 3,2-10’8 7,49 5
3,0-10’8 7,52 6, 54
AgMnO4 [Ag+]-[MnO“] 1,6-10’3 2,79 6, 54
AgN3 [Ag+]-[N;] 2,9-10’9 8,54 6
AgNO2 [Ag+]-[NO-] 1,6-104 3,80 6
6-1О4 3,22 54
Ag2MoO4 [Ag+]2.[MoO!t- ] Комн. 3,2-10’11 10,49 1
25 3,03-10’12 11,52 9,11-Ю’5 3,42-10’3 4
2,8-10“12 11,55 6, 54
AgOH(Ag2O) [Ag+]-[OH~] Комн. 2-Ю’8 7,70 1-ю-4 1,25-10’3 1, 5(25 °C), 71, 9
20 1,52-10’8 7,82 2
25 1,63-10’8 7,79 1,61-Ю-4 2,01-10’3 42
— — 7,6 5
1,6-10’8 7,80 6
1,9510 8 7,71 54
20-25 1,9-10’8 7,82 8
[AgO“][H*] — — 17,7 5
AgOCN [Ag+]-[OCN’] 2,3-10’7 6,64 6, 8, 54
Ag2PO3F [Ag+l2-fPO3F23 — 8,9-1 О’4 3,05 54
Ag3PO4 [Ag*HPO?] 25 1,29-10’20 19,89 4,67-1 О’6 1,96-104 4, 6, 54
18-25 5,4-10’20 19,27 1,55-10’5 6,5-104 71
18-20 1,8-10’18 17,74 5
AgReO4 [Ag+]-[ReO4-] 7,95-10’5 4,10 6, 54
Ag2S [Ag+]2-[S2T Комн. 1,6-1 О'49 48,80 3,4-10’17 8,43-10’16 1,3
18 1,6-1 О'49 48,80 2
25 6,31-Ю’50 49,20 2,51-Ю’17 6,22-10’16 4, 6, 54
20 1-ю-49 49,00 5,9
18-25 1,6-1 О'49 48,80 3,5-10’17 8,7-10’16 71, 10
20-25 6-10’50 49,21 8
Ag2SO4 [Ag+]2-[SO2’] Комн. 6,3-10’5 4,20 1
7,7-10’5 4,11 2,7-10’2 8,42-10’1 3
25 5,02-10’5 4,30 2,32-10’2 0,7244 4
25 8-Ю’5 4,10 5
1,6-10’5 4,80 6, 8, 54
18-25 — — 2,6-10’2 0,80 71
Химические методы количественного анализа
397
Продолжение таблицы 5.1.5
Соединение Ионное произведение t, °C ПР рПР = = -1ёПР S Литература
моль-л 1 г/100 мл
Ag2SO3 [Ag+]2.[SO2~] 25 1,5-10“14 13,82 1,56-105 4,61-10^ 4, 6, 54
AgSCN [Ag+]-[SCN~] Комн. 4,9-10’11 10,31 1
18 0,49-10’12 12,31 2
25 1,16-1012 11,35 1,1-Ю”6 1,8-Ю5 2, 10
25 1-Ю’12 12,00 5,9
1,1-1012 11,96 6, 54
18-25 7,1-10“13 12,15 8,45-10“7 1,4-10 5 71
20-25 1,07-10’12 11,97 8
AgSO3NH2 [Ag+]-[SO3NH2 ] — 1-Ю'1 1 54
AgSeCN [Ag+HSeCN-] 4,0-10“16 15,40 6, 54
Ag2SeO3 [Ag'flSeOf] 9,0-10’16 15,05 6
9,8-Ю’16 15,01 54
Ag2SeO4 [Ag+]2-[SeO2-] 5,6-10“8 7,25 6, 54
AgVO3 [Ag+]-[VO3-] Комн. 1,3-1010 9,89 1
20 5-10 7 6,30 5, 6, 54
Ag2WO4 [Ag-]2.[WO=-J Комн. 5-Ю’10 9,30 1
— 5,5-Ю’12 11,26 6, 54
25 6,15-Ю’12 11,21 1,15-10^ 5,35-103 4
A1(C6H5O2N2)3 (купферонат) [Al3+]-[C6H5O2N2]3 2,8-Ю’19 18,55 4, 213
A1(C9H6ON)3 (оксихинолят) [Al3+]-[Ox~]3 Комн. 5-Ю33 32,3 1,4
1-Ю’19 19,0 И
25 1-Ю’29 29,0 4
1,03-10’29 28,99 12
A1(OH)3 [Al3+]-[OH-]3 25 1,1-10’33 38,96 2,52-10-9 1,97-1О'8 4
— — 32,5 5
18-25 2,0-10’33 32,7 3,3-10'9 2,6-108 71
18-25 — 32,7 9
20-25 4,57-10’33 32,34 8
— 1-10 32 32 6
3,2-10’34 33,5 54
[aio3h;]-[h+] Комн. 1,6-10’14 13,80 1
15 4-Ю’13 12,40 2
18 1,1-10’15 14,96 «
25 3,7-10“15 14,43 «
— — 11,2-13,9 5
[(A10H)2+]-[0H"]2 — 1-Ю’23 23 6
3,2-IO’25 24,5 54
[H+][A1O“] — 1,6-10’13 12,8 6, 54
A1PO4 Комн. 1-Ю”6 6 1
25 5,75-Ю’19 18,24 6,61 -IO’10 8,06-Ю9 4, 6, 54
A1PO4 аморф. [АГ3<Р0‘-] 20-25 6,3-10'19 18,20 8
A1AsO4 [AP‘] [ AsCr- ] 1,6-10’16 15,80 6, 54
Am(OH)3 [Am3+]-[OH~]3 — 2,7-10’20 19,57 6
5,0-10-24 23,3 54
Am(OH)4 [Am4+]-[OIl~]4 — 1-10“56 56,00 6, 54
As2S3 [As3+]2-[S2~]3 Комн. 4-Ю29 28,40 1
398
Новый справочник химика и технолога
Продолжение таблицы 5.1.5
Соединение Ионное произведение /,°С ПР рПР = =-ignp 5 Литера-тура
моль-л 1 г/100 мл
16 4-Ю29 28,40 5
18-25 3,2-10-29 28,49 2-10-6 5-10"5 71
As2S5 [As5+]2-[S2-]5 Комн. 1-Ю30 30 1
18-25 3,7-10“38 37,43 4,5-10-6 1,4-10^ 71
AuBr [Au+]-[Bf] — 5-10-17 16,3 54
AuBr3 [Au3+]-[Br“]3 — 4-10~36 35,4 «
AuCl [Аи+]-[СГ] 2,0-10“13 12,7 «
AuCl3 [Au3+].[C1-]3 3,2-10"25 24,5 «
AuOH [Au+]-[OH] 7,9-10"20 19,1 «
Au(OH)3 [Au3+]-[OFT]3 3,2-10^3 42,5 «
Aul [Аи+]-[Г] 1,6-10"23 22,8 «
Aul3 [Аи3+]-[Г]3 1-Ю^6 46 «
Ba3(AsO4)2 [Ba2+]3-[ AsO34“ ]2 7,8-1051 50,11 6, 54
25 7,76-10"51 50,11 3,73-Ю"11 2,57-10"9 4
Ba(BrO3)2 [Ba2+]-[BrOJ ]2 5,5-10-6 5,26 6, 54
BaCO3 [Ва2+]-[СО2-] Комн. 7-109 8,15 1
16 7-10’9 8,15 2,5
25 8,1-Ю"9 8,09 9- Ю5 1,78-10-3 2,3
25 5,93-10"9 8,23 7,7-105 1,52-10"3 4
4,0-10ч ° 9,40 54
— 5,1-10“9 8,29 6
18-25 8,0-10’9 8,1 8,9-10"5 1,76-10"3 71, 10
20-25 4,9-10-9 8,31 8
18-25 — 8,2 9
BaC2O4 (оксалат) [Ва2*]-[ C2Oj" ] Комн. 1,7-10"7 6,77 1
25 1,49-10"7 6,83 3,85-104 8,68-10’3 4
18 1,6-10-7 6,80 4,0-10^ 9,0-10 3 5, 71, 8, 10
1,1-10"7 6,96 6, 54
ВаС2О4-3,5Н2О [Ва2+]-[С2О2-] 18 1,62-10"7 2
ВаС2О4 • 2Н2О [Ва2+]-[ С2Од" ] 18 1,2-10’7 3,5-10"4 8,55-10"3 2,3
ВаС2О4-0,5Н2О [Ва2+]-[С2О2“] 18 2,18-Ю"7 2
ВаСгО4 [Ва2+]-[СгО2] Комн. 2-1О~10 9,7 1,9
18 1,6-10"10 9,80 2
28 2,4-10"10 9,62 1,5-10“5 3,80-10^ 2
20 2,4-10’10 9,62 1,5-105 3,80-10^ 3
25 1,18-1О10 9,74 1,08-10"5 2,75-1 О'4 4
18 2-10-10 9,7 5
1,2-10’10 9,93 6, 54
20-25 2,4-10’10 9,62 1,55-10"5 3,9-10^ 8, 10
BaF2 [Ba2+].[F~]2 1,1-10^ 5,98 6, 54
18 1,7-10-6 5,77 5
20-25 1,0-10^ 6,0 8
20 1,73-10-6 5,76 7,6-103 1,3310^ 3
Ba2Fe(CN)6 [Ва2+]2-[ Fe(CN)J- ] 3-10'8 7,5 6, 54
18-25 — — 2,1-Ю"3 0,1 71
Ва(Ю3)2 [Ва2+]-[ 1О2 ]2 Комн. 6-10"10 9,22 1
Химические методы количественного анализа
399
Продолжение таблицы 5.1.5
Соединение Ионное произведение t, °C ПР рПР = = -1ёПР 5 Литература
моль-л 1 г/100 мл
25 2,05-10"9 8,69 7,99-10"4 3,89-10"2 4
25 6,5-1О“10 9,19 5
1,50-10'9 8,82 6, 54
Ва(Ю3)2-2Н2О [Ва2+][ЮД2 25 6,5-1О"10 9,19 2
10 8,4-10"11 10,06 2
ВаМпО4 [Ba2+HMnOj~] — 2,5-10’10 9,60 6, 54
ВаМоО4 [Ba2+]-[MoO2“] — 4-10"8 7,40 6, 54
25 5,3-10 8 7,28 2,63-104 7,82-10"3 4
Ba(NO3)2 [Ba2+]-[NO; ]2 4,5 -103 2,35 6
Ва(ОН)2 [Ba2+]-[OH“]2 51О3 2,3 5, 6, 54
18-25 — — 0,20 3,36 7*
[BaO2H"]-[H+] — — 16,5 5
BaPO3F [Ba2+]-[PO3F2"] — 4-10’7 6,40 6, 54
Ва3(РО4)2 [Ba2+]3-[PO3-]2 6,03-10 39 38,22 6, 54
25 3,39-10’23 22,47 1,26-10"5 7,57-104 4
18-25 2,8-10’22 21,55 7
ВаНРО4 [Ba2*]-[HPO2] 20 9,12-10’8 7,04 3,02-104 7,05-10"3 4
Ва2Р2О7 [Ba2+]2-[P2O4“] — З-Ю"11 10,5 6, 54
BaPt(CN)4 [Ba2+]-[Pt(CN)2_ ] — 4-10“3 2,4 «
Ba(ReO4)2 [Ba2+]-[ReO4]2 — 5,25-10"2 1,28 «
BaSO3 [Ba2l[SO2-] — 8,0-10 7 6,1 «
25 1,31-10"6 5,88 1,14-10 3 2,48-10"2 4
BaSO4 [Ba2+]-[SO4“] Комн. 1-Ю"10 10,0 1,5,9
18 О,87-1О"10 10,06 2
25 1,08-10"10 9,97 1,0-10"5 2,33-Ю"4 2,3
50 1,98-1О"10 9,70 2
25 1,05-10 10 9,98 1,02-10"5 2,39-10"4 4
1,1-Ю"10 9,97 1,05-10"5 2,5-104 6, 71, 10, 54
18-25 1,3-Ю"10 9,89 8
BaS2O3 [Ba2+][S2O2~ ] — 1,6-10"5 4,79 6, 54
BaSeO4 [Ba2+]-[SeO2~ ] — 2,8-10"11 10,55 6
5-Ю"8 7,30 54
Be(OH)2 [Be2+]-[OH~]2 Комн. 1-Ю"20 20,0 1
— 4,9-10"22 21,31 54
— — 17,7 5
— 6,3-10"22 21,20 6
[BeOH]-[OH] — 2-Ю"14 13,7 6
1,95-10"13 12,71 54
Be(OH)2 аморф. [Be2+]-[OH“]2 25 1,59-10"21 20,80 7,35-10"8 3,16-10"7 4
Be(OH)2 крист, a [Be2+]-[OH“]2 25 7,94-10"22 21,10 5,83-10”8 2,51-Ю"7 4
Be(OH)2 крист. P [Be2+]-[OH~]2 25 3,16-10"22 21,50 4,29-10'8 1,85-10’7 4
Be(CO3) [Be2+][ CO2" ] 1-Ю"3 3,0 6
BeMoO4 [Be2+]-[ MoO," ] 3,2-10"2 1,49 6
BeNH4PO4-H2O [Be2+] [NH4+] [POJ-] 2,2-10"20 19,66 см. табл. 5.1.11.9.Б 13
400
Новый справочник химика и технолога
Продолжение таблицы 5.1.5
Соединение Ионное произведение t,°C ПР рПР = = -lgIIP S Литера-тура
моль-л 1 г/100 мл
Ве3(РО4)2-6Н2О [Be2+]3-[PO’-]2 1,9-10“38 37,72 см. табл. 5.1.11.9.В 14
BiAsO4 [Bi3+]-[AsO3“] 2,8-10’10 9,55 6, 54
20 4,37-1О“10 9,36 2,09-105 7,27-10^ 4
Bi2(C2O4)3 [Bi3+]2-[C2O2-]3 4-Ю-36 35,4 54
Bi(C6H5O2N2)3 [в^-[с6н,о2н;]3 5,00-1028 27,30 4, 213
Bil3 [Bi3*]-[I-]3 8,1-10’19 18,09 6, 54
BiOCl [BiO+][Cl-] 7-109 8,85 6, 54
Комн. 2-10’9 8,7 1
[Bi3+]-[OH“]2-[Cl“] 1,8-10“31 30,75 6, 54
BiO(OH) [В1О][О1Г] 4-Ю'10 9,4 6, 54
Bi(OH)3 [Bi3+][OH-]3 Комн. 2-10’16 15,7 1
25 4,27-Ю31 30,37 1,12-10~8 2,92-107 4
18-20 4,3-10’31 30,37 5, 15
3,2-10’32 31,49 6
18-25 8,5-10’22 21,07 5,4-10^ 1,4-10^ 71
[BiO3H- ][H+] — — 19,3 5
BiPO4 [Bi3*]-[POJ] Комн. 3,2-10’20 19,49 1
25 1,29-10-23 22,89 3,59-10“12 1,09-10’10 4, 6, 54
20-25 4-Ю23 22,40 8
18-20 1,3-10 23 22,89 16
Bi2S3 [Bi3+].[S2-] Комн. 1,6-1072 71,80 1,5
1-Ю’97 97,0 2,7-10] 5 1,39-10’13 3, 6, 8, 54
25 1,00-10-96 96,0 4
18-25 5,2-1О33 32,28 3,5-10 7 1,8-105 71
3,2-10’31 30,49 17
1,9-10’89 88,70 18
7,1-10^ 60,15 20
Ca3(AsO4)2 [Ca2+]3-[ AsO34- ]2 6,8-10“19 18,17 6, 54
20 — 0,33 1,3-10’2 3
20 6,76-1019 18,17 9,11-Ю”6 3,62-103 4
CaCO3 [Ca2+]-[CO3“ ] Комн. 1,2-108 7,92 1,9
15 0,99-108 8,00 1-ю-4 2,22
25 0,87-10’8 8,06 2
25 4,8-10-9 8,32 8,0-10“5 22
18-25 4,8-10'9 8,32 6,93-105 6,9 10 4 3,7', 10, 23
25 3,26-109 8,49 5,71-105 5,72-10^ 4
20-25 3,8-Ю9 8,42 8, 54
CaC2O4 (оксалат) [Ca2+]-[C2O2-] Комн. 2-10“9 8,7 1
25 2,60-10’9 8,59 4,86-10’5 6,22-104 4,5
2,3-10”9 8,64 6, 8, 54
18-25 — 8,1 9
CaC2O4H2O [Ca2+]-[C2O2-] 18 1,78 109 8,75 2
25 2,57-10“9 8,59 5,1- 10 s 7,45-10^ 2, 3, 22
CaC4H4O6 (тартрат) [Ca2+]-[C4H4O2~] Комн. 7,7-10-7 6,11 8,7-10-4 1, 6, 22, 54
18-25 — 8,5-10’3 1,6-1О2 71
Химические методы количественного анализа
401
Продолжение таблицы 5.1.5
Соединение Ионное произведение °C ПР рПР = = -IgITP S Литература
моль-л 1 г/100 мл
СаС4Н4О6-2Н2О [Са^НСЛО2"] 18 0,77-1 О’6 6,11 2
СаСгО4 [Са2+]-[СгОд“] 7,1-10" 3,15 6, 54
18-20 2,3-10’2 1,64 1,5-10’1 2,32 5, 71, 22
25 3,67-10" 2,44 6,06-10’2 9,45-10" 4
CaF2 [Ca2+]-[F~]2 Комн. 3,4-10’11 10,46 2,0-10" 1,2, 22
26 3,95-Ю"1 10,46 2,1-10" 2, 22
20 4-10’11 10,60 2,2-10" 1,72-10" 3
26 4,70-10’11 10,33 2,27-10" 1,78-10" 4
25 4-10’11 10,40 5, 6, 8, 54
18-25 — 10,5 9
СаНРО4 [Са2+]-[НРО"] 2,7-10”7 6,57 6, 54
25 4,2-10’7 6,38 8,58-10" 1,17-10" 42
Са(Н2РО4)2 [Ca2+]-[H2POJ2 — 1-Ю’3 3,0 6, 54
Ca(NH4)2Fe(CN)6 [Са21[нн; ]2-[Fe<CN)4-] — 4-10-8 7,4 6, 54
20-25 « Ю"2 « 12 ~ 10" «3 71
Са(Ю3)2 [Са2*][Ю3-]2 Комн. 6,5-10’7 6,19 1
7-10" 6,15 6, 54
Са(Ю3)2-6Н2О [Са2*][Ю-]2 10 22,2-10’8 6,65 3,8-10’3 2, 22
18 64,4-10"8 6,19 2
25 1,9-10'6 5,72 5,5-10’3 22
Са(ОН)2 [Са2+]-[ОН~]2 25 3,1-10" 4,51 2,0-10 2 1,48-10" 3
25 7,96-1 О’6 5,10 1,26-10" 9,32-10" 4
18 5,5-10"6 5,26 5, 6,8
— 6,5-10’6 5,19 54
18-25 — — 1,66-10" 0,123 71
18-25 — 5,3 9
[СаОН+]-[ОН~] 1,4-10" 3,85 6
9,1-10 5 4,04 54
Са3(РО4)2 [Са2+]3-[РО3]2 Комн. 1-Ю’25 25,0 1
25 1-Ю’26 26,0 2,47-10’6 7,67-10" 4,8
— 2-10-29 28,7 6, 54
18-25 3,5-10“16 15,46 8,1-10" 2,5-10" 71
CaPO3F [Ca2+]-[PO3F2~] — 4-10’3 2,4 6, 54
Са5(РО4)3ОН [Са2"]5-[РО34“]3-[ОН-] — 1,6-10"8 57,80 6, 54
CaSO3 [Ca2*]-[SO2-] 25 1,59-10’8 7,80 1,26-10" 1,51-10" 4
— 1,3-10" 7,89 6
3,2-10" 6,5 54
CaSO4 [Са2+]-[ SO2’ ] Комн. 6,1-Ю5 4,22 7,8-10" 0,2 1,5 (25 °C), 71, 10
10 1,95-10" 3,71 2
25 2,30-10" 4,64 4,79-10" 6,52-10" 4
— 9,1-10" 5,04 6
2,5-10’5 4,6 54
20-25 2,4-10’5 4,62 8
18-25 — 4,3 9
402
Новый справочник химика и технолога
Продолжение таблицы 5.1.5
Соединение Ионное произведение t, °C ПР рПР = =-ignp S Литера-тура
моль-л1 г/100 мл
CaSeO3 [Ca2+]-[SeOf“ ] — 4,7-10”6 5,33 6, 54
CaSiF6 [Ca2+]-[SiFM — 8,1-10"* 3,09 6, 54
CaWO4 [Ca24][WO2] — 9,0-10’9 8,05 6, 54
18 8,71-IO’9 8,06 9,33-10’5 2,69-10 3 4
Cd3(AsO4)2 [Cd2+]3-[ AsO34- ]2 — 2,2-10"33 32,66 6, 54
Cd(BO2)2 [Ca2+]-[ BO" ]2 — 2,3-IO’9 8,64 54
Cd(CN)2 [Cd2+]-[CN“]2 — 1,0-10-8 8,0 6, 54
CdCO3 [Cd2+]-[COf“ ] — 5,21012 11,28 6
1,0-10“12 12,0 54
25 2,5-10“14 13,60 1,6-10"7 2,76-10“б 3, 5, 7, 8, 9, 22
25 5,25-10’9 2,29106 3,95-106 4
CdC2O4 (оксалат) [Cd2+].[C2O$-] Комн. 1,1-10"8 7,96 1
25 1,96-10’8 7,71 1,40-10"* 2,81-10’3 4
25 2,6-10 9 8,58 5
— 1,5108 7,82 6, 54
18-25 2,9-10’8 7,54 1,7-10"* 3,4-103 7*
CdC2O4-3H2O [Cd2+].[C2O2-] 18 1,53-10“8 7,82 1,2-10"* 3,05-10“3 2, 3, 22
Cd(C9H6ON)2 (оксихинолинат) [Cd2+][C,H6ON-]2 25 1,00-10“22 22,00 4,21
Cd2Fe(CN)6 [Cd2'|2-[ Fe(CN)2- ] — 3,2-10“17 16,49 3,2-10’6 6,35
4,2-10 18 17,38 54
Cd(NH3)6(BF4)2 [Cd(NH3)2* ]-[bf; ]2 — 2-Ю"6 5,7 6,54
Cd(OH)2 [Cd2+]-[OH~]2 25 1,2-10"14 13,92 1,4-10’5 2,04-10"* 3
18-25 5,7-1045 15,24 1,8-10’5 2,6-10"* 71
20-25 2,5-10"14 13,60 8
18 2,4-10’13 12,62 3,9-10“5 22
— — 13,5-14,2 5
[CdO2H-][H‘] — 2-Ю’19 18,7 5, 54
Cd(OH)2 неакт. форма [Cd2+]-[OH“]2 25 3,98-10“15 14,40 1,07-10 5 1,57-10"* 42
Cd(OH)2 свежеосажд. [Cd2+]-[OH“]2 — 2,2-10’14 13,66 6, 54
Cd(OH)2 после старения [Cd2+]-[OH“]2 — 5,9-10’15 14,23 6, 54
Cd3(PO4)2 [Cd2+]3-[PO3-]2 25 2,51 -10~33 32,60 1,18-107 6,24-106 4
CdS [Cd2+]-[S2“] Комн. 4-10"29 28,4 1
18 3,6-10-29 28,56 6,0-1045 8,67-10’14 2, 3, 7, 10
25 7,94-1027 26,10 8,91-10’14 1,151012 4, 6,8
— 1,6-10-28 27,8 54
25 1-Ю’29 29,0 5
18-25 — 28 9
CdSeO3 [Cd2+]-[SeO2“ ] — 1,30-1О’9 8,89 6
5,0-10’9 8,30 54
CdWO4 [Cd2+]-[WO2-] 25 2,7-1043 5,57 1,64-1О3 4,74-10”2 4
— 2-Ю”6 5,7 6, 54
Химические методы количественного анализа
403
Продолжение таблицы 5.1.5
Соединение Ионное произведение t, °C ПР рПР = =-ignp S' Литера-тура
моль-л 1 г/100 мл
Се2(С2О4)3 (оксалат) см. также табл. 5.1.11.1.В [Се3+]2-[С2О2“]3 Комн. 2,6-1О29 28,58 1,5
2,5-10’29 28,60 54
Се2(С4Н4О6)з (тартрат) [Се33]2[С4Н4О2~ ]3 Комн. 9,7-10’20 19,01 1
CeF3 [Се’ЧДТ 25 1,4-10’18 17,85 см. табл. 5.1.11.1.Ж
25 8,1-10“16 15,09 «
25 1,1-10’15 14,96 «
Се(Ю3)з [Се3+]-[Ю3]3 Комн. 3,5-10“10 9,46 1
25 1,15-10“9 8,94 2,56-Ю3 1,70-10"1 4
— 3,2-1О“10 9,49 6, 54
см. табл. 5.1.11.1.И
Се(Ю3)4 [Се4+]-[Ю;]4 — 5-Ю’17 16,3 6, 54
20 3,5-10~10 9,46 4,3 -103 22
Се(ОН)3 [Се3+]-[ОН“]3 см. табл. 5.1.11.1.Д
25 6,32-10“22 21,20 2,21 -106 4,2-105 4
— 1,5-1020 19,82 6
— — 22,3 5
СеО2 [СеО2+]-[ОН“]2 — 1-10"24 24,0 6
1-10-20 20,0 54
СеО2 [Се4н]-[ОН“]4 — 2,5-10“51 50,60 6
1,6-10’48 47,8 54
— — 54,8 5
Ce2(SO4)3 [Ce3+]2-[SO2“]3 — 2-102 1,7 6
Ce2(SeO3)3 [Ce3+]2-[SeOj“ ]3 — 3,7-10"25 24,43 6, 54
Co3(AsO4)2 [Co2+]3-[AsO3-]2 25 7,6-10'29 28,12 2,34-10 6 1,06-104 4, 6, 54
СоСОз [Co:i] [CO; ] 25 1,45-10’13 12,84 3,80-10 7 3,94106 4
25 1-Ю'12 12,0 5
— 1,4-10 13 12,85 6
1,05- Ю10 9,98 54
СоС2О4 [Со2+]-[С2О4“] — 6,3-10“8 7,20 6, 54
25 8,53-10'8 7,07 2,92-104 4,29-103 4
Co(C9H6ON)2 (оксихинолинат) [Co2+]-[C9H6ON“]2 1,59-10’25 24,80 4,21
Co2Fe(CN)6 [Co2+]2-[Fe(CN)3- ] — 1,8-10'5 14,74 1,2-10’5 6,35
4,8-10’38 37,32 54
CoHg(SCN)4 [Co2*H Hg(SCN)4 ] — 1,5-Ю-6 5,82 6, 54
Со(Юз)2 [Со2+][Ю3]2 — 1-Ю"4 4,0 «
Co(NH3)6(BF4)2 [Co(NH3)24]-[BF4~]2 — 4-Ю-6 5,4 «
Co(NH3)6(ReO4)3 [Co(NH3)3*][ReOJ3 — 1,7-1012 11,77 «
Со(ОН)2 [Co2+]-[OFT]2 20-25 2,0-10’16 15,70 3,7-Ю6 3,44-105 3
18-25 3-10’16 15,52 4-Ю-6 3,7105 71
— — 14,2-15,7 5
[СоО2Н].[1Г] — — 19,1 5
Со(ОН)2 голуб. [Co2+]-[OFT]2 25 6,31-Ю’15 14,20 1,16-10’5 1,08-104 4, 6, 8, 54
404
ds Ф Новый справочник химика и технолога
Продолжение таблицы 5.1.5
Соединение Ионное ' Произведение t,°C ПР рПР = = -lgIIP 5 Литера-тура
моль-л 1 г/100 мл
Со(ОН)2 розов., свежеосажд. ^2HU|C(F]-[OH“]2 ;HOkO ; — 2-1 O’15 14,70 6
1,6-1015 14,80 54
25 1,59-10’15 14,80 7,35-106 6,83-10’5 4,8
Со(ОН)2 розов., после старения [Co2+]-[OH~]2 i 25 2-1 O’16 15,7 3,68-10^ 3,42-1 O’5 4, 54
Со(ОН)3 [Co3+]-[0H“]3 — 4-1045 44,4 6, 54
25 I-IO^43 43,0 7,8-1012 8,58-10~8 4
18-25 2,4-10“14 13,62 2,9-10”5 3,2-1 O’4 71
— — 44,5 5
СоНРО4 [Co2+][HPO2- ] 25 7,03-IO’7 6,15 6,73 10 4 1,04-1 O’2 42
Со3(РО4)2 [Co2+]3-[PO3-]2 25 2,00- IO’35 34,70 4,5-1 O’8 l,65-106 4
CoS 18 3-1 O’26 25,52 2 ..
18-25 3,7-10“28 27,43 2-10’14 1,7-10“13 71
CoS а .0(I);nl{C02+]-[S2"] 57c-rL5 7-10’23 22,15 8,4-1 O’12 7,64-1 O’11 3
25 3,98-1 O’21 20,4 6,31-IO’11 5,74-1O“10 4, 6, 8, 54
CoS Р [Co2‘].[S2] 20 2-1 O’27 26,7 4,5-10“14 1,27-1 O’13 3,5
2-1 O’25 24,70 4,47-1 O’13 4,06-1 O’12 4, 8, 54
CoSeO3 CV'[Co2+]-[SeO32’] — 1,6-1 O’7 6,80 6, 54
Cr(AsO4) [Cr’I-tAsOj-] — 7,8-1 O’21 20,11 8,33-10“n 1,69-1 O’9 4, 6, 54
Cr(NH3)6(BF4)3 [Сг(мн3)’*][вр;]3 — 6,2-1 O’5 4,21 6, 54
Cr(NH3)6(MnO4)3 .•CrtNii.jr ! Mno.r — 4,0-10’8 7,40 «
Cr(NH3)6(SO3F)3 [Cr(NHXHSO3F]3 — 4,3-1 O'4 3,37 «
Cr(NH3)6(ReO4)3 [Cr(NH3)3+ ].[ReO4- ]3 — 7,7-IO12 11,11 «
Cr(OH)2 [Cr^HOH-]2 — 1,0-1 O’17 17,00 5, 6, 54
18 2-1 O’20 19,7 5
Cr(OH)3 jCr' HOU f Комн. 1,0-1 O’30 30,0 ; 1
17 5,4-1031 30,27 1,2-1 O’8 1,24-IO7 3,22
25 6,31-10’31 30,20 1,24-10“8 1,27-1 O’7 4, 6, 54
18-25 6,7-1031 30,17 2,9-1 O'8 3-10 7 71
20-25 1-10’31 31,0 8
— — 30 5
[СЮН2+НОН~]2 6,3-10’21 20,20 6
7,9-1 O’21 20,10 : 54
57^ro3H^]-[H+] — 4,0-10~15 14,4 5, 54
CrPO4 зелен. [Cr3+].[po7] 20 2,40-1 O’23 22,62 4,90-1 O’12 7,2-1 O’11 4, 6, 54
CrPO4 фиол. o;v;c(c?thpo3-] 20 1,00-IO"17 17,00 3,16-10 9 4,65-1O’8 «
CsAuCU jc^uci;] — l-10~3 3,0 6, 54
CsBF4 JISyvqcVH 6f; ] — 2-10’5 4,7 5 « '
CsBH4 no^3[Cs+]-[BH;] — 2,5-10“7 6,60 i . <{
CsBrO3 [Cs+]-[BrO3] — 2-102 1,7 «
CsC103 [С8+]-[фЮ,-] — 4-1 O’2 1,4 «
CsC104 ] — 4-1 O’3 2,4
Cs3Co(NO2)6 OTCsTfC^NO^’-] — 5,8-10~16 15,24 * «
CsHgCl3 mtHgCi;] — 2-10’3 2,7 «
Химические методы количественного анализа
405
Продолжение таблицы 5.1.5
Соединение Ионное произведение t, °C ПР рПР = = -lgIIP 5 Литература
моль-л 1 г/100 мл
CsIO3 [Cs+]-[IO3-] — 1-10'2 2,0 6, 54
CsIO4 [Cs+]-[io;] — 4,4-10’3 2,36 «
CsMnO4 [Cs+]-[Mno;] — 9,l-10“5 4,04 «
Cs2PtCl6 [Cs+]2-[PtCl2“ ] — 3-10'8 7,52 «
Cs2PtF6 [Cs]=[ ptp;>- ] — 2,39-10-6 5,62 «
CsReO4 [Cs+]-[ReO4] — 4,0-10"* 3,40 «
Cs2SiF6 [Cs+]2-[SiF62-] — 1,26-105 4,90 «
Cs2SnCl6 [Cs+]2-[SnCl’~ ] — 3,6-10-8 7,44 «
Cu3(AsO4)2 [Cu2+]3-[AsO34-]2 25 7,6-10-36 35,12 9,32-10 8 4,37-10’6 4, 6, 54
CuBr [Cu+]-[Br“] Комн. 4,1-10"8 7,39 1,5,9
18-20 4,15-10“8 7,38 2
25 5,3-10’9 8,28 7,3-10’5 1,05-10-3 3, 8
25 5,25-10’9 8,28 7,24-10-5 1,04-10-3 4, 6, 54
CuCN [Cu+]-[CN-] — 3,2-10’20 19,49 6, 54
CuCO3 [Cu2T[COM 25 2,34-10 10 9,63 1,37-10’5 1,69-10"* 4,8
25 1,4-10-10 9,85 1,2-105 1,4-10"* 5,7'
— 2,5-Ю10 9,60 6, 54
CuC2O4 (оксалат) [Cu27[c:o; ] Комн. 2,9-108 7,54 1,2 (25 °C), 5
25 4,1-Ю’8 7,39 2,02-104 3,06-1 О’ 4
18-25 2,7-10’8 7,57 1,7-104 2,5-103 71
— З-Ю-8 7,5 6, 54
Cu(C6H5N2O2)2 (купферонат) [Cu2+]<c6h5n2o;]2 1,00-1 о16 16,00 4,21
Cu(C9H6ON)2 (оксихинолят) [Cu2+]-[C9H6ON"]2 2,51 • 1О-30 30,60 4,21
CuCl [Cu+]-[CF] Комн. 1 -10 6 6,0 1,5 (18 °C), 9
18-20 1,02-1 О’6 5,99 2
— 1,2-1 О’6 5,92 6, 54
20-25 1,8-10"7 6,74 8
18-25 9-10 s 7,05 3,1-10"* 6,2-103 7*
CuCrO4 [Cu2t][CiO2] — 3,6-1О'6 5,44 6, 54
CuCr(SCN)4(NH3)2 — — 3,67-10’9 8,44 24 (с. 22)
Cu2Fe(CN)6 [Cu+]2.[Fe(CN):-] — 1,3-10-16 15,89 5,1-Ю"6 6, 35, 54
CuHg(SCN)4 [Cu2+]-[Hg(SCN)4~ ] 18 3,3-10"8 27
Cui [Си+нп Комн. 5-10-12 11,3 1,5 (18 °C)
18-20 5,06-10-12 11,30 2,35106 4,38-10-5 2,3
25 1,10-10’12 11,96 1,05-10-6 2,00-10-5 4, 6, 8, 54
1-Ю'8 8,0 24
Cu(IO3)2 [Cu2+][io;]2 Комн. 1,4-10"7 6,85 1, 2(25 °C)
— . 7,4-10"8 7,13 6, 54
CuN3 [Cu+hn;] — 5,0-10’9 8,30 6, 54
406
Новый справочник химика и технолога
Продолжение таблицы 5.1.5
Соединение Ионное произведение t, °C ПР рПР = = -ignp 5 Литера-тура
моль-л 1 г/100 мл
СиОН (Си2О) [Cun-IOJT) 20-25 1-Ю-14 14,0 5, 6, 8, 54
Си(ОН)2 fCu2+]-[OET]2 Комн. 1,4-10’19 18,85 1
25 5,6-1 O’20 19,25 2,4-10-7 2,34-Ю6 3, 5, 71
— 2,2-IO-20 19,66 6
8,3-10’20 19,08 54
20-25 2,6-1 O'19 18,58 8
18-25 19,8 9
— — 19,7 5
1,31-ю-20 19,88 26
[CuOH+]-[OH] 2,2-1013 12,66 6
8,3-Ю-12 11,08 54
[CuO2H]-|H| — 1-10'9 19,0 5, 54
(СиОН)2СО3 [CuOH+]2[CO2- ] — 1,7-1 O’34 33,77 6, 54
Си3(РО4)2 [Cu2+]3-[PO3-]2 25 l,26-10-37 36,90 1,63-Ю-8 6,22-Ю-7 4
Си2Р 2О2 [Cu2*]2-[P;O^] — 8,3-10-16 15,08 6, 54
(8,4 ±2,5)- •10-16 24, 25
CuS [Cu2*J[S2-] Комн. 8,5-10^5 44,07 9,2-Ю-23 8,8-10 22 1, 2(18 °C) 3,5, 7, 9
25 6,31-IO"36 35,20 2,51-Ю-18 2,4-10 17 4, 6, 54
20-25 6-Ю-36 35,22 8
Cu2S [Cu+]2-[S2-] Комн. 2-10^*7 46,7 1,7-10-16 2,71-Ю-15 1,2(16-18 °C), 3, 5 (17 °C)
25 2,51-10^K 47,60 8,56-Ю-17 1,36-Ю-17 4, 6, 54
18-25 2-1O50 49,70 4,5-10-17 7,1-10-16 71
18-25 47 9
CuSCN [Cu+]-rSCN“] Комн. 1,6-ю-11 10,80 1, 2(18 °C) 5,9
— 4,8-10-15 14,32 6, 54
18-25 l,7-10-12 11,77 4,1-Ю-6 5-Ю-5 71
20-25 2-Ю-13 12,7 8
l,38-10-13 12,86 24, 28
CuSe [Cu2-]-[Se!J — l-10^9 49,0 6, 54
CuSeO3 [Cu2+].[SeO2-] — 2,l-10-8 7,68 6
1,7-10-8 7,78 . 54
CuWO4 [Cu2*|.[wo;-] — 1Ю-5 5,0 6,54
25 2,52-10-5 4,60 5,02-10-3 1,56-Ю-1 4
Dy(OH)3 [Dy’ ]-[OH ]’ 25 (1,3-10-26) См. табл. 5.1.11.1.Д
Eu(OH)3 [Eu3+]-[OH-]3 25 2,88-10-27 26,54 1,02-Ю-7 2,06-Ю-6 4
3,4-10-22 21,47 См. табл. 5.1.11.1.Д
0,9-10-23 23,05 «
FeAsO4 [Fe3+]-[ AsO2- ] 25 5,8-10-21 20,24 7,61-Ю-11 1,48-Ю-9 4, 6, 54
FeCO3 [Fe2+].[CO2-] — 3,47-10-11 10,46 2 6,54
18 2,5-IO-11 10,60 5,0-Ю-6 5,79-Ю-5 i - 3, 5
Химические методы количественного анализа
407
Продолжение таблицы 5.1.5
Соединение Ионное произведение t,°C ПР рПР = =-ignp S Литература
моль-л 1 г/100 мл
25 2,09-1 O’11 10,68 4,57-Ю-6 5,30-10'5 4
18-25 10,6 9
FeC2O4 (оксалат) [Fe2+]-[C2OM 25 2-1 O’7 6,7 5, 6, 54
25 2,l-10"7 6,68 4,6-10^ 2, 22
25 3,05-1 O’7 6,52 5,52-1 O^4 7,94-10-3 4
Fe(C6H5O2N2)3 (купферонат) [Fe3+]-[C6H5O2N-]3 l,00-10“25 24,00 4,21
Fe(C9H6ON)2 (оксихинолят) [Fe2+]-[C9H6ON-]2 1,59-10 19 18,80 4,21
Fe4[Fe(CN)6]3 [Fe3+]4-[ Fe(CN)g“]3 3,0-10^' 40,52 6, 8, 54
Fe(OH)2 [Fe2+]-[OH-]2 18 1,64-10 14 13,79 2
20 6,3-1016 15,20 4,9-10-6 4,50-10'5 3
25 7,94-10~16 15,14 1,05-105 9,45-105 42
18 3,2-10 14 13,49 9
— — 15,1 5
— 1-10’15 15,0 6
7,1 10 15 15,15 54
20-25 8-10 16 15,10 8
4,8-1016 15,32 4,9-IO-6 4,4-105 10
[FeOH+]-[OFT] 5-1O’10 9,3 6
2,2-10~H 10,65 54
[FeO2H-].[H+] — 8-10“20 19,1 5, 54
Fe(OH)3 [Fe31-[OH"]3 Комн. l,l-10’36 35,96 1, 2(18 °C)
3,8-1 O’38 37,42 1,9-IO-10 2,03-1О'8 3,7, 10
25 3,72-10 40 39,43 1,8-10~9 1,93-10'8 42
25 4 103x 37,40 5
3,2-1 O’38 37,50 6
20-25 2,5-10“39 38,60 8
18-25 — 37,4 9
Fe(OH)3 свежеосажд. [Fe3+]-[OH“]3 6,3-10’38 37,2 54
Fe(OH)3 после старения [Fe3+]-[OFT]3 3,2-10^° 39,5 54
[FeOH2+]-[OH“]2 2-1 O’26 25,7 6
210 28 27,7 54
[Fe(OH)2 ]-[OFT] 4-1 O’17 16,40 6
6,8-1018 17,17 54
FePO4 [Fe’-Ш'О; ] 25 1,29-IO"22 21,89 1,14-Ю’11 1,71-Ю-10 4
20-25 1,30-1 O’22 21,89 6, 8, 54
FeS [Fe2+]-[S2~] 18 3,8-10’19 18,42 6,1-10 10 5,36-10'9 3
25 5,13-10 18 17,29 2,27-10“9 1,99-108 4
5-1018 17,3 6, 54
18 3,7-1019 18,43 2,5
20-25 6,3-IO"18 17,20 8
18-25 18,4 9
FeS2 [Fe2*]-[sri 6,3-10^31 30,20 6, 54
FeSe [Fe2+]-[Se2-] 1-1026 26,0 «
Fe2(SeO3)3 [Fe3+]2-[SeO2’ ]3 2-10~31 30,7 «
408
Новый справочник химика и технолога
Продолжение таблицы 5.1.5
Соединение Ионное произведение /, °C ПР рПР = =-ignp S Литера-тура
моль-л 1 г/100 мл
Ga4[Fe(CN)6]3 [Ga3+]4-[ Fe(CN)J ]3 1,5-10-34 33,82 1,5-10"5 6, 32, 35
Ga(OH)3 [Ga3+]-[OH~]3 Комн. 110“35 35,0 1,31
7,1-10-36 35,15 6, 29
1,6-1О37 36,8 54
1-Ю"34 34,0 ' 30
[H+]-[H2GaOj ] 2,5-10-1 10,6 54
GeO2 [Ge4+]-[OH“]4 1-Ю’57 57,0 6, 54
GeS [Ge2+]-[S2-] З-Ю’35 34,5 6, 54
Ge(OH)2(C9H12O5)2 (фенилфлуоронат) [Ge(OH)=-][C,ni2O; ]2 20 2,1 -10-28 27,68 33
Ge(OH)2(C]6H13O5)2 [Ое(ОН)3-][С16Н„О;]2 20 8,8-1024 23,06 33
Gd(OH)3 [Gd3+]-[OH~]3 25 2,1 -Ю22 21,68 См. табл. 5.1.11.1.Д
1,8-10"23 22,74 См. табл. 5.1.11.1.Д
GdF3 [Gd3*]-[F-]3 25 6,7-10“17 16,17 См. табл. 5.1.11.1.Ж
Hf(OH)4 [Hf(OH)2+ ]-[OH~]2 25 3,98-10'26 25,40 2,15-Ю 9 5,3110-8 4,6
Hg2Br2 [Hg2’]-[Br-]2 Комн. 1,3-Ю21 20,89 1,2,5
25 5,2-10’23 22,28 2,8-108 1,57-10"6 3
5,75-10“23 22,24 1,38-10"6 7,73-10'5 4
5,8-IO23 22,24 6,34(20 42), 54
3,4-IO22 21,47 7,0 10 8 3,9-10б 71
Hg2CO3 [Hg2+]-[CO2~ ] 20 8,9-10“17 16,05 6, 34, 54
25 9,0-1017 16,05 2,8-10-6 1,29-10"* 3,5
8,91-1017 16,05 2,81-10^ 1,3040"* 4
[Hg+]2-[ co2- ] 18-25 7,3-10-25 24,14 9-10-9 4,1-10“7 7*
Hg2c2o4 [Hg2+].[C2O2-] 20 1-10 13 13,0 3,16-10 7 1,55-Ю-5 4, 6, 34, 54
HgC2O44 [Hg2+].[C2O2-] 18-25 7,8-10 8 7,11 2,8-1 О’4 1,1-Ю-2 71
Hg2C4H40e [Hg2+]-[C4H4O2-] 20 I-IO-10 10,0 34
Hg2(CH3COO)2 [Hg;*][CH3COO']! 20 3,6-10~10 9,44 34
18-25 — 2,9-10-2 0,75 7*
Hg2Cl2 [Hg2+]-[cr]2 Комн. 3,1-10’18 17,51 1,9
25 2-Ю’18 17,7 2,5
20 1,Ь10’18 17,96 1,6-10-5 7,55-10"* 3
25 1,32-Ю18 17,88 6,91-10~7 3,25-105 4, 6, 34(20 °C), 54
18-25 1,2-I019 18,92 4,9-10-7 2,3-10’5 71
Hg2CrO4 [Hg223HQO42-] — 5-Ю’9 8,30 4 (акт.), 6, 34, 54
25 1,13-10~8 8,95 1,06-104 5,5-10 3 4
[HgTl'CrO; ] 18-25 2-Ю’9 8,7 5Ю~5 2,6-10"3 71
Hg2I2 [Hg22+]-[r]2 Комн. 1,2-10-28 27,92 5,2-10-8 3,41-10^ 1, 2(25 °C) 3(25 °C), 5
25 4,47-10-29 28,35 2,24-1 О*10 1,46-108 4
Химические методы количественного анализа
409
Продолжение таблицы 5.1.5
Соединение Ионное произведение t, °C ПР рПР = = -lgnP S Литература
моль-л 1 г/100 мл
4,5-IO-29 28,35 6, 34, 54
3-ю-29 28,52 3,1-IO’10 2 10“8 71
Hgl2 [Нё2+]-[Г]2 18-25 6,8-10~13 12,17 8,8-1 O’5 4,0- IO-3 71
Hgl24 [Hg2+Hn2 Комн. 3,2-10“29 28,49 1
Hg(IO3)24 [Hg2+]-[IO3 ]2 20 3,15-1013 12,50 34
Hg2(IO3)2 [Hg22+]-[IO3 ]2 20 2,45-IO-14 13,35 6, 34, 54
HgN3 [Hg22*]-[N;]2 20 7,1-IO-10 9,15 34
Hg2O [Hg22+]-[OH~]2 20 1,6-10 23 22,80 1,5910 s 6,92-IO-7 4, 6, 34
18 7,8-1024 23,11 5
HgO [Hg2+]-[OH-]2 Комн. 1,4- IO’26 25,85 1,9
25 3-10-26 25,52 1,95-IO-8 4,57-IO’7 4, 6, 34, 54
18 1-1026 26,0 5
Hg2HPO4 [Hg22+]-[HPO2~ ] 20 4-10-13 12,4 6, 34, 54
HgS [Hg2+]-[S2-] Комн. 4-1053 52,4 6,3-IO27 1,47-IO’25 1,3,5
18 4-10-53 -2-IO*49 52,4- -48,7 2
HgS черн. [Hg21-[S2-] 25 1,59-10 52 51,8 4, 6, 8, 54
HgS красн. [Hg2+]-[S2-] 25 3,98-10 53 52,40 4, 6, 8, 54
Hg2S [Hg22+]-[S2-] 20 l-10^7 47,0 6, 34, 54
25 l-10^5 45,0 5
[Hg7[s2n 18-25 l-10^5 45,0 1-1015 4,3-IO’14 7l
Hg2(SCN)2 [Hg22+]-[SCN“]2 3-10-20 19,52 6, 54
[Hg+]-[SCN“] 18-25 4-IO-14 13,4 210-7 5,2-Ю-6 71
Hg2SO3 [Hg22+].[SO|-] 20-25 1-1027 27,0 3,16- IO-14 1,52-Ю’12 4, 6, 34, 54
Hg2so4 [Hg22+].[SO2-] 25 1,11-10^ 5,95 l,05-10-3 5,22- IO-2 4
6,8-107 6,17 4 (акт.), 6, 54
20 4,8-10~7 6,32 5(25 °C), 34
HgSe [Hg2+]-[Se2-] 20 1-1059 59,0 6, 34, 54
Hg2SeO3 [Hg22+]-[SeO3“ ] 20 6,3-IO15 14,20 6, 34, 54
Hg2SeO3*5 [Hg,2l].[SeO,'l 20 l,5-1014 13,82 34
Hg2WO4 [Hg22+]-[ WO2- ] 20 l,l-1017 16,96 6, 34, 54
Ho(OH)3 [Ho3+]-[OH-]3 25 2,82-10 27 (акт.) 26,55 См. табл. 5.1.11.1.Д
In4[Fe(CN)6]3 [In32l4[Fe(CN)l]3 25 1,9-IO"*4 43,72 5,7-10-7 6, 35, 54
In(IO3)3 [In3+][IO-]3 3-10~3 2,5 6, 54
In(OH)3 [In3+]-[OH-]3 Комн. 1-1033 33,0 1,32, 36(25 °C)
25 6,3-1034 33,20 2,2-10“9 3,65-lQ-8 4
5-10-34 33,3 6
1,2-IO-37 36,92 54
[In(OH)2‘][OJT]2 1,2-IO-27 26,92 «
[H+][H2InO3 ] i-io-16 16 «
410
Новый справочник химика и технолога
Продолжение таблицы 5.1.5
Соединение Ионное произведение t, °C ПР рПР = =-ignp S Литера-тура
моль-л 1 г/100 мл
In2Sj [In3+]2-[S2-]3 1-Ю-88 88,0 6
5,75-10 74 73,24 54
1гО2 [Ir4+]-[OH“]4 1,6- ю72 71,80 6, 54
1г20з [Ir3+]2-[OH]-6 2-10 48 47,7 «
IrS2 [Ir4+]-[S2-]2 НО-75 75,0 «
K3A1F6 [kTiaif’-] 25 9,1-IO’9 8,03 5,4-104 0,14 См. табл. 5.1.ПАБ
1,6-1 O'9 8,80 6, 54
kbf4 [K+]-[BF4-] 2-10’3 2,7 6, 54
0 1,67- I04 3,78 l,510~2 0,185 См. табл. 5.1.ПАБ
14 5,62-1 O'4 3,25 2,76104 0,347 «
20 1,36-104 3,87 4,50-Ю4 0,566 «
КВ(С6Н5)4 [K+].[B(C6H5);] 2,25-108 7,65 1, 6, 54
25 5-10’9 8,3 7,l-104 3,6-IO’3 См. табл. 5.1.ПАБ
20 1-Ю’8 8,0 1-104 5,3-IO-3 «
20 1,6 10 s 7,80 1,26-104 6,4-Ю4 «
25 3,3-IO’8 7,48 l,5-104 9,3-IO’3 «
КВН4 [K+]-[BHj 1,3-10’3 2,89 6, 54
KC6H2N3O7 (пикрат) [K+]-[C6H2N3O- ] 0 5,6-105 4,25 8,2-104 0,22 См. табл. 5.1.ПАБ
KC12H4N70i2 (дипикриламинат) [K+]-[C12H4N7O,-] 0 2,7-1 O’7 6,57 5,2-IO4 0,025 См. табл. 5.1.ПАБ
KC12H5N8O12 (монокалийгекса-нитригидразо-бензол) 20 4,2-IO40 9,38 2,05-10“5 0,001 «
КНС4Н4О6 (битартрат) [k+]-[hc4h4o;] Комн. 3-104 3,5 1,7-104 0,32 1.71
0 1,3-ю-4 3,89 1,25-IO4 0,266 См. табл. 5.1.ПАБ
10 2-1 O'4 3,7 1,6-IO4 0,342 «
18 2,2-Ю4 3,66 1,7-102 0,360 «
КС1О4 [K+]-[Cio;] -— 1,1-10 2 1,96 6, 54
0 2,0-10 3 2,70 5,5-IO4 0,76 См. табл. 5.1.ПАБ
14 4,7-10’3 2,33 8,9-IO4 1,24 «
K3[Co(NO2)6] [KT-[Co(NO:)i] — 4,3- Ю40 9,37 6, 54
17 5,4-1042 11,27 1,96-Ю-3 0,089 См. табл. 5.1.ПАБ
K2Ag[Co(NO2)6] [KT[Ag+][Co(NO2)’~] 25 3,9-1048 17,61 3,1-Ю4 1,64-IO’3 «
K2Na[Co(NO2)6] [K+]2-[Na+]-[Co(NO2)3-] 0 3,4-1042 11,47 9,6-Ю4 0,042 «
2,2-IO41 10,66 6, 54
Комн. 2,0-1041 10,70 2,0-IO4 8,7-Ю4 1,71
K2Cu2Fe(CN)6 [K+]2-[Cu+]2-[Fe(CN)4_ ] 2,2-1 O’27 26,66 54
K2GeF6 [K+]2-[GeF62-] — 3-Ю’5 4,5 6, 54
K2HfF6 [K+]2-[HfFM — 2-Ю4 2,7 6, 54
Химические методы количественного анализа
411
Продолжение таблицы 5.1.5
Соединение Ионное произведение t, °C ПР рПР = = -lgIIP 5 Литература
моль-л 1 г/100 мл
К[1п(С2О4)2] [К+]-[1п(С2О4)2] 20 4-105 4,4 6,7-10 3 0,221 См. табл. 5.1.11.4.5
К21гС16 [К*]2-[1гС1М — 6,8-10’5 4,17 6, 54
КЮ4 [к+]-[ю;] — 8,3-10^ 3,08 6, 54
0 4,5-10-5 4,35 7,4-10’3 0,17 См. табл. 5.1.11.4.5
18 2,0-10^ 3,70 1,6-10~2 0,37 «
20 2,5-10^ 3,60 1,8-10’2 0,42 «
KNbO3 [K+]-[Nbo;] 25 7,5-10“7 6,12 8,6- W4 0,0155 «
K2PdC14 [K+]2[PdciM — 1,6-10’5 4,80 6, 54
K2PdCl6 [K+]2-[PdClM 25 6,0-1 О’6 5,22 1,5-10’2 0,596 6, 54, см. табл. 5.1.11.4.5
K2PtCl4 [K+]2-[PtCl2-] 8-103 2,1 6, 54
K2PtCl6 [K+]2-[PtCl2-] 1,110“5 4,96 6, 54
Комн. 1-Ю’5 5,0 1
0 6-10^ 5,22 1,5-102 0,74 См. табл. 5.1.11.4.5
10 1,0-10’5 5,00 1,8-10“2 0,90 «
20 2,0-10’5 4,70 2,3-10’2 1,12 «
K2PtF6 [K+]2-[PtF2-] — 2,9-10’5 4,5 6, 54
KReO4 [K+].[ReO;] — 1,9-10’3 2,72 6, 54
0 1,4-1 О’6 5,85 1,2-10 3 0,036 См. табл. 5.1.11.4.5
30 2,2-10’5 4,66 5,1-Ю’3 0,147 «
50 9,9-10’5 4,00 1,1-10’2 0,221 «
K2SiF6 [K+]2.[SiF2~] — 8,7-1О'7 6,06 6, 54
0 1,1-10’7 6,96 3,5-10“3 0,077 См. табл. 5.1.11.4.5
25 1,1-Ю"6 5,96 8,0-10’3 0,177 «
KTaO3 [K+]-[TaO3] 0 1,9-10’9 8,82 4,3-10’5 1,16-10'3 «
25 2,4-10’9 8,62 4,9-10’5 1,30-10'3 «
K2ThF6 [K+]2-[ThF62“] 25 1,110 й 10,96 1,4-10’6 6-10’5 «
K2TiF6 [K+]2[TiF2-] 5-10^ 3,3 54
K2ZrF6 [K+]2-[ZrFM — 5-10"* 3,3 6, 54
Ьа(ВгОз)з [La3*]-[BrO- f — з-ю3 2,5 6, 54
Ьа2(СОз)3 [La3+]2-[CO3“ ]3 4-Ю’34 33,4 54
Ьа2(С2О4)з [La3+]2-[C2O2-]3 — 2,5-10-27 26,60 6
1-Ю’25 25,0 54
25 1,4-10”28 (акт.) 27,85 См. табл. 5.1.11.1.В
25 2-10’28 27,7 5
Ьа2(С4Н4Об)з (тартрат) [La3+]2-[C4H4O2-]3 2-10’19 18,7 1
LaF3 [La3+]-[F-]3 25 1,4-10-18 17,85 37
См. также табл. 5.1.11.1 .Ж
412
Новый справочник химика и технолога
Продолжение таблицы 5.1.5
Соединение Ионное произведение t,°C ПР рПР = =-ignp S Литера-тура
моль-л 1 г/100 мл
La(IO3)3 [La3+]-[IO;]3 Комн. 5,9-Ю“10 9,23 1
25 1,28-10" 10,89 8,29-10" 5,50-Ю’2 4
— 6,3-1 о"2 11,20 6, 54
См. также табл. 5.1.11.1.3
La2(MoO4)3 [La3+]2-[MoO2’]3 — 4-10’21 20,4 6
2,2-10’21 20,66 54
La(OH)3 [La3+]-[OH-]3 25 1,0-10’19 19,00 См. табл. 5.1.11.1.Д
25 5,0-Ю“21 20,30 «
25 5,5-10’22 21,26 «
25 6,0-1 о-20 19,22 «
25 4,З-Ю'19 18,37 «
25 1,7-10’19 18,77 «
18 1,2-Ю’19 18,92 «
18 1,1-Ю-21 20,96 «
25 0,9-10’21 21,05 «
25 2,00-10’19 18,70 9,27-10" 1,76-10" 4,6
La(OH)3 свежеосажд. [La3+]-[OH-]3 6,5-Ю’20 19,19 54
La(OH)3 после старения [La3+]-[OH”]3 2-Ю’22 21,7 54
Ln(OH)3 [Ln3+]-[OH-]3 3,2-Ю’23-1-Ю’27 22,5-27,0 54
La2S3 [La3+]2-[S2-]3 — 2,0-10’13 12,70 6, 54
La2(SO4)3 [La3+]2-[SO2-]3 — З-Ю’5 4,5 «
Li2CO3 [Li+]2-[CO2-] — 3,98-Ю’3 2,40 «
LiF [Li+]-[F-] — 3,8-Ю’3 2,42 6
1,7-Ю’3 2,77 54
LiOH [Li+]-[OFT] — 4-Ю’2 1,4 6, 54
25 6,25-10’2 1,20 0,3425 0,8202 42
Li3PO4 [Li+]3[PO=] — 3,2-Ю’9 8,49 6, 54
Mg3(AsO4)2 [Mg2+]3-[ AsO3- ]2 20 2,09-Ю-20 19,68 4,54-Ю’5 1,59-Ю’3 4, 6, 54
MgCO3 [Mg2+]-[COj" ] Комн. 2-10" 3,7 1
12 2,6-10’5 4,58 2, 5(12 °C)
20 1,0-Ю’5 5,0 3,2-Ю’3 2,7-Ю’2 3, 4(акт.), 8(20-25 °C)
25 3,08-Ю’5 4,51 5,55-Ю’3 7,68-Ю’2 4
— 2,1-Ю’5 4,67 6, 54
18-25 1,7-Ю-6 5,77 1,3-Ю’3 1,1-Ю-2 71
3,8 9
MgCO3-3H2O 25 3,08-Ю-6 5,51 5,55-Ю’3 7,68-Ю’2 4
MgC2O4 [Mg2+].[C2OJ-] Комн. 8,6-Ю’5 4,07 1,6
18 8,57-Ю’5 4,07 9,2-10’3 1,03-Ю’1 2,3,5
25 1,47-Ю-4 3,83 1,65-10’2 0,1852 4
18-25 — — 6,2-10’3 7-10’2 71
18-25 — 4,1 9
Химические методы количественного анализа
413
Продолжение таблицы 5.1.5
Соединение Ионное произведение t, °C ПР рПР = = -lgITP S Литера-тура
моль-л 1 г/100 мл
Mg(C9H6ON)2 (оксихинолят) [Mg2*]-[C,H6ON-]2 Комн. 2,5-10"17 16,6 1
6,31-IO'16 15,20 4,21
MgF2 [Mg2+]-[F-]2 Комн. 7-10'9 8,16 1
18 7,1-IO"9 8,15 2
27 6,4-1 O’9 8,19 2
25 6-IO’9 8,21 5
— 6,5-1 O’9 8,19 6, 9, 54
Mg(IO3)2 [Mg2+]-[IO3-]2 — 3-10"3 2,5 6, 54
MgK2[Fe(CN)6] (Mg2+]-[K+]2-[Fe(CN)4'] — 5-10 9 8,3 6, 54
Mg(NH4)2[Fe(CN)6] [Mg2i] [NH;]2 [ Fe(CN)4-] — 4-10 8 7,4 6, 54
MgNH4PO4 [Mg2i].[Nii4'|.[po;i Комн. 2,5-10 13 12,60 6,3-io5 8,65-10" 1, 2(25 °C) 3, 5-9, 54
25 2,51-10-13 12,60 6,31-IO’5 8,66-10" 4
Mg(OH)2 [Mg2‘].[OH-]2 Комн. 1,2-10 10,92 1, 2(18 °C) 5,9
20 5-1012 11,30 1,1-Ю-4 6,42-10" 3, 71
20-25 1,8-10" 10,74 8
18-25 11,0 5
[MgOH+][OH“] — 2,3-107 6,64 6
2,6-109 8,59 54
Mg(OH)2 стаб. форма [Mg22HOH-]! 25 l,3210-11 10,88 1,49-1 O'4 8,67-10" 4
Mg(OH)2 свежеосажд. [Mg2T[OH-]2 — 6-1 O’10 9,22 6, 54
Mg(OH)2 после старения [Mg2+].[OH-]2 — 1,8-10’11 10,74 6
7,1-10’12 11,15 54
Mg3(PO4)2 [Mg2+]3-[PO3-]2 — 1-10"13 13 6, 54
2,5-10-16 15,60 1,1-10" 6,4-10" 71
MgSO3 [Mg2+]-[SO2-] — 3-10"3 2,5 6, 54
25 1,88-10’2 1,73 0,14 1,43 4
MgSeO3 [Mg24[SeO2~ ] — 1,30-1 O'5 4,89 6
4,4- IO'6 5,36 54
Mn3(AsO4)2 [Mn2+]3-[ AsO3’ ]2 25 1,9-1 O'29 28,72 7,07-10"7 3,13-10" 4, 6, 54
MnCO3 [Mn2+]-[CO2-] — 8,8-10-11 10,06 9,4-10" 1,08-10" 3
25 5,11-1O"10 9,29 2,26-10" 2,60-10" 4
25 1-1O"10 10 5
— l,810-11 10,74 6, 54
18-25 9,2-1 O’7 6,49 5,7-10" 6,5-IO-3 71
20-25 5-1O10 9,3 8
18-25 10,1 9
MnC2O4 (оксалат) [Mn2+]-[C2O2-] 25 4,88-10" 5,31 3,50-10"3 5,01-IO-2 4
— 5-1 O’6 5,3 6, 54
Mn(C9H6ON)2 [Мп2+]-[С9Н6ОЬГ]2 5-1O"20 19,30 4
Mn2[Fe(CN)6] [Mn24]2-[Fe(CN)64-] — 7,9-1 O'13 12,10 6, 54
MnNH4PO4 [Mn2+]-[NH42}[PO’-] — 1-10"12 12 6, 54
6,3-1 O'12 11,20 1,9-10" 3,110-3 71
414
Новый справочник химика и технолога
Продолжение таблицы 5.1.5
Соединение Ионное произведение t, °C ПР рПР = =-ignp 5 Литера-тура
моль-л 1 г/100 мл
Мп(ОН)2 [Mn2+]-[OH-]2 18 410-14 13,4 2,1-10"* 1,87-10"* 2, 3, 5, 7, 10
25 1,70-1 O'13 12,77 3,49-10“5 3,11-10"* 4
18-25 14,2 9
— l,9-1013 12,72 5, 6, 54
[МпОН+НОН-] — 1,5-109 8,82 6
4,9-IO10 9,31 54
[MnO2H-]-[H+] i-io-19 19,0 5, 54
Мп(ОН)3 [Mn3+]-[OH"]3 36 5
b Ю-56 36 6, 54
Мп(ОН)4 [Mn4+]-[OH“]4 Комн. l-10“56 56 1,54
MnS [Mn2+]-[S2“] 18 1,4-10 15 14,85 3,7-10"8 3,22-10"7 2, 3, 5(20 °C), 7
18-25 14,9 9
MnS розов. [Mn2+]-[S2“] 25 2,51-1O“10 9,60 1,59-105 1,38-10"* 4, 65, 8, 54
7-10“16 15,15 17
l,l-1015 14,96 20
MnS зелен. [Mn2+]-[S2“] 25 2,51-10~*3 12,6 5,01-IO"7 4,36-10-6 4, 65, 8, 54
MnSeO3 [Mn2+]-[SeO2“ ] — 1,26-10“7 6,9 6
5,4-1 O’8 7,27 54
Мо(ОН)4 [Mo4+]-[OH~]4 — I-IO'50 50 6
1-10’56 56 54
(NH4)3A1F6 [nh; ]3-[aif’~] — 1,6-10“3 2,8 6, 54
(NH4)3Co(NO2)6 [NHj^cofNOj; -] — 7,6-1 O’6 5,12 «
(NH4)2IrCl6 [NH;]2[bcf ] — 310 5 4,5 «
(NH4)2PtCl6 [NHJ l2[PtCl;.-] — 9-10”6 5,05 «
Na3AlF6 [NaT-IAlF’- ] — 4,1-IO’10 9,39 «
Na2BeF4 [Na+]2-[BeF4“] — 7-1 O’3 2,15 «
NaIO4 [Na+]-[IO4] — 3-10’3 2,5 «
NaSb(OH)6 [Na+]-[Sb(OH)“ ] — 4-1 O’8 7,4 «
NaH2SbO4 [Na+]-[H+]2-[SbO4“] 18-25 1,0-1 O’12 12,0 1,0-10-3 2,0-102 71
Na2SiF6 [Na+]2-[SiF=] — 2,8-10"* 3,56 6, 54
Ni3(AsO4)2 [Ni2+]3-[ AsO34- ]2 25 3,1-IO’26 25,51 3,l-10-6 1,41-10"* 4, 6, 54
Ni(BO2)2 [Ni2+]-[BO-]2 2-109 8,7 54
Ni(CN)2 [Ni2+]-[CN~]2 — 3-1023 22,5 6, 54
8,77 40
23,2 41
NiCO3 [Ni2+].[CO2~] — 1,3-10’7 6,87 6, 54
25 1,63-1 O’7 6,79 4,03-10"* 4,79-10-3 4
25 1,4-10“7 6,85 5
18-25 6,2-10"7 6,21 7,9-10"* 9,3-10“3 7
20-25 6,3-IO”9 8,80 8
NiC2O4 (оксалат) [Ni2+HC20tJ 25 4,42-Ю-10 9,35 2,11-Ю’5 3,10-10"* 4
Химические методы количественного анализа
415
Продолжение таблицы 5.1.5
Соединение Ионное произведение t,°C ПР рПР = = -ignp 5 Литература
моль-л 1 г/100 мл
— 4-10"° 9,4 6, 54
18-25 4,2-1040 9,38 2,05-105 з-ю" 71
Ni(C4H7O2N2)2 (ди-метилглиоксимат) [Ni:].[C4U,O,N,]2 25 23,66 39
40 23,23 39
— 2,3-10’25 24,64 54
Диоксиматы Ni Cm. табл. 5.1.11.12.E
Ni(C9H6ON)2 (оксихинолят) [Ni2+]-[C9H6ON-]2 1,00-10-26 26,00 4,21
Ni(ClO3)2 [Nuncio,]2 — 1-Ю-4 4 6, 54
Ni2Fe(CN)6 [Ni2+]2-[ Fe(CN)64-] — 1,3-10 й 14,89 1-10 5 6,32, 54
Ni(IO3)2 [Ni2+]-[IO3-]2 — 1,4-1 О’8 7,85 6, 54
Ni(NH3)6(BF4)2 [nhnhXhbf;]2 — 1-10" 6 6, 54
Ni(NH3)6(ReO4)2 [Ni(NH3)2+]-[ReO4-]2 — 5,1-Ю-4 3,29 6, 54
Ni(OH)2 [Ni2+]-[OH“]2 20 4,8-1016 15,32 5,4-10" 5,1-10" 3
1,6-10’14 13,80 2,4-10’5 2,2-10’5 71
25 3,16-1016 15,50 4,4-10”6 4,08-10’5 42
См. также табл. 5.1.11.12.Б
Ni(OH)2 свежеосажд. [Ni2+]-[OFT]2 — 2,0-10~15 14,70 6, 54
Ni(OH)2 после старения [Ni2+]-[OH-]2 — 6,3-10"18 17,20 1,17-10" 1,08-10" 42, 6, 54
[NiO2H’]-[H+] 18,2 5
Ni3(PO4)2 [Ni2+]3-[PO^]2 25 5,0110 31 30,30 3,41-10 7 1,25-10" 4
Ni2P2O7 [Ni2+]2-[P2O4~] — 1,7-1013 12,77 6, 54
NiS [Ni2+]-[S2-] 18 1,4-10’24 23,15 2
18-25 2,2-10’21 20,66 4,7-10'" 7-10’10 71
NiS a [Ni2+]-[S2-] 25 3,16-10 19 18,50 5,62-10 10 5,10-10’9 4
3,2-10“19 18,50 6, 54
18-25 3,0-10“19 18,53 8
См. также табл. 5.1.11.12.А
NiSp [Ni2+]-[S2-] 25 1,00-10-24 24,00 1,00-Ю"2 9,08-10“12 4, 6, 8, 54
20 1-10’26 26 5
См. также табл. 5.1.11.12.А
NiS у [Ni2+]-[S2-] 25 2,00-1О'26 25,70 1,41-10“13 1,28-10“12 4, 6, 8, 54
См. также табл. 5.1.11.12.А
NiSeO3 [Nr'HSeOj ] — 1,0-10-5 5,0 6, 54
Nd2(C2O4)3 [Nd3+]2-[C2O2-]3 25 7,7-10’32 31,11 См. табл. 5.1.11.1.В 42 (акт.)
Nd(OH)3 [Nd3+]-[OFT]3 25 8,71-10~24 23,06 7,54-10" 1,47-10" 4
25 1,9-10’21 20,82 См. табл. 5.1.11.1.Д
1,2-10’24 23,92 «
3,2-10"22 21,49 «
NpO2(OH)2 [ NpO2* НОН’]2 — 2,5-10-22 21,6 6
5,0-1023 21,3 54
Pb3(AsO4)2 [Pb2+]3-[ AsO" ]2 25 4,1-10“36 35,39 3,28-Ю”8 2,95-10" 4, 6, 54
Pb(BO2)2 [Pb2+]-[BO2]2 1,7-Ю’11 10,78 54
416
Новый справочник химика и технолога
Продолжение таблицы 5.1.5
Соединение Ионное произведение t, °C ПР рПР = =—igiip 5 Литера-тура
моль-л 1 г/100 мл
РЬВг2 [Pb2+]-[Br]2 25 7,4-10’5 4,13 2,6-10’2 9,54-10' 3
25 2,76-10“5 4,56 2,73-10’2 1,00 4
25 7,4-10-6 5,13 5
— 9,1-Ю”6 5,04 6, 54
18-25 7,9-10-5 4,10 2,3-10’2 0,842 ’ 7*
РЬ(ВгО3)2 [Pb2+]-[BrOj ]2 — 7,99-10-6 5,10 6, 54
РЬСО3 [Pb2+]-[CO2- ] Комн. 3,3-10’14 13,48 1, 2(18 °C) 5,43
1,510 13 12,82 3,9-10’7 1,04-10"5 3
25 7,41-10-14 13,13 2,71-10 7 7,27-10-6 4
— 7,49-10”14 13,13 6, 54
18-25 4,8-Ю12 11,32 2,2-10-6 6,0-10^ 7*
20-25 8-10~14 13,10 8
18-25 13,5 9
РЬС2О4 [Pb2+] [C2O2 ] Комн. 3,4-10’11 10,47 1
18 2,74-10’11 10,56 2,5
18 3,2-10’11 10,49 5,7-Ю"6 1,68-10"* 3
25 5,32-10’10 9,27 2,31-Ю’5 6,82-10"* 4
— 4,8-10*° 9,32 6, 54
18-25 2,9-1011 10,54 5,4-10"6 1,6-10"* 71
Pb(C5H10NS2)2 (диэтилдитиокарбамат) [Pb2*][C5Hl0NS-]2 2-10’22 21,7 43
Pb(C9H6ON)2 (оксихинолинат) [Pb2+]-[C9H6ON-]2 1-Ю’22 21,00 4,21
Pb(C13H11N4S)2 (дитизонат) [Pb2*][C13H„N4S-]2 25 2-10’24 23,7 3-10’9 43 (вода, насыщ. СС14)
РЬС12 [РЬ2+]-[СГ]2 Комн. 2-10"* 3,7 1
20 2,4-10"* 3,62 3,9-1 О*2 1,08 3,7
25 6,02-10"5 4,22 4,13-Ю’2 1,15 42
25 2-105 4,7 5
— 1,6-105 4,79 6, 8 (18- 25 °C), 9, 54
PbCIF [Pb2+]-[CF].[F-] — 2,8-10’9 8,55 6, 54
РЬСгО4 [Pb2+HCrO2-] Комн. 1,8-10~14 13,75 1,3-107 4,2-10-6 1, 3, 5-8, 43 (18 °C), 54
18 1,77-10“*4 13,75 2
25 1,18-Ю"14 13,93 1,33-107 4,31-Ю”6 4
PbF2 [Pb2+]-[F“]2 Комн. 7-1 О*9 8,2 1, 5(20 °C)
9 2,7-10-8 7,57 2, 6, 8(20- 25 °C), 54
18 3,2-10-8 7,49 2
26,6 3,7-10-8 7,43 2
20 2,7-10’8 7,57 0,64 43
Химические методы количественного анализа
417
Продолжение таблицы 5.1.5
Соединение Ионное произведение °C ПР рПР = =-ignp S Литера-тура
моль-л 1 г/100 мл
Pb2[Fe(CN)6] [РЬ2*]2-[ Fe(CN)M — 3,5-10“15 14,46 1,5-10“5 6, 32
9,55-1019 18,02 54
РЫ2 [РЬ2+]-[Г]2 Комн. 1,3-10~8 7,5 1, 5(20 °C)
15 7,47-10’9 8,13 2
25 1,39-10’8 7,86 2
25 8,7-10’9 8,06 1,ЗЮ’3 5,99-10’2 3,7
25 1,34-10’8 7,87 1,51-Ю"3 6,96-10’2 42
— 1,1-ю-9 8,98 6, 54
20-25 6,5-10’9 8,19 8
РЬ(Ю3)2 [рь2+]-[ю;]2 Комн. 1,2-10“13 12,92 1, 2(18 °C)
9,2 5,3-10“14 13,28 2
25,8 2,6-10]3 12,58 2, 6,43, 54
25,8 2,83-10“13 12,55 4,14-10”5 2,30-10’3 4
РЬМо04 [РЬ2+]-[МоО2-] 25 1,0-10~13 13,0 3,16-1 о7 1,16-10’5 4
— 4,0-10’6 5,4 6, 54
Pb(N3)2 [Pb2+]-[N;]2 — 2,6-10 9 8,59 6, 54
РЬ(ОН)2 [РЬ2+]-[ОН~]2 25 1,0-10 20 20,0 1,36-10’7 3,29-10”4 42
25 2-Ю'16 15,7 5
14,5-15,6 5
1,1-1О’20 19,96 6
18-20 2,9-10“16 15,54 4,0-10’6 9,0-105 71
20-25 8,1-Ю’17 16,09 8
[РЬОН+]-[ОН~] 25 8,7-1044 13,06 2,96-10’7 7,13-Ю’6 42, 6
[РЬО2Н~][Н+] 15-16 5
РЬ(ОН)2 желт. [РЬ2+]-[ОН~12 7,9-Ю16 15,1 54
РЬ(ОН)2 красн. [РЬ2+]-[ОН~]2 5-1016 15,28 «
[РЬОН]-[ОН] 1-109 9,0 «
[Н+][НРЬО-] 3,2-10“16 15,5 «
РЬОНВг [РЬ2+]-[ОН~]-[ВГ] 2-10 15 14,7 6, 54
РЬОНС! [РЬ2+]-[ОН"]-[СГ] 2-Ю’14 13,7 «
РЬО2 [РЬ4+]-[ОН“]4 3-1066 65,5 «
64 5
[РЬО4Н-].[Н+] 32,4 5
РЬ(ОН)4 [РЬ4+].[ОН-]4 6,5-10’71 70,2 6
РЬ3О4 [РЬ2+]2- [РЬО4- ] 5,3-10'51 50,28 6, 54
РЬНРО4 [РЬ2+]-[НРО2~] 25 1,41-Ю"10 9,85 1,19-10-5 3,6-104 4
РЬ3(РО4)2 [РЬ2+]3-[РО3’]2 1,5-10”32 31,82 1,7-107 1,38-10“5 3
25 7,94-1043 42,1 1,49-10’9 1,21-10“7 4, 6, 54
18-25 1,0-10’34 34,0 1,6-10"7 1,3-105 71
РЬ5(РО4)3С1 [РЬ2+]5-[РО3~]3-[СГ] 7,5-10’80 79,12 6, 54
PbPO3F [Pb2+]-[PO3F2“] 1-10“7 7,0 6, 54
PbS [Pb2+]-[S2T Комн. 1-Ю’29 29 1, 5(25 °C)
18 3,4-10’28 27,47 2
1,1-10“29 28,96 3,3-10’15 7,90-1014 3, 71
418
Новый справочник химика и технолога
Продолжение таблицы 5.1.5
Соединение Ионное произведение t, °C ПР рПР = = -ignp S Литера-тура
моль-л 1 г/100 мл
25 2,51-IO"27 26,60 5,04-10"14 1,20-10"12 4, 6, 8, 54
25 8 1028 27,10 43
Pb(SCN)2 [Pb2+]-[SCN~]2 — 2-10"5 4,70 6, 54
PbSO4 [Pb2+]-[SOf] Комн. 1-Ю8 8 1
18 1,06-10"8 7,97 2
2,2-108 7,66 1,5 104 4,55-10"3 3, 71
25 1,96-10 8 7,71 1,40-1 О’4 4,24-10"3 4
25 1,6-10 s 7,8 6, 43, 54
1,66-108 7,78 8
PbS2O3 [Pb2+]-[S2O|] 4-Ю7 6,40 6, 54
PbSe [Pb2+]-[Se2“] 1-Ю’38 38 «
PbSeO3 [Pb2+]-[SeO|“ ] 3-10“12 11,5 «
PbSeO4 [Pb2+]-[SeO2-] 1,45-10"7 6,84 «
PbWO4 [Pb2+]-[WO2-] 4,5-10"7 6,35 «
PoS [Po2+]-[S2-] 5-10"29 28,3 «
Po(SO4)2 [Po4+]-[SO2-]2 2,6-IO7 6,58 «
Pr2(C2O4)3 [Pr3+]2-[C2O^]3 20 1,8-1028 27,74 44 (акт.)
См. также табл. 5.1.11.1.В
Pr(OH)3 [Pr^HOH"]3 25 2,7-1О“20 19,57 См. табл. 5.1.11.1.Д
25 6,4-10”24 23,19 «
25 7,1-10~24 23,15 «
25 6,7 10 22 21,17 «
25 3,16-10"23 22,50 1,04-106 2,00-10 5 4
PtBr4 [Pt4+]-[Br-]4 3-Ю41 40,5 6, 54
PtCl4 [Pt4+]-[CF]4 8,0-10'29 28,1 6, 54
Pt(OH)2 [Pt^HOH-]2 25 1,0-10"35 35 1,23-10"12 2,82-10-11 4, 6, 54
Pt(OH)4 PtO2 [Pt4+]-[OH"]4 25 1,6-10’72 71,8 1,44-10“15 3,79-10-14 4, 6, 54
Pts [Pt2+]-[S2'*] — 8-10"73 72,1 6, 54
Pu(OH)3 [Pu3+]-[OH“]3 — 2-Ю-20 19,7 6, 54
Pu(OH)4 [Pu4+]-[OH~]4 — 1-Ю"52 52 6
3,2-10"50 49,5 54
PuO2(OH) [PuO^HOH-] — 1-Ю"3 3 6
5-10"10 9,3 54
PuO2(OH)2 [PuOt ]2-[О1Г]2 — 3,2-10’21 20,5 6
2,3-10"20 19,64 54
Pu(IO3)4 [Pu4+]-[IO-]4 — 5-1013 12,3 6
Ra(IO3)2 [Ra2*].[IOJ]2 — 8,8-10"10 9,06 6, 54
Ra(NO3)2 [Ra2+]-[NO’]2 — 6,2-10"3 2,21 «
RaSO4 [Ra2*][SOj] — 4,3-10"11 10,37 «
25 4,27-10"11 6,53-1 О’6 2,10-Ю"4 4
RbBF4 [Rb+]-[BF“] — 1-Ю"3 3 6, 54
RbBH4 [Rb+]-[BH;] — 2,5-1 О’4 3,6 54
RbBrO3 [Rb+]-[Bo;] — 2-102 1,7 6, 54
RbC104 [Rb+]-[cio;j — 2,5-10-3 2,6 6, 54
Химические методы количественного анализа
419
Продолжение таблицы 5.1.5
Соединение Ионное произведение t, °C ПР рПР = =-ignp S Литература
моль-л 1 г/100 мл
Rb3Co(NO2)6 [№']’[ CofNOji ] — 1,48-IO15 14,83 6, 54
RbIO4 [Rb+]-[IO4] — 5,5-10"* 3,26 «
RbMnO4 [Rb+]-[MnO4] — 2,9-10 3 2,54 «
Rb2[PtCl6]2 [Rb']:<PtClf] — 9-10 8 7,2 «
Rb2PtF6 [RbTtPtFM — 7,63-IO’7 6,12 «
RbReO4 [Rb+]-[ReOJ — 9,6-104 3,02 «
Rb2SiF6 [Rb+]2-[ SiF62-] — 5-IO-7 6,3 «
Rb2TiF6 [Rb+]2-[TiF62~] — 5,5-10“5 4,26 «
Rh(OH)3 Rh2O3 [Rh3+][OH-]3 25 2-10"*8 47,7 5,21-IO’13 8,02-IO’12 4,6
3,2-10"*° 39,5 54
Ru(OH)3 Ru2O3 [Ru3+][OH-]3 25 l-10~36 36 4,39-IO-10 6,67-10“9 4,6
1-10 38 38 54
RuO2-xH2O [Ru(OH)2+]-[OH“]2 20 4,9-10~23 45
Ru(OH)4 [Ru4+]-[OH-]4 l-10^9 49 47, 54
Sb(OH)3 Sb2O3 [Sb3+]-[OH“]3 25 3,99-10 42 41,4 2,0-IO-5 3,45-10"* 42, 5, 6
1,7-10~38 37,76 54
[SbO+]-[OH~] 25 7,9-10’18 17,1 6
2,5-IO’19 18,61 54
[H2sbo;][H+] 1,3-IO’12 11,9 5, 54
Sb2S3 [Sb3+]2-[S2-]3 — 1,6-1 O’93 92,8 6
18-25 3,1-10’32 31,5 5,0-IQ-6 1,7-10"* 71
[SbO+]3-[H2S~]3 Комн. 3-1027 26,5 1
Sc(OH)3 [Sc3+]-[OH“]3 25 2-1O30 29,7 1,65-Ю-8 1,58-IO-7 4
1-1027 27 6
5,0-Ю37 36,3 54
26,3 5
Sm(OH)3 [Sm3+]-[OH“]3 25 6,8 -IO 22 См. табл. 5.1.11.1.Д
8,4-IO’23 «
Sm(OH)3 свежеосажд. [Sm3+]-[OH~]3 25 8,32-1 O’23 22,08 1,32-IQ-6 2,67-IO-5 4
Sn(C6H5N2O2)4 (купферонат) [Sn4+]-[C6H5N2O2]4 25 7,94-10 35 34,10 4,21
Snl2 [Sn2+]-[r]2 — 1-10"* 4 6
8,3 IO-6 5,08 54
Sn(OH)2 [Sn2+]-[OH~]2 — 6,3-IO27 26,2 5, 6, 54
25 5- IO-26 25,3 2,3-IO’9 3,51-IO’8 3, 5,7
1,41 IO-28 27,85 1,39 10"7 2,12-lQ-6 4
20-25 8-10“29 28,9 8
1,4-lQ-28--5-10"26 48
[SnOH+][OH-] 25 4,6-IO’15 14,3 2,84-106 4,34-IO-5 4,6
2,5-IO’16 15,61 54
[SnO2H"][ H+] — 1,3-IO-15 14,9 5,54
Sn(OH)4 [Sn4+]-[OH“]4 25 l,0-1057 57 l,31-1012 2,45-IO’11 4, 6, 54
25 110~56 56 5,7,8
(1-5) - IO-56 48
420
Новый справочник химика и технолога
Продолжение таблицы 5.1.5
Соединение Ионное произведение t, °C ПР рПР = =-ignp S Литера-тура
моль-л 1 г/100 мл
[SnO,2-]-[H*]2 Комн. 7,8-10-33 32,1 1
8,0-10-33 32,1 48
SnS [Sn2+HS2-] 20-25 1,0- ю-25 25,0 З,16-Ю’13 4,77-Ю”12 4, 6,8
2,5-10“27 26,6 54
25 1-Ю’28 28,0 1-Ю’14 1,51-Ю’13 3,5
18-25 1,4-10’14 13,9 1,2-Ю’7 2-Ю”6 7*
18-25 28 9
8-10’29 28,1 48,49
SnS2 [Sn4+]-[S2’]2 18-25 1,3-Ю18 17,9 1,1-10^ 2-Ю’5 71
Sr3(AsO4)2 [Sr^-tAsO;3]2 20 1,62-Ю’18 17,79 1,09-1 О’4 5,87-10’3 4
1,3-10-18 17,89 6, 54
SrCO3 [Sr^HCO,2-] Комн. 1,6-10’9 8,80 4,0-10’5 5,91-Ю”4 1, 2(25 °C) 3, 5,7,9
25 1,1-Ю’10 9,96 1,05-10’5 1,55-10”4 4, 6, 54
20-25 9,3-10’10 9,03 8
SrC2O4 [S?+]-[C2O2-] Комн. 5-Ю”8 7,3 1, 5(18°С)
18 5,61-10’8 7,25 2,37-Ю”4 4,2-10'3 2,4 (акт.), 6, 71, 8
1,6-107 6,80 54
25 7,61-10’8 7,12 2,41-1 О’4 4,23-Ю’3 4
SrC2O4-H2O [Sr^HC.O2] 18 5,61-10’8 7,25 2,4-1 О’4 4,26-Ю’3 3
SrCrO4 [s^jicro;-] 20 3,5-10“5 4,5 5,9-Ю’3 1,2-10’1 3
25 2,24-10’5 4,65 4,73-10’3 9,63-Ю’2 4,8
18 3,6-10’5 4,44 5, 6, 54
SrF2 [S?1[F-]2 18 2,8-10-9 8,6 2
25 3,37-10"9 8,47 9,44-Ю4 1,19-Ю’2 4
25 З-Ю’9 8,5 5
— 2,5-10’9 8,61 6, 54
2,46-10’9 8,61 8
Sr(IO3)2 [S^MIO-]2 Комн. 2,5-10’10 9,6 1
— 3,3-10’7 6,48 6, 54
SrMoO4 25 3,05-10’7 6,52 5,43 -Ю”4 1,34-10’2 4
2-Ю’7 6,7 6, 54
Sr(OH)2 [Sr^HOH-]2 3,2-10”4 3,50 5, 6, 54
Sr3(PO4)2 [Sr^’-IPOM2 20 4,07-10’23 22,39 1,3-10^ 2,82-Ю’5 4
1-Ю’31 31 6, 54
SrHPO4 [Sr2,]-[HPO3] 20 5,75-1О’7 6,24 7,59-1 О’4 1,39-10”2 4
SrPO3F [Sr^HPCbF2-] З-Ю’3 2,5 6, 54
SrSO3 [S^J-tSO2-] 25 5,14-Ю’8 7,29 2,27-Ю”4 3,81-10’3 4
4-Ю”8 7,4 6, 54
SrSO4 [Sr^HSO’-J Комн. 2,8-Ю’7 6,56 1
2,9 2,77-10’7 6,56 2
17,4 3,81-Ю’7 6,42 2
2,8-Ю’7 6,55 5,3-1 О’4 9,74-10’3 3, 71
Химические методы количественного анализа
421
Продолжение таблицы 5.1.5
Соединение Ионное произведение t, °C ПР рПР = =-ignp 5 Литература
моль-л 1 г/100 мл
25 5,29-Ю7 6,28 7,27-10^ 1,34-102 4
18 3,8-10-7 6,42 5
— 3,2-107 6,49 6, 54
20-25 2,5 10 7 6,42 8
SrSeO3 [Sr2*] [SeO* ] 8,5-10"7 6,07 * 6
4,4-10-6 5,36 54
SrSiF6 [Sr^HSiF2] 1,5-10’2 1,82 6, 54
SrWO4 [s^hwoJ] 25 2,2-10’10 9,77 1,48-10’5 3,98-10 4 4, 6, 54
Те(ОН)2 [Te4+]-[OH~]4 3-1054 53,52 6
2-1058 57,7 5, 54
Th(C2O4)2 [Th«Hc2o;-]2 2,0-10 5 4,70 6
1,1-10“25 24,96 54
См. также табл. 5.1.11.7.Ж-И
Th(IO3)4 [Th4+]-[IO-]4 2,5-1015 14,6 6, 54
1,35-Ю’20 19,87 50
Комн. 1,24-1О"16 15,91 1
См. также табл. 5.1.11.1.3 и 5.1.11.7.3
Th(OH)4 [Th4+]-[OH~]4 25 2,00-10 45 44,70 3,79-10’10 1,14-10-8 4
25 1-1O50 50 5
3,2-10^5 44,5 6
2-1O50 49,7 54
1-1 о42 42 51
1-10 39 39 52
См. также табл. 5.1.11.7.А,Б
[ThO4H”]-[H+] 19,8 5
Th3(PO4)4 [Th44]3-[PO3-]4 — 2,57-10~79 78,59 6
Th(HPO4)2 [Th4+]-[HPO4~ ]2 29 1,20-10’20 19,92 1,44-107 6,11-Ю"8 4
Th(SO4)2 [Th4+]-[SO4’]2 — 4-10 3 2,4 6, 54
См. также табл. 5.1.11.7.В-Е
TiO(OH)2 [TiO2][OH]2 25 1-Ю29 29 1,36-10 10 1,33-10'9 4,6
Комн. 1-1 О'-30 30 1
[TiOOHHOH] 25 3,16-1 о17 16,50 5,62-10’9 5,51 -Ю8 4
Ti(OH)3 [Ti3+]-[OH“]3 Комн. 1-Ю’35 35 1
25 1-10 40 40 5
Ti(OH)4 [Ti41-[OH-]4 6,3-10’52 51,2 54
Соединения Tl См. также табл. 5.1.11.3.А
TlBr [Tl+]-[Br~] — 3,89-106 5,41 6, 54
4,22-106 5,37 2,06-Ю3 5,84-10’2 4
Комн. 4-Ю6 5,7 1, 5(25 °C)
TlBrO3 [Tl+]-[BrO-] — 3,89-10 4 3,41 6
1,7-10^ 3,78 54
Комн. 8,5-Ю5 4,07 1
T12CO3 [Tl+]2-[CO2”] — 4-Ю3 2,4 6, 54
T12C2O4 [П’]'-.[С2О; Д 2 104 3,7 54
T1(C9H6ON)3 (оксихинолят) [Т1+]3-[С9Н6ОЬГ]3 1-Ю'37 37 4,21
422
Новый справочник химика и технолога
Продолжение таблицы 5.1.5
Соединение Ионное произведение t, °C ПР рПР = =-ignp S Литера-тура
моль-л 1 г/100 мл
Т1С1 [Т1+]-[СГ] — 1,7-10"* 3,76 6, 54
Комн. 1,5-10"* 3,82 1
25 2-10"* 3,7 5
Т1С1О4 [Tl+]-[C10J — 4-10-2 1,4 6, 54
T13Co(NO2)6 [T1+]3.[Co(NO2)’~] — 1,0-1016 16,0 «
Т12СгО4 [Tl+]2-[CrO2- ] — 9,8-1013 12,01 «
T14Fe(CN)6 [Tl+]4-[Fe(CN)g“ ] — 5-Ю’10 9,3 «
тп [Т1+]-[Г] Комн. 2,8-10'8 7,55 1, 5(20 °C)
— 6,5-10-8 7,19 6
5,75-10"8 7,24 54
ТПОз [Tl+]-[IO3-] Комн. 2,2-10-6 5,66 1
— 3,1-ю-6 5,51 6, 54
Т1(ОН)3 [Tl3+]-[OH-]3 25 1-10^* 44 5
— 6,3-10-46 45,2 5,6
1,3-Ю"*6 45,9 54
TI3PO4 [ti*]3[poM — 6,7-Ю8 7,18 6, 54
Tl2PtCl6 [Tl+]2-[PtCl2-] — 4-10-12 П,4 6
TlReO4 [Tl+]-[ReOJ — 1,2-10"5 4,92 6, 54
T12S [ti*]2-[s2-] Комн. 4,5-10'23 22,35 1
25 5,0110 21 20,30 1,08-107 4,75-1О"6 4, 6, 54
18 7-10-23 22,2 5
T1SCN [Tl+]-[SCFT] — 1,7-10"* 3,77 6, 54
T12SO3 [Tl+]2-[SO32-] — 6,3-10"* 3,2 «
T12SO4 [Trf-tso:-] — 4-10-3 2,4 «
T12S2O3 [Tl+]2- [S2O2-] — 2,0-1О"7 6,70 «
T1VO3 [Ti+].[vo;] — 1-10’5 5 6
5,5-10-9 8,26 54
T14V2O7 [ti+]4-[ v2o4- ] — 1-Ю’11 11 6
2,6-10-19 18,59 54
UO2CO3 [ио2+исо32-] 1,9-1012 11,73 54
UO2C2O4 [UO2+ ]-[C2O2-] — 4-10"* 3,4 6
2-10"* 3,7 54
25 1,81-Ю’3 2,74 4,25-10-2 1,524 4
(UO2)2Fe(CN)6 [UO2+]2-[Fe(CN)4~ ] — 7-1044 13,15 6
7,1 -Ю14 13,15 54
UO2HAsO4 [UO2+].[HAsO2-] — 3,2-10-11 10,50 6, 54
UO2HPO4 [UO2+]-[HPO2~] — 2,14-10-11 10,67 «
UO2(IO3)2 [uo2+]-[io;]2 — 3-10-8 7,5 «
UO2KAsO4 [UO2+ ]-[K+]-[ AsO3" ] — 2,5-10-23 22,6 «
UO2NH4AsO4 [UO2* ]-[NH4+H AsO’- ] —- 1,7-10-24 23,77 «
uo2nh4po4 [UO2‘ ].[NH,1[ POJ] — 4,4-10-27 26,36 «
UO2NaAsO4 [UO22+ ].[Na+].[ AsO3- ] — 1,3-Ю-22 21,87 «
Химические методы количественного анализа
423
Продолжение таблицы 5.1.5
Соединение Ионное произведение t, °C ПР рПР = = -1ёПР S Литера-тура
моль-л 1 г/100 мл
U(OH)3 [U3+]-[OH-]3 25 1,0-1 O’19 19 7,81-Ю-6 2,26-Ю4 4, 5, 6, 54
U(OH)4 [U4+]-[OH~]4 25 l,0-1045 45 3,30-Ю40 1,01-Ю-8 4, 5,6
6,3-1 о-55 54,2 54
[UO4H~]-[H+] — — 17,8 5
UO2(OH)2 [uo2+]-[oh-]2 25 1,0- ю-22 22 3,98-104 1,21-Ю-2 4,6
4,0-10-23 22,4 54
VO(OH)2 [VO2+]-[OH~]2 25 7,4-10-23 22,13 2,65-10-8 2,67-Ю-7 4,6
l,9-10-24 23,72 54
VO2(OH) (V2O5) [VO2+]-[OH-] 25 1,6- IO45 14,8 3,98-10-8 3,98-10-7 4, 54
(VO)3(PO4)2 [VO2+]3-[PO3-]2 — 8-10’25 24,1 6, 54
W(OH)4 [W4+]-[OH-]4 25 i-io-50 50 3,3-1041 8,31-IO40 4, 6, 54
Y2(C2O4)3 [Y3+].[C2O2~]3 25 5,34-1029 28,27 См. табл. 5.1.1 l.l.B 53 (акт.)
Y(OH)3 [Y3+]-[OH-]3 25 1,59-10-23 22,80 8,75-IO"7 1,23-10-5 4
— 6,3-10“25 24,2 6
8,1-10’23 22,1 См. табл. 5.1.11.1.Д
1,2- 10-24 23,9 «
1,6-10’23 22,8 «
Yb(OH)3 [Yb3+]-[OH-]3 25 2,29-10-27 26,64 9,6-10-8 2,15-Ю-6 4
2,9-1 O’24 23,5 См. табл. 5.1.11.1.Д
2,5-1 O’24 23,6 «
Zn3(AsO4)2 [Zn2+]3-[ AsO3 ]2 25 1,07-1 O’27 26,97 1,58-Ю-6 7,50-IO-4 4,6
1,3-1 O’28 27,89 54
Zn(CN)2 [Zn2+]-[CN-]2 — 2,6-1043 12,59 6, 54
ZnCO3 [Zn24][CO2] Комн. 2-10 8 7,7 1
2,7-1 O’8 1,6-Ю4 2,01 IO-3 3
25 1,45-1 O’11 10,84 3,8-Ю-6 4,77-10-5 4, 6, 54
25 6-Ю41 10,2 5
18-25 6,4-109 8,20 8,0-Ю-5 1,0-10-3 71
20-25 1,7-10” 10,8 8
18-25 10,2 9
Zn(C2O4) [Zn2+]-[C2O2-] 25 1,77-10“9 8,75 2,64-10-5 4,05-IO4 4
— l,5-10-9 8,8 6
2,75-IO"8 7,56 54
1,4-1 O’9 8,9 8-Ю-5 1-Ю-3 71
ZnC2O4-2H2O [Zn2'MC2O; ] 18 1,35-10-9 8,87 2
Zn(C9H6ON)2 (оксихинолят) [Zn2+]-[C9H6ON-]2 3,16-IO’25 24,50 4,21
Zn2Fe(CN)6 [Zn2+]2-[Fe(CN)4-] — 4,1-1046 15,39 7,4-Ю-6 6, 32
2,1-IO"16 15,68 54
ZnHg(SCN)4 [Zn2+]-[ Hg(SCN)2' ] — 2,2-10-7 6,66 6, 54
Zn(IO3)2 [Zn2-][ ro; ]2 — 2-Ю-8 7,7 6, 54
Zn(OH)2 [Zn2+]-[OH"]2 18-20 1,8-IO44 13,7 2
1-Ю47 17 1,4-Ю43 1,49-10-5 3
25 5-1047 16,3 2,0-Ю6 2,0-10-5 5.7’
16,1-16,9 5
2,1-IO46 15,7 8
424
Новый справочник химика и технолога
Продолжение таблицы 5.1.5
Соединение Ионное произведение t,°C ПР рПР = =-ignp S Литера-тура
моль-л 1 г/100 мл
— 7,1-10-18 17,15 6
1,4-10'17 16,86 54
[ZnOH’HOIE] — l,8-10“13 12,75 6
1,4-10'11 10,86 54
[ZnOM[H+]2 — 29,1 5
Zn(OH)2 стаб. форма [Zn2+]-[OH“]2 25 1,2-IO’17 16,9 3,84-10^ 3,82-10’5 4
Zn3(PO4)2 [Zn2+]3-[POM2 — 1,6- Ю24 23,8 6
25 9,12-10-33 32,04 l,53107 5,92-1 O'6 4, 54
ZnS [Zn2+]-[S2-] Комн. 1-10’23 23 1
18 l,21023 22,9 3,5-10“12 3,41-Ю'11 2,3,7
20 7-1 O’26 25,1 5
ZnS а (сфалерит) [Zn2+]-[S2~] 25 1,59-10 24 23,8 l,26-1012 1,23-Ю'11 4, 6, 8, 54
ZnS Р (вюрцит) [Zn2+]-[S2-] 25 2,51-IO’22 21,6 1,59-10’11 1,54-10“'° 4, 6, 54
20-25 5-1025 24,3 8
ZnSe [Zn2+]-[Se2~] — 1-10’31 31 6, 54
ZnSeO3 [Zn23[SeOj] — 2,57-10 7 6,59 6
1,9-10* 7,72 54
Zr(OH)4 [Zr4+]-[OH“]4 25 1 JO-1054 53,96 0,205 3,26 42, 6
7,9-10 55 54,1 54
[Zr(OH)M-[OH-]2 25 3,2-1 O’26 25,5 4,29-10~9 6,83-IO’8 4,6
2,0-10 25 24,7 54
ZrO(OH)2 [ZrO2+][OH“]2 25 2 10“24 23,7 7,93 -IO9 l,12-10“7 4,6
Zr3(PO4)4 [Zr4+]3-[PO^]4 IO’132 132 6, 54
Литература к таблицам 5.1.3-5.1.5
1. Erdey L. Theorie und Praxis der Gravimetrischen Analyse. Bd. III. Budapest: Akademiai Klado, 1964. 274 s.
2. Handbook of Chemistry and Physics. Part 2. A Readyreference Book of Chemical and Physical Data. 37th Edition. Cleveland: Chemical Rubber Publishing Co. 1640 p.
3. Алимарин И.П., Ушакова H.H. Справочные таблицы по аналитической химии. М.: Изд. МГУ, 1960. 22 с.
4. Алимарин И.П., Ушакова Н.Н. Справочные таблицы по аналитической химии. М.: Изд. МГУ, 1977. 32 с.
5. Лазарев А.И., Харламов И.П., Яковлев ПЯ., Яковлева Е.Ф. Справочник химика-аналитика. М.: Металлургия, 1976.138 с.
6. Лурье Ю.Ю. Справочник по аналитической химии. 4 изд. М.: Химия, 1971. 94 с.
7. Бесков С.Д., Слизковская О.А. Аналитическая химия. Качественный и количественный анализ. М.: Учебно-педагогич. изд. Минпросвет РСФСР, 1956. 554 с.
8. Бончев П.Р. Введение в аналитическую химию. Л.: Химия, 1978. 485 с.
9. Кунце У., Шведт Г. Основы качественного и количественного анализа. М.: Мир, 1997. 391 с.
10. Алексеев В.Н. Курс качественного химического полумикроанализа. М.; Л.: Госхимиздат, 1952. 488 с.
11. Тананаев И.В., Виноградова А.Д. // Журн. неорган. химии. 1957. Т. 2. С. 2455.
12. Тиновская Е.С. Химия, технология и применение производных пиридина и хинолина: Сборник. Рига: Изд. АНЛатвССР, 1960. 253 с.
13. Чухланцев В.Г., Алямовская К.В. // Изв. Высших учебных заведений. Химия и хим. технология. 1961. Т. 4. С. 359.
14. Волков Б.В., Несмеянов А.Н. Радиохимия: Сборник / Под ред. Спицына В.И. М.: Изд. МГУ, 1952. 99 с.
15. Bayerle V. // Rec. trav. chim. 1925. V. 44. P. 518.
16. Жаравский Ф.Г. // Труды Комиссии по аналитической химии. 1951. Т. 3(6). С. 101.
17. BrunerL.,ZawadzkiJ.//Z.Anorg.Chem. 191O.Bd.65.S. 145.
18. Kato H. // Sci. Repts. Tohoku Imp. Univ. First Ser. 1940. V. 28. P. 564. Цит. no [19].
19. Бусев А.И. Аналитическая химия висмута. М.: Изд. АН СССР, 1953. 59 с.
20. Капустинский А.Ф. // Докл. АН СССР. 1940. Т. 28. С. 140; Он же // Журн. приклад, химии. 1943. Т. 16. 60 с.
21. Шарло Г. Методы аналитической химии / Пер. с франц, под ред. Лурье Ю.Ю. М.; Л.: Химия, 1965.
22. Лурье Ю.Ю. Расчетные и справочные таблицы для химиков. М.; Л.: Госхимиздат, 1947.
23. Цитович И.К. Курс аналитической химии. М.: Высшая школа, 1964.
24. Подчайнова В.Н., Симонова Л.Н. Медь. (Аналитическая химия элементов). М.: Наука, 1990. 17 с.
Химические методы количественного анализа
425
25. Яцимирский К.Б., Васильев В.П. // Журн. аналит. химии. 1956. Т. 11. С. 536.
26. Аксельруд Н.В., Фиалков Я .А. // Укр. хим. журн. 1950. Т. 16. С. 283.
27. Swinarski A., Ozaki S.M. // Przem. 1955. V. 11. Р. 384.
28. Голуб А.М. // Журн. неорган. химии. 1956. Т. 1. С. 2517.
29. Meites L. Handbook of Analytical Chemistry. New York; Toronto; London: McGraw-Hill Book Company, Inc., 1963.
30. Коваленко П.Н. // Журн. прикл. химии. 1957. Т. 30. С. 52.
31. Moeller Т.» King G.L. // J. Phys. Colloid. Chem. 1950. V. 54. P. 999.
32. Бусев А.И. Аналитическая химия индия. М.: Изд. АН СССР, 1958.
33. Назаренко В.А., Флентикова Г.В., Андрианов А.М. // Журн. неорган. химии. 1967. Т. 12. С. 3072.
34. Гладышев В.П., Левицкая С.А., Филиппова Л.М. Аналитическая химия ртути. М.: Наука, 1974.
35. Тананаев И.В., Глушкова М.А., Сейфер Г.Б. // Журн. неорган. химии. 1956. Т. 1. С. 66.
36. Moeller Т. // J. Am. Chem. Soc. 1941. V. 63. Р. 2625.
37. Kury J.W. // US AEC, UCRL-227. 1953; C. A. 1954. V. 48. 3833.
38. Ringbom A. Solubilities of Sulfides. Report to Analytical Section JUPAC. July 1953.
39. Пешкова B.M., Савостина В.М. Аналитическая химия никеля. М.: Наука, 1966. 23 с.
40. Hume D., Kollhoff J. // J. Am. Chem. Soc. 1950. V. 72. P. 4423.
41. Stary J. // Analyt. chim. acta. 1963. V. 28. P. 132.
42. Crouthamel C.E., Martin D.S., Jr. // J. Am. Chem. Soc. 1951. V. 73. P. 569.
43. Полянский Н.Г. Аналитическая химия свинца. М.: Наука, 1986.
44. Коренман И.М. // Журн. общей химии. 1954. Т. 24. С. 1910.
45. Brandstetr J., Kfivanek М., Vfestal J. // Coll. Czech. Chem. Comm. 1961. V. 26. P. 2596. Цит. no [46].
46. Гинзбург С.И. и др. Аналитическая химия платиновых металлов. М.: Наука, 1972.
47. Bremard С., Nowogrocki G., Tridot G. // Bull. Soc. chim. France. 1968. № 5. P. 1961.
48. Спиваковский В.Б. Аналитическая химия олова. М.: Наука, 1975.
49. Бриттон Х.Т.С. Водородные ионы. Л.: ОНТИ, 1936.
50. Крылов Е.И. // Журн. аналит. химии. 1957. Т. 12. С. 451.
51. Kruyt H.R., Troelstra S.A. // Kolloid-Beihefte. 1943. Bd. 54. S. 262.
52. Latimer W.M. The Oxidation States of die Elements and Their Potentials in Aqueous Solutions. 2nd Ed. New York, 1952. 297 p.
53. Feibush A.M., Rowley K., Gordon L. // Anal. chem. 1958. V. 30. P. 1610.
54. Лурье Ю.Ю. Справочник по аналитической химии. М.: Химия, 1989.
Загрязнение осадков
Погрешности возникают также из-за загрязнения осадка.
Причинами загрязнения являются совместное осаждение и соосаждение примесей. При совместном осаждении реагент-осадитель образует осадки как с определяемым компонентом, так и с примесями. Для предотвращения
этого необходимо проводить предварительное отделение мешающих компонентов или их маскирование.
Причинами соосаждения могут быть
— адсорбция примесей на поверхности осадка;
— окклюзия (захват) быстро растущими частицами осадка сорбированных примесей и маточного раствора;
— изоморфное осаждение;
— послеосаждение, вызванное повышением концентрации осадителя на поверхности осадка за счет адсорбции. В результате произведение концентрации иона-осадителя и примеси превысит величину ПР, и образуется осадок.
Удаление примесей промыванием эффективно только по отношению к адсорбированным примесям. В остальных случаях целесообразно проводить переосаждение осадка, т.е. растворение и повторное осаждение. Удаление части примесей происходит при созревании осадка за счет перекристаллизации и уменьшения поверхности осадка (главным образом, в случае кристаллических осадков).
Высушивание и прокаливание осадков
Выбор оптимальной температуры термообработки осадка для достижения постоянного состава ГФ целесообразно проводить на основе данных термогравиметрии. Продолжительность процедур высушивания или прокаливания определяют экспериментально по достижении тиглем с осадком постоянной массы.
В случае термической нестойкости ГФ высушивание может проводиться при комнатной температуре на воздухе или в эксикаторе над осушителями, а также в вакуум-эксикаторах. В последнем случае осадки обычно промывают спиртом и эфиром.
Специфичность гравиметрического метода
Используемые в гравиметрии реагенты, за небольшим исключением, не являются специфичными. Фактически все неорганические реагенты (H2SO4, НС1, NH4OH (водн.), (NH4)2HPO4, ВаС12 и др.) в гравиметрическом анализе не селективны; их применение требует в большинстве случаев предварительных разделений и маскирования компонентов, сопутствующих определяемому компоненту. Более высокой селективностью, а в ряде случаев и специфичностью обладают органические реагенты. Наиболее широко применяемые из них представлены в табл. 5.1.6.
В аналитической химии органических веществ гравиметрические методы имеют ограниченное применение.
Результаты гравиметрических определений чаще всего выражают в абсолютных величинах или массовых долях (%) к навеске. Для пересчета массы осадка ГФ в массу определяемого компонента вводят гравиметрический фактор или фактор пересчета:
а Мк F =------— ,
b МГФ
где а и b — стехиометрические коэффициенты; Мх и Мгф — молярные массы определяемого вещества и гравиметрической формы соответственно.
426
Новый справочник химика и технолога
Органические осадители [1,14]
Таблица 5.1.6
Осадитель Формула Осаждаемый ион
Диметилглиоксим (реагент Чугаева) СН.-С —С—СН3 ' II II НО—N N—ОН Ni2+, Pd2+, Pt2+
Купферон / \ N=O \У->/ onh4 Fe3+, VO2, Ti4+, Zr4+, Ce4+, Ga4+, Sn4+
8-Оксихинолин (оксин) он Mg2+, Zn2+, Cu2+, Cd2+, Pb2+, Al3+, Fe3+, Bi3+, Ga3+, Th4+, Zr4+, UO2+, TiO2+
Саллицилальдоксим KZal 'Xn—он Хх'/Х^он Cu2+, Pb2+, Bi3+, Zn2+, Ni2+, Pd2+
1-Нитрозо-2-нафтол (реагент Ильинского) О Co2+, Fe3+, Pd2+, Zr4+
Нитрон NC6H5 N— /^\ / N\ «W ^с6н5 no;, cio4, bf;, wof
Тетрафинилборат натрия Na+B(C6H5)4- K+, Rb+, Cs+, NIL, Ag', органические ионы аммония
Щавелевая кислота Й И 1 ° 0—0 II II о о Ca2+
2-Аминобензойная (антраниловая) кислота —соон k<->/L-NH2 Zn2+, Pb2+, Cd2+
Диэтилдитиокарбаминат натрия S // (C2H5)2N—С SNa Многие металлы (кислая среда)
Т етрафини ларсеникумхл орид (C6H5)4As+Cr Cr2O2-, MnO;, ReO;, MoO4”, wo; , cio;, 1;
Литература
1. Основы аналитической химии: В 2 кн. Кн. 2. Методы химического анализа / Под ред. академика Ю.А. Золотова; Изд. 2, перераб. и доп. М.: Высш, шк., 2000. 494 с.
2. Дорохова Е.Н., Прохорова Г.В. Задачи и вопросы по аналитической химии. М.: Мир, 2001. 267 с.
3. Аналитическая химия. Химические методы анализа. Уч. Пособие для ВУЗов. Под ред. О.М. Петрухина. М.: Химия, 1993.
4. Васильев В.П. Аналитическая химия. В 2-х ч. Ч. 1.. Гравиметрический и титриметрический методы анализа. М.: Высш, шк., 1989. 320 с.
Химические методы количественного анализа
427
5. Скуг Д., Уэст Д. Основы аналитической химии: Кн. 1. М.: Мир, 1979. 480 с.
6. Янсон Э.Ю., Путнинь Я.К. Теоретические основы аналитической химии. М.: Высш, шк., 1980. 263 с.
7. Кунце У., Шведт Г. Основы качественного и количественного анализа / Пер. с нем. М.: Мир, 1997.
8. Кузнецов В.В. Применение органических аналитических реагентов в анализе неорганических веществ. М.: МХТИ им. Д.И. Менделеева, 1972. 145 с.
9. Кузнецов В.В. // Успехи химии. 1986. Т. 55. № 9. С.1409-1438.
10. Губен-Вейль. Методы органической химии. М.: Химия, 1967. 1032 с.
11. Лурье Ю.Ю. Справочник по аналитической химии. М.: Химия, 1989. 447 с.
12. Laszlo Erdey Theory und Praxis der Gravimetric Analyse. Budapest: Akademiai Kiado, 1964.
13. Skoog D.A., West M.D., Holler F.J. Analitical Chemistry. An Introduction. 6th ed. Philadelphia etc: Saunders College Publishing, 1994. 612 p.
14. Harris D.C. Quantitative Chemical Analysis. 5th ed. New York, 1999. 899 p.
15. Булатов M.M. Расчеты равновесий в аналитической химии. Л.: Химия, 1984.
5.1.1. Методы гравиметрического определения неорганических ионов
Таблица 5.1.7
Основные методы гравиметрического определения неорганических ионов
Опред. ион, в-во Осаждаемая форма Реагент Среда, условия осаждения Г равиметрическая форма Условия термообработки, t,°C
Li+ LiCl HC1 Кисл. a) LiCl б) Li2SO4 175-600 160-900
Li2SO4 H2SO4 Кисл. Li2SO4 160-900
Li3PO4 Na2HPO4 Аммиачная Li3PO4 450-900
Li-алюминат Алюминат Na Щел. 2Li2O-5Al2O3 470-900
Na+ NaCl Выпар, солянокисл. Р-Ра Свободная от других нелетучих соединений NaCl 400-800
Na2SO4 Выпар, сернокисл. Р-ра Свободная от других нелетучих соединений Na2SO4 650-800
Na{Zn[UO2- •(С2Н3О2)3]з}- •wH2O Zn(C2H3O2)2 + + UO2(C2H3O2)2 Нейтр. a) Na{Zn[UO2-•(C2H3O2)3]3}-6,5H2O 6) Na2U2O7(ZnU2O7)2 105 400-670
Na{Mg[UO2- •(C2H3O2)3]3}- •иН2О Mg(C2H3O2)2 + + UO2(C2H3O2)2 Нейтр. a) Na{Mg[UO2-(C2H3O2)3]3}-6,5H2O 6)Na2U2O7(MgU2O7)2 100-110 400-700
К+ KC1 Выпар, солянокисл. Р-Ра Свободная от других нелетучих соединений KC1 220-600
K2SO4 Выпар, сернокисл, р-ра Свободная от других нелетучих соединений K2SO4 400-900
K2PtCl6 H2PtCl6 Слабо солянокисл. K2PtCl6 100-220
KC1O4 НСЮ4 + СП. Кисл. КСЮ4 70-650
K[B(C6H5)4] Na[B(C<iH5)4] или LiFB(C6H5)4l Слабокисл., нейтр. или щел. a) K[B(C6H5)4] 6) KBO2 100-260 715-825
Rb+ Опр. аналогично К
Rb2[SnCl6] SnCU насыщ. р-р Р-р после отд. К+ Rb2[SnCl6] 110
Cs+ Опр. аналогично Rb+
Cu2+ Cu Электролиз H2SO4, HNO3; NH4OH, KCN Cu Без нагр.
Cu+ CuSCN KSCN + Na2SO3 или FeSO4 Кислая, восстановительная CuSCN <260
Cu2+ CuS H2S или Na2S2O3 Кисл., без NO3 a) Cu2S 6) CuO 400-600 700-900
Cu(OH)2 КОН Щел. CuO 700-900
Cu+ Cui KI + Na2SO3 Кисл. Cui <260
Au3+ Au so2 Кисл. Au 600-900
Au FeSO4 Солянокисл. Au 600-900
Au H2C2O4 Кисл. Au 600-900
428
Новый справочник химика и технолога
Продолжение таблицы 5.1.7
Опред. ион, в-во Осаждаемая форма Реагент Среда, условия осаждения Г равиметрическая форма Условия термообработки, t,°C
Аи3+ Au H2O2 Щел. Au 600-900
Au Формальдегид Щел. Au 600-900
Au Аскорбиновая к-та Кис л. Au 600-900
Ве2+ Ве(ОН)2хН2О NH4OH Слабощел. ВеО 1100-1200
Ве(ОН)2 + таннин NH4OH + таннин Слабощел. ВеО 1J00-1200
Ве(ОН)2хН2О NH4NO2 Нейтр. ВеО 1100-1200
Ве(ОН)2хН2О Гидролиз в присутствии KOCN или бензоата аммония, уротропина, Г + IOJ и др. Нейтр. ВеО 1100-1200
BeNH4PO4-6H2O (NH4)2HPO4 + NH4OH Нейтр. Ве2Р2О7 >700
Mg2+ MgNH4PO4-6H2O (NH4)2HPO4 или Na2HPO4, NaNH4HPO4 NH4C1 + NH4OH a) MgNH4PO4-6H2O б) Mg2P2O7 Без нагр. 600-1000
Mg(C9H6ON)2-• 4Н2О 8-Оксихинолин Аммиачная Mg(C9H6ON)2 200-300
Са2+ СаС2О4Н2О (NH4)2C2O4 или K2C2O4 Уксуснокисл, или аммиачная а) СаС2О4Н2О б) СаС2О4 в) СаСО3 г) СаО д) CaSO4 е) CaF2 100 210-320 440-600 1000-1200 Выпар, с H2SO4 конц.; 800-900 Выпар, с HF; 800-900
CaSO4-2H2O H2SO4 Кисл. + 70% сп. CaSO4 800-900
СаМоО4 (NH4)2MoO4 Нейтр. + сп. СаМоО4 800-900
Sr2* SrC2O4H2O (NH4)2C2O4 Нейтр. a) SrC2O4 H2O б) SrCO3 80 или без нагр. 500-800
SrSO4 H2SO4 Сернокисл, или нейтр. SrSO4 100-300
SrCO3 (NH4)2CO3 Слабоаммиачная SrCO3 500-800
Ва2* BaSO4 H2SO4 или (NH4)2SO4 Кисл. BaSO4 800-1000
BaCrO4 (NH4)2CrO4 Нейтр. BaCrO4 <60
BaC2O4H2O (NH4)2C2O4 Нейтр. a) BaC2O4H2O 6) BaC2O4 в) BaCO3 <76 110-346 >476
Zn2* ZnS H2S pH = 2-3 a) ZnO 6) ZnSO4 в) ZnS 950-1000 400-600 H2S;450-550
ZnNH4PO4 (NH4)2HPO4 pH = 6-7 a) ZnNH4PO4 6) Zn2P2O7 130 900
Zn Электролиз NaOH Zn Без нагр.
Zn[Hg(SCN)4] K2Hg(SCN)4 HNO3 (< 5%) Zn[Hg(SCN)4] 100-110
Zn(C9H6ON)2- •1,5H2O 8-Оксихинолин АсОН; NaOH Zn(C9H6ON)2 180
Zn(C7H6O2N)2 Антранилат натрия Нейтр. Zn(C7H6O2N)2 110
Zn(CioH602N)2- •H2O Хинальдинат натрия Уксуснокисл. Zn(C10H6O2N)2H2O 110
Cd2* CdSO4 H2SO4 Кисл. CdSO4 500-800
Химические методы количественного анализа
429
Продолжение таблицы 5.1.7
Опред. ион, в-во Осаждаемая форма Реагент Среда, условия осаждения Гравиметрическая форма Условия термообработки, t,°C
Cd2' CdS H2S Слабокисл. a) CdS б) CdSO4 130 500-800
CdNH4PO4 (NH4)2HPO4 Слабокисл. a) CdNH4PO4H2O б) Cd2P2O7 Без натр. 700-900
Hg(I,II) HgS H2S Кис л. HgS 110
(NH4)2s Аммиачная HgS 110
Hg2Cl2 HC1 + H3PO3 Кисл. Hg2Cl2 120
Hg а) Аскорбиновая к-та б) Гипофосфит кальция или натрия в) Электролиз г) Fe-порошок Слабокисл. Слабокисл. Слабокисл. Р-ция сухим путем Hg Hg Hg Амальгама Без натр. Без натр. Без нагр. Без нагр.
Hg2(IO3)2 КЮ3 Слабокисл. Hg2(IO3)2 110
В B2O3 СН3ОН, Mg2+ После отгонки (СН3О)3В осажд. Mg2+; прокал. B2O3 + MgO 800-900
c20h16n4-hbf4 Нитрон HF, СН3СООН, охл. c20h16n4-hbf4 105-110
Боросахарат Ba2+ Дикалийсахарат + + ВаС12 Нейтр. Боросахарат Ba2+, f= 0,0201 105
Ba5B2Ci2H8O24 • • 4H2O Винная к-та + ВаС12 Слабоаммиачная Ba2BO6(?), эмпир. /= 0,0287 700-800
А13+ A1(OH)3 a) NH4OH + NH4C1 б) Г + ю; в) NO’ г) (CH2)6N4 pH = 5,5-7,5 pH = 7,6 pH = 4,6-6,0 pH = 5,4 A12O3 ai2o3 A12O3 A12O3 1100-1200 1100-1200 1100-1200 1100-1200
Al(OH)3+xS S2O2’ pH = 4,6 ai2o3 1100-1200
Основной сукцинат алюминия Сукцинат аммония + мочевина pH = 4,2-4,6 ai2o3 1100-1200
Основной бензоат алюминия Бензоат аммония pH = 3,5-4 ai2o3 1100-1200
Основной ацетат алюминия Ацетат аммония pH = 5,2 ai2o3 1100-1200
А1(ОН)3 со2 Сильнощел. A12O3 1100-1200
A1(C9H6NO)3 8-Оксихинолин 4,5-9, тартраты A1(C9H6NO)3 110-130
А1РО4 (NH4)2HPO4 pH = 5 aipo4 1100-1200
Ga3+ Ga(OH)34H2O a) NH4OH б) Гидролиз в присутствии анилина, мочевины, Na2SO3, таннина pH = 4-7 pH = 4-7 Ga2O3 Ga2O3 >550 >550
Ga(C6H5N2O2)3 Купферон H2SO4 (2 н.) Ga2O3 >550
Ga(C9H6ON)3 8-Оксихинолин pH = 7-8 Ga(C9H6ON)3 ПО
Ga(C9H4ONBr2)3 5,7-Дибром-8-оксихинолин Слабокисл, (минеральная к-та) Ga(C9H4ONBr2)3 120
Ga2(CioHi404)3 Камфарная к-та Уксуснокисл, или нейтр. Ga2O3 >550
In3+ In(OH)3 a) NH4OH б) Гидролиз с KOCN Слабощел. pH = 4-6 In2O3 In2O3 850-1000 850-1000
In2S3 H2S 0,03-0,05 н. НС1 или СН3СООН a) In2S3 6) ln2O3 100-200 850-1000
In(C9H6ON)3 8-Оксихинолин СН3СООН + CH3COONa In(C9H6ON)3 100-250
T1(I, Ш) Tl2CrO4 К2СгО4 Нейтр. Tl2CrO4 100-750
430
Новый справочник химика и технолога
Продолжение таблицы 5.1.7
Опред. ион, в-во Осаждаемая форма Реагент Среда, условия осаждения Г равиметрическая форма Условия термообработки, A °C
T1(I, III) TH KI Уксуснокисл, или нейтр. ТП 70-470
Т12О3хН2О a) K3[Fe(CN)6] 6) KOH + Br2 Щел. Щел. Т12О3 Т12О3 100-230 80
Т1[В(С6Н5)4] Na[B(C6H5)4] AcONa Т1[В(С6Н5)4] 105
Sc3+ Sc(OH)3xH2O nh4oh NH4OH, кип. Sc2O3 > 900
NH4OOC(CHOH)2- •COOSc(OH)2 Винная к-та Нейтр. или слабо-аммиачная, нагр. Sc2O3 900
уЗ+ Y2(C2O4)3-7H2O Винная к-та pH >2 a) Y2(C2O4)3-7H2O б) Y2O3 Без нагр., вакуум-эксикатор 800-850
Y(OH)3xH2O NH4OH NH4C1, нагр. y2o3 850-900
Y(PO4)xH2O Na2HPO4 pH = 4,5 ypo4 900
La3+ Ln3+ Ln2(C2O4)3xH2O H2C2O4 НС1 (0,3 н.) a) Ln2O3 6) Ln2(C2O4)3 wH2O 800 Без нагр., вакуум-эксикатор
Се3+ 2Ce(IO3)4KIO3- •8H2O KBrO3 + KIO3 a) HNO3 (25%) б) HNO3 (25%). К ос. добавл. Н2С2О4 2Ce(IO3)4KIO3-8H2O CeO2 40-50 800
Н2С2О4 - см. La3+
NH4OH - см. La3+
С а) Сжигание в О2 при 1000-1100 °C б) H2SO4 + СгО3 в) Ва(ОН)2 Поглощение натронной известью H2SO4 (1:1), поглощение натровой известью а) Поглощение р-ром Ва(ОН)2 б) Поглощение р-ром ВаС12 + NH4OH. Ос. переводят в сульфат (НС1, затем H2SO4) co2 co2 BaCO3 BaSO4 476 800-950
CN“ AgCN AgNO3 Нейтр. или слабоам-миачный a) AgCN 6) Ag НО 520
OCN- AgOCN AgNO3 Нейтр. AgOCN НО
SCN~ AgSCN AgNO3 Нейтр. или слабокисл. AgSCN 115
CuSCN CuSO4 + H2SO3 Нейтр. или слабокисл. (без NO“) CuSCN ПО
Si(IV) SiO2xH2O Кислоты (H2SO4, HC1, HC1O4) а) Выпар, с к-тами б) Осажд. с желатиной SiO2 SiO2 1050-1100 1050-1100
(C9H7N)4-SiO2- 12MoO3-2H2O Хинолин + + (NH4)2MoO4 + HC1 Кисл. (C9H7N)4SiO2- •12MoO3-2H2O ~ 150
Ge(IV) GeS2 H2S H2SO4 (6 н.). Ос. р-ют с Н2О2 и выпар, с H2SO4 GeO2 900
H2GeO3xH2O + таннин Гидролиз в присутствии таннина Кисл. GeO2 900
(C9H7ON)4H4- •[GeCMouCM] (NH4)2MoO4 + + 8-оксихинолин Кисл.; Ge 0,5-1 мг a) (C9H7ON)4H4-•(Ge(Moi204o)] 6) GeO212MoO3 50-115 496-920
Химические методы количественного анализа
431
Продолжение таблицы 5.1.7
Опред. ион, в-во Осаждаемая форма Реагент Среда, условия осаждения Гравиметрическая форма Условия термообработки, t, °C
Ge(IV) (C13H9NH)4- •[Ge(Mo12O40)] (NH4)2MoO4 + акридин HNO3 (1,4 н.) (C13H9NH)4- •[Ge(Mo12O40)] 105
Sn(II, IV) Sn(OH)4 Гидролиз в присутствии NH4NO3 pH = 4 SnO2 950-1000
Sn(OH)4 HNO3 HNO3(1:1) SnO2 950-1000
SnS2 H2S НС1 (1 н.) SnO2 950-1000
Sn Электролиз Н2С2О4 + оксалат аммония Sn Без нагр.
(C6H5N2O2)4Sn Купферон НС1 (0,6-2 н.) или H2SO4 (1,8-5 н.) SnO2 900
(C13HuO2N2)SnCl2 Л'-Бензоилфенил-гидроксиламин НС1 (1-8%), Sn(II) и Sn(IV) a) (C13HnO2N2)SnCl2 6) SnO2 НО 900
Pb2+ PbSO4 a)H2SO4 б) (NH4)2SO4 Выпар, с H2SO4 (конц.) HNO3 (0,01 н.) PbSO4 PbSO4 130-800 130
PbCrO4 KCrO4 СН3СООН PbCrO4 100-900
PbO2 Осажд. на аноде HNO3 PbO2 <200
PbCl2 НС1 НС1 + спирт PbCl2 Без нагр.
Ti(IV) TiO2(H2O)x a) NH4OH б) NH4OH + таннин в) NH4NO2 г) СГ + ВгО; д) NH4OH + сульфосалициловая к-та е) NH4OH + пиридин pH = 6-7 Слабоуксуснокисл. Нейтр. Слабосолянокисл. Сильноаммиачн. pH = 5-6 TiO2 TiO2 TiO2 TiO2 TiO2 TiO2 800-1200 800-1200 800-1200 800-1200 800-1200 800-1200
Ti(C6H5N2O2)4 Купферон Сильнокисл. TiO2 600-1200
Ti(C6H4AsO3)2 и-Оксифениларсоно-вая к-та НС1 (1,5 н.) TiO2 800-1200
TiO(C9H6ON)2- •2H2O 8-Оксихинолин Лимонная или винная к-ты a) TiO2 6) TiO(C9H6ON)2 800-1200 115
TiO(C9H4Br2ON)2 5,7-Дибром-оксихинолин Слабокисл. a) TiO(C9H4Br2ON)2 6) TiO2 160 Н2С2О4; 1200
Zr(IV) ZrO2xH2O a) NH4OH или NH4OH + таннин б) Пиридин pH = 6-7 Нейтр. ZrO2 ZrO2 1000-1200 1000-1200
ZifCeWW Купферон Сильнокисл. ZrO2 1000-1200
ZrO(H2PO4)2 (NH4)2HPO4 Сильнокисл. ZrP2O7 600-1000
Zr(SeO3)2 H2SeO3 Сильнокисл. a) Zr(SeO3)2 6)ZrO2 100 1000-1200
ZrO(H2AsO4)2 NH4H2AsO4 Сильнокисл. ZrO2 1000-1200
Zr(C6H5AsO3)2 Фениларсоновая к-та Сильнокисл. ZrO2 1000-1200
Zr(C8H7O3)4 Миндальная к-та Сильнокисл. a) Zr(C8H7O3)4 6) ZrO2 110 1000-1200
Hf(IV) Cm. onp. Zr (IV)
Th(IV) Th(OH)„X„ n « 3,5; X: СГ, no;, SO2’; m « 0,3-0,8 NH4OH или NaOH, а также гидролиз в присутствии KN3, NaNO2, Na2S2O3, ор-ганич.оснований (анилин, пиридин, хинолин,уротропин) Избыток осадителя (3-, 5-кратный) ThO2 950-1200
Th(C2O4)2-6H2O Н2С2О4 НС1 (1,4%), нагр. ThO2 1100
432
Новый справочник химика и технолога
Продолжение таблицы 5.1.7
Опред. ион, в-во Осаждаемая форма Реагент Среда, условия осаждения Гравиметрическая форма Условия термообработки, t,°C
Th(IV) Th(IO3)4 KIO3 HNO3 (0,5-6 н.) Перевод ос. в гидроксид, ThO2 1100
4Th(IO3)4KIO3- •18H2O KIO3 HNO3 (1:2). Ос. кип. с SO2, осажд. Н2С2О4 ThO2 1100
ThP2O6llH2O H4P2O6 или натриевая соль НС1(10%) a) ThP2O7 б) перевод в оксалат и ThO2 . 1000 1100
ThF4-4H2O HF Кисл. среда, HF, Hg2(NO3)2 a) ThO2 б) перевод ос. в гидроксид, ThO2 1000 1100
Th(C9H6ON)4 (желт.) Th(C9H6ON)4-•C9H7OH (кр.) 8-Оксихинолин pH = 4,4-8,8, осажд. при 50 °C — желт, при 70 °C — кр. ThO2 1000
nh: NH4Hg2I P-в Несслера K2[HgI4] + КОН Щел. NH4Hg2I 150
(NH4)2PtCl6 H2PtCl6 Хлоридный р-р, сп. (NH4)2PtCl6 130
n2h4 Косвенный метод по изб. Ag+ AgNO3 + NH4OH Аммиачный р-р. Изб. Ag+ осажд. СГ AgCl 130-150
HN3 Косвенный метод по количеству осажд. Ag+ (AgN3) AgNO3 Нейтр. или слабокисл, ос. р-ряют в HNO3, осажд. AgCl AgCl 130-150
no; C20H16N4HNO3 Нитрон Слабокисл. (H2SO4 или СН3СООН) C20H16N4HNO3 105-110
Сухие нитраты + + SiO2 (кварц) SiO2 (кварц) Безводная (нейтр.) Убыль веса за счет n2o5 >970
Сухие нитраты + + Na2WO4 Na2WO4 Безводная (нейтр.) Убыль веса за счет n2o5 >950
Сухие нитраты + + К2Сг2О7 K2Cr2O7 Безводная (нейтр.) Убыль веса за счет n2o5 >815
no2 Косвенное опр. по AgBr AgBrO3 СН3СООН, нагр.; затем H2SO4 AgBr 130
SCN’ AgSCN AgNO3 HNO3 AgSCN <224
CuSCN CuSO4 + H2SO3 Кисл. CuSCN <300
BaSO4 Br2, HNO3 (конц.) или H2O2 окисл. до SO4”, осажд. ВаС12 Кисл. BaSO4 600-800
PO’- MgNH4PO4-6H2O Магнезиальная смесь Аммиачная, цитраты a) Mg2P2O7 6) MgNH4PO4-6H2O 560-1000 Без нагр.
(NH4)3P(Mo3O10)4 (NH4)2MoO4 HNO3 a) (NH4)3P(Mo3O10)4 6) P2O5-24MoO3 b) MgNH4PO4-6H2O r) Mg2P2O7 д) PbMoO4 (см. onp. Mo) 180-410 800-850 Без нагр. 560-850
H7[P(Mo2O7)]- •3(C9H7ON)-2H2O (NH4)2MoO4, 8-оксихинолин Слабокисл. H7[P(Mo2O7)]- •3(C9H7ON)-2H2O 105
p2o>- Mg2P2O7 Магнезиальная смесь pH = 4,5 Mg2P2O7 900
Zn2P2O7 Zn(CH3COO)2 pH = 3,8 Zn2P2O7 >610
PO3 Hg2ci2 HgCl2 Кисл. Hg2Cl2 <310
As(III, V) As2S3 H2S НС1 (6 н.) a) As2S3 6) MgNH4AsO4-6H2O в) Mg2As2O7 100-105 Без нагр. 900
Химические методы количественного анализа
433
Продолжение таблицы 5.1.7
Опред. ион, в-во Осаждаемая форма Реагент Среда, условия осаждения Гравиметрическая форма Условия термообработки, t,°C
As(III, V) As2S5 H2S НС1 (6 н.) As2S5 100-105
AS2S3 + 2S H2S + KSCN Солянокисл. a) As2S3 + 2S б) MgNH4AsO4-6H2O в) Mg2As2O7 100-105 Без нагр. 900
MgNH4AsO4-6H2O Магнезиальная смесь Аммиачная a) MgNH4AsO4-6H2O б) Mg2As2O7 Без нагр. 900
Ag3AsO4 AgNO3 СНзСООН + AcONa Ag3AsO4 500
Sb3+ Sb2S3 H2S а) НС1 (0,5 н.) б) НС1 (2,5 н.) в) НО (1,2 н.) оранж. Sb2S3 черн. Sb2S3 Sb2O4 В токе СО2, 270-300 100-110 650-900
SbO(C6H5O3) Пирогаллол Винная к-та SbO(C6H5O3) 100-105
Na[Sb(OH)6] Na2S, тиосоли разрушают H2O2, ос. обрабатывают NaOH и cn. Щел., 50% сп. NaSbO3 650-950
Bi3+ Bi(OH)CO3 (NH4)2co3 NH4OH Bi2O3 900
BiOCl nh4ci Слабоазотнокисл. BiOCl 100
BiOBr a) KBrO3 + KBr 6) NaBrO3 + NaOBr Слабоазотнокисл. Слабоазотнокисл., (NH4)2HPO4 BiOBr BiPO4 110-120 400-900
BiOI KI Слабоазотнокисл. BiOI 105
BiPO4 (NH4)2HPO4 Н3РО4 BiPO4 400-900
Bi(OH)(HCOO)2 hcoonh4 Слабоазотнокисл. Bi2O3 400-900
Bi2s3 H2S Слабосолянокисл. Bi2s3 100
Bi ECHO NaOH Bi 105
Bi Электролиз HNO3 Bi 105
vo3-, VO3+ VO2(C6H8O2N3) Купферон H2SO4 V2O5 700-950
Ag3VO4 AgNO3 NH4OH + AcONH4 Ag3VO4 100-150
AgVO3 AgNO3 CH3COOH AgVO3 60-500
Hg2(VO3)2 Hg2(NO3)2 Нейтр. V2O5 700-950
Nb(V), Ta(V) (Nb2O3 + Та2О3)- H2O a) NH4OH + NH4C1 б) Уротропин + пирогаллол Аммиачный буфер Нейтр. Nb2O5 + Ta2O5 Nb2O5 + Ta2O5 850-1000 1000-1050
(Nb2O5 + Ta2O5) + + таннин Таннин Нейтр. Nb2O5 + Ta2O5 850-1000
(Nb2O5 + Ta2O5) купферонат Купферон Сильносоляно- или сернокисл. Nb2O5 + Ta2Os 700-1000
sot BaSO4 ВаС12 НС1 (0,01 н.) BaSO4 800-900
C12H12N2H2SO4 Бензидин солянокислый, C12H12N2-2HC1 Слабосолянокисл. C12H12N2H2SO4 72-130
sot, S20;, s2or BaSO4 ВаС12 Окисл. Вг2 или Н2О2+ +NH4OH, осажд. в НС1(0,01 н.) BaSO4 800-950
S2" BaSO4 ВаС12 Окисл. Вг2, HNO3 + + НС1, Na2CO3 + NaNO3, Na2O2 + Na2CO3, осажд. вНС1 (0,01 н.) BaSO4 800-950
CdS->CuS CdSO4, CuSO4 Уксуснокисл. CuO <940
CuS CuSO4 Нейтр. CuO <940
s s Экстр. CS2 Испарение экстрагента S < 135
BaSO4 ВаС12 Окисл. Вг2, осажд. в НС1 (0,01 н.) BaSO4 800-950
434
Новый справочник химика и технолога
Продолжение таблицы 5.1.7
Опред. ион, в-во Осаждаемая форма Реагент Среда, условия осаждения Гравиметрическая форма Условия термообработки, t, °C
Se(IV, VI) Se N2H4 H2O или N2H4H2SO4, n2h4hci, nh2ohhci НС1 (14—20%) Se 105
Se SO2 или NaHSO3, Na2SO3, K2S2O5 НС1 (14—20%) Se . 105
Se FeSO4 или FeCl2, SnCl2 НС1 (14-20%) Se 105
SeO2’ SO2 SO2 НС1 (0,5-1 н.) Se 105
SeO2 SO2 SO2 HQ (4 н.) Se 105
Te(IV, VI) Те N2H4 H2O или n2h4h2so4, n2h4hci, nh2ohhci на (7-15%) Те 105
Те SO2 или Na2SO3, NaHSO3, K2S2O5 на (7-15%) Те 105
Те Гипофосфит натрия на (7-15%) Те 105
TeO2 Гидролиз HNO3, нейтр. до pH = 4 ТеО2 120
[(C6H5)4As]2-TeCl6 или [(С6Н5)4Р]2-ТеС16 (C6H5)4AsC1 или (C6H5)4PC1 НС1 (4—5 н.) или H2SO4 (3 н.) [(C6Hs)4As]2-TeCl6 или [(С6Н5)4Р]2-ТеС16 110
Сги Сг(ОН)3хН2О NH3 pH = 6-8 Сг2О3 800-950
Сг(ОН)3хН2О NH4NO2 pH = 4,6-6 Сг2О3 800-950
Сг(ОН)3хН2О KI + KIO3 pH = 7,6 Сг2О3 800-950
Сг(ОН)3хН2О KOCN pH « 9,3 Сг2О3 800-950
CrO2’, Cr2O2’ ВаСгО4 BaCl2 СНзСООН (0,001 н.) ВаСгО4 130
Ag2CrO4 AgNO3 Нейтр. Ag2CrO4 130
РЬСгО4 Pb(CH3COO)2 СНзСООН РЬСгО4 100-120
MoO2’ PbMoO4 (AcO)2Pb Уксуснокисл. PbMoO4 600
MoS3 H2S или тиоуксусная к-та H2SO4(1 н.) МоОз 500-550
Ag2MoO4 AgNO3 HNO3 Ag2MoO4 90-250
BaMoO4 ВаС12 Нейтр. BaMoO4 110-250
MoO2(C9H6ON)2 8-Оксихинолин СН3СООН + AcONH4 a) MoO2(C9H6ON)2 б) МоОз 90-200 <400 (cH2C2O4)
MoO2(C14H12O2N)2 а-Бензоиноксим H2SO4 (5-10%), ацетон (до 20%) MoO2(C14H12O2N)2 105
WO2’ WO3xH2O НС1, HNO3 Сильнокисл. WO3 800
WO3xH2O + цинхонин Цинхонин Сильнокисл. (HNO3) WO3 800
WO2(C9H6ON)2 8-Оксихинолин Уксуснокисл. WO2(C9H6ON)2 120
WO2(C13H10O2N)2 /V-Бензоил фенил гидроксиламин pH > 1, нагр. WO2(C13H10O2N)2 115
[(C4H9)3NH]3- •pw12o40 Три-н-бутиламин НС1 (0,25-2 н.), NaH2PO4 [(C4H9)3NH]3-PW12O40 105-150
UO2+ (NH4)3U2O7 NH4OH или пиридин, бензоат аммония, уротропин, таннин Нейтр. U3O8 750-950
UO2S H2S или (NH4)2S Слабощел. u308 850-950
Химические методы количественного анализа
435
Продолжение таблицы 5.1.7
Опред. ион, в-во Осаждаемая форма Реагент Среда, условия осаждения Гравиметрическая форма Условия термообработки, ?,°C
UO2+ (UO2)(C9H6ON)2- •C9H7ON 8-Оксихинолин Уксуснокисл. a) (UO2)(C9H6ON)2- •c9h7on б) (UO2)(C9H6ON)2 160 250-350
U(C6H5N2O2)4 Купферон Сильнокисл. (H2SO4) u308 800-950
F" PbCIF РЬС12 pH = 4,1-4,8 PbCIF 70-540
CaF2 СаС12 Нейтр. CaF2 400-950
BiF3 Bi(NO3)3 СН3СООН BiF3 50-93
(C6H5)3SnF (C6H5)3SnCl Нейтр. (C6H5)3SnF < 158
СГ AgCl AgNO3 Кисл. (HNO3) AgCl 70-600
Hg2Cl2 Hg2(NO3)2 Кисл. (HNO3) Hg2Cl2 < 130
С1О~ Pb(C102)2 Pb(NO3)2 Слабокисл. (HNO3) Pb(C102)2 <77
сю; C20H16N4HClO3 Нитрон Слабокисл. (H2SO4) C20H16N4HClO3 105
AgCl Восстановитель; AgNO3 Слабокисл. AgCl 130-150
сю; KC1O4 KC1 Солянокисл. KC1O4 110
c20h16n4hcio4 Нитрон Слабокисл. (H2SO4) c20h16n4hcio4 105
вг AgBr AgNO3 Слабокисл. (HNO3) AgBr 130
вго; AgBr Восстановитель; AgNO3 Слабокисл. AgBr 130
г Agl AgNO3 Слабокисл. (HNO3) Agl 60-900
Pdl2 PdCl2 Слабокисл. (НС1) a) Pdl2 6) Pd 80-365 1000, в атм. H2
ю; AgIO3 AgNO3 Нейтр. AgIO3 80^00
Мп2+ MnNH4PO4 (NH4)2HPO4 Аммиачная a) MnNH4PO4 H2O 6) Mn2P2O7 100 950
MnS H2S или (NH4)2S Аммиачная a) Mn3O4 6) MnSO4 в) MnS 1000-1100 450-550 450-550, в атм. Н2
(C10H7O5N4)2Mn Пикролоновая к-та Нейтр. (CioH705N4)2Mn 105
(C26H18O8N4S2)Mn Бриллиантовый желт. АсОН (C26H18O8N4S2)Mn 120
[Mn(C12H8N2)2]- •(SCN)2 о-Фенантролин Слабокисл.; SCN- [Mn(C12H8N2)2]. •(SCN)2 Без нагр.
Re О; C20H16N4HReO4 Нитронацетат Слабокисл. C20Hi6N4HReO4 110
[(C6H5)4As]ReO4 [(C6H5)4As]C1 Сильноаммиачная или сильнокисл. [(C6H5)4As]ReO4 106-185
Re2S7 Na2S или (NH4)2S Щел., затем НС1 (6 н.) Re2S7 Без нагр.
ReCi; или ReBr; (C26H30N4O2H)2- •ReCl6 Диантипирилпропил-метан НС1 (2 н) или НВг (2 н.) (C26H3oN 4O2H)2 • •ReCl6 100-105
Fe2+, Fe3+ FeS H2S или (NH4)2S Аммиачная Fe2O3 850-1100
Fei+ Fe(OH)3xH2O a) NH4OH б)Г+ ю; в) NO; г) Уротропин pH >4 pH = 7,6 pH = 4,6-6,0 pH = 5,4 Fe2O3 Fe2O3 Fe2O3 Fe2O3 850-1100 850-1100 850-1100 850-1100
Основные соли Fe(III) a) AcONa (гор.) б) Бензоат аммония в) Формиат аммония pH = 4,3-5,2 pH = 3,5-4 pH = 3,5-4 Fe2O3 Fe2O3 Fe2O3 850-1100 850-1100 850-1100
Fe(C6H5N2O2)3 Купферон Кисл. (H2SO4) Fe2O3 850-1100
436
Новый справочник химика и технолога
Продолжение таблицы 5.1.7
Опред. ион, в-во Осаждаемая форма Реагент Среда, условия осаждения Г равиметрическая форма Условия термообработки, t,°C
Со2+ CoS H2S pH = 4,3-7 а) Со б) Со3О4 в) CoSO4 H2; 1000 900-950 450-550
Co Электролиз NH4OH + (NH4)2SO4 Со Без нагр.
Co(OH)3 NaOH + K2S2O8 Слабощел. а) Со3О4 б) Со 900-950 H2; 1000
[Co(C5H5N)4]- •(SCN)2 Пиридин + NH4SCN Нейтр.; C5H5N a) [Co(C5H5N)4]-•(SCN)2 б) Со3О4 в) CoSO4 Без нагр. 900-950 450-550
Co(C10H6ONO)3- •3H2O а-Нитрозо-р-нафтол АсОН или НС1 a) Co(C10H6ONO)3 б) Со3О4 в) CoSO4 130 900-950 450-550
K3[Co(NO2)6] kno2 Слабоуксуснокисл. а) Со (электролиз) б) [Co((C5H5N)4]-(SCN)2 Без нагр. Без нагр.
Co(C9H6ON)2-2H20 8-Оксихинолин АсОН Co(C9H6ON)2 180
Ni2+ Ni(C4H7N2O2)2 Диметилглиоксим pH = 4,5-10 a) Ni(C4H7N2O2)2 6)NiO 110-130 900
Ni Электролиз Аммиачная Ni Без нагр.
Ni2+ [Ni(C5H5N)4]-(SCN)2 Пиридин +NH4SCN Нейтр. [Ni(C5H5N)4l- (SCN)2 Без нагр.
Ni(OH)3 NaOH + K2S2O8 или NaOH + Br2 Слабощел. (NaOH) NiO 900
Ni(C9H6ON)2-2H2O 8-Оксихинолин АсОН Ni(C9H6ON)2 200
Ru(III, IV) RuO2xH2O (NH4)2CO3, H2O2 pH «6 Ru Н2; 600
RuO2xH2O NaHCO3 pH = 5-7 Ru Н2; 800
RuCCioHuONSh Тионалид (0,2-0,5 М) Ru Н2; 800
Ru Mg Слабокисл. Ru Н2; 800
Rh(III) (Rh2S3) H2S НС1 (« 5%), NH4C1 Rh Н2; 800
Соед. с тиобарби-туровой к-той (состав не опр.) T иобарбитуровая к-та Кисл. (НС1) Rh Н2; 650-700
Rh(C12H10ONS)3 Тионалид pH ® 3, кип. Rh(C12H10ONS)3 120
Rh Ti2(SO4)3 или TiCl3 Кисл. (H2SO4) Rh Н2; 900
Pd(II) Pdl2 KI HNO3 (0,8 н.) a) Pdl2 6) Pd 110-130 Н2; 500; N2
Pd(C4H7N2O2)2 Диметилглиоксим Кисл. (НС1 или HNO3 « 0,5 М) a) Pd(C4H7N2O2)2 6) Pd ПО Н2; 500; N, или СО2
Pd Муравьиная к-та или гидразинсульфат, СО Слабокисл. (НС1) Pd Н2; 600; N2
Os(IV) OsO2xH2O NaHCO3 pH = 6 Os Н2; 500
Os(C12H10ONS)3 Тионалид НС1 (0,5 н.) Os (по убыли веса) 750
(C12H22O2N2)2H2- •OsC16 (предположительно) Сульфат стрихнина НС1 (Cj2H22O2N2)2H2- •OsC16 /= 0,1771 100-105
Os(C6H4NHN2)3-(OH)3 1,2,3-Бензотриазол Щел., сп. Os(C6H4NHN2)3-(OH)3 100
Ir(III, IV) IrO2xH2O NaBrO3 + NaHCO3 Слабокисл. Ir Н2; 500, затем N2, 700
Химические методы количественного анализа
437
Продолжение таблицы 5.1.7
Опред. ион, в-во Осаждаемая форма Реагент Среда, условия осаждения Г равиметрическая форма Условия термообработки, /,°С
1г(Ш, IV) Оранж, ос., состав не установлен 2-Меркаптобензотиа-зол Ацетатн. буфер 1г H2; 650-700; n2
Ir(C12H10ONS)3 Тионалид 1г(Ш), СН3СООН Ir(Cl2H10ONS)3 115-120
Ir(C7H5N2S)3 2-Меркаптобенз-имидазол 1г(Ш), pH = 3,27-5,37 Ir(C7H5N2S)3 120-140
(C26H30O2N4H)2- •[IrCl6] Диантипирилпропил-метан НС1 (0,1-0,5 н.) (C26H30O2N4H)2- [IrCl6l 100-110
K2IrCl6 КС1 + сп. Слабокисл., СГ; [1гС16]2- Ir Н2; 500
Pt (NH4)2PtCl6 NH4C1 + сп. Слабокисл. (НС1) Pt 800
PtS2 H2S НС1 (0,2-0,6 н.) Pt 800
Pt НСООН pH = 4,5 Pt 800
Pt(C6H5S)2 Тиофенол Слабокисл. Pt 800
Pt(C12H10ONS)2 Тионалид pH ® 3, кип. Pt 800
(C23H24N4O2H)2- [Ptl4] Диантипирилметан НС1 (0,5 н.), KI (C23H24N4O2H)2- [Ptl4] Без нагр.
(C7H6N2S)2[PtCl4] 2-Меркаптобенз-имидазол НС1 (0,1-5%) (C7H6N2S)2[PtCl4] 120-140
Таблица 5.1.8
Принятые сокращения:
Анал. — анализируемый Буф. — буферный
Восст. — восстановители Выдерж. — выдерживают Добавл. — добавляют Изб. — избыток
Мет. — метиловый
Меш. — мешают
Нагр. — нагревают
Н. р. — не растворяется
Опр. — определение, -ют, —ся
Ос. — осадок
Осажд. — осаждение, -ют
Отд. — отделение, -ют
Отстаив. — отстаивают
Переосажд. — переосаждение, -ют Перемеш. — перемешивание, -ют Прокал. — прокаливают Разб. — разбавляют
Сп. — спиртовой
Титр. — титрованный
Ф. б. — фильтр бумажный ф. Г. — фильтр Гуча ф. с. — фильтр стеклянный ЭДТА - Na — натриевая соль этилен-диаминтетрауксусной кислоты Экстр. — экстракт, экстрагируют Эт. — этиловый спирт Эф. — эфир диэтиловый
Основные гравиметрические методики определения неорганических соединений
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература*
I группа Периодической системы
Li+ H2SO4 Анал. р-р, pear. Выпар., высуш. при 200 °C Li2SO4 1,7,Ю
HC1 Анал. р-р (хлориды щ.м.) выпар., смачивают конц. р-вом. Экстр, сухим ацетоном, выпар., прокал. до плавления LiCl 1,7, 10, 11
Na2HPO4 Анал. р-р (щел.), pear. Выпар., раств. в NH4OH. Ф. б. Прокал. при 800 °C Li3PO4 506, 507
Опр. также в виде 2Li3[Fe(CN)6]-K3[Fe(CN)6] 5[(CH2)6N4-6H2O] или в виде LiF [505], [Zn(UO2)3Li(CH3COO)9] [507], [2ZnU2O7 Li2U2O7] [1], 2Li2O-5Al2O3 [507]
Na+ H2SO4 Анал. р-р выпар. 2 раза с pear, прокал. < 875 °C с добавкой (NH4)2CO3 (тв.) Na2SO4 569, 961
Цинк-уранил ацетат Анал. р-р (1 мл, содержащий < 8 мг Na), р-в (10 мл). Ф.с. (промыв, pear., сп., нас. натрий-цинк-уранилацетатом, эф.) Высуш. при 105 °C. Прокал. при 360-670 °C NaZn(UO2)3-(C2H3O2)9-6,5H2O Na2U2O7-2ZnU2O7 514
См. компакт-диск.
438
Новый справочник химика и технолога
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Na+ НС1 Анал. р-р (не более 200 мг Na) добавл. равный объем НС1 (конц.), выпар, на водяной бане в платиновом или фарфоровом тигле до образ, ос., добавл. равный объем НС1 (конц.) и выпар, досуха. К ос. добавл. ок. 0,5 мл НС1 (1:1) и повторно выпар, досуха, ос. нагр. в закрытом тигле 1-2 часа при 130 °C, после чего прокал. не выше 600 °C. Перед концом прокал. крышку тигля периодически поднимают. Li, К, Cs, Rb переводят в перхлораты, экстр, этилацетатом и бутанолом (1:1) Li и Na и осажд. Na р-ром НС1 в бутаноле-1 NaCl 1,6 6,7
Марганецуранил-ацетат 1 мл анал. р-ра (0,1-1,0 мг Na), 6 мл pear., перемеш., обмывают палочку 1 мл pear, и удаляют из р-ра. Через 1 час ос. ф.с., промыв. 6-8 раз по 1 мл сп. (96%, насыщ. натриймарганец-уранилацетатом), затем эф. и высуш. при 50-60 °C. Реагент: 7 г уранилацетата, 35 г ацетата марганца, 6 мл СН3СООН (лед.) и 100 мл Н2О нагрев, до полного раств., охлажд., прибавляют 1/3 объема сп. и немного NaCl. Через 2 суток фильтр, р-р и использ. в качестве pear. /= 0,01437 6,8
1-Нафтиламин-8-сульфоновая к-та Анал. р-р (40-80 мг Na) нейтр., медленно приливают 10 мл pear. (12%), выдерж. 30 мин при 10-15 °C, ф.с., промыв. 3 раза по 2-3 мл насыщ. сп. р-ром натриевой соли pear., 2 раза по 1 мл абсолютного сп. Высуш. при 105 °C. Продолжительность определения 60-80 мин. /= 0,09377 515
Оротовая к-та, соль с Н,№-диметил-этаноламином (0,1 М) Анал. р-р с pH = 6,5-8,0, изб. (20-25 мл) р-ра pear., выдерж. 2 час при 3 °C, ф.с., промыв. 2 раза по 2 мл сп. (70%), высуш. при 105 °C. Не меш.: NIL , Li+, щ. з. м. Меш.: К+ C5H3N2O4Na 9
Опр. также в виде NaMg(UO2)3(CH3COO)9-6H2O [508, 509, 514], {Cs9Na6[Bi(NO2)6]5} [510, 511], NaC104 [512], Na[Sb(OH)6] [513]
к+ (NH4)2SO4 Анал. р-р, p-в. Выпар., прокал. при 400-800 °C с (NH4)2CO3 (тв.) K2SO4
H2[PtCl6] а) Анал. р-р, pear. Выпар., добавл. сп. (80%). Ф. б. (промыв. сп.). Высуш. при 130-200 °C. б) Осажд. K2[PtCl6] (см. п. «а»). Ос. раств. в гор. воде, добавл. HCOONa, выпар., добавл. НС1 (разб.) Ф. б. (промыв. Н2О). Прокал. при 1000 °C K2[PtCl6] Pt 7,11, 524, 525 7, 526, 527
в) Ос. по п. «а» р-ют в гор. Н2О, добавл. HCOONa, по окончании восст. подкисл. HNO3, фильтр, и промыв. К фильтату и промывн. водам добавл. избыток р-ра AgNO3. Ос. AgCl анализируют как при опр. Ag AgCl f= 0,1814 10
Na3[Co(NO2)6] Навеску хлоридов или сульфатов щ.м. р-ют в фарф. чашке в 10 мл воды, подкисл. СН3СООН, и выпар, на водяной бане до малого объема, охлажд., конц. р-р pear, до бур. окр. р-ра. На другой день сливают р-р через ф.с., р-р выпар. до консистенции сиропа, дважды добавляя СН3СООН, охлажд., через тот же ф.с., промыв, хол. водой до бесцветной промывной жидк., сушат при 110 °C 1-2 час « K2NaCo(NO2)6 эмпир./= 0,1721 10,11, 25
НС1О4 Анал. р-р (хлориды щ.м.), pear. Выпар., добавл. Н2О (2-3 мл), бутил, сп. (кипящий, 33 мл на 1 мл Н2О). Ф.с. (промыв. бутил, сп.). Высуш. при 350 °C KC1O4 514, 522, 523
Химические методы количественного анализа
439
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
К+ Na[B(C6H5)4], 3% р-р (0,003% A1C13) Анал. p-p (> 1 мг К, НС1, 0,1 н.), pear. Harp. 15 мин. Ф.с. (промыв, нас. р-ром К[В(С6Н5)4]. Высуш. при 110-130 °C К[В(СбН5)4] 534, 535, 536, 539
NaHC4H40e или Н2С4Н40б Р-р хлоридов Na и К выпар, досуха, раств. в миним. объеме воды; осажд. 2% сп. р-ром Н2С4Н4О6 (или насыщ. битартратом Na); выдерж. 2 час, кип., ф.с., промыв, сп. (96%), высуш. при 80 °C Оптимальный pH осажд. 3,4-3,6. Не меш.: Li, Mg, Na, Са, NO,, СГ, SO^". КНС4Н4О6 10, 537, 538
Битартрат пиридина (или анилина) Анал. р-р (5 мг-мл"1 К), равный объем сп., добавл. мет. оранж. и по каплям при перемет, pear, до желт, окраски р-ра, отстаив. 2 час, ф.с., промыв, сп., высуш. при 80 °C КНС4Н4О6 540
Дипикриламин Анал. р-р (pH = 7-8,8), натр., добавл. гор. pear. Выдерж. 1 сутки. Ф.с. (промыв, нас. р-ром р-ва). Высуш. при 100— 105 °C KN[C6H2(NO2)3]2 Ю, 11, 514, 545
Опр. также в виде: КС1 [516, 517], КЮ4 [518], Кз(МоО3)12РО4 [519], K2Pb[Ni(NO2)6] [533], K2Pb[Cu(NO2)6] [520], KReO4 [348, 521], Pt (восст. магнием [7, 526], ртутью [528], водородом при высокой температуре [528], формальдегидом [529], щавелевой к-той [514] или оксалатом [530]), K2PtF6 [531], K2PtBr6 [532], K2S20g [254, 541], K2SiF6 [219], [K20-2ZrO2-2S03 «H20] [542], Bi2S3 [543], алюмо-калиевых квасцов [544]
Rb+ H2PtCl6 См. опр. К+
НС1 См. опр. К+
(NH4)2SO4 См. опр. К+
Дипикриламин См. опр. К+
Na[B(C6H5)4] См. опр. К+ 546
SnCl4 (нас. р-р) Анал. р-р (хлориды Щ. М. после отделения К+) натр., добавл. кип. p-в. Выдерж. 4 час. Ф.с. Высуш. при 110 °C Rb2[SnCl6] 11
Cs+ (NH4)2SO4 См. опр. К+
H2PtCl6 См. опр. К+
НС1 См. опр. К+
Na[B(C6H5)4] См. опр. К+ 546
SnCU (нас. p-p) См. опр. Rb+
Cs+, Rb+ SnCl4 (нас. p-p) Анал. р-р (хлориды Щ. М.), осажд. К, Rb, Cs хлороплати-натным методом (см. опр. К), Pt осажд. HCOONIL, ф-т выпар. досуха, удаляют соли NIL . Сухой ос. р-ют в нескольких каплях воды, медленно при перемет, добавл. 5 мл смеси (1:2) НС1 (конц.) и сп. Ос. ф.с., промыв, абсолютным сп., высуш. при 110 °C и взвешивают CsCl, Rb2SnCl6 11
Ф-т нагр. до кип., добавл. нагр. до кип. р-р pear., после охл. отстаив. 4 час, ф.с., промыв, абс. сп., высуш. при 110 °C и взвешив. сумму Rb2SnCI6 и CsSnCU. Ос. р-ют в винной к-те (3%), осажд. Sn в виде сульфида, ф-т выпар, досуха и осторожно прокал. (для разрушения винной к-ты). К смеси хлоридов добавл. равное кол-во FeCl3, р-ют в нескольких каплях воды, добавл. 10 мл лед. СН3СООН, нагр. до кип., добавл. 1 мл хол. р-ра SbCl3 (30%) в лед. СН3СООН, нагр. 1 час при 100 °C, отстаив. 12 час. Ос. ф.с., промыв, р-ром SbCl3 (5%) в лед. СН3СООН. Ос. р-ют в НС1 (1:4), осажд. сурьму H2S, ос. фильтр., ф-т выпар, досуха, ос. слабо прокал. и взвешивают CsCl, пересчитывают его на Cs2SnCl6 и по разности опр. Rb2SnCl6
440
Новый справочник химика и технолога
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Cs+, Rb+ (NH4)2SO4 Р-р pear.: 1 г (NH4)2SO4 р-ют в 20 мл воды, добавл. 100 мл сп. (95%) и несколько крист. (NH4)2SO4. Смесь хлоридов К, Rb, Cs р-ют в 0,4 мл воды и нас. газообразным НС1, добавл. 10 мл абсолютного сп. (нас. НС1). Ос. КС1 отфильтр. При необходимости экстракцию повторяют. Р-р содержит RbCl + CsCl + 0,6 мг КО (на каждую экстракцию). Р-р выпар. досуха, ос. р-ют в 0,1 мл (NH4)2SO4 (5%) и добавл. 5 мл р-ра pear. Отстаив. 30 мин, ф. Г., ф-т собирают во взвешенный платиновый тигель. Ос. промыв. 3 раза по 0,5 мл промыв, р-ром (готовят как р-р pear., перед добавл. сп. вносят 0,16 г NHjCl). К р-ру и промыв, водам добавл. немного (NH4)2SO4, выпар, до начала кристаллизации, добавл. 0,2-0,3 мл сп., снова выпар., закрывают тигель стеклом и нагр. для сублимации NH4C1 на стекло, затем (без стекла) до распл. и удаления (NH4)2SO4. Далее нагр. тигель докрасна, охл. в эксикаторе и взвешивают Cs2SO4. Ос. р-ют, р-р выпар, в платиновом тигле, прокал. и взвеш. в виде K2SO4+Rb2SO4. Вводят поправку на К (см. выше) и находят содержание Rb2SO4 (после вычитания расчетного веса K2SO4) Cs2SO4 7
Опр. также в виде CsNa[La(NO2)6], Cs2[Bi2J9], Cs3[Co(NO2)6]H2O [505]
Cu2+ nh4scn Анал. р-р (слабокисл.), нас. SO2, разб. Н2О. Нагр. до 100 °C, добавл. pear. Ф. с. (промыв. NH4SCN, р-ром SO2, 20% сп.). Высуш. при 105-120 °C (< 260 °C) CuSCN 547, 548
NH4SCN (1) и пиридин (2) Анал. р-р, pear. 1 и pear. 2 (до син.). Ф. с. Высуш. при 20 °C под вакуумом [Cu(C5H5N)2]- •(SCN)2 551
Электролиз HNO3 (конц.), добавл. Na-ЭДТА (для маек. Fe3+). Электролиз (0,5-2 а-дм-2) Cu 25,385
Реактив Рейнеке Анал. р-р (H2SO4< 3 н.), SnCl2, pear. Ф.с. Высуш. при 110 °C Меш. только Hg, Ag, Tl Cu[Cr(NH3)2-• (SCN)4] f= 0,1636 7,11, 13, 549, 550
N2H4H2SO4 (нас. р-р) Анал. р-р (> 0,2 мг Cu; H2SO4), pear. Ф.с. Высуш. при 100— ПО °C Cu SO4(N2H4)H2SO4 7, 400
Na2S2O3 В конической колбе анал. р-р (0,1-0,5 г Си, нейтр., без NO3), 5 мл H2SO4 (конц.), разбавл. до 100 мл, добавл. 20 мл Na2S2O3 (10%), закрывают воронкой, нагрев, до кип. Голуб, р-р переходит в желт, и выпадает черн. ос. Кип. до полного обесцвечив. р-ра («15 мин.), ф. б., прокал « 2 час при 850-940 °C CuO 1,11
K2Hg(SCN)4 Анал. р-р (содерж. HNO3 или H2SO4), нагр. до кип., добавл. K2Hg(SCN)4, ф. с., высуш. при 100-110 °C CuHg(SCN)4 14, 553
KI Анал. р-р (< 0,1 г Си, слабокисл., без NO3), 1 г NH4C1, нагрев. до кип., 1 г KI (или Nal) в 5 мл воды, по каплям Na2SO3 (5%) до полного обесцвечивания осадка и еще 10 капель, кип. 1-2 мин. На следующий день ф. с., промыв. 50 мл холодной воды, сушат 2 часа при 130 °C Cui 1,552
8-Гидрооксихи-нолин (3% р-р в сп.) а) Нейтр. или слабокисл. анал. р-р, 3 г крист. AcONa, 10-12 мл СН3СООН (лед.), разб. до 100 мл, нагр. до 60 °C, pear, при перемеш. до пожелтения р-ра, нагрев, до 80-90 °C, ф. с., промыв, гор. водой, высуш. при 105-110 °C Не меш.: Be, Mg, Са, Cd, Pb, As, Mn, PCX . Cu(C9H6ON)2 /= 0,18059 1,556
Химические методы количественного анализа
441
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Си2+ б) Слабокисл. анал. р-р (3-100 мг Си), 3-5 г винной к-ты, NaOH по фенолфталеину и еще 2 мл NaOH (2 М), разбавл. до 100 мл. Осажд. при 20 °C. Нагрев, до 60-70 °C, охлажд., ф. с., промыв, р-ром тартрата Na (1%), водой, высуш. при 105-110 °C. Не меш. по 50 мг Al, Pb, Sn(IV), As(V), Sb(V), Bi, Cr, Fe. в) Возможно осажд. из гомоген. р-ра. Анал. р-р (pH = 8-10) добавл. ацетон, хлороформ, воду, р-р pear, в ацетоне и гликоколевый буфер с pH = 9,8, нагр. при 80 °C до испарения р-телей. Ос. (крупные ромбические призмы) ф.с., промыв, гор. водой, высуш. при 105-110 °C Cu(C9H6ON)2 Cu(C9H6ON)2 1,554 15, 16
Салицилальд-оксим (1% р-р В СП.) Слабокисл. анал. р-р, 20 мл винной к-ты (75%), нейтр. NFUOH (конц.) по мет. кр., СН3СООН (конц.) до изменения цвета, 30 мл р-ра pear., ф.с., промыв, гор. водой 15-20 раз и 1 раз сп., высуш. при 105-110 °C Cu(C7H6O2N)2 17, 18, 25
Хинальдиновая к-та Р-р pear.: 5,64 г хинальдината Na в 150 мл воды Анал. р-р (нейтр., 150-160 мл), 2-5 мл H2SO4 (2 н.), нагрев, до кип., pear, в изб., выдерж. 3-5 мин, ф. с., промыв, гор. водой до отрицательной реакции на Fe2+. Высуш. при 100 °C (C10H6NO2)Cu-H2O 1,555
Опр. также в виде: Си [559] (после восст.: Fe, Al, Zn, Cd, Mg, Na3AsO3, гипофосфитами, соед. V(II) [557], NaBH4 [558], уротропином [560]), Cu2S [1], CuS (осажд. K2CS3) [561], CuC2O4 [562], Cu2[Fe(CN)6 [559], Cu[Fe(CN)5NO]-2H2O (осажд. нитропруссидом Na) [563], Cu(IO3)2 [564], CuSO4 и CuSO4H2O [565], CuO (осажд. щелочами) [1, 559]
Ag+ НС1 Анал. р-р, HNO3 (1%), pear. Нагр. до 70 °C. Оставляют на несколько часов в темноте. Ф. с. (промыв. 0,06% HNO3). Высуш. при 130-150 °C AgCl 1,25, 566
NaBr или КВг Методика аналогична осажд. AgCl. Ос. более чувствителен к свету. Кроме высуш. при 130-150 °C возможно прокал. при 430 °C. В этом случае ос. ф. б., озоляют, металлическое Ag переводят в AgBr действием бромной воды, прокал. при 430 °C AgBr 567
Аскорбиновая к-та Анал. р-р (нейтр., 0,1-0,7 г Ag), 5 мл HNO3 (1:1), разбавл. до 50 мл, нагрев, до 95-100 °C, свежеприготовл. р-р pear. (5%) 5 мл на 100 мг Ag, слабо кип. 15 мин. Непосредственно перед фильтр. 1 мл pear, и гор. ф. т. Промыв. 50 мл гор. и 25 мл хол. воды, высуш. при 100 °C, затем 1 час при 500 °C, поместив тигель с ос. в закрытый фарфоровый тигель Ag 1,592
NaIO3 Анал. р-р (0,1-0,15 г Ag, 70 мл), pH = 2, pear. (5%), выдерж. 2 час, ф. с., промыв. 4—6 раз хол. водой, сп., эф., высуш. при 115-120 °C. Погрешность 0,15% AgIO3 19, 568
(NH4)2s Анал. р-р (слабоаммиачный, 80-250 мг Ag), pear. (1-2 н., приготовленный при низкой температуре), ф.с., промыв, водой, сп., эф. Высуш. 1-2 час в вакуум-эксикаторе. Ошибка определения < 0,3%. Ос. не содержит S0 Ag2S 338
K2CrO4 К изб. р-ра К2СгО4 (разб. до 100 мл) добавл. анал. р-р, ф.с., промыв. 3-4 раза хол. водой, раств. в NH4OH, р-р кип. до удаления NH3. Ос. AgCrO4 тот же ф. с., промыв, хол. во-лой, сп., эф., высуш. при 50-60 °C Ag2CrO4 570
Na2Te (2 н.) Р-р pear.: Н2Те (А12Те3+НС1) пропускают через NaOH (2 н.), изб. Н2Те удаляют током СО2. Анал. р-р, pear, до выпадения ос., ф. с., промыв, водой, сп., высуш. 5 мин, выдерж. в эксикаторе 30 мин Ag2Te 571
442
Новый справочник химика и технолога
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Ag+ [Си^даад-(NO3)2 Анал. р-р (0,02-0,11 г Ag, нейтр., 50-100 мл), изб. KI до раств. ос., нагр. до 70-80 °C, добавл. pear. (нас. р-р), охл., ф. с., промыв, р-ром (0,1 г KI и 0,3 г pear, в 1 л), 1-2 мл этого же р-ра разб. сп. и эф. (1:1), высуш. в вакуум-экси-каторе [Cu(C2H8N2)2]-• [Agl2]2 /= 0,2378 581
1-Амидино-2-тиомочевина (1%) Анал. р-р (NH4OH) натр., изб. pear., NaOH до pH = 10,5, нагр. до коагуляции ос., ф.с., промыв, водой, высуш. при 105-112 °C Ag 585
Бензотриазол (2-5% р-р в NH4OH) Анал. р-р (10-100 мг Ag, нейтр. или слабокисл.), 1-10 г ЭДТА, нагр. до 60-90 °C, изб. pear. (10 мл на 10-100 мг Ag). После коагуляции ф.с., промыв, водой, высуш. при 110 °C AgC6H4N3 /= 0,4774 587, 591
4-Нитро-1окси-1,2,3-бензотриазол (0,4% р-р в 50% сп.) Анал. р-р (нитрат) разб. до 100 мл, нейтр. до pH = 3-4, добавл. 2-кратный изб. pear., выдерж. 30 мин при 100 °C, ф.с., промыв, хол. р-ром pear. (0,06%), сп., высуш. при 110-120 °C. Продолжительность опр. 1 час. Не меш.: Na, К, Zn, Cd, Ni, Со, Ва, Са, Sr, Mg, Pb, Pt, Hg, Cu, Bi, Au, As, Sb, Sn, Fe, Cr, Mn, Al, Tl, Ti, Ce, Be, UOV , Th, Zr AgC6H3O3N4 /= 0,3759 588
2-Метил- меркапто-бензимидазол (0,5% в сп.) Анал. p-p (ok. 50 мг Ag), ЭДТА (10%), тартрат Na, нейтр. (NaOH, CH3COOH) до pH = 8,4-10, разб. до 150 мл, нагр. до 60-70°С, pear. (5 мл на 10 мг Ag), выдерж. 5 мин при 60-70 °C, ф.с., промыв, гор. водой (с добавл. сп.), высуш. при 105-110 °C. Не меш. (в мг): Mn, Са, Sr, Ва, Mg (300); Со, Ni (250); Ti, Th, Zr (200); Cr, Fe (150); Cu, Pb, Cd, As, Bi, Pt (120); Hg, Zn, Tl, Be, Pd, U, W, V (100). Hg(I) окисл. до Hg(II) AgC8H7N2S 589
Диэтилдитиофос-форная к-та Анал. p-p (0,01-0,1 г Ag, NO(, pH < 1,50 мл), добавл. 2-кратный изб. pear., ф.с., промыв. 100-150 мл воды (с добавл. pear.), высуш. 1 час при 105-110 °C. Ошибка опр. < 0,4% Меш.: Hg(II), Pb, Bi AgC4H10O2PS2 f= 0,36819 590
NH4[Cr(NH3)2- •(SCN)4] (соль Рейнеке) Кисл. анал. р-р, нагрев., pear. Возможно определение 5 мг Ag в 100 мл Ag[Cr(NH3)2- •(SCN)4] 19, 20
Нитропруссид натрия Ос. нечувствителен к свету. Анал. р-р (нейтр. или слабокисл.), pear. (0,1 н.), 1-2 г NH4NO3 (крист.), ф.с., промыв. NH4NO3 (3%), водой, сп., эф. Высуш. в вакуум-эксикаторе. В присутствии Zn и Cd осажд. при 50-60 °C Ag2[Fe(CN)5NO] 572
Электролиз Анал. р-р (0,1—0,6 г Ag), 3-4 мл H2SO4 (конц.), упар. до паров H2SO4, разбавл. до 150 мл, нагрев, до 90 °C до раств. ос., электролиз (1,2 В, 0,1 А) Ag 1, 573, 574
Опр. также в виде: Agl [567, 575], AgSCN [576, 577], Ag2WO4 [577], [СгХОН^^гСНгСНг^гМ^^г [578], [Co(NH2CH2CH2NH2)2(SCN)2][Ag(SCN)2] [579], Ag[(C5H4N)2]2-(C104)2 [580], [Cu(NH2C3H6NH2)2][Agl2]2 [582], Ag2C2O4 [586], Ag (после восст. NaOH+H2O2 [583], NaBH4 [584])
Au(III) Гидрохинон Анал. р-р (НС1, 1 н.), pear. Нагр. до 100 °C. Ф. б., промыв, гор. водой. Прокал. при 900 °C Au 7, 428
so2 Анал. р-р (азотнокисл.) упар. до сиропообразн. сост., дважды упар. до сиропообразн. сост. с 5 мл НС1 (конц.), добавл. 25 мл воды, перемеш., еще 125 мл воды, фильтруют. Фильтрат упар. до 75 мл, охл. до 50 °C, добавл. 5 мл НС1 (конц.), 25 мл Na2SO3 (насыщ.). Отстаив. 45 мин, т.ф., промыв, гор. НС1 (0,1 и.), высуш. при 120 °C и прокал. при 900 °C. Ошибка опр. ±0,18% Au 7, 593, 594
Химические методы количественного анализа
443
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Г равиметрическая форма осадка Литература
Au(III) Н2С2О4 (10%) Анал. р-р (без оксидов азота) разб. до 100 мл, добавл. 2 мл НС1 (конц.), 5 капель H2SO4 (конц.), 2-3 мл pear., выдерж. 12 час при 100 °C, ф. б. или ф.т., промыв, водой, прокал. при 900 °C, охл. 15 мин на воздухе, 20 мин в эксикаторе, 20 мин под колпаком весов. Не меш.: Se и Те, применена для опр. Ап в цианистых р-рах Au 7,428
FeSO4 а) Анал. р-р (хлоридный), 20 мл НС1 (конц.), разб. до 200 мл, нагр. до кип., по капллям 20 мл pear. (1,5 г FeSO4-7H2O раств. в 20 мл), кип. 15 мин до коагуляции ос. Отстаив. 24 час при 80 °C, ф. б., промыв, гор. НС1 (10%) до удаления Fe3+ (реакция с SCN~), прокал. при 600 °C Au 7, 454
б) Опр. в присутствии Те: анал. р-р, НС1 до содержания 1,5-2%, разб. водой до 175 мл, нагр. до кип., добавл. 1,5 г FeSO4. Р-р кип. 10-15 мин, ф.Г., промыв, и прокал. по п. «а» Au 602
Н2О2 (3%) Анал. р-р (200 мл), NaOH (10%) до pH = 13 (по универсальной инд. б.), 20 мл pear., покрывают стеклом. Затем кип. до осажд. ос. и просветления р-ра, ф. б., промыв, малыми порциями гор. НС1 (10%), озоляют и прокал. при 600 °C Au 595
Формальдегид (35%) Анал. р-р (200 мл), 3 мл pear., NaOH (10%) до pH = 13 (универсальная инд. б.). Нагр. до кип. и выдерж. 15 мин при 100 °C, в ф-те до осажд. Ап, ф. б., промыв. 50 мл НС1 (10%), трижды сп., озоляют и прокал. при 600 °C Au 1,597
Аскорбиновая к-та (5%, свежеприготовленная) Анал. р-р (без NO,), 2 мл НС1 (конц.), разб. до 50 мл, нагр. до 80 °C, добавл. 10 мл pear, (по каплям, при перемеш.), кип. 5-10 мин до коагуляции ос., охл., ф. б., промыв. 150 мл гор. НС1 (10%), водой, озолляют и прокал. 30 мин при 600 °C Au 598
Диметилглиок-сим (1% В СП.) Анал. р-р (без оксидов азота, < 25 мг Au), разб. до 50 мл, добавл. 0,5 мл НС1 (конц.), 100 мг pear, в виде р-ра в сп., кип. 30 мин, ф. б., далее по методике с гидрохиноном Au 428, 599
NaNO2 (4%) а) Анал. р-р ( АиС14, 20 мл), 50 мл CH3COONa (10%), 5 мл НС1 (6 н.), 25 мл pear., разб. до 150 мл, нагр. до кип. Ос. ф. Г., высуш. при 110 °C. Фильтрат имеет pH = 1,6. б) Осажд. из буферного р-ра, содержащего тартрат K-Na. Возможно опр. в присутствии 5-кратного изб. Те Au Au 7, 600, 601 602
н4р2о6 (0,25 М) Анал. р-р (слабокисл., 50-300 мл), 2 г NaCl, нагр. до кип., добавл. 3-кратный изб. pear., отстаив. при 100 °C до коагуляции (при малых количествах Au на 3-4 часа). Ф. б., промыв. СНзСООН (1%) до удаления СГ. Прокал. при 600-900 °C Au 603
Zn Анал. р-р [Au(CN)2]“ (50 мл), 20 мл РЬ(СН3СОО)2 (нас.), нагр., добавл. цинковую пыль, затем НС1 (до сильнокисл, р-ции). Ос. отфильтр., кип. с водой, отфильтр., фильтр с ос. обрабатывают HNO3 (d= 1,2), отфильтр., кип. 15 мин с HNO3 (конц.), охл., разбавляют водой, ос. Au ф. б., озоляют и прокал. как по методике с SO2 Au 604, 605, 606
NaBH4 (1%) Анал. р-р (100 мг Au), создают среду 0,1 н. НС1 (или HNO3) - 0,5 н. NaOH, по каплям pear., отстаив. 1-2 час при 100 °C, ф. б., промыв, водой, прокал. при 600-900 °C. Меш.: Cu, Ag, Со, Ni, Pd, Pt, Cd, Hg, Sn, As, Sb, Bi Au 558
444
Новый справочник химика и технолога
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Au(III) Си2С12 Анал. р-р (10-85 мг Au, 10-30 мл), НС1 до конц. 2-3 М, медленно добавл. р-р pear., выдерж. при 100 °C, 1 час, охл., ф. с., промыв. НС1 (конц.), гор. водой, сп., высуш. при 110-120 °C. Не меш.: Cu, Zn, Cd, Mn, Со, Ni, Pd, Al, As(III), Bi, Sb(III), Ce, Th, Pt(IV), Sn(IV), Ti(IV), Cr(III, VI). Меш.: Pb, Те, Se, Ag Au 607
CrSO4 (0,12 н.) Анал. p-p (25 мл, хлоридный), медленно добавл. 30-40 мл pear., выдерж. при 100 °C 3 часа, охл., ф.с., промыв, водой, СП., высуш. при 110 °C. Меш.: Hg(I, II), Sb, Bi, Pt, Se, Те Au 176
VSO4 Анал. р-р (0,1 г Au, 20 мл), НС1 до конц. 2-3 М, нагр., малыми порциями при перемеш. р-р pear., выдерж. 2 час при 100 °C, ф.с., промыв, водой, сп., высуш. при 105 °C. Меш.: Cu, Hg, Sb, Bi, Se, Те, Pt, Pd Au 497
CdS Анал. р-р (> 20 мг Au, 10 мл), мет. оранж., НС1 (1:2) до кисл. р-ции и изб. 2-3 мл. Нагр. и кип. 1 мин, добавл. 5-10 мл гор. воды, 0,05-0,1 г порошка CdS, перемеш., кип. 2 мин, ф.т., промыв. 2 раза НС1 (1:2) и 2 раза гор. NH4C1 (1%). Прокал. 3-5 мин при 800 °C в фарфоровом тигле, обрабатывают ос. 1-2 мл HNO3 (1:1), нагр., фиьтр. через тот же фильтр, промыв. гор. водой (4—5 раз). Прокал. в том же тигле при 800-900 °C Au 618
Соль Рейнеке (1% свежеприготовленный Р-Р) Анал. р-р (5-10 мг Au) разб. до 50 мл НС1 (1-2 н.), добавл. 2 мл Na3AsO3 (0,5 н.), нагр. до 100 °C до обесцвечивания р-ра. Добавл. pear. (1,5 мл на 5 мг Au), отстаив. 5 мин в тепле. Охл., ф.с., промыв, теплой НС1 (0,01 н.), высуш. при 110-120 °C Au[Cr(NH3)2-•(SCN)4] f= 0,3825 609, 610
Тиогликолевая к-та (10%) Анал. р-р (6-76 мг Au), НС1 до 6 н. конц., разб. до 50 мл, медленно при перемеш. 10-12 мл pear., выдерж. 20-30 мин при 100 °C, затем 30-60 мин при 20 °C, ф.с., промыв, декантацией 5-6 раз по 10-15 мл воды, высуш. при 110— 120 °C. Ошибка ± 0,5%. Меш.: Pt, Zr, H2SO4. Ос. можно через ф. б. и прокал. до Au C2H3O2Au Au 611
8-Меркапто-хинолин (0,1 М в сп., свежеприготовленный) Анал. р-р (0,2-20 мг Au в виде AuCL, 0,1М КС1), 0,5-1,0 мл pear., ф. с., промыв, водой, высуш. при 80-90 °C HAu(C9H6NS)4 612
Тиофенол (4% в сп., вежеприго-товленный) Анал. р-р (100 мл, 0,1 н. по НС1 или HNO3), изб. pear. (1 мл на 10 мг Au). (Pear, хранить в герметично закрытом сосуде!) Р-р с ос. кип. 2 час, ф.с., промыв, сп., высуш. при 100 °C C6H5SAu 445, 613
Опр. также в виде: Au (после восст. Al-фольгой [614], SbCl3 [619], Mg [620], сахарозой [621], уротропином [615], смесью НСООН+НС1О4 [616], электролизом [385], Au2S3 [617], Au2SeH2Se [465]
II группа Периодической системы
Ве2+ NH4OH Анал. р-р, Na-ЭДТА (15% р-р). Нагр. до 100 °C, добавл. NH4CI, pear, до pH = 9,6. Ф. б. (промыв, гор. 2% р-ром NH4NO3). Прокал. при 1000 °C BeO 622
Мочевина Анал. р-р (30-60 мг Be), 2 г (NH4)2SO4, 10 г NH4C1, 10 г pear., разбавл. до 500 мл, нагр. медленно (1 час) до кип., 2 час кип. (при постоянном объеме), выдерж. 1 час на водяной бане, ф. б., прокал. при 1000 °C. Ос. Ве(ОН)2 плотный, легко фильтр. Абс. ошибка 0,2 мг BeO 21
Химические методы количественного анализа
445
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Ве2+ NH4NO2 (6% р-р) Анал. р-р (10-300 мг ВеО, слабокисл.), разб. до 1 мг ВеО в 1 мл, р-р Na2CO3 до помутнения, просветляют 1-2 каплями НС1, нагр. до 70 °C и при непрерывном продувании воздуха добавл. 50 мл pear, и 20 мл СН3ОН на каждые 100 мг ВеО, выдерж. 30 мин при слабом кип., добавл. 10 мл СН3ОН и выдерж. 1 час на водяной бане, ф. б., промыв, гор. водой, прокал. при 1100 °C ВеО 1,629, 630
Таннин + NH4OH Анал. р-р (10-150 мг ВеО, слабокисл., 300-400 мл), 20-30 г NH4NO3, нагр. до кип., при перемет, добавл. изб. тан-нина (10%), затем NH4OH до полного выделения ос. (Р-р щел. по лакмусу). Ф. б., промыв, гор. водой. При большом содержании Щ. М. ос. переосажд. Ос. высуш. при 100-130 °C, озоляют, смачивают HNO3, прокал. в Pt или кварцевом тигле при 1100-1200 °C, охл. в эксикаторе над H2SO4 или Р2О5, быстро взвешивают ВеО 632, 633
(NaF+BaCl2) 10 мл анал. р-ра (7-22 мг Be), 50 мл р-ра NaF (2,5%), раз-бавл. до 300 мл, 4 г борной к-ты, нейтр. р-ром НС1 (разб.) до pH = 4, нагр. до кип., добавл. 15 мл гор. р-ра ВаС12, выдерж. на кип. водяной бане 2 час, ф.с., промыв. 8-10 раз гор. водой (по 4 мл) до удаления СГ, затем сп. Высуш. при 110-120 °C BaBeF4 22, 23, 631
(NH4)2HPO4, 2 М Анал. р-р, р-в, Na-ЭДТА (15% р-р), NH4OH (10% до помутнения), CH3COONH4 (до pH ® 5). Кип. 3 мин. Переос. Ф. б. (промыв. 1% р-ром NH4NO3). Прокал. при 800 °C ВеР2О7 623, 624
(NH4)2CO3 (I) и [Co(NH3)6]C13 (II) Анал. р-р (3-4 мг Be), pear. I, pear. II (до ор.-желт.), добавл. сп. Выдерж. 1,5 часа. Ф. с. (промыв, сп. + p-в II; сп., эф.). Высуш. в вакуум-эксикаторе [Ве2(СО3)2(ОН)3- (Н2О)3]- [Co(NH3)6-3H2O] /= 0,041 625, 626, 627
2,2-Диметил-гександион-3,5 (нас. р-р) а) Анал. р-р Na-ЭДТА, NH4OH (до pH = 7-8), pear. Выдерж. 4 часа. Ф. с. (промыв, водой). Высуш. при 50-55 °C. б) К р-ру pear. (15 мл на 1 мг Be) добавл. пиридин до pH = = 5,5-6 и 1-2 мл изб., приливают анал. р-р (0,1-1,0 мг Ве/100 мл, всего до 3 мг Be). Крист, ос. ф.с., промыв. 4-5 раз водой, сушат при 50-55 °C. Ошибка 0,1-1%. Мешают все сопутствующие элементы Ве(С8Н13О2)2 Ве[СН3СОСН2-СОС(СН3)3]2 628, 629 24,628
8-Оксихиналь-дин (2% р-р в 1% СН3СООН) Анал. р-р (2-10 мг Be), 1 г NH4C1 в 50 мл, NH4OH (2 н.) до pH « 9, pear, до полного осажд. желт. ос. и еще 30% изб., нагр. 30 мин (60 °C), ф.с., промыв. NH4OH (1:100), высуш. 30 мин при НО °C Be(C10H8NO)2 23, 640
Опр. также в виде: ВеО (после гидролитического осажд. KOCN, а-пиколинэтиламином, метиламином, бензоатом аммония, уротропином, Ю3 + Г, гуанидинкарбонатом) [7, 38, 634-639]
Mg2t (NH4)2HPO4 а) Анал. р-р (слабокисл.), pear., NH4OH. Ф. б. (промыв, хол. 10% р-ром NH4NO3). Прокал. при 1050-1100 °C. б) Осажд. как в п. «а». Ф.с. (промыв. NH4OH, сп.). Высуш. при 20 °C. в) Гомогенное осажд. с помощью РОС13 Mg2P2O7 MgNH4PO4-6H2O 1,7, 25 641- 643 644
8-Оксихинолин а) (2% с добавл. 2 М СН3СООН) а) Анал. р-р, pear. Нагр. до 100 °C, добавл. CH3COONH4, разб. NH4OH (небольшой изб.). Ф. с. (промыв, гор. водой). Высуш. при 250 °C или при 150-160 °C. Прокал. при 1000 °C. Mg(C9H6ON)2 Mg(C9H6ON)2-•2H2O MgO 25
446
Новый справочник химика и технолога
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Mg2+ б) (2% в сп.) в) (2% в сп.) г) (2% р-р в ацетоне) б) Анал. р-р (100 мл), 3 г тартрата натрия, 15-20 мл NaOH (2 М), небольшой изб. pear., нагр. до 80 °C (до перехода ос. в крист, состояние). Охл., ф. с., промыв, хол. р-ром (1% тартрат натрия с добавл. NaOH), затем хол. водой. Высуш. по п. «а». в) Осажд. в присутствии фосфатов. Анал. р-р (0,2-0,5 мг Mg, < 5-кратный изб. РО1 , 50 мл), 1-3 г NH4C1, NH4OH до щел. р-ции, изб. pear., нагр. и кип. 5-10 мин, охл. до 80 °C, ф. с., далее по п. «а» г) Гомогенное осаждение. Анал. р-р (7-16 мг Mg, 10 мл), 9 мл СН3СООН (2 М), 10 мл NH4C1 (0,5 М), 20 мл воды, 30 мл этаноламина (1,66М). Добавл. 80 мл ацетона, 10 мл pear. Р-р с ос. помещают на водяную баню (65-70 °C) на 3 час, ф. с., промыв. NH4OH (1%), высуш. при 102 °C 2 час Mg(C9H6ON)2- 2Н2О /= 0,06975 645 646
Опр. также в виде: MgNH4AsO4-62H2O [647, 648], Mg2As2O7 [649, 650], MgO (после осажд. MgCO3) [651], MgF2 [652], NaMgF3 или KMgF3 [653], MgSO4 [654], MgC2O4-2H2O [655], MgC2O4 [656], MgO [657, 658], комплекс с ЭДТА [659], ос. при действии феррицианида калия в присутствии уротропина [233]
Са2+ (NH4)2C2O4, 4% р-р а) Анал. р-р, pear. Нагр. до 70-80 °C, медленно нейтр. NH4OH. Нагр. еще 30 мин. Через 2 час ф.с. (промыв, разб. (NH^CjO^. Высуш. при 100 °C. Прокал. при 475-525 °C. б) Осажд. СаС2О4 (см. выше). Ф. б. (промыв, разб. (ИЩ^АОД Прокал. при 1000 °C. в) Осажд. СаС2О4 (см. выше). Прокал. при 800 °C, добавл. H2SO4 (1:1), осторожно выпар. Прокал. при 500-600 °C. г) Осажд. СаС2О4 (см. выше), прокал. до СаО в платиновом тигле, добавл. 2-3 мл HF (40%), выпар, на воздушной бане досуха, прокал. 5-10 мин при 800-900 °C. Ос. CaF2 негигроскопичен и не разлагается при плавлении. д) Р-р pear.: 20 г крист, лимонной к-ты и 1 г салициловой к-ты в 1 л воды. Анал. р-р (0,02-0,3 г СаО) нейтр. NH4OH (1:5) до начала выпадения ос., добавл. 25 мл pear, (выпавший ос. должен раств.). Добавл. еще 12-13 мл pear., разбавл. р-р до 200 мл, нагрев, до кип. Добавл. крист. NH4C1 небольшими порциями до прекращения образ, ос., оставляют на ночь. Ос. прокал. до оксида, карбоната или сульфата кальция (см. выше). Не меш.: РО5, Al, Fe, Mg. е) Осажд. по Винклеру. Анал. р-р (< 0,1 г Са) нейтр. NH4OH или НС1 по мет. оранж., разб. до 100 мл, добавл. 3 г NH4C1 и 10 мл СН3СООН (1 н.). К кип. р-ру по каплям добавл. 20 мл pear. (2,5%), к концу добавл. перемеш. р-р, кип. 5 мин, охл. для полного осажд. СаС2О4. Ос. обрабатывают одним из указанных методов. ж) Термографическое опр. см. в разделе «Ва» СаС2О4Н2О СаСО3 СаО CaSO4 CaF2 СаО, СаСОз, CaSO4 СаС2О4Н2О 7, 25, 661, 662 25, 663, 664 666 1,7, 667 1,668 663
H2SO4 (1:1) Анал. р-р (нейтр. или слабокисл.), pear. (10-кратный изб.), сп. (4 объема). Выдерж. 12 час. Ф. б. (промыв. 75% сп.). Прокал. при 500-600 °C CaSO4 1,7, 666
(NH4)2MoO4 (0,4 М, слабо-аммиачный) Анал. р-р (< 0,2 г Са) нейтр. по мет. кр., 1-2 капли NH4OH (конц.) изб., нагрев, до кип. и по каплям pear, до появления изб. pear, (проба с нас. р-ром пирогаллола в СНС13 — бурое окраш.). Пробу делают после 10 мин кип. СаМоО4 1, 669, 670
Химические методы количественного анализа
447
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Г равиметрическая форма осадка Литература
Са2+ P-p охл., разбавл. 1:1 сп., перемеш. и оставл. на ночь. Ф. Г. или ф. т., промыв. 6 раз по 10 мл сп. (70%), сушат при 130 °C и затем медленно нагрев, до 800-900 °C и прокал. 30 мин. Метод применяют при необходимости определения РО*", Са и Mg
Опр. также в виде: Са(Ю3)2 [671], CaWO4 [669], Ca2As2O7 [672], K2Ca[Ni(NO2)6] [673, 674], СаС4Н4О6Н2О [675]
Sr2+ (NH4)2C2O4 (10%) а) Анал. р-р (< 0,15 г Sr), нейтр. по мет. кр., разб. до 100 мл, 1 мл СН3СООН (1 М), нагр. до кип., по каплям pear., оставляют на ночь, ф. с., промыв, сп. (50%) или нас. р-ром SrC2O4, 15 мл сп. (96%). Сушат просасыванием обеспыленного воздуха. б) Ос. по п. «а» можно ф. б. и после промыв, осторожно озоляют, добавл. несколько капель (NH4)2CO3, выпар, и осторожно прокал., медленно повышая температуру до 700 °C, выдерж. 20-30 мин. В токе СО2 (в тигле Розе) можно прокал. при 1000 °C. в) Термогравиметрическое опр. см. в разделе «Ва» SrC2O4H2O /= 0,45248 SrCO3 1,676, 677 678
H2SO4 Анал. р-р (НС1), pear. (10-кратный изб.), сп. Ф. с. или ф. б. (промыв. 75% сп.; разб. H2SO4; сп.). Высуш. при 100-300 °C SrSO4 1,676
(NH4)2CO3 (10%) Анал. р-р (ок. 150 мл, < 0,5 г Sr), NH4OH по фенолфталеину, нагрев, до 60-70 °C, 20 мл pear., нагр. до кип., охл., добавл. несколько капель NI14О11, перемеш. и выдерж. 5-6 час на холоду; ф. б., промыв, хол. водой с несколькими каплями NH4OH, ос. осторожно переносят в тигель, фильтр озоляют над ос., прокал. 20-40 мин при 500-800 °C (не выше 830 °C) SrCO3 679, 680
HNO3 (10%) Анал. р-р по каплям при сильном перемеш. добавл. pear. (4-кратный объем). Ос. отфильтр., высуш., р-ют в 10,0 мл воды, прибавл. из бюретки при перемеш. 26 мл HNO3 (100%), выдерж. 30 мин, ф. Г., промыв. 10 раз по 1 мл HNO3 (80%), высуш. 2 часа при 130-140 °C. При содержании Са более 50 мг проводят переосажд. Меш. Ва и РЬ Sr(NO3)2 7, 681
Опр. также в виде: SrF2 [682, 683], Sr(IO3)2 [648, 685], Sr2As2O7 [686, 687], SrCrO4 [680], SrC4H4O6 H2O [681, 688]
Ba2+ H2SO4 а) Анал. р-р (не более 200 мл, НС1 не более 1 мл в 100 мл р-ра), нагр. до кип., добавл. разб. H2SO4 (до 5 мл на 100 мл р-ра), выдерж. при 100 °C до осажд. ос., ф. б., промыв, гор. водой с добавл. нескольких капель H2SO4, затем водой, ф. б., высуш., обугливают, прокал. при 800 °C, охл., добавл. 1 каплю H2SO4, прокал. при 800-950 °C в течение 1-2 час. б) Ос. по п. «а» ф. с., промыв, сп. (75%), сп. высуш. при 180 °C или в токе фильтрованного воздуха в течение 40 мин в) Опр. в присутствии других катионов (кроме Sr и Са). Анал. р-р, Na-ЭДТА в изб., нейтр. до pH = 4,5-5 (ацетатный буфер: 130 мл конц. СН3СООН, 200 мл конц. NH4OH, 200 мл воды), нагр. до кип. и осажд. изб. кип. р-ра (NH4)2SO4 (10%). Отстаив. 6 час, ф. б., промыв, р-ром ЭДТА (слабокисл.), сжигают и прокал. как указано выше. Для переосажд. ос. на фильтре р-ют гор. 3-5% аммиачным р-ром ЭДТА-Na, промыв, фильтр гор. водой, осажд. BaSO4 добавл. НС1 до кисл. р-ции р-ра (по мет. кр.) BaSO4 BaSO4 BaSO4 7,H, 25, 689 689 690
448
Новый справочник химика и технолога
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Ва2+ Амидосульфоновая к-та Осажд. из гомогенного р-ра. Анал. р-р (1-100 мг Ва, нейтр.) разб. до 100 мл, добавл. 1 г pear., нагр. и выдерж. при 100 °C 30 мин, охл., ф. б., промыв. 25 мл хол. и 25 мл гор. воды. Прокал. при 900 °C. Не меш. РОд”. BaSO4 26
К2СгО4 (10%) Осаждение по Винклеру: Анал. р-р (< 0,15 г Ва) нейтр. по мет. оранж., разб. до 100 мл, нагр. до кип., по каплям 10 мл pear., кип. 2-3 мин. Через сутки ф.с., промыв. 50 мл хол. воды, высуш. 2 час при 130 °C. Рекомендуют также высуш. при <60 °C или при комнатной температуре в токе воздуха ВаСгО4 1,7, 287, 691, 692
(NH4)2[BeF4] (10%) Р-р pear.: см. табл. «Приготовление р-ров». Анал. р-р (25-70 мг Ва, 7-700 мг РЬ, слабокисл.), добавл. Na-ЭДТА (10%) для маскир. всех ионов тяжелых металлов (минимум 10 мл). Р-р разб. до 200 мл и нейтр. NH4OH до pH = 6-7 (по мет. кр.), нагр. до кип. и при перемеш. по каплям добавл. изб. pear., нагр. несколько мин, охл., ф. с., промыв, хол. водой, высуш. при 110 °C (ок. 2 час) Ba[BeF4] /= 0,61770 693, 694
(NH4)2C2O4 а) См. опр. Са с этим pear. б) Термографическое опр. Са, Sr, Ва в форме их оксалатов. Анал. р-р (хлориды Ва, Sr, Са) добавл. 5 мл СН3СООН (1 н.) на 100 мл р-ра (pH * 3), осажд. оксалаты по Винклеру (см. опр. Са), снимают термографическую кривую и ее производную и опр. по ним: Вес смеси крист, оксалатов (А) Потерю крист, воды (А-В) Потерю СО2 (С-Д) Пусть вес СаС2О4 Н2О = х (г) SrC2O4H2O =у (г) ВаС2О40,5Н2О = z (г) Тогда х + у + z = А (г) Вес крист, воды (A-В) (г) Вес СаСО3, эквивалентный СО2= (С-Д) (г) Отсюда х = 3,320147-(С-Д) (А-В)-0,038430-А-0,281759(С-Д) У~ 0,054594 0,093024 • А + 0,00498 • (С - Д) - (А - В) Z~ 0,054594 Метод дает очень точные результаты для Са и Sr и приемлемые для Ва (абс. ошибка при опр. 13,5 мг Ва равна + 0,4 мг) ВаС2О40,5Н2О 663 695
Опр. также в виде: ВаСО3 [696, 697], Ва(Ю3)2 [698], BaS2O3-H2O [699, 700]
Zn2+ H2S (газ) а) Анал. р-р (< 0,2 г Zn), нейтр. по мет. кр., разб. до 150 мл, добавл. 20 мл формиатного буфера, нагр. до 60 °C, пропуск. H2S, продолжая нагр. р-р до кип., а затем охл., закрыв. колбу пробкой, выдерж. 3-4 час, ф. б., промыв, нас. р-ром H2S с добавл. 0,4 мл НСООН (80%) на 100 мл р-ра, тигель с фильтром помещ, в другой тигель, озоляют, охл., ос. смачивают водой, добавл. 1 мл H2SO4 (50%), выпар, досуха и прокал. 20 мин при 400-600 °C. Можно прокал. ос. при 950-1000 °C и взвешивать в форме ZnO. ZnSO4 705
Химические методы количественного анализа
449
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Zn2+ б) Анал. р-р (< 0,3 г Zn, кисл.) нейтр. NH4OH (2 н.) по мет.кр., добавл. 2 мл NH4OH (конц.) и 40 мл лед. СНзСООН, разб. до 250 мл, нагр. до 60-70 °C, пропускают H2S без дальнейшего нагр. 20-40 мин, отстаив. 1-1,5 час, добавл. при сильном перемеш. 50 мг HgCl2, ф. б., промыв. NH4NO3 (1%). Прокал. до ZnO или ZnSO4 (см. п. «а») в) Анал. р-р (< 0,2 г Zn, сернокисл., без СГ) нейтр. по мет. оранж. или мет. кр. р-ром NH4OH, добавл. 3-5 г (NH4)2SO4, 1 мл H2SO4, разб. до 200 мл, нагр. до 60 °C, осажд. ZnS и прокал. как в п. «а». г) Анал. р-р (< 0,3 г Zn) нейтр. NH4OH (2 н.) по мет. кр., добавл. 10 мл монохлоруксусной к-ты (2 н.) до раств. ос., 10 мл СНзСООИа (1 н.), разб. До 150 мл, нагр. до кип., пропускают H2S 15-20 мин, ф. б., промыв. СН3СООН (4%), прокал. до ZnSO4 или ZnO (см. п. «а»). д) Ос. по п.п. «а-г» высуш., переносят в тигель Розе. Фильтр сжигают отдельно, золу добавл. к ос. в тигле, добавл. чистой S (1/3 от ос.), перемеш., покрывают слоем серы, прокал. в токе Н2 при температуре начала покраснения тигля, охл. в токе Н2 ZnO ZnO ZnSO4 ZnSO4 ZnO ZnSO4 ZnO ZnS 1 706 707 7
H2S (газ) и ВаС12 Осажд. ZnS (см. выше), окисл. Опр. SO2” BaSO4
(NH4)2HPO4 а) Анал. р-р (слабокисл.) нагр., добавл. pear. (10-15-кратный изб.). Через 3 час ф. б. (промыв, гор. 1% р-ром (NH4)2HPO4, 50% сп.). Прокал. при 900 °C. б) Анал. р-р (< 0,1 г Zn) нейтр. по мет. оранж., 1 капля H2SO4 (2 н.), 2,0 г NH4C1, нагр. до кип., медленно добавл. 10 мл pear. (20%), оставляют на ночь, ф. с., промыв, нас. р-ром ZnNH4PO4, высуш. 2-3 час при 130 °C Zn2P 2O2 ZnNH4PO4 7, 709 708, 709
8-Оксихинолин (3% в СП. или СН3СООН) а) Анал. р-р (2-150 мг Zn, нейтр. или слабокисл., 100 мл), 105 г CH3COONa или CH3COONH4, НС1 до содержания СНзСООН в р-ре ок. 2-3%. Нагр. до 60 °C, добавл. изб. pear., нагр. до кип., отстаив. до коагуляции ос. (до полного охл.), ф. с., промыв, гор. водой до получения бесцв. ф-та. Высуш. при 160-180 °C. Ошибка 0,2-0,3%. б) Анал. р-р, 2-5 г винной к-ты, нейтр. NaOH (конц.) по фенолфт., добавл. 10-15 мл NaOH (2 н.), разб. до 100— 120 мл, добавл. при перемеш. небольшой изб. pear. Р-р с ос. нагр. до 60°С, ф. с., промыв, гор. водой, высуш. при 160-180 °C. Ошибка опр. 10-70 мг Zn ±1% Zn(C9H6ON)2 Zn(C9H6ON)2 453, 712, 713 453, 712, 713
NH4SCN (1) и пиридин (2) Анал. р-р, pear. 1, pear. 2. Ф. с. Высуш. при 20 °C (под вакуумом) [Zn(C5H5N)2]- •(SCN)2 710, 711
Na2CO3 Анал. р-р нагр. до 100 °C, добавл. pear, (до роз. окраски фенолфт.) В присутствии NH* р-р кип. с pear, до удаления NH3. Ф. б. (промыв, водой). Прокал. при 950-1000 °C ZnO 11
K2[Hg(SCN)2]’ Анал. р-р (р-р ZnS в H2SO4), винная к-та, KSCN, CH3COONa, pear. Через 10-12 час ф. с. (промыв, р-вом 1:400, Н2О). Высуш. при 110 °C Zn[Hg(SCN)4] 7, 11, 702-704
Электролиз Анал. р-р, лимонная к-та (pH = 4-5). Электролиз на Pt катоде (покрытом Си) при 1 А-дм2 Zn 385, 701
* Для приготовления р-ра смешивают равные объемы водных р-ров HgCl2 (27 г-л ') и KSCN (39 гл *).
450
Новый справочник химика и технолога
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Г равиметрическая форма осадка Литература
Zn2+ Антраниловая к-та, натриевая соль (3%) Реагент: 3 г антраниловой к-ты р-ют в 22 мл NaOH (1 н.), нейтр. антраниловой к-той до слабокисл, р-ции, фильтр., разб. до 100 мл. Анал. р-р (10-100 мг Zn, нейтр.), разб. до 100-150 мл, добавл. pear. (20 мл на 100 мг Zn), кип. ок. 1 мин, ф. с., промыв. р-ром pear. (1:20), сп., высуш. 1 час при 100-110 °C. Меш.: Ni, Со, Cd, Мп, Си Zn(C7H6O2N)2 f= 0,19363 715, 716
Хинальдиновая к-та, натриевая соль (3,76%) Анал. р-р (40-100 мг Zn) нейтр., разб. до 150 мл, добавл. 2-5 мл СН3СООН (1 М), нагр. до кип., pear, до небольшого изб., через 5 мин ф. с., промыв, гор. водой, высуш. при 110 °C Zn(C10H6NO2)2- •Н2О f= 0,15285 718, 719
Опр. также в виде: ZnC2O4-2H2O [714]
Cd2+ H2SO4 Анал. р-р (50-500 мг Cd), pear. (0,89 мл 1 М H2SO4 на 100 мг Cd), упар., переносят в тигель и выпар, до удаления паров SO3, прокал. при 500-800 °C. Меш. все нелетучие соединения CdSO4 7, 666
H2S а) Анал. р-р (< 300 мг Cd, нейтр. или слабокисл.), добавл. 1 каплю НС1 (1 М), 3 мл H2SO4 (конц.), нагр. до кип., пропускают H2S (газ). Осажд. начинается при охлажд. р-ра; сосуд помещают в хол. воду и пропускают H2S еще 15 мин, сосуд (коническую колбу) закрывают пробкой и оставляют на ночь. Ф. с., промыв, водой с добавл. неск. капель СН3СООН и H2S, затем 5-10 мл сп. (2:1), высуш. 1 час при 130 °C. б) Анал. р-р (H2SO4, 1:20), pear. К ос. добавл. H2SO4. Выпар. Высуш. при 140-150 °C CdS CdSO4 1,7, 266 1,7, 666
Na2S2O3 Анал. р-р, уксуснокисл, среда, pear., кип. 30 мин; ос. CdS раств. в НС1 (1:3), выпар, с H2SO4 (1:1) и опр., как описано для H2SO4 (см. выше) CdSO4 31
2-о-Оксифенил-бензаксозол (1% сп. р-р) Анал. р-р (тартратн. буф. pH = 9), нагр. до 60 °C, добавл. pear, до pH = 11. Ф. с. (промыв, сп. 1:1. водой); высуш. при 130-140 °C Cd(C13H8O2N)2 30, 726
(NH4)2Mo04 Анал. р-р (кип.), pear., NH4OH. Ф.с. (промыв, гор. водой), высуш. при 120 °C CdMoO4 727
0-Нафтохи-нолин, 2,5% свежеприготовленный р-р в 0,5 н. H2SO4(1)hKI, 10% р-р (2) Анал. р-р (H2SO4, 6%), SO2 (нас. р-р), pear. 2, pear. 1. Ф. с. (промыв, pear. + SO2 + KI); высуш. при 70-80 °C [C13H9NH]2- [CdJ4] f= 0,1147 11
Электролиз Анал. р-р выпар., добавл. H2SO4, нейтр. по фенолфт., добавл. KCN (10% р-р) до раств. ос. Электролиз при 0,6 А-дм”2, затем при 1 А-дм'2. Электрод промыв, сп., эф. высуш. при 100 °C Cd 7, 722, 723
(NH4)2HPO4 См. опр. Mg2+ CdNH4PO4H2O или Cd2P2O7 719- 721
NH4SCN и пиридин См. опр. Zn2+ Cd(C5H5N)2-(SCN)2 724, 725
N2H4(1O%)(1) + KI (10%) (2) Анал. р-р, нейтр. NaHCO3 по мет. оранж., разб. до 100 мл; нагр. до 60-70 °C; 10 мл р-ра 2, по каплям 10 мл р-ра 1; после охл. ф. с., промыв. 100-150 мл KI (0,3%) с N2H4 (0,5%), 10 мл сп., высуш. при 110 °C 1 час Cd(N2H4)2I2 1,728
Хинальдиновая к-та, натриевая соль (см. Zn) Анал. р-р (20-100 мг Cd) нейтр., разб. до 150 мл, нагр. до кип., pear, (изб.), точно нейтр. NH4OH или NaOH. После охл. ф. с., промыв, водой до удаления pear., сушат при 130 °C Cd(C10H6NO2)2 635, 717
Химические методы количественного анализа
451
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Cd2+ 8-Оксихинолин (3% в сп.) а) Анал. р-р (< 250 мг Cd) нейтр. NaHCO3 до помутнения, разб. до 100 мл. Ос. раств. в нескольких каплях 2 н. СН3СООН, нагр. до 60 °C, pear, (в небольшом изб.); кип., ф. с.; промыв, гор., затем хол. водой, высуш. при 240 °C. (При опр. 100-250 мг Cd можно высуш. при 130 °C). Не меш.: Са, Sr, Ва, Mg, РО] и Щ. М. б) Анал. р-р, 2-5 г винной к-ты; нейтр. NaOH по фенолфт., разб. до 100 мл, добавл. 10-12 мл NaOH (2 н.), осажд. pear. (Ос. через 1-5 мин). Далее действуют согласно пункту «а». Метод позволяет отделить Cd от Al, Cr, Fe, As, Sb, Sn, Bi, a также от 50 мг Co, Ni, Mn Cd(C9H6ON)2 Cd(C9H6ON2)2 268 728
Диэтилдитиофосфат никеля В сплавах магния: навеску 0,2-0,5 г (2-3% Cd) раств. в 15 мл H2SO4 (1:5), 15 мл pear. (0,05 н.), отстаив. 1 час, ф. с., промыв. 4-5 раз нас. р-ром диэтилдитиофосфата Cd, высуш. при 105 °C (C4H10O2PS2)2Cd f= 0,2327 25, 731, 732
Дифенилди-тиофосфорная к-та (0,05 М) Анал. р-р прим. 1 н. по H2SO4 (20-100 мг Cd в 100 мл), pear. (2-3-кратный изб.), перемеш., через несколько минут ф. с., промыв, нас. р-ром дифенилдитиофосфата кадмия, высуш. при 80-90 °C [(C6H5O)2PS2]2Cd 27, 28, 730
Опр. также в виде: CdO [31, 721, 733], [Cd(NH3)4][HgI3] [734], [Cd(NH2CSNH2)2]-[Cr(SCN)4(NH3)2]2 [735], [Cu(NH2C2H4NH2)2][CdI4] [736], CdC2O4-3H2O [31], Cd(NH4)2[Fe(CN)6] [31]
Hg(I, П) H2S Анал. р-р (кисл.), pear. Ф. с. (промыв, р-ром H2S, сп., CS2). Высуш. при 105-109 °C HgS 7,31, 266
(NH4)2S Анал. р-р (нейтр. Na2CO3), добавл. pear., NaOH (10% р-р до осветления). Ф. б. или ф. с. Ф-т добавл. NH4NO3 (изб. 25% р-ра), кип. Ф. с. (промыв, р-ром H2S, водой, сп., CS2, сп., эф.). Высуш. при 105-109 °C HgS 11,31, 737
НС1 (I) и H3PO3(II) Р-р pear.: 10 мл РС13 раств. в 200 мл воды, разб. до 250 мл, фильтр, через вату. Анал. р-р, pear. Ф. с. (промыв, водой). Высуш. при 100-105 °C Hg2Cl2 31, 122(6), 219
KIO4, 4% p-p Анал. р-р (< 0,5 г Hg), HNO3 (0,15 н.), H2SO4 (0,1 н.). Нагр. до 100 °C, добавл. pear. Ф. с. (промыв, гор. водой). Высуш. при 100 °C Hg5(IO3)2 7, 739
Соль Рейнеке (нас. водн. р-р) а) Анал. р-р (HNO3) нагр. до 60-70 °C, добавл. pear. Через 30 мин ф. б. (промыв. HNO3, 1:3). Прокал. при 1000 °C. б) Анал. р-р Hg2+ (10-100 мг Hg) нейтр. до 0,5 н. по Н+, нагр., pear, по каплям, ф. с., промыв, гор. водой, сп.; высуш. при 105-110 °C. Не меш. анионы NO,, SOq, СН3СОО“, тартрат. Меш. только Ag, Au, TI Cr2O3 f= 1,319 Hg[Cr(SCN)4- •(NH3)2]2 11,31, 505 2,31, 32, 740, 741
SnCl2 Анал. р-р (НС1), pear. Через 1 час ф.с. (промыв. HCI, 1:1, водой, ацетоном). Высуш. при 20 °C (в токе воздуха) Hg 7, 738
Электролиз а) Нейтр. анал. р-р, 2-4 мл HNO3 (конц.), разб. до 100 мл. Электролиз ведут с золотым или позолоченным электродом. При 2,4-2,5 В; 0,4-0,7 А. Для осажд. 0,3 г Hg требуется 3 час. При 30-40 °C, сильном перемеш., 2,4 В и (3-0,3) А требуется ок. 40 мин. 6) Внутренний электролиз (опр-ся в биологических объектах). Анал. р-р (0,1-0,01 мг-мл1 Hg; р-р после разлож. пробы в смеси конц. HNO3 и H2SO4). Катод — графитовый стержень, анод — цинковая пластина. При нагр. до 80 °C, перемеш. р-ра и вращении электродов время полного осажд. 1 час. Макс, ошибка — 4,2% Hg Hg 31,385, 746 747
452
Новый справочник химика и технолога
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Hg(I, П) Zn Анал. р-р (после кислотного раств. H2SO4, НС1), добавл. цинковые опилки, 25 мл НС1 (1:1). Амальгаму помещают на 24 час в НС1. Шарик Hg отфильтр., промыв. H2SO4 (разб.), высуш. и взвешивают Hg 748
Cu В анал. р-р (нейтр.) нагр. до 30 40 °C, помещают медную пластину. По окончании осажд. промыв, водой, сп. и эфиром. Высуш. на воздухе. Далее удаляют Hg, нагр. в токе воздуха. Содержание Hg опр. по убыли веса. Метод позволяет опр. ок.1 мг Hg Hg 749
Fe Навеску руды (< 0,1 г Hg) смеш. с двухкратным количеством карбонильного Fe, помещ, в тигель, покрывают слоем железа (5-10 мм), закрывают охлаждаемой золотой крышкой и осторожно нагр. дно тигля до темно-кр. каления, выдерж. 30 мин. После охл. промыв, сп., эф., взвешивают Hg в виде амальгамы Hg 753, 754
Fe микроскопический метод а) Ртуть восст. до металла из р-ра солей. Диаметр образовавшейся капли измеряют под микроскопом. Количество Hg (Р) находят по формуле: Р = gp-б'1, где d — диа- метр, g — ускорение силы тяжести, р — плотность. Метод применим для опр. 0,04-10 мкг Hg. б) При опр. 0,3-5 мг Hg опр., измеряя ее объем в капилляре. Малые кол-ва Hg выделяют осажд. в виде HgS на коллекторе CuS, сульфид разрушают прокал. ос. с хроматом свинца, конденсируют Hg в капилляре. В случае малых количеств (мкг) капилляр заполняют Hg с помощью центрифуги Hg 750- 752 751
Аскорбиновая к-та Анал. р-р (0,05-1 г Hg) нейтр. NaOH по мет. оранж., добавл. 5 мл HNO3 (0,2 н.), нагр. до 70 °C, добавл. pear. (1г раств. в небольшом объеме воды), всряхивают, собирая Hg в одну каплю. Р-р отстаив. 4-5 час, ф.с., промыв, хол. водой (50 мл) и сп. (10-12 мл), высуш. над СаС12 1-2 час (или в токе воздуха 15—40 мин) Hg 1
K2Cr2O7 (10%) Слабокисл. анал. р-р Hg (I) нейтр. NaCO3 (2,5%) до начала образ, ос., 10 мл pear, при перемеш. (по каплям), нагр. до перехода кор. ос. в ярко-кр. крист.; охл., ф. с., промыв, хол. водой (50 мл), высуш. при 105 °C прим. 1 час. Меш. все анионы, кроме NO3 Hg2CrO4 744, 745
KIO3 (5%) Слабоазотнокисл. анал. р-р (0,05-0,5 г Hg) разб. до 100 мл, нагр. до кип., по каплям 20 мл pear, при перемеш., отстаив. 2 час, ф. с., промыв, хол. водой (50 мл), сп. (10 мл), высуш. при 110 °C прим. 2 час Hg2(IO3)2 743
NH4SCN (1) + ZnSO4 (2) Pear.: 39 г (1) + 29 г (2) в 1 л. Анал. р-р Hg(II) нейтр. NaOH, 25 мл pear., разб. до 75 мл. Стенки сосуда очищают стеклянной палочкой 5 мин, отстаив. 1 час, ф. с., промыв, разб. pear. (1:90), высуш. 1 час при 102-108 °C ZnHg(SCN)4 31,742
Опр. также в виде: HgO, HgBr2, Hg3(AsO4)2 [31]; {[(NH3)5CoCl2 Co(NH3)2]Cl2-3HgCl2} [755], Co(NH3)2Hg(S2O3)310H2O [756], [Hg(C5H5N)2Cr2O7 [757]; Hgl2 [758, 759]; (Hg2N)2CrO4-2H2O [760]; Hg2WO4 [761, 762]; Hg2(VO3)2 [763]; [Cd(NH3)4][HgI3]2 [764]; [Сп(С2Н8К2)2][ВД [765]; Hg2C2O4 [766]; Co[Hg(SCN)4] [767]; (HgC5H5N)Cl2 [768]
BO', B4o?- CH3OH (1), Mg2+(2) а) Анал. р-р (нейтр.) упар., добавл. 10 мл pear., отгоняют (СН3О)3В при 80-90 °C в р-р (NH4)2CO3. По окончании добавл. точно измеренный объем титр, р-ра Mg(AcO)2, выпар, и прокал. при 800-900 °C. Для опр. кол-ва В2О3 из веса ос. вычитают расчетное кол-во взятого MgO. В2О3» MgO 11
Химические методы количественного анализа
453
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Г равиметрическая форма осадка Литература
ВО;, В4ОГ б) Навеску MgO (или СаО) прокал. в платиновой чашке при 800-900 °C до постоянного веса. К ос. в чашке приливают дистиллят согласно п. «а», выпар, досуха и прокал. при 800-900 °C. Количество В2О3 опр. по привесу ос. В качестве pear. (2) можно применять Mg(CH3COO)2 (р-р ок. 2 г Mg в 1 л) в2о3, MgO 7,11 769-771
ВаС12 + винная к-та Анал. р-р (25 мл), подкисл. НС1, 10 мл NH4C1 (20%), 10 мл винной к-ты (5%), 10 мл ВаС12 (5%). Нейтр. NH4OH (1:1) до pH = 8,6-9,2, охл. до 20 °C, отстаив. 10 мин, ф. б., промыв. водой (с несколькими каплями NH4OH), прокал. при 700-800 °C. Возможно осажд. 5-20 мкг бора. Не меш. (в малых кол-вах мг) Si, Al, Мо, V. ОФ: Ba5B2Ci2H8O24 • 4Н2О Эмпирический Ва2ВО6 (?) /= 0,0287 33, 34, 773, 774
Нитрон + фторид Анал. р-р (125-250 мг Н3ВО3), разб. до 60 мл, 15 мл нитрона (15% р-р в 5% СН3СООН, < 1,3 г HF (48%), отстаив. 10-20 час, затем 2 час на лед. бане, ф. с., промыв, нас. р-ром нит-рон-тетрафторбората 5 раз по 10 мл. Высуш. 2 час при 105— 110 °C. Меш.: NO", СЮ;, Г, SCN", СгО^, сю;, вю, c20h16n4-hbf4 f= 0,02704 35,36
Дикалийсахарат (1) + ВаС12(2) Pear.: 5 г (1), 40 г NH4C1, 15 г (2) р-ют в воде, NH4OH до pH = 8,6-8,8, разб. до 1 л. Анал. р-р (< 50 мкг мл 1 В), pear, (двухкратный изб.), отстаив. 18 час, ф. с., промыв. 2 раза водой, 3-4 раза сп. (25%), высуш. 4 час при 105 °C. Применен для опр. бора в морской воде /= 0,0201 37
Alj+ NH4OH Анал. р-р, NH4CI, мет. кр. Нагр. до 100 °C, добавл. pear, до желт. Ф. б. (промыв. 2% р-ром NH4CI). Прокал. при 1200 °C А12О3 11,776
NH3 (газ) Анал. р-р (pH = 3). Нагр. до 15 °C, пропускают смесь pear, с воздухом. Ф. б. Прокал. при 1200 °C А12О3 505
Бензойная к-та а) Осажд. из гомогенного р-ра, pH = 3,5^4,0 К 350 мл бензоатного буфера (48 г NH4C1 и 18 г бензойной к-ты в 2 л) добавл. 3 г мочевины и 50 мл слабокисл. анал. р-ра (0,01-0,05 г А1), быстро нагр. до кип., 1,5-2 час на паровой бане. Далее как при осажд. NH4OH. б) Анал. р-р (слабокисл., 100 мл) нагр. до 60-80 °C, добавл. 2 г NH4CI, 2 г CH3COONH4 и 1 мл тиогликолевой к-ты, перемеш., вводят 20 мл pear. (10%), бромфеноловый син. и NH4OH (1:1) при перемеш. до изменения окраски р-ра. Кип. 5 мин, выдерж. 15 мин при 100 °C, ф. б., промыв. 5 раз р-ром (1% бензоат + 2% СН3СООН). Ос. р-ют в НС1 (5 н.) и повторно осажд. Прокал. при 1200 °C А12о3 А12О3 41 780
Коричная к-та (5%) Анал. р-р (кисл., ® 60 мл) добавл. 15 г NH4C1, нейтр. NH4OH (1:1) до появления мути, НС1 до ее исчезновения (2-3 капли 6 н. НО), разб. до 80^-90 мл, нагр. до кип. добавляют по каплям 20 мл pear., кип. 1-2 мин, выдерж. при 100 °C 1-1,5 час, ф. б. (декантация), промыв. 2 раза по 20-25 мл гор. р-ром (10 г NH4CI, 20 мл pear. разб. до 100 мл) декантацией и затем на фильтре р-ром из 20 мл pear, и 280 мл воды При исп-ии ф. с. ос. далее промыв, сп. и высуш. при 110 °C При исп-ии ф. б. ос. высуш., озоляют и прокал. до оксида А1(ОН)(С8Н7СОО)2 А12О3 782
C5H5N (20%) Анал. р-р (100 мл) нейтр. до раств. мути (поочередно NH4OH и НС1), 10 г NH4CI, нагр. до кип., по каплям при перемеш. pear, до изменения окраски мет. кр. и изб. 10-15 мл, нагр. до кип., 30 мин на водяной бане. Ф. б., промыв. NH4CI (3%) с несколькими каплями пиридина. Прокал. как при осажд. NH4OH. Мешают SO", ; не мешают Mn, Ni, Со, Zn, Cu, Cd, Mg, Щ. и Щ. 3. М А12О3 38
454
Новый справочник химика и технолога
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
А13+ Мочевина Осажд. из гомогенного р-ра. Анал. р-р (ок. 500 мл) нейтр. NH4OH и НС1 до раств. мути, 10-20 г NH4C1, 1 г (NH4)2SO4, 4 г pear., кип. 1-2 час. Ос. хорошо фильтр., сорбция примесей незначительна. Прокал., как при осажд. NH4OH. Однократным осажд. отд. Са, Mg, Мп и Cd. При отд. от Ni, Со, Zn необходимо переосаждение А12О3 39
NH4NO3(6%) Анал. р-р (содержащий большое кол-во аммонийных солей), 20 мл pear., разб. до 200 мл и кип. до прекращения выделения оксидов азота (ок. 20 мин), нагр. на водяной бане 30 мин. Прокал., как при осажд. NH4OH. Метод отличается высокой точностью А12О3 777
СО2 Анал. р-р (0,1-0,3 г Al), NaOH (свежеприготовленный р-р) до раств. ос. В присутствии Сг добавл. 2-3 мл Н2О2 (30%), кип. до полного разл. Н2О2, 1-2 капли фенолфт., медленно пропускают СО2 до обесцвечивания индикатора и еще 10-15 мин. Крист, ос. ф. б., промыв, гор. водой до отрицательной р-ции на NaOH. Прокал., как при осажд. NH4OH А12О3 778, 779
Янтарная к-та Опр. А1 в железе, стали и шлаке. Анал. р-р, нагр. до кип., доб-т 5 г KHSO4, кип. 1-2 мин, разб. до 400 мл, нагр. до кип., добавл. 5 г янтарной к-ты, 4 г мочевины, 5 г гидрохинона. Кип. 1-2 мин, осторожно добавл. Na-НСО3 до появл. неисчезающего ос., раств. ос. в НО (15%), разб. до 600 мл, нагр. до кип., кип. 45 мин, ф. б., промыв. NH4NO3 (2%). Прокал., как при осажд. NH4OH. Относительная ошибка приб. 2% А12О3 40
(NH4)2HPO4 Анал. р-р (НО, 0,05%). нагр. до 100 °C, добавл. pear. (10-кратный изб.), ацетатн. буфер (pH = 5-5,4). Фильтр, с бум. массой. Ф. б. (промыв. 5% р-ром NI I4NO3), первое. Прокал. при 800-1100 °C А1РО4 7,11, 783, 784
8-Оксихинолин (2,5%) Р-р pear.: 25 г pear., 29 мл НС1 (6 М), разб. до 1 л а) Осажд. из р-ра СН3СООН. Анал. р-р (2-20 мг А1), разб. до 100-125 мл, нейтр. NH4OH и НО (2 М) до исчезновения мути и 5-6 капель изб. Добавл. pear. (0,7 мл на 1 мг А1), медленно 20 мл CH3COONH4 (20%) с pH = 6,5-7,0. Нагр. до кип., 30 мин при 100 °C, охл. до 50 °C, ф.с., промыв < 100 мл р-ра (8 мл р-ра pear. разб. до 0,5 л, 3 капли бромкрезолпурпур. в 20% сп. и NH4OH до pH = = 6 — пурпурная окраска, объем доводят до 1 л, дважды промывают хол. водой (по 5 мл), высуш. при 140-150 °C или ф. б., прокал. при 1200 °C. б) Осажд. из аммиачного р-ра. Анал. р-р (100 мл), 5-кратный (по отнош. к А1) изб. винной к-ты, 1-2 г NH4C1, 8-10 капель бромкрезол пурп., нейтр. NH4OH (1:1) до перехода окр. в пурп., по каплям pear. (2-3% в СН3СООН). Нагр. до кип., выдерж. 1 мин, охл. до 60 °C, ф. с., промыв. 100 мл хол. воды. Ос. обрабатывают по п. «а», в) Осажд. из гомогенного р-ра. Анал. р-р (2-10 мл), 50 мл воды, 60 мл ацетона, 4 мл р-ра pear. (5%), 40 мл CH3COONH4 (2 н.). Р-р выдерж. 3 час при 70-75 °C, охл., ос. ф. с., промыв. 3 раза водой, высуш. при 135-140 °C. Не меш.: Mg, Са, Zn; при pH = 4,7-4,9 - Cd. Меш.: In, Y, Се, Sc A1(C9H6NO)3 А12О3 A1(C9H6NO)3 A1(C9H6NO)3 1,7, 25 785 785 786, 787 788
Химические методы количественного анализа
455
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
А13+ г) Осажд. из гомогенного р-ра. Анал. р-р (25-50 мг А1, 1-2 мл конц. НС1) разб. до 150— 200 мл. На каждые 25 мг А1 добавл. 5-6 мл pear. (10% р-р в 20% СН3СООН) и 5 г мочевины, нагр. до кип., выдерж. 2-3 час при 95 °C. Оранж.-желт. цвет р-ра указывает на конец осажд. Р-р охл., ф. с., промыв, и высуш. как указано в п. «а» 789
NaF (3,5%) а) Анал. р-р, 5-7 мл H2SO4 (конц.), выпар, до паров SO3, раств. при нагр. в 50-70 мл воды, нейтр. NH4OH, ос. р-ют в H2SO4 и добавл. изб. 5-6 капель, маскир. р-ром (смесь 1:1 нас. р-ра (NH4)2C2O4 и 40% цитрата аммония), 60-80 мл pear. Через 20 мин ф. с., промыв. NaF (0,5%), сп., высуш. при 120-130 °C. б) Ос. по п. «а» ф. б. (двойной), 10-15 раз промыв. NaF (0,5%). Ос. р-ют в 50-70 мл кип. смеси (150 мл нас. р-ра Н3ВО3, 250 мл конц. НС1 и 600 мл воды), фильтр промыв, гор. водой, р-р нейтр. NH4OH и НС1 по мет. кр., охл. до 30-40 °C, добавл. 20 мл оксихинолина (2,5% в 4% СН3СООН) и 10 мл CH3COONH4 (20%), нагр. до 60-70 °C, выдерж. 10-15 мин, отстаив. 30 мин, ф. б. (двойной), промыв, р-ром (12,5 мл 20% CH3COONH4, 2 капли NH4OH, разб. до 1 л), озоляют при 400 °C, прокал. при 1100 °C Na3AlF6 А12О3 808- -811 812- 816
Опр. также в виде: А12О3 (после осажд. в форме гидроксида а-пиколином [790], уротропином [791, 792], фенилгидра-зином [793], смесью Na2S2O3 + фенилгидразин + Na2SO3 [794], акридином [795], и-хлоранилином [795], о-фенетидином [796], водн. суспензией ClHgNH2 [797], моноэтаноламином [798], гексаметилендиамином [800], этиленхлоргидрином [801], тиосульфатом [802] и смесью его с Na2SO3 [803] или фенилгидразином [804], смесью KI и КЮ3 [805], карбонатами [806], гидразинкарбонатом [807], а также после осажд. в форме А1С13-6Н2О [7, 819]); Li2O2Al2O3 [817]; ос. с [Co(NH3)6]Cl3 в присутствии NaF [818]
Ga3+ NH4OH Анал. р-р (H2SO4). Нагр. до 100 °C, добавл. pear. Фильтр, с бум. массой. Ф. б. (промыв. 2% р-ром NH4NO3). Прокал. при 1000 °C Ga2O3 7, 820-822
Купферон (6,5%) Анал. р-р (H2SO4, 14%) охл. до 0 °C, добавл. pear. Фильтр, с бум. массой. Ф. б. (промыв. H2SO4 + pear.). Прокал. при 1000 °C Ga2O3 7, 745, 824
NH4HSO3 Pear.: NH4OH (1:4) нас. SO2 Анал. р-р (соляно-, лучше — сернокисл., < 0,1 г Ga) добавл. NII4OII до слабощел. реакции (по лакмусу), 5 мл pear., кип. 5 мин, ф. б., промыв, водой до удаления СГ, далее как при осажд. р-ром NH4OH Ga2O3 473, 822, 823
Пиридин Анал. р-р, NH4C1, нагр. до 100 °C, добавл. pear., фильтр, с бум. массой. Ф. б., промыв. NH4NO3 (2%). Прокал. при 1000 °C. В присутствии большого количества NH4C1 при однократном осажд. ос. содержит ничтожные количества только Со, Zn, Cd, Cu Ga2O3 42
Мочевина Анал. р-р (без СГ, ок. 3 мл H2SO4 конц.), 3 г pear., разб. до 500 мл, по каплям NH4OH до образ, неисчезающей мути, кип. до достижения pH = 4—5,5 (пожелтение мет. оранж.). Ф. б., промыв, хол. водой, прокал. при t > 550 °C. Ос. — основной сульфат галлия (Ga : H2SO4 =8 : 1), плотный, легко переходит в ГФ Ga2O3 43
456
Новый справочник химика и технолога
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Ga3+ Таннин (10%) Анал. р-р (уксуснокисл.), NH4NO3 до получения 2% р-ра, нагр. до кип., по каплям р-р pear, (таннин раств. в нас. на холоду р-ре CH3COONH4). Ф. б., промыв. NH4NO3 (подкисл. СН3СООН) до удаления СГ. Прокалив, t > 550 °C. Метод более чувствителен, не мешает винная и сульфосалициловая к-ты, при переосаждении полностью отделяется от двухвалентных металлов Ga2O3 44
NH4HPO4 (10%) Кисл. анал. р-р (30-150 мг Ga), 10 мл pear., разб. до 50 мл, нагр. до кип., по каплям при перемеш. NH4OH до pH = 3-5, через 10 мин ф. б., промыв. NH4NO3 (5%) до удаления СГ, прокал. при t > 550 °C GaPO4 45
Камфарная к-та а) Анал. р-р (хлорида или нитрата) упар. досуха, ос. р-ют в 100 мл NH4NO3 (2%) или в 100 мл СН3СООН (0,6 н. с 2% NH4NO3), добавл. 1-2 г pear., нагр. 10 мин при 100 °C, ф. б., промыв. 50 мл р-ра (10 мл 6 н. СН3СООН, 20 мл 10% NH4NO3, 80 мл воды), высуш. озоляют сначала ос., затем фильтр, прокал. при t > 500 °C. б) Ос. по п. «а» ф.с., промыв, сп., ацетоном, эф., высуш. при 90-110 °C. в) Осажд. из формиатного буферного р-ра (pH = 3) высуш. при ПО °C Ga2O3 Ga2[C8H14(CO2)2]3 f= 0,189 /=0,213 823, 825, 826 823 826
Бензойная к-та (2%) Анал. р-р (> 15 мг Ga) нейтр. NH4OH и СН3СООН, добавл. ацетата, буфер (pH = 3,72-5,89), нагр. до 80 °C, добавл. 25 мл теплого р-ра pear., нейтр. NH4OH (разб.) по мет. кр. Р-р выдерж. 30-40 мин при 100 °C, ф. с., промыв, водой до удаления СГ, высуш. при 110 °C, прокал. при 1000 °C. Не меш.: Zn, Cd, Hg, Ni, Mn, UO2+, Cu, Щ. 3. M. Ga2O3 827, 828
Фталевая к-та (10% р-р в сп.) Анал. p-p (> 10 мг Ga), нейтр. NH4OH и CH3OHOH, добавл. ацетатный буфер до pH = 3,46,0. нагр. до 60-80 °C, добавл. 20 мл pear., нейтр. NH4OH (разб.) по бромфен. син., выдерж. 30-40 мин при 80-100 °C, ф. б., промыв, гор. водой до удаления СГ, прокал. при 1000 °C Ga2O3 829
8-Оксихинолин а) Анал. р-р (кисл.) разб. до 150-200 мл, добавл. изб. pear. (5% р-р в сп.), нагр. до 70-80 °C, нейтр. NH4OH до сл. запаха, выдерж. 0,5-1 час при 100 °C, охл., отстаив. 2 час, ф. с., промыв. 20 мл гор. воды, хол. водой до обесцвечивания промывных вод, высуш. при 120 °C. б) Анал. р-р (сильнощел.), добавл. изб. pear. (3% р-р в CH3COONH4), нагр. до 70 °C, добавл. инд. (бромтимоловый син.), нейтр. к-той до желт, окраски (pH = 6-8). Ос. обрабатывают по п. «а». Ошибка опр. Ga 3-70 мг ± 0,1 мг Ga(C9H6NO)3 Ga(C9H6NO)3 830 830
Диантипирил-пропилметан (1% в СН3СООН) (1:10) Анал. р-р, pear. (1 мл на 1 мг Ga), равный объем НО (конц.). Ф. с., промыв. НС1 (6 М с 0,05% pear.), НС1 (6 М), высуш. при 100-110 °C. При опр. 3-17 мг Ga ошибка < 2,0%. Не меш.: Mn, Mg, In, Со, Bi, Fe2+, Zn, Cd, Cu, Al. Меш.: Tl3+, Fe3+ C26H30O2N4HGaC14 /=0,1084 831
А-Бензоил-А-фенилгидрокси-ламин Анал. p-p нейтр. до pH = 1,6-3,5, нагр. до 40-50 °C, добавл. прим. 2,5-кратный изб. сп. р-ра pear., выдерж. 45 мин, на ф. с., промыв, сп., высуш. при 110-120 °C. Возможно прокал. до Оа2Оз. Не меш.: Al, Be, Zn, Fe2+, Cu, Th, Ce, U, In. Ошибка ±1% Ga(C13H10O2N)3 832, 833
Химические методы количественного анализа
457
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Ga3+ Электролиз Анал. р-р (сернокисл., 0,05-0,2 г Ga) добавл. 50 мл NH4OH (конц.), 10—40 г (NH4)2SO4, 6 г гидразинсульфата, разб. до 130 мл. Электролиз при 60-80 °C, катод Pt—1г (5% 1г), перемеш. (1200 об/мин), при 3,3--4 В, 5 А требуется 30-60 мин. Эл-ды под напряжением промыв, хол. водой, сп., эф., высуш. в эксикаторе Ga 834, 835
Опр. также в виде: Ga2O3 (после осажд. NaN3 [822], смесью КВ1Ч-КВгО3 [836], CH3COONH4 [836], Na2S2O3 [823], гидроксиламином [837]); Ga4[Fe(CN)6]3 [836, 838, 839]; [Co(NH3)6]GaF6 [840]
In3+ NH4OH Анал. р-р (нитрат или сульфат In) нагр. до 100 °C, изб. ре-аг., кип. до получения плотного ос., гор. р-р ф. б. с бум. массой, промыв, гор. NH4NO3 (5%) до удаления СГ. Прокал. при 850-1000 °C. Иногда рекомендуют прокал. при 1200 °C для получения негигроскопичного ос. При t > 1000 °C возможны потери кислорода In2O3 1,7, 822, 841 842, 841
Мочевина (1) + янтарная к-та (2) + NH4C1 (3) Анал. р-р (слабокисл., 20 мл), 0,25 г pear. (1), 0,15 г pear. (2), 0,5 г pear. (3) или 1 г CH3COONa, нейтр. по мет. кр., разб. до 100 мл, выдерж. 1 час при 100 °C (р-р не должен стать кисл.). Ос. обрабатывают как при опр. с NH4OH. Метод позволяет отд. In от Zn и Cd In2O3 843
KOCN (10%) Слабокисл. анал. р-р, изб. NH4C1, мет. оранж., pear, до изменения цвета, разб. до 200—400 мл, кип., на ф. б., промыв, гор. водой до удаления СГ, прокал. при 850-1000 °C In2O3 841, 842, 844
Пиридин Анал. р-р (солянокисл.) нейтр. NH4OH и НС1 (до просветления), разб. до 150 мл, 15 г NH4C1, бум. массу, нагр. до кип., медленно pear, до желт, окраски (мет. кр.) и изб. 10-15 мл, выдерж. 1-1,5 час при 100 °C, гор. р-р на ф. б., промыв, гор. NH4C1 (3%, с добавл. пиридина), р-ют в НС1 (разб.), повторно осажд. и ф. б., промыв, гор. NH4NO3 (3%) до удаления СГ, озоляют и прокал. при 850-1000 °C. 20-кратный избыток Mn, Со, Ni, Cu, Cd, Zn, Си, Sr, Ва, Mg, К, Na In2O3 38. 790, 845, 846
H2S а) Анал. р-р (14—400 мг In) нейтр. NH4OH, добавл. 30 мл СН3СООН (2 н.) и 10 мл NH4OH (2 н.) на каждые 100 мг In, разб. до 100 мл (pH « 4), нагр. до кип., H2S до охлажд., ф. с., промыв. CH3COON1I4 (0,2 н.), высуш. при 100-120 °C Возможно прокал. при 850-1000 °C, 30 мин. б) Анал. р-р (0,03-0,05 М по НС1,1п>10мгв100мл р-ра), нагр. до 70 °C, пропускают 2 ч H2S, ос. ф. с., промыв, сла-боподкисл. сероводородной водой, высуш. в токе H2S при 350 °C, охл. в струе H2S In2S3 In2O3 In2S3 842, 845 287, 842
(NH4)2s а) Анал. р-р нейтр. NH4OH до появления мути, добавл. pear., ф. с., промыв, гор. водой, высуш. при 120-125 °C в течение 1 час. б) Р-р с ос. по п. «а», подкисл. СН3СООН или НС1 (0,05 н.), нагр. до кип., выдерж. 20 мин, ф. с. и далее как в п. «а». Отд. от сульфидов, р-мых в кисл. среде In2S3 In2S3 859 859
(NH4)2HPO4 (10%) Анал. р-р (минеральнокисл.), 15 мл CH3CONH4 (50%), нагр., 25 мл pear., кип. 15 мин, отстаив., ф. б., промыв, водой. Прокал. 1 час при 1100 °C. Метод дает точные результаты, но ос. медленно фильтр InPO4 847
458
Новый справочник химика и технолога
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
1п3+ [Co(NH3)6]Cl3 (4%) Анал. р-р (< 75 мл), НС1 (конц.) — 20% от объема анал. р-ра, нагр., добавл. pear., охл., отстаив. 12 час, ф. с., промыв. НС1 (1:4, нас. ОФ), сп., эф. Высуш. при 105 °C [Co(NH3)6][InCl6] 847, 848
8-Оксихинолин (5% в сп.) а) Анал. р-р (1-60 мг In, нейтр. или слабо-кисл.) добавл. соответственно, CH3COONa, СН3СООН и воды: если In < 5 мг — 0,5 г; 0,5 мл; до 50 мл In 5-15 мг— 1г; 1 мл; до 100 мл In 15—100 мг — 2 г; 2 мл; до 200 мл. Р-р нагр. до 70-80 °C, добавл. по каплям небольшой изб. pear, при перемеш., охл. и отстаив. 2-3 час, ф.с., промыв, декантацией гор. водой, хол. водой (25-75 мл) до обесцвечивания промывных вод. Высуш. 1,5-2 час при 120 °C. б) Возможно опр. микрометодом (> 1 мг In) с погрешностью ±1,2%. Анал. р-р помещают во взвешенную микрофильтр, трубку, подкисл, каплей конц. НС1, добавл. 0,5 мл pear. (5 г в 12 г СН3СООН и 83 г воды), нагр. до 100 °C., добавл. CH3COONH4 (2 н.) до помутнения р-ра, выдерж. 1 мин, добавл. 0,5 мл CH3COONH4, 10 мин при 100 °C, фильтр., промыв. 4 раза гор. водой, высуш. при 140 °C In(C9H6ON)3 . In(C9H6ON)3 830, 850, 851 849
Диэтилдитио-карбаминат Na (2%) Анал. р-р (< 100 мг In, кисл.), CH3COONH4 до pH = 4 5, небольшой изб. pear., отстаив. 8 час, ф. с., промыв, водой, высуш. при 105 °C. Меш.: Pb, Cd, Zn, Cu, Fe. Ga маек. 10-кратным изб. оксалата (0,4-1,5 г) [(C2H5)2NCS2]3In 847
Внутренний электролиз а) Анал. р-р (1-10 мг In, буферная смесь тартрат натрия — НС1 с pH = 3,5-3,8). Катод — Pt (сетка), анод — Zn (стержень, диаметр 10 мм, покрытый коллодиевой пленкой (получают двукратным погружением в коллодий и высуш. на воздухе). Электроды соединяют медным проводником. Продолжительность электролиза 20-45 мин. Катод, промыв водой, погружают в сп., затем эф., сушат на воздухе, выдерж. 15 мин в эксикаторе. б) Анал. р-р (ок. 30 мг In), 20 мл НС1 (0,1 н.), 15 мл сег-нетовой соли (12%), 10 мл лимонной к-ты, 8 мл малоновой к-ты (5%), 7 мл CH3COONa (6%), 10 г NH4C1. Р-р разб. до 100-150 мл, нагр. до 60-80 °C и подвергают электролизу с электродами Zn-Pt, pH = 3,5-3,7. Катод высуш. по п. «а» In In 852 853
Опр. также в виде: 1п2О3 (после осажд. формиатом или ацетатом натрия [855], салицилатом Na [856], нитритом К [857], уротропином [842], диметиламином [858], Na2CO3 [855, 860], ВаСО3 [855, 860]); In5K[Fe(CN)6]4 [861]; 1п4(Р2О7)3-8Н2О [862]
T1(I, III) К2СгО4 а) Анал. р-р, NH4OH. Нагр. до 80 °C, добавл. pear. Ф. с. (промыв. 1% р-ром К2СгО4, 50% сп.). Прокал. при t < 745 °C. б) Осажд. как в п. «а», р-р охлажд., отстаив. 12 час, ф. с., промыв. К2СгО4 (1%), затем сп. (50%), высуш. при 120— 130 °C 2 час Tl2CrO4 Tl2CrO4 7, 863, 864 46, 863, 864
K3[Fe(CN)6] (8%) Анал. р-р, КОН (изб. 5% р-ра), pear. Через 18 час ф. с. (промыв, гор. водой). Высуш. при 160-80 °C 2 час T12O3 1,46, 865, 866
Химические методы количественного анализа
459
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Т1(1, III) KI Анал. р-р*, СН3СООН. Нагр. до 100 °C, добавл. pear. Через 12 час ф. с. (промыв. KI, СН3СООН, ацетоном). Высуш. при 120-130 °C T1J 1,46, 47, 867, 868
H2PtCl6 Анал. р-р нагр., добавл. изб. pear. Р-р охл., ф.с., промыв, водой, высуш. при 100 °C. Меш.: К+, Rb+, Cs+ Tl2PtCl6 869, 870
КОН или NaOH Анал. р-р, Вг2 (для окисл. ТГ), изб. pear., промыв, гор. водой, высуш. при 70-80 °C Т12О3 46-49
Соль Рейнеке (2,5%) Анал. р-р (1 мл, > 2 мг Tl), H2SO4 до < 2 н., 2-3 мл свежеприготовленного pear., нагр. до 40-50 °C 20 мин, ф.с., промыв. 3-4 раза по 5 мл хол. воды, высуш. при ПО °C. Ошибка опр. 2 мг Т1 < 0,4% Tl[Cr(SCN)4- •(NH3)2] f= 0,3910 51
H2SO4 Анал. р-р (соли с летучими кислотами), добавл. H2SO4, выпар, до прекращения выделения SO3, прокал. при 220-240 °C (< 355 °C) T12SO4 872
H2S Анал. р-р (pH = 5-6 или слабоаммиачный), H2S (газ). Ос. р-ют в HNO3 и опр. с H2SO4 (см. выше) T12SO4 873, 874
Тионалид Анал. р-р (10-20 мл, 25-100 мг Т1), нейтр. NaOH, 2 г тартрата Na, 3-5 г KCN, равный объем NaOH (2 н.), разб. до 100 мл, изб. pear, (на 0,1 г Т1+ ок. 0,4-0,5 г pear, в 10 мл ацетона), нагр. до кип., охл., ф. с., промыв, хол. водой, ацетоном. Высуш. при 100 °C. Не меш.: Ag, Au(III), Pt(IV), Pd, Cu, Pb, Bi, Cd, Sn(II), Zn, Co, Ni, Al, UO3+, Mg, Ca, Sr, Ba и др. Возможно опр. 3-6 мг TI в 1-3 мл р-ра с удовлетворительной точностью T1(C12H10ONS) f= 0,4859 52, 53 875 53
Na[B(C6Hs)4] (2%) Анал. р-р (40 мл), 20 мл CH3COONa (2 н.), изб. pear., отстаив. 2 час Ос. ф. с., промыв. 3 раза по 10 мл воды, высуш. при 105 °C T1[B(C6HS)4] f= 0,3903 50
Меркаптобензотиазол (1% аммиачный р-р) Анал. р-р (ок. 30 мл), большой изб. pear., ф. с., промыв. NH4OH (2,5%), высуш. при 110 °C C7H4NS2T1 f= 0,5515 876- 878
Антипирин (0,1 М) Анал. р-р (9-30 мг Т1, 10-20 мл), 5 мл HNO3 (1:1), р-р КВгО3 (0,1 н.) — 1 мл на 10 мг Т1, 10 мл КВг (2 М). Изб. Вг2 связывают сульфосалициловой к-той (10%). При перемеш. добавл. 10-15 мл pear., отстаив., ф. с., промыв. 2-3 раза р-ром pear., 2-3 раза нас. р-ром ОФ, высуш. при 105-110 °C (CnH12ON2)2-•HTlBr4 f= 0,2265 879
8-Оксихинолин (р-р в СН3СООН) Анал. р-р (ТГ), «бромная вода» до пожелт., изб. Вг2 связывают сульфосалициловой к-той, добавл. винную к-ту, р-р pear, и CH3COONTU Ос. нагр. с NI14ОН (2 н.), разб. водой. Через 2 час ф. с., промыв. М14ОН (разб.) и водой, высуш. при 100 °C T1(C9H6NO)3-3H2O 880
Внутренний электролиз Анал. р-р (слабоазотнокисл., 20 мл, 20-30 мг Т1), разб. до 50 мл, добавл. 1 г NH4NO3, нагр. до 60-70 °C, погружают платиновую сетку (анод), коллодиевую гильзу (катод) и соединяют сетку с РЬО2. На аноде осажд. черн. блестящий ос. Т12О3. Через 30-60 мин анод промыв, водой, сп., эф. и высуш. на воздухе. Катод: целлюлозную гильзу для аппарата Сокслета с толщиной стенки 2 мм погружают на 2 мин в 4% эфирный р-р коллодия, после испарения эфира заполняют кашицеобразной смесью РЬО2 и H2SO4 (конц.). Одной заправки хватает на 50 определений Т12Оз 871
При наличии Т13+ его восстанавливают до Т1+ действием SO2.
460
Новый справочник химика и технолога
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Опр. также в виде: Т1С1 [881, 882]; Т1Ю3 [883]; T14SnS4 [884], [Со(НН3)6][Т1С1б] [885]; Tl2Ca[Fe(CN)6] [886]; Т13[Со(ЫО2)б] и Tl2Na[Co(NO2)6] [887]; Au (после восст. Аи3+ действ. Tl+), Se (после восст. SeOg” действ. Т1+), электролизом в виде Т1 [888] или Т12О3 [889, 890]
Sc3+ NH4OH Анал. р-р (слабокисл.), нагр. до кип., добавл. pear, до щел. р-ции, выдерж. 1 час при 100 °C, ф. б., промыв, гор. водой, прокал. при 900 °C Sc2O3 891, 892
Винная к-та или тартраты Анал. р-р разб. до содержания 1 г оксидов в 100 мл р-ра, нейтр. до почти нейтр. р-ра, добавл. pear, до содержания его прим. 10%. Р-р нагр. до кип., нейтр. NH4OH по мет. кр. (концентр, изб. NH4OH <0,1 н.), выдерж. 1-2 час при 100 °C, отстаив. 8-12 час, ф. б., промыв, р-ром (0,01 н. NH4OH с добавл. тартрата NH4 ), прокал. при 900 °C ОФ: NH4COC(CHOHbCOOSc(OH)2. Отд. от Th, Zr, Hf, Al, Fe, Mn Sc2O3 893, 894
Пиридин + C5H5NHNO3 Анал. р-р (слабокисл.) добавл. буферный р-р (пиридин с его азотнокислой солью) до pH = 5,4. Ос. ф. б., при необходимости переосажд. Отд. от La, Ce(III), Рг, Nd, Sm, Gd, Но, Er, Yb, Y. Ос. обрабатывают как при осажд. NH4OH Sc2O3 895
(NH4)2CO3 Опр. Sc в тортвейтите. Сплавл. 1 г руды с 5-6 г NaOH в Ag- или Ni-тигле при темнокр. калении, охл., выщел. гор. водой. Ос. фильтр., промыв, гор. водой, раств. в H2SO4 (конц.) при нагр., вливают р-р в большое количество хол. воды, добавл. изб. NH4OH, ос. ф. б., промыв., раств. в HNO3. Р-р выпар. досуха, нитраты раств. в хол. воде, осажд. Sc действ. K2SO4, отстаив. 12 час, ф. б., промыв. K2SO4 (нас.), обрабатывают хол. (NH4)2CO3 (25%), ос. отфильтр., промыв, хол. (NH4)2CO3. Ф-т кип. до полного удаления NH3, ос. ф. б., промыв, гор. водой, прокал. при 900-1000 °C Sc2O3 896
Опр. также в виде: [Co(NH3)6][ScF6] [505]; Sc2O3 (после осажд. в виде ScF3 [897, 898], из кипяящего p-paNa2CO3 [898],
в виде двойного сульфата с K2SO4 [7, 900], основного тиосульфата [901])
уЗ+ Н2С2О4 Анал. р-р (< 0,2 н. по НС1), нагр., добавл. pear, до 0,1-0,3 М изб. При осажд. малых количеств (< 0,01 М р-р Y3+) из чистых р-ров р-цию проводят при 0-20 °C с 2-5 кратным изб. pear., pH > 2. Ос. ф. с., высуш. 10 мин в вакуум-эксикаторе Ф. б. Прокал. при 800-850 °C Y2(C2O4)3-7H2O f= 0,31309 y203 f= 0,78746 54, 902 903- 905
NaOH или NH4OH Анал. р-р, небольшой изб. pear., далее как при осажд. Н2С2О4. Прокал. при 850-900 °C Y2O3 f= 0,7846 906- 908
K4Fe(CN)6 Анал. р-р, небольшой изб. pear., сп. до 40-60%, ф.с., высуш. на воздухе или в эксикаторе, Г. Ф. K4Y8[Fe(CN)6]7- •30H2O 909
Na3PO4, Na2HPO4, NaH2PO4 Анал. р-р, изб. pear, до 10“3 М, нейтр. до pH = 4,5, ф. б., прокал. при 900 °C ypo4 /= 0,48354 912
8-Оксихинолин а) Анал. р-р (слабокисл.), pear., мочевина. Р-р нагр. После отстаив. р-р ф. с., высуш. при 110-120 °C или ф. б., прокал. при 700-750 °C. б) Осажд. в присутствии винной, янтарной или лимонной к-т слабокисл. анал. р-р, добавл. pear., медленно нейтр. NII4OH до pH = 8-9, ос. обрабатывают по п. «а» Y(C9H6ON)3 y2o3 Y(C9H6ON) 913 913 914- 916
Химические методы количественного анализа
461
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
La3+ Ln Н2С2О4 а) Анал. р-р (НС1, 0,3 н.)* нагр. до 60-80 °C, добавл. pear. (1 объем нас. р-ра). Ф. б. (промыв. 1% р-ром Н2С2О4). Прокал. при 800 °C см. табл. 5.1.11.1.А,Б,В. б) Анал. р-р (< 0,2 н. НС1), 0,1—0,3 М изб. pear., осажд. из гор. р-ра. Высуш. в вакуум-эксикаторе 6-10 мин с 3-кратным продуванием парами эфира Значение п для: La = 8 Се =10 Рг = 9 Ва20з (кроме Се, Рг, ТЬ) Вп2(С2О4)з‘иН2О /= 0,40499 /= 0,38684 /= 0,39805 25, 54 902
NH4OH Анал. р-р (содержащий NH4C1) нагр. до 100 °C, добавл. pear. Ф. б. (промыв. NH4NO3, NH4OH). Прокал. при 800 °C (см. табл. 5.1.11.1.Д) La2O3 (см. Н2С2О4) 54, 906, 907
Се3' КЮ3 + КВгОз а) Анал. р-р (HNO3, 25%), КВгО3 (0,5 г), КЮ3 (10% в 36% HNO3, 10-15-кратный изб.). Первое. Ф. с. (промыв. КЮ3, сп., эф.). Высуш. 10-15 мин при 40-50 °C. б) Осажд. как в п. «а». К ос. добавл. Н2С2О4, нагр., добавл. Н2О. Через 1-2 час ф. б. (промыв. 1% р-ром Н2С2О4). Прокал. при 800 °C 2Ce(JO3)4KJO3- •8Н2О СеО2 90 278, 917
С Сжигание в токе О2 а) Сжигание в токе О2 при 1000-1100 °C. Образующийся СО2 очищается от пыли, оксидов азота, SO2, SO3, паров воды и поглощается в предварительно взвешенных трубках с натровой известью. б) Определение графита: навеску металла р-ют в HNO3 (1:1) при нагр. на водяной бане, добавл. 1 мл HF, затем добавл. 150 мл гор. воды, фильтр, через прокаленный асбест, промыв, гор. водой, гор. р-ром КОН (2%), НС1 (1:20) и снова гор. водой. Асбест с ос. графита помещают в печь и сжигают по п. «а» СО2 СО2 7, 25, 55 25
H2SO4 (1:1), СгО3 (50%) В колбу для разложения вводят 60 мл р-ра СгОз и 200 мл H2SO4, подключают колбу к установке для опр. С (очистительные трубки, печь при 800-900 °C), пропускают слабый ток воздуха и кип. р-р ок. 1 час. Подключают взвешенную поглотительную трубку, кип. при слабом токе воздуха 2 час, поглотительную трубку взвешивают («холостой опыт»). Привес должен быть < 0,5 мг. Р-р в колбе охл., подключают поглотительную трубку, помещают в колбу навеску образца, постепенно нагр. р-р и кип. не менее 1 час после растворения пробы. Поглотительную трубку взвешивают, вычитают вес «холостой пробы» и опр. вес выделевшегося СО2 СО2 7,918, 919
Ва(ОН)2 Выделенный одним из описанных методов СО2 поглощают Ва(ОН)2. Ос. промыв., прокал. при > 476 °C ВаСОз 920, 921
ВаС12, NH4OH Р-р pear.: 50 г ВаС12-2Н2О, 250 мл NH4OH (плотн. 0,88) разб. до 1 л. Перед применением кип. 1 мин. для осажд. поглощенного ранее СО2, фильтр, гор. через асбест в поглотительный сосуд. Выделенный из пробы СО2 поглощают хол. р-ром pear., продувая струей воздуха, свободного от СО2. По окончании погл. р-р быстро нагр. до кип. и кип. 1 мин в токе воздуха, охл., фильтр., промыв, ос., поглотительный сосуд хол., затем гор. водой, ос. раств. в поглотительном сосуде в НС1 и осажд. BaSO4 (см. опр. Ва2+) BaSO4 922
Для маскировки Tl4+, Ta(V), Nb(V) добавляют Н2О2.
