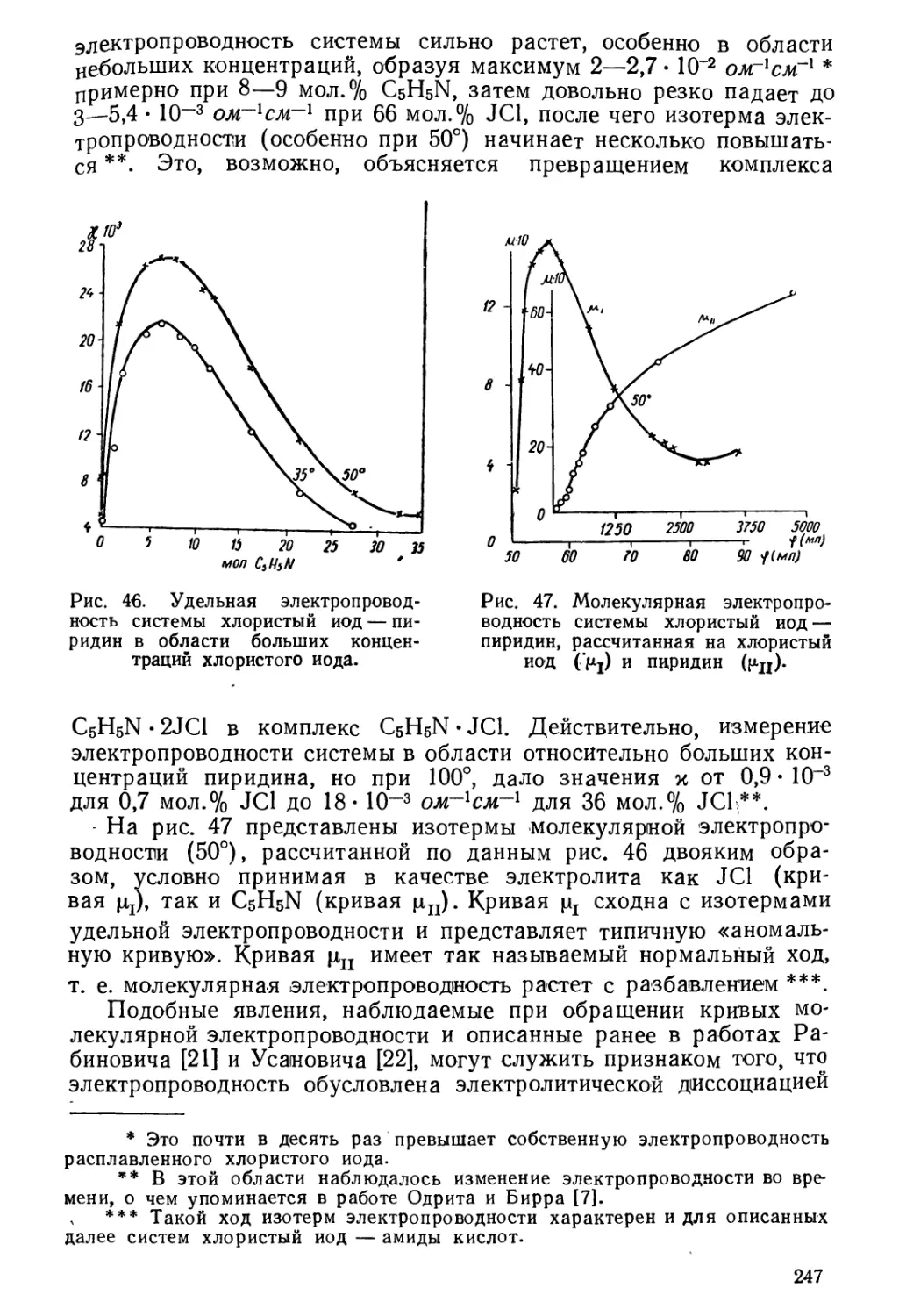
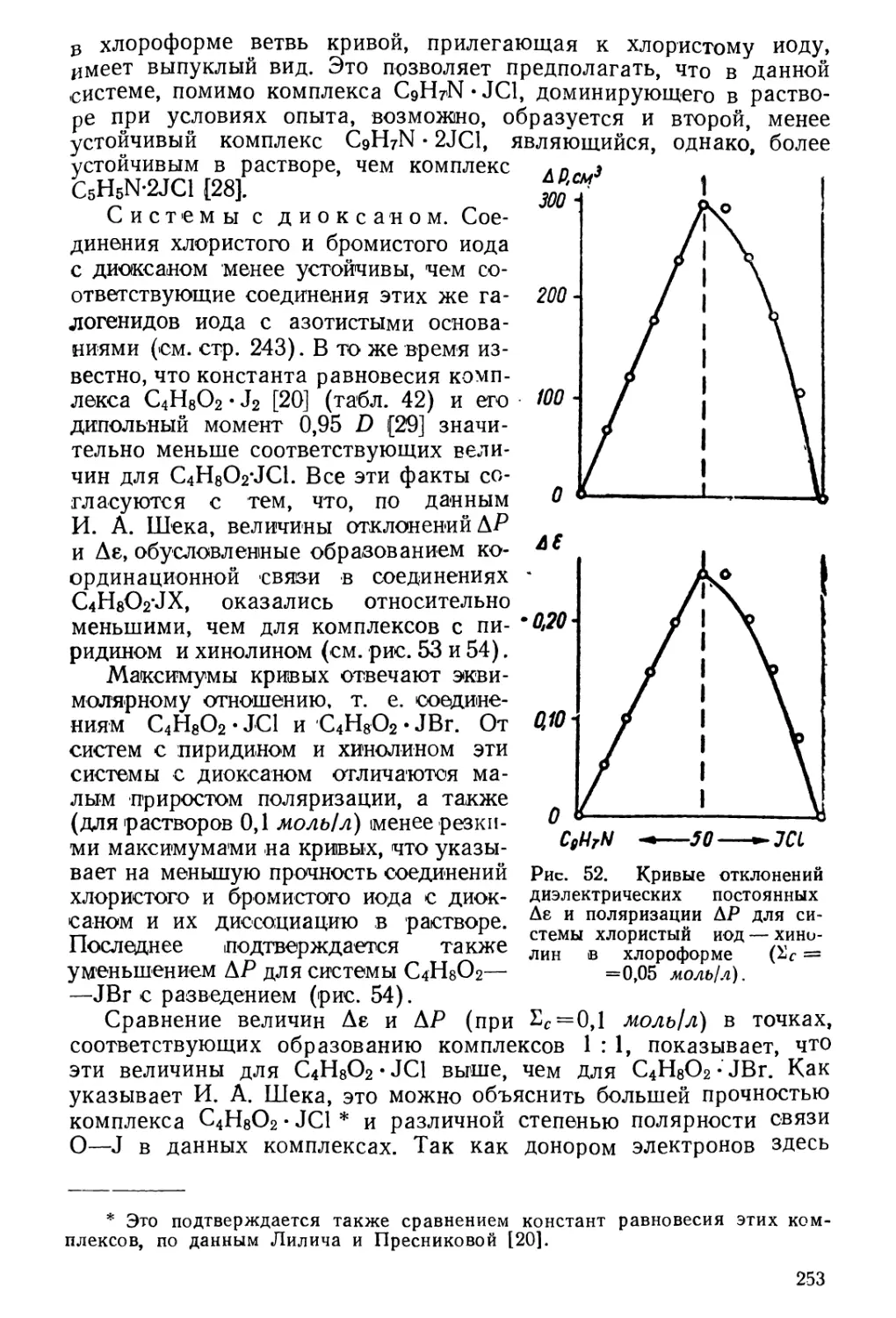
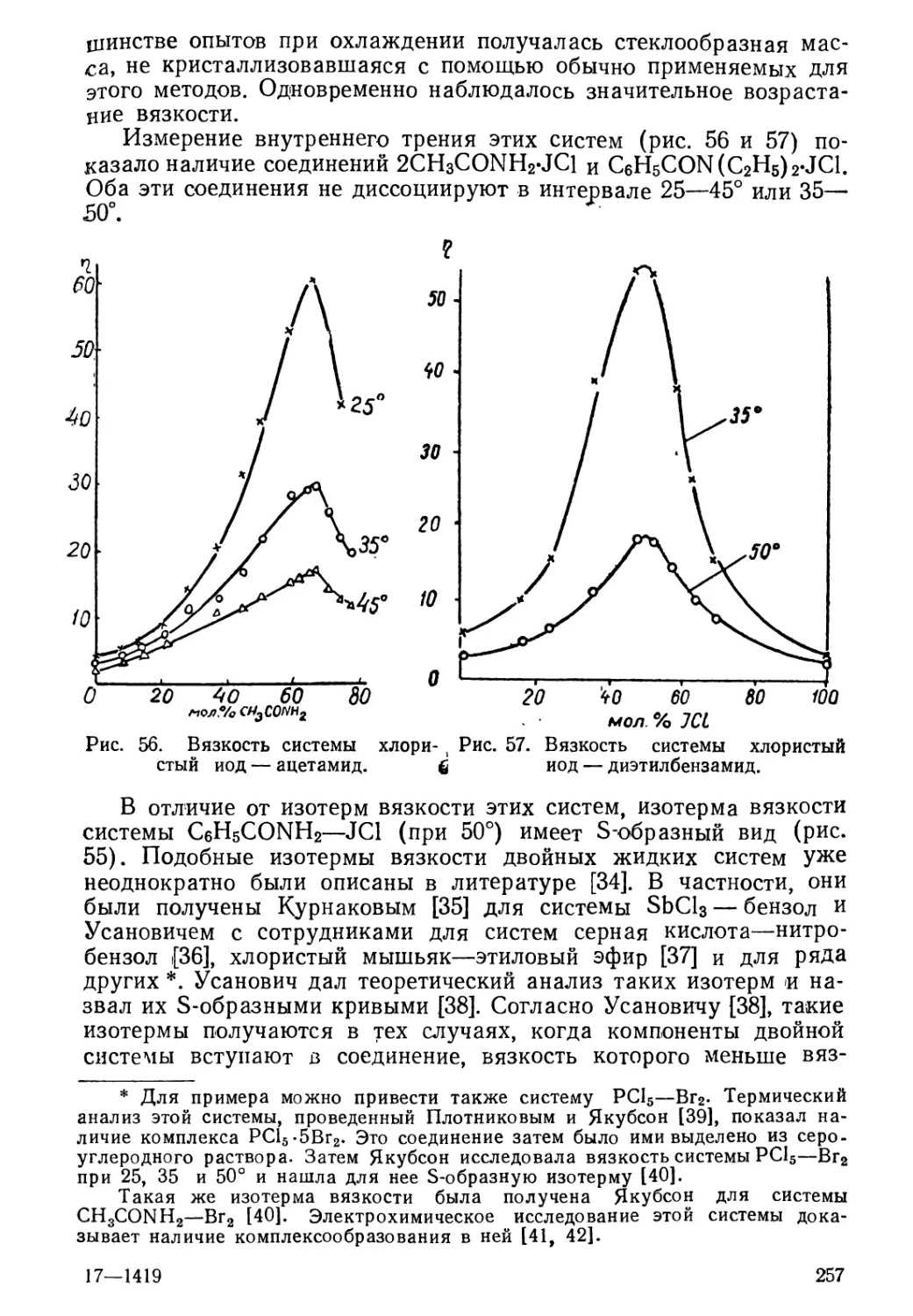
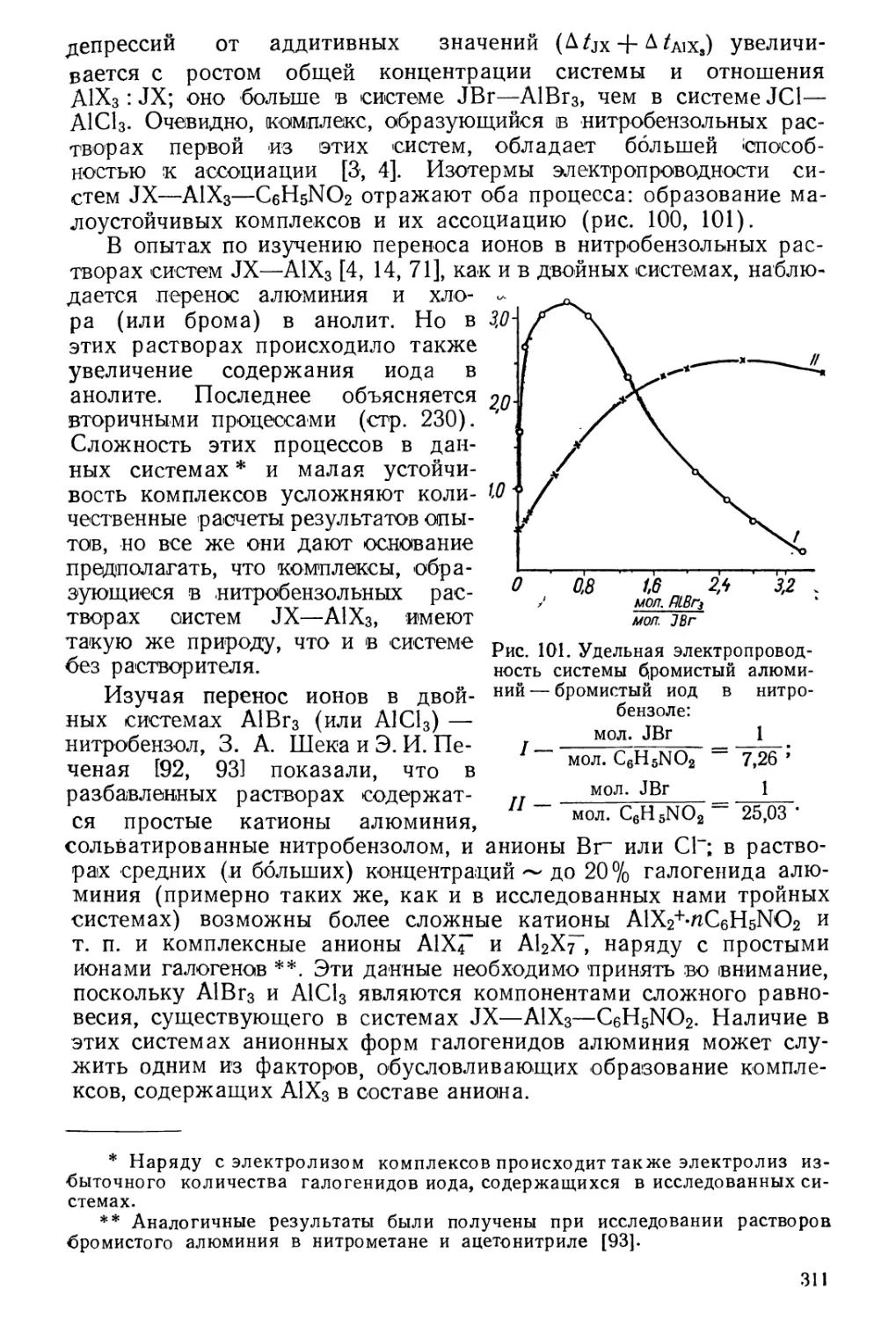
/






































































































































































































































































































































































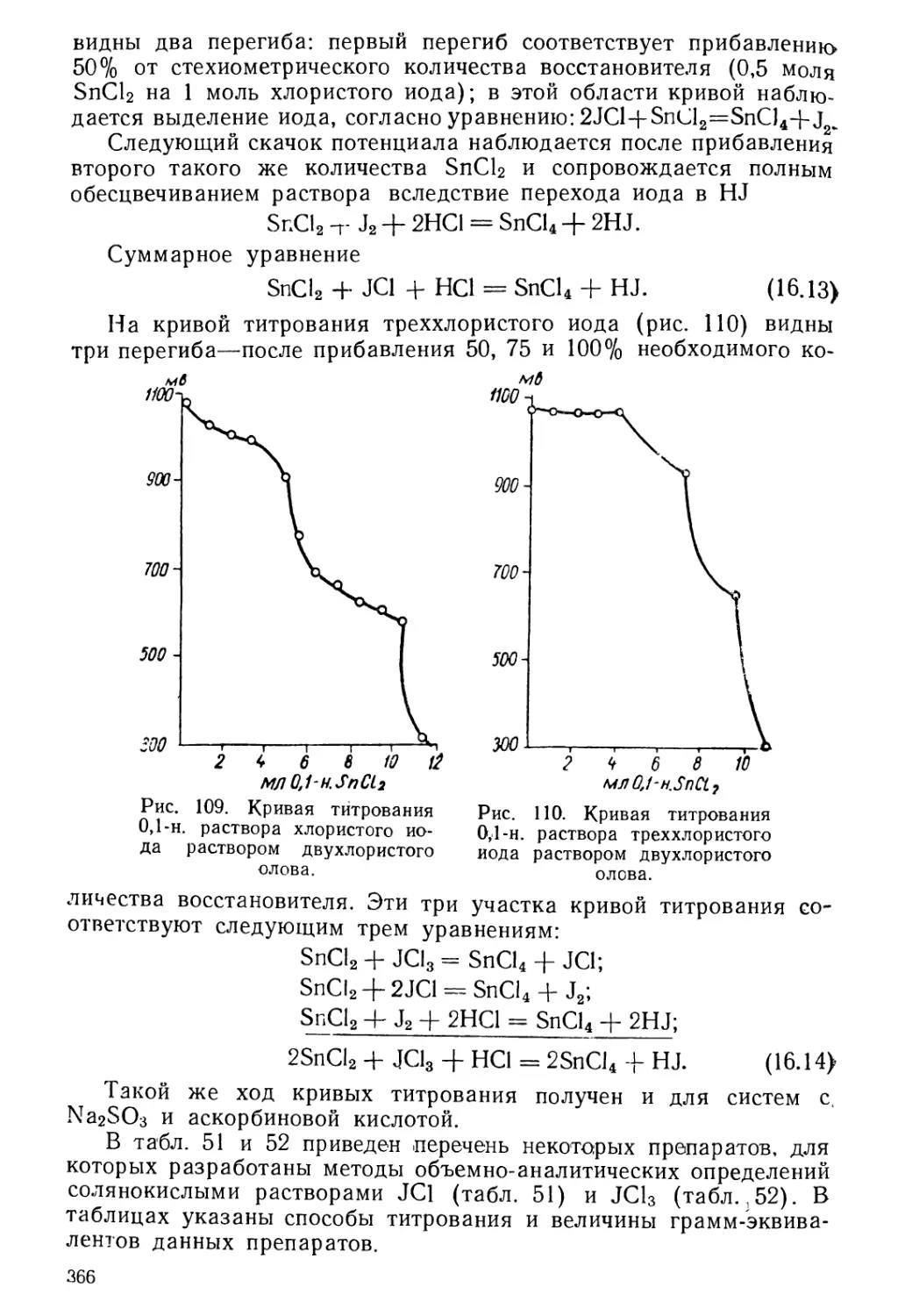


































Текст
^■у\Ж7^^Ж
../;:.. .. •■ •'•...■ -i
Я.ЛлрИАЛКОП
N ГАЛОК
It
I
Ы
АКАДЕМИЯ НАУК УКРАИНСКОЙ ССР
ИНСТИТУТ ОБЩЕЙ И НЕОРГАНИЧЕСКОЙ ХИМИИ
| Я. А. ФИАЛ КОВ |
член-корреспондент АН УССР
МЕЖГАЛОИДНЫЕ
СОЕДИНЕНИЯ
ИЗДАТЕЛЬСТВО АКАДЕМИИ НАУК УКРАИНСКОЙ ССР
КИЕВ—1958
Монография представляет собой отсутствовавший ранее в
литературе обзор физических и химических свойств
межгалоидных соединений и их комплексов с неорганическими и
органическими веществами.
В первых семи главах описаны все известные межгалоге-
ниды, в главах VIII—XII рассматриваются общие физико-
химические, в том числе и электрохимические, свойства межга-
логенидов и формы их применения в практике. Главы XIII—
XV посвящены некоторым теоретическим вопросам,
относящимся к химии межгалоидных соединений а к методам
изучения процессов комплексообразования в неводных системах.
Монография заканчивается главой, в которой описаны
физико-химические основы и методика так называемого иод-
хлор метрического метода объемного анализа неорганических
и органических веществ, основанного на применении
солянокислого раствора хлористого иода.
Книга рассчитана на научных работников,
преподавателей, аспирантов и студентов старших курсов химических и
фармацевтических вузов и факультетов,
инженерно-технических работников.
Ответственный редактор
доктор химических наук И. А. Шека
ПРЕДИСЛОВИЕ
В последнее время интерес к межгалоидным соединениям
значительно повысился. Появился ряд обзорных статей, посвященных
межгалогенидам, особенно фторидам галогенов, как сравнительно
более новым представителям этой группы неорганических
соединений. Увеличилось количество экспериментальных исследований, в
которых изучаются физические и химические свойства межгалоидных
соединений, реакции их взаимодействия с неорганическими и
органическими веществами, применение их для синтеза неорганических и
органических соединений и в химическом анализе.
В литературе отсутствуют работы, в которых были бы
систематизированы и рассмотрены сведения, относящиеся ко всем этим
областям химии межгалоидных соединений.
В настоящей монографии дан обзор физических и химических
свойств межгалоидных соединений. Большое внимание уделено
также комплексам, которые они образуют с неорганическими и
органическими веществами.
В монографии использованы работы, опубликованные, главным
образом, до середины 1956 года.
Книга начинается главой «Общая характеристика межгалоидных
соединений». В главах II—VII описаны все известные
межгалоидные соединения, методы их получения, физические и, более
подробно, химические их свойства, комплексные соединения, реакции с
неорганическими и органическими веществами, пути практического
применения.
В главах VIII—XII рассматриваются некоторые общие для
межгалоидных соединений физико-химические свойства, в том числе и
электрохимические. Основная часть этого раздела посвящена
хлористому, треххлористому и бромистому иоду, как наиболее
изученным представителям межгалоидных соединений. Здесь же описаны
основные типы комплексных соединений, образуемых этими галоге-
'нидамл иода. Наряду с литературными данными здесь рассмотрены
результаты работ в этой области, выполненных в Институте общей
и неорганической химии АН УССР автором с сотрудниками.
Главы XIII—XV посвящены некоторым теоретическим вопросам,
относящимся к химии межгалоидных соединений. Здесь же в связи
с рассмотрением вопроса о методах исследования комплексных
соединений галогенов и межгалогенидов обсуждаются вопросы
изучения процессов комплексообразования в неводных средах.
3
Монография заканчивается главой, написанной совместно с
А. И. Генгриновичем и Ф. Е. Каган, в которой описаны
физико-химические основы, методика и формы применения так называемого
иодхлорметрического метода объемного анализа неорганических и
органических веществ, основанного на применении солянокислых
растворов хлористого и треххлористого иода.
Основной задачей монографии является, помимо обзора
межгалоидных соединений, рассмотрение связанных с этим обзором
некоторых вопросов, относящихся к неорганической химии и химии
комплексных соединений, электрохимии неводных систем и
физико-химическому анализу. При описании химических свойств межгалоидных
соединений было необходимо рассмотреть их реакции с
органическими соединениями и образующиеся при этом продукты. Поскольку
автор не является специалистом в области органической химии, этим
вопросам, возможно, не уделено достаточного внимания, и их
изложение не могло быть достаточно глубоким.
Как уже отмечено, при составлении большой части данной книги
были использованы исследования, проведенные в лаборатории
комплексных соединений Института общей и неорганической химии
Академии наук УССР. В этих многолетних исследованиях
участвовали И. Л. Абарбарчук, К. Я. Каганская, А. А. Кузьменко, И. Д.
Музыка, Ю. П. Назаренко, И. А. Шека, О. И. Шор и др. Автор горячо
благодарит этих лиц за неоценимую помощь в исследованиях
сложной области неорганической химии и химии комплексных
соединений.
Автор выражает благодарность И. А. Шека, 3. А. Шека, А. А. Ка-
гановскому за просмотр рукописи, И. Г. Рыссу за просмотр
главы-XIII и ценные указания, а Н. А. Костроминой — за помощь в
оформлении рукописи и подготовке ее к печати.
Автор будет признателен всем лицам, которые пришлют свои
замечания и пожелания в связи с содержанием данной монографии.
ГЛАВА I
ОБЩАЯ ХАРАКТЕРИСТИКА МЕЖГАЛОИДНЫХ
СОЕДИНЕНИИ
Межгалоидные или межгалогенные соединения (соединения
галогенов друг с другом) еще до сравнительно недавнего времени
можно было считать «обойденными» неорганическими веществами.
Учебные и даже монографические руководства при описании
межгалоидных соединений обычно ограничивались краткими и
отрывочными сведениями о них, отмечая при этом некоторые особенности их
свойств, вообще изученных еще далеко не полно.
В последние два десятилетия были синтезированы некоторые
неизвестные ранее фториды галогенов, более подробно исследованы
физические и химические свойства -межгалоидных соединений и
найдены новые формы их практического применения. В связи с этим
значительно повысился интерес к данной группе неорганических
веществ. В литературе за последние годы появилось несколько
обзоров [1—6,69], посвященных (межгалоидным соединениям, однако ни
один из них не освещает химию межгалоидных соединений с
достаточной полнотой. В большей части этих обзоров описаны лишь
фториды галогенов, правда, являющиеся наиболее многочисленной
группой межгалоидных соединений. Наиболее полный обзор,
содержащийся в статье Н. Н. Гринвуда [6], ограничивается
преимущественно описанием лишь физических свойств межгалоидных
соединений. Между тем химические свойства последних представляют
большой интерес как для теоретического рассмотрения, так и для
использования их на практике.
Еще Д. И. Менделеев [7] обратил внимание на своеобразие
свойств межгалоидных соединений. Он считал, что соединения хлора
с иодом нужно отнести «к числу поучительнейших сложных тел».
Многие особенности межгалоидных соединений обусловлены темт
что они образованы атомами элементов, принадлежащих к одной и
той же группе и подгруппе периодической системы.
За исключением кислорода и серы, образующих соединения
почти со всеми химическими элементами, в том числе и с элементами
подгруппы кислорода (VI группы), известно лишь сравнительно
немного случаев соединения друг с другом элементов, входящих в
одну и ту же группу и подгруппу периодической системы, с образова-
5
нием определенных химически индивидуальных веществ. Таковы,
например, гидриды щелочных металлов, бор иды алюминия,
карборунд, азотистые соединения (нитриды?) фосфора PN, P3N5, P4N6 и
мышьяка AsN * и неустойчивые фосфористые соединения мышьяка
А$2Р, AsP и сурьмы SbP **, окислы серы, селена и теллура,
сульфиды селена и теллура SeS, SeS2, TeS, TeS2 и, наконец,
межгалоидные соединения.
Ряд подобных примеров имеется и среди интерметаллических
соединений (PbSn3, Na2K и др.), хотя и в этой области
неорганических веществ существуют определенные ограничения,
обусловленные положением металлов в периодической системе. Так,
расположенные в больших периодах члены одного и того же ряда аналогов
(например, медь, серебро и золото или цинк, кадмий и ртуть)
обычно не дают друг с другом соединений [8] ***.
Здесь следует напомнить также предложенное Г. Тамманом
правило (не применимое к малым периодам), согласно которому
элементы, стоящие в периодической системе друг под другом, не
вступают между собой в химическое соединение [10]. Приводя это
правило в своем курсе неорганической химии, Ф. Эфраим пишет, что оно
оправдывается в огромном большинстве случаев, в особенности для
металлов, и что единственным исключением из этого правила
являются галогены [11].
Хотя это правило Г. Таммана и суждение Ф. Эфраима нельзя
считать безусловными, все же следует сказать, что способность
элементов одной и той же группы и подгруппы периодической системы
к взаимодействию, проявляющаяся все более значительно по мере
перехода от элементов I группы к элементам VII группы, наиболее
ярко выражена в подгруппе кислорода и особенно в подгруппе
галогенов.
В отличие от того, что известно для значительного большинства
элементов других подгрупп, галогены, вследствие своего положения
в периодической системе элементов, обладают способностью
сравнительно легко соединяться попарно — каждый с любым из трех
остальных — с образованием продуктов различной степени
устойчивости. Более устойчивыми являются соединения, которые
образованы галогенами, дальше отстоящими друг от друга в VII группе.
В настоящее время известны двенадцать межгалоидных
соединений, которые можно разделить на четыре группы—АВ, АВ2, ABs
и АВ7 — по количеству атомов галогена. Межгалоидные соединения,
содержащие нечетное количество атомов галогена или содержащие
три-четыре разных галогена в молекуле, не известны.
Эти схематические формулы отражают только простейший со-
* Азотистые соединения мышьяка, сурьмы и висмута, образующиеся при
взаимодействии тригалогенидов этих элементов с амидом калия в жидком
аммиаке или по реакции As2(NH)3 = 2AsN + NH3, очень неустойчивы.
** В системах мышьяк—■ сурьма и мышьяк—висмут методом термического
анализа не обнаружено образование соединений мышьяка с сурьмой или с
висмутом, хотя имеются указания на получение соединений состава Sb2As, Bi3As4,
BiAs.
*** В системе медь—золото обнаружены соединения CuAu и Cu3Au [9>].
6
став межгалоидных соединений. В действительности же,
молекулярные формулы межгалоидных соединений, вследствие явлений
ассоциации, часто бывают значительно более сложными.
По химическому составу полученные до настоящего времени
межгалоидные соединения можно разделить на три основные
группы:
1. Галогениды хлора
фториды: фтористый хлор C1F, трехфтористый хлор C1F3.
2. Галогениды брома
а) фториды: фтористый бром BrF, трехфтористый бром BrF3,
(пятифтористый бром BrFs;
б) хлориды: хлористый бром BrCl.
3. Галогениды иода
а) фториды: од нофтор истый иод JF, пятифтористый иод JF5,
еемифтористый иод JF7;
б) хлориды: хлористый иод JC1, треххлористый иод JC13;
в) бромиды: бромистый иод JBr.
Межгалоидные соединения можно классифицировать также по
природе более электроположительного элемента, входящего в
состав их молекул. В этом случае также образуются три основные
группы:
1. Фториды галогенов
а) фториды хлора: C1F, C1F3;
б) фториды брома: BrF, BrF3, BrFs;
в) фториды иода: JF, JF5, JF7.
2. Хлориды галогенов
а) хлорид брома: BrCl;
б) хлориды иода: JC1, JC13.
3. Бромиды галогенов
а) бромид иода: JBr.
Эта система классификации использована при описании
отдельных межгалоидных соединений в главах II—VII.
К межгалоидным соединениям примыкают соединения галогенов
с так называемыми «псевдогалогенами» (или «галогеноидами») —
цианом, роданом, оксицианом, азидогруппой — и некоторые другие.
Из межгалоидных соединений раньше всего были известны
хлористый и треххлористый иод. Они были получены И. Л. Гей-Люсса-
ком и Н. Деви еще в 1814 г. Открытие следующих межгалоидных
соединений продолжалось до 50-х годов XX ст. [12—22], (стр. 106).
Все межгалоидные соединения могут быть получены
непосредственно из соответствующих галогенов в определенных условиях.
Высшие формы межгалогенидов (АВ3—АВ7) можно получить также
путем присоединения галогенов к низшим формам. Так, семифто-
ристый иод получают при действии фтора на пятифтористый иод.
Условия синтеза межгалоидных соединений указаны при описании
отдельных их представителей.
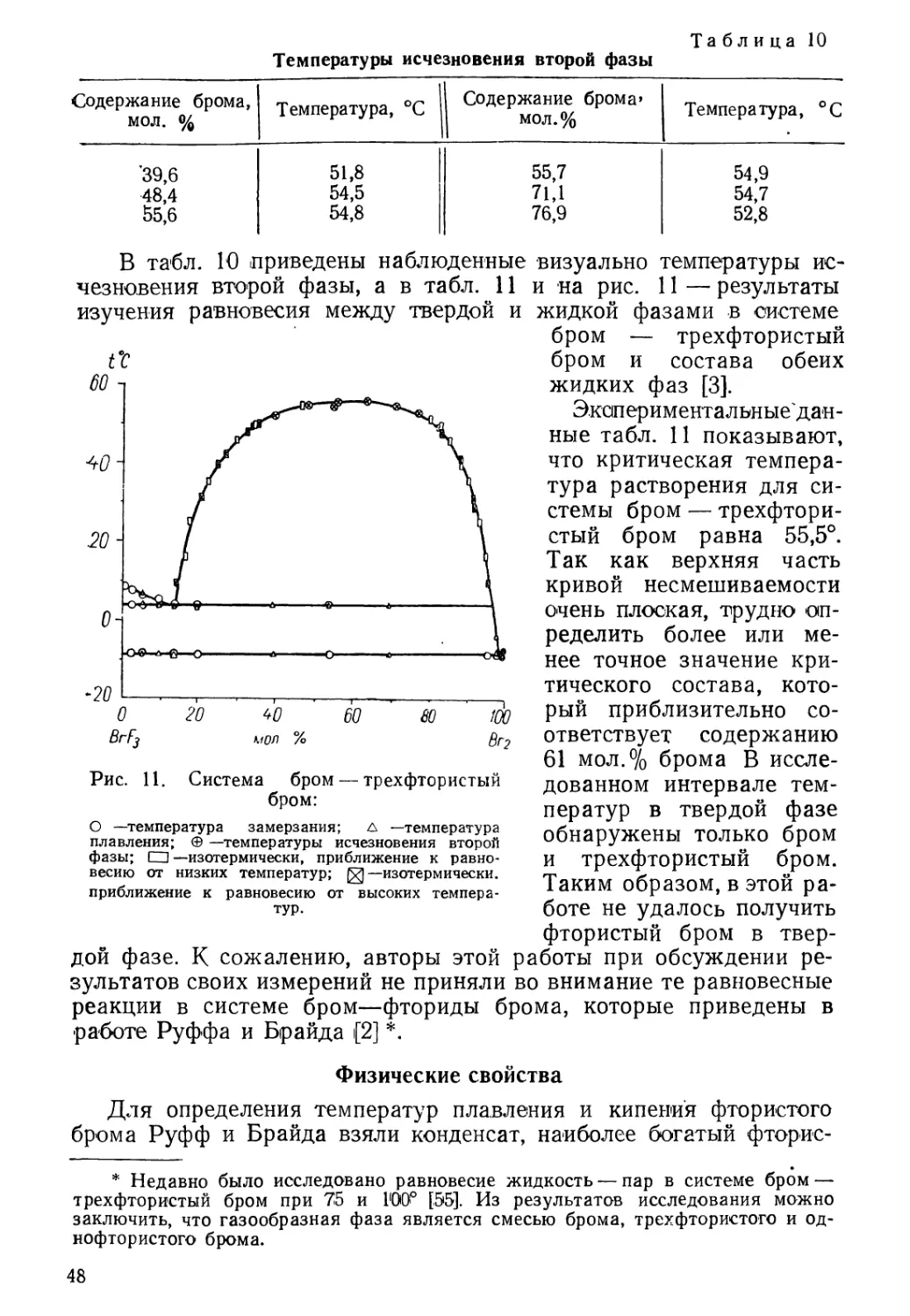
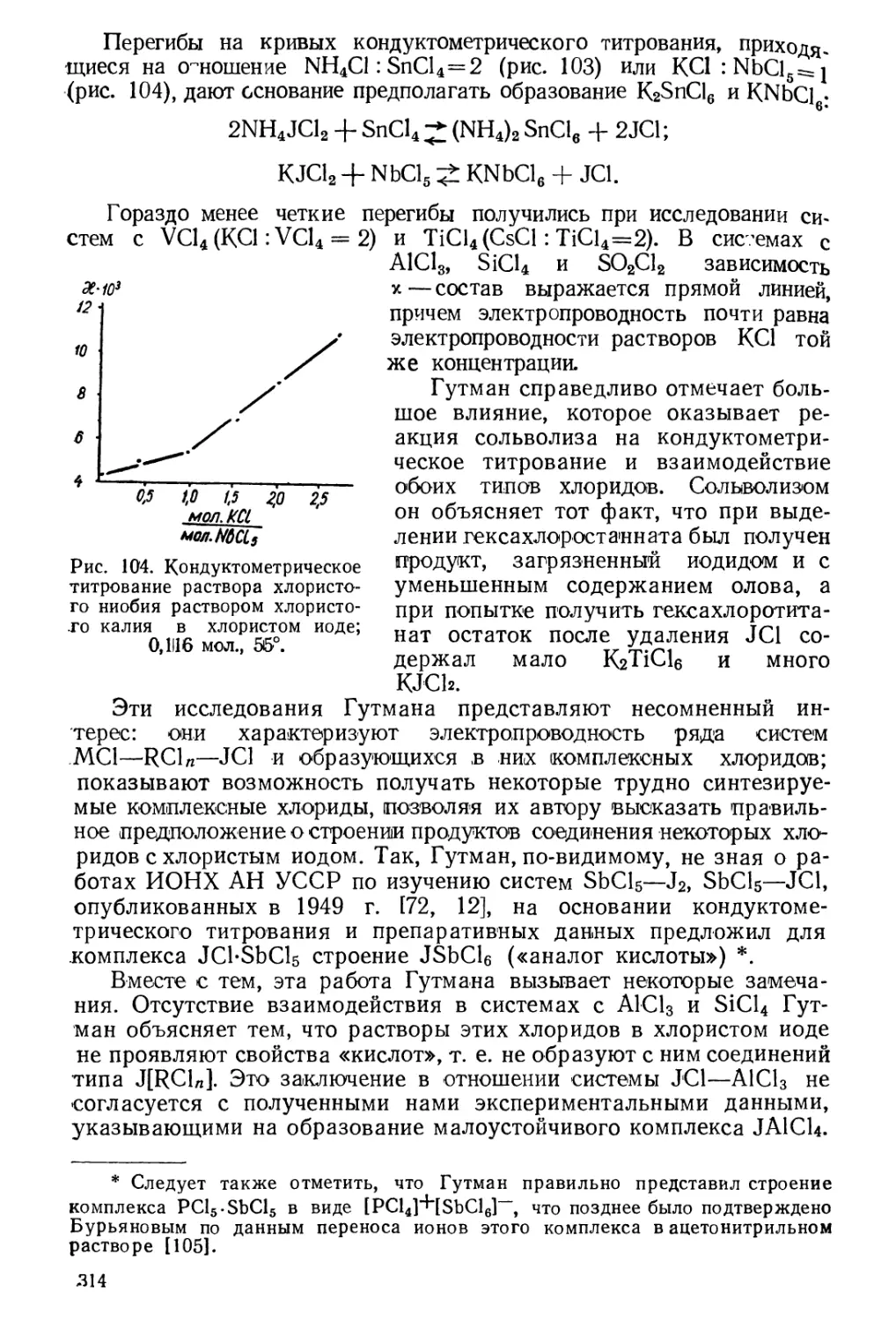
В табл. 1 и 2 и на рис. 1, 2 и 3 * приведены некоторые физиче-
* Рис. 1 и 2 заимствованы из статьи Е. Фессенден [23], а рис. 3 — из статьи
О. Руффа [24].
7
Таблица 1
Некоторые физические свойства межгалоидных соединений
1ежгалоидные
>единения
:> о |
C1F
C1F8
BrF
BrF8
BrF5
JF5
JF7
BrCl
JCla
JClp
JC13
JBr
CO
cO
t?
емпература п.
гния, °С
H ч
— 155,6
-83±0,5
33
8,8
-61.3
9,43
5-6
(под
давлением)
От ~*—66 до
-54
1 27,2
13,9
101 (при
16 атм)
42
пе-
я
емпература к
ия, °С
н я
-100,1
11,75
*^20
127,6+1
40,5±0,5
100,5
4,5
U^ Комнатная
температура
100—102
—
Разлагается
~ 166
° Ь/
о я а.
грегатное со
ние при ком
ой температу
<! к я
Газ
»
Жидкость
ъ
»
Газ
»
Твердое
—
Твердое
1
1г С Я
§ ^ s U
5 акт
Ос
g е &s
Ч О со
(Г) я Он
3-10~9(0°)
8,Ы0~?
9,Ь 10~8
(25°)
1,6-10"5
—
4,40-10~3
—
8,5-Ю-3
4,0-10-4
константа
рутона
«н
28
20,8
20,5 (?)
25
23,7
27,2
<26,4
—
26,7
—
—
—
f-i m
лотность при ■
ературе кипе]
г/см3
С с
1,62
1,85
■
2,515
2,411
2,80
—
—
—
—
—
СО СО -
S сор
=Г O-V^?
тепень диссо!
ии в парооб
ой фазе при 2
%
О д- я
1,2-10-7
5,2*
Очень боль»
шая (полная
при 50°)
Очень слабая
Незначительная**
Незначительная***
Незначительная****
21
0,38
—
Полная
8,8
ские свойства межгалоидных соединений. Данные этих
таблиц *****, а также табл. 4 показывают довольно значительное
различие физико-химических свойств межгалоидных соединений,
обусловленное их химическим составом.
Так, температуры плавления и кипения межгалоидных
соединений ****** повышаются с увеличением атомного веса второго
галогена, т. е. при переходе от соединений хлора к соединениям
брома (C1F и BrF, C1F3 и BrF3) и от соединений брома к
соединениям иода (BrCl и JC1, BrF5 и JF5). To же наблюдается при
сравнении хлористого и бромистого иода, т. е. соединений с общим
«тяжелым» галогеном: фтористый бром обладает более высокой
температурой плавления, чем хлористый бром; однако необходимо
иметь в виду, что оба последних галогенида относятся к наименее
* Эта величина означает степень ассоциации C1F3 в % при Р=1 атм.
** Пятифтористый бром устойчив, по крайней мере, до 460°.
*** Пятифтористый иод начинает разлагаться лишь при температуре выше 400°.
**** Семифтористый иод устойчив, по крайней мере, до 500°.
***** Ссылки на литературу, относящиеся к табл. 1, указаны при описании
отдельных межгалоидных соединений.
****** Необходимо принять во внимание, что зависимость между химическим-
составом межгалоидных соединений и их физико-химическими свойствами
усложняется ассоциацией молекул.
8
Таблица 2
Некоторые физические свойства межгалоидных соединений
соеди- 1
Межгалоидное
нение
C1F
C1F3
C1F3
CIF3
CIF3
BrF3
BrF3
BrF8
BrF5
BrF5
BrF5
BrF5
JF5
JF5
JF5
JF5
BrCI
JCl
JCl
Состояние
Газ
»
»
Жидкость
»
Газ
»
Жидкость
Газ
»
Жидкость
»
Газ
»
Жидкость
»
Смесь
жидких
брома и
хлора
Твердое
1 *
Диэлектрическая
проницаемость е и молярная
поляризация Рм
«Г
о,
а.
0)0
Но
46,1
140,1
0
25,0
142,3
175,0
72,4
156,8
11,7
24,5
119.5
172,8
1 12
40
-60
10
-48
1 —И
8
1,002825
1,001929
4,75**
4,29
1,003748
1,002964
1,006320
1,004378
8,33**
7.91
1,009168
1,007135
38,7**
33,2
4,8
3,5
7,0
1 13,7
-о
о
п
ч
S
24,2
21,7
42,6
37,0
59,6
51,5
97,6
86,9
к
*>>
5 л
Л Он
Й8
[62]
[61]
[61]
[61]
[62]
[62]
[63]
[63]
[63]
[63]
[62]
[62]
[61]
[61]
[64]
[64]
[651
[65]
Молярная
рефракция, см3/моль
[66]
ев"
Он
>ч
Н
оЗ
Он
<D
С
г-1 о
24,0
26,0
53,0
25,0
29,0
к
СЗ
о
ОЗ
2
со
7,62
10,34
12,92****
15,48
19,17
я
О)
о *
S*
я"
Л оЗ
CQ я
7,19
9,89
12,74
15,49
20,70
Молярная
магнитная воепри
имчивость,*****-
Х-106
к
—26,5
—33,9
—45,1
-58,1
—49,4
(12°)
к
СО
я
я
<D
О
я
СГ
—30,1
—39,4
—48,1
—58,4
я
я о.
* £>
5 Й
3 g^
6 8
[68):
[681
[68]!
[681
устойчивым межгалоадным соединениям и что хлористый бром не
удалось получить в индивидуальном состоянии (см. главу V).
Увеличение валентности положительно заряженного центра
координации (т. е. более тяжелого галогена) до трех влечет за
собой повышение температуры плавления и кипения межгалоидных
соединений, что видно при сравнении фтористого и трехфтористого^
хлора, фтористого и трехфтористого брома, хлористого и треххлори-
стого иода. Можно предположить, что причиной этого являются
* В таблицу включены величины е и Рм только для низшей и высшей из
исследованных температур.
** eCiF3 =(4,754—0,0187 r)+0,02; eBrFa = (8,20-0,0117 0;
ejF,=(41,09—0,198 0±0,1.
*** Для атомной рефракции фтора принято значение 1,35; нормальное
значение равно 0,80 см3.
**** По данным [67], Рм для трехфтористого брома равна 13,22 см3, для
пятифтористого брома—15,41 см3.
***** Все эти фториды галогенов изучались в жидкой фазе при комнатной
температуре.
особенности строения молекул межгалогенидов типа АВ3,
обусловливающие их полярный характер. Из межгалогенидов этого типа
дипольный момент определен только для трехфтористого хлора,
причем оказалось, что он несколько меньше дипольного момента
фтористого хлора (табл. 4). Однако физико-химические свойства
трехфтористого брома и треххлористого иода, в частности их способ-
200
150 «1
100 •
50 -
0 '
-50 «
425 ■
-175 .
-225
•А .
/
Шут *JBr
BrFz./
/•BrCL **rf5
л /oCLFj
CL2f « й2
/ *лв
I оЯВт
' 1 1 ' 1 I 1
<tQ 80 120 160 200 W
Молекулярный бес
Рис. 1. Температуры плавления
межгалоидных соединений.
oJF7
40 SO 120 160 200 240
Молекулярный бес
Рис. 2. Температуры кипения
межгалоидных соединений.
кость довольно хорошо проводить ток, могут служить косвенным
подтверждением полярности этих молекул и значительного участия
ионного характера связи.
Следует все же отметить, что при переходе от молекул
межгалогенидов типа АВ к молекулам АВ3 должен проявиться эффект
экранирования центрального атома, снижающий межмолекулярное
сцепление. Для соединений типа АВ3 он, очевидно, еще не играет
значительной роли (за исключением трехфтористого хлора,
характеризующегося сравнительно низкими температурами плавления и
кипения), поэтому результирующая этих факторов направлена в
сторону повышения температур плавления и кипения.
При дальнейшем повышении заряда центральною атома, т. е.
при дальнейшем увеличении количества координированных атомов
галогена до пяти и семи, начинает сильнее влиять эффект
экранирования и изменение характера связи галогенов в сторону ковалент-
ной связи, в результате чего температуры плавления и кипения
межгалоидных соединений начинают понижаться. Такое явление
действительно имеет место при переходе от трехфтористого брома к
ID
CM*
100 л
50
пятифтористому; этим, вероятно, можно объяснить сравнительно
низкую температуру плавления пятифтористого иода и столь же
низкие температуры кипения пятифтористого и семифториетого иода,
несмотря на большие молекулярные веса их.
Связь между молекулярными объемами фторидов галогенов (при
температуре кипения) и их составом наглядно показана на рис. 3:
в каждом из трех рядов —
фторидов хлора, брома и иода —
молекулярные объемы
увеличиваются с ростом
координационного числа,при
сопоставлении же фторидов разных
галогенов оказывается, что
молекулярные объемы фторидов с
одинаковым координационным
числом несколько
уменьшаются при переходе от фторидов
хлора к фторидам брома, а от
последних — к фторидам иода.
Некоторые физические
свойства, например
температуры плавления и кипения,
плотность, окраска тех
межгалоидных соединений, которые
относятся к типу АВ, по своей
величине занимают промежуточное
положение между величинами
тех же свойств для
соответствующих свободных галогенов. Это можно иллюстрировать
данными табл. 3, в которой приведены температуры плавления (верхние
строки) и температуры кипения (нижние строки) галогенов и их
бинарных соединений.
jfr
1 *** П
1 **■ 1
1 "" 1
UV'"
1 *»*
1 **
а%''
** 1
*& "^
| -*Г 1
№ ~ - ' '
ВТГ
35,5
12?
Атомный бес галогеноб
Рис. 3. Молекулярные объемы
фторидов галогенов при температуре
кипения.
Таблица 3
Температура плавления и кипения галогенов и бинарных межгалоидных
соединений, °С
Галоген
Фтор
Хлор
Бром
Иод
Фтор
—219,6
-187,1
— 155,6
-100,1
— -33
—20
—
Хлор
—
— 102
-34,1
От—66 до—54
—Комнатная температура
27,2 (а-форма)
100—102
Бром
—
—
—7,3
58,8
42
— 166
Иод
Z
—
—
114
183
Л
Из сопоставления данных табл. 3 следует, что температуры
плавления и кипения межгалоидных соединений типа АВ всегда ближе
к температурам плавления и кипения галогена с более высоким
атомным весом. Наиболее ярко это проявляется для фтористого
брома, как это видно из величин А , означающих -разницу в
температурах плавления (верхние строки) и кипения (нижние строки)
межгалоидного соединения и соответствующих галогенов.
Фтор Д Фтористый хлор Д Хлор
64,0 53,6
87,0 66,0
Фтор Д Фтористый бром Д Бром
187 25,7
207 38,8
Хлор Д Хлористый иод Д Иод
129,2 86,8
134,0 83,0
Сходство галогенов с простейшими межгалоидными
соединениями по ряду физических и химических свойств неоднократно
отмечалось в литературе. Так, Менделеев, характеризуя хлористый и трех-
хл-ористый иод, писал, что «химические свойства JC1 или JC13,
вполне отвечают свойствам хлора и иода, чего и должно ждать, потому
что здесь произошло соединение по подобию как при образовании
растворов и сплавов» [7]. Эфраим отмечал, что реакции
межгалоидных соединений являются в большинстве случаев ослабленными
реакциями свободных галогенов [11, стр. 338]. По В. Клемму [25],
«дело здесь идет о соединениях с типичными атомными связями в
молекуле, соединения же эти очень сходны с самими галогенами».
«Бромистый иод обнаруживает далеко идущее сходство со своими
компонентами, в отношении температуры кипения и плавления,
окраски, плотности и т. д. он занимает среднее положение между обоими
компонентами, кроме того он образует с ними * смешанные
кристаллы» отмечают авторы в работе [26].
Наряду с таким сходством .межгалоидные соединения проявляют
определенные отличия по сравнению со свободными галогенами, из
которых они образованы. Прежде всего -следует отметить, что все те
межгалоидные соединения, для которых определен дипольный
момент, оказались полярными соединениями. Величины дипольных
моментов указаны в табл. 4.
Различие в электроотрицательное™ галогенов является причиной
того, что в двухатомных молекулах межгалоидных соединений
отрицательный полюс диполей лежит в области более «легкого»
галогена.
О полярном характере межгалоидных соединений
свидетельствуют также и некоторые другие их молекулярные константы. Так,
можно считать установленным, что связь в двухатомных молекулах
межгалоидных соединений не является чисто ковалентной, а имеет
* По данным Меерум-Тервогта [27], бромистый иод образует два
изоморфных ряда смешанных кристаллов с иодом и бромом.
12
Таблица 4
Дипольные моменты межгалоидных соединений
CD
О
д
о д
сз д
(-1 К
О) Ш
^ о
С IF
C1F3
C1F3
BrF
BrF3
BrF5
JF5
JF5
BrCl
JCl
JCl
JBr
JBr
Состояние
Газ
»
Жидкость (0-25°)
Газ
»
»
»
Жидкость (25°)
Жидкость (смесь брома и хлора)
Газ
Раствор в четырех хлористом углероде
и циклогексане
В жидком броме (25°)
В сероуглероде (25°) и в присутствии
бромистого алюминия
Дипольный момент
p. 1018
0,81 ±0,02
0,554 (0,65)
1,00-1,03
1,29
1,19
1,51±0,10
2,18; 2,24±0,10
3,67
г-0,6
1 0,65 (0,5)
1,48
1,21
1,30
к
«=3
сз
Д
>>
СЗ О-
4 £
3?-
Cj£
[24]
[35, 62]
[61]
[6, 63]
[62]
[63]
[62, 61]
[61]
[641
[29, 30]
[31]
[32]
[33. 34]
частично ионный характер. Это следует, например, из того, что одежъ-
ядерные расстояния, определенные электронографически, оказались
меньше, чем полусумма межъядерных расстояний в молекулах
галогенов, из которых образованы молекулы межгалоидных соединений,
т. е. длина связи в молекулах АВ меньше длины ковалентной связи.
Значения межъядерных расстояний указаны в табл. 5, а также при
описании отдельных межгалогенидов.
К такому же выводу в отношении характера связи в
двухатомных молекулах межгалогенидов можно прийти, сопоставляя
величины энергии диссоциации этих молекул с значениями,
вычисленными из энергии диссоциации исходных галогенов по формуле
Дав =о (Da2 +Дв2)- Как показано в табл. 5 [36], величины энергии
диссоциации несколько превышают вычисленные значения
вследствие частично ионного характера связи.
Изучение магнитной восприимчивости некоторых фторидов
галогенов (табл. 2) показало, что все они диамагнитны, причем
наблюденные значения оказались несколько меньше вычисленных из
величин ионной восприимчивости; ,по мнению авторов этой работы [68],
причиной данного 'явления может быть частично ковалентный
характер связи.
Как показывают величины константы Трутона (табл. 1),
большинство межгалоидных соединений в жидком состоянии
ассоциированы. Ассоциация молекул этих соединений, вероятно, отражается
и на величинах их дипольных моментов и на некоторых других
молекулярных константах.
13
Таблица 5
Межъядерные расстояния и энергии диссоциации гал генов и межгалоидных
соединений
Молекула
F2
С12
Вг2
J2
C1F
C1F
BrF
BrF
BrCl
BrCl
JBr
JBr
JC1
JCl
Межъядерное
расстояние,
о
A
1,435
1,989
2,284
2,667
1,625
1,712
—
1,86
(2,U;
2,14
(2,48)
2,48
2,321
2,328
Энергия
диссоциации, эв
2,60
2,481
1,971
1,5422
2,617
2,54
—
2,29
2,26
2,23
1,808
1,76
2,153
2,01
1
Га» эв*~
—
—
—
0,28
—
—
—
0,17
—
0,22
—
0,38
—
Примечания: 1. Вторые значения межъядерных расстояний и энергий
диссоциации, указанные при каждом межгалогениде, вычислены как полусумма тех
же величин для двух соответствующих галогенов
2. А—отклонение от нормальной ковалентной связи, например для хлористого
иода Д=2,153—2,01=0,143 эв; /0143=0,38.
О полярности межгалоидных соединений свидетельствуют также
их способность проводить ток не только в растворах, но и в
индивидуальном (в жидком) состоянии и их способность подвергаться
электролизу с выделением галогенов на электродах. Так, при
электролизе хлористого и бромистого иода как в расплавленном
состоянии, так и в растворах наблюдалось выделение на катоде иода, а на
аноде —хлора или брома (Фарадей [37], Солли [37], Брунер и Бе-
кьер [38"|, Брунер и Галецкий [39], Плотников и Рокотян [40], Сандо-
нини и Бергелло [41], Фиалков и Каганская [42] и др.).
Электропроводность межгалоидных соединений типа АВ обычно
значительно превышает электропроводность составляющих их
галогенов.
Так, в то время как электропроводность жидкого иода (130°)
порядка 10~5, а жидкого брома—10~13, электропроводность
бромистого иода (40°) — порядка Юг4 ом^смг1. Еще в большей степени
выявляется это различие для хлористого и треххлористого иода,
электропроводность которых порядка 10~3 ом^смг1. Из исследованных в этом
отношении межгалоидных соединений наименьшей
электропроводностью — 3-10-9 омг1см~1 (0°)—обладает трехфтористый хлор.
Есть основания полагать, что электропроводность хорошо
проводящих ток межгалоидных соединений преимущественно ионного
типа, чего нельзя сказать об электропроводности иода [43].
Природа ионов — продуктов собственной электролитической
диссоциации межгалоидных соединений — большей частью установлена
14
лишь косвенными путями, на основании явлений, наблюдаемых при
электролизе межгалоидных соединений или на основании их
поведения при химических реакциях.
Процесс электролитической диссоциации межгалоидных
соединений в жидком состоянии и в растворах осложняется тем, что их
молекулы ассоциированы. В простейшем виде — для димерных
молекул— он может быть представлен следующими схемами:
(ЛС1)2^Л++ЛС1Г [41,42];
(BrF3)2;!BrF2+ + BrF4-[44];
(JF5)2 Z JF4"U + JF6~ [6, стр. 458].
В неводных растворах необходимо также принимать во
внимание возможность образования проводящих ток комплексных
соединений со многими растворителями, особенно с теми, которые имеют
характер оснований, например: C5H5N • JC1, C5H5N • 2JC1 [45],
CH3CONH2 • JC1 [46]. Эти соединения образуют комплексные катионы
типа i[C5H5N-J]+ и т. п.
Как будет показано далее, такое состояние межгалоидных
соединений (электролитическая диссоциация ассоциированных
молекул) имеет большое значение в процессах их химического
взаимодействия с галогенидами других элементов. Оно отражается и на
процессах электролиза межгалоидных соединений, вообще
протекающих довольно сложно. Так, например, при электролизе неводных
растворов хлористого иода — в нитробензоле [39, 42], уксусной
кислоте [41] ив некоторых других растворителях — наблюдается
перенос обоих 'галогенов в анодную часть, хотя отношение J : С1 в като-
лите увеличивается, а в анолите — уменьшается.
Электрохимические свойства межгалоидных соединений и их
растворов будут более подробно рассмотрены в главах IX и X.
Химические свойства межгалоидных соединений обусловлены
прежде всего их химическим составом и поэтому они имеют много
общего со свойствами свободных галогенов. Но полярный характер
молекул межгалоидных соединений и их способность
поляризоваться, электроположительное состояние одного из атомов галогенов,
входящих в их состав, придают межгалоидным соединениям ряд
особенностей в их химических свойствах по сравнению с образующими
их галогенами.
Большое влияние на поведение межгалоидных соединений в
химических реакциях оказывает природа растворителя. Влияние
растворителей проявляется в нескольких направлениях:
1. Сольватирование молекул межгалоидных
соединений и образуемых ими ионов
молекулами полярных растворителей. Как и для иода, этот
процесс является одним из основных факторов, обусловливающих
различие в окраске растворов. Так, в тех растворителях, обычно
неполярных или малополярных, которые дают с иодом фиолетовые
растворы (ССЦ, CS2, CHCI3, C6Hi4 и др.), хлористый иод растворяется
с красно-бурой окраской, но он же образует желтые растворы в по-
15
■лярных растворителях (СН3ОН, С2Н5ОН, (С2Н5)20, C5H5N,
СН3СООН и др.), дающих с иодом бурые растворы, в которых
последний находится в сольватированном состоянии. Как будет
показано далее, для хлористого иода удалось установить образование
■соединений с некоторыми растворителями второго типа.
2. Поляризация молекул межгалоидных
соединений под влиянием растворителя. Так, дипольный
момент хлористого иода, в газообразной фазе равный 0,65Д в
четыреххлористом углероде и циклогексане увеличивается до 1,48D
[29, 30]. В связи с этим явлением возможно изменение характера
связи в межгалоидных соединениях в растворах, и связь
галоген—галоген может стать ионной. Увеличение же концентрации
положительно заряженных ионов галогена значительно повышает химическую
активность межгалоидных соединений.
3. Характер диссоциации м е ж та л о и ди ы х
соединений в растворах. Наряду с термической
диссоциацией на молекулы галогенов межгалоидные соединения могут
подвергаться также электролитической диссоциации. Для хлористого
иода эти равновесия можно представить такой схемой:
J2 + Cl2 ^ (JC1)2 ^ J+ + JC12~ ^ 2J+ + 2С1-
Для растворов хлористого иода в неполярных растворителях с
малой диэлектрической постоянной характерна «гомолитическая»
диссоциация, в растворителях же с большой диэлектрической
постоянной, в которых хлористый иод образует желтые растворы,
преобладает электролитическая («гетеролитическая») диссоциация.
Поэтому такие растворы более или менее хорошо проводят
электрический ток и в них можно осуществлять реакции ионного типа. Так,
например, хлористый иод, растворенный в четыреххлористом
углероде, не реагирует с перхлоратом серебра, в нитробензольном же
растворе * быстро осаждается хлористое серебро [47].
Растворитель оказывает сильное влияние также на реакции
замещения водорода в органических веществах (фенолах, аминах
и т. п.) при действии на них межгалоидных соединений. Этот
процесс наиболее хорошо изучен для хлористого иода, причем оказалось,
что, в зависимости от природы растворителя, может преобладать
то реакция хлорирования, то реакция иодирования
органических веществ [48]. В растворителях с большой диэлектрической
постоянной, в которых имеется равновесие (JC1)2 ^12J++2C1",
органические вещества преимущественно иодируются [49]. Более подробно
об этих реакциях и их практическом значении будет сказано далее.
Природа галогенов, из которых образовано данное межгалоидное
соединение, и тип соединения (АВ—АВ7) влияют на характер
химической связи в молекулах межгалоидных соединений и их
полярность, на устойчивость при нагревании и в растворах и на
химические свойства. Поэтому наряду с общими химическими свойствами
отдельные межгалоидные соединения проявляют свойства более ха-
* Дипольный момент нитробензола 3,95D, его диэлектрическая
постоянная Э6Д
16
рактерные для одних и менее — для других представителей этой
группы.
Необходимо также принять во внимание, что химические
свойства некоторых межгалоидкых соединений изучены еще недостаточно
хорошо.
Основные химические свойства межгалоидных соединений
таковы:
а) они являются сильными окислителями по" отношению к
разнообразным неорганическим и органическим веществам. Сюда, в
частности, относятся: реакции с простыми веществами,
сопровождающиеся образованием галогенидов соответствующих элементов;
взаимодействие галогенидов иода с иодидами или галогенидов брома с
бромидами, в результате чего происходит выделение свободного
иода или брома *, например,
JC1 + KJ - КС1 + J2,
JC13 + 3KJ = 3KC1 + 2J2,
BrF3 + 3KBr-3KF-f-2Br2 и т. д;
б) при взаимодействии с различными неорганическими
соединениями— окислами, солями и др. —они часто образуют
соответствующие галогениды — простые или комплексные;
в) некоторые межгалоидные соединения (JC1, JBr, BrCl и др.)
способны присоединяться к непредельным органическим веществам
по месту кратной связи;
г) они обладают способностью галоидировать определенные
органические соединения — фенолы, амины и т. д. — путем замещения
атомов водорода на галогены и соединяются с органическими
азотистыми или кислородными основаниями с образованием
комплексных соединений типа (CH3)3N-JC1, C5H5N-JC1 [45], C5H5N • 2JC1
[45], С4Н8СЫС1 ,[51], C4H802.JF5 152], C5H5NJCI3, C5HnN.JCl
[53] и т. д.;
д) они образуют комплексные галогениды с галоидными
соединениями других элементов, например, КС1 • JC1 [54, 42, 55], PCI5 • JC1
[56, 57], РВЫВг [58], BrF3-KF [59], BrF3-SbF5 [60] и т. д.
Многие другие реакции будут указаны при описании отдельных
межгалоидных соединений.
Уже этот неполный перечень тех типов химических реакций,
которые могут быть осуществлены при помощи межгалоидных
соединений, показывает возможное значение этой группы неорганических
веществ в химической практике.
ЛИТЕРАТУРА
1. N. V. S i d g w i с k, Ann. Rep. Progress Chem., 30, 128 (1933).
2. Л. М. Дубников, Усп. химии, 16, 193 (1947).
3. H. S. Booth, J. Т. Pinks ton, Chem. Rev., 41, 421 (1947).
4. Г. С. Бус, Д. Т. Пинкстон. сб. «Фтор и его соединения», ИЛ, М.,
1953, т. 1, 375.
* Реакции этого типа являются косвенным подтверждением полярности
межгалоидных соединений.
2—1419 17
5. A. G. S h а г р е, Quart. Rev., 4, 115 (1950).
6. Н. Н. Г р и н в у д, Rev. of pure and appl. Chem., 1, 84 (1951); Усп.
химии, 22, 445 (1953).
7. Д. И. Менделеев, Основы химии, изд. 13, М.-Л. (1947), стр. 350.
8. Б. В. Некрасов, Курс общей химии, изд. 9 (1952), стр. 856.
9. Н. С. К у р н а к о в, С. Ф. Жемчужный, М. М. 3 а с е д а-
те л ев ЖРФХО, 47, 871 (1915); Собрание избранных работ Н. С. Курна-
кова, ГОНТИ, т. II, 254 (1939).
10. G. T a m m a n, Z. anorg. Chem., 49, 113, (1906); 55, 289 (1907).
11. Ф. Э ф Р а и м, Неорганическая химия, Госхимиздат, ч. 1 (1932), стр. 59.
12. J. L. G а у - L u s s а с, Ann. chim phys., 91, 5 (1814).
13. Н. Davy, Trans. Roy. Soc. (London), 104, 487 (\8\4).
14. A. J. В a 1 а г d, Ann. chim. phys. 32., 337 (1826).
15. A. J. В a 1 а г d, Ann. chim. phys. 32 , 371 (1826).
16. H. Kam merer, J. pr. Chem., 85, 452 (1862).
17. P. Lebeau, С. г., 141, 1018 (1905); E. В. R. P r i d e a x, J. Chem.
Soc, 89, 316 (1906).
18. O. Ruff, F. A s с h e r, Z. anorg. allg. Chem., 176, 258 (1928).
19. O. Ruff, H. К г u g, Z. anorg. allg. Chem., 190, 270 (1930).
20. O. Ruff, R. F. Keim, Z. anorg. allg. Chem., 193, 176 (1930).
21. O. Ruff, W. Menzel, Z. anorg. allg. Chem., 202, 49 (1931).
22. O. Ruff, А. В г a i d a, Z. anorg. allg. Chem., 214, 81 (1933).
23. E. F e s s e n d e n, J. Chem. Educat., 28, 619 (1951).
24. O. Ruff, Z. ang. Chem., 46, 739 (1933).
25. В. Клемм, Усп. химии, 4, 100 (1935).
26. Abegg's Handbuch der anorg. Chem., B- IV, Abt. 2, 461 (1913).
27. P. С E. M e e r u m - T e r w о g t, Z. anorg. Chem., 47, 209 (1905).
28. D. A. Gilbert, A. Roberts, P. A. G r i s w о 1 d, Phys. Rev.,
76, 1723 (1949).
29. С. Н. Т о w n e s, F. R. Merritt, B. D. Wright, Phys. Rev.,
73, 1334 (1948).
30. K. F. Luf t, Z. Phys., 84, 767 (1933).
31. F. Fairbrother, J. Chem. Soc, 847 (1936).
32. И. А. Ше к а, ЖФХ, 23, 885 (1949).
33. И. А. Шека, ЖФХ, 23, 1180 (1949).
34. И. А. Шека, 3. А. Шека, ДАН, 69, 197 (1949).
35. D. W. M a g n u s о n, J. Chem. Phys., 20, 229 (1952).
36. Г. Г л о к л е р, сб. «Фтор и его соединения», ИЛ, М., 1953, т. 1, 276;
R. A. Durie, A. G. G а у d о n, J. Phvs. Chem., 56, 316 (1952).
37. Цит. по Gmelin — Kraut's Handb., 8, 614, 635 (1931).
38. L. Bruner, E. Bekier, Z. Elcktroch., 18, 368 (1912).
39. L. Bruner, A. Galecki, Z. phys. Chem., 84, 515 (1913).
40. В. А. Плотников, В. Е. Рокотян, ЖРФХО, 47, 723 (1915).
41. С. Sandonini, N. Berghello, Atti d. Reale Accad. d. Lincei,
25 (6), 46 (1937).
42. Я- А. Ф и а л к о в, К. Я- К а г а н с к а я, ЖОХ, 16, 1961 (1946); 18,
289 (1948).
43. М. А. Рабинович, ЖРФХО, 58, 229 (1926).
44. A. A. Banks, H. J. Emeleus, A. A. W о о 1 f, J. Chem. Soc,
2861 (1949).
45. Я- А. Ф и а л к о в, И. Д. Музыка, ЖОХ, 18, 1205 (1948).
46. Я. А. Фиал ков, И. Д. Музыка, ЖОХ, 18, 802(1948).
47. Н. W. Cremer, D. R. Duncan, J. Chem. Soc, 2031 (1932).
48. L. L. Lam bourne, P. W. Robertson, J. Chem. Soc, 1167
(1947).
49. F. W. Ben net, A. G. Sharpe, J. Chem. Soc, 1383 (1950).
50. A. Pictet, G. Kraft, Bull. (3), 7, 73 (1892).
51. H. R h e i n b о 1 d, R. Boy, J. pr. Chem. (2), 129, 273 (1931).
52. A. F. Scott, J. F. В u n n e t t, J. Am. Chem. Soc, 64, 2717 (1942).
53. R. С е г t a n e s с u, M. P о n i, Anal. Acad. Republ. populare Romane.
Ill, 140 (1950).
18
54. J- С о r n о g, Н. Н о г г a b i n, R. К а г g e s, J. Am. Chem. Soc.
60, 429 (1938).
55. Я- А. Фиалков, О. И. Шор, ЖОХ, 19, 1787 (1949).
56. Е. В a u d г i m о n t, С. г., 55, 361 (1862).
57. Я- А. Фиалков, А. А. Кузьме н ко ЖОХ, 19, 812 (1949).
58. А. А. Кузьменко, Я- А. Фиалков, ЖОХ, 19, 1007(1949).
59. A. G. Sharpe, H. J. Emeleus, J. Chem. Soc, 2135 (1948).
60. A. A. Woo If, H. J. Emeleus, J. Chem. Soc, 2865 (1949).
61. M. T. Rogers, H. B. Thompson, J. L." S p e i г s, J. Am.
Chem. Soc, 76, 4841 (1954).
62. M. T. Rogers, R. D. P r u e t t, J. L. S p e i r s, J. Am. Chem.
Soc, 77, 5280 (1955).
63. M. T. Rogers, R. D. P r u e t t, H. B. Thompson, J. L. S p e-
i г s, J. Am. Chem. Soc, 78, 44 (1956).
64. G. Fro h lie h, W. J о s t, Chem. Ber., 86, 1184 (1953).
65. M. C. Ma lone, A. L. F e r g u s s о n, J. Chem. Phys. 2,99(1934).
66. M. T. Rogers, J. G. Malik, J. L. S p e i г s, J. Am. Chem. Soc.,
78, 46 (1956).
67. L. Stein, R. V о g e 1, W. H. L u d e w i g, J. Am. Chem. Soc, 76,
4287 (1954).
68. M. T. Rogers, M. B. P a n i s h, J. L. S p e i r s, J. Am. Chem.
Soc, 77, 5292 (1955).
69. И. Г. Р ы с с, Химия фтора и его неорганических соединений, Госхимиз-
дат, М., 1956, гл. VII.
ГЛАВА II
ФТОРИДЫ ХЛОРА
1. ФТОРИСТЫЙ ХЛОР C1F
Получение
Фтористый хлор был получен Руффом и Ашером в 1928 г. [1].
Этому предшествовал ряд неудачных попыток получить соединение
хлора с фтором. Муасеан [2, 3] смешивал хлор и фтор при
комнатной температуре и показал, что в этих условиях они не реагируют
друг с другом. Лебо [4] пришел к такому же заключению на том
основании, что фтор, действуя на хлориды, выделяет из них хлор, не
соединяясь с ним. Лебо растворял фтор в жидком хлоре при —80°,
но при этом нельзя было заметить образования какого-либо
определенного соединения этих галогенов; при охлаждении этого раствора
до температуры замерзания хлора из него выделялся фтор. Муассан
и Лебо пришли к заключению, что фтор и хлор не соединяются друг
с другом.
Руфф и Цеднер [5] пытались получить соединение фтора с
хлором пропусканием смеси этих газов через вращающуюся
электрическую пламенную дугу с последующим быстрым охлаждением газов
или действием искрового разряда (от индукционной катушки) на
смес^ сжиженных хлора и фтора. При конденсации газов,
подвергшихся действию электрической дуги, Руфф и Цеднер получили
жидкость, окрашенную в бурый цвет. Причина окраски тогда не была
ясна, однако этот факт, как писал Руфф [6], давал основание
считать возможным образование фтористого хлора.
В дальнейшем Руфф решил использовать реакции между
хлором в момент выделения и фтором и, исходя из этого, изучил
действие газообразного хлористого водорода на жидкий фтор [1]. Так
как эта реакция протекает очень бурно, она проводилась при
низкой температуре (сжиженный фтор охлаждался жидким воздухом),
в условиях избытка фтора и медленного течения процесса. При
поступлении хлористого водорода в атмосферу фтора (над жидким
фтором) появляется зеленое свечение и образуются белые хлопья,
медленно осаждающиеся в жидком фторе. Среди продуктов реакции
обнаружены фтористый водород и хлор. В одной из фракций,
кипящей около —100°, содержалось еще одно вещество, которое выделя-
20
ло из йодистого калия иода немного более чем вдвое по сравнению
с рассчитанным для содержания хлора. Это давало основание
предположить, что данное вещество .представляет собой фтористый хлор,
образующийся по реакции
HC1 + F2=HF + C1F.
Но изолировать его не удалось — его количество было очень
малым, к тому же оно взаимодействовало со стеклом и превращалось
в SiF4 и бурый С120.
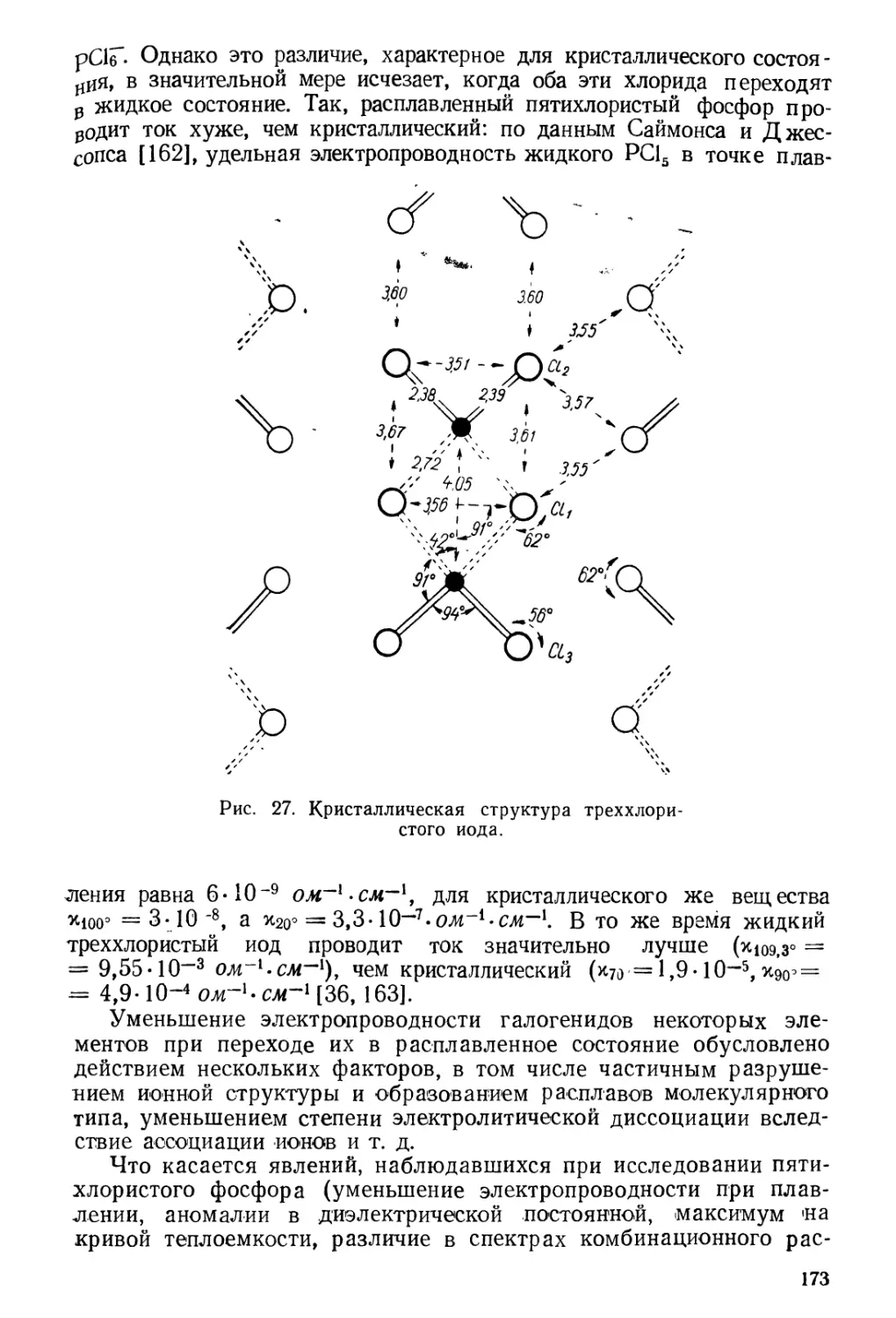
Фтористый хлор удалось получить непосредственным
соединением этих элементов — при горении хлора во фторе с последующей
Рис. 4. Аппарат для получения фтористого хлора.
конденсацией продуктов реакции [1]. При фракционировании в
стеклянных сосудах конденсаты содержали много SiF4, СЬО и С12, что
в некоторых случаях было причиной взрывов. Наличие С120 в
конденсатах свидетельствовало о том, что первично образовавшийся
фтористый хлор взаимодействовал со стеклом, возможно, по
реакции
4ClF + Si02=SiF4 + 2Cl20.
Кварц оказался более устойчивым, но для получения более
чистого продукта реакционный сосуд был изготовлен из меди.
Руфф и Ашер [1] испытали несколько вариантов получения
фтористого хлора, различающихся выбором аппаратуры и методом
отделения от исходных веществ. Так, по одному из вариантов реакция
между фтором и хлором происходила в медном блоке, нагреваемом
на масляной бане до 250°. Этот блок (рис. 4) был соединен с
несколькими ловушками, охлаждаемыми до —100, —150 и —185°, для
разделения продуктов реакции. Схема этой установки приведена на
рис. 4. Сосуды А, В, С, D, Е и F сделаны из меди, сосуд G—из
кварца, а сосуд Н — из обычного стекла. Сосуды А наполнены
фтористым калием для поглощения фтористого водорода,
сопровождающего фтор, идущий из электролизера; В — реакционный сосуд —
медный блок; сосуды С, D, Е и F охлаждаются до —100°, они служат
для улавливания фтористого водорода и хлора; в ловушке G
конденсируется основное количество фтористого хлора, а в ловушке F—
не вошедший в реакцию фтор.
Чтобы обеспечить длительный и без взрыва процесс соединения
фтора с хлором, необходимо было нагреть медный блок до 250°. еще
до введения хлора в наполненную фтором аппаратуру.
21
/
\
TVtVT
Фтористый хлор был выделен фракционированной конденсацией.
В сосуде G конденсировалось большое количество желтоватой
жидкости, которая в жидком.воздухе застывала в
слабо-желтоватого цвета массу. Так как температура плавления хлора составляет
102, а фтора —223° (т. е. хлор при —150° находится в твердом
состоянии, а фтор при —185° еще жидкий), то Руфф и Ашер
заключили, что они имеют дело с новым веществом — фтористым хлором.
Состав этого вещества и его формула C1F были
установлены химическим анализом и
определением молекулярного веса по плотности газа.
Дальнейшая очистка полученного таким
образом фтористого хлора описана в работе Руф-
фа и Лаасса [7].
Методы получения фторидов галогенов, в том
числе и фтористого хлора, были значительно
усовершенствованы Квасником, впервые
опубликовавшим их в «Руководстве по препаративной
неорганической химии» [62]. По Кваснику, C1F
и C!F3 получают в цилиндрическом сосуде из
никеля или монель-металла при 400°.
Реакционные газы проходят через железный холодильник
и две железные ловушки. В первой из них,
погруженной в сухой лед, конденсируются хлор и
C1F3, во второй ловушке, охлаждаемой жидким
кислородом, конденсируется C1F [62, стр. 99].
Реакция образования фтористого хлора из
элементов экзотермична. Горение смеси фтора
с хлором иногда сопровождается сильным
взрывом. Руффу удалось осуществить реакцию
горения смеси этих газов в условиях,
предотвращающих взрыв, в приборе, изображенном на
рис. 5 [8]. По Руффу с сотрудниками [1, 6], эта
реакция имеет равновесный характер, причем
даже при большом избытке фтора, газовая смесь в состоянии
равновесия содержит свободный, не вошедший в реакцию хлор.
Образование фтористого хлора они объясняли действием хлора в
момент выделения на фтор и показали, что можно найти
катализатор, который позволит проводить эту реакцию при более низких
температурах и с лучшими выходами.
Однако Фреденгаген и Крафт [91, исходя из соображения, что
между фтором и хлором должно проявляться значительное сродство
и что фтор, который применяли Руфф с сотрудниками, содержал
значительную примесь кислорода, повторили опыт Руффа. На
основании качественных данных они пришли к заключению, что Руфф
не имел дела с состоянием равновесия.
В дальнейшей работе Руффа, выполненной им совместно с
Кругом, было обнаружено, что в смеси хлора с определенным избытком
фтора (против эквимолярного соотношения) образуется, наряду с
фтористым хлором, также трехфтористый хлор [10]. При взаимодей-
/
Рис. 5. Прибор для
реакции горения
хлора во фторе.
• 22
ствии же эквимолярных количеств фтора и хлора образуется
практически только фтористый хлор. Это следует из опытов Шмитца и
Шумахера [11], которые показали, что в смеси равных объемов хлора
и фтора в пределах 250—350° не происходит изменения упругости
пара по отношению к сумме парциальных давлений фтора и хлора
C12 + F2=2C1F.
Позднее, в 1948 г. Домань и Нейдорфер сообщили, что они
получили фтористый хлор взаимодействием равных объемов фтора и
хлора при 220—230° в аппаратуре из медных трубок. По данным этих
авторов [12], при нагревании смеси фтористого хлора с двумя
объемами фтора до 270—280° образуется трехфтористый хлор по реакции
C1F + F2=C1F3.
В то же время C1F3, реагируя с рассчитанным количеством хлора
при 350°, почти полностью переходит во фтористый хлор [11, 13]
C1F3 + C12=3C1F.
Вследствие этого, а также благодаря большей доступности трех-
фтористого хлора, получение фтористого хлора по последней
реакции легче осуществить, чем путем синтеза его непосредственно из
элементов.
Физические свойства
Физические и химические свойства фтористого хлора впервые
были описаны Руффом и его сотрудниками в ряде работ [1, 6—8, 10,
14—16]. В литературе, посвященной фтористому хлору, имеется ряд
противоречивых сведений и невыясненных вопросов, что
объясняется трудностью точного определения физических и химических
свойств фтористого хлора, вследствие его (высокой химической
активности.
Фтористый хлор при комнатной температуре представляет собой
бесцветный газ с сильным своеобразным запахом, отличающимся от
запаха фтора и хлора. В сосудах из плавленого прозрачного кварца
фтористый хлор имеет оранжевый оттенок, вследствие присутствия
небольших количеств С120, образующегося при взаимодействии
Si02 с фтористым хлором.
При охлаждении фтористый хлор конденсируется в желтоватую
жидкость, в твердом состоянии он представляет собой белое
вещество, плавящееся при —155,6° [16]. Руфф и Лаасс [7] установили, что
зависимость упругости пара (в мм рт. ст.) жидкого фтористого
хлора от абсолютной температуры в пределах от —150 до —105°
выражается таким уравнением:
lgp = 15)738_^+1'538-105-
Т Т2
Температура'кипения (760 мм рт. ст.), определенная
экстраполяцией по кривой упругости пара, оказалась равной —100,1° [7, 16].
23
Плотность, (измеренная при температуре кипения, а4° равна 1,62.
Пользуясь интегрированной формой упрощенного уравнения
Клаузиуса — Клапейрона lgP =^-^^7-^- +С , Руфф и Лаасс [7]
рассчитали теплоту испарения равной 2,27 ккал-моль~г и константу
Трутона—13,17 кал-град'1-моль-1. Как отмечает Гринвуд '[17,
стр. 447], пересчет результатов, полученных Руффом и Лаассом,
показывает, что только точки, соответствующие трем самым низким
температурам, отклоняются от линейной зависимости между lgP и
j, и на этом основании можно получить значение теплоты
испарения — 5,26 ккал-моль'1, характеризующее фтористый хлор как
ассоциированную жидкость. Действительно, Руфф и Круг [10] указали,
что причиной отклонения от линейной зависимости между lgP и
~ является, вероятно, -наличие небольших. количеств примеси C1F3.
Повторив произведенные ранее вычисления, они нашли для теплоты
испарения величину—4,80 ккал^моль"1; константа Трутона, по их
данным, равна 28,0 кал-град~х-моль~х.
Критическая температура фтористого хлора, вычисленная
Руффом и Лаассом [7] из температуры кипения, оказалась близкой к
■—14°. Эти же авторы определили теплоту образования фтористого
хлора. Для этих расчетов они измерили теплоту реакции C1F + H2=
= HC1 + HF, равную 58,6 кал.
CIF + Н2 - НС1 + HF + 58,6 ккал;
I/2F2+1/2H2= HF + 62,3 ккал;
1/2С12 + У2Н2 - HC1 + 22 ккал;
1/2С12 + 1/aF2 - C1F+ 25,7 ккал - моль—х.
Позднее Руфф и Мендель [14], приняв более точные значения для
теплот образования НС1 и HF, вычислили теплоту образования
фтористого хлора: У2 Cl2 +V2F2 = ClF + 27,4 ккал-моль~К
Однако Варгафтиг [18] показал, что вычисленная таким образом
теплота образования фтористого хлора не согласуется с величиной,
принятой для энергии диссоциации фтора на нормальные атомы. На
основании изучения спектров поглощения фтористого хлора в ульт-
о
рафиолетовой и видимой областях (между 3500 и 7000 А и особенно
о
при 4800 А) он пришел к выводу, что теплота образования
фтористого хлора должна быть значительно меньше величины
27,4 ккал-моль~х. Это заключение подтвердили Шмитц и Шумахер
[11], которые вычислили теплоту образования фтористого хлора из
данных, полученных при термохимическом исследовании при 18°
следующих реакций:
NaC1KPHcr + V2F2(r) - NaFKpHCT + ViCl2(r) + (39,5 ± 0,5) ккал. моль-*;
№С1крист + ClF(rr NaFKpHCT + Cl2(r) + (24,5±Q,1) ккал: моль-* ^
1/2Cl2r-h1/2F2r- ClFr + (15 ± 0,5) ккал. моль-*
24
Найденная ими теплота образования фтористого хлора
15 ±0,5 ккал-моль~х довольно близка к величинам 13,4 и
12,0 ккал-моль'1, которые получили эти же авторы [13] и Варгаф-
тиг [181 из спектроскопических измерений, исходя из значений
энергии диссоциации фтористого хлора на возбужденный атом хлора и
невозбужденный атом фтора или наоборот (ом. также стр. 27 и
табл. 12). Близкие, но несколько меньшие величины дали Вике [19]
(11,6 ±0,4 ккал-моль'1) на основании калориметрического
определения теплоты реакции между газообразными фтором и хлором, а
затем Вике и Фриц [19] (11,7±0,5 ккал-моль~х) путем измерения
теплоты, выделяющейся при адиабатическом взрыве смесей фтора
с хлором.
Коунт и Барроу [20] на основании анализа экспериментальных
данных, полученных прежними исследователями, определили
теплоту образования фтористого хлора 12,3±1 ккал-моль~х. Однако
другие литературные данные, подробно обсужденные Рыссом в
обзорной статье, посвященной энергии диссоциации фтора и сродству
фтора к электрону [21], указывают на еще меньшие величины. Поэтому
Рысс, отмечая значительные расхождения данных о теплотах
образования фтористого хлора *, указывает, что даже если отбросить
первые, несомненно грубо ошибочные, определения, то все же
разные значения их колеблются от 2 до 20 ккал-моль~].
Термодинамические свойства фтористого хлора рассмотрены в
ряде работ, включая и цитированные выше. Поттер [22], исходя из
спектроскопических данных, вычислил функцию свободной энергии
(F0—Н°)/Т, абсолютную энтропию и теплоемкость фтористого
хлора для идеального газового состояния (760 мм рт. ст.) и интервала
температур от 298,16 до 2000° К. Эти же термодинамические
свойства, а также функцию теплосодержания (Я0—Н0°)/Т и константы
равновесия реакции диссоциации фтористого хлора и других
межгалоидных соединений для того же интервала температур рассчитали
Коул с сотрудниками [52].
Наиболее поздняя сводка ряда термодинамических свойств
межгалоидных соединений содержится в работе Эванса, Манзона и Ваг-
мана [53], опубликованной в 1955 г. в журнале национального бюро
стандартов США. Эти авторы обработали многочисленные
литературные данные, относящиеся к термодинамической характеристике
ряда газообразных соединений галогенов, в том числе и 10
межгалоидных соединений. Они рассчитали или отобрали наиболее
точные, по их мнению, значения теплоты образования A Я/0, функции
свободной энергии (F0—Но°)/Т, теплосодержания (Я0—Я0°),
функции теплосодержания (Я0—Н0°)/Т, энтропии 5°, теплоемкости Ср>
свободной энергии образования A Ff° и логарифма константы
равновесия реакции образования межгалоидных соединений для
* Такие же расхождения в термодинамических константах известны и для-
других межгалоидных соединений, особенно для фторидов галогенов и
хлористого брома.
25
9S
ел 4*. со to —
о о о о о
о о о о о
1 1 1 1 1
СЛ СЛ СЛ СП СП
J-J CD jD£n^
^Ъо "tO "СП 00
4^ cd ел со оо
ЬЭМЮЮО
00 ОО ОО ОО 00
со со to to —
cd to -j — ел
CD О — CD СО
CD CD CD CD CD
• ^ел ел jt- оо оо
"оо~—Ъ1"оо "о
ооо юосо
оо -^jcooo со
Ю — ОСС 00
ел cd -о оо со
4^ 4^ ел ел cd
ее оо tocooo
о осо toco
<р оо ро оо ро
"О CD О "cD 00
О00 CD CO CD
ILLLL
со со со со со
СП 4^ 4^ 4*- 4^
OCDCD00 00
1 1 1 1 1
1 I 1 ! 1
СЛ СЛ 4^ 4^ 4^
—' о со -<i cd
Ю — ОсО 00
JSDJOJO tO Ю
"ьоЪо"слЪ">"со
О*. OCD —
СО СО СЛ 4^ -41
о со оо --I cd
СО СО СО СО СО
о о о о о
1 1 1 1 1
сл ел сл ел ел
_4ь- GOjOj— О
"•—"to"со"to о
О CD tO -v) 00
-<j о со сл сл
00 ~^^1 ~<1 -4
О CD CD -Ч CD
00 CD О CD СЛ
o~<i to oco
cd CD cd сл ел
JOj— Oj£>j<l
"—"to"to "о"-ч
00 СЛ tO CD 4*.
-4 ^vj СЛ СЛ 4^
00 -vj CD СП 4^
О — CO 4^ СЛ
оо со ю ел со
о -ч —со ел
ОСО CD О*
00 O0CCQO0O
"оо оо^Ъэ'сл
СП ОСО 4^ —
LLLLL
со со со со со
4^ 4^ 4^ -^ 4^
^1 --1 CD CD СЛ
Mill
1 1 i 1 1
4*. 4^- 4^ 4> 4^
ел j^ со to —
~<1CD СЛ 4^ CO
O0O0 OO^OX
"—'"сл^о"^."^-
ОО — tO 4^ 4^
СЛ tO — CD CD
ел 4^ со to to to
ОООСОЧСЯ
ooopowoo
CDCD
1 1 1 1 1 1
4^» 4^»- 4^- 4^ 4^- 4^
00 J-J,4* ^jiwO
"•O "o "cD "CD CO "CD
О 4^ CD tO О -J
tO -4 CD tO О tO
-vj -J -J -4 ^1 ^ О
СЛ СО — — О О
О tO 4^ 4^ СО CD
CD CD 4^ О ООО
сл сл ел ел сл ел
CD ,4=* jOJOj— СО О
"tO Ъо —"о "СО "--4
О ^4 — CD СО СО
00 CD О tO 00 tO
со to to to ^-ь-
^1 CD — — CD -J
СЛ CO 4^ tOCO CD
CO —CO 00 00 СЛ O
О CD tO CO CD О
QO 00 jOj<1 j*4 ^J О
со "ocd"cdui*4^
CO -J 00 -Ч СЛ 4^
I.LLILLL
CO CO CO CO CO CO CO
4^ 4^ 4^ 4^ 4^ 4^ 4^
4^ CO tO tO — —
CO CO 00 CD
1 1 1 1 1 1 1
1 1 1 1 1 1 1
4^ CO CO CO CO CO CO
О CD -vj -o ^j -о ^
— 000 -^14^ tO
— CDCD —
wCD j<| CO О j— j—
~—*Cn"o "o"oXo I
tO CD CO CO OCD 1
CD 4*- CO CO О 4^
— СП О СЛ
о
Я
X *
О» CL»
| J
X
х/ О
х ^
л (\\
1°
li
>
—
a
л-гр
хмс
£ »
сг Ci,
1_
X
^
i
о
о*
Г
_
X ^
1 ^£
X
ккал
£
О*
1
a
^
v
о
. 5а»
О*
►_
0Q
**s
^ 1
о 1
1
^
оо [
^ 1
о 1
1
оо j
?*
Со
о
1
^
о
I
^
оо
Г
>
^
^
о 1
>
° '
о
ш
о
о
н
ш
р
о
S
о»
температур от 0 до 1500° К*. В табл. 6 приведены величины этих
свойств для фтористого хлора по [531 **.
На основании электронографического исследования фтористого
хлора [23] *** установлено расстояние между центрами атомов
о
1,63 ±0,01 А. Спектроскопические исследования дали величины
о
1,625 118] и 1,6281 А [24]. Эти величины очень близки к сумме нор-
о
мальных кшалентных радиусов хлора и фтора (0,64 + 0 99=1,63 А
[26]), но несколько меньше полусуммы расстояний С1—О и F—F в
газообразном состоянии: !/2 (1,894+1,46) = 1,722 А [26] ****.
Последнее указывает <на частично ионный характер связи в молекуле
фтористого хлора.
Помимо тех спектроскопических исследований фтористого хлора,
которые упоминались в связи с его термодинамическими
константами [18, 23, 241, в литературе описаны полосатые спектры [13],
микроволновой [24] и инфракрасный [27] спектры газообразного
фтористого хлора и спектры комбинационного рассеяния жидкого
препарата [27].
Из данных спектроскопических исследований были вычислены
энергия диссоциации фтористого хлора, равная 60,2 и 8,3 ккал-моль'1
при расчете на невозбужденные или, соответственно, на
возбужденные атомы [351, дипольный момент 0,881 ±0,02 (табл. 4),
межъядерные расстояния (см. выше) [24], собственная частота колебаний
772 см~1 для газообразного и 758 слг1 для жидкого фтористого
хлора [27] и некоторые другие молекулярные константы [18, 24, 27,
35, 58].
Химические свойства
Химические свойства фтористого хлора изучены еще весьма
недостаточно; это объясняется значительными
экспериментальными трудностями, обусловленными большой химической
активностью данного межгалоидного соединения. Сведения о
химических свойствах фтористого хлора почти полностью исчерпываются
качественными данными, описанными в работе Руффа и Ашера [1],
имеющей (в этой части) характер предварительного сообщения.
Фтористый хлор — весьма химически активное вещество. С
неорганическими и органическими веществами он реагирует подобно
фтору, а в большом числе случаев даже активнее его. Еще задолго
до открытия фтористого хлора было замечено, что добавление даже
небольших количеств хлора к фтору повышает активность
последнего в реакциях фторирования неорганических соединений. Подобное
явление наблюдали Руфф и Гейнцельман [28] при фторировании
урана или карбида урана. Влияние хлора они объясняли катали-
* Величины термодинамических свойств, вычисленные Поттером и Коулом
с сотрудниками, близки к данным Эванса, Манзона и Вагмана.
** Термические и термодинамические константы межгалоидных соединений
систематизированы также в ряде справочных руководств [54—6-7].
*** Цитировано по Гринвуду [17, стр. 449].
**** По другим данным [17, стр. 449], эта величина равна 1,712 А (см.
табл. б).
27
тическим эффектом. Однако правильнее объяснять это влияние
образованием фтористого хлора, фторирующая способность которого
часто превышает активность элементарного фтора.
Реакции *с участием фтористого хлора в большом числе случаев
протекают бурно, часто с выделением пламени. В более редких
случаях (из описанных в работе Руффа и Ашера), например, при
действии на серу, кварц, стеклянную вату, лробку, фтористый хлор
оказался менее активным, чем фтор.
Реакции с простыми веществами
Фтористый хлор, подобно фтору и другим фторидам галогенов,
реагирует, вероятно, со всеми металлами. Интенсивность этих
реакций на холоду или при нагревании зависит от того, образуется ли на
поверхности металла защитный слой хлорида и фторида и какова
прочность этого слоя *. Так, натрий на холоду образует белый
защитный слой, но при нагревании вспыхивает. Магний и алюминий
горят во фтористом хлоре, если их предварительно сильно нагреть.
Порошкообразные медь и цинк, а также золото и платина — в виде
фольги — реагируют при более слабом нагревании (платина
образует при этом бурый возгон). Железо в порошке уже при комнатной
температуре накаляется и превращается, по-видимому, в смесь
нескольких веществ. Ртуть при комнатной температуре покрывается
оболочкой черно-бурого цвета, но при нагревании реагирует бурно
и образует белый возгон. Мышьяк и сурьма сгорают в атмосфере
фтористого хлора, превращаясь при этом, предположительно, в пяти-
фтористый мышьяк и пятифтористую сурьму.
Аморфный бор, аморфный кремний и красный фосфор сгорают
при комнатной температуре (вероятно, с образованием летучих
фторидов); сера реагирует медленнее, чем со фтором, и без выделения
пламени, водород на холоду не реагирует, но при нагревании горит
во фтористом хлоре. Бром и иод легко реагируют с образованием
трехфтор истого брома и, возможно, пяти фтор истого иода.
Реакция с неорганическими соединениями
Фтористый хлор энергично реагирует с водой, причем
образуются кислород и хлор **.
Окись углерода при нагревании горит во фтористом хлоре. Из
окиси углерода и фтористого хлора был получен карбонилхлорфто-
рид COC1F [29, стр. 120]. Сернистый газ реагирует только при
нагревании, продукты реакции не установлены.
Кварц при сильном нагревании слабо реагирует с фтористым
хлором. Травление стекла происходит уже при комнатной
температуре, но только при наличии следов влаги.
* Поверхностный слой галогенидов, в случае действия фтористого хлора,
состоит из хлорида и фторида данного металла. Большую активность
фтористого хлора Руфф [6] объясняет тем, что защитное действие поверхностного слоя
галогенидов металла исчезает в случае промежуточного расположения хлорида.
** Вероятно, также и фтористый водород.
28
Действие на органические вещества
Фтористый хлор настолько энергично реагирует с органическими
веществами, обычно с выделением пламени, что эти реакции нельзя
было регулировать и контролировать *. Руфф и Ашер испытали
действие фтористого хлора на фильтровальную бумагу, вату, пробку,
пицеин, ацетиленовую сажу, бензол, скипидар, парафиновое масло
и во всех случаях наблюдали явление горения. Особенно
характерным для фтористого хлора является его действие на целлюлозу, с
которой он реагирует на холоду, в то время как фтор — только при
нагревании. По Руффу [6], эту реакцию можно применить для
отличия фтористого хлора от фтора.
По данным Каца и Барра [50], газообразные непредельные
углеводороды (этилен, пропилен, 2-бутилен, изобутилен и хлористый
винил) образуют с фтористым хлором в условиях «газового
титрования» нормальные продукты присоединения. Предельные
углеводороды в этих условиях (комнатная температура, давление 50 мм) не
реагируют.
2. ТРЕХФТОРИСТЫЙ ХЛОР C1F3
Получение
Возможность образования трехфтористого хлора, наряду с
фтористым хлором, при взаимодействии хлора со фтором была
установлена Руффом и Лаассом в их работе по изучению полученного ими
фтористого хлора [7]. С целью определения теплоты образования
фтористого хлора Руфф и Лаасс измерили теплоту реакции
C1F + H2=HC1 + HF и обнаружили, что, если исследования
проводить с одним и тем же препаратом, то теплота реакции со временем
увеличивается. Для объяснения данного факта Руфф предположил,
что фтористый хлор в этих опытах обогащается фтором за счет
отщепления части менее летучего хлора и образования
трехфтористого хлора по уравнению
3C1F^C1F3 + C12.
Проверка этого предположения привела к разработке условий
получения трехфтористого хлора [10].
Фтор реагирует с другими галогенами в широком интервале
температур, причем эти реакции экзотермичны. По литературным
данным, взаимодействие с хлором происходит в интервале от —170 до
400°. При избытке фтора образуется смесь фтористого и трехфто-
* В литературе имеется сообщение о том, что во время второй мировой
войны в Германии было начато промышленное получение фтористого и
трехфтористого хлора, очевидно, в связи с их воспламеняющим действием [17, 30].
Бус и Пинкстон в обзоре, посвященном галоидным соединениям фтора,
сообщают, что в Фалькенхагене был построен завод производительностью более
чем 5 т трехфтористого хлора в день, однако наступление Советской Армии
заставило перенести завод, по-видимому, прежде, чем он проработал сколько-
нибудь значительное время, после чего завод уже не возобновлял работы [31].
29
ристого хлора, причем соотношение этих фторидов зависит от
температурных условий: при более низкой температуре выход
трехфтористого хлора становится относительно большим.
Первое сообщение о получении трехфтористого хлора было
опубликовано Руффом и Кругом в 1930 г. [10]. Высокая химическая
активность этого фторида, в частности в отношении стекла, кварца и
ряда других материалов, весьма затрудняет получение чистого
препарата и уменьшает его выход.
Для получения трехфтористого хлора Руфф и Круг использовали
аппаратуру, изображенную на рис. 4, с тем отличием, что позади
медного цилиндра А (содержащего фтористый калий для
поглощения фтористого водорода) и впереди реакционной камеры В был
помещен кварцевый сосуд G, охлажденный до-температуры — 170°. В
нем конденсировались менее летучие примеси. Три приемника G,
расположенные за медным блоком В (вместо шести приемников на
рис. 4), служили для конденсации трехфтористого и фтористого
хлора и избыточного фтора. Приемник для трехфтористого хлора из
кварцевого стекла был охлажден до —70° и заключен,
предосторожности ради, в медный блок; второй приемник — для фтористого
хлора— также из кварцевого стекла был охлажден до температуры
—150°; третий сосуд из обычного стекла, предназначенный для
улавливания избыточного фтора, охлаждался жидким воздухом.
Аппаратура заканчивалась стеклянной или медной трубкой, содержавшей
фтористый калий для поглощения влаги воздуха.
При пропускании тока фтора — около 1200 еж3 — и тока хлора—
около 800 см2, в час в приемниках за 8 часов было собрано около
5 см3 трехфтористого хлора и 20 см3 фтористого хлора, т. е. сырые
продукты — фтористый и трехфтористый хлор получаются в
соотношении, близком к 4 : 1. Хлор входит в реакцию почти
количественно. Более летучие приме-си, содержащиеся во трехфтористом хлоре,
удаляются выдерживанием препарата в течение 3—6 минут при
давлении 20 мм. Возможность попадания менее летучих примесей,
по данным Руффа и Круга, устраняется в том случае, когда в
установку включен упомянутый выше промежуточный сосуд.
В работе Руффа и Круга [10] отсутствуют прямые указания на
температуру, при которой они получали трехфтористый хлор *. В
выводах к работе Руффа и Круга сообщается, что обратимая
реакция C1F + F2^C1F3 приводит к равновесию, в котором при 250°
количество молекул фтористого хлора во много раз больше количества
молекул трехфтористого хлора.
В дальнейшем метод получения трехфтористого хлора был
упрощен, причем реакция производилась при нагревании до 200—300°.
Банкс, Эмелеус и Вулф [32] получали трехфтористый хлор
пропусканием токов хлора и фтора (21,4 и 13,3 г/час, соответственно)
* Бус и Пинкстон [31] указывают, что Руфф и Круг получали трехфтористый
хлор при —170э. Это ошибочное утверждение основано, очевидно, на том, что
упомингвшийся выше промежуточный сосуд, соединенный с реакционной
камерой, был охлажден до —170°. Однако этот сосуд предназначался для
конденсации примесей, содержавшихся во фторе.
30
через группу реакторов при 280°. В качестве реактора применялась
трубка из никеля, наполненная пластинками серебра и медными
стружками. Трехфтористый хлор собирался в виде бледно-желтой
жидкости в кварцевой колбе при —78° и очищался сперва
пропусканием его паров через металлическую трубку, наполненную
фтористым натрием для связывания паров фтористого водорода, а затем
путем фракционированной конденсации в вакууме, в кварцевой
аппаратуре. Аналогичный метод получения трехфтористого хлора
описали Банкс и Рудж [33] и Гризард с сотрудниками [36].
По данным, изложенным в статьях Лееха [30], Буса и Пинксто-
на (31], в Германии трехфтористый хлор получали в никелевых
аппаратах: сначала, при 200°, образовывался фтористый хлор,
который при 280° превращался в трехфтористый хлор; последний
конденсировался и сохранялся в стальных сосудах. В качестве
катализатора для этой реакции был предложен фтористый никель [59]. Описано
также получение трехфтористого хлора непосредственно из
фтористого хлора и фтора при 270—280° [12].
Бус и Пинкстон [31] сообщают, что в настоящее время
трехфтористый хлор получают смешиванием хлора и фтора в медном
реакторе при 200°, причем реакция проходит мгновенно и количественно.
Продукт реакции может быть очищен фракционированной
перегонкой в медной аппаратуре.
В литературе описана установка из монель-металла для
получения трехфтористого хлора в масштабе крупной лаборатории из сте-
хиометрических количеств фтора и хлора, при температуре 300° [34].
О получении C1F3 по Кваснику см. [62, стр. 100].
Физические свойства
В газообразном состоянии трехфтористый хлор почти бесцветен,
обладает характерным пронзительным запахом, напоминающим
запах фтористого хлора. Твердый трехфтористый хлор бесцветен, при
—190,5±0,05° он испытывает превращение (скрытая теплота
превращения 360,5 ккал-моль~х) [36], плавится в бледно-зеленую
жидкость. Температура плавления, согласно новым данным Николаева
и Малюкова [63], равна —83,0±0,5°, т. кип. —11,75°.
Упругость пара жидкого трехфтористого хлора (в мм рт. ст.), по
Руффу и Кругу [10], находится в следующей зависимости от
абсолютной температуры:
Вычисленная по этой формуле упругость пара при 0° составляет
490 мм, а температура кипения (Р=760 мм) должна быть равна
11,3°. Однако непосредственное определение температуры кипения
трехфтористого хлора дало величину 12,0±0,1 [33] и 11,75° [63].
Гризард, Бернгард и Оливер [36], применяя хорошо очищенный
препарат трехфтористого хлора, содержавший всего 0,04%
примесей, определили для него тройную точку 196,84 ±0,05° К; по их дан-
31
ным, зависимость давления паров трехфтористого хлора от
температуры, в пределах от 226 до 303° К, может быть представлена
уравнением lg Р = 7,37611 —1096,917/(*° + 232,75). Температура кипения,
вычисленная по этому уравнению, равна 11,75°, а теплота
парообразования при температуре кипения составляет 6,58 ккал-моль'1, что
существенно отличается от величины, вычисленной Руффом и
Кругом.
Теплота испарения (у точки кипения), вычисленная по формуле
Руффа и Круга, составила 5,9 ккал- моль*1, критическая
температура 153,5° С, а константа Трутона 20,8 кал • град'1 • моль~]. Шмитц и
Шумахер [35] для константы Трутона приводят величину 24,1, не
указывая, однако, каким образом она получена.
Отмечая, что константа Трутона характеризует трехфтористый
хлор как ассоциированную жидкость, Шмитц и Шумахер указывают,
что их исследования кинетики образования и распада
трехфтористого хлора привели к предположению об ассоциации его молекул
также и в газообразном состоянии при низких температурах.
Изучая степень отклонения трехфтористого хлора от законов идеальных
газов в интервале 300—800 мм рт. ст., эти авторы нашли, что
найденные ими расхождения можно объяснить образованием димерных
молекул (ОРзЬ- Измерения проведены в кварцевом сосуде с
чувствительным манометром, имеющим кварцевую спираль. Величины
константы равновесия Л"Р = ^2cif3 / ^(cif^» приведены в табл. 7.
Тепловой эффект реакций
т а б л и ц а 7 C|F + C1F ^ (C1F8U вычисленный
Константа равновесия реакции а а г , , г,
9Г1р -^(rw \ из температурного коэффи иента ДР,
оказался равным (для комнатной
температуры) 3,3 + 0,5 кк л моль-1.
Из константы равновесия этой реак-
ции были вычислены также следую-
26,9 щие значения: свободная энергия
32,1 образования 2,1 ккагмо7Ь~1 и из-
35'4 менение энтропии 17+1 ккал\
У. град-1-моль-1. Эти величины показывают, что трехфтористый хлор
существует при комнатной температуре преимущественно в моюмерной
форме [i7, стр. 450].
Определение молекулярного веса трехфтористого хлора по
плотности пара [10] дало величины 86,71—87,93, немного меньшие
теоретического значения для мономерных молекул (92,46). Руфф и
Круг объясняют это частичным разложением трехфтористого хлора
в процессе измерений плотности пара и потерей веса вследствие
образования SiF4.
Плотность жидкого трехфтористого хлора, измеренная при точке
кипения d\2, равна 1,77 [161. Банкс и Рудж [33] измерили плотность
жидкого трехфтористого хлора под давлением его паров в
интервале от — 4,07 до 45,63° и установили следующую зависимость
плотности от температуры:
d£ = }>8853 - 2,942-103/ - 3,79- 10-вА
Отклонение от вычисленных значений составляет + 0,0003 г/мл.
2C1F3^(C1F3)2
Температура, °С
9,5
20,0
24,2
/Ср, атм
31:
Плотность трехфтористого хлора при температуре кипения dl42 = 1,8495.
Бус и Пинкстон выражают зависимость между плотностью
жидкого трехфтористого хлора (г/см3) и абсолютной температурой
уравнением d=2J29—0,00307Г [31]; однако они не указывают, для
какого интервала температур применимо это уравнение.
Измерены также поверхностное натяжение и вязкость жидкого
трехфтористого хлора [60]. Поверхностное натяжение, измеренное в
интервале температур от 0,0° (26,6 дин/см) до 49,9° (18,7 дин/см),
описывается уравнением у* =26,7—0,16/±0,2 дин/см. Парахор
равен 111,5.
Вязкость трехфтористого хлора при нагревании от 11,75 до 50,0°
уменьшается от 0,478 до 0,316 сантипуаза. При 25° она равна
0,435 сантипуаза [60].
Шмитц и Шумахер [11] изучили константу равновесия реакции
C1F + F2XC1F3 методом измерения упругости пара в магниевых и
никелевых сосудах с помощью чувствительного кварцевого
манометра. В табл. 8 указаны величины константы равновесия этой реакции:
pclF.p
Кр =—я 1 причем значения температур ниже 250° получены
экстраполяцией.
Теплота образования трехфтористого хлора из элементов по реакц
lci2 + f-F2 = ClF3
вычислена из тепловых эффектов реакций
"2 Clt + 2"F2 = C1F;
C1F+F2 = C1F3
1 3
-o~Cl2 + ~0~^2 = CIF3
Тепловой эффект реакции C1F+F2=*C1F3 был определен тремя
методами, причем были получены следующие величины: 27,0±1,5
[11]; 25±2 [11] и 26,5±0,5 ккал-моль-1 [37].
1. Шмитц и Шумахер [11] для этой цели экспериментально
определили теплоту реакции при 18°
3N*C1KPI1CT + ClF3(r) = 3NaFKpHCT + 2Cl2(r) + 76,5 ккал
и воспользовались термэхямитееки\о даидьши для следующих двух
приведенных далее реакций:
2NaCl + F2 = 2NaF -f- Cl2 + 79,0 ккал ± 0,5;
NaCl + C1F = NaF + Cl2 + 24,5 ккал;
3NaF + 2C12 = 3NaCl + C1F3 - 76,5 ккал
C1F 4- F2 = CiF3 + 27 ккал
2. Теплота этой суммарной реакции, вычисленная из константы
равновесия (табл.8) по уравнению Вант-Гоффа,
dlnKl _ АН0
dT i?r3 '
3—1419
33
оказалась равной 25 ±2 ккал-моль'1 [11, 17]. Эванс с
сотрудниками [53] приводят значение 24,50 ккал-моль*1.
3. Тепловой эффект этой же реакции, рассчитанный из
спектроскопических данных, равен 26,5 ±0,5 ккал-моль*1 [37].
Если для теплоты этой реакции принять значение 24,5, а для
теплоты образования фтористого хлора из элементов значение
13,4 ккал-моль'1, то теплота образования трехфтористого хлора из
элементов
1/2С12(г^ + 3/гр2(Г)= ClF3(r)
будет равна 37,9 ккал-моль"1.
Из констант равновесия для реакции C1F+F2=C1F3 (табл. 8) при
помощи соотношения—Д/7/0 = RTln K% были рассчитаны [17]
значения свободной энергии образования трехфтористого хлора по этой
реакции для*трех температур: 8,45 (523°К); 6,85 (573° К) и 5,26°
(623° К). Изменение энтропии в этой реакции AS°=—31,9±
±0,1 кал-град~имоль~х. Приводя эти данные, Гринвуд [17] отмечает,
что это большое уменьшение энтропии является следствием
уменьшения числа молей газообразных веществ при реакции.
Термодинамические свойства трехфтористого хлора, по данным
[53], приведены в табл. 9.
Величины свободной энергии, энтропии и теплоемкости
трехфтористого хлора (как идеального газа), вычисленные Шерером [38] из
спектроскопических данных для температур от 284,91 (температура
кипения трехфтористого хлора) до 1500° К, очень близки к данным
табл. 9.
Величина энтропии при температуре кипения совпадает с
величиной, указанной в работе Гризарда с сотрудниками [36], равной
66,87 кал-град^1-моль'1*, но она несколько выше (на
1 кал • градгх • моль'1) данных Шефера и Вике [37] **.
Определение термодинамиче-
Т абл ица 8
Значения константы равновесия для
реакции CIF+Fg^ClFg
Температура,
°С
180
200
220
Kv • 104, |
атм |
0,069
0,212
0,63
Температура,
1 °с
250
300
350
Kv • 10*.
атм
2,96
24
143
ских функций и упоминаемые
далее оптические исследования
трехфтористого хлора были
использованы также для суждения
о структуре его молекул.
Решение этого вопроса оказалось
довольно сложным.
Спектр поглощения
трехфтористого хлора был изучен Шмит-
цем и Шумахером [35] в об-
* Энтропия жидкого трехфтористого хлора при температуре кипения равна
43,66 ±0,10 кал.град Кмоаь~* [36].
** Вебер и Феригль [3*9] высказали ряд замечаний в отношении значений
энтропии C1F3, вычисленных Шерером, который исходил из моментов инерции,,
полученных при изучении микроволновых спектров. Они полагают, что в
настоящее время еще не может быть сделано определенного заключения в отношении
термодинамических свойств трехфтористого хлора по спектральным данным, в
частности по той причине, что явление ассоциации молекул его может
обусловить значительные сдвиги частот.
34
Термодинамические свойства C1F3 (г)
Таблица 9
о
0
25о
273,16
298,16
300
400
500
600
700
800
900
1000
1100
1200
1300
1400
1500 1
1 X
s£7
о1 *? |
0
—54,81
-55,76
—56,73
—56,80
—60,24
—63,16
—65,71
—67,97
—70,00
—71,84
—73,52
—75,08
-76,54
—77,90
—79,16 |
—80,34 1
• х
^ 7
о о «з i
| ? 3
S §х
0
10,59
10,94
11,31
11,33
12,59
13,58
14,35
14,96
15,45
15,86
16,20
16,49
16,74
16,95
17,13
17,30
X
7 ~
<3 х
со v* х
0
65,40
66,70
68,04
68,13
72,83
76,74
80,06
82,93
85,45
87,70
89,72
91,57
93,28
94,85
96,29
97,64 1
7
о о S
i 1
0
2647,5
2988,4
3372,2
3399,0
5036
6790
8610
10472
12360
14274
16200
18139
20088
22035
23982
25950
X
7
^7
ос», 5 ^
.0
14,48
15,03
15,55
15,58
17,05
17,92
18,45
18,80
19,03
19,20
19,32
19,41
19,48
19,54
19,59
19,63
1
«о
< *
—37,9
—38,79
—38,79
—38,79
—38,79
-38,72
—38,60
-38,46
—38,31
—38,16
-38,00
—37,84
—37,69
—37,53
-37,37
-37,22
—37,07
7
•о
о
• *
о
< g
-37,9
—30,97
-30,24
—29,46
—29,41
—26.29
—23,19
-20,13
—17,09
— 14,06
—11,05
— 8,05
- 5,08
— 2,14
0,78
3,71
6,65 |
JbjO
27,076
24,193
21,597
21,422
14,363
10,138
7,340
5,332
3,840
2,082
1,176
1,010
0,391
0,132
0,581
0,969
ласти длин волн от 5000 до 2200 А. Спектр поглощения
непрерывный в видимой и в ультрафиолетовой области. Поглощение света
трехфтористым хлором начинается при 4700 А и с уменьшением
длины вол!НЫ возрастает, сперва до длины волны 2600 А (постепенно,
а затем резко, но максимум поглощения не наблюдается. Спектр
не обнаруживает полосатых структур.
Изучение инфракрасного спектра и спектра комбинационного
рассеяния трехфтористого хлора [37, 40] привело к заключению, что
его молекулы имеют симметричную пирамидальную конфигурацию.
Такое представление о строении молекул трехфтористого хлора
подтверждено электронографическим исследованием, причем расстояние
для связи CI—F было найдено равным 1,628А, как и во фтористом
хлоре; угол между связями F—С1—F близок к 86° [17, стр. 451].
Однако это представление о структуре трехфтористого хлора не
согласуется с дальнейшими исследованиями, по которым молекулы
трехфтористого хлора являются полярными. Магнусон [41], измеряя
диэлектрическую постоянную трехфтористого хлора и учитывая
наличие в нем некоторого количества димернои формы и измеренную
ранее Шмитцем и Шумахером /константу равновесия C1F3 + C1F3^
^(C1F3)2 (ом. табл. 8), показал, что общая молярная
поляризация мономера трехфтористого хлора может быть представлена
формулой
1 ЯЯ9
Р = 15,944 + ^щ ± 0,075 с*».
35
Дипольный момент мономерной формы оказался равным 0,554 D
((см. табл. 4). Средняя величина молярной поляризации (C1F3)2
найдена равной 49,8 ±0,5 см?, если предположить, что дипольный
момент димера равен нулю (см. табл. 2).
Наличие дипольного момента в молекулах трехфтористого хлора
исключает плоскостную симметричную модель, подобную -структуре
трехфтористого бора. Если принять для угла связи FC1F
величину 86°, согласно с измерениями электронной дифракции, а для ди-
Рис. 6. Межатомные расстояния и валентные
углы в молекуле трехфтористого хлора.
польного момента фтористого хлора — величину 0,881 D [24], тогда
вычисленный дипольный момент трехфтористого хлора должен быть
равен 1,6 Dy а величина ZFC1F для пирамидальной модели
составила бы 116°. Эти вычисления, как указывает Магнусон [41]* отрицают
возможность симметричной структуры молекул трехфтористого
хлора. Это подтверждено результатами рентгенографического
исследования кристаллического трехфтористого хлора [42].
Предыдущими исследованиями [36] было показано, что трехфто-
ристый хлор имеет одну кристаллическую фазу, устойчивую ниже
—£2,5°. Для рентгеновского структурною анализа был взят
кристалл, сохранявшийся при —120°. Для трехфтористого хлора
характерна орторомбическая система со -следующими константами ячейки:
а = 8,825±0,01 А, Ъ =6,09±0,01 А, с = 4,52±0,01А [42].
Экстраполяция плотности жидкого трехфтористого хлора до
—120° дала возможность заключить, что одна элементарная ячейка
содержит четыре молекулы трехфтористого хлора и что плотность,
определенная рентгенографически, равна 2,53 г/см3.
Структурный анализ [42] показал, что молекулы трехфтористого
хлора -плоскостные, причем расстояние между атомами хлора и од-
о
ним атомом фтора равно 1,621 А, а две другие связи имеют длину
о о
1,716 А; атомы фтора удалены друг от друга на расстояние 2,298 А,
угол связи FC1F равен 86°59' *. Эта модель молекулы трехфтористо-
* В этой же работе предложено объяснение неравноценности связей в
молекуле трехфтористого хлора.
36
f/'ool
Рис. 7. Кристаллическая структура
трехфтористого хлора в проекции вдоль оси с.
CD
Рис. 8. Кристаллическая структура трехфтористого хлора в
проекции вдоль оси а.
го хлора, показанная на рис. 6, удовлетворительно согласуется с
наличием относительно небольшого дипольного момента.
Кристаллическая структура трехфтористого хлора показана в
проекции вдоль оси с на рис. 7, вдоль оси а—на рис. 8. Каждая
молекула трехфтористого хлора находится на расстоянии меньше
о
4 А -с 14 соседними молекулами. Расстояния между атомами
соседних 'молекул обозначены на рис. 7 и 8 [42].
Изучение микроволновых спектров трехфтористого хлора [43]
дало результаты, согласующиеся с данными рентгенографического
исследования, и показало, что молекулы плоскостные и имеют
искаженную Т-структуру с одной короткой (1,598 А) и двумя длинными
(1,698 А) связями С1—F; угол между двумя различными типами
этих связей равен 87°29'. Низкая интенсивность линий спектра
находится в соответствии с малой величиной дипольного момента.
Банкс, Эмелеус и Вулф, измеряя электропроводность жидкого
трехфтористого хлора при 0°, нашли, что она меньше Ю-6 омг1 слг1
[32]. -По данным Банкса, электропроводность трехфтористого хлора
равна 3- 10~9 омгхсм~х [17, стр. 451]. Столь малая
электропроводность этого межгалогенида, очевидно, обусловлена преобладанием
ковалентного характера связи.
Химические свойства
Среди фторидов галогенов трехфтористый хлор отличается
наибольшей химической активностью. Поэтому не только
количественное, но и качественное изучение его химических свойств
сопровождается еще большими затруднениями, чем при изучении фтористого
хлора.
Литературные данные о химических свойствах трехфтористого
хлора очень скудны. Они ограничены главным образом работой
Руффа и Круга [10], в которой очень кратко описано отношение
трехфтористого хлора к ряду химических элементов, неорганических
и органических веществ. Эти авторы отмечают прежде всего
исключительно большое разнообразие химических реакций трехфтористого
хлора.
Реакции с простыми веществами
Литературные данные позволяют предположить, что
трехфтористый хлор способен реагировать со всеми элементами, за
исключением инертных газов, кислорода, азота и, возможно, платины.
Большинство элементов при действии трехфтористого хлора
реагирует с ним со взрывом. Если это не происходит, то в большом числе
случаев достаточно нагреть (зажечь) реагирующее вещество
раскаленным угольком, чтобы началась бурная реакция, часто
сопровождающаяся плавлением и разбрызгиванием продукта реакции.
Замедление реакции обусловлено образованием на поверхности
данного вещества слоя фторида, защищающего его от дальнейшего
действия трехфтористого хлора.
38
По описанию Руффа и Круга, некоторые металлы—калий,
молибден, вольфрам, железо, родий, осмий, иридий — горят в трехфторис-
том хлоре. Большинство же исследованных ими металлов — натрий,
магний, кальций, алюминий, медь, серебро, свинец, цинк, олово —
образуют защитную пленку фторида, которая прекращает реакцию,
но при повышении температуры эти металлы взаимодействуют с
трехфтористым хлором оо взрывом. Более медленно реагирует ртуть.
Водород, кремний, фосфор (красный), мышьяк", сурьма, сера,
селен, теллур, бром воспламеняются при действии на них трехфто-
ристого хлора. Продукты реакции в большинстве случаев точно не
установлены. Сера реагирует с образованием белых паров, запах
которых напоминает S2C12; бром * и иод превращаются, вероятно, в
трехфтористый бром и пятифтористый иод [10]. Селен превращается
в SeF4 [44].
Способность треххлористого «лора фторировать металлы была
использована для переработки облученного урана и разделения
образовавшихся в нем элементов [64].
Реакции с неорганическими соединениями**
Газы — S02, H2S, NH3 — воспламеняются в трехфтористом
хлоре. Вода очень бурно реагирует с C1F3, причем образуются HF, OF2
и С12 [10].
Следующие окислы очень активно реагируют с трехфтористым
хлором, часто с выделением пламени: MgO, CaO, АЬ03, La203, Ti02,
Sn02, Pb02, V2O5, Ta205, Cr203, Сг03 ***, Mo03, W03, Мп02, Р2Об,
В203, As203. Окислы при этом превращаются во фториды и оксифто-
риды соответствующих элементов.
По сообщению Руффа и Круга [10], ZnO, HgO, Si02, Zr02, Th02,
Ге2Оз не реагируют с трехфтористым хлором, однако условия,
необходимые для данных реакций (повышение температуры, наличие
следов воды и т. д.), еще не изучены в достаточной степени. Так, на
совершенно сухое кварцевое стекло и кварцевую вату C1F3 действует
медленно, но в присутствии следов влаги они быстро
разъедаются [10]. Двуокись селена образует SeF4 или SeOF2 [44].
При действии трехфтористым хлором на Со304 95% последнего
превращаются в CoF3. Характерно, что при реакции между Со304
и фтористым хлором образуется только CoF2 [31].
Большой интерес представляет сообщение Попова и Кнудсо-
на [51] о том, что окислы лантана и лантаноидов при действии на
них током трехфтористого хлора в никелевой трубке превращаются
во фториды. Легкость фторирования окислов и скорость реакции
уменьшаются с увеличением атомного номера лантаноида. Так, в
условиях, когда окислы элементов от лантана до самария
фторировались полностью, Gd203 переходила в GdF3 с выходом в 80%, а
* Бром реагирует без выделения пламени.
** О реакциях изотопного обмена трехфтористого хлора со фтором и
фторидами других элементов см. главу XIII.
**■* Образуются бурые пары, возможно, фтористого хромила.
39
ErF3 получался лишь с выходом в 12%. Так как фториды
редкоземельных элементов не растворимы в разбавленных кис/готах, а их
окислы хорошо растворяются, то возможно, что этот метод может
быть использован для «грубого» разделения смесей редкоземельных
элементов.
Водные растворы едких щелочей разлагают трехфтористый хлор
и ускоряют реакцию гидролиза. Руфф и Круг [10] использовали эту
реакцию для количественного анализа трехфтористого хлора — к
щелочному раствору добавляли гидрат гидразина или цинковую
пыль для восстановления соли кислородных кислот хлора. Фтор
определяли в виде CaF2, хлор — в виде AgCl.
Кислоты — серная концентрированная и азотная дымящая —
энергично реагируют с трехфтористым хлором. Такое же явление
характерно для ряда солей — KJ, K2CO3, AgN03 — и карбида
вольфрама *. В условиях опытов, 'произведенных Руффом и Кругом,
не наблюдалась реакция с такими солями: NaCl **, -K2SO4, KNO3,
HgS04, HgCl2 [10]. Хлориды кобальта, никеля и серебра при
действии на них C1F3 превращаются в CoF3 [31], NiF2 и AgF2 [46].
Действие трехфтористого хлора 'на некоторые металлы и их соли
изучалось в связи с разработкой «металлфторидного» метода
фторирования органических соединений.
Непосредственное фторирование органических веществ
элементарным фтором часто осложняется тем, что за счет выделяющейся
при этом энергии происходит разрыв углеродных связей и
разложение органических соединений. Поэтому, а также по другим
причинам, для целей фторирования было предложено применять фториды
ряда металлов, в которых последние находятся в одном <из высших:
валентных состояний, например CoF3. Процесс фторирования может
быть представлен такой, примерно, схемой:
4CoF3 + >СН2 - >CF2 + 4CoF2 + 2HF;
4CoF2 + 2F2 = 4CoF3.
Для металлфторидного метода фторирования органических
веществ чаще всего применяют CoF3. Активными фторирующими
агентами в ряде случаев являются также AgF2, CeF4, MnF3 и SbFs [46].
Эти фториды получали при действии фтора на металлы или на соли
этих же металлов в низших степенях валентности. В последнее
время для этой же цели было испытано действие трехфтористого
хлора. Хюккель [44] получил HgF2, AgF2, CuF2? TiF3, PtF4 и
PbF3 (но не PbF4) нагреванием металлов с трехфтористым хлором
в стальном автоклаве при различных температурах и в течение
определенного времени. Так, для получения HgF2 необходимо было
нагревание в течение 3 часов при 120°. Хром не реагировал с C1F3.
Рохов и Кукин [45] описали метод получения фторидов кобальта,
* Продукты этих реакций не исследованы.
** Однако, как упоминалось выше, Шмитц и Шумахер [11] измерили теплог
вой эффект реакции между трехфтористым хлором и хлористым натрием.
40
никеля и серебра.из хлоридов этих металлов, которые для этой цели
предварительно тщательно обезвоживались при 250°, затем
растирались, просеивались через сито с отверстиями в 30 меш и помещались
з трубку из монель-металла. Процес.фторирования проводили в
реакторе, изображенном на рис. 9. Через реактор сперва пропускали
азот и печь нагревали до 250°, после чего вводили ток трехфтористо-
Рис. 9. Реактор для получения фторидов металлов при
действии трехфтористого хлора на хлориды:
/—насос; 2—реактор; 3—медные ловушки; 4—сухой лед, ацетон; 5—лед.
го хлора. Образующийся при фторировании хлор проходит через,
крайнюю- оправа ловушку, а затем через серную кислоту. Клапан,
ведущий к средней ловушке, должен быть закрыт в течение всего
процесса. Окончание реакции устанавливалось по прекращению
образования пузырьков газа в серной кислоте, так как температура
ловушки должна быть достаточно низкой, чтобы конденсировался
трехфтористый хлор (т. кип. 12°), но не хлор (т. кип. 33,7°).
Оставшийся в приборе трехфтористый хлор выдували током сухого азота.
Продолжительное фторирование посредством C1F3 выше точки
исчезновения хлора у выхода из прибора дало возможность
количественно превратить СоСЬ в GoF3.
Этот же реактор может быть применен для фторирования
углеводородов. Для этой цели кран, ведущий к крайней справа ловушке,,
закрывают, а краны к остальным ловушкам должны быть открыты *.
Образующийся CoF2 окисляется трехфтористым хлором после того,.
* Некоторые детали процесса фторирования углеводородов описаны в
цитируемой работе [45].
