/










































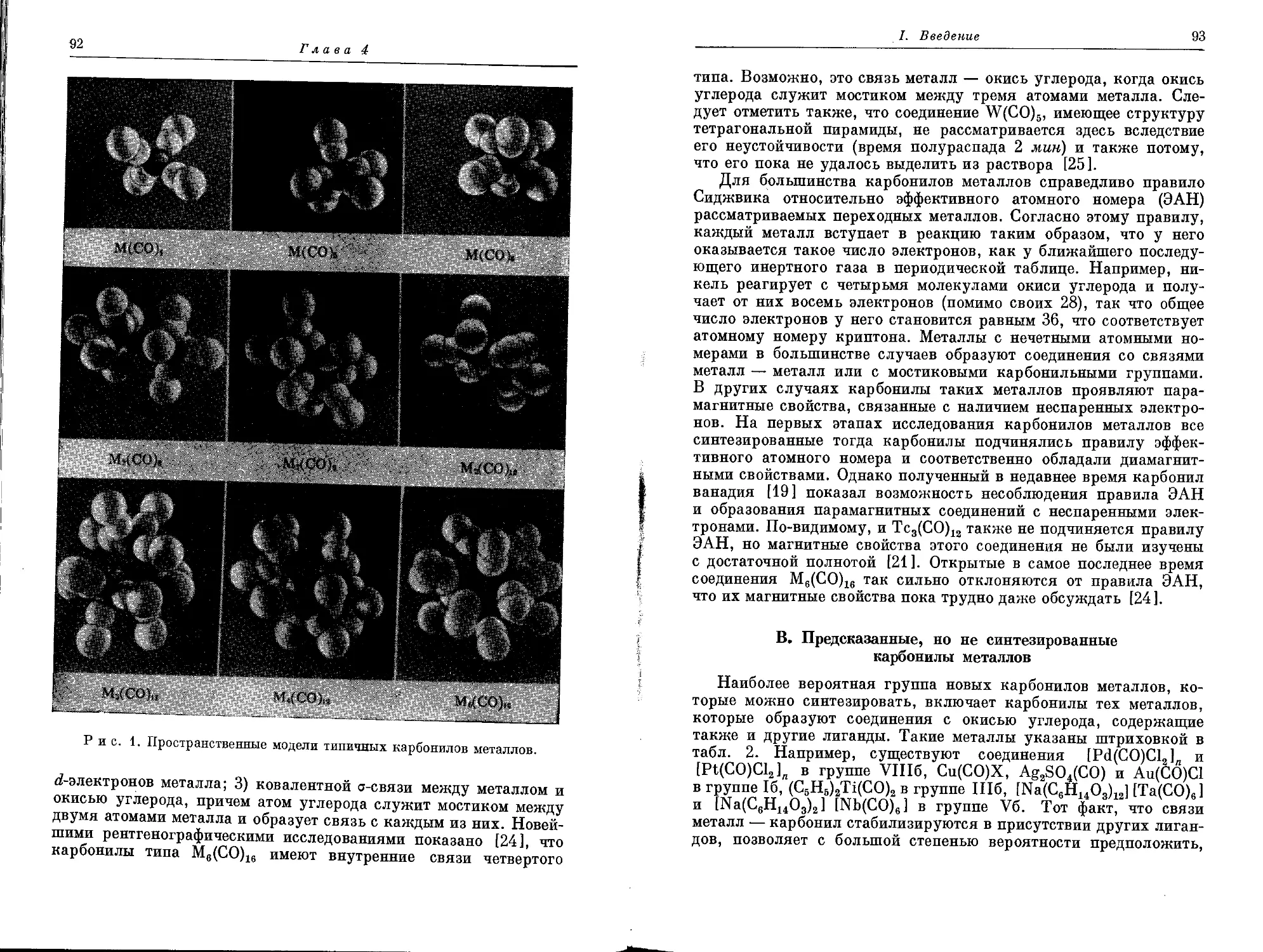


























































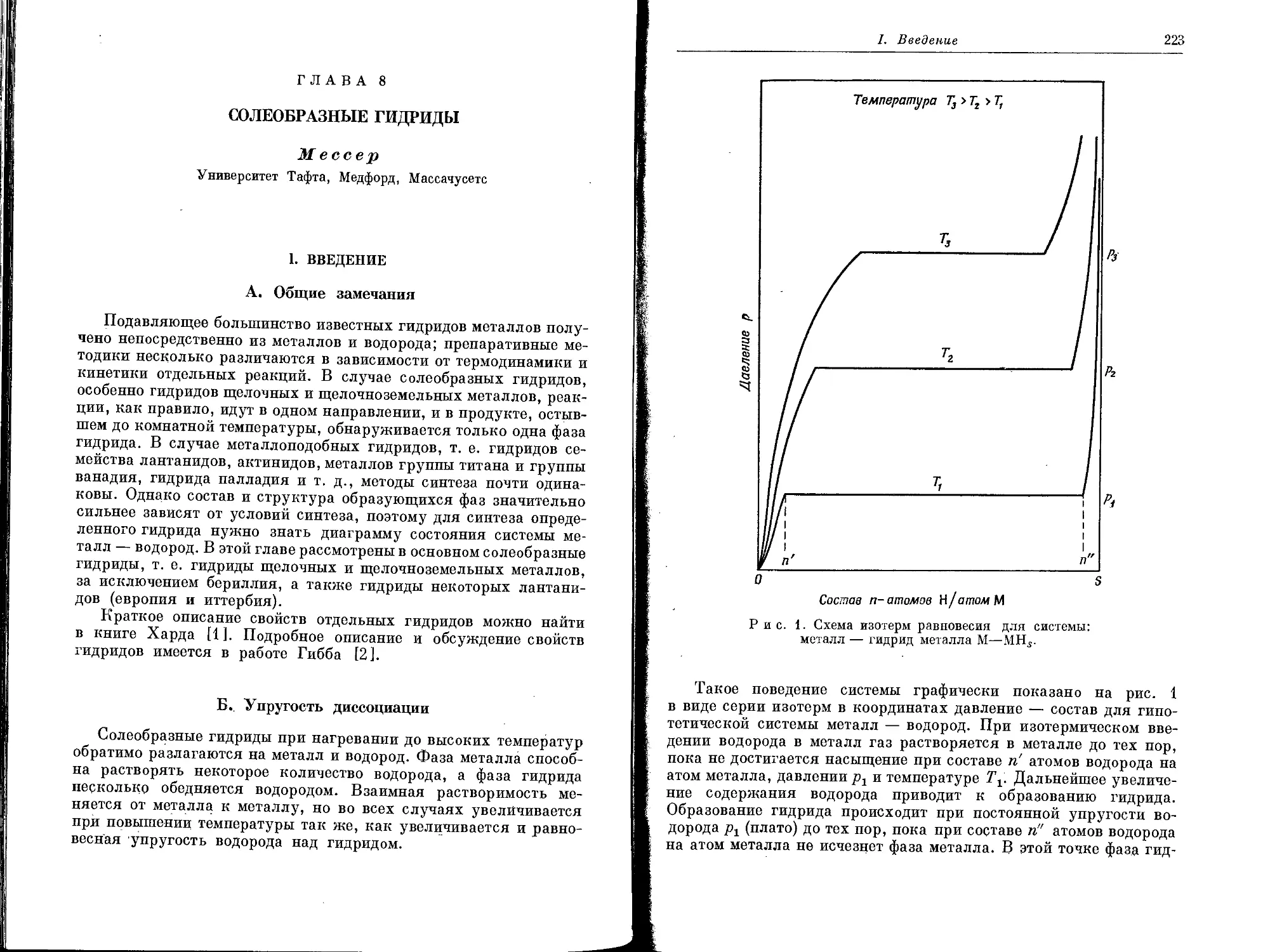









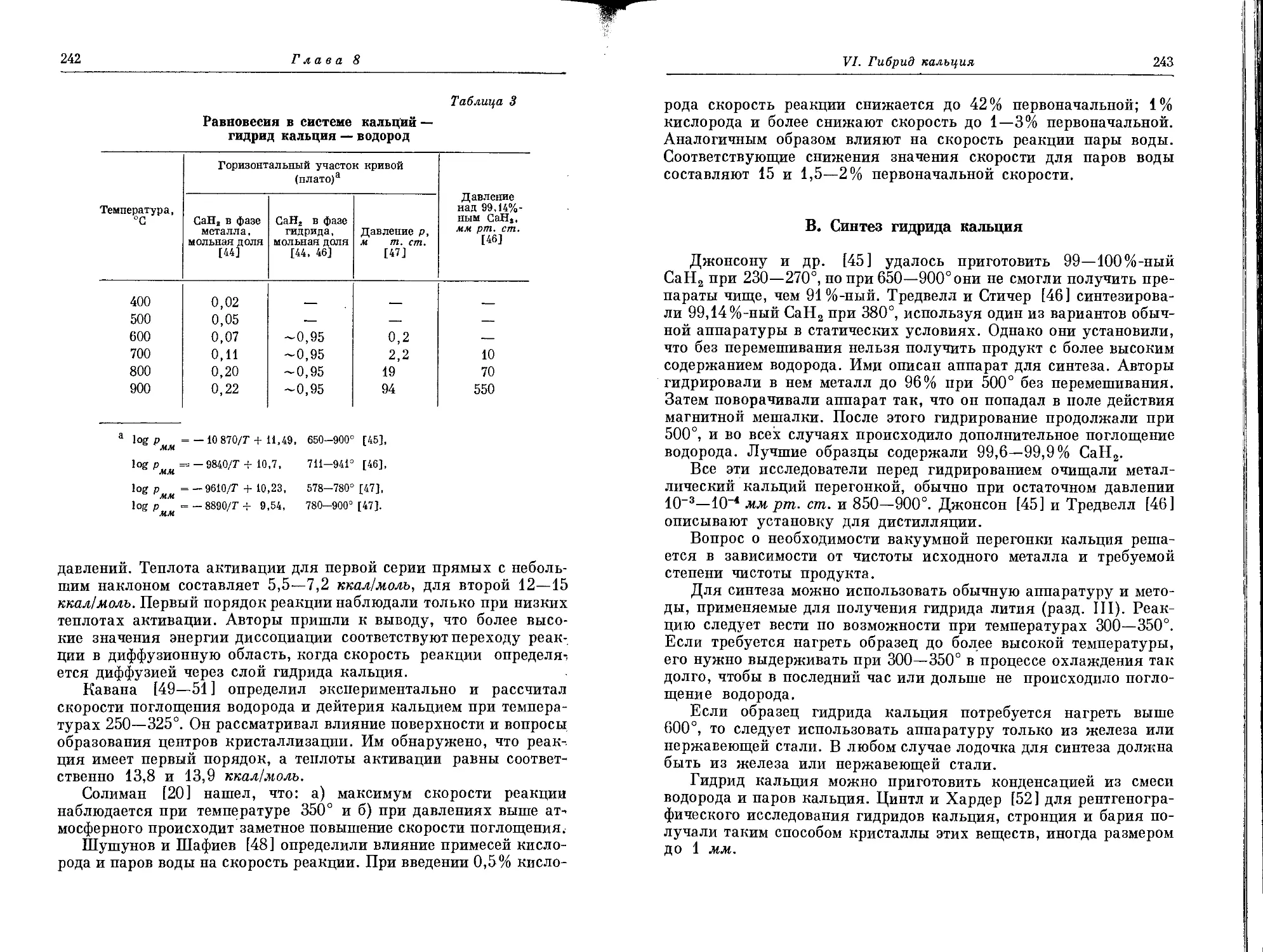



















Текст
СИНТЕЗЫ
НЕОРГАНИЧЕСКИХ
СОЕДИНЕНИЙ
том
I
Больше химической литературы на
vk.com/chemzone
More chemistry books you can find on
vk.com/chemzone
vk.com/chemzone
PREPARATIVE INORGANIC REACTIONS
VOLUME
I
Editor
WILLIAM L. JOLLY
Department of Chemistry University of
California Berkeley, California
1964
INTERSCIENCE PUBLISHERS
a division of John Wiley & Sons
New York»London»Sydney
СИНТЕЗЫ
НЕОРГАНИЧЕСКИХ
СОЕДИНЕНИЙ
Под редакцией
У. ДЖОЛЛИ
том
I
Перевод с английского
Канд. хим. наук А. Д. ВЛАСОВА
канд. хим. наук А. И. ЗАРУБИНА
Под редакцией
академика И. В. ТАНАНАЕВА
ИЗДАТЕЛЬСТВО «МИР»» МОСКВА 196в
УДК 546
Этой книгой начинается новая серия, посвященная
методам синтеза различных классов неорганических
соединений. В отличие от других книг по препаративной
неорганической химии это издание представляет собой
не простое описание препаративных процедур; в нем
дается теория, лежащая В основе метода, и приводится
критическая оценка различных методов синтеза, которая
позволяет в каждом отдельном случае выбрать наиболее
подходящий метод. Этому способствует высокий научный
уровень изложения материала, так как каждая глава
написана видным специалистом в соответствующей об-
ласти синтеза.
Книга предназначена для широкого круга химиков-
неоргаников — работников научно-исследовательских ин-
ститутов и промышленных предприятий.
Инд, 2-5-2
Редакции литературы по химии
ПРЕДИСЛОВИЕ
Предпринятое под редакцией У. Джолли издание по препара-
тивному неорганическому синтезу по своему характеру существен-
но отличается от ранее издававшихся руководств подобного рода.
Так, в книге уделено преимущественное внимание новым классам
неорганических веществ, данные о синтезе которых относительно
мало известны и не обобщены в отдельных изданиях. Сюда отно-
сятся, например, координационные полимеры, карбонилы метал-
лов, безводные нитраты, соединения серы с азотом и фтором, ги-
погалиты и др. Сведения о них разрознены и опубликованы в перио-
дической литературе.
Авторы, как правило, не загромождают текста отдельными кон-
кретными прописями применительно к синтезу того пли иного со-
единения, а приводят общие и теоретические сведения, относящиеся
к рассматриваемому классу в целом. Таким образом, во многих
случаях необходимые условия синтеза могут быть подобраны са-
мим эксперпментатором; соответствующие рекомендации даются
авторами обычно либо в виде примеров, либо в виде прописи для
наиболее сложных и ответственных синтезов. Естественно, что при
таком подходе к изложению авторам удается заинтересовать чи-
тателя и способствовать развитию у него нужных знаний, позво-
ляющих отдельные работы по синтезу проводить в значительной
мере самостоятельно.
В последующих выпусках круг синтезируемых объектов будет
расширен за счет других, в основном новых классов неорганиче-
ских соединений.
И. Тананаев
'I
ПРЕДИСЛОВИЕ РЕДАКТОРА
АМЕРИКАНСКОГО ИЗДАНИЯ
Среди большого числа сведений, которые может сообщить хи-
мик-специалист относительно синтеза того или иного класса сое-
динений, наиболее важным является теория, или то рациональ-
ное, что лежит в основе методов их синтеза. Неспециалист соот-
ветствующие сведения может, вероятно, использовать трояким
образом: он может использовать их для получения еще не синте-
зированных соединений того же класса или же для синтеза совер-
шенно иного класса соединений. При наличии же творческого во-
ображения он может расширить или улучшить саму идею синтеза
и разработать новые принципы, которые приведут коновым мето-
дам синтеза.
В высокоразвитых областях химии принципы синтеза достаточ-
но хорошо установлены, так что они могут быть относительно лег-
ко распространены на новые соединения. В этих областях трудно
усовершенствовать принципы и методы синтеза. С другой стороны,
в новых, малоразвитых областях химии теоретические основы син-
теза представляют собой немногим больше, чем простые рабочие
гипотезы, которые усовершенствуют и видоизменяют по мере на-
копления опыта.
Критический обзор различных методов синтеза индивидуаль-
ных соединений представляет собой иной тип информации, кото-
рую может дать опытный химик. Такие обзоры помогают в выборе
препаративного метода, наиболее отвечающего поставленным
целям.
Таков характер информации, которую предполагается поме-
щать в «Синтезах неорганических соединений». Я полагаю, что
авторы 1-го тома успешно справились со своей задачей.
Уильям Л. Джолли
ГЛАВА 1
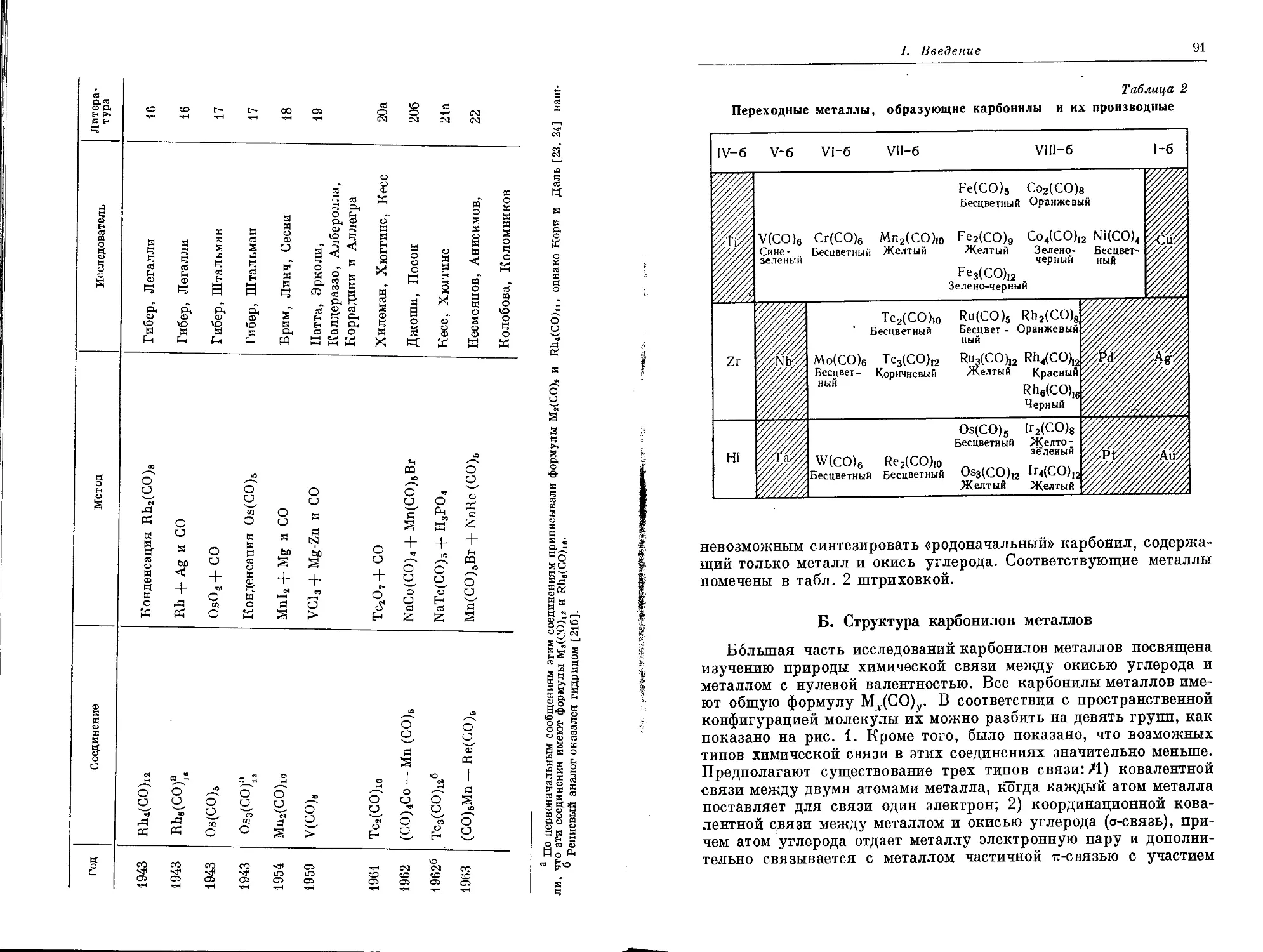
КООРДИНАЦИОННЫЕ ПОЛИМЕРЫ
Бейлар, мл.
Иллинойсский университет, Урбана, Иллинойс
I. ВВЕДЕНИЕ
Для большинства людей слово «полимер» означает пластиче-
ское, гибкое или эластичное органическое вещество, каждая мо-
лекула которого образована соединением множества небольших
молекул. Однако это слово в его первоначальном смысле не имеет
отношения к свойствам материала или его составу, а указывает
только на то, что материал образован повторяющимися структур-
ными элементами. Неорганическая химия имеет много примеров
различных видов полимеров, если говорить о полимерах в широ-
ком смысле этого слова: асбест является линейным полимером,
слюда — плоским, а полевой шпат, каолин и кварц — пространст-
венными полимерами. Если это определение расширить еще
более, то даже такие ионные кристаллические вещества, как хло-
ристый натрий или окись магния, следует рассматривать как
полимеры. Такое расширение значения этого термина можно в не-
которой степени оправдать тем, что эти вещества обладают некото-
рой пластичностью, но вообще неблагоразумно слишком расши-
рять смысл определений, так как они при этом перестают точно
соответствовать конкретной группе сходных явлений.
А. Циано-полимеры
Среди синтетических координационных соединений имеется
много примеров полимерных веществ, но в большинстве случаев
их полимерная структура была обнаружена только в последнее
время. Хлорид палладия (II)
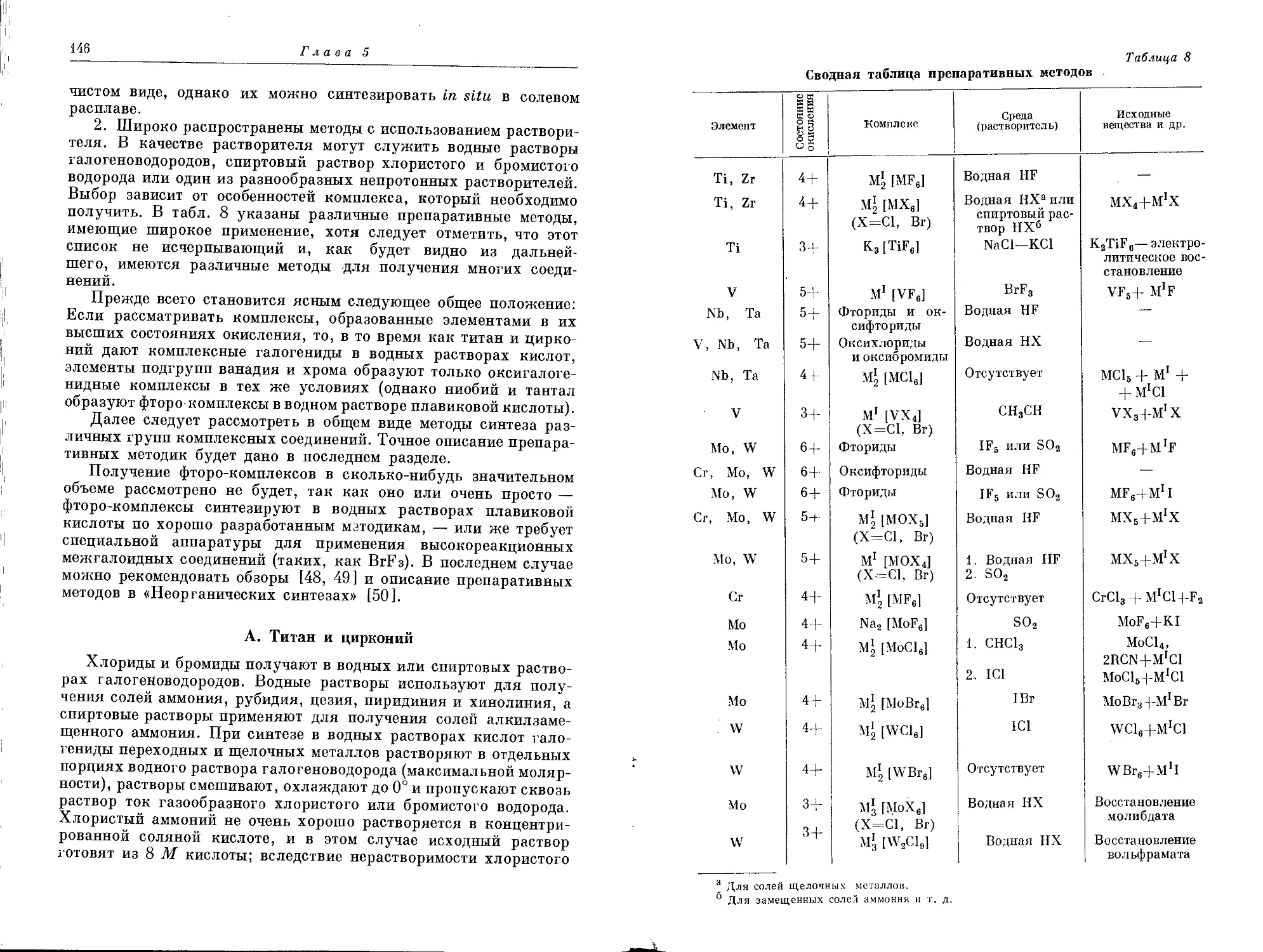
1 Заказ № 547
10
Глава 1
является бесконечным линейным полимером, цианид никеля(П)
=N— Ni—N=C-Ni—teN- Ni—Ne=
является плоским полимером [1, 21, а ферро- и феррицианиды тя-
желых металлов, в которых железо обладает координационным
числом 6, являются бесконечными пространственными полиме-
рами [3]. Каждое из этих веществ имеет интересные и полезные
свойства, но они, как правило, не обладают пластичностью, гиб-
костью или эластичностью. Представляется маловероятным, что-
бы чисто неорганические координационные соединения могли об-
ладать такими свойствами в сколько-нибудь значительной степе-
ни. Валентные связи в таких соединениях жестко ориентированы,
а группы, соединяющие атомы металлов, относительно невелики.
Следует отметить, что в цианиде никеля чередующиеся атомы
никеля имеют неодинаковое окружение, и можно ожидать, что они
будут обладать разными свойствами. Каждый атом никеля, окру-
женный атомами азота, способен присоединить еще две молекулы
аммиака, что увеличивает его координационное число до 6; атомы
никеля, окруженные атомами углерода, не обладают этим свойст-
вом. Молекулы аммиака в аммиакатах располагаются по обе сто-
роны плоскости полимера и раздвигают параллельные плоскости,
благодаря чему появляется возможность образования клатратных
соединений, включающих бензол [1, 2]. Состав клатрата прибли-
жается к Ni(CN)2NH3-CeHe, но в действительности вещество
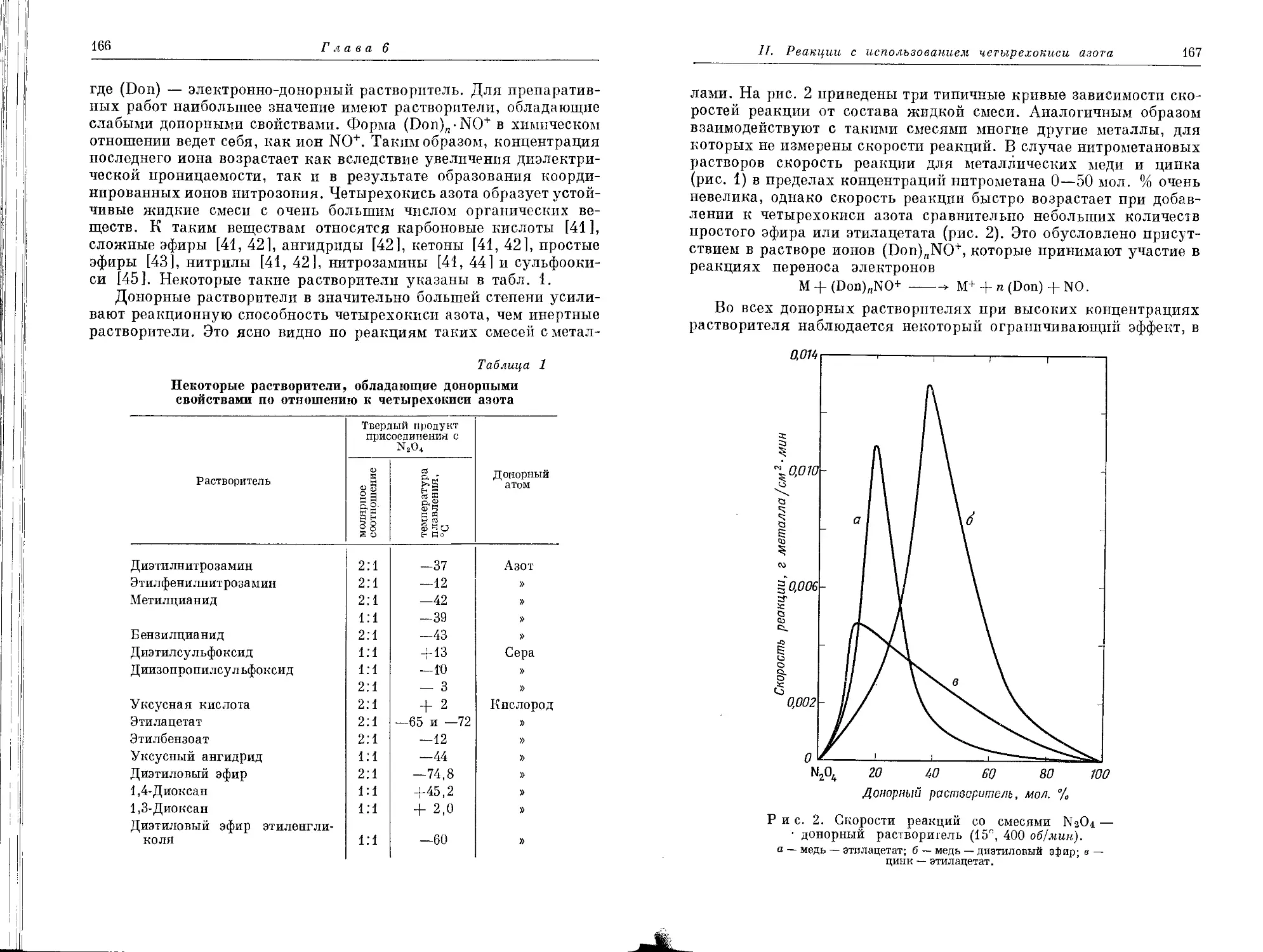
обычно содержит менее 1 моля бензола на каждый моль никеля
[4]. Клатрат легко можно приготовить, встряхивая суспензию
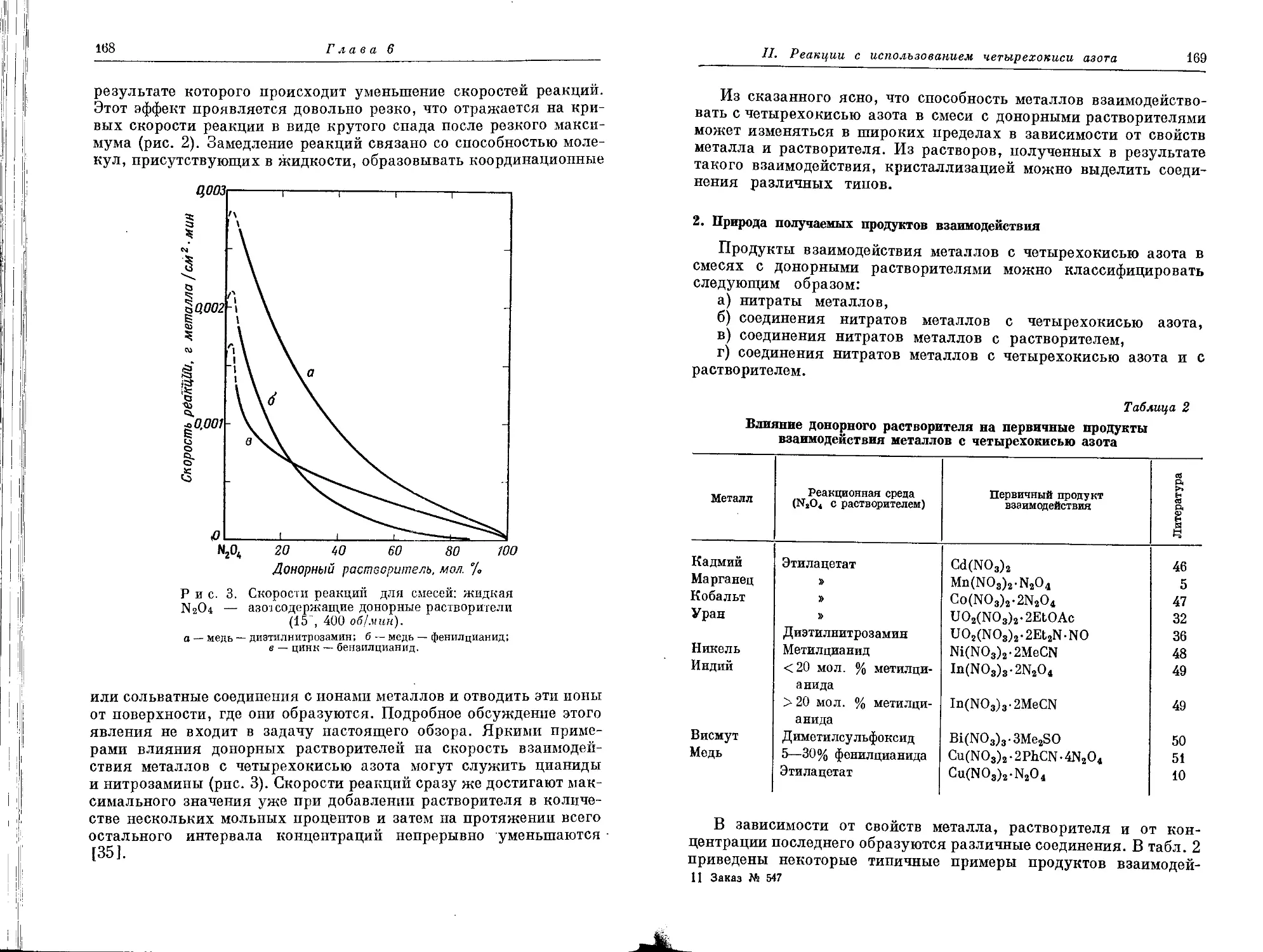
цианида никеля в смеси водного раствора аммиака с бензолом.
Пространственные полимеры берлинская лазурь и турнбулева
синь интересны вследствие их глубоко-синего цвета, который яв-
ляется результатом абсорбционного взаимодействия. Половина
атомов железа в этом полимере находится в двухвалентном состо-
янии, остальные — в трехвалентном. Состав этого соединения
близок к KFeFe(CN)e. Его можно приготовить простым смешива-
нием водных растворов железа(Ш) и гексацианоферрата(П)
/. Введение
11
(ферроцианида) или растворов железа(П) и гексацианоферрата(Ш)
(феррицианида). В этом случае чередующиеся атомы железа ок-
ружены атомами углерода или атомами азота. Несомненно, в
этом соединении легко происходит взаимное окисление и восста-
новление железа, однако следует отметить, что атомы железа,
окруженные атомами азота, склонны оставаться в трехвалентном
состоянии, а атомы железа, окруженные атомами углерода,— в
двухвалентном состоянии, так как координация атомами углерода
стабилизирует более низкие окислительные состояния металлов.
Б. Гидроксо-полимеры
Другой большой класс координационных полимеров составля-
ют гидроокиси металлов, в которых гидроксильная группа слу-
жит связующим мостиком между ионами металла. Потенциомет-
рическим титрованием установлено, что эти соединения обычно
содержат двойные мостики [5], но известны также одинарные и
тройные мостики. Если в системе присутствуют подходящие бло-
кирующие группы, полимеризация может остановиться на ста-
дии димера [6]
[Со (NH3)4 (Н2О)2]з+ + ОН- -> [Со (NH3)4 (Н2О) ОН]2+ ->
г А г
-> (NH3)4Co/ >Co(NH3)4
*- н J
Первая стадия реакции осуществляется в водных растворах, вто-
рая — при нагревании сухой соли до 150°, когда отщепляются
молекулы воды.
В случае акво-ионов, содержащих более двух молекул воды,
процесс оляции может продолжаться до образования ионов высо-
кого молекулярного веса. Этот процесс идет самопроизвольно при
действии оснований на акво-ионы металла в водных растворах и
может продолжаться вплоть до начала выпадения продуктов
реакции в виде осадка. Рост полимера может продолжаться даже
после осаждения, особенно если осадок выдержать в маточном рас-
творе при несколько повышенной температуре. Можно также ос-
тановить рост полимера до начала выпадения осадка. Такого рода
1*
12
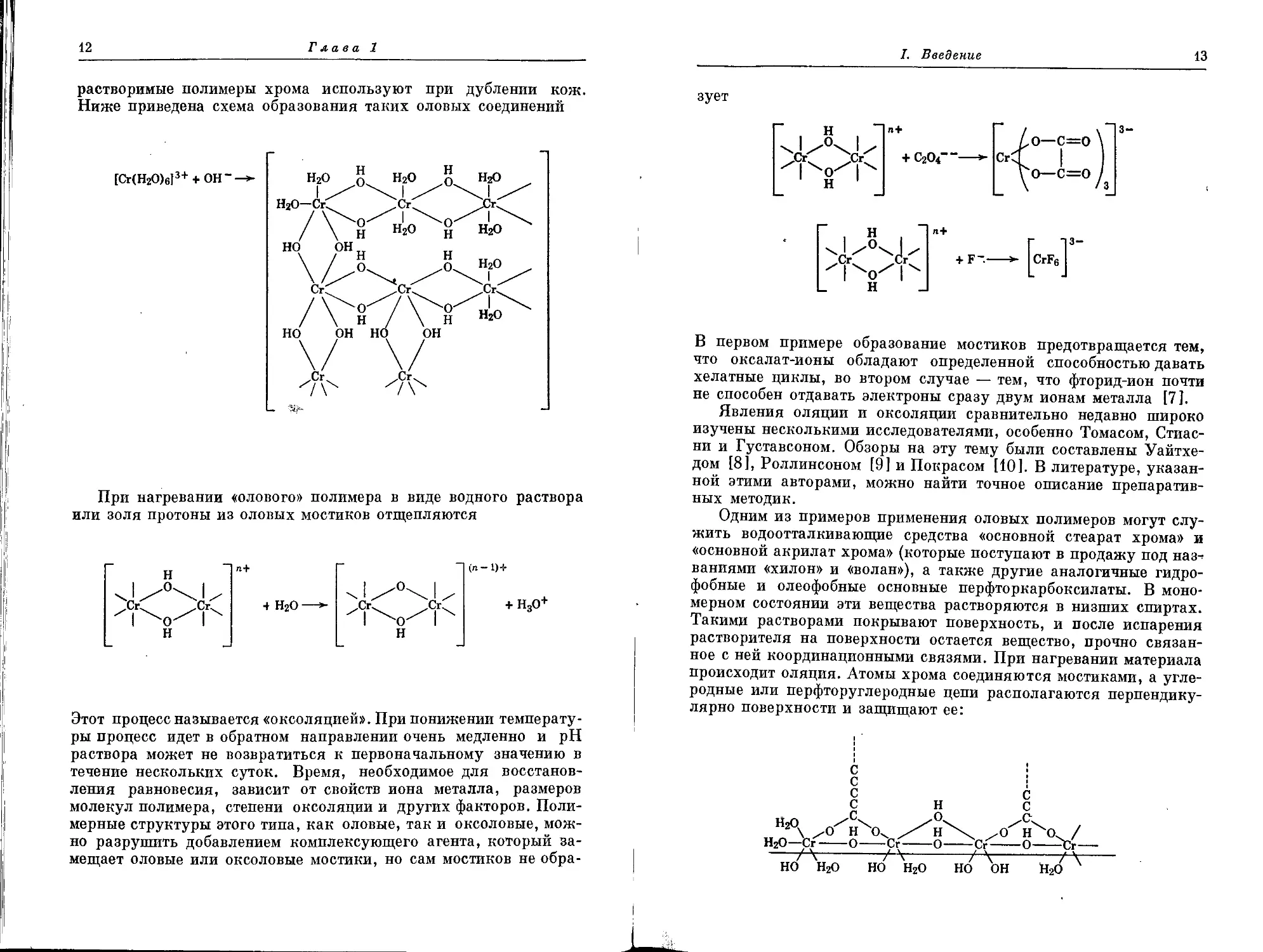
Глава 1
растворимые полимеры хрома используют при дублении кож.
Ниже приведена схема образования таких оловых соединений
[Сг(Н2О)6]3++ ОН~^-
При нагревании «олового» полимера в виде водного раствора
или золя протоны из оловых мостиков отщепляются
Этот процесс называется «оксоляцией». При понижении температу-
ры процесс идет в обратном направлении очень медленно и pH
раствора может не возвратиться к первоначальному значению в
течение нескольких суток. Время, необходимое для восстанов-
ления равновесия, зависит от свойств иона металла, размеров
молекул полимера, степени оксоляции и других факторов. Поли-
мерные структуры этого типа, как оловые, так и оксоловые, мож-
но разрушить добавлением комплексующего агента, который за-
мещает оловые или оксоловые мостики, но сам мостиков не обра-
I. Введение
13
зует
В первом примере образование мостиков предотвращается тем,
что оксалат-ионы обладают определенной способностью давать
хелатные циклы, во втором случае — тем, что фторид-ион почти
не способен отдавать электроны сразу двум ионам металла [71,
Явления оляции и оксоляции сравнительно недавно широко
изучены несколькими исследователями, особенно Томасом, Стиас-
ни и Густавсеном. Обзоры на эту тему были составлены Уайтхе-
дом [8], Роллинсоном [9] и Покрасом [10]. В литературе, указан-
ной этими авторами, можно найти точное описание препаратив-
ных методик.
Одним из примеров применения оловых полимеров могут слу-
жить водоотталкивающие средства «основной стеарат хрома» и
«основной акрилат хрома» (которые поступают в продажу под наз->
ваниями «хилон» и «волан»), а также другие аналогичные гидро-
фобные и олеофобные основные перфторкарбоксилаты. В моно-
мерном состоянии эти вещества растворяются в низших спиртах.
Такими растворами покрывают поверхность, и после испарения
растворителя на поверхности остается вещество, прочно связан-
ное с ней координационными связями. При нагревании материала
происходит оляция. Атомы хрома соединяются мостиками, а угле-
родные или перфторуглеродные цепи располагаются перпендику-
лярно поверхности и защищают ее:
С
С
С
С
С
с
/О Н Ox. / Н /О но/
Н2О—Сг--О---Сг--О---Сг---О---Or
--/А----------------------—АА----
НО Н2О НО Н2О НО ОН Н2О
14
Глава 1
Имеются основательные доказательства того, что в этих полиме-
рах карбоксильные группы, как и гидроксильные, служат мости-
ками между атомами металла.
Шмитц-Дюмон [И] показал, что аммины металлов могут от-
щеплять протоны и молекулы аммиака с образованием полимеров,
формально подобных полимерам, образующимся при процессах
оляции и оксоляции. В этих полимерах мостикообразующими
членами являются группы —NH2—. Основные направления син-
теза таких полимеров рассмотрены в работах [12, 13].
В. Галогено-полимеры
Ионы галогенов проявляют ярко выраженную тенденцию к
образованию мостиков и принимают участие в полимеризации.
Как и в случае гидроокисей, чаще всего встречаются двойные мос-
тики, но известны также одинарные и тройные. Во многих случаях
полимеризация заканчивается на стадии образования димеров и
тримеров, например
Ci сг С1
сТ сК xci
Ск >СЕ .а
Cl— W^-C1-W^C1
сК , ci
но часто образуются и бесконечные полимеры, например PdCl2
и PtCl2. Полимеризация в таких случаях идет самопроизвольно, и,
как правило, оказывается трудным или невозможным выделить
мономерные формы.
Интересные полимерные цепи, образованные «трехвалентной»
платиной с одинарными мостиками, были исследованы рентгено-
графическим методом Броссе [14] и Коэном и Девидсоном [15].
Они содержат чередующиеся атомы Pt(II) и Pt(IV), связанные в
бесконечные цепи
Вследствие абсорбционного взаимодействия эти полимеры окра-
шены в темный цвет. Их можно приготовить обработкой водного
раствора подходящего аммина платины(П) эквивалентным количе-
ством галогена или совместным измельчением эквимолярных ко-
I. Введение
15
личеств сухой тетраамминной соли платины(П) и дигалогентет-
раамминной соли платины(1У)
[PtA,]2 + + [PtA4X2]2+-*+Pt(ii)A4—-X—Pt(iv)A4—Х+——
Несмотря на то что ионы фторида обладают слабой тенденцией
к образованию мостиков, они все же выполняют эту функцию в
комплексе Sn(II)—Ff—Ni(II). При электролизе водных растворов,
содержащих избыток фторида, олово(П) и никель(П) в самых раз-
личных соотношениях, на электроде образуется покрытие в виде
сплава с приблизительно эквимолярным соотношением олова и
никеля [16]. Это покрытие обладает твердостью и подвергается
тонкому шлифованию. В Англии его используют вместо хромиро-
вания. Было показано [17], что в электролите присутствует ком-
плекс, содержащий оба металла в эквивалентном соотношении.
Число фторидных мостиков и форма комплекса неизвестны. Обра-
зование этого иона особенно интересно в связи с тем, что ион ни-
келя сам по себе, по-видимому, не образует комплексного соеди-
нения с фторидом при этих условиях [17].
Г. Тиооксамидо-полимеры
Тиооксамид (рубеановая кислота) и его N-алифатические про-
изводные образуют интересный ряд линейных полимеров. Для
получения таких полимеров органическое вещество смешивают с
водным раствором соли двухвалентного переходного металла и
добавляют эквивалентное количество щелочи. Полимер нераство-
рим в воде и выделяется из раствора в виде порошка
NR=C—SH ,-NR=C—S. ,-NR=C—S'.
MeJ+ + | > Me I Me |
NR=C—SH ' -NR=C—S " ' NR=C—S-''
Образование и свойства таких полимеров изучали Хард с сотруд-
никами [18]. Эти вещества темнеют при нагревании, и поэтому
их используют при копировании и для перевода изображения.
Некоторые из этих соединений (в зависимости от свойств металла
и органической группы) растворяются в неполярных растворите-
лях. Предполагается, что рубеанаты могут быть дезактивирую-
щими агентами для металлов.
Интересный полимер образуется при действии рубеановой кис-
лоты на соли никеля или меди. Этот полимер был изучен Амоном
и Кейном [19], которые формировали его в слое органической
16
Глава 1
пластмассы. По разработанной ими методике лист пластмассы по-
гружают в раствор рубеаната, который пропитывает пластмассу.
Затем лист погружают в раствор, содержащий ионы меди или ни-
келя, в результате в пластмассовом листе образуется рубеанатный
полимер. Если пластмассу затем растягивать в одном направле-
нии, то линейные цепи металл-рубеанатного полимера ориенти-
руются параллельно одна другой, что придает полимеру способ-
ность поляризовать свет.
II. СВОЙСТВА КООРДИНАЦИОННЫХ ПОЛИМЕРОВ
При образовании координационных связей между ионом метал-
ла и лигандом свойства того и другого могут изменяться в различ-
ных случаях по-разному. Часто органический лиганд приобретает
устойчивость по отношению к гидролизу, воздействию химических
реагентов и нагреванию. Например, гидролиз шиффова основа-
ния
протекает значительно быстрее, чем гидролиз хелатного соедине-
ния этого основания с металлом [20]
Этилендиамин быстро разрушается горячей концентрированной
азотной кислотой, но на координационное соединение с кобаль-
том(Ш) азотная кислота не действует даже при нагревании в те-
чение многих часов. бис-Кетоимин
Н3С
НгС
Н3С
:С=О O=Cf
/СН2
£=NCH2CH2N=C\
а СНз
III. Основные структурные закономерности
17
разрушается при слабом нагревании, но его комплексное соедине-
ние с медью
Н3С. ,СН3
о. ^о—сС
НС^ ^-Си\ ^СН
/C=NCH2CH2N=C<
Н3С ^СНз
лишь медленно разрушается при 380° [21 ]. За последнее десяти-
летие было предпринято много попыток придать эти свойства (осо-
бенно термостойкость) органическим пластмассам путем включе-
ния в полимерную цепь координированных ионов металла.
Несмотря на то что ни одна из этих попыток не была вполне ус-
пешной, за это время удалось получить много новых данных, и уже
есть основания полагать, что в конце концов будут получены
новые пластмассы, обладающие ценными свойствами.
III. ОСНОВНЫЕ СТРУКТУРНЫЕ ЗАКОНОМЕРНОСТИ
В препаративной работе по синтезу координационных поли-
меров следует иметь в виду некоторые основные закономерности,
касающиеся структуры и свойств этих соединений. Ниже будут
кратко изложены эти основные закономерности, подробнее они бу-
дут рассмотрены в дальнейших разделах:
1. Непосредственное окружение иона металла не придает или
почти не придает полимеру пластичности, так как обычно связь
металл — лиганд жестко фиксирована. Гибкость или пластич-
ность полимеру могут придать только органические части мо-
лекулы.
2. Ион металла может защитить или стабилизировать только те
участки органического материала, которые находятся в непосред-
ственной близости к нему, поэтому органические части молекулы
полимера должны быть минимальными. Очевидно, этот и предыду-
щий принципы противоречат друг другу, так что здесь следует
искать компромиссные композиции.
3. Термическая устойчивость, устойчивость к окислению и
гидролизу непосредственно между собой не связаны. Например,
один координационный полимер может быть устойчив по отноше-
нию к нагреванию, но гидролитически не устойчив, в то время как
другой может обладать хорошей гидролитической устойчивостью
и плохой термостойкостью. Каждый полимер поэтому можно ис-
пользовать в определенных условиях.
4. Даже те координационные связи, которые обладают наи-
большей степенью ковалентности, все же не чисто ковалентны.
Это значит, что перегруппировки в таких веществах будут про-
18
Глава 1
исходить легче, чем в органических полимерах. Кроме того, ион
металла, даже координированный, несет на себе остаточный поло-
жительный заряд, а лиганд (особенно анион) — остаточный отри-
цательный заряд. Эти заряды создают притяжение между цепями,
что делает полимер более жестким. При разработке координаци-
онных пластмасс следует стремиться использовать материалы, об-
разующие по возможности более ковалентные связи.
5. Соединения, состоящие из ионов, обычно менее пластичны,
чем молекулярные. Межионные силы слишком велики, чтобы ве-
щество обладало пластичностью.
6. Координационные полимеры, состоящие из полимерных ка-
тионов и простых анионов, обычно менее устойчивы по отношению
к нагреванию, чем чисто молекулярные. Многие из обычных ани-
онов сами являются довольно сильными координационными аген-
тами и конкурируют с органическими лигандами за места в коор-
динационной сфере. При повышенных температурах это может
привести к образованию более простых веществ, к деструкции
вещества. Те из обычных анионов, которые не обладают большой
склонностью к комплексообразованию (NO3“, СЮ“4 и т. д.), яв-
ляются сильными окислителями и при нагревании разрушают
лиганды.
7. Образование хелатных циклов увеличивает устойчивость
соединений, и в этих случаях применимы обычные правила отно-
сительно размеров циклов и стереохимических эффектов.
8. Полимер может быть линейным, плоским или трехмерным
в зависимости от соотношения между координационным числом,
стереохимической природой иона металла и природой лиганда,
который присоединяется к иону металла.
9. При синтезе координационных полимеров, равно как и чис-
то органических, необходимо пользоваться чистыми реагентами
и брать их в точно стехиометрических соотношениях, иначе могут
получиться продукты низкого молекулярного веса.
10. Если полимеризацию ведут в присутствии растворителя,
он не должен обладать сильными комплексообразующими свойст-
вами по отношению к иону металла и не должен также выделять
или образовывать реагирующих единиц с сильно выраженными
координационными свойствами, как, например, ОН' в случае при-
менения воды в качестве растворителя.
! За немногими исключениями, работы с координационными по-
лимерами относились к линейным полимерам. Для получения
таких веществ требуется одна из следующих комбинаций реа-
гентов :
а) бис-монодентатный координирующий агент, который не
может образовывать хелатных циклов, и ион металла с координа-
ционным числом 2;
III. Основные структурные закономерности
19
б) бис-бидентатный хелатный агент, в котором две хелатные
группы не могут присоединяться к одному и тому же иону метал-
ла, и ион металла с координационным числом 4;
в) бис-тридентатный хелатный агент, в котором две хелатные
группы не могут присоединяться к одному и тому же иону ме-
талла, и ион металла с координационным числом 6;
г) бис-тетрадентатный хелатный агент, в котором две хелат-
ные группы присоединяются к различным ионам металла, и ион
металла с координационным числом 8;
д) бис-бидентатный хелатный агент (как в пункте «б») и ион
металла с координационным числом 6, две координационные связи
которого блокированы группами, не образующими мостиков.
В любом случае, чтобы удовлетворить изложенным выше тре-
бованиям, нужно тщательно выбирать координационный агент и
ион металла. Во многих случаях большая часть работы по синтезу
координационных полимеров связана с получением органиче-
ской части молекулы. Из четырех возможных комбинаций вариант
«б» изучен больше всех, главным образом вследствие простоты
синтеза. Далее будут подробно рассмотрены примеры полимеров
этого типа и более кратко — других типов.
Большинство полимеров, образованных металлами с коорди-
национным числом 4, содержит в своем составе ионы бериллия,
меди, цинка или никеля. Бериллий обладает некоторыми преиму-
ществами, так как он пе подвергается окислению или восстановле-
нию; его координационное число неизменно равно 4, и связь берил-
лий-кислород приближается к истинно ковалентному типу. С
другой стороны, серьезным препятствием является токсичность
соединений бериллия. Что касается бис-хелатных группировок,
чаще всего используются группировки 1,3-дикетонов, 8-оксихи-
нолинов, шиффовых оснований, фосфинатов и анионов а-амино-
кислот. Почти во всех случаях бис-клешневидные агенты симмет-
ричны, но это лишь вопрос удобства и легкости синтеза. Две хе-
латные группы могут быть соединены любым способом. Например,
в случае р-дикетонов имеются две основные структуры
,R R.
О=СХ /С=О
> ^СН—Y—CH и
О=(Х ^с=о
R' R'
О=С
О=С
R
>С=О
СН2
^^_>С=О
а 8
где R—органический радикал, a Y—связующее звено, например
—СН2—, —СН2СН2— или —С6Н4—. В некоторых случаях две
дикетонные группы бис-хелатного агента связаны непосредствен-
но, например в случае тетраацетилэтана.
20
Глава 1
IV. МЕТОДЫ СИНТЕЗА
А. Полимеры на основе 1,3-дикетонов
Первые исследования полимеров на основе бис-дикетонов были,
очевидно, выполнены Уилкинсом и Виттбекером [22] и продол-
жены Фернелиусом [23], Бейларом [24 ] и Чарльзом с их сот-
рудниками [25], Клуибером и Льюисом [26], Дринкардом, Рос-
сом и Визнером [27 ] и другими. В большинстве ранних работ
пытались получить полимеры теми же методами, которые использо-
вали для получения мономерных комплексов 1,3-дикетонов. По
Оу и Бейлару [24], «метод состоит в смешивании кетона и раство-
римой соли металла в нужной пропорции в водо-органическом рас-
творителе и в нейтрализации раствора до появления осадка. Вслед-
ствие нерастворимости лиганда в воде необходимо добавлять такие
органические растворители, как спирт, диоксан или диметилформ-
амид. Результат синтеза зависит от значения pH, скорости до-
бавления щелочи, скорости перемешивания, температуры и про-
должительности реакции. В некоторых случаях полимер образу-
ется просто при добавлении избытка водного раствора ацетата
металла к раствору бис-ф-дикетона) в диоксане. Нерастворимый
осадок очищают, экстрагируя сначала водой, а затем орга-
ническим растворителем, например спиртом, ацетоном или
бензолом.
«В тех случаях, когда немедленного осаждения не происходит,
pH раствора повышают добавлением аммиака или мочевины.
Если в реакционной смеси даже после длительного выдерживания
не образуется осадка, то смесь медленно выпаривают в вакууме
и остаток собирают. Остаток очищают, экстрагируя сначала во-
дой, а затем органическими растворителями».
На основании химических анализов полученных веществ Оу
п Бейлар показали, однако, что полимеры, которые они получили
таким способом, содержат гидроксильные группы либо в виде оло-
вых мостиков, либо в качестве концевых групп. При наличии та-
ких групп температура плавления полимера повышается, и ве-
щество становится нерастворимым в неполярных растворителях.
В одном из таких случаев в результате реакции между ионом меди
(II) и 1,2-диацетил-1,2-дибензоилэтаном было получено веще-
ство, которое по результатам химического анализа имело следую-
щий состав: 60,94% С, 4,19% Н, 17,68% Си. Теоретический со-
став вещества с формулой (C20HleO4)13 (ОН)2Си14 соответствует
61,17% С, 4,16 Н, 17,48% Си. Продукт реакции не плавится при
250° и не растворяется в неполярных растворителях; он лишь
медленно разлагается при 250°.
IV. Методы синтеза
21
В других случаях, по сообщениям Оу и Бейлара, выпадение
осадка происходит еще до начала образования полимера. При
взаимодействии меди(П) с 1,1,3,3-тетраацетилпропаном они полу-
чили вещество следующего состава: 50,28% С, 6,17% Н, 12,54%
Си. Теоретический состав вещества с формулой (CuH14O4)2Cu-
•2Н2О соответствует: 50,82% С, 6,16% Н, 12,22% Си. Этот ма-
териал размягчается при 140° и растворяется в обычных органиче-
ских растворителях. Он довольно быстро разлагается при 250°.
Интересно, что при температурах ниже 200° наблюдается лишь
небольшая потеря веса, так что молекулы воды, по-видимому,
очень прочно связаны.
Чарльз [25г ] добавлением водного ацетатного раствора меди(П)
к разбавленному раствору лиганда в тетрагидрофуране получил
комплекс меди(П) и тетраацетилэтана.Продукты имели полимерный
характер, и цепи полимера состояли приблизительно из пяти по-
вторяющихся звеньев (м=5). Элементарный анализ и изучение
инфракрасного спектра не показали присутствия воды или гидрок-
сильных групп. Эти полимеры обладали приблизительно такой
же термостойкостью, как и ацетилацетонат меди, но были зна-
чительно менее устойчивы, чем сам лиганд. Однако полимеры маг-
ния, никеля(П) и кобальта(П) обладали несколько более высокой
устойчивостью [25е].
Другой метод, при помощи которого, возможно, удастся прео-
долеть упомянутые трудности, заключается в нагревании ацетил-
ацетонатов металлов с бис-дикетонами и отгонке выделяющегося
ацетилацетона в вакууме. Этим методом Оу и Бейлар получили
полимеры низкого молекулярного веса, т. е. (С17Н18О4)4Ве3, для
синтеза которого использовали кетон 2-фенил-1,1,3,3-тетраацетил-
пропан. Фернелиус [23] этим методом синтезировал комплексы
бериллия и себацилдиацетофенона и получил в различных опытах
полимеры с п, равным 3,8—9,1. Он синтезировал такой же полимер
(м=3,4) нагреванием смеси реагентов с обратным холодильником,
через который удалялся выделяющийся ацетилацетон. Клуибер
и Льюис [26] сочетали две функции дикетонов в одной молекуле
бис-дикетона, в которой две функциональные группы соединены
цепями типа
4 СНг4 > — NH(CH2)^NH — или — СЦСНа)/)—.
Из таких тетракетонов они получили циклические мономеры и
димеры, которые при нагревании перегруппировывались в высоко-
молекулярные полимеры. Вещества со связующими группами
22
Глава 1
-(-снг-)^ показаны ниже:
Мономеры типа I получали из бис-дикетонов, в которых х равно
6 или более, а мономеры типа II — в случаях, когда х равно 6
или менее. Для соединений с х=6 они дают следующие методики
синтеза:
1) Получение циклических мономера и димера из 2,4,11,13-
тетракетотетрадекана. 2,54 г, или 0,01 моля, 2,4,11,13-тет-
ракетотетрадекана и 1,77 г тетрагидрата сульфата бериллия (0,01
моля) перемешивают с 50 мл воды и 10 мл диоксана в течение 1 час,
нейтрализуют едким натром до pH 8 и перемешивают при комнат-
ной температуре еще 16 час. Сырой осадок переносят в пробирку
для сублимации и сублимируют при 200° (0,2 мм рт. ст.) в те-
чение 2 недель. Сублимат кристаллизуется на стенках пробирки
в двух зонах. В нижней зоне (более теплой) собирается 56 мг
вещества с температурой плавления 230° (полимеризуется).
В верхней зоне (более холодной) собирается 379 мг вещества, ко-
торое полимеризуется без видимых признаков плавления при 110°.
Верхняя зона соответствует области, в которой конденсируется
циклический хелатный комплекс бериллия с 2,4,13,15-тетракето-
IV. Методы синтеза
23
гексадеканом. В соответствии с результатами анализа и изучения
инфракрасного спектра, а также по аналогии этому соединению
была приписана структура циклического мономера. Это предпо-
ложение было подтверждено измерениями молекулярного веса
(по понижению температуры замерзания бензола). Измерения
дали величину 227 (расчетная 261). Инфракрасный спектр менее
летучего компонента оказался практически идентичным спектру
мономера, и в соответствии с аналитическими данными, а также
летучестью ему приписана структура димера. Криоскопические
измерения молекулярного веса в бензоле дали величину 555 (рас-
четная 522). Извлеченный из пробирки димер плавился при 215°
(полимеризовался).
2) Полимеризация макроциклов. При нагревании выше темпе-
ратур плавления эти кристаллические макроциклические соеди-
нения самопроизвольно полимеризуются с образованием аморф-
ных (по рентгеноструктурным данным) веществ, которые медленно
растворяются в бензоле. Такие полимеры имеют характеристи-
ческую вязкость в пределах 0,1—2,7, что свойственно полимерам
с высоким молекулярным весом. Высокомолекулярная природа
этих веществ была подтверждена исследованием их механических
свойств. Например, полимер III, где R'=CH3, R=(CH2)8, имеет
характеристическую вязкость 0,9 и обладает гибкостью при ком-
натной температуре (температура перехода в стекловидное состоя-
ние 35°), модулем упругости при 25°, равным 8540 кг!см2, и проч-
ностью на разрыв (100%-ное удлинение в 1 мин), равной 190
кг) см2. Изменение температуры плавления при 198° (ASTM, тест
D 1238—52Т) было в пределах 0,1—2,0 град/мин, что также яв-
ляется признаком высокого молекулярного веса полимера. Анали-
тические данные и инфракрасные спектры полимеров совпадали с
предполагаемой структурой.
Клейн и Бейлар [24] синтезировали полимеры бериллия с
дикетонами совершенно другим методом. Полимеры обладали низ-
ким молекулярным весом, но этот метод должен быть пригоден и
для получения длинных цепей. Их мономерная ячейка соответст-
вует формуле
R. R
Ус—/О—cf
X— ,Ве ^с—X
JX=O o=c:f
R'
где X—группы—СООС2Н3, —ОН,—NH2. Мономеры, содержащие
сложноэфирные группы, превращались в полиэфиры и полиамиды
в расплавах, а мономеры, содержащие гидроксильные или амин-
ные группы, полимеризовались в результате реакции с дифе-
нилдихлорсиланом, терефталоилхлоридом или диизоцианатами.
24
Глава 1
Полиэфиры, полиамиды и некоторые полимеры, полученные из гид-
рокси-мономеров, были стекловидными и растворялись в неполяр-
ных растворителях. Остальные полимеры представляли собой не-
растворимые порошки. Ни одно из веществ, синтезированных
в этой работе, не обладало устойчивостью при температурах
выше 200°.
До некоторой степени аналогичный план был принят в работе
Петерсона и сотрудников [28], которые использовали металл с
координационным числом 6, две связи которого были блокированы
молекулой дикетона. В качестве мономера было взято соединение
сн
НзС—^С—СНз
J О. J)
С2Н5О—С О О ^С— ОСзНз
НС—(f г—сн
СНз
СН3
Мономер полимеризовали реакцией с диолами. Полученное ве-
щество с молекулярным весом 940—2600 нерастворимо в бензоле;
из расплава оно вытягивается в виде длинных волокон. Ни одно
из этих веществ, однако, не обладало устойчивостью при темпе-
ратурах выше 200°.
Берлин и Матвеева [29] также использовали алюминий, у ко-
торого два координационных места были заняты молекулой ди-
кетона. При нагревании мономера
,СН3
Л—с<
(изо-СзНтОЬАБ 4 ^СН
о=сС
•СНз
с различными бис-дикетонами изопропиловый спирт отщеплялся
и происходила полимеризация. Образующиеся вещества имели
низкие молекулярные веса.
Б. Полимеры, содержащие 8-оксихинолины
бис-8-Оксихинолины уже давно используют в качестве осади-
телей в аналитической химии [30]. Нерастворимость осадка, не-
сомненно, во многом обусловлена его полимерной природой. В
IV. Методы синтеза
25
последнее время несколько исследователей пытались получить
полимеры на основе комплексов бис-8-оксихинолинов с металлами
[24, 31,32]. Как и в случае бис-дикетонов, полимеризацию вели
осаждением из водных растворов, в расплавах, а также на гра-
нице раздела фаз. Вещества, полученные осаждением из раство-
ров, вероятно, обладали низким молекулярным весом вследствие
их очень небольшой растворимости и быстрого образования. В од-
ном случае Джудду [24] удалось получить игольчатые кристаллы
при чрезвычайно медленном введении лиганда в раствор иона ме-
талла методом диффузии, но он не обнаружил никаких признаков
пластичности или гибкости этих кристаллов.
Горовиц и Перрос [31 ] использовали для получения бис-хи-
нолиноловых полимеров как термический метод, так и полимери-
зацию из раствора. В их работе имеется следующая методика
получения полимеров:
1) Термическая полимеризация. Координационные полимеры
получали в вакууме при нагревании стехиометрических количеств
лиганда и ацетилацетоната металла при 290°. Реакционный сосуд
погружали в ванну с силиконовым маслом, снабженную электри-
ческим нагревателем. Сосуд и ванна были стеклянные, что позво-
ляло наблюдать за ходом реакции. Образующийся полимер быст-
ро удаляли из бани, чтобы свести к минимуму возможность его раз-
ложения. Продолжительность реакции сокращали от нескольких
часов до нескольких минут, устраняя тем самым трудности, с ко-
торыми встречались другие исследователи при получении коор-
динационных полимеров путем термической полимеризации про-
изводных бис-окспна. Коршак и сотрудники [32], например,
после 3-часового нагревания получили димер соединения никеля
(II) с бис-(8-окси-5-хинолил)-метаном и тример аналогичного
соединения меди(П).
Берг и Алам [30] сообщили, что им не удалось получить по-
лимеры цинка(П) с 8,8'-диокси-5,5'-бпхинолилом путем термичес-
кой полимеризации.
В этих реакциях использовались хелатные ацетилацетонаты.
так как Хоен, Чарльз и Хикам [256] указали, что эти соединения
при нагревании в вакууме разлагаются с образованием летучих
продуктов, состоящих из ацетилацетона, ацетона, уксусной кис-
лоты, метана, окиси углерода и двуокиси углерода. Таким об-
разом, при термической полимеризации в вакууме побочные про-
дукты реакции можно удалить, в то время как металл остается
координированным с бис-лигандом.
Перед каждым опытом реакционный сосуд продували азотом,
очищенным от кислорода. Затем реакционный сосуд, содержащий
реагенты, эвакуировали до остаточного давления приблизительно
10-3 мм pm. cm. и погружали в баню с силиконовым маслом. Пос-
26
Глава 1
ле исчезновения всех видимых признаков реакции продукт из-
влекали из сосуда в виде сыпучего порошка или комков и пере-
носили его в бумажную экстракционную гильзу. В течение ночи
вещество подвергали экстракции диметилформамидом (х. ч.) в
аппарате Сокслета. Сам полимер практически нерастворим в горя-
чем растворителе, но непрореагировавший лиганд, ацетилацето-
нат металла и другие растворимые вещества экстрагируются.