462
Новый справочник химика и технолога
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
CN~ AgNO3 (5%) а) Анал. p-p (20-100 мг CN- нейтр. или слабоаммиачный, ок. 400 мл) добавл. по каплям pear, до появления ос., подкисляют HNO3 (2 н.) и добавляют объем pear., равный уже добавленному с небольшим изб. Ф. с., промыв, хол. водой до отрицательной реакции на Ag+, высуш. 2 час при 110 °C б) Ос. по п. «а» прокал. при 520 °C AgCN Ag 211, 397, 962, 963 962, 963
OCN AgNO3 Анал. р-р (нейтр.), pear. Ф. с., высуш. при 110 °C AgOCN 211
SCN- AgNO3(0,lH.) Анал. р-р (HNO3 разб.), pear. Ф. с. Высуш. при 115 °C AgSCN 211, 576, 577
C11SO4 + H2SO3 Анал. р-р (< 100 мг SCN-, нейтр. или слабокисл., но без NO3, 50-200 мл), 50 мл свежеприготовленного р-ра SO2 или NaHSO3 (10%), при перемеш. 60 мл C11SO4 (0,1 н.), добавл. еще 10 мл р-ра SO2 или NaHSO3, отстаив. 1 час (лучше на ночь). Ф. с., промыв, водой с добавл. SO2 до удаления Си2+ (по р-ции с гексацианоферратом(П)), несколькими мл сп., высуш. 2-4 час при 110 °C CuSCN 964, 965
Окислитель (Br2, HNO3 или H2O2) + BaCl2 Анал. р-р (< 100 мг SCN-, нейтр. или слабокисл.), изб. бромной воды, нагр. при 100 °C 1 час. Р-р нейтр. NH4OH по мет. оранж., добавл. 1 мл НС1 и 1 г тв. NH4C1, затем при кип. 10 мл ВаС12 (5%), кип. еще 2-3 мин, отстаив. 12 час. Ф. б., промыв. 25 мл хол. воды, 25 мл теплой воды, прокал. после сгорания фильтра 10 мин при 620-800 °C. Для получения точных значений вес прокал. ос. умножают на корректирующий фактор 1,0143 (по Винклеру) BaSO4 966
Si Кислоты a) H2SO4. Сернокисл. анал. р-р упар. до паров SO3 и еще 3-4 мин, охл., добавл. 100-150 мл теплой воды и слегка нагр., ф. б., промыв. НС1 (5:95) до удаления Fe3+ (проба с SCN-), затем 8-10 раз теплой водой до удаления СГ (проба с Ag+); ос. прокал. в платиновом тигле при 1050-1100 °C, взвешивают 1-й раз. К ос. добавл. 1-2 капли H2SO4 (1:1) и 3-5 мл осторожно выпар., прокал. и взвешивают 2-й раз. Содержание SiO2 находят по разности 1-го и 2-го взвешиваний. б) НС1. Солянокисл. анал. р-р выпар, досуха, охл., добавл. 25 мл НО (конц.) и повторяют выпар. После появления сухой корки солей нагр. еще 25 мин (или сухой остаток выдерж. 30 мин при 130 °C). После охл. добавл. 20 мл НС1 (конц.) и через 5 мин 150 мл гор. воды, нагр. до 60-70 °C, ф. б. (син. лента), промыв. НС1 (5:100) и гор. водой. Далее как в п. «а». в) НСЮ4. Анал. р-р, 35-40 мл НС1О4, упар. до густых бел. паров и еще 10-12 мин (закрыть стеклом), охл., 15 мл НС1 (конц.) и 100-120 мл гор. воды, раств. соли, ф. б. (бел. лента), промыв. НС1 (5:100) и водой. Далее как в п. «а». г) Желатиновый метод. Солянокисл. анал. р-р упар. до влажных солей, добавл. 3 мл р-ра желатины (2%), раств. соли 50 мл воды (без нагр.), ф. б., промыв, хол. НС1 (5 : 95). Далее как в п. «а» SiO2 SiO2 SiO2 7,11, 25 7,11, 25 7,11, 25, 925, 926 25, 56, 57, 923, 924
Химические методы количественного анализа
463
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Si (NH4)2MoO4 + 8-оксихинолин Анал. р-р (после щел. сплавления пробы), тимоловый син., по каплям НС1 (конц.) до перехода от желт, к кр. затем последовательно при перемеш. 8 мл НС1 (1:9), 5 мл СН3СООН (1:2) и 20 мл р-ра (NH4)2MoO4 (10%), отстаив. 15 мин, добавл. 40 мл НС1 (1:1) и сразу же 60 мл р-ра оксихинолина (14 г р-ют в 20 мл 6 н. НС1 и разб. до 1 л) из бюретки при перемеш.; нагр. при 60 °C в течение 10 мин, ф.с. и промыв, декантацией (2 раза), 10 раз на фильтре нас. р-ром кремнемолибдата оксихинолина и 1 раз 5 мл воды. Высуш. 1 час при 140 °C (4C9H7ONSiO2-12МоО3-2Н2О) 58, 59, 927, 928
(NH4)2MoO4 + хинолин а) Анал. р-р (ок. 5 мг SiO2) в полиэтиленовом стакане разб. до 250 мл, 3 г NaOH (в гранулах), индикатор (смесь тимолового син. с крезоловым кр.), по каплям НС1 до перехода окраски от син. через желт, до кр. Р-р переносят в стеклянный стакан (800 мл), добавл. 10 мл НС1 (1:9), разб. до 400 мл, 50 мл (NHi)2MoO4 (10%), отстаив. 10 мин, 50 мл НС1 (конц.), 50 мл хинолина (2%) по каплям из бюретки при перемеш., нагр. 10 мин до 80 °C и выдерж. 5 мин. Охл. до < 20 °C, ф. с., промыв. 6 раз р-ром хинолина (0,05%), высуш. при 150 °C Рекомендовано для определения SiO2 в силикатах. б) В сталях: навеску 0,5 г раств. в 20 мл H2SO4 (5%), фильтр., промыв. H2SO4, фильтрат разб. до 50-60 мл, добавл. 20 мл (NH4)2MoO4 (5%) с появлением син. окраш. Через 10 мин добавл. 10 мл НС1 (конц.), 30 мл хинолина (2%), нагр. до 70-80 °C. Охл. до 20 °C, ф. с., промыв. 5-6 раз р-ром хинолина (0,05%), высуш. 1 час при 150 °C (SiO212MoO3-•4C9H7N-2H2O) f= 0,02566 См. п. «а» 60 930
(NH4)2MoO4 + диантипирил-метан В силикатах (полумикрометод): 0,05 г пробы (10-50% SiO2) сплавляют с 1 г NaCO3, выщелачивают водой, добавл. 30 мл НС1 (1 н.), разб. до 250 мл. К аликвоте 25 мл добавл. 7 мл НС1 (1 н.), 5 мл (NH4)2MoO4 (10%), нагр. 15 мин, разб. вдвое, 7-14 мл НС1 (конц.), по каплям диан-типирилметан (2% в 0,5 н. НС1); ф.с., промыв, водой (подкисл. НС1), высуш. при 125-130 °C. Ошибка 0,9%, продолжительность 3-3,5 часа /= 0,01986 61
Бензидин + HF В кремнийорганических соединениях: 20-50 мг тетраэтоксисилана помещают в полиэтиленовый стакан, содерж. 50 мл HF (0,1-0,3 н., в сп. или ацетоне), перемеш. 2 мин, добавл. 20 мл бензидина (1% в сп. или ацетоне), ф. с., промыв. охл. сп. или ацетоном, высуш. при 100 °C. Время определения 2-3 час (C12H12N2H2SiF6) 62
Опр. также в виде: SiO212MoO3 [928, 929]
Ge MgSO4 Анал. р-р (20-100 мг Ge, слабокисл.), (NH4)2SO4 (2 н.), ре-аг., NH4OH (изб.). Нагр. до 100 °C, охл. Через 12 час ф. б. (промыв. 3% р-ром NH3). Прокал. при 1000-1100 °C Mg2GeO4 66
Таннин а) Анал. р-р (300 мл 0,07 н. Н2С2О4), pear. (5%, 1 мл на 1 мг GeO2). Ф. б. (промыв. 5% р-ром NH4NO3). Прокал. при 900 °C. б) Анал. р-р (300-400 мл), 5 мл H2SO4 (конц.), 10 г (NHi^SO* нагр. до кип., 10-30 мл pear. (5%), 2 г (NH4)2SO4, охл., ф. б., смывают ос. водой в стакан, добавл. 5 г (NH4)2SO4, 5 мл HNO3 (2 н.), разб. до 100 мл, ф. б. (тот же), промыв, водой, фильтр в тигле смачивают несколькими каплями HNO3 (конц.) и H2SO4 (конц.). Прокал. при 900-1000 °C GeO2 GeO2 960 1
464
Новый справочник химика и технолога
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Ge H2S а) Анал. р-р (> 2,5 мг Ge, H2SO4, 6 н.), pear, (до нас.), отстаив. в закрытом сосуде 12 час, вновь pear, (до нас.), фильтр., промыв. H2SO4 (3-4 н., нас. H2S). Ос. р-ют в NH4OH (10 н.), фильтрат собирают в платиновую чашку, добавл. 25 мл Н2О2 (3%), отстаив. и выпар, на водяной бане досуха. Остаток смачивают H2SO4 (1:1), прокал. при 900 °C. б) Ос. GeS2 по п. «а» кип. с водой до удаления H2S, выпар, досуха, обрабатывают несколькими каплями HNO3, выпар, досуха и прокал. при 900 °C. в) Промытый по п. «а» ос. промыв, сп., высуш. в токе СО2, постепенно нагр. до 350-380 °C, выдерж. 30 мин, охл. в токе СО2, взвеш. ос. GeS2. Возможно опр. не более 0,1 г Ge GeO2 GeO2 /=0,766 7, 63, 64 931 933
3,4-Диокси-азобензол Анал. р-р (5 мл, < 20 мг GeO2), 330 м pear. (1%, в 96% сп.), 6 мл НС1 (конц.), ф. б., промыв. НС1 (1:10), высуш., смачивают HNO3, прокал. медленно до 550-600 °C, затем при 900 °C GeO2 63, 936
TV-Бензоил-фенилгид-роксиламин Анал. р-р (10 мл, 5-20 мг Ge), НС1О4 до 2 н. конц., нагр. на водяной бане, 8-10 мл pear. (5% в сп.), ф. б., промыв. Н2О до удаления С1О4, прокал. при 900 °C GeO2 65
Акридин + (NH4)2Mo04 Анал. р-р (1-10 мг GeO2), 1-2 капли HNO3 (конц.), разб. до 50 мл, 10-20 мл (NH4)2MoO4 (10%), разб. до 80 мл, нагр. 10 мин при 100 °C; добавл. 2 мл HNO3 (конц.), при перемеш. 10-20 мл акридина (1 г раств. в 5 мл лед. СН3СООН и разб. до 1 л), через 1 час ф. с., промыв. 5 раз промыв, р-ром (30 мл HNO3, 50 мл р-ра акридина, 920 мл Н2О), сушат при 105 °C 1 час (C13H9NH)4- •[Ge(Mo12O40)] /= 0,0405 67
Гуанидин + (NH4)2MoO4 Анал. р-р (10-30 мл, 1-30 мг GeO2), нейтр. по тимоловому син., 2 мл (NH4)2MoO4 (5%), H2SO4 (10%) до 0,1-0,2 н. конц.; через 10 мин 2 мл НС1 (конц.), 20 мл сульфата гуанидина (20%) или 20 мл нитрата гуанидина (нас.). Через 12 час ф. с., промыв, нас. р-ром германомолибдата гуанидина, высуш. при 130-140 °C (CH5N3)4-•[Ge(Moi2O40)] /= 0,04897
8-Оксихинолин + (NH4)2MoO4 Анал. р-р (50 мл, 0,5-5 мг Ge, слабокисл.), (NH4)2Mo04 (5%, свежеприготовл.) по 2 мл на 1 мг GeO2 (но не менее 2 мл), 3 мл H2SO4 (10%), разб. до 100 мл; через 5 мин — 9 мл НС1 (конц.), по каплям 20 мл 8-оксихинолина (20 г в 120 мл лед. СН3СООН, Н2О до 1 л). Через 3-12 час ф.с.; промыв, р-ром (7 мл конц. НС1, 25 мл р-ра 8-оксихинолина, вода до 1 л); высуш. при 115 °C. Прокал. при 500-900 °C Эмпирический /= 0,0488 GeO212MoO3 69 176
Опр. также в виде: Mg2GeO4 [932]; сплава cNi (электролиз) [934], сплава с Sn (электролиз) [935]; 1,5А12Оз СеО2 «Н2О [937]; Ba2[Ge(C4H4O6)2]-2H2O [938]
Sn(II, IV) nh4oh а) Анал. р-р (НС1, NH4C1) pear, (по мет. кр.). Фильтр, с бум. массой. Ф. б. (промыв, гор. 2% р-ром NO3). Прокал. при 900 °C. б) Анал. р-р (после раств. навески сплава 0,2-0,5 г в НС1 с Н2О2 и удаления перекиси кип.), добавл. изб. ЭДТА-Na, 10 г NH4NO3, разб. до 100 мл и нейтр. NH4OH до pH = 8-9, отстаив. 18 час, ф. б., промыв. NH4NO3 (1%, нейтр. NH4OH по мет. кр.), прокал. при 900 °C SnO2 SnO2 7 954
Химические методы количественного анализа
465
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Sn(II, IV) H2S а) Анал. р-р (5-15 мл H2SO4, 20-50 мл 20% NH4C1, бум. масса в объеме 200-400 м), насыщ. H2S, отстаив. до осажд. ос., ф. б., промыв. H2SO4 (1:99) с H2S. Прокал. при ® 1000 °C. б) В сталях: навеску 5-10 г раств. в 75—150 мл H2SO4 (1:4), окисл. HNO3 (1:1), разб. до 200 мл, фильтр., промыв. 8 раз гор. НС1 (5:100). К ф-ту добавл. 50 мл винной к-ты (20%), нейтр. NaOH (10%) по бум. конго (серое пятно), нагр. до 60 °C, насыщ. H2S 1 час, охл., фильтр., промыв. 6-8 раз Н28-водой, прокал. при 1000 °C. Ос. переносят в стакан, добавл. 5-8 мл НС1 (1:1) и 3 мл Н2О2 (3%), нагр., добавл. 30 мл воды, ф. б., промыв. 5-6 раз водой, прокал. до SnO2 SnO2 SnO2 70 25
Na2S Анал. р-р (Sn2+ и НС1), бромная вода, нейтр. NH4OH до по-явл. ос., Na2S (2 н.) по 15 мл на каждые 50 мг Sn. Тиосоль разрушают изб. СН3СООН (2-3 н.), отстаив. 10 мин, ф. с., промыв, водой, сп. и эф. Высуш. в вакуум-эксикаторе 1 час SnS2-2H2O 71
HNO3 При раств. навески в HNO3 или при медленном упар. азотнокисл. р-ров при 80-90 °C до малого объема или досуха на водяной бане. Далее добавл. 30 мл HNO3 (1:1), 20 мл NH4NO3 (10%), отстаив. 1 час при 50-60 °C, ф. б. с бум. массой, промыв. HNO3 (1:20), прокал. при 950 °C. Для очистки ос.: в тигле добавл. 3 мл HNO3 и 5 мл НС1, натр., добавл. 15 мл воды, переводят р-р в стакан, нагр. 15 мин, добавл. 30 мл гор. воды, ф. б., промыв. Н2О, прокал. до SnO2 SnO2 72, 73
Купферон (6%) Анал. р-р (H2SO4 или НС1), Н3ВО3 (для маек. F-). Охл. до 0 °C, добавл. pear. Ф. б. (промыв. 1% р-ром НС1). Прокал. при 900 °C. При низком содержании Sn (0,02-0,1%) и для отд. меш. элементов осажд. с коллектором МпО2 SnO2 25
Фениларсоновая к-та Анал. р-р (НС1, 1,5%), pear. Нагр. Ф. б. (промыв. 4% р-ром NH4NO3). Прокал. при 900 °C SnO2 955
8-Оксихинолин (2% в 0,2 н. НС1) Анал. р-р (0,2 н. НС1, 2-10 мг Sn), равный объем pear., отстаив. 2 час при 70-90 °C, ф. с., промыв. НС1 (3 н.), высуш. при 110-120 °C SnCl2(C9H4NO)2 74
V-Бензоил-фенилгидрок-силамин (1% В СП.) Анал. р-р (200 мл, SnCl4), 10 мл НС1 (конц.), по каплям при перемеш. pear. (5 мл на каждые 10 мг Sn и изб. 8 мл). Отстаив. при 0 °C 3 час, ф. с., промыв. 5 мл лед. воды, высуш. при 110 °C. Прокал. до SnO2. Не меш.: Cu, Pb, Zn Sna^nO^ /= 0,1927 75
Электролиз Анал. р-р (25-200 мг Sn), Н2С2О4. Цинковый анод, покрытый коллодием, платиновый катод. 1-3 час (внутренний электролиз) Sn 385
Опр. также в виде: SnSe2 [465]; Se (восст. Sn2+) [956]; Sn (внутренним электролизом) [857-859]
Pb2+ H2SO4(1:1) Анал. р-р, pear. Выпар, до появл. дыма (SO3), добавл. Н2О (до 8% H2SO4). Через 3-4 час ф.с. (промыв. 6% H2SO4, нас. PbSO4), высуш. при 120-130 °C. Или ф. б., прокал. при 300-600 °C PbSO4 939- 941
K2SO4 Анал. р-р, pear, (не менее 0,5 г на 100 мл). Выдерж. 2-3 час. Ф. с. (промыв. 0,03 М K2SO4). Высуш. при 130 °C K2SO4PbSO4 f= 0,4340 76
(NH4)2SO4 Анал. р-р (нейтр. по мет. кр., < 0,5 г Pb), 1 мл HNO3 (1 М), Н2О до 100 мл, нагр. до кип., медленно 10 мл (NH4)2SO4 (10%) так, чтобы р-р не попадал на стенку стакана. Через сутки ф. с., промыв. 50 мл нас. р-ра PbSO4 или сп. (50%), высуш. 2 час при 130 °C PbSO4 1,25, 939-941
466
Новый справочник химика и технолога
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
РЬ2+ (NH4)2MoO4 Анал. р-р (разб. HNO3). Нагр. до 100 °C, добавл. pear., нейтр. NH4OH, добавл. СН3СООН. Ф. с. (промыв. 2% р-ром NH4NO3). Прокал. при 600 °C РЬМоО4 25, 942
Электролиз Анал. р-р (HNO3, 10 объемн. %). Платиновая чашка — анод (« 3 А дм"2) при 70 °C (электрод промыв. Н2О, сп.). Высуш. при 180 °C РЬО2 385, 943- 945
К2СгО4 или К2Сг2О7 Анал. р-р (HNO3,0,3 н.) нагр. до 100 °C, добавл. pear. (4-кратный изб.). Ф. с., высуш. при 140 °C. или ф. б., прокал. при 550-600 °C РЬСгО4 25
СгО42"(Сг3+ + ВгО3") Гомогенное осаждение. Анал. р-р (0,1-0,2 г РЬ), нейтр. NaOH до начала образ, ос., 10 мл ацетатной буферной смеси (6 М СН3СООН в 0,6 М CH3COONa), 10 мл Cr(NO3)3 (10%), 10 мл КВгО3 (2%), нагрев. 30-45 мин при 90-95 °C, после просветления р-ра ф. с., высуш. при 120 °C РЬСгО4 2
НС1 (2% р-р в бутаноле-1) В присутствии больших количеств Bi дает очень точные результаты. Анал. р-р, pear., 5 мин кип., охл., ф. с., промыв. 5-6 раз небольшими порциями осадителя. Высуш. 30 мин при 105-110 °C, затем 10 мин при 250 °C РЬС12 3
Висмутиол II (калиевая соль) Анал. р-р (25-75 мг РЬ), 0,1 М HNO3, 1-2 г NH4NO3, Н2О до 125 мл, нагрев, до 50-60 °C, медленно добавл. свежеприготовленный pear. (0,5%, изб. 3-4 мл, при перемеш.), выдерж. 1 мин и сразу ф. с. Промыв, гор. водой, сушат при 105-110 °C ок. 1 час. В присутствии As(III), Bi, Cu, Sn(II), Sb(III) реагент добавл. в анал. р-р с pH = 3-6,5 с добавл. тартрата. Погрешность < 0,4% (CgH5N2S3)2Pb f= 0,3150 4,5
Пикролоновая к-та (0,01 н.) 50 мл анал. р-ра (ок. 0,1 г РЬ), нейтр. по лакмусу, нагр. до кип., по каплям при перемеш. добавл. 100 мл pear., затем сразу 50 мл. Р-р охлажд. в холодильнике, ф. с., промыв. 30-50 мл лед. воды до бесцветного фильтрата. Ос. крист., высуш. при 130-140 °C около 1-1,5 час (C10H7N4O5)2Pb /= 0,2824 5
Опр. также в виде: PbS [266, 946], РЬ(Ю3)2 [947], РЬ3(РО4)2 [948], РЬСО3 [949], Pb(OH)SCN [950], РЬС2О4 [951], РЬО
[952, 953], РЬС12 3], и менее пригодных форм [5]: РЬНРО4, PbSO3, PbWO4, PbHAsO4, Pb3H4(IO6)2
Ti4+ Купферон (6% р-р) Анал. р-р (НС1 или H2SO4). Охл. до 10 °C, добавл. pear. Ф. б. (промыв, хол. 1% НС1 + pear.). Прокал. при 800-1000 °C (осторожно) TiO2 1,21, 81
Таннин, 10% р-р (1) и антипирин* (20% р-р) (2) Анал. р-р (H2SO4, 6%), pear. 1, pear. 2. Нагр. до 100 °C, добавл. (NH4)2SO4. Прокал. при t > 650 °C TiO2 80
8-Оксихинолин, (2% сп. р-р) Анал. р-р (150 мл), винная к-та (1 г), CH3COONa (0,5 г), СН3СООН (лед.) (1,5 мл), p-в, нагр. до 60 °C, затем при 100 °C. Ф. с. (промыв, гор. Н2О). Высуш. при 110 °C TiO(C9H6ON)2 79
н-Оксифенил-арсоновая к-та Анал. р-р, pear. (4% водн. р-р). Нагр. до 100 °C. Ф. б. (промыв. pear. 0,5% в 0,25 н. к-те). Прокал. при 800-1000 °C TiO2 82
NH4OH или пиридин Анал. р-р, pear, (до pH = 5-6). Ф. б. Прокал. при 800-1000 °C TiO2 1,77, 78
Опр. также в виде Ti3(PO4)4 [505]
Zr4+ NH4OH Анал. р-р, 1-5 г NH4C1, нагр. до 70-80 °C, добавл. разб. NHjOH (до запаха), кип. 2-3 мин, ф. б., промыв. 3-4 раза NH4NO3 (2%), прокал. при 1000-1200 °C. Мешают ионы, осаждаемые NH4OH ZrO2 83
1 -Фенил-2,З-диметилпиразолон-5.
Химические методы количественного анализа
467
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Zr4+ Пиридин (20%) Анал. р-р (солянокисл.), нейтр. (NH4OH, НС1), добавл. NH4CI (5 г на 100 мл р-ра), нагр. до кип., добавл. мет. кр., pear, до перехода окраски из кр. в желт, и изб. 15-20 мл, отстаив. 40 мин при 100 °C. Окраска должна остаться желт. Гор. р-р ф. б., промыв, гор. NH4NO3 (3%) с пиридином, прокал. при 1000-1200 °C ZrO2 38
Уротропин (10%) Анал. р-р (слабокисл.) разб. до 300 мл, добавл. 10 г NH4C1, нагр. до кип., по каплям pear, (с изб. 2-3 мл), отстаив. при 100 °C, ф. б., промыв. NH4NO3 (2%), прокал. при 1000— 1200 °C ZrO2 83,84
Фталевая к-та (2%) В магниевых сплавах: навеску 1 г (0,1-2,5% Zr), 15 мл НС1 (1:1), добавл. 100 мл воды, 30 мл Nl^NO; (нас.), нагр. до кип., 25 мл кип. р-ра pear., кип. 1-2 мин, охл. до 30-40 °C, ф. б. промыв, р-ром (0,1% фталевая к-та в 1% НС1). Прокал. при 800 °C ZrO2 25
Купферон (6% р-р) а) Анал. р-р (H2SO4, 20%); охл. до 10 °C, добавл. pear., фильтр, с бум. массой. Ф. б. (промыв. 3,5% р-ром НС1, охл. до 10 °C). Прокал. при 1200 °C. б) В сплавах с Nb: 0,1 г сплава раств. в 2-3 мл HF с добавл. HNO3, 3-4 мл H2SO4 (конц.), упар. до паров SO3 и еще 15 мин, охл., 25 мл винной к-ты (4%), 10 мл NH4F (2%), переносят из платиновой чашки в стакан, нейтр. NH4OH по феноловому кр. до перехода из желт, в кр., добавл. 2 капли NH4OH, охл., 110-15 мл pear., отстаив. 12 час, ф. б. с бум. массой, промыв. 5-6 раз хол. водой, прокал. до ZrO2 ZrO2 ZrO2 87, 88 86
H2SeO3 (12,5%) Анал. р-р (НС1, 1,5%) нагр. до 100 °C, добавл. pear., фильтр, с бум. массой. Ф. б. (промыв. НС1, 1% р-ром pear.). Прокал. при 1200 °C ZrO2 25, 85, 967, 968
C6H5AsO(OH)2 (10%) Анал. р-р (НС1, 10%), Н2О2 (3% р-р), pear. (10-30 мл), нагр. до 100 °C (2 мин). Ф. б. (промыв. 1% р-ром НС1, 0,1% р-ром pear.). Прокал. при 1000 °C ZrO2 25
(NH4)2HAsO4 Анал. р-р (НС1), pear. (1%). Нагр. до 100 °C, добавл. pear. Ф. б. (промыв. 1 н. НС1, гор. Н2О). Прокал. при 1000 °C ZrO2 11
(NH4)2HPO4 (10%) Анал. р-р (H2SO4, 10 объемн. %), Н2О2, pear. (10-100-кратный изб.). Нагр. до 50 °C. Через 2-24 час ф. б. (промыв. 5% р-ром NH4NO3). Первое, (из плава с Na2CO3). Прокал. при 1000 °C ZrP2O7 /=0,518 11
Миндальная к-та (16%) Анал. р-р (НС1, 20%), pear. Нагр. 25 мин при 85 °C. Охл., ф.с. (промыв. 2% НС1 + pear.; сп.). Высуш. при 110— 120 °C (C6H5CHOHCOO)4Zr /= 0,1772 89
Опр. также в виде: Zr(Cc>H6ON)4 (оксихинолинат), Zr2P$Oi9 (фитинат), 2Zr(IO3)4-KIO3-8H2O, Zr(SeO3)2 [967, 968] или 4ZrO-3SeO218H2O [505]
Hf44 См. опр. Zr4+
Th4+ Н2С2О4 Анал. р-р (НС1, 1,4%) нагр. до 100 °C, добавл. кип. pear. (10% р-р). Выдерж. 10-12 час. Ф. б. (промыв. 2,5% р-ром pear, в 1,2% р-ре НС1). Прокал. при 1100 °C. ThO2 7, 93
468
Новый справочник химика и технолога
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Th4+ КЮ3 (10% в HNO3)(1:2) а) Азотнокисл. (1:2) анал. р-р (20 мл, 4-16 мг Th, 5-6 мл при < 2 мг Th), 5-10 капель Н2О2, равный объем pear., через 30-40 мин ф. с., промыв, р-ром pear. (5%) в HNO3 (1:2), затем р-ром pear. (8 г КЮ3, 50 мл HNO3 конц. в 1 л), 3-4 раза сп., 1-2 раза эф. Высуш. при 40-50 °C в течение 15 мин. б) Осажд. Th как в п. «а». После отстаив. жидкость декантируют через фильтр, переносят на него ос. р-ром КЮз (1%) в HNO3 (1:9), промыв, тем же р-ром, р-ют в 30 мл гор. HNO3, разб. равным объемом воды, переосажд. при добавл. р-ра (4 г КЮ3 в 7 н. HNO3), фильтр, и промыв. как указано выше. Ос. раств. в НС1 в присутствии SO2. В р-ре Th и немного Ti и Zr. Кип. со слабым изб. NH4OH. Гидроокиси р-ют в 10 мл НС1. Осажд. Th в виде оксалата и прокалив, до ThO2. в) Осаждение из гомогенного р-ра: анал. р-р (СЮд, SO,, , NO3; 2-50 мг Th; < 6 мл 6 н. к-ты) упар. до 50 мл, добавл. 40 мл HNO3 (конц.), 14 г КЮ3 в 100 мл Н2О, разб. до 230 мл, охл. до 20 °C. Добавл. 2 мл 0-оксиэтилацетата при перемеш. Через 30 мин добавл. 6 мл Р-оксиэтилацетата. Через 1,5 час добавл. р-р (2 г КЮ3, 9 мл HNO3, 50 мл Н2О). Через 30 мин прекращают перемеш., декантируют через ф. б., промыв. 10 раз р-ром (8 г КЮз, ЮО мл HNO3, 900 мл Н2О). Ос. на фильтре раств. в НС1 (10%, нас. SO2) и собирают в стакане с основной частью ос. Р-р выпар, досуха и сушат 1 час при ПО °C, отфильтр. SiO2, фильтр промыв, гор. НС1 (1:20). Th определяют в ГФ ThO2. Th количественно отделяется от больших количеств TR и фосфатов 4Th(IO3)4KIO3- •18Н2О ThO2 ThO2 7, 90 94 95
Н4Р2Об или натриевая соль Анал. р-р (10% НС1, < 250 мг ThO2), нагр. до кип., добавл. pear., ф. б., промыв. HCI (6%), прокал. до ThP2O7 (при ThP2O7 <10 мг) или переводят в р-р действ. H2SO4 (конц.) или NaOH, осажд. оксалатом и прокал. до ThO2 ThP2O7 ThO2 96, 97 97
Уротропин 100 м анал. р-ра (слабокисл.), NH4C1 (5 г). Нагр. до 30 °C, добавл. pear. (pH = 5,8). Ф. б. (промыв, тепл. 2% р-ром NH4NO3), переос. Прокал. при 750 °C ThO2 100
Себациновая к-та а) Анал. р-р нейтр. при 100 °C, добавл. pear. Ф. б. (промыв, гор. Н2О). Прокал. при 750 °C. б) Осажд. хол. 3% р-ром pear, в сп. ThO2 ThO2 99 98
Na2S2O3 К кип. слабокисл, или нейтр. анал. р-ру медленно добавл. Na2S2O3, ф. б., ос. гидроокиси р-ют в НС1 и осажд. Th в форме оксалата. Используется для опр. Th в присутствии TR ThO2 91
Н2О2 Слабокисл. (1 мл 1,5 н. HNO3 в 50 мл р-ра) анал. р-р, нагр. до 60-80 °C, NH4NO3, pear, и сразу фильтр. Прокал. до ТЪО2. Метод ранее применяли в пристствии TR и в анализе минералов ThO2 92
Опр. также в виде ТЪО2 (после осажд. NH4OH, купфероном или Н2О2), Th(CioH7N405)4H20 (пиколинат), ТЬ(СбН58О2)4 (бензолсульфинат), Th(PO3)4 (осажд. фитином) [505]
Химические методы количественного анализа
469
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
V группа Периодической системы
nh: H2PtCl6 См. опр. К+ (NH4)2PtCl6 \f =Д08094 I VNH4 J или Pt 1 f + = 0,1839 1 V NHt )
Нитробарбиту-ровая к-та (2%, в 50% сп.) 0,1-0,2 г пробы раств. в 25-30 мл воды, добавл. равный объем сп., по каплям pear, до желтоватой окраски р-ра, ф. с., промыв сп., эф. Мешают Cs, Ва, Zn, Mg (К, Rb, Sr). После разлож. по весу ГФ и продуктов разлож. опр. состав бинарных смесей Zn-NIL , Cd-NH; /= 0,0949 (HaNH/) 101, 102
Реактив Несслера Анал. р-р (25 мл), двойной изб. pear., выдерж. 10 час при 20 °C и 3 час при 100 °C, ф. с. (величина пор 1 мк), промыв, водой и высуш. при 150 °C. Для 2-4 мг N ошибка 2,1%. Мешают CN-, SCN-, этилендиамин, пиридин, NH2OH и N2114 NH4HgJ2 f= 0,02584 (naN) 103
n2h4 AgNO3 (косвенное определение) Анал. р-р, изб. р-ра pear, известной концентрации, ос. Ag отделяют, из р-ра изб. Ag осажд. НС1 и взвешивают в виде AgCl AgCl 104, 105
HN3 AgNO3 Анал. р-р (10 мл, ок. 0,03 г HN3), изб. КОН, нейтр HNO3. (1:4) по лакмусу, изб. 1 капля HNO3 (1:100), pear., ос. промыв. декантацией водой, раств. в HNO3 (1:4), кип. для удаления HN3, осажд. Ag действ. НО (изб.). Далее см. определение Ag с НО. (Осторожно! Азиды взрывоопасны! Не высушивать!) AgCl 106
no; Нитрон (10% в 5% СН3СООН) Анал. р-р (80-100 мл, < 100 мг NO;), 1 капля H2SO4 (конц.) или 1 мл лед. СН3СООН, нагр. до кип., 10-12 мл pear., перемеш., отстаив. ок. 1 час, охл. 1,5 час в лед. воде, ф. с., промыв. 5 раз по 2 мл лед. воды, высуш. 1 час при 105 °C c20h16n4- hno3 /= 0,1679 (на HNO3) 7, 107, 108
Дициклогексил-таллия(Ш), карбонат (0,05 н. р-р в 0,2 н. H2SO4) Анал. р-р (50-100 мл, < 100 мг NO;), нагр. до кип., 50-100 мл pear., охл., ф. с., промыв. 2-5 раз по 10 мл лед. воды, высушив. < 150 °C СГ, Вг-, Г предварительно осажд. фторидом или ацетатом серебра C12H22T1NO3 f= 0,1433 (на NO;) 109
no; AgBrO3 Косвенное определение. 1,5 г pear, раств. в 110 мл СН3СООН (2 н.) и 100 мл воды, нагр. до 80 °C, из капельной воронки анал. р-р (200 мл, < 1 г NO;), промыв, воронку, добавл. 30 мл H2SO4 (1:4) при 85 °C, перемеш., нагр. при 90 °C до осветления р-ра, ф. с., промыв. 1 л гор. воды, высуш. при 130 °C AgBr /= 0,7350 (на no;) 110
n2o Fe N2O с газом-носистелем (без О2) пропускают через раскаленную докрасна железную спираль, образующийся О2 реагирует с Fe. Содержание N2O определяют по увеличению веса Fe 106
no CrO3 + HNO3 Р-р pear.: 10 г СгО3 в 15 мл HNO3 (12%) Окись азота полностью поглощается р-ром pear., помещенным во взвешенный сосуд, соединенный со взвешенной хлоркальциевой трубкой. Газ-носитель — чистый СО2 (без примеси воздуха). NO определяют по увеличению веса сосуда NO 969
470
Новый справочник химика и технолога
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
poj- Магнезиальная смесь Анал. р-р (для маек. Fe3+ добавл. лимоннокислый аммоний), pear., NH4OH (25% NH3). Выдерж. несколько часов. Ф. б. (промыв, хол. 1,5% р-ром NH4OH). Прокал. при 900-1000 °C Mg2P2O7 1, 25, 111
(NH4)2MoO4 а) Анал. р-р (> 0,05 мг Р), NH4NO3 (5-10% р-р), HNO3 (5-10 объемн. %). Нагр. до 40-50 °C, добавл. pear. (20-кратный изб.). Через 30 мин ф. с. (промыв. 5 % р-ром NH4NO3). Прокал. при 500-550 °C. Высуш. при 105 °C. Высуш. при 180 °C. б) Ос., полученный как в п «а», раств. в NH4OH (10% NH3), добавл. НС1 и опр. по методу с магнезиальной смесью (см. выше). в) Ос. по п. «а» раств. и осажд. МоО42 в виде РЬМоО4 (см. Мо) НРО312МоО3 (NH4)3P0412Mo03 «= 0,01637) (NH4)3PO4-I2MoO3 (f= 0,01654) Mg2P2O7 PbMoO4 1,25, 111 113 25 112
(HN4)2MoO4, 8-оксихинолин Р-р pear.: 42 мл НС1 (конц.), 42 мл (NH4)2MoO4 (10%) 16 мл р-ра оксихинолина (5%) Анал. р-р (<10 мг Р2О5, слабокислый) разб. до 100 мл, нагр. до 70 °C, при перемеш. по каплям 30 мл pear. Отстаив. 12 ч, ф.с., промыв. 10-12 мл NH4NO3 (1%). Высуш. 4 час при 105 °C H7[P(Mo2O7)]-•3(C9H7ON)-2H2O /= 0,013277 (наР) f= 0,0306 (на P2O5) 1
Опр. также в виде: Ag3PO4 [114], ZnNH4PO4 [115], (UO2)P2O7 [116], MnNH4PO4-H2O [117], BiPO4 [118], [Co(NH3)6]PO4-4H2O или Co(NH3)6HP2O7 и Na[Co(NH3)6]6-(P3Oi0)2 [119], Ba3(PO4)2 [120], (ZrO)2P2O7 [121]
HPO32~ H3PO2 2 о Опр. в виде Hg2Cl2, Mg2P2O7 Опр. в виде Mg2P2O7 Опр. в виде Ag4P2O6 970
As(III) H2S (газ) Анал. р-р (НС1, 30%) нагр. до 15-20 °C, пропускают H2S. Ф. с. (промыв. 24% НС1, нас. H2S, сп., CS2 сп.). Высуш. при 105-110 °C As2S3 1,7
AsO3 H2S (газ) Анал. р-р (НС1, 10 н.) охл. до 0 °C, pear, (пропускают 60 мин). Через 1-2 час ф. с. (промыв. 4 N НС1, Н2О, сп.) Высуш. при 105-110 °C As2S5 1,7, 971, 972
KSCN+H2S Анал. р-р (100 мл, 0,01-0,15 г As2O5), 10 мл НС1 (25%), 10 KSCN (10%), 1 час пропускают H2S, плотно закрывают, отстаив. 12 час, ф. с., промыв. 50 мл р-ра (разб. сероводородная вода с несколькими каплями СН3СООН), высуш. 2-3 час при 100 °C (As2S3+2S) 1, 122
Магнезиальная смесь Pear.: 50 г MgCl2 6H2O, 100 г NH4C1 раств. в 500 мл воды, NH4OH до щелочной реакции, фильтр., разб. до 1 л и подкисл. НС1. Анал. р-р (100 мл, < 0,1 г As, слабокисл.), 25 мл pear, (в присутствии Sn, Sb и Ge добавл. 3 г лимонной к-ты и 50 мл pear.), по каплям NH4OH до щелочной реакции и еще по 5 мл на каждые 100 мл р-ра. Отстаив. 12 час, ф. б., промыв. 2 раза по 20 мл NH4OH (5:95). Ос. р-ют в НС1 (1:2), добавл. 10 мл pear., разб. до 25-100 мл и осажд. NH4OH (по каплям), добавл. изб. 5 мл NH4OH, отстаив. 12 час. а) Ос. ф. б., промыв. NH4OH (5:95), осторожно озоляют, затем прокал. при 850 °C. б) Ос. ф. с., промыв. NH4OH (5:95), высуш. в токе сухого (через СаС12) воздуха Mg2As2O7 MgNH4AsO4-6H2O 1,7, 122, 123
Химические методы количественного анализа
471
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
AsO3” AgNO3 (0,5 н.) Анал р-р (< 100 мг AsO3 ) нейтр. до pH = 5-6 (NH4OH, HNO3), разб. до 50-80 мл, нагр. до кип., по каплям pear, до просветления р-ра, охл. в хол. воде. а) Ф. ф., промыв. 50 мл хол. воды, 10-12 мл сп., высуш. 1,5 час при 170-180 °C и прокалив, при 500 °C 40 мин б) В присутствии СГ, ВГ: ос. как указано выше, ф. б., промыв. NH4OH (5 : 95), ос. Ag3AsO4 раств. в 1 н. HNO3 и осажд. Ag+ в виде AgCl (см. определение Ag) Ag3AsO4 AgCl 1, 123
Опр. также в виде: AS2O5 [124], Ca2As2O7 [125], Ag2TlAsO4 [973], BaSO4 (после осажд. As2S5), Pb2As2O7 [974], BiAsO4 [975]
Sb3+ H2S (газ) а) Анал. р-р (НС1,9%, гор.), pear, (до черн.), Н2О (1 объем), pear. Ф. с. (промыв. Н2О, сп.) Высуш. при 170-290 °C (в атм. СО2). б) Осажд. Sb2S3 (см. выше) как в методе «а». Высуш. при 100 °C, раств. в (NH4)2S. Ф. б. Ф-т упар., добавл. осторожно конц. HNO3, затем нагр.; разб. Н2О, добавл. NH4OH. Упар. до удаления SO3. Прокал. при 800-850 °C Sb2S3 Sb2O4 1,7 7
Пирогаллол Анал. р-р, винная к-та, pear. Ф. с. (промыв. Н2О). Высуш. при 100-105 °C SbO(C6H5O3) 126
Опр. также в виде: Sb (электролиз [976, 977], химическое восстановление [978]); Sb2Ss [979]
ВГ (NH4)2CO3 Анал. р-р (HNO3, > 5 мг Bi), нагр. до 100 °C, добавл. NH4OH, pear. Ф. б. (промыв, гор. Н2О). Прокал. при 1000 °C Bi2O3 7
НС1(1:9) Анал. р-р (< 5 мг Bi), NH4OH (до помутнения), p-в, Н2О (гор.), нагр. 2 час при 100 °C. Ф. б. (промыв, гор. Н2О, сп.). Высуш. при 100 °C. BiOCl 1,7
(NH4)2HPO4 (10% р-р bHNO3)(1:9) Анал. р-р (100 мл, < 250 мг Bi, HNO3), NH4OH (1:1) до помутнения, 5 мл HNO3, нагр. до кип., 30 мл pear, по каплям при перемеш., разб. до 300 мл водой (100 °C), отстаив. 30 мин при 80 °C, ф. б., осторожно обугливают и сжигают при возможно низкой 1°, прокал. при 800 °C BiPO4 7,11, 127, 128
K3[Cr(SCN)6] Анал. р-р (хол., 0,3-1 н. по HNO3 или H2SO4), pear., ф. с., промыв, хол. водой, высуш. при 120-130 °C. Не мешают: Мо, Fe, Al, Cr, Zn, Mn, Ni, Co, Mg Bi[Cr(SCN)6] 7, 130
H2S Анал. p-p (< 0,2 г Bi, без NO3), Ni 14OH (10%) до помутнения, HC1 (10%) до просветления р-ра, добавл. 5 мл НС1 (10%) разб. до 50 мл, добавл. 1 г NH4C1, нагр. до кип., пропускают H2S до охлажд. р-ра, плотно закрывают пробкой и отстаив. 12 час (Осадок защищают от действия света). Ф. с., промыв, гор. водой, дважды сп. по 50 мл и 10 мл сильно просасывают воздух. К ос. добавл. 5 мл CS2, затем промыв. 2 раза по 5 мл сп. и сушат в токе воздуха, прошедшего через р-р СаС12 (нас.) Bi2S3 1, 131
Формальдегид (10%) Анал. р-р (слабокисл., 0,05-0,50 г Bi), 10 мл pear., 10 мл NaOH (10%), разб. до 100 мл, кип. 1 час. Добавл. 10 мл pear., 10 мл NaOH (10%) и в случае больших количеств Bi отстаив. 1 час и вновь добавл. pear, и NaOH. Нагр. 2 час, кип. р-р ф. с., промыв. 150 мл хол. воды декантацией 3-4 раза до исчезновения окраски фенолфталеина, 20-30 мл сп., высуш. 1 час при 105 °C Bi 1, 132
KI (крист.) Анал. р-р (20 мл, 3% HNO3, без СГ, ВГ), pear, до желт, р-ра над черн. ос., 200 мл гор. воды, нагр. при 100 °C до перехода цвета осадка в меднокр. Если р-р желт., добавл. 10 мл гор. воды. Доб. мет. оранж. и по каплям р-р формиата натрия (2,5%) до перехода окраски. Гор. р-р ф. с., промыв. гор. водой до удаления Г, высуш. 1-2 час при 110 °C BiOI 1, 133
472
Новый справочник химика и технолога
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Bi3+ KBrO3+KBr Анал. р-р нагр. до 100 °C, добавл. pear., переос. Ф. с. Высуш. при 110-120 °C BiOBr (/=0,6894) 11
Электролиз Анал. р-р, Fe — анод, коллоидная диафрагма, Pt — катод, 40-50 мин (внутренний электролиз) Bi 11
Опр. также в виде Bi(JO3)3 [134], Bi2(SeO3)3 [135], [2Bi2O3N2O5-H2O] [136] и др. (см. [137])
VO3", vo3+ Купферон (6%) Анал. р-р (0,1-0,4 г V2O5, 200 мл, 10% по H2SO4), охл. до 10 °C, хол. pear, по каплям при постоянном перемеш. Образуется объемный быстро осажд. ос.; появление бел. мути свидетельствует об изб. pear. Ф. б., промыв. H2SO4 (10%, 1,5 гл 1 купферона), осторожно при слабом нагр. высуш. в платиновом тигле, обугливают и озоляют, затем прокал. при 700-800 °C (Гм = 675 °C), обрабатывают несколькими каплями HNO3 (после охл.) и вновь прокал. Для гравиметрического определения ванадия метод наиболее удобен v2o5 1,11, 145, 146
Hg2(NO3)2 Анал. р-р (50-250 мг V, щелочной), HNO3 (2 н.) до кисл. реакции, разб. до прибл. 1 мг-мл 1 V, добавл. pear, при перемеш. до полн. осажд. и изб. 1-2 мл, добавл. 5 мл NH4OH (2 н.) и кип. 3 мин, охл., ф. б., промыв, водой с добавл. ре-аг. В фильтрате проверяют V (с Н2О2), высуш. при 105 °C в платиновом тигле, озоляют и прокал. при 700-800 °C (Осторожно! Выделяются пары ртути!). Pear.: 3-4 г Hg2(NO3)2 (крист.) смачивают 2 каплями HNO3 (конц.), добавл. 10 мл воды, раств. при нагр. После охл. добавл. ок. 5 мл воды v2os 1
AgNO3 а) Анал. р-р (< 0,1 г V, 200 мл), нагр. до кип., нейтр. NaOH или HNO3 до св. желт, р-ра, добавл. NII4OH до обесцвечивания р-ра, 3 г CH3COONI I4 и 0,5 мл N114ОП (конц.), при перемеш. р-р pear., нагр. до кип. и выдерж. при 100 °C 30 мин, проверяют полноту осажд., ф. с., промыв, гор. водой, высуш. при 110 °C. б) Анал. р-р (< 0,1 г V, нейтр. или слабощел.) разб. до 100 мл, СН3СООН (10%) до перехода окраски мет. оранж., pear, в изб., нагр. до кип., 30 мин при 100 °C, ф. с., промыв, гор. водой до удаления Ag+, высуш. 3 час при 150 °C AgVO4 AgVO3 139, 140 1,139
NH4C1, нас. p-p Анал. р-р (конц.), p-в. Ф. б. Прокал. при < 658 °C v2o5 1
Опр. также в виде V2O5 (после осажд. РЬ2+) [141], Ba(VO3)2 [141], (UO2)2V2O7 [142], Mn2V2O7 [143], [Co(NH3)6].(V6O17)3] [144]
Nb(V), Ta(V) Таннин a) (10% p-p) 6) (2%) а) Оксиды Nb или Та (0,25 г), KHSO4 (10 г) сплавл., раств. при нагр. в смеси Н2С2О4 (нас. р-р) + НС1 (конц.) + Н2О (3:1:10). Ф-т, NH4C1, тартрат-ион, Na-ЭДТА, нейтр. нас. NH4C1 до pH = 5-^6, добавл. pear. Кип. 2 мин. Через 30 мин ф. б. (промыв. NH4C1, Na-ЭДТА). Прокал. при 900 °C. б) Ос. пентоксидов Nb и Та спл. с K2S2O7, р-ют в 50-100 мл (NH4)2C2O4 (нас.), разб. до 200 мл водой, нагр. до кип., по каплям 3-5 кратный изб. pear, и далее до появления оранж. ос. (исчезает при перемеш.), 20 мл NH4C1 (нас.), кип. 5-10 мин. Желт. ос. ф. б., промыв. NH4C1 (2%). Ф-т упар., осторожно добавл. pear, и NH4OH (1 н.) до выделения оранж.-кр. ос. оксидов (Nb+Ta). Этот ос. ф. б., прокал. и выделяют дополнительную фракцию Та2О5; танталовые фракции объединяют, прокал. до Та2О5 Объединенные фильтраты, NH4OH до слабощел. р-ции, ос. ф. б., прокал. до Nb2O5. Ta2O5, Nb2O$ Ta2O3 Nb2O5 1,7,11, 25, 147 И, 151, 152 11 11
Химические методы количественного анализа
473
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Nb(V), Ta(V) в) Осажд. в присутствии С2О^ и Na-ЭДТА: в ос. Та, Nb, Ti, а также Sb и Sn. Ионы Pb, Bi, Al, Fe(II, III), Zn, Mn, U(VI), Th, Ca, Mg, TR — не мешают 158
NaH2P2O2(l)H таннин (10% р-р) (2) а) Оксиды Nb или Ta, K2S2O2, сплавл., раств. в (NH4)2C2O4, добавл. NH4CI, pear. 1. Нагр. до 100 °C, добавл. НС1 (25% р-р). Фильтр, с бум. массой. Ос. промыв, гор. Н2О, раств. в Н2С2О4 (6% р-р), добавл. pear. 2, NH4OH, NH4C1, нагр. несколько минут. Ф. б. (промыв. NH4CI), первое. Прокал. при 900 °C. б) Ф-т от Та2О5, добавл. pear. 2, NH4OH, нагр. несколько минут. Ф. б. (промыв. NH4CI). Прокал. при 900 °C Та2О5 11, 150
НС1, H2SO4 1-2 г стали раств. в 25-50 мл НС1 (1:1), HNO3 до прекращения вспенивания, выпар, досуха, 30 мин при 130 °C, охл., 10 мл НС1 (1:1), 100 мл воды, после раств. солей ф. б. Ф-т нейтр. NH4OH, добавл. 10 мл H2SO4 (1:3), 3 г Na2SO3, кип. 30 мин, ф. б. (то же), промыв. H2SO4 (1:200), прокал. в платиновом тигле, к ос. добавл. 3 мл H2SO4, 2-3 мл HF, выпар, до паров H2SO4, добавл. 5 мл H2SO4 и упаривают до 2 мл, охл., переводят в стакан с 50 мл хол. воды, добавл. 250 мл гор. НС1 (2%), 1 г Na2SO4 и кип. 5 мин, отстаив. 25 мин при 60-80 °C, ф. б., промыв, теплым р-ром НС1 (2%). Прокал. при 900-1000 °C Nb2O5+Ta2O5 25,981
НС1О4 1-2 г стали раств. в 30-50 мл НС1 (1:1), HNO3 до прекращения вспенивания, упар. до 10 мл, добавл. 25 мл НС1О4, нагр. до паров НС1О4 и еще 10 мин, охл., добавл. 150 мл гор. воды, слегка нагр., ф. б. с бум. массой, промыв, гор. НС1 (5:95) до удаления Fe3+ и 3-4 раза гор. водой, прокал. при 1050—1100 °C (тигель Pt, добавл. 0,3-0,5 мл H2SO4 (конц.) и 0,3 мл HF и осторожно выпар, до полного удаления H2SO4, прокал. при 1100 °C Nb2O5+Ta2O5 25
NH4OH Эффективное отделение ZrO2. 0,25-0,5 г смеси оксидов (Nb, Та, Zr) в тигле (Pt) спл. с К2СО3 (6-кратный изб.) при 1200 °C 5-10 мин, охл., добавл. 1 г КОН и раств. гор. водой, добавл. бум. массу, ф. б., промыв. К2СО3 (2%), озоляют и повторно спл. с К2СО3. Фильтрат подкисл. НС1 и осажд. пентоксиды Nb и Та NH4OH из кип. р-ра, ф. б., промыв. NH4NO3 (2%), прокал. при 1100 °C Nb2O5+Ta2O5 1
SeOCl2 0,2-0,3 г прокал. смеси пентоксидов тантала и ниобия, 50 мл смеси (1:1) оксихлорида селена и H2SO4 кип. 30 мин, охл., сливают декантацией ф. Г. Ос. в колбе кип. 20 мин с 20 мл смеси, сливают декантацией через тот же ф. Г. (повторяют 3—4 раза), водой переносят ос. на ф. Г. и промыв, водой. Ос. прокал. до Та2О5. Nb2O5 находят по разности Та2О5 7, 153
Фениларсоновая к-та (3%) а) Анал. р-р (1 н. по НС1), нагр., изб. pear. (25 мл на каждые 10 мг оксидов Nb и Та), нагр. 1 час, добавл. бум. массу, отстаив. 12 час, ф. б., промыв, гор. р-ром NH4NO3 (4%, подкисл. HNO3), прокал. при 1000 °C. б) Анал. р-р (0,01-0,09 г Nb2O5 в 0,1 г смеси пентоксидов), H2SO4, Н2О2, pear., нагр. < 40 °C. Ос. прокалив, до Та2О5 Погрешность ± 5% отн. Nb2O5+Ta2O5 И, 148, 149, 155
474
Новый справочник химика и технолога
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Nb(V), Ta(V) Купферон (6%) Анал. р-р (50-60 мг Nb и Та на 100 мл, слабокисл.), H2SO4 (1 : 1) до 10% (по объему), добавл. бум. массу, медленно pear. (10-15 мл на 100 мг оксидов) до полного осажд., отстаив. 5 мин., ф. б., промыв. НС1 (1 : 9) с добавл. pear. Прокал. до пентоксидов Nb и Та Nb2O5;Ta2O5 11, 152, 154
Опр. также в виде Та2О5, Nb2O5 (после осажд. гидролизом в присутствии уротропина и пирогаллола [982], таннина в щавелевокислой среде [983])
VI группа Периодический системы
so;- ВаС12 а) См. опр. Ва2+. Прокал. при 780 °C. Мешают: большой изб. pear., щ. м. и аммоний, нитраты, Cr ', Fe34, Zr(TV), поэтому необходимо длительное (12 час) созревание и/или переосажд. (см. п. «б»). б) Ос. BaSO4 по п. «а» после промыв, раств. на фильтре в гор. р-ре ЭДТА-Na (3-5%, аммиачный), промыв, фильтр гор. водой, 1-2 капли р-ра pear., нейтр. НС1 по мет. кр. Далее как при опр. Ва2+ BaSO4 BaSO4 161
РЬ(СН3СОО)2 См. опр. РЬ2+ PbSO4
S, S2-SO32’, S2O3", s общая Окислители, ВаС12 Окисление до SO4- в ходе разл. навески: а) окислит, сплавление с (Na2CO3+KNO3). В тигле (Pt), 5-6 частей Na2CO3 и 0,1-1 часть KNO3, сплавляют 20 мин, выщелачивают водой, отделяют кремний (см. опр. SiO2) и осажд. BaSO4 (см. опр. Ва2+). Возможно сплавление только с Na2CO3, или Na2CO3+KNO3 (10:1), или Na2CO3+Na2O2. б) Спекание с Na2CO3+KMnO4 (1:1). 0,5-1 г руды, 3^4 г смеси, тигель фарф., медл. нагр. до 700-800 °C и выдерж. 40 мин, охл., выщелачивают гор. водой. в) Спекание с Na2CO3+MgO (или ZnO) (1:4) 0,5-1 г руды, 3-5 г смеси, медленно нагр. до 700-800 °C, выдерж. 1,5 час, далее по п. «а». Окислительное кислотное разложение: г) Навеска 0,3-2 г, 0,5-1,0 г КС1О3, малыми порциями HNO3 (избегать разогрева), отстаив. 12 час, нагр. и выпарив. досуха. Добавл. 2-3 мл НС1 (конц.), 100 мл воды для раств. солей, далее по п. «а». д) HC1+HNO3 (3:1) 10-15 мл на 1 г навески. Далее по п. «г». е) Навеска стали 5 г, 50 мл HNO3 (1:1), по каплям НО (конц.) до раств. навески, 0,5 г Na2CO3, выпар, досуха и выдерж. 15 мин, 5-7 мл НС1 (конц.); 30-40 мл воды, 5 г стружки А1 или Zn, нагр. до почти полного раств. стружки, фильтр. В фильтр, осажд. BaSO4. ж) Микрохимическое опр. S общей. Навеска 3-10 мг в сухом стакане, 1-2 мл р-ра (2 части Вг2 и 3 части ССЦ) выдерж. 15-20 мин (без нагр.), добавл. 2 мл HNO3 (конц.), выдерж. 15 мин., нагр. на водяной бане до конца р-ции и удалении большей части Вг2, выпар, досуха, далее как при опр. SO’ BaSO4 BaSO4 BaSO4 BaSO4 BaSO4 BaSO4 BaSO4 BaSO4 7,11 25 159 160 11 11 11 11 11 7, 11 25 113
S (элемент.) CS2 (экстр.) Навеску породы 0,5-1,0 г в бумажном патроне помещают в аппарат Сокслета, экстр. S в течение 5-6 час (при рассеянном свете) CS2, отгоняют CS2 до остатка ок. 5 мл, который испаряют в вакуум-эксикаторе. В присутствии битуминозных веществ S в сухом остатке окисл. до SO2 («царская водка» с бромной водой) и опр. в виде сульфата бария S BaSO4 11,984 11,985, 986
Химические методы количественного анализа
475
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
S2" НС1, Н2О2 в NH4OH а) Раств. в к-тах сульфиды: навеску руды 0,5-1 г кип. с НС1 (1:1) в токе СО2, H2S поглощают р-ром Н2О2 в NH4OH (2 части 3% Н2О2 и 1 часть NH4OH), сульфат опр. как при опр. Ва2+. б) Нераств. в к-тах сульфиды: оставшийся после отгонки H2S р-р фильтр., ос. промыв, гор. водой (подкисл. НС1), в фильтрате опр. раств. сульфаты («окисленная сера»). На ос. с фильтром действ, гор. HNO3 (Г.1), бум. массу отфильтр., промыв, водой. Фильтрат и промывные воды нейтр. Na2CO3 (частично), выпар, досуха, 1 мл НС1 (конц.) и повторяют выпар. Ос. смачивают НС1, раств. в воде и опр. SO^ , соответствующий сере нерастворимых сульфидов BaSO4 BaSO4 11,986, 987, 988 11
НС1, Cd(CH3COO)2, CuSO4 Навеску стали (5-10 г) раств. в 70-80 мл НС1 (конц.), H2S отгоняют и поглощают последовательно 60 мл Н2О (сосуд 1) и 60 мл р-ра ацетата кадмия (25 г соли, 250 мл Н2О и 250 мл СН3СООН, разб. до 1 л) — сосуд 2. По окончании растворения H2S из сосуда 1 отгоняют и поглощают в сосуде 2, к р-ру в сосуде 2 добавл. 5 мл CuSO4 (60 г CuSO4, 400 мл воды и 60 мл конц. H2SO4), ос. CuS отстаив. и ф. б., промыв, водой (кип. для удаления О2) до удаления Си2+, прокал. до СиО CuO 25, 162
Se(IV, VI) SO2 Анал. р-р (< 0,25 г Se, конц. НС1), добавл. 50 мл р-ра SO2 (нас. р-р в конц. НС1) на холоду, отстаив. до полного осажд. кр. ос. Se, ф. с., промыв, хол. НС1 (конц.), водой до удаления СГ, сп. и эф. Высуш. 3-4 час при 3Q-40 °C, затем 2 час при 120 °C. Те не осажд. Au отделяют переосажд. после растворения Se в HNO3 Se 7, 163, 164
NH2OHH2SO4 (25%) а) Навеску (< 0,5 г оксидов Se и Те, < 0,2 г Те) раств. в 40 мл хол. НС1 (конц.), разб. до 100 мл, 10 мл pear., нагр. 4 час при 90 °C, ф. с., промыв, водой, сп., высуш. при 110 °C. Те не осажд. б) Смесь оксидов раств. в минимальном объеме NaOH, добавл. р-р винной к-ты (25%) до раств. ос. и еще изб. 25 мл, разб. до 100 мл, нагр. до 90 °C, 10 мл pear, и далее по п. «а». Те не осажд. в) Смесь оксидов раств. в минимальном объеме NaOH, разб. до 100 мл р-ром лимонной к-ты (5%), добавл. 10 мл pear., нагр. до 90 °C, далее по п. «а». Те не осажд. Se Se Se 11 7, 163 7, 163
N2H4-H2SO4 (или N2H4-2HC1) Анал. р-р (слабо солянокисл.) нейтр. NH4OH, разб. до 200 мл, добавл. 100-150 мл НС1 (конц.) и 3 г тв. pear, на каждый грамм Se. Р-р с ос. кип., добавл. небольшие порции pear, до перехода кр. Se в черн. (ок. 30 мин), ф. с., промыв. 4-5 раз водой, затем сп., высуш. 1 час при 105 °C Se 1,165
KI (10%) Анал. р-р (НС14 : 100) нагр. до кип., 8 мл pear., нагр. 2 час, отстаив. 12 час, ф. с., промыв. НС1 (1:20) до удаления Г, высуш. при 105-110 °C. Те не осажд. Se 11
Cu2Cl2 (0,1 н. в 2 и. HC1) Анал. р-р (10-30 мл), НС1 до 3-4 н., pear, до полного выделения Se (кр. ос.). Отстаив. 15-30 мин, ф.с., промыв. 2-3 раза конц. НС1, водой до удаления СГ, сп. и эфиром, высуш. при 105 °C Se 164, 166
476
Новый справочник химика и технолога
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Г равиметрическая форма осадка Литература
Se(IV, VI) Z-Аскорбиновая к-та Анал. р-р нейтр. до pH = 1, нагр. до 80-90 °C, добавл. ре-аг., отстаив. до полного выделения Se, ф. с., промыв, водой, высуш. при 105-110 °C Те (< 1 мг) не осажд. Se 167, 168
SeO2- so2 Анал. р-р нейтр. до 0,5-1,0 н. НС1, нагр. до кип., pear., выдерж. в закрытом сосуде, далее как при опр. Se(IV, VI) Se 169, 170
SeO2 so2 Осажд. из НС1 (4 н.), остальное как при осажд. SeO2- Se 169, 170
В качестве восстановителей используют также: для Se(IV) — гидразин (катализ. IC1) [171], Fe(II) [172], гипофосфит Na [173]. Для Se(IV, VI) — SnCl2 [174], Cr(II) [175, 176], V(II) [177] Опр. также в виде сульфидов [178, 179]; электрогравиметрически (Cu-Pt электроды) в виде Cu2Se [180] и Se [192]
Te(IV, vi) SO2 (нас., р-р 1), N2H4-2HC1 (15%, р-р 2) Анал. р-р (ок. 0,2 г. Те, или фильтрат от опр. Se, 50 мл) доводят до 3 н. по НС1, нагр. дол кип., 15 мл р-ра 1, затем 10 мл р-ра 2, добавл. 25 мл р-ра 1, кип. до уплотнения ос. (ок. 10 мин), ф. с., немедленно промыв, гор. водой до удаления СГ, сп., высуш. при 105 °C Те 7,П, 25, 181, 182
Н2О (гидролиз) Анал. р-р (азотнокисл., 50 мл), добавл. Na-ЭДТА (в количестве, необходимом для полного связывания примесей), нейтр. NH4OH до pH = 4. Р-р с бел. ос. нагр. до 100 °C и выдерж. 5-10 мин (переход ос. в крист.), охл., ф. с., промыв. 3 раза хол. водой, затем сп. и эф. Высуш. при 120 °C ТеО2 183- -186
n2h4h2so4 Анал. р-р (без NO,) нейтр. NH4OH, разб. до 100 мл, добавл. 25-60 мл НС1 (конц.), pear. (3 г на 1 г Те), кип. (периодически добавл. pear.), выдерж. 1 час при 95 °C, ф. с., промыв, водой (подкисл. НС1), водой, сп. Высуш. не более 1 час при 105 °C Те 1, 187, 188
Гипофосфит натрия (10%) Анал. р-р (< 0,2 г Те) доводят до 20% по НС1, добавл. 100 мл pear, нагр. на водяной бане при перемеш., выдерж. несколько часов (до уплотнения ос.), ф. с., промыв. 50 мл НС1 (1 н.), 25 мл воды, 10 мл сп., высуш. при 105 °C Те 1,26, 173, 189
Си2С12 Ф-т после отделения Se упар., разб. водой до 2 н. НС1, ре-аг. Опр. заканчивают как описано для Se Те 164, 166
(C6H5)4AsC1 или (С6Н5)4РС1 Анал. р-р (НС1, 4-5 н. и H2SO4, 3 н.), pear. Ф. с. (не мешает Se). Высуш. при 110 °C [(C6H5)4As]2TeCl6 (f= 0,1158) или [(С6Н5)4Р]2-ТеС16 193 194
В качестве восст. используют также: Fe(II) [172], SnCl2 [174], Cr(II) [175, 176], V(II) [177]. Опр. также в виде сульфида [178, 179], [Cr(NH3)6]2(H6TeO6)3 [167], электрогравиметрически [190-192]
Сг3+ NH4OH Анал. р-р (0,2 г Сг, 200 мл), 2 г NH4C1, нагр. до кип., медленно при перемеш. NH4OH (1 : 3) до чисто желт, окраски мет. кр., кип. до исчезновения запаха NH3, ф. б., промыв, декантацией р-ром NH4NO3 (2%, нейтр. про мет. кр.) до удаления СГ. При высокой концентрации солей ос. переосажд. Прокал. при 1000 °C 15 мин. (Есть рекомендация прокал. при 1200 °C в атм. Н2) Сг2О3 1, 195
KOCN Анал. р-р (0,15 г Сг, 200 мл) нейтр. NH4OH до помутнения, 1-2 капли НС1 (2 н.) до просветления, 1 г NH4C1, 1 г KOCN (в виде фильтр, р-ров), медленно нагр. до кип. Р-р с з.-голубым ос. отстаив. 3 час, ф. б., промыв. NH4NO3 (2,5%, нейтр. NH4OH). Ф-т нагр. до кип., подкисл. НС1 и нейтр. NH4OH до появления голубой окраски тимолфталеина. Небольшой ос. ф. б., оба фильтра промыв, до удаления СГ, прокал. в тигле (Pt) при 900-1000 °C 1 час Сг2О3 1,196
Химические методы количественного анализа
477
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Г равиметрическая форма осадка Литература
СгО*-, Сг2О2- ВаС12 (5%) Анал. р-р (0,2 г хромата, 100 мл), 1 мл СН3СООН (0,1 н.), 1 г NaCl, нагр. до кип. и по каплям 10 мл pear., кип. 2-3 мин, отстаив. 12 час. Ф. с., промыв. 50 мл хол. воды, высуш. 2-3 час при 130 °C. (Иногда указывают 60 °C). Хлориды К, Na, Nil], Са занижают, нитраты, ацетаты — завышают результаты ВаСгО4 1, 198, 199
Hg2(NO3)2 (5%) а) Анал. р-р (< 0,3 г хромата) разб. до 100 мл, добавл. 30-50 мл pear., кип. 10 мин, охл., ф. б., промыв. 8-10 раз 10 мл р-ра pear. (2%), высуш. на воздухе, прокал. при 800-1000 °C (Осторожно! Пары ртути!) 30 мин. Иногда указывают прокал. при 1200 °C в атм. Н2. Не меш. только Щ. М. и NO,. б) Р-р по п. «а» после добавл. pear. нагр. до перехода кор. цвета ос. в ярко кр., охл., ф. с., промыв. 50 мл хол. воды, высуш. 1 ч при 105 °C. (см. опр. Hg2+) Сг2О3 Hg2CrO4 1, 197 1
AgNO3 (5%) Нейтр. анал. р-р (< 0,2 г хромата) разб. до 100 мл, нагр. до кип. и по каплям 10 мл pear., отстаив. 12 час. Ф. с., промыв. 50 мл р-ра Ag2CrO4 (нас.). Высуш. 2-3 час при 130 °C. (Внимание! Р-ры и ос. держать в темноте!) Ag2CrO4 1, 199
Pb(CH3COO)2 (7,3%) Анал. р-р нейтр. NaOH (2 н.) или СН3СООН по фенолфталеину до слабороз. окраски, добавл. 2-5 мл СН3СООН (2 н.), разб. до 100 мл, нагр. до кип. и по каплям добавл. pear, до изб., кип. 1 мин, отстаив. 1 час, ф. с., промыв, хол. водой, высуш. при 100-120 °C 2 час. Опр. быстро выполнимо, дает хорошо воспроизводимые результаты. Меш.: SO; PbCrO4 1,200
Опр. также в виде СгРО4 [201], Сг2О3 (осажд. NH4NO2 [202], KI+KIO3 [203])
МоОд” (CH3COO)2Pb (4% в 1% СНзСООН) Анал. р-р нейтр. NH4OH (2 н.) или СН3СООН (2 н.) по лакмусу, добавл. 1-2 мл СН3СООН (1:1), 25 мл CH3COONH4 (50%), разб. до конц. Мо 0,5 мг мл1. Р-р нагр. до кип., по каплям при перемеш. pear, в изб. (до просветления р-ра и осажд. ос.), кип. 2-3 мин, дают ос. осесть, проверяют полноту осажд. (2-3 капли pear.), ф. б., промыв, декантацией гор. NH4NO3 (2%), в присутствии Щ. М. и сульфатов переосажд. (после р-ния ос. НС1 на фильтре), снова ф. б., высуш. при 100 °C, сжигают при возможно низкой /°, прокал. при 600 °C PbMoO4 7,11, 25, 207
Pb(NO3)2 (4%) Анал. р-р (г МоО3, 200 мл), HNO3 до нейтр. или слабокисл, р-ции, нагр. до кип. и к кип. р-ру добавл. по каплям при перемеш. pear, до просветления р-ра, кип. 2-3 мин, дают ос. осесть и проверяют полноту осажд. (Избегать большого изб. pear.). После осажд. добавл. NH4OH (2 н.) по каплям до нейтр. или слабощел. р-ции по лакмусу, затем несколько капель СН3СООН, отстаив. ок. 5 мин, р-р декантируют и промыв, ос. декантацией 3—4 раза по 75 мл гор. NH4NO3 (2%), ос. на фильтре промыв., высуш. при 100 °C, озоляют при низкой f и прокал. при 600 °C. Метод дает точные результаты. Ос. чистый, крист., легко фильтр, и промыв. PbMoO4 204
а-Бензоин-оксим а) Анал. р-р (5-10%, H2SO4), pear. Охл. до 5-10 °C. Ф. б. (промыв. 0,1% р-ром pear.). Прокал. при 500-525 °C. б) Ос. по п. «а» раств. в NH4OH с Н2О2, р-р кип. до разрушения Н2О2, осажд. РЬМоО4 (см. выше). MoO3 PbMoO4 7, 25, 205, 208 209
478
Новый справочник химика и технолога
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
МоОд’ в) Анал. р-р (8-20 мг МоО3,25 мл, 5% H2SO4), добавл. изб. соли Мора, по каплям при перемеш. 10 мл р-ра pear. (0,05 М в смеси ацетон: вода = 1:1), через 10 мин ф. с., промыв, декантацией 5 мл H2SO4 (1%), переносят ос. в тигель водой, промыв. 2 раза по 5 мл смеси (1:1) ацетона и воды. Высуш. при 105 °C MoO2(Ci4H12O2N)2 227
H2S (газ) Анал. р-р (5-200 мг МоО3, ок. 300 мл), H2SO4 до конц. 1 н., нагр. до 80-85 °C, пропускают H2S около 15 мин, отстаив. ок. 1 час в закрытой колбе, ф. б., промыв. H2SO4 (1%, нас. H2S), 15-20 мл сп. Прокал. при 500-600 °C до получения желт, кристал. ос. (ок. 1 час) МоО3 1,H
Na2S (2 н.) Анал. р-р (25 мл), 20 мл pear. (15 мл р-ра pear, на каждные 50 мг Мо), добавл. изб. НС1 (6 н. или 2 н.) до обесцвечивания р-ра и выделения ос. MoS3, нагр. до кип., дают ос. осесть, ф. с., промыв, водой, сп., эф., просасывают воздух 15 мин, высуш. в вакуум-экси-каторе MoS3 210
AgNO3 (5%) Анал. р-р (5-200 мг Мо) разб. до 150 мл, мет. оранж., нейтр. H2SO4 (2 н.) до кр. окраски, 10 мл CH3COONa (10%), нагр. до кип., pear., вновь нагр. до кип., отстаив. 12 час в темноте, ф. с., промыв, pear. (0,5%) 7 раз по 20 мл, 3 раза по 5 мл сп., высуш. 1 час при 110 °C и до постоянного веса при 200 °C. Метод применяют при анализе аммиачных экстрактов МоО4 , а также в анализе сплавов Мо с А1 Ag2MoO4 211
SrCl2, 10% Анал. р-р нейтр. до pH = 8, разб. до 40 мл, добавл. 0,5 мл СН3СООН (1 н.), 10 мл CH3COONa (2%), при кип. pear., ф. с., промыв, сп. (25%), далее как с ВаС12 SrMoO4 212, 213
8-Оксихинолин (4% в 50% сп.) а) Анал. р-р (25-100 мг Мо) разб. до 100-200 мл, NaOH по каплям до pH = 7,3 (по феноловому кр.), нагр. до кип., ре-аг. в изб. 50-100%, кип. 5-10 мин, быстро ф. с. (смывают гор. ф-том), промыв. 5-10 мл гор. воды 3-4 раза, высуш. при 120-140 °C. б) Анал. р-р после отделения кремневой и вольфрамовой к-т, а затем Mn, Сг, Ti, Fe, Nb (NaOH) нейтр. HC1 (по лакмусу), 20-25 мл CH3COONH4 (5%), 15 мл ацетата ортооксихинолина (3%), нагр. до 80 °C, через 30 мин охл., ф. б., промыв. 5-6 раз водой, осторожно озоляют и прокал. при < 400 °C. (Рекомендовано: с Н2С2О4 или с газ. NH3). в) К нейтр. анал. р-ру добавл. 10-кратный изб. Na ЭДТА (считая на присутствующие металлы), 3-5 мл ацетатного буфера (3 части 50% CH3COONH4 и 4 части 50% СН3СООН), разб. до 80 мл, нагр. до кип., pear. (3%, в разб. буфере) до слабожелт, окраски р-ра, нагр. 2-3 мин, ф. с., промыв, гор. водой, высуш. при 130-140 °C. Меш.: WO?4 , VO3. г) Полуколич. определение 0,06-0,5 мкг Мо в 50 мкл р-ра по объему ос. MoO2(C9H<sON)2 MoO2(C9H6ON)2 MoO3 MoO2(C9H6ON)2 213, 214 25 214 215, 216, 217 218
Опр. также в виде: MoS2 [219, 220], МоО2 [222], Мо [221, 223], Hg2MoO4 [221], СаМоО4 [221, 224], CdMoO4 [225], Agl (после окисл. Г до 12 и взаимодействия последнего с Ag металл.) [226], [Cr(NH3)5Cl]MoS4 [989, 990]
Химические методы количественного анализа
479
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Г равиметрическая форма осадка Литература
wo; HNO3 (конц.) а) Анал. р-р подкисл. pear. улар, до 5 мл, добавл. 15-20 мл pear, и повторно упар. до 1,5-2 мл, добавл. 2-3 г NH4NO3, 50 мл гор. воды, нагр. до кип., отстаив. 6-12 час, ф. б., промыв, декантацией р-ром (5% HNO3 с 0,5% NH4NO3), ос. р-ют в гор. р-ре (10% NH4OH с 2% NH4NO3), фильтр, через тот же ф. б. во взвешенную платиновую чашку, осторожно выпар, досуха, нагр. до удаления NH *, охл., смачивают 1-2 мл HF и 5-10 каплями H2SO4, нагр. до удаления к-т. Сухой ос. прокал. при 800 °C. б) Р-р по п. «а» после повторного выпар., добавл. 3-5 мл pear, и 30 мл НС1 (1:1), нагр., добавл. 5 мл р-ра желатины (1%), разб. гор. водой до 150 мл, перемеш. и отстаив. при нагр. до просветеления р-ра. Ос. ф. б. промыв. HNO3 (3%) до удаления СГ, прокалив, при < 800 °C в тигле (Pt), ос. обрабатывают HF и H2SO4 и прокал. при 800 °C. в) Навеску стали (2 г) р-ют в 250 мл НС1 (1:4) при нагр. (избегая кипения и выпар, р-ра); по растворении добавл. HNO3 (конц.) по каплям до прекращения вспенивания и еще 2 мл, отстаив. при слабом нагр. 30-40 мин. В случае зел. ос. добавл. 1-2 мл HNO3 и операции повторяют. Охл. р-р, ф. б. с бум. массой, промыв. 8-10 раз гор. НС1 (5:100) и 3-4 раза NH4NO3 (2%). Ос. прокал. при 750-800 °C. г) Осажд. из гомогенного р-ра: навеску (1-2 г) стали р-ют в 30 мл НС1 (конц.) при нагр., добавл. по каплям 20 мл HNO3 (конц.) до полного удаления НС1, добавл. 50 мл воды, 2 мл Н2О2 (30%) и 50 мл HNO3 (конц.), нагр. 1,5 час при 60 °C, гор. р-р ф. б., промыв. HNO3 (1%) и прокал. 1 час при 800 °C. д) В ос. WO3 и МоОз или в р-ре вольфраматов (до 250 мг) и молибдатов (до 250 мг) осажд. W2O5: анал. р-р разб. до 60-300 мл, нагр. до кип., 20 мл SnCl2 (20% в конц. НС1) на каждые 150 мг WO3, кип. 5 мин., ф. б. промыв, декантацией гор. НС1 (1 : 9) до удаления Мо, син. ос. W2O5 переносят на фильтр, высуш. и прокал. до WO3 WO3 WO3 WO3 WO3 WO3 11,25 11,228, 229, 230 25, 234 231, 232, 233 7, 256
Hg2(NO3)2 (нас. р-р в 0,075 М HNO3) а) Анал. р-р (слабокисл., 25 мл), по каплям HNO3 (3 М) до роз. окраски мет. оранж., нагр. до кип., по каплям 10 мл pear., отстаив., ф. б., промыв, гор. р-ром pear. (1 : 5), прокал. при 850 °C (Осторожно! Пары ртути!). б) Р-р с ос. по п. «а», NH4OH (6 н.) по каплям до перехода желт, в сер. цвет ос., нагр. до кип. (если ос. желт., снова добавл. NH4OH), перемеш. 5 мин, ф. б., промыв, гор. разб. pear. (1: 20), прокал. при 850 °C (Осторожно! Пары ртути!) WO3 WO3 245 1, 246, 247
Цинхонин (5% Р-Р) Анал. р-р (щел.), HNO3, pear., нагр. Ф. б. (промыв, разб. р-ром HNO3). Ос. раств. в NH4OH, первое. Прокал. при 800 °C WO3 235, 236, 241
Р-Нафтохино-лин (2%) Анал. р-р, 5 мл H2SO4 (конц.) дважды упар. до паров H2SO4, охл., добавл. 5 мл воды, нейтр. NaOH (20%) по бум. конго и изб. до р-ния вольфрамовой к-ты, кип., фильтр, и промыв, ос. гидроксидов, ф-т. нейтр. по конго до фиол. окраски, добавл. 0,5 мл Н2О2 (30%), НС1 (конц.) по 7 мл на каждые 100 мл р-ра, 5 мл pear., нагр. до 80 °C, отстаив. 1,5-2 час. Ф. б., прокал. при 750-800 °C WO3 229, 231, 236, 237, 245, 314
480
Новый справочник химика и технолога
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
wo$- Таннин (1) и цинхонин (5% р-р) (2) или Р-нафтохи-нолин (3% р-р) (2) Анал. р-р (щел.), NH4C1, нагр. до 50 °C, добавл. свежеприготовленный pear. 1, НС1, pear. 2. Фильтр, с бум. массой. Ф. б. (промыв. NH4C1). Прокал. при 800 °C WO3 11,231, 239
8-Оксихинолин (4% в сп.) а) Анал. р-р (нейтр. или щел.), изб. pear., нагр. до кип., добавл. СН3СООН до слабокисл, (если исходный р-р кисл., нейтр. 1 н. CH3COONH1). Натр., ф. с., промыв, гор. водой до обесцвечивания промыв, вод. Высуш. при 120 °C. б) Анал. р-р, изб. Na-ЭДТА, буферный р-р, для создания pH = 4,5-5,0, нагр., pear., отстаив. при 50-60 °C, далее по п. «а». Не меш. опр. 5-50 мг W, по 50 мг Al, Си, Со, Сг, Fe, Mn, Ni, Pb, Zn и 10-1200 мг V; Ti и Zr предварительно экстр, из тартратных р-ров WO2(C9H6ON)2 WO2(C9H6ON)2 240, 242, 258 243, 244
А-Бензоил-фенилгидрок-силамин (3% в сп.) Анал. р-р разб. до 200 мл, добавл. 10 мл тартрата Na (10%), создают pH > 1 (в присутствии Ti, V, Мо) или pH = 4 (в присутствии Fe)10 мл pear., нагр. до 80 °C, фильтр, промыв. pear. (1%). Ф-т упар. до 200 мл, добавл. НС1 до конц. 0,1-3 М, 10 мл pear., ф. с., промыв, гор. (60 °C) НС1 (0,06 М), высуш. при 115 °C WO2(C13H10O2N)2 248
Три-н-бутиламмоний-хлорид (2%), NaH2PO4 Анал. р-р (10-200 мг W, 50 мл) добавл. 85 мг Р в форме NaH2PO4, НС1 (6 М) до pH = 2 и еще изб. 4 мл, разб. до 100 мл. Р-р нагр. до кип., вводят при перемеш. в течение 15 мин 10 мл pear., охл., отстаив. 1 час, ф. с., промыв, ре-аг., затем водой, высушив, при 105-150 °C. Опр. 200 мг W не меш.: по 700 мг Са, Сг(Ш), Со, Си, Мп, Ni и 2 г Na [(C4H9)3NH]3- •[PW12O40] 249, 250
Опр. также в виде: Ag2WO4 ем метавольфрамата (pH = 1905], BaWO4 [252, 253], CaWO4 [224, 252], CdWO4 [252], Pb WO4 [230], а также осаждени-2-3,1) и паравольфрамата (pH = 5,1-6,8) пурпуркобальтихлоридом [CoCl(NH3)5]Cl2 [255]
иОг+ NII4OH или таннин а)Анал. р-р (кисл.) нагр. до 100 °C, добавл. pear, (по мет. кр.). Фильтр, с бум. массой. Ф. б. (промыв, гор. 2% р-ром NH4NO3). Прокал. при 750-900 °C. б) Анал. р-р (сернокисл., <1% UO2SO4, без СО2), мет. кр., нагр. до кип., pear, до оранж.-кр. окраски, кип. 10 мин, pear, до желт, окраски, ф. б., далее по п. «а» U3O8 U3O8 7, 260- 264 259
Пиридин (20%) Анал. р-р (содерж. 5-10 г NH4NO3), добавл. pear, до нейтр. р-ции по мет. оранж. и изб. 10 мл, нагр., при необходимости добавл. pear. Ф. б., промыв. NH4NO3 (3% с добавл. pear.), далее по п. «а» U3O8 264
Уротропин (10%) Анал. р-р, NH4OH до начала осажд., НС1 (1 : 3) до р-ния ос., NH4C1 (10 г на 300 мл р-ра), кип. и по каплям pear, до полного осажд. и изб. 2-3 мл, сразу ф. б. с бум. массой, далее по п. «а» с NH4OH. Не меш.: Zn, Со, Ni, Mn, Mg u308 262
Н2О2 Анал. р-р (азотно- или уксуснокисл.), NH4OH до pH = 2-2,5, охл. до 0 °C и при перемеш. Н2О2 (30%, двухкратный изб.), замораживают р-р при -45 °C и выдерж. 1 час, отгаив., ф. б., промыв, р-ром (3% NH4NO3 и 3% Н2О2, pH = 2-2,5), прокал. до U3O8 u308 276
NH4H2PO4 или (NH4)2HPO4 (10%) Анал. р-р нагр., нейтр. NH4OH до помутнения, ос. р-ют HNO3, изб. pear., нагр. до кип., по каплям NH4OH (1 : 1) кип. 30 мин., отстаив., ф. б., промыв. NH4NO3 (5%), прокал. до (UO2)2P2O7 (UO2)2P2O7 276
Химические методы количественного анализа
481
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
ио^ H2S Анал. р-р нейтр. NH4OH до pH = 6, нагр. до 60 °C, 2 г гексаметилентетрамина, нас. H2S (15 мин), нагр. и повторно нас. H2S (15-20 мин), отстаив. 15 мин, ф. б., промыв. NH4NO3 (3% с добавл. NH4OH). Прокал. при 850-950 °C и3о8 7,265, 266
8-Оксихинолин (3% в 4 М СН3СООН) а) Анал. р-р (< 0,3 г U, 200 мл) нейтр. NH4OH, добавл. СН3СООН до содержания 1-2%, 5 г CH3COONH4, нагр. до кип., по каплям pear, до изб. 4-5 мл, NH4OH (1 н., объем равен объему добавл. pear.), кип. 1-2 мин, охл., ф. с., промыв. 50-150 мл pear. (0,03%), высуш. при 120-130 °C Возможно высуш. при 250-300 °C или прокалив, до U3O8. б) Анал. р-р, ацетата, буф. (pH = 5-9), Na-ЭДТА. Нагр. до 100 °C, добавл. pear. Ф. с. (промыв, гор. Н2О). Высуш. при 105-110 °C UO2(C9H6ON)2-•c9h6on UO2(C9H6ON)2 UO2(C9H6ON)2-c9h6on 1, 267, 268 267
Купферон (6%) Анал. р-р (20% H2SO4), КМпО4 до слабороз. окрашивания, хол. pear., фильтр, и промыв, pear. (0,2% в 10% H2SO4). В ф-те восст. U(VI) до U(IV) используя редуктор (Cd или Zn) и повторяют осажд. pear., ос. ф. б., промыв, как ранее, прокал. при 800-950 °C. Первое осажд. отд. Fe, Ti, V, Zr, второе — отд. U(FV) от Al, Cr, Mn, PO3" u308 271, 272
Опр. также в виде: UOF2 [274], U(C2O4)2 [277], UO2 [219], V2O5-2UO3 [279]. U(IV) осажд. в виде: UP2O7 и UP2O6 [275, 276], йодата [280], фторида [273,274]
VII группа Периодической системы
F" Pb(NO3)2 Анал. р-р (нейтр. или щел.), бромфеноловый син., NaCl (10% р-р), Н2О (до 250 мл), нейтр., добавл. pear., CH3COONa. Harp. 30 мин, оставляют на 10-15 час. Ф. с. (промыв, нас. PbCIF, Н2О). Высуш. при 130-140 °C PbCIF 7,11, 281-283
СаС12 (5% р-р) Анал. р-р (pH > 3, без NH4 , SiO^", РО3~), pear. (15 мл на 1 г К), желатина. К ос. добавл. СН3СООН, выпар., добавл. Н2О (15-20 мл). Ф. б. (промыв. 1-2% р-ром NH4OH). Прокал. при 800 °C CaF2 7,11, 284--286
Bi(NO3)3 Анал. р-р (0,2-0,25 г F, 125 мл) нейтр., добавл. 5 капель СН3СООН до pH = 6,5; pear., перемеш. и отстаив. 1-2 час, ф. с., промыв, декантацией 10 мл СН3СООН (2,5%), водой, высуш. на воздухе 1 час и 2 час при 100 °C BiF3 287, 288
(C6H5)3SnCl (р-рвСНС13) Анал. р-р нейтр. до pH = 4-8, pear., перемеш., отстаив., ф. с., промыв, водным, а затем хлороформным р-ром pear., высуш. при 110 °C. Не меш.: РО3 , ВО3 , Al3+, Fe3+ (C6H5)3SnF 289- -291
Опр. также в виде: K2SiF6 [292, 293], BaSiF6 [219,294[, BaF2 [219], SiF4 (по потере веса), UOF2-2H2O->U3O8 [295], LaF3 La2O3 [296]
СГ AgNO3 (5% р-р) а) Анал. р-р (HNO3, разб.), pear., нагр. до 100 °C. Ф. с. (промыв. 0,5% р-ром HNO3). Высуш. при 130-150 °C. б) Восстановление AgCl (см. опр. Ag4) AgCl Ag 7, 11, 297, 298
Hg2(NO3)2 Анал. р-р (HNO3, разб.), pear., нагр. при 70 °C до просветления р-ра, отстаив. в хол. темном месте 10-12 час., ф. с., промыв, хол. водой, 10-15 мл сп., высуш. при просасыва-нии воздуха 30-45 мин или при нагр. < 130 °C Hg2Cl2 299
CIO" Pb(NO3)2 Анал. р-р (слабоазотнокисл.), pear., ф. с., промыв, подкисл, водой, высуш. при < 77 °C Pb(C102)2 300, 301
482
Новый справочник химика и технолога
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
СЮ; Восстановитель; AgNO3 а) Анал. р-р (0,2 г хлората, 100 мл), 50 мл FeSO4 (10%), перемеш. и кип. 15 мин, охл. и осажд. СГ р-ром AgNO3 (см. выше). б) Анал. р-р (слабокисл, или нейтр.), добавл. изб. нас. р-ра SO2 и кип. до удаления SO2, подкисл. HNO3 и осажд. Cl-р-ром AgNO3 (см. выше). в) Анал. р-р, СН3СООН, металлический Zn, кип. 1 час, изб. Zn раств. в HNO3 и осажд. СГ р-ром AgNO3 (см. выше) AgCl AgCl AgCl la la la
Нитрон Анал. р-р (слабосернокисл.), охл. до 0 °C, pear., ф. с. Высуш. при 105 °C. Меш.: no;, no;, С1О4- c20h16n4-hcio3 302
сю; КС1 или СНзСООК Анал. р-р (< 0,4 перхлората, 25 мл) нагр. до 80-90 °C, изб. pear, отстаив. до охл. 1 час, ф. с., промыв, дважды р-ром pear. (0,05 М) и четыре раза безводным этилацетатом, высуш. при 110 °C ксю4 303, 304
Восстановители, AgNO3 Анал. р-р (2 н. по H2SO4, 100 мл) добавл. 1-2 г порошкообразного Cd и 1 г сульфата титана, кип. 1 час с обратным холодильником, разб. до 200 мл, добавл. немного HNO3 для окисл. Ti(III) и определяют СГ как описано выше AgCl la
Нитронацетат (10%) Анал. р-р нейтр. NH4OH, разб. до конц. перхлората 1 мг мл1, подкисл. 1 мл H2SO4 (2 н.), нагр. до кип. и добавл. 10-15 мл pear., отстаив. 2 час в лед. воде, ф. с., промыв. 20-50 мл нас. р-ра ацетата нитрона, 3 раза по 3 мл лед. воды, высуш. 1-2 час при 40-100 °C (Осторожно! Превышение температуры опасно! Осадок легко взрывается!) c20h16n4-hcio4 305, 306
вг AgNO3 (5%) Анал. р-р (0,01-0,7 г бромида, 100 мл), 5 мл HNO3 (1 н.), при перемеш. по каплям pear, в небольшом изб., отстаив. в темноте 1 час, нагр. до коагуляции ос. и оставляют на ночь. Ф. с., промыв. 50 мл хол. воды с 2-3 каплями HNO3, затем 50 мл СН3СООН (1%), высуш. 2 час при 130 °C AgBr 307, 309
Вг2, ВгО, вю; Восстановитель, AgNO3 Определяемые ионы восст. до ВГ (см. определение СЮ;) AgBr 308
сг, вг AgNO3, Cl2 Из анал. р-ра осажд. AgCl и AgBr (А) и определяют как описано выше. Ос. прокал. в токе С12 при 340-450 °C 15 мин, охл. в эксикаторе с КОН и взвешивают в форме AgCl (В). По весу AgCl + AgBr и AgCl рассчитывают содержание AgCl и AgBr: AgBr = 4,2240(4 - В), г; AgCl = А - AgBr, г AgCl la
AgNO3, Н2 Осажд. и определяют ф. Г. суммарный вес (A) AgCl + AgBr. Ос. в тигле нагр. до красного каления в токе Н2 и определяют вес ос. Ag (В). Рассчитывают содержание СГ и ВГ по формулам: СГ= 1,3883 -5 - 0,7975 -А, г; ВГ= 1,7975 -А -2,38835, г Ag la
Cd В аликвотной части анал. р-ра, определяют вес (А) = = AgCl+AgBr. В другой равной аликвоте осажд. AgBr + AgCl, ф. б., ос. смывают в фарфоровую чашку, фильтр, озоляют и присоединяют к ос., добавл. H2SO4 (2 н.) и 1-2 г порошка Cd, нагр. на водяной бане до полного восст. Ag, фильтр., промыв, гор. водой, раств. ос. гор. HNO3 (2 н.) и осажд. NaCl (0,1 н.) в виде AgCl (5). Далее см. определение с С12: AgBr = 4,2240(4 -В), г; AgCl = А - AgBr, г AgCl la
Химические методы количественного анализа
483
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
СГ, Вг“ AgNO3, NH4Br Осажд. и определяют AgCl + AgBr (А) как описано выше, используя ф. Г. Ос. смешивают с 4-кратным количеством NH4Br и прокал. при 500 °C, охл. и взвешивают ос. AgBr (В) Рассчитывают: AgCl = 3,224 • (В - А), г; AgBr = A- AgCl, г AgBr 1
Г AgNO3, 5% р-р Анал. р-р, NH4OH, pear., HNO3 (до 1%). Ф. с. (промыв. 1% р-ром HNO3). Высуш. при 120-150 °C Agl 7,318, 319
PdCb Анал. р-р (НС1, 1%), pear. Выдерж. 1-2 суток при 20-30 °C Ф. с. (промыв. Н2О, сп.), высуш. при 90-95 °C. Или ф. б., прокал. при 1000 °C (в атм. Н2) Pdl2 Pd 7, 307, 320
Г (Вг“, СГ) Косвенное определение каждого иона Отбирают две равные аликвотные части анал. р-ра, в каждой осажд. галогениды р-ром AgNO3 (см. выше). Ос. одной аликвоты ф. с., промыв, и высуш. как описано выше — вес (В). Ос. второй аликвоты фильтр, через асбест, промыв. СН3СООН (1%), к асбесту с ос. в колбе добавл. 2 г К2Сг2О7 и 30 мл H2SO4 (конц.), нагр. при 95 °C 2 час, продувают воздухом до удаления С12 и Вг2, охл., разб. водой до 300-400 мл, отфильтр. асбест и по каплям добавл. нас. р-р NaHSO3 до запаха SO2. Ос. Agl (Q фильтр, и определяют как описано ранее. В ф-те осажд. Agl изб. KI, и взвешивают — вес (£>). Навеска, соответствующая аликвоте, — вес (А). Содержание (% мае.) рассчитывают: СГ= [2,57865 • D - 3,2340(2? - Q] • 24,7384/4; ВГ = [4,22398 • (В - С) - 2,5786/)] • 42,556/4; Г= (54,05 • Q/A 7,310, 311
Г опр. также в виде: Cui [314, 315], ТП [316], РЫ2 [317], Ag, ТН3
Ю", ю;, Восстановители, AgNO3 Восст. Zn или SO2 как описано выше для ВгО3 и осажд. Agl (см. выше) Agl
Ю’ AgNO3 Анал. р-р (нейтр.), pear., ф. с., промыв, водой, высуш. при 80-400 °C AgIO3 312, 313
Мп2+ (NH4)2HPO4 Анал. р-р (слабокисл.), pear., нагр. до 90-95 °C, добавл. NH4OH. Ф. б. (промыв, хол. 10% р-ром NH4NO3). Прокал. при 1000 °C. Ф. с. (промыв. NH4OH, сп. эф.). Высуш. при 20 °C Mn2P2O7 MnNH4PO4H2O 7,H, 325 321
H2S, NH4OH Анал. р-р (0,05-0,30 г Мп, < 0,01 н. П), 5 г NH4C1, разб. до 200 мл, нагр. до 50-60 °C, 15 мин нас. H2S, добавл. 20-30 мл NH4OH (конц.), 50—100 мг HgCl2 на каждые 200 мг Мп, ф. б., промыв. NH4C1 (2,5%). Далее: а) Прокал. при 1000-1100 °C. б) Ос. нагр. до кр. каления, охл., добавл. 2-3 капли НС1 (конц.), 1-2 капли Н2О2 и 1 мл H2SO4 (1:1), осторожно нагр. до удаления H2SO4, прокал. 15 мин при 450-550 °C. в) Ос. прокал. до МпО в тигле Розе, охл., добавл. слой чистой S, осторожно нагр. в токе Н2 и прокал. при 900 °C (в токе Н2) Mn3O4 MnSO4 MnS 266, 322, 323 1,H 324 7
KC1O3 Анал. р-р (азотнокисл.) выпар, до сиропообразного сост., добавл. 50 мл HNO3 (конц.), кип. для удаления оксидов азота, при > 95 °C вводят в течение 10-20 мин 5 г КС1О3 малыми порциями, кип. до прекращения вспенивания, добавл. 40 мл хол. воды, быстро охл., ф. б., промыв, хол. HNO3 и лед. водой. Ос. прокал. при 800-900 °C Mn3O4 11, 160
484
Новый справочник химика и технолога
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Мп2+ Пикролоновая к-та (0,01 н.) Анал. р-р (10 мл, ок. 0,15 г MnSO4), нейтр., добавл. 15 мл pear., отстаив. 1,5-2 час, ф. с., промыв, водой до удаления SO4 , высуш. при 105 °C. Быстрый метод; не меш.: Fe(III), Al, Cr, Ti(IV) (C10H7O5N4)2Mn 327
Бриллиантовый желтый Анал. р-р (10 мл), 7,5 мл СН3СООН (1:50), изб. (1,5 раза) pear., перемеш., отстаив. 1 час. Ф. с., промыв. 1-2 раза водой (подкисл. СНзСООН, 5-10 мл), высуш. при 120 °C. Ос. не гигроскопичен. Не меш.: Fe(III), Al, Cr, Zr, Th, Се(Ш) (C26H18O8N4S2)Mn 328
о-Фенантролин (2%), NH4SCN Анал. р-р (pH = 3-6) разб. до 25-100 мл, нагр. до 95 °C, добавл. 0,3-1,0 г NH4SCN, перемеш., изб. pear., нагр. 2-3 мин, отстаив. 5-10 мин, ф. с., промыв, гор. р-ром (3 г NH4SCN, 10 мл pear. (2%), разб. до 1 л), водой, смесью сп. с эф. (1:3) и эф. Высуш. в эксикаторе. Меш.: Cd [Mn{C12H8N2)2]- •(SCN)2 329
Опр. также в виде: МпС2О4 [330, 331], Мп3О4 (после осажд. из р-ров NH4OH + Br2, (NTLO^Og или Н2О2) [11]
ReO4 Нитронацетат (5% в 3% СН3СООН) Анал. р-р (нейтр., < 0,1 г Re в виде перрената) разб. до 50 мл, 1 мл H2SO4 (2 н.), нагр. до 80 °C, pear, до изб. 0,3-0,4%, охл. до 20 °C, отстаив. в лед. воде 2 час, ф. с., промыв. 10-20 мл лед. pear. (0,3% водным р-ром), 2-3 раза лед. водой, высуш. при 110 °C 2-3 час. Ошибка опр. <0,1 мг Re C20Hi6N4HReO4 7, 332, 333, 334
Тетрафенилар-сонийхлорид (1%) Анал. р-р (5-25 мл, 0,5-100 мг Re, без меш. ионов), NaCl до конц. 0,5 н., pear. (изб.). Объем р-ра не должен превышать 60 мл. Перемеш., отстаив. ночь, ф. с., промыв, лед. водой, высуш. при 110 °C. Возможно определение ультрамикроколичества (C6H5)4AsReO4 7, 335
СН3СООТ1 Навеску 0,3-0,4 г р-ют в 40 мл воды, добавл. 10 мл лед. СН3СООН, pear, (р-р Т12СО3 в СН3СООН), упар. до 4 мл, охл., добавл. 10 мл СН3СООН, ф. с., промыв. СН3СООН, высушив, при 140 °C TlReO4 337
Na2S или (NH4)2S (1-2 н.) Анал. р-р, (NH4)2SO4 (20-25 г), pear, (изб.), НС1 (конц.) до конц. 6 н., нагр. до кип., охл., ф. с., промыв, водой, СП. до удаления СГ, эф. и высуш. в вакуум-эксикаторе 30 мин. Re2S7 338
^(«-хлорбензол)-! -нафтил-метиламин (2%) (1) или ди(1- наф-тилметил)амин (2,5%) (2) Анал. р-р (5-10 мл, 1-10 мг ReO4), нагр. до 80 °C, добавл. гор. р-р pear, на 10 мл р-ра 4,5 мл (1) или 4,0 мл (2), охл. 2 ч на льду, ф. с., промыв. 3 раза по 2 мл лед. водой, высуш. при 110 °C 1: Ci8H17ClN ReO4, 2: C22H20N ReO4 339
Диантипирил-пропилметан (5% в СНзСООН) (1:1) Анал. р-р (15-60 мг Re, ок. 0,1 н. H2SO4), нагр. до 70-80 °C, медленно добавл. 6-7 мл pear., отстаив. без нагр. 1-2 час, ф. с., промыв. 4-5 раз нас. р-ром о.ф., высуш. при 100-110 °C (C26H30N4O2H) • •ReO4 340
Внутренний электролиз а) Анал. р-р (10 мл, 5% H2SO4), анод (стержень Sn, 0 7,5 мм), замыкают электроды медной проволокой, осажд. Re на платиновой сетке, при 75-80 °C, 25-100 мин, периодически добавл. воду (до постоянного объема). Перед окончанием электролиза добавл. 5 мл H2SO4 (1 : 19), сетку промыв. H2SO4 (разб.), водой и ацетоном, высуш. на воздухе, затем 20 с при 105 °C. б) Анал. р-р, 5 мл НС1 (конц.), 2-3 г KI, кип., по каплям Na2S2O3 (0,1 н.) до обесцвечивания иода, разб. до 250 мл, ЭДТА-Na (тв., в десятикратном изб.), NH4OH до pH = 9-10; катод платиновая сетка, анод — Zn, электролиз 2 час. Катод промыв, водой, сп., высуш. при 80-90 °C. Mo(VI) маек. SCN-. После выделения Re можно опр. Pb, Cd, Мо Re Re 341 342, 343
Химические методы количественного анализа
485
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
ReCi2’ (или Re Brig ) Диантипирилпро-пилметан (5% в СНзСООН) (1:1) Анал. р-р (2 н. по НС1 или НВг) медленно добавл. 5-6 мл pear., отстаив. 2 час, ф. с., промыв, нас. р-ром о. ф. в 2 н. НС1 (НВт), 3-4 раза водой, высуш. при 100-105 °C (C26H30N4O2H)2 •ReCl6 344
NX' -Тетраметил-о-толуидин (тетрон) Анал. р-р (солянокисл.), нагр. до 40-50 °C, по каплям pear, до изб., охл. льдом. Ф. с., высуш. при 110 °C. Re Од не осажд. и может быть определен нитроном (см. выше) 345
Опр. также в виде Re (осажд. в виде ReO2 или Re2S7 с последующим восстановлением осадка водородом) [346-348], Re2Se7 [352], электролизом [349-351]
VIII группа Периодической системы
Fe2+ H2S Анал. р-р (0,02-0,3 г Fe, слабокислый, 80-100 мл), добавл. винной к-ты (около 10 г), нейтр. NH4OH (конц.), добавл. 4 мл H2SO4 по каплям при перемеш. 25 мл NH4OH (конц.), пропускают H2S 15 мин, нейтр. NH4OH (конц.) до pH = 9, нагр. до кип., ф. б., промыв, водой с H2S, прокал. 30 мин. при 900 °C Fe2O3 1,7, 266
Опр. как Fe3+ после окисл. Н2О2, Вг2, С12 или HNO3
Fe3+ H2S или (NH4)2s а) Осажд. как Fe2+. Ос. р-ют и осажд. Fe3+ NH4OH. б) Анал. р-р нейтр. до слабокисл., нагр., добавл. 7-8 г уротропина, насыщ. H2S (газ), ос. фильтр, и опр. как при анализе Fe2+ или по п. «а» Fe2O3 Fe2O3 7 354
NH4OH Анал. р-р нагр. до кип., добавл. pear. Ф. б. (промыв. 1% р-ром NH4NO3, декантацией). Прокал. при 1000-1100 °C Fe2O3 1,7
Уротропин (10% р-р) Анал. р-р (слабокисл.), NH4C1, pear. Нагр. до 100 °C. Ф. б. (промыв, гор. Н2О). Прокал. при 1000-1100 °C Fe2O3 38, 353
N2H4H2O Анал. р-р , NH4CI, pear. Нагр. до 100 °C. Ф. б. (промыв. 1% р-ром NH4CI, 1% р-ром р-ва, гор. Н2О). Прокал. при 1000-1100 °C Fe2O3 38
Купферон (6% р-р) Анал. р-р (H2SO4), охл. до 10 °C, добавл. pear. Фильтр, (с бум. массой). Ф. б. (промыв. 3,5% р-ром НС1, 0,15% р-ром pear., NH4OH, Н2О). Прокал. при 1000-1100 °C Fe2O3 1,355, 356, 364
HCOONH4 Анал. р-р (солянокисл.), добавл. 1 г NH4C1, выпар, досуха, разрыхляют ос. и высуш. при 100 °C 5-7 мин. до удаления НС1. Ос. р-ют в воде, добавл. pear. (НСООН нейтр. NH4OH по лакмусу) в изб. (2-3 кратн.), разб. до получения 0,2% р-ра HCOONH4. Гор. р-р нейтр. NH4OH (1-2%) по мет. кр., ф. б., промыв. HCOONH4 (0,1-0,2%), высуш. и прокал. при 850-1100 °C. Возможно регулировать pH гидролизом мочевины Fe2O3 1,361 363
Бензоат аммония (10%) Анал. р-р (слабокисл.), 1 г NH4C1, NH4OH (10%) до начала осажд., добавл. 1 мл СН3СООН и 20 мл pear, на каждые 125 мг Fe, кип. 5 мин., ф. б., промыв, pear. (1% в 2% СН3СООН). Прокал. при 900 °C. Наилучший метод гидролитического осажд. Ос. очень хорошо фильтр. Fe2O3 1,7, 362
Опр. также в виде Ре2Оз после осажд. (I +Ю3) [357], NO2 [358], CIFX'OONa [1, 359], а также в виде FePO4 [360]
[Fe(CN)6]4" и [Fe(CN)6]3- Опр. в виде AgCN (см. опр. Ag+)
486
Новый справочник химика и технолога
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Со2+ H2S а) Анал. р-р (0,2 г Со) нейтр., 34 мл CH3COONH4 (0,4 н.) и 2,8 мл СН3СООН (40%), разб. до 90 мл, нагр. до 90 °C, пропускают H2S, через 5 мин прекращают нагрев и продолжают нас. H2S 1 час, вновь нагр. до 60 °C и прекращают ток H2S. Ф. б., промыв, р-ром (35 мл 0,4 н. CH3COONH4, 3 мл СН3СООН (40%), разб. до 100 мл и нас. H2S), осторожно озоляют, нагр. 45 мин. при 600-750 °C под слоем Н2С2О4, затем 30 мин. при 900-950 °C. б) CoS осажд. и фильтр, по п. «а». Ос. смывают с фильтра, остаток ос. на фильтре раств. в нескольких каплях НС1 (конц.) и HNO3 (конц.), Основной ос. раств. в 10 мл H2SO4 (1 н.) и 1 мл Н2О2 (30%) при нагр., р-ры объединяют и выпарив, до удаления паров H2SO4, затем прокал. при 450-550 °C примерно 1 час, охл., добавл. 1-2 капли Н2О и вновь выпар, и прокал. в) Прокал. ос. по п. «а», далее нагр. в токе Н2 при 1000 °C 15 мин. В случае чистого Со ос. пирофорный. В присутствии Ni получают компактный ос., раств. в HNO3, Ni опр. с диметилглиоксимом и находят содержание Со по разности Со3О4 CoSO4 Со 1,7, 266, 365 1,366, 367 368, 369, 370
(КОН+Вг2), (NaOH+K2S2O8) а) Анал. р-р (слабокисл., ок. 100 мл), добавл. NaOH (свежеприготовленный) до слабощел. р-ции по лакмусу, к хол. р-ру добавл. 2 г K2S2O8, затем еще NaOH до щел. р-ции, отстаив. ночь. Декантируют хол. водой, ф. б., промыв, до нейтр. р-ции, прокал. 1 час при 950-1000 °C. б) Ос. прокал. в токе Н2 (см. опр. с H2S, п. «в») Со3О4 Со 371, 372
(NH4)2HPO4 Анал. р-р (кисл., ок. 100 мг Со), добавл. изб. pear, и постепенно NH4OH до pH = 8-8,5; или: анал. р-р (нейтр.), добавл. CH3COONa, NH4C1, затем небольшой изб. pear. Ос. ф. с., промыв, водой с NH4OH, сп., высуш. при 100 °C. Ос. ф. б., промыв, водой с NH4OH, прокал. при 730 °C. В ф-те может оставаться 0,4—3,0 мг Со CoNH4PO4IJ2O Со2Р2О7 373, 374
8-Оксихинолин (2% в сп.) Анал. р-р (5-60 мг Со, нейтр., 100-200 мл); добавл. 3-4 г CH3COONa до 1-3%, нагр. до 60-70 °C, pear, до небольшого изб., нагр. до кип., выдерж. 15 мин при 100 °C, ф. с., промыв, гор. водой, высуш. при 180 °C Co(C9H6ON)2 375, 376
а-Нитрозо-0-нафтол (р-в Ильинского) (2% в 50% СНзСООН) а) Анал. р-р (< 0,1 г Со, соляно- или сернокисл.), разб. до 200 мл, добавл. НС1 (конц.) до конц. 0,3 н., нагр. до 80 °C, pear. (12,5 мл на каждые 0,01 г Со) и кип. при перемеш. до отделения ос. (Маточный р-р прозрачный и желт.). Добавл. 1-2 мл pear., отстаив. 2 часа, ф. с., промыв, гор. (80 °C) НО, затем гор. водой. Высуш. при 100-110 °C 1 час, озоляют под слоем Н2СгО4 и осторожно прокалив., охл., смачивают ос. HNO3 конц., затем H2SO4, выпар, до удаления паров к-ты и прокал. при 450-550 °C. б) После озоления ос. прокал. при 800-900 °C (см. выше). в) При больших количествах Со ос. оксидов прокал. в токе Н2 (см. выше). г) Анал. р-р (0,03 г Со, соляно- или сернокисл.) упар. до 10-20 мл, охл., добавл. 5-10 капель Н2О2 (30%), затем NaOH (2 н.) до полного осажд. гидроксида Со(Ш), ос. раств. при слабом нагр. в 10-20 мл СН3СООН, разб. гор. водой до 200 мл, 15-20 мл pear., кип. при перемеш. до полной коагуляции ос. Ф. с., промыв. 4-5 раз СН3СООН (33%), 4 раза гор. водой, высуш. при 130 °C. CoSO4 Со3О4 Со Co(C10H6O2N)3 25, 370 370 7,25 377
Химические методы количественного анализа
487
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Со2+ д) Осажд. по методу возникающего pear.: Анал. р-р (10 мл, слабокисл., 1-90 мг Со), 10-15 капель СН3СООН, несколько кусочков льда, 1 г NaNO2, отстаив. 10 мин при перемеш. Затем добавл. 100 мл NH4F (25%) и р-р 2-нафтола (0,5 г pear, в 100 мл гор. 25% СН3СООН). На каждый 1 мг Со необходимо 2 мл такого р-ра + 10 мл изб. Р-р перемеш. 1 мин и отстаив. до полного осажд. (прозрачный маточный р-р). Декантируют через ф. с., оставляя весь ос. в стакане. К ос. добавл. 100 мл НС1 (1:20), нагр. до кип. отстаив. и декантируют к-ту через фильтр, повторяют операцию с NH4OH (1:1), затем с СН3СООН. Ос. промыв, теплой НС1 (1:10) до обесцвечивания ф-та, переносят на фильтр, высуш. при 115 °C. Не меш.: Fe, W. При опр. 10-50 мг Со ошибка ок. 1% Co(C10H6O2N)3- •2Н2О 378, 366
Пиридин + nh4scn Анал. р-р (0,1 г Со, нейтр., 75-100 мл), 0,5-1 rNH4SCN, нагр. до кип., при перемеш. 1-5 мл пиридина. При охл. осажд. мелкокрист. роз. ос. Ф. с., промыв.: 2-4 раза водным р-ром пиридина (0,7%) и NH4SCN (0,5%), затем 10% сп. р-ром пиридина (1,5%) и NH4SCN (0,1%) — 50-60 мл, затем 10 раз эф. с несколькими каплями пиридина, высуш. в вакуум-эксикаторе 15-20 мин Co(C5H5N)4-(SCN)2 379, 380
kno2 Анал. р-р, СН3СООН и CH3COONa (8-10% р-р), pear. (50%, нейтр. СН3СООН). Отстаив. при периодическом перемеш. 24 час, ф. б., промыв. СН3СООК (5%) или KNO2 (5%), подкисл. СН3СООН (0,1%). Далее: а) Промыв, водой с 0,1% СН3СООН переосажд. и высуш. при 105-110 °C. б) Ос. промыв, сп. (95%) до удаления изб. pear., прокал. в фарфоровом тигле, раств. в НО (1:1) и опр. Со в виде CoSO4 как указано ранее. в) Фильтр с ос. переносят в сосуд, где проходило осажд, добавл. 20 мл HNO3, затем 5 мл H2SO4, упарив, до ее паров, охл., обмывают стенки водой, повторно выпар, и опр. Со электролизом K3Co(NO2)6-3H2O или K2NaCo(NO2)6 • •ЗН2О CoSO4 Со 7 381, 382 383, 384 7 7,385
Электролиз Анал. р-р (азотно- или сернокисл., без СГ, 0,05-0,5 г Со) добавл. 5-10 г (NH4)2SO4,30-35 мл NH4OH (конц.), 1 г сульфата гидразина, разб. до 150 мл, нагр. до 70-80 °C и ведут электролиз с электродами сетка - спираль, сила тока 0,25-0,5 А, напряжение 2,2-2,4 В, при перемеш. 0,1 г Со полностью осажд. за 1,5 час. Каплю электролита проверяют на полноту осажд. (с ионом S2-). Электроды под напряжением промыв, водой, спиртом, высушив, на воздухе Со 385- 387
Опр. также в виде Ag3[Co(CN)6] [388],Co(N2H4)2(SCN)2[389], CoHg(SCN)4 [390, 391], [Co(NH3)6] [Co(NO2)6] [392, 393], Co(C5H5N)4 Cr2O7 [394], [Co(NH3)3](HgI3)2 [395], CoWO4-2H2O [396], CoSeO3-2H2O [397]
Ni2+ Диметилглиок-сим (1% в СП.) а) Анал. р-р (< 0,1 г Ni, слабокисл., 150 мл) нагр. до 80 °C, 50 мл pear., ос. р-ют НС1, добавл. (без дополнительного нагр.) NH4OH (1:1) до отчетливого запаха аммиака, отстаив. 2 часа, хол. р-р ф. с., промыв, теплой водой, высуш. 1-2 часа при 110-130 °C. В присутствии Fe(III), А1(Ш) и Сг(Ш) на каждый 1 г металлов добавл. перед осажд. 7 г винной или лимонной к-ты Ni(C4H7N2O2)2 7,11, 25, 399, 400
488
Новый справочник химика и технолога
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Ni2+ б) Ос. по п. «а» ф. б., промыв. NH4NO3; помещают фильтр с ос. в другой фильтр, осторожно высуш. и озоляют ос. в платиновом тигле, прокал. при 900 °C. в) Анал. р-р (0,1 г Ni) нейтр. NaOH до выпадения ос., раств. ос. в нескольких каплях НС1 (1:1), в присутствии Fe(III) добавл. 10 мл нас. р-ра SO2, нагр. до кип., отстаив. 15 мин, добавл. 3-4 г сегнетовой соли, разб. до 150 мл, нагр. до 80 °C, добавл. 50 мл pear., подкисл. 1-2 каплями НС1, добавл. CH3COONa (2-3 г) до pH = 5. Отстаив. 12 час, ф. с., промыв, водой с добавл. CH3COONa и сегнетовой соли (5-6 раз), водой (4-5 раз). Высуш. 1-2 час при 110-130 °C. Осаждение из гомогенного р-ра: а) Анал. р-р (< 0,1 г Ni, слабокисл.) добавл. pear. (1% в пропаноле), мочевина. Р-р нагр., осажд. крупнокрист. ос. б) Анал. р-р, 1,2-бутандион, добавл. гидроксиламин и NH4OH, нагр. Осажд. крист, ос. Высуш. при 110-120 °C NiO Ni(C4H7N2O2)2 Ni(C4H7N2O2)2 Ni(C4H7N2O2)2 1, 404, 408 ' 7,401-403 406, 407 405
Ниоксим (0,5%) (диоксим цик-логександиона-1,2) Анал. р-р 0,5 г ZnSO4 (1,5-3 мг Ni в ZnSO4, 10 мл), 2 г NH4C1, NH4OH (25%) до прозрачного р-ра. Добавл. 5 мл реаг.? ф. с., промыв, водой, высуш. при 110-120 °C Не меш.: Sb, Al, Be, Cd, UO2+, Щ.М., Щ. 3. M. Ni(C6H9N2O2)2 409, 410, 411
Салицилальд-оксим Анал. p-p (< 0,1 мг Ni, нейтр.), pear., pH = 7-8. Ф. с., промыв. хол. водой, высуш. при 105-120 °C Ni(C14HuO2N)2 /= 0,2031 412, 413
а-Бензил-диоксим (р-р в ацетоне) Анал. р-р (150-200 мл, < 20-30 мг Ni), добавл. 30 мл винной к-ты (10%), нагр. до кип., 20 мл ацетона, NH4OH до щел. р-ции (по лакмусу), pear, до полного осажд., ф. с., промыв, гор. водой, высуш. при 120-130 °C. Метод имеет преимущества при опр. малых количеств Ni. Возможно опр. 0,002 мг Ni в 5 мл анал. р-ра с использованием 0,2% р-ра pear, в ацетоне Ni(C14HnN2O2)2 415, 416 414
а-Фурил-диоксим (0,2%) Анал. р-р (0,5 мг Ni, 25-50 мл), нагр., добавл. pear., NH4OH до щел. р-ции (фенолфталеин). После коагуляции ос. (через 10-15 мин), ф. с., промыв, нас. р-ром а-фурил-диоксимата Ni и 1-2 раза водой. Высуш. при 120-130 °C Ni(C10H7N2O4)2 417, 418
Пиридин и NH4SCN Анал. р-р (0,1 г Ni, нейтр., 100 мл), 0,5-1 г крист. NH4SCN, нагр. до кип., добавл. 2 мл пиридина, прекращ. нагр., перемеш. до выпадения небесно-голуб. ос. (10-15 сек), ф. с. Промыв.: а) 4-5 раз р-ром NH4SCN (0,1%) и пиридина (1,5%) в сп. (37%); б) 2-3 раза (ок. 20 мл) р-ром пиридина (1%) в сп.; в) 6-8 раз эфиром с 2 каплями пиридина (ок. 20 мл). Высуш. 15-20 мин в вакуум-эксикаторе. Метод отличается быстротой выполнения Ni(C5H5N)4-(SCN)2 419
NaOH и окислитель (Na2O2, Н2О2, Вг2, S2Og2) Анал. р-р нейтр. NaOH до слабокисл, р-ции, нагр. до кип. и вливают его в 100 мл гор. р-ра NaOH (10%) с добавл. изб. Na2S2O8 (или K2S2O8), кип. 2-3 мин, дают ос. осесть и ф. б. (предварительно фильтр промыв, гор. 5% р-ром NaOH с добавлением Na2SO4), ос. промыв, таким же промыв, р-ром, водой и прокал. при 900 °C. Метод дает завышенные результаты из-за невозможности отмыть весь NaOH (КОН) и может быть использован для отделения Ni от Сг, V, РОд-, U, Zn с дальнейшим опр. в виде диметилглиоксимата или электролизом NiO 7, 420
Химические методы количественного анализа
489
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Ni2+ H2S Анал. р-р (0,2 г Ni, нейтр.), 34 мл CH3COONH4 (0,4 н.), 2,8 мл СН3СООН (6,8 н.), разб. до 20 мл, нагр. до 90 °C (без кип.!), H2S (газ), через 5 мин прекращают нагр. и пропускают H2S в течение 1 час. Нагр. р-р до 60 °C и прекращают пропускать H2S. Ос. ф. б., промыв, буферным р-ром (35 мл 0,4 н. CH3COONH4, 3 мл 6,8 н СН3СООН, разб. до 100 мл и насыщ. H2S), озоляют фильтр при возможно низкой температуре и прокал. до исчезновения запаха SO2, затем 45 мин при температуре красного каления и 30 мин при 1000 °C NiO 1,421
8-Оксихинолин (3% в сп.) Анал. р-р (10-100 мг Ni, нейтр., 100 мл), 1-2 г AcONa, 5 мл СН3СООН, нагр. до 60 °C, добавл. при перемеш. pear., нагр. до кип., отстаив. 15 мин при 100 °C, ф. с., промыв, гор. водой, высуш. при 200 °C Ni(C9H6ON)2 422, 423
Электролиз Анал. р-р (сернокисл.) нейтр. NH4OH, добавл. 20-30 мл NH4OH (конц.), 5-10 г (NH4)2SO4, разб. до 100-150 мл. Электролиз при 70-80 °C и токе 2 А. При этом за 1,5 час осажд. 0,3 г Ni. Электролиз при постоянном нагр. и перемеш. за 1 час осажд. 0,5 г Ni при токе 3-5 А. Полноту осажд. проверяют в отдельной капле электролита р-цией с диметилглиоксимом. Катод промыв, водой, сп., высуш. при обдуве воздухом Ni 7,419, 424
Опр. также в виде: Ni2CO4 [425], NiSO4 после осажд. NiS [см. опр. Со], Ni (после восст. NiO водородом) [421], [Ni(NH3)4]HgI4 [426]
Ru(III, IV) (NH4)2CO3 (2%) Анал. р-р (5-100 мг Ru, солянокисл.), выпар, при 100 °C почти досуха, соли раств. в 100-200 мл воды (100 °C), добавл. бум. массу, pear, до pH = 6, кип. 5 мин, добавл. несколько капель Н2О2 (3%), выдерж. при 100 °C до коагуляции ос. Ф. б., промыв, гор. водой с небольшим количеством сульфата аммония (1%). Фильтр медленно сжигают, затем прокал. ос. при 600 °C на воздухе, затем в токе Н2, охл. в токе СО2 Ru 7, 429, 431
NaHCO3 (5% р-р) Анал. р-р (кисл.), pear, (нейтр. до pH = 5-7 по бромкрезол-пурпур.) Кип. 5 мин. Ф. б. (промыв. 2% р-ром (NH4)2SO4. Прокал. при 800 °C (в атм. Н2) Ru 7, 428, 433, 435
Na2S или (NH4)2s Анал. р-р (слабокисл., pH = 3) добавл. изб. NaNO2 (50%), нагр. 1 час, NaHCO3 (нас.) до щел. р-ции (по лакмусу), кип. до получения желт. р-ра. Ос. ф. б., промыв. NaCl (1%), при необходимости переосажд. К гор. ф-ту добавл. нас. р-р Na2S или (NH4)2S до исчезновения кр. окраски и обр. шоколадно-кор. ос. Р-р кип. 5-7 мин, охл. и нейтр. НС1 до слабокисл, р-ции. Ос. ф. б., промыв, гор. водой, фильтр высуш., медленно озоляют, прокал. и восст. до Ru (см. выше) Ru 427, 434
Тиомочевина Анал. р-р (мкг или мг количества Ru, соляно- или сернокисл.), добавл. H2SO4 (если Ru < 20 мг, достаточно 10 мл к-ты), при отсутствии ионов Щ. М. добавл. 0,1 г Na2SO4. Р-р упар. до паров SO3, охл., разб. до 50 мл, pear. (0,1 г на 5 мг Ru), нагр. до 210-215 °C (не выше) и прекращают нагр. Р-р охл., разб. водой, ф. б., промыв, водой, высуш., прокал. и восст. до Ru (см. выше). Возможно микро- и макроопределение. Ос. не пептизируется, устойчив на воздухе, хорошо фильтр., но имеет переменный состав Ru 439
490
Новый справочник химика и технолога
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Ru(III, IV) Mg Анал. р-р (25-100 мг Ru, слабокисл., 150 мл), добавл. изб. порошка Mg, нагр. до раств. изб. Mg (избегать длительного нагр.). Ф. т., промыв. H2SO4 (0,1 н.), затем водой, высуш. при 100 °C, прокал. в токе Н2 как указано выше. Применен для опр. в медно-никелевых сплавах Ru 428, 436
Zn Анал. р-р (сильнощел.) нагр. с порошком цинка, ф. т., промыв. НС1 (2 н.), высуш. при 100 °C, прокал. при 500 °C и восст. Ru в токе Н2 (см. выше) Ru 428, 436
Тиогликоль-0-аминонафталид (тионалид) Анал. р-р (0,2-0,5 М НС1), pear. Нагр. до 100 °C. Ф. б. (промыв, гор. водой). Прокал. при 800 °C (в атм. Н2) Ru 428, 433, 435
CH2OSO2Na-•2Н2О - Фор-миатосульфо-нат натрия (ронгалит) (250 мг-мли) Анал. р-р (10-70 мг Ru), нейтр. до 0,1-0,2 н. по кисл. (H2SO4, НС1 или HNO3), нагр. до кип., добавл. 4 мл pear., выдерж. при 100 °C при перемеш. 30 мин (до коагуляции ос.), ф. б., промыв, хол. водой, высуш. и прокал. при температуре красного каления. Ос. переносят в стакан, добавл. 25 мл гор. НС1 (1:3), ф. б., промыв, водой, прокал. и восст. в токе Н2 (см. выше). Метод позволяет осаждать Ru, находящийся в форме нитрозосоединений Ru 437
Опр. также в виде: T1Ru2S6 [438], Ru2S3-2H2O [440,441, 442]
Rh (III) H2S (газ) Предварительно сульфатные комплексы Rh переводят в хло-ридные: анал. сернокисл, р-р Rh упар. до 5 мл, добавл. 50 мл NHiCl (20%), выпар, досуха, добавл. 200 мл воды, устанавливают pH = 0,9-1,1. Р-р нагр. до кип., пропускают H2S до полной коагуляции ос., ф. б., промыв. 0,1 н. H2SO4, затем 0,1 М НС1. Ос. прокал. и восстав, аналогично Ru (см. выше) Rh 427, 428
Тиомочевина См. опр. Ru и Ir. 444
NaBrO3+NaBr Анал. р-р (слабокисл., ок. 0,1 г Rh в виде хлоридных комплексов) разб. до 200-400 мл, добавл. 20 мл NaBrO3 (10%), нагр. до 70 °C, по каплям NaBr (10%), нагр. до кип., снова добавл. р-ры бромата и бромида. Прекращение выделения Вг2 указывает на установление равновесия. Добавл. 3-4 капли Na2CO3 (10%), изб. Na2CO3 нейтр. несколькими каплями НС1 (2 М) и кип. р-р. При достаточном изб. Вг- и ВгО3 устанавливается pH = 8. Р-р кип. 1-2 час, отстаив. при 100 °C до коагуляции ос., ф. б., промыв, гор. водой с NH4NO3. Ос. обрабатывают как описано для Ru Rh 443
Ti2(SO4)3 или TiCl3 (20%) Анал. р-р (< 0,1 г Rh, соляно- или азотнокисл.), добавл. H2SO4, упар. до паров SO3, разб. водой и доводят содержание H2SO4 в р-ре до 20-25%. Нагр. до кип., по каплям р-р Ti(III) до коагуляции ос. и появления фиол. окраски р-ра. Р-р кип., разб. водой в 2 раза, ф. б., промыв, гор. H2SO4 (10%) до удаления Ti(IV), затем водой (гор.), высуш., прокал., восст. ос. до Rh (см. опр. Ru) Rh 434
Тиобарбитуро-вая к-та (1,4% в сп.) Анал. р-р (2-25 мг Rh, хлорид, 200 мл), добавл. 3 мл НС1 (конц.), 10 мл pear., нагр. до кип. Через 2-3 мин выпадает кр.-кор. ос. Р-р с ос. кип. 2 час, отстаив., ф. б., промыв, декантацией водой, высуш. и прокал. при 650-700 °C, восст. в Н2 (см. опр. Ru) Rh 445
2-Меркаптобензотиазол (2,0% в лед. СН3СООН) Анал. р-р (5-20 мг Rh), 5 мл NH4C1 (1%), разб. до 50 мл, нагр. до кип., добавл. 3-4 мл pear., выдерж. 2 час при 100 °C, охл., ф. б., промыв. СН3СООН (0,1 М), прокал. на возд., восст. в Н2, охл. в СО2 (см. опр. Ru) Rh 446
Химические методы количественного анализа
491
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Г равиметрическая форма осадка Литература
Rh(III) Гексаммин-кобальт(Ш)-хлорид (нас. р-р) Анал. р-р (0,1-1 г Rh, слабокисл, или нейтр.), добавл. крист. NaNO2 до перехода окраски из кр. в желт., затем по каплям гор. pear, до перехода окраски в оранж. и выпадения крист, ос. Р-р охл., ф. с., промыв, р-ром (5 мл 50% NaNO2, 0,05-0,1 г гексаммина, разб. до 100 мл), затем 45 мл абсолютного сп., 2-4 раза абсолютным эф. Высуш. в вакуум-эксикаторе 10-15 мин. [Co(NH3)6]- •[Rh(NO2)6] 447
Опр. также в виде: Rh (после осажд. магнием [448], Н3РО2 [434], медью [449], сурьмой [450], гидразином [427,434]); после осажд. в виде K3Rh(NO2)6 [1]); TlRh2S4 [438]; Rh2S3-2H2S [428, 440]
Pd(II) KI (1%) а) Анал. р-р (< 25 мг Pd, 150 мл) добавл. 3 мл HNO3 (конц.), изб. pear., осторожно кип. 20 мин, ф. с., промыв, водой, высуш. при 110 °C б) Ос. по п. «а» прокал. и восст. в токе Н2, охл. в СО2 Лучший метод для определения титра стандартных р-ров Pd Pdl2 Pd 428, 991 428
Диметилглиок-сим (водн. р-р) а) Анал. р-р (5-25 мг Pd, 0,25 н., соляно- или азотнокисл.), добавл. изб. pear. (8 мг pear, на 1 мг Pd). Общий объем р-ра 100-200 мл. Р-р перемеш., выдерж. 15 мин при 100 °C, охл. 45 мин (до окончания выделения и коагуляции ос.), ф. с., промыв. 200 мл воды, высуш. при 110 °C. б) Ос. по п. «а» ф. б., промыв, водой, фильтр с ос. помещают в фарфоровый тигель, который помещают в другой тигель и медленно озоляют и прокал. до металла при 500 °C сначала в токе Н2, затем в токе чистого азота в) Осажд. из гомогенного р-ра. См. табл. 5.1.11.11 .В Pd(C4H7O2N2)2 Pd 7, 428 1,7, 428
Диоксимы См. табл. 5.1.11.11.Б и табл. 5.1.11.11.В
Р-Фурфуральд-оксим (10% р-р В СП.) Анал. р-р (> 50 мг Pd в форме комплексного хлорида) разб. до 100 мл, добавл. НС1 до содержания ее ок. 2%, pear, при постоянном перемеш., ф. с., промыв. 50 мл р-ра НС1 (1%), 50 мл воды, высуш. при 110 °C Pd(C5H5O2N)2Cl2 f= 0,2669 427, 451
Монооксимы См. табл. 5.1.11.11.А
Муравьиная к-та или натриевая соль а) Анал. р-р (слабосолянокисл. или нейтр.) добавл. pear., нагр. при 100 °C до прекращения выделения СО2, сразу же добавл. NaCO3 до нейтр. или слабощел. р-ции, нагр. до кип. Ос. ф. б., промыв, гор. водой, влажный фильтр медленно озоляют, прокал., восст. в токе Н2, охл. в токе со2. б) Осажд. Pd по п. «а». Промытый ос. прокал. на сильном пламени горелки Мекера, восст. водородом не требуется. в) Анал. р-р (нейтр. или слабокисл.), добавл. муравьиную к-ту, CH3COONa до щел. р-ции по мет. оранж., нагр. при 100 °C до полной коагуляции ос., ф. б., промыв, гор. водой, прокал. по п. «б» Pd Pd Pd 396, 452 454 455
а-Нитрозо-0-нафтол (нас. р-р в 50% СН3СООН) Анал. р-р (слабокисл.), нагр. до кип., pear. Осажд. объемистый кр.-бур. ос. Pd(Ci0H6NO2)2. Возможно опр. малых количеств Pd (0,001-0,05 мг). а) Ос. ф. б., промыв, гор. водой, прокал. и восст. в токе Н2 как указано ранее. б) Ос. ф. с., промыв, гор. водой, высуш. при 135 °C Pd Pd(C10H6NO2)2 /= 0,2367 456 457 287
492
Новый справочник химика и технолога
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Pd(II) 5-Бром-1,2,3 бензо-триазол (1-2% р-р в СН3СООН) Анал. р-р (0,1-10 мг Pd), НС1 (ок. 10% по объему), pear, до прекращения выделения жел.-бел. ос. и изб. несколько капель, нагр. до 70-80 °C, отстаив. 1 час при 20 °C, ф. с., промыв. гор. водой 6-7 раз, высуш. при 120-130 °C Pd(C6H3BrN3)2Cl2 /= 0,1860 458
Гидролитический метод (NaHCO3, NaBrO3) Анал. р-р (150-200 мл, 15-45 мг хлорида Pd), 10 мл NaBrO3 (10%), при кип. NaHCO3 (10%) до pH ~ 6 (по внешнему индикатору бромкрезоловому пурпурному). При pH = 6 добавл. 5 мл р-ра NaBrO3, кип. 5 мин до коагуляции суспензии, добавл. NaHCO3 до pH = 7,5 по рН-метру или крезоловому кр. (внешний индикатор). Ф. б., промыв. 200 мл NH4C1 (1%), ос. восст. и прокал. как описано выше Pd 428, 470
Опр. также в виде: Pd (после осажд. восстановителями: Hg2Cl2 [459], N2H4 [460, 461], С2Н4 [463]); С2Н2 [462,466]; (NH4)2PdCl6 [464]; Pd(CN)2 [428]; TlPd2S3 [438]; PdSe [465]; Pd[SC(NH2)2]4-[Cr(SCN)4(NH3)2]2 [467, 608]; Pd(NH2CH2-CH2NH2)2HgI4 [468]; [Pd(NH2CH2CH2NH2)2]3[Cr(SCN)6]2 и [Pd(NH2CH2CH2NH2)2]-[B(C6H5)4]2 [429, 469]
Os(IV) Гидролитическое осаждение (NaHCO3) Анал. р-р (дистиллят после отгонки Os в НС1, нас. SO2; 15-50 мг Os) выпар, досуха с 10 мл НС1 (конц.), повторяют трижды. Остаток раств. в 150 мл Н2О, нагр. до кип., добавл. индикатор (бромфеноловый синий, 0,04% р-р) и по каплям NaHCO3 (10%) до появл. голуб, окраски (pH = 4), кип. 5-6 мин. Добавл. 10 мл сп. и нагр. 2 час при 100 °C. Жидкость декантируют через ф. т. К ос. добавл. 25 мл NH4C1 (1%) и 10 мл сп., выдерж. 15 мин при 100 °C и декантируют. Ос. промыв. 4 раза ЫН4С1 (1%) и переносят в ф. т., промыв, сп. Ос. посыпают сверху ЫН4С1, осторожно прокал. в токе Н2 до удаления ЫН4С1, затем в течение часа при 800 °C охлаждают 5 мин в водороде, затем 15 мин в N2, выдерж. 10 мин в эксикаторе над Ca(NO3)2 (нас.) Os 7, 427, 428, 471
(NH4)2S Щел. дистиллят OsO4, добавл. изб. pear, и слабо нагр. до полной коагуляции ос., подкисл, р-р НС1 (разб.), ф. т., высуш. при 80 °C, осторожно прокал. в токе Н2, охлажд. в токе Н2, вытесняют водород углекислым газом Os 427, 472
Тионалид (0,5% р-р в сп.) Анал. р-р (2-30 мг Os, 0,5 по НС1), изб. pear. (ок. 15 мл) по каплям в течение 30 мин, кип. 2 час и отстаив. 1 час при 100 °C, поддерживая постоянный объем р-ра. Ф. т., прокал. в токе Н2 и взвешивают. Затем осмий выжигают на воздухе при 750 °C (тяга!). Снова прокал. в токе водорода и взвешивают. Вес Os находят по разности Os 428, 471
Актидин (2% р-р в СН3СООН) а) Анал. р-р (0,6-5 мг Os, солянокисл., 10-15 мл), изб. pear. Желт, р-р нагр. несколько мин., охл., ф. с., промыв. СН3СООН (разб.), водой, сп., эф. Высуш. 10 мин в вакуум-эксикаторе. Не меш.: Cu, Cr, Со, Fe. б) Тем же способом осажд. [OsBr6]2~. Ос. промыв. СНзСООН, водой. Высуш. при 110 °C H20sC16(C13H9N)2 H20sBr6(C13H9N)2 474, 475
K3[Cr(SCN)6]. • 4Н2О (1), тиомочевина Синтез pear.: Смешивают 15 г KSCN с 12,5 г Cr2(SO4)3-K2SO4-24H2O. Нагр. до 80 °C, выдерж. 2 час, охл., добавл. этанол, фильтр, и упар. до начала кристаллизации K3[Cr(SCN)6]-4H2O Анал. р-р (соляно- или сернокисл, р-р OsO4), добавл. сернокисл. р-р тиомочевины, через 15-20 мин. изб. pear. (1), отстаив., ф. с., промыв, водой, высуш. при 105 °C [Os(CSN2H4)6]- •[Cr(SCN)6] 474
Химические методы количественного анализа
493
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Os(IV) 2-Фенилбензотиазол Анал. р-р (3-25 мг Os, р-р НВг), добавл. 25 мл НВг (4 М) и изб. pear, в 25 мл НВг (4 М). Ос. отстаив. и ф.т., промыв, небольшим объемом НС1 (0,2 М). Ос. прокал. в токе Н2 и выжигают Os (см. опр. с тионалидом) Os 476
1,2,3-Бензо-триазол (2% водный р-р) Анал. р-р (щел. р-р OsO4, ок. 15 мг Os, 10 мл) вливают в колбу с 1 мл сп., добавл. 25 мл pear., нагр. 15 мин при 100 °C, доводят pH р-ром СН3СООН до 3, нагр. 15 мин (до коагуляции ос.). Ф. с., промыв, гор. водой, высуш. при 100 °C Os(OH)3(C6NHN2)3 f= 0,3118 476, 477, 478
Антипирилме-тан (2% в СН3СООН) (1:1) Анал. р-р (5-13 мг Os, 0,1-0,5 н. по НВг, 60-70 мл), медленно при перемеш. добавл. небольшой изб. pear., отстаив. 2-3 час, ф.с., промыв. 4-5 раз р-ром pear. (0,05-0,1%) в НС1 (0,5 н.), высуш. при 100-110 °C Погрешность опр. ±3% (C23H24O2N4H)2-•[OsBr6] /= 0,1314 479
Диантипирилпро-пилметан, (2% в СН3СООН) (1:1) Опр. аналогично опр. с диантипирилметаном, но осажд. из 0,1-0,5 н. НС1 (C26H30O2N4H)2- •[OsC16] 479
Сульфат стрихнина а) Анал. р-р (солянокисл., OsO4), добавл. водн. р-р pear. Стакан накрывают стеклом с отверстием и медленно, по каплям, при перемеш. добавл. NaHCO3 (10%) до нейтр. среды (по индикатору), по каплям добавл. р-р pear., ф. с., высуш. при 100-105 °C. Предполагаемый состав (Ci2H22O2N2)2H2OsC16. б) Анал. р-р (после поглощения OsO4 р-ром НВг) упар., разб., фильтр, и разб. до 50 мл, добавл. изб. pear., нагр. ок. 5 мин до коагуляции ос., охл., отстаив., ф. т., промыв. НС1 (0,02 М). Ос. обрабатывают как при опр. с тионалидом f= 0,1771 Os 427, 476 427, 476
Ir(III, IV) Гидролитические методы: a) NaHCO3 б) NaBrO3, NaHCO3 Анал. р-р (слабо солянокисл.), NaHCO3 (10%) до pH = 6 (по бромфеноловому син.), ф. т., прокал. при 500 °C в токе Н2, охл., промыв. НС1 (0,1 н.), ос. прокал. при 500 °C в токе Н2, затем при 700 °C в токе N2. Анал. р-р (солянокисл.), добавл. 1 г NaCl, выпар, досуха, добавл. 1 мл НС1 (1:1), раств. в 200-300 мл гор. воды. К гор. р-ру добавл. 20 мл NaBrO3 (10%) до прекращения выделения паров Вг2. По каплям добавл. NaHCO3 (5%) до появл. ос., 10 мл NaBrO3 и кип. до коагуляции ос. Нейтрализуют р-ром NaHCO3 по каплям до pH = 7 (универсальная инд. бумага). Энергично кип. 10 мин., снова добавл. NaHCO3 до pH = 8, отстаив. 30 мин при 100 °C, ф. т., промыв. NaCl (10%). Ос. высуш., смачивают р-ром NH4C1 (нас.) и осторожно прокал., обрабатывают при нагр. НС1 (разб.), фильтр., прокал. в токе Н2 (см. п. «а»). Не меш. Pt(IV) Ir Ir 1,7 7,427, 480
(NH4)2s Анал. р-р (Ir в форме гексахлоридиевой к-ты), изб. (NH4)2S, кип. 1,5 час. К кор. р-ру добавл. CH3COONH4 и СН3СООН (pH ~ 4), нагр. 10—15 мин, ф. т., промыв, водой, подкисл. НС1. Ос. высуш. и осторожно нагр. при доступе воздуха до полного выгорания серы. Далее ос. обрабатывают по и. «а». Метод пригоден для опр. Ir после отд. от Rh нитритно-сульфидным методом Ir 427, 440, 441, 478
494
Новый справочник химика и технолога
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Ir(III, IV) КС1 (нас. р-р) Анал. р-р (слабо солянокисл., IrClg”), добавл. Н2О2 (10%), небольшой изб. pear, и равный объем сп., отстаив. 12 час, ф. т., промыв. КС1 (0,1 н.) в 50% сп., прокал. при 500 °C, охл., раств. хлориды Щ. М. в НС1 (0,1 н.), прокал. при 500°С в токе Н2, затем при 700 °C в токе N2 Ir 1
2-Меркапто-бензотиазол (1%р-р В СП.) Анал. р-р (IrClg-, не более 0,01 н. НО, ок. 5 мг 1г, 5 мл), 10 мл лед. СНзСООН, 1 мл CH3COONH4 (20%), 25 мл воды, нагр. до кип., добавл. 10 мл свежеприготовленного pear., кип. 1 час в стакане под часовым стеклом. Стекло обмывают гор. промывным р-ром СН3СООН (2%) и CH3COONH4 (2%) и разб. до 50-70 мл, отстаив. 12 час при 100 °C, ф. б., промыв. 100 мл промыв, р-ра, высуш., осторожно озоляют, прокал. 45 мин при 650-700 °C, далее восст. Н2 как описано выше. Учитывают вес холостой пробы. Возможно опр. 1г в присутствии РЬ Ir 481
Тиомочевина а) Анал. р-р (< 20 мг 1г), 10 мл H2SO4 (конц.), 0,1 г NaCl, выпар, до паров SO3, охл., разб. до 50 мл, добавл. крист, pear. (0,1 г на 5 мг 1г), нагр. до 210-215 °C (термометр). Р-р охл., разб. водой и ф. б., промыв, водой, высуш. и сжигают при доступе воздуха. Ос. прокал. в токе Н2 (см. выше). б) Анал. р-р (после осажд. Ph тиоацетанилидом, 0,5-100 мг Ir), добавл. по 25 мл H2SO4 (конц.) и HNO3 (конц.), нагр. до 250 °C, окисл. 5-7 каплями НС1О4, по окончании р-ции добавл. 2-4 г pear., нагр. до полной коагуляции ос., охл., разб. водой до 250 мл, ф. б., промыв, гор. водой, осторожно прокал. Ос. в платиновой чашке обрабатывают HF, выпар. досуха, смачив. НО (разб.) и смывают в стакан, добавл. равный объем НС1 (конц.) и кип., ф. б., прокал. и восст. до Ir (см. выше) Ir Ir 427, 444 428, 482
и-Амино-фенилдитио-карбаминат аммония (1%) Анал. р-р (ок. 10 мг Ir, 1г NaCl) упар. досуха, добавл. 1-2 мл гор. воды, снова упар. досуха (повторяют до исчезно-вания запаха НС1). Добавл. 10-20 мл воды, нагр. до кип., при перемеш. 4-кратный изб. pear, (свежеприготовленный р-р), разб. до 50-100 мл. Нагр. 3 час при 100 °C, добавл. мет. фиол. (2 мг • мл ') до коагуляции ос., нагр. еще 15 мин. Охл., ф. б., отмыв, от NaCl, медленно сжигают и восст. до Ir (см. выше) Ir 483
Тионалид (2% в лед. СНзСООН) Анал. р-р (Na2IrCl6, 1-20 мг Ir), добавл. сп., 0,1-1 г NaCl, нагр. и выпар, досуха на водяной бане. Сухой ос. раств. в 50 мл воды, нагр. до кип., добавл. 3-кратный изб. pear., кип. 1 час, ф. с., промыв. 10 мл подогретого сп. или лед. СН3СООН, гор. водой до удаления pear, (по иод-крах-мальной бум.), высуш. при 115-120 °C Ir(C12H10ONS)3 f= 0,2293 484
2-Меркапто-бензимидазол (0,5% в 0,5% NaOH) а) Анал. р-р (0,5-2 мг Ir в ацететном буфере, pH = 3,27-5,37), pear, не более 50% изб. (1 мл pear, на 1 мг Ir). Ir(IV) предварительно на холоду восст. 1-2 каплями pear. Р-р нагр. до 100 °C и выдерж. 1-1,5 час, охл., ф. с., промыв. 4-5 раз гор. водой, высуш. при 120-140 °C. б) Анал. р-р (5-10 мл, 1-5% минеральной к-ты по объему), изб. pear. (1 мл на 1 мг Ir), нагр. до 60-70 °C при перемеш. и выдерж. до обесцвечивания р-ра. Р-р охл., ф. с., промыв. НС1 (1% по объему), высуш. при 120-140 °C Ir(C7H5N2S)3 /= 0,3004 Ir(C7H6N2S)3Cl3 f= 0,2567 485 486
Химические методы количественного анализа
495
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
1г(Ш, IV) Тетрафенилар-сонияхлорид (0,04 М) Осадитель необходимо очищать во избежание восст. Ir(IV) до 1г(Ш). Анал. р-р (ок. 5 мг 1г, солянокисл.), несколько капель Н2О2 (30%), 100-200 мг NaCl, упар. досуха при 100 °C, добавл. воды, НС1 до конц. 0,2-1,0 М, pear, при перемеш. до конц. 0,01 М. Ос. отстаив. 1 ч, ф. с., промыв, р-ром осадителя, высуш. при ПО °C. Остальные ПМ не меш. опр. 1г /=0,1647 487
Диантипирилпро-пилметан (2% в СНзСООН) (1:1) Анал. р-р (5-13 мг 1г в форме 1гС1^“ , 0,1-0,5 н. по НС1, 60-70 мл), медленно при перемеш. pear, до прекращения образования ос. и еще 1 мл, отстаив. 2-3 час, ф. с., промыв. 4-5 раз р-ром pear. (0,05-0,1% в 0,5 М НС1), высуш. при 100-110 °C (C26H30O2N4H)2- •[1гС16] /= 0,1523 479
Опр. также в виде 1г при осажд. реагентами: H2S [7], Na2S [427, 478], NH4C1 и КВгО3 [7], ZnO [488], HgO [489,490], NaOH [7]
Pt(IV) H2S (газ) а) Анал. р-р (< 0,5 т Pt, без NO3, нейтр.), 2-3 мл НС1 (конц.) и столько же H2SO4 (конц.) на каждые 100 мл р-ра, нагр. до 95 °C и 45-50 мин пропускают H2S, ф. б., промыв. NH4C1 (1%), озоляют и прокал. на воздухе при 800 °C. Примеси солей Щ. М. можно удалить промыв, прокал. ос. гор. водой и повторным прокал. ос. б) Анал. р-р, добавл. NaCl, выпар, досуха, р-ют в НО (0,5 н.), нагр. до кип., в кип. р-р пропускают H2S в течение 30 мин. Ос. ф. б., промыв. НС1 (разб.), высуш., прокал. при 800 °C. в) Анал. р-р (ок. 10 мг Pt, HC1+HNO3), добавл. по 1 г NH4C1 и NaCl, выпар, досуха при 100 °C, добавл. 10 мл воды. Повторяют 2-3 раза. Добавл. р-р (20 мл 1 н. НС1, 30 мл воды, нас. H2S), выдерж. при 100 °C до осажд. ос. Попеременно подкисл, р-р, кип. и пропускают ток H2S в течение 30 мин. Охл., ф. б., промыв. NH4C1 (1%), осторожно высуш., озоляют и прокал. 1 час при 800 °C. Охл. в эксикаторе над Ca(NO3)2 (нас.). Промыв, р-р снова фильтр., ос. промыв, и при-соед. к основному ос. Pt Pt Pt 1,266 470, 491 428, 491
H2S (газ) в присутствии солей Т1+ Условия осажд. см. выше. Перед введением H2S добавл. необходимое количество соли Т1+ и проводят осажд. как в п. «б» опр. с H2S. Ос. ф. с., промыв, водой, сп., эф., высуш. в вакуум-эксикаторе. Ошибка опр. 0,5-7,7 мг Pt составляет ± (0,02-0,08 мкг) TlPtS3 /= 0,3938 438
(NH4)2S Анал. р-р (0,2-60 мг Pt, < 20 мл), добавл. 5-10 мл H2SO4 (конц.), нагр. до 300-320 °C (или до кип.), охл., разб. водой (при перемеш.) до 50-100 мл, добавл. изб. pear., выдерж. 1 час при 100 °C и отстаив. при 20 °C 14-15 час. Ос. ф. б., промыв, водой (подкисл. H2SO4), высуш. и прокал. до металла (см. выше). Rh и 1г можно определить в фильтрате Pt 492
NH4C1, СП. Анал. р-р (H2PtCl6), нас. р-р NH4C1, выпар, при 100 °C почти досуха. Ос. крист. NH4C1 раств. в небольшом количестве воды, ф. б., промыв. NH4C1 (5-10%), влажным помещают в тигель, покрывают дополнительно фильтром, высуш. и осторожно прокал., постепенно повышая температуру до 800 °C Pt 219, 464
496
Новый справочник химика и технолога
Продолжение таблицы 5.1.8
Определяемое вещество, ион Реагент Методика определения Гравиметрическая форма осадка Литература
Pt(IV) НСООН Анал. р-р (< 0,5 г Pt, слабо солянокисл.) в конической колбе разб. до 100 мл, 6 г CH3COONa (4 г CH3COONH4), 2 мл pear., закрывают воронкой, нагр. при 100 °C до прекращения выделения газа и просветления р-ра. Ф. б. или ф. т., промыв. NH4C1 (1%), озоляют и прокал. при 800 °C Pt 455, 480
Н2С2О4 (нас. р-р) Анал. р-р (< 0,2 г Pt) выпар, с 10 мл H2SO4, добавл. 7 мл HgSO4 (10%) на каждые 100 мг Pt, выпар, до паров SO3 и удаления бел. пленок HgCl2, охл., добавл. 40 мл воды, кип. до р-ния сульфатов, отфильтр. SiO2 и н. р. сульфаты, к ф-ту добавл. 10-15 мл pear., выпар, до начала выделния газа, закрыв, стеклом и нагр. до полного разрушения pear, и коагуляции ос. Охл., добавл. 50 мл воды и кип. для р-ния сульфатов. Ос. Pt ф. б., промыв. H2SO4 (10%), затем водой, озоляют и прокал. при 800 °C Pt 434
Тионалид Осажд. аналогично Rh. Полученный желт. ос. ф. б., промыв. сп., осторожно прокал. при 800 °C. Пригодна для опр. малых количеств (десятые доли мкг • мл-1) Pt 484
Тиофенол (10% в сп.) а) Анал. р-р (10-25 мг Pt, хлоридный, 200 мл), добавл. 4 капли НС1 (конц.), 1 мл pear., кип. 2 час, затем нагр. при 80 °C до полного просветления р-ра (иногда до 12 час), ф. б., промыв, водой, озоляют и прокал. при 800 °C. Прокал. сразу при высокой температуре приводит к завышенным результатам. б) Анализируют по п. «а». После кип. ф. с., промыв. 50 мл НС1 (0,012 М), высуш. при 235-275 °C в течение 1,5-2 часа Pt (C6H5S)2Pt f= 0,4719 445 493
Тиоацетамид (2%) Анал. р-р (12-160 мг Pt, 0,5 н. по НС1), изб. pear. (0,21 мл на 10 мг Pt), нагр. 30 мин при 100 °C, ф. б., промыв. НС1 (разб.) и водой, осторожно озоляют и прокал. при 800 °C в течение 1 час и 10 мин при 900 °C Pt 501, 504
Тиомочевина Анал. р-р, добавл. H2SO4 (5 мл к-ты на 10 мг Pt), выпар, до выделения SO3, далее по методике опр. 1г. Р-р с ос. охл., разб. водой и ф. б., промыв, и прокал. до Pt (см. выше) Pt 427, 444
2-Меркапто-бензимидазол (0,3% в 0,5% NaOH) Анал. р-р (0,2-2 мг Pt, 5-10 мл, 0,1-5,0% минеральной к-ты по объему), изб. pear. (1 мл на 1 мг Pt), нагр. до 70 °C и выдерж. до полной коагуляции оранж. ос. Р-р охл., ф.т., промыв. НС1 (1:100), высуш. при 120-140 °C (C7H6N2S)2PtCl4 f= 0,2082 427
Формамидин-сульфиновая к-та(диоксид тиомочевины) Анал. р-р (комплексные сульфат-, нитрит- или аммиакат-ионы), Н28О4до содержания 5% по объему, нагр. до выделения SO3, охл., разб. водой, pear. (1 г на 1 г Pt), нагр. 2-3 час при 100 °C, ф. б., промыв, гор. водой и прокал. до Pt (см. выше) Pt 494
Диантипирил-метан (0,05 М) Анал. р-р (0,5 н. по НС1, 20 мл) добавл. 0,5-1,0 мл гидразина (25%), 5 мл KI (1,5 М), выдерж. в течение 1 час. Затем добавл. 80 мл НС1 (0,5 н.) и постепенно 10 мл pear. Через 10 мин ф. с., промыв. НС1 (0,1 н.), водой, сп. Высуш. в ва-куум-эксикаторе (C23H24N4O2H)2- •Ptl4 495, 496
Электролиз Осажд. на Pt вращающемся электроде при напряжении 2 В из сернокисл, р-ра при 20 °C. Полное осажд. Pt занимает 30 мин. Катод промыв, водой, сп. и эфиром, прокал. на горелке. Метод предложен для опр. 0,1-50% Pt в урановых сплавах Pt 500
Опр. также в виде: Pt (после осажд. солями Ti(III) и V(I) [497], каломелью [427, 498],хлористой медью (I) [499], фосфорноватистой к-той [434], цинком [428, 502], магнием и алюминием [428, 503]); PtS2-5H2O [428, 440]
Химические методы количественного анализа
497
Таблица 5.1.9
5.1.3. Органические осадители для гравиметрического определения минеральных ионов
Определяемое вещество, ион Реагент Условия осаждения Интервал содержаний Примечание Литература
Li+ Т етра-(и-толил/борат Р-р pear., нас. NaCl Предварительно осажд. К+, Cs+, Rb+ тетрафенилборатом 3,4
Na+ Уранилацетаты, двойные соли с Cu, Mg, Cd, Со, Ni Аналогично осажд. с Zn. Иногда осажд. из водно-сп. р-ров Ос. менее растворим, чем с Zn. Кристаллогидраты с разным количеством воды 57-59, 476
Тетра-(/7-толил)-борат Li ГФ: NaB(C(,H4CH3)4 К, Rb, Cs отделяют тетрафенилборатом лития 60
и-Хлорфенил-а-метоксиуксусная к-та или ее к-соль Осажд. из смеси воды с метанолом (3:1), pH = 3-5, 0-5 °C. Промыв, хол. (0-5 °C) водой « 20 мг Высуш. в вакуум-эксика-торе, промыв, безводным ацетоном, высуш. при 75-90 °C 478
2-Нафтил-а-метоксиук-сусная к-та или ее тетраметиламмонийная соль pH = 3-4, 0,5-5 мг • мл’1 Na. Изб. (75%) pear., отстаив. 90-100 мин, промыв. сп. (нас. ОФ) 93-151 мг NaCl 21—36 мг Высуш. при 105-110 °C /= 0,05066 Погрешность <1%. Не меш.: 2-5-кратные количества NH;, Li, Rb, Cs, Mg, К 479
5 -Бензамидоантрахинон-2-сульфоновая к-та (3% в 50% сп.) Анал. р-р (5 мл), 5 мл. сп., 15 мл pear. Отстаив. 30 мин, ф.с., промыв. СП. (нас. ОФ) и эфиром 1-25 мг Высуш. при 110-120 °C f= 0,05358 Pear, не селективен 477
К+ Диметилпикрат натрия (3%) Ос. промыв, хол. водой Высуш. при 90-150 °C ГФ: KC8H6N3O7 /=0,1324 480
Пикриновая к-та (нас. р-р) Ос. промыв, минимальным объемом хол. воды Высуш. при 100 °C ГФ: KC6H2N3O7 /= 0,1463 481, 489
2-Хлор-З-нитро-толуолсульфокислота, натриевая соль (10%) Анал. р-р (10 мл), 7 мл pear., отстаив. 12 час. при 5 °C, ф.с., промыв, pear., затем хол. водой 0,1-0,2 г Высуш. при 120 °C ГФ: KC7H5C1NSO5 /=0,1349 Меш.: NH; , Rb+, Cs+, Ва2+ 482
5 -Нитробарбитуровая к-та(1% в 50% сп.) Анал. р-р, изб. pear., отстаив. 12 час. при 0 °C, центрифугируют, промыв. сп. (50%) Высуш. при 100 °C ГФ: KC4H2N3O7 /=0,1851 483, 484
Литиевая соль 2,4-ди-нитронафтиламинобен-золсульфокислоты Осажд. из водного р-ра Высуш. при 110 °C Меш.: Mg2+ 485
Нафтоловый желтый S Опр. в присутствии Na 486
3,4-Диоксиазобензол-4 '-сульфокислота (1%) /= 0,119 Меш.: NH;, Rb+, Cs+, Tl+. Соли Ag и Au восст. до металла 487
Тетранитрофентиазин-5,5-диоксид (1% в 0,1 М р-ре LiOH) Высуш. при 110 °C 488
Rb+, Cs+ Тетрафенилборат натрия или лития 0,1 М НС1, pear. (0,6%). NH; маек. НСНО, тяжелые металлы — ЭДТА. Меш.: Hg 2-20 мг Высуш. при 120 °C ГФ: Rb[B(C6H5)4] Cs[B(C6H5)4] 7
498
Новый справочник химика и технолога
Продолжение таблицы 5.1.9
Определяемое вещество, ион Реагент Условия осаждения Интервал содержаний Примечание Литература
Cs+ Тетра(и-фторфенил) борат Р-р слабокислый, 70 °C Высуш. при 110 °C ГФ: Cs[B(C6H4F)4] Отд. от К+ и Rb+ 6
Цианотетрафенилборат рН = 3 20-50 мг Не меш.: К+ до 200-кратного избытка 8
Cu2+ а-Бензоиноксим NH4OH, тартрат > 0,1 мг Высуш. при 105-110 °C ГФ: Cu(Ci4HnO2N) 1,491
Оксим пировиноградной к-ты Быстрое опр. в присутствии Cd. Высуш. при 110 °C ГФ: Cu(C3H4O3N)2 62
Бензоимидазол-1 -карбоновая к-та pH = 3,0-3,5 ГФ: Cu(C8H5O2N2)2 Не меш.: Pd 63
Метилфенилпира-золоноксим 4-16 мг Высуш. при 105-203 °C ГФ: Cu(C10H8N3O2)2 /=0,1355 Погрешность опр. < 2% 494
о-Оксиацетофеноксим pH = 9-10, винная к-та Высуш. при 105-110 °C ГФ: Cu(C12HnNO2) 493
Купферон На холоду, в присутствии фенилгидроксиламина и нитрита натрия 64
2,2-Дипиридил SCN- Микрограв. ГФ: Cu(Ci0H8N2)(SCN)2 /=0,1893 65
1,10-Фенантролин SCNT Микрограв. ГФ: Cu(C12H8N2)2(SCN)2 /= 0,1177 65
1,2,3-Бензотриазол pH = 7-8,5, тартратоаце-татный р-р, ос. сине-зел. ГФ: Cu(C6H4N3)2 496
2(-о-Оксифенил)-бензоксазол pH = 4-11,5 Высуш. при 130-140 °C /=0,1313. ГФ: Cu(Ci3H8O2N)2 497
Р-.п-Хлоранилин-циннамо-«-хлор-анилид или р-и-изомер pH = 6,5-7,0 Высуш. при 100-170 °C ГФ: (C21H15N2OC12)2Cu /=0,0851 Не меш.: Zn, Pb, Mg, Ва 498, 499
Изохинолин pH = 2-10,2 При pH = 3 Си опр. в присутствии Zn 500
Тионалид (0,1% в сп.) < 0,5 н. H2SO4, 10-крат-ный изб. pear., 10% сп. 3-100 мг Высуш. при 80-120 °C ГФ: Cu(C12H10ONS)2 H2O 490
Опр. также с pear.: 1. оксимы: 4,5 диметил-2-гидроксиацетофенона [495], а-кетокислот [501], 1,2-индандиола (моно- и дипроизводные) [492], а также ацетонафтохинондиоксим [502], 2-гидрокси-1-нафтальдоксим [503]; 2. пикролоновая [591] и арсаниловая [592] к-ты; 3. бензоимидазол [504]; 4. бигуанизин и его производные [505]; 5. 8-ацетохинальдин [506]; 6. 2-меркаптобензимидазол, 2-меркаптобензотиазол [491], 2-меркаптометилбензоимидазол [507]; 7. фенилгидра-зиндитиокарбаминовая к-та [508], морфолиндитиокарбамат [509], диаллилдитиокарбамидогидразин [510], Л'-аллилди(тиокарбамил)гидразин [511]; 8. тиосемикарбазоны бензофенона [512], рубеановодородная к-та и тиоацетамид [513], л/-(меркап-тоацетамидо)фенол [514], 2-амино-1,3,4-тиадиазол-5-тиол [515], А-метиламинотиоформил-А-фенилгидроксиламин [516], 1-имино-3-тиоизоиндол [517], тиомочевина [518], сульфид 0-нафтола [519], монотиомоче-винофталевая к-та (гомогенное осажд.) [520].
Ag* 1,2,3 Бензотриазол (2-5% р-р В NH4OH) Слабокисл, р-р, 1-10 г ЭДТА, 60-90 °C. 10-100 мг Высуш. при 110 °C ГФ: AgC6H4N3 46
6-Хлор-4-нитро-1 -окси-1,2,3-бензотриазол pH = 3,2-5,8 ГФ: AgC6H2O3N4Cl Меш.: Pb, Ва, Hg(I,II) 521
2,3-Нафтотриазол pH =10, ЭДТА, тартраты 5-100 мкг ГФ: (C]0H7N3)Ag Меш.: Sb и I 522
Химические методы количественного анализа
499
Продолжение таблицы 5.1.9
Определяемое вещество, ион Реагент Условия осаждения Интервал содержаний Примечание Литература
Ag+ Бензимидазол (1%) Анал. р-р (50 мл), 2-3-кратный изб. ЭДТА, нейтр. до pH = 10, нагр. 10 мин (60-70 °C), изб. pear. (20 мл на 100 мг Ag), нагр. 10 мин, ф. с., промыв, гор. водой 10-100 мг Высуш. при 120 °C ГФ: AgC7H5N2 523
2-Метилбензимидазол NH4OH, pH = 10, ЭДТА ГФ: AgC6H7N2 Не меш.: Cu, Ni, Со, Mg 524
Диазоаминобензол (1% р-р в СП.) Анал. р-р (нейтр.), 2-3-кратный изб. pear., ф. т. 0,03-1 г или 2-20 мг Высуш. при 110 °C, прокал. до Ag (ГФ). Ошибка < 0,4% Ошибка 1,5% 525
и-Аминоазобензол (р-р в эфире) Водно-сп. р-р, pear. ГФ: Ag Меш.: Ni, Cd, Cu. Не меш.: К, Na, Pb 525
Меркаптобензотиазол (каптакс) ЭДТА, тартраты ОФ: AgC6H4NCS2 526
2-Меркапто-5-анилино-1,3,4-тиодиазол (1% р-р В СП.) Анал. р-р (10 мл), 10 мл сп., нагр. до 100 °C, по каплям pear., ф.с., промыв. сп. (50%), сп. (96%), эфиром Высуш. в вакуум-эксика-торе. ГФ: AgC8H6N3S2 Продолжительность опр. 45 мин. 528
Диаллилдитио-карбамидгидразин (даль-зин) (р-р В СП.) рН= 4,7-5,1, ЭДТА, нейтр. Na2CO3 до появл. мути, ее раств. в лимонной к-те, разб. до 200 мл, pear., отстаив. 1 час, промыв. водой, метанолом Высуш. при 75 °C ГФ: AgC8H13N4S2 527
и-Фенилен-ди-( 1 -тетра-золин)-5-тион (2% р-р в ацетоне) Анал. р-р разб. до 50 мл, 5-6 капель HNO3, pear. (2 мл на 1 мг Ag), выдерж. 15-20 мин, ф. с., промыв. 50 мл воды, 25 мл ацетона 1-50 мг Высуш. в вакуум-эксикаторе ГФ: Ag2C8H4N2S2 Продолжительность опр. 1 час. Меш.: Cu, Cd, Au, Rh, Ru и CN- Ошибка « 5%. 529
Висмутиол II (фенилди-тиобиазолонсульфид, калиевая соль) (0,5%) Анал. р-р (< 0,2 н. по HNO3), 1 г ЭДТА, разб. до 125 мл, pH = 5-9, нагр. до 50-60 °C, по каплям реагент Высуш. при 110-120 °C ГФ: AgC8H5N2S2 При кислотности >0,1 н. осажд. только ионы сульфидной группы 71
Тионалид (2% в сп.) 0,5 н. HNO3 (H2SO4), 80 °C, 10-кратный изб. реагента Высуш. при < 105 °C ГФ: Ag(C12H6ONS), но лучше Ag. Не меш.: Pb, Tl, Cd, Fe(II), Со, Ni, Zn, Al, Be, Cr 490
Тиомочевина, тиоацетамид рН> 12 > 0,1 г- л 1 ГФ: Ag2S (крупнокрист.) Опр. Ag в тиосульфатных ваннах 72, 73
Опр. также с pear.: 2-амино-1,3,4 тиодиазол-5-тиол [515], 2-меркаптобензоксазол [530], монофенил- и моноаллилгидра-зодикарбонтиоамид [531], 5-амино-2-и-нитрофениламино-4-тиазолидон [532], о-таллилтиогидантоин [534], сульфат 5-метилтиомочевины (осажд. CH3SAg) [534]; и-диметил-, и-диэтиламинобензилиденроданин [535], бензиловая к-та [536], меркаптофенилтиадиазолон [537, 538], толилтиовиулоровые к-ты [539], фенилгидразиндитиокарбаминовая к-та [540], ,ч-(меркаптоацетамидо)фенол [541], ксантагенат калия [593], танниновая к-та [594], тиомочевина и пикриновая к-та в виде [AgtSC^IUhJCfJLNsO? [595].
500
Новый справочник химика и технолога
Продолжение таблицы 5.1.9
Определяемое вещество, ион Реагент Условия осаждения Интервал содержаний Примечание Литература
Au(I, II) Аскорбиновая к-та (5%) Прим. 0,5 М НС1, нагрев. 80 °C, pear., кип. 15 мин. ~ 100 мг ГФ: Au Прокал. при 600 °C 74
Тиогликолевая к-та 2-10 н. НС1 6-76 мг ГФ: C2H3O2SAu Высуш. при 110-120 °C Ошибка ± 0,5%. Меш.: Pt, Zr, H2SO4 Не меш.: Os, Ir, Ru, Rh, Pd 75
Меркаптобензотиазол (1% р-р в СП.) H2SO4 (1-2%), CN", нагр., ф.б., промыв, водой « 10 мг; 0,04-0,4 мг в 500 мл ОФ: (C7H4NS2)3Au ГФ: Au 76, 542
Тиофенол (4% в сп.) 0,012-0,06 н. НС1 или HNO3, кип. 10-25 мг He меш. Cu, Ni, Fe. 77, 449, 543
л/-(Меркапто-ацетоамидо)фенол (0,5% р-р в 20% сп.) pH = 2-4, KI « 20 мг ГФ: C8H8O2NSAu Высуш. при 110-120 °C При маек. Г, ЭДТА, pH = 3-4 не меш. по 100 мг Щ., Щ.-З., Со, Ni, Fe, Cr, Al, Cd, Zn, Mn, Zr, Th, Tl, VO2+, Cu, Pb, Ag, Bi, Sb. Предварительно отделяют при pH = 2 Pd и Pt 78
Ди-2-тиенилкетоксим, (р-р в диоксане 50 мг на 20 мл) pH = 2,4-3,0, промыв, р-ром диокана (4%) 5-22 мг ГФ: Au(C9H6NOS2)2OH Высуш. при 70 °C He меш. большинство ионов. Помехи от Hg, Ru, Pd, Bi, Sn устраняют предварительным осажд. Au 8-окси-хинолином, р-ют в НС1 + HNO3 и осажд. pear. 79
и-Фенилендиамин-ди-( 1 -тетразолин-5-тион) (1% р-р в ацетоне) рН= 8-11, CH3COONH4, нагр., промыв, ацетоном < 50 мг ГФ: Au(C8H4N8S2)C1 Высуш. при 110-120 °C 80
Триметилфенил-аммония иодид Осадитель: 40 г KI и 25 г pear, в 1 л. Промыв, осадителем (1 :4), толуолом со сп. (2,5%) 5-50 мг Меш. катионы, образующие к-сы с Г 81
Бис-(триметил)-гексаметилендиаммония бромид КВг, pH = 0,5-1,0 > 5 мг ГФ: [(CH3)6N2(CH2)6]- [AuBr4]2 Меш.: Pt, Ir. Не меш. 35-50-кратные кол-ва Ni, Cu, Fe, Zn, Со, Cr, А1 82
Бис-(триметил)-декаметилендиаммония бромид (2%) pH = 0,5-1,0; 0,5 мл КВг (40%), 0,5 мл pear., отстаив. 12 ч в темноте, ф.с., промыв, нас. р-ром ОФ в смеси вода-сп. (1:1) 0,7-2,5 мг Высуш. при ПО °C ГФ: (CH3)3N(CH2)1ON(CH3)3]- •[AuBr4]2 f= 0,3050 Не меш. 50-кратные кол-ва Zn, Со, Mn, А1, Сг, 200-кратные — Ni, Си 545
А-(А-бром-С-те-традецилбетаинил)-С-тетрадецилбетаина, натриевая соль 1-3 н. НВг ГФ: (C38H79N2O4)[AuBr4] Не меш.: Pb, Pd, Bi, Sn(II), Hg(II), Os(VI), Pt(IV), Ir(IV) 544
Химические методы количественного анализа
501
Продолжение таблицы 5.1.9
Определяемое вещество, ион Реагент Условия осаждения Интервал содержаний Примечание Литература
Au(I, II) Тионалид 0,5 н. H2SO4, pear. (1% в сп.), 80 °C Высуш. при 230 °C ГФ: Au(C12H10ONS)3 Не меш.: Pb, Cd, Fe, Ni, Zn, Al, Be, TI 1
Ве2+ 3 - Ацетилгексанон-2 pH = 5,5-6 (устанавливается пиридином или NH4OH) 0,1-3 мг ГФ: Be[CH3COCH(C3H7> •COCH3]2 Высуш. при 50-55 °C Mg до 100 мг не меш. 83,425
Бензоилацетон pH = 8,8-10,5 (в присутствии (NH4)2CO3 3-15 мг ГФ: Ве(С10Н9О2)2 Ошибка 0,5% Меш.: Cu, Ni, Mg, Са, Мп, Fe, Al 84
Ацетоацетанилид pH = 5,5-8 (в присутствии ЭДТА pH =8) 0,5-3 мг ГФ: Be(C10H10O2N)2 Высуш. при 110 °C Ошибка 1-3% Меш.: Fe, Al 85
2-Пиколилфенилкетон CH3COONa 0,5-7 мг ГФ: Be(C13Hi0NO)2 Высуш. при 105-115 °C Меш.: А1 и др. 86
2-Окси-1 -нафтойный альдегид Из аммиачно-сп. р-ра 5-кратным изб. pear, при 70 °C 2-5 мг ГФ: Ве(СцН7О2)2 Высуш. при 110 °C Меш.: Al, Fe, Си 87
Купферон pH = 4,6-6,8 2—4 мг ГФ: Be(C6H5O2N2)2 или ВеО 88
и-Бензоилфенил-гидроксиламин pH = 5,5-6,5; 50-60 °C 0,5-2,8 мг Высуш. при 110 °C. ГФ: Be(Ci3H10O2N)2 или ВеО 89
8-Оксихинолин pH = 8,0; 65 °C ГФ: Be2O(C9H6NO)2 Высуш. при ПО °C 546
Коричнокислый аммоний pH = 3,5-5,0; NH4C1 0,04-36 мг ГФ: (С6Н5-СН=СНСОО)- •ВеОН Высуш. при 110 °C Меш. А1 90
Mg2+ 2-Окси-1 -нафтальдегид pH » 9; 50 °C, 50% сп., промыв. 50% сп. Выдерж. 30 мин. 3-5 мг Высуш. при 130-140 °C ГФ: Mg(C22H14O4) f= 0,06633 93, 547
Бриллиантовый желтый Уксуснокислая среда (pH ® 3,6). Отстаив. 1 час, промыв, водой (подкисл. СН3СООН) 5-26 мг Высуш. при 120 °C /= 0,0403 Не меш. Cu, Be, Cd, Са (5-кратный изб.), Sr (3-кратный изб.). Ошибка 0,4-1% 91,92
А-Бензоилфенил-гидроксиламин pH = 8-9; 55-60 °C; 1,5-кратный изб. pear., отстаив. 15 мин, ф.б., промыв. 0,1% р-ром pear. ОФ: Mg(C13H10NO2)2. Прокал. при 1000 °C ГФ: MgO 94
5 -Нитробарбитуровая к-та Осажд. из водносп. р-ра (1:1), промыв, сп. и эфиром ГФ: Mg(C4N2H2O3 NO2)2- •8Н2О Меш. МГ4, К, Cs, Ва, Zn, Pb 95, 548
Эмбелин (2,5 - Дигидрокси-3 -унде- цил- 1,4-бензохинон) pH = 7-8, 60 °C, выдерж. на паровой бане 30 мин. Промыв. ВОДОЙ, СП. 3-30 мг ГФ: MgCi7H24O4 Высуш. при 110-115 °C Ошибка 1-3% 96
Тропеолин 00 549
502
Новый справочник химика и технолога
Продолжение таблицы 5.1.9
Определяемое вещество, ион Реагент Условия осаждения Интервал содержаний Примечание Литература
Са2+ Пикролоновая к-та (0,27%) Нейтр. р-р, 50 °C Высуш. при 150-170 °C ГФ: Ca(C10H7N4O5)2-•1,5Н2О. Не меш.: Mg., щел. металлы 97
8-Оксихинолин pH = 11,3, тартрат Меш.: ЭДТА, Be. ГФ: Ca(C9H6ON)2 202
8-Г идрокси-5-сульфо-хинолин (лоретин) ГФ: Ca[CQH4NI(OH)SO3]2-•Ю/ЗН2О 202, 550, 551
Zn2+ Хинальдиновая к-та, этиловый эфир pH = 3-5, 80 °C, 1 час (осажд. из гомогенного Р-Ра) 25-100 мг в 100 мл Высуш. при 105-110 °C 10
Акридинсульфат SCNT ОФ: Zn(SCN)2 (Ci3H9N- •HSCN)2 Опр. в мет. Ni ок. 0,001% Zn 552
5-Бромантранилат Na ГФ: Zn(C7H5BrO2N)2 553, 554
Cd2+ Бруцин-бромид (1% р-р в 4 М H2SO4) H2SO4 (1 : 5), КВг, без нагр. 1 час, ф. с., промыв, разб. pear., сп.+эф. (1:7) > 0,2 мг ГФ: H2[CdBr4] [C21H20O2N2-(СН3О)2]2 Высуш. при 130 °C 30 мин. /= 0,0919; Не меш.: Al, Си, Сг, Fe, Mg, Mn, Pb, Zn 98
л-Анизидин Нейтр. р-р, кип., отстаив. 30 мин, промыв. СП. Высуш. при 105-110 °C 20 мин. ГФ: Cd(NH2C6H4OCH3)2SO4 99
Висмутиол II Слабокисл, р-р, нагр. 2-3 мин, отстаив. 1 час, промыв. хол. водой Высуш. при 110-120 °C ГФ: Cd(C8H5N2S3)2 100
Диантипирилметан + бромид H2SO4 (1 : 40), кип., отстаив. 1,5-3 час. Промыв. разб. р-ром pear. Высуш. при 110-120 °C ГФ: H2[CdBr4](C23H24O2N4)2 101, 102
Антраниловая к-та, натриевая соль pH = 5,5-6,5 (5,2), кип. После охлажд. отстаив. 1 час. Высуш. 30 мин при 105-110 °C ГФ: Cd(C7H6O2N)2 557
Гидразид изоникотиновой к-ты + бензоат Нейтр. среда, без нагр. Высуш. в вакуум-эксика-торе. ГФ: [Cd(C6H7ON3)]-(С6Н5СОО)2 103
Диантипирил-о-оксифенилметан H2SO4, без нагр. отстаив. 2 час, промыв, разб. pear., сп. + эф. (1:7) Высуш. при 110-120 °C ГФ: H2[CdBr4][C29H28O3N4]2 104
Салицилальдегидтиосе-микарбазон CH3COONa 4-140 мг Высуш. при 110 °C ГФ: Cd(C8H8ON3S)2 Меш.: СГ, F“, тартрат 14
Диацетил-бис-(5-бром-салицилгидразон) Нейтр. среда, кип., фильтр, без отстаив. Промыв, хол. водой. Высуш. 2 час при 105 °C ГФ: CdC18H20O4N6Br2 f= 0,1712 105
Диацетил-бис-(сали-цилгидразон) То же Высуш. 2 час при 105 °C ГФ: CdC18H22O4N6 f= 0,2254 105
Химические методы количественного анализа
503
Продолжение таблицы 5.1.9
Определяемое вещество, ион Реагент Условия осаждения Интервал содержаний Примечание Литература
Cd2+ 2,2'-Дипиридил + роданид pH = 3-5, нагр., отстаив. до охл., промыв, разб pear., сп., эфиром Высуш. в вакуум-эксикаторе 5-10 мин. ГФ: [Cd(Ci0H8N2)](SCN)2 106
«-(Меркаптоацета-мидо)фенетол pH = 5,5-6,5, нагр., отстаив. 30 мин. Промыв. гор. сп. 4 раза Высуш. 60 мин при ,110 °C /=0,2111 99
Меркаптобензотиазол NH4OH, без нагр., промыв. водой с добавл. NH4OH Высуш. при 110-120 °C, 30-60 мин. ГФ: Cd(C7H4NS2)2 ГФ: Cd(NH3)2(C7H4NS2)2 99 555
Р-Нафтохинолин + иодид 2 н. H2SO4 + SO2, ох-лажд. водой со льдом, 20-30 мин. Высуш. при 70-80 °C ГФ: H2[CdI4](C13H9N2)2 107
(NH4)2C2O4 Нейтр. среда, 50% р-р сп. Отстаив. 15 мин. Высуш. в вакуум-эксикаторе. ГФ: CdC2O4-3H2O 108
2-Оксинафтилаль-2-ами-нопиридин pH = 3,8-4, 50% сп., отстаив. 35 мин. Высуш. при 104 °C 20-25 мин. ГФ: CdCl2 (C16H12ON2)2 /=0,1653 109
Пиридин + хлорид Нейтр. среда, нагр., отстаив. 24 час, промыв, сп. + эф. (3:1), (1:1), эф. Высуш. при 110-120 °C 3-5 час. ГФ: [Cd(C5H5N)]Cl2 108
Пиридин + роданид ГФ: [Cd(C5H5N)(SCN)2 558, 559
Салицилгидроксамовая к-та Слабокисл, среда, кип., затем NH4ON, промыв, хол. водой Высуш. при 110 °C ГФ: CdC7H5O3N 110
Тиокапролактам Нейтр. среда, кип., затем NaON, отстаив. 10 мин при 100 °C, промыв, водой, СП. Высуш. при 110-120 °C ГФ: CdS 111
Тиомочевина + соль Рейнеке 1 н. кисл., без нагр., отстаив. 30-60 мин в воде со льдом, промыв, тиомочевиной (1%), СП. Высуш. при 110-120 °C ГФ: [Cd(SCN2H4)2]-•[Cr(SCN)4-(NH3)2]2 108, 556
Трифенилметиларсоний иодид Нейтр. среда, кип., отстаив. 1,5 час, промыв. KI (0,5%) Высуш. при 105-110 °C ГФ: [CdI4][AsCH3(C6H5)3]2 /= 0,0890 112
1,10-Фенантролин + роданид pH = 3-6, кип., отстаив. 30 мин. Высуш. в вакуум-эксикаторе 10 мин. ГФ: [Cd(Ci2H8N2)2](SCN)2 113
л/-Фенил ен-ди-( 1 -тетразо лин-5 -тион) pH = 5,5-7, нагр. 5 мин на водяной бане, промыв. водой (100 мл), затем ацетоном (25 мл) 0,5-50 мг Высуш. при 110-150 °C 45 мин. ГФ: Cd(C8H4N8S2) f= 0,2892 114
1,2-Ди-(2-пиридил)этилен pH =1,3-2,7 ГФ: [C12H10N2H2][CdI4] Не меш.: 100-кратные кол-ва Zn 561
Хиноксалин-2-карбоновая к-та pH = 4,0-7,3 10-50 мг 562
504
Новый справочник химика и технолога
Продолжение таблицы 5.1.9
Определяемое вещество, ион Реагент Условия осаждения Интервал содержаний Примечание Литература
Cd2+ 2-Хинолилфосфорная к-та pH = 4—7 ОФ: CdC9H6O3NPH2O Высуш. при 220-225 °C ГФ: CdC9H6O3NP Прокал. при 800-900 °C ГФ: Cd2P2O7 560
Тионалид 0,5 н. H2SO4, СГ, 80-85 °C 0,025-1 г в 250 мл Высуш. при 105 °C ГФ: Hg(C10H6NO2)2 Не меш. Fe(II), Ni, Со, Zn, Cd, Pb, TI, Mn, Al, Be, Cr 1, 115
Тиомочевина pH = 4,4-6,8, K4Fe(CN)6 ГФ: [Hg(H2NCSNH2)3]2- •[Fe(CN)6] 117
Диэтилдитиофосфат Из нейтр. и слабокисл, р-ров (pH < 1) ГФ: Hg[(C2H5)2PO4]2 118
2-Аминобензотиазол Сп. р-р pear., pH = 5,8 Высуш. при 100-110 °C ГФ: HgC7H4N2S 119
2-Меркаптобензотиазол В присутствие ЭДТА ГФ: HgC7H5NS2 120
2-Меркапто-5-анилин-1,3,4-тиадиазол Нейтр. или слабокисл, р-р, HNO3<0,2-0,3 н. ГФ: (SC(NHC6H5)NNCS)2Hg 116
Моноаллилдитиокарба-мидогидразин pH = 6-7 ГФ: Hg(C5H8N4S2) 121
Диаллилдитиокарбами-догидразин, натриевая соль pH = 3-3,5 ГФ: Hg(C8H12N4S2) 122
5-Фенил-1,2-дитиол-З -тион Нейтр. р-р, HgCl2. ГФ: 2C9H6S3-HgCl2 123
Тиооксин + KI В сернокисл, среде. Промыв. H2SO4 (4 н.), водой Высуш. при 60-65 °C ГФ: [Hg(C9H6NSH)2I2] 124
Адипиновая к-та Из нитратных р-ров Hg(I),pH = 2,8 ГФ: (HgOOCC2H4)2 125, 563
Феноксиуксусная к-та Из нитратных или перхлоратных р-ров, pH = 3 ГФ: C6H5OCH2COOHg(I) и (C6H5OCH2COO)2Hg(II) 126
Фенилборная к-та Из нейтр. или слабокисл, р-ров Высуш. при 70 °C ГФ: Hg(C6H5)2 127
Фталевая к-та Уксуснокисл, р-р Hg(I) Высуш. при 130 °C ГФ: C8H4O4Hg2 128
Антраниловая к-та Нейтр. р-р Hg(NO3)2, на холоду ГФ: Hg(C7H6O2N)2 116
Бензимидазол (1%) Изб. тартрата, pH = 8,4-10,0 Ос. устойчив до 340 °C ГФ: Hg(C7H5N2)2 129
а-Нафтиламин (4% р-р в сп.) Хлоридные р-ры Hg(II), содержащие SCN" ГФ: Hg(Ci0H7NH2)2(SCN)2 116
Купферон Слабокисл, р-р Hg(I) Бел. тяжелый ос. 130
Предложены также pear.: 2-меркаптометилбензимидазол [507], 2-меркаптобензоксазол [564], «-(меркаптоацеталеидо)ацетанилид [565], меркаптоуксусная к-та [566], 1 -алкил-1 -фенил-1 -нафтил-2,5-дитиомочевина [567], бензидин [568], гидразид изоникотиновой к-ты [569], сульфантрол [570], анилин [571], оксимы (изатиноксим, ванилиноксим, камфораоксим) [572]
АГ 8-Оксихинолин См. табл. «Методики...» а) pH = 4,7-9,8 б) pH = 5,2 (ацетатный буфер) в) Na2SO3, NH2OH, 1,10-фенантролин В железных рудах В силикатах Не меш. Fe 12, 13, 131
а-Пиколин рН= 7,0 Не меш.: Mn, Zn, Со, Ni 132
Химические методы количественного анализа
505
Продолжение таблицы 5.1.9
Определяемое вещество, ион Реагент Условия осаждения Интервал содержаний Примечание Литература
А13+ Коричная к-та, аммонийная соль (5%) pH = 5,0-5,1, NH4C1, кип., 1-1,5 час. на водяной бане 25-50 мг Высуш. при 110 °C ГФ: (С6Н5СН=СНСОО)3- •А1(ОН) Или прокал. до А12О3 133
Купферон pH = 4,6 ГФ: А12О3 Не меш.: Mn, Mg
Л’-Бензоилфенил-гидроксиламин pH = 3,6-6,4, нагр. до 65 °C, ос. крист. Высуш. при ПО °C /= 0,04064 134
Стирилкупферон pH = 7, NH2OHHC1, нагр., смесь сп. с пиридином (4 : 1,15% от общего объема) 4-12 мг Высуш. при 110 °C ГФ: (C14HnO2N2)3Al /= 0,03627 Ошибка < 1,5%. Ос. легко фильтр. 135
Эмбелин pH = 4—4,5 Высуш. при 105-110 °C ГФ:А12(С17Н24О4)3С17Н26О4 Прокал. до А12О3 Не меш. Be 136
Салицилгидроксамовая к-та Cu, Fe, Ti, V, Мо экстр, из р-ров H2SO4 с pear. Анал. р-р, pear., нейтр. до pH = 6-7,5 ГФ: A1(C7H5O3N)(OH) Не меш. РО3’ 137
Таннин Слабокисл., нейтр. или аммиачный р-р. Ос. переменного состава Прокал. при 1200 °C ГФ: А12О3 Легко фильтрующийся, но объемный осадок
Дифенил-2,2'-ди-карбоновая к-та, аммониевая соль pH = 3,2-5,0, NH4C1 или NII4NO3 (0,15 г на 0,03 г А1), осажд. при 100 °C, ф. б. при 40-45 °C ОФ: основная соль А1. ГФ: А12О3 Не меш.: Со, Fe(II), Ni, Mn. Zn отд. переосажд. 573
Бис-2-пиридилгликоль pH = 5,2-8,0 ГФ: А12О3 Меш.: Be, U(VI) 574
Ga3+ Коричная (фенилакрило-вая) к-та Слабокисл, р-р, NH4C1, промыв, гор. pear. Ос. неопределенного состава, плотный. Прокал. до Ga2O3. Не меш.: Mn, Ni, Со, Zn, SOt, NO’ 133, 140
Бензойная к-та (2%) pH = 3,72-4,0, нагр. до 80 °C, pear., нейтр. NH4OH по мет. кр. Отстаив. 30 мин. > 15 мг Высуш. при 110 °C ГФ: Ga(OH)(C6H5COO)2 Метод используется для отделения от большого кол-ва элементов. Возможно прокал. ос. до Ga2O3 141
5,7-Дибром-8-оксихи-нолин НС1 (разб.), ацетон Высуш. при 100-224 °C ГФ: Ga(C9H4Br2NO)3 Немеш.: 1000-кратный изб. А1. Меш.: Fe3+, In3+ 138, 575
Пирролидиидитиокарба-мат натрия pH = 3,8-4,0, винная к-та 0,5-10 мг Высуш. при 110-120 °C ГФ: Ga(C5H8NS2)3 Ошибка ±0,05 мг 143, 144
Салицилгидроксамовая к-та pH = 5,5-6,5, 20-кратный изб. pear., отстаив. 2 час. Высуш. при 110 °C. ГФ: Ga(C7H5O3N)(OH) 145
506
Новый справочник химика и технолога
Продолжение таблицы 5.1.9
Определяемое вещество, ион Реагент Условия осаждения Интервал содержаний Примечание Литература
Ga’1 Диантипирилпропилме-тан, 1% р-р в СНзСООН (1:Ю) 5,5-6 М НС1, промыв. 6МНС1 3-17 мг Высуш. при 100-110 °C ГФ: С2бНзо02Н4 НОаС14 f= 0,1084 Селективный метод. Меш.: Tl3+, Fe3+ 146
А-Бензоил-А-фенилгидроксиламин (р-р В СП.) pH = 1,6-3,5, р-ры НС1 или НС1О4, 45 мин при 100 °C Высуш. при 110-120 °C ГФ: Ga(C13Hio02N)3 или прокал. до Ga2O3. f= 0,0987 Не меш.: Al, Be, Zn, Fe(II), Cu, Th, Ce, U, In 147
Азелаиновая к-та pH = 4-5,5. Белый крист, ос. > 15 мг Высуш. при 110 °C ГФ: (CH2)7(COO)2GaOH 148
Себациновая к-та pH = 4-6. Белый крист, ос. > 15 мг Высуш. при 110 °C ГФ: (CH2)8(COO)2GaOH 148
In3+ Диметил анилин Слабосолянокисл. р-р, нагр., нейтр. NH4OH, добавл. изб. NH4CI. Ос. промыв. NH4CI, водой ГФ: In2O3. Отд. от Mg 576
Метилвиолет (6%) Анал. р-р (250 мл, 0,2-0,5 н. H2SO4, 35 г NH4I, 10 мл pear. Ф. б., р-ют в 1-2 мл H2SO4 6 н. и первое. « 0,15 мкг Из 0,5 М NH4I иодидный компл. In на 90-97% со-осажд. с иодидом pear.; в присутствие (по 15 мг) РЬ, Fe, Hg, Cd, Bi, Sn, Sb, Cu и 30 мг Zn их отд. осажд. из 0,01 MNH4I 577
Алкилксантогенаты (циклогексил-ксантогенат калия) Нейтр. или слабокисл, р-р ОФ: св. желт, алкилксантогенаты In. Ос. устойчив на воздухе, но разл-ся при нагрев. 578
Tl Дибромоксихинолин Т13+ осажд. 8-оксихино-лином. Ос. р-ют в H2SO4 (2 н.), добавл. Вг2, затем сульфосалициловую и винную к-ты, CH3COONH4 И NH4OH до запаха, отстаив. 1 час при 5 °C, промыв, водой Высуш. при 100 °C ГФ: Т1(С9Н4Вг2ЬЮ)з /= 0,1841 149
Меркаптобензотиазол (1% аммиачный р-р) Изб. pear. Ос. желт, крист, промыв. NH4OH (2,5%) Высуш. при 110 °C ГФ: T1(C7H4NS2) 150, 151
Диантипирилметан Tl3+, HNO3, КВг (2 М). Bi и Fe маек. ЭДТА, Sb — винной к-той 8-45 мг Высуш. при 105-110 °C ГФ: С2зН24О2Н4 Н[Т1Вг4] 152
[(C6H5)4As]C1 (6%) Анал. р-р 0,5-2 н. по НС1, нагр., отстаив. 12 час, ф. с., промыв. НС1 (1 н.) Высуш. при 110 °C ГФ: [(C6H5)4As][T1C14] /= 0,2802 153
Тиомочевина Т1(1) Отд. от Ag, Hg, Cu, Cd, Fe, Ni, Co, Zn, Mn, Al, Cr, Ba, Sr, Ca 1
2-Метил-8-оксихинолин ГФ: T1(C10H8NO)3 579
Химические методы количественного анализа
507
Продолжение таблицы 5.1.9
Определяемое вещество, ион Реагент Условия осаждения Интервал содержаний Примечание Литература
Sc3+ 8-Оксихинолин ГФ: Sc(C9H6NO)3C9H7NO 202, 580
2-Метил-8-оксихинолин ГФ: Sc(C10H8ON)3 C10H9NO 158
уЗ+ 5,7-Дигало-8-оксихи-нолин (C9H4X2ON) При комнатной температуре, добав. CH3COONH4 ГФ: Y(C9H4X2ON)3 ГФ: Y2O3. Прокал. для Х = С1 при 580-810 °C X = Рг при 520-760 °C Х = 1 при 350-590 °C 159
Купферон pH = 3,0-4,0, 0,01-0,02 М р-р Y3+, 0,15 М изб. pear. Высуш. при 110-120 °C ГФ: Y(C6H5O2N2)3 Прокал. при 450-600 °C ГФ: Y2O3 160 160, 161
Неокупферон Аналогично купферону; температура < 50 °C Прокал. при 540 °C. ГФ: Y2O3 161, 163
La3+ и лантаноиды (Ln) 8-Оксихинолин а) Гомогенное осажд. из ацетатного буфера, мочевина, нагр. б) винная или янтарная к-ты, pH = 7,1-9,4. в) pH = 4,4-5,8 Высуш. при 110-120 °C. ГФ: Ln(C9H6ON)3 Прокал. при 800 °C (см. табл. 5.1.11.1.А) ГФ: оксиды. См. п. «а». Отд. Th при pH = 3,9 154 156 155
8-Оксихинальдин (2-ме-тил-8-гидроксихинолин) pH > 8,5, опасность неполного осажд. Прокал. при 700-800 °C Позволяет отд. Ln от А1 (см. табл. 5.1.11.1.А) 157, 158
5,7-Дигало-8-оксихи-нолин При 20 °C, добав. CH3COONH4 ОФ: Ln(C9H4X2ON)3 Ln = La - Sm, Y, кроме Се (см. табл. 5.1.11.1.А) 159
Купферон pH = 3,0-4,0; 0,15 М изб. pear. 0,01-0,02 М р-р Ln Высуш. при 110-120 °C ГФ: Ln(C6H5O5N2)3 Ln = La - Sm, Y. Прокал. до Ип20з при 450-650 °C (см. табл. 5.1.11.1.А) 160, 161, 162
Неокупферон См. «Купферон»; нагр. <50 °C ОФ: Ln(Ci0H7O2N2)3 ГФ: оксиды (см. табл. 5.1.11.1.А) 162, 163
А'-Бензоил-фенилгидро-ксиламин pH = 7 ацетатный буфер, нагр., осажд. Се3+ ОФ: Ce(C13H10O2N)3 164
Дибензоилметан Осажд. Се, нагр., pH = 8 (медленно нейтр. NH3 в метаноле), О2 не мешает ОФ: Се(С15НпО2)4 Остальные Ln не осажд. 165
Пирокатехин Этилендиамин, (пиперидин, хинолин), ос. плотный ОФ: (Амин)3[Еп(СбН4О2)з]; с этилендиамином: (Амин)зМС6Н4О2)з]2 Ln = La, Gd, Er, Y 166
Салициловая к-та pH = 3-7 (при нейтр. NH4OH) ОФ: Еп(С7Н5Оз)з-пН2О Высуш. при 100-105 °C. ГФ: Еа(С7Н5Оз)з-1,5Н2О /= 0,24064 167, 168
Диоксивинная к-та Нейтр. и слабокисл, р-ры, в присутствие СНзСООН — другой состав ос. ОФ: 4Ln2O3-5C4H6O8-24H2O Ln = La, Gd, Er, Y. При pH = 0,7-1,0 количественно отд. Th 169
508
Новый справочник химика и технолога
Продолжение таблицы 5.1.9
Определяемое вещество, ион Реагент Условия осаждения Интервал содержаний Примечание Литература
La3+ и лантаноиды (Ln) <7-Камфорная к-та Нейтр. р-р, 30-35 °C, сп. ОФ: Ln2(CioHi404)3; Ln = La, Се, Pr, Nd 170
Фенил- и нафтиларсоновые к-ты pH = 1-3, 15-кратный изб. pear., осажд. Y, La, Се 0,02 М Бел. ос. 171
Морин pH = 5,5-7 0,1-3% Отд. TR от Fe, Са, РО^ 15
о-Оксифенилфлуорон NH4OH, тартрат, NH2OH 0,07 тМ (Sc) Опр. суммы РМ; меш. Fe, Al и др. 172
Таннин Сл. кисл. -> NH4OH тиМ Опр. суммы лантаноидов; тартрат не меш. 173
Si(IV) Пиридин (NH4)2MoO4, щел. расплав или р-р, нейтр. до pH = 2,5. Pear, добавл. к 2,5 М по НС1 р-ру 0,1-100 мг SiO2 Высуш. при 120 °C, затем прокал. при < 500 °C ГФ: SiO212MoO3 174
Опр. также в виде: оксихинолината кремневольфрамовой к-ты [583]; кремне-12-молибдата амидопирина [584], триметиламина [582], 2,4-диметилхинолина [585], уротропина [586]. Обзор применения орг. оснований — см. 175, 581, 582
Ge 5,6-бензохинолин (2%) + Н2С2О4 (1%) Нейтр. среда, изб. Н2С2О4, отстаив. 12-16 час, промыв. разб. Н2С2О4 + pear. 20-100 мг GeO2 Прокал. при 900-950 °C (тяга!). ГФ: GeO2 Меш. СГ 176
Уротропин + (NH4)2MoO4 pH = 3, среда HNO3 Прокал. 440-813 °C ГФ: GeO2-12MoO3 176, 177
Пиридин + (NH4)2MoO4 Кисл. среда (H2SO4, НС1), отстаив. 3-24 ч 0,5-15 мг GeO2 Высуш. 3 час при 150-160 °C 176, 177
5,7 - Дибром-8-оксихи-нолин 3-^ М НС1 0,5-2 мг Высуш. при 90 °C Эмпирический f = 0,03296 (на GeO2) 179
Тетрафениларсония хлорид + (NH4)2MoO4 0,1-0,2 н. H2SO4, после осажд. Na2CO3 до pH = = 7,2-7,4 0,1-10 мг Высуш. при 130 °C ГФ: [Ge(Moi2O40)]- •[(C6H5)4As]4 Прокал. при 580-700 °C ГФ: GeO2-12MoO3 180
Тетрафениларсония хлорид + Na2WO4 pH = 5-6, 100 °C, изб. осадителя. Промыв, разб. HNO3 0,4—6 мг Высш, при 110 °C ГФ: [GeOVuO^-KC^Msh Меш. СГ, SO^ 181
Фениларсоновая к-та 95% р-р СН3СООН, изб. pear. 100 мг - л1 Высуш. при 120°С ГФ: (C6H5AsO3)2Ge. Прокал. при 900-950 °C ГФ: GeO2 181, 182
п- Диметиламиноазофениларсоновая к-та (3%) НС1 (3-4 N), промыв. 3 н. НС1, метанолом Высуш. при 120-130 °C ГФ: [(CH3)2NC6H4N = = NC6H4AsO3]2Ge, /=0,1363 (на GeO2). Допустимо присутствие As, Se и Sb в 100-, 50-, 25-кратном количестве 182
Пирокатехин (1) + орто-ф енантро лин-кадмиевый комплекс (2) pH = 3,5-4,5, Na2SO4 (4-40 г- лг), р-р (1) (10%), р-р (2) (0,01 М), отстаив. 30 мин. 0,5-20 мг Высуш. при 100-110 °C ГФ: [Ge(C6H4O2)3][C12H8N2] /= 0,0835 183
Химические методы количественного анализа
509
Продолжение таблицы 5.1.9
Определяемое вещество, ион Реагент Условия осаждения Интервал содержаний Примечание Литература
Ge Галловая или пирогал-лол-4-карбоновая к-та + диантипирилметан или бриллиантовый зеленый pH = 1-3, отстаив. 2 час 0,5-25 мг Высуш. при 105-110 °C ГФ строго определенного состава: [Ge(C7H4O5)3] •(C23H24N4O2-H)2; [Ge(C7H4O5)3]-(C27H33N2)2 184
Винная к-та + ВаС12 Р-р pear.: 240 г NH4C1, 14 г винной к-ты, 13 г ВаС12-2Н2О. Осажд. в р-ре NH4OH; ацетон после осажд. 5-10 мг - мл 1 Высуш. 1 час при 110 °C. ГФ: Ba2[Ge(C4H4O6)2]-2H2O 185
Pb Антраниловая к-та, натриевая соль (3%) pH = 6 25-100 мг ГФ: Pb(C7H6O2N)2 f= 0,4322 Погрешность 0,1% 47
Щавелевая к-та pH = 3-7 587
8-Оксихинолин pH = 8,5-9,5 5-10 мг 588- 590
Триметилфенил-аммониия иодид Осажд. р-ром 60 г KI + 25 г pear. 4-60 мг Не меш. Ва, Sr, Zn; легко устраняяются помехи Hg, Sb, Se, Sn. Погрешность 0,5%. ГФ: [(CH3)3(C6H5)N]2Pb2I6 f= 0,2861 48
е-Капролактам Р-р pear. (25%) в бензоле 220-500 мг He меш. Cd, Cu, Bi, Fe, Hg(II), Mn, Sb(III). ГФ: (C6H11ON)4-H2PbCl6 f= 0,2369 49
о-Фенантролин 2% р-р pear, в 1% HNO3, NH4SCN 20-200 мг ЩМ, Al, Be, Mg и небольшие количества Ba, Nl-Ц не меш. 50
Тионалид 2% р-р pear, в сп. 255-45 мг Не меш. Ag, Al, As(III), Со, Cu, Fe(II), Ni, Zn. ГФ: Pb(C12H10ONS)2 Погрешность < 0,2% 51
Диаллилдитиокарбами-догидразин 46-76 мг He меш. Щ.З.М. ГФ: C8H12N4S2Pb. Погрешность < 0,6% 53
1 -Имино-3 -тио-изоин-долин, натриевая соль Меш. только: Ag, Au, Co, Cu, Hg(II), Ni, Pd, Pt, Rh, Ru, Tl. ГФ: (C8HsN2S)2Pb 52
5 -Меркапто-2-амино- 1,3,4-тиадиазол 1% р-р pear. 20-70 мг He меш.: Bi, Co, Ni, Zn (в присутствии cn.); Ag, Cd, Cu (в среде NH3); Hg в присутствии KCN. ГФ: (C2H2N3S2)2Pb 54
Меркаптобензотиазол 1% р-р pear, в NH4OH (2,5%) 2-8 мг He меш. большие количества Ba. ГФ: C7H4NS2PbOH 55
Морфолин-А-дитиокар-боксилат морфолиния 0,01 М р-р pear. Ок. 10 мг He меш. многие металлы, кроме Ag, Bi, Cd, Co, Cu, Fe(II), Fe(III), Mn, Ni, UO22+ , Zn. ГФ: C10H16N2O2S4Pb 56
510
Новый справочник химика и технолога
Продолжение таблицы 5.1.9
Определяемое вещество, ион Реагент Условия осаждения Интервал содержаний Примечание Литература
Ti Сульфосалициловая к-та + NH4OH Сильноаммиачная среда, осажд. TiO2 (Н2О)„ Прокал. при 1000-1200 °C ГФ: TiO2 1
5,7-Дибромоксихинолин Слабокисл.среда Высуш. при 160 °C ГФ: TiO(C9H6Br2ON)2 Прокал. (ос. + Н2С2О4) при 1000 °C. ГФ: TiO2 187
Zr Таннин (10%) Ацетатный буфер, NH4NO3, 100 °C, изб. pear. > 0,6 мг • мл 1 ZrO2, но <100 мг. Высуш. при 100 °C и прокал. при 1000-1200 °C. Возможно отд. U и Ti от Zr 188, 189 190, 191
Бензол- и нафталинселе-ниновые к-ты НС1 или HNO3 (1 н.). Ос. ZrO(RSeO2)2 аморфный 2-10% Zr в стали Прокал. при 1000-1200 °C ГФ: ZrO2. Погрешность ± 0,01%. Большинство элементов не меш. Меш.: Sn(IV), Ti(IV), Nb(V), Ta(V). Три последних маек. Н2О2 192
.V-Бензоил фенил- гидроксиламин (4%, в сп.) 0,5 н. сернокисл, р-р, нагр. 20 мин 100 °C, изб. pear. (< 400 мг на 100 мл р-ра), 1 час при 100 °C, промыв, хол., затем гор. водой 3-30 мг Высуш. при 110 °C ГФ: Zr(C]3H10O2N)4 193
(Zr, Hf) «-Бромминдальная к-та Осажд. аналогично миндальной к-те Высуш. при 120 °C ГФ: гг(СбН8О3Вг)4 и НДСбН8О3Вг)4. Прокал. до НЮ2 и ZrO2. Результаты используют для расчета Zr:Hf 194
1 -Нафтилгликолевая к-та НС1 или H2SO4 (< 2 н.), нагр. 20 мин при 85 °C, промыв, разб. pear, и сп. Прокал. до ZrO2. Более избирательна, чем купферон 195
Циклогексанол-1 -карбоновая к-та (2,5%) НС1 (< 0,4 н.) (или HNO3, НС1О4), нагр. до кип., промыв. НС1 (0,1 н.) с pear. « 12 мг Прокал. при 1000 °C ГФ: ZrO2, HfO2. Синтез pear. см. [196] 196
Zr Тартразин (1%) pH = 0,5-1,0, без отстаив., нагр. до 80 °C, промыв, гор. водой 10-100 мг Высуш. при 100 °C или прокал. до ZrO2. Ос. образуют только Th, Ва, Hg. Меш. также 8Од", тартраты, цитраты 197
Флавазин (0,5%) pH = 0,5-1,0, без натр., отстаив., промыв, хол. водой 10-100 мг См. тартразин 197, 198
Фумаровая к-та НС1 (< 0,35 н.) Прокал. до ZrO2. Применена для опр. Zr в цирконе 199
Хинальдиновая к-та (2%) pH = 3^4, нагр. 10-200 мг Прокал. до ZrO2. Отд. от TR. Меш. Th 200
8-Оксихинолин pH = 5,2-10, Н2С2О4 Прокал. до ZrO2. Высуш. при 110 °C ГФ: Zr(C9H6ON)4 202 201
Дибром-8-оксихинолин Соосаждение с pear, при разб. их р-ров в H2SO4 (< 0,2 н.), 0-20 °C 40 мкг, 1 мл Прокал. до ZrO2 203
Химические методы количественного анализа
511
Продолжение таблицы 5.1.9
Определяемое вещество, ион Реагент Условия осаждения Интервал содержаний Примечание Литература
Zr о-Фенилендиоксиди-уксусная к-та (2%) НС1 (< 0,4 н.), 100 °C, 5 г NH4NO3 (на 100 м р-ра), изб. pear., промыв. 0,1% р-ром pear. 2-100 мг Прокал. до ZrO2 204
лг-изомер НС1(<0,1 н.), 100 °C, NH4NO3 2-100 мг Прокал. до ZrO2 204
Цирконон Избирательное осажд. из НС1 (5%) 205
о-Крезотиновая к-та Ацетатный буфер, pH = = 4,3, натр., промыв, гор. водой < 10 мг ГФ: ZrO2 206
4-Аминосалициловая к-та pH = 1,4-3,3, 80 °C 2,5-250 мг ГФ: ZrO2 206
Т иосалицил овая к-та pH = 2, 80 °C, изб. pear., кип. 5 мин. 6—60 мг ГФ: ZrO2 207
2,5 - Д иокси-и-бензохинон НС1 (2 н.), 0,4 г pear., кип. 1-2 мин, промыв, pear. (0,2% в 1 н. НС1) Ок. 50 мг ГФ: ZrO2. Не меш.: 15-кратный изб. Ьа2О3, Се2О3, ПзО^, TiO2 208
Тиогликолевая к-та (5%) pH = 4,5; CH3COONH4, нагр. 5 мин при 90 °C, отстаив. 1 час, промыв. NH4NO3 (1%) ГФ: ZrO2. Метод малоизбирателен 209
Тиояблочная к-та (2%), тиомалоновая к-та НС1 (0,1 н.) или HNO3 (0,2 и.), нагр. 30 мин, 90 °C, отстаив. 1 час. 5-60 мг ГФ: ZrO2. Меш.: Bi 210
Меркаптобензотиазол, натриевая соль (10%, pH = 8-8,5) Слабокисл, р-р, промыв. 200 мл воды 5-60 мг ГФ: ZrO2 211
Мапеониловые к-ты (1% р-р В СП.) pH = 2,4-3,0, 80 °C, CH3COONH4,15 мин при 60 °C 3 мг ZrO2 ГФ: ZrO2. Не меш.: Се-группа и Са, А1, Zn, Mg, Mn, Bi, Cu и Ti. Меш.: Th, F, Pb, Hg и платиновые металлы 212
Флавиановая к-та (конц.) НС1 (0,5 н.), нагр. до 100 °C, ф. б. 20-70 мг ГФ: ZrO2. Погрешность 0,1-0,2% 213
Th .м-Нитробензойная к-та (0,4%) Нейтр. р-р, NO;, 60-80 °C, переосаждают, сжигают влажным 0,1-0,2 г ThO2 Прокал. до ThO2. Метод длителен, но дает хорошие результаты 214
Фумаровая к-та (нас. р-р) Нейтр. р-р (150-200 мл), 40% сп., реагент, нагр. до кип., промыв, сп. (40%), р-ют 30 мл гор. НС1 (1:1), упар., переосажд. 0,1-0,2 г Прокал. до ThO2 214
Пикролоновая к-та СНзСООН (2-3%), pH = = 2-3,2; при кип. 0,5-100 мг Переводят в оксалат и прокал. до ThO2. Пригоден для микровесового опр. Th 214, 215
Фениларсоновая к-та pH = 1-3, ацетатный буфер, ос. фениларсоната переводят в оксалат Прокал. до ThO2 214
Феррон pH « 3, ацетатный буфер, CH3COONH4, 60-80 °C, pear. (0,2%), отстаив. 30 мин, ф. б., промыв, р-ром pear. Сжигают и прокал. при 900 °C ГФ: ThO2 216
512
Новый справочник химика и технолога
Продолжение таблицы 5.1.9
Определяемое вещество, ион Реагент Условия осаждения Интервал содержаний Примечание Литература
Th Бензолсульфиновая к-та (или ее Г и Вг производные) Из гор. р-ров HNO3 (0,5 н.), изб. pear., ф.с. 1—4 мг Высуш. при 110-120 °C ГФ: Th(C6H5SO2)4, Th(C6H4BrSO2)4. Отд. Th от 100-кратного изб. TR, Be, Al 214, 215
8-Оксихинолин pH = 4,4-8,8, ацетатный буфер, нагр. > 70 °C, ф.с. 20-100 мг Высуш. на воздухе. ГФ: Th(C9H6ON)4C9H7ON 202, 218
Купферон Хол. р-р, СН3СООН, pH «4 Прокал. до ThO2 214
Таннин Винная к-та, NH4CI. Ос. хорошо фильтр. Прокал. до ThO2 219
Фенилфосфиновая к-та pH = 0,5 Высуш. при 140-180 °C ГФ: Th(C6H5PO3)2-3H2O Пригоден для опр. Th при ThO2:In2O3«10:l. Прокал. 800-1000 °C. ГФ: Th(HPO4)2 220
«-Аминосалициловая к-та pH = 4-5,6, CH3COONH4 (0,2 г), гор. р-р pear., промыв, кип. водой Ос. подсуш. и прокал. до ThO2 221
Стеариновая к-та pH не влияет на результаты, кип. < 0,2 г ThO2 Опр. Th в чистых р-рах. Прокал. до ThO2 222
Пирогаллол pH = 5-6, CH3COONH4 То же 222
Фенилуксусная к-та (то же с нафтилуксусной и фенилпропионовой) pH = 4,4—4,8, кип. CH3COONH4, для переосажд. р-ют в НС1 (6 н.) Th > 0,02 г ГФ: ThO2 Отд. TR, Ва, Mg, Mn, Al, Ti 229
о-Толуиловая к-та (5%) pH = 3—4, кип. Th> 10 мг ГФ: ThO2 Отд. от TR 230
7 -Оксикумарин-4-уксусная к-та pH = 2,5-3,2, кип. > 1 мг ГФ: ThO2 Отд. от TR цериевой подгруппы, Са, Со, Ni, Mn, А1 231
2,4-Дихлорфенок-сиуксусная к-та (1%) pH = 2,8, гор. р-р pear., отстаив. 5 мин, промыв, pear. (0,2%), ф. с. или ф. б. Высуш. при 105-110 °C ГФ: Th(C8H5O3Cl2)OH-2H2O или прокал. до ThO2. Отд. от TR при ThO2:Ln2O3 = 1:40; Co,Ni, Сг отделяют переосажд. 232
Коричная к-та (0,4%) pH = 2-2,6; кип. р-р, кип. pear., 100 °C в течение 20 мин, промыв, р-р (0,01 н. HNO3 + pear. 1 г• л ') > 0,3 мг ГФ: ThO2 Отд. от TR цериевой подгруппы при Th:TR = 1:2000, а также от Са, Sr, Ва, Mg, Zn, Cd, Со, Ni, Mn, Cu, Pb, Be 233
Бензойная к-та (2%) Анал. р-р, 10 г NH4CI, разб. до 100 м, pH = 2,2-2,6, нагр. до кип., 100 мл кип. р-ра pear., отстаив. 10 мин, ф.б. промыв, pear. (0,25%) < 100 мг ГФ: ThO2 Отд. от TR при ThO2:(Ln)2O3 = 1:160 234, 235
о-Хлорбензойная к-та pH = 2,8, кип., pear. (0,75 г в 75 мл), кип. 3 мин, отстаив. 30 мин, ф. б. < 150 мг ThO2 ГФ: ThO2 Эффективное разд. Th и TR, метод прост 236
См. также: осажд. орг. к-тами [223, 224, 225], с орг. основаниями [226], с комплексонами [227]. Условия прокаливания различных соединений тория [228].
Химические методы количественного анализа
513
Продолжение таблицы 5.1.9
Определяемое вещество, ион Реагент Условия осаждения Интервал содержаний Примечание Литература
nh; Тетрафенилборат натрия pH = 2, 100% изб. pear., ф. с., промыв. СП. Высуш. при 70 °C ГФ: NН4[В(С6115)4] 237, 238
NO; Динафто диметиламин (10% в 50% СН3СООН) Анал. р-р (150-200 мл) нагр. до кип., 2-5 мл H2SO4 (10%), 10 мл pear., охл., ф.с., промыв, хол. водой Высуш. при 110 °C ГФ: C22H19NHNO3 Мешают: NO;-, С1О3-, BrO3-, СЮ; -галогенид-ионы 239, 240
w-T о л илизомочевина, ацетат (0,75 н.) Анал. р-р (10 мл), 10 мл H2SO4 (2 н.), 20 мл pear., 30 мин в лед. воде, ф.с., промыв, разб. pear, и однократно лед. водой, ОХЛ. СП. < 100 мг NO3 Высуш. при 110-120 °C ГФ: [C7H7SC(NH)(NH2)]3- • H2SO4HNO3 / 0,0955 (на HNO3) Мешают: СГ (удаляют), ВГ, Г, СЮ;, МпО;, SCN-, гек-сацианоферрат(П) Не мешают: РО4", CN-, гек-сацианоферрат(П) 240, 241
Цинхонамин сульфат (насыщ.) Нейтр. р-р («0,1 г NO;, без СГ), СНзСООН (1 капля), нагр. до кип., теплый pear., отстаив. 12 час, промыв, pear., водой Ок. 100 мг NO; Высуш. при 100 °C ГФ: Ci9H24N2O HNO3 /=0,1753 (на HNO3) 240
А'-(4-Хлорбензил)-нафтилметиламин С1О4 предварительно осажд. хлоридом тетра-фенилфосфония 1-50 мг NO3 Ошибка определения <1%. Определение NO; в смесях с СЮ; (перхлорат осажд. хлоридом тетрафенилфос-фония) 242
Sb(III) 8-Оксихинолин ГФ: [Sb(C9H6ON)3] 243
Тионалид ГФ: [Sb(C12H10ONS)3] 244
Этилендиамин ГФ: [Cr(C2H8N2)3][SbS4]-H2O 245
BiJ+ Купферон (6%) 1 н. НС1 (или HNO3), хол. р-р, по каплям 5 мл pear, на 0,1 г Bi, промыв, р-ром pear. (0,1%) в 0,1-1,0 н. кислоте Прокал. при 700 °C, смачивают HNO3 и повторно прокалив. при 700 °C ГФ: Bi2O3 или высуш. ГФ: Bi(C6H5O2N2)3 246, 247
Хинальдин + KI H2SO4 (1 :9) Bi > 0,3 мг о.ф.: адкснз-нвщ 248
Меркаптобензотиазол ГФ: Bi(C7H4NS2)3 247, 251
Г ексаметилентетрамин ГФ: [(CH2)6N4I]3 252, 253
8-Оксихинолин pH = 4(6-11), тартрат 0,1-0,2 мкг ГФ: Bi(C9H6ON)3 H2O или Bi(C9H6ON)3 Отделение от Mg, Са, РЬ, (осажд. из гомогенной среды) 23, 250
Тиоацетамид Кисл. среда Осажд. Bi2S3; отделение от остальных элементов 24
Тионалид 2 н. HNO3, иод (титров.) ГФ: Bi(C12H10ONS)3 H2O Не меш. Со, Zn, Cd, TI, Mn, Al, Cr, Ca, Ba, Mg 490
514
Новый справочник химика и технолога
Продолжение таблицы 5.1.9
Определяемое вещество, ион Реагент Условия осаждения Интервал содержаний Примечание Литература
Bi3+ 1,2,3 Триоксибензол 0,1 н. HNO3 ГФ: BiO3C6H3 Мешает Sb; не мешают РЬ, Cu, Cd, Zn 246, 247, 249
V(IV) Бензоат аммония (0,37%) 0,6 н. НС1, кип. Прокал. при 800 °C ГФ: V2O5 Не меш.: Cu, Al, Mg, Mo(VI), W(VI), Ti(IV) Меш.: Fe, Cr 254
5,7-Дибром-8-оксихи-нолин (1%, в ацетоне) pH = 4—5 Высуш. 150-200 °C ГФ: VO(C9H4ONBr2)2 Меш. Al, Zn, Co, Ni, Mn 255
5,7-Дииод-8-оксихинолин (1%, в ацетоне) pH = 4-5 Высуш. 170-180 °C ГФ: VO(C9H4ONI2)2 Меш. аналогичные 255
2-Оксинафтил-1 -метиленэтиламин (2% в сп.) pH « 3, нагр., аскорбиновая к-та Высуш. при 110 °C ГФ: VO(C13H12ON)2 Меш.: Си, РЬ Не меш.: Fe 256
2-Оксинафтил альдоксим (2% в сп.) pH « 3, нагр., аскорбиновая к-та Высуш. при 110 °C ГФ: VO(CuH8O2N)2 Меш.: Ni, Cu, Ti Не меш.: Fe 256
Бензоил-и-нитро-ацетанилид pH = 4,0-5,5, CH3COONH4 (5%), нагр. Высуш. при ПО °C ГФ: УО(С15НпО^2)2 Не меш. Ag, Tl 257
Циклопентанон-2-карбоксианилид (0,3% водно-сп. р-р) pH = 5-6, CH3COONH4 (5%), нагр. Высуш. при 110 °C ГФ: VO(CI2H12O2N)2 Не мешают: Cu, Со, Ni, Al, Zn, Fe(III), Ce(III), Zr(IV); в присутствии Г-Hg 257
Таннин Слабокисл, р-р, (NH4)2C2O4 винная к-та Прокал. до V2O5 Отделение от Ti, Zr, Nb, Ta, W 596
V(V) Диантипирилфенилметан (1% В СП.) Слабокисл, р-р, без нагр. Высуш. при 105-110 °C ГФ: [(СцНцО^гСНСда-H4V6O17 258
о-Оксиацетофеноксим (2% в сп.) pH = 2,2-4,8, нагр. до 35-40 °C, изб. pear. (2,5-кратн.) Высуш. при НО °C ГФ: VO(OH)(C8H8O2N)2 Меш.: Cu, Pd, Ti Не меш.: Fe(III) в присутствии F", С2О<’, тартрата 259
8-Оксихинолин (4% в сп.) pH > 4,5, нагр., ацетатный буфер 5-100 мг V2O5 Высуш. при 120-140 °C ГФ: VO(OH)(C9H6NO)2 а также ГФ V2O3(C9H6ON)4 Прокал. при 800 °C ГФ V2O5 260- 262 1
Nb(V), Ta(V) А'-Фенилбензо-гидроксамовая к-та рН= 1,HF Ок. 0,1 мг Отд. от Al, Со, Cr, Fe, Ni, Sn(IV), V(IV) 263
Nb Галловая к-та ЭДТА Анализ нерж, сталей 26
Дибромгалловая к-та — — — 264
о-Оксифенилфлуоран НС1 (3 н.); 3,3'-диметок-ca-NJ^-дисалицилиден-бензидин 3-100 мкг Анализ силикатов 27
Химические методы количественного анализа
515
Продолжение таблицы 5.1.9
Определяемое вещество, ион Реагент Условия осаждения Интервал содержаний Примечание Литература
Nb 8-Оксихинолин а) pH = 6, тартрат аммония б) NH4OH (изб.), Н2О2 ГФ: (Nb2O5,Ta2O5) отд. от Та. ГФ: (Nb2O5) отд. А1 1, 202, 265, 267
Пирогаллол (1%) (или 5% в 1 н. НС1) Пиросульфатный плав, выщелачивают реагентом (100 мл), NH4OH до запаха, нагр. до кип., нейтр. НС1 по конго, добавл. НС1 до 1 н., кип. 40 мин, ф.б., промыв, р-ром pear. ГФ: (Nb2O5,Ta2O5) ' 266
so;- Бензидин pH = 2,5-3,0 1,5-30 мг Предварительная сорбция меш. ионов на катионите 1,268, 269
Se(IV, VI) Тиомочевина Кисл. р-р, нагр., без большого изб. pear. 0,2-2 мг ГФ: Se Те не мешает 270, 271
Молочный сахар pH = 10-11, кип., винная к-та ГФ: Se Быстрое выделение и высокая точность опр. 272
Т етраэтилтиурамдисуль-фид ГФ: Se Те не восст. 273
SeO32- 1 -Амидино-2-тиомоче-вина НС1 (3,5 н.), 40 °C ГФ: Se Мешает Те, Ag 274
Глюкоза Нейтр. или слабощел р-р, промыв. СП. Высуш. при 80-90 °C 275
Тиосемикарбазид Солянокисл, р-р; промыв. водой, сп., эфиром Высуш. в вакуум-эксикаторе 30 мин ГФ: Se 276
Диэтилдитиокарбамат натрия НС1 или H2SO4 (2,5-4 н.), кип. 15 мин. ГФ: Se 277
Меркаптобензимидазол Солянокисл, р-р, нагр. ГФ: Se Мешают: Ag, Pb, Cu, Hg, Au, Bi, Те 278
2,3-Диаминонафталин Na-ЭДТА, NaF, Na2C2O4 1-10 мг ГФ: Ci0H6N2Se. Высуш. при ПО °C Не мешают: Те, Fe, Cu, Zn, Al Мешают: Се, Ti 281
ТеО^- 1 -Амидино-2-тиомоче-вина НС1 (3,5 н.), 40 °C ГФ: Те Меш.: Se, Ag 274
Тиосемикарбазид Солянокисл, р-р; ос. обрабатывают смесью CS2 и сп. (1 : 1) ГФ: Те Возможно образ. TeS 276 279
(СбН5)48ЬС1 НС1 (4-5 н.) Высуш. при 110 °C ГФ: [(C6Hs)4Sb]2TeCl6 Не меш. Se. Меш. NO3 279
Тиомочевина, соль Рейнеке Кисл. р-р, тиомочевина, затем соль Рейнеке ГФ: [Te(SCN2H4)2]-•fCr(SCN)4(NH3)2] 280
Cr3+ 8-Оксихинолин pH = 5,7-6,0, осажд. из гомогенного р-ра, pH регулируется гидролизом мочевины 0,5-2 мг ГФ: Cr(C9H6ON)3 Не меш.: Са, Mg 30, 282
516
Новый справочник химика и технолога
Продолжение таблицы 5.1.9
Определяемое вещество, ион Реагент Условия осаждения Интервал содержаний Примечание Литература
Сг3+ 5,7-Дибром-8-оксихи-нолин pH = 4,5-8 Опр. Сг в морской воде 29
МоО^ А-Бензоилфенил-гидроксиламин (2-4% в сп.) Анал. р-р (300 мл), НС1 (10 н.) до конц. 0,01— 2,5 н., нагр. до 60 °C, pear, в изб. 0,03%, нагр. 30 мин при 60 °C, ф.с., промыв, теплой НС1 (0,02 н.), 10 мл воды 0,03-0,08 г Высуш. при 110-115 °C ГФ: (Мо02)(С1зН1о02М)2 283
Р-Нафтохинолин (2%) Слабокисл, р-р (по лакмусу), pear., ф. с., промыв. водой Прокал. при 400-450 °C ГФ: МоОз 284
2,2'-Дипиридил Отделение МоО": от Re Од 285, 286
Ванилиденбензидин (3% в 50% СН3СООН) Анал. р-р (20-80 мл), 100 °C, 5-30 мл pear., кип. 30 сек, отстаив. 3-5 час, ф. б., промыв, разб. р-ром pear (1:25) 8-400 мг молибдата Щ. М. Прокал при 400 °C ГФ: МоОз 287
4-Амино-4-хлор-дифенил pH = 1,8-2,8 Прокал. до МоОз 288
WOJ- а-Бензоиноксим H2SO4 (5%) осажд. при 5 °C < 200 мг Анализ стали. Прокал. при < 525 °C ГФ: WO3 289, 290, 299
Р-Нафтохинолин + таннин Опред. 10-35% W в сплаве W-Mo ГФ: WO3 291
Р-Нафтохинолин + хинин 1-5 мг ГФ: WO3 Не меш.: 10-кратный изб. Мо 292
Пирамидон Н2С2О4, НС1 (2 М), нагр. до кип., отстаив. 12 час, промыв, pear. (0,5% в 2% НС1) < 200 мг ГФ: WO3 Осажд. в присутствии равных количеств Мо 292, 293, 305, 314
Риванол (2%) W осажд. кисл. гидролизом, H2WO4 раств. в NH4OH, нейтр. до pH = = 2-3. Pear. (5 мл на каждые 2,5% W) Ок. 100 мг ГФ: WO3 Не меш.: большие количества Ti, Zr, V, Cr, Ni, Co, Fe, Zn, Ce, Be, Nd, Mn, Mo 292, 294
Сульфамидо-2,4-диаминоазобензол (кр. стрептоцид) 10-40 мг ГФ: WO3 Ос. загрязнен Mo 295
Акрихин (1%) НС1 (0,5-1 М). (Полуторные окислы отд. изб. щелочи). Ос. промыв, pear. Опр. больших количеств W в сульфидных рудах. ГФ: WO3 296
Метиленовый голубой (1%) Анал. р-р (15 мл), 15 мл HNO3 (конц.), выпар, до 15 мл, охл., добавл. 20 мл pear., отстаив. ночь, ф. б., промыв, pear. В присутствии больших количеств щ.м. добавл. таннин 38-159 мг ГФ: WO3 Нормальные и паравольфраматы осажд. на 100%, мета-вольфраматы на 99% 297
Химические методы количественного анализа
517
Продолжение таблицы 5.1.9
Определяемое вещество, ион Реагент Условия осаждения Интервал содержаний Примечание Литература
wo$- Вариалионовый синий (1% в 0,01 М СНзСООН) pH = 5. Анал. р-р (нейтр., 40-200 мг W), 10 мл СНзСООН (1%), разб. до 100 мл, 30 мл pear., отстаив. 15 мин, ф.б., промыв, р-ром (0,03% pear, в 0,03 М СНзСООН) 40-200 мг Прокал. при 600-700 °C ГФ: WO3 Не меш.: галогениды, SO; , NO;, SCN“, SiOj”, Щ. M., Cu, As(III), Co, Ni, Al, Zn, Mn, nh; 298
Алкалоиды (бруцин, хинин, стрихнин) 25-700 мг WO3 ГФ: WO3 300 301
Хинин, AsO: Осажд. вольфрамо-мышьяковой к-ты, отстаив. 12 час. 0,02-0,21 г WO3 V восст. до V(IV) действием NH2OH 311
Нитрон ГФ: WO3 302
Антипирин, таннин ГФ: WO3 303
6-Толухинальдин (или 8-толухинальдин) Осажд. из 4 М НС1 или 6 М H2SO4, нагр. до 80 °C ГФ: WO3; опр. в стали (1-19%W). Не меш.: Мо, Сг и др. компоненты стали 292, 304
Таннин 306, 307, 313
Бензидин (2% в 2,5% НС1) Анал. р-р (нейтр., 100 мл), 10 мл H2SO4 (0,1 н.), нагр. до кип., pear, в изб., ф. б., промыв, pear. 50-100 ГФ: WO3; прокал. 700-750 °C Меш.: РО;-Наиболее удобен из ряда изученных производных 1,31, 309
Бензидин, NaH2PO4 Осажд. фосфорновольфрамовой к-ты 310, 313
1,8-Диаминонафталин 3-102 мг WO3 ГФ: WO3; прокал. 720-850 °C 312
Цинхонин Гор. НС1(1 : 10) 100-500 мг Прокал. при 750-800 °C ГФ: WO3 Меш.:As, F-, Мо, Nb, Та, Pt, V, Fe(III) 33, 313, 314
ио*+ Изатин-Р-оксим (1% в 50% сп.) Тартратный р-р, CH3COONa, NH4SCN, отстаив., промыв, pear. (0,05%) 2-100 мг ГФ: [UO2(C8H5N2O2)2] 315, 316
Таннин (5%) Аустативный буфер, NH4C1 < 200 мг ГФ: U3O8 320
Н2С2О4 ИЛИ (NH4)2C2O4 U(IV), НС1 (2-3 н.) перемеш. 1 час, промыв. 100 мл Н2С2О4(1%) в 3 н. НС1 ГФ: U(C2O4)2, U3O8 317
Хинальдиновая к-та или натриевая соль (5%) Аналогично 8-оксихино-лину. Добавл. NH4C1 (5-7 г на 150 мл р-ра), pH = 6-7, кип., промыв, р-ром уротропина (5%) и NH4NO3 (5%) ГФ: U3O8 318, 319
СЮ; сю; Динафтодиметиламин (ди(нафтил-1 )метиламин) ГФ: (C10H7CH2)2NHHClO3; (CioH7CH2)2NHHC104
518
Новый справочник химика и технолога
Продолжение таблицы 5.1.9
Определяемый элемент Реагент Условия осаждения Интервал содержаний Примечание Литература
Мп2+ Т етрафенил фосфоний нитрат Мп2+ окисл. (NH4)2S2Og + Ag+ до MnO4, pear., отстаив. ночь, промыв, водой до удаления SO2 Высуш. в эксикаторе над H2SO4 (конц.). ГФ: [(С6Н5)4Р](МпО4) Не меш.: окислители 321, 322
5-Нитробарбитуровая к-та (2% в 50% сп.) Анал. р-р (нейтр., 25 мл), 0,5 мл СН3СООН (конц.), 15 мл сп., 25-30 мл pear., ф.с., промыв. 5 раз по 3 мл водой, сп. (10 раз по 2 мл), эфиром (5 раз по 1-2 мл, безводного) 0,1-0,2 г Высушив, в вакуум-эксикаторе. ГФ: (C4H2O5N3)2Mn-8H2O Fe3+ связывают сульфосалициловой к-той. Ошибка 0,3-10% 323, 324
а,а'-Дипиридил SCN-, винная к-та ГФ: [Mn(C10H8N2)2].(SCN)2 325
Пиридин SCN-, pear., ф.с. Высуш. в эксикаторе. ГФ: [Mn(C5H5N)2]-(SCN)2 326, 329, 331
8-Оксихинолин pH = 7-9, NH4OH, тартрат аммония ГФ: Mn(C9H6ON)2H2O или Мп3О4 327, 332
и-Окси-л/-нитро-фенилар-соновая к-та или о-нитро-фениларсоновая к-та СНзСООН ГФ: Mn[(C6H4O2N)AsO3H] Меш.: Fe(II), Pb(II) 328, 330
Re Антипирин; диантипи-рил-, диантипирилметил-и диантипирилфенилме-тан а) Осажд. ReO4; ок. 0,1 н. H2SO4, 70-80 °C, добавл. pear., отстаив. 1-2 час б) Осажд. ReCi2”, ReBr2” : ок. 2 н. НС1 или НВг, pear., отстаив. 2 час, промыв, нас. р-ром ОФ в 2 н. НО (НВг), водой 15-60 мг Высуш. при 110 °C ГФ: H(C26H30O2N4)ReO4 Не меш.: Щ.М., Щ.З.М., Zn, Al, Cd, Fe(II), СГ, SO2” и др. Высуш. при 100-110 °C ГФ: (C23H24N4O2H)ReBr6; (C26H30N4O2H)ReCl6 34, 333 34, 333
Ди-(нафтил-1 )-метиламин 597
Fe3+ Неокупферон ГФ: Ре20з 334
А-Бензоилфенил-гидроксиламин [А-(фенил)бензо-гидроксамовая к-та] ГФ: Fe2O3 335
а-Нитрозо-р-нафтол Слабокисл, среда. ГФ: Fe2O3 336, 337
8-Оксихинолин CH3COONH4 1-50 мг в 100 мл Высуш. при 120-125 °C ГФ: Fe(CqIl6NO)3 338- 340
5,7-Дибром-8-оксихино-лин 0,01 М НС1, винная к-та, 20% ацетона 0,4 мг в 100 мл Высуш. при 120-140 °C ГФ: Fe(C9H4Br2NO)3 1
Купферон < 10% H2SO4 или < 20% НС1 < 100 мг Высуш. при 98 °C ГФ: Fe(C6H3N2O4)3 или прокал. до Fe2O3 1
Пиридин (20%) pH « 4, NH4C1, нагр. Отделение от Mn, Zn, Ni, Со и Щ.З.М. Осажд. вместе Al, Ti, РО^” ГФ: Fe2O3 1,132, 341
Химические методы количественного анализа
519
Продолжение таблицы 5.1.9
Определяемый элемент Реагент Условия осаждения Интервал содержаний Примечание Литература
Со2+ Щавелевая к-та pH « 5, уксуснокисл, р-р, изб. pear., промыв, сп. (50%) Высуш. при 100-105 °C ГФ: СоС2О4-2Н2О 342, 343
Антраниловая к-та Анал. р-р (pH = 3,4-4,4), предварительно отд. Zn, Mn, Pb, Hg, Ni, Cd, Cu Микроопр. [345, 346] (0,1-4 мг), макроопр. [344] Высуш. при 105-110 °C ГФ: Co(NH2C6H4CO2)2 Прокал. >1000 °C, ГФ: Со3О4 347, 348
5-Бромантраниловая к-та Аналогично антраниловой к-те ГФ: Со(НН2СбН3ВгСО2)2 349, 350
3 -Амино-2-нафтойная к-та, натриевая соль (3%, в 0,06 М NaOH) Анал. р-р, нагр. до кип., 15 мл pear., ф.с., промыв, р-ром pear. (0,01%), сп. 30-40 мг Высуш. 30 мин при 130 °C ГФ: (H2NC]0H6CO2)2Co 351
Бензимидазол (1%, водный) Анал. р-р, 150 мл воды, 2 г K2SO4, 100% изб. ре-аг., NH4OH до pH = 10, перемеш. и выдерж. 30 мин при 100 °C. Ф.с., промыв, гор. водой до удаления СГ 0,01-0,2 г Высуш. при 105 °C. Меш.: осажд. при pH « 10 гидроксиды 352
5,6-Бензохинальдиновая к-та pH > 3,25 Высуш. при 110-115 °C ГФ: Со(С13Н8СОО)2-2Н2О; Высуш. при 150-155 °C ГФ: Со(С13Н8СОО)2 Меш.: Zn, Ni, Mn 353
1 -Нитрозо-2-нафтиламин (1% В СП.) а) СНзСООН, кип., небольшой изб. pear. б) Анал. р-р, Br2, NaOH, раств. в СНзСООН (без изб.), сразу добавл. pear. Макроопр. Микроопр. Высуш. 2 час при 110 °C ГФ: Co(C10H7ON2)3 Высуш. при 110 °C ГФ: Co(Ci0H7ON2)3 354 355
1 -Нитрозо-2-окси-З -нафтойная к-та Возможно осажд. в присутствии преобладающих количеств Ni Высуш. при 130 °C ГФ: Co(ChH6O4N)2- •(CnH7O4N)2 356
Изонитрозо димедон Микроопр. Высуш. < 260 °C 357
Изонитризотиокамфара (1%) Анал. р-р, CH3COONa, нагр. до кип., изб. pear., ф. с., промыв, гор. водой; р-ром NaOH. гор. р-ром НС1 и гор. водой. В присутствии Ni к р-ру после осажд. Со добавл. НС1 (2 н.) Высуш. при 105-110 °C ГФ: Co(C]0H14ONS)3 /= 0,0910 358
2-Нафтальдоксим (р-р В СП.) Высуш. при 110—130 °C ГФ: Со(СцН8ОЫ)2и Co(CnH8ON)3 359
Фенилтиогидантоиновая к-та Анал. р-р, 8 г лимонной к-ты, нейтр. NH4OH, нагр. до 35 °C, добавл. 0,7-1 г pear, (p-в в 30 мл сп.), нагр. до кип., фильтр, и промыв, р-ром цитрата аммония (0,5%) « 25 мг Прокал. при 1050 °C ГФ: Со3О4 или опр. в виде CoSO4. Не меш. Fe (до 1 г), а также As, U, V, Ti, W, Мо, Zn, Мп, Сг, Al, Mg, Са 360
520
Новый справочник химика и технолога
Продолжение таблицы 5.1.9
Определяемый элемент Реагент Условия осаждения Интервал содержаний Примечание Литература
Со2+ Диэтилдитиокарбаминат натрия (10%) pH » 6, тартрат натрия, нагр. до 40 °C, pear., ф. с., промыв, водой Высуш. при 90 °C. ГФ: [(C2H5)2NCS2]3Co 361
Т еноилтрифторацетон (р-р В СП.) pH = 4,0-7,8, цитраты. Оранж, крист, ос. непостоянного состава. Ос. переводят в CoSO4. При pH = 7 не меш. Fe, Cr, Th, Zr, U. Меш.: Cu, Ni, Мп 362
Ni2+ Гептоксим (1,2-цикло-гептандиона диоксим) pH = 3,4 (2,7-12,4) 0,5-5 мкг Си маскируют NH4SCN при pH = 3,5 363, 364
Николлокс (оксалами-доксим) pH = 7-8 (количественное осаждение). Начало осажд. при pH = 3,78±0,04 365
о-Г идроксиацето-фенок-сим pH - 4,8-8,5 Высуш. при 105 °C. Си осажд. при pH = 2-3 366
Резпропиофеноксим 7-30 мг 367
4-Метил- и 4-изопропил-ниоксимы pH = 3-7; Со и Си маек. CN” и SCN”; Al, Sb(III), Bi, Fe — тартратами Ос. высокой чистоты. Не меш.: СН3СООН, оксалаты, цитраты, сульфосалицилаты 368
Бис-биацетил-монооксимэтилендиамин Слабоаммиачный р-р; Со маек. CN”, Fe — тартратами, Си — ацетатами 369
Бис-биацетил-моноок-сим-о-фенилендиамин Те же 370
Антраниловая к-та Нейтр. и слабокисл, среда ГФ Ni(NH2C6H4CO2)2 376, 377
Бромантраниловая к-та Те же ГФ Ni(NH2C6H3BrCO2)2 378
Диациандиамидин ГФ Ni(C2H5ON4)2-2H2O 379
Диаминоглиоксим ГФ Ni(C2H5O2N4)2.2H2O 380
Нитроаминогуанидин ГФ NiO[HN C(NHNO2)-•NH-NH2]2 381
3-Гидрокси-1,3-дифенилтриазин pH = 4,4-7,0; ацетатный буф. Р-Р- Меш.: Cu, Pd и др. 382
р-Оксинафтойный альдегид pH = 7,2-7,5 Pd осажд. при pH = 2,5-3 383
р-Нитрозо-а-наф-тиламин ГФ: Ni(C10H7ON2)2 384
о-Фенантролин + SCN- См. опр. с пиридином и NH4SCN 385
а,а'-Дипиридил + SCN- См. опр. с пиридином и NH4SCN 386
В качестве осадителей предложены также: 1,2-циклопентандион [371]; 1,2-цикг метилэтилдиоксим [373], фенилдиоксим [374]; бензоилметилдиокс дисалицилалпропилендиамин [598, 599 юдекаидион [372]; метилдиоксим [373], им [375]; салицилаламин и
Ru(III, IV) Тиосалициламид Слабосолянокисл. р-р, pH = 4,5-5,5, Ru(III,IV) переходят в Ru(II) 6-12 мг Высуш. при 110-120 °C. ГФ: Ru(C7H6ONS)2 Ошибка ок. 0,01 мг Меш.: CN-; тартраты маек. Fe, Ni, Со, U; ЭДТА маек. Cu; F маек. Ti(IV). Не меш.: Ir, Au, Os и др. 36, 387
Химические методы количественного анализа
521
Продолжение таблицы 5.1.9
Определяемый элемент Реагент Условия осаждения Интервал содержаний Примечание Литература
Ru(III, IV) Бензидин (1%) Солянокисл, р-р, pH = = 1-2, CH3COONa, нагр. 22-56 мг ГФ: Ru, ошибка ± 0,4%. Высуш., прокал. в токе Нг 388
8-Оксихинолин (C6H5)3CH3AsI Меш.: Os, Pd 35
Rh(III) Формамидинсульфино-вая к-та (двуокись тиомочевины) НС1 (0,2 н.) 0,01-30 мг Восст. в токе Н2. ГФ: Rh Не меш.: Ni, Fe 389
Тионалид (р-р в лед. СН3СООН) Хлоридный р-р, pH = 3; 4-кратный изб. pear., осажд. при кип. 1-20 мг Высуш. при 120 °C ГФ: Rh(Ci2H10ONS)3 390
2-Меркаптобен-зими-дазол а) СНзСООН, pH = 4,27, при кип. б) Азотнокисл, р-р 0,85-1,7 мг 0,42-0,85 мг ГФ: Rh(C7H5N2S)3 Ошибка ± 0,01-0,04 мг ГФ: Rh(C7H6N2S)3Cl3 Ошибка 0,01-0,02 мг 391
Тиосалицил амид Буф. р-р pH = 4,5-5,5 4-12 мг ГФ: Rh(C7H6ONS)3 Не меш.: Ir, Cu, Fe, Со, Ni, Zn 387
и-Аминофенилдитиокарбамат натрия или аммония (1%) Нейтр. хлоридн. р-р, нагр. до кип., 4-кратный изб. pear., отстаив. 1 час при 100 °C, добавл. р-р мет. фиол. (0,002 г • мл'1) в количестве, равном Rh, нагр. 15 мин. « 10 мг Прокал. в токе Н2 ГФ: Rh 392
1 -Нитрозо-2-нафтол pH = 4,85-5,6, изб. pear., гор. р-р Прокал. при 880 °C ГФ: Rh2O3 393
2-Меркаптобензоксазол (р-р в лед. СН3СООН) NH4C1, при кип. 1 час, (см. опр. с меркаптобенз-тиазолом) 5-20 мг ГФ: Rh 394
Pd(II) Диоксимы см. табл. 5.1.11.1.Б
Монооксимы см. табл. 5.1.11.1.А
1,2,3-Бензотриазол а) pH = 2-5,3 (ацетатный буфер), ЭДТА б) Сильнокисл, р-р Высуш. при 110-150 °C ГФ: Pd(C6H4N3)2Cl2 Меш. Au, Pt ГФ: 2C6H4N3 PdCl2 396, 397 395
2(2-Оксифенил)-бензок-сазол Водно-сп. р-р, 20 °C, промыв. СП. Высуш. при 110 °C ГФ: Pd(Ci3H8NO2)2 Не меш.: Fe, Cu, Ni, NO3, SO4', ПМ. Меш.: Аи 398
А'-Фенил-А'-фени-лазогидроксиламин рН = 1,6-8, кип. Отд. от Си при pH = 2-2,5 ГФ: (C12H10N3O)2Pd 399
Висмутиол I pH = 3,5-8,5 Отд. от: Ag, Pb, Hg (pH = 6-8, KI); Au, Ir, Os (pH = 6-7, Na2S2O3); Bi, Sn, Fe(III), Cu, Pb, ПМ (pH = 3,5-8,5, винная и лимонная к-ты) 400
Висмутиол II pH = 6-8 10-18 мг ГФ: Pd(C8H5N2S3)2 Высуш. при 110 °C. Меш.: Sn и СЬГ 400
522
Новый справочник химика и технолога
Продолжение таблицы 5.1.9
Определяемый элемент Реагент Условия осаждения Интервал содержаний Примечание Литература
Pd(II) 2-Меркаптобензимидазол а) pH = 4-7 (ацетатный буфер), винная и лимонная к-ты, ЭДТА-Na, KI б) Сильнокисл, среда 7-13 мг Высуш. при 110 °C ГФ: (C7H5N2S)2Pd Селективный метод опр. Pd Меш.: Pt ГФ: 4C7H6N2-PdCl2 391 391
2-Меркаптометил-бензимидазол (0,5% в сп.) CN-; осажд. pear. Pt(IV), Ag, Hg, Cu. Затем разрушают комплекс Pd с CN-и осажд. Pd pear. ГФ: (C8H7N2S)2Pd 401
2-Меркаптобензотиазол pH = 5,6-10,2 ЭДТА, винная и лимонная к-ты, KI и Na2S2O3 Высуш. при 110 °C ГФ: Pd(C7H4NS2)2 Меш.: Pt(IV), Sn(II), CN- 400
Бензимидазол-1 -карбоновая к-та pH =1,6-2,5 ГФ: Pd(C8H5O2N2)2 Не меш.: 10-кратные количества Ni, Со, Zn, Cd, Pb, Mn, As(V), Ca, Ba, Mg, Ag 403
2-Меркаптобензоксазол pH = 4-10 ГФ: Pd(C7H4NOS)2 H2O He меш.: Pb, Cu, Co, Ni, Fe(III), Al, Ti, Th, Zr, UO^+ , трехвалентный As, Sb, Bi, a также Mo(VI), V(V), Pt(IV) 402
а-Пиколиновая к-та рН= 3-7. Анал. р-р нейтр. NH4OH, добавл. ацетатный буфер, маек, винной к-той, ЭДТА, затем добавл. pear. ГФ: Pd(C6H4NO2)2 Ос. устойчив до 380 °C. He меш.: Cu, Ca, Pb, Sr, Fe(III), Cr(III) 408
р-Аминопиколиновая к-та (2-карбокси-З-амино-пиридин) 1 h.HCI, pH = 12,5 6-25 мг Высуш. < 200 °C ГФ: Pd(C6H7N2O2)2 Реагент селективен 404
8 -Меркаптохинолин, натриевая соль 1-1,5 h.HCI Промыв, водным р-ром метанола (1 : 1) 2-100 мг Высуш. при 120 °C Маск.: тиомочевина - Au, Hg, Os, Sb (pH < 1-2); Bi, Ru, Pb(IV) (pH < 2-4); Cu (pH < 6-7) 405
4-Иодоизонитрозоаце-танилид рН= 1-3 Селективный pear. Onp. Pd в электролитических ваннах (Ni, Pt, Au, Cr, Li) 406
8-Оксихинолин а) СНзСООН, CH3COONH4 б) Слабосолянокисл. р-р < 50 мг Pd на 100 мл 6-25 мг Прокал. до Pd Высуш. при 120-140 °C ГФ: Pd(C9H6NO)2 407
Хинальдиновая к-та pH = 3-7, гор. р-р, NH4CI и винная к-та ГФ: Pd(C10H6NO2)2 Устойчива до 353 °C Меш.: Си, Аи 408
Пиридин а) pH = 3-6, pear., KI (0,2 М), нагр. 3-5 мин при 50-60 °C. б) SCN" в) С г. О? 1-10 мг 10-200 мг 10-200 мг Высуш. при 110 °C ГФ: Pd(C5H5N)2I2 Высуш. в вакуум-эксика-торе ГФ: [(C5H5N)2(SCN)2Pd ГФ: [(C5H5N)4Pd][Cr2O7] Не меш.: Ir, Rh, Au, Ni 37 410 435 436
Химические методы количественного анализа
523
Продолжение таблицы 5.1.9
Определяемый элемент Реагент Условия осаждения Интервал содержаний Примечание Литература
Pd(II) Хинолин SCN 10-200 мг ГФ: [(C9H7N)2(SCN)2Pd Высуш. в вакуум-эксикаторе 435
2,3-Пиридиндикарбоновая (хинолиновая) к-та pH = 2,1; 0,25 МНС1 или HNO3 Высуш. при 110 °C ГФ: Pd(C7H4NO4)2 409
а, 0-Диоксимидоацетоацетанилид ГФ: (Ci0H9ClN3O3)2Pd 411
а,0-Диоксимино- ацетоацет-о-хлоранилид Желт. ос. при р Н О,4-10,1 ГФ: (Ci0H9ClN3O3)2Pd Меш.: NO, Не меш.: Fe, Cu, Ni 440
З-Окси-1 -(и-толил)-З-фенилтриазин pH = 2,0-3,0, нагр. 1 час при 100 °C 10-20 мг Ос. может служить ГФ 413
Производные окситриа-зена Pear, хорошо p-мы в воде ® 12 мг Избирательное ос. в присутствии преобладающих количеств Pt, Ni, Со, Fe. Au и Ag восст. до металлов 412- 415
Диаллилдитио-карбамид-гидразин (дальцин) (р-р В СП.) Цитратный буфер, pH = 3,1-4,5, промыв, гор. водой,ацетоном 30-50 мг Высуш. при 105 °C ГФ: (C8H12N4S2)Pd H2O Не меш.: ПМ, Ni, Zn. Меш.: Cu, Hg, Ag 416, 417
и-Этилсульфонил-бензальдегидтио-семикарбазон 0,4 М НС1, гор. р-р Высуш. при 110-120 °C ГФ: (C10H12N3O2S2)2Pd 418
Диэтилдитиофосфорная к-та pH = 5, NaHSO3 (маек. Pt); pH = 9, ЭДТА и тартраты (маек. Fe, Си, РЬ) Высуш. при 105 °C ГФ: [(C2H5O)2PS2]2Pd 419
а-Диаминодифеновая к-та 3-15 мг Высуш. при 100-105 °C /= 0,2367 Реагент не адсорбируется на ос., избирателен. Не меш.: Pt, Ni, Со, Fe, Au. Опр. Pd в Pt-Pd концентрате 420
Тиовиолуровая к-та pH = 0,3-4,0, маек. ЭДТА, лимонная и винная к-та ~ 10 мг ГФ: Pd(C4H2N3O3S)2 421
1 -(2-Пиридилазо)-2-нафтол (ПАН) Осажд. в широком интервале кислотности. В НС1 (1 н.) — только Pd Высуш. при 115 °C /= 0,2500 422
0-Окси-а-нафтойный альдегид рН= 2,5-3 ® 5 мг Не меш.: Ni и Си 423
3,4-Бензо-1,2,5-селенодиазол (пиаселе-нол), р-р в метаноле Слабокисл, р-р, кип. Промыв, водой, метанолом Полумикро-опр. Высуш. при 105 °C ГФ: (C6H4N2Se)2PdCl2 Не меш. соизмеримые количества Pt, Ni, Со, Pb 424
и-Роданилин pH = 2,5. В присутствии посторонних эл-тов — длительное отстаив. 15-30 мг Метод дает точные и воспроизводимые результаты. Опр. Pd в сплаве Pd-Cu-Fe 447
Салицилгидразид Винная к-та 2,3 мг Не меш. преобладающие количества Cu, Ni, Fe(III), Ti, Rh, Ru, Pt, Au, Co, Hg 426
Т иосалициламид Кисл. р-р ГФ: Pd(C7H7ONS)4Cl2 Отделение от Rh и Ru и небольших количеств Pt и Au 427
524
Новый справочник химика и технолога
Продолжение таблицы 5.1.9
Определяемый элемент Реагент Условия осаждения Интервал содержаний Примечание Литература
Pd(II) 1,10-Фенантролин (0,5% водный р-р) НС1 (5-10% по объему) 0,2-2,0 мг Ос. устойчив до 389 °C. Высуш. при 110 °C ГФ: Pd(C12H8N2)Cl2 Не меш.: Pt. Возможно прокал. до Pd. Не меш.: Rh 428, 429
Формамидинсульфино-вая к-та (диоксид тиомочевины) (нас. р-р) Слабокисл, р-р, нагр. до 40 °C, pear, до превращения возрастания окраски. Добавл. HgCl2 (5%) до прекращения осажд. черн. ос. Ф. б., промыв, водой (подкисл. НС1) Прокал. до Pd в токе Н2 и охл. в токе СО2. Не меш.: Rh, Ir, Ru. Меш.: Pt, Au, Se, Те (осажд. количественно) 427, 430
5-Метил-8-оксихинолин Гор. р-р, изб. ацетата. Осажд. оранж. ос. Высуш. при 105 °C ГФ: Pd(C10H8NO)20,5H2O 431
1,3-Диметил-4-имино-5-оксиминоаллоксан pH = 1-3 Кр. хлорокомплекс Ос. хорошо фильтр. Высуш. при 100-110 °C 432
а, Р-Бис-(оксимино)- ацетоацет-о-толуидид pH = 0,1-10; желт. ос. Меш.: ПМ 433
л-Аминосалициловая к-та pH = 3,7-4,2 Отд. от Pt, Au при /° < 45 °C 434
«-Аминоацетофенон Желт. ос. Высуш. при 80 °C ГФ: (NH2C6H4COCH3)2PdCl2 или прокал. до Pd 437, 438
Производные ароматических аминокислот pH = 2-4,5, желт, или оранж. ос. Не меш.: Cu, Ni Меш. Fe(III) 439
Г идразид-.м-нитро-бензойной к-ты Кислые NO., СГ или SO*~ р-ры Прокал. до Pd. Меш.: только Аи 450
Этилендиамин [I lgl4]2 . Ос. хорошо фильтр. Высуш. при 115 °C ГФ: [Pd(C2H4N2H4)2][HgI4] Меш. многие элементы 428
Ксантогенаты натрия 0,5% кисл. (НС1, HNO3 или H2SO4). Ос. ярко- желт. ОФ: Pd(ROCSS)2 Отд. от Cu, Ni, Fe, соосаждения нет. Отд. Pd от Ir, Rh 429, 441 442
Диметил- и диэтилди-тиофосфорные к-ты Ос. желто-оранж., крист. Ос. могут служить ГФ: Pd[(C2H5O)2PS2]2 419
Оксимидобензо-тетроновая к-та pH = 5,1; 0,75 н. НС1. Ос. кор-кр. Ок. 10 мг ГФ: Pd(C9H4NO4)2 Не меш.: ПМ, Fe, Ni, Со, Ag, Au, оксалат-, фторид-, ацетеат-, тартрат-, цитрат-и фосфат-ионы 443
Бензидин Разб. р-р НО, ос. желт. Ru, Rh, Pt в этих условиях не pear. 444
Опр. также с 6-нитрохинолином и др. замещенными хинолинами [445,446], тиоцианатанилином [447], виолуровой к-той [448], тиокислотами [449], аскорбиновой к-той [451], фенилгидразин о-гидроксиацетофеноном [452], тиомочевиной [453], «-аминосалициловой к-той [454], 1-фенилтетразолин-5-тионом [455], гид раз и н-w-бензойной к-той [456].