45
как через всю систему тщательно пропущен ток азота *.
Прохождение C1F3 «ад CoF2, в течение 5 минут обусловливает окисление более
90% последнего до CoF3.
Способность трехфтористого хлора образовывать комплексные
соединения почти вовсе не исследована. Лишь недавно установлено
образование молекулярного соединения C1F3 • HF [61], возможно,
представляющего собой комплекс типа двойного галогенида (см.
главу XIII). Этот комплекс, по всей вероятности, очень неустойчив;
так, он не обнаруживается на диаграмме плавкости системы
C1F3—HF. Николаев и Малюков [63] исследовали эту систему
методом политермического анализа и обнаружили в ней лишь одну
эвтектику состава 44% C1F3 и 56% HF с т. пл. — 110,7°.
Реакции с органическими соединениями
Органические вещества, если они фторируются, бурно реагируют
с трехфтористым хлором с выдепением пламени. Капля жидкого
C1F3, вылитая на дерево, бумагу, ткани, тотчас производит
воспламенение. Таково же поведение бензола, эфира, уксусной кислоты,
четъгрежлористого углерода **, парафина, шерсти, хлопка,
светильного газа, графита [11].
В связи с такой химической активностью трехфтористого хлора
в отношении органических веществ процесс непосредственного
фторирования последних трехфтористым хлором трудно
контролировать. Обычно получаются смеси различных продуктов фторирования
и разложения. Условия взаимодействия органических соединений с
фтористым и трехфтористым хлором с получением определенных
продуктов фторирования еще мало изучены.
Б литературе имеются краткие сообщения *** о том, что при
действии трехфтористого хлора на органические соединения в среде
фтороуглеводородов [47] или четыреххлористого углерода [48], или
в безводном фтористом водороде [34] ***■* происходит замещение
атомов водорода на фтор и хлор. Растворитель сильно ослабляет
экзотермический эффект реакции и разрешает вести ее в жидкой
<фазе.
Полное замещение водорода, однако, затруднено. Например, при
реакции с углеводородами, содержащими восемь и больше атомов
углерода в молекуле, около 15% водорода не замещается на
галогены, даже если конечная температура реакции достигает 150° [47].
Аналогичный метод описан Леех и Бурнет [48] — трехфтористый
хлор, полностью обезвоженный и свободный от фтористого водоро-
,да, довольно хорошо растворяется в четыреххлористом углероде
(или в C7FI4). Фторирование производится в никелевом реакторе,
снабженном охлаждающим кожухом. Лучше сперва приготовить
* Клапан, ведущий к средней ловушке, должен быть закрыт, а к крайней
справа ловушке — открыт.
*'* Четыреххлористый углерод реагирует при нагревании.
*** Без указания условий опыта.
**** Авторы этой работы отмечают, что реакции фторирования растворами
трехфтористого хлора в безводном фтористом водороде не сопровождаются
эффектом разложения органических веществ, что часто наблюдается при
фторировании элементарным фтором.
42
10%-(НЫЙ раствор углеводорода в четыреххлористом углероде и
пропускать через него газообразный трехфтористый хлор.
Образующийся в процессе реакции фтористый водород удаляется отсасыванием.
Процесс следует проводить в области температур от —22° до +50°.
Чтобы ослабить интенсивность реакции, можно вводить смесь
трехфтористого хлора с азотом.
Реакцию между трехфтористым хлором и бензолом в жидкой
фазе — в четыреххлористом углероде и в присутствии различных
катализаторов — исследовали Эллис и Мусгрев [49]. В качестве
катализаторов были взяты галогены и галогениды некоторых металлов (J2>
HgCl2, CoCl2, CoF2, AgF), которые применялись как катализаторы
также при реакциях прямого фторирования органических
соединений. Из результатов опытов 'следует, что основным процессом,
независимо от присутствия катализатора, является реакция замещения
с образованием фторбензола и хлорбензола. Был также изучен
продукт присоединения, который по своему составу приближается к
хлорфторциклогексану, -циклогексену и -циклогексадиену, но
только в одном из опытов он был получен в сравнительно большом
количестве. Из высококипящих фракций можно выделить небольшие
количества хлорфтордифенила.
Из приведенных катализаторов наибольший выход дает CoF2.
Увеличение количества катализатора и растворителя и,
следовательно, уменьшение относительного количества C1F3 не влияет на выход
продуктов замещения, но повышает выход продуктов присоединения.
Эта авторы предположили, что начальным процессом в реакциях
фторирования, происходящих без участия катализатора, является
диссоциация C1F3 по схеме C1F3^C1F + F++F~. В присутствии же
катализаторов, например CoF2, последний принимает участие в этой
реакции:
C1F3 + 2CoF2 -* 2CoF3 + C1F;
CIF3 + C0F3 ->F+ + CoF4~ + C1F.
Полученные результаты дали этим авторам основание высказать
еще одно предположение: трехфтористый хлор легче, чем
элементарный фтор, продуцирует положительный ион фтора, играющий
важную роль в реакциях замещения водорода ароматического ядра.
Образование хлорбензола авторы объясняют хлорирующим
действием фтористого хлора, возникающего в системе как вследствие
диссоциации трехфтористого хлора, так и в результате побочного
процесса — взаимодействия трехфтористого хлора с растворителем.
C1F3 + ССЦ = CCI3F + 2C1F,
CIF3 + GCI3F = CC12F2 + 2C1F и т. д.
Наряду с реакциями в жидкой фазе производились также
немногочисленные опыты фторирования органических веществ в
газообразной среде. Так, Шарп в своем обэдре [29, стр. 128] сообщает
о том, что Гасцельдин, действуя трехфтористым хлором на бензол и
толуол в парообразной фазе, установил образование не только про-
43
дуктов присоединения фтора и хлора, но и продуктов замещения
атомов водорода. Кац и Барр [50] показали, что этилен в условиях
«газового титрования» количественно реагирует со смесями
трехфториетого хлора (или фтористого хлора) с фтором. При
непосредственном взаимодействии трехфториетого хлора с газообразными
непредельными углеводородами в этих же условиях образуются
преимущественно продукты присоединения с относительно небольшим
количеством продуктов замещения. Состав продуктов реакции не
указан.
Все эти пока еще немногочисленные сообщения о реакциях
между трехфтористым хлором и органическими соединениями приводят
к заключению, что, в противоположность существовавшему ранее
мнению, возможно разработать условия контролируемого процесса
галоидирования органических веществ с помощью трехфториетого
хлора.
ЛИТЕРАТУРА
1. О. Ruff, E. A s с h е г, Z. anorg. allg. Chem., 176, 258 (1928).
2. Н. М о i s s a n, Ann. chim. phys. (6), 24, 224 (1891).
3. H. M о i s s a n, Le fluor et ses composes,* Paris, 1900, стр. 123.
4. P. Lebeau, С. г., 141, 1018 (1905); Bull. Soc. chim., 35, 1158 (1906).
5. O. Ruff, J. Zedner, Ber., 42, 1037 (1909).
' 6. O. R u f f, Z. ang. Chem., 41, 1290 (1928).
7. O. R u f f, F. L a a s s, Z. anorg. allg. Chem., 183, 214 (1929).
8. O. Ruff, Z. ang. Chem., 42, 807 (1929).
9. K. F r e d e n h a g e n, О. Т h. Kraft, Z. phys. Chem., 141, 221 (1929).
10. O. Ruff, H. К r u g, Z. anorg. allg. Chem-, 190, 270 (1930).
11. H. Schmitz, H. Schumacher, Z. Naturforsch., 2a, 362 (1947).
12. L. Do mange, J. N e u d о r f f e г, С. г., 226 920 (1948).
13. H. Schmitz, H. Schumacher, Z. Natui orsch., 2a, 359 (1947);
H. Schumacher, H. Schmitz, P. Broderson, Anales
asoc quim. Argentina, 38, 98 (1950); С. А., 45, 2309 (1951).
14.0. Ruff, W. Menzel, Z. anorg. allg. Chem., 198, 375 (1931).
15. O. Ruff, F. E b e r t, W. Menzel, Z. anorg. allg. Chem., 207, 46
(1932).
16. O. Ruff, Z. ang. Chem., 46, 739 (1933).
17. H. H. Г р и н в у д, Усп. химии, 22, 445 (1953).
18. A. L. W a r h a f t i g, J. Chem. Phys., 10, 248 (1942).
19. E. Wicke, Nachr. Akad. Wiss. Gottingen, 89 (1946); E. W i с k e,
H. Fritz, Z. Elektroch., 57, 9 (1953). |
20. A. D. С a u n t, F. R. Barrow, Nature, 164, 753 0949).
21. И. Г. Рысс, Усп. химии, 21,857(1952;.
22. R. L. Potter, J. Chem. Phys., 17, 957(1949).
23. M. R о g e г s, A. W a r h a f t i g, V. S с h о m a k e r, Abstr. Atlantic
City Meeting, Am. Chem. Soc, 1947, Apr., 21 p.
24. D. A. Gilb'ert, A. Roberts, P. A. G r i s w о 1 d, Phys. Rev.,
76, 1723 (1949); D. A. Gilbert, A. Roberts, Phys. Rev., 77, 742
(1950).
25: Справочник химика, Госхимиздат, 1951, т. I, 286.
26. Л. П а у л и н г, Природа химической связи, Госхимиздат, 1947, стр. 167.
27. Е. Jones, Т. Parkinson, Т. Burke, J. Chem. Phys., 18, 235
(1950).
28. О. Ruff, A. H e i n z e 1 m a n n, Z. anorg. Chem., 72, 63 (1911).
29. A. G. Sharp e, Quart. Rev., 4, 115 (1950).
30. H. R. Leech, Quart. Rev., 3, 22 (1949).
31. Г. С. Бус, Д. Т. П и н к с т о н, сб. «Фтор и его соединения», ИЛ>
М., 1953, т. 1, 163; Chem. Rev., 41, 422 (1947). [
44
32 A. A. Banks, H. J. E m e 1 е u s, A. A. W о о 1 f, J. Chem. Soc,
ч 2861 (1949).
33. A. A. Banks, A. J. R u d g e, J. Chem. Soc, 191 (1950).
34. F. R- Lawdermilk, R. G. Danehower, H. С. М i 1 1 о г,
* J. Chem. Educat, 28, 249 (1951).
35. H. Schmitz, H. Schumacher, Z. Naturforsch., 2a, 363 (1947).
36. J. W. Grisard, H. A. Bernhardt, G. D. Oliver, J. Am.
Chem. Soc, 73, 5725 (1951).
37. K. Schafer, E. Wicke, Z. Electroch., 52; 205 (1948).
38. M. D. S с h e r e r, J. Chem. Phys., 20, 924 (1952).
39. A. Weber, S. M. Ferigle, J. Chem. Phys., 20, 1497 (1953).
40. E. A. Jones, T. F. Parkinson, R. B. Murray, J. Chem.
Phys., 17, 501 (1949).
41. D. W. M a g n u s о n, J. Chem. Phys., 20, 229 (1952).
42. R. D. Burba nk, F. N. В e n s e y, J. Chem. Phys., 21, 602 (1953).
43. D. F. Smith, J. Chem. Phys., 21, 609 (1953).
44. W. H u с к e 1, Nachr. Akad. Wiss.. Gbttingen, Math. phys. Klasse, 36
(1946).
45. E. G. Roc how, J. К u к i n, J. Chem. Soc, 74, 1625 (1952).
46. Т. Д ж. Б р и к, Сб. «Фтор и его соединения», т. 1, 358.
47. Н. J. Emeleus, Nature, 165,224 (1950).
48. Н. R. Leech, R. L. Burnett, Britt. Pat., 633, 678, Dec. 19, 1949;
С. А., 1951, 3588 h.
49. J. F. Ellis, W. K. Musgrawe, J. Chem. Soc, 3608 (1950).
50. S. К a t z, J. T.-Barr, Anal. Chem., 25, 619 (1953).
51. A. J. Popov, G. E. Knudson, J. Am. Chem. Soc, 76, 3921 (1954).
52. L. G. Cole, M. F a r b e r, G. W. Elverum, Jr., J. Chem. Phys.,
20, 586 (1952); L. G. Cole, G. W. Elverum, Jr., J. Chem. Phys.,
20, 1543 (1952).
53. W. H. Evans, T h. R. M u n s o'n, D. D. W a g m a n, J. of
Research Nat. Bureau Standards, 55, 147 (1955).
54. Э. В. Б р и ц к е, А. Ф. К а п у с т и н с к и й, Термические константы
неорганических веществ, АН СССР, М.—Л., 1949.
55. Т. R. В i с h о w s k у, F. D. Rossini, The Thermochemistry of
Ithe Chemical Substances, N. Y., 1936.
56. F. D. Rossini, D. D. W a g m a n, W. H. Evans, S. Levine,
J. J a f f e, Selected values of chemical thermodynamic properties,
Washington, 1952.
57. Landolt-Bornstein, Phys.-chemische Tabellen.
58. A. H. Nielsen, E. A. Jones, J. Chem. Phys., 19, 1117 (1951).
59. Химия фтора, Сб. 1, ИЛ, М., 1948, стр. 207.
60. A. A. Banks, A. D a v i e s, A. J. R u d g e, J. Chem. Soc, 732
(1953).
61. J. P. Pemsler, D. F. Smith, J. Chem. Phys., 22, 1834(1954).
62. Руководство по препаративной неорганической химии, под редакцией
Г. Брауэра, ИЛ, М. 1956, стр. 99—103.
63. Н. С. Николаев, И. М. М а л ю к о в, Ж- неорг. химии, 2, 1587
(1957).
*64. R. A. Gustison a. al., Progr. Nuclear. Energy, 1956, Ser. 3, I; РЖХим,
|1957, реф. 26048.
ГЛАВА III
ФТОРИДЫ БРОМА
1. ФТОРИСТЫЙ БРОМ BrF
Получение
Существование низшего фторида брома наряду с пятифтористым
и трехфтористым бромом предположили Руфф и Мендель [1],
наблюдавшие, что при действии брома на пятифтористый бром
образуется вещество с более высокой упругостью пара и более сильным
разъедающим действием на кварцевое стекло, чем трехфтористый и
пятифтористый бром и бром.
Руфф и Брайда [2] поставили ряд опытов по получению
фтористого брома, но им не удалось получить его в чистом виде. Было
лишь установлено, что в полученном ими веществе на 1 г-атом
брома приходится меньше 3 г-атомов фтора; но так как молекулы всех
известных фторидов галогенов (равно как и все межгалогениды)
содержат четное число атомов галогенов, то Руфф и Брайда
заключили, что они получили фтористый бром в смеси с высшими фторидами
брома, и объяснили этот факт неустойчивостью его. Они
предположили, что образование фтористого брома и последующее
превращение его может быть представлено такими уравнениями:
Br2+F2->Br2F2^2BrF;
Br2F2 + BrF$BrF3 + Br2;
BrF3+Br2F2^BrF5 + Br2.
Бром и фтор непосредственно взаимодействуют с выделением:
значительного количества тепла. Если продукт реакции
конденсировать в кварцевом сосуде, охлажденном до —150э, то на нижней,,
более холодной, части выделяется фтористый бром в виде
блестящих оранжево-красных, напоминающих бихромат, кристаллов.
Выше наблюдаются темно-красно-бурые кристаллы брома и еще
выше— светлые зеленовато-желтые иглы трехфтористого брома.
Между ними распределен пятифтористый бром.
Скорость реакции при 0° очень 'мала, в интервале от +50 до 60°
образуются только трехфтористый и пятифтористый бром, но не
фтористый бром. Поэтому опыты получения фтористого брома Руфф
и Брайда проводили при +10°, разбавляя азотом реакционную смесь
46
в установке, изготовленной из кварца (рис. 10). Продукт реакции
подвергался фракционированному охлаждению при температурах
+ 10, —20, —50, —120 и —185° (жидким воздухом). Разделение
брома и фторидов брома основано на их различной упругости
пара — по возрастающей упругости паров их можно расположить в
такой ряд: BrF3<Br2<BrF5<BrF.
Наибольшее количество фтористого брома .конденсируется при
—120°. На основании результатов химического анализа *, измерения
плотности и давления пара
Руфф и Брайда заключили, ^==^ ,рм-4**>
что смесь продуктов,
конденсирующаяся при — 120°,
содержит около 50%
фтористого брома. Эти расчеты,
однако, нельзя считать
безупречными.
Результаты этого
исследования показали, что при
взаимодействии брома и
фтора образуется сложная
система, компоненты
которой—бром и фториды
брома—находятся в равновесии.
Оно может быть выражено
уравнениями, приведенными на стр. 46. Эта система является
гетерогенной. По Руффу и Брайда [21, фториды брома в жидком
состоянии полностью смешиваются друг с другом, но очень мало
растворимы в броме; поэтому жидкие смеси брома и фторидов брома
образуют два несмешивающихся слоя: нижний, более тяжелый
слой состоит преимущественно из брома, верхний слой — из
фторидов брома.
Для более детальной характеристики фаз, образующихся в
гетерогенных системах бром — фториды брома, а также для выяснения
вопроса об областях существования фтористого брома представляют
интерес результаты исследования фазовых равновесий в системе
бром—трехфтористый бром методом растворимости и термического
анализа [3] **. Это исследование подтвердило, что в жидком
состоянии в данной системе образуются два несмешивающихся слоя,
(причем область несмешиваемости пересекает кривую растворимости
трехфтористого "брома.
•В интервале температур от 9 до 53,9° содержание брома в
верхней фазе увеличивается от 13,9 до 46,8, а в нижней фазе
уменьшается от 95,4 до 80 мол. % ***. Точка смешиваемости обеих фаз
'соответствует температуре 55±0,5° и содержанию 60±5,0 мол. % брома
Рис 10.
Прибор для получения
фтористого брома.
* Продукты реакции подвергали действию 5%-ного раствора 'едкого натра.
Кипячением с избытком гидрата гидразина восстанавливали гипобромит и бро-
мат; избыток гидразина разрушали перекисью водорода. Фтор определяли в виде
CaF2, а бром — в виде AgBr.
** Опыты проводились в сосудах из полихлортрифторэтилена.
*** При температуре Э0,'8°.
47
Та блица 10
Температуры исчезновения второй фазы
Содержание брома,
мол. %
'39,6
48,4
55,6
Температура, °С | ^РД бР0Ма'
51,8
54,5
54,8
55,7
71,1
76,9
Температура, °С
54,9
54,7
52,8
В табл. 10 приведены наблюденные визуально температуры
исчезновения второй фазы, а в табл. 11 и на рис. 11 — результаты
изучения равновесия между твердой и жидкой фазами в системе
бром — трехфтористый
60 -,
обеих
бром и состава
жидких фаз [3].
Экспериментальные
данные табл. И показывают,
что критическая
температура растворения для
системы бром —
трехфтористый бром равна 55,5°.
Так как верхняя часть
кривой несмешиваемости
очень плоская, трудно
определить более или
менее точное значение
критического состава,
который приблизительно
соответствует содержанию
61 мол.% брома В
исследованном интервале
температур в твердой фазе
обнаружены только бром
и трехфтористый бром.
Таким образом, в этой
работе не удалось получить
фтористый бром в
твердой фазе. К сожалению, авторы этой работы при обсуждении
результатов своих измерений не приняли во внимание те равновесные
реакции в системе бром—фториды брома, которые приведены в
работе Руффа и Брайда [2] *.
Физические свойства
Для определения температур плавления и кипения фтористого
брома Руфф и Брайда взяли конденсат, наиболее богатый фторис-
* Недавно было исследовано равновесие жидкость — пар в системе бром —
трехфтористый бром при 75 и 100° [56]. Из результатов исследования можно
заключить, что газообразная фаза является смесью брома, трех фтор истого и од-
нофтористого брома.
Рис. 11. Система бром — трехфтористый
бром:
О —температура замерзания; л —температура
плавления; Ф —температуры исчезновения второй
фазы; □—изотермически, приближение к
равновесию от низких температур; ^—изотермически,
приближение к равновесию от высоких
температур.
48
Таблица 11
Равновесие между жидкими фазами и жидкой и твердой фазами в системе
бром—трехфтористый бром
Содержание
брома,
мол. %
0,00
2,58
5,36
9,80
13,88
19,87
39,61
54,08
69,55
95,22
97,65
99,20
100,0
унивариантная
Кривая
замерзания
8,7
7,4
6,3
4,7
—
—
—
—
—
—
—
—8,0
—7,3
Кривая
плавления
8,6
7,4
6,3
5,0
—
—
—
—
—
—
—3,7
-7,9
-7,1
Температура, °С
инвариантная
Кривая
замерзания
—
3,3
—
3,8
3,6
—
3,8
—
—
—
—
Кривая
плавления
Г~
I 3,4
1 3,6
3,6
3,6
3,5
3,7
3,8
3,5
—
—
—
—
эвтектическая
Кривая
замерза-"
ния
—8,8
—8,6
1 —
—9,0
—9,1
—9,0
—8,8
—8,7
—8,8
—8,8
—8,8
—
Кривая
плавления
—
—8,6
1 -9,0
-8,9
—
-8,9
-8,8
—
—8,6
—8,6
—•
Твердая
фаза
I BrFs
! BrF,
BrF3
BrF3
BrFs
BrF,
BrF3
BrFs
BrFs
BrF3
BrF3
Br2
Br2
тым бромом *. Если охлаждать этот конденсат, он разделяется на
твердую фазу, богатую бромом, и фазу, содержащую фтористый
бром с примесью преимущественно пятифтористого брома.
Последняя фаза расплавилась при нагревании ее до —35°. На этом
основании авторы заключили, что температура плавления фтористого
брома близка к —33°. Расплавленный фтористый бром темно-красного
цвета, в газообразном состоянии он красновато-бурый.
Измеряя изменение упругости пара этого же образца фтористого
брома с температурой, Руфф и Брайда путем экстраполяции до
760 мм рт. ст. определили температуру около 23°. Однако они
приняли температуру кипения фтористого брома равной 20° на том
основании, что примеси, содержавшиеся в исследованном препарате,
несколько повысили температуру кипения.
Измеренная кривая упругости пара соответствует формуле
и молярной теплоте испарения б ккал-моль'1. Однако эти величины,
так же как и вычисленную из них константу Трутона, равную 20,5,
нельзя считать более или менее точными в силу указанных выше
причин.
Термическая устойчивость фтористого «брома быстро уменьшается
с температурой, и уже при 50° он полностью разлагается и превра-
* Соотношение брома и фтора в этом конденсате было равно 1 :2,48. По*
вышенное содержание фтора обусловлено главным образом наличием
пятифтористого брома.
4—1419
49
щается в трехфтористый -и пятифтористый бром с одновременным
выделением брома. Облучение солнечным светом также уменьшает
устойчивость фтористого брома и ускоряет его превращение в
высшие фториды.
Была сделана попытка рассчитать энергию диссоциации
фтористого брома путем анализа полосатых спектров. В спектре системы
бром—фтор в видимой области [4, 5] ясно видны полосы фтористого
брома, но вовсе не проявляются полосы брома. При анализе
полосатого спектра было получено значение энергии диссоциации
фтористого брома «а нормальные атомы BrF=Br+F, равное 59,6±
±0,2 ккал-моль'1.
Дюри [49] также из спектроскопических данных получил по два
значения энергии диссоциации монофторидов хлора, брома и иода,
в зависимости от того, диссоциируют ли молекулы XF на
возбужденный атом фтора F(2Pv2 ) и нормальный атом второго галогена
X(2P«/f ) или наоборот. Эти значения приведены в табл. 12.
Таблица 12
Энергия диссоциации монофторидов хлора, брома и иода
Энергия диссоциации, моль'
XF
C1F
BrF
JF
XF^X(2P3/f) + F(2p1/2)
эв
2,616
2,58
2,87
ккал-
60,3
59,5
66,2
XF^X(«Pt/j + F(*Pi/f)
эв
2,57
2,16
1,98
ккал
59,3
49,8
45,7
Эванс с сотрудниками [50], считая для фтористого брома более
вероятной диссоциацию на возбужденный атом фтора и принимая
для энергии диссоциации более высокое значение, рассчитали
теплоту образования фтористого брома по реакции 1/2Bi2(r),+1/2F2(r) =
=BrF(r) и нашли ее равной 18,3±0,5 ккал-молъ~х (см. также [56]).
Реакция образования фтористого брома из элементов экзотер-
мична. Если принять энергии диссоциации брома и фтора
соответственно равными 45,5 и 39,8 ккал-моль~х, то теплота
образования BrF из газообразных брома и фтора равна ~ 17 ккал-моль~1
[5, стр. 451]. Это значение находится в зависимости от того, какую
величину принять для энергии диссоциации фтора, однако даже этот
приближенный расчет показывает довольно значительную экзотер-
мичность процесса образования фтористого брома. На этом
основании можно полагать, что способность фтористого брома легко
превращаться в высшие фториды объясняется большей устойчивостью
высших фторидов, а не непрочностью связи Вг—F во фтористом броме.
Из полосатых спектров фтористого брома было вычислено, что
о
межъядерное расстояние равно 1,74 А [4]. Изучение микроволновых
50
Таблица 13
Термодинамические свойства BrF(r)
:*;
^Г
0
250
273,16
298,16
300
400
500
600
700
800
900
1000
1100
1200
1300
1400
1500
. X
^ьг«7
* !$
J 41
Ь ?х
0
—46,206
' —46,839
—47,470
—47,514
—49,625
—51,307
-52,713
—53,924
—54,989
—55,940
—56,790
—57,584
—58,305
-58,974
-59,596
1 —60,178
о"о ''О |
1 ^ ^
1 ? о
£,§х
0
1 7,128
7,176
. 7,230
7,234
7,446
7,633
7,789
7,918
8,027
8,118
8,196
8,264
8,323
8,376
8,423
1 8,465
х ]
1
го
2 ^
ьс о
00 Х
0
53,334
54,015
54,700
54,748
57,071
58,940
60,502
61,842
63,016
64,058
64,995
65,848
66,628
67,350
68,019
1 69,643
On ^
^? О
1 ^
5: § J
0
1782,0
1960,2
2155,7
2170,2
1 2978,4
3816,5
4673,4
5542,6
6421,6
7306,2
8196,0
9090,4
9987,6
10888,8
11792,2
1 12697,5
х |
7
0
7,63
7,76
7,88
1 7,88
8,25
8,49
8,64
8,74
8,82
8,88
8,92
8,96
8,99
9,С2
9,04
1 9,07
о
£"1
< § !
-18,3 '
— 18,353
— 18,355
-18,361
-18,302
1 —18,37
—18,38
—18,38
— 18,38
— 18,38
—18,38
—18,38
-18,39
—18,39
— 18,39
-18,39
1 —18,39
1
о f
?
^ 3
< §
—18,3
-18,649
— 18,676
—18,705
—18,707
—18,82
—18,93
—19,04
-19,15
-19,26
— 19,37
— 19,48
— 19,59
—19,70
—19,81
—19,92
1 —20,03
Igff/
I 16,3028
14,9419
13,7107
13,6279
10,283
8,275
6,936
5,980
5,2п2
4,704
4,258
3,892
3,588
3,330
3,109
| 2,918
спектров дало более точное значение—1,759 А [61. Эта величина
очень близка к сумме нормальных ковалентных радиусов брома и
фтора: 1,14 + 0,64= 1,80 [7]. Но, как и в случае фтористого хлора
(стр. 27), она немного меньше аддитивного значения межъядерных
расстояний — полусуммы расстояний Вг—Вг и F—F в газообразном
о
состоянии и равна V2 (2,284+1,435) = 1,859 А. Отсюда следует, что
связь Br—F в молекуле фтористого брома имеет частично ионный
характер. Действительно, молекулы BrF полярны, их дипольный
момент, определенный из данных микроволновой спектроскопии, равен
1,29 D (см. табл. 4).
Термодинамические свойства фтористого брома, по данным [50],
приведены в табл. 13.
Неустойчивость фтористого брома значительно затруднила
получение его физико-химической характеристики.
Химические свойства
По своим химическим свойствам фтористый бром очень сходен с
другими фторидами брома, но он очень мало исследован в этом
отношении.
Руфф и Брайда [2] отмечают, что BrF более активен, чем другие
фториды брома, и объясняют это его способностью к превращениям.
По данным этих авторов, фтористый бром энергично разъедает пла-
51
тину и золото при условиях, в которых на них не действуют ни
бром, ни даже фтор. Аналогично этому и кварц быстро разрушается
фтористым бромом. Вообще же трудно разграничить химическое
действие фтористого брома, трехфтористого, пятифтористого брома
и брома, с которыми он находится в равновесии.
2. ТРЕХФТОРИСТЫЙ БРОМ BrF3
Получение
Взаимодействие фтора с бромом впервые кратко описал Муае-
сан [8]. Он наблюдал, что фтор на холоду энергично соединяется с
парами брома с появлением светящегося пламени, однако без
выделения большого количества тепла. Если вводить фтор в жидкий
сухой бром, то также происходит непосредственное соединение обоих
галогенов, но без появления пламени. Кроме того, Муассан
наблюдал соединение фтора с бромом при разложении фтором бромидов
калия, цинка, хрома. Эту реакцию можно выразить, например, таким
уравнением:
KBr+2F2 = KF+BrF3.
Трехфтористый бром впервые получил Лебо в 1905 г. [9]. Он
установил его состав и описал некоторые его физические и химические
свойства. Этот новый фторид в том же году независимо от Лебо
получил Придо [10].
Лебо получал трехфтористый бром, пропуская фтор под
поверхность сухого жидкого брома, помещенного в платиновый тигель,
охлаждаемый сухим льдом; температура поддерживалась немного
выше точки замерзания брома (—7,2°). Пары трехфтористого брома
конденсировались в тигле и образовывали верхний слой, почти не
растворимый в жидком броме. Трехфтористый бром в свою очередь
растворяет немного брома, образуя раствор желтого цвета.
Руфф и Менцель [11] наблюдали образование трехфтористого
брома при взаимодействии брома с фтором отри 200° (в процессе
получения пятифтористого брома). Руфф и Брайда [12] описали
метод получения трехфтористого брома путем одновременного ввода
брома и фтора в медный Т-образный сосуд. Бром вводился в виде
паров в токе азота. Образовавшийся трехфтористый бром стекал в
охлаждаемый водой никелевый приемник и затем перегонялся при
20° в высоком вакууме *.
В настоящее время трехфтористый бром получают в медном
реакторе с несколько наклоненным трубчатым водяным
холодильником. Бром подают в верхний конец реактора, а фтор вводится около
нижнего конца, из которого отбирается BrF3. Применяемое
охлаждение достаточно для поглощения тепла, выделяющегося в резуль-
* Трехфтористый бром уже при 30° заметно реагирует с кварцем с
образованием брома, кислорода и четырехфтористого кремния, поэтому перегонку
трехфтористого брома из кварцевых сосудов необходимо вести при возможно
более низких температурах.
52
тате реакции (около 70 ккал/'моль BrF3), -и для поддержания
температуры в пределах 15—50° [13].
Аналогичный метод получения трехфтористого брома подробно
описан Квасником [59].
Имеется также указание о получении трехфтористого брома в
железном аппарате в условиях 10% избытка фтора и при
температуре 80—100°, с выходом 98% [14].
Трехфтористый бром можно сохранять в никелевых или стальных
сосудах. Непосредственно перед применением его следует перегнать
в вакууме, чтобы очистить от фторидов металлов, образующихся в
результате действия трехфтористого брома на материал сосуда.
Физические свойства
По данным Руффа и Брайда, трехфтористый бром представляет
собой при комнатной температуре слабоокрашенную желто-зеленую
жидкость [12], сильно дымящую на воздухе. Однако еще Лебо [9]
описал трехфтористый бром, как бесцветную жидкость, которая при
контакте с воздухом окрашивается в желто-оранжевый цвет.
Твердый трехфтористый бром, полученный из жидкой фазы, образует
блестящие иглы до 20 мм длины и 1 мм толщины. Из газовой фазы
он получается в виде призм [15]. По Лебо, твердый трехфтористый
бром бесцветен. Другие авторы отмечают, что он имеет такую же
окраску, как и жидкий препарат [5, стр. 452].
По более новым данным [12, 15], т. пл. BrF3=8,8°. Плотность
жидкого трехфтористого брома при температуре плавления
^8.8 _ 2843 г/мл, а твердого препарата при той же температуре
3,23 г/мл. Зависимость плотности жидкого трехфтористого брома от
температуры может быть (выражена уравнением [12] *:
й\ = 3,623 — 2,77- 10-*Г или &\ = 2,867 - 2,77-10~3/°.
Молярный объем жидкого трехфтористого брома, таким образом,
составляет 48,16 при температуре плавления.
Руфф и Брайда [15] определили давление паров BrF3 в интервале
температур от —5 до 80° ** и полученные ими значения выразили
в виде следующего уравнения:
lgp = 8,41454-2220,2/7.
Температура кипения трехфтористого брома, рассчитанная из
этого уравнения 127,6° (или 127±1°), оказалась немного ниже той
величины (130—140°), которую определил Лебо [9] при непосред-
* Авторы не указали, в каком температурном интервале соблюдается это
уравнение, но даже при температуре кипения оно дает величину,
согласующуюся с опубликованным значением, равным 2,515 [16].
** До 20° измерения можно было проводить в кварцевой аппаратуре. При
более высоких температурах давление паров измерялось по Смит-Менцесу в
платиновом сосуде.
53
ственном измерении*. Теплота испарения, вычисленная из
приведенного выше уравнения, составляет 10,15 ккал-моль~К Константа
Трутона оказалась равной 25,3 кал-град^-моль'1, что указывает на
ассоциацию молекул трехфтористого брома в жидком состоянии.
Приближенное значение критической температуры (по правилу
Гульдберга) равно 328°.
Термодинамические свойства трехфтористого брома еще
недостаточно изучены. Оливер и Гризард [17] уточнили некоторые данные,
полученные Руффом с сотрудниками и расширили эти исследования.
По их измерениям, молярная теплоемкость трехфтористого брома в
интервале температур 15,32—316,27° К изменяется от 1,279 до
30,00 кал-град"1. Среднее значение теплоты плавления (из трех
измерений) равно 2874,6 ±3,0 кал-моль'1. Измерив давление пара
BrF3 в пределах от 38,72 (17,67 мм рт. ст.) до 154,82° (1808 мм рт. ст),
Оливер и Гризард предложили следующую зависимость:
lgp = 7,74853 - 1685,8 / (/ + 220,57).
Более значительные расхождения между наблюденными и
вычисленными величинами давления пара при относительно высоких
температурах (Рнаблюд. —^вычисл.=—2,5 мм) для 154,82° эти авторы
объясняют частичным разложением трехфтористого брома. Теплота
испарения, вычисленная по уравнению
AHv = 7714 [(t + 273,16) / (t + 220,57)]2,
оказалась равной при температуре кипения 10,235, а при 25°
11,370 кал'моль'1] теплота плавления в тройной точке (8,77°) равна
2874,6 ±3 кал-моль'1; энтропия парообразования 38,14 кал-град~1Х
Хмоль~1\ энтропия жидкого трехфтористого брома (25°) 42,57±0,10
и энтропия идеального газа (1 атм, 25°) 71,57 кал-град^-моль-1.
Величины диэлектрической проницаемости, молярной
поляризации, рефракции, магнитной восприимчивости и дипольного момента
трехфтористого брома указаны в табл. 2 и "4.
Описаны также некоторые физические свойства: показатели
преломления, плотность, парциальные моляльные объемы системы
BrF3—BrF5 (и исходных компонентов) при нескольких
температурах. Предложены формулы для расчета состава смесей по
рефрактометрическим данным и плотности [53].
Эмелеус и Вулф [18] предложили определить теплоту
образования трехфтористого брома на основании реакции
3Si02 + 4BrF3 = 3SiF4 + 302 + 2Br2.
Однако эта величина еще не установлена.
Трехфтористый бром имеет непрерывный спектр поглощения, не
дающий указаний на вибрационную структуру [19]. При нагревании
* Руфф и Брайда объясняют это расхождение тем, что Лебо производил
свои измерения, поместив термометр в пары кипящего трехфтористого брома,
которые при этом действовали на стекла термометра (см. сноску на стр. 52) с
выделением тепла. *
54
трехфтористого брома, содержащего свободный бром, до 70°
абсорбция, обусловленная бромом, ослабляется и появляется новая полоса
поглощения в пределах 3000—3600 А с максимумом около 3250 А;
при охлаждении появляется первоначальный спектр. Этот факт
можно объяснить образованием фтористого брома (BrF3 + Br2^I 3BrF)
и его последующим разложением на исходные вещества [19] *.
Микроволновой спектр трехфтористого брома еще не изучен.
Более ранние исследователи [10] полагали, что молекулы BrF3 плоские,
с атомом брома в центре равностороннего треугольника и тремя
атомами фтора на вершинах [20]. Результаты электронографического
изучения дали основание принять для -молекулы BrF3
пирамидальную модель с межъядерным расстоянием для связи Br—F, равным
1,78 А и углом FBrF, равным —86°, как и в трехфтористом
хлоре [21].
Трехфтористый бром довольно хорошо проводит ток в жидком
состоянии. Банкс, Эмелеус и Вулф [22] измерили
электропроводность хорошо очищенного препарата в пределах температур от 5 до
60° в кварцевом сосуде, который наполнялся непосредственной
перегонкой препарата под вакуумом **. Электропроводность
твердого трехфтористого брома при повышении температуры от 4 до 8°
увеличивается от 2,3 • 10~5 до 17,3 • 10~5 ом^см'1. Электропроводность
жидкого препарата (рис. 12) в интервале температур 9—20° почти
постоянна (8,13—8,09-10~3); с, дальнейшим ростом температуры
электропроводность немного уменьшается и при 60° достигает
значения 6,75-10-3 ом~1 см'1 ***.
Большое различие (примерно в 100 раз) в электропроводности
трехфтористого брома оз твердом и жидком состоянии указывает на
то, что электропроводность его имеет ионный характер. Это
обстоятельство, а также некоторые особенности в химическом поведении
трехфтористого брома дали основание считать, что его
электропроводность обусловлена диссоциацией димерных молекул на ионы по
схеме
(BrF3)2^BrF2+ + BrF4-.
Подтверждением этой схемы электролитической диссоциации
могут служить также явления, наблюдавшиеся при длительном
электролизе трехфтористого брома. При этом жидкость в анодном
пространстве оставалась бесцветной, а католит становился коричне-
* Об инфракрасном спектре трехфтористого брома см. [54].
* * Трехфтористый бром, полученный из элементов, очищался перегонкой при
атмосферном давлении в стальной аппаратуре; собиралась фракция, кипящая
при 126-—128°. Эту фракцию затем перегоняли в восстановительной атмосфере
при комнатной температуре. Все возможные примеси: Br2, HF, BrF, BrFs — более
летучи, чем BrF3, кроме того, их удаление сопровождается изменением цвета
жидкости от оранжевого до светло-желтого. Для окончательной очистки
жидкость перегонялась в вакууме в сосуд для измерения электропроводности.
*** Следует отметить, что трехфтористый бром, переохлажденный до 4,4°,
показал электропроводность 8,12 • 10~3 оленем-*, характерную для жидкого
состояния.
55
8,0
Т.ОА
вым [22]. Эти явления можно объяснить электродными процессами,,
сопровождающимися реакциями диспропорционирования:
2BrF4~" — 2е = BrF3 + BrF5;
2BrF2+ + 2е = BrF + BrF3.
(Пятифтористый бром — бесцветен, об окраске однофтористого
брома см. стр. 46).
Тот факт, что трехфториетый бром проводит ток в твердом
состоянии и притом с положительным температурным коэффициентом,
позволяет предположить [22], что BrF3 имеет ионную
кристаллическую решетку. Он проявляет в этом
*'10> х /}. отношении аналогию с пятихло-
^* ^--^#х ристым фосфором, кристалличе-
^■^* екая решетка которого состоит
^"\ из ионов РСЦ+ и РС16~. Так как
эти ионы в кристаллах пятихло-
7о Jo То То Jo во г ристого фосфора упакованы та-
~ 1П __ ким же способом, как ионы Cs+
Рис. 12. Электропроводность трех- ^,_ .
фтористого брома (двух образцов- и С1 в кристаллах хлористого це-
А и В). зия [23], то было высказано
суждение, что в кристаллической
решетке трехфтористого брома ионы одного знака, например BrF\f,
окружены восемью ионами другого знака (BrF2+) [20]. Однако это
предположение должно быть подтверждено экспериментальными
данными.
Отрицательный температурный коэффициент электропроводности
жидкого трехфтористого брома, очевидно, связан с термической
диссоциацией самого трехфтористого брома или его ионов. Повторное
измерение электропроводности BrF3 при 20°, после его
предварительного нагревания до 60°, дало первоначальную величину.
Зависимость силы тока от напряжения подчиняется закону Ома
и имеет линейный характер. Обнаружить напряжение разложения
не удалось, что вообще характерно для некоторых других межга-
логенидов и их комплексных соединений. Этот вопрос, как и вопрос
о других электрохимических свойствах межгалогенидов, будет
рассмотрен в главе IX.
Строение ионов, на которые диссоциирует трехфториетый бром,
представляется в следующем виде: для катиона BrF^"1" более
вероятно не линейное, а угловое расположение связей: анион BrF4~ —
плоский квадрат со стороной 4,84 А [14, 20].
Однако недавно произведенное рентгенографическое
исследование KBrF4 показало, что четыре атома фтора тетраэдричеоки
расположены вокруг брома; расстояние Вг—F равно 1,81, F—F равно
2,81 и 3,03, К — 8F равно 2,84 А [60].
Химические свойства
Трехфториетый бром относится к числу наиболее активных в
химическом отношении межгалоидных соединений. Он проявляет
сильное окислительное действие и обладает весьма значительной способ-
56
ностью фторировать неорганические и органические вещества,
будучи близок в этом отношении к свободному фтору. Известные
преимущества применения трехфтористого брома для указанной цели
(особенно для введения фтора в неорганические соединения)
заключаются в следующем: реакции фторирования можно проводить в
жидкой фазе и при комнатной температуре; в трехфтористом броме
как растворителе можно осуществлять реакции ионного типа;
реакции фторирования часто протекают количественно — избыток BrF3
можно легко и почти без потерь удалить. Кроме того, трехфтористый
бром можно легко транспортировать и сохранять в жидком виде в
металлических сосудах.
Необходимо, однако, отметить, что работы с трехфтористым
бромом сопровождаются экспериментальными трудностями и требуют
соблюдения ряда предосторожностей: попадание незначительных
количеств воды вызывает бурные, взрывного типа реакции;
необходимо пользоваться сильной (и сухой) тягой, работать в асбестовых
перчатках и иметь наготове вату и четыреххлористый углерод для
смывания разбрызгиваемых капель. Реакции проводят чаще всего
в кварцевых сосудах, причем при добавлении трехфтористого брома
в сосуд последний необходимо охлаждать жидким воздухом. Однако
и в этих условиях некоторые реакции протекают очень бурно и с
выделением пламени. Предварительное разбавление реакционной
смеси сухим бромом позволяет значительно умерить интенсивность этих
реакций. При попадании трехфтористого брома на кожу образуются
такие же ожоги, как и при действии фтористого водорода [24].
Реакции с неорганическими соединениями
Уже первые опыты Лебо показали большую химическую
активность трехфтористого брома в 'отношении ряда металлов и
металлоидов. Так, по данным Лебо [9], бор, кремний, мышьяк в порошке,
сурьма и висмут сгорают в жидком трехфтористом броме с
выделением тепла и пламени. Иод реагирует даже с твердым
трехфтористым бромом, с жидким трехфтористым бромом реакция протекает
бурно, причем образуются пятифтористый иод и пары брома;
реакция сильно экзотермична. Сера вспыхивает в жидком трехфтористом
броме, образуя смесь фторидов и бромида серы.
Трехфтористый бром энергично реагирует с водой, причем
образуются 02, HF, НВгО и НВгОз. Первичную реакцию Лебо
представляет следующим уравнением [91:
2BrF3 + 4H20 = 2HBrO + 6HF+02 *.
Аналогично протекает реакция взаимодействия трехфтористого
брома с водными растворами щелочей, например [9]:
2BrF3+8NaOH = 2NaBrO-f 6NaF+4H20 + 02.
* Шарп и Эмелеус [25] полагают, что первичная реакция взаимодействия
трехфтористого брома с водой выражается уравнением
BrF3 + 2Н 20 = НВг02 + ЗН + + 3F"
5Г
Эмелеус и его сотрудники, изучавшие в последние годы реакции
трехфтористого брома с неорганическими веществами, показали, что
он способен превращать простые вещества, окислы, галогениды,
соли во фториды различных элементов, которые могут соединяться с
избытком трехфтористого брома и образовывать комплексные
фториды.
Для того чтобы иметь представление об условиях и некоторых
особенностях этих реакций, необходимо обратить внимание на
отношение фторидов различных элементов к трехфтористому брому [24].
По своему отношению к трехфтористому брому фториды других
элементов можно разделить на четыре группы.
а. Фториды, реагирующие с трехфтористым
бромом. Легко растворимые в трехфтористом броме фториды,
натрия, калия, рубидия, цезия, серебра, бария и некоторых других
элементов образуют с ним комплексные соединения — двойные
фториды *. Так были получены следующие комплексные фториды:
MxBrF4 (М1 = Na, К, Ag) [25], BaBr2F8 [251, AuBrF6 [26], SnBr2F10 [27], NbBrF8,
TaBrFe [28], SbBrF8 [27], BiBrF8 [28], PdBrFG, PtBr2F10 [29] и т.п. (см. табл. И).
Некоторые комплексные фториды были получены таким путем
лишь в виде растворов в трехфтористом броме, а именно: RbBrF4,
CsBrF4, CaBr2F8 [25], TiBr2F10 [30], VBrF8 [31], S03BrF3 [32] и т. д.
Для соединений щелочных, щелочноземельных металлов и
серебра Эмелеус и его сотрудники принимают строение типа
M(BrF4) n, где /г=1 или 2. Фторидам же иных элементов они придают
строение типа [BrF2]m[RFn]—с дифторобромониевьгм катионом,
например, [BrF2][AuF4l; [BrF2][SbF6]; [BrF2]2[SnF6j и т. п.
б. Фториды растворимые и летучие, не
реагирующие с трехфтористым бромом. К этой группе
относятся лишь немногие перечисленные далее фториды, так как
большинство растворимых в BrF3 фторидов образует с ним соединения
(группа а). Здесь идет речь главным образом о легколетучих
соединениях (в скобках указаны температуры кипения): MoF6 ( + 35°),
WF6 ( + 19°), UF6 ( + 56°), SF6(—64°, сублимируется), SeF6
(-34,5°), TeF6 (-35,5°), ReF6 ( + 47°), C1F3 ( + 12°), JF5 ( + 98°).
в. Трудно- и малорастворимые фториды. Такими
являются фториды следующих катионов: лития, меди (II),
серебра (II), бериллия, магния, цинка, кадмия, ртути (HgF2), алюминия,
галлия, индия, таллия (III), церия (III и IV), циркония, тория, свин-
* Шефт, Хайман и Кац [46] количественно определили растворимость
фторидов ряда элементов в трехфтористом броме при 25° (в граммах металла на
100 г раствора). По их данным, растворимость фторидов щелочных (Li, Na, К,
Cs) и щелочноземельных (Са, Ва) металлов резко возрастает с увеличением
атомного веса металла —от LiF (0,12^0,003 г) к CsF (18,8±l,0i г) и от
€aF2 (0,017±0,009 г) к BaF2 (3,53±0,2 г). Растворимость AgF составляет
3,22±0,3 г серебра на 100 г раствора. Фториды Al, La, Zr, Th, Ni2+ и Cu2+ очень'
мало растворимы. В отличие от них NbF5 растворим очень хорошо —15,7 г
металла на 100 г раствора, что объясняется образованием комплекса
BrF2+NbF6.
58
ца (II и IV), висмута (III), хрома (III), марганца (II и III),
железа (III), кобальта (II и III), никеля (II).
г. Фториды, окисляемые трехфтористым б р о-
м о м. Сюда относятся фториды элементов с переменной
валентностью, которые в трехфтористом броме переходят в высшие
фториды: SnF2, TiF3, PF3, AsF3, SbF3, VF3,VF4, MoF3, WF4, UF3, UF4.
Фториды двухвалентных свинца, железа и кобальта лишь
неполностью переходят в высшие фториды. Некоторые из перечисленных
здесь фторидов (фосфора, мышьяка, сурьмы, олова, ванадия) после
окисления могут давать комплексные соединения с трехфтористым
бромом, характерные для высших степеней валентности фторидов
этих элементов (см. выше).
Фторирование неорганических соед и нен и й. Для
получения простых и комплексных фторидов действуют
трехфтористым бромом на металлы или металлоиды, окислы, галогени-
ды или соли кислородных кислот. Эти реакции протекают
количественно в тех случаях, когда образующиеся фториды растворимы в
трехфтористом броме. Обычно получаются фториды элементов в их
высших степенях валентности. Исключение представляют лишь те
фториды — AgF2, CeF4, BiFs, CrF4, которые получаются при
действии элементарного фтора и при повышенной температуре.
Образование на поверхности металла слоя фторида, не
растворимого в трехфтористом броме, препятствует дальнейшей реакции
фторирования.
Реакции с металлами. Ряд металлов, как-то: золото [26],
олово [27], ванадий [31], ниобий и тантал [28], молибден, вольфрам,
рений, цирконий [24], рутений [48] и некоторые другие — энергично
(иногда с выделением пламени) и количественно реагируют с
трехфтористым бромом. На металлическую платину BrF3 не действует.
Для первых пяти из перечисленных выше металлов и рутения
установлено образование комплексных фторидов (см. стр. 58 и
табл. 14).
Реакции с окислами. Из 33 исследованных окислов [18,
24] лишь 15 количественно реагировали с жидким трехфтористым
бромом. Эти реакции контролировались по количеству выделенного
кислорода, а также по количеству и химическому анализу продукта,
остающегося после отгонки избыточного BrF3. Так, в условиях
реакции CuO, B203, T1203, Si02, Ge02, Ti02, U03, J205 превратились во
фториды этих элементов той же валентности [18]. В качестве
примера можно привести уравнения реакций с Si02 (стр. 54) и J205i
6J2O6 + 20BrF3 = 12JF5+1502+ 10Br2.
Мышьяковистый и селенистый ангидриды превращаются в AsF5
и SeF6 [18]. Окись сурьмы Sb203 [18, 27], а также Nb205 и Та205 [28]
полностью перешли в комплексные фториды типа [BrF2]+[MF5] (где
M=Sb, Nb, Та).
Растворяя серный ангидрид в трехфтористом броме с
последующей отгонкой последнего, Вулф [32] получил белое твердое клейкое
вещество, содержащее «а 1 моль S03 ~ 0,7 моля BrF3, которое, по
59
его мнению, является продуктом частичного разложения фторсуль-
фоната дифторобромошия [BrF2]+ [S03F]".
Многие из исследованных окислов металлов не реагировали
количественно с трехфтористым бромом — после отгонки BrF3
получалась смесь фторида с исходным окислом, соотношение между
количествами которых зависело от условий опыта. Такие результаты
были получены для окислов: BeO, MgO, ZnO, CdO, HgO, CaO, A1203,
Ce02, Sn02, PbO, Pb02, Zr02, Th02, Bi203, Mn02, Fe203 [18, 24].
Пятиокиси ванадия и висмута (70% Biv) превратились в VOF3 и
смесь ранее неизвестного BiOF3 с BiF3 и неизменившимся Bi203 [28].
Однако, по данным Шефта с сотрудниками [60], кипящий трех-
фтористый бром количественно фторирует ряд перечисленных выше
окислов, а именно: BeO, HgO, Ce02, PbO, Zr02, Bi203, Мп02, а
также Та2Об, Со203 и NiO. Но такие окислы, как CuO, MgO, ZnO, CdO,
А1203, La203, Сг203, Fe203, не реагируют количественно с
трехфтористым бромом даже при длительном нагревании при 300°.
В заключение этого раздела следует упомянуть о получении
смешанных фтор ангидридов угольной, серной и сернистой кислот. Так,
действием трехфтористого брома на СО, S02 (при одновременном
действии брома) и SOCl2 были получены карбонилбромфторид
COBrF, сульфурилбромфторид S02BrF и тионил хлор фторид SOC1F
([14], стр. 120).
'Реакции с галогенидами* [18, 25—29, 31]. Реакции
взаимодействия галогенидов различных элементов с трехфтористым
бромом, в условиях избытка последнего, можно разделить на
несколько типов в зависимости от природы элемента, его валентного
состояния в данном галогениде, растворимости фторида этого
элемента в BrF3, его температуры кипения и способности соединяться
с BrF3.
1. Превращение хлоридов, бромидов и иодидов элементов с
постоянной валентностью в не растворимые в BrF3 фториды; так,
например, реагируют LiCl, CdJ2, А1С13 и т. д.
6L1C1 + 2BrF3 = 6UF + Вг2 + ЗС1а;
3CdJ2 + 2BrF3 = 3CdF2 + Вг2 + 3J2**.
2. Превращение галогенидов элементов низших степеней
валентности— CuCl, T1C1, РЬС12, PbBr2 и ряда других — в смесь
фторидов низших и высших валентностей: CuF и CuF2> TIF и T1F3, PbF2
и PbF4 и т. д. ***.
3. Образование летучих фторидов элементов в их высших
степенях валентности: MoF6, WF6, UF6, SF6 и т. д. (см. стр. 58).
4. Образование комплексных тетрафторобромиатов щелочных,
щелочноземельных металлов и серебра M(BrF4) .
* Об отношении фторидов к BrF3 см. стр. 58.
** Приведение уравнения реакций имеют предположительный характер.
*** Галогениды трехвалентного висмута не превращаются трехфтористым
бромом в BiF5, вероятно, потому, что образующийся сперва BiF3 не растворим
в трехфтористом броме [18, 28].
60
5. Образование комплексных фторидов с дифторобромониевым
катионом [BrF2]+ [RFJ-. Такие реакции описаны, например, для AuCl [26],
SnCl2 и SnCl4, SbF3 [27], VC13 [311, PdCl2, PtCl4, PtBr4 [29] и т. д.
Реакции четвертого и пятого типов, вероятно, протекают в два
этапа: сперва образуются «простые» фториды, которые затем
соединяются с избыточными молекулами BrF3. Вопрос о причинах
образования комплексных фторидов с анионом.BrF4~или катионом
BrF2+ будет рассмотрен в главе XIII.
Реакции с солями кислородных кисло т. Многие
соли кислородных кислот сравнительно легко реагируют с трех-
фтористым бромом, однако эти реакции протекают более сложным
путем, чем предыдущие. Часто при этом образуются комплексные
фториды, катионом которых является катион исходной соли, а
центральный атом последней (кислотообразующий элемент)
переходит в комплексный фторидный анион. Так, например, при
действии избытка BrF3 на метафосфаты образуются гексафторофосфа-
ты— KPF6, AgPF6 и т. п.
Чтобы объяснить природу реакций образования таких ком-
ллексных фторидов, можно предположить, что сперва образуются
«простые» фториды, например KF и PF5,
2КР03 + 4BrF3 = 2KF + 2PF5 + 2Вг2 + ЗОа,
которые затем соединяются друг с другом. Однако Эмелеус и
сотрудники полагают, что молекулы первично образовавшихся
фторидов сперва соединяются с молекулами BrF3 и превращаются
(применительно к данному примеру) соответственно в KBrF4 и
BrF2PF6, которые затем взаимодействуют по ионному механизму
K+BrFi" + BrF^PFr = KPF6 + 2BrF3.
Согласно Эмелеусу, подтверждением такого представления о
механизме образования комплексных фторидов этого типа при
взаимодействии солей кислородных кислот с BrF3 может быть тот
факт, что эти же реакции можно осуществить, смешивая отдельно
приготовленные растворы M(BrF4)„ с [BrF2]m[RFj или внеся в
BrF3 вещества, которые, реагируя с последним, дают в
отдельности эти два типа бромфторидных комплексов. Так, например, при
взаимодействии металлического серебра и золота с BrF3
получается AgAuF4 [26]. Очевидно, вначале в бромтрифторидном
растворе образуются AgBrF4 и BrF2AuF4, которые затем реагируют
друг с другом по уравнению
Ag+BrFr + BrF^AuFr = AgAuF4 + 2BrF3.
В тех случаях, когда в исходной соли кислородной кислоты
количества катиона и центрального атома отличаются от того
стехиометрического отношения, которое необходимо для
образования комплексного фторида, часто получается смесь последнего
с бромтрифторидным комплексом. Так, например, ортофосфат
натрия при реакции с BrF3 дает смесь 1 моля NaPF6 и 2 молей
61
NaBrF4; Ag3As04 превращается в смесь AgAsF6 и AgBrF4 в таком
же молярном отношении и т. д.
Если анион исходной соли не способен реагировать с трехфто-
ристым бромом и превращаться в комплекс типа [BrF2]m[RFn], та
получаются только тетрафторобромиат данного катиона M[BrF4]n;
непременным условием является растворимость фторида этого
катиона в трехфтористом броме. Поэтому при взаимодействии
карбонатов с BrF3 карбонат натрия дает NaBrF4, а карбонаты
лития и бериллия — только LiF и BeF2. Аналогичные же явления
наблюдаются в реакциях иодатов с трехфтористым бромом: в
системах с KJ03 или NaJ03 образуются KBrF4 или NaBrF4. В то
же время иодаты ртути, таллия (I), марганца (II), свинца (II) и
кобальта (II) в этих же условиях дают только фториды этих
металлов — HgF2, T1F3, смеси MnF2 и MnF3, PbF2 и PbF4, CoF2
и CoF3 [18}. Реакция с хлоратом калия протекает таким
образом, что наряду с KBrF4 образуется фтористый хлорил [47]'
6KClO3+10BrF3=6KBrF4+2Br2+3O2+6ClO2F.
В реакциях с солями кислородных кислот серы были получены
следующие результаты: пиросульфат и персульфат калия
превращаются в фторсульфонат калия KS03F; из дитионата натрия был
получен NaBrF4; сульфаты калия и серебра дают смесь эквимо-
лярных количеств KBrF4 и KS03F [18].
Все эти факты Эмелеус и Вулф объясняют, исходя из указанного
выше представления о реакциях образования комплексных фторидов
в трехфтористом броме как растворителе — первичными
продуктами реакции являются KBrF4 и BrF2S03F, которые затем
взаимодействуют между собой.
Эти реакции, применительно к сульфату калия, Вулф [32]
изображает следующими схемами:
K2S04 -*- 32KBrF4 + [BrF2] S03F;
KBrF4 + [BrF2] SO3F+ KS03F + 2BrF3.
Однако с этой точки зрения еще нельзя объяснить, почему в
реакциях с тиосульфатом и пиросульфатом калия, содержащих
равные количества атомов калия и серы, так же как и в случае
сульфата калия, образуется смесь (1:1) KBrF4 и KS03F [18].
Хотя Гутман и Эмелеус считают доказанным образование
комплекса BrF2B:F6 [28], а для ванадия имеется предположение о
существовании BrF2VF6, при реакции между BrF3 и виомутатами или
ванадатами не удалось получить гексафторвисмутатов или гекса-
фторванадатов *.
Все эти факты показывают, что реакции между солями
кислородных кислот и трехфтористым бромом в ряде случаев протекают
более сложно, чем это следует из изложенного выше представления
о механизме1 таких реакций.
* Эти комплексы получены иным путем [2в, 31].
62
Комплексные соединения трехфтористога
брома с фторидами других элементов
В предыдущем разделе показано, что с участием BrF3 как
реагента и растворителя можно осуществить реакции получения
разнообразных комплексных фторидов, которые следует
разделить на две основные группы: продукты соединения BrF3 с
фторидами других элементов и двойные фториды, не содержащие в
своем составе трехфтористого брома.
Рассмотрим сперва соединения первой из этих групп.
Примеры комплексных соединений BrF3 с фторидами других
элементов — тетрафторобромиатов M(BrF4)n и дифторобромониевых
[BrF2]m[RFn]—приведены выше (стр. 58). Эти комплексные
фториды получаются при непосредственном взаимодействии BrF3 с
простыми веществами, окислами, галогенидами, солями
кислородных кислот, с последующим испарением избытка трехфтори-
стого брома. Среди этих комплексов имеются представители,
значительно различающиеся по термической устойчивости. Поэтому
ряд таких соединений, обладающих сравнительно большой
упругостью диссоциации, не удалось выделить этим способом в
индивидуальном состоянии и об их образовании в трехфтористом
броме, а также об их составе в этом растворе можно было
судить только косвенным путем, изучая реакции комплексообразо-
вания и двойного обмена в трехфтористом броме как
растворителе.
Комплексные соединения фторидов ряда элементов с BrF3
образуют в трехфтористом броме проводящие ток растворы [27].
Эмелеус и сотрудники рассматривают растворы этих комплексов
в трехфтористом броме с точки зрения теории сольвосистем [33].
Исходя из того, что собственная электролитическая диссоциация
трехфтористого брома [22] выражается схемой 2BrF3 ^BrF2++
+ BrF4~, и полагая, что комплексные фториды, о которых здесь
идет речь, могут быть разделены на соединения с дифторобромо-
ниевым катионом [BrF2]+, например [BrF2] [SbF6], и с тетрафторобро-
миатным анионом [BrF4]-, например KBrF4, эти авторы считают
комплексы первого типа кислотами, а комплексы второго типа —
основаниями по отношению к трехфтористому брому *.
Взаимодействие же растворов этих двух типов комплексов они
уподобляют реакции нейтрализации, в результате которой образуются
новые комплексные фториды —продукты соединения фторидов двух
элементов. Таковы, например, следующие реакции:
Ag+BrF^ + (BrF2)+SbF6-=AgSbF6 + 2BrF3; [27]
2K+BrF^ + (BrF2)24SnF2-=K2SnF6 4 4BrF3. [24]
Образование таких комплексных фторидов было установле-
* Комплексы первого типа обладают катионом, а комплексы второго
типа — анионом, общим с растворителем — трехфтористым бромом.
63
tio не только препаративным путем (изолирование комплексов
после испарения избытка трехфтористого брома), но и методом
кондуктометрического титрования, причем на кривой
титрования наблюдался перегиб, соответствующий определенному сте-
хиометрическому отношению компонентов реакции. Так, при
титровании раствора AgBrF4 в трехфтористом броме раствором
комплекса BrF2SbF6 в том же растворителе минимум
электропроводности наблюдался при эквимолярном соотношении
компонентов. Если же титровать раствор KBrF4 раствором (BrF2)2SnF6 в
трехфтористом броме, минимум проводимости соответствует
соотношению этих компонентов 2:1, что иллюстрируется
уравнениями реакций, приведенными выше.
Такой ход кривой кондуктометрического титрования
объясняется тем, что в процессе титрования происходит замещение бо-
©
лее подвижного иона BrF4 с меньшим ионным радиусом (2,39 А)
ионами SbF7 и SnFJc большими ионными радиусами (rsbF7 =
о
= 2,64А). Следует также отметить, что в первой из этих систем
после достижения точки эквивалентности наклон изотермы
электропроводности такой же, как и для растворов BrF2SbF6 в
трехфтористом броме.
Результаты этих кондуктометрических определений были
использованы Эмелеусом и его сотрудниками для того, чтобы
составить суждение о строении комплексных соединений BrF3 с
фторидами других элементов. Так как в данных случаях имеют
место реакции ионного типа, в результате которых образуются соли
AgSbFe, K^SnFe и т. п., то, следовательно, ионы, входящие в их
состав, содержались в исходных комплексах. Это дает основание
приписать тетрафторобромиатньгм комплексам строение типа
Mn+ [BrF4]n, а дифторобромониевым соединениям — строение типа
IBrF2l+[RF„p-
Способность фторидов различных элементов образовывать
комплексы того или иного типа можно объяснить их относительной,
по сравнению с BrF3, способностью проявлять в этих реакциях
свойство донора (NaF, KF, BaF2, AgF) или акцептора фтора
(AuF3, SnF4, SbF5, VF5, PtF4, BiF5 и т. п.) *.
Это представление о строении комплексных фторидов,
образованных трехфтористым бромом, 'можно считать в известной мере
обоснованным результатами упоминавшегося выше
кондуктометрического титрования, а также характером собственной
электролитической диссоциации трехфтористого брома и донорной или
акцепторной (в отношении ионов фтора) способностью фторидов,
реагирующих с трехфтористьгм бромом. Все же необходимо
непосредственное доказательство строения этих комплексов и природы
ионов, на которые они диссоциируют, методами изучения переноса
ионов, кристаллохимическими данными и т. д.**.
* См. также главу XIII.
** Сказанное относится также к комплексным соединениям пятифтористого
иода (см. главу IV).
€4
Таблица 14
Комплексные соединения трехфтористого брома с
фторидами других элементов
Исходное вещество
/(кроме BrF3)
Продукт реакции
Температура
разложения
комплекса,
°С
Ссылка на
литературу
Ag; AgF; AgCl; AgN08
BaCl2. Ba(NU8)8
Na2< 03; Na2S206
KF; KC1; KN03
KJ03
Au
T:F4
Sn; SnCl2; SnCl4
Sb2u3; SbF3**
VC13; V
Nb; Nb205
Та; Та205
BiFb
PdF3
PtGU;
S03
Ru
PtBr4; PtF4
A^BrF4
Ba(BrF4)2
NaB F4
KBrF4
KBrF4
(Brh2)AuF4
(BrF2)TFG*
(BrF2).SnFe
(BrF2)S F6
(BrF2)VF6(?)
(BrF2)NbF6
(BrF2)TaF6
(B;F2)BiF6 1
(BrF2)PdF4
(BrF2)2PtF6
(BrF2)(S03F)
(BrF2)2RuF6 |
—
—-
—
—
—
180
—
190
—
—
150—180***
150—180***
70***
—
180-200
—
120**
[25, 36]
[25, 35]
[18|
[25, 35]
(181
[23]
[3>]
[27]
[27, 60]
[31]
[28]
|28)
[28!
[29]
[29]
[32]
[48]
В табл. 14 показано, при взаимодействии каких веществ с трех-
фтористым бромом можно получить оба типа комплексных
соединений последнего с фторидами других элементов****.
Из тетрафторобромиатных комплексов изучены калиевая,
серебряная и бариевая соли. Соли других металлов не были
выделены в достаточно чистом состоянии. Очистка этих солей
затрудняется тем, что еще не найден растворитель для их
перекристаллизации и отделения от фторидов. Изученные тетрафторобромиа-
ты — белые кристаллические вещества. Они устойчивы при
комнатной температуре и разлагаются лишь при нагревании, причем
выделяется BrF3 и получается фторид данного катиона. Калиевая
соль — наиболее стабильная из солей щелочных металлов тила
M(BrF4). Порядок устойчивости этих солей, в зависимости от
природы катиона, такой: K>Na>Rb>Cs.
Упругость диссоциации KBrF4 в интервале температур 158—356°
описывается уравнением: lg Р (мм) = 3,38—890/Т. Соль плавится
при 330°, после чего упругость диссоциации резко растет с тем-
* Это соединение получено только в растворе (в трехфтористом броме).
** Изучение системы BrF3 -SbF5 показало присутствие в ней двух инкон-
груэнтно плавящихся соединений 3BrF3-SbF5 и 3BrF3-2SbF5 и двух конгруэнтно
плавящихся BrF3.SbF5 (т. пл. 129,8±0,5°) и BrF8-3SbF6 (т. пл. 33,5±0,5°) [57].
*** В вакууме.
**** Примеры, приведенные в табл. 14, не исчерпывают все возможные
пути получения этих комплексов.
5—1419
65
пературо-й [60]. О строении иона BrF4~ уже упоминалось на
стр. 56.
Химическая активность солей типа M(BrF4)„ меньше, чем
свободного BrF3. Так, например, BrF3 очень бурно реагирует с
СС14, ацетоном, диоксаном; в то же время KBrF4 не действует на
эти вещества. Серебряная и бариевая соли несколько более
активны, чем калиевая,— они не реагируют с СС14, но воспламеняют
этиловый эфир, ацетон, диоксан, бензин. Водой KBrF4 (как и
соли других металлов) быстро разлагается, но реакция
протекает менее бурно', чем с BrF3. Продуктами реакции являются
бром, бромноватая и плавиковая кислоты, кислород. Первая
стадия реакции, вероятно, выражается уравнением [25]
BrF4- + 2H20 = HBr02 + 3H++4F-
Однако в то время как BrF3, даже кипящий, не действует на
платину, KBrF4 при нагревании разрушает ее, превращая в PtF4
и K2PtF6.
Бурная реакция происходит также между KBrF4 и бромистым
и йодистым калием, при этом выделяются бром или иод.
Количество выделившегося иода меньше требуемого по уравнению
BrF4~+4J-=Br--b4F-+2J2. Вместо 4 г-атомов иода выделяется
2,5—3,5 г-атома.
Дифторобромониевые комплексы (BrF2)m RF6 или (BrF2)RF4
также представляют собой твердые вещества, большей частью
бесцветные или желтые. Температуры разложения некоторых из
этих комплексов указаны в табл. 14.
Комплекс BrF2SbF6 — вещество канареечно-желтого цвета,
устойчивое на воздухе, плавится при 130°. Упругость диссоциа-
. D , , паа 3030
ции выражается уравнением lg Р (мм) = 7,66 =— в интервале
225—350° [60].
Как и тетрафторобромиаты M(BrF4)„, комплексы типа
(BrF2)m RF6, например BrF2SbF6, менее активны, чем свободный
BrF3, и менее энергично реагируют с водой, СС14 и некоторыми
другими органическими жидкостями. Некоторым исключением в
этом отношении является BrF2AuF4* он очень быстро разлагается
водой, энергично реагирует с четыреххлористым углеродом и
бензолом, взрывает при контакте со спиртом.
По данным Шефта с сотрудниками [60], KBrF4 и BrF2SbF6
энергично фторируют окислы ряда металлов (MgO, CaO, А1203,
Ce02. Nd203, Zr02, Th02, Мп02, Fe203, NiO), «©которые соли
Na2C03, BaS04, силикаты (полевой шпат), при 350—550° до
фторидов и кислорода.
Применение трехфтористого брома
для получения комплексных фторидов
В предыдущих разделах этой главы уже неоднократно
отмечались реакции с участием BrF3, в результате которых
образуются продукты соединения фторидов двух элементов. В частности
66
указывалось, что /такие комплексные фториды можно получать
путем взаимодействия в BrF3 как растворителе двух типов
комплексов—H(BrF4)n и (BrF2)m(RF„) *. Общим методом получения
двойных фторидов, о которых идет речь в данном разделе,
является применение двух реагентов, один из которых образует с BrF3
комплексы типа M(BrF4)«, а другой — дифторобромониевые
комплексные соединения; последующее взаимодействие комплексов
этих двух типов приводит к получению двойных фторидов. Так,
например, из смеси стехиометрических количеств КС1 и Sb203 с
избытком BrF3 можно получить KSbF6 [18], из серебра и треххло-
ристого ванадия — AgVF6 [31], из смеси серебра и золота —
AgAuF4 [26], из смеси карбоната или галогенида натрия и
металлического тантала — NaTaF6 [28] и т. д.
Во всех этих примерах подразумевается, что оба названных
в каждом случае реагента взяты в стехиометрическом
отношении, a BrF3 — в избытке против того количества, которое
необходимо, чтобы образовать с исходными реагентами «первичные»
комплексы, причем первый из каждой приведенной выше пары
реагентов дает комплекс типа M(BrF4)n, а второй — комплекс
типа (BrF2)m(RFrt) —см. табл. 14.
Для получения комплексных фторидов, например AgSbF6, нет
необходимости готовить отдельно растворы в трехфтористом
броме «первичных» комплексов и затем сливать эти растворы в
определенных соотношениях. Для указанной цели можно использовать
непосредственное взаимодействие соответствующих реагентов с
трехфтористым бромом, который в таких случаях играет роль
также и растворителя. Избыток BrF3 затем удаляют испарением
в вакууме.
Таким образом были получены (или установлены в растворе —
в трехфтористом броме) многие комплексные фториды: ранее
неизвестные тетрафтораураты AgAuF4 и др. [26], гексафторвисмутаты
MBiF6(M = Na, К, Rb, Ag)** [28], а также гексафторванадаты MVF€
[31, 34], гексафторниобаты и танталаты [28] M(NbF6)n и M(TaF6)n,
где М — щелочной металл, -кальций или барий, тетрафторбо-
раты, гексафторфосфаты, -арсенаты, -антимонаты [35], гекеафгор-
станнаты Г27], гексафторсиликаты [351, комплексные фториды
платиновых металлов M2PtF6(M = Na, К, Ag), Na2RhF6, Na3RhF7,
[29, 36], M(RuF6)n, где M = K, Cs, Ag, Ca, Sr, Ba [48], KMnF5 [34]
и некоторые другие ***.
* Т. е. «основной» и «кислотной» форм бромфтаридных комплексов, по
терминологии авторов этих работ [34].
** Гексафторвисмутаты щелочных металлов содержат некоторое
количество сольватированного BrF3, что Шарп объясняет явлениями сольволиза
[37]. В виде сольватов с BrF3 получены также гексафторниобаты (и танталаты)
кальция и бария и многие другие из перечисленных далее фторидных
комплексов.
*** Комплексные фториды молибдена, вольфрама, урана, меди, железа,
никеля и кобальта не удалось получить таким методом [24].
67 и
Таблица 15
Комплексные фгорит,'я, пэлученные при помощи трехфтористого брома*
Исходные вещества (кроме BrF3)
Продукт реакции
Ссылка
на
литературу
a) Ag+Au; б) AgCl+AuCl
а) МС1+Аи; б) MCl+AuCl (M=Na, К)
As'BrF4 -f-(BrF2)AuF4
a) Na2B407; б) МС1+В203 (M^Na, К)
KCA+SI
а) MCl-hSnCl2 (M=K,Ag)
б) 2KBr4-f(BrF2)2SnF6
BaCl2-fSnCl2
а) MP03, б) МГал+РГал5 (M-Na, К, Rb, Cs, Ag)
б) \'a3P04
Ag И -И AgCl-рРкрасный
ВаС12-|-Ркрасный
M или MCl-bAs205 или As203 (M=K, Ag)
BaCl24-As205
Ag3As04
M(BrF4)m-f BrF2SbF6 (M-K. Ag, Ba)
a) MCl-bSb203; 6) M2C03 + Sb205 (M=Li,K)
Ag4BiF5; AgBrF4 1 BrF2BiF6
MCI или M2C03+biF5 M-Na,K, Rb)
МГал или M2C03+Nb(M-Li,Na,K, Rb.Cs)
Ag или AgCl-fNb; Nb205
CaF2 или BaCl2-h2Nb
KBrF4-fBrF2NbF6
Ag или AgCl-l-VCl3
BaCl2 f VC13
NaV03
MC1+V или VCl3(M=Li-Cs, Ag)
МГал или M2C03+-Ta (M=Li, Na, К, Rb)
Ag или AgCl+Ta
CaF2 или BaCl2-f-2Ta
а) КС14Сг203; б) K2Cr207*******
•с) Ag2Cr207
MoO+MF(M=K,Rb,Cs)
KMn04
KU + KMn04
KCl+Mn J '3)2
JV\BrF4+BrF2PdF4 (M =K, Ag)
AgAuF4
MAuF4
AgAuI 4
MBFj
K2Si(-G**
M2SnF0
BaSnFe
M F6
NaPFe***
AgPFe
Ba (PF6)2
MAsFrf
Вт (AsF6)2
AgAsFe****
M (SbFe)n
MSbFe
AgBiFe
MBiFe*****
MNbF0
AgNbFG
M(-NbFe)a
KNbFe
AgVF6
Ba(VF6)2
NaVFe******
KVFG
MTaF6
AgTaF6
M(TaFe)a
KCrOF4
AgCrOF4
M2MoF8
KA^nF5
K2MnFG
KMnF5
K2MnF6
MPdF4
* Привзценные примеры не исчерпывают все возможные реакции
получения комплексных фгоридов в трехфтористом броме.
** В данной реакции образуется смесь зквимолярных количеств K2$iFe
и KBrF4.
*** Одновременно образуются 2 моля NaBrF4.
**** Одновоеменно образуются 2 моля A^BrF4.
***** Гексафгорвисмутаты, гексафгорниобагы и гексафтортанталаты часто
получаются загрязненными трехфгористым 6domom (сольволиз?).
****** Состав продукта реакции колеблется от NaVOF4 до NaVF6 в
зависимости от длительности кипячения раствора NaV03 в BrF3. Если немедленно
отогнать избыток BrF3 в вакууме при 45°, состав продукта реакции будет близок
к NaVOF4, после кипячения в течение 30 минут и отгонки BrF3 останется NaVFe.
******* При взаимодействииХ2СЮ4 с BrF3 образуется смесь KCrOF4 и KBrF4
(1:1).
68
Продолжение табл. 15
Исходное вещество (кроме BrF3)
Продукт реакции
K2PdF5
KhdF4
(Agi-dF4
I Ag,PdF5*
M2PclF6
M2PiFe
K21 tF6
M2P1F6
MRhr6
Na.,RhF7
Mi uF6
M(RuF6)2
Ссылка
на
литературу
[29]
129]
[36]
[36]
[29,25]
[29]
[29][61]
[29]
[48J
148]
K2PdGb
KCl+PdCl2
AgCl-tPdCl2
M2PdCl6 (M=K, Rb. Cs)
M2PtCl6 (M-Rb Cs)
a) K2PtBr6; 6) KBrF44-(BrF2)2PtF6; в) Pt-4-KBrF4
MC14-PtCl4 (M - Na, Ag)
2MCl+RhCl3 (M=Na,Cs)
3NaCl+RhCls
Ru+KBr (AgBr или CsCl)
Ru+M(Br08)2 (M=Ca, Sr, Ba)
В табл. 15 приведены некоторые исходные вещества
(реагенты), применяемые для синтеза соответствующих им комплексных
фторидов с металлическим катионом.
С о л ь в о л и з комплексных фторидов. Табл. 15
показывает разнообразие путей получения комплексных фторидов.
Но одновременно следует отметить, что возможности синтеза
таких фторидов ограничены явлениями сольволиза, в результате
чего в ряде случаев были получены продукты, состав которых
формально соответствует сольватированным трехфтористым бромом
комплексным фторидам, например K(MFm)n-A'BrF3.
Шарп [37], исходя из того, что вследствие сольволиза в
данных системах реакции между тетрафторобромиатами и дифторо-
бромониевыми комплексами не доходят до конца, показал, что
подобные «сольваты» в ряде случаев содержат примесь
исходного тетрафторобромиата.
Процесс сольволиза Шарп изучал на примере реакции
2KBrF4 + (BrF2)2 TiF6^ K2TiF6 + 4BrF3.
Если взять эквивалентные количества КВг и ТЮг, а избыток
растворителя (BrF3) удалить испарением в вакууме при
комнатной температуре, то получается продукт состава K2TiF6 • 0,95BrF3.
Вещество примерно такого же состава K2TiF6 • l,lBrF3 получается,
если растворить K^TiFe ** в трехфтористом броме и затем удалить
растворитель таким же путем. Оба продукта, по данным
рентгенографических исследований, содержали примесь KBrF4 [37].
Поэтому Шарп правильно отмечает, что в тех случаях, когда
происходит сольволиз и природа образующегося комплекса неизвестна
(т. е. если не получается чистая соль), необходима осторожность
в интерпретации результатов исследования [29].
* Эти фтогопалладаты получены в виде сольватов с BrF3.
**Чистый K^TiFe для этой цели был приготовлен из двуокиси титана,
плавиковой кислоты и фтористого калия.
63
Комплексные фториды с нитрониевым
и нитрозильным катионами
При посредстве трехфтористого брома удалось значительно
расширить область соединений с нитрониевым (NOj) и
нитрозильным (NO+) катионами и получить ряд соответствующих фторид-
ных комплексов.
Нитрониевые фторидные комплексы. Действуя
трехфтористым бромом на нитраты калия, серебра и бария, Вулф
и Эмелеус [35] получали тетрафторобромиаты этих металлов. Но
если в реакции участвует также вещество, способное реагировать
с BrF3 и давать при этом комплекс типа [BrF2]m [RFn ], то
одновременно с M(BrF4)n образуется соединение общей формулы
[N02]mRFn, которое можно рассматривать как продукт
соединения фтористого нитрония NO2F с фторидом какого-либо
элемента.
Вулф и Эмелеус [35] предложили получать нитрониевые
фторидные комплексы при взаимодействии N2O4, BrF3 и такого
вещества, которое в трехфтор'истом броме превращается <в дифторо-
бромониевый комплекс. Так, например, для получения тетрафторо-
бората нитрония к борному ангидриду в стеклянной колбе
прибавляют жидкий N204 (в избытке), бром — для замедления реакции и
затем медленно вливают BrF3. При последующем охлаждении
реакционной смеси выделяется (N02)BF4 в виде белого
кристаллического порошка с выходом 88% (считая на В2О3).
Аналогичным путем были получены (N02)AuF4, (N02hSnF6,
(N02)PF6, (N02)AsF6 и (N02)SbF6, причем в качестве второго
компонента реакции, помимо N204, были взяты соответственно:
Au, SnF4, PBr5, As203, SbF3 или Sb205 [35].
Нитрониевые фторидные комплексы образуются, очевидно, в
результате таких последовательно протекающих процессов: N204,
реагируя с трехфтористым бромом, превращается сперва в NO2F,
а затем в [N02l[BrF4]. Второй из компонентов реакции (В203, SnF4
и т. д.) при этом образует комплексный фторид дифторобромония
[BrF2]m[RF„]. Конечная реакция выражается такой схемой:
т NO,; BrF4 + (BrF2);RF- = (N02)mRF6 + 2m BrF3.
Относительно химизма этих реакций можно высказать
следующее предположение (на примере образования N02BF4),
борный ангидрид и N204, реагируя в отдельности с трехфтористым
бромом, превращаются в (BrF2)BF4 и N02F; последний
соединяется с BrF3 в комплекс [NC^lfBrFY!; конечная реакция ;вьгражается
уравнением
[N02]+ [BrFJ- + [BrF2]+ [BF4]- = [NO.J [BFJ + 2BrF3.
Это предположение подтверждается тем, что, по литературным
данным [38], фторид нитрония можно получать при
взаимодействии фтора с нитритом натрия, причем эта реакция включает
присоединение фтора к двуокиси азота
2NaN02 4- F2 = 2NaF + 2N02;
2N02 + F2 = 2N02F.
70
Было также показано [38], что N02F способен реагировать с
BrF3 с образованием желтого вязкого вещества. Авторам не
удалось изолировать продукт реакций, но они приписали ему
формулу N02BrF4. Существование этого неустойчивого комплекса,
как промежуточного вещества в реакциях образования нитро-
ниевых фторидных комплексов, предложили также Вулф и Эме-
леус [35]^
Нитрозильные фторидные комплексы. Вулф [32]
показал, что эти комплексы можно получать по реакции,
аналогичной реакции образования соответствующих нитрониевых
соединений, если только вместо N204 взять окись азота. Нужно
полагать, что эта реакция включает, в качестве промежуточного
продукта, образование фтористого нитрозила. Однако из-за
экспериментальных затруднений и легкой окисляемости окиси азота до
двуокиси, следствием чего является загрязнение продуктов реакции
солями нитрония, Вулф [32] предложил < получать нитрозильные
комплексы из хлористого нитрозила. Сжиженный NOC1 (40%
избытка) при помощи вакуум-перегонки переводят в стеклянную
реакционную колбу, содержащую второй компонент,
растворенный в трехфтористом броме; сосуд охлаждают жидким воздухом
и добавляют BrF3; смеси дают нагреться до комнатной
температуры и затем нагревают до окончания реакции. Избыток
растворителя отгоняют в вакууме.
Таким путем были получены (NO)AuF4, (NO)BF4, (NO)2GeF6,
(NO)2SnF6, (NO)PF6. В качестве второго компонента реакции
были взяты: золото, В203, Ge02, SnCl2, PBr5. Соответствующие
силикатный и титанатный комплексы нельзя было получить с
хорошим выходом или в чистом состоянии [32].
Гексафтороарсенат и -антимонат—(NO)AsF6 и (NO)SbF6 —
были получены иным путем из As203 и Sb203. В качестве
источника NO-группы применялся пиросульфат нитрозила,
образующийся при взаимодействии N02 и S02.
Нитрозильные фторидные комплексы были получены также
действием BrF3 на хлоридные комплексы. Так, например,
(NO)2SnCl6 трехфтористым бромом превращается в (NO)2SnF6.
Аналогичная реакция была использована [36] для превращения
гексахлороплатинатов и гексахлоропалладатов в гексафторо-
соединения (см. табл. 15).
Происходящие при этом реакции, очевидно, заключаются в
действии BrF3 на продукты первичной диссоциации хлоридных
комплексов, что можно представить такими схемами (на
примере (NO)2SnCl6):
(NO)2SnCl6 2: 2NOC1 + SnCl4;
2NOC1 Bj£32NOFB-!^32 N OBrF4;
BrF3
SnCJ4 -► (BrF2)2SnF6;
2NOBrF4 + (BrF2)2 SnF6 = (NO)2SnFc + 4BrF3.
71
Косвенным подтверждением этих схем является тот факт,
что превращение хлоридных комплексов во фторидные таким
путем не удается в случаях, когда промежуточный «простой»
фторид, например BiF3, не растворяется в BrF3.
Ионный характер нитрозильных фторидных комплексов
подтверждается тем, что они увеличивают электропроводность трех-
фтористого брома.
Фторсульфонаты. Применение трехфтористого брома
дало возможность получить ряд фторсульфонатов [32]. Так, при
взаимодействии серебра, пиросульфата нитрозила (МО)25207
и BrF3 образуется фторсульфонат серебра AgS03F. Реакция между
серным ангидридом, сульфатом калия, взятыми в эквимолярных
количествах, и BrF3 дает KSO^F. Эту реакцию Вулф изображает
такими схемами:
K2S04Bi^32KBrF4 + (BrF2) S03F;
SO.
BrF,
*(BrF2)S03F;
2KBrF4 + 2(BrF,)S03F = 2KS03F + 4BrF3.
Фторсульфонаты нитрония N02S03F и нитрозила NOS03F
были получены: первый — при действии BrF3 на смесь N'02 и S03,
второй — при действии BrF3 на (NO)2S207 [32].
Схемы получения нитрониевых, нитрозильных комплексных
фторидов и фторсульфонатов и взаимная связь этих реакции
показаны на рис. 13.
"As208 + BrF4 -> (NO)AsF
(NO)S03F-
|BrF8
AgS03F*
Ag + BrFa
KS03F
K2S04 + BrFa
(N02)S03F
N02+BF8
- (NO)2S207
t
—S03« S02
ко.
NOC1-
[_Sb203 + BrF3 - (NO)SbFe
— B203 + BrF3 - (NO)BF4
- PBr5 + BrF3 - (NO)PF6
—Ge02 + BrF3 - (NO)2GeFe
_SnF4 + BrF3 - (NO;2SnFs
SnCl4
(NO)2SnC!6/
Au + BrF3 B203 -f- BrF3
(N02)AuF4 (N02)BF4
SnF4 + BrF8 PBr5+BrFa As203-|-BrF3 Sb2p3+ BrFs
(N02)2SnF6 (N02)PF6 (N02)AsF6 (N02)SbF6
Рис. 13. Схемы получения нитрониевых и нитрозильных фторидных и фтор-
сульфонатных комплексов.
72
Реакции трехфтористого брома
с органическими веществами (фторирование
органических соединений)
Еще Лебо [9], а также Руфф и Брайда [12] наблюдали, что
трехфтористый бром бурно реагирует со многими органическими
веществами. По данным Лебо, эфир, бензол и скипидар
воспламеняются при соприкосновении с трехфтористым бромом даже на
холоду *. Хлористый метил сначала растворяет трехфтористый
бром, а затем реагирует с ним бурно со взрывом. '
По вопросу о применении трехфтористого брома для
препаративных целей в органической химии—для фторирования
органических соединений и т. д.—в литературе пока имеется мало
сведений. Литературные данные ограничены главным образом
несколькими патентами и посвящены почти исключительно
действию трехфтористого брома на галоидпроизводные. Однако и эти
сравнительно скудные сведения показывают возможность
нахождения условий, при которых можно контролировать реакции
фторирования органических соединений и получать продукты болео
или менее определенного состава.
Наттинг и Петри [39] описали метод получения хлорфтормета-
нов с помощью реакции между трехфтористым бромом и четырех-
хлористым углеродом. Реакции между BrF3 и тетрагалогензаме-
щенными метана изучали Бэнкс, Эмелеус и др. [40]. По их данным,
реакция между трехфтористым бромом и четыреххлористым
углеродом протекает бурна; в условиях низкой температуры
образуется смесь хлорфторметанов, преимущественно CCbF и CC12F2,
"с выходам до 96%. При повышенном давлении (в автоклаве) в
основном получается CC1F3.
По вопросу о механизме таких реакций эти авторы высказали
предположение, что в данном случае происходит ступенчатое
замещение атомов хлора
СС14 + BrF3 -* BrF2* + CC13F;
BrF2 + CCI3F ->BrF + CC12F2 и т. д.**.
Реакция между СВг4 и BrF3 протекает спокойно. При
проведении реакции в бромном растворе были получены
преимущественно CBr3F и CBr2F2. В отсутствие растворителя (брома) основным
продуктом реакции является CBrF3; выход достигает 94%.
Четырехиодистый углерод быстро реагирует с BrF3; из газовой
смеси, образующейся при этой реакции (0°), были выделены
CBr2F2 и CF4; иодфторметаны не были получены. Остаток в
реакционном сосуде содержал иод, JBr и JF5.
* Графит не изменяется на холоду при действии на него трехфтористого
брома; сажа адсорбирует трехфтористый бром и при нагревании слабо сгорает
со вспышкой [9].
** Эти реакции, по-видимому, сопровождаются выделением свободного
хлора.
73
Макби, Линдгрен и Лигетт [41] сообщили, что при действии
BrF3 на галоидзамещенные ароматических углеводородов
образуются алициклические галоидпроизводные, содержащие бром и
фтор: из гексахлорбензола СбСЦ и из пентахлор (трифторметил)-
бензола C6CI5CF3 были получены омеси, по составу
приблизительно соответствующие формулам СбВг2С14Рб и С6Вг2С1зРб(СРз).
Эти же авторы (в патентах) кратко описали условия
получения некоторых других фторхлорбромзамещенных углеводородов.
Исходное вещество Продукт реакции
Смесь C6BrCl4F7 и C6Br2CI4F6
Смесь CF3C6BrCl3F7 и CF3C6Br2Cl3F6
Cl0Br2Cl6Fl0
Cj2BrCl8Fi3
(CF3)2C6BrCl3F6.
В результате последовательной обработки C6CI5CF3 трехфто-
ристым бромом, пятифтористой сурьмой, а затем цинковой пылью
и спиртом была получена смесь производных циклогексадиена
CF3C6Cl2F7 и CF3C6C13F6.
Бус и Пинкстон {13, стр. 171] сообщили, по данным Шенка, что
трехфтористый бром действует на разбавленные растворы
некоторых керосиновых фракций в дихлорметане и может служить
эффективным агентом для частичного замещения атомов водорода
на фтор и бром без заметных побочных реакций. При попытке
более полного замещения водорода органического соединения
путем нагревания его с трехфтористым бромом в отсутствие
растворителя необходимо соблюдать осторожность, так как подобные
смеси детонируют.
Из приведенных выше данных следует, что трехфтористый
бром представляет большой интерес как реагент для синтеза
неорганических (а возможно, и органических) фторидов.
Применение трехфтористого брома делает более доступным получение
фторидов ряда элементов и фторидных комплексов, которые
могут быть использованы в практике, но синтез которых ранее был
очень затруднен. Таковы, в частности, комплексные фториды
ниобия, тантала *, платиновых металлов и некоторых других
элементов.
Своеобразие BrF3 как реагента заключается в том, что он в
ряде случаев почти одинаково действует на различные формы
состояния данного элемента'; независимо от того, находится ли
последний в виде простого вещества (например, металла), окисла»
галогенида, соли, трехфтористый бром превращает его сперва в
простой фторид, а затем, если этому благоприятствуют
физические условия, и в комплексный фторид. Примеры подобных
реакций приведены, в частности, в табл. 15. В качестве еще одного'
примера можно указать на то, что при действии жидкого трех-
* Они используются для электролитического получения этих металлов.
74
С6С16
CeCl5CF3
С10С18
(С6С16)2
C6Cl4(CF3)i>
^гористого брома на большинство соединений урана образуется
[етучий шестифтористый уран [42].
Возможность получения при помощи трехфтористого брома
сравнительно легко летучих фторидов ряда элементов в их
высших степенях валентности создает благоприятные условия для
отделения этих элементов от сложных смесей, встречающихся в
природных объектах. В этом отношении заслуживает внимания
опубликованная Эмелеусом и сотрудниками статья [43], в которой
сообщается, что трехфтористый бром быстро и количественно
реагирует при температуре ниже его точки кипения с большинством
урановых соединений, превращая их в летучий UF6. При этом
было установлено, что в таких же условиях ни один из других
встречающихся в природе элементов с атомным весом, большим 80
(исключая радон), не улетучивается количественно. Таким
образом, как отмечают авторы, эта реакция может быть
использована при анализе смесей радиоактивных веществ, а также для
отделения тория от урана и для получения протактиния.
В связи со сказанным выше есть основания предполагать, что
трехфтористый бром может найти применение также в химическом
анализе — для разложения минералов и разделения элементов.
Возможные формы применения трехфтористого брома в
практике— для препаративных работ в неорганической и органической
химии, в химическом анализе и т. д.— еще далеко не исчерпаны
примерами, приведенными в этой главе. Необходимо
дальнейшее изучение трехфтористого брома в этом отношении, равно как
и исследования физико-химического характера — для выяснения
химизма,7 механизма и кинетики химических реакций,
происходящих с участием трехфтористого брома, для изучения комплексных
соединений трехфтористого брома с фторидами других
элементов — физических и химических свойств этих соединений, их
устойчивости, превращений и т. д.
3. ПЯТИФТОРИСТЫЙ БРОМ BrF5
Получение
Пятифтористый бром впервые получили Руфф и Менцель в
1931 г. [И]. Исходя из опыта получения высшего фторида иода JF7
из пятифтористого иода и фтора [44], Руфф и Менцель
разработали метод получения пятифтористого брома, при действии
фтора на трехфтористый бром. Они применяли для этой цели
платиновый аппарат, сконструированный для синтеза семифтористого
иода (гл. IV). Сперва бром превращали в трехфтористый бром,
который затем «напревали до 90—100°; горячие пары его
смешивали с рассчитанным количеством фтора и эту смесь нагревали до
200° в платиновой трубке. Реакционную смесь BrF3 + F2^BrF5
подвергали фракционированной конденсации в кварцевых
приемниках: в первом приемнике, охлаждаемом до —60°, собирался
жидкий пятифтористый -бром и небольшое количество твердого
75
Br2+N2
трехфтористого брома; во втором приемнике с температурой—193°
конденсировался избыточный фтор.
Однако впоследствии Руфф и Мендель отказались от
двухступенчатого процесса Br2~*BrF3-*BrF5 и разработали более
удобный метод непосредственного получения пятифтористого брома из
брома и фтора при 200° в аппаратуре (рис. 14) из меди, которая'
оказалась более устойчивой, чем
платина (в процессе реакции медный сосуд
внутри покрывается слоем CuF и CiiF2).
Бром вводился в виде паров (с помощью
тока азота) со скоростью 6 смъ1мину
фтор — со скоростью 35 см3/мин, т. е. в
количестве, немного превышающем сте-
хиометрическое. Выходящие из реактора
газы конденсировались в кварцевых
приемниках, как описано выше. Очистка
полученного таким образом сырого
продукта производилась путем
фракционированной дистилляции в кварцевой
аппаратуре, описанной в работе Руффа и
Менцеля *. Для получения чистого
бесцветного продукта необходимо, по
возможности, исключить применение
органических веществ — пицеина, жиров для
смазывания кранов и т. п.
Рис. 14. Аппарат для
получения пятифтористого
брома:
1—асбест; 2—обмотка; 3—термо-
Физические свойства
Пятифтористый бром в чистом состоянии представляет собой
при комнатной температуре совершенно бесцветную жидкость,
сильно дымящую на воздухе. Продукты разложения,
образующиеся при действии влаги воздуха и растворяющиеся в пятифтори-
стом броме, окрашивают его, в зависимости от концентрации
примесей, в светло-желтый и темно-красный цвета. Т. пл.
пятифтористого брома —61,3°, т. кип. + 40,5±0,5°**.
Молекулярный вес, определенный по плотности пара при
330 мм давления и 29°, оказался равным 174,7. Хорошее
совпадение этой величины с теоретическим значением (174,92)
показывает отсутствие ассоциации молекул пятифтористого брома в
газообразной фазе в условиях опыта.
* По Бурке и Джонсу [45], для этой цели может быть использована
стальная аппаратура. В Квасник [59] разработал метод получения пятифтористого
брома в железном реакционном сосуде.
** Роджерс и Спирс [62] нашли для пятифтористого брома темп,
замерзания —60,5±0,1°, темп, кипения 40,76±0,05°; lg Р мм = 7,9727—1598,2/Т±
±\ мм в области 400—760 мм; теплота и энтропия парообразования
соответственно 7,31 ккал-моль—' и 23,3 энтропийных единицы.
76
Упругость пара пятифтористого брома измерялась в интервале
температур от —88° ^до +24°, причем температурная зависимость
может быть выражена следующим уравнением:
lgP = 8,0716— 1627,7/Г.
Значения Р, вычисленные для жидкого пятифтористого брома,
очень близки к экспериментально найденным, величинам.
Температура кипения, найденная путем экстраполяции, равна 313,6° К,
или 40,5° С. Теплота испарения в точке кипения равна
7,443 ккал-моль"1. Величина константы Трутона 23,7 указывает на
ассоциацию молекул пятифтористого брома в жидком состоянии.
Приближенное значение критической температуры 313,6 • 3А> равно
470,4° К, или 197,3° С.
Плотность твердого пятифтористого брома при температуре
плавления d4"~6b3=3,09, а жидкого вещества d~61-3 =2,763.
Температурная зависимость плотности жидкого пятифтористого брома
d£ =3,496—0,00346-Г*.
Некоторые другие физические свойства пятифтористого
брома— электропроводность [63], диэлектрическая постоянная,
молярная поляризация, рефракция, магнитная восприимчивость, ди-
польный момент приведены в табл. 1, 2 и 4.
Спектроскопическим исследованиям пятифтористого брома
посвящены работы Бурке и Джонса [45], Стефексона и
Джонса [51]. В спектре комбинационного рассеяния жидкого
пятифтористого брома (при комнатной температуре) было найдено
девять частот в пределах 684—241 см~1. Измерение инфракрасных
спектров производилось для газообразного вещества и при
низком давлении; в пределах 700—400 см~1 найдены четыре
полосы [45].
Недостаточное количество экспериментальных данных для
инфракрасных спектров при низких основных частотах и
поляризация линий спектров комбинационного рассеяния не дали
возможности получить определенное представление о структуре
молекул BrFs. Однако из двух наиболее вероятных моделей —
тетрагональной пирамиды и тригональной бипирамиды — Бурке и
Джонс, сопоставляя количество найденных частот в спектрах
комбинационного рассеяния с предполагаемым, отдают
предпочтение первой структуре [45]. При том принято, что атомы фтора
занимают четыре угла основания и вершину тетрагональной
пирамиды, а центральный атом брома находится внутри пирамиды,
но, возможно, не в плоскости ее основания [201.
Стефенсон и Джонс [51], также исследовавшие раман-спек-
тры пятифтористого брома, отмечают, что полученные ими
результаты согласуются с такой структурой BrF5 (симметрия С^), причем,
по их данным, расстояние Br—F равно 1,78 А, четыре угла между
связями атомов фтора в основании пирамиды равны 86,4°, а углы
* По новым данным [58], d[l = 2,559— 3,484- 10""3t—3,45-1 СП"6 I2 (от —15 до
+76°).
77
в
03
н
о
ч
со
О
о\
о
О
о
та
СО
К
S
я i
о to —
-1 '
о со
*-. ~«
ж to
ся 3
03 X
^ *т
>£ to| сл
со __,
I—I to J
СД Я"
■о
Я
-о
о
]я
Я
Я
ТЗ
о
н
о
Я
чз
ГО
оэ
я
я
ячз
'- ГО
Я
я w
я
я
Яс
* Е
я
п
я
я
чз
я
Я OVi^
Я со ("D
^ *§* 3
я н ^
Л о <т>
ГО <Т>
J3
Я П>
я
я
я
я
я
н
я
43 ^
я я . .
£ о « ^
я о ж
е ° к
я
Я« ^\ Л
ч S
*>< ГО &Э
.. я
^ <Т> ^ ~
Я Я ° ,5
я я *•<
2 " *° s
Я ^Э ^
£з О w х •<;
гртз g я-в^
Я -J
ОЭ ОЭ
о
я
Я
я
я
о
Я* о
я
HtIO
Я ОЭ Я«
я о н
ь^ Я
ГО СО
X
я
я
я
я
-о
го
)я
я
о
5з
о
я s
о
о w
чз<<:
оэ X
я я
£ я
8-rt
я
я
ОЭ >©*
2 н
я о\
О) у-
« Й я
я 5 я
X
S
S
S
о
о
я
о
о
р
ОЭ
S g
я я
я ч
я
CD H
^ °
3*
* я
ст> я
о
я
о
о\
о
)я
я
я
я
о
я
я
я
тз
я
Я "
о
н
Е
я=
о ov
rt> ^
о
ГО
Я "
я
я <т> 2
я
я
■о я сг
п> го , .
я • ^
о
Я <ъ
5^
tr я
о
OS
ov
Я о
я н
*а я
•< о
Я ft)
О -з
"О Я
я я
2 -
о я
2 я
3 s
Я чд
W
W
я
||
-S ^^
о
is
Я "-о
Я £
я
я
го
W
2
я
я
£ Я ^Tj
(Ъ Я
я
я
н слсо
Я о S
я
о
. я
я _
Е ?
о
ХЗ "
я g
го Е
ГО Я
gs
(X)
я
н
43
о
я
я
я
я
я
я
я
*о
я
I
ё
Е
S
я
я
S
от
я
СЛ4^СОЮн-ОС£>00^0)СЛ^СлЭЮЮЮ
ооооооооооооооосооо
! I ' I I I I I ! 1 I I I II I
сос£>сосооооооооо-<1-<1--]а^спст5а>сл
а) 4*. ю^оро слойр^^j^P5r^J~"J-5-00 °
"а^ ^ai'o^'cn'oo'co'co сл"сл "^- V "to сл^"—"cd
-NltOOiOOCncn-OCTiO^ — W 00 СО СД СО
lotototototototototo^—* — ^^-^-
О) О) О) pi СЛ 4^ j£» Ой JS0 J— ^CD^У* СЛ^ СлЭ О
Id а>"to оо ль. оо"»— Ти^'КэЪ^Ъ^'о o"to 4^
Ст)**сооо— cncDocotoooooa^^*^oo
JLO — 00 Gi 00 О J>1 00 .CD^Cn OWG5p*.JOO
"*• lo "to"►&>» Vj Ъо сл "со "соЪо о 1о сд '^ "со "^
oocтlcлcг>cт)ocт)a^coa^cr>,— cDh^cd»—
^COOOOOtOtOfO*— ►— ^-
0--34^ н-^]4^и— ООСДЮСО^^^СОСО
4^КЭ — ОЮОО^О^^^ОООСЛ^ООСО
J^CD-vJUlCnUl-vlNDOW*. -0^—-JCO-<I
OCJ)vlO^Ob-O-N3Ob000UlOOO
cocooooococococototototototototo
со оо "ю"— ЪГоо "ф "со 1о Ъо "►**. Ъо "о "ex? "cd Vj
oo^-^^oo-qai-oaja)'—tooooooo
I I
I I
^- ►— tototototototototototototototo
J^^OjOj—j—^—jO^tOCO COCO *» 4^^^ tO
CO "^ - Ъ О "^ "ОО W 4 ^ СЛ 00 О О О О
I I 1 I I I I I I I I I I I I ^ I
■— ЮСОСО^СЛСЛО^^ЧООсДООООКЭ
O^ O0 Oj<)^^—OOpljOjOj-s]^^ и- ZO СЛ tO
"cooo VToi слЪт ui'aiVj oo ^-V bo coVicn
jsdjw at p^ рон-^ ^jo cd oo^ 4^4^ jjo to i
Гд ч "-оо оо к) "ю со Ь о о сл^ЬЬЪ I
*S — СТ)4^*.СЛР'— CDOOC75SWOOO
^
(F0-H°Q)/T,
кал-град~~{ х
хмоль' 1
Н°—Н°0)/Т,
кал-град~{ х
5°, кал-град *х
х М0ЛЬ~~
Н°-И°0,кал-моль~1
С^кал-град 1х
хмоль
-1
Д///°, ккал-моль"
Д/7/0, /с'/сал -леоль
-1
ig/r/
^ительно лишь в относительно небольшом числе случаев. Тем
не менее эти сведения показывают высокую и разнообразную
химическую активность пятифтористого брома, похожего в этом
оТношении на другие высшие фториды галогенов.
Экспериментальные данные, полученные Руффом и Менделем,
дают основание полагать, что пятифтористый бром может
реагировать со всеми химическими элементами, за исключением
инертных газов, азота и кислорода, и с большинством неорганических,
соединений, исключая фториды.
Реакции с простыми веществами
Пятифтористый бром энергично реагирует с металлами; в
большинстве случаев эти реакции сопровождаются раскаливанием
металлов или появлением пламени. хемпература, при которой
начинаются эти реакции, и их интенсивность в значительной
степени зависят от того, насколько образующийся на поверхности
металла слой фторида защищает его от дальнейшего действия
BrF5. Из исследованных Руффом и Менцелем металлов это
защитное действие фториднои пленки особенно сильно проявилось
в случае магния; реакция прекращается и не протекает даже
при сильном прокаливании.
Литий в порошке, барий, цинк и ртуть *, мышьяк и сурьма,
молибден и вольфрам, селен и теллур, порошкообразные железо
и кобальт, а также иридий ** быстро реагируют с пятифтори-
стым бромом при комнатной температуре. Золото, алюминий в
порошке, висмут, марганец, порошкообразные никель и родий
требуют предварительного слабого нагревания. Более высокие
температуры необходимы для реакции пятифтористого брома с
оловом (после плавления последнего), свинцом (300°), натрием,
кальцием, кадмием, титаном в порошке, хромом, платиновой
жестью (темно-красное каление).
Водород после предварительного зажигания бурно, со
взрывом взаимодействует с пятифтористым бромом. Порошкообразный
бор, аморфный углерод, сера в порошке и красный фосфор
немедленно реагируют с пятифтористым бромом с выделением
пламени и со вспышкой (в случае серы).
При нагревании смесей пятифтористого брома с хлором в
медном аппарате при 300° образуется трехфтористый бром,
возможно, по реакции BrF5 + Cl9 = BrF3 + 2C1F ***. При взаимодействии
пятифтористого брома с бромом при 250° образуется
трехфтористый бром, а при более низкой температуре,— возможно, и фто-
* При реакции на холоду ртуть покрывается бурой пленкой; с кипящей
ртутью образуются белый бромид и желтый фторид.
** Иридий в порошке постепенно растворяется при комнатной
температуре в жидком пятифтористом броме с образованием бурого раствора.
*** Руфф и Мендель пишут, что по техническим причинам они не могли
обнаружить присутствие фтористого хлора. Однако в их работе нет также
доказательств образования при этой реакции трехфтористого брома.
79
ристый бром. Иод реагирует с пятифтористым бромом при
комнатной температуре; продукты этой реакции не
охарактеризованы *.
Реакции с неорганическими соединениями
Пятифтористый бром бурно, иногда со взрывом, реагирует с
водой (при вливании пятифтористого бпома в воду). В случае
введения небольшого количества воды (например, при пропускании
влажного азота) в пятифтористый бром последний окрашивается
сперва в желтый, оранжевый, а затем в красный цвет.
Продукты реакции не установлены, доказано лишь, что весь кислород
добавленной воды освобождается. Водные растворы едких
щелочей энергично реагируют с пятифтористым бромом, причем
наряду с реакцией
BrF5 + 6NaOH = 5NaF + NaBr03 + 3H20
происходят и другие процессы, сопровождающиеся выделением
бурых паров, образованием бромида и кислорода.
Многие окислы — MgO, CaO, B203, P205, As205, Cr03, Мо03,
W03, J2O5— быстро реагируют с BrFs. Эти реакции экзотермичны.
Для реакции с кремнеземом (кизельгуром) и со стеклом
необходимо предварительно^ умрг>ениор нагревание** [11]. При реакции
с Со304 образуется CoF2 (90%) [13].
Окись углерода и сернистый газ горят при взаимодействии с
BrF5 (после зажигания), сероводород и аммиак быстро и с
выделением пламени реагируют с п^м ***. Концентрированные
серная и азотная кислоты взаимодействуют с BrFs с выделением
тепла (в случае H2S04) или газов (при реакции с HN03). x
Галогениды—NaCl, NH4C1, KBr, KJ, а также NaN02 и KN03
быстро реагируют с BrF5, выделяя пламя или газы; при реакции
с NaCl или КВг наблюдалось выделение хлора или брома [11].
Пятифтористый бром, по данным термического анализа, не
образует соединений с фтористым водородом. В системе
обнаружена лишь одна эвтектика, содержащая 95,2 мол.% HF и
плавящаяся при —8561°. Измерены также плотность и давление
паров. Удельная электропроводность этой системы при 25°
увеличивается с ростом концентрации HF и достигает значений
7,8-10~5 (—60°) и 1,25-10"4 ом~хсм-х (25°) при содержании HF
8,84 мол/л [63].
Реакции с органическими соединениями
Руфф и Менцель сообщили, что при действии BrFs на метан,
бензол, этиловый спирт, уксусную кислоту, пробку, хлопок, бума-
* Руфф и Менцель полагают, что при этой реакции образуется
пятифтористый иод.
** При обыкновенной температуре BrF5 медленно действует' на сухое
•«текло, кварцевое стекло практически не реагирует с BrFs-
*** При реакции между газообразным BrFo и F2S выделяется сера.
80
гу, ткани, жир, резину, пицеин происходят быстрые и бурные
реакции, сопровождающиеся в ряде случаев (СН4, С2Н5ОН, С6Нб)
взрывом и появлением пламени. Однако при взаимодействии
газообразных BrFs и СбНб реакция протекает спокойно.
ЛИТЕРАТУРА
1. О. Ruff, W. Menzel, Z. anorg. allg. Chem., 202, 60 (1931).
2. O. Ruff, A. Bra i da, Z. anorg. allg. Chem., 214, 81 (1933).
3. J. Fischer, R. K. Steunenberg, R. С V о g e 1, J. Am.
Chem. Soc, 76, 1497 (1954).
4. P. H. Broderson, H. J. Schumacher, Z. Naturforsch., 1947,
\ 2a, 358; Anales asoc. quim. argentina, 38, 52 (1950) — цит. по [5, стр. 451].
5. H. H. Г р и н в у д, Усп. химии, 22, 445 (1953).
6. D. F. Smith, M. T i d w e 1 1, D. V. P. Williams, Phys.
Rev., 77, 420 (1950).
7. Справочник химика, Госхимиздат, Л.—М., 1951, т. 1, стр. 286.
8. Н. М о i s s a n, Le fluor et ses composes, Paris, 1900, p. 123.
9. P. L e b e a u, С. г., 141, 1018 (1905); Ann. chim. phys. (8), 9, 241 (1906);
\ Bull. Soc. chim., 35, 148 (1906). ;
10. E. B. R. Prideaux, J. Chem. Soc, 89, 316 (1906).
11. O. Ruff, W. Menzel, Z. anorg. allg. Chem., 202, 49 (1931).
12. O. Ruff, А. В г a i d a, Z. anorg. allg. Chem., 206, 63 (1932).
13. Г. С. Бус, Д. Т. Пинкстон, Галоидные соединения фтора, сб.
«Фтор и его соединения», ИЛ, М., 1953, стр. 164.
14. A. G. Sharp e, Quart. Rev-, 4„ 115 (1950).
15. О. Ruff, А. В г a i d a, Z. anorg. allg. Chem., 214, 91 (1933).
16. О. Ruff, Z. ang. Chem., 46, 739 (1933).
17. G. D. Oliver, J. W. Grisa r d, J. Am. Chem. Soc, 74, 2705 (1952).,
18. H. J. E mel e us, A. A. W о о 1 f, J. Chem- Soc, 164 (1950).
19. С F. White, С F. Goodeve, Trans. Faraday Soc, 30, 1049 (1934).
20. E. Fessenden, J. Chem. Educat., 28, 619 (1951).
21. M. T. Rogers, A. L. W a r h a f t i g, V. S с h о m a k e r, Abstr.
Atlantic City Meeting, Am, Chem. Soc, 1947, Apr. 21 p. — цит. по [5,
стр. 452].
22. A. A. Banks, H. J. Emeleus, A. A. W о о 1 f, J. Chem. Soc,
2861 (1949).
23. Б. Ф. О р м о н т, Структуры неорганических веществ, Гостехтеорет-
издат, М.-Л., 1950, стр. 761.
24. V. Gutmann, Z. ang. Chem., 62, 312 (1950).
25. A. G. Sharpe, H. J. Emeleus, J. Chem. Soc, 2135 (1948).
26. A. G. Sharp e, J. Chem. Soc, 2901 (1949).
27. A. A. Woo If, H. J. Emeleus, J. Chem. Soc, 2865(1949).
28. V. Gutmann, H. J. Emeleus, J. Chem. Soc, 1046 (1950).
29. A. G. Sharpe, J. Chem. Soc, 3444 (1950).
30. A. G. Sharpe, J. Chem. Soc, 2907 (1950).
31. H. J. Emeleus, V. Gutmann, J. Chem. Soc, 2979 (1949).
32. A. Wool f, J. Chem. Soc, 1053 (1950).
33. А. И. Шатенштейн, Теории кислот и оснований, Госхимиздат,
М.—Л., 1949, стр. 108, 116, 205, 216.
34. A. G. Sharpe, A. A. W о о 1 f. J. Chem. Soc, 798 (1951).
35. A. A. Woo If, H. J. Emeleus, J. Chem. Soc, 1050 (1950).
36. A. G. Sharpe, J. Chem. Soc, 197(1953).
37. A. G. Sharpe, J. Chem. Soc, 2907 (1950).
38. E. E. А у n s 1 e y, G. H e t h e r i n g t о n, P. L. Robinson,
\ J. Chem. Soc, 1119 (1954).
39. H. S. Nutting, P. S. Petrie, Патенты США, 1,961, 622 (1934).
40. A. A. Banks, H. J. Emeleus, R. N. H a s z e 1 d i n e, V. К е r-
kr i g e n, J. Chem. Soc, 2188 (1948).
6—1419
81
41. Е. Т. МсВее, V. V. L i n d g г е n, W. В- L i g e t t, Ind. Eng-
Chem., 39, 378 (1947); Патенты США, 2, 432, 297 (1947); 2, 489, 969 (1949)t
2, 480, 1080 (1949); Chem. Abstr., 1950, 2019, 2020.
42. Б. В. Некрасов, Курс общей химии, изд. 9, Госхимиздат, М.—Л.
1952, стр. 594.
43. Н. J. Erne le us, A. G. M a d d о с k, G. L. Miles, A. G. S h а г-
ре, J. Chem. Soc, 1991 (1948).
44.0. Ruff, R. Keim, Z. anorg. allg. Chem., 193, 176 (1930).
45. T. G. Burke, E. A. Jones, J. phys. Chem., 19, 1611 (1951).
46. J. She ft, H. Ну man, J. К a t z, J. Am. Chem. Soc, 75,5221(1953).
47. A. A. W о о 1 f, J. Chem. Soc, 4113 (1954); Chemistry a. Ind., 346(1954).
48. M. A. H e p w о r t h, R. D. Peacock, P. L. Robinson,
J. Chem. Soc, 1197 (1954). R.—D. Peacock, Rec, 75, 576(1956).
49. R. A. D u r i e, Proc Roy. Soc, 207, 388 (1951).
50. H. E v a n s, T. R. M u n s о n, D. D. W a g m a n, J. of Research
Nat. Bureau of Standarts, 55, 147 (1955).
51. С V. Stephenson, E. A. Jones, J. Chem. Phys., 20, 1830 (1952).
52. L. S 1 u t s к у, S. H. Bauer, J. Am. Chem. Soc, 76 270 (1954).
53. L.St e in, R. С Vogel, W. H. L ude w i g, J. Am. Chem. Soc, 76, 4287
(1954).
54. H. M. H a e n d 1 e г and joint authors, J. Chem. Phys. 22, 1939 (1954).
55. J. Fischer, J. В i n g 1 e, R. C. Vogel, J. Am. Chem. Soc, 78,
902 (1956).
56. P. H. Bro de г son, J. E. Si с re, Z. Phys., 141, 515(1955); P. H.Bro-
derson, S. Mayo, Z. Phys., 143, 477 (1955).
57. J. Fischer, R. L i i m a t a i n e n, J. В i n g 1 e, J. Am. Chem.
Soc, 77, 5849 (1955).
58. A. A. Banks, J. J. M e d о с к, J. Chem. Soc, 2779 (1955).
59. Руководство по препаративной неорганической химии, под редакцией
Г. Брауэра, И. Л, Москва, 1956, стр. 100—101.
60. J. S h e f t, A. Martin, J. K a t z, J. Am. Chem. Soc, 78, 1557
(1956).
61. B. Cox, D. N. A. Sharpe, A. G. Sharpe, J. Chem. Soc,
876, 1242 (1956).
62. M. T. Rogers, J. L. S p e i г s, J. Phys. Chem., 60, 1462 (1956).
63. M. T. Rogers, J. L. S p e i г s, M. В. Р a n i s h, J. Am. Chem.
Soc, 78, 3288 (1956).
ГЛАВА IV
ФТОРИДЫ ИОДА
Из соединений фтора с иодом до настоящего времени
получены только пятифтористый и семифтористый иод JF5 и JF7.
Попытки получить низшие фториды иода JF и JF3 непосредственно из.
элементов и другими реакциями или установить их наличие как
промежуточных веществ при взаимодействии фтора с иодом, а
также в продукта^ термической диссоциации JF5 и JF7 или в
продуктах взаимодействия JFs с иодом оказались безрезультатными.
1. ПЯТИФТОРИСТЫЙ ИОД JF5
Получение
Пятифтористый иод был первым представителем фторидов
галогенов, полученным в индивидуальном состоянии. Соединение
иода со фтором пытался получить Каммерер в 1862 г. [1]
нагреванием сухого фторида серебра с иодом в запаянной трубке при
70—80° в течение 14 часов до исчезновения цвета иода.
Газообразная часть продукта реакции поглощалась водным раствором
едкого кали. Так как полученный раствор не давал реакции на
ионы иода, Каммерер ошибочно полагал, что выделившийся газ
представлял собой фтор.
Природу реакции между фтористым серебром и иодом
разъяснил Горе (1871 г.), указав, что при этом образуются йодистое
серебро и фторид иода [2] *.
Эта реакция протекает легко лишь при температуре красного
каления. Ее можно выразить следующим уравнением:
5AgF+3J2 = 5AgJ+JF5.
Аналогичную реакцию применил также Руфф [3]. Пропуская
пары иода над фторидом свинца и калия PbF4 • 3KF, он получил
небольшое количество летучего вещества, которое при пропускании1
в воду образует раствор, дающий реакции на HJ03. Только на
этом основании Руфф полагал, что полученное им летучее
вещество должно быть идентично с JFs.
/ * В этом же сообщении Горе описал и некоторые свойства пятифтористого
иода.
83
Пятифтористый иод впервые был точно идентифицирован и
мэписан Муассаном, который получил его (1891 г.)
непосредственно из элементов [4] и показал при этом, что иод способен гореть
во фторе бледно-голубым пламенем с образованием JF5. Для
получения пятифтористого иода Муассан действовал током фтора на
^од в платиновой лодочке, помещенной в стеклянную трубку. Для
угой же цели может быть использована реакция между фтором
(взятым в избытке) и газообразным йодистым водородом
(причем наряду с JF5 образуется и фтористый водород), а также
реакция между фтором и некоторыми иодидами: KJ, СаЛг, PbJ2,
Сщ32- Во всех этих случаях сперва выделяется иод, который
затем соединяется с избыточным фтором [4].
Придо [5] изучил условия взаимодействия иода с жидким
фтором. Оказалось, что иод не растворяется в жидком фторе при
атмосферном давлении и не реагирует с ним. Но при нагревании
в запаянной трубке реакция протекает с воспламенением.
Пятифтористый иод конденсируется в трубке в виде бесцветной
жидкости.
Метод получения JFs из элементов был усовершенствован Руф-
фом с сотрудниками, описавшими несколько модификаций этого
метода [6, 7, 8]. Руфф и Кайм [6] для получения JF5 как
промежуточного продукта в процессе синтеза JF7 применяли аппаратуру
из платины. В охлаждаемый сосуд, содержащий иод, пропускали
фтор в рассчитанном количестве или до тех пор, пока жидкость в
реакционном сосуде не станет бесцветной или лишь слабо-желтой,
что указывает на окончание реакции. '
В дальнейшем эти же авторы [7] получили JF5 в медной
аппаратуре. Их аппарат состоял из широкой трубки, соединенной при
помощи другой трубки меньшего диаметра с кварцевой спиралью
конденсатора и кварцевыми приемниками. Иод помещают в
широкую трубку и пропускают над ним фтор. Реакция наступает
тотчас и протекает с выделением тепла; медные трубки охлаждаются
водой. Основной продукт реакции JF5 конденсируется в приемнике,
охлаждаемом льдом. Для конденсации более летучих веществ
(JF7, SiF4 и избыточного фтора) присоединяется еще один
приемник, который охлаждают жидким воздухом. Он соединен с
атмосферой кварцевой или медной трубкой, содержащей хорошо
высушенный фтористый калий. Пятифтористый иод, собранный в
первом приемнике, обычно содержит немного свободного иода.
Поэтому после того как весь иод в медной трубке прореагировал,
продолжают пропускать фтор до обесцвечивания жидкости в
первом приемнике. Полученный по этому способу сырой
пятифтористый иод часто содержит небольшое количество CuJ2 и поэтому
слабо окрашен в желтый цвет. Его очищают перегонкой при
пониженном давлении' и температуре 5° из кварцевых сосудов в
кварцевый же приемник, охлажденный до —20°.
Руфф и Брайда [8] предложили получать JF5 в кварцевой
аппаратуре при действии на иод смеси фтора и азота (например, в
отношении 1:3). Иод помещают в достаточно широкую и охлаж-
84
даемую кварцевую трубку и пропускают разбавленный азотом
фтор. Реакция происходит без выделения^иламени. Если фтор
содержал примесь кислорода или температура реакции была
слитом высокая *, образуется JOF3, кристаллизующийся при
охлаждении сырого JF5. Последний может содержать также примесь
JF7 **. Продукт реакции очищают перегонкой его в кварцевом
аппарате при давлении 10 мм рт. ст.; собирают фракцию при 10°.
Эта фракция представляет собой чистый JFs. Остаток состоит
из JF5 и JOF3.
Вулф [9] получал пятифтористый иод из элементов при
комнатной температуре в никелевой аппаратуре. Сырой продукт был
очищен перегонкой при 20° и 5 мм рт. ст., собиралась средняя
фракция ***.
По литературным данным, JFs образуется также при действии
BrF3 на J2O5 [10] и Mn (J03h [11], причем эти реакции
сопровождаются выделением кислорода, например:
6J205 + 20BrF3 = 12JF5 + 10Вг2 + 1502.
Большой интерес представляет метод получения пятифтористого
иода, описанный Квасником [44].
Физические свойства
Физические свойства пятифтористого иода описаны в работах
Горе [2], Муассана [4], Придо [5]. Наиболее полно физические
константы его описали Руфф и Брайда [8].
Пятифтористый иод — бесцветная жидкость, замерзающая, по
данным Руффа и Брайда, при 9,6 и кипящая при 98°. Недавно
Роджерс с сотрудниками, работавшие с более чистыми
препаратами, определили для пятифтористого иода температуру
замерзания 9,43±0,01° [31].
Плотность твердого препарата при 0° d°0 = 3,754, а при —193°
она равна 4,07 г/см3. Зависимость плотности жидкого
пятифтористого иода от температуры в интервале от 0 до 35° может быть
представлена уравнением d\Q = 3,29—0,004 t ****.
Упругость пара твердого пятифтористого иода (в мм рт. ст.)
находится в такой зависимости от температуры (°К):
lgP= 11,764—3035/Т.
Для жидкого вещества в интервале 10 — 70°
lgP = 8,83-2205/7,
* При этом кварц начинает разрушаться.
** JF? можно перевести в JF5 путем добавления иода до ясно-желтого
окрашивания.
*** Другие варианты этого способа получения пятифтористого иода из
элементов приведены на стр. 88 и 93.
**** Или d = 4,38—0,004 Т (для °К).
85
Экстраполяция по кривой упругости пара дает т. кип. 98±1J>°
[8].
По данным Роджерса с сотрудниками [31], зависимость
упругости пара от температуры выражается уравнением
lgP = 8,6591 — 2159/7,
согласно которому температура кипения пятифтористого иода
равна 100,5°.
Из измерений упругости пара пятифтористого иода были
вычислены скрытая теплота возгонки 13,89 ккал • моль'1 и испарения
10,09 ккал -моль'1 *; таким образом, скрытая теплота плавления
JFs равна 3,80 ккал • моль*1. Константа Трутона 27,2 характерна
для ассоциированных жидкостей.
Вязкость пятифтористого иода, по данным Хеттерингтона и
Робинсона [45], может быть описана уравнением ч\ = ч{\+М+
+ В/2). Значения констант в интервале от 14,55 до 69,30° таковы:
А =0,04231; В =0,000014; г]в= 0,4325 пуаз.
Пятифтористый иод относится к числу наиболее термически
устойчивых межгалоидных соединений. Он разлагается лишь при
нагревании выше 400° [4]. При нагревании до 500° он частично
превращается в иод и JF7
7JF5 = 5JF7 + J2.
Теплота образования JF5 была определена Вулфом [9] на
основании термохимических исследований реакции гидролиза
пятифтористого иода и его взаимодействия с раствором едкого кали
при 18° С. Уравнения этих реакций [9] приводятся в том виде, как
они приведены в работе Эванса с сотрудниками [37]:
JF5()K) +ЗН20(жГ 5HF(250H2O) +HjO3(1250H2O);
лр5(ж)+6КОН (220Н2О) = 5KF(250H2O; + KJO3(1250H2O) f ЗН20(ж).
Теплота образования жидкого пятифтористого иода
1'А[тъ) + ^F^r) = JF5(>Kb
вычисленная Вулфом по данным перкой и второй из
приведенных выше реакций, равна соответственно 205,3 и 204,2; в
среднем 204,7 ккал-моль'1. Теплота образования газообразного
пятифтористого иода из элементов ра:вна 194,6 ккал- моль'1.
Эванс, Мунзон и Вагман[37], исходя из экспериментальных
данных Вулфа, приведенных к температуре 298,16° К, вычислили
теплоту образования жидкого и газообразного пятифтористого
иода, соответственно равную 212,4 ±1,5 и 200,6±1,6 ккал - моль-1.
* Теплота испарения пятифтористого иода, по [31], равна 9,88, а по [37] —
9,85 ккал-моль"1 для 2.5°.
86
Термодинамические свойства пятифтористого иода, включая и
теплоту образования (стр. 25), тто Эвансу с сшлзудниками [37],
приведены в табл. 17. ^
Таблица 17
и:
о
Е-Г
0
250
273,16
298,16
300
400
500
600
700
800
900
1000
1100
1200
1300
1400
1500
X
1 5 *
о СЗ v
0
-60,74
-61,95
—63,32
-63,41
—68,19
—72,38
-76,13
-79,50
-82,56
—85,36
—87,94
—90,33
—92,56
—94,64
—96,60
—98,45
Термодинамические свойства JF
- х
i ?|
^ §х
0
13,89
14,59
15,31
15,37
17,85
19,77
21,26
22,44
23,39
24,17
24,83
25,38
25,85
26,25
26,61
26,92
X
7
сз
5) х 1
0
74,63
76,54
78,63
78,78
86,04
92,15
97,39
101,94
105,95
109,53
112,77
115,71
118,41
120,89
123,21
125,37
О О О
1 ^
S: §
0
3472
3985
4565
4611
7140
9885
12 756
15 708
18 712
21753
24830
27918
31020
34 125
37 254
40 380
X
7
сз
8-
«* 7
СЗ Л
<=> 3- w
0
21,65
22,65
23,70
23,77
26,57
28,19
29,18
29,82
30,26
30,57
30,79
30,96
31,09
31,19
31,27
31,34
* (Г)
7
«^
о
о
< *
—200,68
—202,59
—202,59
—202,6
—202,59
—202,43
-202,15
-201,80
—201,41
—201,02
—200,61
—200,19
—199,78
—199,36
— 198,95
—198,53
—198,121
7
<£
—200,68
-184,19
-182,47
—180,6
— 180,51
—173,17
— 165,88
-158,66
-151,50
—144,40
-137,34
—130,33
—123,36
-116,44
—109,54
— 102,67
— 95,84
**•*
*<
JW)
161,017
145,987
132,41
131,498
94,613
72,505
57,792
47,301
39,447
33,351
28,483
24,510
21,206
18,415
16,028
13,964
Спектр поглощения пятифтористого иода в интервале
температур 16—60° непрерывный в видимой и ультрафиолетовой
областях [13], что указывает на отсутствие вибрационной структуры.
Вопросу о строении молекул пятифтористого иода было
посвящено несколько работ. Броун и Пинов [141 на основании
исследований электронной дифракции заключили, что молекула JF5
имеет .строение тригональной бипирамиды (Dzh). Однако
рассчитанное ими, исходя из этой модели, среднее расстояние всех связей
J—F 2,56 А оказалось значительно больше суммы ковалентных
атомных радиусов (2,05А), что лишает достоверности
представления Броуна и Пинова о строении пятифтористого иода.
Повторное изучение пятифтористого иода методом
электронной дифракции [15] также не дало удовлетворительных результатов
из-за малой рассеивающей способности атомов фтора по сравнению
с атомами иода, но все же оно показало, что для строения JF5 не
может быть принята модель тригональной бипирамиды *.
* Более вероятным казалось искаженное октаэдр и чес кое расположение
атомов фтора вокруг иода, если принять три различных расстояния J—F.
87
Вопрос о строении молекул JF5, по-видимому, правильно
решен в работе Лорда, Линча, Шомба и Славинского [16]. Эти
авторы изучили спектры комбинационного рассеивания JFs при
комнатной или несколько более низкой температуре, а также
инфракрасные абсорбционные спектры в газообразной фазе в интервале
250—3300 см~1 и при относительно низком давлении паров, причем
они столкнулись с рядом затруднений, обусловленных высокой
химической активностью JFs. Результаты измерений дали
основание принять для молекулы пятифтористого иода строение
тетрагональной пирамиды (симметрия C4v) с четырьмя атомами фтора
в углах квадрата; пятый атом фтора и атом иода расположены
на оси, перпендикулярной к основанию. Если предположить, что
все расстояния J—F равны и что ZFJF равен 105°, то вычисленное
(приближенное) межъядерное расстояние J—F оказывается равным
1,75 А, то Эвансу с сотрудниками [37], — 1,83 А *.
Электронная конфигурация пятифтористого иода рассмотрена
в. работах Кимбалла [17] и Скотта 1181.
Диэлектрическая постоянная пятифтористого иода
сравнительно велика. Измерения показали, что ее величина, в зависимости
от температуры (в пределах 12—42°), может быть выражена
уравнением е^=41,09—0,198 t. Таким обр азом, е25°
= 36,2 [321**. Диполь-
ный момент жидкого пятифтористого иода, равный 3,67 D (для 25°
см. табл. 4), характеризует его как полярное вещество.