Раствор в экстракционной колбе обычно сильно окрашен, и в нем,
как правило, присутствует твердое вещество. Аппарат был скон-
струирован так, что твердое вещество из гильзы не могло попасть
в колбу. После этого в течение нескольких часов полимер экстра-
гировали абсолютным спиртом для удаления следов свободного
диметилформамида и других неполимерных примесей, раствори-
мых в спирте. Полимер не проявлял признаков растворения в
спирте, хотя некоторые вытяжки были окрашены. После экст-
ракции полимер обычно высушивали над пятиокисью фосфора при
140°, но некоторые образцы дополнительно просушивали при 190°.
Судя по данным элементарного анализа, признаков разложения
полимера при этих температурах не наблюдалось.
2) Полимеризация в растворе. Гидратированный ацетат двух-
валентного металла, растворенный в диметилформамиде, медленно
добавляли при перемешивании к раствору лиганда в диметилфор-
мамиде, поддерживая при этом температуру 120°. Во всех случаях
осаждение происходило почти сразу же после смешивания двух
реагентов. Содержимое реакционной колбы охлаждали в течение
ночи до комнатной температуры, после чего продукт собирали,
подвергали экстракции и высушивали аналогично тому, как это
делали после термической полимеризации.
Горовиц и Перрос тщательно исследовали факторы, влияющие
на термическую устойчивость полимеров бис-(8-окси-5-хинолил)-
метана и обнаружили, что она тесно связана с такими периодиче-
скими свойствами металлов, как потенциал ионизации и электро-
отрицательность. Некоторые из приготовленных ими полимеров
были совершенно устойчивы при 500° в вакууме. Эта температура
сопоставима с температурой разложения мономерных хелатов
8-хинолинола, которая была определена Чарльзом и сотрудни-
ками [33]. В случае полимеров, полученных координированием
ионов металлов шиффовыми основаниями, было обнаружено,
что полимеры, в которых связующей группой является группа
—SO2—, значительно более устойчивы к нагреванию по сравнению
с полимерами, в которых связующей группой служит группа
—СНг—. Если эти соотношения сохранятся и для хинолиноловых
комплексов, это позволит получать бис-хинолиноловые полимеры
с очень высокой термической стойкостью.
IV. Методы синтеза
27
В. Фосфинатные полимеры
Блок с сотрудниками 134] показали, что ион дифенилфосфи-
ната может служить мостиковой группой, связывающей два иона
металла; это может привести к образованию полимеров. Они впер-
вые сообщили о реакциях дифенилфосфиновой кислоты с ацетил-
ацетонатом бериллия и ацетилацетонатом хрома(Ш).В результате
такой реакции образуются димеры, в которых две дифенилфосфи-
натные группы связывают два иона металла. Комплексное соеди-
нение хрома имеет структуру
Во второй статье, посвященной этому же вопросу, они пока-
зали, что можно получить полимерные вещества, вытесняя дифе-
нилфосфинатом вторую молекулу ацетилацетона из каждой мо-
лекулы ацетилацетоната хрома(1П). Они писали: «несмотря на
то что мы получили такие вещества различными методами, наи-
более удобным является прямое взаимодействие ацетилацетоната
хрома(Ш) и дифенилфосфиновой кислоты при температурах 175—
250° и медленном пропускании азота». Нагревание вели до тех
пор, пока в отходящем газе не исчезали следы ацетилацетоната,
что устанавливали реакцией с ионами окисного железа. Остаток
затем делили на фракции последовательными экстракциями этано-
лом, бензолом и хлороформом в аппарате Сокслета. После такой об-
работки вещества, образовавшегося в результате взаимодействия
10,5 г Сг(АсСНАс)з и 13,1 г РйгР(О)ОН, осталось 2,7 г нераство-
римого остатка, если реакцию вели при 175°; 5,8 и 9,4 г, если ре-
акцию вели при 200 и 250° соответственно. Выход полимера за-
метно возрастал с повышением температуры. Фракция, нераство-
римая в этаноле, но растворимая в бензоле, содержала 9,4% Ст,
9,3 Р, 58,5 С и 4,9% Н. Фракция, нерастворимая в этаноле и
бензоле, но растворимая в хлороформе, содержала 8,6% Сг,
10,8 Р, 59,6 С и 4,8% Н. Фракция, нерастворимая во всех трех
растворителях, содержала 8,6% Сг, 10,3 Р, 59,1 С и 4,9% Н. Со-
став последних двух фракций хорошо согласуется с теоретическим
составом [Cr(AcCHAc)(OPPh2O)2]„: 8,88% Сг, 10,58 Р, 59,49 С
и 4,65% Н, а первой фракции — с теоретическим составом
28
Глава 1
(AcCHAc)lCr(AcCHAc)(OPPh2O)2]4Cr(AcCHAc)2: 9,66% Сг, 9,21 Р
58,46 С и 4,83 % Н.
«Эбулиоскопические измерения в бензоле для среднего молеку-
лярного веса фракции, растворимой в бензоле, дали величину
19-40—2633 и для фракции, растворимой в хлороформе,— до
10 870. Растворимая в бензоле фракция, таким образом, содержит
в среднем четыре-пять хромсодержащих элементарных звеньев на
один сегмент полимера, если считать, что в качестве концевых
групп функционируют либо ацетилацетонат, либо дифенилфос-
финат. Растворимая в хлороформе фракция содержит около 18
таких звеньев. Нам не удалось определить молекулярный вес не-
растворимой фракции, но, поскольку некоторые растворимые и
нерастворимые фракции имели состав, соответствующий бесконеч-
ному полимеру, разница между фракциями, вероятно, обусловле-
на именно молекулярным весом, и нерастворимые фракции обла-
дают более высоким молекулярным весом».
Блок и сотрудники приписали этому полимеру следующую
структуру:
и почти не остается сомнения, что эта формула верна, если иметь
в виду выделение димера.
В третьей статье Блок сообщил о получении полимеров с хо-
рошей термической устойчивостью из дифенилфосфината и ионов
двухвалентных металлов с координационным числом 4 и тетра-
эдрическим расположением связей (например, из Вег+ и Zn2+).
В его работе приведена следующая методика:
«Мы получали полимерные фосфинаты металлов различными
способами, однако для каждого примера будет указана только
одна типичная методика получения; [Be(OPPh2O)2]v получали
нагреванием в течение 3,5 час до 100° в микроперегонном аппарате
при остаточном давлении 1 мм рт. ст. тщательно перемешанных
5,0 г Ве(АсСНАс)2 и 21,0 г РЙ2Р(О)ОН. Смесь охлаждали, еще раз
измельчали и снова нагревали в тех же условиях. Эту операцию
проводили еще два раза. Продукт подвергали экстракции этило-
вым спиртом, после чего было получено 8,6 г продукта, содержа-
IV. Методы синтеза
29
щего 2,25% Be, 14,09 Р, 65,43 С и 4,60% Н. Теоретический состав
соединения Be(OPPh2O)2: 2,03% Be, 13,91 Р, 65,01 С и 4,55% Н.
Полимер [Zn(OPPh2O)21v был получен путем межфазной полиме-
ризации смеси 100 мл водного раствора, содержащего 2,195 г
Zn(OAc)2-2H2O и 200 мл бензольного раствора, содержащего
4,36 г Ph2P(O)OH. Полимеризацию вели в смесителе Уоринга.
Смесь перемешивали в течение часа, затем фильтровали, тщатель-
но промывали этанолом и сушили. Было получено 4,1 г сухого
продукта, содержащего 13,4% Zn, 12,14 Р, 57,61 С и4,18% Н. Тео-
ретический состав соединения Zn(OPPh2O)2: 13,08% Zn, 12,40 Р,
57,68 С и 4,03% Н. Полимер [Zn(0P(Ph)(Me)02]x получали в ре-
зультате взаимодействия 1,56 г Ph(Me)P(O)OH и 1,10 г Zn(OAc)2-
• 2Н2О в 300 мл абсолютного спирта. Смесь фильтровали после
часового перемешивания при комнатной температуре. После трех
промываний этанолом было получено 1,12 а белого осадка, содер-
жащего 17,7% Zn, 16,14 Р, 44,88 С и 4,56% Н. Теоретический со-
став Zn[OP(Ph) (Ме)О]2: 17,40% Zn, 16,49 Р, 44,77 С и 4,29% Н.
По эбулиоскопическим измерениям в бензоле средний молекуляр-
ный вес продукта составил 5600 (15 мономерных единиц). Белое
твердое вещество превращается в стекловидную массу при тем-
пературе приблизительно 150° и размягчается приблизительно
при 200°. При комнатной температуре из расплава или из про-
дукта, смоченного бензолом, можно вытянуть длинные гибкие
волокна.
«Структура этих полимеров, вероятно, близка структуре, пред-
положительно приписанной полимеру [Сг(АсСНАс)(ОРРЬ2О)2]Л.
с двумя фосфинатными связями между атомами металла, т. е.
Г. Органические полимеры в качестве лигандов
«Основной ацетат бериллия» Ве4О(СгНзО2)в является чрезвы-
чайно устойчивым комплексом, в котором четыре атома бериллия
связаны с центральным атомом кислорода и расположены вокруг
него по четырем углам тетраэдра. Карбоксильные группы ацетат-
ионов в виде мостиков связывают атомы бериллия, образуя шесть
30
Глава 1
ребер тетраэдра. Марвел и М. М. Мартин [35] получили аналогич-
ный комплекс, в котором 2/3 ацетат-ионов заменены ионами сукцина-
та. В результате этого образуется пластичный полимер, устойчивый
до 450°; к сожалению, с течением времени этот комплекс диспро-
порционирует, превращаясь, очевидно, в основной ацетат и силь-
но сшитый основной сукцинат.
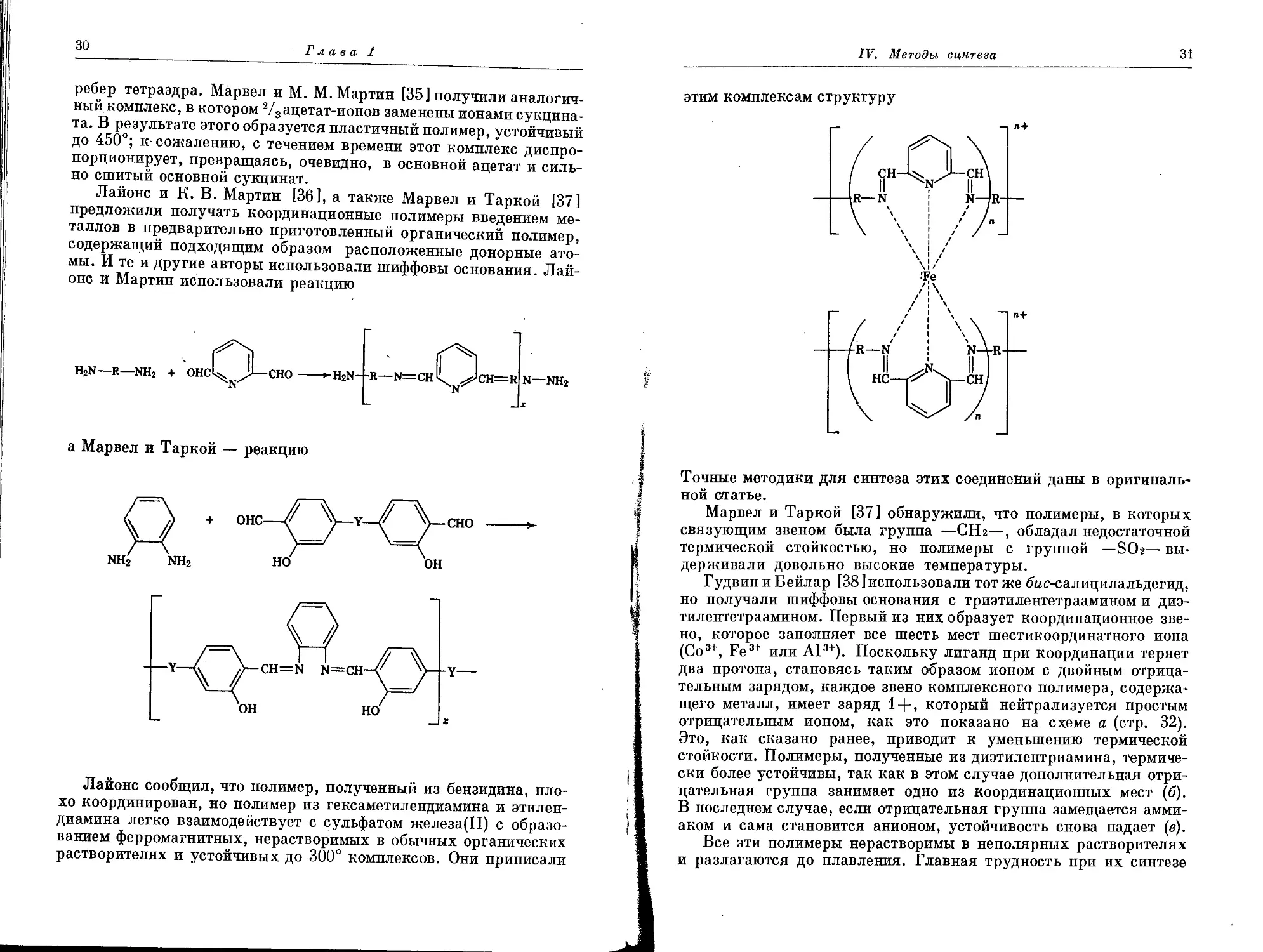
Лайонс и К. В. Мартин [36], а также Марвел и Тарной [37]
предложили получать координационные полимеры введением ме-
таллов в предварительно приготовленный органический полимер,
содержащий подходящим образом расположенные донорные ато-
мы. И те и другие авторы использовали шиффовы основания. Лай-
онс и Мартин использовали реакцию
h2n—r—nh2 + онсЦ^ >—сно
N—NH2
а Марвел и Тарной — реакцию
Лайонс сообщил, что полимер, полученный из бензидина, пло-
хо координирован, но полимер из гексаметилендиамина и этилен-
диамина легко взаимодействует с сульфатом железа(П) с образо-
ванием ферромагнитных, нерастворимых в обычных органических
растворителях и устойчивых до 300° комплексов. Они приписали
IV. Методы синтеза
31
этим комплексам структуру
Точные методики для синтеза этих соединений даны в оригиналь-
ной статье.
Марвел и Тарной [371 обнаружили, что полимеры, в которых
связующим звеном была группа —СНг—, обладал недостаточной
термической стойкостью, но полимеры с группой —SO2— вы-
держивали довольно высокие температуры.
Гудвин и Бейлар [38 ] использовали тот же бис-салицилальдегид,
но получали шиффовы основания с триэтилентетраамином и диэ-
тилентетраамином. Первый из них образует координационное зве-
но, которое заполняет все шесть мест шестикоординатного иона
(Со3+, Fe3+ или А13+). Поскольку лиганд при координации теряет
два протона, становясь таким образом ионом с двойным отрица-
тельным зарядом, каждое звено комплексного полимера, содержа-
щего металл, имеет заряд 1-|-, который нейтрализуется простым
отрицательным ионом, как это показано на схеме а (стр. 32).
Это, как сказано ранее, приводит к уменьшению термической
стойкости. Полимеры, полученные из диэтилентриамина, термиче-
ски более устойчивы, так как в этом случае дополнительная отри-
цательная группа занимает одно из координационных мест (б).
В последнем случае, если отрицательная группа замещается амми-
аком и сама становится анионом, устойчивость снова падает (в).
Все эти полимеры нерастворимы в неполярных растворителях
и разлагаются до плавления. Главная трудность при их синтезе
32
Глава 1
заключается в том, что исходные органические полимеры раство-
римы в неполярных растворителях, но нерастворимы в воде, в
а 6
то время как ионы металла, наоборот, нерастворимы в органиче-
ских растворителях и растворимы в воде. Поэтому образование
полимера происходит всегда в двухфазной системе, вследствие че-
го трудно ввести ионы металла в каждое координационное звено.
Поскольку именно металл придает полимеру устойчивость, важно,
чтобы координация была полной. Возможно, что в дальнейшем
удастся найти комплексы металлов, растворимые в неполярных
растворителях, из которых органический полимер будет вытеснять
олеофильные группы.
Д. Синтез полимеров из карбонилов металлов
Гоан него сотрудники [39] недавно обнаружили, что соедине-
ния, содержащие активный водород, могут реагировать с карбо-
нилами металлов под воздействием ультрафиолетового света:
Av
М (СО)„ + 3LH---MLS + пСО + 3/, Н2 (1)
IV. Методы синтеза
33
(для металлов с координационным числом 6). Они пытались ис-
пользовать эти реакции для получения координационных поли-
меров и проводили эксперименты с дифенилфосфиновой кислотой,
бис-р-дикетонами и бис-8-оксихинолинами [40]. Пентакарбонил
железа и гексакарбонил хрома легко реагируют с дифенилфосфи-
новой кислотой в растворе тетрагидрофурана или толуола. Про-
дукт реакции с карбонилом хрома несколько растворим в толуоле.
Структура этих соединений, по-видимому, соответствует форму-
лам
Ниже приведена одна из методик синтеза: раствор гексакарбонила
хрома и дифенилфосфиновой кислоты в толуоле с соотношением
реагентов 1 : 2 нагревают с обратным холодильником и подвер-
гают воздействию ультрафиолетового света в течение 72 час.
После этого светло-зеленое твердое вещество отделяют от темно-
зеленого фильтрата. Из твердого вещества получены фракции с
молекулярными весами 4670, 6500 и 7130. Фракция с самым вы-
соким молекулярным весом устойчива до 360°.
Е. Фталоцианиновые полимеры
Очень высокая устойчивость фталоцианиновых соединений
металлов позволяет полагать, что удастся получить полимерные
вещества этого типа, если вместо фталевой кислоты использовать
пиромеллитовую кислоту или ее производные. При таком синтезе
должны получиться плоские полимеры. Этот вопрос изучали не-
сколько исследователей. Дринкард и Бейлар [41 ] получили по-
лимеры, состоящие только из нескольких элементарных звеньев,
при этом они использовали методику, применяемую для получе-
ния простых фталоцианинов. Дальнейшему росту полимеров пре-
пятствовала нерастворимость вещества. Размер и форму полиме-
ров удалось оценить по результатам элементарного анализа и
титрования периферийных карбоксильных групп.
4 Заказ № 547
34
Глава 1
По неизвестным причинам пластинки полимера группирова-
лись не в двух направлениях, а преимущественно в одном, обра-
зуя удлиненные кристаллы.
Дринкард и Бейлар приводят следующее описание типичного
синтеза: смесь 20 г диангидрида пиромеллитовой кислоты (0,092 М),
8 г безводного хлорида меди(П) (0,06 М), 108 г мочевины
(1,8 М) и небольшого количества молибдата аммония (катализа-
тор) нагревали при 160° в течение 30 мин. Продукт промывали
6 н. соляной кислотой, растворяли в 200 мл концентрированной
серной кислоты и повторно осаждали, разбавляя раствор 3 л
воды. Осадок декантировали 24 л воды (3-литровыми порциями),
фильтровали и сушили на воздухе. Продукт был сине-зеленого
цвета.
Марвел и М. М. Мартин [42] предприняли очень близкие экс-
перименты со смесью фталевого и пиромеллитового ангидридов с
целью получения линейных цепей. Их полимер состоял только из
нескольких элементарных звеньев. Обе группы исследователей
отметили, что термическая устойчивость этих полимеров значитель-
но меньше, чем мономерного соединения меди со фталоцианином.
Если будут найдены способы получения полимеров более вы-
сокого молекулярного веса, эти полимеры, вероятно, будут обла-
дать полезными свойствами, в том числе полупроводимостью, вслед-
ствие высокой степени конъюгации двойных связей.
Другие исследователи [43] пользовались иными исходными
материалами (нитрилами вместо кислот и ангидридов) и полу-
чали полимеры несколько иными методами, но их результаты мало
чем отличаются от рассмотренных выше.
Литература
35
Берлин, Матвеева и Шерле [44] получили полимер аналогич-
ного типа, используя в качестве органического хелатирующего
агента тетрацианоэтилен. Они нагревали в вакууме до 160—300°
смесь 1 моля ацетилацетоната меди(П) с 2 молями тетрацианоэти-
лена. При нагревании ацетилацетон отщеплялся и оставался не-
плавкий черный полимер, нерастворимый в органических раство-
рителях, щелочах и разбавленных кислотах. Инфракрасный
спектр соединения показал, что его структура соответствует фор-
муле
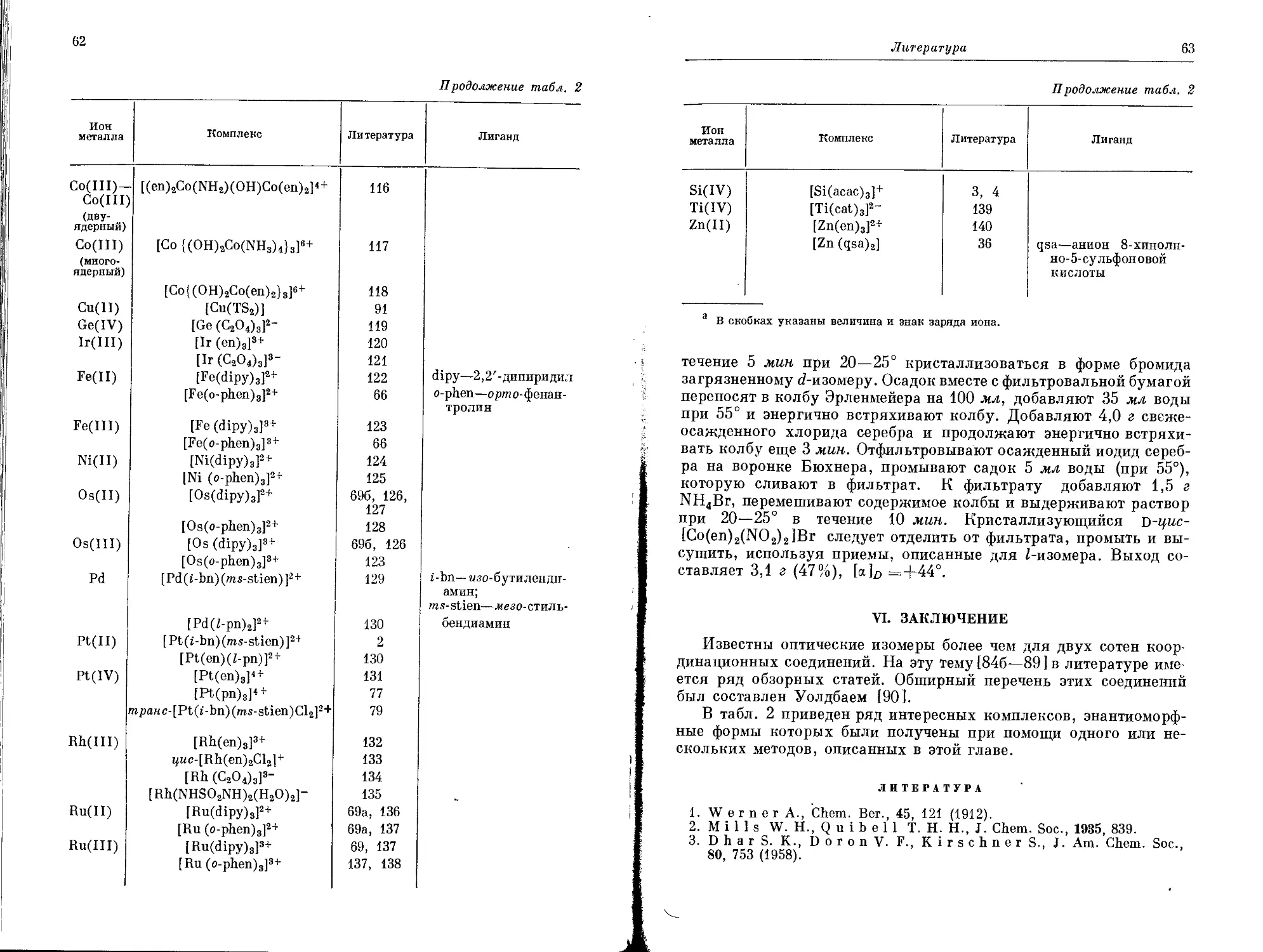
ЛИТЕРАТУРА
1. Powell Н. М., Rayner J. Н., Nature, 163, 566 (1949).
2. Rayner J.H., Powell Н. М., J. Chem. Soc., 1952, 319.
3. Keggin J.F., Miles F. D., Nature, 137, 577 (1936).
4. Drago R. S., personal communication.
5. See for example, Kilpatrick M., P о k r a s L., J. Electrochem.
Soc., 100, 85 (1953); S i 1 1 e n L. G., Proceedings of the Symposium on
Coordination Chemistry, Danish Chemical Society, 1954, p. 74.
6. Werner A., Chem. Ber., 40, 284 (1907).
7. M a r i о n S. P., T h о m a s A. W., J. Colloid Sci., 1, 221 (1946).
8. Whitehead T. H., Chem. Rev., 21, 113 (1937).
9. Роллинсон К., в кн. «Химия координационных соединений», под
ред. Бейлара, мл., и Буша, ИЛ, М., 1960, гл. 13.
10. Р о kr as L., J. Chem. Educ., 33, 152, 223, 282 (1956).
11. For a review of this work see S с h m i t z D umo n t O., “Hochpolymere
Koordinationsverbindungen von Ubergangselementen”, a chapter in Inor-
ganic Polymers, An International Symposium, The Chemical Society,
Special Publication No. 15, London, 1961. This article gives references to
Schmitz-Dumont’s experimental work.
12. S c h m i t z - D u m о n t O., R a a b e F., Z. anorg. allgem. Chem., 277,
297, (1954).
13. Schmitz-Dumont O., Schulte H., Z. anorg. allgem. Chem-,
282, 253 (1955).
14. BrossetC., Arkiv Kemi, Mineral. Geol., 25A, № 19 (1948).
15. Cohen A., Davidson N-, J. Am. Chem. Soc., 73, 1955 (1951).
16. Cuthbertson J.W., Parkinson N., R ooksby H. P., J.
Electrochem. Soc., 100, 107 (1953).
4*
36
Глава 1
17. R a u R. L., В a i 1 а г J. С., Jr., J. Electrochem. Soc., 107, 745 (1960).
18. Hurd R. N., D eLaM at er G., McElheny G.C., Turner
R. J., Wallingford V. H, J. Org. Chem., 26, 3980 (1961).
19. A m о n W. F., Jr., К a n e M. W. (to Polaroid Corp.), пат. США 2505085
(April 25, 1950).
20. Eichhorn G. L., Marchand N. D., J. Am. Chem. Soc., 78, 2688
(1956).
21. I nt err ant e L. V., personal communication; С о m h e s A., Compt.
Rend., 108, 1252 (1889); M о r g a n G. T., Smith J. D. M., J. Chem.
Soc., 1926, 912, indicate that the substance is decomposed slowly at red
heat, but this seems to he overly optimistic.
22. Wilkins J. P., W i t t b e с к e r E. L. (to E. I. du Pont de Nemours
Co.), пат. США 2659711 (Nov. 17, 1953).
23. F e r n e 1 i u s W. C., Wright Air Development Center Report W. A. D. C.
56-203, Part I, Oct. 1956; Part II, Sept. 1957; Part III, Sept. 1958.
24. Bailar J. C., Jr., Drinkard W. C., J udd M.L., W. A. D.C.
Technical Reports 57—391, Sept. 1957; В a i 1 a r J. C., Jr., M c Le an J.,
Judd M. L., W. A. D. C. Technical Report 58—51, April 1958; Bailar
J. C., Jr., M artin К. V., J udd M. L., M c Lean J., W. A.D.C.,
Technical Report 57—391, Part II, August 1958; Bailar J. C., Jr.,
J udd M.L. , McLean J.,W.A.D.C. Technical Report 58—51, Part II,
May 1959; Bailar J. C., Jr., W. A. D.C. Technical Report 59—427,
Jan. 1960; Bailar J.C.,Jr., Goodwin H.A., Moraghan M.,
McLean J., Fujikawa C., Chen L.-C., W. A. D. C. Technical
Report 58—51, Part III, April I960; Klein R., Bailar J.C., Jr., in
press; О h J. S., В a i 1 a r J. C., Jr., J. Inorg. Nucl. Chem., 24, 1225 (1962).
25a. Charles R. G., P awlikowski M. A., J. Phys. Chem., 62, 440
(1958); 6. VonHoene J., Charles R. G., H i с к a m W. M., ibid.,
62, 1098 (1958); в. C h a r 1 e s R. G., H i с к a m W. M., Von H о e n e J.,
ibid., 63, 2084 (1959); r. Cha'rles R. G., ibid., 64, 1747 (1960); 65,
568 (1961); д. C h a r les R. G., J. Polymer Sci., Al, 267 (1963): e. Char-
les R. G., J. Inorg. Nucl. Chem., 25, 45 (1963).
26. К 1 u i b e r R. W., L e w i s J. W., J. Am. Chem. Soc., 82, 5777 (1960).
27. Drinkard W. C., Ross D., Wiesner J., J. Org. Chem., 26, 619
(1961).
28. Patterson T. R., P a vl i к F. J., В a 1 d oni A. A., Frank R.L.,
J. Am. Chem. Soc., 81, 4213 (1959).
29. Б e p л и н А. А., Матвеева H. Г., Успехи химии, 29, 119 (1960).
30. For. a recent example see В e rg E. W., Alam A., Anal. Chim. Acta, 27,
454 (1962).
31. Horowitz E., P e г г о s T. P., J. Inorg. Nucl. Chem., 26, 139 (1964).
32. КоршакВ. В., Виноградова С. В., БабчинитсерТ.М.,
Высокомолекулярные соединения, 2, № 4, 344 (1960).
33. С h а г 1 е s R. G., Langer A., J. Phys. Chem., 63, 603 (1959). С h a r-
1 е s R. G., J. Inorg. Nucl. Chem., 20, 211 (1961); Anal. Chem. Acta, 27,
474 (1962); Charles R.G., PerrottoA., Dolan M. A., J. Inorg.
Nucl. Chem., 25, 45 (1963); Westinghouse Research Laboratories, Scientific
Research Paper 123-4000-P7 (Nov. 16, 1961).
34. Block В. P., RothE. S., Schaumann C. W., OconeL.R.,
Inorg. Chem., 1, 860 (1962); В 1 о с k В. P., S i m k i n J., О с о n e L. R.,
J. Am. Chem. Soc., 84, 1749 (1962); В 1 о с k В. P., Rose S. H., Scha-
uma n n C. W., RothE. S., Joseph Simkin, J. Am. Chem.
Soc., 84, 3200 (1962).
35. M a r v e ГС. S., M a r t i n M. M., J. Am. Chem. Soc., 80, 619 (1958).
36. LionsF., MartinK. V., J. Am. Chem. Soc., 79, 2733 (1957).
37. M ar v e 1 C. S., T a r k о у N., J. Am. Chem. Soc., 79, 6000 (1957).
Литература 37
38. Goodwin Н. A., Bailar J. С., Jr., J. Am. Chem. Soc., 83, 2467
(1961).
39. G о a n J. С., H e u t h e г С. H., P о d a 1 1 H. E., in press.
40. P о d a 1 1 H. E., I a p a 1 u с c i T. L., J. Polymer Sei., Bl, 457 (1963).
41. D r i n к a r d W. G., В a i 1 a r J. C., Jr., J. Am. Chem. Soc., 81, 4795
(1959).
42. Marvel C. S., Martin M. M., J. Am. Chem. Soc., 80, 6600 (1958).
43. Sprague Electric Co. Final Report, Contract No. DA 36-039-SC-87
(U. S. Army Signal Corp.), 1952; L a w t о n E. A., McRitchie D. D.,
W. A. D. C. Technical Report 57-642, November 1957.
44. Берлин А. А., Матвеева H. Г., Шерле А. И., Изв. АН
СССР, ОХН, 1959, 2261.
ГЛАВА 2
ОПТИЧЕСКИ АКТИВНЫЕ КООРДИНАЦИОННЫЕ
СОЕДИНЕНИЯ
Киршнер
Уайнский государственный университет, Детройт,
Мичиган
I. ВВЕДЕНИЕ
Доказательство оптической активности координационных сое-
динений путем разделения рацемической смеси их энантиоморф-
ных форм или другими способами часто имеет большое значение
для определения структуры координационных соединений. Опти-
чески активные комплексы, синтезированные в лаборатории, об-
разуют обычно рацемические модификации, содержащие как пра-
во-, так и левовращающие изомеры, а не одну энантиоморфную
форму какой-либо одной конфигурации. Разработаны методы раз-
деления рацемических смесей на составляющие их энантиоморф-
ные формы. Это разделение служит доказательством асимметрии
(или диссимметрии) комплексов, а также позволяет делать выводы
о природе и механизме реакций, об абсолютных конфигурациях
комплексов, их составе и т. д. Кроме того, разработаны прямые
методы получения отдельных энантиоморфных форм без приме-
нения методов разделения.
Одним из примеров, показывающих важность разделения ра-
цемических модификаций асимметричных комплексов, является
разделение рацемических катионов игрис-(этилендиамин)-кобаль-
та(Ш), DL-[Со(ен)з]3+ (формула I), проведенное Вернером [1].
Доказательство оптической активности этого комплексного
иона подтверждает предложенную для него октаэдрическую кон-
фигурацию I. Так были исключены из рассмотрения возможные
конфигурации этого иона в форме плоского шестиугольника или
тригональной призмы, так как они обладают плоскостной симмет-
рией и, следовательно, не могут проявлять оптическую актив-
ность.
I. Введение
39
-1 з +
Другим важным примером, подтверждающим значение доказа-
тельства оптической активности, является работа Миллса и Куи-
белла [2], которые показали путем разделения рацемических
катионов (л«езо-стилбендиамин)(изо-бутилендиамин)-платина(11)
DL-[Pt(ms-stien) (i-bn) ]2+ (формула II), что ион платины(П) не
обладает тетраэдрической конфигурацией
В этой работе дано подтверждение предполагаемой плоской форме
иона; эта форма не обладает плоскостной симметрией и может про-
являть оптическую активность, в то время как тетраэдрическая
конфигурация (формула III) имеет плоскость симметрии и, таким
образом, оптическая активность ее исключена.
Третьим примером, доказывающим важность разделения раце-
мических модификаций, служат работы Дара, Дорона и Киршнера
40
Глава 2
[3,4], в которых было доказано, что кремний(1У) может иметь
координационное число 6. Эти исследователи разделили ра-
цемические катионы щрнс-(ацетилацетонато)-кремния(1У)
2+
Плоскость симметрии проходит через
изобутилендиамин и центральный ион металла
DL [81(асас)з]+ на его оптические энантиоморфные формы, подтвер-
див таким образом, что структура данного комплексного иона
подобна структуре I. Все другие возможные структуры (напри-
мер, тетраэдрическая, плоская, гексагональная, призматиче-
ская и т. д.), которые можно предложить для этого комплекса,
обладают плоскостной симметрией и, таким образом, не могут
проявлять оптической активности.
Другие примеры важности разработки методов разделения
рацемических модификаций комплексов можно найти в работах
по оптической вращательной дисперсии [5—9] и по изучению
механизма реакций [10,11].
П. ОПТИЧЕСКАЯ АКТИВНОСТЬ, ПОЛЯРИМЕТРИЯ
И СПЕКТРОПОЛЯРИМЕТРИЯ
А. Оптическая активность
Явление оптической активности стали изучать еще в начале
XIX в., когда Араго [12] исследовал поведение солнечных лучей,
поляризованных кристаллами кварца и других минералов. Бьо
[13] детально изучил это явление и заметил, что плоскость поля-
ризации линейно поляризованного света вращается кристаллами
кварца и растворами некоторых природных веществ, например
камфоры, сахара и т. п. Дальнейшие работы привели к выяснению
связи между способностью некоторых веществ Вращать плоскость
II. Оптическая активность, поляриметрия и спектрополяриметрия 41
поляризации света и их асимметрической конфигурацией и
позднее к тонкому различию асимметрических конфигураций от
«диссимметрических». Строго говоря, полное отсутствие элементов
симметрии в молекуле не является необходимым условием опти-
ческой активности. Молекулы, в которых нет центра симметрии и
плоскости симметрии, обычно проявляют оптическую активность,
хотя некоторые элементы симметрии в них имеются (например,
ось симметрии). Такие молекулы называют «диссимметричными»,
и этот термин следует применять по отношению к большинству
оптически активных комплексов, за исключением, конечно, та-
ких, в которых симметрия полностью отсутствует.
Теоретические основы оптической активности с момента ее
открытия стали предметом многочисленных исследований и не-
скольких монографий [14—17]. Важно отметить, что доказатель-
ство оптической активности соединений обычно служит подтверж-
дением отсутствия в молекулах плоскостной симметрии. Это имеет
важное значение для стереохимических и структурных иссле-
дований. Метод вращения плоскости поляризации очень часто
применяют при проведении структурных исследований.
Б. Поляриметрия
В связи с возрождающимся интересом к методу вращения пло-
скости поляризации вообще и к методу оптической вращательной
дисперсии (зависимость оптического вращения от длины волны)
в частности стало доступным большое число приборов (поляри-
метров) для измерения вращения плоскости поляризованного
света различными веществами. Имеются большие различия в типах
приборов (визуальные, фотоэлектрические, фотографические и
т. д.), в принципах их работы (прямое снятие показаний, эффект
Фарадея, использование абсорбции и т. д.), в универсальности
(для одной длины волны, для серии длин волн, спектрополяри-
метры и т. п.), в точности измерений и в стоимости. В ряде статей
[8, 14, 17а] подробно описаны многие из этих приборов, в част-
ности их работа, достоинства и недостатки.
Большинство визуальных определений оптического вращения
при одной определенной длине волны было проведено при помощи
D-линии натрия (5890 А)- Однако часто используют и зеленую
линию ртути (5460 А)- Последнюю предпочитают D-линии натрия
вследствие гораздо большей чувствительности глаза к зеленому
цвету, чем к желтому. Пробу обычно готовят в виде раствора,
который помещают в кювету (длиной 1—2 дм) поляриметра при
температуре 20—25°. Оптическое вращение, как правило, изме-
ряют в единицах «удельного вращения» [а] или «молекулярного
3 Заказ № 547
Глава 2
вращения» [а]д/, выраженных в градусах
[а] = a/lc, (1)
[а^ = М [а]/100, (2)
где I — длина кюветы (дм), с — концентрация пробы раствора
(г/мл раствора), М — молекулярный вес пробы и а — наблюдае-
мый угол в градусах (температуру обозначают справа вверху
и длину волны справа внизу от квадратных скобок).
В литературе имеются разногласия относительно определения
молекулярного вращения [а ]м, так как некоторые исследователи
[18, 19] отбрасывают цифру 100 в выражении (2). Однако в боль-
шинстве исследований по поляриметрии авторы исходят из опре-
делений (1) и (2) [14].
Практически величина оптического вращения, установленная
на визуальном приборе для одной длины волны, представляет со-
бой среднее арифметическое из нечетного числа отдельных оп-
ределений (обычно по крайней мере пяти) и указывается вместе
с отклонением от средней величины. Оператор, работающий с
визуальным прибором высокой точности, при благоприятных ус-
ловиях может достигнуть отклонения от средней величины ±0,003°.
Усовершенствованные электронные приборы превосходят эту
точность на порядок. Для соединений, которые сильно поглощают
свет в области линий натрия и ртути, рекомендуется разбавлять
пробы или, если это неосуществимо из-за небольшого угла вра-
щения, источник света пропускают через фильтры или монохро-
маторы, позволяющие использовать различные длины волн.
В. Спектрополяриметрия
В связи с повышенным интересом к методу оптической враща-
тельной дисперсии было разработано несколько типов приборов
для этих определений [8, 17]. Кривая оптической вращательной
дисперсии данного вещества выражает зависимость оптического
вращения от длины волны (рис. 1).
Если кривая оптической вращательной дисперсии вблизи ка-
кой-либо полосы поглощения имеет форму, показанную на рис. 1,
то эту полосу поглощения называют «оптически активной полосой
поглощения», а форму кривой «аномальной вращательной дис-
персией», или «эффектом Коттона» [14]. Следует отметить, что
при определенных длинах волн оптическая вращательная диспер-
сия отдельной энантиоморфной формы некоторого оптически ак-
тивного комплекса может проходить через нуль. Таким’образом,
при этих длинах волн данная энантиоморфная форма не будет да-
вать оптического вращения.
II. Оптическая активность, поляриметрия и спектрополяриметрия 43
Рис. 1.
I — оптическая вращательная дисперсия D-K3[Cr(C2O4)3]; II — оптический
спектр поглощения D-K3[Cr(C3O4)3],
Для сравнения оптического вращения различных координа-
ционных соединений иногда полезно использовать «эквивалентное
молекулярное вращение» ([а]е, или [М]е, или [7?]). Это позво-
ляет не принимать во внимание нехарактерные различия в моле-
кулярном вращении, возникающие вследствие различий в моле-
кулярном весе оптически неактивного иона, связанногос оптически
активным комплексным ионом. Подобные различия возникают
при сравнении оптических вращений (например, при сравнении
оптического вращения [Со(еп)з12(8О4)з и [Со(еп)з1С1з). Эквива-
лентное молекулярное вращение определяется следующим обра-
зом [8 ]:
[М]е = М [а]/100п = /п, (3)
где п — число оптически активных комплексных ионов в молекуле
координационного соединения.
3*
44
Глава 2
III. МЕТОДЫ ПОЛУЧЕНИЯ ОПТИЧЕСКИ АКТИВНЫХ
КООРДИНАЦИОННЫХ СОЕДИНЕНИЙ
Существует несколько общих методов получения оптически
активных координационных соединений. Методы, наиболее часто
используемые, носят название методов разделения и состоят в
разделении энантиоморфных форм рацемической смеси изомеров
противоположной (зеркально-отраженной) конфигурации. Мож-
но, однако, также получить оптически активные комплексы пря-
мыми методами, исключающими разделение смеси, которая со-
держит нужную энантиоморфную форму. Строго говоря, процессы
разделения представляют собой процессы выделения обеих энан-
тиоморфных форм в рацемической смеси. Между тем многие ав-
торы, которые сумели выделить данным способом только одну
энантиоморфную форму, часто характеризуют этот способ как
метод разделения. В этой главе термин метод разделения отно-
сится только к такому методу, при помощи которого в принципе
можно выделить (хотя бы неполностью) обе энантиоморфные фор-
мы рацемической смеси.
А. Методы получения путем разделения
Со времени ранних работ Пастера в области оптической актив-
ности разработано множество методов разделения рацемических
модификаций. Большую часть этих методов применяют для разде-
ления комплексных неорганических соединений. Многие из ме-
тодов предназначены для полного разделения энантиоморфных
форм, но некоторые из них позволили достигнуть лишь частичного
разделения, которого обычно достаточно для установления опти-
ческой активности и асимметрии (или диссимметрии) исследуемых
комплексов.
1. Механическое выделение антиподов
Первый метод разделения рацемической смеси был разработан
Пастером в 1848 г. [20]. Метод состоял в отборе кристаллов с раз-
личными гемиэдрическими гранями, обычно при помощи микро-
скопа или лупы. Пастер разделил рацемическую смесь энантио-
морфных форм аммонийнатрийтартрата. Метод предполагает
устройстводля выращивания кристаллов и возможность идентифи-
кации гемиэдрических граней и применим только в случае раце-
мической смеси кристаллов (но не к рацемическим соединениям,
сдвоенным кристаллам и т. п.). В настоящее время метод Пастера
не применяют, так как он очень трудоемок, требует точных мани-
пуляций и не всегда приводит к удовлетворительным результатам.
111. Получение оптически активных координационных соединений 45
2. Самопроизвольная кристаллизация рацемических смесей
В 1919 г. Егер [21J показал возможность превращения раце*
мического соединения в рацемическую смесь путем его кристалли-
зации выше определенной температуры, которая зависит от при-
роды изучаемого комплекса. Вариантом метода Пастера является
кристаллизация соединения Кз [Со(С2О4)з] из водного раствора
при температуре выше 13,2° с выделением смеси правых и левых
энантиоморфных форм, разделяемых затем механически. Это пре-
вращение можно представить следующим уравнением*:
13,2°
2К3 [Со (С2О4)3]-3,5Н2О^Г d-K3[Co(C2O4)3]-H2O +
+ ь-К3 [Со (С2О4)3] Н2О + 5Н2О.
Самопроизвольной кристаллизацией ниже 48° Егер [6] полу-
чил рацемическую смесь комплекса циклопентандиамина
[Rh(cptn)3] (СЮ4)з-12НгО и сумел разделить эти изомеры меха-
ническим способом. Кроме уже указанных в разд. 1 недостатков
метода, для его осуществления необходимо определить оптималь-
ную температуру кристаллизации смеси энантиоморфных форм, а
не рацемата. В связи с этим данный метод также мало используют
в настоящее время.
3. Селективная кристаллизация при помощи изоморфной затравки
Основная цель при разделении рацемической смеси комплексов
состоит в выделении энантиоморфных форм. Интересный способ
решения этой проблемы был разработан Вернером и Босхартом
[22а] на примере рацемической смеси ионов [Со(еп)2(С2О4) ]+,
[Сг(еп)2(С2О4) ]+ и [Со(еп)г(КО2)2]+. Эти исследователи установи-
ли, что если затравочный кристалл соединения, содержащего ион
D-[Со(еп)2(С2О4).]+, ввести в раствор рацемической смеси этих
ионов, то кристаллизуется селективно одна из энантиоморфных
форм. После введения в раствор затравочного кристалла следует
добавить некоторое количество смеси этилового спирта и эфира,
чтобы вызвать кристаллизацию. После осаждения одного изомера
фильтрат значительно обогащен противоположной энантиоморф-
ной формой.
Данный метод разделения использовали относительно мало
вследствие того, что оказались эффективными некоторые другие
методы, описанные ниже. Однако этот метод открывает большие
* Прописные буквы D и L относятся к знакам вращения комплексных
ионов при данной длине волны, строчные буквы d и I— к знакам вращения
лигандов; абсолютные конфигурации этих веществ не указаны, кроме спе-
циально оговоренных случаев.
46
Глава 2
возможности для исследователей, так как для его применения тре-
буется только затравочный кристалл, изоморфный по отношению
к выделяемой энантиоморфной форме, а не кристалл выделяемого
изомера [22 в].
Этот метод также можно использовать для определения абсо-
лютных конфигураций оптически активных комплексных ионов.
Наконец, метод дает дополнительное подтверждение абсолютных
конфигураций комплексных ионов, которые были успешно крис-
таллизованы затравочными кристаллами известной конфигу-
рации.
4. Образование «активных рацематов»
Делепэ [23, 24а] разработал метод разделения, основанный
на том факте, что рацемические модификации в твердом состоя-
нии существуют в трех формах, которые иногда совершенно от-
личны от энантиоморфных форм, из которых они образованы.
Эти три формы известны под названиями: рацемические смеси, ра-
цемические соединения и рацемические твердые растворы.
Рацемические смеси получают из асимметрических соединений,
которые образуют кристаллы с гемиэдрическими энантиоморф-
ными гранями. Это такой тип смеси, которая поддается разделе-
нию при механическом отборе кристаллов, содержащих только
одну энантиоморфную форму. Рацемические соединения образуются
тогда, когда две энантиоморфные формы объединяются с обра-
зованием вещества, все кристаллы которого совершенно идентич-
ны и каждый кристалл содержит одинаковые количества каждой
энантиоморфной формы. Такие кристаллы обладают свойствами,
отличными от свойств кристаллов индивидуальных энантиоморф-
ных форм. Рацемические твердые растворы могут кристаллизо-
ваться без образования соединения, если обе энантиоморфные
формы оказываются также изоморфными.
Делепэ установил, что подобные асимметрические (или дис-
симметрические) комплексные ионы имеют одну и ту же относи-
тельную конфигурацию, несмотря на то что их оптическое враще-
ние при данной длине волны совершенно различно. Комбинация
такого иона с «противоположной» энантиоморфной формой дру-
гого иона может, таким образом, давать рацемическое соединение
или рацемический твердый раствор, а не рацемическую смесь.
В этом и состоит метод «активных рацематов». Например, если
выпаривать раствор, содержащий эквимолярные количества D- и
L-K3 [Со(С2О4) з ], то кристаллизуется рацемическая смесь. При
растворении кристаллов в воде полученный раствор не прояв-
ляет оптической активности. Однако если выпаривать раст-
вор, содержащий эквимолярные количества D-K3 [Сг(С2О4) з ] и
III. Получение оптически активных координационных соединений VI
L-Кз [Со(С2О4)з], то образуются кристаллы, которые дают оптиче-
ски активный раствор (активный рацемат), так как комплекс
хрома не обладает оптическим вращением, точно равным оптиче-
скому вращению комплекса кобальта. Если активный рацемат
образует рацемическую смесь, то относительные конфигурации
энантиоморфных форм различны, но если активный рацемат ока-
зывается рацемическим соединением или рацемическим твердым
раствором, то тогда относительные конфигурации компонентов
одинаковы, и в таких случаях имеется возможность разделения.
Делепэ удалось установить, что активные рацематы (рацеми-
ческие соединения или рацемические твердые растворы) в дейст-
вительности образуются из следующих пар:
L-Кз [1г(СгО4)з] и D-K3[Rh(C2O4)3],
D-Кз Пг(С2О4)з] и L-Кз [Со(Сг04)з],
D-Кз [1г(С2О4)з] и Ь-К3[Сг(С2О4)з].
Поэтому, если взять в качестве примера последнюю пару соеди-
нений, метод разделения D,L-K3[Сг(С2О4)з] будет состоять в об-
работке раствора рацемической смеси соединением D-Кз Цг(С2О4)з]
в выпаривании раствора для кристаллизации рацемического сое-
динения (активного рацемата) и в выделении D-Кз[Сг(С2О4)з1
из оставшегося раствора
D-Кз [1г (С2О4)з1 + d,l-K3 [Сг (С2О4)3[ =
= D-K3 [1г (С2О4)3[- ь-К3 [Сг (С2О4)3] -[- d-K3 [Сг (С2О4)3].
Активный рацемат
Последующее выделение активного рацемата при помощи дру-
гих методов даст другую энантиоморфную форму и завершит про-
цесс разделения.
5. Методы, основанные на эффекте конфигурационной активности (асим-
метрические или диссимметрические превращения первого порядка) [24в]
Дуайер с сотрудниками [25, 261 использовали следующее яв-
ление: если оптически неактивные электролиты добавлять к раст-
вору рацемической смеси оптически активного комплексного сое-
динения, то происходит одинаковое изменение активности каждой
энантиоморфной формы; добавление же оптически активного иона
влияет несколько различно на активность каждой энантиоморфной
формы. Различия в растворимости (по Дуайеру) изомеров D- и
L- [Ru(o-phen)3] (С104)2* в разбавленном растворе d-бромо-тс-кам-
форасульфоната аммония или сегнетовой соли составляют до
* o-phea — орто-:[)енантролип.
48
Глава 2
3,5%. Он наблюдал подобные явления и при изучении рацемиче-
ских смесей оптически активных комплексов [Ni(dipy) з ]12* и
[Со(асас)з]**. Дуайер приписывал этот эффект конфигурацион-
ной активности различному взаимодействию электронных полей
различных энантиоморфных форм с полем оптически активной
среды, полученной при добавке оптически активного вещества.
При высоких концентрациях этот эффект менее заметен, вероятно,
вследствие того, что при высокой ионной силе нормальная актив-
ность оказывается сильнее относительно слабой конфигурацион-
ной активности. Этот эффект наблюдали также в других системах
[И].
6. Биохимические методы
Этот метод также был разработан Пастером [27 ], который уста-
новил, что при добавлении дрожжей или плесени Penicillium gla-
исит для брожения аммонийной соли рацемической винной кис-
лоты превращения претерпевала в основном соль природной вин-
ной кислоты (правый изомер), а соль левого изомера винной
кислоты могла быть выделена в оптически чистой форме из бро-
дильной жидкости. Энзимы также катализируют подобные реакции.
Например, действие ацилазы I на ацилированную рацемическую
аминокислоту в водном растворе до завершения гидролиза по-
ловины ацильных групп приводит к образованию аминокислоты,
производной искусственного изомера (d) и свободной аминокисло-
ты природной (1) энантиоморфной формы [28, 29]. В принципе
должны существовать ферменты или бактерии, которые могут воз-
действовать на каждую энантиоморфную рацемическую смесь.
Тем самым теоретически возможно по крайней мере частичное
разделение.
Креспи [30] предпринял попытку осуществить частичное раз-
деление D, L-[Со(еп)з]3+, питая элементарные организмы одной
энантиоморфной формой. Затем бактериям давали для питания ра-
цемическую смесь комплекса. Возможность разделения комплек-
сов этим методом еще не доказана, хотя метод может оказаться
полезным для разделения других веществ, особенно комплексов,
проявляющих заметную биологическую активность.
7. Образование и дробная кристаллизация диастереоизомеров
(асимметрические или диссимметрические превращения
второго порядка)
Это наиболее важный и наиболее часто употребляемый метод
разделения рацемических соединений. Метод заключается в обра-
* dipy — 2,2'-дипиридил.
** асас — ацетилацетонат-ион.
III. Получение оптически активных координационных соединений 49
ботке рацемической смеси некоторого ионного комплекса простой
энантиоморфной формой оптически активного «разделяющего
агента» с противоположным зарядом и в образовании незеркаль-
ного диастереоизомера. Например,
D-[Co (en)3] С13 + L-[Co (en)3] С13 + 2Na2d-tart =
= D-[Co (en)3J Cl d-tart -f- L-[Co(en)3] Cl d-tart -f- 4NaCl.
Обычно эти диастереоизомеры выделяют дробной кристалли-
зацией, так как структура кристалла особенно чувствительна к
малейшим изменениям в строении молекулы диастереоизомеров.
Применяли также другие методы для выделения диастереоизоме-
ров: дистилляцию [31], хроматографию, зонную плавку и т. д.,
однако дробная кристаллизация остается наиболее эффективным
методом. Некоторые из этих методов будут описаны ниже (vide
infra).
В табл. 1 приведены оптически активные ионы, наиболее часто
используемые в качестве разделяющих агентов для рацемических
Таблица 1
Некоторые разделяющие агенты, применяемые для разделения
рацемических комплексов неорганических соединений
Катионы для комплексных анионов Анионы для комплексных катионов Адсорбенты для не ион- ных и ионных комплексов
Стрихнин (гидро-кати- он) Тартрат Кварц (оптически актив- ный)
Бруцин (гидро-катион) Виннокислый антимонил NaC103 (оптически ак- тивный)
Цинхонин (гидро-кати- он) а-Бромокамфора-л-суль- фонат Крахмал
а-Фенилэтиламин (гид- Дибензоил-d-тартрат Окись алюминия с по-
ро-катион) [32, 33] крытием
Цинхонидин (гидро-ка- тион) Камфора-л-сульфонат d-Лактоза гидрат
Хинин (гидро-катион) Нитрокамф оронат Целлюлоза
Хинидин (гидро-катион) Диацетилтартрат Глюкоза
Морфин (в форме гид- ро-катиона) ifuc-[Co(en)2(NO2)2]+ [35] [Со(еп)3]з+ [Ni(o-phen)3]2+ [Co(edta)]" [34] Сахароза Винная кислота Глутаминат натрия Бумага Силикагель с покрытием
50
Глава 2
смесей комплексных ионов. Перечислены также некоторые адсор-
бенты, которые использовали для разделения оптически актив-
ных внутрикомплексных (неионных) соединений, таких, как ион-
ные комплексы.
Этот метод образования диастереоизомеров нельзя непосред-
ственно использовать для разделения рацемической смеси внутри-
комплексных соединений, если они не обладают функциональной
группой, способной к взаимодействию с разделяющим агентом.
Примером разделения таких комплексов при помощи стрихнина
является разделение цинката(П) рацемической бис-(8-хинолино-5-
сульфоновой кислоты) [36].
Для полного разделения необходимо удалить разделяющий
ион после выделения диастереоизомеров, чтобы получить энан-
тиоморфные формы в виде отдельных веществ. Разработаны раз-
личные методы для проведения этого процесса. Например, если
в качестве разделяющих агентов используют алкалоиды, то ком-
плексные диастереоизомеры растирают с твердым иодидом калия
или перхлоратом натрия и затем смесь обрабатывают водой для
удаления солей щелочных металлов из разделяемого комплексного
аниона. В осадке остается относительно нерастворимый иодид или
перхлорат алкалоида.
Тот же диастереоизомер можно обработать основанием и полу-
чить свободный алкалоид, который обычно относительно нераст-
ворим в воде. Разделяющие катионы часто удаляют методами
осаждения или обработкой минеральной кислотой. В последнем
случае разделяющий агент осаждается в виде относительно не-
растворимой свободной кислоты. Новейшие методы удаления раз-
деляющих ионов включают использование ионообменных смол
[3, 4], однако в этом случае следует добавить катион для обес-
печения полного удаления разделяющего иона смолой.
8. Методы вытеснения (кинетика асимметрических или диссимметрических
превращений)
В 1952 г. Бейлар с сотрудниками [37, 38] изучали влияние
конфигурации на кинетику замещения карбонатного иона в сое-
динении [Со(г-рп)2СОз]С1* рацемическими винной, а-хлоропро-
пионовой и молочной кислотами. В 1955 г. Дуайер с сотрудника-
ми [39 ] сообщили, что этилендиамин замещает ион этилендиамин-
тетраацетата в анионе D- [Go(edta) ]“ с сохранением оптической
активности. Киршнер с сотрудниками [40] использовали это яв-
ление для частичного разделения D, L- [Co(edta) ]“ избирательным
замещением иона этилендиаминтетраацетата из одной энантиоморф-
* Z-pn — левовращающий пропилепдиамин.
111. Получение оптически активных координационных соединений 51
ной формы по реакции с левовращающим пропилендиамином
D-[Co (edta)]- -J- L-[Co (edta)]- + 3Z-pn =
= L-[Co (Z-pn)3]3+ + D-[Co (edta)]- + edta4-.
Дас Сарма и Бейлар [41], воздействуя на D,L-yuc-
[Co(trien)C12]Cl* d-антимонилтартратом серебра, получили со-
единение L-[Co(trien) (d-SbOtart)2] (d-SbOtart), которое после пре-
вращения в хлорид дает L-^uc-[Co(trien)C12]Cl; однако степень
оптической чистоты этого соединения не была определена.
Этот метод очень интересен. Его изучали другие исследова-
тели для разделения иных комплексов.
9. Фотохимические методы
В зависимости от направления поляризации (право- и левопо-
ляризованный свет) энантиоморфные формы с противоположной
конфигурацией поглощают в разной степени поляризованный по
кругу свет. Отсюда следует, что эти две энантиоморфные формы
рацемической смеси будут с различной скоростью разлагаться
при облучении поляризованным по кругу светом с определенной
длиной волны.
Кун с сотрудниками [42, 43] успешно применили этот метод
к органическим соединениям, но опыты Егера и Бергера с ионом
D,L-[Co(C2O4)3]3-дали отрицательные результаты [44а]. Цухида
с сотрудниками [446], однако, сообщили о частичном разделении
О,Ь-Кз[Со(СгО4)з] в водном растворе при помощи избиратель-
ного разложения правовращающей энантиоморфной формы поля-
ризованным по кругу светом. Этот метод разделения комплексов
требует дальнейшего изучения. Новейшие достижения в области
приборостроения должны облегчить экспериментирование в этой
области.
10. Методы избирательной адсорбции
а. Порошкообразный оптически активный кварц. Исследова-
тели, интересующиеся разделением рацемических смесей неион-
ных внутрикомплексных соединений, разработали один важный
и интересный метод, основанный на способности некоторых
веществ к избирательной адсорбции (т. е. поглощению или
большего количества, или с большей скоростью) одной энан-
тиоморфной формы рацемической смеси. Цухида с сотрудника-
ми [45] применили этот метод для частичного разделения
* trien — триэтилентетраамин.
52
Глава 2
D,L-[Co(dmg)2(NH3)Gl]* путем взбалтывания раствора этого
соединения с порошком левовращающего кварца. Этот метод ис-
пользовали несколько других исследователей [46]. До сих пор
не был подтвержден ни один случай полного разделения этим ме-
тодом (см., однако, [46 г]). Все же метод оказался очень полезным
для доказательства оптической активности нескольких комплекс-
ных соединений, как ионных, так и неионных. Этот метод имеет
то преимущество, что он исключает химические операции над оп-
тически активными системами (например, образование диастерео-
изомеров) и допускает в случае относительно нестойких веществ
быстрый перенос оптически активной энантиоморфной формы к
поляриметру для быстрого определения активности.
б. Порошкообразный оптически активный хлорат натрия.
Помимо кварца, для разделения рацемических смесей (vide infra)
использовали несколько других оптически активных адсорбентов.
За немногими исключениями, эти адсорбенты являются оптически
активными вследствие присутствия одного или нескольких асим-
метрических атомов углерода, а не потому, что они обладают моле-
кулярной асимметрией (или диссимметрией) без асимметрических
атомов углерода, как, например, оптически активный кварц.
Однако, по сообщению Феррони и Чини [47 ], возможно частичное
разделение О,L-[Be(bzac)2]* путем избирательной адсорбции как
на право-, так и на левовращающем порошкообразном хлорате
натрия. При кристаллизации из водных пересыщенных растворов
этой соли за несколько суток вырастали оптически активные круп-
ные монокристаллы. Хлорат натрия оптически активен благодаря
наличию асимметрической молекулярной конфигурации в крис-
таллах, как и у кварца. Имеется несколько других веществ, ко-
торые обладают в твердом состоянии естественной оптической ак-
тивностью, обусловленной строением кристаллических решеток.
Некоторые из этих веществ изучали с целью применения их в ка-
честве оптически активных адсорбентов при разделении рацеми-
ческих смесей комплексов методом избирательной адсорбции.
в. Порошок крахмала. Кребс с сотрудниками [48] испытывали
крахмал в качестве адсорбента для разделения энантиоморфных
форм и добились частичного разделения нескольких рацемических
смесей, в том числе D,L-K3[Сг(С2О4)3] и D,L-a-[Co(glycinate)3].
г. Окись алюминия с покрытием. Карагунис с сотрудниками
[49] с целью разделения рацемических смесей оптически актив-
ных соединений приготовили адсорбент из окиси алюминия(1 II),
покрытый различными оптически активными веществами. Нес-
колько из этих попыток оказались успешными. Кребс и Раше
* dmg — диметилглиоксимино-анион.
* bzac — анион бензоилацетоната.
111. Получение оптически активных координационных соединений 53
[48а 1 и Пайпэ [50 ] также применили эту методику для разделения
рацемических смесей комплексных соединений.
д. Гидрат d-лактозы. Меллер с сотрудниками [51 ] и Колман
с сотрудниками [10] использовали этот адсорбент для разделения
нескольких внутрикомплексных соединений, например трис-
ацетилацетонатов трехвалентных металлов.
е. Методики с применением паровой фазы. Карагунис и Лип-
польд [63] использовали метод газовой хроматографии для раз-
деления рацемических смесей оптически активных органических
соединений, а Сиверс с сотрудниками [64] — для разделения не-
органических соединений. Сиверс использовал колонну, содержа-
щую порошкообразный (7-кварц, и разделил D,L-[Сг (гексафтор-
ацетилацетонат)3] хроматографически по схеме газ — твердое
вещество.
ж. Другие адсорбенты. С целью разделения рацемических
смесей оптически активных соединений в качестве адсорбентов
было применено несколько других веществ. Не все выбранные ве-
щества оказались пригодными для этого, но и не все вещества бы-
ли испытаны. Кребс с сотрудниками [48г] и Мейер и Мергер [52]
применили целлюлозу; Кребс с сотрудниками [48г] и другие ис-
следователи [53, 54] использовали ионообменные смолы с опти-
чески активными группами; Карагунис и сотрудники [49] при-
меняли (7-фенхон, (7-глюкозу, (7-винную кислоту, Z-глутамат на-
трия и (7-лактозу; Бюшар и Сюши [55] использовали метод
хроматографии на бумаге для комплексов, а другие исследователи
[56, 57] — для органических соединений; силикагель, покрытый
оптически активными веществами, был использован Кюрти и Ко-
ломбо [58] и Клеимом и Ридом [59]; и, наконец, сахароза была
применена Карассити [60]. Эта методика была также использо-
вана Джемисоном и Тарнером и другими исследователями [61 ].
Другие подобные методики находятся на стадии разработки и
должны дать плодотворные результаты. Из последних следует от-
метить избирательное внедрение простой энантиоморфной формы
в оптически активную «клетку», которая, по-видимому, образу-
ется при кристаллизации мочевины, а также разложение простых
энантиоморфных форм катализаторами, содержащими оптически
активные субстраты, например (7-кварц.
11. Методы зонной плавки
Дорон и Киршнер [62] применили зонную плавку для частич-
ного разделения при низкой температуре смесей диастереоизоме-
ров и рацемических комплексов. Растворителем для первых слу-
жила вода, для вторых — смесь воды и диоксана, содержащая
оптически активную соль. Эта соль непосредственно не взаимо-
54
Глава 2
действовала с энантиоморфными формами. Таким образом, в по-
следнем случае для разделения использовали эффект конфигура-
ционной активности Дуайера. Аппаратура была почти целиком
размещена в низкотемпературной холодильной установке и не
требовала особого внимания в процессе выполнения большинства
операций. Сущность метода заключается в следующем: смесь ди-
астереоизомеров (или энантиоморфных форм в оптически активной
среде) растворяется с неодинаковыми скоростями в расплавленной
зоне, которая движется вдоль колонки замерзшего раствора.
Вследствие этого расплавленная зона обогащается веществами,
которые наиболее быстро переходят в расплав. В результате один
конец колонки с замерзшим растворителем обогащается одними
веществами, другой конец — другими.
Б. Методы получения без разделения
1. Равновесные методы получения энантиоморфных форм
а. Различия в растворимости. В опытах по разделению неко-
торых рацемических смесей было найдено, что в ряде случаев ме-
тод осаждения позволял выделить практически 100% одной энан-
тиоморфной формы, несмотря на то что исходная смесь содержала
только 50% этого изомера и 50% его зеркально отраженной фор-
мы. Это происходило вследствие наличия динамического равнове-
сия между энантиоморфными формами в растворе. Осаждение од-
ной энантиоморфной формы в виде диастереоизомера вызывает
быстрое смещение равновесия в сторону образования этой формы.
Снова проводят осаждение добавочной порцией разделяющего
агента. Таким образом достигают полного превращения в данную
энантиоморфную форму, которая образует менее растворимую
соль с данным разделяющим агентом, чем ее антипод.
При проведении опытов по разделению D, L-K3[Сг(С2О4)3]
стрихнином Вернер [65] отмечал, что в растворе этилового спир-
та образуется только правовращающая энантиоморфная форма,
в то время как в воде образуется только левовращающий изомер.
Без сомнения, это объясняется различной растворимостью солей
стрихнина этих энантиоморфных форм в двух данных растворите-
лях. Дуайер и Гьярфас [66] писали, что раствор D,L- [Fe(o-phen)3]2+,
содержащий избыток rf-KSbOtart, дает осадок, в котором все комп-
лексные железосодержащие соединения представлены только
L-[Fe(o-phen)3]-tZ-SbOC4H4O6-4H2O. Это объясняется тем, что
эффект динамического равновесия сочетается со значительно мень-
шей растворимостью в воде этого диастереоизомера по сравнению
с растворимостью левовращающей формы данного железосодер-
III. Получение оптически активных координационных соединений 55
жащего комплекса. В литературе имеются также другие аналогич-
ные примеры получения энантиоморфных форм [67 ].
б. Окислительно-восстановительные процессы. Другие анало-
гичные методы, при использовании которых оказалось возможным
получение энантиоморфных форм с «выходом» больше ожидаемого,
были описаны Бушем [68]. Он обрабатывал D,L-[Со(еп)3]3+ из-
бытком d-тартрата, что приводило к осаждению фракции право-
вращающего изомера этого комплекса. Дальнейшую рацемизацию
фильтрата осуществляли добавлением [Со(еп)3]2+, что приводило
к дополнительному образованию d-изомера, осаждаемого далее
d-тартратом. В конечном счете 3/4 кобальтсодержащего комплекса
было получено в форме d-изомера.
Дуайер и Гьярфас [66, 69а ] разработали метод получения опти-
ческих энантиоморфных форм восстановлением. Например, В- и
L-[Ru(o-phen)3](ClO4)2 независимо окисляются нитратом це-
рия(1У) до трехвалентного состояния рутения (с сохранением оп-
тической активности). Таким путем все четыре энантиоморфные
формы были получены с высокой степенью оптической чистоты.