Os(IV) Бутил идендиантипирин 0,1-0,5МНС1 Высуш. при 100-110 °C. Ошибка 3% 41,42
Химические методы количественного анализа
525
Продолжение таблицы 5.1.9
Определяемый элемент Реагент Условия осаждения Интервал содержаний Примечание Литература
Os(IV) Соли: Тетрафенилфосфония (1), Бензилтрифенилфосфо-ния (2), тетрафенил арсония (3) Свежеприготовленные р-ры pear. Р-ры НС1 — для 2 и 3. Р-ры НВт — для 1. 1-15 мг Высуш. при 110 °C Os осажд. в виде соли [OsCl6]2- или [OsBr6]2“ с органическим катионом 43
Опр. также с pear.: тиооксин [458], замещенные тиазолы [459], тиомочевина и ее производные [428, 459].
1г(Ш, IV) Бензидин Солянокисл, р-р, доб. CH3COONa 22-55 мг Ошибка опр. ±0,5% 444
Бензилтрифенилфосфо-ния хлорид 0,2-1 М НС1, отстаив. 1 час Высуш. при 110 °C 43
4,4'-Бутилиденди-антипирин 0,1-0,5 н. НС1, р-р pear, в СН3СООН Высуш. при 110 °C Меш.: Ru, Rh, частично Pd, Pt 41
Pt(IV) Т етрафенил арсония бромид (0,02 М) Гор. р-р, НВг (0,2 М), pear. Охл., ф.с., промыв. НВг и pear. 5-20 мг Высуш. при 105-110 °C ГФ: [(C6H5)4As]PtBr6 460, 461
Диметилфенилбензи-ламмония хлорид НВг (на 10 мг Pt 4 мл к-ты). Конц. НВг < 30%. Разб. до 100 мл, pear. (5 мл + 1 мл на каждые 10 мг Pt). Отстаив. 3 час, ф. с., промыв, pear. (1%) 1-10 мг Высуш. при 80 °C ГФ: (C15H18N)2PtBr6 Не меш.: Cu, Fe, Ni, Cr 462
Фенилтиосемикарбазид (50% р-р в сп.) pH = 6-7, 150 мл р-ра, нагр. при 100 °C 30 мин. 2-15 мг Прокал. при 900 °C ГФ: Pt Меш.: Au, Pd 463
л/-Меркаптобензотиазол а) pH = 2-2,5 (СН3СООН) б) pH = 10,3, кип. 1,5 час в) НС1 (0,1-12 М) 6-7-кратн. изб. pear., нагр. 10-15 мин г) 0,1-0,35 М НС1, нагр. до кип. 1,3-2,6 мг 3-150 мг ОФ: Pt(C7H4S2N)4 Прокал. до Pt ОФ: Pt(C7H4S2N)2H2O ОФ: Pt(C7H5S2N)(C7H4S2N)2- •С12 Прокал. до Pt 457 44
jw-Меркаптоацет-амидофенол Pt(IV) восст. до Pt(II) Ок. 20 мг ГФ: Pt(C8H8NSO2)2 Ошибка ± 0,07 мг Меш.: Au, Pd, Ag 464
[(CH3)3N(CH2)6N- •(СН3)3]С12 (5% водный р-р) Отстаив. 8 час 8-40 мг ГФ: [(CH3)3(CH2)6N(CH3)3]. •[PtCl6] 466
[Cu(NH2CH2CH2NH2)2]- •(NO3)2-3H2O (крист.) Анал. р-р (> 1 мг • мл 1 Pt) KI (10%), pear, (крист.) 1-21 мг Высуш. 10 мин в вакуум-эксикаторе ГФ: [Cu(NH2CH2CH2NH2)2]- •[Ptl6] 465
Тиомочевина (1) и соль Рейнеке (2) Кисл. р-р (0,8-5 н. НС1 или 1-2,33 н. HNO3) Pt(IV) восст. до Pt(II) при действ, pear. (1). Добавл. pear. (2) < 75 мг ГФ: [Pt(NH2CSNH2)4]-•[Cr(SCN)4(NH3)2]2 467
Опр. также с pear.: производные тиооксина [468, 469], производные диэтилдитиофосфорной к-ты [470], фенотиазин [471], тиоформамид [472], тиоуксусная к-та и др. [473, 474], а-фурилдиоксим [475].
526
Новый справочник химика и технолога
Таблица 5.1.10
Стехиометрические факторы (факторы пересчета)
Гравиметрическая форма Искомое соединение Фактор пересчета
Ag Ag3AsO4 1,4292
AgBr 1,7408
AgCN 1,2412
AgCl 1,3287
Ag2CrO4 1,5377
Agl 2,1764
AgNO3 1,5748
Ag2O 1,0742
Ag3PO4 1,2935
Ag4P2O4 1,4031
Ag2S 1,1486
AgSCN 1,5384
Ag2SO4 1,4452
AgVO3 1,9172
Ag3VO4 1,3552
CN 0,24118
KCN 0,60362
AgBr Ag 0,57445
AgCl 0,76326
Agl 1,2503
As 0,13296
Br 0,42555
BrO3 0,68114
HBr 0,43091
KBr 0,63375
KBrO3 0,88935
NH4Br 0,52161
NaBr 0,54797
Ag2C2O4 (Оксалат) Ag 0,71025
AgCl 0,94368
Ag(C3H2ONS2) (Роданинат) Ag 0,44937
AgCl 0,59707
Ag(C6H4ON3) (Бензотриазолат) Ag 0,47734
AgCl 0,63423
Ag(C6H4ONCS2) (Меркаптобен-зотриазолат) Ag 0,39354
AgCl 0,52288
Ag(C12H10ONS) (Тионалидат) Ag 0,33279
AgCl 0,44217
AgCl Ag 0,75263
Ag3AsO4 1,0757
AgBr 1,3102
AgCN 0,93416
Ag2CrO4 1,1573
Agl 1,6380
AgNO3 1,1852
Ag3PO4 0,97350
Ag4P2O7 1,0560
Ag2S 0,86449
AgSCN 1,1579
Ag2SO4 1,0877
As 0,17420
BaCl2 0,72652
Cl 0,24737
C1O3 0,58224
C1O4 0,69387
HC1 0,25440
HC1O4 0,70090
Продолжение таблицы 5.1.10
Гравиметрическая форма Искомое соединение Фактор пересчета
AgCl KC1 0,52015
KC1O3 0,85503
KC1O4 0,96665
NH4C1 0,37323
NaCl 0,40777
NaClO3 . 0,74264
NaClO4 0,85427
P2O6 0,27549
Ag2CrO4 Ag 0,65033
AgCl 0,86407
CrO4 0,34967
Agl Ag 0,45947
AgCl 0,61049
AgBr 0,79985
HI 0,54482
I 0,54053
KI 0,70706
nh4i 0,61736
Nal 0,63845
Ag3PO4 Ag 0,77312
PO4 0,22688
P2O5 0,16955
Ag2S Ag 0,87061
AgCl 1,1568
S 0,12939
AgSCN Ag 0,65002
AgCl 0,86366
BaSO4 1,4065
KSCN 0,58558
nh4scn 0,45868
SCN 0,34998
AgVO3 Ag 0,52159
V 0,24634
v205 0,43973
Ag3VO4 Ag 0,73791
V 0,11617
V2O5 0,20737
A1(C6H5O2N2)3 (Купферонат) Al 0,061550
ai2o3 0,11630
A1(C9H6ON)3 (Оксихинолинат) Al 0,058722
A12O3 0,11096
Al(C9H4ONBr2)3 (Дибромоксихи-нолят) Al 0,028921
A12O3 0,054647
A12O3 Al 0,52923
A1C13 2,6157
A1C13 • 6H2O 4,7362
A1F3 1,6473
A1(NO3)3 4,1782
A1(NO3)3 9H2O 7,3587
ai2o3h2o 1,1767
A12O3- 2H2O 1,3534
A1(OH)3 1,5301
A1PO4 2,3922
A12(SO4)3 3,3558
A12(SO4)3 • 18H2O 6,5363
KA1(SO4)2 • 12H2O 9,3056
(NH4)A1(SO4)2 • 12H2O 8,8923
A1PO4 Al 0,22123
Химические методы количественного анализа
527
Продолжение таблицы 5.1.10
Гравиметрическая форма Искомое соединение Фактор пересчета
А1РО4 А1С1з 1,0934
A1(NO3)3 1,7466
А12О3 0,41802
A12(SO4)3 1,4028
РО4 0,77881
Р2О5 0,58198
Ag3AsO4 As 0,16195
As2O3 0,21384
As2S3 0,26594
Mg2As2O7 0,33560
AS2O5 As 0,65190
As2S3 As 0,60898
Ag3AsO4 3,7603
AsCl3 1,4737
AsH3 0,63353
AsO3 0,99920
AsO4 1,1293
As2O3 0,80409
AS2O5 0,93416
As2O7 1,0642
AS2S5 1,2607
As2Se3 1,5718
H3AsO4 1,1539
Mg2As2O7 1,2619
Mg(NH4)AsO4 • 6H2O 2,3524
AS2S5 As 0,48306
AsO3 0,79258
As2O3 0,63782
As2O5 0,74100
As2S3 0,79322
Mg2As2O7 1,0010
BiAsO4 As 0,21531
As2O3 0,28430
Mg2As2O7 As 0,48257
Ag3AsO4 2,9798
AsCl3 1,1678
AsH3 0,50203
AsO3 0,79179
AsO4 0,89487
As2O3 0,63718
AS2O5 0,74026
AS2O7 0,84333
As2S3 0,79243
AS2S5 0,99900
As2Se3 1,2456
H3AsO4 0,91435
Mg(NH4)AsO4 • 6H2O 1,8641
Mg(NH4)AsO4 • 6H2O As 0,25887
As2O3 0,34181
As2S3 0,42509
]\4g2As2O7 0,53644
Au AuBr3 2,2170
AuCl 1,1800
AuCN 1,1321
AuCl3 1,5399
Au2(SO4)3 1,7315
Au2(SO4)3 • H2O 1,7772
НАиСЦ 1,7251
KAu(CN)4 1,7268
Продолжение таблицы 5.1.10
Гравиметрическая форма Искомое соединение Фактор пересчета
Au KAu(CN)4 Н2О 1,8182
В2О3 В 0,31074
во2 1,2298
во3 1,6893
В4О7 1,1149
нво2 1,2588
н3во3 1,7760
kbf4 3,6163
Na2B4o7 1,4450
Na2B4O7 • ЮН2О 2,7386
Ba[BeF4] Ва 0,61770
ВаО 0,68966
BaSO4 1,0497
ВаСО3 Ва 0,69595
ВаСгО4 1,2837
ВаО 0,77702
BaSO4 1,1827
ВаС2О4 (Оксалат) Ва 0,60946
ВаС2О4 • Н2О (Оксалат) 1,0800
ВаСгО4 1,1242
BaSO4 1,0357
ВаС2О4 • Н2О (Оксалат) Ва 0,56434
ВаСгО4 1,0410
BaSO4 0,95900
ВаСгО4 Ва 0,54213
Ba[BeF4] 0,87765
ВаСО3 0,77898
ВаС2О4 (Оксалат) 0,88953
ВаС2О4 • Н2О 0,96065
ВаС12 0,82200
ВаС12 2Н2О 0,96424
Ва(Ю3)2 1,9228
Ba(NO3)2 1,0316
ВаО 0,60528
Ва(ОН)2 0,67640
Ва(ОН)2 • 8Н2О 1,2452
BaSO4 0,92126
Ва(Ю3)2 Ва 0,28195
ВаСгО 0,52007
BaSO4 0,47912
BaSO4 Ва 0,58847
Ba[BeF4] 0,95266
ВаСО3 0,84556
ВаС2О4 (Оксалат) 0,96556
ВаС2О4 • Н2О 1,0428
ВаС12 0,89225
ВаС12 • 2Н2О 1,0467
ВаСгО4 1,0855
Ва(Ю3)2 2,0871
Ba(NO3)2 1,1198
ВаО 0,65701
Ва(ОН)2 0,73421
Ва(ОН)2 • 8Н2О 1,3516
BaS2O3 • Н2О Ва 0,51350
BaSO4 0,87260
ВеО Be 0,36033
ВеСО3 2,7595
ВеСО3 • 4Н2О 5,6406
ВеС12 3,1954
528
Новый справочник химика и технолога
Продолжение таблицы 5.1.10
Гравиметрическая форма Искомое соединение Фактор пересчета
ВеО ВеС12 • 4Н2О 6,0766
BeF2 1,8795
Be(NH4)PO4 4,8788
Be(NH4)PO4 • 6Н2О 9,2004
Be(NO3)2 5,3184
Be(NO3)2 • 4H2O 8,1995
Be2P 2O7 3,8375
BeSO4 4,2007
BeSO4-4H2O 7,0816
Ве2Р 2О7 Be 0,093897
BeCO3 0,71909
BeCO3 • 4H2O 1,4699
BeCl2 0,83268
BeCl2 • 4H2O 1,5829
BeF2 0,48978
Be(NH4)PO4 1,2713
Be(NH4)PO4 • 6H2O 2,3975
Be(NO3)2 1,3859
Be(NO3)2 • 4H2O 2,1367
BeO 0,26058
BeSO4 1,0946
BeSO4 • 4H2O 1,8454
Bi Bi 2O3 1,1148
BiPO4 1,4544
BiAsO4 As 0,21531
AsO4 0,39927
Bi 0,60073
B12O3 0,66971
BiPO4 0,87372
Bi(C6H3O3) (Пирогаллат) Bi 0,62935
Bi2O3 0,70162
BiPO4 0,91534
Bi(C9H6ON)3 (Оксихинолят) Bi 0,32582
BiO3 0,36323
BiPO4 0,47388
Bi(C9HeON)3 • •Н2О (Оксихинолят) Bi 0,31692
Bi2O3 0,35331
BiPO4 0,46093
Bi(C12H10ONS)3 • •Н2О (Тионалид ат) Bi 0,23862
Bi203 0,26602
BiPO4 0,34705
Bi[Cr(SCN)6] Bi 0,34289
Bi2O3 0,38227
BiPO4 0,49871
Bi203 Bi 0,89700
BiAsO4 1,4932
BiCl3 1,3535
BiF3 1,1416
Bi(IO3)3 3,1491
Bi(NO3)3 1,6954
Bi(NO3)3 • 5H2O 2,0820
BiOBr 1,3087
BiOCl 1,1179
BiOI 1,5103
Bi(OH)3 1,1160
BiPO4 1,3046
Bi2S3 1,1034
Bi2(SeO3)3 1,7143
KBiI4 3,2435
Продолжение таблицы 5.1.10
Гравиметрическая форма Искомое соединение Фактор пересчета
BiOBr Bi 0,68543
Bi2O3 0,76413
BiPO4 0,99690
BiOCl Bi 0,80243
Bi203 0,89457
BiPO4 - 1,1671
BiOI Bi 0,59390
Bi2O3 0,66210
BiPO4 0,86379
BiPO4 Bi 0,68756
BiAsO4 1,1445
BiCl3 1,0375
BiF3 0,87507
Bi(IO3)3 2,4138
Bi(NO3)3 1,2995
Bi(NO3)3 • 5H2O 1,5959
Bi2O3 0,76651
Bi(OH)3 0,85540
BiOBr 1,0031
BiOCl 0,85685
BiOI 1,1577
Bi2S3 0,84576
Bi2(SeO3)3 1,3141
KBiI4 2,4862
PO4 0,31244
p2o5 0,23349
Bi2S3 Bi 0,81294
Bi2O3 0,90630
BiPO4 1,1824
CO2 BaCO3 4,4846
BaO 3,4846
C 0,27291
CO3 1,3635
C2O4 1,0000
CaCO3 2,2742
CaO 1,2742
K2CO3 3,1404
KHCO3 2,2749
K2O 2,1404
Li2CO3 1,6789
Li2O 0,67892
MgCO3 1,9161
MgO 0,91613
MnCO3 2,6118
MnO 1,6119
Na2CO3 2,4083
NaHCO3 1,9088
Na2O 1,4083
(NH4)2co3 2,1833
SrCO3 3,3546
Ca2As2O7 As2O7 0,76560
Ca 0,23440
CaCO3 0,58536
CaC2O4 (Оксалат) 0,74917
CaC2O4 * H2O 0,85455
CaO 0,32797
CaCO3 CO2 0,43971
Ca 0,40044
Ca2As2O7 1,7084
Химические методы количественного анализа
529
Продолжение таблицы 5.1.10
Гравиметрическая форма Искомое соединение Фактор пересчета
СаСО3 СаС2 0,64044
СаС2О4 (Оксалат) 1,2798
СаС2О4 • Н2О 1,4599
CaCN2 0,80035
СаС12 1,1089
СаС12 • 6Н2О 2,1889
CaF2 0,78010
Са(Ю3)2 3,8955
СаМоО4 1,9985
Ca(NO3)2 1,6395
Ca(NO3)2 • 4Н2О 0,23595
СаО 0,56029
Са3(РО4)2 1,0330
Са(Н2РО4)2 2,3385
СаНРО4 • 2Н2О 1,7195
CaSO4 1,3603
CaSO4 • 2Н2О 1,7203
CaWO4 2,8774
СаС2О4 (Оксалат) Са 0,31288
СаСО3 0,78134
СаС2О4 • Н2О 1,1407
СаО 0,43778
CaSO4 1,0628
СаС2О4 • Н20 (Оксалат) СО2 0,60240
с2о4 0,60240
Са 0,27430
C3.2AS2O7 1,1702
СаС2 0,43869
CaCN2 0,54823
СаСО3 0,68498
СаС2О4 0,87668
СаС12 0,75958
СаС12 • 6Н2О 1,4994
CaF2 0,53436
Са(Ю3)2 2,6684
СаМоО4 1,3689
Ca(NO3)2 1,1230
Ca(NO3)2 • 4Н2О 1,6162
СаО 0,38379
Са3(РО4)2 0,70761
Са(Н2РО4)2 1,6018
СаНРО4 0,93117
CaSO4 0,93177
CaSO4 • 2Н2О 1,1783
CaWO4 1,9710
СаС4Н4О6 • Н20 (Тартрат) Са 0,19440
СаО 0,27201
Ca(C9H6ON)2 (Оксихинолят) Са 0,12205
СаСО3 0,30479
СаС2О4 • Н2О (Оксалат) 0,44495
СаО 0,17077
Ca(C10H7O5N4)2 • • 7Н2О (Пикролонат) Са 0,057870
СаСОз 0,14452
СаС2О4 • Н2О (Оксалат) 0,21098
СаО 0,080972
CaF2 Са 0,51332
СаСОз 1,2819
СаС2О4 (Оксалат) 1,6406
СаС2О4 • Н2О 1,8714
Продолжение таблицы 5.1.10
Гравиметрическая форма Искомое соединение Фактор пересчета
CaF2 СаО 0,71824
CaSO4 1,7437
СаМоО4 Са 0,20037
СаСОз 0,50037
СаС2О4 (Оксалат) 0,64040
СаС2О4 • Н2О' 0,73049
СаО 0,28036
МоОз 0,71964
СаО СО2 0,78479
Са 0,71469
Ca2As2O7 3,0490
СаС2 1,1430
CaCN2 1,4284
СаСОз 1,7848
СаС2О4 2,2842
СаС2О4 • Н2О 2,6056
СаС12 1,9791
СаС12 • 6Н2О 3,9067
Са(Ю3)2 6,9526
СаМоО4 3,5669
Ca(NO3)2 2,9262
Ca(NO3)2 • 4Н2О 4,2111
Са(ОН)2 1,3213
Са3(РО4)2 1,8437
Са(Н2РО4)2 4,1737
СаНРО4 2,4262
CaSO4 2,4278
CaSO4 • 2Н2О 3,0703
CaSiO3 2,0715
CaWO4 5,1355
CaSO4 Са 0,29438
СаСОз 0,73515
СаС2О4 0,94087
СаС2О4 • Н2О 1,0732
СаО 0,41190
CaWO4 Са 0,13917
СаСОз 0,34753
СаС2О4 (Оксалат) 0,44479
СаС2О4 Н2О 0,50736
СаО 0,19472
WO3 0,80507
K2Ca[Ni(NO2)6] Са 0,088469
СаО 0,12379
Cd CdO 1,1423
Cd2P2O7 1,7737
CdS 1,2852
CdSO4 1,8546
Cd(C5H5N)2(SCN)2 (Пиридинтио-цианат) Cd 0,29063
Cd2P2O7 0,51549
CdS 0,37351
CdSO4 0,53898
Cd 0,20626
Cd2P2O7 0,36585
CdS 0,26509
CdSO4 0,38253
Cd(C9HeON)2 (Оксихинолят) Cd 0,28052
Cd2P 2O7 0,49757
CdS 0,36053
CdSO4 0,52024
530
Новый справочник химика и технолога
Продолжение таблицы 5.1.10
Гравиметрическая форма Искомое соединение Фактор пересчета
Cd(C9H6ON)2 • • 0,5Н2О (Оксихинолят) Cd 0,26280
Cd2P2O7 0,46613
CdS 0,33775
CdSO4 0,48737
CdCCjoIWO, (Хинальдинат) Cd 0,24611
Cd2P 2O7 0,43654
CdS 0,31631
CdSO4 0,45643
Cd(NH3)2 (C7H4NS2)2 (Меркаптобензо-тиазолат) Cd 0,23469
Cd2P2O7 0,41627
CdS 0,30163
CdSO4 0,43524
Cd(NH4)PO4 • • Н2О Cd 0,46175
Cd2P 2O7 0,81903
CdS 0,59345
CdSO4 0,85635
Cd(N2H4)2I2 (Дигидразин-кадмийиодид) Cd 0,26122
Cd2P2O7 0,46333
CdS 0,33572
CdSO4 0,48445
CdO Cd 0,87540
Cd2P2O7 1,5527
CdS 1,1251
CdSO4 1,6235
Cd2P2O7 Cd 0,56378
Cd(C2H3O2)2 (Ацетат) 1,1561
CdCl2 0,91943
Cdl2 1,8368
Cd(NH4)PO4 • H2O 1,2210
Cd(NO3)2 1,1858
Cd(NO3)2 • 4H2O 1,5472
CdO 0,64403
Cd(OH)2 0,73441
CdS 0,72458
CdSO4 1,0456
CdSO4 8/3H2O 1,2865
CdS Cd 0,77809
Cd (эмпирич.) 0,7781
Cd(C2H3O2)2 (Ацетат) 1,5955
CdCl2 1,2689
Cdl2 2,5350
Cd(NH4)PO4 • H2O 1,6851
Cd(NO3)2 1,6365
Cd(NO3)2 • 4H2O 2,1353
CdO 0,88884
Cd(OH)2 1,0136
Cd2P2O7 1,3801
CdSO4 1,4430
CdSO4 • 8/3H2O 1,7755
CdSO4 Cd 0,53921
Cd(C2H3O2)2 (Ацетат) 1,1057
CdCl2 0,87936
Cdl2 1,7568
Cd(NH4)PO4 • H2O 1,1678
Cd(NO3)2 1,1341
Cd(NO3)2 • 4H2O 1,4798
CdO 0,61596
Cd(OH)2 0,70240
Cd2P2O7 0,95642
Продолжение таблицы 5.1.10
Гравиметрическая форма Искомое соединение Фактор пересчета
CdSO4 CdS 0,69300
CdSO4 • 8/ЗН2О 1,2305
Се2(С2О4)3 (Оксалат) Се 0,51488
СеО2 0,63246
СеО2 Се 0,81409
Се(С2Н3О2)3 (Ацетат) . 1,8431
Се2(С2О4)3 (Оксалат) 1,5811
Се2(С2О4)3 • 9Н2О 2,0521
СеС13 1,4321
Се(С1О4)4 3,1253
Ce(NO3)3 1,8948
Ce(NO3)3 • 6Н2О 2,5228
Се2О3 0,95352
СеРО4 1,3659
Ce(SO4)2 1,9303
Ce(SO4)2 • 4Н2О 2,3490
Ce2(SO4)3 1,6512
Ce2(SO4)3 • 8Н2О 2,0699
Се2О3 Се 0,85377
СеО2 1,0487
AgCN Ag 0,80568
CN 0,19432
HCN 0,20185
KCN 0,48633
NaCN 0,36605
NH4CN 0,32905
SCN 0,34998
CuSCN AgSCN 1,3646
BaSO4 1,9192
SCN 0,47757
HSCN 0,48586
KSCN 0,79905
NH4SCN 0,62590
NaSCN 0,66661
BaSO4 SCN 0,24884
HSCN 0,25316
KSCN 0,41635
NPUSCN 0,32613
NaSCN 0,34734
c20h16n4hcio4 (Нитронпер-хлорат) C1O4 0,24091
HC1O4 0,24335
KC1O4 0,33562
NaClO4 0,29660
Со CoCl2 2,2031
Co(NO3)2 3,1041
Co(NO3)2 • 6H2O 4,9381
CoO 1,2715
Co3O4 1,3620
Co(OH)2 1,5772
Co2P 2O7 2,4757
CoSO4 2,6300
CoSO4 • 7H2O 4,7696
K3[Co(NO2)6] 7,6737
Co(C5H5N)4(SCN)2 (Пиридинтиоцио-нат) Co 0,11991
CO3O4 0,16332
CoSO4 0,31537
Co(C9H6ON)2 (Оксихинолят) Co 0,16973
CO3O4 0,23117
CoSO4 0,44639
Химические методы количественного анализа
531
Продолжение таблицы 5.1.10
Гравиметрическая форма Искомое соединение Фактор пересчета
Co(C9H6ON)2 • 2Н2О (Оксихинолят) Со 0,15378
Со3О4 0,20944
CoSO4 0,40443
Co(C10H6ONO)3 (а-Нитрозо-Р-нафтол ат) Со 0,10243
Со3О4 0,13950
CoSO4 0,26938
Co(NH2 C6H4 • • СО2)2 (Антранилат) Со 0,17796
CO3O4 0,24237
CoSO4 0,46802
CO3O4 Со 0,73424
CoSO4 1,9310
Со2Р2О7 Со 0,40393
CO3O4 0,55014
CoSO4 1,0623
COSO4 Со 0,38023
СоС12 0,83769
Co(NO3)2 1,1803
Co(NO3)2 • 6Н2О 1,8776
СоО 0,48345
Со3О4 0,51786
Со(ОН)2 0,59970
Со2Р2О7 0,94133
CoSO4 • 7Н2О 1,8136
K3[Co(NO2)6] 2,9179
K3[Co(NO2)6] Со 0,13032
CO3O4 0,17748
CoSO4 0,34272
Ag2CrO4 Сг 0,15677
СгО3 0,30144
Сг2О3 0,22910
СгО4 0,34967
Сг2О7 0,32556
BaCrO4 Сг 0,20527
СгО3 0,39472
Сг2О3 0,30000
СгО4 0,45787
Сг2О7 0,42629
Cr(C9H6ON)3 (Оксихинолят) Сг 0,10735
Сг2О3 0,15689
Сг2О3 Ag2CrO4 4,3648
ВаСгО4 3,3334
Сг 0,68425
СгС12 1,6172
СгС13 2,0837
Сг1С13 • 6Н2О 3,5059
СгО2С12 2,0382
СгО3 1,3157
СгО4 1,5262
Сг2О7 1,4210
Сг(ОН)3 1,3555
СгРО4 1,9338
CrSO4 1,9481
CrSO4 • 7Н2О 3,6072
Cr2(SO4)3 2,5799
Cr2(SO4)3-18H2O 4,7131
K2CrO4 2,5551
К2Сг2О7 1,9354
KCr(SO4)2 • 12H2O 6,5705
(NH4)Cr(SO4)2 • 12H2O 6,2936
Продолжение таблицы 5.1.10
Гравиметрическая форма Искомое соединение Фактор пересчета
Cr2O3 PbCrO4 4,2523
CrPO4 Cr 0,35385
Cr2O3 0,51713
PbCrO4 Cr 0,16091
Cr (эмпирич.) 0,1593
CrO3 0,30942
Cr2O3 0,23516
CrO4 0,35892
Cr2O7 0,33417
AgCl CsCl 1,1746
CsC104 Cs 0,57198
Cs2PtCl6 1,4495
Cs3[Co(NO2)6] • •H2o Cs 0,53041
Cs2PtCl6 1,3442
Cs2PtCl6 Cs 0,39460
Cs2CO3 0,48368
CsCl 0,49987
CsC104 0,68988
Cs3[Co(NO2)6] • H2O 0,74394
CsNO3 0,57870
Cs2O 0,41835
Cs2SO4 0,53721
Cs2[SnCl6] 0,88660
Cs2SO4 Cs 0,73453
Cs2PtCl6 1,8615
Cs2[SnCl6] Cs 0,44506
Cs2PtCl6 1,1279
Cu Cu(C2H3O2)2 (Ацетат) 2,8585
CuCl 1,5580
CuCl2 2,1160
Cu2[Fe(CN)6] • 7H2O 3,6603
Cu3[Fe(CN)6]2 3,2239
Cui 2,9973
Cu(IO3)2 6,5055
Cu(NO3)2 2,9518
Cu(NO3)2 • 3H2O 3,8024
CuO 1,2518
Cu2O 1,1259
Cu(OH)2 1,5353
CuS 1,5047
Cu2S 1,2523
CuSCN 1,9141
CuSO4 2,5120
CuSO4 • 5H2O 3,9297
CuSO4 • 4NH3 • H2O 3,8677
Cu(C5H5N)2(SCN)2 (Пиридинтиоцианат) Cu 0,18803
CuO 0,23538
Cu(C6H5O2N2)2 (Купферонат) Cu 0,18811
CuO 0,23548
Cu(C7H6O2N)2 (Антранилат) Cu 0,18922
CuO 0,23686
Cu(C7H6O2N)2 (Салицилальдок-симат) Cu 0,18922
CuO 0,23686
Cu(C9H6ON)2 (Оксихинолят) Cu 0,18059
CuO 0,22606
Cu(C10H6O2N)2 • •H2o (Хинальдинат) Cu 0,14919
CuO 0,18676
532
Новый справочник химика и технолога
Продолжение таблицы 5.1.10
Гравиметрическая форма Искомое соединение Фактор пересчета
Cu(C12H10ONS)2 (Тионалидат) Cu 0,12808
CuO 0,16033
Cu(C13H8O2N)2 (Бензоазолат) Cu 0,131129
CuO 0,16435
Cu(C14HnO2N) (Бензоиноксимат) Cu 0,22002
CuO 0,27543
Cui Cu 0,33363
CuO 0,41764
Cu(IO3)2 Cu 0,15372
CuO 0,19242
CuO Cu 0,79884
Cu(C2H3O2)2 (Ацетат) 2,2835
CuCl 1,2446
CuCl2 1,6903
Cu2[Fe(CN)6] • 7H2O 2,9240
Cu3[Fe(CN)6]2 2,5754
Cui 2,3944
Cu(IO3)2 5,1969
Cu(NO3)2 2,3580
Cu(NO3)2 • 3H2O 3,0375
Cu2O 0,89942
Cu(OH)2 1,2265
CuS 1,2020
Cu2S 1,0004
CuSCN 1,5291
CuSO4 2,0067
CuSO4 • 5H2O 3,1392
CuSO4 • 4NH3 • H2O 3,0897
Cu2S Cu 0,79851
CuO 0,99959
CuSO4 2,0058
CuSO4 • 5H2O 3,1379
CuSCN Cu 0,52243
CuO 0,65398
CuSO4 1,3123
CuSO4 • 5H2O 2,0529
CuSO4 Cu 0,39810
CuO 0,49834
CuSO4 • 5H2O 1,5644
BaF2 CaF2 0,44526
F 0,21670
Ba[SiF6] CaF2 0,83822
F 0,40794
BiF3 CaF2 0,44030
F 0,21429
(C6H5)3SnF CaF2 0,10579
F 0,051488
CaF2 BaF2 2,2459
Ba[SiF6] 1,1930
BiF3 2,2712
F 0,48668
HF 0,51250
H2[SiF6] 0,61521
kbf4 0,80638
KF 1,4882
K2[SiF6] 0,94045
NI^F 0,94877
Na3[AlF6] 0,89632
NaF 1,0756
Продолжение таблицы 5.1.10
Гравиметрическая форма Искомое соединение Фактор пересчета
CaF2 PbCIF 6,7025
SiF4 0,66656
SiF6 0,60660
U3O8 3,5955
K2[SiF6] CaF2 1,0633
F .0,51750
LaF3La2O3 CaF2 0,17727
F 0,086275
PbCIF CaF2 0,14920
F 0,072611
U3O8 CaF2 0,27813
F 0,13536
Fe(C9H6ON)3 (Оксихинолят) Fe 0,11437
Fe2O3 0,16352
Fe2O3 Fe 0,69944
FeC2O4 (Оксалат) 1,8018
FeC2O4 • 2H2O 2,2530
Fe2(C2O4)3 2,3529
Fe(CN)6 2,6545
FeCO3 1,4510
FeCl2 1,5875
FeCl2- 4H2O 2,4900
FeCl3 2,0316
FeCl3 • 6H2O 3,3853
Fe3[Fe(CN)6]2 1,4815
Fe4[Fe(CN)6]3 1,5373
Fe(NH4)2(SO4)2 • 6H2O 4,9112
Fe(NO3)2 2,2525
Fe(NO3)2- 6H2O 3,6063
Fe(NO3)3 3,0291
Fe(NO3)3 • 6H2O 4,3828
Fe(NO3)3- 9H2O 5,0597
FeO 0,89981
Fe3O4 0,96660
Fe(OH)2 1,1254
Fe(OH)3 1,3384
FePO4 1,8889
FeS 1,1010
FeSO4 1,9025
FeSO4 • 7H2O 3,4819
Fe2(SO4)3 2,5041
Fe2(SO4)3 • 9H2O 3,5194
K3[Fe(CN)6] 4,1235
K4[Fe(CN)6] 4,6132
№ЦРе(8О4)2 • 12H2O 6,0390
FePO4 Fe 0,37030
Fe2O3 0,52942
Ga(C9H4Br2ON)3 (Дибромокси-хинолят) Ga 0,071461
Ga2O3 0,096061
Ga2O3 Ga 0,74392
GaCl3 1,8789
Ga(NO3)3 2,7288
Ga(OH)3 1,2883
Ga2S3 1,2570
Ga2(SO4)3 2,2814
G3.2S3 Ga 0,59180
Ga2O3 0,79552
GeO2 Ge 0,69407
Химические методы количественного анализа
533
Продолжение таблицы 5.1.10
Гравиметрическая форма Искомое соединение Фактор пересчета
GeO2 GeCl4 2,0500
GeK2F6 2,5315
GeS2 1,3071
НЮ2 Hf 0,84798
Co[Hg(SCN)4] Hg 0,40784
HgS 0,47301
Hg Hg(CN)2 1,2594
Hg(CN)2 • HgO 1,1696
HgCl2 1,3535
Hg2Cl2 1,1768
Hgl2 2,2653
Hg2MoO4 1,3987
Hg(NO3)2 1,6182
Hg(NO3)2 • 1/2H2O 1,6631
Hg2(NO3)2 1,3091
Hg2(NO3)2 • 2H2O 1,3989
HgO 1,0798
HgS 1,1598
Hg(SCN)2 1,5790
Hg2(SCN)2 1,2895
HgSO4 1,4788
Hg2so4 1,2394
Hg2wo4 1,6178
Hg3(AsO4)2 Hg 0,68417
HgS 0,79351
Hg2C2O4 (Оксалат) Hg 0,82009
HgS 0,95115
Hg(C5H5N)Cl2 (Пиридинхлорид) Hg 0,57215
HgS 0,66358
HgCC^NCS^ (Меркаптобензо-тиазолат) Hg 0,37630
HgS 0,43645
Hg2(C6H5O2N2)2 (Купферонат) Hg 0,59399
HgS 0,68894
Hg(C7H6O2N)2 (Антранилат) Hg 0,42423
HgS 0,49203
Hg(CI2H10ONS)2 (Тионалидат) Hg 0,31683
HgS 0,36746
Hg2Cl2 Hg 0,48979
HgS 0,98560
Hg2CrO4 Hg 0,77571
HgS 0,89968
Hgl2 Hg 0,44145
HgS 0,51200
Hg2(IO3)2 Hg 0,53422
HgS 0,61959
HgS Hg 0,86221
Hg(CN)2 1,0858
Hg(CN)2 • HgO 1,0084
HgCl2 1,1670
Hg2ci2 1,0146
Hgi2 1,9531
Hg2MoO4 1,2059
Hg(NO3)2 1,3952
Hg(NO3)2 • 1/2H2O 1,4939
Hg2(NO3)2 1,1287
Hg2(NO3)2 • 2H2O 1,2061
HgO 0,93098
Hg2O 0,89657
Продолжение таблицы 5.1.10
Гравиметрическая форма Искомое соединение Фактор пересчета
HgS Hg(SCN)2 1,3615
Hg2(SCN)2 1,1118
HgSO4 1,2751
Hg2SO4 1,0686
Hg2wo4 1,3949
Zn[Hg(SCN)4] Hg 0,40256
HgS 0,46690
In(C9H6ON)3 (Оксихинолят) In 0,20980
In2O3 0,25365
In2O3 In 0,82711
InCl3 1,5934
II12S3 1,1736
In2S3 In 0,70477
In2O3 0,85208
Ag Agl 2,1764
I 1,1764
Agl AgIO3 1,2044
Cui 0,81115
HIO3 0,74926
IO3 0,74496
IO4 0,81311
i205 0,71089
I2O7 0,77904
KH(IO3)2 0,83038
KIO3 0,91150
KIO4 0,97964
NaIO3 0,84289
Pbl2 0,98179
Pdl2 0,76711
TH 1,4110
AgIO3 Agl 0,83026
I 0,44878
Cui Agl 1,2328
I 0,66637
Pbl2 Agl 1,0185
I 0,55055
Pdl2 Agl 1,3036
I 0,70462
IO3 0,97113
i205 0,92671
TH Agl 0,70869
I 0,38307
C6H2(NO2)3 • OK (Пикрат) К 0,14635
KC1 0,27902
K[B(C6H5)4] (Тетрафенил-борат) к 0,10911
KC1 0,20806
K2SO4 0,24315
KBO2 к 0,47729
KC1 0,91012
KC1 к 0,52443
K2(A1O2)2 • 3H2O 1,6780
KA1(SO4)2 • 12H2O 6,3630
К[В(СбН5)4] (Тетрафенилборат) 4,8063
kbf4 1,6889
KBO2 1,0988
KBr 1,5963
KBrO3 2,2401
K2C2O4 1,1147
534
Новый справочник химика и технолога
Продолжение таблицы 5.1.10
Гравиметрическая форма Искомое соединение Фактор пересчета
КС1 KCN 0,87341
K2CO3 0,92688
KHCO3 1,3429
KC1O3 1,6438
KC1O4 1,8585
K2CrO4 1,3024
K2Cr2O7 1,9731
KCr(SO4)2 • 12H2O 6,6987
KF 0,77927
K3[Fe(CN)6] 1,4721
JQFeCCN),] 1,2352
KHQFUOe (Тартрат) 2,5240
KI 2,2266
KIO3 2,8704
KH(IO3)2 5,2299
KNO3 1,3561
K2O 0,63173
KOH 0,75255
K3PO4 0,94905
K2HPO4 1,1681
KH2PO4 1,8253
K2PtCl6 3,2595
K2SO4 1,1687
KHSO4 1,8264
K2S2O8 1,8129
KSCN 1,3035
K2SiF6 1,4773
КС1О4 C1O4 0,71781
к 0,28219
KC1 0,53810
K2O 0,33993
K3[Co(NO2)6] • • ЗН2О к 0,23166
KC1 0,44174
кнСдНА (Тартрат) к 0,20778
KC1 0,39619
КЮ4 к 0,16999
KC1 0,32415
K2PtCl6 к 0,16089
KC1 0,30680
K2SO4 к 0,44874
KC1 0,85567
Pt к 0,40084
KC1 0,76433
K2O 0,48285
L32O3 La 0,85269
LaCl3 1,5056
LiCl Li 0,16369
Li2O 0,35238
Li2SO4 1,2966
Li2O 5A12O3 Li 0,048739
Li2SO4 0,38607
Li3PO4 Li 0,17980
LiCl 1,0984
Li2SO4 1,4242
Li2SO4 Li 0,12624
LiBr 1,5800
Li2CO3 0,67207
LiCl 0,77123
LiNO3 1,2542
Продолжение таблицы 5.1.10
Гравиметрическая форма Искомое соединение Фактор пересчета
Li2SO4 LiNO3 3H2O 2,2374
Li2O 0,27177
LiOH 0,43563
Li3PO4 0,70213
Zn(UO2)3Li • (CH3COO)9 (Цинкуранил-ацетат) Li 0,0049083
Li2SO4 0,038879
ZnU2O7- Li2U2O7 Li 0,0072706
Li2SO4 0,057592
СО2 MgCO3 1,9161
MgO 0,91613
Mg2As2O7 As2O7 0,84333
Mg 0,15667
Mg(NH4)PO4 • 6H2O 1,5811
MgO 0,25974
Mg2P2O7 0,71697
MgC2O4 • 2H2O (Оксалат) Mg 0,16391
Mg(NH4)PO4 • 6H2O 1,6541
MgO 0,27175
Mg2P2O7 0,75010
Mg(C9H6ON)2 (Оксихинолят) Mg 0,077792
Mg(NH4)PO4 6H2O 0,78505
MgO 0,12897
Mg2P2O7 0,35600
Mg(C9H6ON)2-• 2H2O Mg 0,69752
Mg(NH4)PO4 • 6H2O 0,70392
MgO 0,11564
Mg2P2O7 0,31921
Mg(NH4)AsO4 • • 6H2O AsO4 0,48004
Mg 0,084045
Mg(NH4)PO4 6H2O 0,84815
Mg(NH4)PO4 • 6H2O MgO 0,13934
Mg2P2O7 0,38461
Mg 0,099091
Mg2As2O7 0,63248
MgCO3 0,34360
MgC2O4 • 2H2O (Оксалат) 0,60455
MgCl2 0,38801
MgCl2 • 6H2O 0,82846
MgCl2 • KC1 • 6H2O 1,1322
MgF2 0,25392
Mg(NH4)AsO4 • 6H2O 1,1790
MgO 0,16428
Mg(OH)2 0,23771
Mg2p2o7 0,45347
MgSO4 0,49051
MgSO4 • 7H2O 1,0044
p205 0,28919
MgO Mg 0,60317
Mg(NH4)PO4 • 6H2O 6,0871
Mg2P2O7 2,7603
Mg2p2o7 Mg 0,21852
Mg2As2O7 1,3948
MgC2O4 • 2H2O (Оксалат) 1,3332
MgCO3 0,75772
MgCl2 0,85565
MgCl2 • 6H2O 1,8269
MgCl2 • KC1 • 6H2O 2,4969
Химические методы количественного анализа
535
Продолжение таблицы 5.1.10
Гравиметрическая форма Искомое соединение Фактор пересчета
Mg2P2O7 MgF2 0,55995
Mg(NH4)AsO4 • 6Н2О 2,6000
Mg(NH4)PO4 • 6Н2О 2,2052
MgO 0,36228
Mg(OH)2 0,52419
MgSO4 1,0817
MgSO4 • 7H2O 2,2148
p205 0,63772
со2 MnCO3 2,6119
MnO 1,6119
МпС2О4 (Оксалат) Mn 0,38430
МП2Р2О7 0,99268
MnSO4 1,0563
Mn(C9H6ON)2 (Оксихинолят) Mn 0,16006
Mn2p 2O7 0,41345
MnSO4 0,43993
Mn(NH4)PO4 • Н2О Mn 0,29542
Mn2P2O7 0,76310
MnSO4 0,81199
Мп3О4 Mn 0,72030
Mn2P2O7 1,8606
MnSO4 1,9798
Мп2Р2О7 Mn 0,38713
MnCO3 0,81000
MnC2O4 (Оксалат) 1,0074
MnCl2 0,88683
MnCl2-4H2O 1,3946
Mn(NH4)AsO4 • 6H2O 2,2548
Mn(NH4)PO4 • H2O 1,3104
MnO 0,49988
MnO2 0,61262
Mn2O3 0,55624
МП2О7 0,78174
Mn3O4 0,53746
Mn(OH)2 0,62683
MnPO4 1,0564
MnS 0,61309
MnSO4 1,0641
MnSO4 • 4H2O 1,5719
MnS Mn 0,63145
МП2Р2О7 1,6311
MnSO4 1,7356
MnSO4 Mn 0,36383
MnCO3 0,76123
MnC2O4 (Оксалат) 0,94673
MnCl2 0,83344
MnCl2 • 4H2O 1,3107
Mn(NH4)AsO4 • 6H2O 2,1190
Mn(NH4)PO4 • H2O 1,2315
MnO 0,46978
MnO2 0,57574
Mn2O3 0,52276
МП2О7 0,73467
Mn3O4 0,50510
Mn(OH)2 0,58909
MnPO4 0,99278
Mn2P2O7 0,93980
MnS 0,57618
MnSO4 • 4H2O 1,4772
Продолжение таблицы 5.1.10
Гравиметрическая форма Искомое соединение Фактор пересчета
Ag2MoO4 Mo 0,25538
MoO3 0,38314
ВаМоО4 Mo 0,32373
MoO3 0,48417
СаМоО4 Mo 0,47968
MoO3 0,71964
CdMoO4 Mo 0,35229
MoO3 0,52853
MoO2(C9H6ON)2 (Оксихинолят) Mo 0,23050
MoO3 0,34582
МоО3 Ag2MoO4 2,6100
BaMoO4 2,0654
CaMoO4 1,3896
H2MoO4 1,1252
Mo 0,66655
MoO4 1,1111
MoS2 1,1121
MoS3 1,3348
(NH4)2MoO4 1,3618
(NH4)3PO4 • 12MoO3 1,0863
PbMoO4 1,5506
РЬМоО4 Mo 0,26133
MoO3 0,39206
C20H16N4 • HNO3 (Нитроннитрат) HNO3 0,16787
KNO3 0,26934
NaNO3 0,22642
NO3 0,16518
N2O5 0,14387
BaSO4 (NH4)2SO4 0,56612
NH4[B(C6H5)4] (Тетрафенилборат) NH3 0,050498
(NH4)2PtCl6 0,65807
(NH4)2PtCl6 N 0,063112
NH3 0,076736
nh4 0,081277
(NH4)2co3 0,21646
nh4ci 0,24102
NH4NO3 0,36065
nh4oh 0,15791
(NH4)2SO4 0,29769
Pt NH3 0,17461
(NH^PtCl* 2,2754
в2о3 Na2B4O7 1,4450
Na2B4O7 10H2O 2,7385
BaSO4 Na2S 0,33436
Na2S 9H2O 1,0290
Na2SO3 0,53999
Na2SO3 • 7H2O 1,0803
NaHSO4 0,51436
NaHSO4 • H2O 0,59154
Na2SO4 0,60854
Na2SO4 • 10H2O 1,3803
CO2 Na2CO3 2,4083
Na2CO3 • 10H2O 6,5019
Na2O 1,4083
Mg2As2O7 Na2HAsO3 1,0945
Na2HAsO4 1,1976
Mg2P2O7 Na3PO4 1,4732
NaH2PO4 1,0782
Na2HPO4 1,2757
536
Новый справочник химика и технолога
Продолжение таблицы 5.1.10
Гравиметрическая форма Искомое соединение Фактор пересчета
Mg2P2O7 Na2HPO4 • 12H2O 3,2184
Na4P2O7 1,1947
NaCl Na 0,39336
Na3AlF6 1,1974
Na2B4O7 1,7217
Na2B4O7 • 10H2O 3,2629
NaBr 1,7607
NaBrO3 2,5819
NaC2H3O2 (Ацетат) 1,4036
Na2C2O4 (Оксалат) 1,1549
NaCN 0,83852
Na2CO3 0,90673
Na2CO3 • 10H2O 2,4479
NaHCO3 1,4373
NaC103 1,8212
NaC104 2,0950
Na2CrO4 1,3858
Na2Cr2O7 2,2413
NaF 0,71843
Na2HAsO4 1,5903
Nal 2,5647
NaIO3 3,3859
NaNO2 1,1805
NaNO3 1,4543
Na2O 0,53023
Na2O2 0,66711
NaOH 0,68435
Na3PO4 0,93501
Na2HPO4 1,2145
NaH2PO4 2,0528
Na4P2O7 1,1374
Na2S 0,66767
Na2S • 9H2O 2,0547
Na2SO3 1,0783
Na2SO3 7H2O 2,1571
NaHSO3 1,7805
NaHSO4 2,0542
Na2SO4 1,2152
Na2SO4 • 10H2O 2,7564
Na2S2O3 1,3526
Na2S2O3'5H2O 2,1232
NaSbO2 3,0241
Na{Mg[UO2(CH3COO)3]3)- •6,5H2O 25,767
Na2U2O7(MgU2O7)2 15,903
Na{Zn[UO2(CH3COO)3]3} • • 6,5H2O 26,470
Na2U 2 O7(ZnU 2O7)2 16,606
Na2SO4 Na 0,32371
Na2CO3 0,74618
NaCl 0,82293
Na2O 0,43635
Na{Mg[UO2 (CH3COO)3]3} • • 6,5H2O Na 0,015266
NaCl 0,038809
Na2U2O7 • (MgU2O7)2 Na 0,024734
NaCl 0,062880
Na{Zn[UO2- • (CH3COO)3]3}- • 6,5H2O Na 0,014861
NaCl 0,037779
Продолжение таблицы 5.1.10
Гравиметрическая форма Искомое соединение Фактор пересчета
Na2U2O7(ZnU2O7)2 Na 0,023688
NaCl 0,060220
Nb2O5 Nb 0,69904
Nd2O3 Nd 0,85737
Ni NiO 1,2725
Ni(C5H5N)4(SCN)2 (Пиридинтиоцианат) Ni , 0,11950
NiO 0,15207
NiC8H14N4O4 (Диметилглиок-симат) Ni 0,20319
NiCO3 0,41088
Ni(NO3)2 0,63240
Ni(NO3)2 • 6H2O 1,0065
NiO 0,25856
Ni(OH)2 0,32092
NiSO4 0,53566
NiSO4 • 6H2O 0,90977
Ni(C9H6ON)2 (Оксихинолят) Ni 0,16918
NiO 0,21529
Ni(NH2-•C6H4CO2)2 (Антранилат) Ni 0,17738
NiO 0,22573
NiO Ni 0,78584
OsO4 Os 0,74823
(C9H7ON)3 • • H7[P(Mo2O7)6 • • 2H2O P 0,013277
p2o5 0,030423
P2O5 (эмпирич.) (Хинолинфосфомолибдат) 0,0306
BiPO4 P 0,10190
Mg2P2O7 0,36613
p2o5 0,23349
Hg2Cl2 H3PO3 0,17368
PO2 0,13338
PO3 0,16727
Mg(NH4)PO4 • •6H2O Mg2P2O7 0,45347
p 0,12621
p205 0,28919
Mg2P 2O7 Ag3PO4 3,7613
A1PO4 1,0958
Ca3(PO4)2 1,3935
Ca(H2PO4)2 1,0515
CaHPO4 1,2225
FePO4 1,3552
H3PO3 0,73677
KH2PO4 1,2228
Mg(NH4)PO4 • 6H2O 2,2052
(NH4)3P(Mo3Oio)4 16,861
Na3PO4 1,4731
Na2HPO4 1,2756
NaH2PO4 1,0781
P 0,27831
PO3 0,70960
PO4 0,85336
p2o5 0,63772
P2O5 • 24MoO3 16,159
p2o7 0,78148
Zn2P2O7 1,3689
(NH4)3P(Mo3Oio)4 Mg2P2O7 0,059310
p 0,016507
P (эмпирич.) 0,01639
Химические методы количественного анализа
537
Продолжение таблицы 5.1.10
Гравиметрическая форма Искомое соединение Фактор пересчета
(NH4)3P(Mo3O10)4 РО4 0,050613
РО4 (эмпирич.) 0,05025
Р2О5 0,037823
Р2О5 (по Финкенеру) 0,03755
Р2О5 (по Лоренцу) 0,03295
Р2О5 • 24МоО3 Mg2P2O7 0,061886
Р 0,017224
Р2О5 0,039466
Zn2P2O7 Mg2P2O7 0,73050
р 0,20331
р205 0,46585
РЬСО3 РЬ 0,77543
РЬО 0,83530
PbSO4 1,1349
РЬС2О4 (Оксалат) Pb 0,70185
РЬО 0,75605
PbSO4 1,0272
Pb(C7H4NS2)OH (Меркаптобензо-тилзолат) Pb 0,53067
РЬО 0,57165
PbSO4 0,77670
РЬ(С7Н5О3)2 (Салицилат) Pb 0,43039
РЬО 0,46363
PbSO4 0,62993
Pb(C7H6O2N)2 (Антранилат) Pb 0,43216
РЬО 0,46553
PbSO4 0,63252
РЬ(С8Н4О4) (Фталат) Pb 0,55802
PbO 0,60111
PbSO4 0,81673
Pb(C9H6ON)2 (Оксихинолят) Pb 0,41817
PbO 0,45046
PbSO4 0,61204
Pb(C9H4ONBr2)2 (Дибромоксихи-нолят) Pb 0,25545
PbO 0,27518
PbSO4 0,37388
Pb(C10H7N4O5)2 (Пикролонат) Pb 0,28245
PbO 0,30426
PbSO4 0,41340
Pb(C12H10ONS)2 (Тионалидат) Pb 0,32388
PbO 0,34888
PbSO4 0,47403
РЬС12 Pb 0,74503
PbO 0,80256
PbSO4 1,0904
РЬСгО4 Pb 0,64108
Pb (эмпирич.) 0,6378
PbO 0,69058
PbSO4 0,93830
РЬ(Ю3)2 Pb 0,37199
PbO 0,40071
PbSO4 0,54445
РЬМоО4 Pb 0,56436
PbO 0,60704
PbSO4 0,82601
РЬО Pb 0,92832
PbSO4 1,3687
РЬО2 Pb 0,86623
PbO 0,93311
PbSO4 1,2678
Продолжение таблицы 5.1.10
Гравиметрическая форма Искомое соединение Фактор пересчета
Pb3(PO4)2 Pb 0,75595
PbO 0,82509
PbSO4 1,1211
PbS Pb 0,86599
PbO 0,93286
PbSO4 1,2675
PbSO3 Pb 0,72129
PbO 0,77699
PbSO4 1,0557
PbSO4 Pb 0,68324
PbCO3 0,88111
(PbCO3)2 • Pb(OH)2 0,85254
Pb(C2H3O2)2 (Ацетат) 1,0726
Pb(C2H3O2)2 • 3H2O 1,2508
Pb(C2H5)4 (Тетраэтил) 1,0665
PbC2O4 (Оксалат) 0,97348
PbCl2 0,91707
PbCrO4 1,0658
Pb(IO3)2 1,8367
PbMoO4 1,2106
Pb(NO3)2 1,0922
PbO 0,73600
PbO2 0,78875
Pb3o4 0,75358
Pb3(PO4)2 0,89201
PbS 0,78897
PbSO3 0,94724
PbWO4 1,5005
Pd K2PdCl6 3,7344
Pd(CN)2 1,4891
PdCl2 1,6665
PdCl2 • 2H2O 2,0051
Pdl2 3,3855
Pd(NO3)2 2,1656
PdO 1,1504
Pd(C7H6O2N)2 (Салицил альдок-симат) Pd 0,28099
Pd(C8H14O4N4) (Диметилглиок-симат) Pd 0,31607
Pd(C10H6ONO)2 (а-Нитрозо-0-нафтолат) Pd 0,23606
Pd[C10H6O(NO2)2 (а-Нитро-р-наф-толат) Pd 0,22041
Рг2О3 Pr 0,85447
K2PtCl6 Pt 0,40139
(NH4)2PtCl6 Pt 0,43948
Pt H2PtCl6 2,1008
H2PtCl6 • 6H2O 2,6549
K2PtCl6 2,4913
(NHthPtCh 2,2754
PtCL» 1,7270
PtCl4 • 5H2O 2,1887
PtS2 1,3287
PtS2 Pt 0,75260
AgCl Rb 0,59636
538
Новый справочник химика и технолога
Продолжение таблицы 5.1.10
Гравиметрическая форма Искомое соединение Фактор пересчета
AgCl RbCl 0,84372
Rl^PtCl* Rb 0,29537
Rb2CO3 0,39906
RbHCO3 0,50622
RbCl 0,41789
Rb2O 0,32302
Rb2SO4 0,46135
Rb2SO4 Rb 0,64024
Rb2PtCl6 2,1675
BaSO4 FeS2 0,25700
H2S 0,14601
H2SO3 0,35167
H2SO4 0,42018
K2SO4 0,74656
(NH4)2SO4 0,56612
Na2S 0,33436
Na2S • 9H2O 1,0290
NaHSO3 0,44582
Na2SO3 0,53999
Na2SO3 • 7H2O 1,0803
Na2SO4 0,60854
Na2S2O3 0,33868
Na2S2O3 • 5H2O 0,53163
NaS2O4 0,37295
S 0,13737
so2 0,27446
so3 0,34300
so4 0,41155
S2O3 0,24019
s204 0,27446
C]2H]2N2 h2so4 (Бензидинсульфат) BaSO4 0,82680
s 0,11358
CuO BaSO4 2,9347
H2S 0,42849
s 0,40314
CuS s 0,33540
Sb Sb2O4 1,2628
Sb(C9H6ON)3 (Оксихинолят) Sb 0,21969
Sb2O4 0,27743
Sb(C12H10ONS)3 (Тионалид ат) Sb 0,15800
Sb2O4 0,19953
(SbOH)C6H3O3 (Пироталлат) Sb 0,46498
Sb2O4 0,58719
Sb2O4 KSbO • C4H4O6 • 1/2H2O (Тартрат) 2,1719
Na3SbS4 • 9H2O 3,1292
Sb 0,79188
Sb (эмпирич.) 0,7922
SbCl3 1,4837
SbCl5 1,9449
SbOCl 1,1265
SbOCl3 1,5877
Sb2O3 0,94797
Sb2O3 (эмпирич.) 0,9484
Sb2S3 1,1047
Sb2S3 (эмпирич.) 1,105
Sb2S5 1,3132
Sb2(SO4)3 1,7291
Продолжение таблицы 5.1.10
Гравиметрическая форма Искомое соединение Фактор пересчета
Sb2S3 Sb 0,71683
Sb (эмпирич.) 0,7173
Sb2O4 0,90522
Sb2S5 Sb 0,60300
Sb2O3 0,72185
Sb2O4 , 0,76147
Sc2O3 Sc 0,65197
Se H2SeO3 1,6334
H2SeO4 1,8361
Na2SeO3 2,1902
Na2SeO4 2,3929
SeO2 1,4053
SeO3 1,6079
PbSeO4 Se 0,22549
(сдаода! • • (Mo3Oi0)4 (Хинолинмо-либдосиликат) Si 0,012003
SiO2 0,025678
K2SiF6 Si 0,12751
SiO2 0,27278
SiO2 BaSiF6 4,6505
H2SiO3 1,2998
K2SiF6 3,6660
Na2SiO3 2,0315
Si 0,46747
SiF4 1,7322
SiO3 1,2663
SiO4 1,5325
Si2o7 1,3994
Sn SnO2 1,2696
SnO2 (NH4)2SnCl6 2,4388
Sn 0,78766
SnCl2 1,2582
SnCl2 • 2H2O 1,4973
SnCl4 1,7288
SnO 0,89383
Sr2As2O7 Sr 0,40098
SrSO4 0,84056
SrC2O4 (Оксалат) Sr 0,49888
SrSO4 1,0458
SrC2O4 • H2O Sr 0,45248
SrSO4 0,94851
SrCO3 Sr 0,59353
SrSO4 1,2442
SrC4H4O6 • 4H2O (Тартрат) Sr 0,28473
SrSO4 0,59686
SrCrO4 Sr 0,43032
SrSO4 0,90206
SrF2 Sr 0,69752
SrSO4 1,4622
Sr(IO3)2 Sr 0,20032
SrSO4 0,41992
SrSO4 SO3 0,43586
Sr 0,47704
Sr2As2O7 1,1897
SrC2O4 (Оксалат) 0,95621
SrCzO4H2O 1,0543
SrCl2 0,86308
SrCl2-6H2O 1,4515
Химические методы количественного анализа
539
Продолжение таблицы 5.1.10
Гравиметрическая форма Искомое соединение Фактор пересчета
SrSO4 SrCrO4 1,1086
SrF2 0,68390
Sr(IO3)2 2,3814
Sr(NO3)2 1,1522
Sr(NO3)2 • 4H2O 1,5445
SrO 0,56414
SrS 0,65160
SrS2O3 1,0875
Та2О3 Ta 0,81896
TaCl5 1,6213
РЬТеО4 Те 0,31997
Те PbTeO4 3,1253
H2TeO3 1,3919
H2TeO4 1,5173
H2TeO4 • 2H2O 1,7997
TeO2 1,2508
TeO3 1,3761
Th(C9H6ON)4 (Оксихинолят) Th 0,28695
ThO2 0,32652
Th(C10H7O5N4)4 • •Н2О (Пикролонат) Th 0,17811
ThO2 0,20267
ThO2 Th 0,87881
ThCl4 1,4159
Th(NO3)4 1,8181
Th(NO3)4 4H2O 2,0911
Th(SO4)2 1,6064
TiO2 Ti 0,59950
TiCl3 1,9308
TiCl4 2,3746
Ti3(PO4)4 2,1844
Ti2(SO4)3 2,4030
TiO(C9H6ON)2 (Оксихинолят) Ti 0,13600
TiO2 0,22685
TiO(C9H4Br2ON)2 (Дибромоксихи-нолят) Ti 0,071724
TiO2 0,11964
T1C7H4NS2 (Меркаптобензо-тиазолат) TI 0,55145
TH 0,89386
T1C12H10ONS (Тионалидат) TI 0,48586
TH 0,78754
Т1С1 TI 0,85216
TH 1,3813
Т12СгО4 TI 0,77894
Til 1,2626
ТП TI 0,61693
T12CO3 0,70750
T1C1 0,72397
T1C13 0,93800
T1C13 • 4H2O 1,1555
Tl2CrO4 0,79202
T1IO3 1,1449
T1NO3 0,80410
T1(NO3)3 1,1784
T1(NO3)3 • 3H2O 1,3416
T12O3 0,68938
T1OH 0,66827
T1(OH)3 0,77094
T13PO4 0,71249
Продолжение таблицы 5.1.10
Гравиметрическая форма Искомое соединение Фактор пересчета
Til Tl2PtCl6 1,2324
T12SO4 0,76192
T1IO3 TI 0,53886
Til 0,87345
T12O3 Tl 0,89492
Til 1,4506
Tl2PtC16 Tl 0,50058
Til 0,81140
U(C2O4)2 (Оксалат) U 0,57489
u3o8 0,67792
UO2(C9H6ON)2 (Оксихинолят) и 0,42636
u308 0,50277
UO2(C9H6ON)2 • C9H7ON (Оксихинолят) и 0,33839
u308 0,39903
uof2 и 0,81511
u308 0,96120
u3o8 Na2U2O7 1,1294
U 0,84802
U(C2O4)2 (Оксалат) 1,4751
uf4 1,1187
uof2 1,0439
uo2 0,96200
UO2(C2H3O2) 2H2O 1,5110
UO2(NO3)2 • 6H2O 1,7888
(UO2)2P2O7 1,2718
UO2SO4 • 3H2O 1,4967
(UO2)2P2O7 и 0,66678
u308 0,78628
AgVO3 V 0,24634
v205 0,43973
Ag3VO4 V 0,11617
V2O5 0,20737
Ba(VO3)2 V 0,30394
v205 0,54256
V2O3(C9H6ON)4 (Оксихинолят) V 0,14026
V2O5 0,25037
V2O5 AgVO3 2,2741
Ag3VO4 4,8223
Ba(VO3)2 1,8431
HVO3 1,0990
NH4VO3 1,2863
V 0,56020
VC12 1,3399
VOC12 1,5158
VOC13 1,9057
vo4 1,2639
V2O2 0,73612
V2O3 0,82408
v204 0,91204
V2O7 1,1759
V2S3 1,0890
BaWO4 w 0,47729
wo3 0,60189
CaWO4 w 0,63854
wo3 0,80524
PbWO4 w 0,40403
wo3 0,50950
WO3 BaWO4 1,6614
540
Новый справочник химика и технолога
Продолжение таблицы 5.1.10
Гравиметрическая форма Искомое соединение Фактор пересчета
WO3 CaWO4 1,2419
H2WO4 1,0777
Na2WO4 1,2673
NaWO4 • 2H2O 1,4227
PbWO4 1,9627
W 0,79298
WO2 0,93099
ws2 1,0696
WO2(C9H6ON)2 (Оксихинолят) w 0,36468
wo3 0,45988
y2o3 Y 0,78746
YC12 1,7295
Y(NO3)3 2,4349
Y(NO3)3 3H2O 2,6742
Y(OH)3 1,2393
Y2(SO4)3 2,0636
Zn ZnO 1,2447
Zn(C7H6O2N)2 (Антранилат) Zn 0,19363
ZnO 0,24102
Zn(C9H6ON)2 (Оксихинолят) Zn 0,18485
ZnO 0,23009
Zn(C10H6O2N)2 •H2O (Хинальдинат) Zn 0,15285
ZnO 0,19026
Zn[Hg(SCN)4] Zn 0,13120
ZnO 0,16331
Zn(NH4)PO4 Zn 0,36649
ZnO 0,45618
ZnO Zn 0,80339
Zn(C2H3O2)2 (Ацетат) 2,2545
Zn(C2H3O2)2 • 2H2O 2,6973
ZnC2O4 (Оксалат) 1,8850
ZnCl2 1,6748
Продолжение таблицы 5.1.10
Гравиметрическая форма Искомое соединение Фактор пересчета
ZnO ZnCl2 1/2H2O 1,7855
Zn(NH4)PO4 2,1921
Zn(NO3)2 2,3273
Zn(OH)2 1,2214
Zn2P 2O7 1,8721
ZnS 1,1974
ZnSO4 1,9839
ZnSO4 • 7H2O 3,5335
ZnjPjO? Zn 0,42913
ZnO 0,53415
ZnS Zn 0,67094
ZnO 0,83513
ZnSO4 Zn 0,40497
ZnO 0,50407
Zr(C6H5CHOHCOO)4 Zr 0,13110
ZrO2 0,17709
ZrfBrCfftCHOHCOO). Zr 0,090189
ZrO2 0,12183
ZrO2 Zr 0,74030
ZrCl4 1,8913
Zr(NO3)4 2,7532
Zr(NO3)4 • 5H2O 3,4843
Zr(OH)4 1,2924
ZrP2O7 2,1520
Zr(SO4)2 2,2996
Zr(SO4)2 • 4H2O 2,8844
Zr(SeO3)2 2,8010
ZrP2O7 Zr 0,34401
ZrO2 0,46468
Zr(SeO3)2 Zr 0,26430
ZrO2 0,35701
5.1.2. Важные для гравиметрии свойства отдельных элементов
Таблица 5.1.11.1
Редкоземельные элементы и иттрий
А. Основные гравиметрические формы при определении TR [1]
Осадитель Условия осаждения Условия термической обработки Гравиметрическая форма Фактор пересчета на Ln Литература
H2C2O4 0,1-0,3 М избыток осадителя в кислой среде (до 0,2 и. по НС1). Осаждение из горячего р-ра Высуш. в вакуум-экси-каторе 6—10 мин с 3-кратным продуванием парами эф. Ln2(C2O4)3 • пН2О, где Ln: La (n = 8) Се (n = 10) Рг(п = 9) Y(« = 7) 0,40499 0,38684 0,39805 0,31309 2
Прокаливание при 360-400 °C 790-800 °C 730-750 °C 550-730 °C СеО2 РГбОц ТЬ4О7 La2O2CO3 0,81409 0,82772 0,85021 0,75122 3 3 4 3,5,6
Не менее 800 °C Ln2O3 для всех РЗЭ, кроме Се, Рг иТЬ 0,85269 (La) 0,87939 (Lu) 0,78746 (Y) 3, 4, 6, 7
Химические методы количественного анализа
541
Продолжение таблицы А
Осадитель Условия осаждения Условия термической обработки Гравиметрическая форма Фактор пересчета на Ln Литература
Едкие щелочи или аммиак K4[Fe(CN)6] Небольшой изб. осадителя То же, в присутствии 40-60% сп. 850-900 °C Оксиды (см. Н2С2О4) См. Н2С2О4 8,9
Высуш. на воздухе или в эксикаторе KLn[Fe(CN)6] • иН2О, где Ln: La (п = 7) Се3+ (п = 2) Nd (п = 5) 0,26912 0,32800 0,29721 10, 11, 12
Na3PO4, Na2HPO4 или NaH2PO4 10-3-10“4 М избыток осадителя при pH = 4,5 900 °C LnPO4, где Ln — все TR, кроме Се4* La —0,59394 Lu —0,64820 Y —0,48354 13
ПО °C Се3(РО4)411Н2О 0,42099 14
500 °C Се3(РО4)4 0,52524 14
Na2P2O7 или Na2H2P2O7 10~3—10-4 М избыток осадителя при pH = 4,5 110 °C Nd4(P2O7)3-6H2O 0,47810 13
KIO3 0,25 М избыток осадителя из 4-5 М HNO3 10-15 мин при 40- 45 °C 2Се(Ю3)4- КЮ3-8Н2О 0,13754 15
NH4IO3 0,05-0,25 М избыток осадителя из 0,5-1 н. HNO3 в присутствии 3 г персульфата или 1-3 г бромата на 25 мг Се 800-900 °C СеО2 0,81409 16, 17
KIO4 Избыток при pH = 2-7 Высуш. в вакуум-эк-сикаторе над H2SO4 и NaOH при 100-110 °C 3-4 часа при нормальном давлении СеНЮ6Н2О 0,36677 18, 19
102-130 °C СеНЮ6 0,38492 20
700-800 °C СеО2 0,81409 21
2-3 час при 110 °C Се(Ю4)4 0,15505 22
8-Оксихино-ЛИН См. табл. 5.1.9 110-120 °C Ln(C9H6ON)3 La —0,24313 Lu — 0,28807 23,24 25, 26
800 °C — La 700-750 °C — Nd, Sm, Gd, Y 420 °C — Ce 575 °C — Pr Оксиды (см. Н2С2О4) См. H2C2O4 26
8-Оксихиналь-ДИН То же 650 °C — La, Nd, Gd 610 °C — Sm 570 °C — Pr, Y 380 °C — Ce Оксиды (см. Н2С2О4) Cm. H2C2O4 27
5,7-Дигало-8-оксихинолин То же 580-810 °C для Cl-npo-изводного 520-760 °C для Br-производного 350-590 °C для I-про-изводного То же To же 28
542
Новый справочник химика и технолога
Продолжение таблицы А
Осадитель Условия осаждения Условия термической обработки Гравиметрическая форма Фактор пересчета на Ln Литература
Купферон То же 110-120 °C Ln(C6H5O2N2)3 Ln = La, Ce, Pr, Nd, Sm, Y La —0,25245 29
450-600 °C—La, Sm,Y 610 °C —Но 570 °C — Eu 550 °C — Tb, Dy, Er 520 °C —Yb Оксиды (см. H2C2O4) См. H2C2O4 29, 30
Неокупферон То же 750 °C —Nd 660 °C — Pr, Sm 610 °C —La 570 °C — Eu, Tb 540 °C — Dy, Er, Y 490 °C — Ce, Gd, Yb 450 °C — Ho To же To же 30,31
Салициловая к-та То же 100-105 °C La(C7H5O3)3l,5H2O La — 0,24064 32,33
Б. Состав и термические свойства нормальных оксалатов TR, Y и Sc [1] (см. также [3-7])
Исходный гидрат оксалата Продукты термического разложения гидратов оксалатов и область их устойчивости (в °C )
Начало разложения • лН2О Ln2(C2O4)3 Ln2O3 или другие 1
La2(C2O4)310H2O 2 55 380 800
Се2(С2О4)3ЮН2О 50 (300) 360 (СеО2)
Рг2(С2О4)3ЮН2О 40 420 790(Рг6Оп)
Nd2(C2O4)310H2O 50 445 735
Sm2(C2O4)310H2O 45 300-410 735
Eu2(C2O4)310H2O 60 320 620
Gd2(C2O4)310H2O 45 120 (6Н2О) 315-375 700
Tb2(C2O4)310H2O 45 1403 (5Н2О) 265 (Н2О) 435 725 (ТЬ4О7)
Dy2(C2O4)310H2O 45 1403 (4Н2О) 220-295 (2Н2О) 745
Ho2(C2O4)3-10H2O 40 200-240 (2Н2О) 400 735
Er2(C2O4)3-6H2O 40 175-265 (2Н2О) 395 720
Tu2(C2O4)3-5H2O 55 195-335 (2Н2О) 730
Yb2(C2O4)35H2O 60 175-325 (2Н2О) 730
Lu2(C2O4)3-6H2O 55 190-315 (2Н2О) 715
Y2(C2O4)3-9H2O 45 180-260 (2Н2О) 410 735
Sc2(C2O4)3-6H2O 50 185-220 (2Н2О) 635
Примечания.
1 Минимальная температура разложения оксалатов.
2 При 550-735 °C устойчиво соединение Ln2O3 • СО2.
3 Из-за неясного характера кривых термической диссоциации количество гидратной воды, приведенное в таблице, носит приближенный характер.
Химические методы количественного анализа
543
В. Растворимость оксалатов TR в воде при 25 °C и их произведения активностей
Метод определения Растворимость, мг безводной соли • л Литература
La Се Рг Nd Sm Yb Y
Г равиметрический 2,14 0,96 1,09 1,49 1,48 1,48 34, 55
По электропроводности 2,06 0,62 0,80 2,15 1,98 1,37 34
Объемный 2,08 52
Колориметрический 1,95 44
г 2 3 Произведения активностей L, „ (r А >. = ат 3+ • я г Cn2(C2O4)3 Ln C20j
1,4-1028* 2,2-10’28* 1,8-10’28 * 56
5,9-10’30 7,7-IO32 1,24-10"29 57,58
5,34-10~29 59
(3±1) -1026** 60
Примечания.
* Произведения активностей, полученные пересчетом из произведений растворимости, измеренных при 20 °C.
** Произведение растворимости, полученное при 20 °C.
Г. Растворимость * нормальных оксалатов TR в растворах минеральных кислот ив их смеси с Н2С2О4 при 25 [34]
Кислота Нормальность кислоты Растворимость, г безводной соли • л
La Се Рг Nd Sm Gd
НС1 0,1000 0,206 0,130 0,097 0,075 0,052 0,024
0,2500 0,551 0,366 0,272 0,212 0,176 0,096
0,500 1,393 0,840 0,628 0,445 0,235 0,331
1,000 3,20 2,18 1,60 1,256 0,740 0,930
1,500 5,12 3,68 2,75 2,30 1,50 1,62
2,000 7,02 5,72 4,25 3,44 2,37 2,54
3,000 12,08 10,58 7,85 — — —
4,000 14,23 — — — — —
5,200 14,00 — — 16,6 — —
HNO3* 0,250 0,360 0,361 0,293 0,242 0,191 0,222
2,000 9,94 7,30 5,46 4,58 3,64 2,97
4,000 29,9 25,4 18,5 15,0 11,80 10,03
H2SO4 0,100 0,258 0,165 0,120 0,106 0,105 0,100
0,500 1,300 0,866 0,648 0,525 0,538 0,508
1,000 2,70 1,88 1,40 1,123 1,127 1,068
1,000** 2,80 1,88 1,38 1,204 0,897 1,046
2,000 5,90 4,46 3,26 2,64 2,57 2,37
2,600 7,36 6,34 4,52 3,98 — 3,19
В присутствии 0,1 н. раствора Н2С 2О4
НС1 1,000 0,552 0,282 0,133 0,085 0,063 0,063
2,000 3,10 2,19 1,203 0,772 0,434 0,542
3,000 7,26 6,43 3,88 2,61 1,457 1,75
4,000 11,10 12,48 7,63 6,07 3,50 4,12
HNO3 2,00 4,17 3,37 1,38 1,212 0,963 0,818
3,00 13,32 10,02 6,08 4,92 3,09 3,00
4,00 17,8 19,8 12,00 — 7,17 6,22
544
Новый справочник химика и технолога
Продолжение таблицы Г
Кислота Нормальность кислоты Растворимость, г безводной соли • л
La Се Рг Nd Sm Gd
В присутствии 0,5 н. раствора Н2С 2O4
НС1 1,00 0,063 0,050 0,027 0,021 0,010 0,011
2,00 — 0,351 0,180 0,116 0,063 0,038
3,00 1,340 1,452 0,668 0,429 0,214 0,263
4,00 3,46 4,26 2,13 1,154 0,656 0,745
HNO3 2,00 1,386 0,563 0,313 0,209 0,144 0,137
3,00 3,86 2,77 1,41 0,862 0,538 0,492
4,00 8,22 9,04 4,54 2,88 1,82 1,59
В присутствии насыщенного раствора Н2С2О4
НС1 1,50 0,063 0,074 0,049 0,035 0,033 0,028
4,00 1,98 3,47 1,094 0,860 0,316 0,528
6,00 — 2,13 1,70 2,07 1,330 2,19
6,20 — 1,84 1,53 2,04 — —
HNO3 4,00 9,02 4,72 0,779 1,140 — 0,449
6,00 16,2 16,5 11,30 4,22 — 1,515
Примечания.
* Цифровой материал таблицы пересчитан с процентного содержания на концентрацию. Плотности растворов вычислены на основе плотностей растворов отдельных компонентов (минеральная кислота, Н2С2О4, соль) в предположении, что они складываются аддитивно.
** Растворимость оксалатов Dy, Er и Yb равна соответственно 1,437; 2,00 и 1,313 г • л 1 [61].
Д. Значение pH начала осаждения и произведения растворимости гидроксидов TR при 25 ЯС [1]
Пояснение. Приведенные в таблице значения произведений растворимости получены методом титрования растворов солей TR щелочью. Величину произведения растворимости рассчитывали из значения исходной концентрации соли TR и pH начала образования осадка гидроксида [1, с. 2].
Элемент pH начала осаждения Произведение растворимости [Ln3+][OIT]3 Литература Произведение активностей [35, 36, 37]
сю; no; СГ sot
La 7,82 7,41 1,0-10 19 38 2,010-22
8,03 7,61 5,010 21 (СГ)* 39
5,5-10’21 (SO2)* 39
8,35 6,01 O’20* 40
8,71 4,3-10~19 41
8,10 1,7-IO-19 42
1,2-ю-19** 43
1,Ь10"21** 43
0,9-IO-21 44
Се 7,60 7,35 1,5- IO-20 38 6,3-1022
8,1 0,8-IO-20 41
7,41 40
7,07 1,3-Ю"23* 39
8,04 45
7,32 45
Pr 7,35 7,17 2,7-10-20 38 8,32-10’23
7,05 6,4-10 24 * 40
6,98 7,l-10’24* 39
7,40 6,7-10"22 42
Nd 7,31 6,95 1,9-10’21 38 (8,7±4,7)’10-24
7,02 5,4-10’24 (NO;)* 40 1,3-10’24
Химические методы количественного анализа
545
Продолжение таблицы Д
Элемент pH начала осаждения Произведение растворимости [Ln3+][OH~]3 Литература Произведение активностей [35, 36, 37]
сю; no; СГ SO2"
Nd 7,00 5,2-10’24 (Cl")* 40
6,73 1,2-10’24* 39
7,30 3,2-10’22 42
Sm 6,92 6,70 6,8-10’22 38 1,3-10-26
6,83 40
7,13 8,4-10’23 42
Eu 6,82 6,68 3,4-10’22 38 2,88-10-27
6,91 0,9-1023 42
Gd 6,83 6,75 2,1-10“22 38 1,3-10"27
6,84 1,8-10’23 42
Tb 4,90-10’27
Dy 1,3-10’26
Ho 2,82-10’27
Er 6,76 6,50 1,30-10’23 38 2,69-10’27
6,61 4,1-10’24 42
Tu 6,40 6,21 3,3-10’24 38
Yb 6,30 6,18 2,9-10’24 38 2,29-10~27
6,16 39
6,45 2,5-10’24 42
Lu 6,30 6,18 2,5-10”24 38 1,02-10’27
6,45 1,9-10'24 42
Y 6,95 6,83 8,1-1023 38 3,2-10'25
6,78 1,2-10"24* 40
7,39 41
6,81 1,6-10 23 42
Примечания.
* Произведение растворимости вычислено И. М. Коренманом для 18 °C [46].
** Данные получены при 18 °C.
Е. Растворимость карбонатов TR в растворах карбонатов калия, натрия и аммония при 19-23 °C *[1, 47]
Избыток осадителя, н. Растворимость Ln2O3, ммоль-л 1
La Nd Y Er
К2СО3
0 0,27 0,18 0,97 1,15
1 0,46 0,50 1,19 3,24
2 0,55 0,56 13,68 14,30
4 0,89 3,24 69,57 74,47
4,5 — 4,13 73,03 >81,30
6 1,56 > 80,00 > 82,80 >81,30
16 25,20 > 80,00 > 82,80 >81,30
Na2CO3
0 0,031 0,148 0,31 0,65
1 0,061 0,208 0,97 1,38
2 0,092 0,237 9,83 11,58
3,05 0,123 0,416 12,88 37,55
(NH4)2CO3
0 0,12 0,18 0,00 0,10
Продолжение таблицы Е
Избыток осадителя, н. Растворимость Ln2O3, ммоль л 1
La Nd Y Er
1 0,12 0,26 0,49 0,60
2 0,12 0,44 0,66 0,89
4 0,09 — — 1,09
6 0,24 0,50 — 6,90
9,2 0,24 1,07 20,19 26,85
Примечание. * В результате обменной реакции (18 ч) в равновесном растворе присутствовал также 1 н. КС1.
Ж. Произведения растворимости соединений LnF3 при 25 °C
Элемент Метод определения ПР Литература
La Потенциометрия (1,4 ± 0,1)1018 48
Ce To же (1,4 ± 0,2)-10"18 48
Радиометрия (8,1 ± 1,1)-10"’6 49
Кондуктометрия (1,1 ± 0,5)-1015 49
Gd Потенциометрия (6,7 ± 0,2)-10’17 48
546
Новый справочник химика и технолога
3. Растворимость нормальных йодатов TR и Th при комнатной температуре
Концентрация HNO3, н. Растворимость йодатов, ммоль Метод определения Литература
Th La Се3+ Y
7,0 5,0-10’3 Соосажд ение на А1(ОН)3 50
4,0 1,5-10"3 То же
3,0 6,3-104 То же
2,0 6,0104 То же
1,0 517 16180 51*
0,0032 7,1 87 Радиометрия
0,0 2,82 2,46 Элек-гропрово дность ** 52
Примечания.
* Измерения проведены при 20 °C.
** Измерения проведены при 25 °C.
К. Растворимость СеР2О7 в H2SO4 при 25,0%! [53]
Концентрация H2SO4, н. Растворимость, гл1
0,0 0,00007 ± 0,00005
0,114 0,026 ± 0,005
0,228 0,046 ± 0,005
0,342 0,073 ± 0,005
0,684 0,207 ±0,010
Л. Растворимость гипофосфатов TR в растворах НС1 при 25,0 % [54]
Гипо-фосфат НС1, н. Растворимость, г(безводной соли)-л 1 Гипо-фосфат НС1, н. Растворимость, г(безводной соли) л1
0,05 0,0116 Y4(P2o6)3 0,20 0,251
0,10 0,0504 0,30 0,458
0,20 0,168 1,00 2,23
0,30 0,210
1,00 1,52
2,00 3,65
М. Влияние pH раствора на растворимость La(OH)3 при 25% [61]
И. Состав осадка йодатов церия
Пояснение. Приведены условия, соответствующие образованию одной формы без примеси других [1].
Состав Концентрация HNO3, н.
Се(Ю3)4-5Н2О 5
Се(1О3)4-4Н2О 0,5-1,0
Се(Ю3)з(ОН)-2Н2О 0,1-0,5
Се(Ю3)2(ОН)2Н2О 0,015
Се(ОН)3Юз <0,01
рн Растворимость La(OH)3, мг(Ьа2О3) л 1 pH Растворимость La(OH)3, мг(Ьа2О3) • л 1
7,66 63,4 9,20 0,127
7,86 20,35 9,66 0,126
8,07 19,65 9,88 0,057
8,37 4,69 10,17 0,086
8,43 3,64 10,30 0,084
9,00 0,402 10,44 0,005
Н. Растворимость фосфата Се4+ в кислотах при 100 % (г -л ') [14] (Соединение не растворимо в воде и СНзСООН)
Фосфат Се3(РО4)4 • иН2О НС1 H2SO4 HNO3
1,4 н. (5%) 2,9 н. (10%) 1,0 н. (5%) 2,2 н. (10%) 0,8 н. (5%) 1,7 н. (10%) 13 н. (60%)
Свежеосажденный 0,2 0,5 0,86 1,5 1,2 1,9 Растворяется
Высушенный при 110 °C 2 3 0,8 1,6 1 1,6 То же
Прокаленный Не растворяется 1,4
О. Растворимость ЬаРО4 • пН2О в избытке фосфорной кислоты [63] (концентрации пересчитаны с масс. % Ьа2О3 и Р2О5 в молярные для Н3РО4 и LaPO4 [1])
Н3РО4, М Растворимость безводной соли при 20-23 °C, (М • л"1) • 103 Н3РО4, М Растворимость безводной соли при 80 °C, (М л4) • 103
1,58 2,71 1,60 0,664
3,56 8,03 3,46 1,51
5,87 12,9 5,75 2,76
8,48 16,5 8,28 4,58
Химические методы количественного анализа
547
П. Растворимость оксалатов TR (в г безводной соли нал*) в HNO3 и смесях HNO3 + 0,8 н. Н2С2О4 при 90 °C[64]
Нормальность HNO3 La Pr Nd Sm Dy Y
Растворимость в HNO3
0,799 7,35 0,38
1,25 14,3
1,558 20,6 17,8
2,337 30,5
2,50 32,1 30,4 29,2 25,8
3,75 77,7
5,00 173 113,5 111,3 91,8 76,6 59,0
Растворимость в HNO3 + 0,8 н. Н2С2О4
0,799 0,503 0,458
1,25 1,50
1,558 5,13 2,70
2,337 12,5 11,43
2,50 И,1 8,82 6,65 8,43
3,75 58,7
5,00 156 86,3 84,2 67,2 54,0 28,4
Примечание.
*В работе [64] растворимость дана в граммах на 100 мл раствора. По мнению авторов статьи [64] величины растворимостей, особенно при высоких концентрациях HNO3, могут быть несколько ошибочны, поскольку равновесие в системах не всегда достигалось, вероятно, из-за медленного перехода оксалатов в оксалато-нитраты [1].
Р. Константы устойчивости комплексных оксалатов TR при 25 °C
Пояснение. Константы получены приближенным расчетом.
— пересчита-
Кс с 0 — вычислена из концентрации ионов, К
v'2'-z4 /2
на исходя из активностей. Все остальные константы вычислены исходя из активностей.
Элемент ^LnC,O4 К Ln(C2O4 )2 К Ln(C2O4 )3 Литература
Ce 3,3106 9,0-103 6,7 58
La (1,1 ±0,4)-106 (3,0-103) — 60
Nd l,6-107 2,0-104 < l,l-10“2 58
Yb 2,0-107 3,8-104 < l,l-10“2 57
Y «3-106 «1-1О10 2,910й 59
С. Состав осадков TR с ферроцианид-ионом в присутствии ионов одновалентных металлов и растворимость соединений в воде (в молях) при 25 °C [10—12, 65]
Пояснение. Для гравиметрического анализа в качестве осадителя наиболее удобен ферроцианид калия как с точки зрения единообразия состава осадков, так и в смысле полноты осаждения [1]. Кристаллогидраты, высушенные на воздухе или в эксикаторе [68], могут служить непосредственно гравиметрической формой для определения TR.
Элемент Li+ Na+ K+ ** Rb+ Cs+ тГ
La LiLa[Fe(CN)6]- •7H2O* 3-lff4 NaLa[Fe(CN)6]-•иН2О 5,2-10"* KLa[Fe(CN)6]-•7H2O 2,76-10"* RbLa[Fe(CN)6]- 7H2O CsLa[Fe(CN)6]- иН2О TlLa[Fe(CN)6]-•4H2O 2, 1-10 5
2-10"* 4,2-10"* 2,28-10"*
Ce Ce4[Fe(CN)6]3- •20H2O Ce4[Fe(CN)6]3- 2OH2O KCe[Fe(CN)6]- •2H2O*** RbCe[Fe(CN)6]- 2H2O CsCe[Fe(CN)6]- •2H2O TlCe[Fe(CN)6]- •2H2O
548
Новый справочник химика и технолога
Продолжение таблицы С
Элемент Li+ Na+ к+ ** Rb+ Cs+ ТГ
Се 1,3-10 4 при избытке [FetCN),]4-: Na4Ce8-[Fe(CN)6]7- •иН2О 2,1-10"* 2,4-1 0"* 7,1-105 1,7-10"*
Nd Nd4[Fe(CN)6]3- •иН2О Nd4[Fe(CN)6]3-•иН2О при избытке [Fe(CN)6]4?NaNd-[Fe(CN)6] 9,3-105 10,8-10’5 KNd[Fe(CN)6]- •5Н2О 2-104 2,4-1 О'4 RbNd[Fe(CN)6]- •4Н2О 610 5 CsNd[Fe(CN)6]- •иН2О
Y Осадок не образуется Осадок не образуется K4Y8[Fe(CN)6]7- •зон2о 6,2-1 О’6 RbY[Fe(CN)6]-•2Н2О 1,8-104 CsY[Fe(CN)6]-•2Н2О 5,2-10’5
Примечание.
* Количество гидратной воды дается для соли, высушенной на воздухе.
** Состав KLn[Fe(CN)6]«H2O подтвержден также и для Рг, Sm, Gd, Dy, Er и Y [65], хотя имеются противоречивые данные по отношению к La [67].
*** По другим данным, соль содержит 4 молекулы Н2О, а после высушивания при 100 °C — ЗН2О [68].
Литература
1. Рябчиков Д.И., Рябухин В.А. Аналитическая химия редкоземельных элементов и иттрия. М.: Наука. 1966.
2. Ballou N.E. Radiochemical Studies of the Fission Products. Div. IV. V. 9. N.-Y: McGraw-Hill, 1951. P. 1673.
3. Wendlandt W.W. I I Anal. Chem. 1958. V. 30. P. 58.
4. Wendlandt W.W. I I Anal. Chem. 1959. V. 31. P. 408.
5. Caro P., Loriers J. 11 J. Rech. Centre Natl. Recherche Sci., Lab. Bellevue. 1957. V. 52, № 39. P. 107.
6. Padmanabhan V.M., Saraiya S.C., Sundaram A.K. // J. Inorg. Nucl. Chem. 1960. V. 12. P. 356.
7. Duval С. 11 Anal. Chim. Acta. 1954. V. 10. P. 321.
8. Морозов И.С. II Журн. неорг. химии. 1956. T. 1. С. 791.
9. Moosath S.S. И J. Inorg. Nucl. Chem. 1959. V. 11. P. 286.
10. Тананаев И.В., Глушкова M.A., Сейфер Г.Б. Химия редких элементов: Сборник. Вып. 1. / Под ред. Тана-наева И.В. М.: Изд. АН СССР. 1954. С. 58.
11. Тананаев И.В., Левина М.И. Химия редких элементов: Сборник. Вып. 3. М.: Изд. АН СССР. 1957. С. 28.
12. Тананаев И.В., Сейфер Г.Б. И Журн. неорг. химии. 1956. Т. 1.С. 53.
13. Buyers A.G., Giesbrecht Е., Audrieth L.F. И J. Inorg. Nucl. Chem. 1957. V. 5. P. 133.
14. Atanasiu J. A., Babor M. I I Bull. Sect. Sci. Acad. Rou-maine. 1938. V. 20. № 1-3. P. 32.
15. Чернихов Ю.А., Успенская T.A. 11 Завод, лаб. 1940. T. 9. С. 276.
16. Willard Н.Н., Yu S. T’say // Anal. Chem. 1953. V. 25. P. 1754.
17. Редкоземельные металлы: Сборник статей. М.: Гос-атомиздат, 1962. С. 247.
18. Ray Choudhuiy Р.С. И J. Indian Chem. Soc. 1941. V. 18. Р. 335.
19. Venkataramaniah М, Raghavarao Bh. S.V. И Current Sci. (India). 1949. V. 18. P. 248.
20. Пуздренкова И.В., Алимарин И.П., Фролкина B.A. // Вести. МГУ. Серия мат., мех., астр., физ. и химии. 1958. Т. 13, №2. С. 183.
21. Александров Г.П., Тихонова В.С. // Укр. хим. журн. 1956. Т. 22. С. 379.
22. Venugopalan М., George K.J. // Naturwiss. 1956. Bd. 43. S. 348.
23. Berg R., Becker E. 11Z. Anal. Chem. 1940. Bd. 119. S. 1.
24. Hagiwara Z. 11 Technol. Repts. Tohoku Univ. 1953. V. 17. P. 83.
25. Hagiwara Z. // J. Chem. Soc. Japan, Ind. Chem. Sect. 1953. V. 56. P. 566.
26. Wendlandt W.W. //Anal. Chim. Acta. 1956. V. 15. P. 109.
27. Wendlandt W.W. I I Anal. Chim. Acta. 1957. V. 17. P. 274.
28. Wendlandt W.W. 11 Anal. Chim. Acta. 1957. V. 17. P. 428.
29. Wendlandt W.W. H Anal. Chem. 1955. V. 27. P. 1277.
30. Wendlandt W.W. 11 Anal. Chim. Acta. 1959. V. 21. P. 116.
31. Wendlandt W.W., Bryant J.M. I I Anal. Chim. Acta. 1955. V. 13. P. 550.
32. Звягинцев O.E., Судариков Б.Н. И Журн. неорг. химии. 1956. Т. 1.С. 69.
33. Фиалков Я.И., Ермоленко В.И. // Журн. неорг. химии. 1959. Т. 4. С. 359.
34. Sarver L.A., Brinton Р.Н.М.Р. // J. Am. Chem. Soc. 11927. V. 49. P. 943.
35. Аксельруд H.B. Редкоземельные элементы: Сборник/ Под ред. Рябчикова Д.И. М.: Наука. 1963. С. 75.
36. Аксельруд Н.В., Спиваковский В.Б. // Журн. неорг. химии. 1959. Т. 4. С. 56.
37. Аксельруд Н.В., Спиваковский В.Б. И Журн. неорг. химии. 1960. Т. 5. С. 327, 340, 348, 547.
Химические методы количественного анализа
549
38. Moeller Т., Kremers Н.Е. // Ind. Eng. Chem., Anal. Ed. 1945. V. 17. P. 798.
39. Bowles J.A., Partridge H.M. // Ind. Eng. Chem., Anal. Ed. 1937. V. 9. P. 124.
40. Britton H.T.S. // J. Chem. Soc. 1925. P. 2142.
41. Oka Y. // J. Chem. Soc. Japan. 1938. V. 59. P. 971.
42. Moeller T., Fogel N.//J. Am. Chem. Soc. 1951. V. 73. P. 4481.
43. Sadolin E. // Z. Anorg. Allg. Chem. 1927. Bd. 160. S. 133.
44. Kolthoff J.M., Ehnquist R. // J. Am. Chem. Soc. 1931. V. 53. P. 1217.
45. Pan Kuan, Hseu Tong Ming // Bull. Chem. Soc. Japan. 1955. V. 28. P. 309.
46. Коренман И.М. // Ж. общей химии. 1955. Т. 25. С. 1859.
47. Скляренко Ю.С. Дис. ... канд. хим. наук. М. ГЕОХИ АН СССР. 1953.
48. Kury J.W. // US АЕС. UCRL-2271. 1953; С.А. 1954. V. 48. 3833.
49. Weaver J.L., Purdy W.C. // Anal. Chim. Acta. 1959. V. 20. P. 376.
50. Stine C.R., Gordon L.E. // Anal. Chem. 1953. V. 25. P. 1519.
51. Шведов В.П., Мусаев Ш.А. // Радиохимия. 1959. Т. 1. С. 465.
52. Rimbach Е., Schubert А. // Z. Physik. Chem. 1909. Bd. 67. S. 183.
53. Brantley J.C., Huizenga J.R. // J. Am. Chem. Soc. 1952. V. 74. P. 6101.
54. Рябчиков Д.И., Беляева B.K., Ермаков А.Н. // Журн. неорг. химии. 1956. Т. 11. С. 658.
55. Hauser О., Meyer R.J. Die Analyse der Seltenen Erden und der Erdsauren. Stuttgart. Verl. Enke F. 1912. S. 61.
56. Коренман И.М. // Журн. общей химии. 1954. Т. 24. С. 1910.
57. Crouthamel С.Е., Martin D.S., Jr. // J. Am. Chem. Soc. 1950. V. 72. P. 1382.
58. Crouthamel C.E., Martin D.S., Jr. //, J. Am. Chem. Soc. 1951. V. 73. P. 569.
59. Feibush A.M., Rowley K., Gordon L. // Anal. Chem. 1958. V. 30. P. 1610.
60. Бабко A.K., Дубовенко Л.И. // Журн. неорган. химии. 1957. Т. 2. С. 808.
61. Bodlander Е. Dissertation. Berlin. 1929. S. 63. Цит. по [1].
62. Lewis D.C. // Chem. and Ind. 1957. P. 1238.
63. Василенко H.A., Чепелевецкий М.Я. // Журн. неорган. химии. 1957. Т. 2. С. 2486.
64. Neckers J.W., Kremers Н.С. // J. Am. Chem. Soc. 1928. V. 50. P. 950.
65. Тананаев И.В., Глушкова M.A. // Журн. неорган. химии. 1957. Т. 2. С. 2475.
66. Prandt W., Mohr S. // Z. Anorg. Allg. Chem. 1938. Bd. 236. S. 243.
67. Шемякин Ф.М., Волкова B.A. // Журн. общей химии. 1937. Т. 7. С. 1328.
68. Spacu Р. // Z. Anal. Chem. 1936. Bd. 104. S. 28.
Таблица 5.1.11.2
Кадмий
Электрогравиметрическое определение кадмия. Осаждение на платиновом и других электродах [1—3]
Количество кадмия, г Объем раствора, мл Среда Температура, °C Напряжение, В Сила или плотность тока Скорость вращения электродов, об мин 1 Продолжительность электролиза, мин
0,2-0,3 200 10-15 мл НС1 (уд. в. 1,19) + + 2 г NH2OHHC1 70 1,4-1,8 1,0 А 35
0,2 100 0,5 н. H2SO4 18 2,8 600-800 20
0,2 200 0,5 мл H2SO4 + 4-6 г (NH4)2SO4 60 6-7 ЗЛА (5 А на 100 см2) 25
0,15-0,3 120 1-3 мл H2SO4 (1:10) * 8-9 5 А 600 10-15
0,05-0,8 65-75 12 капель H2SO4 (1:4) 20 8 6-9 А на 100 см2 600 35
0,2-0,4 120-200 2 мл H2SO4 (уд. в. 1,8) + NaOH (до помутнения) + 6-12 г KHSO4 18 3,8 2,4 А 300 40
0,1 85 2 мл H2SO4 (уд. в. 1,84) + 3-4 г NaOH + 1,25 мл СН3СООН 80 1,12-1,20 0,4-2,0 А 1200 10-15
0,1-0,2 90-120 3 г CH3COONa + 0,25 мл СН3СООН * 9 5 А 600 4
0,05-0,08 60-65 0,5 г CH3COONa + 1 г K2SO4 20 8 3 А на 100 см2 600 15-20
0,08 70 1-1,5 г NaOH+ 0,5-1 г KCN 20 8 7,5 А на 100 см2 600 30-35
0,2 Нейтрализация разбавленным раствором NaOH до помутнения и еще 2-3 мл + KCN до растворения осадка + 0,5 г KCN 20 3-4 А на 100 см2 600-800 20-30
Примечание.
* Горячий раствор.
550
Новый справочник химика и технолога
Литература
1. Kolthoff I.M., Elving P.J. Treatise on Analytical Chemistry. Part II. V. 3. New York; London, 1961. P. 197.
2. Schleicher A. Elektroanalytische Schnellmethoden. Stuttgart. 1947. S. 111.
3. Щербов Д.П., Матвеец M.A. Аналитическая химия кадмия. М.: Наука, 1973. 61 с.
Таллий
А. Растворимость и произведение растворимости соединений таллия [13]
Таблица 5.1.11.3
Соединение Формула Мол. масса, г-моль 1 Температура, °C Растворимость Коэффициент активности Произведение растворимости Литература
моль-л 1 г-л 1 катион анион
Азид T1N3 246,4 0 6,9- Ю3 1,7 0,91 0,91 3,9-10’5 1
25 1,42-102 3,5 0,87 0,87 1,5-10^ 1,2
Тетрафенил бортал-лий Т1[В(С6Н5)4] 523,2 20 9,5-10’5 5-Ю’2 — — 9,0-10’9 3
Бромат Т1ВгО3 332,4 20 1,04-10’2 3,46 0,90 0,90 8,9-105 4
Бромид Т1Вг 284,3 0 8,45-10^ 0,24 — — 7,1 -107 5
20 1,7-10’3 0,48 0,96 0,96 2,6-10"6 1,4, 6,7
25 1,98-10'3 0,57 0,96 0,96 3,6-10'6 1,7, 8,9
40 3,3-10’3 0,95 0,94 0,94 9,7-10~6 7
60 6,6-10’3 1,9 0,92 0,92 3,6-105 7
Висмутио-ДИД Т12[ВП5] 634,6 20 4-10’5 0,05 — —- 2,5-10 13 10, 11
Иодат Т1Ю3 379,3 0 6,5-104 0,24 — — 3,98-IO’7 12
20 1,5-10 3 0,58 0,96 0,96 2,0-10’6 1,4
25 1,84-10’3 0,68 0,96 0,96 3,0-1 О’6 1, 12, 14, 15
40 3,04-10’3 1,15 0,93 0,93 8,1-Ю"6 12
Иодид T1I 331,3 0 6,0-10’5 0,02 — — 3,9-10’9 7, 16, 17
10 9,0-10’5 0,03 — — 8,1-Ю’9 4
20 1,9-10^ 0,063 — — 3,6-10’8 4, 6, 7, 18-20
40 4,5-1О4 0,15 — — 2,0-10’7 21
60 1,0-10“3 0,35 0,97 0,97 9,4-10’7 21
80 2,1-103 0,70 0,96 0,96 4,0-10"6 21
100 3,6-10'3 1,20 0,94 0,94 1,1-10~5 21
Кальций-ферроцианид Tl2Ca[Fe(CN)6] 660,7 25 3,4-10^ 0,22 0,94 0,78 0,38 1,4-10-14 22
Гексанит-рокобальтат T13[Co(NO2)6] 936,1 10 7,5-10’5 0,071 — — 8,5-Ю’16 23,24
20 8,5-10’5 0,080 — — 1,4-1015 23
30 1,25-10’5 0,118 — — 6,6-1015 23
Лантан-ферроциа-нид TlLa[Fe(CN)6]. •4Н2О 627,1 25 2,1-Ю5 0,0116 — — 9,3-1015 25
Магний-ферроцианид Tl2Mg[Fe(CN)6] 644,1 25 2-Ю*4 0,13 0,96 0,82 0,45 2,Ы015 22
Метавана-данат T1VO3 303,3 11 2,9-10’3 0,87 0,94 0,94 7,5-10^ 17, 26
100 7-10’3 2,1 0,91 0,91 4,0- Ю5 26
Оксихино-линат T1(C9H6NO)3 636,4 — — — — — 6,3-10’28 27
Химические методы количественного анализа
551
Продолжение таблицы А
Соединение Формула Мол. масса, г-моль1 Температура, °C Растворимость Коэффициент активности Произведение растворимости Литература
моль-л 1 г-л 1 катион анион
Ортованадат T13VO4 728,1 15 1,37-10~5 0,01 — — 9,5-10 49 17
Перренат TlReO4 454,7 0 2,53-10"3 1,15 0,95 0,95 5,8-1 О'6 28, 29
20 3,52-10’3 1,6 0,94 0,94 1,1-10'5 28, 29
30 6,55-103 2,98 0,92 0,92 3,6-10"5 28, 29
50 1,22-10’2 5,55 0,90 0,90 1,2-10"* 28,29
Пикрат Т1СбН2К3О7 432,5 0 3,1-10’3 1,35 0,94 0,94 8,5-10'6 7, 26
15 8,2-10’3 3,57 0,91 0,91 5,6-10'5 7,30
20 8,8-10’3 3,8 0,91 0,91 6,4-105 7,31
Пикролонат tic10h7n4o5 467,4 20 1,49-10 4 0,07 — — 2,2-10"8 32
Пированадат T14V2O7 1031,4 15 1,9 10 4 0,2 0,96 0,45 2,4-1017 17
100 2.5-10' 2,6 0,83 0,1 1,2-1012 26
Рейнекеат Tl[Cr(SCN)4- •(NH3)2] 522,7 18 8,7-10'6 1,76- •10’3 — — 7,5-10’11 33,34
Роданид T1SCN 262,4 20 1,2-10’2 3,16 0,89 0,89 1,1-10"* 4, 7, 9, 21,35
25 1,5-10“2 3,92 0,87 0,87 1,7-10"* 7, 9, 21, 36
40 2,8-10 2 7,32 0,82 0,82 5,3-10"* 9,21,26
Селенид Tl2Se 487,7 20 1,2-109 5,8- •10’7 — — 6,9-102 14
Стронций-ферроцианид Tl2Sr[Fe(CN)6] 707,8 25 3,6-10’3 2,55 0,77 0,5 0,07 1,4-10’11 22
Таллихло-рид T13[T1C16] 1030,3 15 2,52-10’3 2,6 0,87 0,33 2,3-1010 26, 37
25 3,29-10 3 3,4 0,85 0,28 5,4-10’10 26, 37
Теллурид Tl2Te 536,4 20 2,9- Ю9 1,5- •10”6 — — 9,5-10’26 14
Тиокарбонат T12CS3 517,0 20 1,93-105 0,01 — — 2,9-10-14 38
Тиосульфат T12S2O3 520,9 25 3,7-10’3 1,92 0,90 0,66 1,08-10’7 39, 40
Ферроцианид Tl4[Fe(CN)6- •2H2O 1067,4 18 3,6-10’3 3,7 0,77 0,07 3,8-10“14 7, 26, 30
Фосфат T13PO4 708,2 20 7,06-10’3 5,0 0,79 0,20 6,6-10 9 1, 19, 26
100 9,46-103 6,7 0,90 0,15 2,3-10'8 1, 19, 26
Хлорид T1C1 239,8 0 7,1-10“3 1,7 0,91 0,91 4,1-105 5,21,35, 41,42
10 1,0-10’2 2,4 0,90 0,90 8,110 s 21,43,44
20 1,4-10’2 3,4 0,87 0,87 1,5-10"* 6, 8,45- -47
40 2,1-Ю’2 5,2 0,85 0,85 3,1-10"* 48-50
60 3,3-Ю2 8,0 0,81 0,81 7,1-10"* 37, 48
80 5,0-102 12 0,75 0,75 1,4-10'3 37
100 7,5-Ю2 18 0,69 0,69 2,7-10-3 17
Хлороплатинат Tl2[PtCl6] 816,7 15 7,8-10’5 0,064 — — 1,9-10”12 7, 19
100 6,2-10 4 0,51 0,97 0,82 7,3-10'10 7, 19,51
Хромат Tl2CrO4 524,8 20 8,0-10 5 0,042 — — 2,0-10’12 52, 53
60 5,7-10"* 0,3 0,97 0,82 5,7-Ю10 26, 54, 55
100 4,3-10’3 2,28 0,88 0,65 1,7-107 19, 26, 55
552
Новый справочник химика и технолога
Б. Коэффициент активности иона Т1+ [56]
Ионная сила раствора, ц 0,001 0,002 0,005 0,01 0,02 0,05 0,10
Коэффициент активности, f 0,97 0,96 0,93 0,90 0,85 0,75 0,64
В. Константы гидролиза иона Т13+ [13]
Реакция Температура, °C К -IgK Литература
Т13+ + Н2О Т1ОН2+ + Н+ — 0,072 1,14 ' 57
Т13+ + Н2О Т1ОН2+ + Нг 25 3,2 -0,50 58
Т13+ + Н2О Т1ОН2+ + IT 32,2 5,3 -0,72 58
Т13+ + Н2О <=> Т1ОН2+ + нг 41,8 6,9 -0,84 58
Т1ОН2+ + Н2О «> Т1(ОН)2+ + н+ — 0,032 1,49 57
T1(NO3)3 + ЗП2О<=> Т1(ОН)3 + 3HNO3 — 13,6 -1,13 59
Литература
1. Справочник химика. Т. 3. М.: Госхимиздат, 1952.
2. Suzuki S. // J. Chem. Soc. Japan, Pure Chem. Soc. 1952. V. 73. P. 150.
3. Geilmann W. // Angew. Chem. 1954. Bd. 66. S. 454.
4. Bottger W. // Z. phys. Chem. 1903. Bd. 46. S. 602.
5. Химические реактивы и препараты / Под ред. В.И. Кузнецова. М.: Госхимиздат, 1953. С. 148.
6. Kohlrausch F. // Z. phys. Chem. 1903. Bd. 44. S. 197; 1905. Bd. 50. S. 355.
7. Seidell A. Solubilities. New York, 1940. V. 1. P. 105, 1424, 1538, 1540, 1542, 1543, 1552.
8. Егоров А.М. // Ж. неорг. химии. 1957. Т. 2. С. 460.
9. Abegg R. Handbuch der anorganischen Chemie. Bd. 3. Leipzig, 1906. S. 418,424.
10. Бусев А.И. Аналитическая химия висмута. М.: Изд. АН СССР, 1953.215 с.
11. Canned G., Реппа G. // Gazz. chim. ital. 1922. V. 52, I. P. 245. Цит. по [10].
12. Bell R.P., George J.H. // Trans. Faraday Soc. 1953. V. 49. P. 619.
13. Коренман И.М. Аналитическая химия таллия. М.: АН СССР, 1960.
14. Латимер В. Окислительные состояния элементов и их потенциалы в водных растворах / Под ред. К.В. Астахова. М.: ИЛ. 1954. С. 168-170.
15. Мег V.K., Goldman F.H. // J. Am. Chem. Soc. 1929. V. 51. P. 2632.
16. Сендэл Е.Б. Колориметрическое определение следов металлов / Под ред. А.С. Комаровского. М.: Госхимиздат, 1949. С. 190-192.
17. Pascal Р. // Traite de chimie minerale. Paris. 1933. T. 9. P. 370, 375, 378, 394.
18. Чин C.C. // Журн. физ. химии. 1952. T. 26. С. 960.
19. Crookes W. // Chem. News. 1863. Bd. 8. S. 159, 243, 255;
1864. Bd. 9. S. 1, 37; Z. Analyt. Chem. 1865. Bd. 4. S. 110.
20. Kohlrausch F. // Z. Phys. Chem. 1908. Bd. 64. S. 129, 168.
21. Мельников H.H. // Успехи химии. 1935. T. 4. С. 255.
22. Тананаев И.В., Глушкова М.А. И Журн. неорг. химии.
1957. Т. 2. С. 280.
23. Коренман И.М. и др. // Журн. неорг. химии. 1958. Т. 3. С.1188.
24. Rosenbladt Th. // Вег. 1886. Bd. 19. S. 2515.
25. Тананаев И.В., Глушкова М.А. И Журн. неорг. химии. 1957. Т. 2. С. 2475.
26. Handbook of Chemistry and Physics / Ed. C.D. Hodman. 1955. P. 384, 608.
27. Borell M., Paris R. // Anal. Chim. Acta. 1952. V. 6. P. 389; C.A. 1952. V. 46. 7850.
28. Jaeger, Beintma. // Proc. Konikl. nederl. akad. wet. 1933. Bd. 36. S. 523; Цит. no [29].
29. Smith W.T. // J. Am. Chem. Soc. 1951. V. 73. P. 77.
30. Lami A. et Des Clouseaux // Ann. Chim. Phys. 1869. Bd.(4)17. S. 310, 344; Z. Anal. Chem. 1870. Bd. 9. S. 384.
31. Rabe O. // Z. Phys. Chem. 1901. Bd. 338. S. 175.
32. Anderson J.R. // Roy. Austral. Chem. Inst. 1950. V. 17. P. 120.
33. Багбанлы И.Л. И Докл. АН АзербССР. 1956. T. 12. C. 459.
34. Воскресенская H.T. // Журн. аналит. химии. 1955. T. 10. С. 222.
35. Noyes А.А. // Z. Phys. Chem. 1890. Bd.6. S. 249; 1892. Bd. 9. S. 603; 1895. Bd. 16. S. 132.
36. Ripan R., Popper E. // Z. anal. Chem. 1943. Bd. 125.
S. 269; 1944. Bd. 127. S. 172.
37. Spenser J.F., Le Pla M. // J. Chem. Soc. 1908. Bd. 93. S. 858.
38. Welcher F. // Organic Analytical Reagents. New York, 1947. V. 2. P. 416; 1948. V. 4. P. 147.
39. Новаковский M.C., Рязанцева А.П. // Уч. зап. Харь-ковск. ун-та, тр. ин-та химии и хим. ф-та. 1954. Т. 11. С. 89.
40. Nilsson R.O. // Arkiv Kemi. 1958. V. 12. Р. 371; РЖ Хим. 1958. №24. 106.
41. Bray W.C., Winninghoff W.J. // J. Am. Chem. Soc. 1911. V. 33. P. 1633.
42. Hill A.E. // J. Am. Chem. Soc. 1910. V. 32. P. 1089.
43. Abegg R., Bodlander // Z. anorg. allgem. Chem. 1899. Bd. 20. S. 453.
44. Berkeley // Phil. Trans. 1904. P. 203, 211; Цит. no [17].
45. Geffcken G. // Z. Phys. Chem. 1904. Bd. 49. S. 257, 296.
Химические методы количественного анализа
553
46. Noyes A.A., Abbot C.G. // Z. Phys. Chem. 1895. Bd. 16. S. 125.
47. Randall M., Chang K.S. // J. Am. Chem. Soc. 1928. V. 50. P. 1535.
48. Лурье Ю.Ю. Расчетные и справочные таблицы для химиков. М.: Госхимиздат, 1947. С. 98, 105, 236.
49. Hill А.Е. // J. Am. Chem. Soc. 1917. V. 39. P. 218.
50. Hill A.E., Simmons J.P. // J. Am. Chem. Soc. 1909. V. 31. P. 821.
51. Carnot A. // Traite d’analyse des substances minerales. Paris. 1922. V. 4. P. 433.
52. Коренман И.М., Ганина В.Г., Лебедева Н.П. // Журн. неорган. химии. 1958. Т. 3. С. 1265.
53. Moser L., Brukl А. // Monatsh.. Chem. 1926. Bd. 47. S. 709.
54. Browning Ph.E., Hutchins G.P. // Amer. J. Sci. 1899. V. (4)8. P. 460.
55. Rupp E. // Z. anorg. und allgem. Chem. 1903. Bd. 33. S. 156.
56. Комарь Н.П. Основы качественного химического анализа. Харьков: Изд. Харьковского ун-та, 1955. С. 13, 30, 392,415.
57. Bidermann G. // Arkiv Kemi. 1953. V- 5. Р. 441.
58. Harbottle С., Dodson R.W. // J. Am. Chem. Soc. 1951. V. 73. P. 2442.
59. Spencer J.F. // Z. Anorg. Allgem. Chem. 1905. Bd. 44.
S. 379.
Таблица 5.1.11.4
Калий
А. Растворимость солей калия и натрия (г/100 г воды) некоторых органических нитросоединений при 30 °C [1,2]
Нитросоединение Соль натрия Соль калия ^сольИа °сольК
Метилпикриновая кислота 3,15 3,78 0,83
Хлорпикриновая кислота 31,2 1,82 17,15
Стифниновая кислота 8,43 1,54 5,47
Флавиановая кислота 9,8 1,03 9,51
Пикриновая кислота 5,6 0,755 7,41
Нитраниловая кислота 0,72 0,567 1,27
Пикролоновая кислота 0,28 0,338 0,83
Дипикриламин 11,6 0,146 79,45
Дилитуровая кислота 1,03 0,086 11,97
Б. Растворимость в воде и произведения растворимости солей калия
Соль Температура, °C Растворимость Коэффициент активности Произведение растворимости Литература
моль-л 1 г-л 1 катион анион
K[BF4] 0 1,5 102 1,85 0,88 0,88 1,67-10^ 3
14 2,76-10“2 3,47 0,86 0,86 5,62-10 4 3
20 4,50-10-2 5,66 0,82 0,82 1,36-10 4 4
к[В(СбН5)4] 25 7,1-10“5 0,036 — -— 5-104 5,6
20 1,0-1 О’4 0,053 — — 1-108 7,8
20 1,26-10^ 0,064 — — 1,6-10 s 9
25 1,8-10^ 0,093 — — 3,3-10~8 10
kc6h2n3o7 (пикрат) 0 8,2-103 2,2 0,91 0,91 5,6-10'5 11-14
20 1,4-10-2 3,7 0,89 0,89 1,5-10^ «
KC12H4N7O12 (ди-пикриламинат) 0 5,2-10^ 0,25 — — 2,7-10’7 15-19
15 2,0-103 0,95 0,95 0,95 6,7-Ю-6 1,20- -23
25 2,5-103 1,19 0,95 0,95 1,0-10'5
35 3,1 -103 1,48 0,94 0,94 1,4-10’5
KC12H5N8O12 (монокалийгексанитрогидразобензол) 20 2,05-10"5 0,01 4,2-10"10 24, 25
554
Новый справочник химика и технолога
Продолжение таблицы Б
Соль Температура, °C Растворимость Коэффициент активности Произведение растворимости Литература
моль-л 1 г-л 1 катион анион
КНС4Н4О6 (битартрат) 0 1,25 Ю 2 2,66 0,90 0,90 1,3-10^ 26
10 1,6-10"2 3,42 0,88 0,88 2,0-104 26
18 1,7-10"2 3,60 0,88 0,88 2,2-104 27
КС1О4 0 5,5-10"2 7,6 0,81 0,81 2,0-103 ' 28, 29
14 8,9-10"2 12,4 0,77 0,77 4,7-103 30
К[1п(С2О4)2] 20 6,7-10 3 2,21 0,91 0,91 4,0-105 31
KJO4 0 7,4-10-3 1,7 0,91 0,91 4,5-1О5 32,33
18 1,6-102 3,7 0,88 0,88 2,0-1 О’4 34
20 1,8-102 4,2 0,87 0,87 2,5-1 О’4 32, 33
KNbO3 25 8,6-10^ 0,155 — — 7,5-10-7 35
КТаОз 0 4,3-105 1,16-10"2 — — 1,9-10"В. 9 36
25 4,9-10"5 1,30-10"2 — — 2,4-10’9 36
KReO4 0 1,2-10"3 0,36 0,97 0,97 1,4-10-6 37
30 5,1 -Ю3 1,47 0,93 0,93 2,2-10"5 37
50 1,1-Ю'2 2,21 0,90 0,90 9,9-10"5 37
K2Ag[Co(NO2)6] 25 3,1 -105 1,64-102 — — 3,9-10“18 38
K2Na[Co(N02)6] 0 9,6-104 0,42 — — 3,4-10"12 39-12
20 2,1-Ю3 0,93 0,95 0,64 4,2-10"11 39—12
K2[PdCl6] 25 1,5-10"2 5,96 0,88 0,58 6,0-1 О’6 31,43, 44
K2[PtCl6] 0 1,5-10"2 7,4 0,88 0,58 6,0-1 О’6 5, 39,40, 45-18
10 1,8-10"2 9,0 0,87 0,57 1,0-10"5
20 2,3-102 11,2 0,86 0,56 2,0-10"5
K2[ThF6] 25 1,4-1 О’6 6-104 — — 1,1-10“17 49
K2[SiF6] 0 3,5- Ю2' 0,77 0,94 0,75 1,1-10"7 46, 50
25 8,0-10 3 1,77 0,91 0,67 1,1-Ю-6 46, 50, 51
K3[A1F6] 25 5,4-10"3 1,4 0,93 0,51 9,1-Ю"9 52
K3[Co(NO2)6] 17 1,96-103 0,89 0,92 0,47 5,4-10"12 53
В. Растворимость сульфата калия в смесях воды
и этанола при 15 °C [43, 54, 55]
Д. Растворимость (г/100 г растворителя) соединений калия в смесях воды и этанола [43, 54]
Этанол, масс, доля, % Растворимость K2SO4, г/100 г раствора Этанол, масс, доля, % Растворимость K2SO4, г/100 г раствора
0 10,4 30 0,55
10 3,9 40 0,21
20 1,46
Г. Растворимость нитрата калия в смесях воды и метанола при 25 °C [43, 56]
Этанол, масс, доля, % KNO3 (20 °C) КС1О3 (30 °C) КОН (30 °C)
10 20,0 — —
20 16,0 4,7 —
40 5,25 2,4 —
50 — — 46
60 2,04 1,02 42,7
70 — — 38
80 0,7 0,24 35,7
90 0,3 0,06 34,8
Метанол, масс, доля, % Растворимость, моль • кг-1 растворителя Метанол, масс, доля, % Растворимость, моль кг”! растворителя
0 3,77 59,9 0,31
4,98 3,05 70,0 0,19
9,45 2,50 78,4 0,11
21,0 1,52 89,5 0,06
40,3 0,71
Е. Растворимость перхлората калия в водных растворах хлорной кислоты при 25 °C [57]
НС1О4, н. Растворимость, г/100 г раствора НС1О4, н. Растворимость, г/100 г раствора
0 2,085 0,10 1,485
0,01 1,999 1,00 0,527
Химические методы количественного анализа
555
Ж. Растворимость перхлората калия в смесях воды и этанола при 14 ЧС [30]
Этанол, масс, доля,% Растворимость, г/л раствора Этанол, масс, доля, % Растворимость, г/л раствора
0 12,4 42,4 3,9
7,1 9,2 58,5 2,6
13,2 7,8 94,7 0,15
27,3 5,7
Примечание. Данные о растворимости перхлората калия в смесях воды и этанола см. также [43, 58-63].
3. Произведение растворимости (ПР) перхлората калия в смесях воды и этанола при 20 Т* [64]
Вода, масс, доля, % ПР105 Вода, масс, доля, % ПРЮ5
0,35 0,099 10 2,00
3 0,165 12 2,99
5 0,52 15 3,18
7 0,65
И, Растворимость перхлората калия (мг/100 мл растворителя) в этаноле, содержащем хлорную кислоту, при комнатной температуре [20, 57]
Этанол, объемн. % НС1О4 (масс, доля, %) в этаноле
0 0,2 0,3 0,75 1 1,5
98 Н,9 2,4 — — — —
96 16,4 3,4 — 2,7 2,4 1,5
94 21,9 6,4 5,9 — 2,9 —
К. Растворимость перхлората калия в разных растворителях при 25 °C [60, 65-68]
Растворитель Растворимость, г/100 мл раствора Растворитель Растворимость, г/100 мл раствора
Вода 2,04 Изобутанол 0,005
Метанол 0,083 Этилацетат 0,0013
Этанол 0,0094 Амилацетат 0,014
и-Пропанол 0,008 Ацетон 0,118
и-Бутанол 0,0036
Л. Растворимость фторосиликата калия в смесях воды и этанола при 14 %! [30]
Этанол, масс, доля, % Растворимость, г/л раствора Этанол, масс, доля, % Растворимость, г/л раствора
0 0,9 27,3 0,09
8,7 0,46 42,4 0,05
15,6 0,29 94,7 0,0096
М. Растворимость хлороплатината калия (г /100 г растворителя) в смесях воды и метанола или этанола при 20 [69]
Спирт, масс, доля, % Метанол Этанол Спирт, масс, доля, % Метанол Этанол
0 0,774 0,774 50 0,062 0,049
5 0,535 0,491 60 0,032 0,026
10 0,412 0,372 70 ' 0,018 0,013
20 0,264 0,218 80 0,012 0,0085
30 0,183 0,134 90 0,0038 0,0025
40 0,116 0,076 100 0,0027 0,0009
Примечание. Данные о растворимости хлороплатината калия в смесях воды с метанолом или этанолом см. также [30,47, 72-75].
Н. Растворимость битартрата калия в воде [70, 71]
t, °C Растворимость, г/100 г раствора Плотность насыщенного раствора гем-3 t, °C Растворимость, г/100 г раствора Плотность насыщенного раствора <7', г-см 3
0 0,265 1,00145 18 0,489 1,00120
5 0,297 1,00224 20 0,534 1,00100
10 0,366 1,00180 25 0,641 1,00079
14 0,430 1,00172 30 0,762 1,00026
О. Растворимость битартрата калия (г /100 г раствора) в водных растворах винной кислоты [70, 76]
Концентрация винной кислоты, г/100 г раствора Температура, °C
0 10 25
0 0,231 0,358 0,641
0,025 0,216 0,346 0,622
0,05 0,208 0,335 0,606
0,1 0,196 0,320 0,584
0,2 0,184 0,300 0,556
0,4 0,168 0,271 0,524
0,6 0,160 0,250 0,498
0,8 0,150 — 0,472
П. Растворимость битартрата калия (г/л) в смесях воды и этанола [26]
Этанол, объемн. % Температура, °C
5 10 14 18 20 25
0 2,66 3,42 3,73 4,64 5,10 5,68
5 2,26 2,66 3,12 3,59 3,85 4,51
10 1,56 1,97 2,31 2,78 2,91 3,42
15 1,16 1,53 1,84 2,16 2,33 2,73
25 0,65 0,92 1,11 1,30 1,43 1,68
Примечание. Данные о растворимости битартрата калия в смесях воды и этанола см. также [30, 71, 77-82].
556
Новый справочник химика и технолога
Р. Растворимость калий-бортетрафенила в смесях воды и ацетона при 28 °C [83]
Ацетон, объемн. % Растворимость, мг • мл 1 Ацетон, объемн. % Растворимость, мг • мл1
16 0,4 60 18,4
25 0,6 70 27,0
32 1,2 80 40,6
40 2,5 90 53,0
44 4,9 95 59,8
50 10,5 100 41,8
С. Растворимость пикрата калия (г /100 мл раствора) в смесях воды с органическими растворителями при 25 °C [84, 85]
Растворитель, объемн. % Метанол Этанол Ацетон
0 0,645 0,645 0,645
10 0,542 0,559 0,726
20 0,470 0,450 0,876
30 0,444 0,472 1,140
40 0,422 0,533 1,155
50 0,411 0,582 2,106
60 0,410 0,574 2,615
70 0,396 0,485 3,09
80 0,332 0,326 3,34
90 0,254 0,174 3,08
100 0,274 — 1,08
Примечание. Данные о растворимости пикрата калия в органических растворителях и их смесях с водой см. также [12, 14, 86-88].
Литература
1. Dermer О.С., Dermer V.H. // J. Am. Chem. Soc. 1939. V. 61.P. 3302.
2. Коренман И.М. Аналитическая химия калия. М.: Наука, 1964.
3. Рысе И.Г., Хордас И.С. // Журн. неорган. химии. 1958. Т. З.С. 1410.
4. Boer J.H., Liempt J.A. // Rec. trav. chim. 1927. V. 46. P. 124.
5. Пршибил P. Комплексоны в химическом анализе. М.: ИЛ, 1960. С. 148, 188.
6. Rudorff W., Zannier Н. // Angew. Chem. 1952. Bd. 64. S. 613.
7. Левина Н.Д., Пантелеева Л.И. // Завод, лаб. 1957. Т. 23. С. 285.
8. Geilmann W., GebauhrW. // Z. anal Chem. 1953. Bd. 139. S. 161.
9. Havir J. I I Chem. listy. 1958. Bdd. 52. S. 1274; РЖ Хим.
1959. 49218.
10. Rudorff W., Zannier H. // Z. Naturforsch. 1953. Bd. 86. S. 611.
11. Designolle N. // Dingl. polyt. 1871. Bd. 192. S. 71. Цит. no [2].
12. Frish K. // J. prakt. Chem. 1867. Bd. 100(1). S. 229.
13. Jander G., Wendt H. Lehrbuch der analytischen und praparativen anorganischen Chemie. Leipzig, 1958. S. 65, 72, 76, 89.
14. Payen A. // Dingl. polytechn. J. 1869. Bd. 192. S. 67.
15. Gericke S. 11 Analytische Chemie der Diingemittel. Stuttgart, 1949. S. 104, 160.
16. Koszegi D. H Acta Sci. Regiae Univ. Hung. 1927. Bd. 11. S. 211; C.A. 1929. V. 23.791.
17. Monnier D., Besso Z., Wenger P.E. // Helv. Chim. Acta. 1951. V. 34. P. 433.
18. Riedler K., Schreiner L. // Chem. Techn. 1959. Bd. 11. S. 593.
19. Valkenburg H.B., McDaniels W.B. // J. Colo-Wyo. Acad. Sci. 1930. V. l.P. 44.
20. Портнов M.A., Афанасьев C.K. // Завод, лаб. 1937. Т. 6. С.1442.
21. Kielland J. // J. Am. Chem. Soc. 1937. V. 59. P. 1675; Ber. 1938. Bd. 71. S. 220.
22. PrzibillaC. //Kali. 1908. Bd. 2. S. 401; 1912. Bd. 6. S. 473.
23. Tredwell W., Hepenstrick H. // Helv. chim. acta. 1949. Bd. 32. S. 1903.
24. Черкесов А.И. H Tp. Комиссии по аналитической химии. 1960. T. 11. С. 137.
25. Черкесов А.И., Кульберг Л.М. // Завод, лаб. 1952. Т. 18. С. 131.
26. Иванов Б.В. // Виноделие и виноградарство СССР. 1947. Т. 7. С. 17.
27. Тредвелл Ф.П., Голл В.Т. Качественный анализ. М.; Л.: Госхимиздат, 1946. С. 38, 309, 331.
28. Calzolari F. // Gazz. chim. ital. 1912. Bd. 42. S. 11, 85.
29. Noyes A.A., Sammet G.V. // Z. phys. Chem. 1903. Bd. 43. S. 530.
30. Pierrot M. // Compt. Rend. 1921. Bd. 172. S. 1041.
31. Комарь Н.П. Основы качественного химического анализа. Харьков: Изд. Харьковск. ун-та, 1955. С. 388, 419,431.
32. Карякин Ю.В., Ангелов И.И. Чистые химические реактивы. М.: Госхимиздат, 1955. С. 185, 210, 423.
33. Hill А.Е. // J. Am. Chem. Soc. 1928. V. 50. P. 2678.
34. Pedersen K. // J. Kgl. Danske Videnskab. Selskab. Math.-fys.Medd. 1941. Bd. 18. S. 26.
35. Лапицкий A.B., Шишкина Л.Н., Пчелкина M.A., Степанов Б.А. // Журн. общей химии. 1955. Т. 25. С. 1862.
36. Лапицкий А.В., Степанов Б.А., Пчелкина М.А. // Журн. общей химии. 1955. Т. 25. С. 1866.
37. Друце И. Рений. М.: ИЛ, 1951. С. 56, 73.
38. Cumings J.N. // Biochem. J. 1939. V. 33. Р. 642.
39. Блок И.И. Качественный химический анализ. М.; Л.: Госхимиздат, 1952. 170 с.
40. Клячко Ю.А., Шапиро С.А. Курс химического качественного анализа. М.: Госхимиздат, 1960. 240 с.
41. Gmelins Handbuch der anorganischen Chemie, System-Nummer22. Kalium. Berlin. 1936-1938.
42. Remy H., Lehrbuch der anorganischen Chemie. Leipzig, 1957. Bd. 1. S. 201, 221, 239, 432; 1957. Bd. 2. S. 271, 282,432, 593, 770.
43. Справочник химика. M.: Госхимиздат, 1952. Т. 1. С. 154, 434, 491, 670; Т. 2. С. 8, 68, 76; Т. 3. С. 77, 194, 362,413,521.
44. Wellmann Н.В. // J. Am. Chem. Soc. 1930. V. 52. Р. 985.
45. Анализ минерального сырья / Под ред. Ю.Н. Книпович и Ю.В. Морачевского. Л.: Госхимиздат, 1956.
Химические методы количественного анализа
557
46. Лурье Ю.Ю. Расчетные и справочные таблицы для химиков. М.; Л.: Госхимиздат, 1947. С. 74, 88, 229, 264.
47. Меншуткин Н.А. Аналитическая химия. М.; Л.: Госхимиздат, 1931. С. 28, 219, 356.
48. Петрашень В.И. Качестввенный химический анализ. М.; Л.: Госхимиздат, 1948. С. 155, 164, 191, 555.
49. Спицин В.И. // ЖРФХО. 1917. Т .49. С.357.
50. Carter R.H. // Ind. Eng. Chem. 1930. Bd. 22. S. 886.
51. Rees A.G., Huddleston L.J. // J. Chem. Soc. 1931. S. 1648.
52. Тананаев И.В., Талипов Ш. // Журн. общей химии. 1939. Т. 9. С. 1155.
53. Rosenblatt Th. // Вег. 1886. Bd. 19. S. 2515.
54. Schiff Н. //Z. Anal. Chem. 1862. Bd. 1. S. 65.
55. Schneider H. // Z. Anal. Chem. 1952. Bd.135. S. 191.
56. Akerlof G., Turck H.E. // J. Am. Chem. Soc. 1935. V. 57. P. 1746.
57. Thin R.H., Cumming A.G. H J. Soc. Chem. 1915. V. 107. P. 361.
58. Baxter G.P., Kobayashi M. // J. Am. Chem. Soc. 1917. V. 39. P. 249; 1920. V. 42. P. 735.
59. Kolarov N. // Z. Anal. Chem. 1941. Bd. 122. S. 399.
60. Rothmund V. // Z. phys. Chem. 1909. Bd. 69. S. 539.
61. Russel O.J. H Brit. J. appl. Physics. 1952. V. 3. P. 47.
62. Seward R.P., Schumb W.C. // J. Am. Chem. Soc. 1930. V. 52. P. 3962.
63. Smith G.F., Shead A. 11 J. Am. Chem. Soc. 1931. V. 53. P. 947; 1932. V. 54. P. 1772.
64. Novak V., Krajina A. // Chem. listy. 1950. Bd. 52.
S. 1506; Collec. czechoslov. chem. communs. 1959. Bd. 24. S. 1808.
65. Ганчев H. // Изв. Хим. ин-т Българ. АН. 1958. Т. 6. С. 149.
66. Smith G.F. // J. Am. Chem. Soc. 1925. V. 47. P. 762.
67. Welcher E.J. Organic Analytical Reagents. V. 1-4. N.Y., 1948.
68. Willard H.H., Smith G.F. // J. Am. Chem. Soc. 1922. V. 44. P. 2816. 1923. V. 45. P. 286.
69. Archibald E.H., Wilcox W.G., Buckby B.G. // J, Am. Chem. Soc. 1908. V. 30. P. 747.
70. Carpenter D.C., Mack G.L. // J. Am. Chem. Soc. 1934. V. 56. P. 311.
71. Paul Th. H Z. Electrochem. 1917. Bd. 23. S. 65; Arb. Reichsgesundh. Amt. 1926. Bd. 57. S. 94.
72. Fresenius R. // Lieb. Ann. 1846. Bd. 59. S. 117.
73. Heigh L.D. // Калий. 1935. № 2. S. 26.
74. PeligotM. // Z. anal. Chem. 1897. Bd. 36. S. 322.
75. Precht H. // Z. anal. Chem. 1879. Bd. 18. S. 509; Chem. Ztg. 1896. Bd. 20. S. 209; 1897. Bd. 36. S. 315.
76. Richert P.H. // J. Am. Chem. Soc. 1930. V. 52. P. 2241.
77. Левензон И.Л. Винная кислота и ее соли. М.; Л.: Снаб-техиздат, 1934.
78. Chancel G. // Compt. rend. 1865. Bd. 60. S. 409.
79. Heidenhain H. // Z. anal. Chem. 1888. Bd. 27. S. 689.
80. Kissel E. I IZ. anal. Chem. 1869. Bd. 8. S. 410.
81. Roelofsen J.A. // Amer. Chem. J. 1894. V. 16. P. 467.
82. Wenger W.H. I I Amer. Chem. J. 1892. V. 14. P. 624.
83. Scott A.D., Hunziker H.H., Reed M.G. // Chemist-Analyst.
1959. V. 48. P. 11.
84. Фишер B.M. //ЖРФХО. 1914. T. 46. C. 1250.
85. Fischer W.M. // Z. phys. Chem. 1916/17. Bd. 92. S. 581.
86. Bolliger A. // Z. anal. Chem. 1933. Bd. 94. S. 403; J. biol. Chem. 1934. Bd. 107. S. 229; C.A. 1934. V. 28. 71; 1935. V. 29. 1031.
87. Hager H. //Pharm. Zentralhalle. 1881. Bd. 22. S. 224.
88. Hager H. // Z. anal. Chem. 1882. Bd. 21. S. 408, 561.
Таблица 5.1.11.5
Золото Отделение золота химическим способом без носителей
Отделяемый элемент Реагент для отделения
Щелочные и щелочноземельные, Mg, Мп CH2O [1], SO2 [2]
Си СН2О [1], СаНРО4 [3], амальгамы Zn, Pb, Bi [4], SO2 [2]
Ag CaHPO4 [3], NaNO2 [5], SO2 [2]
Zn Na2S [6], CH2O [1], SO2 [2]
Hg CH2O [1], см. также обзор [7]
Tl Осаждение на Fe(OH)3 при pH = 5,4-7,0 [8]
Ge Осаждение GeO2 xH2O [9]
Zr Na2S [10]
Pb Na2S [11], (NaPO3)6 [12], CH2O [1], SO2 [2]
Sn, As, Sb CH2O [1], N2H4H2SO4 [13], SO2 [2]
Se Аскорбиновая кислота [14], гидрохинон [15]
Те Аскорбиновая кислота [14], гидрохинон [15], NaNO2, FeSO4 [16], (NaPO3)6 [12], производные пиразолона [17]
Re SO2 [2], Н2С2О4, гидрохинон, сульфитгидрохинон [18]
Ni Амальгамы Zn, Pb, Bi [4], SO2 [2]
Ru SO2 [19]
Rh, Ir SO2 [19], KBr + KBrO3 [20], H2C2O4 [21]
Pd KBr + KBrO3 [20], гидрохинон [22], (C2H5)4NC1 [23], диметилглиоксим, H2C2O4[21, 24], SO2 [19]
Pt Гидрохинон [22], (C2H5)4NC1 [23], H2O2 + + OH“ [25], H3PO2 [26], SO2 [19], H2C2O4 [21]
Литература
1. Аверкиев Н. // ЖРФХО. 1902. Т. 34. С. 828.
2. Карабаш А.Г. и др. Материалы X Всесоюзного совещания по спектроскопии: Физический сборник. Львов: Изд. Львовского ун-та. 1958. Т. 4(9). С. 556.
3. Морачевский Ю.В., Зайцев В.Н. // Уч. зап. ЛГУ. № 297. Хим. науки. 1960. № 19. С. 90.
4. Тананаев И.В., Левина М.И. // Изв. Сектора платины ИОНХ АН СССР. 1948. Вып. 22. С. 114.
5. Звягинцев О.Е., Кулак А.И. // Журн. неорган. химии. 1957. Т. 2. С. 1687.
558
Новый справочник химика и технолога
6. Taimni J.K., Tandon S.N. // Anal. Chim. Acta. 1959. V. 21. P. 110.
7. Stammreich H. // Z. anorg. allgem. Chem. 1925. Bd. 148. S. 93.
8. Новиков А.И., Пирогова T.A. // Радиохимия. 1968. T. 10. С. 82.
9. Шуба И.Д., Разумова Г.Н., Васильев И.Я. И Радиохимия. 1968. Т. 10. С. 501.
10. Salaria G.B.S. //Z. anal. Chem. 1961. Bd. 180. S. 358.
11. Salaria G.B.S. // Anal. Chim. Acta. 1957. V. 17. P. 392.
12. Ripan R., Stanisav C., Marcu G., Vancea M. // Rev. chim. (RPR). 1958. Bd. 3. S. 13.
13. Christensen A. // Z. anal. Chem. 1915. Bd. 54. S. 158.
14. Ripan R., Pop Gh. // Rev. chim. (RPR). 1963. Bd. 14. S. 413.
15. Seath J., Beamish F.E. // Ind. Eng. Chem., Anal. Ed. 1937. V. 9. P. 373.
16. Lenher V., Smith G.B., Knowles D.C. // Ind. Eng. Chem., Anal. Ed. 1934. V. 6. P. 43.
17. Бусев А.И., Бабенко Н.Л. // Журн. аналит. химии. 1964. Т. 19. С. 926.
18. Geilmann W., Bode Н. И Z. anal. Chem. 1951. Bd. 133. S. 177.
19. Hoffmann L., Kriiss G. // Annalen. 1887. Bd. 238. S. 66.
20. Zachariasen H., Beamish F.E. // Taianta. 1960. V. 4. P. 44.
21. Душин И.М., Воронков М.П., Шипулин H.B., Миронов А.В., Мухина П.П. Аффинаж платиновых металлов. М.;Л.:ОНТИ. 1937.
22. Beamish F.E., Russel J.J., Seath J. // Ind. Eng. Chem., Anal. Ed. 1937. V. 9. P. 174.
23. Maynard J.L. // Ind. Eng. Chem., Anal. Ed. 1936. V. 8. P. 368.
24. Wittenbach A. // Helv. Chim. Acta. 1966. V. 49. P. 2555.
25. Vanino L., Seemann L. // Ber. 1899. Bd. 32. S. 1968.
26. Moser L., Niessner M. // Z. anal. Chem. 1923. Bd. 23.
S. 240.
Таблица 5.1.11.6
Натрий
Растворимость (г/100 мл) хлоридов лития, натрия и калия в органических растворителях [1, с. 52]
Растворитель LiCl NaCl КС1
Этанол 2,54 0,0649 0,0294
«-Бутанол 12,68 0,0050 0,0030
и-Бутанол + 6% НС1 + 0,5% НС1О4 10,61 0,0007 0,0005
Изобутанол 10,57 0,0005 0,0008
Пентанол 9,03 0,0020 0,0007
Изопентанол 7,30 0,0016 0,0006
и-Гексанол 7,20 0,0010 0,00005
2-Этилгексанол 3,70 0,0001
Бензиловый спирт 1,43 0,0170 0,0160
Ацетон 3,86
Пиридин 13,39
Диоксан 2,8
Таблица 5.1.11.7
Торий
Л. pH начала осаждения Th(OH)4 0,1 и. раствором
NaOH при 15 °С[1]
Концентрация раствора Th(NO3)4, моль-л 1 pH
0,001 3,60
0,002 3,58
0,010 3,57
Б. pH начала осаждения Th(OH)4 из » 0,01 и. растворов различных солей тория при 17-18 °C [1, 2]
Соль Осадитель pH
Th(NO3)4 NaOH 3,57
ТЬСЦ NaOH 3,51
Th(SO4)2 NaOH 3,53
Th(NO3)4 NH4OH 3,57
В. Растворимость сульфатов тория Растворимость сульфатов тория в хлороводородной кислоте при 30 °C [3]
Соединение HC1, % Растворимость, Th(SO4)2 г/100 г раствора Соединение HC1, % Растворимость, Th(SO4)2 г/100 г раствора
Th(SO4)2- •8H2O 4,55 3,541 Th(SO4)2- •8H2O 18,33 2,199
Th(SO4)2- •8H2O 12,14 2,811 Th(SO4)2- •4H2O 20 2,110
Th(SO4)2- •8H2O 15,71 2,360 Th(SO4)2- 4H2O 23,9 1,277
Г. Растворимость сульфата тория в азотной кислоте при 30 [3]
Соединение HNO3, % Растворимость, Th(SO4)2 г/100 г раствора Соединение HNO3, % Растворимость, Th(SO4)2 г/100 г раствора
Th(SO4)2- •8H2O 5,17 3,68 Th(SO4)2- •8H2O 28,51 3,88
Th(SO4)2- •8H2O 16,68 4,84 Th(SO4)2- •4H2O 33,17 3,34
Th(SO4)2-•8H2O 21,99 4,47 Th(SO4)2- •4H2O 38,82 2,51
Химические методы количественного анализа
559
Д. Растворимость сульфата тория в серной кислоте при 25 °С[3]
Соединение Концен-трация H2SO4, h. Растворимость, Th(SO4)2 г/100 г раствора
считая на оксид считая на безводный сульфат
Th(SO4)2-9H2O 1,1 1,14 1,831
Th(SO4)2-9H2O 2,16 0,9265 1,488
Th(SO4)2-9H2O 4,32 0,545 0,8751
Th(SO4)2-9H2O 6,68 0,2685 0,4312
Th(SO4)2-8H2O 9,68 0,0651 0,1045
Th(SO4)2-8H2O 10,89 0,0396 0,0636
Th(SO4)2-4H2O 15,15 0,0192 0,0308
Е. Растворимость Th(SO4)2 • 8Н2О в растворах сульфата аммония при 25 °C [4]
Продолжение таблицы Ж
Растворимость Th(C2O4)2-6H2O в щавелевой кислоте при 25 °C [5]
Концентрация Н2С2О4 Содержание ThO2, г/1000 г раствора
1 и. 0,0015
Раствор одновременно насыщен щавелевой кислотой и оксалатом тория 0,0029-0,0030
Растворимость оксалата тория в оксалате аммония [7]
Концентрация (NH4)2C2O4, н. Содержание ThO2, г/100 мл раствора
0,01 0,004
0,1 0,22
0,5 1,76
Насыщенный раствор 14,83
(NH4)2SO4, г/100 г раствора Th(SO4)2, г/100 г раствора
0 1,85
3,3 3,50
6,6 5,58
13,2 7,83
26,4 16,24
Ж. Растворимость оксалатов тория
Растворимость Th(C2O4)2-6H2O в НС1 при 25 °C [5]
Содержание НО, % Содержание ThO2, г/1000 г раствора
24,8 0,100
37,0 3,450
37,6 3,492
Растворимость Th(C2O4)2-6H2O в H2SO4 при 25 °C [6]
Концентрация H2SO4, н. Содержание ThO2, г/1000 г растворителя
1 0,0258
2 0,039
2,9 0,045
4,32 0,11
6,175 0,1513
6,685 0,1794
8,45 0,2473
3. Растворимость йодата тория [9]
Растворитель Концентрация растворителя Темпера-тура, °C Растворимость Th(IO3)4
мг-л"1 М-л1
Н2О — 0 138
н2о — 25 203,1 2,17-10^
Н2О — 100 660
КЮз 0,01 м 7,83 •104’
Спирт 95% Не растворим
Эфир 95% То же
Примечание. Произведение растворимости йодата тория [Th4+]{lO~]4 = 1,35 1О“20 [10].
И. Растворимость пикролината тория [11]
Растворитель Температура, °C Содержание ThO2, г/1 л растворителя
Н2О 0 0,09
Н2О 20 0,16
0,005 н. раствор пикроло-новой кислоты 20 0,31
3% СНзСООН + 0,005 н. C10H8N4O5 0 10,12
0,1 н. HNO3 20 19,50
Растворитель, соответствующий условиям осаждения Практически не растворяется
К. Термические свойства пикролината тория [11]
Температура, °C 20 105 125 150 175 200 225 250
Время, час 4 4 20 10 10 10 10
Уменьшение массы, мг 1,1 2,0 11,8 11,8 12,0 12,3 13,8 Разлагается
Уменьшение массы, % 0,15 0,27 1,59
Изменение цвета Нет Слабо-ярко-ранжевый Нет Почти нет Нет Коричневый
560
Новый справочник химика и технолога
Литература
1. Britton H.T.S. // J. Chem. Soc. 1925. V. 127. Р. 2110.
2. Britton H.T.S. Hydrogen Ions. V. 2. London. Champan Hall Ltd. 1942.
3. Koppel L., Holtkamp H. // Z. anorg. Chem. 1910. Bd. 67.
S. 266.
4. Rosenheim A., Zuckerman J. // Z. anorg. Chem. 1932. Bd. 208. S. 95.
5. Hauser O„ Wirth F. // Z. anorg. Chem. 1912. Bd. 78. S. 75.
6. Wirth F. // Z. anorg. Chem. 1912. Bd.76. S. 174, 176.
7. Hauser O., Wirth F. // Z. angew. Chem. 1909. Bd. 22. S. 484.
8. Moeller Th., Fritz N.O. // Chem. Rev. 1948. V. 42. P. 63.
9. Рябчиков Д.И. Гольбрайх E.K. Аналитическая химия тория. М.: Изд. АН СССР, 1960.
10. Крылов Е.И. // Ж. аналит. химии. 1957. Т. 12. С. 451.
11. Hecht F., Ermann W. // Z. anal. Chem. 1935. Bd. 100. S. 87.
12. [1] Полуэктов H.C., Мешкова С.Б., Полуэктова E.H. Аналитическая химия лития. М.: Наука, 1975.
Таблица 5.1.11.8
Цирконий
А. Константы (К) для последовательных реакций гидролиза иона Zr4+ (при ионной силе раствора »2) [1]
Zr4 + Н2О о Zr(OH)3+ + Н+ Ki = 0,6 ± 0,05
Zr(OH)3+ + Н2О « Zr(OH)22+ + Н+ К2 = 0,24 ±0,01
Zr(OH)22+ + Н2О « Zr(OH)3+ + Н+ К3 = 0,09 ± 0,01
Zr(OH)3+ + Н2О о Zr(OH)4 + Н+ К, = 0,068 ± 0,06
Б. Ионное состояние циркония в минеральных кислотах [2]
Кислота Концентрация кислоты, М Ионы циркония Метод Литература
НС1О4 >2 Zr4+ Экстракция 3
2-0,2 Zr(OH)3+ То же 3
0,2 Zr(OH)2+ То же 3
1-2 Zr4+ То же 4
1-2 Zr4+ Ионный обмен 5
0,2-2 Zr(OH)<4")+ Экстракция 6
>2 Zr4+ То же 6
3-6 Почти нет анионов Анионный обмен 5
> 1 Доминируют катионы Электромиграция 7
<0,5 Только катионы То же 7
НС1 0,1 Zr(OH); Ионный обмен 8
0,5-1,5 Zr(OH)2+ Электрофорез 8
2 Zr(OH)3+ То же 8
0,5-2 Катионы То же 9
6 Нейтральные комплексы То же 9
7-12 Анионы Ионный обмен 8, 10
8-12 Значительное количество анионов Электрофорез 11
3-10 Частично анионы Электрофорез, ионный обмен 11, 12
HNO3 pH < 4,2 Катионы Десорбция 9
pH >4,2 Коллоиды Электрофорез 9
pH = 0-1 Zr4+, Zr(OH)3+, Zr(OH)J, Zr(OH)°, мономеры — 13
рН<0 Zr4+, Zr(OH)3+, мономеры — 13
0,001-2 Преобладают катионы, есть анионы и нейтральные частицы Ионный обмен 14
0,2-1 Коллоиды То же 15, 16
0,03-2 Катионы Электромиграция, ионный обмен 9, 12
0,1-2 Катионы, частично нейтральные молекулы Ионный обмен 3,14, 17
Химические методы количественного анализа
561
Продолжение таблицы Б
Кислота Концентрация кислоты, M Ионы циркония Метод Литература
HNO3 <0,5 Катионы Электромиграция 7
>1,5 Анионные и нейтральные комплексные формы То же 7
0,3-2 Катионы, нейтральные молекулы Ионный обмен 8
1-2 Zr(OH)', Экстракция 3
>3 [ZrCOllMNOO# Самодиффузия 9
>4 [ZrtNO,)2J2- Электромиграция 9
H2SO4 <0,1 Катионы Ионный обмен 8, 14
>0,2 Анионы То же 14
0,05-2 То же То же 9, 12, 18
4 То же То же 8
>4 Анионы, нейтральные частицы То же 8
В. Гравиметрическое определение циркония и гафния [2]
Осадитель Цирконий Гафний
Гравиметрическая форма Предел температуры, °C Гравиметрическая форма Предел температуры, °C
Аммиак* ZrO2 >400 ню2 >350
Анилин* ZrO2 >972 — —
Диэтиланилин* ZrO2 >223 — —
Таннин ZrO2 >880 ню2 665
Селенистая кислота* ZrO2 >900 ню2 680
Миндальная кислота* Zr(C6H4CHOHCOO)4 60-188 Hf(C6H4CHOHCOO)4 90-260
То же ZrO2 >959 ню2 >500
н-Оксифениларсоновая кислота ZrO2 >969 HfO2 >660
Двузамещенный фосфат аммония ZrP 2O7 >850 HfP2O7 >750
Купферон ZrO2 >745 HfO2 >670
Пиридин ZrO2 >850 — —
Иодат калия — — — —
Фениларсоновая кислота ZrO2 >969 — —
Нитробензойная кислота ZrO2 1000 — —
Хинолин ZrO2 >878 — —
Примечание.
* Осадок пригоден для автоматической термогравиметрии.
Литература
1. Соловкин А.С. // Журн. неорган. химии. 1957. Т. 2. С. 611.
2. Елинсон С.В., Петров К.И. Аналитическая химия Zr и Hf. М.: Наука, 1965.
3. Connick R., Me Vey // J. Am. Chem. Soc. 1949. V. 71. P. 3182.
4. Connick R., Reas W. // J. Am. Chem. Soc. 1951. V. 73.
P. 1171.
5. Larsen E., Wang P. // J. Am. Chem. Soc. 1960. V. 82. P. 22.
6. Larsen E., Gamill A.. // J. Am. Chem. Soc. 1950. V. 72. P. 3615.
7. Набиванец Б.И. // Журн. неорган. химии. 1961. Т. 6. С. 1150.
8. Белявская Т.А., Му Бин-вень // Вести. Моск, ун-та, сер. хим. 1959. № 4. С. 207.
9. Lister В., McDonald L. // J. Am. Chem. Soc. 1952. V. 74. P. 4315.
10. Краус К.А., Нельсон Ф. Химия ядерного горючего. М.: Госхимиздат, 1956. С. 353.
11. Алимарин И.П., Белявская Т.А., Му Бин-вень // Радиохимия. 1959. Т. 1. С. 645.
12. Bunney L. and oth. // Anal. Chem. 1959. V. 31. P. 58.
13. Старик И.Е., Скульский ИА.. // Радиохимия. 1959. Т. 1. С. 379.
14. Парамонова В.И., Воеводский А.С. // Журн. неорган. химии. 1956. Т. 1. С. 1905.
15. Бабко А.К., Гридчина Г.И. // Журн. неорган. химии. 1962. Т. 7. С. 889.
16. Schubert I. // Nucleonics. 1949. V. 4. Р. 6.
17. Парамонова В.И., Сергеева А.Н. // Журн. неорган. химии. 1958. Т. 3. С. 215.
18. Korkisch L, Farag А. // Z. anal. Chem. 1959. Bd. 166. S. 81.
562
Новый справочник химика и технолога
Таблица 5.1.11.9
Бериллий
А. Гигроскопичность оксида бериллия [1, 3]
Температура прокаливания, °C Адсорбция воды, %
Непрокаленный оксид 34,27
900 3,27
1100 2,13
1300 0,21
1500 0,18
Б. Влияние температуры и кислотности раствора на растворимость NH4BePO4- Н2О [1, 2]
Температура, °C Растворимость, г/1000 г раствора
19,8 9,5-10
36,8 1,14103
66,5 1,5-10 3
100 2,5-10-3
pH Растворимость, г/1000 г раствора (20 °C)
4,0 5,7-102
5,1 2,6-10-2
6,1 4,4-10’3
7,1 0,82-103
В. Влияние pH на растворимость Ве3(РО4)2-6Н2О при 19-20 °C [15]
рн [Ве2+], моль-экв.-л 1
4,15 7,7-10^
3,2 4,7-10’3
2,9 6,5-10-3
2,7 1,2 102
Г. Возможные реагенты для осаждения гидроксида бериллия [1]
Соединение Величина pH, создаваемая реагентом в растворе Температура образования ВеО, °C Литература
Сульфид аммония — 592 4,5
Нитрит аммония — 1000 5,6
Гексаметилентетрамин 8,0 1000 7-9
а-Пиколин 7,0 1000 10, 11
Этиламин и метиламин 8,5 780 5, 12-14
Литература
1. Новоселова А.В., Бацанова Л.Р. Аналитическая химия бериллия. М.: Наука. 1960.
2. Волков Б.В., Несмеянов А.Н. Сб. Радиохимия: Сборник / Под ред. В.И.Спицина. М.: Изд. МГУ. 1952. С. 99.
3. Будников П.П., Звягильский А.А. // Огнеупоры. 1961. Т. 11. С. 525.
4. Bleyer В., Boshart К. // Z. anal. Chem. 1912. Bd. 51. S. 748.
5. Dupuis T. // Compt. rend. Acad. Sci. 1950. V. 230. P. 957.
6. Akiyama T. // Japan Analyst. 1952. V. 1. P. 133.
7. Ray P., Chattopadhya A. // Z. anorg. allg. Chem. 1928. Bd. 169. S. 99.
8. Ray P. П Z. anal. Chem. 1931. Bd. 86. S. 13.
9. Akiyama A. // J. Pharm. Soc. Japan. 1936. V. 56. P. 893; Japan Analyst. 1953. V. 2. P. 116.
10. Остроумов Э.А., Иванов-Эмин В.И. И Завод, лаб. 1945. Т.Н. С. 386.
11. Остроумов Э.А. Применение органических оснований в аналитической химии. М.: Изд. АН СССР. 1959.
12. Akiyama Т. // J. Pharm. Soc. Japan. 1940. V. 60. Р. 309.
13. Welcher Т. Organic Anal. Reagents. N.Y., 1948. P. 379.
14. Fresenius R., Jander G. Handbuch der analytischen Chemie. Teil III. Bd. Ila. 1940. S. 5.
15. Чухланцев В.Г., Алямовская K.B. // Изв. Высш, учебных заведений. Химия и хим. технол. 1961. Т. 4. С. 359.
Таблица 5.1.11.10
Вольфрам
А. Анализ вольфрама. Отделение и концентрирование посторонних элементов
Отделяемый элемент Реагент и условия отделения Литература
Zn, Cd Метиловый фиолетовый, KI, NH4SCN, винная кислота, pH = 3-4 16
Ti Купферон, винная кислота 17
Zr Купферон, винная кислота, pH = 6,8 5, 17
Th Растворимые фториды 18
Pb Метиловый фиолетовый, KI, NH4SCN, винная кислота, pH = 3—4 16
Si H2SO4 + HNO3, Н2О2, Н2С2О4 6
V NaOH 19
А-Бензоилфенилгидроксиламин, тартрат, pH > 1 20
Nb MgSO4, NH4CI, NH3 21,22
Купферон, щавелевая кислота 10
Купферон, фторид 23
P MgCl2, NH4C1, nh3 24
Bi Метиловый фиолетовый, KI, NH4SCN, винная кислота, pH = 3-4 16
Mo Формиат аммония, винная кислота, H2S, pH = 2,9 25
Тионалид, pH = 1-3 26
Mo, U, Fe А-Бензоилфенилгидроксиламин, рН>1 20
Pd а-Пиколиновая кислота, pH = 3-7 27
Хинальдиновая кислота, pH = 3-7 28
Химические методы количественного анализа
563
Б. Определение примесей и компонентов в вольфраме и его соединениях гравиметрическим методом
Определяемый элемент Реагент или осаждаемая форма Анализируемый объект Литература
Na Na2SO4 Боровольфрамат натрия 2
К Тетрафенил-борат калия Вольфрамосиликат калия 3
Си Электролизом (Си) WO3 4
Mg NH4MgPO4 WO3 4
Zr Купферон Сплавы на основе вольфрама 5
Si SiO2 WSi2 6
SiO2 Сплав W-Co 7
SiO2 Сплавы Si-W-Cr-Fe, Si-W-Cr-Al, Si-W-Cr-Ni 8
Sn Электролизом (SnO2) Смеси W-Sn 9
Nb Купферон Сплавы W-Si-Nb 10
Nb, Ta Таннин Вольфрамит 11
S BaSO4 Отработанные растворы 12
Mo МоО3 Вольфрам 12
Mn NH4MnPO4 WO3 4
Co 1-Нитрозо-2-нафтол Сплавы W-Co 7
Ni Диметилдиоксим Вольфрам и его спавы 13
То же Вольфрам 14, 15
То же WO3 4
Литература
1. Бусев А.И., Иванов В.М., Соколова Т.А. Аналитическая химия вольфрама. М.: Наука, 1976.
2. Савальский С.Л., Перельмутер К.С., Бурцева К.Г. Сб.: Материалы н.-т. конференции Северо-кавказского гор-нометаллург. ин-та. 1968: Сборник. Орджоникидзе: Изд. СКГИ, 1970. С. 70.
3. Сесиашвили К.М. // Сообщения АН ГрузССР. 1972. Т. 65. С. 321.
4. Steinbach S., Herring R. // Z. Chem. 1972. Bd. 12. S. 33.
5. Шескольская А.Я. // Журн. аналит. химии. 1962. Т. 17. С. 949.
6. Копылова В.П., Назарчук Т.И. Химические свойства и методы анализа тугоплавких соединений: Сборник. Киев: Наукова думка, 1969. С. 153.
7. Паюсте О.Ю. Сб. н.-т. статей Проектно-технол. НИИ. Таллин. 1968. Вып. 8. С. 177.
8. Юхименко Е.В., Котляр Е.Е. // Завод, лаб. 1971. Т. 37. С. 1052.
9. Коган Ф.И., Коваленко П.Н., Иванова З.И. // Журн. аналит. химии. 1965. Т. 20. С. 329.
10. Пономарев А.И., Шескольская А.Я. // Журн. аналит. химии. 1959. Т. 14. С. 67.
11. Tschemichov J.A., Karsajewskaja М.Р. И Z. anal. Chem. 1934. Bd. 98. S. 97.
12. Талалаевский М.Б. И Н.-технич. сборник ВНИИ источников света. 1968. №3. С. 145.
13. Элвелл В.Т., Вуд Д.Ф. Анализ новых металлов. Ti, Zr, Hf, Nb, Ta, W и их сплавы. М.: Химия. 1970.
14. Постогвард Г.И. // Журн. аналит. химии. 1970. Т. 25. С. 970.
15. Das А.К. // Sci. and Cult. 1971. V. 37. Р. 306.
16. Борчева Т.А., Николаева Т.Ф., Харламов И.П. Сб. Определение микропримесей: Сборник. № 2. М.: Изд. МДНТП им. Дзержинского, 1968. С. 59.
17. Попова О.И., Корнилова В.И. // Журн. аналит. химии. 1961. Т. 16. С. 651.
18. Столярове К.П., Фирюлина В.В., Губайдулин И.З. И Завод, лаб. 1972. Т. 38. С. 664.
19. Мальцев В.Ф., Мартынов А.П., Новак В.П., Боговина В.И. Производство труб: Сборник. Вып. 22. М.: Металлургия. 1969. С. 259.
20. Kaimal V.R.M., Shome S.C. // Anal. Chim. Acta. 1964. V. 31.P. 268.
21. Быковская Ю.И. И Труды Комиссии по аналит. химии АН СССР. 1958. Т. 9(12). С. 323.
22. Голубцова Р.Б. И Журн. аналит. химии. 1951. Т. 6. С. 357.
23. Буданова Л.М., Гаврилова К.Д. // Завод, лаб. 1949. Т. 15. С. 7.
24. Мизецкая И.Б. Сб. Химия редких элементов: Сборник. Вып. 2. М.: Изд. АН СССР. 1955. С. 115.
25. Yagoda Н, Fales Н.А. // J. Am. Chem. Soc. 1936. V. 58. Р. 1494.
26. Мухина З.С., Ильина Л.И., Кондукова Н.С. Химические свойства и методы анализа тугоплавких соединений: Сборник. Киев: Наукова думка, 1969. С. 108.
27. Majumdar А.К., Sen Gupta J.G. // Z. anal. Chem. 1958. Bd. 161. S. 100.
28. Cm. [27]. S. 104.
Таблица 5.1.11.11
Платиновые металлы
А. Гравиметрическое определение Pd с моноксимами в качестве реагентов [1—3]
Реагент Условия определения Гравиме-трическая форма и фактор пересчета F Влияние других элементов Область применения Литература
Кислотность раствора Маскирующие средства Условия термообработки Не мешают Мешают
Салицил-альдоксим Кисл. среда (< 1%НС1, < 5% H2SO4) Винная кислота, Fe(III) Прокал. в токе Н2 Металлический Pd В присутствии 1 мл конц. HNO3-AunPt и 0,5 мл 30% Н2О2 - Ru, Ir, Fe, Ni, Pb Cu Опр. макро- и микроколичеств палладия 2
564
Новый справочник химика и технолога
Продолжение таблицы А
Реагент Условия определения Гравиме-трическая форма и фактор пересчета F Влияние других элементов Область применения Литература
Кислотность раствора Маскирующие средства Условия термообработки Не мешают Мешают
Высуш. при ПО °C (C7H6O2N)2Pd 0,2817 Опр. палладия в свинцовых корольках 3
а-Бензоин-оксим Слабо-кисл, среда (0,14% НС1) Прокал. в токе Н2 Металлический Pd Au, Ni, Си, Fe, Pb, платиновые металлы Опр. микро*-количеств палладия 2
р-Фурфур-альдоксим Кисл. среда (2% НС1) Винная кислота в присутствии Fe(III) Прокал. в токе Н2 Металлический Pd Pt-металлы, Fe, Со, Ni, Си Аи, РЬ Опр. больших количеств палладия в присутствии меди и никеля 2
Высуш. при 110-120 °C (C5H4O2N)2Pd F= 0,2669
Б. Гравиметрическое определение Pd с диоксимами в качестве реагентов [1-3]
Реагент Условия определения Гравиметриче-ская форма и фактор пересчета F Влияние других элементов Область применения Литература
Кислотность раствора Маскирующие средства Условия термообработки He мешают Мешают
Диметилдиоксим Кисл. среда (2 н. НС1) F- при опр. Pd в присутствии Fe(III) и NaNO2 для отд. Pd от Au, Ag и основных металлов -ЭДТА, винная кислота Прикал. в токе Н2 Металлический Pd Pt-металлы, Ni, Co, Cu, Zn, Sn и др. — Опр. в присутствии Ni, Cu, Fe; Отделение Pd от Pt, Ir, Rh, Ru, Os, Pb, Bi, Cd, TI, Cu, Co, Ni, Sn Опр. палладия в объектах, содержащих Pt, Au; Анализ сплавов Au-Ag-Pd 3,4 5 3 6
Высуш. при 110-120 °C (C4N2O2H7)2Pd
Дифенилдиоксим pH = 5-6 — Высуш. при 110-120 °C (C14HnO2N)2. •Pd Нет сведений — — 7,8
Бензоилме-тилдиоксим Кисл. среда — То же (C10H9O3N2)2. •Pd Pt — — 3,9, 10
1,2-Цикло-гександи-ондиоксим pH = 0,5-8,0 Комплексон III в присутствии Fe(III) Высуш. при 110 °C (C6H9O2N2)2- •Pd F= 0,2743 Другие Pt-металлы, U, Be, Al, Щ.М. и Щ.З.М., Ni, Cu, Fe, Pb, Se, Те Большие количества Fe 5-25 MrPd И, 12
а-Фурил-диоксим Кисл. раствор, pH = 0,2-8, pH = 2-3, не > 0,5% НС1 по весу [2] Прокал. в токе Н2; высуш. при 110-120 °C Металлический Pd (Cl0H7O4N2)2. •Pd F- 0,1957 Опр. 30-35 мг Pd 2, 13, 14
Химические методы количественного анализа
565
Продолжение таблицы Б
Реагент Условия определения Гравиметриче-ская форма и фактор пересчета F Влияние других элементов Область применения Литература
Кислотность раствора Маскирующие средства Условия термообработки He мешают Мешают
4-Метил-1,2-цикло-гександи-ондиоксим pH = 0,7-5,0 Винная кислота Высуш. при 105 °C (C7HUO2N2)2-•Pd F= 0,2558 cr, no;, сю;, so4_, C4H40g , Pt, Rh, Ru, Co, Fe(II) и др. Ir, Ni, Nb, Au, Al 15
4-Изопро-пил-1,2-циклогек-сандионди-оксим pH = 1-1,5 То же То же (C9H15O2N2)2- •Pd F= 0,2555 To же To же 15
В. Осаждение Бис-(диоксимато)палладия из гомогенных растворов [1]
Осаждаемая форма Компоненты реагентной смеси Содержание палладия, мг Условия осаждения Влияние других элементов Литература
Бис-(Диметилди-оксимато)палладий Биацетил, гидроксиламин солянокислый 1-200 pH = 0,4, время стояния 13-14 час. Не меш. 75 мг Pt 16, 17
Бис-(1,2-Циклогек-садиондиоксимато)-палладий 1,2-Циклогексан-дион, гидроксиламин солянокислый 1-50 (обычный метод 6-30 мг-мли) 0,3-1 М НС1* Не меш. 25 мг Cu, Ni, Со, Pt** 18
Бис-(Р-Фурфуральд-оксимато)палладий Фурфурол, гидроксиламин солянокислый 30-40 Время стояния 3 час Меш. Аи(Ш) 19
Бис-(Индан-1,2-ди-оксимато)палладий Индан-1-ОН-2-ОК-сим, гидроксиламин солянокислый 5 pH = 2,0-2,5, нагр. в течение 2,5-3 час при 65 °C Меш. Pt, Аи; не меш. Со, Си, Fe(III) в присутствии ЭДТА 20
Бис-(Салицил-альдоксимато)пал-ладий Салициловый альдегид, гидроксиламин солянокислый 3-50 2,0-2,5 объемн. % H2SO4 21
Примечания.
* Результаты точны, если раствор быстро доводят до 100 °C и затем кипятят в течение 5 мин.
** В присутствии платины должно строго дозироваться количество 1,2-циклогександиона.
Г. Реагенты, применяемые для восстановления комплексных хлоридов Ru(III), Os(IV), Rh(III), Ir(III), Pd(II) « Pt(II) до металла [1]
Реакция Реагент Условия*
Pd(II) -> Pd H2, Zn, Mg и другие металлы, стоящие перед Н2 в ряду напряжений; N2H4OH, Hg(I), Cu(I), Cr(II), Sn(II), Na2S2O4, гипофосфит, CO, предельные и непредельные углеводороды, С2Н5ОН, НСООН, глицерин и т.п. Легко и быстро восстанавливается в солянокислых растворах
Pt(II) -у Pt Н2, Zn, Hg(I), Cu(I), Сг(П), Ti(III), аскорбиновая кислота, HCOONa, NH2OH, N2H4OH Восстановление идет медленнее
Rh(III) -> Rh Cr(II), V(III), Ti(III), Zn, Mg, Cu, Sb, H2 Требуются жесткие условия и сильные восстановители. Соли Cu(I) и Hg(I) восст. только при высокой кислотности р-ра. Аскорбиновая к-та и гипофосфит осажд. металл только в присутствии HgCl2
566
Новый справочник химика и технолога
Продолжение таблицы Г
Реакция Реагент Условия*
Ir(III) -> Ir H2 под давлением при высокой температуре; Zn и Mg (не количественно) Требуются еще более жесткие условия. Даже кип. кисл. р-ров с Mg не выделяет иридий количественно
Ru(ni) -> Ru Zn, Mg, Ti(III), Н2 под давлением —
Os(IV) -> Os Fe(II), HCOONa, Cu, Zn Достаточно действия мягких восстановителей
Примечание.
* Легкость восстановления до метала в р-рах галогеноводородных кислот увеличивается при переходе от НС1 к НВг и HI.
Д, Состав продуктов акватации и гидролиза хлоридных комплексов Pt (IV) в состоянии равновесия* ]22]
3. Состав летучих при высоких температурах оксидов платиновых металлов [1]
Концентрационные условия Состав комплекса
> 3 М НС1 [Ptci6]2-
0,1-3 МНС1 [PtCl6]2-, начало гидролиза
0,05 М НС1 20% [РЮНС15] 2~; 80% [PtCl6]2’
0,01 М НС1 [PtOHCl5]2", [Pt(OH)2C14]2’
pH = 7-13 [Pt(OII)5Cl]2
0,1 М NaOH [Pt(OH)6]2-
Примечание.
* В отличие от остальных платиновых металлов все продукты гидролиза платины(ТУ) растворимы в воде, даже при кипячении щелочного раствора, что используется в анализе для отделения платины(ГУ) от других платиновых металлов (Pd, Rh, Ru, Ir), которые в тех же условиях образуют нерастворимые продукты гидролиза. Нерастворимая двуокись PtO2-4H2O или H2[PtCl6] выделяется в твердую фазу только при нейтрализации растворов уксусной или серной кислотой [1].
Металл Состав летучего оксида Литература
Ru RuO* или RuxO 25
RuO2 26
Os OsO2 26
OsO4
Ir IrO3 25, 27, 28
IrO2 29
Rh RhO2 25
RhxO2 27, 28
Pt PtO2 25-28
Примечание.
* Существование сомнительно.
Е. Реакции разложения оксидов родия при постоянном давлении (760 мм. рт. ст.) [23]
Оксид Температура разложения, °C Реакция
Rh2O3 1113 Rh2O3 = 2RhO + 0
RhO 1121 2RhO = Rh2O + 0
Rh2O 1127 Rh2O = 2Rh + 0
Ж. Скорость потери в весе платиновых металлов при прокаливании образцов фольги (мг • см"2- час1) [24]
Металл Температура, °C
1000 1200 1400
Ru 0,58 12 120
Os (1350) (8Ю) 1240
Ir 0,67 1,6 3,1
Rh См.* 8,5-Ю-4 6,8-10’3
Pt Cm.** 1,7-103 9,6-10~3
Примечание.
* При 1000 °C на поверхности металлического родия образуется защитная пленка оксида.
** Слишком малое изменение в весе.
Литература
1. Гинзбург С.И. и др. Аналитическая химия платиновых металлов. М.: Наука, 1972.
2. Пшеницын Н.К., Некрасова Г.А. // Журн. аналит. химии. 1957. Т. 12. С. 205; Изв. сектора платины ИОНХ АН СССР. 1955. Т. 30. С. 126, 156.
3. Бимиш Ф.Е. Аналитическая химия благородных металлов. Ч. 1, 2. М.: Мир, 1969.
4. Gilbert Н., Ayres G.H., Borg E.W. // Anal. Chem. 1953. V. 25. P. 980.
5. Salaria G.B.S. // Anal. chim. acta. 1959. V. 20. P. 122.
6. Gotthard E., Lenk M. // Metall. u. Erz. 1935. Bd. 32. S. 95.
7. Dwyer F.P., Mellor D. I I J. Am. Chem. Soc. 1935. V. 57. P. 605.
8. Wawrzyezek W., Majkowska H., Soboszynska J. // Z. anal. Chem. 1962. Bd. 188. S. 88.
9. Щекочихина Р.Л., Пешкова B.M., Шленская В.И. // Изв. МГУ. Серия хим. 1962. С. 38.
10. Hanus J., Jilev A., Lukas J. // Chem. News. 1925. Bd. 131. S. 401; 1926. Bd. 132. S. 1.
11. Пшеницын H.K., Ивонина O.M. И Журн. неорган. химии. 1957. Т. 2. С. 121.
12. Voter R.C., Banks С.К., Diehl Н. И Anal. Chem. 1948. V. 20. Р. 652.
13. Koji Jshikawa // J. Chem. Soc. Japan. Pure Chem. Sect. 1965. V. 86. P. 1166.
14. Read A., Banks C.V. // Proc. Jowa Acad. Sci. 1948. V. 55. P. 267.
15. Banks C, Uooker D. И Anal. Chem. 1956. V. 28. P. 79.
16. Firsching F.H. 11 Adv. Anal. Chem. and Instrument. 1965. V. 4. P. № 1.
Химические методы количественного анализа
567
17. GordonL.,EllefcenP.R.,WoodG.//Taianta. 1966.V. 13.P.551.
18. Velazquez J., Hileman O. // Taianta. 1968. V. 15. P. 269.
19. Pino-Perez F., Burriel-Marti F., Mertinez Conejero L. // An. Real. soc. esp. fisc, у quim. 1959. V. 55. P. 579.
20. Bark L.S., Brandon D. // Taianta. 1963. V. 10. P. 1189.
21. Anil de K., Chittaranjan S. // Separat. Sci. 1967. V. 2. P. 11.
22. Blasius E., Preetz W., Schmitt R. // J. Inorg. Nucl. Chem.
1961. V. 19. P. 115.
23. Федоров И.А. Родий. M.: Наука, 1966.
24. Krier С.А., Jaffe R.J. // J. Less-Common Metals. 1963.
V. 5. P. 411.
25. Allcock C.B., Hooper G.W. // Proc. Roy. Soc. (London). 1960. V. 254A. P. 551.
26. Phillips W. // Trans. Am. Soc. Met. 1964. V. 57. P. 33.
27. Schaffer H., Heitland H.J. // Z. anorg. allg. Chem. 1960. Bd. 304. S. 249.
28. Schafer V.H., Tebben A. // Z. anorg. allg. Chem. 1960. Bd. 304. S. 317.
29. Талипов Ш.Т., Нигай К.Г., Хакимходжаев H. // Научи, труды Ташкентского ун-та. 1967. Вып. 288. С. 100.
Таблица 5.1.11.12
Никель
А. Свойства сульфида никеля [1]
Соединение Температура, °C рПР Литература Кислоты, в которых растворяется сульфид Литература
NiSa — 20,5 6 1-2 н. НС1 на холоду 2
25 20,5 7
18-25 20,5 8
25 18,5 3
NiSp — 26,0 6 6 н. НС1 + Н2О2 2
18-25 26,0 8
25 24,0 3
NiSy — 27,7 6 2 н. НС1 + Н2О2 3
18 23,85 8 2 н. НС1 + КСЮз
18 24,16 3 СН3СО3ОН + Н2О2 4,5
18-25 27,7 9 HNO3 конц. при нагревании 2,3
20 29,96 10 Смесь HNO3 и НС1
25 20,7 11, 12
Б. Произведение растворимости Ni(OH)2 [1]
Температура, °C рПР Литература
17 18,06 13
18 14,87 14
20 15,0 15
25 13,81 16
25 14,5 17
25 15,21 18
25 18,06 7
25 17,19 19
25 15,5 20
28-30 16,0 21
75 16,2 22
В. Растворимость (S) диоксиматов никеля в воде [1]
Реагент Температура, °C р£ Литература
Диметилдиоксим 25 6,01 (0,05 М) 23
25 5,68 244
25 5,98 25
30 5,86 25
Продолжение таблицы В
Реагент Температура, °C р£ Литература
35 5,74 25
40 5,622 25
45 5,51 25
Ниоксим 25 5,80 26
Гептоксим 25 6,05 26
З-Метилниоксим — 5,51 27
4- Метилниоксим — 6,02 27
З-Этилниоксим — 4,75 27
4-Изопропилниоксим — 6,81 27
а-Фурилдиоксим 25 6,20 26
а-Бензилдиоксим 25 6,15 26
Г. Растворимость (ь-диоксимов в хлороформе и бензоле
Реагент Температура, °C Растворитель pS Литература
Диметилдиоксим 25 СНС13 3,35 28
20-22 СНС13 3,40 23
20-22 СбНб 3,49 23
25 СНС13 3,41 ' 29
Ниоксим 20 СНС13 2,08 30
Гептоксим 20 СНС13 1,72 30
а-Фурилдиоксим 20 СНС13 2,32 30
568
Новый справочник химика и технолога
Д. Значения рПР диоксиматов никеля fp, = 0) [1]
Реагент Температура, °C рПР Литература
Диметилдиоксим 25 23,66 26
— 22,70 23
40 23,23 26
— 23,37 31
— 23,27 32
Этилметилдиоксим 25 23,27 33
Ниоксим 25 28,39 26
40 26,96 26
3 -Метил ниоксим 25 27,62 26
40 26,68 26
4-Метил ниоксим 25 28,25 26
40 27,20 26
Диэтилдиоксим 40 24,21 33
Гептоксим 25 26,64 26
40 25,74 26
4-Метилфенилдиоксим 25 27,84 33
Бензоилметилдиоксим — 21,53 32
Е. Условия выделения диоксиматов никеля и палладия [1]
Реагент Кислотность раствовров при выделении диоксиматов
Никель Палладий
Диметилглиоксим pH = 6,4 НС1 (2 н.)
Никосим pH = 9,3 НС1 (5 н.)
а-Фурилдиоксим pH = 6,0 pH = 0,5
Литература
1. Пешкова В.М., Савостина В,М. Аналитическая химия никеля. М.: Наука, 1966.
2. Алимарин И.П., Архангельская В.Н. Качественный полумикроанализ. М.: Госхимиздат, 1952.
3. Блок Н.И. Качественный химический анализ. М.: Госхимиздат, 1952.
4. Крешков А.П. Основы аналитической химии. Т. 1. М.: Госхимиздат, 1961.
5. Кертман Л. Качественный химический полумикроанализ. М.: Госхимиздат, 1949.
6. Stability constants. London. The Chemical society. Burlington House. 1957.
7. Надеинский Б.П. Теоретические основания и расчеты в аналитической химии. М.: Советская наука, 1956.
8. Ringbom A. Solubilities of Sulfides. Report to Analytical Section. JUPAC. July 1953.
9. Brunner L., Lawadzki J. П Z. anorg. allg. Chem. 1910. Bd. 65. S. 136.
10. Cm. [9]. S. 454.
11. Rossini F., Nagman D., Evans Wm.H. I I Nat. Bur. Stand. Cirkular. 1952. № 500. P. 1266; C.A. 1952. V. 46. 5417f.
12. Moser L., Behr M. // Z. anorg. allg. Chem. 1924. Bd. 134.
S. 49.
13. Britton H.T.S. //J. Chem. Soc. 1925. V. 127. P. 2110.
14. Аксельруд H.B., Фиалков Я.А. // Укр. хим. журн. 1950. Т. 16. С. 283.
15. Cuta F., Ksandr Z., Hejtmanek M. H Chem. Listy. 1956. Bd. 50. S. 1064.
16. Dewijs H.J. 11 Rec. trav. chim. 1925. V. 44. P. 663.
17. Oka Y. H J. Chem. Soc. Japan. 1938. V. 59. P. 971; C.A. 1939. V. 33.2061.
18. NasanenR. //Ann. Acad. Sci. Tennicae. 1942. V. 59A. № 2.
19. Gayer K., Garret A. H J. Am. Chem. Soc. 1949. V. 71. P. 2973.
20. Schwab G., Polydoropoulos. H Z. anorg. allg. Chem. 1953. Bd. 274. S. 234.
21. Jena P., Prasad В. II J. Ind. Chem. Soc. 1956. V. 33. P. 122.
22. Доброхотов Г.Н. II Журн. прикл. химии. 1954. T. 27. С. 1056.
23. Бабко А.К., Михельсон П.Б. И Укр. хим. журн. 1955. Т. 21.С. 388.
24. Barker М. И Chem. News. 1925. V. 130. Р. 99.
25. Fleischer D., Freiser H. H Phys. Chem. 1962. V. 66. P. 389.
26. Banks C.V., Barnum D.V. H J. Am. Chem. Soc. 1958. V. 80. P. 3579.
27. Hooker D, Banks С. I I USA F.C. report 1955. Jsc-597. Yowa state College J.S.C. 1955. 597; C.A. 1956. V. 50. 11878.
28. Christopherson H., Sandell E. 11 Anal. Chim. Acta. 1954. V. 10. P. 1.
29. Dyrssen D., Hennichs M. 11 Acta Chem. Scand. 1961. V. 15.P.47.
30. Савостина B.M., Астахова E.K., Пешкова B.M. // Журн. неорган. химии. 1964. Т. 8. С. 80.
31. Charles R., Freiser Н. // Anal. Chim. Acta. 1954. V. И. P. 101.
32. Пешкова B.M., Зозуля А.П. И Тр. Комиссии по аналит. химии. 1960. Т. И. С. 69.
33. Banks C.V., Anderson S.J. И Inorg. Chem 1963. V. 2. Р. 112.
Таблица 5.1.11.13
Алюминий
Потери А1 при осаждении его в виде оксихинолината алюминия fl]
Условие. Осажд. 5-30 мг алюминия при 50 °C изб. (10 мл) р-ра оксихинолина (2,5%). Общий объем 150 мл.
рн Масса А1 в фильтрате, мкг pH Масса А1 в фильтрате, мкг
3,8 1000 4,8 30
4,0 75 4,9 28
4,3 50 5,0 26
4,5 41 5,3-7,0 22-20
4,7 31
Литература
1. Classen A., Bastings L. И Analyst. 1967. V. 92. Р. 618. Цит. по [2].
2. Тихонов В.Н. Аналитическая химия алюминия. М.: Наука, 1971. С. 33.
Химические методы количественного анализа
569
Таблица 5.1.11.14
Галлий
Отделение галлия от некоторых элементов методом осаждения [1]
Метод Содержание, мг Литература
Ga Отделяемые элементы
Осаждение галлия аммиаком в присутствии NH4C1 Щелочные металлы, Mg, щелочноземельные металлы, Cd, Ti, Mn, Ni 2-8
Осаждение галлия карбонатом бария Mn 2
Fe(II) 9
Осаждение галлия при нейтрализации солянокислых растворов (pH = 3), содержащих фосфат аммония Mg, Ca, Zn, Cd, Pb, Mo, Mn, Co - 5; Cu, V-3 10, 11
Выделение галлия в виде комплексного фторида Cu, Zn, Cd, Mo, W, Mn, Co, Ni 12
Осаждение галлия кипячением с Си2О или Си(ОН)2 в присутствии SO2 Be, Zn, Cd, Al, TR, Tl, Pb, U, Mn, Fe, Co, Ni 2, 8, 13-16
Осаждение галлия ферроцианидом калия из раствора НС1 (1:3) Be, Mg, щелочноземельные металлы, Cd, Hg(II), H3BO3, Al, Se, Y, TR, Tl, Pb, H3PO4, Sb, Bi, Cr, Mn, Co 2, 8,15
Ru, Rh, Ir 8, 17-24
Выделение галлия в виде Ga(OH)3 гидролизом галлата (индикатор метиловый красный) F 25
Выделение галлия избытком СН3СООН Zn, Al, In, Fe 26
Гидролитическое выделение галлия
сульфитом из раствора NH4OH 15-47 Zn - 350; Ca 2, 9, 27-30
мочевиной из раствора H2SO4 (pH = 4-5,5) Ca; Zn < 1103; Mn, Cu, Al 31
анилином в присутствии Na2S2O3 4,6^46 FeCl3 - 5 103 - 5-104 32, 33
фенилгидразином в присутствии NH4HSO3 Fe 34
Осаждение галлия оксихинолином
из горячего раствора NH4OH (5 : 95) (pH = 6-8) Щелочные металлы, V, Mo, W 35
при pH = 3,5 (бифталатный или ацетатный буфер) 5-30 Mg, Ca, Cd, Pb-7; Mn-20; U-0,5 10,36
бифталатный буфер Al 10
в присутствии тиомочевины Cu-5 10
в присутствии аскорбиновой кислоты и гидроксиламина V-l-2 Fe-20 10
Осаждение галлия 5,7-дибром-8-оксихинолином из кипящей 0,06 н. НС1 в присутствии ацетона Ga: Al = 1:1000 37,38
Осаждение галлия купфероном: из 2 н. H2SO4 из 1 н. Н2С2О4, содержащей (NH4)2C2O4 3,2-56,2 Щелочные металлы, Cu, Mg; щелочноземельные металлы, Zn; Al - 10-103; Se, Y, TR, In, As, Cr, U, Mn, Co, Ni 33-35,41- 43
Ti, Zr, Th < 100 35
Осаждение галлия таннином из кипящего 1-2% раствора СН3СООН, содержащего NH4NO3 Щелочные металлы, Be, Mg; щелочноземельные металлы, Zn, Cd, Tl, Mn, Co, Ni 33, 39,40, 42
Осаждение галлия пиридином в присутствии солей аммония Щелочные металлы, Cu, Mg, щелочноземельные металлы, Zn, Cd, Mn, Co, Ni 43
Осаждение галлия камфарной кислотой или камфоратом натрия
570
Новый справочник химика и технолога
Продолжение таблицы 5.1.11.14
Метод Содержание, мг Литература
Ga Отделяемые элементы
из СН3СООН из СН3СООН в присутствии Na2S2O3 Щелочные металлы; Be, Mg, щелочноземельные металлы 44-46
1-100 Zn-1-500; Cd; А120з < 10; Sc, Y, TR, TI, Ge
1-100 Pb -1-200; Th, V, Cr, U, Mn, Co, Ni
Ga2O3 -1103 4-1,5105 Fe2O3 -1-500 47
Выделение галлия в виде основных коричнокислых солей Zn, Mn, Co, Ni 48,49
Осаждение галлия диантипирилметаном из 5,5-6 н. НС1 Cu, Mg, Zn, Cd, Al, In, Bi, Mn, Co, Ni 50
Осаждение меди избытком Na2S из 4 М NaOH или сероводородом из раствора H2SO4 Cu — небольшие количества 51,52
Осаждение серебра в виде AgCl или Ag2S Ag 8, 17,18, 53
Выделение золота восстановлением H2S в растворах НС1, а также при помощи H2SO3 или Си0 Au 8, 17, 18, 53
Осаждение щелочноземельных металлов в виде сульфатов в присутствии спирта Ca, Sr, Ba 5,8
Осаждение цинка избытком Na2S из 4 М NaOH Основа Zn — небольшие количества 9,51
Осаждение цинка роданомеркуриатом калия из разбавленных растворов H2SO4 или HNO3 гп<50мгв100мл 27, 54
Осаждение ртути сероводородом из раствора НС1 Hg 8, 17, 18, 53
Восстановление ртути до металла медью Hg
Выделение ртути при действии кипящей НС1 на смесь окислов ртути и галлия Hg 55
Осаждение алюминия из горячего раствора избытком NH4OH (ограниченное применение) Al - незначительные количества 33,56-58
Выделение алюминия из раствора НС1 в виде А1С13-6Н2О ацетилхлоридом в ацетоне, газообразным НС1 в присутствии ацетона или эфира Al - несколько граммов 27, 29, 41, 59-61
Осаждение Sc, Y и TR из кипящего раствора избытком NaOH или КОН Sc, Y, TR 5, 8, 62
Осаждение индия в виде 1п(ОН)3 карбонатом натрия In 47
Осаждение индия избытком NaOH или КОН In 27, 63
Осаждение индия гексаминокобальти-хлоридом в виде [Co(NH5)6)]InCU из 2,5 н. НС1 In 64
Осаждение индия избытком водного раствора гексаметилендиамина In 65
Химические методы количественного анализа
571
Продолжение таблицы 5.1.11.14
Метод Содержание, мг Литература
Ga Отделяемые элементы
Осаждение индия в виде диэтилдитио-карбамината pH = 3-5, в присутствии избытка оксалата pH = 7-11, в присутствии тартрата или цианида
In : Ga = 2 : 1 66
In:Ga=l:10 67
Осаждение Т1(1) в виде Tl2PtCl6 T1(I) 8, 17, 18, 53
Осаждение Т1(П1) гексаминокобальти-хлоридом в виде [Co(NH3)6)]TlCl6 из 0,5 н. НС1 Tl(III) 64
Выделение SiO2«H2O многократным выпариванием с НС1 Si 8, 14, 68, 69
Осаждение титана и циркония фениларсоновой кислотой из горячей 2 н. H2SO4 Ga2O3 — 10-130 TiO2 — много ZrO2 — 100-2300 35, 70
Осаждение Ti, Zr и Th кипящим раствором щелочи Ti, Zr, Th 5,8, 16,71
Осаждение тория щавелевой кислотой из раствора НС1, не содержащего суль-фат-ионов Ga2O3 — 10-130 ThO2 — 120-2400 35
Осаждение олова сероводородом Sn 8,41,52, 72, 73
Осаждение олова в виде SnO2 Sn 8, 55, 72, 73
Осаждение свинца избытком Na2S из 4 М NaOH или сероводородом из раствора H2SO4 Основа Pb — небольшие количества 51,52
Осаждение свинца в виде PbSO4 выпариванием раствора с H2SO4 Pb 9
Осаждение фосфора в виде фосфоро-молибдата H3PO4 8, 68, 69
Осаждение сурьмы сероводородом из раствора H2SO4 Sb 9, 52
Осаждение висмута сероводородом с многократным переосаждением осадка Bi 9
Выделение висмута соосаждением с основным карбонатом железа Bi 74
Выделение ниобия и тантала гидролизом сернокислых растворов Nb, Ta 9
Выделение ниобия и тантала обработкой пиросульфатного плава окислов ниобия, тантала и галлия кипящей водой Nb, Ta 8, 75, 76
Осаждение селена и теллура действием восстановителей (H2S, H2SO3) Se, Те 8,14,21,24, 68, 77
Осаждение WO3 выпариванием с НС1 W 8,14, 68, 69
Осаждение урана избытком NaOH U 3, 4, 6-8
Осаждение железа из горячего раствора избытком NH4OH (ограниченное применение) Fe 33, 56-58
Осаждение железа избытком NaOH с обязательным переосаждением осадка 10 Fe — 1000 5, 8, 9,47, 57, 78, 79
Осаждение железа в виде FeS в присутствии сульфосалициловой кислоты Ga2O3-13:54 Fe2O3 — 88-590 33
Осаждение железа а-нитрозо-Р-нафтолом из раствора СН3СООН Ga2O3 - 25 Fe2O3 — 4~44 80
572
Новый справочник химика и технолога
Продолжение таблицы 5.1.11.14
Метод Содержание, мг Литература
Ga Отделяемые элементы
Осаждение Ru, Rh, Pd, Os и Pt сероводородом из раствора НС1 Платиновые металлы 8, 17-24
Восстановление родия и палладия металлической медью
Осаждение иридия кипячением сернокислого раствора с КОН
Литература
1. Дымов А.М., Савостин А.П. Аналитическая химия галлия. М.: Наука, 1968.
2. Гиллебранд В.Ф., Лендель Г.Э., Брайт Г.А., Гофман Д.И. Практическое руководство по неорганическому анализу. М.: Госхимиздат, 1957.
3. Lecoq de Bosbaudran И Chem. N. 1882. Bd. 46. S. 152.
4. Cm. [3]. S. 165.
5. Cm. [3]. Compt. rend. 1882. Bd. 94. S. 1227.
6. Cm. [5]. Bd. 95. S. 410.
7. Cm. [5]. Bd. 95. S. 503.
8. Cm. [3]. Ann. chim. Phys. 1884. Bd. (6)2. S. 176.
9. Fresenius R., Jander G. Handbuch der analytischen Chemie. III. Quantitative Analyse. Bd.III. Elemente der dritten Gruppe. Berlin. Springer-Verlag. 1942. S. 531.
10. Черкашина T.B. // Сб. научных трудов Гиредмета. 1959. Т. 2. С. 202.
11. Einecke Е. // Die Chemie. 1942. Bd. 55. S. 40.
12. Тананаев И.В., Баусова Н.В. Химия редких элементов: Сборник. Вып. 2. М.: Изд. АН СССР, 1955. С. 12.
13. Crookes W. Selected Methods in Chemical Analysis. Longsman, Green a. Co. London; New York, 1894.
14. Kriesel F. W. // Chem. Ztg. 1924. Bd. 48. S. 962.
15. Cm. [5]. S. 1439.
16. Cm. [5]. S. 1625.
17. Cm. [5]. Bd. 95. S. 1332.
18. Cm. [3]. 1883. Bd. 47. S. 16.
19. Cm. [18]. S. 100.
20. Cm. [18]. S. 299.
21. Cm. [3]. Bd. 48. S. 15.
22. Cm. [5]. 1893. Bd. 96. S. 152.
23. Cm. [22]. S. 1696.
24. Cm. [22]. S. 1838.
25. Hannebohn O., Klemm W. // Z. anorg. allg. Chem. 1936. Bd. 229. S. 337.
26. Еремин Н.И. Галлий. M.: Металлургия, 1964.
27. Dennis L.M., Bridgman J.A. // J. Am. Chem. Soc. 1918. V. 40. P. 1531.
28. Dupuis T., Duval C. // Anal. chim. acta. 1949. V. 3. P. 324.
29. Hillebrand W.F., Lundell G.E.F., Bright H.A., Hoffman J.I. Applied Inorganic Analysis. Chap. 30. Gallium. 2nd ed. New York: Willey. 1953.
30. Porter L.E., Browning P.E. // J. Am. Chem. Soc. 1919. V.41.P. 1491.
31. Willard H.H., Fogg H.C. // J. Am. Chem. Soc. 1937. V. 59. P. 1197, 2422.
32. Moser L., Brukl A. // Monatsh. Chemie. 1928. Bd. 50. S. 331.
33. Cm. [32]. 1929. Bd. 51. S. 325, 328.
34. Weiner E. // J. Am. Chem. Soc. 1934. V. 56. P. 348.
35. Brukl A. // Monatsh. Chemie. 1929. Bd. 52. S. 253.
36. Черкашина T.B. // Сб. научн. трудов Гиредмета. 1959. Т. 2. С. 189.
37. Gastinger Е. // Z. anal. Chem. 1943. Bd. 126. S. 373.
38. Moeller T., Cohen A.J. // Anal. Chim. Acta. 1950. V. 4. P. 316.
39. Gastinger E. // Z. anal. Chem. 1953. Bd. 140. S. 244.
40. Cm. [32]. S. 181.
41. Scherrer J.A. // J. Res. Nat. Bur. Stand. 1935. V. 15.
P. 585.
42. Черкашина T.B. // Завод, лаб. 1956. T. 22. С. 276.
43. Иванов-Эмин Б.А., Остроумов Э.А. // Завод, лаб. 1946. Т. 12. С. 674.
44. Ato S. // Sci. Pap. Inst. Phys. Chem. Research. 1930. V. 12. P. 225. Chem. Zbl. 1930. V. 1. 3467.
45. Cm. [44]. 1931. V. 15. P. 289; Chem. Zbl. 1931. V. II. 1033.
46. Cm. [44]. 1943. V. 40. P. 228.
47. Cm. [44]. 1934. V. 24. P. 270, 277.
48. Остроумов Э.А., Волков И.И. // Труды Ин-та океанологии АН СССР. 1964. Т. 67. С. 141.
49. См. [48]. Журн. аналит. химии. 1963. Т. 18. С. 52.
50. Бусев А.И., Типцова В.Г. // Журн. аналит. химии. 1960. Т. 15. С. 698.
51. Яценко С.П. Труды Ин-та химии, Уральский филиал АН СССР. 1963. Т. 7. С. 151.
52. Klein Р., Skrivanek V. // Chem. Prumysl. 1963. Bd. 13. S. 250.
53. См. [5].Bd. 95. S. 157, 1192.
54. Lundell G.E.F., Nai Kim Bee // Trans. Am. Inst. Metals. 1914. V. 8. P. 146.
55. Putnam P.C., Roberts E.M., Seichow D.H. // Am. J. Sci.
1928. V. (5)15. P. 425.
56. Einecke E., Harms J. // Z. anal. Chem. 1934. Bd. 98. S. 432.
57. Fricke R. // Z. anorg. Chem.. 1925. Bd. 144. S. 267.
58. Fricke R., Blencke W. // Z. anorg. allg. Chem. 1925. Bd.
143. S. 183.
59. Cm. [44]. 1930. V. 14. P. 36.
60. Browning P.E., Porter L.E. // Am. J. Sci. 1917. V. 44. P. 221.
61. Steinbach J.F., Freiser H. // Anal. Chem. 1954. V. 26. P. 375.
62. Коренман И.М., Назарова Г.В. // Труды по химии и хим. технологии. 1965. Вып. 2. С. 156.
63. Тананаев И.В. // Изв. АН СССР. ОХН. 1957. № 12. С. 1421.
Химические методы количественного анализа
573
64. Takashima Y. // Nippon Kagaku Zasshi. 1959. V. 80. P. 626.
65. Коренман И.М., Челышева С.Ф. И Труды по химии и хим. технологии. 1965. Вып. 1. С. 35.
66. Бусев А.И., Жолондковская Т.Н., Кузнецова З.М. // Журун. аналит. химии. 1960. Т. 15. С. 49.
67. Patrovsky V. // Chem. Listy. 1954. Bd. 48. S. 1047.
68. См. [5]. 1883.Bd.97. S.66.
69. Cm. [5]. 1883.Bd.97. S. 521.
70. Rice A.C., Fogg H.C., James С. H J. Am. Chem. Soc. 1926. V. 48. P. 895.
71. Cm. [5]. 1883. Bd. 97. S. 623.
72. Cm. [3].S.211.
73. Cm. [5]. Bd. 95. S. 703.
74. Jaskolska H., Minczewski J. // Acta chim. Acad, scient. Hung. 1962. Bd. 32. S. 9.
75. Cm. [3]. 1883. Bd. 48. P. 197.
76. Cm. [71]. S. 730.
77. [21]. S. 148.
78. Swift E.H. 11 J. Am. Chem. Soc. 1924. V. 46. P. 2377.
79. Wada J., Ishii R. П Sci. Pap. Inst. Tokyo. 1937/38. V. 34.
P. 787.
80. Papish J., Hoag L.E. 11 J. Am. Chem. Soc. 1928. V. 50.
P. 2118.
5.2. Титриметрический анализ (ТА)
Несмотря на подавляющее применение физико-химических и физических методов анализа, титриметрические методы анализа не потеряли своей актуальности и значимости [1-60]. Они по-прежнему широко применяются в самых различных областях науки и производства. Особенно это касается анализа сырья, различных материалов, сертификации готовой продукции, арбитражных анализов и аттестации стандартных образцов на основные компоненты (1-100% масс.).
Обусловлено это следующими обстоятельствами:
— титриметрические методы характеризуются высокой точностью: при аккуратной работе они дают относительную погрешность определения 0,5-0,1 % при массе анализируемого вещества 0,1-0,5 г, а кулонометрическое титрование — порядка 0,01-0,001%, что недостижимо другими методами;
— конец титрования (конечную точку титрования) можно регистрировать как визуально, так и инструментально (в амперометрическом, кулонометрическом, кондуктометрическом, флуориметрическом, высокочастотном,
нефелометрическом, потенциометрическом, радиометрическом, спектрофотометрическом, термометрическом и турбидиметрическом титрованиях) [16];
— возможностью автоматизации; высокой скоростью определений, простотой, низкой стоимостью; широким ассортиментом используемых реакций, позволяющих определять количественно практически все ионы металлов и многие анионы.
Теоретические основы всех титриметрических методов анализа хорошо изложены в общедоступных отечественных и переводных учебных пособиях [1-15; 30; 31; 39], а также в зарубежных учебниках и монографиях [19-29, 32]. Конкретные методики определения элементов рассмотрены в монографиях [56-58], справочниках [54, 59], руководствах [60-61] и т. д.
В настоящем разделе основное внимание уделено принципам, терминам титриметрических методов анализа, концепции эквивалентов, традиционно связанной с теорией и практикой титриметрии, практическому применению ТА с визуальной регистрацией конечной точки титрования (КТТ).
СПИСОК ОСНОВНЫХ СОКРАЩЕНИЙ
АА — ацетилацетон ДГТА — Р,Р' -диаминодиэтилгликолевый эфир N,
АДА — аитранилдиуксусная кислота N, №, А-тетрауксусной кислоты
АК — аскорбиновая кислота ДДТКМа — диэтилдитиокарбаминат натрия
АОП — 4S-1 -(2 -амино-3 -окси-4 -пиридилазо) ДМГ — диметилглиоксим
бензол-4-сульфокислота ДМГП — димеркаптогликольпропионат
ATM — А-аллилтиомочевина ДМП — 2,3-димеркаптопропанол
АЦГ — ацетонциангидрин домк — 7,8-диокси-4-метилкумарин
БАЭС — бмс-(2-аминоэтил)сульфид ДТАМР — 4-(4,5-диметил-2-тиазолилазо)-2-
БТА —1,2,3-бензотриазол метилрезорцин
БФС — бромфеноловый синий ДТКАА — дитиокарбаминоацетат аммония
ВК — винная кислота ДТКПА — дитиокарбаминопропионат аммония
ГА ГМДТА — гидроксиламин — гексаметилендиаминтетрауксусная ДТПА ДФК — диэтилентриаминпентауксусная кислота — дифенилкарбазон
кислота ДЩК — дитиощавелевая кислота
ГТС — глицинтимоловый синий ДЭГ — диэтанолглицин
ДВК — дитиовинная кислота KA4SN — кислый ализариновый черный SN
ДГМТД — диперхлорат 5,7,7,12,14,14-гекса-метил- ККК — кальконкарбоновая кислота
1,4,8,11 -тетраазоциклотетрадека-4,11-диена КММЯК — карбоксиметилмеркаптоянтарная кислота
574
Новый справочник химика и технолога
КО — ксиленоловый оранжевый
КоК — койевая кислота
кхтс — кислотный хром темно-синий
кхчг — кислотный хром чисто-голубой
кэмяк — карбоксиэтилмеркаптоянтарная кислота
ЛАНАС — 2-(2-лепидилазо)-1-нафтол-4-
сульфокислота
ЛК — лимонная кислота
МДТКАА — А-метилдитиокарбаминоацетат аммония
МК — молочная кислота
МПАНД — 3(5)-метил-1н-пиразол-5(3)-азо-1,2-
нафтол-3,6-дисульфокислота
МТС — метилтимоловый синий
МФ — металлфталеин
НТА — нитрилотриуксусная кислота
ОХК — оксихинолинат калия
ОХСК — 8-оксихинолин-5-сульфокислота
ОЭДТА — А-оксиэтилэтилендиаминтриуксусная
кислота
(ОЭ)2ДТКА — бмс-(2-оксиэтил) дитиокарбаминат
аммония
(ОЭДДТК- — <5мс-(2-оксиэтил)дитиокарбаминат
(ОЭ)2А диэтаноламмония
ПААК — пиридилазоаминокрезол
ПАМ — полиэтиленполиамины
ПАН — 1-(2-пиридилазо)-2-нафтол
ПАОХ — 7-(2-пиридилазо)-8-оксихинолин
ПАР — 1-(2-пиридилазо)резорцин
ПАСК — п-аминосалициловая кислота
ПГК — пирогаллоловый красный
ПКФ — пирокатехиновый фиолетовый
ПОС — припой оловянно-свинцовый
ПТМ — потенциометрическое титрование
СК — салициловая кислота
сск — сульфосалициловая кислота
ТА — тионалид
ТАК — тиазолилазо-п-крезол
ТАР — 4-(2-тиазолилазо)резорцин
ТГК — тиогликолевая кислота
Тетра — бмс-(4-натрий-5-
тетразолилазо)этил ацетат
Тио — тиомочевина
Тир — тирон
ТМК — тиомолочная кислота
Трен — 2,2 ',2 "-триаминотриэтиламин
ТСК — тиосемикарбазид
ТТГА — триэтилентетрамингексауксусная
кислота
ТФМА — трифенилметановые красители
ТФТ — тимолфталексон
ТЭА — триэтаноламин
ТЭАМ — тиоэтаноламин
ТЯК — тиояблочная кислота
Унитиол — 2,3-димеркаптопропансульфонат натрия
ФК — феноловый красный
ХАЗ — хромазурол S
ХАФ — 1-(2-хинолилазо)-2-фенантрол
ХТС — хром темно-синий
ЦДТА — транс-1,2-циклогександиамин-
N, N, N', N -тетрауксусная кислота
ЦПХ — цетилпиридинийхлорид
ЦТА — цетилтриметиламмоний
ЦТАБ — цетилтриметиламмонийбромид
ЧАС — четвертичные аммониевые соли
ЩК — щавелевая кислота
ЩЗЭ — щелочноземельные элементы
ЭБОФГ — N, А-этилен-бмс-[2-(о-оксифенил)]
глицин
ЭДА — этилендиамин
ЭДТА — этилендиаминтетраацетат натрия
ЭДТП — этилендиаминтетрапропионовая кислота
ЭК — этилксантогенат калия
ЭХКВ — эриохромовый красный В
ЭХЦ — эриохромцианин
ЭХЧ — эриохромовый черный Т
ЯК — яблочная кислота
5.2.1. Общие положения
5.2.1.1. Сущность ТА. Точка эквивалентности или точка стехиометричности
Количественные определения, осуществляемые титрованием, объединяются под общим названием титримет-рический анализ или сокращенно — титриметрия.
Титриметрический анализ основан на точном измерении количества реагента, затраченного на реакцию с определяемым веществом в ходе титрования.
Титрование — это процесс постепенного добавления небольшими порциями стандартного раствора реагента (рабочего раствора) к раствору определяемого вещества. Стандартный или рабочий раствор — это раствор с точно
известной концентрацией активного вещества (реагента R) или с точно известным титром.
Стандартный (рабочий) раствор, который используется при проведении титрования, называется титрантом.
Существует два способа введения титранта в исследуемый раствор. В классическом варианте он подается механически, небольшими порциями из стеклянной градуированной бюретки, которая позволяет тщательно измерить объем титранта.
В методе кулонометрического титрования известное количество иона-титранта генерируется в процессе протекания вспомогательной электрохимической реакции на электроде, погруженном в анализируемый раствор. Обра
Химические методы количественного анализа
575
зующийся титрант механически размешивается, реагируя с определяемым ионом.
В классическом титровании масса определяемого вещества рассчитывается исходя из концентрации и объема титранта, затраченного на титрование. При добавлении каждой порции титранта в титруемом растворе протекает химическая реакция между определяемым (титруемым) веществом А и введенным в раствор реагентом R, содержащимся в титранте, и в системе устанавливается равновесие.
Эту реакцию называют реакцией титрования, ее уравнение можно записать в общем виде:
#A + rR <...> продукты реакции. (5.2.1)
Титрование продолжают до тех пор, пока не будет достигнуто стехиометрическое соотношение между количеством определяемого компонента и количеством реагента.
Точка, соответствующая стехиометрическому соотношению реагирующих веществ, называется точкой стехио-метричности (ТС) или точкой эквивалентности (ТЭ).
Если реакция титрования описывается уравнением (5.2.1), то в ТЭ количества стехиометрично прореагировавших веществ А и R относятся как стехиометрические коэффициенты:
откуда
h(A) = -h(R). (5.2.2')
г
Здесь и(А) и h(R) — соответственно количества вещества А и R, измеряемые в молях или ммолях.
Б соответствии с рекомендациями ИЮПАК количественные расчеты в АХ проводятся исходя из уравнений (5.2.2), (5.2.2') через количество вещества, молярную массу, молярную концентрацию.
Большинство расчетов в титриметрии основаны на использовании двух пар простых уравнений, которые вытекают из определений: моль, ммоль, молярная концентрация.
Для вещества X (A, R и др.) можно записать:
. х 1V1C4WC4 /М1 1
количество Х(моль) =--------------------—-------тр ,(5.2.3)
молярная масса Х(г • моль )
количество Х(ммоль) =
___________масса Х(мг)_________ (5 2 3') ммолярная масса Х(мг - ммоль-’)
Вторая пара вытекает из определения молярной концентрации:
количество X (моль) = с(Х) (моль • л-1) • И (л), (5.2.4)
количество X (ммоль) = с(Х) (ммоль • мл-1) -V (мл). (5.2.4')
Уравнениями (5.2.3) и (5.2.4) пользуются, когда объем измеряется в литрах, а масса в граммах, а уравнениями (5.2.3')и (5.2.4'), когда объем измеряется в миллилитрах, а масса в миллиграммах.
Обычно по результатам тетрирования вычисляют массу определяемого вещества от (А) в граммах. Используя уравнения (5.2.2'), (5.2.3), (5.2.4) и подставляя А вместо X в уравнение (5.2.3) и R вместо X в уравнение (5.2.4), с учетом совместимости единиц измерения получают:
^( = -c(R)-K(R)10-J,
А/(А) г
откуда
от(А) = —c(R)• K(R) • M(A) - IO-3 .* (5.2.5)
г
На практике во многих случаях расчет результатов тит-риметрических определений удобнее проводить на основе принципа эквивалентности. При этом стехиометрию кислотно-основных и окислительно-восстановительных реакций выражают в эквивалентной форме, а эталоном химического взаимодействия служит протон или электрон (см. раздел 2.1 и табл. 2.1; 2.2). Все расчеты ведутся через число моль эквивалентов вещества, молярную массу эквивалента и молярную концентрацию эквивалента (нормальную концентрацию**).
Согласно принципу эквивалентности, в любом титровании в точке эквивалентности число моль (ммоль) эквивалентов стандарта точно равно числу моль (ммоль) эквивалентов реагирующего с ним вещества (вещества реагируют между собой в эквивалентных количествах).
Для уравнения (5.2.1) можно записать:
и(МА)А) - T?(f3KB(R)R), (5.2.6)
где /?(^КВ(А)А) и /?(/,Kli(R)R) — символы числа моль или ммоль эквивалентов веществ А и R***.
Исходя из уравнений (5.2.2') и (5.2.6) в табл. 5.2.1 представлены уравнения количественных соотношений реагирующих веществ в молекулярной и эквивалентной формах для типичных титриметрических реакций 1-17 табл. 2.2 (раздела 2.1).
Из примеров 1, 2, 5, 10, 17 табл. 5.2.1 следует, что в случаях, когда стехиометрические коэффициенты реагирующих веществ равны единице, количественные соотношения реагирующих веществ, записанные в молекулярной и эквивалентной формах, тождественны. В этих случаях оперируют терминами количество вещества, молярная масса, молярная концентрация вещества.
По аналогии с уравнениями (5.2.3-5.2.4') из определений «число моль (ммоль) эквивалентов» и «молярная кон
* В тех случаях, когда расчеты ведутся исходя из уравнений (5.2.3') и (5.2.4'), где масса от(А) выражается в мг, а объем K(R) в мл, переводной множитель 10-3 в формуле (5.2.5) опускается.
** Нормальный раствор, нормальность — термины, не рекомендуемые ИЮПАК.
*** Моль эквивалентов, как и моль условных частиц, содержит 6,02 • 1023 эквивалентов.
576
Новый справочник химика и технолога
центрация эквивалента» (раздел 2.1 (2.7-2.8)) можно записать следующие две простые пары уравнений.
Для вещества X (A, R и др.):
число моль эквивалентов X (моль) =
масса X (г)
------------------------------------т-, (5.2.7)
молярная масса эквивалента X (г моль )
число ммоль эквивалентов X (ммоль) =
=___________масса Х(мг)__________ 2
ммолярная масса Х(мг • ммоль1)
Таблица 5.2.1
Количественные соотношения реагирующих веществ в реакциях 1-17 табл. 2.2 (раздела 2.1)
№ п/п В молекулярной форме1 В эквивалентной форме 2
1 п (НС1) = п (NaOH) п (НС1) = п (NaOH)
2 п (Na2CO3) = п (НС1) п (Na2CO3) - п (НС1)
3 2 у п (Na2CO3) = п (НС1) п (l/2Na2CO3) = п (НС1)
4 2 yw (H2SO4) = п (NaOH) w(l/2H2SO4)-«(NaOH)
5 п (Н3РО4) = п (КОН) п (Н3РО4) = п (КОН)
6 2 у п (Н3РО4) = п (КОН) п (1/2Н3РО4) = п (КОН)
7 з Y п (Н3РО4) = п (КОН) п (1/ЗН3РО4) - п (КОН)
8 2 Y п (Н2С2О4) = п (КОН) п (1/2Н2С2О4) = п (КОН)
9 2 (Ва(ОН)2) = п (НС1) и(1/2Ва(ОН)2) = и(НС1)
10 w(FeCl3) = «(Na2H2Y) п (FeCl3) = п (Na2H2Y)
11 | п (КМпО4) = п (Н2С2О4) п (1/5КМпО4) = = и (1/2Н2С2О4)
12 у ц (КМпО4) = и (FeSO4) п (1/5КМпО4) = п (FeSO4)
13 - п (КМпО4) = п (Н2О2) ц(1/5КМпО4) = = п (1/2Н2О2)
14 з -л(К2Сг2О7) = п(12) п (1/6К2Сг2О7) = п (1/212)
15 2 -п (I2) = п (Na2S2O3) w(l/2I2) = «(Na2S2O3)
16 * п (K2Cr2O7) = п (FeCl2) и(1/6К2Сг2О7) = w(FeCl2)
17 п (SnCl2) = п (Fe2(SO4)3) п (l/2SnCl2) = = и (l/2Fe2(SO4)3)
Примечания.
1 Согласно уравнениям (5.2.2), (5.2.2').
2 Согласно уравнению (5.2.6).
Вторая пара вытекает из определения молярной концентрации эквивалента:
Число моль эквивалентов X (моль) =
= С (/экв(Х)Х) (моль- л’1) -Г(л). (5.2.8)
Число ммоль эквивалентов X (ммоль) =
= с (4кв(Х)Х) (ммоль• мл4) • V(мл). (5.2.8')
Используя уравнения (5.2.6)-(5.2.8) и подставляя А вместо X в уравнение (5.2.7) и R вместо X в уравнение (5.2.8), с учетом совместимости единиц измерения получают:
т(А) М4.(А)А)
= c(/3KB(R)R) P(R) -10”3,
откуда:
™(А) = c(/3KB(R)R)-F(R)-M(/3KB(A)A)-10~3 .* (5.2.9)
Если требуется вычислить другой параметр определяемого вещества А (<о, с(А), Г(А) и др.) или даны другие единицы измерения концентрации стандартного раствора реагента R, то пользуются соответствующими формулами сводной таблицы соотношений физических величин (табл. 5.2.2).
Таблица 5.2.2**
Соотношения физических величин, применяемых для расчетов в титриметрии
Уравнения реакций №
а А + rR <-> продукты реакции (5-2.1)
и(А) = — w(R) г (5.2.2')
w(/3KB(A)A) = «(4KB(R)R) (5.2.6)
w(A) = - c(R) • A/(A) • P(R) • 10’3 r (5.2.5)
^(A) = c(/3KB(R)R)-F(R)-A/(/,kb(A)A)-10-3 (5.2.9)
и(/экв(А)А) - J - z(A) • «(A) /экв(А) (5.2.10)
<„(R)R)=-^-=z(R)XR) JЭКВ (5.2.11)
„(A)=-^ v A/(A) (5.2.12)
M(/Ja)A) (5.2.13)
* В тех случаях, когда расчеты ведутся исходя из уравнений (5.2.7') и (5.2.8'), где масса ?и(А) выражается в мг, а объем K(R) в мл, переводной множитель 10”3 в формуле (5.2.9) опускается.
Табл. 5.2.2 составлена на основе формул (2.1) - (2.24) раздела 2.1 применительно к разделу 5. Символ А приписан любому определяемому (титрируемому) веществу. Для стандартного раствора реагента R (титранта) в приведенных формулах, кроме (5.2.5), (5.2.9), (5.2.11), (5.2.28) - (5.2.33), (5.2.39) - (5.2.45) символ А следует заменить на символ R.
Химические методы количественного анализа
577
Уравнения реакций №
Ш„(А)А)= 7т(А)-М(А)- д(А) (5.2.14)
с(А) = V(X) (5.2.15)
и(А) = с(А) • К(А) (5.2.16)
с(Гэ„(А)А)^^М^ (5.2.17)
(5.2.18)
= ДА) • с(Л) /экв (А) (5.2.19)
ти(А) р(А)= —— (г-л ) К(А) (5.2.20)
с(А)=-^- М(А) (5.2.21)
сб<экв(А)А) = ——-м (А. (А) А) (5.2.22)
c«3KB(R)R) = —’ W(A,(R)R) (5.2.23)
Т(А)= (г-мл4) V 7 К(А) (5.2.24)
ДАНО^ М(А) (5.2.25)
^(Лкв(А)А) (5.2.26)
т(А) = ДА) • И(А) (5.2.27)
T(R/A) = (г-мл1) ' E(R) (5.2.28)
c«„(R)R) = = Г(К)М(/„(А)А) М(/,„(А)А) (5.2.29)
C(R)=L..<(F/A)dtf а М(А) (5.2.30)
r(R/A) = C(/„(R)R)x хЛ/(/„(А)А)-10-’ - (5.2.31)
7(R/A)= ^(R).JH(A)103 = (5.2.32)
m(A)=T(R/A)-V(R) = = (R)R) • HR) • M (/„ (A)A) 1 O’’ (5.2.33)
w(A) - T( R/A) • K(R) = - c(R) • V(R) • ЛДА) • 1 O'3 r (5.2.34)
co(A)= ^400 % "’нав (5.2.35)
(ДА) m(A)= th 100 (5.2.36)
Уравнения реакций №
A)=<o(A)-p-io АДА) (5.2.37)
с(/экв(А)А) = -^-Р'.10 M(/„(A)A) (5.2.38)
r(R) = mSl (5.2.39)
c(R) = (5.2.40)
c«3KB(R)R) = — M(/„(R)R)rK (5.2.41)
= r ffl(S)-!Q- C s M(SyV(R) (5.2.42)
.. tfjOSYlO3 c(£kb(R)R) = 7 7 M(/„(S)S)K(R) (5.2.43)
5 Гк M(5)-K(R) (5.2.44)
cyuR)R)=Ik. M(S')io1 VK M(/„(S)S)-K(R) (5.2.45)
Примечание.
1. См. также табл. 2.1 раздела 2.
2. В формулах 5.2.40-5.2.45: т (мг); V(мл).
5.2.1.2. Кривые титрования. Конечная точка титрования (КТТ) и способы ее регистрации [1—13,15, 19, 29]
В процессе титрования изменяются равновесные концентрации вещества титранта и продуктов реакции. При этом пропорционально концентрациям этих веществ изменяются свойства раствора. Например, при окислительно-восстановительном титровании изменяются равновесные концентрации окислителя и восстановителя и, следовательно, потенциал; при изменении концентраций компонентов кислотно-основной реакции изменяется pH раствора. График зависимости параметра системы, связанного с концентрацией титруемого вещества, титранта или продукта, от состава раствора в процессе титрования называют кривой титрования.
Кривые титрования помогают выбрать индикатор, оценить погрешность, наглядно проследить за ходом титрования. При построении кривых по осям координат можно откладывать разные величины. Если по оси ординат отложить логарифм концентрации (или отношения концентраций) или величину, пропорциональную этому логарифму, получаются логарифмические кривые титрования. Если же по оси ординат откладывать концентрацию или физико-химический параметр, пропорциональный концентрации, получаются линейные кривые титрования.
На оси абсцисс можно откладывать объем добавленного титранта (раствора реагента) E(R). Однако для проведе
578
Новый справочник химика и технолога
ния математического анализа кривых титрования более удобно на оси абсцисс откладывать отношение этого объема к объему титранта, содержащему стехиометрическое количество реагента E(RcTex.).
= K(R) w(R) _ и(А)
V(Rcrex.) «(R стех.) П(Астех.) ’
где n(R) — добавленное количество реагента; и(А) — прореагировавшее количество титруемого вещества; «(R^) — количество реагента, необходимое для достижения точки стехиометричности; п(Астех) — количество определяемого вещества, содержащееся в титруемом растворе.
Отношение т называют либо степенью оттитрованно-сти, либо титровальной долей. До точки стехиометричности т < 1 (< 100%), в точке стехиометричности т = 1 (= 100%) и после точки стехиометричности т > 1 (> 100%).
В некоторых случаях продукты реакции титрования практически не влияют на равновесные концентрации А или R. Например, при титровании сильной кислоты сильным основанием, при осадительном титровании, при комплексонометрическом титровании продукты (Н2О, малорастворимое соединение или комплекс) практически выводится из реакции. В этих случаях при построении кривых титрования по оси ординат можно отложить логарифм концентрации А (или R). Такие кривые называют моноло-гарифмическими (например, рис. 5.2.1).
Рис. 5.2.1. Монологарифмическая кривая титрования [1]
Если же продукты реакции каким-то образом влияют на А или R, то при расчете А следует учитывать равновесные концентрации продуктов; например, при титровании слабых кислот (или оснований) и окислительно-восстановительном титровании получаются сопряженные вещества: при титровании слабой кислоты — сопряженное основание, при титровании окислителя — его восстановленная форма. Переменный параметр (pH или Е) будет определяться отношением концентраций сопряженной пары. Кривые, по оси ординат которых отложен логарифм отношения концентраций (или пропорциональная ему величина), называют часто билогарифмическими (рис. 5.2.2).
На логарифмических кривых титрования ТЭ соответствует точке перегиба кривой титрования.
Как видно из рис. 5.2.1 и 5.2.2, логарифмические кривые титрования имеют две пологие ветви, между которыми
находится крутой участок, называемый скачком титрования. Величина скачка титрования определяется значением константы равновесия реакции титрования, концентрациями титранта и тируемого вещества, а также выбором точки начала и точки конца скачка титрования.
Рис. 5.2.2. Билогарифмическая кривая титрования [1]
Если полнота протекания реакции обеспечена не менее чем на 99,9%, то выбор этих точек определяется значением допустимой систематической погрешности при данном титриметрическом определении.
Если допускаемая систематическая относительная погрешность измерения объема титранта, пошедшего на тит-AE(R)
рование -100%, равна 0,1%, то считают, что точка
начала скачка титрования соответствует т = 99,9% (недо-титровано 0,1% определяемого вещества). Точка конца скачка титрования в этом случае соответствует т = 100,1%, т.е. добавлено 0,1% избытка реагента от стехиометрического количества (рис. 5.2.1, 5.2.2). Тогда скачок титрования соответствует диапазону т = 99,9 -100,1%.
При более грубых определениях (измерениях объемах титранта) допускают относительную погрешность
-100% = 1%, и тем самым величина скачка титрова-E(R)
ния возрастает. В этом случае скачок титрования соответствует диапазону т = 99 - 101%.
Подробно расчет кривых титрования и их вид в различных титриметрических методах рассматривается практически во всей учебной и монографической литературе (например, [1-15, 19-27, 39-41]).
Чтобы при титровании погрешность не превышала заданного значения, титрование надо прекратить в пределах скачка титрования. Другими словами, КТТ — точка кривой титрования, при которой прекращается титрование, должна находиться в пределах скачка титрования.
Момент окончания титрования (КТТ) можно установить при помощи химической реакции или по изменению некоторого физико-химического свойства раствора. В классических вариантах титриметрии чаше всего используют индикаторы — органические красители.
Индикаторами называют вещества, которые при изменении концентрации определяемого вещества или титранта
Химические методы количественного анализа
579
изменяют свою окраску, степень люминесценции или образуют осадок в ТЭ или вблизи нее. Присутствуя в достаточно малых концентрациях, они не требуют ощутимого количества титранта в процессе титрования [16].
Если проба или титрант сами по себе окрашены, специальный индикатор может не потребоваться (см. ниже). Цветные индикаторы должны сами проявлять свойства, положенные в основу соответствующей титриметрической реакции, т.е. представлять собой в зависимости от конкретного метода кислотно-основную либо окислительно-восстановительную пару, быть хелатообразующим лигандом и т.п.
Различают следующие типы визуальных индикаторов: одноцветные, двухцветные, кислотно-основные, адсорбционные, хемилюминесцентные, экстракционные, флуоресцентные, металлохромные, металлофлуоресцентные, смешанные, окислительно-восстановительные, осадительные [16]. Индикаторы характеризуются интервалом перехода (окраски индикатора) [16; 18]. Речь идет о минимальных пределах концентрации ионов водорода, металла или другого вещества, в которых человеческий глаз способен различать оттенки интенсивности окраски, степень флуоресценции или другого свойства визуального индикатора, обусловленные изменением соотношения (концентраций) участвующих в процессе двух форм этого индикатора. Указанные пределы обычно выражают в виде отрицательного логарифма концентрации (например, pH). Для окислительно-восстановительных индикаторов интервал перехода выражается пределами окислительно-восстановительного потенциала.
Для нахождения КТТ следует выбирать такой индикатор, который дает сигнал (изменение цвета, степени люминесценции, появление осадка и т.д.) в пределах скачка титрования. КТТ и ТЭ обычно несколько не совпадают, что обусловливает систематическую погрешность; в случае применения индикатора ее называют индикаторной погрешностью (ошибкой) [16, 18]. Индикаторная погрешность в зависимости от правильности выбранного индикатора может колебаться в диапазоне от сотых до нескольких процентов. В общем случае интервал перехода окраски индикатора должен находится в пределах скачка титрования и как можно ближе к ТЭ кривой титрования, а в оптимальном варианте — перекрывать ТЭ. Методики и формулы расчета индикаторных погрешностей при кислотноосновном, комплексонометрическом и окислительно-восстановительном титровании широко представлены в учебной [1-13, 19-23, 39] и научной [14, 43-44] литературе. При визуальной регистрации добавление реагента прекращают, достигнув конечной точки титрования. При инструментальной регистрации титрант обычно добавляют и после конечной точки (примерно до двойного стехиометрического количества), определяя затем КТТ графически из кривой титрования. Основные методы регистрации КТТ приведены ниже.*
* Перечень составлен на основе табл. 5 [12] и рекомендаций ИЮПАК [18].
Химические (визуальная регистрация) Физические (инструментальные)
Органические красители (индикаторы): • кислотно-основное титрование, • осадительное титрование (адсорбционные индикаторы — способ Фаянса), • комплексонометрия, • окислительно-восстановительное титрование. Неорганические окрашенные или малорастворимые соединения: • осадительное титрование: • хлоридов по Мору, • ионов серебра, • галогенид ионов по Фольгарду, • окислительно-восстановительное титрование* Электрометрические: • потенциометрическое, • кондуктометрическое, • амперометрическое, • хронопотенциометрич еское, • высокочастотное тит- рование. Оптические: • спектрофотометрическое, • флуорометрическое, • турбидиметрическое, • нефелометрическое титрование. Радиометрическое титрование. Т ермометрическое: • термометрия, • дилатометрия
5.2.2. Классификация титриметрических методов анализа
5.2.2.1. Классификация по способам титрования
Различают три способа титрования: прямое, обратное и титрование заместителя. Второй и третий применяют, когда не выполняется одно из требований, предъявляемых к реакции прямого титрования.
Прямое титрование — наиболее простой и точный способ, когда анализируемый раствор непосредственно титруют стандартным раствором. Здесь могут быть использованы разные типы реакций, если они удовлетворяют следующим требованиям:
1. Взаимодействие титруемого вещества со стандартным раствором должно быть стехиометричным и специфическим. Побочные реакции посторонних веществ, находящихся в анализируемом растворе, с реагентом титранта и с титруемым компонентом должны быть исключены или их влияние должно быть ничтожным.
2. Реакция титрования должна протекать количественно (т > 99,9%). Это означает, что константа равновесия реакции титрования должна быть достаточно большой.
3. Реакция должна протекать быстро, состояние равновесия после добавления очередной порции титранта должно достигаться практически мгновенно, чтобы можно было проследить за процессом титрования.
4. Имеется подходящий индикатор, позволяющий четко фиксировать конец титрования.
Для реакции титрования общего вида (5.2.1) результат вычисляют, раскрывая основные уравнения (5.2.2') или (5.2.6), в зависимости от поставленной задачи пользуются уравнениями (5.2.3)-(5.2.5); (5.2.40).
* Индикатором является избыток титранта.
580
Новый справочник химика и технолога
В табл. 5.2.3 представлены формулы и исходные данные для вычислений результатов титриметрических определений большого количества веществ.
Таблица 5.2.3
Вычисление результатов титриметрических определений [46]
Пояснение. 1 мл титранта (титрующего раствора) оттитровы-вает <3KB(R)R) • Л/(4КВ(А)А) мг определяемого вещества, где c(4KB(R)R) — молярная концентрация эквивалента (нормальная концентрация) титранта. — молярная масса эквива-
лента определяемого вещества и f3KB — фактор эквивалентности приведены в таблице. Если g— навеска анализируемого материала (в мг), V — объем титранта, пошедшего на титрование, то содержание (массовая доля) определяемого вещества ®(А) в % равно:
<»(А) = c(C(R)R) • W,„(A)A) • V 100/g
Если g выражено в граммах, то:
®(А) = c«3KB(R)R)-W3KB(A)A).R.10/g.
А. Кислотно-основные титрования
Применяемые титранты и концентрации их нормальных растворов:
Формула Концентрация 1 н. р-ра (гл
Кислоты
НС1 36,461 H2SO<KB = 1/2) 49,040 HNO3 63,013 Н2С2О4 • 2Н2О (£кв = 1/2) 63,033 Щелочи NaOH 39,9971 КОН 56,1056 Ва(ОН)2-8Н2О (/экв 1/2) 157,73
Определяемое вещество 7экв(А) МГэга(А)А)
А1 1/3 8,99385
В (титрование Н3ВО3 с фенолфталеином в присутствии маннита или глицерина) 1 10,811
Ва(ОН)2 1/2 85,67
Ва(ОН)2-8Н2О 1/2 157,73
ClbCOO 1 59,045
со2 1/2 22,005
со3 1/2 30,005
СаСО3 1/2 50,044
Са(НСО3) 1/2 81,056
СаО 1/2 28,039
Са(ОН)2 1/2 37,046
Н3ВО3 (с фенолфталеином в присутствии маннита или глицерина) 1 61,833
НВг 1 80,912
НСНО2 (муравьиная) 1 46,026
НС2Н3О2 (уксусная) 1 60,052
Н2С4Н4О4 (янтарная) 1/2 59,045
Н2С4Н4Об (винная) 1/2 75,044
Продолжение таблицы 5.2.3 А
Определяемое вещество /экв(А) Шкв(А)А)
Н3С6Н5О7 (лимонная) 1/3 64,042
Н3С6Н5О7Н2О 1/3 70,047
НС7Н5О2 (бензойная) 1 122,12
Н2С2О4 1/2 45,018
Н2С2О4-2Н2О 1/1 63,033
НС1 1 . 36,461
НС1О4 1 100,459
HF 1 20,00634
HI 1 127,9124
НЮ3 1 175,911
HNO3 1 63,013
Н3РО4 (с метиловым оранжевым, или метиловым желтым, или бромкрезоловым синим) 1 97,995
Н3РО4 (с тимолфталеином, или фенолфталеином, или тимоловым синим в присутствии NaCl) 1/2 48,9976
Н3РО4 (с фенолфталеином в присутствии СаС12) 1/3 32,6650
Н3РО4 (титрование фосфоро-молибденового осадка) 1/23 4,2607
H2SO4 1/2 49,040
К2СО3 (с тимоловым синим или фенолфталеином) 1 138,206
К2СО3 (с метиловым желтым, или метиловым оранжевым, или бромфеноловым синим, или бромкрезоловым синим) 1/2 69,103
КНСО3 1 100,115
КНС4Н4О6 (гидротартрат) 1 188,178
КНС8Н4О4 (гидрофталат) 1 204,22
КН(Ю3)2 1 389,912
КОН 1 56,1056
Li2CO3 (с тимоловым синим или фенолфталеином) 1 73,891
Li2CO3 (с метиловым желтым, или метиловым оранжевым, или бромфеноловым синим, или бромкрезоловым синим) 1/2 39,946
MgCO3 1/2 42,157
MgO 1/2 20,152
N (по Кьельдалю) 1 14,0067
6,25N («белок») — 87,542
6,37N («казеин») — 89,223
5,55N («желатин») — 77,737
NH3 1 17,0305
NHt 1 18,0385
ища 1 53,492
(NH4)2SO4 1/2 66,07
Na [титрование щелочью с фенолфталеином осадка NaZ«(UO2)3(C2H3O2) • 6Н2О] 1/10 2,299
Na2B4O7‘ 10Н2О 1/2 190,69
Химические методы количественного анализа
581
Продолжение таблицы 5.2.3 А
Определяемое вещество /экв(А) Шкв(А)А)
Na2CO3 (с тимоловым синим или фенолфталеином) 1 105,989
Na2CO3 (с метиловым желтым, или метиловым оранжевым, или бромфеноловым синим, или бромкрезоловым синим) 1/2 52,995
Na2CO3- 10Н2О 1/2 143,071
NaHCO3 1 84,007
NaOH 1 39,9971
Р (титрование фосформолиб-датного осадка) 1/23 1,3467
РО3" (титрование фосформо-либдатного осадка) 1/23 4,1292
Б. Методы окисления-восстановления (пермангана-тометрия, хроматометрия, иодометрия, броматометрия, це-риметрия и др.)
Применяемые для титранты и концентрации их нормальных растворов:
Формула Концентрация 1 н. р-ра (гл ;
Окислители
KMnO4(f3KB = 1/5) 31,6068
Ce(NH4)4 • (SO4)4 • 2Н2О 632,55
(Лкв= 1)
Ce(SO4)2-4H2O(/.;KB-l) 404,30
12(Гэкв= 1/2) 126,9047
КВгО3(4кв= 1/6) 27,833
K2Cr2O7 (f3m= 1/6) 49,031
К1О3(/экв=1/6) 33,6668
Са(С1О)2(Гэкв=1) 35,746
NH4VO3(f3KB= 1) 116,9782
Восстановители
Na2S2O3 • 5Н2О (/эКВ = 1) 248,19
Fe(NH4)(SO4)2 6Н2О 392,14
(Гэкв=1)
FeSO4-7Н2О (4кв= 1) 278,01
С6Н8О6 (аскорбиновая 88,065
кислота) (fJKB = 1)
Hg2(NO3)2H2O«3KB=l) 280,61
Определяемое вещество /экв(А) Шкв(А)А)
А1 (после осаждения оксихинолином) 1/12 2,24846
As (As3+ — As5+) 1/2 37,4608
Ва (после осаждения в виде ВаСгО4) 1/3 45,776
Bi (после осаждения оксихинолином) 1/12 17,4150
Вг 1 79,904
ВгО, 1/6 21,317
Продолжение таблицы 5.2.3 Б
Определяемое вещество /экв(А) Шкв(А)А)
С6Н5ОН (фенол по Коппешаару) 1/6 15,685
CN~ (иодометрически и по Шулеку) 1/2 13,009
Са (после осаждения в виде СаС2О4) 1/2 20,039
Cd (после осаждения оксихинолином) 1/8 14,051
Се (после осаждения оксихинолином) 1/12 11,677
С1 (активный) 1 35,4527
С12 1/2 35,453
СЮ 1/2 25,726
сю; 1/6 13,909
Со (после осаждения в виде K3Co(NO2)6) 1/11 5,3576
Со (после осаждения оксихинолином) 1/8 7,3667
Сг (Сг2О72‘ -> 2Сг3+) 1/3 17,3320
СЮ2- 1/3 38,6645
СгЮ; 1/6 35,9980
Си (идометрически) 1 63,546
Си (титрование перманганатом осадка CuSCN) 1/6 10,5910
Си (после осаждения оксихинолином) 1/8 7,9433
Fe (Fe3+ Fe2+) 1 55,847
Fe (после осаждения оксихинолином) 1/12 4,6539
Fe (CN)6[Fe(CN)36- >Fe(CN)^] 1 211,953
Fe(NH4)2(SO4)2-6H2O 1 392,14
FeO 1 71,846
Fe2O3 1/2 79,846
FeSO4 1 151,91
FeSO4-7H2O 1 278,01
Ga (после осаждения оксихинолином) 1/12 8,103
HCN (иодометрически по Шулеку) 1/2 13,513
Н2С2О4 (щавелевая) 1/2 45,018
Н2С2О4-2Н2О 1/2 63,033
HI 1 127,9124
hno2 1/2 23,5067
Н2О2 1/2 17,0073
H2S (иодометрически) 1/2 17,04
H2S (броматометрически и перманганатометрически) 1/8 4,260
HSCN (перманганатометрически) 1/6 9,848
HSCN (иодометрически по Руппу и Шидду) 1/8 7,386
H2SO3 1/2 41,040
H2SO4 (через хромат бария) 1/3 32,693
I 1 126,9047
Г (после окисления до иода нитритом) 1 126,9047
582
Новый справочник химика и технолога
Продолжение таблицы 5.2.3 Б
Определяемое вещество /экв(А) Шкв(А)А)
Г (после окисления до Ю3 бромом) 1/6 21,1508
Ю3 1/6 29,1505
In (после осаждения оксихинолином) 1/12 9,5683
КВгО3 1/6 27,833
КС1О3 1/6 20,425
К2СгО4 1/3 64,730
К2Сг2О7 1/6 49,031
K3Fe(CN)6 1 329,25
K4Fe(CN)6 1 368,35
K4Fe(CN)6-3H2O 1 422,039
KH(IO3)2 1/12 32,4926
KIO3 1/6 35,6668
KMnO4 1/5 31,6068
kno2 1/2 42,5519
Mg (после осаждения оксихинолином) 1/8 3,0381
Mn (висмутатным методом) 1/5 10,9876
Мп (методом Фольгарда) 3/10 16,4814
Мп (методом Форд — Вильямса или Гампе) 1/2 27,4690
Мп (после осаждения оксихинолином) 1/8 6,8673
МпО2 в пиролюзите (обработка FeSO4 —КМпО4) 1/2 43,4684
Мо (иодометрически) 1 95,94
Мо (после восстановления цинком) 1/3 31,980
Мо (после осаждения оксихинолином) 1/8 11,993
NH2OH 1/2 16,5149
NO- 1/2 23,0028
Na (растворение осадка NaZn(UO2)3 (С2Н3О2)8-6Н2О, восстановление цинком и титрование) 1/6 3,8316
Na2C2O4 1/2 67,000
NaCl 1/2 37,221
NaNO2 1/2 34,4977
Na2S (S2- > S°) 1/2 39,023
Na2SO3 1/2 63,023
Na2S2O3 1 158,11
Na2S2O3-5H2O 1 248,19
Nb 1/2 46,4532
Nb (эмпир.) — 49,9
Ni (после осаждения оксихинолином) 1/8 7,336
О («активный кислород») 1/2 7,9997
О3 1/2 23,9991
Р (титрование фосфоромолибдат-ного осадка перманганатом после восстановления цинком) 1/36 0,86038
РЬ (после осаждения РЬС2О4) 1/2 103,6
РЬ (после осаждения РЬСгО4) 1/3 69,1
Продолжение таблицы 5.2.3 Б
Определяемое вещество /экв(А) Шкв(А)А)
РЬ (после осаждения оксихинолином) 1/8 25,9
S (S2~ S0) 1/2 16,033
S(S2- >SO2-) 1/8 4,0083
SCN- (перманганатометрически) 1/6 9,6806
SCN- (иодометрически по Руппу и Шидду) 1/8 7,2605
SO2 1/2 32,032
SO2- 1/2 40,032
SO2- (через хромат бария) 1/3 32,021
S2O3-(2S2O3- >S4Og) 1 112,13
S2O2-(S2O2- >2SO2-) 1/8 14,016
Sb (Sb3+-> Sb5+) 1/2 60,875
Sb (после охлаждения оксихинолином) 1/12 10,146
Sn (Sn2+ -> Sn4+) 1/2 59,355
Th (после охлаждения оксихинолином) 1/16 14,5024
Ti 1 47,88
Ti (после охлаждения оксихинолином) 1/8 5,985
U(U4+ >UO22+) 1/2 119,014
U (после охлаждения оксихинолином) 1/12 19,8357
V(VO2+ >VO3) 1 50,9415
V (после охлаждения оксихинолином) 1/8 6,3677
Zn (после охлаждения оксихинолином) 1/8 8,1738
Zr (после охлаждения оксихинолином) 1/16 5,7015
В. Методы осаждения и комплексообразования
Применяемые титранты и концентрации их нормальных растворов:
Формула
AgN63 Hg(NO3)2H2O
KSCN NaCl
NH4SCN Hg(NO3)2 • 2H2O K2CrO4(f3KB = 1/2)
Концентрация 1 н. раствора (гл 1), (ЛкВ=1) 169,8713 171,31 97,18 58,443 76,122 280,61
97,095
Определяемое вещество Лкв(А) Шкв(А)А)
Ag 1 107,8682
AgNO3 1 169,8731
Ва (прямым титрованием) 1/2 68,665
Вг- 1 79,904
Химические методы количественного анализа
583
Продолжение таблицы 5.2.3 В
Определяемое вещество /экв(А) Шкв(А)А)
CN” (по Мору, Фольгарду, Фаянсу) 1 26,018
CN (по Либиху, Дениже) 2 52,035
С1” 1 35,453
F~ (после осаждения в виде PbCIF) 1 18,9984
НВг 1 80,912
HCN (по Мору, Фольгарду, Фаянсу) 1 27,026
HCN (по Либиху, Дениже) 2 54,052
НС1 1 36,461
HI 1 127,9124
HSCN (по Фольгарту) 1 59,092
Hg (с роданидом) 1/2 100,295
I 1 126,9045
КВг 1 119,002
KCN (по Мору, Фольгарду, Фаянсу) 1 65,116
KCN (по Либиху, Дениже) 2 130,232
КС1 1 74,551
KI 1 166,0028
NH4CI 1 53,491
NaBr 1 102,894
NaCl 1 58,443
Nal 1 149,8943
SCN (по Фольгарту) 1 58,084
Г. Методы титрирования ЭДТА (этилендиаминтетрацетатом натрия, комплексоном III, трилоном Б)
Определяемое вещество /экв(А) Жкв(А)А)
Ag [после добавления K2Ni(CN)4l 2 215,7364
Al 1 26,9815
As (в виде MgNH4AsO4) 1 74,9216
AsO’ (в виде MgNH4AsO4) 1 138,9192
Ва 1 137,33
Bi 1 208,9804
Вг (через AgBr) 2 159,808
CN~ (после добавления соли Ni2+) 4 104,071
Са 1 40,078
Cd 1 112,41
Се 1 140,12
Cl (через AgCl) 2 70,906
Со 1 58,9332
Сг 1 51,9961
Си 1 63,546
F (после добавления соли Са2+) 2 37,9968
F (через PbCIF) 1 18,9984
Fe 1 55,847
Ga 1 69,723
Hg 1 200,59
Продолжение таблицы 5.2.3 Г
Определяемое вещество /экв(А) Шкв(А)А)
I (через Agl) 2 253,8090
In 1 114,82
Ir 1 192,22
К (в виде NaK2Co(NO2)6) 2 78,1966
La 1 138,9055
Mg 1, 24,305
Mn 1 54,9380
Mo (в виде CaMoO4) 1 95,94
Na (в виде NaZn(UO2)3(C2H3O2)9-6H2O) 1 22,9898
Ni 1 58,69
Р (в виде MgNH4PO4) 1 30,9738
РО4’ (в виде MgNH4PO4) 1 94,9714
Pb 1 207,2
Pd (после добавления K2Ni(CN)4) 1 106,42
Pt (после добавления K2Ni(CN)4) 1 195,08
S (в виде BaSO4) 1 32,066
SCN (через AgSCN) 2 116,168
SO2- (в виде BaSO4) 1 96,064
Sn2+ 1 118,710
Sr 1 87,62
Th 1 232,0381
Ti 1 47,88
Tl 1 204,383
U 2 476,0578
V(IV) 1 50,9415
W (в виде CaWO4) 1 183,85
Zn 1 65,39
Zr 1 91,224
Обратное титрирование — титрование избытка стандартного раствора, добавленного к анализируемому раствору. Его называют также титрованием остатка или титрованием по остатку.
Обратное титрование обычно применяют в случае малой скорости прямой реакции, когда отсутствует подходящий индикатор или если определяемое вещество летучее.
При обратном титровании к анализируемому веществу добавляют точно известный избыточный объем первого стандартного раствора R (т.е. известное количество первого реагента). Если реакция протекает медленно, ее ускоряют нагреванием. По завершении реакции А + RH36 > (продукты реакции) остаток первого стандартного раствора титрируют вторым стандартным раствором R2: Roct. + R2 —> продукты реакции.
Пример
Определить содержание карбоната кальция в образце кислотно-основным титрованием.
СаСО3 малорастворим в воде, а гетерогенная реакция его с разбавленным раствором кислоты протекает очень медленно. Прямое титрование невозможно. Поэтому к ана-
584
Новый справочник химика и технолога
лизируемому веществу добавляют точный объем стандартного раствора хловодородной кислоты в заведомом избытке (точно известное количество НС1). Раствор нагревают и ждут завершения реакции
СаСОз + 2НС1 изб. = СаС12 + 2Н2О + СО2,
в ходе которой расходуеся часть кислоты «'(НО) = = 2 и(СаСОз). Оставшуюся часть НС1 (остаток) «"(НС1) титруют стандартным раствором, например, NaOH:
индикатор
NaOH + HCl = NaCl + H2O,
«''(НС1) = «(NaOH).
Количество NaOH известно по результатам титрования «(NaOH) = c(NaOH) • K(NaOH).
Исходя из стехиометрии первой реакции и стехиометрии второй реакции, в точке эквивалентности можно записать:
«(НС1) = «'(НС1) + н"(НС1) = 2«СаСО3 + «(NaOH),
где «(НС1) — исходное количество НС1.
Решим уравнение относительно определяемой величины
«(СаСО3) = | [«(HCl)-«(NaOH)].
В эквивалентной форме проще. Фактор эквивалентности для СаСОз /экв = (СаСОз принимает 2 протона, z* = 2). Следовательно
«(1/2 СаСОз) = «(НС1) - «(NaOH).
Если требуется вычислить массу СаСО3 в образце, а молярные концентрации стандартных растворов с(НС1) и c(NaOH) и их объемы Е(НС1)и E(NaOH) известны по данным анализа, то, пользуясь формулами (5.2.12), (5.2.13) и (5.2.16), получают расчетные выражения в обычной и эквивалентной формах
^а-С°з) = — [с(НС1) • Е(НС1) - c(NaOH) • E(NaOH)]
М(СаСО3) 2
или
—= с(НС1). Г(НС1)_c(NaOH) • К(NaOH) .
М(1/2 СаСО3)
Значения молярных масс Л/(СаСО3) и М(1/2 СаСОз) находят в справочных таблицах (например, в табл. 5.2.3).
Титрование заместителя. Способ имеет и другие названия: титрование по заместителю, косвенное титрование.
Титрование заместителя применяют, например, если невозможно определить КТТ при прямом титровании, при работе с неустойчивыми веществами или когда прямая
реакция нестехиометрична в связи с протеканием побочных реакций. В этом случае к анализируемому раствору добавляют избыток вспомогательного реагента D, с которым определяемое вещество образует стехиометричное количество нового соединения — заместителя С. Полученный заместитель должен легко определяться прямым титрованием: l)A + D->C + E;2)C + R-> продукт реакции.
Пример
Прямым перманганатометрическим титрованием невозможно определить ион кальция, который не реагирует с КМпО4. Поэтому ион кальция осаждают сначала в виде оксалата кальция, применяя избыток осадителя:
Са + С2О4 (изб} = СаС2О4 Ф .
При этом «(Са) = | «(СаС2О4). (5.2.46)
Осадок отмывают от избытка осадителя и растворяют в избытке серной кислоты:
СаС2О4 + H2SO4 (ИЗб.)= CaSO4 + Н2С2О4,
«(СаС2О4) = j «(Н2С2О4). (5.2.47)
Выделившуюся щавелевую кислоту титруют стандартным раствором КМпО4 в кислой среде
5Н2С2О4 + 2МпО;+ 61-Г б = ЮСО2+ 2Мп2++ 8Н2О.
В точке эквивалентности, согласно стехиометрическому отношению (уравнение 5.2.2'), имеем:
^£^4 = I =* и(НЛ°.) = |«(КМпО4). (5.2.48) «(КМпО4) 2 2
Подставляем значение «(Н2С2О4) из уравнения (5.2.48) в выражение (5.2.47), далее полученное значение
«(СаС2О4) = «(КМпО4) подставляем в формулу
(5.2.46) и выводим уравнение для расчета результата анализа (количество КМпО4 известно по данным титрования):
«(Са) = i «(KMnO4). (5.2.49)
Для получения эквивалентной формы расчетного выражения определим факторы эквивалентности веществ. В реакции титрования КМпО4 принимает 5 электронов (z = 5, /«в = 1/5); Н2С2О4 отдает 2 электрона (z = 2, /экз = 1/2). В реакциях растворения и образования осадка СаС2О2 на один ион Са2+ приходится один ион С2О4- (/экв = 1/2). Таким образом
«(|са) = «(1/5КМпО4). (5.2.50)
* z — см. раздел 2.
Уравнения (5.2.49) и (5.2.50) учитывают стехиометрию всех реакций замещения.
Химические методы количественного анализа
585
Для вычисления результатов анализа раскрывают уравнения (5.2.49) или (5.2.50) в зависимости от поставленной задачи и исходного параметра стандартного раствора КМпО4.
Если требуется вычислить массу элемента Са при известной молярной концентрации КМпО4, то, используя формулы (5.2.12) и (5.2.16) для обычной формы (5.2.49), получим выражение:
^^ = |С(КМпО4)-К(КМпО4). (5.2.49’)
Для заданной молярной концентрации эквивалента применяем уравнения (5.2.13) и (5.2.18) эквивалентной формы (5.2.50):
МЛ/2Са) =С(1/5КМП°4) Г(КМПО,)’ <5'2'50,)
где К(КМпО4) — объем стандартного раствора КМпО4, пошедший на титрование выделевшейся щавелевой кислоты.
Формулы для расчета результатов прямого, обратного титрования и титрования заместителя приведены в табл. 5.2.4.
Таблица 5.2.4
Формулы для расчета результатов титриметрических определений [8]
Способ расчета Способ титрования
Прямое Обратное Титрование заместителя
По молярной концентрации эквивалента стандартного раствора (нормальной концентрации) ,А. qK7(R) w(A) = —— х 1000 ,,М(/„(А)) Гобд 1000 ка [c,K,lz(R,)-e2K2lz(R2)] т(А) =1 х 1000 ..^(/,„(А)А) 1000 и C]K7(R) m(A) = ——2 x 1000 ,,M(X.(A)A) Кш 1000 Ka
По титру стандартного раствора ^А)=етмх М(/эга(В)В) V х Общ. К Г 7(R)K(R) _ жте r(R2)K(R2) 1 W,kb(R2)R2)J к T(R)K(RWM(A)A)x W,kB(R)R) у X^to К
По титру, выраженному по определяемому веществу w(A) = T(R/A)x xK(R)^H-К m(A) = [r(R/A)R(R)- -T(R2/A)K(R2)]x^b a щ(А) = T(R/A)K(R)x V x^^ к
Примечание.
т(А) — масса определяемого ввещества, г, С\ c(/?JKB(R)R), с2 — c(Zjkb(R2)R2) — стандартные молярные концентрации эквивалента (нормальные концентрации) реагентов (титрантов) R и R2 (см. обратное тирование); T(R), 7(R2), E(R), F(R2) — титры и объемы стандартных растворов R и R2; ДК/А), ДR2/A) — титры
стандартных растворов R и R2 по определяемому веществу А, (гем1); Кобщ. — вместимость мерной колбы, см3; Ка — объем анализируемого раствора, взятый для титрования (объем аликвоты, см3); Ki и К2 — поправочные коэффициенты к нормальным концентрациям сх и с2 (см. 5.2.4.6).