Пятифтористый иод, как и некоторые другие межгалоидные
соединения, проводит электрический ток. Электропроводность
жидкого пятифтористого иода в интервале температур 10—40°
измерили Бэнкс, Эмелеус и Вулф [19]. Для этой цели они получили
пятифтористый иод прямым фторированием дважды возогнанного
иода в охлаждаемой водой стеклянной колбе. Фтор пропускали
до обесцвечивания жидкости. Для измерений электропроводности
применяли свежеперегнанный препарат. Сосуды для измерений
изготовлялись из кварцевого стекла или из стекла пирекс.
Результаты измерений для трех образцов препарата приведены в табл.
18 ,и на рис. 15. Измерения образцов А и В проделаны с
платиновыми, а образца С — с никелевыми электродами.
Из данных табл. 18 следует, что электропроводность
пятифтористого иода в интервале 10—40° порядка Ю-5 ом~{ см~\
Температурный коэффициент электропроводности положительный.
Измерение электропроводности более тщательно очищенного
препарата пятифтористого иода, произведенное Вулфом, дало несколько
меньшую величину при 25°—1,53 • 10"5сш -^с-м-1 [20] ***. Прибавление
иода к пятифтористому иоду увеличивает электропроводность по-
* По Бауэру, связи J—F0 в пятифтористом иоде неравноценны и равны
1,85 и 2,17 А [43].
** См. также табл. 2, в которой указаны и некоторые другие
молекулярные константы пятифтористого иода.
*** Роджерс с сотрудниками, измеряя электропроводность чистого
пятифтористого иода, перегнанного непосредственно в ячейку из тефлона, нашли
еще меньшую величину 0,54—0,57-Ю-5 ом~~} см~1 (при 25°) [46].
88
\ Таблица 18
Электропроводность пятифтористого4 иода
Образец
препарата
В
А
С
С
А
с
А
Температура, °С
9,8
10,0
10,2
13,5
15,0
19,0
20,0
Электропроводность,
ом~ *см~~
х-105
1,78
1,61
1,91
2,05
1,89
2,28
2,08
Образец
препарата
А
В
С
А
В .
А
А
Температура, °с
25,0
25,0
25,0
31,0
35,0
39,0
40,0
Электропроводность,
ом~* • см~1
х.105
2,30
' 2,45
2,55
2,52
3,12
2,78
2,81
следнего 1191. В табл. 19 показаны величины электропроводности
растворов SbFs, BF3, S03 в пятифтористом иоде, по данным Вулфа
[201. Сведения об электропроводности системы HF—JF5 приведены
на стр. 93.
Эквивалентная электропроводность растворов KJ03 в JF5 в
интервале концентраций от 0,020 до 0,005 моль/л изменяется от
28,5 до 32,0 олг1 • см2 пропорционально корню квадратному из
концентрации. Экстраполированное значение для бесконечного
разведения составляет 35,5 ом-1 • см2. При электролизе растворов KJO*
в JFs на катоде выделяется иод.
Увеличение электропроводности JF5 при растворении в нем
указанных в табл. 19 веществ, особенно резкое в растворах пятифто-
ристой сурьмы, очевидно, обусловлено химическим
взаимодействием компонентов раствора и образованием комплексных
фторидов (стр. 93).
Кривые зависимости силы тока от напряжения показывают,,
что закон Ома не соблюдается, но получить воспроизводимые
величины потенциалов разложения не удалось из-за непрерывного
Т а б л и ц.а 19
Электропроводность растворов в пятифтористом иоде
Растворенное
вещество
Чистый JF5
BF3
SbF5
SbF5
S03
KJ03
Температура, eC
25
25
12,5
25
25
25 ;
Концентрация JF5
Насыщенный раствор
Q.045 моль/кг
0,045 моль/кг
1,71 моль/кг
0 0107—0,0428 моль/л
Эл ектропроводность
омГ -смГ
1,53-Ю^5
3,55-10~5
93-Ю-5
114.Ю-5
8,5-10~5
(3,07—10,75). Ю-4'
89
изменения электропроводности в процессе измерений. При
разности потенциалов в несколько вольт на катоде выделяется иод,
собирающийся затем на стенках и на дне сосуда.
Электрохимические свойства жидкого пятифтористого иода
указывают на то, что он подвергается электролитической
диссоциации. Имеющиеся в настоящее время и
описанные выше экспериментальные
данные не дают оснований сделать
определенные заключения о природе ионов, на
которые диссоциирует JFV Однако по
аналогии с трехфтористым бромом и
принимая во внимание высокое значение
константы Трутона, можно
предположить, что ионизации подвергаются
ассоциированные, в простейшем случае ди-
мерные, молекулы JFs по схеме
*0 t°
(JF5)2^JFt + JFr.
Рис. 15. Электропровод- Это предположение подтверждается
ность пятифтористого иода изучением химических свойств пятифто-
(трех образцов—Л, В, С). ристого иода>
«&*
Химические свойства
Химические свойства пятифтористого иода еще сравнительно
мало изучены. Литературные данные по вопросу о взаимодействии
его с металлами и металлоидами, окислами и солями, а также с
органическими веществами показывают значительное сходство в
поведении пятифтористого иода в этих реакциях с описанными
выше фторидами других галогенов. Особенно много общего
проявляется в поведении пятифтористого иода и трехфтористого брома в
реакциях фторирования неорганических и органических веществ,
при получении продуктов присоединения фторидов других
элементов, при синтезе комплексных фторидов, однако с тем отличием,
что пятифтористый иод менее активен.
Реакции с простыми веществами [2, 4, 7]
Щелочные металлы, погруженные в пятифтористый иод при
комнатной температуре, покрываются поверхностной пленкой гало-
генидов, которая предотвращает дальнейшее действие
пятифтористого иода на эти металлы. Однако если их нагреть до
температуры плавления, то реакция протекает очень энергично, почти
со взрывом [4].
Пятифтористый иод не действует на платину даже при
температуре красного каления [2]. Медь, серебро, магний, ртуть, хром
и железо весьма .незначительно реагируют с пятифтористым иодом
90
даже при длительном соприкосновении. В \ противоположность
этому, молибден и вольфрам горят в нем после нагревания [4, 7].
Бор, кристаллический кремний, мышьяк и сурьма легко реагируют
с пятифтористым иодом, часто с выделением пламени, образуя
иод и фторид соответствующего элемента [4]. Аналогично этому
кадмий превращается в CdF2 [21].
Пятифтористый иод не изменяется при перегонке с водородом
и не реагирует с кислородом при 100°. В результате реакции между
серой и теплым жидким JF5 образуется SF6 и выделяется иод.
Красный фосфор энергично горит в JF5, образуя PF5 и иод. Хлор
на холоду не реагирует с JFs, но при повышении
температуры жидкость окрашивается в желтый цвет. Природу этой
реакции Муассан не выяснил. Бром также не реагирует с JFs
на холоду, но при нагревании образуются BrF3 и JBr; можно
предположить, что эта реакция протекает по уравнению
3JF5 + 4Br2 = 5BrF3 + 3JBr [4]. Иод растворяется в пятифтористом
иоде, но не реагирует с ним [4].
С древесным углем JFs реагирует на холоду без
воспламенения, причем образуются CF4 и иод [41.
Реакция с неорганическими соединениями
Пятифтористый иод энергично гидролизуется водой и водными
растворами щелочей, солей и т. п. с образованием HF, Ш03 или
их солей. Вследствие гидролиза пятифтористый иод дымит на
воздухе, особенно в присутствии влаги, и раздражает
дыхательные пути.
Руфф и Брайда [8] обнаружили, что при осторожном
прибавлении воды к JFs сперва образуется окситрифторид иода *
JF5 + Н20 = JOF3 + 2HF.
При дальнейшем действии воды в условиях бурно
протекающей реакции JOF3 превращается в йодноватый ангидрид:
2JOF3+3H 20 = J А+Ш F;
5JOF3^J205+3JF5.
По мнению Руффа и Брайда [8], последняя из этих реакций
обратима; при (взаимодействии J2Os с JF5 не удается получить
чистого JOF3 и реакция доходит до состояния равновесия.
Однако Эйнслей с сотрудниками [21] показали, что при
нагревании JOF3 до 110° в присутствии сухого азота он превращается в JF5
и фторид иодила JO2F по уравнению
2JOF3 = J02F + JF5.
* Это вещество образуется в процессе, получения JFs из элементов, если
фтор содержал примесь кислорода и.^ч при проведении этой реакции в
кварцевой аппаратуре [8].
91
Окситрифторид иода образуется также при растворении J204 &
пятифтористом иоде [21].
Эти же авторы изучали взаимодействие JFs с рядом
кислотных окислов (Р205, As205, Sb205, V205, Сг03, Mo03, W03, N02)
и обнаружили, что при этих реакциях образуются такие вещества:
1) фторангидриды кислот фосфорной POF3, ванадиевой VOF3 *
и хромовой Cr02F2, причем реакция с фосфорным ангидридом
протекает по уравнению
Р205 + 3JF5 = 2POF3 + 3JOF3;
2) фториды—SbF5-3J02F; 3) продукты соединения окислов с пятифто-
ристым иодом —2Mo03-3JF5, W03-2JF5 **, N02-JF5.
Мышьяковый ангидрид не растворяется в пятифтористом
иоде, но дает с ним объемистое белое твердое вещество
неопределенного состава.
Шарп в своем обзоре сообщил, что при действии JF5 на
хлористый тионил был получен хлорфторид тиоиила SOC1F, а при
действии на окись углерода — иодфторид «арбонила COJF [22,
стр. 120]. Однако, по данным Эйнслея с сотрудниками [21], СО,
а также S02 не реагируют с пятифтористым иодом.
Пятифтористый иод на холоду медленно реагирует с
кремнеземом, но при нагревании реакция протекает более активно с
образованием SiF4. Вообще же соединения кремния энергично
реагируют с JF5. Стекло, даже сухое, при обыкновенной температуре
медленно разъедается пятифтористым иодом, при нагревании же
образуются SiF4 и J2O5. Силициды кобальта, железа и ванадия
горят в легко нагретом JFs [4].
По наблюдениям Муассана [4], гидрид калия загорается в
пятифтористом иоде, продуктами реакции являются HF, KF, KJ и
иод. Карбид кальция при легком нагревании в пятифтористом иоде
накаливается добела. Продукты этой реакции не установлены.
При вливании JF5 в концентрированную серную кислоту он
не растворяется в ней, осаждается на дно сосуда и затем
реагирует с выделением пузырьков газа — фтористого водорода. С
крепкой азотной кислотой JFs смешивается без заметного
химического взаимодействия, бурно реагирует с соляной кислотой, при
этом выделяется фтористый водород [4].
Относительно реакций между пятифтористым иодом и
хлоридами или бромидами других элементов в литературе имеется
очень мало сведений. Известно, что СоС12 при взаимодействии
с JFs превращается в CoF2 [23]. По данным Эмелеуса и Шарпа
[24], кипящий JF5 не реагирует с хлоридами натрия, калия и
серебра.
По данным термического анализа пятифтористый иод не
образует соединений с фтористым водородом [46]. Эта система
* Был получен продукт состава 2VOF3-3JOF3-
** Авторы этой работы не дали доказательств того, что все эти
полученные ими продукты присоединения JOF3, J02F и JFs представляют собой
индивидуальные вещества.
92
относится к простым, эвтектическим; эвтектика отвечает
-99,5 мол.% HF и температуре —83,49°. Роджерс V сотрудниками
«одновременно определили и ряд физических свойств системы
JF5—HF: давление пара при 15°, плотность при 25° и
электропроводность.
Удельная (и молекулярная) электропроводность системы
JF5—HF с ростом концентрации фтористого водорода сначала
уменьшается, затем растет, достигая максимального значения
1,15 • Ю-3 ом~1смгх при содержании 23,7 моль/л HF (25). При
дальнейшем увеличении концентрации HF электропроводность снова
уменьшается.
Моляльное понижение точки замерзания для растворов JF5
в жидком фтористом водороде оказалось равным 2,35°,
рассчитанная величина = 1,53°, что, по мнению авторов, свидетельствует о
частичной диссоциации JF5 в разбавленных растворах [461.
Комплексные соединения пятифтори-
стого иода
Известные до настоящего времени комплексные соединения
пятифтористого иода можно разделить на две группы:
комплексы, состоящие из JF5 и фторидов других элементов (т. е. двойные
фториды), и продукты присоединения JFs к молекулам
неорганических и органических веществ.
Комплексные соединения пятифтористого иода
с фторидами других элементов
Для JF5 известно всего лишь несколько соединений этого типа
(в противоположность тому, что описано для BrF3). Образование
таких соединений можно было ожидать при взаимодействии JFs
с галогенидами других элементов. Однако эти реакции еще очень
мало исследованы, а имеющиеся немногие экспериментальные
данные указывают на относительно слабое фторирующее действие
JFs при таких реакциях.
Эмелеус и Шарп [24] пытались получить комплексные
фториды этого типа путем растворения фторидов металлов в пятифто-
ристом иоде *. Фториды серебра и бария почти не растворимы
в кипящем JFs **. Фтористый калий растворяется в таких
условиях (около 1%). После охлаждения этого раствора выделяется
белое кристаллическое вещество; избыток растворителя удаляется
испарением в вакууме при 2—5 мм давления. Химический анализ
показал, что это вещество представляет собой KJF6. Этот
комплекс был получен также при длительном кипячении
азотнокислого калия с (Пятифтористым иодом (при этом выделялись лары
* Пятифтористый иод для этой цели был приготовлен путем пропускания
фтора через иод, помещенный в U-образную кварцевую трубку, охлаждаемую
частичным погружением в воду; трубка непрерывно встряхивалась, чтобы
предотвратить образование поверхностного слоя пятифтористого иода.
** По данным Вулфа [20], фторид олова также не растворяется в
пятифтор истом иоде.
93
N02) с последующим испарением избытка JFs в вакууме
при 20° [21].
Вода разлагает KJF6 соответственно уравнению
JF^~ + ЗН20 = 6F" + ЮТ + 6Н+.
Так же действуют водные растворы щелочей. При нагреваний
до плавления при 200° KJF6 разлагается, теряя JFs и оставляя
KF. Четыреххлористый углерод не действует на KJF6, а диоксан
производит лишь незначительное разложение [24]. Эти свойства
KJFe свидетельствуют об относительной прочности его.
Помимо KJF6, известен еще один гексафтороиодат— N02JF6.
Гексафтороиодат нитрония был получен [25] непосредственным
взаимодействием фтористого нитрония с пятифтористым иодом
(N02F+JF5 = N02JF6) или с иодом* в виде белого твердого
вещества **, устойчивого при низких температурах и уже при
комнатной температуре значительно диссоциирующего на NO2F и JFs.
Вода разлагает его по уравнению
(N02) JF6 + 4Н20 = HN03 + HJO3 -f- 6HF.
Условия получения и некоторые химические свойства гекса-
фтороиодатав калия и нитрония (см. также стр. 97) дают
основание полагать, что они содержат комплексный анион JF6~.
Однако относительно строения этого аниона еще нет достоверных
сведений.
Выше уже отмечалась аналогия в свойствах BrF3 и JFs,
проявляющаяся, в частности, в предположительных схемах
электролитической диссоциации (стр. 90). Для трехфториегаго брома
известны два ряда комплексных фторидов с катионом BrF2+ и
анионом BrFr. Поэтому можно было ожидать, что и пятифто-
ристый иод образует два таких же ряда комплексов.
Из комплексов первого типа до настоящего времени
получено в индивидуальном состоянии только одно соединение
[JFJ^SbFVT [20]. Пятифтористая сурьма легко растворяется .в
горячем пятифтористом иоде. После удаления избытка растворителя
в вакууме (5 мм рт. ст.) было получено гигроскопическое белое
вещество, плавящееся при 103° с незначительным разложением.
Серный ангидрид легко растворяется в пятифтористом иоде и
увеличивает электропроводность последнего (табл. 19), очевидно,
вследствие образования соединения S03 с JFs. При попытке
выделить это соединение путем перегонки смеси S03 и JF5 была
получена постоянно кипящая смесь состава JFs- 1,17S03. Вулф [20]
предположил, что раствор S03 в пятифтористом иоде содержит
фторосульфонат JF4" S03F~~.
Трехфтористый бор также увеличивает электропроводность
пятифтористого иода, что позволяет предположить образование
* При действии иода на N02F сперва образуется JF5, а затем N02JF6.
** Результаты анализа полученного продукта показали содержание в нем
около 7 мол.% избыточного JF5.
94
фторбората JF4 BF4 . Это предположение косвенно
подтверждается синтезом KBF4 при взаимодействии BF3 и KF в пятифтористом
цоде [20]. Попытки получить или установить образование в
пятифтористом иоде продуктов соединения JF5 с фтористым
водородом (JF^HF^T или H2F+JF6~) были безуспешны.
Продукты присоединения JF5 к неорганическим
или органическим веществам
При изучении реакции взаимодействия JFs с кислотными
окислами (стр. 92) были получены вещества состава 2Мо03 • 3JFs и
W03 • 2JF5, однако условия получения этих продуктов вызывают
сомнение в том, являются ли они химически индивидуальными
веществами; так, Мо03 не растворяется в пятифтористом иоде,.
но увеличивается в объеме, остаток после удаления JF5 (белого
цвета) имеет состав 2Мо03 • 3JF5. Вольфрамовый ангидрид легко
растворяется в кипящем JFs. После удаления избытка JF5
остается светло-желтое твердое вещество, в котором отношение JFs: WO3
при повышении температуры уменьшается и при 160° состав этого
продукта приближается к W03-2JF5 (с некоторым избытком
иода) [21].
Более убедительные данные получены в отношении комплекса
N02*JF5. Он кристаллизуется при пропускании N02 в пятифто-
ристый иод, при дальнейшем пропускании N02 жидкость почти
полностью затвердевает; избыток JFs удаляют отсасыванием
с последующим нагреванием до 100°. Продукт реакции состава
N02 • JF5 представляет собой зернистый порошок кремового
цвета. Это вещество возгоняется при умеренном нагревании, при
более высоких температурах разлагается, освобождая N02; бурно
реагирует с разведенным раствором КОН, образуя KJ03 и KFr
при этом выделяется двуокись азота [21].
В литературе описано также получение продуктов
присоединения JFs к солям кислородных кислот методом растворения солей
в пятифтористом иоде * и испарения избытка растворителя. Вулф
(20] таким путем получил вещества такого состава: КР03 • 2,3JFs **;
K2S2O8.1,02JF5; KJ03. ( ~ 0,5) JF5 ***. Аналогичные вещества—
KJ04-JF5 и Ba(N03)2-2,5JF5 — получили Эйнслей с
сотрудниками [21].
Слабая изученность таких веществ не дает возможности
получить представление об их химической природе и о формах связи
* Не растворимые или труднорастворимые в пятифтористом иоде соли
выдерживаются в нем в течение некоторого времени.
** Вулф отмечает, что продукт реакции не давал осадка с нитроном,
поэтому в нем не содержались ни KPF6, ни KP02F2.
*** Иодат калия растворяется в пятифтористом иоде без выделения
кислорода. Из этого раствора нельзя было выделить чистый KJ03 даже
многочасовым нагреванием в вакууме при 200° остатка после испарения
растворителя — продукты всегда содержали заметное количество JFs- Следует также
отметить, что твердый КЛОэ легко абсорбирует пары JF5 при комнатной
температуре.
9S
-JF5 с солями или окислами. Отсутствие в ряде случаев
определенных стехиометрических отношений и характер изменения
состава от температурных условий указывают на то, что такие
вещества нельзя считать комплексами определенного состава.
Можно было бы предположить, что они являются сольватами. в
.которых JF5 соединен с катионом или анионом соли. Подобного
типа соединения известны, например, для HF или BF3: [HFK] Н2Р04,
[HFK]2S206 [26]; K2SO4 • BF3, K3PO4 • 3BF3, K2P207 4BF3 [27].
Наличие неподеленной пары электронов в молекуле JFs создает
условия для образования продуктов присоединения к веществам,
содержащим акцепторы электронов. Однако возможность
образования таких соединений должна быть подтверждена
экспериментально.
Нельзя исключить также предположения о том, что в данных
случаях мы имеем дело с солями, содержащими продукты их соль-
волиза пятифтористым иодом.
Из продуктов присоединения JF5 к молекулам органических
веществ пока известен только диоксанат пятифтористого иода
С4Н802 • JFs [28]. При добавлении избытка JF5 к диоксану
осаждаются бесцветные кристаллы диоксаната, плавящиеся с
разложением и выделением HF и паров иода. Температура плавления от
84 до 112° (и несколько выше), в зависимости от условий
нагревания. Этот комплекс очень легко гидролизуется на воздухе и
даже в эксикаторе над серной кислотой; одним из продуктов
гидролиза является HJ03.
Руфф и Кайм [7] сообщили, что JFs образует с пиридином
продукт присоединения, но не привели сведений о составе и
свойствах этого комплекса.
Применение пятифтористого иода
для получения комплексных фторидов
Здесь имеются в виду реакции взаимодействия в пятифтори-
стом иоде как растворителе двух типов комплексов, один из
которых содержит катион JF^» а другой — анион JFe~. Общая
простейшая схема этих реакций такова:
М (JF6) + JF4(RFn) = MRF„ + 2JF5*.
До настоящего времени получены лишь единичные комплексные
соединения этих двух типов, что ограничивает возможность
использования этих реакций для синтеза комплексных фторидов MRFn,
M(RF„)2 и т. д. Поэтому в литературе описаны реакции
получения лишь KSbF6 и KBF4 [20]. Первый из этих комплексных
фторидов "был получен путем нагревания эквимолярных количеств
KF и SbF5 в пятифтористом иоде, который затем был удален отгон-
* Реакции этого тила более подробно описаны в главе III (о трехфтори
стом броме).
96
кой ffi-ри 8 мм рт. ст. 'И три комнатной температуре. Остаток
представлял собой продукт состава KSbF6 • 0,23JFs.
По Вулфу [20], фториды калия и сурьмы с пятифтористым иодом
образуют комплексы KJF6 и JF4SbFG, которые затем реагируют друг
с другом:
K+JF6 + JF4+SbF^ - KSbF6 + 2JF5.
Аналогичным путем объясняется получение тетрафторобората
калия
К+JF^ + JFlBFr = KBF4 + 2JF5.
Реакции с органическими соединениями
Муассан [4], изучая свойства JFs, наблюдал, что с
пятифтористым иодом бурно реагируют многие органические вещества:
спирт, эфир, этилацетат, ацетон, бензин, скипидар, сероуглерод,
хлороформ, тетрахлорэтан, трихлорэтилен, тетрахлорэтилен и
некоторые другие. Реакции с водородсодержащими веществами
обычно протекают бурно и сопровождаются выделением
фтористого водорода и появлением пламени. Реакции с серу- и хлорсо-
держащими соединениями при комнатной температуре протекают
более умеренно и характеризуются выделением серы [4, 7].
Руфф и Кайм [7] изучали действие JF5 на бензол при
комнатной температуре *. Основными продуктами реакции являются
фтористый водород, иодбензол, небольшие количества ди- и три-
иодбензола C6H4J2 и C6H3J3. Фторбензол и фториодбензол
образуются лишь в незначительных количествах. Часть бензола осмо-
ляется и обугливается.
Большее (внимание было уделено изучению реакций JF5 с
галоид производными метана [7, 29, 30]. При длительном действии
JF5 на сухой СС14 (при 30—35°) был получен газ, состоящий
главным образом из CCI3F и небольших количеств CCI2F2.
Одновременно образуются 'JO, JC13 а также J2O5 и фторосиликаты
(вследствие разъедания стекла сосуда). Трихлорфторметан CCI3F с
большим трудом .подвергается дальнейшему фторированию при
действии на него «пятифтористым иодом. Так, когда CC13F
пропускали в кипящий JF5 ** и образовавшуюся смесь паров
нагревали сперва до 125, а затем до 225°, то только несколько процентов
CC13F превратилось в CC12F2 [7].
Саймоне с сотрудниками [29] установили, что при действии JFs
на йодоформ в медной аппаратуре реакция начинается при
температуре смеси льда с солью, затем температура постепенно
повышается до 80—90°. Продуктами реакции являются фтороформ и
дииоддифторметан в соотношении приблизительно 15:1. В
аналогичных условиях CJ4 дает гексафторэтан C2F6. Однако Бэнкс с
* Эти авторы отмечают, что при проведении данной реакции необходимо
избегать сильного нагревания, так как при 50° реакция становится очень
бурной, поэтому температура не должна превышать 35°.
** Эту реакцию Руфф и Кайм проводили в аппарате для получения семи-
фтористого иода (стр. 99).
7—1419
97
сотрудниками [30] обнаружили, что если эту реакцию проводить
в стекле, то образуется трифториодметан CJF3 с хорошим
выходом; очевидно, последний, реагируя с медью, превращался в C2F6k
Тетраиодэтилен CJ2 = CJ2 при взаимодействии с JF5 дает иод-
пентафторэтан C2JF5.
В реакции с четырехбромистым углеродом JF5 оказался
более слабым фторирующим реагентом, чем ВгРз [30]. При
комнатной температуре JF5 не реагирует с СВг4. Реакция протекает при
нагревании примерно до 90° и выдерживании при этой
температуре в течение 3 часов. Газообразные продукты реакции
состояли преимущественно из CBr2F2 (83% выхода) и небольших
количеств CBr3F и CF4. Из остатка в реакционной колбе было
выделено немного CBr3F. В результате всех этих исследований Бэнке
с сотрудниками пришли к заключению, что JF5 является удобным
реагентом для фторирования иодпроизводных углеводородов.
2. СЕМИФТОРИСТЫЙ ИОД JF7
Получение
Семифтористый иод—единственное межгалоидное соединение
типа АХ7, содержащее восемь атомов галогена в молекуле. Он
. был получен Руффом и Каймом [6] в 1930 г. Эти авторы,
перегоняя сырой пятифтористый иод, полученный действием фтора на
иод или на нагретый J2O5, обнаружили наличие вещества, более
летучего, чем JF5, содержащего иод и фтор. Они
предположили, что это вещество представляет собой высший фторид
иода, образовавшийся при действии фтора на JF5. Это
предположение подтвердилось дальнейшими опытами.
При пропускании фтора в жидкий JF5 при комнатной
температуре не было замечено образования этого летучего соединения.
Но оно было получено при нагревании смеси фтора с парами
JF5 и оказалось семифтористым иодом, образовавшимся в
равновесной реакции
JF5 + F2^JF7.
По данным Руффа и Кайма, выход JF7 ограничен скоростью
установления равновесия и его состоянием в газовой фазе.
Вследствие экспериментальных затруднений они не смогли получить
точных значений состояния равновесия и влияния скорости
реакции, но показали, что вьпход JF7 растет с температурой пв
интервале 100—270°. Дальнейшее повышение температуры было
невозможно, так как уже при 300° наблюдалось быстрое
разрушение платиновой аппаратуры смесью фтора и фторида иода.
Руфф и Кайм описали два метода получения JF7 — действием
фтора на JF5 или непосредственно на иод *, которые
различаются также своим аппаратурным оформлением. Далее будет описан
второй из этих методов, по данным этих авторов, более
целесообразный. На рис. 16. изображен примененный для этой цели
аппарат, изготовленный из платины.
* В этом случае реакция проходит в два этапа: сперва образуется JF5r ■*
который затем превращается в JF7-
98
Через тубус Т\ вносят взвешенное количество иода и затем
ерез трубку R\ пропускают фтор для превращения иода в JFs,
^пи этом охлаждают нижнюю часть аппарата (#i). Окончание
Рис. 16. Аппарат для
получения семифтористо-
го иода по Руффу и
Кайму.
этой реакции определяют
взятием пробы жидкости
при помощи стеклянного
капилляра, который вводится
через Т\. Жидкость должна
быть бесцветной или лишь
Рис. 17. Аппарат для получения
семифтористого иода по Шомбу и
Линчу:
1—реакторная колонка с никелевой
трубкой; 2—шар из монель-металла диаметром
12,7 см; 3—вентиль; 4—прокладка из
мягкой меди; 5—камера для конденсации
длиной 30,5 см и медная трубка 6,35 см; 6—
никелевые трубка и пластинка;
7—прокладка из тефлона; 8—никелевая камера для
конденсации; 9—трубка для вхождения*
фтора; 10— отвод для входа и выхода
воды; 11—медная трубка, присоединенная к
шару из монель-металла; 12—никелевая;
зажимная пластинка с шестью болтами..
светло-желтого цвета. После
того как весь иод превратился в пятифтористый иод, #i
нагревают до 90°, а трубку Н2 — до 250—270°. Смесь паров
JFs и фтора поступает в Я2, где и происходит реакция
образования JF7. He вошедший в реакцию JF5 конденсируется в холо-
99
дильнике В и стекает снова в Яь Пары семифтористого иода
через Т2 переходят в два стеклянных приемника, первый из которые
охлаждается примерно до —100°, а второй — жидким возду.
хом *. В первом приемнике конденсируется JF7 в виде белой или
желтоватой твердой массы с примесями JF5, J2O5, SiF4 и фторо-
силикатов. Второй приемник служит для конденсации смеси
избыточного фтора с кислородом и азотом (из воздуха).
Таким путем удалось получить JF7 с выходом в 83%, считая
на взятое количество иода. Для очистки сырого продукта из него
сперва удаляли SiF4 отсасыванием при —90°, затем для
освобождения от JF5 достаточна одна перегонка из охлаждаемого льдом
сосуда в приемник с температурой —100° при исключении доступа
воздуха **.
Шомб и Линч [33] для получения очень чистого
семифтористого иода несколько изменили метод Руффа и Кайма, главным
образом в отношении аппаратурного оформления, некоторых
деталей процесса и способа очистки препарата. Примененный ими
реактор (рис. 17) состоял из меди, никеля и монель-металла. Сперва
в этом же приборе получают JF5 из иода и фтора ***. Пятифторй-
стый иод вносят через 3. Фтор из электролизера пропускают
через 9 со скоростью 0,4 моля в час****. Когда поглощение
фтора заметно уменьшается, температура колонки повышается
до 280—290°, тогда скорость протекания фтора уменьшают до
0,2—0,3 моля в час; колба из монель-металла охлаждается
до 70—80° 'посредством водяной бани. Семифтористый иод
конденсируется в ловушке, охлаждаемой до —78° смесью твердой
углекислоты и трихлорэтилена. Выход JF7, из расчета на
израсходованный иод, почти количественный. Очистка препарата
производится фракционированной возгонкой в специальном
аппарате*****. Чистота препарата контролировалась по инфракрасному
.спектру отдельных фракций.
Физические свойства [6]
Чистый семифтористый иод при комнатной температуре
представляет собой бесцветный газ, обладающий затхлым и кислым
раздражающим запахом. На воздухе, вследствие взаимодействия
* Этот второй приемник сообщается с внешней атмосферой при помощи
трубки, наполненной фтористым калием для поглощения влаги воздуха.
** В работе Руффа и Кайма описаны и другие детали процесса очистки
препарата.
*** Для получения JF5 в прибор через 4 вносят иод, удаляют
конденсатор и через центр колонки до дна шара из монель-металла 2 помещают
никелевую мешалку.
**** Фтор очищают от примеси фтористого водорода, пропуская его через
медную ловушку, охлаждаемую до —78°, и через медную трубку, содержащую
фтористый натрий и нагретую до 100°.
***** Этот аппарат и метод очистки семифтористого иода подробно описаны в
статье Шомба и Линча.
Более упрощенный метод получения семифтористого иода предложен
Квасником [44].
100
влагой, он образует туман. При охлаждении, в зависимости от
Условий, он переходит в подвижную бесцветную жидкость или
нежно-белый рыхлый порошок или в бесцветные кристаллы.
Температура плавления (в запаянных кварцевых трубочках, при
давлении 2 атм) 5—6°.
Упругость пара твердого семифтористого иода, измеренная
Б интервале от —63 до 0° (в мм рт. ст.), изменяется с
абсолютной температурой согласно уравнению
\gPMM = 8,6604-1602,6/7.
Вычисленные по этому уравнению значения упругости пара
довольно хорошо совпадают с экспериментальными данными^
Путем экстраполяции было найдено, что упругость пара твердого
семифтористого иода достигает значения 760 мм рт. ст. при 4,59,
следовательно, эту температуру можно считать температурой
кипения семифтористого иода (слегка переохлажденного).
Молекулярный вес газообразного семифтористого иода,
определенный по плотности пара, был найден равным 259,5—260,8,
средняя величина 260,2 (теоретическое значение 259,9).
Руфф и Кайм, пользуясь уравнением lgP= 70 —fC, вы-
4 • о I о • 1
числили теплоту испарения семифтористого иода и нашли ее
равной 7,33 ккал • моль"1, а константу Трутона — равной 26,4. Из
этого они заключили, что семифтористый иод в газообразном и
жидком состоянии образует ассоциированные молекулы. Однако
Гринвуд [341 отмечает, что эти значения ошибочны, так как Руфф
и Кайм не учли, что примененное ими уравнение дает скрытую
теплоту возгонки твердого семифтористого иода, которая
отличается от теплоты испарения на величину скрытой теплоты
плавления. Поэтому истинное значение константы Трутона должно
быть значительно меньше, чем 26,4.
Плотность жидкого семифтористого иода под давлением его
пара (в запаянных трубках) относительно воды при 4е d\ = 2,8
d\ --- 2,7.
Бернштейн и Кац [38] измерили равновесие диссоциации
газообразного семифтористого иода между 450 и 550°К. Для реакции JF7(t) =
= JF5(r) + F2(r), по данным этих авторов, A//°(JF7) = 28,5 ± 2 и
изменение энтропии AS0 равно 43,5+ 3,0 ккал-моль~~х. Эванс, Манзон
и Вагман [37], исходя из этих данных, вычислили A#°298,i6(JF7)
равным 29,1 + 0,5 ккал • молъ~л9 а комбинируя эту величину с теплотой
образования JF5(r) AHf°29s,\e, равной 202,60 ккал-моль-1 (табл. 19),
вычислили для реакции V^tr) +7/2F2(r) = JF7(r) величину A#/°298.i6 JFz(r)
231,7+ 1,8 ккал-моль-1. Термодинамические свойства, JF7, по данным
авторов, приведены в табл. 20.
Лорд с сотрудниками [16] исследовали инфракрасные
спектры поглощения газообразного семифтористого иода ib
широком интервале давлений (2,8—760 мм рт. ст.) и спектры комби-'
нацио'нного рассеяния жидкого семифтористого иода при темпе-'
101
Таблица 20
Термодинамические свойства JF7(r)
К
о
е-Г
0
250
273,16
298,16
300
400
500
600
700
£00
900
1000
^ X
о о ^ !
1 * §
о' ^ ^
0
-61,14
—62,64
—64,23
—64,34
—70,32
-75,74
—80,66
—85,12
-89,21
—92,96
—96,44
X
^7_
О О Q '
© s ^
£ §х ,
0
16,40
17,55
18,96
18,81
22,77
25,77
28,07
29,87
31,31
32,48
33,46
X
i
*7
«" *
8> х
0
1 77,54
80,19
, 82,73
83,15
93,00
101,51
108,73
114,99
120,52
125,44
129,90
7
° ь §
1 ч;
0
4100
4794
5585
5643
9108
12885
16842
20909
25048
29232
33460
X
i
а
з U
~ ^
ОХ
0
29,1
30,8
32,3
32,4
36,5
38,8
40,2
41,1
41,7
42,1
42,4
7
о
о
35 §
<g
—228,69
—231,73
—231,72
—231,7
—231,70
—231,37
—230,85
—230,25
—229,61
—228,94
-228,26
—227,56
7
о
о
^ S
< g
—228,69
—202,27
— 199,53
—196,6
-196,37
— 184,64
—173,02
—161,51
— 150,10
— 138,791
-127,55
— 116,41
V
к
ър
__
176,820
159,641
144,10
143,056
100,881
75,625
58,830
46,862
37,915
30,974
25,440
ратуре, близкой к комнатной. Анализ результатов этих
исследований привел авторов к заключению, что
для семифтористого иода структуры
низшей симметрии должны быть исключены;
из структур высшей симметрии
возможны только две: плоскостной семиугольник
(симметрия D7h) и Пентагон ал ьна1Я битти-
рамида (симметрия D5h), но для JF7
следует принять вторую из этих структур.
Атом иода расположен в центре
правильного пятиугольника, образованного пятью
атомами фтора, остальные два атома
фтора находятся иа равном расстоянии
от плоскости, на оси, проходящей через
атом иода (рис. 18) *.
Даффи [35], применив метод
направленных валентных связей к молекулам
или ионам типа АХ7 (JF7, ZrF?-, NdF?-,
TaF7~), рассчитал волновые функции
орбит для пентагональной бигоира'миды
(считая ее наиболее вероятной моделью
для JF7) и показал, что D5h орбиты в
JF7 можно представить конфигурацией 5s5p35ds. Соображения об
электронной конфигурации иода в семифтористом иоде имеются
также в работе Скотта [18].
* По Бауэру [43], пять связей J— F, направленных к вершинам
основания бипирамиды, имеют длину 1,83 А, а две другие связи, направленные к
вершинам бипирамиды, — 1,94 А.
102
Рис.
18. Структура
семифтористого иода.
Химические свойства
Руфф и Кайм [6], работа которых является, по сути,
единственным сообщением о химических свойствах семифтористого
иода, отмечают, что в этом отношении он очень похож на трех-
фтористый хлор, но' реагирует менее активно. Со многими
веществами семифтористый иод взаимодействует таким образом,
что два атома фтора (очевидно, расположенные на оси,
проходящей через центр пятиугольника) очень легко отщепляются.
Образуется пятифтрр истый иод, который или выделяется, или вступает
в дальнейшие реакции фторирования, сопровождающиеся уже
выделением иода.
Сведения о химических свойствах семифтористого иода и о его
реакциях с неорганическими и органическими веществами имеют
сугубо качественный характер и в большом числе случаев дают
лишь приближенное представление о природе и продуктах
реакций. Приведенные далее данные из работы Руффа и Кайма
относятся к действию газообразного 'семифтористого иода.
Реакции с простыми веществами [6]
Действие семифтористого иода на металлы в качественном
отношении не отличается значительно от действия других
фторидов галогенов. Реакции JF7 с натрием, калием и барием протекают
бурно-и сопровождаются появлением пламени. Магний, алюминий и
олово сперва покрываются коркой, а затем сгорают при нагревании.
Хром, железо и кобальт реагируют при температеое красного
каления. Молибден и вольфрам при нагревании превращаются
в летучие-соединения MoF6 и WF6. Медь и серебро слегка тускнеют
при нагревании их с JF7. Такая же устойчивая пленка
образуется на поверхности ртути. На платину JF7 не действует.
При (Комнатной температуре водород не реагирует с JF7, но
при нагревании происходит бурная, со взрывом, реакция —
образуются HF, HJ и немного иода. Бор, кремний — аморфный и
кристаллический—'реагируют при комнатной температуре с
образованием BF3 и SiF4. Так же реагируют фосфор, мышьяк и сера;
и в этих случаях образуются, вероятно, фториды данных
элементов. Но их состав, а также и природа других продуктов реакции
еще не установлены. Селен и сера реагируют только при
нагревании.
Реакция между хлором и JF7 протекает при нагревании, причем
образуются JC1, JC13 и, очевидно, C1F. Для брома не наблюдалась
реакция с JF7, иод же образует JF5.
Реакции с неорганическими соединениями [6]
Газообразный JF7 спокойно растворяется в воде, часть его
проходит через воду, не разлагаясь; в водном растворе, вследствие
гидролиза, обнаружены ионы J04~ и F-. В водных растворах
юз
едкого натра JF7 растворяется с разогреванием * и образованием
NaF и NaJ04, часть периодата натрия при этом отщепляет кислород,
и превращается в иодат.
Смесь предварительно нагретой окиси углерода с парами JF7
сгорает бледно-желтым пламенем, при этом наблюдается
выделение иода. Окись азота образует при комнатной температуре
едкие газы (NOF, N02F), JF5 и другие продукты. Сернистый
ангидрид реагирует с JF7 при нагревании с образованием JFs и
других веществ невыясненного состава.
Кварц только слегка разъедается семифтористым иодом, стекло
же реагирует с ним гораздо сильнее. Нагретые Мо03 и WO3
реагируют с JF7 с выделением иода и образованием, вероятно, окси-
фторидов, но Р2О5 даже после нагревания слабо реагирует.
В реакции JF7' с бромистым калием наблюдается выделение
брома, более сильное при нагревании, очевидно, вследствие
превращения КВг в KF.
По данным Шомба и Линча [33], 'хлористый рубидий при
взаимодействии с избытком JF7 и последующем испарении
последнего (при комнатной температуре) лишь частично
превращается ,в RbF. Фториды «атрия, калия и рубидия в этих
условиях не растворяются в семифтористом иоде и не соединяются с
ним. На этом основании Шомб и Линч заключили, что при
комнатной температуре не образуется устойчивое соединение типа
MMFe.
Реакции с органическими соединениями [6J
Чистый или активированный древесный уголь быстро
воспламеняется в JF7, при этом выделяется иод. Смесь метана с парами
JF7 взрывает при нагревании; в месте горения наблюдается
выделение иода и HF, но не заметно выделения углерода (возможно,
что образуются фторзамещенные метана). Бензол и бензин сразу
воспламеняются в JF7. Так же ведут себя спирт и эфир. Другие
кислородсодержащие вещества — как ацетон, уксусная кислота и
этилаиетат—при реакции с JF7 разогреваются до 'кипения,
причем образуются J2O5, иод, HF и летучие органические продукты
(фторзамещенные).
Парафиновое масло, смазка для кранов, вата и целлюлоза, в
зависимости от концентрации и количества JF7, реагируют с ним
медленно или с выделением пламени.
При действии JF7 на пиридин образуется твердое
красно-желтое вещество, возможно, продукт присоединения фторида иода
к пиридину.
Галоидпроизводные углеводородов—СНС1Я, CCU, тетрахлорэтан,
тетрахлорэтилен и трихлорэтилен — экзотермично реагируют с
семифтористым иодом. Образуются смеси фторзамещенных
продуктов (например, CFC13, CF2C12 и другие), HF и JC13.
* Так же, по-видимому, реагирует JF7 с водным раствором карбоната
натрия.
104
Шомб и Линч [33] изучали действие JF7 на фреоны CCI2F2 и
CCUFV Эти исследования представляют интерес в том отношении,
qTo наличие в молекуле по меньшей мере двух атомов фтора при
одном из атомов углерода делает инертным атомы другого
галогена (хлора, брома), содержащегося в молекуле [36]. Оказалось,
что JF7 относительно медленно действует на CC12F2 *, 'превращая
некоторую часть его в CCIF3. Реакция значительно ускоряется,
если ее проводить в присутствии HgF2 как катализатора.
3. НИЗШИЕ ФТОРИДЫ ЙОДА (ФТОРИСТЫЙ ИОД)
Из шести возможных двухатомных межгалоидных соединений
до настоящего времени не был известен только фтористый иод JF.
Как упоминалось в начале этой главы, попытки получить низшие
фториды иода JF и JF3 или обнаружить их в качестве
промежуточных продуктов при синтезе JF5 и JF7 были безрезультатны. Так,
по сообщению Руффа и Брайда [8], измерение упругости пара
и температур плавления смесей пятифтористого иода с иодом
не дало никаких доказательств образования низших фторидов
иода. Фракционная перегонка этих смесей всегда давала только
исходные продукты — пятифтористый иод и иод [81.
Эйнслей, Никольс и Робинзон [21] сообщили, что при
растворении двуокиси иода Jo04 в пятифтористом иоде получается
светло-синяя жидкость (по окраске напоминающая водный раствор
тетраммиаката солей меди), содержащая JOF3 и небольшие
количества иода. Окраска исчезает при добавлении незначительного
избытка фтора. Так как растворы иода в пятифтористом иоде шо-
коладно-бурого цвета, то эти авторы предположили, что
наблюденную ими окраску следует объяснить образованием некоторого
количества низшего фторида иода, возможно трехфтористого иода*
Однако эти экспериментальные данные нельзя считать
достаточным обоснованием такого предположения.
Существование фтористого иода было обнаружено при
спектральных исследованиях. В 1950 г. Дюри [40, 41], исследуя
эмиссионные спектры зеленовато-желтого пламени, появляющегося при
действии газообразного фтора (или трехфтористого хлора) на
твердый иод или на органические соединения иода, получил
доказательства образования в этих условиях молекул фтористого иода **.
Спектр фтористого иода состоит из системы полос в интервале
435—690 ммк.
Спектральные данные (вибрационный анализ, расчеты по
Берджу — Спонеру и другие) дали возможность вычислить
энергию диссоциации (табл. 12), силовые константы [40, 41] и
некоторые термодинамические свойства фтористого иода в газообразном
состоянии [37].
* Семифтористый иод сильнее действует на CC12F2, чем фтор; Шомб и Линч
полагают, что это является следствием каталитического действия иода на
реакцию замещения хлора фтором.
** Аналогичным путем в пламени, образующемся при реакции между
фтором и бромом, обнаружен фтористый бром.
105
По Дюри [41], энергия диссоциации JF (г) для основного со-
стояния равна 1,98±0,04 или 2,87±0,04 эв в зависимости от того
диссоциирует ли он на возбужденный иод и нормальный фтор или
наоборот. По мнению Дюри, если принять более высокое из этих
значений энергии диссоциации, то фтористый иод должен быть, по
крайней мере, умеренно устойчивым веществом и даже более
устойчивым, чем фтористый хлор и фтористый бром. Между тем
фтористый иод не удалось изолировать. Поэтому Дюри считает
более правильным для него низшее значение энергии диссоциации
(1,98 эв)у меньшее то сравнению с энергиями диссоциации
фтористого хлора и фтористого брома (табл. 12). В дальнейшем
Дюри и Гейдон [42] сделали некоторые оговорки, но все же они
полагают, что химические данные говорят в пользу низшего
значения.
Однако Слуцкий и Бауэр [39], отметившие некоторые ошибки
в вычислениях Дюри и Гейдона, а также Эванс с сотпудниками [37]
не согласны с этим заключением и считают более обоснованным
высшее значение энергии диссоциации. Исходя из этой величины,
Эванс с сотрудниками вычислили теплоту образования
фтористого иода по реакции 1/2^'г) + V2F2(r) = JF(r). равной 30,0±
±0,05 ккал -моль~х. Другие термодинамические константы
газообразного фтористого -иода (стр. 25), по [37], приведены в табл. 21.
Малая изученность фтористого иода не дает еще основания считать
эти данные достаточно хорошо обоснованными.
Таблица 21
Термодинамические свойства JF (г)
о
к
0
250
273,16
298,16
300
400
500
600
700
800
900
1000
1100
1200
1300
1400
1500
X
| ? §
Ъ § X
0
—47,858
—48,496
—49,133
—49,177
-51,308
—53,007
—54,427
—55.648
—56,722
—57,680
—58,546
—59,335
—60,060
—60,732
—61,358
—61,943
X
ОО q |
О* ^ 2*
£§х
0
7,177
7,234
7,293
7,297
7,521
7,710
7,862
7,987
8,090
8,177
8,250
8,314
8,369
8,418
8,461
8,500
X
1
^7
5 '*
% X
0
55,035
55,730
56,426
56,474
58,829
60,717
62,289
63,635
64,812
65,857
66,796
67,649
68,429
69,150
69,819
170,443
7
об ^
i I
0
1794,2
1976,0
2174,5
2189,1
3008,4
3855,0
4717,2
5590,9
6472,0
7359,3
8250,0
9145,4
10042,8
10943,4
11845,4
12750,0
X
7
си -
* о
0
7,76
7,88
8,00
8,01
8,35
8,56
8,69
8,78
8,84
8,89
8,93
8,96
8,99
9,01
9,03
9,05
7
о
о
< S
—30,0
—30,080
—30,082
—30,089
—30,090
—30,10
—30,10
-30,10
—30,10
—30,10
-30,10
—30,11
—30,11
—30,11
—30,11
1 —30,12
—30,12
7
.о
5
о
о
< S
—30,0
—30,354
—30,378
—30,405
—30,407
—30,51
-30,61
—30,72
—30,82
—30,92
—31,03
—31,13
-31,23
—31,33
—31,43
—31,53
1 —31,63
ч-^.
к
ъс
26,5349
24,3047
22,2868
22,1514
16,670
13,381
11,189
9,622
8,448
7,534
6,803
6,205
5,706
5,284
4,923
4,609
106
ЛИТЕРАТУРА
1. Н. Kam merer, J. prakt. Chem., 85, 452 (1862).
2. G. Gore, Chem. News, 24, 291 (1871); Phil. Mag. (4), 41, 309 (1871).
3. O. Ruff, Z. anorg. Chem., 98, 27 (1926).
4. H. Mo i ss a n, Ann. chim. phys., (6), 24, 240 (1891); С. г., 135, 563 (1902);
Bull. Soc. chim. ,(3), 29, 6 (1903).
5. E. B. R. P r i d e a u x, J. Chem- Soc, 89, 316 (1906).
6. O. Ruff, R. Keim, Z. anorg. allg. Chem.', 193, 171 (1930).
7. O. Ruff, R. Keim, Z. anorg. allg. Chem, 201, 245 (1931).
8. O. Ruff, А. В г a i d a, Z. anorg. allg. Chem., 220, 43 (1904).
9. A. A. Woo If, J. Chem. Soc, 231 (1951).
10. H. J. Emeleus, A. A. W о о 1 f, J. Chem. Soc, 164 (1950).
11. A. G. Sharpe, A. A. W о о 1 f, J. Chem. Soc, 798 (1951).
12. O. Ruff, А. В г a i d a, Z. anorg. allg. Chem., 214, 91 (1933).
13. С F. White, С F. G о о d e v e, Trans. Faraday Soc, 30, 1049 (1934).
14. H. В r a u n e, P. P i n n о w, Z. phys. Chem., В., 35, 239 (1937).
15. M. T. Rogers, A. L. W a r h a f t i g, V. S с h о т а к е r, Abstr.
Atlantic City Meeting, Am. Chem. Soc, 1947, Apr., 21 p.
16. R. С Lord, M. A. Lynch, W. С S с h u m b, E. J. Slowin-
s к i, J. Am. Chem. Soc, 72, 522 (1950).
17. G. E. Kimball, J. Chem. Phys., 8, 188 (1940).
18. R. L. Scott, J. Chem. Phys., 18, 1420 (1950).
19. A. A. Banks, H. J. Emeleus, A. A. W о о 1 f, J. Chem. Soc,
2861 (1949).
20. A. Wo о If, J. Chem. Soc, 3678 (1950).
21. E. E. Aynsley, R. Nichols. P. L. Robinson, J. Chem.
Soc, 623 (1953).
22. A. G. Sharpe, Quart. Rev-, 4, 115 (1950).
23. Г. С. Бус и Д. Т. П и н к с т о н, Галоидные соединения фтора,
сб. «Фтор и его соединения», ИЛ, М., 1953, стр. 163.
24. Н. J. Emeleus, A. G. Sharpe, J. Chem. Soc, 2206 (1949).
25. E. E. Aynsley, G. Hetherington, P. L. Robinson,
J. Chem. Soc, 1119 (1954).
26. R. F. W e i n 1 a n d, J. Alpha, Z. anorg. Chem., 21, 43 (1899).
27. P. В a u m g a r t e n, H. H e n n i g, Ber., 72, 1743 (1939).
28. A. F. Scott, J. F. В u n n e t, J. Am. Chem. Soc, 64, 2727 (1942).
29. J. H. Simons, R. L. Bond, R. E. M с A r t h u r, J. Am. Chem.
Soc, 62, 3477 (1940).
30. A. A. Banks, H. J. Emeleus, R. N. H a s z e 1 d i n e, V.
Kerrigan, J. Chem. Soc, 2188 (1948).
31. M. T. R о ge г s, J. L. S p e i r s, H. B. T h с m so n, M. В. Р a-
n i s h, J. Am. Chem. Soc, 76, 4843 (1954).
32. M. T. Rogers, H. B. Thompson, J. L. S p e i r s, J. Am.
Chem. Soc, 76, 4841 (1954).
33. W. С Schumb, M. A. Lynch, Ind. Eng. Chem., 42, 1383 (1950).
34. H. H. Г р и н в у д, Усп. химии, 22, 458 (1953).
35. G. H. Duf fey, J. Chem. Phys., 18, 943 (1950).
36. A. E. Ч и ч и б а б и н, Основные начала органической химии, Госхим-
издат, М. — Л., 1953, т. 1, 188.
37. Н. Evans, Т. R. M u n s о n, D. D- W a g m a n, J. of Research
Nat. Bureau Standards, 55, 147 (1955).
38. R. B. Bernstein, J. J. К a t z, J. Phys. Chem., 56, 885 (1952).
39. L. S 1 и t s k y, S. H. Bauer, J. Am. Chem. Soc, 76, 270 (1954).
40. R. A. D и г i e, Proc Phys. Soc, 63. A, 1292 (1950).
41. R. A. D и г i e, Proc Roy. Soc, [A] 207, 388 (1951).
42. R. A. D uric, A. G. G а у d о n, J. Phys. Chem., 56, 316 (1952).
43. E. Bauer, J. Phys. Chem., 56, 343 (1952).
44. Руководство по препаративной неорганической химии, под редакцией
Г. Брауера, ИЛ, М., 1956, стр. 101—102.
45. G. Hetherington, P. L. Robinson, J. Chem. Soc, 3681 (1956).
46. М. Т. Rogers, J. L. Spe i rs, M. В. Pani sh, H. В. Thompson, J. Am.
Chem. Soc, 78, 936 (1956).
ГЛАВА V
ХЛОРИСТЫЙ БРОМ BrCl
Начиная со времени открытия хлористого брома Баларом в
1826 г. [1], вопрос о существовании этого соединения обсуждался
в литературе в течение многих лет. В ряде случаев в аналогичных
исследованиях были получены противоречивые результаты.
Причиной этого является значительная неустойчивость хлористого
брома.
Исследования, посвященные доказательству существования
хлористого брома (и высших хлоридов брома), выяснению условий
его образования и устойчивости в индивидуальном состоянии и
в растворах, можно разделить на такие группы:
а) работы препаративного характера,
б) изучение физико-химических свойств • системы бром — хлор,
в) оптические исследования системы бром — хлор в
органических растворителях,
г) изучение реакций смесей брома и хлора с органическими
веществами.
В процессе этих исследований, доказавших существование
хлористого брома, единственного и весьма неустойчивого продукта
взаимодействия этих двух галогенов, были также установлены
некоторые его физико-химические и термодинамические
характеристики.
1. ПОПЫТКИ ПОЛУЧИТЬ ХЛОРИСТЫЙ БРОМ ИЛИ ЕГО ГИДРАТЫ
По описанию Балара, хлористый бром получается при условии
пропускания хлора в бром при обыкновенной температуре и
конденсирования выделяющихся при этом '-паров охладительной
смесью в виде красной (но окрашенной слабее, чем бром)
жидкости, легко подвижной и летучей, с сильным запахом и
слезоточивой [2].
В дальнейшем в литературе встречаются лишь единичные
указания на возможность получения хлористого брома путем
непосредственного взаимодействия хлора с жидким бромом. По Бор-
неману [3], для получения хлористого брома необходимо
действовать хлором на бром при 0°. Образующаяся при этом красная
жидкость имеет состав, близкий к тому, который соответствует
108
формуле BrCl. Эта жидкость начинает кипеть при 13°;
охлажденная до —80°, она обычно растворяет хлор, становится красно-
оранжевой и при охлаждении из нее выделяются кристаллы.
Содержание хлора в кристаллах, в зависимости от температуры
кристаллизации, соответствовало составу BrClM7, BrCli,67, BrGi,72.
Томас и Дюпуи [4] сообщили, что жидкий хлор реагирует с
бромом и образует сперва хлористый бром, который способен
затем присоединять хлор и переходить, вероятно, в ВгС13*.
Получается красная масса, затвердевающая вблизи —75°; при немного
более высокой температуре она плавится в красно-оранжевую
жидкость с т. кип. —19°. Этот продукт разлагается при
температуре значительно ниже 0°, выделяет часть хлора и
превращается в хлористый бром, замерзающий при охлаждении до — 39°
в твердую красную массу. Необходимо, однако, заметить, что -в
цитированных выше работах отсутствуют убедительные
доказательства образования продуктов непосредственного соединения брома
с хлором определенного состава.
То же можно оказать относительно работы Эндрюса и Карл-
тона [5], которые, измеряя плотность жидких смесей хлора и
брома, обнаружили явление незначительной (около 2%)
контракции и на этом основании пришли к выводу, что в данной
системе образуется соединение этих двух галогенов, хотя и в
ограниченной степени.
Совершенно не обоснованное экспериментальными данными
утверждение содержится в работе Крутвига [6], согласно которому
при действии сухого хлора на AgBr03 при 50° образуются BrCl
и AgCl и выделяется кислород.
Были произведены и такие попытки получить хлористый бром:
взаимодействием хлора и брома в водных растворах;
пропусканием хлора -в охлажденный льдом раствор брома ,в смеси равных
объемов хлорноватистой кислоты и воды, причем образуется
солянокислый раствор хлористого брома; электролизом смеси растворов
NaCl и NaBr с угольными электродами [2, стр. 340]; фракционной
перегонкой смесей хлора и брома [50].
Еще в 1829 г. Левиг сообщил о получении оранжево-красного
гидрата хлористого брома BrCl • 5Н20 при пропускании хлора
в бромную воду, охлажденную до 0° [2]. Борнеман повторил опыты
Левига и пришел к заключению, что полученные кристаллы имеют
состав ВгС! • 10Н2О, но, что вероятнее, они представляют собой
смесь С12-10Н2О и Вг2-10Н2О [3]. По наблюдениям Энвор-Ул-
ла [7], хлор и бром взаимно повышают свою растворимость в воде,
что, по его мнению, может служить доказательством образования
хлористого брома в водном растворе. Он пропускал хлор в жидкий
бром под слоем воды до насыщения и затем оставлял на
несколько часов при 18°, после чего начинали выделяться кристаллы
состава BrCl • 4Н26.
Все эти сравнительно немногочисленные работы препаратив-
* Анализ полученных продуктов не производился.
109
ного характера не дали прямых доказательств образования
хлористого брома, как индивидуального вещества, так как
полученные твердые фазы могли представлять собой смеси (твердые
растворы) хлора и брома или их системы с хлористым бромом
(то же и в случае гидратов).
Безуспешность попыток изолировать хлористый бром
объясняется его довольно значительной неустойчивостью, а также, по-
видимому, тем, что скорость установления равновесия
термической диссоциации хлористого брома на бром и хлор очень велика.
Еще менее убедительные результаты дали попытки доказать
образование или получить высший хлорид брома ВгС13.
2. ФИЗИКО-ХИМИЧЕСКОЕ ИССЛЕДОВАНИЕ СИСТЕМЫ БРОМ — ХЛОР
(В ОТСУТСТВИЕ РАСТВОРИТЕЛЯ)
Лебо [8] определял температуры плавления смесей брома с
хлором для решения вопроса, образуется ли продукт соединения
этих двух галогенов. По данным Лебо, кривая плавкости от точки
плавления брома (—7,3°) до точки плавления хлора (—102,5°)
имеет линейный характер без какого-либо заметного максимума.
Из этих данных Лебо заключил, что при кристаллизации смесей
брома с хлором образуются лишь смешанные кристаллы и что
«так называемое соединение брома с хлором соответствует
растворимости хлора в броме при 0°».
Более детальный термический анализ системы бром—хлор
произвел Карстен [9], который определил кривые плавления (и
замерзания) и кривые кипения, а также состав отдельных фаз —
жидкой и твердой или жидкой и газообразной, в зависимости от
исходной концентрации системы, с целью установить
существование и устойчивость возможных соединений брома с хлором.
Анализ кривых плавления и замерзания (рис. 19) и кривых
кипения (рис. 20) * и тот факт, что кривые не сближаются в
области концентраций, соответствующей 50 ат.% хлора, а кривые
точек на диаграмме кипения даже наиболее далеко отстоят друг
от друга в этой области, привели Карстена к заключению, что
хлор и бром не образуют между собой соединений ни в одном из
агрегатных состояний; в твердом же состоянии они образуют лишь
непрерывный ряд смешанных кристаллов.
Многочисленные экспериментальные данные Карстена,
установившие температуры плавления, замерзания и кипения
смесей брома и хлора, не согласуются с соответствующими
значениями, приведенными в работах Борнемана [3], Томаса и Дю-
пуи [4] и относящимися к смесям состава 1 : 1 или 1 : 3. Это дало
основание Карстену опровергнуть выводы этих авторов, в част-
* Все полученные Карстеном кривые состава жидкой и твердой фаз (рис. 19)
или газообразной и жидкой фаз (рис- 20) оказались непрерывными без
разрывов сплошности, без максимумов и минимумов. Такая форма кривых
характерна для многих двухкомпонентных систем, не образующих соединений, но
полностью смешивающихся в твердом и жидком состояниях.
ПО
ности утверждение Томаса и Дюпуи о существовании BrCI
.„ ВгС1з.
Основные выводы работ Лебо и Карстена (отсутствие
соединений в системе бром — хлор, образование смешанных
кристаллов) подтвердили Бутлер и Мак-Интош [2,10]* определением
Рис. 19. Кривые плавления и замерза- Рис. 20. Температуры кипения 'и со-
ния системы бром—хлор. став жидкой и газообразной фаз
системы бром — хлор.
кривых замерзания, а также определением молекулярного веса
брома в жидком хлоре.
К противоположному заключению пришел Луке [11] на
основании того, что пары, выделяющиеся из смеси брома и хлора под
давлением Р, содержат больше брома, чем это следует из
предельного отношения Вг/С1 = Ры/Р — Рвг (Ры — давление паров
чистого брома при температуре бани). По мнению Лукса,
это указывает на существование соединения, более летучего, чем
бром, которое он пытался получить фракционированной
конденсацией паров. Состав фракции при —90° соответствовал экви-
атомному отношению брома и хлора, в твердом виде она
представляла собой желтое, как охра, вещество, плавящееся при
—54° в темно-оранжево-красную жидкость.
На основании результатов физико-химического анализа
системы бром — хлор, приведенных в цитированных выше работах,
нельзя вовсе исключить возможность образования соединений
брома с хлором не только в жидкой и газообразной фазах, но и
в твердой фазе. Установленная Карстеном форма кривых (рис. 19
и 20) не отрицает предположения о наличии в этой системе
* С содержанием работ Бутлера и Мак-Интоша мы имели возможность
познакомиться только по справочной и реферативной литературе.
111
неустойчивого соединения, более или менее значительно
диссоциированного и образующего в твердом состоянии непрерывный
ряд твердых растворов с обоими исходными компонентами. Таков^
же, например, форма кривых плавления и кипения для системы
иод—бром, в которой твердо установлено образование
соединения— бромистого иода (глава VII). Кроме того, для решения
вопроса об образовании соединений в жидкой фазе, в частности
в двойных жидких системах, нельзя ограничиться
физико-химическим анализом данной системы только по одному какому-либо
свойству; необходимо изучение системы несколькими методами
физико-химического анализа и сопоставление полученных при
этом кривых состав — свойство. В литературе по
физико-химическому анализу имеется довольно много примеров, когда
соединение, образующееся в данной системе, не проявляется на
кривых плавкости, но оно обнаруживается, например, на
изотермах вязкости. Такова система трифенилметан — трехбромистая
сурьма, исследованная по плавкости Меншуткиным [131, по
вязкости— Курнаксвьгм с сотрудниками [14], а та,кже системы,
состоящие из фенола и алкиланилинов (метил-, диметил-, этил- и
диэтиланилина), исследованные Виноградовой, Ефремовым и
Тихомировой [15, 16] *.
В качестве примера можно привести также результаты
термического анализа многих систем иод — иодиды, не показавшие
образования полииодидов целого ряда катионов, хотя
существование таких полииодидов в жидкой фазе и особенно в
растворах было убедительно доказано разнообразными методами
исследования [17], в частности реакциями изотопного обмена [18].
Из сказанного становится вполне очевидной необходимость
более разностороннего исследования системы бром — хлор в
различных агрегатных состояниях и в разнообразных растворителях
для суждения о возможных формах взаимодействия этих
галогенов.
Такой подход к исследованию различных систем очень (важен
в тех случаях, когда в них могут образовываться более или
менее неустойчивые соединения, что имеет место в системе бром —
хлор и что подтверждается описанными далее результатами
исследования диэлектрических и оптических свойств этой системы.
Для решения вопроса, образуется ли хлористый бром в
смеси жидких брома и хлора, Фрелих и Иост [50] определили
диэлектрические проницаемости таких смесей для всей области
концентрации (0—100% хлора) в интервале температур от +10
до—60° через каждые 10°. Экспериментальные значения
диэлектрической проницаемости (е) значительно превышают величины е,
вычисленные по правилу смешения, и это различие возрастает с
понижением температуры.
Кривые величин е в зависимости от состава имеют максимум
в области 50 ат.% хлора, который лучше выражен при низких
* Ряд других аналогичных примеров приведен в монографии Аносова и
Погодина [12, глава XVI и др.].
112
температурах. Эти факты указывают на образование в смеси
^идких хлора и брома соединения состава BrCl *.
фрёлих и Иост сообщают, что на основании температурной
зависимости диэлектрической проницаемости и результатов
измерения плотности смесей жидких хлора и брома и отдельных ком-
п0нентов можно было приближенно вычислить величину диполь-
лого момента хлористого брома. Она равна ~ 0,6 D. (Расчетные
данные в работе не приведены).
3. ОПТИЧЕСКИЕ ИССЛЕДОВАНИЯ СИСТЕМЫ БРОМ —ХЛОР
Уже давно, начиная с работы Балара, было замечено, что
смеси брома и хлора в жидком и газообразном состояниях имеют
окраску более слабую по сравнению с чистым бромом. Это
наблюдение служило одним из доказательств образования соединения
брома с хлором.
В количественном отношении это явление впервые было
изучено Песковым [19] по истечении почти 90 лет после его открытия.
Определяя коэффициенты поглощения хлора, брома и их смесей,
Песков установил, что газообразная смесь брома и хлора
подчиняется закону Бера, и на основании этого заключил, что в ней
отсутствует химическое взаимодействие. В дальнейшем Добсон
[2, 20], применяя эту смесь в качестве светофильтра для
наблюдения полос озона в атмосфере, обнаружил, что ослабление
окраски протекает во времени (порядка минуты) и что
поглощение в ультрафиолете смесей брома и хлора не имеет аддитивного
характера.
Баррат и Стейн [20] развили эти наблюдения Добсона и на
основании спектрофотометрических исследований высказали
следующие соображения в пользу доказательства существования
хлористого брома в растворах смеси брома и хлора в четырех-
хлористом углероде:
а) кривая поглощения этой системы значительно отличается
от кривой поглощения раствора брома в ССЦ; константа
равновесия, рассчитанная по данным спектрофотометрических
определений свободного брома в системе бром — хлор — четыреххлористый
углерод, имеет приблизительно постоянную величину при
условии образования хлористого брома;
б) в смеси обоих галогенов обнаружена новая полоса
поглощения с максимумом при 3700 А;
в) изменение окраски смесей бром — хлор в четыреххлористом
углероде протекает во времени, в эфире и хлороформе реакция
протекает моментально, даже при —78°.
* Метод физико-химического анализа растворов, заключающийся в
измерении диэлектрической постоянной и поляризации систем из двух компонентов
в каком-либо растворителе, был разработан И. А. Шека [51]. Состав комплекса
в растворе устанавливался по максимуму отклонения экспериментально
найденной величины диэлектрической поляризации или диэлектрической
постоянной от вычисленной по правилу смешения.
8—1419
113
Джилам и Мортон [21] в общем подтвердили наблюдения пре,
дыдущих авторов; они нашли максимум поглощения при 3800 Д
и показали, что для растворов, в которых отношение брома ц
хлору равно Вг : ЗС1, характерно наличие полос поглощения хло*
ристого брома и хлора. Грей и Стил [22] и Иост [23], изучавшие
коэффициенты экстинкции газообразных смесей брома и хлора,
также пришли з заключению о существовании хлористого брома \
Л. С. Лилич [24] в работе, поовя-
0Л Х^Ч щенной спектрофото'метрическому ис~
р'^л/^ХЧ следованию взаимодействия галогенов,
0^_j j / N(*4 растворенных в четыреххлористом
/У 4*S углероде, указал на некоторые недо-
02 л j/ \ статки предыдущих оптических иосле-
// Ч\ дований систем, состоящих из двух
°!2 г. ' 2\ ■ 1 ' \ i \ i \ галогенов в растворах, в частности си
Вг9
-02
Ч^^ 6 4 2^' стемы бром — хлор. Он правильно
ч^"о-^^_^^г^- отметил, что в разрешении вопроса 6
^v^ jy**' взаимодействии галогенов в растворах
-0,4-J // более определенные результаты долж-
п П1 ^ но дать применение физико-химическо-
Рис. 21. Оптическая плотность r ^
системы бром—хлор 2С = го анализа.
=0,06 моль/л Для решения поставленного вопро-
1-х=ь2о ммК;'ii-i=5oo мм*; т- са применительно к системе иод —
-\=380 ммк; IV-\=370mmk. ХЛОр, ИОД— брОМ И брОМ — ХЛОр Ли-
лич в качестве исследуемого свойства
измерял на спектрофотометре оптическую плотность растворов
этих систем в четыреххлористом углероде, применяя ,при этом
«метод непрерывных изменений» [25—28]. Как известно, этот метод
состоит в изучении свойств смесей растворов при условии
постоянства суммарной концентрации и объема. Положение экстремальной
точки (максимума) на кривых отклонения свойств от
аддитивности, в зависимости от молярного соотношения реагирующих друг
с другом компонентов систем, указывает на их стехиометрическое
отношение в реакции, происходящей в системе.
Во всех исследованных Лиличем системах максимум
приходится на эквимолярное соотношение галогенов, что доказывает
образования 'соединений типа XiXn по схеме реакции
(Xi)2 + (Хц)2 ^ 2ХтХп.
Измерения системы бром—хлор в четыреххлористом углероде
проводились при концентрациях 0,06—0,02 и 0,01 моль/л. В качестве
примера на рис. 21 приведены результаты этих исследований для
раствора Ис = 0,06 моль/л. На оси ординат нанесена величина функции у =
= dc — dXj — dxiv выраженная в единицах оптической плотности (/ =
= IocBd ) с соответствующим знаком; по оси абсцисс нанесено
соотношение объемов или концентрации растворенных галогенов. Величина
* Результаты этих спектрофотометрических исследований были
использованы также для расчета константы равновесия (см. стр. 118).
114
dc получается в результате измерений системы (Xj.)2 — (Хп)2 — СС14,
а°величины dXl и dXu—из результатов определения молярного
коэффициента поглощения соответствующих двойных систем (Х^— СС14 и
(Хп)2-СС14.
Для сравнения на рис. 22 и
ные диаграммы состав — свойство
у
23 приведены
для систем
аналогич-
иод — хлор
12
ом
олл
а2
-еН
-0,8
-1.2
А
/ \_
/А \
// ^
% %
лр 2 * 6 8 \
V \л а а л. о ж ,
asl
0.6 А
С4-
0,2-
1¥л
2 ,6i
II
Я
Ч
-0,21
-ол!
-0.6 1
*^-*
\>^~
£-
% * /
Рис. 22. Оптическая
плотность системы иод—хлор:
1с=0,045жоль/л-, /~Х=580 ммк;
П—\=Шммк; III—\=400 ммк;
/у—а =360 ммк.
Рис. 23. Оптическая плотность
системы иод—бром:
1с-= 0,0105 моль\л\ /—Х=600 ммк; II—
—Х=440 ммк; 111—1=400 ммк; TV—X=
= 380 ммк; V—Х=360 ммк.
(£с=0,045 моль/л)— рис, 22 и иод —бром (2С=0,0105 моль/л) —
рис. 23. Экстремальные точки на этих диаграммах находятся на
пересечении двух «прямых, т. е. они более резко выражены, чем
в системе бром — хлор (даже при большей суммарной
концентрации последней). Это следует объяснить значительно большей
прочностью хлористого иода и бромистого иода по сравнению с
хлористым бромом (ом. табл. 1).
Таким образом, в работе Лилича впервые убедительно
доказано существование хлористого брома в системе бром — хлор — че-
тыреххлористый углерод, показано также большое значение
физико-химического анализа расгаорш в том варианте, который был
использован для решения вопроса о наличии и составе неустойчивых
соединений.
4. ИЗУЧЕНИЕ РЕАКЦИЙ СМЕСЕЙ ХЛОРА И БРОМА С ОРГАНИЧЕСКИМИ
ВЕЩЕСТВАМИ
Как уже упоминалось, эти реакции служили одним из
доказательств существования хлористого брома в смеси брома и хлора
и в растворах смеси этих галогенов в четыреххлористом углероде
или в других растворителях.
115
Прежде всего следует указать на реакции одновременного
присоединения брома и хлора к непредельным соединениям с
двойной связью. Уже давно было замечено образование этилен-
хлорбромида (1-бром-2-хлорэтана) при действии на этилен смеси
паров брома и хлора [29] или их раствора в разбавленной соляной
кислоте или в этиленхлорбромиде [30].
Вальден [31], действуя раствором хлора и брома в
хлороформе на малеиновый ангидрид, получил ангидрид хлорбромян-
тарной кислоты. Хансон и Джемс [32] нашли, что эквимолекулярная
смесь брома и хлора в четыреххлористом углероде
присоединяется к ненасыщенным кислотам гораздо легче, чем каждый
галоген в отдельности, причем образуются хлорбромпроизводные; так,
из коричной кислоты получается смесь двух стереоизомерных форм
Р-хлор-а-бром-р-фенилпропионовой кислоты СбНбСНС1—СНВг—
—СООН. Такой же опыт с фенилпропиоловой кислотой ов СС14 дал
два геометрических изомера кислоты СбНбСС1 = СВгСООН. Эти
реакции имеют бимолекулярный характер.
Реакции присоединения хлористого брома в последнее время
исследовали Баклес и Лонг [33]. Хлористый бром они получали
действием соляной кислоты :на N-бромацетамид
CH3CONHBr + HC1 = BrCl + CH3CONH2.
Этилен превращается в СН2С1 — СН2Вг, стирол—в С6Н5СНС1 — СНВг,
транс-коричная кисЛота—-в С6Н5СНС1СНВгСООН, из транс-стильбена
Br C1
I I
был получен С6Н5 —С—С — С6Н5 (эритро-а-бром-а'-хлордибензил), из
i i
н н
Br H
I I
цис-стильбена —СвН5 — С — С — C6HS (трео-а-бром-а'-хлордибензил)*.
I I
Н С1
Таким образом, присоединение BrCl происходит в
соответствии с правилом Маркокникова. Структура полученных хлорбро-
мидов, по мнению этих авторов, подтверждает полярный механизм
присоединения BrCl, а именно: Вг+ перемещается к двойной
связи, образуя комплекс,— ион бромония,— который затем
взаимодействует с ионом,-С1~ и превращается в конечный продукт. Для
цис-стильбена эти процессы выражаются следующей схемой:
I
С6Н5 - С - Н CH3CONH2Br+ С6Н5 - С - Н г,- Н — С — Вг
|| * | >Br+Xi I
С6Н5—С—Н С6Н5 —С—Н С1-С — Н
* В последних двух реакциях наряду с указанными веществами образуются
и другие продукты.
116
Другого типа реакции с участием BrCl изучали Тейлор и Фор-
и [34]. Они нашли, что эквимолярная смесь брома и хлора очень
быстро реагирует с диазоуксусноэтиловым эфиром (или с бен-
зоилфенилдиазометаном СбН5СЫ2СОСбН5), причем азот
замещается на галогены. Наряду с дихлор- и дибромпродуктами получается
также хлорбромпроизводное, которое при действии водного
раствора аммиака превращается в хлорбромацетамид
QH5OCOCHN2 + V2Br2 + V2C12 -* QH5OCOCHBrCl;
QH5OCOCHBrCl + NH3 ->ClBrCHCONH2 + C2H5OH.
Авторы полагают, что в этой и аналогичных реакциях
происходит смещение равновесия Вг2 + С12?2ВгС1 в правую сторону.
Определяя количества полученных дихлор-, дибром- и хлорбром-
производных, они приближенно установили относительное
содержание молекул хлористого брома и рассчитали константу
равновесия (К = 0,015). Следует, однако-, иметь в виду, что эти расчеты
имеют весьма приближенный характер.
Интересное доказательство образования хлористого брома
в водном растворе содержится в работе Н. П. Каняева и Е. А.
Шилова [35]. Изучение кинетики реакции бромирования ж-анизолсуль-
фоната натрия ж-СН3ОСбН450з№ при действии смесью бромнова-
тистой и соляной кислот привело к представлению, что в
данной системе активным бромирующим агентом является хлористый
бром, образующийся по уравнению
НОВг + НС11 BrCl + Н20.
При таком допущении кинетика реакции бромирования
получает удовлетворительное объяснение.
5. ФИЗИЧЕСКИЕ СВОЙСТВА И ТЕРМОДИНАМИЧЕСКИЕ КОНСТАНТЫ*
В литературе приведены весьма расходящиеся значения для
температур плавления и кипения хлористого брома: т. пл. от
—66 [36] до —54° [И]; т. кип. от +5° [36] до «комнатной темпе-
ратуры» [37]. Эти расхождения объясняются значительной
неустойчивостью хлористого брома. Как правильно отметил Гринвуд [37,
стр. 453], значения, которые были получены по кривым
плавления, замерзания и кипения для эквиатомных соотношений брома
и хлора, должны рассматриваться -не как константы чистого
соединения, а скорее как равновесные температуры для фазового
изменения типа (твердый раствор брома и хлора в хлористом
броме) — (жидкий раствор брома и хлора в хлористом броме). Поэтому
значения температур плавления и кипения, указанные в литерату-
* Физические свойства хлористого брома уже частично рассматривались
в предыдущих разделах этой главы.
117
ре, дают лишь приближенное представление о температурах,
между которыми «нечистый» хлористый бром существует в жидкой
фазе. Так, Карстен [9] нашел, что смесь равных атомных коли-
честв брома и хлора кипит при —5°, но для того, чтобы
газообразная фаза приняла тот же состав, требуется температура
около +36°.
Константа диссоциации хлористого брома.
Вследствие малой устойчивости хлористого брома выявилась
необходимость количественной оценки степени диссоциации его в
зависимости от температуры, растворителя и некоторых других
условий. Этому вопросу был посвящен ряд работ, в которых константа
диссоциации определялась на основе спектрофотометрических,
химических и других исследований.
По данным Баррата и Стейна [20], кривые поглощения брома
и хлористого брома столь различны, что фотометрированием в
желтой спектральной области можно определить концентрацию
свободного брома в системе бром — хлор — хлористый бром.
Большинство исследователей, например Грей и Стил [221 и Иост [23],
принимая это допущение, вели измерения экстинкции при длине
волны 579 ммк (максимум полосы поглощения' брома) и
рассчитывали константу равновесия по уравнению
_ [Вг2] [С12]
А [BrCl]2 '
Константы равновесия, полученные этими и некоторыми другими
авторами, приведены в табл. 22. Однако Веспер и Роллефсон [38]
отметили, что предположение о том, будто BrCl практически не
поглощает света при этой длине волны, не обосновано. Они пересчитали
данные Грея и Стила и графическим путем показали, что если
поглощение BrCl составляет хотя бы 1% от поглощения Вг2 (т. е. если
авгС1/авг2 = 0,01), то значения константы К изменяются от 0,132 и 0,125
(по Грею и Стилу) до 0,121 и 0,114; в случае же, когда осБгС1/авг2 =■
= 0,0124, для обоих опытов получается одно и то же значение /(=0,111.
Веспер и Роллефсон, применяя свет зеленых линий гелиевого
разряда ( ~ 0,502 ммк), измерили коэффициенты экстинкции
газообразных смесей брома и хлора при 28° и, принимая, что
при этой длине волны aBrci /авг2 составляет около 0,1, получили
совпадающие значения А=0,107±0,002. Брауэр и Виктор [39],
проводя измерение смеси газообразных хлора и брома в
соотношениях от 3 : 1 до 1 : 3 в присутствии посторонних (инертных)
газов, получили для К. несколько более высокое значение
0,144 + 0,003 (для комнатной температуры).
Блейр и Иост [40] из измерений парциальных давлений хлора
и брома в ССЦ получили для константы равновесия величины
0,12—0,14.
Еллинек и Шютца [41] на основании изучения равновесия
между бромом, хлором, хлористым и бромистым калием (и другими
парами хлоридов и бромидов) нашли, что константа равновесия
реакции 2ВгС1(г) ^Г Вг2(Г) + С12(г) при 800° равна 0,12. Шютца [42]
118
з
подобных же опытов определил для К величину 0,1325 при 500
% 140±0,005 при 800°. Дальнейшая попытка определить константу
Равновесия диссоциации хлористого брома путем изучения
химических реакций содержится в работе Бизона и Иоста [43]. Эти
авторы изучали равновесие газообразной системы, состоящей из
окиси азота, брома, хлора, хлористого и бромистого нитрозила
и хлористого брома в интервале температур 372—492° К. В этой
системе происходят следующие обратимые реакции (в газовой
.фазе):
2NOBr ^ 2NO + Вг2;
2N0C1^2N0 + C12;
2ВгС1 ^ Вг2 + С12.
Приданной температуре парциальные давления брома и хлора
определяли на основании 'Изучения диссоциации NOC1 или NOBr.
Зная общее давление системы, можно было рассчитать концетра-
цию BrCl. После внесения поправки на отклонение от закона
идеального таза Бизон и Иост получили значение /С = 0,148 при
462° К.
Сводка цитированных выше данных о величинах константы
равновесия реакции 2ВгС1 = Вг2 + С12 приведена в табл. 22.
Величины /С, рассчитанные различными авторами, находятся в
пределах от 0,1 до 0,2, причем заметно не оказывается влияние
температуры от комнатной до 800°, что, возможно, находится в связи
с малой величиной теплоты реакции образования хлористого
брома (см. стр. 123).
Форбес и Фуосс [44] для определения константы равновесия
измерили э.д.с. цепи РЩ2|НС1|НС1|НС1 + С12+ВгС1 + Вг2+Вг"|Р«г
при 25° и получили величину (3,2±0,2) • 10~4 (в 4- и 6-н. соляной
кислоте), значительно отличающуюся от величин, приведенйых в
табл. 22. Эти авторы ^предположили, что BrCl образует устойчивый
комплекс с ионом С1~ и что в исследованной ими системе
происходят и другие процессы, кроме того (2ВгС1 = Вг2+С12), 'который
лежит в основе расчета константы равновесия.
Более глубоко к решению вопроса о константах диссоциации
BrCl подошел К- В. Бутков [45, 46]. На основании произведенного
им анализа спектральных данных (полосатого спектра), учета
межъядерного расстояния и т. д. Бутков изучил сложное
равновесие молекул BrCl, Cl2, Br2, C1 и Вг в температурном интервале
от 298,1 до 3000° К. Он показал, что при температурах выше
1000° уже заметна диссоциация BrCl на атомарные бром и хлор.
Поэтому, чтобы представить термическую устойчивость хлористого
брома, необходимо учесть два равновесия и соответствующие им
константы *:
BrCl^Va Br2 + V2 Cl2;
* Также равновесия диссоциации Вг2 и С12 на атомы.
119
/Ci =
BrCl = Br + CI;
BrCl,
I<2=T
^Br'^Cl
BrCl
Таблица 22
Константы равновесия для реакции 2BrCLrj ^t Вг2/Гч + С12/г) *
Метод определения
Поглощение света . . .
Поглощение света . . .
Поглощение света . . .
Поглощение света . . .
Поглощение света . . .
Измерение парциальных
Измерение парциальных
давлений
гетерогенных систем КС1(ТВ|,
КВг(тв)' ВГ2(г]'С12(г)'
ВгС1(гч и другие пары
Тот же метод для систем
AgCl(TB), AgBr(TB),
Вг2(г)» СЦГ)» ЁгС1(Г) •
Также РЬС1а и РЬВг2
и другие пары солей
Измерение парциальных
давлений системы N0—
-Br2—Cl2—NOCl—
—NOBr—BrCl ....
Масс-спектрометр ический
анализ смеси брома и
Среда
Газ
»
»
»
СС14
СС14
—
—
Газ
»
Температура, °С
Комнатная
»
28
Комнатная
25
—
800
500
800
189
Комнатная
[Вг2 ] [С12 ]
Д~ [BrCl]2
0,132; 0,125**
0,10—0,22
0,107±0,002
0,144±0,003
0,145±0,006
0,12—0,14
0,12
0,1325
0,140
0,148
0,15—0,16
Ссылка на
г литературу
Грей,
Стил [22]
Иост [23]
Веспер, Рол-
лефсон [38]
Брауэр,
Виктор [39]
Попов,
Мэньон [52]
Блейр,
Иост [40]
Еллинек,
Шютца [41J
Шютца [42J
Бизон,
Иост [43]
Метрю с
сотрудниками [53];
В табл. 23 приведены вычисленные Бутковым значения
констант диссоциации и приведенных изобарных термодинамических
потенциалов BrCl ***.
* В литературе, например в обзорной статье Гринвуда [37], встречаются
значения констант равновесия, по данным приведенных в таблице авторов, но
рассчитанные по уравнению
BrCI^V.Br, + V,Q,f т. е. К = [BrCl]l2] •
** Результаты двух опытов.
*** Метод расчета значений этих констант подробно описан в работе Бут-
кова.
120
Таблица 23
Константы диссоциации и приведенные изобарные термодинамические
потенциалы хлористого брома [46]
Темпера- j Приведенный изобарный
тура, °К
3000
2000
1500
1000
900
800
700
600
500
400
298,1
потенциал
69,145
65,567
63,066
59,605
58,759
57,734
56,632
55,381
53,904
52,141
49,884
Константа диссоциации К\ при изменении температуры от 29&
до 3000° К увеличивается всего приблизительно в 3 раза. Зато
/С2, величина которой при температурах 298—1000° К очень мала
(хотя и увеличивается в 1027 раз), затем очень быстро растет
с температурой. Исходя из этих данных, можно считать, что при
2000° К BrCI диссоциирует преимущественно на атомы. Сравнение
значений К\ с приведенными в табл. 22 «показывает [37], .что
величины К\ (если их рассчитать по уравнению BrCI ^/гВгг+'/гСЬ),
равные 0,35 [41] и 0,36 [42] при 800°С (1073° К), близки к тем
значениям, которые найдены Бутковым из спектроскопических
данных.
Значение константы диссоциации хлористого брома при 25° С,
полученное Бутковым, соответствует степени диссоциации 21% [37].
Блейр и Иост [40] приводят для степени диссоциации BrCI в ССЦ
величину 52,4%, а Попов и Мэньон [52]—43,2±1% (также при
25°). Хотя эти значения различаются в 2—2,5 раза, они достаточно
ярко характеризуют значительную неустойчивость BrCI,
обусловленную непрочностью связи Вг—С1.
Кинетика реакции образования хлористого
брома него некоторые термодинамические
функции. Реакция образования BrCI из элементов в газовой фазе
происходит без изменения объема. Таким образом, ее следует
выразить уравнением Вг2 + С12 ^2ВгС1, предусматривающим
образование мономерных молекул [46].
Фотометрическое изучение процесса соединения брома с
хлором при 575 ммк дало основание Иосту [47] сделать вывод, что
эта реакция гомогенная и что она протекает по простейшему
бимолекулярному механизму. Константа скорости в среднем равна
7-10~4 секг\ а теплота активации—14 ккал. Развивая эти
исследования, Веспер и Роллефсон выяснили, что на скорость этой реак-
0,42
0,40
0,37
0,33
0,31
0,30
0,28
0,25
0,22
0,18 1
0,13 1
1 ■ 68,7
0,65
6,3-10-3
7,1-Ю-7
3,4-Ю~8
8,2-Ю-10
6,7-10~12
1,Ы0~14
1,5-Ю"18
2,4-Ю-24
3,3-Ю-34
121
ции большое влияние оказывает освещение—в темноте реакция
между элементами длится от 16 до 60 часов до достижения
равновесия, и тогда она имеет бимолекулярный характер, на свету
реакция значительно ускоряется и при освещении ртутной дугой
равновесие достигается в течение нескольких минут. Реакция ©
темноте катализируется стенками сосуда, так как после очистки
сосуда ее скорость возрастает. Это явление не наблюдается при
освещении, поэтому световая реакция, вероятно, является
гомогенной.
Наконец, Брауэр и Виктор [39], изучая эту же световую
реакцию, пришли к заключению, что она имеет цепной характер и
начинается, вероятно, диссоциацией Вг2 на атомы; квантовый выход
1—2-Ю3. Следующие схемы показывают течение реакций и их
теплоты:
Br2+Av -* Br+Br;
Br+Cl2 -> BrCl+Cl—5,7 кал;
Cl+Br2 -* BrCl+Br+6,0 кал;
Cl+BrCl ->Cl2+Br+5,7 кал;
Br+BrCl -> Br2+Cl—6 кал;
Br+Br -*> Br2+45,2 кал.
Литературные данные по теплоте реакции образования
хлористого брома противоречивы и, очевидно, не могут претендовать на
достаточную точность. Возможно, что причиной этого является
малая «величина теплоты образования его.
Имеющиеся в литературе данные по теплоте образования (или
теплоте диссоциации) хлористого брома в газообразной фазе
[22, 23, 38, 39, 43, 45, 54 и др.] различаются в несколько раз—от
0,105 [39] до 0,78 [45] ккал- моль'1. Так Бизон и Иост [43] из
равновесия реакции Вгад + Оад <^ 2ВгС1(Г) рассчитали три значения
теплоты образования А Я/0: 0,0; —0,27 и —0,55 ккал.
Сопоставление 'прежних кинетических и термохимических данных с
результатами спектроскопических 'исследований привело их к
заключению, что наиболее вероятное значение AHf° для этой реакции
при 25°—0,465 ккал* (или 0,233 ккал • моль*1). Иост же [23] из
температурной зависимости константы равновесия рассчитал
теплоту образования ВгС1(Г) из элементов и нашел ее равной 0,75 ±
±0,5 ккал • моль'1. В обзорной статье Гринвуда [37, стр. 470],
(приведены следующие данные (без ссылок на литературные источники):
1/2Вг2(Г) + 1/2С12(г) = ВгС1(Г) -f- 0,78 ккал-моль~{;
1/2Вг2(Ж| + VjCljtr) = ВгС1(г) — 2,93 ккал-моль-У
Эванс, Манзон и Вагман [54], пересчитав данные ряда авторов
[|22, 23, 38, 39, 43], приняли для Д#/° наиболее вероятное, по их
* См. также [55].
122
**-«-
*
ьс
7
^ о
< ±
о
*
У*
7
1
<5
5" ч
<3
*
*
х
7 •
• ^ 5
о р. Q ^
^Г ^
^ х
а
*
-7
о о л
^ 5
i £
1 ^
о
* §
к
X
7 Т
L л
со g о
§х.
*
~ X
^ "-
^т и
о?^ *
5: § х
w ^
i г- X
1 ^^ -
"^7 1
*: §.5
£ s x
W *
и:
°
ж
^
i
о
°1
о*
1
о
о*
1
о
о
о
о
о
1
J о
^
00
cs
-*&
ю
CS
1
со
о"4
1
t-~
""^
00
о
cd
^
00
«—*
^н
00
00^
ю
CD
CD
°Я.
t^T
ю
00
"^
ОС*
1
о
ю
CN
CD
00
CD
о"
CD
00
ю
о"
1
CN
о*
1
г>-
°1
оо*
"*«
cd
со
о
CN
о
СО^
CD"
ю
CD
CD
"^
h-Г
"tf
ч*
с*
|
CD
СО"
Ь-
CN
Tf
Tf
LO
о"
О
СО^
|
CN
с*
i
CD
СО
00*
ю
ь-
rt«
CN
CN
00
СО
СО^
N*
ю
00
СО
Ю
t^
о
о
°Чч
с*
|
CD
ОО*
CD
CN
LO
СО
ю
о"
СО
CN
СО^
|
CN
о"
1
h-
°°~
со*
CD
CN
CD
CN
CN
О
CD
co^
ю
со
^
lo
t^
t-
Tf
оо^
стГ
1
о
о
со
ю
5Л
о*
CD
о4
1
о"
1
^
СО^
осГ
CN
СО
1—1
СО
3
00^
cd*
Ю
СО
ОС
t^
с^."
,-н
1Л
о
cn~
lo
1
о
о
■^
cn
CD
С0^
С*
О
CD
о"
1
1
^1
о"
!
-^
^
оо"
ю
»—t
00
CD
СО
^
t^
t^
CD
'"О
CD
CD^
t>
00
о
00
со"
|
О
о
ю
CD
СО
с$
СО
1 •
о"
I
СО
°Я.
00
о
о
CD
ОО
-^
CN
t^
СО
СО
CD
О
О
СО
CN
t^
CN
ю"
1
о
о
CD
ю
CD
СО^
о"
t^
1
'
°1
о4
1
00
00^
СО*
CD
ю
Tf
t--
ю
CD
СО
CD
00
О
°1
СО*
00
(N
Ю^
CD*
|
О
о
с--
t^-
ю
СО^
о"
С0^
1
1
сГ
1
CN
°1
СО*
о
CD
СО
CD
CD
CD
CI
CD
CD
Ю
°^
CN
00
^H
CO
CD
t>"
1
о
о
00
о
со
о*
-^
^
1
1
°1
о*
1
ю
°1
СО*
^
CD
CN
Ю
t^
^
N.
CD^
CD
CD
CD
CO
00
CO
CD
00*
LO
|
О
о
CD
CD
CO^
<D
00
lO^
1
1
CN
o*
1
00
°l
CO*
о
CD
CN
-^
00
•^
CN
CD*
CD
CD
CN
^
00
oo
CD
4.
CD*
Ю
|
О
О
о
¥_н
со
о*
С^1
1>^
1
1
CN
сГ
I
о
о
CD*
г>
^
(N
СО
CD
О
со
00*
СО
Г^
1^
"*•
00*
СО
О
°я
о*
CD
|
О
о
^^
00
СО
со
(S
CD
00
1
1
CN
1
CN
О
CD*
CN
Ю
CN
CN
О
-^
CD
Ю
CD*
CD
T—•
CN
to
00
CO
•^
O^
»—t
CD
|
О
О
CN
Ю
CO
CO
o*
CD
CD
1
1
CN
°i
O*
1
со
q
CD*
O^
oo"
<N
y^
CD
00
CN
C*
N-
О
CD
LO
00
CD
CN
t-
»—<
CD
|
О
О
со
CN
CO
CO
o*
CO
CN*
1
1
CN
o*
1
LO
o^
CD*
co^
^■^
CO
О
CN
CO
LO
CD
O*
r-
^
Я1
to
00
CN
CD
CO^
CN*
CD
I
О
О
"^
о
CO
со
сГ
со
CN
CN*
1
1
CN
О*
1
СО
ол
CD*
LO
t:
СО
°l
CN
s
t^-
LO
CN
CO
00
CD
LO
°l>
CN*
CD
|
О
О
Ю
123
мнению, значение 0,20±0,05 ккал-моль~1, которое согласуется с за-
ключением, сделанным Бизоном и Иостом [43]. Термодинамические
свойства газообразного хлористого брома (стр. 25), по данным этих
авторов, приведены в табл. 24 *.
6. ХИМИЧЕСКИЕ СВОЙСТВА**
По этому вопросу в литературе имеются лишь очень
недостаточные сведения. Они относится, вследствие значительной
неустойчивости хлористого брома, к равновесной смеси эквимолярных
количеств брома и хлора или к системам, в которых можно
предположить образование хлористого брома.
Так, на основании работы Каняева и Шилова [48] можно
полагать, что хлористый бром в воде легко гидролизуетсяс
образованием бромноватистой и соляной кислот. Как уже упоминалось
(стр. 117), эти авторы, исследуя кинетику бром'ирования м-гш-
золсульфоната, показали, что одним из бромирующих агентов в
водном растворе является BrCl, образующийся в смеси НОВг и
НС1 по реакции
-* НОВг + Н+С1 *=» BrCl + Н20.
Основываясь на том, что образование BrCl сопровождается
исчезновением из раствора соляной кислоты, Каняев и Шилов
применили измерение электропроводности для изучения
гидролитического равновесия и определили константу гидролиза BrCl,
равную 2,94-10-* при 0°.
Все межгалоидные соединения типа Ti" Г~ имеют полярный
характер (см. табл. 4). Зто обусловливает, в частности, их способность
вступать в реакцию окисления — восстановления с соответствующим
галогенидом другого элемента R+Г", сопровождающуюся выделением
свободного галогена (ri)2 по схеме
Г^ГЙ г R+ГГ = (Г,)2 + R+rH.
Иост [23] предположил аналогичную схему реакции между
хлористым бромом и бромистым водородом
ВгС1 + НВг = Вг8 + НС1.
Предположение о полярном характере хлористого брома или
о его сравнительно легкой поляризуемости подтверждается
описанными выше особенностями реагирования BrCl с
непредельными органическими соединениями.
Изучение электростатических свойств молекул хлористого
брома (дипольного момента, поляризуемости и т. д.) и электрохи-
* Следует еще упомянуть о работах Блейра и Иоста [40], Попова и Мэньона
[52], в которых термодинамические характеристики BrCl рассчитывались
параллельно для газообразного состояния и для раствора в ССЦ.
** Химические свойства хлористого брома уже частично
рассматривались в предыдущих разделах этой главы.
124
^ических свойств * системы Вг2—С12—ВгС1, учитывая также
влияние растворителей, должно представлять несомненный интерес для
дальнейшей характеристики этого межгалогенида. Следует так-
#<е продолжить изучение характера взаимодействия хлористого
брома (системы Вг2—С12—ВгС1) с 'неорганическими и
органическими веществами.
В частности, большой интерес представляет получение и
изучение комплексных соединений хлористого брома типа двойных
галогенидов. Таковы, например: CsBrCl2, который описали Кремер
и Дункан, или соли диазония [C6H5N2] ВгС12 (т. пл. 68°) и т. п.,
которые получил Ганч [56], действуя сухим хлором на суспензию три-
бромида диазония в хлороформе. Эти комплексные галогениды
хлористого брома хорошо растворимы в воде, но разлагаются ею с
выделением хлора.
ЛИТЕРАТУРА
.A. J. В а 1 а г d, Ann. chim. phys. (2), 32, 371 (1826); Pogg. Ann., 8,
466 (1826).
2. Gmelin's Handbuch der anorgan. Chem., 7, 339—342 (1931).
3. W. В о г n e m a n n, Lieb. Ann., 189, 183 (1877).
4. V. Thomas, P. Dupuis, С. г., 143, 282 (1906).
5. L. W. Andrews, H. A. Carlton, J. Am. Chem. Soc, 29, 688
(1907).
6. J. Krutwig, Ber., 14, 304 (1881).
7. S. A n w a r - U 1 1 a h, J. Chem. Soc, 1176 (1932).
8. P. Lebeau, С. г., 143, 589 (1906).
9. В. J. Karsten, Z. anorg. Chem., 53, 365 (1907).
10. K- H. Butler, D. Mcintosh, Proc Trans. Nova Scot. Inst. Sci.,
17, Pt. 1, 23 (1927); С. А., 543 (1928).
11. H. Lux, Ber., 63, 1156 (1930).
12. В. Я- Аносов, С. А. Погодин, Основные начала
физико-химического анализа, АН СССР, М-—Л., 1947, стр. 197, 524 и др.
13. Б. Н. Меншуткин, ЖРФХО, 43, 1805 (1911).
14. Н. С. К у р н а к о в, Д. К р о т к о в, М. О к с м а н, ЖРФХО, 47,
558 (1915).
15. А. Д. Виноградова, Н. Н. Ефремов, Изв. АН СССР, серия
химическая, № 1—3, 143 (1937).
16. А. М. Тихомирова, Н. Н. Ефремов, Изв. АН СССР, серия
химическая, № 1—3, 157 (1937).
17. Я- А. Ф i а л к о в, Доопдження в галуз! комплексних сполук
галогенов алюмЫю i пол!галоген1д1в, АН УРСР, 1947, 50—54; Усп. химии,
15, 506—511 (1946).
18. Я- А. Ф и а л к о в, Ю. П. Н а з а р е н к о, Укр. хим. ж., 19, 356
(1953).
19. Н. П. Песков, ЖРФХО, 47, 928 (1915).
20. S. В а г г a t, С P. Stein, Proc. Roy. Soc, 122, 582 (1929). ~
21. E. G i 1 1 a m, R.A.Morton, Proc. Roy. Soc, 124, 615 (1929).
j* Электрохимические свойства хлористого брома или содержащих его
систем почти вовсе не изучались. Имеются лишь краткие сведения о том, что
раствор брома в жидком хлоре или раствор «хлористого брома» в бензоле не
проводит тока. Броун [49] сделал" попытку определить потенциал разложения
(между платиновыми электродами) сильно гидролизованного водного раствора
хлористого брома. Найденные им значения ^0,011 в вряд ли можно считать
потенциалом разложения BrCl. К электрохимическим исследованиям можно
отнести и работу Форбес и Фуосс [44] (см. стр. 119).
125
22. L. Т. М. G г а у, D. W. G. S t у 1 e, Ргос Roy. Soc, 126, 603 (1930^
23. W. J о s t, Z. phys. Chem., 153, 143 (1931). J%
24. Л. С. Л и л и ч, ЖОХ, 22, 730 (1952).
25. И. О с т р о м ы с л е н с к и й, Вег., 44, 268 (1911).
26. P. Job, Ann. chim., 9, ИЗ (1928).
27. А. К. Бабко, ЖОХ, 16, 33 (1946); 18, 816 (1948); Зап. 1н-ту xiM
8, 3 (1946); 9, 345 (1947); Изв. СФХА, 19, 236 (1950).
28. Н. П. Комар ь, ЖФХ, 26, 686 (1952); ЖФХ, 5, 139 (1950); ДАН СССР
72, 535 (1950); Труды Научно-исследовательского института химии ХГу
8, 37 (1951); 12, 5—76 (1954).
29. М. Simpson, Рг. Roy. Soc, 27, 118 (1878); J. W. James, J. Chem
Soc, 43, 37 (1883).
30. M. D e 1 ё p i n e, L. V i 1 1 e, С. г., 170, 1390 (1920); Bull. Soc. chim.
(4), 27, 673 (1920).
31. П. Вальден, Вег., 30, 2883 (1897).
32. N. W. Hanson, Th. С J a m e s, J. Chem. Soc, 1955, 2979 (1928).
33. R. E. Buckles, J. W. Long, J. Am. Chem. Soc, 73, 998 (1951).
34. T. W. J. Taylor, L. A. F о r s с e y, J. Chem. Soc, 2272 (1930).
35. H. П. К а н я е в, Е. А. Шилов, Труды Ивановского ХТИ, в. 3.
52 (1940).
36. N. V. S i d g w i с k, The chemical elements and their compounds, Oxford,
1950, 1148.
37. H. H. Г р и н в у д, Усп. химии, 22, 446 (1953).
38. Н. G. Vesper, G. К- R о 1 1 е f s о n, J. Am. Chem. Soc, 56, 62Ф
(1934).
39. G. В r a u e г, Е. Victor, Z. Elektrochem., 41, 508 (1935).
40. C. M. Blair, D. M. Jos t, J. Am. Chem. Soc, 55, 4489 (1933).
41. K. J e 1 1 i n e k, H. Schiitza, Z. anorg. allg. Chem.,, 227, 52 (1936).
42. H. Schutza, Z. anorg. allg. Chem., 239, 245 (1939).
43. С. М. В e e s о n, D. M. J о s t, J. Am Chem. Soc, 61, 1432 (1939).
44. G. S. Forbes, R. M. F u о s s, J. Am. Chem. Soc, 49, 142 (1927).
45. К. В. Б утков, Rec trav. chim., 67, 551 (1948).
46. К. В. Бутков, ЖФХ, 23, 232 (1949).
47. W. J о s t, Z. phys. Chem., B. 14, 413 (1931).
48. H. П. К а н я е в и E. A. HI и л о в, Труды Ивановского ХТИ, в. 3^
69 (1940).
49. J. Brown, Phyl. Mag. (5), 31, 457 (1891).
50. G. Frohlich, W. J о s t, Chem. Ber., 86, 1184 (1953).
51. И. А. Шека, Изв. сектора платины ИОНХ АН СССР, 26, 189 (1951).
52. A. I. Popov, J. J. Mann ion, J. Am. Chem. Soc, 74, 222 (1952).
53. H. С Mattraw, С F. P а с h u с k i, N. J. Hawking, J. Chem.
Phys., 22, 1117 (1954).
54. H. Evans, I. R. M u n s о n, D. D. W a g m a n, J. of Research
Nat. Bureau Standards, 55, 147 (1955).
55. Э. В. Брицке, А. Ф. Капустинский, Термические константы-
неорганических веществ, АН СССР, М.—Л., 1949.
56. А. Н a n t z s с h, Ber., 28, 2754 (1895).
ГЛАВА VI
ХЛОРИДЫ ИОДА
1. ХЛОРИСТЫЙ ИОД JC1
Хлористый иод был открыт в 1814 г. Гей-Люссаком [1]* и,
независимо от него, в том же году, Деви [2]. Хлористый иод, а также
треххлористый и бромистый иод, открытые раньше других
межгалоидных соединений, довольно подробно описаны в справочной и
монографической литературе [3, 4, 5 и др.], в которой
систематизированы литературные данные примерно до середины 30-х годов,
текущего столетия.
Физико-химический анализ системы иод — хлор
Эта система впервые была изучена Штортенбекером [6]
методом термического анализа. На рис. 24 изображена диаграмма
плавкости системы иод — хлор [6], показывающая, что в данной
системе образуются две модификации хлористого иода, стабильная
а- и нестабильная |3->модификация, а также треххлористый иод
JC13.
Другие соединения хлора с иодом, кроме хлористого иода и
треххлористого иода, не были обнаружены ни Штортенбекером, ни
другими авторами.
Анализируя полученные им данные, Штортенбекер обоснованно
заключил, что хлористый иод существует также в жидкой фазе,
причем в жидком состоянии при температуре плавления он
диссоциирует очень мало, в отличие от треххлористого иода.
Определение состава газовой фазы при 30 и 80° подтвердило термическую
устойчивость хлористого иода в парах; так, при 30° найдено 51,1,
а при 80° — 52,1 ат.% хлора.
Карстен [7] исследовал температуры кипения смесей иода и
хлора (большей частью при давлении 740 мм рт. ст.) и определил
содержание хлора в жидкой и газообразной фазах. Эти данные,
приведенные на рис. 25, показали, что хлористый иод устойчив
даже при 100° в обеих фазах. В то же время пары треххлористого
иода полностью диссоциированы на хлористый иод и хлор.
* Одновременно им был открыт треххлористый иод.
127
Систему иод—хлор в растворе четыреххлористого углерода
впервые исследовал Лилич [8] методом физико-химического ана~
лиза— путем измерения оптической плотности (см. стр. 114).
Диаграмма состав — свойство, изображенная на рис. 24, показывает
образование хлористого
иода при взаимодействии иода
и хлора также в среде
четыреххлористого углерода.
та
Рис. 24. Диаграмма плавкости
системы иод—хлор.
Рис. 25. Температуры кипения и состав
жидкой и газообразной фаз системы
иод—хлор.
Получение
Хлористый иод образуется в результате разнообразных
окислительно-восстановительных реакций [3].
1. Непосредственным взаимодействием галогенов—действием
сухого хлора на твердый или газообразный иод: J2 + C12 = 2JC1;
избыток хлора приводит к образованию треххлористого иода. Таким
же путем можно получить хлористый иод в неводных растворах.
2. Действием хлора на растворы иодидов: KJ + C12 = KC1+JC1;
в ©одной среде хлористый иод гидролизу ется (см. стр. 144).
3. Действием окислителей в присутствии ионов С1~на растворы
иодидов или на иод, например
5KJ + 2КМп04 + 16НС1 = 5JC1 A- 7KCI + 2МпС12 + 8Н20.
4. Взаимодействием хлоридов с кислородными соединениями
иода в кислой среде
KJ08+2KJ-f-6HCl-3JCl+3KCl+3H20;
KJ03+2J2+-6HC1=5JC1+KC1+3H2Q.
128
5. Окислением иода соединениями хлора:
а) нагреванием гипохлорита кальция со спиртовым раствором иода;
б) перегонкой смеси иода с бертолетовой солью по реакции
2КС103 + J2 = 2KJ03 + Cl2; J2 + С12 = 2JC1;
в) по реакции иода с хлоридами некоторых элементов—РС15, SbCl5,
РЬСЦ, HgC]2, проявляющими в данном случае свойства окислителей:
PCl5+J2 = PCl3+2JCl*;
SbCl6+J.2 = SbCl3+2JCl*;
PbCl4+J2«PbCl2+2JCl [71]**;
S02C12T-J2 = 2JC1+S02 [72]***,
HgCl2+2J2 = HgJ2+2JCl (в спиртовом растворе).
6. Реакцией между хлорамином [156] или дихлорамином [157] и
иодидами в уксуснокислом растворе
> NCI + HJ = > NH + JC1****
Возможно, что в этой реакции сперва образуются иод и хлор,
которые затем соединяются друг с другом [156];
> NCl+2HJ-NH+HCl+Ja;
>NC1+HCU>NH+C12;
J2 + C12 = 2JC1.
7. При гидролизе треххлористого иода (стр. 163) или
восстановлении его иодом: JCl3-fJ2=3JCl.
Некоторые из этих и другие аналогичные реакции можно
использовать для получения хлористого иода в неводных растворах
или в водной солянокислой среде. Кристаллический же препарат
получают почти исключительно путем непосредственного
взаимодействия хлора с иодом.
Для этой цели обезвоженный хлор пропускают над твердым
иодом до полного разжижения реакционной массы, которую затем
перегоняют и собирают фракцию при 100—120°. При этом, однако,
нельзя избежать образования некоторого количества
треххлористого иода. Выход хлористого иода составляет лишь 35% от ко-
* Хлористый иод, образующийся в этих реакциях, может соединяться
с избытком РС15 или SbCI5; при этом получаются двойные галогениды PC15-JCI
191 или [PC14][JC12] [10] и SbCl5-/iJCl [11] или JSbCl6.mJCl [12].
** Ф. Я- Кульба измерил теплоту экзотермической реакции
взаимодействия РЬСЦ с иодом, растворенным в ССЦ- Тепловой эффект оказался
равным 19941±64 кал [71].
*** Эта реакция происходит при участии катализаторов А1С13 или AU3
{см. стр. 159).
**** Бродфильд с сотрудниками [156] полагают, что при взаимодействии
хлорамина с бромистым водородом в уксусной кислоте образуется хлористый
бром по такой же схеме реакции.
9—1419
129
личества иода. Для повышения выхода (до 85%) Корног и Кард
жес [13] перегоняли хлористый иод в токе хлора. Так как в эт^
условиях возможно образование также и треххлористого иода, то
полученный после перегонки препарат анализируют и прибав.
ляют к нему такое количество иода, которое необходимо для пре.
вращения избыточного хлора (т. е. треххлористого иода) в
хлористый иод.
Корног и Карджес предложили также метод получения хлори-
стого иода путем взаимодействия иода с жидким хлором.
Взвешенный сосуд Дьюара, охлаждаемый смесью твердой углекислоты
с эфиром, заполняют немного более половины объема жидким
хлором, прибавляют иод в количестве, составляющем приблизительно
0,5 моля на 1 моль хлора, после чего содержимое сосуда
замораживают. Затем сосуд вынимают из охладительной смеси и
оставляют на некоторое время при комнатной температуре для
испарения не вошедшего в реакцию хлора. После этого сосуд,
содержащий смесь хлористого иода и треххлористого иода,
взвешивают. Зная вес сосуда и вес прибавленного иода, по разности
определяют количество хлора и прибавляют рассчитанное количество
иода, необходимое для получения продукта, состав которого точно
соответствует формуле JC1. Затем сосуд закупоривают и его
содержимое оставляют на 24 часа в жидком состоянии. Полученный
хлористый иод очищают перекристаллизацией (1—2 раза). Для
этого его расплавляют и постепенно охлаждают, пока не
затвердеет около 80%, после чего сливают жидкую часть.
Гутман [60], получая хлористый иод по методу Корнога и Кард-
жеса, внес в него некоторые изменения: операции проводились в
токе азота, отделение от треххлористого иода и очистка
хлористого иода производилась путем многократной
перекристаллизации расплавленного препарата.
Фиалков и Шор [14] получали хлористый иод в колбе с
асбестовой пробкой, сквозь которую были пропущены две трубки: для
ввода хлора (с большим расширением в виде воронки у дна
сосуда) и для вывода избытка его. В колбах помещают иод,
очищенный повторной возгонкой с KJ и ВаО и затем высушенный в
течение нескольких суток над твердым едким кали; пропускают струю
хлора, предварительно промытого водой и обезвоженного
концентрированной серной кислотой и хлористым кальцием. Образование
JC1 сопровождается небольшим экзотермическим эффектом и
разжижением реакционной массы.
Хлор пропускают до достижения эквимолярного соотношения
иода и хлора, что определяют путем периодического взвешивания
колбы с содержимым и химическим анализом пробы полученного
продукта. Если молярное отношение J2 : С12 меньше единицы, то
добавляют рассчитанное количество иода, после чего колбу слегка
подогревают на водяной бане при 45—50°. При охлаждении ^вся
масса застывает при одной температуре. Полученный таким путем
препарат представляет собой чистый a-JCl.
Чистота полученного хлористого иода устанавливается по тем-
130
ературе плавления (или по температуре замерзания), удельной
пл£ктропроводности и результатами химического анализа — коли-
9еСТВенным определением иода и хлора.
Физические свойства
Хлористый иод — единственное межгалоидное соединение, для
которого обнаружено явление полиморфизма *'. В
кристаллическом состоянии он существует в двух ^онотропных формах:
устойчивой а-форме, кристаллизующейся в виде рубиново-красных игл
/т. пл. 27,2°) и неустойчивой р-форме, образующей
красно-коричневые пластинки (т. пл. 13,9°). Обе модификации плавятся в
красно-коричневые жидкости, вполне идентичные по своим свойствам
и кипящие (с разложением) при 100—102°. Пары хлористого иода
имеют цвет брома.
Свежеперегнанный хлористый иод, особенно если он содержит
свободный иод, остается жидким при комнатной температуре в
течение длительного времени; это же явление наблюдается при
охлаждении жидкого хлористого иода в запаянных трубках до 0°.
Прибавление треххлористого иода или кристалликов хлористого
иода способствует кристаллизации препарата в а-форму [3, стр. 607,
609]. Неустойчивый (3-JC1 образуется при быстром охлаждении
жидкого препарата до — 5° (причем при кристаллизации
происходит повышение температуры до 13—14°) или при выдерживании
его между +5 и —10°. По Штортенбекеру [6], (З-JCl может
существовать лишь несколько дней в температурном интервале от
0 до —10° и затем самопроизвольно превращается в a-JCl,
превращение протекает значительно быстрее при внесении кристалла
a-формы. Обратный переход a-JCl в P-JC1 весьма затруднен.
Условия устойчивости p-JCl и его перехода в а-модификацию
подробно изучил Танатар [17]. Он установил, что наличие следов
треххлористого иода препятствует образованию p-JCl. Поэтому
если хлористый иод подвергнуть длительному кипячению в сосуде с
оттянутым горлом для полного удаления JC13 с парами хлористого
иода и затем тотчас же запаять сосуд и охладить его до
комнатной температуры или до —10°, то образуется р-модификация JC1,
которая в этих условиях довольно устойчива (сохраняется в
течение года). Но при охлаждении до —15° или —20° Она переходит:
в a-JCl. Если же a-JCl расплавить при 40° и охладить только до
— 10°, то снова образуются кристаллы р-формы.
Теплота перехода p-JCl в a-JCl в твердом состоянии
составляет 203 кал)моль, по Штортенбекеру [18] и 273 кал/моль, по Та-
* Очевидно, Хра/Пгп [15], получивший хлористый иод не только в
кристаллическом состоянии, но и в виде красно-коричневой жидкости, первый имел
дело с двумя модификациями JC1. Этот факт в свое время подчеркнул
Менделеев в «Основах химии» [16] Ссылаясь на работу Штортенбекара, Менделеев
писал, что современные проверенные сведения о хлористом иоде вполне
подтверждают многие наблюдения Траппа, оправдывая его указания о
существовании двух модификаций этого вещества.
131
Таблица 25
Некоторые физические константы хлористого иода
Свойство
Агрегатное
состояние
или
модификация
Значение констант
Температура плавления, °С
» »
Температура кипения, °С
Плотность dj
Плотность d4
Молекулярный объем
Теплота плавления
Молярная теплоемкость Ср
Молярная теплоемкость Cv
Молярная теплота
испарения
То же
Константа Трутона
Теплота возгонки
Криоскопическая
константа (вычисленная из
теплоты плавления)
Энтропия 5° 98 16
» Sc '
со
298,16
298,16
Вязкость (-rj)
Магнитная
восприимчивость *
a-JCl
P-JC1
Жидкость
a-JCl
P-JC1
[ЖИДКОСТЬ Х)
Жидкость
a-JCl
p-Jci
Жидкость
a-JCl
p-JCl
a-JCl
p-JCl
Жидкость
Газ
Жидкость
a-JCl
a-JCl
NO
a-JCl
Жидкость
Газ
Жидкость
Жидкость
27,2
13,9
100±3(94,7; 97',4°)
3,86(0°)
3,66 (0°)
3,18223(1+9,1590-10-4+
+8,3296-10 ~7t2+
+2,7500-Ю-9/3)-1
3,10
42,1 (0°)
44,3 (0°)
52,3 (29*)
—г^бв22);
—2,319^ ккал-моль"1
-2,2672а);
—2,32226) ккал-моль"1
13,44 кал-град~х -моль~*1 >
(от —13,5 до +15°)
^13,76 кал-град~х -моль"1
(от —10 до 0°)
25,58 кал*град~~1 -моль"1
(от 15 до 77°)
0,0512 кал-град
(от 20 до 100°)
0,0389 кал-град"1 -моль~*
1,3173); 1,303)
10,1 — 10,5 ккая-моль"1
9,95; 9,49 ккал-моль~1
27,0-28,0
11,8 ккал-моль~{
109,8 \ из расчета на 100 г
117,6 J хлористого иода
24,6 кал-град~~1 -моль~1
33,46 кал-град~]-моль~~1
59,15 кал-град~]-моль~1
(1 атм)
5,82 (15°); 4,18 (28,4°)
сантипуаз
-49,4-Ю-6 CGSM (12°)
-56,8 10~6 CGSM
1 моль 1
132
Продолжение табл. 25
Свойство
Агрегатное
состояние
или
модификация
Значение
Ссылка
на
литературу
диэлектрическая
постоянная
Электропроводност ь4)
Дипольный момент5)
Межъядерное расстояние
Твердое
Жидкость
»
Газ
В интервале от —48° до
— 11° изменяется от 7,0 до
13,7
(4,58±0,01)-10~3 ом~* -см~]
(35°)
4,52.Ю-3ом-' - см-ЧЖ)
4,59-10-3 ом~1 • см~1 (34,45°)
0,65D (0,5D)
2,32070 А6)
2.321 А?)
2.322 А8)
[351
[13]
[14J
[36]
[26, 27]
[26, 28J
[29, 30]
[32]
Пояснения: 1) Плотность жидкого хлористого иода измеряли также
Танатар [17] и Генней [31]. По Танатару, <Л6° = 3,2856; №' = 3,2402. Генней
для d16° определил значение такое же, как Танатар, но cfi® 9 по его данным, равна
3,180; d45°=3,132, d98°=2,958.
2а) По Штортенбекеру [18]; 26) По Танатару [17].
3) Величина Ср/Cv, равная 1,317, установлена Штрекером [22] путем
определения скорости звука в газообразном хлористом иоде. Вильсон [23] из
спектроскопических данных получил значение 1,30.
4) О зависимости электропроводности хлористого иода от температуры см.
стр. 203.
5) Величина дипольного момента 0,65D была определена микроволновым
методом [26]. Дипольный момент 0,5Д найденный Люфтом [27] из
температурной зависимости диэлектрической постоянной паров хлористого иода, можно
считать менее точным [19]. О дипольном моменте хлористого иода в растворах
см. табл. 4 и стр. 141.'
6) По данным микроволновой спектроскопии.
7) Из полосатых спектров.
8) Сумма ковалентных радиусов иода и хлора.
натару [17]. Превращение лабильной формы хлористого иода в
стабильную сопровождается явлением контракции [20], что видно
из сравнения величин молярных объемов (табл. 25).
О природе полиморфизма хлористого иода нет определенных
представлений.
Структуру а-модификации хлористого иода недавно
исследовали Босвейк с сотрудниками рентгенографически. Были
определены параметры решетки, координаты атомов и межатомные
расстояния. Структура хлористого иода состоит из двух видов
кристаллографически неэквивалентных молекул [178].
В табл. 25, заимствованной из обзорной статьи Гринвуда [19,
стр. 460] и дополненной нами, приведены некоторые физические
константы хлористого иода.
133
Упругость пара
Еще Трапп [15] установил, что хлористый иод летуч как в жид,
ком, так и в твердом состоянии и что он даже при низких
температурах обладает заметной упругостью пара. Корног и Бауэр [34]
измерили динамическим методом упругость пара твердого a-JQ и
Башли, что в интервале от —15° до температуры плавления она
возрастает от 1,2 до 33,2 мм рт. ст. На основании этих данных
зависимость упругости пара от абсолютной температуры может быть
выражена таким уравнением [19]:
lgP=10,108—2578/Г.
Упругость пара жидкого хлористого иода, по Корногу и Кард-
жесу [13], в интервале от 35 до 70° изменяется от 48,0 до 261,7 мм
рт. ст. *.
В дальнейшем Корног и Бауэр [34] измерили упругость пара
хлористого иода в интервале от 10 до 50°. Полученные ими
значения (от 12 до 107,1 мм рт. ст.) приводят к следующему уравнению
[19]:
lgP = 8,761 — 2174/Г**.
Еще ранее Мак-Моррис и Иост [24] для температурного
интервала 0—70° приняли, что
lg P = 8,5038 — 2079,7/Г.
Это уравнение дает несколько более высокие значения упругости
пара (на 1—10 мм), чем уравнения Корнога и Бауэра, что
уменьшает теплоту испарения до 9,52 ккал-моль~х, а константу Трутона
до 25,5 ккал-гра&-х-моль-х ***.
Термическая диссоциация
При нагревании хлористый иод может диссоциировать на иод
и треххлористый иод (3JC1 ^J2+JC13) или на-иод и хлор (2JC1^
^ J2 + C12). Как упоминалось выше, по данным Штортенбекера [6]
и Карстена [7], хлористый иод очень мало диссоциирован в
интервале температур до 100° как в жидкой, так и в газообразной
фазе.
В некотором противоречии с этими данными находится вывод,
к которому пришли Корног и Карджес [13] при изучении упругости
паров: они заключили, что скорость разложения хлористого иода
при температурах выше 70° становилась столь большой, что не-
* Молярная теплота испарения хлористого иода и константа Трутона,
вычисленные Корногом и Карджесом, приведены в табл. 25.
** Из этих данных следует, что теплота испарения хлористого иода равна
9,95 ккал.моль-\ теплота плавления 1,85 ккал.моль—1,а константа Трутона
26,7 ккал.град—^-моль—1. v
*** Гринвуд [19], отмечая, что в статье Мак-Морриса и Иоста отсутствуют
экспериментальные данные, подтверждающие уравнение зависимости
упругости пара от температуры, справедливо полагает, что следует отдать
предпочтение значениям Корнога и Бауэра.
134
о3можно было получить постоянных результатов. Диссоциация
3 пористого иода при нагревании до кипения (при атмосферном
давлении) являлась, по мнению Корнога и Карджеса, причиной
того, что температуру кипения хлористого иода нельзя было точно
определить (см. табл. 25).
Фиалков и Шор [14] исследовали поведение хлористого иода
при нагревании методом термографии. При исследовании
применялся саморегистрирующий
прибор системы Н. С. Курнакова с
одновременной записью
дифференциальной и простой
термопарой. По кривой нагревания (рис.
26), записанной
дифференциальной термопарой, видно, что при %
65—70° имеет место
эндотермический эффект, указывающий на
процесс термической
диссоциации хлористого иода в этой
температурной области. Этот
факт находится в согласии с
данными Корнога и Карджеса,
полученными при измерении
упругости паров.
Подтверждением этого вывода могут
служить также результаты измерений электропроводности
хлористого иода: на кривой температурной зависимости
электропроводности (рис. 29 и 30) виден пологий максимум в области 45—
70° (по данным Фиалкова и Шор [14]) или при 40—45° (по данным
Гринвуда и Эмелеуса [36]).
Термическая диссоциация хлористого иода обратима: если
нагреть хлористый иод до температуры выше чем 65—70°
(например, до 97°) в сосуде для измерения электропроводности,
запаянном во избежание улетучивания хлора и иода, а затем охладить
до определенной температуры (например, до 35°), то получаются
значения электропроводности, соответствующие этой температу-
ре [14].
Количественная характеристика термической диссоциации
хлористого иода имеется в работе Мак-Морриса и Поста [24]. Эти
авторы в работе, посвященной вычислению свободной энергии и
энтропии хлористого иода на основе определения констант
равновесия J2+CI2 ^ 2JC1 в газовой фазе, указывают в выводах, что
степень диссоциации газообразного хлористого иода на газообразные
иод и хлор равна 0,42% при 25° и 1,58% при 100° *.
Рис. 26.
Кривая нагревания
хлористого иода.
* Следует заметить, что эти величины приведены лишь в выводах к статье
Мак-Морриса и Иосга. Гринвуд [19, стр. 462], пользуясь приведенными в
статье этих авторов термодинамическими функциями, рассчитал степень дис-
.социации газообразного хлористого иода при 25 и 100°, соответственно равной
0,38 и 1,1%.
135
Блейр и Иост [37], вычисляя некоторые термодинамические кон^
станты JCl, JBr и BrCl в растворе СС14, определили, что степень,
диссоциации хлористого иода в ССЦ при 25° равна 0,25%.
Теплота образования
Для определения теплоты образования хлористого иода были:
использованы различные методы.
Некоторые из полученных значений приведены в табл. 26.
Таблица 26
Теплота образования хлористого иода 1/2J2+1/2Cl2 = JCI
Исходное состояние
VA (г), V2CI2 (г)
VA (г). V2CI2 (г)
VA (тв), V«C1« (г)
1/2J2 (г), V2CI2 (г)
VA («), V2CI2 (г)
VA (тв), V2C12 (г)
VA(bCC14), гШ* (г)
Конечное
состояние
JC1 (г)
JC1 (г)
JCla (тв)
1 JC1 (тв)
JCla (тв)
JC1 (ж)
1 JC1 (в СС14)
Теплота
образования,
—1
икал* моль
3,311)
3,462)
6,9; 6,6)
12,1 /
8,043)
5,825
3,197
Ссылка на
литературу
[19]
[24,174]*
[38а]
[19]
[386]
[71]
Пояснения: 1) Это значение вычислено [19] из теплоты диссоциации
газообразного хлористого иода на нормальные атомы, по спектроскопическим данным
Вильсона [23].
-J+C1; А #298.16 " 49,65 ккал-моль~~1 **.
JCb
2) Теплота образования вычислена [24] по термохимическим данным для
реакций
2NOC1 + Jo = 2NO + 2JC1;
2NO + С12 = 2NOC1.
3) Для вычисления теплоты образования» JC1 использованы значения
теплоты возгонки иода (14,13 ккал.моль—1) [34] и хлористого иода (табл. 26).
Термодинамические свойства
Свободная энергия, теплосодержание и энтропия хлористого
иода вычислены из спектроскопических и термохимических данных,
причем были получены удовлетворительно совпадающие значения.
Эти значения приведены в табл. 27, заимствованной из работы
Мак-Морриса и Иоста [24], впервые вычисливших
термодинамические константы хлористого иода. Более полная
термодинамическая характеристика JC1 (г), по данным Эванса с сотрудниками
[175], приведена в табл. 28.
* Брицке и Капустинский [174] считают это значение для JCl(r) наиболее
точным. Для кристаллического JCla они приводят значения Д#°:—8,35;
—8,46 и —8,69 ккал-моль-\
** По спектроскопическим данным Кордеса [39], эта величина равна
47,13 ккал-моль-1. Полученное Дербишером [40] значение 65,68 ккал • моль-1
относится к диссоциации преимущественно на возбужденные атомы.
136
Таблица 27
Термодинамическая характеристика системы 1/2^2(г)+1/2С12(г. = JGM*) при
298,16° К
Термодинамическая константа .
Свободная энергия (термохимич.)
» » (спектроскопич.)
Теплосодержание (термохимич.)
» (спектроскопич.)
Энтропия (термохимич.)
» (спектроскопич.)
Значение константы
—3653 кал-моль~~1
—3879 кал-моль"
—3125 кал-моль
— 3461 кал-моль
59,6 кал-град -моль~~]
59,2 кал-град~~ • моль"
Изменение свободной энергии при реакции *Ы2 (Г> +(1/2С1г (Г) =
=JC1 (г) Мак-Моррис и Иост вычислили по следующим
формулам:
AFf°= —3125— 1,77/Т (из термохимических измерений),
AFf°= — 3441 — 1,40/Т (из спектроскопических измерений),
которые дают, например, для температуры 298,16° К значения
соответственно 3,65 и 3,88 ккал-моль~х.
Гринвуд [19], ссылаясь на работу Блейра и Иоста [37],
указывает, что значения термодинамических функций, имеющиеся в
статье Мак-Морриса и Иоста, содержат некоторые ошибки. Он при-
водит следующие величины, полученные при пересчете данных
последних авторов:
а) Абсолютная энтропия хлористого иода при 25° и при
давлении 1 ат 5^8Л6 = 59,15 кал-град~имоль~19 откуда изменение
энтропии ,в приведенной выше реакции (с газообразными компонентами)
равно:
А^298,16
59,15 — V2(62,29+ 53,31) = 1,35 кал-град'1-моль
—1
Из термохимических данных получено значение 1,77 кал-град~1Х
X моль~К
б) Изменение свободной энергии при этой же реакции для
298,16е К равно 3,71 ккал-град~кмоль~1. Эта величина очень
близка к тем значениям, которые вычислены по формулам,
предложенным Мак-Моррисом и Иостом.
Блейр и Иост [37] отмечают, что значения энтропии для
твердого и жидкого хлористого иода, данные Мак-Моррисом и Иостом,
ошибочны. Они пересчитали эти значения и получили следующие
результаты:
Энтропия твердого a-JC1 при 25° равна 24,6 кал-град"1-моль—1.
Энтропия жидкого JC1 при 25° равна 33,4 кал-град'1-моль*-1.
Изменение энтропии в реакции V2J2 (тв) + V2C12 (г) = JC1 (тв) равно 16,0
* В работе Мак-Морриса и Иоста приведены значения этих же
термодинамических констант для газообразного, жидкого и твердого хлористого
иода, полученного из газообразного хлора и твердого иода.
137
8С1
ел
о
о
|
сп
^
"~4
CD
CO
С»
сп
00
о
-<1
СО
V
-4
со
^^
со
о
ю
!0,0
JD
"о
СО
/
■
1
СлЭ
СО
ел
1
1
Сл
4^
оо
4*.