Далее оказалось возможным (с сохранением оптической актив-
ности) восстановить рутений в энантиоморфных формах сульфа-
том железа(П) и получить двухвалентный рутений в этой молекуле
[70]. Им и Буш [71] показали, что оптически активные пропи-
лендиаминтетраацетатные комплексы кобальта(П) и (III) можно
получить при помощи окислительно-восстановительных реакций,
аналогичных описанным выше.
2. Асимметрический и диссимметрический синтез
а. Абсолютный (общий) асимметрический синтез. Нет единого
мнения в отношении определения терминов асимметрический
синтез (диссимметрический синтез, если образующееся оптиче-
ски активное соединение содержит элементы симметрии), аб-
солютный асимметрический синтез и частично асимметрический
синтез [72]. Фишер [73] и Марквальд [74] рассматривают асим-
метрический синтез как процесс, в котором оптически активные
вещества синтезируют из оптически неактивных веществ с исполь-
зованием оптически активных реагентов, выполняющих роль
только промежуточных звеньев, и без использования методик раз-
деления. Абсолютный асимметрический синтез обычно рассмат-
ривают как процесс, в котором оптически активные молекулы по-
лучают из оптически неактивных веществ без использования как
методов разделения, так и оптически активных реагентов. Однако
при этом не исключается использование таких явлений, как кру-
говая поляризация света и т. п. Несмотря на то что в понятие
абсолютного асимметрического синтеза для органических соедине-
56
Глава 2
ний [75] входит асимметрическая деструкция последних поля-
ризованным по кругу светом [43, 44в], для координационного сое-
динения считают, что в этих условиях достигается абсолютный
асимметрический синтез.
б. Частично асимметрический синтез и частично диссимметри-
ческий синтез. Частично асимметрический синтез (частично дис-
симметрический синтез для оптически активных соединений, со-
держащих элементы симметрии) обычно рассматривают как син-
тез оптически активных молекул из оптически неактивных моле-
кул без использования какой-либо методики разделения, но с
использованием оптически активных реагентов, которые входят
в состав молекулы продукта. Сущность синтеза заключается в
том, что оптически активные реагенты приводят к образованию
преимущественной оптически активной конфигурации в молекуле
продукта, частью которой они сами становятся, например комп-
лексное соединение платины (IV), содержащее пропилендиамин
[Pt(Z-pn)3]4+. По-видимому, на конфигурацию (и вращение) ка-
тиона в целом может влиять присутствие оптически активных ли-
гандов, и часто образуется только один изомер комплекса [76,77],
если присутствует лишь один энантиоморф лиганда.
Другим примером служит реакция между кобальтом(П) и
<7-нг/?анс-1,2-диаминциклопентаном, который (после окисления)
дает практически только энантиоморфный L-[Co(d-cptn)3]3+[78].
3. Методы замещения
Определенные реакции замещения оптически активных энан-
тиоморфных форм комплексов протекают с сохранением (или ин-
версией) оптической активности, что дало особый метод синтеза
оптических изомеров. Дуайер с сотрудниками [39] сообщали о
получении D-[Со(еп)3]3+ после обработки D- [Co(edta) ]“ этилендиа-
мином. Киршнер и сотрудники [40] сообщали о синтезе
L-[Co(Z-pn)3]3+ по реакции D-[Co(edta)]~ с Z-пропилендиамином.
Мессинг и Басоло [79] окислили L- [Pt(wis-stien) (Z-bn)]2+* и по-
лучили L-[Pt(wis-stien) (Z-bn)Cl2]Cl2, которое после восстановле-
ния дало первоначальный комплекс платины(П) с его первона-
чальным оптическим вращением.
Интересный пример применения этого метода представляют
исследования Бейлара и Отена [80], которые наблюдали, как
обработка L-[Co(en)2Cl2]+ карбонатом калия приводила к образо-
ванию D-[Co(en)2CO3]+, а обработка карбонатом серебра — к об-
разованию либо D-, либо L- [Со(еп)2С03]+ в зависимости от усло-
вий проведения реакции. Это свидетельствует о том, что в некото-
рых из этих реакций происходит превращение Вальдена.
* ms-stien — .иезо-стильбендиамип; i-bn — изо-бутилендиамин.
IV. Оптическая чистота
57
IV. ОПТИЧЕСКАЯ ЧИСТОТА
А. Определения
Оптическую чистоту частично разделенного материала опреде-
ляют как избыток одной энантиоморфной формы, выраженный в
процентах от общего количества оптически активного вещества.
Для случая, когда правовращающий энантиоморф находится в из-
бытке, имеем
О. Р. =(£,/7’)-100 = [(D — L)/(D + L)]-100, (4)
где О. Р.— оптическая чистота (%), Е — избыток одной энантио-
морфной формы, Т — общее количество оптически активного ма-
териала, DHL — количества право- и левовращающих энантио-
морфных форм соответственно. Если в паре d и L количества
обеих форм одинаковы, то оптическая чистота равна нулю. Для
полностью разделенного вещества «избыток» энантиоморфных
форм равен по весу общему количеству оптически активного ма-
териала, и оптическая чистота составляет 100%.
Б. Критерии оптической чистоты
Наиболее часто используемым критерием оптической чистоты,
указывающим на полную оптическую чистоту кристаллического
вещества, является неизменность температуры плавления и опти-
ческого вращения вещества после его повторной кристаллизации.
Но этот критерий не следует принимать в качестве безошибочного,
так как некоторые твердые вещества могут образовывать частич-
но разделенные смеси, у которых после рекристаллизации тем-
пература плавления и оптическое вращение остаются неизмен-
ными. Другой часто используемый критерий заключается в полу-
чении энантиоморфных форм, имеющих равные удельные враще-
ния с противоположными знаками. Однако этот критерий следует
принимать с осторожностью до тех пор, пока одни те же энантио-
морфные формы, обладающие одними и теми же удельными вра-
щениями, не будут получены двумя или несколькими различными
методами.
В. Определение оптической чистоты
Существуют три основных метода определения оптической чис-
тоты оптически активных энантиоморфных форм. Ни один из этих
методов не был использован для определения оптической чистоты
неорганических комплексных соединений. Но в принципе эти
методы применимы и для неорганических соединений.
58
Т* л л в л 2
1. Биологический метод
Если какие-либо ферменты, бактерии или другие организмы
селективно взаимодействуют с одной энантиоморфной формой ра-
цемической пары, то тогда контакт противоположного изомера с
этой биологической культурой не вызовет реакции. Наличие ре-
акции укажет на нечистоту изомера. Для каждой системы следует,
конечно, разработать метод обнаружения реакции.
2. Метод сопряженных реакций
Этот метод позволяет определить минимальную степень оп-
тической чистоты полученной энантиоморфной формы при том
условии, что имеется выделенное оптически активное вещество
с известной оптической чистотой, например D-lM(abcd)]. Пусть,
например, следует получить оптически чистый D-[M(abce)], ко-
торый можно превратить химически в D-[M(abcd)]. Степень опти-
ческой чистоты первого соединения должна по меньшей мере быть
равной степени оптической чистоты второго соединения, из кото-
рого первое было получено. D-[M(abce)] может обладать большей
степенью оптической чистоты по сравнению с D-[M(abcd)], так
как в процессе химической реакции может происходить некоторая
рацемизация [81 ].
3. Метод изотопного разбавления
Этот метод заключается в смешивании в растворе предположи-
тельно чистой энантиоморфной формы (например, D-изомера) и
радиоактивного (или иным способом меченного) рацемического ве-
щества. Затем рацемическое вещество разделяют и сравнивают
рассчитанный фактор (радиоактивного) разбавления с найденным
экспериментально. Заниженная величина наблюдаемого фактора
разбавления по сравнению с рассчитанным (исходя из предположе-
ния о чистой исходной D-форме) указывает на примесь противо-
положной формы, количество которой можно вычислить [82, 83].
Смешивание чистой D -формы с меченым рацемическим веществом
приведет после повторного разделения рацемического вещества
к понижению радиоактивности только в D-изомере, но не в L-изо-
мере. Ожидаемый фактор разбавления можно рассчитать по ве-
совым количествам исходной энантиоморфной формы и добавлен-
ного меченого рацемического материала. Если исходное вещество
состояло из немеченых D- и L-форм, то его смешивание с меченым
рацемическим веществом приведет к разбавлению обеих энантио-
морфных форм. Фактор разбавления возрастает по сравнению с
V. Препаративные методики
59
прежним. В математической форме эти соотношения можно пред-
ставить следующим образом:
С± = [а Со] (а + В)/(2В + а — В) (а + /?)], (5)
где С±— активность повторно разделенной рацемической смеси,
Со— активность добавленного рацемического материала, а —
вес добавленного рацемического материала, В — вес отделенной
энантиоморфной формы (оптическая чистота которого неизвестна),
ранее смешанного с а, и В — вес некоторого рацемата (если та-
ковой имеется) в В. Решение уравнения (5) относительно В по-
зволит вычислить оптическую чистоту (%) при помощи следующе-
го выражения:
О. Р. =[(В — 7?)/B]-100, (6)
V. ПРЕПАРАТИВНЫЕ МЕТОДИКИ
Большая часть упоминаемой в предыдущих разделах литера-
туры содержит экспериментальные данные и указания, необходи-
мые для выполнения описываемых процедур. Кроме того, в по-
следнем разделе этой главы приведен список некоторых оптически
активных комплексных неорганических соединений, которые по-
лучают в виде энантиоморфных форм. Приведена литература по
практическому проведению синтезов. Рекомендуемая литература,
помещенная ниже, облегчит читателю выбор статей, в которых
освещаются экспериментальные подробности синтезов многих
энантиоморфных форм координационных соединений.
Особого внимания среди рассмотренных методик заслуживают
некоторые экспериментальные процедуры, используемые при
осуществлении наиболее часто применяемых методов синтеза.
Для метода избирательной адсорбции в одной из последних ста-
тей [10] приведена подробная инструкция по изготовлению перио-
дически действующей адсорбционной колонки, наполненной гид-
ратом d-лактозы. Детальное описание метода конфигурационной
активности приведено в статьях Дуайера с сотрудниками [25, 26].
Подробности осуществления равновесных методов синтеза энан-
тиоморфных форм содержатся в ряде статей [66, 68, 69, 71] и
методов замещения — [39, 40, 79, 80].
Наиболее часто используемый метод состоит в образовании
диастереоизомеров. Большую часть указанной литературы можно
использовать для ознакомления с экспериментальными деталя-
ми синтеза. Описание нескольких совершенных препаративных
процедур для осуществления этого метода имеется в различных
томах «Неорганических синтезов». Интересным примером такого
разделения является модификация метода Дуайера и Гарваиа
60
Глава 2
[35], примененная для разделения рацемического иона ^ис-динит-
ро-бис-(этилендиамин)-кобальт(1П). Ниже дано описание мето-
дики.
Разделение Г),ъ-цис- [Co(en)2(NO2)2]+: 12,0 г в,к-цис-
[Co(en)2(NO2)2]NO2 [84а] растворяют в 50 мл воды при темпера-
туре 60° при энергичном перемешивании в 250-миллилитровой
колбе Эрленмейера. Далее в эту колбу быстро добавляют (при
энергичном встряхивании) раствор, состоящий из 7,0 г антимони-
ла-^-тартрата калия d-KSbOC4H4Oe в 40 ли воды при 75°. Смесь
быстро охлаждают до 25° водопроводной водой и неоплавленным
концом стеклянной палочки, служащей для перемешивания, скре-
бут внутренние стенки колбы ниже поверхности жидкости. Осажда-
ется диастереоизомер k-цис- [Co(en)2(NO2)2](d-SbOC4H4O6) в виде
мелких желтых кристаллов. Раствору дают отстояться в течение
точно 10 мин (при 25°) при периодическом встряхивании и затем
быстро отсасывают на воронке Бюхнера. Фильтрат (I) сохра-
няют до извлечения правовращающей энантиоморфной формы.
Кристаллы диастереоизомера промывают на воронке Бюхнера пор-
циями по 20 мл сначала 50%-ным водным раствором этилового
спирта, затем 100%-ным спиртом и наконец ацетоном. Кристаллы
сушат на воздухе при комнатной температуре. Выход составляет
5,5 г, [a]=+47,4°.
К 5,5 г диастереоизомера добавляют 30 мл воды и растирают
его в стеклянной ступке стеклянным пестиком. Добавляют 7,0 г
Nal и растирают смесь в течение 2 мин. Отфильтровывают слабо
растворимую L-tyuc[Go(en)2(NO2)2]I форму на воронке Бюхнера
и отмывают фильтрат 15 мл воды при 0°. Осадок и фильтроваль-
ную бумагу переносят в 100-миллилитровую колбу Эрленмейера,
добавляют 30 мл воды, нагретой до 55°, и энергично встряхивают
колбу. Добавляют 4,0 г свежеосажденного хлорида серебра и про-
должают энергично встряхивать колбу еще 3 мин. Иодид серебра
отфильтровывают на воронке Бюхнера и промывают осадок 5 мл
воды (при 55°), которая поступает непосредственно в фильтрат.
В ледяной бане добавляют к фильтрату 3,0 г бромида аммония.
Затем, чтобы вызвать кристаллизацию L-цис- [Co(en)2(NO2)2]Br,
скребут неоплавленным концом стеклянной палочки, служащей
для перемешивания, внутренние стенки колбы ниже поверхности
жидкости. Продукт отфильтровывают на воронке Бюхнера и про-
мывают последовательно 10-миллилитровыми порциями 50 % -ного
раствора вода — этанол, 100%-ного этанола и ацетона и высуши-
вают на воздухе. Выход составляет 3,1 г (47%) для продукта с
[аЬ =—44°.
Для выделения d-изомера тотчас же после извлечения 1-изо-
мера к фильтрату (I) добавляют 2,0 г бромида аммония, скребут
стенки колбы стеклянной палочкой (как описано выше) и дают в
Таблица 2
Оптически активные изомеры некоторых координационных соединений
Ион металла Комплекс Литература Лиганд
А1 (!!!) [Al (TS2)1+ 91 TS2— бис- (салицилаль- дегид) -триэтилентет- раамин (2—)а
As(V) К [As (cat)3]-H2O 92 cat—катехин (2—)
Ве(П) [Be (bzac)2] 46b bzac — анион бензоил- ацетоната
В(Ш) [B (sal)2J- 93 sal—анион салицила- та (2—)
Cd(II) [Cd (en)8] Cl2 92 еп—этилендиамин
Cr(III) [Cr (en)3]3+ tyuc-[Cr(en)2Cl2]+ 94 95
i{uc-[Cr(Z-pn)2Cl2]+ K3 [Cr(C2O4)3] K[Cr (en)(C2O4)2]-2H2O 96 21, 65, 97 97 Z-pn—левовращающий пропилендиамин
[Cr (acac)3] 51a асас—анион ацетилаце- тоната
Co(II) [Co (Z-pn)3]2+ 98
[Co (d-glut)2J 99 d-glut—анион d-глута- мата (1—)
[Co (pdta)]2" 71 pdta—анион пропилен- диаминтетраацета- та (4—)
Со(Ш) [Co (en)3]3+ i{uc-[Co(en)2(NH3)2]3+ i{uc-[Co (en)2(H2O)2]3+ цис-[Co(en)2(NH3)(NO2)]2+ ipc-[Co(en)2Cl2]+ Co (en)2CO3]+ [Co (en)(C2O4)2[- [Co (pn)3]3+ ' i{uc-[Co(Z-pn)2(NO2)2]+ 1, 62, 68, 100, 101 102, 103 104 105 106, 107 108 109 40, 76, 110 111
[Co (glyc)3] 48a glyc—анион глицината
[Co (edta)]- [Co (acac)3] [Co (C2O4)3]3- 35, 39, 40, 83, 112, 113 26a, 51a, 62 34, 114, 115 edta—анион этиленди- аминтетраацетата (4-)
62
П родолжение табл. 2
Ион металла Комплекс Литература Лиганд
Со(Ш)— Со(Ш) (дву- ядерный) [(en)2Co(NH2)(OH)Co(en)2]4+ 116
Со(Ш) (много- ядерный) [Co {(OH)2Co(NH3)4}3]6+ [Co{(OH)2Co(en)2}3]e+ 117 118
Cu(ll) [Cu(TS2)] 91
Ge(IV) [Ge (C2O4)3p- 119
Ir(III) [Ir (en)3]3+ [Ir (C2O4)3]«- 120 121
Fe(II) [Fe(dipy)3]2+ 122 dipy—2,2'- дипиридил
[Fe(o-phen)3]2+ 66 o-phen—о pm о-фенан- тролин
Fe(III) [Fe (dipy)3]3!- [Fe(o-phen)3]3+ 123 66
Ni(II) [Ni(dipy)3]2+ [Ni (o-phen)3]2+ 124 125
Os(II) [Os(dipy)3]2+ [Os(o-phen)3]2+ 696, 126, 127 128
Os(III) [Os (dipy)3]3+ [Os(o-phen)3]3+ 696, 126 123 i-bn— изо-бутилендп- амин; ms-stien—лезо-стиль-
Pd [Pd(i-bn)(ms-stien)]2+ 129
[Pd(Z-pn)2]2+ 130 бендиамин
Pt(II) [ Pt(Z-bn) (ms-stien) ]2+ [Pt(en)(Z-pn)]2+ 2 130
Pt (IV) [Pt(en)3p + [Pt(pn)3p + mpa«c-[Pt(Z-bn)(ms-stien)Cl2]2+ 131 77 79
Rh(III) [Rh(en)3]3+ i{uc-[Rh(en)2Cl2l+ [Rh (C2O4)3]3- 132 133 134
[Rh(NHSO2NH)2(H2O)2]- 135
Ru(II) [Ru(dipy)3]2+ [Ru (o-phen)3]2+ 69a, 136 69a, 137
Ru(III) [Ru(dipy)3]3+ [Ru (o-phen)3]3+ 69, 137 137, 138
Литература
63
Продолжение табл. 2
Ион металла Комплекс Литература Лиганд
Si(IV) [Si(acac)3]+ 3, 4
Ti(IV) [Ti(cat)3r 139
Zn(II) [Zn(en)3]2+ 140
[Zn (qsa)2] 36 qsa—анион 8-хинолп- но-5-сульфоновой кислоты
а В скобках указаны величина и знак заряда иона.
течение 5 мин при 20—25° кристаллизоваться в форме бромида
загрязненному с?-изомеру. Осадок вместе с фильтровальной бумагой
переносят в колбу Эрленмейера на 100 мл, добавляют 35 мл воды
при 55° и энергично встряхивают колбу. Добавляют 4,0 г свеже-
осажденного хлорида серебра и продолжают энергично встряхи-
вать колбу еще 3 мин. Отфильтровывают осажденный иодид сереб-
ра на воронке Бюхнера, промывают садок 5 мл воды (при 55°),
которую сливают в фильтрат. К фильтрату добавляют 1,5 а
NH4Br, перемешивают содержимое колбы и выдерживают раствор
при 20—25° в течение 10 мин. Кристаллизующийся т>-цис-
[Co(en)2(NO2)2]Br следует отделить от фильтрата, промыть и вы-
сушить, используя приемы, описанные для Z-изомера. Выход со-
ставляет 3,1 г (47%), [а]д =+44°.
VI. ЗАКЛЮЧЕНИЕ
Известны оптические изомеры более чем для двух сотен коор-
динационных соединений. На эту тему [846—89] в литературе име-
ется ряд обзорных статей. Обширный перечень этих соединений
был составлен Уолдбаем [90].
В табл. 2 приведен ряд интересных комплексов, энантиоморф-
ные формы которых были получены при помощи одного или не-
скольких методов, описанных в этой главе.
ЛИТЕРАТУРА
1. Werner A., Chem. Вег., 45, 121 (1912).
2. М i 11 s W. Н., Q u i b е 11 Т. Н. Н., J. Chem. Soc., 1935, 839.
3. D h а г S. К., D о г о n V. F., KirschnerS., J. Am. Chem. Soc.,
80, 753 (1958).
64
Глава 2
4. D h а г S. К., D о г о n V. F., KirschnerS.,1. Am. Chem. Soc.,
81, 6372 (1959).
5. Mathieu J. Р., Bull. Soc. Chim. France, 6, 873, 1258 (1938).
6. Jaeger F. M., Optical Activity and High Temperature Measurements,
McGraw-Hill, New York, 1930.
7. MatoushW. R., BasoloF., J. Am. Chem. Soc., 78, 3972 (1956).
8. Albinak M. J., Bhatnagar D. C., Kirschner S., Son-
ne s s a A. J., Can. J. Chem., 39, 2360 (1961); Advances in the Chemistry
of the Coordination Compounds, Kirschner S., ed., Macmillan, New York,
1961, pp. 154И.
9. Brushmiller J. G.,Amma E. L., Douglas В. E., J. Am.
Chem. Soc., 84, 111 (1962).
10. Collman J. P., Blair R. P., Marshall R. L., Sla-
d e S., Inorg. Chem., 2, 576 (1963).
11. К i r s c h n e r S., J. Am. Chem. Soc., 78, 2372 (1956); Lut.tring-
h a u s А., В e г r e r D., Tetrahedron Letters, 1959, № 10, p. 10.
12. Arago E., Memoir de 1’Inst., 12, Part I, 93 (1811).
13. В i о t J. B., Memoir de 1’Inst., 13, Part I, 1 (1812).
14. Heller W., Fitts D., in Physical Methods of Organic Chemistry,
3rd ed., Weissberger A., ed., of the Technique of Organic Chemistry Series,
Vol. I, Part 3, Interscience, New York, 1960, Chap. 33.
15. Lowry T. M., Optical Rotatory Power, Longmans, Green, London,
1935.
16. M о с к о в и ц, в книге Джерасси К. Дисперсия оптического вра-
щения, ИЛ, Москва, 1962.
17. Джерасси К., Дисперсия оптического вращения, ИЛ, Москва, 1962.
Kauzmann W., Walter J., Eyring H., Chem. Rev., 26, 339
(1940); Condon E., Rev. Mod. Phys., 9, 432 (1937); Kuhn W.,
Ann. Rev. Phys. Chem., 9, 417 (1958); Moffitt W., J. Chem. Phys.,
25, 1189 (1956); Powell H. M., Endeavour, 15, 20 (1956); В о л ь-
кенштейн М., Журн. эксп. теорет. физ., 20, 342 (1950); Servant
R., J. Phys. Radium, 3, 90 (1942); К i г k w о о d J., J. Chem. Phys., 5,
479 (1937); Born M., Proc. Roy. Soc. (London), A150, 84 (1935); A153
339 (1936); Betti M., Trans. Faraday Soc., 26, 337 (1930); W о Id-
fa у e F., Acta Chem. Scand., 13, 2137 (1959).
18. Moeller T., Inorganic Chemistry, Wiley, New York, 1952, p. 265.
19. Уилки н, Уильямс, в книге «Современная химия координацион-
ных соединений» под. ред. Льюиса Дж. и Улкинса Р., ИЛ, Москва, 1963.
20. Р a s t е u г L., Ann. Chim. Phys., 24, 442 (1848).
21. J а е g е г F. М., Rec. Trav. Chim.,38, 250 (1919).
22a. Werner A., Bosshart J., Chem. Ber., 47, 2171 (1914); 6. S e-
c о r R. M., Chem. Rev., 63, 297 (1963).
23. Del epine M., Bull. Soc. Chim. France, 29, 656 (1921); 1, 1256
(1934).
24a. D el ep in e M., CharonnatR., Bull. Soc. Franc. Mineral., 53, 73
(1930); 6. J a m i s о n M. M., T u r n e r E. E., J. Chem. Soc., 1942, 437.
25. D w у er F. P., G у a r f a s E. C., O’D w у e r M. F., Nature, 167, 1036
(1951).
26. D w у e r F. P. G у a r f a s E. C., Nature, 168, 29 (1951); Davies
N. R., D w у e r F. P., Trans. Faraday Soc., 50, 24 (1954).
27. P a s t e u r L., Compt. Rend., 46, 615 (1858).
28. P r i с e V. E., G r e e n s t e i n J. P., J. Biol. Chem., 175, 969 (1948).
29. GreensteinJ. P., ’’The Resolution of Racemic a-Amino-Acids”, in
Advances in Protein Chemistry, Vol. 9, M. L. Anson, Bailey K., Edsall
J. T., eds., Academic Press, New York, 1954, pp. 121—202.
30. CrespiM. J. S., Dissertation Abstr., 20, 4512 (1960).
Литература
65
31. В a i 1 е у М. Е., Н a a s Н. В , J. Am. Chem. Soc., 63, 1969 (1941).
32. В u 11 е г С. L., С г е t с h е г L. Н., J. Am. Chem. Soc., 55, 2605 (1933).
33. Z etzsche F., Hubacher M., Helv. Chim. Acta, 9, 291 (1926).
34. D wy er F. P., Sarges on A. M., J. Phys. Chem., 60, 1331 (1956).
35. D wy er К. P., G ar v an F. L., in Inorganic Syntheses Vol. 6, Rochow
E. G., ed., McGraw-Hill, New York, 1960, p. 195.
36. L i u J. С. I., В a i 1 a r J. C., Jr., J. Am. Chem. Soc., 73, 5432 (1951).
37. В a i 1 a r J. C., Jr., J onassen H. B., Gott A. D., J. Am. Chem.
Soc., 74, 3131 (1952).
38. G о 11 A. D., В a 11 a r J. C., J. Am. Chem. Soc., 74, 4820 (1952).
39. D w у e r F. P., G у a r f a s E. С., M e 11 о r D. P., J. Phys. Chem.,
59, 296 (1955).
40. К i r schn er S., W ei Y. К., В a i 1 a r J. C., Jr., J. Am. Chem. Soc.,
79, 5877 (1957).
41. Das Sarma В., В a i 1 a r J. C., Jr., J. Am. Chem. Soc., 77, 5480
(1955).
42. Kuhn W., В r a u n E., Naturwissenschaften, 17, 227 (1929).
43. Kuhn W., Knopf E., Z. Physik. Chem. (Leipzig), 7, 292 (1930);
Naturwissenschaften, 18, 198 (1930).
44a. J a eg er F. M., Berger G., Rec. Trav. Chim., 40, 153 (1921);.
Tsuchida R., Nakamura A., Kobayashi M., J. Chem.
Soc. Japan, 56, 1335 (1935).
45. Tsuchida R., Kobayashi M., Nakamura A., Bull. Chem.
Soc. Japan, 11, 38 (1936); К u г о у a H., Tsuchida R., Bull. Chem.
Soc. Japan, 15, 427 (1940); N akahar a A., Tsuchida R., J. Am.
Chem. Soc., 76, 3103 (1954).
46a. В a i 1 a r J. C., Jr., Peppard D. F., J. Am. Chem. Soc., 62, 105
(1940); 6. Busch D. H., В a i 1 a r J. C., Jr., J. Am. Chem. Soc., 75,
4574 (1953); 76, 5352 (1954); в. I r v i n g H., G i 1 1 J. B., Proc. Chem.
Soc., 1958, 168; r. Kuebler J. R., Jr., В a i 1 a r J. C., Jr., J. Am.
Chem. Soc., 74, 3535 (1952); д. D a s S а г m а В., В a i 1 a r J. C., Jr.,
J. Am. Chem. Soc., 77, 5476 (1955); e. Schweitzer G. К., T a 1-
b о t С. K., J. Tenn. Acad. Sci., 25, 143 (1950).
47. F e г г о n i E., C i n i R., J. Am. Chem. Soc., 82, 2427 (1960).
48a. Krebs H., R asche R., Z. Anorg, Allgem. Chem., 276, 236 (1954);
Naturwissenschaften, 41, 63 (1954); б. К r e b s H., Die waldJ., Wag-
fa e r J., Angew. Chem., 67, 705 (1955); в. KrebsH., DiewaldJ.,
Ar lot H., Wagner J., Z. anorg. allgem. Chem., 287, 98
(1956); r. Krebs H., D i ewa Id J., Rashe R., Wagner J.,
Die Trennung von Racematen auf Chromatographischen Wege, West-
deutscher Verlag, Cologne, 1956.
49. Karagounis G., Char bonnier E., Floss E., J. Chroma-
tog., 2, 84 (1959).
50. Piper T. S., J. Am. Chem. Soc., 83, 3908 (1961).
51a. Moeller T., Gulyas E., J. Inorg. Nucl. Chem., 5, 245 (1958); 9,
82 (1958); б. H s e u T. M., M a r t i n D. F., M о e 1 1 e r T., Inorg. Chem.,
2, 587 (1963).
52. Mayer W., M e r g e r F., Ann. Chem., 644, 65 (1961).
53. Arnstein H. R. V., Morris D., Biochem. J., 76, 318 (1960);
Jaeger M., Iskric S., Wickerhauser M., Croat. Chem.
Acta, 28, 5 (1956).
54. Truboyama S., Yanagita M., Sci. Papers Inst. Phys. Chem.
Res. (Tokyo), 53, 245 (1959).
55. В u c h a r E., S u c h у К., Magy. Кет. Folyoirat, 64, 45 (1958).
56. Kikkawa I., Yakugaku Zasshi, 81, 732 (1961).
6 Заказ № 547
66
Глава 2
57. К о t а к е М., S а к a n Т., Nakamura N., S е п о h S., J. Am.
Chem. Soc., 73, 2973 (1951).
58. CurtiR., Colombo U., J. Am. Chem. Soc., 74, 3961 (1952); Gazz.
Chim. Ital., 82, 491 (1952).
59. Klemm L. H., Reed D., J. Chromatog., 3, 634 (1960).
60. С a r a s s i t i V., Ann. Chim., 47, 1337 (1957); 48, 167 (1958); J. Chim.
Phys., 55, 120 (1958).
61. Jamison M. M., T u r n e r E. E., J. Chem. Soc., 1942, 611; G i 1 -
A v E., N u г о к D., Proc. Chem. Soc., 1962, 146.
62. D о г о n V. F., К i r s c h n e r S., Inorg. Chem., 1, 539 (1962).
63. Karagounis G.,Lippold G., Naturwissenschaften, 46,145(1959).
64. Sievers R., Moshier R. W., Morris M. L., Inorg. Chem., 1,
966 (1962); G о 1 d b e r g G., R о s s W. A., Chem. Ind. (London), 1962,
657.
65. W e r n e r A., Chem. Ber., 45, 3061 (1912).
66. D wy er F. P., Gy arf as E. C., J. Proc. Roy. Soc., N. S. Wales, 83,
263 (1950).
67. Jonassen H. B., BailarJ.C., Jr., Huffman E. H., J. Am.
Chem. Soc., 70, 756 (1948).
68. В u s c h D. H., J. Am. Chem. Soc., 77, 2747 (1955).
69 a. Dwyer F. P., G у a r f a s E. C., J. Proc. Roy. Soc. N. S. Wales, 83,
170, 174 (1949); 6. Nature, 166, 481 (1950).
70. Brandt W. W., Dwyer F. P., G у a r f a s E. C., Chem. Rev., 54, 997
(1954).
71. I m Y. A., В u s c h D. H., J. Am. Chem. Soc., 83, 3362 (1961); Busch
D. H., С о о к e D. W., Swaminathan K., Im Y. A., in Advances
in the Chemistry of the Coordination Compounds, Kirschner S., ed., Mac-
millan, New York, 1961, p. 139.
72. И л и e л Э., Стереохимия соединений углерода, изд-во «Мир»,
Москва 1965
73. Fischer Е., Chem. Вег., 27, 3189, 3231 (1894).
74. Marckwald W., Chem. Ber., 37, 1368 (1904).
75. D a v i s Т. L., Н е g g i е R., J. Am. Chem. Soc., 57, 377 (1935).
76. TschugaeffL., Sokoloff W., Chem. Ber., 42, 55 (1909).
77. Smirnoff A. P., Helv. Chim. Acta, 3, 177 (1920).
78. J aegerF. M., BlumendalH. B., Z. anorg. allgem. Chem., 175,
161 (1928); Jaeger F. M., Koninkl. Ned. Akad. Wetenschof. Proc.,
40, 108 (1937).
79. Messing A. F., В a s о 1 о F., J. Am. Chem. Soc., 78, 4511 (1956).
80. BailarJ.C., Jr., A u t e n R. W., J. Am. Chem. Soc., 56, 774 (1934).
81. Burwell R. L., S h i e 1 d s A. D., Hart H., J. Am. Chem. Soc.,
76, 908 (1954).
82. В e r s о n J. A., Ben-EfraimD. A., J. Am. Chem. Soc., 81, 4083
(1959).
83. GraffS., Rittenberg D., К о s t e r G. L., J. Biol. Chem., 133,
745 (1940).
84a . Holtzclaw H. F., Sheetz D. P., McCarty B. D., in Inor-
ganic Syntheses, Vol. 4, Bailar J. C., Jr., ed., McGraw-Hill, New York,
1953, p. 176; б. В u s c h D. H., in Cobalt, Young R. S., ed., Reinhold,
New York, 1960, p. 94.
85. В a s о 1 о F., Chem. Rev., 52, 459 (1953).
86. M a r t e 11 A. E., С a 1 v i n M., Chemistry of the Metal Chelate Com-
pounds, Prentice-Hall, Princeton, New Jersey, 1952.
87. Уилкин и Уильямс, в книге «Современная химия координа-
ционных соединений» под. ред. Лыоиса Дж. и Уилкинса Р., ИЛ,
Москва, 1963.
Литература
67
88. Б а с о л о, в книге «Химия координационных соединений» под ред.
Бейлара и Буша, ИЛ, Москва, 1960.
89. В a s о 1 о F., Pearson R. G-, Mechanisms of Inorganic Beactions,
Wiley, New York, 1958, p. 258.
90. Wo idby e F., ’’Technique of Optical Rotatory Dispersion and Circu-
lar Dichroism”, in Technique of Inorganic Chemistry, Vol. IV, Jonassen
H. B., Weissberger A., eds., Interscience, New York, to be published.
91. Das Sarma B., Bailar J. C., Jr., J. Am. Chem. Soc., 77, 5476
(1955).
92. W e i n 1 a n d R. F., H e i n z 1 e r J., Chem. Ber., 52, 1322 (1919);
Rosenheim A., Plato W., Chem. Ber., 58, 2000 (1925).
93. F a u 1 k n e r I. J., L о w г у T. M., J. Chem. Soc., 127, 1080 (1925).
94. Werner A., Chem. Ber., 45, 865 (1912).
95. Werner A., Chem. Ber., 44, 3138 (1911); S e 1 b i n J., BailarJ.C.,
Jr., J. Am. Chem. Soc., 79, 4285 (1957).
96. R о 1 1 i n s о n C. L., BailarJ.C., Jr., J. Am. Chem. Soc., 66, 641
(1944).
97. В u s h r a E., J ohnsonC. H., J. Chem. Soc., 1939, 1937.
98. W о 1 d b у e F., Optical Rotatory Dispersion of Transition Metal Comp-
lexes, European Research Office, U. S. Army, Frankfurt a. M., 1959; Re-
cord Chem. Progr. (Kresge-Hooker Sci. Lib.), 24, 197 (1963).
99. Lifschitz I., Schouteden F. L. M., Rec. Trav. Chim., 58,
411 (1939).
100. D о u g 1 a s В. E., J. Am. Chem. Soc., 76, 1020 (1954).
101. Broomhead J. A., Dwyer F. P., Hogarth J. W., in Inorga-
nic Syntheses, Vol. 6, Rochow E. G., ed., McGraw-Hill, New York, 1960,
p. 183.
102. Werner A., S h i b a t a Y., Chem. Ber., 45, 3287 (1912).
103. Archer R., Bailar J. C., Jr., J. Am. Chem. Soc., 83, 812 (1961).
104. Mathieu J. P., Bull. Soc. Chim. France, 4, 687 (1937).
105. M a t h i e u J. P., Bull. Soc. Chim. France, 3, 476, 463 (1936).
106. W e r n e r A., Chem. Ber., 44, 3279 (1911).
107. Б e й л a p Дж., мл., в книге «Неорганические синтезы», сб. 2, Издат-
инлит, Москва, 1951.
108. Werner A., Cutcheon М., Chem. Вег., 45, 3281 (1912).
109. Dwyer F. Р., Reidl. К., G а г v a n F. L., J. Am. Chem. Soc., 83,
1285 (1961).
110. Dwyer F. Р., Gar van F. L., Shulman A., J. Am. Chem. Soc.,
81, 290 (1959).
111. О’Brien T. D., McReynoldsJ. P., BailarJ.C., Jr., J. Am.
Chem. Soc., 70, 749 (1948).
112. В uschD. H., В ailar J. C., Jr., J. Am. Chem. Soc., 75, 4574 (1953).
113. WeakliemH. A., H о a r d J. L., J. Am. Chem. Soc., 81, 549 (1959).
114. D elepine M., Bull. Soc. Chim. France, 1, 1256 (1934).
115. Jaeger F. M., Thomas W., Koninkl. Ned.Akad. Wetenschof.
Proc., 21, 693 (1918).
116. Werner A., Chem. Ber., 47, 1961 (1914).
117. W e r n e r A., Chem. Ber., 47, 3087 (1914); Compt. Rend., 159, 426 (1914).
118. Goodwin H. A., G у a r f a s E. С., M e 1 1 о r D. P., Australian J.
Chem., 11, 426 (1958).
119. M о e 1 1 e r T., N i e 1 s e n N. C., J. Am. Chem. Soc., 75, 5106 (1953).
120. Werner A., Smirnoff A. P., Helv. Chim. Acta, 3, 472 (1920);
M a t h i e u J. P., J. Chim. Phys., 33, 78 (1936).
121. D e 1 ё p i n e M., Compt. Rend., 159, 239 (1914); 175, 1409 (1922).
122. W e r n e r A., Chem. Ber., 45, 433 (1911); Dwyer F. P., Gyarfas
E. C., J. Proc. Roy. Soc. N. S. Wales, 85, 126 (1951).
6*
G8
Глава 2
123. DwyerF. Р., G а у г f a s Е. С., J. Am. Chem. Soc., 74, 4699 (1952).
124. Morgan G. Т., Burstall F. H., J. Chem. Soc., 1931, 2213; J a e-
g e r F. M., van D i j к J. A., Z. anorg. allgem. Chem., 227, 304 (1936);
Schweitzer G. K., Lee J. M., J. Phys. Chem., 56, 195 (1952).
125. D wyer F. P., Gy arf as E. C., J. Proc. Roy. Soc., N. S. Wales, 83,
232 (1949).
126. D wyer F.P.,Gy arf asE.C., J. Am. Chem. Soc., 73, 2322 (1951).
127. Burstall F. H., DwyerF. P., G у a r f a s E. C., J. Chem. Soc.,
1950, 953.
128. D w у e r F. P., G у a r f a s E. C., GibsonN. A., J. Proc. Roy. Soc.,
N. S. Wales, 84, 68 (1950).
129. L i d s t о n e A. G., M i 1 1 s W. H., J. Chem. Soc., 1939, 1754.
130. TchugaelfL., Sokol of fW., Chem. Ber., 40, 3461 (1907).
131. W er n e r A., Vierteljahressch. Naturforsch. Ges. Zuerich, 62, 553 (1917);
MathieuJ.P., Bull. Soc. Chim. France, 6, 1258 (1939); J. Chim. Phvs.,
33, 28 (1936).
132. W e r n e r A., Chem. Ber., 45, 1228 (1912).
133. An d er s en S., В as о 1 о F., J. Am. Chem. Soc., 82, 5953 (1960).
134. W e r n e r A., Chem. Ber., 45, 1954 (1914).
135. M a n n F. G., J. Chem. Soc., 1933, 412.
136. Burstall F. H., J. Chem. Soc., 1936, 173.
137. D w у e r F. P., G у a r f a s E. C., Nature, 163, 918 (1949).
138. D wy er F. P., Gy arf as E. C., J. Proc. Roy. Soc., N. S. Wales, 83,
170 (1949).
139. Rosenheim A., Raibmann B., Schendel G., Z. Anorg.
Allgem. Chem., 196, 168 (1931).
140. N e о g i P., Muker j ее G. K., J. Indian Chem. Soc., 11, 681 (1934).
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
Химия координационных соединений под ред. Бейлара и Буша, ИЛ, Москва,
1960.
BasoloF., Pearson R.G., Mechanisms of Inorganic Reactions, Wiley,
New York, 1958.
В a t e s F. J., et al., Polarimetry, Saccharimetry and the Sugars, U. S. Bureau
of Standards, Circular C440, U. S. Government Printing Office, Washing-
ton, D. C., 1942.
de la MareP. B. D.,Kline W., eds., Progress in Stereochemistry,
Vol. Ill, Butterworths, London, 1962.
Джерасси К., Дисперсия оптического вращения, ИЛ, Москва, 1962.
Илиел Э., Стереохимия соединений углерода, изд-во «Мир», Москва,
1965.
«Неорганические синтезы», сб. 2, Издатинлит, Москва, 1951.
Jaeger F. М., Optical Activity and High Temperature Measurements,
McGraw-Hill, New York, 1930.
К i r s c h n e r S., ed., Advances in the Chemistry of the Coordination Com-
pounds, Macmillan, New York, 1961.
KlyneW.,de la M a r e P. B. D., eds., Progress in Stereochemistry, Vol. II,
Butterworths, London, 1958.
К 1 у n e W., ed., Progress in Stereochemistry, Vol. I., Academic Press, New
York, 1954.
«Современная химия координационных соединений», под ред. Льюиса Дж.
и Уилкинса Р., ИЛ, Москва, 1963.
L о w г у Т. М., Optical Rotatory Power, Longmans, Green, London, 1935.
Martell A. E., С a 1 v i n M., Chemistry of the Metal Chelate Compounds,
Prentice-Hall, Englewood Cliffs, New Jersey, 1952.
Литература 69
Rochow Е. G., ed., Inorganic Syntheses, Vol. 6, McGraw-Hill, New York,
1960.
Weissb erger A., ed., Physical Methods of Organic Chemistry, 3rd Ed;
Vol. I, Part 3, of Technique of Inorganic Chemistry Series, Interscience,
New York, 1960.
Уэллс А. Ф., Строение неорганических веществ, Издатинлит, Москва,
1948*.
Вернер А., Новые воззрения в области неорганической химии, ОНТИ,
Химтеорет., Ленинград, 1936.
W о 1 d Ь у е F., “Technique of Optical Rotatory Dispersion and Circular
Dichroism”, in Technique of Inorganic Chemistry, Vol. IV, Jonassen
H. B., Weissberger A., eds., Interscience, New York.
Y о u n g R. S., Cobalt, Reinhold, New York. 1960.
* Имеется английское издание этой книги 1962 г.
ГЛАВА 3
СОЕДИНЕНИЯ МЕТАЛЛОВ С КЕТИМИНАМИ
И АЛЬДИМИНАМИ
Д. Ф. Мартин
Иллинойсский университет, Урбана, Иллинойс
I. ВВЕДЕНИЕ
При конденсации аминов с карбонильными соединениями обра-
зуются шиффовы основания RR'C=NR". Шиффовы основания
могут быть альдиминовые и кетиминовые в зависимости от того,
какое карбонильное соединение подвергается конденсации с ами-
ном — альдегид или кетон
RCHO + R"NH2 RCH=NR" + Н2О,
RR'CO + R"NH2 RR'C=NR" + H2O.
Эти вещества привлекли внимание в качестве модельных соедине-
ний для теоретических исследований и исходных веществ для син-
теза гетероциклических соединений. Шиффовы основания особенно
интересны своей способностью давать координационные соеди-
нения, которые стимулировали исследования в различных обла-
стях физической химии (исследование магнитных свойств, инфра-
красных и ультрафиолетовых спектров, дипольных моментов и
т. д.), в стереохимии, химии координационных полимеров, а так-
же изучение механизма химических реакций. В связи с повышен-
ным интересом к соединениям металлов с кетиминами и альдимина-
ми целесообразно рассмотреть методы их получения.
А. Номенклатура
Следует кратко рассмотреть номенклатуру, которая исполь-
зуется в настоящем обзоре.
Часто для продуктов конденсации будут применены наимено-
вания, производные от названий аминов и карбонильных соеди-
нений. Так, [CH3COCH2C(CH3)=NCH2]2—продукт конденса-
ции ацетилацетона и этилендиамина — будет называться бис-
ацетилацетонэтилендиамином, хотя по систематической номен-
клатуре МХС его называют 4,4'-(этилендинитрило)-2-пентан-2-
I. Введение
71
-он. Аналогичным образом будут называться шиффовы основания,
полученные из замещенных салицилальдегидов. Система-
тическая номенклатура будет применяться для наименования
р-кетоиминов типа RCOCH2C( = NR)R". Так, соединение
CH3COCH2C( = NH)CH3 будет называться 4-иминопентан-2-оном, а
его соединение с медью(П) — бис-(4-иминопентан-2-оно)-медь(П).
Следует отметить, что многие исследователи предпочитают ука-
зывать, что лиганды существуют в кетоаминной форме СН3СОСН =
=C(NH2)CH3. В этом случае лиганд следует называть 4-амино-З-
пентен-2-он, а его соединение с медью(11) — бпс-(4-амино-3-пен-
тен-2-оно)-медь(11).
Б. Побочные реакции
Взаимодействие кетиминов и альдиминов с ионами металлов
может сопровождаться некоторыми побочными реакциями: гид-
ролизом, аминным обменом и реакциями расщепления.
В качестве одной из побочных реакций следует рассмотреть
гидролиз, поскольку эту реакцию можно представить как обрат-
ную по отношению к реакции образования шиффова основания
/ С = N + Н2О = О + RNH2.
'R
Шиффовы основания обладают различной склонностью к гидроли-
зу. Так, бпс-ацетилацетонэтилендиамин [CH3COCH2C(CH3)NCH2]2
можно перекристаллизовать из водного раствора [1], а его
тетраметиленовый аналог неустойчив в воде [2]. Из-за основных
свойств аминов равновесие реакции образование — гидролиз
шиффова основания в присутствии кислот сдвигается в сторону
гидролиза. Поскольку при взаимодействии шиффова основания
HCh с ионом металла может выделиться ион водорода
п HCh + Мл+ —► MCh„ + п Н+,
необходимо регулировать значение pH раствора.
Б зависимости от свойств лиганда акт координации может либо
стабилизировать, либо ослабить шиффово основание в отношении
гидролиза. Интересно, что чувствительность к гидролизу исполь-
зуют при синтезе соединений металлов с р-дикетонами для соз-
дания гомогенной системы [3 ]
, н2о
2RCOCH = С (R") NR2 + МХ2---> (RCOCH = COR")2 М + 2R2 NH2X.
72
Глава 3
Часто гидролиз удается предотвратить, используя буферные
растворы или добавляя в раствор слабое основание, например
аммиак. Если для регулирования pH в раствор вводят аммиак,
то может произойти аминный обмен. Например, взаимодействие
бмс-ацетилацетонтриметилендиамина с ионом меди в присутствии
большого избытка аммиака приводит к образованию [4] бис-(4-
аминопентан-2-оно) -меди( II).
,СН3
О=Су
^СН2 + Cu2+ + 2NH
N= Су
\сн8
з
----> [Н3ССОСНС (=NH) СН3]2 Си + H3N (СН2)3 NH|+.
Удалось наблюдать также расщепление связи N—R в шиффо-
вых основаниях. Пфейффер и сотрудники [5] сообщили о неудаче
попыток получения соединений типа
RCHCOOC2H5
Им удалось выделить из реакционной смеси только кристалличе-
ский бш?-салицилальдиминоникель(Н). Предполагают, что в дан-
ном случае происходит окислительное расщепление, так как в от-
сутствие воздуха реакция не идет. Такими же неудачными были
попытки получить [СНзСОСНС(=КСНз)СНз12Сп взаимодейст-
вием лиганда и ацетата меди. В присутствии иона ацетата образо-
вывался продукт расщепления [СНзСОСНС(=КН)СНз]гСп [4].
В. Получение лигандов
Как правило, лиганды салицилальдиминного типа не выделя-
ются в чистом виде, но все же некоторые исследователи изолиро-
вали такие лиганды [6,7] и определили их физические постоян-
ные. Такие соединения обычно получали прямым взаимодейст-
вием альдегида и амина в присутствии растворителя или без него.
Иногда невозможно получить лиганд прямым взаимодействием.
Например, взаимодействие салицилальдегида и триэтилентетра-
П. Получение соединений металлов с альдиминами и кетиминами 73
амина не приводит к образованию желаемого шиффова основания;
вместо него образуется [8]
HOC6^CH=N(CH2)2N—CH2CH2^^(CH2hN=CHCeH4OH
СбЩОН
Методы получения р-кетоиминов обычно рассматривают в свя-
зи с получением их соединений с металлами [10, 11]. Кромвел [9]
рассмотрел методы получения р-кетоиминов и сделал обзор лите-
ратуры по этому вопросу.
П. МЕТОДЫ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ МЕТАЛЛОВ
С АЛЬДИМИНАМИ И КЕТИМИНАМИ
А. Непосредственное взаимодействие
Для получения координационных соединений прямым взаимо-
действием иминного лиганда и иона металла важное значение име-
ют два момента: выбор растворителя и выбор иона металла. Здесь
уместно рассмотреть эти два вопроса порознь в связи с тем, какие
преимущества или недостатки для синтеза дает выбор того или
иного агента.
1. Выбор растворителя
Осуществление взаимодействия имина и соли металла в вод-
ном растворе обычно ограничивается слабой растворимостью боль-
шинства иминов в воде. Кроме того, некоторые имины в воде гид-
ролизуются. Однако имеются некоторые существенные исключе-
ния. Если имин содержит один или несколько протонов, он может
растворяться в щелочных растворах, а в таких растворах скорость
гидролиза значительно уменьшается. Например, бис-ацетилаце-
тонэтилендиамин растворим в воде, и его соединение с кобаль-
том(П) получают прямым взаимодействием в водном растворе с
использованием эквивалентного количества гидроокиси натрия
[12]. Так, прямым взаимодействием в ацетатном буферном раст-
воре получают также бис-(К,К'-дисалицилэтилендиимин)-р-акво-
дикобальт(П) [13].
Прямая реакция в водном растворе возможна также в том слу-
чае, если имин содержит способствующие растворению группы,
например в случае некоторых тридентатных лигандов (ter), про-
изводных пиридин-2-альдегида и 2-аминометилпиридина [14].
При добавлении такого лиганда к водному раствору сульфата же-
леза(П) образуется соединение типа [Ее(1ег)г]2+ [14], а при до-
5 Заказ № 547
74
Глава 3
бавлении к раствору перхлората натрия осаждается соединение
комплексного иона с перхлоратом.
Затруднения, связанные с плохой растворимостью в водных
растворах и с гидролизом, обычно устраняют, используя смешан-
ные растворители или различные растворители для разных компо-
нентов. Ряд соединений меди с р-кетоиминами получают смеши-
ванием спиртового раствора лиганда и аммиачного раствора аце-
тата или нитрата меди [4, 14, 15]. Аналогичный метод используют
для получения бис-(4-иминопентан-2-оно)-никеля(П) [16, 18]-
Аммиачный раствор предотвращает гидролиз и сдвигает равнове-
сие комплексообразования. Нэдостатко^ этого метода является
введение постороннего иона. Если концентрация аммиака доста-
точно велика, может произойти аминный обмен. Это затруднение
можно преодолеть, если использовать небольшие количества гид-
роокиси аммония. Взаимодействие СНзСОСНгС(=МСНз)СНз с
нитратом меди при добавлении стехиометрических количеств гид-
роокиси аммония приводит к образованию ожидаемого продукта
реакции [19], но при введении большого избытка аммиака обра-
зуется продукт аминного обмена — бис-(4-иминопентан-2-оно)-
медь(И) [4].
Использование неводных растворителей, в которых раствори-
мы лиганд и соединение металла, позволяет избежать гидролиза,
введения посторонних ионов и возникновения нежелательных рав-
новесий. Так, реакцию между ацетатами металлов и [3-кетоимина-
ми можно проводить при нагревании растворов этих веществ в
диоксане [1], этаноле [2, 20] или метаноле [21]. Продукт либ(>
выделяют ^при охлаждении, либо осаждают при разбавлении
водой. . -
2. Выбор исходного соединения металла
Как правило, в качестве источника ионов металла берут'соли
(ацетаты, галогениды, нитраты или сульфаты). Применение аце-
татов дает некоторые преимущества, связанные с контролем pH,
так как при нейтрализации образуется ацетатный буфер.
Ряд хелатных соединений металлов с р-кетоиминами получали
кипячением с обратным холодильником растворов лиганда в аце-
тоне над свежеприготовленными гидроокисями двухвалентных
металлов [22]. Метод использовали для синтеза соединений ме-
ди(П), никеля (II) и кобальта(П), но он не был использован для по-
лучения соединений цинка(П) или кадмия(П) [22]. Реакция в
такой гетерогенной системе может протекать довольно медленно.
Недостатком этого метода является то, что иногда продукт реак-
ции осаждается непосредственно на гидроокиси и затрудняет даль-
нейший ход реакции; обычно продукт растворим в ацетоне, и его
II. Получение соединений металлов с альдиминами и кетиминами 75
легко можно отделить от непрореагировавшей гидроокиси ме-
талла.
Рассмотрение методов получения соединений металлов с 1,3-
дикетонами [23 ] подтверждает мысль, что для синтеза полимеров
можно использовать также другие соединения металлов. Напри-
мер, галогениды многих металлов (такие, как GeCl4, ZrCl4, ThCl4,
ВС1з и А1С1з) растворимы в инертных растворителях; в результате,
реакций с протонсодержащими лигандами они образуют нераст-
воримые галогеноводороды. О таких реакциях, по-видимому,
еще нигде не сообщалось. Однако для получения соединений типа
Cr [RCOCHC(=NR')R"]3 использовали сольват трихлоро-трис-
(тетрагидрофуран)-хром(Ш). При этом сольват добавляли к
эквимолярной смеси калия и лиганда в шрелг-бутаноле [10].
Б. Соединение компонентов
Этот метод заключается в смешивании в определенном порядке
амина, альдегида или кетона, основания и иона металла в подхо-
дящем растворителе. Метод удобен тем, что не требует специаль-
ного синтеза лиганда. Он необходим в тех случаях, когда лиганд
слишком нестоек и не может быть выделен и очищен, а также тог-
да, когда лиганд в отсутствие иона металла подвергается нежела-
тельной конденсации. Однако метод имеет очевидный недостаток,
заключающийся в том, что продукт может быть загрязнен избыт-
ком одного из реагентов, что обычно и наблюдается. Так, при по-
лучении бис-(М,М/-дисалицилэтилендиимино)-[т-акводикобальта(П)
методом прямого взаимодействия образуется более чистый про-
дукт по сравнению с методом соединения компонентов [13].
По-видимому, результат синтеза зависит от порядка смешива-
ния реагентов. Нанес и Эйчхорн [24] изучали скорость образо-
вания соединений типа
/CH=N— сн,
/=<^ I )с=°
<\ /)—О Ni—О
X-J I
Н2О
в 50%-ном (по объему) растворе диоксан — вода. Образование
комплекса замедляется в том случае, если ионы меди или никеля
взаимодействуют сначала лишь с одним из компонентов шиффова
основания (салицилальдегидом или глицином). Если же ионы
меди или никеля добавляют уже к смеси компонентов шиффова
основания, комплекс металла с шиффовым основанием образуется
немедленно, хотя концентрация комплекса металла в этом слу-
5*
1
76
Глава 3
чае практически та же, что и при равновесии. По-видимому, в
этом случае шиффово основание образуется без участия ионов
металла.
Метод соединения компонентов мало используют для синтеза
соединений металлов с р-кетоиминами, хотя потенциальные воз-
можности метода достаточно широки. Это упущение, вероятно,
обусловлено тем, что соединения металлов с (3-дикетонами менее
растворимы и более устойчивы, чем аналогичные соединения р-ке-
тоиминов. Имеется сообщение [25] о синтезе бис-ацетилацетон-
этилендииминомеди(П) из ацетилацетона, этилендиамина и иона
меди. Однако попытки конденсации гексафторацетилацетона и
этилендиамина в присутствии ацетата меди оказались безуспеш-
ными [1].
Для получения соединений металлов с салицилальдиминами
используют две основные методики. Метод Пфейфера [26] заклю-
чается в кипячении с обратным холодильником раствора в мета-
Ноле бис-(салицилальдегид)-меди (II), ацетата натрия и амина при
соотношении реагентов 1:1:1 (по весу). По методу Чарльза
[27, 28] водный раствор ацетата металла добавляют к раствору
салицилальдегида, амина и ацетата натрия в метаноле или в
разбавленном метаноле и затем кипятят смесь с обратным холо-
дильником. Вертер и Фрост [29] считают, что салицилальдимины
производные анилина лучше получать методом Чарльза, а про-
изводные аминоэфиров — методом Пфейфера.
Хорошим примером применения метода соединения компонен-
тов для получения производных нейтральных иминов является
получение щрмс-(биацетил-бмс-метилимин)-кобальта(П) и подоб-
ных ему соединений [30].
Соединения типа [о-ОС6Н4С(СНз)=ХН]2М были получены
смешиванием компонентов в растворе или воздействием аммиака
(R=H) на бис-(о-оксиацетофенон)-никель(П) [31—33].
В. Аминный обмен
Метод заключается в вытеснении иминной группировки RN
амином
М М
\ Т \ 1
C=NR + R'NH2 — C=NR’ + RNH2
Этот метод использовали Вертер и Фрост [29] и Муто [34]
для получения различных соединений типа
[o-OC6H4CH=NR]2Cu.
II. Получение соединений металлов с алъдиминами и кетиминами 77
Методика синтеза проста: готовят суспензию лиганда или
хелатного соединения с медью в амине и смесь нагревают. Часто
продукт удается выделить разбавлением реакционной смеси водой.
Для твердых аминов можно использовать растворитель (этанол,
хлороформ, бензол, толуол, эфир или ацетон) [34]. Метод удобен
тем, что позволяет из одного и того же хелатного соединения меди
получить различные салицилальдиминовые соединения металлов
при использовании различных аминов. Реакцию ведут в восста-
новительных условиях, поэтому окислительное состояние металла
может измениться.
Несмотря на то что салицилальдимины и их соединения с ме-
дью (II) легко подвергаются аминному обмену, этого нельзя ска-
зать о р-кетоиминах и их соединениях с медью(П). Вис-(4-имино-
пентан-2-оно)-медь(П), по-видимому, не реагирует с этилендиа-
мином [4]. Не наблюдается также признаков взаимодействия
4-иминопентан-2-она с этилендиамином в 95 %-ном этаноле, но в
присутствии иона меди этот ^-кетоимин претерпевает аминный
обмен с этилендиамином. Аминный обмен представляет.собой по
существу вариант метода последовательного синтеза.
Имеются некоторые предположения [4, 29] относительно меха-
низма аминного обмена. Важной особенностью реакции является
поляризация азометеновой связи в результате координации, од-
нако это, очевидно, не единственное условие протекания реакции,
так как ряд салицилальдиминов претерпевают аминный обмен в от-
сутствие иона металла [34].
Атом углерода с недостатком электрона, вероятно, может взаимо-
действовать с амином
78
Глава 3
При этом, по-видимому, большое значение имеет прочность
связи металл — азот: чем прочнее связь, тем менее вероятен амин-
ный обмен. Это согласуется с тем фактом, что в аммиачных раст-
ворах соединение СНзСОСНгС(=ХСвН5)СНз способно к аминному
обмену в присутствии никеля (II) и не способно в присутствии иона
меди(П) [4 ]. Полагают [4], что аминный обмен с участием р-кето-
иминов включает реакцию монохелатной формы, которая может
образоваться непосредственно или в результате диссоциации.
Движущей силой реакции, вероятно, является тенденция к обра-
зованию более устойчивых металл-хелатных соединений. Равно-
весие реакции может сдвигаться также в результате удаления ле-
тучего амина. Более подробное обсуждение аминного обмена с
участием р-кетоиминов можно найти в работах [4, 29].
Г. Хелатный обмен
Этот метод можно определить как вытеснение одного хелат-
ного агента HCh из координационной сферы иона металла другим
хелатным агентом HCh':
MCh„ + nHCh' MCh^ + nHCh.
Экспериментально метод заключается в нагревании смеси ли-
ганда и хелатного соединения металла в подходящем растворите-
ле. Часто степень превращения удается определить по цвету раст-
вора. Нужный продукт кристаллизуют или выделяют в осадок
добавлением другого растворителя. Метод хелатного обмена мож-
но использовать для получения соединений металл — р-дикар-
бонил [35, 36]. Распространение этого метода на получение
металлических производных салицилальдиминов [34 ] или р-кетои-
минов [16, 17] дает определенные преимущества. Побочные ре-
акции, такие, как гидролиз и аминный обмен, исключаются; про-
дукт, вероятно, будет чище, так как загрязнение посторонними
ионами сводится к минимуму; препаративные операции просты,
а кроме того, по-видимому, этим способом удастся получать сме-
шанные комплексы типа MChCh' [37].
Метод имеет также несколько недостатков. Равновесие хелат-
ного обмена может неблагоприятно влиять на получение нужного
соединения, и оно в этом случае образовываться не будет. Для
выделения продукта нужно, чтобы хелатное соединение металла
было наиболее растворимым из всех присутствующих соединений
в растворе. Обычно эти требования удовлетворяются. Наконец,
для синтеза соединений этим методом требуются и лиганд, и хе-
латное соединение металла. Недостатком метода можно считать
II. Получение соединений металлов с альдиминами и кетиминами 79
также большие затраты времени по сравнению с методом прямого
взаимодействия, хотя эти затраты и не очень велики.
Однако при синтезе [CeHsCOCHC^NCeHsJCHshCu затраты
времени оправдываются, так как при синтезе методом прямого
взаимодействия с использованием аммиака в качестве основания
образуется продукт аминного обмена [CeHCOCHC(=NH) CH3]2Cu.
Напротив, хелатный обмен происходит легко и с хорошим выхо-
дом продукта [16].
Более серьезным недостатком метода, чем затраты времени,
является появление нежелательного равновесия в системе. Это
затруднение можно преодолеть, добавив избыток лиганда, но по-
следнее может вызвать трудности при выделении продукта. Если
необходимо, можно варьировать исходное хелатное соединение.
Можно также использовать различные исходные хелатные со-
единения. Например, [CH3COCHC(CH3)=NCH2CH(CH)SN=
=С (СНз) СНСОСНз] Ni образуется с выходом 88%, если в ка-
честве исходного соединения для синтеза использовать £мс-(4-ими-
нопентан-2-оно)-никель(П), и с выходом 30%, если при синтезе
исходить из бис-(К-бутилсалицилдиимино)-никеля(П) [37].
Механизм реакции хелатного обмена еще не ясен. Мур и Янг
[35] определили константы равновесия для обмена с участием
р-дикетонов и соединений меди(П) с p-дикарбонилами. Сделаны
также некоторые предварительные кинетические измерения для
Р-кетоиминов и соединений р-кетоиминов с медью(П) [48].
Получение оптически активных хелатных комплексов меди
методом асимметрического синтеза [38] позволяет предположить,
что хелатный обмен может играть важную роль в биологических
процессах. Синтез этих соединений может быть полезным для вы-
яснения механизма хелатного обмена. Хелатное соединение меди
СНСбН4\
HN(CH2>2N^
(СН2) Си
I X \
HN(CH2)2N\ >0
^СНСбН/
может существовать в виде трех изомеров: двух энантиоморфных
форм и .мезо-формы, так как два бензольных кольца не могут рас-
полагаться в основной плоскости комплекса и структура соедине-
ния несимметрична. Z-Форма соединения получена реакцией d-тар-
трата меди и гексадентатного лиганда (образующегося in situ);
d-форма получена из натрий-бис-(1-глутамо)-меди (II), а мезо-сое-
динение не выделено [38].
80
Гл а в а 3
Д. Обмен металла
Этот метод заключается в том, что хелатное соединение метал-
ла смешивают с солью другого металла в растворе, в результате
чего образуется более устойчивый хелатный комплекс металла
МСЬ„ + м' л+ м' Ch„ + мл+.
Преимущество этого метода заключается в простоте операций
и в возможности получения различных соединений из одного хе-
латного соединения металла. Недостаток — в том, что при взаимо-
действии образуется обязательно более устойчивое хелатное сое-
динение металла, если нет способов обойти состояние равновесия.
Кроме того, если даже равновесие благоприятно для образования
нужного соединения, продукт может быть загрязнен ионами ме-
таллов или анионами.
Возможности метода обмена металла в препаративных целях
еще не используют. Потенциальную ценность метода мож-
но уяснить на примере успешного синтеза различных сое-
динений типа Cr[RCOCHC(=NR/)R//]3 из Cr(THF)3Cl3 и
К[RCOCHC(=NR')R//] [10].Так, Пфейфери сотрудники [391 ис-
пользовали этот метод для определения относительных устойчи-
востей соединений типа бис-салицилальэтилендииминометалл(П) и
определили порядок вытеснения: Cu>Ni>Zn>Mg.
Е. Темплатный синтез
Хелатное соединение металла или координационная сфера
иона металла могут играть роль шаблона (темплата) и ориентиро-
вать различные лиганды так, чтобы получилось удобное для кон-
денсации или комплексообразования расположение. Короче го-
воря, ион металла организует реагенты и располагает их в по-
рядке, удобном для комплексообразования. Имеется несколько
примеров такого рода синтеза, которые могут показать потенци-
альные возможности метода.
При конденсации а-дикетонов и меркаптоэтиламидов образу-
ются главным образом тиазолины, а не тетрадентатные лиганды,
но в присутствии иона никеля(П) конденсация приводит к образо-
ванию нужных веществ с хорошим выходом [40]:
R /СНгСНг^
I
C=N^
R ГНгСНг^
II. Получение соединений металлов с альдиминами и кетиминами 81
Атомы серы в этом комплексе алкилируются бензилбромидом
с образованием мономерного соединения; алкилирование а.,а.'-
дибромо-о-ксилолом приводит к образованию макроциклического
соединения [41].
В этом случае координационное соединение играет роль шаблона
(темплата).
На этом же принципе основан синтез соединений типа
Ni [RCOCR/C(R")=NCH2]2 аминокаталитической конденсацией
р-дикетонов (или замещенных салицилальдегидов, или о-окси-
ацетофенонов) с хлоридом бис-этилендиаминникель(П) [42]. По-
следний представляет собой димер с хлоридными мостиками [43].
Высказано предположение [42], что небольшие количества спо-
собного к координации амина, например пиридина (ру) (но не 2,6-
диметилпиридина) будут разрушать мостики с образованием
[Ni(en)2(py)2]C12. После этого координированные молекулы пи-
ридина могут быть замещены атомами кислорода двух различных
молекул р-дикетона. Эти молекулы р-дикетона могут конденсиро-
ваться с прилегающими аминогруппами. Очевидно, для протека-
ния реакции необходимо разрушение мостиков и образование не-
устойчивого соединения [Ni(en)2(py)2]C12, так как в отсутствие
подходящего амина р-дикетоны не конденсируются с [Ni(en) з 1 С1г.
Для объяснения реакции конденсации ацетона с [Ni(en)3] (С1О4)г
[44] приходится принять другой механизм. Продукт кон-
денсации представляет собой координационное соедине-
ние никеля(П) с двумя, тремя или четырьмя присоединенными
группами изопропилидена (СзН4) и имеет структуру плоского
квадрата.
Эти примеры иллюстрируют возможности темплатного синтеза.
82
Глава 3
Ж. Другие методы
Наконец, заслуживают упоминания некоторые потенциально
полезные методы, которые не попадают в указанные выше кате-
гории.
1. Взаимодействие растворимых солей шиффовых оснований и растворимых
солей металлов
. Этот метод был использован для синтеза соединений металлов
с р-дикетонами [23], но мало применяется для синтеза соединений
металлов с кетиминами и альдиминами.
2. Реакции функциональных групп
Потенциально полезным методом синтеза может быть исполь-
зование реакций координированных лигандов. Этот метод прак-
тически еще не используют, но он может включать реакции функ-
циональных групп или введение функциональных групп в коор-
динационное соединение.
Были проведены реакции переэтерификации и амидирования
соединения бис-(салицилальдимин)-медь(П); попытка переэтерифи-
кации бутилмеркаптаном не дала искомого результата [29].
Потенциально полезным методом синтеза является введение
функциональных групп в координированные р-кетоимины. На-
пример, некоторые соединения р-кетоимина с хромом(Ш) легко
реагируют с N-бромсукцинимидом (NBS). Вероятно, появятся
и другие примеры такого рода реакций.