5.2.2.2. Классификация титриметрических методов по типам реакций титрования
Титриметрические методы классифицируются по реакциям титрования. Они могут быть реакциями перехода протонов, обмена электронами, образования малодиссо-циированных (комплексных) частиц или образования малорастворимых электролитов. Соответствующие группы
титриметрических методов называют кислотно-основным титрованием (протолитометрией), окислительно-восстановительным титрованием (редоксметрией), комплексометрическим титрованием (комплексометрией) и осадительным титрованием (седиметрией). Отдельные титриметрические методы называются по реагентам, применяемым в этих методах (табл. 5.2.5).
586
Новый справочник химика и технолога
Таблица 5.2.5
Классификация титриметрических методов по типам реакций титрования [5]
Метод титрования, тип реакции Подгруппы методов Отдельные методы Вещества, применяемые для приготовления титрантов Вещества, определяемые прямым титрованием
Кислотно-основное титрование, протоли-тометрия Н3О+ + 011 = 2Н2О Ацидиметрия НС1, H2SO4 Основания
Алкалиметрия NaOH Кислоты
Окислительно-восстановительное титрование, редоксметрия oOxi + Z?Red2 = aRedi = = bOxi Оксидиметрия Перманганатометрия KMnO4 Восстановители
Иодометрия 12 То же
Дихроматометрия К2Сг2О7 То же
Броматометрия КВгОз То же
Иодатометрия КЮ3 То же
Цериметрия Ce(SO4)2 То же
Ванадатометрия NH4VO3 То же
Редуциметрия Титанометрия TiCl3 Окислители
Хромометрия СгС12 То же
Аскорбинометрия НО—С=С—ОН о— С С— СН— СН2ОН 0 ОН То же
Комплексометрическое титрование, комплексометрия М + L = ML Меркуриметрия Hg(NO3)2 Г алогениды и псевдогалогениды
Цианидометрия KCN Ионы нике-ля(П), кобаль-та(П), алюминия, цирко-ния(1У) и тория(ГУ)
Хелатометрия Комплексонометрия Комплексон III* Ионы металлов
Осадительное титрование, седиметрия М + X = МХФ(тв.) Аргентометрия AgNO3 Галогениды и псевдогалогениды
Меркурометрия Hg2(NO3)2 Хлориды
Примечание. * Комплексон III — двунатриевая соль этилендиаминтетрауксусной кислоты (Na2H2Y • 2Н2О), где Y — символ аниона этилендиаминтетрауксусной кислоты.
5.2.3. Посуда мерная лабораторная: бюретки, пипетки, колбы, цилиндры, мензурки [6,12, 41-53]
В титриметрических методах анализа воспроизводимость и правильность конечного результата в очень большой степени определяются точностью приготовления стандартных растворов и точностью измерения объемов титранта и титруемого вещества. Для точного измерения объемов употребляются бюретки, пипетки и мерные колбы двух классов точности измерений* различной вместимости и модификаций, изготавливаемые промышленностью в соответствии с требованиями ГОСТов и калиброванные
’1-й класс — для более точных изделий, 2-й — для менее точных.
при стандартной температуре 20 °C. Номинальная вместимость мерной посуды не всегда соответствует ее истинной вместимости. Это отражается на точности титриметрических определений — калибровку посуды при точных определениях необходимо проверить.
За единицу объема в метрической системе мер принимают «истинный литр», т.е. объем, занимаемый массой воды в 1 кг при температуре ее наибольшей плотности (т. е. при 3,98 °C), взвешенной в безвоздушном пространстве. В титриметрическом анализе за единицу принят 1 мл, равный 0,001 л.
В качестве стандартной температуры при калибровании мерной посуды в настоящее время принята температура 20 °C.
Химические методы количественного анализа
587
«Нормальный литр» равен объему такого количества вещества, которое занимает при 20 °C объем истинного литра.
Мерную посуду калибруют или проверяют, определяя массу чистой воды, содержащейся в ней, или массу воды, вылитой из нее при определенной температуре; по массе воды рассчитывают вместимость посуды. Такие расчеты производят по специальным таблицам (табл. 5.2.6).
Таблица 5.2.6
Приведение объема воды к объему, занимаемому ею при 20 °C [47]
/, °C Давление
1,01- 105Па (760 мм рт. ст.) 0,99- 105Па (740 мм рт. ст.) 0,96 105 Па (720 мм рт. ст.) 0,93- 105Па (700 мм рт. ст.)
Масса воды в г, соответствующая объему 1000 мл
9 998,43 998,46 998,49 998,52
10 998,39 998,42 998,45 998,48
11 998,31 998,34 998,37 998,40
12 998,23 998,26 998,29 998,32
13 998,14 998,17 998,20 998,23
14 998,04 998,07 998,10 998,13
15 997,93 997,96 997,99 998,02
16 997,80 997,83 997,86 997,88
17 997,65 997,68 997,71 997,73
18 997,51 997,54 997,57 997,59
19 997,34 997,37 997,40 997,42
20 997,18 997,21 997,24 997,26
21 997,00 997,03 997,06 997,08
22 996,80 996,83 996,86 996,88
23 996,61 996,64 996,67 996,69
24 996,39 996,42 996,45 996,47
25 996,18 996,21 996,23 996,26
26 995,94 995,97 995,99 996,02
27 995,70 995,73 995,75 995,78
28 995,45 995,48 995,50 995,53
29 995,19 995,22 995,24 995,27
30 994,92 994,95 994,97 995,00
В табл. 5.2.6 для температур от 9 до 30 °C дается масса воды в граммах, которую нужно отвесить, пользуясь латунными разновесами при температуре опыта и соответствующем барометрическом давлении с тем, чтобы занимаемый ею (в стеклянной посуде) объем был равен при 20 °C точно 1000 мл (при калибровании сосудов меньшей емкости берут соответствующую часть массы воды).
Если принятая за нормальную температура ниже 20 °C, надо к массе, указанной в табл. 5.2.6, прибавить 0,025 г на каждый градус, а если принятая температура выше 20 °C, то массу соответственно уменьшить на 0,025 г. Например, при приведении массы 1 л воды к 15 °C надо все значения масс в табл. 5.2.6 увеличить на 0,125 г. При калибровке температура окружающего воздуха принята равной температуре взвешиваемой воды.
Если давление при проведении опыта не совпадает ни с одним из приведенных в табл. 5.2.6, принимают данные,
относящиеся к ближайшему значению давления. Точное значение может быть найдено интерполированием, однако практически в этом нет необходимости, так как возможная при этом ошибка настолько мала, что ею можно пренебречь.
При калибровании применяют обычную дистиллированную воду. Посуду и воду, предназначенную для ее заполнения, предварительно выдерживают не менее 1 ч в весовой комнате, чтобы они приняли температуру окружающего воздуха. Температуру водй измеряют термометром с погрешностью не более 0,5 °C.
Проверка, калибровка мерной посуды и правила работы с ней подробно описаны в [47]; некоторые моменты затронуты в [46, 48,49].
Бюретки применяются для измерения точных объемов при титровании и при других операциях. Все они предназначаются для измерения вылитой из них жидкости, поэтому калиброваны на выливание. Бюретки представляют собой цилиндрические трубки с суженным концом, к которому приваривается стеклянный кран, либо при помощи резиновой трубки к ним присоединяют оттянутую стеклянную трубочку.
Существуют макро- и микробюретки. Употребляемые в макроанализе бюретки на 50 мл отградуированы на миллилитры и доли миллилитра с ценой наименьшего деления 0,1 мл, а бюретки на 25 мл отградуированы либо аналогично, либо с ценой деления 0,05 мл. Отсчет сотых долей миллилитра производится на глаз. Микробюретки имеют вместимость 1, 2, 5, 10 мл с ценой наименьшего деления 0,01-0,02 мл. Конструкции и рабочие характеристики выпускаемых бюреток представлены в [48, 50] и в табл. 5.2.7.
Таблица 5.2.7
Характеристика бюреток при 20 °C [50]
Номинальная вместимость, мл Цена наименьшего деления, мл Пределы погрешности, мл
1-го класса 2-го класса
1 0,01 ±0,1 ±0,02
2 0,01 ±0,1 ±0,02
5 0,02 ±0,1 ±0,02
10 0,02 ±0,02 ±0,05
0,05 ±0,02 ±0,05
25 0,05 ±0,03 ±0,05
0,1* ±0,05 ±0,1
50 0,1’ ±0,05 ±0,1
100 0,2 ±0,1 ±0,02
Примечание. * Точность отсчетов при работе с такими бюретками составляет 0,02-0,03 мл.
Пипетки. Пипетки служат для отмеривания и переноса точного объема раствора из одного сосуда в другой, бывают двух видов: цилиндрические и с расширением (пипетки Мора с одной меткой) вместимостью от 1 до 100 мл. Существуют микропипетки вместимостью 0,1-0,2 мл.
Цилиндрические пипетки представляют собой узкий стеклянный цилиндр с делениями, соответствующими де
588
Новый справочник химика и технолога
сятым или сотым долям миллиметра. Они менее точны, чем пипетки с расширением; их конструкции и характеристики приведены в [47,48, 50].
Пипетка с расширением — узкая стеклянная трубка с расширением в средней части. На верхней части нанесена круговая черта, до которой полагается заполнить пипетку раствором, чтобы получить объем, соответствующий ее емкости. На стенке пипетки дается маркировка объема, температуры калибрования, класса точности. Пипетки калибруют на выливание. Объем свободно вытекающей жидкости, которой предварительно заполнена пипетка, является номинальным объемом. Согласно ГОСТ 29169-91 (ИСО 648-77), пределы допускаемой погрешности номинальной вместимости не должны превышать значений, указанных в табл. 5.2.8.
Внимание! Засасывания жидкости ртом следует всячески избегать, чтобы исключить опасность контакта с токсичными (кислотами, щелочами и т.д.) или биологическими активными веществами. Для засасывания жидкости следует применять различные вспомогательные устройства, например, резиновые груши или механические микронасосы с ручным управлением.
Таблица 5.2.8
Пределы допускаемой погрешности номинальной вместимости пипеток с одной отметкой [52]
Номинальная вместимость, мл Предел допускаемой погрешности, мл
1-й класс 2-й класс
0,5 ±0,005 ±0,01
1 ±0,008 ±0,015
2 ±0,01 ±0,02
5 ±0,015 ±0,03
10 ±0,02 ±0,04
10,77* ±0,02 ±0,04
20 ±0,03 ±0,06
25 ±0,03 ±0,06
50 ±0,05 ±0,1
100 ±0,08 ±0,15
200 ±0,1 ±0,2
Примечание. * Для пипеток, используемых в быту.
Мерные колбы, мерные цилиндры, мензурки. Мерные колбы применяют для приготовления растворов строго определенной концентрации, точного отмеривания объемов растворов, разбавления растворов и т.п. Этот плоскодонный стеклянный сосуд с длинным узким горлом (шейкой), на которой в определенном месте нанесена кольцевая метка, калиброван при 20 °C на вливание. Если при 20 °C налить воду так, чтобы нижний мениск ее находился на уровне метки, то объем находящейся в колбе воды будет равен номинальной вместимости колбы. На стенке колбы указываются номинальный объем колбы, температура калибрования, класс точности. При любой другой температуре объем воды, налитый до метки, будет больше или
меньше обозначенного. Мерные колбы изготавливают вместимостью 5-2000 мл.
Мерные цилиндры и мензурки применяют при отмеривании примерных объемов растворов реактивов (с погрешностью, не превышающей 1%), объемы которых не учитываются при вычислении результатов.
Эти сосуды имеют сравнительно толстые стенки, тем не менее в них нельзя смешивать реагенты, взаимодействующие с выделением большого количества тепла, т.к. сосуд может лопнуть.
Мерные колбы, мерные цилиндры, мензурки изготавливают в соответствии с ГОСТ 1770-74 (ИСО 1042-83, ИСО 4788-80). Их допустимые погрешности от номинальной вместимости при температуре 20 °C не должны превышать указанных в табл. 5.2.9.
Таблица 5.2.9
Пределы допускаемой погрешности номинальной вместимости колб, цилиндров и мензурок [53]
Номина-льная вместимость, мл Допустимая погрешность, мл
Цилиндры Мензурки Колбы
1-го класса 2-го класса 1-го класса 2-го класса
5 0,10 0,10 — 0,025 0,05
10 0,10 0,20 — 0,025 0,05
25 0,25 0,50 — 0,04 0,08
50 0,25 1,00 2,50 0,06 0,12
100 0,50 1,00 5,00 0,10 0,20
200 — — — 0,15 0,30
250 1,25 2,00 5,00 0,15 0,30
300 — — — 0,20 0,40
500 2,50 5,00 12,50 0,25 0,50
1000 5,00 10,00 25,00 0,40 0,80
2000 10,00 20,00 — 0,60 1,20
Некоторые общие правила работы с мерной посудой.
1. Мерная посуда должна быть чистой, что характеризуется равномерным смачиванием стенок растворителем и раствором.
2. Во избежание ошибок натекания: а) недопустимо быстрое сливание жидкости из бюретки, т.к. она не успевает стекать со стенок и, как следствие, объем измеряется неточно. Время ожидания после окончания титрования до отсчета показаний должно быть не менее 15-20 секунд; б) недопустимо выдувание растворов из пипеток по указанной выше причине. Время ожидания после слива в титровальную колбу или другой сосуд до вынимания пипетки зависит от ее типа и вместимости, но должно быть не менее 3 секунд. Каплю жидкости, оставшуюся в кончике пипетки, ни в коем случае не выдувают.
3. Ввиду того, что калибровка посуды осуществляется при стандартной температуре 20 °C, измерение объемов, по мере возможности, необходимо проводить при комнатной температуре. Для обычных аналитических работ разница в несколько градусов принципиального значения не имеет.
Химические методы количественного анализа
589
5.2.4. Способы приготовления стандартных растворов [2,3, 6, 47, 54, 55]
Стандартным (рабочим, титрованным) называют растворы с точно известной концентрацией. Их готовят либо растворением точно известного количества первичного стандарта в мерной колбе известной вместимости, получая первичный стандартный раствор, либо растворяют приблизительно известное количество вещества и концентрацию полученного раствора определяют титрованием этим раствором точно отмеренного количества другого реагента. Полученный раствор называют вторичным стандартным раствором, а саму процедуру нахождения точного значения концентрации — стандартизацией раствора.
По техническим условиям погрешность в определении концентрации стандартного раствора обычно не должна превышать 0,1% (отн.). В связи с этим концентрацию стандартных растворов принято указывать с точностью четырех значащих цифр. Например, с(НС1) = 0,2020 моль-л1, Т(КМпО4) = 0,009312 г-мл’1.
Правильность результатов дальнейших титриметрических определений (анализов) существенно зависит от первичного стандарта, применяемого для приготовления первичных и вторичных стандартных растворов.
5.2.4.1. Приготовление первичных стандартных растворов
Первичным стандартным раствором называют раствор, приготовленный по точной навеске специального вещества — первичного стандарта. Стандартами называют вещества, состав которых точно соответствует химической формуле. В качестве первичных стандартов может быть использовано ограниченное число веществ, которые легко очищаются от примесей (содержат примеси не более 0,05% по массе), устойчивы на воздухе и в растворе по отношению к кислороду и углекислому газу, не гигроскопичны и не гидролизуются. Для этих целей обычно используют препараты марки х.ч. («химически чистый») или ч.д.а. («чистый для анализа») после высушивания или прокаливания.
Стандартные вещества, которые наиболее широко применяют в кислотно-основном титровании и в редокс-титровании, представлены соответственно в табл. 5.2.10, 5.2.11.
В комплексонометрии для стандартизации раствора ЭДТА (комплексона III) в качестве первичных стандартов применяют металлический цинк, ZnSO4, MgSO4, а в аргентометрии для стандартизации растворов AgNO3 применяют NaCl.
Таблица 5.2.10
Стандартные вещества в кислотно-основном титровании [1]
Вещество Реакции Индикатор Эквивалент Молярная масса эквивалента, г • моль 1 Примечания. Условия подготовки
Карбонат натрия Na2CO3 Na2CO3 + НС1 = NaHCO3 + NaCl Na2CO3 + 2HC1 = H2CO3 + 2NaCl Фенолфталеин Метиловый оранжевый Na2CO3 l/2Na2CO3 106,00 53,00 Прокаливание при 260 °C
Тетраборат натрия(бура) Na2B4O7 • ЮН2О Na2B4O7 + 7H2O = 2NaB(OH)4 + + 2B(OH)3 NaB(OH)4 + HC1 - B(OH)3 + + NaCl + H2O То же l/2Na2B4O7 • • ЮН2О 190,7 Высушивание на воздухе перелопачиванием
Оксалат натрия Na2C2O4 Na2C2O4 = Na2CO3 + CO (титруют образовавшийся Na2CO3) То же Na2C2O4 l/2Na2C2O4 137,00 67,00 Прокаливание до Na2CO3
Иодат калия КЮ3 KIO3 + 5KI + 6HC1 = 3I2 + + 3H2O + 6KC1 Крахмал 1/6KIO3 35,67 Редокс-титрование
Бензойная кислота С6Н5СООН C6H5COOH + NaOH = = C6H5COOHNa + H2O Фенолфталеин C6H5COOH 122,12 Спиртовый раствор
Бифталат калия КНС8Н4О4 KHC8H4O4 + NaOH = = KNaC8H4O4 + H2O То же KHC8H4O4 204,23 —
Щавелевая кислота Н2С2О4- 2Н2О H2C2O4 + 2NaOH = = Na2C2O4+2H2O То же 1/2H2C2O4- • 2H2O 63,03 Высушивание на воздухе
590
Новый справочник химика и технолога
Таблица 5.2.11
Методы окислительно-восстановительного титрования и стандартные вещества [1]*
Метод (титрант) Первичное стандартное вещество Реакция Эквивалент Молярная масса эквивалента, г • моль1** Индикатор
Перманга-натометрия (КМпО4) КМпО4 MnO4 + 5e + 8H+ = = Mn2+ + 4H2O l/5KMnO4 31,6068 КМпО4
Na2C?O4 C2O2’ - 2e = 2CO2 l/2Na2 C2O4 67,0000
K4Fe(CN)6 Fe(CbC -e = = Fe(CN)3’ K4Fe(CN)6 368,35
As2O3 H3ASO3 - 2e + H2O = = H2AsO; + 3H+ 1/4As2O3 49,46
(NH)4Fe(SO4)2 • 6H2O (соль Мора) Fe2+-e = Fe3+ (NH4)2Fe(SO4)2- •6H2O 392,14
Fe(Fe + 2НС1 = FeCl2 + H2) Fe2+-e = Fe3+ Fe 55,847
Иодомет-рия (№28203 • • 5Н2О) Na2S2O3 • 5H2O 2S2O32-2e= S4O; Na2S2O3 5H2O 248,19 Крахмал
Ь I2 + 2e = 2Г 1/2I2 126,905
К2СГ2О7 Cr2O2'+6e+ 14H+ = = 2Cr3+ + 7H2O 1/6 K2Cr2O7 49,031
KIO3 IO3 + 5Г + 6H+= 3I2 + + 3H2O (3I2= IO;) 1/6 KIO3 35,667
KB1O3 BrO3 + 6e +6H+ = Br“ + + ЗН2О 1/6КВЮ3 27,833
K3Fe(CN)6 Fe(CN)3’ + e -= Fe(CNt K3Fe(CN)6 329,25
Дихрома-тометрия (К2Сг2О7) К2СГ2О7 Cr2O2” + 6e + 14H+ = = 2Cr3+ + 7H2O l/6K2Cr2O7 49,031 Дифениламин и другие ре-докс-индика-торы
Бромато-метрия (КВгОз) KB1O3 BrO3 +6e + 6H+ = = Bf- + 3H2O 1/6КВЮ3 27,833 Метиловый оранжевый, нейтральный красный, хинолиновый желтый и др.
Цериметрия Ce(IV) + e = Ce(III) Ce(IV) Комплекс Fe(II) с 1,10-фенантроли-ном (ферроин) и производными 1,10-фенантролина
Na2C2O4 AS2O3 C2O4“ -2e = 2CO2 H3 АзОз -2e + H2O = = h2AsO; + зн+ l/2Na2C2O4 1/4As2O3 67,0000 49,46
Примечания.
* В таблице приведены первичные и вторичные стандарты. Вторичный стандарт, как правило, дает название методу титрования.
** Данные взяты из [46].
Химические методы количественного анализа
591
При растворении навески (взятой с точностью ±0,0002 г) стандартного препарата массой w(R) в мерной колбе вместимостью Кк можно вычислить любую из концентраций — титр T(R), молярную c(R) или нормальную концентрации и титр стандартного раствора R по определяемому веществу A T(R/A) по формулам (5.2.39-5.2.43):
T(R) = ^1 (г-мл-1), "к
w(R)103 c(R) = —------- (моль • л ),
с(/экв (R)R) = —---------- (моль л’1),
7XR/A) = с(/экв (R)R) • M(f3KB (А) А) 1(Г3 (г мл’1).
Последний особенно удобен в производственных условиях, т.к. он позволяет по затраченному объему титранта (R) сразу рассчитать массу определяемого компонента (вещества) исходя из (5.2.33).
Для облегчения работы промышленность выпускает стандарт-титры (фиксаналы), представляющие собой запаянную ампулу, содержащую точно отмеренное количество твердого реагента или его раствора. Чаще всего это количество составляет 0,1 моль эквивалентов вещества (£,KB(R)R). Растворение содержимого такой ампулы в мерной колбе вместимостью 1 л позволяет быстро приготовить 0,1 н. раствор вещества. Это наиболее часто используемая концентрация стандартного раствора.
Варьируя вместимостью мерной колбы и подбирая соответствующий номинал стандарт-титра, можно приготовить стандартный раствор с другим значением концентрации вещества.
Однако, чтобы застраховаться от случайных ошибок, следует стандартизировать даже приготовленные растворы и в любом случае периодически повторять стандартизацию, если только неизвестно, что раствор совершенно устойчив.
Выпускают стандарт-титры НС1, H2SO4, NaOH, солей кальция, цинка, магния, КМпО4 и других веществ.
5.2.4.2. Приготовление вторичных стандартных растворов (вторичный стандарт)
Вторичным стандартным раствором (вторичным стандартом) называют раствор, характеристики которого установлены по подходящему первичному стандарту.
Сертификаты многих реактивов имеют малодостоверные сведения о содержании основного (главного) компонента. Например, для «дымящей» хлороводородной кислоты квалификации х.ч. указывается массовая доля НС1 35-37%. Такие неточные исходные данные приводят к значи
тельной неопределенности величины концентрации приготовленного раствора до 5-7% вместо допустимой 0,1%. Препараты NaOH и КОН сильно поглощают влагу и углекислый газ и имеют достаточно неопределенный состав. Низкая устойчивость растворов препаратов Na2S2O3-5H2O, КМпО4,12 и других требует их стабилизации после растворения, в результате чего возникает значительная неопределенность в величине концентрации приготовленного раствора.
Из таких препаратов сначала готовят раствор приблизительно нужной концентрации, и при необходимости раствор стабилизируют. На этом этапе приготовления растворов допустимо использование грубой мерной посуды (мерных цилиндров или стаканов) и взвешивание на технических весах.
Стандартизацию стабилизированного раствора проводят различными способами:
1) титрованием точных навесок первичных стандартных препаратов (установочных веществ) — см. разделы 5.2.4.1 и 5.2.4.3;
2) титрованием известных объемов другого стандартного раствора (стандартизация по вторичному стандартному раствору) — см. раздел 5.2.4.4;
3) титрованием точных навесок стандартных образцов (СО) контролируемых объектов, с соблюдением общих правил выполнения титриметрических определений — см. разделы 5.2.4.3; 5.2.4.5.
При работе с тестированными бюретками при обычных определениях считываемые по шкале бюретки значения объемов, пошедшие на титрование, являются действительными, которые и подставляются в формулы раздела 5.2.4.3.
При точных аналитических работах калибровка и тестированной мерной посуды должна быть проверена [3, 46, 47]. При несовпадении действительной и номинальной вместимости бюретки строится график поправок к измеряемому объему. По этому графику находят действительный объем, который и подставляют в нижеприведенные расчетные формулы.
5.2.4.3. Стандартизация растворов по точным навескам первичных стандартных (установочных) веществ: способ отдельных навесок, способ пипетирования
Для стандартизации растворов в этих случаях применяют два способа: титрование отдельных навесок и способ пипетирования.
Способ отдельных навесок. В этом способе берут серию (3-5) близких навесок первичного стандартного (установочного) вещества массой m(S) с точностью 0,0002 г, переносят их количественно в чистые конические колбы для титрования, растворяют в небольшом произвольном объеме дистиллированной воды и титрируют с индикатором по соответствующей методике стандартизируемым раствором до изменения окраски индикатора. В каждом случае измеряют затраченный объем титранта K(R) по шкале бюретки с максимальной точностью.
592
Новый справочник химика и технолога
Исходя из соотношений - или n(f(S)S) = n(S) s
= n(^KB(R)R) для точки эквивалентности (для реакции индикатор
титрования sS + rR о продукты реакции) вычисляют молярную концентрацию c(R) или молярную концентрацию эквивалента (нормальную концентрацию) c(/3kb(R)R) по формулам (5.2.42-5.2.43):
_ г ffl(S)-103 С~ s ' M{S}-V(Ry
c(f ^(57)-103
(АД ) M(/3kb(5)5)-K(R)’
где г и 5 — стехиометрические коэффициенты реакции титрования; m(S) — масса первичного стандарта (R). Символы s, S относятся к первичному стандарту, a R — к стандартизируемому раствору.
Далее проводят статистическую обработку результатов параллельных определений c(R) или c(/'1KB(R)R), полученных при титровании*, находят среднее значение концентрации и доверительный интервал с достоверностью до четвертой значащей цифры.
Приблизительную навеску первичного стандарта m(S) рассчитывают, задаваясь объемом титранта (стандартизируемого раствора) — K(R) , который желательно израсходовать (для достижения минимальной относительной погрешности измерения объема > 2/3 вместимости бюретки**), и учитывая ожидаемую концентрацию титранта:
^- = -c(R)K(R) (5.2.51)
m(S) г
и m(S) = — A/(S)c(R)K(R) (мг) при K(R) в мл. (5.2.52) г
Способ пипетирования. Точную навеску первичного стандарта (установочного вещества) m(S)*** переносят в мерную колбу вместимостью Ик, растворяют в небольшом объеме дистиллированной воды, доводят до метки водой и перемешивают многократным переворачиванием закрытой колбы. Затем 2-3 мл этого раствора ополаскивают мерную пипетку с одной меткой вместимостью Еп, заполняют пипетку раствором и дозируют содержимое пипетки в коническую колбу для титрования. Эту порцию раствора, содер-у
жашую долю растворенной навески массой m(S)-^~, называют аликвотной частью (долей) навески или просто аликвотой.
*В простейшем случае как результатов прямых равноточных m(S) определений (см. раздел 2.3); более строго — как результатов косвенных определений, например [2, 5, 38].
“В макрометоде используют бюретки вместимостью 25 и
50 мл, и для достижения погрешности измерения = 0,1% объем расходуемого титранта должен быть не менее 15-20 мл.
‘‘‘Погрешность взвешивания +0,0002 г.
Всего отбирают 3-4 аликвотных части и титрируют их. По результатам титрования вычисляют среднее арифметическое значение измеренных по шкале бюретки объемов титранта****. При работе с бюреткой, номинальная и действительная вместимость которой отличаются, для нахождения действительного эквивалентного объема K(R) пользуются графиком поправок к бюретке. Далее вычисляют молярную концентрацию или молярную концентрацию эквивалента (нормальную концентрацию) по формулам (5.2.44-5.2.45):
c(f = ^)-ю
аджм’
при K(R) в л и m(S) (г) переводной множитель 103 опускают.
Первоначальную приблизительную массу навески m(S) рассчитывают исходя из приготовленной приблизительной концентрации c(R) и приблизительно выбранного объема K(R), который должен пойти на титрование одной аликвотной части:
V И
т(5) = • c(R) • K(R) M(S) -103, (5.2.53)
»>(S) = ^c(/,„(R)R) • V(R) . (5.2.54)
’ll
Все члены правой части формул (5.2.53) и (5.2.54) известны, следовательно, можно рассчитать массу навески первичного стандарта.
Взвешиваемая с точностью ±0,0002 г навеска стандарта не должна превосходить значение m(S), т.к. раствор приготавливается с приближенно известной концентрацией. Если фактическая концентрация раствора окажется выше предполагаемой, то объема бюретки может не хватить на титрование аликвотной части и необходимо взять меньшую навеску m(S).
Способ пипетирования более оперативен, чем способ отдельных навесок.
5.2.4.4. Стандартизация по другому стандартному раствору
В ряде случаев стандартизацию титранта проводят по другому стандартному раствору. Например, нужно стандартизировать раствор NaOH c(NaOH), если имеется стандартный раствор хлороводородной кислоты, приготовленный предварительно по 5.4.2.1.
Для получения надежных результатов рекомендуется и способом пипетирования титровать несколько навесок. Полученные результаты определений обрабатывают методами математической статистики (см. раздел 2.3 и, например, [2, 5, 38]).
Химические методы количественного анализа
593
Рассчитывают и приготавливают из концентрированного раствора гидроксида натрия с концентрацией c(NaOH)KOH4., раствор NaOH с концентрацией c(NaOH)pa36, приблизительно равной значению с(НС1):
«(NaOH)KOHll — «(NaOH)pa36,
c(NaOH)KOHU • K(NaOH)KOHlI = c(NaOH)pa36.
^общл
где И(Н2О) = Иобщ. - Г(МаОН)конц..
Поскольку значения c(NaOH)KOHU и c(NaOH)pa36. приблизительно известны, а Кобщ. задается, то в колбу переносят мерным цилиндром рассчитанный объем щелочи K(NaOH)KOini и дистиллированной воды: К(Н2О) = Кобщ., -- K(NaOH)KOHU. Раствор тщательно перемешивают.
Возможен и другой вариант, когда приготавливают раствор NaOH из сухой навески. В этом случае расчет ведут по схеме:
мшпт = с( Na0H V -(/<Na0H> (5.2.55)
Л/(NaOH)
Отсюда рассчитывают приготавливаемый объем NaOH с концентрацией c(NaOH)pa36. = с(НС1).
Для стандартизации раствора NaOH из бюретки, предварительно промытой дистиллированной водой, стандартным раствором НС1 и заполненной этим раствором, дозируют в конические колбы для титрования несколько (4-5) разных точных порций раствора НС1 (для получения минимальной относительной погрешности определения отбирают объемы не менее 2/3 вместимости бюретки) и вводят в растворы индикатор, например, метиловый оранжевый. Затем последовательно промывают бюретку дистиллированной водой, приготовленным раствором NaOH, заполняют ее раствором NaOH и титруют отобранные порции НС1 до перехода окраски индикатора в желтый цвет.
Далее для каждой пары Г(НС1)- K(NaOH) рассчитыва-
F(HC1)
ют отношение —------—, сопоставляют полученные для
K(NaOH)
всех проб эти отношения на предмет обнаружения и отбраковки промахов (см. раздел 2.3.). Вычислив среднее ( К(НС1) значение —------— , из соотношении
^r(NaOH)
«(NaOH) = «(НО),
c(NaOH) • K(NaOH) = c(HCl) • Г(НС1)
вычисляют точное значение концентрации щелочи:
c(NaOH) = с(НС1)| *. (5.2.56)
\K(NaOH)J
* Следует помнить, что число значащих цифр в результате должно соответствовать числу значащих цифр в наименее достоверном числе.
5.2.4.5. Стандартизация растворов по стандартным образцам
В ходе реального анализа часто выполняют операции растворения анализируемой навески, отделения мешающих примесей и другие. При этом в определении могут иметь место те или иные систематические погрешности. Для их устранения (исключения) в ряде случаев целесообразно использование стандартных образцов (СО)**, близких по составу к анализируемым образцам, но с точно заданным содержанием компонентов.
Стандартный образец состава и свойств веществ и материалов — средство измерения в виде вещества (материала), состав и свойство которого установлены при аттестации.
СО предназначены для обеспечения единства и требуемой точности измерений посредством:
• градуировки, аттестации и поверки средств измерений;
• аттестации методик выполнения измерений;
• контроля правильности результатов измерений;
• измерения состава и свойств веществ и материалов методами сравнения.
СО подразделяются на стандартные образцы состава и стандартные образцы свойств.
Основными метрологическими характеристиками СО являются значения аттестуемой характеристики и его погрешности. В зависимости от установленного порядка утверждения СО подразделяются на следующие категории:
— государственные (ГСО),
— отраслевые (ОСО),
— стандартные образцы предприятий (СОП).
Приготовление первичных стандартов растворов по СО проводится по методике 5.2.4.1, а стандартизация растворов по методикам 5.2.4.3 или 5.2.4.4.
Пользуясь СО, часто (особенно в производственных условиях) устанавливают титр раствора по определяемому элементу, содержание которого в СО точно известно. Например, при определении хрома в сталях для установки титра раствора соли Мора используют стандартный образец стали с известным содержанием хрома. Концентрацию титрованного раствора в этом случае выражают титром по определяемому элементу (Гсоли мора/cr), и он показывает, сколько граммов определяемого элемента оттитровывается 1 мл раствора.
5.2.4.6. Расчет концентрации титрованных растворов с помощью поправочного коэффициента [47]
При выполнении серийных анализов ГОСТ или ведомственная инструкция обычно предусматривают применение раствора заданной концентрации или заданного титра. Приготовленные же растворы титрантов часто имеют отклонения от заданного значения концентрации. В этих случаях концентрацию или титр рассчитывают с помощью поправочного коэффициента (коэффициента поправки) — К.
** ГОСТ 8.315-97. Стандартные образцы. Основные положения.
594
Новый справочник химика и технолога
Поправочный коэффициент выражает отношение фактической (действительной) концентрации раствора к заданной (теоретической): во сколько раз фактическая концентрация или титр стандартного раствора больше или меньше заданного ГОСТом или инструкцией значения:
К Л = . (5.2.57)
c(R), T(R), T(R/A),
Коэффициент поправки (К) показывает:
1) на сколько нужно умножить заданную концентрацию раствора, чтобы найти его фактическую концентрацию: Сф Кс3,
2) на сколько нужно умножить титр раствора точно заданной концентрации, чтобы найти фактический титр данного раствора:
Т(Я)ф = KT(R)3; Т(К/А)ф = KT(R/A)3.
Зная поправочный коэффициент, можно рассчитать содержание определяемого компонента (вещества) по заданной концентрации или по заданному титру. Если необходимо вычислить массовую долю со (в %), то во все формулы добавляется множитель 100/тиНав., где /иНав. — масса анализируемой навески.
Например, ГОСТ предусматривает использование 0,1000 н. раствора КМпО4 (^и =1/5) при определении железа. При этом условии T(KMnO4/Fe) с учетом табличного значения A/(Fe) = 55,85 г • моль-1 по формуле (5.2.31) должен быть равен: T(KMnO4/Fe) = 0,0001-55,85 г мл Л
Пусть приготовленный стандартный раствор имеет фактическую (действительную) концентрацию с(1/5 КМпО4) = = 0,09920 моль • л-1. Тогда К = - 09920 _ q , и расчет 0,1000
результатов определений (результата анализа) должен производится по формуле:
m(Fe) = 0,005585 • 0,9920 • К(КМпО4) =
= 0,005540 • ЦКМпО4),
где действительный (фактический) T(KMnO4/Fe) = = 0,005540 г • мл’1.
Общие правила при определении коэффициента поправки. Для определения коэффициента поправки к заданной концентрации раствора берут обычно не менее трех навесок исходного вещества, взвешивают их с погрешностью не более 0,0002 г или три разных объема стандартного раствора, например, 20, 30, 35 мл, отмеривая их пипетками или бюретками. При взятии масс навесок менее 0,05 г при титровании с помощью полумикробюреток используют микровесы, которые обеспечивают погрешность при взвешивании не более 0,002-0,003 мг. Если микровесы отсутствуют, массу навески взвешивают на обычных весах и растворяют ее в воде в откалиброванной мерной колбе. Затем, чтобы определить поправку, берут аликвотные объемы раствора, соответствующие по концентрации содержанию исходного определения. Взятие масс навесок рекомендуется производить по «методу взвешивания по разности».
1. Для предупреждения ошибок при титровании исходные вещества берут в таких количествах, чтобы на их титрование расходовались примерно следующие объемы стандартизируемого (титрованного) раствора:
30—40 мл, если вместимость бюретки 50 мл 20-25 мл, если вместимость бюретки 25 мл 8-10 мл, если вместимость бюретки 10 мл
2. Величину массы навески исходного вещества в граммах, которую необходимо взять для определения коэффициента поправки, рассчитывают по следующим формулам*:
т = 40c(f3KB(R)R)- ’ если вместимость бю-
ретки 50 мл;
т = 23c(^ra(R)R)- если вместимость бю-
1000
ретки 25 мл;
т = 9c(^KB(R)R)- ^^экв если вместимость бю-1000
ретки 10 мл.
Например, рассчитывают массу навески Na^CO,, которую нужно взять для установления титра 0,5 н. раствора при титровании из бюретки вместимостью 50 мл и А/(1/2 Na2CO3) = 53 г • моль’1
тм = 40 • 0,5~——— — 1,06 г .
1000
Итак, масса навески Na2CO3 должна примерно равняться 1 г.
3. Взятую массу исходного вещества растворяют в дистиллированной воде.
4. Вся применяемая посуда должна быть тщательно вымыта.
5. Мерную посуду (бюретки, пипетки и мерные колбы) обязательно проверяют на правильность калибровки.
6. Точность, с которой выполняют титрование, измерения объемов и последующие вычисления, должна соответствовать допускаемой погрешности.
7. Коэффициент поправки К вычисляют сначала на основании данных титрования каждой отдельной массы исходного вещества или объема раствора. Эти поправки не должны отличаться друг от друга больше, чем на 0,0015 при титровании из обычных бюреток и не больше, чем на 0,003 при титровании из полумикробюреток вместимостью до 10 мл. Затем из вычисленных коэффициентов берут средний, он должен быть в пределах 1 ± 0,02. Если коэффициент поправки выходит из указанных пределов, то раствор соответственно концентрируют или разбавляют.
8. Если стандартизируемый раствор устанавливают и применяют при разных температурах, то следует водить температурную поправку.
9. Необходимо помнить, что изменение температуры на 10 °C изменяет коэффициент поправки на 0,02.
* Здесь 40, 23, 9 — объемы, рекомендуемые для расхода стандартизируемого раствора на одно титрование.
Химические методы количественного анализа
595
10. При длительном хранении раствора периодически проверяют коэффициент поправки, имея в виду сроки устойчивости растворов при хранении.
Поправки на температуру при пользовании титрованными растворами. При выполнении особо точных работ в титриметрическом анализе необходимо помнить, что водные растворы расширяются при повышении температуры и
сжимаются при охлаждении, что приводит к изменению концентрации титрованного раствора.
Кубический коэффициент расширения всякого водного раствора зависит от концентрации растворенного вещества. Для воды и для 0,1 н. водных растворов он практически одинаков.
Таблица 5.2.12
Приведение объема воды и некоторых водных растворов к объему при 20 °C [47]
t, °C Поправки Р (в мл) на объем 1000 мл
вода и 0,1 н. растворы 1 Н. НС1 1 н. (СООН)2 1 н. H2SO4 (Гэкв =1/2) 1 Н. HNO3 1 н. Ка2СОз(4в=1/2) 1 Н. NaOH
5 +1,36 +2,23 +2,38 +3,24 +3,30 +3,32 +3,51
6 +1,36 +2,15 +2,30 +3,09 +3,14 +3,16 +3,32
7 +1,35 +2,07 +2,21 +2,93 +2,98 +2,98 +3,13
8 +1,32 +1,97 +2,10 +2,76 +2,80 +2,79 +2,93
9 +1,28 +1,85 +1,99 +2,58 +2,61 +2,60 +2,72
10 +1,22 +1,73 +1,86 +2,39 +2,41 +2,40 +2,51
11 +1,16 +1,60 +1,72 +2,19 +2,21 +2,19 +2,29
12 +1,09 +1,45 +1,57 +1,98 +1,99 +1,98 +2,06
13 +0,98 +1,30 +1,40 +1,76 +1,76 +1,76 +1,83
14 +0,88 +1,14 +1,23 +1,53 +1,53 +1,53 +1,58
15 +0,76 +0,97 +1,05 +1,30 +1,30 +1,29 +1,33
16 +0,63 +0,79 +0,85 +1,06 +1,05 +1,05 +1,08
17 +0,49 +0,61 +0,65 +0,81 +0,80 +0,80 +0,82
18 +0,34 +0,41 +0,44 +0,55 +0,54 +0,56 +0,55
19 +0,17 +0,21 +0,23 +0,28 +0,27 +0,27 +0,28
20 ±0,00 ±0,00 ±0,00 ±0,00 ±0,00 ±0,00 ±0,00
21 -0,19 -0,22 -0,24 -0,28 -0,28 -0,28 -0,29
22 -0,36 -0,44 -0,49 -0,56 -0,57 -0,56 -0,59
23 -0,59 -0,67 -0,75 -0,85 -0,87 -0,85 -0,90
24 -0,80 -0,91 -1,02 -1,15 -1,17 -1,15 -1,21
25 -1,03 -1,17 -1,29 -1,46 -1,48 -1,46 -1,52
26 -0,26 -1,43 -1,57 -1,78 -1,80 -1,77 -1,84
27 -1,51 -1,70 -1,85 -2,11 -2,13 -2,09 -2,17
28 -1,71 -1,92 -2,14 -2,45 -2,46 -2,41 -2,50
29 -1,99 -2,26 -2,44 -2,79 -2,80 -2,75 -2,87
30 -2,30 -2,55 -2,77 -3,13 -3,14 -3,09 -3,19
В табл. 5.2.12 приводятся поправки к объему воды и некоторых водных растворов, находящихся в стеклянных сосудах, к объему при 20 °C, которая в титриметрическом анализе принимается за нормальную температуру. Пользуются формулой
К20=И(1+0,001Р),
где И20 — искомый объем раствора при 20 °C;
Ki — объем раствора, измеряемый при температуре опыта; Р — поправка (берется из табл. 5.2.12 со знаком + или -) при той температуре, при которой измерен объем.
На практике удобнее, учитывая температурную поправку, пересчитывать не объем раствора, а его коэффициент поправки по следующей формуле:
Ki = К [1-0,001 (Р-А)]
где К — коэффициент поправки раствора при температуре t в день установки титра;
Ki — коэффициент поправки раствора при температуре t\ в день использования раствора;
Р, Р\ — поправки, взятые для соответствующих температур t и Z] из табл. 5.2.12.
Пример
Определить коэффициент поправки 0,1 н. раствора при температуре t\ = 24 °C, если он был установлен при t = 15 °C и в этих условиях был равен 1,000:
К24 = 1,000 [1 - 0,001 (0,76 + 0,80)] = 0,9984.
Для 0,1 н. растворов поправки на температуру могут быть взяты непосредственно из табл. 5.2.13. При этом вы
596
Новый справочник химика и технолога
водят алгебраическую разность между поправками, найденными в табл. 5.2.12 для температур t и t\. Абсолютную величину этой разности прибавляют к коэффициенту поправки, установленному при температуре t, если t > t\, и вычитают, если t <
Пример
Для t = 15 °C и 6 = 24 °C
К24 = 1,000 - [(-0,0008) - (+0,0008)] =
= 1,000-1,0016 = 0,9984.
Таблица 5.2.13
Поправка на температуру для коэффициентов поправки 0,1 и. растворов
/, °C Величина поправки /,°С Величина поправки t, °C Величина поправки
5 -0,0014 14 -0,0009 23 +0,0006
6 -0,0014 15 -0,0008 24 +0,0008
7 -0,0013 16 -0,0006 25 +0,0010
8 -0,0013 17 -0,0005 26 +0,0013
9 -0,0013 18 -0,0003 27 +0,0015
10 -0,0012 19 -0,0002 28 +0,0018
И -0,0012 20 ±0,0000 29 +0,0020
12 -0,0011 21 +0,0002 30 +0,0023
13 -0,0010 22 +0,0004
5.2.5. Общие правила выполнения титриметрических определений [3, 47]
Для получения правильных результатов при приготовлении вторичных стандартов и при титриметрических определениях необходимо придерживаться ряда привил.
• Строго соблюдать пропись выполнения работы, правила измерения массы и объемов, пользоваться реактивами квалификации не ниже «чистый для анализа».
• Предварительно ознакомиться с метрологическими характеристиками мерной посуды и овладеть грамотной техникой работы с ней для достижения минимальной погрешности измерения объемов титранта и титрируемого вещества [47]. При работе с тестированными бюретками при обычных количественных определениях значения объемов, считываемые по шкале, соответствуют действительным, которые и представляются в расчетные формулы табл. 5.2.2.
• При точных определениях калибровку тестированной мерной посуды необходимо проверить [3, 47], т.к. истинная и номинальная вместимость, приводимые в табл. 5.2.7-5.2.9, могут не всегда совпадать. При расхождениях, больше допустимых в табл. 5.2.7-5.2.9, такую посуду отбраковывают или систематически учитывают поправки. При необходимости пользования такой нестандартной бюреткой строят график поправок объемов к бюретке, которым и пользуются для нахождения действительных объемов, затраченных на титрование. Полученные действительные значения объемов затем и подставляются в расчетные формулы табл. 5.2.2.
• Способ пипетирования при предварительном соизмерении вместимости мерной колбы по используемой пипетке позволяет значительно уменьшить погрешность отбора аликвотной части анализируемой пробы.
• При определении КТТ с помощью индикатора, последний добавляют всегда в количестве, которое позволяет четко фиксировать изменение окраски титрируемого раствора. Это количество должно быть всегда минимальным и одинаковым при параллельных титрованиях, поскольку некоторая часть титранта расходуется на взаимодействие с индикатором. Кроме того, при большом количестве индикатора изменение окраски титрируемого раствора уловить гораздо сложнее из-за двух окрасок. Обычно в прописях указывается концентрация индикатора и конкретное количество капель индикатора, которое рекомендуется добавлять в титрируемый раствор. Для более точной регистрации конечной точки титрования и снижения систематической погрешности рекомендуется сравнивать цвет титрируемого раствора с цветом раствора «свидетеля», который помещают рядом и до окраски которого ведется титрование. Человеческий глаз лучше сравнивает окраски, чем оценивает их. «Свидетель» готовят следующим образом: наливают в титровальную колбу объем дистиллированной воды, равный объему аликвотной части анализируемого раствора плюс объем расходуемого титранта, добавляют столько же индикатора, как при титровании, и одну или две капли раствора титранта.
• Цвет титрируемого раствора лучше всего наблюдать на белом фоне.
• Скорость выливания раствора из пипетки и бюретки должна быть всегда одинакова.
• Быстрое выливание раствора из пипетки и бюретки обусловливает значительную «погрешность натекания», поскольку часть раствора не успеет стечь со стенок пипетки или бюретки на момент измерения объема. В конце титрования подача титранта должна осуществляться долями капли во избежание «перетитровывания».
• Для достижения минимальной погрешности титрования объем, затрачиваемый на титрование, должен составлять не менее 2/3 вместимости бюретки.
• При считывании масс по шкале аналитических весов и объемов по бюретке в расчетах и при записи конечного результата грамотно фиксировать число значащих цифр, пользоваться правилами округления.
• При стандартизации растворов и при определении содержания заданного компонента необходимо проводить несколько параллельных титрований. При этом при титровании одинаковых аликвотных частей должно быть не менее трех значений объемов, сходящихся в пределах 0,1-0,2% (отн.).
• Для строгих расчетов воспроизводимости и правильности конечного результата титрования (объема, концентрации, массы), полученного из нескольких параллельных титрований для оценки случайных и систематических погрешностей, применяют методики статистической обработки результатов [1-3, 5, 38] — см. также раздел 2.
Химические методы количественного анализа
597
• Периодически необходимо проверять концентрацию стандартных растворов, т.к. она несколько меняется во времени. Даты приготовления и проверок стандартных растворов должны фиксироваться.
• В рабочем журнале каждая работа должна быть полностью оформлена с указанием даты, цели, сущности работы, уравнений химических реакций, результатов титрования, расчетных уравнений, самих расчетов и выдачей метрологических характеристик результата анализа. Значения конечных результатов: концентраций, титров или масс — должны быть записаны с точностью 4-х значащих цифр, например, с = 0,2108 моль-л1; т = 125,6 мг; ГА - = 0,004237; погрешности — с точностью 1-2 значащих цифр.
5.2.6. Практическое применение титриметрических методов анализа
5.2.6.1. Кислотно-основное титрование. Общая характеристика и возможность метода
В основе метода лежит протолитическая реакция [1]: SH2 +S“ < . >2SH в частности, в водных растворах:
Н3О+ + ОН = 2Н2О.
Прямым титрованием стандартными растворами щелочей и сильных кислот определяют содержание растворимых в воде сильных и слабых кислот и оснований, включая соли, образованные сильными и слабыми кислотами и др.
Обратным титрованием определяют малорастворимые в воде оксиды и карбонаты, аммонийные соли, некоторые сложные эфиры RC(O)ORb
Титрованием заместителя определяют многие вещества, обладающие слабовыраженными йли практически не обладающие кислотно-основными свойствами. Например, катионы, анионы, (соли) можно определить, используя реакцию ионного обмена с катионитом или анионитом. Выделяющиеся в результате ионного обмена эквивалентные количества Н+ или ОН - ионов определяют прямым титрованием стандартными растворами оснований или кислот соответственно.
Способами кислотно-основного титрования определяют, в частности N, S, С, Cl(Br), F, Р, которые входят в состав важных органических и биологических систем. Путем предварительной обработки элементы переводят в неорганическую кислоту или основание, которые затем титруют (табл. 5.2.14).
Таблица 5.2.14
Схемы определения N, S, С, Cl(Br), F, Р кислотно-основным титрованием [21]
Элемент Аналитическая форма Продукт поглощения или осаждения Титрование
N* NH3 NH3 +Н3О+ >NH^+H2O (г) (НС1) Избыток НС1 титруют р-ром NaOH (обратное титрование)
S SO2 SO2 +Н2О2 >H2SO4 (г) Р-ром NaOH
С СО2 СО2 + Ва(ОН)2 >ВаСО3 + Н2О (г) (тв) Избыток Ва(ОН)2 титруют р-ром НС1 (обратное титрование)
С1(Вг) НС1(Вг) НС1+Н2О > СГ +Н3О+ (г) 5 Р-ром NaOH
F SiF4 SiF4+H2O >H2SiF6 (г) То же
Р Н3РО4 12Н2МоО4 + 3NH4+H3PO4 -> -> (NH4)3PO4 12МоО3 +12Н2О+ЗН+ (ТВ) (NH4)3PO4 12МоО3 +26ОН -> (тв) -> НРО4+12МоО4+14H2O+3NH3 (г) Избыток NaOH титруют р-ром НС1 (обратное титрование)
Примечание.
*Азот входит в состав многих неорганических и органических веществ, представляющих большой интерес, например, в состав аминокислот, протеинов, синтетических лекарств, взрывчатых веществ, красителей, растворителей и т.д. В какой бы степени окисления он не был в исходном образце, его предварительно переводят в степень окисления -3(NH3). Для разложения органических соединений применяют метод Кьельдаля (подробнее см. [1]).
Индикаторы
Момент эквивалентности, вернее КТТ большей частью определяют визуально с помощью индикаторов. Цветные индикаторы в кислотно-основном титровании представляют собой слабые органические кислоты или основания,
протонированные и непротонированные формы которых различаются по структуре и окраске. Существуют одноцветные (например, фенолфталеин) и двухцветные (например, метиловый оранжевый) индикаторы. Окраска каждого из индикаторов изменяется в пределах опреде
598
Новый справочник химика и технолога
ленного узкого интервала значений pH, его называют интервалом перехода окарски индикатора. Для двухцветных индикаторов он рассчитывается по формуле
ДрН = рКа±1,
где Ка — константа кислотности индикатора.
В пределах интервала перехода наиболее резкое изменение окраски индикатора наблюдается при определенном значении pH, которое называется показателем титрования и обозначается рТ. Величина рТ находится приблизительно в середине интервала перехода и фактически отождествляется с КТТ.
Выбирают индикатор для титрования так, чтобы интервал перехода окраски индикатора (или рТ) ближе всего совпадал бы с pH титруемого раствора в ТЭ. Поэтому для более правильного выбора индикатора необходимо проследить характер кривой титрования, величину скачка титрования и установить pH, соответствующее ТЭ (см. например, рис. 5.2.3).
Линия эквивалентности
20 40 60 80 100 120 140
V(NaOH), мл
Рис. 5.23. Кривая титрования 100,0 мл 0,1 М НС1 (сплошная линия) и 0,01 М НС1 (пунктир)
Для сужения интервала перехода и получения более резкого перехода окраски применяют смешанные индикаторы, которые составляют из индикатора и красителя. При определенном значении pH цвет красителя является дополнительным к цвету индикатора. В результате в этой точке окраска будет серой, а переход от окрашенного раствора к серому — контрастным.
Так как в кислотно-основных титрованиях обычно в качестве титранта применяют либо сильную кислоту, либо сильное основание, при выборах индикатора для титрования кислот и оснований можно пользоваться следующим общим указанием [47]:
1. При титровании сильных кислот и сильных оснований (примерно 0,1 н.) можно применять любой индикатор с pH интервала перехода в пределах 4,3-9,7 (от метилового оранжевого до фенолфталеина). При титровании более концентрированных растворов, чем 0,1 н., можно применять любой индикатор с pH интервала перехода 3,3-10,7.
2. При титровании слабой кислоты сильным основанием конец титрования наблюдается в интервале pH = 7,74—10. В этих случаях можно использовать, например, индикаторы: феноловый красный, тимоловый голубой, фенолфталеин, тимолфталеин.
3. При титровании слабого основания сильной кислотой конец титрования наблюдается в интервале pH = 6,26-4. В этих случаях можно применять, например, индикаторы: «-нитрофенол, метиловый красный, метиловый оранжевый.
4. При титровании многоосновных кислот или многокислотных оснований нужно рассчитать значения pH для каждой ТЭ и подобрать соответствующий индикатор. Например, при титровании фосфорной кислоты первая ТЭ соответствует pH = 4,33; вторая — 9,57; третья — 12,72. Следовательно, для титрования до NaH2PO4 можно применять метиловый оранжевый, до Na2HPO4 — фенолфталеин, до №зРО4 — тимоловый голубой в присутствии солей кальция.
Наиболее широко применяемые в кислотно-основном титровании индикаторы и интервалы их перехода представлены на рис. 5.2.4 и в табл. 5.2.15-5.2.19.
Химические методы количественного анализа
599
Метиловый фиолетовыйг (первый переход)
Крезоловый красный® (первый переход) Тимоловый синий® (первый переход) Тропеолин 00а Метиловый фиолетовыйг (второй переход) Демитиловый желтый® Бромфеноловый синий® Метиловый оранжевый® Ализарин Sr Бромкрезоловый зеленый® Метиловый красный® и-Нитрофенолг Бромтимоловый синий® Феноловый красныйг Нейтральный красныйг Крезоловый красный® (второй переход) Тропеолин 000а
Тимоловый синий® (второй переход) Фенолфталеин6 Тимолфталеин6 Ализариновый желтый® Тропеолин 0а сгш-Тринитробензойная ки-слотаг Лакмус
желтый синий
красный 10,2-1,81 желтый
красный |1,2-2,8| желтый
красный 11,3-3,2| желтый
синий |1,5-3,2| фиолетовый
красный |2,9-4,0| желтый
желтый |3,<М,6| синий
красный |3,1-4,4| желтый
желтый |3,7-5,2| фиолетовый
желтый |3,8-5,4| синий
красный |4.4-6,2| желтый
бесцветный |4,7-7,9| желтый
желтый |б,0-7,б| синий
желтый |б.4-8.2| красный
красный 16,8-8,01 желтый
желтый [7,2-8,8| красный
желто- 17,6-8.91 красный
коричневый
желтый |8,0-9,б| Синий
бесцветный |8,2-10,01 Красный
бесцветный |9,4-10,б| Синий
желтый |10,0-12,0| Красный
желтый |11,0-12,7| оранжево-коричневый
бесцветный |12,-13,5| Оранжевый
красный И Синий
Рис. 5.2.4. Интервалы перехода (ДрН) окраски важнейших кислотно-основных индикаторов: а— азокраситель; 6— фталеин;в — сульфофталеин; г— прочие
Важнейшие кислотно-основные индикаторы (в порядке возрастания интервалов pH перехода окраски) [46]
Таблица 5.2.15
№ пп Индикатор Формула Молярная масса Концентрация, % Интервал pH перехода и окраска индикатора
1 Метиловый фиолетовый; 1-й переход (см. №№ 7, 15) С=<^ />= N(CH,)2Cl" CH,NH^ 0,1 и 0,05 в воде 0,13-0,5 желтая — зеленая
600
Новый справочник химика и технолога
Продолжение таблицы 5.2.15
№ пп Индикатор Формула Молярная масса Концентрация, % Интервал pH перехода и окраска индикатора
2 а-Нафтол-бензеин; 1-й переход; (см. № 58) \ / он V _j ОН Z \ С (f \—он 0 392,45 0,05 в 70% спирте 0,0-1,0 зеленая — желтая
3 Пикриновая кислота он n°2 no2 229,11 0,05 в воде 0,0-1,3 нет — желтая
4 Метиловый зеленый (СНз)^^^- .^Й(СН3)2СГ Ф +N(CH3)2C1~ 458,47 0,05 в воде 0,1-2,0 желтая — зеленоголубая
5 Крезоловый красный; 1-й переход (см. № 48) Y1 ГТ НзСГ^^^ сн3 J^.so3n 382,44 0,04 в 50% спирте 0,2-1,8 красная — желтая
6 Малахитовый зеленый; 1-й переход (см. № 70) (CH,)N N(CH3)2C1” 364,92 0,1 в воде 0,13-2,0 желтая — голубовато-зеленая
7 Метиловый фиолетовый; 2-й переход (см. №№ 1, 15) См. № 1 — 0,1 и 0,05 в воде 1,0-1,5 зеленая — синяя
8 Метаниловый желтый (виктория желтый, тропеолин Ж) н \ /—N==N—у—N—Z у NaO3S 375,38 0,1 в воде 1,2-2,3 красная — желтая
Химические методы количественного анализа
601
Продолжение таблицы 5.2.15
№ пп Индикатор Формула Молярная масса Концентрация, % Интервал pH перехода и окраска индикатора
9 Бензолазодифениламин tк tн Г к / \ / \ 1 / \ \ /—N==N—К /—N—\ / 272,34 0,01 в 50% спирте с добавлением 1 мл 1 мнет на 100 мл раствора 1,1-2,8 пурпурная — желтая
10 л-Крезоловый пурпурный (н-крезол-сульфофталеин; 1-й переход (см. № 51) н,с сн3 SO3H J 382,44 0,04 в 20% спирте 1,2-2,8 красная — желтая
11 Тимоловый синий (тимолсульфофталеин); 1-й переход (см. № 54) Н3С XX Н3С сн3 ,Х\^° сн3 SO3H J 466,60 0,01 в а) 20% спирте б) в воде с добавлением 4,3 мл 0,05 н. NaOH на 100 мл раствора 1,2-2,8 красная Хмакс. 544 нм* — желтая ^макс. 430 НМ
12 Ксиленоловый синий (л-ксиле-нол-сульфофта-леин; 1-й переход (см. № 55) Н3С Н3С сн3 сн3 . SOJI 410,49 0,05 в а) 20% спирте; б) в воде с добавлением 5,3 мл 0,05 М NaOH на 100 мл индикатора 1,2-2,8 красная — коричнево-желтая
13 Пентаметоксикрасный осн3 осн3 Х1 XX Н3ССГ^^^ осн3 ^Д^осн3 410,47 0,1 в 70% спирте 1,2-3,2 краснофиолетовая — нет
Здесь и далее в таблицах приводятся значения ^макс. поглощения.
602
Новый справочник химика и технолога
Продолжение таблицы 5.2.15
№ пп Индикатор Формула Молярная масса Концентрация, % Интервал pH перехода и окраска индикатора
14 Тропеолин 00 (оранж IV; анилин-гельб, дифенил-оранж) — NH— N= N SO3Na 375,38 1,0; 0,1 и 0,01 в воде 1,3-3,2 красная — желтая
15 Метиловый фиолетовый; 3-й переход (см. №№ 1, 7) N(CH,),cr CHjNH^ 0,1 в воде 2,0-3,0 синяя — фиолетовая
16 Ализариновый желтый Р; 1-й переход (см. № 66) °2N—— N= N—— ОН COONa 309,21 0,1 в воде 1,0-3,3 красная — желтая
17 Бензиловый оранжевый Щ-НН-^~у-Т^ N-^^- SO3K 405,52 0,05 и 0,01 в воде 1,9-3,3 красная — желтая
18 Бензопурпурин 4Б; 1-й переход (см. № 75) NH2 Н3С сн3 N= N—N= N NH;—SO,Na SO3Na —NH, 724,73 0,1 в воде и 0,5 в 90% спирте 1,3-4,0 синефиолетовая — оранжевая
19 2,6-Динитрофенол (р-динитрофенол) ОН no2 184,11 0,1; 0,05 и 0,04 в воде 1,7-4,4 нет — желтая
20 2,4-Динитрофенол; (а-динитро-фенол) он no2 181,11 Нас. и 0,04 в воде и 0,1 в спирте 2,0-4,7 нет — желтая
21 Метиловый желтый (диметилгельб) Н3С z=\ /=« \ / \ /\ N ( ,) N=N G ,) / \ / \ / Н3С У У 225,29 0,1 и 0,01 в 90% спирте 2,9-4,0 красная — желтая
Химические методы количественного анализа
603
Продолжение таблицы 5.2.15
№ пп Индикатор Формула Молярная масса Концентрация, % Интервал pH перехода и окраска индикатора
22 Метиловый оранжевый (метилоранж, гелиантин, оранж III) (CH3)2N— N= N SO3Na 327,33 0,1 в воде 3,1-4,0 красная (^макс. 522 НМ) оранжево-желтая (^макс. 464 НМ)
23 Бромфеноловый синий (тетра-бромфенолсуль-фофталеин) Вг Вг Y1 ( Y Вг ^J^^SO3H 669,97 0,1 в а) 20% спирте б) воде с добавлением 3,0 мл 0,05 М NaOH на 100 мг индикатора 3,0-4,6 желтая (^макс. 436 НМ) — СИНЯЯ (^-макс. 592 НМ)
24 Бромхлорфено-ловый синий Вг Вг Y X f Т Of 581,06 0,04 в а) 20% спирте; б) в воде с добавлением 3,2 мл 0,05 М NaOH на 100 мг индикатора 3,0-4,8 желтая — пурпурная
25 Конго красный N= N—y— N= N H;N—SNaOj NaO3S—NH2 696,07 0,1 и 1,0 в воде 3,0-5,2 (сине-красная — фиолетовая
26 Ализариновый красный С 1-й переход, см. №62 0 OH Г X l I ’ H2° YYX/X/^ SO3Na 0 360,27 0,1 в воде 3,7-5,2 желтая — фиолетовая
27 п- Этоксихризои-дин гидрохлорид h2n C2H5O—N= N—NH2. NCI 292,77 0,04 и 0,2 в воде 3,5-5,5 красная — лимонно-желтая
604
Новый справочник химика и технолога
Продолжение таблицы 5.2.15
№ пп Индикатор Формула Молярная масса Концентрация, % Интервал pH перехода и окраска индикатора
28 Бромкрезоловый синий (бромкрезоловый зеленый) Вг Вг но cyJ\^° Вг^^^^4 Вг J^^SO3H 698,02 0,1 в а) 20% спирте; б) в воде с добавлением 2,9 мл 0,05 М NaOH на 100 мг индикатора 3,8-5,4 желтая (^•макс. 444 нм) — синяя (^•макс. 617 НМ)
29 а-Нафтоловый красный % II 1 247,30 0,1 в 70% спирте 3,7-5,7 фиолетовая — коричневато-желтая
30 2,5-Динитрофенол (у-динитрофенол) ОН j^^no2 X J 184,11 0,1 и 0,025 в воде 4,0-5,4 нет — желтая
31 Лакмоид (резорциновый синий) С12Н9О3 215,21 0,2 и 0,5 в 90% спирте 4,0-6,4 красная — синяя
32 Метиловый красный ноос Н3С /=х >=\ \ / \ / \ N 6 5 N=N < ,) / \ / \ / Н3С '' V__7 269,30 0,1 и 0,2 в 60% спирте 4,2-6,2 красная (^-макс. 530 Нм) желтая (каке. 427 НМ)
33 Хризоидин N= N NH2 • НС1 nh/ 248,71 0,1 в воде 4,0-7,0 оранжевая — желтая
34 Иодэозин (тет-раиодфлуорес-цеин) I I H0\X\z 0 XXXI CY ^^^COONH, 852,93 0,1 в воде 4,5-6,5 нет — красная
Химические методы количественного анализа
605
Продолжение таблицы 5.2.15
Ns пп Индикатор Формула Молярная масса Концентрация, % Интервал pH перехода и окраска индикатора
35 Гематоксилин Т г \ '° Д Дк ^с—сн2 но^^^сД 1 Н2СХ ОН 302,28 0,5 в 90% спирте 5,0-6,0 желтая — фиолетовая
36 Хлорфеноловый красный (дихлорфенол-сульфофталеин) С1 ^^SO3H 423,27 0,1 в а) 20% спирте; б) в воде с добавлением 4,7 мл 0,05 М NaOH на 100 мг индикатора 4,8-6,6 желтая — пурпурная
37 Бромфеноловый красный (диб-ром-фенолсульфоф-талеин) НО\<Д:\ ^l^SO3H 512,18 0,1 и 0,04 в а) 20% спирте; б) в воде с добавлением 3,9 мл 0,05 М NaOH на 100 мг индикатора 5,0-6,8 желтая — красная
38 2-Нитрофенол (о-нитрофенол) он 139,11 0,1 в 50% спирте 5,0-7,0 нет — желтая
39 Бромкрезоловый пурпурный; (дибром-о-крезолсульфо-фталеин) сн3 сн3 НО^^к. вг 540,23 0,1 в а) 20% спирте; б) в воде с добавлением 3,7 мл 0,05 М NaOH на 100 мг индикатора 5,2-6,8 желтая (Лмакс. 433 НМ) — пурпурная (Лмакс. 591 нм)
40 Нитразин желтый Na o2n он \ no2 so3Na 542,37 0,1 в воде 6,0-7,0 желтая — синефиолетовая
606
Новый справочник химика и технолога
Продолжение таблицы 5.2.15
№ пп Индикатор Формула Молярная масса Концентрация, % Интервал pH перехода и окраска индикатора
41 «-Нитрофенол НО——N°2 139,11 0,1 в воде 5,6-7,6 нет — желтая
42 Бромтимоловый синий (дибром-тимолсульфоф-талеин) Вг Вг Н0\_гХХС (СНз^НСГ*4^^ СН(СН3)2 ^L^SO3H QT 624,39 0,05 и 0,1 в а) 20% спирте б) в воде с добавлением 3,2 мл 0,05 М NaOH на 100 мг индикатора 6,0-7,6 желтая (Хмакс. 433 нм) — синяя 0“макс. 617 НМ)
43 Розоловая кислота (аурин; метил-аурин; желтый корал-лин; кораллин-фталеин) НО.^^ он 304,35 0,2 в 50% спирте 6,8-8,0 желтая — красная
44 Нейтральный красный (ней-тральрот) СНз 1111 ’НС1 (H3C)X^^^?r^^NH2 288,78 0,1 в 60% спирте 6,8-8,4 красная — янтарно-желтая
45 Феноловый красный (фенолсульфофталеин) ХХх8°зН ГТ 354,38 0,1 в а) 20% спирте б) воде с добавлением 5,7 мл 0,05 М NaOH на 100 мг индикатора 6,8-8,4 желтая (Хмакс. 433 нм) — красная (Чакс. 558 нм)
46 Хинолиновый синий (цианин) CSH, — N=1 CH—N— CSH„ I 562,54 1,0 в 50% спирте 7,0-8,0 нет — фиолетовая
47 л/-Нитрофенол no2 но 139,11 0,3 в воде 6,6-8,6 нет — желтая
Химические методы количественного анализа
607
Продолжение таблицы 5.2.15
№ пи Индикатор Формула Молярная масса Концентрация, % Интервал pH перехода и окраска индикатора
48 Крезоловый красный (о-крезолсуль-фофталеин); 2-й переход (см. № 5) См. № 5 382,43 0,04 в 50% спирте 7,0-8,8 желтая (^макс. 434 НМ) пурпурная (^макс. 572 нм)
49 А-Нафтол-фталеин НО О OQ 6о J^^COOH 418,44 0,1 и 1,0 в 70% спирте 7,4-8,6 почти нет — зеленовато-синяя
50 Этил-бмс (2,4-динитрофенил) ацетат COO—С2Н5 o2n—— сн—— no2 no2 o2n 420,29 0,1 в спирте 7,4-9,0 нет — синяя
51 jw-Крезоловый пурпурный, 2-й переход, см. № 10 См. № 10 7,4-9,0 желтая — пурпурная
52 Тропеолин ООО НО N= N SO,На 350,32 0,1 и 1,0 в воде 7,6-8,9 желто-розовая — зеленая
53 Куркумин; 1-й переход; см. № 63 ОСН, со— сн= сн—Z у— он Н2С Х/ со— сн= сн—— он осн3 368,39 0,1 в воде 7,4-9,2 желтая — буровато-красная
54 Тимоловый синий; 2-й переход (см. № 12) См. № 12 8,0-9,6 желтая — синяя
608
Новый справочник химика и технолога
Продолжение таблицы 5.2.15
№ пп Индикатор Формула Молярная масса Концентрация, % Интервал pH перехода и окраска индикатора
55 Ксиленоловый синий; 2-й переход (см. № 12) См. № 12 8,0-9,6 желтая — синяя
56 о-Крезолфталеин СН3 СН, 1 1 НО^Х\^ ^\^СООН 346,38 0,2 и 0,02 в 90% спирте 8,2-9,8 нет — красная
57 Фенолфталеин ^х^^соон 318,33 0,1 и 1,0 в 60% спирте 8,2-10,0 нет — пурпурная ^-макс. 553 НМ
58 а- Нафтолбензеин 2-й переход (см. № 2) См. № 2 8,4-10,0 желтая — синяя
59 и-Ксиленолфта-леин XI Х1 З^^соон 374,44 0,1 в 40% спирте 9,3-10,5 нет — синяя
60 Тимолфталеин Н3С сн3 Н3С сн3 чсн чсн Н3С I Снз ^Х/соон 430,54 0,1 в 90% спирте 9,3-10,5 нет — синяя каке. 598 нм
61 Нильский голубой (с2но2Х/\/ NHz z Xj Cl 353,85 0,1 в воде 10,1-11,1 синяя — красная
Химические методы количественного анализа
609
Продолжение таблицы 5.2.15
№ пп Индикатор Формула Молярная масса Концентрация, % Интервал pH перехода и окраска индикатора
62 Ализариновый красный С; 2-й переход (см. № 26) См. № 26 10,0-12,0 фиолетовая — бледно-желтая
63 Курку мин; 2-й переход (см. № 53) См. № 53 10,2-11,8 буро-красная — оранжево-желтая
64 Р-Нафтоловый фиолетовый NaO3S N= N NO, NaO,S ОН 497,37 0,04 в воде 10,0-12,1 оранжевожелтая — фиолетовая
65 Ализариновый желтый ЖЖ (салициловый желтый; протравной желтый) но——n=n—с COONa NO2 309,21 0,1 в воде 10,0-12,1 светло-желтая — темно-оранжевая
66 Ализариновый желтый Р; 2-й переход (см. № 16) См. № 16 10,1-12,1 желтая — коричнево-красная
67 Ализариновый желтый PC НО7 \ N=N / \ NO, COONa7 SO3Na 411,26 0,1 в воде 10,1-12,1 светло-желтая — коричнево-красная
68 Ализариновый синий БС (али-заринбляу SA) О | || || | •2NaHSO3 ° о 499,39 0,05 в воде 11,1-13,0 оранжевожелтая — зеленосиняя
69 Тропеолин 0 (золотистожелтый; хризо-ин; резорциновый желтый)Р НО N= N SO3Na ОН 316,27 0,1 в воде 11,1-13,0 желтая — оранжево-коричневая
70 Малахитовый зеленый; 2-й переход (см. № 6) См. № 6 11,5-13,2 голубоватозеленая — нет
610
Новый справочник химика и технолога
Продолжение таблицы 5.2.15
№ пп Индикатор Формула Молярная масса Концентрация, % Интервал pH перехода и окраска индикатора
71 2,4,6-Тринитротолуол СН3 NOi NO, 227,13 0,1 и 0,5 в 90% спирте 11,5-13,2 нет — оранжевая
72 Оранжевый Ж НО N=N NaO3S / \ SO3Na 452,38 0,1 в воде 11,5-14,0 желтая — красная
73 Индигокармин (индигосульфо-нат натрия) Na°3S\^\C° SO3Na 422,38 0,25 в 50% спирте 11,6-14,0 синяя — желтая
74 1,3,5-Тринитро-бензол no2 213,11 0,1 и 0,5 в 90% спирте 12,2-14,0 нет — оранжевая
75 Бензопурпурин 4Б; 2-й переход (см. № 18) См. № 18 13,0-14,0 оранжевая — красная
Таблица 5.2.16
Солевые поправки для важнейших индикаторов при разной ионной силе растворов
(Ионная сила буферных растворов, применяемых для сравнения, равна 0,1) [46]
Индикатор Солевые поправки при значениях ионной силы
0,0025 0,005 0,01 0,02 0,05 0,1 0,5 (КС1) 0,5 (NaCl)
Бромкрезоловый синий +0,21 +0,18 +0,16 +0,14 +0,05 0,00 -0,12 -0,16
Бромтимоловый синий +0,14 +0,12 +0,11 +0,07 +0,04 0,00 -0,20 -0,28
Бромфеноловый синий +0,15 +0,14 +0,14 +0,13 +0,10 0,00 -0,10 -0,18
Метиловый красный 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00
Метиловый оранжевый -0,04 -0,04 -0,02 0,00 0,00 0,00 0,00 0,00
Тимоловый синий:
1-й переход — — 0,00 0,00 0,00 0,00 0,00 0,00
2-й переход — +0,16 +0,12 +0,09 +0,05 0,00 -0,12 -0,19
Тимолфталеин — — +0,11 +0,09 +0,05 0,00 -0,19 —
Феноловый красный +0,14 +0,12 +0,11 +0,07 +0,04 0,00 -0,20 -0,29
Фенолфталеин — +0,18 +0,12 +0,10 +0,05 0,00 -0,16 -0,21
Хлорфеноловый красный — +0,15 +0,13 +0,12 +0,05 0,00 -0,16 -0,19
Химические методы количественного анализа
611
Таблица 5.2.17
Некоторые смешанные индикаторы* [46]
Показатель титрования (pH)** Компоненты Соотношение объемов Окраска индикатора Примечание
в кислой среде в щелочной среде
3,25 Метиловый желтый, 0,1% раствор в этиловом спирте Метиленовая синь, 0,1% раствор в этиловом спирте 1:1 Синефиолетовая Зеленая Зеленая при pH = 3,4; сине-фиолетовая при pH = 3,2; сохранять в темной склянке
4,1 Метиловый оранжевый, 0,1% раствор в воде Индигокармин, 0,25% раствор в воде 1:1 Фиолетовая Зеленая Особенно удобен для титрования при искуст-венном освещении; сохранять в темной склянке
4,3 Бромкрезоловый синий, натриевая соль, 0,1% раствор в воде Метиловый оранжевый, 0,2% раствор в воде 1:1 Желтая Сине-зеленая —
5,1 Бромкрезоловый синий, 0,1% раствор в воде Метиловый красный, 0,2% раствор в этиловом спирте 3:1 Виннокрасная Зеленая Желтая при pH = 3,5, зеленовато-желтая при pH = 4,05; слабо-зеленая при pH = 4,3. Очень резкое изменение окраски
5,4 Метиловый красный, 0,2% раствор в этиловом спирте Метиленовая синь, 0,1% раствор в этиловом спирте 1:1 Краснофиолетовая Зеленая Красно-фиолетовая при pH = 5,2; грязно-синяя при pH = 5,4; грязно-зеленая при pH = 5,6; сохранять в темной склянке
6,1 Бромкрезоловый синий, натриевая соль, 0,1% раствор в воде Хлорфеноловый красный, натриевая соль, 0,1% раствор в воде 1:1 Желто-зеленая Синефиолетовая Сине-зеленая при pH = 5,4; синяя при pH = 5,8; сине-фиолетовая при pH = 6,2
6,7 Бромкрезоловый пурпуровый, натриевая соль, 0,1% раствор в воде Бромтимоловый синий, натриевая соль 0,1% раствор в воде 1:1 Желтая Синефиолетовая Желто-фиолетовая при pH = 6,2; фиолетовая при pH = 6,6; сине-фиолетовая при pH = 6,8
7,0 Нейтральный красный, 0,1% раствор в этиловом спирте Метиленовая синь, 0,1% раствор в этиловом спирте 1:1 Фиолетовосиняя Зеленая Фиолетовая при pH = 7,0 сохранять в темной склянке
7,2 Нейтральный красный, 0,1% раствор в этиловом спирте Бромтимоловый синий, 0,1% раствор в этиловом спирте 1:1 Розовая Зеленая Розовая при pH = 7,0
7,5 Бромтимоловый синий, натриевая соль, 0,1% раствор в воде Феноловый красный, натриевая соль, 0,1% раствор в воде 1:1 Желтая Фиолетовая —
8,3 Крезоловый красный, натриевая соль, 0,1% раствор в воде Тимоловый синий, натриевая соль, 0,1% раствор в воде 1:3 Желтая Фиолетовая —
8,9 а-Нафтолфталеин, 0,1% раствор в этиловом спирте Фенолфталеин, 0,1% раствор в этиловом спирте 1:3 Бледно-розовая Фиолетовая Бледно-фиолетовая при pH = 8,2; ярко-фиолетовая при pH = 8,4
612
Новый справочник химика и технолога
Продолжение таблицы 5.2.17
Показатель титрования (рнГ Компоненты Соотношение объемов Окраска индикатора Примечание
в кислой среде в щелочной среде
9,0 Тимоловый синий, 0,1% раствор в 50% этиловом спирте Фенолфталеин, 0,1% раствор в 50% этиловом спирте 1:3 Желтая Фиолетовая От желтой через зеленую к фиолетовой
9,9 Фенолфталеин, 0,1% раствор в этиловом спирте Тимолфталеин, 0,1% раствор в этиловом спирте 1:1 Нет Фиолетовая Розовая при pH = 9,6; фиолетовая при pH = 10
10,2 Тимолфталеин, 0,1% раствор в этиловом спирте Ализариновый желтый Р, 0,1% раствор в этиловом спирте 2:1 Желтая Фиолетовая Фиолетовая при pH = 10
Примечания.
‘Индикаторы хранят в склянках из темного стекла.
**Показатель титрования — значение pH, при котором наблюдатель отчетливо отмечает изменение окраски индикатора и признает титрование законченным. Это несколько условная величина, неодинаковая у разных лиц, проводящих титрование.
Таблица 5.2.18
Универсальные индикаторы [46]
Действие Окраска pH
В 500 мл 96% спирта рас-творяют 100 мг фенолфта-леина, 200 мг метилового красного, 300 мг метило-вого желтого, 400 мг бромтимолового синего и 500 мг тимолового синего, затем прибавляют 0,1 М р-р гидроксида натрия до появления чисто-желтой окраски (pH = 6) Красная 2,0
Оранжевая 4,0
Желтая 6,0
Зеленая 8,0
Синяя 10,0
Смешивают 15 мл 0,1% р-ра метилового желтого, 5 мл 0,1% р-ра метилового красного, 20 мл 0,1% р-ра бромтимолового синего, 20 мл 0,1% р-ра фенолфта-леина и 20 мл 0,1% р-ра тимолфталеина Розовая 1,0
Красно-оранжевая з,о
Оранжевая 4,0
Желто-оранжевая 5,0
Лимонно-желтая 6,0
Желто-зеленая 7,0
Зеленая 8,0
Сине-зеленая 9,0
Фиолетовая 10,0
Продолжение таблицы 5.2.18
Действие Окраска рн
В 100 мл 50% спирта рас- Оранжево-красная 0,2
Красно-оранжевая 0,3
Оранжевая 0,4
Желто-оранжевая 0,5
Оранжево-желтая 6,0
Желтая 6,5
Зелено-желтая 7,0
Зеленая 8,0
Зелено-голубая 9,0
Сине-фиолетовая 9,5
Фиолетовая 10,0
Краснофиолетовая 12,0
В 500 мл 96% спирта рас-творяют 100 мг метилово-го красного, 100 мг бром-тимолового синего, 100 мг а-нафтолфталеина, 100 мг фенолфталеина и 100 мг тимолфталеина Красная 4,0
Оранжевая 5,0
Желтая 6,0
Зелено-желтая 7,0
Зеленая 8,0
Сине-зеленая 9,0
Сине-фиолетовая 10,0
Краснофиолетовая 11,0
Химические методы количественного анализа
613
Таблица 5.2.19
Флуоресцентные индикаторы для ацидиметрии и алкалиметрии [42, т. 2]
Название Формула Изменение окраски Область pH Раствор индикатора
Бензофлавин сн’ Желтая/зеленая 0,3-1,7 1% в этаноле
3,6-Диоксифталимид ОН J^^^co Г |Г NH он Синяя/зеленая Зеленая/желто-зеленая 0-2,4 6,0-8,0 То же
Эозин (тетрабром-флуоресцеин) Вг Вг o^J^^ он Br c/Z\X\ Br J^^COOH Не флуоресцирует/ зеленая 0-3,0 1% водный раствор соли натрия
4-Этоксиакридон О II H OC2H5 Зеленая/синяя 1,2-3,2 1% в этаноле
3,6-Тетраметилдиами-ноксантон О (CH,)2N^^^O^^^N(CH2)2 1,2-3,4 То же
Эскулин ттт Голубая/ярко-синяя 1,5-2,0
Антраниловая кислота NH2 ^^^^СООН Не флуоресцирует/ голубая Голубая/темно-синяя Темно-синяя/ не флуоресцирует 1,5-3,0 4,5-6,0 12,5-14 0,1% в 50% этаноле
614
Новый справочник химика и технолога
Продолжение таблицы 5.2.19
Название Формула Изменение окраски Область рн Раствор индикатора
3-Амино-1 -нафтойная кислота соон He флуоресцирует/ зеленая Зеленая/синяя Синяя/не флуоресцирует 1,5-3,0 4,0-6,0 11,6-13,0 0,1% раствор сульфата в 50% этаноле
1 -Нафтиламино-6-сульфамид nh2 XXlj Не флуоресцирует/ зеленая Зеленая/не флуоресцирует 1,9-3,9 9,6-13,0 0,05% в 90% этаноле
1 -Нафтиламино-7-сульфамид nh2 Не флуоресцирует/ зеленая Зеленая/не флуоресцирует 1,9-3,9 9,6-13,0 То же
2-Нафтил амино-6-сульфамид X XX H2NO2S'^^/^x^ Не флуоресцирует/ темно-синяя Темно-синяя/не флуоресцирует 1,9-3,9 9,6-13,0 0,05% в 90% этаноле
2-Нафтиламино-8-сульфамид so2nh2 Не флуоресцирует/ голубая Голубая/не флуоресцирует 1,9-3,9 9,6-13,9 То же
1 -Нафтиламино-5-сульфамид nh2 so2nh2 Не флуоресциру-ет/желто-оранжевая Желто-оранжевая/не флуоресцирует 2,0-4,0 9,5-13,0 То же
1-Нафтойная кислота hooc Не флуоресцирует/ синяя 2,5-3,5 Раствор соли натрия в дистиллированной воде
Салициловая кислота l X ^^^COOH Не флуоресцирует/ темно-синяя 2,5-Л,0 0,5% водный раствор соли натрия
Химические методы количественного анализа
615
Продолжение таблицы 5.2.19
Название Формула Изменение окраски Область pH Раствор индикатора
Флоксин ВА экстра (тетрахлортетрабром флуоресцеин) Вг Вг 1°Н 1 ° Ck^Jl^C=O ci^^^ci Cl Не флуоресцирует/ темно-синяя 2,5-4,0 0,1% в этаноле
Эритрозин В (тетра-иодфлуоресцеин) 1 I он 1 0 А^с=о Не флуоресцирует/ светло-зеленая 2,5-4,0 0,2% раствор соли натрия в дистиллированной воде
2-Нафтиламин Не флуоресцирует/ фиолетовая 2,8-4,4 0,5% в этаноле
Магдала красный иЭ Не флуоресцирует/ пурпурная 3,0-4,0
я-Аминофен и л бензосульф амид h2n SO2NH2 Не флуоресцирует/ голубая 3,0-4,0 0,05% в 90% этаноле
2-Окси-З -нафтойная кислота ОН Голубая/зеленая 3,0-6,8 0,1% раствор соли натрия в дистиллированной воде
Хромотроповая кислота но он HOjS/^^^^X' SO,H Не флуоресцирует/ голубая 3,1^1,4 5% водный
616
Новый справочник химика и технолога
Продолжение таблицы 5.2.19
Название Формула Изменение окраски Область рн Раствор индикатора
1-Нафтионовая кислота „ Z X Я g // \ О % С Э Не флуоресцирует/ синяя Синяя/желто-зеленая 3,0-4,0 10-12 Раствор соли натрия в воде
1-Нафтил амин nh2 Не флуоресцирует/ синяя 3,4—4,8 0,5% в этаноле
5 - Амино салициловая кислота ОН ^^^соон Не флуоресцирует/ светло-зеленая 3,1-4,4 Свежий 0,2% в этаноле
Хинин Н N СН2 ОН— С— СН 4 (СН2)2^ сн— сн= сн2 сн2— сн ГТ Т Синяя/светло-фиоле-товая Светло-фиолетовая/ не флуоресцирует 3,0-5,0 9,5-10,0 0,1% в этаноле
о-Метоксибензальде-гид сно Jx^^OCH, Не флуоресцирует/ зеленая 3,1-4,4 0,2% водный
о-Фенилендиамин 1X Зеленая/не флуоресцирует 3,1-4,4 0,2% в 70% этаноле
«-Фенилендиамин nh2 nh2 Не флуоресцирует/ оранжево-желтая 3,1-4,4 То же
Морин (2',4', 3,5,7-пентаоксифлавон) ОН О X X X /°н он Не флуоресцирует/ зеленая Зеленая/желто-зеленая 3,1—4,4 8-9,8 0,2% в дистиллированной воде
Химические методы количественного анализа
617
Продолжение таблицы 5.2.19
Название Формула Изменение окраски Область рн Раствор индикатора
Тиофлавин S Прямой желтый 7. Метилированный и сульфированный примулин Темно-синяя/голубая 3,1-4,4 0,2% в дистиллированной воде
Флуоресцеин Я О X Розово-зеленая/зеленая 4,0-4,5/6,0 1% раствор соли натрия в дистиллированной воде
Эозин + ксиленоловый цианол FF Не флуоресцирует/ зеленая 4,0-5,0 1% в этаноле и и-бутаноле
Дихлорфлуоресцеин я / ь < Я Сине-зеленая/зеленая 4,0-6,6 1% в этаноле
0-Метилэскулегин ач/° о Не флуоресцирует/ синяя Синяя/светло-зеленая 4,0-6,2 9,0-10,0 То же
Хининовая кислота соон Желтая/синяя 4,0-5,0 Насыщенный водный
0-Нафтохинолин Синяя/не флуоресцирует 4,4-6,3 0,05% в 50% этаноле
Резоруфин (7-оксифеноксазон) Желтая/оранжевая 4,4-6,4 Совсем слабо флуоресцирует
Акридин Зеленая/фиолетовая 5,2-6,6 0,1% в этаноле
3,6-Диоксиксантон 0 II HO'^X^^OH Не флуоресцирует/ сине-фиолетовая 5,4-7,6 1% в этаноле
618
Новый справочник химика и технолога
Продолжение таблицы 5.2.19
Название Формула Изменение окраски Область pH Раствор индикатора
5,7 - Диокси-4-метилку-марин НО О || с=0 ЧАс-сн ОН СНз Голубая/темно-синяя 5,5-5,8
Динитрил-3,6-диокси-фталевой кислоты ОН 1 I ОН Синяя/зеленая 5,8-8,2 1% в этаноле
1,4-Диоксибензо-дисульфокислота ОН ho3S'x^4Vx^ он Не флуоресцирует/ голубая 6-7 0,1% раствор соли калия в дистиллированной воде
Люминол nh2 Jx^^co—NH ^^^CO—NH Не флуоресцирует/ синяя 6-7
2-Нафтол-6-сульфо-кислота(кислота Шеффера) HO3S/X^4^x^X'^ Не флуоресцирует/ синяя 5/7—8/9 Водный раствор соли натрия
Хинолин Синяя/не флуоресцирует 6,2-7,2 Насыщенный водный
1 -Нафтол-5-сульфо-кислота(кислота Клеве) OH SO3H Не флуоресцирует/ зеленая 6,5-7,5 Водный раствор соли натрия
Умбеллиферон Не флуоресцирует/ синяя 6,5-8,0
8-Оксихинолинат магния ?? ' - Mg ®l/2 Не флуоресцирует/ желтая 6,5-7,5 0,1% раствор комплекса в 0,01 М НС1
Химические методы количественного анализа
619
Продолжение таблицы 5.2.19
Название Формула Изменение окраски Область pH Раствор индикатора
Орсинаурин ОН J—СН3 НЧ СН3 He флуоресцирует/ зеленая 6,5-8,0 0,03% в дистиллированной воде
Диазобриллиантовый желтый He флуоресцирует/ синяя 6,5-7,5
Кумариновая кислота он Js^^CH= сн—соон Не флуоресцирует/ зеленая 7,2-9,0 1% в этаноле
Р-Метилумбеллиферон 1 °\ 0 СН3 Не флуоресцирует/ синяя >7,0 0,3% в 50% этаноле
Гармин z Z // \ я # \\ и г—я и я" Синяя/желтая 7,2-8,9
2-Нафтол-6,8-дисуль-фокислота(G-кислота) SO3H JL^^/^^OH Синяя/голубая 7,5-9,1 Водный раствор соли натрия
Салицил альдегидсемикарбазон он СН= N • NH CONH; Желтая/синяя 7,6-8,0 0,1% спиртовый
1 -Нафтол-2-сульфо-кислота OH ^чЛ/8о3н Т емно-синяя/го лу бая 8,0-9,0 Водный раствор соли натрия
Салицил альдегидаце-тилгидразон OH CH= N NH COCH, Не флуоресцирует/ зелено-синяя 8,3 0,1% в этаноле
620
Новый справочник химика и технолога
Продолжение таблицы 5.2.19
Название Формула Изменение окраски Область pH Раствор индикатора
Салицилальдегидтио-семикарбазон он сн= N • NH • CSNHj Не флуоресцирует/ сине-зеленая 8,4 То же
1 -Нафтол-4-сульфо-кислота (кислота Невиля — Винтера) ОН SO3H Темно-синяя/голубая 8,2 Водный раствор соли натрия
Нафтол AS Продукт соединения тетраазотированного о-дианизидана с анилидом З-окси-2-наф-тойной кислоты Не флуоресцирует/ желто-зеленая 8,2-10,3 То же
2-Нафтол Не флуоресцирует/ синяя 8,5-9,5 0,1% в этаноле
Акридиновый оранжевый (CH3)2N^^ Не флуоресцирует/ желто-зеленая 8,4-10,4 1% в этаноле
Орсинсульфофталеин Не флуоресцирует/ желтая 8,6-10,0
2-Нафтол-3,6-дисульфокислота (R-кислота) .х^^^^ОН SO3H Темно-синяя/голубая 9,0-9,5 Водный раствор соли натрия
Этоксифенилнафто-стильбазония хлорид /==\ / \— 'n • сГ н,с,о^ '^=/ Зеленая/не флуоресцирует 9-11 1% в этаноле
о-Оксифенил бензотиазол ( Уч 1 J он Не флуоресцирует/ сине-зеленая 9,3 0,1% в этаноле
о-Оксифенил бензоксазол он Не флуоресцирует/ сине-фиолетовая 9,3 То же
Химические методы количественного анализа
621
Продолжение таблицы 5.2.19
Название Формула Изменение окраски Область pH Раствор индикатора
о-Оксифенилбензими-дазол < Hl J он He флуоресцирует/ сине-фиолетовая 9,9 То же
Кумарин А 1 II с=0 Не флуоресцирует/ светло-зеленая 9,5-10,5
6,7-Диметоксиизохи-нолин-1 -карбоновая кислота X. 1 ,N СН3(Г COOH Желтая/синяя 9,5-11,0 0,1% в глицерине, разбавленном равным объемом этанола и 9 объемами дистиллированной воды
1 -Нафтиламино-4-сульфамид NH; SO;NH; Темно-синяя/светло-синяя 9,5-13,0 0,05% в 90% спирте
1 -Амино-8-оксинафта-л ин-5,7-дисул ьфокис-лота OH nh2 SO3H Фиолетовая/желто-зеленая 10-12 Водный раствор соли натрия
Котарнин П . CH=CH—Nil CH\ 1 СНз о^^^И\СН0 Желтая/белая > 12,5
2-Нафтионовая кислота SO,H Синяя/ фиолетовая 12-13 1% водный раствор соли натрия
2-Нафтиламино-6,8-дисульфокислота SO3H НОзЗ^4^^^^ Синяя/желто-розовая 12-14 1% водный раствор соли натрия
622
Новый справочник химика и технолога
Погрешность титрования [1-3, 5,14,38]
При титровании возможны случайные погрешности, которые связаны с измерением объема и массы навески, и систематические, связанные с несовпадением ТЭ и КТТ. Случайные погрешности обрабатываются по законам математической статистики. Систематические погрешности могут быть положительными (перетитрование) и отрицательными (недотитрование). Оценку систематической погрешности проводят, как правило, расчетным способом [1-3, 4, 14]. Ее величина обычно колеблется от сотых до нескольких десятых процента в зависимости от природы, концентрации титранта, титрируемого вещества и интервала перехода индикатора (точнее, значения рТ).
Однако при неправильно выбранном индикаторе величина систематической погрешности может составить целые проценты [3].
Для уменьшения систематической погрешности рекомендуется использовать контрольный раствор (называемый также «холостым» или «свидетелем») (см. 5.2.5).
Стандартные растворы
Общие схемы, по которым проводится стандартизация растворов, независимо от метода титрования, изложены в 5.2.4. В практическом руководстве [47] даны методики стандартизации растворов кислот и оснований и расчет их концентраций с помощью поправочного коэффициента К. Ниже рассматриваются часто применяемые варианты, взятые из [3].
В методе кислотно-основного титрования в качестве титрантов обычно применяют разбавленные растворы сильных кислот и оснований 0,01-0,1 М, которые готовят, разбавляя концентрированные растворы. Они должны быть стандартизированы с помощью первичных стандартов.
Для стандартизации растворов сильных кислот (НС1, HNO3, H2SO4, НС1О4) применяют карбонат натрия Na2CO3, тетраборат натрия Na2B4O7 • ЮН2О и 3-(гидроксиметил) аминометан (СН2ОН)3—CNH2. При титровании растворов этих первичных стандартов протекают следующие реакции:
Na2CO3 +2НС1 > 2NaCl+H2O+CO2,
Na2B4O7 +2НС1+5Н2О 4Н3ВО3 +2NaCl,
(CH2OH)3CNH2 +НС1 <--> (CH2OH)3CNH3C1.
Для стандартизации растворов сильных оснований NaOH, КОН, Ва(ОН)2 используют о-гидрофталат калия ЮДСООДСДЦ, бензойную кислоту СбН5СООН, щавелевую кислоту Н2С2О4-2Н2О, сульфаминовую кислоту NH2SO3H, янтарную кислоту С2Н4(СООН)2.
Твердый гидроксид натрия (или калия) всегда содержит влагу и до 2% карбоната и хлорида натрия. Поэтому сначала готовят раствор гидроксида натрия приблизительной концентрации, а затем стандартизируют его. Не обязательно стандартизировать и кислоту, и щелочь по первичному стандарту.
Приготовление 0,1 М раствора кислоты. Приготовление стандартных растворов является очень ответственной операцией. В качестве стандартного раствора кислоты используют преимущественно 0,1 М раствор НС1, реже — H2SC>4 и HCIO4. Исходной обычно является хлороводво-родная кислота марки «ч.д.а» 30-33% (масс.), плотностью 1,15-1,19 гем 3. Точную навеску кислоты из-за летучести НС1 взять затруднительно. Поэтому сначала готовят раствор приблизительной концентрации, который' затем стандартизируют по первичному стандарту.
Ниже приведен расчет количества хлороводвородной кислоты, необходимого для приготовления 1 л 0,1 М раствора НС1.
Измерить ареометром плотность кислоты (предположим, р = 1,16 г мл-1) и по справочной таблице найти соответствующую процентную концентрацию НС1 (для нашего случая со% = 32,147). Объем исходной кислоты КНсь необходимый для приготовления 1 л 0,1 М раствора НС1, рассчитывают по уравнению
Л(НС1) = С(НС1)‘Гнс1 = Гнс1 •₽•«>%
1 ; 1000 М(НС1)100’
откуда
v _с(НС1)Гнс1М(НС1)100 нс1- со%-1000
Для нашего случая
V =
*НС1
0,1 1000 36,46 100 (32,14-1,16-1000)
= 9,77 мл.
Рассчитанный объем кислоты отмерить мерным цилиндром и вылить в склянку, содержащую 250-300 мл дистиллированной воды (под тягой), затем долить дистиллированную воду до 1 л и, закрыв пробкой, перемешать.
Стандартизацию (установку характеристик) приготовленного раствора НС1 проводят при помощи установочных веществ. Установочные вещества должны удовлетворять требованиям, предъявляемым к первичным стандартам, а также реагировать с НС1 стехиометрически однозначно и практически необратимо; предпочтительно вещество с большим эквивалентом.
Расчет навески Na2B4O710H2O, необходимой для приготовления в объеме мерной колбы (например, 100 мл) стандартного раствора (например, 0,1000 М), исходя из стехиометрии протекающей реакции:
A/(l/2Na2B4O7 -10H2O) = — ^ 37 =190,68,
n(l/2Na2B4O7 -ЮН2О) = и(НС1),
______^На-гВдО, 10Н2О _ С(НС1)Кнс1
A/(l/2Na2B4O7-ЮН2О)" 1000 ’
Химические методы количественного анализа
623
откуда
_ с(НС1) ГНС1 • A/(l/2Na2B4O7 10Н2О)
^Na2B4O7 • 10H2O — 1mn
0,1 100 190,68
1000
= 1,9068 г.
Взятие навески «по разности»: чистый сухой бюкс взвесить на технических весах, затем на правую чашку весов положить разновески по массе, равной массе бюкса + т, насыпать в бюкс шпателем приблизительно т г тетрабората натрия, поставить бюкс на левую чашку весов, включить весы и, постепенно присыпая в него тетраборат, уравновесить их. Затем взвесить бюкс с навеской на аналитических весах (т i), перенести навеску тетрабората натрия в мерную колбу, для чего опрокинуть бюкс над воронкой с укороченным носиком, вставленной в горло мерной колбы. Приподнять воронку, не вынимая ее из колбы, и горячей дистиллированной водой из промывалки смыть с воронки не просыпавшийся в мерную колбу тетраборат натрия, тщательно промыть внутреннюю поверхность и носик воронки. Долить в мерную колбу воды приблизительно до половины. Перемешивая круговым движением колбы ее содержимое, добиться растворения Na2B4O7 • 10Н2О. После остывания раствора до комнатной температуры довести его объем водой до метки, закрыть колбу пробкой и перемешать.
Взвесить на аналитических весах пустой бюкс (щ2). Разность т\ — т2 дает точную массу Na2B4O7 10Н2О, находящегося в мерной колбе. Нет необходимости добиваться того, чтобы масса навески совпадала с расчетной величиной т, как и не следует стремиться к тому, чтобы тетраборат натрия до последней крупинки высыпался из бюкса в мерную колбу. Необходимо не допускать потери даже самого малого количества навески и важно знать ее точную массу.
По взятой навеске тетрабората натрия и вместимости мерной колбы рассчитать с и Т раствора тетрабората натрия.
Характеристики стандартного раствора НС1 рассчитывают по результатам титрования аликвотных частей раствора Na2B4O710H2O раствором НС1.
Для титрования из мерной колбы отобрать пипеткой аликвотные части раствора в чистые конические колбы (пипетка предварительно должна быть ополоснута этим же раствором), добавить 1-2 капли раствора индикатора метилового красного и титровать раствором НС1 до четкого изменения цвета индикатора (желтый — оранжевый).
Из числа приведенных в справочной таблице индикаторов метиловый красный является наиболее подходящим. При титровании 0,1 М раствора тетрабората натрия в момент эквивалентности в растворе будут находиться NaCl и Н3ВО3, для которой Ki = 5,8-1 (Г10, К2иК3 — еще меньше, поэтому диссоциацией борной кислоты по 2-ой и 3-й ступеням можно пренебречь, тогда в точке эквивалентности
[н+] = 7к^с=75,8-1°_10-0,1 = 7,6-10'6 и pH = 5,12.
Отсчет расхода титранта по бюретке должен быть сделан с точностью до сотых долей миллилитра. Это необходимо потому, что погрешность в отсчете объема при расходе титранта 10 мл, равная 0,01 мл, соответствует 0,1%, а объем капли, вытекающей из бюретки, равен 0,03-0,04 мл.
Необходимо протитровать 5-7 проб. Разница в объемах любой пары титрований не должна превышать 0,05 мл. Результаты всех титрований записать в виде таблицы с графами: ^а2в4о7 юн2о» ^нсь По результатам титрования найти средний объем расхода титранта РНС|, рассчитать характеристики стандартного раствора НС1. На основе закона эквивалентности n(HCl) = n(l/2 Na2B4O7 -ЮН2О)
_ c(l/2Na2B4O7 -10H2O)KNaB 10Н2О z» 1 Hl Л I — ----------—----------------.
j, _ ^Na2B4O710H2O^Na2B4O710H,O^(HCl)
"С1 ' [Г„С]Л/(1/2№2В4О, -10Н2О)] '
Для проверки правильности расчета вычислить
с(НС1)М(НС1)
1000
который должен совпадать с вычисленным ГНа по Т
2Na2B4O7 ЮН2О •
В рабочем журнале должны быть записаны: название работы, на чем основано определение, уравнения реакций, краткая методика выполнения, названия всех операций, для каждой операции— расчетные выражения в общем (формулы) и числовом виде, результаты измерений.
Приготовление 0,1 М раствора щелочи
В качестве стандартного раствора щелочи используют преимущественно 0,1 М раствор NaOH (или КОН), реже — Ва(ОН)2.
Стандартный раствор щелочи не может быть приготовлен как первичный стандарт, так как она очень гигроскопична и поглощает СО2 из атмосферы.
Удобнее всего готовить раствор NaOH (или КОН) из его концентрированного (приблизительно 40%) раствора, который практически не содержит карбоната, т.к. в нем Na2CO3 нерастворим, и если карбонат содержался в исходном реактиве, то он выпадает в осадок.
* По аналогичной схеме проводится стандартизация раствора по безводной соде марки х.ч. или ч.д.а, которая предварительно высушена до постоянного веса. В этом случае реакция титрования: Na2CO3 + 2НС1 = 2NaCl + СО2 + Н2О и, согласно принципу эквивалентности:
n(l/2Na2CO3) = n(HCl), откуда с(НС1) = •
^НС1
Титрование проводят в присутствии индикатора метилового оранжевого до четкого изменения окраски от желтой к оранжево-розовой.
624
Новый справочник химика и технолога
В этом случае раствор NaOH готовят аналогично приготовлению раствора НС1.
Внимание! При работе с концентрированным раствором NaOH необходимо надеть защитные очки, не допускать попадания раствора на кожу во избежание ожога. В случае попадания раствора на кожу немедленно промыть водой пораженный участок, затем разбавленным (3%) раствором уксусной кислоты и снова водой.
Приготовленный раствор NaOH стандартизируют по установочным веществам.
Раствор первичного стандарта в этом случае готовят методом, аналогичным приготовлению раствора для стандартизации раствора НС1. Кроме стандартизации «способом пипетирования» можно использовать «способ отдельных навесок». В этом случае берут 3-5 навесок установочного вещества, переносят их непосредственно в колбы для титрования, растворяют в 10-15 мл воды, добавляют индикатор и титруют. По результатам каждого титрования рассчитывают характеристики раствора и находят их среднее значение. Способ отдельных навесок точнее, но длительней. Например, стандартизуют 0,1 М раствор щелочи, установочное вещество — бензойная кислота, желательно затрачивать на титрование навески 10 мл раствора NaOH.
Исходя из стехиометрии реакции:
С6Н5СООН + NaOH <~z± C6H5COONa + Н2О,
/;КВ(С6Н5СООН) = 1, М(С6Н5СООН) = 122,1, тогда
и(С6Н5СООН) = «(NaOH),
w с6н5соон _ с ( NaOH ) CNa0H
Л/(С6Н5СООН) 1000
откуда
_ C(NaOH) FN,o„ М(С6Н5СООН) _
т с6н5соон- 1ппп
Чтобы выбрать индикатор, нужно рассчитать скачок титрования и pH раствора в момент эквивалентности.
В отдельных случаях характеристики раствора NaOH можно установить по результатам титрования аликвотных частей раствора щелочи стандартным раствором НС1 см. раздел 5.2.4.4. Но в этом случае погрешность в характеристиках раствора NaOH будет больше.
Кислотно-основное титрование в неводных средах [1-32, 47]
Теория, техника, многочисленные примеры кислотноосновного титрования в неводных средах подробно изложены в монографиях [20, 30-32].
Эти методы имеют следующие достоинства [1,47]:
1. Титрование в неводных растворах позволяет определять органические и неорганические вещества, а также смеси, состоящие из различных компонентов (смесей солей, кислот и оснований), которые при титровании в водной среде не дают четких конечных точек титрования.
2. В неводных средах можно титровать соединения, которые нерастворимы в воде, разлагаются водой или образуют с ней стойкие, нерасслаивающиеся эмульсии.
3. В неводных средах можно титровать как бесцветные, так и окрашенные растворы.
4. Титрование неводных растворов можно осуществлять индикаторным, потенциометрическим, кондуктометрическим, амперометрическим и другими физико-химическими методами. Их использование возможно без предварительного отделения анализируемых веществ от сопутствующих примесей.
Однако для применения титрования в неводных средах необходимы тщательное обезвоживание реактивов и защита реактивов и приготовленных растворов от попадания влаги как при титровании, так и при их хранении.
В неводном титровании применяют те же индикаторы, что и при обычном титровании.
Для титрования слабых кислот рекомендуют основные растворители — этилендиамин, бутиланин, пиридин, ди-метилформамид, смесь метанола с бензолом, третичный бутанол.
Титрантом может служить основание, достаточно сильное в данном растворителе. Как правило, берут гидроксид тетрабутиламмония (C4H9)4NOH, хорошо диссоциирующий и растворимый во многих растворителях.
Для титрования слабых оснований применяют растворители с кислотными свойствами. Наиболее часто используемым растворителем является ледяная (безводная) уксусная кислота. Титрантом служит хлорная кислота в уксусной кислоте или диоксане. В ледяной СН3СООН титруют не только основания, но и амфотерные соединения, например, аминокислоты.
И кислоты, и основания можно титровать в универсальных инертных растворителях, таких как метилизобу-тилкетон и метилэтилкетон.
В инертных растворителях проявляется дифференцирующий эффект, что позволяет раздельно титровать вещества с близкими константами. Титрантами служат гидроксид тетрабутиламмония, метилат натрия и т.д.
Для раздельного определения веществ в смеси рекомендуют также титрование в смешанных средах: вода-ацетон, вода-этанол.
Для фиксирования ТЭ применяют визуальные методы (табл. 5.2.20-5.2.22), но преимущественно — потенциометрические методы. Интервалы переходов окраски цветных индикаторов сильно изменяются в неводных средах (табл. 5.2.21).
Химические методы количественного анализа
625
Таблица 5.2.20
Цветные индикаторы для неводных средств [1]
Титруемое вещество Растворитель Индикаторы Изменение окраски
Основание Уксусная кислота (безводная), ацетонитрил, диметил-формамид Метиловый фиолетовый Метиловый красный Фиолетовый/ сине-зеленый Фиолетовый/ сине-зеленый
Кислота Бензол, СНСТ спирты, этилендинамин, диметил-формамид Фенолфталеин, Тимолфталеин, тимоловый синий о-нитроанилин Желтый/синий Желтый/оран-жевый
Таблица 5.2.21
Сравнение интервалов перехода индикаторов в ацетоне, содержащем 10% воды, и в чистой воде
Индикатор Интервалы перехода, pH
в ацетоне в воде
Феноловый красный 11,0-13,0 6,4-8,0
Бромтимоловый синий 11,4-12,8 6,2-7,6
Розоловая кислота 10,5-12,5 6,8-8,0
Бромкрезоловый пурпуровый 9,6-11,1 5,2-6,8
Бромкрезоловый зеленый 8,3-9,8 3,8-5,4
Бромфеноловый синий 6,5-8,3 3,0-4,6
.w-Крезоловый пурпуровый 2,8-4,5 1,2-2,8
Тимоловый синий 2,4-4,0 1,2-2,8
Метиловый красный 1J-3,7 4,4-6,2
Метиловый оранжевый 1,0-2,7 3,1-4,4
Метиловый желтый 0,5-2,5 2,9-4,0
Таблица 5.2.22
Важнейшие индикаторы, применяемые для кислотно-основного титрования в неводных растворах [55]
Индикатор Растворитель Концентрация, % Изменение окраски Определяемые вещества
Азофиолетовый [4-(и-нитрофенил)-азорезорцин] Диметилформамид, пиридин, бензол 0,1 Оранжевая/ голубая Фенолы, енолы, имиды
и-Аминоазобензол Хлороформ 0,1 Желтая/красная Резорцин
Бензоилаурамин Уксусная кислота 0,1 Желтая/серо-фиолетовая Аминокислоты
Бриллиантовый крезоловый синий То же 0,1 Красная/синяя То же
Бромкрезоловый зеленый Хлороформ 0,5 Зеленая/бесцветная Первичные и вторичные аминокислоты
Бромтимоловый синий Пиридин, уксусная кислота, акрилонитрил 0,1 Желтая/голубая Органические кислоты
Бромфеноловый синий Хлорбензол, хлороформ, этанол 0,1 Фиолетовая/желтая Амины, алкалоиды, сульфамидные препараты
Конго-красный Диоксан, метанол 0,1 Красная/синяя Амины
Кристаллический фиолетовый Уксусная кислота 0,1; 0,2 Г олубая/зеленая Алкалоиды, аминокислоты
Малахитовый зеленый То же 0,1; 0,5 Бесцветная/голубая Алкалоиды
Метиловый желтый Хлороформ 0,1 Желтая/красная Амины, никотин
Метиловый красный Хлороформ, ацетонитрил (насыщенный раствор), метанол, уксусная кислота 0,05-0,1 Желтая/красная Кофеин, кофеин-фосфат
Метиловый оранжевый Гликоль, углеводороды, ацетон, метанол 0,1; 0,25 Желтая/малиновая Наркотики группы морфина
Метиловый фиолетовый Уксусная кислота, хлорбензол 0,1-1,0 Зеленая/желтая Производные антипирина
2-Нафтолбензеин Уксусная кислота, изопропиловый спирт, бензол - метанол 0,1-0,5 Желтая/зеленая Аминокислоты, хинин
Нильский голубой Бензол - метанол 0,1 Красная/синяя Дифенилфосф ат
Нитрофенолы Хлорбензол, анизол 0,1 Желтая/ бесцветная Основания
626
Новый справочник химика и технолога
Продолжение таблицы 5.2.22
Индикатор Растворитель Концентрация, % Изменение окраски Определяемые вещества
н-Оксиазобензол Ацетон 0,1 Бесцветная/желтая Карбоновые кислоты
Пикриновая кислота Этилендиамин 0,1 Желтая/бесцветная Фенолы
Тимоловый синий (тимолсульфофталеин) Диметилформамид, метанол, этиленгликоль 0,1; 0,3 Желтая/синяя Соли аммония, ацетилсалициловая кислота, алкалоиды, найлон
Тимолфталеин Пиридин 0,1 Светло-желтая/голубая Амины, ацетилен
Тропеолин 00 Уксусная кислота; метанол; ацетон + пиридин 0,1-1,0 Желтая/фиолетовая Алкалоиды, кофеин, салицилаты
Феноловый красный Этанол 0,1 Красная/желтая Муравьиная кислота
Фенолфталеин Этанол + бензол; этанол + пиридин 0,1 Бесцвет-ная/малиновая Жирные кислоты, сульфамиды, барбитуровая кислота
Четкость перехода окраски при неводном титровании зависит от концентрации индикатора, силы кислот и оснований, концентрации ионов и от изменения состава растворителя в процессе титрования.
В неводном титровании находят применение смешанные индикаторы. Например, растворы 0,1% метилового синего и 0,2% хинальдинового красного в метаноле с соотношением 1 : 1 (при титровании в смеси нитрометана с бензолом из щелочной среды в кислую переход окраски: пурпурная —* синяя —> зеленая).
5.2.6.2. Комплексонометрическое титрование
Основы метода
Теоретические основы изложены в учебниках и монографиях [1-6, 14, 33-37, 39], а практическая сторона в [33-36, 54-61]. Особо следует отметить две работы: монография Р. Пршибла [34] посвящена прикладной комплексоно-метрии, в монографии Юрист И.М. и Талмуд М.М. [36] систематизирован огромный материал по комплексоно-метрии, обсуждены методы селективного комплексонометрического титрования с применением металлохромных индикаторов, дано их теоретическое обоснование и приводятся практические примеры анализов.
Именно этот материал, прежде всего [36], положен в основу практической части настоящего раздела.
Титрование с использованием полидентатных органических реагентов называется комплексонометрией.
Комплексонометрия является основным методом экспрессного и точного количественного определения почти всех ионов металлов и ряда анионов при достаточно высоких содержаниях их в анализируемых объектах.
Комплексонами называют органические реагенты, содержащие одну или несколько аминодикарбоксильных групп— N(CH3COOH)2. Среди многочисленных коплек-сонов для метода комплексонометрического титрования важнейшим является этилендиаминтетрауксусная кислота (комплексон II)
ноосн2с^__СН2—СН2_ + ^сн2соо "ООСН2С^ СН2СООН
Анион обычно обозначают символом Y или Y4, а формулу кислоты и анионов соответственно: H4Y, H3Y~, 1TY2, HY3~, Y4 . Относительное содержание каждой формы зависит от pH раствора. Доминирующими формами* в зависимости от pH являются:
H4Y <=> H+ + H3Y“ <=> 2Н+ + H2Y2~ <=> ЗН+ + pH 2-3 3-6
+ HY3'
7-9
4Н+ + Y4
>10
На практике используют хорошо растворимую соль Na2H2Y • 2Н2О (комплексон III, ЭДТА).
С ионами металлов, начиная с двухзарядных, ЭДТА образует циклические комплексы соединения— хелаты, называемые комплексе нотами.
Схематические реакции комплексона III с ионами металлов разного заряда в умеренно кислой среде (pH = 4-5) можно представить уравнениями:
H2Y2~ + М2+ <—» MY2 + 2Н+,
H2Y2~ + М3+ MV + 2Н1,
H2Y2 + М4' <=» MY + 2Н+,
а в умеренно щелочных растворах:
Mz+ + HY3 <=» MY*2-4* + Н+.
Из приведенных уравнений следует, что ЭДТА с ионами металлов всегда реагирует в молярном отношении 1:1.
При pH < 2 доминируют протонированные формы этилендиаминтетрауксусной кислоты Н5Y и H6Y2 .
Химические методы количественного анализа
627
Для обеспечения полноты протекания реакции ионы водорода связывают буферным раствором, который предварительно добавляют к титруемому раствору. При титровании в кислых и близких к нейтральным растворам обычно используют ацетатный, а при титровании в щелочных средах — аммиачный буферный раствор. Конечную точку титрования регистрируют по изменению окраски индикатора, добавленного после введения буферного раствора или инструментально.
Устойчивость комплексонатов металлов характеризуется константой устойчивости (константой образования).
В общем случае реакция образования комплекса ме-талл-ЭДТА записывается уравнением:
Мг+ + Y4 <=> МУ(г^)+ (5.2.58)
или упрощенно:
М + Y = MY. (5.2.58')
Константа устойчивости комплексоната металла рассчитывается из уравнения:
₽MY=[m’*][y4]
или без указания зарядов:
в
Pmv ММ'
В таблице 5.2.23 приведены значения логарифмов концентрационных констант устойчивости комплексонтов металлов, которые отвечают pHmjn, используемым при определении этих катионов на практике.
Таблица 5.2.23
Изменение 1g pMY и pHmill (при 1g pvMY* = 8) комплексонатов в группах периодической системы элементов (I = 0,1; 18-25 °C) [36]
(5.2.59)
(5.2.59')
Подгруппа Катион ЭДТА
IgP pHmin
1Б Си(П) 18,8 2,8
ПА Mg 8,7 10
Са 10,7 7,6
Sr 8,6 9,8
Ва 7,8 12
ПБ Zn 16,5 4,0
Cd 16,5 3,9
Hg(H) 21,8 1,6
IIIA Al 16,1 4,2
Ga 20,3 2,4
In 24,9 1,3
Т1(1П) 22,5 2,0
1ПБ Sc 23,1 1,7
Y 18,1
Продолжение таблицы 5.2.23
Подгруппа Катион ЭДТА
IgP pHmin
П1Б La 15,5 3,3
Ce(III) 16,0 3,1
Pr 16,4 4,2
Nd 16,6 4,2
Gd 17,4
Yb 19,5
Th 23,2 2,6
IVA Sn(II) 18,3
Sn(IV) 39,8 0,18
Pb(II) 18,0 4,3
IVB Ti(IV) 17,7
TiO(II) 17,3
TiO(H2O2)(II) 20,4
Zr 29,5 1
VA Bi 27,9 1
VE VO(II) 18,8 3,1
VO2(I) 18,1
VIE Cr(III) 23,0 1,2
VIIБ Mn(II) 14,0 5,2
Mn(III) 17,4
VIII Fe(II) 14,3 4,7
Fe(III) 25,1 0,9
Co(II) 16,3 4,0
Co(III) 40,7 1
Ni(II) 18,6 2,8
Pd(II) 24,5
Примечание.
* Pmy — условные константы устойчивости.
Кривые титрования рассчитывают теоретически из формул (5.2.59) или (5.2.59') и строят в координатах рМ = =ЛУэдта) или рМ = fix), где рМ = -lg[M], а [М] — равновесная концентрация титруемого иона.
Реальные кривые могут значительно отличаться от теоретических. Обусловлено это тем, что в реальных условиях на равновесие образования комплексонатов (5.2.58), (5.2.58') почти всегда влияют побочные (сопряженные) конкурирующие равновесия: анионы Y4" в зависимости от pH раствора в большей или меньшей степени протонируются, а ионы металлов Mz+ могут образовывать комплексы с подходящими лигандами. Эти побочные равновесия препятствуют образованию комплексонатов, снижают их выход и ослабляют относительную устойчивость. Поэтому в реальных условиях анализа для оценки относительной устойчивости комплексоната металла и получения объективной информации о равновесной концентрации ионов металла используют условную константу устойчивости.
628
Новый справочник химика и технолога
Условная константа устойчивости характеризует равновесие образования комплексоната металла в конкретных реальных условиях при определенном pH раствора и определенной концентрации побочных (вспомогательных) лигандов L.
Влияние концентрации ионов водорода и образования комплексов со вспомогательными лигандами количественно оценивают с помощью молярной доли незакомплексованных ионов Y4- или Мг+. Молярную долю (ау4.) для ионов Y4- рассчитывают по уравнению:
[Y-]
ау4.=Ц-А (5.2.60)
Су
где Су — общая концентрация незакомплексованной ЭДТА. С помощью ау4 вычисляют условную константу устойчивости MY(z при различных значениях pH:
в - Iму] 1му] - |» J. ”У-[М][У]-|М]ау..с'Ч5“'ау
Рму — Рмуау4“ ’ (5.2.61)
Часто при pH, необходимом для проведения титрования, определяемый ион частично или полностью осаждается в виде гидроксида или основных солей. Чтобы предотвратить осаждение, в титруемый раствор добавляют вспомогательный слабый комплексообразующий реагент L. Например, ионы цинка, кадмия, никеля и ряда других металлов титруют в аммиачных растворах. Аммиачный буфер (раствор NH4OH + NH4CI) поддерживает постоянное определенное pH, а аммиак, связывая эти металлы в комплексы, препятствует осаждению гидроксидов. При этом равновесие (5.2.58), (5.2.58') сдвигается влево. В подобных системах полнота протекания реакции титрования, а следовательно, и качество регистрации КТТ зависят не только от pH раствора, но и от концентрации лиганда L, например, от концентрации cNH,.
Для количественной оценки влияния побочного процесса комплексообразования на равновесие (5.2.58), (5.2.58') рассчитывают а — молярную долю свободных (незакомплексованных) ионов металла:
Гм“+1
ам=^_^} (5.2.62)
см
где — общая концентрация металла, не связанного с ЭДТА.
Условную константу устойчивости с учетом влияния комплексообразования металла с вспомогательным лигандом можно рассчитать, умножив ам на константу устойчивости комплекса MY^-4^:
[MY] [MY] , 1
Pmy “ rAxirvi = > Ivl = Рму ’ Рму =Рмуам • (5-2.63)
[Mj[Yj aMcM[Y] ам
Комбинируя уравнения (5.2.61) и (5.2.63), находим выражение для условной константы устойчивости*, описывающей равновесие при определенном значении pH и при определенной постоянной концентрации вспомогательного лиганда:
, [MY]
Рму = ~ ~ ~ aY4” ' ^MY
СМСУ
(5.2.64)
На рис. 5.2.5 показана зависимость логарифмов условных констант устойчивости комплексонатов некоторых металлов от pH раствора с учетом влияния pH на величину ау4_ (левые ветви кривых) и образования гидроксоком-плексов и комплексов с NH3, т.е. из-за уменьшения ам (правые ветви кривых).
0 1 2 3 4 5 6 7 8 Ч 10 II 12 13рН
Рис 5.2.5. Зависимость условных констант устойчивости некоторых комплексонатов от pH (суммарная концентрация [NH3]+[NH;] = l,0M)[l]
В общем виде влияние pH и комплексообразования с компонентами буферных растворов (характеризуемое соответственно ам и ау) на форму кривой титрования показано на рис. 5.2.6; видно, что побочные процессы протонирования и комплексообразования существенно уменьшают величину скачка титрования.
Кривую титрования строят для правильного выбора индикатора, который должен изменять свою окраску в пределах скачка титрования (как можно ближе к ТЭ).
* В уравнении (5.2.64) и в данных табл. 5.2.28 приведенные значения условных констант устойчивости комплексантов не учитывают влияние образующихся смешанных комплексантов (MHY, MOHY, MLY). Расчет полных условных констант устойчивости комплексантов металлов с учетом всех конкурирующих побочных реакций приведен в монографии [14].
Химические методы количественного анализа
629
Рис. 5.2.6. Влияние aM(L) и ау<н) на форму кривой комплексонометрического титрования (сплошная линия). Штриховой линией показана идеальная кривая титрования, когда нет взаимодействия М с L и Y с Н [ 10]
Индикаторы. Принцип их действия
Для регистрации КТТ в комплексонометрии обычно используют металлохромные индикаторы — органические красители, образующие с ионами металла интенсивно окрашенные комплексные соединения (см. табл. 5.2.24). Металлохромные индикаторы должны удовлетворять следующим требованиям:
1) цвет комплекса металла с индикатором Mind должен отличаться от цвета самого индикатора Ind;
2) устойчивость Mind должна быть ниже устойчивости комплексоната MY:
PMInd
где P'MY и PYnild соответственно условные константы комплексоната металла и комплекса металла с индикатором.
Интервал изменения цвета металлохромного индикатора (интервал перехода) определяется из уравнения:
ApM=lgPUd±l- (5.2.66)
Так как P'M[nd зависит от pH раствора, то изменения pH вызывают смещение интервала перехода.
Индикатор может быть использован, если рМтэ попадает в интервал АрМ. Разберем это на примере наиболее распространенного индикатора в комплексонометрии — эри-охромового черного Т (ЭХЧ-Т). ЭХЧ-Т— это натриевая соль сложной трехосновной сульфокислоты, сокращенно NaH2Ind, которая диссоциирует в водном растворе:
NaH2Ind <=> Na+ + H2Ind“
ЭХЧ-Т проявляет свойства pH индикатора и в зависимости от кислотности среды может присутствовать в растворе в различных ионных формах:
H2Ind-красная (pH = 0-6,3)
Hind2' < > Ind3"
синяя желто-оранж.
(pH = 6,3-11,2) ' (pH >11,2)
Большинство титрований с ЭХЧ-Т проводят в аммиачном буферном растворе при pH = 8-10. Основной формой индикатора при этих значениях pH является Hind2 синего цвета, а титрант преобладает в форме HY3-.
С ионами металлов, например, двухзарядными (Zn2+, Mg2+) индикатор образует комплексы вино-красного цвета:
M2++HInd2- <------->MInd +Н+ .
синяя винно-
красная
(5.2.67)
При титровании ЭДТА реагирует сначала со свободными ионами металла (реакция титрования):
М2+ + HY3- <---->MY2 +Н+ , (5.2.68)
бесцв.
а затем и с теми, которые входят в состав комплекса Mlnd-(реакция в КТТ):
Mind" +HY3’ <t >MY2 + Hind2- . (5.2.69)
винно- бесцв. синяя
красная
В результате реакции (5.2.69) индикатор оказывается в свободном состоянии, и в КТТ окраска раствора изменяется от винно-красной (цвет комплекса металла с индикатором) к синей (цвет свободного индикатора).
Реакция (5.2.69) протекает, поскольку устойчивость комплексного соединения Mind- меньше устойчивости комплексоната металла (P^d <0^).
При титровании ионов Zn2+ и Mg2+ с ЭХЧ-Т при pH « 10 наблюдается резкий переход окраски; при титровании Са2+ — нечеткий переход, т.к. рМтэ не попадает в интервал перехода индикатора. Хорошие результаты получаются при титровании Са2+ с ЭХЧ-Т при pH = 11 или с другими индикаторами — например, мурексидом при pH = 12.
Ионы некоторых металлов (Cu2+, Al3+, Fe3+ и др.) образуют с ЭХЧ-Т очень прочные комплексы. Происходит «блокирование» индикатора: реакция типа (5.2.69) не протекает или вдет очень медленно, прямое титрование становится невозможным.
Пригодность данного индикатора для титрования раствором ЭДТА можно определить по изменению рМ вблизи точки эквивалентности, если 0'Mln(i известна. Расчет погрешности комплексонометрического титрования см. [1—3, 5, 14,43].
Таблица 5.2.24
Наиболее распространенные металлохромные индикаторы в комплексонометрии [42,43,45,46]
Индикатор, концентрация Применение: определяемые ионы, условия определения, изменение окраски титруемого раствора
Ион металла pH Изменение окраски Примечание
Ализариновый красный S 1,2 -Диоксиантрахинон-3 -сульфокислота Синонимы: ализариновый S; TR(III) 4—4,5 Красная/желтая Красная/зеленая Ацетатный буфер, нагревание Смешанный индикатор с метиленовым голубым
ализариновый пунцовый; ализаринсульфокислота Sc(III) 2 То же Смешанный индикатор с индигокармином, горячий р-р
О он Th(IV) 4,5-6,5 Розовая/желтая
II /ОН 1,5-3,8 Красная/желтая Р-р HNO3 или ацетатный буфер
III II I Y(III) 5 Розовая/желтая
II о Применяется также при определении А1 (pH = 3-5), Bi (pH = 2-2,8), дартным р-ром Th(IV) обратным титрованием стан-
Рабочий р-р: 0,05 0,2% водный
Ализаринкомплексон Ва(П) 10 Синяя/красная Аммиачный буфер
3-[Ди-(карбоксиметил) аминометил]-1,2- Са(П) 10 То же То же
диоксиантрахинон Cd(II) 10 То же То же
Синонимы: ализаринметилиминодиуксусная кислота; Cu(II) 4,3 Красная/желтая Ацетатный буфер
ализаринфлуориновый синий In(II) 4 То же Ацетатный буфер, нагревание
Pb(II) 4,3 То же Ацетатный буфер
О ОН Sr(II) 10 Синяя/красная Аммиачный буфер
II 1 II II 1 СН2СООН ch2nz чсн2соон Zn(II) 4,3 Красная/желтая Ацетатный буфер
Рабочий р-р: 0,5% водный
Алюминон Ауринтрикарбоновая кислота Синонимы: хромовый фиолетовый; ультрафиолете- Al(III) 4,4 Красная/сине-фиолетовая Ацетатный буфер, нагревание
вый Ca(II) 8,5-9,9 Красно-фиолетовая/желтая Аммиачный буфер
Fe(III) 1-2 Фиолетовая/бесцветная Ацетон'
Mg(II) 8,5-9,9 Красно- Аммиачный буфер
Cu(II) 8,5-9,5 фиолетовая/желтая
630 Новый справочник химика и технолога
Продолжение таблицы 5.2.24
Индикатор, концентрация Применение: определяемые ионы, условия определения, изменение окраски титруемого раствора
Ион металла pH Изменение окраски Примечание
ноос соон НО ^Y^'^COO’ он Рабочий р-р: 0,5% водный
Арсеназо 2-(о-Арсенофенилазо) -1,8-ди оксинафталин- -3,6-ди сульфокислота Синонимы: неоторин; неоторон; уранон AsO3H2 ОН ОН П—N=N— so; Рабочий р-р: 0,1% водный и 0,5% водный Са(П) Mg(II) Pu(IV) TR(III) Th(IV) U(IV) Y(III) 10 10 0,l-0,2MHCl 5,5-6,5 6,5 1,7-3,0 1,7 5,5-6,5 Фиолетовая/красно-оранжевая То же Сине-фиолетовая/розовая Фиолетовая/красно-оранжевая Фиолетовая/оранжевая Фиолетовая/красно-оранжевая Аммиачный буфер То же Пиридин Уротропин Пиридин
Бромпирогаллоловый красный 3', 3-Дибромсульфогаллеин ОН ОН °Н Рабочий р-р: 0,05% в 50% спирте Bi(III) Cd(II) Mg(II) Mn(II) Ni; Co(II) Pb(II) TR(III) 2-3 9,3 10 10 9,3 5-6 7 Винно -красная/желтая Синяя/красная Синяя/фиолетовая То же Синяя/красная Сине-фиолетовая/красная Синяя/красная Р-р HNO3 Аммиачный буфер То же То же То же Ацетатный буфер CH3COONH4
Применяется также при обратном титровании Со, Cu, Ga, In, Pb, Th, Tl, VOZ+ стандартным р-ром Pb(II) при pH = 5 и Bi, Te(III), Ga, In, Tl стандартным р-ром Bi(III) при pH = 2-3
Химические методы количественного анализа
Продолжение таблицы 5.2.24
Применение:
Индикатор, концентрация определяемые ионы, условия определения, изменение окраски титруемого раствора
Ион металла pH Изменение окраски Примечание
Вариаминовый синий В Cd(II) 5 Фиолетовая/бесцветная Ацетатный буфер, Fe(II)/Fe(III)
К-(п-Метоксифенил)-п-фенилендиамин Си(П) 5,5 Фиолетовая/светло-синяя Ацетатный буфер, NH4SCN
1 |1 II 1 Fe(III) 1,7-3 Фиолетово-синяя/желтая Хлор ацетатный буфер
1 JI II J Pb(II) 2-5 Фиолетовая/бесцветная Ацетатный буфер
Zn(II) 5 То же То же Fe(II)/Fe(III)
Рабочий p-p: 0,1-1% водный или 1 : 300 NaCl (HC1 — соль) V(V) 1,7-2 Синяя/бесцветная P-pH2SO4
Галлеин Al(III) 7 Фиолетовая/красная Уротропин, спиртовый горячий
4,5 - Диоксифлуор есцеин р-р
Синонимы: пирогаллол фталеин; галлоцианин Bi(III) 1-2,3 Синяя/желтая Р-р HNO3
Cd(II) 6,8-10 Фиолетовая/красная Аммиачный буфер
ОН ОН Co(II) 7 То же Уротропин
он Ga(III) 2,8 Синяя/красная Р-р СНзСООН
1 II 1 La(III) 5,5-6,5 Фиолетовая/красная Уротропин
Mn(II) 8 То же Аммиачный буфер, аскорбино-
вая к-та
с=о Ni(II) 7 То же Уротропин
1 1 Pb(II) 6 Сине-фиолетовая/красная Ацетатный буфер
10 Фиолетовая/пурпурная Аммиачный буфер
Th(IV) 2,3 Фиолетовая/желтая Р-р HNO3
Рабочий р-р: 0,1-1% в спирте Zn(II) 7 Фиолетовая/красная Уротропин
Zr(IV) 1 Оранжевая/желтая Р-р НС1
V(IV) 3,5 Синяя/розовая Аскорбиновая к-та
Гематоксилин Al(III) 5-6 Пурпурная/желтая Ацетатный буфер, горячий р-р
3,7,8,5 ',6 -Пентокси-[индено-2', 1 ',3,4-хромен]- Bi(III) 1-2 Кр асная/желтая Р-р HNO3
дигидрид- -(3,4) Синонимы: кампешевый краситель; синий сандал Cu(II) Th(IV) 6 2 Ф (зеленая) Оранжевая/желтая Пиридин Р-р HNO3
Zr(IV) 1-1,5 Красная/желтая
Новый справочник химика и технолога
Продолжение таблицы 5.2.24
Индикатор, концентрация Применение: определяемые ионы, условия определения, изменение окраски титруемого раствора
Ион металла pH Изменение окраски Примечание
Н Н Х(/ ОН ох СН2
1 1 1 Н() JL Л YT
он
Рабочий р-р: 0,8% в СН3СООН; 1% спиртовый
Дитизон 2-Фенилгидразид-фенилазотиомуравьиная кислота Синоним: дифенилтиокарбазон II । N HN II S Рабочий р-р: 0,03% в этаноле Bi(III) Cd(II) In(III) Ni(II) Pb(II) Zn(II) 2,5-5 4,5 4-5 4,5 4,5 5 4,5 3,5-6,4 4,5-5 6-7 4,5 4-6 Красная/зе л еная Красная/зеленовато-желтая Розовая/желтая Красная/желто-зеленая Розовая/желтая Красная/же лто-з е леная Розовая/желтая Красная/зелено-фиолетовая Розовая/зеленовато-желтая 20% пиридин 60% спирт 20% пиридин 60% спирт 60% спирт 20% пиридин 60% спирт 20% пиридин или уротропин 40% спирт 20% пиридин 50% спирт 50% ацетон
Дифенилкарбазон 2-Фенилгидразидфенилазомуравьиная кислота Сл jC ^^^^NH HN^^^ Hg(II) Pb(II) V(V) 1 5-6 4,5-6,5 4,0-6,0 Сине-фиолетовая/ бесцветная Фиолетовая/бесцветная Красная/бе сцветная Фиолетовая/бесцветная Р-р НС1 Уротропин Ацетатный буфер То же
I I NH HN
II 0 Рабочий p-p: 0,2% спиртовый
Химические методы количественного анализа
Продолжение таблицы 5.2.24
Индикатор, концентрация Применение: определяемые ионы, условия определения, изменение окраски титруемого раствора
Ион металла pH Изменение окраски Примечание
Кальцихром Цикло-тирмс-7-(1 -азо-8-оксинафталин-3,6-дисульфокислота) Синонимы: Н-индикатор; кальцион ИРЕА SO; so; _fj ilL ,—L Л JL J—so; / OH ho^ -o3s—( / | \ / ,N OH II 1 1 so; Рабочий p-p: 0,001 M водный Са(П) 13 11-13 Красная/синяя Ярко-розовая/синяя
Кислотный ализариновый черный SN 1 - [ 1 -Окси-6-(2-оксинафтил азо)- -4-сульфо-2-фенилазо]-2-нафтол-6-сульфокислота Синонимы: алмазный ализариновый черный SN; антраноловый прочный черный SM; кислотный хромовый черный NSN; палатиновый хромовый черный S он он но —N— N—l^^jl—N= N \ Z so; \ Z so; Рабочий р-р: 0,1% водный Ва(П) Са(П) Cd(II) Mn(II) Ni(II) Th(IV) Zn(II) 11,5 11,5 8,5 10 10 4 11,5 Красная/синяя То же Красная/синяя Пурпурная/синяя То же Красная/оранжевая Пурпурная/синяя Аммиачный буфер То же Буфер с бурой Аммиачный буфер, гидрокси-ламил Аммиачный буфер Ацетатный буфер Аммиачный буфер
634 Новый справочник химика и технолога
Продолжение таблицы 5.2.24
Индикатор, концентрация Применение: определяемые ионы, условия определения, изменение окраски титруемого раствора
Ион металла pH Изменение окраски Примечание
Кислотный хромовый синий К 7-(2-Окси-5-сульфофенилазо)- Са(П) 12 Красная/синяя
1, 8-диоксинафталин-З,6-дисульфокислота. Синоним: Cd(II) 10 Красно-фиолетовая/синяя Аммиачный буфер
хромовый прочный флотский синий R Mg(II) 10 Красная/синяя То же
ОН ОН ОН Рабочий р-р: 0,1% спиртовый Pb(II) Zn(II) 10 10 Красно-фиолетовая/синяя То же Аммиачный буфер, тартрат Аммиачный буфер
Кислотный хромовый темно-синий 2-(2-Оксифенилазо)-1,8-диоксинафталин-З,6--дисульфокислота Ca(II) Mg(II) 12 10 Красная/сине-фиолетовая Красная/синяя Р-р NaOH Аммиачный буфер
Синоним: кислотный хромовый синий Т Pb(II) 10 Красно-фиолетовая/синяя Аммиачный буфер, тартрат
ОН ОН ОН /tn=nYjl\ “ O3S^^^^ so3_ Рабочий р-р: 0,1% спиртовый Окраска комплексов красная или красно-фиолетовая Zn(II) 10 То же Аммиачный буфер
Ксиленоловый оранжевый Bi(III) 1-3 Кр асная/желтая Р-р HNO3
3, 3 -Бис — [АГ, А'-Ди (карбоксиметил) аминометил]- Ca(II) 10,5 Сине-фиолетовая/ серая Аммиачный буфер
-о-крезол сульфофталеин Cd(II) 5-6 Розовая/желтая Уротропин
НООСН2С СН2СООН 4nh2c ch2nz Co(II) 5-6 Красно-фиолетовая/желтая Уротропин, о-фенантролин
НООСН2С CH2COUH ho^jl Ж/он т X X Т Cu(II) 4-6 Красно-фиолетовая/ желтая (зеленая) Уротропин или ацетат
Fe(III) Hg(II) 1-1,5 5-6 Сине-фиолетовая/ желтая Пурпурная/желтая Р-р HNO3, нагревание Уротропин
НзСГ4^^ СНз 1п(П1) 3-4,5 Красно-фиолетовая/желтая Ацетатный буфер
Рабочий р-р: 0,1% в разбавленном спирте Mg(II) Mn(II) Pb(II) 10,5 10,5 5-6 Красная/бледно-серая Фиолетовая/ бледно-серая Красно-фиолетовая/желтая Аскорбиновая к-та Ацетатный буфер
Химические методы количественного анализа
Продолжение таблицы 5.2.24
Индикатор, концентрация Применение: определяемые ионы, условия определения, изменение окраски титруемого раствора
Ион металла pH Изменение окраски Примечание
TR(III) Sc(III) Th(IV) Tl(III) U(IV) V(V) Y(III) Zn(II) Zr(IV) 4,5-6 2,2-5 1,6-3,5 4-5 1,7-2,2 1,8 4,5-6 5-6 1 M HC1 Красная/желтая To же Розовая/желтая Красная/ желтая То же То же То же Красная/ желтая То же То же Р-р HNO3 или ацетатный буфер Р-р HNO3 Ацетатный буфер Уротропин Ацетатный буфер Горячий р-р
Применяется также при обратном титровании Al, Bi, Tl(III), Ga, In, Pd, Sn, Th, VO2+ стандартным р-ром Th(IV) при pH = 2-3; Ni, Th (pH = 2), Ti, Zn (pH = 4,5) стандартным р-ром Bi(III); Al и Cu (pH = 5) и Sn(IV) (pH = 2,5-3,5) стандартным р-ром Pb(II)
Метомегахромовый синий 2RL Хлор-2-окси-З (2-окси-1 —нафтилазо)-бензолсульфокислота Синонимы: кислотный хромовый синий 2R; магнезон; хромовый прочный синий RLL; эриохромаловый синий 2RL он н° o,s. JL // \ |1—N=N \ у С1 \ f Ca(II) Cd(II) Ni(II) Sr(II) 11,5 11,5 4 12,5 Красная/синяя То же Красная/оранжевая Красная/синяя Аммиачный буфер То же Ацетатный буфер, горячий р-р Диэтиламин
Металлофталеин 3,3 -Диметил-5,5 '-[N, N-ди (карбоксиметил) аминометил] фенолфталеин Синонимы: о-крезолфталеинкомплексон; метилфта-лексон; фталеинкомплексон; фталеиновый пурпуровый Ba(II) Ca(II) Cd(II) Mg(II) Sr(II) 10,5-11 10-11 10 10-11 10,5-11 Красная/розовая Красная/бесцветная Красная/розовая Розовая/бесцветная Красная/розовая То же Красная/бесцветная Аммиачный буфер Аммиак + этанол Аммиачный буфер Аммиачный буфер, этанол Аммиачный буфер То же Аммиак + этанол
Новый справочник химика и технолога
Продолжение таблицы 5.2.24
Индикатор, концентрация Применение: определяемые ионы, условия определения, изменение окраски титруемого раствора
Ион металла pH Изменение окраски Примечание
НООСН2С СН2СООН
xnh2c ch2nz
hooch2cz чсн2соон
он Tf II II
1 II II 1 сн3
C=o
Метилтимоловый синий Ва(П) 10-11 Синяя/серая Аммиачный буфер
3, 3 -Бис-[М TV7-Ди (карбоксиметил) аминометил] Bi(III) 1-3 Синяя/желтая Р-р HNO3
тимол Са(П) 12 Синяя/серая NH3 или NaOH
-сульфофталеин Cd(II) 5-6 Сине-фиолетовая/желтая Уротропин
НООСН2С СН2СООН 12 Синяя/серая Аммиачный буфер
nh2c ch,nz Cu(II) 11,5 Синяя/бесцветная То же
hooch2cz 4 СН2СООН Fe(II) 4,5-6 Синяя/желтая Уротропин
Hg(II) 6 То же То же
In(III) 3^1 То же Ацетатный буфер
Т X f т Mg(II) 10-11,5 Синяя/серая Аммиачный буфер
СН(СН3)2 Mn(II) 6-6,5 Синяя/желтая Уротропин
Pb(II) 6 То же То же
so; TR(III) 6 То же То же
Sc(III) 2,2 или 6 То же HNO3 или пиридин
Sn(II) 5,5-6 То же Пиридин + ацетат
Th(IV) 1-3,5 То же Р-р HNO3
Рабочий р-р: 1 % водный, 1-100 К NO , Zn(II) 6-6,5 То же Уротропин или ацетат
Zr(IV) 0-2,3 То же Хлорацетатный буфер,
горячий р-р
Tl(III) 7-10 Красная/синяя Тартрат
Мурексид Ag(I) 8,5
5,5-Нитрилодибарбитуровая кислота, аммониевая
соль Ca(II) > 10 Розовая или крас- Р-р NaOH, 25% спирт
Синоним: MX ная/фиолетовая
Co(II) 8-10 Желтая/фиолетовая Аммиачный буфер
Химические методы количественного анализа 637
Продолжение таблицы 5.2.24
Индикатор, концентрация Применение: определяемые ионы, условия определения, изменение окраски титруемого раствора
Ион металла pH Изменение окраски Примечание
Н Н Си(П) 4 Оранжевая/красная Ацетатный буфер
7-8 Желтая/фиолетовая Аммиачный буфер
Мп(П) 10 Оранжевая/красная То же
II II Ni(II) 8,5-11,5 Желтая/пурпурная То же
N С N 1Г СГ н II II Sc(III) 2,6 3-7,5 Желтая/фиолетовая То же Р-р НС1
О о Th(IV) 2,5 Желтая/розовая
Рабочий р-р: 1 % водный или 1 : 500 сахарозы Zn(II) 8-9 > 11,5 Розовая/фиолетовая Аммиачный буфер, моноэта-ноламин
Нафтилазоксин Cd(II) 5,5-6,5 Желтая/красная Ацетатный буфер
7-(1 -Нафтилазо)-8-оксихинолин- Cu(II) 4-6,5 То же То же
5-сульфокислота Pb(II) 6-6,5 То же Ацетатный буфер, тартрат
Синоним: а-нафтилазоксинсульфокислота Zn(II) 6-6,5 То же Ацетатный буфер
In(III) 2,5-3 Желтая/пурпурная 70-80 °C
// \ S°’ Ga(III) 2,2-2,8 Желтая/розово-фиолетовая 70-80 °C
/ \ N=N—L JI J \ // jr У ОН Рабочий р-р: 1 % в диметилформамиде или в 4% диоксане Tl(III) 4-4,5 Желтая/красная Тартрат
Нафтоловый фиолетовый 4-(4-Нитрофенилазо)-2-Бис-(карбоксиметил) аминометил-1-нафтол Bi(III) 1-3 Красно-фиолетовая/красно-оранжевая Р-р HNO3
Cd(II) 10-11 Красно-фиолетовая/синяя Аммиачный буфер
СН2СООН Co(II) 10-11 То же Аммиачный буфер, 40-50 °C
ch2n Cu(II) 10-11 То же Аммиачный буфер
г / СН2СООН Mg(II) 10-11 То же То же
— N= N—( у— он Mn(II) 10-11 То же Аммиачный буфер, гидроксиламин
o2n/^^ / \ Используется смесь 1 : 100 NaCl Zn(II) 10-11 То же Аммиачный буфер
Новый справочник химика и технолога
Продолжение таблицы 5.2.24
Индикатор, концентрация Применение: определяемые ионы, условия определения, изменение окраски титруемого раствора
Ион металла pH Изменение окраски Примечание
Омегахромовый черно-синий G Са(П) 11,5 Красная/синяя Аммиачный буфер
5-Хлор-3-(8-ацетамидо-2-окси- Cd(II) 10 To же То же
1-нафтилазо)-2-оксибензолсульфокислота Синонимы: метомегахромовый серый GL; Mg(II) n,5; 10 To же То же
хромовый прочный серый GL; эриохромовый серый SGL Mn(II) 10 To же Аммиачный буфер, аскорбиновая к-та
ОН Ni(II) 4,5 Красная/оранжевая Ацетатный буфер, горячий р-р
“O,S X / \ Pb(II) 10 Красная/синяя Аммиачный буфер, тартрат
3 2 Sr(II) 11,5 То же Аммиачный буфер
CH3COHN / \ С1 \/ Рабочий р-р: 0,1% водный Zn(II) 10 То же То же
o-PAN Bi(III) 1-3 Красная/желтая
1 -(2-Пиридилазо)-2-нафтол Cd(II) 6 То же Ацетатный буфер
Cu(II) 2,5 То же То же
10 Фиолетовая/желтая Аммиачный буфер
—N=N In(II) 2,5 Красная/желтая Ацетатный буфер
Ni(II) 4 Розовая/желтая Ацетатный буфер, нагревание
НО Рабочий р-р: 0,01-0,1% спиртовый Th(IV) 2-3,5 Красная/желтая Р-р HNO3
UO2(II) 4,4 То же Уротропин, изопропиловый спирт
Окраска комплексов красная Zn(II) 5-7 Розовая/желтая Ацетатный буфер
PAR Bi(III) 1-2 Красная/желтая Раствор HNO3
4-(2-Пиридилазо) резорцин Cd(II) 8-11 То же Аммиачный буфер, уротропин
Cu(II) 5-11 Красная/желтая (зеленая) То же
Hg(II) 3-6 Красная/желтая Уротропин
L — N= N 1 J In(III) 2,5 То же Р-р СН3СООН, нагревание
ОН Mn(II) 9 То же Аммиачный буфер, аскорбиновая кислота
Рабочий р-р: 0,05% Ni(II) 5 Красная/ желтая Ацетатный буфер, нагревание
или 0,2% водный Pb(II) 5-9 То же Аммиачный буфер, уротропин
Окраска комплексов красная TR(III) Th(IV) Tl(III) 6 2,3-2,8 1,7 Красная/желтая То же То же Уротропин
Zn 6-11 То же Аммиачный буфер, уротропин
Химические методы количественного анализа
Продолжение таблицы 5.2.24
Индикатор, концентрация Применение: определяемые ионы, условия определения, изменение окраски титруемого раствора
Ион металла pH Изменение окраски Примечание
Пирогаллоловый красный Пирогаллолсульфофталеин Bi(III) 2-3 Красная/оранжево-желтая Синяя/красная Р-р HNO3
Синоним: сульфогаллеин Со(П) 9,3 То же Аммиачный буфер
ОН ОН Ni(II) 9,3 Фиолетовая/красная То же
он ГТ Pb(II) 5-6 Ацетатный буфер
Рабочий р-р: 0,05% в 50% спирте
Пирокатехиновый фиолетовый Bi(III) 2-3 Синяя/желтая Р-р HNO3
3,3 ',4 -Триоксифуксон-2-сульфокислота Синонимы: катехиновый фиолетовый; пирокатехин- Cd(II) 10 Синяя/красновато-пурпурная Аммиачный буфер
сульфофталеин Со(П) 9,3 То же То же
он он Cu(II) 5-6,3 Синяя/желтая Ацетатный буфер
HO^J< 6-7 Синяя/желто-зеленая Пиридин
11 | II 9,3 Синяя/пурпурная Аммиачный буфер
1 II + 1 1| Fe(II) 3-6 Синяя/желтая Ацетатный буфер
Ga(III) 3,8 То же То же
In(III) 5-6 То же То же
Mg(II) 10 Синяя/красновато-пурпурная Аммиачный буфер
Рабочий р-р: 0,1% водный Mn(II) 9,3 То же Аммиачный буфер, гидроксиламин
Ni(II) 8-9,3 Синяя/красновато-фиолетовая Аммиачный буфер
Pb(II) 5,5 Синяя/желтая Уротропин
Th(IV) 2,5-3,5 Красная/желтая Р-р HNO3
Zn(II) 10 Синяя/красновато-фиоле-товая Аммиачный буфер
Применяется также при обратном титровании Al, Bi, Fe(III), In стандартным р-ром Cu при pH = 6-7 и Bi, Fe(III), Ga, In, Ni, Th, Tl стандартным р-ром Bi(III) при pH = 2-3
Новый справочник химика и технолога
Продолжение таблицы 5.2.24
Индикатор, концентрация Применение: определяемые ионы, условия определения, изменение окраски титруемого раствора
Ион металла pH Изменение окраски Примечание
Прочный флотский 2R Са(П) 8,5 Винно-красная/синяя Аммиачный буфер
2-(5 -Хлор-2-окси-3 -сульфофенилазо)-1 -нафтол-5 - Cd(II) 8,35 To же CH3COONH4, тартрат
сульфокислота Mg(II) 8,5 To же Аммиачный буфер
Синонимы: алмазный прочный темно-синий RRL; калькохромовый синий RRL; Mn(II) 8,35 To же СН3СООМН4, тартрат, аскорбиновая к-та
кислотный хромовый синий RRA; Ni(II) 8,35 To же CH3COONH4, тартрат
омегахромовый синий 2R; Pb(II) 8,35 To же То же
солохромовый строчный флотский R2; суперхромовый синий 2R; хромовый синий 2R; хромовый прочный синий ALB; эриохромовый фиолетовый; эриохромовый фиолетовый SB ОН ОН “O3S. X JL в—N=N—1 II 1 Zn(II) 8,35 To же То же
Cl so;
Солохроматовый прочный синий В Cd(II) 10 Красная/пурпурная Аммиачный буфер
Ацетамидо-2-(2-оксн-3,5,6-трихлорфенилазо)-1 - 6,8 Красная/синяя Диэтилбарбитурат
нафтол-5-сульфокислота Mg(II) «10 Розовая/синяя Аммиачный буфер
Синонимы: кислотный монохромовый синий 3; кислотный хромовый сине-черный; Mn(II) 10 Красная/пурпурная Аммиачный буфер, аскорбиновая к-та
метахромовый бриллиантовый синий BL; метомегахромцианин BLL; 6,8 Розовая/синяя Диэтилбарбитурат, аскорбиновая к-та
эриохромовый флотский синий BRL Pb(II) 6,8 Розовая/синяя Диэтилбарбитурат
Zn(II) «10 Красная/синяя Аммиачный буфер
ОН ОН NHCOCH3 СК X. JL Д. ljrN=N4 ci so; 6,8 Розовая/синяя Диэтилбарбитурат
Химические методы количественного анализа
Продолжение таблицы 5.2.24
£
Индикатор, концентрация Применение: определяемые ионы, условия определения, изменение окраски титруемого раствора
Ион металла pH Изменение окраски Примечание
Тимолфталексон 3,3 '-бис — |А,А'-Ди-( карбокси метил) аминоме-тил]тимофталеин Синоним: тимолфталеинкомплексон НООСН2С СН2СООН 4nh2c ch2nz hooch2cz 4 СН2СООН (СН3)2НС^^^\ СН(СН^ Используется в смеси 1 : 100 с KNO3 Ag(I) Ва(П) Са(П) Мп(П) Sr(II) Th(IV) 10-11 10-11 10,5-12 10 10-11 Синяя/светло-зеленая Синяя/зелено-желтая Синяя/бесцветная Синяя/светло-розовая Синяя/зелено-желтая Красная Аммиачный буфер, диметиловый желтый NaOH или NH3 То же Аммиачный буфер, гидроксиламин NaOH или NH3
Применяется также при определении Ва, Mn, Sr обратном титровании стандартным р-ром Са(П) при pH = 12
Торин 2-(2-Окси-3,6-дисульфо-1-нафтилазо) бензоларсоно-вая кислота Синонимы: нафтарсон; торон; торонол; APANSP AsO3H2 № zS°3 —N=N— so3 Рабочий р-р: 0,5% водный Bi(III) Th(IV) U(IV) Sc(III) Y(III) 2-3 1-3 1-1,8 4,5-6,5 6 Красная/желтая Фиолетовая/желтая Розовая/оранжево-желтая Розовая/ желтая То же Р-р HNO3 То же Р-р НСЮ4, 30 °C
Новый справочник химика и технолога
Продолжение таблицы 5.2.24
Индикатор, концентрация Применение: определяемые ионы, условия определения, изменение окраски титруемого раствора
Ион металла pH Изменение окраски Примечание
Хромазурол S 3-Сульфо-2,6-дихлор-3,3 -диметил-4-оксифуксон-5,5 -ди-карбоновая кислота Синонимы: альберон; галлохромовый бриллиантовый синий BLD; политроповый синий В; солохромовый бриллианто- вый синий В; хромоксановый ярко-синий BLD; эриохромазурол S СН3 СН3 X Х.Х X с-^^ соон X X Рабочий р-р: 0,1-0,4% водный А1(Ш) Са(П) Си(П) Fe(III) Mg(II) Ni(II) TR(III) Th(IV) V(IV) 4 11 6-6,5 2-3 10-11 8-11 8 2-3 4,8 4 Фиолетовая/желто-оранжевая Красная/желтая Сине-фиолетовая/желтая (зеленая) Синяя/ оранжевая Красная/желтая Синяя/желтая Фиолетовая/желтая Краснофиолетовая/ оранжевая Фиолетовая/оранжевая Сине-фиолетовая/оранжевая Ацетатный буфер, нагревание Аммиачный буфер Ацетатный буфер Хлорацетатный буфер Аммиачный буфер То же То же Р-р HNO3 Ацетатный буфер То же
Окраска комплексов красная, синяя и фиолетовая
Хромовый зеленый G 5- {3 - [а-(3 -Карбокси-5 -метил-4-оксоциклогекс адиен-2,5 -илиден-1 )-3 -карбокси-4-окси-5-метилбензил]-4-хлорфенилазо} -2,3-крезотиновая кислота Синоним: хромоксановый зеленый GG НООС СООН /0Н сн, 3 1 ’ соон х ^Ln==n—L \снз Ca(II) Cu(II) Mg(II) Ni(II) Th(IV) V(IV) 11 8 11 11 4,8 4 Красная/зеленая Красная/оранжевая Красная/зеленая То же Фиолетовая/красная То же Аммиачный буфер
Химические методы количественного анализа
Продолжение таблицы 5.2.24
Индикатор, концентрация Применение: определяемые ионы, условия определения, изменение окраски титруемого раствора
Ион металла pH Изменение окраски Примечание
Цинкон (о- {2-[а-(2-Окси-5-сульфофенилазо) бензилиден] гидразино} бензойная кислота 0Н НООС\Х-ХЧ. II 1 N N < A Zn(II) 7,8 9-10 Синяя/желтая Триэтаноламин Аммиачный буфер
Рабочий раствор: 0,1% в абсолютном спирте
Эриохромовый красный В Са(П) 9-10 Красная/желтая Аммиачный буфер
4-(2-Окси-4-сульфо-1-нафтилазо)-3-метил-1-фенил-2- Cu(II) 2—4,5 Розовая/желтая Хлорацетатный буфер
пиразолинон-5 Синонимы: омегахромовый красный В; понтахромовый прочный красный Е; Мп(П) 8-10 Красная/желтая Аммиачный буфер, аскорбиновая к-та
солохромовый красный ER; Ni(II) 4-6 Розовая/желтая Ацетатный буфер
хромовый красный В; 7-11 То же Аммиачный буфер, нагревание
хромовый прочный красный В Pb(II) 10 Красная/желтая Аммиачный буфер, тартрат
ОН Н3С J П N—N—Z \—so; N JL ^он / \ Рабочий р-р: 0,1% в этаноле, 0,04% водный, 1 : 100 NaCl Zn(II) 6,5 То же Уротропин
Zn(II) 10 Красная/желтая Аммиачный буфер
644 Новый справочник химика и технолога
Продолжение таблицы 5.2.24
Индикатор, концентрация Применение: определяемые ионы, условия определения, изменение окраски титруемого раствора
Ион металла pH Изменение окраски Примечание
Эриохромовый сине-черный В 1 -(1 -Окси-2-нафтилазо)-2-нафтол-4-сульфокислота Синонимы: омегахромовый сине-черный В; понтахромовый сине-черный R; солохромовый черный 6BN; хромовый прочный цианин G; хромовый сине-черный В ,он он ~O3S N= N Рабочий р-р: 2% в метаноле, 0,5% в этаноле и воде Са(П) Cd(II) Mg(II) Mn(II) U(IV) Zn(II) Zr(IV) 11,5 11,5 10 10-11,5 1-2 10 0,1-0,5 M HC1 Винно-красная/синяя Пурпурная/синяя Винно-красная/синяя Красная/синяя Синяя/красная Винно-красная/синяя Синяя/красная Аммиачный буфер То же То же Аммиачный буфер, аскорбиновая к-та Р-р НС1, нагревание Аммиачный буфер Р-р НС1, нагревание
Эриохромовый сине-черный В 1 -(2-Окси-1 -нафтилазо)-2-нафтол-4-сульфокислота Синонимы: алмазный сине-черный В; калькой; кислотный хромовый сине-черный; солохромовый темно-синий В; хромовый сине-черный RC он но "O,S N= N Рабочий р-р: 0,5% в метаноле или этаноле или соль цинка (цинкхром R) Ca(II) Cd(II) Mg(II) Mn(II) Zn(II) 11,5 12,3-13 11,5 10 10 10 Розовая/синяя То же То же Розовая/синяя Красная/синяя Розовая/синяя Аммиачный буфер Цинхром Этиламин Поливиниловый спирт Ацетилацетон Аммиачный буфер То же Аммиачный буфер, аскорбиновая к-та Аммиачный буфер
Эриохромовый синий SE 2-(5-Хлор-2-оксифенилазо)-1,8-диоксинафталин-З ,6-дисульфокислота Синонимы: кислотный хромовый флотский синий В; коринфский СА; омегахромовый прочный синий В; Ca(II) Cd(II) Mg(II) > 12 11,5 10 10 10 Красная/синяя То же То же Розовая/синяя Красная/синяя Раствор NaOH Смешанный индикатор Аммиачный буфер То же То же
Химические методы количественного анализа
Продолжение таблицы 5.2.24
Индикатор, концентрация Применение: определяемые ионы, условия определения, изменение окраски титруемого раствора
Ион металла pH Изменение окраски Примечание
плазмокоринфский В; хромовый прочный синий 2В ОН ОН ОН ^tn=№Vy\ Cl Рабочий p-p: 0,1% в 25% метаноле, 1 : 200 КС1 Мп(П) Ni(II) Pb(II) Zn(II) 10 10 10 10 To же Фиолетовая/синяя Розовая/синяя То же Аммиачный буфер, аскорбиновая к-та Аммиачный буфер Аммиачный буфер, тартрат То же
Эриохромовый черный А (2-Окси-1 -нафтилазо)-6-нитро-2-нафтол-4-сульфокислота Синонимы: калькохромовый черный АС; омегахромовый черный Р; понтахромовый черный А; солохромовый черный AS; хромовый прочный черный А; хромогеновый черный EAG ОН НО O,S — N= N o2n Рабочий р-р: 0,1% спиртовый Cu(II) Mg(II) Mn(II) Ni(II) Pb(II) Zn(II) 10 10 10 10 10 10 Красная/синяя Винно-красная/синяя Красная/синяя То же Винно-красная/синяя То же Аммиачный буфер, 80 °C NH3H2O NH3-H2O, аскорбиновая кислота NH3-H2O, 80 °C NH3-H2O, тартрат nh3h2o
Эриохромовый черный PV 2-(1,5-Диокси-2-нафтилазо)-1 -оксибензол-4-сульфокислота Синонимы: алмазный черный PV; калькохромовый черный PV; омегахромовый черный PV; понтахромовый черный PV; Ca(II) Cd(II) Fe(II) Fe(III) Mg(II) Mn(II) 11,5 10 4 4 10 10 Красная/синяя Красно-фиолетовая/синяя Оранжевая/красная Желтая/красная Красная/синяя То же Аммиачный буфер То же Ацетатный буфер, нагревание То же Аммиачный буфер Аммиачный буфер, гидроксиламин
646 Новый справочник химика и технолога
Продолжение таблицы 5.2.24
Индикатор, концентрация Применение: определяемые ионы, условия определения, изменение окраски титруемого раствора
Ион металла pH Изменение окраски Примечание
поттинг черный PV; солохромовый черный PV; суперхромовый черный PV; хромовый прочный черный PV; хромовый черный PV ОН ОН i^^j]—n=n— so3~ он РЬ(П) Zn(II) 10 10 Красно-фиолетовая/синяя To же Аммиачный буфер, тартрат Аммиачный буфер
Эриохромовый черный Т 1 -(1 -Окси-2-нафтилазо)-6-нитро-2-нафтол-4-сульфокислота Синонимы: калькохромовый черный Т; омегахромовый черный Т; понтахромовый черный ТА; солохромовый черный Т (или WDFA); хромовый черный ТК; хромогеновый черный ЕТ /Он ОН ~O3S \ N= N o2n Рабочий р-р: 0,05-0,5% спиртовый, 1 : 200 NaCl Ва(П) Cd(II) In(III) Mg(II) Mn(II) Pb(II) TR(III) Zn(II) Zr(IV) 10,5 6,8-11,5 8-10 10 8-10 10 8-9 6,8-10 0,5-2 M HC1 ?/зеленая Красная/синяя То же То же Красная/синяя То же То же То же Сине-фиолетовая/розовая Аммиачный буфер, родизонат натрия Аммиачный буфер Аммиачный буфер, горячий тартрат Аммиачный буфер Аммиачный буфер, аскорбиновая кислота Аммиачный буфер, тартрат То же Аммиачный буфер Раствор НС1, 100°С
Применяется также при определении Sr, Bi, Ca, Hg(II), In, Mn, Ni обратным титрованием стандартным р-ром Mg(II) или Zn(II) при pH = 10. Стандартный р-р Mg(II) в качестве титранта применяют для определения Си (pH = 10), Ga (pH = 6,5-9,5), Sn(IV) (pH = 9), Tl (pH = 8). Стандартный p-p Zn(II) в качестве титранта применяют для определения А1 (pH = 7-8), Сг (pH = 9-10), Fe(III) (pH = 7), Ga (pH = 8-10), Mo(V) (pH = 10), Pd (pH = 10), лантанидов (pH = 8-9), Sn(IV) (pH = 9), Tl (pH = 8)
Эриохромцианин R 2»-Сульфо-3,3 -диметил-4-оксифуксан-5,5 -дикарбоновая кислота Синонимы: ализаролцианин RC; омегахромцианин GR; понтахромовый синий ECR; Al(III) Ca(II) Cu(II) Fe(III) 5-6 11,5 10 2-3 Пурпурная/желтая Фиолетовая/желтая Фиолетовая/желтая (зеленая) Фиолетовая/оранжевая Ацетатный буфер Аммиачный буфер То же Хлорацетатный буфер, горячий раствор
Химические методы количественного анализа
Продолжение таблицы 5.2.24
Индикатор, концентрация Применение: определяемые ионы, условия определения, изменение окраски титруемого раствора
Ион металла pH Изменение окраски Примечание
L хромоксанцианин RA Mg(II) 10 Фиолетовая/желтая Аммиачный буфер
Th(IV) 2-2,5 Пурпурная/розовая Р-р HNO3
сн3 сн3 НООСХ^55^^^ соон J\^so3" Ql Рабочий р-р: 0,1-0,4% водный или 0,1% спиртовый Zr(IV) 1,3-1,5 Розовая/бесцветная Р-р НС1
HPHN 2-(2-Оксифенилазо)-1,5-диоксинафталин Ca(II) 11,5 Красно-фиолетовая/синяя Аммиачный буфер
Cd(II) 10 То же То же
ОН ОН Mg(II) 10 Красно-фиолетовая/синяя То же
N=N ^><^1 Mn(II) 10 То же Аммиачный буфер, гидрокси-ламин
О XI/ он Zn(II) 10 То же Аммиачный буфер
SPADNS 2-(4-Сульфофенилазо)-1,8-диоксинафталин-З ,6-дисульфокислота Th(IV) 2,5-3,5 Сине-фиолетовая/ярко-красная Ацетатно-солянокислый буфер
Синоним: сульфаниловокислый азохромотроповый ОН ОН ' O3S'^4^^ "O3S^4=^X^'^!^ so, Рабочий р-р: 0,02-0,4% водный Zr(IV) 1,5-2,5 Малиноворозовая/ оранжево-красная Р-р НС1, нагревание
Новый справочник химика и технолога
Продолжение таблицы 5.2.24
Индикатор, концентрация Применение: определяемые ионы, условия определения, изменение окраски титруемого раствора
Ион металла рн Изменение окраски Примечание
PADNS 1 -(2-Пиридилазо)-2,3 -диоксинафталин-6- Cd(II) 8-9 Винно-красная/оранжевая Уротропин
сульфокислота Cu(II) 3,3 Красная/желто-зеленая Формиат
SO3 Pb(II) 8-9 Винно-красная/оранжевая Уротропин
—N=N HCi ОН Zn(II) 8-9 То же То же
Таблица 5.2.25
Металлофлуоресцентные индикаторы [42]
Название Формула Титруемые ионы Область рн Изменение флуоресценции Раствор индикатора
Морин (2',4',3,5,7-пен-таоксифлавон) ОН О Г jT jT ,он он Ga3+ In3+ 3,8 5,0 Зеленая/не флуоресцирует Зеленая/не флуоресцирует 0,5% в метаноле или 0,2% в смеси Этанол + вода (1:1)
Салицил альдегидаце-тилгидразон СН= N— NH— СОСН3 ОН Zn2+ 4,5 (ацетат) Синяя/не флуоресцирует 0,1% в этаноле
Сал ицил идеи-о-аминофенол СН= N он но7 А13+, титрование раствором ЭДТА или F 4-5 (ацетат) Желто-зеле-ная/не флуоресцирует Свежий 0,05% в этаноле
Химические методы количественного анализа 649
Продолжение таблицы 5.2.25
Название Формула Титруемые ионы Область pH Изменение флуоресценции Раствор индикатора
3 -Окси-2-нафтойная кислота ^^^^^^соон Г т т А13+ 3,0 Синяя/зеленая 0,1% водный раствор соли натрий
8-Оксихинолин Ga3+ 2,5-3,5 (фталат) Желто-зеле-ная/не флуоресцирует 0,05% в 0,5% уксусной кислоте
Кальцеин ноос—сн2 I 1 сн2-соон N—СН2 1 Я Н2С—-N 1 1 \ 1 ноос—сн2 А%>-со сн2—соон Cu2+ Mn2+ Са2+ Cr3+, Со2+, обратное титрование +Си2+ 10 (NH3H2O + NH4C1) 8-10 (пиридинацетат или NH4OH) > 12 (КОН) 10 (NH3H2O + NH4CI) Не флуоресци-рует/желто-зеленая То же Желто-зеленая/ не флуоресцирует То же 1% дисперсия в порошке КС1
Кальцеиновый синий ноос—сн2 „„ | СН3 N— СН2 1 НООС—СН2 || "А Си2+ Са2+, Ва2+, Sr2*, Cr3*, Ni2*, Со2*, Fe3*, Ti4* 4,8 (ацетат-СН3СООН) >12 (КОН) Не флуоресци-рует/синяя Синяя/не флуо-ресци-рует 0,1% водный
650 Новый справочник химика и технолога
Продолжение таблицы 5.2.25
Название Формула Титруемые ионы Область pH Изменение флуоресценции Раствор индикатора
Al3+, Mn2+, Zn2+, Hg2+, обратное титрование +Си2+ 4,8 (ацетат-СНзСООН) То же
Ксантонкомплексон О сн2- "^^^он сн2—соон N 1 сн2-соон Свойства подобны свойствам кальцеинового синего
о-Дианизидин-АА/У^тУ-тетрауксусная кислота НООС—СН2 N / 1 НООС—СН2 ) осн3 Н3СО сн3—соон -N 1 сн2—соон Си2+ Hg2+ Bi3+, Cd2+, Се3+, Fe2+, Fe3+, In3+, La3+, Ni2+, Pb2+, Th4+, Tl3+, Sn2+, Zr4+, обратное титрование + Cu2+ 4-10 4 6 Не флуоресци-рует/синяя То же Синяя/не флуоресцирует 1% дисперсия соли натрия в порошке KNO3
о-Дифенетидин-ХУуУ^У-тетрауксусная кислота НООС—сн2 N—/ ноос—сн2 ОС2Н5 Н5С2О сн2—соон -N сн2—соон Cu2+ Hg2+ 4-10 4 Не флуоресци-рует/синяя То же То же
3,3 Диокси-бензидин-У^У^У'.У'-тетрауксусиая кислота НООС—сн2 N / НООС—СН2 он но сн2—соон -N сн2—соон Cu2+ Pb2+ Cd2+, Co2+, Mn2+, Zn2+, в смеси с родамином 6-8 6 6 То же То же Розовая/синяя То же То же
Химические методы количественного анализа
Продолжение таблицы 5.2.25
Название Формула Титруемые ионы Область pH Изменение флуоресценции Раствор индикатора
3,3 '-Дикарбо кси-бензидин-Л'',Лт,..'У',Л''-тетрауксусная кислота НООС—Н2С 1 N- НООС—Н2С СООН НООС сн2—соон -N 1 сн2—соон Са2+ 10-13 He флуоресцирует/ зеленая То же
4,4-Диамино-стильбен-Л',Лт,Лт',Л'"-те'грауксусная кислота НООС—сн2 N—/ НООС—сн2 \-сн=сн—/ сн2—соон т—n / 1 сн2—соон Си2+ 6 Не флуоресци-рует/синяя
Bi3\ Со2+, Fe2+,Ni2+, Pb2+, In3+, V4+, обратное титрование + Си2+ 6 Синяя/не флуоресцирует 0,1% дисперсия соли натрия в порошке KNO3
4,4'-Диамино-стильбен- 2,2'-дисульфо-Л',Лт,2У',2У'-тетрауксусна51 кислота НООС—сн2 N—/ 1 \ НООС—сн2 у-сн=сн—Z SO3H SO3H сн2—соон \ 1 y-N z 1 сн2—соон Си2+ 6 Не флуоресцирует/синяя То же
Bi3+, Со2+, Fe2+, Ni2+, Fe2+, Ni2+, Pb2+, In3+, V4+, обратное титрование + Cu2+ 6 Синяя/не флуоресцирует То же
652 Новый справочник химика и технолога
Химические методы количественного анализа
653
5.2.6.3. Техника и применение комплексонометрического титрования
Согласно [33] комплексы металлов с ЭДТА можно разделить на три группы (табл. 5.2.26). В первую группу входят комплексы с константами устойчивости lgPMY >20: все четырех- и трехвалентные ионы металлов (за исключением А1 и TR), а также Hg(II) и Sn(II). Все эти элементы можно титровать при pH = 1-3. Вторая группа содержит комплексы с константами устойчивости lg PMY =12-19; сюда относятся ионы TR и двухвалентных металлов, за исключением щелочноземельных — их можно определять при pH = 5-6. В третью группу входят комплексы щелочноземельных металлов и серебра с константами устойчивости IgpMY =7-11, их определяют обычно при pH =8-10. Определению катионов первой группы не мешает присутствие катионов третьей группы; катионы второй группы не мешают лишь в малых концетрациях. Определению катионов второй группы не мешают катионы третьей группы, а все остальные титруются совместно. Все катионы первой и второй групп мешают определению катионов третьей группы. Определению индивидуальных катионов внутри каждой группы мешают остальные члены этой группы, если не применять маскирующие реагенты [33].
Таблица 5.2.26
Классификация комплексов металлов с ЭДТА [33]
Группа I Группа II Группа III
Катион lg₽ Катион IgP Катион lg₽
Hg(II) 21,8 Cu(II) 18,8 Ag 7,3
Sc 23,1 Zn 16,5 Be 9,8
Ga 20,3 Cd 16,5 Mg 8,7
In 24,9 Al 16,1 ' Ca 10,7
Т1(1П) 21,5 La 15,5 Sr 8,6
Th 23,2 Lu 19,8 Ba 7,8
Sn(II) 22,1 Y 18,1 Ra 7,1
Ti(IV) 20,4 Pb(II) 18,0
Zr 29,5 TiO(II) 17,3
Bi(ni) 27,9 VO2(I) 18,1
V(III) 25,9 VO(II) 18,8
Сг(1П) 23 Mn(II) 13,8
U(IV) 25,5 Fe(II) 14,3
Fe(III) 25,1 Co(II) 16,3
Co(III) 36 Ni 18,6
Комплексонометрическое титрование применяют в вариантах прямого, обратного, вытеснительного, заместительного и косвенного титрования.
Рис. 5.2.7 показывает, что более 70 элементов периодической системы можно определять, применяя ЭДТА: штриховыми линиями ограничены элементы, определяемые непосредственно прямым или обратным титрованием, сплошными — элементы, определяемые косвенными методами.
VIIA
Рис. 5.2.7. Распределение элементов, определяемых комплексонометрически, в периодической системе Д. И. Менделеева
Прямое титрование
Прямое титрование возможно только при быстром и количественном протекании реакции (IgP^y -8)- Им определяют около 30 элементов, в том числе: Mg, Са, Zn, Cd, Cu, Fe, In, Zr и др.
Сущность прямого титрования. К анализируемому раствору добавляют буферный раствор (или кислоту) для создания необходимой величины pH, вводят индикатор и титруют раствором комплексона Ш до изменения окраски раствора.
Исходя из стехиометрии реакции (1 моль комплексона взаимодействует с 1 моль ионов металла) можно записать, что в ТЭ:
и(М) = «(Na2H2 Y), (5.2.70)
откуда
ш(М) = c(Na2H2Y) • K(Na2H2Y) • ЛДМ). (5.2.71)
Обратное титрование
Обратное титрование применяется, если невозможно прямое титрование. Например, когда:
а) ионы металла взаимодействуют с комплексом Ш медленно (например, Со3+, А13+ и др.);
654
Новый справочник химика и технолога
б) в интервале pH титрования образуются осадки гидроксидов металлов;
в) нет подходящего индикатора для фиксации КТТ;
г) соединения не растворимы в Н2О, но растворимы в комплексоне III (PbSO4, СаС2О4 и т.д.).
Сущность обратного титрования. К анализируемому раствору добавляют точно измеренный избыток раствора комплексона III, а затем остаток комплексона оттитровы-вают стандартным раствором второго металла — М . При выборе М руководствуются следующим требованием: величина условной константы устойчивости комплексоната М Y должна быть меньше соответствующей величины для комплексоната определяемого металла, но больше, чем 107:
Pmy > Pm*y > Ю7 • (5-2.72)
Условие (5.2.72) будет выполняться практически всегда, если в качестве М использовать магний, комплексо-нат которого отличается сравнительно невысокой устойчивостью lg₽MgY =8,7.
На практике для обратного титрования чаще всего применяют стандартный раствор магния, который удобен еще и тем, что на ионы магния имеется хороший индикатор — ЭХЧ-Т.
Если комплексонат титруемого металла очень устойчив, в роли М могут выступать и другие металлы — например, цинк и медь, которые пригодны для титрования в кислых растворах.
При определении ионов металлов, реагирующих с комплексоном III слишком медленно (А13+, Со3+ и др.), в качестве М можно использовать ионы металлов, комплексона-ты которых более устойчивы, чем комплексонат определяемого металла. Например:
А13+ + 1LY2 ' >A1Y + 2Н+,
избыток
H2Y2~+Fe3+<z ~°вая > FeY" +2Н+ (pH = 6-6,5).
кислота значительно устойчивее A1Y"
В ТЭ л(А1) = w(Na2H2 Y) -«(Fe) , (5.2.73)
откуда
ти(А1) = [c(Na2H2 Y) F(Na2H2 Y) - c(Fe) • F(Fe)] • M(A1)
(5.2.74)
Селективное комплесонометрическое титрование*
Широкая универсальность ЭДТА (см. рис. 5.2.7) часто препятствует определению того или иного элемента в сложной анализируемой смеси. Поэтому возникает вопрос
Текст и таблицы данного подраздела (кроме табл. 5.2.34) взяты из монографии [36] — с сохранением нумерации литературных ссылок. Список литературы из [36] приведен на компакт диске.
о селективном комплексонометрическом титровании. Селективность комплексонометрического титрования без отделения мешающих катионов достигается главным образом выбором диапазона pH среды (последовательное титрование), использованием подходящих маскирующих реагентов, в некоторых случаях титрованием по методу вытеснения, а также применением других аминополикарбоновых кислот.
Селективное комплексонометрическое определение элементов путем последовательного титрования
Этилендиаминтетраацетаты металлов с высокими значениями условных констант устойчивости остаются достаточно прочными в сильнокислой среде, в то время как другие, менее устойчивые комплексонаты частично или полностью диссоциируют.
Правильный выбор pH среды позволяет избирательно титровать в кислой среде один катион или группу катионов в присутствии других и затем, после повышения pH, следующую группу катионов, комплексонаты которых менее устойчивы. Условия последовательного титрования с точностью 0,1% двух катионов М] и Мп, из которых Mi образует более прочный комплексонат, следующие: М] нужно титровать при таком минимальном значении pH раствора (pHmin), при котором выход его комплексоната будет не менее 99,9%, а выход комплексоната Мц не превысит 0,1%. При таком pH должны быть обеспечены оптимальный выход окрашенного комплекса Mi с индикатором, максимальная контрастность перехода окраски в конечной точке титрования и отсутствие взаимодействия Мп с индикатором. Избирательное титрование катиона Mi в присутствии катиона Мп возможно при соблюдении неравенства [50] (заряды опущены):
Igp^+lgc^ -(lgp^nY +lg<) > 4-21g A, (5.2.75) где P^y и Pmdy — условные константы устойчивости комплексонатов; и — общие концентрации ионов металлов в растворе; А— относительная погрешность, обусловленная присутствием мешающего иона, %.
Если концетрации ионов металлов равны (с£, = ) и
А = 1%, то неравенство (5.2.75) принимает вид
PmjY ~ 1g Рмпу -
а при А = 0,1 % разность логарифмов условных констант устойчивости комплексонатов должна быть
lg Pm,Y ~ 1g Рмпу -
Кроме того, при титровании Mt должны соблюдаться условия lgp^iY > 7, a 1g 0^ <3 [126].
При последующем титровании Мц необходимо повысить pH раствора так, чтобы значение IgP^Y - 7; при этом
Химические методы количественного анализа
655
комплексонах М(У не должен гидролизоваться и блокировать индикатор. Таким образом, при титровании иона металла NT Н -ионы маскируют ион металла Мц, а при титровании иона металла Мп комплексон-титрант маскирует ион металла Мр Наиболее рациональным будет такое последовательное титрование, когда при соответствующих значениях pH оба иона М, и Мц дают окрашенные соединения с одним и тем же индикатором. Если же Mj нужно титровать с одним индикатором, а Мц — с другим, то последний, введенный после повышения pH, не должен реагировать с комплексонатом MjY.
Избирательное титрование можно выполнять, не только последовательно повышая, но и понижая pH: сначала титруют сумму катионов, затем понижают pH и оттитровыва-ют ЭДТА, выделившуюся из менее прочного комплексона-та. И, наконец, при работе с несовместимыми индикаторами проводят определение в двух аликвотных частях раствора. Рассматривая ионы металлов по группам периодической системы и сравнивая логарифмы констант устойчивости с соответствующими значениями pHmm при
lgР' = 8 (см. табл. 5.2.23), можно заключить, что большинство катионов можно титровать раствором ЭДТА уже в кислой среде.
Высокие значения IgP и соответственно низкие pHmin еще не определяют легкость взаимодействия иона металла с ЭДТА. Ионы Al, Ge(IV), Sn(IV), Ti(IV), V(IV), Cr(III), Rh(III), Pt(IV) в кислых растворах при комнатной температуре медленно реагируют с ЭДТА вследствие малой скорости замещения молекул воды на анион комплексона в их аквакомплексах [137].
Подробно кинетика образования комплексонатов (лабильность и инертность комплексов) освещена в монографиях [104, 126, 249, 939-941, 943].
Принимая во внимание, что при последовательном титровании катионов должны соблюдаться условия IgP^Y >7 и lgPJ^nY <3, можно с помощью табл. 5.2.27 предположительно оценить, возникнут ли помехи при титровании того или иного катиона (при том или другом значении pH) от катионов, сопутствующих ему.
Таблица 5.2.27
Значения lg Р' этилендиаминтетраацетатов при различных pH (7= 0,1; 18-25° С) [36]
Подгруппа MY" 4 cM,M 1 2 3 4 5 6 7 8 9 10 11 12 13 14
1Б CuY2’ 0,1 2,80 6,34 8,50 10,44 12,28 13,31 12,80 11,80 10,80 9,66 8,15 6,71 5,36 2,80
0,001 2,80 6,34 8,50 10,44 12,30 14,04 14,65 13,80 12,80 11,66 10,07 8,15 5,35 2,80
AgY3“ 0,90 1,84 2,90 4,04 5,00 6,00 6,84 7,18 6,93 5,83 3,99
ПА MgY2 0,53 2,23 4,00 5,40 6,40 7,38 8,03 6,83 5,22 4,02 2,99
CaY2 0,42 2,35 4,20 6,00 7,40 8,40 9,40 10,25 10,62 10,59 9,63 9,16
SrY2 0,45 2,13 3,90 5,30 6,30 7,30 8,15 8,53 8,56 8,30 6,34
BaY2 0,10 1,35 3,12 4,50 5,50 6,50 7,35 7,73 7,77 7,55 6,24
ПБ ZnY2 0,50 4,00 6,20 8,14 10,00 11,80 13,20 14,11 13,88 12,75 11,14 9,22 6,90 4,17
CdY2’ 0,40 3,95 5,90 8,10 10,00 11,80 13,20 14,19 15,12 15,53 14,60 12,91 10,34 7,42
HgY2 5,90 9,42 11,32 11,72 11,61 11,80 10,80 9,83 9,05 8,60 7,93 6,99 5,99 4,80
ША A1Y~ 3,21 5,61 7,71 9,51 10,54 8,93 6,90 4,90 2,75 0,19
GaY 3,08 6,94 9,70 10,14 10,34 10,83 11,07 10,28 8,54 6,26 2,97
InY 7,09 11,38 14,27 16,12 16,54 16,40 15,81 14,85 13,91 12,72 10,82 8,12 5,16 2,16
T1Y 5,53 7,98 8,70 8,75 10,02 10,66 10,51 9,59 8,61 7,46 5,84 3,91 1,91
ШБ ScY 0,1 5,10 9,60 12,44 13,35 13,50 12,30 12,70 11,70 10,71 9,50 7,80 5,83 3,83 1,83
0,001 5,10 9,60 12,50 14,63 15,47 15,30 14,70 13,53 11,96 9,99 7,88 5,84 3,83 1,83
LaY 0,1 0,84 4,60 7,30 9,40 11,30 13,10 14,50 14,95 15,68 15,00 13,25 10,64 7,30 4,30
CeY 0,40 4,90 7,80 10,00 11,90 13,70 15,10 16,10 16,96 17,06 16,69 13,20 10,26 7,27
PrY 2,70 5,60 7,80 9,70 11,50 12,89 13,80 14,28 13,59 11,77 9,38 6,00 3,00
NdY“ 3,12 5,52 7,60 9,50 12,70 13,67 14,40 14,20 12,64 10,32 7,05 4,05
ThY~ 1,20 5,20 9,69 12,18 12,60 12,46 11,26 8,92 6,56 4,56 2,36
0,001 3,27 5,20 9,70 12,57 14,23 13,46 11,26 8,92 6,57 4,56 2,36
SnOHY 0,1 19,76 21,65 21,62 20,82 19,92 18,52 16,92 14,92 10,77 8,15 5,21
PbY2 1,77 5,15 6,76 7,77 8,71 9,51 9,91 9,93 10,08 10,52 10,82 10,88 10,76 8,96
656
Новый справочник химика и технолога
Продолжение таблицы 5.2.27
Подгруппа МУ"4 См, M 1 2 3 4 5 6 7 8 9 10 И 12 13 14
0,001 1,80 5,36 7,58 9,42 10,65 11,44 11,91 11,93 12,08 12,52 12,88 12,88 11,38 8,96
BiY 0,1 9,80 13,86 15,88 17,10 18,00 18,77 17,40 14,40 11,41 8,37 5,25 2,20
СгУ 0,1 7,32 10,98 13,43 15,36 15,97 17,04 16,57 16,30 15,47 14,35 12,73 10,79 8,80 6,80
МпУ2 1,60 4,15 5,65 7,50 9,30 10,70 11,70 12,70 13,55 13,03 12,20 10,66 8,56
FeY2 0,93 6,41 8,80 9,80 11,13 12,21 12,76 12,43 11,67 10,55
FeY 0,1 7,64 11,15 12,11 12,30 12,20 12,01 11,51 10,99 10,99 10,19 10,90 10,04
0,001 7,65 11,67 13,85 14,30 14,20 14,01 13,51 12,84 12,27 11,71 10,99 10,04
CoY2" 0,1 0,40 3,93 6,05 7,95 9,80 11,60 13,00 14,00 14,93 15,41 14,87 12,99 10,98 8,98
NiY2 2,82 6,32 8,43 10,28 12,13 13,92 15,32 16,32 17,09 17,16 16,28 14,58 12,59 10,59
Последовательное титрование катионов II и других групп
последовательного титрования катионов II группы применяют метод вытеснения с одновременным осаждением. Сначала титруют сумму катионов Mg + Ва; Са + Ва; Zn (Cd, Mn) + Ва или Mg + Са + Sr + Ва; затем комплексо-нат В а разрушают, вводя MgSO4, и оттитровывают избыток ионов Mg. Если кроме комплексоната Ва в растворе образуется и комплексонат Sr, то его затем разрушают ZnSO4 и оттитровывают избыток ионов Zn. Содержание Mg, Са, Zn или Mg + Са определяют по разности. Метод основан на различной устойчивости участвующих в реакциях ком-плексонатов и применяется для анализа природных материалов; ионы РОд и SO2 мешают определению, a Cu(II), Fe(III), Со(П), и Ni(II) можно маскировать [882, 902].
Комплексонаты катионов подгруппы ПБ значительно устойчивее комплексонатов подгруппы ПА; на этом основано последовательное титрование при разных значениях pH следующих пар: Zn(Cd) и Mg [112, 158, 483, 484], Zn и Ва [360].
Метод последовательного титрования применяют для анализа электролитов гальванических ванн, смесей сульфидов, селенидов и теллуридов, а также красящих пигментов.
В табл. 5.2.28-5.2.33 показаны условия избирательного титрования катионов II—VIII групп: пределы pH титрования, применяемые индикаторы, метод определения, объекты анализа, sr — относительное стандартное отклонение (см. раздел 2.3.4).
Таблица 5.2.28
Последовательное титрование катионов П группы [36]
Определяемые катионы pH титрования Индикатор Метод определения Объект анализа зг Литература
Mg, Са, Zn, Cd или Мп(Н)+Ва 10-11 ЭХЧилиМТС Прямое титрование (в присутствии Мп(П) добавляют аскорбиновую к-ту + ТЭА Искусственные р-ры 902
Ва 10 То же Титрование избытка Mg после взаимодействия Ba-ЭДТА с MgSO4 902
Mg + Са + + Sr + Ва 10 ЭХЧ + титановый желтый + нафтоловый зеленый В Прямое титрование Природные материалы 882
Ва 10 То же Титрование избытка Mg после взаимо- действия Ba-ЭДТА с MgSO4 0,002-0,003 882
Sr 10 То же Титрование избытка Zn после взаимодействия Sr-ЭДТА с ZnSO4 0,007-0,038 882
Химические методы количественного анализа
657
Продолжение таблицы 5.2.28
Определяемые катионы pH титрования Индикатор Метод определения Объект анализа Sr Литература
Zn или Cd Mg Cd Mg Zn Ba 5,5-6,8 10 6,8 10 7 10 МТС или ЭХЧ То же ЭХЧ + Zn-ЭДТА То же ЭХЧ То же Прямое титрование Прямое титрование То же То же То же Смеси халькогенидов, р-ры для фосфатирования металлов Искусственные р-ры То же Пигменты То же 0,0024 .0,0032 112, 158, 243, 483, 510 484 484 360 360
Таблица 5.2.29
Последовательное титрование катионов III и других групп [36]
Определяемые катионы pH титрования Индикатор Метод определения Объект анализа sr Литература
Определение в одной аликвотной части р-ра
Sc 2 KO Прямое титрование Смеси оксидов 0,01 105, 778
TR или Al 6 6 To же To же То же Обратное титрование раствором Zn(NO5)2 после кипячения с избытком ЭДТА 0,01
Sc Gd 2 5,5 KO To же Прямое титрование Шихта Ga—Sc—Gd-граната после отделения Ga 0,01 0,004 301 301
Ga 2 ПКФ Прямое титрование при 100 °C Смеси оксидов 0,007 301
Gd 5,8-6,0 KO Обратное титрование избытка ЭДТА р-ром ZnSO4 301
Y 5-6 KO Прямое титрование после предварительного отделения Fe(III) экстракцией Гранаты и ферриты 0,004 171
Ca 10 ЭХЧ Обратное титрование избытка ЭДТА р-ром ZnSO4 171
Y или Yb 5-6 Арсеназо I Прямое титрование Сплавы оксидов 0,017 35
Mg, Ca, Sr или Ba 10 To же То же 35
In 2 3,5-Дибром--ПААК Прямое титрование Сплавы 0,016 92
Zn 5,2 To же То же 92
Ce(IV) 0,25 M H2SO4 Ферроин То же Искусственные р-ры — 599
Ce(III) + La 5 ПГК Прямое титрование 599
Т1(Ш) + РЬ(П) 3-6 КО Обратное титрование ЭДТА р-ром РЬ(П) Сплавы 0,04 505
Т1(Ш) 5-6 То же Титрование выделившейся ЭДТА после восстановления Т1(Ш) до Т1(1) 505
Tl(III) + Bi 2-4 ПАН или ПАОХ Прямое титрование Искусственные р-ры 0,005 56, 57
658
Новый справочник химика и технолога
Продолжение таблицы 5.2.29
Определяемые катионы pH титрования Индикатор Метод определения Объект анализа зг Литература
Т1(Ш) 2-Л Титрование выделившейся ЭДТА р-ром Cu(NO3)2 после восстановления Т1(Ш) до Т1(1) 56, 57
Th(IV) 2 Арсеназо I Прямое титрование Тоже — 166
TR 5 To же То же • 166
Th(IV) 2 ПКФ Прямое титрование Тоже 0,02 233
Ce(III) 10 Мурексид Обратное титрование избытка ЭДТА р-ром NiCl2 233
Th(IV) 3,5 КО Прямое титрование Искусственные р-ры 619
U(IV) 3,5—4,0 То же То же после восстановления U(VI) до U(IV) 619
Определение в двух аликвотных частях р-ра
La, Sc или Y 4,5-6 Арсеназо I Прямое титрование Смеси и сплавы оксидов 0,009-0,02 212,213
Mg + La, Sc или Y 10 ЭХЧ То же 0,009-0,012 (Mg) 212,213
Y 5-6 Арсеназо I То же Смеси оксидов — 212
Ca + Y 10-11 Мурексид Обратное титрование избытка ЭДТА р-ром NiSO4 [212]
Таблица 5.2.30
Последовательное титрование катионов IV и других групп [36]
Определяемые катионы pH титрования Индикатор Метод определения Объект анализа 8Г Литература
Определение в одной аликвотной части р-ра
Ti(IV) 1 KO Обратное титрование избытка ЭДТА р-ром Bi(NO3)3 в присутствии Н2О2 Силикатные материалы после отделения Fe(III) экстракцией; окрашенные силикатные стекла 187,315
Al 5 6 To же То же р-ром соли цинка 0,009 187,315
или Се(Ш)+А1 5,8 To же То же 0,013 187,315
Zr 0,5 M HNO3 To же Прямое титрование при 70-90 °C Сплавы, специальные стекла, керамика, цементы, шлаковая корка электрода 64, 153, 194, 344, 347, 348, 778-780, 798
Zn 5 To же То же, 20 °C 0,013
Al 5 6 To же Обратное титрование р-ром Zn(II) после кипячения с избытком ЭДТА
TR 5-6 To же Прямое титрование 778
Th(IV) 1,5-3 To же То же 0,01 64
Ti(IV) 1-2 То же в присутствии Н2О2 или обратное титрование избытка ЭДТА р-ром Bi(NO3)3 0,005 798
РЬ(П) 5-6 МТС Прямое титрование Сплавы, смеси силикатов 0,03 136, 151, 152
Ca 10-11 To же То же 152
In 3 КО Прямое титрование, 90 °C Сплавы 0,003 401
Pb(H) 5 To же То же, 20 °C
Химические методы количественного анализа
659
Продолжение таблицы 5.2.30
Определяемые катионы pH титрования Индикатор Метод определения Объект анализа Sr Литература
Определение в двух аликвотных частях р-ра
Ti(IV) 0,1 M H2SO4 KO Обратное титрование избытка ЭДТА р-ром Bi(NO3)3 в присутствии Н2О2 Алюминид титана, катализаторы 0,008 212,714
Ti(IV) + Al 5,5-6,0 To же То же р-ром Bi(NO3)3 или ZnCl2 0,002 (Al) 212,714
Ti(IV) 1,3 KO Обратное титрование избытка ЭДТА р-ром Bi(NO3)3 в присутствии Н2О2 Титанаты 0,01 301
Ti(IV) + Al 5,5-5,8 To же То же 301
Ge(IV) + La 5-6 Обратное титрование р-ром Zn(II) после кипячения с избытком ЭДТА Германид лантана — 212
La 7 Арсеназо I Прямое титрование
Zr 1 M H2SO4 KO Прямое титрование Алюминид циркония — 212
Zr + Al 5,5 To же Обратное титрование р-ром ZnCl2 после выдержки с избытком ЭДТА 212
Zr 0,16 M H2SO4 To же Обратное титрование избытка ЭДТА р-ром Bi(NO3)3 Сплавы на основе Nb и других металлов 0,003 106-109
Zr + Ti(IV) 0,08 M H2SO4 To же То же в присутствии Н2О2 106-109
Pb(II) Mo(V) + Pb 5,5-6 8 KO ЭХЧ Прямое титрование Обратное титрование избытка ЭДТА р-ром соли Zn Молибдат свинца 0,001 0,009 (Мо) 948 948
Таблица 5.2.31
Последовательное титрование катионов V и других групп [36]
Определяемые катионы pH титрования Индикатор Метод определения Объект анализа Sr Литература
Определение в одной аликвотной части р-ра
V(V) + Pb(II) 5-6 Дифенил-карбазон Прямое титрование Ванадат свинца 0,005 165
V(V) КХТС Титрование р-ром соли ЭДТА, выделившейся из уо2-эдта То же 165
Sb(III) + Zn, Pb(II) или Bi 2,5-3,0 ПАР + Cu(II) -ЭДТА Прямое титрование Сплавы 0,01 402
Sb(III) 4,5-5 То же Нагревание до кипения и титрование р-ром CuSO4 ЭДТА, выделившейся из Sb-ЭДТА 402
Bi 2MHNO3 ПКФ Прямое титрование Искусственные р-ры 0,005 528
Cu(II) 5 ПАН То же 0,002 528
Bi 0,5-2,0 КО Прямое титрование Сплавы, специальные стекла, концентраты 0,003 29, 62, 182, 376, 387, 396, 605, 653, 685, 729
Zn, Cd, РЗЭ или Pb(II) 5-6 То же То же
660
Новый справочник химика и технолога
Продолжение таблицы 5.2.31
Определяемые катионы pH титрования Индикатор Метод определения Объект анализа Sr Литература
Bi 1,5-2 То же То же Сплавы Bi—Pb—Sn после отделения Sn экстракцией 0,003 422
Pb 5 То же То же То же 0,006 422
Bi 1-2 МТС То же Сплавы, медицинские препараты 0,007 . 130, 456
Zn, Cd, Pb(II) или Fe(II) 5-6 То же То же
Bi 1,4-2,5 ТАК То же Искусственные р-ры 0,02 77
Pb 5,5-6,0 То же То же То же 0,04 77
Определение в двух аликвотных частях р-ра
Bi 1-2 КО Прямое титрование Искусственные р-ры 734
Bi + Ti(IV) 5 То же Обратное титрование избытка ЭДТА р-ром Bi(NO3)3 в присутствии Н2О2 То же 734
Таблица 5.2.32
Последовательное титрование катионов VI и других групп [36]
Определяемые катионы рн титрования Индикатор Метод определения Объект анализа зг Литература
Определение в одной аликвотной части р-ра
Zn 5,5 КО Прямое титрование 50 °C Промышленные катализаторы, гальванические покрытия 0,002 9
Сг(Ш) 2 КО + БФС Обратное титрование р-ром Bi(NO3)3 после кипячения с избытком ЭДТА 9
А1 + Сг(Ш) 5 ССК Обратное титрование р-ром FeCl3 при 40 °C после кипячения с избытком ЭДТА Дубильные р-ры 707
А1 1 То же Обратное титрование р-ром FeCl3 при 60 °C ЭДТА, выделившейся при подкислении 707
А1 5-6 Тирон Обратное титрование р-ром FeCl3 после нагревания при 50 °C с избытком ЭДТА То же 0,016 708
Сг(Ш) 5-6 То же То же после кипячения с избытком ЭДТА 708
Sn(IV) 2 КО Обратное титрование р-ром Bi(NO3)3 после нагревания до кипячения с избытком ЭДТА Сплавы, гальванические покрытия 0,006 9
СДШ) 4-5 То же То же после кипячения с избытком ЭДТА 9
Fe(III) 1-2 ССК Прямое титрование при 45 °C Электролиты для хромирования после отделения СгОДП) 0,02 14
Химические методы количественного анализа
661
Продолжение таблицы 5.2.32
Определяемые катионы pH титрования Индикатор Метод определения Объект анализа sr Литература
Сг(Ш) 5,5 КО Обратное титрование р-ром Zn(CH3COO)2 после кипячения с избытком ЭДТА 14
Fe(III) 1-2 сск Прямое титрование Руды, сплавы — 706
Al 5-6 То же Обратное титрование р-ром FeCl3 после выдержки 15 мин с избытком ЭДТА 706
Cr(III) 5-6 То же То же после кипячения с избытком ЭДТА 706
Fe(III) + Ni(II) 5-6 КО Обратное титрование избытка ЭДТА р-ром Pb(NO3)2 Сплавы 0,004 787
Cr(III) 5-6 То же То же после кипячения с избытком ЭДТА 787
Fe(III) 1 ССК Прямое титрование Искусственные р-ры 0,06 532
Zn 5-6 ПАН То же 532
Cr(III) 5-6 То же Обратное титрование р-ром CuSO4 после кипячения с избытком ЭДТА 532
Fe(III) 1 ССК Прямое титрование Нержавеющие стали 0,008 530
Ni(II) 8 Мурексид То же 0,015 530
Cr(III) 4-5 ССК Обратное титрование р-ром FeCl3 после кипячения с избытком ЭДТА 0,003 530
Определение в двух аликвотных частях р-ра
Zn или Си(И)+ + Сг(Ш) 5-6 КО Обратное титрование р-ром ZnSO4 после кипячения с избытком ЭДТА Катализаторы, смеси оксидов 0,002 134, 771
Zn или Cu(II) 5-6 То же Прямое титрование 134, 771
Cr(III) + Co(III) 0,5-1,0 ПКФ Обратное титрование р-ром Bi(NO3)3 после кипячения с избытком ЭДТА и окисления Со(И) до Со(Ш) Искусственные р-ры 582
Co(III) 0,5-1,0 То же То же без кипячения 582
Fe(III) + Cr(III) 5 ПКФ Обратное титрование р-ром CuSO4 после выдержки с избытком ЭДТА в присутствии катализатора (аниона или растворителя) Катализаторы для конверсии оксида углерода, нержавеющие стали после отделения Ni 0,01 611,769
5 или 10 Мурексид То же р-ром NiSO4 611,769
Fe(III) 2 ССК Прямое титрование 611,769
Mo(V) 5 КО Обратное титрование избытка ЭДТА р-ром Zn(NO3)2 после восстановления Mo(VI) до Mo(V) Молибдат стронция или бария 0,005 115
Sr или Ba 10 ЭХЧ + Mg--ЭДТА Прямое титрование 0,002
Mo(V) + Zn, Cd или Pb(II) 5 КО Обратное титрование избытка ЭДТА р-ром Zn(NO3)2 после восстановления Mo(VI) до Mo(V) Молибдат Cd, Pb или Zn 0,006 (Мо) 115
Zn, Cd или Pb(II) 5 То же Обратное титрование избытка ЭДТА р-ром Zn(NO3)2 0,007 (Zn, Cd), 0,003 (Pb) 115
662
Новый справочник химика и технолога
Последовательное титрование катионов VIII и других групп [36]
Таблица 5.2.33
Определяемые катионы pH титрования Индикатор Метод определения Объект анализа sr Литература
Fe(III) 1-2 сск Прямое титрование при 50-70 °C Объекты разные 27, 47,48,51, 131, 151, 158, 192, 195, 197, 200-202, 254, 267, 289, 321, 380, 385, 393, 410, 433, 524, 527, 533, 598, 682, 705, 798
В том же р-ре в зависимости от анализируемого объекта
Zn 6 МТС Прямое титрование Искусственные р-ры — 682
Al 5-6 сск Обратное титрование р-ром FeCl3 после кипячения с избытком ЭДТА Глины, тальк, металлургические шлаки, сплавы 0,01 47, 151, 195, 321,393,410
Al или Al + Ti(IV) 5-6 ко То же раствором Zn(II) или РЬ(И) Минеральное сырье, сплавы, электролиты, огнеупорные материалы 0,01 27, 158, 197, 200, 385, 527
Al + V(IV) или V(IV) 5 То же Обратное титрование р-ром ZnSO4 после восстановления V(V)floV(IV) Ванадат железа, феррованадий, отложения на котлах, зола 47, 151, 192
TR 5 То же Прямое титрование Сплавы — 151,152
Pb(II) 5 То же То же РЬ-шлаки 0,002 202
5 ПАН + Си(П)-ЭДТА То же Искусственные р-ры — 201,202
5 КО То же Силикат свинца — 152
Mg 10 кхтс То же
Mn(II) 5-6 сск Обратное титрование р-ром FeCl3 Марганцевые руды — 533
4,8-5 Тиояблочная к-та То же р-ром CuSO4 То же 0,006 512
6 ПАН То же р-ром CuSO4 при 90 °C Бронзы после отделения А1 и Си — 289
Mn(II) 6,5 МТС Прямое титрование Искусственные р-ры — 682
10 ТФТ Прямое титрование в присутствии винной к-ты + NH2OHHC1 или аскорбиновой к-ты Ферромарганец 0,0016 380
Mn(II) 6 КО То же в присутствии NH2OH-HC1 Силикат марганца — 151
Mg 10 хтс То же То же 151
Fe(II) 5-6,5 МТС или ТГК Прямое титрование Руды — 131,682
Fe(II) 1,0-1,5 ССК То же после окисления Fe(II) до Fe(III) Лечебная грязь 20
Co(II) 5,5 КО Обратное титрование р-ром соли Zn Сплавы 0,003 51
6-7 Мурексид Прямое титрование Искусственные р-ры 0,01 524
Al 4-5 ССК Обратное титрование р-ром FeCl3 после кипячения с избытком ЭДТА 524
В другом р-ре, в зависимости от анализируемого материала
Химические методы количественного анализа
663
Продолжение таблицы 5.2.33
Определяемые катионы pH титрования Индикатор Метод определения Объект анализа sr Литература
Fe(III) + Zn 8 Мурексид Обратное титрование жствором N1SO4 Сплавы на основе железа — 254
Fe(III) + Al 4,5 Дитизон Обратное титрование р-ром ZnSO4 после кипячения с избытком ЭДТА Каолин 0,01 598
3,5 Ализарин S То же р-ром Th(NO3)4 Цемент, доменные шлаки — 705
5,5 КО То же р-ром ZnCl2 или Bi(NO3)3 Огнеупорные материалы, сплавы, электролиты — 48, 267,433
Fe(III) + Al + + La 5,5 То же То же р-ром ZnSO4 Алюминаты и силикаты TR — 259
Fe(III) + Ti(IV) 1-2 ССК Прямое титрование в присутствии Н2О2 при 40 °C Отходы титанового производства, силикаты — 151
Fe(III) + V(V) 2 То же То же Сплавы Fe—Al—V 0,003 151, 198
Fe(III) + Al 5,5 То же Обратное титрование раствором FeCl3 после нагревания до 80 °C с избытком ЭДТА 151, 198
Определение в одной аликвотной части р-ра
Fe(III) 2 Салициловая к-та Прямое титрование Огнеупорные материалы, руды, минералы 0,01 17, 19
Al 5 КО Обратное титрование избытка ЭДТА р-ром Zn(CH3COO)2 17, 19
Fe(III) 3,5 ЭХЦ Прямое титрование Металлургические шлаки 0,01 899
Al 3,5 То же То же при 100 °C 899
Fe(III) 2-3 KSCN Прямое титрование Доломит 0,01 851
Al 5 3,3-Диметил-нафтидин + ферро-, феррицианид Обратное титрование избытка ЭДТА р-ром ZnSO4 851
Fe(III) 2 о-Дианизидин Прямое титрование Ферромарганец, стали 0,01 446
Mn(II) 7 ЭХЧ То же 446
Co(III) 0,5-0,7 ПКФ Обратное титрование избытка ЭДТА р-ром Bi(NO3)3 с фотометрической индикацией Искусственные р-ры 0,004 582
Cu(II), Zn, Cd, Pb(II) или Mn(II) 5,5 То же Прямое титрование 582
Co(II) + Ni(II) 9 Мурексид Прямое титрование То же 0,006 (Ni) 585
Co(II) 2 ПКФ Титрование выделившейся ЭДТА р-ром Bi(NO3)3 при 0°С
Ni(II) + Cd или Zn 9,7 Мурексид Прямое титрование Активная масса никелевого электрода 0,01 307
664
Новый справочник химика и технолога
Продолжение таблицы 5.2.33
Определяемые катионы pH титрования Индикатор Метод определения Объект анализа Sr Литература
Cd или Zn 2,5 КО Титрование выделившейся ЭДТА р-ром Bi(NO3)3 при 0°С 0,05 307
Ni(II) 2,5-2,8 ПАН + Си(П)-ЭДТА Прямое титрование при 90 °C Смеси оксидов 0,01 403
Mn(II) 9 « То же 403
Определение в двух яликвотных частях р-ра
Fe(IIl) 2-3 Тирон или салициловая к-та Прямое титрование Керамические материалы, руды, минералы 0,01 18,847
Fe(III) + Al 5 КО или салициловая к-та Обратное титрование избытка ЭДТА р-ром Zn(CH3COO)2 или FeCl3
Fe(III) + Ti(IV) 2-3 Салициловая к-та Прямое титрование в присутствии Н2О2 при 50-60 °C 208
Co(III) 0,5-0,7 ПКФ Обратное титрование избытка ЭДТА р-ром Bi(NO3)3 Искусственные р-ры 582
Co(III) + Ni(II) 2 То же То же при 0 °C 582
Co(II) + Mn(II) 5 КО Обратное титрование избытка ЭДТА р-ром Pb(NO3)2 при 95 °C Ферриты после отделения железа 0,01 788
Co(II)+Mn(II)+ + Mg 10 ТФТ То же р-ром MgCl2 в присутствии аскорбиновой к-ты
Ni(II) 5 ССК Обратное титрование избытка ЭДТА р-ром FeCl3 Электролиты, соли 0,005 112, 158, 305,418
Ni(II) 5 ПАН То же р-ром CuSO4 при 90 °C
Ni(II) + Mg 10 Мурексид Прямое титрование
Pd(II) 5,5 КО Обратное титрование избытка ЭДТА р-ром Zn(II) Р-ры, сплавы и отходы благородных металлов — 388,419,420
Pd(II) + Pt(IV) 5,5 То же То же после кипячения с избытком ЭДТА 388, 419, 420
Селективное комплексонометрическое титрование с применением маскирующих реагентов
Селективное комплексонометрическое титрование одного катиона в присутствии других возможно, главным образом, благодаря применению маскирующих реагентов (МР). Маскирующими называют те реагенты, которые устраняют действие мешающих ионов без их выделения из системы. Маскирующие вещества, применяемые в ком-плексонометрии, — это соединения, препятствующие образованию тех или иных комплексонатов металлов. МР понижает концентрацию свободного маскируемого иона и уменьшает величину условной константы комплексоната. С этой точки зрения рассматриваются следующие виды маскирования.
1. Маскирование изменением pH среды (см. выше).
2. Маскирование осаждением. Иногда возможно титрование в присутствии осадка, образованного осадителем и маскируемым катионом. Для этого осадок не должен реагировать с титрантом и индикатором, а также должен ничтожно адсорбироваться.
3. Маскирование окислением или восстановлением. Возможно в тех случаях, когда катион в одном из своих валентных состояний не реагирует с комплексоном-титрантом в заданных условиях.
4. Кинетическое маскирование. Основано на замедленной скорости взаимодействия какого-либо катиона с титрантом в данных условиях. Так, можно титровать Fe(III) [725], Zn или Sn(IV) [9] в присутствии Сг(Ш), практически не реагирующего с ЭДТА при комнатной температуре.
Химические методы количественного анализа
665
5. Самый важный и наиболее распространенный вид маскирования — комплексообразование, когда МР селективно образует с мешающим катионом комплекс, более прочный, чем комплексонат определяемого элемента.
В комплексонометрии к МР предъявляются следующие требования — их комплексы с маскируемыми катионами должны быть настолько устойчивыми, чтобы не реагировать с комплексоном-титрантом и применяемым индикатором. Они также не должны быть интенсивно окрашены.
При использовании МР следует различать два основных метода маскирования.
Прямое маскирование — титрование определяемого элемента при маскировании мешающих элементов, а также его разновидность — титрование двух аликвотных частей раствора, в одной из которых маскируют определяемый элемент, а затем определяют его по разности.
Обратное маскирование — титрование всех связываемых комплексоном ионов металлов, избирательное разру-
шение комплексоната определяемого металла соответствующим МР и титрование выделившегося комплексона.
Второй метод, как правило, дает более точные результаты, предоставляя возможность титровать два катиона в одной аликвотной части раствора или же определять какой-либо катион в присутствии других, не реагирующих с маскирующим реагентом.
При выборе МР для массовых анализов важны эффективность маскирования, а также отсутствие токсичности, устойчивость растворов МР и их доступность. В последние годы стало практиковаться одновременное применение двух и более МР, т.е. одновременное маскирование различных катионов при титровании одного определяемого катиона.
В табл. 5.2.34-5.2.42 из [36, 46] приведены неорганические и органические реагенты, применяемые в качестве МР в комплексонометрии.
Таблица 5.2.34
Маскирующие реагенты [46]
Маскирующий реактив Маскируемые ионы Титруемые ионы Условия титрования Индикатор
Аскорбиновая к-та Сг3+ Ca2+, Mn2+, Ni2+ Добавление реактива к кислому р-ру, кипячение (образование Сг3+-комп.), подщелачивание, обр. титр, солью Са2+ Тимолфталексон
Си2+ Zn2+ Аммиачный р-р + реактив до обесцвечивания, прям. титр. Мурексид
Аскорбиновая к-та + CN “ CN “-группа*, Fe3+ Ba2+, Ca2+, In3+, Mg2+, Pb2+, TR, Sr24 Прям титр., pH = 8-10 Эриохром черный Т
Аскорбиновая к-та + CN + диэтилдитиокарбаминат + Г CN “-группа, Cu2+, Fe3+, Pb2+ Mn2+ Прям, титр., pH = 10 То же
Аскорбиновая к-та + CN ~ + F +Г + S2O2“ CN “-группа, Al3+, Cu2+, Fe3+ Mn2+ Прям, титр., pH = 10, 50-60 °C То же
Аскорбиновая к-та + CN “ + Г CN -группа, Cu2+, Fe3+ Al3+ + Mn2+ + Pb2+ Обр. титр, избытка ЭДТА солью Мп2+, рН= 10 То же
Аскорбиновая к-та + CN " +1 + сульфосалициловая к-та CN -группа, Fe3+, Al3+, Cu2+ Mn2+ Прям, титр., pH = 10 То же
Аскорбиновая к-та + CN“ + тайрон CN -группа, Fe3+ малые к-ва Al3+nTi(IV) Mn2+, Pb2+ Добавление аскорбиновой кислоты, нейтр. до pH = 10, ЭДТА в избытке, нагрев до обесцвечивания NH j-буфер (pH =10) + тайрон, обр. титр. Mg2+ То же
Аскорбиновая к-та +1 Cu2+ (осаждение Cui) Mn2+ Обесцвечивание 1г добавлением S2O2 прям, титр., pH = 10 Эриохром черный Т
Аскорбиновая к-та ~ I или SCN“ Cu2+ (осаждение Cui или CuSCN “) Zn2+ Прям, титр., гексаметилентетрамин, pH = 6 Ксиленовый оранжевый
Аскорбиновая к-та + тиокарбамид Cu2+ (образование комплекса Cui c тиокарбамидом) Pb2+, Zn2+ Прям, титр., pH = 5,5 ПАР
Гидрокарбонат-ионы UO2+ Zn2+ Прям, титр., pH = 8-8,5 Мурексид
666
Новый справочник химика и технолога
Продолжение таблицы 5.2.34
Маскирующий реактив Маскируемые ионы Титруемые ионы Условия титрования Индикатор
Димеркапрол (2,3-димеркаптопропанол; BAL) As3+, Bi3+, Cd2+, Hg2+, Pb2+, Sb3+, Sn(IV), Zn2+, мал. ко-ва Co2+Ni2+. Если есть Fe, добав. TEA Mg2+, Ca2+, Mn2+ Добавление реактива, подщелачивание, pH = 10 (если есть Мп2+, предв. ввод. TEA и NH2OH), прям. титр. Эриохром черный Т
Димеркапрол + CN Cd2+, Co2+, Cu2+, Hg2+, Ni2+,Zn2+H мал. кол-ва Bi3+, Fe2+ и Pb2+ Sc3+ Прям, титр, буфер pH = 8, 70 °C Мурексид
Димеркапрол + ОН” + TEA Al3+, Bi3+, Fe3+, Mg2+, Pb2+ Ca2+ Прям, титр., pH > 12 Мурексид
Диэтилдитиокарбаминат (купраль) Иодид-ионы Cd2+ (осажд.) Zn2+ Прям, титр., pH = 10, 60 °C Эриохром черный Т
Pb24+ (осажд.) Mn2+ Прям, титр., pH = 10 То же
Pb2+ Ca2+ Прям, титр., pH > 12 Мурексид и др. индикаторы Са2+
Fe3+ Ca2+ + Mg2+ Прям, титр., pH = 10 Эриохром черный Т
Hg2+ Cu2+ Прям, титр., pH = 7, 70 °C ПАН
2,4-Пентандион (ацетилацетон) Al3+, Fe3+, Pd2+ (осаждение) Pb2+, Zn2+ Прям, титр., pH = 5-6, гексаметилентетрамин Ксиленоловый оранжевый
Mo(VI) (осаждение) Bi3+ После осажд. Мо приводят pH к 1-1,5, прям. титр. То же
2,4-Пентандион + нитробензол Fe3+, UO2+ Pb2+, Zn2+ Если есть Fe2+, прибавляют Н2О2, не отделяя слоя нитробензола, прям. титр, при pH = 5-6 Ксиленоловый оранжевый
Пероксид водорода Ti(IV) Mg2+, Zn2+ и др. К кислому р-ру прибавляют Н2О2, подщелачивают до pH = 10, быстро прям. титр. Эриохром черный Т
W(VI) Cu2+, Ni2+ Прям, титр., горячий р-р, pH = 3-4 ПАН
W(VI) Cd2+, Fe3+, Zn2+ Прям, титр., горячий р-р, pH = 3-6 Си-ПАН
W(VI) Th(IV) К р-ру НСЮ прибавляют избыток NH3, Н2О2, НСЮ4, ЭДТА, обр. титр, солью Th(IV) Ализариновый красный С
Пирофосфат-ионы Fe3‘,Cr’' Co2+ Прибавляют Na4P2O7, NH3 до pH = 8, NH4SCN, равный объем ацетона, прям. титр, до исчезновения окраски nh4scn
Роданид-ионы Hg2+ Bi3+ Прям, титр., pH = 0,7-1,2 Метилтимоловый синий
Сульфат-ионы Ba2+ (осаждение) Mn2+, Zn2+ Прям, титр., pH = 10 Эриохром красный Б
Pb2+ (осаждение) Sn2+ и Sn(IV) Р-р H2SO4 (1 : 1), кипячение, охлаждение, добавление ЭДТА в избытке, NH4C2H3O2 до pH = 2-2,5, немного твердого Bi(NO3)3, прям. титр. ЭДТА Ксиленоловый оранжевый
Th(IV) Bi3+, Fe3+ Прям, титр., pH = 1,5 Тиокарбамид
Th(IV), Ti(IV) Zr(IV) Обр. титр, избытка ЭДТА солью Bi3+ То же
Химические методы количественного анализа
667
Продолжение таблицы 5.2.34
Маскирующий реактив Маскируемые ионы Титруемые ионы Условия титрования Индикатор
Сульфид-ионы Fe3+, мал. к-ва др. тяж. мет. Ca2+, Mg2+, Ca2+ + Mg2+ Прям, титр., щел. р-р Эриохром черный Т, мурексид (на Са2)
Сульфид-ионы + гидро-ксид-ионы Mg2+, Мп2+ Ca2+ К нейтр. р-ру прибавляют Na2S + +NaOH, через 3-5 мин прям, титр. Любой индикатор на Са2+
Сульфосалициловая к-та А13+ Mn2+ Прям, титр., pH = 10, аскорбиновая к-та Эриохром черный Т
А13+ (малые кол-ва) Zn2+ Прям, титр., pH = 10 То же
Тайрон А13+ Zn2+ Прям, титр., pH = 5,2 Ксиленоловый оранжевый
Al3+, Ti(IV) Mn2+ Обр. титр, избытка ЭДТА солью Мп2+, pH = 10, аскорбиновая к-та Эриохром черный Т
Тартрат-ионы или винная к-та А13+ Zn2+ Прям, титр., pH = 5,2 Ксиленоловый оранжевый
Al3+, Fe3+ Ca2+, Mg2+ Прям, титр., pH = 10 (аскорбиновая к-та при определении Мп2+) Си-ПАН
Al3+. Fe3+, малые K-eaTi(IV) Ca2+ Прям, титр., pH > 12 Кальцеин или другой индикатор на Са2+
Nb(V), Ta(V) Zr(IV) Обр. титр, избытка ЭДТА солью Bi3+, pH = 2-2,2, НС1 Тиокарбамид
Sb3+, Sn(IV), Zr(IV) Bi3+ Прям, титр., pH = 1,5-2 Тиокарбамид
Sb3+, UO24 Cd2+, Co2+, Cu2+, Ni2+, Zn2+ Прям, титр., pH = 5-6, горячий р-р ПАН или Cu-ПАН **
Ti(IV) Ni2+ Прям, титр., +NH3 Мурексид
Mg2+, Mn2+, Zn2+ Прям, титр., pH = 9-10, NH2OH (аскорбиновая к-та при определении Мп2+) Эриохром черный Т
UO2+ Cd2+, Cu2+, Ni2+ Прям, титр., pH = 5,3-5,9 Cu-нафтилазоксин *
W(VI) Cd2+, Fe3+, V5+, Zn2+ Прям, титр., pH = 3-4, горячий р-р Cu-ПАН *
Co2+, Ni2+, Fe3+ Обр. титр, избытка ЭДТА солью Си2+, pH = 3,5-3,8, горячий р-р ПАН
Ti(IV) Обр. титр, избытка ЭДТА солью Си2+, добавление нескольких капель 30% Н2О2, pH = 4,5 Нафтил азоксин
Тартрат-ионы + тиосуль-фат-ионы Cu2+, W(VI) Ni2+ Прям, титр., pH = 4, 70 °C ПАН
Тартрат-ионы + TEA Al3+, Fe3+, Ti(IV) Ca2+ + Mg2+ Прям, титр., pH = 10 Эриохром черный Т
Тетраэтиленпентамин (tetren) Cd2+ Cu2+, Hg2+, Zn2+ Mg2+ Прям, титр., pH = 10 То же
Cd2+, Zn2+ Ba2+ Прям, титр., pH = 12 Метилтимоловый синий
Hg2+, Ni2+, Zn2+ Pb2+ Прям, титр., pH = 12, тартрат NH3 То же
Тиогликолевая (меркап-тоуксусная) к-та Ag\ Cd2+, Cu2+, Pb2+, Zn2+ Mn2+, Ni2+ К кислому р-ру приливают реактив, NH3 до растворения осадка и ЭДТА, обр. титр, солью Са2+ (при определении Мп2+ аскорбиновая к-та) Т имол фталексон
668
Новый справочник химика и технолога
Продолжение таблицы 5.2.34
Маскирующий реактив Маскируемые ионы Титруемые ионы Условия титрования Индикатор
Тиокарбогидразид (тиокарбазид) Си2+ Sn(IV) Обр. титр, избытка ЭДТА солью Th(IV), pH = 2 Ксиленоловый оранжевый
Тиокарбамид Си2+ Fe3+ К слабокислому р-ру прибавляют немного NH4F (маскирование Fe3+), тиокарбамид, ЭДТА в избытке, нейтр. до pH = 5-5,5 гексаметилентетрамином, обр. титр, солью РЬ2+ То же
Си2* Ni2+, Sn(IV) Обр. титр, избытка ЭДТА солью Th(IV), pH = 2 или (для Ni2+) pH = 4-5 То же
Си2+ Pb2+ + Sn(IV) Обр. титр, избытка ЭДТА солью Th(IV), гексаметилентетрамин, pH = 6 Ксиленоловый оранжевый
Тиосемикарбазид Hg2+ Bi3+, Cd2+, Pb2+, Zn2+ Прям, титр., pH = 5-6,5, для определения Bi3+, pH = 1-2 То же
Cd2+, Pb2+, Zn2+ Прям, титр., pH = 5 ПАН
Тиосульфат-ионы Си2+ Cd2+, Zn2+ Прям, титр., pH = 5-6 ПАН
Ni2+ Прям, титр., pH = 4, 70 °C. Добавление Na2S2O3 к нейтр. р-ру, ацетатного буфера, pH = 8,5-9, прям. титр. Мурексид
Pb2+ К нейтр. р-ру прибавляют Na2S2O3 до обесцвечивания, прям, титр., pH = 5 Ксиленоловый оранжевый
Триэтаноламин (TEA) А13+ In3+ Прям, титр., pH = 10 или обр. титр, избытка ЭДТА Эриохром черный Т
Mg2+ Прям, титр., охлаждение р-ра, pH = 9-10 Эриохром черный Т
А13+ (мал. к-ва) Zn2* Прям, титр., pH = 10 То же
Al3+, Cr34, Fe3+ Ca2+ К слабокислому р-ру прибавляют TEA, затем NH3, кипятят, охлаждают ЭДТА в избытке, разбавление, обр. титр, солью Са2+ Тимолфталексон или метилтимоловый синий
Al3+, Fe3+ Mn2+ К кислому р-ру прибавляют TEA, NH2OH, подщелачивают, прям, титр. Тимолфталексон
Al3+, Fe3+ и мал. к-ваМп2+ Ca2+ Прям, титр., pH > 12 Мурексид, любой индикатор на Са2+
Al3+, Fe3+, Ti(IV)H малые к-ва Мп2+ Ni2+ Прям, титр., NH3 Мурексид
Al3+, Fe3+, Sn(IV), Ti(IV) Cd2+, Mg2+, Mn2+, Pb2+, Zn2+, TR3+ К кислому р-ру прибавляют TEA, NH3 до pH = 10 (NH2OH, при определении Мп2+), прям. титр. Эриохром черный
Триэтилентетрамин (trien) Cu2+, Hg2+ Pb2+, Zn2+ Прям, титр., pH = 5, гексаметилентетрамин Ксиленоловый оранжевый
Унитиол Bi2+, Cd2+, Hg2+, Pb2+, Sn2+, Zn2+ Ba2+, Sr2' Прям, титр., NH3, pH = 11 Фталеинкомлексон
Bi3+, Cd2+, Pb2+, Sn2+, Zn2+ Mg24, Ca2+ + Mg2+, Ni2+ Прям, титр., pH = 10 Эриохром черный Т
Химические методы количественного анализа
669
Продолжение таблицы 5.2.34
Маскирующий реактив Маскируемые ионы Титруемые ионы Условия титрования Индикатор
1,10-Фенантролин Cd2+, Со2+, Си2+, Mn2+, Ni2+, Zn2+, uo2+ In3+ Прям, титр., pH = 3, 50-60 °C Ксиленоловый оранжевый
Cd2+, Co2+, Mn2+, Ni2+, Zn2+ Al3+ К кислому р-ру прибавляют ЭДТА в избытке, кипятят 2 мин, охлаждают, приливают индикатор и гексаметилентетрамин до бледно-оранжевой окраски, затем фенантролин, обр. титр, солью РЬ2+* То же
Cd2+, Zn2+ Pb2+ Прям, титр., pH = 5-6, гексаметилентетрамин То же
Фосфат-ионы W(VI) Cd2+, Fe3+, V(V), Zn2+ Прям, титр., pH = 3-6, горячий р-р Си-ПАН
Cu2+, Ni2+ Прям, титр., pH = 3—4, горячий р-р ПАН
Co2+, Mo(VI) Обр. титр, избытка ЭДТА солью Си2+, pH = 4-5 То же
Фторид-ионы Al3+ Cd2+, Cu2+, Pb2+ Прям, титр., pH = 6,8 (для Cd2+), pH = 5-6 (для Сп2+, РЬ2+). При определении Cd2+ и РЬ2+ вводят Си-ЭДТА Нафтилазоксин
Cu2+ Прям, титр., pH = 3-3,5 pH = 4-5 Нафтил азоксин. Пирокатехиновый фиолетовый
Ga3+ Прям, титр., pH = 1,6-2, кипячение pH = 4,5-6 ПАН Морин при УФ-освещ.
In3+ Обр. титр, избытка ЭДТА солью Zn2+, пиридин Эриохром черный Т или пирокатехиновый фиолетовый
Zn2+ Прям, титр., pH = 5-6 Ксиленоловый оранжевый
Zr(IV) Прям, титр., 0,01-0,5 М НС1, 50% метиловый спирт, горячий Р-Р Эриохром синечерный Т
Al3+, Ca2+, Mg2 Ni2+ Обр. титр, избытка ЭДТА солью Мп2+, аскорбиновая к-та, pH = 10 Эриохром черный Т
Al3+, Fe3+ Cu2+ Прям, титр., pH = 6-6,5 Хромазурол С
Al3+, Ti(IV) Cu2+ Прям, титр., pH = 6 Пирокатехиновый фиолетовый
Fe3+ Cd2+, Cu2+, Zn2+ Прям, титр., pH = 5-7 ПАН
Nb(V), Ta(V) Bi3+ Прям, титр., pH = 1,5-2 Тиокарбамид
Nb(V),Ta(V), Ti(IV) Cu2+, Zn2+ Прям, титр , pH = 6-6,4 Нафтилазоксин или ПАН
Sn(IV) Sn2+ Прям, титр., pH = 5,5-6, пиридин + ацетат Метилтимоловый синий
In3+ Прям, титр., pH = 2,5, горячий р-р Си-ПАН
670
Новый справочник химика и технолога
Продолжение таблицы 5.2.34
Маскирующий реактив Маскируемые ионы Титруемые ионы Условия титрования Индикатор
Фторид-ионы Pb2+ Прибавляют H2O2 для окисления всего Sn2+ до Sn(IV), затем F“, кипячение, охлаждение, обр. титр, избытка ЭДТА солью Zn2+, pH = 10 Эриохром черный Т
W(VI) Cd2+, Fe2+, V(V), Zn2+ Прям, титр., pH = 3-6, горячий р-р ПАН
Co2+Mo(VI) Обр. титр, избытка ЭДТА солью Си2+, pH = 4—5, горячий р-р ПАН
Cu2+, Ni2+ Прям, титр., pH = 3-4, горячий р-р То же
Фторборат-анионы А13+ Прям, титр., pH = 3,8 Пирокатехиновый фиолетовый
Цианид-ионы CN “-группа Ba2+, Sr2+ Прям, титр, pH = 10,5-11, 50% метиловый спирт Фталеинкомплексон
Ca2+ Прям, титр, pH > 12 Любой индикатор на Са2+
In3+ Прям, титр., pH - 8-10, Nlh-буфер Эриохром черный Т
Mg2+, Mn2+, Mg2+ + Ca2+, Pb2+ Прям, титр., pH = 10 (аскорбиновая к-та при определении Mg2+) или обр. титр, избытка ЭДТА солью Mg2+ То же
Cu2+, Zn2+ Mn2+, Pb2+ Прям, титр., pH = 10, аскорбиновая к-та при определении Мп2+, тартрат при определении РЬ2+ Эриохром красный Б
Цианид ионы + ОН - + TEA CN “-группа*, А13+ Ca2+ К кислому р-ру прибавляют TEA, NaOH до pH - 12, KCN, прям, титр. Любой индикатор на Са2+
Цианид-ионы + тирон CN-rpynna*, Al3+, Ti(IV) Mg2+ Обр. титр, избытка ЭДТА солью Mg2+, рН= 10 Эриохром черный Т
Цитрат-ионы + лимонная к-та А13+ (мал. количества) Zn2+ Прям, титр., pH = 8,5-9,5, 30 °C Эриохром черный Т
Fe3+ Cd2+, Cu2+, Pb2+ Прям, титр., pH = 8,5, 50% ацетона, при определении Cd2+ и РЬ2+ вводят немного Си-ЭДТА Нафтил азоксин
Mo(VI) Cu2+ Прям, титр., pH = 9 То же
Sn2+, Th(IV), Zr(IV) Zn2+ Прям, титр., pH = 6,4 То же
Cd2+, Co2+, Cu2+, Ni2+, Zn2+ Прям, титр., pH = 5-6 ПАН или Си-ПАН
Zr(IV) Cd2+, Co2+ Прям. титр. pH = 6,5 Нафтил азоксин
Cu2+ Обр. титр, избытка ЭДТА солью Си, pH = 9 То же
Примечание.
* CN-rpynna-Ag+, Cd2+, Со2+, Cu2+, Fe2+, Hg2+, Ni2+, Zn2+, Pd2++ и другие двухзарядные ионы Pt-металлов.
** Cu-ПАН, Cu-нафтилазоксин и т. и. — соответствующие индикаторы с добавлением небольших количеств комплекса Си2+ с ЭДТА.
Химические методы количественного анализа
671
Таблица 5.2.35
Маскирование магния щелочами и определение кальция в различных материалах [36]
Маскирующий реагент pH Индикатор Метод титрования Объект анализа зг Литература
КОН 12,0-12,3 ККК Прямое титрование Карбонатные минералы природные воды 0,01 683,851
12,3-12,5 Калькой + метанило-вый желтый То же после отделения полуторных оксидов Железные руды 423
12 Кальцеин + тимолфталеин Прямое титрование Руды, минералы, шлаки, стекла, эмали 0,02 37, 545, 623, 879
12-13 Кальконалид I То же Пищевая соль 0,02 311
12 Кальцеин + тимолфталеин + титановый желтый То же в глицериноводной среде Магнезит, доломит, асбест 0,02 547
> 12 МТС Титрование Са + Mg и титрование раствором Ca(NO3)2 ЭДТА, выделившейся из Mg-ЭДТА после введения КОН Силикатные материалы 478
NaOH > 13 Антипирилазо III Прямое титрование Доломит, глины, известняк 0,007 487, 739
0,1 М Mg(OH)2-nannaflna3O- Са То же раствором ДГТА Искусственные растворы 437
12 Мурексид Прямое титрование Горные породы после отделения мешающих катионов ДДТККа 951
Таблица 5.2.36
Маскирование цианидом натрия или калия при определении элементов в сложных объектах [36]
Маскируемые катионы Определяемые катионы pH Индикатор Метод титрования Объект анализа sr Литература
Zn, Cd Pb ~9 ДТАМР Прямое титрование в среде винной кислоты Искусственные растворы 858
Zn, Cd, Co Pb « 10 ЭХЧ То же То же 0,01 829
Тяжелые металлы Mg « 10 To же Прямое титрование при 2 °C Mg-Al-шпинели <0,01 606
Тяжелые металлы Ca 12,8 Кальцеин + метиленовый синий Прямое титрование Материалы с большим содержанием алюминия 630
Mn(II) Mg + Ca « 10 Крезолфталексон + + нафтоловый зеленый Прямое титрование при 30—40 °C Руды, шлаки, стекла, эмали <0,01 545, 879
Ca > 12 Кальцеин + тимолфталеин Прямое титрование Руды, шлаки, стекла, эмали <0,01 545,879
Co(II) TR 7,5-8,5 Арсеназо III То же Со-сплавы 0,008 684
Со(П) и Ni(II) Co(II) и Ni(II) « 10 Мурексид Прямое титрование Со + Ni, взаимодействие с KCN при 10 °C и титрование раствором Mg ЭДТА, выделившейся из Со-ЭДТА, далее то же при 50 °C, и титрование ЭДТА, выделившейся из Ni-ЭДТА Искусственные растворы 511
672
Новый справочник химика и технолога
Продолжение таблицы 5.2.36
Маскируемые катионы Определяемые катионы pH Индикатор Метод титрования Объект анализа Sr Литература
Ni Ca «10 ЭХЧ + Mg-ЭДТА Прямое титрование Катализаторы гидрогенизации 0,005 567
Ni, Zn, Cu(II) Pb «10 ПАР То же в присутствии тартрата Na, К Бронзы 0,001 612
Fe(II), Zn, Cu(II) Pb « 10 ЭХЧ То же Концентраты руд 440
Таблица 5.2.37
Прямое маскирование фторидами при определении различных элементов в минеральном сырье, сплавах и производственных смесях [36]
Маскируемые катионы Определяемые катионы pH Индикатор Метод титрования Объект анализа Sr Литература
Be, Al, U(VI) Cu(II) 5,5-8,5 Трополон Прямое титрование Сплавы 0,001 652
Mg, Са Mn(II) 9-10 ЭХЧ или ТФТ То же в присутствии NH2OHHC1 Силикатные материалы, агломераты руд, шлаки 0,03 381,494, 589
Ca Zn, Cd 10-11 ЭХЧ Прямое титрование Г идроксилапатит, Са—Zn-стеарат 0,006 356, 871
Pb « 10 То же То же в присутствии винной к-ты Г идроксилапатит 830
Ca, Al Ni 10 Мурексид То же Катализаторы 0,005 567
Al Zn 5,5 КО То же Руды 898
Hg(II) «7 ХАФ То же Искусственные р-ры 920
Cr(III), Fe(III) 6,5 ПТМ Обратное титрование избытка ЭДТА р-ром Hg(II) Хромистые руды 655
Mn(II) 8-9 МТС Обратное титрование избытка ЦДТА р-ром соли Zn в присутствии NH2OHHC1 Природные материалы, шлаки 651
Al, Ti(IV) V(IV), Fe(III) 5-6 КО или ПТМ Обратное титрование избытка ЭДТА р-ром соли Zn, Pb(II) или Hg(II) Ильменит, зола, шлаки 0,01 192, 621, 656
Al, Ti(IV), Zn Fe(III) 2,7-2,9 ПАР + Cu(II)- -ЭДТА Прямое титрование Силикатные материалы 0,006 751,755
Al, Th, U(VI) Zr 1 M H2SO4 КО То же Сплавы, циркон, технологические р-ры 0,01 163
Al, Zn, Fe(III) Cu(II) 2-4 МП АНД То же Сплавы 377
Al, Fe(III) Cu(II) 5,5 Тетра То же Электролиты для меднения 158
Cu(II) 5,1-5,5 Ализариновый красный S Прямое титрование Искусственные смеси 1 875
Sc Cu(II) «4 ПАН То же Технологические р-ры 0,03 234
Gd Ga «2 ПАН + Cu(II)- -ЭДТА То же Ga—Gd-гранаты 33
Sn(IV) Pb « 10 ЭХЧ или суль-фарсазен То же в присутствии винной к-ты Электролиты для осаждения ПОС сплавов 158
«5 КО То же Sn—РЬ-сплавы 912
Fe(III) Zn 5,5 КО Прямое титрование Концентраты руд, электролиты для латунирования 158, 186
Химические методы количественного анализа
673
Продолжение таблицы 5.2.37
Маскируемые катионы Определяемые катионы pH Индикатор Метод титрования Объект анализа Литература
Fe(III) Ni 8-11 Мурексид То же Файнштейн 188
Al, Ti(IV), Fe(III),Mg, Ca Zn 5-6 ПАН То же Катализаторы конверсии СО 0,009 608
Ti(IV), Zn, Nb, Ta Ni 6,5-6,9 КО Обратное титрование избытка ЭДТА р-ром Zn(NO3)2 Сплавы 540
Таблица 5.2.38
Комплексонометрическое определение различных элементов в условиях обратного маскирования фторидами [36]
Маскируемый и определяемый катионы Маскирующий реагент pH Индикатор Освободившийся комплексон, титрант Объект анализа Sr Литература
А1 NaF 5-6 KO ЭДТА, р-р ZnCl2 или Zn(NO3)2 Катализаторы, содержащие А1—Zn—Си, А1— Ni—Си, А1— Со—Мо, Al—Ni— Мо, Al—Ni; ферросплавы 0,01 2,3, 110, 334, 339
NaF 5-6 KO ЦДТА, р-р ZnCl2 или Zn(NO3)2 Ферровольфрам, силикатные материалы 0,01 328, 481
NaF 4,5 Кальцеин ЭДТА, р-р CuSO4 при 60 °C Искусственные р-ры 565
NaF 6 ГТС или АОП ЭДТА, р-р CuSO4 Ферросплавы, руды 0,02 333, 335, 610
NaF 5,5-6,5 XA3 + БЦТА ЭДТА или ДТПА, р-р CuSO4 Ферротитан после отделения Ti(IV) и Fe(III) 0,015 343
NaF 6-7 ЛАПАС ЭДТА, р-р ZnSO4 Руды 0,008 658
NaF 5 ПАН ЭДТА, р-р CuSO4 при 90 °C Ванадистые шлаки после восстановления V(V) до V(IV) 0,01 179
NaF 5,3 КО ЭДТА, р-р соли Zn Искусственные р-ры 693
KF 5-6 КО ЭДТА, р-р ZnSO4 Al-Y-гранаты после отделения Y(OH)3 121
NH4F 5-6 То же ЭДТА, р-р соли Zn или РЬ Сплавы на основе Cu, Zn или Nb; кианит 0,02 216,513
nh4f 5-6 КО ДТПА, р-р ZnCl Ферровольфрам 0,02 339
nh4f 5-6 ПАН ЭДТА, р-р Cu(NO3)2 при 90 °C Сплавы цветных металлов 0,02 1
nh4f 3-6 ПАН или ПАР То же Искусственные р-ры 338
NH4F + H3BO3 5-6 КО ЭДТА, р-р соли Zn или РЬ Ферросплавы 0,02 323,338
nh4f + H3BO3 «3 То же ЭДТА, р-р соли In или Bi Мартеновские шлаки, ферромарганец 0,01 324, 325, 338
nh4f + H3BO3 3-4 ПКФ + БЦТА ЭДТА, р-р In(NO3)3 Антифрикционный сплав 0,01 342
Sc nh4f «4 ПАН или ПАР ЭДТА, р-р CuSO4npH 90 °C Технологические р-ры 0,03 234, 338
nh4f + H3BO3 2,5 КО ЭДТА, р-р соли In или Bi Искусственные р-ры 325, 338
La KBF3OH 10 ЭХЧ ЭДТА, р-р Zn(NO3)2 То же 736
TR nh4f + H3BO3 4-6 КО ЭДТА, р-р соли Zn, In или РЬ То же 0,005 338
674
Новый справочник химика и технолога
Продолжение таблицы 5.2.37
Маскируемый и определяемый катионы Маскирующий реагент pH Индикатор Освободившийся комплексон, титрант Объект анализа Литература
TR 5-6 ПАН или ПАР ЭДТА, р-р CuSO4 То же
Ti(IV) nh4f 5-6 КО ЭДТА, р-р соли Zn или РЬ То же 0,009'
5-6 ПАН или ПАР ЭДТА, р-р соли Cu(II) То же
Zn NH4F 5,5 КО ЭДТА, р-р соли Zn То же 0,005 338
Sn(IV) NaF или KF 5-6 То же ЭДТА, р-р Pb(NO3)2 Сплавы РЬ—Sn; РЬ— Sn—Sb; Sn—Pb—Se(Te); Sn—Pb—Cu; электролиты Sn—Pb—Cu 0,01 32,469, 602, 650
Fe(III) NaF 5-6 То же То же Халькопириты 440
Таблица 5.2.39
Маскирующие свойства неорганических маскирующих реагентов [36]
Маскирующий реагент Диапазон pH Маскируемые катионы Определяемые катионы
Пероксид водорода <2-6 >7 V(V), Nb(V), Та, Mo(VI), W(VI), U(VI) Ti(IV), V(V) Cu(II), Zn, Cd, Al, Th, Zr, Hf, Y, Fe(III), Ni Mg, Ca, Sr, Cu(II)
Гидроксид натрия или калия 12-13 10,5±0,2 Mg, Al Zn, Al, Ga, Pb Ca Ca, Cd
Цианид натрия или калия 12-13 10-12 5-6 Co(III) Cu(II), Zn, Cd, Hg(II), Tl(III), Mn(III), Fe(II), Co(II), Ni, Pd(II) Cu(II), Hg(II), Co(II), Ni, Pd(II) Ni Mg, Ca, Sr, Ba, РЗЭ, In, Pb, Mn(II) Zn, Cd
Тиоцианат калия <7 Cu(II), Hg(II), Pd(II) Zn, Sc
Аммиак 10-11 5-6 Co(III) Pd(II) Ni Pb
Гидроксиламин гидрохлорид 0-1,6 Ce(IV), Fe(III) Th, Zr
Фосфаты 9-10 <7 Mg, Ba, Pb, Cr(III), Fe(III) Pb, Ti(IV), Fe(III), W(VI) Co(II), Ni Ca, Zn, Cd, Al, V(V), Co(II), Ni
Сульфат натрия или аммония 1-2 Th Sc, Ti(IV), Zr, Bi
Тиосульфат натрия 4,5-9,5 Ag, Cu(II), Hg(II) Mg, Ca, Zn, Cd, Al, РЗЭ, Mn(II), Pb, Ni
Иодид калия <7 >7 Cu(II), Cd, Hg(II) Hg(II) Zn, Sc Cd
Фторид натрия, калия или аммония ^7 Be, Mg, Ca, Al, Sc, РЗЭ, Th, Ge, Ti(IV), Zr, Hf, Sn(IV), Sb(III), Nb, Ta, Mn(II), W(VI), Fe(III) Cu(II), Zn, Cd, Hg(II), Ga, In, Sn(II), Pb, Bi, V(V), Cr(III), Mo(V), Mn(II), Co, Ni, Fe(III)
Таблица 5.2.40
Определение элементов в сложных объектах при использовании маскирования винной кислотой и ее солями [36]
Маскируемые катионы Определяемые катионы pH Индикатор Метод титрования Объект анализа Литература
Al Ca, Ni 9-10 Мурексид Прямое титрование Катализаторы 0,005 567
Zn, Cd, Hg(II), Pb «9 ДТАМР То же Искусственные 858
Химические методы количественного анализа
675
Продолжение таблицы 5.2.40
Маскируемые катионы Определяемые катионы pH Индикатор Метод титрования Объект анализа Sr Литература
Ti(IV) Al 5,5 KO Титрование р-ром соли Zn после кипячения с избытком ЭДТА Сплав ВТ-5 173
Ti(IV) 5,3-5,8 To же Определение Al+Ti обратным титрованием избытка ЭДТА р-ром соли Zn, разрушение Т1(ГУ)-ЭДТА винной кислотой и титрование освободившейся ЭДТА Ферротитан и другие сплавы 0,007 323, 546
Ti(IV), Zr Ba «12 МТС Прямое титрование Титанат бария, титанилоксалат бария, сплавы 0,003-0,008 83
Ti(IV), Nb(V), W(VI) Co(II) 4,5 ПАН Обратное титрование р-ром CuSO4 при 80 °C Твердые сплавы для металлорежущих инструментов 98
Ti(IV), Ta(V) Cr(III) 3,2 КО Титрование р-ром Bi(NO3)3 после кипячения с избытком ЭДТА Бориды Cr—Ti—В, Сг—Та—В 0,007 230
Sn(IV) Zn 5,5 То же Прямое титрование Электролит для лужения 0,01 296
Sn(II) 2-4 То же То же Электролит для кислого лужения 158
Pb 5-6 То же То же Электролит для осаждения ПОС 0,01 296
9-10 ЭХЧ или сульфарсазен То же То же 158
Sn(IV) Ni 9-10 Мурексид Прямое титрование Электролит для осаждения сплава Sn—Ni 0,01 158,296
Sn(IV), Sb(III) Pb 5-6 КО То же Сплавы Sn—Sb—Pb и Sn—Pb—Se(Te) 0,003 32, 602
Sb(III) Pb 6-7 ПАР+Си(П)-ЭДТА То же Искусственные Р-ры 0,01 717
Nb(V) Al + Ni 5-6 То же Титрование р-ром соли Zn после кипячения избытком ЭДТА Сплавы на основе Nb 0,02 216
Nb(V), Mo(VI), W(VI) Al, Zr 5,5 Ко То же Сплавы Nb—Zr—W; Nb—Al—Мо—W 0,006(Zr) 0,003(А1) 346
Nb(V), W(VI) Ti(IV) «2 КО Титрование избытка ЭДТА р-ром Bi(NO3)3 в присутствии Н2О2 Сплавы Nb—W—Ti 98
Ta(V) Mn(II) 9,5-10 ЭХЧ Титрование избытка ЭДТА р-ром соли Zn Танталаты марганца 102
Hf < 1 КО Прямое титрование при«100 °C Бориды Hf—Та—В 0,014 231
Bi, W(VI) Cu(II) >7 МПАНД Прямое титрование Искусственные р-ры 377
676
Новый справочник химика и технолога
Продолжение таблицы 5.2.40
Маскируемые катионы Определяемые катионы pH Индикатор Метод титрования Объект анализа Sr Литература
Mo(VI) Cd 5,5 KO То же То же 370
W(VI) Ga, In 5,2-5,8 To же Титрование р-ром соли Zn после кипячения с избытком ЭДТА Вольфраматы Ga и In 0,005 397
Fe(III) Ca «12 KKK Прямое титрование Силикатные материалы 0,002 ' 589
Fe(III) Мп(П) 9-10 ЭХЧили ТФТ То же в присутствии NH2OH • НС1 Агломераты, шлаки 0,03 381,589
Cd « 10 Дитизонат кадмия Прямое титрование Электролиты для осаждения Cd—Fe-губки 0,006 538
Таблица 5.2.41
Комплексоиометрическое определение элементов в сложных объектах с использованием маскирования триэтаноламином [36]
Маскируемые катионы Определяемые катионы pH Индикатор Метод титрования Объект анализа Sr Литература
Hg(H) Cu(II) 9-10 МПАНД Прямое титрование Искусственные растворы 377
Al Cu(II), Zn 9-10 Мурексид То же Al-Zn-Cu-катализаторы 0,002 3
Cu(II), Ni 9-10 To же То же Al-Ni-Cu-катализаторы 0,002 2
Al Mg 10,5 ЭХЧ То же Фармацевтические препараты 716
« 10 To же Прямое титрование при 2 °C Al-Mg-шпинели 626
Al + Fe(III) Ca « 12 Кальцеин или ККК Прямое титрование Магнезит, доломит 0,01 656, 851
Mg + Ca « 10 МТС или ЭХЧ То же Силикатные материалы, доломит 0,01 38, 851
Al + Fe(III) Ca > 12 Кальцеин + тимолфталеин Прямое титрование Силикатные материалы 38
> 12 ККК Прямое титрование в р-ре метилэтилке-тона То же 0,002 589
Mn(II) « 10 ЭХЧ или ТФТ Прямое титрование в среде метанола и в присутствии NH2OH • НС1 То же 0,002
Al + Fe(III) Mn(II) « 10 МТС Прямое титрование раствором ЦДТА в присутствии NH2OH • НС1 Природные материалы, шлаки 651
Ni « 10 Мурексид Прямое титрование Al-Ni-Mo-катализаторы 0,01 386
Cr(III) La «7 ЭХЧ То же Хромит лантана 0,02 223
Fe(III) Mg « 10 МТС или ТФТ Прямое титрование Карбонатные минералы, природные воды, глины 0,01 683, 879
Fe(III) Mg + Ca « 10 о-Крезолфта-лексон + нафтоловый зеленый В Прямое титрование Руды, шлаки, керамические материалы 0,01 545, 623, 879
Химические методы количественного анализа
677
Продолжение таблицы 5.2.41
Маскируемые катионы Определяемые катионы pH Индикатор Метод титрования Объект анализа Sr Литература
Fe(III) Ca > 12 Кальцеин + тимолфталеин То же То же 0,02
>12 Кальцеин Обратное титрование избытка ЭДТА раствором СаС12 Латексы после озоления 0,005 429
Fe(III) Ca 12-12,3 ККК Прямое титрование Карбонатные минералы, природные воды 0,01 683
12,3-12,5 Калькой + метаниловый желтый То же Fe-руды 423
Fe(III) Ca > 12 Антипирилазо III Прямое титрование Глины, доломит 0,02 487
Ba « 10 ПАР + Cu(II)-ЭДТА То же Барит 0,005 758
Fe(III) Cd »10 Дитизонат кадмия Прямое титрование Электролиты для осаждения Cd и Cd-Fe-губки 0,006 538
Ni 8-9 Мурексид То же Ni-Fe-сплавы 47, 114, 857
Co(III) Cd + Ni 9,7 То же То же Активная масса Ni-электрода 0,01 307
Pd(III) Ni 8-9 То же То же Ag-Pd-Ni-сплавы 0,008 244
Органические маскирующие реагенты [36]
Таблица 5.2.42
Реагент Диапазон pH Маскируемые катионы Определяемые катионы
Глицерин 2,5-3,5 А1 Cu(II), Sn(II)
5-6 Сг(Ш) Zn, Sn(IV), Pb
>9 А1, Сг(Ш), Fe(III) Zn, Cd, Hg(II), Pb, Mn(II), Ni
12-13 Al, Ti(IV), Cr(III), Fe(III) Cu(II), Mg, Ca, Sr, Ba, Zn, Cd, Hg(II), Pb, Mn(II), Co, Ni
Ацетилацетон 5-6 Be, Al, Ga, Th, U22+, MoO2, Fe(III), Pd(II) Zn, Cd, TR, Sn(II), Pb, Bi, Co, Ni
Щавелевая к-та <7 Ca, Al, Sn(IV) Cu(II), Zn, Pb, Cr(III)
Молочная к-та 5-6 Sn(IV), Ti(IV), Sb(III) Cu(II), Zn, Cd, Al, Pb, Zr, Bi, Fe(III), Co, Ni
Яблочная к-та 5 La, Pr, Nd Tm, Yb, Lu
Винная к-та 2,5-6 Sn(IV), Ti(IV), Zr, Sb, Nb, Cr(III), W, Fe(III) Cu(II), Zn, Cd, Ga, In, Pb, Bi, Mn(II), Ni
>7 Al, Ti(IV), Zr, Sn, Sb, Bi, Cr(III), Fe(III), MoO2”, UO2+ Mg, Ca, Ba, Zn, Cd, Ga, In, Mn(II), Ni
Лимонная к-та <3 W Ti(IV), Zr, Hf
<7 Th, Sn(IV), Ti(IV), Zr, As(III), Mo(VI), W Cu(II), Zn, Cd, Al, Pb, TR, In, Bi, Mn(II), Co, Ni
>7 Al, Ti(IV), Zr, Mo(VI), Fe(III) Cu(II), Ba, Zn, Cd, Pb
Аскорбиновая к-та ^7 Cu(II), Hg(II), Ce(IV), Cr(III), Fe(III), Мп(П1, IV, VII), Pu(IV) Mg, Ca, Zn, Cd, Al, Sc, Ga, In, TR, Sn, Pb, Zr, Sb(III), Bi, Fe(II), Mn(II), Co, Ni
Этилендиамин >7 Cu(II), Co, Ni Zn, Cd, Pb, Mn(II)
Полиэтилен-полиамины >7 Cu(II), Zn, Cd, Hg(II), Co, Ni Mg, Ba, Pb
678
Новый справочник химика и технолога
Продолжение таблицы 5.2.42
Реагент Диапазон pH Маскируемые катионы Определяемые катионы
Триэтаноламин >7 Hg(II), Al, Sn, Ti(IV), Sb(III), Bi, Cr(III), Mn(III), Fe(III), Co(III), Pd(II) Cu(II), Mg, Ca, Ba, Zn, Cd, TR, In, Pb, Mn(II), Co(II), Ni
Диэтанолглицин >7 Cu(II), Ti(IV), Bi, Fe(III) Mg, Ca, Sr, Ba, Co(II)
Ацетонциангидрин >7 Cu(II), Zn, Cd, Hg(II), Ni Mg, TR, Pb, Mn(II)
Диметилглиоксим 5-6 Pd(II) Cu(II), Zn, Pb, Fe(III), Co, Ni
8-Окснхинолинат калия 10 Mg Ca
1,10-Фенантролин 5-6 Cu(II), Zn, Cd, Hg(II), UO; , Mn(II), Co, Ni Al, TR, In, Th, Pb
8-9 Cu(II), Co, Ni Mn(II)
10-10,5 Pd(II) Ni
Салициловая к-та 1-2 Al Ti(IV), Fe(III)
Сульфосалициловая к-та 2-5 Al, Th, Ti(IV), Zr Cu(II), TR, Fe(III)
>7 Al, Fe(III) Mg
Пирокатехин-3,5 -дисульфокислота (тирон) >7 Al, Ti(IV) Mn(II)
8-Оксихинолин-5-сульфокислота 10-13 Al, Ti(IV), Fe(III) Ca
Тиомочевина 1-6 Cu(II), Hg(II), Pd(II) Zn, Cd, Al, TR, Y, Th, Sn, Pb, Ti(IV), Zr, Bi, Cr(III), Fe(III), Co, Ni
Т иосемикарбазид 2-5 Hg(II) Zn, Cd, Th, Pb, Bi
N- Аллилтиомочевина 9-10 Hg(H) Mg, Ca, Cd, Pb, Mn(II), Fe(III), Co, Ni
5-6 Hg(II) Cu(II)
Этилксантогенат калия 5,5 Cd Zn
2,4—3,4 Hg(II), Pb, Sb(III), Ni Th
Диэтилдитиокарбаминат натрия 9-11 Cd, Pb Zn
Дитиоглицерин (2,3-димеркаптопропанол) >7 Zn, Cd, Hg(II), Sn, Pb, As, Sb, Bi Mg, Ca
2,3 - Димеркаптопро-пансульфонат натрия (унитиол) >7 Cu(II), Zn, Cd, Hg(II), Ga, In, Tl(III), Sn, Pb, Sb, Bi, Fe(III), Co Mg, Ca, Sr, Ba, TR, Mn(II)
Тиоглико левая к-та >7 Ag, Cu(II), Zn, Cd, Hg(II), In, Tl(III), Sn, Pb, Bi Mg, Ca, Mn(II), Co, Ni
Тиомолочная к-та ^7 Cu(II), Hg(II), Sn, Pb, Bi, Fe(III), Co Mg, Ca, Zn, Al, Mn(II), Ni
Тиояблочная к-та <7 Bi, Fe(III) Th
Дитиовинная к-та ~ 10 Cu(II), Cd, Pb, Co, Ni Mg, Ca, Sr, Ba
« 6 Cu(II), Cd, Hg(II) Zn
Тиоэтанол амин « 10 Cu(II), Zn, Cd, Hg(II), Co, Ni Mg, Ca, Sr, Ba, Mn(II)
Р-Меркаптоаланин (цистеин) 5,5 Cu(II), Cd, Hg(II), Tl(III) Zn, Al, Pb, Fe(III), Co, Ni
Тиосалиииловая к-та «10 Hg(II) Zn, Cd
Тионалид <7 Cu(II), Hg(II), Bi Zn, Th
>7 Cu(II), Cd, Hg(II), Pb, Bi Mg, Ca
Димеркаптогликольпро-пионат « 10 Cu(II) Co, Ni
Комплексонометрическое титрование по методу вытеснения (титрование заместителя)
При титровании по методу вытеснении к определяемому катиону М добавляют избыток комплексоната вспомогательного металла Мь Образующий относительно более
устойчивый комплекс с ЭДТА катион М вытесняет из комплексоната катион Mj по обменной реакции:
M + MjY<=> М, + MY.