о
о
1
СП
v.4^
►—»
CO
СЛ
00
СП
сл
to
^1
1°
Ъо
4^
-4
, -
to
Ъо
CD
ъ
С5
1
чР0
Ьо
сл
1
1
сл
"to
-4
Ъо
to
со
со
о
о
1
СП
jjo
"сл
сл
сл
со
СП
to
-^4
i°
-<1
СП
ь-Ь
>—*
to
о
Ъэ
i°
"о
сл
1
5°
Ъо
сл
1
1
4^
CD
Ъо
СП
со
to
о
о
1
СП
J°
"ОО
СП
СП
оо
сл
ОО
СП
-4
4^
сл
to
^
о
СО
о
со
"to
i°
ъ>
4=ь
1
1
со
со
сл
1
1
"о
о
CD
"СО
о
о
о
1
СП
J°
>—'
to
00
сл
4*-
сп
-1
о
СП
СП
-4
CD
4*
о
CD
Ъп
СО
"о
to
1
1
со
со
сл
1
1
Ъо
СП
о
"со
СП
СП
о
о
о
1
СП
Ъо
о
СО
оо
4^
со
СО
СП
СО
8
00
0О
4^
CD
CD
ъ
CD
"о
о
1
Ъо
4S*
1
1
*4^
Vj
to
"о
со
to
СО
о
о
1
СП
о
~4s*
СП
00
4^
4^
СЛ
СП
00
00
СП
<1
СП
о
"сл
00
"со
00
1
1
со
со
4^
1
I
4^
СЛ
00
со
00
о
о
1
СЛ
СО
4^
to
СЛ
00
со
00
о
СП
^4
00
о
сл
СП
^4
о
4*>
"о
s°
"cD
сл
1
со
со
4*>
1
4^
V
СЛ
"to
СЛ
-^
О
о
1
СЛ
S°
"со
ОО
СО
о
ст>
СП
СП
to
СЛ
00
CD
Vi
2°
~ID
ю
1
jjo
ъ^
4^
1
4^
Ъо
"со
-и.
сл
СП
о
о
~
Сл
J*4
о
со
СО
00
to
о
СП
сл
*°
4*.
О
4^
СО
to
JD
$°
Ъо
~<1
1
со
Ъо
4*-
1
^1
Ъл
СД
о
о
1
сл
Vх
"сл
сл
сл
ОС
о
-4
00
СП
со
СП
to
00
£ь
о
со
СП
"сл
Ос
Ъс
1
со
со
сл
1
"о
со
Vl
СП
со
4^
о
о
i
сл
со
Vi
-4
со
J-4
Id
CD
to
СП
СП
^4
сл
со
СП
JD
"оо
V.00
Vi
о
1
со
CO
СЛ
1
со
"со
о
i°
to
со
й
о
о
1
сл
^
сл
со
4^
^4
СП
СП
СЛ
СЛ
CD
CD
CD
to
to
со
jO
00
сл
о
1
со
со
сл
to
1
CO
V]
СП
о
1°
со
СО
00
to
СО
ро
СП
1
сл
^
4^
00
СП
-<1
СП
с^>
о
41
СО
и^
4^
СП
к
to
00
со
"со
s°
сл
о
1
1
со
со
сл
to
1
со
"-4
сл
^4
to
сл
со
СО
to
-4
JjO
СП
1
сл
^о
Ъо
СО
--4
СЛ
ОО
СП
сл
до
V
о
сл
$2
о
-4
"to
оо
V
to
1
со
со
сл
1
^со
"-4
to
со
to
^о
-4
СО
о
to
сл
о
1
ф-
0О
*д
СО
ОО
^4
сл
оо
сл
СП
»—'
»—'
•—»
ОО
-^
"to
J30
\о
со
1
со
со
СЛ
1
"со
о
JO
Ъо
00
сл
о
о
о
о
о
о
1
со
СО
СО
1
Ъо
со
1
-^
Q
^
х ^ Ъ \
СУ С1)00
1 l^r
X "
Hi
1 Cbi О
X |
1| <2
о* О >.
-1
X
^
g 4
\к
"Л
^ х
1$
1
л^
^ >
о *^?
о»
1
<КГ
^ал-град"х-моль~х, а отсюда изменение свободной энергии А/7!#816 =
=: 3,25 ± 0,1 ккал-моль-{.
Эти же авторы [37] рассчитали термодинамические константы для
растворов JC1 в СС14.
Молекулярное состояние и молекулярная структура
Литературные данные, относящиеся к этим вопросам, в ряде
случаев»содержат противоречивые выводы.
По данным Корнога и Карджеса [131, константа Трутона для
хлористого иода равна 27—28 кал-град~кмоль~1. Эти же авторы
рассчитали изменение энтропии (AHIT) при испарении хлористого
иода (при температуре, когда концентрация равна 0,00507 мольЫ)
и получили значение 33,4, очень близкое к величинам, которые
получены при этом условии для ассоциированных
жидкостей—аммиака (32,4), воды (32,0), этилового спирта (33,4) и т. п. Все это
дало основание Корногу и Карджесу характеризовать хлористый
иод как ассоциированную или полярную жидкость. Вместе с тем
в литературе имеются суждения противоположного характера,
основанные на том, что определение молекулярного веса хлористого
иода в различных растворителях—ледяной уксусной кислоте [18],
хлорокиси фосфора [41, 42], пятихлористой сурьме [43], бромофор-
ме [42], нитробензоле [44] — дало значения, соответствующие
мономерным формам. Однако, исходя из этих экспериментальных
данных, нельзя отрицать наличие ассоциированных форм хлористого
иода по следующим причинам:
а) В растворах возможен распад ассоциированных молекул до
мономерных форм. Так, например, для хлористого и бромистого
алюминия установлено, что как в парообразном, так и в жидком
и кристаллическом состояниях они образуют удвоенные молекулы
[45—47]. В то же время для растворов хлористого алюминия в
эфире [48] и пиридине [49] найден -молекулярный вес, близкий к
теоретическому для молекул А1С13. Доказано, что в разведенных
бензольных растворах бромистого алюминия наряду с удвоенными
молекулами А12Вгб существуют и молекулы А1Вг3 [50].
б) При определении молекулярного веса хлористого иода
необходимо учитывать способность последнего образовывать с рядом
растворителей проводящие ток системы, вследствие чего
получаются величины молекулярного веса, средние для ассоциированных
и мономерных форм и продуктов их электролитической
диссоциации. Таковы, например, системы хлористого иода с перечисленными
выше растворителями: СН3ОООН [51], SbCl5 [12], C6H5N02 [44,
52], причем элеетропроводность таких систем обусловлена не только
собственной диссоциацией хлористого иода (J2Cl2=J+JCl7 и т. д.),
но также диешциа'цией образующихся в них комплексов —
продуктов соединения хлористого иода с растворителем *.
в) В некоторых растворителях наблюдается повышение
молекулярного веса хлтэристого иода с ростом концентрации. Так, на-
* Этот вопрос освещен в главах IX и XI.
139
пример, в бромоформе в интервале Cjci от 0,18 до 0,98%
молекулярный вес увеличивается от 166 до 182 (Л4теор = 162,4) [42]. В
то же время в нитробензольных растворах, в пределах
концентраций от 2 до 6 мол.%, по данным Фиалкова и Каганской [52], не
обнаружена зависимость молекулярного веса (182—196) от
концентрации. Возможно, это объясняется тем, что некоторый рост
степени ассоциации хлористого иода с концентрацией
компенсируется одновременным увеличением электропроводности,
(^условленной электролитической диссоциацией ассоциированных молекул
(J2C12) или продуктов их соединения с растворителем.
Структура и полярность молекул
Гримм и Зоммерфельд [53] вычислили теплоту образования Q'
хлористого иода из расчета «а ионный характер связи J+C1~ и
сравнили с теплотой образования Q, рассчитанной по
экспериментальным данным. Для вычисления Q' эти авторы приняли, что
энергия решетки «полярной соли» J+C1~ приблизительно равна
таковой для Cs+ Cl~, так как иод и цезий являются соседями
одного и того же инертного газа Хе в периодической системе и в этих
соединениях оба они имеют заряд +1. В зависимости от
применяемых значений исходных величин (весьма различающихся у
разных авторов), Гримм и Зоммерфельд нашли для Q' величину
33 ккал*моль~1 либо 6 ккал-моль~\ Из теплоты плавления и
теплоты испарения хлористого иода они рассчитали теплоту образования
Qjci из атомов 59,3—60,8 ккал-моль~1—величины, значительно
превышающие теоретические. Из этого Гримм и Зоммерфельд
сделали вывод, что полярная система J+C1~ должна перейти в
неполярное соединение JC1 с выделением Q—Q' —30 или 54 ккал.
Затем эти авторы рассчитали теплоту диссоциации газообразного JC1
на атомы и показали, что найденная величина 49 ккал-моль~1
находится между величинами теплот диссоциации J2 и CU на атомы,
соответственно равными 36 и 54 ккал-моль~\ На основании этих
данных Гримм и Зоммерфельд пришли к заключению о
неполярной структуре молекул хлористого иода, обусловленной
ковалентной связью между атомами иода и хлора.
Такой же характер связи в хлористом иоде (а также в JBr и BrCl)
принимают Паулинг и Иост [54], основываясь на постулате об
аддитивности энергий нормальных ковалентных связей, согласно которому
энергия нормальной ковалентной связи между атомами А и В равна
среднему арифметическому энергий связи А—А и В —В, т. е. DA-b=
=— (Да—а 4 £>в-^в). Незначительное отклонение энергии связи от
аддитивной величины, вычисляемое по уравнению А = Д\_в ~-(Da—b+
+ DB-b), характеризует данную связь А—В как ковалентную. Веди-
чины для четырех межгалоидных соединений [32, стр. 57] такоЕы:
J-Cl J—Br Br-Cl CI —F
A 4 1,7 0,7 25,7 ккал-моль-1.
НО
По Паулингу, малая величина Д для первых трех
межгалогенидов объясняется небольшим различием электроотрицательностей
хлора, брома и иода *. По этой же причине молекула BrCl
больше ДРУГИХ межгалогенидов «приближается к нормальному кова-
лентному типу», особенно по сравнению с фтористым хлором.
Свойства C1F указывают на то, что тип связи в этом соединении
незначительно отличается от ковалентной.
В гла©е I (стр. 11 и др.) были приведены и другие
литературные данные, в которых отмечается сходство молекулярных
структур межгалоидных соединений типа АВ (JCl, JBr и т. п.) и
галогенов.
В то же время ряд экспериментальных данных показывает, что
многие свойства хлористого (и бромистого) иода сближают их с
полярными солеобразными соединениями. Здесь необходимо
принять во внимание довольно значительную электропроводность JC1
(табл. 25) и JBr [56, 57] в индивидуальном состоянии, результаты,
лолученные при электролизе этих галогенидов иода как в
расплавленном состоянии, так и в растворах—выделение иода на
катоде, а хлора или брома — на аноде, способность хлористого и
бромистого иода вступать в реакцию двойного обмена с галогенидами
и цианидами металлов (см. стр. 145).
Электронная конфигурация хлористого иода, равно как и
других межгалогенидов этого типа, аналогична таковой для молекул
галогенидов :J — С1:, но в данном случае благодаря различию
иода и хлора в сродстве к электрону и в поляризационных
свойствах, связь J—О является полярной с распределением
электронной плотности, соответствующим схеме J+—С1~.
Дипольный момент хлористого иода сравнительно небольшой —
0,65 D [26]. Однако Фэрбразер [58] нашел, что дипольный момент
хлористого иода в циклогексане равен 1,47 D и в четыреххлори-
стом углероде 1,49 D. Повышение дипольного момента хлористого
иода в этих неполярных растворителях, по сравнению с его
значением в газообразном состоянии, Фэрбразер объясняет
увеличением ионного характера связи в растворах. Это заключение он
подтверждает сравнением кривых — приближенно рассчитанной
потенциальной энергии для гомеополярного и ионного состояний
хлористого иода с приближенной величиной потенциальной энергии
образования сольватированных ионов. В полярных растворителях
проявляется более или менее значительная электролитическая
диссоциация хлористого иода. Одновалентно-положительный ион
иода является акцептором электронов, и поэтому он образует
координационную связь с молекулами, содержащими доноры
электронов. Это благоприятствует дальнейшей ионной диссоциации
хлористого иода.
* Электроотрицательности галогенов, отнесенные к
электроотрицательности лития в качестве единицы, равны [55]: F CI Br J
4,0 3,1 2,8 2,5
141
Химические свойства
Благодаря большей доступности хлористого иода по
сравнению с фторидами галогенов, его относительной устойчивости,
возможности проводить работы в стеклянной аппаратуре, хлористый
иод в химическом отношении * изучен сравнительно полнее, чем.
описанные выше межгалоидные соединения — фториды галогенов,
и хлористый бром.
Хлористый иод проявляет все общие химические свойства
межгалоидных соединений,-перечисленные во «Введении» (стр. 17).
Реакции с простыми веществами
Хлористый иод очень энергично взаимодействует со многими
элементами. Эти реакции экзотермичны и в ряде случаев
сопровождаются воспламенением и взрывом. По качественным опытам,
проведенным главным образом Геннеем [31] и отчасти Христома:
носом [59], продуктами реакции являются иодиды и хлориды
соответствующих элементов. Так реагируют: калий (очень бурно),
натрий (реакция протекает медленно), магний в опилках и ртуть
(реагируют без выделения тепла), алюминий (реакция протекает
с выделением пламени). При реакции с порошком меди, оловом,
мышьяком, сурьмой, висмутом наблюдалось также выделение
паров иода. Изучались также реакции с серой, селеном, теллуром,
фосфором, кремнием.
Однако эти исследования были ограничены небольшим числом
элементов; продукты реакции в большом числе случаев не были
выделены и устанавливались предположительно.
В последнее время Гутман [60] вновь воспроизвел и расширил
эти опыты, изучив реакции хлористого иода с 39-ю элементами.
Опыты производились количественно. Избыток хлористого иода и
образующийся при реакции иод удаляли при помощи четырех-
хлористого углерода.
Некоторые элементы: калий, алюминий, титан, олово, фосфор
(белый), сурьма—реагируют с хлористым иодом очень бурно^
иногда со взрывом, и количественно. Полностью (100% выхода
хлорида) реагируют с избытком хлористого иода и такие
элементы: осажденное золото, ртуть, алюминий, германий, мышьяк,
ванадий, сера, селен и теллур (в виде порошков). Медь, серебро,
цинк, барий, висмут, железо реагируют с выходом 45—65%;
значительно слабее действует хлористый иод на кальций (30%),
кобальт (25%), никель (7%) и индий (1%). Не наблюдалась реак-
ция при действии хлористого иода на аморфный бор,
активированный уголь, кремний (аморфный и кристаллический), кадмий
и свинец (в зернах), цирконий, ниобий, тантал, хром, молибден,
вольфрам (в виде порошков), платину (жест^>).
* Сказанное относится отчасти также к треххлористому и бромистому
иоду.
142
Продуктами реакции являются хлориды элементов в высшей
степени валентности и свободный иод.
M-f-nJCl = MCln + ^-J2.
Только в единичных случаях получались хлориды элементов
низших степеней валентности: VC13, AsCl3, S2C12> MnCl2, CoCl2, NiCl2>
реС12 в смеси с FeCl3.
Течение реакции .(как и при действии фторидов галогенов)
определяется в значительной степени растворимостью продукта
реакции в хлористом иоде — образование поверхностного
защитного слоя хлорида, не растворимого в хлористом иоде, препятствует
дальнейшей реакции. При образовании летучих хлоридов реакция
обычно доходит до конца. Как отмечает Гутман, для
препаративных целей могут быть использованы только реакции с золотом,
ртутью, ванадием и теллуром, с прочими же элементами реакция
протекает бурно или не идет до конца.
По Гутману, калий при реакции с хлористым иодом переходит
в комплексное соединение KJCb. Следует, однако, отметить, что
комплексные (двойные) галогениды с хлористым иодом могут
давать и другие указанные в работе Гутмана хлориды ряда
элементов: А1С13 [62], РС15 [9, 10],. SbCl5 [11, 12]. Трудно объяснить,
почему Гутман не заметил образования, например, комплекса
PCWC1 (или PC14+-JC]~), трудно растворимого в четыреххло-
ристом углероде [10].
Образование хлоридов при реакциях элементов с хлористым
иодом Гутман объясняет тем, что хлористый иод в отсутствие
растворителя, вследствие его диссоциации на иод и хлор, всегда
действует хлорирующим образом. Поэтому если хлористый иод
взят в значительном избытке (как в опытах Гутмана), то должен
получаться преимущественно хлорид данного элемента.
Однако в тех случаях, когда нет избытка хлористого иода*
а данный элемент более или менее легко реагирует с
элементарным иодом, возможно образование смеси иодида и хлорида (что
и наблюдалось в опытах Геннея), по схеме
2M + nJCl = MJ„ + MClrt*.
Избыток хлористого иода, взаимодействуя с иодидом,
превращает его в хлорид
MJ„ + /zJCl = MClrt 4 /zJ2.
Реакция между хлористым иодом и водородом является
фотохимической. Изучению этой реакции посвящен ряд работ [63, 64].
* Имеются основания предполагать, что так же могут протекать реакции
в таких неводных растворах, в которых хлористый иод подвергается
электролитической диссоциации с образованием положительных ионов иода J-*",
обладающих значительным окисляющим и иодирующим действием.
на
Боннер, Горе и Иост приняли, что реакция H2-f2JCl = 2HCl+J2
(для газообразных веществ) протекает в два этапа:
Н2 + JC1 = HJ + НС1 (медленная реакция),
HJ + JC1 = НС1 + J2 (быстрая реакция).
По их данным, энергия активации медленной реакции (для
205—240°) равна 33,9 ккал моль~] *.
Более вероятно, что эта реакция имеет цепной механизм [64]:
JC1 + hv -* J + CI;
Cl-f-H2-*HCl + H;
H+JC1 -* HC1+J;
C1+JC1->C12+J;
J+J+-*J2+.
Реакция с неорганическими соединениями
Вода разлагает хлористый иод по уравнению
5JC1 + 3H20 ^ 2J2 + HJ03 + 5HC1 [ 176].
Первым этапом реакции гидролиза является образование
кислот — иодноватистой и соляной — JCl-f-H2O^HOJ + HCl. Иод-
новатистая кислота быстро превращается в йодноватую кислоту
и иод: 5НО1=Ш03 + 2Л2 + Н^0-
Выделяющаяся при гидролизе соляная кислота образует с еще
не разложившимся хлористым иодом комплексную кислоту НС1 • JC1
или HJCb, вследствие чего устанавливается равновесие
реакции гидролиза. Доба1Вледое соляной .кислоты смещает
состояние равновесия в сторону JC1, при этом наблюдается
растворение выделившегося иода [65]. Процесс гидролиза ускоряется при
увеличении рН и в растворах щелочей протекает до конца, с
образованием соответствующего хлорида, иодата и иода (или иодида —
лри достаточном количестве щелочи):
5JC1 + 6NaOH = NaJ03 + 2J2 + 5NaCl + 3H20;
3JC1 +6NaOH = NaJ03 + 2NaJ + 3NaCl + 3H20.
Гидролиз хлористого иода можно значительно ослабить или
практически вовсе предотвратить добавлением достаточного
количества хлористого натрия. Так, по Генгриновичу [177], 4-мол. водный
раствор хлористого иода в насыщенном растворе хлористого
натрия оказался весьма стойким.
Взаимодействие окислов металлов с хлористым иодом
сопровождается выделением кислорода и образованием иодида и
* Шерман и Ли рассчитали энергию активации для этих обеих реакций
соответственно равной 39 и 41 ккал [63].
144
,лорида данного элемента, часто в его высших валентностях
[3, СТР* ^15].
Большой интерес представляет взаимодействие хлористого иода
с солями различных металлов, которое может протекать как
реакции двойного обмена, комплекоообразования и окисления.
По данным Корнога с сотрудниками [33], нитрат и сульфат
калия полностью превращаются в хлориды, если их смешать с
хлористым иодом и выпарить досуха. Цианиды NH4, К, Cs, Cu, Ag,
Cd, Pb, цианаты К и РЬ, роданиды К, NH4, Ag и Pb реагируют с
хлористым иодом по следующим уравнениям (п — валентность
катиона):
М (CN)„ + «JCl=MCln + aiJCN;
ЗМ (OCN)n + 3nJCl = ЗМС1л+пЛ (0CN)3+/2J2;
3M (SCN)„+3n JC1 = 3MCIn -f nJ (SCN)8 + n h.
Трицианат иода J(OCN)3 и трироданид иода J(SCN)3 впервые
были получены при помощи этих реакций.
Сульфиды серебра, цинка, кадмия и свинца взаимодействуют
с хлористым иодом, причем образуются иодид металла,
хлористая сера S2Cl2 и сера по следующей реакции (на примере
CdS):
CdS + 2JC1 -* Cd J2+ S2C12 + S*.
Исследование некоторых из этих реакций в уксусной кислоте
как растворителе пока;зало, что цианиды и роданиды (но не
цианаты) реагируют с хлористым иодом в уксуснокислой среде
так же, как и в отсутствие растворителя.
Реакции между ацетатами тяжелых металлов и хлористым
иодом в уксусной кислоте не всегда дают ясные результаты. При
этом получаются продукты следующего состава: РЬ(С2Н302)2 • JC1,
Cd3(C2H302)2-2JC1 и Си2(С2Н302)2 • JC1. Структура этих
соединений не выяснена [33].
Окислительные свойства хлористого иода— расплавленного
или в неводных растворах —'- по отношению к неорганическим
соединениям еще очень мало изучены.
Окислительное действие хлористого иода «проявляется, в
частности, в его реакциях с иодидами. Так, расплавленный хлористый
иод реагирует с иодидами калия и серебра с образованием
хлоридов и иода:
JC1 + KJ = KC1 + J2 [31,66];
JCl + AgJ = AgCl + J2 [56].
Хлористый иод легко окисляет РС13 до РС15 ** (РС13 + 2JC1 =
= PC15+J2), последний, соединяясь с избытком JC1, образует ком-
* Эта схема не отражает все процессы, происходящие в данной реакции.
Так, авторы допускают, что, кроме S2C12, образуются и другие хлориды серы.
** Следует, отметить, что Вальден, исследуя электролитные свойства
раствора JC1 в треххлористом фосфоре, не обнаружил этого явления и темно-
фиолетовую окраску этого раствора приписал хлористому иоду [67,- стр. 425].
10—1419
145
плекс PC16J (или PCl+JC^ )[68]. В то же время JC1 не окисляет Нь
SbCl3 [69], ни AsCl3 [70]. *
Значительно шире в качестве окислителя был использован со.
лянокислый раствор хлористого иода, в котором он содержится
в виде ШС12. С помощью 'такого раствора, значительно более
удобного в качестве реагента, чем расплавленный JC1 или ег0
неводные растворы, удалось количественно окислить различные вое.
становители и использовать этот раствор также для других
реакций. А. И. Генгринович предложил применять солянокислый рас-
твор хлористого иода в качестве титрованного раствора для це-1
лей объемного анализа. Этому вопросу посвящена глава XVI.
Реакции галоидирования органических
соединений*
Хлористый иод может взаимодействовать с органическими
веществами по реакциям окисления, двойного обмена — превращение
алкилиодидов в соответствующие хлориды [3? стр. 616] **,
галоидирования (замещение атомов водорода на галогены),
присоединения по кратной связи и комплексообразования.
В этом и следующем' параграфах будут описаны лишь
реакции галоидирования и присоединения по двойной Связи.
Комплексные соединения хлористого иода с органическими веществами
будут рассмотрены в главе XI. Примеры реакций окисления
солянокислым раствором хлористого иода приведены в главе XVI.
Известно большое число реакций хлористого иода с
органическими веществами, в которых он проявляет галоидирующее
действие — способность замещать атомы водорода фенолов, аминов
и' т. п. соединений на атомы галогенов. При этом хлористый иод
в одних условиях выступает как иодирующий агент, в других —
как хлорирующий. В литературе описано образование иод- и
хлорпроизводных органических веществ при действии на них
хлористым иодом — парообразным, жидким или растворенным в
различных растворителях.
Так, например, при действии хлористым иодом на фенол Шют-
ценбергер и Сеньевальд [74] получили моно- и дииодфенол.
Михаэль и' Нортон [75], действуя хлористым иодом на растворы
анилина и ацетанилида в ледяной уксусной кислоте, получили
* Этот и следующий параграф данной главы написаны совместно с
Ф. Е. Каган.
** Кифёр и Эндрюс [73] изучали реакции между хлористым иодом и
бромистым или йодистым бензилом или йодистым изопропилом в четыреххлори-
стом углероде как растворителе. Была исследована кинетика этих реакций
спектрофотометрически при 25°. По данным этих авторов, реакции с галидами
бензила протекают как реакции обмена — с выделением иода или бромистого
иода С6Н5СН2Х + JC1 = С6Н5СН2С1 + JX. Реакция с йодистым изопропилом
протекает более сложно: 1 моль иода выделяется на каждые 2 моля хлористого
иода, вошедших в реакцию; одновременно наблюдается выделение НС1. Авторы
объясняют механизм этих реакций промежуточным образованием комплексов-
с хлористым иодом.
146
месь моно- и дииоданилина и я-иодацетанилида, а из смеси хло-
с0формных растворов п-нитроанилина и хлористого иода выдели-
^ дииодзамещенный продукт. Они же, действуя хлористым
* Одом на растворы ароматических аминов в разбавленной соляной
^слоте, получили дииодметанитроанилин, моноиодпаранитроани-
лин, дииодпаратолуидин.
Натриевые соли бензойной и нитробензойной кислот при
взаимодействии с хлористым иодом превращаются в иодбензол и,
с0ответственно в иоднитробензол. Реакция сопровождается де-
карбоксилированием [74].
При действии жидкого хлористого иода на бензол в
присутствии хлористого алюминия был получен моноиодбензол и другие
продукты иодирования бензола [76].
Бродфильд и сотрудники [156], действуя раствором хлористого
иода в уксусной кислоте (стр. 129) на анилин и его производные,
получили ряд продуктов иодирования. В реакциях с простыми
анилинами они применяли рассчитанное количество реагента;
процесс велся при комнатной температуре. Для иодирования
замещенных анилинов требовался избыток реагента и нагревание в
водяной бане.' Соединения иода осаждались при разведении
водой.
Аналогичным путем Джонс и Ричардсон [157], применяя
раствор хлористого иода в уксусной кислоте, получили, с хорошим
выходом, продукты иодирования ряда фенолов, оксибензойных и окси-
нафтойных кислот и ^простых арил-алкиловых зфиров.
Иодированные продукты были получены также при действии
водных растворов хлористого иода на некоторые органические
соединения. Так, при нагревании водного раствора хлористого
иода с ацетоном был получен дииодацетон в виде тяжелого
масла [77], а из продуктов взаимодействия жидкого хлористого иода
с водным раствором резорцина выделен трииодрезорцин [78].
Как указано выше, в ряде случаев при действии хлористого
иода на органические вещества, наряду с продуктами
иодирования, образуются также продукты хлорирования, а иногда
получаются исключительно хлорзамещенные продукты. Этим способом
были приготовлены хлорпроизводные жирного и ароматического
рядов, хлоруксусная [79] и хлормасляная [80] кислоты, а также
различные продукты замещения хлором водорода в бензоле,
толуоле и ксилоле [81]*.
Первую попытку объяснить различные направления реакции
(иодирование или хлорирование) предпринял Зернов [82]. На
основании литературных данных он пришел к заключению, что
при взаимодействии хлористого иода с ароматическими
соединениями преимущественно получаются иодпроизводные, а с
соединениями жирного ряда — хлорпроизводные. Однако Зернову уда-
* Все эти хлорпроизводные получались при пропускании хлора в смесь
соответствующего вещества с иодом- При этом получался хлористый иод (а при
Избытке хлора — и JC13), который и хлорировал органическое соединение.
147
лось выделить также иодпроизводные жирного ряда. Он полуци
иодизовалериановую и иодизомасляную кислоты при действии
хлороформного раствора хлористого иода на соответствующую
кислоту в присутствии пятихлористого фосфора, например,
RHCOOH + РС15 + JC1 = RJCOC1 + РОС13 + 2НС1*.
Экспериментально доказав образование иодизовалериановой
и иодизомасляной кислот, Зернов сделал вывод о том, что и соеди.
нения жирного ряда при взаимодействии с хлористым иодом об*
разуют иодзамещенные продукты. По мнению Зернова, иодпроиз*
водные жирного ряда при действии хлористого иода ранее не
удавалось получить потому, что продукты иодирования в болы
шинстве случаев неустойчивы и их выделение очень затруднено.
В растворе же происходит обменная реакция между иодпроизвод-.
ным и хлористым иодом, согласно уравнению
RJCOOH + JC1 = RC1COOH+J2.
Таким образом, конечным продуктом реакции оказывается
хлорпроизводное соединение.
Накопленный в дальнейшем экспериментальный материал
показал, что на течение реакции галоидирования, происходящей под
действием хлористого иода, оказывают влияние различные
факторы. Так, например, фенол в зависимости от условий по-разному
реагирует с хлористым иодом, а именно: в отсутствие
растворителя происходит главным образом хлорирование фенола, в
растворах — иодирование его.
Для правильного объяснения различных направлений реакции
галоидирования хлористым иодом необходимо учитывать как
свойства вещества, подвергающегося галоидированию (его строение
и полярность), так и природу среды, в которой протекает реакция.
Ламборн и Робертсон [83] указывают, что при иодировании
хлористым иодом имеет большое значение строение молекулы
вещества, подвергающегося иодированию. По мнению этих авторов,
всякие структурные затруднения, а именно — наличие
заместителей в кольце, могут замедлять иодирование в тех случаях, когда
сравнительно большому по размерам иону иода трудно
расположиться в молекуле органического соединения. Например, при
действии хлористого иода в уксуснокислой среде на ацетанилид
90% последнего иодируется, а 10% хлорируется, а при действий
на пантаметилбензол в этих же условиях 70% его иодируется, а
30% хлорируется. Из приведенных примеров действительно
следует, что направление реакции галоидирования зависит от
структуры органических молекул. Однако стерические факторы сами
по себе имеют не самостоятельное значение, а сопутствующее
важнейшим факторам — химической природе вещества (его
полярности) и среде.
* Уравнения реакций приводятся в том же виде, что и в работе Зернова-
148
Ламборн и Робертсон [83] обратили внимание на диэлектриче-
кие свойства растворителя. Они указывают, что при замене
укосной кислоты (диэлектрическая постоянная ДП = 7,1)
хлорбензолом <{ДП=5,8) процесс хлорирования значительно
облегчается — для анизола, например, процент хлорпроизводного
возражает от 20 (в среде уксусной кислоты) до 50 (в хлорбензоле)' *.
Дальнейшее развитие этого положения мы находим в работе
^енетта и Шарпа [84], согласно которым хлористый и бромистый
иод в растворителях с малыми величинами ДП подвергаются
преимущественно гомолитической диссоциации — на молекулы
галогенов; растворители с большими значениями диэлектрической
постоянной благоприятствуют гетеролитической (ионной)
диссоциации. Характер диссоциации галогенидов иода влияет на
направление процесса галоидирования. Так, основной реакцией
между кристаллической салициловой кислотой и парами
хлористого иода является хлорирование; в четыреххлористом
углероде (ДП = 2,23) преобладает иодирование, в нитробензоле
(ДП = 36,4) происходит исключительно иодирование салициловой
кислоты.
Подтверждением влияния растворителя, по данным Бенетта
и Шарпа, может служить также реакция между хлористым иодом
и перхлоратом серебра: в четыреххлористом углероде осаждение
хлористого серебра протекает чрезвычайно медленно, в то время
как в нитробензоле осадок хлористого серебра образуется
мгновенно.
Как уже упоминалось, хлористый иод — вещество мало
полярное. Растворение в тех или иных растворителях приводит к
возрастанию диполького 'момента хлористого иода (стр. 141), а его
растворы в полярных растворителях (в нитробензоле, уксусной
кислоте и др.) хорошо проводят ток.
Хлористый иод в расплавленном состоянии или из неполярных
растворителях подвергается преимущественно гомолитической
диссоциации;
2JCI;£Vf Cl2
или
3JC1 + JC13 + J2; JC13 = JC1 + Cl2.
Этим и объясняется тот факт, что при взаимодействии
хлористого иода со многими органическими веществами в отсутствие
растворителя идет процесс хлорирования — хлор является более
активным галоидирующим средством, чем молекулярный иод.
В то же время сольватация хлористого иода молекулами
полярных растворителей способствует диссоциации его на ионы. Поэтому
в растворах, содержащих ионы J+, идет процесс иодирования.
Имеются, однако, примеры, когда реакция галоидирования
протекает не согласованно с приведенными выше соображениями.
* Такое различие в галоидирующем действии хлористого иода в отношении
анизола в среде этих двух растворителей вряд ли можно объяснить
небольшой разницей в диэлектрических постоянных уксусной кислоты и
хлорбензола.
149
Так, даже в таких малополярных и неполярных растворителях, ^а,
например, четыреххлористый углерод, такие полярные вещества*
как фенол (jut = 1,18 ZD) и салициловая кислота, преимущественн '
иодируются и лишь в незначительной степени хлорируются. °
Но здесь, очевидно, решающее влияние оказывает природ»
и строение вещества, подвергающегося галоидированию. Поляр,
ный характер фенола и салициловой кислоты и наличие элек-
тронно-донорной ОН-группы, приводящей к повышению электрон-
•ной плотности в орто- и пара-положениях, способствуют
образованию комплексов этих веществ с хлористым иодом, дальнейшей
поляризации молекул последнего и их диссоциации на ионы J+
и С1~. Наличие катионов иода, в свою очередь, создает
благоприятные условия для процесса иодирования, например
он
1
1 H+J+C1--
он
1 J
И || +НС1
Поэтому если для реакций галоидирования применять
хлористый иод в виде комплексных соединений, обладающих
значительным дипольным моментом, обусловленным поляризацией
хлористого иода, то можно ожидать, что будет происходить процесс
иодирования даже в неполярных растворителях. Действительно,
Терентьев, Беленький и Яновская [85] при взаимодействии фенола,
Р-нафтола, салициловой кислоты и резорцина с комплексом
хлористого иода и диоксана C4H802-JC1 [86]* в растворе четырех-
хлористого углерода (|х=0) или диоксана (ДП —2, |я=0 или
~ 0,4 D) выделили продукты иодирования: n-иодфенол, 5-иодса-
лициловую кислоту, моно- и 2,4,6-трииодрезорцин ** и т. д.
В еще большей степени должно способствовать иодирующему
действию хлористого иода создание условий, благоприятствующих
его ионной диссоциации. Поэтому для целей иодирования весьма
пригодными оказались солянокислые растворы хлористого иода,
в которых, последний содержится в виде комплексной кислоты
H[JC12], частично диссоциирующей с образованием положительно
заряженных ионов иода JCl2~^J+-b2Cl~" (см. главу XVI).
Следует, однако, подчеркнуть, что приведенные выше
соображения о факторах, обусловливающих характер галоидирующего
действия хлористого иода, нельзя считать исчерпывающими. Так,
например, с ними не согласуется работа Годбира с
сотрудниками [88]. При взаимодействии JC1 с растворами ряда гидроксил-
содержащих органических соединений (2,3—НО—С10Н6СООН,
(р-НОСеНОг, ализарин) <в ледяной уксусной кислоте получены
Б
I * Дипольный момент этого комплекса неизвестен, но можно полагать,
что он должен быть больше, чем дипольный момент комплекса диоксана с
иодом C4H802.J2 (0,95 D) [87].
** В этих опытах был взят 1 моль хлористого иода на 1 моль галоидируе-
jvioro вещества.
150
г10рпроизводные, содержащие лишь незначительные количества
**0да *.
Для решения поставленного выше вопроса необходимы даль-
ейшие разносторонние исследования процесса галоидирования с
Омош.ью хлористого (и бромистого) иода,* охватывающие большой
руг объектов и растворителей.
Галоидирование соединениями электроположительного иода,
главным образом хлористым и треххлористым иодом, уже нашло
применение в химическом анализе и для синтеза. Так, Генгринович
и др. [89, 90] применили солянокислый раствор хлористого иода
для объемно-аналитических определений разнообразных фенолов
и ароматических аминов и для получения ряда их иодпроизводных
(глава XVI). Он же показал возможность применения для этой
цели водного раствора хлористого иода, содержащего хлористый
натрий (стр. 144); такой раствор количественно превращает
фенолфталеин, растворенный в спирте, в тетраиодфенолфталеин.
Уксуснокислый и солянокислый растворы хлористого иода были
применены для иодирования ароматических аминокислот с целью
получения лекарственных рентгеноконтрастных препаратов,
причем были синтезированы следующие дииодзамещенные продукты:
I. р-[4-амино-3,5-дииодфенил]-а-фенилпропионовая кислота
J
I
HaN"MI
j—I J—сн2_сн-соон
с6н5
[I. а-[4-амино-3,5-дииодфенил]-р-фенилпропионовая кислота
C6H5 - CH2 - СН - СООН
I
/\
J-1 H-J
NH2
III. В-[5-амиио-2,4-дииодфенил]-а-фенилпропионовая кислота
NH2
И_Сн2—СН— СООН
j С6Н5
Преаараты I и III были получены путем иодирования
хлористым иодом в растворе уксусной кислоты, а для получения прега-
* Предлагаемое этими авторами объяснение полученных ими результатов
основано на предположении, что хлористый иод в уксусной кислоте
диссоциирует не только на J + и С1~,но и на J- и С1+ и что связь С—С1 устойчивей, чем
связь С—J. С этим связано преимущественное хлорирование.^ Та кое толкование
нельзя считать удовлетворительным. К сожалению, с данной работой мы
знакомы лишь по реферату.
151
рата II иодирование проводилось хлористым иодом в двухфазной
системе, состоящей из 6-н. соляной кислоты и симметричного тет-
рахлорэтана или хлороформа [91, 92].
Для целей рентгенографии был синтезирован также ряд
жирных кислот, замещенных в а-положении амидом иодированной
n-аминобензойной кислоты или иодированным сульфаниламидом.
Иодирование проводилось хлористым иодом в солянокислой или
уксуснокислой среде {93].
Реакции присоединения хлористого иода к
непредельным органическим соединениям сдвой-
ной связью
Способность непредельных углеводородов и некоторых других
органических веществ присоединять хлористый иод по месту
двойной связи известна уже давно. Так, Сими сон еще в 1863 г. [94}
описал реакцию присоединения хлористого иода к этилену и
пропилену. Исследования Танатара [17] показали, что сухой этилен
жадно поглощается охлажденным хлористым иодом.
В литературе имеется ряд работ по вопросу о присоединении
галогенидов иода, в основном хлористого иода, к непредельным
соединениям, главным образом, в связи с .применением неводных
растворов JC1 и JBr для определения йодного числа жиров и
масел. Для таких определений галогениды иода имеют явные
преимущества перед галогенами [95]. Для определения йодного числа
применяются опиртовый раствор хлористого иода (раствор Гюбля),
раствор хлористого иода в ледяной уксусной кислоте (раствор
Вийса), спиртовый раствор хлористого иода, насыщенный
хлористым водородом (раствор Валлера).
Большой интерес представляет вопрос о том, происходит ли
присоединение галогенидов иода к непредельным соединениям в
соответствии с известным правилом Марковникова. Еще в 1870 г.
Сорокин [96] изучал порядок присоединения хлористого иода к
этилену и пропилену. Для хлориодпропилена Сорокин принял
строение СНз—СНС1—CH2J на основании того, что при обработке
спиртовым раствором щелочи этот продукт превращается в р-хлор-
пропилен СНз—СС1=СН2.
По экспериментальным данным Михаэля [97], в этой реакции
(применялся солянокислый раствор хлористого иода) на каждые
четыре молекулы а-иод-р-хлорпропилена СН3—СНС1—CH2J
образуется одна молекула а-хлор-р-иодпропилена СН3—CHJ—CH2CI.
Ингольд и Смит [98] при взаимодействии пропилена с 0,05-мол.
раствором JC1 в разведенной соляной кислоте также получили
смесь двух продуктов: 69% СН3—СНС1—CH2J и 31% СН3—OHJ—
СН2С1.
Истомин [99], изучая присоединение хлористого иода к изобу-
тилену, показал, что строение хлориодизобутилена таково:
(CH3)2CC1CH2J.
Таким образом, эти исследования, установившие, что хлор
(хлористого иода), как более электроотрицательный элемент, присое-
152
диняется по месту двойной связи преимущественно к наименее
гидрогенизированному углеродному атому, подтвердили, что
основное направление этих реакций подчиняется правилу Марковни-
кова.
Подтверждением этого может служить также реакция
присоединения хлористого иода к монохлортрифторэтилену, в
результате которой образуется 1,2-дихлор-1,1,2—трифтор-2-иодэтан [100]:
CF2=CFC1 + JC1=CC1F2 — CFC1J.
Однако при взаимодействии с более сложными органическими
соединениями на присоединение хлористого иода оказывают
влияние различные заместители, имеющиеся в молекуле.
Так, Темникова, Баскова и Хаимова [101] при взаимодействии
хлороформных растворов дифенилпропилена и хлористого иода
получили а, а-дифенил-р-хлорпропилен, по-видимому, за счет
первоначального образования а-иод-а, а-дифенил-|3-хлорпропана,
отщепляющего в момент реакции HJ по схеме *
С6Н5 С6Н5 СбН5 С6Н5 С6Н5СбН5
\ У \ / \ /
С +JC1 * CJ * С +HJ
II I II
СН СНС1 СС1
I I I
СН3 СНз СН3
Присоединением хлористого иода к а, а-дифенилэтилену (в этих:
же условиях) получают а-хлор-а, а-дифенил-р-иодэтан — в
соответствии с правилом Марковникова
С6Н5 СбН5 СбН5СбН5
\ / \ /
С + JC1 » СС1
I
СН-я CH2J
Такое различие в порядке присоединения JC1 к непредельным:
жирноароматическим углеводородам -авторы объясняют влиянием
метильной группы.
Своеобразно протекают также реакции хлористого иода с диал-*
коксиэтиленом, сопровождающиеся выделением иода и
образованием 1,2-дихлор-1,2-диалкоксиэта.на [158]
RO-CH ROCHC1
II + 2JC1 = | Н- J2
RO-CH ROCHC1
Однако если проводить эту реакцию в четыреххлористом
углероде, то при добавлении первых порций хлористого иода
наблюдается сперва обесцвечивание и лишь затем — выделение иода. Это*
дает основание предполагать, что первично образовавшийся про-
* Аналогично же, по данным Хаимовой [101], протекает реакция
взаимодействия хлористого иода с 1,1-ди-п-толилпропеном-1 (СН3С6Н4)2С=СН —СН3-
153
дукт присоединения хлористого иода реагирует с избытком
последнего
RO-CHJ ROCHC1
| + JC1 = | + J2
RO—CHC1 ROCHC1
Если же эту реакцию проводить в среде пиридина, то
образуются эквимолекулярные количества хлорида и иодида N, N'-(1,2-
диалкоксиэтан)-бие-пиридиниума [158]
RO-CH-NC5H5] X
I (Х=С1 или J).
RO-CH—NCeHsJX
Подобного же типа соединения получил Хейс [159] при
взаимодействии хлористого иода с циклогексеном в пиридиновом
растворе
С1
\ \/
|| +JC1 + C5H5N > |
NC5H5]J-
В отсутствие пиридина, в среде уксусной кислоты образуется
продукт присоединения хлористого иода — 2-хлор-1-иодциклогек-
сан [160].
Вопрос о порядке присоединения хлористого иода к
непредельным органическим соединениям, равно как и более широкий
вопрос о характере (механизме) взаимодействия хлористого иода с
последними, требует дальнейшего разностороннего изучения с
учетом не только химического состава, но и ряда других факторов.
Очень важно влияние растворителя и его отношение к
хлористому иоду. Так, изучение реакций присоединения хлористого иода
в водно-спиртовой или в водно-солянокислой среде в ряде случаев
усложняется тем, что вследствие гидролиза образуются продукты
присоединения иодноватистой кислоты. Поэтому, изучая реакцию
взаимодействия реактива Гюбля с жирами, Вийс [102] пришел к
выводу, что по месту двойной связи присоединяется, в первую
очередь, не хлористый иод, а продукт его гидролиза—иодновати-
стая кислота. Она образуется в растворе Гюбля в результате
побочной реакции JC1 + H20 ^ JOH + HC1. В качестве
доказательства такой точки зрения автор приводит известный факт о том,
что прибавление к раствору Гюбля соляной кислоты уменьшает
скорость реакции присоединения галогенов к жирам. С
прибавлением соляной кислоты равновесие реакции гидролиза хлористого
иода смещается в сторону образования JC1. Поэтому Вийс
считает, что уменьшение скорости реакции галоидирования
обусловлено уменьшением концентрации JOH и является доказательством
того, что к жиру присоединяется в первую очередь JOH. Если бы
к жиру присоединялся хлористый иод, то, по мнению этого автора,
с повышением концентрации соляной кислоты, а следовательно,
и концентрации JC1 в растворе, скорость галоидирования жира
увеличилась бы.
354
Эти соображения не могут служить серьезным доказательством
в пользу представления о первичном присоединении JOH. Как
известно, соляная кислота связывает хлористый иод в комплексное
соединение H[JCb], и этим также можно объяснить уменьшение
скорости реакции в присутствии НС1. Это подтверждается также
работой Уайта и Робертсона [103], изучавших кинетику
присоединения хлористого иода к олефиновым соединениям и влияние
хлористого водорода на эту реакцию. Эти авторы показали, что
соляная кислота значительно уменьшает скорость присоединения JC1
к непредельным кислотам и правильно объяснили это образованием
H[JC12].
Ряд других исследователей [104—106] обоснованно принимает,
что JC1 непосредственно присоединяется по месту двойной связи,
причем образовавшиеся иодхлорсоединения далее могут
превращаться в лактоны и иодгйдрины.
Так, при взаимодействии эфирного раствора JC1-HC1 с корич-.
ной кислотой получается а-иод-р-хлор-р-фенилпропионовая
кислота С6Н5—СНС1—CHJ—СООН [104]. Водный раствор HCbJCl
образует с коричной кислотой а-иод-р-гидроксифенилпропионовую
кислоту.
Эрленмейер считает, что и в водном растворе первичным
продуктом реакции является иодхлорфенилпропионовая кислота,
последняя же образует р-лактон в качестве промежуточного
продукта
С6Н5 - СНС1 - CHJ - СООН £ С6Н5 — СН — CHJ — СО + HCI,
который затем разлагается водой, превращаясь в иодгидрин
с6н5—сн - chj —со+н2о -^ с6н5 — снон—chj—соон.
Непосредственное разложение хлориодфенилпропионовой кислоты
водой
С6Н5СНС1СШСООН + Н20 ^C6H5CHOHCHJCOOH + НС1,
по мнению Эрленмейера, не представляет
действительного течения реакции. Автор подтверждает эту мысль тем, что при
взаимодействии эфира коричной кислоты с хлористым иодом
(когда исключена возможность образования лактона) получается фе-
нилхлориодпропионовый эфир, а не иодгидрин.
Образование лактонов подтверждается также данными Бэзе-
кена и Гельбера [105]. Эти авторы пришли к выводу, что только
те иодхлориды разлагаются водой, для которых возможно
образование лактона как промежуточного вещества. Они считают, что в
процессе реакции устанавливается равновесие между иодидхлори-
дом и р-лактоном, почти полностью смещенное в сторону иодид-
155
хлорида. Равновесие это нарушается водой, которая переводит
небольшие количества р-лактона в иодгидрин, что способствует
полному превращению иодхлорсоединений в иодгидрины.
Образование лактонов Бэзекен и Гельбер показали на примере
с бензилиденпропионовой кислотой. После определения йодного
числа этой кислоты, по Вийсу, был получен у-иодлактон, поэтому
они считают, что реакция протекает следующим образом:
c6h5ch=chch2cooh+jci -+ c6h5chcichjch2cooh -
I
- с6н5-сн-сн j-ch2 + на
I I
о со
Если верно предположение о разложении иодхлорпроизводных
водой, то реакция образования лактона должна была бы пройти че>
рез стадию получения иодгидрина, т. е.
C6H5CH=CHCH2COOH+JCl -> C6H5CHC1CHJCH2C00H -+
-* НС1 + C6H5-CH-CHJ-CH2
I I
о со
Доказательством в пользу схемы, предложенной Бэзекеном,
является образование того же уиодлактона в растворе четыреххло-
ристого углерода, т. е. при полном отсутствии воды.
Ингль [106], изучая взаимодействие стильбена и стирола с
реактивами Вийса, Гюбля, Валлера, также пришел к выводу, что во
всех случаях по месту двойной связи сначала присоединяется JC1.
Продукты присоединения бол^е или менее легко разлагаются
водой, содержащейся в реакционной смеси, причем в зависимости от
природы вещества выделяются HJ и НС1 или только НС1 и
образуются соответствующие оксисоединения по схеме
— СН — СНС1 Н20 — СНОН НС1
II —^ I + —> I +
— СН -CHJ Н20 —СНОН HJ
По данным Иятля, стильбен, растворенный в разведенной
уксусной кислоте, образует с реактивом Вийса иодхлорстильбен
СбН5СНС1СШСбН5. В этих же условиях из стирола получен иод-
хлорстирол СбН5СНС1СН2Л *.
При взаимодействии с реактивом Гюбля стильбен превращается
в диоксидифенилэтан, а стирол образует фенилиодоксиэтан.
Образование этих продуктов Ингль выражает следующими
уравнениями:
С6Н5СНС1СШСбН5 + 2Н20=С6Н5СНОНСНОНС6Н5 + НС1 + Н J;
C6H5CHC1CH2J + H20 = CeH5CHOHCH2J + НС1.
* Характер реакции стирола с хлористым иодом был подтвержден Инголь-
дом и Смитом [98].
156
Ингль объясняет более прочную, по сравнению со стильбеном,
связь иода .в стироле тем, что в стироле иод соединен с атомом
углерода, не находящимся под непосредственным влиянием
электроотрицательной фенильной группы.
С реактивом Валлера (см. стр. 152) стильбен не реагирует,
стирол же дает йодное число 224,3, т. е. взаимодействует почти
количественно. Автор и этот факт объясняет наличием в молекуле
стильбена двух фенильных радикалов, что затрудняет
присоединение хлористого иода, связанное с предварительным разрушением
соединения JC1-HC1, которое образуется в реактиве Валлера.
Известны случаи, когда органические соединения, содержащие
двойные связи, не взаимодействуют с реактивами, применяемыми
для определения йодного числа, или когда эти реакции не имеют
количественного характера, известно также, что определение
йодного числа одного и того же продукта различными методами часто
приводит к разноречивым результатам. Поэтому Бэзекен и Гель-
бер [105] предприняли изучение взаимодействия хлористого иода
(реактив Вийса) с различными органическими соединениями. При
этом они пришли к заключению, что присоединение теоретического
количества хлористого иода к непредельному соединению, т. е.
получение точного значения йодного числа, зависит от двух причин:
от скорости присоединения хлористого иода к непредельным
соединениям и от устойчивости образующихся иодхлорсоединений.
Первая зависит главным образом от строения вещества: так
называемые «отрицательные» группы, как, например, СООН, С6Н5,
СООС2Н5, СОСНз, СОСбН5? замедляют присоединение, так что для
достижения правильного значения йодного числа соединений,
содержащих указанные группы, необходимо не менее 8 часов.
Присоединение хлористого иода к таким непредельным
соединениям представляет собой равновесный процесс, например,
С6Н5- СН = СН - СОС6Н5 -f JC1 ^ ССН5—СНС1— СНJ — СОС6Н5-
В связи с этим точность получаемых йодных чисел обусловлена
устойчивостью образующихся иодхлорпродуктов. Последняя в
свою очередь зависит: а) от строения органического соединения—
присутствие в молекуле так называемых «отрицательных» групп
способствует отщеплению галоида; б) от растворителя, в котором
протекает реакция,— в полярных растворителях (ацетон, спирт,
уксусная кислота и др.) состояние равновесия сдвигается влево, а
в неполярных и малополярных (ССм, СНС13) образуются иод-
хлорсоединения; в) от растворимости образующихся
иодхлорпродуктов.
Присоединение хлористого иода к непредельным соединениям
с конъюгированными двойными связями затруднено. Реакция
обычно сопровождается выделением иода, что находит следующее
объяснение [103]: присоединение протекает по принципу Тиле —
одна двойная связь насыщается только хлором, при этом выде-
157
ляется иод
1234 быстоо V 2 3 4/
— С = С-С=С- + 2JC1 оьютро _с_с==с с__ + j2.
1111 i ■ ' Ь
образующаяся двойная связь в положении 2,3 затем медленно
присоединяет хлористый иод
\ / медленно \ /
>с-с = с~С\ -+- jci—-:—* >с-с = с-С\
' I I I I I I I I .
С CI CI CI J С1
Способность хлористого иода присоединяться по месту кратной
связи была использована для химического анализа непредельных
органических соединений. Растворы хлористого иода в
органических растворителях: спирте, ледяной уксусной кислоте и др.
уже давно применяются в анализе жиров. В последнее время [89,
90] для объемно-аналитического определения ряда принадлежащих
к этой группе органических лекарственных препаратов был
использован солянокислый раствор хлористого иода (см. главу XVI).
2. ТРЕХХЛОРИСТЫЙ ИОД JC13
Треххлористый иод был открыт Гей-Люссаком в 1814 г. [1].
Он относится к наименее устойчивым межгалоидным соединениям,
легко диссоциирует на хлор и хлористый иод, и поэтому его
поведение в химических реакциях часто аналогично поведению
хлористого иода. Наряду с этим JCU по своему составу, строению и
свойствам проявляет определенное своеобразие в ряду
межгалоидных соединений.
Получение
Треххлористый иод, (Представляя собой продукт присоединения
хлора к хлористому иоду, образуется при тех же химических
реакциях, что и хлористый иод, но при наличии избытка хлора. Таковы,
например, следующие реакции:
1. Действие хлора на некоторые соединения иода: иодиды,
йодистый водород — газообразный [59] и жидкий [3, стр. 257],
летучие йодистые алкилы [108]
RJ+2C12 = RC1+JC13,
расплавленное иодноватокислое серебро [109]
2 AgJ03+4С12 = 2 AgCl + 2JC13 + 302.
2. Окислительно-восстановительное взаимодействие некоторых
соединений иода и хлора, например,
Н J03 + 5HC1 = JCI3 + Cl2 + ЗН20;
2PCl5 + J2°5 - 2JClo + 2С12-ЬР2С>5 [3, стр. 619];
158
3S02Cl2+J2=2JCl3+3S02 [72]*.
Треххлористый иод получают преимущественно
непосредственным синтезом из элементов **. Известно несколько вариантов
такого метода получения треххлористого иода.
1. Сухой хлор пропускают над твердым иодом, который при
этом превращается сперва в хлористый, а затем в треххлористый
иод [ПО].
2. Возгоняют иод в токе газообразного хлора; избыток хлора
удаляют током С02 [4, стр. 121].
3. Действуют сухим хлором на газообразный йодистый водород
[59]. Оба газа пропускают в сосуд в таких количествах, чтобы
хлор был в избытке против стехиометрического количества
HJ + 2Cl2 = HCl+JCl3. При недостатке хлора образуется
хлористый иод HJ+JC13 = HC1+2JC1.
По Бирку [111], для получения чистою JC13 с количественным
выходом пропускают хлор над сухим иодом, охлажденным смесью
С02 и спирта (—79°), до тех пор, пока становятся заметными
капли жидкого хлора; для завершения реакции сосуд оставляют еще
на несколько часов в охладительной смеси, затем испаряют
избыток хлора.
Эти методы не гарантируют получение чистого продукта, так
как плотная оболочка треххлористого иода, образующаяся на
поверхности кристалликов иода или хлористого иода, препятствует
дальнейшему действию на них хлора, в результате чего JC13
содержит примеси иода и хлористого иода. По этой причине заслуживает
внимания следующий способ (получения JC13 [112].
4. Пропускают струю хлора над определенным количеством
иода со скоростью 80—90 см3/мин при комнатной температуре до
тех пор, пока иод не превратится в жидкую массу (хлористый
иод). Затем температуру повышают до 100°, а скорость
прохождения струи хлора увеличивают до 170 см3/мин. При этом JCI
превращается в JC13, который затем перегоняют в охлажденный
льдом '.приемник ***.
5. Указанные выше затруднения можно предотвратить, если
получать JC13 по Томасу и Дюпуи действием иода на жидкий хлор,
взятый в избытке [113, 114]. Газообразный хлор из баллона
проходит через предохранительную склянку и затем конденсируется
в большой пробирке или колбе, охлаждаемой смесью твердой СОг
с ацетоном или эфиром. Медленно, небольшими количествами,
вносят измельченный иод, который тотчас же превращается в JC13,
постепенно оседающий на дно сосуда. Оставшийся хлор испаряют.
* Реакция между хлористым сульфурилом и иодом была изучена Руффом.
Иод растворяется в хлористом сульфуриле, не реагируя с ним. Если к этому
раствору прибавить катализатор — хлористый или йодистый алюминий, фиолетовая
окраска раствора быстро изменяется в бурую и при осторожном кипячении
раствора начинается возгонка JC1 или JC13. Последний образуется только при
значительном избытке хлористого сульфурила.
** Об образовании треххлористого иода в системе иод—хлор см. стр. 128.
*** В оригинальной работе подробно описана аппаратура для получения
треххлористого иода по данному методу.
, 159
В последнее время Манн [115] предложил получать JC13 дейек
вцем иода на бертолетову соль в солянокислом растворе по реак^
ции
KC103+J2+6HC1 = KC1+2JC13 + 3H20.
В коническую колбу емкостью в 1,5 л помещают 250 г хорошо
измельченного КСЮз, затем вносят 500 г растертого иода и 250 мл
воды, после чего в течение 45 минут приливают 950 мл
концентрированной соляной кислоты небольшими порциями при постоянном
помешивании и температуре 40°. Охлажденный раствор фильтруют
через стеклянный фильтр; выделившийся JC13 перекристаллизовьь
вают из спирта и высушивают в вакууме над хлористым кальцием.
Выход (по иоду) 75%.
Физические свойства
Треххлористый иод при комнатной температуре—твердое,
кристаллическое вещество. Возогнанный препарат образует длинные
лимонно-желтые иглы. При застывании расплавленного треххло-
ристого иода образуются большие красновато-бурые ромбические
пластинки.
Как уже отмечалось, треххлористый иод термически очень
неустойчивое вещество. Измеряя плотность паров, Меликов [116]
показал, что пары JC13 полностью диссоциируют на хлористый иод
и хлор при температуре около 77° и атмосферном давлении, даже
при избытке хлора. Упругость пара твердого треххлористого иода
уже при 64° составляет 1 атм [59]; он заметно испаряется при
пропускании сухого воздуха даже при —12°, а при пропускании С02—
при 15° [116]*.
Термическая неустойчивость JC13 очень затрудняет точное
определение его температуры плавления. Поэтому в старой литературе
встречаются различные данные о точке плавления JCl3 — в
зависимости от внешних условий [3, стр. 621].
Треххлористый иод при нагревании в запаянных капиллярах
плавится под давлением собственных насыщенных паров 16 ат при
ЮГ в красно-бурую жидкость. Температуру плавления при этих
условиях считают константой для JC13 [6, 18].
На воздухе, особенно влажном, JC13 быстро разлагается, но
в атмосфере сухого хлора и при комнатной температуре он
может долго сохраняться, не изменяясь.
Плотность твердого JC13 d\b = 3,1107 (путем взвешивания под
хлором или С02 [59]); d~40 = 3,203 [20]. Молярный объем равен 72,84 при
— 40°. Сравнивая это значение с аддитивной величиной S(J-f3Cl),
Бирк [20] пришел к заключению, что при образовании JQ3 происходит
незначительная контракция.
Теплота образования треххлористого иода из элементов
V2J2(ib) + 3/2С12(г) = JC13(tb), (6-D
* Упругость пара твердого JC13 в равновесии с жидкой фазой
определенного состава определена Штортенбекером [6].
160
Томсену [386], равна 21,5, а по Бертло [38a]f—15,5 ккал- моль~{*.
Яйс и И°ст f117^ подтвердили данные Томсена —для теплоты
образования JC13(tb) из иода и хлора в их стандартных состояниях они нашли
лИчиНУ 21,15 ккал-моль—К
^е По Бертло [38а], тепловой эффект реакции
VaJ2(r) + 3/2С12(Г) = JC13(TB) (6.2)
равен 21,7 ккал-моль~1. В этой же работе Бертло указывает, что для
Теплоты образования JC13(<tb) из JC1(tb) и С12(г) им найдена величина
g 5 ккал-моль"1.
Нис и Иост [117], изучая тензиметрически равновесие
JC13(tb) ^ JCl(r) -f- С12(Г), (6.3)
определили константу равновесия: Кр = Pjci • Рс\2 = 1,09 • 10-3 атм2
Лри 25° и 4,36-10 ~3 атм2 при 35°. Предположив, что газообразные JC1
и хлор проявляют свойства идеальных газов, эти авторы вычислили
теплоту образования JCl3(TB) по этой реакции 25,3 и его свободную
энергию 4,04 ккал-моль-1. По Гринвуду [19], эти значения приводят к
изменению энтропии при данной реакции А 5298,16= —71,34 кал • моль-1 • град~К
Сочетание этой величины с энтропиями газообразных JC1 и С12 дает
стандартную абсолютную энтропию JC13(tb) :5°98Л6 = 41,1 ±0,5 кал -
• моль-1-град'1 [19]. Эту же величину S°9816(JC13(TB)) Нис и Иост
вычислили из термохимических данных для реакции образования JC13 из
элементов по уравнению (6.1).
Энергия образования связи J—С1 в треххлористом иоде, равная
42,7±0,8 ккал, меньше энергии образования связи J—С1 в
хлористом иоде — 51,0 ккал [5]. Подобное уменьшение энергии связи с
ростом ковалентности известно и для других неорганических
соединений, например для треххлористого и пятихлористого фосфора:
энергия образования каждой связи Р—С1 в РС1з равна 75 ккал,
при дальнейшем присоединении двух атомов хлора на каждую
новую овязь Р—О приходится лишь 39 ккал.
Спектр поглощения паров ЛС1з такой же, как паров JC1, но он
существенно отличается от спектра паров иода. Это явление
объясняется диссоциацией паров JC13. Действительно, спектр
поглощения раствора треххлористого иода в четыреххлористом углероде
выражается кривой, представляющей собой сочетание кривых
поглощения света для раствора хлора и хлористого иода в ССЦ
[118], [рис. 37, стр. 217].
Криосколические исследования растворов треххлористого иода
в хлорокиси фосфора [119], нитробензоле [44], уксусной кислоте
[18] показали значительно уменьшенный молекулярный вес JC13—в
разведенных растворах он почти вдвое меньше формульного. Это
можно объяснить диссоциацией ЛСЬ в растворах на хлористый
иод и хлор. Однако Бекман и Юнкер [120] получили для молекуляр.
* Термохимическое исследование реакции растворения JC13 в водном
растворе сернистой кислоты [38а] дало теплоту образования JC13(tb) 17,1 ккал-моль—1.
11—1419
161
ного веса ЛСЦ <в фосгене величину 233; совпадающую с
теоретическим значением (233,4). Пониженный молекулярный вес JC13 в
растворах следует приписать также электролитической диссоциации*
трехх лор истого иода *.
Химические свойства
Реакции с неорганическими веществами
Реакции треххлористого иода с неорганическими веществами
относятся почти исключительно к окислительно-восстановительным,
процессам.
Действие треххлористого иода .на металлы и металлоиды
изучено весьма недостаточно. Так, по данным Кристоманоса, водород
при комнатной температуре не реагирует с треххлористым иодому
умеренно нагретый водород реагирует без выделения пламени,
причем образуются НС1 и JC1
JC13+H2=2HC1+JC1.
С сильно нагретым водородом происходит бурная реакция,
сопровождающаяся выделением НС1, HJ и иода. Фосфор и калий
воспламеняются при взаимодействии с треххлористым иодом [59]. С
иодом JCI3 реагирует с образованием монохлорида
JC13+J2 = 3JC1.
Нужно полагать, что расплавленный треххлористый иод
должен действовать на металлы и металлоиды подобно хлористому
иоду, может быть лишь с тем отличием, что в случае JC13 более
вероятно образование хлоридов тех или иных элементов в их
высших степенях валентности.
Столь же мало описаны реакции треххлористого иода с
неорганическими соединениями. При взаимодействии с избытком
аммиака JCU образует йодистый азот, хлористый и йодистый
аммоний [59]. Хлорноватистым ангидридом треххлористый иод
медленно окисляется, согласно уравнению
2JCl3+5Cl20=J206+8Cl2 1121].
Окись ртути реагирует с треххлористым иодом при слабом
нагревании по 'следующему (предположительному) уравнению [ПО]:
2HgO + 2 JCI3 = HgCl2 + Hg J2 + 2C12 + 02.
Треххлористый иод окисляет сероуглерод, причем образуются СС14,
CSC12, хлористая сера и выделяется иод [31,59]
4CS2 + 6 JCI3 = 2СС14 + 2CSC12 + 3S2C12 + 3 Jr
Несколько больше изучены окислительные свойства
треххлористого иода в водных и солянокислых растворах.
* Электропроводность треххлористого иода в расплавленном состоянии
и в растворах, равно как и некоторые другие его электрохимические свойства,
описаны в главах X и XI.
162
Реакция гидролиза треххлористого иода протекает довольно
сложно: сперва он разлагается на хлористый иод и Хлор [59, 122];
гидролиз хлористого иода дает HJ03, HC1 и иод (стр. 144),
последний, реагируя с хлором, снова образует JCL Поэтому реакцию
гидролиза треххлористого иода можно выразить следующим
уравнением:
2 JC13 + ЗН,0 £ Н Ю3 + 5НС1 + JC1
или 2 JC13 + ЗН20 £ H JOs + 4HC1 + HCl. JCL
По данным Солдатова и Вознесенского [123], для растворов,,
содержащих 1 моль JC13 в 15—115 л воды, степень гидролиза
треххлористого иода составляет соответственно от 76,0 до 83,5%.
В солянокислых растворах равновесие гидролиза смещается в
противоположную сторону, а при достаточной концентрации HCI
треххлористый иод образует комплексную кислоту H[JC14], которую
удалось выделить в виде гидрата ШСЦ^НгО [124].
Едкие щелочи действуют .на растворы JC13 так; же, как и н;а
растворы монохлорида, с образованием хлоридов, иодидов, иодатов
и иода, например
5JC13 + 18КОН - 3KJ03 + J2 + 15КС1 + 9Н20.
Если щелочь взята в избытке, иод не выделяется:
3JC13 + 12КОН - 2KJ03 + К J + 9КС1 -f 6H20.
Возможны также побочные реакции, приводящие к
образованию гипохлоритов, хлоратов и перхлоратов *.
Каган [125] показала, что ib присутствии бикарбоната натрия
треххлористый иод гидролизуется, частично превращаясь в
хлористый иод,
2 JC13 + 6NaHC03 = JC1 + Na J08 + 5NaCl + 6C02 + 3H20.
Водные и солянокислые растворы треххлористого иода были
успешно использованы в качестве окислителя в реакциях с
неорганическими (и органическими) веществами. Так, серебряная
фольга превращается в водном растворе ЛС1з в хлористое и
йодистое серебро. Окись серебра реагирует с избытком JC13 по
уравнению
2JC13 + Ag20 + 2Н20 = Ш03 + 2AgCl + JC1 + ЗНС1.
Если окись серебра взята в избытке, образуется AgJ03, а при!
кипячении—AgJ04 по уравнению
2JC13 + 4Ag20 = Ag J04 + 6AgCl + Ag J.
Водный раствор треххлористого иода окисляет сероводород с
образованием HCl, HJ (или иода) и серы ** [126] и превращает фос-
* В спиртовом растворе едкое кали образует с треххлористым иодом
следующие продукты: КС1, KJ, KJ03 и СШ3 [4, стр. 121].
** Мак-Ивор полагает, что в этой реакции образуется йодистая сера
состава S3J2, но не приводит подтверждающих доказательств.
163
фористую кислоту в фосфорную [123]. Солянокислые растворы трех-
хлористого иода были применены для растворения золота, хорощ0
раздробленной и коллоидной серы [111]*, пирита, медного
колчедана, сернистого мышьяка и некоторых других сернистых
минералов, которые ори этом были окислены в сульфаты. Таким
путем можно было количественно определить FeS2 — после его
окисления по иону Fe3+, а также по иону S042" [127]. Дальнейшие
формы применения солянокислого раствора треххлористого иода
для количественного — объемного анализа, основанного на
реакциях окисления, будут приведены в главе XVI.
Реакции г а л о и д и ро в а н и я органических
соединений
Сведения по этому вопросу в литературе почти вовсе
отсутствуют. Известно лишь, что JCU весьма энергично реагирует с
углеводородами, хлорируя их; например, бензол превращается в СбС16
[113]. Это можно объяснить тем, что JC13, как уже отмечалось,
легко разлагается на хлористый иод и хлор, a JC1 в отсутствие
растворителя действует хлорирующим образом (см. стр. 149).
Для галоидирования органических соединений весьма
пригодными оказались солянокислые растворы треххлористого иода.
Реакции галоидирования с участием этих растворов протекают
количественно и довольно быстро, что дало возможность применить
их для количественных объемно-аналитических определений
большого числа фенолов, аминов и их производных (глава XVI).
Реакции с непредельными соединениями
Литературные данные по этому вопросу ограничиваются почти
исключительно работами А. Н. Несмеянова и сотрудников,
изучавших взаимодействие треххлористого иода с непредельными
соединениями с двойной и тройной связью.
Несмеянов и Фрейдлина [128] получили хлорвинилиодидхло-
рид из ацетилена и раствора JC13 в 15 и 3%-ной соляной кислоте
СН =СН + JC13 -> C1CH = CHJC12.
Фрейдлина, Брайнина и Несмеянов [129] получили также
С1СН = СН
(cich = ch)2jci, ch)jci
и их комплексные соединения с HgCl2
С1СН = СН
(С1СН = СН)2 JC1. 2HgCl2 и \ jci. HgCl2.
с6н5
* Для этих реакций был взят *^ 20%-ный раствор треххлористого иода в
концентрированной соляной кислоте.
164
Фрейдлина и Несмеянов [130] осуществили реакции арилиро-
Бания треххлористого иода (в солянокислом растворе) при
помощи олово- и ртутьорганических соединений:
C6H5SnCl3+JCl3 = C6H5JCl2H-SnCl4;
2С6Н55пС1з+ЛС13=(СбН5ус1+25пС14;
(C6H5)2Hg+JCl3 = (C6H5)2JCl+HgCl2;
2C6H5HgCl+JCl3=(C6H5)2JCl4-2HgCl2.
Брайнина и Фрейдлина [131], применяя водные растворы
треххлористого иода с хлористым натрием, содержащие комплексную
соль NaJCl4, получили продукты взаимодействия JC13 с ацетиленди-
карболовой кислотой (I), с диметиловым эфиром этой
кислоты (II) и с фумаровой кислотой (III), причем были получены
р-хлорзамещенные иодозосоли:
I. Н 02СС=СС02Н + JCl3 ~* ClC—СО
|| >o+HCl + CO
HC-JC1
II. СН302ССееСС02СН з f JC13 -* ClC- СО
II >0 + СН3С1
СН302С—С—JC1
III. 2HOOC-CH = СН—СООН + JCl3 -> С1НС-СО
I >о
НоС—JOCOCH = СМ СООН +
+С02 +~2НС1
Попытки присоединить треххлористый иод к малеиновой
кислоте *, к диметиловому эфиру фумаровой кислоты и к фенил
ацетилену были безрезультатны [131].
Работы Несмеянова с сотрудниками в этой области
представляют интерес не только в том отношении, что они показали новые
возможности применения треххлористого иода для синтеза
органических иодхлорпроизводных, но и потому, что они помогают
уяснить строение треххлористого иода, подтверждают
представление о его ступенчатой диссоциации и показывают аналогию в
поведении треххлористого иода и пятихлористого фосфора.
Строение треххлористого иода
Решение вопроса о строении треххлористого иода в первую
очередь сводится <к выяснению валентного состояния иода в этом
межгалогениде.
Стенлей [132], пытавшийся одним из первых рассмотреть
строение треххлористого иода, допускал для него две возможные струк-
* По мнению Брайниной и Фрейдлиной, JC13 хлорирует малеиновую
кислоту.
165
туры, в одной из которых иод одновалентен, а в другой — трех^
валентен (в обоих случаях — с трехвалентными атомами хлора):
Cl Cl — Cl
V; \J /
Cl I \ | / II
Cl
Однако это мнение Стенлея не нашло поддержки, и в
литературе распространилось представление о треххлористом иоде, как о
соединении трехвалентно-электроположительного иода, с которым
непосредственно связаны три одновалентных атома хлора. Такое
СЧ
строение }J—Cl принимал, в частности, Вернер [133].
СК
Объясняя строение соединений типа M[JC14], где М — щелочной
металл, Вернер рассматривает их как продукты присоединения
МС1 к JCI3, исходя из того, что при действии четыреххлористым
углеродом можно извлечь из них треххлористый иод. Поэтому
данным талогеносолям, так называемым тетрахлороиодиатам,
Вернер придает такое же строение, как и тетрахлороауриатам, а
именно:
С1 1
CUC1
Cl
R
Г с1
С AuCl
Cl
Как видно из приведенной формулы, в тетрахлороиодиатах
имеется комплексный анион, в котором иод играет роль
центрального атома с координационным числом 4.
В недавно опубликованных работах Спаку мы находим
экспериментальное подтверждение взглядов, высказанных Вернером.
Спаку исходил из предположения, что комплексный анион гало-
генидов типа MJC14 в водных растворах разлагается по схеме
JCl4-^JCl2- + CL,
Для обоснования этого предположения Спаку и Попеа [134]
провели следующие эксперименты: они вводили твердый
NH4JCI4 • ЗН20 в раствор комплексных аммиакатов, например
[Cl2Co(NH3)4]Cl, и при этом получили комплексы типа
[Cl2Co(NH3)4][JCl2], которые оказались более устойчивыми, чем
соли щелочных металлов MfJCb].
Образование комплексных соединений типа [комплексный катион]-
• [JC12] по реакции
NH4 [JC1J + [С12Со (NH3)J Cl = [С12Сэ (NH3)4] [JC12] + NH4C1 + Cl2
может иметь место при условии диссоциации иона [ JC1J— по
приведенному выше уравнению. Поэтому авторы решили проводить синтез этих
комплексных соединений, пропуская в раствор струю газообразного
хлора, чтобы сместить равновесие [JC14]-^[JC12]-+ С12 влево, если
такое равновесие действительно существует. Опыт оправдал
предположение Спаку и Попеа. При таких условиях синтеза были получены
166
тетрахлороиодиаты комплексных катионов, например [С12Со (NH3)4 ] [ JC14]
'}1 некоторые другие.
Получение устойчивых комплексных соединений типа R[JC14],
где R — комплексный катион, является дополнительным
доказательством строения аниона JC17, предложенного Вернером и
основанном на представлении о трехвалентности иода в треххлористом
иоде. Стабилизирующая роль комплексных катионов с "большим
лонным радиусом понятна из поляризационных представлений.
В пользу представления о трехвалентности иода в
треххлористом иоде говорит также существование соединений типа ArJC^,
как, например, фенилиодидхлорида СбНбЛСЬ, о- и я-тол-илиодидхло-
рида СНз—СбНЦЛСЬ, иодидхлорида о-иодбешойной кислоты [135].
Эти соединения можно рассматривать как производные
треххлористого иода RJCb, в частности, потому, что они получаются или
путем присоединения хлора к арилиодидам или по методике,
'разработанной Несмеяновым с сотрудниками,— при 'взаимодействии
солянокислых растворов треххлористого иода с олово- и ртутьорга-
ническими соединениями (см. стр. 165).
Рассматривая JC13, как соединение
электроположительно-трехвалентного иода, Вальден [67], на основании своих измерений
электропроводности JC13 в неводных растворах (глава X), принял следующую
схему электролитической диссоциации: JC13^ J3+ + 3C1~. Оддо [119],
желая объяснить пониженную величину молекулярного веса JCl3 в
хлорокиси фосфора (134—173 вместо 233,4), по криоскопическим
данным, допускал, что JC13 диссоциирует на JCI^hQ-.
Однако эти схемы ионной диссоциации JC13 не имели
достаточного экспериментального обоснования. Кроме того, пользуясь
этими схемами, нельзя объяснить разнообразия реакций
треххлористого иода в различных условиях.
Более убедительные выводы о существовании ионов J3+ в
растворах треххлористого иода дали электрохимические
исследования: определение окислительно-восстановительного потенциала
системы J/JC13 и получение /—1/-кривых для (водных растворов
треххлористого иода [136]. Окислительно-восстановительный
потенциал J/JC13 оказался равным 1,032 е.
Из этого окислительно-восстановительного потенциала был
рассчитан потенциал разложения для водного раствора
треххлористого иода вычитанием из электродного потенциала аниона
соответствующего значения для катиона:
ejci8 = еа - еу, еа = 1,366 в\ еЗС\л = 1,366 - 1,032 = 0,334 в.
Таким образом, потенциал разложения водного раствора
треххлористого иода оказался равным 0,334 в. На /—У-кривой для
того же раствора было действительно установлено наличие
перегиба в точке, соответствующей потенциалу 0,33 в, что, по мнению
автора [136], является потенциалом разложения JCU и служит
.убедительным доказательством существования ионов J3+.
Нам кажется, что это доказательство нельзя считать строгим,
так как очень трудно предположить, что <в водном растворе трех-
167
хлористого иода действительно был измерен окислительно-воостацо~
вительный потенциал системы J/JC13, а не более сложной системы
что, как будет показано нами далее, связано с процессом гидр0*
лиза. То же самое относится и к анализу /—У-кривых.
Хотя приведенные сомнения и не опровергают возможности
существования ионов J3+ в водном растворе, однако они указываю^
на то, что результаты этих экспериментальных исследований не*
дают однозначного решения данного вопроса.
Перейдем теперь к рассмотрению тех соображений, которые
высказывались в пользу представления об одновалентности иода
в треххлориетом иоде. Это представление основывалось на том, что
треххлористый иод — непрочное вещество, очень легко
разлагающееся, даже в твердом состоянии, на JC1 и хлор. Это дало
основание предположить, что JCU является продуктом присоединения
молекулы хлора к хлористому иоду JC1 • С12.
Филипп [137] следующим образом обосновывал такое
представление о природе молекул треххлористого иода: если на водный
раствор хлористого иода действовать избытком окиси серебра, то
образуется иодноватокислое серебро согласно уравнению
3 JC1 + 3Ag20 = AgJ03 + 3AgCl + 2AgJ.
При последующем действии хлора (при кипячении) получается
иоднокислое серебро, AgJ04. Так как при кипячении избытка
окиси серебра с треххлористым иодом также было получено
иоднокислое серебро, то Филипп пришел к выводу, что
треххлористый иод является продуктом присоединения молекулы хлора
к хлористому иоду JC1 + СЬ=JC1 • СЬ.
Другим аналогичным доказательством может служить тот
факт, что при взаимодействии треххлористого иода с водными
растворами щелочей, наряду с образованием обычных продуктов
гидролиза, т. е. иодатов, хлоридов и иода (или иодидов),
согласно уравнениям, приведенным на стр. 163, образуются также гипо-
хлориты, хлораты и перхлораты [138]. Можно предположить, что
эти соли получаются в результате реакции щелочей с хлором,
образующимся при диссоциации треххлористого иода.
Если принять, что треххлористый иод является соединением
одновалентноположительного иода, то его строение можно
представить двояким образом. Так, Госселин [139], считая
маловероятным разрыв прочной молекулы хлора для образования столь
малоустойчивого соединения, как треххлористый иод,
рассматривал JC13 как продукт присоединения молекулы хлора к
хлористому иоду, в котором центральным атомом является иод. Тогда
треххлористому иоду следует приписать структуру [Cl2J]+Ch со
сложным катионом. Такое строение согласуется с мнением [140]
о существовании полигалогенидов, в которых центром
координации молекул галогенов является положительный ион иода.
Подобным соединением можно считать гипотрииодитную кислоту
состава [J2-J]OH, полученную лишь в водных растворах в условиях,,
когда JOH образуется в присутствии иода [122, 141, 142].
168
Рассматривая треххлористый иод как продукт присоединения
хлора к хлористому иоду, можно принять для этого межгалогени-
да также структуру анионного полигалогенида J+[C1 • С12]~ с
комплексным анионом [С1з]~. Такой вывод был сделан Плотниковым
и Рокотяном [143] на основании электрохимического исследования;
растворов треххлористого иода в броме.
Плотников и Рокотян определяли электропроводность
растворов треххлористого иода в жидком броме и установили, что
молекулярная электропроводность носит так называемый аномальный
характер, т. е. падает с разведением. Уменьшение молекулярной
электропроводности с разведением эти авторы объясняли
распадом комплексных ионов. Если принять, что JC13 ■— бинарный
электролит с одновалентным электроположительным иодом, то его*
электролитическая диссоциация выразится уравнением
JC13^J+ + [C13]-
Комплексный анион [С13]~ с разведением распадается по реакции
[cy-^ci- + ci2.
Образовавшиеся в растворе ионы J+ и С1~ частично
соединяются в молекулу хлористого иода. Таким образом, с разведением
раствора треххлористый иод превращается в менее проводящий
хлористый иод, что и вызывает падение молекулярной
электропроводности.
Правда, авторы отмечают, что так как сольватация, обычная1
при растворении, может иметь место и в данном случае, то весьма
вероятно, что уменьшение молекулярной электропроводности с
разведением происходит не столько вследствие распадения иона
[С1з]~, сколько вследствие распадения более сложных комплексов,,
проводящих ток.
Если считать иод в молекуле треххлористого иода
трехвалентным, то уравнение электролитической диссоциации примет
вид
JC18;£J+ + + + 3C1-
Однако проведенный Плотниковым и Рокотяном электролиз
бромных растворов треххлористого иода с серебряными
электродами показывает, что количество выделившегося на аноде хлора,,
определенное по привесу анода, значительно превышает то
количество его, которое, в соответствии с показаниями кулонометра,
рассчитано для выделения С1~, если исходить из предположения о
диссоциации JC13 на J3+ и ЗС1~. Этот факт, по мнению авторов,
подтверждает приведенное ранее уравнение диссоциации JC13 на
J+hC1^.
Однако вследствие того, что не был проведен анализ продуктов,
выделяющихся на аноде, этот вывод можно оспаривать, исходя
из следующих соображений. На катоде не наблюдалось выделения
иода, это может быть следствием взаимодействия иода,
выделившегося на катоде, с бромом с образованием бромистого иода
(J2 + Br2 ^2JBr). Но в таком случае дальнейший электролиз бро-
169
чистого иода должен 'был сопровождаться выделением брома на
аноде, чем можно было бы объяснить увеличение количества
выделенного галогена по сравнению с рассчитанным для С1~.
Указанные соображения не могут, однако, служить основанием
JK тому, чтобы отвергнуть схему диссоциации треххлористого иода,
предложенную Плотниковым и Рокотяном. Следует учесть, что
аналогичное явление наблюдали также Брунер и Бекьер [56] при
электролизе бромистого иода в присутствии избытка брома, когда
на серебряном аноде осаждался бром в количестве,- большем
1 г-экв Вг на 1 F. Брунер и Бекьер объяснили этот факт
образованием ионов Вг~ и их разрядом одновременно с ионами Вг~.
Рассмотрение литературных данных о строении треххлористо-
го иода в сопоставлении с его свойствами приводит к заключению,
что не следует противопоставлять два мнения, имеющиеся в
литературе по этому вопросу: 1) треххлористый иод является
соединением электроположительно-одновалентного иода и 2) JC13 —
соединение электроположительно-трехвалентного иода. Можно
лишь предположить, что растворы (или расплавы) треххлористого
иода представляют собой систему, в которой обе эти формы JCI3
я J[C1 • С12] или [СЫ]С1 находятся в равновесии *.
Аналогичное явление известно и для некоторых других
неорганических веществ, например для T1J3. Трехиодистый таллий в
некоторых отношениях весьма похож на полииодиды щелочных
металлов: он легко отщепляет молекулу иода при обработке
органическими растворителями, изоморфно кристаллизуется с три-
иодидами рубидия и цезия. В то же время он легко образует с
иодидами щелочных -металлов комплексы типа MjTUJ, в которых
таллий должен быть трехвалентным.
Эти факты, а также исследование растворов трехиодистого
таллия приводят к заключению, что в растворах существует
равновесие
T1J82:TI[J-J2]^T1J + J2>
которое при добавлении иода или растворимых иодидов смещается
влево, а при извлечении иода органическим растворителем —
в сторону распада полигалогенидного комплекса и его
превращения в T1J. «Таким образом, T1J3 представляет собой
интересный пример перехода между обычными валентными и
комплексными соединениями» [144].
Следует также отметить определенную аналогию между треххло-
ристым иодом и пятигалогенидами фосфора. Последние способны
образовывать комплексные анионы типа [РХб]~~, например, PFe" (в виде
HPF6) и РС1^~ [145-147]. В то же время РС15 и РВг5 при реакции
с галогенидами некоторых других элементов могут образовывать ка-
* Приведенные выше экспериментальные данные говорят в пользу поли-
галогенидной формы с комплексным анионом С1^~, которую мы дальше и
приводим. Но все же для решения этого вопроса необходимы дальнейшие
исследования.
J 70
тионы [РС14]+ и [РВг4]+, например, в комплексных соединениях с га-
логенидами иода [PX4]+[JX2]- (X = С1 или Вг) [10, 148] и с
хлоридами металлов [РСЦ]+ [МСЦ]- (М = Al.Fe), [PC14]2 [SnClJ, [PCl4][SbCl6]
[149]. Пятибромистый и пятихлористый фосфор образуют также
комплексы типа полигалогенидов, например, РС15-5Вг2 [150], или
[РС!4] [СЦВг^], РВг7,РВги[151],или [PBr4] [Br (Br2)J, где п = 1-6;
РС13Вг4-РС13Вг18 [152] или [РС13Вг][Вг(Вг2)п], где п = 1 —8.
При взаимодействии с некоторыми органическими
непредельными соединениями пятихлористый фосфор реагирует таким
образом, что группа РСЦ присоединяется к одному (наиболее гидро-
генизированному) атому углерода, а хлор — к другому по месту
кратной связи [153, 154], например:
СвН5СН = СН2 + 2РС15 = C6H5CHClCH2PCl4-PCl5;
СбН5С = СН + РС15 = С6Н5СС1 = СНРС14.
С восстановителями же PCls и РВгб обычно реагируют в
форме РХ3 • Х2, т. е. отщепляют молекулу хлора или брома.
Аналогично этому треххлористый иод образует комплексные
анионы ЛС1Г, диссоциирует на [JCU] и С1, как, например, в
реакции присоединения к ацетилену (см. стр. 164) * или по схеме
JC1 • С12 — в реакциях с восстановителями **.
Поэтому, говоря о строении и молекулярном состоянии трех-
хлористого иода в растворах или в расплавах, нужно учитывать:
а) равновесие между обычной валентной и полигалогенидной
формами (по аналогии с T1J3):
С1 С1
\ /
J ->J[Cl.Cl2]
б) способность треххлористого иода реагировать то в форме
q/J— С1 с последующим переходом, например, в комплексный анион
JCfcf или присоединяться к азотистым основаниям, то в форме JC1-C12
(при реакциях окисления и галоидирования), то в форме [JC12]C1.
Равновесие JCl3~^ JC1 • С12 в растворах, по мере удаления
молекулярного хлора или связывания JC1, например в реакциях галоидирования,
смещается вправо, а при добавлении хлора, соляной кислоты или
хлоридов—влево. Вводных растворах с достаточной концентрацией соляной
кислоты образуется комплексная кислота — производное трехвалентного
иода JC13 -f HC1 ^ HJC14—которую удалось выделить в виде гидрата
HJC14 • 4Н20 [124]. Однако, как следует из величины константы диссоциа-
* Или также на JC1 + 2С1— при'реакциях галоидирования (глава XVI).
** Отличие от пяти хлор истого фосфора здесь проявляется^ в том, что
первичный продукт диссоциации — хлористый иод обладает свойствами
окислителя.
171
ции, рассчитанной для треххлористого иода в 4— 6-н. растворе соляной
кислоты, даже в таких солянокислых растворах происходит диссоци»
ция JC13 на JC1 и С12 [155]
[JC13] ~7>z'w •
Такая величина константы диссоциации JC13 указывает на
то, что около 4,5% треххлористого иода диссоциирует на
хлористый иод и хлор [71].
Повышая концентрацию хлора в реакционной смеси, можно»
согласно работам Спаку, сдвинуть равновесие [JCl4]- ^ [JC12]--|,
-f- С1а влево, т. е. в сторону образования .-*. комплексного иона, в
котором иод является трехвалентным.
Таким образом, состояние треххлористого йода в солянокислом
растворе можно представить следующей схемой:
С1 НС1
J+[Cl-Cl2]-^\ J-Cl Z H[JC14] 2TH++[JC14]-
Cl
Представления о строении треххлористого иода в системах, в
которых он находится в виде отдельных молекул, необходимо
дополнить данными о его структуре в кристаллическом состоянии.
Этому вопросу посвящена лишь одна работа, в которой Бисвейк и
Вибенга [161] на основании рентгенографических исследований
показали, что кристаллическая решетка треххлористого иода
(рис. 27) состоит из почти плоских димерных молекул J2CI6 или
С1 С1
С1 С1
С1
<
С1
На рис. 27 показаны также некоторые параметры
кристаллической решетки треххлористого иода — межатомные
расстояния * и валентные углы. Эта структура указывает на то, что при
образовании двухъядерного галогенида J2Cle — при димеризации
молекул в процессе кристаллизации — происходит стабилизация
трехвалентного состояния иода.
Таким образом, проводя отмеченную выше аналогию между гало-
генидами фосфора и кода, нужко сказать, что треххлористый иод в
отношении кристаллической структуры отличается от пятихлористого
фосфора, кристаллическая решетка которого состоит ш конов PCLT ж
* Расстояние J—С1 в кристаллах, равное 2,38—2,39 А, лишь немного
' О О
превышает сумму ковалентных радиусов иода и хлора 1,33 А + 0,99 А =
= 2,32 А.
172
pCliT- Однако это различие, характерное для кристаллического состоя-
нля, в значительной мере исчезает, когда оба эти хлорида переходят
р жидкое состояние. Так, расплавленный пятихлористый фосфор
проводит ток хуже, чем кристаллический: по данным Саймонса и Джес-
сопса [162], удельная электропроводность жидкого РС13 в точке плав-
JD.
Рис. 27. Кристаллическая структура треххлори-
стого иода.
ления равна 6-Ю"9 ом~~1 -см—{, для кристаллического же вещества
*юо° = 3-10 8, а Х20* = 3,3-Ю-7, ож-1.^^-1. В то же время жидкий
треххлористый иод проводит ток значительно лучше (хюэ.з0 =
= 9,55 -10"3 ом^-см—х), чем кристаллический (к7о =1,9- Ю-5, щ0- =
= 4,9- Ю-4 ом-1-см"1 [36, 163].
Уменьшение электропроводности галогенидов некоторых
элементов при переходе их в расплавленное состояние обусловлено
действием нескольких факторов, в том числе частичным
разрушением ионной структуры и образованием расплавов молекулярного
типа, уменьшением степени электролитической диссоциации
вследствие ассоциации ионов и т. д.
Что касается явлений, наблюдавшихся при исследовании пяти-
хлористого фосфора (уменьшение электропроводности при
плавлении, аномалии в диэлектрической постоянной, максимум на
кривой теплоемкости, различие в спектрах комбинационного рас-
& ъ
3,60
N»,-
3.60
\ 3J5'
а
3.67 Щ 361
Q1° ''' *'
Q
173
сеяния света для твердого и жидкого РС15 и некоторые дру^
гие [164]), то их можно объяснить тем, что при плавлении его
кристаллов ионная структура исчезает, или же тем, что равновесие
между ионами РС1^и РС1?и отдельными молекулами, имеющими
форму тригональной бипирамиды, почти полностью смещено в
сторону последних *.
При плавлении треххлористого иода происходит явление
противоположного характера. Значительное увеличение
электропроводности при нагревании,
очевидно, является следствием того, что»
димерные молекулы J2CI6
частично диссоциируют на ионы [163],.
возможно по схеме J2Cl6^:JCl?-f
+JC17. При этом нельзя
исключить и предположения о том, чта
одновременно образуются также
простые молекулы Л2С1б ^ 2JC13>.
далее диссоциирующие на ионы.
Аналогичное явление известно*
п «о п также для галогенидов некото-
Рис. 28. Строение димерных молекул м м
галогенидов алюминия. Рых других элементов, например
алюминия. Электронографиче-
ские исследования, произведенные Пальмером и Эллиотом [165],
показали, а позднейшие исследования [166] подтвердили, что
расположение атомов в димерных 'молекулах галогенидов алюминия
может быть представлено в виде двух деформированных
тетраэдров, соединенных двумя общими вершинами (рис. 28) [47]. Эта
структура подтверждена исследованием спектров
комбинационного рассеяния света галогенидов алюминия в твердом и жидком
состояниях [167].
Результаты этих, а также рентгенографических исследований
[45,46] дают полное основание принять для димерных молекул гало-
С1 С1 С1
\ у ч /
генидов алюминия, например для А12С16, строение у^\ у^\
С1 ЧС1 СГ
двухъядерного галогенида, аналогичное строению молекул J2C16.
По данным Бильца и Фойгта [168], кристаллический
хлористый алюминий имеет положительный температурный
коэффициент электропроводности, как и треххлористый иод. При плав-
лении (188°) электропроводность уменьшается, а затем при
дальнейшем нагревании начинает расти. Так, удельная
электропроводность хлористого алюминия при 180° равна 4,8-Ю-6, при
200° — 0,56 • КГ6, а при 245 ° — 1,1 • 10^ омгх. см~\
* При растворении пятихлористого фосфора в некоторых полярных
растворителях, например в ацетонитриле, наблюдается обратное явление:
стабилизация ионного состояния вследствие сольватации ионов [145}.
174
Неводные растворы хлористого, бромистого и йодистого
алюминия в ряде растворителей часто обладают значительной
электропроводностью (порядка 10~3—Ю-2 om~{cm~1) [169]. Последнее явление
объясняется образованием в таких системах ионов, сольватированных
молекулами растворителей, например А13+, [А1П>]+, [А1Г4]~, [А12Г7]- и
других (3. А. Шека [170,171], Н. Н. Лебедев [172]*). В то же время
известно, что димерные молекулы галогенидов алюминия в ряде
растворителей, с которыми они образуют соединения, распадаются на
простые молекулы (см. стр. 139) А12Г6 + mS^2[Air3(S)m] **, в
дальнейшем подвергающиеся электролитической диссоциации.
Таким образом, сопоставление треххлористого иода с галоге-
нидами некоторых других элементов показывает, что треххлори-
стый иод занимает промежуточное положение между галогенида-
ми типа М2Г6 (например, А12С1б) с близкой структурой димерных
молекул и аналогичным поведением в растворах и галогенидами
некоторых элементов в их высших степенях валентности, более
или менее легко отщепляющими часть галогена (PCI5, T1J3 и др.)-
Новые данные о кристаллической структуре треххлористого
иода, состоящей из димерных молекул ^С16, не противоречат
представлениям о строении отдельных молекул JC13 и
изображенной выше (стр. 172) схеме равновесий, существующих в
солянокислых растворах треххлористого иода. Эту схему можно лишь
дополнить равновесием диссоциации димерных молекул на
простые J2Cl6^2JCl3 и на шродукты электролитической диссоциации
последних.
ЛИТЕРАТУРА
1. J. L. G а у - L u s s а с, Ann. chim. phys., 91, 5 (1814).
2. Н. Davy, Trans. Roy. Soc. (London), 104, 487 (1814).
3. Gmelin's Handbuch der anorganische Chemie, B. 8, 1933.
4. J. W. Mel lor, Compr. Treat, on inorg. and theoret. Chemistry,,
v. II, 1927.
5. N. W. S i d g w i с k, The Chemical Elements and their Compounds,
Oxford, 1950, v. II, 1151—1154.
6. W. Stortenbeker, Rec. trav. chim., 7, 152 (1888); Z. phys. Chem.,
3, 11 (1889).
* По Лебедеву [172], димерные молекулы галогенидов алюминия,
реагируя с злектронодонорными растворителями, например с кетонами,
диссоциируют по схеме
Al2Cl6-2R2CO^r [(R2C0)2A1C12]+ + [AlCl4]-,
однако г/га схема отражает, вероятно, лишь первичный процесс диссоциация
молекул А12Гб. Она не учитывает дальнейшей ступенчатой диссоциации ионов
А1Гз", сольватации анионов и ряда других данных, полученных 3. А. Шека
при исследовании неводных растворов галогенидов алюминия [170, 171] и.
треххлористого таллия.
** S — молекулы растворителя.
175..
7. В. J. Karsten, Z. anorg. Chem., 53, 381 (1907).
8. Л. С. Л и л и ч, ЖОХ, 22, 730 (1952).
9. Е. Boudrimont, С. г., 55, 361 (1862).
10. Я. А. Фиалков, А. А. К у з ь м е н к о, ЖОХ, 19, 812 (1949)
11.0. Ruff, J. Zed пег, L. Н е с h t, Ber., 48, 2068 (1915).
12. Я- А. Фиалков, И. Л. Абарбарчук, Укр. хим. ж., 15, 176
372 (1949).
13. J. Co r nog, R. A. Karges, J. Am. Chem. Soc, 54, 1882 (1932).
14. Я- А. Фиалков, О. И. Шор, ЖОХ, 18, 14(1948).
15. Ю. К- Трапп, J. рг. Chem., 63, ПО (1854); Bl. Acad. Petersb., 13, 14
(1855).
16. Д. И. Менделеев, Основы химии, изд. 13, т. 1, стр. 619.
17. С. Т а н а т а р, ЖРФХО, 25, 97 (1893).
18. W. Stortenbeker, Z. phys. Chem., 10, 183 (1892).
19. Н. Н. Г р и н в у д, Усп. химии, 22, 445 (1953).
20. Е. В i r k, Z. anorg. allg. Chem., 172, 385 (1928).
.21. Т. Е. Thorpe, J. Chem. Soc, 37, 174(1880).
22. К- S t г е с к е г, Wied. Ann., 17, 93 (1882).
23. E. D. Wilson, Phys. Rev., 32, 611 (1928).
24. J. McMor r i s, D. M. J о s t, J. Am. Chem. Soc, 54, 2247 (1932).
'25. К- Beck, Z. phys. Ch., 48, 641 (1904).
26. С H. Townes, F. R. M e r r i t t, B. D. Wright, Phys. Rev.,
73, 1334, (1948).
27. K. F. L u f t, Z. phys., 84, 767 (1933).
28. R. T. We i d ne r, Phys. Rev., 73, 254 (1948).
29. W. E. Curtis, J. Patkowski, Trans. Roy. Soc (London), 232 A,
395 (1934).
30. H. M. Hulburt, J. O. H i rsc h f e 1 de r, J. Chem. Phys., 9, 61
(1941).
31. J. B. Hannay, J. Chem. Soc, 26, 815 (1875); Chem. News, 37, 224
(1878).
32. Л. П а у л и н г, Природа химической связи, Госхимиздат, М.—Л., 1947,
стр. 167.
33. J. С о г п о g, H. W. Н о г г a b i n, R. A. Karges, J. Am. Chem.
Soc, 60, 429 (1938).
34. J. С о r n о g, E. Bauer, J. Am. Chem. Soc, 64, 2620 (1942).
35. M. G. Ma lone, A. L. Ferguson, J. Chem. Phys., 2, 99 (1934).
36. N. N. Greenwood, H. J. E m e 1 e u s, J. Chem. Soc, 987 (1950).
37. С M. Blair, D. M. J о s t, J. Am. Chem. Soc, 55, 4489 (1933).
38a. M. В e r t h e 1 о t, С. г., 90, 841 (1880); 6. J. T h о m s e n, Ber., 15,
3021 (1882).
39. H. Cordes, Z. phys., 74, 34 (1932).
40. O. D a r b у s h i r e, Phys. Rev. (2), 39, 162 (1932); 40, 366 (1932).
41. G. Oddo, Atti Line (5), 10, II, 54 (1901); Gazz. chim. ital., 31, II,
146 (1901); G. Oddo, M. Tealdi, Gazz. chim. ital., 33, II, 427 (1903).
42. П. Вальден, Z. anorg. Chem., 68, 307 (1910).
43. E. Mole s, Z. phys. Chem., 90, 70 (1915).
44. L. Bruner, A. G a 1 e с k i, Z. phys. Chem., 84, 548 (1913).
45. E. Томас, Безводный хлористый алюминий в органической химии,
ИЛ, М., 1949, гл. 3.
46. В. Е. Л а ш к а р е в, Z. anorg. allg. Chem., 193, 270 (1930).
-47. Б. Ф. О р м о н т, Структура неорганических веществ, Госхимиздат,
М-, 1950, стр. 421—425.
48. Е. Beckmann, Z. phys. Chem., 46, 853 (1903).
49. A. Werner, Z. anorg. Chem., 15, 1, 24 (1897).
50. В. А. П л о т н и к о в, И. А. Ш е к а, 3. А. Я н к е л е в и ч, ЖОХ,
9, 368 (1939).
51. С. Sandonini, N. В е г g h е 1 1 о, Atti. Acad, dei Lincei, 25 (6),
46 (1937).
52. Я- А. Фиалков, К. Я- К а г а н с к а я, ЖОХ, 16, 1961 (1946).
53. Н. Grimm, A. Sommerfeld, Z. Phys., 36, 58 (1926).
176
54. L. Pauling, D. J о s t, Pr. nat. Acad. Washingt., 18, 415 (1932);
L- Pauling, J. Am. Chem. Soc, 54, 3570 (1932).
55- В. А. К и р е е в, Курс физической химии, Госхимиздат, М.-Л., 1951,
стр. 65.
56. L. Bruner, Е. В е k i e r, Z. Elektroch., 18, 368 (1912).
57. Я- Ф i а л к о в, Зап. 1н-ту xiM., 6, 223 (1940).
58. F. F a i r b г о t h e r, J. Chem. Soc, 847 (1936).
59. А. С. Christomanos, Ber., 10, 434, 782, 1007 (1870).
60. V. Gutmann, Z. anorg. allg. Chem., 264, 169 (1951).
61. Я. А. Фиал ков, К. Я- К а г а н с к а я, ЖОХ, 18,289(1948).
62. Я- А. Фиал ков, О. И. Шор, ЖОХ, 19, 1787(1949).
63. G. К- R о 1 1 е f s о n, F. С. L i n d q u i s t, J. Am. Chem. Soc, 52,
2793 (1930); 53, 1184 (1931); T. D. Iredale, D. P. Mellor, J. Am.
Chem., Soc, 53, 2802 (1931); W. D. В о n n e r, W. L. G о r e, D. M. J о s t,
J. Am. Chem. Soc, 57, 2723 (1935); A. S h e r m a n, N. Li, J. Am. Chem.
Soc, 58, 690 (1936).
64. L. E. Hofer, E. O. W i i g, J. Am. Chem. Soc, 67, 1441 (1945);
G. G. Palmer, E. O. W i i g, J. Am. Chem. Soc, 74, 2785 (1952).
65. W. Borne ma nn, Lieb. Ann., 184, 183 (1877).
66. Я• А. Ф и а л к о в, А. Б. П о л и щ у к, Зап. 1н-ту xiM., 8, 25 (1941).
67. П. В а л ь д е н, Z. phys. Chem., 43, 385 (1903).
68. Я- А. Фиал ко в, А. А. К у з ь м е н к о, ЖОХ, 19, 1645(1949).
69. Я- А. Ф и а л к о в, И. Л. А б а р б а р ч у к, Укр. хим. ж., 15, 116,
194 (1949).
70. И. Л. А б а р б а р ч у к, Укр. хим. ж., 19, 365 (1953).
71. Ф. Я- К У л ьба, Ж- прикл. х., 23, 339 (1950); ЖОХ, 24, 1700 (1954).
72.0. Ruff, Ber., 34, 1749 (1901).
73. R. М. К е е f e r, L. J. Andrews, J. Am. Chem. Soc, 75, 543 (1953).
74. P. Sc h u t ze n b e r ge r, Sengenwald, С. г., 54,197(1863);
J. prakt. Chem., 88, 1 (1863).
75. A. Michael, L. M. Norton, Ber., 11, 107 (1878).
76. W. H. Greene, С. г., 90, 41 (1880).
77. M. Simpson, Bull. Soc chim., 8, 349 (1867).
78. A. Michael, T . H, Norton, Ber., 9, 1752 (1876).
79. H. Mu 1 le r, Lieb. Ann., 133, 156 (1865).
80. В. M a p к о в н и к о в, Lieb. Ann., 153, 241 (1870).
81. F. Beilstein, P. Geitner, Lieb. Ann., 139, 333(1866).
82. B. 3 e p н о в, ЖРФХО, 32, 804 (1900).
83. L. J. Lam bourne, P. W. Robertson, J. Chem. Soc, 1167
(1947).
84. F. В e n n e t, A. Sharpe, J. Chem. Soc, 1383 (1950).
85. А. П. Терентьев, Л. И. Беленький, Л. А. Яновская,
ЖОХ, 24, 1265 (1954).
86. Н. Rheinbold, R. Boy, J. pr. Chem., 129, 273 (1931).
87. Я- К. Сыркин, К. М. Анисимова, ДАН, 59, 1457 (1948).
88. А. М. G a u d b h i r, A. L. F о u s e с a, F. R e b e 1 1 o, A. N. К о t-
h a r e, V. V. Nadkorny, Current Sci. (India), 19, 380 (1950).— цит.
no Ch. Abstr., 482 (1952).
89. Ученые записки Киевского института усовершенствования провизоров,
Госмедиздат УССР, 1950.
90. А. И. Г е н г р и н о в и ч, Ф. Е. Каган, Я- А. Ф и а л к о в,
Труды Комиссии по аналитической химии АН СССР, т. 5 (8), 237 (1954).
91. Т. R. Lewis, M. G. Pratt, E. D. Н о m i 1 1 е г, В. Т. Т и 1-
1 а г, S. А г с h е г, J. Am. Chem. Soc, 71, 3749 (1949).
92. D. Pope, H. В r i e ge r, E. Sc hwend, V. Peterson, J. Am.
Chem. Soc, 72, 4906 (1950).
•93. S. Archer, J. О. Н о p p e, T. R. Lewis, M. N. Haskell,
J. Am. Pharm. Ass., 40, 143 (1951).
94. M. Simpson, Lieb. Annal, 127, 372 (1863).
■95. Я- А. Фиалков, Методы исследования лекарственных веществ,
Медгиз, М., 1945, стр. 222.
12—1419
177
96. В. Сорокин, ЖРХО, 3, 192 (1871).
97. А. М i с h а е 1, J. pr. Chem. (2), 60, 448 (1899).
98. С h. К- In go Id, H. G. Smith, J. Chem. Soc, 2742 (1931).
99- А. В. Истомин, ЖРФХО, 36, 1199 (1904).
100. J. T. Barr, J. D. Guson, R. H. L a f f e г t y, J. Am. Chem
Soc, 73, 1352 (1951).
101. Т. И. Т е м н и к о в а, 3. А. Б а с к о в а, М. А. Хаимова
Сб. статей по общей химии, АН СССР, М.—Л., 1953, т. II, 874; М. А. X а*
и м о в а, ЖОХ, 25, 387 (1955).
102. J. A. Wijs, Z. ang. Chem., 11, 291 (1898).
103. Е. P. White, P. W. Robertson, J. Chem. Soc, 1509 (1939)
104. E. Erlenmeyer, Lieb. Annal., 289, 259 (1896).
105. J. Boseken, E. G e 1 b e r, Rec, 46, 158 (1927); 48, 377 (1929).
106. H. Ingle, J. Soc Chem. Ind., 21, 587 (1902); 23, 422 (1904).
107. А. Баландин, Z. phys. Chem., 116, 123 (1925).
108. L. 1 1 о s v a y, Chem. Ztg., 31, 609 (1907).
109. J. Km twig, Ber., 14, 304 (1881).
110. О. В r e n k e n, Ber., 8, 487 (1875).
111. E. В i r k, Z.' ang. Chem., 41, 751 (1928).
112. E. C. Trues dale, F. С Beyer, J. Am. Chem. Soc, 53, 164
(1931).
113. V. Thomas, P. Dupuis, С. г., 143,282(1906).
114. Неорганические синтезы, ИЛ., М., 1951, сб. 1, стр. 162.
115. G у Mann, Magyar, Kemiai Folyoirat, 57, 143, (1951) — цит. по
Chem. Zbl-, 349 (1953).
116. П. Me л и к о в, Вег., 8, 490 (1875).
117. N. P. Nies, D. M. Jost, J. Am. Chem. Soc, 57, 306 (1935).
118. A. E. Gillam, R. A. Morton, Proc Roy. Soc, A 124, 604 (1929).
119. G. О d do, Gaz. Chim. Hal., 31, II, 151 (1901).
120. E. Beckmann, F. Junker, Z. anorg. Chem., 55, 375 (1907).
121. H. Basset, E. Fielding, Chem. News, 54, 205 (1886).
122. A. Skrabal, F. Buchta, Chem. Ztg., 33, 1184 (1909).
123. Б. В. С о л д а т о в, С. А. Вознесенский, Сб. исследоват.
работ Акад. химич. защиты им. Ворошилова, 1940, стр. 21; Химический
рефер., ж. 4, № 4, 32 (1941).
124. V. С a g 1 i о t i, Atti Line (6), 9, 563 (1929).
125. Ф. E. Kara н, Диссертация, Московский фармацевтический институт,
1952.
126. R. W. E. Mclvor, Chem. News, 86, 6 (1902).
127. Е. D 6 г f u r t, E. Wolf, Z. anorg. allg. Chem., 185, 333 (1930).
128. А. Н.Несмеянов, P. X. Фрейдлина, Изв. АН СССР, ОХ Н,
152 (1943).
129. P. X. Фрейдлина, Э. М. Б р а й н и н а, А. Н.
Несмеянов, Изв. АН СССР, ОХН, 647 (1945).
130. Р. X. Фрейдлина, А. Н. Несмеянов, ДАН, 29, 566 (1929).
131. Э. М. Брайнина, Р. X. Фрейдлина, Изв. АН СССР, ОХН,
314 (1950).
132. Н. Stanley, Chem. N., 85, 134 (1902).
133. А. В е р н е р, Новые воззрения в области неорганической химии,
ОНТИ, Химтеоретиздат, Л , 1936, стр. 103.
134. P. S р а с ti, F. Р о р е a, Anal. Acad. Republ. populare Romane, III,
81 (1950); III, 396 (1950).
135. С W i 1 1 g e г о d t, J. pr. Chem., 33, 154 (1886).
136. В. Финкельштейн, Z. phys. Chem., 124, 285 (1926).
137. J. P h i 1 i p p, Ber., 3, 4 (1870).
138. M e 1 1 о r, Compr. treat, on inorg. and theoret. Chemistry, v. II, 121
(1927).
139. A. G о s s e 1 i n, M. G о s s e 1 i n, J. Chim. phys., 26, 350 (1929).
140. Я- А. Ф и а л к о в, ЖОХ, 18, 1741 (1948).
141. Е. Lenssen, J. Loewenthal, J. pr. Chem., 86, 219 (1862).
142. R. Lang, Z. anorg. Chem., 122, 332 (1922).
143. В. А. Плотников, В. Е. Рокотян, ЖРФХО, 47, 723 (1915).
178
t44 В. В. Некрасов, Курс общей химии, изд. 9, Госхимиздат, М.—Л.,
1 1952, стр. 824.
145. D. S. Payne, J. Chem. Soc, 1052 (1953).
146. Я- А. Фиалков, Я- Б. Бурьянов, ЖОХ, 26, 1003(1956).
147- Я-А. Фиалков, А. А. Кузьменко, Н. А. К о с т р о м и н а,
Укр. хим. ж., 21, 556 (1955).
148. А. А. Кузьменко, Я- А. Фиалков, ЖОХ, 19, 1007 (1949);
21, 473 (1951).
149. Я- А. Фиалков, Я- Б. Бурьянов, ДАН СССР, 102, 585 (1953);
ЖОХ, 25, 2391 (1955).
150. В. А. Плотников, С. И. Я к у 6 с о н, ЖРФХО, 60, 1505, 1513
(1928).
151. W. В i 1 t z, К- Jeep, Z. anorg. allg. Chem., 162, 32 (1927).
152. Я- А. Фиалков, А. А. Кузьменко, ЖОХ, 21, 433 (1951).
153. M. И. К а б а ч н и к, Усп. химии, 16 403 (1947).
154. К. Н. Анисимов, А. Н.Несмеянов, Изв. АН СССР, ОХ Н,
610 (1954); К- Н. Анисимов, Н. Е. Колобов, А. Н.
Несмеянов, Изв. АН СССР, ОХН, 799 (1954); К- Н. Анисимов,
Изв. АН СССР, ОХН, 803 (1954).
155. С. S. Forbes, S. W. Glass, R. M. F u о s s, J. Am. Chem. Soc,
47, 2892 (1925).
156. A. E. Bradfield, К- О г t о n, J. С h. Roberts, J. Chem.
Soc, 782 (1928).
157. B. Jones, E. N. Richardson, J. Chem. Soc, 713 (1953).
158. H. В a g a n z, Chem- Ber., 87, 1373 (1954).
159. F. N. Haves, H. K- Suzuki, D. E. Petersen, J. Am.
Chem. Soc, 72, 4524 (1950).
160. L. Brunei, Ann. chim. (8), 6, 284 (1905).
161. K. H. Boswijk, E. H. Wiebenga, Acta crystall., 7, 417
(1954).
162. J. H. Simons, G- Jessop, J. Am. Chem. Soc, 53, 1263 (1931).
163. С G. Vonk, K- H. Boswijk, E. H. Wiebenga, Rec, 74,
897 (1955).
164. H. Moureu, M. M a g a t, G. Wetroff, С. г., 203,257(1936);
205, 545 (1937).
165. К. Palmer, N. Elliott, J. Am. Chem. Soc, 60, 1852 (1938).
166. R. L. Harris, R. E. Wood, H. L. R i t t e r, J. Am. Chem.
Soc, 75, 3151 (1951).
167. H. Gerding, E. Smith, Z. phys. Chem., B, 50, 171 (1941).
168. W. В i 1 t z, A. V о i g t, Z. anorg. allg. Chem., 126, 39 (1923).
169. В А. Плотников, С. И. Я к у б с о н, Неводные растворы.
I. Таблицы электропроводности, Зап. 1н-ту xiM., 5, 271 (1938).
170. 3. А. Шека, Зап. 1н-ту xiM., 9, 193 (1947); Укр. хим. ж., 14, 112
(1948).
171.3. А. Шека, Э. И. Печеная, Укр. хим. ж., 15, 493 (1949); 16,
337 (1950).
172. Н. Н. Лебедев, ЖОХ, 21, 1788(1951); Усп. химии, 21, 1399(1952).
173.3. А. Шека, Э. И. Печеная, Укр. хим. ж., 21, 149 (1955).
174. Э. В. Б р и ц к е, А. Ф. К а п у с т и н с к и й, Термические
константы неорганических веществ, АН СССР, М.—Л., 1949.
175. Н. Evans, Т. R. Mun.son, D. D. W a g m a n, J. of Research
Nat. Bureau Standards, 55, 147 (1955).
176. F. A. P h i 1 b r i с k, J. Am. Chem. Soc, 56, 1257 (1934).
177. А. И. Г е н г р и н о в и ч, Н. Г. С и м х а е в, Медицин, пром., в. I,
48 (1957).
178. К. Н. Boswijk, J. H e i d e, A. V о s, E. H. Wiebenga,
Acta crystallogr, 9, 274 (1956).
ГЛАВА VII
БРОМИСТЫЙ ИОД JBr
Первое сообщение о соединении иода с бромом было
опубликовано Баларом [1] в 1826 г., в год открытия им же брома. Балар
и некоторые другие авторы [2, стр. 630] полагали, что наряду с
бромистым иодом простейшего состава JBr возможно
существование также высших бромидов. Это мнение высказывалось не
только в старой литературе (XIX в.), но и в позднейшие годы.
Так, Вальден [3, стр. 422] «а основании измерения
электропроводности смеси растворов иода и брома в треххлористом мышьяке
пришел к выводу о существовании JBr3 в растворе. Форбис и
Фауль [4] доказывали образование JBr3 (или его комплексного иона
JBr Г) результатами потенциометрического титрования иода (в
водных растворах НВг) бромом *.
Лабат полагал [5], что в водных растворах, содержащих иод
и бром в соотношении 1 :3, образуется JBr3, на основании того,
что сероуглерод или хлороформ из этих растворов не извлекают
ни иода, ни брома. Однако этот факт необходимо объяснить
окислением иода с образованием кислородных соединений.
Борнеман [6], еще в 1877 г. установил, что так называемый
трехбромистый иод представляет собой смесь бромистого иода и
брома. Такое же заключение сделали Джилам и Мортон [7] из
результатов изучения спектров поглощения брома, иода,
бромистого иода и смеси брома с бромистым иодом, растворенных в
четыреххлористом углероде. И. А. Шека [8], измеряя диоольный
момент бромистого иода в бромном растворе, сделал вывод о
невозможности существования высших бромидов иода в смеси
иод — бром. В качестве такого доказательства приводят также
тот факт, что неизвестны комплексные соединения, содержащие
анионы JBrT, в то время как JBr образует комплексные ионы
JBr Г [2, стр. 631], [9].
* Форбис и Фауль определяли изменение э. д. с, наблюдаемое при
прибавлении брома к исходному раствору, содержащему меньше трех молей брома
на один моль иода.
180
ФИЗИКО-ХИМИЧЕСКИЙ АНАЛИЗ СИСТЕМЫ ИОД — БРОМ
Меерум-Тервогт [10] определил температуры застывания рас-
плавленных смесей иода и брома в зависимости от соотношения
компонентов и изучил равновесие между твердой и жидкой
фазами. Оказалось, что начальные и конечные температуры
затвердевания и построенные на их основании кривые ликвидуса и соли-
дуса (рис. 29) совпадают в точке, соответствующей эквимоляр-
яому отношению брома и иода. Это указывает на то, что в
системе иод—бром при содержании 50 ат. % иода существует бромистый
иод, лишь сравнительно мало дис-
tc 1 социированный на свои
компоненты.
Его
40 60
ат. %
В/- 0 20 W 60 80 100 1
ат. %
Рис. 29. Температуры затвердевания Рис. 30. Температуры кипения и
системы иод—бром. состав жидкой и газообразной
фаз системы иод—бром.
Характер кривых дает основание считать, что бромистый иод
образует как с бромом, так и с иодом два ряда изоморфных
смешанных кристаллов.
Эти данные работы Тервогта недавно были подтверждены
Денисовой, исследовавшей систему бром—иод термографическим,
визуально-политермическими методами и химическим
анализом [55]. 1
Измерения удельного объема системы иод — бром [10] в
жидком и твердом состояниях при 0, 10, 42 и 50° показали, что
контракция системы увеличивается с ростом концентрации иода.
Наибольшее отклонение величин удельного объема от аддитивных
приходится на область концентраций, близкую к экви-
зйачений *
* Имеются в виду величины удельного объема, которые соответствуют
точкам на прямой, соединяющей на концентрационной диаграмме значения
удельного объема иода и брома.
181
атомным отношениям иода и брома, что может служить
доказательством образования бромистого иода в жидкой и твердо^
фазах.
Об образовании межгалоидного соединения в растворе иода в
броме свидетельствуют также обнаруженная Плотниковым и Ро-
котяном [11, 12] способность растворов иода в броме проводить
электрический ток и значительное увеличение электропроводности
системы иод—бром с увеличением концентрации иода (см.
главу IX).
Меерум-Тервогт [10] определил также температуры кипения
системы иод — бром и состав жидкой и газообразной фаз при
различных температурах (рис. 30) и установил, что содержание
иода в жидкой фазе всегда больше, чем в газообразной. Кривые,
характеризующие содержание иода в парах и в жидкой фазе,
немного сближаются в области эквиатомного отношения иода и
брома, что свидетельствует о значительной диссоциации бромистого
иода в газообразном состоянии при температурах опыта.
Существование бромистого иода в газообразном состоянии —
в равновесии с парами брома и иода — подтверждается также
измерением упругости пара [13] при содержании иода 32,5—
92,9 ат. % и температурах соответственно 19—100°,
уменьшением степени термической диссоциации иода на атомы при
добавлении брома [14], а также результатами измерения "равновесия
реакции J2 + Br2^2JBr (см. стр. 185).
Физико-химический анализ (по измерениям оптической
плотности) системы иод — бром в четыреххлористом углероде,
выполненный Лиличем [15], при суммарной концентрации 0,005 и
0,0105 моль/л доказал образование бромистого иода в
неполярном растворителе даже в таких разведенных растворах (см.
рис. 23). На основании кривых поглощения иода и бромистого
иода в ССЦ было найдено [16], что степень диссоциации
бромистого иода в этом растворе при 25° составляет 5,9±1,5%.
ПОЛУЧЕНИЕ
Бромистый иод получают непосредственным соединением обоих
галогенов. Для этого смесь эквивалентных количеств брома и иода
нагревают в тщательно закупоренном * или лучше в запаянном
сосуде до разжижения всей массы и затем охлаждают. Так как
при отвешивании брома возможна потеря некоторого количества
его, то Борнеман [6] предложил брать брома немного больше
эквивалентного количества и избыток его удалять током сухого
углекислого газа при температуре 50°.
Брунер и Бекьер [17] очищали бромистый иод от возможных
нелетучих примесей повторной перегонкой его над окисью
кальция и фосфорным ангидридом. Более рациональный метод
получения чистого бромистого иода предложил Гутман [18]. Смесь
* Применение корковых и резиновых пробок должно быть исключено-
182
эквивалентных количеств хорошо (измельченного иода и сухого
брома в круглодонной колбе нагревают в токе азота в течение
нескольких часов при 45°, расплавленную массу медленно
охлаждают, предохраняя ее от влаги воздуха, и после кристаллизации
большей части продукта сливают оставшуюся жидкость. Эту
операцию повторяют несколько раз до получения постоянной
величины удельной электропроводности, как критерия чистоты
препарата. Эта методика, хотя и гарантирует получение чистого
бромистого иода, несколько усложнена.
Свободный от примесей бромистый иод можно получать [19] в
приборе для определения температуры застывания при
термическом анализе [20], применяя предварительно тщательно
очищенные бром и иод. Бром берут в количестве, несколько большем
эквивалентного, и после расплавления всей смеси определяют в
этом же приборе температуру застывания; обычно она бывает
ниже 40°. Тогда в прибор вносят небольшие количества растертого
иода, слегка нагревают при 45° до растворения последнего и
скова определяют температуру застывания. Так повторяют два-
три раза до получения препарата с температурой застывания в
пределах 40—41°, что, по данным Меерум-Тервогта, соответствует
содержанию 50 ат.% иода в препарате.
Генней [21] приготовлял бромистый иод взаимодействием
хлористого иода и бромистой серы
' 2JC1 + S2Br2 = 2JBr + S,C12.
Однако этот метод значительно сложнее непосредственного
синтеза бромистого иода из элементов и затрудняет получение
чистого препарата.
В работе [22] исследована потенциометрически реакция
между бромом и йодистым калием в водных растворах и
обнаружено два резких перегиба, соответствующих образованию иода и
бромистого иода. Так как бромистый иод очень легко гидроли-
зуется, то получение более или менее устойчивых водных
растворов его возможно лишь в кислой среде. В бромистоводородной
кислоте бромистый иод образует комплексную кислоту ШВгг,
которая, как и аналогичная кислота ШСЬ, устойчива в водных
растворах при достаточной концентрации галоидоводородной кислоты.
Водные растворы бромистого иода, вернее растворы ШВг2, можно
получать, действуя окислителями на иод или иодиды или
восстановителями на иодаты — в обоих случаях в присутствии бромидов и
в кислой среде [2, с~р. 635]. Так, Б. Сингх и А. Сингх [23]
показали, что если к водному раствору различных восстановителей
(J", As203, Sb203> Sn2+, Hg!+, SO|"f Fe2+, SCN~, N2H4 и др.)
прибавить концентрированную соляную кислоту, КВг и СС14 и затем
титровать этот раствор раствором KJ04, то точка эквивалентности
соответствует образованию JBr~, например,
3 J- + JO- + 8Н+ + 8Вг- = 4JBr- + 4Н20;
183
3As203 + 2J0- + 4H+ + 4Br- = 3As205+2JBi^- + 2H20.
Для получения водных растворов HJBr2 можно
воспользоваться первой из этих реакций.
ФИЗИЧЕСКИЕ СВОЙСТВА
Бромистый иод — кристаллическое вещество темно-серого
цвета, в проходящем свете — красного [6], по внешнему виду
напоминает иод, при температуре плавления переходит в
красно-бурую жидкость, обладающую резким запахом брома. Медленной
возгонкой бромистого
иода можно получить
кристаллы длиной 2 см.
Бромистый иод
плавится при 40—41° [10]*
или 42° [24]. Для
температуры кипения в
литературе указаны величины:
выше 100° [6], 116° [251 и
119° [18]. Точное
определение этой величины
оказалось невозможным
вследствие термической
диссоциации бромистого
иода не только в
газообразной, но и в жидкой фазе. Так, на кривой нагревания
бромистого иода, по данным Шор** (рис. 31), наблюдается
эндотермический эффект, начало которого ~~ 40° — почти совпадает с
температурой плавления бромистого иода, а минимум приходится на
50°. Некоторая растянутость эффекта может быть объяснена
наложением второго эндотермического эффекта, соответствующего
термической диссоциации бромистого иода.
По данным Меерум-Тервогта, упругость пара бромистого иода
достигает 1 атм при 116°, но при этом состав газовой фазы
(17 ат.% иода) резко отличается от состава жидкой фазы.
Эквивалентное отношение иода и брома в газовой фазе (50 ат.% иода)
достигается лишь при 159—160°.
Плотность бромистого иода: твердого d°4 4,4157; d\° 4,4135; жидкого
df 3,7616; df 3,7343 [10].
Удельная теплоемкость газообразного бромистого иода была
определена Штрекером [26] по скорости звука в газе в интервале от 20
до 100° : Ср = 0,039, a Cv -0,029 кал-ггад-1-моль~\ CP,CV = 1,34.
Как и для хлористого иода (см. табл. 25), величина Cp/Cv оказалась
Рис. 31. Кривые нагревания бромистого
иода.
* По данным Меерум-Тервогта, этот температурный интервал
представляет температуру застывания бромистого кода. >
** Из диссертации О. И. Шор «Физико-химическое исследование систем
галогениды иода — галогениды калия или аммония», выполненной под
руководством автора, ИОНХ АН УССР, К., 1951.
184
,еньше тех, которые характерны для двухатоуных молекул газов,
JfTo свидетельствует о значительном отклонении этих галогенидов иода
оТ „идеальною" состояния. Кр^е -ого, необходимо принять во
внимание термическую диссоциацию бромисою иода.
Предполагая, что в жидком бромистом иоде существует равновесие
[27]
2JBr^J2 + Br2, (7.1)
jVleepyM-Тервогт [10] приближенно рассчитал, что при 50,2°
константа равновесия для жидкого бромистого иода равна 0,014, а
степень диссоциации составляет 20%. Константа термической
диссоциации газообразного бромистого иода оказалась в 7,5 раз
больше таковой в жидкой фазе.
Несмотря на значительную термическую неустойчивость
бромистого иода, равновесие его диссоциации обнаружено и
исследовано в газообразной фазе даже при сравнительно высоких
температурах. По данным Мюллера [28] *, константа равновесия при
304,8° (577,9° К) КР = [J2][Br2]74[JBr]2 = 0,013 **, а степень
диссоциации равна 20%. Боденштейн и Шмидт [14] на основании своих
тензиметрических измерений вычислили Кр = [J2}[Br2]/4[JBr]2 при
1222° (1495°К) и нашли ее равной 0,093***, что, по их мнению,
согласуется с /СР, найденной Мюллером для 304,8°. Однако Грин-
ВУД [29] отмечает, что данные Боденштейна и Шмидта вполне
согласуются с величиной, определенной спектроскопически при той
же температуре (см. табл. 29), а значение Кр ,полученное
Мюллером, меньше величины, рассчитанной из спектроскопических
измерений для 300°
У^ = 0,149=(6,70)-1.
Мак-Моррис и Иост [30], исследуя реакцию между иодом и
системой из СиВг и СиВг2 в интервале от 115 до 176°, рассчитали
величины константы равновесия. Значения /Ср для более высоких
температур отклонялись от спектроскопических значений,
приведенных в табл. 29 по данным Цейзе [31], для реакции
lj2 + ^Br2 = JBr. (7.2)
Межъядерное расстояние для бромистого иода было принято
равным средней величине межъядерных расстояний в броме и иоде.
Наиболее вороятными значениями степени термической
диссоциации газообразного бромистого иода следует считать величины,
вычисленные из константы равновесия для реакции (7,2) Иостом
с сотрудниками [32]: при 25° степень диссоциации равна 8,8%, при
100° она составляет 13,4%, а для растворов бромистого иода в
* Эти данные основаны на изучении тормозящего влияния иода на
реакцию Н2+Вг2«_2НВг, обусловленного образованием бромистого иода.
** ]/Ж~р- 0,114 или (8.77Г1.
*** \ZKV = 0,305 или (3,48)_1.
185
четыреххлористом углероде при 25° равна 9,5% (см. такжр
стр. 182) *.
Таблица 29
Константы равновесия бромистого иода Кр = ^JBr/V^j *^Вг
Температура °К 298,1 400 600 800 1000 1200 1400 1600 1800 2000
/Ср, % 20,66 11,42 6,37 4,80 3,99 3,58 3,29 3,12 2,98 2,84
Теплота образования и термодинамические
константы бромистого иода
Бертло [33], принимая во внимание теплоту возгонки бромистого
иода, равную 11,87 ккал-моль-1 **, определил теплоту образования
JBr по реакции
-yJ2 (тв)+ "о-ВГ2(ж) = JBr(TB)
и нашел ее равной 2,47 ккал-моль-1 ***. Такую же величину теплоты
образования JBr (тв) рассчитали Мак-Моррис и Иост [3j] из данных
по равновесию. Эти же авторы для реакции -^ h(Ti )+~к Вг2(Ж) =
= JBr(r) нашли значение AHf JBr(r)=9,96 ккал-моль-1.
Наибольшее количество данных имеется для теплоты образования
газообразного бромистого иода из газообразных брома и иода по
реакции
~2 J2 (г) + -g- ВГ2 (г) = JBr(r).
Мак-Морис и Иост [30] из термохимических исследований (при
'25°) получили величину 1,27±0,05 ккал. Близкие значения дали
спектроскопические исследования [34, 35, 49, 51]. Из анализа
спектров поглощения газообразного бромистого иода найдена теплота
диссоциации JBr на нормальные атомы иода и брома, равная
41,4±0,7 [51] или 41,89 [29] ккал • моль"1. Из этих данных и из
величин теплот диссоциации исходных галогенов на атомы
рассчитана теплота образования JBr(r 1,38—1,37 ккал-моль'1.
Такую же теплоту образования JBr(r) определил и Цейзе [31],
исходя из энергии диссоциации бромистого иода ^o(JBr) 1,81 эв или
4,17 ккал-моль"1, рассчитанной им на основании данных
Брауна [49].
Из результатов кинетических исследований — определения
константы равновесия — Бодвдштейн и Шмидт [14] и Мюллер [28]
рассчитали по приближенной Формуле Нернста теплоту образова-
* Эти же авторы показали, что упругость пара бромистого иода над
«го^раствором в четыреххлористом углероде соответствует закону Генри.