(BuSfeCu
III. Экспериментальные методы
83
Н3С
НС\ Сг/З-^^д
\ -z vrlUi £л
/C=N
H3CZ \
Н3С
хР—о.
Вг—С Сг/3
^C=N^
H3CZ R
(R = CeH5, л-СН3СбН4, о-СНэСвЩ)
III. ЭКСПЕРИМЕНТАЛЬНЫЕ МЕТОДЫ
А. Непосредственное взаимодействие
1. Бис -(N, N' -дисалицилэ тилендиимин) -р -акводикобальт( 11)
Синтез лиганда (выход 95%) и синтез хелатного соединения
металла (выход 90%) изложены в сборнике «Неорганические син-
тезы» [13].
2. Вис-(4-иминопентан-2-оно)-никель(П) [СНзСОСНС(=ЫН)СНз]Ы1
Синтез хелатного соединения металла при обработке раствора
лиганда в этаноле аммиачным нитратом никеля изложен в сбор-
нике «Неорганические синтезы» [16]. Методика аналогична мето-
дике синтеза соединений меди (II). Синтез соединения никеля (II)
описан независимо Арчером [18].
Б. Соединение компонентов
Весьма полезным оказался метод, разработанный Чарльзом
[27, 28]. Метод применим для синтеза соединений меди, никеля,
цинка и кобальта(П) с N-алкил салицил ал ьдиминами.
1. Бис- [Ы-(и-бутил)-салицилальдимино]-медь(11)*
[o-OCeH4CH(=NCH2CH2CH2CH3)]2Cu
К 100 мл метанола последовательно добавляют салициловый
альдегид (6,1 г, 0,05 моля), н-бутиламин (3,8 г, 0,05 моля) и 50 мл
1 М раствора едкого натра. Спустя 15 мин добавляют раствор
♦ Этот синтез проверен Страссом [37].
84
Глава 3
тригидрата нитрата меди(П) (6,05 г, 0,025 моля) в 100 мл воды
и смесь энергично перемешивают. После охлаждения в ледяной
воде отфильтровывают сырой продукт. После перекристаллиза-
ции из 95%-ного этанола получается около 8,6 г темно-зеленых
кристаллов. Еще 1,1 г продукта можно получить, добавив к кипя-
щему этаноловому фильтрату (около 70 мл) для индуцирования
кристаллизации приблизительно 35 мл воды. Общий выход 9,7 г
(91%), температура плавления 79—81°. Согласно Чарльзу [27],
температура плавления вещества равна 80,5—81°.
2. Бис- [Ы-(м-бутил)-салицилальдимино]-кобальт(11)*
[o-OC6H4CH(=NCH2CH2CH2CH3)]2Co
К 200 мл метанола последовательно добавляют салициловый
альдегид (12,2 г, 0,10 моля), н-бутиламин (7,6 г, 0,10 моля) и 100 мл
1 М раствора едкого натра. Через раствор в течение 15 мин про-
пускают азот, после чего колбу закрывают и оставляют стоять.
Тем временем через 200 мл воды в течение 5 мин пропускают азот,
после чего в воде растворяют гексагидрат нитрата кобальта(П)
(14,7 г, 0,05 моля). Этот раствор добавляют к раствору салицилово-
го альдегида. Через смесь в течение 15 мин пропускают азот. Ма-
точный раствор отделяют при помощи фильтровальной трубки, а
продукт перекристаллизовывают из ацетона в атмосфере азота.
Вещество имеет вид красных игольчатых кристаллов, выход
16,2 г (75%). Температура плавления 151—153°. Анализ: для
СггНгвОгМгСо рассчитано (%) 64,20 С, 6,81 Н; найдено 63,91 С,
6,85 Н.
В. Аминный обмен
Предложен метод синтеза по способу, описанному Муто [34].
Соединение было приготовлено другим методом [45], но данные
о температуре плавления не приведены.
1. 2>ис-салицилальэтилендииминомедь(11) (о-ОСбН4СН=ХСН2)2Си
К раствору перекристаллизованной бис-(М-н-бутилсалицилаль-
диимино)-меди(П) добавляют безводный этилендиамин (0,6 г,
5 л4Л1оль).Цвет раствора меняется от темного до светло-зеленого.При
нагревании на паровой бане в течение 5 мин или менее образуется
осадок продукта, который отделяют фильтрованием и высушивают
в вакууме. Выход 0,7—0,8 г (85—97%) светло-зеленых чешуек;
* Этот синтез разработан Страссом [37].
III. Экспериментальные методы
85
температура плавления 315° (разлагается). Анализ (неперекрис-
таллизованное вещество); рассчитано для Cl6H14O2N2Cu (%);
58,27 С, 4,28 Н, 8,50 N; найдено: 57,94 С, 4,42 Н, 8,33 N.
Г. Хелатный обмен
1. Лис-(3-фенилимино-1-фенилбутано-1-оно)-медь(П)
[СвН5СОСНС(=МСвН5)СНз12Си
Синтез этого хелатного соединения изложен в сборнике «Не-
органические синтезы» [16]. Этот метод, по-видимому, можно ис-
пользовать для синтеза соединений р-кетоиминов с медью (II) и
никелем(П).
Д. Обмен металла
1. 77>яс-(4-толуидино-3-пентен-2-оно)-хром(111)
[CH3(4-CH3C6H4NH)C= СНСОСНз]зСг [10]
Получение этого соединения изложено в сборнике «Неоргани-
ческие синтезы» [46]. Соединение получают взаимодействием ка-
лиевой соли р-кетоимина и трихлоро-пг/шс-(тетрагидрофуран)-
хрома(Ш). Реакцию, по-видимому, можно использовать для син-
теза различных соединений хрома(Ш) с р-кетоиминами.
Е. Темп латный синтез
1. Общий метод
К раствору хлорида бис-этилендиаминникель(П) в воде (2,49 г
на 20 мл воды) добавляют 2 капли пиридина [47]. В раствор вво-
дят р-дикетон или салицилальдегид (0,02 моля) либо в чистом ви-
де, либо в виде раствора в 20 мл метанола. Смесь кипятят с обрат-
ным холодильником в течение 2 час. За это время продукт выкрис-
таллизовывается. Кристаллы отфильтровывают. Часто получается
вещество аналитической чистоты, а выход почти количественный.
2. £ис-ацетилацетонэтилендииминоникель( 11)
[СН зСОСЩСН 3)C=NCH 3]г№
Синтез ведут по изложенной выше общей методике, исполь-
зуя 1,98 г ацетилацетона. Получаются оранжево-коричневые крис-
таллы с выходом 82%; температура плавления 198°; по литера-
турным данным [42], температура плавления равна 198, 200°.
86
Гл а в а 3
3. 5ис-(о-оксиацетофеноп)-этилендииминоникель(11) [421*
[o-OC6H4C(CH3)=NCH2]2Ni
К раствору хлорида бис-этилендиаминникеля(П) (2,49 г)
в 20 мл воды добавляют 2 капли пиридина. Окраска раствора долж-
на сразу же измениться от синей к зеленой. Затем добавляют
2,76 г (0,02 моля) о-оксиацетофенона и смесь нагревают с обратным
холодильником в течение 2 час. За это время образуются корич-
невые кристаллы, их отфильтровывают. Выход продукта 81%,
температура плавления 290°. Аналитические данные указаны в
работе [42].
ЛИТЕРАТУРА
1. М а г t е 11 А. Е., В е 1 f о г d R. L., С а 1 v i n M.,J. Inorg. Nucl. Chem.,
5, 170 (1958).
2. Hovey R. J., O’C onnell J. J., Martell A. E., J. Am. Chem.
Soc., 81, 3189 (1959).
3. Gash V. M., Abstracts of Papers, 138th National Meeting of the Ameri-
can Chemical Society, New York, Sept. 11—16, 1960, p. 58P.
4. Mart inD.F., Advan. Chem. Ser., 37, 192 (1963).
5. Pfeiffer P., Offermann W., Werner H., J. Prakt. Chem.,
159 , 313 (1941).
6. Eichhorn G. L., В a i 1 a r J. C., Jr., J. Am. Chem. Soc., 75, 2905
(1953).
7. В a k e r A. W., S c h u 1 g i n A. T., J. Am. Chem. Soc., 81, 1523 (1959).
8. Das Sarma В., В a i 1 a r J. C., Jr., J. Am. Chem. Soc, 76,4051 (1955)..
9. Cromwell N. H., Chem. Rev., 38, 83 (1946).
10. С о 1 1 m a n J. P., К i t t 1 e m a n E. T., Inorg. Chem., 1, 499 (1962).
11. Martin D. F., J anusonisG. A., Martin В. B., J. Am. Chem.
Soc., 83, 73 (1961).
12. M о r g a n G. T., S m i t h J. D. M., J. Chem. Soc., 1925, 2030.
13. Д и e л, Г a x, в сб. «Неорганические синтезы», сб. 3, Издатинлит, Моск-
ва, 1951.
14. G о о d w i n Н. A., L i о n s F., J. Am. Chem. Soc., 81, 6415 (1959).
15. H о 11 z с 1 a w H. F., Jr., Collman J. P., Alice R. M., J. Am.
Chem. Soc., 80, 1100 (1958).
16. S t r u s s A. W., M a r t i n D. F., in Inorganic Syntheses, Vol. 8, Holtzc-
law H. F., Jr., ed., McGraw-Hill, New York,
17. H s e u T. M., Martin D. F., Moeller T., Inorg. Chem., 2, 587
(1963).
18. A r c h e r R. D., Inorg. Chem., 2, 292 (1963).
19. H о 11 z с 1 a w H. F., Jr., private communication.
20. H о 1 m R. H., J. Am. Chem. Soc., 82, 5632 (1960).
21. L i о n s F., M a r t i n К. V., J. Am. Chem. Soc., 80, 3858 (1958).
22. McCarthy P. J., HoveyR. J., UenoK., MartellA. E., J.
Am. Chem. Soc., 77, 5820 (1955).
23. FerneliusW. C., Bryant В. E., in Inorganic Syntheses, Vol. 5,
Moeller T., ed., McGraw-Hill, New York, 1957, p. 105.
24. Nunez L. J., Eichhorn G. L., J. Am. Chem. Soc., 84, 901 (1962).
25. Martell A. E., Calvin M., Chemistry of the Metal Chelate Com-
pounds, Prentice-Hall, Englewood Cliffs, New Jersey, 1953, p. 424.
* Детали этого синтеза разработаны Е. Олыпевски.
Литература
87
26. Pfeiffer Р., Hesse Th., P f i t z n e r H., Scholl WT h i e-
1 e r t H., J. Prakt. Chem., 149, 219 (1937).
27. CharlesR. G., J. Org. Chem., 22. 677 (1957).
28. C h a r 1 e s R. G., J. Inorg. Nucl. Chem., 9, 145 (1959).
29. V ert er H. S., Pros t A. E., J. Am. Chem. Soc., 82, 85 (1960).
30. Figgins P. E., В u s c h D. H., J. Am. Chem. Soc., 82, 820 (1960).
31. DelepineM., Bull. Soc. Chim. France, 21, 943 (1899).
32. S c h i f f H., Ann. Chem., 150, 193 (1869).
33. Pfeiffer P., Buchholz E., Bauer 0., J. Prakt. Chem., 129,
163 (1931).
34. Muto Y., Nippon Kagaku Zasshi, 76, 252 (1955); through Chem. Abstr.,
51, 17559 (1957).
35. Moore T. S., Young M. W., J. Chem. Soc., 1932, 2694.
36. W о 1 f L., В u 11 e r E., W e i n e 11 H., Z. anorg. allgem. Chem., 306,
87 (1960).
37. S t r u s s A. W., M a r t i n D. F., J. Inorg. Nucl. Chem., 25, 1409 (1963).
38. DasSarma В., В a i 1 a r J. C., Jr., J. Am. Chem. Soc., 77, 5476
(1955).
39. Pfeiffer P., Theilert H., Glasser H., J. Prakt. Chem., 152,
145 (1939).
40. Thompson M. C., Busch D. H., J. Am. Chem. Soc., 84, 1762 (1962).
41. Thompson M. C., Busch D. H., Abstracts of Papers, 142nd
National Meeting, American Chemical Society, Atlantic City, New Jersey,
Sept. 1962, p. 13 N.
42. Olszewski E. J., BoucherL. J., Oehmke R. W., В a i 1 а г
J. C., Jr., M a r t i n D. F., Inorg. Chem., 2, 661 (1963).
43. Анчишкина А. С., Порай-Кошиц M. А., ДАН СССР, 143,
105 (1962); Chem. Abstr., 57, 2954 (1962).
44. В 1 i g h t M. M., CurtisN.F., J. Chem. Soc., 1962, 1204.
45. P f e i f f e г P., В r e i t h E., L ii b b e E., T s u m a k i T., Ann. Chem.,
503, 84 (1933).
46. Collman J. P., Kittleman E. T., in Inorganic Syntheses, Vol.
8, Holtzclaw H. F., Jr., ed., McGraw-Hill, New York, to be published.
47. S t a t e H. M., in Inorganic Syntheses, Vol. 6, Rochow E. G., ed., McGraw-
Hill, New York, 1960, p. 198.
48. M a r t i n D. F., Chem. Ind. (London), 1963, 1528.
ГЛАВА 4
КАРБОНИЛЫ МЕТАЛЛОВ
Хиле ман
Эль-Каминский колледж, Тарране, Калифорния
I. ВВЕДЕНИЕ
Эта глава написана для химиков-специалистов, у которых
на определенной стадии исследовательской работы может возник-
нуть необходимость в получении карбонилов металлов, но которые
не обладают достаточным опытом для синтеза соединений подоб-
ного типа. В тех случаях когда автор предполагал, что практиче-
ский опыт читателя ограничен, методические вопросы рассмат-
ривались очень подробно; однако в большинстве случаев считали,
что читателю известны обычные препаративные методы.
В данной главе подробно изложены препаративные методики
только для тех карбонилов металлов, которые не поступают в
продажу. Однако указания на оригинальную литературу включа-
ют получение всех карбонилов металлов. В качестве дополнения
приведены также несколько обзорных статей по карбонилам ме-
таллов [1—5а].
А. Известные карбонилы металлов
Со времени открытия Мондом“в 1890'г. карбонила никеля вни-
мание химиков неизменно привлекают соединения окиси углеро-
да и переходных металлов с нулевой валентностью. Табл. 1, в ко-
торой указаны известные карбонилы металлов в порядке их от-
крытия, дает некоторое представление о настойчивости, с которой
проводили исследования в области карбонилов металлов.
Развитие работ в области синтеза карбонилов металлов можно
наглядно иредставить, используя периодическую систему. Из
табл. 2 следует, что синтезированы карбонилы большинства
переходных металлов. Были получены также соединения, в ко-
торые окись углерода входит как составная часть вместе с дру-
гими лигандами, в том числе и в тех случаях, когда оказалось
и
я
ф
1=5
Я
е
сб
и
и
к
История открытия карбонилов металлов
co
00
о
co хО со
о о
СМ СМ СМ
см
см
Исследователь Гибер, Легалли Гибер, Легалли Гибер, Штальман Гибер, Штальман
- о СО Ф 5 « « 1=3 Он - О ф я сх. Ф О И ф ч и о s 5 "! Ь и О - й я 2 8 g О’ о ~ s? о F и « g § * к £ ч ° « В и . р. tr S я X - -« ф со S д S 2 « Он ф S * s |С ч р, ч g 8 □и со со ф а й ф и к к и х «к
п
о
S3
S
§
я
(а
§
S
о
о
и
ч
л
tt
S
S
к
о
X
те
к
«
I. Введение
91
Таблица 2
Переходные металлы, образующие карбонилы и их производные
IV-6 v-б vi-б VII-6
Viil-б 1-6
Fe(CO)5
Бесцветный
Со2(СО)8
Оранжевый
V(CO)6 Сг(СО)б Мп2(СО)ю Fe2(CO)9
Сине- Бесцветный Желтый Желтый
зеленый
Fe3(CO)12
Зелено-черный
Тс2(СО)ю
Бесцветный
Мо(СО)6 Тс3(СО)12
Бесцвет- Коричневый
нын
Ru(CO)5 Rh2(CO)8
Бесцвет - Оранжевый
ный
Ru3(CO)12 Rh4(CO),2
Желтый Красный
Rh6(CO),6
Черный
Оз(СО)5
Бесцветный
W(co)6 Re2(CO)|0 Л
Бесцветный Бесцветный vS3(CO)|2
Желтый
ir2(CO)8
Желто-
зеленый
1г4(СО)12
Желтый
Со4(СО)12 Ni(CO)4
Зелено- Бесцвет-
черный ный
невозможным синтезировать «родоначальный» карбонил, содержа-
щий только металл и окись углерода. Соответствующие металлы
помечены в табл. 2 штриховкой.
Б. Структура карбонилов металлов
Большая часть исследований карбонилов металлов посвящена
изучению природы химической связи между окисью углерода и
металлом с нулевой валентностью. Все карбонилы металлов име-
ют общую формулу Мг(СО)у. В соответствии с пространственной
конфигурацией молекулы их можно разбить на девять групп, как
показано на рис. 1. Кроме того, было показано, что возможных
типов химической связи в этих соединениях значительно меньше.
Предполагают существование трех типов связи: /1) ковалентной
связи между двумя атомами металла, когда каждый атом металла
поставляет для связи один электрон; 2) координационной кова-
лентной связи между металлом и окисью углерода (a-связь), при-
чем атом углерода отдает металлу электронную пару и дополни-
тельно связывается с металлом частичной тс-связью с участием
92
Глава 4
Рис. 1. Пространственные модели типичных карбонилов металлов.
d-электронов металла; 3) ковалентной о-связи между металлом и
окисью углерода, причем атом углерода служит мостиком между
двумя атомами металла и образует связь с каждым из них. Новей-
шими рентгенографическими исследованиями показано [24], что
карбонилы типа М6(СО)16 имеют внутренние связи четвертого
I. Введение
93
типа. Возможно, это связь металл — окись углерода, когда окись
углерода служит мостиком между тремя атомами металла. Сле-
дует отметить также, что соединение W(CO)5, имеющее структуру
тетрагональной пирамиды, не рассматривается здесь вследствие
его неустойчивости (время полураспада 2 мин) и также потому,
что его пока не удалось выделить из раствора [251.
Для большинства карбонилов металлов справедливо правило
Сиджвика относительно эффективного атомного номера (ЭАН)
рассматриваемых переходных металлов. Согласно этому правилу,
каждый металл вступает в реакцию таким образом, что у него
оказывается такое число электронов, как у ближайшего последу-
ющего инертного газа в периодической таблице. Например, ни-
кель реагирует с четырьмя молекулами окиси углерода и полу-
чает от них восемь электронов (помимо своих 28), так что общее
число электронов у него становится равным 36, что соответствует
атомному номеру криптона. Металлы с нечетными атомными но-
мерами в большинстве случаев образуют соединения со связями
металл — металл или с мостиковыми карбонильными группами.
В других случаях карбонилы таких металлов проявляют пара-
магнитные свойства, связанные с наличием неспаренных электро-
нов. На первых этапах исследования карбонилов металлов все
синтезированные тогда карбонилы подчинялись правилу эффек-
тивного атомного номера и соответственно обладали диамагнит-
ными свойствами. Однако полученный в недавнее время карбонил
ванадия [19] показал возможность несоблюдения правила ЭАН
и образования парамагнитных соединений с неспаренными элек-
тронами. По-видимому, и Тс3(СО)12 также не подчиняется правилу
ЭАН, но магнитные свойства этого соединения не были изучены
с достаточной полнотой [21 ]. Открытые в самое последнее время
соединения М6(СО)16 так сильно отклоняются от правила ЭАН,
что их магнитные свойства пока трудно даже обсуждать [24].
В. Предсказанные, но не синтезированные
карбонилы металлов
Наиболее вероятная группа новых карбонилов металлов, ко-
торые можно синтезировать, включает карбонилы тех металлов,
которые образуют соединения с окисью углерода, содержащие
также и другие лиганды. Такие металлы указаны штриховкой в
табл. 2. Например, существуют соединения [Pd(CO)Cl2]n и
[Pt(CO)Cl2]n в группе VIII6, Cu(CO)X, Ag2SO4(CO) и Ап(СО)С1
в группе 16, (C5H5)2Ti(CO)2 в группе Шб, [Na(C6H14O3)12] [Та(СО)61
и [Na(C6H14O3)2] [Nb(CO)6] в группе V6. Тот факт, что связи
металл — карбонил стабилизируются в присутствии других лиган-
дов, позволяет с большой степенью вероятности предположить,
94
Глава 4
что имеются такие экспериментальные условия, в которых могут
существовать и соответствующие карбонилы. Подалл [26] пока-
зал, что термическая устойчивость карбонилов уменьшается слева
направо в рядах периодической таблицы в следующем порядке:
Для четных Z-. Cr (СО)6 > Fe (СО)5 > Ni (СО)4
Для нечетных Z: Мп2 (СО)10 > Со2(СО)8.
Точно так же склонность к образованию связи металл—карбонил,
судя по скорости реакции металла с окисью углерода, изменяется
в следующем порядке:
Для четных Z (по ряду): Ст < Fe < Ni
Для четных Z (по группе): Cr < Mo < W
Для нечетных Z (по ряду): V < Мп < Со
Следует отметить, что карбонил меди, если он будет синтези-
рован, должен иметь формулу Си2(СО)6 либо со связью Си—Си,
либо с комбинацией связи Си—Си и мостиковых карбонильных
связей. Существование карбонила цинка состава Zn(CO)3 считают
маловероятным из-за устойчивости конфигурации электронов
и отсутствия у лигандов СО вокруг атома цинка возможности
гибридизации в конфигурацию р 3. Образование катиона Zn(CO)42+
считают более вероятным, чем образование соответствующего
карбонила.
Металлы групп 1116 и 1V6 более склонны в образованию ме-
талл-карбонильных анионов. Эти анионы более устойчивы, чем
соответствующие карбонилы, вследствие большей обратной коор-
динации rf-электронов к лигандам СО. Исходя из термодинамиче-
ских соображений, Ормонт [27] предполагает, что металлы груп-
пы Пб могут образовывать трикарбонилы, а карбонилы Sn, Ge,
Mg, Са, Sr и Ва должны быть неустойчивы. Поспехов [28] разра-
ботал формулу, расширяющую правило ЭАН. На основании этой
формулы были успешно предсказаны синтезированные в дальней-
шем полимерные карбонилы металлов. Поспехов предполагает
также возможность синтеза Ag2(CO)6 и Си2(СО)6.
Другая возможность синтеза новых карбонилов связана с
тем фактом, что некоторые известные в настоящее время карбо-
нилы составляют «гомологические ряды». Например, три карбо-
нила железа Fe(CO)5, Fe2(CO)9 и Fe3(CO)12 составляют ряд, в ко-
тором каждый более сложный член является производным про-
стейшего. Представляется вероятным, что существуют еще более
сложные члены этого ряда, хотя, по-видимому, их трудно отде-
лить от низших членов возгонкой или другим принятым способом.
Митлин, Вендер и Штернберг [29а ] обнаружили, что Со2(СО)8 при
I. Введение
95
высоких давлениях адсорбирует СО, но образующееся соединение
оказалось недостаточно устойчивым для его выделения. Гибер
[296] сообщил о существовании аниона Fe4(CO)132-, который соот-
ветствует невыделенному карбонилу Fe4(CO)14. Такие соединения
с высокими молекулярными весами плохо растворимы в обычных
растворителях и поэтому не могут быть идентифицированы обыч-
ным методом инфракрасной спектроскопии. Как и на ранних ста-
диях истории изучения органических полимеров, теперь химикам-
неорганикам, вероятно, придется исследовать осадки, образую-
щиеся на дне реакционных сосудов.
Рутений и осмий также образуют ряды карбонильных соеди-
нений, и на протяжении многих лет существование Ru2(CO)9 и
Os2(CO)9 считалось само собой разумеющимся аналогично сущест-
вованию Fe2(CO)9. Однако тщательные рентгеноструктурные ис-
следования Кори и Даля [23] привели к тому, что соединениям,
считавшимся ранее нонакарбонилами, пришлось приписать фор-
мулы Ru3(CO)12 и Os3(CO)12. Сразу же возник вопрос, имеются ли
соединения рутения и осмия, соответствующие Fe2(CO)9. И если
при обычном синтезе пентакарбонилы рутения и осмия Ru(CO)5
и Os(CO)5 превращаются непосредственно в додекакарбонилы, то
можно ли при помощи более сложных методик получить нонакар-
бонилы, существование которых является теперь проблематичным?
Джоши и Посон [206] достигли удивительного успеха в син-
тезе карбонилов металлов, когда они получили димерный карбо-
нил, содержащий два металла (СО)4Со—Ми(СО)3. Это, однако,
был только первый шаг, и вскоре после появления соединения с
Со и Мп Несмеянов и др. [22] сообщили о синтезе лимонно-желтого
вещества (СО)5Мп—Re(CO)5. Пока нет оснований считать, что
возможен синтез только димеров или только соединений, содер-
жащих лишь два металла.
Возможен также синтез новых карбонилов элементов, отно-
сящихся к семействам актинидов и лантанидов. Известна лишь
одна серьезная попытка приготовить карбонил урана, окончивша-
яся неудачей [30а]. Трудно теоретически предсказать способность
нижних /-орбиталей участвовать в образовании обратнодонорных
тс-связей, а также их влияние на несколько электронов верхних
с?-орбиталей, но некоторые условия для образования связей ме-
талл—карбонил, по-видимому, и в этих случаях имеются.
В Тилденской лекции 1960 г. Нихолм [306] оценил факторы
устойчивости связи металл—лиганд, и его соображения были ис-
пользованы для «открытия» новых карбонилов металлов. Были
выдвинуты предположения о том, что: «а) атом металла должен
иметь электроны на rf-орбиталях, способные к образованию связи.
Следовательно, для несвязанной конфигурации dn— число воз-
можных двойных связей — уменьшается по мере приближения п
96
Глава 4
к’нулю (этим можно объяснить тот факт, что непереходные метал-
лы не образуют карбонилов); б) лиганд должен иметь незанятые
<7-(как в случае R3P) или р-орбитали (как в случае СО), способные
принять эти (7-электроны; в) размеры орбиталей должны быть та-
кими, чтобы обеспечивалось эффективное перекрывание». Нихлом
далее утверждает, что способность элементов образовывать
к-связи будет изменяться в следующем порядке:
Ni° » Pt» > Pd°,
Au+ > Cu+ > Ag+,
Металлы нулевой валентности> двухвалентные>трехвалентные.
Неудачные попытки получить Pt(CO)4 или Pd(CO)4 можно объ-
яснить высокими теплотами атомизации, затратами энергии для
перехода в спин-параллельное состояние и относительной лег-
костью удаления электрона из несвязывающей оболочки dВ * 10.
II. ОБОРУДОВАНИЕ
"И
А. Высокое давление
В основном оборудование высокого давления для синтеза кар-
бонилов металлов напоминает оборудование для гидрогенизации.
Фирмы поставляют большую серию приборов и вспомогательных
устройств. Ниже будет приведено несколько примеров исполь-
зования этих аппаратов и приборов для работы одного экспери-
ментатора. Однако эти методы не единственные, и вполне надеж-
ных результатов можно достигнуть при помощи других методов и
другого оборудования.
На рис. 2 приведены типичные автоклавы с внешним устрой-
ством для перемешивания. Преимущества реакторов высокого
давления с внешним устройством для перемешивания заключают-
ся в том, что они позволяют выбирать вспомогательные устройст-
ва. Пробы можно отбирать в процессе перемешивания; вспомога-
тельные внутренние охлаждающие змеевики предотвращают раз-
гон реакции. В этих автоклавах можно использовать стеклянные
внутренние стаканы для жидких растворов; меняя стаканы,
можно варьировать рабочую емкость автоклава. (Ограничению
подлежит только максимально допустимое давление в аппа-
рате, которое в зависимости от температуры и от завода-из-
готовителя меняется в пределах 70—420 ати. Поскольку макси-
мальное давление в баллонах с окисью углерода равно 68 ат, для
многих экспериментов можно в качестве источника высокого дав-
ления использовать баллон с сжатым газом. Однако если для
Рис. 2. Автоклавы высокого давления с внешним устройством для перемешивания,
автоклав емкостью 300 см’ фирмы «Autoclave Engineers, Inc.»; б — аппарат для реакций под высоким давлением
фирмы «Рагг Instrument Со.» серии 4500.
8 Заказ Аз 547
Рис. 3. Качающиеся автоклавы высокого давления.
а — качающийся автоклав высокого давления емкостью 500 или 1000 с№
фирмы «Рагг Instrument Со.» серии 4000; б — качающийся автоклав вы-
сокого давления емкостью 43—400 см3 фирмы «American Instrument Со.»
микросерия.
II. Оборудование
99
наполнения автоклава используют полный баллон давлением 68 ат
при комнатной температуре, а потом автоклав нагревают, то при
этом давление может превысить допустимый предел. В таких
случаях внешнее перемешивание может оказаться невозможным,
и тогда приходится использовать другую крышку автоклава — без
мешалки. Таким образом, некоторые автоклавы с внешним уст-
ройством для перемешивания не позволяют экспериментатору
Рис. 4. Насосы высокого давления.
а — ручной насос для нагнетания газа фирмы «American Instrument’’’Со.»
б — насос для нагнетания газа с электродвигателем фирмы «American_Inst
rument Со.»
полностью использовать все преимущества наиболее дешевого
источника высокого давления — баллона с окисью углерода.
Автоклавы для более высоких давлений устроены таким обра-
зом, что для перемешивания содержимого они либо покачиваются,
либо вращаются. Качающиеся автоклавы показаны на рис. 3.
В литературе имеется описание качающегося автоклава, который
Гибер использовал для большинства своих работ [ЗОв]. В зави-
симости от температуры автоклавы этого типа обычно выдержи-
вают давления, значительно превышающие давления, необходи-
мые для синтеза карбонилов. Но большая часть возможностей
этих автоклавов останется неиспользованной, если не применять
8*
100
Глава 4
вспомогательные насосы для нагнетания окиси углерода в авто-
клав и для повышения первоначального давления газа. Для под-
качки небольших автоклавов можно пользоваться ручными на-
сосами, а для автоклавов емкостью 1—2 л имеются насосы с элек-
тродвигателями. Два таких насоса показаны на рис. 4. Для
давлений до 2100 ати имеются также легкие компрессоры диаф-
рагменного типа.
В руководстве для лабораторных работ Джолли [31 ] подроб-
но рассмотрены качающиеся автоклавы и вспомогательные насосы
для нагнетания газа, а также многие другие вопросы техники син-
теза при высоких давлениях. В литературе нет сообщений о син-
тезе карбонилов металлов при давлениях порядка 7000 ати, од-
нако при таких высоких давлениях, возможно, удалось бы син-
тезировать некоторые наиболее «трудные» карбонилы. При таких
условиях объем реакционного пространства должен быть неболь-
шим. В продаже имеется оборудование с объемом рабочего про-
странства 100 мл для рабочих давлений до 7000 ати.
В последнее время появились прессы (рис. 5) для давлений
свыше 70 000 ати, но Пет данных об их использовании для син-
теза карбонилов металлов. По-видимому, на этих прессах можно
работать только с твердым материалом; превращение одного твер-
дого карбонила в другой в условиях высоких давлений представ-
ляется заманчивой перспективой. Объем рабочего пространства
таких прессов примерно равен 0,5 см3.
Для химиков-экспериментаторов, не имеющих достаточного
опыта, могут быть полезны некоторые советы относительно работы
с автоклавами высокого давления. Большинство качающихся авто-
клавов завинчивают пробкой так, что она прижимает медную про-
кладку к корпусу автоклава. Важно отжечь медную прокладку,
чтобы она могла деформироваться и заполнить зазор. Чтобы от-
жечь медь и в то же время не дать ей покрыться хрупкой окали-
ной, ее удобно нагревать до красного каления в пламени горелки
Фишера и после этого погрузить в ацетон так, чтобы жидкость по-
крыла прокладку. (Ацетон обладает восстановительными свойст-
вами, и образование окалины сводится к минимуму.) Отожженную
прокладку можно использовать много раз, если она не становится
слишком тонкой. Иногда в качестве прокладок используют дру-
гие металлы — серебро или нержавеющую сталь.
Заслуживают внимания несколько соображений относительно
безопасности работы. Для защиты манометров и самого баллона с
окисью углерода в системе обычно ставят серебряные разрывные
диски, особенно в тех случаях, когда для нагнетания газа исполь-
зуют насосы. Разрывные диски должны быть расположены так,
чтобы возможный взрыв направлялся в сторону от оператора.
Вследствие сжимаемости окиси углерода (или другого газа) нару-
Р ИС. 5. Пресс для давлений до 170 000 ати Р и с. 6. Передвижной экран для обору-
фирмы «Tem-Press Research 1пс.» дования высокого давления фирмы
«Autoclave Engineers Inc.»
102
Глава 4
шение герметичности аппарата приводит к более сильному взрыву,
чем в случае гидравлических жидкостей. Многие автоклавы скон-
струированы так, что оператор может управлять двигателями и
нагревателями, регулировать температуру и давление, находясь
на расстоянии от автоклава. На рис. 6 показан передвижной за-
щитный экран для работы с автоклавами высокого давления.
Не следует погружать стальную бомбу автоклава в жидкий
азот для конденсации реагентов в самом автоклаве. Полагают,
что при таких низких температурах может произойти кристалли-
зация стали, в результате чего даже после нагревания до комнат-
ной температуры прочность стали может понизиться. Следует
помнить также о том, что окись углерода чрезвычайно токсична
и огнеопасна. В книге [32] указаны приемы работы с окисью
углерода.
Б. Вспомогательное оборудование для синтеза
карбонилов металлов
Многие операции при синтезе карбонилов металлов требуют
применения вакуумной техники. В задачу этой главы не входит
подробное описание методов работы с вакуумным оборудованием,
но все же здесь следует упомянуть о некоторых специальных ме-
тодах и приемах.
Высокое давление паров окиси углерода при температуре жид-
кого азота (400 мм рт. ст. при —196°) не позволяет полностью
перевести окись углерода в вакуумную линию без специального
насоса. Чаще всего используют насос Теплера с автоматической
электронной системой контроля [31]. Для переноса неконденси-
рующихся газов применяют также автоматический насос, разра-
ботанный Барчота и др. [33]. Благодаря возможности количест-
венно откачать объем оставшегося газа при помощи этих насосов
удается определить содержание окиси углерода в карбонилах ме-
таллов, которые при нагревании разлагаются на металл и окись
углерода.
Для разделения и очистки карбонилов металлов широко ис-
пользуют метод фракционной сублимации при пониженном дав-
лении. На рис. 7 схематически показан простой и недорогой аппа-
рат для сублимации, который был описан Кингом и Стоуном [34].
Для многих операций, связанных с получением чувствитель-
ных к присутствию воздуха веществ, используемых для синтеза
карбонилов, а в некоторых случаях для синтеза самих карбони-
лов, которые не выдерживают контакта с воздухом, очень полезен
«сухой бокс». Термин «сухой бокс» относится к различным лабо-
раторным устройствам, сильно различающимся по эффективности
и стоимости. Самые простые из них — это дешевые полиэтилено-
II. Оборудование
103
Рис. 7. Устройство для
сублимации [34].
1 — пробирка, наполненная
льдом (диаметр 22 мм); 2 — стан-
дартный шлиф 34/45.
вые мешки, наполненные азотом, с содержимым которых опери-
руют снаружи. Дешевые мешки такого рода описаны Шором [351
и Оконе и СимйинОм [36]. Имеются в продаже также недорогие
плексигласовые перчаточные боксы, несколько более сложные,
чем простые мешки. Один из таких боксов показан на рис. 8,а.
Наиболее эффективные «сухие боксы» точнее следует назвать «ла-
бораториями с инертной атмосферой». В одном из устройств та-
кого типа, показанном на рис. 8,6, происходит нзпрерывная
циркуляция инертного газа. Газ прохо-
дит над раскаленной медью и затем че-
рез молекулярное сито, благодаря че-
му концентрация кислорода поддержи-
вается на уровне ниже 1 ч. на млн., а
объемная концентрация водяных па-
ров — нйже 0,1 ч. на млн. Если не-
обходимо, внутри «сухого бокса» мож-
но установить весы и другое оборудо-
вание.
Иногда необходимо вести реакцию в
атмосфере окиси углерода при высоком
давлении, но так, чтобы другие реаген-
ты не соприкасались с металлическими
стенками бомбы.;В некоторых случаях
пригоден стальной стакан, вставляю-
щийся в автоклав. Этот стакан имеет-
ся в комплекте1 автоклава. В других
случаях удобен видоизмененный аппа-
рат Гросса [371. Аппарат Гросса со-
стоит из пробирки со спиралью из ка-
пиллярной трубки такого малого сече-
ния, чтобы не происходила диффузия
веществ из пробирки во время опыта.
Для этого диаметр капилляра должен быть меньше среднего про-
бега молекул при высоком давлении. На рис. 9 показан аппарат
Гросса для бомбы емкостью 1 л. При помощи такого аппарата про-
изводили синтез Тс2(СО)10 из ТсО2 и синтез галогенопроизвод-
ных этого карбонила [38]. С успехом применяют также более
простые формы1 пробирки Гросса с U-образной капиллярной
трубкой.
Кобаяши и др. [40а] усовершенствовали прибор для опреде-
ления молекулярного веса карбонилов металлов таким образом,
что его теперь можно использовать в вакуумной системе. В этом
приборе сравнивается давление паров растворителя над раство-
ром, содержащим исследуемое вещество, и над чистым раствори-
телем. Главная часть прибора приведена на рис. 10. В систему
II. Оборудование
105
включается, кроме того, термостатическая ванна и катетометр.
Известно, что криоскопический метод в применении к некоторым
карбонилам металлов дает ошибку, достигающую почти 100%.
Рис. 9. Видоизменен-
ный аппарат Гросса для
реакций с окисью азота
при высоких давлениях
[39].
1 — капиллярная трубка вн.
0 3 мм, внутр 0 0,5 мм.
г — стандартный шлиф
10/30.
К манометру
К глубокому вакууму
Рис. 10. Высоковакуумный тензиметрический
аппарат для измерения молекулярного веса кар-
бонилов металлов.
1 — колба для пробы и мешалка; 2 — колба с чистым
растворителем; 3 — тензиметр, давление паров раство-
рителя; 4 — ртутный затвор.
Тензиметрический метод позволяет определить молекулярный вес
наиболее сложных соединений в вакууме с точностью до 10% при
очень небольшой пробе.
7 Заказ № 547
106
Глава 4
III. МЕТОДЫ ПОЛУЧЕНИЯ КАРБОНИЛОВ
МЕТАЛЛОВ
А. Литература по синтезу карбонилов металлов
В литературе имеются сведения о многих синтезах карбонилов
металлов, которые не будут рассмотрены в связи с методиками,
изложенными в данной главе. Это относится главным образом к
синтезам, описанным в патентной литературе, или к промышлен-
ным синтезам. Методы, которые использовали для обнаружения
каждого карбонила металла, указаны в табл. 1, но во многих
случаях эти методы в дальнейшем заменяли более эффективными.
Иногда разработка методов, описанных в научной литературе,
определялась имеющимся в распоряжении экспериментаторов обо-
рудованием, желанием определить пригодность той или иной ме-
тодики или наличием какого-либо реагента. Для более подроб-
ного изучения проблем синтеза карбонилов металлов в табл. 3
приведены ссылки на научную литературу по методам синтеза
карбонилов. В таблице собраны все методы (хорошие, плохие и
посредственные), о которых известно автору. Для удобства пов-
торены также ссылки, упомянутые в табл. 1. Табл. 3 включает
ссылки на статьи в книгах, посвященных специально вопросам
синтеза, например в сборнике «Неорганические синтезы».
Б. Получение карбонилов железа
В связи с тем что в продаже имеется пентакарбонил железа,
сравнительно легко синтезировать и другие карбонилы железа —
понакарбонил Fe2(CO)9 и додекакарбонил Fe3(CO)12. Для этого
даже не требуется карбонилизации при высоком давлении. Для
многих синтезов формы более высокого молекулярного веса пред-
почтительны по сравнению с пентакарбонилом. Имеется очень
мало данных о термодинамических свойствах карбонилов, содер-
жащих два или три атома железа в молекуле. Большинство тер-
модинамических данных относится к пентакарбонилу Fe(CO)5.
В некоторых исследованиях, посвященных изучению механизмов
реакций с участием Fe(CO)5, рассмотрены также и карбонилы бо-
лее высокого молекулярного веса. В табл. 4 собраны данные о
свойствах карбонилов железа. ,
Полагают, что при образовании Fe2(CO)9 из Fe(CO)5 происхо-
дит предварительное образование очень активной формы Fe(CO)4.
Несмотря на то что соединение Fe(CO)4 никогда не было выде-
лено из раствора, Штольц и др. [25] в инфракрасных спектрах
Таблица 3
Литература по синтезам карбонилов металлов
Карбонил Метод Литература
Со2 (СО)8 Со + СО 10
СоСО3 + Н2 + СО 31, 406
Со(СО)4Н + нагревание 41
СоО + СО 42
Co(NH3)eCl2 + СО 43
Со2О3 + синтетический газ 44
Соли кобальта + Cu(Ag) + СО 30b
НСо(СО)4 + олефин + СО 45
Со4(СО)12 Со2(СО)3 + нагревание 10, 30b, 46—48
Сг(СО)в СгС13 + реактив Гриньяра + СО 11, 41, 49—56
СгС13 + Et3Al + СО 57, 58a, 586
СгС13 + бензофенонкетил натрия 4- 59
+СО
СгС13 +Mg +12 + СО 18, 60
Ацетилацетонат хрома(Ш) + Mg+ 61, 616
+ СО
СгС13 + А1 + А12С1в + СО 62, 63
Na2 [Сг3(СО)14] + СО 64
СгС13 + Na + СО 65—68, 69a, 696
СгС13 + LiAlH4 + СО 70
Fe(CO)6 Fe + CO 7, 8
FeSO4 4-NH3 + СО 43
Fe(CO)4I2 + Си 41, 71a
Fe2(CO)9 Fe(CO)5 + освещение 8, 41, 47, 716
Fe3(C(J)i2 Fe2(CO)B + нагревание 9
HFe(CO) + «активная» MnO2 41, 71b
HFe(CO) + окислители 72, 73
H2Fe3(CO)u-NR3 + кислота 74, 75
1г2(СО)18 1гС13 + Си + CO 14
1г4(СО)12 Ir2(CO)8 + нагревание 14
Мп2(СО)щ Mnl2 + Mg (Cu) + CO 18, 76
Mn(C2H3O2) + бензофенонкетил нат- 77
рия + CO
Mn(C2H3O2)2 + Et3Al + CO 57
MnCl2 + фeнилMgX + CO 78
MnCl2 + Na + CO 79
it-CH3CsH5Mn(CO)3 + Na + CO 80, 82
Mn(CO)5NO3 + нагревание 81
Мо(СО)в Mo + CO 10
7*
Продолжение табл. 3
Карбонил Метод Литература
MoCl6 + реактив Гриньяра + CO 41
MoCl5 + Et3Al +CO 57, 58a, 586
MoCl6 4- Na 4- CO 65, 696
MoCl6 + Zn + CO 54, 83
MoCl6 4- Al 4- CO 84
MoCl6 +Fe(CO)6 85
Cr(CO)e 4- продукты деления U3O8 86
Na2Mo3(CO)14 4- CO 64
Na2Mo2(CO)10 + CO 87
Ni(CO)4 Ni + CO 6, 88, 89
Ni(NH3)e Cl2 + CO 43
H2Ni2(CO)e + кислота 90
H2Ni2(CO)e + основание 91
(C3H8NiRr)2 + CH3OH + CO 92
NiSe + основание -f- CO 93
NiS или Ni (CN)24-CO 94, 95
Соли никеля(Н) + восстановитель + 41, 96—98, 99a
+ CO 996
Os(CO)5 OsO4-f-CO 17
0s3 (CO)i2 Os(CO)6 4- нагревание 17, 23, 100
He2(CO)10 Re2O7 + CO 15, 38
Re2S7 + CO 15
KReO4 4- CO 15
HRe(CO)5 4- нагревание 101
Rh2(CO)8 Rh+ CO 16
Rh4(CO)12 Rh2(CO)8 + нагревание 16
Rhe(CO)ie RhCl3 4- Cu(Ag) 4- CO 16, 24
Ru (C0)5 Rul3 + Ag + CO 13
Ku3(C(J)i2 Ru(CO) 5 4- нагревание 13, 23
Tc2(CO)10 Tc2O7 4- CO 20
TcO2 4- CO 38
Tc3(CO)12 NaTc(CO)6 4- H3PO4 21
V(CO)e VC13 4- Mg-Zn 4- CO 19, 102
Mg[V(CO)e] I24-HC1 103
[Na(diglyme)2] [V(CO)8] 4- H3PO4 104
Дитолуолванадий 4- CO 105
W(CO), WCle 4- реактив Гриньяра 12, 41, 54
WC1, 4- Zn 4- CO 83
WCle 4- Et3Al 4- CO 57, 58a, 586
WCle + Fe(CO)5 106
WCle4-Fe(CO)64-H2 107
Галогенид вольфрама(У) 4- Na 4~CO 65, 696
III. Методы получения карбонилов металлов
109
Свойства карбонилов железа Таблица 4
Свойство Fe(CO)s Fe2(CO)» Fe3(CO)12
Цвет Молекулярный вес .... Температура плавления, °C Температура кипения, °C Температура и давление сублимации Температура разложения, °C . . . , Плотность, г/см3 Длина связи М—М, А . . Длина связи М—С, А . . Бесцветный3 196 —20 103 130 1,52 1,82 Желтый 364 Разлагается 100 2,08 2,46 1,9 Черный 504 Разлагается 60°; 0,1 мм pm. cm. 140 2,00
Длина связи С—О, А . . . 1,14 1,15 1,3 —
I —182,6 — —
&Н разл, ккал ...... 138,Зб — —
ЛЯ °сгор, ккал —385,9 — —
У , 1 &.Н°К(.п,ккал Окислительно-восстановите 3Fe (CO)2"=F( а В соответствии с большей ч что, вероятно, объясняется присут 6 Для реакции Fe(CO)6 8,9 льные потенциа 3з(СО)12 + 6е_, . астью данных цве1] ствием следов жел газ ^^газ “Ь лы Е = —0,74 в. Fe(CO)5 от соломе того Fe2(CO)2. согаз- иного до желтого,
растворов Fe(CO)6, облученных ультрафиолетовым светом, на-
блюдали следы очень активного Fe(CO)4
Fe (СО), 4- ультрафиолетовый свет —► Fe (СО), + СО.
но
Глава 4
Вероятно, Fe(CO)4 может образовываться также в результате
разложения карбонилгидрида железа
Fe (СО)4Н2----► Fe (СО)4 + Н2.
Считают, что Fe2(CO)9 образуется в результате последующего
взаимодействия Fe(CO)4 и Fe(CO)5
Fe(CO)4 + Fe (СО)5--->Fe2(CO),.
Однако исследование изотопного обмена окиси углерода, предпри-
нятое Кили и Джонсоном [471, позволяет предполагать, что про-
цесс фотодиссоциации не является основным в реакции образова-
ния нойакарбонила. Авторы предлагают другой механизм реакции
с участием активированной молекулы Fe(CO)5*:
Fe (СО)5 + ультрафиолетовый свет-> Fe(CO)5 ,
Fe (СО)* + Fe (СО)6 --> Fe2 (СО), + СО.
Механизм реакции образования Fe3(CO)12 не ясен. Процесс обра-
зования этого соединения при нагревании Fe2(CO)9 может отли-
чаться от процесса его образования в водных растворах путем
окисления Fe(CO)4H2 или Fe(CO)42-. При термическом разложении
Fe2(CO)9 в органических растворителях при 95° могут возникать
такие же активированные промежуточные формы, как и при реак-
ции Fe(CO)6, и тогда общую реакцию можно выразить в виде урав-
нения [1]
. 3Fe2 (СО),---- Fe3 (СО)12 + 3Fe (СО)5.
В водных растворах анионы карбонилата железа претерпева-
ют взаимное превращение, определяющееся температурой и pH
раствора. При pH более 10 проявляется тенденция к устойчивому
существованию Fe^COlg2-, а в пределах pH 5—10 образуется анион
Fe:i(GO)2-n. <
В работе Кили и Джонсона [47] описана методика получе-
ния Fe2(CO)9, по которой пробу Fe(CO)5 вводят в ампулу из стекла
пирекс и ампулу запаивают. Нижнюю часть ампулы (жидкость)
затемняют, а верхнюю (газообразную фазу) подвергают действию
солнечного света в течение 2 час. На освещаемых стенках ампулы
йостепённо накапливается твердый Fe2(CO)9; процесс идет до тех
пор, пока жидкая фаза не исчезает. Таким образом, выход Fe2(CO)9
почти количественный. Поскольку этот метод синтеза очень прост,
а в литературе можно найти и другие такие же эффективные и
111. Методы получения карбонилов металлов
111
простые методики (табл. 3), нет нужды дольше останавливаться
на методах синтеза Fe2(CO)9. Однако следует предостеречь экспе-
риментатора относительно токсичности Fe(CO)5, а в ампуле следует
оставлять достаточно места для выделяющейся СО во избежание
чрезмерного увеличения давления.
i При получении Fe3(CO)12 также не требуется карбонилизации
при высоком давлении. В «Неорганических синтезах» изложена
испытанная методика Кинга и Стоуна, где уделено особое внима-
ние приготовлению очень активной формы двуокиси марганца,
которая служит окислителем, а также хранению чувствительного
к воздействию воздуха карбонила металла. Указано, что реакция
протекает по приведенным ниже уравнениям без каких-либо заме-
чаний относительно механизма реакций
Fe (СО)5 + ЗОН"---> HFe (СО)" + СО|" + Н2О,
3HFe (СО)* -I- ЗМпО2 ф- ЗН2О-> Fe3 (СО)12 ф- ЗМп2+ + ЭОН”.
Для синтеза пользуются обычной лабораторной техникой:
вытяжным шкафом, поддоном и, по-видимому, защитным экраном.
Колба емкостью 2 л снабжается мешалкой, трубкой для ввода
азота и обратным холодильником. В продутую азотом колбу поме-
щают 60 г (42 мл) Fe(CO)5 (технической чистоты), 170 мл метанола
и 135 г водного раствора, содержащего 45 г гидроокиси натрия.
Смесь перемешивают в течение 30 мин. Реакция идет с выделением
тепла. После этого в колбу добавляют 125 мл насыщенного ра-
створа хлорида аммония.
Продажный МпО2 не годен для окисления забуференного
раствора HFe(CO)4“, поэтому специально готовят активную форму
МпО2 осторожной обработкой раствора 67 г КМпО4 в 300 мл воды
95%-ным этанолом (100 мл). Иногда при перемешивании начи-
нается энергичная реакция; иногда для ее возбуждения смесь
немного подогревают. Красно-фиолетовый раствор КМпО4 превра-
щается в коричневую пасту МпО2.
В колбу, в которой ведут синтез карбонила, добавляют пасту
МнО2, перемешивают и оставляют на 2 час. Реакция сопровожда-
ется выделением тепла. Затем в колбу вводят раствор 40 г Fe2SO4-
•7Н2О в 250 мл 1 М серной кислоты. Соць железа(П) восстанав-
ливает избыток МпО2. К образовавшемуся красному раствору
добавляют 300 мл 9 М серной кислоты и смесь перемешивают до
тех пор, пока полностью не выделится твердый Fe3(CO)12 и не
останется светло-зеленый раствор. Отфильтрованное вещество
промывают горячей 1 М серной кислотой, затем 95 %-ным этано-
лом, а после этого низкокипящим углеводородом парафинового
ряда, например пентаном. Образуется до 45 а продукта (выход
112
Глава 4
90% в расчете на пентакарбонил железа). Продукт достаточно
чист для большинства применений. Во избежание окисления воз-
духом Fe3(CO)12 хранят в атмосфере азота. Устойчивость вещества
повышается, если в нем оставить следы растворителя. Додекакар-
бонил железа Fe3(CO)12 высокой чистоты получают либо в виде
черного сублимата при 60° и остаточном давлении 0,1 мм рт. ст.,
либо экстрагированием пентаном под азотом в аппарате Сокслета.
При нагревании выше 60° Fe3(CO)12 разлагается с выделением
маталлического железа. Известно, что при длительном хранении
вещества высокой степени чистоты образуются пирофорные про-
дукты разложения.
Имеется и другой метод синтеза, основанный на подкислении
амминных комплексов H2Fe3(CO)u (табл. 3). При синтезе этим ме-
тодом получают Fe3(CO)12 с очень хорошим выходом.
В. Получение карбонилов кобальта
Несмотря на то что Со2(СО)8 и Со4(СО)12 в продаже нет, их
можно довольно легко получить, не проводя карбонилизацию при
высоком давлении. Более высокий выход в расчете на кобальт
получается, однако, в том случае, если Со2(СО)8 получают, поль-
зуясь сжатой смесью водорода и окиси углерода. Поэтому ниже
будет изложен метод, разработанный Бендером и др. [406] и ви-
доизмененный Джолли [31 ]. Этот метод может служить как бы
введением в технику синтеза с применением высоких давлений.
Другие методы, не требующие высокого давления, можно найти
по литературным ссылкам, приведенным в табл. 3.
Октакарбонил кобальта Со2(СО)8 готовят прямой карбони-
лизацией металла при высоком давлении. Предполагают, что ме-
ханизм процесса карбонилизации подобен механизму карбонили-
зации железа и никеля. Однако Подалл считает, что карбонилы,
содержащие два атома металла в молекуле с карбонильными мос-
тиками или с непосредственной связью металл—металл, требуют
специального рассмотрения. Термодинамическая устойчивость
связи металл — карбонил может иметь меньшее значение, чем
факторы, влияющие на скорость образования первой связи ме-
талл — окись углерода. Исследование обмена карбонила, про-
веденное Кили и Джонсоном [47 ], не позволило выяснить меха-
низм реакции, так как на свету и в темноте все группы СО обме-
ниваются настолько быстро, что скорость процесса не поддается
измерению. Совершенно по-другому протекает восстановительная
карбонилизация иона кобальта(П) с образованием НСо(СО)4
и с последующим превращением карбонилгидрида в Со2(СО)8 при
нагревании. Однако подробности этого процесса неизвестны,
III. Методы получения карбонилов металлов
113
поскольку этому вопросу посвящено мало работ. Физические и тер-
модинамические данные, имеющиеся в литературе, собраны в
табл. 5.
Таблица 5
Свойства карбонилов кобальта
Свойство СоДСО), СоДСОЩ
Цвет Оранжевый Зеленовато-черный
Молекулярный вес 342 572
Температура плавления, °C 51 Разлагается
Температура кипения, °C Разлагается Разлагается
Температура и давление сублимации 45°; 10мм рт. ст. —
Температура разложения, °C 52 60
Плотность, г/см3 1,73 —
Длина связи М—М, А 2,54 2,50
ДЯобр, ккал — 33 000а
Окислит ельно-восстановителы зыи потенциал
2Со(СО)“ =Со2(СО)34-2е-, £ = — 0,4 в.
а Для образования из Сог(СО)«, а не из элементов [48].
Синтез Со2(СО)8 при высоком давлении основан на реакции
2СоСО3 + 2Н2 + 8СО--------► Со2 (СО)8 + 2Н2О + 2СО2.
В качающийся автоклав емкостью 300 мл загружают 7,5 г
СоСО3 и 75 мл низкокипящего растворителя (парафинового ря-
да); автоклав продувают окисью углерода для удаления воздуха,
после чего вводят смесь окиси углерода и водорода (1 : 1) при дав-
лении 240 ати. Смесь газов составляют следующим образом: в
автоклав нагнетают окись углерода до давления 120 ати, после
чего нагнетают водород до конечного давления в автоклаве
240 ати. Качающийся автоклав нагревают до 150—160°, выдержи-
вают при этой температуре в течение 3 час, охлаждают до комнат-
ной температуры и выпускают газ в вытяжку. Образовавшийся
темный раствор фильтруют через бумагу и выдерживают при тем-
пературе около —10° в течение ночи. Декантируют растворитель
от темно-красных кристаллов. Кристаллы сушат в токе сухого
114
Глава 4
азота. Выход продукта около 6 г, что соответствует приблизитель-
но 55% исходного количества кобальта.
Со4(СО)12 получают с количественным выходом простым нагре-
ванием Со2(СО)8 в атмосфере азота до температуры около 50°.
Никаких сведений о механизме реакции нет. Результаты рентге-
нографического изучения неупорядоченных кристаллов [107]
не позволили надежно установить структуру этого соединения
2Со3 (СО)8 -]- нагревание->• Со4 (CO)i2 + 4СО.
Г. Получение карбонила технеция
Поскольку соответствующие термодинамические данные от-
сутствуют, методика получения Тс2(СО)10 [20а ] была разработана
в полной аналогии с методикой Рибера и Фухса [15] для полу-
чения Ве2(СО)10. Однако в случае технеция дело осложняется тем,
что единственным изотопом Тс, который можно было получить в
значительных количествах, был "Тс, слабый ^-излучатель
(0,29 Мэв) с периодом полураспада 2,12-105 лет. Несмотря на то
что достаточной защитой от такого слабого излучателя могут слу-
жить стенки любого контейнера или сосуда, все же он довольно
активен, и требуется постоянный контроль при помощи счетчиков
Гейгера с тонким окном. Любое загрязненное оборудование и все
остатки синтеза должны быть захоронены в море.
Изотоп "Тс можно было получить в Окриджской лаборатории
в различных формах. Для рассматриваемого синтеза в качестве
исходного вещества используют пертехнетат аммония в виде на-
сыщенного водного раствора. После упаривания бесцветного ра-
створа остается белый NH4"TcO4.
Пробу сухого пертехнетата аммония (0,85 г) отвешивают в
лодочку для сжигания, которую проталкивают к запаянному кон-
цу трубки из стекла викор длиной 30 см и диаметром 25 мм. Труб-
ку укрепляют в зажимах так, что при передвижении трубчатой
печи лодочка попадает в печь. Холодный конец трубки через
ловушку, охлаждаемую сухим льдом с ацетоном, соединяют с ис-
точником азота давлением 1 атм. Затем трубку продувают азотом,
после чего холодную печь быстро разогревают приблизительно до
400°. При этом происходит пиролиз пробы и образуется черный
ТсО2. Образующиеся при разложении газы откачивают до остаточ-
ного давления 1 мм рт. ст. через ловушку, охлаждаемую смесью
СО2 с ацетоном, где конденсируются вода и синяя N2O3. Большая
часть ТсО2 образуется на горячих стенках трубки или остается в
лодочке. После очистки ловушки трубку соединяют с источником
кислорода давлением 1 атм и температуру печи повышают до
600°. Образуются желтый Тс2О7 и следы оранжевого вещества
III. Методы получения карбонилов металлов
115
(предположительно НТсО/ Оба веЩества яаСТИЧЯ° М“ГР?’РЗТ \3
печи и конденсируются на родных стенках трубки сразу же по
выходе из печи После охлажДения системы трубку перев0ДЯТ в
вертикальное положение и
стеклодувной горелкой пере-
гоняют полосу желтой Тс2О7
к нижнему концу трубки.
Стальную бомбу емкостью
350 мл, плакированную
медью, продувают сухим азо-
том. Не прекращая тока
азота, конец трубки поме-
щают в ступку и разбивают
молотком так, чтобы оскол-
ки стекла и лодочки вме-
сте с Тс2О7 падали в бомбу.
Бомбу «прополаскивают»
химически чистой окисью
углерода при давлении
55 ати и комнатной темпе-
ратуре, после чего нагнета-
ют окись углерода до давле-
ния 204 ати при этой же
температуре. Бомбу нагрева-
ют до 220° и выдерживают в
течение 20 час без перемеши-
вания. Давление в бомбе в
этот период составляет 350
ати. После охлаЩдения'ав-
токлава до комнатной темпе-
ратуры окись углерода вы-
пускают через ловушку, ох-
лаждаемую СО2 с ацетоном.
Ловушка предотвращает вы-
деление технеция-99 в атмо-
сферу.
Автоклав промывают по-
Р и с. 11. Аппарат для оттопки раство-
рителя при пониженном давлении.
1 — ловушка с раствором двуокиси углерода —
пентан; 2 — дефлегматор с Ледяным охлажде-
нием; 3 — спуск для ледяной воды; 4 — водя-
ная баня.
следовательно тремя порция-
ми диэтилового эфира по 50 мл, который растворяет Тс2(СО)10.
Осколки стекла, лодочку и черный остаток переносят в. раствор
6 М азотной кислоты для извлечения остатков 89Тс, а бомбу бы-
стро промывают разбавленной азотной кислотой. Ее внутрен-
ние стенки остаются заметно радиоактивными.
Эфир отгоняют из раствора Тс2(СО)10 при пониженном давле-
нии, используя в качестве аспиратора водяной Маностат. Предва-
116
Глава 4
рительные опыты с Сг(СО)6, Мп2(СО)10 и Re2(CO)10 подтвердили,
что при таком методе удаления растворителя вместе с ним отго-
няется некоторое количество карбонила металла, если пары не
пропускать через дефлегматор с температурой 0°, показанный
на рис. 11. Остаток после отгонки эфира сублимируют при 50°
и давлении 10-2 мм рт. ст. в аппарате, приведенном на рис. 7.
Выход при этом составляет около 0,10 г бесцветных кристаллов,
что соответствует приблизительно 10% исходного количества "Тс.
Карбонил технеция Тс2(СО)10 плавится при 159—160° в запаян-
ном капилляре.
Разработан также другой метод синтеза Тс2(СО)10 [38]. В этом
случае используют азотнокислые растворы остатков, получаю-
щихся при выделении Тс2О7. Осторожным выпариванием с сер-
ной кислотой почти досуха вытесняют азотную кислоту. При
этом надо следить, чтобы не улетучивался НТсО4. Раствор после
этого нейтрализуют 1 М раствором NaOH с фенолфталеином до
перехода окраски, добавляют избыток ацетальдегида и выдержи-
вают раствор теплым в течение нескольких часов до коагуляции
ТсО2. Осадок двуокиси технеция отфильтровывают, сушат при
комнатной температуре и карбонилизируют в аппарате Гросса
(рис. 9).
Для опыта обычно берут 0,50 г ТсО2, при комнатной темпера-
туре нагнетают в автоклав окись углерода до давления 20,4 ати,
нагревают автоклав до 250° и выдерживают без покачивания в те-
чение 24 час. Из бомбы извлекают около 0,50 г Тс2(СО)10, что соот-
ветствует приблизительно 50%-ному выходу по металлу.
Д. Получение карбонила марганца
Предложенный для синтеза Мп2(СО)10 метод значительно от-
личается от остальных методов, рассмотренных в этой главе. Для
получения хорошего выхода потребовалось сочетать воздействие
восстановителя, координирующего растворителя и карбонилиза-
ции при высоком давлении. Подалл называет весь процесс «вос-
становительной карбонилизацией». Метод «восстановительной кар-
бонилизации» пригоден также для синтеза некоторых других
карбонилов, например V(CO)6 и Сг(СО)6. Его особенности будут
показаны на примере синтеза Мп2(СО)10.
Первый синтез Мп2(СО)10, выполненный Бримом, Линчем и
Сесни [18] с использованием реакции Гриньяра, а также синтез
Сг(СО)в, выполненный Фишером и Хафнером с использованием
реакции Фриделя—Крафтса (табл. 3), можно рассматривать как
варианты «восстановительной карбонилизации», и во всех случаях
основной этап этого синтеза — образование металлорганичес-
III. Методы получения карбонилов металлов
117
ких промежуточных соединений с металлом в низком окисли-
тельном состоянии. Лиганд и координированные молекулы раст-
ворителя вытесняются затем окисью углерода при высоком дав-
лении. Имеется несколько различных методов синтеза Мп2(СО)10,
которые дают хороший выход (табл. 3). Один из методов — вос-
становление триэтилалюминием,— разработанный Подаллом, Дан-
ном и Шапиро, будет подробно рассмотрен в этой главе.
Физические и термодинамические свойства Мп2(СО)10 указаны
в табл 6.
Таблица 6
Свойства Мп3(СО)10
Свойства
Мп,(СО),,
Цвет
Молекулярный вес
Температура плавления, °C
Температура кипения, °C
Температура и давление сублимации
Плотность, г/см3
Длина связи М—М, А
Д//Обр, ккал
Желтый
470
151
Разлагается
50°; 10~2 мм pm. cm.
1,81
2,93
—400,9
Окислительно-восстановительный потенциал
2Мп (СО)- = Мп2 (СО)10 + 2е~, Е=~ 0,68 в.
Предполагаемый механизм образования карбонила металла
при использовании алкилалюминия [57] можно представить сле-
дующей схемой:
ГалогеииЗ металла
+ EtjAl Промежуточное соединение (I)
Промежуточное соединение
Металл
Промежуточное соединение (п)
Промежуточное соединение
EtgAl/CO „ .
(II)______-_________> Карбонил металла
ki
118
Глава 4
Авторы предположили, что относительные константы скорости
уменьшаются в порядке Zc3>Zc2>^i- Если это справедливо, то
скорость всего процесса определяется скоростью образования
промежуточного соединения I — предположительно соединения
металл — этил. Полагают, что образование промежуточного сое-
динения! I происходит быстро, на что указывают небольшие коли-
чества образующегося свободного металла. Стадия, соответству-
ющая /с4, более подвержена влиянию стерических эффектов по
сравнению со стадией, соответствующей кг. В действительности
стадия, соответствующая Zc4, может состоять из нескольких после-
довательных стадий.
Механизм синтеза карбонилов металлов, по Гриньяру, рас-
смотрен в обзорах нескольких авторов [1—3, 51], но они не при-
шли к окончательному согласию. Однако имеется согласие отно-
сительно образования промежуточного соединения металл—угле-
водород, предшествующего гидролизу. Все авторы сходятся в том,
что, по крайней мере в случае Сг(СО)6, карбонил металла образу-
ется только после гидролиза реакционной смеси Гриньяра.
Для синтеза Мп2(СО)10 берут 49 г тетрагидрата ацетата мар-
ганца^ I) (0,20 моля) и сушат его путем азеотропной отгонки со
100 мл бензола в аппарате с ловушкой для отделения воды. Если
смесь не перемешивать во время отгонки пластинчатой механиче-
ской мешалкой, безводный ацетат марганца слипается в кусок.
Попытки использовать магнитную мешалку оказались безуспеш-
ными. Такой кусок легко разрушается пестиком в ступке, пока он
еще смочен бензолом. Ацетат марганца отфильтровывают и следы
бензола удаляют в вакуумном эксикаторе при разрежении 0,01
мм рт. ст. Сухое вещество хранят в эксикаторе над осушающим
веществом драеритом.
Для приготовления суспензии безводного ацетата марганца
берут 150 мл диизопропилового эфира, предварительно высушен-
ного перегонкой в вакууме из смеси с алюмогидридом лития. Сухой
эфир используют также для приготовления раствора триэтилалю-
миния (ТЭА), содержащего 120 мл ТЭА (0,80 моля) в 300 мл
диизопропилового эфира.
При растворении ТЭА в диизопропиловом эфире выделяется
тепло, и растворение следует вести при 0° в атмосфере азота.
Следует соблюдать чрезвычайную осторожность, так как ТЭА
обладает пирофорными свойствами. Для соединения сосуда с
ТЭА и колбы с диизопропиловым эфиром следует применять
трубку из тигона и смешивание вести очень медленно. [Следует
упомянуть о возможности использования триизобутилалюминия
(ТИБА) вместо ТЭА. Первые исследователи синтеза обнаружили,
что ТИБА дает несколько больший выход, а, поскольку он менее
пирофорен, с ним легче работать.]
III. Методы получения карбонилов металлов
119
Суспензию безводного ацетата марганца вводят в круглодон-
ную трехгорлую колбу емкостью 1 л. Колба должна быть снабже-
на мешалкой, соединена с источником азота и колбой, содержащей
раствор ТЭА в эфире, и помещена в ледяную ванну с температу-
рой 0°.
К суспензии безводного ацетата марганца при перемешивании
медленно в течение 2 час добавляют раствор ТЭА. Образующуюся
темно-коричневую смесь под сильным током азота переносят в
2-литровую плакированную внутри медью бомбу высокого дав-
ления. Бомбу предварительно продувают сухим азотом (первые
исследователи этого синтеза переносили в бомбу смесь ацетата
марганца с ТЭА и эфиром в сухом боксе, заполненном азотом).