Затем прямым титрованием определяют количество вытесненного катиона М| (заместителя), экивалентное количест
Химические методы количественного анализа
679
ву определяемого иона М. Условная константа обменного равновесия определяется отношением условных констант устойчивости комплексонатов определяемого и замещаемого металлов:
к, р k][M,Y] ₽м/
Чтобы эта реакция протекала количественно, необходимо обеспечить образование комплекса MY не менее чем на 99,9%, поэтому при 100% избытке комплексенанта вспомогательного металла М( ^[MjY]«^cm ^отношение условных констант устойчивости комплексонатов должно составлять не менее 103.
Этот прием титрования используют в тех случаях, когда невозможно прямое титрование либо потому, что реакция определяемого иона с комплексоном протекает очень медленно, либо потому, что не удается подобрать хороший индикатор, позволяющий достаточно точно установить конец титрования. Если при прямом титровании реакция образования комплексоната определяемого металла протекает медленно, то и обменная реакция протекает, как правило, тоже медленно. Поэтому необходимо ждать, пока процесс пройдет полностью или даже ускорить его нагреванием раствора, прежде чем начать титрование «осовободившегося» катиона Мь При этом обменная реакция обязательно должна пройти количественно. Если прямое титрование определяемого иона затруднительно не из-за скорости реакции, а только из-за отсутствия хорошего индикатора, то степень протекания обменной реакции особого значения не имеет, поскольку для наблюдения изменения окраски индиатора достаточно лишь незначительного количества замещаемого металла. В этом случае соотношение условных констант устойчивости комплексонатов оказывает влияние лишь на количество добавляемого комплексоната вспомогательного металла [14].
В ряде случаев благодаря комплексонометрическому титрованию по методу вытеснения устраняется гидролиз легкогидролизующихся катионов, понижается pH титрования, в раствор переходят катионы из малорастворимых солей, селективно связываются некоторые катионы, без применения маскирующих реагентов.
Иногда при прямом титровании иона М из-за отсутствия хорошего индикатора (когда Ig^in^ 3) применяют «чужой» индикатор, чувствительный к вспомогательному катиону Mi; чтобы избежать дополнительного расхода титранта, вспомогательный катион Mi добавляют в виде комплексоната. Так, если при установлении конечной точки прямого титрования раствором комплексона идет реакция (заряды опущены)
Mind + Y MY + Ind, (5.2.76)
то при титровании по методу вытеснения имеют место две реакции — (5.2.77) или (5.2.78) и (5.2.79):
М+ M.Y-i Ind «=> M lnd+MY, (5.2.77) р; р; Рз
(ион М не реагирует с индикатором);
MInd+ MTY MY+ М, Ы,, (5.2.78)
pi Р; р; р;
(ион М реагирует с индикатором);
MjInd + Y^MjY + Ind.
(5.2.79)
Смещение равновесия реакций (5.2.77) и (5.2.78) вправо зависит от величины константы равновесия реакции, т.е. насколько сумма логарифмов условных констант Р' в правой части уравнения больше этой суммы в левой части.
Следовательно, основным условием возможности комплексонометрического титрования по методу вытеснения будет
(ig₽;+igp;)«(ig₽;+ig₽;) или
igK = (ig₽; +ig₽;)-(igp;+ig₽;) »o.
Условие это выполняется, если комплексы MY или Mjlnd достаточно устойчивы.
Часто имеют место реакции, в которых обрузуется ком-плексонат MY, менее прочный, чем введенный в реакционную среду более прочный вспомогательный комплексо-наг MjY .
В табл. 5.2.43 даны примеры реакций вытеснения [412], в которых более прочной комплексонат MjY разрушается с образованием менее прочного комплексоната MY благодаря тому, что одновременно образуется весьма устойчивый комплекс с индикатором M,Ind. Во всех приведенных примерах рассчитанные логарифмы констант равновесия реакции
lgK = lg₽3+lgp4-lg₽1-lg₽2*
имеют значительную положительную величину, и чем она больше, тем полнее идет реакция вытеснения.
* В данных табл. 5.2.44 во многих случаях, когда мольное отношение концентраций комплексоната вспомогательного металла Мг и титруемого иона М меньше единицы I сХ4 V / сХ4 <11 вытеснение незначительного количества вспо-\ MjY м /
могательного иона Mj используется только для четкого изменения окраски индикатора при косвенной индикации конечной точки прямого титрования [37].
680
Новый справочник химика и технолога
Таблица 5.2.43
Реакции вытеснения М + М, Y+ Ind -> MY+ M.Ind или Mind + М, Y -> MY+ M.Ind [36]
Pl ₽4 ₽2 Pl ₽’ ₽4
Определяемый катион М Введенный комплексонат MjY pH Индикатор IgP, lgP2 lgp3 lgP4 IgX
Ва Mg-ЭДТА 10 ЭХЧ 8,0 1,4 7,3 5,4 3,3
Mg Си(П)-ЭДТА 10 ПАН 9,7 — 8,0 13,8 ' 12,1
Al Си(П)-ЭДТА 3 То же 8,5 — 5,6 6,8 3,9
Zn или Cd Си(П)-ЭДТА 6 Нафтил азоксин 13,3 — 11,8 11,5 10,0
La Си(П)-ЭДТА 6 То же 13,3 — 13,1 11,5 11,3
Mn(II) Си(П)-ЭДТА 7 То же 12,8 — 10,7 11,5 9,4
Co(II) Си(П)-ЭДТА 6 То же 13,3 — 11,6 11,5 9,8
Примером вытеснительного титрования может служить определение ртути, свинца и др. Например:
Pb2+ + MgY2 PbY2’ + Mg2+.
Магний, выделившийся в количестве эквивалентном количеству свинца, оттитровывают стандартным раствором ЭДТА в присутствии индикатора ЭХЧ:
Mg2+ +11Y3 MgY2’ + Н+.
В ТЭ реакция (5.2.79) и
и(РЬ) = «(Mg) = w(Na2H2Y), а
ш(РЬ) = c(Na2H2Y) • K(Na2H2Y) • М (Pb).
Таким образом, расчеты при вытеснительном титровании аналогичны расчетам при прямом титровании.
Практические приложения комплексонометрического титрования по методу вытеснения представлены в табл. 5.2.44.
Таблица 5.2.44
Комплексонометрическое титрование катионов по методу вытеснения — реакция (5.2.77) или (5.2.78) [36]
Определяемый катион Введенный комплексонат Индикатор рн Дополнительные условия Мольное отношение, [MjY]/[M]
Mg(II) Си(П)-ЭДТА Нафтилазоксин 6С 10 В 80% этаноле 0,01-0,06
To же ПАН 10 50 °C 0,01-0,05
Ca(II) Mg-ЭДТА ЭХЧ 10 0,1
To же МТС 11 5-6
Си(П)-ЭДТА ПАН 10 50 °C 0,01-0,05
То же Нафтилазоксин 6С 10 в 50-75% ацетоне + ТЭА 0,01-0,03
Zn-ЭДТА Нафтилазоксин 8-9 0,03
Zn-ДГТА Цинкон 9,5-10 Титрование р-ром ДГТА 0,1
То же ПАН 8 Обратное титрование избытка ДГТА р-ром соли Са 0,03
То же Калькой 9
уо2-эдта ДФК 6,6-6,8 В 50% этаноле
Sr(II) или Mg-ЭДТА ЭХЧ 10 0,1-6
Ba(II) То же МТС И Титрование при 6-8 °C (анализ титаната) 0,8-7,5
Mg-ДПТА ЭХЧ 10 >1,4
Zn-ЭДТА То же 11 0,1-1
Химические методы количественного анализа
681
Продолжение таблицы 5.2.44
Определяемый катион Введенный комплексонат Индикатор pH Дополнительные условия Мольное отношение, [ MjY ] / [М]
Ва(П) Mg-ЭДТА ХТС 10
Си(И)-ЭДТА ПАР 10-11 Обратное титрование избытка ЭДТА р-ром Zn(CH3COO)2, 70-80 °C >
To же То же 11,5
Zn-ЭДТА ЭХЧ 10
To же То же 11 Обратное титрование избытка ЦДТА р-ром СаС12
А1(Ш) Си(П)-ЭДТА ПАН 3; 3,6 Кипящий р-р 0,25-0,5
То же ПАР 3,6 «
То же КХЧГ 1^1 « 0,03-0,17
VO2 уо2-эдта ДФК 4,5-6 В 50% этаноле + ССК, при 80 °C
Ga(III) Си(П)-ЭДТА ПАН 3-3,5; 3-5 0,01-0,05
То же То же 1,6-2 Горячий р-р,« 70 °C
То же ТАР 3,5-4 50-70 °C 0,5-10
Се(Ш)-ЭДТА КО 5,6 ± 0,2 1,7-16
In(III) Си(П)-ЭДТА ПАН 2,5 0,01-0,05
То же Нафтилазоксин 4С 3-3,5 0,04-0,75
Си(П)-ЭДТА ТАР 3,5-4,5 50-70 °C 0,3-5
Се(Ш)-ЭДТА КО 5,6 ± 0,2 1,7-16
Tl(III) Mg-ЭДТА ЭХЧ 10
Sn(IV) Си(П)-ЭДТА ПАН 1,8-2,2 90 °C 0,06-0,1
То же ПАР 2,5-3 0,11-0,17
Zn-ЭДТА КО 5-6 2,5
Pb(II) Mg-ЭДТА ЭХЧ 10 0,1
Си(П)-ЭДТА ПАН 3-3,5 Горячий р-р 0,01-0,05
То же ПАР 2,5-3 0,17-0,5
То же ТАР 4-5 50-70 °C 0,3-5
То же Нафтилазоксин 5,5-6,5 0,03
То же Нафтилазоксин 6С 5,5-6 0,03
Zn-ЭДТА Цинкон 9,5-10 0,5
То же ЭХЧ 10
ВДП)-ЭДТА Цинкон 5,5-7 0,5
уо2-эдта ДФК 5-6
То же ПКФ 5-6
Се(Ш)-ЭДТА КО 5,6 ± 0,2 1,7-16
Sb(III) Си(П)-ЭДТА ПАР 2,5-3 0,25-1
Bi(III) Си(П)-ЭДТА ПАР 2,5-3 0,2-0,4
Се(Ш)-ЭДТА КО 5,6 ± 0,2 1,7-16
уо2-эдта ДФК 3 0,05-1
Cu(II) Се(Ш)-ЭДТА Арсеназо 5,6 ± 0,2 1,7-16
Zn(II) или Cd(II) Mg-ЭДТА ЭХЧ 10 0,1; 0,4-4,5
Си(П)-ЭДТА ПАН 4,5 0,01-0,05
То же ТАР 5-6,5 50-70 °C 0,3-5
То же Нафтилазоксин 5,5-6,5 0,03
То же Нафтилазоксин 6С 6 0,01-0,03
Се(Ш)-ЭДТА КО 5,6 ± 0,2 1,7-16
уо2-эдта ДФК 4-5,5 0,025-2
То же ПКФ 5-6
682
Новый справочник химика и технолога
Продолжение таблицы 5.2.44
Определяемый катион Введенный комплексонат Индикатор pH Дополнительные условия Мольное отношение, [ Mj Y ] / [М]
Zn(II) Мё(П)-ЭДТА ПАР 2,5-3 0,07-0,2
Cd(II) Mg-ЭДТА МТС 11
Zn-ЭДТА Цинкон 9,5-10 0,5
Нё(П)-ЭДТА То же 5,5-7 0,5
Hg(II) Mg-ЭДТА ЭХЧ 10
Mg-ЭДТА или Са-ЭДТА КАЧ SN и ЭХК В 10,5-11,5 0,17
Си(П)-ЭДТА ПАН 3-3,5 В нагретом р-ре 0,01-0,05
То же ТАР 5,5-6,5 50-70 °C 0,3-5
Се(Ш)-ЭДТА КО 5,6 ± 0,2 1,7-16
Sc(III) Си(П)-ЭДТА ТАР 3,5-5 50-70 °C 0,3-5
То же Нафтилазоксин 6С 4 0,01-0,03
Се(Ш)-ЭДТА КО 5,6 ± 0,2 1,7-16
Y(III) Си(П)-ЭДТА Нафтилазоксин 5,5-6,5 0,03
То же Нафтилазоксин 6С 6 0,01-0,03
TR Сг(П)-ЭДТА Нафтилазоксин 5,5-6,5 0,03
Си(П)-ЭДТА Нафтилазоксин 6С 6 0,01-0,03
Zn-ЭДТА Нафтил азоксин 5,5-6,5 0,03
уо2-эдта ДФК 3-4,5
Th(IV) Си(П)-ЭДТА Нафтил азоксин 3-3,5 0,03
Zn2-TTTA КО 5,5
уо2-эдта ДФК 3-4,5
Pu(III, IV, V, VI) Си-ЭДТА ПАН 2,5-3 100 °C 0,01-0,06
Am(III) Се(Ш)-ЭДТА КО 5,6 ± 0,2 1,7-16
Zr(IV) Се(Ш)-ЭДТА КО 5,6+0,2 1,7-16
VO2+ Си(П)-ЭДТА ПАН 3,5 В присутствии аскорбиновой к-ты 0,03
Мп(П)-ЭДТА ЭХЧ 9-10 То же
Mo(VI) Си(П)-ЭДТА ПАН 3,5 0,8
уо2-эдта ДФК 5,2-5,6 В 50% этаноле 0,005-0,5
Mn(II) Mg-ЭДТА ЭХЧ 10 В присутствии аскорбиновой к-ты или NH2OH • НС1
Са-ЭДТА Калред 13 В среде ТЭА + аскорбиновая к-та
Си(П)-ЭДТА ПАН 10 50 °C, в присутствии аскорбиновой к-ты 0,01-0,05
То же ТАР 5,5-6,5 50-70 °C 0,5-3
Mn(II) То же Нафтил азоксин 6,5-7 0,03
То же Нафтилазоксин 6С 8,5 В присутствии аскорбиновой к-ты 0,01-0,03
Се(Ш)-ЭДТА КО 5,6 ± 0,2 1,7-16
уо2-эдта ДФК 6-6,5 0,02-1
Fe(III) Си(П)-ЭДТА ПАН или ПАР 3; 3,6 В ацетатном или тартратном кипящем р-ре 0,05
То же ПАР 2,7-2,9
То же ТАР 3-4 В нагретом р-ре 0,1
То же Нафтил азоксин 3-3,5 0,03
Се(Ш)-ЭДТА КО 5,6 ± 0,2 1,7-16
Химические методы количественного анализа
683
Продолжение таблицы 5.2.44
Определяемый катион Введенный комплексонат Индикатор pH Дополнительные условия Мольное отношение, [MjY]/[M]
Со(П) или Ni(II) Си(П)-ЭДТА ПАН >3 Со — в присутствии аскорбиновой к-ты Ni в нагретом Р-Ре 0,01-0,05
То же Нафтил азоксин 5,5-6,5 0,03
То же Нафтилазоксин 6С 5,5-6 0,01-0,03
Се(Ш)-ЭДТА КО 5,6 ± 0,2 1,7-16
уо2-эдта ДФК 4-5,5 0,025-1
Ni(II) Си(П)-ЭДТА ТАР 3-6,8 80-100 °C 0,15-3
5.2.6.4. Косвенные комплексонометрические методы определения анионов неорганических кислот и катионов
Комплексонометрическое определение анионов неорганических кислот
Анионы не реагируют непосредственно с комплексонами, поэтому их определению предшествуют вспомогательные реакции, например:
осаждение в виде малорастворимых солей катионами, которые затем титруют комплексоном.
Даже косвенные комплексонометрические методы определения большинства анионов имеют большое практическое значение благодаря своей простоте, быстроте и точности; они значительно менее трудоемки, чем гравиметрические методы, и широко применяются для макро- и микроопределений [275, 393, 458, 725]. Эти методы во многих случаях более точны, чем непосредственное осадительное или оксидиметрическое титрование. Комплексонометрия выгодно отличается от других титриметрических методов тем, что для определения всех анионов применяется единый весьма устойчивый титрант.
Методы осаждения
В косвенном комплексонометрическом определении анионов наибольшее распространение получил метод осаждения их ионами металлов в виде малорастворимого осадка с последующим титрованием иона металла в растворенном осадке или избытка его в фильтрате.
Примеры:
Определение SO4”.
a) SO2’ + Ва2+ * ** < BaSO4 (осадок отделяют)
>Вяу2- (рН = П) остаток в фильтрате
«(SO2’) = и(Ва2+) - «(Na2H2 Y)
б) SO2’ + Ва2+ <—± BaSO4 Ф
избыток
* Точно измеренный избыточный объем стандартного раствора Ва2+.
** Титрование остатка Ва2+ стандартным раствором ЭДТА.
BaSO4 + Y4’ *** < > BaY2’ + SO2’ (pH = 12 -13)
осадок
Y4’(HY3’)+Mg2+**** <..ЭХЧ~Т >MgY2’ (pH = 10) остаток
«(SO2’) = «(Na2H2Y)-«(Mg2+)
Определение PO4 .
PO3 > MgNH.PO. Ф
Осадок растворяют в кислоте, ионы Mg2+ определяют комплексонометрически:
Mg2+ + HY3’ < ЭХЧ~Т > MgY2’ + Н+ (pH = 10)
и(РО3") = «(Mg2+) = «(Na2H2Y).
Основные требования, предъявляемые к методу осаждения,— быстрое образование осадка стехиометрического состава, достаточно малая его растворимость и чистота. Если растворимость осадка в растворе комплексона ничтожна, можно титровать избыток осаждающего иона металла непосредственно в растворе без отделения осадка. Такой способ дает большой выигрыш во времени, но он возможен только в тех случаях, когда в растворе отсутствуют или могут быть замаскированы мешающие катионы. Более избирательным, но и более длительным, является выделение осадка, растворение его и титрование катиона.
В табл. 5.2.45 приведены малорастворимые соединения, используемые для косвенного комплексонометрического определения анионов, сгруппированные по способам титрования катионов: в растворенном осадке, фильтрате или непосредственно в реакционной жидкости без отделения осадка.
Комплексонометрическое определение одновалентных металлов
Все методы определения одновалентных металлов при помощи ЭДТА являются косвенными и заключаются в следующем:
1. Количественное осаждение малорастворимых комплексных солей известного состава, содержащих катио-
*** Растворение осадка BaSO4 точно измеренным избыточным объемом стандартного раствора ЭДТА.
**** Титрование остатка ЭДТА стандартным раствором Mg +.
684
Новый справочник химика и технолога
ны— определяемый и способный связываться с ЭДТА; после чего последний определяют комплексонометрическим титрованием в осадке или в избытке осадителя (см. табл. 5.2.46).
2. Замещение определяемого катиона другим, способным давать устойчивые комплексы с ЭДТА (см. табл. 5.2.47).
Таблица 5.2.45
Осаждаемые формы для анионов [36]
Анион Малорастворимые соединения
Катион титруют в растворенном осадке или фильтрате
В(ОН)" Ba5B2C]2HgO24 • НгО
СО; СаСО3; SrCO3; ВаСО3
[Fe(CN)6]^ Na2Zn[Fe(CN)6]; K2Zn[Fe(CN)6]; Pb2[Fe(CN)6]; Cu2[Fe(CN)6]
РО3- MgNH4PO4; ZnNH4PO4; A1PO4; FePO4;
ГМ4 BiPO4; Ca3(PO4)2
Р О4 Zn2P2O7; Mn2P2O7
AsO3- MgNH4AsO4; ZnNH4AsO4; BiAsO4
so2- BaSO4; PbSO4
S2’ CuS; ZnS; CdS
CrO2", Cr,O; BaCrO4; PbCrO4
F“ CaF2; Ba[BeF4]; PbCIF
СГ (C16H12ON2)2 PbCl2
Br“ (C16H12ON2)2PbBr2
Катион титруют в фильтрате
SCN- CuSCN; [CuPy2](SCN)2; [NiPy4](CSN)2
po3- ZnO2 • P2O5; Th3(PO4)4; LaPO4
Продолжение таблицы 5.2.45
Анион Малорастворимые соединения
(Ро3)Г СибС6(ОН)6(РО3)6
MoO; РЬМоО4
wo2- PbWO4
сг BiOCl
Катион титруют в растворенном осадке
РО3’ MnNH4PO4
SiO2" CoSi4O9
РО5-r3w10 Zn2HP3Oio
NbO, ZnNb2>63Ox
SeO2- PbSeO4
Se2’ Cu2Se
Те2’ Cu2Te
MoO; CaMoO4
wo2- CaWO4
Г TH
ю. PbIO3
Re О; TlReO4
Катион титруют в реакционной жидкости
псо, SrCO3
so2- BaSO4 (в водно-этанольной среде)
po3- LaPO4; Th3(PO4)4; BiPO4 (в водно-этанольной среде)
AsO3 BiAsO4
F" LaF3; CeF3; SmF3
Таблица 5.2.46
Косвенное комплексонометрическое определение одновалентных металлов в различных объектах, основанное на реакциях осаждения [36]
Определяемый катион Формула осажденной соли Титруемый катион осадка pH титрования Индикатор Метод титрования Объект анализа Определяемые количества Sr Литература
Li LiK[FeIO6] Fe(III) « 1 NH4SCN Прямое титрование Горные породы, специальные стекла 0,3-148 мг 0,0013 263, 731
То же Искусственные р-ры; вода 2-8 мг 0,02 225, 392, 574, 868
Na NaZn(UO2)3 • • (CH3COO)9 • 9H2O Zn 8-9 ЭХЧ Прямое титрование в 50% этаноле Силикаты, биологические объ- екты SO-318 мкг 0,02 624
5-6 5,3 Дитизон КО Прямое титрование Силикаты; алюминаты натрия 40 мг 0,003 883
Химические методы количественного анализа
685
Продолжение таблицы 5.2.46
Определяемый катион Формула осажденной соли Титруемый катион осадка pH титрования Индикатор Метод титрования Объект анализа Определяемые количества Sr Литература
Na NaCo(UO2)3 • • (CH3COO)9 • xH2O U(IV) 2-3 To же Обратное титрование избытка ЭДТА раствором Th(NO3)4 Минеральная вода, физиологический р-р, соли органических кислот 10- 250 мкг <0,01 560
NaMg(UO2)3 • • (СН3СОО)9 • хН2О U(IV) «2 То же, раствором Bi(NO3)3 Сплавы на основе железа после отделения Fe(III) 0,2-1 мг 0,07 6
Na NaCo(UO2)3 • • (СН3СОО)9 • хН2О Co(II) 8 Мурексид Прямое титрование Искусственные р-ры 10 мг 888
К K2Na[Co(NO2)6] Co(II) 7-8 NH4SCN Прямое титрование в среде 50% ацетона Соли калия 10 мг 0,01 868
8-9 Мурексид Прямое титрование Почвенные р-ры, биологические объекты 0,2-30 мг 0,015 306, 907
5-6 ПАН + + Cu(II)-ЭДТА То же в горячем р-ре Соли калия 65- 650 мкг 0,02 625
» 10 ЭХЧ Обратное титрование избытка ЭДТА р-ром ZnSO4 То же 27-54 0,01 770
K2Pb[Cu(NO2)6] Cu(II) + + Pb 5-6 ПКФ Прямое титрование Соли калия 1,5-3 мг 0,03 141
5Cd2 [Fe(CN)6] • • 4Кд [Fe(CN)6] Cd » 10 ЭХЧ То же Природные соли калия 8-10 мг 0,003 238
m = nh4+, К, Rb, Cs, T1(I) M2Ag [Co (NO2)6] Co(II) 7-8 nh4scn Прямое титрование в 50% ацетоне 1,5-53 мг 0,01 867
Rb Rb2Na [Co(NO2)6] Co(II) 7-8 То же То же Соли рубидия 10-105 мг 0,02-0,008 535
Cs Cs3[Co(NO2)6] Co(II) 7-8 То же То же Соли цезия 9-228 мг 0,01- 0,004 536
M = NH4, K, Rb, Cs M2Pb[Cu(NO2)6] Pb » 10 ЭХЧ Прямое титрование в присутствии винной кислоты Соли аммония в варочных р-рах целлюлозно-бумажного производства 30-40 мг 0,006 537
M = Rb, Cs M2Sr[Cu(NO2)6] Cu(II) 8-9 Мурексид Прямое титрование Соли рубидия или цезия 40-200 мг 0,005 472
M Rb, Cs M2Ca[Fe(CN)6] Ca « 12 ККК То же То же 20-200 мг 0,005 473
Cs Cs12Mg8[Fe(CN)6]7 Mg 10 ЭХЧ То же Соли цезия 140- 300 мкг 0,01 474
Cs3[Bi2I9] Bi 4,5 Bii; То же То же 29-87 мг 0,003 516
686
Новый справочник химика и технолога
Таблица 5.2.47
Косвенное комплексонометрическое определение одновалентных металлов в различных объектах, основанное на применении реакций замещения и восстановления
Определяемые катионы Титруемый ион Реакция pH титрования Индикатор Метод титрования Объект анализа Диапазон определяемых содержаний зг Литература
Na, К Mg Ионный обмен 9-10 ЭХЧ Прямое титрование Морская вода, отложения морских солей 0,2-50 мг Na 0,12-50 мг К 0,02 0,01 449, 597
К IIY3 Взаимодействие К[В(С6Н5)4] с HgY2- 6-6,3 ПАН + метиленовый синий Титрование раствором ZnCl2 Силикатные материалы, удобрения, природные и сточные воды, фармацевтические препараты, пищевые продукты 0,55-11 мг 0,005 155, 393, 586, 777, 852
То же «10 ЭХЧ Титрование раствором Mg(NO3)2 0,5-18 мг 0,005
Hg(H) Взаимодействие К[В(С6Н5)4] с избытком HgCl2 « 10 То же Обратное титрование избытка ЭДТА р-ром ZnSO4 Природные воды 1-8 мг 0,05 224
nh; Взаимодействие NH4[B(C6H5)4] с HgY2 « 10 То же Титрование р-ром Mg(NO3)2 Соли аммония 0,5-7 мг 0,01 852
H+ Взаимодействие NH^ с HgY2’ <6,8 Бромтимоловый синий Титрование р-ром NaOH до pH = 6,8 То же 0,85-17 мг 0,004
Li, К, Cs Ni Выделение хлоридов, осаждение AgCl и обмен с [Ni(CN)4]2 «8 Мурексид Прямое титрование Соли Li, К или Cs 10- 500 мкг Li2O 5-200 мг (К2О или Cs2) 0,02 0,01 549, 554, 555, 557
Cs Ca Экстракционный обмен с дипикрилами-натом Са « 10 ЭХЧ Прямое титрование Соли Cs 0,1-2 мг 0,02 694
Ag Ni Взаимодействие с [Ni (CN)4]2’ «8 Мурексид То же Сплавы, фотоматериалы, электролиты для серебрения 15-54 мг 0,01 86, 95, 47, 676, 695, 887
Ag Ni То же 7-10 ПАН Обратное титрование избытка ЭДТА р-ром Pb(NO3)2 Фталоцианиновый комплекс серебра 9-17 мг 0,001 506
Zn То же с [Zn(CN)4]2- «9 Мурексид Прямое титрование Соли серебра 5-136 мг 0,013 921
Химические методы количественного анализа
687
Продолжение таблицы 5.2.47
Определяемые катионы Титруемый ион Реакция pH титрования Индикатор Метод титрования Объект анализа Диапазон определяемых содержаний sr Литература
Ag Cd To же c [Cd(CN)4]2~ «9 ЭХЧ То же То же 6-22 мг 0,006 710
Cu(II) Обмен с суспензией Си(П)-ДДТК 8,5 Мурексид То же То же 3 мг 14
Экстракционный обмен с Си(П)-ДДТК 4-5 ПАН То же То же 746
Взаимодействие с CuS 4-5 ПАР + метиленовый синий То же То же 10-40 мкг 0,03 604
Ag Cu(II) Восстановление медью «8 Мурексид То же То же 22-108 мг 0,005 620
Hg(H) Восстановление ртутью « 10 ЭХЧ Обратное титрование избытка ЭДТА раствором соли Zn Растворы AgNO3 66-132 мг 0,01 866
Zn Восстановление амальгамой 10 То же Прямое титрование То же И мг п мкг 0,002 0,02 445
Bi Восстановление амальгамой 1-3 КО или ПКФ То же То же 66-132 мг 0,01 445, 866
T1(I) Zn Восстановление амальгамой Zn «9 ЭХЧ То же Соли Т1(1) 149- 298 мг 0,006 866
5.2.6.5. Окислительно-восстановительное титрование [1 -12, 20, 21, 26, 27, 39, 42, 44, 54, 56]
Основы метода [1]
Окислительно-восстановительным титрованием или редоксметрией называют количественное определение веществ титрованием стандартными растворами окислителей или восстановителей.
В основе метода лежит изменение потенциала окислительно-восстановительной системы при изменении соотношения концентраций окисленной и восстановленной форм в процессе титрования.
В окислительно-восстановительной реакции
п2Ох} + пх Re d2 «]Ох2 + п2 Re dj
участвуют две редокс-системы — титруемого вещества (1) и титранта (2), каждая из которых описывается уравнением Нернста:
^/Red =^/Red +^^lg—, (5.2.80)
«2 aRedj
^O^/Redj — ^Ox^Redj + 4? (5.2.81)
«1 «Red,
где ^Oi/Red — стандартный потенциал; п — число электронов, участвующих в реакции; «Охи aRed— соответственно активность окисленной и восстановленной форм, t = 25 °C.
В реальных условиях, т. е. при определенных ионной силе, pH и концентрации комплексообразующих веществ, следует вместо использовать формальный (условный) потенциал £°, рассчитанный или измеренный для конкретных условий. При этом, как правило, влиянием ионной силы пренебрегают, полагая у = 1.
Титрант (или титруемое вещество) может быть как окислителем, так и восстановителем.
Методы титрования стандартными растворами окислителей называют оксидиметрическими, а стандартными растворами восстановителей — редуциметрическими.
Кривые титрования строят обычно в координатах Ebx/Red ./(т), исходя из уравнений (5.2.80)-(5.2.81)*. Общий вид кривой окислительно-восстановительного титрования представлен на рис. 5.2.8.
* Строго все расчеты следует проводить используя значение £°’. Однако, как правило, они неизвестны. Поэтому обычно используют справочные значения Е?, понимая, что результаты в определенной степени носят оценочный характер.
688
Новый справочник химика и технолога
E,S
1,75 •
1,5 •
1,25 •
1,0-
0,5 1,0 1,5 х
Рис. 5.2.8. Кривая окислительно-восстановительного титрования
Потенциал в ТЭ рассчитывают из уравнения:
_п.Е\+п2Е\
^ТЭ
«1+«2
(5.2.82)
Полнота протекания окислительно-восстановительной реакции зависит от разности стандартных (или формальных) потенциалов систем, поскольку константа равновесия связана величинами стандартных потенциалов:
[OV-tRedJ”’ (£“-£>•«,
1g К —------------—-------------- ( j.Z.oj )
[OxjHRedJ”1 0,059
При «1 - П2 = П‘.
lgK=(£'>r.^0).:«. (5.2.84)
0,059
Знание стандартных потенциалов позволяет выбрать окислитель (или восстановитель), титрование которым обеспечивает заданную погрешность титрования.
Вычисленные на основе уравнения (5.2.83) минимальные разности стандартных потенциалов, при которых окислительно-восстановительные реакции протекают количественно, представлены в табл. 5.2.48.
Таблица 5.2.48
Минимальная разность между Д° - Е2°, при которой окислительно-восстановительная реакция протекает количественно (100,1% > т > 99,9%)
п = П\ • «2 (Е^-е°2), в
для пары Ох! + п\ ё Redi( Е^) для пары Ох2 + niQ Red2(Е2)
1 1 +0,354
1 2 +0,266
2 2 +0,177
2 3 +0,148
Примечание. Принято Е^ > Е2 , т. е. Oxi — окислитель;
Red2— восстановитель.
Расчет погрешности, обусловленной несовпадением Етэ и Ектт, см. [1, 5, 44].
Способы обнаружения конца титрования
Для обнаружения КТТ используют: 1) исчезновение или появление окраски титранта или титруемого вещества, 2) окислительно-восстановительные и специфические индикаторы; 3) инструментальные методы (потенциометрическое титрование и др.).
При титровании раствором КМпО4 с концентрацией не менее 0,02 М раствор окрашивается в розовый цвет при введении минимального избытка титранта (одной капли). Можно ввести поправку на избыток КМпО4, проводя контрольный опыт с раствором, содержащим те же количества воды, кислоты и электролитов, какие были в анализируемом растворе.
Специфические индикаторы — это вещества, которые образуют интенсивно окрашенное соединение с одним из компонентов окислительно-восстановительной системы. Например, при титровании иода используют специфический индикатор — крахмал, образующий темно-синее соединение с 13 — ионами. При титровании Fe(III) раствором соли Ti(III) в качестве индикатора используют тиоцианат-ионы, которые образуют с Fe(III) комплексы, окрашенные в интенсивно-красный цвет; конечную точку титрования определяют по исчезновению окраски.
Окислитепьно-восстановительные редокс-индикаторы — это соединения, в основном органические, способные к окислению и восстановлению, причем их окисленная и восстановленная формы имеют разную окраску. В качестве редокс-индикаторов применяют также комплексы органических лигандов с металлами, способными изменять степень окисления.
Для сопряженной редокс-пары
In0.v + пе InRed
по уравнению Нернста (при 25 °C)
£ = £о +0^059 ig (5.2.85)
п
lnKed
В конкретных условиях следует использовать формальный потенциал Е°. Если пренебречь влиянием ионной силы, можно заменить активности равновесными концентрациями.
Приняв предельные соотношения окисленной и восстановленной форм индикатора, при которых еще заметна окраска одной из форм, равными 10:1 и 1:10, область перехода окраски можно охарактеризовать как область значений потенциала:
де(5.2.86)
Д£ = <,„„.±^. (5-2.87)
Так же, как и при кислотно-основном титровании, интервал перехода окраски индикатора должен лежать внут
Химические методы количественного анализа
689
ри скачка титрования. Например, для дихроматометрического определения Fe(II) в 1 М H2SO4 пригодны индикаторы, изменяющие окраску в интервале 0,85-1,25 В, а в присутствии 1 М Н3РО4 в интервале 0,75-1,25 В, в том числе и широко известный дифениламин (£° = 0,76 В) или дифениламиносульфокислота (£° = 0,85 В).
Окислительно-восстановительная реакция с участием индикаторов-комплексов обратима. Таким индикатором является ферроин (комплекс Fe(II) с 1,10-фенантролином), переходящий при окислении в комплексе Fe(III):
Fe(Phen)2+ Fe(Phen)2+ + е. красный голубой
Оба комплекса очень устойчивы (lgfJ3 = 21,3 и 14,1 соответственно), не разрушаются в сильнокислых средах и в присутствии фосфорной кислоты. Используются также другие производные фенантролина, например, комплекс Fe(II) с 5-нитро-1,10-фенантролином (£° = 1,25 В, 1 М H2SO4) применяется при титровании восстановителей стандартным раствором Ce(IV) в азотнокислой и хлорнокислой средах.
Важнейшие окислительно-восстановительные индикаторы представлены в табл. 5.2.49.
К группе редокс-индикаторов относятся так же красители, разрушающиеся необратимо при определенном потенциале, например, нейтральный красный, метиловый красный, метиловый оранжевый, которые используются в броматометрии.
Практическое применение [1]
Стандартные вещества. В табл. 5.2.11 приведены первичные и вторичные стандарты. Вторичный стандарт, как правило, дает название методу титрования.
Предварительное окисление и восстановление. Элементы могут находиться в растворе в разных степенях окисления. До начала титрования нужно количественно перевести определяемое вещество в одну степень окисления, подходящую для взаимодействия с титрантом. Для этого используют окислители или восстановители более сильные, чем определяемые вещества, при этом избыток этих вспомогательных реагентов должен быть удален из раствора полностью и быстро.
Наиболее подходящие окислители:
1) персульфат аммония, переводящий в кислой среде в присутствии ионов серебра (катализатор) ионы Мп(П), Сг(Ш) и V(IV) в МпО“, Сг2О2- и VO" — ионы и разрушающийся при нагревании:
2S,O~ + 2Н2О = 4SO; + О2Т + 4Н+;
2) висмутат натрия, окисляющий Мп(П) до Мп О4; избыток его можно удалить фильтрованием;
3) пероксид водорода или натрия в кислой или щелочной среде; избыток разрушается при нагревании
2Н2О2 = 2Н2О + О2Т;
4) перманганат калия, окисляющий Сг(Ш) до Сг2О2 и V (IV) до VO3; избыток реагента удаляют кипячением с НС1, при этом получается МпО2, который отфильтровывают.
В качестве восстановителей применяют: 1) металлы (Zn, Cd, Ag, Bi) и амальгамы металлов; их используют в виде гранул, стержней, порошка. Более удобны редукторы-колонки, заполненные гранулами или порошком металлов, через которые пропускают анализируемый раствор (например, редуктор Джонса, заполненный амальгамированным цинком); 2) SnCl2, используемый для восстановления Fe(III); избыток реагента удаляют добавлением HgCl2:
SnCl2’ + 2HgCl2 = SnClJ" + Hg2Cl2;
3) газообразные восстановители — диоксид серы, сероводород.
Перманганатометрия [1]. Рабочий раствор КМпО4 неустойчив из-за реакции с водой, катализируемой диоксидом марганца на свету:
4МпО; + 2Н2О = 4МпО2Ф + ЗО2Т + 4ОН”.
Поэтому растворы перманганата калия следует готовить, используя чистую воду (органические примеси в воде могут реагировать с Мп О4 и давать МпО2, ускоряющий разложение реагента), отфильтровать от диоксида марганца и хранить в темных склянках; раствор следует выдержать несколько недель для окончания протекания всех процессов. Очевидно, что раствор следует стандартизировать, для чего используют оксалат натрия и другие восстановители (см. табл. 5.2.11). Реакция
2МпО. +5С2О4 +16Н+ = 2Мп2+ +10СО2 +8Н2О автокаталитическая, поэтому, несмотря на большую раз-
МпО; С2О2’
ность потенциалов пар и ' > ДЛЯ ускорения
процесса раствор следует нагреть.
Титрование раствором перманганата используют для определения множества веществ-восстановителей и окислителей (с предварительным восстановлением).
Перганганатометрия — один из лучших способов определения железа в разных объектах. После растворения образца (руды минерала, сплава) железо оказывается в степени окисления +3, поэтому его предварительно переводят в Fe(II) SnCl2 или металлами. При растворении часто используют НС1, поэтому титрование приходится осуществлять в присутствии хлорид-ионов. При этом возможно протекание индуцированной реакции окисления СГ-ионов перманганат-ионами. Поэтому титрование проводят в присутствии защитной смеси Рейнгарда-Циммермана. Эта смесь состоит из серной и фосфорной кислот и сульфата марганца. Фосфорная кислота и сульфат марганца, обеспечивающие понижение потенциала системы Мп(Ш)/Мп(П), предотвращают окисление хлорид-иона, фосфорная кисло
690
Новый справочник химика и технолога
та необходима также для связывания в бесцветный фосфатный комплекс образующегося Fe(III), раствор которого имеет желтую окраску. H2SO4 обеспечивает необходимую кислотность раствора.
Таким же образом прямым титрованием, возможно, определять V, Мо, W, U, Ti, Nb, Sn, Sb (см. табл. 5.2.50) после растворения образцов эти элементы получаются в высшей степени окисления. Перед титрованием их восстанавливают обычно пропусканием анализируемого раствора через подходящий редуктор.
Расчет массы определяемого элемента можно провести исходя из соотношения числа моль (ммоль) эквивалентов (вТЭ):
«(/ЭИ>(М)М) = «(1/5КМпО4), (при/ЭКВ(М) = 1)
т(М) = с(1/5 КМпО4) • К(КМпО4) • М(/экв (М)М).
Перманганатометрически можно определять ионы, образующие малорастворимые оксалаты (Са, Mg, Zn, Ва, Pb, Ag, Sr, Co, Th) способом обратного титрования или титрования заместителя (см. табл. 5.2.50).
В первом случае количественные расчеты можно провести, исходя из соотношения (в ТЭ):
»(Г_(М)М) = h{1/2(NH4)2C2O4}-«(1/5 КМпО4);
7И(М) =
c{l/2(NH4)2C2O4}-K(NH4)2C2O4
-с(1/5 КМпО4) • Е(КМпО4)
•^(ЛВ(М)М)
Во втором варианте:
«(/,„(М)М) = И(1/5КМпО4).
Для элементов II группы (М):
«(1/2 М) = «(1/5КМпО4) ,
т (М) = с (1/5 КМпО4) • V (КМпО4) • М (1/2 М),
где М(1/2М) — молярная масса эквивалента; с(КМпО4) — молярная концентрация эквивалента (нормальная концентрация КМпО4); К(КМпО4) — объем КМпО4> пошедший на титрование.
Окислители определяют перманганатометрически способом обратного титрования. К анализируемому раствору добавляют точно измеренный избыток стандартного раствора восстановителя и затем титруют его остаток раствором КМпО4. Например, хроматы, персульфаты, хлориты, хлораты и другие окислители можно определять, добавив к анализируемому раствору избыток стандартного раствора Fe2+, непрореагировавший остаток которого оттитровывают стандартным раствором КМпО4:
Cr,O; + 6Fe2+ +14Н+ = 2Сг3+ + 7Н2О + (ре2+остаток), 5Fe2+ + MnO4 +8Н+ = 5Fe3+ +Мп2+ +Н2О (остаток).
Расчетное уравнение можно записать в виде:
«(l/6K2Cr2O2) = «(Fe) - «{1/5 КМпО4),
т«(К2Сг2О7) =
c(Fe2+)-V(Fe2+)-
- с (1/5 КМпО4) • V (КМпО4)
•М(1/6К2Сг2О7)
Дихроматометрия. Достоинством метода является то, что рабочий раствор можно приготовить по точной навеске, поскольку К2Сг2О7 удовлетворяет всем потребностям первичного стандарта. Раствор К2СГ2О7 очень устойчив. Его применяют для определения железа (после предварительного восстановления) и органических компонентов вод или почв (окисляемость по дихромату) Индикатором обычно служат редокс-индикаторы — дифениламин и его производные. Титруют Fe(II) в присутствии фосфорной кислоты. Механизм реакций с участием ионов Сг,О7 сложен. Скорость реакции зависит от многих факторов, в том числе от концентрации Fe(II), поэтому вблизи ТЭ титрование следует вести медленно. Замедленность некоторых реакций (например, с органическими соединениями) заставляет часто прибегать к обратному титрованию: раствор определяемого вещества кипятят с избытком К2Сг2О7, а избыток оттитровывают растворам соли Мора (так определяют окисляемость воды).
Броматометрия. Реакция титрования броматом калия протекает до ТЭ (в присутствии избытка восстановителя) с образованием бромид-ионов:
ВгО" + 6Н+ + бе = Вг“ + ЗН2О.
При добавлении лишней капли бромата протекает реакция
ВгО3“ + 5Вг“ + 6Н+ = ЗВг2 + ЗН2О.
Образующийся бром может бромировать органические соединения, например краситель метиловый оранжевый и нейтральный красный (необратимые редокс-индикаторы) или хинолиновый желтый (бромируется обратимо). По исчезновению окраски красителей судят о конечной точке титрования. Реакцию проводят в кислой среде (pH = 1). Достоинством метода является устойчивость и чистота бромата калия. Броматометрия — лучший метод определения сурьмы и олова:
SbCl3’ - 2е = SbCl". о о
Метод применяют также для определения мышьяка, железа и органических соединений. Скорость реакции с восстановителями ускоряется в присутствии солей ртути(П).
Полиметрия, иодометрия. Реакция окисления — восстановления с участием иода обратима:
12 + 2е^2Г
(е«лг =0,62 в).
Химические методы количественного анализа
691
Иод плохо растворим в воде, но в присутствии иодид-ионов образуется комплекс 12, поэтому при титровании протекает реакция
I’ + 2е ЗГ = 0,54 в).
Стандартные потенциалы многих восстановителей меньше Е^. , а многих окислителей — выше, а поэтому система служит для определения окислителей и восстановителей.
При прямом определении восстановителей рабочим раствором служит раствор иода, который готовят растворением смеси 12 (очищенного сублимацией) с KI в очищенной от ионов металлов воде (реакция иодида с кислородом ускоряется в их присутствии). Хранить раствор следует в темной склянке во избежание окисления иодида и улетучивания образовавшегося иода. Можно приготовить раствор по точной навеске, но можно проверить его концентрацию по первичному стандарту, например AS2O3.
Метод титрования раствором иода иногда называют иодиметрией. Его используют для определения As(III) и As(V) после предварительного восстановления его до As(III). Для восстановления добавляют к соляно-кислому раствору As(V) восстановитель (Zn, CuO и др.) и отгоняют мышьяк в виде AsCl3. Реакция I2 с As(III) протекает в щелочной среде, но не следует создавать pH среды выше 10 во избежание взаимодействия 12 с ОН с образованием ги-поиодида
12 + 2011 = 10 + Г + Н2О,
а в более щелочной среде и йодата. Обычно при титровании добавляют NaHCO3, (pH « 8). Вместо NaHCO3 рекомендуют также (если раствор сильнокислый) тетраборат натрия или дигидрофосфат натрия.
Прямая реакция восстановления иода идет быстро, но обратная реакция окисления иодида протекает медленнее. Поэтому использовать раствор иодида для определения окислителей путем прямого титрования невозможно. К тому же растворы иодида (например KI) неустойчивы, поскольку иодид окисляется кислородом воздуха. Поэтому используют заместительное титрование — добавляют к окислителю избыток иодида, а выделившийся иод оттит-ровывают стандартным раствором тиосульфата натрия. Этот метод называют иодометрией. Индикатором, так же как и в ио диметрии, служит крахмал.
Раствор тиосульфата натрия является вторичным стандартным раствором. Его готовят из пентагидрата Na2S2O3 • •5Н2О с добавкой карбоната натрия. Раствор неустойчив. В нем возможно, с одной стороны, окисление кислородом воздуха S20f“ до SO2- и S (при этом концентрация S2O2 уменьшается); с другой стороны, при pH = 5 (а такова среда в воде, поглотившей диоксид углерода) происходит реакция
S2O2’ + Н+ = HS2O3~ = HSO’ + S i.
Образующийся HSO3 реагирует с иодом, но f3KS -\/2 (в отличие от S20j“, имеющего в реакции с иодом Лкв = 1)’ поэтому, несмотря на уменьшение концентрации S2O2”, количество иода на реакцию увеличивается, что равноценно повышению концентрации раствора тиосульфата натрия. Добавление Na2CO3 и предохранение его от СО2 с помощью хлоркальциевой трубки стабилизирует раствор. Рекомендуют также добавлять немного фенола или хлорамина для уничтожения серных бактерий, способствующих разложению реагента.
Для стандартизации раствора обычно используют дихромат калия:
Cr2O2’ + 6Г + 14Н+ = 312 + 2Сг3+ + 7Н2О .
Выделившийся через несколько минут иод титруют раствором тиосульфата:
282ОГ +12 = 2Г +S4O3-.
Конечную точку титрования фиксируют по обесцвечиванию иод-крахмального комплекса (крахмал добавляют, когда титруемый раствор приобретает светло-соломенный цвет).
Цепочку расчетных уравнений можно записать в виде: и(1/6К2Сг2О7) = w(Na2S2O3). Откуда:
^(K2Cr2O7) zN s 0 \ r/Na s 0 \ и
M(l/6K2Cr2O7) V 2 2 3 7 V 2 2 3 7
/xjj, ем __________w(K2Cr2O7)________
2 2 3 7 M(l/6K2Cr2O7)-K(Na2S2O3)’
Примеры йодометрического определения некоторых катионов (Pb2+, Ва2+, Sr2’) и т. д. — см. табл. 5.2.51.
Иодометрия — лучший и самый точный метод определения сравнительно больших количеств меди (в сплавах, рудах, высокотемпературных сверхпроводниках):
2Си2+ + 4Г = 2CuI| +12.
Реакция протекает слева направо количественно, поскольку восстановленная форма системы Cu2+/Cul+ (Е° = = 0,15 В) связана в малорастворимый иодид меди, что приводит к образованию системы Cu2+/Cul (Е° = 0,86 В). Титрование следует проводить при pH « 2-4; в более щелочных средах образуются СиОН~ — ионы, в кислых средах возможно окисление иодида кислородом (особенно в присутствии оксидов азота, которые могут остаться в растворе после растворения образцов в азотной кислоте).
Из мешающих элементов отметим Fe(III), также реагирующий с иодидом с выделением иода. Его влияние устраняют добавлением фторида аммония (или фосфорной кислоты).
Важнейшие окислительно-восстановительные индикаторы (в порядке уменьшения их стандартных окислительных потенциалов) А. индикаторы, мало зависящие от pH и ионной силы раствора [46]
Таблица 5.2.49
Индикатор Окисленная форма соединения Молярная масса Раствор £°, В Окраска
окисленной формы восстановленной формы
2,2-Дипиридил (комплекс с рутением) Ru3+ ( Z \у 1 \ —N N—V /з 569,63 В разбавленной HNO3 +1,33 Нет Желтая ^макс. = 418 НМ
Нитро-о-фенантролин (нитроферроин), комплекс с Fe2+ Fe3+ / /°2 \ N N V 1 3 731,47 0,025 М в воде +1,25 Бледно-голубая Красная ^макс. 510 НМ
1,10-Фенантролин (ферроин), комплекс с Fe2+ Fe 1 / \ / \ 1 И— N //3 596,47 0,025 М: 1,624 г гидрохлорида фенантролина и 0,695 г FeSO4-7H2O в 100 мл воды +1,06 Бледно-голубая То же
А-Фенилантраниловая кислота (2-дифениламин-карбоновая кислота) СООН NH 213,24 а) 0,2% в воде б) 0,1% в 0,2% р-ре Na2CO3 +1,00 Фиолетовокрасная ^макс. = 524 НМ Нет
5,6-Диметил-1,10-фенан-тролин (комплекс с Fe2+) Fe3+ 1 Н3С сн3 3 680,63 0,025 М в воде +0,97 Желто-зеленая Красная ^-макс. 520 НМ
2,2'-Дипиридил (комплекс с Fe2+) \ Г~\ / \ N N V / 3 524,41 В разбавленной HCI или H2SO4 +0,97 Бледно-голубая Красная ^макс. 522 НМ
Дифениламинсульфонат натрия (или бария) NH SO3Na 271,27 0,05% в воде +0,84 Синяя ^-макс. = 593 НМ Нет
692 Новый справочник химика и технолога
Продолжение таблицы 5.2.49 А
Индикатор Окисленная форма соединения Молярная масса Раствор В Окраска
окисленной формы восстановленной формы
3,3'-Диметоксибензидин (о-Дианизидин) н3со ОСН3 HN—— N 244,29 В разбавленной HCI +0,78 Красная ^макс. 510 НМ То же
Дифениламин NH 169,23 1% в концентрированной H2SO4 +0,76 Фиолетовая ^макс. 565 НМ То же
Дифенилбензидин NH—NH— 336,44 То же +0,76 Фиолетовая Какс. = 560 нм То же
и-Этоксихризоидин С2Н5О—N= N—NH2 НС1 H2N 292,77 0,05-0,2% в воде +0,76 Бледно-желтая Красная
Вариаминовый синий H2N NH °СНЗ 214,27 0,5% раствор HCI-соли +0,71 Фиолетовая ^макс. 570 НМ Нет
Химические методы количественного анализа
Б. Индикаторы, чувствительные к изменению pH и ионной силы раствора
Индикатор Окисленная форма соединения Молярная масса Раствор £°, В при pH Окраска
0 7 окисленной формы восстановленной формы
2,6-Диброминдофенол (2,6-дибромбензолиндофенол), Na-соль Вг О=^ ^=N ONa Вг 378,98 0,02% в воде +0,668 +0,216 Синяя Нет
Продолжение таблицы 5.2.49 Б
Индикатор Окисленная форма соединения Молярная масса Раствор Е°, В при pH Окраска
0 7 окисленной формы восстановленной формы
2,6-Дихлориндофенол (2,6-дихлорфенолиндофенол) Na-соль CI О=^ N—— ONa СГ 290,08 То же +0,668 +0,217 То же То же
о-Крезолиндофенол, Na-соль сн3 0=^ />===N ONa 235,22 +0,62 +0,19 То же То же
Толуиленовый синий сн3 (CH3)2N=^ N NH2 nh/ 255,34 0,05% в 60% спирте +0,601 +0,115 То же То же
Тионин (диаминофенотиазин; фиолетовый Лаута) nh2 228,30 То же +0,563 +0,064 Фиолетовая То же
Тиазиновый синий (CH^N^4^^ N(C2H5)2 312,46 +0,537 +0,027 Синяя То же
Толуидиновый синий ch3 (СН3)2ЬГ^Ч=^Х^ NN2 270,38 0,05% в 60% спирте +0,534 +0,034 То же То же
Тионол 229,26 То же +0,53 +0,037 То же То же
Новый справочник химика и технолога
Продолжение таблицы 5.2.49 Б
Индикатор Окисленная форма соединения Молярная масса Раствор Е°, В при pH Окраска
0 7 окисленной формы восстановленной формы
Метиленовый синий (метиленовый голубой) (ch3)2nx4^x^ SxX4^X^N(CH3)2 319,85 0,05% в воде +0,532 +0,011 То же То же
Индиго-5,5',7,7'-тетра-сульфокислота О О НОз&^|Хх^| | II ,^/SO:11 HO3S SO3H 582,52 То же +0,365 -0,046 Синяя ^макс. 590 НМ Нет или бледно-желтая
Индиго-5,5-7-трисульфокислота О О И so3h SO3H 502,46 То же +0,332 -0,081 Синяя ^макс. 596 НМ То же
Индиго-5,5'-дисульфо-кислота (индиго-кармин)но О "XcLJ 3 SO3H 422,40 0,05% в воде +0,291 -0,125 Синяя ^макс. 602 НМ То же
Индиго-5-моносульфо-кислота с HN^ ) 0 342,33 То же +0,262 -0,157 Синяя ^макс. 608 НМ То же
Феносафранин <111: Cl 322,80 То же +0,280 -0,252 Красная Нет
Химические методы количественного анализа
Продолжение таблицы 5.2.49 Б
Индикатор Окисленная форма соединения Молярная масса Раствор Е°, В прн pH Окраска
0 7 окисленной формы восстановленной формы
Сафранин Т СНэ 1Nil, сГ 350,85 То же +0,235 -0,289 Фиолетовокрасная То же
Нейтральный красный СИз 1 1 1 1 нс1 (NH2 0,01% в 60% спирте +0,240 -0,325 Красная То же
Таблица 5.2.50
Примеры определения некоторых неорганических и органических соединений перманганатометрическим методом [3]
Определяемое соединение (ион) Реакции, используемые в ходе анализа Условия анализа
Sb(III) 5Sbin + 2MnO; + 16Н+ = 5Sbv + 2Mn2+ + 8Н2О Прямое титрование. Среда— 1 моль-л'1 НС1
Sn2+ 5Sn2+ + 2MnO; + 16Н+ = 5Sn4+ + 2Mn2+ + 8H2O Среда — 1 моль-л'1 H2SO4, исключить доступ О2
Ti3+ 5Ti3+ + MnO' + 8H+ - 5Ti4+ + Mn2+ +4H2O Среда — 1 моль-л'1 H2SO4
W3+ 5W3+ + 3MnOJ + 241Г = 5W6+ + 3Mn2+ + 12H2O «
u4+ 5U4+ + 2MnO4 + 16H+ = 5U6+ + 2Mn2+ + 8H2O «
v4+ 5V4+ + MnO; + 8H+ = 5V5+ + Mn2+ + 4H2O «
Br' 10 Br' + 2MnO4 + 16H+ = 2Mn2+ + 8H2O + 5Br2 Титрование в 2 моль-л'1 H2SO4 при кипячении для удаления Вг2
CH3OH ch3oh + 6Mno;+8OH' = co2' + 6MnO2' + 6H2o+(Mno;) изб. OCT. 2MnO4 + HCOO' + ЗВа2 + ЗОН' = Ф 2BaMnO4 + BaCO3 + 2H2O OCT. Обратное титрование:остаток непрореагировавшего МпО4 после добавления соли бария титруют раствором формиата натрия
Новый справочник химика и технолога
Продолжение таблицы 5.2.50
Определяемое соединение (ион) Реакции, используемые в ходе анализа Условия анализа
Са2+, Mg2+, Zn2+, Со2+, La3+, Th4+, Ва2+, Sr2*, Pb2+, Ag* М2+ + С2О2" = Ф МС2О4 + (С2О4) изб. ост. 5С2О2"+2МпО; +16Н+ = ЮСО2 +2Мп2+ +8Н2О ОСТ. Осадок МС2О4 отделяют и отбрасывают, фильтрат и промывные воды титруют при 70-80° С раствором КМпО4 в среде 2 моль л-1 H2SO4. Обратное титрование
м2* +с2о2- =Фмс2о4 МС2О4 + 2Н+ = М2+ + Н2С2О4 5С2О2" + 2МпО; + 16Н+ = 10СО2 + 8Н2О + 2Мп2+ Осадок МС2О4 отделяют, промывают, растворяют в 2 моль л-1 H2SO4 и титруют, как указано выше
Таблица 5.2.51
Примеры определения некоторых неорганических и органических соединений йодометрическим методом [3]
Определяемое соединение (ион) Реакции, используемые в ходе анализа Условия анализа
Титрование по замещению, в результате предварительной реакции выделяется 12, который титруют раствором Na2S2O3
CIO3 ,Вг3",Юз ВгОз + 6Г + 6Н+ = Вг" + 312 + ЗН2О Среда - 0,5 моль л 1 НС1, реакция замедлена
Вг2, С12 С12 + 21" = 2СГ + 12 То же
О2 О2 + 4Мп(ОН)2 + 2Н2О = 4Мп(ОН)3 2Мп(ОН)з + 61Г + 2Г = 2Мп2* +12 + 6Н2О Добавление Мп2+ и NaOH, затем подкисление 2 моль л"1 НС1 до pH = 4—5, введение KI
Н2О2 Н2О2 + 2Г + 2Н* = 2Н2О +12 Среда — 1 моль л-1 H2SO4, катализатор — NH4MoO3
МпО; 2MnO, +101" + 16Н+ = 2Мп2+ + 512 +8Н2О Среда — 0,1 моль-л-1 H2SO4
МпО2 МпО2 + 2Г + 4Н* = Мп2* +12 + 2Н2О Среда — 0,5 моль-л"1 H3SO4
NO- 2NO2 + 2Г + 4Н+ = I2 + 2NO + 2Н2О Среда — 1 мольл-1 H2SO4
Fe3* 2Fe3* + 2Г = 2Fe2* + 12 Среда — конц. раствор НС1, реакция замедлена
Се4* 2Се4* + 2Г = 2Се3* + 12 Среда — 1 моль-л-1 H2SO4
S2O2- S2O2" +21 = 2SO2" + 12 Среда нейтральная, перед титрованием подкислить раствором НС1
Химические методы количественного анализа
Продолжение таблицы 5.2.51
Определяемое соединение (ион) Реакции, используемые в ходе анализа Условия анализа
Fe(CN)’~ 2Fe(CN)3’ + 2Г = 2Fe(CN)*’ +12 Среда — 1 моль-л’1 НС1
As(V) H3AsO4 + 2Г + 2Н+ = HAsO2 + 2Н2О +12 Среда — 5 моль-л’1 НС1
Sb(V) sbci; + 2Г = sbci; + 2сг +i2 Среда 6 моль-л-1 НС1
Pb2+, Ba2+, Sr2+ М2' I CrO2 = i МСгО4 2МСгО4 + 2Н+ = 2М2+ + Сг2О2” + Н2О Сг2О2” + 6Г + 14Н+ = 2Сг3+ + 312 + 7Н2О Осадок МСгО4 растворяют в 2 моль-л-1 НС1 в присутствии избытка KI
ROOH (органические пероксиды) ROOH + 2Г + 2Н+ = ROH + 12 + Н2О Среда — 0,1 моль-л-1 НС1
RCOON (карбоновые кислоты) 6R-COOH + 5KI + КЮ3 = 6R-COOK + 312 + ЗН2О В присутствии избытка Г и Ю3
Обратное титрование
SO2- SO2 +12 + Н 2О = SO2 + 21 + 2Н+ + (12) изб. ост. Остаток 12 в слабокислой среде титруют раствором Na2S2O3
S2’ S2’+I2 = S + 2I-+(I2) OCT. То же
Cd2+, Zn2+ м2+ + h2s = ;ms + 2Н+ MS + 2H+ = H2S + M2+ H2S+I2 = S + 2H++2Г+(l2) изб. OCT. Осадок MS растворяют в 3 моль-л’1 НС1, добавляют изб. 12, остаток которого титрируют раствором Na2S2O3
CS(NH2)2 (тиомочевина) CS(NH2) + 4I2 + 5H2O = CO (NH2 )2 + SO2’ + 8Г + IOH+ + (l2) изб. ост. Остаток 12 в нейтральной среде титруют раствором Na2S2O3
NH2NHCONH2 (семикарбазид) NH2NHCONH2 + I2 + H2O = N2 + CO2 + NH4 + 41’ + 3H+ + (I2) изб. OCT. То же
Новый справочник химика и технолога
Химические методы количественного анализа
699
5.2.6.6. Осадительное титрование
Основы метода [1]
Осадительное титрование объединяет титриметриче-ские методы, основанные на реакциях осаждения. В основном используют реакции титрования, при которых ионы титана и титруемого компонента взаимодействуют в молярном отношении 1:1. Осадительное титрование имеет ограниченное применение, что связано с неколичественным и нестехиометрическим протеканием многих реакций. Требованиям, которые предъявляются к реакциям титрования, удовлетворяют реакции осаждения галогенидов и тиоцианата серебра (аргентометрия), а также хлорида одновалентной ртути (меркурометрия). В аргентометрии кривые титрования строят обычно в координатах рГ-т, где Г-СГ, Вг или Г. Общий вид кривых титрования для каждого ониона в отдельности приведена на рис. 5.2.9. Кривые титрования симметричны относительно ТЭ.
Рис. 5.2.9. Кривые осадительного титрования растворов галогенидов раствором нитрата серебра
Способы фиксирования конечной точки титрования и практическое применение.
Метод Мора. Индикатором служит хромат-ион, который образует красно-кирпичный осадок Ag2CrO4, более растворимый, чем галогениды серебра
S»g!c«. = = 6,5-10"5М, = 1,3.10-М, -КГ’М
(при С°г = 0,1 М ).
На практике обычно создают концентрацию СгО2’, равную 0,01-0,005 М.
Титрование по методу Мора следует проводить в слабокислой среде. В кислой среде необходимо учитывать протолитическую реакцию
СгОд” + FT НСгО: и 2НСгО; Сг2О2’ + Н2О.
В щелочной среде следует иметь в виду возможность протекания реакций
Ag+ -OH-AgOHj,
2AgOH = Ag2O + H2O.
Метод Мора непригоден для определения иодид- и тиоцианат-ионов.
Метод Фольгарда основан на титровании раствора ионов Ag+ раствором KSCN в присутствии ионов Fe(III):
Ag+ + SCN’ = AgSCNJ.,
Fe3+ + «SCN’ = Fe(SCN)(nM+ .
После оттитрования ионов Ag избыток титранта дает с ионами Fe'' красный комплекс. Обычно создают концентрацию Fe3+ около 0,01 М.
Для определения анионов этим методом (СГ, Вг’, CN’, СО2’, СгО2”, S2”, РО3’) используют обратное титрование. К раствору титруемого иона добавляют избыток стандартного раствора нитрата серебра. После образования осадка оттитровывают избыток стандартным раствором KSCN в присутствии раствора FeCF.
При титровании Вг’, Г, AsO3” удаление соли серебра в процессе титрования не требуется. В случаях титрования анионов COf”, СгО2”, CN”, СГ, С2О2’, PO3”, S2’ требуется удаление соли серебра перед обратным титрованием избытка Ag+.
При титровании одновалентных анионов расчетное уравнение записывается в виде:
«(МА) = «(AgNO3) - «(KSCN), откуда при известных c(AgNO3), K(AgNO3), c(KSCN) и K(KSCN) находят массу «?(МА) (при известном Г(МА)).
Метод Фаянса (титрование с адсорбционными индикаторами). При титровании анионов в присутствии адсорбционных индикаторов КТТ устанавливают по реакции, которая протекает на поверхности осадка AgA, где А — анион.
Ag+ + А’ -> AgA Ф — реакция титрования,
(стандартный раствор AgNO3)
Ag+ + AgA + Ind -> AgA: Ag+ Ing реакция в КТТ. Окраска I Окраска П
700
Новый справочник химика и технолога
Примеры титрования с адсорбционными индикаторами приведены в табл. 5.2.52.
Таблица 5.2.52
Адсорбционные индикаторы, классифицированные по механизму их действия [46, 55]
Тривиальное название Химическое название или формула Титруемый ион (титрант) Изменение окраски
Индикаторы для титрования по методу осаждения
Ализариновый красный Ализаринсульфокислота О ОН SO3H О SCN”(Ag+) Желтая/красная
Бриллиантовый орсейль С NH т/ XN O2N N= N—fip4] NaOaS^^^^^^ SO3Na СГ, Вг”, Г (Ag4), Ag4 (Вг) Красная/синевато-зеленая
Бромфеноловый синий Тетрабромфоно л сульфофталеин Дибром (R) сульфофталеин Дибром (R) флуоресцеин Дииод (R) сульфофталеин Диметил (R) дииод R флуоресцеин Диметил (R) флуоресцеин Дихлор (Р) флуоресцеин Дихлор (R) сульфофталеин Дихлор (R) флуоресцеин СГ, F(Ag+) СГ, Вг”, Г, SCN” (Ag4) Вг”, I”(Ag+) СГ, Вг”, Г, SCN” (Ag4) IW) СГ, Вг”, Г (Ag4) СГ, ВГ, Г (Ag4) СГ, ВГ, Г SCN” (Ag4) СГ,Вг”, I”(Ag+) Желтовато-зеленая/зеленая и желтовато-зеленая/синяя Желтая/густорозовая Желтовато-красная/кр асновато-фиолетовая Желтая/красновато-фиолетовая Желтовато-красная/ красноватофиолетовая Зеленовато-желтая/розовая То же Желтая/розовая Зеленовато-желтая/красная
Метаниловый желтый 4-Анилиназобензол-jw сульфокислота N= N NH SO3H СГ, Вг” I”(Ag+) Желтая/красная и красная/синяя
Примулин СГ, ВГ, Г SCN” (Ag4) Голубая флуоресценция; избыток галогенида
Розовый бенгальский Дихлор (Р) тетраиод (R) флуоресцеин r(Ag+) Красная/синевато-красная
Сульфофлуоресцеин Резорцинсульфофталеин СГ, Вг”, Г, SCN” (Ag4) Желтая/розовая
Химические методы количественного анализа
701
Продолжение таблицы 5.2.52
Тривиальное название Химическое название или формула Титруемый ион (титрант) Изменение окраски
Тартразин SO3Na ii ii NaOOC—С С—N=N Г SO3Na Г + СГ(АвЭ Ag* (СГ, Вг", Г, SCN") Бесцветная/ зеленая
Флоксин Дихлор(Р)тетрабром(К)флуоресцеин Вг“, Г (Ag+) Желтовато-красная/красновато-фиолетовая
Флоксин ВА экстра Тетрахлор(Р)тетрабром(Н)флуоресцеин ВГ, F(Ag+) То же
Флуоресцеин Резорцинфталеин СГ, ВГ, Г, SCN (Ag+) Зеленовато-желтая/розовая
Флуоресцеин-ком-плексон бмс-[ЛуУ-Ди (карбоксиметил) аминометил] флуоресцеин НООС СН НОх /0Н гн rnni- N—Н2С—1 || || ксн,-т/ ноос-сн/ С Но— COOF СХ/° ВГ, Г, SCN” (Ag+) Желтая/розовая
Хромотроповый F4B он N=N— SO,Na SO,Na ВГ,Г(Аё+) Розовая/серовато-зеленая
Эозин (0,5% водный раствор или 0,1% в 60-70% этаноле) Тетрабром(К)флуоресцеин ВГ, I-(Ag+) Желтовато-красная/красновато-фиолетовая
Эритрозин Дииод(К)флуоресцеин I (Ag ) То же
Эритрозин В Тетраиод(К)флуоресцеин HAg+) Красная/ красноватофиолетовая
702
Новый справочник химика и технолога
Продолжение таблицы 5.2.52
Тривиальное название Химическое название или формула Титруемый ион (титрант) Изменение окраски
Поверхностные кислотно-основные индикаторы
Метаниловый желтый 4'-Анилиноазобензол-л/-сульфокислота СГ, Вг~ P(Ag+) Желтая/красная и красная/синяя
Метиловый красный Диметиламиноазобензол-о-карбоновая кислота (CH3)2N^ —N= N—— СООН Zn [Fe(CN)^| Розовая/ желтая
Годамин 6 G NMCJh • НС1 JL^COOC2H5 Ag+(Br) Желтовато-красная/красно-фиолетовая
Фенилазо-1 -нафтиламин N— N——NH2 СГ, ВГ, Г. SCN’ (Ag+) (pH = 4-5) Оранжевая/ фиолетовая
Феносафранин NH О " СГ, ВГ (Ag+) Ag+ (ВГ) Красная/синяя
4-(2'-Этилфенилазо)-1 -нафтиламин с2н5 —N=N——NH, SCN-(Ag+) Фиолетовая/ желтая
л-Этоксихризоидин Н5С2О^ N= N NH2 nh/ I (Ag+) Желтая/рыжевато-красная
Поверхностные окислительно-восстановительные индикаторы
о-Дианизидин H2N——nh2 С Н Ю ()СН3 Zn [Fe(CN)*'],Pb [в присутствии гекса-цианоферрата(Ш)] Фиолетовая/бесцветная
Химические методы количественного анализа
703
Продолжение таблицы 5.2.52
Тривиальное название Химическое название или формула Титруемый ион (титрант) Изменение окраски
Дифенил аминовый синий (1% водный раствор) Дифениламин NH СГ, ВГ, Г, SCN (Ag+) (в присутствии иода или ванадата) Фиолетовая/ зеленая
Ксиленоловый синий VB SO3Na L J SO, Zn [Fe(CN)] [в присутствии гекса-цианоферрата(Ш)] Розовая/зеленая
(С2Н5)2К N(C2H5)2
Нейтральный красный ^СН3 4NH, Вг , Г (Ag+) Фиолетово-красная/ желтовато-красная
Патентованный синий V SO3Ca HO^^^JL f TTX (C2H5)2N'X^^ SO3 Ч(С2Н5)2 Zn (Fe(CN) ] [в присутствии гекса- цианоферрата(Ш)] Розовая/зеленая
Поверхностные комплексометрические индикаторы
л-Диметиламинобензилиденроданин 11—N C=O SC^ C= CH / \ N(CH3)2 HgJ'Hgl lB, ) Фиолетово-красная/голубая
Дифенилкарбазид (0,1% раствор в этаное) NH—NH—/ \ ос \=/ \ll—Nil— Hg (Cl + Br),Cl-(Hg2) Синевато-фиолето-вая/бесцветная
Дифенилкарбазон (0,2-0,3 % раствор в этаноле) N=N—/ \ ОС ^NH-NH-^ СГ, ВГ, Г, SCN~ (Ag4) (в присутствии HgCl2) Фиолетовая/голу-бая
704
Новый справочник химика и технолога
Продолжение таблицы 5.2.52
Тривиальное название Химическое название или формула Титруемый ион (в скобках указан титрант) Изменение окраски
PAN 1'-(1 -Пиридилазо)-2-нафтол 0—N==N •он Г (Ag+) или Си (ЭДТА) (в присутствии Си2+) Красновато-фиолето-вая/бледно-зеленая
Поверхностные флуоресцентные индикаторы
Ауразин Диаминодиметил акоидинформиат нсоон сн3 Ag+(SCN~) Зеленая флуоресцен-ция/оранжево-красная флуоресценция
Индигозоль синий IBC HN— Схдр SO3H SO3H Вг, Г, SCN~ (Ag+) Зеленовато-желтая флуоресценция при избытке галогенида
Лактофлавин CH2- CHOH- CHOH- CHOU- CH,OH 1 CPU N. N. if c=o JL JL 1 nh CH< b II 0 Ag+ (SCN") (Ag+) Зеленовато-желтая флуоресценция при избытке SCN”
Магдала красный CbuQ NH2 Соломенно-желтая флуоресценция/ фиолетовая
Примулин NH2 H3cr ^C1 СГ, ВГ, Г, SCN (Ag+) Голубая флуоресценция при избытке галогенида
Тиофлавин S NHz Г N >] N II К JL c Us XX c Us Jk H3C ^^XSO,Na СГ, ВГ, Г, SCN (Ag+) То же
Химические методы количественного анализа
705
Продолжение таблицы 5.2.52
Тривиальное название Химическое название или формула Титруемый ион (титрант) Изменение окраски
Трипановый красный и,и'-Диаминодифенил-л/-сульфокислота-диазо-бис-2-нафтиламин-3,6-дисульфокис лота SO3Na SO3Na N N II II N N Na(X,SX^^ SO3Na СГВг“, Г, SCN (Ag+) To же
Трипафлавин NH2.1 CH3 h/n^^^ 4C1 •2 СГ HC1 SCN(Ag) Зеленая флуоресценция
Хинин CH A CH2 CH2 CH-CH=CH2 III HO— CH- CH— N- C H2 СГ, Br~, Г, SCN (Ag4) Желтовато-зеленая флуоресценция
Этоксидиаминоакридинлактат H HOOCCHOH CH, OC,H, СГ, Br“, Г, SCN (Ag+) Зеленоватая флуоресценция при избытке серебра
Литература
1 Основы аналитической химии: В 2 кн. Изд. 2-е перераб. и доп. / Под ред. акад. Ю.А. Золотова. М.: Высш, шк., 2000. Кн. 1. Общие вопросы. Методы разделения. 351 с. Кн. 2. Методы химического анализа. 494 с.
2. Васильев В.П. Аналитическая химия: Т. 1. Гравиметрический и титриметрический методы анализа. М.: Высш, шк., 1989. 320 с.
3. Аналитическая химия. Химические методы анализа/ Под ред. проф. О.М. Петрухина. М.: Химия, 1993.397 с.
4. Харитонов Ю.Я. Аналитическая химия. Аналитика в 2-х книгах. М.: Высш, шк., 2001. Кн. 1. Общие теоре
тические основы. Качественный анализ. 615 с. Кн. 2. Количественный анализ. Физико-химические (инструментальные) методы анализа. 559 с.
5. Янсон Э.Ю. Теоретические основы аналитической химии. Изд. 2-е. М.: Высш, шк., 1987. 364 с.
6. Скуг Д., Уэст Д. Основы аналитической химии / Под ред. чл.-корр. АН СССР Ю.А. Золотова; Пер. с англ. М.: Мир, 1979. Т. 1. 480 с. Т. 2. 438 с.
7. Пилипенко А.Т., Пятницкий И.В. Аналитическая химия: В 2-х кн. М.: Химия. 1990. 846 с.
8. Посыпайко В.И., Козырева Н.А., Логачева Ю.П. Химические методы анализа. М.: Высш, шк., 1989. 448 с.
706
Новый справочник химика и технолога
9. Посыпайко В.И., Васина Н.А. Аналитическая химия и технический анализ: Учебн. пособие для вузов. М.: Высш, шк., 1979. 384 с.
10. Фритц Д.Ж., Шенк Г. Количественный анализ / Под ред. чл.-корр. АН СССР Ю.А. Золотова; Пер. с англ. М.: Мир. 1978, 557 с.
11. Лайтинен Г.А., Харрис В.Е. Химический анализ. Изд. 2-е, перераб. / Под ред. проф. Ю.А. Клячко; Пер с англ. М.: Химия, 1979. 624 с.
12. Кунце У., Шведт Г. Основы качественного и количественного анализа. 4-е изд., перераб. / Пер. с нем. А.В. Гармаша. М.: Мир, 1997. 424 с.
13. Попадич И.А. и др. Аналитическая химия. М.: Химия, 1989. 240 с.
14. Булатов М.И. Расчеты равновесий в аналитической химии. Л.: Химия, 1984. 184 с.
15. Белявская Т.А. Практическое руководство по гравиметрии и титриметрии. М.: МГУ, 1986. 158 с.
16. Номенклатурные правила ИЮПАК по химии: Т. 1. Полутом 2. М.: ВИНИТИ, 1979. 660 с.
17. Номенклатурные правила ИЮПАК. Аналитическая химия. Т. 4. М.: ВИНИТИ, 1985.
18. Inezedy J., Lenquel Т., Ure A.M. Compendium of Analytical Nomenclature. Definitive rules 1997 3d Ed. Oxford: Blackwell Science, 1998. ISBN 0865426155.
19. Treatise on Analytical Chemistry / Ed. Kolthoff I.M. and Elving Ph. J. 2nd ed. Part I. Theory and Practice. London; New York: Wiley and Sons. 1978. V. 1. 1979. V. 2. 1983. V. 3.
20. Treatise on Analytical Chemistry. / Ed. Kolthoff I.M. and Ph. J. Elving. Part I. Theory and Practice. V. 11. New York: Willey and Sons, 1975.
21. Skoog D.A., West D.M., Holler F.Y. Analytical Chemistry: an Introduction. Saunders Golden Sunburst Series. 7th ed. Fort Worth. London: Saunders College, 2000. 773 p. ISBN/ISSN 0030202930.
22. Christian G.D. Analytical Chemistry. 5th ed. New York: Willey and Sons, 1994. 812 p.
23. De Levie R. Principles of Quantitative Chemical Analysis. New York: McGraw-Hill, 1997. 737 p. ISBN/ISSN 0071142886; 0070163626.
24. Handbook of Analytical Techniques / Ed. by Gunzler H. and Williams A. New York: Wiley; VCH; Weinheim, 2001. 1182р. QD 75. 22. H36 2001 54321; GBA 1 - Z8392.
25. Official and Standartized Methods of Analysis. 3d ed. / Ed. by Watson C. Cambridge; The Royal Society of Chemistry, 1994. 778p. ISBN 0-85186-441 -4.
26. Harris D.C. Quantitative Chemical Analysis 5th ed. New York.: W.H. Freemen and Company, 1999. 899p, + Appendix + CD ROM. ISBN 0-7167-2881 -8.
27. Vogel's Textook of Quantitative Chemical Analysis I Ed. by Jeffery G.H. et al. Pull.: Longman Scientific and Technical Copublished in the United States. New York: Wiley and Sons, 1989. 877 p.
28. Cooper D. and Doran C.; N.B. Chapman. Classical methods. Vol. 1. Chichester: Published on behalf of ACOL London by Wiley. 1987. 373 p. ISBN 0 471 91362.
29. Wilson's Comprehensive Analytical Chemistry. Classical analysis. Titrimetric Analysis. Organic Quantitative Analysis. Elsevier. Amsterdam etc., 1960. 878 p.
30. Крешков А.П., Быкова Л.Н., Казарян Н.А. Кислотноосновное титрование в неводных растворах. М.: Химия, 1967. 192 с.
31. Денеш И. Титрование в неводных средах. / Под ред. д. х. н. И.П. Белецкой; Пер. с англ. М.: Мир, 1971.413 с.
32. Wilson a. Wilson's. Comprehensis Analytical Chemistri. Ed. by G Svehla. Vol XXII. Titrimetric Analysis in Organic Solvents. By L. Safafik and Z. Stansky. Elsevier. Amsterdam etc., 1986. 531 p.
33. Пришибил P. Комплексоны в химическом анализе. 2-е изд. перераб. и расш. / Под ред. д. х. н. Лурье Ю.Ю.; Пер. с чешек. М: ИЛ, 1960. 580 с.
34. Pribil R. Applied Complexometry By R. Pribil; translated by R. Pribil and M. Stulikova. Col. R.A. Chalmers. Series: Pergamon series in analytical chemistry; V. 5. Oxford: Pergamon, 1982. 410 p. ISBN/ISSN 0080262775.
35. Шварценбах Г., Флашка Г. Комплексонометрическое титрование / Пер. с нем. Ю.И. Вайнштейна. М.: Химия, 1970. 360 с.
36. Юрист И.М., Талмуд М.М. Селективное комплексонометрическое титрование. М.: Наука, 1993. 232 с.
37. Инцеди Я. Применение комплексов в аналитической химии / Пер с англ. д. х. н. О.М. Петрухина, к. х. н. Б.Я. Спивакова. М.: Мир, 1979. 376 с.
38. Дёрффель К. Статистика в аналитической химии / Под ред. к. т. н. Ю.П. Адлера; Пер. с нем. М.: Мир, 1994. 268 с.
39. Дорохова Е.Н., Прохорова Г.В. Задачи и вопросы по аналитической химии. М.: Мир, 2001. 267 с.
40. Толстоусов В.Н., Эфрос С.М. Задачник по количественному анализу. Л.: Химия, 1986. 160 с.
41. Меркушева С.А. Методика решения задач по аналитической химии. Минск: Высш, шк., 1985. 223 с.
42. Индикаторы: В 2-х т. / Под ред. Э. Бишопа; Пер. с англ. под. ред. д. х. н. Марова И.Н. М.: Мир, 1976. Т. 1.495 с. Т. 2.446 с.
43. Hulanicki A., Gtab S., Ackermann G. Compleximetric Indicators: Characteristics and Application (Commission on Analytical Reactions and Reagents // Pure a. applied Chemistry, 1983. V. 55, № 7. P. 1137-1230.
44. Hulanicki A. and Gtab S. Redox Indicators, Characteristics and Applications. (Commision on Analytical Reactions and Readents) // Pure a. applied Chemistry, 1978. V. 50, №5. P. 463-498.
45. Хольцбехер 3. и др. Органические реагенты в неорганическом анализе / Пер. с чешек, д. х. н. 3.3. Высоцкого. М.: Мир, 1979. 752 с.
46. Лурье Ю.Ю. Справочник по аналитической химии. 6-е изд. перераб. и доп. М.: Химия, 1989. 448 с.
47. Сусленникова В.М., Киселева Е.К. Руководство по приготовлению титрированных растворов. Изд. 6-е. перераб. Л.: Химия, 1988. 336 с.
48. Правдин П.В. Лабораторные приборы и оборудование из стекла и фарфора: Справочное изд. М.: Химия, 1988. 336 с.
49. Кульский Л.А. и др. Справочник по свойствам, методам анализа и очистке воды. Ч. 1. Киев: Наукова думка, 1980.
50. Посуда лабораторная стеклянная. Бюретки. Ч. 1. Общие требования. ГОСТ 29251-91 (ИСО385/1-84).
Химические методы количественного анализа
707
М.: Комитет стандартизации и метрологии СССР, 1992. 20 с.
51. Посуда лабораторная стеклянная. Пипетки градуированные. Ч. 1. Общие требования. ГОСТ 29227 (ИСО 8351/1-81) М.: Комитет стандартизации и метрологии СССР. 1992. 14 с.
52. Посуда мерная лабораторная стеклянная. Пипетки с одной отметкой. ГОСТ 29169-91 (ИСО648-77) М.: Комитет стандартизации и метрологии СССР, 1992. 13 с.
53. Посуда мерная лабораторная стеклянная. Цилиндры, мензурки, колбы, пробирки. Технические условия. ГОСТ 1770-74 (ИСОЮ42-83). М: Госстандарт России, 1992. 25 с.
54. Коростелев П.П. Титриметрический и гравиметрический анализ в металлургии: Справочник. М.: Металлургия, 1986. 320 с. Он же: Фотометрический и комплексонометрический анализ в металлургии: Справочник. М.: Металлургия, 1984. 272 с.
55. Лазарев А.И. и др. Справочник химика-аналитика. М.: Металлургия, 1976. 184 с.
56. Серия: Аналитическая химия элементов. М.: Наука, 1980-1990.
57. Wilson a. Wilson's. Comprehensive Analytical Chemistry. V. 1c. Classical analysis: Gravimetric and Titrimetric Determination of the Elements. Amsterdam etc: Elsevier. 1962. 728p.
58. Treatise on Analytical Chemistry Ed. Kolthoff I.M. and Elving PJ. Pt. 2. Analytical Chemistry of Elements. Wiley, New York etc. V. 1. 1961. V. 10. 1978.
59. Котик Ф.И. Ускоренный контроль электролитов, растворов и расплавов: Справочник. М.: Машиностроение, 1978. 191 с.
60. Руководство по анализу в производстве фосфора, фосфорной кислоты и удобрений / Под ред. И.Б. Мойжес. Л.: Химия, 1973.216 с.
61. Огнеупоры и огнеупорные соединения: Ч. 3. М.: Изд. Стандартов, 1986. 424 с.
Раздел 6
ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Авторы-составители:
проф., акад. РАЕН, д.х.н. Ю.Г. Власов, проф., д.х.н. Ю.Е. Ермоленко,
к.х.н. В.А. Демин, н.с. В.В. Колодников
СИМВОЛЫ, ОБОЗНАЧЕНИЯ
МВИ — методика выполнения измерений ПАВ — поверхностно-активное вещество ТМ — тяжелые металлы
ЭМА — электрохимические методы анализа
МЕТОДЫ
ВА — вольтамперометрия
ИВ — инверсионная вольтамперометрия
ИАдсВА — инверсионная адсорбционная вольтамперометрия
ИДИВ — инверсионная дифференциально-импульсная вольтамперометрия
ИКВВ — инверсионная квадратно-
волновая вольтамперометрия
ИП — импульсная полярография
ИПРВ — инверсионная переменно-
токовая вольтамперометрия
ИПТВ — инверсионная постоянно-
токовая вольтамперометрия
ИХП — инверсионная хронопотонцио-метрия
КВВ — квадратноволновая вольтамперометрия (полярография)
кул. — кулонометрия
ОП — осциллографическая полярография
ПРП — переменно-токовая вольтамперометрия (полярография) (АС)
ПТП —- постоянно-токовая вольтамперометрия (полярография) (DC)
ХА — хроноамперометрия
ХП — хронопотонциометрия
ЭЛЕКТРОДЫ РАЗЛИЧНОГО
НАЗНАЧЕНИЯ
ГЭ — графитовый электрод (из углеродного материала)
ЗГЭ — золото-графитовый электрод
ИСЭ — ионоселективный электрод нас. к. э. — насыщенный каломельный электрод
н. в. э. —- нормальный водородный элек-
трод
х. с. э. — хлорид-серебряный электрод РГЭ — ртутно-графитовый электрод РКЭ — ртутный капающий электрод РПЭ — ртутный пленочный электрод СРКЭ — стационарный ртутный капельный электрод («висящая капля»)
СУ — стеклоуглерод
СУЭ — стеклоуглеродный электрод
УПЭ — угольно-пастовый электрод
Ag-Э, Аи-Э, Pt-Э — серебряный, золотой, платиновый электроды соответственно
а,—активность определяемого иона, моль-л 1
а,— активность постороннего (мешающего) иона, моль-л 1
А — площадь поверхности электрода, поверхность раздела фаз, см2
bv — полуширина пика тока, мВ
72
с — концентрация, моль-л '
сн — нижняя граница определяемой кон-
центрации
с, — концентрация иона в растворе, моль-л 1
D — коэффициент диффузии, см2-с 1
Е — измеряемый потенциал сенсора, В
Е$ — константа, в которую входят стандартный потенциал сенсора, потенциал электрода сравнения и жидкостный потенциал, В
ЕУ1
— потенциал полуволны, В
Е3 — потенциал электролиза
Ец — потенциал относительно нормального водородного электрода, В
Еп — потенциал максимума пика тока, В f— коэффициент активности иона
F — число Фарадея (96487 Кл-моль”1 [1]) i — плотность тока, А-см”2
I— сила электрического тока, А; для раздела 6.1 — ионная сила раствора, моль-л”1
/д — диффузионный ток, А
1С — емкостный ток, А
/ост. — остаточный ток, А
/ф — фарадеевский ток
Kj (Kj/j, КА/В) — коэффициент селективности
L — лиганд
М— полярная, ионная или атомная масса вещества;
М — молярная концентрация определяемого элемента
т — масса вещества
Ме(и) — ион металла в валентном состоянии п
п — число электронов, участвующих в реакции
Q — количество электричества, Кл
R — универсальная газовая постоянная (8,31470±0,00034 Дж-(К-моль) 1 [2])
5 — крутизна электродной функции, В
Т — абсолютная температура, К
t — текущее время, с
Т,л — время (длительность) электролиза
V — объем, см3
w — скорость развертки потенциала, мВ-с”1
zt — заряд определяемого иона
Zj — заряд постороннего (мешающего) иона
0 — константа кондуктометрической ячейки, см 1
ср — фазовый угол
т]а — перенапряжение водорода на металле при i = 1 А-см”2
р — удельное сопротивление, Ом-м
т — переходное время
х — удельная электропроводность, См-см”1
со — частота переменного тока
710
Новый справочник химика и технолога
КЛАССИФИКАЦИЯ ЭЛЕКТРОХИМИЧЕСКИХ МЕТОДОВ
Электрохимические методы анализа (ЭМА) основаны на исследовании процессов, протекающих на поверхности электрода или в приэлектродном пространстве. Аналитическим сигналом служит электрический параметр (потенциал, сила тока, сопротивление и др.), функционально связанный с концентрацией определяемого компонента раствора и поддающийся правильному измерению.
Классификация ЭМА, предлагаемая ИЮПАК, за последние десятилетия претерпела определенные изменения, в нее внесены уточнения (пояснения) и дополнения [3-8].
Существенное внимание уделяется электрохимическим ячейкам и датчикам аналитического сигнала (электродным системам, различным электрохимическим сенсорам), именно эти первичные электрохимические преобразователи определяют аналитические возможности любого метода. В настоящее время не представляет проблемы самая совершенная и быстрая обработка сигнала от датчика, расчет статистических характеристик как исходного сигнала, так и результатов всего анализа в целом [8]. Именно поэтому важно получить достоверный исходный сигнал, чтобы прокалибровать его в единицах концентрации.
Согласно общей классификации, предложенной ИЮПАК, ЭМА подразделяются на методы, в которых возбуждаемый электрический сигнал постоянен или равен нулю и на методы, в которых возбуждаемый сигнал меняется во времени. Исходя из этой преамбулы, в [8] выделены следующие методы (области, fields) ЭМА:
вольтамперометрические - voltammetry, 7# 0; Е = f(t)\ потенциометрические - potentiometry, (/ = 0);
амперометрические - amperometry 0; Е = const); хронопотенциометрические, Е = f(t)', I = const;
импедансные, или кондуктометрические — измерения, использующие наложение переменного напряжения малой амплитуды;
другие, комбинированные (например, спектроэлектрохимические).
Литература
1. Плэмбек Дж. Электрохимические методы анализа. / Пер. с англ. М.: Мир, 1985. 496 с.
2. Краткая химическая энциклопедия. М.: Советская энциклопедия, 1964. Том 1. А-Е. 758 с.
3. Классификация и номенклатура электрохимических методов И Журн. аналит. химии. 1978. Т. 33, вып. 8. С.1647-1665.
4. Recommended Terms, Symbols and Definitions for Elec-troanalytical Chemistry // Pure & Appl. Chem. 1979. Vol. 51. P. 1159-1174.
5. Об использовании понятия «химический эквивалент» и связанных с ним величин: Журн. аналит. химии.
1989. Т. 44, вып. 4. С. 762—764; Журн. аналит. химии.
1982. Т. 37, вып. 5. С. 946; Журн. аналит. химии. 1982. Т. 37, вып. 5. С. 947.
6. Нейман Е.Я. Терминология современной аналитической химии и ее формирование // Журн. аналит. химии. 1991. Т. 46, вып. 2. С. 393-405.
7. Представление результатов химического анализа (Рекомендации ШРАС 1994 г.) // Журн. аналит. химии. 1998. Т. 53. № 9. С. 999-1008.
8. Compendium of Analytical Nomenclature (Definitive Rules 1997). 3rd ed., IUPAC, Blackwell Science, 1998. 8.1-8.51 (Electrochemical Analysis).
6Л. Химические сенсоры. Потенциометрия
Определение сенсора как аналитического инструмента
Сенсор — это первичное устройство, реагирующее (откликающееся) на определенные свойства окружающей среды и позволяющее регистрировать этот отклик в виде соответствующего электрического (оптического и др.) сигнала [1].
Химические сенсоры — это датчики, дающие прямую (т.е. без фиксированного отбора пробы и подготовки) информацию о химическом составе окружающей среды, обычно в непрерывном режиме и с малым временем отклика [2].
6.1.1. Типы сенсоров и физические принципы их функционирования
По типу преобразователя все химические сенсоры можно разделить на четыре группы.
Электрохимические. Это потенциометрические (ион-селективные электроды — ИСЭ, ионселективные полевые транзисторы — ИСПТ) и вольт- и амперометрические сенсоры, в том числе твердые электролитические газовые сенсоры. Полупроводниковые газовые сенсоры также могут быть включены в эту категорию, хотя механизм их действия не включает химическую реакцию.
Оптические. В оптических сенсорах спектроскопическое определение связано с химической реакцией. Оптические сенсоры часто называют оптодами, и в будущем применение оптических волокон будет повсеместным. Оптические измерения используются во многих биосенсорах. В зависимости от типа оптических сенсоров в них измеряют поглощение, отражение или люминисценцию.
Масс-чувствительные. Этот тип сенсоров основан на использовании пьезоэлектрического эффекта. Сюда включают такие устройства, как поверхностные акустоволно-вые сенсоры (ПАВ-сенсоры), основанные на использовании пьезоэлектрического эффекта и особенно полезные в качестве газовых сенсоров.
Теплочувствительные. Сенсоры, относящиеся к этой группе, часто называют калориметрическими. Их действие основано на регистрации с помощью преобразователя — например, термистора или платинового термометра — теплового эффекта химической реакции с участием аналита. На этом же принципе может быть основано действие противопожарных газовых сенсоров.
Биосенсоры. Действие биосенсоров основано на важнейших химических реакциях, от которых, без преувеличения, зависит сама жизнь. Реакции: антитело - антиген, фермент - субстрат, рецептор - гормон— используются для
Электрохимические методы анализа
711
получения высокоселективных и чувствительных сенсоров на конкретные определяемые вещества. Для объяснения принципа действия биосенсоров часто используют схему, подобную представленной на рис. 6.1, хотя она достаточно универсальна и применима к любым типам сенсоров, в которых реагент обладает сродством к индивидуальному веществу. Для иллюстрации высокоселективных реакций, протекающих между биологическими молекулами, предложен механизм, получивший название «ключ-замок».
Соединение
Раствор амаяита. wviepoumS определяемое •». (к-шдаж.
с прес&ражватежм
Слой, адсржаашй спеакфвчйекий ршкпнжнций реагент
Рис. 6.1. Биосенсор
В биосенсорах узнающим реагентом обычно является макромолекула, иммобилизованная внутри мембраны либо химически связанная с поверхностью, контактирующей с раствором определяемого вещества. Между реагентом и определяемым веществом проходит специфическая химическая реакция. Это может быть либо прямое взаимодействие реагента с определяемым веществом, как в случае реакции антиген - антитело, либо каталитическое взаимодействие иммобилизованного фермента с определяемым веществом с образованием легко определяемого продукта. В качестве трансдьюсеров (трансдьюсер — преобразователь сигнала сенсора в электрический сигнал) могут использоваться любые из упомянутых выше.
Виды потенциометрических сенсоров
Химические сенсоры способны селективно откликаться на изменение концентрации какого-либо компонента (ион, молекула) в жидкой или газовой фазах. Приведенная ниже (рис. 6.2) классификация потенциометрических химических сенсоров [3] показывает многообразие их типов. В классификации химических сенсоров датчики с твердотельными кристаллическими мембранами занимают центральное место не только по числу определяемых компонентов (более 20 различных ионов) и по селективности (сенсоры на ионы серебра и фтора), но и по той роли, которую они играют в качестве базовых объектов для изучения таких сенсорных механизмов, как селективность, предел обнаружения, быстродействие, влияние pH, Red/Ox и др.
При классификации сенсоров учитывают
- тип определяемых частиц: электроны, ионы, молекулярные соединения — это верхний уровень классси-фикации;
- тип используемой мембраны: жидкая или твердая, гомогенная или гетерогенная (средний уровень);
- вид (структура) материала мембраны: стеклянная, кристаллическая, ионообменная (нижний уровень).
В отдельный подуровень выделены ионоселективные полевые транзисторы (ИСПТ) и химически чувствительные полупроводниковые устройства, отличающиеся рядом
особенностей, обусловленных во многом спецификой микроэлектронной основы и возможностью применения в качестве мембран диэлектрических пленок.
Среди химических сенсоров pH-сенсоры были разработаны одними из первых и в настоящее время являются наиболее широко используемыми при научных исследованиях и в промышленности. Для правильного измерения величины pH в растворах приводим в сокращенном виде последние (2000 г.) рекомендации ИЮПАК «Измерение pH. Определения, стандарты и процедуры». Они разработаны на основе рекомендаций Национального бюро стандартов США (NBS, ныне NIST — Национальный институт стандартизации) еще в 50-60-е гг. XX века под руководством Р. Бейтса (R.G. Bates) и приняты во многих странах, включая СССР, в качестве Государственного стандарта (ГОСТ), имеющего силу закона.
Главным в этих рекомендациях является набор первичных стандартных (эталонных) буферных растворов, pH которых точно определен на основе некоторого «первичного метода». К веществам, составляющим эти буферные растворы, и к их буферным свойствам предъявляются чрезвычайно высокие метрологические требования: высокая буферная емкость; стойкость к разбавлению; слабая зависимость pH от температур; низкий остаточный диффузионный потенциал; низкая ионная сила. Кроме того, реактивы и вода, используемые для приготовления первичных буферных растворов, должны иметь высший метрологический сертификат чистоты и стабильности при хранении. Этим требованиям удовлетворяют семь буферных растворов (табл. 6.1.).
Таблица 6.1
Состав и pH первичных стандартных буферных растворов при 25 °C
Состав pH
Насыщ. КНСдНдОб (моногидрат тартрата калия) 3,557
0,05 моль-кг1 КН2С6Н5О7 (дигидрат цитрата калия) 3,776
0,05 моль-кг1 КНС8Н4О4 (моногидрат фталата калия) 4,005
0,025 / 0,025 (моль-кг1) Na2HPO4/KH2PO4 6,865
0,03043 / 0,008695 (моль-кг’1) Na2HPO4/KH2PO4 7,413
0,01 моль-кг’1 Na2B4O7 10 Н2О 9,180
0,025 / 0,025 (моль-кг1) NaHCO3/Na2CO3 10,012
Первичные стандарты со значениями pH, установленными с высокой точностью (табл. 6.1), нужны, в основном, метрологическим учреждениям. На их основе устанавливают вторичные стандарты, предназначенные (с округленными до 0,01 ед. значениями pH) для широкого употребления в научных, медицинских, заводских лабораториях и др. Требования к веществам и буферным свойствам растворов в этом случае не столь строгие, как в случае первичных стандартов. Составы и значения pH некоторых вторичных буферных растворов приведены в табл. 6.2.
712
Новый справочник химика и технолога
Рис. 6.2. Классификация химических сенсоров
Таблица 6.2
Состав и pH вторичных стандартных буферных растворов при 25 °C
Состав рн
0,05 моль-кг4 КН3С4О8 (К-тетраоксалат) 1,68
0,05 моль-кг1 NaH-дигликолят 3,49
0,1/0,1 (моль-кг4) CH3COOH/CH3COONa 4,65
0,01/0,1 (моль-кг4) CH3COOH/CH3COONa 4,72
0,02 моль-кг4 пиперазин фосфат 6,29
0,05/0,01667 (моль-кт !) Трис • НС1 [Трис - трис(гидроксиметил)аминометан] 7,70
0,05 моль-кг4 Na2B4O7-10 Н2О 9,19
Насыщ. Са(ОН)2 12,45
Описанные буферные растворы широко применяются для градуировки стеклянных электродов, для поддержания pH производственных растворов, микробиологических питательных сред, физиологических растворов и т.п. Для различных применений разработаны специальные буферные растворы. Например, для многих технических целей не так важна точность указания pH, как более высокая буферная емкость. Для медицинских лабораторий разработан набор из шести буферов на основе системы KH2PO4/Na2HPO4 + NaCl в узком интервале pH = 6,8-7,3 с точным (до 3-го знака) указанием pH (NaCl вводится для придания буферному раствору изотоничности с сывороткой крови при 25 и 37 °C). Для биологических лабораторий разработаны наборы буферов, не содержащие ионов, принимающих участие в клеточном метаболизме. Для низких (до -10 °C, с добавлением этанола) и высоких (до 150 °C) температур, для тяжелой воды (pD) стандартизованы специальные буферы.
Следует отметить, что шкалы pH, разработанные для водных растворов, исходящие из определенных значений Kw, лишь с поправками применимы для растворов в смешанных растворителях, и тем менее, чем меньше в них воды. Для перехода разработаны специальные процедуры. Официально признанных международных стандартов pH для неводных растворителей пока не существует.1
6.1.2. Интеллектуальные сенсорные системы {«электронный нос» и «электронный язык»)
Решение задачи применения неспецифических (неселективных) сенсоров было заимствовано из биологии, в результате чего появились системы типа «электронный нос» [193], «сенсоры вкуса» [194] и «электронный язык» [187]. Они реализуют мультисенсорные системы на основе неспецифических (неселективных) сенсоров с последующей обработкой результатов измерений методом распознавания образов с применением, например, искусственных нейронных сетей. Такой подход характеризует новый этап в истории развития химических сенсоров.
Разработка систем «электронный нос» и «электронный язык» стимулируется желанием смоделировать и расширить возможности, а в некоторых случаях заменить такие человеческие способности, как обоняние и восприятие вкуса. Устройство таких сенсорных систем основано на принципах организации биологических систем — массивов неспецифичных рецепторов с последующим распознаванием образов нейронной сетью головного мозга человека. Поскольку в сенсорных системах используются многие методы обработки данных высокой размерности и нейрокомпьютерные подходы, то «электронный нос» и «электронный язык» можно рассматривать как специальную
1 Сведения по применению pH-сенсоров любезно предоставлены профессором СПбГУ А.А. Белюстиным.
Электрохимические методы анализа
713
ветвь развития искусственного интеллекта и «электронного мозга». Образное представление о работе электронного языка дано на рис. 6.3.
Рис. 6.3. Образное изображение функционирования «электронного языка» при распознавании вкуса напитков (пива и др.) (Analyst, 1993. V. 18. 1-я стр. обложки)
«Электронный язык» представляет собой аналитическое устройство для качественного и количественного анализа многокомпонентных растворов различной природы, состоящее из массива (набора) неспецифических (неселективных) химических сенсоров, обладающих перекрестной чувствительностью. Для обработки сигналов от данной мультисенсорной системы используются различные математические методы распознавания образов (искусственные нейронные сети, анализ по главным компонентам, нечеткая логика и т.п.).
6.1.3. Основные характеристики потенциометрических сенсоров
6.1.3.1. Уравнение Никольского
Независимо от природы сенсора возникающий на нем потенциал подчиняется уравнению Нернста:
£ = £0 ±2,3—-Iga-, (6.1)
z.F 1
где Eq — стандартный потенциал сенсора; R — газовая постоянная; Т — температура; z, — заряд определяемого иона; F — число Фарадея; а, — активность определяемого иона, ±— знак «+» относится к катионселективным, а «-»— к
„ RT анионселективным сенсорам. Величина 2,3--называется
z;F крутизной электродной функции и обозначается обычно как 5; a, = Cjf (с, — концентрация определяемого иона; f— коэффициент активности иона).
Уравнение Нернста справедливо в случае, когда в растворе присутствуют только потенциалопределяющие ионы (например, раствор одной соли). Если в анализируемом растворе также присутствуют ионы, которые более или менее влияют на потенциал химического сенсора (раствор нескольких солей), то используется уравнение Никольского:
E = E0±2,3^1g а,+^Кюа‘‘ z,r
(6.2)
где — коэффициент селективности; а7 — активность мешающего иона; z7 — заряд мешающего иона.
Влияние мешающих ионов на потенциал химического сенсора количественно характеризуется коэффициентом селективности. Чем меньше значение коэффициента селективности, тем более селективен сенсор к определяемому иону.
6.1.3.2. Крутизна электродной функции
Крутизна электродной функции, определяющая чувствительность химического сенсора, — это параметр, выражающий связь между величинами ЭДС и логарифма кон-ценрации (активности) определяемого вещества (рис. 6.4). Нернстовский угловой коэффициент наклона зависит от температуры; при 25 °C и десятикратном изменении активности определяемого иона, он равен 59,16 мВ для однозарядных ионов и 29,58 мВ для двухзарядных ионов. Однако обычно угловой коэффициент наклона меньше теоретического значения, что обусловлено присутствием мешающих примесей или старением жидкостного электрода. Угловые коэффициенты наклона, величины которых превышают теоретическое значение, наблюдаются довольно редко; они указывают на то, что при измерениях происходит больше одного электродного процесса (см. уравне-
Рис. 6.4. Типичная электродная функция (на примере фторид-селективного электрода) [4]
6.1.3.3. Предел обнаружения
Предел обнаружения наименьшей концентрации иона или газа, которую можно определить данным методом, для большинства электродов определяется растворимостью материала мембраны, в состав которого входит потенци-алопределяющий ион или вещество. Величина кажущейся
714
Новый справочник химика и технолога
концентрации определяемого иона соответствует ЭДС электродной цепи при его нулевой концентрации в анализируемом растворе. Величину фона получают экстраполяцией линейного (нернстовского) участка калибровочного графика (рис. 6.5). Отклик электрода, помещенного в холостой раствор, возникает вследствие частичного растворения электродноактивного соединения. Фоновой концентрации соответствует, таким образом, концентрация потен-циалопределяющего иона в растворе, контактирующем с мембраной ионоселективного электрода. Следует иметь в виду, что соответствующий ион можно количественно определять при концентрации как минимум на один порядок меньшей, чем фоновая концентрация, так как наклон графика даже в этом случае имеет постоянную величину.
Рис. 6.5. Графический способ определения фоновой концентрации электрода (с — концентрация определяемого элемента, обусловленная растворимостью ионочувствительной мембраны) [4]
Произведение растворимости — константа, характеризующая растворимость соединения. Равняется произведению концентраций двух образующих это соединение ионов, взятых в степени, соответствующей стехиометрическим коэффициентам. Так, произведение растворимости (ПР) соединения АхВу равно [A]v-[Bf . Термин «произведение растворимости» применим только к насыщенным растворам, находящимся в равновесии с избытком нерастворенного твердого вещества. Чем меньше растворимость вещества, тем меньше произведение растворимости.
Образование осадка начинается после того, как произведение концентраций соответствующих ионов в растворе превысит величину ПР независимо от их относительной концентрации и источника ионов в растворе. Поэтому растворимость ионных соединений в растворителе, содержащем один из входящих в это соединение ионов, меньше, чем в чистом растворителе (эффект одноименного иона).
При построении градуировочной зависимости в растворах «свободных» потенциалопределяющих ионов нернстовская
область, как правило, наблюдается до 10 5 10 7 моль-л'1. Применение буферных растворов позволяет расширить область теоретической зависимости, описываемой уравнением Нернста. Так, например, для Ag2S-3neKTpofla отклик на ион Ag+ сохраняет теоретическое значение до величины pAg = 25. Эти результаты представляются закономерными, поскольку в буферном растворе компенсируется как выведение небольших количеств потенциалопределяющих ионов за счет адсорбции, так и появление тех же ионов, вносимых с реактивами, за счет реакций компонентов раствора с поверхностью мембраны.
Следует указывать экспериментальные условия, имеющие место при определении величины предела обнаружения, так как на эту величину влияют многие факторы, например: состав раствора (наличие мешающих компонентов, вымывание ионов из мембраны, загрязнение примесями реагентов, наличие специально введенных фоновых электролитов), «история» и предварительная обработка электрода.
Вообще говоря, пределом обнаружения считается концентрация определяемого иона, начиная с которой калибровочная кривая отклоняется от линейной зависимости в пределах точности измерения. По предложению ИЮПАК пределом обнаружения при использовании ионочувствительной измерительной техники по аналогии с другими физико-химическими методами считается такая концентрация определяемых ионов, при которой аналитический сигнал становится вдвое больше фонового. Это происходит в том случае, если отклонение от уравнения Нернста становится равным 18/г мВ т.е. (59,1/z • lg2 = 18/г, при 25 °C).
6.1.3.4. Селективность
Селективностью сенсора называют его способность различать ионы различного вида, присутствующие в растворе. Потенциал идеально селективного, или специфичного, сенсора не зависит от концентрации любого примесного иона, содержащегося в растворе, кроме того иона, относительно которого сенсор обладает селективностью (специфичностью).
Относительно применения терминов «константа селективности», «отношение селективности» и «коэффициент селективности» существуют различные мнения. Обычно рекомендуется употреблять два последних термина, поскольку эти параметры зависят от применяемого метода измерения и характера раствора.
Для использования в аналитической практике селективность к определяемым ионам должна быть по возможности велика, с тем чтобы нернстовская зависимость потенциала от ионной активности сохранялась в наибольшем диапазоне активностей. Это условие выполняется, если коэффициент селективности для других ионов достаточно мал. В этом случае соотношение для одновалентных ионов
ah ^А/В^В (6-3)
выполняется даже при большом избытке посторонних ионов по сравнению с определяемым ионом.
Электрохимические методы анализа
715
Все предложенные методы определения коэффициента электродной селективности являются приближенными, что связано с применением полуэмпирических уравнений для мембранного потенциала и с использованием при расчетах условных активностей отдельных ионов или просто концентраций. Для систем, содержащих ионы различных зарядов, коэффициенты селективности обычно определяют по уравнению Никольского.
Приближенность определяемых экспериментально величин Kj/j (Ад/в) проявляется в том, что они часто зависят от состава раствора, концентрации обменника в мембране и от метода определения [75]. Для простоты при описании методов определения коэффициентов селективности рассмотрим два иона одинакового заряда А+ и В+. Коэффициент Хд/в может быть рассчитан на основании измерения ЭДС элемента с мембраной в растворах, содержащих либо один электролит (чистые растворы), либо смесь электролитов (смешанные растворы). В соответствии с этим имеется две группы методов:
первая — определение коэффициента селективности на основе чистых растворов, один из которых содержит ионы А+, а другой — ионы В+;
вторая — определение с помощью смешанных растворов.
Первая группа методов. Метод 1а.
В данном случае применяют гальванический элемент типа:
Ag AgCl, KCl(m) Мембрана Исследуемый раствор АХ, ВХ Электрод сравнения
Мембранный электрод
ЭДС этого элемента подчиняется уравнению
Е = Е0+51ё(аА+^А/Вав)5 (6-4)
которое для случаев содержания в исследуемом растворе только АХ (дв = 0) принимает вид:
Е = ЕХ =E0+51gaA. (6.5)
Эта зависимость представлена графически на рис. 6.6.
(прямая 7). Если раствор содержит только ВХ (аА = 0), то
Е = Е2 =E0 + SlgXA/B + 51gaB. (6-6)
(рис. 6.6, прямая 2).
Вычитая (6.5) из (6.6), получим:
£,-£,= S(lgJrA/B + lgaB-lgaA)= Slg^B-. (6.7)
«А
Если аА = «в, то:
(6.8)
Рис. 6.6. Графическая иллюстрация определения коэффициента селективности методами 1а и 16'.
1 — зависимость Е от -lgaA (А — основной ион);
2 — зависимость Е от -Igag (В — влияющий ион)
Вычисление KKiV> по уравнению (6.8) требует определения ЭДС соответствующего элемента или потенциалов мембранного электрода в чистых растворах основного и постороннего иона. Этот метод обычно называют методом биионных потенциалов (БИП).
Метод 16. Если Е\ = Еъ то из уравнения (6.8) получим:
И
К^=^. (6.10)
аъ
Иными словами, коэффициент селективности равен отношению активностей ионов в чистых растворах, в которых электрод имеет одинаковый потенциал (ЭДС). Если же ионы имеют различные значения зарядов, например, посторонний ион двузаряден, то
ал=АГл/в(ав)12- (6-11)
Вторая группа методов. Метод 2а.
Этот метод наиболее надежен в случае оценки электродной селективности при не очень различающейся избирательности ионов. Метод основан на измерении потенциалов (ЭДС) в смешанных растворах с постоянным содержанием постороннего иона В+ (фоном) и переменной концентрацией основного иона А+. По мере того как активность основного иона падает, нарастает влияние постороннего иона и кривая зависимости E=/(lg аА) (рис. 6.7) становится горизонтальной, т. е. Е остается постоянной при дальнейшем изменении аА. Это означает, что электрод приобрел функцию постороннего иона (В4} и потерял функцию основного иона (А ). Если прямую, соответствующую А+-функции электрода, продолжить до пересечения с горизонтальной прямой, то точке пересечения будет отвечать условие Ei = Ег. Коэффициент селективности вычисляется по формуле Кмз = а^!аъ.
716
Новый справочник химика и технолога
В некоторых случаях требуемую величину «А можно находить по точке на экспериментальной кривой, в которой разность ЛЕ между экспериментальной кривой и нернстовской прямой равна (по вертикали) 18/zA (мВ). Активность ионов А+ в этой точке (ах) равна искомому значению, входящему в уравнение (6.10).
Рис. 6.7. Графическая иллюстрация определения коэффициента селективности при закрепленной концентрации влияющего иона: ЛЕ = (59,16/z) 1g 2 = 18/z [5]
Метод 26. В этом методе также используют данные для смешанных растворов. Из уравнений (6.4) и (6.5) получаем уравнение:
ЛЕ = Е-Е} =51gaA+^A/BaB , (6.12)
«А
откуда
Значение Ei рассчитывают для значения аА, соответствующего экспериментальному значению Е в смешанном растворе. Можно для расчета использовать уравнение (6.13), но более надежные данные получают графическим способом, который включает серию измерений. Первоначально измеряют Е в заданном растворе, содержащем ион А+ с активностью <тА. Затем частями добавляют известный объем раствора, содержащего ионы В+, и измеряют Е в смешанных растворах.
В результате получают серию значений Е в смешанных растворах, каждое из которых отвечает определенной активности ионов А+ и В+. Откладывая в соответствии с уравнением (6.13) по оси ординат произведение (1()ЛЛ5”1) • «д, а по оси абсцисс ав, находим К^, равную тангенсу угла наклона прямой. Этот способ удобен при невысокой селективности электрода. Если соответствует высокой селективности электрода по отношению к иону А+, то потенциал электрода не будет чувствителен к небольшим изменениям активности иона В+. В этом случае в растворе поддерживают высокий фон постороннего иона, и в этот раствор постепенно добавляют основной ион.
Различия в коэффициентах селективности, определяемых различными методами, могут быть значительными (до порядка величины), особенно при сравнении результатов пер
вой и второй группы методов. Большинство исследователей предпочитает методы на основе смешанных растворов.
6.1.3.5. Рабочая область pH
Влияние pH анализируемого раствора. Величина pH служит мерой активности ионов Н+ (водорода) (и косвенно ОН -ионов) в растворе. Ионы Н+ или ОН могут взаимодействовать с определяемыми ионами, уменьшая таким образом концентрацию свободных ионов, определяемых данным электродом. В итоге результаты исследования будут искажены. Ионы водорода и гидроксид-ионы могут менять электродную функцию, являясь причиной возникновения дополнительного электродного потенциала, что также приводит к ошибкам. Контроль pH необходим и в тех случаях, когда ионы или газы переводят в определенную форму, к которой чувствителен данный электрод.
На рис. 6.8 представлена зависимость показаний свинцового селективного или Pb-электрода от pH исследуемого раствора. Пунктирной линией показаны максимально допустимые значения pH, при которых еще возможно определение соответствующей концентрации свинца. При большей основности раствора начинает осаждаться гидроксид свинца, что приводит к уменьшению содержания определяемых ионов в анализируемом растворе. Чтобы избежать осаждения, необходимо строго контролировать pH анализируемого раствора. Несмотря на мешающее влияние ионов водорода при низких концентрациях свинца (<10 3М), свинцовый электрод можно применять в довольно широком диапазоне pH.
Рис. 6.8. Влияние pH на ЭДС элемента, составленного из свинец-селективного электрода и электрода сравнения [4]
Получение заниженных результатов вследствие влияния ОН~-ионов. Медь(П)-селективный электрод используют для определения меди(П) в растворе при pH = 12,0. Чтобы результаты анализа не оказались заниженными из-за образования Си(ОН)2, pH раствора снижают до 8,0, добавляя хлороводородную кислоту. На рис. 6.9 приведены
Электрохимические методы анализа
717
графики, с помощью которых можно установить максимальные концентрации ионов металлов, при которых начинается осаждение гидроксидов при данном pH.
Перевод ионов или газов в формы, удобные для определения. Газочувствительный электрод для измерения концентрации диоксида углерода используют при определении общего содержания карбоната (СО2, Н2СО3, НСО3, СО2-) в растворе при pH = 8,6. Чтобы такое определение было возможным, необходимо перевести все формы карбоната в газообразный диоксид углерода. С этой целью раствор подкисляют до pH = 4,8 и добавляют буферный раствор, чтобы pH оставался постоянным (рис. 6.10).
Получение завьииенных результатов вследствие влияния ионов водорода. Для определения концентрации ионов натрия в растворе при pH = 4,0 используют натрий-селективный электрод. Этот электрод чувствителен не только к ионам натрия, но и к ионам водорода, поэтому результаты определения натрия при таком значении pH получаются завышенными. Повышая pH раствора до 8,5,
можно уменьшить влияние ионов водорода.
помощью данного электрода. Поэтому, чтобы результаты анализа оказались правильными, pH раствора необходимо повысить, освободив, таким образом, фторид-ионы, связанные с ионами водорода (рис. 6.11).
Рис. 6.11. Влияние pH на точность определения фторид-ионов: pH должен быть выше 5,6, чтобы избежать образования HF и HF7 [4]
Рис. 6.9. Максимальные концентрации металлов, при превышении которых начинается осаждение гидроксидов [4]
6.1.3.6. Время отклика
Время отклика — время, по истечении которого потенциал электрода принимает постоянное значение при перемещении электрода из одного анализируемого раствора в другой, отличающийся по концентрации определяемого иона. Время отклика зависит от типа электрода, присутствия мешающих ионов, температуры, разности концентраций в указанных растворах, а также от характера изменения концентрации — увеличения или уменьшения. После смены анализируемого раствора величина ЭДС постепенно приближается к истинному значению. В большинстве случаев уже через одну минуту фиксируемая ЭДС составляет > 90% конечной величины (рис. 6.12).
520 Г £ мВ
10 ’-io 2MAgNO?
Рис. 6.10. Графическая иллюстрация нахождения общего содержания карбоната: все его формы переводят в диоксид углерода, который определяют, используя соответствующий газочувствительный электрод [4]
Получение заниженных результатов вследствие влияния ионов водорода. Для определения фторид-ионов в растворе при pH = 2,6 используют фторид-селективный электрод. При таком значении pH, а следовательно, и достаточно высокой активности иона водорода (1() 2'6) образуются HF и HF2 -ионы, которые нельзя определить с
Ю-3-10"4 М AgNO3
10 3-10 5 М AgNO3
10 3-Ю 6MAgNO3
200
। . ।_<_I_1__L
0 12 3 4
Время, мин
Рис. 6.12. Типичные электродные функции, наблюдаемые при ступенчатом изменении концентрации исследуемого раствора AgNO3 (полученные при использовании Ag-селективного электрода) [4]
Для сравнения времени отклика для различных сенсоров используется величина т ® Z63 или кратные величины Зт ® /95 и 5т « /99 (время, за которое потенциал сенсора достигает 63, 95 и 99% от величины равновесного значения потенциала соответственно).
718
Новый справочник химика и технолога
6.1.4. Таблицы потенциометрических сенсоров [5—7]1
Пояснения к таблицам. Все типы сенсоров полностью твердотельные, с рабочим диапазоном температур 0-80 °C.
* Крутизна электродной функции сенсоров XC-Fe и XC-Cr(VI) является нелинейной величиной и составляет 15-50 mV/pXв зависимости от концентрации определяемых ионов.
* * В скобках указаны допустимые кратные количества мешающих ионов по отношению к определяемому. Для сенсоров 1-8 допустимо присутствие в пробе 104-106-кратных количеств щелочных и щелочноземельных металлов: Zn2+, Ni2+, Cu2+, Mn2+, La3", Al3.
' Таблица 6.3
Жидкостные мембранные электроды [5, 6]
Основная функция электрода Материал электрода Определяемые ионы А Диапазон определяемых концентраций, моль-л”1 Коэффициенты селективности Кд/В Интервал pH Температурный интервал, °C Электрическое сопротивление, МОм
рСа Са-соль дидецилфосфорной кислоты в диок-тилфенилфосфа-те Zn2+, Са2+, Fe2+, Pb2+ 1-10”5 Zn2+ ~ 3,2; Са2+ « 1,0; Fe2+« 0,8; РЬ2+« 0,63; Си2+ « 0,27; Ni2+ « 0,080; Sr24 « 0,017; Mg2+« 0,014; Ва2+ « 0,010; Na+, К+ « 10”3 5,5-11 0-60 25-500
Са-соль дидецилфосфорной кислоты в диок-тилфенилфосфа-те в поливинилхлоридной матрице Zn2+, Са2+, Fe2+, Pb2+ 1-Ю”5 Zn2 ' « 1-5; Са2+« 1; А13+ « 0,90; Мп2+ « 0,38; Си2+« 0,070; Fe2+« 0,045; Со2+ « 0,042; Mg2+ ~ 0,032; Ва2+ « 0,020; Li+ « 10”4; Na+ « 10”5; К+ « 10”6 5,5-11 0-60 2-25
p(Ca+Mg) Са-соль дидецилфосфорной кислоты в деканоле Zn2+, Fe2+, Cu2+, Ni2+, Ca2+, Mg2+, Ba2+, Sr2+ 1-10”5 Zn2+, Fe2+«3,5; Cu2+«3,1; Ni2+« 1,35; Ca2+« 1,0; Mg2 , «1,0; Ba2+«0,94; Sr2+ ~ 0,54; Na+, K+ « 0,01 5,5-11 10-60 25-500
рС1 Крупный четвертичный амин фирма «Orion» (США) cio;,r, HS , NO3\ Br”, OH”, cr 1-10”5 C1O4« 32; I” « 17; HS” « 7,5; NO; « 4,2; Br« 1,6; OH” «1,0; ПРО2 =0,97; CH3COO” « 0,32; SO2” «0,14; F”«0,10 2-10 0-50 «25
фирма «Coming» (США) r, no;, Br”, cr, OH” 0,1-10”5 I” «15; CIO; «5; NO3”, Br”«2,5;Cl”« 1; OIL«0,4; CH3COO”«0,22 1-12 10-50 «100
рС1О4 Крупный четвертичный амин cio4 0,1-10”5 Г « 1,2 10’2; HCO3= 1,5-10”3; Br”«5,6- W4; CH3COO”«5,1 • IO-4; HCO; « 3,5 • 10”4; F« 2,5 • 104; Cl” «2,2- 10"4; SO2” «1,6- 10^ 4-11 0-50 «25
1 Некоторые данные в таблицах п. 6.1.4 обновлены (уточнены, исправлены, дополнены) авторами раздела.
Электрохимические методы анализа
719
Продолжение таблицы 6.3
Основная функция электрода Материал электрода Определяемые ионы А Диапазон определяемых концентраций, моль/л Коэффициенты селективности ^А/В Интервал pH Температурный интервал, °C Электрическое сопротивление, МОм
pNO3 Nr-Фенантро-линовый комплекс в о-нитро-цимоле сю;, г, сю;, no; 0,1-10-5 СЮ4103; Г - 20; С1О3 = 2; Вг" - 0,9; S2~ ~ 0,57; NO;~6- 10-2;CN~2- 10-2; НСО;~2- 10“2; С1 ~6 - 10-3; сНзСОО", со2-, s,o; , SO/-6- 10 3; Г' ~ 9 • 10 4; SO2-гб.Ю-4; Н2РО4, ро2 ~з - юЛ нро;~8- ю-5 2-12 0-50 -25
pBF4 Nr’-Фенантро-линовый комплекс в о-нитро-цимоле г, bf4 0,1-10-5 Г ~ 20; NO; - 0,1; ВГ ~ 4 • 10 2; CIPCOO , нсо;~ 4 • 10-3; F", сг, sor = I03 2-12 0-50 -25
рК Валиномицин фирма «Orion» Cs", Rb", К+ 1-Ю’5 Cs" - 1,0; NH4“ - 3 • 10 2; Na+, Н" - 1 • 10“2; Ag" - 1 • 103; Li"~ 10 4 2-11 15-50 >25
фирма «Coming» Cs", Rb", K+ 1-1 о45 Cs" « 20; Rb" - 10; NH4+ ~ 2 10 2; Na" ~ 10-2; Са2" = 5 • 10 3; Mg2+ « 3 • 10-3; Li"-4- 10"4 2-11 0-50 = 100
Валиномицин (0,009 М) в дифениловом эфире Rb", K+, Cs" 1-Ю-6 Rb ’ - 1,9; Cs"- 0,38; NH4+ - 10-2; Na" - 2,6 • Ю4; Li+ ~ 2,1 ЮЛ Са2+~ ЮЛ Н+ - 5,5 • 10-5 2-11 0-50 2-5
pNH4 Нонактин - мо-нактин в три-(2-этилгексил)-фосфате NH/ 0,1-10-5 К ~ 0,12; Rb’-4,3 • I0 3; Н"- 1,6 • 10~2; Cs’ -4,8 • 10"3; Li" - 4,2 • 10 3; Na" - 2 • 10-3; Са2" - 1,7 • 10-4 2-8 0-50 < 1
Таблица 6.4
Гомогенные электроды с твердыми мембранами [5,6]
Основная функция электрода Материал электрода Определяемые ионы А Диапазон определяемых концентраций, моль-л-1 Коэффициенты селективности Интервал рн Температурный интервал, °C Электрическое сопротивление, МОм
pAg Ag2S поликри-сталлический (прессованный) Ag", S2- 1-107; < 10-23 Си2+ » 1045; Pb2+ ~ 10-7; ме- шают ионы Hg2"; после длительного контакта с растворами, содержащими Hg2", необходима обработка поверхности 2-9 -5-100 <0,1
Ag2S монокристаллический Ag", S2- 1-Ю-7 Cu2+~ 10 s; РЬ2" ^ 10 л; Н" - НГ7; мешают ионы Hg2" — — 0,1
720
Новый справочник химика и технолога
Продолжение таблицы 6.4
Основная функция электрода Материал электрода Определяемые ионы A Диапазон определяемых концентраций, моль-л"1 Коэффициенты селективности Интервал рн Температурный интервал, °C Электрическое сопротивление, МОм
рСи CuS-Ag2S Agl-, S2. Cuz+, Hg2+ 1-10-8; < Ю’17 (буферный раствор) Fe3+ ~ 10; Cu+ ~ 1; ионы СГ, Вг“ мешают при высоких концентрациях; ионы Ag+, Hg2+ должны отсутствовать 0-14 0-100 <0,1
CuSe монокристаллический Cu+, Cu2+ 1-1О"6; < 10“'7 (буферный раствор) Pb2+ ~ Ю"4; Cd2+ = 10“5; ОТ ~ Ю11; Ag+~ Ю6; Hg2 ~ Ю4 0-14 -5-60 <0,01
pCd CdS-Ag2S Ag+, S2’, Cu2+, Cd2+ 0,1-Ю’7; < Ю’10 (буферный раствор) Fe2+, ТГ ~ 120; Pb2+~ 6; Мп2+ ~ 3; ионы Ag+, Hg2+, Cd2+ должны отсутствовать 1-14 0-100 < 0,001
рРЬ PbS-Ag2S Ag+, S2+, Cu2+, Pb2+ 0,1-10’7; < Ю’10 (буферный раствор) Fe3+*, Cd2+ ~ 1; ионы Ag+, Hg2+, Cu2+ должны отсутство- вать 2-14 0-100 <0,001
pS Ag2S поликри-сталлический (прессованный) Ag+, S2- 1-Ю"6; < 10"'2 (буферный раствор) После длительного контакта с растворами, содержащими ионы Hg2+, необходима обработка поверхности 13-14 -5-100 <0,1
Ag2S монокристаллический Ag+, S2’ 1-1 оЛ < Ю’12 (буферный раствор) Си2+~ Ю“5;РЬ2+~ ПГ6 13-14 -5-100 <0,1
pF LaF3 монокристаллический F’ 1-Ю"6 ОН’ ~ 0,1; СГ, Вг’, Г, NO3’, СО’, SO2’^10’3 4—8 -5-100 0,2-5
pCI AgCl-Ag2S (прессованная смесь) Ag+, СГ 1-10-5 Br~ 102;I’~ 104; ОН ’ ~ 10 “2; CN ’ = Ю’1; ионы S2’ должны отсутствовать 2-12 0-100 <0,01
AgCl монокристаллический Ag+, СГ 1-Ю’5 CN’8; Вг“~ Ю2;Г~ 104; NH3-10’1; ОН’~ Ю’2 0-14 -5-60 ~ 10
AgCl (фирмы Philips; Schott and Gen.; Tacus-sel) — — CN’~4- Ю’2;Г=Ю02; SO2 ~ 60; Вг“~ 1;ОН’~ Ю2; СО2- 10’3 1-10 0-100 < 1
pBr AgBr-Ag2S (пресссованная смесь) Ag+, Br 1-3 • ю 6 Г~5- 103;CN’~102; СГ~5 • Ю’3; ОН - Ю’5; ИОНЫ S должны отсутствовать 2-12 0-100 <0,01
монокристаллический AgBr Ag+, Br“ 1-Ю’6 Г~2; CN’~1;C1’~5- Ю’3; NH3~4- 10’3; О Н -~ Ю 4 0-14 -50-60 <25
(фирмы Philips; Schott and Gen.; Tacussel) Ag+, Br’ 1-1 О’6 CN’~25, Г~20; SO2” ~ 1,5; СГ~6- Ю’3; СО2~2- Ю’3 ОН- Ю’3 1-11 0-50 < 1
Электрохимические методы анализа
721
Продолжение таблицы 6.4
Основная функция электрода Материал электрода Определяемые ионы A Диапазон определяемых концентраций, моль-л 1 Коэффициенты селективности Интервал pH Температурный интервал, °C Электрическое сопротивление, МОм
Pl AgI-Ag2S (прессованная смесь) Ag+, S2 , Г 1-5 • IO 8 SO2 ~ 3 • Ю 2; CN' « 10 2; ВГ, SCN ~ 10 4; NH3~3 • 10"5; СГ-10Л ОН 1(Г7 2-12 0-80 <0,01
Agl (Philips; Schott and Gen.; Tacussel) Ag+, Г 1-5 • IO 8 CN ~0,34; СгО2~4- 1СГ3; S2O32' ~ 7 • КУ4; ВГ ~ 6 • 1(Г5; СГ ~ 6 1 (Г7; ионы S2- должны отсутствовать 2-12 0-50 <0,5
pSCN AgSCN-Ag2S прессованная смесь Ag+, SCN" 1-5 • КГ6 Г~ 103; Br ,CN-~ 102; NH3~10; СГ~ 101; ОН" ~ 10 2; ионы S2- должны отсутствовать 2-11 0-95 <0,01
pCN AgI-Ag2S прессованная смесь Ag+, Г, CN’ ю2-ю6 Г~ Ю2; Вг—10-4; СГ-КГ6; ОН- ~ ПГ8; ионы S2- должны отсутствовать 11-13 0-80 <0,01
Agl (Philips; Schott and Gen., Tacussel) Ag+, Г, CN’ Ю2106 Г«3; СгО2 ~ ПГ2; SO2- ~ 10 3; СО2 ; Вг ~ 10"*; СГ~ 1(Г5 10-12 0-50 0,5
Таблица 6.5
Гетерогенные электроды с твердыми мембранами [6] на основе полимерных матриц*
Основная функция электрода Материал электрода Определяемые ионы A Диапазон определяемых концентраций, моль-л-' Коэффициенты селективности Кл/в Интервал pH Температурный интервал, °C
pAg Ag2S Ag+, S2- 1-Ю7 1-Ю-17 в буферных растворах Мешают ионы Hg2+ 2-8 0-80
pCd CdS Ag+, Hg2+, Cd2+ 10--Ю-6 Pb2+ =; 1; Zn2+, Со2+, Ni2+ ~ Ю-3 1-8 0-50
pCu CuS Ag+, Hg2+, Cu2+ 1-Ю6 Pb2+, Cd2+, Со2+, Ni2+, Zn2+ ~ 10 3 1-14 0-50
pHg Agl Ag+, Hg2+ Ю^-1 о-6 СГ, ВГ ~ 102 1-6 0-50
pPb PbS Ag+, Hg2+,Pb2+ 10-1О6 Fe2 ' - Ю2; Cd2-1 2-7 0-50
pS Ag2S Ag+, S2- 1-Ю7 Мешают ионы Hg2+ 13-14 0-50
pCl AgCl Ag+, S2, Г, Вг-СГ 1-5-105 CN" ~ 400; Г ~ 86,5; SO2’ ~ 60; Вг’ ~ 1,2; 01Г - 2 • 10 2; SO2~6 - 103 2-12 0-50
pBr AgBr Ag+, S2~,r,Br~ 1-Ю6 CN" » 25; Г ~ 20; SO2" ~ 1,5; NO ~l;Cr~6- Ю 3 2-12 0-50
* Электрическое сопротивление данного типа мембран « 1 МОм
722
Новый справочник химика и технолога
Продолжение таблицы 6.5
Основная функция электрода Материал электрода Определяемые ионы А Диапазон определяемых концентраций, моль-л’1 Коэффициенты селективности Ка/в Интервал рн Температурный интервал, °C
Pl Agl Ag , S2’, Г, ВС 1-5-108 SO^~1;CN~~0,34 3-12 0-50
pCN Agl Ag+, S2-, Г, СЬГ ю-2—ю-6 Г, SO: ~3 11-13 0-50
pSCN AgSCN Ag+, S2", Г , Вг 1-105 Г~70;ВГ^1 2-12 0-50
Таблица 6.6
Пленочные мембранные электроды с поливинилхлоридной матрицей [7]
Основная функция электрода Компоненты электродной мембраны Диапазон определяемых концентраций моль-л 1 Интервал pH Угловой коэффициент dE/д lga к(а) Тип мембраны Литература
рСа Ди(октилфенил)фосфат кальция + диоктилфенилфосфонат ПГМО’1 ~ (4—10) 30 Селектрод [8]
рСа Различные ди- и моноэфиры фосфорной кислоты + ди(2-этилгек-сил)фосфонат ~ (10 5 10 2) ~(4-9) 30 Проволока с покрытием [9]
Теноилтрифторацетон + трибутилфосфат (тригексилфосфат) 2-10 4101 (2-10^-10°) 3,5-10 28-20 Пленка [Ю]
рВа Мембрано-активный комплексон в о-нитродифениловом эфире 105-10' 2-9 28 То же [Н]
pNa Na-лиганд + дибензиловый эфир io4-nr' 2-9 58 Проволока с покрытием [12]
Na-лиганд + дибутил фтал ат 10^-10° 4-9 57 Пленка [13]
Na-лиганд + дибутил фтал ат + + NaSCN 103 3 4-9 56-57 То же [14]
рК Валиномицин + бис-(2-этил-гексил)адипинат 510’5-10ч 4-9 58,5 То же [15]
Валиномицин + дибутилфталат 5 104-5 10* 0-9 57-58 То же [16]
Валиномицин + дипентилфталат 104-1(Г 2-10 57 Проволока с покрытием [17]
Тетра-и-хлорфенилборат + 3-нит-рооксилол 104 101 3-12 57 То же [15]
pUO2 Уранильный комплекс ди(2-этил-гексил)фосфорной кислоты + ди-амилфосфонат или три(2-этил-бутил)фосфат 10410’ 2-4 25-26 Пленка [18]
Алкилсульфоксид (диоктилсульфоксид) lO^-lO"1 2-4 28-29 То же [19]
pNO3 Нитрат тридодецилгексадецил-аммония 10 4-1 о1 2,5-8 57 То же [20]
Нитрат тетрадециламмония + дибутилфтал ат ю^-г 0-10 58 То же [21]
рСЮ4 Перхлорат тетрадециламмония + и-нитроцимол 105-104 0-10 59 То же [22]
Электрохимические методы анализа
723
Продолжение таблицы 6.6
Основная функция электрода Компоненты электродной мембраны Диапазон определяемых концентраций моль-л1 Интервал pH Угловой коэффициент dE/д lga к(а) Тип мембраны Литература
рС1О4 Перхлорат фенантролинового комплекса Fe(II) (1:10) + н-нитроцимол зю^-ю1 — 60 Проволока [23]
pSCN Роданид тетралауриламмония + дибутилфталат 10510° 2-10 57 Пленка [Ю]
Роданид тетрадецилдиметилбен-зиламмония + диоктилфталат 10“3,5-10-1 2-10 59 Тоже [24]
pSal Салицилат тетрадециламмония + н-нитроцимол Ю^-5 101 5-10 58 То же [25]
Салицилат метилтрикаприлам-мония + диоктилфталат 10 310' 5-10 59 То же [24]
pBenz Бензоат тетрадециламмония + дибутил фталат lO^-S-lO"1 5-10 58 То же [25]
Бензоат метилтрикаприламмония + изодеканол 10"3-10-1 5-10 50-59 То же [26]
Таблица 6.7
Характеристики халькогенидных стеклянных (ХС) химических сенсоров для определения ионов тяжелых металлов [8]
Тип сенсора Диапазон определяемых концентраций, моль-л 1 Крутизна mV/pX Рабочий диапазон pH Сопротивление мембраны, кОм Селективность, мешающие ионы
1. XC-Ag-01 1 • 10’7-1,0 58-59 -0,5-9 <50 Hg2+(100) **
2. XC-Cu-01 1 • 10’7-1,0 28-29 0-8 <330 Fe3+H<2(0,2-0,3); Fe^>3 (102 -104); Ag+, Hg2+ — отсутств.
3. XC-Pb-01 3 • 107-1,0 28-29 1-8 <10 Cd2+(100); Fej~.1,Cu2l(O,1-0,01); Ag+, Hg2- — отсутств.
4. XC-Cd-01 3 • 1071,0 26-28 1-8 < 100 Cu2+, (0,01); Pb2+(0,1); Ag+, Hg2+ — отсутств.
5. XC-Fe-01 5 • Ю^-Ю"2 * 0-2 <100 Cu2+, Cr(VI)(5); Ag+, Hg2+ — отсутств.
6. XC-Hg-01 1 • 10 61,0 40-45 0-2 <20 Ag+(1)
7. XC-T1-01 1 • 10 б-1,0 56-58 1-11 <500 Cu2+, Fe3H<2 (1 - 10); Ag+, Hg2+ — отсутств.
8. XC-Cr(VI)-011 1 • 10’7-103 * 0-2 <350 Cu2+, Fe3+(100); Ag+, Hg2+ — отсутств.
9. XC-S-01 1 • 10’7-1,0 28-29 11-14 < 10 no;, cio;, so2-(io6)
10. xc-nh4-oi (поликристалл) 1 • 10 6-1,0 58-59 2-9 < 100 Li+(20); Na+(10); K+(3); Ca2+, Mg2+(5)
Все типы сенсоров полностью твердотельные, с рабочим диапазоном температур 0-80 °C.
Крутизна электродной функции сенсоров XC-Fe и XC-Cr(IV) является нелинейной величиной и составляет
15-50 mV/pXв зависимости от концентрации определяемых ионов.
В скобках указаны допустимые кратные количества мешающих ионов по отношению к определяемому. Для сенсоров 1-8 допустимо присутствие в пробе 104-10б-кратных количеств щелочных и щелочноземельных металлов: Zn2+, Ni2+, Cu2+, Mn2+, La3+, Al3+.
724
Новый справочник химика и технолога
6.1.5. Методы аналитического определения
6.1.5.1. Прямая потенциометрия
Как метод анализа потенциометрия обладает целым рядом достоинств, связанных, несмотря на разнообразные конкретные условия аналитических определений, с весьма простым приборным оформлением. С одной стороны, этот метод позволяет определять содержащиеся в растворах вещества в широких пределах изменения их концентрации с использованием одного и того же оборудования при незначительном изменении методики измерения. С другой стороны, потенциометрические измерения могут проводиться как в стационарных, так и в полевых условиях, причем они удобны для непрерывного и дистанционного контроля за концентрацией определяемого вещества с помощью автоматического оборудования, например, на промышленных установках и в случае мониторинга окружающей среды.
К потенциометрическим методам анализа относятся следующие методы: метод прямой потенциометрии (ПП), методы стандартной добавки (МСД) и стандартного удаления (МСУ) и их модификации, метод многократных добавок (ММД) и его модификации и потенциометрическое титрование (ПТ) с использованием в качестве индикаторов ионоселективных электродов. Описание этих методов дано в различных трудах [4, 5, 27-30] и научных статьях по развитию и усовершенствованию методов потенциометрии. Метод прямой потенциометрии хорошо известен и широко применяется в аналитической практике.
Прямая потенциометрия является наиболее простым методом определения, но обладает рядом недостатков. В частности, она позволяет определять только свободные ионы, требует постоянного контроля за величиной стандартного потенциала и крутизны электродной функции, кроме того, желательно, чтобы стандартные растворы имели фон, близкий по составу фону исследуемой пробы; фон же часто либо неизвестен, либо меняется от пробы к пробе, а добавление какого-либо буфера может привести к большим ошибкам в определении особенно низких концентраций из-за присутствия в реактивах следов определяемого вещества или мешающих ионов.
Достоинством метода стандартной добавки (МСД) является то, что все измерения проводятся в присутствии всех компонентов пробы, кроме того, он позволяет при определенных условиях находить суммарную концентрацию вещества, что и требуется практикам-аналитикам.
Идеальные условия для применения этого метода возникают тогда, когда в процессе измерения: а) коэффициенты активности не меняются, б) количество свободных, незакомплексованных ионов остается постоянным, в) крутизна электродной функции не меняется, г) величина диффузионного потенциала постоянна. И хотя условия трудно выполнимы в принципе, однако на практике можно добиться, чтобы эти изменения были незначительны.
Теория метода
Когда к известному объему пробы Vx неизвестной концентрации сх иона А добавляют известный объем Va стан
дартного раствора с известной концентрацией са, тогда потенциал ионоселективного электрода, чувствительного к иону А, в исходном растворе определяется уравнением:
Е = Ео + 51g/AaAcx +Ej-Eref , (6.14)
где Eq — стандартный потенциал электрода; S — крутизна электродной функции; fA — коэффициент активности иона; аА = [А]/сх — отношение концентрации свободных ионов [А] к общей концентрации; Ej — диффузионный потенциал; Eref— потенциал электрода сравнения.
Из уравнения (6.14) следует:
Е, = Е, + Slg/A + XlgaA + Slgc, + Е, - Er<f . (6.15)
После добавки стандартного раствора имеем:
Ег = E0 + S4g/; + S4ga; + S4gc;+E'-E^. (6.16)
Для успешного применения метода необходимы следующие приближения:
S « S; Ej « Е' ;/А« Д; a * a'. (6.17)
Предположим, что эти приближения верны, тогда из уравнений (6.15) и (6.16) получим:
Е2-Е} = Slgcx±S}gcx, (6.18)
а так как
, (^Xcx + Vca} i _ х х а а
vx + va J’
(6.19)
Ег+Д =Slg
сх(Ех + Еа)_
(6.20)
откуда получаем, что:
(6-21)
Если выполняется условие Va Vx, то уравнение (6.21) упрощается:
(6-22)
Влияние различных факторов в МСД на точность определения концентрации подробно обсуждается в работе [31].
Объемные ошибки. Ошибка в определении сх зависит V с V
от отношения — и от отношения Ф = -2-а-. Минималь-ные ошибки наблюдаются при Ф = 1-2, причем для того,
Электрохимические методы анализа
725
чтобы выполнялось уравнение (6.22), необходимо выпол-V
нение соотношения — < 0,01.
Vx
Угловой коэффициент. На практике величина 5 определяется эмпирически. Можно рекомендовать три способа ее определения: калибровка известным разбавлением; калибровка по стандартным растворам, определение крутизны в анализируемом растворе с известным содержанием определяемого вещества.
Рассмотрим указанные выше способы определения S.
1. После добавки и измерения Е2 добавляют известный объем VD разбавителя и измеряют Е2.
E3=Eq+ S'lgf; + ГIga; + S’lgc: + Е”-Eref (6.23)
Делая те же предположения (6.17), получим:
^-.E^Slgc.-Slgc,'.
(6.24)
Так как
,= +
* vx + va + vD>
(6.25)
то
(у +у +у А
Ч (6.26)
откуда можно рассчитать S.
Разбавитель должен иметь состав, близкий к составу пробы и добавки. Разница Е2 - Е2 должна быть ~ 20 мВ для однозарядных ионов и ~ 10 мВ для двухзарядных. Разбавитель должен иметь ту же температуру, причем разница в температуре в 1 °C дает ошибку в концентрации 0,5%.
2. Калибровку по стандартным растворам следует проводить в растворах с добавлением всех реагентов, присутствующих в анализируемой пробе.
3. Угловой коэффициент можно определить, находя ДЕ в стандартном растворе с известным содержанием определяемого вещества, затем вычисляя 5 по уравнению (6.21).
Ионная сила анализируемого раствора и раствора добавки должны быть близки друг к другу. Для постоянства показателей коэффициентов активности желательно, чтобы ионная сила раствора создавалась, в основном, за счет посторонних ионов, а не определяемого иона.
Влияние комплексообразователей. Успешное применение МСД зависит от обоснованности допущения о постоянстве а (моль доля свободных катионов). В растворах, не содержащих комплексообразующих агентов, a = 1 до и после добавки. В общем случае, однако, a = l/fl + 2₽,[i]], где [L] — равновесная концентрация конкурирующего лиганда L, а р, — общая константа устойчивости комплекса MLt (М — определяемый ион);
[ML]
Р; = —---. Следовательно, чтобы а оставалась практи-
№1
чески постоянной, значения L не должны сильно изменять
ся после добавки. Для выполнения последнего условия введение добавки не должно приводить к заметному разбавлению пробы; кроме того, количество лиганда, необходимое для связывания в комплекс вводимого с добавкой определяемого вещества, должно быть незначительным, т.е. bfi < 10-3. Если константа устойчивости комплекса имеет большую величину, например (5, > 103, то последнее условие означает: в анализируемом растворе концентрация лиганда в 1000 раз превышает общую концентрацию определяемого вещества. Если же количество лиганда в пробе недостаточно для того, чтобы после введения добавки величина а оставалась постоянной, то перед измерением Е) в пробу следует добавить некоторое количество этого лиганда.
С целью уменьшить влияние диффузионного потенциала при работе с ионоселективными электродами, следует использовать электрод сравнения, заполненный насыщенным или концентрированным (3 моль-л1) раствором КО. При измерениях в области малых концентраций с постоянной ионной силой (например, 1 = 0,1 моль л-1 KNO3) часто используют электролитический ключ, заполненный таким же раствором. В ряде случаев можно в качестве электрода сравнения использовать твердофазные ионоселективные электроды (ИСЭ) с обязательным введением в раствор потенциалопределяющих ионов для электрода сравнения. Например, при определении фторидов с помощью F-ИСЭ в качестве электрода сравнения был взят I-ИСЭ с добавлением в пробу определенного количества иодида калия [32].
Для уменьшения ошибки, вызванной растворимостью мембраны, необходимо, чтобы измеряемая концентрация сх была существенно больше, чем концентрация потенциалоопределяющего иона, которая образуется вблизи поверхности мембраны из-за ее растворимости, т.е.
должно выполняться условие > 10, где ПР — произве-
дение растворимости.
Как правило, случайные ошибки вызваны неточностями в измерении величины потенциала, углового коэффициента, объемов Vx и Va и концентрации cs. Обычно ошибки в определении объемов и концентрации малы. Точность определения величины потенциала должна быть < 0,1 мВ.
МСД является наиболее популярным методом определения концентрации веществ в различных средах. Примеры его применения представлены в табл. 6.8.
Таблица 6.8
Примеры применения метода стандартных добавок
Электрод Определяемое вещество Объект исследования Литература
С1-ИСЭ Хлориды Пищевые продукты [33-35]
Фармацевтические препараты [36, 37]
Горные породы [38—40]
Промышленные растворы [41-43]
726
Новый справочник химика и технолога
Продолжение табл. 6.8
Электрод Определяемое вещество Объект исследования Лите- ратура
С1-ИСЭ Хлориды Гальванические ванны [44]
Вг-ИСЭ Бромиды Пищевые продукты [45]
1-ИСЭ Йодиды Воздух [46]
Пищевые продукты [47,48]
Горные породы и почвы [49]
Селен [50]
Биологические растворы [51]
CN-ИСЭ Цианиды Гальванические ванны [52, 53]
s-исэ Сульфиды Цемент [54]
Зубная паста [55]
Сероводород Воздух [56]
F-ИСЭ Фториды Зубная паста [57]
Пищевые продукты [35, 58, 59]
Горные породы [39,40, 60-62]
Растения [63, 64]
Лекарства [65]
Уголь [66]
Селен [50]
Цинковые концентраты и растворы [67]
Гальванические и травильные растворы [68, 69]
Стеклокристаллиты [70]
Природные и промышленные воды [32,42, 71,72]
Гидроксид натрия [73]
Соединения урана [74]
Серная кислота [75]
nh4-ИСЭ Аммиак Пищевые продукты [76-78]
Азот Уголь [79]
NO3-исэ Нитраты Почва [80]
Вода [81]
Продолжение табл. 6.8
Электрод Определяемое вещество Объект исследования Лите- ратура
NO3-исэ Нитраты Травильные растворы [82]
Фосфорная кислота [83]
Пищевые продукты [84, 85]
Нитраты и нитриты Пищевые продукты [86, 87]
bf4-исэ Бор Промышленные растворы [88]
к-исэ Калий Лекарства [89]
Объекты стекольной промышленности [90]
Na-ИСЭ Натрий Пищевые продукты [91]
Полимеры [92]
Поваренная соль Маргарин [93]
РЬ-ИСЭ Свинец Яйца [94]
Бронзовые сплавы [95]
Cd-ИСЭ Кадмий Сточные воды [96]
Си-ИСЭ Медь Пищевые продукты [97, 98]
Ag-ИСЭ Серебро Руды [99]
Гальванические ванны [52]
Метод стандартного удаления (МСУ) отличается от МСД тем, что реагент, добавляемый в пробу, не повышает, а понижает концентрацию свободных определяемых ионов. К известному объему пробы Vx определяемого вещества А с концентрацией сх добавляют известный объем Va реагента Д с концентрацией сА. Ионоселективный электрод чувствителен к иону А. Из МСД следует, что потенциал ИСЭ в исходном растворе дается уравнением (6.14). Если х молей определяемого вещества реагирует с у молей вещества Д, то новая концентрация определяемого вещества будет равна
, Vc xVac
х y.+Va yV+V'
х a J х а
(6-27)
Потенциал ИСЭ после добавки реагента дается уравнением (6.16); при этом предполагая, что условия (6.17) выполняются, из (6.15) и (6.16) получим (6.18). Подставив (6.17) в (6.18), имеем уравнение:
£2±£,=51g
vxcx-~vact У
сх(Ух + Уа)
(6.28)
Электрохимические методы анализа
727
которое можно преобразовать относительно сх:
Не вдаваясь в подробности математических преобразований, предлагаем соответствующее уравнение для МДА:
сх
Х jr ---VaCa
______У________
—fv
К 10s -2- + 1 -1 к
(6.29)
— f v
ю5 -2-+1 -1
U J
(6.31)
для МУА:
Если Va Vx> уравнение (6.29) можно записать:
— Vaca
С
х ( ЬЕ
К 10 s -1
(6.30)
Растворами реагента могут быть вещества, осаждающие определяемое вещество, переводящие его в комплексы, или редокс-реагенты. В работе [100] изучено влияние различных факторов на точность определения этим методом.
Для реагентов, осаждающих определяемое вещество, с2
главным фактором (Ф) является отношение где про-
изведение растворимости ПР = СхСМ. Минимальная ошибка с2
достигается при > 4. В случае комплексообразовате-лей величина сх должна быть больше 20. Соотношение Ф должно быть равно 0,5-1. Другие факторы, влияющие на точность, такие же, как для МСД.
МСУ гораздо менее распространен в практике ионометрических определений, в качестве примеров его использования можно привести следующие: определение сульфидов в промышленных растворах бумажной промышленности [101], сульфатов в строительных материалах [102], серебра в фотографических материалах [103].
Известны различные модификации МСД и МСУ. Метод добавок анализируемого раствора к стандартному раствору (МДА) основан на прибавлении точно измеренного объема пробы к стандартному раствору определяемого иона, а в методе уменьшения концентрации анализируемого раствора при его добавлении к стандартному раствору (МУА) пробу прибавляют к раствору, содержащему ион, который стехиометрически взаимодействует с определяемым ионом и специфически определяется данным ИСЭ. Эти методы имеют преимущества над другими методами добавок при анализе проб малого объема, которые в противном случае приходится анализировать с использованием микроэлектродов. При применении МДА и МУА нет необходимости регулировать pH и маскировать мешающие вещества в пробе, так как все эти операции можно заблаговременно провести в стандартном растворе. Более того, МУА расширяет возможности ионометрии, поскольку этим методом можно определять ионы (для которых нет ИСЭ) и даже неионогенные соединения различных типов, если они вступают в стехиометрическую реакцию с ионом, находящимся в стандартном растворе.
АЕ
10s
(6.32)
где Vx — объем стандартного раствора; cs — концентрация стандартного раствора; Vs — объем анализируемой пробы; сх — концентрация пробы.
Для этих методов влияние различных факторов на точность определения такое же, как в случае МСД и МСУ [100, 104].
Примеры применения МДА и МУА представлены в табл. 6.9.
Таблица 6.9
Примеры применения МДА и МУА
Электрод Определяемое вещество Объект исследования Литература
С1-ИСЭ Хлориды Пищевые продукты [Ю5]
Вг-ИСЭ Гипохлорит Отбеливающие растворы [Ю6]
F-ИСЭ Фториды Криолит [Ю7]
Вода [Ю8]
Кровь [Ю9]
Алюминий Водопроводная вода [1Ю]
М14-ИСЭ Аммоний Дрожжевое производство [1И]
ЫОз-ИСЭ Нитраты Сточные воды [112]
Травильные растворы [ИЗ]
К-ИСЭ Калий Пищевые продукты [Ю5]
Na-ИСЭ Натрий Пищевые продукты [Ю5]
Технологические растворы в бумажной промышленности [П4]
РЬ-ИСЭ Сульфаты Вода и донные отложения [И5]
Свинец Г альванические ванны [П6]
Ag-ИСЭ Хлориды Природные воды [Н7]
Мышьяк Горные породы, минералы и почвы [И8]
728
Новый справочник химика и технолога
Методы многократных добавок (ММД) служат для дополнительного повышения точности измерений. Для исходного раствора справедливо уравнение (6.16), после и-й добавки потенциал электрода будет равен:
Е, = Е„ +S'lg/; + S'lg< + S'lgc' +E't-E„, (6.33)
Га+ТУл'] v.+ty, n J
Предполагая, что условия (6.17) верны, мы можем записать общее уравнение:
En=EF+Slg
где Ер включает все постоянные члены. Беря разность потенциалов, получим:
Таблица 6.10
Примеры применения метода двойной стандартной добавки
(634)
(6.35)
Электрод Определяемое вещество Объект исследования Литература
С1-ИСЭ Поваренная соль Производные крахмала [Н9]
Хлориды Природные воды ,[125]
CN-ИСЭ Цианиды Алкогольные напитки [126]
F-ИСЭ Фториды Вода [120, 125]
№н4-исэ Азот Уголь [127]
К-ИСЭ Калий Удобрения [128]
Природные воды [125]
Na-ИСЭ Натрий Природные воды [125]
£_-£, = Sig
(6.36)
Отсюда, строя зависимость величин 10/ s от объема, получают график Грана. При применении линеаризации по Грану в ММД уравнение (6.35) преобразуется в следующую форму:
Частным случаем ММД является метод двойной стандартной добавки (МДСД). При двух добавках отношение Еу-Е} к Er-Е] дает некое выражение R. Значения R приведены в таблице в работе [27]. Величина S в данном случае сокращается.
Созданы компьютерные программы для МДСД [119-123]. Влияние различных факторов на точность определения концентрации МДСД обсуждается в работе [101] и практически аналогично МСД.
Для получения результатов с минимальной ошибкой у
необходимо, чтобы =0,01, Г2 = 2Fj и погрешность в
определении ДЕ не превышала бы ±0,1 мВ. Примеры применения МДСД представлены в табл. 6.10.
МДСД не дает существенного выигрыша в точности определения, поэтому следует отдавать предпочтение ММД. Чаще всего неизвестную концентрацию определяемого вещества по ММД находят методом итераций на ЭВМ (см. [129-136]). В качестве примеров применения ММД можно привести определение фторидов в пищевых продуктах [137, 138]; калия в эритроцитах [139].
В потенциометрии нашел широкое применение метод Грана, при котором концентрацию определяемого иона выражают графически в виде функции объема добавляемого стандартного раствора [140]. Математически линеаризация зависимостей достигается путем перевода уравнения Нернста в антилогарифмическую форму:
10£/s= 10£°/s -c = fc. (6.37)
+ н!'о) 10£"|,s = 10 е' • (с,К, + исаКа), (6.38)
которая является уравнением прямой линии
у = ЬУа + а, (6.39)
где y = (Vx + пУа) 10£-1/5; b = 10£°/s пс; а = 10£°/5 • схУх. Следовательно, в точке пересечения прямой с осью абцисс (при у = 0) соблюдается условие:
ncaVa = - cxVx,
и отсюда
(6.40)
(6-41)
сх
-ПСУа К
где Vx — объем прибавляемого стандартного раствора, отвечающий концентрации определяемого вещества сх.
Разновидностью данного метода является графический метод, в котором строится зависимость 10v;s от добавляемого объема [141]. Он несколько упрощает построение графика, так как в исходном растворе величина АЕ равна 0, следовательно, первая точка на оси F будет равна объему пробы (рис. 6.13). В работах [142, 143] обсуждаются факторы, влияющие на точность определения концентрации данным методом. Показано, что число добавок должно быть около 10, а погрешность определения S не должна превышать 2%. Предлагаются компьютерные программы для ММД по Грану [144, 145]. Применение этого метода представлено в табл. 6.11.
Электрохимические методы анализа
729
Рис 6.13. Графический вариант метода многократных добавок по Грану: 1-F= 10&; 2-F =
Необходимость точно знать величину углового коэффициента (что не всегда возможно) ограничивает возможности применения ММД по Грану. Недавно Ли [162] предложил способ построения графика Грана при неизвестной крутизне электродной функции. Опуская математические выкладки, приводим конечную формулу:
ДЕ
(Гх+^)105' -Vx
+ L____________
HL H
(6.42)
где St — теоретическое значение углового коэффициента;
Таблица 6.11
Примеры применения метода многократных добавок по Грану
ДЕ/ С С
Я = АЕ(Их + И$.)10 А; l =
Электрод Определяемое вещество Объект исследования Литература
1-ИСЭ Йодиды Молоко [146]
F-ИСЭ Фториды Воздух [147]
Морская вода [148]
Силикатные породы [149]
Пробы смешанного состава [150]
NH4-HC3 Аммиак Гальванические ванны [145]
NO3-HC3 Нитраты Газовая сажа [151]
К-ИСЭ Калий Морская вода [152]
Пищевые продукты [153, 154]
Na-ИСЭ Натрий Пищевые продукты [153, 155]
Поваренная соль Рыба [156]
Са-ИСЭ Кальций Молоко [154]
РЬ-ИСЭ Свинец Нефть [157]
Си-ИСЭ Медь Гальванические растворы [158]
Водопроводная вода [159]
Ag-ИСЭ Сера Хром и феррохром [160]
Цианиды Сточные воды [161]
с V.
График функции У = относительно
ДЕ
(Гл^Ю5 -vx
будет линеен с наклоном, равным
V 1g
сх, при этом ось У пересечется в точке , откуда может быть найдена реальная величина S.
6.1.5.2. Потенциометрическое титрование
Расчет результатов анализа в методах потенциометрического титрования сводится к нахождению значения эквивалентного объема Кэкв Широко распространены графические методы нахождения Иэкв. по графикам зависимости некоторой функции потенциала электрода от объема титранта. Эта зависимость может быть представлена в интегральном виде E=flV) и дифференциальном виде: dE d2E
=ЛУ) или = ЛЛ). Точку эквивалентности (ТЭ) находят экстраполяцией на ось абцисс точки максимального изменения потенциала, точки экстремума его первой производной по объему или точки, в которой меняется знак второй производной потенциала по объему (см. рис. 6.14).
Эти методы достаточно просты и оперативны, с их помощью можно получить точное значение Кэкв. на симметричных кривых титрования, когда определяемый ион и титрант взаимодействуют в соотношении 1:1. В графических методах надежность фиксирования конечной точки титрования тем выше, чем больше разница в изменении потенциала в ТЭ и вблизи от нее.
Титриметрия — совокупность методов количеств анализа, основанных на измерении кол-ва реагента, необходимого для взаимодействия с определяемым компонентом в растворе или газовой фазе, а процесс постепенного добавления этого реагента (титранта) к анализируемому раствору — титрование.
730
Новый справочник химика и технолога
Рис. 6.14. Интегральные и дифференциальные кривые потенциометрического титрования: l-E=f(V); - 5-^»’
Однако достаточно часто кривая титрования асимметрична относительно ТЭ, т.е. наблюдается несовпадение ТЭ и конечной точки титрования. Это происходит в тех случаях, когда
1) однозарядный ион титруется «-зарядным или, в общем случае, стехиометрия реакции отлична от соотношения 1:1;
2) ионоселективный электрод обратим лишь к одному из титруемых ионов или крутизна его электродных функций по отношению к титруемому иону и иону-титранту различна;
3) константа комплексообразования для ионов, образующих титрационную систему, имеет небольшое значение.
Потенциометрическое титрование успешно применяется для определения органических веществ (табл. 6.12).
Таблица 6.12
Определение органических веществ, которые образуют комплексы с ионами металлов
Органическое вещество Химический сенсор Литература*
Титрование с солями Си+2
Аминофенолы, алифатические диамины и полиамины Си2+ 102-104
Аминокислоты (глютаминовая, аспаргиновая, глициновая) Си2+ 105
Аминокарбоновые кислоты (NTA) Си2+ 106
Фенилгидразин Си2+ 107
8-Оксихинолин и его производные Си2+ 108-111
Лимонная кислота Си2+ 106,111,112
Тиосемикарбазоны Си2+ 113
Сульфонамиды Си2+ 114
Тиоацетамиды Си2+ 115
6-Метил-2 -тиоурацил Си2+ 116
Аминоиндолы Си2+ 252
Сульфамиды и метилксантины Си2+ 265
* Литературу к табл. 6.12 см. на компакт-диске.
Продолжение табл. 6.12
Органическое вещество Химический сенсор Литература
Пестициды Си2+ 278
Титрование с солями Ag+
Тиомочевина, дитиомочевина и их производные Ag+-S2- 117-120
Тиобарбитураты Ag+-S2’ . 121
Триазины с -SH группой Ag+-S2’ 122
Цистеин и тиолы Ag+-S2- 123
Сульфонамиды и сульфонимиды Ag+-S2- 114, 124
Амины Ag+-S2“ 125
Имиды и замещенные урацилы Ag+-S2“ 259
Монозамещенные производные ацетилена Ag+-S2~ 260
Тиамин Ag+-S2- 262
Сульфонамиды и метилксантины Ag+ 265
Пестициды Ag+-S2’, Br 278
Титрование с солями РЬ2+
Щавелевая кислота Pb2+ 126, 127
NTA, диэтилдитиокарбамиды Pb2+ 127
Диэтилентриаминпента-уксусная кислота Pb2+ 128
Катехинолы Pb2+ 129
Пеницилламин Pb2+ 130
Пестициды Pb2+ 278
Титрование с солями Cd2+
8-Оксихинолин и его производные; пиридилазо красители (PAN, PAR), аминокарбоновые кислоты (NTA, EDTA, DCTA), диэтилдитиокарбамиды и др. Cd2+ 131
Пестициды Cd2+ 278
Титрование с солями 11g2
Аминокислоты Hg2* 132
Аминокислоты и другие аминопроизводные Ag+-S2- 133,134
Тиолы Ag+-S2- 135-137
Титрование с солями Са2 ’
Щавелевая кислота Ca2+ 138, 139
Аминокарбоновые кислоты (NTA, EDTA, DCTA, HEDTA, EGTA) Ca2+ 139
Мыла (пальмитаты, стеараты, олеаты) Ca2+ 139
Электрохимические методы анализа
731
В 1950 г. Гран [163] предложил использовать в качестве
ЛК
кривой титрования зависимости от К В этих координатах на графике получаются две линейные зависимости, точка пересечения которых лежит на оси абцисс и соответствует ТЭ (рис. 6.15).
Точками, расположенными около ТЭ, можно теперь пренебречь и, таким образом, добиться повышения правильности и точности определения, особенно при построении соответствующих прямых с использованием метода линейной регрессии.
В потенциометрии с использованием ИСЭ особенно широкое применение нашел, однако, другой метод Грана [140]. В этом методе концентрацию определяемого иона выражают графически в виде функции объема добавляемого титранта, что позволяет получать зависимости линейного вида. Для каждого титрования имеются две функции: одна из них справедлива до конечной точки, а другая — после нее. Обзор этих функций дан ниже: в каждом случае функцию F наносят на график в зависимости от объема добавляемого титранта V, а эквивалентный объем находят как точку пересечения графиком оси объемов.
Рис. 6.15. Типичный пример использования метода Грана — титрование хлорид-ионов (25 мкг/мл) 210 3 М раствором AgNO3: а — обычная кривая титрования; б — график Грана;
— точка эквивалентности
Во всех дальнейших уравнениях приняты следующие обозначения:
Vx — объем титруемого раствора;
Е — наблюдаемый потенциал ИСЭ;
5 — крутизна электродной функции.
А. Титрование сильная кислота — сильное основание
1. Перед ТЭ
(Vx + И)Ю£/5 ИЛИ F = (Vx + ГЖрН
2. После ТЭ
F=(VX + V)\0~E/s или F= (Vx + ЮЮрН-
Б. Титрование слабая одноосновная кислота — сильное основание
1. Перед ТЭ
F = ИО*'5 или F- V10 рН.
2. После ТЭ
F=(VX+ Г)ПГ£/5илиГ’=(Кс+ ПЮрН.
В. Титрование одноосновное слабое основание — сильная кислота
1. Перед ТЭ
F=(VX + Р)10£/5или^=(Кх+Г)-10"рН.
2. После ТЭ
F= Г 10/;5илиГ= К10рН.
Г. Осадительные титрования хА +уВ = АХВЛ.
а) Электрод реагирует на ионы А .(определяемое вещество):
1. Перед ТЭ
F=(VX + V)'l^ls.
2. После ТЭ
F=(KX+ Г) 10-^.
б) Электрод реагирует на ионы В (титрант):
1. Перед ТЭ
F=(KX+ Г) 10^.
2. После ТЭ
F=(VX + Vytffs.
Д. Комплексонометрические титрования хА +у В = АХВ,.
а) Электрод реагирует на ионы А (определяемое вещество):
1. Перед ТЭ
F=(VX + V)WEIS.
2. После ТЭ
F= (Vx + р)1-^) П)-^5.
б) Электрод реагирует на ионы В (титрант):
1. Перед ТЭ
р= (кх+ V1^ 10~yE/xS.
2. После ТЭ
F=(VX+ VylO®3.
Е. Окислительно-восстановительные титрования
а) Окисление определяемого вещества
«АВаг HqK0X + /7дВте6/.
1. Перед ТЭ
f=i0-«a^
2. После ТЭ
F = 10"A^.
б) Восстановление определяемого вещества
«вА0Х + ^дВгее/ — ИвАгее/ + /7дВ0Х.
1. Перед ТЭ
F = K-10”a£/\
2. После ТЭ
F = 1(T”a/?/\
Уравнения графиков Грана часто записывают в форме, отличающейся от приведенных выше. Это обусловлено введением в исходные уравнения произвольных постоянных для облегчения расчетов. Так, Е может быть заменено на (Е + Ес), где Ес — произвольная постоянная. Хотя для получения каждой точки на графике Грана требуется больше расчетов, чем при построении кривой титрования или дифференциальной кривой титрования, тем не менее можно обойтись лишь несколькими точками. Результаты
732
Новый справочник химика и технолога
анализа, полученные с помощью графиков Грана, не включают в себя ошибок, связанных с тем, что точку, где первая производная максимальна, принимают за ТЭ, так как такие графики полностью скорректированы на эффекты разбавления; более того, графики Грана можно использовать для осадительных титрований, даже если осадок образован ионами с разными по величине зарядами.
Вывод уравнения Грана не является очень строгим, так что в некоторых случаях графики могут быть искривлены, т.к. они не учитывают:
• изменения коэффициентов активности.
• влияния побочных процессов при установлении равновесия в ходе титрования, что проявляется в виде искривления графика в очень разбавленных растворах или когда реакция между определяемым веществом и титрантом протекает сравнительно вяло.
Улучшению графиков Грана посвящен ряд работ [164—173]. На основе метода Грана предложен автотитратор [174]. Применение данного способа нахождения ТЭ представлено в табл. 6.13.
Таблица 6.13
Примеры применения потенциометрического титрования с нахождением ТЭ по Грану
Электрод Определяемое вещество Объект исследования Литер атура
1-ИСЭ Хлориды, бромиды и иодиды Водоросли [175]
S-ИСЭ Полисульфиды Нефть [176]
F-ИСЭ Торий и редкоземельные элементы Монацит [177]
СО3-ИСЭ Карбонаты Щелочи [178]
Ва-ИСЭ Сульфаты Природные воды [179]
РЬ-ИСЭ Диоксид серы Дымовые газы [180]
Сера Сплавы [181]
Сульфаты Технологические растворы бумажной промышленности [182]
Cd-ИСЭ Сера Воды, химические реактивы, железо и сплавы [183]
Си-ИСЭ Цитраты Химические реактивы [184]
Медь Ванны химического меднения [185]
6.1.5.3. Другие методы («электронный язык»)
«Электронный язык» был успешно применен для качественного, количественного и одновременно качественного и количественного анализов жидких сред различной природы [31].
Качественный анализ пищевых продуктов позволяет распознавать различные напитки и устанавливать или сопоставлять их сорта и качество. Были проанализированы кофе (натуральный, растворимый), чай, пиво, некоторые сорта итальянских вин, минеральные воды (боржоми, итальянские воды), прохладительные напитки, фруктовые соки [186, 187]. Одним из важных результатов при проведении исследований было то, что удалось обнаружить возможность применения химических сенсоров с неорганическими мембранами (например, халькогенидных стеклянных сенсоров) для анализа напитков различной природы. Так, из рис. 6.16 видно, что девять различных халькогенидных стеклянных сенсоров дают различные отклики в зависимости от сорта пива.
□ Свеиое Ц Темное
Рис. 6.16. Типичный отклик массива халькогенидных стеклянных сенсоров в различных сортах пива.
Все сенсоры имеют различные составы и различную перекрестную чувствительность
Количественный анализ был проведен для установления концентрации тяжелых металлов (Cu, Pb, Zn, Cd, Сг и др.) в загрязненных природных водах (р. Нева, Санкт-Петербург), для обнаружения урана в водах заброшенных урановых шахт (Германия, Россия), содержания солей в грунтовых водах (земля Брауншвейг, Германия), тяжелых металлов в дыме мусоросжигательных заводов (проточный анализ, Дания), для определения Са2+, Mg2+, фосфатов и др. ионов в крови и плазме крови человека [187, 188, 189-192]. Во всех указанных случаях применение мультисенсорной системы — «электронного языка» давало новые возможности для химического анализа, который нельзя было бы провести с помощью единичных сенсоров или другими методами. Например, таким образом решалась проблема недостаточной селективности сенсоров по отношению к ионам Са2+ и Mg2+ (анализ крови), или открывалась возможность определения концентрации ионов (например, Zn2+, Fe3+ или UO2+), для которых не существует хороших селективных электродов (рис. 6.17). Некоторые результаты количественных определений приведены в табл. 6.14 и 6.15.
Электрохимические методы анализа
733
Таблица 6.14
Определение содержания урана(М), железа(П) и железа(Ш) в растворах, моделирующих воду в затопленных урановых шахтах, с помощью «электронного языка»
Компонент Введено, моль-л 1 Найдено, моль-л 1
Fe(II) 1 • ж7 2,1 • 107
Fe(II) 1 • 10^ 1,6 - 106
Fe(II) 5 • 10^ 4,3 • 10^
Fe(II) 1 • IO5 8,0 • 10 6
Fe(II) 1 • ю4 1,0 • 107
Fe(III) 1 • 10~7 1,0 10’7
Fe(III) 1 • Ж 1,4 • 106
Fe(III) 1 10^ 4,8 • 10^
Fe(III) 1 • 10"5 1,8 105
Fe(III) 1 • 104 1,0 • 104
UO2+ 1 • 10’6 3,0 • 10^
UO2+ 1 • 105 2,6 • 10’5
Таблица 6.15
Определение содержания кальция, магния, гидрокарбоната и фосфатов в растворах, близких по составу к плазме крови, с помощью «электронного языка»
Ион Введено, моль-л 1 Найдено, моль-л 1
Mg2+ 5 • Ж 4,9 • Ю 4
Mg2+ 7,5 • 10 4 7,5 • Ж
Mg2+ 1 • ю3 1 • io3
Ca2+ 8 • Ю4 8,9 10 4
Ca2+ 1,1 • 10’3 1,1 • 10’3
Ca2+ 1,5 10’3 1,5 • 10 3
HCO; 3 • 102 3,4 • Ж2
hco; 3,5 • IO’2 3,6 • 10’2
hco; 4 10“2 3,9 • 10’2
HPO2- 9 • 10 4 9,0 • 10 4
HPO7 1,4 • 10~3 1,4 • Ю3
hpo24- 1,8 • 103 2 • 10’3
h2po; 1,4 • Ю4 1,4 • 104
h2po; 1,7 104 1,8 • Ю4
h2po; 2- 10 4 2,0 • 10 4
«Электронный язык» может быть применен с использованием описанных выше методов для одновременного качественного и количественного анализов. Так, нами были проведены предварительные работы по установлению сортов вин и содержания в них алкоголя, кислотности, сухого остатка, минеральных солей.
Рис. 6.17.
Определение концентрации железа в растворах сложного состава с помощью «электронного языка». Мультисенсорная система (массив сенсоров) не содержит сенсоров,селективных к ионам железа:
1— увеличение концентрации железа;
2 — увеличение концентрации меди
Указанные примеры применения «электронного языка» показывают новые перспективы в использовании химических сенсоров. «Электронный язык» позволяет определять качество и вкус напитков и решить проблему недостаточной селективности сенсоров и отсутствия сенсоров на отдельные ионы или частицы в растворах, при этом оказывается возможным с помощью стабильных твердотельных сенсоров с неорганическими мембранами обнаруживать как неорганические, так и органические компоненты в растворах.
6.1.6. Список фирм, изготавливающих оборудование
для потенциометрического анализа
1. Activation Glass Ltd., Halstead, Essex CO9 2EX, UK.
2. Balsbaugh Laboratories Inc., 25 Industrial Park Rd., S. Hingham, Mass. 02043, USA.
3. Beckman Instruments Inc., 2500 Harbor Blvd., Fullerton, Calif. 92634, USA.
4. Coleman Instruments, 2000 York Rd., Oakbrook, Ill.
60521, USA.
5. Coming Glass Works, Houghton Park, Coming, N.Y. 14830, USA.
6. Electrofact N.V., P.O. Box 163, Radium Weg 20, Amer-foort, Netherlands.
7. Electronic Instruments Ltd., Hanworth Lane, Chertsey Surrey KT 16 9LF, UK.
8. The Foxboro Company, Neponset Ave., Foxboro, Mass. 02035, USA.
9. Instrumentation Laboratory Inc., 113 Hartwell Ave., Lexington, Mass. 02173, USA.
10. Leeds and Northrup Co., Sumneytown Pike, North Wales, Pa. 19454, USA.
11. Metrohm A.G., CH-9100 Herisau, Switzerland.
12. Orion Research Inc., 380 Putnam Ave., Cambridge, Mass.
02139, USA.
13. N.V.Phillips’Gloeilampenfabriken, Teinhoven, Netherlands.
14. Radiometer A/S, Emdruprej 72, DK-2400 Copenhagen NV, Denmark.
15. Simac Instrumentation Ltd., Lyon Rd., Hersham, Walton-on-Thames, Surrey KT 12 3PU, UK.
16. [Schott] Janaer Glaswerk Schott and Gen., Hatten-bergstrasse 10, Postfach 24 80, Mainz, Germany.
17. Tacussel electronique, Solea, 72-78 me d’Alsace, 69100 Villeaurbanne, France.
734
Новый справочник химика и технолога
Литература
1. Власов Ю.Г. // Журн. аналит. химии. 1992. Т. 47. С. 114.
2. Основы аналитической химии: В 2 кн./ Под ред. акад. Ю.А. Золотова. М.: Высшая шк., 1999. 494 с.
3. Власов Ю.Г., Легин А.В. // В сб. Академик Б.П. Никольский. Жизнь. Труды. Школа. СПб.: Изд. СПбГУ, 2000. С. 267.
4. Справочное руководство по применению ионоселективных электродов / Под ред. О.М. Петрухина. М.: Мир, 1986. 231 с.
5. Никольский Б.П., Матерова Е.А. Ионоселективные электроды. Л.: Химия, 1980. 239 с.
6. Cammann К. Das Arbeiten mit ionenselektiven Elektroden. Berlin; Heidelberg; New York: Springer Verlag, 1977. 230 s.
7. Ion-selective Electrodes in Analytical Chemistry I Ed. by H.Freiser. New York: Plenium Press, 1978. V. 1. 439 p.
8. Ruzicka J., Hansen E.H., Tjell J.C. H Anal. Chim. Acta. 1973. V. 67. P. 155.
9. Cattrail R.W., Drew D.M. П Anal. Chim. Acta. 1975. V. 77. P. 17.
10. Матерова E.A., Грекович А.Л., Дидина C.E. И Электрохимия. 1976. Т. 12. С. 723.
11. Jaber A.M.J., Moody G.J., Thomas J.D.R. H Analyst. 1976. V. 101. P. 179.
12. Morf W.E., Ammann D., Simon W. 11 Chimia. 1974. V. 28. P. 65.
13. Алагова 3.C., Матерова E.A., Шумилова Г.И. И Вестник ЛГУ. 1978. Вып. 22. С. 112.
14. Алагова З.С., Матерова Е.А., Шумилова Г.И. И Вестник ЛГУ. 1980. Вып. 22. С. 134.
15. Davies J.E.W., Moody G.J., Price М.М., Thomas J.D.R. // Lab. Practice. 1973. V. 22. P. 20.
16. Никольский Б.П. и др. I Никольский Б.П., Матерова Е.А., Грекович А.Л., Юринская В.Е. // Журн. аналит. химии. 1974. Т. 24. С. 205.
17. Cattrail R.W., Tribuzio S., Freiser H. H Anal. Chem. 1974. V. 46. P. 2223.
18. Manning D.L., Stokely J.R., Magouyrk D.W.// Anal. Chem. 1974. V. 46. P. 116.
19. Ионный обмен и ионометрия. I Под ред. Никольского Б.П.. Л.: ЛГУ, 1979. Вып. 2. С. 134.
20. Davies J.E.W., Moody G.J., Thomas J.D. H Analyst. 1972. V. 97. P. 87.
21. Матерова E.A., Грекович А.Л., Шекина Г.И. И Электрохимия. 1974. Т. 10. С. 342.
22. Барт Т.Я., Матерова Е.А., Измайлова О.Г. // В сб. Электрохимия ионитов. Вып. 2. Куйбышев: Изд. Куйбышевского ун-та, 1977. С. 32-37.
23. Rohm T.J., Guilbault G.G. И Anal. Chem. 1974. V. 46. P. 590.
24. Ishibashi N., Jyo A., Matsumoto K. // Chem. Lett. 1973. P. 1297.
25. Матерова E.A., Овчинникова C.A., Смекалова C.A. И Электрохимия. 1978. Т. 14. С. 71.
26. James Н., Garmack G., Freiser Н. И Anal. Chem. 1972. V. 44. Р. 586.
27. Камман К. Работа с ионселективными электродами. М.: Мир, 1980. 283 с.
28. Байулеску Г., Кошофрец В. Применение ион-селек-тивных мембранных электродов в органическом анализе. М.: Мир, 1980. 230 с.
29. Мидгли Д., Торренс К. Потенциометрический анализ воды. М.: Мир, 1980. 516 с.
30. Методические рекомендации по определению различных элементов (ионов) с использованием ионоселективных электродов / Под ред. Р.Р. Тарасянца. Черкассы. 1990. 112 с.
31. Midgley D. // Analyst. 1987. V. 112, № 5. Р. 557.
32. Xie S. // Analyst. 1987. V. 112, № 4. P. 543.
33. Anders U., Hailer G. // Ditsh. Lebensm. Rundsch. 1975. Bd. 71, №6. S.208.
34. Bernal J.L. et al. 11 An. Bromatol., 1981. V. 33, № 2. P. 219.
35. Gonzalez M.A. et al. // Electroanalysis. 1991. V. 3, № 4-5. P. 439.
36. Speights R.M., Brooks J.D., Barnard A. J. 11 J. Pharm. Sci. 1971. V. 60, №15. P. 748.
37. Qiu Z., Chen Z. // Yaowu Fenxi Zazhi. 1984. V. 4, № 3. P. 159.
38. Haynes S.J., Clark A.H. H Econ. Geol. 1972. V. 67, № 3. P. 378.
39. Haynes S.J. H Taianta. 1978. V. 25, № 2. P. 85.
40. Rice T.D. 11 Taianta. 1988. V. 35, № 3. P. 173.
41. Korhonen J., Lumme P.O. // Pap. Puu. 1977. V. 59, № 9. P. 558.
42. Midgley D. 11 Analyst. 1985. V. 110, № 7. P. 841.
43. Midgley D., Gatford C. // Microchem J. 1990. V. 42, № 2. P. 225.
44. Sun L. 11 Yejiu Fenxi. 1988. V. 8, № 1. P. 57.
45. Graf J.E., Vaughn T.E., Kipp W.H. 11 J. Ass. Off. Analyt. Chem. 1976. V. 59, № 1. P. 53.
46. Buchberger W., Huemer P. // Mikrochim. Acta. 1985. V. l,№5-6. P.421.
47. Crecelius Е.А. П Anal. Chem. 1975. V.47, № 12. P. 2034.
48. Miles P. H J. Ass. Off. Analyt. Chem. 1978. V. 61, № 6. P. 1366.
49. Ficklin W.H. П J. Res. U.S. Geol. Surv. 1975. V. 3, № 6. P. 753.
50. Westerlund-Helmerson U.// Anal. Chem. 1971. V. 43, № 8. P. 1120.
51. Gottardi W. H Laboratoriumsmedizin. 1992. Bd. 16, № 9. S. 283.
52. Lapatnick L.N. 11 Anal. Chim. Acta. 1974. V. 72, № 2. P. 430.
53. Zhou J., Wang X., Zhu P. // Huaxue Shijie. 1987. V. 28, №6. P. 261.
54. Bemal J.L. et al. П Mater. Constr. 1988. V. 38, № 209. P. 5.
55. Sim Y. // Fenxi Huaxue. 1988. V. 16, № 3. P. 288.
56. Asano Y., Ito S. // Nippon Kagaku Kaishi. 1980. № 10. P. 1494.
57. Sara R., Waenninen E. П Taianta. 1975. V. 22, № 12. P. 1033.
58. Postel W., Goerg A., Guvenc U. 11 Brauwissenschaft. 1976. Bd. 29, №5. S. 132.
Электрохимические методы анализа
735
59. Clifford G., Kirk D. И Analyst. 1981. V. 106, № 1269. Р. 1341.
60. Bodkin J.B. П Analyst. 1977. V. 102, № 1215. P. 409.
61. Troll G., Farzaneh A., Cammann К. 11 Chem. Geol. 1977.
V. 20. P. 295.
62. Рокота H. // Chem. Prum. 1978. V. 28, № 5. P. 238.
63. Baker R.L. // Anal. Chem. 1972. V. 44, № 7. P. 1326.
64. Villa E. //Analyst. 1979. V. 104, № 1239. P. 545.
65. Wang J., Xi D. // Yaoxue Tongbao. 1985. V. 20, № 12. P. 727.
66. Thomas J., Gluskoter H.J. I I Anal. Chem. 1974. V. 46, №9. P. 1321.
67. Raghavan R., Gupta B.L., Raha S. 11 Taianta. 1992. V. 39, № 3. P. 243.
68. Кириллов В.Н. //Завод, лаб. 1986. Т.52, № 1. С. 7.
69. Кириллов В.Н., Клетеник Ю.Б. И Завод, лаб. 1991. Т. 57, №7. С. 14.
70. Данилевич Т.И., Абрамов В.В., Андреев П.А. // Завод, лаб. 1983. Т.49, № 5. С. 23.
71. Hino Т., Nakanishi S., Nagashima К. И Bunseki Kagaku.
1992. V. 41,№ 10. Р. 125.
72. Shan W., Yin Z. I I Analyst. 1993. V. 118, № 12. P. 1537.
73. Rice T.D. H Anal. Chim. Acta. 1983. V. 151, № 2. P. 383.
74. Shelton B.J. // Rep. Natn. Inst. Metall. 1975, № 1722. P. 4.
75. Brown D.K., Parker A.G.// Analyst. 1980. V. 105, № 1257. P. 1208.
76. Wisk T.J., Siebert K.J. // Proc. Amer. Soc. Brew. Chem. 1973. P. 26.
77. Drawert F., Nitsche T. // Brauwissenschaft. 1976. Bd. 29, № 10. S. 299.
78. Helaine E. // Ind. Aliment. Agric. 1977. V. 94, № 6. P. 581.
79. Rice T.D. et al. I Rice T.D., Sweeney Y., Semitekolos R., Rhyder G.J. // Taianta. 1984. V. 31, № 8. P. 607.
80. Li S., Smith K.A. I I Commun. Soil Sci. Plant Anal. 1984. V. 15, № 12. P. 1437.
81. Hulanicki A., Lewandowski R., Maj M.// Anal. Chim. Acta. 1974. V. 69, № 2. P. 409.
82. Burman J.O., Johansson G. // Anal. Chim. Acta. 1975. V. 80, №2. P. 215.
83. Логинова Л.П., Маслий О.Г., Помочева Г.В. // Вестник Харьковского ун-та. 1988. № 319. С. 36.
84. Pfeiffer S.L., Smith J.J. // Ass. Off. Analyt. Chem. 1975. V. 58, № 5. P. 915.
85. Kaneda Y., Kanamori T., Iwaida M. // Eisei Kagaku.
1977. V. 23, №5. P. 301.
86. ChoiKK,FungKW.//Analyst 1980. V. 105,№ 1248. P. 241.
87. Zhou X., Zhang T., Liu J. // Wuxi Qinggongye Xueyuan Xuebao. 1991. V. 10, № 2. P. 19.
88. Gulens J., Leeson P.K. // Anal. Chem. 1980. V. 52, № 13. P. 2235.
89. Nobile L. et al. // Phannazie. 1989. V. 44, № 1. P. 66.
90. Данилевич Т.Н. // Журн. аналит. химии. 1987. Т. 42, №1.С.92.
91. McNemey F.G. //J. Ass. Off. Analyt. Chem. 1976. V. 59, №5. P. 1131.
92. Lu W., Zhang Q. // Fenxi Huaxue. 1988. V. 16, № 9. P. 815.
93. Миронова И.В. и др. I Миронова И.В., Штерман В.С., Фролова Т.Т., Росин И.В. И Изв. ВУЗов. Пищ. технология. 1978. №5. С. 147.
94. Leng Z., Xie К., Wang J. И Fenxi Huaxue. 1987. V. 15, № 12. P. 1142.
95. Yan F., Lu X. // Lihua Jianyan. Huaxue Fence. 1988. V. 24, № l.P. 34.
96. Lin S. et al. I Lin S., Wang X., Wang Y., Zeng Y. // Fenxi Huaxue. 1987. V. 15, № 9. P. 861. .
97. Fung Y.S., Fung K.W. // Analyst. 1978. V. 103, № 1223. P. 149.
98. YuH., YangZ. //Fenxi Huaxue. 1987. V. 15, № 1. P. 12.
99. Tong S. // Fenxi Huaxue. 1982. V. 10, № 5. P. 297.
100. Midgley D., Analyst. 1988. V. 113, № 7. P. 997.
101. Swartz J.L., Light T.S. // TAPPL 1970. V. 53, № 1. P. 90.
102. Valentova M., Sucha L., Fischerova H.// Sb. Vys. Sk. Chem.-Technol. Praze. // Anal. Chem. 1982. V. Hl7. P. 43.
103. Берестецкий В.И., Федорук Л.И., Ленгрен Л.Г. И Техн, кино и телевидения. 1991. № 7. С. 43.
104. Midgley D. // Analyst. 1989. V. 114, № 1. Р. 1.
105. Chapman В.К., Goldsmith I.R. П Analyst. 1982. V. 107, № 1278. P. 1014.
106. Масса C., Fresenius Z. // Anal. Chem. 1987. Bd. 326, № 3. S. 559.
107. Головнев H.H. и др. // Журн. аналит. химии. 1994. Т. 49, №3. С. 1010.
108. Kissa Е. И Anal. Chem. 1983. V. 55, № 8. Р. 1445.
109. Kissa Е. И Clin. Chem. 1987. V. 33, № 2, Р. 253.
110. Trojanowicz М., Hulanicki А. // Mikrochim. Acta. 1981. V. II, № 1-2. Р. 17.
111. Головнев Н.Н. и др. // Журн. аналит. химии. 1994. Т. 49, № 11. С. 1237.
112. Yan Y., Tu J., Yana X. // Shanghai Huanjing Kexue. 1991. V. 10, №2. P. 35.
113. Graggs A., Moody G.J., Thom. J.D.R. // J. Chem. Educ. 1974. V. 51,№8.P. 541.
114. Lenz B.L., Mold J.R. // TAPPL 1971. V. 54, №12. P. 2051.
115. Wilson B.L., Schwarzer R.R., Mafoti R. // Microchem. J. 1984. V. 29, № l.P. 74.
116. Frant M.S. П Plating. 1971. V. 58, № 7. P. 686.
117. Hara H., Okazaki S.// Analyst. 1984. V. 109, №10. P. 1317.
118. Tan K. // Fenxi Shiyanshi. 1988. V. 7, № 10. P. 27.
119. Polasek M., Zavesky R. П Cesk. Farm. 1985. V. 34, № 10. P. 425.
120. Bond A.M. et al. H Anal. Chim. Acta. 1990. V. 237, № 2. P. 345.
121. Bond A.M. et al. // Anal. Chim. Acta. 1988. V. 208, № 1-2. P. 195.
122. LiH. //Analyst. 1987. V. 112, № 11. P. 1607.
123. Wang J. // Analyst. 1990. V. 115, № 1. P. 53.
124. Midgley D. //Analyst. 1993. V. 118, № 11. P. 1347.
125. Qian G, Wu G. // Fenxi Huaxue. 1987. V. 15, № 10. P. 929.
126. Budimir M.V., Sak-Bosnar M., Jovanovic M.S. I I Anal. Chim. Acta. 1987. V. 196. P. 293.
736
Новый справочник химика и технолога
127. Rice T.D. et al. // Taianta, 1984. V. 31, № 8. P. 607.
128. Diaz C. et al. // Microchem. J., 1989. V. 39, № 3. P. 289.
129. Brand M.J.D., Rechnitz G.A. // Anal. Chem. 1970. V. 42, № 11. P. 1172.
130. Ariano J.M., Gutknecht W.F. // Anal. Chem. 1976. V. 48, №2. P. 281.
131. Horvai G. et al. // Anal. Chem. 1978. Bd. 292, № 2. S.132.
132. Wang J. // Fenxi Huaxue. 1986. V. 14, № 5. P. 384.
133. Wang J. et al. // Fenxi Huaxue. 1986. V. 14, № 6. P. 401.
134. Huang J. // Fenxi Huaxue. 1986. V. 14, № 8. P. 579.
135. Comor J., Polin C. // Lab. Pract. 1991. V. 40, № 7. P. 23.
136. Cruz G.V. // Tec. Lab. 1992. V. 14, № 169. P. 128.
137. Garcia C. et al. // Food Chem. 1992. V. 45, № 5. P. 365.
138. Martin et al. // Food Chem. 1993. V. 46, № 1. P. 85.
139. Mangubat E.A., Hinds T.R., Vincenzi F.F. // Clin. Chem.
1978. V. 24, № 4. P. 635.
140. Gran G. // Analyst. 1952. V. 77, № 920. P. 661.
141. Campbell A.D., Graham P.B. // N.Z. J. Sci. 1983. V. 26, № 4. P. 433.
142. Buffle J., Parthasarathy N., Monnier D. // Anal. Chim. Acta. 1972. V. 59, № 3. P. 427.
143. Buffle J. // Anal. Chim. Acta. 1972. V. 59, № 3. P. 439.
144. Chaudhuri N.K., Sawant R.M. // Anal. Lett. 1991. V. 24, №9. P. 1605.
145. Martyak N.M. // Plat. Surf. Finish. 1992. V. 79, № 2. P. 60.
146. Sucman E., Sucmanova M., Synek O.Z. // Lebensm. Unters. Forsch. 1978. Bd. 167, № 1. S. 5.
147. Oehme M., Stray H., Fresenius Z. // Anal. Chem., 1981. Bd. 306, № 5. P. 356.
148. Anfalt T, Jagner D.// Anal. Chim. Acta. 1971. V. 53, № 1. P. 13.
149. Jagner D., Pavlova V. H Anal. Chim. Acta. 1972. V. 60, № l.P. 153.
150. Котова B.H., Губанова B.H. // Завод, лаб. 1992. Т. 58, №2. С. 10.
151. Perez-Olmos R. et al. // Analyst. 1994. V. 119, № 2. P. 305.
152. Anfalt T., Jagner D.// Anal. Chim. Acta. 1973. V. 66. № l.P. 152.
153. Perez-Olmos R., Echevarria J. // Food Chem. 1989.
V. 32, №3.P. 201.
154. Al-Hitti I.K., Thomas J.D.R.// Anal. Lett. 1985. V. 18, № A8. P. 975.
155. Comer J. // Lab. Pract. 1988. V. 37, № 8. P. 61.
156. McNemey F.G. // J. Ass. Off. Analyt. Chem. 1974. V. 57, №5. P. 1159.
157. Farroha S.M., Habboush A.E., Issaq N. // Analyst. 1984.
V. 109, № 12. P. 1531.
158. Hulanicki A., Sokalski T., Lewenstam A. // Chem. Anal.
1987. V. 32 №4, P. 601.
159. Smith M.J., Manahan S.E.// Anal. Chem. 1973. V. 45, № 6. P. 836.
160. Bozon H., Bozon S. // Analusis. 1978. V. 6, № 6. P. 243.
161. Frant M.S., Ross J.W., Riseman J.H.// Anal. Chem.
1972. V. 44, № 3. P. 2227.
162. Li H. // Anal. Lett. 1991. V. 24, № 3. P. 473.
163. Gran G. // Acta Chem. Scand. 1950. V. 4, № 4. P. 559.
164. McCallum C., Midgley D. // Anal. Chim. Acta. 1973. V. 65, № l.P. 155.
165. Midgley D., McCallum C. // Taianta. 1976. V. 23, № 4. P. 320.
166. Still E. // Anal. Chim. Acta. 1979. V. 107. P. 377.
167. Ivaska A. // Taianta. 1980. V. 27, № 2. P. 161.
168. Gran G., Johansson A., Johansson S.// Analyst. 1981. V. 106, № 1267. P. 1109.
169. Magallanes J.F., Carid A.F.// Anal. Chim. Acta. 1981.
V. 133, №2. P. 203.
170. Li H. // Analyst. 1987. V. 112, № 11. P. 1605.
171. Масса C., Bombi G.G.// Analyst. 1989. V. 114, №4. P. 463.
172. Cantallops J., Estela J.M., Cerda V. // Anal. Chim. Acta. 1985. V. 169. P. 397.
173. Масса C., Merkoci F.// Taianta. 1994. V. 41, №12, P.2033.
174. Rigdon L.P. et al. // Anal. Chim. Acta. 1979. V. 112, № 4. P. 397.
175. Whyte J.N.C., Englar J.R.// Analyst. 1976. V. 101, № 1207. P. 815.
176. Farrona S.M. etal. // Analyst. 1987. V. 112, № 12. P. 1773.
177. Chang F.-C., Tsai H.-Т., Wu S.-C. // J. Chin. Chem. Soc. Taipei. 1975. V. 22, № 4. P. 309.
178. Vesala A.J. // Chem. Educ. 1992. V. 69, № 7. P. 577.
179. Lutze O., Rob B., Cammann K., Fresenius J. // Anal. Chem. 1994. V. 350, № 10-11. P. 630.
180. Young M., Driscoll J.N., Mahaney K. // Anal. Chem.
1973. V. 45, № 13. P. 2283.
181. Li Y. // Huaxue Shijie. 1990. V. 31, № 5. P. 220.
182. Korhonen J., Lumme P.O. // Pap. Puu. 1978. V. 60, № 2. P. 81.
183. Chakraborti D., Adams F. // Anal. Chim. Acta. 1979. V. 109, № 2. P. 307.
184. Olin A, Wallen B.// Anal. Chim. Acta. 1983. V. 151, № l.P. 65.
185. Власов Ю.Г. и др. // Журн. аналит. химии. 1982. Т. 37, № 12. С. 2155.
186. Legin A.V. et.al. // Sensors and Actuators В. 1997. V. 44. P.291.
187. Vlasov Yu.G. , Legin A.V. Fresenius J. // Anal. Chem. 1998. V. 361. P. 255.
188. Власов Ю.Г. и др. // Журн. аналит. химии. 1997. Т. 52, № 11. С. 1199.
189. Власов Ю.Г., Легин А.В., Рудницкая А.М.// Журн. аналит. химии. 1997. Т. 52, № 8. С. 837.
190. Di Natale С. et al. // Sensors and Actuators B. 1997.
V. 44. P. 423.
191. Власов Ю.Г. и др. // Журн. прикл. химии. 1998. Т. 71. С.1483.
192. Мортенсен Дж. и др. // Журн. прикл. химии. 1999.
Т. 72. С. 535.
193. Persaud К, Dodd G.H. //Nature. 1982. V. 299. Р. 352-355.
194. Toko Т., Murata Т., Matsuno Т. et al. // Sensor. Mater. 1992. V. 4. P. 145-151.
Информация о новых методиках анализа и изготовителях химических сенсоров: sensor 2000 @ vk 5346 spb. edu.
Электрохимические методы анализа
737
62. Кулонометрия [1,2,3]
Кулонометрический анализ заключается в определения количества электричества, расходуемого в ходе электрохимической реакции. Количество электричества Q равно произведению силы тока I на время его прохождения через раствор t: Q = It.
Единицей количества электричества является кулон (Кл), равный количеству электричества, протекающего через поперечное сечение проводника в течение 1 секунды при силе тока 1 А. Электрохимический эквивалент — это масса вещества, выделившегося на одном электроде (или растворившегося, с другого электрода) в процессе электролиза при протекании единицы количества электричества, т. е. 1 Кл.
Прямая кулонометрия. В прямой кулонометрии определяемое вещество реагирует непосредственно на поверхности электрода, поэтому этот метод пригоден для определения только электроактивных веществ. Расчеты, проводимые при применении прямой кулонометрии, не отличаются сложностью. В их основе лежат законы Фарадея, которые формулируются следующим образом:
1. Масса вещества, выделившегося на электроде, прямо пропорциональна количеству электричества Q, прошедшего через электролит.
2. Массы различных веществ, выделенных или растворенных при прохождении одного и того же количества электричества, пропорциональны их электрохимическим эквивалентам.
Суть законов Фарадея заключается в том, что для превращения 1 грамм-эквивалента любого вещества необходимо пропустить через раствор одно и то же количество электричества. Эта величина, равная 96487 Кл/моль, была названа впоследствии постоянной Фарадея — F.
пт
Величину прошедшего количества электричества Q находят методом интегрирования или (для кулонометрии при постоянном токе) путем умножения силы тока на время.
Кулонометрический анализ при постоянной силе тока. В большинстве случаев при кулонометрическом определении можно менять (контролировать) ток или потенциал электрода один независимо от другого. Выбор независимого параметра определяется характером аналитической методики или имеющимся оборудованием.
Кулонометрия при постоянной силе тока является более простым, но менее селективным способом. В этом случае контролируется сила тока, протекающего через электролитическую ячейку. Измерить силу постоянного тока и время его прохождения, т. е. определить количество прошедшего электричества (Q - It), несложно, поэтому данный способ анализа применяется уже в течение долгого времени.
Недостатки метода:
-для проведения количественной реакции требуется длительное время;
-потребляемый ток (особенно по мере завершения электролиза) может частично расходоваться на прохождение не представляющей интереса побочной реакции, в результате чего выход по току снижается и становится менее 100%.
Добиться 100% выхода по току какого-либо вещества, присутствующего в растворе, очень трудно, так как необходимо обеспечить окисление или восстановление на электроде только этого вещества.
В ходе проведения кулонометрического анализа при контролируемом (постоянном) потенциале ток больше не остается неизменным, поэтому требуется проводить интегрирование по времени измеряемых значений мгновенного тока. Такое интегрирование можно осуществить с помощью кулонометра (химического, механического или электронного) или же расчетным путем (компьютерная обработка данных с помощью аналого-цифрового преобразования измеряемого тока). Точность кулонометрического анализа при постоянном потенциале в значительной степени определяется не точностью электронного интегратора, а погрешностью химической процедуры анализа; в настоящее время вполне возможны измерения с погрешностью менее 0,5%. Концентрация вещества, установленная этим методом, меньше отличается от истинной концентрации определяемого вещества в растворе, чем при кулонометрическом анализе при постоянном токе. В этом случае поддержание постоянного потенциала исключает протекание побочных реакций, которые характерны для кулонометрии при постоянной силе тока в условиях изменяющегося (при изменении концентрации) потенциала.
Косвенная кулонометрия
Кулонометрическое титрование
В методах косвенной кулонометрии определяемое вещество не участвует в реакции, протекающей непосредственно на электроде. В результате электрохимической реакции, проходящей на электроде, генерируется промежуточный реагент, который взаимодействует с определяемым веществом в объеме раствора. Косвенная кулонометрия применяется значительно чаще, чем прямая, как способ введения в раствор некоторого количества электричества, необходимого для осуществления титрования. В методах анализа, не связанных с титрованием, этот способ используют редко, поскольку анализируемый раствор должен содержать подходящий промежуточный реагент. Единственным недостатком косвенной кулонометрии по сравнению с прямой является необходимость количественного протекания реакций на обоих этапах косвенного кулонометрического анализа. Образование промежуточного реагента должно происходить с выходом, равным 100% по фарадеевскому току, а реакция генерированного титранта с определяемым веществом должна быть и быстрой, и количественной. Косвенная кулонометрия характеризуется также большей скоростью, поскольку концентрация веще
738
Новый справочник химика и технолога
ства, из которого электрохимически генерируется промежуточный реагент, может значительно превышать концентрацию определяемого вещества, что, как правило, и имеет место на практике.
Наиболее важной и фактически единственной областью применения косвенной кулонометрии в аналитической химии является кулонометрическое титрование.1
Титрант выбирают таким образом, чтобы он взаимодействовал с определяемым веществом быстро и стехио-метрически, по достижении точки эквивалентности процесс титрования немедленно прекращают.
Кулонометрические методы отличаются от других видов титрования только способом добавления титранта в исследуемый раствор. Электрохимические способы обнаружения конечной точки рассматриваются в разделе «Титриметрические методы электроаналитической химии».
Метод кулонометрического титрования был предложен впервые в 1938 г., широкое применение он нашел с 1948 г. В настоящее время кулонометрическое титрование проводят в большинстве случаев при постоянном токе, в то время как другие формы кулонометрического анализа обычно проводят при контролируемом (постоянном) потенциале.
Кулонометрический способ обладает следующими преимуществами перед другими способами введения в раствор титранта:
1. Нет необходимости в приготовлении стандартных растворов, так как собственным первичным стандартом здесь служит постоянная Фарадея.
2. Простота точного измерения небольших количеств электричества, вплоть до 1 мКл.
3. Реагенты, которые трудно хранить или стандартизировать, могут быть получены in situ и стандартизованы кулонометрическим способом, их можно использовать при проведении количественного анализа; например, медь(1), титан(П), молибден(У), бром.
Кислотно-основное титрование. Реакции кислотноосновных титрований протекают с потерей или присоединением протонов и, следовательно, с кулонометрическим генерированием ионов Н+ или ОН-, как правило, из воды или из водной составляющей смешанного растворителя.
1 Определение титрирования см. п. 6.1.5.2
Иначе говоря, кулонометрическая реакция может служить для связывания Н+ или ОН-, образующихся в ходе титрования. В любом случае обычно применяют инертные (платиновые) электроды с большой площадью поверхности (табл. 6.16).
Осадительное титрование. Наиболее широко распространенным видом кулонометрического осадительного титрования является титрование, в котором при анодном растворении электрода образуется ион Ag+, служащий титрантом для галогенид-ионов. Ход реакции титрования легко контролируется потенциометрическим датчиком, чувствительным к иону Ag+ или галогенид-иону. Этот способ позволяет определять лишь суммарное содержание галогенид-ионов (табл. 6.16, 6.17).
Окислительно-восстановительное (редокс-титрова-ние). Реакции титрования редокс-типа часто можно эффективно использовать для кулонометрического генерирования титранта. Окисление бромид-ионов с образованием брома, вероятно, является наиболее широко используемой реакцией кулонометрического генерирования титранта. В этих же целях широко применяется генерирование иода, железа(П) и олова(И). Электрогенерированный ион брома используют и для кулонометрического титрования ненасыщенных связей жирных кислот в пропиленкарбонате (табл. 6.16, 6.17).
Комплексометрическое титрование. При кулонометрическом титровании, основанном на реакциях комплексообразования, комплексообразующие агенты, как правило, являются электрохимически неактивными. Комплексообразующий агент в этом случае генерируется путем кулонометрического восстановления комплекса, в состав которого он входит.
Для кулонометрического генерирования комплексообразующих агентов необходимо, чтобы комплекс, образующийся при титровании, был менее устойчив, чем исходный комплекс, и при этом не участвовал в реакции восстановления. В этих целях можно использовать некоторые комплексы ЭДТА и родственных ей соединений. Известны также случаи, когда при кулонометрическом титровании используется более одного типа реакций; так, определение пенициллинов и пеницилламинов, включает и образование нерастворимого сульфида ртути, и хелатирование.
Электрогенерированные кулонометрические титранты
Таблица 6.16
Титрант Вспомогательный реагент Реакция генерации титранта на электроде Применение
Кислотно-основное титрование
ОН Н2О 12ОН-+Н2 Титрование кислот
РГ Н2О Титрование СГ, Вг-, Г, органических серосодержащих веществ
Осадительное титрование
Ag* Ag-анод Ag<=> Ag++e Титрование С1, Вг , Г-, органических серосодержащих веществ
Электрохимические методы анализа
739
Продолжение таблицы 6.16
Титрант Вспомогательный реагент Реакция генерации титранта на электроде Применение
Окислительно-восстановительное титрование
Мп3+ MnSO4 Мп2+ <=> Мп3+ + е Титрование Fe(II) Н2С2О4
Вг2 Вг Вг2 + 2е 2Вг“ Титрование As(III), Г, фенолов
СиС13 СиС12 Си2+ + ЗСГ + е CuCl" Титрование Cr(VI), Ю3
С12 КС1 2СГ <=» С12 + 2е Титирование Г, As(III)
ъ KI 2Г <=Н2+2е Титрование S2O3-,As(III)
Таблица 6.17
Некоторые типичные примеры кулонометрического титрования [4]
Принятые обозначения. А — амперометрический; И — индикатор; П — потенциометрический.
Генерируемый титрант Состав электролита Титруемое вещество Нахожде ние КТТ
Реакции нейтрализации
Н Сульфат натрия (0,2 М) ОН ; органические основания П
ОН Сульфат натрия (0,2 М) Н^; органические основания П
Redox-реакции
С12 НС1 (2,0 М) As(III); Г А
Вг2 КВг (0,2 М) Sb(III); T1(I); U(IV): Г; SCN"; NH3; ВД; NH4OH А
KI (0,1 М, фосфатный буфер, pH = 8) As(III); Sb(III); S2O3; S2“; аскорбиновая кислота А, П,И
Ce(IV) Ce2(SO4)3 (0,1 М) Fe(II): Ti(III); U(IV); As(III); Fe(CN)3 П
Мп(Ш) Mn SO4 (0,5 М), H2SO4 (1,8 М) Fe(II), As(III); щавелевая кислота П
Ag(I) AgNO3 (0,1 М), HNO3(5 М)* As(III);Ce(III); V(IV) П
Cu(II) CuSO4 (0,1 М) V(V); Cr(VI); IO^;Br2 П
Fe(II) Fe2(SO4)3, №)2, SO4 (0,3 М), H2SO4 (2 М) V(V); Cr(VI); MnO4 П
Ti(III) Ti(l У)-сульфат (0,6 М), H2SO4 (6 М) Fe(III); Ce(IV); V(V); U(VI) П
Реакции осаждения
Ag(I) KNO3 (0,5 М)** CI ; Br ; I; меркаптаны П
Hg(I) НС1О4 (0,5 М)*** СГ; Вг“ П
НС1О4 (0,1 М), KNO3 (0,4 М) Г П
Fe(CN)^ K3Fe(CN)6 (0,2 М), H2SO4 (0,1 М) Zn П
Реакции комплексообразования
ЭДТА Hg(NH3)-conb ЭДТА (0,1 М); NH4NO3, (о,1М),н-8,з; Са(П); Cu(II); Zn(II); Pb(II) п
Разнородные (разнообразные) реакции замещения
Вг2 КВг (0,2 М) Ароматические амины, фенолы, оксины А
Реакции дополнения
Вг2 КВг (0,2 М)**** Ненасыщенные углеводы, например, алкены; циклогексан А
Примечания. *3олотой анод. ** Серебряный анод. *** Ртутный анод. **** В качестве катализатора добавлены следы ацетата ртути(П) растворенного в смеси метанола и уксусной кислоты; * Ртутно-ЭДТА-реагент: приготовить рабочий раствор, содержащий 8,4 г нитрата Hg(II) и 9,3 г динатриевой соли ЭДТА в 250 мл (каждого реагента по 0,1М), Смешать 25 мл этого рабочего раствора с 75 мл раствора нитрата аммония (0,1М) и довести до pH 8,3 концентрированным раствором аммиака.
740
Новый справочник химика и технолога
6.3. Вольтамперометрия [1-5]
6.3.1. Классические методы
Вольтамперометрия — это группа электрохимических методов анализа, в которых контролируемый параметр — потенциал индикаторного электрода — меняется во времени, а измеряемой величиной является ток, протекающий через индикаторный электрод. Классическая вольтамперометрия объединяет вольтамперометрические методы, в которых потенциал меняется во времени достаточно медленно, так что наблюдаемые явления могут быть количественно описаны на основании равновесных или квазиравно-весных теорий. Вольтамперометрический анализ, включает в качестве составной части полярографию.
Полярография — это часть вольтамперометрии, в которой в качестве индикаторного электрода используется жидкий металлический электрод в виде растущей, вытекающей из капилляра, капли (обычно ртутный).
Ртуть, служащая катодом, вытекает с определенной скоростью из тонкого стеклянного капилляра. Отрываясь от последнего, ртутная капля уносит с собой выделившийся на ней металл, и процесс восстановления продолжается уже на свежей капле, появляющейся в устье капилляра тотчас же после отрыва предыдущей капли. Это постоянное обновление поверхности является достоинством ртутного капельного электрода. Кроме того, на ртути велико перенапряжение для выделения водорода, что позволяет восстанавливать ионы электроотрицательных металлов (свинца, цинка, кадмия и т. п.). В положительной области потенциалов применение ртутного электрода ограничено окислением самой ртути (потенциал, при котором происходит это окисление, зависит от состава раствора).
Принципиальная схема полярографа чрезвычайно проста. Полярографическая ячейка состоит из ртутного капающего электрода (капилляр диаметром 0,03 мм), который соединен с резервуаром, дно которого заполнено ртутью. Конец капилляра опущен в исследуемый раствор, образующиеся капли ртути падают сквозь раствор на дно сосуда. Современный ртутно-капающий электрод представляет собой высокотехнологичное изделие, обеспечивающее полную герметизацию ячейки и рабочего объема ртути. Стряхивание растущих капель — принудительное и осуществляется молоточком, управляемым электронным устройством. Стоимость такого комплекта весьма высока. Напряжение, приложенное между капельным электродом и электродом сравнения (донной ртутью), вызывает ток, который приводит к поляризации электродов (изменению поверхностного потенциала). Влияние проходящего тока на величину потенциала поляризуемого электрода соответствует площади его поверхности. При этом электрод сравнения, обладая существенно большей площадью, практически не поляризуется. Принимая его потенциал равным нулю (точка отсчета в эксперименте), можно записать, что приложенное напряжение поляризации близко к потенциалу ртутно-капельного электрода Е = -Ет.
Кривая зависимости силы тока от потенциала (полярограмма) состоит из трех характерных участков: остаточно
го тока, участка крутого подъема тока («волна») и предельного тока (рис. 6.3.1).
Рис. 6.18. Полярографическая волна [5]
При значениях приложенного потенциала, недостаточных для разряда определяемых ионов, сила тока возрастает по мере увеличения напряжения. Остаточный ток включает фарадеевский ток /ф, возникающий за счет электроразряда примесей, имеющихся в растворе, и емкостный ток 1с, обусловленный емкостью двойного электрического слоя на свежеобразующейся поверхности ртутной капли (вклад 1с в величину остаточного тока обычно значительно превышает /ф). Остаточный ток ограничивает предельную чувствительность метода. Как только потенциал разряда определяемого иона достигнут, начинается электрохимическая реакция, и сила тока резко возрастает. При этом в приэлектродном слое наблюдается снижение концентрации электрохимически активного вещества. При дальнейшем увеличении напряжения на ячейке сила тока достигает своего предельного значения потому, что концентрация в приэлектродном слое становится практически равной нулю. Такая кривая носит название «полярографическая волна».
Потенциал полуволны Ev соответствует потенциалу /2
при силе тока, равной половине диффузионного тока /д. В отличие от потенциала восстановления, Е,7не зависит от /2
концентрации электровосстанавливающих ионов в растворе (рис. 6.19). По значению предельного диффузионного тока /д судят о концентрации вещества в растворе, а по потенциалу полуволны о природе иона.
Ориентировочно величину Ev можно найти, опустив /2
перпендикуляр из середины полярографической волны на ось потенциалов. Более точным способом нахождения потенциала полуволны является графический (компьютер-
1g / ный) метод в координатах —=—- f(E) . Поскольку при
постоянстве состава раствора, pH и температуры величина потенциала полуволны определяется природой деполяри-
Электрохимические методы анализа
741
затора, то по найденной величине Ev можно установить, /2
какой ион находится в исследуемом растворе.
Рис. 6.19. Сдвиг потенциала восстановления
(Ев) кадмия при различной его концентрации
в растворе
= const j
Таким образом, нахождение величины потенциала полуволны является основой качественного полярографического анализа. Площадка диффузионного тока хорошо различима на полярографической кривой, если потенциалы полуволн веществ отличаются друг от друга более, чем на 0,1-0,2 В. В этом случае на одной полярограмме можно получить хорошо выраженные волны нескольких веществ. Такая кривая носит название полярографического спектра. Например, полярографический спектр смеси веществ, содержащей определяемые катионы, снимается в диапазоне от 0 до -2,0 В, так как в этом интервале происходит восстановление почти всех катионов. Если разница потенциалов полуволн двух ионов меньше 0,1-0,2 В, две волны сливаются в одну. Для повышения разрешающей способности полярографического метода используют различные способы обработки сигнала (см. 6.5.4.7) или варьируют состав фонового раствора таким образом, чтобы изменить взаимное расположение потенциалов полуволн соседних веществ (см. 6.5.4.6).
Уравнение зависимости силы тока от концентрации деполяризатора (уравнение Ильковича). Величина тока, проходящего через полярографическую ячейку в условиях ограниченного подвода ионов деполяризатора к электроду, подчиняется законам диффузии. Исходя из закона Фарадея для тока, обусловленного электрохимической реакцией на ртутном капельном электроде, можно написать
_ nF • dM di
dM где-------число молей деполяризатора, которое подхо-
dt
дит к электроду за единицу времени и вступает в электрохимическую реакцию. Если деполяризатор доставляется к электроду только за счет диффузии, то, согласно законам
Фика, можно вычислить величину---, а, следовательно,
dt
и величину диффузионного тока.
В 1938 г. Д. Илькович вывел уравнение, описывающее зависимость диффузионного тока к сферическому электроду (ртутной капле) от концентрации деполяризатора, оно известно как уравнение Ильковича [1,3]:
(-If36лУ '2 2 1
/А = ~ л 2 nFcD2m3t6. (6.45)
W {р )
В нем константой пропорциональности является коэффициент диффузии D, представляющий собой число молей вещества, которое диффундирует через единицу площади за единицу времени, при градиенте концентрации, равной единице. У поверхности такого электрода в стационарных условиях величину диффузионного тока определяет градиент концентрации, который так же, как и градиент концентрации в любой области раствора, является функцией времени.
Обычно уравнение Ильковича записывается в форме, учитывающей численные величины входящих в него констант, а также плотность ртути (13,534 кг/м3 при 25 °C). Это выражение для средней величины тока за период жизни ртутной капли, полученное интегрированием уравнения для мгновенного тока по времени жизни капли имеет вид:
12]
- 606,9ncD2m3t6', (6.46)
где в соответствии с СИ ток выражается в амперах, с — в молях на кубический метр, D — в квадратных метрах в секунду и t — в секундах.
При выводе уравнения Ильковича были сделаны следующие допущения:
1. Скорость истечения ртути не меняется в течение всего периода жизни капли. Это не совсем справедливо, особенно в начальный период жизни капли.
2. Капли ртути имеют сферическую форму. Фотографии показывают, что форма капель отклоняется от строго сферической; однако это допущение приемлемо для маленьких капель.
3. Центр симметрии каждой капли не меняет своего положения. В действительности же по мере роста капли он постепенно снижается.
4. Экранирующее действие нижнего среза капилляра капающего электрода не принимается во внимание.
5. Концентрация восстанавливающих веществ падает до нуля у поверхности электрода и остается постоянной в глубине раствора. Уменьшение объемной концентрации в результате электродной реакции считается пренебрежимо малым, что для малых электродов вполне допустимо.
6. Предполагается, что перемешивание полностью отсутствует.
7. Используется теория линейной, а не сферической диффузии.
742
Новый справочник химика и технолога
Уравнение Ильковича справедливо для диффузионноограниченного тока к расширяющейся сфере в любой момент времени. Кривая изменения тока во времени показана на рис. 6.20. Когда сфера (капля) достигает своего максимального размера, что для ртутного капающего электрода соответствует моменту отрыва капли, ток достигает максимальной величины (которая также определяется уравнением Ильковича).
тока. Диаграмма работы и вид полярограммы приведен на рис. 6.23 (условия съемки идентичны приведенным на рис. 6.22).
Рис. 6.22. Вид полярограмм при различной скорости капания ртути [6]
Рис. 6.20. Ток на расширяющемся сферическом электроде [1]: максимальное значение тока 7макс достигается в момент (т) 7 отрыва капли; этот ток равен — среднего 6
за весь период жизни капли тока
При решении практических задач (учитывая, что для одного и того же капилляра, из которого истекает ртуть, 2 1
щ3т16 = const) обычно используют линейную зависимость величины диффузионного тока от концентрации [2]:
такт
измерение (выборка)
молоточек
Вкл. развертки
►-время
1^1 где к — константа Ильковича, равная 607и D2v3t6; с — концентрация деполяризатора в растворе.
На рис. 6.21 показана временная диаграмма напряжений, налагаемых на ячейку, на рис. 6.22 — примеры кривых при различной скорости капания ртутно-капельного электрода.
временная диаграмма
Рис. 6.21. Диаграмма напряжений линейной развертки в постоянно-токовой полярографии
Для уменьшения колебаний тока, характерных для момента отрыва капель, в полярографах реализован т. н. таст-режим. В этом случае отсчет значения тока производится в короткое время перед принудительным (с помощью молоточка) сбросом капли. Измеренное значение запоминается электронной схемой полярографа до следующего отсчета
Рис. 6.23. Таст-режим регистрации полярограмм : а — временная диаграмма; б — полярограмма [6]
Ток, протекающий через полярографическую ячейку, является суммой фарадеевского тока I®, обусловленного разрядом деполяризатора на электроде (аналитический сигнал) и емкостного тока 1с, вызванного заряжением изменяющейся в процессе съемки полярограммы емкости двойного электрического слоя индикаторного электрода (помеха). При низких концентрациях деполяризатора соотношение сигнал-помеха ухудшается и становится трудно выделять аналитический сигнал на фоне помехи.
Электрохимические методы анализа
743
Способы улучшения соотношения фарадеевский ток -емкостный ток
Известны три практических способа улучшения соот-
4 ношения —:
4
1) увеличение уровня сигнала /ф;
2) уменьшение емкостного тока 1С\
3) инструментальное разделение /ф и 1С.
Первый способ успешно реализуется в осциллографической и инверсионной полярографии, второй — в импульсной и квадратно-волновой, третий — в синусоидальной переменно-токовой полярографии.
Современные разновидности полярографии [1,3,6-8]
Осциллографическая полярография — вольтамперометрия с быстрой линейной разверткой потенциала.
В этом методе поляризующее постоянное напряжение, изменяющееся по линейному закону, подают в отличие от классической полярографии с высокой скоростью (0,1-1 В/с). Развертку потенциала от некоторой начальной величины включают в определенный момент жизни капли.
Высокая скорость развертки потенциала позволяет зарегистрировать всю полярограмму за время жизни одной капли. Общий вид полярограммы отличается от классической наличием спада тока (рис. 6.24). Это объясняется тем, что поверхность не обновляется и ток, достигнув максимума, уменьшается из-за размывания диффузионного слоя и уменьшения градиента концентрации. Но, поскольку за короткое время регистрации полярограммы фронт диффузии не успевает отойти далеко от поверхности электрода, градиент концентрации высокий. Поэтому /макс на осциллополярограмме заметно выше предельного тока на классической полярограмме.
Рис. 6.24. Сигналы в осциллополярографии: а — временная диаграмма «пилообразной» развертки потенциала; б — осциллополярограмма [3]
Так как за время регистрации полярограммы поверхность остается практически постоянной (электрод можно dA
считать стационарным, ~ 0), емкостный ток значи-
тельно ниже, чем при регистрации «многокапельной» классической полярограммы. Этим и обусловлена более высокая чувствительность осциллографической полярографии: Смин ~ п • 10ч’М. Величина /Макс зависит от скорости развертки потенциала w:
3 £ 2
/макс = kn2AD2w2c,
(6.48)
здесь п — число электронов; А — площадь поверхности электрода; D — коэффициент диффузии, w — скорость развертки потенциала.
С повышением скорости развертки емкостный ток растет
у быстрее фарадеевского (Ic ~ kw в то время, как Лр ~ k w2), форма полярограммы резко ухудшается, поэтому скорость развертки подбирают эмпирически.
Если в какой-то момент времени изменить направление развертки и вернуть потенциал к исходной величине, то вместо пилообразной получают циклическую развертку потенциала (рис. 6.25). То есть за время жизни одной капли регистрируется не только процесс восстановления исходного деполяризатора, но и процесс окисления продукта, полученного при развертке в прямом направлении.
Рис. 6.25. Циклическая полярограмма [8]
Симметричность катодной и анодной ветвей циклической полярограммы указывает на обратимость окислительно-восстановительной системы. Это простой, достаточно надежный и наиболее часто употребляемый способ оценки обратимости (см. 6.5.4).
744
Новый справочник химика и технолога
Импульсная полярография. Для уменьшения 1с поляризующее постоянное напряжение налагают на индикаторный электрод (на каждую образующуюся каплю) отдельными кратковременными импульсами (~ 50 мс), а ток измеряют в конце наложения импульса. В первое мгновение после наложения импульса резко возрастают 1с (за счет заряжения емкости электрода) и 7Ф (из-за начала разряда деполяризатора). Емкостный ток спадает быстрее, чем фарадеевский, поэтому через 20-40 мс после наложения импульса он практически равен нулю, фарадеевский же ток изменяется значительно медленнее. Выбор длительности импульса задержки отсчета от момента его приложения 4 позволяет оптимизировать отношение ~.
Существует два способа наложения импульсов и соответственно две разновидности импульсной полярографии — нормальная и дифференциальная.
Нормальная импульсная полярография. Индикаторный электрод поляризуют линейно увеличивающимися импульсами постоянного напряжения, налагаемыми на постоянный начальный потенциал. Каждый импульс подают на новую каплю, и через 50 мс потенциал возвращается к исходной величине (рис. 6.26а). Нормальная импульсная полярограмма (рис. 6.266) имеет ту же форму, что и классическая (рис. 6.27).
Действующее
молоточек I I________________Г
(обрыв капли)
Рис. 6.26. Временная диаграмма и соответствующая ей нормальная импульсная полярограмма [3, 6]
Зависимость предельного тока от концентрации описывается уравнением
1 2 _1 2
= 460/Ю2 т3 t~2 t3 <6-49)
где п — число электронов; D — коэффициент диффузии; т — скорость вытекания ртути; t3 — время с момента подачи импульса до измерения; /в — время выдерживания электрода при Енач. до подачи импульса. Длительность воздействия приложенного импульса на диффузионный слой электрода здесь существенно меньшая, чем в постояннотоковом режиме (особенно при относительно редких импульсах), что снижает вклад от диффузионного тока более электроположительных компонентов в процессе развертки (возрастания амплитуды налагаемых импульсов). Этот метод особенно эффективен при работе с твердыми индикаторными электродами (из платины, графита и т. п.), когда непрерывная развертка оставляет на твердой поверхности все больше разрядившихся на ней компонентов (слой металлического осадка). Электрод большую часть времени находится под минимальным потенциалом и лишь при наложении импульсов смещается в область разряда компонентов.
Рис. 6.27. Реальные кривые: 7 — нормальная импульсная и 2 — постоянно-токовая (DC) полярограммы, снятые в таст-режиме в тех же условиях, что и кривые на рис. 6.22 и 6.23 [6]
Для нормальной импульсной полярографии величина сн ~ 5-10”7 М, разрешающая способность та же, что у
классической полярографии
= 0,1-0,2В
Дифференциальная импульсная полярография. В этом методе традиционно на линейно увеличивающееся постоянное напряжение (5 мВ/с) через равномерные промежутки времени (в примере на рис 6.28в - 56,7 мс) налагают одинаковые (20-100 мВ) импульсы в течение ~ 20 мс при частоте сети питания 50 Гц и 16,7 мс при 60 гц (рис. 6.1а, в).
Измерение тока проводят дважды: до подачи импульса и в конце действия импульса. Зависимость разности токов
Ы от линейно увеличивающегося постоянного напряжения
выражается кривой с максимумом
^4акс “ j
и называ-
ется дифференциальной импульсной полярограммой (рис. 6.286). Величина сн зависит от обратимости электродного процесса: для обратимых процессов ~ ПГ8 М, для
Электрохимические методы анализа
745
необратимых заметно выше — 5 • ПГ8 М. Разрешающая способность весьма высока, при разности потенциалов пиков 0,04-0,05 В они достаточно хорошо разделяются. На рис. 6.29 для сравнения показаны нормальная (без «таст-сглаживания») и дифференциально-импульсная вольтам-перограммы реальных растворов органических веществ.
б
-0,5 -1,0 Е, В
Рис. 6.28. Временная диаграмма (а) и вид дифференциально-импульсной полярограммы (б)
В современных компьютеризированных дифференциально-импульсных измерениях длительность и частоту наложения импульсов варьируют в широких пределах: /имп. = 1-Ю0 мс, Периода= 10-200 мс. В случае твердых электродов выбор обычно определяется площадью их рабочей поверхности.
Переменно-токовая полярография Известны две разновидности переменно-токовой полярографии: синусоидальная и квадратно-волновая. В первой из них улучшение соотношения — достигается за счет фазовой селекции
токов, во второй — за счет временной селекции. В методе синусоидальной переменно-токовой полярографии поляризующее напряжение является суперпозицией линейно увеличивающегося постоянного напряжения (Es) и пере-
менного напряжения синусоидальной формы с фиксированной частотой (» 50 Гц) и амплитудой (ДЕ ~ 10 мВ):
Е - Es + АЕ sin wt.
(6.50)
О -0,1 -0,2-0,3-0,4-0,5-0,6-0,7-0,8-0,9-1,0-1,1-1,2-1,3-1,4-1,5
Потенциал [Е vs S. С. Е.]
—।----1—।—I—।—।—।—।—।—।—।—।—I—।—
О -0,1 -0,2-0,3-0,4-0,5-0,6-0,7-0,8-0.9-1,0-1,1 -1,2-1,3-1,4 -1,5
Потенциал [Е vs S. С. Е.]
Рис. 6.29. Сравнение дифференциально-импульсной полярограммы с постоянно-токовой (без таст-сглаживания), снятых в идентичных условиях [7]
В результате через ячейку протекает и постоянный, и переменный ток. Возникновение переменного тока обусловлено периодическими изменениями концентрации окисленной и восстановленной форм деполяризатора вслед за изменением потенциала электрода (AEsincoZ) относительно номинального значения; за один полупериод увеличивается концентрация восстановленной формы, а за другой — окисленной формы. Аналитическую информацию в данном случае несет только переменный ток. Протекающий через ячейку переменный ток имеет ту же частоту, что и переменное модулирующее напряжение, но сдвинут по фазе на угол 9:
=/(E)sin(W + 9), (6.51)
причем 9 зависит от степени обратимости электродного процесса и для обратимых процессов 9 = 45°. Только в последнем случае возможно четкое разделение фарадеевского и емкостного токов.
Амплитуда переменного тока модулирована по постоянному напряжению, и при соответствующей обработке
746
Новый справочник химика и технолога
сигнала ток плавно изменяется вслед за линейным изменением постоянного напряжения. График зависимости амплитуды переменного тока от величины линейно ме
няющегося постоянного поляризующего напряжения называют переменно-токовой полярограммой (рис. 6.30 иллюстрирует вышесказанное для обратимого электродного процесса). Амплитуда переменного тока достигает максимальной величины при потенциале полуволны на классической полярограмме. Характеристиками переменнотоковой полярограммы являются потенциал пика Ет ширина пика на половине высоты и ток I или высота
пика. Высота пика линейно зависит от концентрации де
поляризатора. Величина Ev
72
зависит от числа электронов
90
и для обратимого электродного процесса Е}/ = — мВ
(25 °C); Для необратимого электродного процесса 90
Еу > — мВ, пик несимметричен и плохо выражен (ток
в случае обратимого восстановления деполяризатора в методе квадратно-волновой полярографии на порядок ниже (5-10 8М), чем в синусоидальной переменно-токовой полярографии. Разрешающая способность АЕР ~ 50 мВ.
мал), в этом случае переменно-токовая полярография не имеет преимуществ перед классической. Минимальная определяемая концентрация при обратимом восстановлении деполяризатора порядка 5 • 10“7 М. Разрешающая способность метода АЕР ~ 50 мВ.
/, мкА
I, мкА
Рис. 6.30. Принцип переменно-токовой полярографии.
Индексы ас и de относятся соответственно к переменно-токовой и постоянно-токовой составляющим сигнала
-1,65 2,4 1,6 0,8
в Содержание цинка, мг
б
Рис. 6.31. Трехмерные сплайн-экстраполяции вольтамперограмм серебра(1) и золота(Ш)
Современная компьютерная обработка переменнотоковых полярограмм позволяет строить трехмерные зависимости 1-Е-$ (т. е. формы 7-Е-пиков от фазового угла (рис. 6.31) [9, 10] и с их помощью выбирать условия регистрации переменнотоковых вольтамперограмм). Оптимальное значение фазового угла для условий съемки вольтамперограммы (например, в процессе разработки методики анализа) можно выбрать, сравнивая сечения, перпендикулярные оси ф.
В методе квадратно-волновой переменно-токовой полярографии линейно изменяющееся постоянное напряжение модулируют прямоугольными импульсами переменного напряжения. Как и в методе импульсной полярографии, используют временную селекцию фарадеевского и емкостного тока, измеряя ток в конце действия импульса (рис. 6.32). Временная селекция токов дает лучшие результаты, и поэтому минимальная определяемая концентрация
Ввиду токсичности ртути и в связи с новейшими достижениями в области электрохимического концентрирования вещества на поверхности индифферентных твердых электродов, традиционные полярографические методы анализа вытесняются вольтамперометрическими на твердых электродах. Прямая вольтамперометрия методически и инструментально аналогична полярографии. Индикаторный электрод изготавливают из индифферентного электропроводящего материала (платины, золота, серебра, графита и других углеродных материалов).
Наиболее радикальный способ улучшения соотношения фарадеевский ток - емкостный ток реализуется в методе инверсионной вольтамперометрии. Существенное (многократное) увеличение фарадеевского тока происходит за счет электроконцентрирования определяемых компонентов раствора на поверхности или в объеме (в случае стационарной ртутной капли или амальгамного покрытия) индикаторного электрода (см. п. 6.5).
Электрохимические методы анализа
747
Рис. 6.32. Форма налагаемого переменного напряжения (а) и изменение фарадеевского и емкостного тока (б), общий вид развертки потенциала (в) в методе квадратно-волновой полярографии
6.3.2. Хронопотенциометрия
Методы электрохимического анализа, основанные на определении зависимости величины электрического сигнала от времени относят к так называемым хронометодам. В настоящее время в аналитической практике получила распространение хронопотенциометрия [1].
В методе хронопотенциометрии потенциал индикаторного электрода измеряют как функцию времени, при этом ток, протекающий через ячейку, поддерживают постоянным (либо изменяющимся во времени по заданному закону). Теория хроно потенциометрических измерений была разработана Сэндом в начале XX в. Этот метод находился в центре внимания исследователей с 1953 по 1970 г.; в настоящее время популярностью пользуется автоматический анализ на основе инверсионнной хронопотенциометрии (см. 6.5.3).
Преимущество хронопотенциометрического метода состоит в том, что измеряемой величиной в нем является время— фактически длительность волны потенциала, по форме сходной с полярографической волной. Поскольку время легко оцифровывается, то процесс измерения легко автоматизировать, даже промежуточные результаты эксперимента можно без труда передавать по современным линиям связи.
Теория хронопотенциометрии
Уравнения, описывающие процессы, происходящие при хронопотенциометрических измерениях, выведены из предположения о постоянстве силы тока, протекающего через ячейку, и допущения, что массоперенос осуществляется только путем полубесконечной линейной диффузии к плоскому электроду с постоянной площадью поверхности. В этих условиях в результате диффузии у поверхности электрода возникает изменяющийся во. времени градиент концентраций, приводящий к изменению потенциала индикаторного электрода.
Если для хронопотенциометрических измерений применяют не плоские электроды, а, например, цилиндрическую проволоку или сферический электрод в виде висящей капли ртути, то для учета кривизны поверхности электрода используют теорию полубесконечной цилиндрической или сферической диффузии. Для реакции восстановления обычными граничными условиями являются следующие:
CRed,x,0 0’ СОх.х,0 СОх’ CRcd,V °’ ПРИ * притих,
(6.52)
Эти граничные условия приводят к дифференциальному уравнению, решенному Сэндом и другими исследователями
£ = £,z+RTln
/zF ,
(6.53)
где параметр Р определяется выражением
(6.54)
;t2zFJD20x
а значение является обычным полярографическим по-
/2
тенциалом полуволны. Теоретически наблюдаемый потенциал должен стремиться к бесконечности, как только числитель подлогарифмического выражения будет приближаться к нулю. Это произойдет в момент времени, отвечающий так называемому переходному времени т, равному
т = (6.55)
Вводя в уравнение (6.53) вместо сОх соотношение, полученное из выражения (6.55), получаем
RT
(6.56)
zT^ln т2 — t2 \!t2
748
Новый справочник химика и технолога
Это уравнение по форме аналогично уравнению Гейров-ского-Ильковича, которое представляет потенциал электрода как функцию диффузионного тока и фактического тока для одного и того же электродного процесса.
Объем: [Cd(II)] = [РЬ(П)] cd(II)
Время —
б
Рис. 6.33. Соотношение полярограммы а и хронопотенциограммы б [1 ]
Взаимосвязь между полярограммой и хронопотенцио-граммой становится ясной из рис. 6.33. По прошествии времени, большего, чем переходное время т, потенциал не возрастает до бесконечности, как предсказывает теория для одностадийного процесса. Потенциал электрода при восстановлении фактически увеличивается только до тех пор, пока на электроде после восстановления не начнется некоторый другой процесс, например восстановление растворителя или индифферентного электролита.
Для упрощенного случая, когда концентрация окисленного вещества у поверхности электрода становится равной нулю (с = 0), из вышеприведенных уравнений Сэндом было выведено уравнение
t~ логарифмический член уравнения (6.56) равен нулю.
В полярографии два электрохимических процесса, аналогичных приведенным на рис. 6.33, дают на полярограмме две волны. Первая из этих волн продолжается в течение всего процесса измерения, т. е. вторая волна накладывается на первую. Диффузионно-ограниченный ток второй волны получают путем вычитания диффузионно-ограниченного тока первой волны из наблюдаемого суммарного диффузионного тока, зафиксированного на полярограмме.
В хронопотенциометрии положение полностью анало-
2 гично наблюдаемому в полярографии, поскольку т2, как и /д, пропорционально концентрации диффундирующих частиц в объеме раствора. На хронопотенциограмме также имеются две волны, как это показано на рис. 6.33.
Ситуация существенно изменяется, если измерительной гальваностатической стадии предшествует стадия предварительного электролиза — накопления определяемых компонентов раствора на поверхности индифферентного электрода (см. п. 6.5.3). В этом случае каждая последующая ступенька-волна хронопотенциограммы начинается только после полного израсходования вещества предыдущего компонента на электроде. При этом переходное время т прямо пропорционально количеству электроактивного компонента на поверхности электрода, которое в свою очередь, прямо пропорционально его концентрации в растворе.
6.3.3. Потенциалы полярографических полуволн в различных фоновых электролитах
Ix^zVAc(nDT2
2
(6-57)
6.3.3.1. Потенциалы полярографических полуволн неорганических веществ
В табл. 6.18-6.40 приведены потенциалы полуволн Е,,
/2
которое является хронопотенциометрическим аналогом уравнения Ильковича. Уравнение Сэнда можно преобразовать таким образом, чтобы его левая часть содержала 2
т2
член — , который известен как константа переходного с
т
времени. При подстановке значения г= в уравнение (6.56)
находим, что поверхностная концентрация деполяризатора равна точно половине его объемной концентрации. Следовательно, потенциал этой точки Ех/ соответствует поляро-уч
графическому потенциалу полуволны для реакций
восстановления на ртутных электродах, поскольку при
полярографического восстановления или окисления неорганических веществ на ртутном капельном электроде (в
специально оговоренных случаях — на платиновом элек
троде) при 25 °C. Величины Еу
/2
выражены в вольтах от-
носительно насыщенного каломельного электрода. В первой графе (столбце) указаны определяемые ионы, во второй — число электронов, участвующих в электродном
процессе: положительные значения свидетельствуют о катодном, отрицательные (-ан.) — об анодном процессе. Сокращение «не восст.» означает, что в данном растворе определяемый ион восстанавливается позже вещества фо
на.
Значения Еу
/2
для небольших полярографических волн
помещены в скобках.
Электрохимические методы анализа
149
Таблица 6.18
Фоновый электролит НС1 (1 М)
Определяемое вещество (ион) n
А13+ He восст. —
As(III) 3 0,43
6 -0,6 (макс.)
As(V) He восст. —
Bi3+ 3 -0,09
Cd2+ 2 -0,64
Ce4+ 1 +0,1
Cr3+ 1 -0,99
3 -1,26
Cr(VI) 3 -0,78
Cu2+ 1 +0,04
2 -0,22
Fe3+ 1 >0
Ga3+ 3 -1,2 (0,001 МНС1)
Ge2+ 2 -0,35
Ge(IV) He восст. —
Hg2+ 2 >0
Hg2+ 2 >0
In3+ 3 -0,56 (0,1 МНС1)
Mo(VI) 1 -0,14 (0,3 МНС1)
3 -0,53
Nb(V) (no;) Восст. (no;) -0,76
O2 2 -0,05
4 -0,94
Pb2+ 2 -0,44
Pu 1 0,40
Re(IV) 1 -0,53 (2,4 МНС1)
Re(Vn) -0,40 (2,4 МНС1)
SO2 -2 (ан.) -0,6
2 -0,37
Sb(III) 3 -0,15
Sb(V) 2 >0
5 -0,35
4 -0,55
Se(IV) He восст. —
Se(VI) -2(ан.) ИЛ)
Sn2+ 2 -0,47
2 -0,1
Sn(IV) 4 -0,47
Ta(V) 1 -1,16 (0,9 МНС1)
Ti(IV) 1 -0,81 (0,1 МНС1)
Продолжение таблицы 6.18
Определяемое вещество (ион) п
Т1+ 1 -0,48
Т13+ 2 >0,3 (0,6МНС1)
3 -0,45
U(VI) 2 -0,20 (2МНС1)
3 -0,9 (2МНС1)
V(V) 1 >0(0,1МНС1)
3 -0,80
W(VI) Не восст. —
Таблица 6.19
Фоновый электролит НС1 (12 М)
Определяемое вещество (ион) n ЕУ
Ag4 1 >0
As(III) 3 -0,55
As(V) 5 >0
8 -0,52
Bi3+ 1 -0,45
3 -0,57
Cd2+ He восст. —
Ce4+ 1 -0,68
Cr3+ He восст. —
Cr(VI) 3 -0,61
Cu2+ 2 -0,71
Fe2+ He восст. —
Fe3+ 1 >0
3 -0,65
In3+ 3 -0,77
Mn2+ He восст. —
Mo(VI) 2 -0,56
Nb(ni) -2 (ан.) -0,32
Nb(V) 1 -0,40
-0,67
Ni2+ 2 -0,80
o2 2 0
4 -0,7
Os(VIII) 5 >0,11
Pb2+ 2 -0,90
Ru(IV) 1 >0
2 -0,51
3 -0,62
4 -0,75
Sb(IH) 1 -0,52
3 -0,66
750
Новый справочник химика и технолога
Продолжение таблицы 6.19
Определяемое вещество (ион) n ЕУ
Sb(V) 2 >0
-0,48
-0,57
Sn2+ 2 -0,83
Sn(IV) 2 -0,50
4 -0,83
Te(VI) 1 >0
6 -0,43
-0,79
ТГ He восст.
U(IV) 1 >0
2 -0,62
-0,75
U(VI) 2 >0
3 -0,63
W(VI) 1 >0
3 -0,54
Zn2+ He восст. —
Таблица 6.20
Фоновый электролит НС1О4 (0,1 М) или NaClO4
Определяемый ион n ЕУг
Ag+ 1 >0
Bi3+ 3 0,22
Cd2+ 2 -0,64
Ce4+ 1 >0(0,001 MHC1)
Cr3+ 1 -1,46 (0ДМНС1)
Cu2+ 1 0,01 (0,001 MHC1)
Eu2+ -1 (ан.) -0,47
Eu3+ 1 -0,66
io; 2 >0
io; 6 -0,04
Mo(VI) 1 -0,03
-0,24
-0,77
Ni2+ 2 -1,01
Np(IV) 1 -0,10(1 MHC1O4 или NaClO4)
Os (VHI) 5 >0,10(1 МНСЮ4 или NaClO4)
Pb2+ 2 -0,38(1 МНСЮ4 или NaClO4)
Re3+ 1 -0,28
3 -0,46
Продолжение таблицы 6.20
Определяемый ион n ЕУ
Re(VII) 3 -0,40
Ru(IV) 1 >0
2 -0,21
Sb(V) He восст. —
Sn2+ -2 (ан.) 2 +0,13(1МНСЮ4 или NaClO4) -0,43
Sn(IV) He восст. —
Ti(IV) 1 -0,81
ti+ 1 -0,46
Ti3+ 2 >0(1 МНСЮ4 или NaClO4)
3 -0,48 (1 МНСЮ4 или NaClO4)
U(IV) 1 -0,93
1 -0,18 0,5 MNaClO4 + 0,01 М НС1О4
U(VI) 3 -0,84
Zn2+ 2 -1,00(1 МНСЮ4 или NaClO4)
Таблица 6.21
Фоновый электролит Н3РО4 (73 М)
Определяемый ион n ЕУг
А13+ He восст. —
As(III) 3 -0,46
6 -0,71
As(V) He восст. —
Bi3+ 3 -0,15
Cd2+ 2 -0,77
Ce3+ He восст. —
Ce4+ 1 >0
Co2+ 2 -1,20
Cr3+ 1 -1,02
Cr (VI) 3 >0
Cu2+ 2 -0,09
Fe2+ He восст. —
Fe3+ 1 +0,06
Mn2+ He восст. -—
Ni2+ 2 -1,18
o2 2 -0,2
4 -0,6
Os(VIII) >0,6
Pb2+ 2 -0,53
Электрохимические методы анализа
751
Продолжение таблицы 6.21
Определяемый ион n ЕУ
SO32' 1 -0,67 Na2HPO4 + + KH2PO4; pH = 6; 1-я волна отвечает восстановлению SO2 до S2O2-, 2-я до S2O2
2 -1,23
Sb(HI) 3 -0,29
Sb(V) He восст.
Sn2+ 2 -0,58
Sn(IV) (-0,65)
Te(VI) -0,87
ti+ 1 -0,63
U(VI) 1 -0,12
-0,58
V(V) 1 >0
5 -0,54
-0,91
W(VI) 1 -0,59
Zn2+ 2 -1,13
Таблица 6.22
Фоновый электролит НС1 (2 М) + NaH2PO2 (0,1 М)
Определяемый ион n ЕУг
Ag+ 1 >0
Al3+ He восст.
Bi3+ 3 -0,11
Cd2+ 2 -0,72
Со2+ He восст. —
Cu+ 1 -0,20
Cu2+ (в присутствии NaH2PO4 восстанавливаются до Cu+) 1 -0,20
Fe2+ He восст. —
Fe3+ He восст. —
Ga3+ He восст. —
Ge2+ -2 (ан.) -0,18 (4 М НзРО2)
2 -0,57 (4 М Н3РО2)
In3+ 3 -0,65
Mn2+ He восст. —
Ni2+ -0,67
Pb2+ 2 -0,48
Sb(III) -2 (ан.) -0,57
3 -1,25
Продолжение таблицы 6.22
Определяемый ион n Ey,
Sb(V) He восст. —
Sn2+ 2 -0,5
Sn(FV) -0,3
Th4+ He восст. —
Ti(IV) 1 >0
Tl+ 1 -0,52
Zn2+ He восст. —
Таблица 6.23
Фоновый электролит NaOH (1 М)
Определяемый, ион n ЕУ
Al3+ He восст. —
As(III) -2 (ан.) -0,26 (0,5 M KOH)
3 -1,8
As(V) He восст. —
Au(III) 1 -0,2 (2MKOH)
2 -0,4 (2MKOH)
3 -1,1 (2MKOH)
Bi3+ 3 -0,6
Bro; 6 -1,78
Cd2+ 2 -0,78
C1O2 4 -1,02
C1O3 He восст. —
cio; He восст. —
Co2+ 2 -1,46 (1 MKOH)
Cr2+ -1,9
Cr(VI) 3 -0,85
Cu2+ 2 -0,41
Fe2+ -1 (ан.) -0,9 (добавляют маннит)
Fe3+ 1 -0,9 (0,1 MNaOH)
3 -1,5
Ga3+ He восст. —
H2O2 -2 (ан.) -0,18
2 -1,0
In3+ 3 -1,09
jo; 6 -1,20
Mn2+ 2 -1,70
Mo(VI) He восст. —
o2 2 0,17
4 -1,2
752
Новый справочник химика и технолога
Продолжение таблицы 6.23
Определяемый, ион n
Os(VI) 2 -0,61
3 -1,54
Os(VIII) 4 -0,63
5 -1,54
Pb2+ 2 -0,76
-2 (ан.) -0,50
Pd(II) 2 -1,41 (2 М NaOH)
Re(VII) -1,5
Sb(III) -2 (ан.) -0,45
3 -1,15
Sb(V) He восст. —
Se(IV) He восст. —
Se(Vl) He восст. —
Sn2+ -2 (ан.) -0,73
2 —
Sn(IV) He восст. —
Ta(V) He восст. —
Te2+ ... (ан.) -0,42
... (ан.) —
Te(IV) 6 -1,19
Te(IV) 6 -1,19
Te(VI) 8 -1,57
Ti(IV) Не восст.
Tj+ 1 -0,48 (С Pt-электродом)
V(III) -2 (ан.) -0,38
V(IV) -1 (ан.) -0,43
V(V) 3 -1,7
W(VI) Не восст. —
Zn2+ 2 -1,53
Таблица 6.24
Фоновый электролит КС! (0,1 М) или LiCl, NH4C1, [(CH3)4N]C1
Определяемый, ион п
А13+ 3 -1,75
Ва2+ 2 -1,92
Ве2+ 2 -1,8 (Без фонового электролита)
вго; 6 -1,78
Са2+ 2 -2,20
Cd2+ 2 -0,60
Се3+ 3 -2,0
Се4+ 1 >0
Продолжение таблицы 6.24
Определяемый, ион n ЕУг
СЮ' 2 >0
сю; He восст. —
сю; He восст. —
Со2+ 2 -1,20
Сг24 -1 (ан.) -0,5
Сг3+ 1 -0,60
3 -0,80
-3 (ан.) -1,50
Cr(VI) -0,3
3 -1,0
4 -1,55
6 -1,8
Cs+ 1 -2,1 (0,1 М [(CH3)4N]C1)
Cu2+ 1 +0,04
2 -0,22
Dy3+ 3 -1,85
Er3+ 3 -1,85
Eu2+ -1 (ан.) -0,54
Eu3+ 1 -0,69
3 -2,47 (Без фонового электролита)
Fe2+ 2 -1,3
[Fe(CN)6]4' -1 (ан.) >0
Ga3+ 3 1,1
Gd3+ 3 1,75
H+ 1 -1,58 (0,007 М LiCl, pH = 3)
H2O2 2 -0,94
Hg -1 (ан.) -0,2
Hg2+ 2 >0
Hg2* 2 >0
Ho3+ 3 -1,85
In3+ 3 -0,56 (0,1 М [(CH3)4N]C1)
jo; 6 -1,2
K+ 1 -2,13
La3+ 3 -1,9
Li+ 1 -2,35
Lu3+ -1,8
Mg2+ 2 -2,30
Электрохимические методы анализа
753
Продолжение таблицы 6.24
Определяемый, ион n ЕУ
Мп2+ 2 -1,51
Mn(VII) 5 >0
Mo(VI) He восст. —
NH/ 1 -2,21
[N(C2H5)4]+ -2,67 (Без фонового электролита)
NO3 (см. также Th4+и U VI) -2,10
Na+ 1 -2,12 (0,1 М [(CH3)4N]C1)
Nb(V) 1 -1,28
-0,80 (LiCl или [(CH3)4N]I; 0,001 н. H2SO4)
Nd3+ 3 -1,83
Ni2+ 2 -1,1
o2 2 -0,06
4 -0,94
OH’ -1 (ан.) -0,08
Pb2+ 2 -0,40
Pr3+ 3 -1,8
Pt(II) 2 >0
Pt(IV) 1 -0,1
2 -i,o
Ra2+ 2 -1,84
Rb+ 1 -2,03
Re(VII) 3 -1,7
[SbS4]3’ 5 -1,34
Sc3+ 3 -1,80
Se(IV) -1,8 (LiCl или [(CH3)4N]I; 0,001 н. H2SO4
Se(VI) He восст. —
Sm3+ 1 -1,8
3 -1,97 (LiCl или [(CH3)4N]I; 0,001 н. H2SO4
Sr2* 2 -2,10
Te(VI) (-1,1)
2 -1,5
Tb3+ 3 -1,85
Th4* Восст. NO3 -i,o
Продолжение таблицы 6.24
Определяемый, ион n Ey,
ti* 1 -0,46
Tl3* 3 >0
Tu3* 3 -1,85
U(IV) 1 -0,93
U(IV) Восст. NO3 -1,0
W(VI) He восст. —
Y3* 3 -1,8
Yb3* 1 -1,17
3 -2,0
Zn2* 2 -1,00
Zr4* 4 -1,65
Таблица 6.25
Фоновый электролит СаС12 (5 М)
Определяемое вещество (ион) n ЕУ
BrO3 6 -1,52
Cd2* 2 -0,80
Co2* 2 -0,82
Cr3* 1 -0,51 (насыщ. р-р СаС12)
Cu2* 1 >0
Eu2* 2 -0,33
-1 (ан.) -0,54
Fe3* 1 >0
3 -1,20
H+ 1 1,608 (0,5 М СаС12; РН = 3)
io; 6 -0,9
Mn2* 2 -1,45
Mn(VII) 7 -1,5
Ni2* 2 -0,56
NO3“ -1,74
Pb2+ 2 -0,53
Ti(IIl) -1 (ан.) -0,12 (насыщ. р-р СаС12)
Ti(IV) 1 -0,12 (насыщ. р-р СаС12)
754
Новый справочник химика и технолога
Таблица 6.26
Фоновый электролит К2СО3 (2 М); pH = 11
Определяемое вещество (ион) n ЕУ
As(III) He восст. —
Bi3+ 3 -0,64
Cd2+ 2 -0,74
Се3+ -1 (ан.) -0,16
Се4+ 1 -0,16
Со2+ He восст. —
Сг3+ He восст. —
Cr(VI) 3 -0,47
Cu2+ 2 -0,24
Eu3+ -1 (ан.) -0,53
Fe3+ 1 -0,86
Ga3+ He восст. —
Gd3+ He восст. —
Ge(VI) -1,44
Hg2* 2 >0
In3+ 3 -1,24
Mn2+ -1 (ан.) -0,1
Mo(VI) Не восст. —
Nd3+ Не восст. —
Ni2+ 2 -1,2
Pb2+ 2 -0,66
Pr3+ Не восст.
Sb(III) 3 -1,1
[SbS4]3 5 -1,30
Sc3' Не восст.
Sm3+ Не восст.
Sn(IV) -1,15
Te(VI) 8 -1,37 (pH = 8,5)
Th4+ Не восст.
Ti(IV) 1 -1,5
Tl+ 1 -0,47
U(VI) -1,01
V(V) 1 -1,08
V(VI) -1 (ан.) -0,16 (0,1 MNaHCO3)
W(IV) Не восст.
Zn2+ 2 -1,20
Таблица 6.27
Фоновый электролит
NH4OH (1 М) + NH4C1 (1 М) + желатина (0,005%)
Определяемое вещество (ион) n ЕУ
Ag* 1 >0
As(III) -1,41
-1,63
Au(III) 3 >0
BrO3 6 -1,72
Cd2+ 2 -0,81
Co2+ 2 -1,29
Co3+ 1 -0,54 (2,5 MNH4OH + 0,lMNH4Cl)
3 -1,36
Cr3+ 1 -1,43
3 -1,71
Cr(VI) 3 -0,2
6 -1,6
Cu+ -1 (ан.) -0,22
1 -0,50
Cu2+ 1 -0,24
2 -0,51
Fe2+ -1 (ан.) -0,34
2 -1,49
Ga3+ 3 -1,6
Ge(IV) 2 -1,45 (0,5 MNH4OH + 1 М NH4CI)
1г(Ш) и (IV) He восст. —
Mn2+ 2 -1,66
Mo(VI) 1 -1,71
Ni2+ 2 -1,10
Pd(II) 2 -0,75
Pt(II) He восст. -—
Rh(III) 2 -0,93
Ru He восст. —
Se2 -2 (ан.) -0,84
Se(IV) 6 -1,53
Se(VI) He восст. —
Sn(IV) 2 >0,3
4 -0,52
Те2 -2 (ан.) -1,1
Te(IV) 4 -0,67
Te(VI) 8 -1,21
ti+ 1 -0,48
U(VI) 1 -0,8
3 -1,4
Электрохимические методы анализа
755
Продолжение таблицы 6.27
Определяемое вещество (ион) n ЕУ,
V(IV) -1 (ан.) -0,32
2 -1,28
V(V) 1 -0,96
3 -1,26
W(VI) He восст. —
Zn2+ 2 -1,35
Таблица 6.28
Фоновый электролит
N2H4 (1 М) + NH4OH (1 М) + NH4C1 (1 М)
Определяемое вещество (ион) n Ey
Bi3+ 3 -1,54
Cd2+ 2 -0,84
Со2+ 2 -1,29
Сг2+ 1 -1,40
Fe2+ 2 -1,47
Fe3+ 1 -1,09
3 -1,49
In3+ -1,15
Mn2+ He восст. —
Mo(VI) He восст. —
Ni2+ He восст. —
Pb2+ 2 -0,77
Sb(III) 3 -1,21
Sb(V) He восст. —
Sn2+ -2 (ан.) -0,851
2 -1,219
-1,57
Sn(IV) He восст. —
Te(VI) 1 -1,21
ТГ 1 >0
U(VI) -0,98
V(V) He восст. —
W(VI) He восст. —
Zn2+ 2 -1,52
Таблица 6.29
Фоновый электролит
C5H5N (0,1 М) + C5H5N • НС1 (0,1 М)
Определяемое вещество (ион) n ЕУ
Ag+ 1 >0
As(III) -0,90
As(V) He восст. —
Продолжение таблицы 6.29
Определяемое вещество (ион) n ЕУг
Bi3+ 3 -1,00
Cd2+ 2 -0,62
Ce4+ He восст. —
Co2+ 2 -1,06
Cr(VI) 3 >0
4 -1,20
Cu2+ 1 >0
2 -0,22
Eu2+ -1 (ан.) -0,49
Fe2+ He восст. —
Fe3+ He восст. —
In3+ He восст. —
Mn2+ He восст. —
Ni2+ 2 -0,75
Os(VII) 2 >0,3
Pb2+ 2 -0,40
Pd(II) 2 -0,31 (1 MC5H5N + I M KC1.)