** По более новым данным [30], теплота возгонки твердого бромистого
иода равна 12,433—12,545 ккал-моль—1-
*** Теплота образования ЛВг(ТВ) из твердых иода и брома, по Бертло [33],
равна 2,34 ккал-моль—1-
186
ei
и
CO
5<
tuo
£ ^
^
о
*
*
1 ""■
1
p ^
< 4
Q
*
g
x-
7 1
1 л
„ ^ «S
<= о, а о
<^ &*
• x
"*
о
*
^^ 1
* 1
1 3
о ~
a: §
«
x_
~7 1
! о
со §_:*
? x
^
a
*
^-X7
^~ *
00 ^ че
• ? X
£_ ^
^ S
^ «
fcC x„
1 ^^" T I
i o"o *
1 ? X
\ ? ^
^- a
| w *
^
■1
1
CM
^
1
I
CM
rh
1
0
0
0
0
0
0
1
00
CO
LO
CO
t^
|
1
CM
T^
1
CM
CO
00"
CM
^
Ю
CD
CO
0
CO
0
CO
Ю
0
00
Is-"
, 1
0
10
CM
Ю
1
1
О
Ю
CM
cd
CO
CO
Tf
CO
cd
t>^
1
\
0
CM
Tf
|
00
CO
00
t-
^J4
Ю
CM
CO
Is-
О
CO
Is-
Is-
00^
Is-"
CO
cd
CO
ю
1
1
CO
CO"
Is-
см
t-
ri«
CO
О
CO
00
1
1
CM
CM
Tt^
|
CM
t^
00"
CM
cd"
CO
CO
CM
Ю
CO
00
CO
CO
^
0>
r>*
CD
00
00
CO*
1
1
CO
ex)
CD
CM
Ю
CO
CO
CO
CO
°q.
[
1
CM
CM
^
|
CM
Is-
00
CO
LO"
00
CO
см
cd
00
00
CO
T—1
LO
°i
l>"
00
CO
°1
CO
LO
1
I
О
О
CO
CO
Is-
О
Is-
°l
1
I
CM
^h
|
-*
00
00"
-*
rh"
CO
см
CO
Is-
r—(
Tf
3
*—1
CO
*~1
00
CO
LO
CM
со"
LO
1
1
0
0
TF
CM
CD
0
,—1
—!
CM
1
CM
r^
I
0
CD
00
0
см"
LO
^
Is-
CD
CO
CO
CO
^
0
°°,
00
CO
CT>
00*
LO
1
1
0
0
LO
00
00
0
«*
CM
cm'
1
1
CM
-*
1
Tj«
CD
00*
CM
Tt
^
0
LO
-*
CM
О
00
CO
Is-
О
t£
OO*"
Is-
cq
CD
LO
I
1
0
0
CO
CO
^
Is-
0
00
CO
cm"
1
1
CM
Ч*1
1
Is-
°l
00"
CM
0"
Tf
CD
LO
tJ«
О
■*
CD
CO
CO
OO
"<*
00"
00
1—(
CD
cd"
CO
1
I
0
0
Is-
00
00
CO
0
CM
Ю
cm"
1
1
CM
^
1
CD
CD
00
-*
OO"
CO
CD
CO
•*
О
CO
0
r-
00
**
LO
OO"
CO
LO
CD
CM*
CO
1
О
О
00
^h
4*
CO
0
CO
CO^
cm"
1
1
CM
Tt>
|
.—1
CD
cd"
•^
cd"
CO
1^
t^.
LO
CO
CO
t^-
CD
CD
LO
00*
CO
CO
О
CO*
CO
1
О
О
CD
О
CO
О
CD
tr^
CM
1
1
CM
"*!
|
CO
CD
CD
О
^
-*
CO
00
LO
CO
CM
t--
r—(
-*
CO
00"
^h
Is-
CD
CO"
CO
1
1
О
О
О
CM
00
LO
0
CO
CD
cm"
1
1
CM
"^
1
Ю
CD^
cd"
t^
Tt*
-*
LO
CD
CO
b-
"*
CO
l>
^
t-
CO^
00"
CD
CD
CO
I
1
О
О
^
CD
LO
LO
0
Is-
0
CO
I
1
CM
4.
1
CO
0
CD*
CO^
cd"
Tj«
-*
0
'—
^
CO
CM
^
t--
00
0
l^
00
CO
LO
Ю
LO"
CO
1
1
0
0
CM
00
CO
LO
0
0
°i
со"
1
1
CM
^1
1
o-
0
CD
00^
со"
LO
CO
'—'
0
CD
CD
Tj*
Г-
CO
CO
t^
00"
T^
10
CM
CO"
CO
1
0
0
CO
CM
Ю
0"
-*
CO
CO
1
1
CM
^1
1
CD
0
CD*
"*
LO"
CO
CM
CM
—'
CO
CO
CO
LO
Is-
^—1
CO
!>;
00"
CM
0
CD
CO*
CO
1
1
0
0
rf
Is-
0
LO
0
00
"*
со"
1
CO
^
1
0
G>
Ю
r^
CO
'—'
0
CD
CM
CO
r-
C0
00
Is-
OO"
r-
0
LO
l>"
CO
1
1
0
0
LO
187
ния JBr (Г) соответственно 1,84 (для 1495° К) и 2,49 (дЛ5Г
577,8° К) ккал - моль-1.
Эванс, Манзон и Вагман [50] рассчитали теплоту образования
бромистого ' иода из данных двух последних авторов равной
—1,48 и —1,73 ккал - моль*1. Исходя из данных Мак-Морриса
Иоста [30] и Брауна [49], они получили AHf° =—1,42, или —1,41 V
±0,06 ккал-моль~\ Последнее значение они считают наиболее
точным (см. табл. 30) *.
Теплота образования бромистого иода (а также хлористого иода
и хлористого брома) из элементов в четыреххлористом углероде как
растворителе, например,
-i J2(b CC14) + -i С12(в СС14) = JC1 (вСС14),
по данным Блейра и Иоста [35], равна: для JC1 3970 ± 30, для JBf
1630 ± 30, для BrCl 378 ± 10 кал-моль-1.
Таким образом, по величине теплоты образования бромистый
иод в ряду этих межгалоидных соединений (занимает среднее
место — впереди хлористого брома. Такое же положение занимает
бромистый иод в отношении степени диссоциации в
индивидуальном состоянии ив растворах четыреххлористого углерода.
Термодинамические константы (стр. 25) бромистого иода, по
[50], приведены в табл. 30 **.
Изменение энтропии в реакции образования JBr(r) по
данным [30], AS298,i6= 1,75, а из спектроскопических данных —
1,37 кал-град~ -моль~\
Таблица 31
Стандартные абсолютные энтропии S298 ]6 галогенов и бинарных межгалоидных
соединений (кал-град~х -моль~~1)
F2
45,48
C1F
52,06
С12
53,29
BrF***
54,7
BrCl
57,39
Вг2
58,65
JF
56,43
JC1
59,15
JBr
61,84
J2
62,28
* В отличие от этих значений теплоты образования бромистого иода,
Биховский и Россини [52] указывают, что из данных Боденштейна и Шмидта
[14] и Мюллера [28] для равновесия "у ^2(г)^Т^г(2(г))^-*^г(г) ими была Рас"
считана теплота образования—9,6 ккал. Они же отмечают, что
спектроскопические данные Кордеса и Спонера [51] для энергии диссоциации JBr(r) на
нормальные атомы иода и брома приводят к теплоте образования бромистого
иода, равной 10,3 ккал. Расчетные данные не указаны. Такие же величины
теплоты образования бромистого иода приведены в книге Брицке и Капустин-
ского с сотрудниками [53].
** Некоторые термодинамические константы бромистого иода для
растворов в СС14 приведены в работе Блейра и Иоста [35].
*** Гринвуд [29] указал, что величины энтропии таких межгалогенидов
несколько превышают среднее значение для исходных галогенов. Он
рассчитал, что абсолютная энтропия фтористого брома должна быть <*** 54,9 кал X
Хград~1 • моль" *.
188
Значения энтропии газообразных JBr, JC1 и BrCl находятся
между значениями энтропии для соответствующих галогенов. Это
положение можно распространить и на другие двухатомные меж-
галогениды, что показано в табл. 31.
Молекулярное состояние и структура молекул
По вопросу о молекулярном состоянии бромистого иода и
структуре его молекул можно высказать те же соображения, что
приведены при описании хлористого иода. Константа Трутона для
бромистого иода не установлена вследствие его значительной
термической диссоциации. Электрохимические свойства бромистого
иода в расплавленном состоянии и в растворах (глава IX), его
поведение как растворителя и криоскопические исследования
(глава X) дают основание предположить, что бромистый иод, как и
хлористый иод, ассоциирован в жидком состоянии и в ряде
растворов.
Соображения, которые приводят Гримм и Зоммерфельд, а
также Паулинг и Иост в пользу неполярного характера связи
в хлористом иоде, относятся также к бромистому иоду. Однако
наличие хотя и небольшого дипольного момента (см. табл. 4),
способность бромистого иода проводить ток * и его поведение при
химических реакциях свидетельствуют о том, что связь J—Br в
некоторой степени полярна, хотя, вероятно, в меньшей мере, чем
связь J—С1, вследствие меньшего различия в величинах
электроотрицательности иода и брома.
ХИМИЧЕСКИЕ СВОЙСТВА
По химическим свойствам бромистый иод во многом
напоминает хлористый иод, но его активность несколько меньше. Следует,
однако, отметить, что химические свойства бромистого иода
изучены мало. Так, например, в т. 8 справочного руководства Гмелина
[2], где собраны литературные данные до июня 1933 г.,
сообщается о действии бромистого иода лишь на воду, водород
(образуется смесь бромистого и йодистого водорода [28]), бор,
серебро, йодистое серебро и бертолетову соль. Систематические
исследования химических реакций бромистого иода с
неорганическими веществами начаты лишь недавно [36, 37].
Р е а к ц и и с простыми веществами
Гутман [37] исследовал взаимодействие 39 элементов ** с
бромистым иодом, взятым в значительном избытке. Реакции
проводились сперва на холоду ***, а под конец при нагревании с об-
* Удельная электропроводность жидкого бромистого иода пооядк
10—4 ом—1 см— 1 (см. главу IX).
** Были взяты те же элементы, что и при изучении реакции с хлористым
иодом [38].
*** В случае бурно протекающих реакций—с калием, алюминием, оловом
и белым фосфором — необходимо было охлаждение.
189
ратным холодильником. Избыток бромистого иода и выделившийся
иод удалялись четыреххлористым углеродом. По данным этого
автора, бромистый иод по химической активности занимает сред,
нее положение между -иодом и хлористым иодом. Продуктами
реакции являются бромиды соответствующих элементов, в ряде
случаев в высших степенях валентности (CuBr2, AuBr3, HgBr2,
SnBr4, PBr5, TeBr4, FeBr3).
Наиболее активно (~ 100% выхода) реагировали калий, медь,
золото, ртуть, алюминий, олово, титан, фосфор белый и красный,
мышьяк, сурьма, висмут, сера, селен, теллур, железо; с выходом
в 10—50% реагировали серебро, кальций, барий, цинк, германий,
ванадий, кобальт, никель; очень слабо реагировали натрий,
магний, индий, марганец. Вовсе не реагировали с бромистым иодом
кадмий, бор, активный уголь, аморфный кремний, свинец,
цирконий, ниобий, тантал, хром, молибден, вольфрам, платина.
Реакции элементов с бромистым иодом Гутман выражает следующей
схемой:
ttJBr+M = MBrn+-|j2.
Таким образом, и в этом случае образуются соединения
элементов с наиболее электроотрицательным галогеном.
Бромистый иод в реакциях с металлами и металлоидами
проявляет много общего с хлористым иодом, но он реагирует несколько
менее активно. В ряде случаев реакции с хлористым иодом
протекают с большим выходом и более энергично. Обращает на себя
внимание весьма различное отношение некоторых близких по
химическим свойствам элементов к хлористому и бромистому иоду.
Подобные примеры приведены в табл. 32.
Таблица 32
Реакции некоторых металлов с хлористым и бромистым иодом [37]
Металл
Na (кусочки;
К
Си (порошок)
£g
Аи (осажденное)
Zn (порошок)
Cd (зерна)
Hg (жидкая)
Sn (порошок)
Pb (зерна)
Ti (порошок)
Zr
Продукт
реакции
NaCl
KJC12
CuClo
AgCl
AUC13
ZnCl2
HgCl2
SnCl4
He pea
TiCl4 |
He pea
Хлористый иод
Выход,
%
2-6
- 100
45
55
100
50
He реап
100
100
гирует
100
гирует
Характер
реакции
При
незначительном
нагревании
Со взрывом
Энергично
Со вспышкой
При нагревании
Бурно
<грует
При нагревании
Очень бурно
Очень бурно
Бромистый иод
Продукт Вы-
реакции ход, %
NaBr
КВг+
+KJBr2
CuBr2
AgBr
AuBr3
ZnBr2
I
100
98
35
100
10
Характер
реакции
При
незначительном
нагревании
Со взрывом
Спокойна
Энергично
Спокойно
м
He реагирует
HgBr2
SnBr4
TiBr4 (?)
100 1 Энергично
100 1 Бурно
He реагирует
100 | Энергично
He
реагирует
190
Возможность использовать реакции бромистого иода с
элементами для получения галогенидов последних менее
благоприятна, чем в случае хлоридов иода, и особенно фторидов хлора, бро-
lVja и иода. Причины этого таковы: меньшая химическая активность
бромистого иода, трудность отделения многих бромидов от
избытка бромистого иода и от большого количества одновременно
образующегося иода (и других побочных 'продуктов реакции).
Поэтому Гутман [37] считает возможным использовать для
препаративных целей (из изученных им реакций) лишь реакции
получения таких бромидов: HgBr2, BiBr3, TeBr4, FeBr3.
Реакции с неорганическими соединениями
Описанные в литературе реакции бромистого иода с
неорганическими соединениями относятся почти исключительно к
процессам окисления и комплексообразования *.
Бромистый иод медленно разлагается водой с постепенным
выделением иода. Гидролиз бромистого иода происходит,
вероятно, по схеме, аналогичной с реакцией гидролиза хлористого иода:
JX + НОН Z НХ + HOJ (X = С1 или Вг);
5НО J = Н J03 + 2 J2 + 2Н20.
Аналогичное явление наблюдается и при изучении влияния
ионов Н+ и ОН~ на процесс гидролиза. С водными растворами
щелочей, взятыми в достаточном избытке, бромистый иод
реагирует по уравнению
3JBr + 6KOH = ЗКВг + 2KJ + KJ03 + ЗН20.
Химические свойства бромистого иода как окислителя в
реакциях с неорганическими соединениями еще очень мало
исследованы. Описаны лишь единичные реакции. С иодидами металлов
бромистый иод в расплавленном состоянии или в растворах
реагирует быстро с выделением иода [17]:
JBr + KJ = KBr + J2;
JBr-l-AgJ = AgBr+ J2.
С синильной кислотой бромистый иод (в растворах бромисто-
водородной кислоты) реагирует так же, как и хлористый иод (в
соляной кислоте)
JX + HCN = JCN 4- НХ (X = С1 или Вг).
Но в случае бромистого иода одновременно, хотя и в
значительно меньшей степени, происходят реакции, приводящие в
конечном итоге к выделению иода [39]:
2JBr+ 2HCN = JCN + HJ + HBr+ BrCN;
* Реакции комплексообразования с участием бромистого иода описаны
в главах XI и XII.
19]
BrCN + 2HJ = J2 + HCN + HBr;
JCN + HJ = J2+HCN.
Этот же раствор бромистого иода окисляет гидразин {39]
4JBr + N2H4 = 2J2 + 4Br~ + 4H+ + N2.
Бромистый иод в расплавленно \ состоянии и в неводных растворах
легко окисляет тр.>хбр шистый фосфор до пятибромисого и затем
соединяется с последним в комплекс PBr6J или PBr+JBr" [40,41]:
PBr3 + 2JBr^PBr5 + J2;
PBr5 + JBr = PBr6J.
Реакции с органическими соединениями
1. Реакции галоидирования (замещения водорода).
Способность бромистого иода замещать атомы водорода в органических
соединениях была замечена при определении йодного числа жиров
и масел по методу Хануса раствором бромистого иода в уксусной
кислоте.
В отличие от хлористого иода, бромистый иод оказался лишь
Хромирующим средством. Это явление наблюдалось не только в
неполярном растворителе — четыреххлористом углероде, но и в
полярных растворителях — уксусной кислоте и нитробензоле.
Реакция бромирования происходит по схеме
RH-f JBr=RBr+HJ; (7.3)
HJ+JBr = HBr + J2; (7.4)
RH + 2JBr = RBr + HBr + J2. (7.5)
Так, по данным Милитцера [42], фенол быстро реагирует с
бромистым иодом (в СС14), образуя с хорошим выходом я-бром-
фенол. При реакции с анилином в том же растворителе
осаждается- и иодистоводородная соль /г-броманилина; из 1-нафтола (в
уксусной кислоте) образуется 4-бром-1-нафтол; нафталин (в ССЦ)
превращается в 1-бромнафталин. По Беннету и Шарпу [43], фенол
и салициловая кислота в нитробензольном растворе быстро бро-
мируются бромистым иодом, одновременно выделяется большое
количество иода.
Недавно Б. Сингх и М. Сингх [54] показали возможность
количественного определения ряда фенолов и аминов (и нафталина)
путем потенциометрического титрования бромистым иодом в
уксусной кислоте. В этих условиях фенол, гидрохинон, резорцин,
пирогаллол, флороглюцин, |3-нафтол, о- и я-крезол, анилин,
^-толуидин, антраниловая кислота, я-аминобензойная кислота
превращаются в трибромзамещенные; тимол, а-нафтол, о- и я-ни-
трофенол, о- и я-толуидин дают дибромзамещенные; а-нафтил-
амин, ацетанилид и нафталин дают однозамещенные; салициловая
кислота превращается в трибромфенол.
192
Хотя такие факты и немногочисленны, они требуют
объяснения того, почему в растворах бромистого иода, молекулы которого
по своей структуре и полярности мало отличаются от хлористого
иода, не наблюдалась «конкуренция» разных направлений
процесса галоидирования — иодирования и бромирования — и их
зависимость от природы растворителя, как это известно для
хлористого иода. Предположение о том, что бромистый иод в реакциях
замещения реагирует в форме J~Br+, нельзя считать обоснованным.
Ламбурн и Робертсон [44], а затем Беннет и Шарп [43]
предложили следующее объяснение: бромистый иод значительно
сильнее подвергается гомолитической (термической) диссоциации на
молекулы галогенов, чем хлористый иод (см. стр. 149);
бронирование же фенола и аналогичных соединений бромом протекает
значительно быстрее, чем иодирование электроположительным
иодом, вследствие чего равновесие диссоциации бромистого иода
настолько быстро смещается вправо, что практически происходит
только бромирование, причем бромистый иод [44] или иод [43]
каталитически ускоряет этот процесс
J++ Br-^ JBr «=± -I J2 + 1 Вг2.
Пирсон и Росс [45] выдвинули другое объяснение, согласно
которому при взаимодействии фенола с бромистым иодом в
первую очередь происходит иодирование, а образовавшийся я-иод-
фенол затем быстро реагирует с бромистым водородом
СбН5ОН + JBr = JC6H4OH + НВг; (7.6)
2JC6H4OH + НВг = ВгСбН4ОН + С6Н5ОН + Ja. (7.7)
Эти авторы показали, что реакция (7.7) в ССЦ заканчивается
через 85 минут при 45° или через 8—10 часов при комнатной
температуре. При таких же условиях протекает описанная Ми-
литцером [42] реакция бромирования 1-нафтола бромистым иодом
в ССЦ. Однако Шарп в более поздней работе [46] отвергает это
объяснение на том основании, что реакция между фенолом и
бромистым иодом в ССЦ происходит значительно быстрее — при
комнатной температуре уже через 10 минут половинное
количество брома (но не иода) вступает в ароматическое кольцо.
Поэтому он вновь подтвердил объяснение этой реакции, основанное на
представлении о термической диссоциации бромистого иода.
В то же время Шарп показал, что в смеси растворов анилина
и избытка бромистого иода в ССЦ наряду с очень быстрым бро-
мированием происходит и более медленное иодирование анилина,
что дало ему основание представить эту реакцию следующим .
уравнением:
2C6H5NH2 + 2JBr = C6H4BrNH2 + C6H4JNH2 + НВг + HJ.
Таким образом, вопрос о механизме галоидирования фенолов,
аминов и других органических соединений нельзя считать
разрешенным (см. главу VI).
13—1419
193
2. Реакции 'Присоединения по кратной связи. Способность
бромистого иода присоединяться к непредельным органиче.
ским соединениям с двойной связью используется при опр^
делении йодного числа жиров и жирных масел. Для этой цели
обычно применяют предложенный в 1901 г. Ханусом [47] раствор
бромистого иода в ледяной уксусной кислоте *, которым действуют
на жиры и масла, растворенные в хлороформе или четыреххлори-
стом углероде. При этом происходит присоединение бромистого
иода к непредельным жирным кислотам (по месту двойных
связей) и превращение последних в иодбромпроизводные. Так, по
данным Хольде и Горгаса [48], из эрукоозой кислоты С21Н41СООН
при действии реактивом Хануса была получена иодбромбегено-
вая кислота C2iH4iJBrCOOH, а из линолевой Ci7H3iCOOH —
дииоддибромстеариновая кислота СпНзьЬВггСООН.
Метод Хануса нашел широкое применение, и в ряде стран он
является стандартным при анализе жиров и аналогичных
продуктов. Что же касается реакций бромистого иода с другими
непредельными органическими соединениями (кроме жирных кислот
и их эфиров), то литературные данные по этому вопросу весьма
скудны.
ЛИТЕРАТУРА
1. A. J. В а 1 а г d, Ann. chim. phys., 32, 337 (1826).
2. Gmelin's Handbuch der anorg. Chem., 8, (1931).
3. П. В а л ь д е н, Z. phys. Chem., 43, 385 (1903).
4. G. S. Fo г bes, J. H. F a u 1 1, J. Am. Chem. Soc, 55, 1820 (1933).
5. A. Lab at, Bull. Soc. Chim. (4), 9, 503 (1911).
6. W. Bornemann, Ann., 189, 202 (1877).
7. A. E. G i 1 1 a m, R. A. Morton, Proc. Roy. Soc, A 124, 607 (1929).
8. И. А. Ш е к а, ЖФХ, 23, 885 (1949).
9. T h. H. R e a d e, J. Chem. Soc, 125, 148 (1924).
10. P. С E. M e e г u m - T e г w о g t, Z. anorg. Chem., 47, 209 (1905).
11. В. А. Плотников, ЖРФХО, 35, 794 (1903).
12. В. А. П л о т н и к о в, В. Е. Р о к о т я н, ЖРФХО, 45, 193 (1913).
13. Н. R. К г u g t, W. D. H e 1 d е г m a n, Z. phys. Chem., 93, 89 (1919).
14. M. В о d e n s t e i n, A. S с h m i d t, Z. phys. Chem., 123, 28 (1926).
15. С. Л. Л и л и ч, ЖОХ, 22, 730 (1952).
16. A. J. P о р о v, К- С. В г i n k е г, L. С a m p a n а г о, R. W. R i n-
chart, J. Am. Chem. Soc, 73, 514 (1951).
17. L. Bruner, E. В е к i e r, Z. Elektroch., 18, 368(1912).
18. V. Gutmann, Monatsh., 82, 156 (1951).
19. Я- А. Ф1алков, Зап. 1н-ту xiM., 6, 223 (1940).
20. Я- А. Ф и а л к о в, А. А. К у з ь м е н к о, ЖОХ, 6, 265 (1936).
21. J. H anna у, J. Chem. Soc, 26, 815 (1873).
22. Н. Т. S. В г i t t о n, H. G. В г i t t о n, J. Chem. Soc, 3879 (1952).
23. B. S i n g h, A. S i n g h, Analyt. Chim. Acta, 9, 1, 22 (1953).
24. A. E. G i 1 1 a m, Trans. Faraday Soc, 29, 1132 (1933).
25. N. V. S i d g w i с k, The Chemical Elements and their Compounds,
Oxford, 1950.
26. K. Strecker, Wied. Ann., 17, 93 (1882).
27. J. Laar, Z. phys. Chem., 47, 129 (1904).
* Этот реактив получают, растворяя эквивалентные количества иода и
брома в уксусной кислоте.
194
og. W. Miiller, Z. phys. Chem., 123, 1 (1926).
од. Н. H. Гринвуд, Усп. химии, 22, 445 (1953).
«О. J- McMorris, D. M. J о s t, J. Am. Chem. Soc, 53, 2625 (1931).
31. H. Zeise, Z. Elektrch., 41, 267 (1935).
32. D. M. J о s t, T. F. A n d e r s о n, F. S к о о g, J. Am. Chem. Soc,
55, 552 (1933).
33. M. Berthelot, Ст., 90, 841 (1880).
34. R. M. В a d g a r, D. M. J о s t, Phys. Rev.. 37, 1548 (1931).
35. С h. M. В 1 a i r, D. M. J о s t, J. Am. Chem. Soc, 55, 4489 (1933).
36. V. G u t m a n n, Mnnatsh., 82, 156 (1951).
37. V. G u t m a n n, M( natsh., 82, 280 (1951).
38. V. Gutman n, Z. an rg. allg. Chem., 264, 169 (1951).
39- R.Lang, Z. an^rg. allg. Chem., 142, 229 (1925).
40. А. А. Кузьменко, Я- А. Ф и а л к о в, ЖОХ, 19, 1007 (1949).
41. Я- А. Ф и а л ко в, А. А. Кузьменко, ЖОХ, 19, 1645 (1949).
42. W. M i 1 i t z е г, J. Am. Chem. Soc, 60, 256 (1938).
43. F. W. В е n n e t, A. G. S h а г p e, J. Chem. S^c, 1383 (1950).
44. L. J. L a m bo u r n e, P. W. Robertson, J. Chem. Soc, 1167(1947).
45. D. E.Pearson, С J. R о s s , J. Am. Chem. Soc, 74, 2933 (1952).
46. A. G. S h а г p e, J. Chem. Soc, 3713 (1953).
47. J. H a n u s, Z. Nahr. u. Genupmit., 4, 913 (1901).
48. D. H о 1 d e, A. G о г g a s, Ber., 58, 1071 (1925); 59, 113 (1926).
49. W. G. Brown, Phys. Rev., 42, 355 (1932).
50. H. E v a n s, T. R. M u n s ^ n, D. D. W a g m a n, J. of Research Nat.
Bureau Standards, 55, 147 (1955).
51. H. Cnrdes, H. S p о n e r, Z. Phys., 63, 334 (1930); H. С о г d e s,
Z. Phys., 74, 34 (1932).
52. T. R. В i с h о vv s k y, F. D. Rossini, The Thermochemistry of the
Chemical Substances, N. Y.f 1936.
53. Э- В. Брицке, А. Ф. Капустинский и сотрудники,
Термические константы неорганических веществ, АН СССР, М. —Л., 1949.
54. В. S i n g h, M. S i n g h, Res. Bull. Panjab Univ., 73, 73 (1955).
55. Л. Ф. Денисова, Ж- неорг. химии, 1, 758 (1956).
ГЛАВА VIII
СОПОСТАВЛЕНИЕ НЕКОТОРЫХ ХИМИЧЕСКИХ СВОЙСТВ
МЕЖГАЛОИДНЫХ СОЕДИНЕНИЙ
Из описанных выше химических реакций межгалогенидов с
неорганическими веществами наиболее характерными и важными
в практическом отношении являются реакций окисления,
обусловленные электроположительным состоянием одного из галогенов.
В предыдущей главе закончено рассмотрение химических свойств
отдельных межгалогенидов (исключая реакции комплексообразова-
ния). Здесь целесообразно сравнить межгалогениды как окислители
по отношению к неорганическим веществам *. Необходимо,
однако, иметь в виду, что такое сравнение очень затруднено.
Большинство литературных данных относится только к качественной
характеристике окислительных реакций межгалогенидов и к
характеристике их взаимодействия с металлами, металлоидами,
окислами,, солями и т. д. (а также с органическими веществами) и часто
основано на недостаточно полном изучении этих реакций. Так,
для фторидов галогенов, вследствие их большой химической
активности, во многих реакциях не была Точно установлена даже
природа всех образующихся продуктов реакции. Кроме того,
имеющиеся в литературе сведения в ряде случаев относятся к
различным условиям проведения реакций (дисперсность
восстановителей, количественные соотношения, температура и т. д.).
Имеет значение также различие в типах межгалогенидов (АВ,
АВ3 . . . АВ7 — см. главу I). Однако это различие в ряде случаев
сглаживается тем, что высшие галогениды в реакциях окисления
часто диссоциируют ступенчато, в то время как некоторые низшие
межгалогениды, например фтористый бром, вследствие реакций
диспропорционирования находятся в равновесии с высшими
формами.
Из межгалогенидов наиболее активными окислителями
являются фториды галогенов. Несмотря на отмеченные выше
затруднения и некоторую условность в характеристике их окисляющего
действия, фториды галогенов по реакционной способности можно
расположить приблизительно в такой ряд: ClF3>BrF5>JF7>ClF>
* Окислительные свойства хлористого и треххлористого иода в соляно-
кислых растворах будут описаны в главе XVI.
196
^BrF3>JF5>BrF [1]. Далее следует поместить треххлористый й
гористый иод и бромистый иод *.
Фториды галогенов реагируют при определенных условиях поч-
тц со всеми металлами **, а также с большинством металлоидов,
исключая благородные газы, азот и кислород. В то же время
хлористый и бромистый иод не действуют на многие металлы: Cd, Pb,
Zr, Nb, Та, Сг, Мо, W, Pt; не реагируют с бором, углем, кремнием.
Течение реакции межгалогенидов с элементами в значительной
степени зависит от свойств продуктов реакции и от их отношения
к исходным межгалогенидам. Реакции обычно доходят до конца
Б тех случаях, когда образующиеся галогениды летучи в условиях
реакции, хорошо растворимы в избытке межгалогенида или
соединяются с ним в растворимый комплекс. Наоборот, реакция
замедляется или даже прекращается, если на поверхности металла
образуется слой галогенида, прочный при температуре опыта,
играющий роль защитной пленки и препятствующий дальнейшему
действию межгалогенида на металл. Это создает возможность
использовать при работе с фторидами галогенов такие металлы, как
медь (до 400°), никель (до 750°), мягкую сталь (приблизительно
до 250°) Ш.
Значение этого фактора можно показать на следующем
примере [2]: железо очень медленно реагирует с трехфтористым бромом,
так как FeFq трудно растворяется в последнем; с хлористым иодом,
в котором FeCl3 заметно растворимо, железо реагирует с выходом
в 65%; с наименее активным бромистым иодом реакция доходит
почти до конца (выход 98%) по той причине, что FeBr3 хорошо
растворимо в нем.
Однако растворимость галогенидов не является единственным
фактором, определяющим отношение металлов к межгалогенидам.
Имеются и другие еще далеко не выясненные причины различия
в 'поведении металлов в среде межгалогенидов. Так, поверхность
хрома вовсе не изменяется после нагревания в трехфтористом
броме, хлористом и бромистом иоде. Тантал хорошо реагирует с
трехфтористым бромом, но не с хлористым и бромистым иодом, хотя
ТаВгб хорошо растворяется в бромистом иоде [2]. В последнем
случае решающее значение имеет, вероятно, различие в
окислительном действии этих межгалогенидов.
Все реакции между металлами и межгалогенидами протекают
таким образом, что металлы соединяются с более
электроотрицательным галогенидом; тот галоген, который составляет
электроположительную часть межгалогенида и является
окислителем в этих реакциях, выделяется в свободном состоянии, что
может быть выражено такой схемой:
т АВ„ + лМ = /zMBm + -!j A2.
* Вследствие значительной термической нестойкости хлористого брома
его химические свойства почти не изучены.
** Трехфтористый бром и следующие за ним в приведенном выше ряду
фториды галогенов не действуют на платину.
197
Реакции с высшими межгалогенида-ми (/г>3) могут протекать
и по более сложному механизму. Так, Гутман [2] полагает, что
трехфтористый бром в реакции с молибденом вначале восстанав-
ливается до монофторида, который затем превращается в трифТ(>
рид и свободный бром:
3BrF3 + Mo= MoF6 + 3BrF;
3BrF = Br2 + BrF3;
2BrF3 + Mo = MoFe -r Br2.
Как уже отмечалось в предыдущих главах, подобные схемы
реакций возможны при условии более или менее значительного
избытка межгалогенида, в противном случае образуются соединения
металлов с обоими из галогенов.
Кроме того, необходимо предусмотреть возможность комплек-
сообразования между исходным межгалогенидом и одним из
первичных продуктов реакции.
Все сказанное относится также к реакциям межгалогенидов с
металлоидами с тем лишь отличием, что последние обычно не
образуют на своей поверхности защитного нерастворимого слоя га-
логенида. л
Несколько своеобразно протекают реакции межгалогенидов со
свободными галогенами — при взаимодействии галогенидов иода,
брома и хлора соответственно с иодом, бромом и хлором
происходит восстановление высших межгалогенидов до низших форм.
Так, семифтористый иод переходит в JF5; пятифтористый бром—в,
BrF3, а затем в BrF; трехфтористый хлор—в C1F ; треххлористый
иод — в JC1. Некоторые из этих реакций имеют равновесный
характер вследствие диспропорционирования. В то же время
некоторые фториды галогенов (C1F, BrF, BrF3, JF5) и хлористый иод
при действии на них соответственно фтора или хлора окисляются
до возможных высших форм.
Взаимодействие межгалогенидов с галогенами, не входящими
в их состав, например реакция между фторидами иода и хлором
или бромом, имеет более сложный характер и в ряде случаев еще
не изучено не только количественно, но даже качественно. Так,
например, при реакции семифтористого иода с хлором образуются
JC1, JC13 и C1F; продукты взаимодействия C1F3 или BrFs с цодом
или бромом не охарактеризованы [1].
С водородом межгалогениды реагируют энергично, часто с
воспламенением (хотя для этого обычно необходимо предварительное
нагревание водорода), причем образуется смесь соответствующих
галоидоводородов, например JBr + H2 = HJ-fHBr.
Все межгалогениды более или менее легко гидролизуются.
Одним из продуктов гидролиза является галоидоводородная кислота:
HF — при гидролизе фторидов галогенов, НС1 — при гидролизе
хлористого иода и т. д. Второй из галогенов, находящийся в
электроположительном состоянии, образует кислородную кислоту,
которая затем диспропорционируется, как, например, иодноватистая
198
Кй'С«л°та (продукт гидролиза хлористого и бромистого иода), или
разлагается с выделением кислорода и галогена. Возможны и
другие вторичные процессы.
В табл. 33 приведены некоторые из продуктов гидролиза
межгалогенидов.
Таблица 33
Продукты гидролиза межгалоидных соединений
Межгалогенид
Фтористый хлор
Трехфтористый хлор
Тре>фтористый бром
Пят1.фтор истый бром
Пятифтористый иод
Семифтористый иод
Хлористый бром *
Хлористый иод
Трех хлористый иод
Бромистый иод
Продукты гидролиза
HF, 02, 03, С12
HF, Cl2, OF2
HF, НВгОиНВЮ3,02
HF, BrOF3 (?)
HF, HJ03
HF, HJO4
HCI, HOBr
HC1, HJO3, J2
HCI, HJO3, JC1
HBr, HJ03, J2
Повышение рН усиливает гидролиз межгалогенидов и
благоприятствует смещению равновесия реакций диспропорционирова-
ния низших кислородных кислот галогенов. Реакция гидролиза
межгалогенидов, за исключением хлоридов иода и бромистого иода,
в количественном отношении еще очень мало исследованы. Для
последних двух межгалогенидов установлено, что в растворах
соляной или бромистоводородной кислот гидролиз отсутствует
вследствие образования комплексных кислот ШСЬ, ШС14 и НЛВгг.
Взаимодействие фторидов галогенов с окислами металлов и
металлоидов превращает последние в соответствующие фториды (в
этих случаях выделяется кислород окислов) или оксифториды;
хлористый иод превращает окислы металлов в хлориды и иодиды.
Таковы только первичные реакции, возможны также реакции ком-
плексообразования, подобные описанным в главе III, раздел 2.
Межгалогениды окисляют также другие группы неорганических
соединений (водородные соединения, соли и др.), причем
окисление фторидами галогенов, как правило, сопровождается
фторированием.
Общей для всех межгалоидных соединений является их
окислительно-восстановительная реакция с галогенидами других
элементов, содержащими галоген, общий с электроположительной
частью межгалогенида; этот галоген выделяется в свободном
состоянии. Схема этих реакций такова:
ткВп + nRkm = пЯВт+ {—^^ К
(например,
2JC13 + ЗСа J2 = ЗСаС12 -f 4J2.
199
Этим объясняется тот факт, что при действии избытка межга-
логенида на неорганические вещества всегда получается галогенид^
более электроотрицательного из галогенов, образующих данный
мсжгалогенид.
Сопоставление окислительных свойств межгалогенидов
требует знания их окислительно-восстановительных потенциалов. Однако,
эта количественная характеристика определена только для
хлористого, треххлористого и бромистого иода [3]. Потенциалы
реакции восстановления JC1 и JC12~ до иода и хлорида равны
С1-
1
1 к ^2 — Л^1(водн) ~\~ в \ tL -
1,19*;
2С1- + ± J2 = JCl^ + ег\ Е° =- - 1,06 **.
Потенциалы реакции восстановления JBr и JBr2~ составляют
соответственно —1,02 и —0,87 е.
Для треххлористого иода:
2С1- + JC1(1B) - JC13(tb) + 2е~; £° =- - 1,31 в.'
ЗС1- + -i J2= JC13(TB) + Ъе~\ £° = — 1,28 в.
Отсутствие таких же данных для других межгалоидных
соединений в аналогичных условиях позволяет провести лишь весьма
приближенное сравнение их окислительных свойств путем
сопоставления результатов реакций с одним и тем же веществом в
одинаковых условиях. Так, например, Бус и Пинкстон [1] сопоставляюг
реакции некоторых фторидов галогенов с закись-окисью кобальта
и хлористым кобальтом (табл. 34), взятыми в твердом виде.
Таблица 34
Реакции некоторых фторидов галогенов с Со304 и СоС12
Фторид галогена
Трехфтористый хлор
Хлористый фгор
Пятифтористый бром
Пятифтористый иод
Продукты реакции
с Со304
Более чем 95% CoF3
. 98% CoF2
. 90% CoF2
Не реагирует
с СоС12
100% CoF3
55% CoF3+45% CoF2
45% CoF3+55% CoF2
72% CoF2
Из табл. 34 видно, что только трехфтористый хлор полностью»
или почти полностью окисляет закись-окись и хлорид закиси
кобальта, превращая их в CoF3.
* См. также главу XVI.
200
В заключение этого раздела необходимо резюмировать
высказывавшиеся выше при описании отдельных межгалогенидов
суждения об их применении для препаративных целей — для синтеза
неорганических и органических соединений. Положительной
особенностью межгалогенидов в этом отношении является
их большая химическая активность и связанная с этим
возможность получать галогениды, в особенности фториды многих
элементов, в их высших степенях валентности, которые ранее
относились к малодоступным веществам. В предыдущих главах
показано, как с помощью межгалогенидов удалось синтезировать большое
число комплексных галогенидов, в том числе и новые, неизвестные
ранее соединения.
Большой интерес могут представить реакции межгалогенидов
с органическими соединениями для получения различных галоид-
производных путем замещения водорода в фенолах, аминах,
углеводородах * и т. д. или путем присоединения по кратной связи.
Наконец, следует также упомянуть о комплексах—продуктах
соединения межгалогенидов с органическими веществами, которые, в
свою очередь, можно использовать для синтеза галоидпроизводных-
Но межгалоидные соединения как реагенты для препаративных
работ часто обладают рядом значительных недостатков, как-то:
бурное протекание реакций и трудность их контролирования по
этой причине, затруднения в разделении продуктов реакции,
сильное физиологическое действие, особенно фторидов галогенов:
раздражение глаз и слизистой оболочки (слезоточивое действие,
раздражение дыхательных путей, кашель и т. п.), ожоги при
попадании на кожу или сильное раздражение кожи.
Все эти недостатки нельзя считать непреодолимыми. Их можно
более или менее значительно ослабить или устранить, в частности,,
проводя реакции в среде какого-либо растворителя, инертного по
отношению к данному межгалогениду или образующего с ним
комплексные соединения. Это позволяет делать более спокойными
бурно протекающие реакции и управлять ими. Примером в этом
отношении могут служить результаты исследований, проведенных с
солянокислыми растворами хлористого и треххлористого иода.
Наконец, нетрудно применять такие приемы работы с межгалогени-
дами, которые могут значительно ослабить их вредное
физиологическое действие.
ЛИТЕРАТУРА
1. Г. С. Б у с, Д. Г. П и н к с т о н, сб. «Фтор и его соединения», ИЛ, М.,
1953, 168.
2. V. Gutmann, Monatsh., 82, 280 (1951).
3. Б. Л а т и м е р, Окислительные состояния элементов и их потенциалы
в водных растворах, ИЛ, М., 1954.
* Особый интерес представляет получение фторзамещенных
углеводородов.
ГЛАВА IX
ЭЛЕКТРОХИМИЧЕСКИЕ СВОЙСТВА ХЛОРИСТОГО,
ТРЕХХЛОРИСТОГО И БРОМИСТОГО ИОДА
Хлористый, треххлористый и бромистый иод в
электрохимическом отношении изучены лучше, чем другие межгалоидные
соединения.
Первые сведения об электрохимических свойствах хлористого
и бромистого иода появились >в литературе уже давно — в работах
Фарадея [1] и Солли [2], которые установили способность этих
межгалогенидов проводить электрический ток. Фарадей же
показал, что при электролизе жидкого хлористого иода между
платиновыми электродами на катоде выделяется иод, а на аноде — хлор.
Электролиз хлористого и бромистого иода в расплавленном
состоянии более подробно исследовали Брунер и Бекьер [3],
применяя серебряные электроды. При электролизе хлористого иода (при
40°) оба электрода покрываются слоем хлористого серебра.
Количество хлористого серебра, образовавшегося на аноде (выход
хлора), близко к теоретическому—1 г-экв хлора на 1 F (особенно
при избытке иода в препарате). Образование хлористого серебра
на катоде можно объяснить тем, что первичный продукт реакции—
йодистое серебро — реагирует с хлористым иодом
AgJ+JCl=AgCl+J2.
Аналогичные явления наблюдались этими авторами при
электролизе бромистого иода; на аноде выделялся бром, постепенно
превращая его в бромистое серебро. Если добавлять иод к
расплавленному бромистому иоду, то выход брома на аноде повышается
и становится равным теоретическому из расчета на разряд ионов
Вг~. При наличии избытка брома, он разряжается в количестве,
большем теоретического. Для объяснения этого факта Брунер и
Бекьер (предположили, что в расплаве находились, кроме ионов
Вг~, также ионы Вг3~*, разряд которых, даже в небольшом
количестве, увеличивает выход брома. На катоде же вначале
образуется йодистое серебро, которое затем превращается в бромистое
* Брунер и Бекьер предположили, что в системе JBr—Br2 образуется
полибромид JBr-Br2 или J[Br3], диссоциирующий на ионы J4" и Вгз\ Разряд
ионов Bi\j~ увеличивает теоретический выход брома, рассчитанный на Вг~.
Подобный факт обнаружили Плотников и Рокотян [4], при электролизе
бромных растворов треххлористого иода.
202
54
^
30
—г"
50
—г-
70
90 t°
Рис. 32. Удельная электропроводность
хлористого иода при разных
температурах, по данным Фиалкова и Шор.
/VgJ+JBr=AgBr+J2. В процессе электролиза бром переносится в
янолит.
Для хлористого иода величина электропроводности при 35 рав-
на 4,52—4,59-10~3 ом-1слС{ (см. табл. 25). Зависимость
электропроводности хлористого иода от температуры исследовали Фиалков
и Шор [5] в интервале темпе-
ратур 35—97°, Гринвуд и Эме- fW
леус [6] — !в интервале 26,75—
122,0°. Результаты этих
измерений, приведенные на рис. 32 и
33, показывают, что удельная
электропроводность хлористого
иода с повышением
температуры сперва растет, загем в
интервале 45—70° (по данным
Фиалкова и Шор) или 40—45°
(по данным Гринвуда и Эме-
леуса) величина ее
(практически не изменяется, лосле чего электропроводность уменьшается и
достигает значений меньших, чем при начальной температуре.
В табл. 35 приведены, по данным этих авторов, величины
электропроводности, соответствующие лишь начальным и конечным
температурам
измерений и температурам,
при которых
наблюдались максимальные
значения
электропроводности.
Такой же характер
температурной
зависимости
электропроводности наблюдается для
треххлористого иода,
по измерениям
Гринвуда и Эмелеуса [6], и
для бромистого иода,
по измерениям
Фиалкова [7].
Электропроводность
треххлористого иода
измерена в интервале
температур 70—146,6°,
причем измерения до
ЮГ проводились с твердым сплавленным препаратом.
При повышении температуры электропроводность твердого
треххлористого иода быстро растет —от 1,9-10-5 ом~1 см-1 при 70°
до ~ 8,4-10—3 при температуре плавления (ЮГ). В интервале
температур 103,7—115,0° электропроводность изменяется всего от
9,13 до 9,3410~3 ом~х смг1 (максимум 9,6-10_3 вблизи 110—111°);
WQV
Рис. 33. Удельная электропроводность
хлористого иода при разных температурах, по данным
Гринвуда и Эмелеуса:
/—неочищенный
очищенный;
препарат; //—препарат, неполностью
///—высокоочищенный препарат.
203
Таблица 35
Зависимость электропроводности хлористого
иода от температуры
Температура, °С
35
45
50—70
97
х-103,
ом~1см~~1
[51
4,52
5,11
5,14
3,49
Температура, °С
34,45
39,9
45,1
122,0
х-103,
ом~хсм~~{
[6]
4,593
4,637
4,610
1,817
после чего при дальнейшем нагревании до 146,5° она уменьшается
до величины 0,36-10-3 (рис. 34).
Треххлористый иод (даже твердый) обладает большей
электропроводностью, чем жидкий хлористый иод, при одинаковых
температурах, что видно, например, из данных табл. 36.
Таблица 36
Сравнение электропроводности хлористого и треххлористого иода
Температура, °С
26,75
(<^т. пл.)
97,0
122,0
Хлористый иод
Агрегатное
состояние
Жидкость
Жидкость
Жидкость
х.103,
ом~~1см~х
4,40 [6]
3,49 [5]
1,82 [6]
Треххлористый иод
Темпера-
тура, °С
97,7
101,0
(т. пл.)
122,9
Агрегатное
состояние
Твердое
Жидкость
Жидкость
%.ю3,
омГ1смГт{
1 6,87 [6]
! 8,40 [6]
8,15 [б]
Этот факт заслуживает взимания, так как в большинстве
случаев электропроводность галогенидов одного и того же
элемента (металла) при температурах плавления уменьшается с
увеличением его валентности (например, электропроводность SnCl2 и
SnCl4, РЬС12 и РЬСЦ, InCl, InCl2 и InCl3, T1C1 и Т1С13).
Возможно, что на электропроводность треххлористого иода
большое влияние оказывает своеобразие его строения (глава VI).
По мнению Вонка, Босвейка и Вибенга [19], высокая
электропроводность жидкого треххлористого иода объясняется тем, что
плоские молекулы Л2С1б с ковалентными связями частично
диссоциируют на ионы при высоких температурах. Эти авторы
отмечают, что, в отличие от данных Гринвуда и Эмелеуса, их измерения
температурной зависимости электропроводности треххлористого
иода показали прерывность вблизи точки плавления (вблизи 100°).
Это явление наблюдается, если предотвратить переход твердого
треххлористого иода в жидкость и газ при более низких темпера-
204
турах. Как следует из фазовой диаграммы системы иод—хлор,
это можно осуществить, прибавляя избыток хлора к образцу
препарата в сосуд для измерения электропроводности. При этом
условии треххлористый иод отчетливо плавится при 98,5° (без
предварительного частичного превращения в жидкость).
Электропроводность полученной таким образом жидкости при 100° равна
8,5-10"3 ом~1 см~К При охлаждении, как только появляются первые
кристаллы, наблюдается
уменьшение
электропроводности. Если при 98°
вещество полностью
кристаллизуется, электропроводность
резко уменьшается в 103—
105 раз.
Способность бромистого
иода проводить ток была
обнаружена Солли в 1836 г
[2]. Затем Плотников в
1903 г. [8] сообщил, что
растворы иода в броме
проводят ток.
Электропроводность этих растворов была
позднее подробно
исследована им же совместно с Ро-
котяном [9], причем
оказалось, что эта система
начинает заметно проводить ток
лишь при довольно
значительных концентрациях
иода, вернее — бромистого
иода. Электропроводность быстро растет с концентрацией; так, в
интервале концентраций иода от 12,7 (20,9% JBr) до 44,5% (72,3%^
JBr) удельная электропроводность при 25° возрастает от 0,32 • 10~6
до 12,6 • 10~4 ом-1см~1. Молекулярная электропроводность
увеличивается с концентрацией, т. е. ее концентрационная зависимость
имеет «аномальный» характер. С повышением температуры
электропроводность уменьшается.
Электропроводность бромистого иода впервые определили Бру-
нер и Бекьер [3]. Она оказалась равной при 16,3° (для твердого
препарата) 6,51 • 10~6, а при 40,6° (для расплавленного
препарата) — 3,08 • Ю-4 омг1см-1. При дальнейшем повышении
температуры (44—50°) электропроводность начинает уменьшаться.
Зависимость электропроводности бромистого иода от температуры в
пределах 17—110°, по данным Фиалкова [7], показана на рис. 35:
при повышении температуры до 65° электропроводность
повышается, а затем падает *.
-2,0^
-з,о-
-# J
!50
1—г™
130
т
110 90
■ i
~о©-од>£Г
—i 1 1—
го
i^
2ft
2fi 2.9
2.5 2,6 27
10ZIT (°Ю.
Рис. 34. Удельная электропроводность
треххлористого иода при разных
температурах (стрелка показывает температуру
плавления препарата).
* Электропроводность жидкого бромистого иода при 45° равна 6,5-10"
при 50°— e^-lO-W-'c*-1 [Ю].
205
Электропроводность бромистого иода при температуре плав-
ления оказалась в 10 раз меньше электропроводности хлористого
иода.
Кривые зависимости силы тока от напряжения, полученные для
расплавленного хлористого и треххлористого иода, имеют
прямолинейный характер, т. е. они не пают
отклонения от закона Ома [6].
Длительный электролиз
хлористого иода в сосуде с диафрагмой
вызывает обратную э. д. с, величина
которой зависит от количества
прошедшего тока [6]:
х-ю*
125-
100
75
504
25-\
J
25 50 100 С
Рис. 35. Удельная
электропроводность бромистого иода при
разных температурах (стрелка
показывает температуру
плавления препарата).
Продолжительность
электролиза в минутах 30 55 178
Количество тока ,/МО4 0,868 1,59 5,15
Обратная э. д. с, мв 7 8,5 И
Теперь рассмотрим некоторые
вопросы, вытекающие из обзора
электрохимических свойств хлоридов и
бромида иода.
Причина максимума на кривых температурной
зависимости электропроводности
Это явление можно объяснить тем, что хлористый, треххло-
ристый и бромистый иод при нагревании выше определенной
температуры подвергаются заметной термической диссоциации:
2JC1^J2 + C12 или 3JC1^JC13 + J2;
JCl3^JCl + C]2;
2JBr^J2 + Br2,
причем продукты диссоциации понижают электропроводность
исходных межгалогенидов. С повышением температуры
термическая диссбциация увеличивается и вследствие этого температурный
коэффициент электропроводности, сперва положительный,
становится равным нулю, а при более высоких температурах — и
отрицательным. Это объяснение подтверждается следующими
данными:
а) кривые зависимости электропроводности хлористого и
бромистого иода от температуры (рис. 32, 33, и 35) согласуются с
кривыми нагревания (см. рис. 26 и 31), согласно которым
термическая диссоциация этих межгалоидных соединений становится
заметной в том же температурном интервале, в котором
электропроводность не изменяется или даже уменьшается;
б) изменение электропроводности хлористого и бромистого
206
лода с температурой имеет обратимый- характер, как и
термическая диссоциация [5, 7];
в) электропроводность бромистого иода понижается от
добавления как брома [3, 9], так и иода [7]. Необходимо также принять
Бо внимание, что электропроводность иода значительно меньше
электропроводности хлористого иода.
Гринвуд и Эмелеус [6] объясняли наличие максимума на
кривой температурной зависимости хлористого иода * уменьшением
ионизации г повышением температуры, но это утверждение
основано на весьма приближенных расчетах, согласно которым
степень ионизации хлористого иода при 95° равна 1,2%. Расчеты для
других температур в работе этих авторов не приведены. В
последней работе Гринвуд [И, стр. 463] отмечает, что это .уменьшение
числа ионов обусловлено их термической неустойчивостью **, т. е-.
он дает, по сути, то же объяснение максимума, которое было
ранее предложено нами [5, 7].
Природа ионов, образуемых галогенидами иода
в расплавленном состоянии
Природа ионов, на которые диссоциируют галогениды иода,
JC1, JBr, JCI3 (а также JFs и ВгРз), в значительной степени
обусловлена молекулярным состоянием последних. Как отмечалось
в главах VI и VII, ряд физико-химических свойств хлористого и
бромистого иода в расплавленном состоянии характеризует их как
ассоциированные жидкости. Поэтому можно полагать, что
существующие в таких средах ассоциаты молекул (автокомплексы)
диссоциируют с образованием как сложных, так и простых ионов,
по следующим схемам ***:
(JX)w^^J+ + [Jm_rtXwr-;
(JX)m ^ [JmXm_J"+ + пХ~ (X = С1 или Вг).
В простейшем случае т = 2, /г=1.
Криоскопическое и электрохимическое исследования растворов
хлористого и бромистого иода в некоторых растворителях [12, 13]
(см. главу X) показали, что эти галогениды иода находятся в
них в виде димерных молекул, диссоциирующих по схеме
(JX2^J+ + JX^;
JX^" zljX + Х"\
* Этот максимум сохраняется и после введения поправки на изменение
числа молей между электродами, вызванной термическим расширением
жидкости.
** Такое же объяснение Гринвуд [11, стр. 453] предлагает для
отрицательного температурного коэффициента проводимости трехфтористого брома.
*** В наиболее общем виде эта схема может быть п|едставлена 1аким об| азом:
(JX)m ^ Um_„xm_;.] '»-">++ [j„xpl ("-">-.
207
Эти факты наводят на мысль, что такая же схема ионизаццй
может быть перенесена и на расплавленное состояние хлористого
и бромистого иода. Так, Гринвуд [11, стр. 463] пишет, что на осно.
вании исследований Фиалкова и Каганской [13] ионизация расплав,
ленного хлористого иода может быть представлена таким обра,
зом: 2JC1^J++JC1^ .
Приводя эти схемы, вероятность которых не вызывает
сомнения, необходимо все же указать, что в расплавленном хлористом
и бромистом иоде вполне возможно наличие и более сложных
ионных групп, распадающихся при нагревании или растворении.
Еще более сложен вопрос о природе ионов в расплавленном
треххлористом иоде, вследствие значительной термической
диссоциации на хлористый иод и хлор и некоторых особенностей его
строения (см. главу VI).
Для треххлористого иода можно предположить такую же общую
схему ионизации, которая с большим или меньшим вероятием принята
для других межгалогенидов (Алп)2 — [AX„_i] г н- [AX//41]~.
Применительно к треххлористому иоду эта схема принимает вид: (JC13)2 ZE JOls?" +
+ ЛС1Г- Однако в то время как способность JC!3 образовывать
комплексные анионы хорошо доказана, нет убедительных данных
о существовании в расплавленном треххлористом иоде или в его
растворах катионов JCI2". Точно так же лишено достаточного
экспериментального обоснования мнение Оддо [14] о том, то JC13 диссоциирует на
ионы JCI2" и С1_, равно как и более раннее мнение Вальдена [15]
о диссоциации JC13 на J3+ и ЗС1~. Таким образом, вопрос о природе
ионов, образуемых треххлористым иодом в расплаве, остается
открытым.
Продукты электролиза межгалоидных
соединений
Исходя из некоторых экспериментальных данных и
представлений о природе ионов в жидких межгалогенидах, процессы,
происходящие при электролизе, можно представить следующим образом
(табл. 37).
В действительности же эти схемы не исчерпывают возможного
разнообразия электродных процессов, происходящих при
электролизе жидких межгалогенидов, прежде всего вследствие большей
сложности молекулярного состояния последних в таких системах.
Кроме того, эти схемы не предусматривают возможного катодного
восстановления иода (стр. 230).
Процесс электролиза межгалогенидов и электродные
процессы в неводных растворах упрощаются вследствие уменьшения
степени ассоциации. Но в то же время необходимо принять во
взимание возможность образования комплексных соединений
межгалогенидов с молекулами многих растворителей, особенно с теми,
которые имеют характер оснований, например CsHsN'JCl,
C5H5N-2JC1 и т. д. (глава XI), вследствие чего появляются новые
208
Таблица 37
Электродные процессы при электролизе межгалогенидов
Межгалогениды
Катодные процессы
Анодные процессы
^пористый иод
бромистый иод
Трехфтористый бром
[16]*
Пятифтористый иод**
2J+ + 2е = J2
2J+ + 2е = J2
2BrF++2e = BrF + BrF3
3BrF = BrF3 + Br2
2JC1J-—2e = 2JCl + Cl2
JCl + Cl2:£JCl3
2JBr- —2e = 2JBr + Br2
2BrF~— 2e =BrF3+BrFr5
2JF7—2e=JF5+JF7(?)
виды ионов, участвующих в переносе тока и в электродных
процессах. Эти вопросы будут рассмотрены в главе X.
Потенциал разложения
Ни для одного из межгалогенидов, проводящих ток ***, не
удалось определить потенциалы разложения на кривых зависимости
силы тока от напряжения. Это явление можно объяснить,
предположив, что в жидких межгалогенидах, вследствие термической
диссоциации, присутствует небольшое количество тех веществ,
которые являются продуктами термической диссоциации или
продуктами электролиза и действуют деполяризующим образом.
Наиболее подробно это явление изучено для хлористого иода. В связи
с этим рассмотрим на примере этого межгалогенида те
соображения, которыми Гринвуд [11, стр. 463] обосновывает приведенное
выше объяснение отсутствия отклонения от закона Ома.
Термическая диссоциация хлористого иода, а также процесс
электролиза приводят к следующему равновесию:
3JC1^J2 (в JC1) -t-JC1» (в JC1).
Изменение свободной энергии в этой реакции, по данным Грин-
вуда, для 35° равно 2,8 ккал\ это соответствует степени диссо-
* При электролизе трехфтористого брома было замечено, что жидкость
вокруг катода становится коричневой, в то время как цвет ее у анода не
изменяется,- что было объяснено следующим образом [16]: катион BrF^~,
разряжаясь, превращается в бледно-желтый BrF3 и коричневый BrF, анион же
BrF^~ дает BrF3 и бесцветный BrF5-
** Для пятифгористого иода анодный процесс представлен как
предположительный. Для катодного процесса нельзя принять такую же схему, как для BrF3,
требующую превращения JF^f" в JF3 и JF5, вследствие отсутствия или весьма
значительной неустойчивости JF3. Возможно, что в этом случае наряду с JF5
образуется элементарный иод: 5JF^ + Ъе =« 4JF5 + V2-b-
*** За исключением, может быть, лишь пятифтор исто го иода (см. стр. 89).
14—1419
209
циации 0,5%. При этих условиях электролиз изменяет локально
концентрацию иода и треххлористого иода у электродов и вознц-
кает концентрационная э. д. с, которая изменяется с количеством
протекающего тока. Величина такой э. д. с, даже при длительном
электролизе, значительно меньше, чем обычные значения для об.
ратной э. д. с, и в этом заключается причина отсутствия потен-
циала разложения на кривых зависимости силы тока от
напряжения. Количество протекающего тока в системе
исключительно мало, и поэтому можно пренебречь концентрационной
поляризацией. Возможно, заключает Гринвуд, что подобные
соображения могут объяснить отсутствие потенциала разложения во
многих наводных растворах межгалоидных соединений.
Природа электропроводности галогенидов
иода
По характеру температурной зависимости электропроводности
галогениды иода (JC1, JBr, JC13) проявляют аналогию с
элементарным иодом, обладающим (положительным температурным
коэффициентом электропроводности в твердом состоянии (что
характерно для ионной проводимости) и отрицательным
температурным коэффициентом в жидком состоянии (что указывает на
наличие электронной проводимости) [17]. По порядку величины
электропроводности при температурах плавления (жидких
веществ) можно построить следующий ряд:
J2 JBr JC1 JCl3
10~5 10""4 10~3 \0-2олГ1см'1
Коэффициент разрыхления кристаллической решетки для иода,
измеряемый отношением электропроводностеи твердого и жидкого
иода при температуре плавления (113°), сравнительно велик и
составляет 12,7 • 10^7: 5,35- Ю-5 = 0,024 [17]. Для трехфтористого
брома получено значение 1,73 • Ю-4: 8,13 • 10~3 ~ 0,021. Такие
величины указывают на наличие ионной проводимости или на
значительное преобладание ее. Для галогенидов иода (JC1, JBr, JCI3)
электропроводность в твердом и жидком состояниях * одного и
того же порядка.
Как известно, иод обладает смешанной электронной и ионной
проводимостью [17]. Для галогенидов иода, в отличие от
элементарного иода, по-видимому, характерно наличие только. ионной
проводимости или преобладание последней. Отсутствие потенциала
разложения на кривой зависимости силы тока от напряжения
не может служить доводом в пользу электронной проводимости.
Это явление, как описано выше, объясняется иными
.причинами.
Большое значение для решения поставленного вопроса имеет
* При температуре плавления.
210
г0т факт, что величины электропроводностей твердых треххлори-
сТого и хлористого иода того же порядка, что и ионная
проводимость кристаллов галогенидов щелочных металлов вблизи их
температуры плавления. Вместе с тем нельзя вовсе исключить
возможность проявления межгалоидными соединениями, и в том
ццсяе галогенидами иода, свойств полупроводников. Для
решения вопроса о механизме проводимости тока необходимы
дальнейшие количественные исследования — определение переноса ионов
в кристаллах и в расплавах и другие.
Сравнение элек т р опроводностей хлоридов
иода и хлоридов некоторых других элементов
Бильц и Клемм [18] систематизировали величины
электропроводности расплавленных хлоридов большого числа элементов при
температурах плавления, исходя из положения их в
периодической системе Д. И. Менделеева. Как показано в широко известной
таблице этих авторов (табл. 38), в шести «главных»
подгруппах * хорошо проводящие ток хлориды резко отделяются от
непроводящих ступенчатой линией. Эквивалентная электропроводность
хлоридов закономерно уменьшается во всех горизонтальных рядах
с увеличением порядкового числа элементов **.
В подгруппах элементов с увеличением порядкового числа
электропроводность хлоридов (за исключением хлоридов
щелочных металлов) увеличивается. Систематика Бильца и Клемма
относится только к элементам малых периодов и четных рядов
больших периодов. Хлориды элементов нечетных рядов в общем
проводят ток несколько хуже, причем в горизонтальных рядах
наблюдается такое же уменьшение электропроводности хлоридов с
возрастанием валентности элементов, как и в главных подгруппах.
Хлориды иода представляют некоторое исключение из этой
систематики Бильца и Клемма. Хлористый и треххлористый иод
по величинам удельной и эквивалентной электропроводности (хотя
эти величины часто значительно меньше электропроводности
хлоридов многих металлов при соответственных температурах
плавления) занимают место выше ступенчатой линии, отделяющей
проводящие ток хлориды от непроводящих, если продолжить ее
до седьмой группы периодической системы.
Что касается связи между электропроводностью самих меж-
галогенидов и их составом, то пока не представляется возможным
* Принятые Бильцем и Клеммом главные и побочные подгруппы
элементов больших периодов не являются общепризнанными.
** Антипин [20] в соответствии со взглядами других авторов [21]
отмечает произвольность деления расплавленных солей (хлоридов) на хорошие
и плохие проводники тока и предлагает различать следующие четыре группы:
1) хорошо проводящие ток хлориды: хлориды щелочных и
щелочноземельных металлов, LaCl3;
2) хлориды с пониженной электропроводностью: MgCl2, SeCl3, YC13;
3) малопроводящие хлориды: ВеС12, А1С13;
4) непроводящие хлориды: ВС13> SiCl4 и др.
211
Таблица 38
Эквивалентная проводимость при точке плавления
НС1 1
~10"6 1
LiCl
166
NaCI
133,5
KCl 1
103,5
RbCl
78,2 ,
CsCl
66,7
I BeCl2
0,086
MgCl2 1
28,8 1
CaCl2
51,9
SrCl2
55,7
BaCl2
64,6
■ |
BC13
0
AICI3
15-10"6
ScCl3
15
YCI3
9,5
ССЦ
0
SiCl4
0
TiCl4
0
ZrCl4
LaCl3 j HfCl4
29,0 j
ThC14
16
РСЦ
0
VC14
0
1 NbCl5
j * = 2.10~7
1 TaCl5
M0CI5
x=l,8.10"-6
WCle
| y.=2-10"6
UC14
x=0,34
сделать общее заключение по данному вопросу из-за отсутствия
экспериментальных данных для ряда межгалогенидов: C1F, BrF,
BrF5> JF7, BrCl *.
Все же можно сопоставить (по величине электропроводности)
два межгалогенида, взятые ори температуре плавления и имеющие
один общий галоген или различающиеся по валентности одного
из -галогенидов (см. табл. 1):
BrF3>ClF3; JCl>JBr; JCl3>JCl.
Экспериментальные данные для первых двух пар показывают,
что большей электропроводностью обладают те межгалогениды,
которые составлены из галогенов, дальше отстоящих друг от
друга в седьмой группе. Влияние валентности пока можно
проследить только на хлоридах иода.
ЛИТЕРАТУРА
1. Ostwalds Klassiker, № 86, Leipzig, 1897, S. 43.
2. E. Solly, Phil. Mag., (3), 8, 130, 400 (1836).
3. L Bruner, E. В e k i e r, Z. Elektroch., 18, 368 (1912).
4. В. А. Плотников, В. Е. Рокотян, ЖРФХО, 47, 723 (1915).
5. Я- А. Ф и а л ко в, О. И. Ш о р, ЖОХ, 18, 14 (1948).
6. N. N. Greenwood, H. J. Emeleus, J. Chem. Soc, 987 (1950).
7. Я- А. Ф и а л ко в, Зап. 1н-ту xiM., 6, 223 (1940).
8. В. А. Плотников, ЖРФХО, 35, 794 (1903); Z. phys. Chem., 48,
220 (1904).
* Необходимо принять во внимание значительную термическую
неустойчивость фтористого и хлористого брома.
212
9. В. А. П л о т н и к о в, В. Е. Р о к о т я н, ЖРФХО, 45, 193 (1913V
Z. phys. Chem., 84, 365 (1913).
10. Я- А. Ф и а л к о в, ЖОХ, 11, 910 (1941).
Ц. Н. Н. Г р и н в у д, Усп. химии, 22, 445 (1953).
12. L. В г и п е г, A. G а 1 е с k i, Z. phys. Chem., 84, 515 (1913).
13. Я- А. Ф и а л к о в, К- Я- К а г а н с к а я, ЖОХ, 16, 1961 (1946).
14. G. Oddo, Gazz., 31, И, 146 (1901).
15. П. В а л ь д е н, Z. phys. Chem., 43, 385 (1903).
16. A. G. Sharpe, Quart, rev-, 4, 115 (1950).
17. M. А. Рабинович, ЖРФХО, 58, 223 (1926); Z. phys. Chem., 119,
79 (1926).
18. W. Biltz, W. Klemm, Z. anorg. allg. Chem., 152, 267(1926).
19. С G. V о n k, K. H. Boswijk, E. H. W i e b e n g a, Rec, 74,
897 (1955).
20. Л. Н. Антипин, Усп. химии, 25, 633 (1956).
21. Б. Ф. Марков, Ю К. Д е л и м а р с к и й, Укр. хим. ж., 19, 255
(1953).
ГЛАВА X
НЕВОДНЫЕ РАСТВОРЫ ГАЛОГЕНИДОВ ИОДА
Физико-химические свойства неводных растворов
межгалоидных соединений изучены почти исключительно для хлористого,
треххлористого и бромистого иода, так как фториды галогенов
более или менее агрессивно действуют на большинство
растворителей, особенно органических, а растворы хлористого брома очень
неустойчивы. Поэтому в данной главе будут описаны только
растворы хлоридов и бромида иода.
Эти растворы представляют не только научный интерес для
характеристики физико-химических свойств межгалоидных
соединений, но они имеют также и практическое значение. Известно
их использование для галоидирования органических соединений
и количественного определения последних, для определения
йодного числа жиров и других аналогичных веществ (см. главы VI
и VII).
ЦВЕТ РАСТВОРОВ ХЛОРИСТОГО И БРОМИСТОГО ИОДА
Цвет растворов этих галогенидов иода в неорганических и
органических растворителях так же, как и цвет растворов иода,
зависит от природы растворителей. В этом отношении большинство
растворителей можно разделить в основном на две группы,
образующие с иодом растворы фиолетового и бурого цвета * [1].
Растворители первой группы образуют с хлористым иодом
растворы бурого **, а с бромистым иодом—красного цвета. С
растворителями второй группы хлористый иод дает светло-желтые^
а бромистый иод—-желто-оранжевые растворы*** [2, 3].
В табл. 39 приведены примеры растворителей, в которых
проявляется дихроизм иода, хлористого и бромистого иода.
* Выделяют также группу растворителей, с которыми иод дает растворы
красного цвета [1]. Однако ряд авторов относит большинство этих растворителей
(бромоформ, бромистый этилен, бензол, толуол, ксилолы и другие) к первой
группе, образующей фиолетовые растворы.
** Такого же цвета пары хлористого иода.
*** В ряде случаев наблюдаются исключения из этой схемы. Так,
например, растворы бромистого иода в жидком сернистом ангидриде и в четырех
хлористом углероде бурого цвета. Кроме того, существенное значение имеет
чистота растворителей и концентрация растворов.
214
Таблица 39
Растворители, в которых проявляется дихроизм иода, хлористого
и бромистого иода *
Первая группа
растворитель
Цетыреххлористый
углерод
Хлороформ
#-Гексан
Циклогексан
Бензол
Толуол
а-Ксилол
jw-Ксилол
п- Ксилол
Сероуглерод
fi.101*
0
1,15
0
0
~0
•0,4
0,55
0,4
0
0
е
2,23
5,1
1,89
2.05
2.23
2,29
2,57
2,38
—
2,65
Вторая группа
Растворитель
Метиловый спирт
Этиловый спирт
Этиловый эфир
Этилацетат
Амилацетат
Муравьиная кислота
Уксусная кислота
Уксусный ангидрид
Ацетон
Пиридин
fx-1018
1,69
1,70
1,18
1,81
1,80
1,5-2,1
1,73
—
2,95
2,25
е
33,7
25.8
4,34
6,1
4,81
58,5
6,29
20,5
21,4
12,5
В моп.
Как видно из данных табл. 39, растворители первой группы —
вещества неполярные или малополярные,— с малыми величинами
диэлектрической постоянной (ДП). Ко второй группе
растворителей относятся полярные вещества,
содержащие атомы азота, кислорода;
многие из них проявляют свойства
оснований, в частности в реакциях с
галогенидами иода; за малыми
исключениями для них характерны
достаточно большие величины ДП **.
Для примера на рис. 36 приведены
спектры поглощения растворов
хлористого иода в безводной уксусной
кислоте (кривая /) и в четыреххлориотом
углероде (кривая //).
Максимум поглощения для -бурых
растворов хлористого иода лежит
вблизи 460 ммк, для желтых он
приходится на область 360—350 ммк. Молярный коэффициент
экстинкции, соответствующий максимуму поглощения, для обоих типов
растворов примерно одинаков и составляет 150—160 еж-1 [2]. В
солянокислых растворах хлористого иода максимум поглощения еще
более смещен—до ~ 340 ммк, но коэффициент экстинкции в два раза
больший. Увеличение концентрации НС1 от 0,2-н. до 2-н. не
изменяет положения максимума или интенсивности поглощения.
3000 W00 5000
Рис. 36. Спектр
поглощения хлористого иода в
уксусной кислоте (/) ив че-
тыреххлористом углероде
* Дипольные моменты (jjl) даны в единицах Дебая; величины диэлек
трических постоянных (г) относятся к температуре, близкой к комнатной.
** Как будет показано далее, различие в окраске этих двух групп раство -
ров хлористого и бромистого иода нельзя объяснить только величинами диполь-
ных моментов растворителей.
215
Такая же зависимость положения максимумов поглощеНи
от природы растворителя наблюдается и для растворов бромистп*
го иода. С целью сопоставления приводим положение полос поглсГ
щения в ммк растворов иода, хлористого и бромистого иода
неполярном и полярном растворителях [4].
Растворитель Иод Бромистый иод* Хлористый иод
Четыреххлори- 520 494 460
стый углерод
Спирт 447 390 355
А 73 104 105
Как и в случае растворов иода, добавление к растворителям
первой группы иногда даже небольших количеств растворителей
второй группы изменяет цвет растворов хлористого и бромистого
иода и придает им окраску, характерную для растворов в растворив
телях второй группы. Например, максимум поглощения растворов
хлористого иода в хлороформе (~460 ммк) при добавлении
следов этилового спирта смещается почти на 100 ммк в
коротковолновую область.
Аналогия с растворами иода проявляется также во влиянии
температуры на цвет растворов: так, желтые растворы хлористого
иода в уксусной кислоте при нагревании буреют, бурые же
растворы в четыреххлористом углероде при сильном охлаждении
приобретают более светлую окраску.
Обе эти группы растворов хлористого и бромистого иода
различаются по устойчивости **, по способности проводить ток и
поведению при химических реакциях. Такое различие
физико-химических свойств этих двух трупп растворов хлористого и бромистого
иода объясняется, как и в случае растворов иода, тем, что в
растворителях второй группы происходит образование сольватов и
комплексных соединений.
В настоящее время можно считать доказанным, что в бурых
(коричневых) и красных растворах иода образуются
молекулярные соединения, главным образом состава RJ2 [5]***. Хлористый и
бромистый иод образуют соединения состава RJX и реже R(JX)2-
* По данным Джилама и Мортона [2], максимум поглощения растворов
бромистого иода в четыреххлористом углероде лежит при 487 ммк.
** Некоторые растворители второй группы образуют менее устойчивые
растворы; таковы, например, желтые растворы хлористого иода в этиловом
спирте или в этиловом эфире, что в данных случаях обусловлено
окислительным действием хлористого иода.
*** Убедительные доказательства эквимолярного состава таких соединений
иода дали Щукарев и Лилич [63] путем физико-химического анализа — спек-
трофотометрическим методом — систем, состоящих из иода и органического
вещества в четыреххлористом углероде, в растворах изо молярных
концентраций 0,002—0,06 моль/л. В качестве второго компонента были исследованы
пиридин, хинолин, диэтил-, дипропил-, дибензил-, дифенилсульфид, дипро-
пилдисульфид, изоамилселенид, диоксан, бутиловый спирт. Во всех
системах обнаружены соединения состава 1:1.
216
Так, например, доказательства обра;зования таких соединений
состава 1 : 1 дали спектрофотометрические исследования Кифера и
Эндрюса [6] систем из хлористого иода и бензола и некоторых его
производных (толуола, ксилолов, хлорбензола, бромбензола,
третичного бутилбензола) в четыреххлористом углероде, а также
спектрофотометрические исследования Лилича и Пресниковой [7\
систем JC1 (JBr) — диок-
сан (или метиловый
спирт) в среде СС14.
Кольтгоф с
сотрудниками [8] для
исследования системы JC1 —
пиридин в хлороформе или
нитробензоле применили
метод спектрофотометри-
ческого титрования. Они
определили величины эк-
стинкции в зависимости
от молярного отношения
Ру : JC1. Растворы
хлористого иода (б • 10-3-мол.)
титровали пиридином,
причем перегиб на кри-
3000
то
5000
6000
Рис. 37. Спектры поглощения растворов,
хлора (/), иода (//), хлористого иода (///)
и треххлрристого иода (IV) в
четыреххлористом углероде.
вых титрования приходится на отношение Ру : JC1=1 : 1.
Таким образом, спектрофотометрические исследования *
показали, что оптические (а также электрохимические) свойства
растворов хлористого и бромистого иода в растворителях второго
типа обусловлены образованием в них молекулярных соединений.
Большое значение имеет, однако, не только наличие комплексов,
но и их строение и состояние в растворах.
Что касается оптических свойств растворов треххлористого
иода, то они в значительной мере обусловлены тем, что JC13 в
растворах диссоциирует на JC1 и СЬ. Поэтому растворы JCl^ в
отношении окраски напоминают растворы хлористого иода.
Джилам и Мортон [2] показали, что кривая поглощения растворов
треххлористого иода в четыреххлористом углероде представляет собой
как бы сумму кривых поглощения растворов хлористого иода и
хлора (рис. 37).
МОЛЕКУЛЯРНОЕ СОСТОЯНИЕ ГАЛОГЕНИДОВ ИОДА В НЕВОДНЫХ
РАСТВОРАХ
Для исследования растворов галогенидов иода в неводных
растворителях, для выяснения природы ионов в этих растворах и
для решения некоторых других вопросов необходимо знать
молекулярное состояние галогенидов иода в неполярных и полярных
* Способность галогенидов иода образовывать соединения с органическими
веществами, принадлежащими к растворителям второго типа, установлена
также другими методами физико-химического анализа [9,10, 41], описанными
в следующей главе.
211
-40
растворителях. В качестве неполярного растворителя все
исследователи обычно применяли ССЦ.
В этом разделе приведены основные результаты сравнительно
немногочисленных исследований фазовых равновесий, а также
температур замерзания и кипения ряда двойных систем, содержащих
галогениды иода.
Корног и Олъсон [11] исследовали системы JU— >LU4 и
JC1—СНзСООН, изучив состав жидкой фазы, .находящейся в
равновесии с твердой фазой, в зависимости от температуры. Система
JC1—ССЦ оказалась очень
сложной, хотя в «ей и нет
химического взаимодействия.
В этой системе образуется
ряд твердых растворов;
кривая ликвидуса АС В имеет
S-образный характер, соли-
дус представлен кривой AD.
Эвтектика с т. пл. —26,4°
содержит 98,2 мол. % ССЦ.
Компоненты этой системы
образуют метастабильную
систему из двух жидких
слоев (кривая EFG) с
критической температурой
около 14° (рис. 38). В то же
время в системе JC1—
—СНзСООН обнаружена
лишь одна эвтектика,
содержащая 60 мол.% СНзСООН,
т. пл. равна —35,5° (рис. 39).
Чтобы показать, что характер взаимодействия хлористого иода
в таких системах определяется не только дипольным моментом
второго компонента, приводим иа рис. 45 диаграмму плавкости
системы JC1—пиридин, по данным Фиалкова и Музыки [9]. Пиридин
образует с хлористым иодом два соединения: довольно прочный
комплекс CsHsN-JCl и гораздо менее прочный комплекс
C5H5N.2JC1* (см. стр. 246).
Криоскопические исследования растворов хлористого иода в
уксусной кислоте [12], хлорокиси фосфора [13, 14] и бромофор-
~ме [14] дали величины молекулярного веса, соответствующие
мономерным формам. Однако для суждения о действительном
молекулярном состоянии хлористого (и бромистого) иода в этих и других
растворах необходимо знать также электрохимические свойства
последних.
Так, Брунер и Галецкий [15], Брунер и Бекьер [16] на
основании криоскопических измерений в нитробензоле пришли к
заключению о том, что хлористый иод мономолекулярен в этом
растворителе. Фиалков и Каганская [17] установили, что величина моле-
* Если судить по характеру кривой плавкости.
60
мол. % са+
Рис. 38. Система хлористый иод — четырех-
хлористый углерод, по данным Ольсона.
218
кулярного веса хлористого иода в нитробензоле больше теоретик-
ческой (162,4), а в интервале концентраций 2—6 мол.% (13
измерений) среднее значение молекулярного веса оказалось равным
189,6, причем не была обнаружена зависимость этой величины
ют концентрации. Так как эти растворы проводят ток (х25# ^
^Ю-4 ом—1смг1)9 то найденная величина молекулярного веса
является средней для .ионизированных и неионизированных молекул.
МОЛ.У.ЩС00И
Рис. 39. Система хлористый
иод—уксусная кислота по данным Корнога и
Ольсона.
Х/0г
1000 -л
800
Ш ]
2001
Рис. 40. Удельная
электропроводность растворов
хлористого иода и треххлори-
стого иода в уксусной
кислоте.
Поэтому можно было* предположить, что электролитической
диссоциации подвергаются, по-видимому, ассоциированные димерные
молекулы (JC1)2. Это заключение, вероятно, можно распространить
также на растворы хлористого иода в уксусной кислоте, если
принять во внимание результаты их электрохимического исследования
(стр. 221—222) *.
В литературе имеются и явно ошибочные суждения о
состоянии хлористого иода в растворах, основанные на том, что авторы
этих работ не учитывали химического взаимодействия. Так, Валь-
ден [18] указывает, что растворы хлористого иода в треххлористом
фосфоре фиолетового цвета и не проводят тока. Между тем, по
данным Фиалкова и Кузьменко [19], в системе JC1—РС13 происхо-
* Для бромистого иода подобные исследования еще не производились.
Однако большая аналогия в свойствах хлористого и бромистого иода
позволяет предположить, что молекулярное состояние этих двух межгалогенидо в
в растворах не различается в качественном отношении.
219
дят окислительно-восстановительные процессы, приводящие к
образованию комплекса РС15 • JC1:
PC!3 + 2JC1^PC15 + J2;
РС15 + JC1 = PC16J;
РС18 + 3 JC1 — PC16J + J2.
Фиолетовая окраска принадлежит одновременно
образующемуся иоду. Высокая электропроводность этой системы, достигающая
при 45° значений 10~2 ом~1см-\ обусловлена тем, что комплекс
PCbJ или [РСЦН^СЫ" имеет характер сильного электролита [20].
С повышением содержания РС13 электропроводность системы
уменьшается. Вероятно, этим обстоятельством следует объяснить
то, что Вальден [18], исследовавший лишь растворы малых
концентраций JC1 в РС13 не обнаружил электропроводности этой
системы.
Подобную ошибку в отношении системы JC1-—SbCl5 допустил
также Молес [21], по криоскопичееким исследованиям которого JCI
в пятихлористой сурьме мономолекулярен; причем Молес не
обнаружил в этой системе химического взаимодействия. Руфф с
сотрудниками [22] методом термического анализа системы JCI—SbCl*
обнаружили в ней два соединения SbCls • 2JC1 и SbCl5 • 3JC1,
образующие непрерывный ряд смешанных кристаллов и
диссоциирующих в расплаве (см. рис. 76). В дальнейшем Фиал-ков и Абарбар-
чук [23] показали, что система SbCIs—JCI хорошо проводит ток.
Изотермы удельной электропроводности при 55 и 65° имеют
максимум в области ~ 10 мол% SbCl5—1,25- Ю-2 ом~1см~1. В области
больших концентраций SbCU электропроводность резко падает —
до ~ \0~6 ом~1см—1 (см. рис. 92). Эти же авторы выделили
комплекс SbCb • 3JC1 и установили, что он имеет строение двойного
галогенида с иодом, как катионом (стр. 304).
Криоскопические исследования растворов треххлористого иода
в хлорокиси фосфора [13], уксусной кислоте [12], нитробензоле [15]
дали величины молекулярного веса, примерно вдвое меньшие
теоретического значения для JC13 (233,4). В отличие от этих фактов
криоскопические и эбулиоскопические исследования растворов
треххлористого иода в фосгене [24] дали величину, очень
близкую к 233.
Пониженный вдвое мол. вес, казалось бы, легко объяснить
тем, что треххлористый иод, вследствие значительной
термической неустойчивости, почти полностью диссоциирует на JC1 и хлор.
Однако это априорное объяснение требует экспериментального
подтверждения применительно к каждому растворителю.
Необходимо принимать во внимание также электрохимические
свойства растворов треххлористого иода и то понижение
молекулярного веса, которое обусловлено электролитической диссоциацией.
Нельзя также исключить предположения о том, что сольватация
треххлористого иода и продуктов его электролитической
диссоциации и образование комплексных молекул и ионов может повысить
его термическую устойчивость.
220
ЭЛЕКТРОХИМИЧЕСКИЕ СВОЙСТВА РАСТВОРОВ ГАЛОГЕНИДОВ
ИОДА
Электропроводность
Способность хлористого и бромистого иода проводить ток в
неводньтх растворах —в эфире — впервые обнаружил Солли [25]*.
Первые количественные исследования электропроводности
растворов галогенидов иода принадлежат Вальдену [18], измерившему
электропроводность растворов хлористого, бромистого и треххло-
ристого иода в жидкой двуокиси серы, хлористом сульфуриле и
треххлористом мышьяке. По данным Вальдена, эти
растворы—относительно слабые электролиты, с повышением температуры
электропроводность которых уменьшается, ттри низких температурах
эквивалентная электропроводность их увеличивается с разведением.
Позднее Брунер и Бекьер [161 повторили измерение
электропроводности этих галогенидов иода в жидком S02 и нашли
величины, значительно меньшие (до 100 раз), чем Вальден **: по их
данным, удельная электропроводность растворов хлористого
иода—порядка Ю-7—Ю-6 (в зависимости от концентрации), а
бромистого и треххлористого иода — порядка Ю-6 ом~1см~\
В дальнейшем была измерена электропроводность этих гало-
тенидов иода в нитробензоле [15, 17], уксусной кислоте [26];
бромистого и треххлористого иода в броме [27, 28] и треххлористого
иода в дихлоруксусной кислоте [28] ***, жидком сероводороде
{29]****, метилэтилкетоне [30]*****. Одрит и Бирр [31]
обнаружили электропроводность растворов хлористого иода в пиридине.
Для характеристики электролитных свойств растворов гало-
тенидов иода приведем более подробные данные для некоторых
из перечисленных выше систем.
На рис. 40 и 41, заимствованных из работы Брунса [26],
приведены изотермы удельной электропроводности растворов хлористого и
треххлористого иода и молекулярной электропроводности раствора
JC13 в уксусной кислоте. Растворы треххлористого иода оказались
лучшими проводниками тока, чем растворы хлористого иода. Как
правильно отмечает Брунс, этот факт (Противоречит мнению о полной
диссоциации треххлористого иода в этом растворе на хлористый
иод и хлор, так как, по данным этого же автора, растворы хлора
в уксусной кислоте не проводят тока.
Для сравнения электропроводности хлористого и
треххлористого иода в различных растворителях на рис. 42 приведены изотермы
* По данным Солли, растворы бромистого иода в сероуглероде и од-
нохлористой сере не проводят тока.
** Это расхождение с данными Вальдена Брунер и Бекьер объяснили
большей чистотой примененного .ими сернистого ангидрида.
*** Для 0,31%-ного раствора (v = 56 л) при 25° % = 0,6-10-6 олГ1 слГК
**** Для насыщенного раствора JCl3 при температуре твердой углекислоты
-х. = 13,42 • 10~7 олГ1 см-\
***** В интервале концентраций 1,95*10 —7,5-Ю-3 моль/ л при 25° т.
равна 3,9-Ю-5 — 8,7-10 ом"1 см~{. Электропроводность растворов изменяется во
времени вследствие реакции JCl3 с растворителем (происходит, вероятно, галоиди-
рование метилэтилкетона).
221
логарифм разведения б моль/п
Рис. 41. Молекулярная
электропроводность системы треххлористый иод —
уксусная кислота.
удельной электропроводности * растворов хлористого иода в нитро-
бензоле [17] и уксусной кислоте [32], треххлористого иода в броме
[28] и уксусной кислоте [32]. Отдельно на рис. 43 приведены изотер,
мы электропроводности
систем JC13—СНзСООН [261
и JC13 — C6H5N02 [15] для
интервала сравнительно
небольших концентраций **.
Хотя Брунер и Галецкий
получили для системы JQ3—
СбН5ГТО2 расходящиеся
данные для трех серий опытов,
тем не менее из рис. 43
видно, что нитробензольные
растворы треххлористого
иода, как и растворы хлористого иода (рис. 42), лучше проводят
ток, чем растворы в уксусной кислоте.
lOOOi
600
№
200
О
8
28
12 Ю 20 2+
мол % XL или М3
Рис. 42. Удельная электропроводность растворов
хлористого и треххлористого иода в нескольких
растворителях:
/—раствор треххлористого иода в броме, 2—раствор хлористого
иода в уксусной кислоте, 3—раствор хлористого иода в
нитробензоле, 4—раствор треххлористого иода в уксусной кислоте.
Величина удельной электропроводности растворов
треххлористого иода в трех растворителях соответствует в данном случае
последовательности их диэлектрических постоянных:
Br2 < CHgCOOH < C6H5N02
3,2
6,29
36,45
* С целью сопоставления электропроводность всех этих систем
рассчитана по данным цитированных здесь работ [15, 17, 28, 32] как функция
концентрации, выраженной в мол.%.
** Электропроводность системы JC13—Вг2 измерена при 25°, прочих си-
стем — при 18°.
222
Электропроводность растворов хлористого и треххлористого
иода в жидком -сернистом ангидриде, по данным Брунера и Бекье-
ра [16], имеет примерно одну и ту же величину*, несколько
меньшую, чем для системы JBr—-SO2.
Величины же электропроводности этих трех галогенидов иода
в жидком состоянии при температурах их плавления
располагаются в следующий ряд: JCl3>JCl>JBr (см. главу IX).
Попов и Скелли [66] измерили электропроводность растворов
хлористого, бромистого и цианистого иода в ацетонитриле. По
величине электропроводности эти системы располагаются в
следующий ряд:
JC1 > JBr > JCN
Концентрация, моль /л 6-10
Порядок
электропроводности 10
i-б
6-10"
10"
,-з
0,448
ю-6
Электропроводность этих систем немного увеличивается во
времени. Причины этого явления рассмотрены ниже (см. стр. 231).
К сожалению, небольшое
Ж-Ю9 . *
24
20 i
16 \
12
количество экспериментальных
данных по электропроводности
систем галогенидов иода с
различными растворителями и
отсутствие таких сведений для
растворов других
межгалоидных соединений не позволяет
сделать выводы общего
характера о зависимости
электропроводности таких систем
от природы межгалогенида и
растворителя.
Потенциал разложения
Для неводных растворов
хлоридов и бромида иода, как
и при исследовании
индивидуальных межгалогенидов в
жидком состоянии,
зависимость силы тока от
наложенного потенциала выражается
на /—1/-кривых прямыми
линиями без какого-либо
перегиба. Так, (потенциал
разложения не был обнаружен в
растворах треххлористого
иода в уксусной кислоте [32], в растворах JC13 или JBr в броме,
жидком сернистом ангидриде, треххлористом мышьяке, уксусной
кислоте [33], а также в наших исследованиях нитробензольных
растворов хлористого иода. Эти факты, очевидно, можно объяснить
* Немного большую для растворов хлористого иода. •
Н
0 0,4 Ofi 12 1,6мап.%ЛХ*
Рис. 43. Удельная
электропроводность растворов треххлористого иода
небольших концентраций:
/—раствор в нитробензоле, 2—раствор в
уксусной кислоте.
223
так же, как и аналогичные, описанные в предыдущей главе
явления, наблюдавшиеся при исследовании индивидуальных межга"
лоидных соединений.
Природа ионов в неводных растворах галогенидов иода и процесс
электролиза
Как уже отмечалось в предыдущих главах, для межгалоидных
«соединений характерна электролитическая диссоциация
ассоциированных молекул. Сопоставление данных по электропроводности
талогенидов иода с результатами определения молекулярного
состояния дает основание перенести это представление и на неводные
растворы межгалогенидов, в данном случае на хлориды и бромид
иода. Однако в литературе по вопросу о природе ионов в невод-
;ных растворах межгалогенидов и, в связи -с этим, о процессе
электролиза последних имеются 'Противоречивые суждения.
Так, Вальден [18] предполагал, что электропроводность
галогенидов иода и их растворов обусловлена диссоциацией на ионы в
■мономерной форме, например JC1 ^ J++ С1~ или JC13^J3+ + ЗС1~.
Другие авторы, исходя из опытов по электролизу, полагают, что
электролитической диссоциации подвергаются продукты
соединения галогенидов иода с молекулами растворителя.
Решение этих вопросов (представляет интерес при исследовании
не только межгалогенидов и их растворов, но и большой группы
■систем, образованных галогенами (бромом, иодом) или галогени-
дами иода 'и различными растворителями (большей частью
органическими веществами). Таковы, например, следующие системы:
ацетамид — бром [34—37], бензамид—бром [38], пиридин — бром
[39], эфиры * — бром [40], нитробензол — бром (иод) [15],
пиридин— хлористый иод [9, 10, 41], ацетамид (или бензамид)
—хлористый иод [42], бензамид — бромистый иод [43], уксусная
кислота — треххлористый иод [32] и др. (см. главу XI).
Наличие комплексных соединений в этих системах доказано
получением соответствующих кристаллических продуктов, например
СбН*С01ЧН2 • Вг2 (комплекс Лорана) [441, C5H5N • Вг2 HC6H5N-Br4
[45], (СНз)20-Вг2 [46, 47] и (С2Н5)2О.Вг3 [47] или результатами
физико-химического исследования.
Все эти системы оказались очень хорошими проводниками
электрического тока, с удельной электропроводностью,
достигающей в большинстве случаев значений 10~3—\0~~2 омгхсмгх, как,
например, в системах с ацетамидом, бензамидом, эфирами и
некоторыми другими веществами.
Для выяснения природы электролитов в этих системах большой
интерес представляют результаты, полученные в опытах по
электролизу. Оказалось, что молекулы органического вещества
переносятся к катоду, а галогены (в том числе иод и хлор, входящие
* Исследовались системы с эфирами: метиловым, этиловым, пропиловым,
>изоамиловым, дихлорэтиловым и метилалем.
224
0 состав JC1 и JiCl3), (выделяются на аноде. Так, три электролизе
бромных растворов ацетамида на серебряном аноде выделялся
бром (с отклонением от закона Фарадея на 10—30%), а на
катоде выделились кристаллы ацетамида. Кроме того, при измерении
э. д. с. концентрационного элемента CH3CONH2—Br2 оказалось, что
ацетамид переносится в направлении тока, т. е. образует ионы с
положительным зарядом [34].
Аналогично ведет себя система CeHsCONHs—Вг2. При
электролизе в нитробензольном растворе [38] на аноде выделяется бром
приблизительно в соответствии с законом Фарадея, а на
катоде—кристаллы бензамида. При электролизе системы C5H5N—Br2
обнаружено, что пиридин переносится из анодного в катодное
отделение, причем на 1 F переносятся 2 моля пиридина [39]. При
электролизе бромо-эфирных растворов на аноде выделяется бром
в соответствии с законом Фарадея [40].
При электролизе нитробензольных растворов брома или иода
эти галогены выделяются на аноде, причем, по данным Брунера
и Галецкого, выход соответствует почти точно 1 г-экв брома или
иода на 1 F. При длительном электролизе весь галоген осаждается
на аноде, а нитробензол остается неизменившимся [15].
Подобные явления наблюдались также при исследовании
систем с галогенидами иода. Электролиз нитробензольных растворов
хлористого, бромистого и треххлористого иода впервые изучали
Брунер и Галецкий [15]. По данным этих авторов, весь галоген —
иод и хлор или иод и бром — переносится к аноду и выделяется
на нем. При этом сперва выделяется лишь хлор или бром,
образуя с серебром анода AgCl или AgBr, затем в электролитическом
процессе начинает принимать участие также иод, выделяясь на
аноде и образуя на нем AgJ, причем в обеих стадиях на 1 F
выделяется 1 г-экв галогена. В конце концов, раствор может быть
полностью освобожден от галогенов. На серебряном катоде
выделение галогенов не происходит.
Аналогичные факты обнаружены Брунером и Галецким при
изучении подвижности галогенов и их соединений — JC1, JBr,
JC13—в нитробензольных растворах, в этих опытах также
наблюдался перенос хлора и иода (или брома и иода) к аноду.
Количественная характеристика результатов этих опытов для растворов
JC1 не является вполне определенной, о чем пишут авторы этого
исследования. В отличие от опытов по электролизу JC1, в которых
прохождению 1 F соответствовало выделение 1 г-экв галогенов, в
данных опытах 1 F соответствует перенос 1,92—2,35 г-экв
галогенов.
Электролиз растворов бромистого и треххлористого иода в
жидком сернистом ангидриде с серебряными электродами,
произведенный Брунером и Бекьером [16], показал полную аналогию
этих систем «с нитробензольными растворами галогенидов иода.
По данным Брунса [32], при электролизе растворов
треххлористого иода в уксусной кислоте на серебряном аноде образуется
хлористое серебро лишь со следами йодистого серебра, причем
15—1419
225
осаждались 2 г-экв хлора на 1 F. Серебряный катод весьма незна.
чительно увеличивается в весе.
Авторы некоторых указанных выше работ [15, 32, 34, 40]
приходят к заключению, что комплексное соединение, образую,
щееся в исследованных системах и являющееся © них электро-
литом, 'подвергается электролитической диссоциации на
составляющие его компоненты, т. е. на молекулы растворителя и рас-
творенного вещества, но уже несущие каждая противоположный
электрический заряд. При этом молекулы ацетамида, бензами-
да, эфиров, нитробензола, сернистого ангидрида и т. п. являются
катионами, а молекулы брома или галогенидов иода образуют
анионы, как, например,
C6H5CONH2Br2^:C6H5CONH2++ + 2Br- [38];
JC13-лСН3СООН Z (СН3СООН)п + + JCI— f32^
JC13.C6H5N02 ^CeH6NO++ + JC1— [15].
Такая схема электролитической диссоциации комплексов
впервые была предложена Вальденом в отношении систем: третичный
амин (хинолин) —сернистый газ или триарилметил (С6Н5)зС—S02
[18, 48]. На возможность электролитической диссоциации подобного
рода указывал также Плотников на основании измерений
электропроводности системы этиловый эфир—бром [491 применительно к
комплексу Шютценбергера (СгНбЬО-Вгз или [(С2Н5)20- Br3k
образующемуся в указанной системе; при электролизе растворов
этого же комплекса в хлороформе наблюдалось выделение брома на
аноде в соответствии с законом Фарадея [50] *.
Аналогичное суждение было высказано Брунером и Галец-
ким [151, Брунером и Бекьером [16] и Брунсом [32, 40]
применительно к исследованным ими системам, в которых, как указывают эти
авторы, образуются оксониевоподобные соединения галогенидов
иода с молекулами растворителя. По мнению этих авторов,
приведенная выше схема электролитической диссоциации, согласно
которой в исследованных системах отсутствуют катионы галогенов,,
объясняет явления, наблюдавшиеся ими при электролизе
растворов галогенидов иода — отсутствие выделения иода на серебряном
катоде и др. В частности, тот факт, что анодный осадок при
электролизе растворов ЛС1з в уксусной кислоте состоит
преимущественно из хлористого серебра (2С1 на IF), Брунс объясняет следующим
образом: первичный разряд ионов ЛС1з должен дать на аноде
3AgCl + AgJ (на 2 Т7), но йодистое серебро в результате реакции с
хлористым иодом превращается в хлористое.
Однако дальнейшее электрохимическое изучение неводных
растворов хлористого иода дало основание иначе представить
состояние галогенидов иода в растворах и по-иному объяснить
электродные процессы, происходящие при электролизе этих растворов. Так,
* В этой работе Плотников, однако, высказывает также предположение,
что не весь бром этого комплекса переходит в ионизированное состояние и
что часть брома может входить в состав катиона.
226
5лектролиз растворов хлористого иода изучали также Сандонини
и Бергело [511, применяя в качестве растворителя ледяную
уксусную кислоту и нитробензол. Электролиз производился в сосуде с
диафрагмой из пористого иенского стекла; при этом определялось
не только общее количество галогенов в анодном и катодном
распорах, но ,и отношение J: О (в грамм-атомах) до и (После элек-
тролиза.
При электролизе растворов хлористого иода в ледяной уксусной
кислоте оказалось, что анолит обогащается обоими галогенами,
но отношение J : С1 уменьшается сравнительно с начальным, в ка-
толите же это отношение, увеличивается. Если электролизу
подвергались достаточно концентрированные растворы, то на катоде
(и на стенках) выделялся кристаллический осадок чистого иода.
Прохождение 1 F соответствовало обогащению анолита
приблизительно 3 г-экв галогена (2 г-экв хлора и 1 г-экв иода).
Полученные результаты Сандонини и Бергело объясняют тем,
что хлористый иод в растворе находится в виде автокомплекса
JfJCb], хотя и в небольшом количестве, так как, по данным Штор-
тенбекера [12], мол. вес JC1 в уксусной кислоте почти равен
теоретическому. В первый период электролиза в катодном пространстве
разряжаются положительно заряженные ионы иода.
Выделившийся иод может далее переноситься к аноду.
Аналогичные явления наблюдали Сандонини и Вертело "при
электролизе растворов хлористого иода в нитробензоле, но с тем
отличием, что (прохождение 1 F соответствовало обогащению анолита
приблизительно 2 г-экв галогенов — немного меньше 1 г-экв для
иода и немного больше 1 г-экв для хлора.
Таким образом, работа Сандонини и Бергело не подтвердила
всех выводов, сделанных Брунером и Галецким [15], и указывает
на более сложный механизм электролитической диссоциации и
электролитического разложения хлористого иода в неводных
растворах (в частности, в нитробензольном растворе).
Такой же вывод следует из опытов Фиалкова и Каганской [52]
по электролизу раствора хлористого иода в нитробензоле. В
процессе электролиза происходило уменьшение содержания
электроположительного иода вследствие его восстановления до ^2- В анодром
осадке на серебряном аноде, приготовленном пр ррунеру, найдены
AgCl и AgJ. При прохождении 1 F выделяется, около i,5 г-экв
хлора. Количество же иода, выделившееся на аноде, значительно
меньшее, чем 1 г-экв J «a 1 F.
Эти результаты показывают, что при электролизе нитробензоль-
ных растворов хлористого иода на аноде осаждаются оба
галогена—хлор и иод, как это наблюдали Брунер и ТалёЦкий [15].
Однако схема электролиза, данная Брунером и Галецкййй.заключаю-'
щаяся в том, что на аноде сначала выделяется хлор, а затем,
позднее, и иод, причем в обеих стадиях на 1 F выделяется I, г-экв
галогена, нашими опытами не подтвердилась: результаты анализа
анодных осадков и раствора после электролиза показали,1 что зто вре-}
мя как в растворе еще есть хлор, на аноде начинает выделяться
также и иод.
227
Эти данные, а также результаты криооколического иоследо;ваНй
системы хлористый -иод—нитробензол (см. стр. 219) [17] 'Подтв^^
ждают мнение Сандонини и Бергело и дают основание считать
что ассоциированные молекулы хлористого иода в этой системе (и*
в других аналогичных растворителях) диссоциируют на ионы J+
и JC12~ *, вероятно, сольватированные молекулами растворителя
Это представление можно распространить и на другие рассма-ь
риваемые здесь галогениды иода.
Общее решение вопроса о природе ионов в данных системах ц
о процессах, происходящих при электролизе, возможно лищь в
связи с представлением о механизме образования комплексных
соединений указанного выше типа ** и о поведении при этом мс>
лекул галогенов или галогенидов иода.
В качестве одного из компонентов этих систем выступает
соединение, содержащее атомы со свободными электронными парами
(доноры электронов), как, например, амины, гетероциклические
азотистые основания, карбоновые кислоты, эфиры и другие
соединения. Молекулы этих веществ обычно тюлярны. Вторым
компонентом этих систем являются сравнительно легко
поляризующиеся молекулы галогенов — брома и иода — или дшольные, хотя
и с небольшим дипольным моментом, молекулы галогенидов иода
JC1, JBr, JC13 (см. табл. 4).
Хотя молекулы галогенов в индивидуальном состоянии неш>
лярны, они обладают способностью легко поляризоваться,
особенно под влиянием химических факторов, в том числе и комплексо-
образования, причем этот процесс может привести к диссоциации
молекул галогенов на положительно и отрицательно заряженные
ионы, например:
J2 £ J+ -f J~ (ит:и J2 ^ : J + -f : J:").
Подобное предположение было высказано еще Вальденом [18]
для объяснения электропроводности растворов иода и М. А.
Рабиновичем [54] для объяснения природы электропроводности брома
и иода в индивидуальном состоянии.
Одним из наиболее убедительных доказательств
электролитической диссоциации молекул галогенов являются работы
М. И. Ушакова с сотрудниками [55, 56]. Они получали нитраты
галогенов при взаимодействии галогенов с нитратом серебра в
спиртовых растворах, например,
J2 + AgN03 = AgJ + JNO3 ***.
* Такое же мнение высказывает Шарп [53], основываясь на работах
Сандонини и Бергело, Фиалкова и Каганской.
** Доказательство образования комплексных соединений этого типа,
природа связи в них, сравнительная характеристика их устойчивости и другие
свойства будут описаны в следующей главе.
*** Фиалков и Каганская доказали ^наличие катионов иода в спиртовых
растворах, получаемых по этой реакции," а также способность галогенидов
иода JC1 и JBr реагировать с нитратом серебра аналогично иоду
JCl+AgN03=AgCl+JN03 [57].
228
Аналогичное заключение позволяют сделать работы Файгля
[58, 59] по получению сахарината иода и работы Биркенбаха [60]
с сотрудниками по получению кислородных солей иода.
Галогениды иода, имеющие полярный характер и довольно
значительную электропроводность даже в индивидуальном
состоянии, способны подвергаться дальнейшей поляризации и
электролитической диссоциации в значительно большей степени, нежели
молекулы галогенов, образуя при этом катион иода и
соответствующий анион второго галогена, например:
JC1—JT+ :C1:~
или
(JC1)2^J+ + JC!-
Вполне возможно, что такая же диссоциация галогенов и гало-
генидов иода на положительно и отрицательно заряженные ионы
происходит также под влиянием поляризующего и сольватирующе-
го действия указанных выше молекул органических соединений.
Этот процесс должен привести к образованию комплексных
соединений таких молекул с молекулами галогенов или галогенидов
иода, так как положительно заряженные ионы галогенов, будучи
акцепторами электронов, присоединяются к донорам электронов,
содержащимся в молекулах растворителей, и образуют с ними
комплексный катион. В последнем координационно-ковалентная
связь осуществляется по типу «ониевых» соединений, как это
обычно бывает, когда молекулы аминов, эфиров и т. п. соединений
выступают в качестве одного из компонентов комплексных
молекул, а вторым компонентом являются акцепторы электронов.
Анионами образующихся таким образом комплексных молекул
являются отрицательно заряженные атомы (анионы) галогенов.
Если молекулу органического соединения выразить через RA:
(А — донор электронов — атом азота или кислорода), а молекулы
галогенов или галогенидов иода — соответственно через Х2 или JX,
то образование и электролитическая диссоциация комплексных
молекул этих компонентов могут быть представлены следующим
образом:
RA: + Х2 = [RA :Х:] :Х:■£ IRAXJ+ + X"
или
RA: + JX = [RA :J:] :Х: ^ [RAJ]+ + X"
Так, например, для хорошо проводящей ток системы ацет-
амид — бром и для комплексных соединений пиридина C5H5N • Вг2
и C5H5N • JBr эта схема принимает следующий вид:
CH3CONH2 + Br2 ^ [CH3CONH2Br]+ + Br~; [36]
[C5H5N :Вг:] :Вг: £ [C6H6NBr]+ f Br" *;
* Следует отметить, что еще Ганч и Граф [61] рассматривали полученные
ими соединения аминов с бромом (CH3)3N-Br2 и ВгС6Н4Ы(СНз)2-Вг2 как гало-
гензамещенные аммонийные соли формулы [R3NBr]Br, т. е. как производные
аммониевых соединений, в которых атом водорода замещен на галоген.
229
[C5H5NJ] Br ^[C5H5NJ]+ + Br-.
При электролизе подобных комплексных соединений анион га.
логена разряжается на аноде (Br~—e=V2Br2), комплексный катион
переносится к катоду, где и разряжается, в результате чего
освобождаются как молекулы органического вещества, так и атомы
галогенов, например,
[C5H5N- Br]+ + е -> C5H5N + -[ Вг2;
[C6H6NJ]++e^QH6Nf -2"J2.
Выделившиеся у катода галогены не осаждаются на
серебряном катоде (не образуют с ним галогенида серебра), но могут
вновь соединяться с растворителем, образуя комплекс RA • Х2 (или
[RA • Х]Х), <в котором половинное количество «катодного»
галогена является анионом и переносится к аноду, где и осаждается
так же, как и первоначальные анионы этих комплексных молекул.
Если подвергать электролизу систему, содержащую галогенид
иода, например систему нитробензол — хлористый иод, то,
согласно приведенным здесь соображениям, простейший состав
первичного комплекса выражается формулой [C6H5N02 • J]C1, вторичный
комплекс представляет собой C6H5N02 • J2. Поэтому в процессе
электролиза сперва должен выделяться на аноде лишь хлор, а
затем, по истечении некоторого времени, и иод, что действительно
наблюдалось Брунером и Галецким и подтверждено Фиалковым и
Каганской. Эти же соображения согласуются с наблюденным Сан-
донини и Бергело [51] фактом увеличения при электролизе
системы уксусная кислота — хлористый иод отношения J : С1 в като-
лите и уменьшения этого отношения в анолите одновременно с
обогащением последнего обоими галогенами.
Возможно и иное объяснение указанных фактов,
заключающееся в том, что атомы галогенов, образовавшиеся у катода при
разряде комплексного катиона, вследствие большого сродства
к электронам, принимают от катода еще один электрон *, после
чего начинают переноситься к аноду, где и разряжаются. Исходя
из этого, разряд комплексного катиона можно представить
следующей общей схемой: [RX]++2e->- R + X~.
Такое катодное восстановление иода было доказано Ф. Д.
Шевченко [62] ** при изучении электролиза и переноса ионов в раство-
Аналогично этому Плотников и Михайловская [39] указывают, что
комплексное соединение пиридина с бромом C5HsN-Brn подвергается электролитической
диссоциации по схеме C5H5NBrn= CsHsNBr^ + Br~.
* Этим можно объяснить тот факт, что разряжающиеся на серебряном
катоде атомы галогена не взаимодействуют с ним.
** В диссертационной работе, выполненной под руководством автора-
230
pax полииодидов калия и аммония или комплексных катионов
(аммиакатов ряда металлов). '
Таким образом может быть объяснена отмеченная выше
особенность рассматриваемых здесь систем, заключающаяся в том,
что при их электролизе молекулы растворителя переносятся к
катоду, а галогены полностью выделяются лишь на аноде.
Предложенное нами объяснение природы ионов во всех
системах указанного выше типа более вероятно, нежели та схема
электролитической диссоциации, которая заключается в ионизации
комплекса на положительно заряженные молекулы растворителя
и отрицательно заряженные галогены или галогениды иода, так
как последняя схема требует перехода электронной пары от
молекул растворителя к молекулам галогенов; этот процесс
энергетически менее вероятен вследствие того, что потенциал ионизации
аминов, эфиров, нитросоединений с отщеплением от них
электронов должен быть весьма значительным.
Для суждения о природе ионов в неводных растворах
хлористого и бромистого иода, проводящих ток, большой интерес
представляют недавно опубликованные результаты спектрофотометриче-
ских исследований растворов хлористого иода [64—€6],
подтвердившие наличие в этих растворах ионов JC1~ ^-^ заключение сделано
на основании того, что максимум поглощения 350—360 ммк,
характерный для растворов хлористого иода в растворителях, с
которыми он образует соединения, очень близок к максимуму поглощения
дихлороиодаатов M+JC1^ во многих растворителях.
В то же время спектр поглощения растворов хлористого
иода в .нитрилах (CH3CN, C2H5CN, C4H9CN) и в спиртах
(С2Н5ОН, (СНз)зСОН) имеет максимум также вблизи 225—
229 ммк, характерный для ионов JCl^, но не для самого JC1.
Высота этого максимума растет во времени в случае растворов в
CH3CN, C2H5CN и (СНз)зСОН.
По Баклесу и Милсу [65], эти факты дают основание полагать,
что в таких системах хлористый иод реагирует с молекулами
растворителей или с содержащимися в них электронодонорными
примесями по такой схеме:
В + JCl^BJCl;
2BJC1 Z BJ+ + JCli".
Подобное же изменение (во времени) оветопоглощения растворов
хлористого иода в ацетонитриле наблюдали Попов и Скелли [66].
Одновременно, измеряя электропроводность растворов галогенидов
иода (JC1, JBr, JCN) в ацетонитриле, они обнаружили увеличение
электропроводности во времени; например, для раствора
1,52- 10~2 моль/л JC1 электропроводность изменилась от 2,01 • 10~5
до 11,36- Ю-5 ом~1см-х в течение 82,5 часов. Измерения в
запаянной аппаратуре показали значительно меньшее увеличение
электропроводности во времени (для 3,65 • 10~2-мол. раствора JC1 от
1,18-Ю"5 до 4,52-Ю-5 ом-1см~х за 210 часов). Такие же явле-
23!
ния наблюдались для растворов бромистого и цианистого иод^
(рис. 44).
Попов и Скелли склонны объяснять результаты этих спектро-
фотометрических измерений и определения электропроводности
взаимодействием галогенидопв иода с примесями (влагой), но не
образованием соединений с растворителем. Однако можно полагать,
что это мнение недостаточно-
обосновано, по крайней мере, &
тех случаях, когда измерения
(Производились в условиях,
предотвращающих попадание влаги.
Необходимо принять во
внимание, что эти явления
наблюдались в растворителях, молекулы
которых обладают электронодо-
норными свойствами, что создает
возможность присоединения к
ним ионов J+ — акцепторов
электронов. Подобные же явления
изменения электропроводности во
времени наблюдались в ряде
других неводных систем, в которых
доказано образование
комплексов, причем удалось проследить
кинетику процесса и вычислить
(приближенно) константу
скорости реакции комплексообра-
зования 167—7UJ. В частности, это относится к системе пиридин—
иод, физико-химические свойства которой — электропроводность
порядка Ю-5— Ю-4 омг1см-х и % ^ 130—132 ом'1см2 [31], а
также дипольный момент, равный 4,17D [71],— подтверждают
образование в этой системе высокополярного комплекса [C5H5N—J]+J~
или [CsHsN-JftJar.
Одрит и Бирр [31] полагают, что в системе пиридин — иод
возможны такие равновесия:
Ру + J2iPy-J2^:JPy+ + J~ ;J Ру"-++ 2J"; (ЮЛ)
2[PyJ2]^JPy+ + JPyJ^ (Ю-2)
Высокое значение X ^ эти авторы объясняют образованием
тройного комплекса (Py^^J-) по схеме (10.1). Но нельзя
исключить объяснения, основанного на предположении о большой
подвижности ионов иода (сольватированных пиридином) в этой
системе, обусловливающей высокое значение Я^, что делает
излишним предположение о существовании ионов Ру*4" в системе
иод — пиридин. .
Столь подробное обсуждение работ Баклеса и Милса, Попова
и Скелли вызвано тем, что на основании этих работ можно
объяснить сбразование в растворах хлористого и бромистого иода ионов
хю5
40 80 120 №
Время, час
Рис. 44. Изменение удельной
электропроводности ацетонитриль-
ных растворов хлористого и
бромистого иода во времени:
1 — раствор бромистого иода 5,95-10—3
моль /л; 2 — раствор хлористого иода
1,54-Ю-2 моль/л; 3— раствор
хлористого иода в запаянной аппаратуре
3,65 1О-2 моль/л. Значения ординаты
для кривой 1 нужно уменьшить в
10 раз.
232
JCb и ЛВгг не только наличием в растворе димерных молекул и их
диссоциацией по схеме (JX)2 ^ J++ JX7, но и иным путем,
включающим взаимодействие с растворителем*. Для решения этого вопроса
и дифференциации обоих путей образования ионов JX<T в растворах
хлористого и бромистого иода в зависимости от природы растворителя
необходимы дальнейшие исследования.
То обстоятельство, что в растворах хлористого и бромистого
иода, наряду с ионами С1~ и Вг~, возможны и комплексные
анионы JX2~,He изменяет сущности приведенной выше схемы
электродных процессов. (Процесс на серебряном аноде в этом случае
должен выразиться схемой
JX7 - е + 3Ag -* AgJ -f 2AgX,
объясняющей осаждение на аноде иода и хлора при электролизе
нитробензольных растворов хлористого иода.
Что касается природы ионов в неводных растворах треххло-
ристого иода и процессов, происходящих при их электролизе, то
по этому вопросу пока еще трудно высказать вполне
определенные суждения. Причиной этого является сложность равновесий,
существующих в растворах треххлористого иода, обусловленная
его легкой термической диссоциацией на хлористый иод и хлор,,
своеобразием его строения, возможностью ступенчатой
электролитической диссоциации и диссоциации ассоциированных
молекул. Эти факторы создают возможность разнообразия ионных
форм в растворах треххлористого иода, наличия в них ионов JCI2V
J3+, J+, Cl~, JC1 Г»[Оз]~. Кроме того, нужно принять во внимание и
возможность хлорирующего действия треххлористого иода в
отношении ряда растворителей, что /приводит к образованию НС1 и:
ионов ЛС1Г-
Экспериментальные данные по вопросу об электрохимических
свойствах растворов треххлористого иода еще недостаточны, и.
на их основании нельзя составить более или менее четкое
представление о природе ионов, согласованное с количественными
результатами опытов электролиза. Так, Брунер и Галецкий [15]
отмечают, что при электролизе растворов JCU в нитробензоле на
аноде выделяется сперва хлор — 1 г-экв на 1 F. Длительный
электролиз приводит к полному выделению растворенного JCb, причем
процесс в конце концов сводится к электролизу хлористого иода и
иода.
Предлагаемая Брунсом [32] схема электролитической
диссоциации и электродных процессов при электролизе системы
СНзСООН—JC13 (стр. 226), удовлетворительно сочетается с его
экспериментальными данными (выделение на аноде 2 г-экв О
на 1 F)y но эти данные можно объяснить и иным путем, принимая,
* К сожалению, для исследованных этими авторами систем (кроме системы*
с уксусной кислотой) отсутствуют криоскопические данные.
233.
что на аноде разряжаются не молекулярные ионы JC1 з , а ионы
С1- и [С13]-.
Дальнейшие исследования строения и молекулярного
состояния треххлористого иода в растворах и электрохимических свойств
последних, вероятно, внесут ясность в этот вопрос.
ЛИТЕРАТУРА
1. Gmelin's Handb. d. anorg. Chem., В. 8, 111, 616, 637 (1931).
2. A. E. G i 1 1 a m, R. A. Morton, Proc. Roy. Soc, 124, 604 (1929)-
132, 152 (1931).
3. A. E. G i 1 1 a m, Trans. Farad. Soc, 29, 1132 (1933).
4. N. W. S i d g w i с k, The chemical elements and their compounds,
Oxford, v. II, 1950, 1151, 1154, 1157.
5. A. H. T e p e и и н, Усп. химии, 24, 121 (1955).
6. R. Keefer, L. J. A n d r e w s, J. Am. Chem. Soc, 72, 4677, 5170
(1950).
7. Л. С. Л и л и ч, О. Е. П р е с н и к о в а, Уч. зап. ЛГУ, сер. хим.
наук, вып. 12, 3 (1953).
8. I. М. К о 1 t h о f f, D. S t о с е s о с а, Т. S. L e e, J. Am. Chem. Soc,
75, 1834 (1953).
9. Я- А. Ф и а л к о в, И. Д. М у з ы к а, ЖОХ, 18, 1205 (1948).
10. Я- А. Ф и а л к о в, И. Д. Музыка, Изв. сект, физ.-хим. анализа
ИОНХ АН СССР, 19, 314 (1949).
П. J. Co r nog, L. Olson, J. Am. Chem. Soc, 62, 3328 (1940).
12. W. S t о r t e n b e k e r, Z. phys. Chem., 10, 183 (1892).
13. G. Oddo, Gaz. chim. ital., 31, II, 146 (1901).
14. П. Вальден, Z. anorg. Chem., 68, 307 (1910).
15. L. В rune r, A. G a 1 e с k i, Z. phys. Chem., 84, 515 (1913).
16. L. В r u n e г, Е. В e k i e r, Z. phys. Chem., 84, 548 (1913).
17. Я. А. Ф и а л к о в, К. Я- К а г а н с к а я, ЖОХ, 16, 1981 (1 946)
18. П. В а л ь де н, Z. phys. Chem:, 43, 385, 425 (1903).
19. Я- А. Ф и а л ко в, А. А. Кузьменко, ЖОХ, 19, 1645 (1949).
20. Я- А. Ф и а л к о в, А. А. К у з ь м е н к о, ЖОХ, 19, 812 (1949).
21. Е. Moles, Z. phys. Chem., 90, 70 (1915).
22. О. R u f f, J. Z e d n e r, L. H e с h t, Ber., 48, 2068 (1915).
23. Я- А. Фиалков, И. Л. Абарбарчук, Укр. хим. ж., 15, 176,
372 (1949).
24. Е. Beckmann, F. J u n k e r, Z. anorg. Chem., 55, 371 (1907).
25. E. Solly, Phil. Mag., (3), 8, 133 (1836); J. pr. Chem., 7, 415 (1836).
26. Б. П. Брунс, Z. phys. Chem., 118, 89 (1925).
27. В. А. П л о тн и ко в, В. Е. Рокотян, ЖРФХО, 45, 193 (1913).
28. В. А. Плотников, В. Е. Рокотян, ЖРФХО, 47, 723 (1915).
29. G. N. Q u a m, J. A. W i 1 k i n s о n, J. Am. Chem. Soc, 47, 989 (1925).
30. Е. J. Birr, Z. phys. Chem., 165, 311 (1933).
31. L. F. A u d r i e t h, E. J. В i r r, J. Am. Chem. Soc, 55, 668 (1933).
32. Б. П. Б р у н с, Z. phys. Chem., 118, 89 (1925).
33. B.C. Финкельштейн, Z. phys. Chem., 115, 303 (1925).
34. В. О. П л о т н i к о в, С. I. Я к у б с о н, Зап. 1н-ту xiM., 1, 185 (1934).
35. С. И. Я к у б с о н, М. А. А б р а м о в а, Укр. хим. ж., 15, 136 (1949).
36. М. С. А ш к и н а з и, ЖОХ, 2, 790 (1932).
37. Е. Я- Г о р е н б е й н, П. И. Смоленцев, Сб. статей по общей
химии, 2, 809 (1953).
38. В. С. Финкельштейн, Z. phys. Chem., 121, 46(1926); 131, 338(1928).
39. В. О. П л о т н i к о в, В. I. Михайловська, Зап. 1н-ту xiM., 7,
в. 1, 85 (1940).
40. Б. П. Б р у н с, Z. anorg. allg. Chem., 163, 120 (1927).
41. И. А. Шека, ЖФХ, 30, 109 (1956).
42. Я- А. Ф и а л ко в, И. Д. М у з ы к а, ЖОХ, 18, 802 (1948); 19, 1416
(1949).
234
43. И. Д. Музыка, Укр. хим. ж., 16, 331 (1950).
44. Beilst. Hanub., В. 9, 200 (1926).
45. P. F. Т г о w b r i d g е, О. С. D i e h e, J. Am. Chem. Soc, 19, 558
(1897); Beilst. Handb., B. 20, 189 (1935).
46. D. Mcintosh, J. Chem. Soc, 87, 789 (1905); J. Am. Chem. Soc, 33,
71 (1911).
47. P. S с h u t z e n b e г g e г, С. г., 75, 1511 (1872); Ann. Chim., 187, 86
(1873).
48. П. Вальде н, Электролиты и растворители, Труды юбилейного
Менделеевского съезда, 1, 528 (1936).
49. В. А. Плотников, ЖРФХО, 38, 1096 (1906).
50. В. А. Плотников, ЖРФХО, 44, 1919 (1912).
51. С. S a n d о n i n i, N. В е г g h е 1 1 о, Atti d. Reale Acad. d. Lincei,
25 (6), 46 (1937).
52. Я- А. Ф и а л к о в, К. Я- К а г а н с к а я, ЖОХ, 18, 289 (1948).
53. A. G. Sharpe, Quart. Rev., 4, 115 (1950).
54. М. А. Р а б и н о в и ч, Z. phys. Chem., 119, 78 (1926).
55. М. И. Ушаков, ЖОХ, 1, 1260 (1931); 4, 194 (1934).
56. М. И. Ушаков, В. С. Чистов, ЖОХ, 5, 1387 (1935); 7, 253
(1937).
57. Я- А. Ф и а л к о в, К. Я- К а г а н с к а я, ЖОХ, 14, 3 (1944).
58. F. F e i g 1, Е. С h а г g о f f, Monatsh., 49, 417 (1927).
59. F. F e i g 1, А. В о n d i, Monatsh., 53/54, 508 (1929).
60. L. Birkenbach u. a., Ber., 63, 2544 (1930); 64, 961 (1931); 65,
395, 1949 (1932); 66, 1280, 1571 (1933); 67, 917, 1420, 1729 (1934).
61. A. Han tsch, W. Graf, Ber., 38, 2154 (1905).
62. Ф. Д. Шевченко, Полииодиды комплексных неорганических
катионов, Дис, К., Ун-т, 1951.
63. С. А. Щ у к а р е в, Л. С. Л и л и ч, Уч. зап. ЛГУ, сер. хим. наук,
вып. 11, 3 (1952).
64. R. Е. В иск les, J. F. Mills, J. Am. Chem. Soc, 75, 552 (1953).
65. R. E. В и с к 1 е s, J. F. Mills, J. Am. Chem. Soc, 76, 4845 (1954).
66. A. J. Popov, N. E. Skelly, J. Am. Chem. Soc, 77, 3722 (1955).
67. В. О. П л о т н i к о в, Е. Б. Г i т м а н, Зап. 1н-ту xiM., 7, 351 (1940).
68. А. В. Б е р н ш т е Й н, ЖОХ, 11, 901 (1941).
69. В. О. П л о т н i к о в, С. Й. Я к у б с о н, Зап. 1н-ту xiM., 2, 99 (1935).
70. Е. Я- Горенбейн, ЖОХ, 6, 1230 (1936); 9, 1369 (1939).
71. Я. К. С ы р к и н, К- М. А н и с и м о в а, ДАН СССР, 59, 1457 (1948).
ГЛАВА XI
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ ГАЛОГЕНИДОВ ИОДА
(JC1, JBr, JC18) С ОРГАНИЧЕСКИМИ ВЕЩЕСТВАМИ
Межгалогениды, в том числе и галогениды иода, образуют
комплексные соединения преимущественно двух типов:
а) продукты присоединения молекул, главным образом
органических веществ, содержащих доноры электронов — атомы
кислорода, азота и др.;
б) продукты -присоединения галогенидов других элементов —
комплексы типа двойных галогенидов.
Примеры этих двух типов комплексных соединений
межгалогенидов приведены в конце главы I и более подробно — при
описании трехфтористого брома (для которого известны только
комплексы второго типа) и пятифтористого иода.
В данной и следующей главах будут описаны комплексные
соединения, образованные хлористым, бромистым и треххлори-
стым иодом, поскольку они известны в относительно большем
количестве и более изучены. В этих же главах будут рассмотрены
основные результаты работ Института общей и неорганической
химии АН УССР в этой области.
Анализ экспериментальных данных, относящихся к
комплексным соединениям межгалогенидов, даст возможность рассмотреть
некоторые вопросы, охватывающие более широкую область
комплексных соединений.
Комплексные соединения галогенидов иода с молекулами
органических веществ обычно относят к так называемым молекулярным
соединениям. Они имеют много общего с продуктами
присоединения галогенов к молекулам тех же или аналогичных органических
веществ в отношении механизма образования, природы связи и
физико-химических свойств.
В предыдущей главе отмечалось, что особенности
физико-химических свойств многих неводных растворов галогенидов иода
объясняются образованием в них соединений последних с
молекулами растворителей, доказательством чему могут служить спек-
трофотометрические и электрохимические методы исследования.
В данной главе будут рассмотрены описанные в литературе
комплексные соединения галогенидов иода с органическими
веществами, полученные как в твердом виде, так и установленные в двои-
236
кых жидких системах или в растворах физико-химическим
анализом или другими методами исследования. В индивидуальном
состоянии получены лишь наиболее устойчивые представители данной
группы. Это преимущественно продукты соединения галогенидов
иода с азотистыми основаниями — гетероциклическими и
алифатическими аминами *.
Эти комплексы получаются чаще всего путем
непосредственного взаимодействия компонентов. Синтез удобно осуществлять в
среде какого-либо растворителя (воды, спирта, эфира,
хлороформа, четыреххлористого углерода), в котором образующиеся
комплексы не растворимы (или трудно растворимы). Другие способы
их получения будут указаны при описании отдельных комплексов.
1. КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ ХЛОРИСТОГО И БРОМИСТОГО
ИОДА С АМИНАМИ
Наиболее подробно изучались комплексные соединения
пиридина и его аналогов.
Пиридин, хинолин и другие аналогичные азотистые основания,
как известно, являются очень хорошими комплексообразователями
и дают большое число комплексных соединений с различными
неорганическими и органическими веществами. Их комплексообра-
зующие свойства, несомненно, связаны с наличием в молекуле
донора электронов — третичного аминного азота со свободной парой
электронов.
В литературе описаны следующие соединения пиридина и его
солянокислой соли с хлористым и бромистым иодом:
C5H5N.JC1 и C5H5N-HC1-JCI II— 3];
C5H5N-JBr [4] и C5H5N-HBr-JBr [5].
Аналогичные же соединения описаны и для хинолина [1, 5],
C9H7N-JC1, C9H7N-JBr и для пиперидина [2] C5HnN.JCl.
Соединение пиридина с хлористым иодом CsHsN-JCl впервые
было получено, очевидно, Остермайером [6] ** и затем Дитмаром
[1]. Ряд авторов получали этот комплекс из дихлориодида пириди-
ния C5H5N • НС1 • JC1 (или [C5H5NHIJCI2). Последнее соединение
получают из пиридина и водного солянокислого раствора
хлористого иода, содержащего комплексную кислоту HJC12 [1,3], или, по
Пикте и Крафту [2], действуя на водный раствор пиридина трех-
хлористым иодом:
2JC13 + 3H20 = JCl + 5HC1 + HJ03;
C5H5N + HCl -f JCl = C5H5N - HCl • JCl.
* Ароматические амины при взаимодействии с галогенидами иода обычно
подвергаются реакции галоидирования.
** С оригиналом работы Остермайера мы не имели возможности
ознакомиться.
237
Дихлороиодаат ггиридиния затем превращают в C5H6N-JC1C
помощью щелочи [2] обработкой водой [1, 9]* или
перекристаллизацией из разведенного спирта [3]. Таким же путем были получены
аналогичные соединения хинолина C9H7N • JC1, пиперидина
C5HUN-JC1 и триметиламина (CH3)3N.JCl [2].
Подобные комплексы были получены также реакциями в
неводных средах. Так, Одрит и Бирр [7], добавляя пиридин к раствору
хлористого иода в эфире, получили CsHsN-JCl. Цаппи и Фер-
нандец [8] осуществили эту же реакцию в СС14. Для получения
комплексных соединений хинолина или триметиламина они
добавляли иод к растворам этих аминов в ССЦ и затем пропускали
хлор. При этом сперва образовывались комплексы с иодом типа
RN-J2, которые затем превращались в RN-JC1.
Удобный метод получения дигалидов пиридина применил
Уильяме [15]. Он медленно приливал рассчитанный объем 1-мол.
раствора галогена (или межгалогенида) в СС14 к 1-мол. раствору
пиридина в том же раеттюрителе; дигалиды пиридина быстро
осаждались, выход 80—90%'.
Деляби и Шарона [12], действуя на 8-оксихинолин,
растворенный в хлороформе, 1 или 2 молями JC1, получили продукты состава
CgHyON-JQ и C9H70N-2JC1, которые при нагревании в воде
или абсолютном спирте превращались в б^-дииод-в-оксихинолин
C9H5J2ON.