Бомбу «прополаскивают» окисью углерода под давлением 55 ати
и заполняют окисью углерода до давления 204 ати (при комнат-
ной температуре). При покачивании в течение 1 час температуру
бомбы доводят до 100°. Вначале считали, что бомбу (при объеме
1 л) нужно выдерживать при температуре 80—85°, пока не про-
изойдет энергичная начальная реакция ТЭА и Мп(ОАс)2. Однако
По дал л, Данн и Шапиро не наблюдали такой реакции при непре-
рывном нагревании автоклава до 100° и выдержке при этой тем-
пературе в течение 6 час. Возможно, более массивная колба ем-
костью 2 л играет роль теплового буфера и снижает эффект вне-
запного выделения энергии.
После охлаждения бомбы до комнатной температуры из нее
выпускают избыток окиси углерода. Содержимое переносят в токе
азота в 2-литровую круглодонную трехгорлую колбу с мешалкой.
Колбу подсоединяют к источнику азота через ловушку с пальцем,
охлаждаемым двуокисью углерода. Колба должна быть снабжена
также капельной воронкой на 500 мл с выровненным давлением.
Сначала колбу помещают в лед и в смесь медленно добавляют
400 мл ледяной воды, а затем 100 мл концентрированной соляной
кислоты. Потом колбу нагревают на водяной бане и при снижен-
ном давлении из смеси отгоняют диизопропиловый эфир. Для
этого используют ледяную колонку для отгонки растворителя,
приведенную на рис. И. Из водной суспензии после отгонки эфи-
ра извлекают Мп2(СО)10 путем перегонки с паром. Паровой кон-
денсат фильтруют, а Мп2(СО)10 нагревают до 40° и сублимируют
при давлении 0,01 мм рт. ст. Сублимацию проводят в аппарате,
показанном на рис. 8; поверхность конденсации охлаждают до 0°.
Вначале авторы сообщали, что они получили выход 54% (21 г),
т. е. почти такой же, какой достигается в только что рассмотрен-
ной методике. После сублимации с охлаждаемой пробирки сни-
мают хорошо образованные золотисто-желтые кристаллы, которые
хранят в атмосфере азота в банке с завинчивающейся крышкой.
Температура плавления 153—155° (не проверено).
120
Глава 4
Е. Получение карбонила ванадия
При получении карбонила важное значение для выхода имеет
выбор растворителя. Например, Натта и др. [61] нашли, что вы-
ход Сг(СО)в можно увеличить более чем до 80% при использо-
вании пиридина в качестве комплексообразующего раствори-
теля.
Это согласуется с общим представлением о «восстановительной
карбонилизации», рассмотренном в разделе Д. Итальянские хи-
мики при синтезе карбонилов пользуются растворителями типа
пиридина. Этим методом они впервые синтезировали V(CO)e
[19]. Изложенная ниже методика рассчитана на использование
азотистых оснований и в точности соответствует методике синтеза
V(CO)e по Эрколи и др. [102].
В качающийся автоклав из нержавеющей стали емкостью
500 мл помещают 9,9 г безводного VC13, 220 г сухого пиридина и
16,0 г активированной смеси металлов. Активированная смесь
металлов состоит из 2 г иода (активирующий агент) и 14 а смеси
порошков магния и цинка в соотношении 1 : 2,7 (3,8 г магния и
10,2 г цинка). Смесь реагентов в автоклаве энергично перемеши-
вают и при перемешивании нагнетают туда окись углерода до дав-
ления 135 ат. Автоклав нагревают до 135°, выдерживают при этой
температуре в течение 8 час и после этого выпускают окись угле-
рода. После 8 час нагревания давление с 208 ат уменьшается до
160 ат.
Остаток после реакции вымывают из бомбы пиридином и остав-
ляют на сутки. В течение всего этого времени вещество выдержи-
вают в атмосфере азота. Отстоявшийся раствор перегоняют в ва-
кууме без нагревания при остаточном давлении около 1 ммрт. ст.,
а оставшуюся твердую массу смешивают с 200 мл воды и 400 мл
«очень чистого» диэтилового эфира. Смесь охлаждают и, время от
времени встряхивая, подкисляют 300 мл 4 М соляной кислоты
(при этом выделяется водород). Эфирный слой быстро отделяют
от водного слоя, промывают разбавленной соляной кислотой и
затем водой, после чего в течение ночи сушат с MgSO4. Эфирный
раствор разгоняют в вакууме. Образуется коричневый концентри-
рованный раствор, который выделяет водород. При дальнейшем
удалении эфира образуется кристаллическое вещество, которое
сублимируют при 40—50° и давлении 15 мм рт. ст. Сублимат
представляет собой влажный V(CO)e. Его сушат над Р2О5 в ат-
мосфере азота и еще раз сублимируют, соблюдая предосторож-
ности против потери вещества из-за улетучивания или чрезмер-
ного нагревания. При синтезе получают 5,3 г сине-зеленого
продукта, что соответствует выходу 38% в расчете на металличе-
IV. Идентификация карбонилов металлов
121
ский ванадий. Карбонил ванадия V(CO)e разлагается без плавле-
ния в заполненной азотом запаянной ампуле для определения
температуры плавления. Разложение происходит при 60— 70°.
Карбонил ванадия чувствителен к воздуху, поэтому его нужно
хранить в атмосфере азота.
! IV. ИДЕНТИФИКАЦИЯ КАРБОНИЛОВ МЕТАЛЛОВ
А. Инфракрасные спектры
^Обычно для идентификации карбонилов металлов используют
инфракрасные спектры. Частота валентных колебаний связи угле-
род—кислород и характеристика карбонильной группы довольно
сильно зависят от металла и свойств симметрии молекулы карбо-
нила металла, поэтому большинство соединений легко можно иден-
тифицировать. Форма, волновое число и интенсивность максимума
поглощения часто хорошо согласуются с теоретическими представ-
лениями о молекулярной структуре. Относительно инфракрасных
спектров карбонилов металлов имеется обширная литература
[108]. Вообще все карбонилы металлов имеют по крайней мере
один максимум поглощения в области 2000 см-1, который отража-
ет взаимодействие металл — СО. Если в соединении имеется мости-
кообразующая карбонильная группа, неизменно наблюдается
полоса валентных колебаний при 1800 см-1, но обратное не всегда
справедливо, так как возможны различные типы резонансного
взаимодействия. Точное расположение максимумов несколько
зависит от типа растворителя, используемого при снятии спектра,
наиболее четкие формы получаются в случае неполярных углево-
дородов. Общая «картина» спектра определенного карбонила ме-
талла зависит также от оптической системы прибора., Следова-
тельно, если требуется сравнивать экспериментальные спектры
со спектрами, имеющимися в литературе, целесообразно исполь-
зовать сравнимые растворители и оптические системы,;
Для иллюстрации того, насколько велика разрешающая спо-
собность инфракрасных спектров, на рис. 12 приведены спектры
карбонилов металлов группы VI16, снятые на двухлучевом при-
боре с оптикой из фторида лития. В качестве растворителя ис-
пользовали четыреххлористый углерод.
В табл. 7 приведены литературные данные относительно мак-
симумов поглощения карбонильных групп в карбонилах металлов
для разных растворителей и разных оптических систем. Неко-
торые старые данные содержат неточности, но и они приведены в
таблице, если нет более поздних данных для сравнимых раство-
рителей и оптических систем.
Таблица 7
Максимумы поглощения инфракрасных спектров карбонильных групп в карбонилах металлов
Соединение Максимумы3 частот валентных колебаний, слг-1 Оптическая система Растворитель или состояние Литера- тура
Со2(СО)8 2070 (с) 2025 (с) 1858 (ср) NaCl н-Гексан 109
2068,8 (ср) 2058,7 (сл) 2042,4 (с) 2030,7 (ср) LiF н-Гептан НО
2022,7 (ср) 2001,7 (оч. сл) 1991 (оч. сл) 1866,0 (сл)
1857,2 (сл) 2071 (ср) 2069 (ср) 2059 (ср) 2044 (с) Решетка Гексан 111
2031 (ср) 2022 (ср) 1866 (сл) 1857,5 (сл)
2071 2042 2005 1845 Решетка Хлороформ 112
2072 (ср) 2069 (ср) 2059 (сл) 2042 (с) » СС14 111
2030 (ср) 2022 (ср) 1865 (сл) 1851 (сл)
Со 4(СО)12 2063,2 (с) 2054,5 (с) 2037,9 (оч. сл) 2027,5 (оч. сл) LiF н-Гептан НО
1867,0 (сл) 2103 (оч. сл) 2063 (с) 2055 (сл) 2037,5 (сл) Решетка Гексан 111
2027 (сл) 2018 (оч. сл) 1990 (оч. сл)
Сг(СО)в 1987,5 (с) 1955 (оч. сл) » » 111
2122 (оч. сл) 1967,5 (сл) 2089 (оч. сл) 2020 (оч. сл) 2000 (с) LiF Газообразное 113
1983,5 (с) 1953 (оч. с л) Решетка С2Н2С14 111
2000,1 LiF Газообразное 114
1984 ? Циклогексан 115
Fe(CO)5 2084 (ср) 2028 (с) 1994 (ср) 1935 (сл) NaCl Газообразное 116
2022,9 (ср) 2000,3 (с) 1963,6 (сл) LiF н-Гептан 110
2034,4 2013,5 LiF Газообразное 114
2022 (ср) 2000 (с) 1964 (оч. сл) Решетка Гексан 111
2034 2014 ? ? 117,118
2020 (ср) 1997 (с) 1961 (оч. сл) Решетка C2H2C14 111
2019 1998 Ъ Хлороформ 112, 115
2021 2000 ? Циклогексан 115
F е^ССОэ 2087 2023 1831 ? ? 1
2080 (ср) 2034 (с) 1828 (с) NaCl Твердое И6
Fe3(CO)i2 2046 (с) 2023 (ср) Решетка Гексан 111 /
2047 (с) 2024 (ср) 1865 (оч. с л) 1834 (оч. сл) » С2Н2С14 111
2043 (с) 2020 (с) 1833 (ср) NaCl Толуол 119
2043 (с) 2020 (с) 1997 (ср) 1858 (оч. сл) LiF Дисульфид угле- 120 -
рода 121
1826 (оч. сл)
Mn2(CO)I0 2033 2028 1997 ? ?
2074 2015 1972 ? ? 11/
2044 (ср) 2013 (с) 2002 (сл) 1983 (ср) LiF Циклогексан 38
2044 (ср) 2012 (с) 1918 (ср) LiF СС14 20
2043 2013 1982 Решетка Циклогексан 112
2043 2010 1980 » Хлороформ 112
Мо(СО)в 1989,5 (с) 1957 (оч. сл) » Гексан 111
2002,6 LiF Газообразное 114
1985,5 (с) 1955 (оч. сл) Решетка С2Н2С14 111
2115 (сл) 2110 (сл) 2021 (ср) 1983 (с) LiF Хлороформ 122
1984 ? Циклогексан 115
Ni(CO)4 2109 (оч. сл) 2060 2046 2057 2084 (оч. сл) 2020 (оч. сл) 2004(c) 1971(сл) LiF NaCl ? ? Газообразное » Циклогексан ? ИЗ, 122 123 115 117, 118
2043 (с) - 2004 (оч. сл) Решетка С2Н2С14 111
2045,7 (с) 2007,0 (оч. сл) LiF н-Гептан ПО
2057,6 LiF Газообразное 114
2057 (с) 2122 (оч. сл) 1990 (оч. сл) 2018 (оч. сл) LiF » 124
Продолжение табл.
сильные; ср — средние, сл — слабые; оч. ел — очень слабые.
IV. Идентификация карбонилов металлов
125
Рис. 12. Инфракрасные спектры карбонилов металлов
VII6 группы [5].
Максимумы валентных колебаний СО (см~1) : ---- Мп: 2044,
2013, 1981; ---Тс:2064, 2016, 1981;--Re : 2070, 2013, 1975.
Б. Рентгеноструктурный анализ
Структурная идентификация карбонилов металла обязательно
включает тщательное рентгенографическое исследование монокри-
сталла с подробным анализом данных, часто при помощи вычисли-
тельных устройств. В некоторых случаях возникали эксперимен-
тальные трудности, связанные либо с летучестью карбонила
Структура кристаллов карбонилов металлов
IV. Идентификация карбонилов металлов
127
металла, либо со способностью рентгеновских лучей разлагать ве-
щество. Иногда для сравнения общей картины известных карбо-
нилов с новым, имеющим предположительно такую же кристал-
лическую структуру, используют метод порошковой рентгено-
графии, (например, при установлении октаэдрической симметрии
V(CO)e по аналогии с Сг(СО)в [127]).
Рентгеноструктурные данные для карбонилов металлов собра-
ны в табл. 8.
В. Различные методы анализа
Интересны магнитные свойства карбонилов металлов, особенно
в случае вновь открытых соединений. В присутствии парамагнит-
ных веществ, растворенных в углеводороде, пики сигнала ядер-
ного магнитного резонанса углеводородов сдвигаются. Величину
этого сдвига можно рассчитать теоретически. В случае карбони-
лов металлов оказался эффективным [38] метод коаксиальных
трубок, разработанный Эвансом [137]. Однако иногда, особенно
если карбонил металла слабо растворим в углеводороде, более
эффективным может оказаться метод баланса Гуи.
Рис. 13. Положение и относительные интенсивности полос ва-
лентных колебаний в инфракрасных спектрах соединений
М2(СО)8Х2 металлов VII6 группы. Растворитель ССЦ, оптика
из LiF.
128
Глава 4
Важнейшее значение для синтеза карбонилов металлов имеет
измерение молекулярного веса. Криоскопический метод в неко-
торых случаях дает большую ошибку [127], но, как правило,
если используется подходящий растворитель, метод дает хорошие
результаты. Метод тензиметрического измерения давления паров
в аппарате, показанном на рис. 10, дал хорошие результаты для
Тс2(СО)10 [38]. В настоящее время разрабатывается метод диффе-
ренциального измерения температуры чистого растворителя по
сравнению с раствором карбонила металла в этом же раствори-
теле. Для этого используют прибор типа осмометра «Мечролаб».
При промышленном производстве карбонилов обычно прово-
дят элементарный анализ для определения эффективности, но для
определения содержания углерода (и, следовательно, окиси угле-
рода) можно использовать обычную методику определения СО2.
Металл можно определить, если это необходимо, обычными мето-
дами количественного анализа. Рассмотренное ранее использова-
ние насоса Тендера показывает, что для определения содержа-
ния металла и окиси углерода могут применяться и другие ме-
тоды.
Определение состава и структуры вещества часто основыва-
ется на реакциях образования производных карбонилов метал-
ла, иногда в сочетании с исследованием ИК-спектров. Например,
считали, что карбонил технеция должен быть похож на Мп2(СО)10
и Re2(CO)10 по структуре и реакциям с галогенами. Инфракрас-
ные спектры галоидных производных также близки. Схемы спек-
тров, приведенные на рис. 13, позволяют сделать вывод о том,
что формула карбонила технеция действительно Тс2(СО)10.
С аналитической точки зрения интересна также недавняя ра-
бота Лундквиста и Кайса [138], посвященная ультрафиолетовым
спектрам поглощения и коэффициентам экстинкции Fe(CO)s
Cr(CO)e и Мп2(СО)10. Имеются также и более ранние работы по
исследованию ультрафиолетовых спектров, но большинство из
них носит преимущественно качественный характер.
Наконец, для анализа карбонилов металлов и подобных ве-
ществ можно использовать данные электронного спинового резо-
нанса. Штольц и др. [25] использовали электронный спиновый
резонанс для определения осколков Fe(CO)4 в облученном ультра-
фиолетовым светом растворе Fe(CO)5, а Ригер и Франкель изучали
электронный спиновый резонанс радикалов карбонильных анио-
нов [139].
В литературе есть указания на работы, посвященные исследо-
ванию инфракрасных спектров низкой частоты, но эти работы от-
носятся скорее к структурным исследованиям, чем к анализу (см.,
например, работу Эджелла и др. [140]).
Литература
129
ЛИТЕРАТУРА
1. Чатт, Паусон, Венанзи, в книге «Химия металлоорганических
соединений» под ред. Г. Цейсса, ИЛ, Москва, 1960, гл. 16.
2. Emeleus Н. J., Anderson J. S., Modern Aspects of Inorganic
Chemistry, 3rd ed., D. Van Nostrand Company, Princeton, New Jersey,
1960.
3. Маттерн Дж., Г u л л С., в книге «Химия координационных соеди-
нений», под ред. Дж. К. Бейлара, мл., ИЛ, Москва, 1960, гл. 16.
4. Durrant Р. J., Durr ant В., Introduction to Advanced Inorganic
Chemistry, Wiley, New York, 1962.
5. H i 1 e m a n J. C., «Carbonyls», in Kirk-Othmer Encyclopedia of Che-
mical Technology, 2nd ed., Vol. IV, Standen A., ed., Interscience, New
York, to be published.
5a. A b e 1 E. W., Quart Rev., 17, 133 (1963).
6. M о n d L., Langer C., Quincke F., J. Chem. Soc., 57, 749
(1890).
7. MondL., Quincke F.,. J. Chem. Soc., 59, 604 (1891).
8. D e w a г J., J о n e s H. O., Proc. Roy. Soc. (London), A76, 558 (1905).
9. D e w a r J., J о n e s H. O., Proc. Roy. Soc. (London), A79, 66 (1906).
10. Mond L., Hirtz H., С о w a p M. D., J. Chem. Soc., 97, 798 (1910).
11. Job A., C a s s a 1 A., Compt. Rend., 183, 58, 392 (1926).
12. J о b A., R о u v i 1 1 о i s J., Compt. Rend., 187, 564 (1928).
13. M a n c h о t W., M anchot W. J., Z. anorg. allgem. Chem., 226, 385
(1936).
14. Hieber W., Legally H., Z. anorg. allgem. Chem., 245, 321 (1940).
15. H i e b e r W., Fuchs H., Z. anorg. allgem. Chem., 248, 256 (1941).
16. H i e b e r W., Legally H., Z. anorg. allgem. Chem., 251, 96—113
(1943).
17. H i e b e r W., Stallman FL, Z. Elektrochem., 49, 228 (1943).
18. В r i m m E. O., Lynch M. A., Jr., S e s n у W. J., J. Am. Chem.
Soc., 76, 3831 (1954).
19. N a 11 a G., E г co И R., Calderazzo F., Alberola A., Cor-
r a d i n i P., A 1 1 e g r a G., Atti Accad. Naz. Lincei. Rend., Classe Sci.
Fis. Mat. Nat., 27, 107 (1959); through chem. Abstr., 54, 16252 (1960).
20a . Hileman J. C., Huggins D. К., К aesz H. D., J. Am. Chem.
Soc., 83, 2953 (1961); 6. J о s h i К. К., P a u s о n P. L., J. Chem. Soc.,
1962, 336.
21a . К a e s z H. D., Huggins D. K., Can. J. Chem., 41, 1250 (1963);
б. К a e s z H. D., private communication, 1964.
22. Несмеянов A. H., Анисимов К. H., Ко Лобов a H. Е.,
Кол омников И. С., Изв. АН СССР, ОХН, 1963, 193.
23. С о г е у Е. R., D a h 1 L. F., J. Am. Chem. Soc., 83, 2203 (1961).
24. С о г e у E. R., D a h 1 L. F., Beck W., J. Am. Chem. Soc., 85,1202
(1963).
25. S t о 1 z I. W., Dobson G. R., S h e 1 i n e R. K., J. Am. Chem. Soc.,
84, 3589 (1962).
26. P о d a 11 H. E., Isolated Bond Strength vs. Molecular Stability for the
Metal Carbonyls, Melpar Inc., Falls Church, Virginia, 1962, pp. 1—10.
Presented in part before the Division of Chemical Education, 141st Nati-
onal American Chemical Society Meeting, Washington, D. C., March 21—
29, 1962, Abstracts of Papers, P. 8—F.
27. О r m о n t B., Acta Physicochim. URSS, 21, 413 (1946).
28. П о с n e x о в Д. А., Ж0Х, 18, 2045 (1948).
29a. Met lin S., Wonder I., Sternberg H. W., Nature, 183, 457
10 Заказ № 547
130
Глава 4.
(1959); б. Н i е b е г W., Angew. Chem., Intern. Ed. Engl., Sample Issue,
65—73 (May 1961).
30a. Гринберг А. А., Птицын Б. H., Филинов Ф. M,., Лав-
рентьев В. H., Tp. Радиевого инет. АН СССР, химия и геохимия,
7, 14 (1956); б. N у h о 1 m R. S., Proc. Chem. Soc., 1961, 273; в. Hie-
b e r W., S c h u 11 e n H-, Marin R., Z. Anorg. Allgem. Chem., 240,
261 (1939).
31. lolly W. L., Synthetic Inorganic Chemistry, Prentice-Hall, Englewood
Cliffs, New Jersey, 1960.
32. Matheson Gas Data Book, The Matheson Co., East Rutherford, New Jer-
sey, 1961.
33. Bartocha B., Graham W. A. G., S t о n e F. G. A., J. Inorg. Nucl.
Chem., 6, 119 (1958).
34. К i n g R. B., S t о n e F. G. A., Inorg. Syn., 7, 99 (1963).
35. S h о г e S. G., J. Chem. Educ., 39, 465 (1962).
36. OconeL. R., Simkin J., J. Chem. Educ., 39, 463 (1962).
37. F e i s e r L. F., Experiments in Organic Chemistry, 3rd ed., Heath D. C.,
Boston, Massachsetts, 1955.
38. H i 1 e m a n J. С., H u g g i n s D. К., К a e s z H. D., Inorg. Chem.,
1, 933 (1962).
39. G г о s s e A. V., J. Am. Chem. Soc., 60, 212 (1938).
40a. Kobayashi G., Huggins D. К., К a e s z H. D., private com-
munication, 1962; 6. Wenderl.,Sternberg H. W.,MetlinS.,
Orchin M., Inorg. Syn., 5, 190 (1957).
41. Seel F., «Carbonyls and Nitrosyls», in Handbook of Preparative Inor-
ganic Chemistry, Vol- II, Brauer G., ed., Ferdinand Enke Verlag, Stutt-
gart, 1962.
42. Arthur P., Jr., England D.C., Pratt В. C., Whitman
G. M., J. Am. Chem. Soc., 76, 5364 (1954).
43. R e p p e W., and co-workers, Ann. Chem., 582, 116 (1953).
44, I w a n a g a R., Bull. Chem. Soc. Japan, 35, 865 (1962).
45. Kirch L., Orchin M-, J. Am. Chem. Soc., 81, 3597 (1959).
46. Hi eb er W., Muhlbauer F., Ehmann E. A., Chem- Ber.,
65, 1090 (1932).
47. Keeley D. F., J ohnson R. E., J. Inorg. Nucl. Chem., 11,33 (1959).
48. E г с о 1 i R., В arbieri-Hermitt e F., Atti Accad. Naz. Lincei
Rend. Classe Sci. Fis., Mat. Nat., 16, 249 (1954); through Chem. Abstr.,
48, 10408 (1954).
49. Job A., C a s s a 1 A., Bull. Soc. Chim. France, 41, 814 (1927).
51. Cumming W. M., H о r n J. A., Ritchie P. D., J. Appl. Chem.,
2, 624 (1952).
52. О w e n В. В., E n g 1 i s h J., Jr., C a ssi dy H. G., D un d о nC. V.,
J. Am. Chem. Soc., 69, 1723 (1947).
53. О w e n В. В., E n g 1 i s h J., Jr., Cassidy H. G., DundonC. V.,
Inorg. Syn., 3, 156 (1950).
54. А и и с и м о в К. H., Несмеянов А. Н., Изв. АН СССР, 26,
58 (1940).
55. Н i е Ь е г WRomberg Е., Z. Anogr. Allgem. Chem., 221, 321 (19.35).
56. Windsor М. М., Blanchard A. A., J. Am. Chem. Soc., 56, 823
(1934). ,
57. P о d a 11 H. E., D u n n J. H., Shapiro H., J. Am. Chem. Soc.,
82, 1325 (1960).
58a . Захаркин Л. И., Гавриленко В. В., Охлобыстин
О. Ю., пат. СССР, 111382 (25 июня 1958); б. 3 а х а р к и я Л. И.,
Гавриленко В. В., Охлобыстин О. Ю-, Изв. АН СССР,
ОХН, 1958, 100.
Литература
131
59.
60.
61а.
62.
63.
64.
65.
Rabizzoni A.,
ErcoliR., С a 1-
66.
67.
68.
69а.
70.
ClossonR. D., BuzbeeL. R., Е с k е G. G., J. Am. Chem. Soc.
80, 6167 (1958).
Brimm E. O., Lynch M. A., Jr., Sesny W. J., пат. США
2803525 (August 20, 1957).
N a 11 a G., E г с о 1 i R., Calderazzo F.,
J. Am. Chem. Soc., 79, 3611 (1957); 6. Nat taG..
d e r a z z о F., итал. пат. 578713 (July 1, 1958).
Fischer E. O., Hafner W., Z. Naturforsch., 10b, 665 (1955).
Fischer E. O., Hafner W., герм. пат. 1007305 (May 2, 1957).
Behrens H., Haag W., Chem. Ber., 94, 320 (1961).
Podall H. E., Prestridge H. B., Shapiro H.,
Chem. Soc., 83, 2057 (1961).
Несмеянов A. H., Михеев E. П., Анисимов
Филимонова H- IL, ЖНХ, 4, 1958 (1959).
Podall H. E., пат. США 2952521 (Sept. I960).
Podall H. E., пат. США 2952523 (Sept. 1960).
Podall H. E., Prestridge H. В., Shapiro H.,
Chem. Soc., 83, 2057 (1961); 6. S h a p i г о H. ~
Inorg. Nucl. Chem., 24, 925 (1963).
Несмеянов A. H., Анисимов К. H., Волков В.Л..
Е. П., Медведева А. Б..
ЖНХ, 4, 1958 (1959).
1 2952521 (Sept. I960).
I. Am-
К. Н.,
J. Ат.
Р б d а 11 Н. Е., I.
Несмеянов А. Н.,
Фридепберг А.Э., Михеев
ЖНХ, 4, 1827 (1959).
71а. Н i е b е г W., Legally Н., Z. Anorg. Allgem. Chem., 245, 295
(1940); б. Speyer Е., Wolf Н., Chem. Ber., 60, 1424 (1927); в.
К i n g R. В., Stone F. G. A., Inorg. Synth., 7, 193 (1962).
г
г
Z. anorg. allgem. Chem., 204, 165 (1932).
Brendel G., Z. anorg. allgem. Chem., 289, 324 (1957)
SedlmeierJ., WernerR., Chem. Ber., 90, 278
72. Hi eb e
73. H i e b e
74. H i e b e
(1957).
75. H e i n t
76. L у n c h M. A., Jr.; пат. США 2825631 (March 4, 1958).
77. С 1 о s s о n R. D., В u z b e e L. R., E c k e G. C., J. Am. Chem. Soc.,
80, 6167—6170 (1958).
78. H n i z d a V., пат. США 2822247 (Feb. 4, 1958).
79. P о d a 1 1 H. E., пат. США 2952522 (Sept., 1960).
80. P о d a 11 H. E., G i г a i t i s A. P., J. Org. Chem., 26, 2587 (1961).
81. Addison С. C., Kilner M.,
1961, 4839.
Cordes B.J.F.,
Волков В. Л.,
сееваП. Е.,Г
Hurd D. Т., пат. США 2554194 (Мау 22, 1951).
е с
о л
а и
(1961).
87. Behrens Н.,
W.,
W.,
W.,
z
е 1 о
г М., герм. пат. 928044 (1955).
Wojcicki A., J. Chem. Soc.,
82.
83.
84.
85. H
В
86. В
м e я
ков
m g a r t
N е u b а и е г D., Z. Naturforsch., 17b, 791 (1962).
Михеев Е.
Валуева 3. П.
Н., Анисимов К. Н., Е
ЖНХ, 3, 2433 (1958).
п о
В. Л., В а л у е в а 3. П., ЖНХ, 4, 503 (1959).
в А. Н., Михеев Е. П., Анисимов К.
пег F.,
Reichold Р., Z. Naturfor;ch., 16а,
Haag W., Вег., 94, 319—9 (1961).
л и-
Н,
946
88. S t a m m е г i с h Н., К a w a i К., S а 1 а О., J. Chem. Soc., 35, 2168
(1961).
89. Джилмонт, Бланчард, в сб. «Неорганические синтезы», сб. 2,
ИЛ, Москва, 1952.
90. В е h г е n s Н., L о h о f е г F., Вег., 94, 1391 (1961).
91. Behrens Н., Zizlsperger Н., Rauch R., Chem. Ber. 94,
1497 (1961).
92. F i s с h е г Е. О., В и г g е г G., Z. Naturforsch., 17b, 484 (1962).
10*
132
Глава 4
93. Behrens Н., von TaueffenbachG., Z. anorg. allgem.
Chem., 315, 259 (1962).
94. Manchot W., Gall H., Chem. Ber., 62, 678 (1929).
95. В 1 a n c h а г d A. A., R a f t e г J. R., A d a m s W. B., Jr., J. Am.
Chem. Soc., 56, 16 (1934).
96. H i e b e r W., Fischer E. 0., Z. anorg. allgem. Chem., 269, 292
(1952).
97. H i e b e r W., Fischer E. O., Z. anorg. allgem. Chem., 269, 308
(1952).
98. H i e b e r W., Bruck R., Z. anorg. allgem. Chem., 269, 28 (1952).
99a. H i e b e r W., Angew. Chem., 69, 469 (1962); 6. Fischer E. 0.,
В о с к 1 у Е., Z. anorg. allgem. Chem., 269, 308 (1952).
100. С о г е у Е. R., D a h 1 L. F., Inorg. Chem., 2, 521 (1962).
101. Beck W., Hi eb er W., Braun G., Z. anorg. allgem. Chem.,
308, 23 (1961).
102. E г с о 1 i R., Calderazzo F., Alberola A., J. Am. Chem.
Soc., 82, 2966 (1960).
103. Calderazzo F., Ercoli R., Chim. Ind. (Milan), 44, 990 (1962).
104. W e r n e r R. P. M., P о d a 1 1 H., Chem. Ind. (London), 1961, 144.
105. Pruett R. L., Wyman J. B., Chem. Ind. (London), 1960, 119.
106. Несмеянов A. H-, Анисимов А. К., Михеев E. IL,
Волков В. Л., Валуева 3. IL, ЖНХ, 4, 249 (1959).
107. CorradiniP., J. Chem. Phys., 31, 1676 (1959).
108. Huggins D. К., К a e s z H. D., «Use of Infrared and Raman Spect-
roscopy in the Study of Organometallic Compound», in Progress in Solid
State Chemistry, Vickery R. C., ed., Pergamon Press, London, 1963.
109. С a b 1 e J. W., NyholmR. S., Sheline R. K., J. Am. Chem.
Soc., 76, 3373 (1954).
110. В о r G., M а г к о L., Spectrochim. Acta, 36, 1105 (1960).
111 Noack K., Helv. China. Acta, 45, 1847 (1962).
112. Barraclough С. C., Lewis J., Nyholm R. S., J. Chem. Soc.,
1901 2582
113. Jones L. H., Spectrochim. Acta, 18, 329 (1963).
114. Bor G., Spectrochim. Acta, 18, 585—594 (1962).
115. Nyholm R. S., Proc. Chem. Soc.., 1961, 273.
116. Sheline R. K., Pitzer K. S., J. Am. Chem. Soc., 72, 1107
(1950).
117. Engel 1 W. F., (Huff J., Thomas J., Lehman H., An-
gell C., Asa to G., J. Am. Chem. Soc., 82, 1254 (1960).
118. H i e b e r W., Beck W., Braun G., Angew. Chem., 72, 795 (1960).
119-/S h e 1 i ne R. K.., J. Am. Chem. Soc., 73, 1615 (1951).
420. (Cotton F. A., Wilkinson G., J. Am. Chem. Soc., 79,752 (1957).
121. Cotton F. A., Liehr A. D., Wilkinson G., J. Inorg. Nucl.
Chem., 2, 141, (1956).
122. Jones L. H., J. Chem. Phys., 36, 2375 (1962).
123. F r i e d e 1 R. A., Wender I., Shut 1 er S. L., Sternberg
H. W., J. Am. Chem. Soc., 77, 3951—3958 (1955).
124. Jones L. H., J. Chem. Phys., 28, 1215 (1958).
125. H i e’b e r W., H e r g e t C., Angew. Chem., 73, 579 (1961).
126. Hi eb er W., Pe terhans J., Winter E. (in collaboration
with Beck W.), Chem. Ber., 94, 2572 (1961).
127. Calderazzo F., C i n i R., CorradiniP., E г с о 1 i R.,
N a 11 a G., Chem. Ind. (London), 1960, 500.
128. D a h 1 J. F., R u n d 1 e R. E., Acta Cryst., 16, 419 (1963).
129. R u d о r f f W., Hoffmann U., Z. Pbysik Chem. (Leipzig), B28,
351 (1935).
Литература
133
130. Powell Н. М., Ewens R. V. G., J. Chem. Soc., 1939, 286.
131. H a n s о n A. W., Acta Cryst., 15, 930 (1962).
132. Dahl L.F., Rundle R. E., J. Cbem. Phys., 26, 1751 (1957).
133. DahlL. F., Ishishi E., Rundle R. E., J. Chem. Phys., 26,
1750 (1957).
134. Ladell J., Post B., Frankuchen I., Acta Cryst., 5, 795
(1952).
135. Corey E. R., Dahl L. E., Inorg. Chem., 1, 521 (1962).
136. Wallach D., Acta Cryst., 15, 1058 (1962).
137. Evans D. F., J. Chem. Soc., 1959, 2003.
138. Lundquist R. T., Cais M., J. Org. Chem., 27, 1167 (1962).
139. R e i g e r P. H., Fraenkel G. K., J. Chem. Physics, 37, 2811
(1963).
140. E d g e 1 1 W. F., Helm С. C., Anacreon R. E., J. Chem.
Phys., 38, 2039 (1963).
ГЛАВА 5
ГАЛОГЕНИДНЫЕ И ОКСИГАЛОГЕНИДНЫЕ
КОМПЛЕКСЫ ЭЛЕМЕНТОВ ПОДГРУПП
ТИТАНА, ВАНАДИЯ И ХРОМА
Фау лс
Саутгемптонский университет, Англия
В разд. 1 этой главы рассмотрены галогенидные и оксигалоге-
нидные соединения элементов подгрупп титана, ванадия и хрома
и кратко описаны их свойства, и структура. В разд. 11 приведены
сравнительный анализ применяемых препаративных методов и
практические приемы синтеза характерных представителей этих
соединений.
1. ГАЛОГЕНИДНЫЕ И ОКСИГАЛОГЕНИДНЫЕ КОМПЛЕКСЫ
ЭЛЕМЕНТОВ ПОДГРУППЫ ТИТАНА
Большинство заслуживающих доверия работ, выполненных в
этой области, относится к синтезу гексагалогено-соединений че-
тырех- и трехвалентного титана и четырехвалентного циркония.
В отличие от элементов двух других подгрупп титан и цирконий
легко образуют соединения этого типа, которые в настоящее время
хорошо изучены.
А. Для четырехвалентного состояния
В случае титана(1У) были получены безводные комплексы с об-
щей формулой М| [TiXe], где X — F, С1 и Вг, а М1 — различные
щелочные металлы или одновалентный органический катион (на-
пример, пиридиний). Трудности синтеза этих соединений возра-
стают в ряду F, G1, Вг,1, а гексаиодные соединения до сих пор не
выделены. Несмотря на то что соли такого типа с иодом не были
получены, темно-красная окраска раствора, образованного ио-
дидом титана (IV) в концентрированном растворе йодистого водо-
рода, по-видимому, обусловлена присутствием аниона [Tile]2".
Известно большое число гексафторо-солей [1 ], которые до-
вольно устойчивы при нагревании и очень медленно гидролизу-
ются. Несколько менее изучены гексахлоро- и гексабромо-соли;
I. Галогенидные и оксигалогенидные комплексы элементов подруппы Ti 135
только в самое последнее время они были получены в безводном
состоянии [2, 3]. В одной из ранних работ, например, сообщается
о синтезе бромида [4] в виде дигидрата аммонийной соли
(NH4)2[TiBre] • 2Н2О, хотя расчет по опубликованным данным
анализа дает отношение Ti : Вг«1 : 4,5, что, пожалуй, указывает
на наличие гидролизованного, а не гидратированного продукта.
Было синтезировано также несколько тетрахлородибромотитана-
тов [6]. Меньше пока известно о соединениях циркония, особенно
о бромо-производных. Так, в настоящее время получен целый ряд
соединений с хлором [7], в то время как сведения об аналогичных
соединениях с бромом ограничены соединениями пиридиния и хи-
нолиния [8], и снова расчет по данным химического анализа дает
отношение Zr : Вг=1,0 : 4,9 (для соли пиридиния), что указывает
на заметный гидролиз продуктов. В табл. 1 приведены хлоро- и
бромо-производные, которые были получены для четырехвалент-
ных титана и циркония.
Таблица 1
Гексахлоро- и гексабромо-соли титана(ГУ) и циркония(1¥)
Соединение Цвет М1
mJ [Tici,] Желтый К, Rb, Cs, NH4, (CH3)4N, (CH3)3NH, (CH3)2NH2, C6HeN, C9H8N
mJ iTiCi4Br2] Красный (C2H6)4N, (C2H6)3NH, (C2H5)2NH2i C6HeN, C9H8N
mJ [TiBr,] Темно-красный NH4, C5H8N, C9H8N
mJ [Zrci,] Белый Na, Rb, Cs, NH4, (CH3)3NH, (C2H5)3NH, (CH3)2NH2, (C2H6)2NH2, c2h6nh3, C6HeN, C9H8N
mJ [ZrBre] C6HeN, C9H8N
Все эти гексагалогено-соединения склонны к гидролизу, а
также к аммонолизу под действием жидкого аммиака [7, 9, 10];
аммонолизу подвержены одна связь цирконий—хлор и две связи
136
Глава 5
титан—хлор или титан — бром при 33,5°. Были изучены реакции
бис-(алкиламмоний)-гексахлороцирконатов(1У) с алифатическими
аминами [7]. Было показано, что с третичными аминами эти сое-
динения не реагируют, в то время как со вторичными аминами
протекает медленный сольволиз и, наконец, один атом хлора
полностью замещается в результате реакции с этиламином. Гек-
сахлорцирконаты устойчивы при нагревании и не обнаруживают
признаков разложения в вакууме при температурах до 200°.
Гексагалогено-анионы имеют форму октаэдра, за исключением
некоторых солей [ZrFe]2- в твердом состоянии [48]. Растворы,
образованные хлоро-соединениями в концентрированной соляной
кислоте, дают пики поглощения при 44 800 см-1 для титана [111
и 46 500 см"1 для циркония [7]. Эти пики приписывают переходу
тс-электронов хлора на орбиту атома металла.
Б. Для трех- и двухвалентных состояний
Очень мало известно о галогенидных комплексах, образован-
ных цирконием в низших валентных состояниях. Однако за по-
следние десять лет проведено много исследований систем с тита-
ном. Брайт и Вурм [12] электролитическим восстановлением соот-
ветствующих четырехвалентных фторидов получили комплексные
фториды M3[TiFe] (где М—Na и К) в виде твердых веществ фиоле-
тового цвета. В случае хлоридов гексахлоро-соединения M3TiGle,
по-видимому, не были выделены в чистом виде, несмотря на то
что в ранних работах [13, 14] имеются сообщения о получении
солей M2TiCl5-H2O (где М—Rb и Сз), в которых молекула воды
может завершать октаэдрическую группировку вокруг атома
титана. Изучение [15] систем NaCl—TiCl3, КС1—TiCl3 и КВг—
TiBr3 показало присутствие гексагалогенотитанов(Ш) в каждой
из этих систем. Грун и Макбет [16] исследовали абсорбционный
спектр Ti3+ в эвтектике хлористый литий — хлористый калий при
400° и наблюдали широкие полосы с максимумом при 13 000 см-1
и ступенькой при 10 000 см"1; новая полоса при 7000 слС1 воз-
никла при повышении температуры. Грун и Макбет полагают,
что спектральные свойства объясняются лучшим образом при до-
пущении равновесия
[TiCLl3~ ;-- [TiClJ- + 2С1-.
Полоса при низких волновых числах обусловлена переходом к
аниону в форме тетраэдра.
II. Галогенидные и оксигалогенидные комплексы элементов подгруппы V137
11. ГАЛОГЕНИДНЫЕ И ОКСИГАЛОГЕНИДНЫЕ
КОМПЛЕКСЫ ЭЛЕМЕНТОВ ПОДГРУППЫ ВАНАДИЯ
А. Для пятивалентного состояния
В табл. 2 указаны основные типы известных галогенидных и
оксигалогенидных комплексов. Анион отмечен знаком **, если
соединение выделено в чистом виде и измерены его характеристики,
и знаком *, если соединение идентифицировано только по диаграм-
ме состояния или по данным измерения электропроводности в
подходящем растворителе. В литературе есть указания только на
более современные исследования. Относительно ранних работ сле-
дует обращаться к классическим справочникам (например,
Sidwick N. V., Chemical Elements and Their Compounds,
Oxford, 1950).
Таблица 2
Комплексы элементов подгруппы ванадия
Ванадий Ниобий Тантал
Фториды [MFJ- **[17, 18]
[MF,]2- — **
[MF8p- — — **
Оксифториды [MOFJ- ** — —
[MOF5]2- —
[MOFe]3- — **
[MOF7]«- — ** —
Хлориды [MCle]- — *[19—21] *[21]
Оксихлориды [MOCU]- ** ** —
[MOC1J2- — ** **
Оксибромиды [MOBrJ- — ** —
[MOBr5]2- —• **
Все три элемента образуют комплексные фториды MUMFe],
однако аналогичные хлоро-соединения установлены только по
диаграммам состояния. Так, было показано [21], что в системах,
образованных хлоридами ниобия(У) и тантала(У) с хлоридами
всех щелочных металлов (за исключением системы LiCl—NbCU),
присутствуют соли MHNbClel и МЧТаСЫ. Гексафторованада-
ты(У) очень легко гидролизуются и дымят на влажном воздухе;
комплексные фториды ниобия и тантала менее склонны к гид-
ролизу. Соединения тантала меньше подвержены гидролизу, чем
аналогичные соединения ниобия. Например, K2TaF7 можно пе-
9 Заказ № 547
138
Глава 5
рекристаллизовать из разбавленной плавиковой кислоты, однако
в этих же условиях образуется K2NbOFs.
Известна структура некоторых комплексных ионов; структура
ряда других комплексных ионов предполагается достаточно
близкой. Так, гексагалогено-анионы имеют октаэдрическое строе-
ние, и очень вероятно, что в анионах [MOXs]2~ кислород занимает
место одного атома галогена.
Это, конечно, характерно и для соединений молибдена и вольф-
рама, которые будут рассмотрены в следующем разделе
о
Х^||/Х
Xх I X
X
Структура анионов [МОХ4]_ неизвестна. Возможно, эти анио-
ны образуют пирамиду с квадратным основанием (в качестве про-
изводных ионов [MOXs]2-, образующихся в результате потери
одного X-, находящегося в транс-положении к кислороду) или
тригональную бипирамиду (подобно пентагалогенидам в паро-
вой фазе).
Высшие фториды интересны тем, что они являются простейши-
ми соединениями, в которых металл обладает координационным
числом более 6. В одном из первых сообщений [22] указано, что
анионы [MF7]2- построены в виде гранецентрированных триго-
нальных призм, однако эта работа нуждается в проверке, посколь-
ку было показано, что аналогичный ион [ZrF,]3- построен в виде
пентагональной бипирамиды с атомами фтора вокруг металла.
Комплексный октафторид-ион [TaF8]3~ имеет структуру антиприз-
мы с квадратным основанием [23].
Б. Для низших валентностей
В табл. 3 приведены типы известных соединений. Условные
обозначения те же, что и в табл. 2.
Как видно из табл. 3, для ниобия и тантала в низших состоя-
ниях окисления было проведено мало исследований. Единствен-
ными соединениями, о которых есть указания в литературе, яв-
ляются гексахлоро-производные четырехвалентных элементов, и
соли [ХЬгС19]3-. С другой стороны, было синтезировано большое
число соединений с ванадием; присутствие ряда других комплекс-
ных ионов было предположено на основании физических изме-
рений. Так, изучение электропроводности показало [20], что в
растворе монохлорида иода хлорид калия и хлорид ванадия(1У)
II. Галогенидные и оксигалогенидные комплексы элементов подгруппы V13!)
Таблица 3
Комплексы элементов подгруппы ванадия для состояний
окисления менее 5-}-
Состояние окисления Комплексный анион Ванадий Ниобий Тантал
4+ [MOF4]2- ** — —
[МС1в]2- *[20] **[24] **[24]
[МОС14]2- ** — —
34- [MF,]’- ** — —
[MC1J3- *[25] — —
[МС14]- **[25, 26]
[М2С19]з- **[24] **[24] —
[МВг4]~ **[26]
2+ [МС1,]4- *[25] — —
[МС14]2- *[25, 27] — —
[МС13]- *[27] — —
реагируют с образованием комплекса Кг[УС1в]. Имеются сообще-
ния о нескольких хлоро-анионах для трехвалентного элемента;
спектроскопическое исследование [25] природы катиона V3+ в
эвтектике LiCl — КС1 указывает на равновесие
[VC16]3- 7--- [VC14]_ + 2С1-.
Аналогичное равновесие
[vcie]<- 7--* [vci4]2 - + 2С1-
предполагают в подобных растворах V2+. Изучение диаграмм со-
стояния [27] указывает на существование соединений KVCI3,
K.2VC14 и CsVCb в системах хлорид ванадия(П) — хлориды
калия и цезия.
Гексагалогено- и тетрагалогено-производные имеют соответ-
ственно октаэдрическую и тетраэдрическую конфигурации; струк-
тура оксигалогенидных комплексов и анионов [MaClg]3- и [VCI3 ]~
неизвестна.
S*
140
Глава 5
111. ГАЛОГЕНИДНЫЕ И ОКСИГАЛОГЕНИДНЫЕ
КОМПЛЕКСЫ ЭЛЕМЕНТОВ ПОДГРУППЫ ХРОМА
А. Для состояний окисления 6+ и 5-J-
В табл. 4 приведены основные типы соединений, которые были
идентифицированы для элементов в данных состояниях окис-
ления. ;
Таблица 4
Галогенидные и оксигалогенидные комплексы элементов
подгруппы хрома для состояний окисления 6+ и 5
Состояние окисления ' Анион Хром Молибден Вольфрам
6+ [MF8]2' — **[28, 29] **[29, 30]
[MF7]- — **[28] **[30]
[MO3FJ- ** — —
[MO3FS]2' — * * **
[MO3F3p- — **[31, 32] **[33]
[MO2F4]2" — ** **
[MOF6J- — **[34] **[28]
[MO3C1]~ ** — —
[MO3Br]- — —
5+ [MFJ- — **[34] **[34]
[MFgp- — **[28] **[30]
[MOF5]2- — ** —
[MCI6]- — *[35] —
[МОСЦ]- * * **[36] **[36]
[MOCIJ2- ** **[36] **[36]
[MOBr4]~ — **[36] **[36]
[MOBr5]2- — **[36] **[36]
Значительный прогресс был достигнут в изучении свойств
комплексных фторидов. Высшие фториды MMMFg] и М1 [MF7],
где М — молибден или вольфрам, представляют собой белые
твердые вещества, легко гидролизующиеся, но устойчивые в су-
хом воздухе. Быди синтезированы также многие оксифториды.
За исключением производных хрома МЧМОзХ], сведений о син-
тезе хлоро- или бромо-комплексов этих элементов для состоя-
ния окисления 6+ нет.
III. Галогенидные и оксигалогенидные комплексы элементов подгруппы Сг 141
Пятивалентные молибден и вольфрам образуют гексафторо- и
октафторо-соединения. Эти соединения были изучены более под-
робно. Было обнаружено, что анионы солей калия имеют иска-
женную тетрагональную структуру, наблюдаемую также для анио-
на ниобия [NbFel", а именно структуру с четырьмя длинными и
двумя короткими связями металл — фтор. При комнатной темпе-
ратуре соединения К [MoFe]- и К [WFв]- имеют магнитные момен-
ты соответственно 1,24 и 0,51 магнетона Бора. Оба соединения
очень чувствительны к следам влаги, но устойчивы при нагревании
вплоть до 250°.
Аналогичные соединения хлора МЧМС1в]не были выделены.
Однако раствор хлорида молибдена(У) в расплавленном хлориде
калия имеет спектр, предсказанный для аниона [MoCle ]”.
В последнее время проведено много работ по получению и
изучению структуры оксигалогенидных комплексов пятивалент-
ных молибдена и вольфрама. Особое внимание было уделено ком-
плексным анионам [MOXs]2~, где X — С1 или Вг. Несмотря на
то что эти анионы имеют по существу октаэдрическую форму,
кратная связь металл — кислород обусловливает значительную
симметрию с соответствующим расщеплением d-линий спектра.
Область этих <71-линий представляет значительный теоретический
интерес. Обзор полученных величин и обсуждение различных
точек зрения по этому вопросу приведены в работе [36].
Соединения хлора и брома этих двух металлов имеют соответ-
ственно зеленую и коричневую (или желто-коричневую) окраску.
Все они легко гидролизуются, но устойчивы на воздухе. Аналогич-
ные соединения хрома более чувствительны к влаге, кроме того,
они склонны к диспропорционированию с образованием произ-
водных хрома(Ш). Соли аниона [МоОС15]2- реагируют с диме-
тиламином [37 ] с сольволизом одной связи молибден — хлор и
с образованием МоОС1г(ХМе2); NHMea находится в смеси с соля-
нокислым диметиламином.
Основной интерес к соединениям с анионом [МОХ4]_ вызван
способом их получения и причинами именно их преимуществен-
ного образования по сравнению с анионом [MOXs]2-. Обсуждение
этих соединений проведено в разд. II рассматриваемой главы.
В табл. 5 приведены основные соли обоих типов.
Б. Для состояний окисления 4-(-, 3-(- и 2±
В табл. 6 суммированы все данные, имеющиеся по этим соеди-
нениям. Обозначения те же, что и в других таблицах.
Ранее из комплексов типаМММХв] для элементов подгруп-
пы хрома были получены гексафторохроматы(1У). В настоящее
время синтезированы соответствующие фторо-соединения молиб-
Таблица 5
Известные комплексные оксигалогениды пятивалентных
молибдена и вольфрама
Тип комплекса м1
Мг [МоОС15] NH4, К, Rb, Cs, C5H6N, CeHgN,
М1 [МоОС14] М2 [МоОВг5] М1 [МоОВг4] М2 [WOC15] М1 [WOCI4] М2 [WOBr5] М1 [WOBr4] (CH3)3NH, (CH3)2NH2, CH3NH3 Rb, Cs, C5H6N, C9HsN, (C2H5)2NH2 NH4, K, Rb, Cs, C5HeN, CeHgN C5H6N, C9H8N NH4, Rb, Cs, CgHgN, C6H5NH3, (CH3)3NH C5H6N, C9HgN NH4, Rb, Cs C3H6N, C9H8N, изо-CgHgN
Таблица 6
Комплексы элементов подгруппы хрома для состояний
окисления 4+, 3+ и 2+
Состояние окисления Анион Хром Молибден Вольфрам
4+ [MF6]2- **[38] **[39] —
[MCI6]2- — **[36,40,41] **[42, 43]
[МВг6р- — **[43] **[43]
3+ [MF4]- — * * —
[MFe]3- * * — —.
[MC16]3- ** * * —
[MBr6]3- — ** —
[M2C1B]3- — **[24] * *
[M3C114]5 — — **[44]
2+ [MF3]- **[45] — —
[MC14]2- *[16] — —.
[MC16]3- *[46] . — —
III. Галогенидные и оксигалогенидные комплексы элементов подгруппы Ст 143
Таблица 7
Комплексы типа mJ [МХ6]
(М — молибден или вольфрам)
Комплекс M1 Магнетон Бора, p. Цвет
mJ [CrF6] K, Rb — Телесный
mJ [MoF6] Na — Темно-коричневый
mJ [MoClfi] К 2,28 Темно-зеленый
Rb 2,24 »
Cs 2,24 »
c5h6n 2,36 Желтый
c9h8n 2,30
(C2H5)2NH2 2,30 »
mJ [МоВгв1 Rb 2,18 Оливково-зеленый
Cs 2,08 »
mJ [wci6] К 1,43 Красный
Rb 1,48 »
Cs 1,48 »
(CH3)3NH 1,55 Желтый
(C2H5)3NH 1,34 Коричневый
(C2H5)2NH2 1,59 Розовый
mJ [WBr6] к 1,50 Оливково-зеленый
Rb 1,43 . »
Cs 1,72 Травянисто-зеленый
дена, а также хлоро- и бромопроизводные молибдена и вольфрама.
В табл. 7 приведены соединения этого типа, полученные в настоя-
щее время. В таблице указаны окраска комплексов и их ма: нит-
ный момент (при комнатной температуре).
Значения магнитных моментов этих соединений интересны по
той причине, что они показывают, как по мере увеличения атомно-
го номера в пределах одной подгруппы возрастает роль таких фак-
торов, как спаривание спинов, и уже перестают быть правильны-
ми формулы с неспаренным спином. Это подробно рассмотрено в
работе Пикока и сотрудников [41]. Эти соединения молибдена и
вольфрама не так легко гидролизуются, как другие комплексы
этих элементов в четырехвалентном состоянии. Соли рубидия и
цезия труднее гидролизуются, чем соли других элементов. Они
144
Глава 5
нерастворимы в растворителях, с которыми они не реагируют, за
исключением галогенидных растворителей, например монохло-
рида иода. Гексахлоромолибдат и гексабромомолибдат калия реа-
гируют с жидким аммиаком несколько неожиданным образом:
по-видимому, происходит диспропорционирование и аммонолиз с
образованием нерастворимых комплексов трехвалентного молиб-
дена КзМоаХд и растворимых галогеномолибдатов(У) аммония.
В связи с нерастворимостью гексахлоро-комплексов изучение
спектров было проведено только для твердых веществ, и поэтому
нет уверенности в том, являются ли три наблюдаемые полосы в
области 21 000—28 000 сдгх полосами поля лигандов или полоса-
ми, возникающими в связи с перераспределением заряда.
Доказано образование комплексных анионов типа [MX в]3’
трехвалентными хромом и молибденом. Напротив, вольфрам обра-
зует только комплексные анионы, содержащие два или более ато-
мов вольфрама, соединенных мостиками из атомов хлора. В ани-
оне ГУУгСУэ!3-, например, три атома хлора соединяют две группы
WCls. Было установлено [47], что МзГ^гСД] реагирует с пири-
дином и анилином с образованием замещенных производных
(например, pysW^Cle), являющихся неэлектролитами.
Грун и Макбет [16] изучали спектры Сг3+ и Ст2+ в эвтектике
LiCl — КС1 и установили только незначительные изменения,
появляющиеся в интервале температур 400—1000°; эти изменения
показали присутствие только одного типа частиц для каждой
системы, а именно [СгС16]3- — для хрома(Ш) и [СгС14]2- — для
хрома(П). По-видимому, здесь не имело место равновесие ионных
форм октаэдр — тетраэдр, как это наблюдалось для растворов
галогенидов титана и ванадия. На основании изучения диаграммы
состояния системы NaCl — СгСЬ Шилов [46] сообщил об обра-
зовании соединения NasCrCls.
Интересна структура кристаллов комплексных фторидов, об-
разованных двухвалентным хромом КСгБз, так как каждый атом
хрома окружен шестью атомами фтора, расположенными в верши-
нах искаженного октаэдра. Наблюдаемые межатомные расстояния
(четыре длинных и два коротких) для связей хром — фтор были
предсказаны на основании теории поля лигандов.
IV. МЕТОДЫ ПОЛУЧЕНИЯ ГАЛОГЕНИДНЫХ
И ОКСИГАЛОГЕНИДНЫХ КОМПЛЕКСНЫХ СОЛЕЙ
Можно использовать два общих метода получения этих комп-
лексных соединений.
1. Прямая реакция без растворителя, обычно при высокой тем-
пературе, например
ЗМ’Х-рМХз----->М|[МХ6].
IV. Получение галогенидных и оксигалогенидных комплексных солей 145
2. Прямая реакция в подходящем растворителе.
1. В известном смысле такое разделение является искусствен-
ным, поскольку реакции, протекающие в первую очередь, могут
использовать один из исходных компонентов реакции (обычно
МТХ) в качестве растворителя, т. е. в виде расплавленной соли.
Однако первый тип реакций полезно рассмотреть отдельно, так
как такие методы получения наталкиваются на специфические
экспериментальные трудности. Так, вообще трудно гарантировать
образование чистых соединений, так как одно из реагирующих
веществ обычно взято в небольшом избытке и во многих случаях
не может быть удалено (когда оно нелетуче и нерастворимо в раст-
ворителях, инертных по отношению к продукту). В некоторых слу-
чаях, конечно, реагирующие вещества можно смешать точно в
стехиометрическом соотношении без особых экспериментальных
затруднений, однако необходимо быть уверенным, что продукт
является действительно индивидуальным соединением, а не
смесью.
В общем уравнении реакции, приведенном выше, автор пред-
положил, что 3 моля галогенида щелочного металла реагируют с
1 молем галогенида переходного металла с образованием 1 моля
комплекса М*[МХб]. Но ведь продукт мог оказаться смесью,
например, 2М1Х-(-М1 [МХ4]. Поэтому необходимы расширенные
исследования по идентификации продукта '(изучение фазовых
диаграмм, рентгеновские исследования), прежде чем данный
метод может быть использован для получения соединений, которые
не были ранее охарактеризованы. Применение метода синтеза без
растворителя обычно ограничено получением солей щелочных ме-
таллов, поскольку при высоких температурах галогениды аммония
и замещенного аммония диссоциируют с образованием аммиака
или аминов, которые могут воздействовать на галогениды переход-
ных металлов. Это может быть показано на примере соединения
(NH4)3 [TiClв], которое нельзя получить прямой реакцией между
NH4C1 и TiCl4 в герметичной ампуле; в этом случае обязательно
происходит аммонолиз.
Способ расплавленной соли был применен для получения солей
щелочных металлов на основе нескольких анионов комплексных
галогенидов, например [Tide]3- и [ZrCle]2"; во многих случаях,
однако, эти соединения можно получить проще и более чистыми,
используя растворитель. Этот способ имеет особое значение для
получения комплексных галогенидов некоторых элементов (на-
пример, Nb и Та) в низших состояниях окисления вследствие
того, что простые галогениды часто нерастворимы в большинстве
обычных растворителей при комнатной температуре, но раство-
римы в расплавленных галогенидах щелочных металлов. Более
того, простые галогениды не всегда можно легко получить в
146
Глава 5
чистом виде, однако их можно синтезировать in situ в солевом
расплаве.
2. Широко распространены методы с использованием раствори-
теля. В качестве растворителя могут служить водные растворы
галогеноводородов, спиртовый раствор хлористого и бромистого
водорода или один из разнообразных непротонных растворителей.
Выбор зависит от особенностей комплекса, который необходимо
получить. В табл. 8 указаны различные препаративные методы,
имеющие широкое применение, хотя следует отметить, что этот
список не исчерпывающий и, как будет видно из дальней-
шего, имеются различные методы для получения многих соеди-
нений.
Прежде всего становится ясным следующее общее положение:
Если рассматривать комплексы, образованные элементами в их
высших состояниях окисления, то, в то время как титан и цирко-
ний дают комплексные галогениды в водных растворах кислот,
элементы подгрупп ванадия и хрома образуют только оксигалоге-
нидные комплексы в тех же условиях (однако ниобий и тантал
образуют фторо комплексы в водном растворе плавиковой кислоты).
Далее следует рассмотреть в общем виде методы синтеза раз-
личных групп комплексных соединений. Точное описание препара-
тивных методик будет дано в последнем разделе.
Получение фторо-комплексов в сколько-нибудь значительном
объеме рассмотрено не будет, так как оно или очень просто —
фторо-комплексы синтезируют в водных растворах плавиковой
кислоты по хорошо разработанным мэтодикам, — или же требует
специальной аппаратуры для применения высокореакционных
межгалоидных соединений (таких, как BrFa). В последнем случае
можно рекомендовать обзоры [48, 49] и описание препаративных
методов в «Неорганических синтезах» [50].
А. Титан и цирконий
Хлориды и бромиды получают в водных или спиртовых раство-
рах галогеноводородов. Водные растворы используют для полу-
чения солей аммония, рубидия, цезия, пиридиния и хинолиния, а
спиртовые растворы применяют для получения солей алкилзаме-
щенного аммония. При синтезе в водных растворах кислот гало-
гениды переходных и щелочных металлов растворяют в отдельных
порциях водного раствора галогеноводорода (максимальной моляр-
ности), растворы смешивают, охлаждают до 0° и пропускают сквозь
раствор ток газообразного хлористого или бромистого водорода.
Хлористый аммоний не очень хорошо растворяется в концентри-
рованной соляной кислоте, и в этом случае исходный раствор
готовят из 8 М кислоты; вследствие нерастворимости хлористого
Таблица 8
Сводная таблица препаративных методов
Элемент Состояние окисления Комплекс Среда (растворитель) Исходные вещества и др.
Ti, Zr 4+ м' [MF„] Водная HF —
Ti, Zr 4+ М2Г [MXe] (Х=С1, Вг) Водная НХа или спиртовый рас- твор НХ6 МХ44-М1Х
Ti 3+ K3[TiFeJ NaCl—КС! K2TiF6— электро- литическое вос- становление
V 5+ м1 [VF6] BrF3 VF54- m'f
Nb, Ta 5+ Фториды и ок- сифториды Водная HF —
V, Nb, Ta 5+ Оксихлориды и оксибромиды Водная НХ —
Nb, Ta 4± М| [MCIJ Отсутствует МС15 + М1 4- 4-м’с1
V 3+ М1 [VXJ (Х=С1, Вт) СН3СН VXg-f-M’X
Mo, W 6+ Фториды IF5 или SO2 MF64-M’f
Cr, Mo, W 6+ Оксифториды Водная HF —
Mo, W 6+ Фториды IF5 или SO2 MF^M1!
Cr, Mo, W 5+ М’ [МОХ5] (Х=С1, Вг) Водная HF MX54-MjX
Mo, W 5+ М1 [МОХ4] (Х=С1, Вг) 1. Водная HF 2. SO2 МХ54-М!Х
Cr 44- м!2 [MF6] Отсутствует СгС13 4- M’Cl-l-Fa
Mo 4+ Na2 [MoFel so2 MoF64-KI
Mo 4+ Mj [МоС16] 1. СНС13 2. IC1 MoCl4, 2RCN4-MIC1 MoClj-f-M^l
Mo 4+ M| [MoBr6] IBr MoBr34-MIBr
W 4+ М* [WC16] IC1 WClj+M’ci
w 4+ М| [WBre] Отсутствует WBr64-M'l
Mo w 3+ 34- М| [Д1оХ6] (Х=С1, Вг) М’ IW2C19] Водная НХ Водная НХ Восс та нов ление молибдата Восстановление вольфрамата
* Для солей щелочных металлов.
0 Для замещенных солея аммония и т. д.
148
Глава 5
натрия в соляной кислоте, следуя этой методике, трудно получить
соли натрия. Другие соли осаждаются довольно хорошо, и они
совершенно устойчивы, пока смочены концентрированной кисло-
той, однако требуется большая осторожность на конечной стадии
выделения безводной соли. Так, если следы кислоты удаляются не-
посредственно в вакууме, то галогеноводород улетучивается пер-
вым, остается нераздельно кипящая кислота, а при таких концен-
трациях г ало г еноводорода комплексные анионы гидролизуются.
Эту трудность преодолевают в первую очередь отмыванием
водного раствора кислоты. В случае хлоро-комплексов промывание
осуществляют 5%-ным раствором тионилхлорида в эфире с после-
дующим промыванием безводным эфиром. Тионилхлорид особенно
подходит для этой цели, так как его продукты гидролиза SO2
и НС1 не могут реагировать с гексахлоро-анионами. Было бы,
однако, неосторожным использовать чистый тионилхлорид в ка-
честве промывной жидкости, так как его следы нелегко удалить из
комплексной соли. По-видимому, аналогичным образом можно
использовать тионилбромид на конечной стадии выделения ком-
плексных бромидов, но вследствие того, что тионилбромид не всег-
да имеется в распоряжении, его обычно заменяют бромом.
В случае комплекса титана (NH4)2[TiBre] бром используют в ка-
честве растворителя и в качестве промывной жидкости, поскольку
бромид титана(1У) можно получить in situ прямой реакцией метал-
лического титана в виде стружки с бромом.
Методика получения в этиловом спирте совершенно подобна
методике в водном растворе, за исключением того, что тионилхло-
рид уже непригоден в качестве конечной промывной жидкости, так
как возможно вытеснение катиона амина. Удовлетворительные ре-
зультаты дает последовательное промывание безводным этиловым
спиртом.
Б. Ванадий, ниобий и тантал
В последнее время было проведено очень мало работ по изу-
чению оксихлоридных и оксибромидных комплексов этих элемен-
тов. Для ванадия, например, указывают, что оксихлоридные ком-
плексы пяти- и четырехвалентного V, а именно M’iVOClJ и
MJjVOClJ, настолько хорошо растворимы, что имеются сведения
только о синтезе соединений пиридиния и хинолиния; последние
были получены в этиловом спирте.
За последние 50 лет также мало внимания было уделено ком-
плексным оксихлоридам и оксибромидам. Соединения ниобия
типа M’tNbOCls], где M!=Rb и Cs, образуются довольно лег-
ко смешиванием водных солянокислых растворов, содержащих
хлорид щелочного металла и хлорид ниобия(У) или окси-
IV. Получение галогенидиых и оксигалогеиидных комплексных солей 149
трихлорид ниобия(У). Соли пиридиния и хинолиния с анионом
[NbOClJ- были получены с использованием аналогичных ре-
акций в растворах хлористого водорода в воде и в этиловом спир-
те. Интересно отметить, что в том случае, когда образуются соли
двух таких типов, щелочные металлы обычно дают соли типа
МЦМОХа], а большие органические катионы дают соли второго
типа, МЧМ0С14].
Имеется очень мало сведений о комплексных галогенидах ни-
обия и тантала в их низших валентных состояниях, хотя и очевид-
но, что такие соединения можно получить сплавлением. Так, взаи-
модействие NbCl4 и КС1 или NaCl ведет к образованию комплек-
сов М( [NbCle], и те же соединения можно получить восстановле-
нием раствора NbCh в MiCl щелочным металлом.
Одна или две соли с тетраэдрическими анионами тетрахлоро-
и тетрабромованадатов(Ш) были получены реакцией в растворе
метилцианида. Трихлорид и трибромид растворяются в метилциа-
ниде (и других алкилцианидах) при кипячении с обратным холо-
дильником, хотя образующиеся комплексы VX3-3RCN лишь не-
значительно растворимы в избытке алкилцианида при комнатной
температуре. В присутствии галогенидов алкиламмония, таких,
как хлорид тетраэтиламмония, образуется соль
MeCN
VC13------> [Et4N] [VClJ-2MeCN---> [EtjN] [VC14].
Et.NCl
К сожалению, точное описание методики синтеза еще не опуб-
ликовано и поэтому не может быть приведено в следующем разделе.
В. Хром, молибден и вольфрам
Для этих элементов в пятивалентном состоянии имеются два
типа оксихлоридных и оксибромидных комплексов, как и для нио-
бия: М’ [MOXsIh М1 [МОХ4]. В табл. 5 приведен список получен-
ных солей. Все комплексы первого типа получают в водных раст-
ворах соляной или бромистоводородной кислоты реакцией раст-
вора М!Хс другим раствором, содержащим молибден или вольф-
рам в пятивалентном состоянии. Последний раствор можно приго-
товить электролитическим восстановлением растворов молибдатов
и вольфраматов, однако для осуществления этого процесса необ-
ходимо конструировать подходящую электролитическую ячейку.