Rh(lII) 2 -0,80
Sb(III) -0,47
Sb(V) He восст.
Sn2+ 2 -1,12
Sn(IV) He восст. —
Te(VI) He восст. —
ti+ 1 -0,451
U(VI) 1 -0,23
2 -0,45
3 -0,98
V(V) 1 -0,11
-0,43
3 -0,77
W(VI) He восст. —
Zn2+ 2 -1,03
Таблица 6.30
Фоновый электролит
СН3СООН (2 М) + CH3COONH4 (2 М) + желатина (0,01%)
Определяемое вещество (ион) n ЕУ
Ag+ 1 >0,1
Al3+ He восст. —
As(III) 3 -0,92
As(V) He восст. —
Au(III) >0,5
756
Новый справочник химика и технолога
Продолжение таблицы 6.30
Определяемое вещество (ион) n ЕУг
Bi3+ 3 -0,25
Cd2+ 2 -0,65
Со2+ 2 -1,19 (0,1 M CH3COONa; pH-5)
Ст3+ 1 -1,2
Си2+ 2 -0,07
Еи3+ 1 -0,81 (0,1 M CH3COONa; PH = 5)
Fe2+ He восст. —
Fe3+ 1 >0
Ga3+ He восст. —
Hg2+ 2 >0
In3+ 3 -0,71
Ir(IV) 3 >0(0,1 M CH3COONa; pH = 5)
IO3’ 6 -0,5
Mn2+ He восст. —
Mo(VI) -0,6
-1,1
-1,2
Ni2+ 2 -1,1
Pb2+ 2 -0,50
Pd(II) 2 -0,6
Pt(II) 2 >0,4 (0,1 M CH3COONa; PH = 5)
Pt(IV) > 0 (0,1 M CH3COONa; pH =5)
Rh(III) 3 >0(0,1 M CH3COONa; pH = 5)
Ru(III) 1 >0(0,lM CH3COONa; pH-5)
so2- -0,54
Sb(III) -0,40
3 -0,59
Sn2+ -2 (ан.) -0,16
2 -0,62
Sn(IV) 2 -1,1
Te(IV) 4 -0,48 (pH = 4,4)
Te(VI) 8 -1,18
Продолжение таблицы 6.30
Определяемое вещество (ион) n
Ti(IV) 1 -0,85 (0,2 M CH3COOH; pH = 5,1)
—1,50
ti+ 1 -0,47
U(IV) -1 (ан.) -0,12 (pH = 4,4)
1 -0,45
1 -1,2
V(V) 1 >0
W(VI) (-0,70)
Zn2+ 2 -1,1
Zr(IV) He восст. —
Таблица 6.31
Фоновый электролит NaF (1 М) + желатина (0,01%)
Определяемое вещество (ион) n ЕУ
Ag+ 1 >0
Au(IIl) 3 >0
Bi3+ 3 -0,11 (pH = 0,7)
Cd2+ 2 -0,64
Co2+ 2 -1,3 8 (pH = 3-6)
Cr(VI) 1 -0,26 (pH=9,3)
3 -1,1
Cu2+ 2 ±0,00 (pH = 5)
Eu2+ -1 (ан.) -0,10
Eu3+ 1 -1,1 (насыщенный раствор NaF)
Fe2+ -1 (ан.) +0,11 (0,1 M KF)
Fe3+ 1 -0,74 (pH = 7)
3 -1,44
Ga3+ 3 -1,38
Ge(IV) He восст. —
Hgf 2 >0
Hg2+ 2 >0
Ю 2 -0,6
Ir(IV) -1,4
Mn2+ 2 -1,55 (pH = 3-6)
Nb(V) 1 -1,90 (3MNH4F)
Ni2+ 2 -1,12
Pb2+ 2 -0,41 (pH= 13)
Электрохимические методы анализа
757
Продолжение таблицы 6.31
Определяемое вещество (ион) n
Re’ ...(ан.) * +0,03
...(ан.) * -0,26
...(ан.) * -0,42
...(ан.) * -0,54
Rh(III) 2 -1,0
Sb3+ 3 (-0,68)
Se(IV) -0,80
Sn2+ -2 (ан.) -0,20 (pH = 4-6)
2 -0,73
Sn(IV) Не восст. —
Ta(V) -0,95 (насыщенный раствор NaF)
Te(IV) 4 -0,89
T|+ 1 -0,50
U(VI) 1 -0,37
2 -0,94
3 -0,21 (0,01 MHF)
Zn2+ 2 -1,14 (pH = 4-6)
Примечание* Нет точных данных.
Таблица 6.32
Фоновый электролит Н2С2О4 (0,25 М) и Na2C2O4 или (NH4)2C2O4; pH = 3-4
Определяемое вещество (ион) n
As(III) 6 -0,79
Cd2+ 2 -0,63
Cr3+ 1 1,0 (0,1 M K2C2O4; pH = 5,1)
2 -1,15 (0,1 M K2C2O4; pH = 5,1)
Cu2+ 2 -0,15
Fe2+ -1 (ан.) -0,23
Fe3+ 1 -0,23
Ge(IV) He восст. —
Mo(VI) -0,35
Nb(V) -1,53 (pH = 7)
Pb2+ 2 -0,50
Sb(III) -0,51
Sn2+ -0,7
Sn(IV) 4(?) -1,51
Продолжение таблицы 6.32
Определяемое вещество (ион) n
Ta(V) 1 -1,40 (0,5 M K2C2O4); pH = 0,5-3)
Te(VI) 8 -1,23 (насыщенный раствор (NH4)2C2O4)
Ti(lV) 1 -0,37
ti+ 1 -0,46
U(VI) 2 -0,13 (0,5 M H2C2O4)
-0,44
V(V) 1 > 0 (1 M K2C2O4; pH = 4,6)
3 -1,33
V(IV) -1 (ан.) -0,1
Zn2+ 2 -1,3
Таблица 6.33
Фоновый электролит Н2С4Н4Об (винная кислота) (0,25 М) или KNaC4H4O6; pH = 3-4
Определяемое вещество (ион) n
Al3+ 3 -1,79
As(III) -0,62
6 -1,0
As(V) He восст. —
Bi3+ 3 -0,14
Cd2+ 2 -0,59
Co2+ -1,58
Cr3+ 1 -1,00
Cu2+ 2 -0,03
Eu3+ 1 -1,08
Fe2+ -1 (ан.) -0,12
Fe3+ 1 -0,12
3 -1,50
Ga3+ He восст. —
Ge(IV) He восст. —
ln3+ 3 -0,58
Nb(V) -1,96 (1 M К2С4Н4О6; pH = 6,5)
Ni2+ 2 -1,13
Pb2+ 2 -0,42
758
Новый справочник химика и технолога
Продолжение таблицы 6.33
Определяемое вещество (ион) n
Sb(in) 3 -0,56
Sn2+ -2 (ан.) -0,18
2 -0,49
Sn(IV) He восст. —
Ta(V) 1 -1,57 (0,1 М К2С4Н4О6; pH = 3-5)
Th4+ He восст. —
Ti(IV) 1 -0,61
ti+ 1 -0,45
W(VI) Каталитическая волна водорода -1,63
Yb3+ Не восст. —
Zn2+ 2 -1,20
Таблица 6.34
Фоновый электролит Na2C4H4O6 (0,5 М);
желатина (0,01%), pH - 9
Определяемое вещество (ион) n
As(III), (V) He восст. —
Bi3+ 3 -0,70
Cd2+ 2 -0,64
Со3+ 1 -0,31 ([Co(NH3)6]3+ в 1 М К2С4Н4О6)
3 -1,32
Cu2+ 2 -0,30 (pH = 10)
Fe2+ -1 (ан.) -0,6
2 -1,6
Fe3+ 1 -1,20
3 -1,73
Mn2+ -1 (ан.) -0,4
2 -1,7
Mn(IV) 1 -1,1
2 -1,3
4 -1,7
Mo(VI) He восст. —
Pb2+ 2 -0,7
Rh(III) He восст. —
Sb(III) 1 -1,0
3 -1,2
Sn2+ -2 (ан.) -0,33
2 -0,92
Sn(IV) Не восст. —
Zn2+ 2 -1,15
Таблица 6.35
Фоновый электролит Н3СбН5О7 (лимонная кислота) или К3С6Н5О7, или (NH4)3C6H5O7; pH = 3-4
Определяемое вещество (ион) n Е/
As(III) -0,75
Bi3+ 3 -0,19
Cd2+ 2 -0,59
Со2+ He восст. —
Cu2+ 2 -0,03
Eu2+ -1 (ан.) -0,9
Eu3+ 1 -1,16
Fe2+ -1 (ан.) -0,05
Fe3+ 1 0,05
H2O2 —2 (ан.) -0,18
2 -1,0
jo; 6 -0,65 (pH = 5-6)
Mn2+ Не восст. —
Ni2+ Не восст. —
Pb2+ 2 -0,43
Sb(in) 3 -0,64
Sn2+ -2 (ан.) -0,21
2 -0,54
Sn(IV) Не восст. 0,25 М Н3СбН5О7; pH = 5
Ti(IV) 1 -0,73
ti+ 1 -0,45
U(1V) Двойная волна -0,38 (0,1 М КзСбН5О7 + 0,1 М Н3С6Н5О7)
Zn2+ 2 -1,04
Таблица 6.36
Фоновый электролит NajCJEIsO? (1 М) + NaOH (0,1М)
Определяемое вещество (ион) n Ey
Ag* 1 >0
As(III) -2 (ан.) -0,31
As(V) He восст. —
Bi3+ 1 -0,8
3 -0,9
Cd2+ 2 -1,46
Ce4+ 1 -0,3
4 -0,8
Co2+ 2 -1,45
Cr3+ 1 -1,45
Электрохимические методы анализа
759
Продолжение таблицы 6.36
Определяемое вещество (ион) n ЕУ
Cr(VI) -0,23
3 -0,83
4 -1,49
Cu2+ 2 -0,50
Fe2+ -1 (ан.) -0,30
2 -0,49
Fe3+ 1 -0,93
3 -1,70
Mn2+ He восст. —
Ni2+ He восст. в цитратно-аммиачн. буф. Е^ =-1,09 и-1,39 Л (АеГ = 2)
Pb2+ 2 -0,78
Sb(III) -2 (ан.) -0,44
3 -1,24
Sb(V) He восст. —
Sn2+ -2 (ан.) -0,91
2 -1,12
Sn(IV) -1,22
Te(VI) -1,54
ti+ 1 -0,56
U(VI) -0,98
V(IV) -1 (ан.) -0,47
2 -1,76
V(V) 3 -1,70
W(VI) Не восст. —
Zn2+ 2 -1,43
Таблица 6.37
Фоновый электролит Na-ЭДТА (0,05 М) + СНзСООН (0,8 М); pH ~ 2
Определяемый ион n ЕУ,
Bi3+ 3 -0,5
Cd2+ 2 -0,65
Со2+ He восст. —
Cu2+ 2 -0,17
Fe2+ -1 (ан.) -0,12 (pH = 4)
Fe3+ 1 -0,12
Mn2+ He восст. —
Mo(VI) -0,63 0,1 h. Na-ЭДТА + 0,1 h. CH3COOH + 0,1 н. CH3COONH4
Продолжение таблицы 6.37
Определяемый ион n Ey
Nb(V) 1 -0,61 (pH-3)
-1,05
Ni2+ He восст. —
Pb2+ 2 -0,75
Sb(TII) 3 -0,62
Sn2+ = 0,7
Ti(IV) 1 -0,22 (pH- 1,0-2,5)
ТГ 1 -0,50
W(VI) He восст. —
Zn2+ He восст. —
Таблица 6.38
Фоновый электролит Na-ЭДТА (0,05 М) + CH3COONH4 (0,8 М); pH = 7
Определяемый ион n ЕУ
As(III) = -1,0
Bi3+ 3 -0,66
Cd2+ 2 -1,27
Co2+ He восст. —
Cu2+ 2 -0,31
Eu3+ 1 -1,17 (Na-ЭДТА (0,1 н.); pH = 6-8
Fe3+ 1 -0,15
Ge(IV) 4 -1,30 (Na-ЭДТА (0,1 h.); pH = 6-8
Mn2+ He восст. —
Ni2+ He восст. —
Pb2+ 2 -1,37
Sb(IH) 3 -0,79
Sn2+ 2 -1,26
-2 (ан.) -0,20
ti+ 1 -0,50
U(VI) 1 -0,37
V(V) 3 -1,22 Na-ЭДТА (0,05 M) + CH3COONa; pH = 9,5
Zn2+ He восст. —
760
Новый справочник химика и технолога
Таблица 6.39
Фоновый электролит KSCN (1 М)
Таблица 6.40
Фоновый электролит KCN (1 М)
Определяемый ион n
Аи(П1) 1 -1,16 (0,1 M KSCN + 0,lMNH4OH (Au(III)—>Au(I))
Cd2* 2 -0,65
Ce(IV) -0,53
Со2* 2 -1,08
Сг2* -0,80
Сг3* 1 -1,05 (двойная волна)
Cr(VI) (>0)
(-0,46)
4 (одна ступень) -0,94
1 >0
Си2* 2 -0,54
Fe2* 1,52 1 MKSCN + 0,001 M H2SO4
Fe3* 1 >01MKSCN + 0,001 MH2SO4
In3+ -0,72
-1,73
Ir(IV) -1,6
Mn2* 2 -1,54
Mo(VI) He восст. —
Ni2* 2 -0,69
Pb2+ 2 -0,45
Pd(II) >0(0,1 MKSCN + 0,1 MNH4OH (Au(III)—>Au(I))
Rh(III) -0,46
Re(VII) -1,2
Sn2* 2 -0,46
Sn(TV) 4 -0,50 (1 M KSCN + 0,02MHCl)
Te(VI) -1,46
Ti(IV) 1 -1,46 (0,IM KSCN + 0,1М№С,ОН (Au(III) —> Au(I))
ti* 1 -0,52
U(VI) 1 -0,26
2 -1,32
V(V) 3 -1,26
Zn2* 2 -1,06
Определяемый ион n
Ag+ 1 -0,3 (IM KCN и 0,001 M)
Au* 1 -1,46
Au(III) 2 >0
3 -1,46
Cd2* 2 -1,18
Co2* 1 -1,3
Co3* 2 -1,45
Cr2* -1 (ан.) -1,38
Cr3* 1 -1,38
3 -1,45
Cu2* He восст. —
Fe2* He восст. —
Fe3* 1 -0,7
Ga3* 3 -1,29
Ir(III) He восст. —
Ir(IV) He восст. —
Mn2* 1 -1,33
Ni 2 -1,36 (0, IM KCN и 0,1 MHC1)
Os(VI) 3 >0,5
4 -0,81
Pb2* 2 -0,72
Pd(II) 2 -1,77
Pt(II) He восст. —
Rh(III) 1 -1,47
Ru(IV) He восст. —
Sb(III) 3 -1,13
TI* 1 >0
Te(VI) -1,36
V(III) 1 -1,2
W(VI) He восст. —
Zn2* He восст. —
Электрохимические методы анализа
761
6.3.3.2. Потенциалы полярографических полуволн органических соединений
Таблица 6.41
Потенциалы полярографических полуволн органических соединений
Принятые обозначения. a-Pt — платиновый анод; a-Hg — ртутный анод; в остальных случаях ртутный катод — к-Hg.
Определяемое Фоновый электролит Элек- Е ,
вещество состав рн трод У
Азобензол Буф. в 48% сп. 4,0 9,0 к-Hg « -0,20 -0,57
0,1 н. LiCl в 79% СП. — « -0,91
Антраниловая кислота 0,05 н. [(CH3)4N]J — « от-1,52 до — 1,60
Антрахинон Ацетатн. буф. в 40% диоксане 7,4 « -0,54
0,1 н. H2SO4 в 80% мет. сп. — « -0,36
Антрацен 0,175 М [(СЩ^Ьф в 75% диоксане — « -1,94
0,1 М [(C4H9)4N]I в 75% диоксане — « -2,09
/)Л-Арабиноза Фосфатн. буф. 6,5 7,0 7,5 « « « -1,54 -1,57 -1,57
Аскорбиновая кислота Фосфатн. буф., 1,5% р-р НРО3 3,41 a-Hg 0,17
Бифталатн. буф., 3% р-р НРО3 2,71 « 0,24
0,1 М CdSO4 — a-Pt -0,8*
Аурин Фосфатн. буф. в 30% сп. (60 °C) 7 к-Hg -0,76 -1,20
Ацетальдегид 0,1 н. LiOH — « -1,73
Фосфатн. буф. 6,8 « -1,89
0,1 h.NH4C1 —- « -1,60
Ацетилацетон 0,1 н. LiCl — « -1,07**
0,01 н. НС1, 0,1 н. КС1 — « -1,20**
0,1 н. NH.tCl — « -1,69**
Ацетон 0,025 М [(CH3)4N]J — « -2,20
0,05 М [(CH3)4N]Br в 75% диоксане; 2,5 н. NH4OH; 2,5 н. (NH4)2SO4 9,3 « « -2,46 -1,52
Продолжение таблицы 6.41
Определяемое Фоновый электролит Элек Е,.
вещество состав рн трод У
Ацетофенон Цитратно-фосфатн. буф. 1,3 4,9 8,4 11,3 « « « « -1,14 -1,33 -1,58 -1,60
Бензальдегид Цитратно-фосфатн. буф. 2,2 8,0 11,0 « « « -0,96; -1,32 -1,33; -1,41 -1,44
Бензил (дибензоил) Цитратно-фосфатн. буф. 1,2 7,2 11,3 « « « -0,27 -0,64 -0,75
Бензойная кислота 0,05 М [(CH3)4N]Br в 75% диоксане 5 « -1,90; -2,42 0,046
Бензофенон Цитратно-фосфатн. буф. 1,5 « -0,93; -1,28
4,8 « -1,10; -1,29
7,9 « -1,33
11,0 « -1,43
Бензохинон-1,4 Цитратно-фосфатн. буф. 5,3 7,0 11,0 « « « 0,04 -0,11 -0,35
Бромбензол 0,05 М [(CH3)4N]Br в 80% сп. — « -2,22
Бромоформ 0,05 М [(CH3)4N]Br в 75% диоксане — « -0,60; -1,47
Бутадиен 0,05 М [(C2H5)4N]Br в 75% диоксане — « -2,59
Винилацетилен 0,05 М [(C2H5)4N]Br в 75% диоксане « -2,40; -2,58
Витамин В] (тиамин) 0,1 н. КС1 7,2 « « -1,25 -1,30
Боратн. буф. 11 « -0,38
Витамин В2 (рибофлавин, лактофлавин) Фосфатн. буф. 7,2 « -0,40
Витамин Вб (фолиевая кислота) Буф- 7,6 9 « « -0,73 -0,87
D-Галактоза Фосфатн. буф. 7 « -1,56
762
Новый справочник химика и технолога
Продолжение таблицы 6.41
Определяемое вещество Фоновый электролит Элек-трод
состав рн
Галловая кислота Цитратно-фосфатн. буф. 6,0 11,2 a-Hg « « 0,49 0,41 0,34 0,28 0,04
0ДМКС1 — к-Hg -1,60
0,1 н. H2SO4 — a-Hg -0,57
а-Гексахлорц-иклогексан 0,1 М [(C2H5)4N]J в 80% сп. — к-Hg -2,02
Р-Гексахлор-циклогексан 0,1 М [(C2H5)4N]J в 80% сп. — « -2,15
у-Гексахлор-циклогексан 0,1 М [(C2H5)4N]J в 80% сп. — « -1,61; -2,58
З-Гексахлор-циклогексан 0,1 М [(C2H5)4N]J в 80% сп. — « -2,08
Гексахлорэтан 0,05 М [(CH3)4N]Br в 75%-ном диоксане — « -1,84
ГидроКСил-амин Ацетата. -фосфатн.-боратн. буф. 4,6 9,2 « « -1,42 -1,65
Гидрохинон Ацетата, буф. 5,4 a-Hg -0,136
Фосфата, буф. 8,0 « -0,024
Гликолевая кислота 0,1 н. LiOH — к-Hg -1,40
Глиоксаль 0,1 н. NH4CI — « -1,50
D-Глюкоза Фосфата, буф. 7 « -1,55
Диацетил Фосфата, буф. 6,9 « -0,70; -1,75
Диацетилен 0,05 М [(C2H5)4N]Br в 75% диоксане -— « -2,27
Дибромуксусная кислота Фосфатно-цитратн. буф. 3,8 5,8 « -0,24; -0,77
« -0,32; -1,10
NH4OH + NH4CI 8,6 « -0,37; -1,21
Дивинилацетилен 0,05 М [(C2H5)4N]Br в 75% диоксане — « « -2,07 -2,55
о-Динитро-бензол Буф. в 80% сп. 7,4 « -0,41; -0,75
jw-Динитро-бензол Буф. в 80% сп. 7,4 « -0,46; -0,71
п- Динитробензол Буф. в 80% сп. 7,4 « -0,35; -0,83
Продолжение таблицы 6.41
Определяемое вещество Фоновый электролит Элек-трод ЕУг
состав рн
Дифенил 0,175 М [(C4H9)4N]J в 75% диоксане — «
1,2-Дихл op-этан 0,1 н. КО « -1,32
Инден 0,175 М [(C4H9)4N]J в 75% диоксане — « -2,77
Индигокармин Буф. 2,6 « -0,20
Иодбензол 0,05 М [(CH3)4N]Br в 80% сп. — « -1,58
Йодоформ 0,05 М [(CH3)4N]Br в 75% диоксане — « -0,45; -1,05; -1,46
Карбазол 0,1 М [(СДШф в 75% диоксане — « -2,77
Коричная кислота 0,1 М [(C4H9)4N]J в 50% диоксане — « -1,89; -2,02
1 н. НО — « -1,13
КС1, LiCl в 50% диоксане — « -1,5; -2,0
Коричный альдегид Буф. 4,9 7,2 « « -0,82; -1,26 -0,94; -1,26
Кротоновая кислота 0,1М LiOH 13 « -1,90
Кротоновый альдегид Ацетата, буф. в 50% диоксане 5 « -1,09
Малеиновая кислота Ацетата, буф. 7,8 « -1,15; -1,34
D-Манноза Фосфата, буф. 7 « -1,53
Масляный альдегид Водный раствор [(CH3)4N]J — « -1,88
Меркапто-бензтиазол Фосфатно-боратн. буф. 7 a-Hg -0,25
Метиленовый синий Фосфатно-боратн. буф. 6,7 9,2 к-Hg « -0,22 -0,41
Метил-салицилат [(C4H9)4N]OH в 30% монометиловом эфире этиленгликоля — « -2,15
Р-Метил-стирол 0,175 М [(C^NJJ в 75% диоксане — « -2,54
Метил этил-кетон 0,025 М [(CH3)4N]J — « -2,25
Электрохимические методы анализа
763
Продолжение таблицы 6.41
Определяемое Фоновый электролит Элек- Et/
вещество состав pH трод X
Монобромуксусная кислота Буф- 7,2 11,1 « « -1,30 -1,32
Муравьиная кислота 0,1 н. КС1 — « -1,38
Нафталин 0,175 М [(C4H9)4N]J в 75% диоксане — « -2,50
Нафтионовая кислота 0,05 М [(CH3)4N]J — « от-1,42 до — 1,52
Нейтральный красный Фосфатно-боратн. буф. 7 10 « « -0,57 -0,72
Никотиновая кислота 0,1 н. НС1 — « -0,83
Нитробензол Фосфатно-боратн. буф. 7 10 « « -0,47 -0,60
Нитрометан Фосфатно-боратн. буф. в 30% мет. сп. 4,6 11,6 « « -0,85 -0,90
а-Нитро-нафталин 0,1 н. НС1 в 50% СП. — « -0,22
о-Нитрофенол Фосфатно-боратн. буф. в 80% сп. 7,0 11,0 « « -0,73 -0,97
8-Окси-хинолин Фосфатн. буф. 8 « -1,18; -1,71
Боратн. буф. 10 « -1,46; -1,81
Пикриновая кислота Буф. (СН3СООН + LiOH) 4,2 11,7 « « -0,34 -0,36 -0,54 -0,96 -1,0
Пирен 0,175 М [(C4H9)4N]J в 75% диоксане — « -2,10 -2,46 -2,68
Пиридин Ацетатн. буф. 5,4 6,3 7,2 « « « -1,35 -1,76 -1,77
Пировиноградная кислота Цитратно-фосфатн. буф. 2,8 4,0 5,8 « « « -0,64 -0,91 -0,72 -1,07 -0,80 -1,18
Пирогаллол Фосфатно-боратн. буф. 4 7 9 a-Hg « « -0,18 -0,35 -0,47
Пропионовый 0,1 н. LiOH — к-Hg -1,76
альдегид 0,1 н. NH4CI — « -1,60
Продолжение таблицы 6.41
Определяемое вещество Фоновый электролит Элек-трод
состав pH
Рибоза Фосфатн. буф. 7,0 « -1,77
Салициловая 0,1 н. КС1 — « -1,29
кислота [(C4H9)4N)OH — « -2,39
Стильбен 0,175 М [(C4H9)4N]I в 75% диоксане — « -2,14
0,1 М [(C4H5)4N1I в 75% диоксане — « -2,17
Стирол 0,175 М [(С4Н9)4М]1 в 75% диоксане — « -2,35
Стрептомицин 3% р-р [(CH3)4N]OH 13,8 « -1,41
Стрихнин Буф. 6,0 8,0 « « -1,64** -1,55**
Сульфазол 0,1 н. [(CH3)4N]OH — « -1,67
Тиомочевина 40% сп. р-р 6 н. НС1 — « -0,3 -0,58
1,3,5-Три-нитробензол Буф- 6,4 12 « « -0,26; -0,36; -0,48 -0,31; -0,44 -0,60
Тринитротолуол Фталатн. буф. 7,4 « -0,31 -0,48 -0,62
Трихлоруксусная кислота NH4OH, NH4C1 6,8- 8,8 « -0,84 -1,56
Фенантрен 0,175 М [(C4H9)4N]I в 75% диоксане — « -2,45 -2,69
Фенол Фосфатн. буф. 9,0 a-Pt -0,515
Фенолфталеин Бифталатно- 2,5 к-Hg -0,67
фосфатн. буф. в 6,9 « -0,86
25% сп. 10,1 « -1,01 -1,37
Флуорен 0,175 М [(C4H9)4N]I в 75% диоксане — « -2,65
Флуоресцеин Буф. 7,1 9,0 10,8 « « « -0,80 -1,07; -1,38 -1,20
Формальдегид 0,2 н. LiOH — « -1,59
0,05 н. LiOH -— « -1,56
0,05 М [(CH3)4N]Br в 75% диоксане — « -0,45; -1,05; -1,46
764
Новый справочник химика и технолога
Продолжение таблицы 6.41
Определяемое Фоновый электролит Элек- Е„
вещество состав pH трод У
Фруктоза 0,1 н. LiCl — « -1,76
Фталевая кислота Буф- 2,9 4,1 « « -1,24 -1,35
0,03 М ВаС12 — « -1,50
Фталимид Буф- 1,9 4,2 « « -0,86 -1,05; -1,45
Фумаровая кислота Ацетатн. буф. 5,9 « -1,19
Фурфурол 4,0 6,5 « « -1,24 -1,52; -1,72
Хинальдин 0,2 н. НС1 — « -0,70
0,1 MNH4OH + 0,1 MNH4CI в 50% сп. 9,9 « -1,51
Хинальдиновая кислота Фосфатн. буф. 8 « -1,00; -1,16
Хинидин Цитратно-фосфатно-боратн. буф- 12 « -1,55
Хинин Цитратно-фосфатно-боратн. буф. 12 « -1,56
Хинолин Ацетатн. буф. в 50% сп. 3 4 5,4 7,2 « « « « -1,25 -1,25; -1,71 -1,0; -1,30; -1,73 -1,32; -1,71
Хлоральгидрат 0,1 н. КС1 в 50% СП. — « -1,51
Хлороформ [(CH3)4N]Br в смеси СН3ОН + Н2О(1:1) — « -1,68
Хлоруксусная кислота 0,1 н. NaOH — « -1,56
а-Хлорэтил-метилкетон 0,01 н. НС1 + 0,01 н. КС1 в 80% сп. — « -1,26
Циклогексанон 0,05 М [(CH3)4N]Br в 75% диоксане — « -2,42
2,5 н. NH4OH + 2,5 н. (NH4)2SO4 9,3 « -1,50
Цинхонин Буф- 12 « -1,61
Четыреххлористый углерод 0,1 М [(CH3)4N]Br в смеси СН3ОН + Н2О (2:1) — « -0,75; -1,70
Продолжение таблицы 6.41
Определяемое вещество Фоновый электролит Элек-трод ЕУг
состав pH
Щавелевая 0,1 н. КС1 — « -1,31
кислота MgCl2 (или (CH3COO)2Mg) 8,25 « -1,9
Этилмер каптан 0,025 н. H2SO4 в 85% сп. — a-Hg 0,02
Этилнитрат LiOH — к-Hg -0,94
Этилнитрит LiOH — — -0,65
Примечания. Еу измерена относительно Cd-катода.
** Потенциал начала полярографической волны.
6.4. Амперометрическое титрование
Непосредственно связан с прямой вольтамперометрией метод амперометрического титрования. Он основан на измерении величины диффузионного тока, который проходит через электролитическую ячейку, состоящую из поляризующегося индикаторного электрода (ртутный капельный или вращающийся твердый электрод) и электрода сравнения (каломельного, хлорид-серебряного) (при постоянном значении потенциала). Величину потенциала с целью повышения чувствительности метода выбирают таким образом, чтобы титрование проводилось при предельных токах восстановления или окисления веществ. Для фиксирования точки эквивалентности в методе амперометрического титрования используют появление или исчезновение диффузионного тока на поляризующемся электроде. В основе метода лежит пропорциональность между величиной диффузионного тока и концентрацией вещества, участвующего в электрохимическом процессе на электроде и обусловливающего наблюдаемый диффузионный ток. Для применения метода амперометрического титрования к какой-либо реакции, используемой для объемных определений, необходимо, чтобы одно из реагирующих веществ восстанавливалось или окислялось на индикаторном электроде и потенциал должен быть таким, чтобы величина диффузионного тока была бы пропорциональна концентрации
/д=кс. (6.58)
Следовательно, метод амперометрического титрования представляет собой видоизменение полярографического метода анализа, поскольку он основан на измерении величины диффузионного тока восстановления или окисления реагирующих веществ. Непосредственную связь между методами амперометрического титрования и полярографическим можно показать графически. На рис. 6.34а приведены полярографические кривые растворов соли цинка различной концентрации. Если, исходя из приведенных полярограмм, построить график зависимости величины диффузионного тока от концентрации цинка в растворе, получим ряд точек на прямой линии (рис. 6.346). Эта прямая отвечает уравнению /д = кс. Подобную прямую мож
Электрохимические методы анализа
765
но получить и экспериментальным путем. Если к раствору, содержащему ионы цинка, прибавлять какой-нибудь осадитель, то снижение концентрации ионов цинка в растворе вызывает соответствующее уменьшение диффузионного тока.
Рис. 6.34.
Графическое отображение связи между полярографией и амперометрическим титрованием: а — полярограммы растворов соли цинка различной концентрации; б— изменение силы тока в растворах соли цинка при титровании [2]
При амперометрическом титровании после каждого добавления реактива (в данном случае осадителя) нет необходимости снимать всю полярограмму, достаточно лишь определить величину предельного тока. Предварительно устанавливают потенциал индикаторного (рабочего) электрода таким, чтобы он находился в пределах площадки предельного тока, например, -1,65 В (относительно нас. к. э.). Этот потенциал поддерживают постоянным в течение всего процесса осаждения цинка. По мере прибавления осадителя, например ферроцианида калия, предельный ток цинка будет уменьшаться (серия полярографических кривых на рис. 6.34а).
Зависимость величины предельного тока от количества добавленного реактива представляет собой типичную кривую амперометрического титрования. После полного осаждения цинка ток остается постоянным (рис. 6.34а). Для построения кривой титрования достаточно иметь по 3-4 точки для каждой ветви кривой. Точку эквивалентности находят экстраполяцией. Метод амперометрического титрования отличает от полярографического то, что в полярографическом методе анализа сам определяемый ион должен восстанавливаться (или окисляться) на электроде. Для метода амперометрического титрования это не является обязательным: достаточно, чтобы на электроде мог восстанавливаться (или окисляться) хотя бы один из двух участвующих в титровании реагентов или продукт их реакции, т.е. электроактивным может быть определяемое вещество, титрант или образующийся продукт. Для проведения метода амперометрического титрования необходимо установить на индикаторном электроде потенциал, отвечающий области диффузионного тока того вещества, которое участвует в электродном процессе и концентрация которого изменяется в процессе титрования. Для этой цели можно использовать реакции осаждения (например, титрование цинка ферроцианидом калия), реакции окисления - восстановления (например, титрование ванадата солью Мора) и реак
ции комплексообразования (например, титрование при помощи различных комплексонов).
Основные формы кривых амперометрического титрования
Необходимым условием для применения метода амперометрического титрования является предварительное полярографическое исследование реагирующих веществ, а иногда и продуктов реакции. Для титройания выбирают потенциал индикаторного электрода, который соответствует области диффузионного тока иона, дающего электродную реакцию. В этом случае прямолинейный ход кривой титрования сохраняется в широких пределах концентрации. Если же потенциал не соответствует области диффузионного тока, то на кривой титрования появляется «загиб», свидетельствующий о том, что прямолинейная зависимость /д = кс не выдерживается. Если реакция амперометрического титрования протекает по схеме А + В = АВ, то возможны следующие случаи:
1. В электродную реакцию вступает только одно вещество А, т. е. оно электроактивно. Тогда в зависимости от того, какое вещество добавляется в раствор в процессе титрования, возможны два вида кривых. Если А — определяемое вещество, а к нему добавляется титрующее вещество В, как, например, при титровании свинца сульфатом натрия, кривая соответствует кривой а на рис. 6.35а. Если же В — определяемое вещество, а титрантом является элек-троакгивное вещество А, то предельный ток вещества А появляется и возрастает после точки эквивалентности (ТЭ) по мере добавления избытка реактива (рис. 6.356).
2. В электродную реакцию вступают оба реагирующих вещества А и В. В большинстве случаев потенциалы восстановления или окисления веществ А и В различны, поэтому при титровании могут быть получены кривые различного вида в зависимости от того, при каком потенциале электрода проводится амперометрическое титрование. В случае, когда титрование проводят при потенциале, достаточном для достижения предельного тока обоих веществ, получаются кривые, аналогичные рис. 6.32в. До ТЭ ток обусловлен восстановлением или окислением вещества А, так как добавляемое к раствору вещество В сразу вступает в реакцию. После ТЭ ток обусловлен восстановлением или окислением избыточного вещества В в растворе. Если же одно из веществ при данном потенциале будет восстанавливаться, а другое окисляться, то ток после конечной точки титрования (КТТ) изменит свое направление (рис. 6.32г).
3. Если в определяемом растворе присутствуют разные ионы, способные давать соединения с одним и тем же титрантом и один из этих ионов электроактивен при данном потенциале, а другой — нет, то имеем кривую, аналогичную рис. 6.356. Возьмем, например, титрование свинца и бария бихроматом с использованием в качестве индикаторного электрода ртутного капельного. По мере титрования свинца ток уменьшается, и, достигнув минимального значения, остается постоянным во время титрования бария.
766
Новый справочник химика и технолога
После ТЭП ток, вследствие восстановления бихромата, возрастает.
4. Возможны и такие случаи, когда при заданном значении потенциала индикаторного электрода электрохимически неактивны как определяемое вещество, так и титрант. Электрохимически активное вещество образуется в процессе титрования и, восстанавливаясь (или окисляясь) на электроде, дает диффузионный ток (рис. 6.32е).
При выполнении амперометрического титрования в качестве индикаторного электрода чаще всего используют вращающийся твердый электрод.
Амперометрическое титрование применяют при анализе разбавленных растворов (10“2-10“5 моль-л"1). Ошибка метода при количествах определяемого вещества 1-0,1 мг не превышает обычно ± 2%. От других электрохимических объемных методов амперометрическое титрование отличается высокой точностью определения малых количеств веществ, быстротой выполнения, в ряде случаев возможностью выполнения анализа в присутствии большого количества посторонних веществ.
7 Л
д
Рис. 6.35. Различные формы кривых амперометрического титрования [2]
Таблица 6.42
Условия амперометрического титрования некоторых веществ
Принятые обозначения а — анодный ток окисления к — катодный ток восстановления
Формы кривых титрования
II Ш IV
Ион или молекула, дающая диффузионный ток; характер электродной реакции Потенциал платинового индикаторного микроэлектрода относительно электрода сравнения Состав фонового раствора Форма кривой титрования Титруемое вещество Титрующий реактив Примечание
МИЭ,В нас.к.э., В
Ag(I); к 0 -0,25 HNO3; KNO3; CH3COONa I Ag(I) Г, Вг", СГ и другие, образующие малорастворимые осадки с Ag(I)
Электрохимические методы анализа
767
Продолжение таблицы 6.42
Ион или молекула, дающая диффузионный ток; характер электродной реакции Потенциал платинового индикаторного микроэлекгрода относительно электрода сравнения Состав фонового раствора Форма кривой титрования Титруемое вещество Титрующий реактив Примечание
МИЭ,В нас.к.э., В
Ag(I); к 0 -0,25 HNO3; KNO3; CH3COONa II СГ Вг-, Г, SCN-и другие, образующие малорастворимые осадки с Ag(I) Ag(I)
+0,40 +0,15 H2SO3 I Ag(I) Г и другие, образующие малорастворимые осадки с Ag(l) Титрование можно проводить в присутствии Cu(II)
+0,40 +0,15 H2SO4 П СГ и другие, образующие малорастворимые осадки с Ag(I) Ag(I) То же
отО до +0,40 от -0,25 до +0,15 NH3+NH4CI 11 POp;Ag(I)
As(III); а +1,30 +1,05 HC1, H2SO4 I As(III) ВгО-
Au(III); к отО до +0,40 от -0,25 до +0,15 HC1; H2SO4; KNO3; NaNO3 I Au(lII) 2- S2O з и различные органические соединения, например меркаптотиазол, гидрохинон, тиокарбамид Титрование Аи(Ш) во сстановителями удобнее проводить по току их окисления, например тиокарбамида при потенциале микроэлектрода, равном +0,8 В (МИЭ). В этом случае кривая титрования имеет форму II
+0,40 +0,15 KNO3; H4NO3; H2SO4 I Au(IlI) Fe(CN);-
Bi(TII); а +0,1 -0,15 HC1O4; pH--l,2 HNO3; pH=l,5 II PO4 Bi(ni)
Вг2;к +0,45 +0,20 NaHCO3 + KBr II As(IIl), NH3 ВгО-, СЮ-
+0,40 +0,15 2н. HC1, 4h. H2SO4, +СГ II As(III), Sb(III) BrOg Если в испытуемом растворе присутствуют Hg(II), Cu(II), Ag(I) и другие, то титрование проводят при потенциале микро-электрода, равном +0,70 В (МИЭ)
Вг-; а +1,30 +1,05 2h. H2SO4 II Tl(III) Вг-
768
Новый справочник химика и технолога
Продолжение таблицы 6.42
Ион или молекула, дающая диффузионный ток; характер электродной реакции Потенциал платинового индикаторного микроэлектрода относительно электрода сравнения Состав фонового раствора Форма кривой титрования Титруемое вещество Титрующий реактив Примечание
МИЭ,В нас.к.э., В
Ce(IV); к +0,75 +0,50 H2SO4 II Оксалат, V(IV)’Fe(II), Т1(1) Ce(IV) Если в этих условиях восстановитель дает анодный ток, то кривая титрования имеет форму Ш с обратным ходом
+0,45 +0,20 НС1 II Sn(II) Ce(IV)
+1,00 +0,75 H2SO4 III Различные органические соединения (гидрохинон, метол, пирокатехин, п-аминофенол и др.) Ce(IV) До ТЭ — ток окисления органических соединений, за ТЭ — ток восстановления Ce(IV)
Се(Ш); а +1,60 +1,35 CH3CO2Na + этанол II РО34" Ce(III)
С12; к +0,70 +0,45 НС1 — — — Ток восстановления С12 используется для определения хлора в воде
С1; а +1,50 +1,25 НС1 — — — В среде НС1 ток окисления хлорида может мешать другим анодным процессам
C(VI); к +0,40 +0,40 +0,70 +0,90 +0,15 +0,15 +0,45 +0,65 HC1, < 6 н. H2SO4, 4-6 н H2SO4, 8-12 h. H2SO4, > 12 h. Различные восстановители, например, Fe(ll), Sb(III), As(III) и др. Cr2O" Форма кривой титрования зависит от титруемого восстановителя и установленного на микроэлектроде потенциала
Сг(П) +0,65 +0,40 H2SO4 II Mo(VI), W(VI) Cr(II)
Cu(II) < +0,30 < +0,05 Индифферентные разбавленные кислоты В амперометрическом титрировании ток восстановления Cu(II) не используется, но может мешать при определении других веществ
Fe(III); к -0,15 -0,40 H2SO4 i(NH4)2SO4 II Ti(III) Fe(IH)
0 -0,25 HC1,~1,2 h. I Fe(III) Аскорбиновая кислота
0 -0,25 В зависимости от определяемого вещества V Zr(I), А1(Ш), Be(II) F“ Титрование проводят в присутствии Fe(III) (индикаторный метод)
Электрохимические методы анализа
769
Продолжение таблицы 6.42
Ион или молекула, дающая диффузионный ток; характер электродной реакции Потенциал платинового индикаторного микроэлектрода относительно электрода сравнения Состав фонового раствора Форма кривой титрования Титруемое вещество Титрующий реактив Примечание
МИЭ,В нас.к.э., В
Fe(II) + 1,25 +1,00 H2SO4 II Mn(VII), Cr(VI), V(V), Ce(IV) Fe(Il) Если в этих условиях окислитель дает катодный ток, то кривая титрования имеет форму Ш
+1,10 +0,85 H2SO4, НС1 I Fe(Il) МпО4;Сг2О^; VO3; Ce(IV) То же
Fe(CN)tK +0,05 -0,20 NH3+NH4C1 II Co(Il) Fe(CN)^ Можно титровать Fe(CN)6 стандартным раствором Со(П), тогда кривая титрования имеет форму I
+0,20 -0,05 NaOH 11 T1(I) Fe(CN)^ Титрование гексацианоферратом (III) проводят в присутствии катализатора — OsO4
Fe(CN)‘- ;а от +0,70 до 1,00 от +0,45 до +0,75 — — Zn(II), Pb(II), Cd(H), Ca(II), Cu(II), Mn(II), Zn(lII), Ag(I)n др., образующие малорастворимые осадки, Au(III) — реакцией восстановления Fe(CN)‘“ Потенциал микроэлектрода, состав фона и, следовательно, форма кривой титрования зависят от определяемого вещества
Hg(n);K +0,40 +0,15 NaNO3; NH4NO3; H2SO4 I Hg(Il) Г, сг
+0,40 +0,15 NaO3; NH4NO3; H2SO4 II СГ Hg(II)
Hg(I) +0,40 +0,15 NaNO3; NH4NO3 II Пирофосфат, Mo(VI), W(VI) Hg(i)
H2O; к Начало восстановления — — — Ток восстановления воды ограничивает катодную область электродной реакции
-0,70 -0,40 0 -0,95 -0,65 -0,25 Щелочная среда Нейтральная среда Кислая среда — — —
770
Новый справочник химика и технолога
Продолжение таблицы 6.42
Ион или молекула, дающая диффузионный ток; характер электродной реакции Потенциал платинового индикаторного микроэлектрода относительно электрода сравнения Состав фонового раствора Форма кривой титрования Титруемое вещество Титрующий реактив Примечание
МИЭ, В нас.к.э., В
Н2О; а Начало окисления
+0,80 +1,20 +1,50 +0,55 +0,95 +1,25 Щелочная среда Нейтральная среда Кислая среда — — — Ток окисления воды ограничивает анодную область электродной реакции
12; к +0,20 -0,05 НС1>9н. IV As(V), Se(IV) Г
0 -0,25 CH3CO2Na I I2 s2o32- Этой реакцией пользуются для определения Cu(II), Fe(IIl), As(III) после прибавления к испытуемому раствору избытка Г
+0,45 +0,20 Сегнетова соль + NHCO3 II Sn(II), As(III), Sb(III) I2
+0,40 +0,15 НС1, 2 н.; H2SO4, 2 н. + КС1 11 Sb(III), TH ю;
Г; а +1,00 +0,75 H2SO4, pH = 1-2 II Ag(I), Hg(II), Pb(II) г
+1,00 +0,75 KNO3, NH4NO3 II T1(I) г
+1,00 +0,75 НС1 > 6 н. II Se(IV) г
Ir(IV); к +0,70 +0,45 HC1 I Ir(IV) Аскорбиновая кислота, гидрохинон
Mn(VII); к +0,80 +0,55 H2SO4 < 8 н. — Различные восстановители, например оксалат, гексацианоферрат (II), V(IV), Fe(II), As(III), SbIII, Sn(II) МпО4 Форма кривой титрования зависит от титруемого вещества
+0,75 +0,50 HC1, 1 h. II T1(I) Мпо; —
+0,40 +0,15 CH3CO2Na + ZnO II Mn(II) Мпо; —
+0,40 +0,15 k4p2o7. pH-6 7 II Mn(II) МпО4 —
+0,40 +0,15 NaOH, 1 h. II Te(IV) мпо; —
Электрохимические методы анализа
771
Продолжение таблицы 6.42
Ион или молекула, дающая диффузионный ток; характер электродной реакции Потенциал платинового индикаторного микроэлектрода относительно электрода сравнения Состав фонового раствора Форма кривой титрования Титруемое вещество Титрующий реактив Примечание
МИЭ,В нас.к.э., В
Мп(Ш); к +0,90 +0,65 H2SO4 > 9 н. I Мп(Ш) Н2С2О4 Этой реакцией пользуются для определения РгО2 после прибавления к испытуемому раствору избытка Мп(П)
+0,90 +0,65 Н3РО4>Юн. I Мп(Ш) Fe(II) Можно титровать Мп(Ш) по току окисления Fe(II) при потенциале микроэлектрода, равном +1,3 В. В этом случае кривая титрования имеет форму II
Мп(П); а +1,20 +0,95 Кислая, нейтральная, CH3CO2Na — — — В амперометрическом титровании ток окисления Мп(П) не используется, но может мешать при определении других веществ
no; От +0,40 до +1,00 От+0,15 до +0,75 — — — В амперометрическом титровании ток восстановления не используется, но может мешать при определении других веществ, если концентрация HNO3, применяемой в качестве фона, > 5 н.
в зависимости от концентрации HNO3
NO;; а +1,30 +1,05 H2SO4, 0,05 н. I no; Сильные окислители, например МпО4, Ce(IV) и др., сульфаниловая кислота Для амперометрического титрования NO; на фоне цитрата аммония (pH = 4-4,5) можно использовать в качестве титранта также хлорамин Т по току его восстановления при потенциале микроэлектрода, равном +0,3 В
772
Новый справочник химика и технолога
Продолжение таблицы 6.42
Ион или молекула, дающая диффузионный ток; характер электродной реакции Потенциал платинового индикаторного микроэлектрода относительно электрода сравнения Состав фонового раствора Форма кривой титрования Титруемое вещество Титрующий реактив Примечание
МИЭ,В нас.к.э., В
О2; к Начало восстановления +0,10-0,15 +0,40 +0,15 +0,80 +0,55 Щелочная среда Нейтральная среда Кислая среда Ток восстановления О2 используется для полярографического определения растворенного кислорода. При амперометрическом титровании ток восстановления О2 сказывается при определении других веществ
РЬ(П); к -0,60 -0,85 CH3CO2Na, CH3CO2NH4 I РЬ(П) Cr2O ; MoO4 ;WC>4~ —
+ 1,80 +1,55 ch3co2nh4 или CH3CO2Na, 0,5 н. II SO4“ Pb(II) В качестве титранта используется раствор соли ацетата свинца, содержащий 4-5% уксусной кислоты
Sn(II); а -0,20 -0,45 Щелочной раствор ЭДТА III Sn(II) Hg(II) В качестве титранта используется раствор соли хлорида ртути(П). После ТЭ ток восстановления Hg(II)
Te(IV); к +0,60 +0,35 H2SO4, 6 н. I — — Для амперометрического титрования ток восстановления Te(IV) не используется, он может мешать при определении других веществ
Ti(III); а +0,75 +0,50 H2SO4 + + (NH,), so4 I Ti(III) Cr2O2,’ —
+1,00 +0,75 H2SO4, 10 н. II V(V), Cr(VI) Ti(III) —
+1,00 +0,75 H2SO4, 1 н. + + H4p2o7 II U(VI) Ti(IlI) —
Т1(Ш); к +0,50 +0,25 KNO3, CH3CO2Na и др. I Tl(III) Различные восстановители, например тиокарбамид Амперометрическое титрование Т1(Ш) тиокарбамидом удобнее проводить по току окисления последней при потенциале микроэлектрода, равном +0,80 В (МИЭ). В этом случае кривая титрования имеет форму П
Электрохимические методы анализа
ТГ5
Продолжение таблицы 6.42
Ион или молекула, дающая диффузионный ток; характер электродной реакции Потенциал платинового индикаторного микроэлектрода относительно электрода сравнения Состав фонового раствора Форма кривой титрования Титруемое вещество Титрующий реактив Примечание
МИЭ,В нас.к.э., В
тцт>;к -0,60 -0,85 CH3CO2Na I T1(I) Г Для амперометрического титрования ток окисления ТГ не используется, но может мешать при определении других веществ
Т1(1); а +1,40 +1,15 KNO3, MH4NO3, H2SO4
U(III); а +0,20 -0,05 H2SO4 III U(III) le(III) После ТЭ ток восстановления Fe(III)
V(V); к +0,50 +0,25 H2SO4, 12-16 н. 11 Различные восстановители, например Fe(II) vo:
+1,00 +0,75 H2SO4, 18-24 h. III V(V) Fe(Il) После ТЭ ток окисления Fe(II)
V(IV); а +0,85 +0,60 CH3CO2H + + CH3CO2Na, pH = 4 II ЭДТА vo; Этой реакцией пользуются для определения А1(Ш), Zr(II), Th(lV) и других веществ после прибавления к испытуемому раствору избытка ЭДТА
V(II); а +0,85 +0,60 H2SO4 + H3PO4 II Mo(VI), V(IV) V(H)
+0,45 +0,20 H2SO4 II Ti(IV) ,V(V) v(n)
6.5. Инверсионная вольтамперометрия [13-19]
6.5.1. Сущность метода инверсионной вольтамперометрии (ИВ) [14,15,17]
Метод ИВ основан на электрохимическом концентрировании электроактивных компонентов раствора (металлов) при постоянном потенциале на поверхности индикаторного электрода и последующем растворении полученного концентрата при заданной скорости изменения потенциала.
Главными отличиями ИВ от классической полярографии (прямой вольтамперометрии) являются:
1) наличие стадии накопления (электроконцентрирования) определяемого вещества;
2) применение стационарных (обычно твердых) электродов вместо капающих.
Электролиз проводят при потенциале предельного тока восстановления или окисления вещества при энергичном перемешивании раствора. Для полного выделения вещест
ва из раствора понадобилось бы бесконечно большое время, что непригодно для анализа, поэтому электролиз ведут в течение какого-то строго контролируемого времени. При этом выделяется пропорциональная часть анализируемого вещества, хорошо воспроизводящаяся, если выдерживать постоянные условия эксперимента.
Процесс измерений включает несколько стадий. В наиболее распространенном варианте метода - анодной ИВ, реализуют обычно четыре стадии.
1. Электрохимическая регенерация поверхности индикаторного электрода: потенциал регулируется в пределах 0-650 мВ (номинальное значение для хлоридного фона Е\ = +50 мВ), длительность ее постоянна и находится в пределах 20-30 с.
2. Электроконцентрирование — электролитическое накопление определяемых металлов на поверхности индикаторного электрода в режиме предельного тока при вращении электрода и поддержании отрицательного потенциала
774
Новый справочник химика и технолога
Еэ в пределах от 0 до -1500 мВ (номинальное значение Е2 от -1100 до -1200 мВ). Длительность стадии накопления составляет 30-2500 с. На протяжении 2-й стадии раствор перемешивается (вращается индикаторный электрод).
Например, при определении следов кадмия на ртутнографитовом электроде (см. ниже п. 6.5.6) в ртутном микро-капельном покрытии образуется амальгама
Cd(II) + 2е + Hg -> Cd(Hg).
3. Успокоение раствора перед съемкой вольтамперной кривой. Длительность стадии постоянна и составляет обычно 10-20 с. Потенциал электрода поддерживается близким (равным) потенциалу стадии накопления (£3 = Е2)-
4. Измерительная стадия. Потенциал линейно изменяют от Е3 до Е\. Скорость развертки задается в пределах 100-500 мВ/с. Форма налагаемого напряжения и способ обработки аналитического сигнала аналогичен рассмотренным для различных разновидностей полярографии.
На временной диаграмме рис. 6.36 показано изменение потенциала и тока во времени при проведении этих стадий.
Инверсионная вольтамперограмма имеет характерный вид кривой с пиками (рис. 6.37)
Рис. 6.36. Диаграммы изменения задаваемого потенциала индикаторного электрода в процессе анализа Еэя =f(t) и тока, протекающего через электрод / на 2-й стадии ток «зашумлен» из-за переменного сопротивления щеток токосъема при вращении индикаторного электрода (описание стадий см. в тексте)
Рис. 6.37. Инверсионная вольтамперограмма, снятая в режиме линейной развертки потенциала (см. 4 стадию на рис. 6.36) [14]
Аналитическим сигналом является высота анодного тока (/п), пропорциональная концентрации определяемых ионов в растворе при постоянстве всех условий опыта, а потенциал анодного пика Еп характеризует природу химического вещества в анализируемых условиях.
6.5.2. Аналитический сигнал в методе ИВ
Аналитический сигнал в ИВ — ток растворения продукта электролиза с электрода — имеет форму пика, характеризуется высотой, потенциалом пика, шириной полупика. Высота характеризует чувствительность к определяемому элементу, потенциал — его природу, а ширина — меру селективности, или разрешающей способности.
Высота пика прямо пропорциональна концентрации определяемого вещества или иона. Форма пика (при анодном растворении амальгамы металла) объясняется следующим образом. При достижении равновесного потенциала системы амальгама/ионы металла и снижении потенциала в положительную сторону через электрод протекает анодный ток, величина которого возрастает в соответствии с законами электрохимической кинетики. Однако при уменьшении концентрации частиц на поверхности электрода лимитирующей стадией процесса может стать доставка (например, диффузия атомов металла к поверхности), а не ионизация, и ток будет уменьшаться, по мере электроокисления металла практически падая до нуля. Очевидно, что площадь под анодным пиком равна количеству электричества, затраченного на восстановление металла и, по закону Фарадея, связана с количеством растворенного металла. Для оценки концентрации ионов металла можно измерять и площадь под пиком, и высоту пика. Величина и форма аналитического сигнала зависят от формы поляризующего напряжения в перечисленных выше вариантах вольтамперометрии.
Ниже рассматриваются факторы, влияющие на величину аналитического сигнала в постоянно-токовой ИВ. Кроме протекания тока, связанного с растворением концентрата, на электроде протекают другие побочные электрохимические процессы, объединяемые понятием «остаточный ток» (рис. 6.38).
а б
Рис. 6.38. Остаточный ток: а) фарадеевский:
кривая 1 — в растворе фона, содержащем кислород; кривая 2 — после удаления кислорода;
б) емкостная составляющая тока при линейном изменении потенциала в катодную и анодную сторону без учета фарадеевской составляющей [17]
Остаточный ток характеризует процессы на электроде; когда в растворе отсутствуют определяемые ионы, его можно измерить в той же ячейке. Он состоит главным образом из двух составляющих: емкостной и фарадеевской. Емкостный ток связан с образованием на электроде двойного электрического слоя (ДЭС) и равен:
Ic=ACdw, (6.59)
Электрохимические методы анализа
775
где А — поверхность электрода, Cd — дифференциальная емкость ДЭС при заданном потенциале.
Как видно, емкостный ток тем больше, чем больше поверхность электрода, скорость изменения потенциала и Cd (рис. 6.386). Другая составляющая остаточного тока — фарадеевская (рис. 6.38а), то есть вызванная восстановлением (кислорода, ионов водорода, воды и др.) или окислением компонентов раствора или электрода (например, ртути).
Для снижения предела обнаружения в ИВ увеличивают полезный сигнал и уменьшают остаточный ток за счет снижения как его емкостной, так и фарадеевской составляющих. Ниже рассмотрены некоторые способы и приемы увеличения полезного сигнала и снижения фарадеевской составляющей остаточного тока.
6.5.3. Инверсионная хронопотенциометрия (ИХП)
Инверсионный хронопотенциометрический анализ включает в себя стадию предварительного накопления и гальваностатического (режим заданного тока 1 = const) растворения осадка. Традиционно наибольшее распространение получила анодная ИХП тяжелых металлов. В ней электролитическое накопление производится при отрицательном (катодном) потенциале на индикаторном электроде, а на измерительной стадии задается анодный ток (+), смещающий потенциал индикаторного электрода в положительную область. Потенциал изменяется быстро вплоть до начала растворения электроактивного компонента, далее в процессе окисления компонента, осажденного на электроде, скорость изменения потенциала замедляется. На хронопотенциограмме возникает ступенька-волна (см. рис. 6.336), длительность которой прямо пропорциональна количеству растворенного с электрода компонента (и, согласно теории ИХП, концентрации этого же компонента в анализируемом растворе).
В анодной ИХП последовательность возникновения волн соответствует расположению определяемых металлов в ряду напряжений, а длительность задержек — переходное время т — прямо пропорциональна концентрации их ионов в растворе.
6.5.4. Факторы, влияющие на величину и форму аналитического сигнала
6.5.4.1. Тип индикаторного электрода
Индикаторным электродом обычно служит вращающийся платиновый, углеродный (пирографитовый, углеси-талловый, стеклоуглеродный) или золотой электрод. В инверсионной вольтамперометрии применяют также стационарный ртутный электрод (висящая ртутная капля) и пленочные ртутные электроды (слой амальгамы на серебряной подложке). Твердые индикаторные электроды отличаются от капающего ртутного электрода, во-первых, тем, что они имеют другой интервал поляризации, и, во-вторых, тем, что их поверхность во время регистрации вольтамперограммы не возобновляется (подробнее см. п. 6.5.6).
6.5.4.2. Состав фонового раствора
Полярографический фон или индифферентный фоновый электролит, выполняет несколько функций в ИВ-анализе: - уменьшает сопротивление раствора;
- образует комплексы с определяемыми или мешающими ионами (карбоновые, оксикарбоновые кислоты, аммиак, щелочи, хлориды и др.);
- создает буфер для постоянства pH раствора (NH4OH +
+ NH4CI и др.);
- предотвращает гидролиз многовалентных ионов, в частности при введении кислот HC1O4j H2SO4;
- предотвращает гидролиз и образует комплексы (НС1, НВг, Н3РО4).
Химический состав фона влияет на £п и равновесные потенциалы пар Ме(и)/Ме, если фон содержит лиганды L и образует комплекс МеЛр: при увеличении концентрации L зависимость I = ДЕ) смещается в сторону отрицательных потенциалов.
Кроме того, химический состав фонового электролита влияет на обратимость электродного процесса; вязкость раствора и коэффициент диффузии ионов, адсорбцию ио-нов/молекул фона или комплекса МеЛр на поверхности электрода, а, следовательно, на величину остаточного тока и полезного сигнала; ширину рабочей области потенциалов рабочего электрода (рис. 6.39).
Рис. 6.39. Зависимость остаточного тока на Hg-электроде от состава фона [17]
6.5.4.3. Потенциал электролиза и его длительность
Потенциал электролиза обычно соответствует потенциалу предельного тока и расположен на 0,2-0,3 В отрицательнее потенциала полуволны. На практике строят график I- Еэ и выбирают область предельного тока.
Длительность электролиза зависит от концентрации металла в растворе: чем она меньше, тем необходимо больше Гэл для получения хорошо измеряемого сигнала. Обычно время электролиза не превышает 10 мин. Зависимость Ia =f (Гэл) легко получить из опыта. При постоянстве всех факторов, влияющих на катодный и анодный процессы, т. е. на выбранном фоне, постоянных Еэ, /э, и с в отсутствии истощения раствора и других осложнений (адсорбция, гидролиз и др.) эта зависимость — прямолинейна и выходит из начала координат.
6.5.4.4. Обратимый и необратимый электродный процессы в ИВ
Если в последовательности стадий электрохимической реакции самой медленной (лимитирующей) является диффузия, а остальные стадии протекают относительно быстро
776
Новый справочник химика и технолога
(с большой константой скорости электрохимического процесса), то последний процесс называется обратимым. В этом случае при равновесном потенциале в единицу времени на границе электрод — раствор обменивается так много электронов (большой «ток обмена»), что протекание небольшого внешнего тока от разряда вещества не скажется на равновесии. Говорят, что потенциал мало отличается от равновесного, или перенапряжение при протекании тока мало.
Наоборот, если процесс разряд-ионизации ионов протекает с малой скоростью (константа скорости мала), то процесс называют необратимым, в нем относительно быстро протекают все стадии, кроме электрохимической, лимитирующей процесс. При протекании тока отклонение потенциала от равновесного значения велико. Для практической реализации метода важны следующие выводы и положения теории ИВ:
1. Обратимые пики более узкие, высокие и Еп близки к Ev.
/2
2. Разница в положении пиков анодного и катодного процессов тем меньше, чем больше число электронов п участвует в электродной реакции (рис. 6.40).
Рис. 6.40. Влияние обратимости электродного процесса на положение и форму пиков:
а) обратимый; б) необратимый катодный и анодный процессы [17]
3. Факторы, влияющие на изменение , влияют и на Еп. (Например, при введении в состав фонового раствора комплексообразователя, с увеличением концентрации лиганда сдвигается Еу и, следовательно, Еп.). Степень обратимости зависит как от природы ионов, фона и электрода, так и от скорости изменения потенциала. Чем больше скорость изменения потенциала W, тем сильнее проявляется необратимость, тем сильнее сдвиг катодного Е» в отрицательную, а анодного Еп — в положительную сторону потенциалов.
4. Для накопления металла на электроде (в амальгаме) необходимо задать Еэ отрицательнее ЕП в области предельного тока.
5. Так как потенциал Еп зависит от многих факторов, для идентификации электроактивного вещества, дающего пик, необходимо сделать несколько стандартных добавок
искомого элемента предпочтительно в той же форме, в какой он находится в растворе.
6.5.4.5. Растворенные окислители
Присутствующие в анализируемом растворе окислители — молекулы (О2, Н2О2 NO2), катионы Hg(II) или анионы фона (NO3) — восстанавливаются в рабочей области потенциалов и вызывают возникновение дополнительного тока, который маскирует ток аналитического сигнала. Предложено много методов удаления их из раствора.
Химический состоит в добавлении к раствору соответствующего восстановителя: в кислой среде — аскорбиновой кислоты, в нейтральный и щелочной — сульфита натрия. Однако эти реактивы редко бывают достаточно чистыми для определения следов примесей в методе ИВ. Физический — понижение парциального давления кислорода над анализируемым раствором и как следствие — удаление О2 из раствора. Для этого чаще всего пользуются деаэрацией, т.е. пропусканием через раствор инертного газа: азота, аргона, гелия, реже водорода и СО. Вакуумирование раствора или его замораживание требуют специального оборудования и более продолжительны. Фотохимический — дезактивация кислорода путем фотохимической реакции с радикалами, полученными при введении в раствор фотоак-тивного вещества (фона) и облучении этого раствора УФ-светом. Электрохимический — прекращение перемешивания раствора по окончании стадии электронакопления. На стадии успокоения раствора при потенциале, близком к Е,. происходит обеднение приэлектродного слоя частицами окислителя, разряжающимися на индикаторном электроде.
6.5.4.6. Наложение пиков элементов. Образование интерметаллических соединений
Соседние пики элементов, расположенных близко по потенциалам, могут накладываться, искажаться по форме и затруднять определение высот. Используют ряд приемов для устранения этих помех.
Выбор потенциала электролиза, при котором один элемент выделяется на электроде, а другой — нет. Это возможно, если в катодном процессе «волны» восстановления достаточно раздвинуты (рис. 6.41).
Еэ1 Еэ2 Еэ1 Еэ2
Рис. 6.41. Выбор потенциала электролиза для разделения двух сигналов [17]
Подбор подходящего фона, содержащего лиганд. Для маскировки одного из ионов Е^ и Еп сдвигаются в отрицательную сторону потенциалов тем сильнее, чем более прочный комплекс МеЛр и чем больше концентрация лиганда L. Удаляется элемент путем комплексообразования, осаждения, восстановления до металла, электролизом и т. д. На рис. 6.42 приведены фоны для разделения Т1, РЬ, Sn (известный пример из [18]).
Электрохимические методы анализа
777
PbSnTl । । । НС1
Т1 1 РЬ । Sn । НС1 + en
Т1 1 РЬ 1 Sn i HCI +NH;
Т1 1 р.ь SnNH3 +NajCit
-6,4 1 -0,6 -6,8 -i,o
Рис. 6.42. Разделение пиков свинца, олова и таллия путем изменения состава фона [18]
Смена электролита после электролиза. Накопление ведут на одном фоне (например, Pb, Sn на фоне НС1), а анодное растворение — на другом (на ацетатном буфере эти элементы дают раздельные анодные пики). При этом во время замены электролита контакт между рабочим электродом и электродом сравнения не должен прерываться.
Уменьшение скорости изменения потенциала приводит к сужению пиков, их раздвижению по оси потенциалов, хотя при этом уменьшаются высоты пиков.
Остановка потенциала после получения пика более электроотрицательного элемента позволяет провести анодное растворение 1-го пика без мешающего влияния второго, затем развертка потенциала продолжается и записывается пик второго элемента (рис. 6.43) [10,17].
Рис. 6.43. Прием остановки потенциала для разделения налагающихся пиков:
а) без остановки; 6) с остановкой [10, 17]
Управление аналитическим процессом с помощью компьютера позволяет оптимизировать условия осаждения и последующей съемки кривых, а путем обработки регистрируемых сигналов разделять налагающиеся соседние пики [19].
При совместном выделении нескольких металлов на электроде могут образовываться сплавы или интерметаллические соединения (ИМС). Сигналы анодного растворения таких соединений могут отличаться от сигналов отдельных элементов (быть меньше или больше), что искажает информацию о содержании этих элементов в исследуемом растворе. Так, медь образует ИМС с цинком; с золотом взаимодействуют Cd, Sn, Mn, Zn, As и др. Для устранения этого нежелательного явления на графитовом электроде в анализируемый раствор вводят соль Hg(II) в концентрации в 100 и более раз превышающей определяемые металлы. Осаждаясь вместе с металлами при электролизе, ртуть образует амальгаму — раствор металлов в ртути (о ртутно-графитовом электроде см. п. 6.5.6) в результате сигналы металлов не искажаются. Если в ртути образуются малорастворимые ИМС, например, CuZn, то
сигналы цинка и меди искажаются: пик цинка уменьшается, так как скорость растворения цинка из осадка ИМС меньше скорости диффузии цинка, а пик меди увеличивается за счет совместного разряда меди и ИМС. Избежать образования ИМС можно, используя различные приемы:
— уменьшить концентрации меди и цинка в амальгаме путем разбавления анализируемого раствора или путем увеличения концентрации ионов ртути в растворе и, как следствие, увеличения массы микрокапель амальгамы ртути на РГЭ;
— вести электролиз при потенциале, обеспечивающем разряд ионов меди и выделение ее в амальгаму, в то время как ионы цинка остаются в растворе (-0,8 В в хлоридном фоне);
— внести в раствор ионы элемента, образующего в выбранных условиях опыта более прочное интерметаллическое соединение с одним из реагирующих компонентов (например, галлий образует с медью более прочное ИМС чем цинк; в известной методике [20] поддерживают концентрацию галлия существенно большую, чем меди).
Интерметаллические соединения не всегда являются вредным фактором в методе ИВ: в аттестованных методиках определения мышьяка в различных объектах сигнал As(IIT) получают при растворении его ИМС с золотом (на золотом или золото-графитовом электроде), см. п. 6.5.6.
6.5.4.7. Поверхностно-активные вещества
Поверхностно-активные вещества (ПАВ) — природного (гумусовые) или техногенного происхождения (синтетические ПАОВ, ОП-10 и др.) оказывают разнообразное мешающее влияние на определение тяжелых металлов (ТМ) методом ИВ. При этом: изменяются высоты пиков (чаще уменьшаются); смещаются £п в катодную или анодную сторону; нарушается линейность градуировочных графиков.
Исследование механизма влияния ПАОВ на анодные пики элементов показывает, что наблюдаемые искажения связаны:
— с адсорбцией ПАОВ на поверхности электрода, в результате чего уменьшается свободная поверхность, доступная разряду ионов;
— с образованием комплексов ПАОВ с ТМ в растворе, которые с трудом разряжаются на электроде (необратимые процессы);
— с образованием комплексов ПАОВ с ТМ на поверхности электрода, что может привести к ускорению разряда или увеличению поверхностной концентрации металла (пик может при этом увеличиваться).
Для обеспечения правильности результата анализа методом ИВ используют следующие способы устранения влияния ПАОВ.
1. Деструкция ПАОВ:
— мокрое озоление [21],
— сухое сожжение[21],
— электрохимическое окисление[22],
— УФ- разрушение [23].
2. Введение в раствор неионогенного ПАВ, обладающего большей энергией адсорбции, чем у любого из ПАВ и
778
Новый справочник химика и технолога
ПАОВ, присутствующих в анализируемом растворе [24, 25]. Такое ПАВ, например «Тритон Х-100» [26], ОП-10 (алкилфениловый эфир полиэтиленгликоля) [25] обеспечивает постоянство поверхностных свойств индикаторного электрода и воспроизводимость аналитического сигнала, хотя и искаженного присутствием ПАВ.
6.5.4.8. Катодная инверсионная вольтамперометрия. Адсорбционная ИВ [27]
Рассмотренный выше вариант метода называют анодной инверсионной вольтамперометрией.
Существует другой вариант метода — катодная инверсионная вольтамперометрия. В этом случае вещество концентрируют на электроде в виде продукта окисления. Например, марганец можно концентрировать в виде МпО2 при потенциале предельного тока окисления Мп(П) до Mn(IV). Включив развертку потенциала в направлении более отрицательных потенциалов, регистрируют катодную инверсионную вольтамперограмму восстановления полученного продукта.
Большой интерес для аналитиков представляет современная разновидность инверсионной вольтамперометрии — адсорбционная инверсионная вольтамперометрия. Этот метод основан на предварительном адсорбционном концентрировании определяемого компонента на поверхности электрода и последующей регистрации вольтамперограм-мы полученного продукта. Таким способом можно концентрировать многие органические соединения, а также ионы металлов в виде комплексов с органическими лигандами, особенно азот- и серосодержащими. Концентрирование
проводят в течение контролируемого времени при потенциале максимальной адсорбции. В качестве индикаторных электродов пригодны и стационарный ртутный электрод, и электроды из угольных материалов. Особенно хороши для этих целей химически модифицированные электроды: вследствие наличия реакционно-способных групп, закрепленных на электроде, определяемое вещество концентрируется исключительно на поверхности электрода и в результате чувствительность определения повышается. Адсорбционная инверсионная вольтамперометрия обладает превосходными метрологическими характеристиками: при времени предварительного накопления 60 с и при использовании дифференциального импульсного режима регистрации вольтамперограммы удается достичь пределов обнаружения на уровне Ю10-Ю 11 моль л-1. Метод пригоден для определения многочисленных органических и неорганических веществ, которые не могут быть сконцентрированы электролитически, но способны сильно и воспроизводимо адсорбироваться на электроде.
6.5.4.9. Техника работы в методе ИВ
Техника работы подробно изложена в целом ряде руководств и учебных пособий [2, 5, 17, 28]. Подробное описание лабораторных приемов, способов разведения растворов, измерительные процедуры на приборах содержат также все аттестованные методики анализа (см. Приложение 2). В помощь экспериментатору в Приложении 3 приведены примеры типичных вольтамперограмм для различных объектов анализа.
6.5.5. Электрохимические характеристики процесса разряда-ионизации элементов и условия анализа некоторых материалов методом инверсионной вольтамперометрии (реактивы и вещества высокой степени чистоты, материалы металлургического производства, природные и сточные воды, жидкие и твердые продукты питания)
Экспериментальные значения Еп, /„ и Ьх
для некоторых металлов
Таблица 6.43
в различных основных электролитах [13]
Условия: V- 5 мл, tei - 3 мин, w = 0,4 В/с, концентрация металла ЗЮ8 г/мл, концентрация основного электролита 0,02 моль л *,0,1 и
0,5 г • экв.-л 1 (приведенные значения Еп, 1П и Ь,, являются средними, найденными для трех указанных концентраций электролита). /2
Металл £n,B Itt- 108,A 6X,mB E В D In- 108,a bv, мВ /2 £”п, В In- 108,A , мВ /2
NaCl (NH4)2SO4 KOH
Zn -0,97 8,0 50 -1,00 2,0 50 -1,07 26 40
Cd -0,61 7,0 35 -0,59 3,3 35 -0,63 7,5 30
In -0,46 5,6 7 -0,43 0,6 17 -0,66 8,0 17
Sn -0,44 6,0 15 -0,44 1,5 15 -0,44 0,8 17
Tl -0,50 20,0 150 -0,47 6,0 120 -0,51 13 90
Pb -0,41 6,5 35 -0,40 2,7 40 -0,49 7,7 33
Sb -0,22 18,0 30 -0,18 +0,1 1,4 0,6 17 -0,29 5,2 33
Cu -0,12 3,5 52 +0,06 2,6 50 -0,05 15 35
Bi 0 13,0 20 +0,04 2,4 17 -0,14 9 23
Электрохимические методы анализа
779
Продолжение таблицы 6.43
Металл En,B 4’ 108,A Ьуг , мВ fi’n, В 4- ю8,а b,7, мВ 72 £П,В 4- Ю8,А b,,, мВ 72
HC1 HNO3 Н3РО4
Zn -0,91 7,5 40
Cd -0,64 8,5 35 -0,62 6,0 30 -0,53 2,5 50
In -0,59 0,13 16 0,50 10,3 17
Sn -0,49 1,0 13 -0,49 0,2 12
Tl -0,54 13,5 127 -0,58 14,0 НО -0,55 13,0 НО
Pb -0,44 10,0 30 -0,46 6,0 30 -0,38 6,5 45
Sb -0,18 7,3 28 -0,14 7,0 27 -0,12 8,0 47
Cu -0,16 5,0 57 -0,005 8,0 40 -0,04 8,0 65
Bi -0,06 10,0 17 -0,02 10,0 17 -0,04 5,3 27
Щавелевая кислота Лимонная кислота nh4f
Zn -1,0 0,75 55 -1,02 13 40
Cd -0,60 3,2 47 -0,59 2,3 47 -0,62 8,0 30
In -0,65 1,4 30 -0,53 1,7 20 -0,60 13 23
Sn -0,60 0,2 18
Tl -0,58 13,0 НО -0,48 9,0 140 -0,51 24 93
Pb -0,47 6,7 55 -0,42 3,0 40 -0,43 9,0 30
Sb -0,22 6,6 33 -0,18 0,7 30 -0,05 9,0 14
Cu -0,10 11,5 37 0 7,5 45 -0,07 6,0 47
Bi -0,10 2,3 26 -0,03 2,3 23 -0,08 14 25
Na4P2O 7 LiOH HBr
Zn -1,10 8,0 77
Cd -0,64 3,7 55 -0,65 4,8 33 -0,70 12,5 30
In -0,84 2,2 13 -0,62 0,1 17
Sn -0,51 0,4 15
Tl -0,56 6,7 130 -0,43 12 120 -0,59 13,0 НО
Pb -0,52 7,7 65 -0,55 4,7 42 -0,48 12,5 32
Sb -0,17 0,4 23 -0,35 4,8 40 -0,17 И,7 33
Cu -0,15 4,0 45 -0,18 5,3 37
Bi -0,11 14,5 23 -0,21 9,0 27
H2SO4 НС1О4 Винная кислота
Zn
Cd -0,56 4,5 40 -0,63 5,5 37 -0,58 4,7
In -0,52 2,6 27
Sn -0,61 -i,o 27
Tl -0,55 12,5 100 -0,56 13,0 105 -0,55 13,0 130
Pb -0,40 6,5 35 -0,45 7,2 37 -0,42 5,5 45
Sb -0,13 6,3 27 +0,08 П,7 40 -0,11 9,0 40
Cu -0,015 13,3 30 +0,03 7,1 44 -0,01 9,0 37
Bi +0,005 5,8 17 -0,01 5,3 22 -0,04 4,3 21
O,1M NH4F в 10% С2Н5ОН
Zn -1,02 17,0 45
Cd -0,60 12,0 30
In -0,51 1,6 17
Sn
Tl -0,58 12,0 80
Pb -0,44 14,0 30
Sb -0,18 0,6 13
Cu -0,09 4,7 55
Bi -0,07 10,7 20
780
Новый справочник химика и технолога
Таблица 6.44
Электрохимические характеристики разряда-ионизации металлов на графитовых электродах в некоторых электролитах (с0 = 5 • 1(Г7 М; v = 1 В/мин: /э = 2 мин) [15]
Пояснение. Потенциалы приведены относительно нас. к. э.
Ион Фоновый электролит в Еэ, В £п,В
Си2+ 1 MKNO3 +0,35 -0,6 o.i
1 M KSCN (pH = 2) -0,3 -0,7 -0,25
0,02 M винная кислота + 0,02 M NH4C1 + NaOH (pH = 9) -0,8 -0,4
1 MNaSCN + 1 MKOH -1,3 -0,4
1 M KOH + 0,1 M винная кислота -1,6 -0,5
1 M KOH + 0,1 M этилендиамин -1,2 -0,5
Ag+ 1 MKNO3 +0,05 -0,4 +0,1
1 М KSCN -0,3 -0,6 -0,15
0,2 М H2SO4 +0,02 -0,6 +0,15
Au3+ 1 МНС1 +0,5 -0,2 +0,5
1 M HNO3 +0,75 -0,2 +0,8
1 M KSCN (pH = 8) +0,4 -0,6 +0,4
Zn2+ 1 M KOH + 0,1 M этилендиамин -1,6 -1,3
1 M KSCN (pH = 5) -1,02 -1,3 -1,05
1 M KSCN (pH = 6) -1,1 -1,6 -1,1
Cd2+ 0,1 MHC1 -0,72 -i,o -0,75
1 M KC1 (pH = 2) -0,78 -1,0 -0,72
1 M KSCN -1,2 -0,78
1 M KNO3, (pH = 3) -1,1 -0,64
1 MNH4C1+ 1 MNH3 -0,81 -1,1 -0,82
Hg2‘ 0,1 MKNO3 -0,1 -0,4 +0,1
1 M KSCN -0,25 -0,6 -0,12
In3+ 0,1 M KC1 (pH = 3) -0,9 -1,4 -0,72
1 MKBr -0,8 -1,2 -0,76
1 M nh4scn -0,9 -1,3 -0,70
Продолжение таблицы 6.44
Ион Фоновый электролит в Еэ, В £П,В
Ti+ O,2MNH3 + O,1MNH4C1 (pH = 8) -0,95 -1,4 -0,48
0,1 MNaSCN -1,2 -0,47
0,1 MKC1 -1,2 -0,47
Sn2+ 2MHC1 -0,6 -1,1 -0,55
1 M CH3COOH + 1 M CH3COONa -i,o -0,68
1 M C2H2O4 -i,o -0,48
Pb2+ 0,1 M KNO3 0.4 -0,8 -0,43
0,1 MHC1 -0,48 -i,o -0,55
1 M CH3COOH + 1 M CH3COONa -i,o -0,48
1 M NH4C1 + 20% лимонная кислота 0,85 -0,55
Sb3+ 1 MHC1 -0,25 -0,6 -0,22
Bi3+ 1 M NH4C1 -0,2 -0,5 -0,15
0,1 MHC1 (pH= 1) 0.2 -0,4 -0,08
1 M KOH + 0,25 M этилендиамин -0,6 -0,18
Te4+ 1 MHC1 -0,6 +0,37
1 M KBr (pH = 2) -0,6 +0,25
Fe2+ 1 M KOH + 0,01 M винная кислота -1,4 -1,7 -0,8
0,05 M тартрат Na (pH = 5) -1,6 -0,6
Co2+ 0,2MNH3 -1,2 -0,4
0,1 MK2SO4 -1,4 -0,1
1 M KOH + 0,1 M винная кислота -1,6 -0,6
0,1 M KSCN -1,2 -0,5
Ni2+ 0,1 MNH4C1 + O,1 mnh3 -1,1 -1,2 —0,5
1 M KNO3 + 0,001 M HNO3 -1,1 -1,2 -0,1
0,1 M KSCN -1,2 -0,5
Pd2+ 1 M H2SO4 -0,6 +0,4
1 M HC1 + 1 M KC1 -0,6 +0,4
1 M HC1O4 -0,4 +0,35
Таблица 6.45
Потенциалы полуволны различных металлов в некоторых средах
Принятые обозначения, f — волна сливается с волной растворения ртути; —> — волна отсутствует в данной области потенциалов; [-] малая растворимость в данной среде.
Состав основного раствора Потенциалы полуволны Е}/ , В для /2
Си" Си2+ Bi3+ Sb3+ тг РЬ2+ Sn2+ Cd2+ In3+ As3" Zn2’
Нейтральный (КС1, NaCl и т. д.) -0,2г1 -0,03 — — -0,50 -0,43 — -0,63 -0,572 — -1,02
Кислый — — -0,03 -0,343 — — -0,394 — -0,63 -0,07 —
Электрохимические методы анализа
781
Продолжение таблицы 6.45
Состав основного раствора Потенциалы полуволны Ev , В для /2
от Cu2+ Bi3+ Sb3" Tl+ Pb2+ Sn2+ Cd24 In3+ As3+ Zn2+
1 М щелочь — [-0,5] [-0,6] -1,30 -0,50 [-0,80] -1,26 [-0,80] -1,13 — -1,53
1 MNH3+1 MNH4CI -0,54 -0,25 — — -0,52 [-] — -0,85 [-] — -1,38
1 MKCN —> — — -Щ7 t -0,74 — -1,15 — — —>
1 М KCNS -0,43 т — — — — — — — — -1,11
0,5 М Na2F2 -0,43 — -0,11 -0,72 -0,53 -0,45 -0,77 -0,67 — — -1,17
0,2 М пиридин +0,1 М НС1 -0,29 — — — — — — — — — —
0,05 М ЭДТА (pH 7) + NaAc — -0,31 -0,66 -0,79 -0,50 -1,37 -1,26 -1,27 — (-1,0) —
Кислый тартрат (pH 4,5) — -0,13 -0,33 -0,84 -0,49 -0,52 -0,58 -0,68 — -1,04 -1,27
0,5 М тартрат натрия — -0,16 -0,74 -1,04 -0,49 -0,54 -0,96 -0,68 0,8 — -1,19
Щелочной тартрат (pH 13) — -0,4 -1,04 -1,36 -0,50 -0,79 -1,20 -0,83 — — -1,46
СаС12 — -0,32 -0,2 — — -0,52 -0,46 -0,7 — — -1,08
0,3 М ТЭА + 0,1 М КОН -— -0,50 -0,78 —> — -0,80 -1,43 -0,81 — — -1,49
0,3 М ТЭА + 0,5 М NH3 + NH4C1 -0,54 -0,25 — — — -0,59 — -0,85 — — -1,40
Примечания. 1В 1 М хлориде. 2 В 0,1 М KI.3 В 0,5 М H2SO4 4 В 2 М НС1О4; в 0,5 М КС1.
Таблица 6.46
Примеры определения галогенидов методом инверсионной постоянно-токовой ВА
Определяемый ион Основной электролит Материал электрода Интервал определяемых концентраций Анализируемый материал Г.В Еп, В Примечания
СГ 0,1 MKNO3 + + 0,1 М лимонная кислота Hg 0,210"-2Ю"% (UO2)SO4 +0,4 ~ +0,32
СГ 0,1 MHNO3 Hg 2-10”6— 2-105 моль-л" +0,4 Метод хроноамперометрии
СГ 20% Н3РО4 Hg 10"-10"% Кислоты +0,32 +0Д51
СГ 30% НСЮ4 или 20% H2SO4 Hg 0,210"-110"% Кислоты
СГ 0,1 MKNO3 или 0,05 М H2SO4 Hg 105 моль-л" Природные воды, соли 0в —0,152
СГ 0,06 М KNO3+ + 0,1 MHNO3 Hg 5-10" моль-л" +0,35
СГ НС1О4 Hg > 10 5 моль-л" +0,42 от +0,2 до +0,222
СГ 0,1 MCH3COONa + + 1 М СН3СООН + + 8% этанола (pH = 4,7) Ag 10"мольл" +0,35 -0,05
сг HNO3 + винная кислота Hg 10"-5-10" % CaWO4, SrWO4 -0,05 от-0,25 до -0,35
сг 0,1 MHNO3 или 0,1 MKNO3 Hg 3 10~5- 10" моль-л" +0,4 Конвективный ртутный электрод
сг Н3РО4 Ag 8-10"-2-10" % Н3РО4 и ее соли
782
Новый справочник химика и технолога
Продолжение таблицы 6.46
Определяемый ион Основной электролит Материал электрода Интервал определяемых концентраций Анализируемый материал £э,В Еп, В Примечания
СГ 5MHF + + 1 М HNO3) Ag Микрограммовые количества Та (металлич.) +0,4 Замена основного раствора на 0,1 М KNO3
ВГ 0,1 М KNO3 Hg I0 6 моль-л-1 Природные воды, вода, соли -0,152 -0,282
ВГ 0,1 М СНзСООН Hg > 10-6 моль-л-1 +0331 +0,17
Вг- 0,06 М KNO3 Hg 10 6 моль-л1 +0,35 +0,05
г 0,1 М KNO3 Hg > 5-10 7 моль-л-1 -0,22 -0,52
г 0,1 М CH3COONa + + 0,1 М СНзСООН (рн = 4,7) Ag 4-10-8 моль-л-1 +0,18 -0,22
г 0,1 MNaNO3 Hg 10-7 моль-л-1 +0,25* -О,!!1
г 0,1 MKNO3 Hg 5-10-7 моль-л-1 Соли, природные воды +0,152 -0,522
Г 0,2 М НС1 + + 5 -10-5 М рода-мин В Графит 10-7% +0,8 Окисление и осаждение Щ12С1]
г HNO3 Hg 5-10-8- 3-10-7 моль-л-1 СаСО3, ВаСО3, SrCO3 -0,3 Не мешают 5000-кратный избыток СГ и 50-кратный избыток Вг-
СГ +ВГ Hg ~ 10-6 моль-л-1 +0,33 +0,22 (СГ + +ВГ)2Вг- [СГ]:[Вг-] = = 0,29
Примечания. 1 Хлорсеребряный электрод сравнения. 2 Меркурсульфатный электрод сравнения.
Таблица 6.47
Концентрирование элементов в ИВ в видемалорастворимых оксидов, гидроксидов и солей [15]*
Определяемый элемент Электродная реакция Состав электролита Еэ, В Еп, В Смин. Ю7, М
Т1 тГ + зно-<=» Т1(ОН)3 + 2Г 0,35 М (NH4)SO4 + + NH4(pH = 7) +1,0 -1,2 +0,3 5
Се Се3+ + 4Н2О Се(ОН)4 + е- 0,1 М СНзСООН + + 0,1 М CH3COONa +0,7 +1,0 +0,3 100
РЬ РЬ2+-2е'РЬО2 Буферные растворы (pH = 1-13) от-1,4 до -0,5 от-1,6 до-0,7 от-0,8 до 0,0 2
Сг СгО2- + 4Н2О + 3 е Сг(ОН)3 + 5НО- 0,4 М NH4C1 + 0,1 MNH3 -0,45 -0,7 +0,6 0,4
V V5+ + 2e"^V2O3 0,1 М NH4C1 + NH3 (pH = 8) -0,9 -0,25 0,1
В табл. 6.47-6.49 потенциалы приведены относительно нас. к. э.
Электрохимические методы анализа
783
Продолжение таблицы 6.47
Определяемый элемент Электродная реакция Состав электролита £Х,Б Еэ, В £П, В СМин. • 107,М
Мо MoO2 +2е Мо02 0,5 М (NH4)2SO4 + + 0,05 М ЭДТА -1,4 -0,15 0,3
Мп Мп2+ + 4НО Мп(ОН)4 + 2е~ 2 М (NH4)2SO4 + + H2SO4 (pH = 5) +0,75 +0,9 +0,25 5
Мп Мп2+-2е~ + 4Ю; «=» Мп(Ю4)4 0,02-0,2 М HNO3 + + 3 • 10’3КЮ4 +1,3 +1,4 +1,1 4
Re ReO4 + ЗН2 + Зе~ & ReO2 + ЗН2О 4 5 M H3PO4 -0,8 -0,9 -0,25 0,1
Fe Fe2+ +ЗНО Fe(OH)3 + е~ Н3ВО3 + NaOH (pH = 8) -0,05 -0,5 8
Fe3+ + 2НО^+ Fe(OH)2 - е~ 0,05 М лимонная кислота + NaOH (pH =10) -1,0 -0,7 20
Таблица 6.48
Концентрирование ионов металлов в ИВ в виде малорастворимых соединений с органическими реагентами [15]
Принятые обозначения. RH — 1-нитрозо-2-нафтол; ДДТК — диэтилдитиокарбаминат-ион; Rd+ — катион родамина С; Kt+ — катион трифенилметанового красителя; Ft — фитин; КЛМ —каприлактам; АМП — 1 -фенил-2,3-диметил-4-диметиламинопиразолон-5 (амидопирин); АНТ— 1-фенил-2,3-диметилпиразолон-5 (антипирин).
Определяемый ион Электродная реакция Состав электролита £Э,В Ea, В Смнн • 108, M
Се2+ Ce3+ + Ft«z> [Ce4+-Ft] + e“ (0,1-0,3) M HNO3 + 0,1% Ft +1,2 +0,8 0,5
Sb3+ [SbCl6]3’ + 2Rd+ Rd2[SbCl6]“ + 2e~ 1,5 M KC1 + 0,5 M H2SO4 + 4 • 104 M Rd +0,8 +0,35 4
Sb3+ [Sb(OH)„Cl6_„]3- + 2Kt+^ <=» Kt2[Sb(OH)„Clfr_„]’ + 2e~ 1,2 M HC1 + 5,10 4 M Kt+ +0,8 +0,4 0,2
Мо6+ Mo6+ + 4КЛМ + 5 SCN’ F* Mo(SCN)4(KJ]M)4SCN-e 1 M HC1 (0,5 M H2SO4) + 0,03 M SCN’ + 0,2MKJIM -0,2 +0,1 5
Мо6+ Mo6+ + ЗАМП + 3SCN + 2HO MoO(SCN)3(AMH)3 + H2O-e“ 0,5 M HC1 (0,25 M H2SO0 + + 0,1 M АМП + 0,03 M SCN’ -0,3 +0,1 0,5
Мо6+ Mo6+ + ЗАНТ + 3SCN + 2HO“ f* F* MoO(SCN)3(AHT)4 + H2O-e 0,5 M HC1 (0,25 M H2SO4) + + 0,05 M AHT + 0,03 M SCN’ -0,3 +0,1 0,05
Г 2Г + СГ + Rd+ [RdI2Cl] + 2e~ 0,1 M H2SO4 + 0,1 M KC1 + 5 • 10’5 M Rd+ +0,8 +0,35 0,5
Со2+ Co2+ + 3RH f* CoR3 + 3H+ + e~ 0,4 M NH3 + 0,05 M NH4C1 + + 1,5 • 10’3MRH -0,5 -0,7 1
Со2+ Co2+ + ЗДДТК Со(ДДТК)3 + e~ 0,5 M NH3 + 0,5 M NH4CI + + 5 10’5МДДТК -0,5 -1,1 5
Ni2+ Н1(ДМГ),г + 2HO“ F* Н1(ОН)2(ДМГ),ь2 + 2ДМГ + e~ 0,03 M KOH + 1 • IO6 M ДМГ +0,8 +0,4 0,2
784
Новый справочник химика и технолога
Таблица 6.49
Адсорбционное концентрирование в методе ИВ органических веществ, лекарственных препаратов и ионов металлов в виде комплексов с органическими веществами [15,16]
Определяемое вещество Комплексообразующий реагент Индикаторный электрод Состав электролита Е3, В Еп, В Смин Ю , м
Аскорбиновая кислота УПЭ ацетатный буфер (pH = 5,1) + + 0,2 М тетраэтилацетат -0,45 -0,85 2
Атетрин СРКЭ, УПЭ 0,1 М NaCl + 0,2 М ЭДТА -0,30 -0,75 2
Атропин СРКЭ 0,05 М КОН -0,70 -1,35 20
Диазепам РГЭ ацетатный буфер (pH = 4,5) -0,30 -0,62 20
Додецилсульфат СРКЭ 1 М NaOH -0,70 -1,20 10
5 М NaOH -1,05 -1,20 2
Допамин СРКЭ ацетатный буфер (pH = 4,3) -0,30 -0,90 2
Интеркаин СРКЭ 0,05 М КОН -0,70 -1,25 1
Кодеин СРКЭ 1 М NaOH -0,70 -1,12 10
Кокаин СРКЭ 0,05 М NaOH -0,70 -1,20 10
Лаурилсульфат СРКЭ 1 М NaOH -0,90 -1,30 60
Нитразепам СРКЭ ацетатный буфер (pH = 4,3) -0,70 -1,35 0,7
Папаверин СРКЭ 1 М NaOH -0,70 -1,40 1
Перкаин СРКЭ 0,4 М КС1 + 0,01 М H2SO4 -0,50 -0,82 0,1
Политрин РГЭ 0,1 М КС1 + 0,2 М ЭДТА -0,30 -0,45 2
Рибофлавин СРКЭ фосфатный буфер (pH = 8,3) -0,35 -0,90 4
9,10-Фенан-трахинон УПЭ ацетатный буфер (pH = 5,1) + + 0,2 М тетраэтилацетат -0,40 -0,90 2
Цистеин Cu2+ РГЭ буфер Бриттона-Робинсона (pH = 3,4) -0,40 -0,95 0,02
Си2+ KSCN СРКЭ 1 МКС1 -0,20 -0,6 0,04
Mg, Са, Sr, Ва солохромфиолетовый RS СРКЭ аммиачный буфер (pH = 9,5) -0,80 -1,05-0,08 0,003-0,006
А1 солохромфиолетовый RS СРКЭ 0,2 М ацетатный буфер (pH = 4,5) -0,45 -0,65 0,005
Ti3+, Fe3+ солохромфиолетовый RS СРКЭ 0,2 М ацетатный буфер (pH = 5,1) -0,40 -0,54 (Ti) -0,61 (Fe) 7 0,4
La, Се, Pr солохромфиолетовый RS СРКЭ 0,1 М аммиачный буфер (pH = 9,4) -0,40 -1,1 0,006
Y, Dy, Ho, Yb солохромфиолетовый RS СРКЭ ацетатный буфер (pH = 5,1) -0,75 -1,0 0,01
Zr4+ купферон РГЭ 0,5 М щавелевая кислота -0,60 -1,1 0,06
Ge солохромфиолетовый RS СРКЭ ацетатный буфер (pH = 4,7) -0,50 -1,25 0,05
Mo5+ 8-гидроксихинолин УПЭ фосфатный буфер (pH = 7,6) -0,65 -0,95 0,02
Ni2+, Co2+ диметилглиоксим РГЭ 0,2 М NH4C1 + 0,1 М тетраэтил ацетат -0,75 -0,94 (Ni) -1,04 (Со) 0,1 0,4
Электрохимические методы анализа
785
Таблица 6.50
Определение меди, кадмия, цинка и свинца в различных объектах [13]
Опреде ляемый металл Анализируемый материал Основной электролит £э,в £П,В Электрод Предел обнаружения Метод Примечания
Си РЬ (ме-тал-лич.) HNO3; замена на 0,1 М винную кислоту + 0,25 М NII4(CH3COO) -0,35 СРКЭ 10"7-10”8% иптв В присутствии Вг, Sb
Си Сталь 0,2 М ЭДТА СРКЭ 10-8 молы л-1 иптв В присутствии Hg, Pb, Bi, Мп
0,1 М HNO3; замена на 0,25 М Na(CH3COO ) + 0,1 М винная кислота -0,35 СРКЭ 10^% В присутствии Bi
HNO3 -0,25 СРКЭ 10’5% Потенциометрия; -0,36 В Одновременно определяется Bi
0,3 М ТЭА + 0,3 М NaOH -i,o РПЭ 5 • 10"*% ИПТВ
Си Са3(РО4)г, СаСОз 0,5 М NH4(CH3COO“)+ 0,1 н. винная кислота -0,05 4 • 10“7 % ИПРВ
Си HC1O4 0,2 н. тартрат аммония 1,6 10"5-10’7% ИПРВ Экстракция аце-тилацетоната Си вСНОз
Си Активированный уголь 1 MNH3+ 1 MNH4C1 -0,6 РПЭ 10"5% ИПТВ
Азид свинца 0,25 М NH4(CH3COO“) + 0,1 М винная кислота РПЭ Одновременно определяется Ag
Cd Пищевые продукты 0ДМНС1 СРКЭ 3 • 10^% ИПТВ
-0,8 СРКЭ 10“7 моль-л'1 ИПТВ Одновременно определяется Т1
Cd Си (металлич.) KNO3 -i,o гэ 105%
Cd РЬ, сплавы РЬ СРКЭ 10’7% иптв
Cd Стали 1 МНС1 СРКЭ 4,4- 10"5 % иптв Cd отделяется на дауэкс 50-Х8
Zn Силикаты НАс + Na(CH3COO) (pH = 4,5) -1,2 СРКЭ иптв
Zn Морская вода -1,15 РГЭ 109 моль л’1 иптв Трубчатый электрод
Pb Bi (чистый) HNO3, НС1 РПЭ 10^ % иптв
Pb А1 (чистый) 0,1 МКС1 или 0,01 MHC1 -i,o РПЭ 10"5 % иквв
Pb А1С13 A1C13 + HC1 -1,9 РПЭ 10"6 % оп
Pb А1 (чистый) HC1 (pH = 3) Hg (Au) 10^% иптв Cd не влияет (образуется соединение Cd-Hg-Au)
Pb Zn, Ga (металлич.) 0,16 MHC1 СРКЭ 10"5 % иптв
Pb Моча 10 мл пробы + 1 мл HC1 СРКЭ 5 • 10"8 моль л"1 иптв
786
Новый справочник химика и технолога
Продолжение таблицы 6.50
Определяемый металл Анализируемый материал Основной электролит £Э,В Eni В Электрод Предел обнаружения Метод Примечания
РЬ Сыворотка крови 20% СС13СООН РПЭ 10”8% ИПТВ
РЬ Соединения Cu, Fe 1 МНС1 -0,6 РПЭ 5 • Ю^/о Одновременно определяется Sb
РЬ Расплав ZnSO4 + KCl + NaCl (11:5:4) W (диск) 10 5 моль-л-1
РЬ Сплавы Fe-Cr, Fe-Mn 1 МНС1 -0,8 гэ 10”5%
РЬ Cd (металлич.) 0,4 М НС1О4 +1,4 (РЬО2) Pt 0,1 мкг Хронометрия Растворение в ацетатный буферный раствор
0,1 М NH4NO3 (pH 6) +1,3 (РЬО2) Pt 0,05 мкг Кул.
0,1 МНС1О4 -1,5 РПЭ 3 нг ИПТВ Электролиз из 0,02 мл раствора
0,1 МКС1 -1,0 СРКЭ 10”7 моль л"1
(РЬО2) SnO Кул.
РЬ РЬ, Си Дождевая вода H2SO4 (РЬО2) СУ иптв 10”7%
0,2 М НС1 + NaCl -0,2 (Си); -0,4 (РЬ) СУ, РПЭ 10”5-10”6% ИПТВ
Си, РЬ НС1, H2SO4 -0,7 СРКЭ 10”7% ИХП
Си, РЬ Zn, ZnS hno3 , на -0,8 СРКЭ 106 моль-л1 ИПТВ
0,5 М NaCl -1,0 РГЭ иптв
Zn, Cd As, GaAs од мка СРКЭ ю х109 моль л'1 ИХП
Zn, Cd Al (металлич.) 0,5 М КОН + 1 MN2H4 РПЭ: Hg (Ag) 6- 10”5%Zn 2- lO^/oCd иптв
Zn, Cd Морская вода Оксалат, цитрат, SCN” -1,1 (Zn) РПЭ 10-8 моль л-1 иптв
Zn, Pb Bi 0,1 М НС1 + 1% пиридин -1,25 СРКЭ
Pb, ZnSO4, ZnCl2, Zn(NO3)2, 7 М NH3 + 2% лимонная кислота -1,0 СРКЭ 10”8 моль-л1 ПРП Одновременно определяется Т1
NH4C1, NaBr + ЭДТА РПЭ 5 • 10”6 моль-л"1 иптв Одновременно определяется Bi, Tl
Раствор, содержащий ЭДТА СРКЭ 1(Г6 моль-л1 ПРП Одновременно определяется Т1
Pb, Cd V 0,1 МКС1 + 0,1 МНС1 -1,0 РПЭ 10”5% ПРП
Cu, Pb, Cd Zn (металлич.) на СРКЭ
Cu, Pb, Cd Минеральные воды NaCl, KNO3 СРКЭ 10”8 моль-л”1 иптв
Электрохимические методы анализа
787
Продолжение таблицы 6.50
Определяемый металл Анализируемый материал Основной электролит £Э,В ЕП,В Электрод Предел обнаружения Метод Примечания
Си, РЬ, Zn HC1, NaOH pH = 1-2 -1,3 СРКЭ 10’5% ИПТВ
Си, РЬ, Zn Силикаты 0,3 М ТЭА + 0,3 М NaOH СРКЭ иптв
Си, РЬ, Zn Sn 0,1 М КОН -1,7 СРКЭ Ю^/о иптв
Си, РЬ, Cd, Zn As, GaAs НС1 СРКЭ 10~6% иптв
Си, РЬ, Cd, Zn HNO3 0,1 М винная кислота -1,0 или -1,2 РПЭ 10’8%
Си, РЬ, Cd, Zn Минеральные кислоты, воды 0,1 М винная кислота -1,6 РПЭ 10’5% иптв
Си, РЬ, Cd, Zn Почва, растения НС10,1 М -0,8 (-1,26 Zn) РПЭ 10х моль-л-1 иптв
Си, Cd, Pd, Zn Al, Mg, Ga 0,05 М НС1 -1,5 РПЭ 105% иптв
Си, Pb, Cd, Zn Вращ. РПЭ 106 моль-л 1 иптв
Cu, Cd, Pb, Zn Hg 0,1 МНС1+ 1 % пиридин -1,25 СРКЭ иптв
Cu, Cd, Pb, Zn Mg, Be, HF 0,1 1,0h5MHF По-стоян ный ток/ РПЭ: Hg (Ag) оп
Cu, Cd, Pb, Zn Минеральные кислоты 0,1 М винная кислота -1,6 СРКЭ 10 8 моль-л-1 иптв
Cd, Pb, Zn Cu и соединения Cu 0,1 М КС1 -1,4 СРКЭ 10“9 моль-л1
Cd, Pb, Zn NH3, NH4CI -2,2 РПЭ: Hg (Ag)
Cu, Pb, Zn In 0,1 М винная кислота + 0,25 М NH4(CH3COO ) -0,20 (Си) -0,45 (РЬ) -0,96 (Zn) СРКЭ 10“8 моль-л1
Ga 2 М КВг (pH 2)
Cu, Pb, Cd, Zn Воды Ацетатный буферный раствор, 0,1 М КС1, 0,02 М КС1 СРКЭ 10“8 моль-л-1 иптв
Cu, Pb, Cd, Zn Воды 0,4 М КС1 + 0,3 М НС1 -1,2 СРКЭ 10“8% квв
Cu, Pb, Cd, Zn In, соли In НС1 или NaOH -1,3 СРКЭ 10^% иптв
Cu, Pb, Cd, Zn Воды NaNO3 СРКЭ 106-]07 мольл1 иптв
788
Новый справочник химика и технолога
Продолжение таблицы 6.50
Определяемый металл Анализируемый материал Основной электролит £Э,В £П,В Электрод Предел обнаружения Метод Примечания
Си, РЬ, Cd, Zn Атмосферный воздух 0,1 HNO3 + 0,1 KNO3 -1,35 СРКЭ иптв Минерализация смесью HNO3 + + НС1О4 или HNO3+H2SO4
Таблица 6.51
Примеры практического использования вращающихся дисковых электродов в вольтамперометрии [31]
Определяемый элемент (вещество) Концентрация, М Фоновый электролит Вариант метода ВА Тип вращающегося дискового электрода
Cd, Си, РЬ 10^-10^ 0,1 М KNO3 ВА Торцовый (d= 0,14 мм) микроэлектрод из амальгамируемого графита
Cd, Си, Pb, Zn 10'7-10’s 0,1 МНС1; 0,1 MNH4C1 + NH3 ИВ Стеклоуглеродный и углеситаловый
Cd, РЬ п 10’9 Морская вода идив Дисковый стеклоуглеродный, покрытый пленкой пиролитического графита
С12 Ацетатный буферный раствор с pH = 3,6 ВА Золотой
Си, Mn, Zn п 107 Питательные растворы гидропоники ИВ Стеклоуглеродный типа «отражающая стенка» и кольцо, покрытые пленкой ртути
Fe3+/Fe3+ п 10^ 0,01 М НС1 ВА Платиновый
Fe(CN)’- 0,05 М H2SO4 ВА Диск и кольцо из керамики, покрытой электропроводящим материалом
Fe(CN)’- Fe(CN)'- п • 10-6 KCN ВА Железный со сменными дисками и кольцо из платины или золота
pH (pH 7,0) ВА Золотой и золотое кольцо, электролитически покрытые висмутом
H2O, H2O2 Водные растворы ВА Диск (освещаемый) из TiO2, и кольцо
Hg(II) 2 • 1094 • 10’7 0,1 МНСЮ4 + + 0,0025 М НС1 ИДИВ Золотой, обработанный фотоокислением
Ga(III) п • IO 7 0,5 NaOH ИВ Стеклоуглеродный
Sn(IV) в присутствии Pb(II) п • Ю8 1 МНС1; 1 МНВгвСНзОН ив Стеклоуглеродный и кольцо; пленочный ртутный и кольцо
Те, H2Te 0,1 М НС1О4 ВА Золотой и теллуровый с кольцами из золота и серебра
Zn(II) комплексы и ионные формы Zn «10^ NaNO3+NaCl ИВ Стеклоуглеродный, покрытый ртутной пленкой
Оксианионы халькогенов п- IO 7 Нейтральные и кислые среды ВА Оловянный, свинцовый, никелевый, медный, ртутный
Флюоресцеин п- 106 Водные растворы ВА Медный и кольцо, электролитически покрытые пленкой ртути
Электрохимические методы анализа
789
Таблица 6.52
Условия анализа воздуха, почв, растений, биологических объектов с помощью ИВ [15,16]
Пояснение. Концентрации выражены в нг/дм3; остальные содержания даны в %.
Объект анализа Определяемый компонент Метод Электрод Состав электролита £7 В Метод выделения сп
Воздух атмосферный Hg ИПТВ сэ 10% KI + 2% 12 + 20 г/л аскорбиноваякислота -0,35 Прокачка воздуха через раствор фонового электролита 0,03*
Pb иптв РГЭ 0,2 М КС1 + 0,5 М СНзСООН + 4 • 10 5 М Hg(NO3)2 -1,3 То же 0,10*
рабочей зоны H2S иптв Hg(Pt) 0,4 М NaOH+ 6 • Ю5М Hg(NO3)2 -0,5 То же 0,02*
Аэрозоли
ПЫЛЬ (по данным ФРГ) Cd СРКЭ 0,02 М HNO3 + 2 М винная кислота 0,9 Улавливание фильтром из стекловолокна 0,02*
(по данным США) Cd, Pb, Zn СРКЭ 0,1 М виннокислый аммоний -1,2 Улавливание тефлоновым фильтром 0,15*
Pb, Cu идив РГЭ 0,1 MNaCl + 2- Ю5М Hg(NO3)2 -1,0 Улавливание мембранным фильтром 0,5*
туман (по данным ФРГ) Pb, Cd иптв РГЭ 0,5 М НСЮ4 + 4 • Ю4 М HgCl2 -1,2 Улавливание фильтром из стекловолокна 0,4*
Почвы
США (западные районы) Cu, Pb, Cd, Zn идив СРКЭ 0,01 М HNO3 (pH = 2,2-2,4) -1,3 Озоление пробы HNO3 под давлением 1 • 10~5
ФРГ (кислые) Tl идив СРКЭ 0,2 М НС1 -0,7 Экстракция Т1Вг4-диэтиловым эфиром 2- 10"4
0,7 Ионообменная хроматография 1 • 10’5
(чернозем) Fe3+ ИХП Аёэ 1 М КОН + 0,1 М винная кислота -1,8 Оксалатная вытяжка 1 • 104
Растения Se идив (катодная) РГЭ 1 М NH4C1 (pH = 3,5) -0,05 Сжигание в токе О2 и улавливание поглотителем 2 • 10’5
Se ИПТВ АиЭ 0,05 М НС14 -0,8 Ионообменная хроматография 4- 106
Дентин Cu, Cd, Pb, Zn ИДИВ РГЭ 0,3 М CH3COONH4 -1,2 Разложение пробы смесью HNO3 + + H2SO4 + НСЮ4 2 • 10 7
Кровь и моча Cu, Pb, Cd ИДИВ СРКЭ 0,01 МНС1 -0,9 То же Си-0,5* РЬ-0,02* Cd-0,01*
Se идив (катодная) СРКЭ 0,01 М НС1 -0,3 То же Se-0,1*
Sb ИПТВ РГЭ 6 М НС1 + 1 М NH3 + + 1 м nh4ci -0,45 10*
790
Новый справочник химика и технолога
Продолжение таблицы 6.52
Объект анализа Определяемый компонент Метод Электрод Состав электролита ЕЭ,В Метод выделения Сп
Кровь и моча Bi ИПТВ РГЭ 1,4% Са(СН3СОО)2 + + 3 • 10 3 % Hg(NO3)2 -0,3 Разрушение органических комплексов 1% СгС13 5*
Ni, Со ихп РГЭ 0,2 М КОН + 2 • 10’3 М диметилглиоксим -0,8 Озоление HNO3 при 500 °C и повышенном давлении 10*
Морские организмы, планктон Cd, Pb идив СРКЭ 0,1 М СН3СООН + + 1 М CH3COONa -1,0 Озоление пробы при 500 °C 1,2 • 10°
As идив СРКЭ 0,1 MHCl + 0,1 М Cu (NO3)2 + метанол -0,4 Окислительное растворение пробы 1 • 10 4
Таблица 6.53
Условия анализа природных и сточных вод методом инверсионной вольтамперометрии без предварительного химического концентрирования примесей [14]
Анализируе мый объект Определяемый элемент Тип электрода Еэ, В ЕП,В Индифферентный электролит Нижняя граница определяемых концентраций, М
Морская вода Bi РГЭ, СУ -0,25 -0,1 0,05 М НС1 + 2 • 10“5 М HgCl2 2 • 10’11
Bi -0,9 -0,09 0,02 М НС1 + 4 • 10“5 М HgCl2 2 109
Cu -0,24
Pb -0,48
Cd -0,69
Zn -1,3 -1,05 0,002 М НС1 + 4 • 10 5 М HgCl2
Cd -1,1 -0,68 0,05 М НС1 1 • 10’11
Pb -0,55
Cu -0,30
Zn -1,3 -1,0 0,001 МНС1
Cd -1,0 -0,75 0,5 М NaCl + 6,6 • 10 5 Hg(NO3)2 5 • 10 ’9
Pb -0,5
Cu -0,3
Cu -1,0 -0,25 0,001 М НС1 + 1 • Ю 5 М HgCl2 4 • 10 8
Pb -0,50
Cd -0,65
Zn -1,3 -1,0 0,25 М NaCl + 0,5 М MgCl2 + + 1 • Ю5 HgCl2 2 • 10’7
Cu -0,9 -0,06 0,016 М ацетатный буферный раствор + + 6 • 10’5 М Hg (II) (pH = 4,8) 3 • 10’9
Hg гэ -0,5 0,1 0,025 М HNO3
Hg Au-Э -0,2 +0,6 0,1 М НС1О4 + 3 • 10’ М NaCl 2 • 10’’°
Se Pt-Э +0,35 ИХП 0,6 М HNO3 + 2 • 10"4 М Hg(II)
Природные воды Zn РПЭ -1,4 -1,05 0,1 М СН3СОО + 0,01 М НС1 (pH = 6) 8 • 10~9
Cd -0,65
Pb -0,48
Tl РПЭ -1,0 -0,5 0,005 М KNO3 + 2 10’5 М Hg(NO3)2 6 • Ю7
Pb -0,43
Cu -0,20
Электрохимические методы анализа
791
Продолжение таблицы 6.53
Анализируе мый объект Определяемый элемент Тип электрода Еэ, В Еп, в Индифферентный электролит Нижняя граница определяемых концентраций, М
Природные воды Cd РПЭ -1,0 0,65 НС1(рН = 2,1) 1 • ю9
Pb -1,0 -0,5 5 • 10’8
Cu -1,0 -0,1 5 10*
Zn -1,33 -1,0 НС1 (pH = 3,5) 5 • 10~7
TI -1,2 -0,6 1 М CH3COONa + 0,2 М ЭДТА (pH = 4,7) 6 Ю’10
Ag СУ+Pt -0,4 +0,3 0,05MH2S02
In РПЭ -1,3 -0,60 0,4М НС1 1,5 • Ю9
As Pt-Э -0,2 +0,5 0,24 М НС1 + 1 • 10“5 М Hg(II)
Природные воды, содержащие медь Hg УПЭ -0,5 -0,02 0,1 М KSCN + 0,025 М НС1
Природные воды, кровь, моча, плазма Cd РПЭ -1,2 -0,65 1,7М СН3СООН + 1,2 М CH3COONa 4 10’9
Pb -0,8 -0,5 2 • 10“9
Речная вода Cd То же -0,7 -0,67 0,1 М KNO3 + (1-3) • 10 5 М Hg(NO3)2 1 • 10~9
Pb -0,46
Cu -0,04
Питьевая вода Cu СУ -0,8 -0,45 0,01 М НС1 + 4 • 10’5 М HgCl2 2 • Ю’10
Pb -1,0 -0,5
Cd РГЭ -1,0 -0,7
Сточные воды цинкового производства Ag, As ГЭ -0,65 +0,3 1 М H2SO4 + 2 М НС1 + 2 Ю 5 М CuSO4
Примечание. СУ — диск, Pt — кольцо.
Таблица 6.54
Условия анализа природных и сточных вод методом инверсионной вольтамперометрии с предварительным химическим концентрированием примесей [14]
Анализируемый объект Определяемый элемент Тип электрода Еэ, в £и,В Индифферентный электролит Нижняя граница определяемых концентраций, М Дополнительные операции
Природные воды Ag ГЭ -0,6 -0,2 1 М KSCN+ 0,1 М ЭДТА 5 • 10’11 Экстракция дити-зоната Ag
Au ГЭ -0,2 + 0,5 4-0,6 1 М КВг + 0,03 М НВг Экстракция дити-зонатов металлов
Ag
Hg
In РПЭ -0,8 -0,61 Ацетатно-бромидный электролит (pH = 4,6) 5 10'11 Соосаждение In с Fe(OH)3
Sb РГЭ -0,8 -0,16 0,6 М НС1 + 4 • 10 5 М HgCl2 1 • Ю’10 Соосаждение Sb с Mg(OH)2
TI РГЭ -1,1 -0,62 5 М НС1 + аскорбиновая кислота + 4 • 10’5 М HgCl2 5 • 10’11 Соосаждение Т1 с Mg(OH)2
792
Новый справочник химика и технолога
Продолжение таблицы 6.54
Анализируемый объект Определяемый элемент Тип электрода Еэ, В Еа, В Индифферентный электролит Нижняя граница определяемых концентраций, М Дополнительные операции
Природ-ные и сточные воды Sb РГЭ -0,6 0.2 1 М НС1 + 4 • 10 5 М HgCl2 1 10~п Экстракция диэтилдитиокарбаматов
Bi РГЭ -0,6 -0,2
Природные воды, промстоки Bi РГЭ -0,6 -0,2 3 М НС1 + 4 • 10'5 М HgCl2 1 • 10п Экстракция диэтилдитиокарбамата
Cu РГЭ -1,0 -0,19 0,1 М НС1 + 1 • 10’5 М HgCl2 1 • 10’5 Экстракция избытка меди
Pb -0,42
Речная вода, снег Ag ГЭ -0,6 +0,2 1 М KNO3 4 • 1012 Ионный обмен
Таблица 6.55
Условия анализа природных материалов методом инверсионной вольтамперометрии [14]
Анализируемый объект Определяемый элемент Тип электрода Е3, в Ел, в Индифферентный электролит Нижняя граница определяемых концентраций Дополнительные операции
Горные породы и минералы Au ГЭ 0.7 +0,5 1 М КВг + 0,03 М НВг 1 • 108% Экстракция золота
Минералы Au ГЭ -0,3 +0,85 0,1 МНС1 5 • 10’8% Сорбция на анионитах
Аэрозоли Cd РПЭ 0,9 М H2SO4 5 • 10’9М
Воздух Pb РГЭ 0,4 М НС1 + 1 М СНзСООН И • 104 М HgCl2 4- 10“8М
Морские водо-росли, рыбы и организмы Bi РГЭ 0,2-0,6 М НС1 + + 4 • 10 х М Hg(NO3)2 2 • 10’8М
Cu
Pb
Cd
Zn
Зола растений Au ГЭ 1 МНС1
Таблица 6.56
Условия анализа реактивов и веществ особой чистоты методом инверсионной вольтамперометрии [14]
Анализируемый объект Определяемый элемент Тип электрода £Э,В Еп, В Индифферентный электролит Нижняя граница определяемых концентраций, %
Хлоро-водородная кислота Cd РГЭ -1,0 -0,65 0,5 М НС1 + 1- 104 М HgCl2 1 • IO7
Cu + Bi -0,25
Pb -0,55
Au ГЭ -0,5 +0,5 0,1 МНС1
Cu СУ -1,0 -0,35-0,15 1МНС1
As ГЭ -0,8 -0,26 1 М НС1 + 2 • 10 5 Cu (NO3)2
Электрохимические методы анализа
793
Продолжение таблицы 6.56
Анализируемый объект Определяемый элемент Тип электрода Еэ, В Еп, В Индифферентный электролит Нижняя граница определяемых концентраций, %
Азотная кислота Си РГЭ -1,0 -0,3 0,1 М HNO3 + 1 • 10~5 М Hg(NO3)2 1,5 • 10г*
РЬ -0,5
Си РПЭ -0,8 -0,15 0,1 MKNO3
As ГЭ -0,7 +0,25 1М HNO3 + 1 • 10“5 М Cu (II)
Pd ГЭ -0,6 +0,2 2МКС1 10’5
2МКВг
0,2М НС1
Хлорная кислота Pb СУ -1,0 -0,5 0,1 МНС1О4
Au СУ -0,6 +0,7 0,1 МНСЮ4
Серная кислота Си СУ -1,0 -0,35-0,15 1 М H2SO4
As ГЭ -0,7 +0,25 1 М H2SO4
Лимонная и винная кис-лоты Ni ГЭ -1,0 +0,7 0,1 М NaOH + 1 • I0 5М диме-тилглиоксим 1 • 10"5
Co ГЭ -1,6 -0,75 0,5 М цитрат натрия (тартрат натрия) + NaOH (pH = 9-11) 1 • 10~5
Хлорид калия Pb ГЭ -0,9 -0,45 1 МКС1
Cd ГЭ -1,0 -0,70 0,0002 М КС1 1,2 • I0 7
Sb ГЭ +0,8 +0,35 2,5 М KCl t 4 - 10 ' М родамин С 3 • 10’6
Хлориды и перхлораты щелочных металлов Ag СУ -0,6 +0,05 0,1 MHCl + 0,1 MNaClO4
Хлорид кальция Co ГЭ -0,5 -0,75 1 М СаС12 + 0,1 М NH4C1 + + 2 • Ю ! М а-нитрозо-0-нафтол (pH = 8) 1 • 10"6
Нитрат калия Ag ГЭ -0,39 +0,25 1 MKNO3 2 • 108
Pb -1,0 -0,5 1 • 106
Tl -1,25 -0,4 4 • 10“*
Pb ГЭ -0,9 -0,45 1 MKNO3
Ag СУ 0,2MKNO3; 1MKNO3 2,5 • 10^
ГЭ -0,4 +0,27
ГЭ -0,2 +0,25 1 M раствор анализируемой соли 4 • 107
Ni Pt-Э -1,2 -о,з 0,1 MKNO3+0,01 М 1 10’5
Аи-Э KSCN
Нитрат натрия Cu СУЭ -1,0 -0,35-0,15 1 М NaNO3
Нитрат алюминия Ni ГЭ +1,0 +0,8 0,2 М A1(NO3)3 + 0,5 М КОН + + 1 • 10“5 М диметилглиоксим 1 • 10 4
Нитрат свинца Ag ГЭ 0 +0,25 Раствор Pb(NO3)2 1 • 10"
Перхлорат натрия Cu СУ -1,0 -0,35-0,15 1 М NaClO4
Hg СУ -1,0 -0,05 0,1 MNaClO4
Сульфат лития Hg ГЭ -0,5 -0,1 1,8 М KSCN или 0,1 М KSCN
794
Новый справочник химика и технолога
Продолжение таблицы 6.56
Анализируемый объект Определяемый элемент Тип электрода Еэ, В Еп, В Индифферентный электролит Нижняя граница определяемых концентраций, %
Роданид калия Hg ГЭ пропитанный воском -0,6 0.1 0,1 М KSCN 8 • 10’7
Си СУ -1,0 -0,35-0,15 1 М раствор анализируемой соли
Ацетат свинца Ag ГЭ 0 +0,25 Раствор анализируемой соли 1 10’5
Сульфиды кадмия и цинка Си ГЭ -0,6 -0,25 CdCl2 ZnCl2 3 • 10~5
Вольфрамат натрия Ni ГЭ +1,0 +0,7 0,2 М КОН + 1 • 10’5 М диме-тилглиоксим 1 • 10’5
Мо ГЭ +0,4 при выключенной ячейке -0,25 Ацетатный буферный раствор + ди этилдитиокарбамат 5 • 10^
Молибдат аммония Со ГЭ -0,5 -0,7 1,25М NaSCN + 2,5 М КОН 1 10^
Си ГЭ -0,7 -0,4 1 М КОН + 0,04 М винная кислота + 0,1 М NH4C1 1 105
Fe ГЭ -1,5 -0,9 1,5 М КОН + 0,005 М винная кислота + 0,001 М Na2S 1 • 10’5
Молибдаты РЗЭ Fe ГЭ -1,5 0.9 1М КОН + 0,05 М винная кислота 1 • 10"4
Со ГЭ -1,6 -0,8 0,25 М лимонная кислота + +0,4 М NH4OH + 0,05 М NH4CI + + NaOH (pH = 8) 3 • 10’5
Фосфор, борный ангидрид Zn РГЭ -1,4 -1,0 1 МКС1 + 4 • 104MHgCl2 10’6
Соли щелочных и щелочноземельных элементов Ag ГЭ -0,4 +0,25 1 М раствор анализируемой соли 1 • 10‘5
Тиокарбамид То же ГЭ 0 +0,25 0,5 М CH3COONa + + HNO3 (pH = 4) 5 • 10’6
Аи ГЭ -0,8 +0,5 0,1 МНС1 + 0,5МКВг 1 • 105
Реактивы высокой чистоты Zn РГЭ -1,3 -1,05 1МКС1 1 • ю-7
Cd -1,0 -0,75 (1 М НС1) + 2 • 10’5 HgCl2
TI -1,1 -0,65
Pb -1,0 -0,55
Sb -0,6 -0,25
Си СУ -1,6 -0,5 1 М КОН + 1 М винная кислота
Se +0,25 -0,2 0,1 М раствор реактива + + 1 • 10’5 М Cu(II)
Электрохимические методы анализа
795
Таблица 6.57
Условия анализа полупроводниковых материалов
методом инверсионной вольтамперометрии [15,16]
Анализируемый объект Определяемый элемент Электрод £^э, В £п, В Индифферентный электролит Нижняя граница определяемых концентраций, % Дополнительные операции
Арсенид галлия (пленки) Те ГЭ -1,1 -0,2 2М НС1 1 • 10'5 Послеэлек-тролиз
Арсенид галлия и мышьяк Ag ГЭ -0,4 +0,05 GaBr3+ 0,01 MNH4OH + + 0,001 М NH4NO3 2 • 10'5 Удаление As в виде AsBr3
Арсенид галлия и галлий Au ГЭ 0-(0,40) +0,85 20% лимонная кислота + + 0,4 М НС1 3 • 10-5
Антимонид индия (пленки) Те ГЭ -1,1 -0,2 2МНС1 5 • 10^ После-электролиз
CdxHg!_xTe Те, Cd ГЭ -1,0 Растворы НС1+ 4 10“5 Hg(NO3)2 5 • Ю5
Ge-Sn-P Ag ГЭ -0,4 +0,08 1 м nh4ci + 1 м nh4oh 5- 10^
SnC>2-ZnS • Mn-SiC>2 (пленки) Zn РГЭ -1,3 -1,0 0,1 MNaCl «2- Ю 5
Mn -1,9 -0,8 0,1 MNaCl
Sn -1,0 -0,6 ЗМ НС1
Cu -1,0 -0,35 ЗМНС1
Cd -1,0 -0,75 ЗМНС1
Условия анализа руд и получаемых из них металлов методом инверсионной вольтамперометрии [15,16]
Таблица 6.58
Анализируемый объект Определяемый элемент Тип электро да £Э,В Ян, В Индифферентный электролит Нижняя граница определяемых концентраций, % Дополнительные операции
Руды Ag СУ -0,6 +0,07 0,1 MKNO3; 1 МНС1 5 • 10^ Экстракция дитизоната Ag
ГЭ -0,6 -0,2 1 М KSCN + 0,1 М ЭДТА 5 • 10'9 Экстракция дитизоната Ag
ГЭ -Ю,4 +0,4 0,02 М NH4CI + 1 М NH4OH 5 • 10^ Экстракция Ag дибутилсуль-фидом
СУ -0,1-1,2 +0,3-0,1 0,5 М KNO3, дитизон bO,25MNH4SCN Экстракция дитизоната Ag
ГЭ -0,1 +0,15 1 м nh4oh + 1 М NH4NO3
Au СУ -1,0 Хлордиметилтриоктиламмония + метанольный раствор 5 • Ю"1 Экстракция АиСЦ и дитизоната Au(III)
ГЭ -0,20 +0,5-0, 6 1 М КВг + 0,03 М НВг 1 • 10'8 Экстракция дитизоната Au(III)
ГЭ -1,0 +0,4 Смесь (1:3) HCI + HNO3, разбавленная (1:9) 2,5 • 10~6
Руды суль-фидные Bi ГЭ -1,0 -0,15 1 М NH4CI + 0,05 М лимонная кислота Соосаждение BinTl с Fe(OH)3
Tl -0,6
796
Новый справочник химика и технолога
Продолжение таблицы 6.58
Анализируемый объект Определяемый элемент Тип электрода £П,В Индифферентный электролит Нижняя граница определяемых концентраций, % Дополнительные операции
Руцы и продукты их обогащения РЬ ГЭ -0,55 ихп 1 МНС1
Руды, сплавы на медной основе, катодный порошок Pd ГЭ -0,5 +0,60 2 М H2SO4 + 0,4 М КС1 + 4 • 10’5 HgCl2 2- 1045 Экстракция Pd(II)
Медь Cd РГЭ -1,1 -0,65 1 МКС1 + 4- 10 'М Hg(II) 1 • 10’5 Отделение Си электролизом
Pb ГЭ -1,0 -0,5 1 М КС1 pH = 6 1 • 10’5 Соосаждение Pb с Fe(OH)3
Sb ГЭ -0,45 -0,25 1 М НС1 + 4% ли-монная кислота + 2 • 10'5 М HgCl2 5 • 10 4’ Соосаждение Sb и Bi с Fe(OH)3 и La(OH)3
Bi ГЭ -0,45 -0,19
Те ГЭ -0,35 +0,25 1 МНС1 4- 1045 Соосаждение примесей с Fe(OH)3
Bi -0,35
Sb РГЭ -1,0 -0,2 1 М НС1 + аскорбиновая кислота + + 2 • 10 5 М Hg(II) 1 • 106
Pb
Ag ГЭ +0,5 -0,1 0,01 М аммоний-нонитратный буферный раствор 4- 10^ Сорбция Ag на анионите
Катодная и вайербар-совая медь, продукты медного производства Au ГЭ -0,3 +0,5 0,1МНС1 1 • IO’5 Экстракция и послеэлектролиз
Никель Pb ГЭ -0,5 -0,5 0,25-1,0 МНС1
Bi -1,0 -0,2
Bi РГЭ -0,40 -0,10 1 МНС1 2 • 10 5 Сорбция примесей на анионите АВ-17
Zn -1,3 -0,95 1 М КС1 + 2 • 10’5 Hg(II) 4- 104
Cd -1,3 -0,68 5 • 10 5
Pb -1,3 -0,52 1 • 10 4
Mn ГЭ +1,4 +0,9 0,02-0,2 М HNO3+ + 3 • 10“3 М перйодат калия 1 • 10’5
Кадмий Ag СУ -0,65 +0,10 1 М CdCl2 + 1 М НС1 5 • 107 Послеэлектролиз
ГЭ -0,6 -0,2 IM KSCN+ 0,1 М ЭДТА 5 • Ю9 Экстракция дитизоната Ag
-0,2 + 0,2 (NO3)2 + HNO3 (рН = 1) 2 • 10 s
Sb РГЭ -0,5 -0,25 CdCl2 + 2 М НС1 + + 1 • 10 4 М HgCl2 5 • Ю5
As ПЭ или Аи-Э -0,7 + 0,25 1 М НС1 + 1 • 10 5 М Си(П) и 1 М НС1 4- 10 6 Отгонка Cd в вакууме
Свинец Ag ГЭ -0,2 + 0,2 Pb(NO3)2 + HNO3 (рН= 1) 2 • 10’5
Электрохимические методы анализа
797
Продолжение таблицы 6.58
Анализируемый объект Определяемый элемент Тип электрода £Э,В £п,в Индифферентный электролит Нижняя граница определяемых концентраций, % Дополнительные операции
0.4 -0,05 1 MNH4SCN или 0,05 М KSCN 1 • 10’8
Sb РГЭ -0,5 -0,25 6 М НС1 + 0,06 М Pb(NO3)2 + 11 О’4 MHgCl2 5 • Ю5
Олово Ag ГЭ -0,3 +0,25 Sn(SO4)2 + 5 М H2SO4 1 • ю4
Sb РГЭ -0,6 -0,12 1 М H2SO4 + 0,5 М НС1 1 • 104
Bi РГЭ -0,6 -0,10 0,02 М НО+ 4- 10’5 MHg(NO3)2 1 • 10 4
Цинк Cd РГЭ -0,7 0,5 М НС1 + 4 • 105 М Hg(NO3)2 2 • 104
Pb -0,9 -0,55
Cu 0,3
Cd РГЭ -0,9 -0,65 0,1 М СН3СООН + + 0,1 М CH3COONa + 1105 HgCl2 1 • 104
Pb -0,48
Cu -0,15
Sb ГЭ -0,8 -0,25 4% винная кислота
Сурьма Au ГЭ -Ю,2 +0,5 1 МНС1 5 • 104
+0,20 +0,85 1 МНС1 1 • 104 Отгонка Sb
Серебро Sb ГЭ -0,6 -0,2 1 МНС1 1 • 10 6 Экстракция Ag дибутил-сульфидом
Ниобий Cu СУ -0,6 0,1 М Н2С2О4 + 0,4 М Н2С4Н4О6 1 • 104
Тантал Ag СУ -0,2 +0,2 1 М HNO3 + 1 М HF 2 • 104
Cu СУ -1,0 -0,15 0,1 МНС1 + 0,5М HF 2 • 105 С переносом электрода
Алюминий Ga РГЭ -1,5 -0,85 1 М КС1 или 1 М KSCN + 1 • 104 HgCl2 1 • 104 Экстракция Ga хлорексом
Бериллий Al ГЭ 0,0 +0,87 0,2 М CH3COONa + 100 мкг-мл4 соло-хромфиолетовый RS 4- 104
Хром Sb ГЭ +0,8 +0,4 1,5МНС1 + 4- 104 М малахитовый зеленый 3 • 10 6
Хром и железо Bi РГЭ -0,3 -0,2 1 М NH4C1 + 0,2 М лимонная кислота + 4- 10"5MHgu (pH *2) 2 • 10 "7
Pb РГЭ -0,9 -0,5 1 М НС1 + 4 • 10 5 HgCl2
Железо, медь и висмут Sb ГЭ -0,5 -0,17 1 МНС1 Экстракция Sb, ионный обмен
798
Новый справочник химика и технолога
Продолжение таблицы 6.58
Анализируемый объект Определяемый элемент Тип электрода Е3, В £П,В Индифферентный электролит Нижняя граница определяемых концентраций, % Дополнительные операции
Сплавы
Медные (бронзы и латуни) Sb ГЭ + 0,8 + 0,35 1 М НС1 + 1 • 10 3 М родамин С 5 • 10 4 С переносом электрода
Свинцовые Au ГЭ -0,30 + 0,85 0,1 МНС1 5 • Ю5
Цинковые Cd РГЭ -0,9 -0,65 0,1 МСН3СООН + 0,1 М NaCH3COO + 1 • Ю 5 М HgCl2 1 • 10"*
Pb -0,48
Cu -0,15
Бериллиевые Al ГЭ 0,0 + 0,87 0,2 М CH3COONa + 100 мкгмл1 солохромфиолетовый RS
Жаропрочные Pb РГЭ -1,0 -0,45 1 М НС1О4 + 1 М Н3РО4 + + 2 • 10~5 М HgCl2 2 • 10~5
Sn РГЭ -1,1 -0,6 2 М НС1 + 5 10“5 MHgCl2 2 • 10’6
Ферроспла-вы, содержащие хром Bi РГЭ -0,5 -0,15 1 М NH4CI + 20% лимонная кислота + 4 • 10 5 М HgCl2 2 • 10’6
Pb -1,0 -0,55
Ферромарганец Sb РГЭ -0,55 -0,18 1 М НС1 + 2 • 10’5 М HgCl2 4 • 10’5 Соосаждение МпО2
Ферро-ниобий Bi РГЭ -0,35 -0,18 1 М NH4CI + 20% лимонная кислота + 4 • 10 5 М HgCl2 1 • ю6
Pb -0,9 -0,57 1 • 10 s
Ферробор Pb РГЭ Раствор анализируемого вещества + аскорбиновая кислота + + 4 10’5 М HgCl2 2-10"*
Медно-ни-келевые концентраты, растворы аффинажа Pt ГЭ СУ +0,2 -0,36 2 М НС1 + 2 • 10“2 М ЭДТА + 0,03 М Sn(IV) 6 • 10’7
Гальвани-ческие покрытия Au ГЭ +0,20 +0,85 1 МНС1 1 • 10^
Ag ГЭ -0,4 +0,05 0,01 MNH4OH + + 0,001 м NH4NO3 (рн = 9)
Катализаторы Pd ГЭ +0,2 (+0,6) Хлоридные растворы 1 • 10"*
Растворы, содержащие Fe(III), тиокарбамид, цианистые растворы Au ГЭ -0,8 +0,5 0,1 М КВг + + 0,5 М НВг 1 10”6 экстракция дитизоната Au(III)
Цианистые растворы ГЭ -0,80 +0,58 0,1 МНС1 + + 0,2 М КВг
Электрохимические методы анализа
799
Таблица 6.59
Условия анализа некоторых материалов металлургических производств с помощью ИВ [14]
Принятые обозначения. ХН — химическое накопление; ТМ — тиомочевина.
Определяемый элемент Метод Фоновый электролит £э, В £П,В Электрод Анализируемый материал сн 104, %
Ag ИПТВ 0,1 MKNO3 + 0,01 м ЭДТА -0,3 + 0,26 УПЭ Медь и медные сплавы 5
1 М KNO3 (pH = 1-2) 0,0 + 0,38 ГЭ Никель 1
Ag, Си ИПТВ HF + HNO3 (предэлектро-лиз) -0,2 (Ag) -1,0 (Си) СУ Тантал 0,5 (Ag) 0,1 (Си)
0,1 М НС1 (электрорастворение) -0,25 (Ag)
+ 0,15 (Си)
Ag ИПТВ 1 М Ga(NO3)3 0,0 + 0,30 СУ Галлий 0,02
Ag ИПРВ 0,05 г/л CdCl2 -0,65 + 0,12 ГЭ Кадмий 0,2
Ag ИПТВ 0,1 М L1CIO4 в ацетонитриле -0,3 + 0,18 СРКЭ Ртуть 2,0
As ИПТВ 2 М НС1 + 0,35 М Hg2+ + + 0,4 М KI 0-4 -0,7 РГЭ Железные руды 0,01
As ИПТВ 3 М H2SO4 -0,7 -0,15 Аи-Э Кремний
As ИПРВ ЗМНС1 +0,015 HI + 1 5 • I0"1 М Си2+ -0,4 -0,72 СРКЭ Галлий 0,03
As, Sb ИПТВ 2 М H2SO4 -0,3 (As) -0,05 (Sb) -0,35 + 0,21 + 0,34 + 0,04 згэ Медь Медь 0,1 0,5
As ихп 7МНС1
7МНС1+ 10MN2H5+ -0,35 + 0,04 згэ Железо, никель, хром, медь, олово, свинец 0,5
As ИПТВ 4МНС1 -0,5 + 0,4 АиЭ Олово 0,5
Au ИПТВ 0,1 МНС1 -0,3 + 0,45 УПЭ Медь и продукты медного производства 0,1
0,5 М Н2СЮ4 + 4 М НС1 +0,5 + 1,16 УПЭ Кремний 0,01
0,2 М КС1 + 5 М НС1 -0,6 + 0,38 ГЭ Сурьма, железные руцы 0,01
1 МНС1 -0,4 + 0,42 ГЭ Железо, никель, хром, олово 0,1
Bi, Sb ИПТВ 1,1 МНС1О4 + 0,2 М ТМ + 2,1 М H2SO4 ХН 0,0 (Sb) + 0,1 (Bi) СРКЭ Медные сплавы 1,0
Bi, Pb ИПРВ 6% NH4C1 + 20% лимонная кислота + 1 • 10^ М Hg2+ -0,9 -0,57 (Pb) -0,18 (Bi) ГЭ Ферросплавы, железо, ниобий 0,1 (РЬ) 0,02 (Bi)
Bi, Pb, Zn ИПТВ 1 М НС1 (Bi) 0,5 М H2SO4 (Pb, Zn) -0,3 (Bi) -0,18 -0,92 СРКЭ Алюминий 0,1 (Bi, Pb) 0,5 (Zn)
-1,2 (Zn, Pb) -0,52
Bi, Cu ИПТВ 0,25 М НС1 + п М NiCl2 -0,45 (Си) -0,3 (Cu) Никель
-0,30 (Bi) -0,12 (Bi)
800
Новый справочник химика и технолога
Продолжение таблицы 6.59
Определяемый элемент Метод Фоновый электролит г„в £П,В Электрод Анализируемый материал сн • 104, %
Bi, Sb, Pb ИПТВ 1 М НС1 + 5 • Ю 4 М Hg2+ -1,0 -0,55 (РЬ) ГЭ Медь 0,1 (РЬ)
-0,28 (Sb) 0,03 (Sb, Bi)
-0,15 (Bi)
Bi иптв 0,75МНС1+1 • Ю4М Hg2+ -0,8 -0,19 ГЭ Оксид сурьмы 2
Bi, Sb, Те иптв 0,2 М НС1 (Те) -0,45 (Те) + 0,37 ГЭ Аффинированное серебро 0,05 (Bi, Sb)
0,2 М НС1 + 1 • Ю4 Hg2+ (Sb, Bi) -0,5 -0,28 (Sb) 0,1 (Те)
-0,15 (Bi)
Bi, Sb ИПРВ 1 МНС1 -0,28 -0,22 (Sb) СРКЭ Кадмий и цинк 1,2 (Sb) 0,5 (Bi)
-0,15 (Bi)
Bi, Cu иптв 1 M HF + 1 M HNO3 (предэлектролиз) -1,0 -0,24 (Cu) СУ Цирконий 0,01
5 M НС1 (электрорастворение) -0,17 (Bi)
Bi, Sb иптв 1 МНСЮ4 + 0,1 МТМ** + + 0,2 М этилацетат ХН 0,1 (Sb) 0,0 (Bi) СРКЭ Олово 10 (Sb) 4 (Bi)
Cd, Pb, Cu, Zn иптв, идив тартрат Na (pH = 3,5) -1,2 -0,98 (Zn) -0,65 (Cd) -0,50 (Pb) -0,05 (Cu) СРКЭ, РГЭ Цирконий, оксид циркония 2,0 (Zn) 0,05 (Cd) 0,08 (Pb) 0,06 (Cu)
6.5.6. Свойства индикаторных электродов [13—17, 28-33]
Требования к электродам для инверсионной вольтамперометрии можно сформулировать следующим образом [13]:
1. Электрохимическая инертность в широкой области потенциалов.
2. Высокое перенапряжение выделения водорода и кислорода.
3. Низкий ток емкости двойного слоя (отсутствие пор и сильно выраженной шероховатости поверхности).
4. Низкое омическое сопротивление.
5. Возможность достаточно просто воспроизводить поверхность.
Все это должно обеспечить необходимые метрологические характеристики анализа: высокую чувствительность и воспроизводимость результатов, низкий предел обнаружения.
Типы индикаторных (рабочих) электродов
Стационарные ртутные электроды нашли широкое применение благодаря своим выгодным электрохимическим свойствам и особенно благодаря широкой катодной области рабочих потенциалов. Перенапряжение водорода на ртути в катодной области составляет около -1,000 В, достигая в щелочных растворах -2,6 В (н.к.э.). С понижением pH потенциал выделения водорода сдвигается к по
ложительным величинам (на 0,059 В на единицу изменения pH). Положительная область потенциалов ограничена растворением ртути при +0,4 В (н. к. э.) в некомплексообразующей среде. В настоящее время (особенно за рубежом) используют стационарные ртутные капельные электроды (СРКЭ). Именно на них проведена значительная часть цитируемых работ по инверсионнным электроанали-тическим методам. Различают два вида СРКЭ:
а) висящий ртутный капельный электрод со стеклянным капилляром (рис. 6.44);
б) стационарный ртутный капельный электрод на металлической подложке (рис. 6.45).
У висящих СРКЭ ртутная капля «подвешена» на ртутном столбике в капилляре с внут-ренни диаметром 0,15-0,50 мм. Ртуть выдавливается в капилляр поршнем (иглой) с помощью микрометрического винта.
Каякд
Hg
Микрометрическая школа
Резиновое уплотнение
Статной
Рис. 6.44. Конструкция висящего стационарного ртутно-капельного электрода с капилляром [13]
Электрохимические методы анализа
801
У СРКЭ на металлической подложке инертные контакты выполняют чаще всего из золота, серебра или платины. Одной из наиболее удачных и отработанных конструкций является электрод У ндеркоф л ера-Шейна [13].
Его отличительной особенностью является незначительная утопленность поверхности металлической подложки от среза стеклянного капилляра, что позволяет лучше удерживать капельку ртути на кончике капилляра, допуская даже вращение электрода (платина травится «царской водкой» так, чтобы впаянный конец Pt-проволочки стал на ~ 0,1мм ниже поверхности стекла капилляра).
Рис. 6.45.
Конструкция висящего стационарного ртутно-капельного электрода на металлической подложке [13]
Твердые вращающиеся электроды имеют различное конструктивное исполнение, но включают в себя сходные по функциям элементы (рис. 6.46).
Внешний токосъем
Проводник
Изолятор (фторопласт, пента пласт)
Электродный материал (СУ, углеситалл, Au, Ag.Pt)
Рис. 6.46. Конструкция вращающегося твердого электрода
Ртутный пленочный электрод (РПЭ) на Ag-субстрате— представляет собой тонкую пленку ртути (20-100 мкм), нанесенную на отшлифованную серебряную проволоку или путем погружения ее в чистую ртуть, или путем электролиза. Серебряная подложка крепится в инертном материале — стекле, фторопласте или полиэтилене. Поверхностная пленка ртути тщательно растирается (калькой, фильтром) по серебру, для предотвращения контакта серебра с раствором.
Импрегнированный графитовый электрод (ГЭ) Получают путем пропитки под вакуумом заготовок из спектрального угля специальными составами: воском, смесью парафина с полиэтиленом, парафина с полистиролом, или эпоксидными смолами. Полученные заготовки, например,
запрессовывают в стержень из инертного материала и шлифуют торец для получения рабочей поверхности (0,1-0,2) см2. ГЭ имеют ограниченное применение, так как при электролизе на них металлы взаимодействуют между собой и аналитические сигналы искажаются. ГЭ применяют наряду со стеклоуглеродным электродом (СУЭ) СУ-850, СУ-2000, СУ-2500 для определения никеля, кобальта, ртути, металлов благороднее ртути.
Ртутно-графитовый электрод (РГЭ) имеет более широкое применение чем ГЭ. Его получают, нанося на подложку ГЭ пленку Hg (фактически мельчайшие капельки ртути, рис. 6.47) путем электролиза — заранее, или in situ, т. е. непосредственно в анализируемом растворе. Для этого в раствор вводят соль Hg(II) в концентрации п-105 моль-дм-3 и проводят электронакопление ртути вместе с определяемыми металлами.
Рис. 6.47. Микрофотография РГЭ, сделанная методом «лагуны» (сквозь раствор): на поверхности полированного стеклоуглерода видны микрокапли ртути размером 3-5 мкм, одиночные дефекты поверхности — 7 и след переместившейся при вращении электрода капли Hg — 2, увеличение х 600
Золотографитовый электрод (ЗГЭ) получают нанесением тонкой пленки золота на поверхность ГЭ путем электролиза раствора НАиС14 концентрации 100-200 мг-дм-3. Электролиз проводят из перемешиваемого раствора от любого источника постоянного тока (Е = -0,2 —0,4 В, время порядка 30-300 с).
ЗГЭ дает наивысшую чувствительность при определении мышьяка за счет образования ИМС. Одновременно определяют Hg, As и др. благородные металлы. Применяется также золотой электрод в форме диска для определения Hg, As.
У льтр амикроэл ектроды
Описанные выше традиционные твердые электроды имеют линейные размеры (диаметр торца, длина) 1-5 мм; микроэлектроды (термин введен Кольтгофом) — 0,1-0,2 мм; ультрамикроэлектроды (УМЭ) — это твердые электроды с линейными размерами менее 10-20 мкм.
Первые публикации по ультрамикроэлектродам УМЭ появились в 1985-86 годах. В последующие годы были предложены теоретические модели, описывающие их свойства и особенности применения УМЭ [33].
УМЭ могут представлять собой торец микропроволочки из платины или золота запаянной в химическое стекло (рис. 6.48), торец углеродного волокна диаметром 6-2 мкм заполимеризованного в инертный пластик, боковая поверхность волоска из металлической проволоки или угольного
802
Новый справочник химика и технолога
волокна, линейчатый срез металла (прокатанной золотой фольги) толщиной 5-15 мкм между двумя фторопластовыми пластинами и т. п.
Наиболее перспективными считается так называемый анасамбль электродов, в котором УМЭ отстоят друг от друга на расстояние превышающее толщину диффузионного слоя, обычно не менее, чем на 0,5 мм. Ансамбль может состоять из 6 65 100 и более электродов (рис. 6.49).
Основные проблемы при изготовлении УМЭ заключаются в:
— обеспечении качества полировки поверхности;
— обеспечении качества герметизации микровставок;
— устранении влияния возможных механических и химических примесей вблизи рабочего пятна электрода.
Рис. 6.48. Схематическое изображение торца экранированного платинового микроэлектрода впаянного в стекло: 1 — микропроволока, полированный торец которой представляет собой УМЭ;
2 — макропроволока с оттянутым носиком, к которому подпаян УМЭ;
3 — сетка-экран (электрически связанная с экраном кабеля);
4 — стеклянный изолятор
Рис. 6.49. Схематическое изображение ансамбля из 16-ти платиновых УМЭ: 1 — один из сегментов ансамбля (торец Pt-микропроволоки);
2 — электрический вывод (оттянутый «носик» Pt-проволоки);
3 — место общего контакта (спая);
4 — стеклянный изолятор
Визуальный и электрический контроль трудно реализуем и не гарантирует высокие эксплуатационные характеристики электрода.
Преимущества аппаратуры, использующей УМЭ:
— гидродинамика процесса осаждения (стадия накопления в инверсионной вольтамперометрии) на УМЭ аналогична таковой для вращающегося макроэлектрода;
— мал вклад емкостного тока, что обеспечивает весьма незначительное искажение сигнала, практически «идеальная» форма кривых (уровни шумов и утечки зависят от аппаратуры); возможна съемка очень быстрых кривых «ток-потенциал»;
— из-за крайне низких значений токов невелик вклад от падения напряжения при протекании тока через анализируемый раствор, что позволяет проводить анализ в пресной воде, т. е. в природном объекте, не используя фоновые электролиты (хотя последние исследования показали что IR-составляюшая падения напряжения в приэлектрод-ной «микрообласти» может оказаться значительной);
— возможность размещать микроэлектродные датчики в проточных, инжекционных системах, не заботясь при этом о необходимости перемешивания раствора (новейшие детекторы для жидкостной хроматографии, контроллеры на сточных коллекторах, на буях в чистых реках и океане);
— возможность получения аналитической информации с помощью портативных, мало потребляющих (без использования электропривода), автономных устройств наподобие иономеров.
Однако применение УМЭ до настоящего времени ограничено, в основном, исследовательскими лабораториями. Сложность задачи заключается в том, что уровни токов на УМЭ на 4 порядка ниже, чем на традиционные макроэлектроды (т. е. доли наноампер), а аппаратура, оставаясь простой, должна быть малопотребляющей и малогабаритной. Уровень шума зависит от качества экранирования ячейки, УМЭ, присоединительных проводов. Некоторый запас по шумам и утечкам регистрирующей аппаратуры дают ансамбли электродов (сигнал возрастает кратно используемому количеству входящих микроэлектродов).
Проблемой остается обеспечение быстродействия системы регистрации при уровне токов < 0,1 нА. (Время заряжения емкости 100 пФ током 0,1 нА составляет 1 секунду). Для минимизации шумов от УМЭ входной усилитель располагают как можно ближе к источнику сигнала, т. е. к рабочей поверхности электрода, а проводник, связывающий вход усилителя с сенсором, по возможности экранируют (включая корпус УМЭ).
Используют высококачественные малошумящие усилители малых токов с автономным питанием, включаемые непосредственно на входе токоизмерительной цепи.
В таблицах 6.5.7, 6.5.8 приведены физико-химические свойства материалов индикаторных электродов, рабочие области потенциалов ИЭ, способы изготовления и примеры использования таких электродов в инверсионной вольтамперометрии.
6.5.7. Физико-химические свойства материалов индикаторных электродов (табл. 6.60-6.71)
Таблица 6.60
Растворимость металлов в ртути [13]
Металл Температура, °C Растворимость
масс. % ат. %
Li 25 0,048 1,34
Na 25 0,57 4,8
К 25 0,395 2,0
Rb 25 1,37 3,15
Cs 25 4,0 6,0
Cu 20 0,003 0,006
Ag 20 0,035 0,066
Au 20 0,1306 0,1329
Be 100 106 2 • 10’5
Электрохимические методы анализа
803
Продолжение таблицы 6.60
Ме- талл Температура, °C Растворимость
масс. % ат. %
Mg 17 0,31 2,5
Са 25 0,30 1,48
Sr 20 1,04 2,34
Ва 20 0,33 0,48
Zn 20 1,99 6,4
Cd 20 5,3
Al 20 0,002 0,015
Ga 22 1,13 3,19
In 20 57 70,3
Tl 20 42,8 42,6
La 25 0,0092 0,0133
Ce 20 0,016
Th 20 0,016 0,014
U 20 0,005 0,0042
Pu 20 0,0154 0,0127
Si 20 (0,001) 0,007
Ge 25 1 • 10’6
Sn 20 0,6 1,26
Pb 20 1,1 1,1
Ti 20 5 • 10-6 2. 105
Zr 20 0,002 0,007
Sb 20 2,9 • 10’5 4,7-10“5
Bi 20 1,1 1,1
V 20 5 • 105 2-IO4
Nb 20 (0,001) 0,002
Cr 20 3,1 • 10-11
4 • 10’7
Mo 20 2 • Ю5 4- 10’5
W 20 10"5 10’5
Mn 20 0,0018 0,0066
Fe 20 1,15 • IO’17
7 • 10’5
Co 20 8 • IO 5
Ni 20 4,8 • 10~5
Ru 20 0,353 0,694
Rh 20 0,16 0,311
Pd 20 0,006 0,012
Ir 20 0,001 0,001
Pt 24 0,09 0,10
Таблица 6.61
Коэффициенты диффузии металла в ртути [13]
Металл Температура, °C £)Hg • 105,cm2/c
Li 25 0,93
Na 25 0,86
Продолжение таблицы 6.61
Металл Температура, °C • 105,см2-с1
К 20 0,66
Rb 25 0,61
Cs 25 0,65
Cu 25 1,06
Sn 25 1,5
Bi 25 1,5
Ag 25 1,0
Au 25 0,73
Ca 10,2 0,62
Sn 9,4 0,54
Ba 7,8 0,60
Zn 25 2,4
Pb 25 2,1
Mn 20 1,84
Cd 25 2,0
Hg 20 1,55
Ga 20 1,64
In 20 1,42
Tl 25 1,18
Ge 20 1,71
Sb 20 1,47
Ni 25 2,0
Таблица 6.62
Произведения растворимости некоторых интерметаллических соединений в ртути при 20 °C [13]
Соединение Произведение растворимости
AuZn 2,5 • IO’12*
AuCd 2,5 • 10’9
Auln 1,8 • IO-6
AgZn 3 • 106
AgCd 7 • 106
CuZn 4- 10 6
Cu3Sn 3 • IO’12
CuSn 4- 10“7
CuGe3 8,4 • IO13
CuGa 2 IO 6
CuSb 3,2 • 10’7
SbZn 2 • IO'9
SbCd 1 • 10~8
Sbln 2 • 10 х
MnCd3 5,7 • IO11
MnSn3 7 • 10’9
GaCo 2,6 • IO46
GaNi 3,9 • 10’16
Примечание. * При 90 °C.
804
Новый справочник химика и технолога
Таблица 6.63
Свойства металлов, используемых в качестве контактов в ртутных электродах
Металл Растворимость в Hg, масс. % т]а,В Металлы, с которыми образуются интерметаллические соединения
Pt 0,03 0,1 Zn, Sb, Sn
Ag 0,05 0,95 Cd, Zn
Au 0,01 0,8 Cd, Sn, Mn, Zn
Примечание. т|а — перенапряжение водорода на соответствующих металлах при i = 1 А-см-2.
Таблица 6.64
Основные характеристики некоторых амальгамных электродов
Металл D, см/с Основной электролит Яо, В (н. в. э.) (кат.), В (н. в. э.)
Li 9 • Ю2 0,1 M [(CH3)4N]OH -3,05 -2,10
Na 0,4 0,1 M [(CH3)4N]C1 -2,93 -1,88
К 0,1 0,1 M [(CH3)4N]C1 -2,92 -1,88
Rb 0,2 0,1 M [(CH3)4N]C1 -2,92 -1,85
Cs 9 10’2 0,1 M [(CH3)4N]C1
Tl 1,2 1 M HC1O4 -0,33 -0,23
Zn 1,7 • 103 1 М NaC104 -0,76 -0,77
Cd 1,8 1 М NaClO4 -0,40 -0,33
In 105 1 М NaC104 -0,34 -0,75
In 1 1 МНС1 -0,34 -0,36
Sn NaC104 + HC1O4 -0,14 -0,16
Pb 1,1 1 M НС1О4 -0,13 -0,13
Bi 3 103 1 МНС1 +0,32 +0,28
Cn 10 4 1 М НС1О4 +0,34 +0,27
Mn 0,6 М Mg(C104)2 -1,18 -1,26
Fe < 10° 1 М НС1О4, -0,44 -1,02
Co <10 6 1 М K2SO4 -0,28 -1,18
Ni < 106 1 М НС1О4 -0,25 -0,85
Таблица 6.65
Рабочие области потенциалов платиновых электродов
Среда ЯН,В
анодный катодный
6МНС1 +0,97 -0,30
0,1 МНС1 +1,10 -0,30
Ацетатный буфер, pH = 4 +0,90 -0,50
Фосфатный буфер, pH = 7 +0,94 -0,70
0,1 MNaOH, pH = 12,9 +0,72 -0,91
0ДМКС1 +1,0
Таблица 6.66
Рабочие области потенциалов золотых электродов
Среда Ян, В
анодный катодный
1 М НС1О4 +1,5 -0,2
Ацетатный буфер, pH = 4,0 -0,88
Фосфатный буфер, pH = 7,0 -1,19
0,1 MNaOH, pH = 12,9 ' -1,28
0,1 М NaC104 (небуферированный), pH = 7,0 -1,13
Таблица 6.67
Предельные рабочие анодные потенциалы для электродов из хрома и карбида вольфрама при различных pH
pH Ян, В
хрома карбида вольфрама
1,0 0,9 0,95
4,2 0,7 0,8
10,0 0,35 0,7
Таблица 6.68
Рабочие области потенциалов для графитовых электродов, импрегнированных парафином
Среда Ян, В
анодный катодный
НС1+КС1, pH = 1,2 +1,32 -1,28
Фталатный буфер, pH = 3,95 +1,33 -1,30
Фосфатный буфер, pH = 7,02 +1,35 -1,38
Боратный буфер, pH = 9,80 +1,04 -1,61
Таблица 6.69
Рабочие области потенциалов для различных углеродных электродов в 0,2 М KNO3
Материал электрода Ян, В
Графит, импрегнированный парафином от +0,25 до 0,0
Углерод, импрегнированный парафином от +0,50 до 0,0
Угольная паста от +0,85 до -0,25
Пиролитический графит от +0,90 до-0,75
Стеклоуглерод от+1,0 до -0,75
Таблица 6.70
Предельные рабочие анодные потенциалы угольных настовых электродов при 1=2 мкА
Среда Ян, В
1 МКС1 +1,10
0,1 MH2SO4 +1,30
0,2 М NaOH +0,87
Электрохимические методы анализа
805
Таблица 6.71
Рекомендуемые «третьи» элементы, для некоторых бинарных систем в методе ИВ
Определяемый элемент Бинарная система «Третий» элемент Электрод
Zn Zn—Си Ga, Ge РГЭ
Zn—Ag Ga РГЭ
Cd Cd—Си Sn, Hg ГЭ
Cd—Sb Cu РГЭ
Продолжение таблицы 6.71
Определяемый элемент Бинарная система «Третий» элемент Электрод
In In—Cu Те РГЭ
Sb Sb—Se Hg ГЭ
Sb—Те Hg ГЭ
Se Se—Sb Cu ГЭ
Pb Pb—Cu •Hg ГЭ
Те Те—Cu Hg ГЭ
6.5.8. Способы изготовления и примеры использования электродов в ИВ
Таблица 6.72
Способы изготовления и примеры применения ультрамикроэлектродов в вольтамперометрии
Материал электрода Диаметр волокна, мкм Способ изготовления и обработки электрода Метод исследования Определяемый элемент (вещество) область применения Фоновый электролит
Графитовые волокна со стеклоуглеродной поверхностью 10-15 Вклеивание в стекло ВА Тяжелые металлы Водные растворы КС1, NaNO3
Золотая проволока 0,9 Электрополировка в НС1 и HNO3 импульсом тока КВВ Исследование кинетики быстрых электрохимических процессов Водные растворы электролитов
Золотая проволока 0,1-0,9 Вклеивание в стекло; литография КВВ, ИДИВ, ПТП Ферроцен 0,1 М ацетонитрил в СН3ОН
Золотая, платиновая проволока в виде кольца (диаметр 2 см) 50 Вакуумное напыление на стеклянную трубку, полировка ВА Cd(II), Pb(II) Водные растворы КС1, НС1
Золотая, платиновая, угольная полоса (ширина) 5 х 2300 (длина) Полировка ВА Ru(NH3X+ 0,1 М фосфатный буферный раствор
Иридиевая проволока 127(длина) Вакуумное напыление в стеклянную микропипетку; электролитическое покрытие ртутью ИВ, ВА с треуг. разв. Cd(II), Pb(II) НС1
Платиновая лента 2 (ширина) Впаивание в стекло Циклич. ВА Ферроцен Ацетонитрил
Платиновая проволока 10 Впаивание в стекло Циклич. ВА Органометаллические соединения (C4H9)4NPF6 в CH3CN, ДМФА, ТГФ
1-5 Впаивание в стекло Циклич. ВА Ферроцен Ацетонитрил
5-10 Впаивание в стекло; электронакопление ртути до полусферы Циклич. ВА Pb(II) НС1, КС1
2-25 Вклеивание; электронакопление пленки ртути на торце; поляризация ИВ Комплексы Hg(I) NaNO3, NaBr, Na2C2O4, NaOH, NaSCN
806
Новый справочник химика и технолога
Продолжение таблицы 6.72
Материал электрода Диаметр волокна, мкм Способ изготовления и обработки электрода Метод исследования Определяемый элемент (вещество) область применения Фоновый электролит
Платиновая проволока <2 Впаивание в стекло; покрытие пленкой ртути ВА Ферроцен Бензол, толуол, ксилол
10 Впаивание в стекло Циклич. ВА 1,1 -диметилбиантрон 0,6 M(C2H5)4xNC1O4 в ДМФА .
Платиновая, вольфрамовая проволока, углеродные волокна 2-10 Впаивание в стекло; полировка среза ВА Hg Водные растворы nh4scn
Углеродные волокна 8 Вклеивание в стекло ВА Fe(CN)’“ фенол, антрацен, фенантрен (C4H5)NC1O4 в хлорбензоле
8 Впаивание в стекло ВА Fe(CN);-, Fe(CN)^
0,4-1,5 Впаивание в стеклянный капилляр; полировка ВА Аскорбиновая кислота; метронидазол, антипирин Биологические среды+0,1 МНС1
Углеродные волокна в виде кольца 1 Пиролиз метана в кварцевой микропипетке ВА с треуг. разв. Допамин Аскорбиновая кислота
Углеродные волокна, платиновая проволока 11 Вклеивание в стекло ВА Трис (2,2-дипиридил) рутения (II)
Углеродные волокна 8 Электронакопление пленки ртути КВВ, циклич. ВА РЬ(П) Ацетатный родани-вый буферный раствор
10 Впаивание или вклеивание в стеклянный капилляр; покрытие пленкой ртути ИВ, КВВ Cd(II), Hg(II) 0,1 М KSCN
7 Вклеивание в стекло; электронакопление пленки ртути ИВ Металлы Неводные растворители + (C2H5)4NC1 + + НС1
Углеродные моноволокна 1-10 Термообработка при 1000-2800 °C ВА Металлы Водные растворы КС1, NaNO3
Углеродные нити 1-32 Вклеивание в стекло ВА Ферроцен; гидрохинон Ацетонитрил, этиленгликоль, вода
Угольное волокно 0,5 Электрополировка в КОН КВВ Изучение кинетики быстрых электрохимических процессов
10 Вклеивание в стекло; со скошенной поверхностью; полировка ВА Допамин Биологические среды + КОН (НС1)
10 Электрохимическое покрытие пленкой ртути ИДИВ Cd(II), Pb(II) НС1
Угольные волокна; платиновая золотая проволока 7-10 Электроосаждение ртути на поверхности ИВ Cd(II), Pb(II) Водные растворы КС1, НС1
Примечание. ДМФА — диметилформамид; ТГФ — тетрагидрофуран.
Электрохимические методы анализа
807
Таблица 6.73
Электроды, модифицированные предварительно
Электрод Способ модифицирования Определяемый элемент или вещество Сминл М
СУЭ Прививка групп О Si(CH2)3NHCOCOOH Си2+ 1,5 Ю8
СРКЭ Адсорбция пирокатехина из буферного раствора (pH = 7,7), содержащего 0,1 М пирокатехина Сии 1,5 • Ю'10
СУЭ Адсорбция хингидрона из раствора 0,5 М H2SO4, содержащего хингидрон Аскорбиновая кислота 1()2
УПЭ Введение в УПЭ амберлита А-2 Аи3+ 5 • 10^
СУЭ Образование пленки цианидов Ru(III) и Ru(II) из раствора 2 • 1()3 М RuCl3 + 2 • 10~3 М K4Fe(CN)6 + 0,5 М NaCl (pH = 2) в результате циклической поляризации в интервале 0,35-0,85 В (нас. к. э.) As3+ 5 • 10 6
УПЭ Введение в УПЭ катионообменной смолы типа Дауэкс Cu2+ 2- 10 6
СРКЭ, СУЭ Адсорбция триоктилфос-финоксида (ТОФО) из раствора 5 • 10 3 М ТОФО и6+ 10’9
Адсорбция поли-4-винил-пиридина (ПВП) из раствора 0,4% ПВП в метаноле Сг6+ п 10~8
РПЭ Адсорбция дитизона из фосфатного буферного раствора (pH = 4) Cu6+, Cd2+ 10’7
Адсорбция аллиламина из 0,5% раствора аллиламина Ферро-ценкар-бокси-альдегид 10 7
Таблица 6.74
Электроды, модифицированные in situ
Электрод Состав раствора для модифицирования Определяемые ионы или вещество £минл M
СРКЭ 0,1 М NaClO4 или 0,01 М ацетатный буфер + + 1 • 10"4 М тиомочевина Си2+ IO'7
РПЭ O,2MNH4C1 + 1% диметилглиоксима (ДМГ) Со2+ 8 • 108
Ni2+ 5 • 107
Au 0,001-0,8 М H2SO4 + + 5 • Ю2 г/л тетра-этиламмониййодида Cd2+ 2 • 10~8
СРКЭ 0,1 М аммиачный бу-ферный раствор + + 5 • Ю 7 М о-крезол-фталексон La3+ 1,2 • 10~10
Се3+ 1,7 • 10~l°
Pr3+ 1,4 • 10~10
СРКЭ 10-3 М пирролидиндитиокарбамат Zn2+ 3 • 10“
УПЭ 104% раствор гематеина в диоксане Sn4+ 8,4 • 108
УПЭ Раствор солохром-фиолетового RS Al3+ —
ГЭ 0,1 М H2SO4 + 106 М трифенилметановый краситель (кристаллический фиолетовый, метиловый фиолетовый, малахитовый зеленый) Sb3+ 3 • 10’8
ГЭ 1 МНС1 + 6- Ю 4М родамин С Sb3+ 5 • 10"8
ГЭ 1,5 М NH4SCN + 0,03 М (C2H5)4NI + 0,3 М (CH2NH)2 Co2+ 10 6
ГЭ 0,01 МНС1 + 0,05 М [(C2H5O)2PSS]2Ni Sn4+ 2 • 10-7
ГЭ 0,1 М раствор КС1, насыщенный коричной кислотой Cr3+ 2 • 10 7
ГЭ 0,1 MNH4C1 + O,1 м NH4OH + 0,04% 1 -нитрозо-2-нафтола Co2+ 1 • IO"8
808
Новый справочник химика и технолога
Таблица 6.75
Примеры вольтамперометрического определения ионов металлов, неорганических и органических веществ с помощью химически модифицированных электродов
Определяемый элемент (соединение) Субстрат (электрод) Модификатор (иммобилизатор) Способ модификации (иммобилизации) Фоновый электролит Вариант метода BA Диапазон определяемых содержаний (М); объект анализа
Неорганические вещества
Ag Графитовый Полиакрилами-доксим Комлексообразова-ние и осаждение Водная среда ИВ 1 • 1081 • 105
Стеклоуглеродный Хелатный полимер, содержащий иминодикарбоновые группы Электрохимическая восстановительная полимеризация HNO3 ИВ; цик-лич. ВА Ю~7-10^
Стеклоуглеродный вращающийся Адсорбированный слой Н2 Электровыделение при -0,8 В 0,15MNH4NO3 + 0,75 М NH3 ВА 8 • 10 7-5 • Ю”6; осадочные породы, руды
Стеклоуглеродный Комплексоны: ими-но диуксус -ная кислота; 3,6-диоксиоктан-1,8-диамин- N, У,У,7У-тетра-уксусная кислота; ЭДТА Комплексообразование 0,1 MKNO3 ВА цик-лич. п- 10”6
Угольно-настовый Цеолит и вазелин Растирание и упаковка 0,05 М NaNO3, pH =12,4 ИВ Смин. 0,08 мг/л
Ag, Cd. Hg, Pb, Tl, Zn Угольно-настовый Воск; бромнаф-талин; бромоформ; эпоксидная смола Втирание 0,1 MKC1 ИВ
Ag2O, AgO Композитный серебряный Порошки серебра (1 мкм) и политрихлорэтилена (100 мкм) Гомогенизация в СН3ОН, высушивание и прессование в таблетки под вакуумом 0,1 MNaOH ВА
As Стеклоуглеродный RuCl3 + K4Ru(CN)6 Адсорбция металло-комплексов 0,5 NaCl, pH = 2,0 ВА 5 • 10^-1 • 10"*
Угольно-настовый Аморфный и кристаллический мышьяк Втирание в пасту HC1 ВА
As, Bi, Sb Из хлопкового волокна Меркаптогруппы Хемосорбция Водные растворы ИВ
As, Cu, Те Графитовый Неорганические комплексные ионы Электрополимеризация in situ HCl + Hg(NO3)2 ИВ Смин.: 3 • 10 9 (As); 5 • 10~10 (Те); природная вода
Электрохимические методы анализа
809
Продолжение таблицы 6.75
Определяемый элемент (соединение) Субстрат (электрод) Модификатор (иммобилизатор) Способ модификации (иммобилизации) Фоновый электролит Вариант метода ВА Диапазон определяемых содержаний (М); объект анализа
Au Графитовый пастовый Дитизон Адсорбция 0,01 М НС1 ИДИВ катод н. 0,01-1 мкг/мл
Угольно-пастовый Амберлит LA-2 Втирание в пасту НС1, НВг ВА, ИДИВ п • 10-4 г/л
Угольно-пастовый Фосфороорганические соединения (триоктил-фосфиноксид или трибутилфосфат) Втирание в пасту Водные растворы КВг, КС1 ИДИВ
Bi Ртутный пленочный Оксид три-и-ок-тил-фосфин в ПВХ Нанесение пленки 0,5МНС1 + 0,4М Н3РО4 ИВ 0,002-0,5%; медь и медные сплавы
Bi, Na, Pb Филломанг анатный МпО2 Обмен; хемосорбция 9МКОН Циклич. ВА
CO2 Стеклоуглеродный вращающийся Металл офталоци аниды (Со, Ni, Fe, Mg, Mn, Zn) Вакуумное напыление с плазменной обработкой 0,5 М Na2SO4 + + Na2CO3, pH = 6,65 ВА
Cd Ртутный угольно-пастовый Коллоидальный графит, полиметилметакрилат, растворитель Нанесение пленки лака в виде аэрозоля и образование пленки ртути in situ KNO3 + Hg(II) ИДИВ с 1О“10 ‘-'МИН. 1 v
Cd, Cu, Pb Ртутно-стеклоуглеродный Полимер нафион-125 Нанесение мембраны KNO3, pH = 4,5-5,0 ИВ 2 10 7, моча
Cd, Cu, Pb, Zn Ртутно-стеклоуглеродный Полимер нафион-125 Нанесение мембраны p < 0,1, водные растворы ИДИВ, ИВ 2 • 10’8-2 • 10-6
Cd, In, Pb Графитовый; ртутнографитовый Натриевая соль полистиролсульф окислоты или хлорид поли(диметилди-аллил) аммония у-облучение и электролиз ртути Ацетатный буферный раствор с pH = 4,7 ИВ, циклич. ВА п- 10-6
Co(II) Угольно-пастовый 1,10-Фенант-ролин Втирание в пасту 0,1 МКС1, KNO3 циклич. ВА, ИДИВ 1 • 10^-3 • IO 4
Co(II), Cu(II), Fe(III) Графитовый Сополимер винилпиридина с винилферроце-ном; полимер комплекса Ru (2+) с 4-винил-4-метил-2,2 -бипиридином Электроосаждение; погружение в раствор Природные воды ВА, циклич. ВА, ИДИВ Природные воды (определение физико-химических форм)
810
Новый справочник химика и технолога
Продолжение таблицы 6.75
Определяемый элемент (соединение) Субстрат (электрод) Модификатор (иммобилизатор) Способ модификации (иммобилизации) Фоновый электролит Вариант метода ВА Диапазон определяемых содержаний (М); объект анализа
Cr(VI) Платиновый Адсорбированный 12 Доннановская диффузия; электроокисление lai 1 МНС1 ИВ 6 • 10^-2 10 '
Cu(I) Угольно-пастовый 2,9-диметил- 1,10-1,10-фенант-ролин Введение в порошок Водные растворы Циклич. ВА, ИДИВ Смин. 3 • 10-7, вода
Cu(II) Ртутнопленочный Щавелевая кислота Электронакопление ртути; электрополяризация 0,1 МН2С2О4 + + 7 • IO-6 М Hg(II) ИДИВ Смин. 0,01 мкг/л
Cu(II) Углеродный Полипиррол-У-дитиокарбаминат Электрополимеризация в среде ацетонитрила Водные растворы Циклич. ВА 1 • 1 ОМ • 10 s
Угольно-пастовый Катионообмен-ник Дауэкс Втирание в пасту KNO3, pH = 9,1-7,4 ИДИВ 1-80 мг-мл'1; фармацевтические препараты
Платиновый Полипиррол-У-карбодитионат Электроокисление в ацетонитрильном растворе Раствор соли Циклич. ВА п • 10 6
Из оксида индия 3-(2-аминоэтил-ами-но)пропил-триметоксисилан и гемоглобин Погружение в растворы Пиридин ВА
Cu, Hg, Ni, Pb Платиновый, стеклоуглеродный Дитиокарбами-натные лиганды Электросинтез полимеров из эфиров пиррола и обработка CS2 Водные растворы ВА п 10’7
Cu, Hg, Ni, Pb, ионы платиновых металлов Графитовый Дитиокарбами-натные лиганды; полипиррол; А'-этиламин Комплексообразование Водные растворы ВА и - 106
Cu и другие ионы металлов Ртутнопленочный на серебряных и графитовых подложках Три-н-октил-фосфин-оксид Нанесение покрытия в летучем растворителе и нагревание; электролиз 0,1 М НС1 ацетатный буферный раствор Циклич. ВА п 10’7
Платиновый, танталовый Полипиррол Электрохимическое нанесение пленки из ацетонитрильных растворов Водные растворы ВА
Fe(II), Fe(III) Платиновый Аденозин-5'-монофосфат Комплексообразование Хлорацетатный буферный раствор, pH = 3,5 ИВ катодн. Ю’МО-6
Электрохимические методы анализа
811
Продолжение таблицы 6.75
Определяемый элемент (соединение) Субстрат (электрод) Модификатор (иммобилизатор) Способ модификации (иммобилизации) Фоновый электролит Вариант метода ВА Диапазон определяемых содержаний (М); объект анализа
Fe(II), Fe(III) Угольно-пастовый 1,10-фенантролин Втирание в пасту 1 МКС1, pH 0-5 Циклич. ВА 106103
Цеолит на платиновом Ионы железа, тетрагидрофуран с полистиролом Пропитка в FeSO4 pH = 1,5; обработка тетрагидрофураном 0,1 М NaCl+ 0,1 МНС1, pH = 1,5 Циклич. ВА
Fe(II), Fe(CN)3; Fe(CTC Графитовый Поли(4-винил-пиридин), частично поперечно-сшитый бато-фенантролинди-сульфоновой кислотой Полимеризация СНзСООН ВА с треуг. разв. пот. 1 • 10-1 • 107
Fe(III), Ni(II) Графитовый Т етрацианохино-диметан; поливи-нилферроцен Адсорбция 0,2 М NaNO3, фосфатный буферный раствор Циклич. ВА
Fe(CN)36-> Fe(CN)^ Угольно-пастовый Амберлит LA-2 Втирание в пасту НС1 ИДИВ 3 • 10’7-6 • 10’3 г-л”1
Набор платиновых проволок диаметром 0,1-0,5 мкм Поликарбонат-ный полимер Электроосаждение платины и иммобилизация пленкой полимера Ацетонитрильная среда ВА
Fe3O Угольно-пастовый Fe3C Втирание в пасту НС1, глюконатный раствор ВА с треуг. разв. пот.
Fe3O4 Угольно-пастовый Магнезит Втирание в пасту 0,1 МНС1 ВА сМин. ~ 3 мг; магнезит
Fe3O4, Fe(OH)2 Стеклоуглеродный, платиновый, золотой Fe(OH) Химическое осаждение МеОН + Me2SO4 Me: Li, Na, К Циклич. ВА
Платиновый Ru (2,2-дипири-дил)3С12; хлоранилин; о-фенилдиамин Адсорбция; электролиз; погружение в раствор Неводные среды ВА Биологические жидкости
HNO3 Угольно-пастовый Наполнители Втирание в пасту 0,1 М CH3COONH4 ВА
H2O2 Стеклоуглеродный Полимер нафион с диспергированной в нем платиной Нанесение покрытия в летучем растворителе, нагревание и электролиз для платины K2PtCl4 ВА 10’6-10’2
Угольно-пастовый Фталоцианин Fe (2%): парафин Растирание с графитом Фосфатный буферный раствор с pH = 3-9 ВА 5 • 10 5 6 • 104
812
Новый справочник химика и технолога
Продолжение таблицы 6.75
Определяемый элемент (соединение) Субстрат (электрод) Модификатор (иммобилизатор) Способ модификации (иммобилизации) Фоновый электролит Вариант метода ВА Диапазон определяемых содержаний (М); объект анализа
Н2О2 Угольно-пастовый Связующие компоненты Втиряние в пасту 2МКС1 ПТП (•-мин. 2,3 мг-л
Hg24 Угольно-пастовый Цеолит Растирание с пастой KNO3 ВА с треуг. разв. пот. 0,1 -2,2 мкг-мл-1
Угольно-пастовый Вазелиновое масло + дибензо-18 -краун-6 Растирание с пастой Ацетатный буферный раствор с pH = 4,0 Циклич. ВА Смин. 2 • Ю 6
Г, Sb(III) Графитовый Трифенилметановые красители Адсорбция in situ Водные растворы ИВ смин, 4 • Ю 9(1); 3 • 10 s (Sb)
Ir(IV), ItCf Угольно-пастовый Амберлит А-2: хлорид три-каприлметил-аммоний Втирание в пасту и адсорбция 0,1 5МНС1 Циклич. ВА, ИДИВ Смин. 2 • 10-5%
к Стеклоуглеродный Цеолитоподобная пленка, содержащая Ag (I) и Mo(IV) Электроосаждение 0,1 MHNO3, 0,1 MKH3NO3 Циклич. ВА
к Стеклоуглеродный, посеребренный Mo(CN)^ Электроокисление ВА
К, Li, Ni Платиновый Гексацианоферрат V4[Fe(CN)6]5 Электрохимическое нанесение пленки K2SO4, Li2SO4, n2so4 циклич. ВА
Ni Угольно-пастовый Диметилглиок-сим Втирание в пасту и погружение в раствор Аммиачный буферный раствор с pH = 8,0 ИДИВ, ИВ 5 • 10“8; природная вода; уголь, речные отложения
о2 Стеклоуглеродный Полимеризованный протопорци-рин железа Электрохимическое нанесение пленки Фосфатно-цитрат-ные буферные растворы Циклич. ВА
Стеклоуглеродный Тетрафенил пор-финат железа Адсобция и термообработка в азоте Буферные растворы с pH = 3,0 Циклич. ВА
Стеклоуглеродный 9,10-фенантра-хинон; 5,8-ди-гидрокси-1,4-нафтохинон; 5-гидрокси- 1,4-нафтохинон Адсорбция из растворов ДМФА и поляризация ВА
Стеклоуглеродный, и-УаР, RuS2 Пленка нафиона с внедренными халькогенидными кластерами Осаждение H2SO4 Циклич. ВА
Электрохимические методы анализа
813
Продолжение таблицы 6.75
Определяемый элемент (соединение) Субстрат (электрод) Модификатор (иммобилизатор) Способ модификации (иммобилизации) Фоновый электролит Вариант метода ВА Диапазон определяемых содержаний (М); объект анализа
о2 Золотой, платиновый, из SnO2 Полипиррол с мезотетракис-(4-карбокси-фенил)порфири-нат Мп(П1), Со(1П), Fe(HT) Покрытие пленкой 0,1 МНС104 Циклич. ВА
О2, растворенный в жидкости Угольно-пастовый Парафин Образование пасты 2МНС1 ВА 0,2-28 мкг/мл
РЬ в присутствии In Ртутнопленочный на стеклоуглероде Ацетилцеллюлоза Погружение в раствор и обработка КОН Ацетатный буферный раствор с pH = 4,5 ИВ 107-105
Pb, Se Угольно-пастовый 5% Pb(NO3)2 + 4% Se Растирание с пастой 0,1 МНС1, НС1О4 Циклич. ВА 0,01-10%; селенид свинца
Pb и другие ионы металлов Стеклоуглеродный 12-Молибдо-фосфат-ионы Циклическая поляризация H2SO4 Циклич. ВА
Ti(IV) Стационарный ртутный Солохромфиолет овый RS Комплексообразование Ацетатный буферный раствор с pH —5,1 ИВ с 7 • 1О“10- *--МИН. ' 1V 5 природная вода
Т1(1) Стеклоуглеродный Пленка полимера нафион, содержащая дициклогексил-1 8-краун-6 Адсорбция 0,1 MLiC104 ИВ Смин. 2 • 10’12
U(VI) Стеклоуглеродный Триоктилфосфи-ноксид Нанесение на поверхность Ацетатный буферный раствор ВА Смин. 1 • Ю-6; морская вода
V(II), V(III), V(IV), V(V) Угольно-пастовый V2O5, VOSO4, NaVO3, NaVO2, v2o3 Втирание в пасту 1 МНС1 ВА
W(V), W(VI) Графитовый Вольфрамовые бронзы (WO3 • 2Н2О) Na2WO4 + H2SO4 Циклич. ВА 1-60%; вольфрамовые бронзы
Органические вещества
Адриамицин Пирографи товый Фосфоролипид-ные мономолеку-лярные мембраны Адсорбция при погружении в раствор Ацетатный буферный раствор с pH = 4,5 Циклич. ВА
Амины Угольно-пастовый Поли-4-винил-пиридин с противоионами [Fe(CN)5L]3-, L: пиридин-4-карбоксальдегид Введение полимера в виде раствора в СН3ОН; конденсация СН3ОН ВА 3 • 1О'-1,4• 1(Г2
814
Новый справочник химика и технолога
Продолжение таблицы 6.75
Определяемый элемент (соединение) Субстрат (электрод) Модификатор (иммобилизатор) Способ модификации (иммобилизации) Фоновый электролит Вариант метода ВА Диапазон определяемых содержаний (М); объект анализа
Анилин Угольно-пастовый Сепиолит Смешивание с маслом Nyiol и графитом 0,1 MKNO3, pH = 6,5 ИДИВ смин. 15 нг/мл; вода, водка, молоко
Аскорбиновая кислота, гидрохинон, пирокатехин Стеклоуглеродный Диспергированные оксиды металлов Химическое осаждение Циклич. ВА
Аскорбиновая кислота, допамин, ксантин, мочевая кислота, 6-мер капто-пурин, 6-тио-ксантин Угольно-пастовый Проводящие органические полимеры Втирание в пасту Буферные растворы, pH = 7,8 Циклич. ВА 10 5-70'3
Аскорбиновая кислота, ни-котинамида-денинди-нуклеотид Углеродный адА’Л'-тет-раметил-н-фени-лендиамин Нанесение пленки и поляризация Водные растворы Циклич. ВА 5 10“5-5 • 10“3
Бензохинон, гидрохинон Пирографи товый Поливиниловый спирт с лакказой Нанесение пленки ВА
Платиновый Полиуретан с лакказой Нанесение пленки ВА
Бутоксианизол, хлорпромазин Угольно-пастовый Мочевая кислота Втирание в пасту Водные растворы ИДИВ
Везикул яичный лецитин Ртутный Ферредоксинкла-стер [Fe4S4(CR4)]2’R= — С6Н4-п—н—CgHi7 Адсорбция на ртути Буферные растворы с pH = 5,7-11,5 Циклич. ВА
Гетерополиоксиметаллаты, Н2 Стеклоуглеродный, золотой, золотортутный Г етерополиокси-металлаты Нанесение покрытия 0,5 М H2SO4 То же
Глицерин, глюкоза, рибоза, сахароза Серебряный Оксид серебра Электронакопление in situ Щелочные растворы ИВ
Глюкоза Углеродный Фермент на основе ферроцена Адсорбция ВА (0^10) • 10~6
Платиновый Оксид в присутствии слоя электроосажден-ного таллия или свинца Электроосаждение Циклич. ВА 0-0,6
Электрохимические методы анализа
815
Продолжение таблицы 6.75
Определяемый элемент (соединение) Субстрат (электрод) Модификатор (иммобилизатор) Способ модификации (иммобилизации) Фоновый электролит Вариант метода ВА Диапазон определяемых содержаний (М); объект анализа
Глюкозо-оксидаза Стеклоуглеродный Аминофенилборная кислота Комплексообразование Водные растворы Циклич. ВА, ИДИВ
Дианизидин Углеродный волоконный Полиакрилонитрил; ПВХ-смола Полимеризация, промывка в ацетоне Неводные среды Циклич. ВА, ВА
Допамин; эпинефрин Стеклоуглеродный Перфорированный иономер на-фион и ацетилцеллюлоза Физическое нанесение двухслойного покрытия 0,07 М КОН ВА 7,5 • 10’6
НАДН Стеклоуглеродный, покрытый оксидами олова и индия Гидрохинон Образование по реакции Манниха НАД+, pH = 8,0 ВА
Катализатор (порошок) Угольно-пастовый Политетрафторэтилен + катализатор Втирание в пасту Водные растворы ВА
Катализаторы рения на Ni-Al и Си-А1 основах Угольно-пастовый Вазелиновое масло + катализатор Втирание в пасту Водные растворы ВА
Комплексы металлов Титановые пленки Полимерные металлоорганические комплексы, включающие ацетилацетонаты Адсорбция; комплексообразование Водные растворы ВА
Красители Бислойные липидные мембраны Красители Адсорбция Циклич. ВА
Нитрофенол Угольно-волокнистый Триметилсилан То же Водные среды, pH = 4,0 То же 10"-10л
о-Нитрофе-нол; изомеры анилина Стеклоуглеродный Полимер «Нафион» Нанесение пленки а-циклодекстрин ВА
Органические вещества Углеродный Берлинская лазурь Адсорбция 1 М NaC104 в ДМФА Циклич. ВА
Платиновый Поли[Ии (2,2'-ди-миридил)2-(4-ви-нилпиридинг]2+ Электрополимеризация Ацетонитрил КВВ, циклич. ВА
Органические соединения Никелевый Тетрацианохино-диметан; поливинил ферроцен Адсорбция Неводные растворы Циклич. ВА
816
Новый справочник химика и технолога
Продолжение таблицы 6.75
Определяемый элемент (соединение) Субстрат (электрод) Модификатор (иммобилизатор) Способ модификации (иммобилизации) Фоновый электролит Вариант метода ВА Диапазон определяемых содержаний (М); объект анализа
Редокс-частицы Стеклоуглеродный Редокс-частицы, Ru(bpy)j*, Ru(NH3)’+, Cu(NH3)+, ферроцен, гидрохинон, рибофлавин Электрохимическое окисление и выдерживание в растворах Водные среды Циклич. ВА
Ru(NH3)’+; а2 = К^Р 2 W17МоО62 Стеклоуглеродный Гетерополи-и изополиоксиметаллаты Нанесение покрытия 0,5 М H2SO4 Циклич. ВА
[Ru(dipy)2Cl2] PF6 Платиновый Тозилат полит-рис-[5,5-бис(3-акрилил-1-про-покси)карбонил]-2,2-дипиридил рутения (2+) Термическая полимеризация Циклич. ВА
Ru(bpy);', Ru(NH3r, Cu(NH3X+ Стеклоуглеродный Редокс-частицы Электрохимическое окисление; циклическая поляризация H2SO4, Н3РО4, Na2CO3 ВА
Хиноны Стеклоуглеродный, пирографитовый, платиновый, SnO2 Моно- и диамино-производные п-бензохинона и /7-нафтохинона Силанизация и нуклеофильное присоединение хинонов Неводные растворы Циклич. ВА
Хиноны, фенолы Графитовый, угольно-пластовый Хиноны, фенолы Прессование порошка 0,1 MNaC104 рН = 1,2; ацетатный буферный раствор с pH 5,0 Циклич. ВА
Хлорид ацетилхолина ПВХ-гель Нитробензол Полимеризация Неводные среды ИДИВ 5 • 10^-10^
Хлорпромазин Стеклоуглеродный Ацетилцеллюлоза Нанесение на поверхность Фосфатный буферный раствор с pH = 7,4 ИВ, ИДИВ Смин. 1,3 • 108; моча
Циклические соединения Платиновый Полипиррол-№(П)-пиридин-порфирин Циклирование при электролизе ВА
Ферредоксин Золотой Мостиковый переносчик ди(4-пиридил)-дисульфид Химическое осаждение Раствор белка ВА 5 • 10“7; раствор белка
Электрохимические методы анализа
817
6.6. Кондуктометрия и кондуктометрическое титрование (по данным Г.В. Прохоровой) [3]
Кондуктометрический метод анализа основан на измерении удельной электропроводности анализируемого раствора. Электропроводностью называют величину, обратную электрическому сопротивлению R. Единицей измерения электропроводности является Ом1, или См (сименс). Растворы электролитов, являясь проводниками II рода, подчиняются закону Ома. По аналогии с сопротивлением проводников I рода сопротивление раствора прямо пропорционально расстоянию между электродами d и обратно пропорционально площади их поверхности А:
Я = р4, (6.60)
А
где р — удельное сопротивление (Ом • см). При d = 1 см и А = 1 см2 имеем R = р, следовательно, удельное сопротивление равно сопротивлению 1 см3 раствора. Величину, обратную удельному сопротивлению, называют удельной
электропроводностью х = ~. Удельная электропроводность (См • см ’) численно равна току (в А), проходящему через слой раствора с поперечным сечением, равным единице, под действием градиента потенциала 1 В на единицу длины.
Электропроводность разбавленных растворов электролитов зависит от числа ионов в растворе (т. е. от концентрации), числа элементарных зарядов, переносимых каждым ионом (т. е. от заряда иона), и от скорости движения одинаково заряженных ионов к катоду или аноду под действием электрического поля. С учетом всех этих факторов электропроводящие свойства ионов характеризуют эквивалентной ионной электропроводностью (подвижностью). Она равна произведению абсолютной скорости движения иона на константу Фарадея и имеет размерность См • см2 (моль • экв). Под эквивалентом здесь подразумевается частица с единичным зарядом, например К+, СГ, Mg2+, j Al3+.
Эквивалентная электропроводность (подвижность) уменьшается с повышением концентрации раствора. При повышении концентрации электролита увеличивается ионная сила, а скорость движения ионов уменьшается за счет межионных взаимодействий. При нулевой концентрации (бесконечное разбавление) подвижности ионов становятся постоянными и максимальными и эквивалентная электропроводность раствора электролита А. при бесконечном разбавлении равна сумме подвижностей ионов, отнесенных к единичному заряду:
А,°=Х°+А,°_. (6.61)
Так как при концентрации электролита с моль • экв • л’1 в 1 см3 содержит 1 • 10 3 с моль • экв, то удельная и эквивалентная электропроводность электролита А. связаны соотношением
х=110'3сА,, (6.62)
следовательно, удельная электропроводность, определяемая суммой вкладов всех ионов, равна
х=1-10”3—У А.;с;, (6.63)
d
где с, — концентрация i- иона, моль-экв/л; А, — эквивалентная электропроводность i- иона, См-см2 (моль-экв); А — площадь поверхности электродов, см2; d — расстояние между электродами, см. Величины А* полученные экстраполяцией при бесконечном разбавлении и отнесенные к одному заряду, приводят в справочных таблицах. В табл. 6.76 даны величины А для некоторых ионов при 25 °C.
А
Величину, обратную отношению называют константой кондуктометрической ячейки 0, ее размерность — см-1, а величина зависит от геометрии ячейки.
Таблица 6.76
Эквивалентная электропроводность ионов при бесконечном разбавлении и 25 °C [СМ’СМ2/(МОЛЬ’ЭКВ)]
Катион А.° Анион А.°
Н+ 349,8 ОН' 199,0
К+ 73,5 80,0
nh; 73,5 Вг" 78,1
-рь2+ 2 68,5 Г 76,8
—Fe3+ 3 68,0 СГ 76,4
—Ва2+ 2 63,9 NO; 71,4
Ag+ 61,9 —СО2' 2 3 69,8
—Са2+ 2 59,5 сю; 67,3
Na+ 50,1 нсо; 44,5
Ячейка для измерения электропроводности состоит из двух платиновых электродов Э1 и Э2, впаянных в стеклянный сосуд, в который помещают анализируемый раствор (рис. 6.50). Электрический эквивалент этой ячейки (рис. 6.51) состоит из активного сопротивления раствора между электродами 7?р, сопротивлений проводников R\ и /?2, а также параллельно включенными емкостями двойного слоя на электродах Q и С2 и межэлектродной емкостью Ср. Ячейку подключают к источнику переменного тока частотой порядка 1000 Гц. При достаточно большой площади поверхности электродов (для этого их платинируют, т.е. покрывают платиновой чернью) заметного изменения концентрации не происходит, и поэтому сопротивлениями
и R можно пренебречь.
818
Новый справочник химика и технолога
Рис. 6.50. Ячейка для кондуктометрических измерений
Так как площадь поверхности электродов А велика, а расстояние между обкладками молекулярного конденсатора мало (порядка радиуса иона), то из соотношений
<6-64)
/ ®с
где С — емкость, а со — частота переменного тока, следует, что емкость С большая, a Rc мало. Поэтому можно пренебречь и емкостями С. и С2 ,тогда из электрического эквивалента ячейки остаются только Rp и Ср. Если сопротивление раствора не очень велико, электроды можно расположить на значительном расстоянии друг от друга. В этом случае емкость Ср будет мала, а сопротивление этого конденсатора Rc будет велико по сравнению с сопротивлением раствора Rp. Поэтому ток будет проходить только через Rp, и при таких условиях ячейка пригодна для измерения активного сопротивления раствора, следовательно, его удельной электропроводности.
Активное сопротивление Rp можно измерить с помощью моста Уитстона (рис. 6.51).
Рис. 6.51. Мост Уитсона: 1 — источник переменного тока; 2 — индикатор тока (осциллограф, наушники, усилитель переменного тока); 3 — ячейка
Он состоит из четырех сопротивлений: измеряемого сопротивления Rp, калиброванного магазина сопротивлений R и двух сопротивлений на концах проволочного реохорда Г] и г2. Величины сопротивлений R, и г2 можно подобрать такими, чтобы индикатор тока показал отсутствие тока в диагонали моста. При этом условии
R „ Rr2 - = — и R6= —
Г1 Г2 Г1
(6.65)
Сопротивления Г\ и г2 берут постоянными, a R подбирают в процессе измерения. Для компенсации реактивного сопротивления параллельно сопротивлению R включают
емкость С. Звуковой генератор служит для питания моста переменным током с частотой порядка 1000 Гц.
Электропроводность растворов зависит от температуры и увеличивается на 1-2% при повышении температуры на 1 °C, поэтому измерения рекомендуется проводить в термостатированной ячейке. Удельная электропроводность и измеренное активное сопротивление связаны соотношением
(6.66)
поэтому нужно знать константу ячейки 0. Ее можно определить, измерив сопротивление раствора с известной удельной электропроводностью. Для этого можно использовать, например, 1 М раствор КС1, для которого при 18 °C х = 0,09827 См • см”1.
Различают прямую и косвенную кондуктометрию, или кондуктометрическое титрование.
Прямая кондуктометрия находит в аналитической химии ограниченное применение. Причина этого в том, что электропроводность является величиной аддитивной и определяется присутствием всех ионов в растворе. Прямые кондуктометрические измерения можно использовать для контроля качества воды, применяемой в химической лаборатории, и современные установки для перегонки или деминерализации воды снабжаются кондуктометрическими датчиками — кондуктометрами для измерения удельной электропроводности растворов. Следует напомнить, что детекторы по электропроводности применяются в таком современном и перспективном методе анализа, как ионная хроматография.
Для кондуктометрического титрования пригодны кислотно-основные или осадительные реакции, сопровождающиеся заметным изменением электропроводности вследствие образования малодиссоциирующих или малорастворимых соединений.
Для примера рассмотрим кондуктометрическое титрование сильной кислоты НС1 сильным основанием. По мере добавления титранта NaOH в анализируемый раствор
Н+ + СГ + (Na+ + ОН”) -> Na+ + СГ + (Н2О) (6.67)
удельная электропроводность начнет резко снижаться, так как ионы водорода будут замещаться гораздо менее подвижными ионами натрия (табл. 6.76). После того как вся кислота будет оттитрована, удельная электропроводность начнет повышаться за счет появления избытка гидроксидионов. Восходящая ветвь на кривой титрования, изображенной на рис. 6.52, обусловлена суммарной электропроводностью ионов натрия и гидроксид-ионов. Точка пересечения соответствует конечной точке титрования.
Несимметричность ветвей обусловлена различием в подвижностях ионов Н и ОН” (см. табл. 6.76). При кондуктометрическом титровании можно применять ячейки с незакрепленными электродами и вводить их в раствор перед титрованием. Константу ячейки знать не нужно.
Электрохимические методы анализа
819
Рис. 6.52. Кривая кондуктометрического титрования соляной кислоты раствором гидроксида натрия и вклад отдельных ионов в электропроводность раствора
К достоинствам метода кондуктометрического титрования относится возможность проводить измерения с высокой точностью даже в очень разбавленных растворах. В термостатированной ячейке погрешность определения для 1'10^ М растворов не превышает 2%. В отличие от титриметрических методов с применением визуальных индикаторов кондуктометрическое титрование пригодно для анализа окрашенных или мутных растворов. Графический способ нахождения конечной точки титрования позволяет избежать трудностей, возникающих из-за замедления реакции вблизи конца титрования и снижающих точность фиксирования конечной точки. Иногда с помощью кондуктометрического титрования можно проводить последовательное определение компонентов смеси, например, титровать кислоты с различающимися константами диссоциации.
В таблице 6.77 приведены рабочие растворы, применяемые для определения кондуктометрическим методом важнейших неорганических веществ (ионов), и указаны условия титрования. Во втором столбце диапазон определяемых концентраций М (моль л-1) приведен в единицах -IgM. В последнем столбце приведены приблизительные значения погрешности данного определения.
Таблица 6.77
Кондуктометрическое титрование [11]
Определяемое вещество (ион) -IgM Рабочий раствор Условия титрования Погрешность, отн. %
Н" кислоты: сильные рК<2 1-5 NaOH Отсутствие СОг 0,1-1
слабые рК = 2-6 1-4 NH4OH То же 0,1-1
очень слабые рК<7 1-4 LiOH Отсутствие СО2; водонерастворимые кислоты титрование в 75% сп. 0,5-5
Продолжение таблицы 6.77
Определяемое вещество (ион) -IgM Рабочий раствор Условия титрования Погрешность, отн. %
ОН основания: сильные 1-5 НС1 Отсутствие со2 0,1-1
слабые 1-4 СНзСООН То же 0,1-1
очень слабые <3 НС1 Отсутствие СО2; водо-нераств. основания титруются в 75% сп. >0,5
Н2О2 1-3 КМпО4 0,05 М H2SO4 1
I группа Периодической системы
К+ = 2,3 pH “ 5-10 0,5
Си2+ 1-2 Li2C2O4 Нейтр. р-р 0,1
1-3 Na-ЭДТА pH = 5 (разб. ацетатн. буф.) 0,2
3-5 H2S, водный раствор (0,001 М) Нейтр. р-р; отсутствие со2 1-5
Ag+ 1—4 NaCl или LiCl Нейтр. или слабокислый р-р 0,2
1-3 Na2CrO4 или Li2C2O4 Нейтр. р-р 0,5
4-6 H2S, водный р-р (0,001 М) Нейтр. р-р; отсутствие СО2 1-5
II группа Периодической системы
Ве2+ 2-4 NaOH Хлориды или нитраты; отсутствие СО2; 80 °C. В кислом р-ре два перегиба: 1 нейтрализация к-ты; 2 осажд. Ве(ОН)2 2-3
Mg2+ 1-2 NaOH pH- 10 (аммиачн. буф.) 1
Na-ЭДТА 0,2
Са2+ 1-2 Na-ЭДТА То же
820
Новый справочник химика и технолога
Продолжение таблицы 6.77
Определяемое вещество (ион) -IgM Рабочий раствор Условия титрования Погрешность, отн. %
Sr2+ 2 Na-ЭДТА То же 0,2
1-2 Ы2С2О4 Нейтр. р-р 1
Ва2+ 1-3 Li2SO4 или Li2c2o4 30% сп.; ожидать несколько минут после каждой порции рабочего р-ра до установления постоянной электропроводности 0,1-1
Zn2+ 2-3 NaOH В кислом растворе два перегиба: 1 нейтрализация к-ты; 2 осажд. Zn(OH)2 0,5
Cd2+ 2 Na-ЭДТА pH -5 (ацетата, буф.) 0,2
1 Li4[Fe(CN)6] Не меш. РЬ2+ 0,5
1 Na-ЭДТА pH -5 (разб. ацетата. буф.) 0,2
4-6 H2S, водный р-р (0,001 М) Нейтр. р-р; отсутствие со2 1-5
Hg2+ 1-2 NaOH Рабочий р-р титр, анализ, р-ром (HgCl2) 1
4-6 H2S, водный р-р (0,002 М) рН = 3; отсутствие со2 0,5-5
III группа Периодической системы
А13+ 2-3 NaOH В кислом р-ре три перегиба: 1 нейтрализация к-ты; 2 осажд. А1(ОН)3; 3 образование А1О2 1
Продолжение таблицы 6.77
Определяемое вещество (ион) -IgM Рабочий раствор Условия титрования Погрешность, отн. %
Т1+ ~2,3 Na2CrO4 Нейтр. р-р 0,2
2 Na[B(QH5>] То же 0,5
1-2 KSCN То же 0,5
IV группа Периодической системы
CN 1-3 В нейтр. р-ре два перегиба: 1 образование [Ag(CN)2]“; 2 осажд. AgCN. Положение 1-го перегиба определяется с большей точностью 0,2
2 Hg(C104)2 В нейтр. р-ре два перегиба: 1 образование [Hg(CN)4)]2” (можно в присутствии СГ); 2 осажд. Hg(CN)2 0,1
CNO 1-2 AgNO3 Нейтр. р-р 0,5
СО2” 2^4 HC1 При титровании. три скачка нейтрализации: 1 ОН”, 2СО3, 3 НСО3. Положение 3-го перегиба определяется более точно 0,5-1
2-3 BaCl2 Медленное титрование 0,1
SCN 1-5 AgNO3 Нейтр. или слабокислый р-р (при титровании очень разбавл. р-ров добавл. сп. до 90%) 0,2- 10
Электрохимические методы анализа
821
Продолжение таблицы 6.77
Определяемое вещество (ион) -IgM Рабочий раствор Условия титрования Погрешность, отн. %
РЬ2+ 1-4 Li2C2O4 или K4[Fe(CN)6], или Na2CrO4 Нейтр. р-р 0,2
4-6 H2S, водный p-p (0,001 M) Нейтр. р-р; отсутствие со2 1-5
2 Na-ЭДТА pH = 5 (разб. ацетата. буф.) 0,2
V группа Периодической системы
no; 0,3 Нитрон в СНзСООН Слабокислый р-р 0,5
ро; 1-3 BiOClO4 НС1О4 (0,3 М); отсутствие As(V)
2-3 Уранилацетат CH3COONa (0,01 М) 0,3
AsO3” 4-5 Иод, сп. раствор НСОз”, гладкий Pt-электрод 3-12
Bi3+ 4-6 H2S, водный раствор (0,001 М) Нейтр. р-р; отсутствие СО2 1-5
V(I11) 2-3 КМпО4 H2SO4 (разб. р-р) 1
V(V) 2 AgNO3 Нейтр. р-р 1
2-2,3 [СЬ№Шз Нейтр. р-р 1
VI группа Периодической системы
SO2” W (СН3СОО)2Ва и СНзСООН (1%р-р) 20% сп. р-р (меш. большие к-ва NO;) 0,2-2
S2O32” 1-2 Pb(NO3), Нейтр. р-р 0,5
SeO2” 2-3 Pb(NO3)2 или AgNO3 Нейтр. р-р 0,5-1
SeO2” 1-2 Pb(NO3)2 или ВаС12 50% сп. р-р 0,5
Cr2O2” 3 Fe(NH4)2 (SO4)2-6Н2О H2SO4 (0,2 М) 0,1
CrO2” 2 ВаС12 Нейтр. р-р 0,2
U(IV) 2-3 КМпО4 H2SO4 (0,2-0,5 М) 1
Продолжение таблицы 6.77
Определяемое вещество (ион) -IgM Рабочий раствор Условия титрования Погрешность, отн. %
VII группа Периодической системы
F’ 1-3 А1С13 30-50% сп. р-р; 50-кратный избыток NaCl+ 1-3
СГ 1-5 AgNO3 При титровании очень разбавл. р-ов добавл. сп. до 90% 0,2- 10
1^1 Hg(C104)2 Нейтр. или слабокислым р-ром 0,1-1
Вг” 1-5 AgNO3 Нейтр. или слабокисл. р-р; при титр, очень разб. р-ров добавл. сп. до 90% 0,2- 10
Г 1^1 AgNO3 NH4OH (2%NH3) 0,2-1
ю; 2-4 HC1 Нейтр. р-р; KI и Na2S2O3 (в небольшом избытке по ю;) 0,5
VIII группа Периодической системы
Fe2+ 2-3 K2Cr2O7 H2SO4 (0,1 М) 0,2
2 KMnO4 H2SO4 (0,1 М) 1
[Fe(CN4F 1-2 AgNO3 Нейтр. р-р 1
P^CN,)]4” 1-2 Pb(NO3)2 Нейтр. или слабокисл. р-р титр, при 100 °C 0,1
ZnCl2 Нейтр. р-р** 0,2
Со2+ 2 Na-ЭДТА pH = 5 (ацетата. буф.) 0,2
2 Li3[Fe(C3N)6] Слабокисл. Р-Р 1
822
Новый справочник химика и технолога
Продолжение таблицы 6.77
Определяемое вещество (ион) -IgM Рабочий раствор Условия титрования Погрешность, отн. %
Ni2+ 2 Диметилглиоксим, сп. р-р Аммиачн. буф.; добавл. избыток рабочего р-ра и титр. анал. р-ром 1
Примечание. * При титровании образуется NaaAlF6;
** При титровании осаждается K2Zn[Fe(CN)6].
В таблице 6.78 приведены данные, дающие общую характеристику условий выполнения титрования отдельных соединений. На вид кривой высокочастотного титрования, на его чувствительность и точность более всего влияют частота, на которой ведут титрование и электрические характеристики ячейки.
Таблица 6.78
Высокочастотное титрование
Определяемый ион (соединение) Рабочий раствор Область концентраций (количеств) определяемого соединения Относительная ошибка определения, % Примечание
соединение концентрация
Н+ (сильные кислоты) NaOH 0,001-1,0 М 0,0002-0,02 М 0,01-0,5
Н+ (слабые кислоты) NaOH 0,02-1,0 М 0,002-1,0 М 0,1-1,0 У многоосновных кислот конец титрования слабо выражен
Н+ (смесь сильных и слабых кислот) Метилглю камин (1 -дезокси-1 -метиламиносорбит) 0,01 М ~5-10 мг ==-1,0
ОН (основания) НС1 0,002-1,000 М 0,002-1,0 М 0,2-1
Ag* NH4SCN или KSCN 5% <2,5-104М Титрование проводят в 1/300 М HNO3
А13+ NaOH или NaF 0,2 М 0,01-0,1 м См. также Ионы металлов и Mg2+
(NH4)2HPO4 или 8-оксихинолин 2,7-7,0 мг В водно-спиртовом р-ре при pH = 4,1; см. также Cd2+ и Mg2+
Ва2+ К2Сг2О7 0,1 М 0,01-1 М См. также Ионы металлов
Ве2+ NaOH 0,5 М 10-40 мг ~1
Вг (см. СГ)
CN'(cm. СГ)
СО’ (см. ОН)
с2о2- СаС12 или (NH4)2HPO4 >2-10'3М = 1,8
Са2+ (NH4)2C204 0,25 М ~ 100 мг = 1 Титрование проводят в 50% мет. сп. См. также Ионы металлов
Cd2+ (NH4)2HPO4 22,5-35,7 мг 0,5 Титрование проводят в водно-спиртовом р-ре при pH = 6,0. При других значениях pH можно опр. ионы: Mg2+ (рН = 10,3), Мп2+(pH = 6,5), Zn2+(pH = 7,2), А13+ (pH -4,1); см. также Ионы металлов
Электрохимические методы анализа
823
Продолжение таблицы 6.78
Определяемый ион (соединение) Рабочий раствор Область концентраций (количеств) определяемого соединения Относительная ошибка определения, % Примечание
соединение концентрация
СГ AgNO3 или Hg(NO3)2 0,001-0,1 м 0,0002-0,02 М Так же можно титр. CN", SCFT, ВГ, Г
Со2+ 8-Оксихинолин 1,5% в 50% сп. ~ 0,1 мг См. также ионы металлов и Mg2+
Си2+ (см. Ионы металлов)
Диметилглиоксим NiSO4 или CoSO4 0,005 М -0,0001 м Титрование проводят в аммиачной среде
F“ (CH3COOH)3La или А1(МО3)3 0,03 М 0,01-0,03 м ~ 1
Th(NO3)4 0,02 М 4-6 мг -0,2
(СН3СОО)2Са 0,025 М -1 Опр. во фторполимерах после сожжения в кислороде и поглощения оксида фтора 0,5% CH3COONa
Fe3+ Купферон 0,15 М Изменяя пределы pH- осажд. (даны в скобках) можно опр.: Fe3+ (2,3-3,4), Sn2+ (2,75-3,16), Sn4+ (2,74-3,00), V3+ (1,78-1,92), VO2+ (4,2-4,45), Ti4+ (2,45-2,65) и ZrO2+ (3,0-3,4 при 5-10 °C).
Fe2+ КМпО4 0,002-0,02 М 0,0001-0,02 М Титр, в кислом р-ре. См. также ионы металлов
Hg2+ NaCl или KSCN 0,005-0,1 М 0,001-0,1 М 0,1-0,5 См. титр. Ag+
Ионы металлов Na-ЭДТА 0,02-0,1 М 0,0002-0,005 М Титр. Al3+, Ва2+, Са2+, Cd2+, Со2+, Fe2+, лантаниды, Mg2+, Mn2+, Ni2+, Pb2+, Sr24, Th4+, U(VI), Zn2+
Г (см. СГ)
Ю3 НС1 0,04 М 0,04 ммоль 0,5 Титрование в присутствии изб. KI и Na2S2O3
К+ МВ(СбН5)4] 0,1 М ~ 8 мг
Лантаниды (см. Ионы металлов)
Mg2+ 8-Оксихинолин 1,5% в 50% сп. 0,1 мг Изменяя пределы pH- осаждения (даны в скобках), можно опр.: Mg2+ (8,2-9,2), Со2+ (4,2-5,6), А13+ (5,5-6,2), Т13+ (3,7-4,6), Th4+ (4,8-7,7), V(V) (4,6-6,2), Mo(V) (3,3-7,6), U(V)(4,6-8,2). См. также Ионы металлов
(NH^HPCU 0,75 pH = 10,3. См. также Cd2+
Мп2+ (см. Ионы металлов и Cd2+)
Mo(VI) 8-Оксихинолин ~ 0,1 мг См. также Mg2+
Ni2+ Диметилглиоксим
824
Новый справочник химика и технолога
Продолжение таблицы 6.78
Определяемый ион (соединение) Рабочий раствор Область концентраций (количеств) определяемого соединения Относительная ошибка определения, % Примечание
соединение концентрация
Нитрилтриуксусная к-та СиС12 0,05 М ~ 0,1 г
Окситетрациклин Na-ЭДТА 0,01 М ~ 0,1 г При pH = 2 добавл. изб. 0,01 М FeCl3 и обратно титр, непрореагировавший Fe3+ р-ром Na-ЭДТА
РЬ2+ (см. Ионы металлов)
SCN (cm. СГ)
SO4~ (СН3СОО)2Ва 0,01 М 0,1-0,8 мг Титр, в 50% р-ре диоксана
Sr24" (см. Ионы металлов)
Sn2+ и Sn4+ Купферон 0,15 М См. также Fe3+
Th4+ Th(NO3)4 0,025 М ~ 0,5 ммоль Добавл. изб. Сг2О 4 и проводят обратное титр. Прямое титр, можно производить с NaF
8-Оксихинолин ~ 50 мг pH = 4,8-7,7. См. также Mg2+
Ti4+ Купферон 0,15 М pH = 2,45-2,65. См. также Fe3+
ti+ Na[B(C6H5)4] 0,2 М 0,05-0,2 ммоль 1 Титрование не мешают Cd2+, Cu2+, СН3СООН, H2SO4, Na+, Zn2+
8-Оксихинолин См. также Mg2+
UOf+ + (см. Ионы металлов и Mg24)
V(V) 8-Оксихинолин ~ 0,1 мг pH = 4,6-6,2. См. также Mg2+
Купферон 0,15 М См. также Fe3+
Zn2+ (см. Ионы металлов и Cd2+)
ZrO2+ Купферон См. Fe3+
Литература к 6.2 - 6.6
1. Плэмбек Дж. Электрохимические методы анализа. Пер. с англ. М.: Мир, 1985. 496 с.
2. Краткая химическая энциклопедия. Том 1. А-Е. М.: Советская энциклопедия, 1964. С. 758.
3. Классификация и номенклатура электрохимических методов (рекомендации утверждены в 1975 г.) // Журн. аналит. хим. 1978. Т. 33, вып. 8. С. 1647-1665.
4. Recommended Terms, Symbols and Definitions for Elec-troanalytical chemistry // Pure & Appl. Chem., 1979, Vol. 51. P. 1159-1174.
5. Об использовании понятия «химический эквивалент» и связанных с ним величин // Журн. аналит. хим. 1989. Т. 44, вып. 4. С. 762-764; по этому же вопросу Журн. аналит. хим. 1982. Т. 37, вып. 5. С. 946; Журн. аналит. хим. 1982. Т. 37, вып. 5. С.947.
6. Нейман Е.Я. Терминология современной аналитической химии и ее формирование // Журн. аналит. хим. 1991. Т. 46, вып. 2. С. 393-405.
7. Представление результатов химического анализа (Рекомендации ГОРАС 1994 г.) // Журн. аналит. хим. 1998. Т. 53, № 9. С. 999-1008.
8. Compendium of Analytical Nomenclature (Definitive Rules 1997). 3d ed. IUPAC, Blackwell Science, 1998, 8.1-8.51 (Electrochemical Analysis).
9. Брайнина X.3., Нейман Е.Я., Слепушкин B.B. Инверсионные электроаналитические методы. М.: Химия, 1988. 240 с., ил.
10. “EG&G Princetone Applied Research Analitik mit Polarogra-phie und Voltammetrie’7 Katalog fur Polarographische und Voltammetrische Anwendungsmoglichkeiten und Gerate. Al-chema-85. Frankfurt, 1985.
Электрохимические методы анализа
825
11. В помощь аналитику-практику: виртуальные приборы в электрохимическом анализе / Под ред. В.А. Демина. Спб.: Алтей, 1997. 111 с.
12. Богданов Э.В., Борцов Н.Н., Шартуков О.Ф. Электрохимические методы анализа: Уч. пособие / Л.: ЛТИ им. Ленсовета, 1987. С. 93.
13. Основы аналитической химии: В 2 кн. Кн. 2. Методы химического анализа: Учеб, для вузов / Ю.А. Золотов, Дорохова Е.Н., Фадеева В.И. и др.; Под ред. Золотова Ю.А.. М.: Высш, шк., 1996. 461 с.
14. Вольтамперометрия. Методические указания к лабораторным работам. / Сост. Л.С. Надежина. Л.: ЛПИ им. М.И. Калинина, 1983. 44 с.
15. Е 100 Universal DC Polarograph. Karlsruhe-Rheinstetten: Bruker-physik AG, 1972. 30 p.
16. Flato J.B. The Renaissance in Polarographic and Voltammetric Analysis // Analyt. Chem. 1972, V. 44. P. 75A-87A.
17. Румянцев А.И. Компьютеризованный многокомпонентный вольтамперометрический анализ / Автореф. на со-иск. уч. ст. к. х. н. М.: МГУ, 2000. 18 с.
18. Лушов К.А. Компьютеризованный вольтамперометрии-ческий анализ на модифицированных атомами серебра и золота электродах / Автореф. на соиск. уч. ст. к. х. н. М.: МГУ, 2001.20 с.
19. Инверсионная вольтамперометрия: Методич. указания и практ. руководство по физ. химии для студентов химических специальностей / Сост. Э.А. Захарова, Н.П. Пикула. Н.М. Мордвинова. Томск: ТПУ, ВНПФ «ЮМХ», 1998. 65 с.
20. Брайнина Х.З., Нейман Е.Я. Твердофазные реакции в электроаналитической химии. М.: Химия, 1982.264 с.
21. Neeb R., Kiehnast I. / Z. Anal. Chem., 1968. V. 241. P. 142.
22. Kiyger L. Microcomputers in Electrochemical Trace Elemental Analysis // Analytica Chimica Acta. 1981. V. 133. P. 591-602.
23. Методика выполнения измерения содержания цинка в водных растворах методом инверсионной вольтамперометрии / Свид. о метрологической аттестации 2420/473-95/0611 от 02.08. 1995 г. СПб, 1995.
24. Бок Р. Методы разложения в аналитической химии. М.: Химия, 1984, 316 с.
25. Чернышова Н.Н., Свинцова Л.Д. Изучение возможности восстановления градуировочных характеристик при ИВ-определении металлов в электрохимически обработанных растворах СПАВ. Тезисы IV конференции «Электрохимические методы анализа (ЭМА-94)». Ч. 1 // Завод, лаб., 1991. Т. 57, №8. С. 151.
26. Немодрук А.А., Безрогова Е.В. Фотохимические реакции в аналитической химии. М.: Химия, 1992. 169 с.
27. Jagner D. Potentiometric stripping analysis in non-deaera-ted samples // Anal. Chem., 1979. V. 51. P. 342-345.
28. Галинкер Э.В., Маковецкий А.Л., Гороновский И.Т. Хронопотенциометрический способ определения свинца в растворе / Авт. свид. СССР 485973. Бюлл. изобр. 1975. № 36.
29. Barradas R.G., Kimmerle / Effect of Highly Surface-Active Compounds on Polarographic Electrode Processes. Part I. Electrochemical Adsorption and Structure at the Dropping Mercury Electrode-Solution Interface // J. Electoanal. Chem. 1966. V. 11. P. 128-136.
30. Нейман Е.Я., Драчева Л.В. Адсорбционная инверсионная вольтамперометрия // Журн. аналит. химии. 1990. Т. 45, вып. 2. С. 222-236.
31. Справочник химика Т. IV. Аналитическая химия, спектральный анализ, показатели преломления. М.; Л.: Химия, 1965. 920 с.
32. Выдра Ф., Штулик К., Юлакова Э. Инверсионная вольтамперометрия. М.: Мир, 1980. 280 с.
33. Harris D.C. Quantitative chemical analysis. 5th ed. New York: W.H. Freeman & Company, 1999. 899 p.
34. Van der Linden W.E., Dieker J.W.Glassy Carbon as Electrode Material in Electroanalytical Chemistry // Analytica Chimica Acta, 1980. V. 119. P. 1-24.
35. Trace Element Analysis/ Method and Program Specification: Radiometer A/S. Copenhagen, 1990.
36. Добош Д. Электрохимические константы: Справочник для электрохимиков. М.: Мир, 1980. 365 с.
37. Справочник по электрохимии / Под ред. А.М. Сухотина. Л.: Химия, 1981. 488 с.
38. Корыта И. Ионы, электроды, мембраны / Пер. с чешек. М.: Мир, 1983.264 с.
39. Вольтамперометрия органических и неорганических соединений / Под ред. П.К. Агасяна, С.М. Жданова. М.: Наука, 1985.248 с.
40. Каплан Б.Я., Пац Р.Г., Салихджанова М. Ф. Вольтамперометрия переменного тока: Сер. Методы аналитической химии. М.: Химия, 1985. 264 с.
41. Галлай З.А. и др. Применение анодной вольтамперометрии для количественного определения органических соединений (Обзор) // Журн. аналит. химии. 1986. Т. XLI, вып. 5. С. 773-787.
42. Ханина Р.М., Татауров В.П., Брайнина Х.З. Электроды в инверсионной электроаналитической химии (Обзор) // Завод, лаб. 1988. Т. 54, № 2. С. 1-13.
43. Каплин А.А., Пикула Н.П., Нейман Е.Я. Электроды в вольтамперометрии // Журн. аналит. химии. 1990. Т. 45, вып. 11. С. 2086-2120.
44. Электрохимические методы // Журн. аналит. химии. 1994. Т. 49, № 9. С. 901-1040.
Дополнительная литература к 6.2 - 6.6
1. Special issue Papers presented at the Fifth European Conference on Electroanalysis (ESEAC'94), Jesolo Lido, Ve-nece, Italy, May 22-26, 1994. // Analitical Chimica Acta. 1995. V. 305, № 1-3, p. 1-359.
2. Bard A. J. Larry R. Electrochemical Methods: Fundamentals and Applications. 2nd ed. New York: Wiley, 2001. 833 p.
3. Brainina Kh. Z. Neyman E. Electroanalytical stripping methods. New York: Wiley, 1993, 198 p.
826
Новый справочник химика и технолога
4. Electroanalytical Chemistry: Vol. 18 / Ed. by Bard A.J. New York: Dekker, 1993. 416 p.
5. Gaius Z. Fundamentals of Electrochemical Analysis. 2 nd ed. New York: Ellis Horwood, 1991. 520 p.
6. Hart J.P. Electroanalysis of biologically important compounds. -New York etc.: Ellis Horwood, 1990. 213 p.
7. Hulanicki A., Glab S. Present and Future Status of Organic Analytical Reagents. Part I // Pure and Appl. Chem. 1990. V. 62. P. 2147. Part II // Pure and Appl. Chem. 1990. V. 62, P. 2323. Part III. Organic Analytical Reagents in Electroanalysis // Pure and Appl. Chem. 1991. V. 63, № 12, P. 1805-1826
8. Klug O. New Developments in Conductimetric and Oscillometric Analysis. Amsterdam etc.: Elsevier. 1988. 313 p.
9. Modem Techniques in Electroanalysis / Ed. by Vanysek P. New York: Wiley, 1996. 369 p.
10. Monk P. Fundamentals of Electro-Analytical Chemistry. New York: Wiley, 2001, 384 p.
11. Riley T., Watson A. Polarography and Other Voltammetric Methods /Ed. by James A. M. Chichester etc.: Wiley, 1987. - XXII, 283 с.: ил. (Analytical chemistry by open learning).
12. Svehle G. Comprehensive Analytical Chemistry. V. XXVII: Analytical Voltammetry / Eds. by Smyth M.R., Vos J.R. Amsterdam; London; New-York; Tokyo: Elsevier Science Publ., 1992, 578 p.
13. Wang J. Selectyvity Coefficients for Amperometric Sensors // Taianta, 1994. V.41, № 6. P. 857-864.
Приложение 1
Перечень ионометрических методик
Принятые обозначения. ХС — халькогенидные стеклянные химические сенсоры; ГЭТМ — гомогенные электроды с твердыми мембранами; ЖМЭ — жидкостной мембранный электрод
№ Определяемый ион Объекты анализа Марка электрода (фирма изготовитель) Методики КХА Пределы определения моль-л”1
1 Ag+ Угли активные (каменные, древесные, импре-гнир. серебром) ЭК-13.01.01 ЭК-13.01.02 XC*-Ag-01 ГК СЭН РФ. Свидет. об аттестации № 08-47/096 по реестру метрологической службы ГСЭН РФ МО”1- 5-107
2 Ва2+ ЭК-11.01.01 ЭК-11.01.02 МО-1-5-10”5
3 Вг” Вода ЭК-22.01.01 ЭК-22.01.02 ГЭТМ** рВг фирм Philips; Schott ond Gen.; Tacussel Мидгли Д. Потенциометрический анализ воды. М.: Мир, 1980 МО”1-МО”6
4 Са2+ Воды минеральные питьевые лечебные, лечебно-столовые и природные столовые ЭМ-08.01.02 ЭМ-08.01.02 ГОСТ 23268.5-78 Воды минеральные питьевые лечебные, лечебно-столовые и природные столовые. Методы определения ионов кальция и магния. ГОСТ 26449.1-85. Установки дистилляционные опреснительные стационарные. Методы химического анализа соленых вод. Методы определения кальция МО”1-МО”5
Воды соленые Унифицированные методы анализа качества вод. Т. 1.4.1.Методы химического анализа вод. М.: СЭВ, 1987. РД 52.24.45-91
Воды грунтовые и поливные МСХП РФ. МУ по ионометрическому определению катионно-анионного состава грунтовых и поливных вод. (Утв. 14. 04. 1994 г.) М.: ЦИНАО, 1995. (Ионометрическое определение кальция С. 35-40)
Электрохимические методы анализа
827
Продолжение таблицы
№ Определяемый ион Объекты анализа Марка электрода (фирма изготовитель)* Методики КХА Пределы определения моль-л-1
4 Са2+ Молоко Методика количественного ионометрического анализа молока на содержание ионов хлора, в т. ч. для выявления анормального молока (аттестация проведена ГП ВНИИМ им. Менделеева), М., 1999
5 Cd2+ Природные и сточные воды ЭК-20.01.01 ЭК-20.01.02 XC-Cd-01 1-Ю’1-1-Ю-6
6 СГ 1 Почвы ЭК-21.01.01 ЭК-21.01.02 ГОСТ 26425-85. Почвы. Методы определения ионов хлорида в водной вытяжке 1 10'-З-Ю'5
Вода ЭК-21.01.03 ЖМЭ *** рС1 фирм Orion, Coming Унифицированные методы анализа качества вод. Т. 1.4. 1. Методы химического анализа вод. М.: СЭВ, 1987. РД 52.24.7-83 0,6 -3-1О'3
Воды грунтовые и поливные МСХП РФ. МУ по определению хлоридов, нитратов и аммония в водах. (Утв. 14.04. 1994 г.) М.: ЦИПАО, 1996. (Ионометрическое определение хлора С. 5-9) МСХП РФ. МУ по ионометрическому определению катионно-анионного состава грунтовых и поливных вод. (Утв. 14.04. 1994 г.) М.: ЦИПАО, 1995. (Ионометрическое определение ионов хлорида С. 14-18) Методические указания по определению pH, нитратов, хлоридов и фторидов на многоканальном поточном измерителе с использованием ионселекгивных электродов. М.: ЦИ-НАО, 2000
Молоко Методика количественного ионометрического анализа молока на содержание ионов хлора, в т. ч. для выявления анормального молока (аттестация проведена ГП ВНИИМ им. Менделеева), М., 1999
7 сю; ЭМ-09.01.01 ЭМ-09.01.02 ЖМЭрС1О4 фирм Orion, Coming
8 CN" Вода ЭК-15.01.01 ЭК-15.01.02 ЭМ-CN-Ol Мидгли Д. Потенциометрический анализ воды. М.: Мир, 1980 1-Ю'2-1-Ю'5
9 СО32' Воды грунтовые и поливные ЭМ-10.01.01 ЭМ-10.01.02 МСХП РФ. МУ по ионометрическому определению катионно-анионного состава грунтовых и поливных вод. (Утв. 14.04.1994 г.) М.: ЦИНАО, 1995. (Нитриметрическое определение ионов карбоната и бикарбоната с помощью pH-электрода С. 7-10) 1-Ю'3-1-Ю'7
10 Cr(VI) XC-Cr(VI)-01 1-10'7- 103
828
Новый справочник химика и технолога
Продолжение таблицы
№ Определяемый ион Объекты анализа Марка электрода (фирма изготовитель)* Методики КХА Пределы определения моль-л 1
11 Си2+ Природные и сточные воды ЭК-19.01,01 ЭК-19.01.02 ХС-Си-01 140"1 - 110^
12 F” Воды минеральные питьевые лечебные, лечебно-столовые и природные столовые ЭК-12.01.01 ЭК-12.01.02 ЭК-12.01.03 ГОСТ 23268.18-78. Воды минеральные питьевые лечебные, лечебно-столовые и природные столовые. Методы определения фторид-ионов. ГОСТ 4386-89.Вода питьевая. Методы определения массовой концентрации фтора. Унифицированные методы анализа качества вод. Т. 1.4. 1. Методы химического анализа вод. М.: СЭВ, 1987. 1-Ю"1 - НО'5
Вода питьевая Природные воды, сточные воды производства полимеризационных пластмасс, редкометаллической отрасли промышленности. Черкассы, 1990. 1 - МО’5
Анализ соленых вод ГОСТ 26449.1-85. Установки дистилляционные опреснительные стационарные. Методы химического анализа соленых вод. Методы определения сульфат-ионов. РД 52.24.6-83 1-10 2 1-ю5
Воды грунтовые и поливные МСХП РФ. МУ по ионометрическому определению катионно-анионного состава грунтовых и поливных вод. (Утв. 14.04. 1994 г.) М.: ЦИПАО, 1995. (Определение иона фторида С. 43-47) 1-10 “2- 110-5
Корма, комбикорма, растительная продукция МСХП РФ. МУ по ионометрическому определению содержания фтора в растительной продукции, кормах и комбикормах. (Утв. 14.04. 1995 г.)М.: ЦИПАО, 1995. Методические указания по определению pH, нитратов, хлоридов и фторидов на многоканальном поточном измерителе с использованием ионселективных электродов. М.: ЦИПАО, 2000
Молоко Методические указания по определению фторидов в молоке потенциометрическим методом. М.: ЦИПАО, 1993
13 Fe3+ XC-Fe-01 5-10-6 -1,0
14 FT Вода питьевая ЭСЛ-ИГ-04 ЭСЛ-11 Г-05 ЭСЛ-41 Г-04 ЭСЛ-41Г-05 ЭСЛ-45-07 ЭСЛ-43-07 ГОСТ 2874-82. Вода питьевая. Гигиенические требования и контроль за качеством. ГОСТ 26449.1-85. Установки дистилляционные опреснительные стационарные 0-14 ед. pH
Воды природные и сточные ЭКОМ-рН Методика количичественного химического анализа вод и водных растворов для определения pH потенциометрическим методом с помощью ионоселективного электрода «ЭКОМ-pH». М., 1997. Per. № 001-73-97 (Утв. ГНМЦ «ВНИИФТРИ» 03.06. 1997)
Электрохимические методы анализа
829
Продолжение таблицы
№ Определяемый ион Объекты анализа Марка электрода (фирма изготовитель)* Методики КХА Пределы определения моль-л-1
14 Н' Воды поливные ЭСЛ-63-07 ЭСЛ-15-11 ЭСЛ-45-11 МСХП РФ. МУ по ионометрическому определению катионно-анионного состава грунтовых и поливных вод. (Утв. 14.04. 1994 г.) М.: ЦИНАО, 1995. (Определение pH С. 6-7)
Вода соленая Методы химического анализа соленых вод. Электрометрический метод определения pH. ГОСТ 26449.1-85. Установки дистилляционные опреснительные стационарные. Методы химического анализа соленых вод. Титриметрические методы определения общей ще лочности
Молочные продукты 5М2.840.019 5М2.840.074 ЭС-01.08.04 ЭС—71—11 ГОСТ 30305.3-95. Консервы молочные сгущенные и продукты молочные сухие. Титриметрические методики выполнения измерений кислотности
СТП. Методика определения кислотности молока и молочных продуктов путем измерения pH. ВНИМИ, 1995. Методика выполнения измерений pH молока и молочных продуктов (аттестация проведена ГП ВНИИМ им. Менделеева), М., 1999
Почвы ГОСТ 26483-85. Почвы. Приготовление солевой вытяжки и определение ее pH по методу ЦИНАО. ГОСТ 26484-85. Почвы. Метод определения обменной кислотности. ГОСТ 26212. Определение гидролитической кислотности почв pH-метрическим методом
Лесохимические продукты Продукты лесохимические. Метод определения кислотного числа потенциометрическим титрованием. ГОСТ 26423-85. Почвы. Методы определения удельной электрической проводимости, pH и плотного остатка водной вытяжки
Нефтепродукты ГОСТ 11362-76. Нефтепродукты. Метод определения числа нейтрализации потенциометрическим титрованием (введен 04.02.1976, СТ СЭВ5025-85, проверен в 1986 до 1994 г.) Методические указания по определению pH, нитратов, хлоридов и фторидов на многоканальном поточном измерителе с использованием ионселективных электродов. М., ЦИНАО, 2000
15 Hg2+ ЭК-18.01.01 ЭК-18.01.02 XC-Hg-01 1-Ю-1- 1'10 6
830
Новый справочник химика и технолога
Продолжение таблицы
№ Определяемый ион Объекты анализа Марка электрода (фирма изготовитель)* Методики КХА Пределы определения моль-л-1
16 Г Вода ЭК-23.01.01 ЭК-23.01.02 ЭМ-1-01 ГЭТМ** р! фирм Philips; Schott ond Gen.; Tacussel Унифицированные методы анализа качества вод. Т. 1.4. 1. Методы химического анализа вод. М.: СЭВ, 1987 1 -101 - 1-ю6 1-Ю1-1-10 5
17 К+ Вода ЭМ-05.01.02 ЭМ-05.01.02 ЖМЭ фирмы Orion, Coming Унифицированные методы анализа качества вод. Т. 1.4. 1. Методы химического анализа вод. М.: СЭВ, 1987.РД 52.24.69-91 1-Ю"1-1-Ю'5
Почва МСХП РФ, ГУ химизации и защиты растений с госхимкомиссией МСХ РФ, ЦИНАО: МУ по определению легкоподвижного калия в почвах ионометрическим методом (Утв. 14.04. 1994 г.). М.: ЦИНАО, 1996 5-500 мг-дм'3
18 Li+ ЭС-03.01.01 1 -1-Ю"4
19 Na+ Комбикорма, комбикормовое сырье ЭС-04.01.01 ЭС-04.01.02 ЭСЛ-51Г-04 ЭСЛ-51Г-05 ЭСП-05-06 ЭС-10-07 ГОСТ 13496.1-89. Комбикорма, комбикормовое сырье. Методы определения натрия и хлористого натрия МО-1-1-Ю-4
Вода соленая ГОСТ 26449.1-85. Установки дистилляционные опреснительные стационарные. Методы химического анализа соленых вод. Методы определения натрия РД 52.24.14-91
Воды грунтовые и поливные МСХП РФ. МУ по ионометрическому определению катионно-анионного состава грунтовых и поливных вод. (Утв. 14.04. 1994 г.) М.: ЦИНАО, 1995. (Ионометрическое определение натрия С. 27-30)
Молоко Методика количественного ионометрического анализа молока на содержание ионов натрия, в т. ч. для выявления его фальсификации содой (аттестация проведена ГП ВНИИМ им. Менделеева), М., 1999
20 nh; Вода ЭМ-06.01.01 ЭМ-06.01.02 XC-NH4-OI Унифицированные методы анализа качества вод. Т. 1.4.1. Методы химического анализа вод. М.: СЭВ, 1987. РД 52.24.46-91 МСХП РФ. МУ по определению хлоридов, нитратов и аммония в водах. (Утв. 14.04.1994 г.) М.: ЦИНАО, 1996. (Ионометрическое определение аммония С. 21-28). З-Ю’1 -110-5
Корма, растения МСХП РФ. МУ по ионометрическому определению аммиачного азота в кормах и растениях. (Утв. 14.04. 1995 г.) М.: ЦИНАО, 1995
Электрохимические методы анализа
831
Продолжение таблицы
№ Определяемый ион Объекты анализа Марка электрода (фирма изготовитель)* Методики КХА Пределы определения моль-л-1
20 Молоко Методика количественного ионометрического анализа молока на содержание ионов аммония, в т.ч. для выявления фальсификации гидроокисью аммония (аттестация проведена ГП ВНИИМ им. Менделеева), М., 1999
21 NO' Корма, растения МСХП РФ, ГУ химизации и защиты растений с госхимкомиссией МСХ РФ, ЦИНАО: МУ по определению нитритов в кормах и растениях ионометрическим методом (Утв. 10.01. 1996 г.). М.: ЦИНАО, 1996 5-101 - 5105
Мясные продукты Количественный химический анализ мясных продуктов. Методика выполнения измерений содержания нитрит-ионов в мясных продуктах, а также рассолах и посолочных смесях потенцио-метрическим методом с использованием нитрит-селективного электрода «ЭЛИТ -071»
22 no; Продукция растеневодства ЭМ-02.06.04 ЭМ-02.06.05 Методические указания по определению нитратов и нитритов в продукции растеневодства. М., 1989 г 5-10' 5105
Почвы ГОСТ 26951-86. Почвы. Определение нитратов ионометрическим методом. ГОСТ 27753.7-88. Грунты тепличные, методы определения нитратного азота
Комбикорма, комбикормовое сырье ГОСТ 13466.19-93. Комбикорма, комбикормовое сырье. Методы определения содержания нитратов и нитритов. ГОСТ Р 0465. Корма
Воды минеральные питьевые лечебные, лечебностоловые и природные столовые ГОСТ 23268.8-78. Воды минеральные питьевые лечебные, лечебно-столовые и природные столовые. Методы определения нитрат-ионов. Унифицированные методы анализа качества вод. Т. 1.4.1. Методы химического анализа вод. М.: СЭВ, 1987. МСХП РФ. МУ по определению хлоридов, нитратов и аммония в водах. (Утв. 14.04.1994 г.). М.: ЦИНАО, 1996. (Ионометрическое определение нитратов С. 10-14). РД 52.24.8-83
Вода питьевая Воды грунтовые и поливные МСХП РФ. МУ по ионометрическому определению катионно-анионного состава грунтовых и поливных вод. (Утв.14.04.1994 г.). М.: ЦИНАО, 1995. (Ионометрическое определение нитратов С. 48-52) Методические указания по определению pH, нитратов, хлоридов и фторидов на многоканальном поточном измерителе с использованием ионселективных электродов. М.: ЦИ-НАО, 2000
23 РЬ2+ Природа, и сточные воды ЭК-18.01.01 ЭК-18.01.02 ХС-РЬ-01 Природные и сточные воды. Черкассы, 1990 1-Ю’1-1-10^
832
Новый справочник химика и технолога
Продолжение таблицы
№ Определяемый ион Объекты анализа Марка электрода (фирма изготовитель)* Методики КХА Пределы определения моль-л1
24 SCN Вода ЭК-16.01.01 Унифицированные методы анализа качества вод. Т. 1.4. 1. Методы химического анализа вод. М.: СЭВ, 1987 1-Ю"1-1-Ю"5
25 S2” Вода ЭК-23.01.01 XC-S01 Унифицированные методы анализа качества вод. Т. 1.4. 1. Методы химического анализа вод. М.: СЭВ, 1987 1-Ю’1-1-Ю’5
26 so42-no; Вода Почва, грунтовые воды РД 52.24.107-91 Методические указания по ионометрическому определению иона сульфата в засоленных почвах и грунтовых водах. М.: ЦИНАО, 1996
27 ti+ XC-TI01 1 • ПГ6 -1,0
28 Сахар Вино, виноматериалы, коньяки Методика «Количественный химический анализ вина, виноматериалов, коньяков, продуктов переработки винограда на содержание массовой концентрации инвертного сахара и сухих виноградных вин на содержание массовой концентрации остаточного сахара потенциометрическим мето-дом». МУ 08 47/081 по реестру метрологической службы ГСЭН РФ
Примечание* ХС — халькогенидные стеклянные химические сенсоры; ** ГЭТМ — гомогенные электроды с твердыми мембранами;*** ЖМЭ — жидкостной мембранный электрод.
Приложение 2
Перечень аттестованных методик вольтамперометрического анализа
№ п/п Определяемый компонент Объект анализа Аттестация Диапазон определяемых концентраций, мг-кг-1 или мг-дм-3
Номер свидетельства Организация
1 Ag Уголь каменный, древесный, активирован, (потенциометр) 08-47/096 УНИИМ 150-1000
2 As Молоко и молочные продукты 08 47/051 УНИИМ, ГКСЭН 0,04-1,0
3 As Молоко и молочные продукты 08 47/094 УНИИМ, ГКСЭН 0,004
4 As Алкогольные и безалкогольные напитки, вода ГОСТ Р-51823-01 вниим
5 As Вода питьевая и минеральная, напитки алкогольные и безалко- гольные 08 47/058 УНИИМ, гксэ 0,01-2,0
6 As Напитки 08 47/093 УНИИМ, ГКСЭН 0,001
7 As Напитки алкогольные и безалкогольные 08 47/124 УНИИМ, ГКСЭН 0,002
8 As Вода природная, питьевая и сточная 08-47/125 УНИИМ, ГКСЭН 0,005
9 As Вода природная, питьевая и сточная Е 131/98 УНИИМ
10 As Вода природная и очищенная сточная ПНДФ 14.1:2.94-97 ГК РФ по ООС 0,01-0,1
11 As Вода природная, питьевая и очищенная сточная 35-01 вниимс 0,001-0,2 0,02-1,2
Электрохимические методы анализа
833
Продолжение таблицы
№ п/п Определяемый компонент Объект анализа Аттестация Диапазон определяемых концентраций, мг-кг-1 или мг-дм~3
Номер свидетельства Организация
12 As Поверхностные воды суши и очищенные сточные воды 371 РД 52.24.378-95 Госкомгидромет 0,01-01
13 As Рыба и рыбопродукты 08-47/061 УНИИМ, ГКСЭН . 0,03-10,0
14 As Рыба и продукты ее переработки 08-47/095 УНИИМ, ГКСЭН 0,03
15 As Яйцо. Мясо и продукты их переработки 08-47/062 УНИИМ 0,002-3,0
16 As Сахар, свекла 08-47/063 УНИИМ 0,001-2,0
17 As Комбикорма и продукты переработки 08-47/065 УНИИМ 0,007-7,0
18 As Пробы пищевых продуктов и продовольственного сырья Е 133/98 УНИИМ
19 As Hg Овощи, фрукты и продукты их переработки 08-47/097 УНИИМ 0,04-0,9 0,01-0,1
20 As, Zn Cd, Pb Cu Посуда и посудохозяйственные товары 08-47/085 УНИИМ 0,01-0,1 0,002-0,2 0,0001-0,2 0,001-0,2
21 Bi Вода природная и очищенная сточная ПНДФ 14.1:2.11-95 Мин ООС и ПР РФ 0,0001-0,1
22 Bi Вода питьевая, природная и очищенная сточная 2420/146-98; ПНДФ 14.1:2:4.152-99 То же
23 Bi Вода природная, питьевая, очищенная сточная 08-48/018 УНИИМ, ГКСЭН, ПНДФ 0,0001-0,1
24 Bi Вода природная, питьевая, очищенная сточная 08-48/103 УНИИМ, ГКСЭН 0,0001
25 Bi, Zn, Cd, Pb, Cu Почвы, осадки, донные отложения 2746/74-2001
26 Витамин Bi Овощи, фрукты, детское питание 08-47/053 УНИИМ, ГКСЭН 0,01-100
27 Витамин в. Корма, витаминные подкормки, премиксы 08-47/088 УНИИМ 10-3500
28 Витамин в2 Корма, витаминные подкормки, премиксы 08-47/054 УНИИМ, ГКСЭН 0,02-500
29 Витамин В2 Корма, витаминные подкормки, премиксы 08-47/089; ФР. 1.31.2001.00247 ФР. 1.31.2001.00248 50-3000
30 Витамины Bi в2 С Е кварцетин Биологически активные добавки (БАД) 08-47/115 УНИИМ 0,005-1,0 0,01-0,3 1,0-20,0 0,2-3,0 0,01-15,0
31 Витамин С Соки, напитки овощные и фруктовые, детское питание 08-47/040 УНИИМ, ГКСЭН ФР. 1.31.2001.00251 30-2500
32 Cd, Pb, Cu Атмосферный воздух Воздух рабочей зоны 28-99 вниимс 0,1-5 мкг-м-3 (в воздухе) 0,2-5000
834
Новый справочник химика и технолога
Продолжение таблицы
№ п/п Определяемый компонент Объект анализа Аттестация Диапазон определяемых концентраций, мг-кг”1 или мг-дм“3
Номер свидетельства Организация
33 Cd, Pb, Cu Природные воды Очищенные сточные воды 16-99 ВНИИМС 1,0-200 1,0-200
34 Cd, Pb, Cu Питьевые, природные воды и сточные воды 1-95 2-95, 3-95; ПНД Ф 14.1:2:4.63-96 УНИИМ
35 Cd, Pb, Cu Поверхностные воды суши и очищенные сточные воды 371 РД 52.24.371-95 Госкомгидромет 0,5-30
36 Cd Pb Cu Косметика 08 -47/059 УНИИМ, ГКСЭН 0,2-200 0,2-5 0,1-10
37 Cd Pb Cu Биологические объекты (кровь, моча) 08 -47/073; ФР. 1.39.2001.00216 УНИИМ 0,004-0,2 0,008-0,2 0,015-0,25
38 Cd, Pb Sb Se As Hg Детские игрушки 08 -47/083 УНИИМ 0,03-50,0 0,005-50,0 0,5-250 5,0-50,0 5,0-150
39 Cd Pb, Zn Cu Корма растительные для сельскохозяйственных животных 08 -47/075 УНИИМ 0,02-2,0 0,1-2000 2,0-1200
40 Co Напитки алкогольные 08 47/030 УНИИМ, ГКСЭН 0,2-3,0
41 Co Молоко и молочные продукты 08 47/032 УНИИМ, ГКСЭН 0,7-3,0
42 Cr Вода, природная, питьевая и сточная 08-47/122 УНИИМ, ГКСЭН 0,008
43 Cr Вода питьевая, природная, и сточная 4-95 ПНДФ 14.1:2:4.72-96 УНИИМ
44 Cr(VI) Поверхностные воды суши 82; РД 52.24. 428-95 Госкомгидромет 0,5-6,0
45 Fe Вода природная, питьевая и очищенная сточная 08 47/104 УНИИМ 0,005
46 Fe As Жировые продукты: масла растит, и продукты их перераб. — маргарин, майонез; масла живота, сливочное масло, жиры 08 47/077; ФР. 1.31.2001.00237 УНИИМ 0,9-70 0,04-1,10
47 Fe As Напитки алкогольные и безалкогольные 08 47/078 УНИИМ 5,0-50 0,01-1,0
48 Hg Вода природная питьевая Вода очищенная сточная 57-00 ВНИИМС 0,005-10 0,2-100
49 Hg Вода природная, питьевая, очищенная сточная 01.09.327/2002 ВНИИМС
50 Hg Вода природная питьевая Вода очищенная сточная 08-47/036 УНИИМ, ГКСЭН 0,000015-0,4
51 Hg Вода природная, питьевая, очищенная сточная 08 47/127 УНИИМ, ГКСЭН 0,0001
52 Hg Вода питьевая и природная неминерализованная (определение на анализаторе АВС-1 с эл. хим. датчиком «Модуль ЕМ-04») 2430/553-97/0553 0,25-500
Электрохимические методы анализа
835
Продолжение таблицы
№ п/п Определяемый компонент Объект анализа Аттестация Диапазон определяемых концентраций, мг-кг-1 или мг-дм“3
Номер свидетельства Организация
53 Hg Вода питьевая, природная и очищенная сточная 2420/143-98; ПНД Ф 14.1:2:4.150-99
54 Hg Напитки алкогольные и безалкогольные 08—47/037 УНИИМ, ГКСЭН . 0,0006-0,1
55 Hg Косметика 08-47/051 УНИИМ, ГКСЭН 0,002-1,0
56 Hg Рыба и рыбопродукты 08-47/060 УНИИМ, ГКСЭН 0,008-10,0
57 Hg Рыба и продукты ее переработки 08-47/126 УНИИМ, ГКСЭН 0,01
58 Hg Биологические объекты (кровь, моча) 08—47/074; ФР. 1.39.2001.00217 УНИИМ 0,001—0,45
59 Hg Сахар 08-47/076 УНИИМ 0,003-0,1
60 Hg Молоко и кисломолочные продукты 08-47/081 УНИИМ 0,0025-0,05
61 I Напитки безалкогольные, воды питьевые и минеральные 08-47/112; ФР. 1.31.2001.00214 УНИИМ Воды 0,08-0,20 — ПТ 0,005-1,3 —ИВ
Хлеб 0,2-2,3 —ИВ
Соль поваренная 1,0-60 —ИВ
62 I Молоко и молочные продукты 08-47/121 УНИИМ 0,05-10,0
63 I Био. объекты (моча) 08-47/128 УНИИМ 0,02-10,0
64 Mn Вода природная, питьевая, очищенная сточная 08 48/017; ПНД Ф 14.1:2:4.17-95 УНИИМ, ГКСЭН, ПНДФ 0.03-6.0
65 Mn Вода природная, питьевая, очищенная сточная 08-48/101 УНИИМ, ГКСЭН 0,005
66 Mn Вода природная, питьевая, очищенная сточная Е 132/98 УНИИМ
67 Mo Вода природная, питьевая, очищенная сточная Е 130/98 УНИИМ
68 Ni Напитки алкогольные 08-47/031 УНИИМ, ГКСЭН 0,3-2,0
69 Ni Вода питьевая, природная и очищенная сточная 2420/145-98; ПНДФ 14.1:2:4.150-99
70 Ni Вода питьевая, природная и очищенная сточная 6-95 ПНД Ф 14.1:2:4.73-96 УНИИМ
71 Pb Бензины автомобильные 08-47/114; ФР. 1.31.2001.00252 УНИИМ 0,01-0,03 Свыше 0,03-0,1 Свыше 0,1-20,0
72 Pb, Cu Cd, Zn Сахар 08-47/067 УНИИМ 0,1-10 0,02-0.5
73 Sb Вода природная, питьевая, очищенная сточная 08-48/021 УНИИМ, ГКСЭН, ПНДФ 0,0001-0,03
74 Sb Вода природная, питьевая, очищенная сточная 08 48/102 УНИИМ ГКСЭН 0,0001
75 Se Вода питьевая, минеральная 08-47/082 УНИИМ 0,003-5,0
836
Новый справочник химика и технолога
Продолжение таблицы
№ п/п Определяемый компонент Объект анализа Аттестация Диапазон определяемых концентраций, мг-кг-1 или мг-дм-3
Номер свидетельства Организация
76 Sn Методика выполнения измерений массовой концентрации ионов олова в галлии по ГОСТ 13637.5 -93.
77 Sn Pb Соки, напитки овощные и фруктовые, детское питание 08-47/039 УНИИМ, ГКСЭН 0,5-500 0,1-5
78 Sn Pb Продукты консервированные 08-47/050 УНИИМ, ГКСЭН 0,2-400 0,1-5,0
79 Sn, Pb Продукты консервированные, расфасованные в жестяную тару Е 133/98 УНИИМ
80 U Биологические объекты (моча) 08-47/105; ФР. 1.39.2001.00215 УНИИМ 0,0001-0,005
81 Zn Вода природная, питьевая, очищенная сточная 14-00 вниимс 1,0-10000 вкл
82 Zn Поверхностные воды суши 373; РД 52.24. 373-95 Госкомгидромет 4,0-50
83 Zn, Cd, Pb, Cu Вода природная, питьевая, очищенная сточная 08 47/008 УНИИМ, ГКСЭН 0,0006-1,0
85 Zn, Cd, Pb, Cu Вода природная, питьевая, технологическая, очищенная сточная 08-47/120 УНИИМ, ГКСЭН 0,0002-1,0
86 Zn, Cd, Pb, Cu Питьевая, природная и очищенная сточная вода 2420/144-01; ПНДФ 14.1:2:4.149-99
87 Zn, Cd, Pb, Cu Продукты пищевые и продовольственное сырье ГОСТ Р 51301-99
88 Zn, Cd, Pb, Cu Пищевые продукты и продовольственное сырье 203/001808-2001
89 Zn, Cd, Pb, Cu Пищевые продукты и продовольственное сырье Е 135/98 УНИИМ
90 Zn Cd Pb Cu Молоко и молочные продукты 08-47/027 УНИИМ, ГКСЭН, гост 0,2-50 0,005-1,5 0,02-2,5 0,1-15
91 Zn Cd Pb Cu Жиры, маргарины, масла 08-47/123 УНИИМ, ГКСЭН 0,1 0,003 0,01 0,05
92 Zn Cd Pb Cu Крупа, зерно, мука, кофе, какао, мясо, рыба и продукты 08-47/042 УНИИМ, ГКСЭН, гост 1,0-100 0,05-50 0,04-10 0,05-30
93 Zn Cd Pb Cu Крупы, зерно, какао, мука, кофе, чай, конфеты, мясо, рыба, овощи, фрукты, другие продукты 08-47/092 УНИИМ, ГКСЭН 0,5 0,0015 0,01 0,05
Электрохимические методы анализа
837
Продолжение таблицы
№ п/п Определяемый компонент Объект анализа Аттестация Диапазон определяемых концентраций, мг-кг-1 или мг-дм’3
Номер свидетельства Организация
94 Zn Cd Pb Cu Se As Fe Биологически активные добавки (БАД) 08—17/118 УНИИМ 0,5-10,0 0,001-0,5 0,01-1,0 0,1-200 0,1-50 0,05-1,0 20-2000
95 Zn Cd Pb Cu Напитки алкогольные и безалкогольные 08—17/043 УНИИМ, ГКСЭН, ГОСТ 0,01-20 0,001-0,02 0,004-0,2 0,02-2,0
96 Zn Cd Pb Cu Напитки алкогольные и безалкогольные 08—17/071 УНИИМ, ГКСЭН 0,1 0,003 0,03 0,05
97 Zn Cd, Pb Cu Hg Средства гигиены полости рта (зубные пасты, эликсиры и т. д.) 08-47/084; ФР. 1.39.2001.00253 УНИИМ 0,03-1,0 0,01-0,5 0,1-2,0 0,1-2,0
98 Zn, Cu, Cd Pb Почвы 08—17/056 УНИИМ, ГКСЭН 5.0—100 0,1-50
99 Zn, Cd, Pb Cu Воздух рабочей зоны и пром-выбросы № 2746/73-2001
100 Zn, Cu, Cd Pb Сахарная свекла 08-47/064 УНИИМ 3,0-30 0,1-6,0
101 Анилин Вода природная, питьевая, очищенная сточная 08-17/066 УНИИМ 0,00005-5,0
102 Левомицетин Молоко и молочные продукты 08—17/086 УНИИМ 0,003-0,030
103 Левомицетин Яйцо, мясо и продукты их переработки 08—17/106; ФР. 1.31.2001.00252 УНИИМ 0,006-0,10
104 Мочевая кислота Сыворотка крови 08—17/028 УНИИМ 0,001
105 Сахар Напитки алкогольные (рН-метрия) 08-47/080 УНИИМ 0,2-20 г/100г
106 Тетрациклин Левомицетин Таблетки, порошки, капсулы 08—17/111; ФР. 1.39.2001.00249 УНИИМ 40-550 0,24-0,55
107 Фенол Вода природная, питьевая, очищенная сточная 08—18/020 УНИИМ. ГКСЭН, ПНДФ 0,0001-0,4
838
Новый справочник химика и технолога
Приложение 3
Типичные вольтам перо граммы (графики, иллюстрирующие методики определения неорганических и органических компонентов)
As
Рис. А. Пики анодного тока растворения мышьяка при его определении на уровне «-10 мкг-л-1 в водных объектах (см. п.п. 4—12 Приложения 2)
Рис. Б. Пики анодного тока растворения кадмия, свинца и меди при их определении на уровне единиц мкг-л-1 в различных объектах (см. п.п. 32-37 Приложения 2)
Сг
Рис. В. Прямая вольтамперограмма хрома в растворе 0,15М H2SO4 + 5-10-6 М дифенилкарбазида + 1 мкг-л-1 Cr(VI) [34]
Рис. Г. Пики анодного тока растворения висмута с поверхности РПЭ при анализе вод (см. п.п. 23-24 Приложение 2)
Электрохимические методы анализа
839
Рис. Д. Пики анодного тока растворения ртути и меди при их определении на уровне единиц мкг-л-1 в водных объектах (см. п.п. 47-54 Приложения 2)
Рис. Ж. Пики анодного тока растворения селена при его определении на уровне десятков мкг-л-1 в водных объектах (см. п.75 Приложения 2)
Hg, Си
Рис. 3. Пики анодного тока растворения ртути и меди с поверхности золотого микроэлектрода в растворе 0,1М KSCN + 0,01М НСЮ4 + (1... 10) 10 8 Hg(II) [35]
Рис. Е. Пики катодного тока растворения осадка иода с поверхности РПЭ при его концентрации в растворе на уровне единиц мкг-л1 (см. п.п. 61-63 Приложения 2)
Мп
Рис. И. Пики анодного тока растворения марганца при его определении на уровне десятков мкг-л-1 в водных объектах (см. п.п. 64-66 Приложение 2)
840
Новый справочник химика и технолога
Sb, Bi
-1.00 -0.80 -0,60 -0.40 -0.20 -0.00
Рис. К. Определение в морской воде ряда цветных металлов, показано влияние концентрации [NaCl] = (а) 0 М, (б) 0,5М, (в) 1,0М, (г) 1,5М, (д) 2,ОМ и (е) 2,5М [36]
Рис. Л. Совмещенный график, полученный наложением вольтамперограмм свинца в слабокислом фоновом растворе и олова в кислом растворе при их последовательном определении (см. п.п.77-79 Приложения 2)
W
Рис. М. Пики катодного тока десорбции урана с поверхности СРКЭ при его концентрации в растворе 2.510 4 М хлоранилиновой кислоты на уровне единиц мкг-л \ метод ИАдсВА [37]
Рис. Н. Дифференциально-импульсная вольтамперограмма вольфрама (VI) в растворе 0,9М H2SO4 + 0,066 М NH4CNS+ 0,0265 М антипирина + 5 мкг-л“! W(VI) без деаэрации [34]
Рис. О. Дифференциально-импульсные вольтамперограммы: а) витамина D2; б) витамина А; в) смеси витаминов А и D2 1:5 в растворе фонового электролита 0,1М Bu4NPF6 [38]
Рис. П. Дифференциально-импульсная вольтамперограмма мочевой кислоты в диапазоне концентраций 8-10”5- 110 3 М на стеклоуглеродном электроде на фоне 0,05 М фталевокислого калия
Раздел 7
МАСС-СПЕКТРОМЕТРИЧЕСКИЙ МЕТОД АНАЛИЗА
Автор-составитель: доц., к.х.н. В.Ф. Теплых
Научный редактор: к.х.н. В.И. Мосичев
Масс-спектрометрический (МС) анализ используется в различных областях науки и техники. С помощью МС-анализа были открыты изотопы и впоследствии установлен изотопный состав всех элементов периодической системы; измерены с высокой точностью массы атомов, молекул и обнаружены их дефекты; выявлена тождественность изотопного состава элементов в земной коре и в космических объектах; определены периоды полураспада некоторых радиоактивных изотопов; по накоплению в природных материалах изотопов свинца, стронция и аргона измерен абсолютный возраст геологических образований.