В табл. 40 приведены комплексные соединения хлористого и
бромистого иода с аминами и диоксаном и указаны их внешний
вид и температуры плавления. В работе Дитмара [1]
упоминается также о получении комплексов хлористого иода с а-окси-
хинолином, цинхониновой кислотой и с большим числом
алкалоидов, причем из последних указан состав только для комплекса с
хинином C20H24O2N2 • 2JC1. Кофеин и теобромин не дают
аналогичных соединений, что следует поставить в связь с очень слабыми
основными свойствами этих алкалоидов.
Приведенные в табл. '40 комплексы содержат в своем составе
главным образом такие амины, которые трудно подвергаются
реакциям галоидирования. Что касается тех аминов, которые,
вследствие особенностей своего состава и строения, способны
галоидироваться (8-оксихинолин, антипирин и др.), то при их
взаимодействии с хлористым иодом возможны такие случаи
(иногда в зависимости от условий проведения реакции):
а) сразу же образуются продукты галоидирования (иодзаме-
щенные);
б) сперва образуются продукты присоединения хлористого-
иода, которые затем превращаются в продукты иодирования;
в) происходит присоединение хлористого иода к продукту
иодирования, как, например, в случае антипирина.
* По данным Глея и Ягемана [9], дихлороиодааты пиридиния, хинолиния
и изохннолиния при промывании водой переходят в иодхлорпроизводные
RN-JCL Но аналогичные соединения акридина [C13H9NH]JC12 и о-фенантро-
лина [Ci2H8N2H]JCl2 при обработке водой не отщепляли хлористый водород.
238
Комплексные соединения аминов с бромистым иодом известны
в значительно меньшем количестве. Они описаны главным
образом в работе Мунейра [4], который получал их осаждением из
водных растворов пиридина, хинолина, гексаметилентетрамина и
алкалоидов при добавлении к ним бромистого иода *.
Состав и свойства комплексов с пиридином и хинолином
указаны в табл. 40. Уильяме [15] получил CsHsN-JBr реакцией в
четыреххлористом углероде. Этот же автор получил единственный
описанный в литературе бромхлорид пиридина C5H5N • ВгС1 (см.
табл. 40), менее устойчивый сравнительно с иодхлоридом и иод-
бромидом пиридина комплекс, медленно разлагающийся во
влажном воздухе; его не удалось перекристаллизовать.
Помимо комплексов эквимолярного состава для некоторых
аминов, содержащих два или более атомов трехковалентного азота,
известны соединения, в состав которых входит несколько молекул
JC1 или JBr. Таковы, например, комплексы с гексаметилентетр-
амином (уротропином)**: C6Hi2N4 • ftJCl (/i=l, 2, или 3) [4, 10, 13],
C6H12N4-2JBr [4], диацетилпиперазином C4H8N2(COCH3)2-2JCltl2]>
хинином C20H24O2N2 • 2JC1 [4]. В значительно меньшем количестве
известны комплексы, в которых на один атом азота приходится
по две молекулы JC1 или JBr (табл. 40).
Все упоминавшиеся выше комплексные соединения
представляют собой кристаллические вещества, большей частью
бесцветные или желтоватые (иодхлориды) или желтые (иодбромиды).
Структурному анализу был подвергнут только комплекс
C5H5N • JC1, образующий моноклинные кристаллы. По данным
Хасселя и-Ремминга [72], атомы азота, иода и хлора лежат
приблизительно на одной прямой. Межатомные расстояния таковы:
J—С1 2,51; N—J 2,26 А.
Обращает на себя внимание довольно высокая температура
плавления многих комплексов, описанных в табл. 40, по
сравнению с температурой плавления исходных компонентов, что
указывает на их относительно большую термическую устойчивость ***..
* В этой работе Мунейра кратко сообщает о том, что JBr легко
соединяется с большим числом алкалоидов, образуя желтые или желто-коричневые
не растворимые в воде продукты.
Характеристика этих соединений в работе Мунейра отсутствует.
Теобромин и кофеин в этих условиях не дали соединений с бромистым иодом (как и
с хлористым иодом).
** Эти соединения были получены взаимодействием уротропина с
хлористым иодом в среде хлороформа или с водным солянокислым раствором
хлористого иода. Условия получения комплекса C6H12N4-2JBr в работе
Мунейра [4] не указаны. Это вещества белые или желтоватые (комплекс C6H12N4-2JBr
желтый), почти не растворимые в воде и во многих органических
растворителях (спирт, эфир, хлороформ, бензол, ацетон), а также в разведенных
кислотах.
*** Отдельные исключения (табл. 40) относятся к комплексам типа
RN-2JCl(CyH5N-2jCl) или к комплексам, состав и строение которых вызывают
сомнение. Таковы, например, соединения JC1 с 8-оксихинолином или пирролом:
известно, что пиррол и 8-оксихинолин легко иодируются, возможно также,
что эти продукты содержали примеси иода или продуктов окисления аминов.
239
to Таблица 40
5 Комплексные соединения хлористого и бромистого иода с органическими веществами
Амины и другие вещества
Пиридин
Пиридин
Пиперидин
Хинолин
Изохинолин
6-Оксихинолин
5-Аминохинолин
8-Оксихинолин
8-Оксихинолин
а-Метилхинолин
Толухинолин
Р-Нафтохинолин
Оксиметилхинолин****
Пиррол
Иодантипирин
Состав комплекса
C5H5N.JC1
C5H5N.2JCI*
CeHnN.JCl
C9H7N.JC1
C9H7N.JC1
HOC9HeN.JCl**
H2NC9H6N.JC1***
HOC9H0N.JC1
HOC9H6N-2JCi
C10H9N-JC1
Ci3H9N.JCl
C4H5N-JCl
CnHuJON.JCl
Внешний вид
Иодхлориды
Светло-желтые, почти бесцветные
иглы
Мелкие красно-коричневые кристаллы
Белые иглы
Желтые дискообразные листочки
Белые иглы
Буроватый микрокристаллический
порошок
Желтые кристаллы
Темно-желтые иглы
Желтые иглы
Порошок черного цвета
Светло-желтые иглы
Температура плавления,
" °С
1 132 [2,8]; 135 [9]
35
; из
156-157 [14];
159,5 [2]
155
252; при 225 начинается
разложение
148
182—184
<w ]89 с разложением
170
157
1
Ссылка на
литературу
| [К 3, 7, 15]
[16]
[16]
[2]
[1, 5, 8, 9]
[9]
19]
[12]
[12]
IU
11]
IU
[1]
[4]
14
Продолжение табл. 40
Амины и другие вещества
Гексаметилентетрамин
Триметиламин
Диоксан
Пиридин
Хинолин
Гексаметилентетрамин
Диоксан
Состав комплекса
CeH12N4.2JCl****
(CH3)3N.JC1
C4H802-JC1
C5H5N.JBr*****
C9H7N-JBr
C6H12N4.2JBr
C4H802.JBr
Внешний вид
Иодхлориды
Белый, слегка желтоватый порошок
Белые иглы
Маленькие красно-бурые
пластинчатые кристаллы
Иодбромиды
Золотисто-желтые иглы
Желтый порошок
Красно-бурые, блестящие кристаллы
Температура плавления,
°С
161—162 с разложением
77
56-58
115—117
138—140
~65
Ссылка на
литературу
[4, 10, 13]
[2]
[18)
[4, 15]
[4]
[4]
[18]
Пиридин
C5H6N.BrCl
Бромхлориды
Белые кристаллы
107-108
[15]
* Получение и свойства комплекса C5H5N-2JC1 описаны на стр. 246.
** Этот комплекс осаждается из смеси солянокислых растворов обоих компонентов [9].
*** Получается как предыдущий комплекс, но после прибавления ацетата натрия.
**** Известны также соединения CeHi2N4-JCl и CeHi2N4-3JCl (стр. 239).
***** Для пиридина известен еще один комплекс с бромистым иодом C5H6N-2JBr, сбнаруженный в бензольном растворе
[17] (стр. 251).
Эти комплексы нерастворимы в воде, плохо растворяются ^
в неполярных или малополярных жидкостях (ССЦ, СбН6 и т. п.)э
лучше или хорошо растворяются в полярных растворителях, иа
которых их можно перекристаллизовать. Так, например>
CsHsN-JCl растворяется в нитробензоле до 17,5 мол. % при 450>
[16], пиридине, ацетоне, горячем спирте [15]. Они обладают
способностью присоединять галоидводородные кислоты, образуя при
этом хорошо кристаллизующиеся и растворимые в воде
комплексные соли типа [RNHUX2, где Х=С1 или Вг. Крепкая серная
кислота разлагает их с выделением* иода. При действии на них
избытка щелочи они разлагаются с выделением азотистого
основания.
Для всей этой группы соединений Дитмар [1] предложил
отличительную реакцию: при действии на них водного раствора
аммиака они образуют темно-зеленые или черные взрывчатые
продукты — производные йодистого азота.
Все комплексные соединения хлористого и бромистого иода
с аминами содержат иод в активном состоянии (в виде J+), что
было показано выделением иода при действии йодистого калия,
например, на раствор CsHsN-JCl или C6Hi2N4'2JCl в соляной
кислоте *:
C5H5N- JC1 + KJ = QH5N + КС1 + J2;
] C6H12N4.2JC1 + 2KJ = C6H12N4 + 2KC1 + 2J2 [13].
В тоже время все эти комплексы очень медленно разлагаются
водой, хотя хлористый и бромистый иод, отдельно взятые, легко
гидролизуются. Многие из, «их бесцветны или окрашены
значительно слабее, чем исходные-'галогениды иода (табл. 40). Эти и
некоторые другие особенности таких комплексных соединений
хлористого и бромистого иода, например их диэлектрические
свойства (стр.. 252), указывают на относительно большую прочность
связи хлористого и бромистого иода с азотистыми основаниями и
на значительную поляризацию комплексов.
Все сказанное подтверждает, что соединения пиридина и
других- азотистых оснований с хлористым и бромистым иодом
представляют большой интерес для выяснения особенностей всей этой
группы комплексов галогенидов иода с органическими веществами.
2. СОЕДИНЕНИЯ ХЛОРИСТОГО И БРОМИСТОГО ИОДА
С КИСЛОРОДСОДЕРЖАЩИМИ ОСНОВАНИЯМИ
В предыдущей главе (стр. 217) уже упоминались
относящиеся к этой группе комплексов соединения (эквимолярного состава)
хлористого иода с метиловым спиртом и диоксаном,
обнаруженные при спектрофотометрическом исследовании этих систем в
четыреххлористом углероде [20].
* Такое же свойство проявляют соединения хлористого и бромистого иода
с кислородсодержащими веществами и соединения трех хлор исто го иода с
аминами.
242
Последний из этих комплексов, а также аналогичное соединение
бромистого иода удалось. получить в кристаллическом состоянии!
Для этой цели галогениды иода растворили при умеренном
нагревании в диоксане (2 : 10); из охлажденных до комнатной
температуры растворов выделялись кристаллы комплексов. Их
внешние свойства и температуры плавления приведены в табл. 40.
Эти комплексы — продукты соедиренря галогенидов иода с
кислородсодержащими .основаниями, проявляя много общих свойств
с аналогичными соединениями аминов (наличие активного иода,
поляризация связи О—J и т. д.), отличаются от последних яркой
окраской и меньшей устойчивостью в отношении температуры и
влаги. , . " .. о" _
3. СОЕДИНЕНИЯ ТРЕХХЛОРИСТОГО ИОДА С АМИНАМИ
Продукты присоединения треххлористого иода к органическим
основаниям впервые удалось получить в неводных средах. Так,
пропуская хлор в растворы пиридин- и хинолиниодхлорида в
хлороформе, Цаппи и Фернандец [8] получили комплексы: C5H5N • JC13V
т. пл. 195—196° (с разложением) * и GgHyN-JCls — желтые
кристаллы, т. пл. 152—160° (с разложением). Путём пропускания
хлора в раствор (CH3)3N«JC1 в метиловом 'спирте был получен
комплекс (CH3)3N.JC13, Т. ПЛ. 170°. ; :..::iAi:,-.H.: ;;. .;■■;
Цернатеску и Пони [19] получили аналогичные комплексу
реакциями в водной среде. Так как треххлористый иод очень
быстро гидролизуется, вследствие/чего при непосредственном
взаимодействии треххлористого иода и аминов в водных растворах
образуются лишь иодхлориды аминов (стр. ч 237), то румынские
исследователи осуществили эти реакции -таким образом, что
треххлористый иод, образующийся в ^реакционной среде, давал
нерастворимые в условиях опыта продукты. Для этой цели
органическое основание они растворяли в воде или- разведенной соляной
кислоте и прибавляли йодноватую и концентрированную соляную
кислоту**. Иногда, :для того чтобы, выпал осадок, необходимо
было подогреть до кипения раствор'и'потом охладить. Осадки
отмывали раствором соляной кислоты (15—50 %) и высушивали
на воздухе и в эксикаторе над едким кали.
Были получены следующие соединения состава 1:1 (в скобках
указаны температуры плавления): (I) пиридин-JC13 (178—180°);
(II) а-пиколин ■ JCI3; (III) р-пйколйн- JG13:; (IV) 'у-лутиДия- JC13;
(V) хинолин-JCls (168°); (VI) у-леттцкЬ-JG13' (140° с разло*-
жением); (VII) пиперидин • JC13 (160^ 162е); (VIII) никотин- 2JC1&
(IX) спартеи'К JC13 (при нагревании) и (X) спартеин • 2JC13 (Hai
холоду (104—106°); (XI) пилокарпин - JC13 (104—106°) ***.
* Этот комплекс получен также непосредственным взаимодействием
пиридина и треххлористого иода.
** Треххлористый иод образуется по реакции. ' '.' i
HJ08+5HCl=jCl8+Ci2+3H26. !
*** Не реагируют с треххлористым иодом алкалоиды— нарцеин и пури-
новые производные (кофеин, теобромин, теофилин). Некоторые алкалоиды—
бруцин, тебаин, берберин—дают специальные реакции.
243
Все эти комплексы — вещества желтого цвета (различных от^
тенков) со слабым запахом хлора; они растворяются в воде, под^
вергаясь при этом гидролизу, разлагаются щелочами, легко рас.
творимы в спирте и ацетоне. При нагревании разлагаются с
выделением паров иода.
По описанию Цернатеску и Пони, все полученные ими ком-
плексы обладают окислительным и галоидирующим действием
как и хлористый или треххлористый иод; они легко окисляют
серу пирита, в щелочных растворах окисляют соли железа,
марганца и ряда других металлов, причем осаждаются гидроокиси
катионов в высших степенях валентности.
При действии на основания, содержащие ациклический азот,
например на анилин, не происходит осаждения продуктов
присоединения. Эти основания, равно как и фенолы и аминофенолы,
реагируют, часто энергично, с перечисленными выше комплексами.
Так, при взаимодействии C5H5N • JC13 в ацетоновом растворе с а«и-
зиди-нО'М образуется растворимый в ацетоне моноиоданизидин и
осаждается хлоргидрат этого амина.
Строение комплексных соединений ЛС1з с гетероциклическими
аминами Цернатеску и Пони выражают такими формулами (на
примере соединений пиридина (I), а-пиколина (II), хинолина (V)
и спартеина (X).
/Ч /X /\/Ч
II I II I I II 1
N Д1) N W) N (V)
fcis ici jci3
Н2|сн> н>>
N N
1 I
JCI3 ici3
Комплексные соединения треххлористого июда с аминами еще
не подвергались физико-химическому исследованию: 'неизвестно их
молекулярное состояние в растворах, их отношение к
электрическому току, оптические и другие свойства. Недостаточная
изученность этих комплексов не дает возможности более детально
представить их строение и химическую природу, особенно если принять
во внимание, что и вопрос о строении треххлористого иода еще
нельзя считать вполне решенным. В связи с этим дальнейшие па-
|раграфы этой главы будут посвящены главным образом
комплексным соединениям хлористого и бромистого иода.
244
4. ФИЗИКО-ХИМИЧЕСКОЕ ИССЛЕДОВАНИЕ СИСТЕМ ХЛОРИСТЫЙ
(БРОМИСТЫЙ) ИОД —ОРГАНИЧЕСКИЕ ОСНОВАНИЯ
И ОБРАЗУЮЩИХСЯ В НИХ КОМПЛЕКСНЫХ СОЕДИНЕНИЙ*
Имеющиеся в литературе данные о комплексных соединениях
галогенидов иода с органическими азотистыми или
кислородсодержащими основаниями до последнего времени ограничивались лишь
сведениями об их составе в растворах, установленном на основании
результатов оптических исследований, о методах получения более
устойчивых комплексов и их составе, о внешних свойствах,
температурах плавления и некоторых химических реакциях.
Высказывавшиеся в литературе представления о строении этих соединений
основывались на некоторых общих соображениях, на аналогиях,
но не на результатах физико-химических исследований.
Те свойства соединений хлористого иода с аминами, которые
описаны на стр. 242, давали основание предполагать, что эти
комплексы имеют солеобразный характер и являются электролитами.
Действительно, Одрит и Бирр [7], обнаружив, что растворы
хлористого иода в пиридине проводят электрический ток, причем
электропроводность растет со временем, (Приписали это
образованию четвертичной 'пиридиниевой соли, выделенной ими из
этой системы (стр. 238). Однако несколько ранее Уильяме [15]
измерил электропроводность комплекса C5H5NJCI в пиридине при
25° и обнаружил, что эквивалентная электропроводность в
пределах концентраций 1,16-Ю-3 до 17,40 • Ю-3 изменяется от 2,396 до
1,637 омг1см2, Хо = 2,4. Столь малая величина
электропроводности системы C5H5N • JC1 — пиридин, которую Уильяме приписал
действию следов влаги, привела его к отрицанию способности этого
комплекса диссоциировать на ионы. Как мы увидим далее, это
заключение было ошибочным.
Производившиеся ранее исследования таких систем, как
препаративно-аналитические, так и физико-химические (спектрофото-
метрические), не имели систематического характера и -не давали
оснований для обобщений в этой области химии комплексных
соединений.
В связи с этим и принимая во внимание принципиальный
интерес, который могут представлять соединения галогенидов иода
с органическими основаниями, в лаборатории химии комплексных
соединений ИОНХ АН УССР было предпринято исследование
систем из хлористого или бромистого иода и органических веществ
(пиридин, амиды кислот, диоксан) различными методами физико-
химического анализа и электрохимическими методами, а в ряде
случаев также изучение комплексов, образующихся в этих системах.
В данной главе будут приведены лишь важнейшие результаты.
Детали экспериментальных исследований описаны в
оригинальных работах.
* По результатам исследований, выполненных в лаборатории химии
комплексных соединений ИОНХ АН УССР Я-А. Фиалковым, И. Д. Музыкой и И. А. Шека.
245
а. Система хлористый иод — пиридин [16]
Термический анализ этой системы был произведен почти во всем
интервале концентраций, за исключением смесей, содержавших 80
88 мол. % JC1, которые весьма трудно кристаллизуются. На
диаграмме плавкости (рис. 45) выявлены две эвтектики (1 мол. % JC1
т. пл. — 50,2°; 64,9 мол. % JC1, т. пл. — 23,6°) * и две дистекти-
яш, соответствующие соединениям состава C5H5N\JC1, т. пл. 132,5°
и C5H5N • 2JC1, т. пл. ~ 35°. Насколько можно судить по диаграмме
плавкости, второй из этих
комплексов, ранее не описанный в
литературе, должен быть гораздо
менее устойчив.
Состав этих соединений был
подтвержден их получением
непосредственно из компонентов,
взятых в стехиометрических
количествах, и химическим
анализом. Для получения комплекса
C5H5N-2JC1 пиридин следует
прибавлять небольшими порциями к
хлористому иоду с последующим
перемешиванием и охлаждением,
при этом образуются мелкие
красно-коричневые . кристаллы.
Это вещество в воде почти не
растворимо, но медленно (хотя
и быстрее, чем CeHsN-JCl)
разлагается водой с выделением иода. Оно не растворимо также в
спирте, хорошо растворяется в нитробензоле. Щелочи и кислоты
разлагают этот комплекс, при этом выделяется иод **.
Система пиридин—хлористый иод проводит ток, но измерить
электропроводность этой системы во всем интервале концентраций
было затруднительно, так как для этого измерения нужно было
проводить при температурах выше 130°. При малых концентрациях
хлористого иода это могло привести к испарению пиридина, а при
больших концентрациях — к .разложению неустойчивого комплекса
Ру • 2JCl^Py-JCl+JCl. Поэтому сперва была измерена
электропроводность смесей с относительно большим содержанием хлористого
иода, состав которых соответствовал правой части диаграммы
плавкости этой системы, т. е. области существования комплекса
C6H5N-2JC1.
Изотермы удельной электропроводности (35 и 50°) имеют
своеобразный вид (рис. 46): с увеличением концентрации пиридина
1
70-
30-
-10*
-50-
-70
»
/
и
1 —1— 1—1
k&bfrja
1
—1—'
\/\
1 , 1
20
40 60
мол. % Ж
100
Рис. 45. Диаграмма плавкости
системы хлористый иод — пиридин.
* Третья эвтектика находится в еще не исследованной области
концентраций.
** Комплекс C5H5N-JC1, полученный путем добавления хлористого иода
к пиридину, взятому в небольшом избытке против эквимолекулярного
количества,, описан выше (см. стр. 238).
246
электропроводность системы сильно растет, особенно в области
небольших концентраций, образуя максимум 2—2,7 • 10~2 омг1слС1 *
примерно при 8—9 мол.% C5H5N, затем довольно резко падает до
3—5,4 • Ю-3 ом~1см-1 при 66 мол.% JC1, после чего изотерма
электропроводности (особенно при 50°) начинает несколько
повышаться **. Это, возможно, объясняется превращением комплекса
Ю 15 20 25
мол CiU5N
30 35
Рис. 46. Удельная
электропроводность системы хлористый иод —
пиридин в области больших
концентраций хлористого иода.
м-Ю
3750 5000
90 Цмл)
Рис. 47. Молекулярная
электропроводность системы хлористый иод —
пиридин, рассчитанная на хлористый
иод (>j) и пиридин (р.л).
C5H5N • 2JC1 в комплекс C5H5N • JC1. Действительно, измерение
электропроводности системы в области относительно больших
концентраций пиридина, но при 100°, дало значения к от 0,9 • 10~3
для 0,7 мол.% JC1 до 18- 10~3 ом-1см~1 для 36 мол.% JC1,**.
На рис. 47 представлены изотермы молекулярной
электропроводности (50°), рассчитанной по данным рис. 46 двояким
образом, условно принимая в качестве электролита как JC1
(кривая jlxj), так и C5H5N (кривая \iu). Кривая цт сходна с изотермами
удельной электропроводности и представляет типичную
«аномальную кривую». Кривая \хи имеет так называемый нормальный ход,
т. е. молекулярная электропроводность растет с разбавлением ***.
Подобные явления, наблюдаемые при обращении кривых
молекулярной электропроводности и описанные ранее в работах
Рабиновича [21] и Усановича [22], могут служить признаком того, что
электропроводность обусловлена электролитической диссоциацией
* Это почти в десять раз превышает собственную электропроводность
расплавленного хлористого иода.
** В этой области наблюдалось изменение электропроводности во
времени, о чем упоминается в работе Одрита и Бирра [7].
*** Такой ход изотерм электропроводности характерен и для описанных
далее систем хлористый иод — амиды кислот.
247
продуктов комплексообразования компонентов системы, в данном
случае комплексов C5H5N • JC1 и C5H5N • 2JC1.
Для определения природы ионов, на которые диссоциирует/
комплекс C5H5N • JC1, был изучен электролиз и перенос ионов r
его растворах.
При электролизе этого комплекса в нитробензольном и
пиридиновом растворах * в первой стадии электролиза наблюдалось
выделение иода на катоде, что можно было заметить по интенсивному
окрашиванию катодной жидкости в бурый цвет, сперва вблизи
электрода, а затем, по мере накопления иода, и во всей катодной
жидкости. По истечении некоторого времени (1—1,5 уаса)
начинала буреть и анодная жидкость вследствие выделения иода,
перенесенного из катодной части в анодную.
Катодная жидкость, полученная при электролизе нитробен-
зольных растворов комплекса, имела запах пиридина, что может
свидетельствовать об электролитическом переносе пиридина в
католит в составе комплексного катиона и о разряде последнего
с освобождением пиридина.
Перенос ионов изучали в нитробензольных растворах
комплекса CbHsN-JCI, в сосуде с тремя отделениями. Катод применяли
платиновый, анод серебряный, покрытый слоем галогенида
серебра (по Брукнеру). Для этого серебряный электрод
выдерживали в нитробензольном растворе хлористого иода до образования
плотного слоя галогенида серебра на поверхности электрода и
достижения /Постоянного веса электрода **. Продолжительность
Таблица 41
Перенос ионов в нитробензольных растворах
C5H5NJC1
о?
ентрат
%
о -£ о
7,15
6,25
10,93
лилось меди
юметре, г
а *
CQ cq
0,0756
0,0515
0,0312
Католит,
анолит
Католит
Анолит
Католит
Анолит
Католит
Анолит
Должно
быть, г
0,1301
0,1417
0,0845
0,1140
0,7144
0,1294
Хлор
Найдено,
г
0,0532
0,2195
0,0319
0,1658
0,1403
0,1689
Разность,
г
0,0769
0,0768
0,0526
0,0518
0,0341
0,0395
Число
переноса
СГ
0,912
0,911
0,901
0,915
—
0,088
0,087
0,099
0,085
—.
cd
СО
8 ь
О С
1,99
0,77
1,20
0,82
* Опыты электролиза проводились в сосуде U-образной формы с
широким краном в середине, позволяющим отделять катодную жидкость от анодной.
Электроды платиновые.
** Такая же методика применялась при аналогичных исследованиях
других систем, описанных в этом параграфе.
248
электролиза 20—21 час, сила тока 5—7 ма. В этих опытах мы
наблюдали те же явления, что и при электролизе.
Поверхность катода и вес его не изменялись. Анод всегда
увеличивался в весе вследствие электролитического осаждения на
нем галогенида серебра.
Полученные нами результаты, часть которых приведена в
табл. 41, можно кратко изложить следующим образом.
1. При электролизе происходит перенос хлора из катодной
части раствора в анодную. Количество хлора, перенесенное в
анолит, близко к вычисленному по закону Фарадея, если считать,
что прохождение 1 F соответствует переносу 1 г-экв хлора.
2. Первичный перенос иода в катодную часть доказывается
тем, что отношение J : С1 в процессе электролиза изменяется —
увеличивается в католите и уменьшается в анолите (табл. 41).
Аналогичное явление наблюдалось при исследовании систем
хлористый иод—амиды кислот, то же наблюдали Сандонини и
Бергело при электролизе растворов хлористого иода в
нитробензоле или уксусной кислоте [23].
3. Число переноса, вычисленное для С1-ионов (~0,9),
значительно превышает число переноса комплексного катиона ( — 0,1).
Можно предположить, что катион сольватирован нитробензолом.
Результаты электролиза комплекса CsHsN'JCl в нитробензоль-
ном и пиридиновом растворах и изучения переноса ионов
показывают, что строение этого комплекса может быть выражено
формулой [C5H5N-J]C1, и находятся в соответствии с высказанными
на стр. 228—231 представлениями о природе комплексных
соединений галогенов и галогенидов иода с азотсодержащими
органическими молекулами (типа аминов, амидов кислот и других
аналогичных соединений).
Для второго из обнаруженных в системе JC1—C5H5N
комплексов, т. е. для C5H5N-2JC1, можно предположить строение
[C5H5N.J№Cl2;h
б. Системы хлористый (бромистый) иод —пиридин (хинолин,
диоксан) в органических растворителях
Существование и состав комплексов, образующихся в таких
системах, ранее были установлены лишь спектрофотометрически-
ми (и препаративными) исследованиями, причем во всех случаях
были обнаружены комплексы состава 1:1. Для полноты
характеристики этих систем было необходимо исследовать их и
другими методами физико-химического анализа. В качестве таковых
были взяты криоскопический метод в том варианте, который был
предложен Фиалковым и Музыкой [17], и метод, основанный на
определении диэлектрической постоянной и поляризации
растворов, разработанный И. А. Шека [24—28].
249
€f} JBr
€A
-0,2
-0Л
C5H5N
20
*tO 60 мол.%
Рис 48. Криоскопическая кривая системы
бромистый иод — пиридин в бензоле.
0.WA
Q12A
ДОН
/
/
/
0Л 1,2 20
мол. JBr
мол. CsW
а
GJ
0,6
Oh
02
lV
0,9 1.2 2,0
мол. JBr
мол C5U5A/
б
Рис. 49. Криоскопическая" кривая системы пиридин —
бромистый иод:
а—в бензоле, б—в нитробензоле.
Криоскопические исследования
На рис. 48 показана криоскопическая кривая (т. е. кривая
изменения депрессии) растзора бромистого иода в бензоле при
добавлении к нему пиридина; депрессия At * сначала уменьшается
до достижения соотношения C5H5N : 2JBr, после чего начинает
увеличиваться, что указывает на образование комплекса
C5H5N • 2JBr. Пиридин, реагируя с избытком бромистого иода,
образует сперва этот, более богатый бромистым иодом комплекс,
что приводит к уменьшению общего количества молекул. По
достижении соотношения C5H5N : 2JBr добавляемый пиридин реагирует
с этим комплексом по уравнению CsHsN^JBr-fCsHsN ^
^ 2CsH5N-JBr, вследствие чего
увеличивается количество частиц, и депрессия начинает
возрастать.
Если же добавлять бромистый иод к
раствору пиридина в бензоле, то в условиях
взаимодействия бромистого иода с избытком
пиридина создаются благоприятные условия
для образования комплекса C5H5N • JBr
(электролита), что является причиной
увеличения депрессии даже до достижения точки
эквивалентности (рис. 49). После
«превышения эквимолярного соотношения C5H5N : JBr
наблюдается значительно более резкое
увеличение депрессии. Нужно полагать, что
комплекс C5H5N • JBr лишь в малой степени
переходит в менее устойчивое соединение
С5Н^ • 2JBr.
Таким образом, криоскопическое
исследование системы JBr—C5H5N в бензоле (а
также в нитробензоле) показало, что в
зависимости от концентрационных условий в
системе могут существовать два комплекса
C5H5N-JBr и C5H5N-2JBr в равновесии друг
с другом.
Такой же вывод следует из результатов исследования системы
JC1—C5H5N в бензоле или нитробензоле. На рис. 50 показана
кривая изменения депрессии системы JC1—C5H5N — бензол при
внесении пиридина в бензольный раствор хлористого иода. Эта
кривая имеет такой же характер, как и кривая, изображенная на
рис. 48. Она указывает на образование в растворе комплекса
C5H5N-2JC1, обнаруженного при термическом анализе системы
JC1—C5H5N.
Рис. 50.
Криоскопическая кривая системы
хлористый иод — пиридин в
бензоле.
*я!д*в Д*1— AV гДе А^0~~пониженИе температуры замерзания, наблюдаемое
при криоскопии раствора одного из компонентов системы; Д^—депрессия,
наблюдаемая при добавлении второго компонента к исходному раствору.
251
Измерение диэлектрических постоянных
и поляризации
Системы с пиридином и хин о л и ном. Физические
свойства соединений хлористого иода с пиридином и хинолином
давали основание предполагать, что эти комплексы должны быть
высокополярными, во всяком случае значительно более
полярными, чем соединения пиридина с иодом CsHsN-^, дипольный
момент которого, по данным Сыркина и Анисимовой [29], равен
4,171). Это предположение отчасти подтверждается
электрохимическими свойствами системы пиридин — хлористый иод и компле-
Д£ , ДР,см* [ - i
0J0
00-
"CgUsU **—so—** за c5h5n **—50—- за
Рис. 51. Кривые отклонений диэлектрических
постоянных Де и поляризации АР для системы хлористый
иод — пиридин в хлороформе (2с = 0,06 моль)л).
кса C5H5N • JC1. Оно полностью подтвердилось при изучении
И. А. Шека [28] диэлектрических свойств систем хлористый иод —
'Пиридин (или хинолин) в хлороформе большими отклонениями
поляризации и диэлектрических постоянных от вычисленных
величин *.
На диаграммах Ае— и АР—состав (рис. 51 и 52) имеется только
один хорошо выраженный максимум, характеризующий эти
системы как рациональные и соответствующий эквимолярному
соотношению компонентов. Таким образом, этот метод
физико-химического анализа не обнаружил комплекса C5H5N • 2JC1. Это, по-
видимому, объясняется тем, что в то время как присоединение
первой молекулы JC1 к пиридину сопровождается образованием
сильно полярной координационно-ковалентной связи, вторая
молекула JC1 присоединяется к комплексу [C5H5N—J]+Ch в
результате поляризационного взаимодействия, и потому комплекс
[C5H5N—J][C1 • JC1] не проявляется в исследованном интервале
концентраций на кривых АР— и Де—состав [28].
В отличие от системы хлористый иод — пиридин, на
диаграммах Ае— и АР — состав для системы хлористый иод — хинолин
* Так, для системы пиридин — хлористый иод в хлороформе &Р равно
259,6 см3 при экспериментальном значении Р, равном 418,3 см3. Для системы
с хинолином эти величины составляют соответственно 298,5 и 445,4 см3.
252
300
200
в хлороформе ветвь кривой, прилегающая к хлористому иоду,
имеет выпуклый вид. Это позволяет предполагать, что в данной
системе, помимо комплекса C9H?N • JC1, доминирующего в
растворе при условиях опыта, возможно, образуется и второй, менее
устойчивый комплекс CgH7N • 2JC1, являющийся, однако, более
устойчивым в растворе, чем комплекс лп з
C5H5N-2JC1 [28]. й*ш
Системы с д и океаном.
Соединения хлористого и бромистого иода
с джжеаном менее устойчивы, чем
соответствующие соединения этих же га-
логенидов иода с азотистыми
основаниями (см. стр. 243). В то же время
известно, что константа равновесия
комплекса С4Н802 • J2 [20] (табл. 42) и его 100 \
дипольный момент 0,95 D [29]
значительно меньше соответствующих
величин для C4H802*JC1. Все эти факты
согласуются с тем, что, по данным
И. А. Шека, величины отклонений ДР
и Де, обусловленные образованием
координационной связи в соединениях
C4H802\JX, оказались относительно
меньшими, чем для комплексов с
пиридином и хинолином (см. рис. 53 и 54).
Максимумы кривых отвечают экви-
молярному отношению, т. е.
соединениям C4H802-JC1 и C4H802-JBr. От
систем с пиридином и хинолином эти
системы с диоксаном отличаются
малым приростом поляризации, а также
(для растворов 0,1 моль/л) менее
резкими максимумами на кривых, что указы-
0
-0,20
0,10
CgHrN
вает на меньшую прочность соединении Рис. 52. Кривые отклонений
хлористого и бромистого иода с диок- диэлектрических постоянных
саном и их диссоциацию в растворе. Ле и поляризации АР для си-
гт г г стемы хлористый иод — хини-
Последнее .подтверждается также лин в ^ороформе (Яг =
уменьшением ДР для системы С4Н802— =0,05 моль/л).
—JBr с разведением (рис. 54).
Сравнение величин Ае и АР (при £<7 = 0,1 моль/л) в точках,
соответствующих образованию комплексов 1:1, показывает, что
эти величины для C^sC^-JCl выше, чем для C4H802:JBr. Как
указывает И. А. Шека, это можно объяснить большей прочностью
комплекса C4H802-JC1* и различной степенью полярности связи
О—J в данных комплексах. Так как донором электронов здесь
* Это подтверждается также сравнением констант равновесия этих
комплексов, по данным Лилича и Пресниковой [20].
253
Таблица 42
Константы равновесия комплексных соединений галогенов или галогенидов иода
с органическими веществами (1:1) в четыреххлористом углероде
Компоненты комплекса
Органическое вещество
и его дипольный момент
Галоген или
галогенид I
иода
Константа
равновесия
К
Ссылка на.
литературу
Бензол О
Толуол 0,4
о-Ксилол 0,55
ж-Ксилол 0,4
/г-Ксилол 0
Хлорбензол 1,70
Бромбензол 1,52
Иодбензол 1,31
Бензол 0,
Толуол 0,4
о-Ксилол 0,55
ж-Ксилол 0,4
л-Ксилол 0
Хлорбензол 1,70
Бромбензол 1,52
ж-Бутилбензол
1,4-Диоксан 0
1,4-Диоксан 0
1,4-Диоксан 0
1,4-Диоксан 0
Метиловый спирт 1,69
Метиловый спирт 1,69
Метиловый спирт 1,69
Метиловый спирт 1,69
w-Дибутиловый эфир
я-Дибутилсульфид 1,57
Диизоамилселенид
Пиридин 2,25
Вг2
Вг2
Вг2
Вг2
Вг2
Вг2
Вг2
Вг2
JC1
JC1
JC1
JC1
JC1
JC1
JC1
JC1
J2
Вг2
JC1
JBr
J2
Br2
JC1
JBr
J2
J2
J2
JC1*
1,04
1,44
2,29
2,16
2,26
0,90
1,18
1,59
4,76
7,97
15,4
16,0
13,4
2,24
3,43
8,70
10,50
3,75
632
153
3,10
(3)
204
33,6
11,4
1830
22500
0,96
0,69
0,44
0,46
0,44
1,11
0,85
0,63
0,21
0,125
6,5-10-2
6,3-10-2
7,5-10"2
0,44
0,29
0,115
95,10-2
0,27
1,58-10 -3
6,54-10~3
0,32
0,33
4,92.10" ~
■10
-2
2,9
8,8 <10~2
5,46-10-4
4,44-10 ~5
9-10-7
[59a]
[59a]
[59a]
I59aj
[59al
[59a T.
[59a]
[59a]
[596]
[596]
[596]
]596J
[596]
[596}
[596[
[596}
[201
[20]
[20J
[20]
120?
[20|
[20]
[20]
[20]
[20]
[20]
[62]
является кислород диоксана, а акцептором—ион J+, то различие
в полярности обоих соединений обусловлено влиянием атомов
хлора и брома: сродство к электрону у хлора выше,- чем у брома,
поэтому хлор сильнее оттягивает электроны от иода, в результате
*Так как константы равновесия вычислены исходя из отношения [АВ]/[А][В],
где А—органические вещества, а В—галогены или галогениды иода, то
вычисленные нами величины \/К'можЪо считать константами неустойчивости.
** Эту вели шну Кольтгоф с сотрудниками определили путем спектрофэтометри-
ческого титрования JCI пиридином в нитробензольном растворе
iX 25±3—
[Py][JCl]/[Py.JCl].
Так как комплекс Py-JCl представляет собой довольно сильный
электролит [PyJ]+Cl~, то желательно знать также константу неустойчивости
комплексного катиона Л'=[Ру][Л+]/[Ру-Л~|~].
254
чего соединение С4Н8О2 • JC1 оказывается более полярным и
более прочным, чем С4Нв02 • JBr.
Эти же рассуждения можно распространить на иодхлориды и
иодбромиды пиридина и хинолина. Сравнение же величин АР и Де
для соединений хлористого иода, с одной стороны, с пиридином и
хинолином, а с другой стороны, с диоксаном приводит к выводу,
что полярность связи азот—иод в комплексах C5H5NJCI и
CgHrN-JCl выше полярности связи кислород—иод в C4H802-JCI в
соответствии с различием прочности этих
двух типов соединений. Это указывает на
связь между полярностью
координационной связи и прочностью образующихся
при ее участии соединений [28].
LP, см*
30
20
Ю i
в. Системы хлористый (бромистый)
, иод—амиды кислот
Изучение взаимодействия галогенидов
иода с амидами кислот RCONH2
представляет интерес, в частности, в том отно- ^
шении, что в молекулах последних
содержатся два разных донора электронов; это
могло отразиться на процессе комолексо-
образования. Для исследования были
взяты амиды кислот с алифатическим . или
ароматическим радикалом, а именно:
ацетамид CH3CONH2 и бензамид
С6Н5СО]\[Н2. Кроме тою, т^к как для
амидов возможна таутомерная
перегруппировка по схеме
0,10 \
0.05
О К
R-O
NH2
:r-c
/
он
4NH
Рис. 53. Кривые отклонений
диэлектрических постоянных
Ае и поляризации АР для
системы хлористый иод —
диоксан в четыоеххлористом
углероде (2С=0Д моль/л).
представляло интерес выяснить, в какой из
этих двух форм—амидной или имидной—
амиды кислот реагируют с галогенида-
ми иода. С этой целью были исследова-ны
такие производные амидов кислот, в которых' один или оба атома
водорода амидной группы замещены на углеводородные радикалы:
диэтилбензамид CeHsCON^Hsb и ацетанилид (или фенилацет-
амид) CH3CONHC6H5, для которых таутомерные перегруппировки
должны быть очень ослаблены, а для двузамещенных — и вовсе
невозможны.
Были исследованы как двойные системы амидов кислот с
хлористым иодом, так и их растворы в нитробензоле [16, 17, 30—33].
Методом термического анализа удалось исследовать только
систему СеНбСОГШг—JC1, в которой обнаружено соединение
состава 1 : 1 (рис. 55). Попытки получить диаграмму плавкости систем
с ацетамидом и диэтилбензамидом не дали положительных
результатов, так как в средней части интервала концентраций в боль-
255
Рис. 54. Кривые отклонений диэлектрических постоянных
Де и поляризации ДР для системы бромистый иод—ди-
QKcaH в четыреххлористом углероде; а—Ъс=0,\ молы'л
и б—Ъс = 1 моль/л.
20 ¥)
моя. % CsHsCOMj
i i г-
io ' во во too
а б
Рис. 55. Система хлористый иод — бензамид:
а—изотерма вязкости; б—диаграмма плавкости,- /
щинстве опытов при охлаждении получалась стеклообразная мас-
са, не кристаллизовавшаяся с помощью обычно применяемых для
этого методов. Одновременно наблюдалось значительное
возрастание вязкости.
Измерение внутреннего трения этих систем (рис. 56 и 57)
показало наличие соединений 2CH3CONH2-JCl и C6H5C0N(C2H5)2-JC1.
Оба эти соединения не диссоциируют в интервале 25—45° или 35—
50°.
20 40 60
мол?/о CH3WHZ
\0 60 80 100
мол. % JCL
Рис. 56. Вязкость системы хлори-. Рис. 57. Вязкость системы хлористый
стый иод — ацетамид. 6 иод — диэтилбензамид.
В отличие от изотерм вязкости этих систем, изотерма вязкости
системы C6H5CONH2—JC1 (при 50°) имеет S-образный вид (рис.
55). Подобные изотермы вязкости двойных жидких систем уже
неоднократно были описаны в литературе [34]. В частности, они
были получены Курнаковым [35] для системы SbCl3 — бензол и
Усановичем с сотрудниками для систем серная
кислота—нитробензол [36], хлористый мышьяк—этиловый эфир [37] и для ряда
других *. Усанович дал теоретический анализ таких изотерм и
назвал их S-образными кривыми [38]. Согласно Усановичу [38], такие
изотермы получаются в тех случаях, когда компоненты двойной
системы вступают в соединение, вязкость которого меньше вяз-
* Для примера можно привести также систему РС15—Вг2. Термический
анализ этой системы, проведенный Плотниковым и Якубсон [39], показал
наличие комплекса РС15-5Вг2. Это соединение затем было ими выделено из
сероуглеродного раствора. Затем Якубсон исследовала вязкость системы РС15—Вг2
при 25, 35 и 50° и нашла для нее S-образную изотерму [40].
Такая же изотерма вязкости была получена Якубсон для системы
CH3CONH2—Вг2 [40]. Электрохимическое исследование этой системы
доказывает наличие комплексообразования в ней [41, 42].
17—1419
257
2 10*
24
6 1>
.4-.^*-*—*-
л/"0--?""*"^—*
20 40 60
мол. %CHjC0NH2
80
Рис. 58. Удельная электропроводность системы
хлористый иод — ацетамид: / — при 45°; // — при
35°; /// — при 25°.
ВО f
Рис. 59. Молекулярная
электропроводность системы хлористый
иод—ацетамид, рассчитанная на
хлористый иод (/JijJ и ацетамид
20 30 *0 50 W
Рис. 60. Удельная электропроводность
системы хлористый иод—бензамид.
кости одного из компонентов, причем такое соотношение
коэффициентов внутреннего трения возможно лишь при условии, что один
из компонентов—ассоциированная жидкость (в данном случае
таким компонентом является хлористый иод), образующееся же
соединение является жидкостью нормальной.
Изотермы удельной и молекулярной электропроводности всех
исследованных систем с амидами кислот (рис. 58—63) очень по-
г*. \
20
10-
о
30
130
f мл
10 20 30 40 50 60 70
мол. % C6H5C0N(C3U5)2
Рис. 61. Молекулярная электропровод- Рис. 62. Удельная
электропроводность системы хлористый иод — бенз- ность системы диэтил бензамид —
амид, рассчитанная на хлористый иод хлористый иод:
(Y-i) и бензамид (f*n). 7 _ при 5QO
2 — при 35°.
хожи на такие же изотермы системы C5H5N—JC1: Во всех случая^"
электропроводность быстро растет с концентрацией амида кисло-,
ты и достигает максимума 0,02—0,03 ом~1 см~1. В области
сравнительно небольших концентраций амида * — при рассмотрений
изотерм молекулярной электропроводности—обращает на себя
внимание тог факт, что «нормальный ход» кривых наблюдается
в тех случаях, когда и рассчитывалась на тот компонент системы,;
который в индивидуальном состоянии не является электролитом.'
Такой же характер имеют, по данным Музыки [45],
концентрационные зависимости удельной и молекулярной электропроводностк
системы бензамид—бромистый иод.
При электролизе системы CH3CONH2—JC1 наблюдалось вы-
* Аналогичное явление наблюдалось также при исследовании систем
ацетамид — бром [41], бензамид — бром [42], этиловый эфир — бром [44]
и др., что может служить одним из показателей общей природы всех
подобных систем.
259
деление хлора .на аноде и кристаллического иода на поверхности
катода.
Исследование этих же систем в нитробензольных растворах
дало дополнительные сведения о процессе комплексообразования и
состоянии комплексов «в
растворах, их природе и
строении.
Ацета'мид и бензамид,
трудно растворимые в
нитробензоле, хорошо
растворяются в нем в присутствии
хлористого [30, 31] или
бромистого иода [45]. Количество
бензамида, перешедшего в
РЦ^***^ раствор, составляет
примерно 1 моль на 1 моль JC1 или
JBr, а количество
растворившегося ацетамида ^1,7
600 1000 ф2 мл' М0ЛЯ На Х М0ЛЪ xJl°PHCTOro
—, .—22-^ ,— иода, несмотря на довольно
250 ft мл широкий интервал концен-
Рис. 63. Молекулярная электропровод- ТРаЩШ rMOTe™B ™Да В
ность системы диэтилбензамид—хлористый этих опытах.
иод, рассчитанная на хлористый иод (^ Измерения электропро-
и диэталбензамид (н-ц)- водности (рис. 64—67)
показали, что в сравнительно
более концентрированных — по отношению к JC1 —
растворах на изотермах электропроводности систем с ацетамидом
и диэтилбензамидом наблюдается размытый максимум,
приходящийся примерно на эквимолекулярное отношение амида и
хлористого иода. В системе с бензамидом ори увеличении концентрации
мол. JC1 1
хлористого иода в интервале отношения
от
ДО
1,89
мол. C6H5N02 3,39
наблюдается сдвиг максимальных значений электропроводности
от отношения С6Н5СОЫН2: JC1 ~ 1,5 к отношению ~ 0,33. В системе
с ацетанилидом наблюдался ясно выраженный максимум,
соответствующий отношению CH3CONHC6H5 : JCl^l : 2. Таким образом,
измерения электропроводности приводят к заключению, что в ряде
случаев, в зависимости от концентрационных условий, в
нитробензольных растворах возможно равновесие, по крайней мере, между
двумя формами комплексов — с большим и меньшим содержанием
хлористого иода.
Величина электропроводности нитробензольных растворов
исследованных систем — в области максимума — значительно
меньше электропроводности соответствующих двойных систем
(RCONH2—JX), примерно в 10 раз, и имеет порядок 10~3 омг1см~1 *.
* В системе C6H5CON(C2H5)2—JC1— C6H5N02, в более разбавленных
растворах (кривые lull рис. 66), электропроводность порядка \0~~4ом~~ смГ -
260
Интересные результаты дали криоскопические исследования.
Так, при добавлении амидов кислот к нитробензольному раствору
хлористого или бромистого иода депрессия температуры
замерзания растворителя не только не увеличивается, но остается
постоянной, как в системе с бензамидом, или даже уменьшается — до
достижения эквимолекулярного соотношения — в системах с ацетами-
дом, бензамидом и
диэтилбензамидом (рис. 64, 65, 66) или до
соотношения CH3CONHCeH5:
:JC1<- 1 :2 (рис. 67).
О 20 40 60 80
мол.%СН3С0МН2
Рис. 64. Удельная
электропроводность системы ацетамид —
хлористый иод в
нитробензоле: I 11, III —
мол. JC1 __ 1
мол. С6Н5Ш2 ~~ ~2£1~'9
мол. JCI
IV, 1/, VI
мол. JCI
Рис. 65. Удельная
электропроводность
мол. JC1
3,39/
мол. C6H5N02
1
6,01
\ мол. C6H5N02 3,.'
и криоскопия системы бенз-
амид — хлористый иод в
нитробензоле.
Таким образом, исследование
систем из амидов кислот с
хлористым (и бромистым) иодом различными методами
физико-химического анализа дало возможность обнаружить ряд новых
соединений амидов кислот с галогенидами иода. Для бензамида и ди-
этилбензамида всеми методами исследования были обнаружены
комплексы 1:1, для ацетамида — комплексы 1 :2 (методом
измерения вязкости двойной системы) и 1:1*. Ацетанилид (по дан-
* Различие в составе комплексов ацетамида с хлористым иодом,
обнаруженное разными методами исследования, будет обсуждено в главе XV.
261
. ным криоскопии и измерения электропроводности в нитробен^
золе) образует комплекс 1 :2 (т. е. C6H5NHCOCH3 • 2JC1).
Вычисление молекулярного (веса комплексов, по данным тех
опытов, в которых отношение ами-
&Ю\ 35° да кислоты к JX было очень близ-
120\
0,4 0,6 1.2 1.6 2,0
мол. С6Н,Ш(Сги5)2
Рис. 66. Удельная
электропроводность
мол. JC1 1^
11 ~~ мол. C6H5N02 ~~~ 77.0;
мол. JC1
мол. C6H«iN02 =
криоскопия
системы диэтилбензамид —
хлористый иод в нитробензоле.
ОЛ ОЯ V 1,6
мол.СЩЛ/ЩН,
мол. JUL
2.0 2/t
(-■
ш,iv —
= 1ТД")И
Рис. 67. Удельная электропровод-
/ мол, JC1 1 \
ность ( мол. C6H5N02 = "ТД49"]
и криоскопия системы ацетанилид —
хлористый иод в нитробензоле.
ко к составу комплекса, или по
данным криоскопии нитробен-
зольных растворов
синтезированных комплексов *, показало в
большинстве случаев повышенные величины молекулярного веса
против формульного значения для мономерных молекул комплекса,
* Или растворов, в которых амид кислоты и хлористый иод были взяты
в точно стехиометрических количествах.
262
например* C6H5CONH2 • JC1—309,8 (283,4) [31], C6H5CON(C2H5)2X
JC1 —350,9 (329,4) [33] и т. д. Кажущийся фактор ассоциации этих
комплексов относительно невелик, но, если принять во танимаиие,
что электропроводность их нитробензольных растворов, в которых
производилось определение молекулярного веса, довольно
значительна, порядка 10~3 ом~хсм~1, то можно полагать, что
действительный фактор ассоциации близок к 2 **.
Чтобы определить природу ионов, на которые диссоциируют
комплексные соединения амидов кислот с галогенидами иода, и
строение этих комплексов, количественно изучался перенос ионов
в нитробензольных растворах систем амид кислоты — хлористый
(бромистый) иод, содержавших эти компоненты в
приблизительно эквимолекулярных соотношениях. Из результатов этих
исследований можно сделать следующие выводы:
1. Во всех случаях происходит увеличение концентрации
амида кислоты в католите, а иода и хлора — в анолите. В ряде
опытов наблюдалось выделение на поверхности катода кристаллов
ацетамида или бензамида. Одновременно происходит также
разряд ионов J+ у катода, что качественно можно было обнаружить
по углублению окраски катодной жидкости в первый период
электролиза. Следовательно, амиды кислот вместе с иодом образуют
комплексный катион и потому они совместно переносятся ©
католит ***.
2. Тот факт, что концентрация иода увеличивается в анолите,
не противоречит представлению о первичном переносе его ib католит
в составе комплексного катиона, так как во всех опытах в
результате электролиза наблюдается увеличение отношения J : С1 в
католите и уменьшение этого отношения в анолите ****. Это дает
основание считать, что конечный перенос иода в анолит и его
осаждение на серебряном аноде обусловлены вторичными
процессами, рассмотренными в главе X.
3. Количества ацетамида, перенесенные в католит, и
количества хлора, перенесенные в анолит, близки к вычисленным по
закону Фарадея, если принять, что прохождение 1 F соответствует
выделению 1 г-экв CH3CONH2 или хлора.
4. Результаты этих опытов подтвердили тот факт, что в
нитробензольных растворах имеется равновесие между димерными
и мономерными молекулами комплексов. Так, при вычислении
чисел переноса для ацетамида и хлора были получены
следующие значения: для ацетамида ~ 0,8—0,9, для хлора ~ 1,2—1,35 *****.
* В скобках указан теоретический молекулярный вес.
** Лишь для комплекса CH3C0NHC6H5-2JC1 был найден молекулярный
вес 406,3, меньший формульного значения 4598 [33].
*** Подобные явления наблюдались при электролизе бромных растворов
ацетамида [41] и нитробензольных растворов комплекса бромбензамида [43].
**** Точно такое же явление наблюдалось в аналогичных исследованиях
системы пиридин — хлористый иод (стр. 248).
***** Число переноса больше 1 дает основание считать, что хлор
переносился к аноду в составе комплексного аниона [46].
263
Если на основании данных по криоскопии принять, что комплекс
CH3CONH2 • JC1 ассоциирован в нитробензоле и что фактор
ассоциации равен 2, и соответственно этому^ рассчитать числ^,
переноса для ацетамида и ионов хлора, то получаются значения
вдвое меньшие: для ацетамида 0,42—0,46, для хлора 0,59—0,67 \
в сумме ~ 1,0 [30].
В опытах с бензамидом [31] обнаружено, что количества
эквивалентов иода и хлора, перенесенные в анолит, находятся в
отношении от 1 : 1,5 до 1 : 2. Этот факт можно объяснить, если
принять, что димерные молекулы комплекса (QHsCONF^ • JC1)2
диссоциируют на ионы [(СеНбССЖНгЬ- J]+ и [JC12]".
Результаты исследований показали, что системы из
хлористого иода и незамещенных амидов кислот (CH3CONH2^
C6H5CONH2) и комплексы, образующиеся в этих системах,
вполне аналогичны соответствующим системам или комплексным
соединениям, которые состоят из хлористого иода и замещенных
амидов кислот СНзССЖНСбНб и СбНбСОЩСгНбЬ- Так как диэтил-
бензамид не подвергается таутомерной перегруппировке, то он,,
следовательно, реагирует с хлористым иодом лишь в своей амид-
ной форме. Из этого (можно заключить, что и незамещенные амиды
кислот таким же образом реагируют с хлористым и бромистым
иодом.
Исходя из результатов исследований систем галогенидьг
иода — пиридин (хинолин) или диоксан, показавших, что связь
N—J более полярна и прочна, чем связь О—J, можно
предположить, что присоединение хлористого и бромистого иода к амидам
кислот происходит по азоту, но не по кислороду.
5. ОСНОВНЫЕ ТИПЫ КОМПЛЕКСНЫХ СОЕДИНЕНИИ ГАЛОГЕНИДОВ
ИОДА (JCI, JBr, JCI3) С ОРГАНИЧЕСКИМИ ВЕЩЕСТВАМИ, ИХ СТРОЕНИЕ
И ХИМИЧЕСКАЯ ПРИРОДА
Согласно литературным данным и результатам исследований,
проведенных ib ИОНХ АН УССР, к настоящему времени
установлены разнообразные типы комплексов, образуемых галогенидами
иода с органическими веществами.
а. Соединения хлористого и бромистого иода
1. Комплексы типа RA-JX **: C5H5N-JC1, C9H7N-JBr,
C4H802.JC1, C6H5C0NH2-JC1, C6H5CONH2.JBr***.
2. Комплексы типа RA^-zzJX. Здесь имеются в виду такие
соединения, в которых одна молекула органического вещества содержит
несколько атомов—доноров электронов, к каждому из которых при'
* Эти значения чисел переноса для анионов хлора близки к величинам,
определенным в водных растворах многих хлоридов.
** А — донор электронов — атом азота или кислорода, X — С1 или Бг.
*** Другие примеры комплексов этого и других типов можно найти в*
табл. 40.
264
соединяется одна молекула JX. В качестве примеров можно привести
соединения гексаметилентетрамина C6H12N4-/2JC1, где п= 1, 2 или 3;
C6H12N4-2JBr, соединения хинина C20H24O2N2-2JCl и т. п.
3. Комплексы типа RA-2JX: C5H5N-2JC1, C5H5N-2JBr,
CeH6NHCOCH3-2JCl.
4. Комплексы типа 2RA-JX: 2CH3CONH2- JCl [32].
5. Комплексы типа (RA-JX)2: (C6H5CONH2•JC1)2.
На основании результатов, полученных при изучении
переноса ионов, и некоторых других данных можно принять
следующее строение и природу ионов для всех этих типов комплексных,
соединений [47]:
а) Для RA-JX : [^=^ N - л]+С1~; [c6H5CONH2-j]+Br-.
Аналогичное строение можно предположить и для комплексов
типа RAm • /zJX.
б) ДляЯА-2ЛХ : [^=^N-j]+ [jCla]"".
-CH3CONH2 у
в)Для2КА.ЛХ:'
•
LCH3CONH2 J
+
Ci"
г) Для (RA-JX)2:
fC6H5CONH2-| +
V [JCl2p.
LC6H5CONH2 J
В соединениях последних двух типов J+ как акцептор
электронов образует с одной молекулой амида кислоты координацион-
но-ковалентную связь, а с другой молекулой — связь,
по-видимому, за счет поляризационного или ван-дер-ваальсовского
взаимодействия. Эти соединения в отношении их состава и строения
можно уподобить так называемым «аномальным» аммониевым
солям, например [2NH3 • Н]С1 [48]. В обоих случаях акцептор
электронов Н+ или J+ соединен с двумя донорами электронов. Такое
же строение характерно для аналогичной соли ацетамида
2CH3CONH2 • НС1. Для этого комплекса Альбанский [49]
предложил строение [CH3CONH2 ->: Н : <- HsNCOCHsl+Ch с
координационным числом водорода, равным 2.
Таким образом, для ионов J+, находящихся в составе катионов:
[(RA)2J]+, следует принять координационное число 2, равно как
и для центрального атома иода в анионе JX J.
Комплексные катионы типа [(RA)2J]+ могут подвергаться
ступенчатой диссоциации и находиться в равновесии с катионами
менее сложными и даже с катионом J+.
[(RA)2 J]+;£[RA.J]+ + RA^I2RA+J+.
Сольватация ионов иода молекулами растворителя может
способствовать смещению этого равновесия вправо.
265
Комплексы с анионом JX^~ можно представить как продукты
присоединения одной или двух молекул RA к димерным
молекулам (JX)2, например:
C5H5N + J+ [ЛС12Г Z [C5H5N - J]+ [JXJ-.
Возможен, особенно в растворах, и иной механизм
ступенчатого образования таких комплексов, который уже упоминался
в связи с физико-химическим анализом системы пиридин —
хлористый иод в хлороформе.
Для всех этих типов комплексов следует принять схемы
электролитической диссоциации и электродных процессов, которые
лриоведены в главе X (стр. 228 и след.).
б. Соединения треххлористого иода
Для треххлористого иода пока обнаружены соединения только
'С азотистыми основаниями и притом следующих трех типов:
K'N • JC13; RN2 • JCI3 и RN2 • 2JC13, строение которых показано на
стр. 244.
Выше уже неоднократно отмечалось большое сходство
физико-химических свойств комплексных соединений многих
органических веществ с галогенидами иода (JC1, JBr) или молекулами
галогенов. Эта аналогия отражает также общность в механизме
образования и строении этих двух групп комплексов.
По вопросу о природе неводных растворов иода и о механизме
образования и природе связи молекулярных соединений иода с
разнообразными, главным образом органическими, веществами
имеется много литературы. Последний из этих вопросов хорошо
освещен в недавно опубликованной работе Теренина
«Молекулярные соединения и спектр междумолекулярного переноса
электрона» [50], в которой систематизировано и рассмотрено большое
число современных литературных данных *.
Как отмечает Теренин, по вопросу о механизме образования
и природе связи молекулярных соединений иода и других
галогенов существуют, в основном, две точки зрения. Согласно одной
из них, высказанной Бенеши и Гильдебрандом [51] и Малликеном
[52], образование межмолекулярных соединений иода и их
сравнительная стабильность обусловлены донорно-акцепторным
взаимодействием, при котором доля электронного заряда молекулы
органического вещества оказывается сосредоточенной на молекуле
иода. Это можно обозначить применительно к соединениям
ароматических углеводородов, как Ar8+J|-. Подтверждением такого
объяснения может служить наличие дипольного момента в раство-
* Представление о до но рно-акцептор ном характере взаимодействия
органических оснований с молекулами галогенов или межгалогенидов
высказывалось ранее Уильямсом [15], Фиалковым [69], Щукаревым и Лиличем [20,
61].
266
pax иода в бензоле (0,6 D), я-ксилоле (0,9 D), диоксане (1,3^)
я т. п. [53—55].
Другое представление по данному вопросу, известное под
названием «гипотезы междумолекулярного переноса электрона»
J56], заключается в том, что валентно насыщенные молекулы,
например бензол и иод, с полностью укомплектованными
межмолекулярными орбитами, занятыми парами электронов, могут
образовывать слабую межмолекулярную связь ковалентного типа в
том случае, если энергия отрыва электрона от молекулы донора
(«основания») 'может быть скомпенсирована ев некоторой мере
энергией присоединения к другой молекуле — акцептору электрона
(«кислоте»). При этом молекула акцептора должна располагать
свободной, достаточно низкой в энергетическом отношении
молекулярной орбитой *. Перенос части заряда электрона на молекулу
иода создает межмолекулярный дипольный момент и
электростатическое притяжение партнеров, дополнительное к слабому ко-
валентному притяжению [50].
Возможность образования таких молекулярных соединений
и их прочность обусловлены химической природой и строением
молекул — доноров электронов и природой молекул галогенов
(и межгалогенидов), прежде всего в силу энергетических
соображений. Так, нафталин, имеющий более низкий потенциал
ионизации (более низкую работу отрыва электрона), чем бензол,
образует и более прочное соединение («ассоциацию») с иодом.
Заместители в бензольном кольце оказывают такое же влияние. Они
повышают электронную плотность бензольного кольца, а
следовательно, и его электроно-донорные свойства.
Слабая ковалентная связь (или «ковалентное притяжение»),
характерная для комплексов ароматических углеводородов с
иодом, в зависимости, с одной стороны, от природы донора
электронов, а с другой стороны—от способности молекул галогена
поляризоваться в данной системе, может переходить в более ярко
выраженную ковалентную связь с молекулой галогена, приводящую в
пределе к соединениям с частичной ионной связью, ib которых
один из атомов галогена переходит в ионное состояние. Такое
явление имеет место, например, в ряде систем, образованных
кислород- или азотсодержащими донорами электронов. Оно может
быть представлено следующими схемами (применительно к эфирам
или пиридину):
CSH5N-J2^[C5H5N-J]+J-**.
* В статье Теренина [50, стр. 138] приведено модельное изображение
схемы переноса электрона и образования ковалентной связи между молекулами.
Эти новые взгляды на механизм образования подобного рода комплексов
не исключают, а дополняют представления о роли дисперсионного и
поляризационного взаимодействия.
** Рейд и Малликен [71] называют первую из этих форм «внешним
комплексом», вторую форму, с ионной связью, — «внутренним комплексом».
267
Образованием таких соединений объясняется электропровод^
ность многих систем, например: эфиры — бром [44, 57], ацетамид-^
бром [41], бензамид — бром [43]. Еще более ярко выражена
способность к образованию соединений ионного типа в системах с
пиридином: C5H5N-Br2 [58] и особенно С5Н5№Л2 (7), обладающее
высоким дипольным моментом [29].
Все сказанное выше в отношении механизма образования и
характера связи полностью относится также к системам с гало-
генидами иода. Различие между этими двумя типами систем — с
галогенами или с галогенидами иода — имеет количественный
характер. Оно обусловлено полярностью молекул последних и их
большей поляризуемостью по сравнению с молекулами иода и
брома, а также различием в сродстве к электрону.
Как следует из изложенного выше материала, комплексные
соединения хлористого и бромистого иода с органическими
веществами можно большей частью разделить на продукты их
соединения с ароматическими углеводородами, с
кислородсодержащими и азотистыми основаниями. В такой же
'последовательности можно 'расположить эти группы комплексов по их
.прочности.
В табл. 42 приведены величины констант равновесия»
[RA • JX]/[RA][JX]. Для первых двух групп комплексов хлористого
и бромистого иода (состава 1:1) константы равновесия вычислены
Кифером и Эндрюсом [59] и Лиличем и Пресниковой [20] на
основании результатов спектрофотометрического исследования в че-
тыреххлористом углероде как растворителе (при 25°) по методу,
предложенному Бенеши и Гильдебрандом [60] *. Для сравнения
приведены константы равновесия некоторых комплексов с бромом
и иодом.
Из данных табл. 42 наглядно видно, что прочность
-рассматриваемых в ней комплексов возрастает в ряду соединений
Br2<J2<JBr<JCl (с одними и теми же аддендами), т. е. по мере
увеличения поляризуемости галогенов или полярности и
поляризуемости галогенидов иода, обусловленной различием в сродстве
к электрону атомов галогена в молекулах JC1 и JBr.
Заместители, повышающие электронную плотность
бензольного ядра, а следовательно, и его электронно-донорные свойства
(как «основания»)*, например СН3, я-С4Н9, увеличивают
устойчивость комплексов, заместители же, оттягивающие на себя
электроны и уменьшающие электронную плотность бензольного ядра (С1Г
Br, J), оказывают противоположное влияние.
Константы равновесия комплексов хлористого иода с поли-
алкилбензолами в четыреххлористом углероде, вычисленные из.
спектрофотометрических данных, описаны также в работе Очи-
мати, Эндрюса и Кифера [73]. Кроме того, они рассчитали
изменение свободной энергии и теплоты образования. Из результатов
* Этот метод подробно описан в работе Лилича и Пресниковой [20J.
268
этого исследования авторы заключили, что в отсутствие стериче-
«ских затруднений изменение свободной энергии при
взаимодействии хлористого иода с полиалкилбензолами может служить
мерой электронного влияния алкильных заместителей на донорную
-способность алкилбензолов.
Комплексы с кислородсодержащими аддендами более
устойчивы, чем комплексы с ароматическими углеводородами, причем
прочность комплексов увеличивается по мере возрастания элек-
троно-донорных свойств адденда, например при переходе от эфи-
ров к сульфидам и селенидам.
Данные табл. 42 показывают, что комплекс C5H5N • JC1,
несмотря на различие в природе растворителя и в методе расчета
константы равновесия (неустойчивости), значительно прочнее
комплекса хлористого иода с диоксаном.
Сравнение констант прочности аналогичных комплексов в ряду
ароматических углеводородов или в ряду диоксан — метиловый
спирт подтверждает уже высказывавшееся выше мнение о том,
что в рассматриваемых здесь системах нет зависимости между
величиной дипольното момента молекул аддендов и прочностью их
•соединений с галогенами или галогенидами иода [20, 61].
Энергетические факторы, взятые в отдельности, не определяют
прочности рассматриваемых здесь комплексов и характера связи
в них. Так, работа отрыва электрона от молекул этилового эфира
равна 10,2 эв, а от молекул бензола и мезитилена
соответственно 9,24 и 8,1 эв. В то же время в системе эфир—иод образуется
гораздо более прочное соединение, чем в системе бензол — иод,
к тому же система эфир — иод проводит ток. Точно так же диполь-
ный момент электроно-донорной молекулы еще не определяет
прочности связи, что видно из сопоставления констант равновесия
комплексов диоксан — иод, метиловый спирт — иод [20] (см.
табл. 42).
Поэтому наряду с энергетическими факторами (энергия отдачи
и присоединения электронов), полярностью и поляризуемостью
компонентов системы необходимо учитывать состав и строение
органических аддендов, взаимное влияние атомов в молекулах
последних и другие аналогичные химические факторы, влияющие
на их электроно-донорные свойства.
Ряд авторов уподобляет электроно-донорное — акцепторное
взаимодействие галогенов или галогенидов иода с
органическими аддендами кислотно-основному взаимодействию, в котором
молекулы последних играют роль оснований [20, 50, 51, 59, 62, 63].
Эта аналогия исходит как из общих положений теории
обобщенных кислот и оснований Льюиса [64—66], так и из того, что
органические вещества, образующие комплексы с галогенами или
межгалогенидами, в большинстве случаев имеют характер
оснований, причем прочность этих комплексов возрастает в меру
усиления основных свойств органических аддендов, например: пири-
269
дин > диоксан > метиловый спирт *. Кифер и Эндрюс [59]
перенесли это представление и на область соединений галогенов и га-
логенидов иода с ароматическими углеводородами. При этом они
отметили, что способность ароматических соединений
присоединять JC1 весьма близка к той же способности по отношению
к НС1 [59с]**.
Эта аналогия — качественного характера. Она представляет
известный интерес с точки зрения представлений о явлениях амфотер-
ности в реакциях образования комплексных соединений [69].
Продукты присоединения галогенов или галогенидов иода к
органическим основаниям, имеющие солеобразный характер и
свойства электролитов, следует рассматривать как аммониевые,
оксониевые, сульфониевые и аналогичные им соединения. Так,
еще Курнаков и Анисимов [70], обнаружившие на изотермах
вязкости системы этиловый эфир — бром комплекс (С2Н5)2 ОВг3,
считали это соединение оксониевым трибромидом этилового эфира.
Условиями, ограничивающими возможность относить подобные
соединения к оксониевым, являются валентное состояние
центрального атома таких комплексов — трехковалентность кислорода
и серы, четырехковалентность азота и т. д.— и наличие солеобраз-
ных свойств, проявляющихся, в частности, в способности провод
дить ток.
С этой точки зрения выявляется общность в химической
природе продуктов присоединения как галогенов, так и межгало-
генидов к органическим основаниям, если они удовлетворяют
этим условиям. Таковы, например, [C5H5N—JJ+J", {C5H5N—JfCh,
[C6H5CONH2 • BrpBr-, [C6H5CONH2 - JJ+Вг- и. т. д.
ЛИТЕРАТУРА
1. М. D i t t m а г, Вег., 18, 1612 (1885).
2. A. P i с t e t, G. К г a f f t, Bl. Soc. Chim., (3), 7, 72 (1892).
3. W. J. S e 1 1, F. W. D о о t s 0 n, J. Chem. Soc, 75, 979 (1899).
4. A. Mouneyrat, С. г., 136, 1470 (1903).
5. P. F. T г о w b r i d g e, О. С D i с h e, J. Chem. Soc, 19, 560 (1897).
6. Ostermayer, Mitteil. aus d. Chem. Versuchstation in Wiesbaden,.
1883/84.
7. L. F. A u d r i e t h, F. J. В irr, J. Am. Chem. Soc, 55, 668 (1933).
8. E. V. Z a p p i, M. Fernandez, An. Ass. Quim. Argent., 27, 102
(1939); Chem. Zbl., 1940. I, 1503.
9. К- Gleu, W. J age man n, J. prakt .Chim., N. F., 145, 257(1936).
10. M. H о с h n e 1, Arch. Pharm., 237, 692 (1899).
11. E. A. Worker, J. Chem. Soc, 89, 1625 (1906).
12. R. D e 1 a b y, R. С h a r о n n a t, Congr. Intern. Chem. pure apl., 9r
IV, 301 (1934); Chem. Zbl., 1937, II, 231.
* Процесс комплексообразования между органическими основаниями1
и галогенами или галогенидами иода можно рассмотреть также с точки зрения
теоретических представлений, развиваемых Усановичем [67, 68], принимая во
внимание, что образующиеся комплексы часто имеют свойства электролитов.
** Кифер и Эндрюс полагают, что относительные значения этих констант
равновесия являются мерой основности ароматических веществ.
270
i3. А. И. Г е н г р и н о в и ч, Уч. зап. Киев, ин-та усоверш. провизоров
1 1950, стр. 104. F '
14 М. Kohn, A. Klein, Monatsh., 33, 967 (1912).
j5. D. M. Wi 1 1 i a ms, J. Chem. Soc, 2783 (1931).
j6. Я. А. Фиалков, И. Д. М у з ы к а, ЖОХ, 18, 1205 (1948).
17. Я- А. Фиалков, И. Д. М у з ы к а, Изв. СФХА, 19, 314 (1949).
18. Н. R h e i n Ъ о 1 d, R. Boy, J. pr. Chem., 129, 273 (1931).
19. R. С е г п a t e s с u, M. Poni, Anal. Acad. Republ. Populare Roma-
ne, III, 140 (1950).
20. Л. С. Л и л и ч, О. Е. П р е с н и к о в а, Уч. зап. ЛГУ, сер. хим.
наук, в. 12, 3 (1953).
21. А. И. Р а б и н о в и ч, Z. phys. Chem., 99, 434 (1921).
22. М. И. Усанович, ЖРФХО, 59, 14 (1927).
23. С. S a n d о n i n i, N. В е г g h e 1 о, Atti Acad., d. Lincei, 25 (6L
46 (1937).
24. И. А. Ш е к а, Полярные свойства комплексных соединений и метод
физико-химического анализа, основанный на этих свойствах, Докторская
дис, ИОНХ АН УССР, 1952.
25. И. А. Ш е к а, 3. А. Ш е к а, ДАН СССР, 73, 739 (1950).
26. И. А. Шека, Изв. сектора платины ИОНХ АН СССР, 26, 189 (1951).
27. И. А. Ш е к а, К. Ф. К а р л ы ш е в а, Укр. хим. ж., 20, 247 (1954).
28. И. А. Шека, ЖФХ, 30, 109 (1956).
29. Я. К. С ы р к и н, К. М. А н и с и м о в а, ДАН СССР, 59, 1457 (1949).
30. Я- А. Ф и а л к о в, И. Д. Музыка, ЖОХ, 18, 802 (1948).
31. Я. А. Ф и а л к о в, И. Д. М у з ы к а, ЖОХ, 19, 1416 (1949).
32. Я. А. Ф и а л к о в, И. Д. Музыка, ЖОХ, 20, 385 (1950).
33. Я- А. Ф и а л к о в, И. Д. Музыка, ЖОХ, 21, 823 (1951).
34. В. Я- Аносов, С. А. Погодин, Основные начала
физико-химического анализа, АН СССР, М.—Л., 1947, стр. 166.
35. Н. С. К у р н'а к о в, Д. К р о т к о в, М. О к с м а н, ЖРФХО, 47.
558 (1915).
6. М. И. Усанович, Г. Козьмина, В. Тартаковская, ЖОХ,.
5, 701 (1935).
37. Ф. И. Т е р п у г о в, ЖОХ, 2, 868 (1932).
38. М. И. Усанович, ЖОХ, 5, 996 (1935).
39. В. А. П л о т н и к о в, С. И. Я к у б с о н, ЖРФХО, 60, 1513 (1928).
40. С. И. Я к у б с о н, М. А. А б р а м о в а, Укр. хим. ж., 15, 136 (1949).
41. В. А. П л о т н и к о в, С. И. Я к у б с о н, ЖОХ, 5, 1337 (1935).
42. Е. Я- Г о р е н б е й н, П. И. С м о л е н ц е в, Сб. статей по общей
химии, АН СССР, т. II, 802, 1953.
43. В. Финкельштейн, Z. phys. Chem., 121, 46 (1926); 131, 338(1928).
44. В. А. Плотников, ЖРФХО, 38, 1006 (1906); Z. phys. Chem., 57„
502 (1906).
45. И. Д. М у з ы к а, Укр. хим. ж., 16, 331 (1950).
46. 3. А. Шека, Сб. «Работы по химии растворов и комплексных
соединений», ИОНХ АН УССР, 1954, стр. 113—131.
47. И. Д. Музыка, Физико-химическое исследование систем галогениды
иода—амины или амиды кислот, Дис, ИОНХ АН УССР, 1949.
48. А. В е р н е р, Новые воззрения в области неорганической химии, ОНТИ„
Л., 1936, стр. 228.
49. В. Л. А л ь б а н с к и й, ДАН СССР, 67, 77 (1949).
50. А. Н. Т е р е н и н, Усп. химии, 24, 121 (1955).
51. Н. А. В е n e s i, J. H. H i 1 d e b г a n d, J. Am. Chem. Soc, 70, 2832
(1948).
52. R. S. M u 1 1 i k e n, J. Am. Chem. Soc, 72, 600 (1950); J. Chem. Phys.,.
19, 514 (1951).
53. J. W. W i 1 1 i a m s, Phys. Z., 29, 174 (1928).
54. F. Fairbrother, Nature, 160, 87 (1947); J. Chem. Soc, 1051 (1948);.
180 (1950).
55. G. К о г t u m, H. W a 1 z, Z. Elektrochem., 57, 73 (1953).
56. R. S. M u 1 1 i k e n, J. Am. Chem. Soc, 74, 811 (1952).
271
57. Б. П. Б р у н с, Z. anorg. allg. Chem., 163, 120 (1917).
58. В. А. Плотников, В. И. Михайловская, Зап. 1н-ту xim.
7, 85 (1940).
• 59. R. M. Kee f е г, L. J. Andrews, J. Am. Chem. Soc, a) 72, 4677
(1950); в) 72, 5170 (1950); с) 73, 462 (1951).
60. H. Bene si, J. H i 1 d e br a n d, J. Am. Chem. Soc, 71, 2703 (1949).
•61. С. А. Щ у к а р е в, Л. С. Л и л и ч, Уч. зап. ЛГУ, сер. хим. наук,
в. И, 3 (1952); С. А. Щ у к а р е в, Л. С. Л и л и ч, А. Б.
Шейнин, ДАН СССР, 85, 1333 (1952).
62. J. М. Ко 1 t ho f f, D. Stocesoca, Т. S. Lee, J. Am. Chem. Soc,
75, 1834 (1953).
63. R. Sco t t, J. Am. Chem. Soc, 75, 1550 (1953).
64. G. N. Lewis, Valence and Structure of Atoms and Molecules (1923);
J. Frankl. Inst., 226, 292 (1938).
'65. А. И. Шатенштейн, Теории кислот и оснований, Госхимиздат,
1949, гл. 16.
*66. В. Люде р, С. Цуффанти, Электронная теория кислот и
оснований, ИЛ, М., 1950.
'67. М. И. У с а но в и ч, ЖОХ, 9, 182 (1939); Вестн. Каз. ССР, 1947, № 5
(26), 22; 1950, № 9 (56), 56; Изв. АН Каз. ССР, 1951, в. 4, 97.
•68. М. И. У с а н о в и ч, Что такое кислоты и основания, Изд-во АН
Каз. ССР, Алма-Ата, 1953.
69. Я- А. Ф и а л ко в, Усп. химии, 15, 490 (1946).
70. Н. С. К у р н а к о в, П. А. А н и с и м о в, ЖРФХО, 44, 1005 (1912).
71. G. R e i d, R. S. M u 1 1 i k e n, J. Am. Chem. Soc, 76, 3869 (1954).
72. О. Н a s s e 1, С h r. R о m m i n g, Acta, Chem Scand., 10, 696 (1956).
73. N. Ogimachi, L. Andrews, R. M. Keefer, J. Am. Chem.
Soc, 77, 4202 (1955).
ГЛАВА XII
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ ХЛОРИСТОГО,
БРОМИСТОГО И ТРЕХХЛОРИСТОГО ИОДА ТИПА ДВОЙНЫХ
ГАЛОГЕНИДОВ
Рассмотрим комплексные соединения хлористого, бромистого и
треххлористого иода с галогенидами щелочных, щелочно-земель-
ных металлов и некоторых других элементов, азотистых и тому
подобных оснований и других сложных неорганических и
органических катионов. Анионами таких галогенидов являются чаще
всего JC12~, JBr2", CUBr", JC14- *. Как будет показано далее, эти
двойные галогениды проявляют большую аналогию с полигалоге-
нидами.
1. МЕТОДЫ ПОЛУЧЕНИЯ
Методы получения и реакции образования рассматриваемых в
этой главе комплексных соединений можно разделить на
следующие группы:
а. Непосредственное соединение галогенидов иода с другими
галогенидами в растворах и расплавах
Таким лутем были получены комплексные кислоты: ШС12 —
растворением хлористого иода в концентрированной соляной кислоте —
в виде темно-бурой жидкости, содержащей 80% HJC12 и 20% воды [1]:
ШВг2 — растворением бромистого иода в бромистоводороднои
кислоте — в виде вязкой темно-красной жидкости, содержащей
лишь 6% воды. Растворяя бромистый иод в соляной кислоте, Кре-
мер и Дункан [1] получили 83%-ный раствор HJBrCl. Треххлористый
иод в концентрированной соляной кислоте образует раствор HJC14.
Водные растворы солей этих кислот получают растворением
галогенидов иода в крепких растворах хлоридов или бромидов
щелочных металлов. Однако при этом необходимо принять во
внимание гидролиз солей типа MJC12, MJBr2, MJC14. В связи с этим
для получения таких солей удобно пользоваться неводными рас-
* Двойные галогениды других межгалоидных соединений (BrF3, JF5 и др.)
рассматривались в соответствующих главах.
18—1419
273
тв-орителями. Так, Джексон и Дерби получили очень неустойчи*
вую в водных растворах соль NH4JBr2 по реакции между JBr ^
эфирным раствором бромистого аммония [2]. В ИОНХ АН УССР
при физико-химическом исследовании нитробензольных растворов
систем JC1—КО [3], JBr—КВг [4} было установлено образование
соединений KJ02 и KJBr2 (см. стр. 292—296).
Хлористый и бромистый иод в расплавленном состоянии
способны растворять хлориды и бромиды ряда других элементов или
смешиваться с расплавленными галогенидами, причем в таких
системах возможно образование комплексных соединений типа
двойных галогенидов. Так, Корног с сотрудниками [5] установили, что
хлориды лития, натрия, серебра, меди и бария слабо растворяются
в хлористом иоде (менее 0,6%) и не реагируют с ним. В отличие
от них, хлориды калия, рубидия, цезия и аммония образуют с
хлористым подом соединения типа MJC12. Корног и Бауэр [61
получали KJC12, выдерживая смесь равномолекулярных количеств
хлористого иода и хлористого калия в запаянном сосуде в течение
нескольких недель. Эта смесь почти черного цвета медленно
изменялась, приобретая блестящую оранжево-желтую окраску ди-
хлороиодаата калия. Полученный таким путем препарат KJC12
плавился при 195°. Корног и Бауэр выделили из системы JC1—КО
еще один комплекс KJ2O3 или KO-2JC1, кристаллизующийся при
температуре ниже 45° в виде кристаллов почти черных в массе и
рубиново-красных в тонком слое.
В растворах хлоридов или бромидов ряда элементов в
расплавленном хлористом или бромистом иоде было установлено
образование PCl5-JO [7—9], PBr5-JBr [9, 10], SbCl5-2JCl и SbCl5-3JO
[11, 12], AlCl3-2JO ИЗ, 14], AlBr-JBr [15, 16] и некоторых других
комплексов, которые будут описаны далее. *
v
б. Окислительно-восстановительные реакции
В тех случаях, когда в реакции участвуют галогены, сперва
происходит образование JO, JBr, JC13 (или другого межгалогени-
да), которые затем соединяются с избытком исходного галогенида.
Таково, например, получение KJ02 и KJBr2 по схеме [17]:
2KJ + Х2 = 2КХ + J2; (X = С1 или Вг)
J2 -f-Xg = 2JX;
2KX + 2JX = 2KJX2
KJ-t-X2 = KJX2.
Применяя этот метод, Уэлс [181, Уэлс и Уилер [19] получили
из водных растворов дихлсфо- и дибромоиодааты щелочных метал-
* Пропуская хлор в разбавленный раствор JC13 в хлористой сере, Руфф
и Фишер [11] получили комплекс 2JC13-SC14, свойства и строение которого
еще не изучены.
274
лов MJCb (M = K, Rb, Cs), CsJBr2. Так же был приготовлен
^H4JBr2 [20] *.
Этим же методом были синтезированы тетрахлорокодиатная кислота
и ее соли. Пропуская ток хлора в охлажденную льдом суспензию
йода в концентрированной соляной кислоте, Коглиоти [21J выделил
HJC14-4H20:
J2 + ЗС12 + 2НС1 = 2Н JC14.
Эта кислота имеет вид оранжево-желтых пластинок, легко
расплывается на воздухе, малоустойчива.
Соли этой кислоты: калиевую, аммониевую, -магниевую—впервые
получил Фильголь [22], применявший для этого следующие
реакции, которые были использованы и последующими авторами
почти без всяких изменений:
1) KJ03 + 6HC1 = KJC14 + C12 + 3H20.
Этот метод непригоден для получения солей тех катионов,
иодаты крторых труднорастворимы.
2) В концентрированный раствор иодида пропускают хлор до
насыщения (см. выше).
3) К насыщенному раствору хлорида прибавляют иод (в
теоретическом или полуторном количестве) и пропускают хлор до
насыщения:
J2 + 3C12 = 2JC13;
MCln + nJCl3 = M(JCl4)n.
Применяя эту реакцию, Уэлс и Уилер [23] приготовили соли
щелочных метал ов: LiJCI4 4H20; NaJCl4-2H20; K(Rb) (Cs) JC14.
Вайнланд и Шлегельмильх [25] синтезировали ряд солей
двухвалентных катионов общей формулы MTI(JC14)2-8H20, где Мп = Ве,
Мр, Са, Sr, Zn, Mnf Co, Ni, пропуская хлор в суспензию иода в
растворе хлоридов. Аналогичным путем, но работая при 60—70°, Целис
с сотрудниками [26] получили соли М1 JC14 и Mn(JCl4)2, в которых
Мх = и Na, К, Rb, Cs, NH4, а Мп = Be, Mg, Ca, Sr, Mn, Ni, Co,Zn.
При гидролизе треххлористого иода образуется HJ03. Это
Может повлечь за собой образование труднорастворимых иодатов
ряда катионов. Чтобы избежать этого, Вайнланд и Шлегельмильх
в таких случаях (при получении солей Be, Ca, Sr) добавляли к
реакционной смеси соляную кислоту **. Но это ъ свою очередь
затрудняет получение солей тех катионов, хлориды которых, например
ВаС12, CdCl2, PbCl2, труднорастворимы в соляной кислоте***.
Окислительно-восстановительные реакции были использованы
для получения двойных галогенидов йода также и в отсутствие рас-
* Дихлороиодаат аммония NH4JC12 был получен при действии хлора на
водный раствор хлористого аммония, к которому был добавлен иод [24].
** Так же поступали Уэлс и Уилер [23] при получении Cs.JCl4.
*** Соли трехвалентных катионов (Al, Fe, Cr, Bi) не удалось
получить [24].
275
творителя, в расплавах, Так, комплексы PC15-JC1 и PBr5-JBr
были получены при взаимодействии иода с пятихлористым фос~
фором [7, 8] или с пятибромистым фосфором [10], хлористого или
бромистого иода соответственно . с треххлористым или трехбро.
мистым фосфором [27]. Аналогично этому при реакции между пя-
тихлористой сурьмой и иодом были получены комплексы
SbCl5-2JCl и SbCl5-3JCl [11, 12].
Другие примеры окислительно-восстановительных реакций по*
лучения двойных галогенидов иода будут приведены далее.
в. Получение двойных галогенидов в гетерогенных системах
Для этого на кристаллические галогениды действуют
газообразными или жидкими галогенами или межгалогенидами в
течение более или менее длительного времени. Эти так называемые
«сухие методы», которые особенно подробно исследовали Кремер
и Дункан [1, 29] и Рэй [30], можно разделить на две группы:
1) Непосредственное присоединение галогена или межгалоге-
нида к галогениду какого-либо элемента. В этих случаях упругость
диссоциации продукта реакции должна быть меньше давления
паров галогена. Таким путем были получены, например, CsJBr2 из
йодистого цезия и брома, MC1JC1 и MBr-JBr (M=K, Cs, NH4),
KCI.JCI3 [29].
2) Непрямое образование комплексов, протекающее при участии
окислительно-восстановительных и других реакций, например, в
системах KJ + Cl2^KClJCl->KCbJCl3; KJ+ JC1-*KJC12 и т. д.
Так, для получения солей типа MJC12 или MJBr2 Рэй [20]
выдерживал измельченные иодиды в парах хлора или брома до
достижения привеса, соответствующего эквивалентному количеству
хлора или брома, присоединившегося к иодиду. Аналогично
поступали Кремер и Дункан [29]. При этом происходят реакции того же
типа, что и приведенные на стр. 274.
Методы получения двойных галогенидов иода* и полигалогени-
дов в гетерогенных системах длительны: вч зависимости от состава
образующегося комплекса реакция продолжается от нескольких
часов до нескольких десятков дней. Большое значение в этих
реакциях имеют такие процессы, как диффузия галогенов и
галогенидов иода в глубину кристаллов и перекристаллизация
комплексов, образовавшихся на поверхности твердой фазы. Поэтому
тщательное измельчение кристаллов и наличие следов воды
благоприятствуют течению этих реакций.
2. ОСНОВНЫЕ ФОРМЫ ДВОЙНЫХ ГАЛОГЕНИДОВ ХЛОРИСТОГО,
БРОМИСТОГО И ТРЕХХЛОРИСТОГО ИОДА
По природе аниона эти комплексы можно разделить на две
группы: тригалогениды — с анионами JCI2", JBr7, JBrCP, JBrF
и пентагалогениды—с анионами JCl^f, JCI3F~. Наиболее распростра-
276
ценными из них являются дихлоро- и дибромоиодааты и тетрахлоро-
^одиаты; методы получения их описаны выше.
Отдельную группу составляют комплексы, анионы которых
содержат три разных галогена. Из них хлоробромоиодааты MCUBr
(М==К, Rb, Cs, NH4) [18, 19, 28, 29] могут быть получены
указанными ранее способами, в частности действием брома и иода на
хлориды. Так, Чэтейуэй [24] для получения NH4CUBr прибавлял
эквивалентные количества брома и иода к теплому водному
раствору хлористого аммония; по охлаждений выделяются темио-гра-
натово-розовые призмы.
Фторсодержащие галогениды известны лишь в единичном
количестве. Комплексы MFJC13 (M=K, Rb, Cs, NH4) [33] были
получены действием хлора на смесь эквивалентных количеств
фторида и иода или растворением JC13, взятого в небольшом
избытке, в водном насыщенном и подкисленном растворе фторида. Эти
комплексы очень мало растворимы в воде, они не растворимы в
ССЦ и бензоле, но все же медленно реагируют с последним.
Большое разнообразие -проявляют двойные галогениды .иода
в зависимости от природы катиона. Известны такие группы этих
галогенидов:
а. Соли -металлических катионов—щелочных, ще-
лочно-земельных и некоторых других металлов. Сюда же можно
отнести и соли аммония. Методы получения этих солей описаны
выше.
б. Соли комплексных катионов типа гексам'миакатов
или диацидотетрам'миакатов кобальта и хрома, впервые полученные
Спаку и Попеа [31, 32]. Эти авторы путем взаимодействия NH4[JCl4]c
хлоридами таких катионов получили следующие комплексы: [С12СоХ
X (NH8) J JC12 (транс), [Cl2Coen2] JC12 (цис и транс*), [С12СоРу4] JC12
и [CoB^(DH)2] JC12, где В = NH3, Py, C2H5NH2, a DH2 — диметилглио-
ксим.Реакция сопровождается разложением иона JC17 на JCl2~ и х/ор:
NH4 [ JC14] + [С12СоВ4] С1 = [С!2СоВ4] [ JC12] + NH4C1 + Cl2.
Пропускание хлора в реакционную среду дало возможность
сместить равновесие диссоциации ЛСЦ~ и получить тетрахлороиоди-
аты ряда перечисленных выше комплексных катионов аммиакатов
и аминатов кобальта, а также [Соепз]С1[ЛС14]2 и [Cren2]Cll[JCl4]2.
в. Соли аминов и четырехзамещенных аммониевых
оснований. Комплексные соединения этой группы содержат
преимущественно катионы типа [RNH3]+> [R2NH2]+, [R3NH]+,[>NH]\
[R*N]+ и ]^>NR]+, анионами же являются большей частью JCli",
JBri" и JCir. В литературе описано большое число таких комплексов
[32, 34—47]. В частности, они известны почти для всех аминов, при-
. - ,.^!
* Транс-изомеры этих комплексов оказались более устойчивыми чем
цис-изомеры. Так, не удалось получить цис-формы [Co(NH3)4Cl2] JC12, а цис-
изомер комплекса [Coen2 Cl2] JC12 при нагревании с соляной кислотой
превращался в транс-изомер [31], что характерно для перегруппировки ряда и
других виолеосолей в празеосоли.
277
веденных в табл. 40. Для их получения применяют разнообразные
способы:
а) взаимодействие галогенидов иода с солями аминов (в
солянокислом растворе), например,
RN-HCl + JCl„-*[RNH][JCln+i] (л=1 или 3);
б) взаимодействие аминов с растворами галогенидов иода в
соляной или бромистоводородной кислоте, например:
C5H5N + HJC12 = [C5H5NH] JC12;
в) действие хлора или брома на гидроиодиды аминов RN-HJ*;
г) растворение иодхлоридов, иодбромидов и иодтрихлоридов
аминов (табл. 40) соответственно в соляной или бромистоводородной
кислоте
[C9H7N. J] CI + НС1 = [C9H7NH] JC12 и т. д.
д) соли тетрахлороиодиатной кислоты были получены также
действием хлора на дихлороиодааты [RNHJJCb или на комплексы
типа RNJC1 в среде уксусной кислоты или метилового спирта [45].
Для этой же цели можно действовать хлором на суспензию иода
в растворах а^минов в концентрированной соляной кислоте [43].
Тетрахлороиодиаты могут быть превращены в дихлороиодааты
действием двух эквивалентов щелочи, например [40],
[QH5 (С3Н7)3 N] JC14 -f 2NaOH = [С2Н5 (С3Н7)3 N] JC12 + #
[+ NaCl + NaOCl + Н20.
Описанные в литературе и рассматриваемые в этом параграфе
соли весьма разнообразны по своему составу в зависимости от
природы азотистого основания. В их состав входят алифатические
амины—третичные, вторичные и первичные [38, 42, 43 и др.];
диамины—этилендиамин [32, 43], мочевина, гуанидин [43]; ароматические
амины, моно-, ди- и трибензиламин [43]; гетероциклические
амины—пиридин, хинолин и их гомологи [35, 38, 39, 43—45], а-амино-
пиридин [32], акридин, о-фенантролин [44], цинхониновая «кислота
[35], пиридинбетаин [43], пиперазин, кофеин [43], четвертичные
соли пиридиния [39] и т. д. Обращает на себя внимание тот факт,
что даже такие слабые основания, как мочевина, гуанидин,
кофеин, образуют сравнительно устойчивые тетрахлороиодиаты [43].
Соли четырехзамещенных аммониевых оснований содержат чаще
всего низшие алифатические радикалы и фенил.
В отношении физико-химических свойств: окраски,
растворимости, термической устойчивости и некоторых других—соли
аминов проявляют значительное сходство с солями щелочных
металлов.
г. Соли сульфониевых оснований. Доббин с
сотрудниками [37, 48], действуя хлором или бромом на иодид триметил-
* Происходящие при этом реакции могут быть выражены уравнениями,
приведенными на стр. 274.
278
сульфония, получил [(CH3)3S]JCl2 и [(CH3)3S]JBr2 (уравнения
реакций, см. стр. 274). Проводя такую реакцию в уксусной кислоте,
рернер [40] превратил иодиды триметил- и триэтилсульфония в
.соответствующие тетр ахлороиоди аты; при пропускании хлор а
сперва выделяется иод, а затем по мере взаимодействия
последнего с хлором образовывались желтые сульфониевые комплексы
[(CH3)3S]JCi4 с т. пл. 180° [49] и [(C2H5)3S]JC14.
При нагревании или при действии щелочи (2 моля на 1 моль
комплекса) они превращались в дихлороиодааты:
{(CH3)3S]JC12—блестящие тонкие золотисто-желтые пластинки с т. пл. 103° и
[(C2H5)3S]JCl2—вязкая оранжево-красная жидкость. Строение
этих комплексов доказывается также тем, что они были получены
путем взаимодействия хлоридов сульфониевых соединений с
солянокислым раствором хлористого иода:
[R3S]C1 + JC1 = [R3S]JC12.
Окислительно-восстановительное равновесие [R3S] JC14^L[R3S]JC12-|-
-f- Cl2 было подтверждено при изучении спектров поглощения
растворов (CH3)3SJC14 и (CH3)3SJC12 в ацетонитриле [49].
д. Соли дифенилиодония. Как известно, соли дифе-
нилиодония, например [(С6Н5ЫУ, принадлежат к классу «оние-
вых» комплексных соединений. Галогениды дифенилиодония,
подобно галогенидам аммония и сульфония, способны соединяться
с галогенами и галогенидами иода с образованием три- и пентага-
логенидов. Так, Форстер и Шеппи [50], пропуская сухой хлор в
суспензию иодида дифенилиодония, получили тетрахлороиодиат
(СбНб^^СЦ—кристаллы желтого цвета с т. пл. 118—120°. Ацетон
разлагает этот комплекс на [(CeHsbflCl и JC13.
Дихлороиодаат дифенилиодония содержится среди продуктов
разложения тетрахлороиодиата. При охлаждении полученного при
этом прозрачного светло-бурого раствора выпадают желтые
блестки состава [(С6Н5ЫУС12, с т. пл. 136—137°. Этот же комплекс
образуется по реакции
2 [(С6Н5)2 J] JC14 + [(С6Н5)2 J]J3 = 3 [(С6Н5)2 J] JC12 + 2JC1.
Тригалогениды дифенилиодония можно получать теми же
методами, что и тригалогениды щелочных металлов. Так, действием
брома на иодид дифенилиодония был получен [(C6H5)2J]JBr2 в виде
оранжевых пластинок с т. пл. 143°.
Три- и пентагалогениды дифенилиодония не исследованы в
физико-химическом отношении.
е. Соли диазония. Тригалогениды солей диазония
[ArN2]JCl2, [ArN2]JBr2 и (ArN2]CUBr получил Ганч [51]. Общим
способом получения этих комплексов галогенидов иода является
непосредственное взаимодействие хлористого и бромистого иода с
хлоридом или бромидом диазония:
[ArN2] X+JX -> [ArN2] JX2.
279
Таким путем Ганч получил [C6H5N2]JCI2 (т. пл. 86—87°)>
[CGH5N2] JBr2 — бурые кристаллы, разлагающиеся при 77°; [C6H5N2]>(
XCIJBr и [BrC6H4N2]CUBr—■ золотисто-желтые иглы, плавящиеся
соответственно при 80—81° и 111 — 112°*.
Эти тригалогениды диазония малорастворимы в ©оде, в
растворах частично разлагаются. В спирте они более растворимы и
более устойчивы.
Ганч отметил аналогию этих полученных им комплексов с
соответствующими солями щелочных металлов.
3. НЕКОТОРЫЕ ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА ДВОЙНЫХ
ГАЛОГЕНИДОВ ИОДА
Все перечисленные выше формы комплексных соединений
хлористого, бромистого и треххлористого иода имеют много общих
свойств. Наибольшее количество литературных данных относится
к солям щелочных металлов и аммония, на примере которых
будут кратко описаны некоторые физико-химические свойства этой
группы комплексов.
В подавляющем большинстве случаев — это кристаллические
вещества, желтые, оранжево-желтые, желто-бурые.
Рентгенографическому исследованию были подвергнуты
только некоторые тригалогениды и лишь один пентагалогенид KJCU-
Из комплексов типа MJX2 были исследованы CsJCl2, CsJBr2,
(CH3)4NJC12 и NH4CUBr [52—56].
Кристаллическая решетка этих соединений тетрагональная для
(CH3)4NJCl2, ромбоэдрическая для CsJCl2 и ромбическая для
остальных. Анион в решетке этих солей, как и в решетке поли-
иодидов J3~ имеет линейное строение, например, [С1—J—С1"Г в
виде пространственной диагонали, причем центральным атомом
является иод, находящийся на равном расстоянии от Двух других
атомов галогена. Расстояние J—С1 в CsJCl2 равно 2,25 А, в
(CH3)4NJC12—2,35 А. В решетке NH4CUBr (рис. 68) расстояние
J—С1 составляет 2,38 A, a J—Br — 2,50 А.
Тетрахлороиодиат калия резко отличается по своей структуре от
тригалогенидов [57]. Комплексный анион в моноклинной решетке KJC14
образует слегка искаженный квадрат, в центре которого находится
о
атом иода, а в углах — атомы хлора. Расстояние J—С1 равно 2,34 А **.
Б. Ф. Ормонт [56] указывает на то, что строение [JC14P можно
Ск |/С1
представить как октаэдр с двумя незанятыми вершинами yi\ .
CV | ХС1
Л. Паулинг [58], отмечая, что атом иода в этом анионе обладает
* Два последних соединения были получены при действии хлористога
иода на бромид диазония или бромистого иода на хлорид диазония.
** Междуатомные ^расстояния в JC1^~ и в ионах тригалогенидов почти равнь*
сумме ко валентных радиусов соответствующих галогенов.
280
двумя неподеленными парами электронов, полагает, что последние
занимают две вершины ^октаэдра, выше^и ниже экваториальной
плоскости иона JC14.
Аналогично этому Паулинг, исходя из того, что электронная
конфигурация анионов тригалогенидов имеет вид [ :Х:J:X:]~
рассматривает их строение как треугольные бипирамиды с двумя атомами гало-
Рис. 68. Кристаллическая решетка Рис. 69. Модель молекулы
NH4CUBr. трехфтористого хлора.
гена в вершинах бипирамиды и тремя неподеленными парами электронов
в плоскости среднего сечения (т. е. в экваториальной плоскости
бипирамиды).
Представление о том, что неподеленные пары электронов могут
занимать вершины многогранников, которыми выражают строение
отдельных молекул, некоторые авторы [58—60] распространяют
на многие вещества, в том числе на молекулы некоторых межга-
логенидов, например йа пятифтористый иод (октаэдр, одна
-вершина которого занята электронной парой) и на C1F3 (бипирами-
да — рис. 69 — с двумя парами электронов [60] и т. д.). Однако
такое представление о строении молекул и ионов является
предметом дискуссии [61].
Отношение к нагреванию: температуры плавления,
упругость диссоциации и температуры разложения
Температуры плавления определены почти для всех
описанных в цитированной литературе представителей перечисленных
выше форм комплексных соединений хлористого, бромистого и
треххлористого иода с другими галогенными.
281
Определение температуры плавления этих комплексов, за
исключением наиболее устойчивых их представителей (солей
рубидия, цезия и некоторых аммониевых оснований), часто
осложняется рядом факторов — частичной диссоциацией .при нагревании,
взаимной растворимостью продуктов термической диссоциации,
гигроскопичностью многих из этих комплексов и их способностью
удерживать -следы воды — значительно понижающих температуру
п\°*—•— • ■
20 W 60 80 ГС
Рис. 710. Упругость диссоциации KJBr2 и его
гидрата.
плавления. Этими причинами, очевидно, можно объяснить
значительные в ряде случаев имеющиеся в литературе расхождения в
величинах температуры плавления двойных галогенидов иода.
Так, дихлороиодаат калия, по данным Эфраима [17], плавится
около 60°. По данным Кремера и Дункана [28], температура
плавления его несколько ниже 60°, но препарат, сохранявшийся над
Р2О5 в течение пяти месяцев, не плавился даже при нагревании
до 180°; после кратковременного пребывания на воздухе
температура плавления понизилась до 55° вследствие поглощения
кристаллами следов влаги. Корног и Бауэр [6] определили для
безводного препарата KJC12 т. пл. 195°, а для моногидрата
KJC12-H20—43°. Такие же различия в температурах плавления
известны и для дибромоиодаата калия: 58° — для моногидрата и
180° — для безводной соли.
Сравнение рассматриваемых в этой главе тригалогенидов солей
•щелочных металлов и аммония [28] показывает, что по
величинам температур плавчения в зависимости от состава аниона
их можно, в общем, расположить в следующий ряд: JBr~ > JC1~ >
> ClJBr-, а в зависимости от природы катиона—в ряд Cs+>Rb+>
> NH^ > К+. Для тетрахлороиодиатов последовательность температур
ллавления соответствует ряду Cs > Rb >K > NH4.
Упругость диссоциации измерялась в связи с вопросом об
относительной термической устойчивости безводных и гидрат-
ных форм. На рис. 70 показано, по данным Чизмана и
282
Мартина [62], что безводный KJBr2 обладает меньшей
упругостью диссоциации, чем его моногидрат *. Такое же
соотношение упругостей диссоциации обнаружено для KJQ2 и
KJC12 • Н20 [6, 17, 28].
Термическая устойчивость комплексных галогенидов хлористого,
бромистого и треххлористого иода устанавливалась разными
методами, что обусловило как различные критерии устойчивости
этих комплексов, так и расхождение экспериментальных данных.
При нагревании этих комплексов, равно как и полигалогенидов,
они диссоциируют таким образом, что твердый остаток
представляет собой моногалогенид, содержащий наиболее
электроотрицательный из галогенов, входивших в состав комплекса, например:
МО — при разложении MJC12 и MCUBr; МВг — при разложении
MJBr2 и MF — при разложении MFJC13.
Ряд авторов [19, 20, 23 и др.] сравнивал термическую
устойчивость три- и пентагалогенидов по температуре полного побеле-
ния комплекса и превращения его полностью в хлорид, бромид
или фторид данного катиона. Некоторые из имеющихся в
литературе данных приведены в табл. 43; в ней для сопоставления
показаны температуры, 'Соответствующие упругости паров продуктов
диссоциации (при 760 мм рт. ст. [17]).
Таблица 43
Температура разложения некоторых тригалогенидов
Формула комплексов
CsJBr2
RbJBr2
CsJCl2
CsClJBr
RbJCl2
RbCUBr
KJC12
KJBr2
CsBrCl2
RbBrCl2
Температура разложения, °С
по побелению остатка по упругости паров
320
265
290
290
265
200
215
180
150
110
242,5
186,5
209
—
151
—
109**
142**
—
—
Как и следовало ожидать, «температуры побеления» более или
менее'значительно превышают температуры разложения,
определенные по упругости пара и точнее отражающие устойчивость
тригалогенидов щелочных металлов.
Несмотря на расхождение в величинах температур разложения,
определенных различными методами, литературные данные позво-
* Как видно из рис. 70, данные Зфраима [17] по упругости диссоциации
KJBr2 укладываются на кривую для гидрата. То же можно сказать и в
отношении данных Эфраима для упругости диссоциации KJC12.
По-видимому, эти данные относятся к препаратам, представляющим собой гидраты или
содержащим значительное количество последних.
** Эти значения вычислены Зфраимом по формуле ГклХг = ^Rbjci2 " J/V /VRh,
в которой Т — абсолютная температура разложения, а V — атомный объем калия
или рубидия, Х=С1.
283
ляют установить некоторые общие факторы устойчивости этой
группы комплексных соединений галогенидов иода в твердом
состоянии (в отсутствие растворителя). Характерным, как и для
комплексов типа полигалогенидов, является ясно выраженная
зависимость их термической устойчивости от поляризационного
взаимодействия составных частей комплекса.
Так, устойчивость тригалогенидов и тетрахлороиодиатов
щелочных металлов и аммония и комплексов типа MFJC13 возрастает
в ряду катионов Li; Na, К, NH4, Rb и Cs в соответствии с
величинами ионных радиусов. Тетрахлороиодиаты лития, натрия и
двухвалентных катионов были получены в виде гидратов *. Замена
катионов щелочных металлов на комплексные катионы типа
замещенных аммониевых оснований часто приводит к
повышению устойчивости, особенно в 'случаях симметрического
строения последних **. Аммониевые соединения оказались устойчивее
сульфониевых. Устойчивыми оказались и тетрахлороиодиаты амина-
тов кобальта, полученные Спаку и'Попеа [31, 32].
Термическая диссоциация тетрахлороиодиатов щелочных
металлов [20], аммониевых и сульфониевых оснований {40] протекает
ступенчато: сперва отщепляется молекула хлора, а затем при более
высокой температуре — хлористый иод.
RJC14^RJC12 + C12;
RJCla^RCl + JC!.
Так, по данным Рэя [20], CsJCl4 при 23° мед пенно превращается
в CsJCI2, а последний при 116—118° постепенно переходит в CsCl.
Исходя из предположения, что центральным а-омом рассматриваемых
анионов является наиболее электроположительный галоген, и из
некоторых экспериментальных данных, Кремер и Дункан [28] показа пи,
что более устойчивыми должны быть тригалогениды с симметричными
анионами. Устойчивость последних должна возрастать в таком ряду
(в который для сравнения включены также полигалогениды BrJ~ и
J~): Br-J—J < F—J—Br<Cl—J—Br < J-J—J~< Br—J—Br < Cl-
—J—CI.
Эта последовательность термической устойчивости в ряде
случаев не совпадает с экспериментальными данными [63]. Так,
температуры разложения MJC12 (M=K, Rb, Cs) по упругости пара
(табл. 43) всегда ниже температур разложения MJBr2 ***, проти-
* Гидратированные катионы обладают меньшим эффектом
контрполяризации. Гидратация катиона в комплексах типа М (JC14)„ -тН20
подтверждается тем, что аналогичные комплексы, а также тригалогениды, содержащие
комплексные катионы [Соеп2С12]+, [СоРу4С12] + и тому подобные, обычно не гидра-
тируются [31, 32].
** Тригалогениды, содержащие четное число алкильных групп, более
устойчивы.
*** Аналогично этому степень гидролиза JBr^~ меньше степени гидролиза
JC1-" (табл. 44).
284
воположно тому, что следует из представлений Кремера и
Дункана и из температур разложения, определенных по повелению
остатка (табл. 43).
Рис. 71. Кривая нагревания"-и охлаждения KJC12.
В связи с этими расхождениями следует привести результаты
термографического исследования дихлоро- и дибромоиодаата калия,
по данным Шор, полученным в ИОНХ АН УССР [64]. На рис. 71
Рис. 72. Кривая нагревания и охлаждения KJBr2.
и 72 приведены кривые нагревания и охлаждения KJC12 и KJBr2,
записанные показаниями простой и дифференциальной термопары.
Как видно из рис. 71, разложение KJC12 начинается уже при
95°, более интенсивно происходит при 108—110°* и заканчивает-
* Визуальные наблюдения показали, что при этих температурах
происходит "интенсивное разложение KJC12 и KJBr2 с выкипанием образующегося
хлористого или бромистого иода.
285
ся при 135°. Кривая рис. 72 показывает, что частичное разложение
KJBr2 начинается при 120° и выражено небольшим термическим
эффектом; 'полное разложение происходит лри 160°. Эти
эффекты отсутствуют на кривых охлаждения, следовательно, они
отражают не плавление комплексов, а их разложение.
Температуры разложения KJC12 (108—110°) и KJBr2 (160°) у
полученные при термическом исследовании, оказались очень
близкими к данным, рассчитанным Эфраимом (табл. 43).
Отношение к растворителям. Тригалогениды и
тетрахлороиодиаты щелочных металлов и аммония более или
менее легко растворимы в воде. Увеличение размера катиона часто-
уменьшает растворимость этих комплексов. Соли комплексных
катионов типа [Co(NH3)4Cl2Jf, тригалогениды диазония, соли типа
M[FJC13] труднорастворимы.
Растворимые в воде соли, за исключением наиболее устойчивых^
разлагаются в растворе вследствие гидролиза анионов,
сопровождающегося выделением иода и образованием других продуктов,,
например:
5JC12"+ ЗН2 О Z J07+ 10СГ+ 2J2 4- 6Н+П06].
Кремер и Дункан [65] определили степень гидролиза галогени-
дов иода (JCl, JBr, JC13) и их двойных галогенидов в 0,05-мол.
растворах при 25° взбалтыванием их в течение трех дней с че-
тыреххлориетым углеродом ,и извлечением выделившегося при этом
иода. Полученные ими значения приведены в табл. 44.
Таблица 44
Степень гидролиза галогенидов иода и их комплексных соединение
Галогенид
HJC14
KJC14
JCI3
KJBr2
HJBr2
NH4JBr2
HBrJCl
KBrJCl |
1 Степень
гидролиза,
' %
13
13
24
29
30
33
47 1
59 |
Галогенид
NaBrJCI
JBr
HJC12
KJC12
NaJCl2
NH4JC12
JCl
—
Степень
гидролиза, %
61
65
70
83
86
87
100
—
При электролизе водных растворов RbJCl2 (подкисленных
соляной кислотой) [66] или [C5H5NH]JC12 и [С6Н5(СНз)зЫУС12 [67J
было обнаружено выделение иода у катода. Это можно объяснить
разрядом ионов J+, участвующих в сложном ионном равновесии в
этих системах, наряду с продуктами диссоциации и гидролиза.
Неводные растворители, неполярные и с малыми значениями
диэлектрической постоянной, обычно не растворяют двойных
галогенидов иода. Многие из них растворяются лишь в полярных
растворителях, хотя в этом отношении известны довольно
значительные различия как в растворимости их, так и в отношений
286
устойчивости в неводных средах, где возможна диссоциация на
галогенид иода и моногалогенид. Так, например, Доббин и Уокер
[37], определив молекулярный вес эбулиоскопическим методом в
спиртовом растворе дихлоро- и дибромоиодаатов органических
катионов, показали, что их устойчивость увеличивается в ряду
JC1F < JBr7 и в ряду [(СН3)3 S]+ < [(CH3)4 N]+ < [C5H5NCH3]+.
Неводные растворители при действии на твердые двойные
галогениды могут более или менее легко извлекать содержащийся
в них галогенид иода в соответствии с термической устойчивостью
этих комплексов. Так, Кремер и Дункан [28] показали, что
равновесные концентрации галогенида иода в СС14 при действии
последним на твердые тригалогениды уменьшаются в ряду солей
с анионами JBrCl~>J^rr>JCl^~ и с катионами K>NH4>Rb>Cs,.
т. е., по данным этих опытов, термическая устойчивость тригало-
генидов увеличивается в таком же направлении (см., однако,
стр. 282 и 316).
Говоря о равновесиях, существующих в растворах двойных га-
логенидов иода, необходимо отметить, что для тетрахлороиодиатов
характерно также окислительно-восстановительное равновесие по>
схеме JCl^i^JQ^r+Cb. Так, например, Спаку и Попеа [31, 32] на-
блюдали это равновесие в водных растворах солей с комплексными
катионами. Попов и Джесап [49] показали наличие такого же
равновесия, изучая спектры поглощения растворов [(СН3)з$УС14 в аце-
тонитриле. Баклес и Миллс [47] спектрофотометрически
исследовали состояние равновесия (в области 300—400 ммк при 25°)
I(QH9)4N] JC14 - [(C4H9)4N] JC12 + Ci2.
Константа равновесия в ацетонитриле равна (1,43±0,19)-10"4,
а в хлористом этилене (1,85±0,11)-10~4. Состояние равновесия в
растворах 10~3 моль/л достигалось в течение 1—2 часов. Более
разбавленные растворы порядка 10~5 моль/л, а также растворы
[(CH3)4N]JCl4 сильно разлагались даже в темноте.
Электролитическая диссоциация.
Электрохимические свойства растворов двойных галогенидов иода, в частности
их электропроводность, еще мало исследованы. Отчасти это
объясняется значительной термической неустойчивостью многих из этих
комплексов * и их способностью в ряде случаев реагировать с
растворителем (реакции гидролиза или сольволиза, окисления и
т. д.).
Результаты измерения электропроводности систем КО—JC1 и
КВг—JBr в нитробензоле [3, 4] **,а также растворов некоторых три-
* По этой причине измерение электропроводности и ряда других свойств
растворов тетрахлороиодиатов не дает определенных результатов.
** Удельная электропроводность этих растворов, в которых образуются
комплексы KJC12 и KJBr2, порядка Ю"*3 сш-1 см~1.
287
галогенидов аммониевых оснований и цезия [46, 68] характеризуют
эти комплексы как сильные электролиты. Так, по данным Попова
и Скелли [46], предельная эквивалентная электропроводность
[(CH3)4N]JBr2, [(CH3)4N]JBiCl и CsJBr2 в ацетонитриле (ДП = 36,5)
при 25° равна соответственно 193,0—197,2 — 187,1 ом~1 см 1 [46]
Для растворов [(CH3)4N] JBrCl, [(nC3H7)4N] JBr2 и [(rcC4H9)4N] JCl^
вдихлорэтилене(ДП=10,23)Хоо при 25° равна 39,0—36,8—38,3 О-м^слО,
причем ионная подвижность комплексных анионов уменьшается в ряду
ЛВгСГ > JC17 > JBr^".
Измерение спектров поглощения растворов тетраалкиламмо-
ниевых солей этих анионов в ацетонитриле и дихлорэтилене и их
систем с ионами хлора и брома [106] дало основание считать, что
в данных растворах существуют такие равновесия:
JCir^JCl + СГ;
JBrF^ JBr + ВГ;
2JBrCr^JCir + JBrF.
4. ХИМИЧЕСКИЕ СВОЙСТВА И РЕАКЦИИ ДВОЙНЫХ ГАЛОГЕНИДОВ
ИОДА
Наиболее систематические исследования в этом направлении
проведены Кремером и Дунканом [29, 65], изучавшими химические
реакции этой группы комплексов как в отсутствие растворителя,
так и в растворах. Эти реакции представляют интерес также
потому, что они дают представление о строении молекул двойных
галогенидов иода.
Реакции в отсутствие растворителя [29]. Как
отмечают Кремер и Дункан, во многих случаях в реакцию вступают
не непосредственно комплексные галогениды, а продукты их
термической диссоциации. Это возможно в тех случаях, когда комплексы
неустойчивы и обладают большой упругостью диссоциации или
когда в реакции участвуют газы, смещающие равновесие
диссоциации комплексных галогенидов. В подтверждение сказанного
можно привести следующие реакции:
а. Аммиак образует продукты присоединения с дибромоиодаата-
ми RJBr2 • 2NH3, есл'и упругость диссоциации RJBr2 меньше 0,005
мм рт. ст. В противном случае он реагирует с бромистым иодом,
образуя йодистый азот и бромистый аммоний: *
RJBr2;£RBr + JBr;
NH3 + 3 JBr = N J3 + ЗНВг;
NH3+HBr = NH4Br.
* Дихлороиодааты, очевидно, реагируют с аммиаком подобным же
образом [48].
288
б. Озон превращает KJBr2 в иодат калия:
KJBr2 + 03 — KJ034-Br2.
в. Хлор, реагируя с дибромоиодаатами, быстро замещает только
атомы брома, вовсе не замещая иода:
KJBr2 ± С12 - KJC!2 + Вг2.
Эта реакция указывает на различное валентное состояние
атомов галогена в молекуле KJBr2 и подтверждает такое строение
аниона [Br~J+Br~J~.
г. Один галогенид иода — продукт термической диссоциации
комплекса — замещается другим, например:
МJC12 + 2JBr == MJBr2 -f 2JC1.
Эта реакция подтверждает большую устойчивость MJBr2 по
сравнению с MJC12.
д. Галогены могут присоединяться к тригалогенидам
MJC12 + C12;£MJC14.
Однако Кремер и Дункан отмечают, что реакции присоединения
галогенидов иода неизвестны, вероятно, потому, что они должны
сопровождаться изменением знака заряда иона в адденде (т. е.
восстановлением J+ до J~), например:
CsJCl2 + JBr -> Cs
CI"
CPJ+JT
Br-
Этому утверждению противоречит обнаружение и по пучение Корногом
и Бауэром [6] комплекса состава KC1-2JC1. Для этого галогенида
-+ —
можно предположить строение двухъядерного комплекса К [C1JC1...
н—
, ..JCI], образовавшегося по реакции
КС1 -г J [JC12] = К [C1J (JC17)]*.
Разумеется, это предположение должно быть подтверждено
экспериментально.
Реакции в растворах. В растворах реакции протекают
несколько отлично от описанных выше.
а. Действие галогенов проявляется в реакциях присоединения^
замещения и окисления. Так, при пропускании хлора в водный
раствор MJC12 происходят последовательные реакции,
выражаемые такой схемой:
JClj-H2 JCir Cl2+Hg-?J07.
* Это соединение не может быть построено аналогично KJC14, так как
наличие в одной и той же координационной сфере разнозаряженных атомов
иода J+ и J~~ должно вызвать образование молекулярного иода.
19—1419
289
Дибромоиодааты так же реагируют с хлором, только сперва про.
исходит реакция замещения ионов брома: JBr7+ Cl2 = JCljT-f- Вг&
Бром же непосредственно окисляет JBr<T:
ЛВгГ + 2Вг2 + ЗН20 - J03~ + 6Н+ + 6Вг~ [65].
б. Сильные окислители действуют на водные растворы двойных
галогенидов иода таким образом, что происходит выделение хлора или
брома (но не иода) и образование иодатов [66, 67].
в. При взаимодействии JC1<T, JBrJT и JCli" с восстановителями
образуются СР, Вг~ и иод; последний затем восстанавливается до
J—.' Иодиды количественно реагируют с приведенными выше
комплексными анионами по схемам:
JXn + (п - 1) J" = -f J2 + /iX"
например, JC\T + 3J~ = 2J2 + 4СГ [65, 67].
Все эти реакции подтверждают, что в комплексных галогени-
дах иода центральным атомом является J+ или J3+.
5. ФИЗИКО-ХИМИЧЕСКОЕ ИССЛЕДОВАНИЕ СИСТЕМ ХЛОРИСТЫЙ
(БРОМИСТЫЙ) ИОД —ГАЛОГЕН ИДЫ ДРУГИХ ЭЛЕМЕНТОВ
И КОМПЛЕКСНЫХ СОЕДИНЕНИЙ, ОБРАЗУЮЩИХСЯ В ЭТИХ СИСТЕМАХ
Хлористый и бромистый иод как растворители и их системы с
галогенидами других элементов ранее были очень мало
исследованы. Этому вопросу были посвящены лишь насколько работ
Корнога, Руффа, Гутмана и некоторые другие.
О растворимости галогенидов ряда элементов в жидком
хлористом иоде и о способности некоторых из них образовывать с ним
двойные галогениды уже сообщалось выше.
Физико-химическое исследование систем хлористый
иод—хлориды калия и аммония впервые произвел Корног с сотрудниками.
Корног и Бауэр [6] исследовали упругость пара системы JC1—КС1
при 25° и обнаружили образование в ней двух соединений KC1-JC1
и KC1-2JC1. На рис. 73 линия АВ представляет упругость пара
ненасыщенных растворов, в точке В начинается образование твердого
KJ2CI3 и заканчивается у С, где исчезает жидкая фаза. Линия DE
соответствует образованию второго комплекса KJCb (КДгОз + КО^
^2KJC12), а линия GH — равновесию между KJCb и KG (при
постоянном давлении).
Образование комплекса KJ2CI3 подтверждено этими же
авторами [6] изучением растворимости КО в хлористом иоде и
выделением этого комплекса. На рис. 74 отрезок кривой АО
представляет твердую фазу KC1-2JC1, переходящую при 45° в
KC1-JC1 (отрезок кривой ОВ)\ линия СО относится к метастабиль-
ной форме комплекса KC1-JC1.
Корног и Карджес [69], исходя из того, что расплавленный
хлористый иод представляет собой ассоциированную жидкость, к
290
едполагая поэтому, что он должен быть «ионизирующим
растворителем» для галогенидов других элементов, измерили электро-
Р оВодность растворов КС1 и NH4C1 в хлористом иоде при 35°.
Электропроводность быстро растет с концентрацией и для
максимальных из исследованных концентраций — 0,5 моль1л KQ и
1 моль/л NH4C1 — составляет соответственно 1,898 • Ю-2 и
о 418-10-2 ом~1 смт1, т. е. превышает электропроводность
хлористого иода при 35° примерно в 4 и 5,5 раз.
В100 г
JCL
41
Р
30
25
К
\ \в с
20
tf \
10
5
0
\D £
И
0,2 0,4 0,6 0.8 1.0
мол. на
Рис. 73. Зависимость
упругости пара системы хлористый
иод — хлористый калил от
состава при 25°.
25 *5
ГС
85
Рис. 7'4. Кривая
растворимости хлористого калия а
. хлористом иопе.
Более подробные исследования электропроводности хлоридов
щелочных металлов и хлористого аммония были произведены
Гутманом; (J94J По его данным, LiCl и NaCl малорастворимы в
хлористом иоде, в отличие от хлоридов калия, рубидия и цезия,
которые к тому же образуют очень хорошо проводящие ток растворы.
На рис. 75 приведены изотермы эквивалентной
электропроводности этих растворов *.
■Помимо перечисленных выше систем с хлористым иодом,
была исследована также система SbCU—JC1. Своеобразная
диаграмма плавкости этой системы (рис. 76), по данным Руффа с
сотрудниками [11, 70], указывает на образование двух соединений
SbCl5-2JCl и SbCl5-3JCl с температурами плавления между 62 и
63°, непрочных и образующих друг с другом непрерывный ряд
твердых растворов.
Эти немногочисленные сведения о физико-химических свойства^
* По данным Гутмана [94], хлориды серебра и бария лишь незначительно
повышают электропроводность хлористого иода; хлористый сульфурил,
наоборот, понижает ее с ростом концентрации. Такие хлориды, как SiCl4, TiCU,
VC14, NbCl6, SbCl5, в растворах— 0,1 моль/'л оказывают слабое влияние на
электропроводность хлористого иода.
291
систем, состоящих из хлористого (и бромистого) иода *, не могли
дать достаточной характеристики образующихся в них соедине.
ний, возможных типов отих комплексов, их строения,
химической природы и т. д.
10000
20000
30000 V
Рис. 75. Эквивалентная электропроводность
растворов хлоридов щелочных металлов и
аммония в хлористом иоде.
С целью решения этих вопросов в Институте общей и
неорганической химии АН Украинской ССР были исследованы 'системы **,
Рис. 76. Диаграмма плавкости системы пятихлористая
сурьма — хлористый иод.
одним из компонентов которых был хлористый или бромистый иод,
а другим компонентом соответственно КС1 [3, 13/14, 71], КВг
[4, 14-16], AlCls [3, 13, 14, 711, А1Вг3 14, 14—16], РС15 [9], РВг5 [9],
* Бромистый иод как растворитель ранее вовсе не был исследован.
** В выполнении этих работ принимали участие Абарбарчук, Каганская,
Кузьменко, Музыка, Шор.
292
SbCl5 [12], SbCl3 [73, 741, SbBr3 [73, 74], AsCl3 [75]. Эти
двойные системы были исследованы также в нитробензоле. Кроме
того, Для выяснения условий образования некоторых комплексов
были исследованы системы РС15—иод [8], РВг5 — иод [10], РС13—
JC1 и РВгз—JBr [27], SbCl5—иод [72, 76]. Наконец, исследованию
подвергались также системы «смешанного типа»: JC1—КВг [15],
JC1—А1Вг3 [15], РС15—JBr и PBr5—JC1 [76].
Мы рассмотрим сперва те системы (КХ—JX и РХ5—JX), в
которых образуются комплексы анионного типа M[JX2], а затем—-
системы (А1Х3—JX, SbCl3—JC1, SbCU— JC1 и некоторые другие),
в которых обнаружены комплексы типа J[RXj (X=C1 или Вг).
Системы, состоящие из хлористого или бромистого калия и гало-
генидов калия, равно как и их растворы в нитробензоле,
оказались довольно хорошими электролитами: удельная
электропроводность систем КХ—JX при 35—45° порядка 10~2 ож~х см-\
нитробензольных растворов — порядка Ю-3 ом~х см~1 (рис. 77—82).
Изотермы удельной электропроводности двойных систем
показывают примерно одинаковую концентрационную зависимость
(максимум в области ~ 8 мол. % галогенида калия). По величине
электропроводности (в области максимума эти системы располагаются в
следующий ряд: JC1—KCKJC1—KBr<JBr—КВг (рис. 77, 80, 82).
Электропроводность двойных систем MX—JX обусловлена
электролитической диссоциацией образующихся в данных системах
комплексов, имеющих характер довольно сильных электролитов,,
что видно, в частности, из кривых молекулярной (эквивалентной);
электропроводности этих систем (рис. 75, 79) *.
Доказательства образования в системах JX—КХ комплексов
типа KXJX приведены выше. Такие же комплексы образуются и в.
тройных системах, доказательством чему могут служить,
например, такие факты: галогениды калия, нерастворимые в
нитробензоле, растворяются в нитробензольных растворах хлористого и
бромистого иода; при добавлении KG. или КВг к нитробензольным
растворам JC1 или JBr депрессия температуры замерзания не
только не увеличивается, но даже уменьшается, т. е. в растворе идет
процесс, сопровождающийся уменьшением общего количества
частиц.
Криоскопическое исследование систем КС1—JC1 и КВг—JBr в
нитробензоле как растворителе [3, 4] показывает, что образующиеся
в этих растворах комплексы находятся в ассоциированном
состоянии. Кажущийся фактор ассоциации увеличивается с концентрацией
и с ростом молярного отношения КХ : JX, достигая значений ~3—3,5.
При этом в нитробензольных растворах системы JBr—КВг явления
* Наши данные для системы JC1—КС1 [13] в области небольших
разведений — до V = 5300 см3 (рис. 77) хорошо согласуются с данными Гутмана
(рис 75).
Как видно из рис 75, даже при сравнительно больших разведениях не
удается приблизиться к величине Хоо вследствие того, что в этой области
концентраций начинает сильно проявляться электропроводность самого
растворителя [94].
293
ассоциации выражены в несколько большей степени, чем в раство.
pax системы JC1—КО. Возможно, что этим явлением объясняется
тот факт, что для данных растворов наблюдается обратный поря.
4 6 д
мол. %№
Рис. 77. Удельная электропроводность
системы хлористый иод — хлористый калий:
1 — данные Корнога и Карджеса [69], 2—
данные Фиалкова и Шор [13].
0,16 Opt
мол. №
мол. 3CL
Рис. 78. Удельная электропроводность
системы хлористый иод — хлористый калий—
нитробензол
мол. JCl I
II —
мол. CeH5N02
мол. JC1
"мол. C6H5N07
9,7'
1
19,83
док величин электропроводности по сравнению с
соответствующими двойными системами JX—КХ.
Для решения вопроса о составе комплексов и природе ионов,
образующихся в этих системах, большое значение имеют
результаты опытов по изучению переноса ионов. Продуктами электроли-
294
йО
50 J
40 •)
30
20 ]
10
О
WOO 2000 SOOO WQQ 5000
Рис. 79. Молекулярная электропроводность
системы хлористый иод — хлористый калий.
2*68
мол.% ИВг
Рис. 80.. Удельная
электропроводность системы
бромистый иод — бромистый
калий при 45°.
О 0А 0,6 1,2 1,6 2,0 2А 2.8
мол. UBr
мол. JBr
Рис. 81. Удельная
электропроводность системы бромистый иод —
бромистый калий — нитробензол.