Поэтому приведенный здесь метод предусматривает использование
хлорида молибдена(У) (имеющегося в продаже) или оксалатоди-
оксивольфрамата(У) калия (который можно легко приготовить).
150
Глава 5
Соединения хрома несколько менее устойчивы, но их можно полу-
чить в относительно чистом виде почти аналогичными методами.
Исходным соединением хрома служит СгОз; хром восстанавливают
до пятивалентного в растворе хлористый водород — ледяная
уксусная кислота.
Для вольфрама эта методика с применением водного раство-
ра кислоты в некоторых случаях дает комплексы второго
типа, если присутствуют большие катионы: (С Н N) [WOC141;
(CsHeN) [WOBrJ; (C9H8N) [WOBr4]; (w3o-C9H8N) [WOBrJ.
Однако вообще комплексы второго типа лучше получать с
использованием жидкой двуокиси серы в качестве растворителя.
Так, M0CI5 реагирует с М!С1 в жидкой двуокиси серы с образова-
нием солей М1 [МоОС14]; M0CI5 сначала сольватируется с образова-
нием окситрихлорида МоОС1з, который реагирует с М!С1, давая
комплексную соль. Соли пиридиния и хинолиния также можно
получить при обработке солей MJJMoOCls] жидкой двуокисью
серы, в результате чего наступает разложение
М| [МоОС15]---> М1 С1-.М1 [МоОС14].
При получении солей рубидия и цезия наблюдается интересная по-
бочная реакция, так как в обоих случаях образуются два продук-
та: первый — ожидаемый оксихлорид МЧМоОС14] и второй —
гексахлоромолибдат(1У) MJ, [Model. Еще не ясно, как идет это
восстановление и почему оно наблюдается только в реакциях с хло-
ридом рубидия и цезия; тем не менее выход продукта оказался
более 30%.
Производные щелочных металлов М1, [МоС1 е] также получают
реакцией M0CI5 с М*С1 в монохлориде иода. Самый легкий способ
приготовления соответствующих солей с органическими катионами
заключается в реакции М’С1 с комплексным соединением МоС14-
•2СзН7СЫ в растворе хлороформа; комплекс с ?г-пропилцианидом
очень легко получают обработкой MoCh избытком цианида, в ре-
зультате чего пятивалентный молибден количественно восстанавли-
вается до четырехвалентного.
Соли щелочных металлов с аналогичными анионами гексахло-
ровольфраматов(1У) можно получить реакцией в растворе мо-
нохлорида иода; эти и аналогичные им бромо-соединения также
получают реакцией между WCle или WBre с МП в отсутствие
растворителя.
Комплексы трехвалентных молибдена и вольфрама М^ [МоХ6]
и М!,[W2C19]можно приготовить довольно легко восстановлением
шестивалентных элементов (т. е. МоОз и M!,W04) в концентриро-
ванных растворах соляной кислоты. Восстановление молибдена
V. Методики синтеза
151
обычно осуществляют электролитическим путем и вольфрамат вос-
станавливают оловом. Так как подробности получения имеются в
сборнике «Неорганические синтезы» [51], они не будут приведены
в следующем разделе.
V. МЕТОДИКИ СИНТЕЗА
А. Подгруппа титана
1. Cs2TiCl6 [2]
TiCl4 и CsCl растворяют в отдельных объемах концентрирован-
ной соляной кислоты. Концентрации их обычно ниже раствори-
мости, за исключением CsCl, раствор которого делают обычно насы-
щенным. Порции двух отдельно приготовленных растворов сме-
шивают в отношении TiCl4 : CsCl, несколько большем 1 : 2. Затем
раствор охлаждают до 0° и насыщают газообразным хлористым во-
дородом. Образующиеся бледно-желтые кристаллы отфильтровы-
вают и промывают последовательно а) концентрированной соляной
кислотой, б) 5%-ным раствором тионилхлорида в диэтиловом эфи-
ре и в) безводным диэтиловым эфиром. Промывание раствором тио-
нилхлорида продолжают до тех пор, пока не прекратится вспени-
вание. Последняя операция заключается в выдерживании кристал-
лов в вакууме порядка 10'3 мм рт. ст. в течение нескольких ча-
сов при непрерывном откачивании. В процессе последних операций
(т. е. при промывании эфиром и откачивании) важно предохранить
кристаллы от контакта с влажным воздухом; поэтому представ-
ляет известные преимущества такое устройство, когда сосуд для
фильтрования является частью вакуумной системы, так что про-
мывание и откачивание можно осуществлять без переноса продукта.
Соли аммония, калия, рубидия, пиридиния и хинолиния гото-
вят аналогичным способом. Единственное изменение в процедуре
относится к приготовлению раствора М'С1 в соляной кислоте. В тех
случаях, когда трудно получить достаточно концентрированный
раствор в 11 М соляной кислоте, кислоту разбавляют.
2. [(СНз)з1Ш]2Т1С16 [2]
Процедура получения этого соединения подобна указанной в
разд. 1 для CsaTiCle, за исключением того, что в качестве раствори-
теля используют безводный этиловый спирт, насыщенный НС1.
(CH3)3NHC1 получают in situ растворением амина в этиловом спир-
те и пропусканием через раствор газообразного НС1. Продукт про-
мывают безводным этиловым спиртом.
152
Глава 5
Другие соли алкиламмония, например [(CH3)2NH2]2TiCle
и [CH3NH3]2TiCl6 получают таким же способом.
3. [(СН3) 3NH]2ZrCl6 [7]
Это соединение и большое число солей алкиламмония получают
так же, как и аналогичные соединения титана. Белые кристаллы
гексахлороцирконатов не всегда осаждаются тотчас; в этом случае
следует оставить раствор на ночь в холодильнике.
4. IC6H5NH)2TiBr6 12]
TiBr4 (0,01 моля) и C5H5N (0,02 моля) растворяют отдельно в
48 %-ном водном растворе НВт, растворы смешивают, охлаждают
до 0° и насыщают газообразным НВг. Темно-красные кристаллы
отфильтровывают, промывают последовательно безводным бромом
(предварительно перегнанным над пятиокисью фосфора) и безвод-
ным диэтиловым эфиром и затем откачивают в вакууме.
Так как TiBr4 не всегда имеется в лаборатории, иногда предпо-
читают использовать свежеосажденную «гидроокись» титана, раст-
воренную в НВг; кроме того, удобно в качестве промывной жид-
кости использовать тионилбромид (в эфире), а не чистый бром.
Б. Подгруппа ванадия
1. Cs2[NbOCl5|
Это соединение и аналогичное соединение рубидия можно полу-
чить методом, описанным для CsaTiCle (А.1) с использованием
NbOCla или NbCls в качестве исходного вещества. Соответствую-
щие бромиды можно получить в водном растворе НВг (см. метод
А.4).
В. Подгруппа хрома
1. Cs2[CrOCl6J
СгОз растворяют в ледяной уксусной кислоте, насыщенной
хлористым водородом, и раствор выдерживают приблизительно в те-
чение 30 мин. Добавление раствора CsCl в \ЛМ НС1 приводит к
немедленному осаждению комплексной соли в виде тонкого крас-
ного порошка. Продукт отфильтровывают и промывают так же,
как и Cs2TiCle (метод А.1).
V. Методики синтеза
153
2. Cs2[MoOCI5] и CsafWOCls] |36]
Эти соединения и другие соли, перечисленные в табл. 5, полу-
чают методом А.1, но с использованием M0CI5 и WCls в качестве
источников молибдена и вольфрама соответственно. Торговый
M0CI5 иногда загрязнен продуктами гидролиза, но это не имеет
значения, так как в любом случае соляная кислота вызывает гид-
ролиз хлорида молибдена с образованием аниона [MoOCIs]3-.
Поскольку WCls не всегда имеется в распоряжении, иногда исполь-
зуют К [WO2(C2O4)l-a;H2O в качестве исходного соединения пяти-
валентного вольфрама. Это соединение получают следующим
образом.
Водный раствор, содержащий 6,5 г оксалата калия, 3 г щавеле-
вой кислоты и 1,3 г вольфрамата калия, нагревают до кипения и
затем восстанавливают гранулированным оловом при 80°. В начале
восстановления раствор изменяет окраску и становится последо-
вательно голубым, зеленым и наконец красным. Когда восстановле-
ние закончено (обычно в пределах 1 час), через раствор пропускают
H2S для осаждения олова в виде сульфида; раствор отфильтровы-
вают в 500 мл этилового спирта. Медленно осаждаются красные
кристаллы, которые затем отфильтровывают и промывают этило-
вым спиртом.
3. (C6HeN)[MoOCl4] [36]
CsHeNCl (0,022 моля) и M0CI5 (0,01 моля) вводят в ампулу, при-
соединенную к вакуумной линии. В ампулу конденсируют с избыт-
ком двуокись серы и затем ее герметизируют. Комплексная соль
осаждается, и ее можно отфильтровать в вакууме (перед открыва-
нием ампулу охлаждают). Затем соль промывают СНС1з для удале-
ния избытка CsHeNCl. Соединение хинолиния получают аналогич-
ным образом. Соль диэтиламмония очень хорошо растворима в
жидкой SO2; растворитель в этом случае удаляют выпариванием,
и остаток обрабатывают СНС1з для удаления избытка хлористого
водорода.
Соли рубидия и цезия также растворимы в SO2, но они загряз-
нены нерастворимыми солями М*МоС1в. В этих случаях берут не-
большой избыток MoCls, реакционную смесь фильтруют, SOa
выпаривают из фильтрата и остаток обрабатывают СНС13 для уда-
ления непрореагировавшего M0CI5.
4. (C5HeN)2 [МоС16] [36]
Сначала готовят MoC14-2C3H7CN, для чего в ампулу запаивают
M0CI5 с избытком C3H7CN и выдерживают их в течение нескольких
суток. Затем ампулу вскрывают, присоединяют к вакуумной линии
154
Глава 5
и таким путем удаляют избыток СзН7СГ<. Экстракция остатка без-
водным бензолом дает MoC14-2C3H7CN. Растворы MoC14-2C3H7CN
(0,01 моля) и CsHeNCl (0,02 моля) в СНС13 смешивают и светло-
желтый осадок фильтруют в вакууме, промывают СНС1з и затем
откачивают в течение нескольких часов. Соли хинолиния и диэтил-
аммония можно получить тем же способом.
Соли рубидия и цезия лучше получать методом, описанным в
разд. В.З.
5. CeUWCld [43]
Тонкоизмельченный CsI (0,01 моля) помещают в пробирку, кото-
рую выдерживают в печи несколько часов при 130°; затем добав-
ляют WCle (0,0055 моля) или в сухом боксе, или по обычной мето-
дике в вакууме и пробирку герметизируют. Пробирку выдерживают
при 130° в течение 3 суток, затем открывают и подсоединяют к
вакуумной линии. Иод и избыток WCle удаляют нагреванием про-
бы приблизительно до 300°- Соли калия и рубидия получают по-
добным же образом.
При желании можно использовать монохлорид иода в качестве
растворителя.
6. Cs2[WBr6] [43]
Это соединение и аналогичные соли калия и рубидия получают
по методу, описанному в разд. В.5, используя в качестве исходных
веществ CsI и WBre.
ЛИТЕРАТУРА
1. Sidgwick N. V., The Chemical Elements and Their Compounds, Ox-
ford Univ. Press, New York, 1950, p. 645.
2. F о w 1 e s G. W. A., Nicholls D., J. Inorg. Nucl. Chem., 18, 130
(1961).
3. Wernet J., Z. anorg. allgem. Chem., 272, 279 (1953).
4. J a n d e r J., Z. anorg. allgem. Chem., 294, 181 (1958).
5. Rosenheim A., Schutte O., Z. anorg. allgem. Chem., 150, 69
(1925).
6. В у e J., H a e g i W., Compt. Rend., 236, 381 (1953).
7. D r a k e J. E., F о w 1 e s G. W. A., J. Inorg. Nucl. Chem., 18, 136
(1961).
8. Rosenheim A., Frank P., Chem. Ber., 38, 812 (1905).
9. F о w 1 e s G. W. A., Nicholls D., J. Chem. Soc., 1961, 95.
10. Drake J. E., F о w 1 e s G. W. A., J. Less-Common Metals, 3,149 (1961).
11. Jorgensen С. K., Absorption Spectra and Chemical Bonding in
Complexes, Pergamon, New York, 1962, p. 284.
12. В r i g h t N. F. H., Wurm J. G., Can. J. Chem., 36, 615 (1958).
13. Stabler A., Chem. Ber., 37, 4405 (1904).
14. Stabler A., Chem. Ber., 38, 2619 (1905).
15. E h г И c h P., Ku pa G., Blankenstein K., Z. anorg.
Chem., 299, 213 (1959).
Литература
155
16. Gruen D. M.. McBeth R. L., Seventh International Confe-
rence on Co-ordination Chemistry, Butterworths, London, 1963, p. 27;
Nature, 194, 468 (1962).
17. Emeleus H. J., Gutmann V., J. Chem. Soc., 1949, 2979.
18. Sharpe A. G., Wolf A. A., J. Chem. Soc., 1951, 798.
19. П алкин А. П., Чиранов H. Д., ЖНХ, 4, 898 (1959).
20. Gutmann V., Z. anorg. allgem. Chem., 264, 151 (1951).
21. Huber K., Just E., N e u e n s c h w a n d e r E., StuderM.,
Roth B., Helv. Chim. Acta, 41, 2411 (1958).
22. Hoard J. L., J. Am. Chem. Soc., 61, 1252 (1939).
23. Hoard J. L., Martin W. J., Smith M.E., Whitney J.F.,
J. Am. Chem. Soc., 76, 3820 (1954).
24. I j d о D. J. W., private communication.
25. G r u e n D. M., McBeth R. L., J. Phys. Chem., 66, 57 (1962).
26. N у h о 1 m R. S., Croat. Chem. Acta, 33, 157 (1961).
27. Seifert H. J., Ehrlich P., Z. anorg. allgem. Chem., 302, 284
(1959).
28. Hargreaves G. B., Peacock R. D., J. Chem. Soc., 1958, 4390.
29. Cox B., Sharp D. W. A., Sharpe A. G., J. Chem. Soc., 1956,
1242.
30. Hargreaves G. B., Peacock R. D., J. Chem. Soc., 1958, 2170.
31. Schmitz-Dumont O., Heckmann I., Z. anorg. allgem.
Chem., 267, 277 (1952).
32. Schmitz-Dumont O., Opgenhoff P., Z. anorg. allgem.
Chem., 275, 21 (1954).
33. Schmitz-Dumont O., Bruns I., Heckmann I., Z.
anorg. allgem. Chem., 271, 347 (1953).
34. Hargreaves G. B., Peacock R. D., J. Chem. Soc., 1957, 4212.
35. Horner S., Tyree S. Y., private communication.
36. A 1 1 e n E. A., Brisdon B. J., Edwards D. A., F о w-
1 e s G. W. A., Williams R. G., J. Chem. Soc., 1963, 4649.
37. Edwards D. A., F о w 1 e s G. W. A., unpublished observations.
38. Huss E., Klemm W., Z. anorg. allgem. Chem., 262, 25 (1950).
39. Edwards A. J., Peacock R. D., Chem. Ind. (London), 1960,
1441.
40. Allen E. A., Edwards D. A., F о w 1 e s G. W. A., Chem.
Ind. (London), 1962, 1026.
41. Edwards A. J., Peacock R. D., Said A., J. Chem. Soc.,
1962, 4643.
42. Brisdon B.J., Fowles G. W. A., Osborne В. P., J. Chem.
Soc., 1962, 1330.
43. Kennedy C. D., Peacock R. D., J. Chem. Soc., 1963, 3392.
44. Laudise R. A., Young R. C., J. Am. Chem. Soc., 77, 5288 (1955).
45. E d w a r d s A. J., P e a с k о c k R. D., J. Chem. Soc., 1959, 4126.
46. S h i 1 о f f J. C., J. Phys. Chem., 64, 1566 (1960).
47. Jonassen H. B., Cantor S., Tarse A. R., J. Am. Chem. Soc.,
78, 271 (1956).
48. Sharpe A. G., Advan. Fluorine Chem., 1, 29 (1959).
49. Peacock R. D., in Progress in Inorganic Chemistry, Vol. 2, Cot-
ton F. A., ed., Interscience, New York, 1960, p. 193.
50. Прист, в книге «Неорганические синтезы», сб. III, Издатинлит, Мо-
сква, 1952.
51. Lohmann К. Н., Young R. С., in Inorganic Syntheses, Vol. 4,
Bailar J., ed., McGraw-Hill, New York, 1953, p. 97; Laudise R. A.,
Young R. C., in Inorganic Syntheses, Vol. 6, Rochow E. G., ed., McGraw-
Hill, New York, 1960, p. 149.
ГЛАВА 6
БЕЗВОДНЫЕ НИТРАТЫ МЕТАЛЛОВ
Эдисон и Логан
Ноттингемский университет, Англия
1. ВВЕДЕНИЕ
В виде кристаллогидратов известны нитраты многих металлов,
однако безводных нитратов до последних лет получено сравнитель-
но мало. Хорошо изучены безводные нитраты всех щелочных метал-
лов. Безводные нитраты металлов второй группы периодической
системы — кальция, стронция и бария — легко можно получить,
удаляя воду из кристаллогидратов. Известны также безводные
нитраты серебра, свинца и таллия(1). Однако безводные нитраты
большинства металлов не были выделены до самого последнего
времени. Если говорить о методах синтеза, главным препятствием
было настойчивое использование водных систем. Попытки удалить
воду из кристаллогидратов обычно приводили к гидролизу с обра-
зованием основных нитратов, гидроокисей или окислов и с выде-
лением азотной кислоты. Поэтому обычно считали, что безводные
нитраты, особенно нитраты переходных элементов, должны быть
неустойчивыми соединениями. В последнее время многие такие
соединения были синтезированы, и оказалось, что они обладают
высокой термической устойчивостью. Успешный синтез этих сое-
динений является результатом более широкого использования не-
водных растворителей в препаративной неорганической химии.
Интерес к препаративным работам в этой области значительно
возрос после того, как выяснилось, что физические свойства и хи-
мическая активность некоторых безводных нитратов совершенно
отличаются от тех, которые можно было ожидать, исходя из из-
вестных химических свойств кристаллогидратов. Химические
свойства безводных соединений подробно рассмотрены в работе
[1]; в данной главе будут описаны только те, которые имеют не-
посредственное отношение к методам синтеза. В настоящее время
уже совершенно ясно, что связь между атомом металла и нитрат-
ной группой может быть в значительной степени ковалентной и
что нитратная группа может занимать одно или два координа-
ционных места, а также образовывать мостики между атомами ме-
I. Введение
157
таллов. В простых нитратах M(NO3)2, где М — Мн, Ni, Си или
Zn, нитратные группы ковалентно связаны с атомом металла, но
кристаллическая структура нитрата меди(П) [2] показывает,
что в каждой нитратной группе ковалентно связаны с металлом
более одного атома кислорода. Можно доказать, что нитратная
группа занимает одно координационное место только в комплек-
сах типа Mn(CO)5NO3 [3] и Me3SnNO3 [4]. В первом соединении
связь металла и нитрата может сохраняться при многих обменных
реакциях, в которых замещаются карбонильные группы [5].
По два координационных места занимают нитратные группы в ио-
не [UO2(NO3)3]_ [6] и в ди- и тригидратах нитрата уранила [7];
в анионе [M(NO3)4]2“, где М — Си или Ni, атом металла, по-види-
мому, имеет координационное число 6, так что и здесь должна су-
ществовать бидентатная связь [8]. Образование нитратных мости-
ков между атомами металла характерно для структуры основного
нитрата бериллия [9] и твердого нитрата меди(П) [2].
Наличием этой прочной ковалентной связи обусловлено наи-
более важное физическое свойство — заметная летучесть некото-
рых безводных нитратов металлов. Например, соединения
Cu(NO3)2[10], Ti(NO3)4 [И, 16, 59], Zr(NO3)4 [12] и Be4O(NO3)6
[9] могут существовать в газообразном состоянии и, следователь-
но, могут быть очищены возгонкой. При рассмотрении методов син-
теза будет обращено внимание на некоторые химические свойства,
обусловленные наличием ковалентной связи. Многие ковалентные
нитраты [например, Cu(NO3)2, Zn(NO3)2] обладают хорошей раст-
воримостью в полярных органических растворителях. Так, нит-
рат меди(П) лучше растворим в этилацетате, чем в воде, и выкри-
сталлизовывать его из раствора в этилацетате не удается [13].
В водных растворах, как правило (но не всегда), наблюдается обыч-
ная диссоциация соединений на катионы металла и анионы нит-
рата. При растворении безводного нитрата бериллия в воде около
10% нитратных групп в растворе превращаются в ионы нитрита
[14]. Некоторые нитраты с особо прочной ковалентной связью
реагируют (иногда со взрывом) с органическими соединениями.
Так, нитрат меди энергично реагирует с диэтиловым эфиром [15] и
нитрометаном [13, 15], а тетранитрат титана реагирует с додека-
ном, образуя алкилнитраты, нитроалканы и карбоновые кислоты
[16].
Препаративные методы, разработанные для синтеза безводных
нитратов металлов, учитывают физические и химические свойства
каждого отдельного нитрата. Поэтому методы синтеза могут быть
весьма разнообразными. Почти во всех методах применяют невод-
ные растворители. Некоторые растворители в настоящее время ма-
ло используют, но и они будут кратко рассмотрены в этом обзоре,
так как в особых случаях их можно применить. Растворы нитратов
158
Глава 6
металлов можно получить обменной реакцией между нитратом
серебра и растворами галогенидов различных металлов в ацетоне,
однако этим способом, по-видимому, нельзя получить твердые сое-
динения [17]. Вытеснением серебра из раствора нитрата серебра в
ацетоне, фенилцианиде или в жидком аммиаке различными метал-
лами можно получить нитраты металлов в виде сольватов [18].
Попытки получения безводных нитратов с использованием чистой
азотной кислоты в качестве реакционной среды оказывались в
общем безуспешными. В результате взаимодействия чистой азотной
кислоты с некоторыми безводными хлоридами металлов образуют-
ся гидратированные нитраты или окислы [19, 20]. По-видимому,
под действием безводного хлористого водорода и ионов металла
азотная кислота разлагается на ангидрид и воду, которая образует
гидраты нитратов и удаляется из сферы реакции при осаждении
последних. Использование в качестве реакционной среды расплав-
ленной соли еще не изучено в достаточной степени. Основные труд-
ности таких методов синтеза связаны с выделением продукта и с
применением высоких температур. При растворении дигидрата
нитрата ртути(П) в расплавленном бромиде ртути(П) [21] кри-
сталлизационная вода испаряется (238—320°). Окись лантана
Ьа2О3 реагирует с расплавленным нитратом аммония при 170°. Из-
быток нитрата аммония можно удалить отгонкой, после чего ос-
тается безводный нитрат лантана [22]. Этот метод заслуживает
дальнейшего изучения.
В препаративных методах синтеза, которые имеют наиболее
широкое применение, используются четырех- или пятиокись азо-
та, а также их производные. Именно эти методы будут подробно
рассмотрены в данном обзоре.
11. РЕАКЦИИ С ИСПОЛЬЗОВАНИЕМ ЧЕТЫРЕХОКИСИ
АЗОТА
В некоторых странах в продаже имеется жидкая четырехокись
азота, которую легко можно очистить перегонкой. В лабораторных
условиях чистую четырехокись азота легко получают термическим
разложением нитрата свинца [23, 24]. В обычных условиях это
жидкое вещество (т. пл. —11,2°, т. кип. 21,15°), и реакции почти
всегда проводят при комнатной температуре. При работе с жидкой
четырехокисью азота важно защитить ее от влаги. Сосуды для нее
перед использованием следует высушить при температуре 100°.
Жидкость можно хранить и использовать в стеклянной аппарату-
ре, а для проведения реакций раствор следует переливать из од-
ного сосуда в другой через соединительные стеклянные трубки.
Закрытые сосуды с соединительными трубками сообщаются с ат-
мосферой через осушительные трубки, наполненные пятиокисью
II. Реакции с использованием четырехокиси азота
159
фосфора на стеклянной вате. Через осушительные трубки в ат-
мосферу выделяется некоторое количество четырехокиси азота,
так что работать нужно в вытяжном шкафу. Следы влаги в четы-
рехокиси азота можно обнаружить замораживанием жидкости —
это один из наиболее чувствительных способов проверки. Чистая
четырехокись замерзает с образованием бесцветного стеклообраз-
ного вещества, но в присутствии следов влаги образуется трех-
окись азота, которая окрашивает замерзшую четырехокись в зеле-
ный цвет. Перед использованием жидкости для синтеза обычно
рекомендуется охладить ее приблизительно на 10° ниже темпера-
туры кипения, иначе теплота, выделяющаяся при реакции, может
вызвать энергичное кипение и выделение большого количества
ядовитых паров из аппарата.
Жидкая четырехокись азота диссоциирует на ионы в соответ-
ствии с уравнением
N2O4 ;—NO+ -f- NO3.
Это было подтверждено несколькими физическими методами [25—
27 ] и вполне согласуется с химическими свойствами жидкости.
Отсюда следует, что в жидкой четырехокиси азота нитрат-ион иг-
рает ту же роль, какую гидроксил-ион в воде. В чистой четырехо-
киси степень ионизации очень невелика, однако диссоциация об-
легчается в присутствии полярных растворителей. Описанные ниже
методы синтеза подразделяются в соответствии с тем, какая жид-
кая среда используется для проведения реакции. В качестве ис-
ходных веществ для реакции обычно берут металлы, безводные
галогениды металлов, окислы или карбонилы металлов.
А. Четырехокись азота без растворителя
|Галогениды металлов легче подвергаются сольволизу четырех-
окисью азота в присутствии донорных растворителей. Примеры
этому будут приведены в дальнейшем. Нитраты некоторых ланта-
нидов получаются в результате прямого взаимодействия четырех-
окиси азота с окисью металла в герметичной бомбе [28], но этот
метод редко используют, так как реакцию нужно проводить при
высокой температуре. Важное значение имеют реакции четырех-
окиси без растворителя с карбонилами металлов и с металлами.
Ниже приведены примеры каждой такой реакции.
1. Реакции с карбонилами металлов
Чистая жидкая четырехокись азота взаимодействует со всеми
карбонилами металлов, которые были испытаны. Скорость реак-
ции зависит от физического состояния карбонила и от температуры
160
Глава 6
[3]. Так, жидкие карбонилы (тетракарбонил никеля и пентакар-
бонил железа) реагируют с четырехокисью чрезвычайно энергич-
но; твердые Мп2(СО)10 и Fe(CO)5 легко реагируют с жидкой четы-
рехокисью азота при комнатной температуре, но при температуре
0° скорость реакции незначительна.
а. Безводный нитрат марганца (II). Дорогостоящий декакар-
бонил марганца Мп2(СО)10 обычно не применяют в качестве исход-
ного продукта для получения безводного нитрата, но для иллюст-
рации типа реакций, протекающих в подобных системах, ниже
приведен один из таких способов синтеза нитрата марганца.
К твердому Mn2(CO)10 (1 г), находящемуся в стеклянной про-
бирке, сообщающейся с атмосферой через осушительную трубку с
пятиокисью фосфора, при комнатной температуре (20°) добавляют
четырехокись азота (6 мл). Карбонил сначала не реагирует с
N2O4, а растворяется в ней, но приблизительно через 3 мин на-
чинается энергичная реакция с образованием мутного темно-
зеленого раствора
Мп2 (СО)10 + 4N2O4->- 2Mn (NO3)2 + 4NO + 10СО.
Энергичная реакция заканчивается через 30 мин, а выделение газа
и образование желтого осадка продолжаются в течение 20 час.
Спустя еще 24 час осадок собирают на стеклянном фильтре,
который должен быть снабжен осушительной трубкой с пяти-
окисью фосфора. Осадок промывают четырехокисью азота и высу-
шивают в токе азота. Желтый осадок представляет собой
сольват Mn(NO3)2-N2O4. Из него можно получить безводный
нитрат в виде белого порошка нагреванием Mn(NO3)2-N2O4
в течение 12 час при температуре 100° и остаточном давлении
10-2 мм рт. ст. [5].
Если карбонил марганца Мп2(СО)10 растворить при 0° в четы-
рехокиси азота, смешанной с петролейным эфиром, то из получен-
ного раствора можно выделить соединение Mn(CO)5NO3 [3]. В на-
стоящее время это единственный известный нитрат карбонила
металла, а все другие испытанные карбонилы металлов в резуль-
тате такой реакции дают простые нитраты.
Нитрат марганца(П) можно синтезировать в сравнительно
больших количествах растворением порошкообразного металличе-
ского марганца в смеси четырехокиси азота и этилацетата. По-
следующая обработка раствора производится так же, как обработ-
ка раствора для выделения соединения Be(NO3)2 (см. ниже).
Это соединение (и все другие нитраты, рассмотренные в этой гла-
ве) сильно расплывается на воздухе, поэтому контакт с атмосфе-
рой следует исключить.
II. Реакции, с использованием четырехокиси азота
161
2. Реакции с металлами
Реакции M+xN2O4->M(NO3)A.+xNO идут с участием щелочных
металлов [29, 30], цинка [31] и ртути [32], но большинство дру-
гих металлов в компактном состоянии (включая барий, стронций
и кальций [14]) не реагирует с четырехокисью азота. Вследствие
низкой диэлектрической проницаемости чистой четырехокиси азо-
та (2,42) ни одна простая неорганическая соль не растворяется в
жидкой четырехокиси, и продукт реакции образуется в виде не-
растворимой пленки на поверхности металла. Эффективность реак-
ции поэтому зависит от когезионных свойств образующегося нит-
рата. Реакция с натрием при погружении металла в жидкость вна-
чале протекает энергично, но нитрат натрия обладает сильными
когезионными свойствами, и реакция быстро полностью прекра-
щается. Реакция с цинком идет до конца и поэтому изучена более
подробно.
а. Безводный нитрат цинка. Кусочек цинка (5 г) со свежеза-
чищенной поверхностью погружают в жидкую четырехокись азота
(10 мл) при температуре около 20°. В течение 10 мин металл по-
крывается тонким слоем белого мелкокристаллического вещества,
выделяется окись азота и жидкость в непосредственном контакте
с металлом окрашивается трехокисью азота в зеленый цвет. По ме-
ре того как слой продукта становится толще, начинают формиро-
ваться кристаллы сольвата Zn(NO3)2-2N2O4, которые легко от-
деляются от поверхности металла при слабом помешивании. За
24 час образуется около 1—2 г продукта, но его образование в
дальнейшем все труднее поддается регулированию. После удаления
избытка металла четырехокись азота отгоняют выпариванием;
при этом образуется сыпучий мелкокристаллический порошок.
Четырехокись азота слабо связана с нитратом цинка и выделяется
при нагревании порошка. При удалении около 10% присоединен-
ной четырехокиси твердое вещество превращается в жидкость.
Изменение фазового состояния, без сомнения, вызывается пониже-
нием температуры плавления исходного сольвата в результате об-
разования нитрата цинка. Реакция
Zn (NO3)2-2N2O4 ;-Zn (NO3)2 +2N2O4
обратима; полное удаление четырехокиси происходит при нагрева-
нии в течение 6 час при 100° и пониженном давлении. Получаю-
щийся безводный нитрат цинка представляет собой сыпучий белый
порошок [31 ].
Трехокись азота, образующаяся в растворе при взаимодействии
металлического цинка с четырехокисью азота, заметно влияет
на скорость реакции [33]. Если кусок металла неподвижен, ско-
12 Заказ № 547
162
Глава 6
рость реакции постепенно возрастает, но, если раствор перемеши-
вают самим металлом, скорость уменьшается. Такое поведение от-
личает эту систему от обычных гетерогенных процессов взаимодей-
ствия металла с жидкостью, когда у поверхности устанавливается
концентрационный градиент активных форм, и перемешивание уве-
личивает скорость реакции. Это явление объясняют присутствием
трехокиси азота у поверхности металла. Молекулы этого вещества
более полярны, чем молекулы четырехокиси, и поэтому они уве-
личивают диэлектрическую проницаемость среды и концентрацию
активной формы NO+. Присутствие трехокисп у поверхности об-
легчает протекание реакции, а при перемешивании вещество рас-
пределяется в растворе. Трехокись не принимает непосредственно-
го участия в реакции; в противном случае неизбежно происходило
бы образование нитрита металла в результате диссоциации трех-
окисп на NO+и NO2~. Скорость растворения цинка пропорциональ-
на [N2O3 Р*; при температуре 0° скорость реакции в растворе, содер-
жащем 40% трехокиси азота, в восемь раз больше, чем в чистой
четырехокиси азота, но продукт не содержит даже следов нитрита.
Б. Четырехокиси азота в инертном разбавителе
Термин «инертный разбавитель» используют здесь для обозна-
чения такого растворителя, который не взаимодействует в сколько-
нибудь заметной степени с четырехокисью азота и который не об-
разует сольватов с четырехокисью в жидком или в твердом состоя-
нии. Использование смеси инертного разбавителя с четырехокисью
азота дает определенные преимущества по сравнению с одной че-
тырехокисью азота, что можно показать па примере нитрометана,
который наиболее широко применяют для этой цели. Нитрометан
образует устойчивые смеси с четырехокисью азота и смешивается с
ней в любых соотношениях. Сольватов в этой системе не образует-
ся, и фазовая диаграмма имеет кривую ликвидуса с одной эвтек-
тической точкой [34]. Образующиеся при взаимодействии с метал-
лами безводные нитраты растворяются в реакционной смеси и
могут быть выделены кристаллизацией. Диссоциация четырехоки-
си в смеси на ионы облегчается высоким значением диэлектриче-
ской проницаемости нитрометана (е=37), поэтому такая смесь
более реакционноспособна, чем чистая четырехокись. Методы синте-
за с использованием нитрометана еще мало изучены, так как эти
же продукты часто значительно быстрее можно синтезировать в
смесях четырехокиси азота с донорными растворителями, напри-
мер этилапетатом (разд. II.В). Однако в тех случаях, когда нитрат
металла образует прочные сольваты с донорными растворителя-
II. Реакции с использованием четырехокиси азота
163
мп, лучше использовать нитрометан. Так, металлический уран
взаимодействует с указанными смесями следующим образом:
и + N2O4 + EtOAc-----> UO2 (NO3)2-2EtOAc,
U + N2O4+MeNO2-------> UO2(NO3)2-N2O4----» UO2(NO3)2.
Из сольвата с этилацетатом не удается выделить безводного
нитрата уранила [32], а в смеси четырехокиси азота с нитроме-
таном образуется соединение состава 1:1, из которого осторож-
ным нагреванием можно выделить безводный нитрат уранила.
На рис. 1 показана зависимость скорости реакции меди, цинка
и урана от состава смеси. Кривая скорости для меди (которая не
реагирует с чистой четырехокисью азота) имеет резкий максимум,
расположенный приблизительно при концентрации нитрометана
85 мол. % [35]. Аналогичным образом изменяется электропровод-
ность смеси, так что наибольшая скорость реакции соответствует
таким соотношениям компонентов смеси, при которых концентра-
ция NO+ наибольшая. Из этого можно сделать вывод, что главной
реакцией, определяющей скорость процесса, является реакция
Cu-|-NO+-----> Cu+ 4-NO.
Соответствие зависимостей скорости реакции и электропровод-
ности смеси обусловлено тем, что медь устойчива в одновалент-
ном состоянии. Скорость растворения определяется скоростью
передачи одного электрона. Затем происходит окисление меди до
двухвалентного состояния, но эта реакция не определяет скорости
растворения меди. Из раствора можно выкристаллизовать соеди-
нение Cu(NO3)2- N2O4. Его обычно получают из смеси четырехокиси
азота с этилацетатом (разд. II.В), и в этом разделе пет необхо-
димости рассматривать дальнейшую обработку этого вещества.
Кривая скорости реакции для цинка (рис. 1) имеет тот же харак-
тер, что и кривая скорости реакции для меди, но максимум не-
сколько смещен. Максимум на кривой скорости для урана не со-
ответствует максимуму электропроводности. Ни цинк, ни уран не
устойчивы в одновалентном состоянии. Реакция этих металлов с
NO+ имеет высший порядок, и скорость реакции поэтому не долж-
на непосредственно зависеть от его концентрации. При взаимодей-
ствии с цинком образуется уже упомянутое соединение Zn(NO3)2-
• 2N2O4. Чтобы показать возможность использования инертных
разбавителей, в дальнейшем указан метод синтеза безводного нит-
рата уранила. Этот синтез интересен еще и тем, что в соединении
UO2(NO3)2-N2O4 четырехокись азота очень прочно связана с нит-
ратом уранила, и ее можно удалить только при строгом соблюде-
нии условий разложения.
12*
164
Глава 6
Рис. 1. Скорости реакций металлов со смесями N2O4—
CH3NO2 при 15°. (Скорость перемешивания 400 об/мин.
Значения для урана увеличены в 50 раз.)
1. Безводный нитрат уранила
Кусок свежезачищенного металлического урана погружают в
избыток смеси четырехокиси азота и нитрометана (~30 мол. %).
После выдерживания в течение нескольких часов при комнатной
температуре жидкость становится зеленой, что, с одной стороны,
обусловлено образованием трехокиси азота, с другой — наличием
в растворе четырехвалентного урана. Никакого замедления реак-
ции вследствие образования нерастворимых продуктов не проис-
ходит, и реакция протекает спокойно. Когда металл полностью
растворяется, зеленая окраска раствора исчезает, так как четырех-
II. Реакции с использованием четырехокиси азота
165
валентный уран, окисляющийся в растворе под влиянием N2O4
до шестивалентного, уже больше не поставляется металлом. Пос-
ле этого добавляют избыток четырехокиси азота, и из раствора вы-
деляется светло-желтый кристаллический порошок состава
UO2(NO3)2-N2O4. Это соединение можно получить в виде крупных
желтых кристаллов, если пары четырехокиси азота медленно
растворить в концентрированном растворе желтого порошка в
нитрометане [36].
Такое же соединение было получено при взаимодействии с Ки-
слов урана с четырехокисью азота в автоклаве [37]; для этой реак-
ции рекомендуется трехокись урана, полученная нагреванием
UO4- 2Н2О в течение 150 час при температуре 500° [38]; но для пол-
ного взаимодействия трехокиси урана с четырехокисью азота в
запаянной стеклянной ампуле требуется 48—72 час. Другие сое-
динения урана — четыреххлористый уран, пятихлористый уран
и хлорид уранила — быстро превращаются в UO2(NO3)2-N2O4
при их взаимодействии со смесью четырехокиси азота и нитроме-
тана [39].
Очень важно точно соблюдать температуру разложения соеди-
нения нитрата уранила с четырехокисью азота. Чистый нитрат
получают нагреванием сольвата при температуре 163° и давлении
0,1 мм рт. ст. в течение 20 час [36] или при температуре 165°
и давлении 10-5 мм рт. ст. в течение 110 мин [38]. Если темпера-
тура отличается более чем на несколько градусов от 163—165°,
то получить чистый безводный нитрат уранила не удается. Напри-
мер, пробы, продолжительно выдержанные при температуре 160°,
содержали небольшие количества четырехокиси азота, а пробы, на-
гретые до 176°, содержали примесь трехокиси урана [36].
В. Четырехокись азота с донорными растворителями
1. Свойства жидких смесей
Молекула четырехокиси азота склонна к присоединению элект-
рона. Это же характерно для иона NO+, образующегося при диссо-
циации четырехокиси азота. При смешивании четырехокиси азота
с органическими растворителями, содержащими атомы, способные
отдавать электроны, происходит частичное перераспределение элек-
тронов. При охлаждении таких смесей можно выкристаллизовать
соединение четырехокиси азота с растворителем. В жидкой фазе и
N2O4 и NO+ ассоциированы с донорным растворителем, и ультра-
фиолетовые спектры смесей показывают, что в растворе нет от-
дельных молекул этих соединений [40]. Свойства жидких смесей
хорошо выражаются равновесиями
[(Don)„NO]++ NOJ ;---- п (Don) + N2O4 (Don)„N2O4,
166
Глава 6
где (Don) — электронно-донорный растворитель. Для препаратив-
ных работ наибольшее значение имеют растворители, обладающие
слабыми донорными свойствами. Форма (Don)„-NO+ в химическом
отношении ведет себя, как ион NO+. Таким образом, концентрация
последнего иона возрастает как вследствие увеличения диэлектри-
ческой проницаемости, так и в результате образования коорди-
нированных ионов нитрозония. Четырехокись азота образует устой-
чивые жидкие смеси с очень большим числом органических ве-
ществ. К таким веществам относятся карбоновые кислоты [41],
сложные эфиры [41, 42], ангидриды [42], кетоны [41, 42], простые
эфиры [43], нитрилы [41, 42], нитрозамины [41, 44] и сульфооки-
си [45]. Некоторые такие растворители указаны в табл. 1.
Донорные растворители в значительно большей степени усили-
вают реакционную способность четырехокиси азота, чем инертные
растворители. Это ясно видно по реакциям таких смесей с метал-
Таблица 1
Некоторые растворители, обладающие донорными
свойствами по отношению к четырехокиси азота
Растворитель Твердый продукт присоединения с n2o„ Донорный атом
молярное соотношение : температура плавления, °C
Диэтилнитрозамин 2:1 —37 Азот
Этилфенилнитрозамин 2:1 —12 »
Метилцианид 2:1 —42 »
1:1 -39
Бензилцианид 2:1 —43
Диэтилсульфоксид 1:1 + 13 Сера
Диизопропилсульфоксид 1:1 —1'0 »
2:1 — 3 »
Уксусная кислота 2:1 + 2 Кислород
Этилацетат 2:1 —65 и —72
Этилбензоат 2:1 —12 »
Уксусный ангидрид 1:1 —44 »
Диэтиловый эфир 2:1 —74,8 »
1,4-Диоксан 1:1 +45,2 »
1,3-Диоксан 1:1 + 2,0 »
Диэтиловый эфир этиленгли-
КОЛЯ 1:1 —60 »
II. Реакции с использованием четырехокиси азота
167
лами. На рис. 2 приведены три типичные кривые зависимости ско-
ростей реакции от состава жидкой смеси. Аналогичным образом
взаимодействуют с такими смесями многие другие металлы, для
которых не измерены скорости реакций. В случае нитрометановых
растворов скорость реакции для металлических меди и цинка
(рис. 1) в пределах концентраций нитрометана 0—50 мол. % очень
невелика, однако скорость реакции быстро возрастает при добав-
лении к четырехокиси азота сравнительно небольших количеств
простого эфира или этилацетата (рис. 2). Это обусловлено присут-
ствием в растворе ионов (Don)„NO+, которые принимают участие в
реакциях переноса электронов
М ф- (Don)nNO+---> М+ + п (Don) ф- NO.
Во всех донорных растворителях при высоких концентрациях
растворителя наблюдается некоторый ограничивающий эффект, в
Рис. 2. Скорости реакций со смесями N2O4 —
• донорный растворитель (15", 400 об/мин).
а — медь — этилацетат; б — медь — диэтиловый эфир; в —
цинк — этилацетат.
168
Глава 6
результате которого происходит уменьшение скоростей реакций.
Этот эффект проявляется довольно резко, что отражается на кри-
вых скорости реакции в виде крутого спада после резкого макси-
мума (рис. 2). Замедление реакций связано со способностью моле-
кул, присутствующих в жидкости, образовывать координационные
Рис. 3. Скорости реакций для смесей: жидкая
N2O4 — азотсодержащие донорные растворители
(15', 400 об/мин).
а — медь — диэтилнитрозамин; б — медь — фенилцианид;
в — цинк — бензилцианид.
или сольватные соединения с ионами металлов и отводить эти ионы
от поверхности, где они образуются. Подробное обсуждение этого
явления не входит в задачу настоящего обзора. Яркими приме-
рами влияния донорных растворителей на скорость взаимодей-
ствия металлов с четырехокисью азота могут служить цианиды
и нитрозамины (рис. 3). Скорости реакций сразу же достигают мак-
симального значения уже при добавлении растворителя в количе-
стве нескольких мольных процентов и затем на протяжении всего
остального интервала концентраций непрерывно уменьшаются
[35].
II. Реакции с использованием четырехокиси азота
169
Из сказанного ясно, что способность металлов взаимодейство-
вать с четырехокисью азота в смеси с донорными растворителями
может изменяться в широких пределах в зависимости от свойств
металла и растворителя. Из растворов, полученных в результате
такого взаимодействия, кристаллизацией можно выделить соеди-
нения различных типов.
2. Природа получаемых продуктов взаимодействия
Продукты взаимодействия металлов с четырехокисью азота в
смесях с донорными растворителями можно классифицировать
следующим образом:
а) нитраты металлов,
б) соединения нитратов металлов с четырехокисью азота,
в) соединения нитратов металлов с растворителем,
г) соединения нитратов металлов с четырехокисью азота и с
растворителем.
Таблица 2
Влияние донорного растворителя на первичные продукты
взаимодействия металлов с четырехокисью азота
Металл Реакционная среда (N2O4 с растворителем) Первичный продукт взаимодействия Литература
Кадмий Этилацетат Cd(NO3)2 46
Марганец » Mn(NO3)2.N2O4 5
Кобальт » Co(NO3)2.2N2O4 47
Уран » UO2(NO3)2-2EtOAc 32
Диэтилнитрозамин UO2(NO3)2-2Et2N-NO 36
Никель Метилцианид Ni(NO3)2-2MeCN 48
Индий <20 мол. % метилци- анида In(NO3)3-2N2O4 49
>20 мол. % метилци- анида In(NO3)3-2MeCN 49
Висмут Диметилсульфоксид Bi(NO3)3-3Me2SO 50
Медь 5—30% фенилцианида Cu(NO3)2-2PhCN-4N2O4 51
Этилацетат Cu(NO3)2-N2O4 10
В зависимости от свойств металла, растворителя и от кон-
центрации последнего образуются различные соединения. В табл. 2
приведены некоторые типичные примеры продуктов взаимодей-
11 Заказ № 547
170
Глава 6
ствия. Возможность получения нитрата металла из сольвата за-
висит от прочности связи координированных молекул раствори-
теля с металлом. Это в свою очередь зависит от свойств металла и
растворителя, так что каждое соединение представляет собой осо-
бую проблему.
До сих пор были рассмотрены только реакции с металлами.
Следует отметить, что многие галогениды металлов, которые не
реагируют с чистой четырехокисью азота, претерпевают сольволиз
в смесях четырехокиси азота с донорными растворителями, что
составляет еще одно преимущество при использовании таких сме-
сей. Для получения безводных нитратов чаще всего используют
этилацетат и метилцианид. Ниже приведены три методики синтеза
с использованием металлов или галогенидов.
а. Нитрат никеля(11). Металлический никель при комнатной
температуре не реагирует с чистой четырехокисью азота, а также
со смесями четырехокиси азота с этилацетатом или метилцианидом.
Безводный хлорид никеля также не взаимодействует с жидкой че-
тырехокисью азота при комнатной температуре. Поэтому к суспен-
зии хлорида никеля (1 г) в четырехокиси азота (10 мл) добавляют
метилцианид. Количество добавленного метилцианида не имеет
особого значения, но реакция протекает быстрее, если добавляют
сравнительно небольшие количества метилцианида (рис. 2). Сра-
зу же начинается реакция с выделением нитрозилхлорида. Хлорид
никеля медленно растворяется с образованием прозрачного зеле-
ного раствора, и реакция завершается за 48 час при комнатной
температуре. Избыток четырехокиси азота и метилцианида отка-
чивают при комнатной температуре. Раствор из зеленого становится
синим, и из него постепенно выкристаллизовывается темно-синее
вещество состава Ni(NO3)2-3MeCN [48]. При продолжительном
пребывании в вакууме вещество превращается в бирюзово-зеле-
ный твердый Ni(NO3)2-2MeCN. Последний разлагается в вакууме
при температуре 170° с образованием чистого зеленого порошка
безводного Ni(NO3)2.
Нитрат никеля можно получить также сольволизом хлорида
никеля в смеси четырехокиси азота с этилацетатом. При этом обра-
зуется соединение Ni(NO3)2-N2O4 [48].
б. Нитрат меди(П). К смеси четырехокиси азота (20 мл) с
сухим этилацетатом (20 мл) в стеклянной пробирке, закрытой
притертой пробкой с трубкой, наполненной пятиокисью фосфора,
добавляют свежую стружку чистой меди (10 г). При этом начинает-
ся энергичное выделение окиси азота, и при комнатной температу-
ре реакция заканчивается за 4 час. К образовавшемуся синему
раствору добавляют избыток жидкой четырехокиси азота для пол-
ного осаждения Cu(NO3)2-N2O4 в виде сине-зеленого тонкого
кристаллического осадка. Порошок собирают на стеклянном по-
II. Реакции с использованием четырехокиси азота
171
ристом фильтре, промывают несколько раз небольшими порциями
четырехокиси азота и переносят (в сухом боксе) в пробирку раз-
мером 17x3 см, откуда в вакууме удаляют избыток четырехокиси
азота.
Для термического разложения полученного соединения про-
бирку погружают в масляную баню и, поддерживая вакуум, мед-
ленно повышают температуру до 120°. При этом образуется синий
аморфный порошок безводного нитрата меди(П) [10]. В кристал-
лической форме это соединение можно получить сублимацией.
Для этого на поверхность порошка на дне пробирки кладут слой
(толщиной 0,5 см) чистой сухой стеклянной ваты. Пробирку по-
гружают в масляную баню так, чтобы уровень масла был на 3 см
выше стеклянной ваты, и снова откачивают. По достижении тем-
пературы бани 200° нитрат меди быстро сублимируется, причем
сублимат собирается на стенках пробирки примерно на 0,3 см
выше уровня масла. Можно получить также глубоко-синие кри-
сталлы, собирая их на охлаждаемом стеклянном пальце, введен-
ном в пробирку до этого уровня. При вакууме 10-3 мм pm. cm.
процесс сублимации 10 г нитрата меди идет приблизительно 4 час.
Остаток составляет лишь около 5% исходного вещества. Возможна
сублимация при значительно более низких температурах, но она
протекает медленнее. Так, при температуре 130° за 2 час можно
собрать 100 мг вещества, а еще меньшие количества можно соб-
рать даже при температуре 100°. Нитрат меди вначале конденси-
руется в виде красивых игольчатых кристаллов, пригодных для
рентгеновского кристаллографического исследования. На послед-
них стадиях сублимации кристаллы становятся толще и образуют
плотную массу [10].
в. Нитрат бериллия. При получении этого соединения встре-
чаются две трудности: во-первых, высокая растворимость нитрата
бериллия в этилацетате, во-вторых, тенденция нитрата бериллия
к разложению с образованием летучего основного нитрата. Без-
водный хлорид бериллия (2 г), который можно приготовить in
situ из металлического бериллия и хлора, растворяют в этилацета-
те (50 мл), после чего в раствор по каплям добавляют четырехокись
азота. Каждое добавление четырехокиси азота сопровождается
энергичным выделением нитрозилхлорида. Добавление четырех-
окиси азота продолжают до тех пор, пока это выделение не прекра-
тится. Можно также хлорид бериллия размешать в жидкой четы-
рехокиси азота и по каплям добавлять в смесь этилацетат. Выде-
ление нитрозилхлорида прекращается, когда растворяется весь
хлорид бериллия. Следы нитрозилхлорида быстро удаляют из
прозрачного раствора, полученного этим методом, в вакууме.
Если на этой стадии попытаться осадить нитрат бериллия добавле-
нием четырехокиси азота, то потребуются очень большие количе-
11*
172
Глава 6
ства последней. Поэтому раствор нагревают до 40—50° и в вакууме
удаляют большую часть этилацетата, пока не останется вязкая смо-
листая масса. После этого к остатку добавляют 100 мл жидкой че-
тырехокиси азота. Постепенно выделяются бледно-желтые кристал-
лы сольвата Be(NO3)2-2N2O4 [14].
Рис. 4. Термическое разложение Be(NO3)2-2N2O«.
Для определения условий разложения этого соединения с
образованием простого нитрата его нагревали в эвакуированном со-
суде и давление выделяющихся паров по мере повышения темпера-
туры определяли по манометру со стеклянной спиралью. Резуль-
таты (рис. 4) ясно показывают, что термическое разложение про-
исходит в несколько стадий. Излом в точке Б свидетельствует о
том, что в температурном интервале АВ происходит последователь-
ное отщепление двух молекул четырехокиси азота. Участок кривой
ВГ соответствует газовым законам, из чего можно сделать вывод,
что нитрат бериллия устойчив до температуры 125°. При
нагревании выше 125° (участок ГД) происходит быстрое разложе-
ние; выделяется двуокись азота и образуется основной нитрат
бериллия, который конденсируется на холодных частях сосуда
в виде кубических кристаллов.
III. Реакции с использованием пятиокиси азота
173
111. РЕАКЦИИ С ИСПОЛЬЗОВАНИЕМ ПЯТИОКИСИ АЗОТА
Использование пятиокиси азота для получения безводных ни-
тратов металлов изучено не так хорошо, как четырехокиси. Рабо-
тать с чистой пятиокисью азота труднее, так как при температу-
рах выше 0° она медленно разлагается на четырехокись и кисло-
род, а число растворителей, с которыми пятиокись не реагирует,
не так велико, как в случае четырехокиси. В чистом виде окись
обычно получают окислением в потоке двуокиси азота избытком
кислорода (предварительно пропущенного через озонатор) с после-
дующей конденсацией или растворением продукта [53]. Чистая
пятиокись азота плавится при температуре 41°, диссоциируя (по-
видимому, так же как и в растворе) на ионы NO+2 и NO“3, нали-
чием которых характеризуется пятиокись в твердом состоянии.
Для многих синтезов не обязательно, вероятно, пользоваться чи-
стой пятиокисью. Удовлетворительные результаты дает и неочи-
щенный продукт, полученный дистилляцией из смеси азотной кис-
лоты и пятиокиси фосфора. Этот продукт обычно содержит немного
азотной кислоты и четырехокиси азота.
А. Пятиокись азота без добавок
Для иллюстрации методов синтеза с применением пятиокиси
азота в этом разделе рассмотрены лишь некоторые типичные при-
меры. Другие примеры приведены в разд. IV.
1. Нитрат циркония(1У)
Нитрат циркония можно получить прямым взаимодействием
безводного тетрахлорида циркония и безводной пятиокиси азота
ZrCl4 + 4N2O6--> Zr(NO3)4 + 4NO2Cl.
Очищать пятиокись для этого синтеза, по-видимому, не обязатель-
но [12]. Ковалентный тетранитрат циркония летуч, и его можно
очистить возгонкой. На свежеприготовленный тетрахлорид цирко-
ния, внесенный в сосуд, охлаждаемый жидким азотом, перего-
няют избыток пятиокиси азота, полученной из дымящей азотной
кислоты, смешанной с пятиокисью фосфора. Смеси пятиокиси азо-
та с тетрахлоридом циркония дают медленно прогреться до комнат-
ной температуры, а затем нагревают ее до 30°. При этой темпера-
туре неочищенная пятиокись азота плавится. Смесь выдерживают
под обратным холодильником в течение 15 мин; за это время про-
исходит реакция и выделяется нитрозилхлорид. После этого систе-
му откачивают и выдерживают при остаточном давлении 0,1 ми
pm. cm. и комнатной температуре в течение часа для удаления из-
бытка пятиокиси. Остающееся после этого желтое твердое веще-
174
Глава 6
ство имеет состав Zr(NO3)4, 0,4N2O5, 0,6N2O4. Связанные окислы
азота удаляются при нагревании вещества до 100° и остаточном
давлении 0,01 мм рт. ст. в течение 4 час, после чего безводный
Zr(NO3)4 начинает медленно сублимироваться. Его собирают в ви-
де бесцветных кристаллов на охлаждаемом водой пальце.
2. Нитрат титана(1У)
Пятиокись азота является, по-видимому, одним из немногих
реагентов, способных отнимать воду от кристаллогидратов нитра-
тов металлов без разложения последних. Этот метод использовали
для получения безводных нитратов меди, гафния, циркония и ти-
тана, и, возможно, он будет хорошим общим методом получения
безводных нитратов [16].
Для синтеза соединения титана раствор трихлорида титана(Ш)
обрабатывают гидроокисью аммония, и выделившуюся гидрати-
рованную гидроокись титана растворяют в дымящей азотной кис-
лоте. Раствор осторожно выпаривают досуха, в результате чего
образуется гидратированный нитрат титана(1У). Непосредственно
на это вещество, содержащееся в сосуде, охлаждаемом жидким
азотом, перегоняют избыток пятиокиси азота (полученной из сме-
си дымящей азотной кислоты и пятиокиси фосфора). Далее синтез
ведут так же, как синтез нитрата циркония(1У), имея в виду толь-
ко, что нитрат титана (IV) значительно более летуч. После откачи-
вания при комнатной температуре вещество нагревают приблизи-
тельно до 36°. При этой температуре тетранитрат начинает
сублимироваться при остаточном давлении 0,03 мм рт. ст., и его
собирают на охлаждаемом водой пальце в виде крупных кубических
кристаллов. При более высоких температурах сублимация про-
исходит быстрее, и при 100° (остаточное давление 0,02 мм рт. ст.)
можно собрать до 2 г вещества в течение часа [161.
3. Нитрат уранила
Интересная гетерогенная реакция между двумя твердыми фа-
зами происходит при перемешивании трехокиси урана и чистой
пятиокиси азота [53]. Активную форму [37] трехокиси урана
(0,0028— 0,0036 моля), просеянную через сито 325 меш, перемеши-
вают с порошкообразной чистой пятиокисью азота (0,09 моля)
при температуре 0—10°. Смесь следует интенсивно перемешивать,
причем удобно для этой цели пользоваться магнитной мешалкой.
Во время опыта над светло-оранжевой смесью пропускают ток
озона. Приблизительно через 10 мин окраска внезапно меняется на
ярко-желтую, причем не происходит заметного выделения тепла.
В течение последующих 1—2 мин образуется мелкий желтый сыну-
IV. Сравнительная оценка четырехокиси и пятиокиси азота
175
чий кристаллический порошок. После удаления избытка N2O5
при температуре 25—30° и остаточном давлении 10~4 .« рт. ст.
образуется вещество состава UOa(NO3)2-N2O5. Вероятно, реакция
в действительности происходит между ,UO3 и парами пятиокиси
азота, а перемешивание порошков лишь способствует поддержанию
высокой концентрации паров N2O5 у поверхности частиц UO3.
Если полученный сольват UO2(NO3)2-N2Os прогреть прибли-
зительно в течение 200 мин при температуре 125—130° и остаточ-
ном давлении 10~4 л<л1 рт. ст., то получается чистый нитрат
уранила. Особенно интересно то, что соединение UO2(NO3)2-
•N2O5 имеет меньшую термическую устойчивость, чем иОг(КОз)2-
•N2O4, которое при этих условиях диссоциирует неполностью
(разд. II.Б.).
Б. Пятиокись азота в чистой азотной кислоте
Попытки синтезировать безводные нитраты, используя 100%-
ную азотную кислоту без добавок, оказались малоплодотворными,
но для некоторых синтезов вполне подходящей и удобной реак-
ционной средой оказались растворы пятиокиси азота в чистой
азотной кислоте.
1. Нитрат тория(1У)
К 10—20 мл безводной азотной кислоты при 25° добавляют
1—2,5 г ТЬ(КОз)4-4НгО и туда же перегоняют пятиокись азота.
После того как сконденсируется достаточное количество пятио-
киси, смесь разгоняют в вакууме для удаления азотной кислоты
и непрореагировавшей пятиокиси азота. Важно, чтобы в начале
синтеза в реакционную смесь был введен достаточный избыток
пятиокиси азота. Если введено менее 5,5 моля N2O5 сверх того,
что требуется для реакции с водой исходного тетрагидрата, то по-
лучается загрязненный продукт, содержащий четырехокись азота
и некоторое количество воды. При введении 5,5—8,2 моля избы-
точной N2O5 получаются твердые белые гранулы состава
Th(NOs)4-2N2O5 [52]. Это соединение нагревают в течение 4—
5 час при 150—160° и остаточном давлении 10-5 .о рт. ст., причем
N2O5 полностью удаляется и получается чистый Th(NOg)4 в виде бе-
лого порошка.
IV. СРАВНИТЕЛЬНАЯ ОЦЕНКА ЧЕТЫРЕХОКИСИ
И ПЯТИОКИСИ АЗОТА
Подробный обзор известных безводных нитратов металлов
. имеется в работе [1 ]. Однако из сказанного здесь ясно, что во мно-
гих случаях одни и те же вещества можно получить, используя в
176
Глава 6
качестве реагента и N2O5 и N204. В некоторых случаях пятиокись
азота обладает явными преимуществами. Это относится особенно к
случаям, когда металл может существовать в нескольких состоя-
ниях окисления и легко образует оксинитраты; поэтому пятиокись
азота оказалась особенно полезной для синтеза безводных нитра-
тов металлов, стоящих в начале ряда переходных металлов. Если
металл может образовать и нитрат и оксинитрат, часто при реак-
циях с участием пятиокиси легче образуется нитрат. Например,
галогениды титана и циркония реагируют с четырехокисью азота
с образованием оксинитратов TiO(NOa)2 и ZrO(NOa)2 [54], однако
при взаимодействии с пятиокисью азота получаются нитраты
Ti(NO3)4 и Zr(NOa)4, как это было отмечено выше. В тех случаях,
когда могут образоваться несколько оксинитратов, при взаимо-
действии с пятиокисью обычно образуются соединения с более
высоким содержанием нитратных групп. Это будет показано в
дальнейшем на примере оксинитратов ванадия.
Для разработки препаративных методов важное значение име-
ет относительная устойчивость соединений, образующихся в ре-
зультате присоединения четырехокиси или пятиокиси азота к
нитратам металлов. Как правило, продукты присоединения пятио-
киси менее устойчивы, чем продукты присоединения четырехоки-
си, так что безводные нитраты легче получить термическим разло-
жением первых. Это уже было отмечено в связи с рассмотрением
продуктов присоединения окислов азота к нитрату уранила (разд.
III.А), а недавние работы по синтезу нитрата хрома являются яр-
ким примером, иллюстрирующим большую разницу в способности
этих двух окислов азота к комплексообразованию [55, 56].
А. Соединения хрома
Карбонил хрома взаимодействует как с пятиокисью, так и с
четырехокисью азота. Даже в присутствии избытка пятиокиси азо-
та нитрат хрома(Ш) выделяется в чистом виде, без присоединен-
ной пятиокиси [56]. При использовании четырехокиси неизменно
образуется соединение Сг(МОз)з-2КгО4, причем оно 'настолько
устойчиво, что до сих пор не найдено способа удаления присое-
диненной четырехокиси азота без одновременного разложения
нитрата металла [55].
1. Нитрат хрома(Ш)
Пятиокись азота, выделяющуюся из смеси пятиокиси фосфора
и дымящей азотной кислоты, обрабатывают озонированным кисло-
родом для окисления примеси четырехокиси. Ток газа вводят в
100 мл сухого четыреххлористого углерода так, чтобы получился
IV. Сравнительная оценка четырехокиси и пятиокиси азота
177
практически бесцветный раствор, содержащий 3,0—3,5 г пятио-
киси азота. Медленно при перемешивании добавляют раствор кар-
бонила хрома (1 г) в четыреххлористом углероде (100 лм). Сразу
же начинается реакция: выделяются газы и образуется зеленая
суспензия. Сосуд закрывают пробкой с осушительной трубкой,
наполненной пятиокисью фосфора, и оставляют на 12 час для коа-
гуляции и осаждения суспензии. Осадок затем отфильтровывают
на фильтре, защищенном от контакта с атмосферой, и промывают
четыреххлористым углеродом. После удаления в вакууме четырех-
хлористого углерода на фильтре остается светло-зеленый порошок,
имеющий точный состав Сг(КОз)з. По уравнению
Сг(СО)6 + 3N2O6---т Cr(NO3)3 + 6СО + 3NO2
на 1 моль карбонила хрома расходуется 3 моля пятиокиси, а для
синтеза берут 6 молей пятиокиси на 1 моль карбонила хрома.
Несмотря на такой избыток пятиокиси, никаких признаков обра-
зования сольвата нитрата хрома(Ш) с пятиокисью азота ни на
какой стадии реакции не обнаружено. Для окончательного высуши-
вания требуется лишь непродолжительное пребывание вещества
в вакууме при комнатной температуре, так что если перед высу-
шиванием в веществе и имеется некоторое количество сольвата,
его устойчивость должна быть очень невелика.
2. Соединение нитрата хрома(Ш) с двумя молекулами
четырехокиси азота
Тонко измельченный карбонил хрома вводят в избыток жидкой
четырехокиси азота при комнатной температуре. Карбонил мед-
ленно растворяется, и из раствора выделяется серый порошок.
Реакция идет медленно, и реакционной смеси поэтому дают стоять
несколько суток. После удаления четырехокиси азота в вакууме
остается сыпучий серый порошок состава Сг(МОз)з-2К2О4.
Это соединение начинает разлагаться с заметной скоростью при
температуре 40°. Исследование процесса разложения при помощи
химических анализов, измерений давления паров и термограви-
метрического анализа [55] не обнаружило никаких признаков
образования простого нитрата при термическом разложении про-
дукта взаимодействия. Сам по себе нитрат хрома(Ш) при темпе-
ратурах выше 50° неустойчив [56], так что попытки получить это
вещество из сольвата с четырехокисью азота ограничивались низ-
кими температурами, однако устойчивость продукта присоеди-
нения четырехокиси в этих условиях настолько высока, что до сих
пор не найдено эффективного метода удаления из него четырех-
окиси.
178
Глава 6
Б. Соединения ванадия
Нитрат ванадия VfNChJs неизвестен, но выделены два окси-
нитрата УО(КОз)зи VO2(NO3). На примере методов синтеза этих
соединений видно, как состав продуктов зависит от вида окиси
азота, используемой для синтеза.
1. Окситринитрат ванадия
Пятиокись азота взаимодействует с пятиокисью ванадия сле-
дующим образом:
V2O5 + 3N2O5 --2VO (NO3)3.
Окситринитрат желтого цвета плавится при температуре 2°
и кипит при 68—70° в глубоком вакууме [И, 57]. Подробных све-
дений о методе синтеза этого соединения в литературе нет. При
реакции с УОС1з получается тот же продукт.
2. Диоксимононитрат ванадия
Это соединение образуется в результате реакций с участием
четырехокиси азота
V-}-2N2O4(b MeCN)---->- VO2(NO3) + 3NO.
Стружку электролитически чистого ванадия (5 г) вводят в
75—100 мл четырехокиси азота в охлажденной до 0—5° колбе.
При добавлении 5—8 мл ацетонитрила начинается реакция и обра-
зуется осадок. Реакционную смесь оставляют на 5—6 час, после
чего в вакууме отгоняют четырехокись азота и ацетонитрил. Про-
дукт представляет собой кирпично-красный порошок состава
VO2(NO3) [58].
V. НИТРАТЫ ГАЛОГЕНОВ
Несмотря на то что в литературе нет подробных сведений об
опытах с использованием нитратов галогенов для синтеза безвод-
ных нитратов металлов, Шмейсер и Брэндл [59] составили
обзор работ о этому вопросу. Этот метод может быть
особенно ценным в тех случаях, когда необходимо получить
нитраты, устойчивые только при очень низких температурах. По
мере того как синтезируется' все больше нитратов металлов, ста-
новится все яснее, что температурные пределы устойчивости этих
соединений могут очень сильно изменяться в зависимости от таких
факторов, как электронная структура металлов, устойчивые ва-
лентные состояния, число нитратных групп в соединении и способ-
V. Нитраты галогенов
179
ность металла образовывать полимеры вида М—О—М. Это
видно уже при простом сопоставлении температур разложения
нитратов щелочных металлов (475—600°); кальция, стронция и
бария (575—675°); кобальта, никеля, меди и цинка (270—350°),
бериллия (125°) и хрома (50°). Если бы можно было осуществить
реакцию при достаточно низких температурах, вероятно, удалось
бы получить нитраты металлов, неизвестные в настоящее время. С
этой точки зрения нитраты галогенов, особенно нитрат хлора,
обладают определенными преимуществами. Нитрат хлора полу-
чают совместной конденсацией моноокиси хлора и пятиокиси азо-
та при охлаждении жидким воздухом
С12О -f- N2O3---► 2C1NO3,
Для завершения реакции смесь выдерживают в течение нескольких
часов при температуре —80° [60]. Соединение можно получить
также взаимодействием моноокиси хлора с четырехокисью азота
161 ]
2С12О + N2O4----> 2C1NO3 + Cl2.