Современная масс-спектрометрия является одним из наиболее тонких и чувствительных методов анализа вещества и характеризуется самыми низкими пределами обнаружения следов элементов при их одновременной регистрации [1-12] (табл. 7.1). МС-методом определяют элементный и молекулярный состав различных природных и синтезированных веществ [2, 8,13], исследуют кинетику химических реакций и измеряют энергию связи между атомами и между молекулами [14-18], идентифицируют химические соединения и расшифровывают структуру молекул [4, 5, 19, 20], измеряют наличие микропримесей на уровне менее 10“8% в полупроводниковых материалах и металлах [21-23]. Неоценима роль элементной и молекулярной МС-анализ в современной медицине [1, 5, 24], микробиологии [3, 10, 11] и экологии [4, 7, 12, 25, 26]. По темпам развития, по технической оснащенности и по использованию в различных областях науки МС-метод занимает в последнее десятилетие одно из первых мест [1]. Это стало возможным благодаря широкому применению искровой и лазерной масс-спектрометрии [10, 11, 25, 27], сочетанию МС-анализ с газовой хроматографией в режиме on-line [6, 26, 28] и особенно созданию масс-спектрометров, в которых для генерации ионов использованы тлеющий разряд и индуктивно-связанная плазма [2, 7-9, 21, 29-32]. С появлением приборов, работающих на принципе ионно-циклотронного резонанса с Фурье-преобразованием, стало вполне реальным получать разрешение по массе 290000 и более, что дает возможность легко разделять сложные ионы с одинаковыми массовыми числами, не прибегая к стандартным образцам [16,22,33-36].
Таблица 7.1
Пределы обнаружения изотопов некоторых элементов при анализе воды МС-методом, нг • мл-1 [2]
Элемент Масса, а.е.м. Предел обнаружения Элемент Масса, а.е.м. Предел обнаружения
Li 6 0,05 Мо 95 0,01
7 0,005 96 0,01
В 10 0,2 97 0,02
И 0,05 98 0,005
А1 27 0,005 100 0,02
V 51 0,1 Ва 135 0,02
Сг 52 0,005 137 0,01
53 0,05 138 0,002
Мп 55 0,002 и 235 0,03
Со 59 0,002 238 0,005
Си 63 0,005 РЬ 206 0,01
65 0,01 207 0,01
Zn 66 0,02 208 0,005
Rb 85 0,01 Cd НО 0,01
112 0,005
114 0,005
Разрешающая способность масс-спектрометра характеризует возможность раздельной регистрации ионов, близких по массам, и определяется величиной R* = Af/AA/, где АА/ — минимальная разность масс однозарядных ионов, регистрируемых прибором раздельно, вблизи массы М. Величина R* рассчитывается (рис. 7.1) по формуле
/-(Af1+Af2) k~(a + b)(M.-M2)’
(7-1)
где М\ и М2 — массы соседних ионов в а.е.м.; I — расстояние между пиками А4 и М2; а и b — ширины пиков на вы
842
Новый справочник химика и технолога
соте kh {к определяется долей высоты пика: при уровне 10% к = 0,1; при уровне 50% к = 0,5); hj и h2 — высоты пиков (интенсивность линий).
Рис. 7.1. Определение разрешающей способности масс-спектрометра
В соответствии с величиной R10% масс-спектрометры условно подразделяют по разрешающей способности:
Rio% <100 — низкая, 100 < R10o/o < 1000 — средняя и Rio% > 1000 — высокая.
Во многих типах масс-спектрометра разрешение ухудшается по мере увеличения массы ионов. Поэтому показателем качества прибора служит «единичное разрешение»— наибольшая величина массы иона, при которой достигается разделение двух пиков одинаковой высоты. Если этому критерию, например, удовлетворяют массы от 1000 до 1001, то говорят, что такой прибор имеет разрешение 1000. «Единичное разрешение» магнитного масс-спектрометра может достигать 6000, а спектрометра с двойной фокусировкой — 80000 и более.
7.1. Источники ионов
В зависимости от свойств анализируемых веществ и характера решаемой задачи ионы атомов и молекул могут быть получены несколькими способами, в том числе: электронным ударом, бомбардировкой поверхности пробы пучком электронов, ионов или нейтральных атомов, испарением с накаленных металлических поверхностей, фотоионизацией, в газовом разряде, в вакуумном высоковольтном разряде, плазменными методами. Многие из этих способов оформлены в виде конструкций, дающих возможность ионизировать вещества в газовой, жидкой и твердой фазах, обеспечивать более или менее одинаковую ионизацию всех компонентов смеси или селективное усиление ионизации веществ определенной структуры, получать положительные и отрицательные ионы. Разнообразие способов ионизации является одной из сильных сторон масс-спектрометрии, что дает возможность в зависимости от задач исследования и природы веществ менять вид воздействия на молекулы.
От источника ионов в значительной степени зависят такие важные характеристики масс-спектрометра, как чувствительность, пределы обнаружения и определения, правильность анализа и селективность. Чувствительность обусловлена эффективностью ионизации, «свето
силой» прибора и определяется как отношение количества ионов данного типа, достигнувших детектора, к массе молекул этого же типа, помещенных в источник ионов. Наименьшая масса вещества в пробе, при которой наблюдается над фоном аналитический сигнал на выходе прибора, принимается как предел обнаружения. Эта характеристика приводится при оценке возможностей различных типов спектрометров, а также при сравнении методов анализа. Минимальное содержание анализируемого компонента в пробе, которое может быть измерено с заданной погрешностью, характеризует предел определения для данного прибора. Предел определения зависит не только от предела обнаружения, но и от стабильности основных функциональных узлов масс-спектрометра. Такая характеристика, как селективность, зависит от используемого способа ионизации, поскольку эффективность ионизации одних молекул может быть значительно больше, чем других. Селективность определяет величину коэффициента относительной чувствительности (КОЧ).
При проведении элементного анализа различных веществ важнейшим требованием является высокая правильность полученных результатов, которая зависит от дискриминации конкретного метода или прибора и от стабильности КОЧ. Правильность определения оценивается отклонением результата измерений от надежно установленного действительного содержания вещества в пробе и проверяется анализом стандартных образцов, состав которых измерен различными методами.
7.1.1. Источники с электронным ударом (ЭУ)
Одним из первых и наиболее распространенным вплоть до настоящего времени способов ионизации является ионизация электронным ударом (ЭУ) [37]. Источник ионов с ЭУ обычно имеет камеру ионизации, в которую вводят поток паров анализируемого вещества (рис. 7.2.). Перпендикулярно этому потоку камеру пересекает пучок ускоренных до заданной энергии электронов, эмитируемых нагретым рениевым или вольфрамовым катодом. Этот пучок электронов бомбардирует молекулы анализируемого вещества. Если энергия электронов больше потенциала ионизации молекулы, то с определенной вероятностью неупругие соударения приводят к образованию ионов в результате «выбивания» из молекулы одного из электронов: М + е -> М+ + 2е~. Ионизация ЭУ имеет много достоинств: это, прежде всего, простота устройства источника ионов, хорошая воспроизводимость масс-спектров и высокая чувствительность. Абсолютная эффективность ионизации составляет 0,01-1% количества молекул, введенных в источник, а тепловой разброс ионов по энергиям находится в пределах 3-5 эВ, что позволяет достичь высокого разрешения (8000 и более) без применения масс-анализаторов с двойной фокусировкой. Вероятность ионизации ЭУ зависит от потенциала ионизации атомов и молекул (табл. 7.2; 7.3) и сечения ионизации (табл. 7.4).
Масс-спектрометрический метод анализа
843
Рис. 7.2. Схема ионного источника типа Нира:
1 — трубка напуска анализируемого газа;
2 — ионизационная камера; 3 — катоды;
4 — фокусирующий магнит; 5 —ускоряющийэлектрод;
6 — отклоняющие пластины; 7— ионный пучок;
8 — вытягивающий электрод; 9 — электронный пучок;
10 — диафрагма напуска
Среди всех элементов наибольшими потенциалами ионизации обладают благородные газы Не (24,56 эВ), Ne (21,559 эВ) и Аг (15,755 эВ), а наименьшими — такие элементы, как Cs (3,893 эВ), К (4,339 эВ), Ва (5,20 эВ) и Na (5,138 эВ).
Сечение ионизации зависит от энергии бомбардирующих электронов. Начиная от порога ионизации, сечение быстро растет и достигает максимальной величины в области энергий Ее = 50-150 эВ. Из благородных газов наименьшее сечение ионизации имеет Не (0,102-10-16 см2) и наибольшее — Хе (6,42-1016 см2), а из других элементов большие сечения имеют металлы Cs (11,64-Ю-16 см2), Ва (11,941016 см2), La (11,96-Ю"16 см2), Се (И,70-Ю’16 см2). Из этих примеров видно, что при одинаковой концентрации в парах такие элементы, как Cs, Ва, La и Се, будут образовывать большее количество ионов, и их пики на масс-спектрограммах будут выше. Следовательно, для выполнения количественного анализа необходимо проводить калибровку прибора. Кроме того, сравнение масс-спектров, полученных разными авторами, возможно, только если использована одна и та же энергия ионизирующих электронов.
Таблица 7.2
Потенциалы ионизации элементов, эВ [48]
Элемент Состояние ионизации
I II III IV V VI VII VIII
Н 13,595
Не 24,580 54,493
Li 5,390 75,619 122,420
Be 9,320 18,206 153,850 217,66
В 8,296 25,149 37,920 259,298 340,13
С 11,264 24,376 47,854 64,476 391,986 489,84
N 14,54 29,605 47,426 77,450 97,863 551,925 666,8
О 13,614 35,164 54,934 77,394 113,873 138,080 739,133 871,1
F 17,42 34,98 62,646 87,23 114,214 157,117 185,139 953,60
Ne 21,559 41,07 63,43 97,16 126,4 157,91 207,3 239,0
Na 5,138 47,29 71,65 98,88 138,60 172,36 208,444 264,155
Mg 7,644 15,03 80,12 109,29 141,23 186,86 225,31 265,957
Al 5,984 18,823 28,44 119,96 153,77 190,42 241,93 285,13
Si 8,149 16,34 33,46 45,13 166,73 205,11 246,41 303,87
P 10,55 19,65 30,156 51,354 65,007 220,414 263,31 309,26
S 10,357 23,4 34,7 47,29 72,5 88,029 280,99 328,80
Cl 13,01 23,80 39,90 53,3 67,80 96,75 114,27 348,3
Ar 15,755 27,62 40,90 59,8 74,97 91,41 124,20 143,46
К 4,339 31,81 46,1 61,3 82,5 99,88 118,24 154,6
Ca 6,111 11,87 51,21 67,7 84,39 109 128 148,1
Sc 6,56 12,80 24,75 74,3 92,0 111,1 138 159,3
Ti 6,83 13,57 27,47 43,24 99,8 120 140,8 171
V 6,74 14,65 29,31 48 65,2 128,9 151 173,7
Cr 6,764 16,49 30,95 49,6 73,2 90,60 161,2 185
Mn 7,432 15,64 33,69 52 76,1 98 119,24 196,4
Fe 7,896 16,18 30,63 57,1 78 102 128 151,1
844
Новый справочник химика и технолога
Продолжение таблицы 7.2
Элемент Состояние ионизации
I II III IV V VI VII VIII
Со 7,86 17,05 33,5 53 83,5 106 132 161
Ni 7,633 18,15 35,2 56,0 78 110 136 166
Си 7,723 20,29 37,0 58,9 82 106 140 169
Zn 9,391 17,96 39,7 62 86 112 142 177
Ga 6,00 20,53 30,7 64,1 90 118 144 174
Ge 7,88 15,93 34,23 4,7 93,4 113 148 177
As 9,85 20,2 28,1 50,1 62,8 127,5 150 182
Se 9,750 21,4 32,0 42,9 73,1 81,8 155 187
Br 11,84 21,6 35,9 50 60 87 104 193
Kr 13,996 24,58 37,0 52 66 80 110 127
Rb 4,176 27,4 39,3 52 71 86 102 134
Sr 5,692 11,03 43,0 57 72 93 109 126
Y 6,6 12,3 20,5 62 76,9 94 117 135
Zr 6,95 14,03 24,11 33,99 83 98,8 118 143
Nb 6,77 13,5 28,1 38,3 49,5 103 125 145
Mo 7,18 15,2 27,0 40,5 56 72 125 153
Tc 7,45 15 29 43 59 76 94 162
Ru 7,5 16,4 28,6 46,5 63 81 100 119
Rh 7,7 18,1 31,0 45,6 67 85 105 126
Pd 8,33 19,9 33,4 48,8 66 90 110 132
Ag 7,574 122,0 39,7 52 70 89 116 139
Cd 8,991 16,92 38,2 55 73 94 115 146
In 5,785 18,87 27,8 58 77 98 120 144
Sn 7,332 14,63 30,6 39,6 81,1 103 126 150
Sb 8,64 16,7 24,8 44,0 55,8 107,6 132 157
Те 9,01 18,8 30,6 37,9 60,3 72,3 137,1 164
I 10,44 19,5 31,4 41,7 52 77 90 169,9
Xe 12,127 21,2 32,1 45,5 57 68 96 110
Cs 3,893 23,5 35 45,5 62 74 86 117
Ba 5,210 10,01 35,7 48,8 62 80 93 106
La 5,61 11,43 19,2 52 66 80 100 114
Ce 6,9 12,3 20 33,5 70 85 100 122
Pr 5,76 10,55 20,96 89 106 122
Nd 6,3 10,72 20,51 110 128
Pm 5,554 10,90 22,0 135
Sm 5,7 24,0
Eu 5,67 11,3 24,56
Gd 6,16 12,1 21,3
Tb 6,7 11,52 21,02
Dy 6,8 11,67 21,83
Ho 6,018 11,80 22,2
Er 6,101 11,93 22,4
Tm 6,184 12,05 23,9
Yb 6,26 12,11 25,61
Lu 5,425 13,9 23,7
Hf 6,65 14,9 21 31
Ta 7,89 16,2 22,3 33,1 43
W 7,98 17,7 24,1 35,4 48 61
Re 7,87 16,6 26,0 37,7 51 64 79
Os 8,7 17 25 40 54 68 89 99
Ir 9,2 17 27 39 57 72 88 104
Масс-спектрометрический метод анализа
845
Продолжение таблицы 7.2
Элемент Состояние ионизации
I II III IV V VI VII VIII
Pt 8,96 18,56 28,5 41,1 55 75 92 109
Au 9,223 20,1 30,5 43,5 58 73 96 114
нё 10,434 18,761 34,21 46,0 61 77 94 120
TI 6,106 20,43 29,8 50,7 64 81 98 116
Pb 7,415 15,03 32,0 42,3 69,73 84 103 122
Bi 8,3 16,7 25,4 45,3 56,0 94,42 107 127
Po 8,4 18,2 28 38 61 73 112 132
At 9,5 18,2 30 41 51 78 91 138
Rn 10,745 19,9 29,8 43,8 55 67 97 111
Fr 4,0 21,5 32 43 59 71 84 117
Ra 5,277 10,15 34,3 46,4 58,5 76 89 103
Ac 5,17 11,5 18,9 49 62 76 95 109
Th 6,08 11,5 18,7 33,6 65 80 94 115
Pa 5,89 11,3 20,5 36,4 84 100 115
U 6,05 11,6 20,5 36,89 104 121
Таблица 7.3
Вертикальные потенциалы ионизации (ПИ) нейтральных веществ [96]
Вещество (R) ПИ, эв
1. Неорганические вещества
H 13,6
D 13,6
н2 15,42
d2 15,46
He 24,6
Ne 21,8
Ar 15,75
Kr 14,0
Xe 12,13
f2 15,7
Cl2 11,48
Br2 10,53
h 9,28
HF 15,77
HC1 12,74
HBr 11,62
HI 10,38
2. Углеводороды
C 11,3
’ CH3- 9,82
CH4 12,70
cd4 12,88
C2H2 11,40
c2D2 11,42
СД 10,51
c2H6 11,52
Вещество (R) ПИ, эв
C3H4
Аллен 10,2
Пропин 10,38
Циклопропен 9,95
с3н6
Пропен 9,73
Циклопропан 10,1
с3н8 11,14
С4Н2(1,3- Бутадиин) 10,2
с4нб
1,2-Бутадиен 9,57
1,3-Бутадиен 9,08
1-Бутин 10,18
2-Бутин 9,9
с4н8
1 -Бутен 9,58
z/wc-2-Бутен 9,13
транс-2-Бутен 9,13
Изобутен 9,23
Циклобутан 10,58
С4Н10
Изобутан 10,57
С5Н6 (Циклопентадиен) 8,9
Вещество (R) ПИ, эв
с5н8
1,2-Пентадиен 9,42
1,3-Пентадиен 8,68
2,3-Пентадиен 9,26
1,4-Пентадиен 9,58
Циклопентен 9,01
с5н10
1 -Пентен 9,50
z/wc-2-Пентен 9,11
транс-2-Петен 9,06
2-Метил-1 -бутен 9,12
2-Метил-2-бутен 8,67
Циклопентан 10,9
с5н12
Пентан 10,35
Изопентан 10,32
Неопентан 10,35
С6Н4 (Дигидробензол) 9,75
С6Н6 (Бензол) 9,25
C6D6 (Бензол-П6) 9,25
С6Ню (Циклогексен) 8,94
с6н12
1-Гексен 9,45
2,3-Диметил-2-бутен 8,30
Циклогексан 9,88
Вещество (R) ПИ, эв
с6н14
Гексан 10,18
Изогексан 10,12
З-Этилбутан 10,08
с7н8
Толуол 8,82
Циклогептатриен 8,65
С7Н10 (Норборнен) 9,0
С7Н]4 (Метилциклогексан) 9,85
С7Н16 (Гептан) 9,90
С8Н6 (Фенила-цетилен) 8,82
с8н8
Стирол 8,47
Циклооктатетраен 7,99
Кубан 8,74
с8н10
Этилбензол 8,76
о-Ксилол 8,56
jW-Ксилол 8,56
и-Ксилол 8,44
C8Hi8 (Изооктан) 9,86
с9н12 (Триметилбензол)
1,2,3- 8,48
1,2,4- 8,40
1,3,5- 8,39
846
Новый справочник химика и технолога
Вещество (R) ПИ, эв
СюН8
Нафталин 8,12
Азулен 7,43
СюН18 (Декалины)
цис- 9,61
транс- 9,61
Ci2H10 (Бифенил) 8,27
Ci4H)o
Дифенил ацетилен 8,85
Антрацен 7,55
Фенантрен 8,10
3. Соединения с гетероатомами
n2 15,56
NH3 10,15
NO 9,25
n2o 12,89
no2 9,78
ch3nh2 8,97
ch2n2 (Диазометан) 8,99
ch2cn 12,21
c2h5nh2 8,86
Вещество (R) ПИ, эв
(CH3)2NH 8,24
ch2chcn 10,91
c2h2cn 11,84
c2h6n2 (Азометан) 8,65
(CH3)3N 7,82
C4H5N (Пиррол) 8,20
C5H5N (Пиридин) 9,23
c6h5nh2 (Анилин) 7,69
C6H5CN (Бензонитрил) 9,71
C6H5CH2NH2 7,56
(C6H5)2NH 7,25
(C6h5)3n 6,86
O2 12,06
O3 11,7
H2o 12,61
d2o 12,64
co 14,01
co2 13,79
CH2O 10,90
CH3OH 10,85
HCOOH 11,05
CH3CHO 10,21
CH3OCH3 9,96
Вещество (R) ПИ, эв
CH3CH2OH 10,48
CH3COOH 10,35
CH3COCH3 9,69
CH3COOCH3 10,27
CH3CH2CHO 9,98
CH2CHCH2OH 9,67
C4H4O (Фуран) 8,89
(C2HS)2O 9,53
C4H8O (Тетрагидрофуран) 9,42
C4H8O2 (и-Диоксан) 9,13
C6H5OH 8,50
C6H4O2 («-Бензохинон) 9,67
C6HSOCH3 8,20
C6H5CHO 9,51
C6H5COCH3 9,27
(C6H5)2O 8,82
(C6H5)2CO 9,46
hconh2 10,25
ch3conh2 9,77
c6h5conh2 9,4
c6h5nhcoch3 8,39
ch3no2 11,08
Продолжение таблицы 7.3
Вещество (R) ПИ, эв
C6H5NO2 9,92
c6h6n2o2 (и-Нитроанилин) 8,85
c6h5no3 («-Нитрофенол) 9,52
CH3C1 11,26
CH3Br 10,53
CH3I 9,54
c6h5f 9,19
C6H5C1 9,07
C6H5Br 8,98
C6H5I 8,62
(CH3)4Si 9,5
H2s 10,42
cs2 10,08
so2 12,34
CH3SH 9,44
(CH3)2s 8,68
C4H4S (Тиофен) 8,86
C6H5SH 8,32
C6H5SCH3 8,9
При ионизации ЭУ образовавшийся ион находится в возбужденном состоянии и может распадаться на различные фрагменты в зависимости от величины переданной электроном энергии, условий локализации заряда и наличия «слабых мест» в структуре молекулы [13] (рис. 7.3.). Из-за значительной фрагментации масс-спектры некоторых соединений не содержат пика молекулярного иона, что является одним из недостатков ионизации ЭУ. Уменьшение энергии ионизирующих электронов приводит к увеличению относительной интенсивности пиков молекулярных ионов и к уменьшению, а иногда и полному исчезновению пиков осколочных ионов. При этих условиях получаются малолинейчатые спектры, которые используют для выявления пиков молекулярных ионов. Однако при уменьшении энергии бомбардирующих электронов резко снижается эффективность ионизации, и поэтому на практике приходится искать компромисс между требованием уменьшения пиков осколочных ионов и необходимостью сохранения чувствительности прибора.
Таблица 7.4
Максимальные сечения ионизации (Q) химических элементов, 10 16 см2 [48]
Элемент Q Элемент Q Элемент Q Элемент Q
H 0,234 Fe 4,71 Sb 6,94 Os 7,11
He 0,102 Co 4,28 Те 7,31 Ir 7,10
Li 3,50 Ni 3,99 I 6,92 Pt 7,17
Продолжение таблицы 7.4
Элемент Q Элемент Q Элемент Q Элемент Q
Be 1,92 Cu 4,06 Xe 6,42 Au 6,46
В 2,11 Zn 3,17 Cs 11,46 Hg 5,34
C 1,73 Ga 5,12 Ba 11,94 Tl 7,11
N 1,38 Ge 5,20 La 11,96 Pb 7,34
О 1,31 As 4,87 Ce 11,70 Bi 8,40
F 1,02 Se 5,16 Pr 10,91 Po 8,39
Ne 0,820 Br 4,60 Nd 10,68 At 8,36
Na 4,27 Kr 4,12 Pm 10,43 Rn 7,94
Mg 3,44 Rb 8,93 Sm 10,23 Fr 12,04
Al 5,43 Sr 8,88 Eu 9,86 Ra 12,15
Si 4,94 Y 8,64 Gd 9,53 Ac 12,90
P 4,20 Zr 7,90 Tb 9,50 Th 12,91
S 4,10 Nb 8,29 Dy 8,82 Pa 12,01
Cl 3,38 Mo 7,39 Ho 8,62 U 11,62
Ar 2,83 Tc 6,31 Er 8,39 Np 11,24
К 7,67 Ru 7,20 Tm 8,43 Pu 10,00
Ca 7,05 Rh 6,61 Yb 7,80 Am 9,72
Sc 6,56 Pd 6,55 Lu 8,43 Cm 10,19
Ti 5,97 Ag 5,44 Hf 7,76 Bk 9,88
V 5,58 Cd 4,57 Ta 7,41 Cf 8,47
Cr 5,45 In 6,74 W 7,07
Mn 4,56 Sn 7,01 Re 7,25
Масс-спектрометрический метод анализа
847
Рис. 7.3. Масс-спектр этилена (* — линия, обусловленная вкладом изотопа 13С)
Масс-спектрометр с ионизацией ЭУ особенно подходит для анализа органических соединений в органическом синтезе, нефтехимии, медицине, биологии, а также при анализе загрязнений окружающей среды [13, 40], т.к. дает возможность получить общую характеристику неизвестного соединения по масс-спектру, содержащему пики как молекулярных, так и осколочных ионов. Следует отметить, что основной материал по масс-спектрометрии органических соединений разных классов, накопленный и представленный в каталогах, — это, главным образом, масс-спектры, полученные при анализе ЭУ. Поэтому автоматические системы обработки результатов масс-спектрометрических измерений, использующие библиотечный поиск, ориентируются именно на эти данные [81-87].
7.1.2. Источники с поверхностной ионизацией или термоионные источники (ТИ)
В источнике ионов с поверхностной ионизацией или в термоионном источнике анализируемая проба испаряется с раскаленной поверхности [37]. При этом часть вещества испаряется в виде положительных ионов. Термоионная эмиссия описывается известным соотношением Саха-Лангмюра
п+
— = ехр п
-e(W - <р) кТ
(7-2)
где п+/п — степень ионизации (отношение числа заряженных и нейтральных частиц, испаряемых с нагретой поверхности электрода); ф — потенциал ионизации испаряемого атома; Т — абсолютная температура поверхности электрода; W — работа выхода электрона с поверхности электрода при данной температуре Т; е — заряд электрона; к — постоянная Больцмана.
Из приведенного соотношения видно, что наибольшую степень ионизации при испарении с нагретой поверхности имеют элементы с низким потенциалом ионизации, такие, как Na и К, для которых W - ф » к 7'. Степень ионизации также относительно высока для многих элементов I—III и VI-VIII групп и некоторых легкоионизируемых органических соединений (табл. 7.3).
Исследуемые образцы в виде капли раствора обычно помещают на ленточку из рения, вольфрама или тантала и высушивают инфракрасными лучами. При анализе через ленточку пропускают электрический ток, поднимая ее температуру до необходимого значения (500-2500 °C). Некоторые добавки (бораты, силикаты и др.) благоприятствуют образованию положительных ионов. Иногда для получения ионов пробу испаряют с одной ленточки, а ионизацию производят на поверхности другой, более раскаленной ленточки. Такой вариант предпочтительнее, так как ток ионов при повышенных температурах возрастает на несколько порядков. ТИ-источники с двумя горячими ленточками часто используются для анализа трансурановых элементов ввиду очень высокой эффективности ионизации и, следовательно, малого размера пробы, необходимой для анализа. Одним из значительных преимуществ ТИ-источника является отсутствие в нем электронного луча, что приводит к резкому уменьшению числа и интенсивности фоновых пиков в масс-спектрах.
7.1.3. Химическая ионизация (ХИ)
Для снижения энергии возбуждения ионизируемых молекул применяют методы «мягкой» ионизации. Одним из важнейших методов низкоэнергетической ионизации является химическая ионизация [38]. ХИ обычно осуществляется путем ионно-молекулярной реакции между нейтральными молекулами анализируемых веществ и ионами газа-реагента (реактанта), в качестве которого используют водород, метан, пропан, изобутан, аммиак и другие газы (табл. 7.5). Ионы газа-реагента получают бомбардировкой молекул газа электронами с энергией 100-500 эВ при давлении в источнике ионов 10-10 2 Па. Образовавшиеся ионы-реагенты взаимодействуют с нейтральными молекулами этого же газа, что приводит к образованию ионов типа СН5 hC2Hj из метана, C4H^ —из изобутана, М14 —из аммиака. Эти ионы затем вступают в реакции с молекулами анализируемых веществ (М), протонируют их или образуют с ними ионы-аддукты, например: СН5 + М —> СН4 + + (М + Н)+; СН3 +М —> (М + СН3)+. Количество М, как примесь в газе-реагенте, должно быть малым и составлять не более 0,1%. В этом случае можно пренебречь их ионизацией бомбардирующими электронами и считать, что ионы исследуемого газа (и протонированные, и аддукты) образуются только за счет ХИ. Результаты, полученные методами ХИ, показывают, что квазимолеку-лярные ионы не обладают большой избыточной внутренней энергией. Поэтому осколочных ионов в спектре очень мало или они вообще отсутствуют. Это является заметным преимуществом, особенно при анализе биологически важных соединений, таких, как терпены, стероиды, сахара и т.п., которые образуют ионы (М+Н)+. В зависимости от газа-реагента можно изменять картину масс-спектра и наблюдать тонкие различия [14, 38].
848
Новый справочник химика и технолога
Таблица 7.5
Газы-реагенты, применяемые для химической ионизации, их основные ионы и типы взаимодействия [38]
Пояснения, RA(X) — сродство к протону, кДж • моль’1;
RE(X+) — энергия рекомбинации, эВ.
Тип 1 Тип 2
Газ (X) Ионы-реагенты RA(X) Газ (X) Главные ионы RE(X+)
н2 H3+ 418,4 He He+* 24,6
сн4 (СН5)+ 531,4 Ne Ne+* 21,6; 21,7
сн4 (С2н5)+ 665,2 Ar Ar+* 15,8; 15,9
сн4 (С3н5)+ — Ar Ar2+ * 24
С2н4 (С2н5)+ 669,4 n2 15,3
изо-C4Hio трет-(С4Н9)+ 815,9 n2 N+ или (N2)2+ —
изо-с5н12 трет-(С5Нц)+ — n2 n;- —
С6Н14 (С6Н13)+ — Kr Kr4* 14,0; 14,7
С6н18 (С8Н17)+ — Kr Kr24 21
с8н18 (С6Н13)+ — CO co+* 14,0
С8н18 (С5нп)+ — co (co): ’ 11-12
н20 (Н3О)+ 690,4 co2 co: • 13,8
H2S (H3S)+ 711,3 co2 (co): —
СН3ОН (СН3ОН)2+ 716,5 co2 or 9,7-17,0
NH3 (NH4)+ 866,1 Xe Xe+* 12,1-1 3,4
ch3nh2 (CH3NH3)+ 903,7 Xe Xe2+ 18-20
(CH3)2NH [CH3)2NH2]+ 928,8 o2 or 9,7-17,0
(CH3)3N [CH3)2NH]+ 949,8 cf4 CF3+ 9,17
(CH3)4Si [(CH3)Si]+ — NO NO+ 9,25(8,3)
В настоящее время часто применяют комбинированные источники ионов, сочетающие два вида ионизации, например, ЭУ и ХИ (рис. 7.4). В этом случае полученные масс-спектры могут быть чрезвычайно информативными.
Для ХИ используют источник ионов закрытого типа. Область ионизации в нем ограничена камерой, в которой поддерживается относительно высокое давление (иногда атмосферное). Расход газа-реагента составляет 1-10 л • мин1, поэтому применяется эффективная дифференциальная система откачки.
Чувствительность положительной химической ионизации (ПХИ) для многих соединений сравнима с чувствительностью ионизации ЭУ. Однако, применяя ХИ, необходимо учитывать, что отсутствие в масс-спектре пиков осколочных ионов не позволяет пользоваться библиотечным поиском для установления структуры химических соединений.
Рис. 7.4. Комбинированный источник ионов с химической ионизацией и ионизацией электронным ударом: 1 — камера ионизации электронным ударом; 2 — камера химической ионизации; 3 — зонд системы непосредственного ввода проб в источник ионов; 4 — линия ввода проб из газового хроматографа; 5 — устройство перемещения камеры химической ионизации; 6 — линия подачи газа-реагента
При анализе некоторых химических веществ, обладающих высоким сродством к электрону, получила распространение отрицательная химическая ионизация (ОХИ) [41, 42]. Существует три основных процесса образования ионов в ОХИ:
а) ионно-молекулярные реакции с образованием ионов-аддуктов: М + R“ -> (M+R)
б) резонансный захват тепловых электронов с образованием молекулярного иона: М + е —> М ;
в) диссоциативный захват электрона с образованием осколочного иона и нейтральной частицы: АВ + е” —> А + В“.
Масс-спектры, полученные методом ОХИ, обычно более просты, чем при ионизации ЭУ, и иногда они состоят только из молекулярных ионов. ОХИ обладает высокой селективностью и чувствительностью. Такие соединения, как галогенсодержащие углеводороды и нитросоединения, легко анализируются, захватывая электроны, тогда как насыщенные углеводороды в этих условиях почти не ионизируются. Для соединений с высокой степенью сродства к электрону (хлорированные углеводороды и др.) чувствительность ОХИ часто в 100-1000 раз выше, чем при ионизации ЭУ и ПХИ. Предел обнаружения таких соединений при использовании ОХИ достигает 10 13 г.
Одним из эффективных методов ионизации нелетучих и термически нестабильных соединений является десорбционная химическая ионизация (ДХИ). В этом методе пробу из раствора или суспензии наносят на металлическую ленточку (проволоку), которую с помощью зонда вводят непосредственно в область плазмы источника ионов с ХИ. Под действием плазмы происходит образование молекулярных или кластерных ионов даже нелетучих веществ практически без фрагментации. При анализе смеси нескольких соединений через проволоку пропускают ток, постепенно повышая температуру пробы, и таким образом ее фракционируют. Проволоку иногда активируют, обрабатывая бензонитрилом при высокой температуре.
Масс-спектрометрический метод анализа
849
7.1.4. Полевая ионизация (ПИ)
Результатом другого метода «мягкой» ионизации нелетучих соединений — полевой ионизации является образование в основном молекулярных ионов при очень небольшой степени фрагментации. Молекулы анализируемых веществ адсорбируются из газовой фазы на эмиттере и ионизируются под действием электрического поля. Эмиттер изготавливают из вольфрамовой или рениевой проволоки диаметром 10 мкм и активируют при нагревании в парах бензонитрила при температуре ~ 900 °C. Эмиттер при этих условиях активации покрывается тонкими иглами пиролитического углерода длиной 30—40 мкм, на концах которых наиболее эффективно происходит ионизация. Эмиттер находится под высоким напряжением — порядка 7-14 кВ, у его поверхности образуется высокий градиент потенциала (107—108 В • см-1), и удаление электронов из молекул пробы происходит вследствие туннельного эффекта. Чувствительность ПИ в 5-10 раз ниже, чем при ионизации ЭУ, но преимущество ПИ в том, что молекулярные ионы образуют даже те соединения, которые не возникают при ионизации ЭУ.
Большое распространение получил метод полевой десорбции (ПД), в котором не требуется перевода пробы в газовую фазу. Пробу из раствора или суспензии наносят на активированный эмиттер, температуру которого подбирают так, чтобы обеспечить достаточную интенсивность в масс-спектре пика молекулярного иона при минимальной фрагментации (обычно 400-600 К). Иногда нагрев эмиттера с пробой осуществляют с помощью лазера, что оказывается более эффективным для образования молекулярных ионов [43]. В этом методе ионизация и десорбция происходят при наложении между эмиттером и противоэлектро-дом, находящимися на расстоянии 2-3 мм, электрического потенциала около 10 кВ и нагревании пробы. Образуются, как правило, ионы М+, (М+Н)+, (M+Na)+, а вероятность образования осколочных ионов мала. Показано, что добавление в матрицу солей щелочных металлов увеличивает ионизацию полярных молекул, а добавление винной и сульфоновой кислот приводит к увеличению количества ионов (М+Н)+. Порог поля десорбции ионов уменьшается при добавлении таких соединений, как сахароза, поливиниловый спирт, полиэтиленоксид [44]. Если использовать неактивированный эмиттер, то полностью подавляется ионизация летучих продуктов и получаются более простые масс-спектры.
ПД может использоваться с любыми типами анализаторов: магнитными, квадрупольными, времяпролетными и др. Однако широкое энергетическое распределение образующихся ионов обусловливает предпочтение приборам с двойной фокусировкой (тандемные МС).
К недостаткам метода следует отнести зависимость результатов анализа от качества эмиттера и плохую воспроизводимость масс-спектра.
7.1.5. Эмиссия вторичных ионов (ЭВИ)
Бомбардировка поверхности с пробой пучком быстрых ионов инертных газов приводит к эмиссии вторичных ионов анализируемого вещества [23] в основном в виде про
тонированных (М+Н)+ и депротонированных (М-Н)- молекулярных ионов и кластерных ионов (М+катион)+. В роли катиона выступает обычно щелочной металл. Пучок первичных ионов с энергией 2-20 кэВ направляют на поверхность с образцом под определенным углом. Вторичные ионы, эмитируемые с поверхности, отбирают под другим углом и впускают в масс-анализатор. Отношение токов ЭВИ (М+Н)+ / (М-Н)- сильно зависит от химической природы подложки, может меняться на несколько порядков и практически не зависит от массы первичных (бомбардирующих) ионов и их энергии в пределах 2-10 кэВ. Эффективность ионизации в методе ЭВИ очень высока и может достигать 10%, что дает теоретический предел обнаружения ~ 10-19 моль при использовании времяпролетного анализатора и ~ 10-12 моль — для магнитного прибора с двойной фокусировкой. Однако на практике этот предел не может быть реализован главным образом из-за загрязнений пробы. Благодаря низкому пределу обнаружения и возможности получать ионы больших органических молекул термически нестабильных и нелетучих соединений, метод ЭВИ можно использовать для обнаружения следовых количеств органических веществ.
Ионы исследуемых веществ получают и методом ионизации при бомбардировке быстрыми атомами (ББА) [19]. В этом случае пучок ионов инертного газа (обычно ксенона), ускоренных до энергии 3-10 кэВ, нейтрализуют путем захвата электронов или путем перезарядки, превращая его в пучок ускоренных атомов. Пучок атомов под углом от вертикали к поверхности ~ 70 °C направляют на мишень, которая представляет собой глицериновую матрицу, содержащую растворенную или суспендированную пробу. В качестве матрицы применяют также тиоглицерин, тетраэтиленгликоль, диэтиламин и другие жидкости, характеризующиеся низким давлением паров [44, 45]. Такая матрица обеспечивает постоянное обновление молекул анализируемых веществ на поверхности и длительный стабильный ионный ток, что позволяет использовать плотность первичного пучка бомбардирующих атомов более высокую, чем при работе с твердой подложкой, без существенного повреждения образцов [45]. Для получения полного масс-спектра методом ББА достаточно 5 нг вещества. Однако количественные измерения затруднительны, поскольку на результаты оказывает влияние концентрация пробы в гетерогенной смеси, наличие солей и других примесей. Масс-спектры, полученные методом ББА, как и в методе ЭВИ, содержат пики протонированных, молекулярных и осколочных ионов, а также ионы, образовавшиеся при присоединении к молекуле катиона Na+ или К+. Матрица может взаимодействовать с пробой, быть причиной химического шума, обусловленного самой матрицей и ее аддуктами с анализируемым веществом или загрязнителями, например, щелочными металлами. С другой стороны, химические процессы, происходящие в самой матрице, а также при взаимодействии анализируемого вещества с матрицей и примесями, могут повысить чувствительность анализа и дать структурно-информативные ионы.
850
Новый справочник химика и технолога
7.1.6. Искровые ионные источники (ИИИ)
В искровом ионном источнике происходит высоковольтный пробой вакуумного промежутка между электродами, в результате которого исследуемое вещество, нанесенное на электроды, распыляется и частично ионизируется (рис. 7.5) [48]. Электроды соединены со вторичной обмоткой высоковольтного трансформатора, на первичную обмотку которого подается напряжение от генератора высокой частоты. Ионы, образующиеся при пробое вакуумного промежутка, ускоряются напряжением в 15-25 кВ и, пройдя систему коллимирующих щелей, попадают в масс-анализатор. Мощность искры в значительной степени зависит от изменения частоты следования и продолжительности запускающих импульсов. Амплитудное значение напряжения в искре выбирается в зависимости от природы анализируемого вещества и находится в пределах от 20 до 100 кВ.
I +20 кВ Т ।—
Рис. 7.5. Принципиальная схема искрового ионного источника: 1 — высокочастотный генератор; 2 — усилитель мощности;
3 — генератор запускающих импульсов;
4 — регулируемый источник анодного напряжения;
5 — камера источника ионов со щелями;
6 — форма искровых пакетов; 7 — масс-анализатор
Основные недостатки НИИ: во-первых, ионный ток по самой природе вакуумной искры нестабилен; во-вторых, высокое напряжение, приложенное к электродам, приводит к большому разбросу ионов по энергиям, достигающему 1,5-5 кэВ. Такой разброс обусловливает необходимость использования масс-анализаторов с двойной фокусировкой. Кроме вышеуказанных недостатков, необходимо отметить, что вероятность ионизации разных химических элементов в ИИИ различна, что затрудняет получение количественных результатов без использования стандартов.
В отличие от рассмотренного высоковольтного искрового источника в низковольтном источнике после первичного пробоя, напряжение которого регулируется расстоянием между электродами, низковольтный разряд (несколько сотен вольт) поддерживается специальной электронной системой разряда [48]. Электронная температура плазмы в таком источнике составляет 50000-100000 К, что значительно снижает концентрацию многоатомных ионов. Существенным недостатком низковольтного источника является большой вклад дважды и даже трижды ионизованных молекул. В ИИИ ионы образуются одновременно по нескольким механизмам, среди которых можно выделить катодное распыление, автоэмиссию (полевая ионизация), взаимо
действие распыленных частиц с плазмой и термоионную эмиссию. Все эти механизмы образования ионов, рассмотренные ранее, используются в других типах источников ионов. Что касается взаимодействия распыленных частиц с плазмой, то в ней будут происходить химическая ионизация, а также ионизация за счет соударений с ионами большей энергии, приобретенной в электрическом поле.
ИИИ в основном нашли применение при элементном анализе твердых, химически и термически стойких веществ.
7.1.7. Источник с тлеющим разрядом (ТР)
Для элементной масс-спектрометрии [9, 12, 22, 31, 49] простыми и эффективными являются источники ионов с тлеющим разрядом (ТР). Это разряд газа низкого давления, в котором положительные ионы благородного газа, например аргона, притягиваются к катоду, изготовленному из материала пробы, и распыляют атомы с поверхности катода с достаточно высокой эффективностью. Величины оптимального значения напряжения и тока разряда определяют такие факторы, как свойства газа, его давление и конфигурация источника. Этот тип источника ионов пришел на смену импульсному источнику с высоковольтной и высокочастотной искрой, дающему ионы с большим разбросом по энергии.
Источник с тлеющим разрядом представляет собой простое двухэлектродное пространство, заполненное благородным газом при давлении 10-1000 Па. Напряжение, равное нескольким сотням вольт, подаваемое на электроды, вызывает пробой газа и образование ионов, электронов и других частиц. Положительные ионы газа, ускоряясь в электрическом поле, бомбардируют катод, который испускает различные вторичные частицы — ионы и атомы анализируемого вещества. При напряжении 500 В и давлении 100 Па средний свободный пробег атомов находится в пределах 0,1-0,05 мм, что предполагает частые столкновения входящих в катод и выходящих из него частиц. Это приводит к потере энергии ионами аргона, но оставшейся энергии вполне достаточно для распыления большого количества пробы. Относительное количество распыленных нейтральных атомов и молекул больше, чем ионизированных, и они диффундируют в пространство между анодом и катодом, где в электронно-ионной плазме подвергаются ионизации. Тлеющий разряд не только атомизирует твердую пробу, но и представляет собой средство, с помощью которого ионизируются эти атомы.
В основном выделяют два вида ионизации атомов пробы — электронным ударом: М + е —> М+ + 2е” и ионизацией Пеннинга (соударения с метастабильными атомами аргона): М + Аг —> М+ + Аг + е”.
Аргон имеет метастабильные состояния при 11,55 эВ и 11,72 эВ, т.е. величинах, превосходящих почти все потенциалы первой ионизации элементов (табл. 7.2). Считают, что ионизация Пеннинга является преобладающим процессом в тлеющем разряде. Масс-спектры, полученные при ионизации тлеющим разрядом, состоят из однозарядных атомных ионов и только при более высокой чувствитель
Масс-спектрометрический метод анализа
851
ности регистрируются такие ионы, как (М + Аг)4, (М2)+ и (М + О)+, (Fe + Fe)+. Источники ионов с ТР являются экономически выгодными источниками, могут работать в режимах импульсного и постоянного тока, а также высокочастотном режиме. При этом мощность разряда составляет всего 5-10 Вт. Скорость натекания благородного газа можно сделать достаточно низкой (3-5 см3 • мин-1). Давление газа в наиболее распространенных источниках ионов ТР составляет ~ 100 Па, что оказывает на МС нагрузку в виде проточного газа и требует дифференциальной откачки.
Анализ образовавшихся ионов при ионизации ТР проводят как с помощью квадрупольных [9, 31, 32], так и с помощью МС, работающих на принципе ионно-цикло-тронного резонанса [22], и магнитных секторных МС [12].
7.1.8. Источники ионов с лазерной десорбцией (ЛД)
Характер процессов, развивающихся при взаимодействии лазерного пучка с поверхностью вещества в твердой фазе [9, 11, 25, 27, 47], определяется плотностью потока излучения q, которую можно изменять в широких пределах (от 10"3 до 1015 Вт • см"2). При плотности потока лазерного излучения выше порогового значения поглощенная веществом энергия превышает энергию связи атомов и энергию ионизации, в результате чего слой облучаемого вещества превращается в плазму [9, 27]. Сильно ионизированная плазма экранирует поверхность, поглощает поступающее лазерное излучение, что приводит к ее значительному разогреву. Из-за большого градиента давления вещество выбрасывается с облученной поверхности, и происходит газодинамическое расширение плазменного сгустка. Одним из важных параметров лазерной плазмы является электронная температура Ге, которая пропорциональна плотности лазерного излучения: Те ~ q4/<) [63]. Так, абсолютное значение электронной температуры в лазерной плазме в диапазоне плотности потока излучения 109-1011 Вт • см"2 изменяется от 10 эВ до 100 эВ. При этом электронная плотность лазерной плазмы составляет ~ 1019 см"3, а плотность нейтральных атомов ~ 102° см"3. Этот метод ионизации — неселективный, и он не зависит от длины волны лазерного излучения [27]. Используются в основном два варианта этого метода: а) лазерный микрозондовый анализ и б) облучение пробы несфокусированным лазерным пучком (около 1 мм2). В втором случае плотность энергии излучения составляет 106—108 Вт • см"2 при длительности 5-50 нс., что дает возможность применять для анализа ионов времяпролетный масс-анализатор.
Масс-спектры, полученные с источником ЛД, имеют значительно меньший фон, чем в случае ББА, и содержат молекулярные, кластерные и протонированные ионы. ЛД может проводиться и с матрицы, содержащей анализируемое вещество. При этом может идти дополнительная химическая ионизация исследуемых молекул, полученных в результате испарения, за счет взаимодействия их с примесями, имеющимися в матрице и перешедшими в газовую фазу в виде ионов под действием лазерного луча, например с К+ [25]. Усиленная матрицей лазерная ионизация полу
чила краткое название MALDI (Matrik Assisted Laser Desorption and Ionization). Она была разработана в 1988 г. специально для анализа больших молекул [62] и используется для изучения биополимеров в молекулярной биологии и медицинской биохимии. Источники ионов с ЛД, с десорбцией путем бомбардировки быстрыми ионами или атомами дают возможность передать анализируемым молекулам за сравнительно малое время (10"8-10 9 с) значительное количество энергии — достаточное для их испарения, что невозможно сделать путем обычного нагревания пробы, которое приводит к ее термическому разложению и потере информации, необходимой для идентификации.
7.1.9. Источники с экстракцией ионов электрическим полем (ЭРИАД)
Источник с экстракцией ионов из растворов электрическим напряжением при атмосферном давлении предназначен для ионизации и анализа больших молекул белка, ДНК и других биополимеров [15, 56]. Впервые метод электрической экстракции ионов из растворов для ЖХ-МС был предложен и испробован в 1984 г. авторами [58] для системы газожидкостной хроматографии — масс-спектрометрии (ЖХ - МС). Во многом сходный с ним, получивший название электровпрыска, описан в другой работе [59]. В этом методе анализируемое вещество в виде раствора подается насосом через металлический капилляр малого диаметра (меньше 0,2 мм) в область электрогазодинамического распыления, находящуюся при атмосферном давлении. К капилляру прикладывается высокое (несколько кВ) напряжение, вызывающее мелкодисперсное распыление раствора и движение образовавшегося аэрозоля от капилляра к диафрагме-противоэлектроду (рис. 7.6). Отверстие в этой диафрагме, разделяющей объемы с атмосферным и форвакуумным давлением, является звуковым газодинамическим соплом. При движении аэрозоля через плотный газ к звуковому соплу происходит распад микрокапель раствора и образование ионов и ионных кластеров анализируемого вещества, которые затем вместе с потоком газа через звуковое сопло (диафрагму) проходят в область форвакуума. В этом объеме интерфейса формируется газодинамическая струя, в центре которой находятся ионы анализируемого вещества. Выделение ядра струи осуществляется конусным скиммером с отверстием в вершине порядка 0,1 мм. Через отверстие скиммера ионы попадают в высоковакуумную область формирования ионного пучка, общую с масс-аналиатором, где производится анализ пучка ионов по массам. К области интерфейса между соплом (диафрагмой) и скиммером прикладывается регулируемое напряжение (до 1 кВ), позволяющее управлять видом регистрируемого масс-спектра, изменяя его состав от масс-спектра кластеров до масс-спектра фрагментов молекул.
Электровпрыск часто используют в качестве интерфейса между жидкостным хроматографом и масс-спектрометром, что связано прежде всего с очень высокой чувствительностью метода, а также с отсутствием ограничений в молекулярной массе анализируемого вещества.
852
Новый справочник химика и технолога
Рис. 7.6. Схема газодинамического интерфейса ЭРИАД:
1 — капилляр; 2 — сопло; 3 — скимер;
4 — формирующая оптика; 5 — выходная диафрагма интерфейса; 6 — объектная щель масс-анализатора
7.1.10. Источник ионов с ионизацией индуктивносвязанной плазмой (ИСП)
Источник с ионизацией ИСП, предназначенный для многоэлементного и изотопного анализа [2, 6-8, 21, 30, 50, 51, 90-95], отличается простотой конструкции и состоит из распылителя пробы, горелки индуктивно-связанной плазмы (индукционный плазмотрон) и интерфейса для отбора пробы из плазмы и экстрагирования ионов. Раствор пробы (несколько мл) накачивается в распылитель, где он диспергируется до размера частиц, равных ~ 1 мкм. Небольшая часть (~ 1%) распыленной пробы вводится в плазменную горелку в потоке аргона со скоростью 10-15 л • мин1. Газы плазмы собираются конусообразным устройством с отверстием для отбора пробы, которое расположено перед конусообразным скиммером для сбора ионов (рис. 7.7). Для распыления растворов используются ультразвуковые, пневматические и другие распылители. Способ введения жидкой пробы влияет на пределы детектирования. Экспериментально доказано, что ультразвуковое распыление более качественно и при прочих равных условиях обеспечивает на выходе прибора сигнал примерно в 10 раз больший на единицу концентрации, чем пневматическое распыление при анализе проб раствора урана с содержанием несколько нгм в литре [7].
Рис. 7.7 1 — индуктивно-связанная плазма (ИСП);
2 — интерфейс для экстракции ионов с конусом скиммера;
3 — ионопровод; 4 — сопло (жиклер); 5 — (форвакуумный) механический вакуумный насос; 6 — откачка паромасляным насосом; 7 — дифференциальная откачка (1 600 литров • с"1); 8 — квадрупольный масс-анализатор; 9 — ионно-оптическая система для направления ионов в детектор;
10 — электронный умножитель
Применяются три основных типа высокочастотных плазмотронов. Первые два разработаны Грифилдом и Фас-селом [52, 53], третий предложен Сукачем и др. [54]. Все горелки работают при атмосферном давлении аргона на частоте генератора ~ 27 МГц и потребляют мощность 2,0-2,5 кВт. Индуктор охлаждается проточной водой. Скорость подачи анализируемой пробы водного раствора обычно составляет 0,5 мл • мин-1; скорость подачи газа-распылителя (аргона) — 0,7 л • мин-1; газа, охлаждающего плазму, — 15 л • мин-1. Количественный анализ можно проводить без каких-либо затруднений при использовании калибровки по простым стандартным растворам [2]. МС с ионизацией ИСП может использоваться и для анализа органических растворов на содержание микропримесей металлов. При этом условия анализа незначительно отличаются от анализа проб водных растворов солей за исключением того, что в газ-распылитель добавляется 5-7% кислорода для предотвращения накопления углерода на коническом устройстве ввода пробы в интерфейс. В этих условиях для большинства элементов пределы детектирования находятся на уровне нескольких нг на ми-лилитр. При прохождении в течение примерно 2 мс через ядро плазмы, температура которой 7000-8000 К, практически происходит полная диссоциация соединений пробы и при этом элементы с энергией ионизации менее 10 эВ ионизируются с эффективностью 90-100%. Вероятность образования дважды ионизированных ионов в этих условиях мала. Барий и стронций являются единственными элементами, для которых были зарегистрированы двухзарядные ионы. Эти два элемента имеют самые низкие энергии повторной ионизации: Ва2+ (IE = 10,01 эВ) и Sr2+ (IE = 11,03 эВ) (табл. 8.2). Несколько выше этих значений энергии ионизации некоторых редкоземельных элементов: Eu2+ (1Е= 11,3 эВ), Sm2+ (IE = 11,4 эВ) и La2+ (1Е= 14,43 эВ), у которых тоже будут наблюдаться дважды ионизированные атомы, поскольку установлено, что I, имеющий сравнимую с ними энергию ионизации первого электрона (IE = 10,44 эВ), ионизируется на 40%, в то время как Cd (IE = 8,99 эВ) — на 100% [55]. Большинство элементов, например Li, Na, К, V, Cr, Mn, Со, Ni, Cu, Zn, Мо, Ag, Cd, Cs, Hg, Tl, Pb и др., регистрируются в виде одноатомных однозарядных положительных ионов, а вклад ионов типа МО+ незначителен. Однако масс-спектр урана состоит в основном из UO+ и UO;, а не из одноатомных ионов, что связано с недостаточной диссоциацией этих термостойких оксидов в источнике ИСП [55]. Спектр масс основных сопутствующих ионов (скорость счета менее 2 000 имп • сек-1), получаемых из ИСП, при распылении стандартных водных растворов содержит АгН+, Ar+, Н3О+, Н+, НО+, NO+, о;, но;, ArN+ и Аг2Н+, которые не перекрываются с массами большинства определяемых элементов. В табл. 7.6 приведены фоновые пики, появляющиеся при распылении водных растворов некоторых часто используемых кислот в анализе проб методом ИСП-МС [92].
Масс-спектрометрический метод анализа
853
Таблица 7.6
Основные фоновые пики в методе ИСП-МС при распылении водных растворов 5% кислот (азотной, серной и хлороводородной) [22]
м Изотоп (ат. %) HNO3 H2SO4 HC1
1 Н (99,985) ‘H — —
2 Н (0,015) 2H — —
3 — — — —
4 Не (100) — — —
5 — — — —
6 Li (7,5) — — —
7 Li(92,5) — — —
8 — — — —
9 Be (100) — — —
10 В (19,91) — — —
11 В (80,09) — — —
12 С (98,89) 12C — —
13 С (1,11) 13c — —
14 N (99,63) 14n — —
15 N (0,37) 15n — —
16 О (99,76) 16q — —
17 О (0,04) !6OH — —
18 О (0,20) 16OH2 — —
19 F(100) ,6OH3 — —
20 Ne (90,92) 18oh2 — —
21 Ne (0,26) 18OH3 — —
22 Ne (8,82) — — —
23 Na (100) — — —
24 Mg (78,8) — — —
25 Mg (10,15) — — —
26 Mg (11,05) — — —
27 Al (100) — — —
28 Si (92,21) 14n 14n, 12c 160 — —
29 Si (4,7) ,4n14nh, I2C16OH — —
30 Si(3,09) 14n 16o — —
31 P(100) 14n16oh •— —
32 S (95,02) 16q 160 32S —
33 S (0,75) 16O16OH 33S, 32SH —
34 S (4,21) 16018O 34S, 33SH —
35 Cl (75,77) 16O18OH 34SH Cl
36 Ar (0,34) 36Ar 36s 35C1H
S (0,02) — — —
37 Cl (24,23) 36ArH 36SH 37C1
38 Ar (0 06) 38Ar — 37C1H
39 К(93,08) 38ArH — —
Продолжение таблицы 7.6
M Изотоп (ат. %) HNO3 H2SO4 HC1
40 Ar (99,6) 40 Ar — —
Ca (96,97) — — —
К (0,01) — — —
41 К (6,91) 40ArH — —
42 Ca (0,64) 40ArH2 — —
43 Ca (0,14) — — —
44 Ca (2,06) 12c 160160 — —
45 Sc (100) 12Cl6O16OH — —
46 Ti(7,99) 14N,6O16O 32s14n —
Ca (0,003) — —
47 Ti(7,32) — 33s14n —
48 Ti (73,98) — 34s 14n,32s16o —
Ca (0,19) — — —
49 Ti (5,46) — 33S16O 35C1 l4N
50 Ti(5,25) 36Ar14N 34s 160 —
Cr (4,35) — — —
V (0,24) — — —
51 V (99,76) — — 37C114N, 35C1160
52 Cr (83,76) 36s 160 35C116OH
53 Cr (9,51) — —- 37C1160
54 Fe (5,82) 40Ar14N — 37C116OH
Cr(2,38) 40Ar I4NH — —
55 Mn(100) 40Ar ,6O — —
56 Fe (91,66) — — —
57 Fe (2,19) — — —
58 Ni (67,77) — — —
Fe (0,33) — — —
59 Co (100) — — —
60 Ni (26,16) — -— —
61 Ni(l,25) — — —
62 Ni (3,66) — — —
63 Cu (69,1) — — —
64 Zn (48,89) — 32S,6O16O32S32S —
Ni (1,16) — — —
65 Cu (30,9) — 33S 16O16O32S33S —
66 Zn (27,81) — 34S16016O32S34S —
67 Zn(4,ll) — — 35C1160160
68 Zn (18,57) 40Ar!4N14N 36S16O,6O32S36S —
69 Ga (60,16) — — 37C1160,6O
70 Ge (20,51) 40 Ar 14N lbO — —
Zn (0,62) — — —
71 Ga (39,84) — — 36Ar3SCl
72 Ge (27,4) 36Ar36Ar — —
73 Ge (7,76) — — 36Ar37Cl
74 Ge (36,56) 36Ar38Ar — —
Se (0,87) — — —
854
Новый справочник химика и технолога
Продолжение таблицы 7.6
м Изотоп (ат. %) HNO3 H2SO4 HC1
75 As (100) — — 40Ar35Cl
76 Ge (7,77) 36Ar40Ar — —
Se (9,02) — — —
77 Se (7,58) 36Ar40ArH — 40Ar37Cl
78 Se (23,52) 38Ar40Ar — —
Kr(0,35) — — —
79 Br (50,54) 38Ar40ArH — —
80 Se (49,82) 40Ar40Ar — —
Kr (2,27) — 32S 16O 16O16O —
81 Br (49,46) 40Ar40ArH 32S 16O 16OI6OH —
82 Kr (11,56) 40 Ar 40 Ar H2 34S16O 16OI6O —
Se (9,19) — — —
83 Kr (11,55) — 34S16O 16O16OH —
84 Kr (56,9) — 36S 16O 16O!6O —
Sr (0,56) — — —
Поскольку источник ионов ИСП расположен вне вакуумной системы, ионы масла насосов не детектируются. Нет вклада ионов из материалов конструкции источника ионов ИСП, так как ионизация осуществляется бесконтактно. Основные трудности, встречающиеся при анализе, связаны с подавлением степени ионизации некоторых элементов в присутствии высокой (более 0,2%) концентрации легкоионизируемых соединений, например натрия, калия, что обусловлено изменением плотности электронов в плазме. Снижение суммарной концентрации солей в растворе ниже 0,2% дает возможность анализа проб без вышеуказанных помех.
Ввод твердых проб в источник ионизации ИСП можно осуществлять путем лазерной абляции, достигая таких же-пределов определения элементов, как и при использовании растворов солей. Этот метод ввода исключает необходимость применения длительных операций растворения исследуемого образца, тем самым уменьшается вероятность его загрязнения. Для абляции исследуемых проб твердых материалов их размещают в абляционной камере. Луч лазера фокусируется на поверхности пробы, и управляемые лазерные импульсы продолжительностью, равной миллисекундам, испаряют материал пробы. Образующееся облачко пробы, состоящее из микрочастиц, уносится потоком аргона в факел ИСП и затем ионизируется в плазме. При этом обеспечиваются пределы детектирования, превосходящие возможности оптических систем. Размер пятна лазерного луча можно регулировать от 10 до 300 мкм, что дает дополнительную возможность пространственного анализа дискретных характеристик пробы. Особое значение такой прибор имеет для использования в полупроводниковой, ядерной, минералологической и керамической областях, где необходимо быстро определять содержание примесей на уровне менее 10-9-10“8 г-1 без растворения. МС-анализ (с ИСП и лазерной абляцией в совокупности) является единственным методом, который удовлетворяет всем аналитическим требованиям, предъявляемым к ана
лизу следовых количеств примесей в современных химически стойких керамических материалах.
Автоматический режим работы масс-спектрометра с ИСП с возможностью полуколичественного анализа по всему диапазону масс элементов, длящимся менее минуты, и пределами обнаружения, отличающимися по крайней мере на порядок от пределов обнаружения с помощью других методов, обеспечивает широкие возможности применения прибора в анализе окружающей среды [2, 7, 8].
Низкие пределы обнаружения (уран в водном растворе измеряют на уровне нескольких пикограмм на миллилитр) и возможность одновременного определения природного происхождения материала путем измерения изотопных отношений являются отличительными особенностями метода, что имеет решающее значение для ядерной промышленности.
Учитывая возрастающее количество ограничений на использование радиоизотопов в клинических исследованиях человека, МС-анализ с ионизацией ИСП приобретает исключительное значение в медицине, особенно если учесть уникальную способность масс-спектрометров с ИСП к быстрому (за 1-2 мин) определению концентрации стабильных изотопов в жидкостях тела человека.
Возможность анализа больших количеств проб (до 100 в день) с широким ассортиментом элементов при их низком содержании имеет неоценимое значение в геологии. Кроме того, элементы при использовании ИСП имеют прекрасную постоянную чувствительность в большом диапазоне концентраций, что позволяет проводить точную оценку их относительного содержания в пробах. В таблице 8.7 в качестве примера приведены типичные пределы, достигаемые при определении некоторых элементов в геохимических материалах МС-методом с ИСП. В работе [95] приведены пределы обнаружения и соответствующие относительные стандартные отклонения, полученные при анализе этим методом целого ряда элементов (таблица 7.7).
Таблица 7.7
Пределы определения элементов в геохимических материалах МС-методом с ИСП, мкг • г-1 [93, 94]
Элемент Предел определения* Элемент Предел определения* Элемент Предел определения*
Li 0,3 Co 0,3 Sb 0,3
В 4 Ni 3 Те 3
Na 3 Cu 0,2 I 0,7
Mg 0,9 Zn 0,6 Ba 3
Al 1 As 3 La 0,08
Si 33 Se 0,3 W 0,1
К 4 Rb 0,4 Au 0,1
Ca 10 Sr 0,1 Hg 0,2
Sc 0,06 Zr 0,009 Tl 0,03
Ti 2 Nb 0,1 Pb 0,3
V 0,2 Mo 0,7 Bi 0,1
Cr 0,1 Ag 0,3 Th 0,04
Mn 0,3 Cd 0,6 и 0,02
Fe 2 Sn 0,4
* Данные находили по правилу «6 о» холостого опыта.
Масс-спектрометрический метод анализа
855
Таблица 7.8
Пределы обнаружения и точность результатов определения ряда элементов методом ИСП-МС, нг • мл’1 [95]
Определяемый элемент Предел обнаружения* Относительное стандартное отклонение, %**
7Li 0,4 5
9Ве 0,4 4
ИВ 0,5 5
27А1 0,4 2
52Сг 0,1 7
55Мп 0,03 5
59Со 0,06 3
63Си 0,1 2
66Zn 0,4 5
78Se 4 6
88Sr 0,03 4
95Мо 0,2 3
138Ва 0,03 2
205j| 0,1 4
2°8pb 0,3 3
* Для доверительной вероятности 0,95 и длительности интегрирования аналитического сигнала 10 с.
** Величина относительного стандартного отклонения приведена для растворов с содержанием определяемого элемента 50 нг • мл ’.
7.2. Масс-анализаторы
7.2.1. Магнитные секторные анализаторы
Заряженные частицы, ускоренные в электрическом поле, движутся в магнитном поле по траекториям, радиусы которых определяются соотношениями:
Му2 = evB, R Mv2 = eu, 2 (7.3) (7.4)
о 1 \2UM R — * I , (7.5)
где М— масса частицы, кг; В — индукция магнитного поля, Тл; v — скорость частицы, движение которой перпендикулярно магнитному полю, м • с-1; U— ускоряющее напряжение, В; R — радиус траектории, м; е — заряд электрона, Кл.
Из уравнения (7.5) видно, что каждый отдельный ион, характеризуемый конкретной величиной М/е, при данной индукции магнитного поля движется по собственной траектории. Одновременно секторное магнитное поле действует как собирающая линза, фокусируя ионные пучки, отличающиеся угловым распределением (рис. 7.8). Из этого
м r2b2
уравнения также следует, что — =-------, и интервал
е 2U
сканирования масс можно изменять, варьируя либо В, либо U, поддерживая остальные параметры постоянными.
Рис. 7.8. Магнитный секторный анализатор
Разрешение секторного масс-анализатора, достигающее 6000 [39], ограничено разбросом кинетических энергий ионов и невозможностью четкого обозначения границ магнитного поля.
7.2.2. Приборы с двойной фокусировкой (тандемные)
Пучок ионов можно превратить в моноэнергетический с помощью электростатического сектора анализатора А (рис. 7.9). Если пучок ионов движется по части окружности в пространстве между двумя концентрическими электродами цилиндрической формы, то радиус R и напряженность наложенного электрического поля Е определяют энергию частиц, которые могут пройти через анализатор:
М ER
(7.6)
Из данного уравнения следует, что массу ионов, проходящих анализатор, можно контролировать, изменяя напряженность электрического поля между электродами. Этот электростатический анализатор, иногда называемый энергетическим фильтром, используют в сочетании с магнитным анализатором В для создания масс-спектрометра с двойной фокусировкой, поскольку в таких приборах ионы фокусируются и по энергиям, и по массам (рис. 7.9).
Рис. 7.9. Масс-анализатор Нира-Джонсона с двойной фокусировкой: А — электростатический анализатор, В — магнитный анализатор
856
Новый справочник химика и технолога
Анализаторы с двойной фокусировкой имеют высокое разрешение по массам (80000 и более) и часто используются в тех случаях, когда нужно идентифицировать сложные молекулы с одинаковыми массовыми числами, а также когда источник дает ионы с большим разбросом по энергии, например, при искровой ионизации.
7.2.3. Времяпролетный масс-анализатор
Если применить прерывистый ускоряющий потенциал, то пучок ионов можно разбить на отрезки или импульсы. Это дает возможность сортировать ионы по их скоростям, что равносильно сортировке по массам (рис. 7.10.). Именно такой принцип положен в основу времяпролетного масс-спектрометра [36], где пучок электронов ионизирует образец в режиме электронного удара или химической ионизации. К сетке приложен ускоряющий потенциал порядка 2000 В в форме импульсов длительностью 1 мкс или менее, повторяющихся около 20000 раз в секунду. Эти импульсы положительного напряжения сообщают ускорение ионам, которые затем движутся в длинной свободной от полей трубе дрейфа со своими собственными скоростями. Поскольку все ионы приобрели одинаковую энергию, то
( м>\2
скорость каждого из них пропорциональна — и время
прохождения определяется уравнением
где L — длина трубы дрейфа, м; U — ускоряющее напряжение, В.
Если принять L = 1 м, U = 2 кВ, М - Мц = 1,67-1 (Г27 кг, е = 1,6-1019 Кл, то согласно уравнению (7.7) время прохождения ионом водорода пролетного расстояния равно 1,58 мкс. Для других ионов это время составит:
12С2Н3+ - 8,39 мкс; 16О2+ - 9,13 мкс; шХс+ - 18,54 мкс.
Рис. 7.10. Схема времяпролетного масс-спектрометра
В качестве детекторов ионов в таких анализаторах используется электронный умножитель с большим быстродействием (с малой постоянной времени) для того, чтобы регистрировать ионы без просчетов. Времяпролетные масс-спектрометры позволяют анализировать ионы с массой до 7000 а.е.м. [11].
7.2.4. Квадруполъные масс-анализаторы
Квадрупольный масс-анализатор — это прибор, в котором ионы можно разделить в соответствии с величиной Ml е без применения магнитного поля [36]. Анализатор такого рода состоит из четырех абсолютно прямых металлических стержней, расположенных строго параллельно на определенном расстоянии друг от друга — так, чтобы пучок ионов мог двигаться непосредственно вдоль центральной оси (рис. 7.11). В качестве входного отверстия используют не щель, а круглое отверстие диаметром около 0,1 мм. Стержни, расположенные по диагонали друг против друга, соединены электрически, и обе пары присоединены к противоположным полюсам источника постоянного напряжения, а также к генератору радиочастоты.
Рис. 7.11. Квадрупольный масс-спектрометр
Сканирование можно осуществлять, изменяя напряжение постоянного тока и радиочастоты при их постоянном отношении.
На продольное движение ионов оказывает влияние поле квадрупольного конденсатора, вызывая их боковое смещение. При фиксированных значениях частоты и амплитуды переменного напряжения только у ионов с определенным значением М/ е амплитуда колебаний в направлении, поперечном оси анализатора, не превышает расстояния между стержнями. Такие ионы за счет начальной скорости проходят через анализатор, и на выходе из него регистрируются, попадая на коллектор ионов. Сквозь квадруполь проходят ионы, масса которых удовлетворяет условию [36]:
(ЯЛ) (О
где а — постоянная прибора.
Амплитуда колебаний ионов других масс нарастает по мере их движения в анализаторе так, что эти ионы достигают стержней и нейтрализуются. Сканирование можно осуществлять, изменяя величины постоянного напряжения Un и амплитуду радиочастотного напряжения СЬ) при их постоянном соотношешш. Боковое ускорение иона при его движении в квадрупольном электрическом поле вдоль осей х и у (рис. 7.12) можно найти, рассмотрев результирующий потенциал U в любой точке (х,у) в зависимости от времени:
Масс-спектрометрический метод анализа
857
C/ = [lZn + lZr-cos«W)]-i-^-, (7.9)
где £7П — положительный постоянный потенциал; Ua — амплитуда переменного напряжения с частотой со; г — радиальный размер — расстояние от центральной оси до поверхности стержня.
Рис. 7.12. Расположение стержней в квадрупольном анализаторе
Напряженность электрического поля, которая будет действовать на ион в точке х, у, можно найти, дифференцируя выражение (7.9) по х и у:
dU _[Ua+Ua •cos(co/)]2x
ЭЕ/= [£4+^4 •cos(coz)]2y ду г2
Отсюда сила F, действующая на ион с единичным зарядом, будет равна
_cp7n+E4-cos((o/)]2x
„ [^n+^-cosW]2^
Л, =------:------
Ускорение ионов можно выразить как отношение силы
к массе иона:
2[^п +t4 -cos(со/)]х
2[^п + ^4 -COS(top]у
(МЛ 2 --- г
V е )
(7.12)
Из этих уравнений следует, что боковое движение имеет периодическую составляющую с частотой со, пропорциональную Un и Um и обратно пропорциональную массе иона. Если ион при пролете через квадруполь совершает достаточное число высокочастотных циклов ускорения, то разрешение зависит только от отношения UJUa. Подробный анализ уравнений (7.12) показывает, что максимальное разрешение достигается при отношении Е7П/Е4, приближающемся к предельной величине 0,1678 [36].
Современные квадрупольные масс-анализаторы работают в диапазоне масс от 2 до 2000 и имеют минимальную ширину пика 0,3 а.е.м. вплоть до наибольшей массы. Сильной стороной квадрупольных масс-анализаторов являются напряжения с частотой со (рад • с-1); возможности сканирования масс-спектра за доли секунды и быстрого переключения анализатора с одного массового числа на другое в широком диапазоне масс. Квадрупольные масс-анализаторы используются почти со всеми типами источников ионов. Их можно также быстро переключать с анализа положительных ионов на анализ отрицательных ионов, так как в анализаторе нет дискриминации по полярности. Пропускаемая масса линейно зависит от приложенных напряжений Е/п и Ua, вследствие чего имеет место линейная шкала масс.
В качестве детектора ионов на выходе квадрупольного анализатора обычно используется электронный умножитель.
Основным недостатком таких анализаторов по сравнению с магнитными является изменение их пропускания в зависимости от массы иона, что приводит к заметным искажениям масс-спектров в области больших масс.
7.2.5. Омегатронный масс-спектрометр
Еще одним типом масс-анализатора является омегатрон. В нем используется движение ионов под влиянием переменного электрического поля в плоскости, перпендикулярной постоянному магнитному полю (рис. 7.13). Все образовавшиеся при ионизации ионы движутся в этой плоскости по круговой траектории. Время, необходимое для совершения каждого полного оборота, не зависит от направления и начальной скорости ионов, но зависит от их массы и заряда, а также от напряженности магнитного поля:
т 2пМ м В-е ’ е сои
где В — индукция магнитного поля, Тл; ом— циклическая частота движения ионов с массой М на орбите.
Рис. 7.13. Результирующая траектория ионов в омегатроне (магнитное поле направлено перпендикулярно площади чертежа): 1 — траектория ионов; 2 — радиочастотный генератор;
3 — коллектор ионов
Если период вращения иона совпадает с периодом переменного электрического поля, то он ускорится этим полем и начнет движение по спирали Архимеда к коллек
858
Новый справочник химика и технолога
тору. При этом нерезонансные ионы на коллектор не попадут. Таким образом, меняя частоту электрических колебаний, можно регистрировать ионы с разными отношениями массы к заряду. Относительная простота и компактность позволяют использовать омегатрон не только как анализатор масс ионов, но и как течеискатель для вакуумных систем.
7.2.6. Масс-анализатор на основе ионно-циклотронного резонанса (ИЦР)
Масс-анализатор ИЦР, называемый также масс-спектрометр с преобразованием Фурье (МС-ПФ), в последнее время находит все большее применение для аналитических целей [16, 22, 60]. Основным элементом спектрометра ИЦР (с наличием или без Фурье-приставки) является прямоугольная шестиэлектродная ячейка со стороной, равной нескольким сантиметрам, внутри которой создается высокий вакуум и сильное магнитное поле (рис. 7.14). В ней производится ионизация исследуемых молекул импульсным пучком электронов (в течение 1-5 мс) или другим методом. Образовавшиеся ионы движутся в магнитном поле по циклическим траекториям с так называемой циклотронной частотой сом определяемой указанным соотношением (7.13). Ионы удерживаются в ячейке с помощью потенциальной ямы, образованной наложением положительного напряжения (~ 1,0 В) на боковые пластины и отрицательного напряжения (~ -0,5 В) на верхнюю, нижнюю и две торцевые пластины. Разделение по массам достигается в результате подачи переменного радиочастотного поля с частотой со, на верхнюю и нижнюю пластины. Если частота электрического поля совпадает с циклотронной частотой (со, = сом), то ионы будут поглощать энергию и их скорость и радиус траектории увеличатся. Все ионы с отношением М!е будут циркулировать в фазе с радиочастотным возбуждением. Энергию, поглощаемую ионами в резонансе, измеряют с помощью специальной схемы. Однако схема работает только при частоте выше 75 кГц, что ограничивает анализ ионов с большими массовыми числами.
Нижняя пластина (-0.5В)«АДАЛ-'
Балансный конденсатор
К предусилителю
Рис. 7.14. Ячейка ИЦР-масс-спектрометра с Фурье-преобразованием объемом около 10 см 3
В усовершенствованном методе (с приставкой Фурье) проводится быстрое сканирование в пределах всего интересующего диапазона частот (20 кГц до 10 МГц при В = 1-2 Тл) за 1 мс. Это заставляет все ионы в заданном диапазоне массовых чисел циркулировать в фазе, т.е. поглощать энергию, когда их циклотронная частота совпадает с радиочастотой. Как результат такого поглощения энергии при резонансах на верхней и нижней пластинах ячейки индуцируется импульсный ток, который можно регйстрировать, предварительно усилив его электронным усилителем. Величины сигналов обусловлены количеством ионов данной конкретной массы, находящихся в ячейке, циклотронная частота которых совпадает с радиочастотным электрическим полем. Полученные в результате сигналы в измеряемом промежутке представляют собой совокупность импульсов от ионов всех анализируемых масс и, следовательно, содержат всю информацию об образце, которую дает МС рассматриваемого типа. С помощью специального преобразования можно перейти от полученной временной зависимости величин импульсов за определенный отрезок времени к зависимости их от частоты, которая непосредственно связана с массами ионов. В результате такого преобразования получается традиционный масс-спектр анализируемых ионов. Сама процедура перехода к масс-спектрам называется преобразованием Фурье. В МС-ПФ достигнуто рекордное для масс-спектрометрии разрешение 250000-280000 и более [22]. Как следует из соотношения (7.13), в МС-ПФ не надо калиброваться по массам с помощью стандартов, т.к. этот метод дает точное значение масс анализируемых ионов.
Метод МС-ПФ может использоваться в сочетании с источниками химической ионизации при низком давлении (порядка 10 4 Па), а также с газовой хроматографией. Сочетание МС-ПФ со способами ионизации при более высоком давлении (химическая ионизация, лазерная десорбция, тлеющий разряд [22]) осуществляется путем разделения процессов ионизации и анализа по разным ячейкам, в которых дифференциальной откачкой поддерживается необходимое рабочее давление [33]. По некоторым характеристикам МС-ПФ можно сравнить с МС с двойной фокусировкой. Оба прибора дают возможность быстрого сканирования (< 1 с) и обеспечивают высокое разрешение.
Необходимо заметить (см. формулу 7.13), что высокое разрешение при одной и той же величине постоянного магнитного поля МС-ПФ дается только для легких масс. Например, вблизи М = 10-12 а.е.м. разность частот на одну а.е.м. составляет Асо ~ 1,39 МГц при В = 1 Тл, которая легко может быть измерена с погрешностью 1 Гц. В этих же условиях для масс 500 и 501 а.е.м. разность частот составляет всего 61,0 Гц и, следовательно, разрешение при прочих равных условиях будет намного хуже. В таком анализаторе разрешение возрастает пропорционально величине магнитного поля. Так, при В= 10 Тл, которое уже используется на практике, разрешение для одних и тех же масс ионов будет в 100 раз лучше, чем при В = 0,1 Тл.
Масс-спектрометрический метод анализа
859
7.3. Детектирование ионов
Вторичный электронный умножитель (ВЭУ) является одним из основных видов детекторов ионов в современных масс-спектрометрах. Ионы, прошедшие анализатор и имеющие энергию 1-10 кэВ, попадают на коллектор, которым является первый динод ВЭУ. Каждый ион выбивает из первого динода один или большее число электронов. Эти электроны ускоряются разностью потенциалов между первым и вторым динодами (~ 100 В) и выбивают из второго динода следующую порцию дополнительных электронов. Таким образом происходит умножение начального количества выбитых электронов на всех 10-20 ступенях умножителя и на последний динод, на каждый детектируемый ион приходится до 108 электронов. Такие умножители электронов с дискретными динодами отличаются как большим коэффициентом усиления, так и быстродействием и сравнительно малым шумом. Динамический диапазон их достаточно велик — от 101Х (что соответствует одному иону в секунду) до ПГ9 А. Недостатком ВЭУ этого типа является «старение», т.е. изменение характеристик со временем или в результате загрязнения. Другой тип ВЭУ с распределенными динодами (каналтроны) характеризуется большей стабильностью. Каналтроны прочны и устойчивы к внешним воздействиям. Максимальный ток каналтрона значительно меньше, чем у ВЭУ с дискретными динодами (~ КГ11 А).
Наряду с электронным умножителем используется и коллектор Фарадея. Он применяется для регистрации более сильных токов, чем детектируемые ВЭУ, а также при прецизионных исследованиях, например при измерении изотопных отношений.
Получили распространение и способы детектирования ионов с помощью микроканальных пластин, представляющих собой матрицу, состоящую из 104-107 каналтрон-ных электронных умножителей (диаметр каждого из них ~ 12 мкм при расстоянии между центрами соседних элементов ~ 15 мкм).
Следует заметить, что в МС ионно-циклотронного резонанса нет ни ВЭУ, ни коллектора Фарадея, и регистрация ионов осуществляется по величине тока, который идет на увеличение энергии ионов той или иной массы при резонансной частоте переменного напряжения, приложенного к пластинам ячейки анализатора.
Кроме регистрации масс-спектра, в МС предусмотрена регистрация полного ионного тока. Для этого имеется специальный элетрод: он располагается либо в источнике ионов перед выходной щелью, либо между электрическим и магнитным секторами МС с двойной фокусировкой. Сигнал с него служит для оценки условий работы МС и качества юстировки источника ионов, а в ХМС — для регистрации хроматограммы.
В процессе анализа изотопного или элементного состава вещества подачу пробы в источник ионов можно растянуть во времени для того, чтобы многократно записать масс-спектры при плавной развертке тока магнита. При анализе пробы на ХМС это время строго ограничено. Так, например, ширина хроматографического пика, выходящего из капиллярной колонки, может составлять 5-10 с; сле
довательно, для неоднократной записи масс-спектра время одной развертки в заданной области массовых чисел должно быть менее 0,1 от этой величины. Скорость сканирования 0,3 с на 10 массовых чисел обеспечивает МС с двойной фокусировкой и многослойным магнитом. Еще меньшее время на однократную развертку затрачивают квадрупольный и времяпролетный МС. В соответствии с этим временем должна быть обеспечена и скорость регистрации тока ионов. В этих условиях ВЭУ обычно работают в режиме счета ионов, и импульсы с его выхода через быстродействующий усилитель поступают на вход многоканального анализатора. Многократно записанные масс-спектры считываются с анализатора и обрабатываются ЭВМ по соответствующей программе [65].
7.4. Анализ изотопного и элементного состава вещества
Одним из направлений современной масс-спектрометрии является изучение вариаций (часто очень малых) изотопного состава химических элементов. Эти вариации — результат природных физико-химических явлений или искусственного образования изотопов в лабораторных и промышленных условиях.
Для анализа изотопного состава газообразных веществ с погрешностью 0,1-0,02% количество газа в пробе должно быть в пределах 1(Г1О-1(ГП г. Анализ благородных газов (Хе, Кг) с погрешностью 1-3% проводили и с Гораздо меньшим количеством (10“12-10“13 г), применяя ионизацию ЭУ и статический режим работы МС, когда камеру секторного анализатора перекрывали от диффузионных насосов, а высокий вакуум поддерживали с помощью геттерных насосов.
Соли и оксиды многих элементов можно анализировать, нанося вещество в виде раствора или суспензии на рениевую или вольфрамовую ленту термоэмиссионного источника ионов, в котором под действием высокой температуры (1000-2300 К) происходит поверхностная ионизация. Эффективность ионизации значительно возрастает, если использовать две ленты, одна из которых имеет пониженную температуру и является испарителем, а другая — более высокую температуру и служит ионизатором.
Анализ на изотопную распространенность и элементный состав жидких проб — водных или органических растворов и суспензий с диспергированными твердыми нерас-творенными частицами — проводят на МС, в которых в качестве источников ионов используется ИСП. В плазме при воздействии высокой температуры (7000-8000 К) и высокочастотного электрического поля происходит атомизация вещества пробы и образование ионов [2, 7, 8, 21, 30]. Анализаторами ионов для таких источников являются секторные магниты, системы разделения с двойной фокусировкой, анализаторы на основе ионно-циклотронного резонанса, времяпролетные и квадрупольные системы. Выбор анализатора зависит от требуемого разрешения по массам (из-за наложения дублетов ионов в исследуемой области массовых чисел), погрешности измерения изотопного и элементного состава, количества анализов, а также экономических факторов. Наиболее часто встречающиеся в масс-спектрах дублеты приведены в табл. 7.9.
860
Новый справочник химика и технолога
Таблица 7.9
Дублеты, часто встречающиеся в масс-спектрах, и соответствующие им разности масс
Массовое число Ион Распространенность, ат. % Точная масса м/ш
6 12^2+ 98,89 6,000000 400
6Li+ 7,4 6,015121
7 14n2+ 99,63 7,0015366 480
7Li+ 92,6 7,016091
32S4+ 95,0 7,9930182 4000
8 24Mg3+ 78,7 7,9950142 3300
16q2+ 99,76 7,9974571
27ai3+ 100 8,9988440 1600
9 18q2+ 0,20 8,995808 710
9Be+ 100 9,012940
10 3°Sj3+ 3,09 9,991253 460
i°B+ 19,7 10,012940
11 33gj 3+ 0,76 10,9904878 2100
HB+ 80,3 10,9956923
12 24Mg2+ 78,7 11,9925213 1600
12c+ 98,89 12,000000
39j^3+ 93,08 12,9879037 3800
13 26Mg3+ 11,17 12,9912961 1100
13c+ 1,И 13,0033554 3700
12c‘h+ 13,0078246
2SSj2+ 92,21 13,988465 960
14 14n+ 99,63 14,0030732 1100
12c V 14,0156492
45Sc3+ 100 14,9853059 9600
3°Si2+ 3,09 14,986879 1100
15 15n+ 0,37 15,0001088 1400
14N1H+ 15,0108978 1200
12C 1H3+ 15,0234738
32s2+ 95,0 15,9860364 1800
16 160+ 99,76 15,9949141 670
14n’h; 16,0187224 1300
,2c’h; 16,0312984
34s2' 4,22 16,9839314 1100
17 17O+ 0,04 16,9991322 900
’h 160+ 17,0027387 690
14n !h^ 17,0265470
18O+ 0,2 17,9991616 1600
18 ’ H2 16O+ 18,0105633 760
14 №h; 18,0343716
24 24Mg+ 78,7 23,9850426 1600
12q 97,8 24,000000
25Mg+ 10,1 24,9858372 1400
Продолжение таблицы 7.9
Массовое число Ион Распространенность, ат. % Точная масса
25 12,13с2 2,2 25,0033554 5600
25,0078246
52С?+ 83,76 25,9702555 12000
78Se3+ 23,5 25,97246 2600
26 26Mg+ 11,17 25,98259222 1300
12С14^ 98,2 26,0038732 7100
13 с; 0,01 26,0067108 2900
12с’2н; 26,0156492
54Fe2+ 5,82 26,969803 12000
81Br3+ 49,46 26,97214 2900
27 27А1+ 100 26,9815420 1100
13с 14n* 1,1 27,0064286 1600
12с’н2 27,0234738
56Fe2+ 91,7 27,967469 3000
28Si+ 92,2 27,976929 1600
28 12С 16О+ 98,7 27,9949141 2500
14N2 99,3 28,0061464 1100
12с’2н; 28,0312984
58Fe2+ 0,33 28,966657 28000
58Ni2+ 67,71 28,967675 8000
29Si+ 4,7 28,976492 1300
29 13c 160+ 1,П 28,9982695 6500
12c 'h 16o+ 29,0027387 65000
14’15N2+ 0,7 29,0031820 810
12C21H5+ 29,0391230
60Ni2+ 26,16 29,965384 11000
90Zr3+ 51,46 29,96811 5300
30 30Si+ 3,09 29,973758 1200
l4N16O+ 29,9979873 12000
12c1h216o+ 30,0105633 820
“c/iV 30,0469476
63Ni2+ 3,66 30,964169 7000
31 31p+ 100 30,96855 1000
12^i 98,89 30,9984022 3100
12C 'H316O+ 31,0183879
MZn2+ 48,99 31,964571 4300
32 32s+ 95,0 31,972072 1800
16o2+ 99,5 31,9898282 880
12C'H*6O+ 32,026125
33 66Zn2+ 27,81 32,963024 3900
33s+ 0,76 32,9714635
68Zn2+ 18,57 33,962429 6300
34 34s+ 4,22 33,9678628 1300
16’18o; 0,4 33,9940757 580
Масс-спектрометрический метод анализа
861
Продолжение таблицы 7.9
Массовое число Ион Распространенность, ат. % Точная масса М/АМ
34 (I4n'h3V 34,0530940
70Ge2+ 20,51 34,962000 5600
35 105pj3+ 22,23 34,96827 60000
35С1+ 75,53 34,9688531
72Ge2+ 20,51 35,96080 5700
36S+ 0,01 35,9670890 49000
36 lOSpd^4 26,71 35,96782 4100
lH35Cl+ 75,53 35,9766777 1500
2c; 36,0000000 520
(14^н4)2+ 36,0687432
37 37C1+ 24,47 36,9659034 880
12C’H+ 37,0078246
114Cd3+ 28,86 37,96785 6300
38 ‘H37C1+ 24,47 37,9737280 880
12c3 *H2+ 38,0156492
78Se2+ 23,52 38,95870 7800
39 39K+ 93,08 38,9637110 9800
117Sn3+ 7,61 38,96770 680
12C3H3+ 39,0234738
80Se2+ 49,82 39,95824 9200
40Ca+ 96,97 39,9625863 8300
40 120Sn3+ 32,85 39,96740 3200
24Mg l6O+ 78,7 39,9799567 2700
12c216o+ 39,9949141 1100
40,0312984
82Se2+ 9,19 40,95832 12000
41K+ 6,91 40,9618311 6600
41 123Sb3+ 42,75 40,96806 3200
25Mg16O+ 10,13 40,9807513 1900
12c2 'h 16o+ 41,0027387 1100
12c3 'H5+ 41,0391230
42Ca+ 0,64 41,9586259 4500
126Te3+ 18,71 41,96796 4400
42 26Mg16O+ 11,17 41,9775063 1300
12c2 ‘H l6O+ 42,0105633 1200
,2C^H6+ 42,0469476
86Sr2+ 9,86 42,95468 2000
43 27ai 16o+ 99,76 42,9764461 1000
12C *H3160+ 43,0183879 1200
12c3 W 43,0547722
88S?+ 82,56 43,95301 18000
44Ca+ 2,06 43,9554950 2700
44 28Si 16O+ 92,0 43,971843 2400
Продолжение таблицы 7.9
Массовое число Ион Распространенность, ат. % Точная масса М!ЬМ
44 12с 160+ 98,9 43,9898282 1200
12с2 'н 16о+ 44,0262125 1200
12с3 44,0625968
90Zr2+ 51,46 44,95217 12000
45 45Sc+ 100 44,9559177 2900
29Si 16О+ 4,7 44,971406 720
12C2 45,0340371
46Ti+ 8,0 45,9526306 28000
92Mo2+ 19,8 45,95428 3300
138Ba3+ 71,7 45,96829 120000
46 30Si16O+ 3,1 45,968672 4200
23Na2+ 100 45,9795620 4100
14N16O2+ 45,9929014 940
12c2 'h616o+ 46,0418617
47Ti+ 7,28 46,9517571 23000
47 94Zr2+ 17,4 46,95382 3100
141P?+ 100 46,96916
48Ti+ 73,94 47,9479453 11000
96Mo2+ 16,53 47,95248 980000
48 48Ca+ 0,18 47,9525293 3400
32s 160+ 94,8 47,9669868 16000
144Nd3+ 23,85 47,96993 520000
24Mg; 62,4 47,9700852
49Ti+ 5,51 48,9478656 9600
49 98Mo2+ 23,78 48,95299 2700
24’2W2 16,0 48,9708798 7800
147Sm3+ 14,97 48,97150
50Ti+ 5,34 49,9447879 40000
5°cr+ 4,31 49,9460496 45000
50 50y+ 0,25 49,9471634 2500
24’26Mg2+ 17,6 49,9676348 1600
12c19f2+ 98,89 49,9968044
5iy+ 99,8 50,9439769 6500
51 102Ru2+ 31,6 50,95187 2400
,53Eu3+ 52,2 50,9735
52Cr+ 83,8 51,9405109 4600
52 104pd2+ 11,0 51,95164 110000
,04Ru2+ 18,6 51,95211 2400
156Gd3+ 20,5 51,97409
53Cr+ 9,6 52,9406483 4900
53 106pd2+ 27,3 52,95148 2300
159rp^3+ 100 52,974960
862
Новый справочник химика и технолога
Продолжение таблицы 7.9
Массовое число Ион Распространенность, ат. % Точная масса м/ш
53 54Cr+ 2,4 53,9388723 74000
54Fe+ 5,8 53,939606 4500
54 108pj2+ 26,7 53,95174 4800
27ai2+ 100 ^3,963064 4500
162Dy3+ 25,5 53,9752
55Mn+ 100 54,9380549 4000
55 110Cd2+ 12,4 54,95165 93000
110pd2+ 11,8 54,95224 2200
165Ho3+ 100 54,9764
56Fe+ 91,7 55,934938 3400
112Cd2+ 24,1 55,95153 24000
56 28Si2+ 85,0 55,953858 15000
40Ca16O+ 96,7 55,9575004 2900
168Er3+ 27,1 55,9769
57Fe+ 2,2 56,935405 3500
57 114Cd2+ 28,9 56,95178 35000
28>30si; 8,7 56,953421 2200
,7,Yb3+ М,3 56,979
58Fe 0,33 57,933313 19000
58Ni+ 67,71 57,935350 3700
58 2830 si; 5,7 57,950687 14000
116Sn2+ 14,3 57,95110 2400
174Yb3+ 31,84 57,9752
59Co+ 100 58,9331811 3200
59 118Sn2+ 24,03 58,95103 2800
27A116O2 99,5 58,9713602 6000
177Hf3+ 18,56 58,9808
60Ni+ 26,16 59,930768 3100
"Ca’V 2,06 59,9504091 87000
60 120Sn2+ 32,85 59,95110 3800
28 si ,6o; 91,8 59,966757 3900
180эд5+ 35,22 59,9820
61 61Ni+ 1,2 60,931088 1200
183\y3+ 14,4 60,9827
62Ni+ 3,66 61,928338 3200
62 31P2+ 100 61,9475280 1800
18^3+ 28,41 61,9836
63Cu+ 69,09 62,929592 2800
63 126»у 2+ 18,71 62,95194 1800
1890s3+ 16,1 62,9862
64Ni+ 1,16 63,927957 54000
MZn+ 48,99 63,929141 4700
48Ti 16q+ 73,8 63,9428594 50000
Продолжение таблицы 7.9
Массовое число Ион Распространенность, ат. % Точная масса м/ш
64 32q + ^2 90,3 63,9441454 7400
128-р^2+ 31,79 63,85278 7000
32S16О2 94,5 63,9619003 2500
1920s3+ 40,95 63,9874
65Cu+ 30,91 64,927779 2500
65 1 34,48 64,95348 1800
195pt3+ 33,8 64,9887
В работе [97] представлены схемы и описана работа выпускаемых промышленностью масс-спектрометров, где используются источники ионов с ионизацией индуктивносвязанной плазмой. В табл. 7.10 и 7.11 приведены пределы обнаружения элементов в исследуемых пробах. Данные результаты были достигнуты на приборах Elan-500 фирмы «Перкин-Эльмер» (табл. 7.10) и VG Plasma Quad PQ2 Turbo Plus фирмы VG Instruments (табл. 7.11).
Таблица 7.10
Пределы обнаружения элементов, достигаемые методом МС-анализа с ИСП на приборе модели
Elan-5000 фирмы «Перкин-Эльмер», нг • мл [97]
< 0,001 0,001-0,01 0,01-0,1 0,1-1 1-10 >10
Bi Sr Ag Nd в Br 44 Ca 13C
Се Ta Al 60 Ni Be 56 Fe Cl 34 s
Со Tb As Pb Cr К
Cs Nh Au Pd Li P
Er Tl Ba Pt Na Si
Eu Tm Cd Rb Sc
Hf U Cu Ru 82 Se
Но Y Dy Sb Те
In Ga Sm
Ir Gd Sn
La Ge Ti
Lu Hg V
Nb I w
Pr Mg Yb
Re Mn Zn
Rh Mo Zr
Таблица 7.11
Пределы обнаружения элементов, достигаемые методом МС-анализа с ИСП на приборе модели
VG Plasma Quad PQ Turbo Plus фирмы
VG Instruments, нг • мл -1 [97]
< 0,001 0,001-0,01 0,01--0,1 0,1-1 >10
Li Nd V La В Si c
Be Sm Cr Hf Na P F
Масс-спектрометрический метод анализа
863
Продолжение таблицы 7.11
< 0,001 0,001-0,01 0,01--0,1 0,1-1 > 10
Со Eu Mn Ta Mg К S
Ga Gd Ni Re Al Ca Cr
Rb Tb Cu Pt Sc
Sr Dy Zn Au Ti
Y Ho As Fe
In Er Zr Ge
Cs Tm Nb Se
Ba Yb Mo Br
Hg Lu Rh Ru
TI Th Pd Те
Pb U Ag I
Bi Cd W
Ce Sn Os
Pr Sb Ir
Твердые, прочные, плохо растворимые вещества — такие, как алмаз, кварц, карбиды, нитриды, кремний и различные минералы анализируют, используя лазерные и искровые источники ионов или источники ионов с бомбардировкой пробы быстрыми атомами и осколками деления ядер, а также источник ионов с тлеющим разрядом [12, 22, 23, 32, 49, 70]. Ионы, полученные методом искрового разряда, обычно анализируют на приборах с двойной фокусировкой, поскольку такой источник дает ионы с большим разбросом по энергии, достигающим нескольких кэВ. Остальные перечисленные источники ионов могут работать с любыми анализаторами в зависимости от решаемой задачи.
Во всех вышеперечисленных случаях проведения анализа проб на элементный состав используемый масс-спектрометр необходимо предварительно прокалибровать для определения коэффициентов чувствительности прибора к исследуемым элементам.
7.4.1. Идентификация органических соединений и установление их структуры
Количественному анализу молекулярного состава вещества предшествует качественный анализ. В его задачу входит идентификация и установление состава многокомпонентной смеси на основе индивидуальных свойств соединений, проявляющихся при ионизации ЭУ или других видах ионизации. Под идентификацией следует понимать процесс отнесения данного вещества к весьма узкому типу известных веществ.
При анализе на МС органических соединений обычно используются электроны с энергией 50-100 эВ, так как в этом интервале вероятность ионизации наибольшая и от энергии электронов она зависит незначительно. Наряду с ионизацией взаимодействие электронов таких энергий с молекулами приводит к разрыву химической связи в молекулах и образованию «осколочных» ионов (табл. 7.12 и 7.14). Вероятность образования тех или иных осколков обычно коррелирует с прочностью разрываемых связей в
исходной молекуле. Так как каждый спектральный сигнал несет определенную информацию об исследуемой молекуле, то обилие линий в спектре свидетельствует о больших возможностях метода МС, тем более что число пиков в масс-спектре всегда может быть уменьшено либо снижением энергии ионизирующих электронов, либо применением других способов ионизации. По масс-спектрам устанавливают молекулярные веса и основные структурные фрагменты молекулы. Масс-спектр неизвестного соединения можно сравнить с имеющимися масс-спектрами стандартов, и если среди них имеется масс-спектр анализируемого соединения, то его можно таким образом идентифицировать. По масс-спектрам высокого разрешения можно с большой точностью измерить массы молекулярного и осколочных ионов, а по ним, в свою очередь, установить элементный состав этих ионов. Наличие разных методов ионизации («мягкой», сохраняющей молекулярный ион, по которому можно определить молекулярную массу; десорбционной ионизации, позволяющей ионизировать высокомолекулярные, нелетучие и термически нестабильные соединения) делает масс-спектрометрию пригодной для анализа практически любых органических соединений [11, 19, 61].
Таблица 7.12
Положительные ионные фрагменты, часто встречающиеся в масс-спектрах
M/e Ионные фрагменты
14 сн2
15 СНз
16 О
17 ОН
18 н2о, nh4
19 F, Н3О
26 CN, С2Н2
27 С2Н3
28 СО, N2, С2Н4, CHNH
29 С2Н5, СНО
30 NO, CH2NH2
31 СН3, СН2ОН
32 О2
33 SH, ch2f
34 H2s
35 Cl
36 НС1
39 C3H3
40 CH2CN, Ar (арил)
41 C3H5, C2H2NH, CH4CN + H
42 c3H6
43 CH3C0, c2h5n, c3h7
44 CH2CHO + H, co2, ch3chnh2, nh2co, (CH3)2N
45 CH3CHOH, CH2CH2OH, CH2OCH3, CO2H
864
Новый справочник химика и технолога
Продолжение таблицы 7.12
М/е Ионные фрагменты
46 no2
47 CH3S, CH2SH
48 CH3S + H
49 CH2C1
51 chf2
53 с4н5
54 ch2ch2cn
55 с4н7, СН2СНСО
56 С4Н8
57 С4Н9, С2Н5СО, СН3СОСН2
58 СН3СОСН2 + Н, C2H5CHNH2, (CH3)2NCH2, c2h5nhch2, c2h2s
59 CH3OCO, CH2OC2H5, (CH3)2COH, CH3OCHCH3, CH2CH2SH, CH2SCH3
60 CH2CO2H + H, ch2ono
61 СНзОСО + 2H, CH3OCH3SH
65 Циклопентадиенил-катион
67 c5h7
68 (CH2)3CN
69 C5H9, CF3, CH3CHCHCHO, CH2C(CH3)CO
70 c5H10
71 C5Hn, C3H7CO
72 CH2COC2H5, изомеры C4Hl0N, (CH3)2NCO
73 Гомологи ионов c (Mle)= 59 (+ СН2-группы)
74 CH2CO2CH3 + H
75 C2H5OCO + 2Н, CH2SC2H5, СН(ОСН3)2, (СН3)2, (CH3)2CSH
77 с6н5
78 с6н5 + н
79 С6Н5 + 2Н, Вг
80 Пирролил-2-СН2
81 Фуранил-2-СН2, С6Н9
82 СС12, С6Н10, (CH2)4CN
83 С6Нц, СНС12, тиофенил
85 С6Н13, С4Н9СО, CC1F2
86 С3Н7СОСН2 + Н, изомеры C5H12N
87 С3Н7СО2, гомологи ионов с (Mlе) = 73, СН3ОСОСН2СН2
88 Н + СН2СО2С2Н5
89 С3Н7ОСО + 2Н, С6Н5С
90 СбН5СН
91 С6Н5СН2(С7Н7), C6H5N, (СН2)4С1
92 C5H4NCH2 (пиридинил-СН2)
93 СН2Вг, СбН5О (или СбН4ОН), С7Н9
94 2-Пирролоил (C4H4NCO), СбН5О + Н
95 2-Фураноил (С4Н3ОСО)
Продолжение таблицы 7.12
М!е Ионные фрагменты
96 (CH2)5CN
97 C7Hi3, C4H3S - СН2, (тиофенеил-2-СН2)
99 С7Н15, СбНпО
100 С4Н9СОСН2 + Н, изомеры C6H14N
101 С4Н9ОСО
102 С3Н7ОСОСН2 + Н
103 С4Н9ОСО + 2Н, C5HnS, (СН3СН2О)2СН
104 C2H5CHONO2
105 С6Н5СО, С6Н5С2Н4
106 С6Н5СНСН2
107 С6Н5СН2О, НОС6Н5СН2 (о- и П-)
108 Л'-метил-2-пирролоил
111 2-Тиофеноил
119 CF3CF2, С6Н5С3Н6, СН3СбН4С2Н4, СН3С6114СО
/ 0
120 О
НОС6Н5СО, СН2С6Н4ОСН3, С9Н13,
121
123 FC6H4CO
Разные методы ионизации характеризуются селективностью по отношению к некоторым классам соединений, благодаря чему можно выделить даже небольшие их количества на фоне других соединений.
О типе исследуемого соединения в известной степени
можно судить по отношению величины пика молекулярных ионов к полному ионному току т\м= hM/ Yhi- Одно из эмпирических правил состоит в том, что ароматические соединения обладают большим т|м, чем сопряженные диены. Последние устойчивее, чем моноолефины, т]Л/ которых выше т|м алициклических и парафиновых углеводородов. Для соединений, содержащих функциональные группы, можно написать следующий ряд: т|м карбонильных соединений > Т|2И простых эфиров > Т]ти КИСЛОТ > Т|2И спиртов > Г|м
аминов.
Внутри данного типа соединений также наблюдаются
закономерности, качественно связывающие величину молекулярного пика со структурой изомера. Так, молекулярные пики метановых углеводородов нормального строения во много раз интенсивней молекулярных пиков соединений с разветвленными цепями.
Исследование масс-спектров азотсодержащих гетероциклов позволило расширить представления о связи величин пиков молекулярных ионов со структурой. Было установлено,
Масс-спектрометрический метод анализа
865
что для метилзамещенных пиразолов обычно больше у изомеров, где метильная группа соединена с атомом азота.
При структурных исследованиях пользуются определенными эмпирическими правилами, в соответствии с которыми интенсивность пиков основных осколочных ионов определяется перечисленными ниже факторами [37].
1. Повышенной вероятностью отрыва небольших радикалов R от исходной молекулы при разрыве химически слабой связи (например, отрыв метильной группы, находящейся в 0-положении к ненасыщенной связи). При этом образуется интенсивный пик ионов (M-R)+. В ряде случаев гетероатомы N, О, S влияют на распад аналогичной двойной связи.
2. Повышенной вероятностью удаления из исходной молекулы или из образовавшихся осколочных ионов частиц Н2О, СО, С2Н2, С2Н4 и т.д. с образованием интенсивных пиков ионов (М-Н2О)+, (М-СО)+ и т.д.
3. Вероятностью образования заряженной стабильной ненасыщенной системы при отрыве радикала или радикалов от исходной молекулы или от образующихся осколков.
4. Возможностью образования таутомерных структур на первых или последующих стадиях распада молекулярного иона. Корреляция между структурой молекулы и ее масс-спектром проявляется также в том, что для каждого типа углеводородов характерно образование различных гомологических рядов ионов. Например, для парафиновых углеводородов характерны ионы с массовыми числами 43, 57, 71 а.е.м и т.д., для моноолефинов и нафтенов — ионы с массовыми числами 41, 55, 69, 83 а.е.м., для циклоолефинов и диенов — ионы с массовыми числами 67, 68, 81, 82, 95, 96 а.е.м. Имеются аналогичные ряды и для других углеводородов (см. табл. 7.13).
Таблица 7.13
Массовые числа характеристических ионов различных углеводородов [61]
Условные наименования типов соединений Эмпирическая формула Массовые числа характеристических ионов Обозначение характеристических сумм
Алкилбензолы СиН2и-6 91, 105, 119, 133 91
Инданы (татралины) СиН2и-8 117, 131, 145, 159 117
Динафтенбензолы C„H2„-io 129, 143, 157, 171, 185 129
Нафталины С„н2и_12 141, 155, 169, 183 141
Аценафтены С„Н2и-14 153, 167, 181, 195,209 153
Флуорены CrtH.2rt—16 165, 179, 193,207 165
Фенантрацены С„н2п18 191,205,219, 233 191
Нафтено-фенантрены СиН2„_2о 203,217,231, 145, 259 203
Пирены С„н2„_22 215, 229,243,257 215
Хризены С„Н2„_24 241,255, 269, 283 241
Бензтиофены c„H2„_10s 147, 161, 175, 189 147
Дибензтиофены c„H2„_16s 197,211,225, 239 197
Нафтобензтиофены С„н2„_22 247, 261,275, 289 247
После сопоставления полученного масс-спектра со спектрами стандартов, предварительного рассмотрения ряда альтернативных структур, детального анализа гипотетических путей их распада при электронном ударе и других видах ионизации осуществляется выбор той или иной структуры.
7.4.2. Определение индивидуального состава смеси веществ
При анализе молекулярного состава пробы должны выполняться соответствующие условия.
1. Ионные токи отдельных компонентов должны быть пропорциональны содержанию их в пробе, а масс-спектры линейно налагаться и не зависеть от присутствия других компонентов. Для газообразных органических соединений это условие обеспечивается молекулярным режимом натекания исследуемой смеси в источник ионов. При таких измерениях жидкие и твердые органические соединения обычно переводят в парообразное состояние и поддерживают необходимую температуру подогрева системы напус
ка во избежание сорбции вещества на внутренние стенки. Подобным методом могут быть исследованы только такие соединения, упругость паров которых при максимальной температуре системы напуска составляет не менее 20-30 Па и которые не подвержены термическому разложению при рабочей температуре. Анализ смесей, содержащих большие нелетучие органические молекулы, разлагающиеся при повышенных температурах, такие, как белки, дезоксирибонуклеиновые кислоты, протеины и др., проводят с использованием источников ионов с ЛД, ББА (с активацией матрицами или без нее), либо применяя источники с электрораспылением — ЭРИАД [10, 11, 19, 20, 25, 27, 56].
2. Поток анализируемого вещества через МС и соотношение между компонентами этого вещества в пробе должны в процессе анализа оставаться неизменными. В действительности это соотношение при анализе газов может нарушаться. Причины нарушения — более быстрая убыль легких компонентов, и, следовательно, обогащение исходной смеси тяжелыми компонентами в процессе анализа, а также убыль самой пробы. Чтобы эти процессы заметным образом не искажали результатов
866
Новый справочник химика и технолога
анализа газов и паров органических соединений, объем системы напуска и площадь отверстий в напускной диафрагме выбираются так, чтобы убыль наиболее легких компонентов за время анализа не превышала 0,5-1%, а количество пробы в системе уменьшалось не более чем на 3-5%. Процессы сорбции и десорбции в вакуумных системах масс-спектрометров могут привести к влиянию ранее проанализированных проб на масс-спектр последующих анализов («память» прибора). Для уменьшения памяти масс-спектрометров должны быть приняты такие меры, как высокотемпературный прогрев вакуумных частей и их промывка сухими чистыми газами (Аг, N2) или анализируемой пробой.
До определения индивидуального состава смеси прибор калибруют в строго определенных условиях следующим образом:
1) Снимают спектры индивидуальных соединений или их искусственные смеси с внутренними стандартами. При анализе газов и паров — это Аг (арил), н-бутан, бензол; при анализе же нелетучих соединений (с использованием источников ионов ЯД, ББА и электровпрыска) в качестве внутреннего стандарта берут близкие по свойствам соединения, пики которых не перекрываются с пиками ионов анализируемых молекул;
2) Записывают концентрации соответствующих индивидуальных соединений и стандартов в пробах или давление газа (пара) в системе напуска;
3) Интенсивности каждой из зарегистрированных линий индивидуальных соединений и стандартов приводят к одной шкале измерений;
4) Вычисляют относительные интенсивности линий полученного масс-спектра, применяя какую-либо (например молекулярную) линию за 100%;
Используя полученные данные, вычисляют коэффициенты абсолютной чувствительности масс-спектрометра к индивидуальным соединениям и к внутреннему стандарту. Например, для газов
± (7.14)
Р. Ра
где S„ и Sb — абсолютная чувствительность МС к индивидуальному соединению и стандарту соответственно; 1п и k — интенсивности линий индивидуального соединения и стандарта, принятые за 100% (их измеряют в вольтах, количестве зарегистрированных импульсов или других единицах); рп и ро — соответственно парциальные давления газа (пара) анализируемого индивидуального соединения и внутреннего стандарта в системе напуска, Па;
Далее вычисляют Кг — коэффициент относительной чувствительности МС к анализируемому индивидуальному соединению как отношение абсолютных чувствительно-стей МС к нему и к соединению, принятому за стандарт:
$0 Рп Ц
(7-15)
При анализе нелетучих соединений с использованием источников ионов ЛД, ББА и электровпрыска коэффици
ент относительной чувствительности определяется как отношение
<716) Сп If)
где сп и с0 — концентрации индивидуального соединения и стандарта соответственно в активирующей матрице или в распыляемом растворе.
После выполнения калибровки МС в тех же условиях проводят анализ молекулярного состава пробы, предварительно введя в нее стандарты. При анализе смеси пики, получающиеся от одного индивидуального соединения, будут частично совпадать и складываться с пиками других индивидуальных соединений.
На основании полученного масс-спектра можно составить систему уравнений [37].
Пусть масс-спектр, полученный при анализе смеси, содержащей п индивидуальных соединений, содержит линии высотой (интенсивностью) На, Нь, ..., Нт, где а, Ь, ..., т — индексы массовых чисел компонентов масс-спектра. Тогда экспериментальные данные можно записать в виде следующей системы уравнений:
ha)P)+ha2P2+-hanPn =На hb)P)+hb2P2+-hbnPn^Hb
hm)P)+hm2P2+-hmnPn=Hm.
где рп — парциальное давление индивидуального соединения в системе напуска; hmn — относительная интенсивность пика массы т спектра для индивидуального соединения п.
Из приведенных уравнений находят неизвестные значения р\, р2, ...., рп в системе напуска, из которых вычисляют массы индивидуальных соединений в пробе.
При анализе смеси нелетучих соединений с использованием источников ионов ЛД, ББА или электровпрыска в уравнение (7.17) вместо парциальных давлений войдут неизвестные (искомые) концентрации (ci,c2, ...., сп> индивидуальных соединений в пробе.
Решение системы уравнений значительно упрощается, если отобрать свободные от наложения пики и пики с малым числом наложений. При этом уменьшается и погрешность вычислений.
В описанном методе стандартные соединения, использованные при калибровке МС и добавляемые в пробы при анализе состава, служат для снижения погрешностей измерений, вносимых нестабильностью приборов.
Приведенный выше алгоритм анализа состава смеси на МС и последующий расчет, а также методики идентификации органических веществ путем сравнения масс-спектра исследуемого соединения со спектрами стандартных соединений (автоматический поиск) обычно выполняет ЭВМ по разработанным программам [20, 65].
Спектры масс органических и некоторых неорганических соединений приведены в табл. 7.14, куда включены соединения с числом углеродных атомов < 7 и небольшое количество других соединений. Приведены массовые числа и интенсивность (в скобках) пяти важнейшихпиков со
Масс-спектрометрический метод анализа
867
единений — материнского; основного (наиболее интенсивного) и трех последующих в порядке уменьшения их интенсивности. Материнский пик — это пик, соответствующий молекулярному весу соединения; для молекул с содержанием многоизотопных элементов — весу, который отвечает наиболее распространенным изотопам элементов, составляющих молекулу. Интенсивность во всех случаях нормирована по отношению к принятой за 100% интенсивности основного пика бутана с массовым числом т = 43.
При пользовании таблицей нужно учитывать следующее: а) возможно колебание относительной интенсивности в пределах ± 10% в зависимости от применяемого прибора;
б) отношение интенсивности двух пиков в спектре одного соединения при измерении на различных приборах изменяется меньше, чем отношение интенсивности любого пика соединения к основному пику бутана с массовым числом т =43.
Соединения расположены по суммарным формулам в порядке возрастания числа атомов углерода; остальные элементы расположены по начальным буквам их символов в алфавитном порядке. Неорганические .соединения помещены в конце таблицы.
Все спектры получены при величине энергии ионизирующих электронов порядка 70 эВ.
Таблица 7.14
Спектры масс органических и некоторых неорганических соединений [99]
Формула Название Массовое число и (интенсивность — пики)
материнский основной убывающие по интенсивности
CBrClF2 Дифторхлорбромметан 164 (0,23) 85 (86) 87 (27) 129(17) 131 (16)
CBr2F2 Дифтордибромметан 208 (1,7) 129 (70) 131 (68) 79(18) 31 (18)
CC12F2 Дифтордихлорметан 120 (0,07) 85 (33) 87(11) 50 (3,9) 101 (2,8)
CCI3F Трихлорфторметан 136 (0,04) 101 (54) 103 (35) 66 (7,0) 35 (5,8)
ССЦ Четыреххлористый углерод 152 (0,0) 117(39) 119(37) 35 (16) 47(16)
CF3I Трифториодметан 196(51) 196 (51) 127 (49) 69 (40) 177 (16)
cf4 Четырехфтористый углерод 88 (0,0) 69 (57) 50 (6,8) 19 (3,9) 31 (2,8)
CHBrCIF Фторхлорбромметан 148 (5,5) 67(120) 69 (38) 31(13) 111(H)
CHBrF2 Дифторбромметан 130 (13) 51(83) 31 (18) 132(13) 79(13)
CHCI3 Хлороформ 118(1,3) 83 (69) 85 (44) 47 (24) 35 (13)
CHF3 Фтороформ 70 (0,25) 69 (20) 51 (18) 31 (9,9) 50 (2,9)
CHN Водород цианистый 27 (92) 27 (92) 26(15) 12 (3,8) 28(1,6)
CH2C1F Фторхлорметан 68 (48) 68 (48) 33(25) 70(15) 49(11)
CH2C12 Дихлорметан 84 (41) 49(71) 86 (26) 51 (21) 47(13)
CH2F2 Дифторметан 52 (2,7) 33 (26) 51 (25) 31 (7,3) 32 (2,9)
CH2O Муравьиный альдегид 30(19) 29 (21) 28 (6,6) 14 (0,94) 13 (0,92)
CH2O2 Муравьиная кислота 46 (72) 29(118) 45 (56) 28 (20) 17 (20)
CH3 Cl Хлористый метил 50 (66) 50 (66) 15 (54) 52 (21) 49 (6,6)
CH3F Фтористый метил 34 (29) 15(31) 33 (28) 14 (5,3) 31 (3,2)
CH31 Йодистый метил 142 (78) 142(78) 127 (29) 141 (И) 15 (10)
CH3NO2 Нитрометан 61(35) 30 (65) 15 (34) 46(23) 29 (5,3)
CH4 Метан 16 (67) 16 (67) 15 (58) 14(11) 13 (5,5)
CH4O Метиловый спирт 32 (26) 31 (38) 29 (25) 28 (24) 18 (0,7)
CH4S Метилмеркаптан 48 (49) 47 (65) 45 (40) 46 (9,5) 15(8,9)
ch5n Метиламин 31 (30) 30 (53) 28 (47) 29 (87) 27 (8,6)
c2f4 Тетрафторэтилен 100 (20) 31 (47) 81 (34) 50 (14) 12 (3,6)
c2f6 Гексафторэтан 138 (0,14) 69 (95) 119(39) 31 (17) 50 (9,6)
C2F6Hg Гексафтор диметилртуть 340 (0,83) 69(111) 202 (26) 271 (22) 200 (21)
C2H2 Ацетилен 26 (102) 26(102) 25 (20) 24 (5,7) 13 (5,7)
C2H2C1N Хлорацетонитрил 75 (51) 75 (51) 48 (46) 40 (23) 77 (16)
C2H2C12 цис-1,2-Дихлорэтилен 96 (53) 61 (72) 98 (34) 63 (23) 26 (22)
868
Новый справочник химика и технолога
Продолжение таблицы 7.14
Массовое число и (интенсивность — пики)
Формула Название материнский основной убывающие по интенсивности
С2Н2С12 транс-1,2-Дихлорэтилен 96 (49) 61(73) 98 (32) 26 (25) 63 (23)
С2Н2С14 1,1,2,2-Тетрахлорэтан 166 (5,9) 83 (95) 85 (60) 95(11) 87 (9,7)
c2h2f2 1,1 -Дифторэтилен 64 (32) 64 (32) 45 (21) 31(16) 33 (13)
С2Н3С13 1,1,1 -Трихлорэтан 132 (0,0) 99 (37) 99 (24) 61 (19) 117(7,1)
С2Н3С13 1,1,2-Трихлорэтан 132 (3,9) 97 (43) 83 (41) 99 (27) 85 (26)
c2h3f3 1,1,1 -Трифторэтан 84 (0,94) 69(81) 65 (31) 15(13) 45 (10)
c2h3n Ацетонитрил 41 (89) 41(89) 40 (46) 39(17) 38(10)
с2н4 Этилен 28 (66) 28 (66) 27 (43) 26 (41) 25 (7,8)
С2Н4ВгС1 1 -Хлор-2-Бромэтан 142 (7,9) 63 (93) 27 (82) 65 (30) 26 (24)
С2 Н4Вг2 1,2-Дибромэтан 186(1,6) 27 (93) 107 (72) 109(67) 26 (23)
С2Н4С12 1,1-Дихлорэтан 98 (5,7) 63 (89) 27 (64) 65(28) 26 (21)
С2Н4С12 1,2-Дихлорэтан 98 (1,7) 62 (12) 27(11) 49 (4,9) 64 (3,9)
c2h4n2 Диазоэтан 56(16) 28 (27) 27 (25) 26 (21) 41 (5,2)
С2Н4О Уксусный альдегид 44 (30) 29 (66) 43 (18) 42(6,1) 26 (6,1)
С2Н4О Оксид этилена 44 (30) 29 (46) 15(30) 14(12) 43 (7,1)
С2Н4О2 Уксусная кислота 60(19) 43 (37) 45(33) 15(21) 14(8,0)
С2Н4О2 Метилформиат 60 (27) 31 (96) 29 (60) 32 (33) 28 (6,8)
С2Н5Вг Бромистый этил 108(35) 29 (54) 27 (48) 110(33) 26(16)
С2Н5С1 Хлористый этил 64 (36) 64 (36) 28 (32) 29 (30) 27 (27)
c2h5f Фтористый этил 48 (2,4) 47 (24) 27 (8,9) 33 (8,2) 26 (3,0)
c2h5n Этиленимин 43 (31) 42 (56) 28 (44) 15(20) 41(11)
c2h5no2 Нитроэтан 75 (0,0) 29 (85) 27 (74) 30(19) 26(11)
c2h5no3 Этилнитрат 91 (0,01) 46 (95) 29 (42) 30 (29) 76 (23)
c2H6 Этан 30 (26) 28 (99) 27 (33) 26 (23) 29 (21)
c2H6o Этиловый спирт 46(9,7) 31(63) 45 (22) 29 (14) 27 (14)
c2H6o Диметиловый эфир 46 (32) 45(71) 29 (56) 15(41) 14(8,9)
с2н6о2 Пероксид метила 62 (28) 29 (47) 31 (45) 15(16) 30(12)
c2H6s Диметилсульфид (2-тиапропан) 62 (56) 47 (69) 45 (42) 46 (29) 35 (24)
c2H6s Этилмеркаптан 62 (44) 62 (44) 29 (43) 47 (36) 27 (35)
c2H6s2 Диметилдисульфид (2,3-дитиабутан) 94 (95) 94 (95) 45 (59) 79 (56) 46 (34)
c2H6s3 2,3,4-Тритиапентан 126(54) 126 (54) 45 (32) 79 (27) 47 (19)
c2h7n Этиламин 45 (18) 30 (96) 28 (28) 44 (19) 27 (13)
c2h7n Диметиламин 45 (36) 44(71) 28 (48) 15(14) 42 (13)
c2h8n2 Этилендиамин 60 (2,7) 30(11) 18(14) 42 (6,9) 43 (5,9)
C3F6 Гексафторпро пилен 150(16) 31 (56) 69 (44) 131(41) 100 (20)
c3F8 Октафторпропан 188 (0,0) 69(171) 31 (49) 169(42) 50(16)
C3H3N Акрилонитрил 53 (55) 26 (55) 52 (41) 51 (18) 27 (10)
c3H4 Аллен (проподиен) 40 (72) 40 (72) 39 (69) 38 (29) 37 (23)
c3H4 Аллилен (метилацетилен) 40 (79) 40 (79) 39(73) 38 (29) 37 (22)
C3H4C1N [З-Хлорпропионитрил 89(12) 49(68) 54 (54) 51(29) 26 (20)
C3H4O Акролеин 56(16) 27 (25) 26(15) 28(13) 55 (11)
C3H5C1 1-Хлорпропен-1 76 (30) 41(70) 39 (43) 40 (10) 78 (9,6)
Масс-спектрометпрический метод анализа
869
Продолжение таблицы 7.14
Массовое число и (интенсивность — пики)
Формула Название материнский основной убывающие по интенсивности
C3H5CIO Эпихлоргидрин 92 (0,19) 57 (55) 27(53) 29 (40) 31 (21)
С3Н5С1О2 Метилхлорацетат 109 (0,23) 59 (56) 49 (44) 15 (43) 29 (37)
с3 Н5 С13 1,2,3-Трихлорпропан 146 (0,71) 75 (61) 110(22) 77(19) 61 (18)
c3h5n Пропионитрил 55 (8,3) 28 (83) 54 (51) 26(17) 27 (15)
С3н6/ Циклопропан 42 (64) 42 (64) 41 (58) 39(44) 27 (23)
С3н6 Пропилен 42 (39) 41 (58) 39(41) 27 (22) 40 (17)
С3Н6С12 1,1 -Дихлорпропан 112(0,0) 63 (27) 41 (25) 77 (22) 62 (19)
С3Н6С12 1,2- Дихлорпропан 112(2,6) 63 (51) 62 (36) 27 (29) 41 (25)
с3н6о Аллиловый спирт 58(12) 57 (43) 29 (34) 31 (26) 27 (19)
С3н6о Пропионовый альдегид 58 (25) 29 (66) 28 (46) 27 (38) 26 (14)
С3н6о Ацетон 58 (24) 43 (85) 15(26) 27 (5,9) 42 (5,9)
С3н6о Оксид пропилена 58 (19) 28 (44) 29 (30) 27 (28) 26(18)
с3н6о 1,3-Диоксалан 74(3,1) 73 (52) 43 (36) 44 (30) 29 (30)
с3н6о2 Пропионовая кислота 74 (27) 28 (34) 29 (28) 27 (21) 45 (19)
С3н6о2 Этилформиат 74(5,8) 31 (82) 28 (60) 29 (54) 27 (36)
С3н6о2 Метилацетат 74 (22) 43 (148) 29(16) 42 (15) 59(8,4)
С3н6о3 Метил карбонат 90 (3,3) 15 (93) 45 (54) 29 (43) 31 (34)
С3Н7Вг 1-Бромпропан 122 (14) 43 (94) 27 (55) 41(47) 39 (22)
С3Н7Вг 2-Бромпропан 122(11) 43 (100) 27 (50) 41(47) 39 (24)
С3Н7С1 1-Хлорпропан 78 (3,6) 42 (60) 29 (27) 27 (22) 41 (14)
С3Н7С1 2-Хлорпропан 78 (14) 43 (58) 27 (20) 63 (15) 41(13)
C3H7F 2-Фторпропан 62 (1,0) 47 (84) 46 (24) 61 (12) 27 (7,6)
c3h7n 1 -Метилэтиленимин 57 (31) 42 (94) 15 (46) 28 (25) 27(17)
c3h7n 2 -Метилэтиленимин 57 (22) 28 (76) 56 (34) 30 (24) 29 (19)
c3h7no N, А-Диметилформамид 73 (54) 44 (63) 42 (29) 28 (25) 15(24)
c3h7no2 1-Нитропропан 89 (0,0) 43 (68) 27 (67) 41(58) 39 (24)
c3h7no2 2-Нитропропан 89 (0,0) 43 (75) 41 (55) 27 (53) 39 (23)
c3H8 Пропан 44 (25) 29 (85) 28 (50) 27 (33) 43 (19)
c3H8o Пропиловый спирт 60 (7,2) 31 (115) 27(18) 29 (17) 59 (10)
c3H8o в/иор-Пропиловый спирт 60 (0,45) 45(112) 43 (19) 27(18) 29(11)
c3H8o Метилэтиловый эфир 60 (24) 45 (94) 29 (46) 15 (23) 27 (19)
C3H8O2 Метилаль (диметоксиметан) 76(1,6) 45(117) 29 (51) 75 (51) 15(48)
C3H8O2 2-Метоксиэтанол (метилцеллозольв) 76 (7,3) 45(122) 29 (44) 15 (38) 31 (32)
c3H8s Метилэтилсульфид (2-тиабутан) 76 (47) / 61(73) 48 (40) 47 (30) 27 (27)
c3H8s Пропилмеркаптан (пропантиол-1) 76 (30) 47 (43) 43 (34) 27 (34) 41 (32)
c3H8s Изопропилмеркаптан (пропантиол-2) 76 (41) 43 (65) 41 (44) 27 (41) 61 (26)
c3h9n Пропиламин 59 (1,5) 30 (20) 28 (2,5) 27 (1,3) 41 (1,0)
c3h9n Триметиламин 59 (37) 58(95) 42 (44) 15 (32) 30(17)
C3Hi2B3N3 В, В', й"-Три метил боразол 123 (30) 108 (102) 107 (77) 67 (38) 66 (34)
c4f6 Гексафторциклобутен 162 (21) 93 (80) 31 (51) 143 (15) 74(6,9)
c4f6 Гексафторбутадиен-1,3 162 (27) 93 (90) 31 (45) 74 (10) 112(10)
C4 F6 Гексафторбутин-2 162(18) 93 (47) 143 (38) 31(25) 69 (20)
870
Новый справочник химика и технолога
Продолжение таблицы 7.14
Массовое число и (интенсивность — пики)
Формула Название материнский основной убывающие по интенсивности
c4f8 Октафторциклобутан 200 (0,12) 100 (97) 131 (84) 31 (53) 69 (24)
c4f8 Октафторизобутилен 200 (14) 69 (74) 181 (54) 31 (44) 93 (22)
c4f8 Октафторбутен-1 200(11) 131 (122) 31(86) 69 (44) 93 (16)
C4Fio Декафторбутан 238 (0,0) 69(178) 119(33) 31 (22) 100(15)
c4hf7o2 , Гептафтормасляная кислота 214 (0,0) 45 (26) 69 (24) 119(17) 100 (14)
c4H2 Бутадиин-1,3 50(133) 50(133) 49 (57) 48 (14) 25 (12)
C4H4 Винилацетилен 52 (55) 52 (55) 51 (28) 50 (23) 49 (7,2)
C4H4O Фуран 68 (36) 39 (58) 38 (9,7) 29 (9,3) 40 (6,7)
C4H4S Тиофен 84 (93) 84 (93) 58 (56) 45 (49) 39 (24)
C4 H4 s2 2-Тиофентиол 116(68) 116(68) 71(64) 45 (31) 39(11)
C4H5N З-Бутеннитрил 67 (27) 41 (80) 39 (36) 27 (30) 40 (20)
c4h5n Пиррол 67 (67) 67 (67) 39 (46) 41 (42) 40 (36)
c4H6 Бутадиен-1,2 54 (65) 54 (65) 27 (35) 53 (29) 39 (28)
C4H6 Бутадиен-1,3 54 (46) 39 (53) 27 (36) 53 (31) 28 (24)
c4H6 Бутин-1 54 (64) 54 (64) 39 (49) 53 (27) 27 (26)
c4H6 Бутин-2 54 (93) 54 (93) 27 (42) 53 (41) 39 (24)
C4H6C12O2 Этилдихлорацетат 156 (0,12) 29(192) 27 (58) 83 (23) 28 (19)
C4 H6 02 Диацетил 86(13) 43(118) 15 (46) 14(12) 42 (8,6)
c4H6o2 Метилакрилат 86 (2,0) 55 (98) 27 (66) 15 (27) 26 (22)
C4 H7 BrO2 2-Бромэтилацетат 166 (0,03) 43 (158) 27 (35) 106(31) 108 (30)
C4H7C1 2-Хлорбутен-2 90 (27) 55 (68) 27 (21) 39 (21) 29 (18)
C4H7C1O2 2-Хлорэтил ацетат 122 (0,0) 43 (162) 73 (43) 15 (36) 27 (29)
C4H7C1O2 Этилхлорацетат 122 (0,96) 29(130) 27(41) 77 (37) 49 (29)
c4h7n Бутиронитрил (бутаннитрил) 69 (0,15) 41 (112) 29 (70) 27 (38) 28(11)
c4h7n Изобутиронитрил (2-метилпропаннитрил) 69 (1,7) 42 (79) 68 (38) 28 (26) 54 (19)
c4H8 Циклобутан 56 (41) 28 (65) 41 (58) 27 (27) 26 (15)
c4H8 Изобутилен 56 (36) 41 (85) 39 (37) 28(18) 27 (17)
c4H8 Бутен-1 56 (32) 41 (87) 39 (30) 27 (26) 28 (26)
c4H8 г/мс-Бутен-2 56 (36) 41 (76) 39 (27) 27 (25) 28 (24)
c4H8 тиранс-Бутен-2 56 (37) 41 (80) 27 (27) 39 (26) 28 (26)
C4H8C12 1,2-Дихлорбутан 126 (0,30) 41 (39) 77 (35) 27 (20) 76(16)
C4H8C12 1,4-Дихлорбутан 126 (0,03) 55 (87) 41 (29) 27 (24) 90 (23)
C4H8C12 л*езо-2,3-Дихлорбутан 126 (0,95) 63 (64) 27 (57) 62 (54) 55 (31)
c4H8N2 Ацетальдазин 84 (23) 42 (92) 15(47) 28 (46) 69 (38)
c4H8o Масляный альдегид 72(19) 27 (41) 29 (38) 44 (34) 43 (32)
C4H8O Метилэтилкетон 72 (17) 43 (97) 29 (24) 27(15) 57 (6,0)
C4H8O Винилэтиловый эфир 72 (27) 44 (64) 43 (56) 29 (49) 27 (43)
C4H8O г/мс-2,3-Эпоксибутан 72 (3,6) 43 (67) 44 (39) 27 (35) 29 (33)
C4H8O транс-2,3 -Эпоксибутан 72 (3,5) 43 (69) 44 (35) 29 (32) 27(31)
c4H8o Тетрагидрофуран 72 (22) 42 (76) 41 (39) 27 (25) 71 (20)
C4 H8 02 2-Метил-1,3-диоксолан 88 (0,33) 73 (67) 43 (48) 45 (44) 29 (34)
C4 H8 02 1,4-Диоксан 88 (42) 28 (138) 29 (51) 58 (33) 31(24)
Масс-спектрометрический метод анализа
871
Продолжение таблицы 7.14
Формула Название Массовое число и (интенсивность — пики)
материнский основной убывающие по интенсивности
С4 Hg О2 Масляная кислота 88(1,0) 60 (40) 73 (12) 27 (9,6) 41 (9,1)
с4н8о2\ Изомасляная кислота 88(8,1) 43 (77) 41 (33) 27 (26) 73 (19)
с4 н8 О2 Пропилформиат 88 (0,41) 31(123) 42 (89) 29 (38) 27 (36)
с4 н8 02 Этилацетат 88 (7,1) 43(181) 29 (46) 45 (24) 27 (24)
с4 н8 02 Метилпропионат 88(23) 29(110) 57(83) 27 (40) 59 (27)
c4H8s 3 -Метилтиациклобутан 88(42) 46(101) 45 (31) 39 (24) 47 (21)
C4H8S Тиациклопентан (тетрагидротиофен) 88 (44) 60 (82) 45 (29) 46 (29) 47(22)
С4 Н9ВГ 1-Бромбутан 136 (7,0) 57 (86) 41(63) 29 (50) 27 (46)
С4Н9Вг 2-Бромбутан 136 (0,72) 57 (108) 41(65) 29 (61) 27 (36)
C4H9NO2 Пирролидин 71(24) 43 (102) 28 (38) 70 (33) 42 (20)
c4h9no2 Бутилнитрат 103 (0,0) 27 (55) 43 (54) 41 (50) 30 (47)
c4H10Hg Диэтилртуть 260 (12) 29(188) 27 (54) 28 (21) 231(15)
C4Hio Бутан 58(12) 43 (100) 29 (44) 27 (37) 28(33)
С4Ню Изобутан 58 (3,2) 43(117) 41 (45) 42 (39) 27 (33)
с4н10о Бутиловый спирт 74 (0,37) 31 (52) 56 (44) 41(31) 43 (30)
С4 НдоО втирр-Бутиловый спирт 74 (0,30) 45(116) 31 (23) 59 (22) 27 (20)
С4НюО тирети-Бутиловый спирт 74(0,0) 59 (92) 31 (31) 41 (19) 43 (14)
С4НюО Изобутиловый спирт 74(7,5) 43 (84) 31 (56) 42 (48) 41 (47)
С4НюО Диэтиловый эфир 74 (22) 31(73) 59 (34) 29 (29) 45 (28)
с4н10о Метилизопропиловый эфир 74 (8,3) 59(126) 29 (42) 43 (37) 15(32)
С4Н10О2 1,1-Диметоксиэтан (диметилацеталь) 90 (0,06) 59 (93) 29 (52) 15 (37) 31 (37)
C4Hi0O2 1,2-Диметоксиэтан 90(12) 45 (177) 29 (53) 15 (50) 60 (16)
С4н10о2 2-Этоксиэтанол (этилцеллозольв) 90 (0,49) 31(112) 29 (57) 59 (56) 27 (31)
с4н10о2 Пероксид этила 90 (20) 29(116) 15(42) 45 (34) 62 (30)
C4Hi0S Метилизопропилсульфид (3-метил-2 -тиабутан) 90 (41) 41 (49) 75 (47) 43 (41) 48 (38)
c4H10s Метилпропилсульфид (2-тиапентан) 90 (58) 61 (126) 48 (50) 41(43) 27 (43)
c4H10s Диэтилсульфид (3-тиапентан) 90(41) 75 (59) 47(51) 27 (39) 61 (33)
C4Hi0S Бутилмеркаптан (бутантиол-1) 90 (40) 56 (74) 41(65) 27 (42) 47 (31)
C4Hi0S втирр-Бутилмеркаптан (бутантиол-2) 90 (34) 41 (56) 57 (50) 61 (46) 29 (46)
C4HioS тирети-Бутилмеркаптан (2- метил-пропантиол-2) 90 (34) 41(68) 57(61) 29 (44) 39 (21)
C4Hi0S Изобутилмеркаптан (2-метилпропантиол-1) 90 (35) 41 (60) 43 (46) 56 (34) 47 (29)
C4 HioS2 Метилпропилдисульфид (2,3 -дитиагексан) 122 (37) 80 (53) 43 (36) 41 (27) 27 (25)
C4H10S2 Диэтилдисульфид (3,4-дитиагексан) 122 (73) 29(82) 66(81) 27 (57) 94 (53)
С4Н108Оз Этил сульфит 138 (3,3) 29(131) 31 (59) 45 (42) 27 (39)
C4HuN Диэтиламин 73(17) 58(83) 30(81) 28 (30) 27 (24)
c4h„n Бутиламин 73 (12) 30(200) 28(23) 27 (16) 18(12)
C4HuN втирр-Бутиламин 73 (1,2) 44 (170) 18(25) 41(18) 58(18)
C4HuN тирети-Бутиламин 73 (0,25) 58(127) 41 (26) 42 (20) 15(18)
C4HhN Изобутиламин 73 (1,0) 30 (22) 28 (2,0) 41 (1,2) 27(1,1)
C4H12Pb Тетраметилсвинец 268 (0,14) 253(69) 223 (59) 208 (46) 251 (36)
872
Новый справочник химика и технолога
Продолжение таблицы 7.14
Массовое число и (интенсивность — пики)
Формула Название материнский основной убывающие по интенсивности
c5f10 Декафторциклопентан 250 (0,62) 131(173) 100 (41) 31 (40) 69 (28)
c5f12 Додекафтор-2-метилбутан 288 (0,0) 69 (277) 119(45) 131(23) 31 (18)
c5f12 Додекафторпентан 288 (0,08) 69(259) 119(76) 169 (25) 31(24)
c5hf9 Нонафторциклопентан 232 (0,07) 131 (61) 113(49) 69 (34) 31 (19)
c5h5n Пиридин 79(135) 79(135) 52 (95) 51 (48) 50 (35)
c5H6 Циклопентадиен 66 (95) 66 (95) 65 (40) 39 (35) 40 (30)
c5H6 транс-Пентен-2-ин-4 66 (77) 66 (77) 39 (54) 65 (38) 40 (35)
c5h6n2 2-Метилпиразин 94 (81) 94 (81) 67 (48) 26 (33) 39 (30)
C5H6O2 Фурфуриловый спирт 98 (3,4) 98 (3,4) 41 (3,3) 39 (3,3) 42 (2,6)
c5H6s 2-Метилтиофен 98 (68) 97 (125) 45 (26) 39(17) 53(11)
C5H6S 3-Метилтиофен 98(74) 97(138) 45(35) 39 (14) 27(11)
c5H8 Метиленциклобутан 68 (38) 40 (67) 67 (48) 39 (47) 53 (21)
c5H8 Спиропентан 68 (8,9) 67 (58) 40 (56) 39 (52) 53 (23)
c5H8 Циклопентен 68 (41) 67 (99) 39 (36) 53 (23) 41 (19)
c5H8 З-Метилбутадиен-1,2 68 (53) 68(53) 53 (40) 39 (28) 41 (26)
c5h8 2-Метилбутадиен-1,3 (изопрен) 68 (40) 67 (48) 53 (41) 39 (34) 27(23)
c5h8 Пентадиен-1,2 68 (39) 68 (39) 53 (38) 39 (37) 27 (31)
c5H8 z/пс-Пентад иен-1,3 68 (40) 67 (53) 39 (43) 53 (38) 41 (25)
c5H8 /иранс-Пентадиен-1,3 68 (41) 67 (52) 39 (43) 53 (39) 41 (26)
c5H8 Пентадиен-1,4 68 (40) 39 (47) 67 (35) 53 (33) 41 (30)
c5H8 Пентадиен-2,3 68 (62) 68 (62) 53 (42) 39 (36) 41 (31)
c5H8 З-Метилбутин-1 68 (8,5) 53 (74) 67 (45) 27 (35) 39(21)
c5H8 Пентин-1 68 (8,7) 67 (50) 40 (44) 39 (42) 27 (34)
c5H8 Пентин-2 68(67) 68 (67) 53 (61) 39 (32) 27 (27)
c5h8n2 3,5 -Диметилпиразол 96 (47) 96 (47) 95 (37) 39(16) 54(12)
c5H8o2 Ацетилацетон (2,4-пентандион) 100(22) 43 (120) 85(33) 15(23) 27(11)
C5H8O2 2-Пропенилацетат 100 (0,16) 43 (177) 41 (30) 39 (29) 15(28)
c5H8o2 Метилметакрилат 100 (26) 41 (78) 69 (52) 39(31) 15 (16)
C5H9C1O2 Этил-0-хлорпропионат 136(0,70) 27 (65) 29 (62) 91(42) 63 (37)
c5H10 цис-} ,2-Диметилциклопропан 70 (39) 55 (77) 42 (35) 39 (32) 41 (32)
c5H10 транс-} ,2-Диметилциклопропан 70 (42) 55 (79) 42 (34) 41 (33) 39 (30)
c5H10 Этилциклопропан 70 (26) 42 (93) 55 (47) 41 (39) 39 (35)
c5H10 Циклопентан 70 (44) 42 (148) 55 (43) 41(43) 39 (31)
c5H10 2-Метилбутен-1 70 (30) 55(97) 42 (36) 39 (34) 41 (28)
c5H10 З-Метилбутен-1 70 (26) 55(102) 27 (31) 42 (28) 29 (27)
c5H10 2-Метилбутен-2 70(31) 55 (88) 41 (31) 39 (28) 42 (27)
c5H10 Пентен-1 70 (27) 42 (89) 55(53) 41 (39) 39 (31)
c5H10 г/мс-Пентен-2 70 (30) 55 (89) 42 (41) 39 (30) 29 (25)
c5H10 транс-Пентен-2 70 (31) 55(93) 42 (41) 39 (30) 41 (28)
c5H10o Изовалериановый альдегид 86 (3,0) 41 (30) 43 (26) 58 (20) 29 (20)
c5H10o Метилпропилкетон 86(16) 43 (106) 29 (23) 27 (23) 57 (20)
Масс-спектрометрический метод анализа
873
Продолжение таблицы 7.14
Формула Название Массовое число и (интенсивность — пики)
материнский основной убывающие по интенсивности
с5н10о Диэтилкетон 86(15) 57 (87) 29 (87) 27 (32) 28 (9,4)
с5н10о Аллилэтиловый эфир 86 (62) 41 (52) 29 (48) 58(44) 57 (42)
с5н10о Винилизопропиловый эфир 86 (21) 43 (87) 44 (69) 41(46) 27 (45)
с5н10о 2-Метилтетрагидрофуран 86 (8,9) 71 (57) 43 (55) 41(40) 27 (27)
С5 Нюо2 Тетрагидрофурфуриловый спирт 102 (0,02) 71 (8,9) 43 (6,8) 41 (4,8) 27 (3,8)
С5 Н10О2 Винил-2-метоксиэтиловый эфир 102 (3,0) 29 (69) 45 (58) 15 (48) 58 (45)
С5Н10О2 Пивалевая (триметилуксусная) кислота 102(2,0) 57 (83) 41 (38) 29 (27) 39 (12)
с5н10о2 а-Метилмасляная кислота 102 (0,32) 74 (54) 57 (34) 29 (33) 41 (28)
С5н10о2 Бутилформиат 102 (0,27) 56 (80) 41 (48) 31 (47) 29 (42)
С5 Н10О2 етирр-Бутилформиат 102 (0,17) 45 (99) 29 (49) 27 (32) 41 (31)
С5н10о2 Изобутилформиат 102 (0,27) 43 (8) 56 (48) 41 (46) 31 (38)
CsHio02 Пропилацетат 102 (0,07) 43 (176) 61 (34) 31 (31) 27 (26)
CsHio02 Изопропилацетат 102 (0,17) 43 (155) 45 (50) 27 (22) 61 (18)
CsHio02 Этилпропионат 102(10) 29(151) 57 (97) 27 (52) 28 (24)
c5H10o2 Метилбутират 102(1,0) 43 (53) 74 (37) 71(29) 27 (23)
С5Н10О2 Метилизобутират 102 (8,9) 43 (69) 71 (23) 41(19) 59 (17)
c5H10o3 Диэтилкарбонат 118(0,30) 29(114) 45 (80) 31 (60) 27 (46)
c5H10s 2-Метилтиациклопентан 102 (37) 87 (88) 41 (30) 45 (29) 59(18)
c5H10s 3 -Метилтиациклопентан 102 (40) 60 (45) 41 (31) 45 (25) 74 (23)
C5H10S Тиациклогексан 102 (43) 87 (44) 68 (33) 61 (32) 41 (28)
C5H10S Циклопентантиол 102(19) 41 (48) 69 (47) 39 (26) 67(18)
C5HhN Пиперидин 85 (22) 84 (43) 57 (22) 56 (22) 44(17)
C5HhNO Л'-Метилморфолин 101 (4,4) 43 (18) 42 (8,6) 15 (3,4) 71 (2,9)
C5HnNO2 Изоамилнитрит 117(0,0) 29 (75) 41 (68) 57 (43) 30 (42)
c5H12 2,2-Диметилпропан (неопентан) 72 (0,01) 57 (126) 41 (52) 29 (49) 27 (20)
C5H12 2-Метилбутан (изопентан) 72 (4,7) 43 (74) 42 (64) 41 (49) 57 (40)
c5h12 Пентан 72(10) 43(114) 42 (66) 41(45) 27 (39)
c5H12o 2-Метилбутанол 88(0,18) 57 (57) 29 (55) 41(53) 56 (50)
C5H12O З-Метилбутанол-1 88 (0,02) 55 (47) 42 (42) 43 (39) 41 (38)
c5H12o 2-Метил бутанол-2 88 (0,0) 59 (43) 55 (37) 45 (25) 73 (22)
c5H12o Пентанол-1 88 (0,0) 42(41) 55 (30) 41 (25) 70 (23)
C5H12O Бутилметиловый эфир 88(3,1) 45(211) 56 (36) 29 (36) 27 (28)
c5H12o в/ирр-Бутилметиловый эфир 88 (2,0) 59 (142) 29 (50) 27 (27) 41 (25)
c5H12o wpew-Бутилметиловый эфир 88 (0,02) 73 (119) 41 (33) 43 (32) 57 (32)
C5H12O Изобутилметиловый эфир 88(12) 45(186) 41 (30) 29 (30) 15 (27)
C5H12O Изопропилэтиловый эфир 88 (2,6) 45 (143) 43 (46) 73 (40) 27 (24)
c5h1202 Диэтоксиметан (этилаль) 104(2,1) 31(104) 59 (99) 29 (62) 103 (39)
CsHi2O2 1,1 -Диметоксипропан 104(0,05) 75 (84) 73 (62) 29 (43) 45 (37)
c5H12s Бутилметилсульфид (2-тиагексан) 104 (38) 61(77) 56 (50) 41 (39) 27 (33)
C5H12S wpew-Бутилметилсульфид (3,3 -диметил-2-тиабутан) 104(30) 57(83) 41 (62) 29 (42) 39(16)
c5H12s Изобутилметилсульфид (4-метил-2-тиапентан) 104 (37) 41(46) 56 (38) 27 (29) 39 (23)
874
Новый справочник химика и технолога
Продолжение таблицы 7.14
Массовое число и (интенсивность — пики)
Формула Название материнский основной убывающие по интенсивности
c5H12s Пропилэтилсульфид (3-тиагексан) 104 (30) 75 (72) 27 (53) 47 (50) 62(33)
c5H12s Изопропилэтил сульфид (2-метил-З -тиапентан) 104 (82) 89(119) 62 (79) 43 (63) 61 (58)
c5H12s 2,2-Диметилпропантиол-1 104(31) 57 (100) 41 (55) 55 (48) 29 (42)
c5H12s 2-Метилбутантиол-1 104 (28) 41(65) 29 (44) 57 (40) 70 (40)
c5H12s 2-Метилбутантиол-2 104(18) 43 (88) 71 (54) 41 (46) 55 (34)
c5H12s 3 -Метилбутантиол-2 104 (23) 61(73) 43 (55) 27 (33) 55 (28)
c5H12s Пентантиол-1 104 (35) 42 (91) 55 (44) 41 (39) 70 (39)
C5H12S Пентантиол-2 104(28) 43 (72) 61(52) 27 (39) 55 (38)
c5H12s Пентантиол-3 104 (23) 43 (56) 41 (48) 75 (29) 47 (23)
c5H12s2 тирети-Бутилметилдисульфид (4,4-диметил-2,3 -дитиапентан) 136(12) 57 (74) 41 (38) 29 (36) 80(13)
c5H12s2 Изопропилэтилдисульфид (2-метил-З ,4-дитиагексан) 136(20) 94 (49) 27 (46) 43 (39) 66 (37)
C5H14Pb Триметилэтилсвинец 282 (0,64) 223(61) 253 (52) 208 (51) 221 (33)
C6F6 Гексафторбензол 186(95) 186 (95) 117(59) 31 (58) 93 (23)
C6Fi2 Додекафторциклогексан 300 (0,96) 131 (138) 69 (97) 100 (40) 31 (30)
CeFu Тетрадекафторгексан 338 (0,13) 69 (268) 119(74) 169 (51) 131 (37)
C6H5Br Бромбензол 156(75) 77 (98) 158 (74) 51 (41) 50 (36)
C6H5C1 Хлорбензол 112(102) 112(102) 77 (49) 114(33) 51(17)
c6h5no2 Нитробензол 123 (39) 77 (93) 51 (55) 50 (23) 30(15)
c6H6 Бензол 78(113) 78(113) 52 (22) 77 (20) 51 (18)
c6H6 Гексадиин-1,5 78 (58) 39 (65) 52 (38) 51 (32) 50 (26)
c6H6 Гексадиин-2,4 78(108) 78 (108) 51 (55) 52 (38) 50 (31)
C.6H6S Тиофенол 110(68) 110(68) 66 (26) 109(17) 51 (15)
c6h7n Анилин 93 (19) 93 (19) 66 (6,5) 65 (3,6) 39 (3,5)
c6h7n 2-Метилпиридин 93 (86) 93 (86) 66 (36) 39 (28) 51 (16)
c6h7no 1 -Метил-2-пиридон 109(1) 109(1) 81 (49) 39 (34) 80 (29)
c6h8 Циклогексадиен-1,3 80 (53) 79 (92) 77 (35) 39(21) 27(18)
C6H8O 2,5-Диметилфуран 96 (57) 43 (65) 95 (48) 53 (37) 81 (24)
c6H8s 2,3-Диметилтиофен 112(44) 97 (53) 111 (44) 45 (16) 27 (9,4)
c6H8s 2,4-Диметилтиофен 112(27) 111 (36) 97(18) 45 (9,4) 39 (7,0)
еда 2,5-Диметилтиофен 112(67) 111(95) 97 (59) 59 (23) 45 (19)
еда 2-Этилтиофен 112(27) 97 (68) 45 (16) 39 (8,9) 27 (5,4)
еда 3-Этилтиофен 112(54) 97 (147) 45 (38) 39 (20) 27 (12)
c6h9n 2,5-Диметилпиррол 95 (73) 94(127) 26 (52) 80 (22) 42 (19)
c6H10 Изопропенилциклопропан 82 (20) 67 (92) 41 (47) 39 (46) 27 (22)
c6H10 1 -Метилциклопентен 82 (26) 67 (98) 39(21) 81(16) 41 (16)
c6H10 Циклогексен 82 (33) 67 (83) 54 (64) 41 (31) 39 (30)
СбНю 2, 3-Диметилбутадиен-1,3 82(41) 67 (60) 39 (55) 41 (44) 54 (22)
с6н10 2-Метилпентадиен-1,3 82 (23) 67 (48) 39 (30) 41 (26) 27(13)
СбНю Гексадиен-1,5 82(1,3) 41 (98) 67 (80) 39 (60) 54 (52)
Масс-спектрометрический метод анализа
875
Продолжение таблицы 7.14
Массовое число и (интенсивность — пики)
Формула Название материнский основной убывающие по интенсивности
QHio 3,3-Диметил бутин-1 82 (0,57) 67(101) 41(57) 39 (31) 27(11)
СбНю 4-Метилпентин-1 82 (2,3) 67 (82) 41(74) 43 (64) 39 (55)
СбНю Гексин-1 82 (1,0) 67(131) 41(88) 27 (85) 43 (67)
СбНю Гексин-2 82 (56) 67 (58) 53 (50) 27 (39) 41(36)
СбНю Гексин-3 82 (55) 67 (59) 41(55) 39 (37) 53 (20)
С6НюО Циклогексанон 98 (32) 55 (102) 42 (86) 41(35) 27 (64)
СбНюО 4-Метилпентен-3-он-2 98 (40) 55 (82) 83 (82) 43 (64) 29 (38)
СбНюСЬ Гександион-2,5 (ацетонилацетон) 114(4,0) 43 (148) 15(25) 99 (22) 14(14)
СбН10Оз Пропионовый ангидрид 130 (0,0) 57 (190) 29(119) 27 (62) 28 (26)
СбНюОз Этилацетоацетат 130 (8,3) 43 (150) 29 (52) 27 (32) 15 (27)
C6HnN Капронитрил 97 (0,54) 41 (73) 54 (49) 27 (43) 55 (40)
CeHuN Изокапронитрил 97 (0,13) 55 (98) 41 (51) 43 (45) 27 (39)
С6Н12 1,1,2-Триметилциклопропан 84 (38) 41(132) 69 (81) 39 (34) 27 (24)
С6н12 1 -Метил-1 -этилциклопропан 84 (25) 41(78) 55 (58) 69(53) 27 (33)
С6н12 Изопропилциклопропан 84 (2,0) 56(114) 41(84) 39 (30) 43 (28)
С6н12 Этил циклобутан 84 (3,8) 56 (138) 41 (89) 27 (35) 55 (34)
С6н12 Метилциклопентан 84(18) 56(116) 41(74) 69 (37) 42 (33)
С6н12 Циклогексан 84 (58) 56 (75) 41 (44) 55 (25) 42 (21)
С6н12 2,3-Диметилбутен-1 84 (27) 41 (117) 69 (96) 39 (36) 27 (24)
СбН12 3,3-Диметил бутен-1 84 (23) 41(112) 69 (107) 39 (28) 27 (26)
С6н12 2-Этилбутен-1 84 (30) 41(74) 69 (66) 55 (56) 27 (38)
СбН12 2,3 -Диметил бутен-2 84 (32) 41(108) 69 (88) 39 (35) 27 (20)
С6н12 2-Метилпентен-1 84 (29) 56 (91) 41(73) 55 (39) 39 (36)
С6н12 3 -Метилпентен-1 84 (25) 55 (85) 41(67) 69 (60) 27(43)
С6Н12 4-Метилпентен-2 84 (12) 43 (ПО) 41(80) 56 (47) 27(37)
С6н12 2-Метилпентен-2 84 (36) 41 (120) 69(111) 39 (35) 27 (28)
СбН12 3 -Метил-гу пс-пентен-2 84 (37) 41(104) 69 (82) 55 (46) 27 (36)
С6н12 3 -Метил-тиранс-пентен-2 84 (38) 41(102) 69 (81) 55 (47) 27 (35)
С6н12 4-Метил-гупс-пентен-2 84 (35) 41(122) 69(114) 39 (35) 27 (26)
С6н]2 4-Метил-?иранс-пентен-2 84 (34) 41 (123) 69(112) 39 (34) 27 (26)
С6Н12 Гексен-1 84 (20) 41 (70) 56 (60) 42 (52) 27 (48)
С6н12 гупс-Гексен-2 84 (27) 55 (91) 42(51) 41 (45) 27 (45)
С6н12 тиранс-Гексен-2 84 (32) 55(112) 42 (54) 41 (46) 27 (41)
С6н12 гупс-Гексен-З 84 (28) 55 (81) 41 (62) 42 (54) 27 (32)
С6н12 гаранс-Гексен-3 84 (32) 55 (89) 41 (72) 42 (62) 27 (35)
c6H12N2 Ацетоназин (кетазин) 112(31) 56 (99) 15(31) 97(31) 39 (26)
СбН12О Циклопентилметанол 100 (0,02) 41(35) 68 (32) 69(31) 67 (24)
СбН12О Изобутилметилкетон 100(12) 43 (115) 58 (37) 41 (22) 57 (22)
с6н12о Бутилвиниловый эфир 100 (5,7) 29 (80) 41 (59) 56 (45) 57 (35)
СбН12О Винилизобутиловый эфир 100 (5,8) 29 (73) 41(65) 57 (58) 56 (40)
СбН12О2 Диацетоновый спирт 116(0,0) 43 (149) 15(45) 58 (32) 27 (14)
СбН12О2 Бутилацетат 116(0,03) 43 (172) 56(58) 41 (30) 27 (27)
876
Новый справочник химика и технолога
Продолжение таблицы 7.14
Формула Название Массовое число и (интенсивность — пики)
материнский основной убывающие по интенсивности
СвН12О2 Пропилпропионат 116(0,03) 57 (147) 29 (84) 27 (57) 75 (47)
СбН^Ог Изопропилпропионат 116(0,26) 57(116) 43 (88) 29 (54) 27 (46)
CgHi2O2 Метилпивалат (метиловый эфир триметилуксусной кислоты) 116(3,2) 57(85) 41 (32) 29 (24) 56 (21)
СбН12О2 Этилбутират 116(2,2) 43 (50) 71(45) 29 (43) 27 (31)
СбН^Оз 2,4, 6-Триметил-1,3,5-триоксициклогексан 132 (0,12) 45 (196) 43 (107) 29 (35) 89 (23)
C6H12S Метилциклопентилсульфид 116(31) 68 (72) 41(64) 39 (37) 67 (37)
C6H12S г/мс-2,5-Диметилтиациклопентан 116(32) 101(85) 59 (34) 41 (26) 74 (24)
СбН128 транс-2,5-Диметилтиациклопентан 116(32) 101(85) 59 (34) 74 (25) 41(25)
C6H12S 2-Метилтиациклогексан 116(42) 101(81) 41 (37) 27 (32) 67 (30)
C6H12S З-Метилтиациклогексан 116(41) 101 (55) 41 (47) 39 (33) 45 (28)
c6H12s 4-Метилтиациклогексан 116(46) 116(46) 101(44) 41 (40) 27 (39)
c6H12s Тиациклогептан 116(60) 87 (75) 41 (66) 67 (48) 47 (46)
c6H12s 1 -Метилциклопентантиол 116(20) 83 (76) 55(58) 41 (39) 67 (33)
c6H12s г/мс-2-Метилциклопентантиол 116(32) 55 (55) 83 (54) 60 (48) 41 (47)
c6H12s га/?я//с-2-Метил цикло пентантио л 116(28) 67 (48) 55 (46) 41 (42) 83 (40)
c6H12s Циклогексантиол 116(21) 55 (56) 41 (45) 67(35) 83 (32)
C6H13N Циклогексиламин 99 (8,9) 56 (92) 43 (25) 28(13) 30(13)
C6H13N 3 -Метилпиперидин 99 (23) 44 (49) 30 (34) 28 (27) 57 (26)
C6Hl3NO /V-Этилморфолин 115(2,0) 42 (9,8) 57 (7,0) 100 (5,2) 28 (4,3)
C6H14 2,2-Диметилбутан 86 (0,04) 43 (85) 57 (82) 71(61) 41 (51)
C6H14 2,3-Диметилбутан 86 (5,3) 43 (157) 42 (136) 41(49) 27 (40)
СбН14 2-Метилпентан 86(4,4) 43 (147) 42 (78) 41(47) 27 (40)
C6H14 З-Метилпентан 86 (3,2) 57(105) 56 (80) 41(67) 29 (64)
C6H14 Гексан 86(12) 57 (87) 43 (71) 41(64) 29 (55)
C6H14N2 ?/мс-2,5-Диметилпиперазин 114(0,38) 58 (10) 28 (7,7) 30 (4,7) 44(4,2)
C6Hi4O 2-Этилбутанол-1 102 (0,0) 43(114) 70 (40) 29 (39) 27 (38)
с6н14о 2-Метилпентанол 102 (0,0) 43 (ПО) 41 (40) 29 (34) 27 (33)
с6н14о З-Метилпентанол-1 102 (0,0) 56 (26) 41 (20) 29 (19) 55 (18)
C6Hi4O 4-Метилпентанол-2 102 (0,08) 45(111) 43 (34) 41 (17) 27 (14)
СбН14О Гекс анол-1 102 (0,0) 56 (63) 43 (52) 41 (37) 55 (36)
C6Hi4O Бутилэтиловый эфир 102 (3,8) 59 (108) 31 (87) 29 (61) 27 (42)
СбН14О eraqp-Бутилэтиловый эфир 102(1,5) 45(150) 73 (76) 29 (51) 27 (39)
СбН14О Изобутилэтиловый эфир 102 (8,7) 59(124) 31 (95) 29 (53) 27 (38)
СбН14О Диизопропиловый эфир 102 (1,4) 45 (125) 43 (66) 87 (23) 27(19)
СбН14О2 1,1-Диэтоксиэтан (ацеталь) 118(0,0) 45(132) 73 (69) 29 (36) 27 (27)
С6НМО2 1,2-Диэтоксиэтан 118(1,2) 31 (124) 59(88) 29 (72) 45 (53)
СбН14О3 Бис-2-метоксиэтиловый эфир 134 (0,0) 59 (140) 29 (74) 58 (57) 15 (56)
C6H14S Бутилэтил сульфид (3-тиагептан) 118(35) 75 (55) 29(33) 27 (33) 62 (28)
c6H14s вгарр-Бутилэтилсульфид (4-метил-З-тиагексан) 118(195) 89 (585) 29 (343) 27(296) 41 (27)
СбН148 гарега-Бутилэтилсульфид (2,2-диметил-З -тиапентан) 118(33) 57 (147) 41 (70) 29 (54) 27 (40)
Масс-спектрометрический метод анализа
877
Продолжение таблицы 7.14
Формула Название Массовое число и (интенсивность — пики)
материнский основной убывающие по интенсивности
CgHuS Изобутилэтилсульфид (5-метил-З -тиагексан) 118(171) 75 (520) 41 (230) 47(224) 56 (217)
C6H14S Дипропилсульфид (4-тиагептан) 118(47) 43 (86) 89 (74) 41 (57) 27 (55)
C6Hi4S Изопропилпропилсульфид (2-метил-З-тиа-гексан) 118(206) 43 (540) 41 (317) 42 (301) 27(287)
c6H14s Диизопропил сульфид (2,4-диметил-3 -тиапентан) 118(33) 43 (94) 61 (85) 41 (48) 103 (44)
c6H14s 2-Метилпентантиол-1 118(19) 43 (96) 41(51) 56 (32) 27 (31)
C6Hi4S 4-Метилпентантиол-1 118(30) 56 (142) 41 (57) 43 (57) 27 (32)
C6Hi4S 4-Метилпентантиол-2 118(6,3) 43 (68) 69 (61) 41 (56) 84 (42)
C6Hi4S 2-Метилпентантиол-З 118(20) 41(64) 43 (63) 75 (50) 27 (28)
c6H14s Гексантиол-1 118(16) 56 (66) 41 (41) 27 (40) 43 (38)
CeHi4S2 вщо/?-Бутилэтилдисульфид (5-метил-3,4-дитиагептан) 150 (14) 29 (86) 94 (66) 66 (57) 27(41)
c6H14s2 Изобутилэтилдисульфид (6-метил-3,4-дитиагептан) 150(4,9) 29 (42) 66 (40) 122 (30) 94 (29)
C6H14S2 Дипропилдисульфид (4,5-дитиаоктан) 150(44) 43 (167) 27 (65) 41 (64) 108 (35)
СбН1482 Диизопропилдисульфид (2,5-диметил-3,4-дитиагексан) 150 (31) 43(152) 108 (41) 41 (36) 27 (30)
c6h15n Триэтил амин 101 (21) 86(134) 30 (46) 27 (36) 58 (35)
c6h15n Дипропиламин Ю1 (7,1) 30 (89) 72 (70) 44 (36) 43 (28)
c6h15n Диизопропиламин 101 (5,0) 44(171) 86 (52) 58 (24) 42 (22)
C6H16Pb Диметилдиэтилсвинец 296 (0,98) 267(89) 223 (83) 208 (79) 221(44)
C6F14 Тетрадекафторметилциклогексан 350 (0,0) 69(244) 131 (107) 181 (48) 100 (38)
C6Fi6 Гексадекафторгептан 388 (0,0) 69 (330) 119(89) 169 (68) 131 (44)
c7h5n Бензонитрил 103 (246) 103 (246) 76 (80) 50 (42) 51 (24)
C7H7Br о-Бромтолуол 170 (48) 91 (97) 172 (46) 39 (21) 63 (20)
C7H7Br и-Бромтолуол 170 (46) 91(97) 172(45) 39 (20) 65 (19)
C7H7C1 о-Хлортолуол 126(44) 91 (121) 63 (20) 39(19) 89(18)
C7H7C1 ж-Хлортолуол 126 (51) 91 (120) 63 (19) 39(18) 128 (16)
C7H7C1 и-Хлортолуол 126(44) 91(120) 125(19) 63(18) 39(17)
c7h7f .и-Фтортолуол 110(79) 109 (129) 83(17) 57(12) 39(12)
c7h7f и-Фтортолуол 110(73) 109(122) 83 (16) 57(12) 39 (9,3)
c7H8 Толуол 92 (82) 91 (108) 39 (20) 65 (14) 51 (10)
c7H8s Метилфенилсульфид 124 (76) 124 (76) 109 (34) 78 (25) 91(19)
c7h9n 2,4-Диметилпиридин 107 (76) 107 (76) 106 (29) 79 (16) 92 (13)
c7H10s 2,3,4-Триметилтиофен 126 (50) 111 (81) 125 (47) 45 (22) 39(18)
c7H12 Винилциклопентан 96 (13) 67 (118) 39(44) 68 (38) 54 (35)
c7H12 Этилиденциклопентан 96 (40) 67 (180) 39 (44) 41 (30) 27 (30)
c7H12 Бицикло [2,2,1] гептан 96 (12) 67 (64) 68 (50) 81 (44) 54 (30)
c7H12 3 -Этилциклопентен 96 (29) 67(193) 39 (36) 41 (35) 27 (26)
c7H12 1 -Метилциклогексен 96 (32) 81(83) 68 (38) 67 (37) 39 (33)
c7H12 4-Метилциклогексен 96 (28) 81 (84) 54 (50) 39(4) 55 (34)
c7H12 4-Метилгексин-2 96(13) 81(71) 67 (52) 41 (48) 39 (35)
878
Новый справочник химика и технолога
Продолжение таблицы 7.14
Формула Название Массовое число и (интенсивность — пики)
материнский основной убывающие по интенсивности
с7н12 5,6-Метилгексин-2 96 (42) 43 (49) 81(43) 27 (39) 39 (38)
С7н12 Гептин-1 96 (0,44) 41(75) 81(70) 29(65) 27 (47)
с7н14 1,1,2,2-Тетраметилциклопропан 98 (21) 55 (92) 83 (90) 41(69) 39 (41)
С7н14 цис-1,2-Диметилциклопентан 98(19) 56 (85) 70 (77) 41(65) 55 (65)
С7н14 транс-1,2-Диметилциклопентан 98 (25) 56 (93) 41(63) 55 (61) 70 (54)
с7н14 цис-1,3-Диметил циклопентан 98(12) 56 (81) 70 (78) 41(64) 55 (59)
С7н14 транс-1,3 - Д имети лцикл опентан 98 (13) 56 (81) 70 (68) 41(63) 55 (58)
С7н14 1,1 -Диметилциклопентан 98 (6,7) 56 (81) 55 (63) 69 (56) 41 (55)
С7н14 Этилциклопентан 98 (14) 69 (83) 41(78) 68 (60) 55 (46)
с7н14 Метилциклогексан 98 (41) 83 (94) 55(78) 41 (55) 42 (34)
С7н14 Циклогептан 98 (37) 41 (57) 55 (54) 56 (50) 42 (49)
С7н14 2,3, З-Триметилбутен-1 98 (20) 83 (101) 55(83) 41 (61) 39 (33)
С7н14 З-Метил-2-этилбутен-1 98 (22) 41 (71) 69 (71) 55 (62) 27 (38)
с7н14 2,3-Диметилпентен-1 98(13) 41(92) 69 (86) 55 (40) 39 (35)
С7н14 2,4-Диметилпентен-1 98 (9,1) 56(117) 43 (68) 41 (61) 39 (39)
С7н14 3,3 -Д имети лпентен-1 98 (9,4) 69(104) 41(85) 55 (42) 27 (36)
с7н14 3,4-Димети лпентен-1 98 (0,61) 56 (75) 55 (62) 43 (55) 41 (54)
С7н14 4,4-Диметилпентен-1 98 (2,6) 57(161) 41(86) 29 (52) 55 (49)
С7н14 З-Этилпентен-1 98 (19) 41 (116) 69 (91) 27 (43) 39 (37)
С7н14 2,3 -Диметилпентен-2 98 (31) 83 (80) 55(75) 41(63) 39 (34)
С7Н14 2,4-Диметилпентен-2 98 (26) 83 (97) 55 (71) 41(52) 39 (34)
С7н14 3,4-Д иметил-г/мс-пентен-2 98 (30) 83 (87) 55 (82) 41 (52) 27 (32)
с7н14 3,4-Диметил-транс-пентен-2 98 (31) 83 (89) 55 (83) 41 (52) 27 (34)
С7н14 4,4-Диметил-г/ыс-пентен-2 98 (27) 83 (96) 55 (92) 41(62) 39 (35)
С7н14 4,4-Диметил-транс-пентен-2 98 (28) 83 (105) 55 (89) 41 (58) 39(31)
С7н14 З-Этилпентен-2 98 (33) 41 (86) 69 (80) 55 (74) 27 (33)
С7н14 2-Метилгексен-1 98 (4,6) 56 (105) 41(54) 27 (30) 39 (27)
С7н14 3 -Метилгексен-1 98 (7,7) 55 (76) 41 (60) 69 (57) 56 (48)
С7н14 4-Метилгексен-1 98 (4,9) 41(98) 57 (94) 56 (80) 29 (70)
с7н14 5-Метилгексен-1 98(1,6) 56 (91) 41 (75) 55 (47) 27 (42)
С7н14 2-Метилгексен-2 98 (28) 69(113) 41 (99) 27 (36) 39 (33)
С7н14 З-Метилгексен-2 98 (30) 41(95) 69 (90) 55 (42) 27 (36)
С7Н14 4-Метилгексен-2 98 (23) 69(118) 41 (106) 55 (40) 39 (35)
С7н14 5-Метилгексен-2 98 (13) 56 (90) 55 (74) 43 (71) 41(57)
С7н14 2-Метил-лтранс-гексен-3 98 (24) 69 (86) 41 (74) 55 (62) 56 (37)
С7н14 3 -Метил-г/мс-гексен-3 98 (28) 69 (98) 41 (82) 39 (33) 27 (33)
с7н14 3 -Метил-тиранс-гексен-3 98 (28) 69 (97) 41 (86) 55 (63) 39 (35)
с7н14 Гептен-1 98 (15) 41 (91) 56 (79) 29 (64) 55 (54)
С7н14 транс-Гептен-2 98 (27) 55(64) 56 (59) 41 (50) 27 (35)
С7н14 транс- Гептен-3 98 (27) 41(98) 56 (65) 69 (55) 55 (47)
С7Н14О 2,4-Диметилпентанон-З 114(13) 43 (226) 71(62) 27 (49) 41 (42)
С7н14о2 Бутилпропионат 130 (0,03) 57(152) 29 (98) 56 (54) 27 (52)
Масс-спектрометрический метод анализа
879
Продолжение таблицы 7.14
Формула Название Массовое число и (интенсивность — пики)
материнский основной убывающие по интенсивности
С7Н14О2 Изобутилпропионат 130 (0,07) 57(187) 29 (87) 56 (52) 27 (47)
С7Н14О2 Пропилбутират 130(0,05) 43 (96) 71 (90) 27 (54) 89 (48)
С7Н14О3 Дипропилкарбонат 146 (0,02) 43 (171) 27 (61) 63 (55) 41(49)
С7Н14О3 г/нс-2-Метилциклогексантиол 130 (28) 55 (138) 97 (70) 81 (44) 41 (44)
c7h15n 2,6-Диметилпиперидин 113(5,3) 98 (73) 44 (43) 42 (34) 28 (26)
С7н16 2,2,3-Триметилбутан 100 (0,03) 57(110) 43 (84) 56 (67) 41 (64)
С7н16 2,2-Диметилпентан 100 (0,06) 57(130) 43 (95) 41(59) 56 (52)
С7н16 2,3-Диметилпентан 100 (2,1) 43(94) 56 (93) 57 (67) 41 (64)
С7н16 2,4-Диметилпентан 100(1,6) 43 (139) 57 (93) 41 (59) 56 (50)
С7н16 3,3-Диметилпентан 100 (0,03) 43 (166) 71 (103) 27 (38) 41 (36)
С7н16 3-Этилпентан 100 (3,1) 43 (175) 70 (77) 71 (77) 29 (45)
С7н16 2-Метилгексан 100 (5,9) 43 (154) 42 (59) 41 (57) 85 (49)
С7н16 З-Метилгексан 100(4,0) 43(110) 57 (52) 71 (52) 41 (50)
С7н16 Гептан 100 (17) 43 (126) 41(65) 57 (60) 29 (58)
С7Н16О Гептанол-2 116(0,01) 45(131) 43 (29) 27 (25) 29 (23)
с7н16о Гептанол-3 116(0,01) 59 (61) 69 (41) 41 (29) 31 (25)
с7н16о Гептанол-4 116(0,02) 55 (102) 73 (72) 43 (45) 27 (32)
С7Н16О Бутилпропиловый эфир 116(3,7) 43 (120) 57 (102) 41 (51) 29 (49)
С7н16о2 Дипропоксиметан (дипропилформаль) 132 (0,58) 43 (194) 73 (114) 27 (45) 41 (34)
С7н16о2 Диизопропоксиметан (диизопропил-формаль) 132(0,16) 43 (133) 45 (84) 73 (71) 27 (28)
С7Н16О2 1,1 - Диэтоксипропан 132 (0,0) 59 (138) 47 (88) 87 (84) 29 (74)
c7H16s втирр-Бутилизопропилсульфид (2,4-ди-метил-3 -тиагексан) 132 (30) 57(149) 41(74) 29 (35) 43 (32)
c7H16s wpe/и-Бутилизопропилсульфид (2,2,4-три-метил-3 -тиапентан) 132 (30) 61(94) 103 (60) 41(51) 43 (46)
c7H16s Гексилметилсульфид (2-тиаоктан) 132 (34) 61 (73) 56 (53) 27 (46) 41 (44)
C7H16S Гептантиол-1 132 (14) 41 (48) 27 (40) 56 (39) 70 (38)
C7H18Pb Метилтриэтилсвинец 310(0,84) 281 (86) 208 (76) 223 (66) 237 (60)
C7H18Pb Бутилтриметилсвинец 310(0,14) 253 (76) 223 (75) 208 (68) 295 (52)
c7H18Pb в/ирр-Бутилтриметилсвинец 310(1,8) 253 (94) 223 (85) 208 (74) 251 (45)
c7H18Pb /ире/п-Бутилтриметил свинец 310(0,09) 252 (95) 223 (82) 208 (65) 250 (46)
c8H10 «-Ксилол 106 (52) 91 (91) 105 (22) 39(15) 51 (14)
c8H10 jw-Ксилол 106 (58) 91 (93) 105 (26) 39(17) 51(14)
c8H10 и-Ксилол 106 (52) 91(85) 105 (25) 51 (13) 39(13)
c8H10 Этилбензол 106 (45) 91 (146) 51 (19) 39 (14) 65 (12)
B2H6 Диборан 28 (0,13) 26 (54) 27 (52) 24 (48) 25 (30)
B5H9 Пентаборан 64(15) 59 (30) 60 (30) 62 (24) 61 (21)
co Оксид углерода 28 (78) 28 (78) 12 (3,7) 16(1,3) 29 (0,9)
cos Серооксид углерода 60 (83) 60 (83) 32 (48) 28 (6,9) 12 (5,0)
co2 Диоксид углерода 44 (76) 44 (76) 28 (5,0) 16(4,7) 12(1,9)
cs2 Сероуглерод 76(184) 76(184) 32 (40) 44(33) 78 (16)
HC1 Хлористый водород 36 (54) 36 (54) 38 (17) 35 (9,2) 37 (2,9)
880
Новый справочник химика и технолога
Продолжение таблицы 7.14
Формула Название Массовое число и (интенсивность — пики)
материнский основной убывающие по интенсивности
H2S Сероводород 34 (75) 34 (75) 32 (33) 33 (32) 1 (4,1)
n2 Азот 28(65) 28 (65) 14(3,3) 29 (0,47)
NF3 Трехфтористый азот 71 (10) 52 (33) 33 (13) 14 (3,0) 19(2,7)
NH3 Аммиак 17 (32) 17 (32) 16(26) 15 (2,4) 14 (0,7)
n2h4 Гидразин 32 (48) 32 (48) 31 (23) 29(19) 30(15)
NO Оксид азота (II) 30 (76) 30 (76) 14(5,7) 15(1,8) 16(1,1)
no2 Диоксид азота (II) 46 (6,6) 30(18) 16(4,0) 14 (1,7) 47 (0,02)
n2o Оксид азота (II) 44 (60) 44 (60) 30 (19) 14 (7,8) 28 (6,5)
O2 Кислород 32 (54) 32 (54) 16(2,7) 28 (1,7) 34 (0,22)
PH3 Фосфин 34 (59) 34 (59) 33 (20) 31 (19) 32 (7,5)
so2 Сернистый ангидрид 64(47) 64 (47) 48 (23) 32 (4,9) 16 (2,4)
O2 Кислород 32 (54) 32 (54) 16(2,7) 28 (1,7) 34 (0,22)
PH3 Фосфин 34 (59) 34 (59) 33 (20) 31 (19) 32 (7,5)
so2 Сернистый ангидрид 64(47) 64 (47) 48 (23) 32 (4,9) 16(2,4)
Природная распространенность и относительная масса изотопов элементов периодической системы даны в табл. 7.15.
Таблица 7.15
Стабильные изотопы элементов периодической системы [48]
Порядковый номер в периодической системе Элемент Массовое число Природная распространенность, % Относительная масса, а.е.м.
I Н 1 99,9850 1,00797
2 0,0150 2,014
2 Не 3 0,000137 3,016030
4 99,999863 4,002604
3 Li 6 7,42 6,015126
7 92,58 7,016005
4 Be 9 100,0 9,012186
5 В 10 19,61 10,012939
11 80,39 11,009305
6 С 12 98,893 12,000000
13 1,107 13,003354
7 N 14 99,6337 14,003074
15 0,3663 15,000108
8 О 16 99,7587 15,994915
17 0,0374 16,999133
18 0,2039 17,999160
9 F 19 100,0 18,998405
Продолжение таблицы 7.15
Порядковый номер в периодической системе Элемент Массовое число Природная распространенность, % Относительная масса, а.е.м.
10 Ne 20 90,923 19,992440
21 0,257 20,993849
22 8,82 21,991384
11 Na 23 100,0 22,989773
12 Mg 24 78,70 23,985045
25 10,13 24,985840
26 11,17 25,982591
13 Al 27 100,0 26,981535
14 Si 28 92,21 27,976927
29 4,70 28,976491
30 3,09 29,973761
15 P 31 100,0 30,973763
16 S 32 95,0064 31,972074
33 0,760 32,971460
34 4,22 33,967864
36 0,0136 35,967091
17 Cl 35 75,529 34,968854
37 24,471 36,965896
18 Ar 36 0,337 35,967548
38 0,063 37,962724
40 99,600 39,962384
Масс-спектрометрический метод анализа
881
Продолжение таблицы 7.15
Порядковый номер в периодической системе Элемент Массовое число Природная распространенность, % Относительная масса, а.е.м.
19 К 39 93,1082 38,963714
40 0,0118 39,964008
41 6,88 40,961835
20 Са 40 96,97 39,962589
42 0,64 41,958628
43 0,145 42,958780
44 2,06 43,955490
46 0,0033 45,953689
48 0,185 47,952363
21 Sc 45 100,0 44,955919
22 Ti 46 7,93 45,952633
47 7,28 46,951758
48 73,94 47,947948
49 5,51 48,947867
50 5,34 49,944789
23 V 50 0,24 49,947165
51 99,76 50,943978
24 Сг 50 4,31 49,946051
52 83,76 51,940514
53 9,55 52,940651
54 2,38 53,938879
25 Мп 55 100,0 54,938054
26 Fe 54 5,82 53,939621
56 91,66 55,934932
57 2,19 56,935394
58 0,33 57,933272
27 Со 59 100,0 58,933189
28 Ni 58 67,85 57,935342
60 26,22 59,930783
61 1,19 60,931049
62 3,66 61,928345
64 1,08 63,927959
29 Си 63 69,09 62,929594
65 30,91 64,927786
30 Zn 64 48,89 63,929145
66 27,81 65,926048
67 4,11 66,927149
68 18,57 67,924865
70 0,62 69,925348
31 Ga 69 60,4 68,925682
71 39,6 70,924840
Продолжение таблицы 7.15
Порядковый номер в периодической системе Элемент Массовое число Природная распространенность, % Относительная масса, а.е.м.
32 Ge 70 20,52 69,924277
72 27,43 71,921740
73 7,76 72,923360
74 36,54 73,921150
76 7,76 75,921360
33 As 75 100,0 74,921580
34 Sc 74 0,87 73,922450
76 9,02 75,919229
77 7,58 76,919934
78 23,52 77,917348
80 49,82 79,916512
82 9,19 81,916660
35 Br 79 50,537 78,918348
81 49,463 80,916344
36 Kr 78 0,354 77,920368
80 2,7 79,916388
82 11,56 81,913483
83 11,55 82,914131
84 56,90 83,911504
86 17,37 85,910617
37 Rb 85 72,15 84,911710
87 27,85 86,909180
38 Sr 84 0,56 83,913376
86 9,86 85,909260
87 7,02 86,908890
88 82,56 87,905610
39 V 89 100,0 88,905430
40 Zr 90 51,46 89,904320
91 11,23 90,905250
92 17,11 91,904590
94 17,40 93,906140
96 2,80 95,908200
41 Nb 93 100,0 92,906020
42 Mo 92 15,84 91,906290
94 9,04 93,904740
95 15,72 94,905720
96 16,53 95,904550
97 9,46 96,905750
98 23,78 97,905510
100 9,63 99,907570
882
Новый справочник химика и технолога
Продолжение таблицы 7.15
Порядковый номер в периодической системе Элемент Массовое число Природная распространенность, % Относительная масса, а. ем.
44 Ru 96 5,51 95,907600
98 1,87 97,905500
99 12,72 98,906080
100 12,62 99,903020
101 17,07 100,904120
102 31,61 101,903720
104 18,58 103,905530
45 Rh 103 100,0 102,904800
46 Pd 102 0,96 101,904940
104 10,97 103,903560
105 22,3 104,904640
106 27,33 105,903200
108 26,71 107,903920
ПО 11,81 109,904500
47 Ag 107 51,35 106,904970
109 48,65 108,904700
48 Cd 106 1,215 105,905950
108 0,875 107,904000
ПО 12,39 109,902970
111 12,75 110,904150
112 24,07 111,902840
113 12,26 112,904610
114 28,86 113,903570
116 7,58 115,905010
49 In 113 4,28 112,904280
115 95,72 114,904070
50 Sn 112 0,96 111,904940
114 0,66 113,902960
115 0,35 114,903530
116 14,30 115,902110
117 7,61 116,903060
118 24,03 117,901790
119 8,58 118,903390
120 32,85 119,902130
122 4,72 121,903410
124 5,94 123,905240
51 Sb 121 57,25 120,903750
123 42,75 122,904150
Продолжение таблицы 7.15
Порядковый номер в периодической системе Элемент Массовое число Природная распространенность, % Относительная масса, а.е.м.
52 Те 120 0,089 119,904510
122 2,46 121,903000
123 0,87 112,904180
124 4,61 123,902760
125 699 124,904420
126 18,71 125,903242
128 31,79 127,904710
130 34,48 129,906700
53 I 127 100,0 126,904352
54 Хе 124 0,096 123,906120
126 0,090 125,904169
128 1,919 127,903538
129 26,44 128,904784
130 4,08 129,903510
131 21,18 130,905087
132 26,89 131,904162
134 10,44 133,905398
136 8,87 135,907221
55 Cs 133 100,0 132,905090
56 Ва 130 0,101 129,906247
132 0,097 131,905120
134 2,42 133,904310
135 6,59 134,905570
136 7,81 135,904360
137 11,32 136,905560
138 71,66 137,905010
57 La 138 0,089 137,906810
139 99,911 138,906060
58 Се 136 0,193 135,907100
138 0,250 137,905720
140 88,48 139,905280
142 11,07 141,909040
59 Рг 141 100,0 140,907390
60 Nd 142 27,11 141,907478
143 12,17 142,909620
144 23,85 143,909900
145 8,30 144,912160
146 17,22 145,912690
148 5,73 147,916480
150 5,62 149,920710
Масс-спектрометрический метод анализа
883
Продолжение таблицы 7.15
Порядковый номер в периодической системе Элемент Массовое число Природная распространенность, % Относительная масса, а.е.м.
62 Sm 144 3,09 143,911650
147 14,97 146,914620
148 11,24 147,914560
149 13,83 148,916930
150 7,44 149917010
152 26,72 151,919490
154 22,71 153,922010
63 Eu 151 47,82 150,919630
153 52,18 152,920860
64 Gd 152 0,20 151,919530
154 2,15 153,920720
155 14,73 154,922590
156 20,47 155,922100
157 15,68 156,923940
158 24,87 157,924100
160 21,90 159,927120
65 Tb 159 100,0 158,924950
66 Dy 156 0,0524 155,923760
158 0,0902 157,923960
160 2,294 159,924830
161 18,88 160,926600
162 25,53 161,926470
163 24,97 162,928370
164 28,18 163,928830
67 Ho 165 100,0 164,930300
68 Er 162 0,136 161,928780
164 1,56 163,929290
166 33,41 165,930400
167 22,94 166,932050
168 27,07 167,932380
170 14,88 169,935510
69 Tm 169 100,0 168,934350
70 Yb 168 0,135 167,933900
170 3,03 169,934880
171 14,31 170,936460
172 21,82 171,936560
173 16,13 172,938300
174 31,84 173,939020
176 12,73 175,942740
71 Lu 175 97,41 174,940890
176 2,59 175,942740
Продолжение таблицы 7.15
Порядковый номер в периодической системе Элемент Массовое число Природная распространенность, % Относительная масса, а.е.м.
72 Hf 174 0,18 173,940260
176 5,20 175,941650
177 18,50 176,943480
178 27,14 177,943870
179 13,75 178,946020
180 35,24 179,946810
73 Та 180 0,0123 179,947520
181 99,9877 180,947980
74 W 180 0,135 179,946980
182 26,41 181,948270
183 14,40 182,950290
184 30,64 183,950990
186 28,41 185,954340
75 Re 185 37,07 184,953020
187 62,93 186,955960
76 Os 184 0,018 183,952560
186 1,59 185,953940
187 1,64 186,955960
188 13,3 187,955970
189 16,1 188,958250
190 26,4 189,958600
192 41,0 191,961410
77 Ir 191 37,3 190,960850
193 62,7 192,963280
78 Pt 190 0,0127 189,599950
192 0,78 191,961430
194 32,9 193,962810
195 33,8 194,964820
196 25,3 195,964981
198 7,21 197,967530
79 Au 197 100,0 196,966552
80 Hg 196 0,146 195,965822
198 10,02 197,966769
199 16,84 198,968256
200 23,13 199,968344
201 13,22 200,970315
202 29,80 201,970630
204 6,85 203,973482
81 Tl 203 29,50 202,972331
884
Новый справочник химика и технолога
Продолжение таблицы 7.15
Порядковый номер в периодической системе Элемент Массовое число Природная распространенность, % Относительная масса, а.е.м.
82 РЬ 205 70,50 204,974462
204 1,48 203,973069
206 23,6 205,974459
207 22,6 206,975898
208 52,3 207,976644
83 Bi 209 100,0 208,980417
90 Th 232 100,0 232,038211
92 и 234 0,0056 234,040900
235 0,7205 235,043933
238 99,2739 238,050760
7.4.3. Метод изотопного разбавления
Метод используется для определения как элементного состава проб, так и примесей 10-7—10-8% отдельных элементов в анализируемых пробах с погрешностью в пределах 1,5-2% [78].
Определяемое количество элемента оценивается по изменению изотопного состава пробы после добавления известного количества этого же элемента, но с отличным от анализируемой системы изотопным составом. Обычно определяемый элемент в пробе имеет хорошо известный природный изотопный состав, а добавляется тот же, называемый внутренним стандартом элемент, обогащенный в 1,5-2 раза по отдельным изотопам (или изотопу) по сравнению с природным соотношением. Для достижения полного изотопного обмена обычно достаточно прогреть раствор пробы при тщательном перемешивании, но иногда требуется провести цикл окислительно-восстановительных реакций. Из полученного раствора определяемый элемент выделяют методом экстракции и измеряют изотопный состав смеси на масс-спектрометре. В ряде случаев элемент не экстрагируют и для изотопного анализа берут аликвоту полученной смеси.
Рассмотрим общий подход к данной задаче [79]. Пусть необходимо определить в пробе неизвестное количество атомов какого-то элемента, имеющего три изотопа А, В и С с содержанием каждого в процентном соотношении: ап + + + сп = 100%. Добавим в пробу в качестве внутреннего
стандарта известное количество атомов этого же элемента с известным изотопным составом: + Лсг + сст = 100%. Измерим изотопный состав полученной смеси: асм + Ьск + + ссм= 100%. Баланс числа атомов каждого из изотопов дает систему уравнений:
&лНх ^ст Act ^см(Ах + Аст), (А)
6ПАХ + 6СГ Ncv = bCM(Nx + Аст), (В)
СпАх+сстАст Ccm(Nx “I" Act), (С)
где Nx и Act — количество атомов определяемого элемента в пробе и добавленном стандарте соответственно; а, Ь, с —
относительное содержание изотопов А, В и С определяемого элемента в пробе (оно обозначено индексом «п» , в стандарте — индексом «ст» и в смеси — индексом «см»), выраженное в атомных %.
Решая уравнение А относительно Ах, находим
Nx = . (718)
ап ~ асм
Из уравнений (А), (В) и (С) также следует, что
А =А и N = N Sm ~gCT. (719)
Сп~Ссм
Следовательно, имеется возможность проверить правильность определения неизвестного количества элемента в пробе по изменению содержания других изотопов в смеси.
От числа атомов можно перейти к весовому количеству определяемого элемента в пробе
где тх и тСТ — масса элемента в стандарте и анализируемой пробе соответственно; Мх и Мсх — средняя атомная масса элемента природного состава и стандарта.
Концентрацию определяемого элемента в пробе вычисляют в процентах по формуле
п=£к.100%, (7.21)
где т„ — масса пробы.
Если в масс-спектрометрических анализах результаты получены в виде отношения токов ионов отдельных масс элемента к току ионов какого-либо одного изотопа (К), то вычисление неизвестного количества элемента в пробе можно провести по следующей формуле:
Ч(я„-я„)Л+1) -----7------X-------> x(vu<mi)
п г, &СТ г, ^см
где Rn = т—, RCT = -г-, RCM = -т-отношение содержания
Оп ”ст ”см
двух изотопов определяемого элемента в пробе, стандарте и смеси соответственно.
Для повышения надежности определения содержания какого-либо элемента в пробе следует измерять изотопный состав не только в полученной смеси и стандарте, но и в пробе перед добавлением стандарта.
Рассмотренный метод позволяет определять содержание отдельных элементов в твердых веществах — минералах, сплавах, солях, полупроводниковых материалах, почвах, растениях, продуктах питания; в жидких средах, а также при анализе состава благородных газов [80] (например, примесей металлов в нефти, морской воде и грунтовых водах). В табл. 7.16 приведены пределы обнаружения ряда элементов при исследовании плазмы крови методом изотопного разбавления [98].
Масс-спектрометрический метод анализа
885
Таблица 7.16
Пределы обнаружения ряда элементов масс-спектрометрическим методом изотопного разбавления [98]
Изотоп Ожидаемая концентрация, нг • л-1 Предел обнаружения , нг
6Li 450 1
7Li 5550 2
67Zn 40 2
68Zn 190 4
70Zn 6 3
54Fe 60 8
57Fe 20 2
58Fe 3 1
63Cu 700 2
65Cu 300 1
74Se 1 3
77Se 8 4
82Se 9 3
79Br 1500 50
81Br 1500 200
Примечание* При расходе 10 мл образца на анализ
Масс-спектрометрический метод изотопного разбавления применим для элементов, имеющих два и более изотопа. Моноизотопные элементы 19F, 23Na, 27Al, ssMn, 59Co, 79As, 127I, 209Bi и др. (всего 17 изотопов) этим методом не определяются.
7.4.4. Хромато-масс-спектрометр (ХМС)
ХМС в режиме on line — новый самостоятельный метод анализа, обладающий аналитическими возможностями, которые превышают простую сумму возможностей обоих методов, входящих в комплекс [64]. В приборе роль хроматографа состоит в том, чтобы разделить исследуемую смесь на отдельные компоненты, а масс-спектрометр является детектором индивидуальных соединений. Анализ на ХМС применяется для идентификации и выявления в окружающей среде содержания тяжелых металлов и диоксинов [26], компонентов химического оружия и продуктов их разложения [4], для определения в биологических объектах супермалых количеств таких присутствующих в воздухе, воде и почве промышленных районов высокотоксичных веществ, как полихлорвиниловые дибензо-п-диоксины (ПХДД) и дибензофураны (ПХДФ) [41]. При анализе на ХМС изомеров ПХДД и ПХДФ удалось достичь супермалых пределов обнаружения (50-100 фг), благодаря применению источников ионов с отрицательной химической ионизацией. ХМС является незаменимым прибором для определения состава различных природных и искусственных ароматизаторов в пищевой промышленности и в медицине [66], для контроля содержания в питьевой воде, в алкогольных напитках и плазме крови таких вредных и опасных для здоровья веществ, как этиленгликоль и ди
этиленгликоль [6, 67, 68]. Показано, что предел обнаружения диэтилен-гликоля в жидких пробах с помощью ГХ-МС анализа составляет 0,2 мг л-1, а ГХ с плазменно-ионизационным детектором — 0,2 мг • л'1, т.е. чувствительность комплекса ГХ-МС анализа на порядок выше.
Как правило, в хромато-масс-спектрометрах используются серийный газовый (ГХ) или жидкостной (ЖХ) хроматографы, условия их работы идентичны: вид газа-наполнителя, его расход, параметры хроматографических колонок, выбор неподвижных фаз, параметры температурных программ. В ХМС применяются насадочные, но чаще более чувствительные капиллярные колонки, особенно когда анализируются следовые количества определяемых соединений.
Наибольшее распространение получили колонки из плавленого кварца, отличающиеся химической стойкостью и гибкостью. Сверху для защиты поверхности кварцевые колонки покрыты слоем полиамидного материала. Капиллярные колонки имеют малый внутренний диаметр и большую длину, например, диаметр 0,53 мм и длину 15 м [39]; диаметр 0,32 мм и длину 5 и 25 м [6]. На внутреннюю поверхность таких колонок наносят тонкие слои (десятки микрометров) прочно удерживаемых сшитых неподвижных фаз или фаз, привитых непосредственно к поверхности кварцевой колонки. Капиллярные колонки работают при более низкой температуре по сравнению с насадочными, чем снижается вероятность разложения компонентов анализируемой пробы.
Хроматографические газовые колонки
Могут соединяться с масс-спектрометром непосредственно или через интерфейс для сброса большей части газа-носителя. Интерфейс как соединительная часть между выходом хроматографической колонки и источником ионов МС в значительной мере определяет характеристики всей системы. При соединении МС с колонкой через эффузионный сепаратор (интерфейс) последний служит для обогащения исследуемой пробы по отношению к газу-носителю. За счет того, что молекулы газа-носителя обладают большей подвижностью по сравнению с тяжелыми органическими молекулами, они быстрее диффундируют через небольшие отверстия, имеющиеся в стенке сепаратора, в соседнюю откачиваемую камеру. При последовательном соединении двух сепараторов степень обогащения может достигать 100 и более раз.
В большинстве ГХ-МС поток газа-носителя, выходящего из колонки, уменьшают путем пропускания элюата через струйный сепаратор. Такой сепаратор состоит из откачиваемой камеры, в которой находятся на одной оси сопло и диафрагма (вблизи от сопла). Поток газа вырывается из сопла в область низкого давления в виде расширяющейся струи. Более тяжелые газы, благодаря большей инерции, проходят через отверстие, а легкие молекулы отклоняются и откачиваются. Обычно сепаратор двухступенчатый, и первая ступень откачивается до давления 10-20 Па, вторая — до 0,1 Па. Поток элюата, поступающего при этих условиях в источник ионов, уменьшается от 40 мл • мин-1 до 0,025 мл • мин1.
886
Новый справочник химика и технолога
В сепараторе с проницаемой мембраной элюат вводится в камеру, отделяемую от источника ионов МС тонкой (0,025-0,04 мм) мембраной из силиконовой резины, проницаемость которой в ~ 1 000 раз больше для органических молекул, чем для газа-носителя (Не, Н2). Мембранный сепаратор удобен тем, что после него поток газа содержит 10- 50% анализируемых веществ, однако он имеет существенные недостатки — различную проницаемость мембраны в зависимости от природы органического вещества и влияние температуры на проницаемость от элюата.
7.4.5. Жидкостной хромато-масс-спектрометр
(ЖХМС) как прибор в режиме on-line применяется с 1972 г. [57]. Анализ с помощью ЖХМС дает возможность разделять и идентифицировать высокомолекулярные термически нестабильные соединения, поскольку в нем не используются такие высокие температуры, как в ГХ.
Соединение колонок ЖХ с МС осуществляется с помощью различных интерфейсов:
— транспортный интерфейс, в котором элюат из хроматографической колонки переносится в источник ионов движущимся транспортером, причем на пути в источник путем испарения удаляется большая часть растворителя;
— интерфейс с непосредственным вводом жидкости в источник ионов (НВЖ);
— термоструйный интерфейс (ТС).
В интерфейсе с движущимся транспортером транспортер представляет собой ленту из нержавеющей стали или полиамидного полимера. Такой интерфейс способен принять на ленту и испарить поток органического растворителя или водного раствора со скоростью 0,1-0,2 мл • мин4. При этом в источник ионов поступает всего 10 7-10“8 г • с4 растворителя с исследуемым соединением, следовательно, при анализе можно использовать любые методы ионизации (ЭУ, ХИ и десорбционные) [71-75]. К недостаткам такого интерфейса относятся: наличие памяти, неравномерность нанесения пробы и, иногда, частичное разложение термически нестабильных соединений в пробе.
Интерфейс с НВЖ прост и удобен для ввода в МС элюата из микроколонок с малым расходом подвижной фазы (30-150 мкг • мин-1). Такой интерфейс очень удобен, когда пары подвижной фазы одновременно служат газом-реагентом для ХИ. Недостатками интерфейса НВЖ являются: нестабильность, частые засорения и плохая теплопередача в вакууме при испарении растворителя из капель (требуется дополнительное нагревание).
ТС-интерфейсы работают по принципу распыления поступающего из колонки элюата при проходе его через нагретый металлический капилляр, из которого он выходит в виде сверхзвукового потока мелких капель. Если к элюату добавить электролит, например ацетат аммония, то ионы образуются без какого-либо другого внешнего воздействия (термоструйная ионизация). Этот метод позволяет ионизировать без разложения крайне нелетучие соединения. При термоструйной ионизации фрагментация, как правило, очень мала. Иногда при необходимости для повышения
степени ионизации либо увеличивают температуру капилляра, либо используют дополнительный источник ионизации, например ЭУ и электрический разряд. Усовершенствований ТС-интерфейс состоит из генератора моно дисперсного аэрозоля, десольватационной камеры и сепаратора [76]. Генератор создает струю аэрозоля путем пропускания жидкого элюата из хроматографической колонки через стеклянную диафрагму с малым отверстием, при этом поток разбивается на однородные капли, увлекаемые потоком газа-носителя, который вводится под прямым углом к потоку жидкости. При диаметре отверстия около 10 мкм образуются капли размером ~ 20 мкм, а оптимальный расход жидкости составляет 0,1-0,5 мл • мин4.
Стеклянная десольватационная камера, куда поступает аэрозоль, имеет длину 30 см и диаметр 40 мм, она обеспечивает испарение растворителя при атмосферном давлении. Далее аэрозоль с парами растворителя поступает в двухступенчатый сепаратор для отделения растворителя от газа-носителя. При использовании такого интерфейса с генератором аэрозоля и ионизацией ЭУ на масс-спектрометре Varian МАТ112S получены пределы обнаружения: 10 нг для низкомолекулярных и 1 нг для высокомолекулярных соединений. Разработаны и другие конструкции интерфейсов с генерированием струи аэрозоля [77].
Система ЖХМС-анализа более активно начала применяться с появлением интерфейса ЭРИАД (экстракция ионов из раствора при атмосферном давлении) [58]. Суть действия такого интерфейса состоит в том, что под влиянием неоднородного электрического поля высокой напряженности при атмосферном давлении инертного газа происходит распыление раствора, содержащего анализируемые молекулы. При этом часть молекул ионизируется, ионы формируются в пучок и подаются в анализатор ионов (см. рис. 7.5).
Идентификация индивидуальных веществ при использовании ХМС-анализа проводится по времени удержания в хроматографической колонке в том числе и традиционным методом сравнения полученных масс-спектров со спектрами стандартных соединений. Предварительно хроматографические колонки и масс-спектрометр калибруются путем анализа образцов, содержащих исследуемое вещество в смеси с другими известными соединениями. В исследуемые пробы перед анализом обычно добавляют внутренние стандарты с известным временем удержания, масс-спектры которых не перекрываются со спектрами исследуемых соединений. Используются и дополнительные нетрадиционные критерии идентификации органических соединений, связанные с термодинамическими свойствами соединений, обработкой масс-спектров и т.д. [4, 69].
Литература
1. Архипов Д.Б., Галь Л.Н. Современное состояние методологии молекулярной масс-спектрометрии // Журн. аналит. химии. 1999. Т. 54, № 6. С. 585-592.
2. Эпов В.Н., Васильева И.Е., Сутурин А.Н. и др. Определение микроэлементов в Байкальской воде методом масс-спектрометрии с индуктивно-свя-
Масс-спектрометрический метод анализа
887
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
занной плазмой // Журн. аналит. химии. 1999. Т. 54, 14.
№ 11. С. 1170-1176.
Чиванов В.Д., Гребеник Л.И., Еременко В.А. и др.
Обнаружение папаверина в биологических жидкостях с использованием плазменно-десорбционной 15. масс-спектрометрии с ионизацией осколками деления И Журн. аналит. химии 1997. Т. 52, № 9.
С. 992-997.
Бродский Е.С., Киреев А.Ф. Применение масс-хроматограмм по характеристическим ионам и 16. разностям масс-ионов для идентификации и определения компонентов химического оружия и продуктов их разложения // Журн. аналит. химии.
1997. Т. 52, № 8. С. 884-888. 17.
Чиванов В.Д., Гребеник Л.И., Баранова В.М. и др.
Экспресс-обнаружение антибиотиков в мясопродуктах методом времяпролетной плазменно-де- 18
сорбционной масс-спектрометрии И Журн. аналит.
химии. 1997. Т. 52, № 10. С. 1105-1109.
Савчук С.А., Бродский Е.С., Формановский А.А. 19.
Газохроматографическое и хромато-масс-спектро-метрическое определение гликолей в питьевой воде и спиртных напитках И Журн. аналит. химии.
1999. Т. 54, № 8. С. 836-847.
Crain J.S., Mikesell B.L. Detection of Sub-ng/L Acti- 20. nides in Industrial Wastewater Matrices by Inductively Coupled Plasma-mass-spectrometry H Appl.
Spectrosc. 1992. V. 46, № 10. P. 1498-1502.
Barefoot R.R. Distributions and Speciation of Plati- 21. num Group Elements in Environmental Matrices П
Anal. Chem.Ref. Ed. 1999. V. 18. P. 702-707.
Guzowski J.P., Hieftje G.M. Gas Sampling Glow Discharge: a Versatile Ionization Source for Gas Chromatography Time-of-flight Mass Spectrometry H Anal.
Chem. 2000. V. 72, № 16. P. 3812-3820. 22.
Nilsson C.L., Larsson T., Gustafsson E et al. Identification of Protein Vaccine Candidates From Helicobacter Pylori Using a Preparative Two-dimensional Electrophoretic Procedure and Mass Spectrometry. H Anal. Chem 2000. V. 72, № 9. P. 2148-2153.
Koomen J.M.,Russell W.K.,Hettick J.M. et al. Improvement of Resolution, Mass accuracy, and Repro- 23. ducibility in Reflected mode DE-MALDI TOF Analysis of DNA Using Fast Evaporation-overlay er Sample Preparations 11 Anal. Chem 2000. V. 72, № 16.
P. 3860-3866.
Jones D.,Chapman J.,Ryan P. High Resolution GD- 24.
MS For the Qualitative and Quantative Analysis of All Elements in a Single Experiment: [Pap.] 40th ASMS Conf. Mass Spectrom and Allied Top., Washington D.C., May 31- June 5, 1992 // ICP Inf. News- 25. left. 1993. T. 18, № 9. P. 563-564.
Gupta A.K., Raza S.K., Dubey D.K. et al. Mass Spectral Studies on Synthetic Analogues of Organophosphorus Toxin Isolated From Ptychodiscus Brevis.// Phosph, Sulfur and Silicon and Kelat. Elem. 2000. 26.
V. 156. P. 203-212.
Tu Ya-Ping, Holmes John Li. The Chemistry of Solvated Distonicions: Peparation, Isomerization, and Fragmentation H. J. Amer. Chem. Soc. 2000. V. 122, № 23. P. 5597-5602.
Feichtinger Derec, Plattner Dietmar A. Oxygen Transfer to Manganese — Salen Complexes: on Electrospray Tandem Mass Spectrometric Study // J. Amer. Chem. Soc. Perkin Trans. 2000. V. 2, № 5. P. 1023-1028.
Michael L., Brauman Ohn I. Hydrogen-banded Complexes of Methanol and Acetylides. Structure and Energy Correlation. Chabinuc, Brauman Ohn 11 I J. Amer. Chem. Soc. 2000. V. 122, № 22. P. 5371-95378.
Nagao Satoshi, Kato Akiko, Nakajima Atsushi. Multiple-decker Sandvich Polyferrocene Ciasters // J. Amer. Chem.Soc. 2000. V. 122, № 17. P. 4221^222.
Масс-спектрометрия и химическая кинетика. (Сб. статей. Отв. ред. В.Л. Тальрозе. АН. Ин-т хим. физики). М.: Наука. 1985. 344 с.
Yasumoto Takeshi, Igarashi Tomoji, Legrand Anne-Marie et al. Structural Elucidation of Cigutoxin Cougeners by Fast-atom Bombardment Tandem Mass spectroscopy 11 J. Amer. Chem. Soc. 2000, V. 122, № 20. P. 4988^989.
Zhang Wenzhu, Chait Brian T. Profound: An Expert System for Protein Identification Using Mass spec-trometie Peptide Mapping Information H Anal. Chem. 2000. V. 11, № 72. P. 2482-2489.
Saraswati Raj ananda, Coving fon John ICP-MS Analysis of Sub-parts Per Trillon Trace Impurities in Ultra High Purity Chemicals Using in Semiconductor Industry: Abstr. Pittcon 2000. New Orleans. La March 12-17, 2000 11 ICP Inf. Newslett. 2000. V. 25, № 12. P. 939-940.
Watson C., Barshick C., Wronka L. et al. Glow Discharge Ionization on a High Mass Resolution External ion Source. Fourier Transform Ion Cyclotron Resonance Mass Spectrometer: [Pap.], 40th ASMS Conf. Mass Spectrom and Allied Top., Washington D.C., May 31 - June 5, 1992 // ICP Inf. Newslett. 1993. V. 18, №9. P. 561-562.
Witte H.De, Gendt S.De., Douglas M. et al. Evolution of Time-of-flight Secondary Ion Mass Spectrometry for Metal Contamination Monitoring on Si Wafer Surfaces // J. Electrochem. Soc. 2000. V. 147, № 5. P. 1915-1917.
Belu Anna M., Davies Martyn C., Newton J. Mike et al. TOF-SIMS Characterization and Imaging of Con-trolled-release Drug Delivery Systems H Anal. Chem. 2000. V. 72, № 22. P. 5625-5638.
Lazar Alexandru C., Ramsey J. Michael. Laser Desorption in Situ Chemical Ionization Aerosol Mass Spectrometry for Monitoring Tributyl Phosphate on the Surface of Environmental Particles. 11 Anal. Chem. 2000. V. 72, № 9. P. 2142-2147.
Семенов С.Ю., Зыкова Г.В., Смирнов B.H. и др. Оценка уровня загрязнения суперэкотоксикантами
888
Новый справочник химика и технолога
бассейна Средней и Нижней Волги // Анализ объектов окружающей среды: Тез. докл. 4-й Всероссийской конф. «Экоаналитика - 2000». Краснодар 41. 2000. С. 66-67.
27. Demirev Р., Westman A., Reimann С.Т. et al. Matrix-assisted Laser Desorption With Ultra-short Laser
Pulses // Rapid Commun. Mass Spectrom. 1992.V. 6, №3. P. 187-191.
28. Carbonell Silvia Albero, de Aquino Neto, Francisco Radler et al. Rapid Screening of Natural Products by 42. High-resolution High-temperature Gas Chromatography 43. // J. Chromatogr. Sci. 2000. V. 38, № 6. P. 234-240.
29. Zhao Jianguo, Leibman D.M. Detection at Liquid Injection Using on Atmospheric Pressure Ionization 44. Radio Frequency Plasma Source // Anal. Chem 1993. 45.
V. 65, № 7. P. 866-878.
30. Myers D., Hiefic G. Design at a Time-of-flight Mass 46. Spectrometer for a Plasma Source: [Pap.] 40th ASMS Conf. Mass Spectrom and Allied Top., Washington 47. D.C., May 31-June 5, 1992 // ICP Inf. Newslett. 1993.
V. 18, №9. P.557-558. 48.
31. Yang Chenglong, Mohill Maffhew L., Harrison W.W. Grim-type Glow Discharge Ion Source For Time-of-flight Mass Spectrometry: Abstr. Winter Conf. On 49.
Plasma Spectrochemistry, Fort Landerdale, Fla Jan. 10-15, 2000. // ICP Inf. Newslett. 2000. V. 26, № 4. P. 301-302.
32. Jakubowski N., Abell Ian Tanner Scott D., Hamester 50. M et al. New Plasma Source Mass Spectrometers: What are the Challenges for Novel Instrumentation in the Next Millenium? Abstr. Winter Conference on 51. Plasma. Spectrochemistry, Fort Landerdale Fla, Jan.
10-15, 2000 // ICP Inf. Newslett. 2000. V. 26, № 4.
P. 314-322.
33. Chaderi S., Kulkami P.S., Ledford E.B. et al. Chemical Ionization in Fourier Transform Mass Spectrometry // Anal. Chem. 1981. V. 53. P. 428.
34. Daugherty R.C. Negative Chemical Ionization Mass 52. Spectrometry // Anal. Chem. 1981. V. 53. P. 625A.
35. Comisarow M.B., Marshall A.G. Resolution-enhanced Fouriertransform Ion Ciclotron Resonance Spectros- 53. copy // J. Chem. Phys., 1975. V. 62. P. 293.
36. Юинг Г. Инструментальные методы химического анализа. М.: Мир, 1989. 608 с. 54.
37. Полякова А.А., Хмельницкий Р.А.. Масс-спектро-
метрия в органической химии. Л.: Химия, 1972. 368 с.
38. Полякова А.А., Ревельский И.А., Токарев М.Н. и 55. др. Масс-спектральный анализ смесей с применением ионно-молекулярных реакций. Под ред.
А.А. Поляковой. М.: Химия, 1989. 240 с.
39. Лийв М.Э., Мюрисепп А.М., Грюпер Э.Г. Изуче- 56. ние состава этакольного экстракта натурального мумие методом хромато-масс-спектрометрии // Журн. аналит. химии. 1994. Т. 49, № 3. С. 315-318.
40. Recommended operating procedures for sampling and 57. analysis in the verification of chemical disarmament. /
Самсонов Д.П., Кирюхин В.П., Жирюхина Н.П. Хромато-масс-спектрометрическое определение содержания полихлорированных дибензо-и-диок-синов и дибензофуранов в биологических объектах методом химической ионизации с детектированием отрицательных ионов // Журн. аналит. химии, 1994. V. 49, № 8. С. 868-973.
Levsen К. // Org. mass spectrom. 1988, V. 23. 406 р.
Beckey H.D. Principles of Field Ionization and Field Desorbtion Mass Spectrometry. Oxford: Pergamon Press, 1977. 338 p.
Rollgen F.W. // Trends in Anal. Chem. 1982. V. 1. 304 p.
Cochran R.L. // Appl. Spectrosc.Rev. 1986. V. 22. 137 p.
Barber M., Bordoli R.S., Sedgwick D. et al. // J. Chem. Soc. Chem. Common. 1981. P. 325.
Быковский Ю.А., Неволин B.H.. Лазерная масс-спектрометрия. М.: Энергоиздат, 1985. 325 с.
Чупахин М.С., Крючкова О.Н., Рамендик Т.Н. Аналитические возможности искровой масс-спектрометрии. М.: Атомиздат, 1972. 224 с.
Сопрыкин А.И. Повышение эффективности источников ионов радиочастотного тлеющего разряда наложением постоянного магнитного поля И Журн. аналит. химии. 1999. Т. 54, № 7. С. 758-768. Fucsko J., Liloyd A. Cool Plasma Perfomance Axial TOF - ICP-MS and Simultaneous Advantages // ICP Inf. Newslett 2000. V. 25, № 12. P. 937-938.
Ghazi A.M., Durrance E.M., Sandness K.L. et al. ICP/MS Determination of Pb and Pb Isotope Ratios in Bones and Artifaction a Native American Tribe; a Pb Poisoning and Pb Source Study: [Pap.] 40th ASMS Conf. Mass Spectrom and Allied Top., Washington, D.C., May 31-June 5, 1992 // ICP Inf. Newslett. 1993. V. 18, № 9. P. 571-572.
Greenfield S., McGlachin H.D., Smith P.B. Plasma Emission Source in Analytical Spectroscopy. Ill // Taianta. 1976. V. 23, № 1. P. 1-11.
Wendt R.H., Fassel V.A. Induction - Coupled Plasma Spectrometric Excitation Source // Anal. Chem. 1965. V. 37. P. 920-924.
Брицке М.Э., Сукач Ю.С., Филимонов Л.Н. Индукционный ВЧ-разряд и его применение в спектральном разряде // Журн. прикл. спектроскопии. 1976. Т. 25, вып. 1. С. 5-11.
Gray A.L. Inductively Coupled Plasma as an Ion Source for Aqueous Solution Samples, Dynamic Mass Spectrometry. V. 6 / Ed. Price D., Told J. London, 1981. P. 251-266.
Katsuo A., Kentaro Y., Tsuneo I. Electrospray Ionization Mass Spectrometric Observation of the Coordina-tive Interaction of a, 0 - unsaturated Ketones With Cerium III Cation. // Chem. Lett. 2000. № 4. P. 424-425.
Тальрозе В.Л., Скурат B.E., Городецкий И.Г. Стеклянные кварцевые натекатели для капиллярной
Масс-спектрометрический метод анализа
889
системы напуска жидкостей в масс-спектрометр // 80.
Журн. физич. химии. 1972. Т. 46, № 3. С. 788-791.
58. Александров М.Л., Галль Л.Н., Краснов Н.В. и др. Экстракция ионов из растворов при атмосферном 81. давлении — метод масс-спектрометрического анализа биологических веществ // ДАН СССР. 1984.
Т. 77, №2. С. 379-383. 82.
59. Whitehouse C.V., Dreyer R.N., Yamashita М. et al. // Anal. Chem. 1985. V. 57, № 3. P.675-680.
60. Gapeev A., Dunbar R.C. Binding of Alkaline Earth 83. Halide Ions MX+, to Benzene and Mesitylene // J. Phys. Chem. A. 2000. V. 104, № 17. P. 4084-4088.
61. Полякова А.А.. Молекулярный масс-спектральный 84. анализ нефтей. М.: Недра, 1973. 184 с.
62. Karas М., Hilenkamp F. // Anal.Chem. 1988. V. 60. 85.
№ 20. Р. 2299-2305.
63. Бойко В.А., Крохин О.Н., Склизнов Г.В. Исследование параметров и динамики лазерной плазмы при острой фокусировке излучения на твердую мишень // Труды Физ. института АНСССР. 1974. 86.
Т. 52. С. 186-228.
64. Хмельницкий Р.А., Бродский Е.С. Хромато-масс-спектрометрия. М.: Химия, 1904. 216 с.
65. Гуревич А.Л., Русинов Л.А., Могильницкий А.М. 87. и др. Автоматизация обработки масс-спектрометрической информации. М.: Энергия, 1978. 184 с.
66. Машарина Т.А., Головня Р.В., Стражненко Е.С. и 88. др. Сорбционно-структурные и масс-спектрометрические характеристики летучих компонентов 89. модельных систем и ароматизаторов с запахом мяса // Журн. аналит. химии. 1994. Т. 49, № 7.
С. 722-728.
67. Laurence J.F., Chadha R.K., Lan B.P.Y. et al. // 90.
J. Chromatogr. 1986. V. 367. 213 P.
68. Brooks J.B., Basta M.T., Alley C.C. et al. // J. Chromatogr. 1986. V. 309. 269 P. 91.
69. Зинкевич И.Г. Нетрадиционные критерии хроматографической и хромато-масс-спектрометрической идентификации органических соеди- 92. нений // Журн. аналит. химии. 1998. Т. 53, № 8.
С. 828-835.
70. Levsen К. // Org. Mass Spectrom. 1988. V. 23. 406 р. 93.
71. Me Fadden W.H. // J. Chromatogr. Sci. 1980. V. 18.97 p.
72. Stout S.I., da Curha A.R. // Anal. Chem. 1985. V. 57.
1783 p.
73. Voyksener R.D., Bursey J.T. // Anal. Chem. 1984. 94.
V. 56. P. 1582.
74. Aptino P.J. // J. Chromatogr. 1985. V. 323. P. 3.
75. Coley T.R. // Anal. Chem. 1986. V. 58. P. 1451 A.
76. Willoughby R.C., Browner R.F. // Anal. Chem. 1984. 95.
V. 56. P. 2624.
77. Winkles R.F. // Anal. Chem. 1988. V. 60. P. 489.
78. Миллер Ю.М., Чупахин М.С. Журн. аналит. хи- 96. мии. 1968. Т. 23. С. 833.
79. Метод изотопного разбавления (внутреннего стан- 97. дарта) // Труды Радиевого института. 1956. Том VII, вып. 1. 128 с.
Adams F., Gijbels R., Van R. Grieken. Inorganic Mass Spectrometry. University of Antwerp. Department of Chemistry. Antwerpen: Wirijk,1988. 404 p. Справочная литература по молекулярной спектроскопии / Под ред. В.А. Коптюга. Новосибирск: Ин-т органической химии СО АН СССР. 1974. 338 с.
Eight Peak Index of Mass Spectra // Imperial Chemical Industries Ltd., Aldermaston, England, Mass Spectrometry Data Centre. 1970.
A.Comu, R.Massot. Compilation of Mass Spectral Data // Heyden and Son in Cooperation With Presses Universitaires de France. 1966-1968.
Index of Mass Spectral Data // American Sosiety for Testing and Materials. Philadelphia. 1963.
Берендяев Б.Г., Покровский Л.М., Нехорошее С.А.и др. Машинная информационно-поисковая система для масс-спектрометрии // Изв. СО АН СССР. Сер. хим. наук. 1977. № 4, вып. 2. С. 109— 115.
Берендяев Б.Г., Коптюг В.А., Лебедев К.С. и др. Машинная информационно-поисковая система на базе каталога полных масс-спектров // Автометрия. 1979. № 4. С. 3-13.
Heller S.R., Milne G.W.A., Feldmann R.J. A Computer-based Chemical Information System. // Science. 1977. V. 195, № 4275. P. 253-259.
Бейнон Дж. Масс-спектрометрия и ее применение в органической химии. М.: Мир, 1964.
Будзикевич Г., Джерасси К., Уильям Д. Интерпретация масс-спектров органических соединений. Пер. с англ. / Под ред. Н.С. Вульфсона М.: Мир, 1966. 323 с.
Gray A.L. Inductinely Coupled Plasma Source Mass Spectrometry, in Inorganic Mass Spectrometry. New York: Wily etc. 1988.
Date A.R., Dray A.L. Applications of Inductively Coupled Plasma // Mass Spectrometry. Glasgow: Blackie, Ltd. 1989.
Tan S.H., Horlick G. Background Spectral Features in Inductively Coupled Plasma / Mass Spectrometry // Appl. Spectrosc. 1986. V. 40. P. 445-460.
Russ P.G., Bazan J.M. Isotope Ratio Measurements With an Inductively Coupled Plasma Source Mass Spectrometer // Spectrochim. Acta, 1987. V. 42B. P. 49-62.
Jarvis K.E. Applications of ICP-MC in the Earth Sciences. In: Applications of Inductively Coupled Plasma // Mass Spectrometry / Eds. Date A.R. London: Blackie and Son etc. 1989. P. 43-70.
Skogerboe R.K., Grant C.L. Detection Limits and Sensitivities for Spectroscopic Methods // Spectrosc. Lett. 1970. № 3. P. 215-219.
Гордон А., Форд P. Спутник химика. M.: Мир, 1976. 544 с.
Montaser A., Golightly D.W. Inductively Coupled Plasmas in Analytical Atomic Spectrometry. New York: Second Edition, VCH Publishers, Inc, 1992.
890
Новый справочник химика и технолога
98. Schmidt H.L., Foerstel Н., Heinziger К. Stable Isotopes. New York: Elsevier, 1982.
99. Справочник химика. T. 4. Л. , М.: Химия, 1985. 919 с.
100. Adams F.,Gijbels a. Van Grieken R/Eds./ Inorganic Mass Spectrometry. New York.: Wiley, 1988.
101. Date A.R. and Gray A.L. /Eds./ Applications of Inductively Coupled Plasma Mass Spectrometry. Glasgow: Blackie, 1989.
102. Chemical Analisis: Ser. of monogr. on Analyt. Chemistry a. its Applications. New York: Wiley, Vol. 95:
Inorganic Mass Spectrometry / Eds. by F.Adams et al./1988.
103. Holland G. and Eaton A.N. /Eds./ Applications of Plasma Source Mass Spectrometry. Cambridge: RSC, 1991.
104. Jarvis K.E, Gray A.L. a. Houk R.S. Handbook of Inductively Coupled Plasma Mass Spectrometry. Cambridge: RSC, 1991.
105. Jarvis K.E, Gray A.L, Williams J.L. a. Jarvis I. Plasma Source Mass Spectrometry. Cambridge: RSC, 1990.
Раздел 8
ГАЗОВЫЙ АНАЛИЗ
Авторы-составители: проф., д.т.н. В. М. Немец,
к.т.н. А. А. Соловьев
8.1. Определение газового анализа
Газовый анализ (ГА) — направление в аналитической химии, использующее методы определения газовых компонентов и газообразующих элементов в газах, жидкой и твердой фаз в веществах, а также дисперсной фазы в газах.
8.2. Анализируемые объекты и определяемые компоненты в газовом анализе
В роли анализируемых объектов (АО) могут быть газы (чистые газы, газовые смеси, технологические, органические, неорганические и природные газы), жидкости (вода и водные растворы различных веществ, органические жидкости, электролиты), твердые вещества (металлы, сплавы, минералы, полупроводниковые, диэлектрические, органические материалы). Определяемыми компонентами могут быть: газовые примеси в газовых смесях, чистых газах; газовые включения в жидких и твердых веществах; газообразующие примеси (элементы) в газах, жидкостях и твердых веществах; аэрозольные и радиоактивные вещества и частицы в газах.
8.3. Задачи, решаемые с помощью газового анализа
• Контроль технологии производства газов и газовых смесей.
• Входной контроль состава газов и газовых смесей при их применении.
• Аналитическое обеспечение решения экологических и санитарных задач, а также задач поисковой геологии (атомхимические методы ГА).
• Контроль степени и состава газового насыщения жидкостей различной природы и назначения.
• Контроль уровня и состава газосодержания твердых веществ и материалов при проведении научных исследований и в промышленных условиях.
8.4. Основные направления развития газового анализа
• Анализ газов высокой чистоты.
• Анализ сложных газовых смесей.
• Анализ неорганических газов.
• Анализ органических газов.
• Анализ реакционно-активных газов.
• Определение количества и состава дисперсных включений в газах.
• Лабораторный анализ.
• Анализ на технологических линиях.
• Дистанционный анализ.
8.5. Величины, характеризующие химический состав газов
Наименования и определения величин, характеризующих химический состав вещества, установлены международным стандартом (ИСО 31/8-80) «Величины и единицы физической химии и молекулярной физики». Величины, наиболее распространенные в ГА, приведены в таблице 8.1.
Таблица 8.1
Величины, применяемые для количественного описания химического состава в ГА
Величина Определяющее уравнение Формула размерности Обозначение
1. Массовая концентрация компонента В V MIL3 кг/м3, г/м3, мг/м3, мг/дм3
2. Массовая доля компонента В т„ ™в=~ т — %, млн'1
3. Объемная доля компонента В VB <р = — V — %, млн-1
4. Молярная концентрация компонента В Св= — в V N/L3 моль/м3
5. Молярная доля компонента В хв=-п — %, млн-1
892
Новый справочник химика и технолога
Примечания:
1. В таблице приняты следующие обозначения: тв — масса компонента В в смеси; V— объем смеси; т - масса смеси; Ув— объем компонента В в смеси; пв — количество вещества компонента В; п — количество вещества смеси; NB — число молекул (атомов) компонента В; М — масса; L —длина; N — число молей.
2. Следует отметить особенности расчета величины wB в случаях анализа веществ конденсированной фазы:
1) расчет содержания газа осуществляется по формуле
где Го— объем газа при О °C и 760 мм рт. ст.; d— плотность газа при 0 °C и 760 мм рт. ст. (кг/м3); а — масса навески анализируемого вещества (г);
2) расчет содержания определяемого вещества (В) проводится по формуле
где f — коэффициент пересчета (масса определяемого вещества в граммах, соответствующая 1 дм3 образовавшегося газа, приведенного к 0 °C и 760 мм рт. ст.), значения которого приведены в табл. 8.2.
Термин «концентрация» в том смысле, в котором он употреблен в Политехническом словаре 1980 г. и в Физическом энциклопедическом словаре 1983 г., следует понимать как обобщенное наименование величины, количественно характеризующей присутствие определяемого компонента в анализируемом веществе (в отличие от конкретного, но иного смысла этого термина в позициях 1, 4 и 6 табл. 8.1). Можно избежать путаницы в толковании термина «концентрация» (обобщенное или конкретное), если в качестве обобщенной количественной характеристики присутствия определяемого компонента в анализируемом веществе использовать не термин «концентрация», а термин «содержание», как это рекомендовано в ГОСТ 8.505-84 «ГСП: Метрологическая аттестация методик выполнения измерений содержаний компонентов проб, веществ и материалов».
В табл. 8.3. приведены значения коэффициента f при различных значениях температуры и давления, используемых в определениях содержания углерода (С) с выделением диоксида углерода; в табл. 8.4 — соотношения, необходимые для пересчета содержаний газов и паров при переходе от одной размерности к другой.
Таблица 8.2
Значения коэффициента f при 0 °C и 760 мм рт. ст.
В основу приведенных данных положены экспериментально найденные плотности соответствующих газов.
Образовавшийся газ Определяемое в-во f 1g/
со2 C 0,5391 ' -1,73169
со2 CO2’ 2,6955 0,43064
со2 MgCO3 3,7879 0,57840
со2 CaCO3 4,4968 0,65290
С2н2 CaC2 2,8850 0,45018
С2н2 H2O 0,8109 -1,90897
Н2 Al 0,8015 -1,90391
Н2 Fe 2,4899 0,39614
Н2 Zn 2,9145 0,46456
H2S H2S 1,5393 0,18732
n2 n2 1,250 0,09709
n2 n2* 1,2818 0,10782
n2 NH3 1,5200 0,18184
n2 NH3* 1,5582 0,19263
n2 CO(NH2)2 2,6806 0,42824
NO n2 0,6256 -1,79633
NO NO 1,3402 0,12717
NO N2O3 1,6974 0,22979
NO n2o5 2,4120 0,38239
NO NO3 2,7690 0,44238
NO HNO3 2,8143 0,44937
NO NaNO3 3,7963 0,57936
NO KNO3 4,5152 0,65468
NO NaNO2 3,0818 0,48880
NO kno2 3,8008 0,57987
NO nh4no3** 3,5748 0,55325
o2 o2 1,4289 0,15500
o2 of 0,7145 -1,85397
o2 H2O2~** 3,0379 0,48257
o2 H2O2***‘* 1,5189 0,18154
o2 Na2O2 6,9650 0,84294
o2 MnO2*** 3,8817 0,58902
o2 KMnO4*** 2,8230 0,45065
SiF4 f2 3,4280 0,53408
SiF4 CaF2 7,0439 0,84774
В азотометрических методах с бромощелочными растворами.
В нитрометрическом методе.
При реакции с Н2О2.
Каталитическое разложение.
Обработка КМпО4.
Газовый анализ
893
Таблица 8.3
Значения коэффициента f при различных значениях температуры и давления
Темпе-ратура газа, °C Давление, мм рт. ст.
730 735 740 745 750 755 760 765 770 775
10 0,0500 0,0503 0,0507 0,0510 0,0513 0,0517 0,0521 0,0524 0,0527 0,0530
12 0,0496 0,0500 0,0503 0,0506 0,0510 0,0513 0,0517 0,0520 0,0523 0,0527
14 0,0493 0,0496 0,0499 0,0503 0,0506 0,0510 0,0513 0,0516 0,0520 0,0523
16 0,0489 0,0493 0,0496 0,0499 0,0503 0,0506 0,0509 0,0513 0,0516 0,0519
18 0,0486 0,0489 0,0493 0,0496 0,0499 0,0503 0,0506 0,0509 0,0513 0,0516
20 0,0483 0,0486 0,0489 0,0492 0,0496 0,0499 0,0502 0,0506 0,0509 0,0512
22 0,0479 0,0482 0,0486 0,0489 0,0493 0,0496 0,0499 0,0502 0,0506 0,0509
24 0,0476 0,0479 0,0483 0,0486 0,0489 0,0492 0,0496 0,0499 0,0503 0,0505
26 0,0473 0,0476 0,0479 0,0483 0,0486 0,0489 0,0492 0,0496 0,0499 0,0502
28 0,0470 0,0473 0,0476 0,0479 0,0483 0,0486 0,0489 0,0492 0,0495 0,0499
Пример пользования таблицей 8.4.
Дано: а = 10 об. %, Т= 293 К, р - 770 мм рт. ст. Требуется найти х — значение концентрации, выраженное в моль/дм3.
В первой графе находим горизонтальную строку, соответствующую заданным единицам концентрации (строка 4), а в этой строке — вертикальный столбец, соответствующий требуемым единицам концентрации (моль/дм3). На пересечении строки 4 и вертикального столбца находим формулу, по которой определяем значение концентрации в требуемых единицах:
16,04-10-5 ар 16,04-10 5-10-770 . пп.. . 3
х =----------£- = —’------------= 0,0042 моль/дм .
Т 293
Этой же задаче служит номограмма, представленная на рис. 8.1.
Для пересчета находят на графике отрезок радиальной прямой, соответствующий данному газу (или радиальную прямую, соответствующую молярной массе газа М), и при помощи линейки соединяют его с нулевой точкой. Ордината каждой точки полученной прямой выражает концентрацию газа в об. %, а абсцисса — соответствующую величину в г/м3.
При отсутствии на графике радиальной линии молярной массы искомого газа находят любую точку этой линии по одной из следующих формул:
е = 0,1604^ (г/м3);
СТ
V = 6,236— (об. %).
Мр
Соединив найденную точку с нулевой точкой графика, получают прямую, которая и служит для пересчета.
На графике значения V даны только до 1,1, а значения с — до 16. Если заданная концентрация выше этих величин или ее абсолютное значение очень мало, то по графику
находят значение меньшее (большее) в К 10” раз (где К= 1, 2, 3, ..., 9, а п = 0 или ±1, 2, 3...), а затем найденную величину увеличивают (уменьшают) в К • 10" раз.
Рис. 8.1. Номограмма для пересчета концентрации газов и паров
Объем Fq для расчета результата ГА (объем сухого газа при 0 °C и 760 мм рт. ст.) находят по формуле
F0 = F-F,
где V — объем газа, измеренный при заданных температуре и давлении; F — коэффициент пересчета, значения которого приведены в таблице 8.5.
Таблица 8.4
Соотношения для пересчета содержаний газов и паров при переходе от одних единиц к другим
В таблице приняты следующие условные обозначения:
а — численное значение содержания в заданных единицах;
х — численное значение содержания в искомых единицах;
М— молярная масса газа;
р — общее давление газа в мм рт. ст.;
Т— температура газа в К;
z — коэффициент разбавления (отношение общего объема газа к объему соответствующего компонента).
При пользовании таблицей следует иметь в виду соотношения:
1 г / м3 = 1 мг / дм3 = 1 мг / л
1 мг / м3 = 1 мкг / дм3 = 1мкг / л
1 моль / дм3 = 1 моль / л
1 дм3 / м3 = 1 мл / л
1 мм3 / л = 1 мкл / м3
X г/м3 мг/м3 моль/дм3 Об. % Об. %о* см3/м3 ” 3 з *** мм /м Z
г/м3 1 103а 10Л/ М 62,36 Ю^аГ Мр 62,36аГ Мр 62,36-103аГ Мр 62,36 106 аГ Мр 16,04Мр аТ
мг/м3 103б/ 1 Ю^а М 62,36-10 4 аГ Мр 62,36-10~3аГ Мр 62,36аГ Мр 62,36-103 аГ Мр 16,04-103Лф аТ
моль/дм3 \tfaM 10 5аМ 1 62,36 1 02аГ Р 62,36 103 аГ Р 62,36 106аГ Р 62,36 109аГ Р 16,04-10~3Мр аТ
Об. % 16,04 10 2аЛф Т 16,04 -Ю^аЛф Т 16,04-10 5 ар Т 1 10а 104а 107а 102 а
Об. %’ 16,04-10~3аЛ$? Т \6,МаМр Т 16,04-10 6ар Т 10-1а 1 103а 106а 103 а
3/ 3 ** см /м 16,04 -10-6 аМр Т 16,04 10~3аМр Т 16,04-Ю^ар Т 104а 10’3а 1 103а 10б а
3 з ♦** мм /м \6,М-МГ9аМр Т 16,04 10~6аЛф Т 16,04-1012 ар Т 10 7а 10€а 10~3а 1 109 а
Z 16,04Аф zT 16,04-103Мр zT 16,04-10'р zT 102 Z 103 Z 10б Z 109 Z 1
Новый справочник химика и технолога
* Лат. partes pro mille (промилле) - частей на тысячу.
” Лат. partes pro million (ppm) - частей на миллион (1 ppm = 1 млн1).
*** Лат. partes pro billion (ppb) - частей на миллиард (Ippb = 10 3 млн"1).
Газовый анализ
895
Таблица 8.5
Значения коэффициента F для различных значений температуры и давления
Температура газа, °C Давление р, мм рт. ст.
690 692 694 696 698 700 702 704 706 708 710 712
5 0,8915 0,8941 0,8967 0,8993 0,9019 0,9045 0,9070 0,9096 0,9122 0,9148 0,9174 0,9200
6 0,8883 0,8909 0,8935 0,8961 0,8986 0,9012 0,9038 0,9064 0,9091 0,9115 0,9141 0,9167
7 0,8852 0,8877 0,8903 0,8929 0,8954 0,8980 0,9006 0,9031 0,9057 0,9082 0,9108 0,9134
8 0,8820 0,8846 0,8871 0,8897 0,8922 0,8947 0,8973 0,8999 0,9025 0,9050 0,9076 0,9101
9 0,8789 0,8814 0,8840 0,8865 0,8891 0,8916 0,8942 0,8967 0,8993 0,9018 0,9043 0,9069
10 0,8757 0,8783 0,8808 0,8834 0,8859 0,8884 0,8910 0,8935 0,8961 0,8986 0,9011 0,9037
11 0,8727 0,8752 0,8777 0,8803 0,8828 0,8853 0,8878 0,8904 0,8929 0,8954 0,8980 0,9005
12 0,8696 0,8721 0,8746 0,8772 0,8797 0,8822 0,8847 0,8872 0,8898 0,8923 0,8948 0,8973
13 0,8665 0,8691 0,8716 0,8741 0,8766 0,8791 0,8816 0,8841 0,8866 0,8892 0,8917 0,8942
14 0,8635 0,8660 0,8685 0,8710 0,8735 0,8760 0,8785 0,8810 0,8835 0,8861 0,8885 0,8911
15 0,8605 0,8630 0,8655 0,8680 0,8705 0,8730 0,8755 0,8780 0,8805 0,8830 0,8855 0,8880
16 0,8575 0,8600 0,8625 0,8650 0,8675 0,8700 0,8724 0,8749 0,8774 0,8799 0,8824 0,8849
17 0,8546 0,8571 0,8595 0,8620 0,8645 0,8670 0,8694 0,8719 0,8744 0,8769 0,8793 0,8818
18 0,8516 0,8541 0,8566 0,8590 0,8615 0,8640 0,8664 0,8689 0,8714 0,8739 0,8763 0,8788
19 0,8487 0,8512 0,8536 0,8561 0,8586 0,8610 0,8635 0,8659 0,8684 0,8709 0,8733 0,8758
20 0,8458 0,8483 0,8507 0,8532 0,8556 0,8581 0,8605 0,8630 0,8654 0,8679 0,8703 0,8728
21 0,8429 0,8454 0,8478 0,8503 0,8527 0,8551 0,8576 0,8600 0,8625 0,8649 0,8674 0,8698
22 0,8401 0,8425 0,8449 0,8474 0,8498 0,8522 0,8547 0,8571 0,8595 0,8620 0,8644 0,8669
23 0,8372 0,8397 0,8421 0,8445 0,8469 0,8494 0,8518 0,8542 0,8566 0,8591 0,8615 0,8639
24 0,8344 0,8368 0,8392 0,8416 0,8441 0,8465 0,8489 0,8513 0,8537 0,8562 0,8586 0,8610
25 0,8316 0,8340 0,8364 0,8388 0,8412 0,8436 0,8461 0,8485 0,8509 0,8533 0,8557 0,8581
26 0,8288 0,8312 0,8336 0,8360 0,8384 0,8408 0,8432 0,8456 0,8480 0,8504 0,8528 0,8552
27 0,8260 0,8284 0,8308 0,8332 0,8356 0,8380 0,8404 0,8428 0,8452 0,8476 0,8500 0,8524
28 0,8233 0,8257 0,8281 0,8304 0,8328 0,8352 0,8376 0,8400 0,8424 0,8448 0,8472 0,8496
29 0,8206 0,8229 0,8253 0,8277 0,8301 0,8325 0,8348 0,8372 0,8396 0,8420 0,8443 0,8467
30 0,8179 0,8202 0,8226 0,8250 0,8274 0,8297 0,8321 0,8345 0,8368 0,8392 0,8416 0,8439
31 0,8152 0,8175 0,8199 0,8223 0,8246 0,8270 0,8294 0,8317 0,8341 0,8364 0,8388 0,8412
32 0,8125 0,8149 0,8172 0,8196 0,8219 0,8243 0,8266 0,8290 0,8313 0,8337 0,8360 0,8384
33 0,8098 0,8122 0,8145 0,8169 0,8192 0,8216 0,8239 0,8263 0,8286 0,8310 0,8333 0,8357
34 0,8072 0,8095 0,8119 0,8142 0,8166 0,8189 0,8212 0,8236 0,8259 0,8282 0,8306 0,8329
35 0,8046 0,8069 0,8092 0,8116 0,8139 0,8162 0,8185 0,8209 0,8232 0,8255 0,8279 0,8302
36 0,8020 0,8043 0,8066 0,8089 0,8112 0,8136 0,8159 0,8182 0,8205 0,8229 0,8252 0,8275
37 0,7994 0,8017 0,8040 0,8063 0,8086 0,8109 0,8132 0,8156 0,8179 0,8202 0,8225 0,8248
38 0,7968 0,7991 0,8014 0,8037 0,8060 0,8083 0,8106 0,8129 0,8153 0,8176 0,8199 0,8222
39 0,7942 0,7965 0,7988 0,8011 0,8034 0,8057 0,8080 0,8103 0,8126 0,8149 0,8176 0,8195
40 0,7917 0,7940 0,7963 0,7986 0,8009 0,8031 0,8054 0,8077 0,8100 0,8123 0,8146 0,8169
Температура газа, °C Давление р, мм рт. ст.
714 716 718 720 722 724 726 728 730 732 734 736
5 0,9226 0,9251 0,9277 0,9303 0,9329 0,9355 0,9381 0,9406 0,9432 0,9458 0,9484 0,9510
6 0,9192 0,9218 0,9244 0,9270 0,9295 0,9321 0,9347 0,9373 0,9398 0,9424 0,9450 0,9476
896
Новый справочник химика и технолога
Продолжение таблицы 8.5
Температура газа, °C Давление р, мм рт. ст.
714 716 718 720 722 724 726 728 730 732 734 736
7 0,9159 0,9185 0,9211 0,9236 0,9262 0,9288 0,9313 0,9339 0,9365 0,9390 0,9416 0,9442
8 0,9127 0,9152 0,9178 0,9203 0,9229 0,9255 0,9280 0,9306 0,9331 0,9357 0,9383 0,9408
9 0,9094 0,9120 0,9145 0,9171 0,9196 0,9222 0,9247 0,9273 0,9298 0,9324 0,9349 0,9375
10 0,9062 0,9088 0,9113 0,9138 0,9164 0,9189 0,9214 0,9240 0,9265 0,9291 0,9316 0,9341
11 0,9030 0,9055 0,9081 0,9106 0,9131 0,9157 0,9182 0,9207 0,9233 0,9258 0,9283 0,9308
12 0,8999 0,9024 0,9049 0,9074 0,9099 0,9125 0,9150 0,9175 0,9200 0,9225 0,9251 0,9276
13 0,8967 0,8992 0,9017 0,9042 0,9067 0,9092 0,9118 0,9143 0,9168 0,9193 0,9218 0,9243
14 0,8936 0,8961 0,8986 0,9011 0,9036 0,9061 0,9086 0,9111 0,9136 0,9161 0,9186 0,9211
15 0,8905 0,8930 0,8954 0,8979 0,9004 0,9029 0,9054 0,9079 0,9104 0,9129 0,9154 0,9179
16 0,8874 0,8899 0,8923 0,8948 0,8973 0,8998 0,9023 0,9048 0,9073 0,9097 0,9122 0,9147
17 0,8843 0,8868 0,8893 0,8917 0,8942 0,8967 0,8992 0,9017 0,9041 0,9066 0,9092 0,9116
18 0,8813 0,8837 0,8862 0,8887 0,8911 0,8936 0,8961 0,8985 0,9010 0,9035 0,9059 0,9084
19 0,8782 0,8807 0,8832 0,8856 0,8881 0,8905 0,8930 0,8955 0,8979 0,9004 0,9028 0,9053
20 0,8752 0,8777 0,8801 0,8826 0,8850 0,8875 0,8899 0,8924 0,8948 0,8973 0,8997 0,9022
21 0,8722 0,8747 0,8771 0,8796 0,8820 0,8845 0,8869 0,8894 0,8918 0,8942 0,8967 0,8991
22 0,8693 0,8717 0,8742 0,8766 0,8790 0,8815 0,8839 0,8863 0,8888 0,8912 0,8936 0,8961
23 0,8663 0,8688 0,8712 0,8736 0,8761 0,8785 0,8809 0,8833 0,8858 0,8882 0,8906 0,8930
24 0,8634 0,8658 0,8683 0,8707 0,8731 0,8755 0,8779 0,8804 0,8828 0,8852 0,8876 0,8900
25 0,8605 0,8629 0,8653 0,8678 0,8702 0,8726 0,8750 0,8774 0,8798 0,8822 0,8846 0,8870
26 0,8576 0,8600 0,8624 0,8648 0,8672 0,8696 0,8720 0,8745 0,8769 0,8793 0,8817 0,8841
27 0,8548 0,8572 0,8596 0,8620 0,8644 0,8667 0,8691 0,8715 0,8739 0,8763 0,8787 0,8811
28 0,8519 0,8543 0,8567 0,8591 0,8615 0,8639 0,8662 0,8687 0,8710 0,8734 0,8758 0,8782
29 0,8491 0,8515 0,8539 0,8562 0,8586 0,8610 0,8634 0,8658 0,8681 0,8705 0,8729 0,8753
30 0,8463 0,8487 0,8510 0,8534 0,8558 0,8583 0,8605 0,8629 0,8653 0,8676 0,8700 0,8724
31 0,8435 0,8459 0,8482 0,8506 0,8530 0,8553 0,8577 0,8601 0,8624 0,8648 0,8672 0,8695
32 0,8408 0,8431 0,8455 0,8478 0,8502 0,8525 0,8549 0,8572 0,8596 0,8619 0,8643 0,8667
33 0,8380 0,8404 0,8427 0,8450 0,8474 0,8497 0,8521 0,8544 0,8568 0,8591 0,8615 0,8638
34 0,8353 0,8376 0,8399 0,8423 0,8446 0,8470 0,8493 0,8516 0,8540 0,8563 0,8587 0,8610
35 0,8325 0,8349 0,8373 0,8395 0,8419 0,8442 0,8465 0,8489 0,8512 0,8535 0,8559 0,8582
36 0,8298 0,8322 0,8345 0,8368 0,8391 0,8415 0,8438 0,8461 0,8484 0,8508 0,8531 0,8554
37 0,8271 0,8295 0,8318 0,8341 0,8364 0,8387 0,8410 0,8434 0,8457 0,8480 0,8503 0,8526
38 0,8245 0,8268 0,8291 0,8314 0,8337 0,8360 0,8383 0,8407 0,8430 0,8453 0,8476 0,8499
39 0,8218 0,8241 0,8264 0,8287 0,8310 0,8333 0,8356 0,8380 0,8403 0,8426 0,8449 0,8472
40 0,8192 0,8215 0,8238 0,8261 0,8284 0,8307 0,8330 0,8353 0,8376 0,8399 0,8422 0,8444
Температура газа, °C Давление р, мм рт. ст.
738 740 742 744 746 748 750 752 754 756 758 760
5 0,9536 0,9561 0,9587 0,9613 0,9639 0,9665 0,9691 0,9717 0,9742 0,9768 0,9794 0,9820
6 0,9501 0,9527 0,9553 0,9579 0,9604 0,9630 0,9656 0,9682 0,9707 0,9733 0,9759 0,9785
7 0,9467 0,9493 0,9518 0,9544 0,9570 0,9596 0,9621 0,9647 0,9673 0,9698 0,9724 0,9750
8 0,9434 0,9459 0,9485 0,9510 0,9536 0,9561 0,9587 0,9613 0,9638 0,9664 0,9689 0,9715
9 0,9400 0,9426 0,9451 0,9477 0,9502 0,9528 0,9553 0,9578 0,9604 0,9629 0,9655 0,9680
Газовый анализ
897
Продолжение таблицы 8.5
Температура газа, °C Давление р, мм рт. ст.
738 740 742 744 746 748 750 752 754 756 758 760
10 0,9367 0,9392 0,9418 0,9443 0,9468 0,9494 0,9519 0,9544 0,9570 0,9595 0,9621 0,9646
11 0,9334 0,9359 0,9384 0,9410 0,9435 0,9460 0,9486 0,9511 0,9536 0,9562 0,9587 0,9612
12 0,9301 0,9326 0,9351 0,9376 0,9402 0,9427 0,9452 0,9477 0,9503 0,9528 0,9553 0,9578
13 0,9269 0,9294 0,9319 0,9344 0,9369 0,9394 0,9419 0,9444 0,9469 0,9495 0,9520 0,9545
14 0,9236 0,9261 0,9286 0,9311 0,9336 0,9363 0,9386 0,9411 0,9436 0,9461 0,9486 0,9511
15 0,9204 0,9229 0,9254 0,9279 0,9304 0,9329 0,9354 0,9378 0,9404 0,9428 0,9453 0,9478
16 0,9172 0,9197 0,9222 0,9247 0,9271 0,9296 0,9321 0,9346 0,9371 0,9396 0,9420 0,9445
17 0,9140 0,9165 0,9190 0,9215 0,9239 0,9264 0,9289 0,9314 0,9339 0,9363 0,9388 0,9413
18 0,9109 0,9134 0,9158 0,9183 0,9207 0,9232 0,9257 0,9282 0,9306 0,9331 0,9356 0,9380
19 0,9078 0,9102 0,9127 0,9151 0,9176 0,9200 0,9225 0,9250 0,9275 0,9299 0,9324 0,9348
20 0,9046 0,9071 0,9096 0,9120 0,9145 0,9169 0,9194 0,9218 0,9243 0,9267 0,9292 0,9316
21 0,9016 0,9040 0,9065 0,9089 0,9113 0,9138 0,9162 0,9187 0,9211 0,9236 0,9260 0,9285
22 0,8985 0,9010 0,9034 0,9058 0,9083 0,9107 0,9131 0,9155 0,9180 0,9204 0,9229 0,9253
23 0,8955 0,8979 0,9003 0,9028 0,9052 0,9076 0,9100 0,9125 0,9149 0,9173 0,9197 0,9222
24 0,8924 0,8949 0,8973 0,8997 0,9021 0,9045 0,9070 0,9094 0,9118 0,9142 0,9165 0,9191
25 0,8894 0,8919 0,8943 0,8967 0,8991 0,9015 0,9039 0,9063 0,9087 0,9112 0,9135 0,9160
26 0,8865 0,8889 0,8913 0,8937 0,8961 0,8985 0,9009 0,9033 0,9057 0,9081 0,9105 0,9129
27 0,8835 0,8859 0,8883 0,8907 0,8931 0,8955 0,8979 0,9003 0,9027 0,9051 0,9074 0,9099
28 0,8806 0,8830 0,8853 0,8877 0,8901 0,8925 0,8949 0,8973 0,8997 0,9021 0,9044 0,9068
29 0,8776 0,8800 0,8824 0,8848 0,8872 0,8895 0,8919 0,8943 0,8967 0,8990 0,9014 0,9038
30 0,8748 0,8771 0,8795 0,8819 0,8842 0,8866 0,8890 0,8914 0,8937 0,8961 0,8985 0,9008
31 0,8719 0,8742 0,8766 0,8790 0,8813 0,8837 0,8861 0,8884 0,8908 0,8931 0,8955 0,8979
32 0,8691 0,8714 0,8736 0,8761 0,8784 0,8808 0,8831 0,8855 0,8878 0,8902 0,8926 0,8949
33 0,8662 0,8685 0,8709 0,8732 0,8756 0,8779 0,8803 0,8826 0,8850 0,8873 0,8897 0,8920
34 0,8634 0,8658 0,8680 0,8704 0,8727 0,8750 0,8774 0,8797 0,8821 0,8844 0,8867 0,8891
35 0,8605 0,8629 0,8652 0,8675 0,8699 0,8722 0,8745 0,8768 0,8792 0,8815 0,8839 0,8862
36 0,8577 0,8601 0,8624 0,8647 0,8670 0,8694 0,8717 0,8740 0,8763 0,8787 0,8810 0,8833
37 0,8549 0,8573 0,8596 0,8619 0,8642 0,8665 0,8689 0,8712 0,8735 0,8758 0,8781 0,8804
38 0,8522 0,8545 0,8568 0,8591 0,8615 0,8638 0,8661 0,8684 0,8707 0,8730 0,8753 0,8776
39 0,8495 0,8518 0,8541 0,8564 0,8587 0,8610 0,8633 0,8656 0,8679 0,8702 0,8725 0,8748
40 0,8467 0,8490 0,8513 0,8536 0,8559 0,8582 0,8605 0,8628 0,8651 0,8674 0,8697 0,8720
Темпе-ратура газа, °C Давление р, мм рт. ст.
762 764 766 768 770 772 774 776 778 780
5 0,9846 0,9871 0,9897 0,9923 0,9949 0,9975 1,0001 1,0026 1,0051 1,0078
6 0,9810 0,9836 0,9862 0,9888 0,9913 0,9939 0,9965 0,9990 1,0016 1,0042
7 0,9775 0,9801 0,9827 0,9852 0,9878 0,9904 0,9929 0,9955 0,9980 1,0006
8 0,9741 0,9766 0,9792 0,9817 0,9843 0,9868 0,9894 0,9919 0,9945 0,9970
9 0,9706 0,9731 0,9757 0,9782 0,9807 0,9833 0,9859 0,9884 0,9910 0,9935
10 0,9671 0,9697 0,9722 0,9747 0,9773 0,9798 0,9824 0,9849 0,9874 0,9900
11 0,9637 0,9663 0,9688 0,9713 0,9739 0,9764 0,9789 0,9814 0,9839 0,9865
12 0,9603 0,9629 0,9654 0,9679 0,9704 0,9730 0,9754 0,9780 0,9805 0,9830
898
Новый справочник химика и технолога
Продолжение таблицы 8.5
Температура газа, °C Давление р, мм рт. ст.