мол. JBr . 1
~~ " ПТ28 '
1
= 23,99'
/ —
Л —
мол. C6H5N02
мол. JBr
мол. C6H5N02
0,008
Ь S
мол. % Mr
12
Рис. 82. Удельная
электропроводность системы хлористый код —
бромистый калий: /—35°; //—45°.
за систем, -состоящих из галогенидов калия и галогенидов иода
являются на катоде калий и небольшое количество иода'
образующегося в результате электролиза избытка галогенидов"
иода*, а на аноде — хлор или бром. При электролизе нитробен-
зольных растворов этих систем в католите накапливается калий,
а в анолите—иод и хлор или бром. Количественная
характеристика опытов по переносу ионов представлена в табл. 45.
Таблица 45
Числа переноса иоков в системах галогениды калия — галогениды иода **
Элемент
К
J
О или Вг
Система
JC1-KC1 [15]
Опыт 1 J Опыт 2
0,29
0,19
1,00
0,31
0,25
0,91
Система
JBr-KBr [16]
Опыт 1 | Опыт 2
0,43
0,17
1,50
0,42
0,18
1,47
! Система JBr—
KBr-C6H5N02
[4]
Опыт 1
0,59
0,69
1,31
Опыт 2
0,51
0,69
1,04
Анализ количественных результатов, приведенных в табл. 45г
[14] показал, что в системах JX—КХ и в их нитробензольных
растворах электролизу подвергаются комплексы K[JX2],
диссоциирующие на ионы К+ и JX2". Различие состоит лишь в том, что в
двойных системах КХ—JX, содержавших значительный избыток
хлористого или бромистого иода, последние подвергались электролизу
наряду с комплексом. Это приводило к переносу иода в католит,
а брома (или хлора) в анолит. При рассмотрении результатов
опытов с нитробензольными растворами систем JX—КХ
необходимо принимать во внимание также вторичные процессы, которые
обусловили различное поведение иода в системах с растворителем
и без него.
Таким образом, результаты этих исследований подтвердили, что
комплексные галогениды — продукты соединения галогенидов
калия с хлористым и бромистым иодом—содержат катион К+
(в'системах с нитробензолом он, вероятно, сольватирован) и
комплексные анионы JC127 JBr~ CUBr-***.
Следующей группой исследованных в Институте общей и
неорганической химии АН УССР комплексов, содержащих эти анионы,
являются продукты соединения пятихлористого и пятибромисто-
го фосфора с хлористым и бромистым иодом. Для получения и
* В расплавленном хлористом или бромистом, иоде при 35—55° удается
растворить не более 8 мол. % хлористого или бромистого калия.
** Число переноса иона К+ в системе КС1—JC1—C6H5N02 равно 0,49 [71].
Исходя из этих данных, Гринвуд и Эмелеус [78] рассчитали число переноса
для иона JC12~~ в этой же системе. Оно оказалось равным 0,504. Эти числа
переноса ионов К+ и JC12~ очень близки к тем значениям, которые определены
для ионов К+ и С1_ в водных растворах.
Тот факт, что в наших опытах (табл. 45) числа переноса, рассчитанные
для ионов С1~" и Вг~, оказались равными или большими 1, подтверждает,
что хлор или бром переносились в анолит в составе комплексного аниона.
*** Более сложные анионы, например Л2С1з~, этими исследованиями не
обнаружены.
296
исследования этих комплексов были взяты системы, состоящие, с
одной стороны, из три- или пятигалогенидов фосфора, а с другой
стороны—из иода или галогенидов иода (JC1, JBr).
Тригалогениды фосфора РС13, РВг3 (а также PJ3) не дают с
иодом соединения и не образуют с ним электролитных систем [8].
Иначе ведут себя РС1б [8] и РВг5 [10]. Эти пятигалогениды фосфора
окисляют иод до хлористого или бромистого иода,
восстанавливаясь при этом до РС13 или РВг3. Моногалогениды иода
соединяются с непрореагировавшими еще молекулами РХб в комплексы
состава PCUJ или PBrJ, что можно выразить следующими
уравнениями:
PX5 + J2^PX3 + 2JX;
2PX5 + 2JX = 2PX6J
ЗРХ5 + J2 = 2PX6J + РХ3 (X - С1 или Br). У }
Первый из этих комплексов (PCUJ) получил Бодримой [7] в
1862 г., но не исследовал его. Комплекс PBr6J ранее не был
описан.
Комплекс PCUJ Бодримой получил несколькими способами:
взаимодействием пятихлористого фосфора с иодом или с
хлористым иодом; взаимодействием РС13 и РСЬ с треххлористым иодом:
РС13 + JC13 = PC16J;
РС15 + JC13 = PC16J + Clo.
Чтобы подтвердить приведенный выше механизм образования
комплексов РСЫ и PBr6J из пятигалогенидов фосфора и иода,
Фиалков и Кузьменко [27] исследовали системы из РС13 или РВг3 и
соответствующего моногалогенида иода. В этих системах также
происходит окислительно-восстановительная реакция, но обратная
по отношению к показанной выше—образуются иод и РХб,
молекулы последнего, соединяясь с избытком галогенида иода, дают
те же комплексы PC16J или PBr6J [27]:
PX3 + 2JX^PX5 + J2;
РХ5 + JX = РХ6 J
РХ3 + 3JX = PX6J + J2. (12.2)
Наконец, эти же комплексы были получены при непосредственном
взаимодействии РС15 иги РВг5 соответственно с JC1 или JBr [9]
в расплавах или в неводных средах.
Реакции (12. 1) и (12. 2) были изучены не только
препаративным путем, но и физико-химическими методами. Так, на
диаграммах плавкости систем РС15—иод и РВг5—иод (рис. 83, 84)
эвтектические температуры (79,0—79,5° в системе с РСЬ и 13,0—13,5° в
системе с РВгб) можно было проследить только до содержания
РХб" 70 мол.%, т. е. примерно до такого соотношения РХб : Лг,
которое выражается суммарным уравнением (12,1). При содержании
РВгб, большем 76 мол. %, кривая плавкости начинает понижаться.
297
80 PCi5
Рис. 83. Диаграмма плав-
жости системы пятихлорис-
тый фосфор — иод.
г
ffOJ
90-
/о-
SO-
SO-
ю~
J2
' 1—
°\ г
°\ /
1
РВгЛ
Р N
/ ч
/ \
/ 1
/ о—о--
20
hO 60 80 100
мол. % РВг5
Рис. 84. Диаграмма плавкости
системы пятибромистый фосфор — иод.
мол.%
Рис. 85. Диаграмма плавкости
системы пятибромистый фосфор —
бромистый иод.
'XI 30 50
мол.%
70
РС15
Рис. 86. Криоскопическое
исследование системы пягихло-
ристый фосфор — хлористый
иод в нитробензоле.
Максимум на диаграммах плавкости соответствует температурам,
очень близким к температурам плавления комплексов PCls • JC1
(212—214°) и PBr5-JBr (114,5°).
Образование комплексов PXs-JX хорошо доказано также
изучением плавкости системы РВг5—JBr (рис. 85) и криоскопическим
методом — в нитробензоле как растворителе (рис. 86, где Л£
означает изменение депрессии, наблюдаемое при добавлении PCls
к раствору JC1 в нитробензоле [9]).
Комплексы РС1бЛ и PBr6J обладают свойствами сильных
электролитов. Так, все исследованные системы, в которых образуются
эти комплексы, хорошо проводят ток — удельная
электропроводность достигает значений 0,03—0,04 омг1 см~х (рис. 87—89), в то
время как электропроводность расплавленных иода в 1000 раз, га-
логенидов иода в 10—100, а пятихлорйстого фосфора в 107 раз
меньше; тригалогениды фосфора не проводят тока.
Способность комплексов РХб*1 хорошо проводить ток
подтверждается их исследованием в индивидуальном состоянии. Для этой
цели они были получены «путем -сливания неводных растворов PCls
{в ССЦ) или РВг5 ( в CS2) и иода в тех же растворителях (3
моля РХб на 1 моль иода или равномолярные количества РХ5 и JX
[8, 9]).
Комплекс PCUJ или PCls-JCl мелкокристаллический, лимон-
но-желтаго цвета, плавится при 212—214°*. Комплекс PBr6J или
PBrs'JBr был получен в виде иголочек темно-вишневого цвета с
т. пл. 114,6°. Аналогичным путем ** Музыка [76] получил
комплексы PBrs-JCl — игольчатые кристаллы вишнево-красного цвета с
т. пл. 112,5° и PCl5JBr—мелкокристаллический желтый порошок
с т. пл. 140° (с разложением).
Эти комплексы трудно или сравнительно мало растворяются
в неполярных или малополярных органических жидкостях и в
CS2, насыщенные растворы обычно не проводят тока. В полярных
растворителях образуются хорошо проводящие ток растворы;
удельная электропроводность растворов РСЫ и PBr6J в
нитробензоле порядка 10~4—Ю-3, а в ацетонитриле порядка Ю-3 ом~1 см~1,
в хлороформе—10~5 ом~1 см~1 ***. Хорошо проводящие ток
растворы дают PC16J и PBr6J в броме, в хлористом или бромистом иоде.
Изотермы электропроводности (при 25—55°) этих систем имеют
примерно такую же концентрационную зависимость, как и в случае
системы РВГ5—JBr (рис. 89), с максимумом порядка 10г2омг1 смг1
в области 5—15 мол. %.
. Эти комплексы являются хорошими проводниками тока не
только в растворах, но и в расплавленном состоянии. Так,
электропроводность расплавленного PBr6J при 130° достигает 0,05 сш-1 см~К
Этот комплекс проводит ток даже в твердом состоянии.
Криоскопия разведенных растворов в нитробензоле дала ве-
* В запаянном капилляре.
** Реакцией между соответствующими РХ5 и JX и СС14.
*** Более детальные сведения о растворимости PC16J и PBrcJ и
электропроводности растворов см. [8—10].
299
-eg I
To 5Г
ыол. % PCLs
60
Рис. 87. Удельная
электропроводность системы пятихлористый
фосфор—иод при 130°.
~40 То То ТОО
мол.%РВг9
Рис. 88. Удельная
электропроводность системы пятибромистый
фосфор —иод при 130°.
ЭВг Ю 20
мол. %РВг5
30
Рис. 89. Удельная
электропроводность системы пятибромистый
фосфор—бромистый иод при 45 и 55°.
ичины мол. веса, примерно в два раза меньшие, чем теоретиче-
ше, что показано, в табл. 46.
Таблица 46
Молекулярные веса комплексов PX6J по криоскопическим данным 19,76]
Комплекс
PC16J
PBr6J
PBr5JCl
PBr5JCl
Растворитель
Нитробензол
»
Бензол
Молекулярный вес
найденный | теоретический
180,3-189,1
353,9-358,6
318,9
590,8
370,6
637,5
592,9
Столь значительное понижение экспериментально найденных
величин молекулярного веса сравнительно с теоретическими
значениями (примерно в два раза) нельзя объяснить термической
диссоциацией комплексов, так как для PBrs'JCl в бензоле найден
молекулярный вес, практически совпадающий с формульным значением.
Это явление, вероятно, обусловлено электролитической
диссоциацией данных комплексов на два иона.
Природа ионов была установлена изучением переноса ионов в
иитробензольных растворах PCUJ и PBr6J. Результаты этих опытов
дали основание выразить строение комплексов формулами
[PCUHJCbl'H [PBr4]+[JBr2r~, представляющими их как двойные га-
логениды—соли галогензамещенного фосфониевого катиона *.
Комплексам смешанного типа PCls-JBr и PBr5-JCl тогда следует
приписать строение [PCI4]+ [CUBrJ* и [РВг4]+ [BrJClh. Подобное строение
комплексов PX6J было затем подтверждено при спектрофотометри-
ческом исследовании PCleJ и PBr6J в ацетонитрильных растворах
183] и рентгенографическом исследовании кристаллического PCleJ
[84]. При этом было показано, что ион [РСЦ]* имеет тетраэдрическое
строение (как и в пятихлористом фосфоре), а ион
[CUClh—линейное (как и в кристаллах NH4JC12) **.
Переходя ко второй из упомянутых выше групп систем,
рассмотрим сперва некоторые результаты, полученные при
исследовании систем с галогенидами сурьмы, с целью сравнить отношение
галогенидов фосфора и сурьмы к иоду или к галогенидам иода.
Тригалогениды сурьмы не образуют соединений ни с бромом
185, 86], ни с иодом [87]. В противоположность этому в системе
SbCU — иод [И, 70] происходит окислительно-восстановительное
взаимодействие, приводящее к образованию двух комплексов
* Эти данные представляют интерес так;:се в том отношении, что -ими
впервые доказано существование катионных комплексов тетрацидо-типа на
примере солей [РХ4] [JX2] [79]. В дальнейших работах ИОНХ АН УССР было
установлено, что катион [РС14]+, ранее обнаруженный в кристаллической
решетке пятихлористого фосфора, содержится также в комплексных соединениях
РС15 с хлоридами ряда металлов А1С13, FeCl3, SnCl4, SbCl5 [80, 81]. Есть
основания предполагать, что катионы [РС141+, [РВг4]+, [РС13Вг]+ содержатся также
в некоторых полигалогенидах фосфора [82].
** В этой же работе определены межатомные расстояния Р—С1 и J—С1.
301
SbCl5-2JCl и SbCl5-3JCl, выражаемое, по Руффу, следующими урав-
нениями:
SbCl6 + J2 = SbCl3 + 2JC1;
SbCl6+2JCl = SbClg-2JCl;
2SbCl5 +J2 = SbCl6-2JCl + SbCl3; (12.3)
3SbCI5 + 2J2 = SbCl5-3JCl + 2StCl3 + JC1 *. (12.4)
Оба эти соединения были получены из системы SbCls—JC1 {11]
в виде кристаллов сине-черного цвета, дымящих на воздухе,
плавящихся в пределах 62—63° (см. также рис. 76). О структуре этих
соединений и об их химической природе Руфф не дает никаких
сведений. Кроме того, данные Руффа о существовании
соединений SbCb с JC1 не были подтверждены Молесом [88],
исследовавшим методом криоскопии растворы иода или хлористого иода в
SbCl5.
SbCls, как и РС15, неполярен и также реакционно активен.
Является окислителем вследствие своей способности отщеплять
хлор и переходить при этом в SbCl3 и комплексообравователем,
дающим с различными хлоридами комплексный анион SbClg".
Отдельные молекулы пятигалогенидов фосфора и сурьмы имеют
бипирамидальную структуру, но в то время как структура РСЬ и
РВг5 резко изменяется ори их переходе в кристаллическое
состояние и вследствие явлений поляризации, бипирамидальная
структура SbCl5,'очевидно, сохраняется и в кристаллическом
состоянии **.
Было интересно выяснить, как отразится это отличие SbCls от
пятигалогенидов фосфора на характере их взаимодействия с иодом
или с хлористым иодом и на строении образующихся при этом
соединений.
Важнейшие результаты этих исследований таковы:
а. Система SbCl3—J2 [72]. Термический анализ
характеризует эту систему, как и систему РСЬ—Лг 18], как простую, с
одной эвтектикой. Электропроводность ее мало отличается от
электропроводности расплавленной SbCb и то лишь при больших
концентрациях последней (рис. 90).
б. Система SbCls—J2 [72]. На кривой плавкости этой
системы обнаружен весьма пологий максимум. Прекращение вторых
остановок при 66 мол.% SbCls подтверждает наличие в этой
системе нового соединения, для образования которого SbCls
и иод реагируют в отношении 2 : 1. Об этом же свидетельствует
и резкое изменение вязкости данной системы в этой же области
концентрации (рис. 91).
* Образование второго из этих комплексов можно представить также
другим уравнением:
5SbCl5 + 3J2 = 2 (SbClu.3JCl) + 3SbCl3 [72].
** Так, известно, например, что дигалогенидтриметилсурьма (CH3)3Sbr?
и в кристаллическом состоянии представляет собой упаковку бипирамидальных
молекул 189].
302
Измерение электропроводности системы SbCls—J2 также даег
основание считать, что в ней образуется новое вещество, имеющее
характер сильного электролита, так как электропроводность этой
системы имеет максимальное значение порядка 0,02 ом~} см~А при
28 мол. % SbCl5 и 55—65°, в
то время как
SbCls—неэлектролит, а электропроводность
расплавленного иода
порядка 10~5 ом~1 см~К
Хорошо проводит ток также и
нитробензольный раствор
SbCls и иода (*25—35° порядка
10"
ом
см~1).
20 W 60
мол. % Зр_
¥) 60 60
мол.% SbCLs
Рис. 90. Система трелхло- Рис. 91. Система пятихлористая сурьма-
ристая сурьма — иод. иод.
При изучении переноса ионов в нитробензольных растворах
системы SbCls—J2 было обнаружено, что иод переносится к катоду,
а сурьма и хлор — к аноду.
Были использованы два способа разделения продуктов
взаимодействием SbCls с иодом и получения в индивидуальном
состоянии комплексных соединений SbCl5*2JCl и SbCls-3JCl,
представляющих собой темно-бурые хорошо образованные кристаллы,
плавящиеся при 62—63°, сильно дымящие на воздухе.
Эти факты подтвердили предложенное Руффом представление
о природе химического взаимодействия SbCls и иода и показали,
что комплексные соединения SbCls с хлористым иодом имеют
характер сильных электролитов. В этом отношении система SbCl5—J2
напоминает системы из пятигалогенидов фосфора и иода.
308
цВ. Система SbCls—JC1 [82]. Как уже упоминалось, для дан.
ной системы была известна лишь диаграмма плавкости, показав-
шая образование двух соединений SbCl5-2JCl и SbCl5-3JCl с т пл
62 и 63° [11].
На изотерме вязкости этой системы (рис. 92) наблюдается
пологий максимум, приходящийся приблизительно на соотношение
SbCls: 3JC1. Второй из
упомянутых выше комплексов SbCl5-2JCl
не проявился на изотерме
вязкости .
Электропроводность системы
SbCls—JC1 в несколько раз
превышает электропроводность
хлористого иода. Максимум
электроне attoJ
120-
40 60 80
мал. % SbCLs
Рис. 92. Система пятихлорис-
тая сурьма — хлористый иод.
ОЛ 0,6
мол. SbCl5
moaJCI
Рис. 93. Удельная
электропроводность системы пятихлористая
сурьма — хлористый иод —
нитробензол.
мол. JC1 1
мол. CeH5NOa = Щ '
мол. JC1
1
мол. C6H5N02 3,67*
лроводности при 55—65°,
порядка 10~2 ом~1 см~\ приходится на
11 мол. % SbCl5 (рис. 92).
Изучение переноса -ионов в ни- //.
тробензольных растворах этой
системы показало, что катионом образующегося в «ней электролита
является иод, анион же состоит из сурьмы и хлора.
Наконец, были поставлены опыты получения комплексного
соединения SbCl5 с JC1 при взаимодействии их в газообразной
фазе. Хлористый иод был взят в избытке. Полученные при этом (на
стенках колбы) крупные темно-бурые блестящие кристаллы в
виде усеченных пирамид состава SbCls-3JCl плавятся при 62°, на
воздухе легко гидролизуются.
Тройная система SbCls—JC1—.нитробензол проводит ток
лучше, чем отдельно взятые нитробензольные растворы JC1
или SbCls- Изотерма электропроводности при 25° имеет максимум
(4,3-1(Г3 ом~1 см-1) вблизи соотношения SbCl5:2JCl (рис. 93). Крио-
скопические исследования [12] дают основание полагать, что при
добавлении SbCls в нитробензольныи раствор хлористого иода
сперва образуется комплекс SbCls'3JCl, 'переходящий затем в
SbCls - 2JC1.
304
г. Исследование комплекса SbCl5-3JCl [72].
Изучение этого комплексного соединения производилось в нитробензоль-
ном растворе. Удельная электропроводность этих растворов растет с
повышением 'концентрации и достигает значения 5,3-10"3 ож~х смг1
для насыщенного раствора, содержащего ~ 13 мол.% SbCl5-3JCl.
Криоскопические исследования растворов SbCl5-3JCl в
нитробензоле показали значительно уменьшенный 'молекулярный вес
(200 вместо 786), что должно быть объяснено электролитической
диссоциацией этого комплекса, а возможно также и частичным
распадом его на компоненты.
Изучение переноса ионов дало те же результаты, какие уже
наблюдались при исследовании систем SbCls—J2 и SbCls—JC1 в
нитробензоле. Иод переносился к катоду, а сурьма и хлор в
составе комплексного аниона — к аноду. В данном случае с 1 г-ат
сурьмы в анолит переносится~7,5 г-ат хлора. В католит при этом
переносится 2,5—2,9 г-ат иода.
Все эти исследования позволяют охарактеризовать комплексные
соединения SbCls с хлористым иодом как электролиты, ib которых
катионом является иод. К вопросу о строении этих комплексов мы
еще возвратимся, а лака рассмотрим результаты исследования
систем SbCl3—JC1 « SbBr3—JBr.
д. Система SbCl3—JC1 [73, 74]. Методом термического
анализа обнаружено соединение SbCl3-5JCl. Измерения вязкости и
электропроводности, однако, приводят к заключению, что в жидкой
фазе существует соединение SbCl3-4JCl. Это соединение имеет
характер довольно сильного электролита с максимальной удельной
электропроводностью 0,01 ом~1 см-1 при 45° и 16,4 мол,% SbCl3
{рис. 94).
Опыты по переносу ионов привели к результатам, аналогичным
тем, которые были получены для системы SbCl3—JC1, а именно:
иод переносится к катоду, а сурьма и хлор — к аноду. На 1 г-ат
сурьмы, перенесенный в анолит, приходится от 4 до 5 г-ат иода,
перенесенных в католит. Можно поэтому считать, что химическая
природа комплексов SbCls или SbCl3 с JC1 тождественная.
е. Система SbBr3—JBr [73, 74]. В противоположность тому,
что наблюдалось в системе SbCl3—JC1, трехбромистая сурьма не
образует соединений с бромистым иодом. Такое заключение можно
сделать на основании термического анализа системы SbBr3—JBr,
измерения ее вязкости, электропроводности, удельного веса
и на основании криоскопических исследований. Зависимость
всех указанных свойств от состава имеет линейный характер
(рис. 95).
Такое различие между SbCl3 и SbBr3 в их взаимоотношении к
соответствующим галогенидам иода, вероятно, обусловлено
соотношением ионных радиусов сурьмы и соответственно хлора и
брома, вследствие чего сурьма не может координировать вокруг себя
более трех атомов брома (см. также стр. 307).
Перейдем теперь к сопоставлению свойств пятигалогенидов
фосфора и сурьмы. Как показывают экспериментальные данные,
химическое взаимодействие пятигалогенидов фосфора и сурьмы с
20—1419
305
иодом имеет один и тот же характер, а именно: эти пятигалогешь
ды окисляют иод, переходя в тригалогениды фосфора или сурьмы
и в хлористый или бромистый иод, которые затем соединяются с
П-Ю*
не прореагировавшими еще
молекулами SbCU или РХ5. Но
получающиеся при этом продукты
весьма различны по своему
строению.
¥) ВО 80
мол. % SbCL3
Рис. 94. Система треххлористая
сурьма — хлористый иод.
W 60 60
,мол. % S6Br3
100 SbBr3
Рис. 95. Система трехбромнстая
сурьма — бромистый иод.
Комплексные соединения PX5-JX представляют собой двойные
галогениды строения [РХ4] • fJX2], а пятихлористая сурьма SbC'U
может соединяться с двумя и тремя молекулами JC1, причем
образуются комплексные соединения, имеющие, вероятно, такое
строение: J[SbCl6]-ftJCl, где п=\ или 2*. В пользу этого говорят и мно-
* Результаты опытов по переносу ионов в нитробензольных растворах
комплекса SbCl5-3JCl или J[SbCl6] 2JC1 можно объяснить тем, что
одновременно с электролизом J [SbCl6] происходит и электролиз хлористого иода.
Такое же явление имеет место и при изучении переноса ионов в системе
SbCl3—JC1, где образуются комплексы SbClg-rcJCL
306
численные факты, описанные в литературе и указывающие, что
пятихлористая сурьма сравнительно легко образует комплексный
янион [SbCU] ~~, в котором ей свойственно координационное число
шесть [79, стр. 177]. Известны также такие соединения, в которых
на одну молекулу SbCls приходится не только одна, но и
несколько молекул галогенида щелочного металла и аммония, например,
lsfH4Cl-SbCl5 и 3NH4CbSbCl5 [90], которые можно рассматривать
как двойные галогениды тиоа NH4SbCl6 • (NH4Cl)n. Эти
галогениды, вероятно, являются одним из примеров соединений, на
которых можно наблюдать различие между так называемым
аналитическим и истинным координационным числом, как это доказано,
например, для (NH4)3SiF7 или (NH4)2SiF6 • NH4F [91].
Это различие во взаимодействии пятихлористой сурьмы и РС1$
или РВг5 с хлористым или бромистым иодом объясняется,
очевидно, различием их строения.
Пятихлористый и пятибромистый фосфор, как и SbCb, в виде
отдельных молекул имеют строение тригональной бипирамиды. Но
в то время как PCls образует кристаллическую решетку,
состоящую из катионов [РС14]+ и анионов [РС1б]~, и может реагировать
с различными веществами в форме РСЦ-С1, для SbCl5 такие
свойства не обнаружены. Вследствие этого РС15, реагируя с JC1,
образует катион двойного галогенида, в котором JC1 играет, роль гало-
генангидрида (акцептора хлора). В комплексе SbCU-JCl галоген-
ангидридом является уже молекула SbCls, а хлористый иод
является донором хлора.
Эти факты показывают, какое влияние имеют структура
исходного галогенида и его поляризуемость на природу и свойства
образуемых им соединений с другими галогенидами *.
Что касается трихлоридов сурьмы и фосфора, то, хотя по
отношению к иоду они ведут себя совершенно одинаково, их
взаимодействие с хлористым иодом протекает весьма различно.
Как уже упоминалось, реакция между пятигалогенидами
фосфора и иодом обратима: РХ5 + J2 ^ РХ3 + 2JX (X = С1 или Вг).
Тригалогениды фосфора окисляются хлористым или бромистым
иодом, давая исходные пятигалогениды фосфора.
В противоположность этому реакция между SbCU и иодом
необратима. Объясняется это различной восстановительной
способностью РС1з и SbCl3. Как известно, РС13 является значительно
более сильным восстановителем, чем SbCb, что проявляется и в ряде
других реакций. Поэтому SbCl3 в отличие от РС13 не окисляется
хлористым иодом, но соединяется с ним в комплексы общей
формулы SbCl3-nJCl.
Трехбромиетая сурьма не окисляется бромистым иодом, но и не
соединяется с ним, очевидно', также (стр. 305) вследствие меньшей
* Рассматривая зависимость между положением элемента в
периодической системе и его способностью к комплексообразованию с различными адден-
дами, в данном случае с галоидными ионами, необходимо учитывать и этот
фактор.
307
прочности координационной связи Sb — Br по сравнению Со
связью Sb—С1 и одновременно вследствие значительного эфф©КТа
контрполяризации со стороны J+.
Нужно полагать, что отсутствие химического взаимодействия в
системе AsCl3—JC1, которое установил Абарбарчук по данным
измерений электропроводности и ^
криоскопических исследований
[75], объясняется этими же причи-
нами—слабой восстановительной т2\
способностью AsCl3 в отношении
хлористого иода и значительной от]
неустойчивостью комплексов
типа JIAsCU].
Строение комплексных
соединений SbCl3-"4—6JC1 следует
представить так же, как и
аналогичных соединений шятихлористой
от а
о 20. to 60
мол%т3
Рис. 96. Система хлористый
алюминий — хлористый иод *.
¥1 60
мол. % твп
Рис. 97. Система бромистый
алюминий — бромистый иод; / — 45°; // —
55°; III — 100°.
сурьмы, а именно: J[SbCl4] -nJCl, где п = 3 -или 4. Эти соединения
можно уподобить двойным галоидным солям, которые образуют три-
* Электропроводность измерялась при 35 (нижняя изотерма) и 45°.
308
галогениды сурьмы с галогенидами щелочных металлов или ам-
мония, например, KCl-SbCl3 и 3KCl-SbCl3 [90], [91, стр.404].
Систем ы А1Х3—JX (Х=С1 или Вг). При исследовании
этих систем были обнаружены комплексы такого же типа, как и в
системах с галогенидами сурьмы. Образование комплексных
соединений доказано, прежде всего, термическим анализом. На кривой
плавкости системы JC1—А1С13 (рис. 96) обнаружена дистектика с
т. пл. 110°, выраженная «небольшой площадкой, в области
33—36 мол. % А1С13, что говорит о нестойкости комплекса
А1С1з • 2JC1, образующегося в этой системе [13].
50
2Ю
Ш
290 370
f - при ростборителе ffCBo
Рис. 98. Молекулярная электропроводность системы
бромистый иод — бромистый алюминий,
рассчитанная на бромистый иод как электролит.
При исследовании системы JBr-AlBr3 обнаружена дистектика,
содержащая 51 мол. % А1Вг3 с т. пл. 85° (рис. 97), что
.подтверждает образование соединения эквивалентного состава [16].
Этому же отношению JBr : А1Вг3 соответствуют максимум на изотерме
вязкости (100°) системы JBr—А1Вг3 (54 мол. % А1Вг3) * и
максимум на кривой молекулярной электропроводности, рассчитанной
на JBr как электролит (рис. 98) [15].
Системы А1Х3—JX проводят ток. Изотермы
электропроводности имеют максимум (порядка 10"3 ом~1 см~1) в области
сравнительно небольших концентраций галогенида алюминия (10—
12 мол. % А1Х3). Следовательно, образующиеся в этих системах
комплексы хорошо проводят ток в избытке хлористого или
бромистого иода как растворителя [рис. 96, 97, 99]. По величине
электропроводности в области максимума при 45° исследованные
системы находятся в такой последовательности:
JC1— А1С13> JCl-AlBr3> JBr-AlBr3 [13, 15, 16].
Катодный осадок, образующийся при электролизе этих систем,
состоял из иода, в анодных осадках (после удаления избытка
галогенида иода) были обнаружены алюминий и хлор или бром.
Изучение переноса ионов показало, что в католите увеличивается
содержание иода, а в анолите — содержание алюминия и хлора или
* Вязкость измерялась при 100°, чтобы охватить весь интервал
концентраций.
309
брома, т. е. иод переносится к катоду, а алюминий и галоген — ^
составе комплексного аниона — к аноду.
Из этого следует, что комплексные соединения,
образующиеся в системах из галогенидов, иода и галогенидов алюминия, со-
держат в своем составе катион иода J+, анион состоит из алкь
х
0,008-
0№-
' То ' й? ' То so loo
мол. % Шг3
Рис. 99. Удельная электропроводность системы
бромистый алюминий — хлористый иод:
7—35°; //—45 о.
миния и хлора или алюминия и брома, причем, как показывает
анализ количественных данных опытов по переносу ионов в
двойных системах [14], анион, .по всей вероятности, сольватирован хло-
х
20
ю-
s 0,08 6,16 0,24 0,32 QfiO 0/S
мол. RLClt ., %
мол. ж
Рис. 100. Удельная электропроводность
системы хлористый алюминий — хлористый иод в
нитробензоле:
мол. JC1 1
7~~ мол. C6H5N02 = "б^2~;
мол. JC1 1
11 ~ мол. C6H5N02 == Щ&-
ристым иодом. Принимая во внимание результаты
физико-химического анализа систем А1Х3—JX, можно предположить, что в этих
системах образуются малоустойчивые комплексы состава
JtAlXJ-nJX, например J[AlCl4]-JCr, JAlBr4 и т. д.
Образование комплексов в нитробензольных растворах этих
систем доказано измерением электропроводности и криоскопиче-
окими исследованиями. Отклонение экспериментально найденных
310
депрессий от аддитивных значений (A/jx + A ^aix3)
увеличивается с ростом общей концентрации системы и отношения
А1Х3: JX; оно больше в системе JBr—А1Вг3, чем в системе JC1—
А1С13. Очевидно, комплекс, образующийся ов нитробензольных
растворах первой из этих систем, обладает большей
'способностью к ассоциации [3, 4]. Изотермы электропроводности
систем JX—А1Х3—СбНбГ\Ю2 отражают оба процесса: образование
малоустойчивых комплексов и их ассоциацию (рис. 100, 101).
В опытах по изучению переноса ионов в нитробензольных
растворах систем JX—А1Х3 [4, 14, 71], как и в двойных системах,
наблюдается перенос алюминия и
хлора (или брома) в анолит. Но в 3,0]
этих растворах происходило также
увеличение содержания иода в
анолите. Последнее объясняется 20-1
вторичными процессами (стр. 230).
Сложность этих процессов в
данных системах * и малая
устойчивость комплексов усложняют
количественные расчеты результатов
опытов, но все же они дают основание
предполагать, что комплексы,
образующиеся в .нитробензольных
растворах систем JX—А1Х3, имеют
такую же природу, что и <в системе
без растворителя.
Изучая перенос ионов в
двойных системах А1Вг3 (или А1С13) —
нитробензол, 3. А. Шека и Э. И.
Печеная [92, 93] показали, что в
разбавленных растворах
содержатся простые катионы алюминия,
сольватированные нитробензолом, и анионы Вг~ или С Г; в
растворах средних (,и больших) концентраций ~ до 20% галогенида
алюминия (примерно таких же, как и в исследованных нами тройных
системах) возможны более сложные катионы AlX2+-flC6H5N02 и
т. п. и комплексные анионы А1ХГ и AI2X7", наряду с простыми
ионами галогенов **. Эти данные необходимо принять во внимание,
поскольку А1Вг3 и А1С13 являются компонентами сложного
равновесия, существующего в системах JX—А1Х3—C6H5N02. Наличие в
этих системах анионных форм галогенидов алюминия может
служить одним из факторов, обусловливающих образование
комплексов, содержащих А1Х3 в составе аниона.
1,6 2*
мол. П1Вг3
мол. JBr
Рис. 101. Удельная
электропроводность системы бромистый
алюминий — бромистый иод в
нитробензоле:
мол. JBr 1
II
мол. C6H5N02
мол. JBr
мол. C6H5N02
7,26 '
1
25,03 '
* Наряду с электролизом комплексов происходит также электролиз
избыточного количества галогенидов иода, содержащихся в исследованных
системах.
** Аналогичные результаты были получены при исследовании растворов
бромистого алюминия в нитрометане и ацетонитриле [93].
311
Эти комплексные соединения хлористого и бромистого алюми-,
ния с галогенидами иода, равно как и аналогичные соединения
трех- и пятихлористой сурьмы, представляют собой двойные га~
логениды, катионом которых является J+. Эта группа соединений
электроположительного иода ранее не была описана. Ряд таких
двойных галогенидов J2[SnCl6], JrfVCl6], JfSbCl6], J[NbCb] обнару,
жил Гутман [94] при кондуктометрическом титровании растворов
соответствующих галогенидов (SnCU, VCU, SbCls, NbCl5) в
хлористом иоде растворами хлористого калия в том же растворителе
(ом. § 6).
6. РЕАКЦИИ ОБРАЗОВАНИЯ КОМПЛЕКСНЫХ ХЛОРИДОВ В ХЛОРИСТОЕ
ИОДЕ КАК РАСТВОРИТЕЛЕ
В этом параграфе будут описаны преимущественно работы
Гутмана [94], посвященные реакциям между хлоридами различных
элементов в среде хлористого иода. Эти исследования проводились
на основе теории сольвосистем кислот и оснований [95], согласно
которой кислотами являются вещества, образующие катионы,
общие с катионами растворителя, а основаниями — вещества, обра^
зующие анионы, общие с анионами растворителя [96] *. Подобные
«кислоты» и «основания» в таких растворителях, как трехфтори-
стый бром и пятифтористый иод, и реакции между ними описаны
в главах III и IV.
Рассмотрение теории сольвосистем кислот и оснований не
входит в задачу настоящей работы. Можно лишь ограничиться
указанием, что эта теория подверглась обоснованной критике [95, 97—*
100]. Если считать, что для каждого растворителя существуют свои
специфические кислоты и основания, не являющиеся таковыми
в других растворителях, а также в отсутствие растворителя, и
исключить возможность существования кислот и оснований в не-
ионизирующихся растворителях и водородных кислот в апротон-
ных растворителях, то эта теория не согласуется с известными
фактами. Схемы ионизации, вытекающие из теории сольвосистем*
в ряде случаев имеют произвольный характер и не
подтверждаются экспериментальными данными, полученными, в частности, при
помощи реакций изотопного обмена [100].
Вместе с тем, нельзя отрицать и некоторого положительного
значения теории сольвосистем, заключающегося в том, что она
способствовала изучению химических реакций в неводных
системах и помогла в ряде случаев предвидеть поведение многих
веществ и протекание различных реакций в растворителях,
диссоциирующих, хотя бы в малой степени, на ионы по аналогии с
реакциями в воде.
Все эти особенности теории сольвосистем в той или иной степени
проявились в работе Гутмана. Этот автор, исходя из того, что
хлористый иод диссоциирует на ионы по схеме (JC1)2 ^ J+ + JC17, считает
* Иначе говоря, кислоты повышают концентрацию катионов, а
основания — концентрацию анионов растворителя. ]
312
дихлороиодаты щелочных металлов М+ [JC12P „аналогами оснований",
а соединения типа J+ [RCln]~~ — „аналогами кислот". Взаимодействие
комплексов обоих типов в хлористом иоде он уподобляет реакции
нейтрализации:
M+JCI2~ + J+RC17= M [RCln] + 2JC1
vm
MJC12 + RClm = М [RCWi ] + JC1.
В качестве аналогов оснований Гутман применял растворы
хлоридов щелочных металлов (К, Rb, Cs) и аммония в хлористом
иоде, содержащие комплексы MJCb, хорошо проводящие ток.
Методом кондуктометрического титрования он изучал их
взаимодействие с растворами (в том
же растворителе) «аналогов
х-ю3
20
Х103
9\
8
7
6
/ w
12
8 \
/
/
/
/
у
0,5 1.0 15 2,0 2,5 3,0
мол. RbCL f
мол. SbCLs
Рис. 102. Кондуктометрическое
титрование раствора пятихло-
ристой сурьмы раствором
хлористого рубидия в хлористом
иоде; 0,172 мол., 55°.
W
2.0 3,0
мол. Sn СL+
W
Рис. 103. Кондуктометрическое
титрование раствора хлорного
олова раствором хлористого
аммония в хлористом иоде;
0,053 мол., 55°.
кислот» и ансольвокислот — SbCl5 (JSbCl6) и ряда других
хлоридов: TiCU, SnCl4, VC14, NbCl5 и т. п. По минимуму или перегибу на
кривых кондуктометрического титрования, соответствующему
определенному молярному отношению обоих хлоридов, Гутман судил
о составе образующегося в системе комплексного' хлорида (рис. 102,
103, 104).
Согласно этим данным, в системе KCl — SbCl5 — JCl образуется
KSbCl6, что Гутман выражает таким уравнением:
J+SbCl^ + K+JC171 KSbCl6 + 2JC1.
Образование KSbCl6 было доказано его получением после растворения
хлористого иода в СС14.
* RClm — ансольвокислота.
313
/'
Перегибы на кривых кондуктометрического титрования, приходя,
щиеся на отношение NH4C1: SnCl4=2 (рис. 103) или KCI :NbCl5=i
(рис. 104), дают основание предполагать образование Кг5пС16 и KNbQ •
2NH4JC12 + SnCl4 Z (NH4)2 SnCle + 2JC1;
KJC12 + NbCl5 ^ KNbCl6 + JC1.
Гораздо менее четкие перегибы получились при исследовании
систем с VC14(KC1:VC14 = 2) и TiCl4(CsCl: TiCl4=2). В системах с
А1С13, SiCl4 и S02C12 зависимость
х-ю3 у-—состав выражается прямой линией,
*2л причем электропроводность почти равна
электропроводности растворов КС1 той
же концентрации.
s' Гутман справедливо отмечает боль-
' шое влияние, которое оказывает
реакция сольволиза на кондуктометри-
/—**" ческое титрование и взаимодействие
^—j-Q—j5—Yq—Т5— обоих типов хлоридов. Сольволизом
'мол.'ка ' ' он объясняет тот факт, что при выде-
нолМС15 лениигексахлоростанната был получен
Рис. 104. Кондуктометрическое продукт, загрязненный иодидом и с
титрование раствора хлористо- уменьшенным содержанием олова, а
го ниобия раствором хлористо- ПрИ попытке получить гексахлоротита-
хо калия^^ «юристом иоде; нат остаток после удаления JC1
содержал мало K2TiCl6 и много
KJCb.
Эти исследования Гутмана представляют несомненный
интерес: они характеризуют электропроводность ряда систем
МС1—RClrt—JC1 и образующихся в них комплексных хлоридов;
показывают возможность получать некоторые трудно
синтезируемые комплексные хлориды, позволяя их автору высказать
правильное предположение о строении продуктов соединения некоторых
хлоридов с хлористым иодом. Так, Гутман, по-видимому, не зная о
работах ИОНХ АН УССР по изучению систем SbCl5—J2, SbCls—JC1,
опубликованных в 1949 г. [72, 12], на основании
кондуктометрического титрования и препаративных данных предложил для
комплекса JCl-SbCU строение JSbCl6 («аналог кислоты») *.
Вместе с тем, эта работа Гутмана вызывает некоторые
замечания. Отсутствие взаимодействия в системах с А1С13 и SiCl4
Гутман объясняет тем, что растворы этих хлоридов в хлористом иоде
не проявляют свойства «кислот», т. е. не образуют с ним соединений
типа J[RC1«]. Это заключение в отношении системы JC1—А1С13 не
согласуется с полученными нами экспериментальными данными,
указывающими на образование малоустойчивого комплекса JA1CU.
* Следует также отметить, что Гутман правильно представил строение
комплекса PCl5-SbCl5 в виде [PCl4]~nSbCl6]~, что позднее было подтверждено
Бурьяновым по данным переноса ионов этого комплекса в ацетонитрильном
растворе [105].
-314
Возможно, что это расхождение объясняется значительным
различием в концентрации исследованной системы А1С13—JC1. В
работе Гутмана не указана концентрация исследованного им
раствора А1С13 в хлористом иоде *, но если предположить, что для
исследования системы КС1—А1С13—JC1 применялись растворы ансоль-
вокислоты! такой же концентрации, как и для других описанных
в этой работе систем, а именно: 0,05—0,2 моль/л, то вследствие
большой плотности жидкого хлористого иода (rf45 = 3,13) Гутман
имел дело с растворами, концентрация которых не превышала
1 мол. % А1С13. Нами же и О. И. Шор эта система исследована во
всем интервале концентраций или до содержания 30—55 мол. %
А1С1з **. Здесь уместно отметить, что электропроводность систем
JC1—А1С13 [13], JC1—А1Вг3 [15] в области малых концентраций га-
логенида алюминия, до 2—3 мол. % А1Х3, 'меньше
электропроводности хлористого иода (рис. 96).
Следующее замечание к работе Гутмана касается вопроса об
«амфотерности» кохмплекса РС15 • JC1. Гутман, сопоставляя
результаты кондуктометрического титрования (в хлористом иоде как
растворителе (комплекса SbCl5 • JC1 комплексом РС15 • JC1 [94]
(рис. 105) и титрования комплекса PCls-JCl комплексом KC1-JC1
[101] (рис. 106), полагает, что в обоих случаях комплексы
реагируют друг с другом в эквивалентных отношениях по уравнениям:
РСЦ+ЛС1Г + J+SbCir^PCl4+SbCir -Ь 2JC1;
к+те + j+ РС16" :£ kpci6+2jci.
Но если в реакции с JSbCl6 комплекс PCWC1 играет роль
«основания», то во второй реакции он должен проявлять свойства
«кислоты» в хлористом иоде. Поэтому Гутман приходит к выводу,
что комплексу PCU-JCl свойственно двоякое строение: РСЦ JC1J»
установленное Фиалковым и Кузьменко, и J+ PC16~— на основании
данных титрования раствором К^СЬ.
Ошибочность этого представления о строении комплекса
PCWC1 уже отмечалась нами в другой работе [102]. Следует
также указать, что произведенная Гутманом попытка выделить
КРС1б из системы KJC12—PC15-JC1 не дала определенных
результатов [103]. На кривой кондуктометрического титрования этой
системы (рис. 106) не виден столь отчетливый перегиб, как на
кривой титрования системы с SbCl5 (рис. 105); некоторое изменение
в градиенте роста электропроводности после достижения эквимо-
лярного отношения KJC12 : РСЬ • JC1, вероятно, объясняется иными
причинами,, которые Гутман не принимает во внимание: изменение
молекулярного состояния компонентов, вязкость и т. п.
Наконец, следует также отметить, что в более поздней работе
Гутман [104] на кривых кондуктометрического титрования комплек-
* Указаны лишь молекулярные отношения КС1 : А1С13.
** Например, для изучения переноса ионов были взяты растворы 20 —
28 мол.% А1С13.
315
ca PBr5\JBr комплексами NH4JBr2 или CsJBr2 в бромистом иоде не
обнаружил образования нового соединения и поэтому принял, что
комплекс PBr5-JBr .имеет только то строение PBr4+ JBr2", которое
было установлено Кузьменко и Фиалковым [10].
дСШ3
16-
12
№ 4
6 \
/
S'
/
05
W V 2,0.
МОЛ. PCL5
мол. SbCLs
2.5 3,0
Рис. 105. Кондуктометрическое
титрование раствора пятихлористой
сурьмы раствором пятихлористого
фосфора в хлористом иоде;
0,165 мол., 50°.
х-ю*
16 \
1Л
12
to
1ft 20
моя. К J CI 1
MonMl5JCL
Рис. 106. Кондуктометрическое1
титрование раствора
пятихлористого фосфора раствором
хлористого калия в хлористом иоде;
0,053 мол., 50°.
С нашей точки зрения, явление амфотерности в реакциях ком-
плексообразования состоит в том, что одно и то же вещество может
образовывать катионные и анионные формы комплексов, будучи
в первом случае донором, а во втором акцептором определенных
ионов и т. п., как, например, [BrF2F i[JSbF6]~ и К+ [BrF4]" и др. [100].
Этот вопрос рассматривается в следующей главе.
Фаулл [107] изучил количественно равновесие диссоциации анионов
JC17, JBrjr и JBrCI" по схемам:
JXF^JX + X" (X=C1 или Вг),
JBrCf^JBr + Cr
и определил константы равновесия [ JX] [Х~~]/[ ЛХЛ и [JBr] [Cl~]/[JBrCFJ.
По его данным: KJBrF = 0,0027; КЗС1- = 0,006; KJBrC1_ = 0,022.
Еще раньше Форбис и Фаулл [108] определили константы
равновесия термической диссоциации хлористого и бромистого иода»
в водных растворах НС1 или НВг, содержащих анионы JX2".
Константы диссоциации р2] [C12]/[JC1]2 в 2-, 4- и 6-н. растворах НС1
равны 1,62-КГ10, 2,27-1 О^10 и 3,4-10~10, константы диссоциации:
[h] [Br2]/.[JBr]2 примерно в 105 раз больше: в 1-н. НВг — 1,18-Ю-5
и в 4-н. НВг—1,90-ЮЛ
Аналогичные данные получили также Форбис, Гласе и Фуоес
[109], изучая равновесие между треххлористым иодом и продукта-
316
туги его диссоциации (в 4-, 5- и 6-н. растворах соляной кислоты: они
определили константы равновесия:
/CJC1 = [J21 [C12]/[JC1]2 = 7,8. Ю-15
Kjci, = Ш [JCl3]/[ JC1]3 = 1,1 • Ю"11. И
Разделив первую из этих констант равновесия на вторую, можно
рассчитать константу диссоциации JC13 на JC1 и С12, т. е. [JCUx
X[C12]/[JC131 — 7,2-Ю~4. Отсюда следует, как отмечает Кульба [ПО],
что даже в довольно' крепких растворах соляной кислоты,
содержащих JC13 в виде ШС14, около 4,5% JC13 диссоциирует на JC1 и хлор.
Эти количественные данные характеризуют равновесия,
существующие в водных растворах двойных галогенидов хлористого
бромистого и треххлористого иода, и показывают, что увеличение
концентрации Н+ и анионов С1~ или Вгсмещает равновесие
гидролиза и увеличивает концентрацию комплексных анионов.
ЛИТЕРАТУРА
1. Н. W. Creraer,D. R. Duncan, J. Chem. Soc, 1857 (1931).
2. С. L. Jackson, J. H. D e г b y, Am. Chem. J., 24, 15 (1900).
3. Я- А. Ф и а л к о в, К. Я. К а г а н с к а я, ЖОХ, 16, 1961 (1946).
4. Я- А. Ф и а л к о в, О. И. Ш о р, ЖОХ, 19, 1197 (1949).
5. J. Cornog, H. W. Н о г г a.b i n, R. А. К а г g e s, J. Am. Chem.
Soc, 60, 429 (1938).
6. J. Cornog, E. Bauer, J. Am. Chem. Soc, 64, 2620 (1942).
7. E. В a u d г i m о n t, С г., 55, 361 (1862).
8. Я- А. Ф и а л ко в, А. А. К у з ь м е н к о, ЖОХ, 19, 812 (1949).
9. А. А. К у з ь мен ко, Я- А. Ф и а л к о в, ЖОХ, 21, 473 (1951).
10. А. А. К у з ь м е н к о, Я- А. Ф и а л к с в, ЖОХ, 19, 1007 (1949).
И. О. R u f f, J. Z e d n е г, L. H e с h t, Ber., 48, 2068 (1915); O. Ruff,
G. Fischer, Ber., 37, 4519 (1904).
12. Я- А. Ф и а л к о в, И. Л. А б а р б а р ч у к, Укр. хим. ж., 15, 176
(1949).
13. Я- А. Ф и а л к о в, О. И. Ш о р, ЖОХ, 19, 1787 (1949).
14. Я- А. Ф и а л ко в, О. И. Ш о р, ЖОХ, 23, 363 (1953).
15. Я. А. Ф и а л к о в, ЖОХ, 11, 910 (1941).
16. Я- А. Фи а л ко в, О. И. Шо р, ЖОХ, 23, 357 (1953).
17. F. E p h г a i m, Ber., 50, 1069 (1917).
18. Н. Wells, Z. anorg. Chem., 1, 83 (1892).
19. H. W e 1 1 s, H. W h e e 1 e r, Z. anorg. Chem., 1, 442 (1892).
20. W. Rae, J. Chem. Soc, 107, 1286 (1915); 113, 880 (1918).
21. V. С a g 1 i о t i, Atti Lincei, (6), 9, 563 (1929).
22. F i 1 h о 1, J. Pharm., 25, 431, 506 (1839); J. pr. Chem., 18, 457 (1839).
23. H. W e 1 1 s, H. W h e e 1 e r, Z. anorg. Chem., 2, 255 (1892).
24. F. D. Ch a t t a wa y, J. Chem. Soc, 107, 105 (1915).
25. R. F. W e i n 1 a n d, F. S с h 1 e g e I m i 1 с h, Z. anorg. Chem., 30,
I34f(1902).
26. G. de Cells, Ann. Soc. Esp. Fisica Quim., 30, 540 (1932); 33, 203
(1935); Chem. Zbl., 1932, II, 2613; 1936, I, 299.
27. Я- А. Ф и а л ко в, А. А. К у з ь м е н к о, ЖОХ, 19, 1645 (1949).
28. Н. W. С г е me r, D. R. Duncan, J. Chem. Soc, 2243 (1931).
29. H. W. Cremer, D. R. D u n с a n, J. Chem. Soc, 181 (1933).
'30. S. K. Ray, J. Indian Chem. Soc, 9, 259 (1932); 10, 213 (1933); 11,
115 (1934); 13, 456 (19360; Chem. Zbl., 1933, I, 197; 1933, II, 2804; 1934,
II, 582- 1937, I, 304.
317
31. P. S p а с u, F. P о р е a, Analele Acad. Rep. Pop. Romane, Seria Mate-
matica, Fizica, Chimie, t. Ill, mem. 4 (1950).
32. P. S p а с u, F. Pope a, ibidem, t. Ill, mem. 17 (1950).
33. H. S. Booth, С F. Swinehart, W. С. М о г г i s, J. Am. Chem.
Soc, 54, 2563 (1932); J. Phys. Chem., 36, 2784 (1932).
34. Z. Wetzien, Ann., 99, 1 (1856).
35. M. Dittmar, Ber., 18, 1612 (1885).
36. E. Ostermayer, Ber., 18, 2298 (1885).
37. L. Dobbin, J. Walker, Chem. N., 66, 111 (1892).
38. A. Pictet, G. Kraft, Bull. Soc. Chim. Fr., (3), 7, 72 (1892).
39. P. F. T г о w b г i d g e, О. С D i e h e, J. Am. Chem. Soc, 19, 55$
(1897).
40. E. A. Werner, J. Chem. Soc, 89, 1625 (1906).
41. M. Ко h n, A. Klein, Monatsh., 33, 967 (1912).
42. F. D. Ch a t t a w a y, G. Hoyle, J. Chem. Soc, 123, 654 (1923).
43. F. D. С h a t t a w a y, F. L Garton, J. Chem. Soc, 125, 183 (1924)-
44. K. G 1 e u, W. J a g e m a n n, J. pr. Chem., N. F., 145, 257 (1936).
45. E .V. Z a p p i, M. Fernandez, Ann. Assoc, quim. Argent., 27,
102 (1939); Chem. Zbl., 1940, I, 1503.
46. A. I. Po p о v, N. E. Skelly, J. Am. Chem. Soc, 76, 5309 (1954).
47. R. E. В иск les, J. F. Mi 1 Is, J.Am. Chem. Soc, 76, 3716 1954).
48. L. Dobbin, O. M a s s о n, J. Chem. Soc, 47, 67 (1885); 49, 850 (1886).
49. A. J. P о р о v, J. N. J e s s и p, J. Am. Chem. Soc, 74, 6127 (1952).
50. M. O. F о r s t e r, J. H. S с h a e p p i, J. Chem. Soc, 101, 382 (1912).
51. A. Hantzsch, Ber., 28, 2754 (1895).
52. R. W. G. Wy cko f f, J. Am. Chem. Soc, 42, 110 (1920).
53. G. L . Clark, W. D и a n e, Phys. Rev. (2), 21, 380; Chem. Zbl.,
1923, II, 657.
54. R. M. В о z о r t h, L. P а и 1 i n g, J. Am. Chem. Soc, 47, 1561 (1925).
55. R. С L. Moo ney, Z. Kristallogr., 90, 143 (1935); 98, 324 (1938); 100„
519 (1939).
56. Б. Ф. Ормонт, Структуры неорганических веществ, Гостехиздат,
М.—Л., 1950, стр. 670—672.
57. R. С. L. Moo ney, Z. Kristallogr., 98, 377 (1938).
58. Л. П а у л и н г, Природа химической связи, Госхимиздат, М.—Л.,.
1947, стр. 115.
59. А. Ф. Уэллс, Строение неорганических веществ, ИЛ, 1948, стр. 308—309.
60. Е. Fessenden, J. Chem. Educ, 28, 619 (1951).
61. A. G. Sharpe, Quart. Rev., 4, 128 (1950).
62. G. H. С h e e s m a n, J. H. M a r t i n, J. Chem. Soc, 586 (1932).
63. Gmelins Handb. d. anorg. Chem., 8, 414 (1931).
64. О. И. Ill о р, Физико-химическое исследование систем галогениды
иода— галогениды калия или алюминия, Дис, ИОНХ АН УССР, 1951.
65. Н. W. С г е mer, D. R. D u n с a n, J. Chem. Soc, 2031 (1932).
66. Е. С. S и 1 1 i v a n, Z. phys. Chem., 28, 523 (1899).
67. Т. Н. Reade, J. Chem. Soc, 853, (1929).
68. A. J. Po p о v, N. E. S k e 1 1 у, J. Am. Chem. Soc, 77, 5507 (1955).
69. J. С о r n о g, R. А. К а г g e s, J. Am. Chem. Soc, 54, 1882 (1932).
70. O. Ruff, Ber. 42, 4021 (1909).
71. Я- А. Ф и а л к о в, К- Я- К а г а н с к а я, ЖОХ, 18, 289 (1948).
72. Я. А. Ф и а л к о в, И. Л. А б а р б а р ч у к, Укр. хим. ж., 15, 37 2
(1949).
73. Я- А. Ф и а л к о в, И. Л. А б а р б а р ч у к, Укр. хим. ж., 15, 116
(1949).
74. Я- А. Ф и а л к о в, И. Л. А б а р б а р ч у к, Укр. хим. ж., 15, 194
(1949).
75. И. Л. А б а р б а р ч у к, Укр. хим. ж., 19, 365 (1953).
76. И. Д. М у з ы к а, Я- А. Ф и а л к о в, ДАН СССР, 83, 415 (1952).
77. Я- А. Ф и а л к о в, А. А. К у з ь м е н к о, И. Л. А б а р б а р ч у к,
Изв. сектора платины ИОНХ АН СССР, 26, J24 (1951).
78. N. N. Greenwood, H. J. Emeleus, J. Chem. Soc, 987 (1950).
318
79. А. А. Гринберг, Введение в химию комплексных соединений, Гос-
химиздат, Л.—М., 1951, стр. 218.
80. Я- А. Ф и а л к о в, Я- Б. Б у р ь я н о в, ДАЦ СССР, 92, 585 (1953);
ЖОХ, 25, 2391 (1955).
81. Я- Б. Бурьянов, ЖОХ, 26, 1363 (1956).
82. Я- А. Ф и а л ко в, А. А. К у з ь м е н к о, ЖОХ, 21, 433 (1951)-
22, 1290 (1952).
83. A. J. P о р о v, E. H. Schmorr, J. Am. Chem. Soc, 74, 4678 (1952).
84. W. F. Z e 1 e z n у, N. С. В a e n z i g e г, J. Ащ. Chem. Soc, 74, 6151
(1952).
85. В. А. П л о т н и к о в, О. К. К у д р а, ЖРФХО, 62, 365 (1930).
86. N. Pus in, J. Mak u с, Z. anorg. allg. Chem., 237, 177 (1938).
87. M. F. J a e g e r, H. J. D о о г n b о s с h, Z. anorg. Chem., 75, 261
(1912).
88. E. Mo les, Z. phys. Chem., 90, 78 (1915).
89. A. F. Wells, Z. Kristallogr., 99, 367 (1938).
90. R. A b e g g, Handb. d. anorg. Chem., Bd. Ill, 587—591, 615—617.
91. Б. В. Некрасов, Курс общей химии, Госхимиздат, М., 1952, стр. 812,.
ol о.
92. 3. А. Ше к а, Э. И. П е ч е н а я, Укр. хим. ж., 14, 112 (1948).
93. 3. А. Ш е к а, Сб. «Работы по химии растворов и комплексных
соединений» АН УССР, вып. 1, 1954, ИЗ.
94. V. G u t m a n n, Z. anorg. allg. Chem., 264, 151 (1951).
95. А. И. Ш а т е н ш т е й н, Теории кислот и оснований, Госхимиздат„
д\. л# 1949.
96. Н. Р/С a d у, Н. М с Е 1 s е у, J. Chem. Educ, 5, 1425 (1928).
97. А. И. Шатен штейн, Вестн. АН Каз.ССР, 1953, № 12, 32.
98. М. И. Уса но вич, Вестн. АН Каз.ССР, 1950, JSib 9, (56), 56; Изв..
АН Каз. ССР, 1951, в. 4, 97.
99. Н. Го л л, Усп. химии, 10, 719 (1941).
100. Я- А. Ф и а л к о в, Усп. химии, 23, 901 (1954).
101. V. Gutmann, Research, 3, 337 (1950).
102. Я- А. Ф и а л к о в, Дополнения к статье Н. Н. Гринвуда «Физико-
химические свойства межгалоидных соединений», Усп. химии, 22, 475
(1953).
103. V. Gutmann, Monatsch., 83, 583 (1952).
104. V. Gutmann, Monatsch., 82, 156 (1951).
105. Я- Б. Бурьянов, Комплексные соединения пятихлористого фосфора
с хлоридами некоторых элементов, Дис, ИОНХ АН УССР, 1953.
106. A. J. Popov,R. F. Swensen, J. Am. Chem. Soc, 77, 3724 (1955).
107. J. H. F a u 1 1, J. Am. Chem. Soc, 56, 522 (1934).
108. G. S. Forbes, J. H. F a u 1 1, J. Am. Chem. Soc, 55, 1809 (1933),
109. G. S. F о г b e s, S. G 1 a s s, R. F u о s s, J. Am. Chem. Soc, 47,.
2892 (1925).
110. Ф. Я- К у л ь б а, Ж. прикл. х., 23, 339 (1950).
ГЛАВА XIII
МОЛЕКУЛЯРНЫЕ И ИОННЫЕ ФОРМЫ МЕЖГАЛОГЕНИДОВ
И ИХ КОМПЛЕКСНЫХ СОЕДИНЕНИЙ
Приведенные на стр. 7 формулы известных в (настоящее время
межгалогенидов не исчерпывают всего разнообразия их
молекулярных форм. Помимо мономерных молекул, состав которых
выражен этими формулами, известны и димерные молекулы,
например (JC1)2 и т. д., неоднократно упоминавшиеся в предыдущих
главах, и другие ассоциированные формы.
В результате электролитической диссоциации межгалоидных
-соединений, способных проводить ток в жидком состоянии и в
растворах, могут возникать такие ионные формы:
а) элементарные катионы J+ — продукты диссоциации
мономерных или ассоциированных молекул хлористого и бромистого
иода. Анионами в этих системах являются ионы СЬ, Вг~ или
JCk", JBr2"" и т. п. * (см. также стр. 207);
б) в неводных растворах катионы иода, по всей вероятности,
сольватированы молекулами растворителя. Такие сольватирован-
ные «катионы иода, например [CsHbN—JJ*, [Л(С6Н5ССЖН2)2]+ и т. д.,
образуются при электролитической диссоциации комплексных
соединений хлористого и бромистого иода с органическими основа-
киями (глава XI);
в) продукты ступенчатой диссоциации мономерных форм меж-
галогенидов типа АХ3 и AXs, например, JC12+, BrF2+, JF4+;
г) продукты электролитической диссоциации ассоциированных
(димерных) молекул межгалогенидов этих типов, представляющие
собой комплексные катионы АХ2+, АХ4+ и анионы АХ4~, АХб"
и т. п.
Такого же состава ионы (катионы или анионы) образуются при
электролитической диссоциации комплексных соединений
межгалогенидов с галогенидами других элементов. Эти комплексы можно
разделить на две группы — «катионные» и «анионные». Примеры
таких комплексов приведены в табл. 47.
* Возможно, что катионы иода в расплавленных хлористом и бромистом
иоде сольватированы молекулами этих же галогенидов иода и образуют сложные
катионные группы [J(JX)n]~'~. Так, Оддо [1] предположил, что димерные
молекулы хлористого иода диссоциируют по схеме J2C12 ^t J2C1 + + С1~. Однако
доказательства существования таких ионных групп отсутствуют.
320
Еще нельзя сказать вполне утвердительно, ограничиваются ли
такие комплексные формы только теми межгалоидными
соединениями, которые приведены в табл. 47. Так, попытка получить
продукты соединения трехфтористого хлора с фторидами других
элементов была безрезультатной [3]. Однако недавно были
получены доказательства образования таких комплексов.
Таблица 47
Катионные и анионные комплексы галогенидов брома и иода с галогенидами
других элементов
Межгалогенид
Хлористый иод
Бромистый иод
Трехфтористый бром
Треххлористый иод
Пятифтористый иод
Хлористый бром
Катионный комплекс
J[A1C14]-JC1
J[SbCl,].nJCl; (n = 1,2)
JlSbCUl-nJCl; (n=3,4)
[C5H5N-J]C1
[(CCH5C0NH2)2J]C1*
J [AlBr4]
[C6H5CONH2-J]Br
[BrF2][SbF6]
lBrF2]2[SnF6]
[JC12] [SbCl6]**
[JF4][BF4]
[JF4] [SbF6]
Анионный комплекс
К [ JC12]
[PC14] [JC12]
К [CUBr]
К [JBr2]
[PBr4] [JBr2]
K[BrF4]
Ba[BrF4)2
Ag[BrF4]
K[JC!4]
H[JC14].4H20
Cs[JCl3F]
KJF6
Cs [BrCl2]
[C6H5N21 [BrClJ
Изучая реакции изотопного обмена фтора (с «помощью F18)
между фтористым водородом и фторидами галогенов, Роджерс и
Кац [4] обнаружили быстрый и полный обмен при 27° в жидкой
фазе систем HF—C1F3 (или BrF3, BrF5, JF5, SbF5), C1F3—BrF3, a
также в газообразных системах HF—C1F3 (или BrF3, BrFs, JF7),
C1F3—<BF3. В жидких системах обмен заканчивается за 10 минут,
в газообразных — за 3 минуты***. Авторы считают наиболее
вероятным ассоциативный механизм обмена, основанный на
образовании промежуточных относительно устойчивых комплексов и их
последующей диссоциации. Обмен фтора объясняется наличием
* Известны и такие соединения, как [(Cf)H5CONH2)2 J] JC12> в молекуле
которых имеются и катионная и анионная формы межгалогенида, в данном
случае хлористого иода.
** Сведения об этом комплексе ограничены очень кратким сообщением:
в сноске к статье, посвященной электропроводности твердого треххлор истого
иода [2], авторы высказали предположение, что в недавно полученном ими
комплексе JSbCl8, вероятно, содержится ион jClg"-
*** В гетерогенных системах газ — тв. тело C1F3—NaHF^,8 и C1F3—NaF18
обнаружен лишь слабый обмен, 14% и 3—20%, соответственно.
21—1419 321
ионных равновесий, в которых C1F3 * в системах с HF или с BrF3
является донором фтора, например:
HF + BrF3^BrF,++HF-;
HF + ClF3^ClF2+-fHFr;
CIF3 + BrF3^C!F2++BrF4" и т. д.**
Эти реакции изотопного обмена подтверждают существование
ионов BrF2+ и JF4+, установленных и другими методами, и
позволяют предположить образование комплексов, содержащих ионы
C1F2+, BrF4+ и JF6+, в отношении существования которых в
литературе отсутствуют другие доказательства.
Соображения, которые приводят Роджерс и Кац в пользу
принятого ими представления о механизме всех этих реакций
изотопного обмена фтора и о природе и строении промежуточных
комплексов, не лишены вероятия, хотя их и нельзя считать вполне
обоснованными ***.
Наличие хотя и слабого обмена фтора между трехфтористым
хлором и фтористым натрием дает основание предположить, что в
данной системе первый ир них играет роль акцептора фтора, т. е.
здесь он образует промежуточный комплекс анионного типа ****.
Такое же строение, по данным Пемслера и Смита [7], в отличие
от представлений Роджерса и Каца, следует приписать и
комплексу, образующемуся в системе HF—C1F3. Измерение инфракрасных
спектров поглощения смесей фтористого водорода и трехфтористо-
го хлора привело этих авторов к заключению, что в этой системе
образуется комплекс состава HF-C1F3, существующий только в
области низких концентраций; теплота реакции равна —
3,92 ккал/моль. Связь в этом комплексе осуществляется через
фтор, что позволяет предположить для него строение H[F-C1F3].
Для хлористого брома имеются сведения о существовании
комплексов только анионного типа, содержащих анион ВгС12~.
Существование такого комплекса предполагается в солянокислых
растворах хлористого-'брома [8]. К этой группе можно, пока еще
предположительно, отнести такие соединения, как RbBrCl2 и CsBrCl2 [9, 10]
и производные органических катионов, например [C6H5N2] BrCl2
* Также и другие указанные выше фториды.
** По выражению Роджерса и Каца, C1F3 здесь реагирует как основание.
*** Здесь следует отметить, что обмен фтора между фторидами галогенов
(C1F3, BrF5, JF7) и элементарным фтором, наблюдавшийся при температурах
200—300°, Кац с сотрудниками [5, 6] объясняют диссоциативным
механизмом в гомогенной фазе или гомогенно-гетерогенным процессом, в котором
участвуют фториды металлов, покрывающие стенки реакционного сосуда [6],
причем доля гомогенного процесса (основанного на обратимой диссоциации
фторида галогена) уменьшается в ряду JF7>ClF3>BrF5. В этих же
работах [6] рассмотрены вопросы кинетики реакций изотопного обмена фтора.
Энергии активации гомогенных реакций в системах F2—C1F3 и F2—JF7 равны
29 и 40 ккал-моль~~ соответственно. ДляЦгетерогенных же реакций между
фтором и C1F3, BrF5 и JF7 энергии активации равны 24, 23 и \Ъ,Ьккал-моль~х.
**** Аналогично ведет себя пятифтористая сурьма
HF + SbFs^H' +SbF£-.
ооо
[11]. Однако .некоторые авторы изображают эти комплексы как по-
лигалогениды формулой RBr-Cl2 [9, стр. 406].
Катионные комплексы межгалогенидов образуются в результате
соединения последних с азотистыми основаниями и с неполярными
или малополярными галогенидами других элементов. Анионные же
комплексы большей частью являются продуктами соединения
межгалогенидов с высокошлярными галогенидами щелочных или щелоч-
но-земельных металлов, с галогенидами серебра и т. д.
Существование катионных и анионных форм двойных галоге-
нидов, образуемых межгалоидными соединениями, отражает
способность последних проявлять двойственную
функцию—своеобразную амфотерность — в процессах образования комплексных
соединений. Это явление характерно не только для галогенидов, но
и для многих других классов неорганических соединений, если в
результате взаимодействия с другими компонентами, в
зависимости от химической природы последних, они образуют катион
комплекса или входят в состав сложного аниона.
Решающим условием образования данным веществом
комплексов катионного или анионного типа является его донорно-акцеп-
торная функция — в отношении определенных иоков или групп—
при взаимодействии с другими компонентами реакции комплексо-
образования. Так, BrF3 в реакции с SbF5 является донором фтора
и потому дает катионный комплекс [ВгРг]4" [SbF6]~, при
взаимодействии же с фтористым калием он реагирует как акцептор фтора:
KF + BrF3 = K"h[BrF4r.
Это явление амфотерности, проявляющееся в реакциях
образования комплексных соединений, подробно описано нами в
другой работе [12].
В табл. 48 систематизированы все известные межгалоидные
соединения, и соответствующие им ионные формы, установленные до
настоящего времени. Вопросительным знаком (?) обозначены те
виды ионов, в отношении которых еще нет достоверных
доказательств, а имеются лишь более или менее предположительные дан-
ные.
Рассмотрение литературных данных о молекулярных и ионных
формах межгалоидных соединений, а также и образуемых ими
типах комплексов с галогенидами других элементов гоказывает,
что в этой области химии комплексов имеется еще много
невыясненных вопросов.
В данной главе мы рассмотрим лишь те вопросы, которые
связаны с причиной отсутствия или значительной неустойчивости ряда
молекулярных форм межгалоидных соединений, а именно:
а) отсутствие ряда высших галогенидов: хлора—C1F5 и CIF7,
брома —BrF7, BrCl3—BrCl7, иода —JC15 и JC17, JBr3—ЛВг7;
б) весьма значительная неустойчивость или отсутствие низших
фторидов иода: JF и JF3;
в) незначительная устойчивость некоторых межгалогенидов
типа АХ, например хлористого и фтористого брома, и сравнительно
323
большая устойчивость ряда высших межгалогенидов BrFs, JF5
JF7*.
Эти и тому подобные вопросы представляют интерес не только
для химии галогенов и их соединений друг с другом, так как
аналогичные факты об относительной устойчивости соединений низших
Таблица 48
Сопоставление молекулярных и ионных форм межгалоидных соединений
Тип
межгалогенидов и их
ионов
А+
АХ
AXf
АХ3
АХ7
АХ+
АХ5
АХ^"
АХ +
АХ7
Фторид
хлора
C1F
ClF+(?)**
C1F3
ClF4-(^)
—
—
Фторид
брома
BrF
BrF+
BrF3
BrF4"
BrF4+(?)
BrF5
_=
Хлорид
брома
BrCl-
BrCl2
=
.
—
Фторид
иода
JF
—
JF+
JF5
HO)
JF7
Хлорид
иода
J+ 1
JC1
JC1 + (?)
JClg
—
Бромид
иода
J+
JBr
JBr^
—
и высших валентных форм известны и для элементов ряда других
подгрупп периодической системы.
Способность галогенов к соединению друг с другом с
образованием высших молекулярных форм АХ3—АХ7 нередко ставят в
связь с их относительным положением в периодической системе:
чем более галогены удалены один от другого, тем легче получаются
высшие формы межгалоидных соединений. Так, хлор в его
соединениях со фтором проявляет положительную валентность 1 и 3,
бром в его фторидах—валентность 1, 3 и 5, а иод—валентность 5 и 7.
Развивая эту мысль, Дамиен в обзоре, посвященном химии
фтора [13], писал, что при взаимодействии галогенов,
расположенных в периодической системе по соседству, проявляется
положительная валентность лишь 1 и 3. Если галогены отделены одним
периодом, то получаются соединения с валентностями 1, 3 и 5, если
* Относящиеся к этим вопросам литературные данные частично
рассматривались в главах II—VII.
** Николаев и Малюков [23] на основании результатов исследования
системы тр хфтористый хл р—фгористый водород и расчета количества осмотически
действующих частиц пришли к заключению, чтэ диссоциацию C1F3 в жидком
фтористом водороде (в разбавленных растворах) можно представить по схеме:
C1F3 ^ C1F2+ + F-.
324
же они разделены двумя периодами, то валентность может
достигать максимального значения, равного 7. По Дамиену, «на осно-
вэни1; этих наблюдений можно заключить, что увеличение
расстояния в группе между двумя элементами вызывает углубление
различия в свойствах, а следовательно, облегчает взаимное
насыщение».
Представление о том, что на способность галогенов
образовывать друг с другом те или иные, в том числе и высшие,
межгалоидные соединения оказывает влияние различие в свойствах,
обусловленное относительным положением галогенов в
периодической системе, имеет слишком общий характер и поэтому не
может объяснить многие отмеченные выше факты, например: почему
хлор или бром по отношению к фтору проявляют валентность 1+
и 3+, а иод их не проявляет. Хотя различие в свойствах галогенов
в последнем случае наиболее резкое, оно почему-то
благоприятствует образованию только высших (JF5 и JF7), но не низших форм
фторидов иода.
Говоря о различии свойств химических элементов, как об
основном факторе, определяющем состав и устойчивость образуемых
ими соединений, необходимо учитывать, что свойства данного
атома или иона могут более или менее значительно изменяться под
влиянием своих «партнеров», т. е. других компонентов молекулы
(системы). Кроме того, в межгалоидных соединениях одни и те
же галогены могут находиться в различных валентных состояниях,
что усложняет определение «различия свойств». Поэтому при:
обсуждении поставленных выше вопросов необходимо принимать
во внимание ряд количественных характеристик (свойств), и
притом не только исходных галогенов, но и образуемых ими
межгалоидных соединений.
Одной из таких характеристик является геометрический
фактор— соотношение размеров галогенов в их соединениях*. Так,
Сиджвик [141, обозначая молекулы межгалогенидов формулой
АВг7, отмечает: чем тяжелее центральный а том А, — иначе
говоря, чем больше его радиус,—тем больше он может присоединить
атомов В; в то же время чем легче галоген В, тем большее его
количество связывается с данным центральным атомом. «Однако,—
продолжает Сиджвик, — эти очевидные стерические эффекты не
являются единственным фактором стабильности межгалогенидов.
Например, из соединений типа АВ менее стабильны те, в которых,
различие атомных номеров А и В наибольшее; так, BrF гораздо
менее стабилен, чем C1F, a JF вообще не был получен».
Пространственный фактор, как причина отсутствия или
неустойчивости тех или иных соединений в ряду фторидов
галогенов, рассмотрен в одной из работ Руффа [15] Этот автор обсуждает
вопрос о том, может ли обусловливаться отсутствие неко1чэрых
высших форм фторидов галогенов невыгодным соотношением раз-
* Этот фактор, в спою очередь, зависит от валентного состояния
галогенов, электростатических и поляризационных свойств компонентов данной системы
и т. д.
325
мер'ов центрального иона и ионов фтора. Не приводя никаких
числовых данных и расчетов, Руфф утверждает, что это мнение
при проверке не подтвердилось, и поясняет это заключение тем,
что размеры пяти- и семивалентного иона хлора и семивалентного
иона брома достаточно велики, чтобы удержать соответствующие
количества ионов фтора.
Это утверждение Руффа нельзя считать обоснованным, если
принять во внимание имеющиеся в литературе, к сожалению,
далеко не полные сведения о величинах ионных радиусов галогенов в
различных степенях валентности.
Таблица 49
Кристаллические ионные радиусы галогенов в А*
[16, 17, 18J
Таблица 50
Отношение величин
ионных радиусов
Заряд
иона
1-
1 +
5+
7+
Фтор
1,33
1,36
—
—
Хлор
1,81
1,81
—
—
(0,26)**
Бром
1,96
1,95
—
—
(0,39)
Иод
2 20
2Д6
1,30
094
(0,50)
Система
Ап+/Х-
rAn+/rx_
j+/ci-
J+/Br-
0.98
0,72
0,66
J5+/F-
J5+C1~
J5+/Br-
0,71
0,52
0,43
J'+/F-
j7+/cr
J7+/Br "
Br7+/F-
Cl7+/F-
0,38
0,28
0,20
0,29
0,20
Экспериментальные данные имеются
только для одновалентно-отрицательных
ионов галогенов (по Гольдшмидту).
Данные для семивалентно-шлоЖ'Ительных
ионов рассчитаны Паулингом [16].
Величины ионных радиусов галогенов
приведены в табл. 49.
Если, пользуясь данными табл. 49,
вычислить отношения величин ионных
радиусов для различных сочетаний
галогенов и различных степеней валентности
центрального атома межгалоидных соединений, то получим
значения, приведенные в табл. 50.
Приведенные в табл. 50 величины отношений ионных радиусов
следует сравнить с так называемыми критическими отношениями
радиусов, вычисленными Лембертом для различных
координационных чисел и (Применительно к определенным пространственным
конфигурациям. Это значение га/гх для равностороннего треуголь-
* Величины ионных радиусов относятся к координационному числу,
равному 6. Для других координационных чисел необходимы поправки;
последние, однако, не столь велики, чтобы они могли более или менее значительно
отразиться на расчете отношений величин ионных радиусов.
В первой строке даны ионные радиусы, определенные Гольдшмидтом из
рентгенографических данных, в строках 2,4 и 5-ионные радиусы по данным
Паулинга-
** В скобках приведены вычисленные значения ионных радиусов.
326
ника равно 0,15, для тетраэдра—0,22, для октаэдра—0,41, для
куба—0,73, соответственно координационным числам 3; 4; 6 и 8.
Применение этих расчетов к межгалоидным соединениям имеет
условный характер в связи с тем, что они не учитывают такого
важного фактора, как поляризационное взаимодействие (которое
в молекулах межгалоидных соединений должно иметь большое
значение) и сопровождающий его эффект сжатия или отсутствия
аддитивности эффективных радиусов. Поэтому, как пишет
Некрасов, проводимые на основе эффективных радиусов модельные
расчеты максимально возможных для данных комплексообразовате-
лей и аддендов координационных чисел не всегда подтверждаются
опытом [18].
Действительно, эти расчеты для систем J+—Х~ (где X~=F~,
С1~ и Вг~) дали значения, превышающие или близкие к тем
величинам, которые соответствуют координационному числу 8 и
кубической конфигурации. Между тем, в этих системах известны, кроме
хлористого, треххлористого и бромистого иода, лишь комплексные
ионы состава JCb" и JBr2~, имеющие линейное строение в
кристаллической решетке солей типа MJX2.
Для систем J5+—Х~ более или менее удовлетворительные
результаты дает лишь расчет для пятифтористого иода, образующего
комплексные анионы JF6". Можно предположить, что этот ион
имеет октаэдрическое строение, однако критическое отношение
ионных радиусов, вычисленное для октаэдра (0,41), значительно
меньше рассчитанного значения г^/гР-9 равного 0,71.
Согласно этому же расчету ион J5+ должен координировать, по
крайней мере, шесть ионов С1~ или Вг~~, что не подтверждается
фактами.
Для системы J7+—F~ отношение ионных радиусов равно 0,38,
что соответствует (принимая во внимание возможный эффект
сжатия) октаэдрическому иону JF6~ 'существование которого
предполагается в составе 'промежуточного комплекса [JF6] [HF2] (стр. 322).
В то же ©реМ'Я эта величина почти вдвое меньше значения
0,73 для координационного числа 8; это согласуется с тем, что
не удалось получить комплексные фториды типа M^JFs].
Данные табл. 50 не подтверждают мнения Руффа о том, что
семивалентные хлор или бром * могут координировать семь ионов
фтора, если только не предположить, что в этих системах должен
проявиться сильный эффект сжатия.
По отношению к фторидам галогенов выводы, которые могут
быть сделаны из рассчитанных величингап+/гх— сравнительно
удовлетворительно согласуются с фактами существования
определенных молекулярных и ионных форм этих межгалоидных
соединений. Что же касается хлоридов и бромидов галогенов, то в этих
системах, по-видимому, большое значение имеют и другие
факторы, например, поляризуемость ионов С1~ и В г"" положительно
заряженными ионами иода, препятствующая образованию высших
молекулярных и ионных форм.
*^Как известно, для брома валентность 7+ не обнаружена.
327
Здесь необходимо также принять во внимание довольно резкое
уменьшение полярности связи при переходе от фторидов к
хлоридам и бромидам галогенов. Так, полярность связи, вычисленная из
значений электросродства по уравнению Некрасова [19]
Ев — Еа
£б + Аа
дает следующие величины: J — F — 0,30, Br — F — 0,26, 'CI - F—0,24,
J-CI—0, 71, J—Br-0,0:0 и Br—CI-0,022 (см. также табл. 4).
В такой же последовательности уменьшается способность к
образованию высших форм межгалоидных соединений и их
устойчивость.
Руфф в упомянутой выше работе высказал по данному вопросу
еще одно предположение о том, что «плотность валентных
электронов во всей молекуле не может превысить некоторого значения»; это
обстоятельство ограничивает возможность образования высших
форм тех или иных межгалоидных соединений.
Этот фактор (плотность валентных электронов) имеет
существенное значение, так как анионы галогенов, координируясь
вокруг центрального положительно заряженного иона галогена,
создают, в случае высокой валентности последнего, большую
электронную плотность, отражающуюся на устойчивости молекул и
комплексных ионов межгалоидных соединений.
Влияние этого фактора сочетается с характером связей и
структурой молекул. Основываясь на этом, Барг [20] высказал
соображения о причинах различия устойчивости некоторых фторидов
галогенов. При этом он отмечает, что строение и химические
свойства последних связаны с размерами атомов хлора, брома и иода
и их способностью притягивать электроны.
По Баргу, фтористый бром легко диспропорционируется в трех-
фтористый и молекулярный бром (3BrF=Br2 + BrF3), очевидно,
потому, что в результате такого процесса получается более низкое
электронное состояние брома в трехфтористом броме *. Ионный
характер связи увеличивается в ряду CI—F (^20%), Br—F
(~30%) и J—F (^50%), поэтому C1F и C1F3 устойчивы примерно
в равной мере, a C1F3 несколько более реакционноспособен, чем
BrF3. Что касается гипотетического трехфтористого иода, который
должен был быть аналогичен трехфтористому брому, то, если бы
это соединение даже и образовалось, оно вследствие большей
полярности связи J—F должно было бы диспропорционироваться на
иод и JF5.
Следует, однако, отметить, что соображения Барга не
отличаются достаточной четкостью и являются дискуссионными.
Отсутствие высших межгалоидных соединений, например в ряду
* Барг объясняет это следующим образом: молекулы BrF3 имеют строение
тригональнай бипирамиды с двумя непсделёнкыми парами электронов,
расположенными выше и ниже плоскости; каждая же связь Br—F приблизительно
на 1/3 ионная. Это должно привести к снижению электронной плотности вокруг,
атома брома по сравнению с нормальным октетом.
328
хлористого брома, хлоридов и бромидов иода, фторидов хлора,
можно объяснить, в частности, тем, что при увеличении
положительного заряда центрального атома галогена таких межгалоге-
нидов резко возрастает его сродство к электрону и его
поляризующее и окислительное действие на анионы галогенов *.
Однако таким образом нельзя объяснить невозможность
получения низших фторидов иода или незначительную устойчивость
монофторида брома, а также тот факт, что устойчивость моно- и
трифторидов галогенов понижается при переходе от соединений
хлора к соединениям брома и особенно иода.
Эти особенности межгалоидных соединений Руфф [15]
объяснил тем, что в молекулах BrF3 и BrF5, JF5 и JF7 центральный атом
лучше экранирован и поэтому они более устойчивы, чем BrF, JF и
гипотетический JF3.
Эффект экранирования для фторидов галогенов, несомненно,
должен иметь большее значение, чем для хлоридов и бромидов, в
связи с меньшим радиусом иона фтора. Недостаточное
экранирование центрального атома благоприятствует возможности
взаимодействия полярных молекул фторидов галогенов, которое
проявляется в реакциях диспропорционирования, сопровождающихся
переходом положительно заряженных атомов галогенов в высшую
валентную форму и элементарный галоген, например:
3BrF ^ BrF3 + Br2; EBrF3 ^ SBrF5 -f Br2 и т. д.
Можно также предположить, что монофторид иода, первично
образующийся при взаимодействии фтора с иодом, столь быстро
превращается в высший фторид, что невозможно установить
промежуточные формы—JF и JF3.
Отсутствие или незначительную устойчивость упоминавшихся
выше низших фторидов галогенов и превращение их в высшие
фториды в реакциях диспропорционирования Сиджвик [14]
обосновывал термодинамическими соображениями, принимая во внимание
энергии ординарных связей в двухатомных молекулах межгало-
генидов. Эти значения (в ккал/моль) таковы: [16, стр. 61]:
Cl-F Br-Cl J—CI J-Br
86,4 52,7 51,0 42,9
При этом Сиджвик оговаривается, что хотя энергии связей в
высших межгалогенидах (АВ3 и т. д.) в большинстве случаев
неизвестны, все же можно принять, что с возрастанием ковалентнос-
ти энергии связей уменьшаются лишь немного, если нет каких-либо
стерических препятствий образованию таких молекул. Далее
Сиджвик отмечает, что при образовании соединений высшей
валентности общее конечное количество связей увеличивается. Так, в
реакциях типа AB + nB2 = AE2/2+i количество связей растет от п+\
♦Возможности образования высших фторидов галогенов
благоприятствует устойчивость аниона фтора в отношении окислителей и высоковалентных
положительно заряженных ионов галогенов, активных в поляризационном
отношении.
329
до 2/г+1. То же имеет место и в реакциях диспропорционирова-
ния, например, в реакции ЗАВ^А2 + АВ3, количество связей уве~
личивается от 3 до 4. Если, по Сиджвику, принять энергию ордИ_
нарных связей в межгалогенидах в среднем равной 50 ккал, то,
вследствие увеличения количества связей, реакции последнего
типа должны сопровождаться выделением теплоты.
Следовательно, в ряду межгалогенидов проявляется тенденция к образованию
высших форм, если этому благоприятствуют пространственные
факторы.
Эти соображения Сиджвика, основанные на сочетании
термодинамических и пространственных факторов устойчивости
межгалоидных соединений, удовлетворительно согласуются с
экспериментальными данными. В частности, они объясняют, почему
способность к реакциям диспропорционирования и, следовательно,
способность к образованию высших молекулярных форм
усиливается в ряду: фториды хлора <фториды брома<фториды иода.
Большое значение пространственного фактора выявляется
также при сопоставлении соединений иода с хлором и бромом:
хлористый иод превращается в треххлористый, присоединяя хлор, или
в реакции диспропорционирования, происходящей при нагревании в
СС14: 3JC1^JC13+J2. В системе иод—бром не удалось обнаружить
трехбромистого иода.
Эти соображения Сиджвика о факторах устойчивости
межгалогенидов не исключают значения и других рассматривавшихся
выше факторов — полярности связи, поляризационного
взаимодействия и т. д. Кроме того, они лишены строгого количественного
обоснования. Именно в этом направлении должны развиваться в
дальнейшем представления о факторах устойчивости межгалогенидов
и их комплексных соединений. \
Здесь следует отметить, что такие же явления, которые
характерны для фторидов галогенов, широко распространены также
среди галогенидов элементов и других групп периодической системы.
Можно привести ряд примеров того, что при переходе от одного
элемента данной подгруппы к другому с большим атомным
номером способность к образованию высших форм фторидов
увеличивается, причем одновременно наблюдается уменьшение способности
образовывать низшие формы.
Так, в то время как для марганца известны ди- и трифторид
MnF2 и MnF3, рений образует только тетра- и гексафторид (ReF?
не удалось получить).
Аналогично этому для кислорода известны только 02F2 и OF2,
а для селена и теллура — тетра- и гексафториды, но отсутствуют
моно- и дифториды.
В подгруппе хрома мы знаем соединения CrF2, CrF3, CrF4 и
неустойчивый CrF5, для молибдена и вольфрама формы MoF2,
WF2, WF3 неизвестны или очень неустойчивы, в то время как MoFe
и WF6 хорошо охарактеризованы.
Ванадий образует VF3, VF4, VF5, а для ниобия и тантала
устойчивы только пентафториды,
330
Описанные здесь явления, однако, не имеют общего
характера. Так, они не характерны для фторидов элементов подгруппы
галлия. Попытки получить фториды одно- и двухвалентного
галлия были безуспешны. Не удалось получить также монофторид
дадия. Дифторид индия InF2 (или In2F4) получается с большим
трудом. Вообще же в ряду моногалогенидов индия устойчивость в
отношении реакции их превращения в 1пХ3 и металлический индий,
происходящей ори действии воды, возрастает <в ряду InF—InCl—
InBr—InJ. В то же время таллий образует устойчивые T1F и T1F3.
Таким образом, в этой подгруппе наблюдается повышение
устойчивости низшей формы фторидов при переходе от элементов
четвертого периода к элементам пятого и шестого периодов. Такое
же явление наблюдается для фторидов германия, олова и свинца.
Как уже неоднократно отмечалось выше, хлористый и треххло-
ристый иод образуются сравнительно легко, а такие же формы
фторида иода получить не удалось. Аналогичные факты —
отсутствие низших фторидов данного элемента при наличии хлоридов—
широко представлены среди галогенидов большого числа
элементов: рутения, родия, рения, селена, теллура, молибдена, ванадия,
ниобия, тантала, титана, галлия, индия и т. д. В то время как
известен ряд хлорвисмутитов, получен только один двойной
фторид висмута—NH4[BiF4].
Наконец, следует также отметить, что галогениды (особенно
часто фториды) ряда элементов, как и межгалоидные соединения,
способны подвергаться реакциям диспропорционирования *,
например:
2CuF = Cu + CuF2;
2GeF2 = Ge -f GeF4;
2VF4 = VF3 + VF5;
3InCl = 2In + InCI3 и т. д.
Можно полагать, что все эти реакции обусловлены теми же
физико-химическими факторами, которые имеют место и в ряду
межгалоидных соединений.
Это сравнение межгалоидных соединений с галогенидами
других элементов по их химическим свойствам имеет целью показать,
что многие особенности межгалоидных соединений, в частности
те, которые проявляются в значительном различии их устойчивости
в зависимости от состава, не являются характерными только для
межгалогенидов. Они проявляются также в ряду галогенидов
многих других элементов и, очевидно, отражают определенные
закономерности периодической системы.
В заключение нужно рассмотреть данные табл. 47, 48,
относящиеся к ионным формам межгалоидных соединений. Только
для хлористого и бромистого иода, трехфтористого брома и пяти-
* Для осуществления и количественного протекания этих реакций часто
необходимо воздействие повышенной температуры или растворителя.
331
фтористого иода хорошо установлены катионные и анионные фор^
мы, а для треххлористого иода—анионная форма. Как уже
отмечалось, для многих фторидов галогенов имеются только косвенные
доказательства существования их ионных форм (в виде
промежуточных комплексов), основанные на предположении об
ассоциативном механизме реакций изотопного обмена фтора.
Косвенные или пока еще недостаточные доказательства имеются в
отношении катионной формы треххлористого иода (ЛСЬ+) и анионной
формы хлористого брома (ВгСЬ").
Рассмотрение вопроса об отсутствии или значительной
неустойчивости тех или иных ионных форм межгалоидных соединений
затрудняется тем, что произведенные до настоящего времени
электрохимические и тому подобные исследования межгалогенидов и
их комплексных соединений еще далеко недостаточны. Большим
препятствием для подобных исследований является- значительная
неустойчивость таких межгалогенидов, как фтористый и особенно
хлористый бром.
Отсутствие или значительная неустойчивость тех или иных
ионных форм межгалогенидов и их комплексных соединений в
известной степени обусловлены теми же факторами, какие обсуждались
при рассмотрении молекулярных форм межгалогенидов и которые
характерны не только для последних, но и для галогенидов многих
других элементов.
Так, в некоторых случаях отсутствие или трудность
обнаружения комплексных анионов высших межгалогенидов JF8~, BrF6~,
C1F4" и т. п., вероятно, являются следствием неблагоприятного
отношения радиусов составлящих их ионов *, подобно тому, что
известно для пятибромистого фосфора.
Факты, относящиеся к ионным формам треххлористого иода
(наличие JC14~ и трудность обнаружения JC12+) и высших фторидов
брома и иода (наличие ионов BrF4~ и JF6~ и трудность
обнаружения однотипных по составу, но различных по знаку ионов BrF4+
и JF6+), отражают способность галогенидов многих
высоковалентных элементов легче присоединять анионы галогенов с
образованием комплексных анионов, чем переходить в катионные формы.
Таковы, например AuCl3, BF3, галогениды галлия, его аналогов и
элементов четвертой группы, галогениды сурьмы, ванадия, ниобия,
молибдена, вольфрама и ряда других элементов — в их высших
валентных состояниях. Иначе говоря, галогено-акцепторные
свойства всех этих галогенидов более или менее значительно
превышают их галогено-донорную функцию, что несомненно связано с
высоким зарядом центрального атома и с .преобладанием ковалент-
ного характера связи в молекулах этих галогенидов.
ЛИТЕРАТУРА
1. G. О d d о, Gazz. chim, ital., 31, II, 146 (1901).
2. С. G. V о п k, К- Н. В о s w i j k, E. H. Wiebenga, Rec, 74,
879(1955). *
3. H. J. Eraeleus, .A. G. S h a r p e, J. Chem. Soc, 2206 (1949).
* Возможно, также и повышенной электронной плотностью.
332
4. М. Т. R о ge r s, J. J. K a t z, J. Am. Chem, Soc, 74, 1375 (1952)-
5. R. B. Bernstein, J. J. К a t z, J. Phys. Chem., 56, 885 (1952).
6. R.M.Adams, R. В. В e r n s t e i n, J. J. К a t z, J. Chem. Phys.,
22, 13 (1954).
7. J. P. P e ms 1 e r, D. F. S m i t h, J. Chem. Phys., 22, 1834 (1954).
8. G. S. Forbes, R. M. Fuoss, J. Am. Chem. Soc, 49, 142 (1927).
9. H. W. С г e m e r, D. R. Duncan, J. Chem. Soc, 181 (1933).
10. Gmelin's Handb. d. anorg. Chem., 8, 415 (1931).
11/A. Hantsch, Ber., 28, 2754 (1895).
12.'?Я. А. Ф и а л к о в, Усп. химии, 23, 901 (1954).
13.'А. Д а м и е н, Усп. химии, 5, 1428 (1936).
14. N. W. S i d g w i с k, The chemical elements and their compounds, v. II,
1146.
15. O. Ruff, Angew. Chem., 46, 739 (1933).
16. Л. П а у л и н г, Природа химической связи, Госхимиздат, М.—Л., 1947,
.** стр. 337—338.
17. Г. Б. Б о к и й, Введение в кристаллохимию, МГУ, 1952, 120;
А. К- Болдырев, Атомные и ионные радиусы в кристаллах, Труды
Юбилейного Менделеевского съезда, АН СССР, М —Л., 1936, 233.
18. Б. В. Некрасов, Курс общей химии, Госхимиздат, М.—Л., изд. 9,
1952, стр. 613, 626.
19. Б. В. Некрасов, ЖОХ, 16, 983 (1946); ДАН СССР, 59, 255 (1948).
20. А. Б. Б а р г, Летучие неорганические фториды, сб. «Фтор и его
соединения», ИЛ, М., 1953, стр. 72.
21. Н. С. Николаев, И. М. М а л ю к о в, Ж- неорг. химии, 2, 1587
(1957).
ГЛАВА XIV .
О ХИМИЧЕСКОЙ ПРИРОДЕ КОМПЛЕКСНЫХ СОЕДИНЕНИЙ
МЕЖГАЛОГЕНИДОВ С ГАЛОГЕНИДАМИ ДРУГИХ
ЭЛЕМЕНТОВ
Продукты соединения галогенидов ряда элементов, а также
азотистых и других аналогичных оснований с межгалогенидами в
литературе часто относят к полигалогенидам. Такое мнение
высказывают, например, Вернер [1], Гринберг [2], ван Аркель и
де Бур [3], Хейн [4] *, Сиджвик [5], Эфраим {6], Некрасов [7].
Такие комплексные соединения часто рассматривают
совместно с соответствующими по количеству атомов галогена полигало-
генидами. Так, Вернер поместил KJC12 в группу тригалогенидов
совместно с CsBr2Cl, CsBr37 CsJ3; KJC14 — в группу пятигалогенидов
совместно с CsJs и т. д. При этом Вернер различает «чистые» по-
лигалогениды от «смешанных», содержащих атомы нескольких
галогенов.
В пользу представления об общности химической природы по-
лигалогенидов и комплексных соединений межгалогенидов с га-
логенидами других элементов (следует подчеркнуть, что речь идет
по'ка только о комплексах анионного' типа, табл. 47) можно
привести следующие данные:
1а. Эти комплексы, как и полигалогениды, содержат в
комплексном анионе нечетное количество атомов галогена.
б. Некоторые рассматриваемые здесь комплексы межгалогенидов,
например М [ JBr2], можно представить как промежуточные звенья в ряду
„чистых" и „смешанных" комплексов типа полигалогенидов,
образованных определенными галогенами. Таков, например, следующий ряд:
[Вг3]~ или [Вг~-Вг2] — Шг2Г, или [Br- J+ Br]"— [BrJ2P, или [Br~* J2]—
— [J3] , или [J J2].
Оба промежуточных комплекса («смешанного типа»)
различаются по природе центрального атома J+ — в первом и Вг" — во
втором. Этот вопрос, как мы увидим далее, более сложен, чем
кажется на первый взгляд.
2. Хлористый и бромистый иод и другие межгалогениды типа
АВ проявляют значительное сходство со свободными галогенами
(глава I). В отношении ряда физико-химических свойств межга-
* Хейн при этом делает важную оговорку, приведенную далее (см. стр. 338.
334
логениды АВ занимают промежуточное положение между
соответствующими галогенами А2 и В2.
3. Как 'ПолигалО'Гениды, так и комплексные соединения меж-
галогенидов можно получить путем непосредственного
взаимодействия галогенидов с молекулами галогенов или межгалогенидов,
например:
MJ + J2 = M[J3]; (14.1)
МВг +Ja = M[BrJ2];
MBr-f JBr = M [BrJBr]. (14.2)
4. В механизме образования обоих типов комплексов (14.1 и
14, 2) преобладающее значение имеют поляризационные явления,
что дает основание предположить и общий характер связи в них.
Поляризационные явления играют столь значительную роль в
образовании комплексов типа полигалогенидов и оказывают столь
большое влияние на их свойства, что Гринберг [2], отмечая это
обстоятельство, вполне справедливо выделил их в особую группу
комплексов.
5. Термическая устойчивость комплексов обоих типов в
значительной степени зависит от природы катиона ■— от его
поляризационных свойств, как в активном, так и в пассивном смысле.
Например, в ряду полигалогенидов щелочных металлов с одним й
тем же анионом устойчивость значительно увеличивается при
переходе от соединений лития к солям цезия. Так, в системах иодид
щелочного металла—иод (в отсутствие растворителя) образование
полииодида установлено только для йодистого цезия и, возможно,
для йодистого рубидия [8]. Йодистый калий образует полииодиды
только тогда, когда -возможна сольватация иона калия молекулами
растворителя, уменьшающая эффект контрполяризации со
стороны катиона. Йодистый натрий сравнительно мало растворим
в расплавленном иоде и лишь слабо повышает электропроводность
последнего [9].
Аналогичные явления известны и для систем межгалогенидов с
галогенидами щелочных металлов (MX—JX и т. п.) и для
образующихся в этих системах комплексов, например МУХг], где М =
=Li—Cs, a X=C1 или Вг. Комплексные соли лития и натрия
весьма неустойчивы; хлориды (или бромиды) лития и натрия плохо
растворимы в хлористом (или бромистом) иоде,
электропроводность этих систем, в отличие от систем с галогенидами калия,
рубидия, цезия и аммония, лишь незначительно превышает
электропроводность хлористого (или бромистого) иода [10].
Теперь рассмотрим экспериментальные данные, которые, в
отличие от предыдущих, характеризуют различие химической
природы полигалогенидов и комплексных соединений
межгалогенидов с галогенидами других элементов.
1. Межгалогениды типа АВ отличаются от галогенов, в
частности, тем, что их молекулы имеют хотя и небольшой
собственный дипольный момент (табл. 4), «роме того, они сравнитель-
335
но легко поляризуются. Это создает возможность образования-^
при взаимодействии таких межгалогенидов с галогенидами других
элементов — комплексов типа двойных галог^нидов или галогено-
солей (уже относящихся к так называемым ацидокомплексам).
2. Сходство физико-химических свойств межгалогенидов с
соответствующими им галогенами в значительно меньшей степени
или вовсе не распространяется на высшие межгалогениды (типа
АВ3, АВ5), которые также образуют анионные комплексные
соединения с галогенидами щелочных металлов и ряда других
элементов. Межгалогениды такого типа проявляют значительное сходство
€ галогенидами некоторых других многовалентных элементов
металлоидного или промежуточного характера, например с
галогенидами бора, кремния, мышьяка, сурьмы и др. *.
3. В большом числе случаев образование комплексных
соединений межгалогенидов при взаимодействии галогенидов с галогенами
можно только формально представить в виде реакций
непосредственного соединения (как при образовании полигалогенидов). В
действительности же, в таких случаях мы имеем дело с
значительно более сложными реакциями, протекающими сперва как
окислительно-восстановительные процессы и лишь затем—как реакции
комплексообразования (глава XII).
4. Большое значение поляризационных явлений в образовании
обоих типов комплексов и влияние этих явлений на свойства, в том
числе на термическую устойчивость, сами по себе не могут служить
доказательством в пользу общности их химической природы,
поскольку такая же связь известна также для галогеносолей.
5. Системы, образованные галогенидами различных элементов
с галогенами, например KJ—J2, и межгалогенидами, например
КС1—JC1, значительно различаются в электрохимическом
отношении. Это различие проявляется в разном характере
концентрационной зависимости удельной и молекулярной
электропроводности, что видно, например, из сравнения изотермы
электропроводности систем иодиды щелочных металлов—иод (рис. 107) [9] ** и
хлориды щелочных металлов — хлористый иод (см. рис. 75 и 79)
[Ю, 11].
Различие обоих типов систем проявляется также при их
электролизе как в отсутствие растворителя, так и в неводных
растворах (глава IX). Особенность поведения систем с межгалогенидами
и образующихся в них анионных комплексов (KJC12 и т. п.)
обусловлена наличием электроположительных ионов галогенов.
* Попытки обнаружить более сложные по составу молекулы галогенов,
например J4, и представить их в виде J(J-J2) или J+ т<Гне дали
положительных результатов.
** Очень высокая молекулярная электропроводность систем иодиды — иод,
достигающая значений 130—180 ом~1 см2 в области максимума, очевид.
но, объясняется аналогией в состоянии ионов J в среде молекул иода,
обусловленном равновесием J +J2 ^ J»~» и ионов водорода в водных
растворах кислот: Н ^ + Н2О^Н30"1".
336
6. Спектры поглощения спиртовых растворов чистых и
смешанных полигалогенидов цезия CsJ3, CsJ2Br и комплексных соединений
хлористого и бромистого иода CsJCl2, CsJBr2, CsJBrCl различны
Рис. 107. Молекулярная электропроводность
систем иодиды щелочных металлов — иод.
[12, 13]. Как видно из рис. 108, для комплексов первого типа
характерны два максимума, почти совпадающие для J3~ и J2Br~
(3600и 3580 А и 2900 А), на ш^оп.
кривых же для комплексов п
второй группы имеется лишь
по одному максимуму (3460;
3800 и 3840 А,
соответственно). Возможно, что этот факт
является результатом большей
симметрии в строении анионов
второй группы, например
C1JCL
Структура такого типа
характерна также для тетрахло-
роиодиатов; так, строение
плоских ионов JC1~^~, по данным
рентгенографического
исследования, может быть представ-
лено квадратом с трехвалент-
ным ионом иода в центре Л мах (я)
(СМ. стр. 280) И.деформирован- Рис. 108. Спектры поглощения раст«о-
НЫМИ ионами хлора на верши- poBCsJg, CsJ2Br, CsJBr2, CsJBrCl и CsJCl2
нах. Приводя эти данные, в этиловом спирте [121.
В. Некрасов 114] предполагал, что для иона JC14~ возможно
равновесие между структурами двойного галогенида и полигалогенида:
2000
22—1419
CI" +С1-Г
СГ
сг
—
—>
ГС1 СП
1 J I
С1 С1
337
Однако экспериментальные данные не подтверждают такого
равновесия *.
7. Наконец, необходимо принять во внимание, что в системах
с межгалогенидами могут образовываться не только анионные, но
и ранее неизвестные (Катионные комплексы BBriF2], [SbF6], J[AlBr4]
и т. д. Для комплексов типа полигалогенидов катионные комплексы
весьма мало характерны. Представителем их, пожалуй, можно
считать лишь гмютрииодитную кислоту J3OH или [J2*J]OH [15, 16].
Эти и некоторые другие особенности комплексных соединений
межгалогенидов с галогенидами других элементов, отличающие их
от полигалогенидов и сближающие их с ацидокомплексами типа
двойных галогенидоов, побудили некоторых авторов поставить
вопрос о том, насколько' обоснованно представление об общности
химической природы обоих типов комплексов. В этом отношении
очень характерны высказывания Вайнланда и Хейна, которые, как
указывалось выше, рассматривают комплексные соединения
межгалогенидов совместно с полигалогенидами.
Так, Хейн [4, стр. 126], говоря о тригалогенидах, образованных
хлористым и бромистым иодом, писал, что эти галогениды иода,
JC1 и JBr, которые по своему солеобразному характеру подобны
тригалогенидам сурьмы, обладают такими же комплексообразую-
щими свойствами, как AgCl и CuBr. Поэтому строение тригалоге-
нидов М1Х3 можно выразить таким образом:
M4CUCl]~MT[ClAgCl].
Еще более определенно высказался по этому вопросу Вайнланд [17J,
считая, что тетрахлороиодиаты MIJC1J являются настоящими галоге-
носолями, как М [AuQJ и М [SLC1J. Точно так же оч относит к га-
логеносолям и аналогичные комплексы хлористого и бромистого иода
MX-JX, принимая, например, для C^CbJCl, строение
К]
Cs. Что
же касается полигалогенидов, не содержащих иода, например, Q.Cl2Br,
CsBr2Cl, то, по Вайнланду, трудно решить вопрос, являются ли они
галогеносолями строения
нидами
Cs
Вг
ВК
ЧС1
-CI]
^си
Cs и
ВгС
/а
Вг
Cs!
или полигалоге-
и
Cs
СК
-Вг
\Вг.
химической приро-
галогенидами дру-
Анализ литературных данных по вопросу о
де комплексных соединений межгалогенидов с
гих элементов приводит к заключению, что представление о них
* В последующих изданиях «Курса общей химии» [7] Некрасов не
высказывает этого предположения.
** Вайнланд указывает, что доводом против возможности относить эти
комплексы к галогеносолям является отсутствие соединения «первого
порядка» между бромом и хлором. Однако современными экспериментальными
данными доказано существование, хотя и очень непрочного, хлористого брома
(см. главу V). По нашему мнению, только первый из этих комплексов
CsCl2Br можно считать производным хлористого брома.
338
как о двойных галогенидах следует считать более обоснованным.
Это представление распространяется на катионные комплексы,
которые по составу и структуре не могут быть отнесены к
какому-либо другому классу комплексных соединений, в том числе и к по-
лигалогенидам, и на анионные комплексы. Последние имеют очень
много общего с галогеносолями, образуемыми и другими
неметаллическими и многовалентными элементами. Вместе с тем
разностороннее рассмотрение этого вопроса в той его части, которая
относится к анионным комплексам межгалогенидов, не 'следует
ограничивать только противопоставлением охарактеризованных выше двух
точек зрения на их химическую природу.
Классификация химических соединений, тем более комплексных
соединений, представляет собой трудную задачу, отчасти из-за
существования веществ промежуточного типа, свойства которых
являются в той или иной степени характерными для двух или
нескольких типов соединений и которые, в зависимости от данного
свойства, приближаются то к одному, то к другому из соседних
классов.
Сказанное относится и к рассматриваемым здесь комплексным
соединениям. Такой промежуточной формой комплексов являются
анионные комплексные соединения межгалогенидов, причем в
большей степени те из них, которые относятся к тригалогенидам. Если
рассматривать их, исходя из такого важного фактора комплексо-
образования, как поляризационное взаимодействие компонентов,
образующих данный комплекс, то одновременно с полигалогени-
дами и анионными комплексными соединениями межгалогенидов
необходимо вовлечь в сферу рассмотрения также комплексные
соединения галогенов и межгалогенидов с азотистыми,
кислородсодержащими и аналогичными основаниями.
Так, комплексы типа C5H5N-Br2, CsHsN-^ * и т. п. обладают
свойствами довольно сильных электролитов. Совокупность их
свойств дает основание принять для них строение [C5H5N— Х]+Х~ со
связью N—X ковалентного типа. Образование этих соединений
является результатом электроно-донорно-акцепторного
взаимодействия компонентов комплекса и одновременно поляризации
молекул галогена полярными молекулами азотистого основания.
Аналогичными свойствами, строением и природой связи
обладают комплексные соединения этих же оснований с галогенидами
иода (глава XI). И в данных случаях решающими факторами
являются донорно-акцепторное взаимодействие и дальнейшая
поляризация молекул галогенидов иода — эти свойства обусловливают
образование комплексов типа [RN—J] ^Х— и [RN—J]fJX2~**.
* Представление о комплексах типа CbH5N—J2 как о полииодидах было
высказано Вайнландом [17].
** Физико-химические свойства многих комплексных соединений
хлористого иода с азотистыми основаниями (см. табл. 40) — бесцветность или
лишь желтоватая окраска, сравнительно высокие температуры плавления,
электропроводность и т. д. — являются результатом поляризации молекул
хлористого иода, ведущей к образованию комплексного катиона указанного
выше типа.
339
Природа связи в молекулярных соединениях брома, иода,
хлористого и бромистого иода (возможно, и других межгалогенидов
типа АВ) с органическими основаниями одна и та же. Прочность
этих молекулярных соединений, по данным Шукарева и Лилича с
сотрудниками [18, 19], растет в ряду соединений JCl>JBr>J2>Br2)
т. е. с возрастанием электроно-акцепторной способности молекул
галогенов и межгалогенидов * и электроно-донорных свойств
молекул основания.
В этом отношении комплексы типа RA-X2, RA-JX (А—донор
электронов) очень близки к комплексам типа полигалогенидов, в
которых донорами электронов и центрами поляризующего
действия являются анионы галогенидо!В, а также к анионным галогенид-
ным комплексам межгалогенидов **.
Рентгенографическое исследование нескольких простейших
полигалогенидов—трииодидов CsJ3 [20, 21], NH4J3 и (СНзЬШз [22]
+ —
показало, что анион J3~ имеет такое же линейное строение J... J—J,
— + — - + -
как и анионы С1—J—CI, C1—J—Br и т. п. (см. стр. 280). Обычно
принято, что в полигалогенидах, в данном случае в трииодидном
анионе, центральным атомом является анион исходного галогени-
да—центр координации молекул иода. Однако строение трииодного
аниона, по рентгенографическим данным, равноценность всех связей
между атомами иода в этом анионе, по данным реакции
изотопного обмена (23, 24] (косвенно свидетельствующая о значительной
поляризации молекулы иода), дают основание полагать, что
центральным атомом аниона J3~ является, в действительности,
положительно заряженный атом иода J+, как и в анионах JC12~, JBr2~,
CUBr~. Это, в свою очередь, сближает полигалогениды (трига-
логениды) с тригалогенидными комплексными анионами
межгалогенидов. Следует заметить, что такое соображение впервые было
высказано Вальденом [25], который еще в 1903 г. указал на то,
что «полииодиды и полибромиды можно формально считать
двойными солями типа MJ • MJ, где Mi=J+» ***.
Необходимо оговорить, что эти представления, характерные
для тригалогенидных анионов, нельзя в полной мере
распространить на анионы, содержащие большее число атомов галогена. В
то время как тригалогенидные анионные комплексные соединения
межгалогенидов имеют промежуточный характер между поли-
* Лилич и Прёсникова [19], уточняя это положение, отмечают, что
устойчивость этих комплексов увеличивается по мере возрастания асимметрии
распределения внешних электронов в молекулах галогенов и межгалогенидов,
которое достигается либо за счет разнородности сродства к электрону двух
атомов в молекуле межгалогенида, либо за счет различия в - поляризуемости
'Шлекул галогенов.
** С этой точки зрения комплексы типа RN2J2 являются настоящими
полигалогенидами, поскольку их строение можно представить так: [RN—J] ' х
;x[J.J2]~.
5 *** TV-е-, по Вальдену,' можно считать, что' MJ.MXJ = MtJ-M^] или
M[J J J].
340
галогенидами и двойными галогенидами, анионные комплексы
типа MJC14, MBrF4, MJF6 и т. п. по структуре и свойствам можно
считать двойными галогенидами (галогеносолями).
Таким образом, рассматриваемые здесь комплексные
соединения галогенов и межгалогенидов можно расположить в такой ряд:
продукты соединения азотистых и тому подобных оснований с
молекулами галогенов или межгалогенидов, полигалогениды,
анионные комплексы межгалогенидов, двойные галогениды.
ЛИТЕРАТУРА
I. А. В е р н е р, Новые воззрения в области неорганической химии, ОНТИ,
Л., 1936, стр. 101—103.
2. А. А. Гринберг, Введение в химию комплексных соединений, Гос-
химиздат, М.— Л., 1951, стр. 266—268.
3. Ван Аркель, де Бур, Химическая связь, ОНТИ, Л., 1935, стр. 140.
4. F. H e i п, Chemische Koordinationslehre, Leipzig, 1954, 123—128.
5. N. W. S i d g w i с k, The chemical elements and their compounds.
6. Ф. Э ф р а и м, Неорганическая химия, Госхимиздат, Л., 1937, ч. 1,
стр. 209—211.
7. Б. В. Некрасов, Курс общей химии, Госхимиздат, 1952, 823.
8. Я- А. Ф i а л к о в, Досл1дження в гaлyзi комплексних сполук галогенов
алюмшю i пол1галоген1*д1в, Вид-во АН УРСР. 1947, стор.49—50; Усп. х1*мп,
15, 509 (1946).
9. В. А. П л о т н и к о в, Я- А. Ф и а л к о в, В. П. Чалый, ЖОХ,
5, 392 (1935); Z. phys. Chem., 172, 304 (1935).
10. V. G u t m a n n, Z. anorg. allg. Chem., 264, 151 (1951).
II. Я- А. Ф и а л ко в, О. И. Шор, ЖОХ, 19, 1787 (1949).
12. F. F a i г b го t h е г, J. Chem. Soc, 847 (1936).
13. F. G i 1 b е г t, R. Goldstein, T. L о w г у, J. Chem. Soc, 1092
(1931).
14. Б. В. Некрасов, Курс общей химии, Госхимиздат, изд. 7, М., 1945,
стр. 851—852.
15. Я- А. Ф и а л ко в, ЖОХ, 18, 1741 (1948).
16. В. Л. П а в л о в, Ю. Я- Ф и а л к о в, ЖОХ, 36, 1534 (1956).
17. R. W e i n 1 a n d, Einfuhrung in die Chemie der Komplexverbindungen,
Stuttgart, 1924, стр. 355—357.
18. С. А. Ш у к а р е в, Л. С. Л и л и ч, А. Б. Шейнин, ДАН СССР,
85, 1333 (1952).
19. Л. С. Л и л и ч, О. Е. П р е с н и к о в а, Уч. зап. ЛГУ, серия хим^
наук, вып. 12, 3 (1953).
20. G. L. С 1 а г k, W. D u a n e, Phys. Rev., (2), 21, 380; Chem. Zbl.„
1923, II, 657.
21. R. M. В о z о r t h, L. Р а и 1 i n g, J. Am. Chem. Soc, 47, 1561 (1925).
22. R. C. L. Mo о n e y, Z. К г i s t, 90, 143 (1935); 98, 324 (1938); 100,
519 (1939).
23-. А. И. Б р о д с к и й, Химия изотопов, АН СССР, М., 1957, стр. 320.
24. Я- А. Ф и а л к о в, Ю. П. Н а з а р е н к о, Укр. хим. ж., 19, 356
(1953).
25. П. В а л ь д е н, Z. phys. Chem., 43, 385 (1903).
ГЛАВА XV
О МЕТОДАХ ИЗУЧЕНИЯ КОМПЛЕКСНЫХ СОЕДИНЕНИЙ
В НЕВОДНЫХ СИСТЕМАХ
Изучение процессов комплексообразования — выявление
наличия комплексных соединений в данной среде, установление их
состава, определение их свойств, строения и химической природы,
количественная характеристика их устойчивости, а также состояния
равновесия в растворах, определение термодинамических функций
и т. :п. — представляет собой одну из важнейших проблем химии
комплексных соединений.
В более узком смысле под изучением процессов
комплексообразования понимают только первые две из перечисленных задач. Им
посвящена большая литература, в которой рассматриваются общие
пути их решения, обсуждаются применяемые при этом методы
исследования, формы и условия их применения, интерпретация
результатов исследования и т. д.
Общеизвестно исключительно большое значение, которое имеет
физико-химический анализ в решении вопросов, относящихся к
установлению состава комплексных соединений и изучению
условий равновесия в жидкой фазе.
Изучение процессов комплексообразования выдвигает
необходимость учитывать большое количество факторов, в том числе и
влияние растворителя. В обширной литературе по
физико-химическому анализу еще не уделяется достаточного внимания данному
вопросу. Его нельзя свести только к исследованию методами физи-
ко-ки1мического анализа систем, одним из компонентов которых
является данный растворитель. Так, например, трехкомпонентные
системы в ряде случаев рассматривают как системы из двух
компонентов А и В, растворенных в общем растворителе. Такой подход
к трех (много) компонентным системам возможен в относительно
разбавленных растворах * или когда растворитель можно считать
индифферентным по отношению к растворенным в нем
компонентам А и В. Такие случаи раньше почти вовсе не рассматривались
в монографической литературе по физико-химическому анализу
[1, 2]. Аносов и Погодин [2] приводят данные из работы Курнакова и
* Т. е. когда данная система занимает область треугольника состава,
примыкающую к вершине, обозначающей «растворитель».
342
Квята по физико-химическому анализу системы анилин—аллиловое
горчичное масло [3], согласно которым прибавление толуола к
этой системе снижает вязкость ее, не изменяя общего характера
изотерм (т. е. действует так же, как повышение температуры), и
отмечают, что это явление имеет довольно общий характер: на
многих диаграммах третий компонент играет роль, аналогичную роли
температуры [1, стр. 530]. Разумеется, это заключение относится
только к тем системам типа А—В—растворитель, в которых
последний проявляет довольно значительную индифферентность к
исходным компонентам А и В и к продуктам их взаимодействия.
За последнее время появилось много работ по
физико-химическому анализу систем типа А—В—растворитель, изучавшихся по
различным характеристикам: оптическим свойствам,
электропроводности, вязкости, поверхностному натяжению, молекулярному
весу, депрессии температуры замерзания, удельному объему,
показателю преломления света, отклонению поляризации и
диэлектрической постоянной от аддитивных величин и ряду других. Этому
же разделу физико-химического анализа посвящена монография
Бабко [4], в которой, наряду с общими вопросами
физико-химического анализа растворов комплексных соединений, рассматриваются
преимущественно оптические методы исследования.
В связи со сказанным выше необходимо подчеркнуть некоторую
особенность физико-химического анализа систем А—В—неводный
растворитель, вследствие неограниченно большого разнообразия
физических и химических свойств неводных растворителей.
Последнее обусловливает молекулярное состояние растворенных
веществ, образование сольватов и связанную с ним стабилизацию
комплекса Am Bw S* (S—растворитель) или обратное
явление—«конкуренцию» процессов образования комплексов Am Вп, Am Sx или
Вт Sy и т. п., реакции сольволиза, значительное подчас изменение
электрохимических и других свойств системы по сравнению со
свойствами компонентов А и В, окислительно-восстановительные
реакции и т. д.
Наряду с этим при изучении реакций комплексообразования
необходимо принимать во внимание и свойства «главных»
компонентов системы и продуктов их соединения друг с другом.
Все упоминавшиеся выше и другие аналогичные обстоятельства
должны отразиться как на выборе путей (методов) исследования
процессов комплексообразования, так и на толковании
результатов исследования. Сказанное выше относится не только к трех
(много) компонентным системам, в ряде случаев эти факторы
проявляются и при физико-химическом анализе двойных систем.
Исходя из этих соображений, следует рассмотреть методы
изучения реакций образования комплексных соединений галогенов
(брома и иода) и некоторых межгалогенидов (главным образом
хлоридов и бромида иода) с органическими основаниями и гало-
генидами других элементов, принимая при этом во внимание
некоторые особенности таких комплексов (о которых шла речь в
предыдущих главах) и систем, в которых они образуются: значи-
343
тельное, а в большом числе случаев даже преобладающее влияние
поляризационных явлений на образование и устойчивость этих
комплексов, промежуточное положение межгалогенидов между
галогенами и галогенидами многих других элементов, окислительные
свойства галогенов и межгалогенидов и возможность их
окислительно-восстановительного взаимодействия с другими
компонентами систем. Сравнительная оценка .методов изучения реакций
образования таких комплексов может иметь и общее значение.
Анализ литературных данных, а также опыт работ лаборатории
химии комплексных соединений Института общей и неорганической
химии Академии наук УССР дают основание утверждать, что для
возможно более всестороннего изучения процессов комллексооб-
разования необходимо сочетать такие пути исследования:
а) физико-химический анализ систем по нескольким свойствам
и по нескольким параметрам;
б) исследование двухкомоонентной системы несколькими
методами физико-химического анализа также в растворах для полноты
представлений о.химизме системы и о возможных формах
взаимодействия ее компонентов;
в) изучение химических реакций, происходящих в исследуемой
системе, — основной и побочных;
г) физико-химическое исследование системы, в частности,
количественное изучение равновесий при стехиометрическом отношении
компонентов, установленном физико-химическим анализом,
количественное изучение существующих в ией равновесий;
д) синтез соединений, образующихся в данной системе, прежде
всего, тех, которые обнаружены физико-химическим анализом, и
установление их состава;
е) изучение физических и химических свойств синтезированных
препаратов—индивидуальных соединений и их растворов,
установление их количественных характеристик, в частности, показателей
устойчивости.
Разумеется, сказанное не исчерпывает всех возможных и
необходимых в каждом конкретном случае путей исследования * и форм
их применения; с другой стороны, не следует утверждать, что во
всех случаях необходимо полностью выполнить также эту
программу.
Чтобы обосновать необходимость сочетания перечисленных
выше путей исследования процессов комплексообразования,
рассмотрим их, в частности, применительно к системам, содержащим
галогены или галогениды ряда элементов, в том числе и межгалоидные
соединения-
1. ФИЗИКО-ХИМИЧЕСКИЙ АНАЛИЗ СИСТЕМ ПО НЕСКОЛЬКИМ
СВОЙСТВАМ
Работы в области химии комплексных соединений ранее
заключались главным образом в исследовании систем неравновесного
* Так, например, при исследовании систем из катионов металлов и
слабых или умеренно сильных кислот необходимо изучить влияние рН среды.
344
типа, в которых образуются более или менее прочные комплексы.
Поэтому в химии комплексных соединений основным методом
исследования был препаративно-аналитический, сопровождавшийся
изучением превращений комплексов, изучением их химических и
физических свойств и т. п. В дальнейшем в область химии
комплексных соединений были вовлечены—и во все большем
количестве—равновесные системы с содержащимися в них относительно
непрочными комплексами. Это обстоятельство, а также ряд других:
факторов обусловили широкое применение идей и методов физико-
химического анализа для изучения процессов комплексообразова-
ния.
Необходимость исследования систем несколькими методами
физико-химическою анализа и ino нескольким параметрам
(температура, концентрация, если исследуются системы А—В
—растворитель, рН -и т. д.) и сопоставления полученных при этом диаграмм
состав—свойство уже неоднократно отмечалось в литературе.
Химическая природа компонентов системы и характер их
взаимодействия, а также условия равновесия в системе в ряде
случаев по-разному и своеобразно отражаются на концентрационной
зависимости данного свойства, и это приводит к тому, что можно
не обнаружить соединения или обнаружить соединение не того1
состава, который находят при исследовании этой системы другими
методами физико-химического анализа. Такие факты уже
приводились в предыдущих главах, в частности, в связи с сопоставлением
диаграмм плавкости и вязкости некоторых систем. В добавление к
ним следует привести еще несколько примеров из области
рассматриваемых в этой книге объектов.
а. Термический анализ системы пиридин—хлористый иод (см.
рис. 45), как уже упоминалось, показал образование двух
комплексов C5H5NJCI и C5H5N*2JC1 [5]. То же следует из результатов крио-
скопического исследования растворов этой системы в бензоле и в
нитробензоле (см. рис. 50) [6]. Спектрофотометрическое же
исследование системы C5H5N—JC1 в хлороформе или нитробензоле, по
данным Кольтгофа с сотрудниками [7], или измерение отклонений
диэлектрической постоянной Де—состав или поляризация
Др—состав этой же системы от аддитивных величин в хлороформе и диок-
сане (см. рис. 51, 53), по данным И. А. Шека [8], обнаружили лишь
один комплекс эквимолярного состава.
б. Изотерма вязкости системы бешамид—хлористый иод (при
50°) имеет S-образный характер—рис. 55 [9]. Изотермы этого типа
обычно отражают процесс комплексообразования, но не указывают
состава комплекса. На диаграмме же плавкости этой системы (см.
рис. 55), равно как и оз нитробензольных растворах, — по данным
криоскопического исследования (см. рис. 65),—обнаружен
комплекс состава CeHsCONH^-JCl; то же подтверждается и изучением
растворимости бензамида в нитробензольном растворе хлористого
иода, а также электрохимическими исследованиями [10].
Интересно отметить, что при изучении системы диэтилбенза-
мид—хлористый иод были получены изотермы вязкости с острым,
.345.
максимумом (см. рис. 57), не сдвигающимся с температурой (35—
50°) и приходящимся на эквимолярное отношение (XI). Это
различие в поведении C6H5CONH2 и СеНбОСЩСгНбЬ нельзя объяснить
тем, что комплекс CeHsCONr^-JCl образуется только в твердой
фазе.
в. При исследовании системы SbCl3—JC1 термический анализ
показал образование соединения SbCl3-5JGl; в жидкой же фазе, по
данным измерения вязкости, электропроводности и температурного
коэффициента электропроводности, существует комплекс SbCl34JCl
(см. рис. 94) [12]. Комплекс такого же состава обнаружен в нитро-
бензольном растворе этой системы [12]. В то же время заключение
•об отсутствии комплексюобразования в аналогичной системе
SbBr3—JBr было сделано на (основании ее исследования
несколькими методами физико-химического анализа (см. рис. 95) [12].
2. К ВОПРОСУ О ВЛИЯНИИ РАСТВОРИТЕЛЯ НА |ПРОЦЕСС
КОМПЛЕКСООБРАЗОВАНИЯ
(Исследование систем типа А—В—растворитель)
Уже 'приведенные выше примеры показывают, что исследование
двухкомпонентных систем также в среде тех или иных
растворителей расширяет возможности и пути исследования процессов ком-
плексообразования. Влияние растворителя на процессы комплексо-
образования весьма разнообразно. В рамках данной главы можно
коснуться лишь некоторых моментов, связанных с этим столь
значительным для химии комплексных соединений и теорий
растворов вопросом. В связи с этим рассмотрим такие формы влияния
растворителя на процессы комплексообразования:
1) Ступенчатая диссоциация комплекса сперва на простейшие
формы, например C5H5N.2JX^ C5H5№JX+JX (X = С1 или Br),
а затем и на составляющие его компоненты, как это наблюдается
при внесении некоторых пюлигалогенидов в неполярные
растворители.
2) Сольватация комплекса или продукта его первичной
диссоциации. Этот процесс может способствовать как разложению
комплекса, так и стабилизации комплекса или одной из ступеней его
диссоциации. Так, измерение вязкости системы CH3OONH2—JC1
доказало образование в ней комплекса (CH3CONH2)2 • JC1
(см. рис. 56) [9]. В нитробензольных же растворах этой системы ацет-
амид и хлористый иод образуют комплекс с эквимолярным
соотношением компонентовCH3C0NH2-JC1 [13]. Можно полагать, что
равновесие
(CH3CONH2)2. JC1 Z CH3CONH2 • JC1 + CH3CONH2 ^
^2CH3CONH2-f JCl
в нитробензольных растворах смещается вправо вследствие сольва-
тирования молекул комплекса CH3CON1H2JCI или ацетамида
молекулами нитробензола.
.346
3) Стабилизация неустойчивых комплексных форм. Одним из
наиболее убедительных примеров 'подобных явлений могут служить
многочисленные факты из химии полигалогенидов—продуктов
присоединения молекул галогенов к анионам галогенидов.
Образование полигалогенидов в водных и различных '.неводных растворах
хорошо доказано различными методами исследования систем гало-
генид—галоген—растворитель, а также выделением этих
комплексов из растворов, чаще всего в виде сольватов.
В то же время термический анализ большого числа двойных
систем иодиды—иод, бромиды—бром и т. п. не дал доказательства
образования комплексов [14]. Измерение вязкости систем KJ—
иод и T1J—иод также дало отрицательные результаты [15]. В
системах иодиды—иод (в отсутствие растворителя) образование по-
лииодидов * обнаружено лишь в тех случаях, когда катион высо-
кополярного иодида обладает относительно малым поляризующим
действием (большой ионный радиус, низкая валентность), как,
например, в системах иод—иодиды цезия [16], замещенных
аммониевых оснований [17, 18], гексаммиаката никеля, тетраммиакатов
цинка и кадмия [19] и т. п.
Изучение реакций изотопного обмена брома и иода в системах
бромиды—бром [20—24], иодиды—иод [23, стр. 272—73; 21, 25],
особенно в жидких системах, показало довольно быстрый и полный
обмен даже в тех двойных системах, в которых методами физико-
химического анализа не обнаружено образования полибромидов
или полииодидов. Так как ассоциативный (Механизм этих реакций
изотопного обмена наиболее вероятен, то есть основания полагать,
что и в этих системах образуются очень неустойчивые комплексы,
причем в жидкой фазе происходит непрерывное образование
полигалогенидов и распад на исходные вещества—галоген и галогенид.
Сольватация катиона полигалогенидов молекулами
растворителя," увеличивая их объем, ослабляет эффект .контрполяризации в
отношении полигалогенидного аниона и тем самым более или
менее значительно усиливает устойчивость комолексов.