Нитрат хлора представляет собой желтую жидкость с температу-
рой плавления —107°, температурой кипения -[-18° (по экстра-
поляции) [62]. Можно считать, что атом хлора в этом соеди-
нении несет частичный положительный заряд СР +—ONO8 “г,
так что при взаимодействии с другими веществами это соединение
является поставщиком ионов нитрата [И, 59]. Возможным даль-
нейшим преимуществом использования нитрата хлора является то,
что реакции с участием этого вещества не будут осложняться иона-
ми NO+, или NO+a, которые неизбежно присутствуют при реакциях
с участием N2O4 или N2O5.
Вследствие низкой температуры плавления реакции с участием
нитрата хлора, например
TiCl4 + 4C1NO3---> 4С12 + Ti(NO3)4,
можно проводить при температуре твердой двуокиси углерода
(—80°), причем хлор и избыток нитрата хлора можно удалять в
вакууме при этой же температуре [11, 59]. Есть сведения, что этим
способом при указанных температурах можно синтезировать
B(NO3)3 (-78°), A1(NO3)3 (-7°) и Sn(NO3)4 (-60°).
Для получения тетранитрата олова используют также трини-
трат брома, который получают следующим образом:
BrF3 + 3N2O5---->- Br(NO3)3 + 3NO2F
или
BrF3 + 3HNO3-----> Br(NO3)3 + 3HF.
180
Глава 6
Однако тринитрат брома плавится с разложением при 48° и не об-
ладает благоприятными физическими свойствами, присущими три-
нитрату хлора [11, 59].
ЛИТЕРАТУРА
1. Addison С. С., Logan N., Advan. Inorg. Chem. Radiochem., 6,
to be published.
2. Wa] 1 work S. C., Proc. Chem. Soc., 1959, 311; J. Chem. Soc., to be
published; Duffin B., Wall work S. C., Acta Cryst., 16, A33
3. A d d i s о n С. С., К i 1 n e r M., W о j c i c k i A., J. Chem. Soc.,
1961, 4839.
4. C 1 a r k H. С., О ’В r i e n R. J., Inorg. Cbem., 2, 740 (1963).
5. A d d i s о n С. С., К i 1 n e r M., unpublished results.
6. H о a r d J. L., S t г о u p e J. D., Atomic Energy Project Report 1943,
A1229; Zacharia sen W. H., Acta Cryst., 7, 795 (1954).
7. Gatehouse В. M., С о m у n s A. E., J. Chem. Soc., 1958, 3965;
A 1 1 p r e s s J. G., H a m b 1 у A. N., Australian J. Chem., 12, 569
(1959); Вдовенко В. M., Строганов С. В., Соколов А.Р.,
Радиохимия, 1, 97 (1963) и ссылки в этой работе.
8. S t г a u b D. К., Drago R. S., Donoghue J. Т., Inorg. Chem.,
1, 848 (1962).
9. A d d i s о n С. G., Walker A., Proc. Chem. Soc., 1961, 242.
10. A d d i s о n С. C., Hathaway B. J., J. Chem. Soc., 1958, 3099.
11. Schmeisser M., Angew. Cbem., 67, 493 (1955); Schmeisser
M., Brandie K., Angew. Cbem., 69, 781 (1957).
12. F i e 1 d B. 0., Hardy C. J., Proc. Chem. Soc., 1962, 76.
13. Addison С. C., Hathaway B. J., Logan N., Walk er A.,
J. Chem. Soc., 1960, 4308.
14. A d d i s о n С. C., Walker A., J. Chem. Soc., 1963, 1220.
15. A d d i s о n С. C., Advan. Chem. Ser., 36, 131 (1962).
16. F i e 1 d В. O., Hardy C. J., J. Chem. Soc., 1963, 5278.
17. Naumann A., Chem. Ber., 37, 4333 (1904).
18. G u n t z A., Martin M., Bull. Soc. Chim. France, 7, 313 (1910).
19. Hathaway B. J., Underhill A. E., J. Chem. Soc., 1960, 648.
20. J a n d e r G., Wendt H., Z. anorg. allgem. Chem., 258, 1 (1949).
21. J and er G., В г о d e r s e n K., Z. anorg. allgem. Cbem., 262,33 (1950).
22. A u d r i e t h L. F., Jukkola E. E., Meints R. E., Hop-
kins B. S., J. Am. Chem. Soc., 53, 1805 (1931).
23. Addison С. C., Allen J., Bolton H. C., Lewis J.,
J. Cbem. Soc., 1951, 1289.
24. Addison С. C., Thompson R., J. Chem. Soc., 1949, 218.
25. С 1 u s i u s К., V e с c h i M., Helv. Chim. Acta, 36, 930 (1953).
26. Millen D. J., Watson D., J. Chem. Soc., 1957, 1369.
27. G о u 1 d e n J. D. S., Millen D. J., J. Cbem. Soc., 1950, 2620.
28. Moeller T., A f t a n d i 1 i a n V. D., J. Am. Chem. Soc., 76, 5249
(1954); M о e 1 1 e r T., A f t a n d i 1 i a n V. D., Cullen G. W., in
Inorganic Syntheses, Vol. 5, Moeller T., ed., McGraw-Hill, New York,
1957, p. 37.
29. Addison С. C., Thompson R., Nature, 162, 369 (1948).
30. Addison С. C., Thompson R., J. Chem. Soc., 1949, 211.
31. Addison С. C., Lewis J., Thompson R., J. Chem. Soc.,
1951, 2829.
32. Addison С. C., Champ H. A. J., unpublished results.
Литература
181
33. Addison С. С., Lewis J., Thompson R., J. Chem. Soc.,
1951, 2838.
34. Addison C.C., Hodge N., Lewis J., J. Chem. Soc., 1953, 2631.
35. A d d i s о n С. C., Sheldon J. C., Hodge N., J. Chem. Soc.,
1956, 3900.
36. Addison С. C., Hodge N., J. Chem. Soc., 1964, to be published.
37. Gibson G., Katz J. J., J. Am. Chem. Soc., 73, 5436 (1951).
38. J e z о wska-Tr z eb i a t о wska В., К e d z i a B., Bull. Acad.
Polon. Sci., Ser. Sci Chim., 10, 213 (1962).
39. Addison С. C., Hodge N., J. Chem. Soc., 1961, 2490.
40. Addison С. C., Sheldon J. C., J. Chem. Soc., 1958, 3142.
41. A d d i s о n С. C., Sheldon J. C., J. Chem. Soc., 1956, 1941.
42. Addison С. C., Sheldon J. C., J. Chem. Soc., 1956, 2709.
43. Rubin B., S i s 1 e г H. H., Schechter H., J. Am. Chem. Soc.,
74, 877 (1952).
44. Addison С. C., Conduit С. P., Thompson R., J. Chem.
Soc., 1951, 1303.
G. C.,
C. C.,
C. C.,
Sheldon J. С., J. Chem. Soc., 1956, 2705.
Logan N., unpublished results.
Hathaway B. J., Sutton D., unpublished
45. Addis
46. Addis
47. A d d i
results.
48. A d d i
49. A d d i
о
о
о
n
n
n
s
s
n
C. C.,
C. C.,
D. K.,
Johnson B. F. C ., unpublished results.
Smith В. C., unpublished results.
Sisler H. H., R у s c h к e w i t s c h G. E.,
о
son
50. S t r a u b
J. Inorg. Nucl. Chem., 24, 919 (1962).
51. A d d i s о n С. C., Hathaway B. J., unpublished results.
“ ~ - - ” * ’ Gibson G., J. Am. Chem.
52.
Ferraro J. R., Katzin L. I.,
Soc., 77, 327 (1955).
Gibson G., Beintema C. D.,
Katz J. J., J. Inorg. Nucl.
53.
Chem., 15, 110 (1960).
54. Gutmann V., Tannenberger H., Monatsh. Chem., 87, 421
(1956).
55. A d d i s о n С. C., N о r b u г у A. H., unpublished results.
56. A d d i s о n С. C., Chapman D. J., J. Chem. Soc., 1964, 539.
57. Schmeisser M., Liitzow D., Angew. Chem., 66, 230 (1954).
58. Panontin J. A., Fischer A. K., Heintz E. A., J. Inorg.
Nucl. Chem., 14, 145 (I960).
59. Schmeisser M., В r a n d 1 e K., Angew. Chem., 73, 388 (1961).
60. Schmeisser M., Fink W., Brandie K., Angew. Chem., 69,
780 (1957).
61. Martin H., Jacobsen Th., Angew. Chem., 67, 524 (1955).
62. Martin H., Angew Chem., 70, 97 (1958).
ГЛАВА 7
ГАЛОГЕНО- И ГАЛОГЕНОИД-ПРОИЗВОДНЫЕ
СИЛАНОВ
Мак-Дайармид
Пенсильванский университет, Филадельфия
1. ВВЕДЕНИЕ
Силаны можно рассматривать в качестве кремниевых аналогов
алканов. Силаны образуют гомологический ряд соединений об-
щей формулой SinH2n4-2- Несмотря на то что выделены все соеди-
нения этого ряда от п=1 до п=8, изучены были только силан
SiH4, дисилан Si2H6 и трисилан Si3H6 [1—4]. Были также иден-
тифицированы изомеры некоторых высших силанов [5].
Известно большое число галогено- и галогеноид-производных
силанов. В последнее время было синтезировано несколько моно-
галогенодисиланов. Моногалогено-производные силана и дисилана
имеют большое значение, так как они служат исходным материа-
лом для синтеза многих производных, содержащих силил-группу
—SiH3 или дисиланил-группу —SibhSiHj соответственно. Обе
эти группы очень интересны, так как соединения силила можно
рассматривать в качестве кремниевых аналогов соединений мети-
ла, и точно так же соединения дисиланила можно рассматривать
как кремниевые аналоги соединений этила. Соединения силила и
дисиланила не рассматриваются в этой главе. Они описаны в мо-
нографиях [1, 1а] ив обзорных статьях [2, 3].
Многие органические и неорганические соединения кремния
можно рассматривать в качестве производных галогеносиланов.
Например, соединение SiHsI — иодосилан или силилиодид можно
представить как член ряда CHsSiHal, (CHs^SiHI и (СНз)зЗН,
в котором один, два или три атома водорода замещены метильной
группой. Подобным же образом если атомы водорода замещают ато-
мами галогена, например хлора, с образованием соединений
CISiHal, CbSiHI и CisSil, то их можно рассматривать в качестве
производных SiHsI. Эта точка зрения применима и в случае
(CaHsO^SiHCl и других подобных соединений. Отсюда ясно, что
в одной главе невозможно описать множество соединений, которые
можно классифицировать в качестве галогено- и галогеноид-
производных силанов. Поэтому обсуждение ограничено неорга-
I. Введение
183
ническими соединениями, хотя в некоторых случаях будут затро-
нуты и органические соединения кремния*.
В данном разделе рассмотрены только соединения кремния с
галогеном и кремния с галогеноидом, которые, помимо этих свя-
. зей, имеют еще одну или более связей кремний — водород и крем-
ний — кремний. Таким образом, опущена довольно большая груп-
па соединений, которая содержит другие элементы, связанные
с кремнием, например (SiBrs^O [12], [Si(NCO)3]2O [13],
(SiCl3)3N [14], (SiCl3)2S [15] и т. д.
Все соединения, описанные в дальнейшем, склонны к гидроли-
зу (даже во влажном воздухе), а некоторые, например дисиланил-
галогениды, воспламеняются на воздухе. По этой причине, а так-
же потому, что в течение многих лет для синтеза таких соединений,
как SiHsI, использовали силан, который воспламеняется на воз-
духе, синтез многих соединений проводят в вакууме. До послед-
него времени получение относительно больших' количеств этих
соединений, например SH3I, было очень сложным. Для получения
из них других производных силила довольно существенно приме-
нение вакуумной техники. При работе с небольшими количествами
веществ количество материала, необходимого для детального изу-
чения реакции в вакуумной системе, обычно значительно меньше,
чем при работе в обычных условиях. В настоящее время SH3I
можно получить в относительно больших количествах, и, следова-
тельно, теперь можно использовать его в определенных реакциях
с применением обычных лабораторных приемов. Некоторые про-
изводные гидридов кремния, содержащие связи кремний — водо-
род, например SiHCl3, относительно легко можно синтезировать в
больших количествах [16, 17] или купить. Следовательно, не-
смотря на то что для проведения многих операций с производными
силанов требуется применение вакуумной техники, во многих слу-
чаях можно пользоваться обычными химическими препаративными
методами. Некоторые работы по синтезу, которые были проведены
в прошлом с применением вакуумной техники из-за малых коли-
честв имеющегося в наличии материала, теперь можно проводить
без вакуума. Однако при этом необходимо соблюдать определен-
ные меры предосторожности, бывшие необязательными при выпол-
нении работ в вакууме. В частности, в соединениях, содержащих
группу —SiH3, могут присутствовать небольшие количества сила-
на, который может быть причиной сильных взрывов.
Несмотря на то что имеется ряд руководств [18—23], где из-
ложены приемы работы с летучими материалами и способы про-
* Более точное описание неорганических или органических соединений
кремния содержится в ряде книг и обзорных статей [1—4, 6—11], в которых
излагаются различные аспекты этого большого раздела химии.
184
Глава 7
ведения реакций в вакуумной системе, эти методы трудно освоить
без специального инструктажа. Поэтому не рекомендуется присту-
пать к работе с такими взрывоопасными веществами, как силан
или дисилан (при наличии даже следов воздуха в вакуумной си-
стеме), прежде чем оператор основательно не ознакомится и не
освоит на практике все необходимые манипуляции. Настоятельно
рекомендуется при работе со взрывоопасными веществами приме-
нять в качестве хладоагента жидкий азот, а не жидкий воздух или
жидкий кислород. Если ловушка даст трещину при погружении в
жидкий воздух или кислород, вероятность взрыва значительно
возрастает.
11. ОБЩИЕ МЕТОДЫ СИНТЕЗА
Существует несколько общих типов реакций, используя кото-
рые можно синтезировать галогено- и галогеноид-производные
силанов. Эти типы реакций и их применение для синтеза характер-
ных соединений кремния кратко рассматриваются в данном разде-
ле. Менее общие типы реакций описаны в следующем разделе.
Следует помнить, что все галогено- и галогеноид-производные
силанов легко гидролизуются даже влагой, содержащейся в воз-
духе, поэтому нужно всегда принимать надлежащие меры, предот-
вращающие гидролиз. Некоторые соединения со связями кремний —
кремний и кремний — водород самовоспламеняются на возду-
хе. Те из них, для которых это свойство не типично, могут также
содержать или образовывать в результате разложения воспламе-
няющиеся продукты; они требуют поэтому осторожного обращения.
Способность к воспламенению многих соединений, которые были
синтезированы с применением вакуумной техники, не проверена.
Поэтому правильнее рассматривать их как воспламеняющиеся до
тех пор, пока не будет изучена их природа.
А. Из кремния или силицидов
При нагревании все элементарные галогены реагируют с крем-
нием или силицидами металлов с образованием тетрагалогенидов
кремния, а в некоторых случаях с образованием галогенидов выс-
ших силанов.
Летучие соединения со связями кремний —водород можно очень
легко получить с использованием до некоторой степени аналогич-
ных реакций, протекающих при нагревании кремния или силицида
металла в токе соответствующего безводного газообразного га-
логеноводорода. Этим методом с хорошим выходом были получены
тригалогеносиланы: SiHCls [16, 17, 24], SiHBis [25, 26] и SiHIs
[27]. Тетрагалогениды и галогениды высших силанов можно
II. Общие методы синтеза
185
также синтезировать при помощи этих реакций, используя сили-
циды, например ферросилиций [28], силицид меди [1, 29] и т. д.
Если в реакции описанного выше типа галогеноводород находится
в смеси с водородом, то, помимо тригалогеносиланов, могут
образовываться дигалогеносиланы [1 ]. При нагревании кремний
реагирует также с рядом органических галогенидов (в присутст-
вии таких катализаторов, как металлическая медь) с образова-
нием органогалогеносиланов, например (CHsJaSiCh [9, 30] или
CHsSiHCh [31 ] и т. д. Этот способ является основным для промыш-
ленного получения органогалогеносиланов, используемых в боль-
ших количествах для приготовления полисилоксанов.
Б. Из соединений со связью кремний — кремний
В некоторых случаях для получения производных кремния,
содержащих определенные галогены, практикуют расщепление
связи кремний —кремний в ди- и трисиланах. Так, при нагревании
Si2Fe с хлором образуется FgSiCl [32]:
Si2Fe + Gl2---> 2F3SiCl. (1)
Реакция осложнена одновременным образованием других соеди-
нений, например SiFaCh. Главное неудобство метода заключается
в том, что соединения, содержащие связь кремний — кремний,
обычно гораздо менее доступны, чем производные силанов. Кроме
того, применение метода ограничено соединениями, в которых от-
сутствуют связи кремний — водород, так как эти связи будут
галогенироваться одновременно со связями кремний — кремний.
В. Из соединений со связью кремний — водород
Атом водорода, связанный с атомом кремния, можно непосред-
ственно заместить галогеном при реакции гидрида кремния с
элементарным галогеном или с газообразным галогеноводородом
в присутствии галогенида алюминия в качестве катализатора. Эти
типы реакций, особенно последний, были хорошо изучены для всех
галогенов, кроме фтора.
Реакции элементарных галогенов со связями кремний — водо-
род вообще протекают значительно энергичнее и менее контроли-
руемо, чем соответствующие реакции с галогеноводородом и гало-
генидом алюминия [4]. Как и следовало ожидать, энергия взаимо-
действия ослабевает при переходе от хлора к брому и от брома к
иоду. Скорость реакции также снижается с уменьшением числа свя-
зей кремний — водород в молекуле [2, 33]. Реакция хлора и бро-
ма с SiH4 при комнатной температуре протекает очень бурно [34],
однако при низких температурах можно добиться более медлен-
186
Глава 7
ного и контролируемого процесса. Например, при —80° твердый
бром реагирует с SiH4 с образованием SiHsBr и некоторого коли-
чества SilbBn [34]
SiH4+Br2------> SiH3Br + HBr. (2)
При проведении реакции в инертном растворителе [33 ] можно осу-
ществить частичное замещение водорода галогеном в кремний-
органических гидридах
C6H5SiH3 + Вг2----> CeH6SiH2Br + НВт. (3)
Наиболее распространенный метод контролируемого замещения
водорода (в молекуле SiH4) галогеном заключается во взаимодей-
ствии SiH4 с газообразным галогеноводородом в присутствии без-
водного галогенида алюминия в качестве катализатора [2].
Для SiH4 было показано, что реакция не идет даже при нагре-
вании, если отсутствует катализатор [35]. В общем случае хо-
роший выход получают при продолжительности реакции 6—
24 час при 100° и атмосферном давлении или ниже. Изменяя коли-
чественные соотношения SiH4 и галогеноводорода, можно получить
любой из продуктов замещения — SiHsX, SiHzXz, SiHXs или
SiX4— с хорошим выходом; однако всегда будут присутствовать
набольшие количества других продуктов замещения [35, 36].
А1,С1.
SiH4+HCl------> SiH3Cl + Н2. (4)
Подобный тип реакции имеет место в случае кремнийорганиче-
ских гидридов. Этим способом, например, CHeSiHe можно превра-
тить в CHsSiHzCl [35]. Иногда в отсутствие катализатора га-
логенида алюминия галогеноводороды вступают в реакцию с сое-
динениями, содержащими связи кремний — водород. Так, при
120° иодистый водород и С(.Н581Нз, помимо других продуктов,
дают CeHoSiHal 137]. Как показано в разд. II.Ж, в настоящее
время стали доступны более совершенные методы синтеза силил-
галогенидов по сравнению с описанными выше (при которых ис-
пользовали взрывоопасный силан). Однако последний метод яв-
ляется пока единственно известным для получения моногалогено-
производных дисилана.
А121в
Si2H6 + HI----> Si2H5I + H2e (5)
Следует отметить, что дисилан реагирует с HBr, НС1 и HI [38, 39]
при более низкой температуре и с большей скоростью, чем силан.
Наиболее распространенный метод прямого замещения водо-
рода галогеном или галогеноидом по связи кремний — водород
заключается в восстановлении галогенида или галогеноида метал-
II. Общие методы синтеза
187
ла, например HgCh, AgCl, C11CI2, AgNCO и т. д. связью кремний —
водород [40—42]. Соль металла добавляют порциями к избытку
чистого силана при комнатной температуре или при температуре
кипения (с обратным холодильником) или к кипящему раствору
силана в инертном растворителе (например, в четыреххлористом
углероде)
H-C7H15SiH3 4- HgBr2---> H-C7H15SiH2Br Hg + НВт, (6)
цикло-СвНцВхНз + 2AgNCO----> miKno-C6HnSiH2NCO + 2Ag +HNCO. (7)
Этим методом можно заместить более одного атома водорода, если
соль металла взята в большом избытке.
В некоторых аналогичных реакциях органические галогениды
преобразуют связь кремний — водород в связь кремний — гало-
ген. Например, (CzHsJeSiH следующим образом реагирует с
н-СвН13С1 в присутствии катализатора хлорида алюминия при тем-
пературе кипения (с обратным холодильником) [24]:
А12С1в
(С2Н5)3 SiH + я-С6Н13С1 --> (С2Н5)з SiCl + н-СвНм. (8)
Подобные реакции наблюдали также для высших силанов. Напри-
мер, Si3Hs и хлороформ реагируют в присутствии катализатора
хлорида алюминия при 50—70° [43] по реакции
Si3H8 + 4СНС13---> Si3H4Cl4 + 4СН2С12. (9)
Следует отметить, что органогалогеносиланы, например
(СгНа^ЭШС!, были получены смешиванием реактива Гриньяра
с избытком SiHCl3 [44].
Реакции перераспределения, затрагивающие связи кремний —
водород, обсуждаются в разд. II.3.
Г. Из соединений со связью кремний — галоген
Прямое замещение галогена в связи кремний — галоген дру-
гим галогеном, или галогеноидом, или каким-либо другим атомом,
или группой является одной из наиболее употребимых реакций в
химии кремния. Такое замещение можно обычно осуществить при
контакте галогеносилана в газообразном или жидком состоянии с
подходящим галогенидом или галогеноидом металла [2]. В неко-
торых случаях необходим умеренный нагрев, особенно для крем-
нийорганических галогенидов [45—47 ].
Для реакций соединений силила с солями серебра был состав-
лен «ряд превращений», который показывает, в каком порядке
происходит замещение атомов или групп атомов в связях с участи-
ем кремния. Обработка соединения подходящей солью серебра мо-
жет привести к превращению данного соединения в любое другое
188
Глава 7
соединение, стоящее дальше в этом ряду, но ни в одном из пред-
шествующих соединений [3, 45—48, 48а]. Этот ряд составлен на
основании аналогичного ряда, полученного ранее для соединений
триорганосилила [47]. Замещение однотипных соединений силила,
исследованных до настоящего времени, также проводят в этом
порядке.
SiH3I----> (SiH3)2 Se--> (SiH3)2 S---> SiH3Br---->
----> SiH3NCSe----> (SiH3N)2C----> SiH3Cl----> SiH3CN---->
----> SiH3NCS----> SiH3NCO-----> (SiH3)2O----> SiH3F.
Так, SiHsI реагирует c AgCN с образованием SiH3CN [49].
SiH3I + AgCN----> SiH3CN + Agl, (10)
но (SiHg^O не взаимодействует c AgCl с образованием SiH3Cl.
Имеются сведения о подобном ряде превращений для солей рту-
ти(П), однако было изучено лишь несколько реакций [3]. Опре-
деленные превращения имели место в случае взаимодействия крем-
нийорганических соединений с солями ртути(П), свинца(1У),
меди(П) и с солями кадмия [47]. Вполне возможно, что взаимодей-
ствие солей этих и других металлов с неорганическими соедине-
ниями кремния выразится в подобных же превращениях. Несмот-
ря на то что приведенные выше ряды справедливы лишь для соеди-
нений, содержащих группу —SiH3, они кажутся вероятными и
для ди-, три- и тетрагалогеносиланов, а также для дисиланилгало-
генидов. В самом деле, удалось установить, что SiHzH можно
превратить в SiHaCh с довольно хорошим выходом пропусканием
паров силилиодида над хлоридом серебра при комнатной темпе-
ратуре [50]. Дисиланилиодид SizHsI был превращен таким спо-
собом в SiaHsCl [51 ].
Силилиодид является наиболее полезным силилгалогенидом,
так как, по-видимому, его можно превратить в любое другое сое-
динение данного ряда. В самом деле, силилиодид уже был исполь-
зован для синтеза (SiHs^Se [52], SiH3CN [49], SiH3Cl [53],
SiH3NCS [49], SiH3NCO [48], SiH3NCSe [48a], (SiH3N)2C [48a]
и (SiH3)2S [52] с соответствующими солями серебра. Это очень
удобно, поскольку SiH3I теперь синтезируют (разд. III.Г), не
используя силан, который в больших количествах представляет
известную опасность.
Следует иметь в виду, что если в соединениях кремния имеется
связь кремний — водород, то использование фторида серебра для
получения фторида кремния не приводит к удовлетворительным
результатам, так как реакция осложняется окислением по связи
кремний — водород [16]. В этом случае нужно использовать та-
II. Общие методы синтеза
189
кие фториды, как SbFs, SnF4 и TiF4 [16, 54, 55]. Применение
SbFg (с SbCh в качестве катализатора) обеспечивает хороший вы-
ход SiHeF, SiHaFa и SiHF3 из соответствующих хлоридов [16]
3SiH2Cl2 + 2SbF3--> 3SiH2F2 + 2SbCl3. (И)
Однако, как показано в разд. III.А, совершенно чистым продуктом
является только SiHzFz, полученный этим методом; чистый SiH3F
лучше получать другими методами. Трехфтористую сурьму также
можно использовать для частичного замещения галогенов в тетра-
галогенидах кремния. Например, SiCl4 экзотермически реагирует
с недостаточным количеством SbFg в присутствии катализатора
SbCls с образованием смеси, из которой можно выделить SiFClз,
SiF2Cl2, SiF3Cl и SiF4 [56].
В случае азидов возможна замена связи Si — Cl связью Si —
Nз по реакции
(СН3)3SiCl + NaN3---> (СН3)3SiN3 + NaCl. (12)
Для этого достаточно раствор хлорида кремния в азиде натрия на-
греть с обратным холодильником в присутствии небольших коли-
честв катализатора LiAlH4 [57] или азида алюминия [58].
Попытки частичного восстановления галогенидов кремния гид-
ридами, например LiAlH4, всегда приводили к полному восстанов-
лению. Так, из SiCl4 был получен SiH4 и не были выделены такие
соединения, как SiHsCl. Однако частичное восстановление было
осуществлено в некоторых случаях при пропускании паров хлоро-
силана в смеси с водородом над алюминием или цинком, нагретым
до 400° [59]. Этим способом, например, SiCl4 был превращен с 25%-
ным выходом в SiHCl3 и CH3SiCl3 — в CH3SiHCl2:
6SiCl4 + ЗН2 + 2А1--> 6SiHCl3 + А12С16. (13)
В ходе этих реакций могут образовываться промежуточные сое-
динения — гидриды металлов, которые затем вступают в реакцию
обмена с галогенидом кремния; подтверждения такого механизма
получено не было.
Реакции перераспределения связей кремний — галоген об-
суждаются в разд. II.3.
Д. Из соединений со связью кремний — кислород
Соединения со связями кремний — кислород часто вступают в
интересные реакции с некоторыми безводными галогеноводородами
(и плавиковой кислотой), а также с другими ковалентными гало-
генидами с образованием галогеносиланов. Было установлено, что
в некоторых случаях иод реагирует подобным же образом.
190
Глава 7
Расщепление связи кремний — кислород под воздействием пла-
виковой кислоты было показано на примере образования цикло-
CsHjjSilLF при на1ревании (цпкло-СдН,(SiH.,)2O с 48%-ным вод-
ным раствором HF в течение 10 мин при 60° [42]
(nHK.TO-CeH11SiH2)2O 4-2HF--> 2 цикло-СвНц31Н2К Н2О. (14)
Эта реакция совершенно аналогична хорошо известной реакции
SiO* 4- 4HF ;--- SIFa 4- 2Н,0 (15)
Ход этой реакции в значительной степени определяется обра-
зованием очень прочных связей Si—F [60] (связь Si—0 =
= 108 ккал/молъ; связь Si—F=135 ккал/молъ); поэтому аналогич-
ная реакция с хлористым водородом (связь Si—0=91 ккал1 моль)
не протекает с такой легкостью, хотя и можно получить 25%-ный
выход цикло-СвНи81Н2С1, если пользоваться нагретым хлористым
водородом в присутствии Р2О5 (для удаления воды и смещения
равновесия вправо) [42]. Йодистый водород может расщеплять
связь кремний — кислород в (CHsSiFb^O с образованием
CHsSiFhl [61]. Триорганосилилфториды (и хлориды) часто полу-
чают из соответствующего дисилоксана* и фтористого (или хло-
ристого) водорода, приготовленного нагреванием дисилоксана со
смесью концентрированной серной кислоты и галогенида аммония
[62].
Следует отметить, что плавиковую кислоту можно также ис-
пользовать для прямого замещения атомов хлора атомами фтора в
некоторых соединениях кремния. Так, нагретый 48%-ный раст-
вор плавиковой кислоты легко переводит цикло-СдН.^СЬ в
цикло-С6Нп81Гз. Реакция протекает в одну стадию [42]. Этот
тип реакции можно рассматривать как гидролиз хлорида с обра-
зованием промежуточного силоксана (цикло-С6Н118Ю115)1., в ко-
тором затем связи кремний— кислород расщепляются плавиковой
кислотой. Связь кремний — кислород расщепляется некоторыми
ковалентными или полуковалентными галогенидами при низкой
температуре или при умеренном нагревании [62]. Например, при
—78° (SiH3)2O и BF3 реагируют спокойно с образованием SiH3F
[63]
(SiH3)2O + BF3----> SiH3OBF2SiH3F. (1G)
* Термином «силоксан» назван эфир кремния. Так, соединение (SiHa^O
правильнее называть дисилоксаном, хотя его часто называют дисилил-эфир,
особенно, когда желают подчеркнуть его родство с углеродным аналогом
(СНзЖ Сведения по номенклатуре, применяемой в химии кремния, можно
получить из нескольких источников [6, 10, 64].
II. Общие методы синтеза
191
Образующийся SiHsOBFz при комнатной температуре медленно
разлагается
3SiH3OBF2-----> 3SiH3F + В2О3 + BF3. (17)
Подобным же образом некоторые галогениды бора быстро реаги-
руют при низких температурах с другими дисилоксанами, напри-
мер с (СНзЭШг^О, [(CHg^SiHhO или [(СНз)з31]2О с образова-
нием аналогичных продуктов [61]. Алкоксисиланы можно также
расщепить BF3. Так, СНзОЭШз и BF3 реагируют при —96°
с образованием SiHeF [65]
CH3OSiH3+BFs------> SiH3F-J-CH3OBF2. (18)
Расщепление связи кремний — кислород при реакции с BF3
представляет, вероятно, наиболее удобный метод получения аб-
солютно чистых фторидов кремния, например SiHsF, CHsSiFhF
и т. д. Как и следует из рассмотрения стерических факторов [2],
реакция идет лучше с незамещенными соединениями гидрида,
например (81Нз)гО, чем с замещенными производными, например
[(СНз)з8Ц2О. Так, (81Нз)гО реагирует с AKCle при низкой темпе-
ратуре с образованием SiHsCl [66]
2(SiHs)2O + А12С16--> 2SiH3OAlCl2 + 2SiH3Cl. (19)
Образующийся SiHsOAlCH разлагается при низкой температуре с
выделением дополнительного количества SiHsCl. В случае
[CH3(C2Hs)2SiКО реакция протекает быстро только при нагре-
вании до 135—210°, и образуется устойчивое соединение
СНз(С2Н5)28ЮА1С12 [67].
Интересно отметить,что при комнатной температуре иод расщеп-
ляет связь кремний — кислород в некоторых дисилоксанах с обра-
зованием силилиодида. Например, как (81Нз)гО [52], так и
'* CH3OS1H3 [65] дают 81Нз1, и из (цикло-С6Н1181Н2)2О получают
цикло-СвНп8Ш21 [42]
i
J 3 (цикло-СвНцЗШ2)2О 312----» 3 miKno-CgHjjSiHJ
| -Нцикло-СвНпЗШСОз + ЗШ. (20)
V Несмотря на то что силилсульфиды и си ли лее лениды, по-види-
мому, не имеют практического значения для получения больших
количеств силилгалогенидов, следует отметить, что связи Si — S
и Si —Se в (8Шз)г8 и (SiH3)280 расщепляются иодом при комнат-
ной температуре с высоким выходом SiHel, а также серы или селе-
на соответственно [52]. Эти соединения легко вступают в реакцию
I с HI с образованием SiHsI и сероводорода или селеноводорода
[52].
А
192
Глава 7
Е. Из соединений со связью кремний — азот
Силил замещенные амины, как правило, реагируют с безвод-
ными галогеноводородами, или галогеноидами, или ковалентными
и полуковалентными галогенидами так, как и силоксаны, с той
разницей, что реакции протекают легче. Связи кремний — азот
расщепляются без нагревания, часто при низких температурах,
с образованием соответствующих галогенидов кремния. Так, сое-
динение (SiH3)3N [74] активно взаимодействует с газообразным
хлористым водородом [68]
(SiH3)3N + 4НС1---> 3SiH3Cl + NH4C1. (21)
Этот тип реакции, по-видимому, представляет собой удобный
способ превращения одного галогенида кремния в другой, даже и в
том случае, если направление превращения противоположно
«ряду превращений» солей серебра. Так, воздействуя хлоридом
серебра, можно превратить иодид кремния в хлорид кремния, но
нельзя перевести хлорид обратно в иодид реакцией с иодидом се-
ребра. Однако если, например, SiHCl3 сначала перевести в (SiHN)x
или в SiH(NHC6Hs)3 воздействием аммиака или анилина соответ-
ственно, то SiHI3 легко можно получить из этих соединений при
обработке их сухим иодистым водородом [69]
SiHCl3 + 6CeH6NH2 -•-> SiH(NHCeH5)3 + 3CeH5NH2- HC1, (22)
SiH (NHCeHs)3 + 6HI---> SiHI3 + 3CeH6NH2 • HI. (23)
Реакция (23) возможна и при замене HI каким-либо иодидом с ко-
валентной связью, например Р13, так как известно, что реакция
типа
4 (СН3)3 SiNHC6H5 + GeBr4--> 4 (СН3)3 SiBr + Ge (NHCeH5)4 (24)
протекает без затруднений [70]. Интересно отметить, что поведе-
ние NH3 при взаимодействии с соединениями, содержащими свя-
зи кремний — азот, подобно поведению галогеноводородов. Не-
давно NH3 был использован для синтеза SiH3N3 [71 ] и (CH3)3SiN3
[72]. Подобным же образом реагирует с [(CH3)3Si]2NH цианистый
водород, образуя (CH3)3SiCN [73].
По-видимому, приведенные выше типы реакций не были ис-
пользованы в полной мере, особенно при получении соединений,
содержащих связи кремний — водород, кремний — галоген или
кремний — галогеноид.
Реакции, аналогичные реакциям с дисилоксанами, протекают
между ковалентными галогенидами и соединениями, содержащими
связи кремний — азот. Так, (SiH3)3N при —78° реагирует с BF3
и ВС13 с образованием твердых продуктов присоединения
II. Общие методы синтеза
193
(SiH3)3NBF3 или (SiH3)3N-BCl3. При нагревании до комнатной
температуры эти соединения медленно разлагаются с образова-
нием (SiH3)2NBF2 или (SiH3)2NBCl2 и соответствующего силил-
галогенида
(SiH3)3N- BF3---> (SiH3)2NBF2 + SiH3F. (25)
Как (SiH3)2NBF2, так и (SiH3)2NBCl2 при выдерживании в ус-
ловиях комнатной температуры разлагаются с выделением допол-
нительного количества силилгалогенида. В случае (SiH3)2NBF2
возможно образование силилборазина [74, 75]:
3 (SiH3)2 NBF2--► (SiH3NBF)3 + 3SiH3F, (26)
Этот тип реакций также является превосходным методом получе-
ния очень чистого SiH3F [75]. Подобные реакции протекают также
с аналогичными соединениями, например (Si2Hs)3N [76],
(SiH3)2NCH3, SiH8N(CH3)2[75], (CH3SiH2)3N, CH3SiH2N(CH3)2
[77] и [(CH3)3Si]2NH [78, 79] и т. д.
Гексаметилдисилазан [(CH3)3Si]2NH вступает в целый ряд
интересных реакций с ковалентными или полуковалентными гало-
генидами, например BF3, ВС13, РС13, SbCl3, BiCl3, А12С1в, при
температуре ниже комнатной или при кипячении с обратным
холодильником с образованием соответствующего триметилсилил-
галогенида [79]. Например, с РС13 протекает следующая реакция:
](CH3)3Si]2 NH + РС13-> (CH3)3SiNHPGl2 + (CN3)3 SiCl. (27)
Следует отметить, что связь Si—Р в SiH3PH2 расщепляется
при воздействии НВт аналогично связи Si—N с образованием
SiH3Br и РН3 [79а].
Ж. Из соединений со связью кремний — фенил*
Синтез галогенидов кремния при посредстве расщепления свя-
зи кремний — фенил безводным галогеноводородом является од-
ним из наиболее важных последних достижений в неорганической
химии кремния. Без использования на промежуточных стадиях
SiH4 этим методом нельзя получить сразу в довольно больших
количествах такие важные исходные вещества, как SiH3I [37,
80—84]. Расщепление фенилсиланов CeHsSiH3 и некоторых их
производных безводными галогеноводородами происходит без
растворителя и катализатора при низких температурах. Часто
получают количественный выход галогеносиланов. Ниже приве-
дены некоторые типичные реакции
* Верма и Лейтч [Can. J. Chem., 41, 1652 (1963)] описывают существен-
ные улучшения в методе синтеза SiH3Br и SiH3Cl из C«H6SiH3 и соответст-
вующего галогеноводорода.
14 Заказ № 547
194
Глава 7
-40° C6H5SiH3 + HI * SiH3I -J- CeHe. (28)
— 20°
ClCeH4SiH3 + HI -> SiH3I + CeH6Cl, (29)
—40°
(CeH5)2SiH2 + 2HI -> SiH2I2-f 2CeHe, (30)
-78°
CeH5SiH3 + HBr -» SiH3Br CeHe, (31)
—78°
(CeH6)2SiH2 + HC1 -> CeH5SiH2Cl + CeHe. (32)
По-видимому, чем более электроотрицателен галоген, связан-
ный с кремнием, или чем больше атомов галогена связано с крем-
нием, тем медленнее протекает реакция расщепления. Так, соеди-
нение CeH5SiCl3 не расщепляется под воздействием галогеново-
дородов. Точно так же хлористый водород не расщепляет с такой
легкостью связь кремний — фенил, как бромистый или иодистый
водород. Хлорид алюминия каталитически сильно ускоряет реак-
цию хлористого водорода с CeHsSiH3 даже при —78°. Однако об-
разующийся SiH3Cl нельзя выделить в чистом виде, так как хлорид
алюминия вызывает его диспропорционирование.
Безводный фтористый водород не реагирует с CeH5SiH3 при
—60°, возможно, вследствие того, что эти вещества образуют два
несмешивающихся жидких слоя. Однако при 0° происходит коли-
чественная реакция с образованием бензола и летучих фторидов
кремния. Чистый фторид не был выделен в этой реакции. Кажется
вероятным, что SiH3F, который, по-видимому, образуется в первую
очередь, в присутствии фтористого водорода диспропорционирует
на другие фториды, например SiH2F2 и т. д. Это подтверждается
тем фактом, что SiHF3, совершенно устойчивый в чистом виде,
разлагается в присутствии фтористого водорода [85]. В присут-
ствии солей меди, например хлорида меди(П), фторирование свя-
зей кремний — водород легче протекает при 0°; так, CeH5SiH3
дает хороший выход фенилфторосиланов, например CeHsSiF3,
но реакция расщепления затрудняется.
Смешанные галогеносиланы, например SiH2BrI, можно полу-
чить при использовании следующей реакции:
CeH5SiH2I + НВг---> SiH2BrI + С6Н6. (33)
Однако такие соединения не были выделены и охарактеризованы.
Представляется вероятным, что обмен галогенами
SiH2BrI + НВг----> SiH2Br2 + HI . (34)
или диспропорционирование этих соединений может сильно за-
труднить их выделение и очистку.
II. Общие методы синтеза
195
Следует заметить, что если иодистый водород пропускать
через кипящий C6HsSiH3 (т. кип. 120°), то происходит реакция
C.H3SiH3 + HI----> CeH5SiH2I + Н2 (35)
в дополнение к реакции расщепления [реакция (28)1. Реакция
расщепления идет легче при низких температурах и при —40°
дает количественный выход в течение 1 час.
3. Через реакции перераспределения
Уже много лет известно, что связи кремний — галоген и крем-
ний — водород подвержены реакциям перераспределения при
нагревании (при относительно низких температурах) в присут-
ствии галогенида алюминия в качестве катализатора. Силан и
SiH2Cl2, например, выдержанные при 100° в течение 7 суток в
присутствии хлорида алюминия, реагируют с образованием равно-
весной смеси хлоросиланов. Если исходные вещества взяты в
надлежащей пропорции, получают большой выход SiH3Cl [35].
При этом идет следующая реакция:
SiH2Cl2 + SiH4---> 2SiH3Cl. (36)
Точно так же SiH4 и SiCl4 реагируют с образованием SiH2Cl2
при нагревании до 300° в присутствии хлорида алюминия [86].
В подобных условиях любой хлоросилан дает смесь всех возмож-
ных продуктов диспропорционирования.
Другие катализаторы, помимо галогенидов алюминия, также
эффективно влияют на скорость реакции. Например, можно до-
стигнуть хорошего выхода SiH2Cl2 при кипячении с обратным хо-
лодильником SiHCl3 (одного из нескольких имеющихся в продаже
соединений со связями кремний — водород) в течение 5 час в
присутствии дпметилцианамида [87].
2SiHCl3----> SiH2Cl2 + SiCl4. (37)
При 200° в присутствии того же катализатора получают SiH2Cl2
с выходом 15% от взятого SiH3Cl [87]. Дихлоросилан можно так-
же получить с выходом 15% при нагревании SiHCl3 до 300—400°
в присутствии таких катализаторов, как А12С16, Fe2Cle и BF3
[88]. По-видимому, этот метод можно соответственно модифици-
ровать для получения SiH3I из SiHI3, причем последнее соедине-
ние можно получить из SiHCl3 [69].
Аналогичные типы реакций использовали также для синтеза
тетрагалогенидов кремния, в молекуле которых содержались два
различных галогена. Например, SiCl3Br, SiCl2Br2 и SiClBr3
получают с хорошим выходом при пропускании смеси SiCl4 и
196
Глава 7
SiBr4 через трубку, нагретую до 600°, в отсутствие катализатора
[89]. При осуществлении почти аналогичной реакции было уста-
новлено, что при высокой температуре атом галогена из газооб-
разного галогеноводорода может замещать галоген в тетрагало-
гениде кремния. Так, SiICl3 и иодистый водород реагируют при
нагревании в бомбе до 240— 250° в соответствии с уравнением
[90]
SilClg + HI--> SiI2Cl2 + НС1. (38)
111. ПРОИЗВОДНЫЕ СИЛАНА
А. Фторосиланы
SiH3F (т. кип.—88,1°) [75]. Фторосилан (силилфторид) в чистом
виде относительно термически устойчив, однако в присутствии ма-
лых количеств неидентифицированных примесей он разлагается с
заметной скоростью при комнатной температуре [16, 63].
Это соединение можно получить общим методом, включающим
фторирование SiH3Cl трехфтористой сурьмой SbF3 в присутствии
небольших количеств катализатора SbCls. Процесс легко идет в
паровой фазе; имеется сообщение о 79%-ном выходе после реакции,
продолжающейся 24 час при комнатной температуре [16]. Однако
этим методом нельзя получить продукт со степенью чистоты более
98%. Это, возможно, обусловлено медленным диспропорциониро-
ванием в процессе очистки, так как давление паров вещества мед-
ленно возрастает при комнатной температуре.
Как было показано выше, чистый продукт можно легко полу-
чить при низкой температуре действием BF3 на амины [75] или
эфиры [63, 65], содержащие группу SiH3. Поскольку (SiH3)2O
можно получить проще, чем такие амины, как (SiH3)3N (например,
путем почти количественного гидролиза SiH3I [66]), постольку
лучшим практическим методом синтеза SiH3F является, по-ви-
димому, взаимодействие (SiH3)2O с BF3. Ниже изложен метод,
описанный Онищуком [63]. Все манипуляции следует выполнять
в вакууме.
Трифторид бора (0,0581 а, 0,85 ммоль) и (SiH3)2O (0,1010 г,
1,28 ммоль) конденсируют вместе и выдерживают при температу-
ре —78° в течение 12 час. Загрязненный силилфторид перегоняют
затем из реакционного сосуда, который поддерживают при темпе-
ратуре —78°. Окончательную очистку осуществляют дистилляци-
ей SiH3F из ловушки при —132° (выход 0,0378 г, 1,44 ммоль).
Так как количество полученного SiH3F больше того, которое мож-
но получить реакцией (16), то представляется вероятным, что не-
которое количество первоначально образующегося SiH3OBF2
претерпевает разложение с выделением добавочного количества
III. Производные силана
197
SiH3F в соответствии с реакцией (17). Белый твердый SiH3OBF2,
остающийся в реакционном сосуде при нагревании до комнатной
температуры и при перегонке из одной ловушки в другую, постепен-
но разлагается без плавления. Общий выход чистого SiH3F
(0,110 г, 2,18 ммоль', определенный молекулярный вес 50,4, вычис-
ленный 50,1) составляет 85%, если исходить из суммарной реак-
ции
3 (SiH3)2O + 2BF3 —> 6SiH3F + В2О3. (39)
Аналогичные реакции были успешно использованы для син-
теза CH3SiH2F и (CH3)2SiHF из (CH3SiH2)2O и [(CH3)2SiH]2O
соответственно [61].
SiHJ^ (т. пл. —122,0°, т. кип. —77,8°). Дифторосилан являет-
ся относительно устойчивым веществом и не изменяет упругости
паров при выдерживании над ртутью в течение 14 суток при ком-
натной температуре. Однако по истечении 2 лет он полностью дис-
пропорционировал на SiH4 и SiF4 [16].
Дифторосилан можно получить с выходом до 80% при фториро-
вании трехфтористой сурьмой SbF3 (содержащей 10% SbCls в
качестве катализатора) газообразного SiH2Cl2 в течение 24 час
при комнатной температуре. Такие продукты неполного фториро-
вания, как SiH2FCl, могут образоваться в процессе реакции, од-
нако выделены они не были [16].
SiHF3 (т. пл. —131,4°, т. кип. —95,0°). По имеющимся сведе-
ниям [54], трифторосилан диспропорционирует (возможно, на
SiH4 и SiF4)npn очень низких температурах. В более поздней ра-
боте указывается, что он устойчив при низких температурах,
но быстро диспроцорционирует при комнатной температуре [16].
В последнее время было показано,что если это соединение не содер-
жит примеси фтористого водорода, то оно устойчиво длительное
время даже при повышенных температурах [85]. В самом деле,
полное диспропорционирование может наступить только при про-
пускании трифторосилана над нагретым фторидом натрия [85]
4SiHF3 + 6NaF----► SiH4 -)- 3Na2SiFe. (40)
Трифторосилан можно получить взаимодействием SiF4 с водо-
родом при высокой температуре [85] или фторированием SiHCls
трехфтористой сурьмой [54]. Экспериментальные подробности пер-
вого метода не были опубликованы, а второй метод дает малый
выход продукта, так как большая часть исходного вещества всегда
остается непрореагировавшей. В процессе реакции SiHCl3 с SbF3
образуются также соединения SiHFCl2 и SiHF2Cl [54]. Наиболее
практичным методом получения относительно чистого SiHF3 яв-
ляется фторирование SiHCl3B вакууме. Пары трихлоросилана в те-
чение 3 час при комнатной температуре пропускают, меняя направ-
198
Глава 7
ление над SbF3, содержащим около 5% SbCl5 в качестве катализато-
ра [16]. В этих опытах 28% SiHCl3 было извлечено непрореагиро-
вавшим, однако 75% прореагировавшего трихлоросилана было
превращено в SiHF3. При большем времени контакта SiHCl3 реа-
гирует полностью с возможным выходом 91% SiHF3.
SiHFCl2 (т. пл. —149,5°, т. кип. —18,4°). Фтородихлоросилан
можно получить в относительно больших количествах вместе с
SiHF3 и небольшими количествами SiHF2Cl фторированием SiHGl3
трехфтористой сурьмой SbF3 [54]. SbF3 добавляют медленно к
SiHCl3, содержащему 3—7% SbCl5 в качестве катализатора. Реак-
ция имеет бурный экзотермический характер.
SiHF2Cl (т. пл. —144°, т. кип. примерно —50°). Дифторохло-
росилан получают в небольших количествах в процессе синтеза
SiHF3 и SiHFCl2, который описан выше [54].
Б. Хлоросиланы
SiH^Cl (т. пл. —118,1°, т. кип. —30,4°). Хлоросилан (силил-
хлорид) — довольно устойчивое соединение [4,35]. Его можно по-
лучить нагреванием (в течение 30 час при 100°) 10%-ного избытка
SiH4 с хлористым водородом в присутствии небольших количеств
хлорида алюминия в качестве катализатора. Образуются хлоро-
силан и SiH2Cl2 в соотношении 4 : 1 [35] [см. реакцию (4)].
Однако в связи с тем, что в настоящее время SiH3I получают в
больших количествах, более простой метод синтеза заключается в
пропускании паров SiH3I над хлоридом серебра
SiHgl + AgCl---► SiH3Cl + Agl. (41)
Было установлено, что этот метод позволяет получить довольно
хороший выход чистого SiH3Cl [53], если реакцию вести таким же
образом, как и реакцию получения SiH3NGS, описанную в разд.
111.Е.З. Можно вести процесс не в вакууме, с использованием рас-
твора SiH3I в инертном растворителе, чтобы реакция протекала
менее бурно.
SiH2Cl2 (т. пл. —122°, т. кип. 8,3°). Дихлоросилан в чистом
виде — совершенно устойчивое вещество [4, 35]. Его можно полу-
чить нагреванием SiH3Cl с хлористым водородом в присутствии
хлорида алюминия в качестве катализатора или же нагреванием
SiH4 и хлористого водорода (взятых в молярном отношении
1 : 2) в присутствии хлорида алюминия в течение 7 суток при 100°.
Получаемый таким способом продукт представляет собой в ос-
новном SiH2Cl2 [35]. Однако, учитывая существующий в настоя-
щее время простой метод получения SiH2I2 в относительно боль-
ших количествах, может оказаться более выгодным синтезировать
SiH2Cl2 пропусканием паров SiH2l2 над хлористым серебром по
III. Производные силана
199
методике, описанной в разд. III.Е.З для синтеза SiH3NCS.
Этим методом получен довольно хороший выход SiH2Cl2, синте-
зированного из SiH2I2 [50]. Процесс можно осуществить в открытом
опыте с использованием раствора SiH2I2 в инертном растворителе
с целью замедления реакции. В том случае, если требуются боль-
шие количества SiH2Cl2, его, вероятно, лучше получать диспро-
порционированием SiHCl3 в присутствии катализатора, которое
было рассмотрено в разд. II.3 [87, 88]. Само соединение SiHCl3
можно получить в готовом виде или синтезировать в больших
количествах из кремния и хлористого водорода.
SiHCls (т. пл. —126,5°, т. кип. 31,8°). Трихлоросилан — со-
вершенно устойчивое соединение [4, 91]. Несмотря на то что его
можно получить из SiH4 и хлористого водорода при их нагревании
в присутствии хлорида алюминия в качестве катализатора, более
распространенным методом является синтез трихлоросилана из
кремния или ферросилиция и хлористого водорода при повышен-
ных температурах [16, 17, 24, 92]. Метод, описанный Уитмором и
сотр. [24], приведен ниже.
Реакцию проводят в горизонтальной трубке (диаметр 3 см,
длина 45 см) из стекла пирекс с электрообогревом. Вход трубки сое-
динен с осушающей колонкой с серной кислотой и с расходомером
газа. Выход трубки соединен с водоохлаждаемым конденсатором,
который ведет к колбе, охлаждаемой смесью соли и льда. В ка-
честве меры предосторожности против потери вещества колба
соединена с ловушкой, погруженной в смесь ацетона и сухого
льда, которая в свою очередь связана с осушающей колонкой с
серной кислотой. В трубку загружают 375 г ферросилиция (около
90% Si) в виде гранул и систему осушают потоком азота, пропу-
скаемого в течение 24 час при температуре печи около 300°. Затем
пропускают безводный хлористый водород при температуре труб-
ки 350° до того момента, пока в приемнике не начнут конденсиро-
ваться первые капли жидкости. После этого температуру пони-
жают и поддерживают в пределах 290—310°, а поток хлористого
водорода регулируют в пределах 0,6—0,85 моль/час. Приблизи-
тельно через каждые 10 час систему промывают сухим азотом в те-
чение 10 мин. В обычных условиях через 60 час получают 1370 г
жидкого продукта. Фракционная перегонка в колонне, имеющей
приблизительно 15 теоретических тарелок, дает 1045 г SiHCl3
(т. кип. 31,5—32° при 729 мм pm. ст.). Это количество составляет
11% от неочищенного продукта; остаток представляет собой в
основном SiCl4 и небольшие количества хлорополисиланов. В дру-
гих подобных опытах был получен выход 70—77%. Следует учесть,
что при большей скорости потока хлористого водорода и темпера-
туре реакции 360—370° выход понижается до 32%.
200
Глава 7
В. Бромосиланы
SiH3Br (т. пл. —94°, т. кип. 1,9°). Бромосилан (силилбромид)
воспламеняется на воздухе, однако в чистом виде он термически
устойчив [34].
Это соединение можно получить обычным нагреванием силана с
безводным бромистым водородом в присутствии бромида алюминия
в качестве катализатора [94]. Такой метод был выбран для полу-
чения SiH3Br непрерывным способом в любых количествах (пропу-
сканием реагирующих веществ через печь в присутствии катали-
затора) [93]. Однако изготовление цельнометаллической аппара-
туры оправдано лишь при необходимости больших количеств
этого соединения. Небольшие порции SiH3Br можно получить бро-
мированием SiH4 твердым бромом при —80°. Некоторое количество
SiH2Br2 также образуется в этой реакции [34].
Лучший метод получения SiH3Br — взаимодействие избытка
бромистого водорода с CeH5SiH3 при —78° [см. уравнение (31)].
Реакция с количественным выходом заканчивается в течение 3 час
[80, 82]. Следует принимать меры предосторожности в связи с
возможной воспламеняемостью SiH3Br.
ЗШйВгй(т. пл.—70,1°, т. кип. 66°). Дибромосилан—устойчивое
вещество, но иногда воспламеняется на воздухе [34]. Его можно
получить в чистом состоянии реакцией SiH4 с бромистым водородом
при осторожном нагревании в присутствии бромида алюминия в
качестве катализатора [93, 94]. Он также образуется при взаимо-
действии твердого брома с SiH4 при —80° [34].
Если исследователя удовлетворяет раствор SiH2Br2 в бензоле,
то последний можно проще всего получить реакцией CeH5SiH2Br
с избытком бромистого водорода при —78° в течение 6 суток [82]
CeH8SiH2Br + НВг ->SiH2Bra+CeHe, (42)
Необходимый для синтеза CeH8SiH2Br получают из (CeH5)2SiH2
и НВг. Так как бензол имеет точку кипения (80,1°), довольно
близкую к точке кипения SiH2Br2, могут возникнуть затруднения
при разделении веществ, если в распоряжении нет эффективной
ректификационной колонны.
Другой метод получения чистого SiH2Br2 заключается в реак-
ции SiH2l2 с бромистым серебром. На основании «ряда превраще-
ний», который обсуждался выше, можно сделать заключение, что
такая реакция вероятна.
SiHBr3 (т. пл. —73,5°, т. кип. 111,8°). Трибромосилан, —
по-видимому, устойчивое вещество, но при разливке на воздухе
он обычно воспламеняется. Трибромосилан можно получить броми-
рованием SiH4 при низкой температуре элементарным бромом [34],
однако практически удобнее синтезировать трибромосилан нагре-
111. Производные силана
201
ванием кремния при 360—400° в токе бромистого водорода по мето-
дике, аналогичной получению SiHGl3 [25, 26].
Г. Иодосиланы
SiH9I (т. пл. —57,0°, т. кип. 45,4°). Иодосилан (силилиодид) —
относительно устойчивое вещество, и его можно хранить при
комнатной температуре в герметичной ампуле [36]. Силилиодид
можно получить нагреванием SiH4 с иодистым водородом в присут-
ствии иодида алюминия в качестве катализатора при температуре
80—100° в течение 12—18 час [36, 53]. При этом также образуются
соединения с более высоким содержанием иода SiH2I2, SiHIg
и Sil4 в зависимости от относительных количеств используемых
реагентов.
В настоящее время иодосилан можно получить несравненно
проще: путем расщепления либо CeH5SiH3, либо ClCeH4SiHs
иодистым водородом [см. уравнения (28) и (29)]. В первом случае
был достигнут количественный выход (исходя из применяемого
CeH5SiH3) [37], во втором — 60% (исходя из применяемого
CeH5SiCl3) [84].
Ниже приведен метод получения SiH3I из CeH6SiH3, описан-
ный Фрицем и Куммером [37].
Фенилсилан CeH5SiH3 получают медленным добавлением при
постоянном перемешивании торгового CeH6SiCl3 (150 г) к LiAlH4
(23 г), растворенного в 700 мл диэтилового эфира. Смесь нагре-
вают с обратным холодильником в течение 3 час. Раствор, содержа-
щий соли лития и алюминия, отфильтровывают через стеклянный
фильтр без доступа воздуха. Затем при пониженном давлении из
раствора солей отгоняют эфир и C6H6SiH3, принимая меры против
повышения температуры в колбе выше 30°. Если отгонку ведут при
атмосферном давлении, то в присутствии растворенного хлорида
алюминия необходимая для этого более высокая температура мо-
жет вызвать диспропорционирование CeH6SiH3 [95] с образова-
нием SiH4. Последний на воздухе может воспламениться и вызвать
взрыв
4C„H6SiH3----> 3SiH4+(C6H6)4Si. (43)
Раствор C6H5SiH3 без этой соли (А12С16) можно безопасно под-
вергать дистилляции при атмосферном давлении с отгонкой чи-
стого CeH5SiH3 (т. кип. 120°; выход 61 г, 79%).
Затем конденсируют иодистый водород в молярном избытке по
отношению к любому количеству CeH6SiH3. После выдерживания
при —40° в течение 1 час смешивание ускоряют нагреванием и пов-
торной конденсацией веществ. Эту операцию повторяют два или
три раза. Избыток йодистого водорода удаляют возгонкой и про-
дукты выделяют дистилляцией.
13 Заказ № 547
202
Глава 7
Имеется сообщение о видоизмененном варианте этого метода
[84]. В этом варианте вместо GeH6SiH3 используют хлорофенил-
силан ClCeH4SiH3, так как хлоробензол, образующийся в этой
реакции [см. уравнение (29)], имеет более высокую температуру
плавления, чем бензол, и, таким образом, его легче отделить
от SiH3I. Ниже описан синтез, проводимый в вакуумной сис-
теме.
Фенилтрихлоросилан CeH5SiCl3 (25 г) очищают фракционной
перегонкой при —46° и затем переводят в GlGeH4SiGl3 [96, 97].
(Хлорирование идет без затруднений при 60—70° в течение 40 мин
в присутствии 0,5% порошка железа в качестве катализатора
[97].) Затем к желтоватому продукту добавляют 25 мл безводного
диэтилового эфира. Полученный раствор медленно приливают к
смеси LiAlH4 (16 г) и эфира (30 мл), содержащейся в трехгорлой
колбе, заполненной азотом и снабженной обратным холодильником
и предохранительной трубкой. Реакция протекает очень бурно.
После выдерживания в течение ночи все летучие продукты уда-
ляют из реакционной колбы 6-часовым откачиванием. При этом
колбу периодически подогревают. Полученные летучие вещества
перегоняют несколько раз через ловушку (при —64°), где конден-
сируется ClCeH4SiH3. Хлорофенил силан помещают в толстостен-
ную пробирку из стекла пирекс (приблизительно на 100 мл)
и туда же дистиллируют безводный иодистый водород (25 г). После
запаивания пробирку медленно нагревают до комнатной темпера-
туры. По истечении 12 час присутствующие летучие продукты отго-
няют и получают в остатке чистый SiH3I (выход: 11 г, 60%; молеку-
лярный вес: найденный — 158, вычисленный—157,9). Примечание'.
нескольких перегонок через ловушку (при —64°) достаточно
для удаления всего хлорбензола из более летучего SiH3I.
SiHJ2 (т. пл. —1,0°, т. кип. 149,5°). Дииодосилан, по-ви-
димому, устойчивое вещество [36]. Его можно получить из SiH4
и йодистого водорода по методике, описанной для SiH3l [36].
Однако удобнее получать дииодосилан с высоким выходом реак-
цией (CeH6)2SiH2 с иодистым водородом [82, 84] [см. уравнение
(30)]. Метод аналогичен описанному выше для синтеза SiH3I
из CeH6SiH3.
SiHI3 (т. пл. 8°, т. кип. 220°). Трииодосилан — устойчивое
вещество, которое образуется в небольших количествах при реакции
SiH4 с иодистым водородом [36]. Это соединение можно получить
пропусканием йодистого водорода над нагретым кремнием [27]
по методике, которая аналогична применяемой для синтеза SiHGl3
и SiHBr3. Лучший метод синтеза включает использование SiHCl3
в качестве исходного материала [69]. SiHCl3 превращают в
(SiHN)^ под влиянием аммиака или в SiH(NHGeH5)3 воздействием
анилина. Оба соединения легко разлагаются иодистым водородом
III. Производные силана
203
с хорошим выходом SiHI3 [72] [см. уравнения (22) и (23)]. Ниже
приведено описание метода, данное Раффом [69].
Безводный анилин (125 г) растворяют в безводном бензоле
(300 мл) в 1-литровой отсасывающей склянке, боковое горло кото-
рой соединено с осушающей трубкой. Затем трихлоросилан (25 г),
растворенный в безводном бензоле (150 мл), медленно добавляют че-
рез капельную воронку, вставленную через пробку в горло отса-
сывающей склянки. В процессе добавления, занимающего около
1 час, содержимое склянки следует энергично перемешивать встря-
хиванием или магнитной мешалкой. Температуру постепенно
повышают до 50—60° [см. реакцию (22)]. Осадок GeH6NH2-HCl
отфильтровывают (в отсутствие влаги) в атмосфере сухого воздуха,
азота или двуокиси углерода и промывают три раза 50-миллилит-
ровыми порциями бензола.
Затем раствор SiH(NHGeH6)3 в бензоле переводят в другую
отсасывающую склянку, снабженную в боковом горле осушающей
трубкой, и осуществляют медленный барботаж безводного йодистого
водорода через раствор по трубке, введенной через пробку в горло
отсасывающей склянки. Происходит реакция, описываемая урав-
нением (23). Барботаж йодистого водорода прекращают, когда из
осушающей трубки начинается сильное выделение паров йодисто-
го водорода. Осажденный CeH6NH2-HI отфильтровывают в отсут-
ствие влаги. После промывания тремя порциями бензола (по 50 мл
каждая) бензол удаляют дистилляцией приблизительно при 50 мм
рт. ст. и 40—50°. Если окраска отогнанного бензола (обычно свет-
ло-розовая) становится несколько темнее, то это значит, что отгонка
протекала слишком быстро и что вместе с бензолом было отог-
нано некоторое количество продукта. Остаток, окрашенный в тем-
но-красный цвет вследствие присутствия свободного иода, взбал-
тывают с небольшим количеством ртути для удаления иода. Если
иод не удалить, выход SiHI 3 уменьшится из-за протекания реакции
SiHI3 + I2-->SiI4+HI, (44)
которая может начаться при последующей дистилляции. Неочищен-
ный SiHI з помещают в меньшую колбу и перегоняют приблизи-
тельно при 20 мм рт. ст. и 110° (выход: 45—50 г, 60—65%).
Д. Тетрагалогеносиланы
SiFi (т. пл. —90,2° при 1318 мм рт. ст., т. субл. —95,7°).
Тетрафторид кремния — термически устойчивый газ, который обра-
зуется при контакте плавиковой кислоты или фтористого водорода
с веществами, содержащими SiO2. В зависимости от метода полу-
чения SiF4 в большей или меньшей степени загрязнен фтористым
водородом или H2SiFe [98]. Тетрафторид кремния высокой чисто-
13*
204
Глава 7
ты можно получить нагреванием BaSiFe при температуре 300—
500° [98]. BaSiFe в свою очередь получают реакцией хлористого
бария с водным раствором кремнефтористоводородной кислоты.
SiClt (т. пл. —68°, т. кип. 57,0°) [99]. Тетрахлорид кремния —
термически устойчивое вещество, которое можно получить различ-
ными способами. В том случае, если требуются относительно боль-
шие количества, наиболее удобным методом является пропускание
хлора или безводного хлористого водорода над нагретым кремни-
ем или кремниевым сплавом (или силицидом металла) [7,24,
100—103]. Наибольший выход получают при высоких температу-
рах. При низких температурах доля образующихся высших хло-
ридов, например Si2Cle, Si3Cl8 и т. д., возрастает. По-видимому,
первоначально образующиеся соединения наиболее сложны и со-
держат цепи атомов Si — Si, характерные для кремния в элемен-
тарном состоянии. При избытке хлора и высоких температурах
эти цепи разрываются с образованием SiCl4.
SiBri (т. пл. 5,4°, т. кип. 154°). Тетрабромид кремния термически
устойчив. Его можно легко получить пропусканием паров брома в
токе азота над нагретыми гранулами кремния. При подходящей
скорости тока и нагревании в качестве единственного про-
дукта получают SiBr4 высокой чистоты [25, 26, 100, 104—106].
Silt (т. пл. 120,5°, т. кип. 287,5°). Тетраиодид кремния — тер-
мически устойчивое вещество, но он темнеет при выдерживании в
запаянной ампуле на прямом солнечном свету [100, 107]. Инте-
ресно отметить, что в действительности Sil4 более устойчив, чем
его углеродный аналог С14, который разлагается до Са14 как при
нагревании, так и на солнечном свету [108].
Тетраиодид кремния можно легко получить в виде бесцветного
кристаллического вещества пропусканием паров иода в токе двуо-
окиси углерода над нагретым кремнием [106, 107].
SiF3Cl(T. пл. —142,0°, т. кип. —70,0°). Трифторохлоросилан
можно получить фторированием SiCl4 трехфтористой сурьмой
SbF3. SiCl4 смешивают с SbF3, взятой с недостатком, и затем мед-
ленно добавляют к смеси SbGl6 в качестве катализатора. В резуль-
тате этой экзотермической реакции получают хороший выход
SiF3Cl наряду с SiF2Gl2 и SiFGl3 [56, 109]. Это же соединение полу-
чают также реакцией SigFe с хлором. При сильном местном перегре-
ве реакционного сосуда пламенем горелки происходит небольшой
взрыв и образуются как SiF3Gl, так и SiF2Gl2 [32]. Трифторохло-
росилан получают с небольшим выходом (наряду с SiF2Gl2 и SiFGl3)
пропусканием смеси паров SiF4 и SiCl4 через трубку, нагретую до
700° [110].
SiF3Cl3 (т. пл. —139,7°, т. кип. —32,2°). Дифтородихлоросилан
можно получить методом, описанным выше для SiFsGl [32, 56,
109, НО].
111. Производные силана
205
SiFCl3 (т. пл. —120,8°, т. кип. 12,2°). Фторотрихлоросилан
можно получить методом, описанным для SiF3Cl [32, 56,109,110].