762 764 766 768 770 772 774 776 778 780
13 0,9570 0,9595 0,9620 0,9645 0,9670 0,9695 0,9720 0,9745 0,9771 0,9796
14 0,9536 0,9561 0,9586 0,9612 0,9637 0,9661 0,9686 0,9711 0,9736 0,9762
15 0,9503 0,9528 0,9553 0,9578 0,9603 0,9628 0,9653 0,9678 0,9703 . 0,9728
16 0,9470 0,9495 0,9520 0,9545 0,9570 0,9595 0,9619 0,9644 0,9669 0,9694
17 0,9438 0,9462 0,9487 0,9512 0,9537 0,9561 0,9586 0,9611 0,9636 0,9661
18 0,9405 0,9430 0,9454 0,9479 0,9504 0,9528 0,9553 0,9578 0,9602 0,9627
19 0,9373 0,9397 0,9422 0,9447 0,9471 0,9496 0,9520 0,9545 0,9569 0,9594
20 0,9341 0,9365 0,9390 0,9414 0,9439 0,9463 0,9488 0,9512 0,9537 0,9561
21 0,9309 0,9333 0,9359 0,9382 0,9407 0,9431 0,9455 0,9480 0,9504 0,9529
22 0,9277 0,9302 0,9326 0,9350 0,9375 0,9399 0,9423 0,9448 0,9472 0,9496
23 0,9246 0,9270 0,9294 0,9319 0,9343 0,9367 0,9391 0,9416 0,9440 0,9464
24 0,9215 0,9239 0,9263 0,9287 0,9311 0,9336 0,9360 0,9384 0,9408 0,9432
25 0,9184 0,9208 0,9232 0,9256 0,9280 0,9304 0,9328 0,9352 0,9377 0,9401
26 0,9153 0,9177 0,9201 0,9225 0,9249 0,9273 0,9297 0,9321 0,9345 0,9369
27 0,9122 0,9146 0,9170 0,9194 0,9218 0,9242 0,9266 0,9290 0,9314 0,9338
28 0,9092 0,9116 0,9140 0,9164 0,9187 0,9211 0,9235 0,9259 0,9283 0,9307
29 0,9062 0,9086 0,9109 0,9133 0,9157 0,9181 0,9205 0,9228 0,9252 0,9276
30 0,9032 0,9056 0,9079 0,9109 0,9127 0,9151 0,9174 0,9198 0,9222 0,9245
31 0,9002 0,9026 0,9050 0,9073 0,9097 0,9121 0,9144 0,9168 0,9191 0,9215
32 0,8973 0,8996 0,9020 0,9043 0,9067 0,9091 0,9114 0,9138 0,9161 0,9185
33 0,8943 0,8967 0,8990 0,9014 0,9037 0,9061 0,9084 0,9108 0,9131 0,9154
34 0,8914 0,8938 0,8961 0,8984 0,9008 0,9031 0,9055 0,9078 0,9101 0,9125
35 0,8885 0,8908 0,8932 0,8955 0,8978 0,9002 0,9025 0,9048 0,9072 0,9092
36 0,8856 0,8880 0,8903 0,8926 0,8949 0,8972 0,8996 0,9019 0,9042 0,9065
37 0,8828 0,8851 0,8874 0,8897 0,8920 0,8943 0,8967 0,8990 0,9013 0,9036
38 0,8799 0,8822 0,8845 0,8869 0,8892 0,8915 0,8938 0,8961 0,8984 0,9007
39 0,8771 0,8794 0,8817 0,8840 0,8863 0,8886 0,8909 0,8932 0,8955 0,8978
40 0,8743 0,8766 0,8789 0,8812 0,8835 0,8857 0,8881 0,8903 0,8926 0,8949
Значения F можно также вычислить по формуле: Ио = V • F, (8.3)
F = -^--760, (8.1)
1 + а/
где ро - атмосферное давление, приведенное к О °C, мм рт. ст.; а — коэффициент объемного расширения газа, град1; t — температура газа, °C. Для приведения атмосферного давления к О °C с достаточной точностью можно воспользоваться формулой:
ро=А-О,125Г, (8.2)
где pt — давление при температуре t °C, мм рт. ст.; f — температура ртути в барометре, °C.
Объем газа, собранного над водой или водными растворами солей, приводится к нормальным условиям по формуле:
но в этом случае р$, входящая в формулу (5.1), составляет:
= Pt-0,U5t'-p&, (8.4)
где р3 — давление паров воды при соответствующей температуре, мм рт.ст.
При особо точных расчетах к установленному давлению добавляют поправку на капиллярную депрессию ртути. Коэффициент F в этих случаях следует вычислять по формуле:
F = —^—760, (8.5)
1 + d'Z
пользуясь значением а для данного газа. В таблице 8.6. приводятся значения а для некоторых газов в интервале температур от 0 до 100 °C при давлении 760 мм рт.ст.
Газовый анализ
899
Таблица 8.6
Значения коэффициента а
Газ а Газ а
Азот 0,00367 Диоксид углерода 0,00371
Водород 0,00366 Кислород 0,00367
Воздух 0,00367 Оксид углерода 0,00367
Гелий 0,00366 Хлор 0,00383
В соответствии с требованиями ГОСТ 2939-63 при расчете объема газа следует исходить из объема, приведенного к 20 °C и 760 мм рт.ст. Объем газа при этих условиях устанавливают по формуле:
К20 = 1,0733КГ. (8.6)
На рис. 8.2 изображена номограмма для приведения объема газа к условиям 0 °C и 760 мм рт. ст., которой удобно пользоваться для ускорения вычислений при массовых анализах.
Атмосферное
давление, мм рт. ст.
780 г
760
-
750
710
700
Температура, *С
-1,3000
0,800- ? 050
10- Ё- w
Ы i r 150
30] r 200
904 ? 250
50-] г 300
1 & г 350
g 704 -4 Ё- 400
80 *; Ё- 450
SQA 41,9500
-i
еямн ? 550
10 4 - 600
20- г 650
эо] - 700
:- 750
50]
г 800
60-,
- - 850
?oi
8o] г 300
so] ? 950
1,000 J -0,0000
?70 ?
Рис. 8.2. Номограмма для приведения объема газа к 0 °C и 760 мм рт. ст.
Для расчета по номограмме соединяют линейкой точки, соответствующие давлению и температуре, при которых измерен объем газа. В точке пересечения линейки со шкалой, находящейся в правой части номограммы, отыскива
ют коэффициент пересчета F (или его логарифм), на который умножают измеренный объем.
Пример (указан на номограмме пунктиром). При давлении 730 мм рт. ст. и температуре 20 °C получают (для сухого газа): F= 0,895 и IgF =1,9517; для влажного газа F= 0,873 и lgF = T,9410.
В таблице 8.7 приведены соотношения между значениями давления газа, выраженными в различных единицах.
Таблица 8.7
Соотношения между значениями давления, выраженными в различных единицах
мм вод. ст. ммрт. ст. мм вод. ст. мм рт. ст. мм вод. ст. ммрт. ст.
1 0,07 35 2,57 69 5,08
2 0,15 36 2,65 70 5,15
3 0,22 37 2,72 71 5,22
4 0,29 38 2,79 72 5,30
5 0,37 39 2,87 73 5,37
6 0,44 40 2,94 74 5,44
7 0,52 41 3,02 75 5,52
8 0,59 42 3,09 76 5,59
9 0,66 43 3,16 77 5,66
10 0,74 44 3,24 78 5,74
11 0,81 45 3,31 79 5,81
12 0,88 46 3,38 80 5,88
13 0,96 47 3,46 81 5,96
14 1,03 48 3,53 82 6,03
15 1,10 49 3,60 83 6,10
16 1,18 50 3,68 84 6,18
17 1,25 51 3,75 85 6,25
18 1,32 52 3,82 86 6,33
19 1,40 53 3,90 87 6,40
20 1,47 54 3,97 88 6,47
21 1,54 55 4,05 89 6,55
22 1,62 56 4,12 90 6,62
23 1,69 57 4,19 91 6,69
24 1,77 58 4,27 92 6,77
25 1,81 59 4,34 93 6,84
26 1,91 60 4,41 94 6,91
27 1,99 61 4,49 95 6,99
28 2,06 62 4,56 96 7,06
29 2,13 63 4,63 97 7,13
30 2,21 64 4,71 98 7,21
31 2,28 65 4,78 99 7,28
32 2,35 66 4,85 100 7,36
33 2,43 67 4,93
34 2,50 68 5,00
900
Новый справочник химика и технолога
8.6. Физические и физико-химические свойства чистых газов
Газ — агрегатное состояние вещества, в котором его частицы не связаны или весьма слабо связаны силами взаимодействия и движутся свободно, заполняя весь предоставленный им объем. Вещества в газообразном состоянии образуют атмосферу Земли, в значительных количествах содержатся в твердых земных породах, растворены в воде океанов, морей, рек и озер. Солнце, звезды, облака межзвездного вещества состоят из газов — нейтральных или ионизованных. Встречающиеся в природных условиях газы представляют собой, как правило, смеси химически индивидуальных газообразных веществ. Газы целиком заполняют сосуд, в котором находятся, и принимают его форму. В отличие от твердых веществ и жидкостей, объем газов существенно зависит от давления и температуры. Коэффициент объемного расширения газов в обычных условиях (при 0-100 °C) на два порядка выше, чем у жидкостей, и составляет при 0 °C - 0,003663 К1.
Физические свойства газов приведены в табл. 8.8-8.13.
В таблице 8.8 приводятся значения удельной х и объемной (относительной) км магнитной восприимчивости.
Под объемной магнитной восприимчивостью км подразумевается коэффициент пропорциональности между намагниченностью (плотностью магнитного момента*) J единицы объема газа или пара и напряженностью Н магнитного поля в этом объеме, т. е. это коэффициент в выражении J = км Н Поскольку размерности величин J и Н одинаковы, величина км безразмерна.
Удельная магнитная восприимчивость связана с объемной соотношением:
км К = ——, р
где р — плотность газа или пара.
Величина х характеризует магнитную восприимчивость единицы массы вещества. В единицах системы СГС х выражается в см3/г, а в единицах СИ — в м3/кг.
Значения км и х, выраженные в системе СГС, могут быть пересчитаны в единицы СИ с помощью соотношений:
4тс^мйю = и 4л • 103 хсгс = хси.
Над чертой (в «числителе») приводятся значения х • 104 и км • 1010 в единицах системы СГС, под чертой (в «знаменателе») — значения х • 103 и Ам • 109 в единицах СИ.
* Магнитный момент, обусловливающий намагниченность, выражается в единицах, которые определяются через напряженность магнитного поля, создаваемого этим моментом, но не через магнитную индукцию.
Таблица 8.8
Магнитная восприимчивость газов и паров
Газ или пар х при 20 °C км при 20 °C и 760 мм рт. ст.
название формула
Азот n2 -0,43 -5,4 -5,01 -6,30
Аммиак NH3 -1,1 -14 -7,90 -9,93
Аргон Аг -0,48 -6,0 -7,97 -10,0
Ацетилен с2н2 -0,48 -6,0 -5,24 -6,57
Водород н2 -2,0 -25 -1,68 -2,11
Гелий Не -0,48 -6,0 -0,80 -1,0
Диоксид углерода СО2 -0,48 -6,0 -8,77 -11,0
Оксид азота (I) (оксид диазота) n2o -0,43 -5,4 -7,93 -9,97
Кислород о2 +106,2 +1335 +1414 +1777
Метан СН4 -0,76 -9,6 -5,08 -6,38
Неон Ne -0,38 -4,8 -3,19 -4,01
Оксид азота (II) NO +49 +620 +611 +768
Оксид углерода СО -0,42 -5,3 -4,88 -6,13
Сероводород H2S -0,75 -9,4 -10,8 -13,6
Хлор Cl2 -0,59 -7,4 -17,7 -22,2
Хлористый водород НС1 -0,60 -7,5 -9,19 -11,5
Хлористый метил СН3С1 -0,64 -8,0 -14,2 -17,8
Этан с2н6 -0,73 -9,2 -9,24 -11,6
Этилен С2Н4 -0,43 -5,4 -5,05 -6,35
Таблица 8.9
Коэффициенты теплопроводности некоторых газов и паров при различных температурах
Значения коэффициентов теплопроводности А приведены в двух системах единиц: над чертой — в единицах системы СГС, т.е. в кал/(см-с-град.), увеличенные в 105 раз, а под чертой — в единицах СИ, т.е. вт/(м град), увеличенные в 103 раз. Точное соотношение между этими единицами: 1 (кал/см-страд) = 4,1868-102 (вт/мтрад).
Предельная погрешность А для температур до 200 °C составляет ±1%, для более высоких температур она может достигать 3-4%.
Величины А, В и С, значения которых приведены над чертой, — постоянные уравнения А-105 =А+ Bt + С? при пользовании единицами СГС, а под чертой — по-
стоянные уравнения А-103 = А + В + С? при пользовании «единицами СИ. С помощью этих уравнений можно интерполировать (в соответствующих единицах) значения коэффициентов теплопроводности при промежуточных температурах.
Газ пли пар Коэффициент теплопроводности при температуре, °C Постоянные уравнения температурной зависимости коэффициента теплопроводности
название формула 0 25 50 100 150 200 300 400 А В С
Акрилонитрил ch2=chcn — 2,31 9,67 — — — — — — — — —
Аммиак NH3 5,18 21,7 5,90 24,7 6,68 28,0 8,41 35,2 10,36 43,38 12,54 52,50 17,6 73,7 23,5 98,4 5,18 21,7 0,0279 0,117 448-10~7 188-106
Аргон Ar 3,91 16,4 4,21 17,6 4,50 18,8 5,09 21,3 5,69 23,8 6,28 26,3 7,46 31,2 8,65 36,2 3,91 16,4 0,0119 0,0498 —
Ацетилен сн=сн 4,51 18,9 5,20 21,8 5,91 24,7 7,36 30,8 8,88 37,2 10,45 43,75 13,8 57,8 17,35 72,64 4,51 18,9 0,0273 0,114 12 106 50-10 6
Бутадиен (1,3-дивинил) сн2= снсн= сн2 2,90 12,1 3,57 14,9 4,29 17,9 5,83 24,4 7,53 31,5 9,39 39,3 13,6 56,9 18,4 77,0 2,90 12,1 0,0262 0,110 3111О’7 130 -10 6
Бутан СН3(СН2)2СН3 3,05 12,8 3,68 15,4 4,36 - 18,2 5,88 24,6 7,60 31,8 9,50 39,8 14,0 58,6 19,2 60,4 3,05 12,8 0,0243 0,102 403 10 7 169-106
Бутен-1 СН3СН2СН= сн2 3,02 12,6 3,60 15,1 4,24 17,7 5,70 23,8 7,39 30,9 9,31 39,0 13,9 58,2 19,3 80,8 3,02 12,6 0,0222 0,0929 463-10 7 194-Ю’6
Воздух — 5,77 24,2 6,20 26,0 6,64 27,8 7,50 31,4 8,37 35,0 9,23 38,6 10,96 45,89 12,70 53,17 5,77 24,2 0,0173 0,0724 —
Диоксид углерода со2 3,48 14,6 3,92 16,4 4,37 18,3 5,33 22,3 6,34 26,5 7,41 31,0 9,67 40,5 12,04 50,41 3,48 14,6 0,0174 0,0728 П-10’6 46-10’6
1,1 -Дихлорэтил ен СН2= СС12 1,65 6,91 1,92 8,04 2,20 9,21 2,79 11,7 3,41 14,3 4,07 17,0 5,49 23,0 7,04 29,5 1,65 6,91 0,0107 0,0448 686 Ю’8 287-10’7
Изобутан (СН3)3СН 3,16 14,5 3,79 15,9 4,47 18,7 5,98 25,0 7,69 32,2 9,60 40,2 14,02 58,70 19,23 80,51 3,16 14,5 0,0242 0,101 400-10~7 167-Ю’6
Газовый анализ
Продолжение таблицы 8.9
Газ пли пар Коэффициент теплопроводности при температуре, °C Постоянные уравнения температурной зависимости коэффициента теплопроводности
название формула 0 25 50 100 150 200 300 400 А В С
Изопентан (СН3)2СНС2Н5 2,80 11,7 3,35 14,0 3,95 16,5 5,32 22,3 6,90 28,9 8,70 36,4 12,9 54,0 18,0 75,4 2,80 И,7 0,0209 0,0875 430 10 7 180-106
Криптон Кг 1,95 8,16 2,07 8,67 2,20 9,21 2,44 10,2 2,68 И,2 2,90 12,1 3,36 14,1 3,83 16,0 1,95 8,16 0,00469 0,0196 —
Метан сн4 7,11 29,8 8,09 33,9 9,07 38,0 11,15 46,68 13,3 55,7 15,5 64,9 20,25 84,78 25,3 106 7,11 29,8 0,0387 0,162 171-107 716-Ю7
2-Метилпропен (СН3)2С=СН2 3,04 12,7 3,67 15,3 4,34 18,2 5,78 24,2 7,37 30,9 9,11 38,1 13,0 54,4 17,5 73,3 3,04 12,7 0,0244 0,102 295 10 7 124 106
Оксид этилена (СН2)2О 2,47 10,3 2,97 12,4 3,53 14,8 4,82 20,2 6,34 26,5 8,10 33,9 12,3 51,5 17,4 72,9 2,47 10,3 0,0189 0,0791 464-10 7 194-106
Пропан СН3СН2СН3 3,56 14,9 4,26 17,8 5,01 21,0 6,64 27,8 8,46 35,4 10,47 43,84 15,05 63,01 20,4 85,4 3,56 14,9 0,0271 0,113 374-10~7 157-10“6
Пропилен СН3СН= сн2 3,48 14,6 4,14 17,3 4,85 20,3 6,44 27,0 8,24 34,5 10,3 43,1 14,9 62,4 20,4 85,4 3,48 14,6 0,0253 0,106 43 10~6 18-Ю’5
Трихлорэтилен СНС1= СС12 — 0,185 0,775 — — — — — — — — —
Хлористый винил СН2= СНС1 2,21 9,25 2,60 10,9 3,03 12,7 3,95 16,5 4,99 20,9 6,14 25,7 8,78 36,8 11,9 46,4 2,21 9,25 0,0152 0,0637 223-10~7 934 10’7
Хлористый метил СН3С1 2,17 9,09 2,52 10,6 2,90 12,1 3,76 15,7 4,75 19,9 5,87 24,6 8,50 35,6 11,65 48,78 2,17 9,09 0,0132 0,0553 261 107 109-Ю6
Хлористый метилен СН2С12 1,58 6,62 1,77 7,41 1,98 8,29 2,46 10,3 3,03 12,7 3,68 15,4 5,24 21,9 7,15 29,9 1,58 6,62 0,00706 0,0296 171 10 7 716-Ю7
Хлористый этил СН3СН2С1 2,17 9,09 2,58 10,8 3,01 12,6 3,99 16,7 5,09 21,3 6,33 26,5 9,20 38,5 12,6 52,6 2,17 9,09 0,0155 0,0649 263-10~7 110-106
Цианистый водород HCN 2,35 9,84 2,83 11,8 3,30 13,8 4,27 17,9 5,22 21,9 6,18 25,9 8,09 33,9 10,00 41,9 2,35 9,84 0,0191 0,0800 —
Этан СН3СН3 4,23 17,7 5,07 21,2 5,94 24,9 7,82 32,7 9,84 41,2 12,0 50,2 16,80 70,34 22,2 92,9 4,23 17,7 ,0,0329 0,138 298-10 7 125-106
Этилен сн2= сн2 4,18 17,5 4,93 20,6 5,73 24,0 7,46 31,2 9,87 41,3 11,45 47,94 16,1 67,4 21,6 90,4 4,18 17,5 0,0293 0,123 353-10~7 148-10’6
902 Новый справочник химика и технолога
Газовый анализ
903
Таблица 8.10
Скорость звука в газах и парах
Газ или пар Температура, °C Скорость звука с, м/сек Температурный коэффициент скорости звука АС7А/, (м/сек)/град
название формула
Газы
Азот n2 0 334 0,6
Аммиак NH3 0 415 —
Аргон Ar 0 319 0,56
Бромистый водород HBr 0 200 -
Водород н2 0 1284 2,2
Воздух 0 331 0,59
Гелий He 0 965 0,8
Диоксид серы SO2 0 213 0,47
Диоксид углерода CO2 0 259 0,4
Дейтерий d2 0 890 1,6
Оксид азота (I) n2o 0 263 0,5
Йодистый водород HJ 0 157 -
Кислород O2 0 316 0,56
Метан CH4 0 430 -
Неон Ne 0 435 0,8
Оксид азота (II) NO 10 324 -
Оксид углерода CO 0 338 0,6
Сероводород H2S 0 289 —
Хлор Cl2 0 206 —
Хлористый водород HC1 0 296 -
Четырехфтористый кремний SiF4 0 167 —
Этан C2H6 10 308 —
Этилен C2H4 0 317 -
Пары
Ацетон CH3COCH3 97,1 239 0,32
134 251 -
Бензол СбН6 97,1 202 0,3
134 213 -
Бутанон-2 CH3COC2H5 134 223 -
Продолжение таблицы 8.10
Газ или пар Температура, °C Скорость звука с, м/сек Температурный коэффициент скорости звука ДС7А/, (м/сек)/град
название формула
втор-Бутиловый спирт С2Н5СНОНСН3 134 215 —
трет-Бутиловый спирт (СН3)3СОН 134 180 —
Винилацетат Н2С=СНОС(О)СН3 134 203 -
Вода Н2О 134 494 —
Гексан СН3(СН2)4СН3 134 199 —
Диметиловый эфир (СН3)2О 25 246 0,39
97,1 274
Дипропил о-вый эфир (С3н7)2о 97,1 194 -
Дихлорэтан С2Н4С12 97,1 181 0,24
134 190
Диэтиловый эфир (С2н5)2о 97,1 206 0,3
134 217
Изопропиловый спирт СН3СНОНСН3 97,1 255 0,4
134 270 —
Метиловый спирт СН3ОН 97,1 335 0,46
134 352 -
Метилциклогексан СбНиСНз 134 185 -
Пентан СН3(СН2)3СН3 134 220 —
Пропиловый спирт СН3СН2СН2ОН 134 244 -
Сероуглерод cs2 97,1 220 -
Хлористый метилен СН2С12 97,1 204 0,24
134 213 —
Хлороформ СНС13 97,1 171 0,24
134 180 -
Циклогексан С6Н12 97,1 191 о,3
134 202 -
Четыреххлористый углерод СС14 97,1 145 -
134 153 -
Этилацетат С2Н5ОС(О)СН3 97,1 189 0,27
134 199 -
Этиловый спирт СН3СН2ОН 97,1 269 0,4
134 284 -
904
Новый справочник химика и технолога
Таблица 8.11
Физические свойства некоторых чистых газов
Свойство Азот Аргон Водород Воздух Кислород Углекислый газ
Масса (г) 1 моля 28,02 39,94 2,016 28,96 32,00 44,00
Плотность (кг/ м3) при 0 °C и 0,1 МПа 1,2748 1,8185 0,0916 1,3178 1,4567 2,0140
Теплоемкость Су, (кДж/кмоль, 0 °C) 20,85 12,48 275,6 20,81 20,89 30,62
Скорость звука (м/с) при 0 °C 333,6 319,0 1286 331,5 314,8 260,3
Вязкость и. при 0 °C (ц-106 Па с) 16,6 21,2 8,4 17,1 19,2 13,8
Диэлектрическая проницаемость е при 0 °C и 0,1 МПа 2,43 1,62 16,8 2,41 2,44 1,45
Теплопроводность (Х-102 Дж/мсК) 1,000588 1,000536 1,000272 1,000590 1,000531 1,000988
Удельная магнитная восприимчивость х при 20 °C (х-106 на 1 г) -0,43 -0,49 -1,99 - +107,8 -0,48
Таблица 8.12
Абсолютные показатели преломления некоторых газов и паров при нормальных условиях
Газ (Уе-1) ‘ 106 Х=546,1 нм Газ (Уе-1) 106 Х=546,1 нм
Азот 299,14 Йодистый водород 925,8
Аммиак 379 Кислород 272,27
Аргон 283,14 Криптон 428,7
Ацетилен 600,7 Ксенон 706
Ацетон 1096,0 Метан 443,3
Бензол 1835 Неон 67,25
Бром 1185 Оксид азота 295
Бромистый водород 614,9 Оксид углерода 336
Водяной пар 256,9 Озон 520
Воздух (сухой) 292,9 Ртутный пар 940
Водород 140,18 Сернистый газ 664
Гелий 34,9 Сероводород 644
Диоксид углерода 450,6 Хлор 784
Дейтерий 137,9 Хлористый водород 448,0
Дициан 854 Этан 764,8
Оксид азота 510 Этиловый эфир 1509,0
Таблица 8.13
Показатели преломления сжиженных газов
Газ £К пв X, нм
n2 78 1,205 589,3
Продолжение таблицы 8.13
Газ Г, К nD X, нм
н2 20,46 1,1121 546,1
22 1,1093 546,1
24 1,1049 546,1
26 1,0992 546,1
28 1,0937 546,1
30 1,0859 546,1
о2 92 1,221 546,1
8.7. Методы получения и очистки газов
Вне зависимости от назначения чистого газа уровень содержания примесей в нем не должен превышать п-10 4 мол. % и варьируется в диапазоне от и-10-8 до 10 4 мол. %. Особенно жесткие требования предъявляются к чистоте газов, используемых в качестве основы при приготовлении поверочных газовых смесей (ПГС) в ядерной энергетике и в производстве полупроводниковых материалов.
Для очистки газов используются различные методы. Исторически первыми и интенсивно используемыми до настоящего времени являются абсорбционные методы. Они состоят в поглощении удаляемых компонентов жидкими поглотителями (индивидуальными веществами, смесями или растворами) и основаны на физической абсорбции или абсорбции, сопровождающейся химической реакцией с активным компонентом абсорбента. Очистка газов осуществляется чаще всего промывкой газа в барботажных или насадочных противоточных аппаратах. Недостаток абсорбционных методов — загрязнение очищаемого газа парами растворителя. В таблицах 8.14-8.22 указаны возможности различных твердых веществ, жидких растворов и химических процессов к извлечению из газовых смесей их отдельных компонентов.
Таблица 8.14
Возможности избирательной осушки и изъятия отдельных компонентов газовых смесей
Осушка и очистка сложных газовых смесей, в которых определяются один или несколько компонентов, требует избирательных сорбентов. Так, для очистки от сероводорода газовой смеси, содержащей даже небольшие примеси диоксида углерода или других кислых газов, не могут быть применены щелочные сорбенты
(аскарит, ХПИ, щелочи). Фосфорный ангидрид неприменим для осушки галогеноводородов, с которыми в присутствии влаги он взаимодействует с образованием галогенокислородных соединений фосфора. Применение силикагеля ограничивается его высокой сорбционной способностью по отношению ко многим газам. Ан-
гидрон частично поглощает непредельные углеводороды, вследствие чего не может быть использован в качестве осушителя в их присутствии. Кроме того, выбор сорбента в каждом отдельном случае зависит от требуемой степени осушки или очистки газовой смеси.
Анализируемый компонент Среда Удаляемый компонент Сорбент Температура очистки, °C Максимальная объемная скорость, отнесенная к объему сорбента, ч-1 Остаточное содержание удаляемого компонента, мг/л Сорбционная емкость, масс. % от массы сорбента Условия регенерации сорбента
наименование состав зернение, мм
Азот, водород, кислород, инертные газы Азот, воздух Пары воды Силикагель кем 2-5 20-30 40 60 43-59 6 • 10"3 5 • 10 2 1 • 10 1 17-19 6 Многократно регенерируется продувкой воздухом, осушенным ангидроном и Р2О5 при 180-200 °C
ОС Силикагель ШСК, пропитанный СаС12 (25 масс. %) 2-3 20 10 1 • 10 1 49 То же
Хлорид кальция СаС12 плавленый 2-6 20-60 75-240 2 • 101 - Не регенерируется
Ангидрон Mg(ClO4)2 безводный 2-3 20-30 65-160 5 • 10^ 20-60 То же
Фосфорный ангидрид р2о5 Порошок 20-35 - 2 • 10'5 - То же
Аммиак Азот, воздух, нейтральные газы, предельные углеводороды Пары воды Гидрооксид калия КОН Таблетки диаметром 5-7 мм 55-65 2 • 103 — То же
Гидрооксид натрия NaOH То же 75-170 2 • 10’1 - То же
Диоксид и оксид углерода, предельные и ароматические углеводороды Воздух, углеводороды То же ОС Силикагель ШСК, пропитанный СаС12 (25 масс. %) 2-3 20 10 1 • 10 1 49 Многократно регенерируется продувкой воздухом, осушенным ангидроном и р2о5 при 180-200 °C
Хлорид кальция СаС12 плавленый 2-6 20-60 75-240 2 • 10~* Не регенерируется
Ангидрон Mg(ClO4)2 безводный 2-3 20-30 65-160 5 • 10^ 20-60 То же
Газовый анализ
Продолжение таблицы 8.14
Анализируемый компонент Среда Удаляемый компонент Сорбент Температура очистки, °C Максимальная объемная скорость, отнесенная к объему сорбента, ч-1 Остаточное содержание удаляемого компонента, мг/л Сорбционная емкость, масс. % от массы сорбента Условия регенерации сорбента
наименование состав зернение, мм
Непредельные углеводороды Воздух, нейтральные газы Пары воды ос Силикагель ШСК, пропитанный СаС12 (25 масс. %) 2-3 20 10 1 • 101 49 Многократно регенерируется продувкой воздухом, осушенным ангидроном иР2О5 при 180-200 °C
Хлорид кальция СаС12 плавленый 2-6 20-60 75-240 2 • 10 1 - Не регенерируется
Ангидрон Mg(ClO4)2 2-3 20-30 65-100 5 • 10л 20-60 То же
Нейтральные газы, диоксид углерода, углеводороды Воздух или любой из определяемых газов Аммиак Купрамит Глина, пемза или активированный уголь, пропитанные CuSO4 2-3 20-30 13-15 — — Не регенерируется
Азот, водород, предельные углеводороды Воздух Сероводород Тоже То же 2-3 20-30 — Обычными методами не улавливается 7,0 То же
Воздух, водород, предельные углеводороды, оксид углерода Любой из определяемых газов Диоксид углерода ХПИ Смесь СаО (96%) и NaOH(4%) Гранулы диаметром 1,5-5 мм 20-25 — 1,1 25-30 То же
Аскарит NaOH на асбесте - - - До 20 То же
Водород, воздух, предельные и ароматические углеводороды, диоксид и оксид углерода Азот, воздух Оксиды азота (NO, NO2) И-ХПИ ХПИ, пропитанный КМпО4 (4 масс. %) 20-40 120 4 • 10 6 То же
Диоксид углерода и другие кислые газы Азот, воздух, углеводороды Сернистый газ 1-2 % раствор бертолетовой соли КС1О3 - — __ — — То же
Водород, воздух, предельные и ароматические углеводороды, оксид углерода Азот, воздух То же Оксид кальция СаО гранулированный 4-8 — — — — То же
906 Новый справочник химика и технолога
Газовый анализ
907
Таблица 8.15
Растворимость газов в концентрированных растворах хлорида натрия
а — коэффициент абсорбции Бунзена, т. е. объем газа (приведенный к 0 °C и 760 мм рт. ст.), поглощаемый единицей объема жидкости при парциальном давлении газа, равном 760 мм рт. ст.
Газ Концентрация NaCl, масс. % Значения а при температуре, °C
5 10 15 20 25 30
Азот 11,90 0,010 0,0092 0,0081 0,0066 0,0048 —
Ацетилен 26,42 - - - — 0,320 —
Водород 6,10 0,0184 0,0175 0,0164 0,0153 0,0138 —
15,80 0,00699 — —
23,84 - - 0,00595 — — —
Диоксид серы 15,80 - — — — 28,79 —
Диоксид углерода 15,70 1,399 1,205 1,043 0,915 0,816 0,727
18,70 0,577 0,503 0,442 0,393 0,352 0,319
Оксид азота (I) 25,25 — — — — 0,172 —
Кислород 26,4 0,0059 0,0056 0,0054 0,0052 0,0050 0,0048
Сероводород 15,8 — 1,753 0,36 1,354 — 1,138
Хлор 26,4 0,44 0,40 0,36 0,34 0,30 0,28
Таблица 8.16
Очистка и осушка газов при повышенном давлении
Назначение очистки Режим работы Остаточное содержание примесей в очищенном газе, об. % Условия регенерации поглотителя
рекомендуемые поглотители и температурный режим объемная скорость очищаемого газа, отнесенная к объему поглотителя, ч 1
Очистка от кислорода Металлическая медь, восстановленная водородом из оксида, в гранулах диаметром 3-5 мм; 300-350 °C 30-60 Не более 0,001 Восстанавливают оксид меди водородом при 300-350 °C. При очистке водорода регенерации не требуется
Очистка от водорода Гранулированный оксид меди; 300-320 °C 30-60 Не более 0,01 Регенерацию производят воздухом при 300-350 °C
Очистка от оксида углерода Гранулированный оксид меди; 300-350 °C 30-50 Не более 0,001 То же
Очистка от диоксида углерода Натронная известь, диаметр зерен 2-3 мм; комнатная температура 15-30 Не более 0,001 Не регенерируется
Осушка и одновременная очистка газов с температурой кипения ниже -180 °C от примесей кислорода, азота, диоксида и оксида углерода, углеводородов Силикагель марки КСМ, охлаждаемый жидким азотом, диаметр зерен 1-5 мм; предварительно газ подвергают осушке силикагелем при комнатной температуре 60-80 Не более: О2 — 0,0005; СО, СО2 — 0,001; влаги — 0,05 мг/л Вакуумирование при 200-230 °C
Очистка от углеводородов, масел и других органических веществ Активированный уголь марки АГ-2, диаметр зерен 2^4 мм; комнатная температура 60-80 Не более 0,001 Вакуумирование при 100 °C. При очистке от масел регенерацию рекомендуется проводить отгонкой с водяным паром с последующей сушкой
Очистка гелия или аргона от азота Металлический кальций; 650 °C 30-50 Не более 0,005 Не регенерирует
908
Новый справочник химика и технолога
Таблица 8.17
Поглотительные растворы и поглотители для абсорбционного анализа газов
Определяемый газ Поглотитель Приготовление поглотительного раствора
Ацетилен и углеводороды ряда ацетилена Тетраиодомеркуроат калия 25 г Hgl2 и 30 г KI растворяют в 100 мл воды; перед применением реактив подщелачивают КОН
Хлорид меди в аммиачной среде 32 г СиСЬ растворяют в 110 мл 25% водного раствора NH4C1 и добавляют 80-100 мл 25% раствора NH3
Диоксид углерода, хлористый водород, хлор,сероводород, диоксид серы и другие кислые газы Гидрооксид калия 30-35% водный раствор КОН
Гидрооксид калия или натрия Сухой поглотитель в палочках или гранулах
Кислород Пирогаллол 56 г пирогаллола растворяют в 100 мл воды и добавляют 260 мл 33% водного раствора КОН
Триацетилоксигидрохинон 40 г триацетилоксигидрохинона растворяют в 200 мл 38% водного раствора КОН
Гидросульфит натрия Смешивают свежеприготовленные растворы: 1) 50 г Na2S2O4 в 250 мл воды и 2) 30 г NaOH в 40 мл воды
Аммиакат меди Медную проволоку погружают в смесь равных объемов конц. раствора NH3 и насыщенного водного раствора NH4C1
Белый фосфор Сухой поглотитель в палочках
Таблица 8.18
Абсорбционная способность некоторых растворов
В таблице указано число миллилитров газа, абсорбируемого 1 мл раствора. Состав поглотительных растворов см. выше.
Поглотитель Газ
о2 H2S so2 AsH3 РН3 со СО2 COS С2н2 С2Н4
Ацетат кадмия — 50 90 40 0,5 — 0,5 0,2 — -
Гидрооксид калия — 190 190 — o,i — 150 — 0,05 —
Гидросульфит натрия 7 — — — — — — — — —
Пирогаллол 35 — — — — — — — — —
Селенистая кислота — — — — 100 — — — 0,80 —
Сульфат меди (I) с 0-нафтолом — — — — — 18 — - — -
Сульфованадиевый реактив — — — - — — — — — 60
Тетраиодомеркуроат калия — — — — — — — — 20
Триацетилоксигидрохинон 22 — — — — — — — — —
Хлорид меди (I) в аммиачной среде — - - - - 14 — - - -
Таблица 8.19
Давление паров воды над насыщенными растворами хлорида и сульфата натрия
Раствор Температура,^ Давление паров, мм рг. ст. Температура,^ Давление паров, мм рг. ст. Температура, °C Давление паров, мм рт. ст.
NaCl 5 4,9 14 9,1 23 15,9
6 5,3 15 9,7 24 16,9
7 5,7 16 10,3 25 17,9
8 6,1 17 11,0 26 19,0
9 6,5 18 11,7 27 20,2
10 6,9 19 12,4 28 21,4
Продолжение таблицы 8.19
Раствор Температура, °C Давление паров, мм рт. ст. Температура, °C Давление паров, мм рт. ст. Температура, °C Давление паров, мм рт. ст.
NaCl 11 7,4 20 13,2 29 22,7
12 7,9 21 14,1 30 24,0
13 8,5 22 15,0 — —
Na2SO4 16 11,6 24 18,9 32,4 30,8
18 12,9 26 21,4 35 36,0
20 14,7 28 23,8 39 44,6
22 16,7 30 27,2 — —
Газовый анализ
909
Таблица 8.20
Очистка и осушка газов при атмосферном давлении
Газ Удаляемые примеси Поглотитель
Аммиак Влага СаО
СО2 NaOH плавленый
Ацетилен РН3; H2S 10% раствор СгОз в разб. (1:1) H2SO4
Влага КОН плавленый
Диоксид азота NO; N2O Окисление кислородом с последующей конденсацией при -80 °C
Влага р2о5
Диоксид серы Влага Последовательно H2SO4 иР2О5
Диоксид углерода СО Гопкалит при 100 °C
Влага Последовательно СаС12и р2о5
Оксид азота (I) NO Насыщенный раствор FeSO4
СО2; NO2 КОН плавленый
Влага р2о5
Оксид углерода со2 Натронная известь или аскарит
Fe(CO)5; Ni (СО)4 Активная медь при 180— 200 °C
Непредельные углеводороды Влага Последовательно СаС12 и р2о5
Предельные углеводороды о2 Медные стружки, смоченные аммиачным раствором NH4C1
Непредельные углеводороды Бромная вода или пемза, смоченная конц. H2SO4
Влага Последовательно СаС12 иР2О5
Хлор НО; влага Последовательно Н2О, конц. H2SO4, СаО иР2О5
Таблица 8.21
< Растворимость диоксида углерода в некоторых жидкостях при 25 °C
а25 — объем газа (приведенный к 25 °C и 760 мм рт. ст.), поглощаемый единицей объема жидкости при парциальном давлении газа, равном 760 мм рт. ст.
Затворная жидкость «25
Вода 0,759
Глицерин, 50% водный раствор 0,540
Сульфат натрия, 20% водный раствор с добавкой серной кислоты (4—5 об. %) 0,270
Тиосульфат натрия, 4% водный раствор 0,125
Хлорид натрия, 18,7% водный раствор 0,352
Таблица 8.22
Растворимость газов в водном растворе сульфата натрия при 25 °C
Состав раствора: 200 г безводного Na2SO4 + 800 г дистиллированной воды + 40 мл 96% H2SO4.
а — коэффициент абсорбции Бунзена (см. выше); а25 — объем газа (приведенный к 25 °C и 760 мм рт.ст.), поглощаемый единицей объема жидкости при парциальном давлении газа, равном 760 мм рт. ст.
Газ а «25 Газ а «25
Азот 0,0045 0,0049 Кислород 0,0081 0,0089
Ацетилен 0,324 0,343 Метан 0,0085 0,0093
Водород 0,0067 0,0073 Оксид углерода 0,0036 0,0039
Диоксид серы 12,5 13,6 Этан 0,0099 0,0108
Диоксид углерода 0,247 0,270 Этилен 0,022 0,024
Оксид азота (I) 0,146 0,159
Метод конденсации и вымораживания основан на переводе примеси соответственно в жидкое или твердое агрегатное состояние и осуществляется путем охлаждения очищаемого газа. Необходимая для достижения заданной степени очистки температура определяется равновесным содержанием удаляемой примеси над конденсированной фазой в среде очищаемого газа при рабочем давлении. Этим методом можно проводить очистку газов, имеющих низкую температуру конденсации, и только в случаях, когда примесь имеет меньшую летучесть по сравнению с очищаемым газом.
Диффузионный метод очистки основан на различии коэффициентов диффузии и проницаемости через твердую перегородку (мембрану) очищаемого газа и примесей. Используются как металлические, так и полимерные мембраны.
Метод низкотемпературной ректификации является одним из основных промышленных методов, относящихся к многоступенчатым процессам разделения, и основан на многократном повторении акта разделения, обусловленного различием состава жидкой фазы и равновесного с ней пара. Осуществляется в тарельчатых или насадочных ректификационных противоточных колоннах, связанных внизу с кубом-испарителем или вверху — с конденсатором.
Адсорбционные методы основаны на физической избирательной адсорбции примеси поверхностью пористого адсорбента (активные угли, силикагели, алюмогели, природные и синтетические цеолиты) и осуществляются путем пропускания очищаемого газа через слой гранулированного или дробленого твердого адсорбента. Эффективность адсорбционной очистки тем выше, чем ниже температура и ниже давление.
Химические методы основаны на превращении примеси в нелетучие соединения при контакте очищаемого газа с твердыми химическими поглотителями (геттерами) и осу
910
Новый справочник химика и технолога
ществляются при нормальной или повышенной температуре. Поглотители должны иметь развитую и доступную для газа поверхность.
Каталитические методы состоят в том, что на твердом катализаторе протекает реакция превращения трудноуда-ляемой примеси в другую, легко удаляемую тем или иным способом химическую форму.
Методы фильтрования используются для очистки газов от взвешенных частиц — аэрозолей. Хорошо зарекомендовали себя фильтрующие материалы из ультратонких полимерных волокон типа ФПП (фильтры Петрянова), ультра-тонкого стекловолокна, фильтрующие мембраны.
8.7.1. Очистка водорода
Водород газообразный технический согласно ГОСТ 3022-80 выпускается трех марок: А, Б и В. Водород марки А, получаемый из азот-водородной смеси, содержит не менее 99,99 мол. % основного вещества; марки Б, получаемый электролизом воды, высшего сорта — 99,95 мол. %, и первого сорта — 99,8 мол. % основного вещества. Основные примеси — кислород, азот, аргон, оксиды углерода, метан. Содержание влаги в сжатом водороде в баллонах для марок А и Б — не более 0,2 г/м3.
Очистка водорода от примеси кислорода методом каталитического гидрирования по реакции
Н2 + уО2 = Н2О + 243 кДж/моль
позволяет просто снижать содержание кислорода. К наиболее активным катализаторам реакции гидрирования кислорода относятся металлы — платина, палладий, никель, нанесенные на пористый носитель с развитой поверхностью. Применение активного катализатора позволяет проводить процесс очистки при температуре около 100 °C.
Для очистки водорода от примеси кислорода нет необходимости использовать Pd- и Pt-катализаторы, достаточно активен никель-хромовый катализатор (ОСТ 6—03—314—71), который работает в восстановленном состоянии и может обеспечить высокую степень очистки. Поставляется катализатор в виде таблеток 4x4 мм в восстановленном и пассивированном состоянии (ввиду его пирофорности). Недостатки никель-хромового катализатора — невысокие механическая прочность и термостойкость (315 °C), последнее обусловливает возможность его применения при содержании примеси кислорода в водороде не более 1,5-2 мол. %. Для целей, рассматриваемых здесь, никель-хромовый катализатор вполне пригоден.
Для очистки водорода от кислорода недавно предложен никель-медный таблетированный катализатор НКО-2. Рекомендуемый состав: 25% NiO, 10% CuO, остальное — алюминат кальция.
Очистка водорода от кислорода может быть проведена методом химического поглощения последнего восстановленными металлами, например медью, при 180-200 °C. Поглотителями кислорода могут служить также восстановленные металлические катализаторы, например железный промотированный катализатор синтеза аммиака, ни
кель-хромовый катализатор или никель на кизельгуре (50% Ni). При пропускании водорода при комнатной температуре через поглотитель оксисорб (СгО на SiO2) со скоростью 120 л/мин (диаметр реактора 40 мм) также резко снижается содержание кислорода. Имеется практический опыт очистки водорода от кислорода методом низкотемпературной адсорбции на активированном угле. Равновесная адсорбция кислорода активированным углем СКТ при -196 °C характеризуется следующими данными:
Равновесное давление кислорода, гПа 6,7 13 67 133 200
Адсорбция, см3/г 499,4 513,0 612,5 639,0 660,0
Глубина очистки адсорбционным методом зависит от степени предварительной регенерации адсорбента. В варианте адсорбционного способа выделения и очистки водорода процесс адсорбции примесей происходит под давлением 2,0-4,0 МПа, а десорбция проводится сбросом давления до атмосферного, затем вакуумированием и, наконец, продувкой чистым водородом. Установка, состоящая из трех, четырех и более адсорберов с автоматически действующей системой переключения обеспечивает непрерывную работу (из четырех адсорберов один на стадии адсорбции, остальные на различных стадиях регенерации) и позволяет получать водород чистотой 99,9999 мол. %. При наличии в сырье активных сильносорбирующихся примесей перед входом в основную установку рекомендуется включить дополнительный адсорбер. При лабораторных испытаниях адсорбционного способа выделения чистого водорода из метано-водородной фракции пиролиза с использованием в качестве адсорбента активного угля СКТ получают водород с содержанием основы 99,9 мол. %.
Дальнейшую очистку от примесей СО, СО2 и ацетона проводят каталитическим гидрированием, промывкой 8-10% раствором триизобутилалюминия в октане и цеолитом NaX.
Глубокая очистка водорода от примесей азота, оксидов углерода, метана и аргона может быть осуществлена также переводом примеси в конденсированную фазу. При этом степень очистки определяется равновесным содержанием примеси в газе над твердой фазой при данных температуре и давлении. Следует иметь в виду, что фактическое содержание примеси в газовой фазе в этих условиях (высокое давление и низкая температура) значительно выше, чем рассчитанное по законам идеальных газов и равновесному давлению примеси над твердой фазой чистой примеси. При этом имеют место не только количественные различия, но и изменение характера зависимости остаточного содержания примеси в газе от давления в некоторой области р и Т: с ростом давления наблюдается не снижение, а повышение содержания примеси в газе. В соответствии с этим для снижения содержания указанных примесей в водоро
Газовый анализ
911
де методом вымораживания до уровня менее 10^ мол. % необходимо охлаждение до температуры ниже 243 К при давлении не более 0,5 МПа. Непрерывная очистка водорода методом вымораживания может быть осуществлена в реверсивных или парных переключающихся теплообменниках. В связи с тем, что возможна частичная конденсация примесей в объеме газа и водород после вымораживания может содержать примеси во взвешенной дисперсной (аэрозольной) форме, рекомендуется установка фильтров.
Диффузионный способ очистки дает возможность получить водород высокой чистоты с содержанием примесей менее 1(Г* мол. %. Водород отличается уникальной способностью диффундировать через тонкую перегородку (мембрану) из палладия и его сплавов с серебром, никелем; при этом газы-примеси остаются по другую сторону мембраны. Палладиево-серебряные мембраны выполняют в виде тонкостенной трубки или фольги. Фольгой из палладиевого сплава обертывают пористую трубку из мелкопористой металлокерамики. Фольгу используют также в диффузионных ячейках с плоской или спиралеобразной мембраной. Производительность диффузионных водородных очистителей с палладиевыми сплавами возрастает с повышением давления и температуры. Например, диффузионный элемент для очистки технического водорода (содержание Н2 99,7 мол. %) при давлении на входе 1 МПа и на выходе 0,1 МПа при температуре 177 °C пропускает 3,1 л/ч на 1 см2 поверхности. Выделенный водород имеет точку росы -90 °C и чистоту 99,99999 мол. %. В качестве материала для диффузионных элементов рекомендуются сплавы В-1 и В-2.
8.7.2. Очистка оксида углерода
Оксид углерода получают разложением (дегидратацией) муравьиной кислоты или ее солей (формиата натрия или бария) концентрированной серной или фосфорной кислотой: НСООН = СО + Н2О. При применении фосфорной кислоты получается более чистый оксид углерода. Выделяющийся газ отмывают от СО2 и паров кислоты водным 25% раствором КОН. Оксид углерода можно также получить восстановлением диоксида углерода при температуре около 1000 °C по известной реакции
СО2 + С = 2СО
или пропусканием СО2 через слой дисперсного углеродсодержащего материала.
В технике оксид углерода выделяют из продуктов газификации твердых топлив или при конверсии природного газа с водяным паром. Оксид углерода поставляют по ТУ 6-02-7-101-78 с содержанием основного вещества 99,9 мол. % в стальных баллонах под давлением. Следует иметь в виду, что после хранения в стальных баллонах в оксиде углерода обнаруживаются следы СО2 и карбонила железа. Из твердых поглотителей для очистки оксида углерода от диоксида применяется аскарит (плавленый NaOH на асбесте) по ТУ 6-09-4128-75. Применяется и Ф1 — гранулированный химический поглотитель СО2 на основе гидроксидов металлов. Хорошая очистка от СО2 достигается также методом вымораживания в ловушке,
охлаждаемой жидким азотом. Для очистки от кислорода пригодны поглотители, указанные выше для очистки водорода. При очистке путем пропускания через активную медь при температуре 200 °C одновременно происходит разложение карбонила. Очистка от примесей азота, кислорода, метана, водорода достигается многократной фракционной дистилляцией или ректификацией в насадочной колонне, как это практикуется при выделении из СО тяжелого изотопа углерода. Пригоден также адсорбционный способ очистки. В области малых парциальных давлений СО2 в качестве адсорбента наиболее активны цеолиты, а из цеолитов — СаА, СаА-4В и СеЕН-4В. Наиболее чистый оксид углерода, выпускаемый фирмой L'air Liquide (Франция), содержит 99,9972 мол. % основного вещества, примесей азота 10“3, кислорода 10”3, водорода 10“4, метана 10^ мол. %.
8.7.3. Очистка диоксида углерода
В лаборатории газообразный диоксид углерода получают разложением карбонатов кислотами (например, гидрокарбоната калия серной кислотой), а также термическим разложением гидрокарбоната натрия (110-120 °C) или карбоната магния (540 °C). Диоксид углерода в промышленности получают термическим разложением известняка или выделяют чаще всего абсорбционным методом из продуктов горения топлива или из промышленных газов, например так называемых конвертированных газов для синтеза аммиака. Диоксид поставляется в жидком состоянии в стальных баллонах (давление в баллоне 56 МПа) либо в твердом виде (сухой лед). Диоксид углерода пищевой содержит 99,96 мол. % основного вещества. Фирма Mateson (США) выпускает диоксид углерода для исследовательских целей (research grade) чистотой 99,995 мол. %. Простой способ снизить содержание более летучих примесей в баллонном диоксиде углерода состоит в испарении части содержимого баллона.
Глубокая очистка диоксида углерода достигается многократной фракционной возгонкой (температура возгонки СО2 -78,5 °C). Для этого газ из баллона после осушки цеолитом конденсируют в ловушке при температуре жидкого азота, откачивают оставшиеся газы, соединяя ловушку с вакуумной системой, затем дают диоксиду испариться и повторяют описанный процесс замораживания и откачки еще дважды. Затем полученный газ переводят в другую ловушку и снова перегоняют, отбирая в качестве конечного продукта среднюю фракцию. Чистота получаемого таким путем диоксида углерода не менее 99,999 мол. %.
Для очистки СО2 возможно применение адсорбционного метода. Известен вариант адсорбционно-конденсационного метода получения особо чистого диоксида углерода с содержанием примесей влаги на уровне менее ПГ4 мол. %, углеводородов менее КГ6 мол. %, воздуха менее 51 (Г5 мол. %, минеральных масел и кислотных примесей ниже предела обнаружения методики. Для получения особо чистого СО2 достаточно 2-3 циклов переконденсации. Используется экспериментальная установка, выполненная из материалов, имеющих минимальную пористость, газо-
912
Новый справочник химика и технолога
отделение и высокую коррозионную стойкость. Все уплотнения сварные. Полимерные и резиновые прокладки отсутствуют. Коммуникации выполнены из нержавеющей стали 12Х18Н9Т с внутренней полировкой. Установку предварительно обезгаживают в вакууме при 1000 °C в течение 8 ч, запорная арматура с сильфоновым уплотнением и все коммуникации имеют внешний прогрев при 100-150 °C. Предварительная тренировка в вакууме проводится в течение 15-18 ч при давлении 1 Па. Для поглощения использован цеолит КА-ЗМ. Условия регенерации: продувка сухим азотом с последующим вакуумированием (1 Па, 400 °C, 12 ч).
8.7.4. Очистка метана
Метан в настоящее время чаще всего выделяют из природного газа. Метановые фракции получают также при низкотемпературном разделении газов пиролиза и крекинга нефтепродуктов, продувочных газов синтеза аммиака. Метан получают либо каталитическим гидрированием оксида углерода, либо из метилиодида, метилбромида по реакции Гриньяра через магнийиодметил или соответственно магнийбромметил. Дополнительная очистка метана может быть проведена низкотемпературной ректификацией с использованием жидкого азота в качестве хладоагента, а также низкотемпературной адсорбцией. Наиболее чистый метан содержит (мол. %): основного вещества — 99,9995, примесей азота — 210“5, кислорода —0,510“5, водорода — 0,110 5, СО2 — 1 • 10 5 мол. %.
8.7.5. Очистка оксида азота
Оксид азота в больших количествах получается в составе так называемых нитрозных газов в производстве азотной кислоты. Однако содержание NO в этих газах составляет всего 10-12 мол. %. Поэтому, а также вследствие сложности состава нитрозных газов, содержащих кроме NO другие оксиды азота, кислород, пары воды, элементарный азот, выделение чистого NO из этой смеси затруднительно и нерационально. Высокие содержания оксида азота получают восстановлением азотной кислоты диоксидом серы:
2HNO3 + 3SO2 + 2Н2О = 3H2SO4 + 2NO.
Наряду с этим может протекать реакция, приводящая к образованию диоксида азота:
2HNO3 + SO2 = 2NO2 + H2SO4.
Протеканию второй реакции способствует повышение концентрации кислоты, однако при снижении концентрации реакция замедляется. С учетом выхода NO и скорости его образования рекомендуется использовать 4,5-молярный раствор азотной кислоты. Высшие оксиды азота образуются также вследствие окисления NO кислородом в газовой фазе. Полученный описанными выше способами оксид азота подвергают очистке. В качестве первой стадии очистки рекомендуется промывка водой, при которой проис
ходит поглощение высших оксидов с образованием дополнительного количества NO по реакциям:
3NO2 + Н2О = 2HNO3 + NO;
3N2O3 +Н2О = 2HNO3 +4NO.
При этом в целях предотвращения окислительного процесса рекомендуется применять деаэрированную (кипячением или вакуумированием) воду. Для повышения степени очистки и осушки газа, а также исключения кислых стоков промывку оксида азота целесообразно проводить серной кислотой. Дальнейшая очистка и осушка NO осуществляется в двух последовательных колоннах, заполненных силикагелем и работающих при температуре 0 и -83 °C, после чего оксид конденсируют с помощью жидкого азота. Для дальнейшей очистки NO испаряют, пропускают через дополнительную силикагелевую колонку при -83 °C и далее подвергают низкотемпературной ректификации.
Один из лабораторных способов получения и очистки оксида азота состоит в медленном (по каплям) прибавлении 40% водного раствора нитрита натрия к раствору сульфата железа (II). Выделившийся газ очищают промывкой раствором КОН и концентрированной серной кислотой, осушают охлаждением твердым диоксидом углерода и пентаоксидом фосфора и вымораживают с помощью жидкого азота. Неконденсирующиеся газы откачивают вакуумным насосом. Реализован более совершенный способ очистки оксида азота, получаемого с использованием последней реакции в баллонах под давлением 3,4 МПа. Очистку от влаги и диоксида азота проводят методом вымораживания с фильтрованием на металлической сетке при температуре 143 °C. При этом обеспечивается высокая чистота продукта, не ниже 99,9 мол. %, поскольку очистка не связана с применением химических веществ. Инертные газы и азот отделяют методом низкотемпературной сублимации. Фракционная дистилляция и возгонка твердого оксида азота в вакууме дают возможность получить газ с содержанием примесей 104 мол. %.
8.7.6. Очистка кислорода
В настоящее время кислород получают низкотемпературной ректификацией воздуха либо электролизом воды. Технический газообразный кислород первого сорта содержит не менее 99,7 мол. % основного вещества. Кислород особой чистоты по ТУ 6-21-05-22-79 содержит не менее 99,999 мол. % кислорода, не более 104 примеси диоксида углерода, не более 9-104 мол. % (в сумме) примесей азота, аргона, неона, криптона, ксенона и метана. Дальнейшая очистка газообразного кислорода, поставляемого в баллонах или получаемого газификацией жидкого кислорода, может быть осуществлена сочетанием осушки и удаления диоксида углерода и углеводородов сорбционным методом с помощью цеолитов и ректификации. Наиболее трудноотделимой примесью, лимитирующей очистку, является аргон, так как коэффициент разделения его относительно невелик и в области малых содержаний аргона при давлении 1,5 Па составляет 1,65. Очевидно, что все остальные,
Газовый анализ
913
более летучие, чем аргон, примеси эффективно удаляются с дистиллятом. Методами охлаждения или вымораживания удаляют примеси влаги и диоксида углерода. Количество влаги в очищенном газе соответствует точке росы при температуре выхода из теплообменника (табл. 8.23).
Таблица 8.23
Содержание влаги в кислороде при различных температурах
Точка росы (°C) при давлении Равновесное давление пара, Па Влагосодержание
10 кПа 20 МПа г/м3 млн 1
-90 -56 1,45 • 10~2 7,6 • 10’5 0,092
-80 -43 7,92 • 10’2 4,3 • 10^* 0,53
-76 -36 1,49 • 101 0,8 • 10“3 1,01
-70 -27 3,64 • 101 2,1 • 103 2,56
-60 -5 1,45 8,6 • 10’3 10,6
-50 +2 3,89 3,2 • 10 2 38,8
Таблица 8.24
Характеристика фильтровальных материалов
Материал Толщина, мм Пористость, отн. ед. Нормальная тонкость* очистки, мкм Прочность, МПа
Фильтровальная сетка № 685 из стали 12Х8Н10Т 0,138 - 20 -
Пористый фторопласт: ФЭП-10-12; ФЭП-0,5-1,0 6,6-7,0 0,728 0,676 10-12 0,5-1,0 0,82 0,87
Ленточный пористый прокат: ФНС-10 ФНС-2 0,13-0,25 0,1-0,14 0,38-0,44 0,25-0,38 10-12 2-3 Не менее 40
Следует понимать как минимальные размеры задерживаемых частиц.
Недостаток данного метода — возможность образования аэрозоля. При быстром объемном охлаждении очищаемого газа примесь замерзает раньше, чем заканчивается ее транспорт к охлаждаемой поверхности. В таком случае необходимо устанавливать за теплообменником фильтры для взвешенных тонкодисперсных частиц примеси. Основными видами твердых примесей в криогенных системах являются кристаллы замерзшей жидкости, причем они обладают таким абразивным действием на материалы оборудования, что приводят к его преждевременному износу и образованию вторичных твердых примесей в потоках криогенных веществ — частиц металлов и используемых сорбентов. В настоящее время используется ряд материалов для фильтрования криогенных жидкостей и газов (табл. 8.24).
К числу перспективных фильтровальных материалов, позволяющих осуществлять высокую тонкость очистки (до 5-10 мкм), следует отнести многослойные пористые материалы (ПСМ), основные характеристики которых приведены в табл. 8.25. Пакетный и гофрированный фильтры испытаны при очистке жидкого азота и водорода. При перепаде давления 2,5 кПа удельный расход жидкого азота составил 0,2 л/(см2 мин). Для глубокой очистки криопродуктов используют фильтры Петрянова. Однако часть примеси, растворенной в жидком продукте (например, количество диоксида углерода, растворяющееся в жидком воздухе, составляет около 56 см3/м3), не задерживается фильтрами и извлекается другими методами. Используются фильтры из высокочистого диоксида кремния, содержащего примеси металлов в сумме 7-10 6 масс. %, с пористостью 30-60 %, позволяющие снизить концентрацию частиц размером более 0,04 мкм до 103 см-3.
Содержание взвешенных нерастворимых частиц можно существенно снизить также путем медленного испарения жидкой фазы целевого продукта без кипения. Глубокая очистка кислорода перед подачей в колонну низкотемпературной ректификации начинается с очистки воздуха от влаги, диоксида углерода и ацетилена методом адсорбции. Обычно этот процесс проводят комплексно, т. е. одновременно извлекают из потока газа влагу и диоксид углерода на цеолитах. Из промышленных цеолитов рекомендуется цеолит марки NaX, емкость которого по диоксиду углерода при очистке влажного воздуха равна 2,3-3,5%, а динамическая активность по парам воды составляет 2,5-5,5% от массы сорбента при давлении от 2,5 до 20 МПа. Ацетилен и другие углеводороды адсорбируются почти полностью и не оказывают влияния на очистку воздуха от диоксида углерода.
Таблица 8.25
Характеристика многослойных ПСМ
Тип сетки Число слоев в пакете Толщина ПСМ, мм Пористость Средний размер пор, мкм
N685 10 0,87-1,3 0,11-0,3 6,6-22
С 450 10-20 1,5-3,0 0,13-0,32 11-51
С 200 10 3,2-3,8 0,11-0,22 19,9-56,3
С 120 4 1,36-1,45 0,11-0,15 19^2,5
П80 8 2,99-3,69 0,37-0,47 146-191
П60 10 3,2-4,5 0,05-0,3 12-250
0071 10 0,71-1,0 0,24-0,53 27-65
914
Новый справочник химика и технолога
Применение мембранных процессов в разделении газовых смесей — новое перспективное направление, позволяющее в ряде случаев получить значительный экономический эффект, особенно для маломасштабных задач. Однако использование известных в настоящее время полимерных мембран для глубокой очистки газов еще не получило широкого распространения. Поскольку в области малых содержаний резко уменьшается движущая сила диффузии (разность парциальных давлений) молекул примеси, то преимущественно через мембрану проникает основной компонент. Поэтому материал мембраны должен обладать большей проницаемостью по отношению к основному компоненту. Удаление накапливающегося в кислороде метана (несколько долей на миллион) может быть осуществлено путем его выжигания в печах на катализаторах (оксиды меди или алюминия). Оно должно предшествовать очистке кислорода от влаги и диоксида углерода. Примеси криптона и ксенона могут быть удалены из смеси с кислородом методом адсорбции на силикагеле.
8.7.7. Очистка азота
В больших масштабах азот получают на криогенных воздухоразделительных установках методом ректификации. По ГОСТ 9293-74 азот особой чистоты содержит не менее 99,9996 мол. % основного компонента, а примесей водорода, кислорода, углерода (в пересчете на диоксид углерода) менее 1(Г4мол. % каждого компонента и менее 5-10 4 мол. % влаги. Азот высшего качества, выпускаемый за рубежом, содержит от 99,9990 до 99,9996 мол. % основного компонента. Наиболее легко методом ректификации отделяются легкие примеси (водород, гелий, неон). Поэтому обычно азот, получаемый в воздухоразделительных установках, уже не требует дополнительной очистки от легких примесей. Из более тяжелых примесей труднее всего отделить кислород, аргон и оксид углерода, но и в этом случае коэффициент разделения превышает 1,8 (по оксиду углерода при атмосферном давлении), что позволяет получить азот особой чистоты непосредственно в воздухоразделительной установке. Более глубокая очистка может быть достигнута ректификацией в насадочной колонке.
Азот, получаемый в небольших количествах из воздуха методом адсорбции на углеродных молекулярных ситах, содержит гораздо больше примесей — до 2101 мол. % и требует дополнительной очистки. Азот, получаемый диффузионным методом с использованием полимерных мембран, содержит еще больше примесей. Содержание основного вещества в продукте в зависимости от производительности колеблется от 95 до 99 мол. %, т.е. продукт требует дополнительной очистки. От влаги и диоксида углерода азот очищают теми же методами, что и кислород. Очистка технического азота от кислорода проводится путем его химического связывания в слое меди, нагретом до 400-470 °C. Технический азот с добавкой аммиака пропускают над активной медью. При этом кислород соединяется с медью, а оксид меди восстанавливается аммиаком до свободной меди. Такая очистка позволяет снизить содержание кислорода в техническом азоте до 10 5 мол. %. Ка
талитические методы очистки газов от кислорода предполагают присутствие в очищаемом газе добавки водорода, превышающей его стехиометрическое количество для образования воды. Добавление водорода является недостатком метода, так как затем требуется очистка газа от водорода. Этот метод позволяет снизить содержание кислорода в газах с нескольких процентов до нескольких миллионных долей. Адсорбционная очистка азота от СО на цеолитах СаХ, СаА позволяет снизить содержание примеси СО до 104 мол. %, а на угле БАУ при температуре от -76 до -196 °C можно снизить содержание примеси СН4 до 10 5 мол. %.
8.7.8. Очистка аргона
Одним из основных продуктов, получаемых из воздуха методом низкотемпературной ректификации, является аргон. В табл. 8.26 представлены данные по составу примесей, содержащихся в аргоне, производимом в России.
Таблица 8.26
Содержание примесей в аргоне п • 104 мол. %
Компонент Аргон высокой чистоты ТУ 6-21-12-79
по нормативному документу, газообразный, 1 сорт после дополнительной очистки на БКЗ* по ТУ 6-21-30-83
Гелий - 2
Неон - 2
Водород 2 1
Кислород 2 1
Азот 10 3
Оксид углерода - 1
Диоксид углерода 0,2 1
Метан 1 .1
Пары воды 3 2,5
ОАО Балашихинский кислородный завод.
Аргон, производимый за рубежом, относится к наиболее чистым газам. Содержание основного вещества составляет 99,9990-99,99990 мол. %. С помощью адсорбции при комнатной температуре возможна очистка аргона от примесей СО2 , Н2О, NH3 и СН4 до уровня менее 10-4 мол. %. Отбираемый из воздухоразделительной установки сырой аргон содержит около 92 мол. % основного вещества, остальное, главным образом, примеси кислорода и азота.
Первая стадия очистки аргона предусматривает извлечение кислорода. В промышленности применяется метод очистки аргона от кислорода, основанный на каталитическом гидрировании кислорода. Наиболее эффективны катализаторы на основе металлов платиновой группы. Чаще используются платина и палладий, нанесенные на активный оксид алюминия. Содержание активного компонента не превышает 5% (по массе). Температура в контактном аппарате не превышает 500 °C. При стехиометрическом со
Газовый анализ
915
отношении содержаний водорода и кислорода после установления термодинамического равновесия компонентов давление кислорода по расчетным оценкам составляет 1О-10 МПа, а при небольшом избытке водорода давление кислорода можно снизить до 10-25 МПа. На практике глубина очистки аргона от кислорода этим методом составляет менее 1 (Г4 мол. % кислорода. По правилам техники безопасности и условиям эксплуатации стандартного палладиевого катализатора исходное содержание кислорода не должно превышать 2 мол. %. При доочистке аргона от кислорода перед приготовлением газовых смесей используется хромсиликатный сорбент, позволяющий снизить содержание кислорода до уровня менее 210-6 мол. %.
Проводится очистка аргона от кислорода и методом ректификации сырого аргона или аргоновой фракции воздухоразделительной установки. Преимущества этого метода состоят в использовании высокоэффективных контактных устройств — насадок из металлической сетки, позволяющих проводить очистку в колоннах небольшого диаметра, и выражаются в отсутствии каталитического гидрирования кислорода, упрощении технологии очистки аргона и исключении вторичного загрязнения аргона водородом.
Очистку аргона от азота целесообразно проводить методом низкотемпературной ректификации в комплексе с очисткой от кислорода.
8.7.9. Очистка неона
Единственным источником получения неона является атмосферный воздух, в котором содержится 18-10 4 мол. % неона. Неон не сжижается в воздухоразделительной установке, а накапливается вместе с гелием в верхней части конденсатора нижней колонны до содержания от 3 до 10 мол. % (неон + гелий). Основным компонентом сырой неон-гелиевой смеси является азот, присутствуют также водород и следы кислорода. Для увеличения содержания неона и гелия отбираемую при давлении 0,6 МПа фракцию переохлаждают в дефлегматоре кипящим при 0,14 МПа жидким азотом. При этом азот из потока сырой неонгелиевой смеси частично конденсируется, а доля неона и гелия повышается примерно до 50-60 мол. %. По ТУ 6-21-21-77 сырая неон-гелиевая смесь должна иметь состав: неон + гелий — не менее 60, азот — не более 40, водород — не более 3, кислород — не более 0,3 мол. % и влага не более 0,1 г/м3 (102 мол. %). Дальнейшее концентрирование неонгелиевой смеси после каталитической очистки от водорода происходит при ее охлаждении кипящим под вакуумом жидким азотом. Получаемая смесь уже содержит 5-10 мол. % азота, однако при этом теряется часть неона, вследствие его растворения в жидком азоте. Последующая очистка неон-гелиевой смеси от азота производится методом низкотемпературной адсорбции на активированном угле. Такая многоступенчатая очистка неон-гелиевой смеси от азота, не претерпевая принципиальных изменений, применяется повсеместно. Получаемый продукт, согласно ТУ 6-21-4-76, в своем составе содержит: 99,985 мол. %
неона и гелия в отношении 3:1; 0,01 мол. % азота; по 0,001 мол. % кислорода, водорода, оксида и диоксида углерода; 0,0013 мол. % углеводородов и паров воды. Этот продукт служит сырьем для получения неона. В табл. 8.27 приведен состав примесей в неоне. Выпускаемый за рубежом неон содержит основного вещества от 99,995 до 99,9995 мол. %.
Таблица 8.27
Содержание примесей в неоне «104 мол. %
Компонент Неон высокой чистоты ТУ 6-21-09-78
по нормативному документу дополнительно очищенный на БКЗ
Неон 99,994 99,995
Гелий 50 40
Водород 1 1
Кислород 1 1
Азот 3 3
Оксид углерода — о,1
Диоксид углерода 0,1 0,1
Метан 0,1 0,1
Пары воды 2 2
Аргон — —
Криптон — 0,6
Углеводороды - 0,5
Наибольшую трудность представляет отделение примеси гелия. На промышленных установках разделение неонгелиевой смеси и очистка неона осуществляется адсорбционным методом, в том числе на углях БАУ, СКТ, АГ2, методом вымораживания с использованием жидкого водорода, а также конденсационным методом. В первом методе перспективно использование в качестве адсорбента активного угля. Процесс адсорбции смеси протекает при температуре жидкого азота, кипящего под вакуумом. Не-сорбирующийся газ, обогащенный гелием, собирается и компримируется в баллоны, а обогащенная неоном часть десорбируется при температуре 20 °C и также компримируется в баллоны. Регенерация сорбента проводится при температуре 127 °C и остаточном давлении З-Ю4 Па. Установка перерабатывает 2,8 м3/ч сырой неон-гелиевой смеси. Второй метод, связанный с применением жидкого водорода, взрывоопасен и может быть использован лишь на специализированных предприятиях, имеющих резервы жидкого водорода.
Используется и конденсационно-ректификационный способ разделения неон-гелиевой смеси. Установка ТОН-2 для очистки неона от гелия и водорода включает ректификационную отгонную колонну, заполненную нерегулярной насадкой КМС (кольца из металлической сетки) с размером элементов 5,2x5,2 мм, конденсатор, сепаратор, заполненный регулярной насадкой из металлической сетки, неоновый холодильный цикл и теплообменную аппаратуру. Основные аппараты размещены в вакуумном кожухе.
916
Новый справочник химика и технолога
Сырьем служит неон-гелиевая смесь, предварительно очищенная от азота и кислорода адсорбционным методом на активном угле.
Существенным отличием установки является объединение неонового холодильного цикла с кубом ректификационной колонны. Отвальный поток, обогащенный гелием (до 10 мол. % гелия) и содержащий водород, выводится из верха сепаратора. Поток, выходящий из головы колонны, по составу близок к исходному и объединяется с ним, возвращаясь на переработку.
Полученный на установке ТОН-2 неон по качеству является наиболее чистым в стране и представлен на постоянно действующей выставке коллекции высокочистых веществ. Содержание гелия в неоне не превышает 4-1О3 мол. %.
8.7.10. Очистка гелия
Источником получения гелия являются природные газы. Для эксплуатируемых месторождений характерно высокое содержание гелия — от 0,9 до 5,7 мол. %. Помимо гелия природные газы обычно содержат 10-30 мол. % азота, а также метан и незначительные примеси менее летучих углеводородов, углекислоты, влаги, сероводорода, водорода. Так как гелий наиболее летучий из известных газов, то его получение сводится к конденсации всех остальных компонентов смеси и окончательной очистке методом низкотемпературной адсорбции. Извлекается гелий методами глубокого охлаждения, причем процесс осуществляется в две стадии: получение так называемого сырого гелия и последующая его очистка. В таблице 8.28 указан средний состав природного газа, поступающего на переработку, а также состав переработанного газа после извлечения из него гелия.
Таблица 8.28
Состав поступающего и отвального газов
Компонент Содержание в газе, мол. %
поступающем выходящем
Метан 64 67
Этан 3,1 3,3
Пропан 1,9 2,0
н-Бутан 0,6 0,7
Изобутан 0,4 0,4
н-Пентан 0,2 0,2
Изопентан 0,2 0,2
Циклопентан 0,05 0,05
Гексан и менее летучие УВ 0,2 0,2
Азот 26 26
Аргон 0,1 0,1
Водород 0,05 0,05
Сероводород следы —
Углекислота 0,8 -
Гелий 2,1 0,13
Природный газ поступает из месторождения под давлением 30-45 атм. и подвергается очистке от углекислоты и осушке. При удалении углекислоты частично поглощается и влага. Выходящий газ содержит 0,005 мол. % углекислоты и около 0,2 г/м3 влаги. Дальнейшая осушка газа проходит в адсорбере с активированным бокситом и синтетическими цеолитами. Содержание влаги после этого снижается до ®ЗЮ~5 г/м3, что соответствует температуре точки росы -73 °C при давлении ® 30 атм. Очищенный таким образом природный газ поступает в разделительную установку, где получают сырой гелий. Для этого газ охлаждают до -157 °C, что сопровождается конденсацией углеводородов, которые затем выводятся, компримируются и направляются в газопровод для дальнейшего использования. Несконденсиро-вавшийся газ представляет собой сырой гелий, в котором содержится небольшое количество водорода.
Для его удаления к потоку газа добавляется соответствующее количество воздуха и на платиновом катализаторе производится связывание водорода с кислородом. Образующаяся влага поглощается в адсорбере. Содержание водорода в сыром гелии не превышает 0,01 мол. %. Сырой гелий компримируют до 190-200 атм., осушают и подают на дальнейшую очистку. Очистка осуществляется путем дальнейшего охлаждения сжатого газа, сопровождающегося выделением примесей, и поглощения оставшихся примесей активированным углем при низких температурах. Продукционный гелий под высоким давлением (150-200 атм.) поступает в стальные баллоны. Содержание примесей в продукционном гелии лежит в следующих пределах:
Водород 4 : 10"5-210^ мол. %
Неон 4 • 10^-6-10 4 мол. %
Азот 2 • 10“5-5-10 4 мол. %
8.8. Производство газовых смесей
Для нужд различного рода технологий и для решения задач метрологического обеспечения ГА (градуировка и поверка средств ГА) используются специально приготавливаемые газовые смеси. В последнем случае они называются поверочными газовыми смесями (ПГС).
8.8.1. Основные типы ПГС и требования к ним
Существующие ПГС имеют статус либо государственных стандартных образцов (ГСО), либо отраслевых (ОСО). Основные требования к ПГС:
1. Строгая определенность номенклатуры ПГС для каждого типа рабочих средств измерения (РСИ).
2. Состав ПГС должен быть неизменен в течение определенного, известного, срока их хранения.
3. Количество ПГС должно быть достаточным для обеспечения операции поверки.
Наиболее эффективный способ хранения ПГС — в металлических баллонах (со специально подготовленной внутренней поверхностью стенки) под давлением (как правило, 15,0 МПа). Технические требования к выпускаемым промышленностью ПГС определены ТУ-6-16-2956-87
Газовый анализ
917
Состав исходных газов, используемых при производстве ПГС
Таблица 8.29
Газ Содержание компонента в газе, мол. % Сн2О, г/м3
Не Аг Ne Хе N2 о2 Н2 со2 СН4 СО
Не 99,98 1-Ю"3 9-10“3 — 5-10 3 210"3 2,5-10"3 1-Ю"3 ЗЮ"3 — 2-10"3
Ne 2103 - 99,99 — ЗЛО4 1-Ю"4 МО4 1-Ю"5 , 1-Ю"5 З-Ю"3
Аг - 99,99 — — 2-10"3 2404 2-10"4 5-Ю"5 1-Ю"4 — 2-Ю"3
Хе — - - 99,99 МО"3 5-Ю"4 — 1-10 4 МО4 — 4-Ю"3
n2 — — - - 99,999 5-Ю"4 110 4 5-Ю"5 5-10"5 5-Ю"5 5-10"3
О2 - 3-Ю"4 ЗЮ5 З-Ю"5 З-Ю"4 99,999 — 6-10"5 6-10"5 1-Ю"5 7-10"3
Н2 — 3-Ю"4 — — З-Ю4 ЗЛО4 99,999 — — — 2-1 О’4
со2 - - — — — — — 99,8 — — 4-Ю"2
сн4 — — - - З-Ю"2 3-Ю"2 — — 99,9 — З-Ю"2
со - - - - 0,5 0,1 0,3 1 — 98,0 -
«Смеси газовые поверочные — стандартные образцы состава». В зависимости от допускаемой погрешности действительного значения содержания определяемого компонента ПГС делятся на три разряда — нулевой, первый и второй. Изготавливаемые смеси закрепляются за определенным заказчиком.
8.8.2. Условия и особенности приготовления ПГС
Для производства ПГС применяют технические и чистые газы, поставляемые промышленностью в сжиженном или сжатом состоянии в баллонах под давлением. Обычно ПГС состоят из одного или двух определяемых компонентов в среде газа-разбавителя (например, СО и Н2 в азоте, N2 в аргоне, СН4 и другие углеводороды в воздухе и т.п.). При приготовлении ПГС следует учитывать несовместимость некоторых газов, т.е. возможность взаимодействия между ними в обычных условиях или в присутствии того или иного «третьего» компонента. Это в дальнейшем приводит к погрешностям в градуировке средств измерения. Несовместимы: аммиак и галогены, аммиак и галогеново-дороды, аммиак и оксид хлора, ацетилен и хлор, водород и оксид хлора (при освещении), водород и хлор (при освещении), оксид азота и кислород, оксид азота и хлор (в присутствии паров воды), оксид углерода и хлор (при освещении), сероводород и кислород (в присутствии паров воды), сероводород и диоксид углерода (в присутствии паров воды), углеводороды (алифатические) и хлор (при освещении), этилен и хлор.
Не содержащие агрессивных газов ПГС можно хранить в течение полугода без проведения повторных анализов.
Содержащие микроконцентрации агрессивных газов (NO, NO2, SO2, NH3 и др.) ПГС могут храниться в баллонах при соответствующей подготовке последних (отмывка, обработка поверхности стенки, специальные покрытия и т.п.) в течение длительного времени. Однако их необходимо периодически анализировать, т.к. содержание агрессивного компонента постепенно снижается. Смеси с микро- и макросодержаниями сероводорода хранению в баллонах не
подлежат. Состав газов, используемых при производстве ПГС, приведен в табл. 8.29.
8.8.3. Способы приготовления ПГС
1. Способ парциальных давлений, состоящий в использовании закона Дальтона при смешении газов, в соответствии с которым сумма парциальных давлений />, равна дав-
п
лению смеси /?см, т.е. p = ^Pi, где суммирование прово-;=1
дится по п компонентам смеси. Предварительно по заданному конечному давлению смеси и задаваемыми содержаниями Х( компонентов смеси рассчитывают давления Pi каждого компонента по формуле р, = х, • рсм.
С учетом результатов расчетов каждый из компонентов поочередно дозируют в баллон, контролируя по манометру парциальные давления />,. Такой способ приготовления смесей осложнен отклонением законов, описывающих состояние идеальных газов, от законов для случая реальных газов. Величина отклонения для каждого газа определяется коэффициентом сжимаемости
Коэффициент сжимаемости зависит от температуры газа в баллонах. Учесть в процессе производства смесей это обстоятельство (особенно в случае приготовления многокомпонентных смесей) довольно трудно, следствием чего являются отклонения от номинального значения содержания, называемые погрешностью приготовления смеси. Поэтому после приготовления смесь должна пройти аттестацию, т.е. должна быть проанализирована с привлечением образцового средства измерения.
2. Динамический способ, заключающийся в смешении точно измеренных потоков двух и более компонентов при низком давлении с последующим компримированием смеси и подачей ее в баллоны. Расчет потоков смешиваемых компонентов qt производят по заданным содержаниям х, компонентов в смеси и производительности компрессора
918
Новый справочник химика и технолога
Qi- Qi = Qi ’ xi- Высокая точность приготовления смеси в этом случае обусловлена возможностью в ходе процесса смешения осуществлять корректировку потоков компонентов по показаниям газоанализатора. В этом случае полученная смесь также должна пройти аттестацию.
3. Гравиметрический (или весовой) способ заключается в последовательном дозировании компонентов смеси в баллон и проведении взвешивания с целью нахождения массы вводимых компонентов. Массу компонента определяют как разность результатов взвешиваний до и после введения компонента. По результатам взвешивания, с учетом чистоты исходных газов, определяют содержание каждого компонента в смеси (молярную долю):
П{ = qilm^qilmi,
где mi — масса одного моля компонента. На практике используются весы, на которых возможно взвешивание масс до 30 кг с точностью до 3 мг, 50 кг с точностью 3 мг и 100 кг с точностью 10 мг. Ниже приведены значения относительной погрешности (%) приготовления смесей весовым методом:
мол. % Германия Япония Франция
0,0005-0,01 1 0,2 10
0,01-2,0 0,5 0,2 1
2,0-50 0,1 0,05 2
Применение весового способа не требует аттестации полученных смесей. Поэтому он является основным средством поверки образцовых средств измерения состава газовых сред.
8.8.4. Способы перемешивания компонентов в процессе приготовления смесей
Однородность газовой смеси в баллоне достигается в результате действия естественной молекулярной диффузии, принудительного перемешивания компонентов путем вращения баллона и принудительного перемешивания в процессе конвекции. Первый способ заключается в том, что баллон после дозирования в него компонентов выдерживается определенное время в состоянии покоя. За счет молекулярной диффузии компоненты смеси равномерно распределяются по объему баллона. Время т, в течение которого обеспечивается однородность смеси, оценивается по формуле
где L — характерный размер емкости, a D — коэффициент взаимной диффузии компонентов смеси. В общем случае время перемешивания может изменяться от 2 до 30 суток и зависит от свойств смешиваемых компонентов, соотношения их содержаний, давления смеси, геометрии объема, его размеров, температуры газа.
Второй способ перемешивания путем вращения баллона в горизонтальном положении вокруг горизонтальной
оси со скоростью 80 об/мин позволяет сократить время перемешивания до 2 часов для всех газовых смесей.
Третий способ состоит в создании градиента температуры по высоте баллона, вследствие чего возникает конвективный поток газа и происходит принудительное конвективное перемешивание (нагрев нижней и охлаждение верхней части баллона). Для газов разных составов время перемешивания варьируется в пределах от 20 минут до 8 часов.
8.8.5. Источники загрязнения газов и методы его предупреждения
Многочисленные источники возможного загрязнения газов можно разделить на две основные группы: взаимодействие газов с поверхностью твердых тел, ограничивающих объем газа, и негерметичность системы. При соприкосновении газа с поверхностью твердого тела имеют место два противоположных процесса — поглощение газа (сорбция, адсорбция) и газовыделение (десорбция с поверхности, диффузия газа из объема твердого тела, сквозная диффузия из окружающей среды). Так, например, наблюдается заметная диффузия Не, Н2, Ne, Аг, О2 через стекло. Скорость ее зависит от перепада давления. Водород хорошо диффундирует через нагретые палладий, сталь. Легко проницаемы для газов полимеры. Поэтому для снижения роли различного рода загрязняющих процессов необходимо правильно выбирать материалы для систем хранения газов и использовать необходимые в каждом конкретном случае приемы обработки поверхности, контактирующей с газом (шлифовка, полировка, покрытия различного рода, термообработка и т.п.).
8.8.6. Вспомогательные вещества
На различных этапах подготовки газа к анализу и непосредственно в процедуре анализа используются те или иные вещества в качестве затворных жидкостей. Чаще всего для этой цели применяют концентрированные растворы солей (NaCl, Na2SO4 и др.). При работе с такими растворами следует учитывать давление паров воды над ними. Для обеспечения минимальных погрешностей, обусловленных растворимостью газов, наиболее эффективно применение в качестве затворной жидкости очищенной и перегнанной ртути. Давление паров ртути очень мало, и некоторые газы (например, Н2, О2, N2) в ней нерастворимы. В случае анализа сероводорода и других газов, реагирующих со ртутью, в качестве затворных жидкостей применяют смазочные масла, предварительно насыщенные анализируемым газом (табл. 8.30 и 8.31).
В качестве затворных жидкостей в газовом анализе чаще всего применяют концентрированные растворы солей (NaCl, Na2SO4 и др.). При работе с такими растворами следует учитывать давление паров воды над ними.
Погрешности анализа за счет растворимости газов в затворной жидкости и давления паров растворителя над ней могут быть исключены, если в качестве такой жидкости применять очищенную и перегнанную ртуть; давление паров ртути очень мало, и некоторые газы (например, Н2, N2, О2) в ней нерастворимы.
Газовый анализ
919
В случае анализа сероводорода и других газов, реагирующих с ртутью, в качестве затворной жидкости применяют смазочные масла, предварительно насыщенные анализируемым газом.
Таблица 8.30
Затворные жидкости, применяемые в газовом анализе
Затворная жидкость Примечание
Вода Применяется при не очень точных анализах в отсутствие щелочных и кислых газов
Глицерин, 50% водный раствор Заметно растворяет СО2
Ртуть Неприменима в присутствии F2, Cl2, HBr, НС1 и H2S
Серная кислота, 10% водный раствор Применяется в отсутствие NH3 и других щелочных газов
Смазочные масла Применяются в качестве затворных жидкостей для H2S и SO2
Сульфат натрия, 20% водный раствор с добавкой серной кислоты (4-5 об. %) Применяется при температурах выше 16 °C, так как при более низкой температуре из раствора выкристаллизовывается Na2SO4 • ЮН2О
Тиосульфат натрия, 4% водный раствор Применяется в отсутствие газов-окислителей
Хлорид натрия, 22% водный раствор Очень слабо растворяет СО2
Таблица 8.31
Давление паров некоторых смазочных масел
Наименование масла Состав Давление пара при 20 °C, мм рт. ст.
Д-1А Смесь углеводородов 2 • 1(Г9-4 • НГ8
Д-1Б То же 2 • 10’9-4 • 10 8
Г То же 1 • 10^-5 • Ю 5
ВМ-4 Тоже 1 • 106-4 ПГ5
Бутил фталат С6Н4(СООС4Н9)2 ~1 • НГ5
ВКЖ-94А Смесь этилполисилоксанов Не более 5 • 10 х
ВКЖ-94Б То же 5 • 10*1 • НГ6
ПФМС-2 Смесь метилфенил-полисилоксанов 5 • 10’9-7 Ю 7
8.9. Методы газового анализа
8.9.1. Химические методы
Методы химического анализа основаны на преобразовании определяемого элемента в результате селективной химической реакции с реагентом и измерении изменившихся свойств газовой среды или реагента. В зависимости от способа измерения методы могут быть разделены на следующие группы: объемно-манометрические (волюмо-
метрические), титриметрические, термохимические и гравиметрические.
8.9.1.1. Волюмометрический и манометрический методы
Методы основаны на измерении изменения давления анализируемого газа в известном (мерном) объеме а) либо в результате поглощения жидким или твердым реагентом определяемого компонента в процессе химической реакций, б) либо при сжигании компонента на нагретом катализаторе (оксиде меди, палладии).
Анализ проводят с использованием измерительных бюреток, мерных сосудов, катализаторов, компенсационных устройств, манометров. В качестве поглощающих реагентов используются:
• жидкие: С6Н3(ОН)3 (пирогаллол) или Na2S2O4 (гидросульфид натрия); подкисленный раствор хлорида хрома (II); КОН, NaOH (гидроксиды калия или натрия).
• твердые: желтый фосфор; медная стружка; натронная известь; синтетические цеолиты; силикагель; активированный уголь.
Анализ газовых смесей осуществляется путем либо поглощения основного компонента (диоксида углерода, кислорода) и измерения давления остаточной газовой фазы, либо поглощения определяемого компонента и измерения уменьшения давления газа в мерной емкости. Анализ сложных газовых смесей ведется на приборах, содержащих несколько устройств для раздельной избирательной аб-сорбциии различных компонентов. В целом методы позволяют определять микропримеси до 0,01-0,1 мол. %, а содержание основного компонента до 99,9 мол. %.
К приборам такого типа относятся газоанализаторы КГА-1 (определение примеси кислорода, оксида и диоксида углерода), КГА-2 и КГА-3 (анализ промышленных газов на содержание водорода и углеводородов).
8.9.1.2. Титриметрический метод
Метод основан на измерении объема стандартного раствора— титранта, необходимого для проведения реакции с определенным компонентом. Конечную точку титрирования фиксируют по изменению окраски раствора или специального индикатора визуально или с помощью какого-либо инструментального метода применительно к газовому анализу. Титриметрический метод предполагает выделение в конденсированную фазу определяемого компонента или какого-либо его соединения, в которое он предварительно превращается. Для этого используются как химические реакции, так и процессы химической абсорбции. В аналитическом процессе используются различные приемы титрования — прямое и обратное, метод замещения и др.
В качестве индикаторов, фиксирующих конечную точку титрования, используют органические вещества — слабые электролиты, обладающие кислыми или основными свойствами. Наиболее распространенными индикаторами (КС — кислая среда, ЩС — щелочная среда) являются: лакмус фиолетовый (цвет в КС, — красный, а в ЩС, — синий); метиловый оранжевый (цвет в КС — красный, а в
920
Новый справочник химика и технолога
ЩС — желтый); фенолфталеин (цвет в КС — бесцветный, а в ЩС — красный); тимолфталеин (цвет в КС — бесцветный, а в ЩС— синий); ализариновый желтый (цвет в КС — желтый, а в ЩС — сиреневый).
Титриметрические методы используют для определения микропримесей: азота — от 510 3 до 5-10 4мол. %, водорода— от 10“3 до 10^* мол. %, кислорода— от 103 до 10^* мол. %, диоксида углерода — от 10”2 до 10"3мол. %, оксида углерода — от 2,0 до 10 3мол. %, суммы углеводородов — от 10 3 до 5-10^ мол. %, сероводорода — от 5,0 до 5-10~3мол. %. Методы используются также для анализа сложных газовых смесей и воздуха на содержание: диоксида серы — 0,1 мол. %; фосфина — от 10"1 до ИГ2 мол. %, хлороводорода — от 5-10“2 до 10,0 мол. %, хлора — менее 2-3 мол. %.
8.9.1.3. Термохимический метод
Метод основан на измерении теплового эффекта экзотермической реакции с участием определяемого компонента газовой смеси. Метод пригоден только для определения горючих веществ (Н2, H2S, СО, SO2, СН4 и других углеводородов). В аналитической практике используется беспламенное горение на мелкодисперсном катализаторе с развитой поверхностью. Существуют два варианта термохимического метода анализа газов. В первом определяемый компонент сгорает непосредственно на чувствительном элементе, в качестве которого, как правило, применяют терморезистор, служащий одновременно катализатором или покрытый слоем катализатора. Повышение температуры Д7' терморезистора является при этом функцией содержания определяемого компонента. Во втором варианте проба газа пропускается через камеру, где на насыпанном слое катализатора протекает реакция, в результате которой повышается температура катализатора, являющаяся и в этом случае функцией содержания определяемого компонента. Повышение температуры катализатора измеряют термопарой, сравнительный спай которой помещают в потоке газа до камеры, а измерительный спай — в камеру непосредственно в катализаторе.
Идеальным материалом для изготовления термохимических датчиков является платина, поскольку она обладает высокой каталитической активностью, линейной зависимостью температурного коэффициента сопротивления от температуры и, к тому же, является химически инертной. В ряде случаев, когда верхняя граница измеряемого температурного диапазона не превышает 300 °C, возможно применение никеля в качестве материала для изготовления чувствительного элемента. При этом надо учитывать, что этот материал имеет нелинейную зависимость сопротивления от температуры.
Метод позволяет определять горючие и взрывоопасные вещества в диапазоне концентраций от 10“3 до 100 мол. %. Наиболее широкое распространение получили приборы — газосигнализаторы возникновения пожаро- и взрывоопасной ситуаций в воздухе промышленных помещений, горных выработок и т.п. Существующие серии сигнализаторов отличаются уровнем срабатывания сигнала, отсчиты
ваемым от значения нижнего концентрационного предела воспламенения (НКПВ). Так, сигнализаторы типа СТХ-5А, СТХ-ЗУ4, СТХ-6, СВК-ЗМ1, «Щит-1У4», СТМ имеют уровень срабатывания от 5 до 50% НКПВ, а типа СВИ-4, СВИП-1 и СВМП-1 — характеризуются уровнем срабатывания в 20% НКПВ.
8.9.1.4. Гравиметрический метод
Метод основан на измерении изменения массы поглотителя в результате поглощения им определяемого компонента газовой пробы. Используют два варианта метода: в первом поглотитель непосредственно поглощает определяемый компонент, во втором — поглотитель поглощает продукты химической реакции, протекающей с участием определяемого компонента. В качестве поглотителей используются: пентаоксид фосфора или перхлорат магния (поглощение водяного пара); гидроксид калия или натрия (поглощение диоксида углерода).
Первый вариант метода встречается очень редко, в основном для определения влажности воздуха и продуктов его разделения, и позволяет определять пары воды до (1—3)-10 3 мол. %. Более распространен второй вариант с использованием комбинации различных приемов каталитического сжигания и последующего поглощения продуктов химической реакции. Такой комбинированный вариант применяется для определения водорода, оксида и диоксида углерода в газах.
8.9.2. Оптические эмиссионные методы
Эмиссионные оптические методы базируются на измерении интенсивности света, излученного атомом или молекулой, возбуждаемыми за счет энергии газового разряда, потока внешнего электромагнитного излучения или энергии химической реакции, а также рассеянного на частицах (атомах, молекулах, аэрозолях) определяемого компонента.
8.9.2.1. Газоразрядный метод
Метод основан на применении плазмы газового разряда для формирования возбужденных аналитических частиц (атомов, молекул, радикалов) определяемых компонентов газовой пробы и измерении интенсивности свечения аналитических линий атомов или молекулярных полос, являющихся функцией содержания определяемого компонента в газе.
Для возбуждения аналитического спектра используют различные типы газового разряда. Электрический — высокочастотный, импульсный, постоянного тока; микроволновый разряд; лазерная искра. При этом применяются разные устройства — кварцевые трубки с внешними или внутренними электродами; факел индуктивно связанной плазмы в потоке аргона. Аналитические линии или полосы выделяются специальными приборами — монохроматорами, а также многослойными интерференционными фильтрами. Интенсивность спектральных линий и полос регистрируются фотоэлектронными умножителями (ФЭУ) или фотодиодами.
Газовый анализ
921
Эмиссионные методы успешно применяются для анализа бинарных газовых смесей (смесей инертных газов, смесей молекулярных газов), а также для определения примесей молекулярных газов (Н2, О2, N2, СО, СО2 и др.) в инертных. В первом случае возможно определение примесей до 101! О 3 мол. %, во втором — до 10п-10 5 мол. %. При анализе сложных газовых смесей определяют различные примеси диапазоне от 5-10 2 до десятков мол. %.
Пример успешного применения эмиссионного газоразрядного метода — возможность непрерывного определения микропримеси азота в аргоне (до 104мол. %) на приборах типа «Свет» и «Азот-01».
8.9.2.2. Флуоресцентный метод
Метод основан на применении монохроматического (как правило, лазерного) излучения для возбуждения атомов или молекул определяемых компонентов и измерении интенсивности флуоресценции, являющейся функцией содержания определяемого компонента газовой смеси.
Для возбуждения флуоресценции применяются различные лазеры (импульсные и непрерывные, твердотельные, газоразрядные, на растворах красителей), а также ртутные лампы. Для выделения аналитического спектра — монохроматоры и интерференционные фильтры. При определении трудновозбудимых примесей используется их девозбуждение в плазме газового разряда либо комбинация из двух и даже трех лазеров.
Несмотря на значительное развитие лазерной техники метод флуоресцентного анализа не получил пока широкого развития в газоаналитической практике и реализуется только на сложных лабораторных установках. Наиболее заметные результаты в лазерном атомно-флуоресцентном анализе достигнуты при определении содержания неона в гелии » 10-8 мол. %, а в лазерном молекулярно-флуоресцентном — при определении оксида и диоксида азота, диоксида серы ~ 1 (Г7 мол. %. На основе применения импульсных газоразрядных ламп созданы флуоресцентные газоанализаторы для определения диоксида серы и сероводорода до ПГ7 мол. %.
8.9.2.3. Хемилюминесцентный метод
Метод основан на измерении интенсивности излучения частиц, являющихся продуктами химической реакции, т.е. когда молекула, образовавшаяся в результате протекания химической реакции, находится в возбужденном электронно-колебательном состоянии и в процессе релаксации излучает в определенном спектральном диапазоне. Иногда используется обратный процесс, когда аналитическим сигналом является «тушение» определяемыми частицами свечения (фосфоресценции) некоторых органических красителей. Существует два варианта хемилюминесцентного метода — пламенный и не пламенный. В первом случае регистрируется изменение свечения пламени при введении в него продуктов химической реакции, во втором — интенсивность излучения самих продуктов химической реакции.
Выделение нужной спектральной области достигается за счет использования различных оптических фильтров. Интенсивность люминесценции или фосфоресценции регистрируется фотоэлектронными умножителями (ФЭУ) или фотодиодами. В пламенной хемилюминесценции обычно используется водородно-воздушное или кислородное пламя. В качестве газа-реагента применяют озон, этилен, кислород.
Наиболее широко хемилюминесцентные методики применяются при определении диоксида азота (10 4 10 7 мол. %), а при использовании термических конверторов — оксида азота до (10~7 мол. %). Известны методики определения арсина и фосфина (2-10 6-2-10 7 мол. %), а для определения этих примесей в воздухе рабочей зоны используются модификации газоанализатора «Платан». Метод применяется также для определения диоксида серы в воздухе (10-4 мол. %), фосфора в инертных газах (10'4 мол. %). Примером методики «тушения» может служить методика определения кислорода в различных газах, на основе которой создан газоанализатор ФФ5101 с диапазоном измерения (4- Ю^-Ю-5 мол. %).
8.9.2.4. Метод рэлеевского рассеяния
Метод основан на измерении интенсивности упругого взаимодействия (без изменения частоты) рассеянного зондирующего излучения с частицами газовой среды. Существует два варианта метода: нефелометрический, когда измеряется интенсивность рассеянного частицами газовой среды зондирующего излучения; турбидиметрический, когда измеряется ослабление интенсивности прошедшего газовую среду зондирующего излучения за счет рассеяния на частицах газовой среды.
В качестве источника излучения применяются лампы накаливания, а в последнее время — лазеры. Рассеяние регистрируется под некоторым углом (обычно 90°) к зондирующему пучку (первый вариант метода), как в газовой среде, так и в различных растворах, через которые прокачивается анализируемая газовая смесь.
Нефелометрический метод используется, как правило, для контроля запыленности воздуха рабочих зон, управления пылегазовыми потоками и промышленными технологиями дробления твердых веществ. Диапазон определяемых содержаний пыли обычно находится в пределах от 1 мкг/м3 до 100 мг/м3.
Турбидиметрический метод чаще всего применяется для определения концентраций аэрозолей вредных соединений в воздухе рабочих помещений, например тумана серной кислоты в атмосфере сернокислотных цехов в диапазоне 0-1 мг/л. Метод позволяет определять и такие примеси в воздухе, как оксид (10 3 мол. %) и диоксид (и-10~3 мол. %) углерода, циановодород. При определении содержания диоксида углерода регистрация рассеянного излучения осуществляется в насыщенном водном растворе гидроксида бария, оксида углерода — в аммиачном растворе нитрата серебра, циановодорода — в аммиачной суспензии иодида серебра.
Примером воплощения первого варианта метода могут служить нефелометры типа ПФК-2, ИВА-2, ИВА-3, ЛАЗА,
922
Новый справочник химика и технолога
СЭВМП-1Э СПКП-1 — для контроля запыленности атмосферы рабочих зон и технологических процессов в мукомольной, горной и химической промышленности. Второй вариант метода применяется в турбидиметре типа СИДА «Атлас» для контроля мутности атмосферы рабочих помещений химического производства.
8.9.2.5. Метод комбинационного рассеяния
Метод основан на измерении интенсивности рассеянного в результате неупругого (с изменением частоты) взаимодействия фотонов зондирующего излучения с молекулами газовой среды. Регистрация рассеянного излучения ведется на смещенной (относительно зондирующего) длине волны. Существует два варианта метода — спонтанное комбинационное рассеяние (СКР) и когерентное активное комбинационное рассеяние (КАСКР). В первом случае рассеяние происходит на молекулах, находящихся в хаотическом тепловом колебательном и вращательном состояниях, и поэтому является изотропным и некогерентным. Во втором — на молекулах, в которых внутримолекулярные колебания предварительно селективно возбуждены и сфазированы в некотором объеме с помощью двух лазерных лучей, и рассеяние является анизотропным и когерентным.
Возбуждение спектров комбинационного рассеяния осуществляется мощными импульсными лазерными источниками излучения. Для выделения аналитических линий используются монохроматоры. Для подавления засветки на длине волны зондирующего излучения и неселективно рассеянного света применяются специальные фильтры. Для регистрации интенсивности рассеянного излучения используются фотоэлектронные умножители (ФЭУ) или фотодиодные матрицы.
Метод спонтанного комбинационного рассеяния применяется для анализа как сложных газовых смесей и динамики смешивания газов, так и для определения микроконцентраций различных газов в газах и газовых потоках. Известны спектры и сечения рассеяния пяти десятков газообразующих веществ. Возможности СКР при определении примесей в газах составляют: азота— 10 4мол. %, метана— 10 4мол. %, кислорода, оксида и диоксида углерода, аммиака—10 3 мол. %, йода—10-5 мол. %, водорода— 1 (Г3 мол. %. Метод когерентного активного комбинационного рассеяния из-за относительной сложности довольно ограниченно применяется в аналитической практике. Известны методики определения водорода (до 210“ 5 мол. %), диоксида углерода (10“2 мол. %), диоксида азота (10“3мол. %). Следует отметить, что метод СКР широко используется для решения задач дистанционного мониторинга атмосферы промышленных зон с помощью лидар-ных комплексов.
8.9.3. Оптические абсорбционные методы
Оптические абсорбционные методы базируются на измерении ослабления интенсивности зондирующего излучения при прохождении его через анализируемую газовую среду. Поглощение происходит на резонансных частотах, опреде
ляемых в атомах их электронными энергетическими состояниями, а в молекулах — электронно-колебательными энергетическими состояниями. В зависимости от способа регистрации поглощения зондирующего излучения существуют различные варианты метода — измерение прямого поглощения, внутрирезонаторного поглощения, оптико-акустический, термолинзовый и спектрофотометрический методы.
8.9.3.1. Метод прямого линейного поглощения
Метод основан на непосредственном измерении ослабления интенсивности прошедшего поглощающую газовую среду зондирующего излучения. Существует несколько вариантов метода, отличающихся способами формирования и обработки аналитического сигнала. Формирование аналитического сигнала осуществляется либо дифференциальным измерением поглощения, либо модуляцией интенсивности сигнала.
Дифференциальный метод существует в двух формах: — метод двух линий, когда измеряют интенсивность прошедшего анализируемую газовую среду излучения на двух частотах Vi и v2, одна из которых совпадает с максимумом линии или полосы поглощения определяемого компонента, а вторая — попадает в область минимального поглощения. Разность сигналов является мерой концентрации поглощающих частиц;
— метод двух пучков, когда зондирующее излучение разделяется на два луча, один из которых пропускается через кювету с анализируемой газовой смесью, а второй — через кювету сравнения, заполненную газовой смесью известного состава. Разность сигналов от опорного и рабочего каналов — мера концентрации определяемого компонента.
Модуляционный метод существует в трех формах:
— модулируется прошедшее поглощающую газовую среду зондирующее излучение с помощью коррелирующих элементов (масок, газонаполненных кювет, сканирующего интерферометра Фабри - Перо);
— модулируется частота поглощения определяемого компонента за счет помещения рабочей кюветы в переменные магнитное (Зееман-эффект) или электрическое (Штарк-эффект) поля;
— модулируется частота зондирующего излучения за счет помещения источника зондирующего излучения в переменные магнитное или электрическое поля.
Обработка аналитического сигнала осуществляется двумя способами: дифференциальным и интегральным. Первый способ заключается в измерении первой или второй производной от приемника излучения, меняющегося по гармоническому закону сигнала, а второй — в выделении селективных участков спектра, где находятся линии или полосы поглощения определяемых газовых компонентов, и учете их переложений.
В качестве источников зондирующего излучения используются тепловые (глобар, штифт Нернста, лампы накаливания), газоразрядные (водородные, дейтериевые, ртутные, СВЧ-лампы, лампы с полым катодом), когерентные (лазеры, светодиоды) излучатели. В качестве приемников лучистой энергии используются тепловые (термо
Газовый анализ
923
элементы, болометры), фотоэлектрические (фотоэлементы, фотоэлектронные умножители, фотосопротивления) детекторы. Выделение заданных участков спектра осуществляют с помощью монохроматоров или многослойных интерференционных фильтров. Кюветы с анализируемым газом обычно имеют длину от 1 до 500 см и часто конструируются таким образом, чтобы обеспечить многократное прохождение зондирующего излучения через поглощающую газовую среду. Коррелирующими элементами, осуществляющими модуляцию регистрируемого излучения, служат различные устройства: простейшие механические обтюраторы, газонаполненные кюветы, матричные элементы, выделяющие заданные участки спектра, и т. д.
Метод прямого линейного поглощения успешно применяется при анализе бинарных и сложных газовых смесей, а также для определения микропримесей газовых соединений и атомов различных элементов в газовых смесях и воздухе. Метод позволяет определять примеси газовых соединений: SO2 — до 2-10”8 мол. %, NO — до З-Ю-6 мол. %, NO2—до 1(Г7мол. %, N2O—до 1(Г7мал. %, СО—до 1СГ7мол. %, СОг—до 10“5мол.%, Н2О—5-Ю”6мол. %, Оз—до б-Ю^моп.^ СН4 — до б-Ю^мол. %, NH3 — до 510“9 мол. %, H2S — до 5-10 6 мол. %, HF — до 10 6 мол. %, НС1 —до 10’7мол. %, CF2C12 (фреон) — до 10"8мол. %. Метод позволяет определять содержание различных компонентов, находящихся в газах и воздухе в виде пара, аэрозольных частиц и пыли (в сочетании с методиками накопления и атомизации этих соединений). Известны методики определения следующих элементов: Са — до 6-10”8 мол. %, Mg — до 6-10 8 мол. %, Fe — до 4-1 (Г8 мол. %, Си—до КГ8 мол. %, Hg—6- 10 ю мол. %, РЬ — 2-10-9 мол. %, Se — до 3 • 10”9 мол. %, Be—до 1 (Г7 мол. %, As—до 5-КГ9 мол. %.
В настоящее время широко распространен абсорбционный газоанализатор ГИАМ-5, предназначенный для определения в газах оксида углерода в диапазоне от 5 10“3 до 100 мол. %; диоксида углерода — от 10“2 до 100 мол. %, метана — от 2-Ю-3 до 100 мол. %.
8.9.3.2. Метод внутрирезонаторного лазерного поглощения
Метод основан на регистрации изменения спектра генерации лазера и измерении интенсивности его излучения в целом или на отдельных частотах, совпадающих с линиями поглощения определяемых компонентов газовой смеси, помещенной внутрь резонатора лазера. Таким образом, положение провалов в спектре генерации лазера позволяет идентифицировать поглощающие частицы, а измерение глубины и формы провалов — определять концентрацию поглощающих частиц. В зависимости от соотношения ширин линий поглощения определяемых частиц и линии генерации лазера различают два варианта метода: — ширина линии поглощения больше или равна ширине линии генерации. В этом случае наблюдается уменьшение интенсивности излучения лазера при увеличении концентрации поглощающих частиц вплоть до срыва генерации;
— ширина линии поглощения меньше ширины линии генерации. В этом случае меняется структура спектра гене
рации лазера, т.е. происходит «выгорание» мод, совпадающих с линиями поглощения.
Первый вариант метода адекватен линейному поглощению со значительным увеличением эффективной толщины поглощающего слоя (аналог поглощения в многоходовой кювете). Второй вариант имеет принципиально нелинейный характер поглощения, т.к. генерируемые моды, совпадающие с линиями поглощения, испытывают двойное подавление как за счет поглощения определяемым компонентом газовой смеси, так и за счет конкуренции различных мод лазера.
Лазерный внутрирезонаторный спектрометр содержит два основных блока: лазер, с помещенной внутрь резонатора кюветой с анализируемым газом, и систему регистрации и преобразования спектра излучения. В качестве лазера используются лазеры на красителе, рубиновый, неодимовый, гелий-неоновый. В качестве детекторов лазерного излучения применяются фотодиодные матрицы и ПЗС-линейки.
Метод внутрирезонаторного лазерного поглощения пока не получил заметного развития при проведении анализов из-за относительной сложности технических элементов (лазеров и многоканальных детекторов излучения) и в силу достаточно сложного и нелинейного характера зависимости регистрируемого сигнала от концентрации определяемых компонентов (измеряемый сигнал зависит как от параметров линий поглощения анализируемых газов, так и от параметров линии генерации лазера, к стабильности которых в этом методе предъявляются довольно жесткие требования). Тем не менее созданные методики позволяют определять примеси: паров воды — до 10 4мол. %, СО2 — до 3-10~2мол. %, О2 — до 340-1мол. %, N2O — до 240“7мол. %, СН4 — до 6-10Аюл. %, NH3 — до 2 10“5мол. %.
8.9.3.3. Оптико-акустический (ОА) метод
Метод основан на измерении интенсивности акустических колебаний (в поглощающей газовой среде), вызванных преобразованием части поглощенной энергии модулированного зондирующего излучения в тепловую энергию газа (кинетическую энергию поглощающих молекул), вследствие чего в зоне облучения увеличивается давление, которое в виде волн распространяется по объему кюветы.
Существующие в настоящее время оптико-акустические газоанализаторы (ОАГ) можно разделить на две группы: недисперсионные и лазерные. Особенностью недисперсионных ОАГ является использование раздельных элементов: абсорбционной кюветы и оптико-акустического приемника, — в то время как в лазерном ОАГ абсорбционная кювета конструктивно совмещена с микрофонным датчиком акустических колебаний.
В качестве источников зондирующего излучения в недисперсионном ОАГ используются тепловые источники сплошного спектра (нихромовая проволока, нагретая до 700-900 °C; штифт Нернста, нагретый до 1400 °C). В лазерных ОАГ — непрерывные и импульсные лазеры, генерирующие в ИК-, видимом и УФ-областях спектра. Модуляция зондирующего излучения осуществляется с помо
924
Новый справочник химика и технолога
щью обтюратора. Абсорбционные кюветы представляют собой металлический или стеклянный цилиндр с прозрачными для зондирующего излучения торцевыми окнами.
Оптико-акустический приемник (ОАП) в недисперсионном варианте представляет собой двухкамерную кювету, заполненную анализируемым газом или его смесью с непоглощающим зондирующее излучение газом. Лучепри-емная камера (в которой происходит поглощение прошедшего абсорбционную кювету с анализируемым газом модулированного зондирующего излучения) соединена через капилляр со второй, так называемой конденсаторной, камерой, в которой расположен оптико-акустический датчик (ОАД) — плоский конденсаторный микрофон. Такая конструкция обеспечивает компенсацию медленных изменений температуры и давления газа в ОА-приемнике. В лазерном ОАГ оптико-акустический приемник (спектрофон) представляет собой цилиндрическую абсорбционную кювету, совмещенную в единой конструкции с датчиком акустических колебаний — конденсаторным или электретным микрофоном цилиндрической или плоской конфигурации.
Лазерные ОАГ работают по однолучевой схеме и различаются источниками зондирующего излучения, способами его модуляции и типами спектрофонов (резонансным, нерезонансным, с однократным или многократным прохождением излучения). Схемы недисперсионных ОАГ существуют в двух вариантах: одноканальные и двухканальные. В одноканальном варианте зондирующее излучение от источника света, модулированное обтюратором с частотой от нескольких герц до десятков килогерц, проходит фильтрующие элементы (газонаполненные кюветы, интерференционные или стеклянные фильтры) и пропускается через абсорбционную кювету с анализируемым газом, а затем попадает в лучеприемную камеру ОАП. ОА-сигнал пропорционален селективно поглощенной энергии. В двухканальных ОАГ, помимо рабочего, имеется канал сравнения, в котором формируется опорный сигнал сравнения. Существует три основных типа таких ОАГ: — дифференциальный ОАГ с непосредственным отсчетом, когда модулированное зондирующее излучение проходит одновременно рабочую и сравнительную кюветы, а затем попадает в ОАП с общей лучеприемной камерой, который выделяет разность сигналов от рабочего и опорного каналов;
— комбинированный ОАГ с двумя лучеприемными камерами в ОАП, когда модулированное в противофазе (в рабочей и опорной кюветах) излучение попадает в раздельные лучеприемные камеры ОАП, а оптико-акустический датчик фиксирует сигнал разбалансировки рабочего и сравнительного полупериодов;
— компенсационный ОАГ, когда разностный сигнал от рабочего и сравнительного каналов поступает на компенсирующее устройство, на котором предварительно сбалансированы сигналы от обоих каналов.
Оптико-акустический метод широко применяется как при анализе сложных газовых смесей, так и для определения микропримесей в газовых средах. Метод позволяет определять примеси: NO в диапазоне до 10-9мол. %,
NO2 — до 4-10 8 мол. %, NH3 — до 4-10’8 мол. %, СН4 — до 10 6 мол. %, СО2 — до 2-10 5 мол. %, SO2 — до 210-8 мол. %, SF6 — до 109мол. %, Н2О — до 106мол. %, О3 — до 10“б мол. %, СО — до 10“5мол. %, N2O — до 10-4 мол. %, HF — до КГ4 мол. %, Н2 — до 10 5 мол. %.
В настоящее время существует множество типов ОАГ недисперсионного типа, предназначенных для контроля содержания различных газов: для метана, оксида и диоксида углерода — приборы типа ОА; для оксида углерода, аммиака — приборы типа ГИП. Диапазон измеряемых содержаний указанных типов газоанализаторов охватывает интервал от сотых долей до десятков процентов. ОАГ типа «Кедр» позволяет определять примеси метана, оксидов углерода в технологических газах в диапазоне от 5-1 О' до 20-50 мол. %.
8.9.3.4. Термолинзовый метод
Метод основан на регистрации локальных изменений оптических свойств газовой среды, возникающих при нагреве газа в зоне поглощения зондирующего излучения и преобразования части поглощенной энергии в тепловую. Существуют различные способы детектирования «тепловой линзы»: интерферометрический, рефрактометрический, внутрирезонаторный.
Наибольшее распространение получила двухлучевая схема фотометрических спектрометров, когда область фокусировки зондирующего лазерного излучения коллинеарно просвечивается излучением пробного лазера. В качестве пробного лазера обычно используется излучение He-Ne-лазера с X = 632,8 нм. В качестве зондирующего излучения применяется СО2-лазер (X = 10,6 мкм или 9,2 мкм).
Созданные в настоящее время термолинзовые методики позволяют определять содержание SF6 в воздухе на уровне до 10 6мол. %; фреона (CF2C12 в аргоне — до 210“5мол. %; NH3 в азоте — до610“5мол. %; 8О2варгоне — доЗ-ЮАюл. %;NO2 в воздухе —до 810“5мол. %.
8.9.3.5. Колориметрический
и спектрофотометрический методы
Методы основаны на измерении оптической плотности растворов, состоящих из поглощенного сорбентом из газовой фазы определяемого компонента и специально подобранного фотометрического реагента.
Визуальные колориметрические методы основаны на сравнении окраски раствора с неизвестной концентрацией поглощенного определяемого компонента и окраски одного или нескольких растворов, в которых концентрации этого компонента точно известны. Вариантом колориметрического является линейно-колористический метод, который базируется на зависимости длины окрашенного слоя сорбента от содержания определяемого компонента при пропускании через него анализируемого газа.
Если оптическую плотность измеряют при определенной длине волны, то метод называют спектрофотометрическим. В этом методе, в отличие от колориметрического,
Газовый анализ
925
наряду с видимым светом, используют излучение в УФ- и ИК-областях спектра.
Для измерения поглощения используют либо фотоколориметры (зондирующее излучение от источника сплошного спектра), либо спектрофотометры (зондирующее излучение монохроматично). Отбор проб осуществляют в специальных поглотительных сосудах (приборы Петри, Полежаева и Зайцева) аспирационным способом, основанном на пропускании известного количества анализируемого газа через поглощающую среду со скоростью до десятков литров в час.
В качестве поглощающего сорбента используются: твердые (углеволокнистые сорбенты, активированные угли, молекулярные сита, силикагели, пленочные сорбенты на основе тетраметилдипропилентриамина) или жидкие (растворы аминов в спирте и других органических растворителях; растворы иодида калия; нитрата серебра; желатина и аммиака в этаноле и т.п.) поглотители.
В качестве фотометрических реагентов, способствующих образованию поглощающих комплексов, используются: 1,3,5-тринитробензол; смесь спиртового раствора пиперазина и ацетата меди; трифенилметановые красители; У-(1-нафтил эти лен диамин); а-нафтиламин; диметил-и-фенилендиамин и т.п.
Метод применяется для определения в воздухе примесей: сероуглерода — до 10 6 мол. %, диоксида серы — до 210~7мол. %, диоксида азота — до 10 6 мол. %, оксида азота — до 51 (Г6 мол. %, оксида углерода — до 210-6 мол. %, озона — до 5-1 (Г7 мол. %, аммиака — до 1 (Г5 мол. %, сероводорода — до 7-10"4 мол. %, циановодорода—до 3-104 мол. %, хлора—до 2-10-5 мол. %.
Линейно-колористический метод реализуется на основе серии индикаторных трубок, которыми комплектуются газоопределители типа ГХ, предназначенные для определения вредных газов в рудничной атмосфере, и газоанализаторов УГ-2 или УГ-3, предназначенных для контроля воздуха производственных помещений.
Спектрофотометрический метод реализуется на основе ряда моделей спектрофотометров: СФ-46 (рабочий спектральный диапазон 190-1100 нм), СФ-39 (рабочий спектральный диапазон 190-750 нм) и др.
8.9.4. Масс-спектрометрический метод1 *
Метод базируется на измерении ионного тока, получаемого в результате ионизации компонентов газовой смеси и последующего временного или пространственного разделения ионов по отношениям их масс к зарядам. Более подробно принципы и варианты метода изложены в разделе 7.
Масс-спектрометр состоит из пяти основных блоков: системы достижения высокого вакуума, системы напуска анализируемого газа, ионизационной камеры, анализатора
1 Наряду с термином масс-спектрометрический (метод) в
литературе (в том числе в данном издании) допускается употребление синонимов масс-спектроскопический и масс-
спектр альный.
и коллектора ионов. Система вакуумирования обеспечивает разрежение в анализаторе до 10“7 мм рт. ст. В ионизационной камере происходит ионизация атомов и молекул газовой смеси. Способы ионизации могут быть различны (ионизация электронным ударом, ионизация высоким градиентом электрического потенциала, ионизация фотонами, ионизация при атмосферном давлении). В анализаторе осуществляется разделение ионов и формирование ионного спектра. По принципу разделения приборы классифицируются на статические (пространственное разделение ионов в магнитном поле) и динамические (временное разделение ионов). Для анализа газов обычно применяются динамические масс-спектрометры (времяпролетные, квадрупольные, радиочастотные), которые характеризуются быстротой записи масс-спектра и малыми размерами. Регистрация ионных токов осуществляется с помощью вторичных электронных умножителей (ВЭУ).
В целом, масс-спектрометрический метод обеспечивает определение микропримесей на уровне 1(Г3-10“7 мол. %, а с применением специальных приемов концентрирования— до 10“9мол. %. Хотя масс-спектрометр является универсальным газоанализатором, для решения той или иной задачи (анализ высокочистых инертных газов, технологических газов, анализ атмосферы) необходимо выбирать соответствующую модификацию прибора.
Анализ высокочистых газов на содержание основных атмосферных компонентов проводится на масс-спектрометре типа MX 1304, определяющем примеси инертных газов до 10“7 мол. %, углеводородов до 10“5 мол. %, диокисцда углерода до 510 4мол. %, кислорода до 5-10 6 мол. %. Для анализа атмосферного воздуха при геологоразведке на выявление характерных ореолов рассеянных газов применяется радиочастотный масс-спектрометр типа МАГ-80. Для анализа воздуха при медицинских исследованиях органов дыхания применяется времяпролетный масс-спектрометр типа МИ5302 или радиочастотные масс-спектрометры типа МХ6202, МХ6203. Для исследования верхних слоев атмосферы Земли и планет Солнечной системы используют радиочастотный масс-спектрометр типа МХ6401 и его модификации. Анализ технологических газовых смесей, содержащих водород, оксиды углерода, азот, кислород и аргон, выполняется на масс-спектрометре типа МХ1214. Для анализа изотопного состава газообразных элементов применяется магниторезонансный масс-спектрометр типа МИ9302 или масс-спектрометр высокого разрешения типа МХ1310.
8.9.5. Хроматографический метод
Отличительной особенностью газохроматографических методов анализа является наличие этапа разделения, предшествующего детектированию определяемых компонентов пробы. Проба перемещается в потоке газа-носителя вдоль слоя сорбента, заполняющего хроматографическую колонку. За счет различий адсорбционной активности сорбента по отношению к различным компонентам газовой смеси происходит разделение пробы. Более подробно принципы и варианты метода рассмотрены в разделе 4.
926
Новый справочник химика и технолога
Газовый хроматограф состоит из системы регулировки потока газа-носителя, разделительной колонки, детектора разделенных компонентов пробы, а иногда дополняется специальным концентрирующим устройством.
В качестве газа-носптеля используются: азот, аргон, гелий, водород.
Разделительная колонка — стеклянная или металлическая трубка диаметром от 1 до 6 мм и длиной от 0,5 до 6 м.
Сорбенты: активированные угли (ОКТ-2, АГ), силикагели (КСК, КСС, КСМ4 или КСМ5), синтетические цеолиты (NaA, СаА, NaX, СаХ), силохромы, пористые полимеры (паропак Т, Q, Р), полисорбы, хромосилы и др.
Детекторы: по измерению теплопроводности (катарометры); по измерению плотности (плотномеры); ионизационные (электронного захвата, гелиевый, аргоновый); масс-спектрометр.
При определении микропримесей используются различные приемы концентрирования или специальные накопительные устройства (ловушки). В качестве накопительных ловушек применяются трубки, заполненные различными сорбентами и охлаждаемые вплоть до криогенных температур. При определении сильносорбирующихся примесей в слабосорбирующемся газе применяются различные варианты термических методов концентрирования (программирование температуры разделительной колонки во времени, теплодинамический режим нагрева колонки, когда ее температура меняется не только во времени, но и по длине за счет перемещения вдоль колонки специальной нагревательной трубчатой печи). При определении слабо-сорбирующихся примесей в сильносорбируемых газах применяют метод фронтально-адсорбционного концентрирования, когда в откачанной хроматографической колонке на переднем фронте хроматографической зоны концентрируются слабопоглощающиеся сорбентом примеси, которые на выходе разделительной колонки отбираются и направляются в другую, где анализируются обычным методом с дополнительным газом-носителем.
Газохроматографический метод позволяет анализировать сложные газовые смеси и определять микропримеси Не, Ne, Аг, Хе, Kr, Н2, N2, О2, СО, СО2, N2O на уровне 10~310 4 мол. %, а с применением различных приемов концентрирования — до 10“5-10“7 мол. %.
Применяется метод и для определения неорганических соединений серы (SO2, H2S, CS2, COS) в воздухе, технологических и природных газах с пределом определения до КГ-КГ4мол. %. При анализе газов и газовых смесей, содержащих оксиды азота (N2O, NO, NO2, N2O4), применяются многоколоночные системы, разные колонки которых заполняются различными сорбентами.
Для решения различного рода газоаналитических задач используются хроматографы марок «Агат», АХГ-002, «Цвет-500М», «Цвет-610», ХПМ-4, ХПМ-54, «Автохром», «Кристалл-3000», и др.
8.9.6. Электрохимические методы
К электрохимическим относят большую группу методов, которые базируются на процессах, протекающих на электродах или в межэлектродном пространстве специ
альной (электрохимической) ячейки; результатом этих процессов является изменение ряда параметров системы — потенциал, сила тока, количество электричества, полное сопротивление, электрическая емкость, электрическая проводимость, диэлектрическая проницаемость. Более подробно принципы и варианты методов описаны в разделе 6.
Необходимым условием применения электрохимических методов в ГА является насыщение тем или иным способом электролита анализируемым газом. Перспектива использования их при решении газоаналитических задач обусловлена рядом факторов: простота автоматизации; низкие пределы определения (до 10 510 7 мол. %); возможность создания малогабаритных, с низким энергопотреблением измерительных датчиков; возможность использования в широком интервале давлений и температур.
Одно из наиболее перспективных и современных направлений развития метода — создание пленочных металлических, полупроводниковых и полимерных датчиков состава газов-сенсоров.
На базе электрохимического метода разработана серия приборов: «Оникс» — для определения кислорода, водорода и паров воды в азоте и инертных газах в диапазоне 2-10 5 5-10 2 мол. %; «Циркон» — для определения кислорода в инертных газах и азоте в диапазоне от КГ5 до 100 мол. %; «Агат» — для определения кислорода от 5 10 4 до 100 мол. %; «Топаз» — для определения кислорода в диапазоне 15-45 мол. %; «Лазурит» — для определения кислорода и водорода в инертных газах и азоте в диапазоне от I03 до 10"’ мол. %. Создана серия портативных газосигнализаторов с использованием в качестве датчиков электрохимических сенсоров: ИВГ-1 — для измерения микровлажности в азоте, аргоне, воздухе, гелии, кислороде и их смесях до 510 5 г/м3 (-90 °C), ТГС-3 —для контроля содержания метана (модификация ТГС-З-МИ в диапазоне 0-3 об. %), кислорода (модификация ТГС-З-КИ в диапазоне 28-18 об. %), аммиака (модификация ТГС-З-АИ в диапазоне 2-10“4-1-10-2 мол. %).
8.9.7. Методы, основанные на зависимости интегральных свойств газов от содержания определяемого компонента
Методы базируются на измерении интегральных характеристик газовых сред (коэффициента теплопроводности, магнитной проницаемости, плотности, вязкости, коэффициента преломления света), меняющихся при изменении содержания примесных компонентов в газовой смеси. В зависимости от тестируемых свойств газовой среды методики называются: термокондуктометрические, магнитные, механические, акустические, рефрактометрические, интерференционные.
8.9.7.1. Термокондуктометрический метод
Метод основан на измерении изменения теплопроводности газовой среды при изменении ее состава и применяется только для анализа бинарных или псевдобинарных газовых смесей.
Газовый анализ
927
Измерительная камера термокондуктометрического газоанализатора представляет собой цилиндр с расположенными вдоль оси терморезисторами, которые выполняют одновременно роль нагревателя и измерителя температуры. В качестве терморезисторов используются проволочные резисторы на основе платины, вольфрама или никеля. Схема газоанализатора представляет собой различные варианты моста Уитстона. Для уменьшения влияния неопределяемых компонентов применяются двухмостовые дифференциальные схемы.
Вследствие сильного влияния неопределяемых компонентов газовой смеси на ее теплопроводность, метод применяется для анализа бинарных смесей (как правило). При этом возможно определение содержания компонентов в диапазоне от 10 2 до 100 мол. %.
На этом принципе основаны приборы для определения
• водорода: ТКГ — в азот-водородных, водородкислородных смесях, аммиаке, аргоне; ДТ — в азоте, аммиаке, азот-аргоновых смесях; «Диск» — в водород-аммиачных, водород-азот-аргоновых, водород-азот-аммиачных смесях; ВХЛ — в хлорсодержащей смеси; ТП различных модификаций — в воздухе и технологических газовых смесях;
• метана: «Темп» — в метан-азотных смесях.
Газоанализаторы марки 155ТПО-3, -4, -5 предназначены для определения водорода в азоте, азота в диоксиде углерода и кислорода в аргоне.
8.9.7.2. Магнитный метод
Метод основан на различиях в магнитных свойствах газов. Неорганические газы обладают как диамагнитными (инертные газы), так и парамагнитными (кислород, оксиды азота) свойствами. Наиболее заметными значениями магнитной восприимчивости характеризуются парамагнетики, в ряду которых выделяется кислород. Магнитный метод имеет три основные модификации: термомагнитный, магнитомеханический и магнитопневматический.
В термомагнитном варианте для получения аналитического сигнала используется зависимость интенсивности конвекционных потоков (возникающих в газовой смеси при наложении на ее объем определенным образом ориентированных неоднородного магнитного и неоднородного теплового полей) от содержания парамагнитного компонента в газовой смеси.
В магнитомеханическом варианте используется зависимость скорости истечения из отверстия газовой смеси в неоднородном магнитном поле от содержания в ней парамагнитного компонента.
Магнитопневматический вариант основан на взаимодействии потоков анализируемого и сравнительного газов в неоднородном магнитном поле, приводящем к разбалансировке пневматического моста.
Термомагнитный газоанализатор (термоанемометр) представляет собой установленную вблизи полюсов магнитной системы стеклянную трубку, на наружной поверхности которой намотана спираль, состоящая из двух секций. При протекании электрического тока спираль нагре
вается, образуя по длине трубки перепад температур с максимумом в центре. С появлением в анализируемой смеси примеси с парамагнитными свойствами возникает поток газа через зазор между полюсами магнита, и скорость этого потока пропорциональна содержанию примеси. Поток охлаждает первую секцию нагревательной системы и нагревает вторую, смещая тем самым положение максимума температуры. Измеряемая разность сопротивлений секций характеризует содержание примеси в газовой смеси.
Магнитомеханический газоанализатор представляет собой установленную вблизи зазора полюсов магнитную систему на упругой нити стеклянной гантели с закрепленной на ней проволочной рамкой. С появлением в анализируемой смеси примеси с парамагнитными свойствами возникает поток газа через зазор магнитной системы. Давление потока газа вызывает поворот рамки, а величина силы тока, компенсирующая этот поворот, характеризует содержание примеси в газовой смеси.
Магнитопневматический газоанализатор представляет собой измерительную камеру, содержащую пневматический мост, который образован двумя дросселями и двумя парами наконечников (одна пара — полюса магнита — создает магнитный зазор, а вторая — немагнитный). В диагональ моста встроен термоанемометр. Сравнительный газ (обычно азот) подается на вход моста и при отсутствии в газовой пробе компонентов, обладающих парамагнитными свойствами, распределяется двумя симметричными равномерными потоками. С появлением в пробе парамагнитной примеси последняя втягивается в магнитный зазор и увеличивает давление в рабочем плече моста, инициируя поток газа через термоанемометр, что приводит к разбалансировке электрического моста, образованного секциями термоанемометра, и величина появляющегося электрического сигнала характеризует содержание примеси в газовой смеси.
С помощью большинства магнитных газоанализаторов определяют содержание кислорода в диапазоне от 41 (Г2 до 100 мол. %:
Прибор ГТМК-ИМА служит для анализа бинарных и квазибинарных смесей, прибор ГТМК-16 — для анализа кислород-азотных, кислород-гелиевых, кислород-диоксид-углеродных, кислород-метановых смесей, прибор МН-5122-1 — для определения кислорода в воздухе, прибор «Оскар» — для определения кислорода в технологических газах, АКД —в атмосфере пожароопасных участков шахт.
8.9.7.3. Механический метод
Метод основан на измерении плотности или вязкости газовых сред в зависимости от состава и имеет две модификации: аэродинамическую (пневматическую) и аэростатическую. В первом варианте количественной мерой, описывающей изменение вязкости или плотности при вариациях состава смеси, практически всегда является давление газа, формируемое в различных точках чувствительных элементов. Во втором — содержание определяемого компонента находится путем сравнения плотностей анализируемой смеси и стандартного образца (воздуха или чистого
928
Новый справочник химика и технолога
азота) и последующего расчета искомого значения по правилу аддитивности (плотность смеси слагается из произведений плотностей компонентов на их содержание).
В аэродинамических газоанализаторах измерения проводят с использованием формирователей газового потока, которыми являются разного типа гидравлические сопротивления: ламинарные, турбулентные — либо их совокупность. В качестве ламинарного сопротивления обычно используется капилляр, а в качестве турбулентного — турбулентный дроссель, через который с постоянной скоростью выталкивается газ. Часто используются гидравлические элементы на основе комбинации этих сопротивлений: «ламинарное сопло — приемный канал»; схемы с дополнительным турбулентным фактором; струйный элемент «сопло— резонатор» с возбуждением звуковой волны. В аэростатических газоанализаторах измерения проводят на коромысловых газовых весах Штока или пружинных весах Големана, а также используется метод сравнительного взвешивания по Плитцеру.
Механический метод применяется для анализа бинарных смесей. Выпущены аэродинамические газоанализаторы типа: «Ламинар» для определения водорода в азоте в диапазоне 50-80 мол. %; «Поток» — хлора в хлор-газе в диапазоне 50-100 мол. %; «Трель» — кислорода в азоте в диапазоне 0,5-5 мол. %. Аэростатические газоанализаторы позволяют анализировать бинарные смеси с минимальным содержанием одного из компонентов до 10 3 мол. %.
8.9.7.4. Акустический метод
Метод основан на измерении скорости распространения в газовой среде звуковых или ультразвуковых волн, которая зависит от содержания определяемого компонента в анализируемом газе.
Акустические газоанализаторы конструктивно могут быть представлены двумя схемами. В основу первой положен принцип относительного отсчета скорости звука, а второй — измерение сдвига фаз посланной и принятой акустических волн. Практическое значение получил ультразвуковой диапазон частот — десятки килогерц. Поскольку сильное влияние на скорость акустических волн в газовой среде оказывает ее температура, рабочие камеры анализаторов жестко термостатируются.
Газоанализаторы первого типа используются для определения метана в бинарных смесях и воздухе в диапазоне от 2-101 до 5 мол. %, а также при определении оксида углерода, паров воды и кислорода. Газоанализаторы второго типа используются для определения содержаний кислорода до 5-Ю-3 мол. % , водорода до 6-10 4 мол. % и этилена до 8-103 мол. %.
8.9.7.5. Рефрактометрический и интерферометрический методы
Методы основаны на измерении изменения показателя преломления газовой среды при изменении содержания определяемых компонентов и применимы лишь для анализа бинарных газовых смесей.
В газоанализаторах используются две схемы измерения показателя преломления — угловые (рефрактометрия) и интерференционные. В приборах, реализующих первую схему (рефрактометры), измеряется либо предельный угол преломления (когда угол падения излучения на среду близок к 90°), либо угол полного внутреннего отражения. В приборах, реализующих вторую схему (интерферометры), измеряется смещение интерференционных полос вследствие изменения оптической плотности газовой среды при изменении ее состава. Как правило, в газоанализаторах используется двухлучевая схема интерферометра (Рождественского или Рэлея).
Рефрактометры позволяют определять примеси в бинарных смесях на уровне до Ю 2 мол. %. Более распространены интерферометрические газоанализаторы: ГИК-1 — для определения метана, водорода и диоксида углерода в воздухе в диапазоне от 5-10 2 до 5 мол. %; ЛИ-4М — для определения метана и водорода при их совместном присутствии в воздухе в диапазоне от 4-Ю-1 до 12 мол. %; ГАК-1 —для определения оксида и диоксида углерода в колошниковом газе доменных печей в диапазоне от 6101 до 25 мол. %.
8.9.8. Ионизационные методы
Методы основаны на индивидуальном характере степени ионизации различных газов, возникающей под воздействием какого-либо источника ионизации (^-излучения или ультрафиолетового излучения). Существует три варианта метода: электронно-захватный, по сечению ионизации и фотоионизационный. Методы не являются селективными и поэтому применяются либо для анализа бинарных смесей, либо для детектирования выделенных тем или иным способом (например, хроматографическим) компонентов анализируемого газа.
Ионизационные газоанализаторы представляют собой ионизационные камеры с встроенными источниками ионизирующего излучения, плоскими или цилиндрическими электродами и электрометром. В качестве источников ионизирующего излучения используются либо излучение радиоактивных изотопов (^-излучение): трития (3Н), нанесенного на поверхность титановой или циркониевой пленки, никеля (63Ni), криптона (85Кг), — либо ультрафиолетовое излучение водородных газоразрядных ламп низкого давления (А = 121,6 нм). В последнее время появились фо-тоионизационные методики с применением лазерного излучения в качестве ионизирующего.
Ионизационные газоанализаторы позволяют определять в инертных газах примеси H2S, COS, SO2, CS2, NO, NO2, N2O, CO2, H2 и O2 на уровне 10'10 7 мол. %.
8.9.9. Радиоспектроскопический метод
Метод основан на измерении поглощения молекулами излучения в радиочастотном диапазоне (X « 1 мм). В анализе используются два варианта метода: электронный парамагнитный резонанс (ЭПР) и ядерный магнитный резонанс (ЯМР). В первом случае происходит резонансное поглощение радиоволн в веществах, обладающих парамагнитными свойствами, при наложении внешнего стати
Газовый анализ
929
ческого магнитного поля; во втором — в веществах, не обладающих парамагнитными свойствами.
Радиоспектрометр состоит из источника радиочастотного излучения (клистрона), системы передачи излучения (в резонатор и к детектору) и электромагнита. Измеряется изменение добротности резонатора, в который помещена кювета с веществом, являющимся жидким или твердам поглотителем определяемого компонента анализируемого газа. Его содержание в таком экстрагенте определяет величину изменения добротности резонатора. Метод пока имеет ограниченное применение в ГА.
8.9.10. Метод изотопного разбавления
Метод основан на применении редких стабильных изотопов определяемого элемента в качестве как внутреннего стандарта, контролирующего аналитическую процедуру, так и меры для расчета его содержания в анализируемом газе.
Метод предусматривает при анализе проведение ряда процедур: дозированное введение в анализируемую пробу изотопа определяемого элемента в некоторой молекулярной форме, достижение равномерного перемешивания изотопосодержащих молекул по всему объему пробы. Затем аналитическая процедура, в зависимости от поставленной задачи, проводится в двух вариантах. Первый — проведение уравновешивания изотопов определяемого элемента по всем его молекулярным формам, измерение изотопного состава элемента в рабочей смеси и расчет искомой концентрации (схема позволяет определять полное содержание элемента в анализируемой пробе вне зависимости от его молекулярных форм). Второй — выделение определяемой молекулярной формы примеси, измерение изотопного состава в выделенной фракции и расчет искомого содержания (схема позволяет определять содержание той или иной молекулярной формы примесного элемента).
Принципиальными особенностями метода являются, во-первых, отсутствие необходимости в использовании поверочных газовых смесей (стандартных образцов состава) для калибровки аналитических методик и газоаналитических установок, во-вторых, возможность расчета содержания определяемого компонента, используя значение измеренной относительной концентрации изотопов и значения параметров процедуры дозирования изотопа в газовую пробу (величин объемов и парциальных давлений смешиваемых газов).
Измерение изотопного состава определяемых элементов осуществляется методом эмиссионной спектрометрии или масс-спектрометрии. В первом случае используются сканирующие монохроматоры с разрешающей способностью до 0,05 нм. Во втором — масс-спектрометры высокого разрешения. Определение изотопного состава водорода проводится по его атомарному спектру, а азота, кислорода и углерода — по молекулярным спектрам N2, СО, С2 соответственно.
В качестве изотопосодержащих газов используются: D2, D2O, CD4, ND3, 18O2, C18O, c18o2, h218o, 15n2, 15nh3, I5no, 13CO, 13CO2,13CH4 (с обогащением от 65 до 99,9 %).
Уравновешивание по объему осуществляется методами диффузионного или турбулентного перемешивания. Уравновешивание по молекулярным формам — газоразрядное или каталитическое. Используются различные способы разделения анализируемой смеси: селективная диффузия через мембраны и газовая хроматография. Применяются различные приемы концентрирования примесей: криогенная адсорбция в поглотительных ловушках, диффузия через палладий и т.п.
Инструментальные методики, т.е. методики, содержащие процедуры изотопного разбавления, газоразрядного уравновешивания изотопов и возбуждения аналитических спектров, а также расчета искомых содержаний определяемых элементов (Н, С, N, О), позволяют определять примеси в инертных газах (Не, Ne, Аг) водорода на уровне 10 4 мол. %, азота — 5-10-5 мол. %, кислорода — 10-Змол. %, углерода— 10“3 мол. %, а в других молекулярных газах и воздухе — на уровне I О1 мол. %.
Комбинированные методики, т.е. методики, содержащие также различные приемы разделения и концентрирования, позволяют определять примеси Н2, N2, О2, СО, СО2, СН4 в различных газах на уровне 10 610 х мол. %.
8.9.11. Методы изотопного анализа газов и газообразующих элементов
Существует два метода изотопного анализа газообразующих элементов: масс-спектрометрия и эмиссионная спектрометрия. При этом используются различия в масс- и эмиссионных спектрах стабильных изотопов, обусловленные отличиями в массах атомов этих элементов.
Стабильными изотопами газообразующих элементов являются: водород —- *Н, 2Н; углерод — 12С, 13С; азот — 14N, l5N; кислород — 1бО, 18О; гелий — 3Не, 4Не; неон — 20Ne, 21Ne, 22Ne; аргон — 36Аг, 38Аг, 40Аг; криптон — Кг(6 изотопов с массами от 78 до 86); ксенон — Хе (9 изотопов с массами от 124 до 136).
При масс-спектрометрическом определении изотопного состава измеряется отношение токов атомарных или молекулярных ионов на статических масс-спектрометрах высокого разрешения типа МИ 1201 и МИ 1330 либо магниторезонансных типа МИ9301, МИ9302, МИ9303.
При эмиссионном определении изотопного состава измеряются относительные интенсивности в атомарных или молекулярных спектрах: водорода — атомарные линии Бальмеровской серии с длиной волны X = 486,13 нм (изотопное смещение АХ = 0,13 нм); азота — полосы первый положительной системы молекулы N2 с X = 380,49 нм (АХ = 0,83 нм); кислорода — полосы системы Ангстрема молекулы СО с X = 519,8 нм (АХ = 1,96 нм); углерода — полосы системы Свана молекулы С2 с X = 563,6 нм (АХ = 0,9 нм).
Метод масс-спектрометрии позволяет определять изотопный состав всех газообразующих элементов с погрешностью от 0,1 до 2-3%. Метод эмиссионной спектрометрии применим при анализе таких элементов, как Н, С, О, N. Погрешность определения составляет 1-3%.
930
Новый справочник химика и технолога
8.9.12. Методы определения газовых включений и газообразующих элементов в веществах конденсированной фазы
Анализ жидкостей. Как правило, речь идет об определении газов, растворенных в жидкостях (водах различной природы и назначения, нефтепродуктах, растворах и др.). Наибольшее распространение для анализа таких объектов получили электрохимические и хроматографические методы. Электрохимические методы (полярография, амперометрия, кулонометрия) могут быть непосредственно использованы для определения газов в водах, что и определило приоритетность применения этого метода в решении такого рода задач. Газы, содержащиеся в нефтепродуктах (масла, топлива и пр.), обычно определяются газохроматографическими методами. В этом случае аналитическая процедура включает экстракцию газов из пробы анализируемой жидкости.
Анализ твердых неметаллических материалов. Такие объекты включают в себя природные минералы, руды, полупроводниковые вещества и материалы, различного рода стекла. Основу газосодержания перечисленных объектов составляют газовые включения. Задача анализа в этом случае может заключаться как в определении полного газосодержания, так и в определении содержаний отдельных газообразуюших элементов (как правило, кислорода, углерода, серы). Первая задача обычно решается применением вакуумной высокотемпературной экстракции газов из анализируемой пробы с последующим объемно-мано-метрическим измерением количества газа. Условия экстракции (температура, сбор газа и пр.) определяются отдельно для каждой конкретной аналитической задачи. Вторая задача решается на основе применения различных селективных методов анализа — масс-спектрального, спектрального, различных вариантов метода изотопных добавок и др.
Анализ металлов и сплавов. Исторически сложившееся понятие «газы в металлах» включает три элемента — водород, кислород, и азот, которые находятся в твердых и расплавленных металлических системах в виде растворов и избыточных фаз. При определенных условиях эти элементы могу образовывать в металле и истинные газовые включения. Поэтому водород, азот и кислород стали называть «газами в металлах», а также «газообразующими элементами». Сегодня под «газообразующими» понимают более широкий круг элементов, а именно те, определение которых возможно с помощью газоаннаЛизаторов после перевода их из конденсированной фазы в газовую. Водород, азот, кислород и углерод могут быть количественно выделены из металлов в газовую фазу под воздействием высокой температуры, реагентов и при смещении равновесия за счет удаления газообразных продуктов реакций. Эти соединения: Н2, N2, О2, СО, СО2, Н2О, СН4, С2Н2, NH3 — определяют методами анализа газовой фазы. Указанные соединения формируются тем или иным газообразующим элементом самостоятельно или в комбинации с другим газообразующим элементом, служащим при этом реаген
том для выделения первого в газовую фазу. С этой точки зрения к газообразующим элементам относятся также и сера, образующая такие соединения, как SO2, H2S и др. В последние десятилетия в связи с этим углерод и серу также стали относить к «газам в металлах».
Однако следует помнить, что, в отличие от серы, водород, азот, кислород, углерод образуют в металлических системах растворы внедрения. Газы могут находиться в металлах не только в виде твердых растворов внедрения, но и в виде избыточных фаз (как конденсированных, так и газообразных), скоплений вокруг дислокаций, сорбционных слоев на внутренних поверхностях. В реальном металле переход примеси из газовой фазы через поверхность в конденсированную фазу может быть представлен несколькими последовательными стадиями: адсорбция, диссоциация, образование поверхностного раствора, диффузия, растворение, распределение примеси между твердым раствором и дефектами структур, зарождение и выделение избыточных фаз. На различных стадиях получения и технической эксплуатации металлов имеет место перераспределение газообразующих элементов между различными формами их нахождения.
Присутствие газовых примесей в металлах и сплавах сильно влияет на физико-химические свойства и эксплуатационные качества последних. Так, например, известно, что введение элементов внедрения в металл приводит к повышению его жаростойкости, сопротивления ползучести и оказывает сложное влияние на прочность. Имеется возможность регулирования механических свойств сплавов и их поведения при различных температурах путем использования закономерности взаимодействия элементов внедрения с дислокациями и перераспределения примесей по формам нахождения в зависимости от внешних условий. Имеются многие примеры негативного влияния газов на свойства металлов. Так, примеси водорода, кислорода, азота и углерода вызывают переход тугоплавких металлов из пластичного состояния в хрупкое. Можно выделить три основных направления в использовании методов определения газов в металлах.
Первое направление — контроль промышленной продукции и технологических процессов. Во многих случаях известны предельно допустимые содержания газов, превышение которых снижает качество металла или делает его негодным к использованию. Содержание газов нормировано ГОСТами на многие виды продукции металлургической промышленности. В таких технологических процессах, как производство стали, необходим контроль содержаний кислорода и углерода в жидкой ванне по ходу плавки. Для этого созданы экспрессные методы определения этих газов. Определение газов требуется при любых технологических процессах, связанных с воздействием на металл высокой температуры. Наиболее часто при использовании методов определения газов для контроля в промышленности оказывается достаточным определение общего (валового) содержания того или иного газа в образце без разделения по формам его нахождения. Исключение составляют чистые металлы, например медь, выплавленная
Газовый анализ
931
в вакууме, содержание кислорода в которой может находиться на уровне 10“5 масс. %. При таких содержаниях количества кислорода, сорбированного поверхностью образца и содержащегося в его объеме, соизмеримы. Для анализа чистых материалов используют методы, позволяющие определять содержания газа в объеме образца путем устранения или учета влияния поверхностно сорбированного газа на результаты анализа. Повышенная точность анализа материалов промышленного назначения может требоваться в случаях, когда газы служат легирующими добавками в сплавах, повышающими их прочность или коррозионную стойкость (кислород в титане, азот в стали). Высокая точность нужна при определении кислорода в оксидах, например урана и тория, применяемых для изготовления тепловыделяющих элементов (ТВЭЛ) ядерных реакторов. Их свойства в значительной степени зависят от небольших изменений в содержании кислорода.
Второе направление — исследование взаимодействия газов с металлами при изучении термодинамических и макрокинетических параметров кислорода, азота, углерода и водорода в металлах (например, послойное определение содержаний газов).
Третье направление — установление зависимости свойств твердых фаз от их состава и структуры. Исследование корреляции между составом и строением твердых тел, с одной стороны, и их свойствами — с другой, осуществляется путем использования комплекса физических и химических методов определения газов в металлах. При этом, наряду с задачей определения валового содержания того или иного газообразующего элемента, возникает и задача их раздельного определения в разных формах нахождения. Химическая форма и место локализации в металле газовой примеси могут быть различны. Газ может находиться в кристаллической решетке металла в виде раствора внедрения или замещения (в атомном или ионном состоянии); может быть связан в химические соединения (гидриды, нитриды, оксиды и т.д.) как с основным элементом исследуемого материала, так и с различными случайными примесями или легирующими добавками; может быть сорбирован на поверхностях металла (как наружных, так и внутренних) в виде атомов, молекул или химических соединений; может быть «зажат» под большим давлением в пузырьковых дефектах внутри металла в состоянии молекулярного газа; может находиться в составе случайных загрязнений поверхности металла, возникающих в результате небрежного их хранения (влага, тонкие пленки нефтепродуктов и пр.). Совокупность методов определения газов в металлах может быть представлена несколькими основными группами.
Методы вакуумной экстракции: газ выделяется в результате вакуум-нагрева, восстановительного плавления (в металлической ванне или без ванны) в вакууме или в инертной атмосфере, импульсного нагрева и т.д. Анализ экстрагированного газа осуществляется либо химическим разделением компонентов смеси с последующими объемно-манометрическими измерениями, либо с использованием хроматографии, масс-спектрометрии, ИК-спектро-
метрии. Известно применение для этой цели и хромато-масс-спектрометрии. Все методы основаны на полном выделении газов из навески металла, полном их улавливании и количественном измерении каждого компонента газовой смеси.
Химические методы: кислородный при определении водорода; раскисления металла жидким алюминием с последующим определением количества А12О3; вакуумной дистилляции; ртутной экстракции для отделения металла от избыточных фаз, содержащих газовые примеси и т. д. при определении кислорода; метод Кьельдаля и его разновидности при определении азота и др. — предназначены для определения лишь одной газовой примеси.
Ядерно-физические методы основаны на облучении образца элементарными частицами или у-квантами. В результате ядерной реакции образуется радиоактивный изотоп. Число образовавшихся радиоактивных атомов примеси пропорционально ее содержанию в анализируемом образце. Существуют методы определения кислорода, азота и углерода с использованием ядерных реакций на заряженных частицах (протонах, дейтронах, тритонах, гелии-3 и а-частицах), 14 МэВ-нейтронов и тормозного у-излучения. Для повышения чувствительности ядерно-физических методов применяется радиохимическое выделение с использованием восстановительного плавления, дистилляции и т.п.
Спектральные методы реализуются в двух основных направлениях, основанных либо на полной экстракции газов из анализируемой навески с последующим спектральным анализом выделенного газа, либо на частичной экстракции газа и одновременном его возбуждении при локальной поверхностной эрозии образца. Будучи достаточно экспрессными и производительными, методы этой группы нуждаются в использовании стандартных металлических образцов, что ограничивает их возможности.
Масс-спектрометрические методы, например, метод определения кислорода и азота с помощью искрового масс-спектрометра или масс-спектрометра с воздействием на образец лазерного излучения для получения плазменного факела и его последующего масс-спектрометрического анализа. Имеется опыт применения и электронной пушки для экстракции газов из металла и их ионизации.
Группа методов изотопного уравновешивания, обладающих рядом бесспорных достоинств — высокими точностными параметрами, отсутствием необходимости построения градуировочных характеристик на основе применения адекватных стандартных образцов и слабой зависимостью результатов анализа от его условий и состава пробы. Суть подхода состоит в следующем. В обезгажен-ный реакционный сосуд (обменник) помещают анализируемый образец металла. Туда же вводится либо в газообразном состоянии, либо в составе дозировочного металлического образца дозированное количество редкого стабильного изотопа определяемого газообразующего элемента (2D, 15N, 18О, 13С). При достаточно высоких температурах в обменнике осуществляется уравновешивание изотопов, в результате которого в газовой фазе над метал
932
Новый справочник химика и технолога
лом образуется равновесная смесь редкого и распространенного изотопов. По результатам анализа изотопного состава этой смеси (оптического спектрального или масс-спектрального) рассчитывается искомое содержание газа в анализируемом образце.
Методы, основанные на зависимости интегральных физико-химических параметров металлов от их газосо-держания. К этой группе относятся метод пробкового индикатора примесей (выпадение примесей и закупорка ими пористой пластины индикатора при понижении температуры расплава анализируемого металла); метод измерения электросопротивления, которое является функцией содержания примесей, в том числе и газообразующих в металлах; и метод ЭДС с использованием твердого электролита (вариант электрохимического метода). В последнем случае твердоэлектролитные ячейки используют как для определения состава газовой фазы, экстрагированной из металла, так и непосредственно для определения кислорода в металлическом расплаве.
8.9.13. Методы определения дисперсной фазы в газах
Источники формирования дисперсной фазы в газах. К дисперсной фазе, загрязняющей газы, обычно относят пыль, влагу, примеси масел, пары и аэрозоли жидких химических веществ.
Пыль — дисперсная система, состоящая из мелких твердых частиц, находящихся в газе во взвешенном состоянии. Наличие пыли приводит к выходу из строя контрольно-измерительной аппаратуры, к снижению качества изделий, использующих загрязненный газ и т.д. Поэтому содержание пыли в газах является одним из нормируемых параметров, подлежащих контролю.
Влага частично сохраняется в процессе многоступенчатого политропического сжатия газа в компрессоре и осушки его в адсорберах. Присутствие высоких количеств влаги в газах при ее конденсации может быть причиной выхода из строя различного рода датчиков и пневмосистем, работающих на этом газе. Поэтому содержание влаги в газах является также одним из параметров, определяющих их кондиционность.
Масла используются при компримировании газов. Перечень наиболее употребительных при этом масел приведен в таблице 8.32.
Таблица 8.32
Специальные и машинные масла, используемые при переработке газов
Марка масла Температура, °C
вспышки застывания
П-28 285 -10
Авиационное МК-22 230 -14
Компрессорное КС-19 270 -15
Компрессорное К-19 242 -5
Авиационное МС-20С 250 -18
Компрессорное К-12 216 -25
Продолжение таблицы 8.32
Марка масла Температура, °C
вспышки застывания
ДС-8 190 -25
М-36/1-К 195 -60
Авиационное МС-14 200 -30
К-28 275 -ю
Турбинное 46 (т) 195 -10
Турбинное 30 (ут) 180 -10
Турбинное 22 (л) 180 -15
Веретенное АУ 163 -45
ВМ-4 206-213 -15
Индустриальное 50 (машинное-су) 200 -20
Индустриальное 30 (машинное-л) 190 -15
Индустриальное 45 (машинное-с) 190 -10
ДП-11 200 -18
ТАП-10 95 -
ТАП-15 95 -
Минеральные смазочные масла представляют собой сложные смеси углеводородов различных рядов с числом атомов углерода в молекуле от С2о до С40 и более, а также кислородных, сернистых и азотсодержащих соединений. Состав образующихся при термическом окислении и, следовательно, попадающих в газ продуктов, представлен в таблице 8.33.
Таблица 8.33
Состав веществ, образующихся при термическом окислении масла (МС-20)
Вещество Содержание, масс. %
Испарившаяся и сконденсировавшаяся часть масла 31,2
Масло, образовавшее лак 35,1
Кислоты 6,4
Альдегиды 1,4
Спирты 0,33
Газообразные углеводороды 2,9
Диоксид углерода 2,8
Реакционная вода 2,4
Перекись водорода 0,0028
Оксид углерода 0,15
Кислород 0,5
Аммиак и алифатические амины 0,25
Ароматические соединения 0,10
Оксиды азота следы
Газовый анализ
933
Как видно из табл. 8.33, при термоокислительной деструкции смазочного минерального масла образуется сложная смесь паров и аэрозолей, содержащая летучие продукты испарения и окисления масла, сложные эфиры, спирты, органические кислоты, кетоны, альдегиды и оксид углерода. Поэтому агрегатное состояние примесей масла в газах и их содержание требует учета при разработке средств контроля состава газов.
8.9.13.1. Методы определения примесей масла в газах
Особую сложность решения такой аналитической задачи обусловливает агрегатная неустойчивость масла, зависимость его состояния от физико-химических свойств газа, материала поверхностей, ограничивающих его объем, и других факторов (давление, температура, скорость потока газа и др.).
Гравиметрический метод основан на нахождении разности массы аналитического фильтра до отбора (т\) примесей и после отбора (ти2). Предварительно фильтр, например аналитический типа АФА-В-10, АФА-В-18 или ФПП-15, высушивают в течение 20-30 мин при температуре 50-60 °C и взвешивают на весах с ценой деления 0,06 мг. Через фильтр пропускают 1,5 м3 газа в течение двух часов с расходом 12,5 л/мин. Затем фильтр высушивают в том же режиме и определяют массу фильтра. Абсолютное количество Ати масла будет равно ти2 - т\- Тогда содержание масла в газе w = Д/л/К, где V— объем пропущенного через фильтр газа. Помимо фильтров возможно использование высокотемпературных вымораживателей и других охлаждающих систем.
Рефрактометрический метод основан на использовании зависимости показателя преломления какого-либо растворителя, через который пропускается анализируемый газ, от содержания растворенного масла. В качестве растворителя применяют этиловый эфир [п™ = 1,3505), этилацетат (п™ = 1,372б), н-гексан (п2£ =1,3749) и бутилацетат (и70 =1,3961). Используется интерферометр ИТР-1 с кюветой 8-10 см.
Нефелометрический метод основан на ослаблении интенсивности светового луча за счет рассеяния света взвешенными либо в жидкой, либо в газовой среде дисперсными частицами. Метод реализуется либо путем экстрагирования масла с фильтра и образования эмульсии, взвешенные частицы которой рассеивают свет, а, следовательно, ослабляют интенсивность проходящего через раствор луча, либо путем измерения уменьшения интенсивности луча света, непосредственно проходящего через анализируемый газ. Анализ выполняется на нефелометрах при сравнении (визуальном или фотоэлектрическом) интенсивностей прошедшего и рассеянного лучей. Существующие нефелометры позволяют измерять содержания частиц размером 0,1-0,3 мкм, в пределах от 1 до 107 см-3.
Люминесцентный (флуоресцентный) метод описан выше. В данном случае возбуждаемая УФ-излучением люминесценция масел представляет суммарное свечение ряда
компонентов. Содержание люминесцирующих соединений находится в зависимости от основной массы углеводородов, входящих в состав масла. Поэтому выходной сигнал функционально зависит от его содержания. В основном люминесценция масел определяется ароматическими углеводородами и смолами. Метод используется для определения содержания масел в атмосферном воздухе, воздухе рабочей зоны на предприятиях, в кабинах самолетов, рудничном воздухе и т.д.
Абсорбционный спектральный метод с использованием инфракрасных спектров применяется для измерений при условии предварительного концентрирования паров масел в органических растворителях.
Линейно-колористический метод основан на цветных реакциях, протекающих в различных средах. Индикаторные трубки из прозрачного материала заполняют слоем индикаторного порошка, закрепленного с помощью тампонов из стекловолокна. Индикаторный порошок состоит из зерен адсорбента (носителя), на поверхности которого закрепляется слой реагента (индикатора), изменяющего свою окраску при взаимодействии с определяемым компонентом газа. При анализе трубка вскрывается с обоих концов и через нее пропускается определенный объем анализируемого газа. О величине содержания масла в газе судят по длине изменившего окраску слоя индикаторного порошка. Иногда в трубку помещают дополнительные слои различных адсорбентов, назначение которых — удаление из анализируемого газа примесей, мешающих определению искомого компонента. Кроме того, в некоторых случаях в трубке размещают ампулы с раствором реактивов, создающих условия протекания цветной реакции (кислоты, щелочи и т.п.) или выполняющих другие вспомогательные функции.
8.9.13.2. Методы измерения содержания влаги в газах
Для количественной оценки влажности газов используются две группы характеристик.
Первая группа — характеристики, оценивающие содержание водяного пара:
а) абсолютная влажность а, т. е. масса водяного пара, содержащаяся в единице объема газа (г/м3, мг/м3, мг/мл);
б) упругость или парциальное давление водяного пара е, выражаемое в мм рт. ст., миллибарах или паскалях.
Вторая группа — характеристики влажностных отношений:
а) влагосодержание d — отношение массы водяного пара к массе сухого газа в том же объеме, выраженное в безразмерных единицах (массовые доли, процент по массе). Эта характеристика используется для оценки малых содержаний водяного пара. Удобной единицей в этом случае является миллионная доля (международное обозначение «ррт» 1 м.д. = 10 6 = 10 4 % = 1 ррт). Реже используется отношение массы водяного пара к массе влажного газа, называемое удельной влажностью q;
б) объемное влагосодержание х, равное отношению объема водяного пара к объему газа;
934
Новый справочник химика и технолога
в) молярная доля водяного пара s, равная отношению числа молей водяного пара к общему числу молей влажного газа;
г) температура точки росы (т) равна температуре, которую примет влажный газ, если охладить его изобарно до полного перехода водяного пара в состояние насыщения;
д) относительная влажность /, равная отношению действительной влажности газа к его влажности, соответствующей насыщенному водяному пару при данной температуре. Относительная влажность выражается в относительных единицах (0< f< 1) или в процентах (0< f< 100 %).
Метод точки росы. При повышении количества водяных паров в газе при постоянной температуре или при понижении температуры при постоянном количестве водяных паров, последние приходят в состояние насыщения, а затем становятся перенасыщенными и конденсируются, т.е. влага выпадает в виде росы. Температура, при которой пар достигает насыщения и начинается конденсация влаги, называется точкой росы. Основанные на этом принципе измерители влажности (гигрометры) называют конденсационными. Измерив температуру конденсации — точку росы, по таблицам находят абсолютное содержание влаги в газе. Достоинства гигрометров: большой диапазон измерений, охватывающий низкие отрицательные температуры и высокие давления анализируемого газа; удовлетворительная точность во всем диапазоне измерений. В основу конденсационного гигрометра положены три элемента — конденсационное зеркало, охлаждающее устройство и измеритель температуры поверхности зеркала. Выпускаются гигрометры как с визуальной фиксацией точки росы, так и с ее фотоэлектрической индикацией.
Кулонометрический метод основан на поглощении влаги из дозируемого потока газа и ее электролитическом разложении на водород и кислород. О содержании влаги судят по величине тока электролиза. На этой основе созданы и производятся гигрометры семейства «Байкал» — автоматические, непрерывно действующие приборы, предназначенные для измерения объемной доли влаги в газах (азоте, воздухе, водороде, кислороде, инертных газах, в их смесях и других газах, не взаимодействующих с Р2О5). Большое число моделей гигрометров выпускаются зарубежными фирмами.
Сорбционно-частотный метод основан на измерении резонансной частоты колебаний пьезоэлемента, зависящей от массы пленки нанесенного на элемент сорбента, которая, в свою очередь, определяется влажностью окружающего газа. В гигрометрах, построенных на этой основе, в качестве датчика влажности, как правило, используется кристалл кварца, покрытый пленкой сорбента, для изготовления которого используются тонкопористые материалы — цеолиты, оксид алюминия, полиамид, полиэтиленгликоль. В наиболее распространенной модели сорбционно-частотного гигрометра, выпускаемой фирмой «Du Pont» (США), чувствительный элемент находится попеременно в потоке влажного (анализируемого) и «сухого» газов. При этом измеряется изменение частоты колебаний кристалла. Поэтому подводимый к гигрометру газ разделяется на две
линии — линию анализа и контрольную линию. В последней газ проходит через осушитель, где из него извлекается влага. Влажным элементом таких гигрометров является встроенный генератор влажности (сосуд с дистиллированной водой), позволяющий осуществлять быструю проверку калибровки прибора.
8.9.13.3. Методы измерения содержания твердой дисперсной фазы (пыли)
Пыль в подавляющем большинстве случаев является полидисперсной и характеризуется широким спектром размеров аэрозольных частиц (от 10 2 до 102 мкм) и содержаний (от 10 8 до 105 мг/м3), что в сочетании с неустойчивостью системы «твердая дисперсная фаза-газ» не позволяет использовать какой-либо метод в качестве универсального. Кроме того, форма и физико-химические свойства частиц пыли могут быть различными, а зачастую — непостоянными во времени. Поэтому все методы измерения содержания пыли делятся на методы, основанные на предварительном осаждении частиц пыли, и методы без их предварительного осаждения. При малых давлениях газов с успехом могут быть использованы те и другие методы. С повышением давления предпочтительными становятся последние, так как резко возрастают погрешности, связанные с отбором пробы при ее редуцировании. Ниже рассматриваются наиболее употребительные из них.
Гравиметрический (весовой) метод является наиболее точным, надежным и информативным способом определения содержания пыли. Применительно к газам он состоит в прокачке анализируемого газа через предварительно взвешенный фильтр (т() и последующем взвешивании фильтра с осевшей на нем пылью (т2). Тогда масса пыли определится как т2 - т\, а ее содержание в газе как
v-t
где v — объемная скорость прокачки газа через фильтр, а / — время прокачки. Отобранная пыль может быть подвергнута дальнейшему анализу на химический состав (капельный анализ, эмиссионный, абсорбционный, рентгенофлуоресцентный и ядерно-физические методы) или гранулометрическому исследованию. Помимо бумажных или ватных фильтров в гравиметрических измерениях используют электростатическое осаждение пыли на металлических пластинах (например, алюминиевых) пылеемкостью до 60 мг. Пластину взвешивают до и после осаждения пыли. Используется и мокрое пылеулавливание в промывной склянке с использованием смачиваемого спиртом отражателя. После испарения спирта остаток аэрозоля взвешивают. Помимо взвешивания осажденного аэрозоля, возможно использование различных светотехнических методов измерения степени почернения пылевого осадка на фильтре. Приборы для определения содержания пыли разработаны и производятся различными фирмами. В основном они предназначены для контроля запыленности атмосферного воздуха и воздуха рабочей зоны.
Газовый анализ
935
Метод счета частиц по измерению светорассеяния основан на фотоэлектрической регистрации света, рассеянного отдельными аэрозольными частицами заданных размеров при их прохождении через оптическую регистрирующую систему. Такие измерения возможны, поскольку интенсивность рассеянного частицей света зависит от ее размеров и формы, угла светорассеяния, длины волны света и коэффициента отражения света поверхностью частицы. Для конкретной измерительной схемы амплитуда импульса рассеянного света определяется размером частицы и ее коэффициентом отражения. Импульсы светорассеяния регистрируются и суммируются по диапазонам. При этом определяется как счетная концентрация частиц, так и их дисперсный состав. Наибольшее распространение получили анализаторы с углом наблюдения рассеянного света, равным 90°. Анализируемый газ пропускается через узкий капилляр (менее 2 мм). Используемые обычно анализаторы позволяют измерять счетные концентрации от 1 до 107 см-3 с размерами регистрируемых частиц от 0,3 до 10 мкм. Разработаны и производятся различные модели счетных анализаторов твердой дисперсионной фазы в газах.
Акустический метод основан на зависимости скорости звука или акустических потерь от состава газовых смесей. Для измерения концентрации пыли используется разновидность метода, основанная на измерении величины акустических потерь, зависящих от размеров аэрозольных частиц, плотности вещества частиц, их концентрации, частоты ультразвуковой волны и др.
Радиоизотопный метод основан на свойстве частиц радиоктивного излучения оседать на частицах пыли. Анализируемый газ пропускают через фильтр и определяют массу осевшей на фильтре пыли по ослаблению радиоактивного излучения при его прохождении через слой осевшей пыли. Содержание пыли с определяют по формуле
где т — масса пыли на фильтре; v — объемная скорость пропускаемого через фильтр газа; t — время отбора пыли. Массу осевшей на фильтре пыли определяют из зависимости
I = /0 ехр(-цит),
где 1 и /0 — интенсивность прошедшего и падающего радиоактивного излучения соответственно; — массовый коэффициент поглощения радиоактивного излучения, равный а Ет^х (здесь Етах — максимальная энергия радиоактивного излучения; а и b — постоянные, равные -0,05 и -0,36 соответственно. Коэффициент зависит от вида и энергии излучения, а также от условий измерений. Наиболее широко для измерения концентрации пыли используют Р-излучение. Метод может быть реализован и без предварительного осаждения пыли на фильтре. В этом случае источник p-излучения и детектор располагают на противоположных стенках газохода напротив друг друга. О содержании пыли судят по поглощению излучения.
Метод измерения содержания пыли по числу частиц основан на том, что из известного объема газа осаждается на прозрачную поверхность пыль, видимые частицы которой подсчитываются под микроскопом. Осаждение частиц осуществляется следующими четырьмя способами. Первый: пыль задерживается во время прохождения пробы воздуха на фильтре. Второй: поток газа направляется на клейкую прозрачную поверхность, к которой прилипают частицы пыли. Третий: пылевые частицы осаждаются в результате термофореза на охлаждаемых деталях прибора. Четвертый: частицы задерживаются в жидкости при пропускании пробы газа через специальные промывные склянки (отражатели), в которых пузырьки газа (следовательно, и частицы пыли) ударяются о стеклянные поверхности.
Подсчет частиц с помощью микроскопа проводится на прозрачном или непрозрачном субстрате (подложке) в светлом или темном поле. Различимость под микроскопом ограничивается размерами мелких частиц. Трудности возникают уже при подсчете частиц диаметром менее 1 мкм. Результаты подсчета дают число частиц в 1 см3 воздуха. Для пересчета этого показателя в значение концентрации (мг/м3) необходимо иметь данные по плотности вещества частиц и их распределению по размерам. Для приближенной оценки могут быть использованы следующие соотношения:
Число частиц в 1 см3 Содержание пыли, мг/м3
500 2
2000 10
20000 100
Метод, основанный на эффекте Тиндаля, состоит в измерении интенсивности рассеянного света, обусловленного содержанием в газе пылевых частиц. При этом интенсивность рассеянного света, выходящего из освещенной пылевой камеры под углом 30°, соотносится с интенсивностью первичного луча, ослабленного в необходимой степени.
Нефелометрический метод основан на непосредственном измерении ослабления луча света, проходящего через камеру с запыленным газом.
Другие, менее употребительные методы. Электростатический: проба газа пропускается в виде вихревого потока через измерительную камеру. В результате трения частиц пыли о внутреннюю поверхность стенки камеры на последней возникает статическое электричество. Отводимый от камеры ток является мерой содержания пыли.
Ионизационный: проба газа пропускается через камеру, в которой создан с помощью слабого у-излучения ток ионизации. Присутствие пыли ослабляет ток ионизации тем больше, чем выше ее содержание в газе.
В случаях, когда необходим анализ химического состава пылевой составляющей, используются такие методы, как эмиссионный спектральный, атомно-абсорбционный, рентгено-спектральный, нейтронно-активационный, а так
936
Новый справочник химика и технолога
же метод лазерной искры. Последний может быть отнесен к эмиссионному спектральному методу.
8.9.13.4. Методы определения радиоактивных веществ в газах
Задача контроля радиоактивности в основном характерна для атмосферного воздуха, хотя существуют определенные аналитические проблемы и для газовых теплоносителей энергетических установок. Носителями радиоактивности воздуха являются, главным образом, аэрозоли пылевидных частиц размером 0,02-1 мкм. Поэтому необходимым этапом аналитического процесса является количественный отбор пыли на тот или иной фильтр или липкую ленту. Измерение уровня радиации, как правило, проводят несколько раз в течение определенного времени с тем, чтобы обеспечить возможность раздельной оценки естественной быстропадающей и искусственной радиоактивности. Измерению уровня радиоактивности подвергаются пробы пыли непосредственно после их отбора и по истечении двух суток. Для измерений обычно применяются пропорциональные счетчики, импульсы которых позволяют различать а- и [3-излучения и проводить их раздельное измерение. Интенсивность у-излучения измеряется, как правило, с помощью сцинтилляционных счетчиков. При необходимости осуществляется выделение того или иного радионуклида из газовой пробы и его концентрирование методами радиохимии.
8.9.14. Дистанционные методы газового анализа
Эти методы используются в целях контроля атмосферного воздуха, а также для анализа технологических газов в труднодоступных по той или иной причине местах. Особую роль дистанционные методы играют в контроле загрязнений атмосферного воздуха, поскольку именно ими в основном обусловлено глобальное распространение загрязнений на планете. В дифференциальном варианте дистанционных методов определяется содержание с примеси в заданной точке пространства в нужный момент времени. В интегральном варианте определяют количество Q примеси на трассе длиной R = f • c(r)dr. В этом случае получают
статистически достоверные данные о среднем содержании
Q „
сср = —. Существующие дистанционные методы основаны R
на взаимодействии электромагнитного излучения с атомами и молекулами газов, а также частицами дисперсной фазы. Источниками излучения могут быть как естественные объекты (Солнце, Луна, звезды, земная поверхность, непосредственно детектируемые компоненты), так и специально созданные источники света (газоразрядные лампы, лазеры). В соответствии с этим методы контроля могут быть пассивными (источники света — естественные объекты) и активными (специально созданные источники света). Измерения проводятся как на станциях наземного базирования, так и на различных летательных аппаратах (аэростаты, самолеты, ракеты, искусственные спутники Земли).
Пассивные методы включают абсорбционный и эмиссионный варианты. Первый основан на измерении поглощения детектируемыми компонентами прямого излучения Солнца, Луны, звезд, а также рассеянного дневным небом излучения Солнца. Аппаратура с достаточно высоким спектральным разрешением (< 0,01 см ’) дает возможность проводить измерения спектрального пропускания Дсо) или спектрального поглощения Л(со) атмосферного воздуха при оценке фоновых содержаний СО, СО2, NO2, N2O. В основе эмиссионного метода лежит перенос теплового излучения в атмосфере от детектируемых молекул. Поскольку максимум интенсивности их излучения (температура газа обычно лежит в пределах 220-500 К) приходится на спектральный диапазон от 6 до 13 мкм, то измерения эмиссионным методом проводятся в ИК-, а также в микроволновых диапазонах, где интенсивность собственного излучения газов еще достаточно велика (оценка содержаний Н2О, Оз, СО2). К эмиссионным пассивным методам обычно относят и измерения резонансного комбинационного рассеяния на детектируемых молекулах. Это предельный случай КР, когда частота возбуждающего излучения приближается к собственным частотам энергетических переходов молекул детектируемого газа, что приводит к резкому увеличению интенсивности рассеяния. Резонансное рассеяние обычно наблюдается в УФ-диапазоне спектра (например, для молекулы NO — это 200-220 нм), т.е. в области электронных переходов.
Активные методы основаны на использовании процессов поглощения, рассеяния и флуоресценции, возникающих при прохождении излучения от специально созданного источника электромагнитного излучения через газовую среду. Соответственно различают методы— абсорбционный, комбинационного рассеяния и резонансной флуоресценции. Сюда же можно отнести и методы, базирующиеся на «классическом» рассеянии света аэрозольными частицами. Методы этой группы в основном базируются на использовании лидаров (аббревиатура английского термина «Light Detection and Ranging»).
8.10. Метрологические аспекты газового анализа
Рассмотрение метрологических аспектов того или иного метода важно для выявления его реальных возможностей и необходимо для оценки основных направлений и перспектив его развития. По этой причине в последние два десятилетия все более серьезное внимание уделяется метрологическим аспектам анализа веществ в целом и газового анализа в частности.
8.10.1. Понятие «метрологическое обеспечение газового анализа» и его структура
Метрологическое обеспечение анализа в самом общем случае — это совокупность принципов, практических и организационных решений, позволяющих находить искомое содержание определяемого компонента с удовлетворительными показателями точности (сходимости, воспро
Газовый анализ
937
изводимости и правильности). Метрологическое обеспечение включает с себя:
1. Обоснование практической применимости фундаментального принципа, положенного в основу аналитического метода.
2. Нахождение оптимальных условий анализа, обеспечивающих наилучшие (или требуемые) показатели точности и границы диапазона определяемых содержаний.
3. Получение зависимости показателей точности от содержания определяемого компонента и установление на этой основе границ диапазона определяемых содержаний.
4. Непосредственно аналитическую методику как результат перечисленных выше действий.
5. Организационные мероприятия, обеспечивающие единообразие применяемых аналитических приборов и мер (в том числе — стандартных образцов состава).
8.10.2. Метрологический подход в газовом анализе, его основные направления и формы
Метрологический подход к достижению требуемой точности аналитической информации базируется на системном исследовании элементов и стадий аналитического процесса. Современное приборостроение предоставляет в распоряжение аналитиков и технологов как устройства, позволяющие выполнять отдельные стадии и этапы аналитической процедуры, так и устройства, предназначенные для реализации нескольких этапов или процедуры в целом. Среди тех и других имеются устройства, классифицируемые как средства измерений содержания компонентов в газовых средах — анализаторы газов. В зависимости от назначения и области применения анализаторы газов можно разделить на два вида.
К первому относятся анализаторы конкретного (узкоцелевого) назначения. Они используются для решения одной или нескольких конкретных аналитических задач, при этом постановка задачи обычно предшествует созданию анализатора. Такие анализаторы применяются в промышленных отраслях, а также в лабораториях и организациях экологического и медицинского профиля, осуществляющих инспекционные функции. Метрологические характеристики анализаторов этого вида приведены, как правило, к входным сигналам (т.е. к значениям содержания определяемого компонента). При классификации анализаторов, прежде всего учитываются их функциональные особенности (преобразователь, прибор, установка, система) и лишь затем назначение (решаемая аналитическая задача), принцип действия (метод анализа) и конструкция.
Второй вид анализаторов — это средства измерений универсального назначения. Они применимы для решения широкого круга аналитических задач, конкретизируемых при эксплуатации анализаторов. Основная область их применения — научные лаборатории. Метрологические характеристики таких анализаторов приведены к выходным сигналам или «привязаны» к одной-двум типовым (модельным) газоаналитическим задачам. Для решения с помощью таких анализаторов конкретных газоаналитических задач
разрабатывают специальные методики. При классификации анализаторов в качестве основного признака выступает метод анализа (газовая хроматография, интерферометрия и т. п.).
Среди метрологических проблем, относящихся к анализаторам газов, наиболее важной представляется проблема нормирования и определения метрологических характеристик. Суть проблемы — в достижении компромисса между стремлением к подробному описанию возможностей анализаторов и ограничениями технико-экономического характера, действующими при определении метрологических характеристик. Достигаемые компромиссы закрепляются в нормативно-технических документах в виде комплексов нормируемых метрологических характеристик.
Выбор комплексов и методов определения метрологических характеристик непосредственно связан со степенью разработанности математической модели погрешности анализатора. Чем подробнее эта модель, тем больше возможностей для частичной замены экспериментальных методов определения метрологических характеристик на существенно более дешевые расчетные методы.
При создании анализаторов газов конкретного назначения аппаратурные и методические вопросы прорабатываются комплексно. Процедура получения результата анализа описывается в технической документации на анализатор, и, таким образом, у пользователя, как правило, не возникает необходимости в разработке специальных методик анализа. Исключением являются ситуации, когда при использовании анализатора (прибора) необходимо регламентировать процедуру отбора проб.
Иначе обстоит дело, когда применяются анализаторы универсального назначения. В таких случаях проведению анализа предшествует разработка и опробование методики. Методика анализа (методика выполнения измерений содержания компонентов) — это совокупность условий и операций, обеспечивающая решение поставленной аналитической задачи. Документальное оформление методики определяется ее назначением и сферой применения. При приведении разового исследования методика может быть изложена в его программе. При необходимости частого применения методики ее оформляют в виде нормативнотехнического документа: методических указаний, инструкций, разделов технических условий или регламентов и т. п. В случаях массового применения методики (например, при контроле качества продукции, серийно выпускаемой предприятиями нескольких отраслей) методику включают в государственные стандарты. В связи с расширением торговли между странами осуществляется стандартизация отдельных методик на международном уровне. Перечень государственных и международных стандартов, регламентирующих методики анализа неорганических газов, приведен в табл. 8.34.
Стандартизированные методики составляют лишь незначительную долю от общего числа методик газового анализа. Поэтому в качестве основного механизма достижения надлежащего метрологического уровня методик в настоящее время рассматривают их метрологическую ат
938
Новый справочник химика и технолога
тестацию. Метрологическая аттестация — один из этапов разработки методики, особенность которого состоит в том, что он проводится при непосредственном участии специалистов-метрологов, а порядок его проведения четко регламентирован. Метрологическую аттестацию методик выполнения измерений содержания компонентов проводят с целью установления приписанных характеристик погрешности (показателей точности измерений) и проверки их соответствия нормам погрешности. Приписанные характеристики погрешности — это погрешности, соответствующие любому из результатов измерений, полученному при реализации данной методики. Нормы погрешности — это значения характеристик погрешности, задаваемые в качестве требуемых или допускаемых в заданиях на разработку методики.
Таблица 8.34
Отечественные и международные стандарты, регламентирующие методики анализа газов
Обозначение стандарта Название
ГОСТ 20461-75 Гелий газообразный. Метод определения содержания примесей эмиссионным спектральным анализом
ГОСТ 5583-78 Кислород газообразный технический и медицинский. Технические условия
ГОСТ 8050-85 Двуокись углерода газообразная и жидкая. Технические условия
ГОСТ 10219-77 Ксенон. Технические условия
ГОСТ 10218-77 Криптон и криптон-ксеноновая смесь. Технические условия
ГОСТ 3022-80 Водород технический. Технические условия
ГОСТ 9293-74 Азот газообразный и жидкий
ГОСТ 10157-79 Аргон газообразный и жидкий
ГОСТ 14022-78 Водород фтористый безводный. Технические условия
ГОСТ 17.2.4.03-81 Охрана природы. Атмосфера. Иодофе-нольный метод определения аммиака
ГОСТ 17.2.2.05-86 Охрана природы. Атмосфера. Нормы и методы измерения выбросов вредных веществ с отработавшими газами тракторных и комбайновых дизелей
ГОСТ 17.2.2.03-87 Охрана природы. Атмосфера. Нормы и методы измерений содержания окиси углерода и углеводородов в отработавших газах автомобилей с бензиновыми двигателями. Требования безопасности
ГОСТ 17.2.2.04-86 Охрана природы. Атмосфера. Двигатели газотурбинные самолетов гражданской авиации. Нормы и методы определения выбросов загрязняющих веществ
Продолжение таблицы 8.34
Обозначение стандарта Название
ГОСТ 12.1.014-84 Система стандартов безопасности труда. Воздух рабочей зоны. Метод измерения концентраций вредных веществ индикаторными трубками
ИСО 4220-83 Воздух атмосферный. Определение показателя загрязнения воздуха газообразными кислотами титрометрическим или потенциометрическим методом детектирования с двумя индикаторными электродами
ИСО 4221-80 Качество воздуха. Определение массовой концентрации двуокиси серы в окружающем воздухе. Спектрофотометрический метод с применением торина
ИСО 7996-85 Воздух атмосферный. Определение массовой концентрации окислов азота хемилюминесцентным методом
ИСО 6768-85 Качество воздуха. Определение массовой концентрации двуокиси азота. Модифицированный метод Грисса-Зальцмана
ИСО 6568-81 Газ нейтральный. Анализ пробы методом газовой хроматографии
Характерными этапами метрологической аттестации методики являются:
а) выбор формы представления погрешности;
б) формирование математической модели погрешности;
в) анализ источников погрешности;
г) планирование экспериментов по получению исходных данных для расчета составляющих погрешности;
д) проведение экспериментов;
е) расчет составляющих и статистических оценок погрешности;
ж) установление приписанных характеристик погрешности;
з) расчет нормативов контроля характеристик погрешности;
и) сопоставление приписанных характеристик погрешности с требуемыми или допускаемыми.
Традиционная для газоаналитиков проблема сопоставимости результатов анализов трансформируется метрологами в проблему обеспечения единства газоаналитических измерений.
Под единством измерений понимается такое их состояние, при котором результаты выражены в узаконенных единицах и погрешности известны с заданной вероятностью. Достижение единства и требуемой точности газоаналитических измерений в метрологии обеспечивается в трех направлениях (рис. 8.3).
Газовый анализ
939
Обеспечение единства и требуемой точности газоаналитических измерений
V
Создание и эксплуатация специальных технических средств (воспроизведение единиц физических величин, характеризующих состав газовых сред. Передача размеров единиц)
• Поверочные газовые смеси и чистые газы, хранящиеся в баллонах
• Генераторы газовых смесей
• Поверочные установки
• Передвижные поверочные лаборатории
• Образцовые газоаналитические и газосмесительные установки
• Установки внешней точности
• Эталоны (комплексы прецизионной аналитической и смесительной аппаратуры)
Стандартизация (разработка нормативно-технических документов)
Стандартизация (разработка нормативно-технических документов)
• Термины
• Физические величины и их единицы
• Нормы точности измерений
• Формы представления результатов измерений
• Номенклатура нормируемых метрологических характеристик средств измерений
• Методы и средства поверки
• Методики выполнения измерений
• Поверочная схема
• Организация и порядок проведения метрологических работ
• Справочные данные о свойствах газов
• Государственные испытания средств измерений
• Метрологическая аттестация нестан-дартизированных средств измерений
• Поверка средств измерений
• Метрологическая экспертиза документации
• Метрологическая аттестация методик выполнения измерений
• Аттестация аналитических лабораторий
• Проверка соблюдения метрологических правил
• Аттестация стандартных образцов (поверочных газовых смесей)
• Подготовка и повышение квалификации работников метрологических служб
Рис. 8.3. Пуги обеспечения единства и требуемой точности измерений
8.10.3. Метрологические характеристики анализаторов газов
Общие требования к нормируемым метрологическим характеристикам (НМХ) промышленных автоматических газоанализаторов установлены ГОСТ 13320-81. В стандарте приведен перечень НМХ, указаны способы их нормирования и определения. Различные комплексы НМХ присущи газоанализаторам-преобразователям, газоанализаторам-сигнализаторам и показывающим газоанализаторам. Среди последних разные комплексы имеют аналоговые и цифровые анализаторы, приборы с равномерной и неравномерной шкалой. Наибольшие сложности при формировании комплексов НМХ связаны с выбором рациональной номенклатуры НМХ. Номенклатура НМХ газоанализаторов расширяется и уточняется по мере накопления новых конструкторских решений или новых средств градуировки и поверки. В последние годы международные метрологические организации прилагают серьезные усилия для достижения единообразия при нормировании метрологических характеристик. Помимо документов Международной электротехнической комиссии («Выражение работы газоанализаторов» — публикация 1982 г., стандарт «Выражение работы инфракрасных анализаторов качества воздуха» — публикация 1975 г.) следует отметить стандарт ИСО 815885 «Оценка эксплуатационных характеристик газоанализаторов». Этот стандарт распространяется на газоанализаторы, градуировку которых может осуществлять потребитель. Оценивание основной погрешности базируется в стандарте на использовании статистических методов обработки результатов эксперимента, связанного с построением градуировочной характеристики. Междуна
родной организацией законодательной метрологии (МОЗМ) разработана рекомендация 74-1985 «Методы оценки основной погрешности и гистерезиса при проверке газоанализаторов» и проект рекомендации «Методы и средства поверки рабочих приборов для измерения содержании приоритетных газовых загрязнителей в атмосфере. Общие требования» (1988 г.). В этих документах описаны методы, идентичные применяемым в нашей стране. Нормирование метрологических характеристик анализаторов состава газов универсального назначения осуществляют безотносительно к конкретным измерительным задачам. Наиболее детально разработанным может быть признан комплекс НМХ лабораторных аналитических газовых хроматографов. Аналитический лабораторный хроматограф представляет собой индивидуально градуируемую измерительную систему, предназначенную для качественной идентификации компонентов анализируемой смеси веществ и измерения относительного или абсолютного количества каждого из компонентов. Входным сигналом хроматографа является концентрация или масса (объем) определяемого компонента, а выходным сигналом — амплитуда в максимуме хроматографического пика и (или) площадь хроматографического пика и время удерживания определяемого компонента. Комплекс НМХ лабораторного аналитического газового хроматографа включает как характеристики отдельных блоков (термостата, устройства деления выходного сигнала, дозирующего крана, интегратора и др.), так и «совокупные» характеристики, определяемые при подаче на вход хроматографа типового вещества. К последним относятся:
а) уровень флуктуационных шумов нулевого сигнала;
б) относительное среднее квадратическое отклонение выходного сигнала;
940
Новый справочник химика и технолога
в) минимальное допустимое значение амплитуды выходного сигнала в максимуме хроматографического пика при определенном содержании контрольного вещества в детекторе;
г) систематическое относительное изменение выходного сигнала за 48 часов непрерывной работы;
д) относительное отклонение выходного сигнала при изменении напряжения питания хроматографа.
Рассмотренные выше комплексы НМХ соответствуют группам анализаторов, занимающим крайние места на «шкале универсальности». В последние годы все чаще разрабатываются средства измерений, занимающие промежуточное положение. К ним могут быть отнесены потоковые хроматографы, промышленные газоаналитические установки и устройства с конструктивно обособленными системами формирования и подготовки пробы, программми-руемые анализаторы. Подобные средства измерений, обладая определенной универсальностью, могут достаточно быстро настраиваться потребителем на конкретную аналитическую задачу. Для таких средств измерений формируются специфические комплексы НМХ. Специфика, в частности, может состоять в представлении основной погрешности в виде функций таких аргументов, как погрешность средства градуировки, параметры состояния анализируемой и внешней среды, погрешности отдельных блоков и т.п.
8.10.4. Чистые газы как средства обеспечения единства газоаналитических измерений
Являясь материальными носителями информации о содержании компонентов, чистые газы могут быть классифицированы как меры, т. е. средства измерений, предназначенные для воспроизведения физической величины заданного размера. В данном случае под заданными размерами физической величины следует понимать значения объемной (молярной) доли основного компонента — 100 % и примесных компонентов — 0 %. Действительные значения содержания основного и примесных компонентов всегда отличаются от заданных соответственно в меньшую и большую сторону. Оценки значений, найденные на основе эксперимента или расчета, выступают в качестве действительных значений содержания основного Хо или примесного ХП компонента. Недостоверность последних характеризуется погрешностями Л%0 и АХП. При применении чистых газов для градуировки и поверки анализаторов возможны три типичные ситуации:
а) когда необходимо знать только Хо и Л%0;
б) когда необходимо знать только Хп и АХП (например, при регулировании с помощью чистого азота, при регулировании «нулевых» показаний газоанализатора, предназначенного для измерения микросодержаний метана в азоте; в этом случае индекс «п» соответствует метану);
в) когда необходимо знать Хо, АХ0, а также Хп и АХП для группы примесных компонентов (в частности, при приготовлении высокоточных многокомпонентных смесей).
В каждой из перечисленных ситуаций требования к Хо и АХ0 (Хп и АХП) можно варьировать в довольно широких пределах. В большинстве случаев для метрологических
целей могут быть использованы чистые технические газы, выпускаемые промышленностью в баллонах под давлением (азот, аргон, кислород, неон, водород, гелий, оксид и диоксид углерода, аммиак, диоксид серы, метан, пропан, бутан и др.). В некоторых случаях газы (азот, гелий, водород) доочищают перед использованием с помощью фильтров или специальных очистителей. В отдельных случаях используют газы, полученные лабораторными методами в сосудах, а также газы, полученные с помощью динамических генераторов: водород, кислород, хлор, озон и др. В связи с увеличившейся в последние годы потребностью в газах для контроля нулевых показаний анализаторов атмосферного воздуха было организовано промышленное производство «нулевого азота» и «нулевого воздуха». Содержание компонентов в выпускаемых промышленностью чистых газах нормируется путем указания нижней границы интервала допускаемых содержаний основного компонента X* и верхней границы интервала допускаемых содержаний примесного компонента X*. Контроль осуществляется путем сопоставления норм с результатами измерений содержания основного и примесных
компонентов. Должны выполняться условия: для основного компонента
Ли-Д*ои>Лд, (8-7)
для каждого из примесных компонентов
ХПИ + АХПИ<ХП\ (8.8)
Обычно в сертификатах на отдельные образцы технических газов указываются лишь нормы (т. е. Х° и Х„). В связи с этим возникает задача интерпретации сертификатов, т. е. перехода от норм к действительным значениям содержания компонентов (Хо и Х„). Существует несколько решений этой задачи (рассматривается только определение Хоу.
а) Хо принимается равным 100%. Очевидно, что при выполнении условия (8.7) истинное значение содержания основного компонента находится в интервале от 100% до X* . Погрешность действительного значения может быть найдена из выражения:
АХо=100-^од- (8.9).
Так, если Х° = 99,6%, то Л%0 = ± 0,4%. Подобной оценкой погрешности неудобно оперировать при дальнейших расчетах в силу ее несимметричности;
б) Хо принимается равным X*. Погрешность в этом случае чаще всего считают симметричной относительно Хо и равной по модулю |Х0‘ —100|. Для рассмотренного выше примера Л%0 = ± 0,4%. Подобную оценку погрешности следует признать завышенной;
в) Хо рассчитывается из выражения
Л=ЛД + 1ОО~Х°Д- (8.10)
Газовый анализ
941
Погрешность при этом может быть выражена симметричной величиной:
АЛГ0=±
100-2Год 2
(8.И)
В рассмотренном примере Хо = 99,8%, АХ0= ± 0,2%. Последнее решение является наиболее рациональным, но оно корректно только при выполнении изготовителем чистого газа условий (8.7) и (8.8). Если уверенности в этом нет, то целесообразнее пользоваться решением б). При использовании чистых газов для приготовления наиболее точных смесей, а также в других ответственных случаях действительное значение содержания компонента принимается равным значению, полученному при измерении (т.е. Хо = X" и АХо = АХ"). При этом могут быть использованы либо данные, полученные изготовителем газа при выходном контроле, либо данные, получаемые потребителем.
8.10.5. Поверочные газовые смеси
Поверочные газовые смеси (ПГС), приготавливаемые и хранящиеся в баллонах, являются традиционным объектом исследований в аналитической метрологии. В последние годы основные усилия были направлены на стандартизацию ПГС и методов их приготовления, разработку новых типов ПГС, обеспечение необходимого качества ПГС в условиях их промышленного производства. Комплекс характеристик, значения которых указываются в паспорте на ПГС, установлен международным стандартом.
К ним относятся:
а) содержание определяемого компонента, выраженное в единицах молярной, массовой или объемной доли;
б) оценка погрешности (действительная в течение предписанного времени хранения смеси);
в) исходное давление смеси;
г) минимальное рабочее давление;
д) минимальная температура хранения.
Кроме того, в паспорте должны быть указаны наименования неопределяемых компонентов, дата приготовления и срок годности, метод приготовления, а также должны быть даны предупреждения о токсичности и воспламеняемости смеси. При промышленном производстве ПГС значения характеристик каждого экземпляра смеси конкретного типа должны соответствовать нормам, установленным для характеристик этого типа ПГС. Комплекс подобных «типовых» характеристик регламентирован отечественным нормативно-техническим документом. Для типа ПГС при его утверждении устанавливаются: компонентный состав (композиция исходных газов); номинальное (заданное) значение содержания определяемого компонента X" (или интервал подобных значений); пределы допускаемого отклонения действительного значения содержания определяемого компонента от номинального значения D; пределы допускаемой погрешности действительного значения содержания определяемого компонента Ад.
Действительное значение содержания определяемого компонента Х3 устанавливается при выпуске отдельного
экземпляра ПГС и вносится в его паспорт при выполнении условия
рг.-лфо. (8.12)
В паспорт вносятся также значения AD, выраженные в абсолютной форме и симметричные относительно Х3 (как правило, эти значения в 2-6 раз меньше D). Предусмотрено, что при контроле характеристик экземпляра ПГС (установлении X) могут использоваться только те методики выполнения измерений, которые обеспечивают получение результата измерений с погрешностью Аи меньшей или равной Ар. В зависимости от значений Ад серийно выпускаемые ПГС относятся к нулевому, первому или второму разрядам (табл. 8.35). Разрядность ПГС отражается на их стоимости: цена ПГС нулевого разряда в 6-10 раз превышает цену смесей второго разряда. Серийно выпускаемым ПГС придан правовой статус государственных стандартных образцов состава. К началу 1991 г. в Государственном реестре средств измерений было зарегистрировано 477 типов государственных стандартных образцов состава газовых смесей.
Таблица 8.35
Характеристики поверочных газовых смесей
Молярная (объемная) доля компонента, % Допустимая погрешность, %
Разряд 0 Разряд 1 Разряд 2
(1-19) • 10-4 1,0-2,5 3,0-6,0 7,0-20,0
0,002-0,49 0,5-1,6 1,7-4,5 5,0-15,0
0,5-9,9 0,2-0,6 0,7-2,5 3,0-10,0
10-94 0,02-0,06 0,07-0,3 0,4—0,8
95-99 0,01-0,03 0,04—0,10 0,2-0,8
В настоящее время ПГС выпускают более десятка предприятий, расположенных в различных регионах страны. Государственный надзор за выпуском ПГС осуществляется территориальными органами Госстандарта РФ. Наиболее точные ПГС (при отсутствии в них сильнополярных и агрессивных компонентов) удается получать с помощью весового метода. Международный стандарт регламентирует три варианта метода (однократное, двойное и тройное разбавление) и три процедуры измерения массы газов (простое взвешивание, взвешивание по опорному баллону, взвешивание в вакууме). В 1983 г. в стандарт было введено дополнение, в котором детально описаны приемы работы с весами и баллонами, позволяющие подавить (или существенно ослабить) влияние источников различного рода методических (процедурных) погрешностей. Для подтверждения оценок погрешности высокоточных ПГС проводятся их периодические сличения как на национальном, так и на международном уровне. В 1975-1976 гг. в первом цикле международных сличений участвовали три организации из Японии и США. В цикле, проведенном в 1980-1983 гг., приняли участие шесть организаций из Японии, США, Англии, Голландии и Франции. Использовали 30 образцов смесей CO2/N2, CO/N2, C3I1s/N2, С3Н8/воздух, NO/N2, O2/N2 с молярной долей опре
942
Новый справочник химика и технолога
деляемого компонента от 0,0004 до 2%. Сопоставляли данные, полученные при приготовлении смесей весовым методом, и результаты анализа смесей. В 90 % случаев относительные расхождения не превышали 1 %, а в 70 % случаев — 0,5 %.
8.10.6. Генераторы газовых смесей
Под генераторами газовых смесей понимается широкий класс устройств, позволяющих приготавливать газовую смесь с заданным содержанием определяемого компонента и выдавать ее в виде потока (струи). Генераторы незаменимы при создании смесей, включающих агрессивные и легкоконденсируемые газы, а также парогазовых смесей.
Применительно к генераторам газовых смесей полезна следующая классификация:
• по области применения — общепромышленные, лабораторные и специальные;
• по назначению — универсальные, индивидуальные (конкретного назначения) и комбинированные;
• по числу компонентов в смеси — генераторы бинарных и многокомпонентных смесей;
• по исполнению — стационарные, возимые, переносные;
• по конструкции — генераторы с встроенным источником газа, без встроенного источника газа;
• по числу ступеней разбавления — одноступенчатые, многоступенчатые;
• по способу задания (регулирования) значения содержания компонента в смеси — генераторы с непрерывным задатчиком, с дискретным задатчиком;
• по принципу дозирования (методу приготовления смеси) — механические (потоковые), инжекционные, диффузионные, с проницаемыми трубками, электролитические, химические и др.;
• по виду дозирующего органа — капиллярные, крановые, шприцевые, мембранные, диафрагменные, поршневые и т. п.
Существенным шагом в унифицировании способов описания метрологических свойств генераторов и нормирования их характеристик стал комплекс стандартов, разработанных ИСО:
ИСО 6145 Анализ газов. Приготовление поверочных газовых смесей. Динамические объемные методы
ИСО 6145/1-86 Методы калибровки
ИСО 6145/2- Объемные насосы (в стадии разработки)
ИСО 6145/3-86 Периодическая инжекция
ИСО 6145/4-86 Непрерывная инжекция
ИСО 6145/5- Капиллярные устройства (в стадии разработки)
ИСО 6145/6-86 Отверстия со звуковым потоком
ИСО 6145/7- Регуляторы массового расхода газов (в стадии разработки)
ИСО 6145/8- Диффузия (в стадии разработки)
ИСО 6147-79 Анализ газов. Приготовление поверочных газовых смесей. Метод насыщения
ИСО 6349-79 Анализ газов. Приготовление поверочных газовых смесей. Метод проникновения
ИСО 7395-84 Анализ газов. Приготовление поверочных газовых смесей. Метод динамиче-ского взвешивания
Стандарты распространяются на те методы приготовления смесей, которые лежат в основе работы наиболее широко используемых генераторов. Для каждого из методов указаны: область применения; варианты аппаратурного оформления; основные уравнения, описывающие процесс приготовления смеси; источники и способы оценки погрешности содержания определяемого компонента в смеси.
В ИСО 6145/1-86 охарактеризованы возможности различных методов:
Методы Объемная доля компонента, % Относительная погрешность, %
Объемные насосы От 1 до ~ 100 0,5
Периодическая инжекция От 10 4 до 1 1,0
Непрерывная инжекция От 10~3 до 1 5,0
Капиллярный От 10 6 до =100 1,0
Отверстия со звуковым потоком От 10"1 до ~ 100 0,5
Регуляторы массового потока От 10"1 до - 100 1,0
Диффузия От 10’5 до ~ 100 3,0
Проницаемость От 10 3 до 10”2 1,0
Способы нормирования метрологических характеристик генераторов газовых смесей разработаны не так подробно, как, например, способы нормирования характеристик автоматических промышленных газоанализаторов или ПГС. Выделены четыре наиболее распространенных способа.
1. Метрологические характеристики приведены к выходу генератора, т. е. к содержанию определяемого компонента в приготовленной смеси. Нормируется диапазон воспроизводимых значений содержания компонента в смеси, погрешность (погрешность значения содержания компонента в смеси), изменение содержания компонента в смеси за регламентированный интервал времени непрерывной работы генератора. (Другие нормируемые характеристики: допускаемое отклонение от заданного значения содержания компонента, время установления постоянного значения содержания компонента, давление и объемный расход смеси на выходе генератора, время прогрева).
Этот способ используется для генераторов конкретного назначения при условии жесткой регламентации применяемых исходных веществ.
2. Метрологические характеристики не связаны с параметрами исходных веществ. Нормируется диапазон или номинальные значения коэффициентов разбавления (К)
Газовый анализ
943
или отношения расходов (Л), погрешность (погрешность реализуемых значений К или R), характеристики нестабильности и т. д. Данный способ применяется для генераторов (смесителей) универсального назначения. При его использовании погрешность содержания компонента в смеси находят как сумму погрешности генератора и погрешности, с которой установлено содержание определяемого компонента в исходных веществах.
Нормируются метрологические характеристики отдельных элементов генератора универсального назначения. (Способ реализован применительно к серийно выпускаемой установке «Микрогаз».) Установка состоит из термостата, в котором размещена сменяемая ампула со сжиженным газом 1, и устройство для подачи газа-разбавителя 2. Состав смеси, образующейся в результате проникновения газа 1 через стенку ампулы в поток газа 2, регулируется путем изменения температуры и расхода газа 2. Содержание определяемого компонента в смеси рассчитывают на основе информации об исходных газах, объемном расходе газа 2 и скорости диффузии газа 1 из ампулы при заданном значении температуры. Комплекс НМХ включает: диапазон рабочих температур, систематическую и случайную составляющие погрешности установления температуры, погрешность поддержания постоянного значения температуры, диапазон устанавливаемых расходов газа 2, случайную погрешность установления расхода, изменение расхода в течение 8 ч и т. д. Погрешность содержания компонента в смеси устанавливают путем суммирования составляющих, связанных с ампулой, газом-разбавителем, термостатом и газоподающим устройством.
4. Нормируются характеристики, относящиеся как к генератору в целом, так и к его отдельным элементам. Подобный способ реализуется для генераторов конкретного назначения в тех случаях, когда предпочтительна их поэлементная (независимая) поверка в процессе эксплуатации.
В стандарте ИСО 6145/1-86 выделены две группы методов оценивания (контроля) метрологических характеристик генераторов (в стандарте использован термин «метод калибровки»). Одна группа включает методы, опирающиеся на результаты прецизионных измерений расходов исходных газов. Подобные измерения могут осуществляться с помощью мыльнопленочных или поршневых расходомеров, газовых счетчиков, колокольных газометров. Применяются также методы, основанные на определении массы газа, прошедшего через калибруемый расходомер в течение заданного интервала времени, или на определении массы вытесненной воды. Другая группа включает методы, опирающиеся на результаты анализа смеси, проводимого непосредственно после ее приготовления (при этом погрешность результата анализа должна быть меньше погрешности генератора). Одна из возможностей связана со сравнением смеси, приготовленной с помощью исследуемого генератора, с высокоточной ПГС, отбираемой из баллона, или со смесью, приготовленной с помощью другого, более точного генератора.
8.10.7. Использование стандартных образцов чистых газов и газовых смесей для построения градуировочных характеристик анализаторов
Одна из основных метрологических задач в газовом анализе состоит в построении градуировочной характеристики аналитического устройства, позволяющей находить искомое содержание компонента с незначительной (или допустимой) систематической составляющей погрешности. Наиболее прямое и надежное решение задачи состоит в использовании адекватных стандартных образцов (АСО), т.е. образцов, обеспечивающих интенсивность аналитического сигнала, равную ее величине и в анализируемом образце при условии равенства содержаний определяемого компонента в них в заданном диапазоне и одинаковых условиях измерений. Очевидно, идеальные АСО — это образцы, идентичные по составу анализируемому газу. Однако применительно к анализу неорганических газов, особенно сложных газовых смесей неизвестного и неконтролируемо изменяющегося состава, а также газов высокой чистоты, такой путь по ряду обстоятельств либо затруднителен, либо вовсе невозможен. Не меньшую, а во многих случаях — главную роль играют стандартные образцы, неадекватные относительно величины аналитического сигнала (НСО). Именно с их применением в большинстве случаев связывается развитие современных методов анализа неорганических газов. В целом система стандартных образцов и способов их использования в анализе неорганических газов показана на рис. 8.4.
Как видно из рис. 8.4, в целом система представляется двумя основными группами стандартных образцов: адекватными и неадекватными. Безусловно, такое деление стандартных образцов на две группы не очень строгое, поскольку могут быть случаи, когда один и тот же стандартный образец может быть адекватным в одном методе и неадекватным в другом. Так, стандартный образец чистого газа может быть адекватным в газоаналитическом методе, базирующемся на использовании спонтанных спектров комбинационного рассеяния, но неадекватным в любом методе, использующем газоразрядные источники света.
Что касается АСО, то удовлетворить условию адекватности в методах анализа газов могут, прежде всего, стандартные образцы, идентичные по составу анализируемым газам, причем должны быть удовлетворены и весьма жесткие требования к одинаковости и стабильности экспериментальных условий при построении градуировочной характеристики и выполнении анализа. Кроме того, возникают дополнительные затруднения при приготовлении таких образцов, поскольку в этом случае требуется априорная информация о составе анализируемого газа, в частности о составе неопределяемых компонентов. Указанные обстоятельства свидетельствуют о трудностях (в ряде случаев непреодолимых), стоящих на пути развития газоаналитических методов, метрологическое обеспечение которых базируется на использовании идентичных АСО.
Проще обстоит дело лишь в тех методах, в которых для построения градуировочных характеристик возможно ис-
944
Новый справочник химика и технолога
Рис. 8.4. Основные типы стандартных образцов неорганических газов и способы их использования
пользование АСО, неидентичных по составу анализируемым газам, в частности стандартных образцов чистых газов. Такой подход допустим, например, в методе, использующем спектры комбинационного рассеяния света, в котором градуировочная характеристика строится по образцам чистого газа при различных парциальных давлениях.
Если же необходимо найти относительное содержание двух компонентов в анализируемом газе и для них известны сечения рассеяния, то в этом случае вообще отпадает необходимость в использовании каких-либо стандартных образцов, и относительное содержание находят расчетным путем на основе фундаментального соотношения, согласно которому отношение интенсивностей рассеяния с точностью до отношения сечений рассеяния равно отношению содержаний этих компонентов в анализируемом газе. По-видимому, это пока единственный случай, когда решение газоаналитической задачи оптическим спектральным методом не требует применения стандартных образцов газов.
Что касается НСО, то их применение в анализе неорганических газов возможно в нескольких вариантах. Так, в практике оптического спектрального анализа сложных газовых смесей используются в качестве НСО простейшие бинарные смеси, состоящие (в пределах уровня значимости) из основы и определяемого компонента. Метрологическое обеспечение в этом случае базируется на построении с их помощью так называемых стандартных градуировочных характеристик и коррекции реально измеряемого аналитического сигнала с помощью корреляционных связей
между аналитическим и корректирующим сигналами. Такой подход реализован в методе эмиссионного анализа.
В качестве НСО используются также и чистые газы. Первый пример — это построение градуировочных характеристик с помощью доз чистого газа определяемого компонента в хроматографических методах. В этом случае в поток газа-носителя вводят различные количества чистого газа и строят градуировочную характеристику, отражающую зависимость того или иного параметра хроматографического пика от количества чистого газа в дозах. При этом предполагается идентичность условий поступления определяемого компонента в хроматографическую колонку, условий разделения и формирования аналитического сигнала в процессе аналитических измерений и при построении градуировочных характеристик. Такое предположение обычно базируется на результатах специальных исследований, проводимых на этапе разработки методик и определяющих диапазон условий, в котором это предположение оказывается состоятельным. Этот прием может быть использован и в других комбинированных методах, основанных на физико-химическом выделении определяемого компонента.
Второй пример — применение классического метода добавок, в котором дозируемая добавка представлена чистым определяемым компонентом с природным изотопным составом. В этом методе процедура построения градуировочной характеристики сводится к введению в отдельные порции анализируемой пробы различных количеств добавки и к построению градуировочного графика, по которому оценивают содержание определяемого компонента. При
Газовый анализ
945
менение чистых газов в этом случае позволяет получать адекватные градуировочные характеристики при соблюдении определенных требований к условиям введения в анализируемую газовую смесь добавок чистого газа. Эти требования заключаются в том, что желательно, а иногда и необходимо, вводить в анализируемый газ добавку в той же молекулярной форме, в которой в анализируемом газе находится определяемый компонент, а также в том, что чистый газ-добавка должен дозироваться в таких количествах, при которых не менялись бы существенно экспериментальные условия (например, параметры газового разряда), формирующие аналитический сигнал. В используемом чистом газе должно быть известно содержание основного компонента, причем это содержание, как правило, может составлять 98-99 мол.%.
Разновидностью метода добавок является метод изотопных добавок, который, наряду с определенной общностью с рассмотренным выше вариантом метода добавок, принципиально отличается от него в том плане, что введение дозированных количеств редкого изотопа определяемого компонента в анализируемый газ обеспечивает одновременно и меру количества для расчета содержания этого компонента в пробе, и наилучший внутренний стандарт для контроля условий эксперимента. Поэтому в этом методе для нахождения содержания определяемого компонента необходимо введение в анализируемый газ лишь одной добавки. Это выгодно отличает метод изотопных добавок. Используемый при этом чистый газ с отличным от природного изотопным составом должен быть аттестован как по содержанию основного компонента, так и по относительному содержанию его изотопов. При этом требования как к самой чистоте газа, так и к точности его аттестации невелики. Молярная доля основного компонента может варьироваться от 90 до 100 мол.%, а допустимая относительная погрешность определения этого содержания составляет 1-2%. В то же время желательно иметь максимальную степень обогащения газа-добавки редким изотопом от 50 до 99 мол. %, так как в этом случае обеспечивается минимальный уровень определяемых содержаний. Ограничение применения метода изотопных добавок в анализе газов состоит в том, что он без особых трудностей может быть реализован лишь в высокоселективных методах: оптическом спектральном и масс-спектральном.
Резюмируя обсуждение возможностей использования НСО для метрологического обеспечения анализа неорганических газов, следует особо подчеркнуть, что в настоящее время приготовление, аттестация, хранение, транспортировка и применение таких стандартных образцов не составляет особой проблемы, в связи с чем их использование представляется одним из наиболее перспективных направлений дальнейшего развития методов анализа неорганических газов. Это особенно важно в тех случаях, когда отсутствие АСО становится фактором, сдерживающим развитие метода в целом. Главным условием применения НСО является возможность обеспечения требований к точности градуировочной характеристики, и это должно быть во всех случаях надежно установлено. Составляющая погрешности градуировки, обусловленная НСО, оценивается обычно
на этапе разработки методики. Найденные оценки служат основой для доказательства «адекватности» градуировочной характеристики (применимости НСО). На практике используются различные способы и приемы доказательства. К ним можно отнести, во-первых, сопоставление результатов определений при изменении условий анализа или состава и количества неопределяемых примесей, во-вторых, одновременное использование нескольких независимых методов анализа и, в-третьих, применение анализа синтетических смесей по схеме «введено—найдено». Чем шире круг привлекаемых для этой цели способов и приемов, тем надежнее устанавливается адекватность градуировочной характеристики и формируются оптимальные условия проведения анализов.
8.10.8. Государственная поверочная схема для средств измерений содержания компонентов в газовых средах
Поверочной схемой называют утвержденный в установленном порядке документ, устанавливающий средства, методы и точность передачи размера единицы от эталона или установки высшей точности рабочим средствам измерений. Применительно к средствам газоаналитических измерений поверочная схема разработана в виде методики НПО ВНИИМ им. Д. И. Менделеева. Схема представляет собой общее описание системы воспроизведения и передачи размеров единиц физических величин, характеризующих химический состав газовых сред. С помощью схемы решаются задачи классификации существующих и создаваемых средств измерений, выбора рациональных форм их метрологического обслуживания, регламентации прав и обязанностей организаций, специализирующихся в области газоаналитических измерений. Государственная поверочная схема охватывает множество метрологических цепей (вариантов соподчинения средств измерений), соответствующих определенным газоаналитическим задачам и точности рабочих средств измерений (задача характеризуется компонентным составом анализируемой газовой среды, измеряемой физической величиной и диапазоном ее значений). Метрологические цепи могут состоять из различного числа звеньев, иметь различную структуру. Элементами государственной поверочной схемы (звеньями метрологических цепей) являются:
• рабочие средства измерений высокой, средней и низкой точности (РСИ-1, РСИ-2, РСИ-3);
• стандартные образцы состава чистых газов 1-го и 2-го разрядов;
• стандартные образцы состава газовых смесей 0-го, 1-го и 2-го разрядов;
• образцовые генераторы чистых газов и газовых смесей 1-го и 2-го разрядов;
• образцовые газоаналитические и газосмесительные установки 1-го и 2-го разрядов (в том числе установки высшей точности УВТ-1 и УВТ-2);
• рабочие эталоны (РЭ) -— комплексы смесительной и аналитической аппаратуры для приготовления стандартных образцов газовых смесей 0-го разряда;
946
Новый справочник химика и технолога
• эталоны сравнения (чистые газы и газовые смеси в баллонах);
• государственный первичный эталон (ГПЭ) единицы молярной доли компонентов (комплекс смесительной и аналитической аппаратуры).
Единицу физической величины (например, молярной доли /-компонента) для конкретной газоаналитической задачи воспроизводят путем косвенного измерения данной величины в интервале значений, характерных для этой задачи. При измерении используют уравнения, связывающие измеряемую величину с другими физическими величинами, размер которых установлен независимым образом. К последним могут относиться:
• масса, вместимость, давление, объемный расход газа, сила и другие физические величины, размер которых заимствуется из соответствующих государственных поверочных схем;
• физические константы;
• содержание компонента, воспроизводимое средством измерений, не относящимся к данной поверочной схеме (например, государственным стандартным образцом состава жидкого или твердого вещества);
• содержание компонента, воспроизводимое средством измерений, относящимся к другим газоаналитическим задачам (например, молярная доля /-компонента, воспроизводимая стандартным образцом, неадекватным анализируемому газу).
Наряду со средствами измерений конкретного назначения элементами схемы являются анализаторы (генераторы) универсального назначения, используемые на основе методик выполнения измерений (МВИ). В этом случае передача размера единицы осуществляется при градуировке анализатора (генератора) или при контроле точности измерений, выполняемых по МВИ. Воспроизведение единиц содержания компонентов для различных газоаналитических задач осуществляют с помощью средств измерений, находящихся на различных ступенях схемы.
В зависимости от ступени, на которой воспроизводят единицу, газоаналитические задачи подразделяют на четыре группы: А, Б, В и Г. При выборе ступени (классифицировании газоаналитических задач) исходят из технической и экономической целесообразности централизации воспроизведения единицы, степени приоритетности задачи и распространенности средств измерений, предназначенных для ее решения. Предполагается, что в каждом конкретном случае существует оптимальный уровень централизации воспроизведения единицы, позволяющий с наименьшими затратами обеспечить требуемую точность рабочих средств измерений.
С помощью ГПЭ централизованно воспроизводят единицу для задач группы А. В эту группу входят газоаналитические задачи общегосударственного значения; требования к точности измерений в этих задачах, как правило, установлены государственными стандартами или международными соглашениями. Для решения задач, подлежащих государственной поверке (при выпуске и в процессе эксплуатации), используются все возможные и необходимые
средства. Для поверки применяются разнообразные образцовые средства измерений.
С помощью рабочих эталонов воспроизводят единицу для задач группы Б. В эту группу входят газоаналитические задачи межотраслевого характера, связанные с контролем состава распространенных в промышленности газовых сред.
С помощью образцовых средств измерений 1-го и 2-го разрядов воспроизводят единицы для задач групп В и Г. Это задачи ограниченного распространения; решаемые преимущественно в одной отрасли с помощью газоанализаторов низкой и средней точности.
При необходимости централизованного воспроизведения единицы используемым метрологическим средствам придают статус установок высшей точности: УВТ-1 и УВТ-2. Таким образом, при переходе от ГПЭ к рабочим эталонам и далее к образцовым средствам измерения возрастает как число объектов метрологического обслуживания, так и число охватываемых газоаналитических задач. Лишь ограниченное число метрологических цепей имеют своим верхним звеном ГПЭ, большинство из них возглавляется образцовыми средствами измерений. На эталонном уровне воспроизводят единицу молярной доли компонентов, на других уровнях — единицы любых физических величин, характеризующих состав газовых сред. При передаче размера единицы допускается проводить пересчеты значений разных физических величин, характеризующих состав одной и той же газовой среды (например, значения молярной доли оксида углерода в воздухе в значения массовой концентрации оксида углерода при температуре 20 °C и давлении 101,3 кПа).
Регламентируемый схемой порядок передачи размера единицы может быть реализован при испытаниях, аттестации, поверке анализатора либо непосредственно при выполнении измерений. При этом не исключается возможность сочетания различных методов передачи размера единиц: при аттестации может быть использован один метод (например, основанный на применении адекватных стандартных образцов), а при периодической поверке другой (основанный на использовании образцовых средств измерений, заимствованных из других поверочных схем или из других метрологических цепей данной поверочной схемы).
Регламентированы соотношения погрешностей соподчиненных средств измерений: образцовых средств измерений 0-го и 1-го разряда — не более 1 :2, образцовых средств измерений 1-го и 2-го разряда — не более 1 :2, образцовых и рабочих средств измерений — не более 1 : 2,5. При применении поверочной схемы наряду с указанной информацией используют сведения о характеристиках эталонов и образцовых средств измерений, содержащиеся в информационных материалах Госстандарта РФ. Государственный первичный эталон воспроизводит единицы молярной доли компонентов. ГПЭ состоит из четырех комплексов аналитической аппаратуры (хроматографической, электрохимической, оптико-акустической, спектральной) и комплекса газосмесительной аппаратуры (объемной, манометрической, весовой, динамической).
Газовый анализ
947
Использование аппаратуры, реализующей принципиально различные методы, позволяет с большой степенью достоверности выявлять и контролировать источники систематических погрешностей при анализе и синтезе газовых смесей. Применяемые методические решения позволяют обеспечить наивысшую в стране точность измерений.
Получаемая на ГПЭ измерительная информация передается эталонам сравнения, используемым затем для сличений рабочих эталонов или для градуировки отдельных образцовых газоаналитических установок 1-го разряда. Кроме того, эта информация используется при установлении погрешности генераторов чистых газов и газовых смесей (в процессе их испытаний или аттестации).
Структура ГПЭ обеспечивает возможность достаточно оперативного расширения его функциональных возможностей в случае появления новых объектов метрологического обслуживания. ГПЭ эксплуатируется в НПО «ВНИИМ им. Д.И. Менделеева» (Санкт-Петербург). Иерархические системы средств измерений содержания компонентов в газовых средах функционируют и в ряде других стран. С 1974 г. в Японии действует национальная «система соподчинения стандартных газов», ориентированная главным образом на задачи, связанные с контролем состава выхлопов автотранспорта и контролем чистоты воздуха. В США соподчиненные первичные и вторичные стандартные образцы состава газовых смесей являются элементами общей системы обеспечения единства химико-аналитических измерений, включающей также эталонные, образцовые и рабочие методы. Номенклатура первичных стандартных образцов газовых смесей, выпускаемых национальным Бюро стандартов, близка к номенклатуре эталонов сравнения, аттестуемых на ГПЭ (см. табл. 8.36).
Литература
1. Агафонов И.Л., Девятых Г.Г. Масс-спектрометрический анализ газов и паров высокой чистоты. М.: Наука, 1980. 334 с.
2. Альперин В.З., Конник З.И., Кузьмин А.А. Современные электрохимические методы и аппаратура для анализа газов в жидкостях и газовых смесях. М.: Химия, 1975. 184 с.
3. Аманназаров А., Розинов Г.Л., Чубукова Н.М. Методы и приборы для определения водорода. М.: Химия, 1987. 126 с.
4. Аманназаров А., Шарнопольский А.И. Методы и приборы для определения кислорода. М.: Химия, 1988. 147 с.
5. Байбаков Ф.Б., Шарапов В.М. Контроль примесей в сжатых газах. М.: Химия, 1989. 158 с.
6. Бочкова О.П., Шрейдер Е.Я. Спектральный анализ газовых смесей. М.: Физматгиз, 1963. 308 с.
7. Бреслер П.И. Оптические абсорбционные газоанализаторы и их применения. Л.: Энергия, 1986. 64 с.
8. Горелик Д.О., Конопелько Л.А. Мониторинг загрязнения атмосферы и источников выбросов. М.: Изд-во стандартов, 1992. 433 с.
9. Горелик Д.О., Конопелько Л.А., Панков Э.Д. Экологический мониторинг. Оптикоэлектронные приборы и системы. СПб., 1998. Т. 1. 734 с.; Т. 2. 583 с.
10. Гроздовский М.К. Анализ воздуха в промышленных предприятиях. М.: Гос. социально-экономическое изд-во, 1931.288 с.
11. Контроль химических и биологических параметров окружающей среды. И Энциклопедия «Экометрия» / Под ред. проф. Л.К. Исаева. СПб., 1998. 851 с.
12. Лазеева Г.С., Петров А.А., Сирота Л.Б. Спектральноизотопный метод в агрохимии и биологии. СПб.: Изд-во СПбГУ, 1999. 446 с.
13. Лейте В. Определение загрязнений воздуха в атмосфере и на рабочем месте. Л.: Химия, 1980. 344 с.
14. Лидов А.П. Анализ газов. Л.: Научное химикотехнологическое изд-во, 1928. 331 с.
15. Методы анализа неорганических газов / Под ред. проф. В.М. Немца. СПб.: Химия, 1993. 560 с.
16. Муравьева С.И., Кознина Н.И., Прохорова Е.К. Справочник по контролю вредных веществ в воздухе. М.: Химия, 1988. 320 с.
17. Немец В.М.: Петров А.А., Соловьев А.А. Спектральный анализ неорганических газов. Л.: Химия, 1988. 348 с.
18. Перегуд Е.А., Горелик Д.О. Инструментальные методы контроля загрязнения атмосферы. Л.: Химия, 1981. 384 с.
19. Петров А.А. Спектрально-изотопный метод исследования материалов. Л.: ЛГУ, 1974. 328 с.
20. Петров А.Д. Газовый анализ. Анализ и синтез отравляющих веществ. Л.: Госхимтехиздат, ЛО, 1933. 97 с.
21. Симонов В.А., Нехорошева Е.В., Заворовская Н.А. Анализ воздушной среды при переработке полимерных материалов. М.: Химия, 1988. 224 с.
22. Францен Г. Газовый анализ. Пер. с нем. С.П. Гвоздева. М.; Пг.: Изд-во советской нефтяной пром-ти, 1923. 96 с.
23. Шидловский Ф.И. Опыт применения диффузии газов через пористые тела к определению влаги и углекислоты в окружающей среде. Докт. дис. С.-Петербург, 1886. 47 с.
24. Travers M.W. The experimental study of gases. London: Macmillan, 1901. 95 p.
СОДЕРЖАНИЕ
Предисловие......................................3
Раздел 1. Информационные базы и общие вопросы аналитической химии
1.1. Основные печатные и электронные справочные источники по аналитической химии.................7
1.1.1. Рекомендации Международного союза теоретической и прикладной химии (ИЮПАК) по вопросам номенклатуры аналитической химии и их перевод в России......................7
1.1.2. Журналы по аналитической химии...........11
1.1.3. Справочники и руководства общего характера ....12
1.1.4. Серии монографий и обзорная информация по аналитической химии...........................13
1.1.5. Основная учебная литература по аналитической химии...........................21
1.1.6. Интернет: обзор основных сайтов по аналитической химии...........................22
1.1.7. Compendium of Analytical Nomenclature (см. компакт-диск)...............................32
1.1.8. Русско-анлийский и англо-русский словари терминов по аналитической химии
(рекомендации ИЮПАК) (см. компакт-диск)...32
1.2. Структура и методы аналитической
химии (АХ) [1-3].........................33
1.3. Общая схема количественного анализа......35
1.4. Выбор метода анализа [1 ]................36
1.5. Отбор и подготовка проб к анализу
(способы вскрытия) [1-23] (использованы материалы И.Ф. Долмановой [1])...........39
1.5.1. Отбор пробы..............................39
1.5.2. Потери и загрязнения при отборе пробы.
Хранение пробы...........................43
1.5.3. Подготовка пробы к анализу...............44
Раздел 2. Метрологические основы методов количественного анализа
2.1. Количество вещества, эквивалентность, способы
выражения концентраций........................53
2.2. Аналитический сигнал. Методы измерения [4] ....59
2.3. Погрешности количественного химического
анализа. Представление результатов химического анализа...........................61
2.3.1. Общие термины [1, 15-21, 32, 33, 35]..........62
2.3.2. Классификация погрешностей....................63
2.3.3. Систематические погрешности. Способы
их выявления [1,4, 22-24, 32, 36, 38, 47, 48].64
2.3.4. Статистическая обработка результатов прямых
равноточных наблюдений (определений)..........66
2.3.4.1. Термины, относящиеся к параллельным
измерениям. Оценка воспроизводимости..........67
2.3.4.2. Оценка правильности результата измерения (анализа).......................................68
2.3.5. Оценка грубых погрешностей (промахов).........69
2.3.6. Сравнение двух средних [4, 22]...........70
2.4. Расчет уравнения линейного градуировочного графика, его метрологических характеристик и метрологических характеристик результата анализа [4, 22, 23, 25, 56]....................71
2.5. Термины по представлению результатов химического анализа
(в алфавитном порядке) [15]..............75
2.6. Предел обнаружения. Диапазон определяемых содержаний. Нижняя граница определяемых содержаний......................................76
2.7. Значащие цифры и правила округления. Точные и приближенные вычисления................89
Раздел 3. Методы разделения и концентрирования
3.1. Методы разделения, основанные на образовании
выделяемым веществом новой фазы, в зависимости от агрегатного состояния
исходной смеси и выделяемых веществ.......95
3.1.1. Осаждение.................................95
3.1.2. Отгонка из раствора......................103
3.1.3. Отгонка в парах реагента (ОПР)...........104
3.1.4. Упаривание, дистилляция, ректификация....107
3.2. Методы разделения, основанные на различиях
в распределении веществ между фазами....108
3.2.1. Общие характеристики методов, основанных
на различиях в межфазном распределении...109
3.2.2. Сорбционные методы.......................110
3.2.2.1. Молекулярная адсорбция..................НО
3.2.2.2. Ионный обмен...........................113
3.2.2.3. Комплексообразующая сорбция............133
3.2.2.4. Твердофазная экстракция................138
3.2.3. Динамическая сорбция.....................138
3.2.4. Кристаллизационные методы концентрирования................................139
3.2.4.1. Соосаждение............................139
3.2.4.2. Зонная плавка и направленная кристаллизация. 155
3.2.5. Жидкостная экстракция....................156
3.2.5.1. Экстракция по механизму физического распределения...................................156
3.2.5.2. Реакционная экстракция в органические экстрагенты.....................................157
3.2.5.3. Реакционная экстракция в расплаве металлов (пробирная плавка)..............................168
3.2.6. Сверхкритическая флюидная экстракция (СКФЭ)...............................169
3.2.7. Газовая экстракция.......................170
3.2.8. Жидкостная абсорбция.....................174
3.2.9. Хроматография и хроматографические методы
разделения веществ (см. также раздел 4)..176
3.2.9.1. Классификация хроматографических методов разделения веществ. Основные термины............176
Содержание
949
3.2.9.2. Теоретические основы хроматографии....181
3.2.9.3. Практические следствия теоретических
представлений о хроматографическом процессе.... 184
3.2.9.4. Хроматографические методы, являющиеся вариациями хроматографического способа осуществления процесса межфазного
распределения...........................187
3.2.9.5. Варианты хроматографических методов разделения в зависимости от агрегатного состояния фаз и механизма удерживания разделяемых веществ стационарной фазой........191
3.2.9.6. Хроматографические методы анализа....214
3.3. Мембранные методы разделения веществ...214
3.3.1. Внутригрупповая классификация методов..214
3.3.2. Диффузионные мембранные методы.........215
3.3.3. Электромембранные методы...............218
3.3.4. Баромембранные методы (мембранная
фильтрация)............................219
3.3.4.1. Мембранная фильтрация жидких сред....220
3.3.4.2. Мембранная фильтрация воздуха........226
3.4. Методы внутрифазного разделения........241
3.4.1. Электрофорез в растворе................242
3.4.2. Проточное фракционирование в поперечном
поле (ППФ-методы)......................244
3.5. Комбинированные и прочие методы
разделения.............................246
3.5.1. Фотохимические методы..................246
3.5.2. Хроматомембранный массообменный процесс и
хроматомембранные методы...............247
Раздел 4. Аналитическая хроматография и капиллярный электрофорез
4.1. Аналитическая хроматография..................254
4.1.1. Газовая хроматография........................254
4.1.1.1. Параметры удерживания в газовой хроматографии..................................254
4.1.1.2. Параметры хроматографического пика.........255
4.1.1.3. Основные виды размывания полос в колонке...255
4.1.1.4. Основные уравнения в газовой хроматографии.257
4.1.1.5. Влияние экспериментальных параметров на хроматографическое разделение
(на величины a, R и N).......................258
4.1.1.6. Аппаратура для газовой хроматографии.......259
4.1.2. Газо-жидкостная хроматография................270
4.1.2.1. Преимущества и недостатки газо-жидкостной хроматографии..................................270
4.1.2.2. Твердые носители...........................275
4.1.2.3. Процесс приготовления сорбента.............277
4.1.3. Газоадсорбционная хроматография..............279
4.1.3.1. Особенности газоадсорбционной хроматографии..................................279
4.1.3.2. Классификация адсорбентов по химической природе поверхности............................280
4.1.3.3. Классификация адсорбентов по геометрической структуре....................280
4.1.3.4. Методы модифицирования адсорбентов в газовой хроматографии........................281
4.1.3.5. Основные типы адсорбентов....................281
4.1.4. Капиллярная газовая хроматография..............284
4.1.4.1. Особенности капиллярной хроматографии....284
4.1.5. Основные рекомендации по выбору сорбентов и условий хроматографического разделения смесей конкретных веществ........................286
4.1.6. Качественный анализ в газовой хроматографии ..288
4.1.7. Основные методы количественного анализа в газовой хроматографии..........................290
4.1.8. Высокоэффективная жидкостная хроматография (ВЭЖХ).............................300
4.1.8.1. Основные особенности метода и аппаратуры.300
4.1.8.2. Классификация хроматографических систем: сорбент - элюент - сорбат по типам межмолекулярных взаимодействий...................301
4.1.8.3. Общие требования к элюентам в ВЭЖХ...........306
4.1.8.4. Адсорбенты для ВЭЖХ..........................308
4.1.8.5. Динамическое модифицирование адсорбентов.... 314
4.1.8.6. Общая схема жидкостного хроматографа.........315
4.1.9. Ионная хроматография...........................326
4.1.10. Метод тонкослойной хроматографии (ТСХ)........338
4.1.11. Особенности хроматографии с подвижной фазой в сверхкритическом состоянии.....................340
4.2. Капиллярный электрофорез.......................343
4.2.1. Основы метода КЭ...............................343
4.2.2. Способы ввода пробы. Зона пробы в капилляре .. 345
4.2.3. Ведущий электролит.............................346
4.2.3.1. Добавки к ведущему электролиту...............346
4.2.4. Основные варианты капиллярного электрофореза....................................349
4.2.4.1. Капиллярный зонный электрофорез (КЗЭ)........350
4.2.4.2. Мицеллярная электрокинетическая хроматография (МЭКХ)...........................................350
4.2.5. Детектирование в КЭ............................351
4.2.5.1. Зарубежные коммерческие системы капиллярного электрофореза.......................353
4.2.5.2. Отечественные системы капиллярного электрофореза....................................356
4.2.6. Характеристики удерживания.....................356
4.2.7. Количественный и качественный анализ в КЭ......358
4.2.8. Эффективность, разрешение и селективность метода КЭ........................................358
4.2.9. Чувствительность метода КЭ.....................359
4.2.9.1. Селективность................................362
4.2.10. Объекты для анализа методом капиллярного электрофореза....................................364
4.2.11. Подготовка пробы к анализу методом капиллярного электрофореза.......................368
Раздел 5. Химические методы количественного анализа
5.1. Гравиметрические методы..................389
5.1.1. Методы гравиметрического определения
неорганических ионов.....................427
5.1.2. Важные для гравиметрии свойства
отдельных элементов......................540
5.2. Титриметрический анализ (ТА).............573
5.2.1. Общие положения..........................574
950
Содержание
5.2.1.1. Сущность ТА. Точка эквивалентности или точка стехиометричности.....................574
5.2.1.2. Кривые титрования. Конечная точка титрования (КТТ) и способы ее регистрации
[1-13, 15, 19, 29].......................577
5.2.2. Классификация титриметрических методов анализа.................................579
5.2.2.1. Классификация по способам титрования...579
5.2.2.2. Классификация титриметрических методов по типам реакций титрования.....................585
5.2.3. Посуда мерная лабораторная: бюретки, пипетки, колбы, цилиндры, мензурки [6,12, 41-53].........586
5.2.4. Способы приготовления стандартных растворов [2, 3, 6, 47, 54, 55].................589
5.2.4.1. Приготовление первичных стандартных растворов........................................589
5.2.4.2. Приготовление вторичных стандартных растворов (вторичный стандарт)...................591
5.2.4.3. Стандартизация растворов по точным навескам первичных стандартных (установочных) веществ: способ отдельных навесок, способ пипетирования...................591
5.2.4.4. Стандартизация по другому стандартному раствору.........................................592
5.2.4.5. Стандартизация растворов по стандартным образцам.........................................593
5.2.4.6. Расчет концентрации титрованных растворов с помощью поправочного коэффициента [47] ....593
5.2.5. Общие правила выполнения титриметрических определений [3, 47]..............................596
5.2.6. Практическое применение титриметрических методов анализа..................................597
5.2.6.1. Кислотно-основное титрование. Общая характеристика и возможность метода.............597
5.2.6.2. Комплексонометрическое титрование......626
5.2.6.3. Техника и применение комплексонометрического титрования......................................653
5.2.6.4. Косвенные комплексонометрические методы определения анионов неорганических кислот и катионов......................................683
5.2.6.5. Окислительно-восстановительное титрование [1 -12, 20, 21, 26, 27, 39,42, 44, 54, 56]......687
5.2.6.6. Осадительное титрование................699
Раздел 6. Электрохимические методы анализа
6.1. Химические сенсоры. Потенциометрия.......710
6.1.1. Типы сенсоров и физические принципы их функционирования.............................710
6.1.2. Интеллектуальные сенсорные системы («электронный нос» и «электронный язык»)........712
6.1.3. Основные характеристики потенциометрических сенсоров.......................................713
6.1.3.1 Уравнение Никольского..................713
6.1.3.2. Крутизна электродной функции..........713
6.1.3.3. Предел обнаружения....................713
6.1.З.4. Селективность.........................714
6.1.3.5. Рабочая область pH....................716
6.1.3.6. Время отклика............................717
6.1.4. Таблицы потенциометрических сенсоров [5-7]...................................718
6.1.5. Методы аналитического определения..........724
6.1.5.1. Прямая потенциометрия....................724
6.1.5.2. Потенциометрическое титрование...........729
6.1.5.3. Другие методы («электронный язык»).......732
6.1.6. Список фирм, изготавливающих оборудование для потенциометрического анализа.................733
6.2. Кулонометрия [1,2,3].......................737
6.3. Вольтамперометрия [ 1-5]...................740
6.3.1. Классические методы........................740
6.3.2. Хронопотенциометрия........................747
6.3.3. Потенциалы полярографических полуволн в различных фоновых электролитах.................748
6.3.3.1. Потенциалы полярографических полуволн неорганических веществ...........................748
6.3.3.2. Потенциалы полярографических полуволн органических соединений..........................761
6.4. Амперометрическое титрование...............764
6.5. Инверсионная вольтамперометрия [13-19].....773
6.5.1. Сущность метода инверсионной вольтамперометрии (ИВ) [14,15,17]................773
6.5.2. Аналитический сигнал в методе ИВ...........774
6.5.3. Инверсионная хронопотенциометрия (ИХП).....775
6.5.4. Факторы, влияющие на величину и форму аналитического сигнала...........................775
6.5.4.1. Тип индикаторного электрода..............775
6.5.4.2. Состав фонового раствора.................775
6.5.4.3. Потенциал электролиза и его длительность.775
6.5.4.4. Обратимый и необратимый электродный процессы в ИВ....................................775
6.5.4.5. Растворенные окислители..................776
6.5.4.6. Наложение пиков элементов. Образование интерметаллических соединений....................776
6.5.4.7. Поверхностно-активные вещества...........777
6.5.4.8. Катодная инверсионная вольтамперометрия. Адсорбционная ИВ [27]............................778
6.5.4.9. Техника работы в методе ИВ...............778
6.5.5. Электрохимические характеристики процесса
разряда-ионизации элементов и условия анализа некоторых материалов методом инверсионной вольтамперометрии (реактивы и вещества высокой степени чистоты, материалы металлургического производства, природные и сточные воды, жидкие и твердые продукты
питания)...............................778
6.5.6. Свойства индикаторных электродов [13-17, 28-33]...............................800
6.5.7. Физико-химические свойства материалов индикаторных электродов (табл. 6.60-6.71)....802
6.5.8. Способы изготовления и примеры использования электродов в ИВ................805
6.6. Кондуктометрия и кондуктометрическое титрование (по данным Г.В. Прохоровой) [3].... 817
Приложение 1.................................826
Приложение 2.................................832
Приложение 3.................................838
Содержание
951
Раздел 7. Масс-спектрометрический метод анализа
7.1. Источники ионов.........................842
7.1.1. Источники с электронным ударом (ЭУ).....842
7.1.2. Источники с поверхностной ионизацией
или термоионные источники (ТИ)..........847
7.1.3. Химическая ионизация (ХИ)...............847
7.1.4. Полевая ионизация (ПИ)..................849
7.1.5. Эмиссия вторичных ионов (ЭВИ)...........849
7.1.6. Искровые ионные источники (ИИИ).........850
7.1.7. Источник с тлеющим разрядом (ТР)........850
7.1.8. Источники ионов с лазерной десорбцией (ЛД) .... 851
7.1.9. Источники с экстракцией ионов электрическим
полем (ЭРИАД)...........................851
7.1.10. Источник ионов с ионизацией индуктивносвязанной плазмой (ИСП)........................852
7.2. Масс-анализаторы........................855
7.2.1. Магнитные секторные анализаторы.........855
7.2.2. Приборы с двойной фокусировкой
(тандемные).............................855
7.2.3. Времяпролетный масс-анализатор..........856
7.2.4. Квадрупольные масс-анализаторы..........856
7.2.5. Омегатронный масс-спектрометр...........857
7.2.6. Масс-анализатор на основе
ионно-циклотронного резонанса (ИЦР).....858
7.3. Детектирование ионов....................859
7.4. Анализ изотопного и элементного состава
вещества................................859
7.4.1. Идентификация органических соединений
и установление их структуры.............863
7.4.2. Определение индивидуального состава
смеси веществ...........................865
7.4.3. Метод изотопного разбавления............884
7.4.4. Хромато-масс-спектрометр (ХМС)..........885
7.4.5. Жидкостной хромато-масс-спектрометр.....886
Раздел 8. Газовый анализ
8.1. Определение газового анализа............891
8.2. Анализируемые объекты и определяемые компоненты в газовом анализе...................891
8.3. Задачи, решаемые с помощью газового анализа........................................891
8.4. Основные направления развития газового анализа........................................891
8.5. Величины, характеризующие химический состав газов...................................891
8.6. Физические и физико-химические свойства чистых газов...................................900
8.7. Методы получения и очистки газов.......904
8.7.1. Очистка водорода.......................910
8.7.2. Очистка оксида углерода................911
8.7.3. Очистка диоксида углерода..............911
8.7.4. Очистка метана.........................912
8.7.5. Очистка оксида азота...................912
8.7.6. Очистка кислорода......................912
8.7.7. Очистка азота..........................914
8.7.8. Очистка аргона.........................914
8.7.9. Очистка неона...........................915
8.7.10. Очистка гелия..........................916
8.8. Производство газовых смесей.............916
8.8.1. Основные типы ПГС и требования к ним....916
8.8.2. Условия и особенности приготовления ПГС.917
8.8.3. Способы приготовления ПГС...............917
8.8.4. Способы перемешивания компонентов
в процессе приготовления смесей.........918
8.8.5. Источники загрязнения газов и методы
его предупреждения......................918
8.8.6. Вспомогательные вещества................918
8.9. Методы газового анализа.................919
8.9.1. Химические методы.......................919
8.9.1.1. Волюмометрический и манометрический методы........................................919
8.9.1.2. Титриметрический метод................919
8.9.1.3. Термохимический метод.................920
8.9.1.4. Гравиметрический метод................920
8.9.2. Оптические эмиссионные методы...........920
8.9.2.1. Газоразрядный метод...................920
8.9.2.2. Флуоресцентный метод..................921
8.9.2.3. Хемилюминесцентный метод..............921
8.9.2.4. Метод рэлеевского рассеяния...........921
8.9.2.5. Метод комбинационного рассеяния.......922
8.9.3. Оптические абсорбционные методы.........922
8.9.3.1. Метод прямого линейного поглощения....922
8.9.3.2. Метод внутрирезонаторного лазерного поглощения....................................923
8.9.3.3. Оптико-акустический (ОА) метод........932
8.9.3.4. Термолинзовый метод...................924
8.9.3.5. Колориметрический и спектрофотометрический методы........................................924
8.9.4. Масс-спектрометрический метод...........925
8.9.5. Хроматографический метод................925
8.9.6. Электрохимические методы................926
8.9.7. Методы, основанные на зависимости интегральных свойств газов от содержания определяемого компонента......................926
8.9.7.1. Термокондуктометрический метод........926
8.9.7.2. Магнитный метод.......................927
8.9.7.3. Механический метод....................927
8.9.7.4. Акустический метод....................928
8.9.7.5. Рефрактометрический и интерферометрический методы........................................928
8.9.8. Ионизационные методы....................928
8.9.9. Радиоспектроскопический метод...........928
8.9.10. Метод изотопного разбавления...........929
8.9.11. Методы изотопного анализа газов и газообразующих элементов....................929
8.9.12. Методы определения газовых включений и газообразующих элементов в веществах конденсированной фазы.........................930
8.9.13. Методы определения дисперсной фазы в газах... 932
8.9.13.1. Методы определения примесей масла в газах.... 933
8.9.13.2. Методы измерения содержания влаги в газах.... 933
8.9.13.3. Методы измерения содержания твердой дисперсной фазы (пыли)........................934
952
Содержание
8.9.13.4. Методы определения радиоактивных веществ
в газах..................................936
8.9.14. Дистанционные методы газового анализа...936
8.10. Метрологические аспекты газового анализа.936
8.10.1. Понятие «метрологическое обеспечение
газового анализа» и его структура........936
8.10.2. Метрологический подход в газовом анализе,
его основные направления и формы.........937
8.10.3. Метрологические характеристики
анализаторов газов.......................939
8.10.4. Чистые газы как средства обеспечения единства газоаналитических измерений....................940
8.10.5. Поверочные газовые смеси...............941
8.10.6. Генераторы газовых смесей..............942
8.10.7. Использование стандартных образцов чистых газов и газовых смесей для построения градуировочных характеристик анализаторов.......943
8.10.8. Государственная поверочная схема для средств измерений содержания компонентов в Газовых средах.........................................945
Авторский коллектив
Барбалат Юрий Александрович Власов Юрий Георгиевич Демин Владимир Александрович Ермоленко Юрий Евгеньевич Золотов Юрий Александрович Иванов Вадим Михайлович Калинкин Игорь Петрович Карцова Анна Алексеевна Колодников Василий Викторович Мосичев Валентин Иванович Москвин Леонид Николаевич Немец Валерий Васильевич Родинков Олег Васильевич Сайдов Геннадий Викторович Свердлова Ольга Владимировна Соловьев Анатолий Анатольевич Теплых Виссарион Федорович Халонин Александр Сергеевич Якимова Нина Михайловна Яшин Александр Яковлевич Яшин Яков Иванович
аыммд.
4 (0444'14' СПРАВОЧНИК ХИМИКА И ТЕХНОЛОГА
Аналитическая химия
Часть I
Научное издание
Издание подготовлено и отпечатано в АНО НПО «Мир и Семья». 191023, Санкт-Петербург, ул. Садовая, 28-30, корп. 35. Тел.: (факс) 321-67-38; 110-59-91; 110-57-93; 989-03-28. Лицензия ИД № 03298 от 20 ноября 2000 г. Лицензия ПД № 2-69-574 от 14 ноября 2000 г.
mis@npomis.com http ://www.npomis. com
Ответственный за издание: А.А. Полуда Ответственный за выпуск: Н.В. Емельянова Ответственный за подготовку: Л.В. Белканова Научный редактор: В.А. Столярова Редактор-корректор: О.Б. Маршкова, Е.А. Монахова Компьютерная верстка: Т.А. Балбуцкая, Т.А. Бойченко, Н.Н. Грибещенко, Н.В. Коробова Техническое сопровождение: Т.Н. Жадобина
Сдано в набор 16.04.2002. Подписано к печати 12.12.2002. Формат 60x90/8. Бумага офсетная, плотность 80 г/м2 Объем 120,5 п. л. Тираж 1000 экз.
Посвящается 300-летию Санкт-Петербурга — Колыбели отечественной химической науки, и промышленности
|Мир и Семья, Профессионал, СПб
но^вы'й: СПРАВОЧНИК ХИМИКА И ТЕХНОЛОГА
Аналитическая химия
Часть I
НПО "Мир и Семья" Санкт-Петербург 2002
II
III
IV
VI
VII
VIII
8
10
1 H
1,00794(7)
ВОДОРОД
ydro;
1s1
jenium
3 Li
6,941(2) He2s1
ЛИТИЙ 2
lithium
4 Be
9,012182(3)
He 2s2
БЕРИЛЛИЙ
2
2 beryllium
5
6
7
8
9
10,811(7) He 2s22p1
БОР
2 borum
12,0107(8) He 2s22p2
УГЛЕРОД
2 carboneum
14,00674(7) He 2s22p3
АЗОТ
5
2 nitrogenium
15,9994(3) He 2s22p4
КИСЛОРОД
2 oxygenium
18,9984032(5)
He 2s22p5
ФТОР
2 ftorum
nNa
22,989770(2)
Ne 3s1
НАТРИЙ
________natrium
8
12Mg
24,3050(6)" ’
Ne 3s2
МАГНИЙ
19K
39,0983(1) Ar 4s1
КАЛИЙ
8
8
2 kalium
29 Cu
63,546(3) Ar 3d104s1
МЕДЬ
18 8
2
cuprum
37 Rb
85,4678(3) Kr 5s1
РУБИДИЙ
8
18 8
2 rubidium
47 Ag
107,8682(2)
18
Kr4d105s1 18
СЕРЕБРО 2
________argentum
55 CS ;
132,90545(2) 18
Xe6s’ 18
ЦЕЗИЙ
__________cesium
79 л
Au,;
196,96655(2) 32
Xe 4f145d106s1 18
ЗОЛОТО
aurum
87 Fr
[223,0197] Rn 7s1
ФРАНЦИЙ
8 18
32 18
8 2 francium
2 8
2 magnesium
20Ca
40,078(4) Ar 4s2
КАЛЬЦИЙ
2 8
8
2 calcium
30 Zn
65,39(2) 2
Ar 3d104s2
ЦИНК
___________zincum
38 Sr
87,62(1)
Kr 5s2
СТРОНЦИЙ
strontium
2
8
18
8
48 Cd
112,411(8) Kr 4d105s2 КАДМИЙ
2 18 18
8 2 cadmium
56 Ba
137,327(7) Xe 5s2
БАРИЙ
2
8
18
18
8
2
barium
80 ту
Hg,;
200,59(2) 32
Xe 4f145d106s2 18
РТУТЬ 2
hydrargyrum
88 Ra
[226,0254] Rn 7s2
РАДИЙ
2
8
18
32
18
8
2 radium
2 He
4,002602(2)
1s2
ГЕЛИЙ 2
helium
10
20,1797(6)
He 2s22pB
НЕОН
neon
13 26,981538(2) Ne 3s23p1 3 АЛЮМИНИЙ 2 aluminium 14 28,0855(3) Ne 3s23p2 4 КРЕМНИЙ 2 silicium |
21 Sc
44,955910(8)
Ar 3d14s2
СКАНДИЙ
scandium
2
9
8
22 .j.
47,867(1) 2
Ar3d24s2 1®
ТИТАН 2
__________titanium
15
16
17
18
31
32
30,973761(2)
Ne 3s23p3
ФОСФОР
2 ihosphorum
32,066(6) Ne 3s23p4
СЕРА
e
2 sulfur
35,4527(9) Ne 3s23p5
ХЛОР
2 chlorum
39,948(1) Ne3s23p6 8
АРГОН
argon
69,723(1) 3
Ar3d104s24p1
ГАЛИЙ 2
___________qalium
72,61(2) 4
Ar3d104s24p2 13
ГЕРМАНИЙ 2
germanium
39
88,90585(2) Kr 4d15s2
ИТТРИЙ
18 8 2 ttriiim
40 Zr
91,224(2) Kr 4d25s2
ЦИРКОНИЙ
2
10
18
2 zirconium
49
50
114,818(3) Kr 4d1Q5s25p1
ИНДИЙ
18
18
8
2 indium
118,710(7)
Kr 4d105s25p2 18
ОЛОВО 2
stannum
18
57-71 *j
La- •Lu
72 Hf
178,49(2) Xe 4f145d26s2
ГАФНИЙ
10 32
18 8
2
hafnium
81
82
18
32
204,3833(2)
Xe 4f145d106s26p118
ТАЛИЙ 2
___________thallium
89-103 **
Ac—Lr
18
32
1207,2(1)
Xe 4f145d106s26p218
СВИНЕЦ 2
________plumbum
104 Rf
[263,1125]
Rn 5f146d27s2
РЕЗЕРФ0РДИЙ2
rutherfordium
2
10
32
32
23 v
50.9415(1) 2
Ar3d34s2 1g
ВАНАДИЙ 2
________vanadium
24 Cr
51,9961(6)
Ar 3d54s1
ХРОМ
________chromium
13
8
25 Mn
54,938049(9) 2
Ar 3d54s2 13
МАРГАНЕЦ 2
manganum
26 Fe
55,845(2) 2
Ar 3d64s2 14
ЖЕЛЕЗО
ferrum
33
34
35
36
74,92160(2) 5
Ar 3d104s24p3 18
МЫШЬЯК 2
________arsenicum
78,96(3) 6
Ar3d104s24p4 18
СЕЛЕН 2
_________selenium
79,904(1) 7
Ar3d104s24p5
БРОМ
bromum
83,80(1) 8
Ar 3d104s24p6 '?
КРИПТОН 2
krypton
41 Nb
92,90638(2) Kr 4d45s1
НИОБИЙ
51
12
18
8
2
niobium
42 Mo ,
95,94(1) 13
Kr4d55s2
МОЛИБДЕН 2 molybdaenum
43 Tc
[97,9072]
Kr 4d55s2
ТЕХНЕЦИЙ
2 13 18
8 2 technetium
44 Ru,
101,07(2) 15
Kr 4d75s1 18
РУТЕНИЙ 2
ruthenium
52
53
54
121,760(1) Kr 4d1 °5s25p3
СУРЬМА
5 18
18 8
2
stibium
127,60(3)
Kr4d105s25p4
ТЕЛЛУР 2
tellurium
18
126,90447(3) 18
Kr4d105s25p5
ИОД 2
_________iodum
8
18
131,29(2) Kr 4d105s25p6 1®
КСЕНОН
xenon
73 Ta >
180,4979(1) 32
Xe 4f145d36s2 18
ТАНТАЛ
_________tantalum
74 w
183,84(1)
Xe 4f145d46s2
ВОЛЬФРАМ
12
32
18
8
2
wolframium
75 n
Re
186,207(1) 32
Xe 4f145d56s2 1®
РЕНИЙ
rhenium
76 Os,;
190,23(3) 32
Xe 4f145dB6s2 18
ОСМИЙ
osmium
83
5
18
208,98038(2) 32
Xe4f145d106s26p318
ВИСМУТ
_______bismuthum
106 Db .?
32
[262,1144] 32
Rn 5f146d37s2 18
ДУБНИЙ
_________dubnium
84
6
18
32
85
86
[208,9824]
Xe 4f145d106s26p418
ПОЛОНИЙ 2
polonium
106
2
12
32
[266,1219] 32
Rn5f146d47s2 18
СИБОРГИЙ 2
seaborgium
18
32
[209,9871]
Xe 4fl45d106s26p518
АСТАТ 2
astatium
107Bh j
[264,1247] 32
Rn 5f146d57sz 1?
БОРИЙ
borium
8
18
32
[222,0176]
Xe 4f145d106s26p’ 18
РАДОН
radon
_________________
108 Hs
32
[269,1341] 32
Rn 5f146d67s2 18
ХАССИЙ
__________hassium
57 La I 138,9055(2) 18 Xe 5d16s2 18 ЛАНТАН 2 lanthanum 58 Ce , 140,116(1) 20 Xe 4f26s2 18 ЦЕРИЙ cerium 59 Pr , 140,90765(2) 21 Xe4f36s2 18 ПРАЗЕОДИМ 2 praseodymium 60Nd г 144,24(3) 22 Xe4f46s2 18 НЕОДИМ 2 neodymium 61Pm , [144,9127] 23 Xe4fs6s2 18 ПРОМЕТИЙ 2 promethium 62Sm , 150.,36(3) 24 Xe4f66s2 18 САМАРИЙ 2 samarium 63 Eu; 151,964(1) 25 Xe 4f76s2 18 ЕВРОПИЙ г europium 64 Gd > 157,25(3) 25 Xe4f75d16s2 18 ГАДОЛИНИЙ 2 gadolinium 65 Tb J 158,92534(2) 27 Xe 4f96s2 18 ТЕРБИЙ 2 terbium |
18 [227,0277] 32 Rn 6d’7s2 18 АКТИНИЙ 2 actinium 90 Th » 18 232,0381(1) 32 Rn6d27s2 18 ТОРИЙ 2 thorium 91 Pa t 20 231,03588(2) 32 Rn 5f26d’7s2 18 ПРОТАКТИНИЙ 2 protactinium 92 U J 238,0289(1) 32 Rn SfWZs2 18 УРАН 2 uranium 93 Np j [237,0482] 32 Rn Srtd^s2 18 НЕПТУНИЙ 2 neptunium 94 Pu ; i M 24 [244,0642] 32 Rn 5f67s2 18 ПЛУТОНИЙ 2 plutonium 95Am j [243,0614] 32 Rn 5f77s2 18 АМЕРИЦИЙ 2 americium "Cm J [247,0703] 32 Rn 5f76d’7s2 18 КЮРИЙ curium [247,0703] 32 Rn 5f97sz 18 БЕРКЛИЙ 2 berklium |
27 Co I28 Ni
58,933200(9) 2 SB 58,6934(2) 2
Ar3d74s2 Ar3d84s2l|
КОБАЛЬТ 21 {НИКЕЛЬ
cobaltum ' ' niccolum
45
Rh ,
102,90550(2) 16
Kr4d85s1
РОДИЙ
rhodium
46 Pd .
106,42(1) 18
Kr4d10
ПАЛЛАДИЙ 2 palladium
СДПU < ’I И ; \ \ C{ C rrts’il И Л \И С тоН’. *
ПРОФЕССИОНАЛА
www.npomis.com
(812)3216738, 9890328
МОВЫТС
СПРАВОЧНИК
ХИМИКА и ТЕХНОЛОГА
Аналитическая химия
Руководители проекта:
А.А.Полуда, Н.В.Емельянова
Под общей редакцией:
профессора, доктора химических наук
И.П. Калинкина
к.х.н. Ю.А. Барбалат акад. РАЕН, проф., д.х.н. Ю.Г. Власов к.х.н. В.А. Демин проф., д.х.н. Ю.Г. Ермоленко акад. РАН, проф., д.х.н. Ю.А. Золотов проф., д.х.н. В.М. Иванов проф., д.х.н. И.П. Калинкин проф., д.х.н. А.А. Карцова с.н.с. В.В. Колодников к.х.н. В.И. Мосичев
Авторы:
акад. РАЕН, проф., д.х.н. Л.Н. Москвин проф., д.т.н. В.М. Немец доц., к.х.н. О.В. Родинков проф., д.х.н. Г.В. Саидов доц., к.ф.-м.н. О.В. Свердлова с.н.с., к.т.н. А.А. Соловьев доц., к.х.н. В.Ф. Теплых доц., к.х.н. А.С. Халонин доц., к.х.н. Н.М. Якимова проф., лауреат Гос. премии СССР и РФ, д.х.н. Я.И. Яшин к.х.н. А.Я. Яшин
ммп Санкт-Петербург 2002