Интересную и своеобразную форму влияния растворителя на
процесс комплексообразования, обусловленного сольватационными
процессами, (наблюдали И. А. Шека и В. А. Войтович [26] при
криоскопическом изучении систем ZrCl4 или ШСЦ с метиловым
спиртом в нитробензоле. Эти тетрахлориды реагируют с
метанолом с образованием хлористого водорода и метоксихлоридов
МС13(ОСНз) и МС12(ОСН3)2 [27, 28]. По данным Шека и
Войтовича, максимум отклонения депрессии температуры замерзания
системы МСЦ—СН3ОН в нитробензоле приходится на 33—35 мол.%
МСЦ, т. е. соответствует образованию комплексов МС14 • 2СН3ОН,
вероятно сольватированных нитробензолом. Между тем, если
бы в этой системе -происходила реакция алкоголиза,
например, 2гС14+СН3ОН=2гС1з(|ОСН3)+НС1, то температура замер-
* Как известно, -люлииодиды являются наиболее устойчивыми полигало-
генидами. _
347
зания растворов была бы постоянной; так как количество
кинетических частиц не изменяется.
Отсутствие реакции алкоголиза в тройных системах авторы
объясняют тем, что по отношению к тетрахлоридам циркония r
гафния нитробензол не является индифферентным растворителем,,
а сольватирует их, на что указывает положительный тепловой
эффект реакции: ZrCl4+^C6H5NO2=ZrCl4-nC6H5NO2+10,5 ккал [29].
Некоторые другие комплексообразователи вытесняют
нитробензол—полностью или частично—из комплекса с Zr€l4; таким путем
был получен кристаллический комплекс ZrCl4-POCl3 ив раствора'
ZrCl4 в нитробензоле. Аналогичные явления, по-видимому, происхо-
дят и при введении метанола в нитробензол ьные растворы ZrCl4
и HfCl4; эта реакция также происходит с выделением тепла *.
4) Кислотно-основное взаимодействие растворителя с
компонентами двойной системы. Стабилизация комплекса в ряде
случаев может быть следствием кислотно-основного взаимодействия
с растворителем. Такое явление наблюдали Фиалков и Бурьянов
[30], исследуя систему НС1—PCI5 в органических растворителях:
в то время как в нитробензольном растворе этой системы /методом
измерения электропроводности не удалось обнаружить
образования комплекса, в ацетонитрильных растворах ее обнаружен
комплекс НСЬРСЬ, которому, по данным изучения переноса
ионов, можно приписать строение Н+[РС16]~. Ацетонитрил,
проявляя основные свойства по отношению к кислотам [31],
сольватирует ионы Н+, вследствие чего стабилизируется комплекс
[(CH3CN)t2H]+ [PC16K Это предположение подтвердилось при
исследовании системы (CH3)4NC1—PCI5—ацетонитрил, в которой
легко образуется комплекс f(CH3)4N]+ [PC16]~ 132].
5) Изменение молекулярного состояния компонентов системы
и продуктов их взаимодействия. Здесь имеется в виду
возможность превращения ассоциированных молекул компонентов
двойной системы в мономерные (что происходит в растворах) или,
наоборот, способность растворителей, особенно с малыми
величинами диэлектрической постоянной, благоприятствовать
образованию ассоциированных форм.
Как отмечают Измайлов и Парцхаладзе [33], наличие третьего
компонента—растворителя, особенно если он инертен к
компонентам двойной системы, позволяет расчленить суммарный эффект
взаимодействия на эффект изменения степени ассоциации каждого
из компонентов и эффект взаимодействия между ними.
Большое значение имеет также влияние растворителя на
процесс электролитической диссоциации растворенных веществ.
Эти и другие формы влияния растворителей на равновесие в
растворах двойных систем увеличивают разнообразие форм
взаимодействия компонентов системы и более или менее значительно
отражаются на концентрационной зависимости свойств системы,
г следовательно, и на виде диаграмм состав—свойство. Так, на-
* Возможно, что в этом случае некоторое значение имеет и резкое
уменьшение скорости реакции алкоголиза в нитробензольных растворах.
348
/гример, способность хлористого и бромистого иода образовывать
различные типы комплексов с органическими основаниями (см.
главу XI) или с галогенидами других элементов (см. главу XII) в
известной степени обусловлена молекулярным состоянием этих га-
логенидо'в иода и равновесиями между их молекулярными .и
ионными формами,'например, (JX)2^ J+ + JXJ~;=±2J+ + 2Х~.
6) Помимо отмеченных выше особенностей исследования
процессов комплексообразования, в системах типа
А—В—растворитель (сравнительно с исследованием соответствующих двойных
систем А—В) необходимо отметить еще следующие возможности
физико-химического анализа таких систем (по данным работ
Измайлова [33, 341):
а. Изменение состояния системы можно наблюдать также по
изменению свойств растворителя, причем для построения диаграмм
состав—-свойство могут быть использованы любые парциально-
молярные свойства как растворенных веществ, так и
растворителя *.
б. Построение диаграмм состояния на основе
термодинамических свойств растворителя позволяет рассчитать выход реакции
взаимодействия и построения диаграмм выход—состав. На основе
данных о выходе реакции и о составе равновесных растворов
можно рассчитать константы равновесия реакции взаимодействия
компонентов системы (константу нестойкости комплекса) и свободную
энергию реакций взаимодействия в растворах.
Эти работы Измайлова являются развитием идей Степанова по
метрике равновесной химической диаграммы [57],
устанавливающей количественную зависимость между геометрическими
элементами диаграммы и измеримыми свойствами системы.
Многие другие важнейшие вопросы физико-химического
анализа растворов, в частности, равновесие 'Комплексообразования в
растворах, методы определения состава и констант диссоциации
комплексов, метод изомолярных серий, оптические методы
определения состава и равновесий образования комплексов,
рассмотрены в уже упоминавшейся монографии Ба-бко [4], в ряде работ Яди-
мирокого [58], Комаря [60] и других исследователей.
Изучение процессов комплексообразования в растворах имеет
большое практическое значение, поскольку комплексные
соединения применяются в практике чаще всего в растворенном состоянии.
3. ИЗУЧЕНИЕ ХИМИЧЕСКИХ РЕАКЦИЙ, ПРОИСХОДЯЩИХ
,В ИССЛЕДУЕМОЙ СИСТЕМЕ
Физико-химический анализ должен сопровождаться
одновременным химическим (или физико-химическим) исследованием
системы, задачей которого является выяснение природы
взаимодействия ее компонентов. Такое исследование безусловно
необходимо, когда нет определенных данных о том, как реагируют друг
* См. также [35].
349
с другом компоненты системы: протекает ли в ней реакция
аддитивного комплексообразования, двойного обмена, окислительно-
восстановительная и т. д. или происходит несколько типов реакций,
в конечном итоге приводящих к образованию комплекса. Кроме
того, одновременно с основной реакцией в системе возможны и
побочные процессы, протекающие гораздо медленнее и потому часто
не обнаруживаемые в условиях физико-химического анализа.
Отсутствие же точных сведений о природе химического
взаимодействия компонентов системы и о происходящих в ней побочных
процессах может привести к значительным ошибкам в понимании
результатов физико-химического анализа и выводах, которые
могут быть сделаны из диаграмм состав—свойство. Для пояснения
приведем следующие факты.
а. На диаграмме плавкости системы пятибромистый фосфор—
иод (см. рис. 84) [36] обнаружен дистектический максимум,
приходящийся на 75 мол.% РВГб *, что давало основание сделать
вывод об образовании комплекса 3PB15J2. Однако подробное
химическое исследование систем PCls—иод и РВг5—иод показало, что
пятигалогениды фосфора окисляют иод, причем получаются JC1
или JBr, которые затем соединяются с избытком РХ5 в комплекс
РХ5-JX (см. стр.297). Суммарная реакцияЗРХ5+^ = 2(РХ5\1Х)+РХ3
характеризуется стехиометрическими коэффициентами (3:1),
соответствующими квазидистектическому максимуму на диаграмме
плавкости. Химизм реакции образования комплексов PXs'JX (Х=С1
или Вг) подтвержден также исследованием систем РХ3—JX и
РХ5—JX [37, 38].
б. Такой же характер имеет взаимодействие компонентов
системы SbCb—иод. Пологий максимум на диаграмме плавкости в
области 66 мол.% SbCU и прекращение вторых температурных
остановок в этой же области (см. рис. 91) подтверждают наличие
нового соединения в системе, для образования которого SbCU и Лг
реагируют в соотношении 2SbCl5 : J2 [39]. Однако, как показали
Руфф с сотрудниками [40], это соотношение характеризует
суммарную реакцию, состоящую из двух
процессов—окислительно-восстановительного и образования комплекса.
в. Удовенко и Фиалков [41] на изотермах вязкости
систем SiCl4—метилаль или ацеталь обнаружили максимум (при
20, 30 и 40°) в области 33 мол.% четыреххлористого кремния. Но
они же установили, что в исследованных смесях происходят
реакции, в которых 1 моль SiCl4 реагирует с 2 молями органического
компонента:
SiCl4 -Ь 2СН2 (ОСН3)2 = Si (ОСН3)2 С12 + 2СН3ОСН2С1;
SiCl4 + 2СН3СН (ОС2Н5)2 = Si (ОС2Н5)2 С2 + 2СН3СНС1 (ОС2Н5).
Таким образом, изотермы вязкости этих систем отражают
реакцию образования диметокси- и диэтоксидихлорида кремния.
* Аналогичные данные получены также для системы пятихлористый
фосфор —- иод (см. рис. 83) [37].
350
Следующие примеры иллюстрируют влияние побочных
реакций.
г. Как известно, горчичные масла образуют с первичными и
вторичными аминами устойчивые соединения—производные
тиомочевины, например CeH5NH2 + C3H6NCS = SC(NHC3H5) (NHC6H5).
Эти системы были исследованы Н. С. Курнаковым с сотрудниками,
Н. А. Трифоновым, Н. К. Воскресенской и другими авторами
различными методами физико-химического анализа: плавкости,
вязкости, рефракции, поверхностного натяжения, удельного веса,
теплоты смешения, электропроводности [1, 2].
На диаграммах состав—свойство этих систем обычно имеется
сингулярный максимум, приходящийся на эквимолярное отношение
амина и горчичного масла. Лишь на изотермах х—состав этому
отношению компонентов системы соответствует минимум
электропроводности. Однако электропроводность подобных систем,
достигающую значений порядка 10~3 ом~1 смг\ нельзя приписать
производным тиомочевины указанного выше типа, не обладающим
свойствами электролитов. Мискиджьян [42] показал, что в системах
из аминов и горчичных масел, наряду с образованием производных
тиомочевины, например аллилфенилтиомочевины, проходит
побочная реакция с образованием роданистоводородных солей аминов
+ _
[C6H5NH2 - СдН5] SCN и т. п., которые являются электролитами
и служат источником ионов SCN~. Выдерживание смесей и
особенно нагревание благоприятствуют образованию комплексов типа
аммонийных солей и повышению концентрации ионов родана *.
Особый интерес в этом отношении представляет система
пиридин—аллиловое горчичное масло. На основании результатов
измерения вязкости и удельного веса этой системы при 25° Курнаков
и Жемчужный [43] установили отсутствие взаимодействия.
Воскресенская же [44], нашла, что эта система обладает значительной
электропроводностью, возрастающей со временем. Мискиджьян
подверг физико-химическому анализу смеси C5H5N и C3H5NCS
после длительного (9 месяцев) хранения и нагревания и
обнаружил на изотермах вязкости и удельного веса острый максимум при
соотношении 1:1, указывающий на глубокое взаимодействие.
Этот вывод был подтвержден электрохимическими исследованиями
и выделением роданистоводородного аллил-пиридина [C5H5N—
CsHslSCN.
д. Кривая оптической плотности неводных растворов ксанто-
гената кобальта, извлекаемого органическими растворителями из
водных растворов системы C0SO4—KC2H^OCS2 (изомолярные
серии), имеет максимум при соотношении 1 : 2. Из этого был сделан
вывод, что максимуму на кривой оптической плотности отвечает
соединение Со^НяОСЗгЬ [45]. Между тем,
препаративно-аналитические данные [46—49], в том числе и анализ продукта,
извлекаемого неводными растворителями [50], и результаты физико-
* Так, по Мискиджьяну, максимум на изотерме исправленной
"электропроводности (х-т,) системы анилин — аллиловое горчичное масло совпадает
с максимумом концентрации ионов SCN".
351
химического исследования'[50, 51] показали, что устойчивой фор.
мой ксантогената кобальта является препарат состава
Co(C2H5OCS2)3. По данным Шека и Крисе [50], первично
образующийся неустойчивый ксантогенат двухвалентного кобальта
затем окисляется до Co(C2H5OCS2)3. Одновременно в системе
происходит ряд побочных процессов (образование CoS, продуктов
разложения и окисления 'ксантогенатов кобальта и калия и т. д. [52]'*
сочетание которых с основными процессами образования
Co(C2H50CS2)3 обусловливает отношение исходных компонентов
(соли кобальта и ксантогената калия) и соответствующий ему
максимум на диаграмме состав—свойство **.
4. ФИЗИКО-ХИМИЧЕСКОЕ ИССЛЕДОВАНИЕ СИСТЕМ
Приведенные выше экспериментальные данные убедительно
подтверждают, что химическое исследование системы существенно
дополняет физико-химический анализ ее по различным свойствам.
Такое сочетание путей исследования важно также в тех случаях,
когда возможно образование неустойчивых соединений, которые
не удается обнаружить каким-либо методом физико-химического
анализа. В качестве примера можно привести систему хлористый
иод—уксусная кислота, которая по данным Корнога и Олсона [53],
является .простой бинарной системой с одной эвтектикой (см. рис. 39).
В то же время цвет растворов хлористого иода в уксусной
кислоте и, особенно, результаты электрохимических исследований
дают убедительные доказательства образования в этих растворах
соединений—сольватов хлористого иода.
Физико-химическое исследование в ряде случаев дает
представление о молекулярном состоянии комплекса, образующегося
в системе. Так, криоскопическое и электрохимическое
исследования системы бензамид—хлористый иод в нитробензоле привели к
заключению о том, что в этих растворах имеется равновесие
ассоциированных (димерных) и мономерных молекул комплекса
CeHsCONHa-JCl.
5. О СОЧЕТАНИИ ФИЗИКО-ХИМИЧЕСКОГО АНАЛИЗА С СИНТЕЗОМ
КОМПЛЕКСНЫХ СОЕДИНЕНИИ ПРИ ИЗУЧЕНИИ ПРОЦЕССОВ
КОМПЛЕКСООБРАЗОВАНИЯ
Вопрос о сравнительной оценке физико-химического метода
анализа (изучения равновесий) и препаративно-аналитического
метода неоднократно был предметом дискуссии ***. Однако
постановку этого вопроса в такой общей форме, предусматривающей
противопоставление этих двух путей исследования, нельзя считать
обоснованной в свете современных представлений о строении ве-
* Большое влияние на эти процессы оказывает кислотность среды.
** Недавно Коренман с сотрудниками [59J, измерив оптическую плотность
хлороформных экстрактов ксантогената кобальта, образующегося при
взаимодействии водных растворов нитрата кобальта и этил- или бензилксантогената
калия (изомолярные серии), установили, что независимо от рН раствора состав
ксантогенатов кобальта выражается формулой (ROCS^Co. Однако и в этой
работе не принята во внимание возможность побочных процессов.
*** В последнее время этот вопрос был рассмотрен в монографии Бабко [4].
352
ществ в различных агрегатных состояниях и в растворах, о
различных формах комплексообразования, о влиянии растворителя
на этот процесс, о неустойчивых комплексных формах,
возникающих в жидкой фазе и существующих более или менее
кратковременно, о промежуточных комплексах и т. п.
Выбор между этими путями исследования, преимущественное
значение одного из них в каждом конкретном случае или их
взаимное дополнение зависят от задач, которые ставит перед
собой исследователь, от природы компонентов исследуемой системы
и образующихся в них соединений.
Сказанное согласуется с тем заключением, которое сделано
Бабко [4, стр. 17] из рассмотрения данного вопроса: «метод
изучения равновесий и препаративный метод не исключают друг
друга... и не могут заменить один другого» и «в зависимости от
характера изучаемых явлений необходимо выбирать тот или другой
метод как основной метод исследования». Но нельзя согласиться с
там, что «в большинстве случаев (практически бесполезно пытаться
дополнить один метод другим; это может быть целесообразно
только в отдельных конкретных случаях».
Необходимость сочетания физико-химического анализа систем
с синтезом соединений, образующихся в жидкой фазе (и в
растворах), и с их последующим исследованием обосновывается
следующими соображениями:
а. Имеется очень много фактов, говорящих о глубоком
различии структуры веществ в газообразном, жидком и
кристаллическом состоянии и в растворах, о различии структуры вещества в
виде отдельных молекул и в кристаллических образованиях.
Поэтому для возможно более всестороннего и полного познания вещества
его необходимо изучать во всех возможных для него агрегатных
состояниях и в растворах. Для иллюстрации можно привести такой
простейший пример: допустим, что при изучении системы треххло-
рйстый фосфор—хлор в неполярных растворителях мы
ограничились только констатацией факта, что эти компоненты реагируют
друг с другом с образованием соединения простейшего 'состава—ш-
тихлористого фосфора РС1;г и установлением состояния равновесия
в растворе, но не синтезировали продукт реакции — пятихлористый
фосфор—и не исследовали его в твердом и расплавленном
состоянии, не исследовали его растворов в полярных растворителях.
Тогда мы не знали бы многих замечательных особенностей пятихлори-
стого фосфора. Сказанное можно распространить на очень
широкий круг веществ.
б. Как уже отмечалось, изучение процессов
комплексообразования не ограничивается выявлением наличия и установлением
состава комплекса в жидкой фазе и в растворе или только
нахождением стехиометрических отношений исходных компонентов
системы, соответствующих экстремальным точкам на диаграммах
состав—свойство. Но даже такая суженная постановка вопроса
требует в ряде случаев применения как препаративных, так и
физико-химических исследований. Таковы, например, случаи, когда
найденное методами физико-химического анализа среднее отноше-
23—1419
353
ние компонентов комплексных групп (дробная валентность при
окислительно-восстановительных реакциях и т. д.), обусловленное
условиями равновесия, не дает точного представления об
истинном составе продуктов взаимодействия в системе. Выделение этих
продуктов в индивидуальном состоянии и их исследование могут
дать ценные вспомогательные сведения для познания природы
данного раствора.
Современные методы исследования строения молекул в
кристаллах—определение межатомных расстояний, валентных углов,
координационного числа и т. п. — дают новые возможности
характеризовать прочность комплексов, прочность отдельных связей и
предвидеть или объяснить особенности их химического поведения.
В качестве примера можно привести работу Бокия и Кукиной [54]
по рентгеноструктур-ному исследованию солей К^СЦС^Н^НгО
и KIPtBraCsfH^HzO. При этом было обнаружено, что две связи
Pt—С1 и Pt—Вг на координатах CI—Pt—С1 и Br—Pt—Br
являются нормальными и равны 2,26 кХ и 2,42 кХ соответственно.
Третья же длина связи Pt—С1 и Pt—Вг, когда С1 и Вг находятся
в транс-положении к этилену, имеет аномальную величину 2,40 и
2,50 кХ. Последнее объясняется большим транс-влиянием
молекулы этилена, лабилизующим эти связи.
в. Известно, что соединения, выделяющиеся из раствора, не
всегда тождественны по своему составу с комплексными формами,
существующими в жидкой фазе, и что поэтому нельзя
ограничиться выделением комплексов и анализом твердой фазы для
характеристики состава и состояния комплексов в растворах.
В'месте с тем, знание состава твердой фазы, знание состава
продуктов, выделенных из раствора (вообще—из жидкой фазы),
физических и химических свойств раствора (депрессия
температуры замерзания, электропроводность, диэлектрическая
проницаемость, отношение к химическим реагентам и т. д.) дает
возможность предвидеть, предположить природу, а иногда и простейший
состав комплексных групп в растворе.
С другой стороны, оба эти пути
исследования—физико-химический анализ и метод синтеза—имеют свои ограничения областей
применения. Так, в ряде случаев эти методы исследования не могут
дать исчерпывающей характеристики состояния веществ в
растворе и процесса комплексообразования в нем, если речь идет,
например, о степени сольватации ионов в растворах, о составе очень
неустойчивых комплексных форм, о промежуточных формах, о
механизме процесса комплексообразования и т. п. Выше уже
приводились такие примеры (системы галогенид—галоген и др.)- В
таких случаях приходится прибегать также к другим путям
исследования, в частности к .методу меченых атомов и к углубленному
исследованию систем разнообразными физическими и химическими
методами.
■г. Физико-химический анализ растворов незаменим, когда
целью исследования является выявление некоторых важнейших
характеристик процессов комплексообразования (состояние
равновесия и др.), протекающего в данном растворителе, определение
354
показателей устойчивости комплекса в нем, а при наличии
ступенчатого комплексообразования — определение соотношения между
концентрацией отдельных форм в исследуемом растворе.
Для решения этих вопросов большую помощь может оказать
одновременное исследование растворов индивидуальных
комплексных соединений, образующихся в данной системе, причем такие
растворы можно получать путем растворения заранее
синтезированного комплекса или синтезируя его непосредственно в данном
растворителе. Особенностью таких растворов является отсутствие
в ряде случаев посторонних веществ—продуктов реакции
взаимодействия компонентов системы.
д. Сочетание физико-химического анализа растворов с
препаративным методом имеет также большое 'практическое значение.
Синтез комплексов и выделение их из растворов часто лежат в
основе разделения элементов, удаления примесей и получения
веществ в чистом состоянии; «синтез» сплавов, сложных силикатов,
солевых систем типа двойных солей и твердых растворов,
осуществляемый на основе предварительного физико-химического
анализа, и их исследование, с одной стороны, расширяет и углубляет
данные физико-химического анализа, а с другой стороны, дает
практике весьма 'ценные материалы. Многие комплексные
соединения необходимо выделять из растворов, чтобы иметь возможность
применять в различных областях техники, в медицине и т. д.
6. ИЗУЧЕНИЕ ФИЗИЧЕСКИХ И ХИМИЧЕСКИХ СВОЙСТВ
СИНТЕЗИРОВАННЫХ КОМПЛЕКСОВ В ИНДИВИДУАЛЬНОМ
СОСТОЯНИИ И В РАСТВОРАХ
Это исследование важно не только для изучения химической
природы, строения и прочности комплексов, но и для
подтверждения и расширения результатов исследования систем, в которых
образуются эти комплексы. Вообще нельзя строго разграничивать
исследование системы методами физико-химического анализа от
физико-химического исследования комплекса в расплавленном
состоянии или в растворе. Любой метод физико-химического анализа
характеризует то или иное физическое свойство продукта,
образующегося в системе: температуру плавления; электрохимические
свойства—при измерении электропроводности как свойства; мол.
вес—при криоскопическом методе; полярность связи—при
измерении поляризации или диэлектрической проницаемости и т. д. Но
значительно более полные сведения о физических и химических
свойствах комплекса, необходимые для того, чтобы определить
его строение, природу составляющих его ионов, химическую
природу и т. п., можно получить в результате целеустремленного
исследования индивидуального комплекса в растворах (или также в
системах, в которых он образуется) или в расплавленном виде.
Благодаря такому направлению исследований в работах ИОНХ
АН УССР удалось определить не только простейший состав
многих комплексных соединений хлористого и бромистого иода,
например, с галогенидами других элементов, но и установить их ори-
355
надлежность к двум классам двойных галогенидов—катионных
Я+ [JX2]- и анионных J+ [MXnJ- (Х = С1 или Вг), т. е. установить
строение их молекул. Аналогично, при исследовании систем из пя-
тихлористого фофсора с хлоридами некоторых других элементов
их растворов в нитробензоле, ацетонитриле и других растворителях
и комплексных соединений, образующихся в этих системах, было
доказано, что эти комплексы представляют собой галогеносоли
типа [РСЦГ 1МШС14]Г[РС14]2Ь [MIVC16]2"[PC14]+ [MVC16]~ обладающие
характерными свойствами довольно сильных электролитов—солей
четырехгалогензамещенного фосфониевого катиона [55, 56].
В отличие от этих систем, в ацетонитрильных растворах систем
НС1—РС15 [30] и (CH3)4NC1—PC15 [32] с эквимолярным
соотношением обоих галогенидов было установлено образование
комплексных галогенидов типа R4PC16]_, т. е. гексахлорфосфатов. Если бы
исследования ограничились только установлением стехиометриче-
ских коэффициентов, нельзя было бы сделать вполне определенные
заключения о строении этих комплексов, об их принадлежности к
катионному и анионному типам двойных галогенидов пятихлори-
стого фосфора.
Методы исследования комплексных соединений следует
выбирать таким образом, чтобы они отражали наиболее характерные
свойства данной группы комплексов. Так, в связи с тем, что
упомянутые выше группы комплексов являются хорошими
электролитами, при их исследовании необходимо было широко применять
электрохимические методы: измерение электропроводности,
электролиз, изучение переноса ионов, кроме того, производились крио-
скопические измерения в двух растворителях—в одном из них
(например, в нитробензоле) данный комплекс проявляет свойства
электролита, а с другим (например, в бензоле) образует не
проводящие ток растворы.
Все описанные выше пути исследования процесса комплексо-
образования вытекают из изложенного в начале этой главы
представления о том, какие задачи охватывает вся эта проблема.
Сочетание всех этих путей исследования (или некоторых из них,
например, физико-химического анализа с препаративным методом)
и их последовательность зависят от основной задачи, которую
ставит перед собой исследователь, приступая к изучению процесса
комплексообразования в данной системе, а также от физических и
химических свойств отдельных компонентов и системы в целом.
Само собой разумеется, что в конкретных случаях большее
значение могут иметь те или иные из рассмотренных выше путей
исследования, которые нельзя считать равноценными, независимо от
упомянутых выше условий.
ЛИТЕРАТУРА
1. Н. С. К у р н а к о в, Введение в физико-химический анализ, Изд-во
АН СССР, 1940.
2. В. Я- Аносов, С. А. Погодин, Основные начала
физико-химического анализа, Изд-во АН СССР, 1947.
3. Н. С. К у р н а к о в, И. К в я т, ЖРФХО, 46, 1372 (1914).
356
4. А. К- Б а б к о, Физико-химический анализ комплексных соединений
в растворах, Изд-во АН УССР, 1955.
5. Я- А. Ф и а л ко в, И. Д. М у з ьг к а, ЖОХ, 18, 1205 (1948).
6. Я- А. Ф и а л к о в, И. Д. Музыка, Изв. сектора физ.-хим. анализа,
19, 314 (1949).
7. J. М. К о 1 t h о f f, D. S t о с е s о с а, Т. S. Lee, J. Am. Chem.
Soc, 75, 1834 (1953).
8. И. А. Ш е к а, Ж-Ф.Х., 30, 109 (1956).
'9. Я. А. Ф и *а л к о в, И. Д. Музыка, ЖОХ, 20, 385 (1950).
10. Я. А. Ф и а л к о в, И. Д. Музыка, ЖОХ, 19, 1416 (1949).
11. Я- А. Ф и а л ко в, И. Д. Музыка, ЖОХ, 21, 823 (1951).
12. Я- А. Ф и а л к о в, И. Л. А б а р б а р ч у к, Укр. хим. ж., 15, 116,
194(1949).
13. Я- А. Ф и а л к о в, И. Д. Музыка, ЖОХ, 18, 802 (1948).
14. Я- А. Ф i а л ко в, Усп. xiMii, 15, 485, 508 (1946); Досл1*дження в галуз1
комплексних сполук галоген1Д1в алюмшш i пол1галоген!д1в, Вид-во АН УРСР,
1947, стор. 49—50.
15. Я- А. Ф и а л к о в, А. А. К у з ь м е н к о, ЖОХ, 6, 265 (1936); Зап.
1н-ту xiM., 4, 309 (1937).
16. Т. R. Briggs, J. Phys. Chem., 34, 2260 (1930).
17. F. О 1 i v a r i, Atti Acad. Lincei, 17, 717 (1908).
18. Я. А. Ф и а л ко в, Н. В. А к с е л ь р у д, ЖОХ, 13, 753 (1943).
19. Я- А. Ф и а л к о в, Ф. Д. Ш е в ч е н к о, ЖОХ, 20, 1358 (1950); 22,
1101 (1952); 23, 547 (1953).
20. С. Р о г и н с к и й, Н. Г о х ш т е й н, Phys. Z. Sovjetunion, 7, 672
(1939).
21. Н. Е. Брежнева, С. 3. Р о г и н с к и й, Усп. химии, 7, 1503 (1938);
ЖФХ., 14, 49 (1940).
22. Н. Е. Брежнева, С. 3. Р о г и н с к и й, А. И. Ш и л и н с к и й,
ЖФХ., 8, 849 (1936); 9, 296, 367, 752 (1937).
23. «Использование радиоактивности при химических исследованиях», под
ред. А. Воль и Н. Боннер, ИЛ, М., 1954, стр. 263—266.
24. Я- А. Ф и а л к о в, Ю. П. Н а з а р е н к о, Изв. АН СССР, ОХН,
590 (1950); ДАН СССР, 73, 727 (1950).
25. Я- А. Ф и а л к о в, Ю. П. Н а з а р е н к о, Укр. хим. ж., 19, 356
(1953).
26. И. А. Ш е к а, Б. А. Войтович, Ж- неорг. х., 2, 426 (1957)
27. R. S. H a n s е п, К. G u n n а г, A. Jacobs, С. R. Simmons,
J. Am. Chem. Soc, 72, 5043 (1950).
28. D. С. В г a d 1 e у, F. M. H a 1 i m, W. W а г d 1 a w, J. Chem. Soc,
3450 (1950).
29. W. S. Hummers, S. Y. T у г с e, S. Y о 1 1 e s, J. Am. Chem. Soc,
74, 139 (1952).
30. Я- А. Ф и а л к о в, Я- Б. Бурьянов, ЖОХ, 26, 1003 (1956).
31. М. И. У с а н о в и ч, В. Д у л о в а, ЖОХ, 16, 1978 (1946); 17, 669
(1947).
32. Я- А. Ф и а л к о в, А. А. Кузьменко, Н. А. Костромина,
Укр. хим. ж., 31, 556 (1955).
33. Н. А. Измайлов, К- П. П а р ц х а л а д з е, Укр. хим. ж., 22,
156 (1956).
34. Н. А. Измайлов, ЖФХ, 25, 1070 (1950); 27, 807 (1953).
35. Я- А. Ф и а л к о в, В. Б. Ч е р н о г о р е н к о, Укр. хим. ж., 24,
13 (1957).
36. А. А. К у з ь ме н ко, Я- А. Ф и а л к о в, ЖОХ, 19, 1007(1949).
37. Я- А. Ф и а л к о в, А. А. К у з ь м е н к о, ЖОХ, 19, 812, 1645 (1949).
38. А. А. К у з ь м е н к о, Я- А. Ф и а л к о в , ЖОХ, 21, 473 (1951).
39. Я- А. Ф и а л к о в, И. Л. А б а р б а р ч у к, Укр. хим. ж., 15, 372
(1949).
40. О. R u f f, J. Z e d n е г, L. H e с h t, Ber., 48, 2068 (1915).
41. В. В. У д о в е н к о, Ю. Я- Ф и а л к о в, ЖОХ, 28, 814 (1958).
357
42. С. П. М и с к и д ж ь я н, ЖОХ, 23, 1947 (1953); 26, 1046 (1956); 27, 175S
(1957); Ж. физ. хим., 29, 855 (1955).
43. Н. С. К у р н а к о в, С. Ф. Жемчужный, Изв. Петерб. политехи
инст., 18, 1, 115 (1912).
44. Н. К. Воскресенская, Уч. зап. Саратов, ун-та, 6, в. 3, 227
(1927).
45. А. Т. П и л и п е н к о, Н. В. У л ь к о, Ж- анал. х., 10, 299 (1955).
46. A. Rosenheim, A. L. Davidson, Z. anorg. Chem., 41, 23J
(1904).
47. J. V. D u b s k у, J. pr. Chem., 90, 61 (1914).
48. M. D e 1 e p i n, L. С о m p i n, Bui. Soc. Chim., [4], 27, 469 (1920)
49. L. M a 1 a t e s t a, Gazz., 70, 541 (1940).
50. 3. А. Ш е к а, Е. Е. Крисе, Ж- неорг. х., 1, 586 (1956).
51. L. Cambi, L. S z e g 6 e, Ber., 64, 2591 (1931).
52. С. Н. Данилов, Ж- прикл. х., 19, 1059 (1946).
53. J. С о г n о g, L. E. О 1 s о n, J. Am. Chem. Soc, 62, 3328 (1940).
54. Г. Б. Б о к и й, Г. А. К у к и н а, VII Всесоюзное совещание по химии
комплексных соединений, Тезисы докладов, Л., 1956, стр. 24—27.
55. Я- А. Ф и а л к о в, Я- Б. Б у р ь я н о в, ДАН СССР, 92, 585 (1953)-
ЖОХ, 25, 2391 (1955).
56. Я- Б. Бурьянов, ЖОХ, 26, 1363 (1956).
57. Н. И. Степанов, Изв. АН СССР, сер. хим., в. 2, 219, (1936); в. 2,
293 (1938).
58. К- Б. Яцимирский, Усп. хим. 22, 410 (1953); ЖОХ, 24, 1498
(1954); Ж- неорг. хим., 1, 412, 2306 (1956).
59. И. М. К о р е н м а н, Ф. Р. Ш е я н о в а, С. А. Р я б о в а, Ж- неорг.
хим., 2, 65 (1957).
60. Н. П. Комар ь, Ж. неорг. х. 1, 1243 (1956); Труды хим. фак. Харьк. ун-та,
16, 87, 95(1957); 18, 117(1957) и предыдущие сообщения.
ГЛАВА XVI
ПРИМЕНЕНИЕ СОЛЯНОКИСЛЫХ РАСТВОРОВ ХЛОРИСТОГО
И ТРЕХХЛОРИСТОГО ИОДА В КОЛИЧЕСТВЕННОМ
АНАЛИЗЕ
(иодхлорометрический метод объемного анализа) *
В предыдущих главах неоднократно отмечалась способность
межгалоидных соединений, в том числе и соединений
электроположительного иода, галоидировать различные вещества, проявлять
свойства сильных окислителей, образовывать комплексы с гало-
генидами других элементов и с органическими веществами. Эти
особенности межгалоидных соединений можно использовать не
только для препаративных работ, но и для целей количественного
объемного анализа.
Наиболее пригодными в этом отношении межгалогенидами
являются хлористый и треххлор'И'Стый иод. Как было
показано в главе VI, для галоидирования органических соединений
(преимущественно для получения иодпроизводных) ранее
применялись растворы хлористого иода, главным образом в неводных
растворителях—четыреххлористом углероде, уксусной кислоте и
некоторых других.
Для аналитической практики такие растворы имеют ряд
неудобств, и их применение в этом направлении может быть лишь
более или менее ограниченным, например, для определения йодного
числа жиров (см. главу VI).
Значительно целесообразнее оказалось применение
солянокислых растворов хлористого и треххлористого иода, в которых эти
галогениды иода содержатся в виде комплексных кислот HJC12
и HJC14. Так, например, Михаэль и Нортон, действуя хлористым
иодом на солянокислый раствор анилина, получили трииоданилин
[1]. Хейсиг [2] предложил солянокислый раствор хлористого иода
как окислитель для количественного определения хлористого
железа **, а Кубина [3]—для определения мышьяковистого водорода.
Применение солянокислого раствора хлористого иода для
объемно-аналитических целей (в качестве титрованного раствора)
* Эта глава написана совместно с А. И. Генгриновичем и Ф. Е. Каган по
материалам статьи, опубликованной в «Трудах аналитической комиссии»,
т. V, 1954, дополненной новыми экспериментальными данными.
** По Хейсигу, иод, выделяющийся при взаимодействии хлористого иода
с хлористым железом, титруют раствором KJ03 в присутствии хлороформа.
359
б литературе не было описано. Что касается использования трех-
хлористого иода для указанных выше целей, то в литературе
(глава VI) имеется лишь весьма мало сведений о применении
солянокислых растворов треххлористого иода для окисления фосфористой
кислоты [4], для растворения сульфидов [5], коллоидной 'серы и
золота [6]. В качестве иодирующего средства треххлор истый иод не
был исследован.
С солянокислыми растворами хлористого и треххлористого иода
были выполнены те работы, основные результаты которых будут
кратко описаны в данной главе.
Работы по применению солянокислых растворов хлористого
иода для объемно-аналитических определений были начаты Генгри-
новичем еще в 1940 г. и затем продолжались им же и его
сотрудниками, а также Шах, Каган (которой принадлежат работы по
изучению и применению, главным образом, солянокислых
растворов треххлористого иода), Pan апортом и др. Эти исследования
дал,и возможность разработать так называемый иодхлорометриче-
ский метод *, который, как будет показано далее, имеет довольно
разнообразные формы своего приложения в аналитической
практике для объемно-аналитического определения как неорганических,
так и (что следует особо отметить) большого числа органических
соединений.
Задачей настоящей главы является описание основных
положений иодхлорометрического метода и возможных форм его
применения. Так как объемно-аналитические определения с помощью
солянокислых растворов JC1 и JC13 являются, по сути,
разновидностью иодометрического метода объемного анализа, то сперва
необходимо кратко охарактеризовать некоторые недостатки
классического метода иодометрии, основанного на применении
титрованного водного, раствора иода:
а. Летучесть иода, особенно в теплое время года.
б. Для приготовления водных растворов иода приходится
затрачивать большие количества йодистого калия, в 2,5—3 раза
превышающие количество взятого для растворения иода.
в. В случае применения водных растворов иода для
замещения атомов водорода в органических соединениях по схеме
RH+J2^ RJ + HJ только половинное количество иода входит в
состав иодпроизводного, вторая же часть израсходованного иода
образует побочный продукт. Таким образом, при прямом
титровании таких веществ раствором иода рационально используется лишь
14—16% общего количества иода в растворе; при титровании же
по остатку коэффициент полезного использования иода еще
меньше.
г. Йодистый водород, образующийся в процессе реакций
окисления и замещения, часто обусловливает обратимость процесса,
например:
С6Н5ОН + 3J2 ^ С6Н2J3OH + ЗН J.
* Это название предложено Генгриновичем, работы которого послужили
основой для данного метода.
360
Чтобы устранить обратимость таких реакций, приходится
добавлять щелочно-реагирующие вещества для нейтрализации HJ,
что усложняет технику определения; кроме того, в щелочной среде
некоторые органические вещества (например, фенолы) окисляются
иодом, иногда с образованием окрашенных продуктов.
д. Продукты иодирования органических соединений (иодпроиз-
водные) часто адсорбируют иод, что вызывает необходимость
затрачивать довольно значительные количества органического
растворителя (спирта) для растворения адсорбированного иода, как
например, при количественном определении антипирина по методу
Государственной фармакопеи (65 мл спирта на одно определение).
е. Йодистый калий, содержащийся в водном растворе, часто
замедляет и смещает в сторону исходных продуктов состояние
равновесия обратимых реакций иодирования либо растворяет
образующиеся при таких реакциях продукты.
Все эти факты явились причиной того, что водный раствор иода
нашел лишь весьма ограниченное применение для объемного
определения органических соединений.
Иодхлорометрический метод, основанный на (применении
титрованных солянокислых растворов хлористого и треххлористого иода,
как мы увидим далее, лишен этих недостатков.
1. ПРИГОТОВЛЕНИЕ ТИТРОВАННЫХ СОЛЯНОКИСЛЫХ РАСТВОРОВ
ХЛОРИСТОГО И ТРЕХХЛОРИСТОГО ИОДА
Водные растворы хлористого и треххлористого иода весьма
неустойчивы ©следствие процессов гидролиза (см. стр. 144 и 163).
В солянокислой среде при достаточной концентрации НС1 эти
галогениды иода не гидролизуются вследствие образования
комплексных кислот ШСЬ и ШСЦ.
Солянокислые растворы хлористого и треххлористого иода
можно приготовить, растворяя последние в соляной кислоте или
пропуская хлор в суспензию иода в воде. Однако значительно
проще получать эти растворы при взаимодействии KJ03 с KJ (или
с иодом) в солянокислой среде, что выражается следующими
уравнениями:
KJ03 + 2KJ + 6HCI = 3JC1 4- ЗКС1 + ЗН20; (16.1)
KJ03 + 2J2 + 6HC1 = 5JC1 + KC1 + 3H20; (16.2)
2KJ03 + К J + 12НС1 = 3JC13 + ЗКС1 4- 6Н20; (16.3)
3KJ03 + J2 + 18HC1 - 5JCI3 + 3KC1 + 9H20. (16.4)
По данным Ланга [7], раствор хлористого иода в 4-н. соляной
кислоте, приготовленный «по уравнению (16.1), неограниченно
стоек. Генгринович, изучив устойчивость хлористого иода в
растворах соляной 'кислоты различной концентрации, (показал, что 0,1-н.
раствор хлористого иода в 0,4-н. соляной кислоте, приготовленный
по Лангу, устойчив в течение не менее 6—7 месяцев и может быть
использован в качестве титрованного раствора [8] *.
* При недостаточной концентрации соляной кислоты реакция (16,1)
протекает с образованием иода. Это следует, в частности, из опытов Бриттона
с сотрудниками [9]: при потенциометрическом титровании йодистым калием
раствора KJ03 в соляной кислоте <1-н. (а также в 0,7—6,0-н. H2S04) резкий
361
К такому же заключению пришла и Каган [10], изучив
устойчивость 0,1-н. раствора треххлористого иода в 0,4-н. соляной кисло- ~
те, приготовленного по уравнениям (16.3) или (16.4).
Для приготовления таких растворов хлористого и треххлори-
стого иода KJ и KJ03 следует брать в стехиометрических
количествах. Для примера 'приводим метод приготовления ~ 0,1-н.
раствора хлористого иода в 0,4-н. соляной кислоте (1 л).
Отвешивают 5,53 г KJ и 3,55 г KJ03 в склянку с притертой пробкой,
приливают 40—50 мл воды и 40 мл соляной кислоты (уд. вес 1,19) и
взбалтывают до полного растворения образующегося при реакциях
иода, после чего прибавляют 10 мл хлороформа; если последний
окрашивается в фиолетовый ,'цвет *, то приливают по каплям 1 %-ный
раствор KJ03 при сильном взбалтывании до обесцвечивания
хлороформного слоя. После отстаивания водный слой сливают и
добавляют воды до 1 л. Образуется раствор лимонно-желтого цвета.
Чтобы установить титр (коэффициент нормальности) этого
раствора, к 10 мл раствора прибавляют 1 г KJ и иод,
выделившийся по реакции
KJ+HJ'C12=KC1+J2 + HC1 **, (16.5)
титруют раствором Na2S203. К оттитрованному раствору
прибавляют одну-две капли метилового оранжевого и титруют соляную
кислоту раствором едкого натра.
Аналогично можно получить раствор JQ3. Для приготовления
1 л 0,1-н. раствора берут 3,57 г KJ03, 1,38 г KJ и 41 мл соляной
кислоты (уд. вес 1,19). JC13 реагирует с йодистым калием по
уравнению
3KJ + JC13 = 2J2 + 3KC1. (16.6)
Равновесия в солянокислых растворах
хлористого и треххлор истого иода. Комплексные
кислоты, образующиеся в этих растворах, находятся в равновесии с
продуктами диссоциации, что может быть представлено следующими
схемами:
а) HJCl2^H+-f JCV;
ЮГ t JCI + СГ Z J+ + 2СГ;
б) HJCl4^Hf + ЮГ;
Ю7 Z JCIs+СГ ^ ЛС1 + С18 + СГ.
Повышение концентрации соляной кислоты сдвигает эти
равновесия в сторону образования комплексных ионов и значительно
перегиб на кривой титрования приходится на соотношение 5KJ * KJ03 (т. е.
5KJ+KJOg+6HCl=6KCl+3J2-j-3H20). В 1—2-н. соляной кислоте получены два
перегиба: первый — при соотношении 2KJ : KJ03 —соответствует
образованию JC1, а второй соответствует окончанию реакции между JC1 и KJ после
прибавления еще 3 молей KJ-
* Если хлороформ остается бесцветным (вследствие возможного избытка
KJ03), то добавляют каплями 1%-ный раствор KJ до появления фиолетового
окрашивания хлороформного слоя, которое затем обесцвечивают, приливая
одну-две капли раствора KJ03-
** Или J-+JC12- = J2+2C1.
362
ослабляет или полностью устраняет процесс гидролиза, но
одновременно уменьшается и концентрация электроположительных
ионов иода. Этот факт обусловливает влияние концентрации соляной
кислоты на поведение хлористого и треххлористого иода при
химических реакциях.
2. ФОРМЫ ПРИМЕНЕНИЯ ХЛОРИСТОГО И ТРЕХХЛОРИСТОГО ИОДА
В ОБЪЕМНОМ АНАЛИЗЕ
Применение хлористого и треххлористого иода в объемном
анализе основано на следующих химических реакциях:
а) окисление многих неорганических и органических
восстановителей; б) галоидирование органических соединений путем
замещения атомов водорода фенолов, аминов и тому подобных веществ
на галогены; в) присоединение по месту двойной связи с
образованием галоидпроизводных; г) присоединение JC1, JC13 (или
соответствующих им комплексных кислот) к органическим азотистым
и аналогичным основаниям, причем образуются комплексные
соединения определенного состава.
Далее будут приведены примеры применения хлористого и
треххлористого иода для объемно-аналитических определений,
основанных на перечисленных выше химических реакциях.
Титрованные солянокислые растворы хлористого и
треххлористого иода могут быть использованы как для прямого титрования,
так и для титрования по способу остатков или замещения.
Индикатором при прямом титровании является система J2—J"—
крахмал. Так, например, при титровании водного раствора какого
либо иодида (KJ и т. п.) в присутствии хлороформа
солянокислым раствором JC1 [Ц] или JC13 (см. уравнения (16.5) и (16.6)
после прибавления первой капли реактива водный раствор тотчас
синеет, и эта окраска, благодаря наличию J~-hohob, сохраняется до
конца титрования. По достижении точки эквивалентности синее
окрашивание водного слоя исчезает.
В тех случаях, когда при взаимодействии хлористого иода с
восстановителем, например, с аскорбиновой кислотой, образуется
HJ,
0=C-C=C-C-CH0H-CH20H+JC1 = HJ+HC1+
+ 0=С—С-С-С-СН ОН-СН2ОН, (16,7)
II II I
ООН
хлористый иод ino достижении точки эквивалентности реагирует с
йодистым водородом (с ионами J~), причем выделяется иод
HJ+JC1 = HC1+J2 и раствор приобретает синюю окраску.
При титровании органических соединений, атомы водорода ко-
363
торых замещаются на иод по схеме RH+JC1 ~^ RJ + HC1, в
качестве индикатора прибавляют несколько капель разведенного
раствора KJ. Ионы J" в кислой среде восстанавливают
незначительное количество иодзамещенного RJ + HJ^; RH+J2, в результате
чего появляется синяя окраска крахмала. К концу титрования JCI
реагирует с Л~-ионами, превращая их в иод, и окраска раствора
исчезает. Подобное явление наблюдается при титровании
антипирина C11H12ON2 хлористым иодом [121:
НС=С - СН3 J—С = С-СНз
I I +JC1= . | | +НС1;
°=CxN/N-CH3 0=C\N/N-CH3 (16.8)
I I
с6н5 с6н5
C}1HuJON2+HJ ^ СПН12(Ж2+Л2.
Титрование по способу остатков заключается в
том, что к исследуемому раствору добавляют определенный объем
солянокислого раствора JC1 или JCU, избыток последних разлагают
раствором KJ. Выделившийся при этом иод титруют раствором
Na2S203. Такой -метод титрования применяют, например, при
определении фенолов, сульфаниламидных препаратов, ряда
восстановителей. В последнем случае образующуюся в процессе
титрования окисленную форму приходится иногда связывать в комплекс,
во избежание ее взаимодействия с йодистым калием. Так,
например, при определении солей закиси железа после взаимодействия
таковых с избытком JC1 или JCls
2Fe- + 2JC1 = 2Fe- + 2СГ+ Ja; (16.9)
6Fe- + 2JCIs=6Fe- + 6СГ * J2 (16.10)
добавляют раствор сегнетовой соли для перевода солей окиси
железа в комплексное соединение [13].
Титрование по способу замещения. К
исследуемому раствору добавляют избыток солянокислого раствора JC1 и
далее титруют выделившийся при этом иод, для чего его
предварительно извлекают хлороформом*.
Чтобы избежать операции извлечения иода, избыток JC1 можно
связать салициловой кислотой (добавляя салицилат натрия). Как
показал Генгринович [14], при действии солянокислого раствора
JC1 на салициловую кислоту последняя количественно переходит в
дииодсалициловую кислоту
С6Н4 (ОН) СООН + 2JC1 = C6H2J2 (ОН) СООН -л- 2HCI. (16.11)
Образующийся осадок дииодсалициловой кислоты не мешает
титрованию иода раствором Na2S203.
Титрование по способу замещения с помощью солянокислых
растворов треххлористого иода не применяется, так как образую-
* Из солянокислой среды хлороформ извлекает лищь ничтожные
количества HJClo.
364
щийся при этом иод восстанавливает избыток JC13 до хлористого
иода:
JC13 + J2 = 3JC1. (16.12)
Исходя из этих форм применения солянокислых растворов
хлористого и треххлористого иода в объемном анализе, перейдем к
рассмотрению методов определения ряда неорганических и
органических веществ на основе реакций окисления, иодирования,
присоединения по двойной связи, комплексообразования *.
3. РЕАКЦИИ ОКИСЛЕНИЯ
По вопросу об окислительно-восстановительных потенциалах
растворов хлористого и треххлористого иода в литературе не
имеется достаточных сведений, что вызвало необходимость проверить
и дополнить литературные данные.
Каган [10] измерила окислительно-восстановительные
потенциалы водных и солянокислых растворов хлористого и
треххлористого иода, применяя следующую цепь:
Pt
исследуемый I 1-н. раствор I Hg2Cl^
раствор
1-н.КС!
Hg.
КС1
Приводим некоторые из результатов измерений: для 0,35-мол.
водного раствора JC1 найдена величина 1150 мв, для 0,41-мол.
раствора JC13—1190 мв**. Для 0,1-н. раствора JC1 в 0,4-н. соляной
кислоте была найдена величина 1061 мв, а для такого же
раствора JCb—1064 мв. С повышением концентрации НС1 величины
потенциалов понижаются.
По литературным данным, нормальный окислительный
потенциал JC1 в солянокислом растворе 2JCl2~+2^/J2(TBrh 4СГ = 1,06в;
концентрация раствора не указана [16].
На основании результатов этих измерений мы применили
солянокислые растворы хлористого и треххлористого иода для
количественного определения ряда •восстановителей (см. табл. 51, 52) ***.
Для изучения реакций хлористого и треххлористого иода с
некоторыми восстановителями было проведено потенциометрическое
титрование 0,1-н. солянокислых растворов JC1 и JC13 0,1-н-
растворами SnCl2, Na2S03, аскорбиновой кислоты. В качестве
примера приводим кривые титрования растворов JC1 и JC13 раствором
SnCl2 в соляной кислоте.
На кривой титрования раствора хлористого иода (рис. 109)
* Детали техники определения описаны в цитированных работах.
** Результаты этих измерений удовлетворительно совпадают с
литературными данными: для JC1 — 1118 мву для JC13 — 1177 же [15].
****В недавно опубликованных работах Б. Сингх и А. Сингх [17—19]
предложили использовать окислительно-восстановительные свойства системы
JC1—J2 в качестве индикатора при количественном определении ряда
восстановителей (KJ, Na3As03, Hg2Cl2, KSCN, Na2S03, NaHS03, FeS04, N2H2.H2S04).
К|навескеТвосстановителя прибавляют 25 мл воды, 30 мл концентрированной
соляной кислоты и 5 мл 0,02-мол. JCI. Это количество хлористого иода
окисляет лишь незначительное количество восстановителя и превращается в иод.
Затем смесь титруют раствором хлорамина [18, 19] или ванадата натрия [17]
до перехода розовой окраски хлороформного слоя (иод )в светло-желтую (JC1)-
365
видны два перегиба: первый перегиб соответствует прибавлению
50% от стехиометрического количества восстановителя (0,5 моля
SnCl2 на 1 моль хлористого иода); в этой области кривой
наблюдается выделение иода, согласно уравнению: 2JCl+SnCl2=SnG44-J2k
Следующий скачок потенциала наблюдается после прибавления
второго такого же количества S-nCb и сопровождается полным
обесцвечиванием раствора вследствие перехода иода в HJ
SnCI2 -r- J2 + 2HC1 = SnCl4 + 2HJ.
Суммарное уравнение
SnCl2 + JC1 + НС1 = SnCl4 + HJ. (16.13)
На кривой титрования треххлористого иода (рис. ПО) видны
три перегиба—после прибавления 50, 75 и 100% необходимого ко-
4 б в w
Mn0,1'H.SnCL2
Рис. 109. Кривая титрования
0,1-н. раствора хлористого
иода раствором двухлористого
олова.
1100
900
700
500
300
мЬ
2 <t 6 8 10
MjrOj-H.SnCL?
Рис. ПО. Кривая титрования
0,1-н. раствора треххлористого
иода раствором двухлористого
олова.
личества восстановителя. Эти три участка кривой титрования
соответствуют следующим трем уравнениям:
SnCl2 + JC13 = SnCl4 + JC1;
SnCl2 + 2JCl = SnCl4 +J2;
SnCl2 + J2 + 2HC1 = SnCl4 + 2HJ;
2SnCI2 + JCI3 -f HC1 = 2SnCl4 -f HJ. (16.14)
Такой же ход кривых титрования получен и для систем с,
Na2S03 и аскорбиновой кислотой.
В табл. 51 и 52 приведен перечень некоторых препаратов, для
которых разработаны методы объемно-аналитических определений
солянокислыми растворами JC1 (табл. 51) и JC13 (табл. ,52). В
таблицах указаны способы титрования и величины
грамм-эквивалентов данных препаратов.
366
Таблица 51
Титрование восстановителей раствором хлористого иода
Название препарата
Йодистый калий и другие
иодиды
Каломель
Хлористое олово
Сульфит натрия
Мышьяковистый ангидрид
Фосфорноватистая кислота
и ее соли
Соединения закиси железа
(неорганические и
органические)
Аскорбиновая кислота
Анальгин
Тиомочевина
Тиобарбитураты
Тифен *
Способ титрования
Прямое
По замещению
По остатку
»
Прямое
и
и
По замещению
По остатку
По замещению
По остатку
Прямое
„
По остатку
и »
По замещению
г-экв
моль/2
моль/2
моль/2
моль/2
моль/4
моль/4
моль/4
моль/1
моль/1
моль/2
моль/2
1 моль/8
моль/8
моль/12
Ссылка
на литературу
[и]
[20]
[13]
[13]
[21]
[22]
[13, 23]
[20. 24]
[251
[26. 27]
[26, 27]
[58]
Таблица 52
Титрование восстановителей раствором треххлористого иода
Название препарата
Йодистый калий и другие
растворимые иодиды
Двуиодистаа ртуть
Хлористое олово
Сульфит натрия
Соединения закиси железа
(неорганические и
органические)
Фосфорноватистая
кислота и ее соли
Однохлористая ртуть
(каломель)
Способ титрования
Прямой
w
м
V
По остатку
» »
1» П
г-экв
моль/4
моль/8
моль/2
моль/2
моль/1
моль/4
моль/2
Ссылка
на литературу
[28]
[28]
[13]
[13]
[13, 23]
[22]
[13]
К табл. 51 и 52 необходимо сделать следующие пояснения.
1) Растворимые иодиды можно титровать как непосредственно
растворами хлористого и треххлористого иода, так и способами
замещения остатков раствором хлористого иода. Прямое титрование
иодидов хлористым иодом позволяет количественно определять их
в присутствии свободного иода. Иодхлорометрическии метод дает
возможность определять иодиды в присутствии хлоридов.
* Хлористоводородная соль диэтиламиноэтилового эфира дифенилтиоуксусной
кислоты.
367
Для определения двуиодистой ртути навеску 0,03—0,05 г ря
творяют в 15 мл 25% HQ, прибавляют 5 мл хлороформа и титруй
раствором ЛСЬ до обесцвечивания хлороформного слоя г-жв= ^!^
3HgJ2 + 2 JCI8 = 4J2 + 3HgCr2; 8
4J2 + 4JC13= 12 JCl;
HgJ2 + 2JCI3 - HgC!2 + 4JCL
2) Реакции JCl или JC13 с SnCl2 (16.13, 16.14) с ионами
закиси железа (16.9, 16.10) и с аскорбиновой кислотой (16.7) уже
приводились выше.
3) Сульфит натрия определяют прямым титрованием. Реакция
протекает по уравнениям:
Na2S03 + JCl + H20 = Na2S04 + HJ + HCl; (16.15)
2Na2S03 -+ JC13 + 2H20 - 2Na2S04 + HJ + 3HCI. (16.16)
4) Каломель быстро растворяется в солянокислом растворе
JC1 или JC13:
Hg2C!2 + 2JCI = 2HgCl2 + J2;
3Hg2CI2f 2JC13 = 6HgCl2 +J2; (16.17)
JC18 + J2 = 3JG;
Hg2CI2 + JCI3 = 2HgCl2 +JCl. (16.18)
Быстрота растворения каломели является одним из
преимуществ этого метода 'количественного определения, так как при иодо-
метр'Ическом методе определения каломели, по Государственной
-фармакопее, растворение ее происходит очень медленно. Иод-
хлорметрическим методом можно определять каломель в смесях
с сахаром и сантонином.
5) Для солей закиси железа разработаны два варианта:
а) по избытку хлористого или треххлор истого иода — к
раствору 0,1—0,2 г препарата в 10—15 мл воды добавляют двух-
трехкратный избыток титрованного раствора JCl или JC13, затем,
через 3—б минут, 20 мл 10%-ного раствора сегнетовой соли,
оставляют на 5—10 минут, после чего прибавляют 10 мл 10%-ного
раствора KJ и по истечении 5 минут титруют иод 0,1 -н. раствором МагБгОз.
Индикатор—крахмал;
6) по'выделившемуся иоду—к раствору 0,1—0,35 г соли
железа в 5—10 мл воды в делительной воронке прибавляют избыток
раствора JC1, оставляют на 3—5 минут, затем выделившийся иод
количественно извлекают хлороформом и титруют раствором
Na2S203 *.
* Для контроля к жидкости, оставшейся после извлечения иода и
содержащей избыток JC1, прибавляют 20 мл 10%-ного раствора сегнетовой соли и
10 мл 10%-ного раствора KJ; выделившийся иод титруют раствором Na2S203»
В этом случае г-экв солей железа равен моль/2.
368
Такими методами были количественно определены: сульфат
закиси железа, соль Мора, K4'Fe(CN)6* [13].
2K4Fe(CN)6 + 2JC1 = 2K3Fe(CN)G + 2КС1 + J2; (16.19)
6K4Fe (CN)6 + 2JCi3= 6K3Fe (CN)6 4- 6KC1 + J2 (16.20)
молочнокислое железо закисное, углекислое закисное железо с
сахаром, сироп с йодистым железом [23].
При анализе последних трех препаратов нет необходимости
в предварительном сжигании или окислении органических веществ
(что требуется стандартами Государственной фармакопеи),
вследствие чего техника определения значительно упрощается; кроме
того, оказывается возможным определить собственно содержание
только закисной формы, в то время как по Государственной
фармакопее определяется общее количество железа.
6) Фосфорноватистая кислота окисляется хлористым иодом по
уравнению:
Н3Р02 + 4JC1 + 2Н20 = Н3Р04 + 2J2 + 4HC1. (16.21)
Количественное определение можно проводить как по
количеству выделяющегося иода, так и по избытку хлористого иода.
С треххлористым иодом Н3Р02 реагирует по уравнению:
ЗН3Р02 + 4JC13 + 6Н20 = ЗН3Р04 + 2 J2 + 12HCI. (16.22)
Определение ведут по избытку JC13.
7) Мышьяковистую кислоту и ее растворимые соли необходимо
титровать в слабощелочной среде, после предварительного
добавления бикарбоната натрия для нейтрализации HJ и НС1,
образующихся при окислении хлористым иодом:
As203 + 2JC1 + 2Н20 Z As205 + 2HJ + 2НС1.
Титруют солянокислым раствором хлористого иода в
присутствии крахмала до посинения раствора [21]**.
* При определении K4Fe(CN)6 сегнетову соль добавляют для уменьшения
концентрации водородных ионов.
** Недавно Чигалик и сотрудники [54, 55] опубликовали ряд сообщений,
посвященных потенциометрическому титрованию восстановителей (J~, S20^ ,
SOg , [Fe(CN)6]4" и др.) 0,2-н. раствором JC1 в ^2-н. соляной кислоте,
приготовленным по Лангу. По данным этих авторов, реакция с J~ проходит
количественно только в нейтральных и слабокислых ( < 0,05-н. НС1) растворах.
Наличие Вг~ (в соотношении 1 : 10) или СГ (в соотношении 1 : 100) не
мешает. Тиосульфаты определяют косвенным методом, титруя исследуемым
раствором иод, выделившийся из 1 г К J под действием известного количества JC1.
Для определения SO^ исследуемый раствор приливают к раствору JC1,
избыток последнего определяют потенциометрически раствором арсенита. Ферро-
цианиды определяют прямым титрованием. В других сообщениях описаны
условия титрования соединений Asm, Sbni, Snii, цианидов, роданидов, гидразина,
фенилгидразина, гидроксиламина, гидрохинона, метола, л-фенилендиамина,
тиосемикарбазида, димеркаптопропанола, меркаптобензтиазола, тиомочевины,
хинонимина и некоторых других восстановителей.
24—1419
369
8) Анальгин (1 -фенил-2,3-ди'метил-5-пиразолон-4-метила,мин
метиленсернистокислый натрий) окисляется хлористым иодом п
уравнению: °
CH2S03Na Н Н
I \/
H8C-N-C11Hu02N+JCl+2H20=[H3C-N-C11Hi102N] J+HCl+NaHS04+HCOH
Определение анальгина проводится титрованием хлористым
иодом в спиртово-хлороформной среде.
9) При исследовании тиобарбитуратов (производных тиобарби-
туровой кислоты) было установлено [26, 27], что хлористый йод
в солянокислом горячем растворе количественно окисляет серу
группы >C = S в ионы S(V~. Эта реакция может быть использована для
количественного^ определения не только тиобарбитуратов, но и
некоторых других органических соединений, содержащих группу
>C = S, например тиомочевины. В последнем случае реакция
может быть выражена уравнением:
(NH2>2CS+8JCl+5H20=(NH2)2CO+8HCl+H2S04+4J2
4. РЕАКЦИИ ИОДИРОВАНИЯ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
При взаимодействии хлористого и треххлористого иода с
органическими веществами возможно образование не только иод-/-но
и хлорпроизводных. В главе VI были рассмотрены
физико-химические факторы, которые способствуют иодированию органических
веществ: полярность растворителя, в котором происходит
реакция галоидирования, химическая природа, строение и полярность
галоидируемого вещества, условия проведения реакции.
Из рассмотрения этих факторов следует, что, применяя
солянокислые растворы хлористого (и треххлористого) иода,
необходимо создать условия, благоприятствующие электролитической
диссоциации хлористого иода и повышению концентрации ионов J+.
Чтобы проверить это предположение, было исследовано
действие солянокислых растворов хлористого и треххлористого иода
на органические соединения в условиях объемно-аналитических
определений.
Результаты этих исследований подтвердили значение этих
факторов, а также и высказанное выше предположение о
положительных особенностях солянокислых растворов хлористого и
треххлористого иода для целей иодирования различных фенолов, аминов
и их производных.
При этих исследованиях в каждом отдельном случае было
изучено влияние на процесс иодирования избытка хлористого и треХг
хлористого иода, концентрации соляной кислоты, разведения
водой, времени взаимодействия, температуры.
Из полученных данных были сделаны следующие выводы, на
основании которых можно было количественно определять
разнообразные фенолы и амины.
1) Определение фенолов и аминов без разбавления водой или
разбавление недостаточным количеством воды приводит к пони-
370
женным результатам *. Для фенолов требуется десятикратное
разбавление водой, для аминов и их производных разбавлять
необходимо не меньше чем в 30—40 раз, причем горячей водой
(температура 60—70°). .
2) Для получения количественных результатов необходим трех-
четырехкратный избыток реактивов. Больший избыток JC1 или
ЛС1з на результаты иодирования не влияет.
3) Длительность взаимодействия от 20—30 минут до 24 часов
приводит к одинаковым результатам.
При учете всех этих условий иодирования была разработана
техника количественного определения ряда фенолов, аминов и их
производных: к 10 мл приблизительно 0,1-н. раствора
исследуемого препарата, после разбавления соответствующим количеством
воды (в случае аминов и их производных прибавляют горячую
воду), прибавляют двух-четырехкратный избыток (20—40 мл) 0,1-н.
раствора JC1 или трех-четырехкратный избыток раствора ЛС1з>
взбалтывают и оставляют на 10—20 минут, после чего прибавляют
10 мл 10%-ного раствора йодистого калия и выделившийся иод
титруют 0,1-н. раствором Na2S203B присутствии крахмала (табл. 53) **.
Для изучения химизма взаимодействия солянокислых
растворов JC1 и JCU с аминами и фенолами были изолированы продукты
реакции, после чего определяли их температуру плавления и
содержание иода, чаще всего ло Степанову [29]. В продуктах
иодирования хлор не был обнаружен. Кроме того, определяли
количество НС1, выделяющееся при взаимодействии фенолов и аминов
с JC1 и JC13.
Все эти исследования дали возможность установить природу
химических реакций, протекающих в исследованных нами системах-
Для примера приводим некоторые из таких реакций:
ОН ОН
I I
I || +3JC1 —^J | ||~~J +3HC1
Ч/ Ч/
I
J
ОН ОН ,,;„
I I
| H+2JC1 — JH ||~ +2НС1
Ч/ / \/
I I
NOo N02
* Разведением водой достигается уменьшение концентрации соляной
кислоты, в результате чего равновесия диссоциации ШС12 и HJC14 сдвигаются
в сторону увеличения концентрации J +-ионов, что благоприятствует
иодирующему действию JC1 и JCl8. !'r^ ч*.-.!*
** Осадки иодпроизводных не реагируют с йодистым калием или с. иодом,
вследствие чегонет необходимости отфильтровывать их.
,371
он он
I I
I II +3JC1—>J—| || J +3HC1
I II он * —ОН
I
J
NH2 NH2
I I
I H +2JC1—*J | || J+2HCI
\/ %У
S02NHR S02NHR
сульфаниламидные препараты
COOH COOH
А-он f)r0ii
| II +2JClTi || т +2HC1
I I
NH2 NH2
J
I
f || I+2JQ—,J || | +2HC1
I N | N
OH OH
H H
HO-f ^—С—С—{~"Y-OH+4JC1—-
\=/ I I \=/
синэстрол
f H H f
I I I I
— HO-f V_C—C— f V-OH+4HC1
X|=/ I I X=,'
j C2H5C2H5 j
Реакции с треххлористым иодом протекают аналогично, но с
выделением эквивалентного количества хлора, например,
он °н
}\ J-^\-J
f i|+3JCl,--L ])"J+3HC1+3C12
XX
I
J
Реакции иодирования с участием хлористого и треххлористого
иода полностью подчиняются обычным правилам галоидирования
фенолов и ароматических аминов.
372
Таблица 53
Иодхлорметрическое определение фенолов, аминов и их производных
Название препарата
фенол
я-Хлорфенол
п- Нитрофенол
Салициловая кислота
Салол
Тимол
8-Оксихинолин
п-Аминосалициловая кислота
Метиловый и этиловый эфиры
салициловой кислоты
4-Гексшьрезорцин
Синэстрол
Анизол
Фенолфталеин
Цинковая соль сульфофеноловой
кислоты
Анилин
Ацетанилид
Новокаин
о- и я-Толуидин
Анестезин г)
Стрептоцид (белый)
Стрептоцид (белый растворимый)
Сульфидин
Дисульфан 2)
Сульфанилилгуанидин (сульгин)
Ацетилсульфагуанидин
Сульцимид 3)
Сульфацил (альбуцид)4)
Сульфазин (сулгфадиазин)5)
Сульфамеразин 6)
Сульфадимезин ?)
Этазол8)
Антраниловая кислота
Риванол
Количество
атомов иода
в ядре иодпро-
изводного
3
2
2
2
5
2
2
2
2
2
4
1
2
4
3
3
2
2
2
2
2
2
2
2
2
2
2
3
3
2
2
2
2
Ссылка на
литературу
Метод определения
раствором
JC1
[14]
[31]
131]
[14]
[14]
[31]
[14, 56]
[32]
[59]
[31]
[31]
[31]
[53]
[14]
[33]
[33]
[33]
[31]
[33]
[34]
[35]
[34]
[34]
[34]
[36]
[35]
[36]
[37]
[37]
[35]
[57]
[56]
[60]
раствором
•ICL
[30]
[31|
[31]
[30]
[30J
1311
[31]
[31]
[31]
[311
131]
—
[ЗЦ
[31]
[311
[31]
[30]
:—
]30]
[30]
—
—
—
—
—
[57]
[571
Пояснения: г) Этиловый эфир n-аминобензойной кислоты; 2) 4-Сульфанй-
лилсульфаниламид; 3) Сульфанилцианамид; 4) Сульфанилацетамид; 5) 2-Сульфанил-
амидопиримидин. При иодировании сульфазола и сульфамеразина два атома иода
входят в бензольное ядро, а третий — в пиримидиновое; 6) 2-Сульфаниламидо-
4-метилпиримидин; 7) 2-Сульфаниламидо-4,6-диметилпиримидин; 8) 2-Сульфанилами*-
до-2-этил-3,4 -тиодиазол.
В табл. 53 приведен перечень ряда препаратов, для которых
были разработаны методы объемно-аналитических определений.
В таблице указано количество атомов иода, вошедших в молекулу
органического соединения (по данным титрования и химического
анализ?, продуктов иодирования).
Если в молекуле иодпроизводного содержится 1—2—3 атома
373
иода, то г-экв исходного препарата равен моль/2, моль/4, моль/fi
соответственно. '
Пояснения к табл. 53. Из препаратов, растворимых в
воде, приготовляют растворы приблизительно 0,1-н. концентрации'
Прегараты основного характера, не растворимые в воде (амины
сульфаниламидные препараты), растворяют в небольшом
количестве 1-н. соляной кислоты. Некоторые препараты (синэстрол, гек-
силрезорцин) растворяют в 5 мл спирта.
Эфиры салициловой кислоты определяют после
предварительного омыления раствором щелочи,
Ацетанилид и другие амины с ацетилированной аминогруппой
например ацетилсульфагуанидин, предварительно гидролизуют,'
нагревая с 1-н. соляной кислотой [38].
Стрептоцид белый растворимый (10 ма~ 0,1-н. раствора) для
освобождения NH2-rpynnbi кипятят с 2—3 мл разведенной (^16%)
серной кислотой до исчезновения запаха формальдегида и
сернистого газа:
H2NS02 — СбН4 — NHCH2S03Na + H2S04 = H2NS02 — C6H4 - NHa -f
4- HCHO + S02 + NaHS04.
При определении сульфодимезина лучшие результаты
получаются, если титровать избыток хлористого иода после отфильтро-
вания осадка.
В некоторых случаях нельзя использовать солянокислые
растворы хлористого и треххлористого иода для реакции галоиди-
рования некоторых фенолов и аминов, например бетанафтола и
сульфазола, вследствие окисления последних этими растворами.
В связи с этим была изучена возможность иодирования таких
фенолов и аминов растворами хлористого и треххлористого иода в
слабощелочной среде после прибавления бикарбоната натрия.
Треххлористый иод в растворе NaHC03 сначала
подвергается неполному гидролизу с освобождением хлористого иода
2JCI.3 + 6NaHC03 ^ JC1 + NaJ03 + 5NaCl + 6C02 + 2Н20,
последний же вступает в реакцию иодирования. Таким путем
удалось разработать условия количественного определения
сульфазола [37, 24] и р-нафтола [241.
5. РЕАКЦИИ ПРИСОЕДИНЕНИЯ ГАЛОГЕНИДОВ ИОДА
К НЕПРЕДЕЛЬНЫМ СОЕДИНЕНИЯМ
По вопросу о присоединении галогенидов иода, главным
образом хлористого иода, к непредельным соединениям в литературе
имеется ряд работ, особенно в связи с .применением неводных
растворов хлористого и бромистого иода для определения йодного
числа жиров и масел (глава VI). На процесс присоединения
хлористого иода к непредельным соединениям большое влияние
оказывают наличие в молекуле непредельного соединения определен-
374
ных; заместителей, природа растворителя, величина избытка
хлористого иода и некоторые другие факторы.
Таким образом, литературные данные (см. главу VI) указывают
на то, что реакция присоединения хлористого иода к непредельным
соединениям не протекает во всех случаях одинаково. Это
усложняет возможность применения данной реакции для аналитических
целей и требует подробного изучения ее в каждом отдельном
случае.
Еще более сложно протекает реакция между треххлористым
иодом и непредельными соединениями. Как уже отмечалось, ряд
важных работ в этой области принадлежит Несмеянову и его
сотрудникам.
Фиалков и Каган [39] изучили реакции между аллиловым
спиртом, коричной кислотой, петрозелиновой кислотой, диаллилбарбиту-
ровой кислотой (диалом) и солянокислыми растворами хлористого
и треххлористого иода, что дало возможность разработать
следующие условия объемно-аналитического определения этих
препаратов: навеску препарата растворяют в воде или в спирте (петро-
зелиновую кислоту), прибавляют избыток раствора хлористого или
треххлористого иода и оставляют на 20—30 минут; затем
добавляют раствор KJ и выделившийся иод титруют раствором Na2S203.
Одновременно было определено содержание иода в продуктах
иодирования, полученных при действии растворами хлористого
или треххлористого иода *. Продукты иодирования коричной
кислоты и диала не содержали хлора. Это можно было объяснить тем,
что первично образующиеся иодхлорзамещенные затем гидроли-
зовались с выделением хлористого водорода. Действительно, в
случае реакции с диалом оказалось, что на каждый моль вошедшего ъ
реакцию диала выделяется ^ 2 моля HCI.
На основании этих экспериментальных данных указанные выше
реакции можно выразить следующими уравнениями:
СН3 - (СН2)10 - СН = СН — (СН2)4 - СООН + JC1 -^ СН3 — (СН2)10 -
петрозелиновая кислота
- СНС1 — СН J - (СН2)4 - СООН;
С6НД - СН = СН - СООН + JC1+H20 -> С6Н5 — СНОН -
коричная кислота
-CHJ-COOH + HC1;
NH-CO NH-CO
» I ХН2-СН=СН2 I I /CH2-CHOH-CH2J
СО С( +2JC1+2H20—СО СС
I | ЧСН2-СН=СН2 I * \CH2-CHOH-CH2J4 2HC1.
NH-CO NH-CO
диаллилбарбитуровая кислота
* Идентичность полученной в этих опытах а-иод-р-окси-р-феншшропионо-
вой кислоты была установлена также по температуре плавления (141—141,5°),
что совпадает с данными Эрленмейера [41].
375
Реакции с треххлористым иодом протекают так же, но с вы_
делением хлора, например:
С6Н5СН = СН — СООН + JCI3 + Н20 -> С6Н5 - СНОН —
- CHJ - СООН + С12 + НС1.
Г-экв коричной и петрозелиновой кислот в этих реакциях
равен моль/2, а г-экв диала равен моль/4.
Фиалков и Рапапорт [40, 61], продолжив исследование реакции
между диалом и хлористым иодом, подтвердили приведенные выше
данные в отношении химизма этой реакции и показали, что для
получения продукта присоединения 2 молей хлористого иода к
диаллилбарбитуровой кислоте реакцию следует проводить в
отсутствие воды *, причем присоединение хлористого иода
происходит, по всей вероятности, согласно с правилом Марковникова.
При изучении взаимодействия хлористого иода с р-бромаллиль-
ными производными барбитуровой кислоты (содержащими
группу СН2 = СВг—СН2—) в среде соляной кислоты оказалось, что в
этих случаях реакция протекает значительно сложнее. Так, в
реакции с ректоном (вторичный амил-р-бромаллилбарбитуровая
кислота) первично образующийся продукт присоединения постепенно
гидролизуется. При этом быстро отщепляется НС1 и образуется
иодгидрин ректона, который затем превращается в смесь диокси-
бромпроизводного и иодацетонпроизводного [40]:
JC1 Н20
СН2=СВг-СН2 ^CH2J-CBrCl-CH2 ► CH2J—СВгОНСН2—
Н20
- - СН2ОН-СВгОН—СН2—hCH2J- CO-CH2— **.
Такое усложнение реакции не позволяет распространить иод-
хлорметрический метод на (3-бромаллильные барбитураты.
Количественное определение барбитуратов, содержащих цикло-
гексениальную группу (фанадорма, эвипаната натрия), описано в
работе Рапапорта [61].
Интересной формой применения солянокислых растворов
хлористого иода в объемном анализе является разработанный Генг-
риновичем и Крамаренко [42] метод количественного определения
хинина, цинхонина и эухинина в присутствии продуктов их
гидрирования:
•\/\ ™„ _/*Ч.
СН,
r-;
-CHORx-
для эухинина
для хинина
СН,
—сн=сн2
\/
R=OCH3, R,=CO (OC2H5);
R=OCH3, Ri=H;
для цинхонина R = H, Ri = H.
* По методу, который применил Эрленмейер для получения а- ио д-р-хлор -
£-фен и л про пионовой кислоты из коричной кислоты [41].
** В этих схемах показаны только те изменения, которые происходят в>
р-бромал л ильной группе данных барбитуратов.
376
Продажные 'Препараты хинина, цинхонина и эухинина не лред-
ставляют собой индивидуально чистых веществ. В них содержатся
(обычно от 3 до 12%) гидрированные продукты, которые вместо
винильной группы СН2 = СН— содержат этильную группу
СНз—СН2—.
Для отделения хинина и цинхонина от гидрохинина и гидро-
цинхонина были предложены методы, основанные на том, что
эти алкалоиды, содержащие винильную группу, образуют
комплексные соединения с двухлористой медью [43] или с
уксуснокислой ртутью [44, 45]. Эти методы очень трудоемкие и
длительные.
Для количественного определения этих препаратов Генгринович
и Крамаренко использовали способность хлористого иода (из
солянокислого раствора) присоединяться к винильной группе с
образованием радикала —СНС1—СНгЛ. При избытке хлористого
иода он присоединяется также к аминному азоту, однако этот
продукт присоединения хлористого иода непрочный и легко
разлагается при действии йодистого калия.
Техника определения: 0,2—0,3 г соли алкалоида растворяют в
50—70 мл воды и прибавляют при взбалтывании 0,1-н.
солянокислый раствор хлористого иода до появления неисчезающей
мути. Затем прибавляют 5 мл 2%-ного раствора KJ, -взбалтывают
и титруют иод 0,Ьн. раствором Na2S203 (индикатор—
крахмал).
Можно применить также прямое титрование: навеска 0,2—
0,3 г, объем раствора 20 мл, в качестве индикатора применяют
2 кашли 0,2%-но'ГО раствора KJ и 1 мл раствора крахмала, титруют
при взбалтывании 0,1-н. раствором хлористого иода до
исчезновения синей окраски жидкости.
Результаты определений совпали с данными, полученными по
методу Трона и Диршеля [44].
Определение йодного числа жиров. К этой группе
исследований относится разрабатываемый Генгриновичем метод
определения йодного числа жиров и масел в водной среде с 'помощью
солянокислого раствора хлористого иода. Для этой цели
Генгринович и Юдович [46] применили предложенный автором [47] вариант
метода Маргошеса [48] определения йодного числа жиров в водной
среде, для чего навеску жира предварительно эмульгируют, а затем
добавляют спиртовый [47] или водный [49] раствор иода. Навеску
0,04—0,5 г жира (в зависимости от природы жира) эмульгируют
в чашечке * с помощью 0,05—0,25 г абрикосовой камеди в
нескольких каплях воды, постепенно добавляют 10 мл воды и
количественно переносят в колбу, смывая небольшими порциями воды.
Затем приливают 25 мл 0,2-н. солянокислого раствора JC1,
взбалтывают, оставляют на 5 минут, вносят 5 мл 10%-ного раствора KJ и
выделившийся иод титруют 0,1-н. раствором Na2S203 (индикатор—
крахмал). Кислотность смеси при этом не увеличивается; поэтому
* Твердый жир эмульгируют после расплавления.
377
можно полагать, что по месту двойной связи глицеридов
присоединяется JC1, но не JOH—продукт гидролиза хлористого иода.
Генгринович разработал еще один вариант иодхлорметриче-
ского метода определения йодного числа, причем вместо
эмульгирования жиров их растворяют в этиловом эфире [50]: к навеске
жира в склянке с притертой пробкой прибавляют 3 мл эфира («для
наркоза»), 25 мл 0,2-н. солянокислого раствора JC1, взбалтывают
1 минуту и поступают, как указано выше. В случае твердого жира
навеску его растворяют в 6—9 мл эфира и помимо раствора
хлористого иода прибавляют 25—50 мл воды.
Оба эти варианта иодхлорметрического метода определения
йодного числа твердых жиров и масел (невысыхающих,
полувысыхающих и высыхающих) дали величины, очень близкие к
полученным по методу Гюбля.
6. РЕАКЦИИ КОМПЛЕКСООБРАЗОВАНИЯ
Как указывалось на стр. 363, применение хлористого и трех-
ллористого иода в объемном анализе, основанное на реакциях
комплексообразования, заключается в способности галогенидов
иода или их комплексных кислот образовывать комплексные
соединения с азотистыми и тому подобными основаниями, например:
C5H5N.JC1, C5H5N2JC1, [C5H5NH]JC12, [C5H5NHjJCl4 и т. п.
Некоторые из таких комплексов очень прочны, не растворимы
в воде и почти не разлагаются ею. К их числу относятся и
комплексные соединения уротропина с хлористым иодом (стр. 241).
Генгринович [51] показал, что при действии солянокислого
раствора хлористого иода на уротропин в широких пределах
соотношений компонентов реакции (количество хлористого иода
составляло от 25 до 400% от стехиометрического) всегда образуется
одно и то же кристаллическое вещество (CH2)6N4 • 2JC1. Оно не
растворимо в воде и разведенных кислотах, растворяется в крепком
растворе KJ, в растворах щелочей, антипирина и тиосульфата
натрия.
Для количественного определения 0,03—0,15 г уротропина
растворяют в небольшом количестве воды в мерной колбе на 100 мл,
прибавляют 15—50 мл 0,1 -н. солянокислого раствора JC1,
взбалтывают, доводят водой до метки и фильтруют, отбрасывая первые
10 мл фильтрата. К 25 мл фильтрата добавляют 10 мл 10%-ного
раствора KJ и выделившийся иод титруют 0,1-н. раствором Na2S203.
На основании реакции (СН2)6 м4 • 2JC1 + 2KJ = (СН2)6 N4 + 2KCI +
-j- 2J2 г-экв уротропина равен моль/4.
Аналогичный метод применил Вайсман [52] для
количественного определения акрихина, одна молекула которого присоединяет
две молекулы JC1; г-экв акрихина равен моль/4.
Необходимо отметить, что этот раздел иодхлорометрического
метода, основанный на реакциях комплексообразования, еще
мало разработан.
378
ЗАКЛЮЧЕНИЕ
Иодхлорометрический метод объемного анализа, основанный
на применении солянокислых растворов хлористого и треххлори-
стого иода лишен ряда тех недостатков иодометрического метода,
о которых говорилось выше.
1. Хлористый и треххлористыи иод в солянокислых растворах
не летучи.
2. Эти галогениды иода можно применять для разнообразных
аналитических определений, причем они обычно проявляют
значительно большую активность по сравнению с водными растворами
иода.
3. Реакции иодирования органических соединений путем
замещения атомов водорода не являются обратимыми (как при
иодировании молекулярным иодом), так как в этом случае
выделяется HCl (RH+JC1 = RJ + HQ). Кроме того, эти реакции можно
проводить в кислой среде. Продукты иодирования получаются,
как правило, свободными от примеси хлорзамещенных.
Солянокислые растворы галогенидов иода не адсорбируются продуктами
иодирования.
4. Применяя солянокислые растворы галогенидов иода,
удается осуществить реакцию присоединения хлористого иода по
двойной связи с количественным выходом и разработать на основании
этой реакции методы объемного анализа непредельных соединений.
5. Применение солянокислых растворов хлористого и треххло-
ристого иода дало возможность разработать методы объемного
анализа большого числа органических соединений. Эти растворы
можно использовать также для препаративных работ—для
получения иодпроизводных, и притом гораздо более широко, чем это
представлено в литературе.
В заключение необходимо сравнить солянокислые растворы
хлористого и треххлористого иода, имея в виду их применение в
объемном анализе. В реакциях окисления хлористый и
треххлористыи иод, в основном, реагируют одинаково. Применение
хлористого иода имеет то преимущество, что позволяет разработать в
некоторых случаях несколько вариантов методики определения, как
например, для солей закиси железа.
В реакциях замещения водорода на иод хлористый иод OKaj
зался более активным иодирующим средством, чем треххлористыи
иод. Это следует из того, что для количественного определения
фенолов и аминов треххлористыи иод необходимо брать в большем
избытке по сравнению с хлористым иодом примерно в два раза *.
В некоторых случаях меньшая активность треххлористого иода
является его 'преимуществом. Так, например, о- и /г-толуидины
нельзя иодировать раствором хлористого иода, так как наряду с
иодированием происходит и окисление этих аминов, в результате
чего образуются осадки бурого цвета, получаются повышенные
* Это объясняется тем, что содержание иода в растворах треххлористого
иода в два раза меньше, чем в растворах хлористого иода той же нормальности.
379
и несовпадающие результаты; эти явления не наблюдаются при
применении растворов треххлористого иода.
В реакциях присоединения по двойной связи хлористый и трех-
хлористый иод ведут себя одинаково, однако в ряде случаев для
количественных определений необходим больший избыток
треххлористого иода по сравнению с хлористым иодом.
Приведенные выше факты далеко не исчерпывают всех
возможностей иодхлорметрического метода. Однако они дают
основание полагать, что солянокислые растворы хлористого и
треххлористого иода могут найти широкое применение в практике
объемного анализа неорганических и органических соединений.
ЛИТЕРАТУРА
1. A. Michael, L Norton, Ber., 9, 1752 (1876); 11, 107 (1878).
2. G. В. Н е i s i g, J. Am. Chem. Soc, 50, 1687 (1928).
3. H. К u b i n a, Z. anal. Chem., 76, 39 (1929).
4. Б. В. Солдатов, С. А. Вознесенский, Сб. исследов. работ.
Акад. хим. защиты им. Ворошилова, 1940, стр. 21; Хим. рефер. ж., 4,
№ 4, 32 (1941).
5. Е. D б г f u r t, E. Wolff, Z. anorg. allg. Chem., 185, 333 (1930).
6. E. В irk, Z. ang. Chem., 41, 751 (1928).
7. R. Lang, Z. anorg. allg. Chem., 142, 280 (1925).
8. А. И. Г е н г р и н о в и ч, Укр. фарм. ж., в. 2, 27 (1940).
9. Н. Т. S. В г i t t о n, R. Е. С о с k a d а у, J. К- Foreman. JL
Chem. Soc, 3879 (1952).
10. Ф. Е. Каган, Применение треххлористого и хлористого иода для
количественного определения некоторых групп фармацевтических
препаратов, Дис.,Моск. фарм. ин-т, 1953.
11. А. И. Г е н г р и н о в и ч, М- И. Печеный, М. С. Барон,
Ученые записки Киевского ин-та усовершенствования провизоров*, 1, 97 (1950).
12. А. И. Г е н г р и н о в и ч, Укр. фарм. ж., в. 3, 15 (1940).
13. Я- А. Ф и а л к о в, Ф. Е. К а г а н, Укр. хим. ж., 18, 55 (1952).
14. А. И. Г е н г р и н о в и ч, Фармация, в. 6, 5 (1946).
15. В. Финкельштейн, Z. phys. Ch., 124, 285(1926).
16. И. М. К о л ь т г о ф, В. А. С е н д е л, Объемный анализ, Госхим-
издат, М.—Л., 1950, т. I.
17. В. Singh, R. Singh, Analyt. Chim. Acta, 10, 408 (1954).
18. В. Singh, К- С h. S о о d, Analyt. Chim. Acta, II, 303 (1954).
19. A. S i n g h, J. Indian. Chem. Soc, 31, 647 (1954).
20. Консультационные материалы Центр, н.-и. аптечной лаборатории Мин.
здравоохр. УССР, в. 1—2 (1952).
21. А. И. Г е н г р и н о в и ч, Укр. фарм. ж., в. 4, 23 (1940).
22. Я- А. Ф и а л к о в, Ф. Е. К а г а н, ЖОХ, 24, 3 (1954).
23. Ф. Е. Каган, Аптечное дело, в. 5, 10 (1955).
24. Ф. Е. К а г а н, Ц. И. Ш а х, Укр. хим. ж., 23, 537 (1957).
25. Л. И. Р а п а п о р т, М- Л. Ш в а р ц б у р д, Аптечное дело, в. 5,
47 (1954).
26. Л. И. Р а п а п о р т, Качественное и количественное определение
производных барбитуровой кислоты, Дис, Моск. фарм. ин-т, 1954.
27. Л. И. Р а п а п о р т, Я- А. Ф и а л к о в, ЖОХ, 26, 279 (1956).
28. Ф. Е. Каган, Аптечное дело, в. 1, 14 (1957).
29. А. В. Степанов, ЖРФХО, 37, 12 (1905).
30. Я- А. Ф и а л к о в, Ф. Е. К а г ан, Укр. хим. ж., 18, 64 (1952).
31. Ф. Е. Kara и, Укр. хим. ж., 22, 44, 95 (1956).-
* Далее сокращенно обозначается «Уч. зап.».
380
32. А. И. Генгринович, М. С. Барон, Аптечное дело, в. 4, 27
(1952).
33. А. И. Г е н г р и н о в и ч, Я- К- Кадыров, Аптечное дело, в. 1,
46 (1952).
34. А. И. Г е н г р и н о в и ч, Ц. И. Ш а х, Мед. пром., в. 3, 37 (1948).
35. А. И. Г е н г р и н о в и ч, А. Ю. И б а д о в, Аптечное дело, в. 3, 18
(1952).
36. Ц. И. Ш а х, Мед. пром., в. 4, 29 (1951).
37. А. И. Г е н г р и н о в и ч, Ц. И. Ш а х, Уч. зап., 1, 79 (1950).
38. Г. А. В а й с м а н, Укр. фарм. ж., в. 5—6, 109 (1933).
39. Я- А. Ф и а л к о в, Ф. Е. К а г ан, ЖОХ, 27, 2836 (1957).
40. Я- А. Ф и а л к о в и Л. И. Р а п а п о р т, ЖОХ, 25, 2265 (1955).
41. Е. Erlenmeyer, Ann., 289, 259 (1896).
42. А. И. Г е н г р и н о в и ч, В. Ф. Крамаренко, Уч. зап., 1, 87
(1950).
43. A. Cohen, J. Chem. Soc, 996 (1933).
44. H. T h г о n, W. D i r s с h e 1, Lieb. Ann., 515, 252 (1934).
45. H. E. 3 e л и г с о н, А. К- С а н ь к о в с к а я, ЖОХ, 15, 957 (1945).
46. А. И. Г е н г р и н о в и ч, Э. А. Ю д о в и ч, Аптечное дело, в. 5, 17
(1952).
47. Я- А. Ф и а л к о в, Укр. хим. ж., 2, 139 (1926); Хим. фарм. ж., в. 3
(1927).
48. В. М. М а г g о s с h e s, W. H i n n е г, Z. ang. Chem., 37, 202, 334,
982 (1924).
49. Е. С. С к в и р с к а я, Укр. фарм. ж., в. 2, 38 (1936).
50. А. И. Г е н г р.и н о в и ч, Молочная промышленность, 6, 29 (1954).
51. А. И. Г е н г р и н о в и ч, Уч. зап., 1, 104 (1950).
52. Г. А. В а й с м а н, Аптечное дело, в. 6, 25 (1953).
53. А. И. Генгринович, И. Мансурханова, Аптечное дело,
в. 6, 9 (1954).
54. J. С i h а 1 i k, D. V a v r e i n о v a, Chem. listy, 49, 693, 1167, 1176,
1731 (1955).
55. J. С i h a 1 i к, К- Terebova, Chem. listy, 50, 1761, 1768 (1956);
J. С i h a 1 i к, J. R u 2 i с к a, Chem. listy, 51, 264 (1957).
56. J. С i h a 1 i к, К- Terebova, Chem. listy, 51, 272 (1957).
57. Ц. И. Ill a x, Аптечное дело, в. 1, 22 (1957).
58. Л. И. Р а п а п о р т, Научно-консультационные материалы Центр, н.-и.
аптечной лаборатории Мин. здравоохр. УССР, в. 25, 37 (1957).
59. А. И. Генгринович, А. К- Кадыров, Аптечное дело, в. 2,
68 (1957).
60. П. П. Супрун, Мед. пром., в. 2, 49 (1957).
£1. Л. И. Р а п а п о р т, Ж- анал. хим., 12, 415 (1957).
АВТОРСКИЙ УКАЗАТЕЛЬ
Абарбарчук И. Л., 176, 177, 220,
234, 292, 317, 318, 357.
Абрамова М. А., 234, 271
Аксельруд Н. В., 357
Альбанский В. Л., 265, 271
Анисимов П. А., 272
Анисимов К- Н., 179
Анисимова К. М., 177, 235, 252,
270, 271
Аносов В. Я., 112, 125,271,342,356
Антипин Л. Н., 211, 213
Аркель ван, 334, 341
Ашкинази М. С, 234
Бабко А. К., 126, 343, 349, 352,
353, 356
Баландин А., 178
Баскова 3. А., 153, 178
Барг А. Б., 328, 333
Барон М. С, 380, 381
Беленький Л. И., 150, 177
Бернштейн А. В., 235
Бокий Г. Б., 333, 354, 358
Болдырев А. К., 333
Брайнина Э. М., 164, 165, 178
Брежнева Н. Е., 357
Брик Т. Дж., 45
Брицке Э. В., 45, 126, 136, 179,
188, 195
Бродский А. И., 341
Брунс Б. П., 221, 225, 226, 233,
234, 272
де Бур, 334, 341
Бурьянов Я- Б., 179, 314, 318, 319,
348, 357, 358
Бутков К. В., 119—121, 126
Вайсман Г. А., 378, 381
Вальден П., 116, 126, 145, 167, 176,
177, 180, 194, 208, 213, 219—221,
224, 226, 228, 234, 235, 340, 341
Вернер А., 166, 167, 178, 271, 334,
341
Виноградова А. Д., 112, 125
Вознесенский С. А., 163, 178, 380
Войтович Б. А., 347, 357
Воскресенская Н. К., 351, 358
Генгринович А. И., 144, 146, 151
177, 179, 271, 359—361, 364, 376—'
378, 380, 381
Гитман Е. Б., 235
Глоклер Г., 18
Голл Н., 319
Горенбейн Е. Я., 234, 235, 271
Гохштейн Н., 357
Гринберг А. А., 318, 334, 335, 34!
Дамиен А., 324, 325, 333
Данилов С. Н., 358
Делимарский Ю. К-, 213
Денисова Л. Ф., 181, 195
Дубников Л. М., 17
Дулова В., 357
Ефремов Н. Н., 112, 125
Жемчужный С. Ф., 18, 351, 358
Заседателев М. М., 18
Зелигсон Н. Е., 381
Зернов В., 148, 176
Ибадов А. Ю., 381
Измайлов Н. А., 348, 349, 357
Истомин А. В., 152, 17
Кабачник М. И., 179
Каган Ф. Е., 146, 163, 177, 178,
359, 360, 362, 365, 375, 380, 381
Каганская К. Я-, 14, 18, 140, 176,
177, 208, 213, 218, 227, 228, 230,
234, 235, 292, 316, 318
Кадыров Я. К., 381
Каняев Н. П., 117, 124, 126
Капустинский А. Ф., 45, 126, 136,
179, 188, 195
Карлышева К- Ф., 148
Квасник В., 152
КвятИ.,343,356
Киреев В. А., 177
Клемм В., 18
Козьмина Г., 271
Колобов Н. Е., 179
Кольтгоф И. М., 380
382
Комарь Н. П., 126, 349, 358
Коренман И. М., 352, 358
Костромина Н. А., 179, 357
Крамаренко В. Ф., 376, 377, 381
Крисе Е. Е., 352, 358
Кротков Д., 125, 271
Кудра О. К., 318
Кузьменко А. А., 19, 176, 177, 179,
194, 195, 219, 234, 292, 297, 315,
317, 318, 357
Кукина Г. А., 354, 358
Кульба Ф. Я., 129, 177, 316, 319
Курнаков Н. С, 18, 135, 112, 125,
257, 270, 271, 272, 342, 351, 356,
35
Латимер Б., 201
Лашкарев В. Е., 176
Лебедев Н. И., 175, 179
Лилич Л. С, 128, 114, 115, 126,
176, 182, 194, 216, 217, 234, 235,
253, 266, 268, 271, 272, 340, 341
Людер В., 272
Малюков И. М., 31, 42, 45, 324, 333
Мансурханова И., 381
Марков Б. Ф., 213
Марковников В., 116, 177
Меликов П., 160, 178
Менделеев Д. И., 5, 12, 18, 131, 176
Меншуткин Б. Н., 112, 125
Мискиджьян С. П., 351, 358
Михайловская В. И., 230, 234, 272
Музыка И. Д., 18, 218, 234, 245,
259, 271, 292, 318, 358
Назаренко Ю. П., 125, 341, 357
Некрасов Б. В., 18, 82, 179, 318,
327, 328, 333, 334, 337, 338, 341
Несмеянов А. Н., 164, 165, 167,
178, 179, 375
Николаев Н. С, 31, 42, 324, 333
Оксман М., 125, 271
Ормонт Б. Ф., 81, 176, 280, 318
Остромысленский И., 126
Павлов В. Л., 341
Парцхаладзе К. П., 348, 357
Паулинг Л., 44, 176, 280, 281, 318,
326, 333
Песков Н. П., 113, 125
Печеная Э. И., 179, 311, 318
Печеный М. И., 380
Пилипенко А. Т., 358
Плотников В. А., 14, 18, 169, 170,
176, 178, 179, 182, 194, 202 205,
212, 213, 226, 230, 234, 235, 257,
271,272,318,341
Погодине. А., 112, 125,271,342,356
Полищук А. Б., 177
Пресникова О. Е., 217, 234, 253
268,271, 272,340,341
Рабинович А. И.,247, 271
Рабинович М. А., 18, 213, 228, 235
Рапапорт Л. И., 360, 380, 381
Рогинский С. 3., 357
Рокотян В. Е., 14, 18, 169, 170,
178, 182, 194, 202, 212, 213, 234
Рысс И. Г., 19, 25, 44
Рябова С. А., 358
Саньковская А. К., 381
Симхаев Н. Г., 179
Сквирская Е. С., 381
Смоленцев П. И., 234, 271
Солдатов Б. В., 163, 178, 380
Сорокин В., 152, 178
Степанов А. В., 371, 380
Степанов Н. И., 349, 358
Стендел В. А., 380
Супрун П. П., 381
Сыркин Я- К., 177, 235, 252, 271
Танатар С, 131, 133, 152, 176
Тартаковская В., 271
Темникова Т. И., 153, 178
Теренин А. Н., 234, 266, 267, 271
Терентьев А. П., 150, 177
Терпугов Ф. И., 271
Тихомирова А. М., 112, 125
Томас Е., 176
Трапп Ю. К., 131, 134, 176
Трифонов К. А., 351
Удовенко В. В., 350, 357
Улько Н. В., 358
Усанович М. И., 247, 257, 271,
272, 319, 357
Ушаков М. И., 228, 235
Уэллс А. Ф., 318
Фиалков Ю. Я., 341, 350, 357
Фиалков Я. А., 14, 18, 19, 125, 130,
135, 140, 176, 179, 194, 195, 203,
205, 208, 212, 213, 218—220, 227,
228, 230, 234, 235, 245, 266, 271,
272, 294, 297, 315—319, 333, 341,
348, 357, 358, 375, 376, 380, 381
ФинкелыитейнВ., 178,234,271
Фрейдлина Р. X., 164, 165, 17в
Фрумкин А. Н., 26
Хаимова М. А., 153, 178
Хейн, 334
Цуффанти С., 272
Чалый В. П., 341
Черногоренко В. Б., 357
383
Чистов В. С, 235
Чичибабин А. Е., 107
Шатенштейн А. И., 81, 272, 318
Шах Ц. И., 360, 381
Шварцбурд М. Л., 380
Шевченко Ф. Д., 230, 235, 357
Шейнин А. Б., 272, 341
Шека 3. А., 176, 179, 271, 311, 318,
352 358
Шека'И. А., 18, 113, 126, 175, 176,
180, 194, 234, 245, 252, 271, 345,
347, 357
Шеянова Ф. Р., 358
Шилинский А. И., 357
Шилов Е. А., 117, 124, 126
Шор О. И., 19, 130, 135, 176, 177,
184, 203, 212, 285, 292, 294, 315—
317, 341
Шукарев С. А., 216, 235, 266, 272,
340, 341
Юдович Э. А., 377, 381
Эфраим Ф., 6, 12, 18, 334, 341
Якубсон С. И., 179, 234, 235, 257, 271
Янкелевич 3. А., 177
Яновская Л. А., 150, 177
Яцимирский К. Б., 349, 358
Adams R. М., 333
Alpha J., 107
Anderson Т. F., 195
Andrews L. W., 108, 125
Andrews L. J., 147, 177, 217, 234,
268, 270, 272
Anwar-Ullah S., 109, 125
Archer S., 177
Ascher E., 20, 21, 22, 27, 28, 44
Audrieth L. F.f 221, 232, 234, 238,
245,247,270
AynsleyE. E., 81, 91, 92,95, 105, 107
Badgar R. M., 195
Baenziger N. C, 318
Bagans H., 179
Balard A. J., 18, 108, 113, 125, 180,
194
Banks A. A., 18, 30—32, 38, 45,
55, 73, 81, 82, 88, 97, 98, 107.
Barr J. Т., 29, 44, 45, 178
Barrat S., 113, 118, 125
Barrow F. R., 25, 44
Basset H., 178
Baudrimont E., 19, 297, 317
Baumgarten P., 107
Bauer E., 88, 102, 107, 134, 176,
274, 282, 289, 290, 317
Bauer S. H., 78, 82, 107
Beck K-, 176
Beckmann E., 161, 176, 178, 234
BeesonC. M., 119, 120, 121, 124, 126
Beilstein F., 177
Bekier E., 14, 18, 170, 177, 182
194, 202, 205, 212, 218, 221, 223,
225, 226, 234
Benesi H. A., 266, 268, 271, 272
Bennet F. W., 18, 149, 177, " 192,
193, 195
Bensey F. N., 45
Berghello N. N.. 14, 18, 176, 227,
228, 230, 235, 249, 271
Bernhardt H. A., 31, 45
Bernstein R. В., 101, 107, 333
Berthelot M., 161, 176, 186, 195
Beyer F. C, 178
Bichowsky T. R., 45, 188, 195
Biltz W., 174, 179, 211, 213
Bingle J., 82
Birk E., 159, 160, 176, 178, 221, 380
Birkenbach L., 235
Birr E. J., 221, 232, 234, 238, 245,
247, 270
Blair С. М., 118, 120, 121, 126, 136,
137, 176, 188, 195
Bodenstein M., 185, 186, 188, 194
Bond R. L., 107
Bondi A., 235
Bonner W. D., 144, 176
Booth H. S., 17, 29—31, 33, 44, 74,
81, 107, 200, 201, 317
Bornemann W., 108—110, 125, 177,
180, 182, 194
Boseken J., 155—157, 178
Boswijk К. Н., 133, 172, 179, 204,
213,332
Boudrimont E., 176
Boy R., 18, 177, 271
Bozorth R. M., 318, 341
Bradfield A. E., 129, 147, 179
Bradley D. C, 357
Braida A., 46—49, 51—54, 73, 81,
84, 85, 91, 105, 107
Brauer G., 118, 120, 121, 126
Braune H., 87, 107
Brenken O., 178
Brieger H., 177
Briggs T. R., 357
Brinker К. С, 194
Britton H. G., 194
Britton H. T. S., 194, 361, 380
Broderson P. H., 81, 82
Brown J., 126
Brunei L., 179
Bruner L., 14, 18, 170, 176, 177, 182,
194, 202, 205, 212, 213, 218, 221—
223, 225—227, 230, 233, 234
Brown W. G., 186, 188, 195
Buchta F., 178
384
Buckles R. E., 116, 126, 231, 232,
235, 287, 317
Bunnett J. F., 18, 28
Burbank R. D., 45
Burke T. G., 44, 76, 77, 82
Burnett R. L., 42, 45
Butler К. Н., 111. 125
Cady H. P., 318
Caglioti V., 178, 275, 317
Cambi L., 358
Campanaro L., 194
Carlton H. A., 108, 125
Caunt A. D., 25, 44
Celis G., 275, 317
Certanescu R., 18, 245, 271
Chargoff E., 235
Charonnat R., 238, 270
Chattaway F. D., 277, 317
Chesman G. H., 283, 318
Christomanos A. C, 142, 162, 17?
Cihalik J., 369, 381
Clark G. L., 317, 341
Cockaday R. E., 380
Cohen A., 381
Cole L. G., 25, 27, 45
Compin L., 358
Cordes H., 136, 176, 188, 195
Cornog J., 19. 130, 134, 135, 139, 145
176, 218, 234, 274, 282, 289, 290,
294, 317, 318, 352, 358
Сох В., 82
Cremer H. W., 18, 125, 273, 276,
282, 284—289, 316—318, 333
Curtis W. E., 176
Danehower R. G., 45
Darbyschire O., 136, 176
Davidson A. L., 358
Davies A., 45
Davy H., 7, 18, 127, 175
Delaby R., 238, 270
Delepine M., 126, 358
Derby J. H., 274, 316
Diehe О. С, 235, 270, 317
Dirschel W., 377, 381
Dittmar M., 237, 242, 270, 317
Dobbin L., 278, 287, 317
Domange L., 23, 44
Doornbosch H. J., 318
Dootson F. W., 270
Dorfurt E., 178, 380
Duane W., 317, 341
Dubsky J. V., 358
DuffeyG. H., 102, 107
Duncan D. R., 18, 125, 273, 276,
282, 284—289, 316—318, 333
Dupuis P., 108, 110, 111, 125, 159,
178
Durie R. A., 18, 50, 82, 105—107
Ebert F., 44
Elliott N., 179
Ellis J. F., 43, 45
Elverum G. W., 45
Emeleus H. J., 18, 19, 30, 38, 45,
54, 55, 57, 58, 61—64, 70, 71,
73,75, 81, 82, 88, 92, 93, 107, 135,
176, 203, 204, 207, 212, 296, 318,
332
Ephraim F., 282, 283, 285, 317
Erlenmeyer E., 155, 178, 375, 376,
381
Evans W. H., 25, 27, 34, 45, 50, 82,
86—88, 101, 106, 107, 136, 122,
126, 136, 179, 188, 195
Fairbrother F., 18, 141, 177, 271, 341
Farber M., 45
Faull J. H., 180, 194, 316, 319
Feigl F., 235
Fergusson A. L., 19, 176
Ferigle S. M., 34, 45
Fernandez M., 238, 243, 270, 317
Fessenden E., 7, 18, 81, 318
Fielding E., 178
Filhol., 275, 317
Fischer G., 274, 317
Fischer J., 81, 82
Forbes G. S., 119, 126, 179, 180,
194,316, 319, 333
Foreman J. K-, 380
Forscey L. A., 117, 126
Forster M. O., 279, 317
Fouseca A. L., 177
Fredenhagen K., 22, 44
Fritz H., 25, 44
Frohlich G., 19, 112, 113, 126
Fuoss R. M., 119, 126, 179, 316,
319, 333
Galecki A., 14, 18, 176, 213, 218,
222, 225—227, 230, 233, 234
Garton F. L., 317
Gaudbhir A. M., 150, 177
Gaydon A. G., 18, 106, 107, 126
Gau— Lussak J. L., 7, 18, 127, 158,
175
Geitner P., 177
Gelber E., 155—157, 178
Gerding H., 179
Gilbert D. A., 18, 44
Gilbert F., 341
Gillam A. E., 114, 125, 178, 180,
194, 216, 217, 234
Glass S. W., 179, 316, 319
Gleu K., 238, 270, 317
Goldstein R., 341
Goodevoe C. F., 81, 107
Gore G., 83, 85, 107, 144
Gore W. L., 177
Gorgas A., 195
25—1419
385
Gosselin A., 168, 178
Gosselin M., 178
Graf W., 229, 235
Gray L. Т. М., 114, 118, 120, 126
Greene W. H., 177
Greenwood N. N.. 5, 18, 24, 27, 34,
44,81, 101, 107, 117, 120, 122, 126,
133-135, 137, 161, 176, 185, 188,
195, 203, 204, 207—210, 212, 213,
296, 318
Grimm H., 140, 176, 189
Grisard J. W., 31, 34, 45, 54, 81
Griswold P. A., 18, 44
Gunnar K., 357
Guson J. D., 178
Gustison R. A., 45
Gutmann V., 62, 81, 130, 142, 143,
177, 182, 189—191, 194, 195, 198,
201, 290, 291, 293, 312—315, 318,
319, 341
Haendler H. M., 82
Halim F. M., 357
Hannay J. В., 133, 142, 143, 176,
182, 194
Hansen R. S., 357
Hanson N. W., 116, 126
Hantsch A., 125, 126, 229, 235, 279,
. 280, 317, 333
Hanus J., 194, 195
Harris R. L., 179
Haskell M. N.. 177
Hassel O., 239, 272
Haszeldine R. N.. 43, 81, 107
Hawking N. J., 126
Hayes F. N., 179
Hecht L., 176, 234, 317, 357
Heide J., 179
HeinF.,334,338, 341
Heinzelmann A., 27, 44
Heisig G. В., 359, 380
Helderman W. D., 194
Hennig H., 107
Hepworth M. A., 82
Hetherington G., 81, 86, 107
Hildebrand J. H., 266, 268, 271, 272
Hinner W., 381
Hirschfelder J. O., 176
Hochnel M., 270
Hofer L. E., 177
Holde D., 194, 195
Homiller E. D., 177
Hoppe J. O., 177
Horrabin H., 19, 176, 317
Hoyle G., 317
Huckel W., 40, 45
Hulburth H. M., 176
Hummers W. S., 357
Hyman H., 58, 82
Ilosway L., 178
Ingle H., 156, 157, 178
Ingold Ch. K., 152, 178
Iredale T. D., 177
Jackson С L., 274, 316
Jacobs A., 357
Jaeger M. F., 318
Jaffe J., 45
Jagemann W., 238, 270, 317
James Th. C., 116, 126
James J. W., 126
Jeep K., 179
Jellinek К., П8, 120, 126
Jessop G., 173, 179
Jessup J. N., 287, 317
Job P., 126
Jones E. A., 44, 45, 76, 77, 82
Jones В., 147, 179
Jost D. M., 118—120, 121, 122, 124,
126, 134—137, 140, 144, 161, 176,
177, 178, 185, 186, 188, 189, 195
Jost W., 19, 112—114, 118, 120,
121, 124, 126,
Junker F., 178, 234
Kammerer H., 18, 83, 107
Karges G., 19, 130, 134, 135, 139,
• 176, 290, 294, 317, 318
Karsten B. J., 127, 134, 110, 111,
118, 125, 176
KatzJ. J., 58, 82, 101, 107, 321, 322,
333
Katz S., 29, 44, 45
Keefer R. M., 146, 177, 217, 234,
268, 270, 272
Keim R. F., 18, 82, 84, 85, 96—
101, 103, 107
Kerrigen V., 81
Kimball G. E., 88, 107
Klein A., 271, 317
Klemm W., 12, 18, 211, 213
Kohn M., 271, 317
Kolthoff J. M., 217, 234, 254, 272,
345, 357
Kortum G., 271
Kothare A. N., 177
Kraft G., 18, 237, 270, 317
Kraft O. Th., 22, 44
Krug H., 22, 24, 30—32, 38—40, 44
Krugt H. R., 194
Krutwig J., 108, 125, 178
Kubina H., 359, 380
Kukin J., 40, 45
Knudson G. E., 39, 4Й
Kwasnik W., 22, 31, 53, 76, 85,
100, 107
Laar J., 194
Laas F., 22—24, 29, 44
386
Labat A., 180, 194
Lafferty R. H., 178
Lambourne L. L., 18, 148, 149, 177,
193, 195
Landolt-Botnstein, 45
Lang R., 195, 361, 380
Lawdermilk F. R., 45
Lebeau P., 18, 20, 44, 52, 53, 57,
73, 81, 110, 111, 125
Lee T. S., 234, 272, 357
Leech H. R., 31, 42, 44, 45
Lenssen E., 178
Levine S., 45
Lewis G. N., 269, 272
Lewis T. R., 177
Li N.. 144, 177
Ligett W. В., 74, 82
Liimatainen R., 82
Lindgren V. V., 74, 82
Lindquist F. С, 177
Loewenthal J., 178
Long J. W., 116, 126
Lord R. C, 88, 101, 107
Lowry T., 341
Luder W. F., 345
Ludewig W. H., 19, 82
Luft K. F., 18, 133, 176
Lux H., Ill, 125
Lynch M. A., 88, 100, 104, 105, 107
Maddock A. G., 82
Magat M., 179
Magnuson D. W., 18, 35, 36, 45
Makuc J., 318
Malatesta L., 358
Malik J. G., 19
Malone M. C, 19, 176
Mann Gy., 160, 178
Mannion J., 120, 121, 126
Margosches В. М., 377, 381
Martin A., 82
Martin J. H., 283, 318
Masson O., 317
Mattraw H. C, 120, 126
Mayo S., 82
McArthur R. E., 107
McBee E. Т., 74, 82
McElsey H., 318
Mcintosh D., Ill, 125, 235
MclvorR. W. E., 163, 178
McMorris J., 134—137, 176, 185,
186, 188, 195
Medock J. J., 82
Meerum—Terwogt P. С. Е., 12, 18,
181—185, 194
Mellor D. P., 177
Mellor J. W., 175
Menzel W., 24, 44, 46, 52, 75, 76,
78— 80, 81
Merrit F. R., 18, 176
Michael A., 146, 152, 177, 178, 359,.
380
Militzer W., 192, 193, 195
Milles G. L., 82
Millor H. C, 45
Mills J. F., 231, 232, 235, 287 317
Moissan H., 20, 44, 52, 81 84 8r>
91, 92, 97, 107 ' . ° . °o,
Moles E., 176, 220, 234, 302, 318
Mouneyrat A.,' 239, 270
Mooney R. C. L., 318, 341
Morris W. C, 317
Morton R. A., 114, 125, 178 180
194, 216, 217, 234
Moureu H., 179
Muller H., 177
Muller W., 185, 186, 188, 195
Mulliken R. S., 266, 268, 271 272
Munson Tn. R., 25, 27, 45, 86' 101
107, 122, 126, 188, 195
Murray R. В., 45
Musgrawe W. K-, 43, 45
Nadkorny V. V., 177
Neudorffer J., 23, 44
Nichols R., 105, 107
Nielsen A. H., 45
Nies N. P., 161, 178
Norton L. M., 146, 177, 359, 380
Nutting H. S., 73, 81
Oddo G., 176, 178, 208, 213, 234,
320, 332
Ogimachi N.. 268, 272
Olivari F., 357
Oliver G. D., 31, 45, 54, 81
Olson L., 218, 234, 352, 358
Orton K., 179
Ostermayer E., 237, 270, 317
Pachucki C. F., 126
Palmer K-, 179
Palmer G. G., 177
Panisch M. В., 19, 82, 107
Parkinson T. F., 44, 45
Patkowsky J., 176
Pauling L., 140-, 141, 177, 189,
318, 341
Payne D. S., 179
Peacock R. D., 82
Pearson D. E., 193, 195
Pemsler G. P., 45, 322, 333
Peterson D. E., 179
Peterson V., 177
Petrie F. S., 73, 81
Philipp J., 168, 178
Philbrick F. A., 179
Pictet A., 18, 237, 270, 317
Pinkston J. Т., 17, 29-31, 33, 44,
74 81, 107, 200, 201
Pinnow P., 87, 107
387
Poni M., 18, 243, 244, 271
Pope D., 177
PopeaF., 166, 178,277,284,287,317
Popov A. J., 39, 45, 120, 121, 126,
194, 223, 231, 232, 235, 287, 288,
317—319
Potter R. L., 25, 27, 44
Pratt M. G., 177
Prideaux E. B. R., 18, 52, 81, 84,
85, 107
Pruett R. D., 19
Pusin N.. 318
Quam G. N., 234
Rae W., 284, 317
Ray S. K., 276, 317
Reade Т. Н., 194, 318
Rebello F., 177
Reid G., 268, 272
Rheinbold H., 18, 177, 271
Richardson E. N.. 147, 179
Rinchart R. W., 194
Ritter H. L., 179
Roberts A., 18, 44
Roberts J. Ch., 179
Robertson P. W., 18, 148, 149,
156, 177, 178, 193, 195
Robinson P. L., 81, 82, 86, 105, 107
Rochow E. G., 40, 45
Rogers M. Т., 19, 44, 76, 81, 82, 85,
86, 88, 83, 107, 321, 322, 333
Roliefson G. K-, 118, 120, 121, 126,
177
Romming Chr., 239, 272
Rossenheim A., 358
Ross C. J., 193, 195
Rossini F. D., 45, 188, 195
Rudge A. J., 31, 32, 45
Ruff O., 7, 18, 20—24, 27, 32, 38—
40, 44, 46—49, 51—54, 73, 75, 76,
78—82, 83—85, 91, 96—101, 103,
105, 107, 159, 176, 177, 220, 234,
274, 290, 291, 302, 303, 317, 318,
325—329, 333, 350, 357
RuZicka J., 381
Sandonini C, 14, 18,226—228, 230,
235,249,271
Schaeppi J. H., 279, 317
Schafer K., 34, 45
Scheft J., 58, 82
Scherer M. D., 34, 45
Schlegelmilch F., 275, 317
Schmidt A., 185, 186, 188, 194
Schmitz H., 23, 24, 32—35, 44, 45
Schmorr E. H., 318
Schomaker V., 44, 81, 107
Schumacher H., 23, 24, 32—35, 44,
45, 81
Schumb W. C, 100, 104, 105, Ю7
Schiitza H., 118, 120, 126
Schiitzenberger P., 146, 176, 23^
Scott A. F., 18, 107
Scott R. L., 88, 102, 107, 272
Sell W. J., 270
Sengenwald 144, 177
Sharpe A, G., 18, 19, 43, 44, 57, 69
69, 81, 92, 93, 107, 149, 177, 127'
193, 195, 213, 235, 318, 332
Sharpe D. W. A., 82
Schwend E., 177
Sherman A., 144, 177
Sheft J., 58, 60, 66, 82
Sidgwick N. V., 17, 126, 175, 194
234, 325, 329, 330, 333, 334, 341*
Sicre J. E., 82
Simmons C. R., 357
Simons J. H., 97, 107, 173, 179
Simpson M., 126, 152, 177
Singh A., 183, 192, 194, 365, 380
Singh В., 183, 192, 194, 195, 380.
Singh M., 192, 195
Singh R., 380
Skelly N. E., 223, 231, 232, 235, 288,
317, 318
Skrabal A., 178
Skoog F., 195
Slowinski E. J., 88
Slutsky L., 78, 82, 106, 107
Smith D. F., 45, 81, 322, 333
Smith E., 179
Smith H. G., 152, 178
Solly E., 14, 202, 205, 212, 221, 234
Sommerfeld A., 140, 176, 189
Sood K. Ch., 380
Spacu P., 166, 178, 277, 284, 287, 317
SpeirsJ. L., 19, 76, 82, 107
Sponer H., 188, 195
Stanley H., 165, 178
Stein С. Р., 113, 118, 125
Stein L., 19, 82
Steunenberg R. K-, 81
Stephenson C. V., 77, 82
Stocesoca D., 234, 272, 357
Stortenbecker W.,I27, 131, 133, 134,
160, 175, 176, 227, 234
Strecker K-, 133, 176, 182, 194
Style D. W. G., 114, 118, 120, 126
Sullivan E. C, 318
Suzuki H. K., 179
Swensen R. F., 319
Swinehart C. F., 317
Szegoe L., 358
Tamman G., 6, 18
Taylor T. W. J., 117, 126
Terebova K-, 381
Thomas V., 108, 110, 125, 159, 178
Thompson H. В., 19, 107
Thomsen J., 161, 176
388
Thorpe Г. Е. , 176
Thron H., 377, 381
Tealdi M., 176
Tidwell M., 81
Townes С. Н., 18, 176
Trowbridge P. F., 235, 270, 317
Truesdale E. C., 178
Tullar В. Т., 177
Tyrce S. Y., 357
Vavreinova K., 381
Vesper H. G., 118, 120, 121, 126
Victor E., 118, 120, 121, 126
Ville L., 126
Vogel R. S., 19, 82
Voigt A., 174, 179
Vonk С G., 179, 204, 213, 332
Vos A., 179
Wagman D. D., 25, 27, 45, 82, 86, 101,
107, 122, 126, 188, 195
Walker J., 287, 317
Walz H., 271
Wardlaw W., 357
Warhaftig A. L., 24, 25, 44, 81, 107
Weber A., 34, 45
Weidner R. Т., 176
Weinland R.,
Weinland R. F., 26, 275, 317, 338, 341
We Is A. F., 318
Wells H., 274, 275, 317
Werner A., 176
Werner E. A., 279, 317
Wetroff G., 179
Wetzien Z., 317
Wheeler H., 274, 275, 317
White С F., 81, 107
White E. P., 155, 178
Wicke E., 25, 34, 44,45
Wiebenge E. H., 172,' 179,204 213 332
Wiig E. O., 177
Wiys J. A., 154, 178
Willgerodt C, 178
Willians D. M., 238, 239, 266, 271
Williams D. V. P., 81
Williams I. W., 271
Wilson E. D., 133, 136, 176
Wolf E., 178, 380
Wood R. E., 179
Woolf A. A., 18, 19, 30, 38, 45, 54,
59, 62, 70—72, 81, 82, 85, 86,
88, 89, 93—95, 107
Worker E. A., 270
Wright B. D., 18, 176
Wyckoff R. W. C, 317
Yolles S., 357
Zappi E.'V., 238, 243, 244,270,317
Zedner J., 20, 44, 176,234,317,257
Zeise H., 185, 186, 195
Zelezny W. F., 318
ОГЛАВЛЕНИЕ,
Стр.
Предисловие 3
Глава I. Общая характеристика межгалоидных соединений
Литература . . 7
Глава II. Фториды хлора
1. Фтористый хлор C1F . . . .... .... 20
Получение .... .... .... 20
Физические свойства .... .... 23
Химические свойства .... 27
Реакции с простыми веществами .... 28
Реакции с неорганическими соединениями. . . .... 28
Действие на органические вещества .... 29
2. Трехфтористый хлор ClF3 ■ . > . 29
Получение .... 29
Физические свойства . . '. . 31
Химические свойства .... 38
Реакции с простыми веществами .... 38
Реакции с неорганическими соединениями . . .... 39
Реакции с органическими соединениями ... .... 42
Литература ... .... 44
Глава III. Фториды брома
1. Фтористый бром BrF . . . .46
Получение . . 46
Физические свойства . 48
Химические свойства . . . . 51
2. Трехфтористый^бром BrF3 . . . 52
Получение ""....'. . . 52
Физические свойства .... 53
Химические свойства . . 56
Реакции с неорганическими соединениями 57
Фторирование неорганических соединений 59
Реакции с металлами 59
Реакции с окислами 59
Реакции с галогенидами 60
Реакции с солями кислородных кислот 61
Комплексные соединения трехфтористого брома с фторидами других
элементов 63
Применение трехфтористого брома для получения комплексных
фторидов 66
Сольволиз комплексных* фторидов
Комплексные фториды с нитрониевым и нитрозильным катионами 70
390
Реакции трехфтористого брома с органическими веществами
(фторирование органических соединений) 73
3. Пятифтористый бромлВгР5 '. 75
Получение " 75
Физические свойства .... 76
Химические свойства 78
Реакции с простыми веществами 79
Реакции с неорганическими соединениями . 80
Реакции с органическими соединениями -80
Литература 81
Глава IV. Фториды иода
1. Пятифтористый иод JF5 ...... 83
Получение 83
Физические свойства 85
Химические свойства 90
Реакции с простыми веществами 90
Реакции с неорганическими соединениями 91
Комплексные соединения пятифтористого иода 93
Применение пятифтористого иода для получения комплексных
фторидов 96
Реакции с органическими соединениями 97
2. Семифтористый иод JF7 • 98
Получение • 98
Физические свойства 100
Химические свойства *. . . ... . 103
Реакции с простыми веществами .... ... . .103
Реакции о. неорганическими соединениями ... .103
Реакции "с органическими соединениями . ... 104
3. Низшие фториды иода (фтористый иод) .... ... .105
Литература ... 107
Глава V. Хлористый бром BrCl
1. Попытки получить хлористый бром или его гидраты 108
2. Физико-химическое исследование системы бром—хлор (в отсутствие
растворителя) ПО
3. Оптические исследования системы бром — хлор . 113
4. Изучение реакций смесей хлора и брома с органическими веществами 115
5. Физические свойства и термодинамические константы 117
6. Химические свойства 124
Литература . 125
Глава VI. Хлориды иода
1. Хлористый иод JC1 127
Физико-химический анализ системы иод — хлор . 127
Получение 128
Физические свойства 131
Упругость пара 134
Термическая диссоциация 134
Теплота образования 136
Термодинамические свойства }3^
Молекулярное состояние и молекулярная структура . 139
Структура и полярность молекул 140
Химические свойства 142
Реакции с простыми веществами 142
Реакции с неорганическими соединениями 144
Реакции галоидирования органических соединений . 146
391
Реакции присоединения хлористого иода к непредельным
органическим соединениям с двойной связью 152
2. Треххлористый иод JC13 158
Получение 158
Физические свойства 160
Химические свойства • 162
Реакции с неорганическими веществами . 162
Реакции галоидирования органических соединений ... . 164
Реакции с непредельными соединениями .164
Строение треххлористого иода 165
Литература .175
Глава VII. Бромистый иод JBr
Физико-химический анализ системы иод — бром ... 181
Получение 182
Физические свойства 184
Молекулярное состояние и структура молекул . 189
Химические свойства 189
Реакции с простыми веществами 189
Реакции с неорганическими соединениями . . 191
Реакции-с органическими соединениями . . . 192
Литература 194
Глава VIII. Сопоставление некоторых химических свойств
межгалоидных соединений
Литература . . . 201
Глава IX. Электрохимические свойства хлористого,
треххлористого и бромистого иода
Причина максимума на кривых температурной зависимости
электропроводности . . 206
Природа ионов, образуемых галогенидами иода в расплавленном
состоянии 207
Продукты электролиза межгалоидных соединений 208
Потенциал разложения 209
Природа электропроводности галогенидов иода 210
Сравнение электропроводностей хлоридов иода и хлоридов
некоторых других элементов 211
Литература 212
Глава X. Неводные растворы галогенидов иода
Цвет растворов хлористого и бромистого иода 214
Молекулярное состояние галогенидов иода в неводных растворах . . . 217
Электрохимические свойства растворов галогенидов иода 221
Электропроводность 221
Потенциал разложения 223
Природа ионов в неводных растворах галогенидов иода и процесс
электролиза 224
Литература 234
Глава XI. Комплексные соединения галогенидов иода (JC1,
JBr, JC13) с органическими веществами
1. Комплексные соединения хлористого и бромистого иода с аминами . . 237
2. Соединения хлористого и бромистого иода с кислородсодержащими
основаниями 242
392
3. Соединения треххлористого Иода с аминами t t 043
4. Физико-химическое исследование систем хлористый (бромистый) иод —
органические основания и образующихся в них комплексных соедине
ний м 2.15
а. Система хлористый иод — пиридин " ' 24*б
б. Системы хлористый (бромистый) иод—пиридин (хинслин, диоксан)
в органических растворителях 249
Криоскопические исследования 251
Измерение диэлектрических постоянных и поляризации . . . 252
в. Системы хлористый (бромистый) иод — амиды кислот .... 255
5. Основные типы комплексных соединений галогенидов иода (JC1, JBr,
JCI3) с органическими веществами, их строение и химическая природа 261
а. Соединения хлористого и бромистого иода 261
б. Соединения треххлористого иода 266
Литература . 270
Глава XII. Комплексные соединения хлористого, бромистого и
треххлористого иода типа двойных галогенидов
1. Методы получения 273
а. Непосредственное соединение галогенидов иода с другими га-
логенидами в растворах и расплавах 273
б. Окислительно-восстановительные реакции 274
в. Получение двойных галогенидов в гетерогенных системах . . 276
2. Основные формы двойных галогенидов хлористого, бромистого и
треххлористого иода 276
3. Некоторые физико-химические свойства двойных галогенидов иода . . 280
4. Химические свойства и реакции двойных галогенидов иода 288
5. Физико-химическое исследование систем хлористый (бромистый)
иод — галогениды других элементов и комплексных соединений,
образующихся в этих системах 290.
6. Реакции образования комплексных хлоридов в хлористом иоде как
растворителе 312
Литература 317
Глава XIII. Молекулярные и ионные формы межгалогенидов и их
комплексных соединений
Литература . . . 332
Глава XIV. О химической природе комплексных соединений
межгалогенидов с галогеьидами других элементов
Литература .... ... .... 341
Глава XV. О методах изучения комплексных соединений
в неводных системах
1. Физико-химический анализ систем по нескольким свойствам .... 341
2. К вопросу о влиянии растворителя на процесс комплексообразования 346
3. Изучение химических реакций, происходящих в исследуемой системе 349
4. Физико-химическое исследование систем 352
5. О сочетании физико-химического анализа с синтезом комплексных
соединений при изучении процессов комплексообразования 352
6. Изучение физических и химических свойств синтезированных
комплексов в индивидуальном состоянии и в растворах 355
Литература 356
393
Глава XVI. Применение солянокислых растворов хлористого и треххлористого
иода в количественном анализе (йодхлорометрический метод объемного анализ)
1. Приготовление титрованных солянокислых растворов хлористого и
треххлористого иода 361
2. Формы применения хлористого и треххлористого иода в, объемном
анализе 363
3. Реакции окисления • 365
4. Реакции иодирования органических соединений 370
5. Реакции присоединения галогенидов иода к непредельным соединениям 374
6. Реакции комплексообразования 378
Заключение 379
Литература 38©
Фиалков Яков Анатольевич
Межгалоидные соединения
Печатается по постановлению ученого совета
Института общей и неорганической химии АН
УССР
Редактор издательства К. И. Лабезник
Технический редактор Е. К. Сиваченко
Корректоры В. Е. Дементьева, Л. А. Марченко
БФ 21611. Зак. 1419. Изд. № 9. Тираж 2000.
Формат бумаги 60y92i/1ft. Печ. листов 24,75.
Учетно-издат. листов 28,31. Бум. листов 12,375.
Подписано к печати 3/ХН 1958 г.
Цена 15 руб. 65 коп.
Типография Идательства АН УССР, Киев,
Репина, 2.
Стр.
22
31
39
49
86
102
116
129
Строка
5 св.
12 сн.
15 св.
15 сн.
14 св.
И ей.
Схема
17 св.
ОПЕЧАТКИ
Напечатано
102
—11,75°
треххлористого
температуру около
г|==т(1 + а; + в/2)
NdF*~
CH3CONH2Br +
> NC1+2HJ = NH+HC
■1+J
Следует читать
- 102°
11,75°
трехфтористого
температуру кипения около
т],= Ъ/(1 + А* + В/2)
NbFf"
BrCl
> NCl+2HJ=>NH-fHCI+J
1419