SiF3Br (т. пл. —70,5°, т. кип. —41,7°). Трифторобромосилан был
получен медленным добавлением SbF3 (без катализатора) к нагре-
тому SiBr4. Был получен хороший выход SiF3Br наряду с SiF2Br2
и SiFBr3 [111]. Трифторобромосилан можно также получить вме-
сте с SiF2Br2 и SiFBr3 пропусканием смеси паров SiF4 и SiBr4 че-
рез трубку, нагретую до 700° [110].
SiF2Br2 (т. пл. —66,9°, т. кип. 13,7°). Дифтородибромосилан
при хранении разлагается. Это соединение можно легко получить
с хорошим выходом методом, описанным для SiF3Br [110, 111].
Это соединение можно также синтезировать вместе с SiF3Br,
смешивая Si2Fe с избытком брома и при местном нагревании реак-
ционного сосуда на малом пламени горелки. В результате неболь-
шого взрыва происходит образование названных продуктов [111].
SiFBr3 (т. пл. —82,5°, т. кип. 83,8°). Фторотрибромосилан
можно легко получить методом, описанным для SiF3Br [110, 111].
SiFCl2Br (т. пл. —112,3°, т. кип. 35,4°). Фтородихлоробромо-
силан можно получить медленным добавлением SbF3 к SiCl2Br2
[112], или же нагреванием SiFBr3 с хлором в запаянной ампуле в
течение 2 час на небольшом пламени, или нагреванием SiFBr3
с SbCl3 [112].
SiFClBr2 (т. пл. —99,3°, т. кип. 59,5°). Фторохлородибромо-
силан образуется при постепенном добавлении и энергичном пере-
мешивании SbF3 к SiGlBr3 или нагреванием SiFBr3 с SbCl3 [112].
SiF3I (т. пл. около —92°, т. кип. —24°). Трифтороиодосилан не
очень устойчив при перегонке при атмосферном давлении. Его
получают вместе с SiF2I2 и SiFI3 пропусканием смеси паров SiF4
и Sil4 через трубку, нагретую до 700°. Реакция идет без катали-
затора, и атомы различных галогенов распределяются в случай-
ных соотношениях в молекулах галогеносиланов [110].
SiF2I2 (т. пл.около —83°, т. кип. 84,5°). Дифтородииодосилан
получают методом, описанным для SiF3I [110].
SiFI3 (т. пл. около —41°, т. кип. 188°). Фторотрииодосилан
можно получить методом, описанным для SiF3I [110].
StCl3Br (т. кип. 80°). Трихлоробромосилан можно синтезиро-
вать вместе с SiCl2Br2 и SiClBr3: а) пропусканием смеси хлора
и фтора над нагретым до 700° кремнием: б) 48-часовым нагрева-
нием SiBr4 в атмосфере хлора с конденсацией флегмы; в) нагрева-
нием смеси SiBr4 и 8ЬС13или г) пропусканием смеси паров брома и
Si2Cl8 (или Si3Cl8) через трубку, нагретую до 500° [112].
SiCl2Br3 (т. кип. 105°). Дихлородибромосилан можно получить
методами, описанными для SiCl3Br [112].
SiClBr3 (т. кип. 128°). Хлоротрибромосилан можно также полу-
чить методом, описанным для SiCl3Br [112].
206
Глава 7
SiCl3I (т. кип. 113,5°). Трихлороиодосилан представляет собой
тяжелую бесцветную жидкость, медленно разлагающуюся при
комнатной температуре с выделением иода. Разложение ускоря-
ется на свету [901. Это соединение легко можно получить нагре-
ванием SiHGl3 и иода в бомбе при 240—250° в течение 15 час
190, ИЗ].
SiHCl3 + 1а--► SiCl3I + HI. (45)
Одновременно образуется много SiCl212 в соответствии с реакцией
(38). Трихлороиодосилан можно также получить вместе с SiCl2I2
и SiClI3 пропусканием смеси паров йодистого водорода и SiCl4
через нагретую докрасна стеклянную трубку [ИЗ]. В небольших
количествах его получают обработкой SiCl4 расплавленным иоди-
дом калия [89].
SiCl2I2 (т. кип. 172°). Дихлородииодосилан по своим химиче-
ским свойствам подобен SiGl3I. Это соединение получают тем же
методом, что и SiGl3I [90, ИЗ], или пропусканием паров SiCl3I
через нагретую трубку [89].
SiClI3 (т. пл. 2°, т. кип. 234—237°). Хлоротрииодосилан можно
получить из SiCl4 и йодистого водорода [ИЗ] методом, описанным
для SiGl3I, или нагреванием SiGl3I [89].
SiBr3I (т. пл. 14°, т. кип. 192°). Трибромоиодосилан представ-
ляет собой бесцветное твердое вещество, которое можно получить
вместе с небольшими количествами SiBr2I2 и SiBrI3 нагреванием
при 200—250° SiHBr3 и иода в запаянной ампуле. Это соединение
также получают в смеси с большими количествами двух других
бромоиодосиланов, пропусканием паров 1Вг над кремнием при
температуре красного каления [114, 115].
SiBr2I2 (т. пл. 38°, т. кип. 230,5°). Дибромодииодосилан можно
получить методами, рекомендуемыми для получения SiBr3I [114,
115].
StBrI3 (т. пл. 53°, т. кип. около 255°). Бромотрииодосилан по-
лучают методом, описанным выше для SiBr3I [114, 115].
Е. Галогеноидные производные
1. Цианиды'
SiH3CN (т. пл. 32,4°, т. кип. 49,6°) [49]. Было показано, что
силилцианид имеет линейную структуру, однако вопрос о струк-
туре замещенных силилцианидов, например (GH3)3SiCN, остается
неясным [2]. Предполагают, что это соединение в действительности
представляет собой смесь нормального цианида и изоцианида
[73, 119].
Силилцианид обладает неожиданно высокой температурой плав-
ления. При комнатной температуре он представляет собой бесцвет-
III. Производные силана
207
ное кристаллическое вещество, в вакууме легко сублимируется.
В совершенно чистом состоянии пробы SiH3GN можно хранить в
герметичных ампулах при комнатной температуре в течение 6 ме-
сяцев без или с незначительным разложением. Однако в присут-
ствии следов примесей, например конденсированных паров ртути,
за несколько суток при комнатной температуре происходит раз-
ложение.
Силилцианид можно получить с хорошим выходом просасыва-
нием паров SiH3I над цианидом серебра при комнатной темпера-
туре [49] [см. уравнение (10)]. Подобным образом можно синте-
зировать соединения типа CH3SiH2CN [120].
Cl3SiCN (т. пл. —46,2°, т. кип. 73,2°). Трихлороцианосилан
был получен взаимодействием цианида ртути с жидким или га-
зообразным Si2Cle при 100° [121].
2. Изоцианаты
SiHsNCO (т. пл. —88,6°, т. кип. 18,1°). Изоцианат силила,
подобно другим цианатам кремния, по-видимому, имеет такую же
изоструктуру [48]. Он отличается otCH3NCO, но подобен SiH3NCS
тем, что имеет такую же линейную молекулу (симметричный вол-
чок). В чистом виде изоцианат силила устойчив при комнатной
температуре по крайней мере в течение 2 недель, но в присутствии
небольших количеств примесей он быстро разлагается.
Изоцианат силила получают барботированием азота через
SiH3I при —46°. Смесь газообразный азот—SiH3I затем прокачи-
вают через цианат серебра
SiH3I + AgNCO---->• SiH3NCO + Agl, (46)
Интересно отметить, что попытки получить SiH3NCO просасыва-
нием паров одного только SiH3I через цианат серебра окончились
неудачей [49]. Разложение комплекса сопровождается образо-
ванием Si(NCO)4. В настоящее время показано, что цианат серебра
каталитически разлагает SiH3NCO при комнатной температуре,
однако разбавление паров SiH3I азотом в процессе реакции приво-
дит к замедлению разложения, что позволяет выделить SiH3NCO,
хотя выход продукта невелик (до 25%) [48].
SiF3NCO (т. кип. —6,0°). Трифтороизоцианатосилан при ком-
натной температуре неустойчив и имеет тенденцию к диспропор-
ционированию при температуре кипения. Трифтороизоцианато-
силан получают медленным добавлением трехфтористой сурьмы к
кипящему Si(NCO)4 [122]. Реакция протекает без катализатора.
Выход продукта достаточно велик, однако одновременно образуют-
ся относительно большие количества SiF2(NCO)2 и SiF(NCO)s.
Трифтороизоцианатосилан можно также получить вместе с
208
Глава, 7
SiFz(NCO)2 и SiF(NGO)s пропусканием смеси паров SiF4 и Si(NCO)<
через трубку, находящуюся при температуре 700° [110].
SlF^NCO)^ (т. пл. —75,0°, т. кип. 68,6°). Дифтородиизоциа-
натосилан получают вместе с SiFsNGO методом, описанным выше
[НО, 122].
StF(NCO)3 (т. пл. —29,2°, т. кип. 134,3°). Фторотриизоцианато-
силан получают из Si(NCO)4 методом, описанным для SiFsNGO
[110, 122].
SiClsNCO (т. пл. —69°, т. кип. 86,8°). Трихлороизоцианато-
силан можно получить вместе с SiG12(NCO)2 и Si(NGO)4 очень мед-
ленным добавлением цианата серебра к взятому в большом из-
бытке SiCl4, растворенном в смеси органических растворителей.
Реакция протекает с выделением тепла. Это соединение можно
также синтезировать вместе с SiC12(NCO)2 и SiCl(NGO)s по реакции
перераспределения. Тетрахлорид кремния и Si(NCO)4 нагревают в
трубке Кариуса в течение 70 час при 135°. Получают хороший вы-
ход всех указанных продуктов. Вычислено, что для достижения
вполне статистического распределения* необходима выдержка
при 135° в течение 1 недели. Продукты образуются значительно
быстрее, если пары SiCl4 и Si(NGO)4 пропускают через трубку, наг-
ретую до 600° [123].
SiCl3(NCO)3 (т. пл. —80°, т. кип. 117,8°). Дихлородиизоциана-
тосилан получают методами, описанными для SiClsNGO [123].
Это соединение можно также получить реакцией общего типа,
при которой наиболее летучие компоненты отгоняют из системы.
Так, если нагревать Si(NGO)4 и CeHsGOCl (или подобные высоко-
кипящие хлориды), то из смеси можно отогнать SiG12(NGO)2 и
SiCl(NGO)3 [124].
SiCl(NCO)3 (т. пл. —35°, т. кип. 152°). Хлоротриизоцианатоси-
лан получают методами, описанными для SiGlsNGO [123] и
SiC12(NCO)2 [124].
StfNCO'ji (т. пл. 26,0°, т. кип. 185,6°). Изучение инфракрасных
спектров и спектров комбинационного рассеяния показывает,
что это соединение имеет структуру изоцианата [125]. Тетраизо-
цианат кремния легко получают добавлением SiCl4 к суспензии
цианата серебра в бензоле. Реакция заканчивается в течение полу-
часа при нагревании смеси на паровой бане [126]. В противопо-
ложность ранним сообщениям в этой реакции тетрацианата крем-
ния Si(OGN)4 не образуется [127 ]. 81(ХСО)4можно также получить,
воздействуя SiCl4 на цианат калия, растворенный в смеси расплав-
ленных хлоридов лития и калия [128].
* Имеется в виду распределение групп NCO- и С1_ в молекулах хлорои-
зоцианат осилена с образованием SiCl3NCO, SiCh(NCO)2, Si(NC0)4 и т. п.—
Прим, перев.
III. Производные силана
209
3. Изотиоцианаты
SiH3NCS (т. пл. —51,8°, т. кип. 84,0°). Силилизотиоцианат,
подобно другим тиоцианатам кремния, по-видимому, имеет такую
же изоструктуру [129, 130]. Это соединение подобно SiHsNCO,
но отличается от CHsNGS линейной структурой (симметричный
волчок) [129—132]. Силилизотиоцианат разлагается при комнат-
ной температуре в течение 2—3 недель.
Это соединение получают просасыванием паров SiHsI над
тиоцианатом серебра при комнатной температуре
SiH314- AgNCS---► SiHsNCS + Agl. (47)
Метод синтеза, описанный Мак-Дайармидом, приведен ниже [49].
Типичная методика синтеза заключается в следующем: стеклян-
ную реакционную трубку, содержащую тиоцианат серебра (20 г)
в смеси с стеклянной ватой, подсоединяют к вакуумной системе
через ловушку, которую можно затем погрузить в жидкий азот.
Пробирку, содержащую SiHsI (1,1560 г), присоединяют к другому
концу реакционной трубки посредством конусного шлифа с кра-
ном. Систему откачивают в течение 2—3 час для удаления следов
влаги, адсорбированной на тиоцианате серебра и стеклянной вате.
Затем ловушку погружают в жидкий азот и медленно прокачивают
SiHsI через реакционную трубку. Продукты конденсируются в ло-
вушке с жидким азотом. Прокачивание следует вести непрерывно,
в противном случае водород, выделяющийся в процессе реакции
(вследствие разложения), сильно замедлит скорость потока SiHsI
через реакционную трубку. Длительный контакт с солью серебра
обычно уменьшает выход продукта. С целью понижения скорости
разложения в процессе реакции скорость потока SiHsI регулируют
путем установки крана в таком положении, чтобы реакционная
трубка при прикосновении была едва теплой. Время пропускания
SiHsI через реакционную трубку составляет 2 час. Если в процес-
се очистки,описанной ниже, обнаруживают непрореагировавшигт
SiHsI, его можно повторно обработать тиоцианатом серебра, для
чего требуется свежая порция соли серебра. (По-видимому, реак-
ция протекает только на поверхности частиц тиоцианата серебра,
а площадь реакционной поверхности сильно зависит от равномер-
ности распыления соли серебра на стеклянной вате.)
Неочищенный SiHsNGS при температуре —64° несколько раз
перегоняют через ловушку, в которой его конденсируют. Затем
продукт отделяют от менее летучих примесей пропусканием через
ловушку при температуре —48° (выход: 0,4340 г, 66%; найденный
молекулярный вес 89,5, вычисленный 89,2).
SiCl3NCS (т. пл. —75°, т. кип. 129,5°). Трихлороизотиоцианато-
силан относительно термически устойчив, но несколько менее,
210
Глава 7
чем SiCleNCO. При комнатной температуре в течение 4 мес около
35% этого соединения разлагается на Si(NCS)4 и SiCl4, в то время
как при сопоставимых условиях соединение SiCleNCO разлагается
не более чем на 2—3 %. Трихлороизотиоцианатосилан получают до-
бавлением приблизительно эквимолярного количества тиоцианата
серебра к SiCl4 шестью отдельными порциями с промежуточным
(между добавками) получасовым нагреванием (на паровой бане) с
обратным холодильником [133, 142].
Si(NCS)t (т. пл. 143,8°, т. кип. 313,0°) [133]. Тетраизотиоцианат
кремния представляет собой термически устойчивое бесцветное
кристаллическое вещество, которое можно получить добавлением
Sid4 к суспензии тиоцианата свинца в бензоле. Реакцию закан-
чивают нагреванием на паровой бане [143]. Это соединение можно
также получить взаимодействием смеси расплавленных тиоциана-
тов натрия и калия с SiCl4 [128]. Изучение инфракрасных спектров
и спектров комбинационного рассеяния показывает, что это соеди-
нение имеет структуру изотиоцианатов [134].
4. И зоселено цианаты
SiHsNCSe (т. пл.—15,1°, т. кип. ~111°). Силилизоселеноциа-
нат при комнатной температуре медленно полимеризуется даже в
темноте. Нагревание, ультрафиолетовое облучение, наличие
AgNCSe или следов ВЕз ускоряют разложение. Это соединение по-
лучают пропусканием паров SiHsI, разбавленных азотом, над
AgNCSe при комнатной температуре [48а].
5. Азиды
SiHsN3 (т. пл. —81,8°, т. кип. ~28°). Силилазид достаточно
устойчив, чтобы его можно было выделить и изучить его свойст-
ва [71], однако он медленно разлагается в вакуумной системе с вы-
делением SiH4. Интересно отметить, что кремнийорганические ази-
ды проявляют необычную термическую устойчивость [58, 135].
Так, термическое разложение (CHeJsSiNe наступает лишь при
температуре около 500° [58].
Силилазид получают обработкой (SiHs)sN или CaHsN^iHe^
раствором HNs в ди-п-бутиловом эфире [71]:
(SiH3)3N + 4HN3---> 3SiH3N3 + NH4N3. (48)
Si(N Тетраазид кремния — сильно взрывчатые белые кри-
сталлы, чувствительные к влаге. Это соединение растворимо в бен-
золе и простом эфире. Si(Ne)4 получают с небольшим содержанием
примесей нагреванием с обратным холодильником раствора
SiCl4 в бензоле с азидом натрия в присутствии каталитических ко-
личеств ЫА1Н4 [57].
IV. Производные дисилана
211
IV. ПРОИЗВОДНЫЕ ДИСИЛАНА
А. Фтородисиланы
(т. пл. —100,4°, т. кип. —10,0°). Дисиланилфторид,
возможно, воспламеняется на воздухе, но он не обнаруживает
признаков разложения при выдерживании в виде пара в течение
25 час при 0°. Его можно получить с выходом 78% реакцией
(Si2H6)3N с BF3 при -78° [76]
(Si2H6)sN + BF3 -•-> Si2H6F + (Si2H6)2NBF2. (49)
SizF в (т. пл. —18,7° при 780 мм рт. ст., т. субл. —19,1°).
Гексафтородисилан — устойчивое вещество, которое сублими-
руется при нагревании от низких температур. Его можно легко
получить с хорошим выходом при нагревании до 50—60° SiaGle
с безводным фтористым цинком. При этом начинается бурная реак-
ция, которая протекает в течение 1 час или более без дальней-
шего подогрева. Образующийся SiaFe можно без особых трудно-
стей собрать в ловушку, погруженную в смесь ацетона с сухим
льдом [136].
Б. Хлородисиланы
SizIhCl (т. пл. —111,6°, т. кип. 40,1°). Дисиланилхлорид вос-
пламеняется на воздухе и обнаруживает незначительное терми-
ческое разложение при 18°. Однако в жидком состоянии при —24s
он претерпевает быстрое разложение в присутствии следов хлори-
стого водорода или хлорида алюминия [51 ].
Дисиланилхлорид можно получить при комнатной температуре
взаимодействием SiaHe (взятого в избытке) с хлористым водородом
в присутствии хлорида алюминия в качестве катализатора. Эта
реакция аналогична реакции (5) [39]. Но вследствие уже упомя-
нутой способности к разложению выход продукта очень невелик,
и эта реакция не рекомендуется в качестве препаративного метода.
Более удобно осуществить синтез этого соединения (с хорошим вы-
ходом) при прокачивании паров S12H5I над хлористым серебром
[51] по методу, аналогичному получению SiH3NGS, описанному
в разд. III.Е.З.
Si2HхС1в_х. С большим выходом хлорированные дисиланы обра-
зуются в результате реакции Si2He с хлористым водородом, опи-
санной выше; но ни один из них не был идентифицирован [39].
Хлородисиланы также образуются в результате мало изученной
реакции Si2He с четыреххлористым углеродом или хлороформом в
212
Глава 7
присутствии следов воздуха [43]. Соединение Si2Cl6H, по-видимо-
му, образуется, когда водород и пары SiCl4 реагируют в тлеющем
разряде [137].
Si2Cl& (т. пл. 2,5°, т. кип. 147°) [103, 138]. Гексахлородисилан
термически относительно устойчив. Его получают вместе с SiCl4
при пропускании хлора над нагретыми кремниевыми сплавами или
силицидами [103]. Это соединение можно также получить из SiCl4,
используя различные методы создания электрического разряда, в
присутствии акцепторов галогена, например водорода или металла
[103, 138]. С другой стороны, гексахлородисилан образуется в
результате реакции SiCl4 с кремнием при температуре 1000°
[103]. Наилучший метод получения гексахлородисилана одновре-
менно с хлоридами высших силанов заключается в медленном
пропускании хлора над сплавом кремния и кальция («силицид
кальция») при низкой температуре (150°). Более высокие скорости
тока хлора и более высокие температуры приводят к сильному сни-
жению выхода продукта и к увеличению количества SiCl4 [103].
При указанных выше оптимальных условиях полученный продукт
имеет приблизительно следующий состав: 65% SiCl4; 30% SiaCle;
4% Si3Cl8; 1% Si4Cl10, Si5Cl12 и Si6Cl14 [103].
В. Бромодисиланы
SiJI^Br (т. пл. —97,2°, т. кип. 69,5°). Дисиланилбромид воспла-
меняется на воздухе и обнаруживает лишь слабое разложение при
нагревании до 50°. Чистое соединение или смесь его с бромистым
водородом разлагается в очень слабой степени при выдерживании
при 0° в течение 2—3 суток; однако присутствие бромистого алю-
миния вызывает быстрое разложение при 0° [139].
Дисиланилбромид получают прогонкой паров Si2H6I над бро-
мистым серебром [139], но в относительно больших количествах
его лучше получать реакцией Si2He с бромистым водородом в при-
сутствии бромистого алюминия в качестве катализатора (при ком-
натной температуре или при —78°) [39]. Эта реакция аналогична
реакции (5). Вероятно, дисиланилбромид является наиболее под-
ходящим исходным веществом для синтеза соединений, содержащих
группы Si2H6. Его применение имеет преимущества по сравнению
с Si2H6I, так как дисиланилбромид легче возгоняется в вакууме
и может быть синтезирован в больших количествах за определен-
ный промежуток времени.
Ниже приведен метод получения, описанный Абедини и сотруд-
никами [39].
По обычной методике [140] дисилан (7,84 г, 126 ммоль) и бро-
мистый водород (3,4 г, 42 ммоль) конденсируют в боковом отростке
5-литровой реакционной колбы, соединенной с вакуумной систе-
IV. Производные дисилана
213
мой. На внутренних стенках реакционной колбы сублимирован
безводный бромистый алюминий (~0,5 г). Реагирующие вещества
перегоняют в реакционную колбу. После 5 мин выдерживания
при комнатной температуре все летучие вещества откачивают из
реакционной колбы в четыре ловушки, погруженные в жидкий
азот, с целью отделения образующегося водорода. Непрореагиро-
вавший Si2He и бромистый водород (если он имеется) удаляют из
продуктов реакции, прогоняя через ловушку, поддерживаемую
при температуре —96°, в которой конденсируются только продук-
ты реакции. Затем продукты переводят в низкотемпературную
ректификационную колонну (описанную ниже) и после дистил-
ляции получают сначала небольшие количества Si2He (температура
колонны примерно —96°), а затем чистый Si2H6Br (температура
колонны около —64°) (1,08 г, 7,68 ммоль-, выход 18% по бромис-
тому водороду; найденный молекулярный вес 141,4, вычисленный
141,1). Если температура ректификационной колонны несколько
выше, получают слегка загрязненный Si2H6Br (0,72 г, 5,1 ммоль-,
выход 12%; найденный молекулярный вес 145,0). Этот загрязнен-
ный материал можно использовать без очистки для большинства
синтезов с применением Si2H6Br (общий выход 30%). Небольшие
количества не пр среагировавшего бромистого водорода (если он
присутствует) выводят из сферы реакции; расщепления связи
кремний — кремний не наблюдалось.
Низкотемпературная ректификационная колонна, применяемая
для этого синтеза, не обязательна, но она позволяет быстрее осу-
ществить разделение и получить большие количества чистого
Si2H6Br, чем при обычной перегонке из одной ловушки в другую.
Эту колонну применяют и в других системах разделения в вакууме.
Она состоит из трубки с двойными стенками (подобно сосудам
Дьюара); внутренний диаметр трубки 72 льм, внешний диаметр
100 мм, длина трубки 250 мм. Оба конца трубки закрыты больши-
ми пробками. Через пробки пропущена стеклянная трубка дли-
ной 600 мм с внутренним диаметром 15 мм. Небольшая стеклянная
колба (диаметр 30 мм) подсоединена к одному из концов стеклян-
ной трубки так, чтобы расстояние между дном колбы и нижним
концом трубки с двойными стенками составляло 150 мм. К другому
концу стеклянной трубки присоединен притертый переходник, с
тем чтобы систему можно было соединить с вакуумом. Через верх-
нюю пробку пропущены две длинные трубки диаметром 8 мм и
один пентанный термометр. Одна из 8-миллиметровых длинных
трубок присоединена к сосуду Дьюара (емкостью 25—50 л) с жид-
ким азотом. В жидкий азот опущена нагревательная трубка или
небольшой электрический нагреватель. При пропускании тока
через нагреватель азот испаряется, и холодный газообразный азот
поступает в одну трубку и выходит через вторую 8-миллиметровую
214
Глава 7
трубку. Чем больше ток, пропускаемый через нагреватель, тем
ниже температура в трубке с двойными стенками. (50-литрового
сосуда Дьюара достаточно для поддержания температуры в колон-
не примерно —130° в течение 1 час или более с перепадом ±1°.)
Показания температуры по пентанному термометру не точны и за-
висят от положения термометра. Если необходимо, то на дно кол-
бы можно поместить небольшой магнитный сердечник для переме-
шивания содержимого колбы. Так поступают, если дистилляции
подвергают большие количества вещества.
Работа ректификационной колонны начинается с того, что ве-
щества перегоняют в колбу, погруженную в жидкий азот. Затем,
насколько возможно, понижают температуру колонны. Сосуд
Дьюара с жидким азотом удаляют из-под колбы и повышают темпе-
ратуру колонны до тех пор, пока не начнется отгонка. Температу-
ру выдерживают на одном уровне до окончания перегонки. Далее
температуру повышают, пока не начнется дистилляция следующей
фракции, и выдерживают на этом уровне. Эту процедуру выполня-
ют для каждой новой отгоняемой фракции.
SiJHхВг$_х. Большие количества полибромодисиланов были
получены в результате реакции Si2He с бромистым водородом,
описанной ранее [39], но ни одно из этих соединений не было изу-
чено.
Si^Br^ (т. пл. 95°, т. кип. 265°). Гексабромодисилан термически
относительно устойчив. Это бесцветное кристаллическое вещество,
растворимое в различных органических растворителях [105].
Несмотря на то что это соединение можно получить действием брома
на кремний, применение сплава кальций — кремний вместо крем-
ния позволяет понизить температуру реакции и ведет к существен-
ному увеличению выхода продукта [105]. В этом отношении дан-
ный синтез аналогичен синтезу Si2Cl6; отличие состоит в том, что
для переноса паров брома над нагретым кремнием (при 180—
200°) можно использовать кислород. Было установлено, что ок-
сибромиды в этом случае практически не образуются [12, 105].
Г. Иододисиланы
Sl^HJ (т. пл. —86,1°, т. кип. 102,8°). Дисиланилиодид воспла-
меняется на воздухе и при комнатной температуре разлагается в
течение нескольких часов [38]. Вероятно, это разложение ускоряет-
ся в присутствии следов йодистого алюминия, которые трудно уда-
лить из продукта. Дисиланилиодид можно получить взаимодей-
ствием Si2He с иодистым водородом при комнатной температуре в
присутствии йодистого алюминия в качестве катализатора [38]
[см. реакцию (5)].
V. Производные высших силанов
215
SizHxI6_x. Большие количества полииодосиланов получают
реакцией Si2He с иодистым водородом, описанной ранее [38],
но ни один из продуктов не был изучен.
SiJe [т. пл. (разл.) 250°]. Гексаиододисилан начинает разла-
гаться приблизительно при 250°. При медленном нагревании до
350—400° в токе азота это соединение частично разлагается на
Sil4 и твердый (Sil)x [107]. Термическая устойчивость гексагало-
генодисиланов уменьшается при последовательном замещении в
молекуле атомов хлора на атомы иода [141]. Гексаиододисилан
можно получить нагреванием Sil4 с тонко измельченным металли-
ческим серебром в течение 6 час при 280° [100, 107]:
2SiI4-|-2Ag--Si2I6 2AgI. (50)
Д. Галогеноидные производные
Устойчивые неорганические галогеноидные производные диси-
лана или высших силанов не были выделены. Так, попытки полу-
чить Si2(NGO)e оказались безуспешными [13]; Si2H6GN быстро
разлагается при низких температурах [51 ]. Это, однако, не являет-
ся неожиданностью, так как даже (GH3)3SiSi(CH3)2CN разлагается
при нагревании с обратным холодильником [144]:
х (CH3)3SiSi (CH3)2CN-> (х — 1) (СН3)3 SiCN +
+ (CH3)3Si [Si (CH3)2]X CN. (51)
V. ПРОИЗВОДНЫЕ ВЫСШИХ СИЛАНОВ
Относительно мало известно о галогенопроизводных' высших
силанов. Было выделено несколько типов молекул, например
Si4Cl10, которые обладают летучестью или растворимы в органиче-
ских растворителях, но они еще не были изучены в деталях.
Выделено довольно большое число «субгалогенидов» кремния,
например (SiBr)n или (SiBr2)n. Большей частью они представляют
собой аморфные, нерастворимые, неплавкие вещества, и их можно
рассматривать в качестве галогенопроизводных соответствующих
силанов (SiH)„ и (SiH2)n. Соединения типа (SiX2)n являются в дей-
ствительности галогенированными высшими силанами 'с общей
формулой X3Si(SiX2)nSiX3, где п очень велико. Они могут образо-
вывать циклические соединения. Если на один атом кремния при-
ходится менее двух атомов галогена, то можно ожидать образова-
ния поперечной связи между атомами кремния, что приводит к
снижению летучести и растворимости этих соединений в органиче-
ских растворителях [2].
216
Глава 7
А. Фторосиланы
Каких-либо определенных производных высших силанов не
выделено [7].
Б. Хлоросиланы
Si3HiCll был получен из Si3H8 и хлороформа [43] по методу,
описанному ранее [см. уравнение (9)].
SisCls (т. пл. —67°, т. кип. 216°) образуется в процессе получе-
ния Si2Cl8 методом, описанным ранее [7, 103].
£г4С[10 (т. кип. 150° при 15 мм рт. cm.), Si5Cl12 (т. кип. 190°
при 15 мм рт. ст.) и Sifil^ (сублимируется в вакууме при 200°)
получают в процессе синтеза SiaCle, описанного ранее [7, 103].
Эти соединения, вероятно, представляют собой смесь изомеров. Они
не были достаточно хорошо изучены [103]. При нагревании на воз-
духе пары высших хлоридов загораются.
Si5Cl12 [т. пл. (разл.) 341°] был получен нагреванием SieCl14 с
триметиламином при 70°. Вероятно, это соединение имеет структу-
ру типа тетраметилметана [145, 146].
SieCllt (т. пл. 320°) был получен по очень интересной реакции,
в которой триметиламин вызывает диспропорционирование
SiaCle [145, 146], легко протекающее при 0°:
5Si2Cle---> Si6Cl14+ 4SiCl4. (52)
Вероятно, это соединение имеет структуру типа триметилэтилме-
тана [145, 146].
lS'j10CZ22 представляет собой очень вязкое масло, которое мож-
но перегнать при 215—220° в вакууме. Его идентификация была
подтверждена измерением молекулярного веса. Это соединение
не горит, разлагается на SiCl4 и Si2Cl8 при нагревании в вакууме.
Si10Cl22 получают пропусканием смеси водорода и паров SiCl4
над кварцевым стержнем, нагретым до 1000—1100°, с последующей
конденсацией газообразных продуктов при температуре жидкого
воздуха [147].
Si^Clbi является твердым веществом, загорающимся на воз-
духе при трении. Оно растворимо в эфире. Это соединение получают
аналогично Si10Cl22 с той только разницей, что азот заменяют во-
дородом [148].
571ОС72оЯ2 — это бесцветное очень вязкое масло, разлагающее-
ся при нагревании с образованием (SiCl)^. Это соединение получают
интенсивным нагреванием смеси водорода и паров SiCl4 в «холод-
но-горячей» трубке [149, 150].
(SiClz)x представляет собой белое твердое вещество, которое
получают реакцией смеси водорода и паров SiCl4 в тлеющем раз-
Литература
217
ряде [137] или пропусканием хлора над нагретым кремнием при
контролируемых условиях [151].
[5iCZ(o,5—2.6)]Х' Соединения указанного состава могут быть
получены действием тлеющего разряда на SiHCl3 или смесь
водорода и паров SiCl4 [152]. [SiCl2,614 можно получить в виде
вязкого возгоняющегося масла при пропускании паров SiCl4
над кремнием при 1000° [153].
(SiCl)x представляет собой желтое твердое вещество, получаемое
при разложении Si10Cl20H2 в инертной атмосфере при 300° [150,
154]. Оно также образуется при продолжительном нагревании
Si2Cl6 [141].
В. Бромосиланы
Si3Br8 (т. пл. 133°) и 81^г10 [т. пл. (разл.) 185°] получают воз-
действием электрического разряда на пары SiHBr3 [155].
(SiBr^x — коричневое смолоподобное твердое вещество, раст-
воримое в неполярных органических растворителях. Оно воспла-
меняется при температурах выше 100°, а при нагревании до 300°
разлагается на (SiBr)x и Si2Bre. (SiBr2)x получают восстановле-
нием SiBr4 кремнием при 1200° [156].
(StBr)x — желто-коричневое твердое вещество, нерастворимое в
бензоле. Его получают нагреванием с обратным холодильником
Si2Bre при 300° в течение 6—8 час [141]. Его можно также получить
реакцией магния с SiBr4 в эфире [156].
Г. Иодосиланы
(Sil)x — оранжево-красное аморфное твердое вещество, нераст-
воримое во всех органических растворителях. Оно не плавится.
Это соединение получают медленным нагреванием Sial в при 350—
400° в токе азота [107].
ЛИТЕРАТУРА
1. Stone F. G. A., Hydrogen Compounds of the Group IV Elements,
Prentice-Hall, Englewood Cliffs, New Jersey, 1962.
la. E bswort h E. A. V., Volatile Silicon Compounds, Pergamon Press,
New York, 1963.
2. MacDiarmid A. G., «Silanes and Their Derivatives», in Advances
in Inorganic Chemistry and Radiochemistry, Vol. Ill, Emeleus H. J.,
Sharpe A. G., eds., Academic Press, New York, 1961, p. 207.
3. MacDiarmid A. G., Quart. Revs. (London), 10, 208 (1956).
4. Stock A., Hydrides of Boron and Silicon, Cornell Univ. Press, Ithaca,
New York, 1933.
5. Borer K., Phillips C. S. G., Proc. Chem. Soc., 1959, 189.
6. E a h о r n C., Organositicon Compounds, Butterworths, London, 1960.
7. Gmelins Handhuch der Anorganischen Chemie, Vol. 15, Verlag Chemie,
Weinheim, 1959, Part B.
218
Г ла в а 7
8. Gmelins Hanbuch der Anorganischen Chemie, Vol. 15, Verlag Chemie,
Weinheim, 1958, Part C.
9. Rochow E. G. An Introduction to the Chemistry of the Silicones,
2nd ed., Wiley, New York, 1951.
10. Post H. W., Organic Silicon Compounds, Reinhold, New York, 1949.
11. Рохов Ю., Херд Д., Льюис Р., Химия металлоорганических соеди-
нений, ИЛ, Москва, 1963.
12. S с h u m h W. С., Klein С. H., J. Am. Chem. Soc., 59, 261 (1937).
13. Forbes G. S., Anderson H. H., J. Am. Chem. Soc., 69, 3048
(1947).
14. P flugmacher A., Dahmen H., Z. anorg. allgem. Chem., 290,
184 (1957).
15. Panckhurst D. J., Wilkins C. J., Craighead P. W.,
J. Chem. Soc., 1955, 3395.
16. EmeHus H. J., Maddock A. G., J. Chem. Soc., 1944, 293.
17. Booth H. S., S till well W. D., J. Am. Chem. Soc., 56, 1529
(1934).
18. J о 11 у W. L., Synthetic Inorganic Chemistry, Prentice-Hall, Engle-
wood Cliffs, New Jersey, 1960, Chap. 10.
19. Sanderson R. T., Vacuum Manipulation of Volatile Compounds,
Wiley, New York, 1948.
20. Dodd R. E., Robinson P. L., Experimental Inorganic Chemi-
stry, Elsevier, New York, 1954, Chap. 2.
21. Stock A. Hydrides of Boron and Silicon, Cornell Univ. Press, Ithaca,
New York, 1933, Chap. 30.
22. Dushman S., Lafferty J. M., Scientific Foundations of Vacuum
Technique, Wiley, New York, 1949.
23. Farkas A., Melville H. W., Experimental Methods in Gas
Reactions, Macmillan, London, 1939.
24. Whitmore F. С., P i e t r u s z a E. W., Sommer L. H.,
J. Am. Chem. Soc., 69, 2108 (1947).
25. S c h u m b W. C., Young R. C., J. Am. Chem. Soc., 52, 1464 (1930).
26. Ill у м б, в книге «Неорганические синтезы», сб. I, Издатинлит, Москва,
1951.
27. Buff Н., Wohler F., Annalen, 104, 94 (1857).
28. Т а у 1 о г A. G., Walden В. V. de G., J. Am. Chem. Soc., 66, 842
(1944).
29. С о m b e s C., Compt. Rend., 122, 531 (1896).
30. E a b о r n C., Organosilicon Compounds, Butterworths, London, 1960,
pp. 36—45.
31. British Thomson-Houston Co., англ. пат. 626519 (1949); throgh Chem.
Ahstr., 44, 2547 (1950).
32. S c h u m b W. C., Gamble E. L., J. Am. Chem. Soc., 54, 3943
(1932).
33. E a b о r n C., Organosilicon Compounds, Butterworths, London, 1960,
pp. 209—212.
34. Stock A., Somieski C., Chem. Ber-, 50, 1739 (1917).
35. Stock A., Somieski C., Chem. Ber., 52B, 695 (1919).
36. Emeleus H. J., Maddock A. G., Reid C., J. Chem. Soc.,
1941, 353.
37. Fritz G., Kummer D., Z. anorg. allgem. Chem., 304, 322 (1960).
38. Ward L. G. L., M a c D i a r m i d A. G., J. Am. Chem. Soc., 82,
2151 (1960).
39. Ahedini M., Van Dyke C.H., MacDiarmidA. G., J.
Inorg. Nucl. Chem., 25, 307 (1963).
40. Anderson H. H., J. Am. Chem. Soc., 80, 5083 (1958).
Литература
219
41. Anderson Н. Н., Hendifar A., J. Am. Chem. Soc., 81, 1027
(1959).
42. Anderso n H. H., J. Am. Chem. Soc., 81, 4785 (1959).
43. S t о с к A., S t i e b e 1 e r P., Chem. Ber., 56B, 1087 (1923),
44. Emeleus H.J., Robinson S. R., J. Chem. Soc., 1947, 1592.
45. Anderson H. H., Fischer H., J. Org. Chem., 19, 1296 (1954).
46. E a b о r n C., J. Chem. Soc., 1950, 3077.
47. E a horn C., Organosilicon Compounds, Butterworths, London, 1960,
p. 174.
48. E b s w о r t h E. A. V., Mays M. J., J. Chem. Soc., 1962, 4844.
48a. Ebsworth E.A. V., Mays M. J., J. Chem. Soc., 1961, 4879;
1963, 3893.
49. M a c D i a r m i d A. G., J. Inorg. Nucl. Chem., 2, 88 (1956).
50. A b e d i n i M., Ph. D., Dissertation, University of Pennsylvania,
1963.
51. Craig A.D., Urenovitch J. V., MacDiarmid A.G.,
J. Chem. Soc., 1962, 548.
52. Emeleus'H. J., MacDiarmid A.G., Maddock A.G.,
J. Inorg. Nucl. Chem., 1, 194 (1955).
53. A b e d i n i M., MacDiarmid A. G., unpublished results.
54. Booth H.S., Stillwell W. D., J. Am. Chem. Soc., 56, 1531
(1934).
55. В u f f O., Albert C., Chem. Ber., 38, 53, 222 (1905).
56. Booth H. S., S w i n e h a r t C. F., J. Am. Chem. Soc., 57, 1333
(1935).
57. Wiberg E., M i c h a u b H., Z. Naturforsch., 9b, 500 (1954).
58. Connolly J. W., U г г у G., Inorg. Chem., 1, 718 (1962).
59. Hurd D. T., J. Am. Chem. Soc., 67, 1545 (1945).
60. Cottrell T., The Strength of Chemical Bonds, 2nd ed., Butterworths,
London, 1958, pp. 270—283.
61. E me leu s H. J., Onyszchuk M., J. Chem. Soc., 1958, 604.
62. E a born C., Organosilicon Compounds, Butterworths, London, 1960,
pp. 171—173.
63. О n у s z c h u к M., Can. J. Chem., 39, 808 (1961).
64. Crane E. J., Chem. Eng. News, 24, 1233 (1946).
65. Sternbach B., MacDiarmid A. G., J. Am. Chem. Soc.,
83, 3384 (1961).
66. К r i n e r W. A., MacDiarmid A. G., Evers E. C., J. Am.
Chem. Soc., 80, 1546 (1958).
67. О p л о в H. Ф., ДАН СССР, 114, 1033 (1957).
68. Stock A., Somieski C., Chem. Ber., 54, 740 (1921).
69. Ruff O., Chem. Ber., 41, 3738 (1908).
70. Anderson H. H., J. Am. Chem. Soc., 73, 5802 (1951).
71. Ebsworth E.A. V., Jenkins D.R., Mays M.J.. Sug-
den T. M., Proc. Chem. Soc., 1963, 21.
72. В i г к о f e r L., Ritter A., R i c h t e r P., Angew. Chem. Intern.,
Ed. in Engl., 1, 267 (1962).
73. В i t h e г T. А., К n о t h W. H., Lindsey R. V., Jr., Shar-
key W. H., J. Am. Chem. Soc., 80, 4151 (1958).
74. Barg А. В., К u 1 j i a n E. S., J. Am. Chem. Soc., 72, 3103 (1950).
75. Sujishi S., Witz S., J. Am. Chem. Soc., 79, 2447 (1957).
76. A b e d i n i M., MacDiarmid A. G., Inorg. Chem., 2, 608 (1963).
77. Ebsworth E. A. V., Emeleus H J., J. Chem. Soc., 1958, 2150.
78. Sujishi S., Chemistry of Group IV Hydrides, Final Report, Office
of Ordnance Research, Contract No. DA-11-022-0RD-1264, Aug., 1957.
79. Beck e-G6ehring M., Krill H., Ber., 94, 1059 (1961).
220
Глава 7
79а. Fritz G., Z. anorg. allgem. Chem., 280, 332 (1955).
80. F r i t z G., Kummer D., Z. anorg. allgem. Chem., 308, 105 (1961).
81. Fritz G., Kummer D., Z. anorg. allgem. Chem., 310, 327 (1961).
82. Fritz G., Kummer D., Ber., 94, 1143 (1961).
83. Fritz G., Kummer D., Z. anorg. allgem. Chem., 306, 191 (1960).
84. А у 1 e 11 B. J., Ellis I. A., J. Chem. Soc., 1960, 3415.
85. Wolfe J. K., Cook N. C., Abstr. Papers, 128th National Meeting
of the American Chemical Society, Minneapolis, Minn., 1955, p. 48M.
86. Clasen H., Angew. Chem., 70, 179 (1958).
87. Bailey D. L., Wagner G.H., пат. США 2732280—2 (1956).
88. Erickson С. E., Wagner G. H., пат. США 2627451 (1953);
through Chem. Ahstr., 48, 1420 (1954); пат. США 2735861 (1956); through
Chem. Abstr., 50, 13986 (1956).
89. Forbes G.S., Anderson H. H. J. Am. Chem. Soc., 66, 931
(1944).
90. West R., R о c h о w E. G., in Inorganic Syntheses, Vol. 4, Bailar J.,
ed., McGraw-Hill, New York, 1953, p. 41.
91. Stock A., Zeidler F., Chem. Ber., 56B, 986 (1923).
92. Dudani P. G., Plust H. G., Nature, 194, 85 (1962).
93. О p i t z H. E., Peake J.S., Nebergall W. H., J. Am. Chem.
Soc., 78, 292 (1956).
94. Stock A., Somieski C., Chem. Ber., 51, 989 (1918).
95. Speier J.L., Zimmerman R. E., J. Am. Chem. Soc., 77, 6395
(1955)
96. Я к у б о в и ч А. Я., М о ц а р е в Г. В., ДАН СССР, 91, 277 (1953).
97. Якубович А. Я., М о ц а р е в Г. В., ЖОХ, 26, 568 (1956).
98. Hoffman С. J., G u t о w s к у Н. S., in Inorganic Syntheses, Vol. 4,
Bailar J., ed., McGraw-Hill, New York, 1953, p. 145.
99. Gmelins Handbuch der Anorganischen Chemie, Vol. 15, Verlag Chemie,
Weinheim, 1959, Part B, pp. 672—673.
100. Schumb W. C., Chem. Rev., 31, 587 (1942).
101. J ackson К. E., Chem. Rev., 25, 67 (1939).
102. Baxter G. P., Weather ill P. F., Holmes E. O., Jr.,
J. Am. Chem. Soc., 42, 1194 (1920).
103. Шум б, Гэмбл, в книге «Неорганические синтезы», сб. 1, Издат-
инлит, Москва, 1951.
104. Kennard S. М. S., McCusker Р. A., J. Am. Chem. Soc., 70,
1039 (1948).
105. Ш у м б, в книге «Неорганические синтезы», сб. II, Издатинлит, Москва,
1951.
106. Gatterman L., Chem. Ber., 22, 186 (1889).
107. Schwarz R., Pflugmacher A., Chem. Ber., 75B, 1062 (1942).
108. Артур и Симонс, в книге «Неорганические синтезы», сб. III,
Издатинлит, Москва, 1952.
109. Booth Н. S., S w i n е h а г t С. F., J. Am. Chem. Soc., 54, 4750
(1932).
110. Anderson Н. Н., J. Am. Chem. Soc., 72, 2091 (1950).
111. Schumb W. C., Anderson H. H., J. Am. Chem. Soc., 58, 994
(1936).
112. Schumb W. C., Anderson H. H., J. Am. Chem. Soc., 59, 651
(1937).
113. Besson A., Compt. Rend., 112, 611, 1314 (1891).
114. Friedel C., Chem. Ber., 2, 57 (1869).
115. Besson A., Compt. Rend., 112, 788, 1447 (1891).
116. Linton H. R., Nixon E. R., Spectrochim. Acta, 10, 299 (1958).
117. Muller N., Bracken R. C., J. Chem. Phys., 32, 1577 (I960).
Литература
221
118. Sheridan J., Turner A. C., Proc. Chem. Soc., 1960, 21.
119. Seyferth D., Kahlen N., J. Am. Chem. Soc., 82, 1080 (1960).
120. E melons H. J., Onyszchuk M., К u c h e n W., Z. anorg.
allgem. Chem., 283, 74 (1956).
121. Kaczmarczyk A., U г г у G., J. Am. Chem. Soc, 81, 4112 (1959).
122. Forbes G. S., Anderson H. H., J. Am. Chem. Soc., 69, 1241
(1947).
123. Anderson H. H., J. Am. Chem. Soc., 66, 934 (1944).
124. Anderson H. H., J. Am. Chem. Soc., 75, 1576 (1953).
125. Miller F. A., Carlson G. L., Spectrochim. Acta, 17, 977 (1961).
126. Forbes G. S., Anderson H. H., J. Am. Chem. Soc., 62, 761
(1940).
127. Anderson H.H., private communication, 1963.
128. Sundermeyer W., Z. anorg. allgem. Chem., 313, 290 (1961).
129. MacDiarmid A. G., Maddock A. G., J. Inorg. Nucl. Chem.,
1, 411 (1955).
130. Jenkins D. R., Kewley R., Sugden T. M., Proc. Chem.
Soc,, 1960, 220.
131. Jenkins D. R., Kewley R., Sugden T. M., Trans. Faraday
Soc., 58, 1284 (1962).
132. Ebsworth E. A. V., Mould R., Taylor R., Wilkin-
son G. R., Woodward L. A., Trans. Faraday Soc., 58,1069 (1962).
133. Anderson H. H., J. Am. Chem. Soc., 69, 3049 (1947).
134. Carlson G. L., Spectrochim. Acta, 18, 1529 (1962).
135. West R., Thayer J. S., J. Am. Chem. Soc., 84, 1763 (1962).
136. Schumb W. C., Gamble E. L., J. Am. Chem. Soc., 54, 583 (1932).
137. Schwarz R., P i e ts c h G., Z. anorg. allgem. Chem., 232, 249 (1937).
138. Stock A., Brandt A., Fischer H., Chem. Ber., 58, 643 (1925).
139. Ward L. G. L., MacDiarmid A. G., J. Inorg. Nucl. Chem., 20,
345 (1961).
140. В e t h к e G. W., Wilson M. K., J. Chem. Phys., 26, 1107 (1957).
141. Pflugmacher A., Rohrman I., Z. anorg. allgem. Chem.,
290, 101 (1957).
142. Anderson H. H., J. Am. Chem. Soc., 67, 223 (1945).
143. Reynolds J. E., J. Chem. Soc., 89, 397 (1906).
144. Urenovitch J. V., MacDiarmid A. G., J. Am. Chem. Soc.,
85, 3372 (1963).
145a. U г г у G., Nuss J. W., Abstr. of Papers, 144th National Meeting
of the American Chemical Society, Los Angeles, Calif., 1963, p. 16K; 6.
private communication, 1963.
146. Kaczmarczyk A., Millard M., U г г у G., J. Inorg. Nucl.
Chem., 17, 186 (1961).
147. Schwarz R., Meckbach H., Z. anorg. allgem. Chem., 232,
241 (1937).
148. Schwarz R., Danders C., Chem. Ber., 80, 444 (1947).
149. Schwarz R., T h i e 1 R., Z. anorg. allgem. Chem., 235, 247 (1938).
150. Schwarz R., Angew. Chem., 51, 328 (1938).
151. Антипин П. Ф., Сергеев В. В., ЖПХ, 27, 784 (1954).
152. Hertwig К. A., Wiberg Е., Z. Naturforsch., 6b, 336 (1951).
153. Ro chow E. G., D i d t s c h e n к о R., J. Am. Chem. Soc., 74, 5545
(1952).
154. Schwarz R., Gregor U., Z. anorg. allgem. Chem., 241, 395
(1939).
155. Besson A., Fournier L., Compt. Rend., 151, 1055 (1910).
156. Schmeisser M., Schwarzmann M., Z. Naturforsch., 11b, 278
(1956).
ГЛАВА 8
СОЛЕОБРАЗНЫЕ ГИДРИДЫ
Иессер
Университет Тафта, Медфорд, Массачусетс
1. ВВЕДЕНИЕ
А. Общие замечания
Подавляющее большинство известных гидридов металлов полу-
чено непосредственно из металлов и водорода; препаративные ме-
тодики несколько различаются в зависимости от термодинамики и
кинетики отдельных реакций. В случае солеобразных гидридов,
особенно гидридов щелочных и щелочноземельных металлов, реак-
ции, как правило, идут в одном направлении, и в продукте, остыв-
шем до комнатной температуры, обнаруживается только одна фаза
гидрида. В случае металлоподобных гидридов, т. е. гидридов се-
мейства лантанидов, актинидов, металлов группы титана и группы
ванадия, гидрида палладия и т. д., методы синтеза почти одина-
ковы. Однако состав и структура образующихся фаз значительно
сильнее зависят от условий синтеза, поэтому для синтеза опреде-
ленного гидрида нужно знать диаграмму состояния системы ме-
талл — водород. В этой главе рассмотрены в основном солеобразные
гидриды, т. е. гидриды щелочных и щелочноземельных металлов,
за исключением бериллия, а также гидриды некоторых лантани-
дов (европия и иттербия).
Краткое описание свойств отдельных гидридов можно найти
в книге Харда [1]. Подробное описание и обсуждение свойств
гидридов имеется в работе Гибба [2].
Б. Упругость диссоциации
Солеобразные гидриды при нагревании до высоких температур
обратимо разлагаются на металл и водород. Фаза металла способ-
на растворять некоторое количество водорода, а фаза гидрида
несколько обедняется водородом. Взаимная растворимость ме-
няется от металла к металлу, но во всех случаях увеличивается
при повышении температуры так же, как увеличивается и равно-
весная упругость водорода над гидридом.
I. Введение
223
Рис. 1. Схема изотерм равновесия для системы:
металл — гидрид металла М—MHS.
Такое поведение системы графически показано на рис. 1
в виде серии изотерм в координатах давление — состав для гипо-
тетической системы металл — водород. При изотермическом вве-
дении водорода в металл газ растворяется в металле до тех пор,
пока не достигается насыщение при составе п' атомов водорода на
атом металла, давлении рг и температуре Дальнейшее увеличе-
ние содержания водорода приводит к образованию гидрида.
Образование гидрида происходит при постоянной упругости во-
дорода рг (плато) до тех пор, пока при составе п" атомов водорода
на атом металла не исчезнет фаза металла. В этой точке фаза гид-
224
Глава 8
рида имеет равновесный дефицит водорода. Дальнейшее повыше-
ние содержания водорода в твердой фазе сопровождается быстрым
увеличением упругости водорода вплоть до предельного содержа-
ния водорода, соответствующего почти стехиометрическому соста-
ву гидрида (s атомов Н на атом металла). Однако состав гидрида
зависит от используемого давления водорода. По мере увеличе-
ния температуры от через Тч к Тз предельные значения насы-
щения п' и п" сближаются, а предельный состав фазы гидрида
при определенном давлении, например при 1 атм, все более и более
отличается от стехиометрического состава. Поэтому, если необ-
ходимо получить возможно более чистый гидрид, синтез следует
проводить или по крайней мере заканчивать при самой низкой
температуре, которую допускает кинетика процесса гидрирования.
П. АППАРАТУРА И МЕТОДИКА СИНТЕЗОВ
А. Общие замечания
На рис. 2—4 показана аппаратура, пригодная для получения
-небольших количеств гидридов большинства металлов. Аппара-
тура состоит из обычной стеклянной системы высокого вакуума,
системы очистки водорода с приборами для измерения количе-
ства газообразного водорода и реакционной камеры. Последняя
система лишь немногими деталями отличается от обычной.
Металл отвешивают в лодочке в сухом боксе, заполненном ар-
гоном; затем лодочку переносят в контейнер, который закрывают,
вынимают из бокса, присоединяют к вакуумной системе и откачи-
вают. В систему вводят очищенный водород и определяют его ко-
личество, измеряя давление, объем и температуру. Затем нагре-
вают печь для осуществления реакции. Изменение температуры
и время прогрева выбирают в зависимости от свойств гидрида.
Количество непрореагировавшего водорода определяют при неко-
торой определенной температуре печи, что позволяет рассчитать
количество вступившего в реакцию газа. Если необходимо достиг-
нуть максимального поглощения водорода при высоком конечном
равновесном давлении, можно отключить реакционную камеру,
добавить в систему еще некоторое отмеренное количество водорода
и повторить обработку образца.
Б. Реакционная камера
Обычно реакционную камеру (рис. 3) выполняют в виде гори-
зонтальной трубки с вакуумплотным соединением и охлаждаю-
щим змеевиком на конце. Трубка соединяется с линией при помо-
щи шарового шлифа. Между соединениями имеется кран. Для из-
II. Аппаратура и методика синтезов
225
мерения и регулирования температуры в трубке должны быть один
или несколько карманов для термопар. Для синтеза небольших
количеств гидрида реакционную камеру можно изготовить из
трубки диаметром 2,5 см, а для обогрева камеры можно использо-
вать обычную трубчатую печь размерами 30,5x3,2 см.
Гис. 2. Схема вакуумной системы и системы измерения
количества водорода:
1 — к ртутному диффузионному насосу и масляному форвакуум-
ному насосу; 2 — к источнику газообразного водорода (рис. 4);
з — к манометру Мак-Леода или ионизационному манометру; 4 —
к ртутному манометру; S — к насосу Теплера (необязательно);
6 — охлаждаемая ловушка; 7 — калиброванная колба; 8 — шаро-
вой шлиф к реакционной камере и печи (рис. 3).
Рис. 3. Реакционная камера и печь:
1 — лодочка; 2 — охлаждающие змеевики; 3 — печь; 4 — спай металл—
стекло; 5 —кольцевая прокладка; в — проволочная набивка из нержаве-
ющей стали; 7 —образец; 8 —термопара; 9 — кран.
!/„ 16 Заказ № 547
226
Глава 8
Реакционную камеру для синтеза большинства гидридов мож-
но изготовить из кварца. Для синтеза гидрида лития нужна каме-
ра из нержавеющей стали (типа 347 или 316), так как стекло и
кварц быстро разъедаются металлическим литием. Пары некоторых
Рис. 4. Система подачи водорода и его очистки:
1 — ртутный затвор; 2— устройство для каталитической очистки; 3 — осушительная
трубка; 4 — печь; S — манометр; 6 — резьбовое соединение; 7—спай металл—стекло;
S — игольчатый клапан; 9 — проволочная набивка из нержавеющей стали; 10 — ре-
дукторный клапан; 11 — стеклянный кран; 12 — баллон высокого давления; 13 —
металлический уран или титан (геттер).
других металлов также разъедают кварц (автор наблюдал это
явление в случае бария), но весьма медленно.
Для загрузки кварцевой реакционной камеры открывают
коническую притертую пробку, а для загрузки камеры из нержа-
веющей стали снимают муфту с резьбой и с уплотнением в виде
кольцевой прокладки круглого сечения. Если синтез ведется в
больших бомбах из нержавеющей стали, вместо кольцевого уплот-
нения можно использовать фланцевое соединение с плоской
кольцевой прокладкой. На соединительной трубке бомбы из не-
ржавеющей стали ставится вакуумплотный игольчатый клапан.
Можно использовать также вертикальную печь с реакционной
камерой тигельного типа. В таких аппаратах по методу Стокбер-
гера были выращены монокристаллы гидрида лития [3].
II. Аппаратура и методика синтезов
227
В. Очистка водорода
На рис. 4 приведена аппаратура, в которой получают водород
высокой чистоты для синтеза гидрида. Водород из баллона посту-
пает через камеру для каталитической очистки, через осушитель-
ную трубку, минуя ртутный предохранительный затвор, в сталь-
ную трубку, наполненную металлическим титаном или ураном и
способную выдержать высокое давление. Эта трубка помещена в
печь; при нагревании до сравнительно невысоких температур в
трубке происходит поглощение водорода под давлением 1 атм и
образуется гидрид титана или гидрид урана. При более высоких
температурах водород выделяется и поступает в баллон высокого
давления. При проведении синтеза водород из этого баллона про-
пускают через редуктор, мимо второго затвора, в аппарат, где
производится синтез. Если водород загрязнен кислородом или азо-
том, титан или уран действуют по отношению к примесям как
геттеры, так как металлы поглощают их вместе с водородом, а при
нагревании выделяют только водород.
В тех случаях, когда не требуется очень высокой чистоты,
геттер и баллон высокого давления не нужны, и газ можно подавать
непосредственно ко второму затвору, который отделяется кранами
от первого затвора и вакуумной линии.
Г. Материалы и их обработка
Все рассматриваемые металлы и их гидриды легко взаимодей-
ствуют с компонентами воздуха, так что работать с ними прихо-
дится в защитной атмосфере. Для большинства операций необхо-
димо использовать сухой наполненный аргоном бокс, но прежде
для работ небольшого масштаба применяли другое оборудование
[4, 5]. Большой сухой бокс такого рода был описан в работе
Гибба [6]. Для большей части операций можно пользоваться про-
дажным сухим боксом меньшего размера, но тогда необходимо пол-
ностью защитить материалы от воздействия воздуха путем отка-
чивания бокса или пропускных камер. При конструировании обо-
рудования для получения гидридов приходится строго учитывать
размер пропускных камер. При планировании работы важно учесть
необходимость откачивания бокса или пропускных камер — все,
что вносится в бокс, должно подвергаться откачиванию или нахо-
диться в контейнерах, способных противостоять внутреннему дав-
лению, равному 1 ати. Нельзя использовать дерево, бумагу и
другие содержащие влагу материалы.
По возможности металлы для гидрирования следует извлекать
из товарной упаковки в сухом боксе. Если это по какой-либо при-
чине невозможно, металл очищают от пленки окиси и нитрида
!/Я 16*
228
Глава 8
под маслом, промывают безводным эфиром и смоченным в эфире
переносят в сухой бокс. Затем уже в боксе еще раз тщательно от-
скабливают или обрезают металл с поверхности. Большинство ме-
таллов очищают таким образом при помощи ножа или другого
простого инструмента, который можно внести в бокс.
В большинстве случаев металл бывает достаточно чист для
получения из него гидрида, в противном случае его приходится
очищать вакуумной дистилляцией в аппаратуре из нержавеющей
стали. Подобного рода вакуумная реторта описана в работе
Дугласа и др. [7].
Образец металла для синтеза гидрида обычно помещают в ло-
дочку, подобную той, которую применяют для сжигания проб.
Если реакционная камера имеет диаметр 2,5 см, то можно исполь-
зовать стандартную лодочку для сжигания длиной 89 мм, шириной
15,9 мм и глубиной 9,5 мм. Можно использовать лодочки из желе-
за Армко, нержавеющей стали типов 347 и 316 или из молибдена.
Тонкие стальные лодочки можно изготовить методом прессования
или отбортовкой, такие лодочки не пропускают расплавленных
веществ. Для изготовления других лодочек нужно применять свар-
ку или иные методы. Вследствие летучести рассматриваемых метал-
лов их нельзя непосредственно подвергать действию вакуума при
высоких температурах, особенно в отсутствие фазы гидрида.
В нескольких работах по измерению упругости диссоциации рас-
смотрены закрытые контейнеры, ппопускающие водород и не про-
пускающие пары металлов.
Д. Определение количества водорода в образце
Количество водорода, поглощенного образцом, определяют,
измеряя давление, объем и температуру до и после реакции.
В вакуумной системе имеется стеклянная колба известного объема,
предварительно откалиброванная по весу воды. Это позволяет
калибровать объемы остальных частей аппаратуры, измеряя пара-
метры газа после его распределения в измеряемом объеме из пред-
варительно наполненной этим газом колбы. Для этого измеряемые
объемы предварительно эвакуируют. Для повышения точности в
систему можно включить не одну, а несколько колб разных разме-
ров, или газовую бюретку, заполняемую ртутью, или и то и
Другое.
Следует учесть поправку на потери водорода в результате его
диффузии через стенки реакционной камеры, если она изготовле-
на из нержавеющей стали. Диффузия становится заметной при
температурах выше 650°. Скорость диффузии измеряют непосред-
ственно в аппаратуре по изменению давления в холостом опыте.
Для реакционной камеры, приведенной на рис. 3, автор опреде-
11. Аппаратура и методика синтезов
229
лил потерю водорода, равную 1,3-10~6 молъ/мин-атм '/• при тем-
пературе 720° [8].
Для определения количества поглощенного образцом водорода
в конце опыта при охлаждении образца до комнатной темпера-
туры или для определения поглощения водорода в процессе опыта
нужно определить эффективный «горячий объем» реакционной
камеры при различных температурах печи. Для этого наполняют
систему, откалиброванную при комнатной температуре, до давле-
ния несколько ниже 1 атм и определяют зависимость манометри-
ческого давления от температуры печи, после чего рассчитывают
эффективный объем системы, включая реакционную камеру, при
условном допущении, что печь также имеет комнатную темпера-
туру-
Для достижения максимальной точности нужно определять
комнатную температуру в нескольких точках по всей длине ва-
куумной системы и в колбе и для подсчета количества газа в систе-
ме пользоваться средневзвешенной температурой, рассчитанной по
температурам и объемам различных частей системы.
В целях достижения максимальной точности нужно также под-
держивать постоянный уровень охладителя в сосуде Дьюара,
охлаждающего ловушку. Если в качестве охладителя используют
твердую углекислоту, то для теплопередачи следует пользоваться
не ацетоном, а тяжелой хлорированной жидкостью, например
трихлорэтиленом, так как при этом перепады температур в ловуш-
ке будут меньше.
Если аппаратура предназначена также для количественного
удаления водорода из образца или для анализа проб на содержа-
ние водорода, в систему необходимо включить также насос Тенде-
ра. Последние порции водорода удаляются из образцов с большим
трудом,а щелочные и щелочноземельные металлы вносят дополни-
тельные трудности, обусловленные летучестью этих металлов.
Выделение водорода из большинства солеобразных гидридов мож-
но облегчить добавлением металлического олова, так как образо-
вание интерметаллических соединений между оловом и металлом
способствует количественному освобождению водорода.
Е. Анализ
В этом разделе рассмотрены только такие методы, которые
можно использовать для всех или большинства гидридов. Основной
метод анализа гидридов заключается в разложении гидролизом с
выделением водорода и в последующем титровании образующегося
при гидролизе раствора. Колба для отвешивания проб снабжена
краном или стеклянным шлифом с делительной воронкой и боко-
вой отводной трубкой с краном. Жидкость вводят через воронку,
15 Заказ № 547
230
Глава 8
а боковую трубку подсоединяют к измерительной системе так,
чтобы выделяющийся водород вытеснял из баллона воду в сосуд
для взвешивания.
В зависимости от характера пробы для ее разложения вводят
ту или иную жидкость. Это может быть вода, ледяная вода или
чистый сухой диоксан. После разложения в колбу добавляют воду,
если гидроокись металла растворима, или соляную кислоту,
если гидроокись в воде нерастворима. Общий объем добавленной
жидкости должен быть точно известен. Если используют соляную
кислоту, то должна быть точно известна ее концентрация. Кисло-
ту, остающуюся в воронке, нужно смыть в колбу, если раствор
предполагают титровать.
Содержание металла в пробе можно определить титрованием
раствора после гидролиза. Если гидроокись растворима, можно
определить также содержание карбоната. Для этого раствор тит-
руют сначала с бромтимоловым синим до перехода при pH 7, а за-
тем с метиловым оранжевым до перехода при pH 4.
Водород можно определить и иначе, после вакуумного разло-
жения пробы с использованием насоса Теплера (см. выше).
В гидридах обычно бывают две основные примеси: нитридный
азот и ацетилидный углерод. Первый можно определить по методу
Кьельдаля в щелочном растворе после гидролиза. Углерод лучше
всего определяется колориметрически аммиачно — СиС1(1)-мето-
дом [9]. При гидролизе образца ацетилен выделяется вместе с
водородом и образует красный ацетилид одновалентной 'меди,
который определяют колориметрически.
111. ГИДРИД лития
А. Введение
Для получения гидрида лития пользуются обычной методикой.
Чистый гибрид лития представляет собой крупнокристаллическое
твердое вещество белого или голубовато-серого цвета. Голубой
цвет обусловлен небольшим количеством коллоидного лития, кото-
рый может выделятся при охлаждении в том случае, если при вы-
соких температурах гибрид имеет дефицит водорода [10]. Этого мо-
жно избежать, если обращать особое внимание на чистоту веществ
и очень медленно охлаждать гибрид, чтобы обеспечить максималь-
ное поглощение водорода.
Гибрид лития слабо чувствителен к свету при комнатной тем-
пературе и весьма чувствителен к ультрафиолетовому свету и ио-
низирующей радиации. Можно наблюдать также и другие окраски
вещества [11]. Дефицит водорода в веществе обычно настолько
III. Гидрид лития
231
мал, что его нельзя обнаружить аналитически. Белый цвет веще-
ства восстанавливается при нагревании гидрида в водороде.
Загрязненные образцы могут иметь форму более мелких кри-
сталлов.
Чистый гидрид лития плавится при температуре 688° в атмос-
фере водорода и давлении Нг 1 атм [8]. Это единственный гид-
рид, который плавится без разложения при указанном давлении
водорода. Двухфазная система металл — гидрид плавится при
монотектической температуре 685° и упругости водорода около
20 мм рт. ст. [8].
Свойства двухфазной равновесной смеси при нескольких темпе-
ратурах приведены в табл. 1.
Таблица 1
Фазовые равновесия в системе литий — гидрид лития
Темпера- тура, °C Гидрид лития, мольная доля Плато,
жидкая фаза металла твердая фаза гидрида жидкая фаза гидрида Р, а чм
625 0,13 [8] 0,99+ — 3,5 [12]
685 0,26 [8] 0,98—0,99 [8] 0,06—0,97 [13] 20 [13]
700 0,26 [13] — 0,96 [13] 28 [13]
750 0,30[13] — 0,94 [13] 71 [13]
а log р = - 9600/7" 4- 11,227 [12], 500-650°.
log рлл = - 8224/Г + 9,926 [13], 700-800°.
Б. Кинетика образования гидрида лития
Изменение скорости поглощения водорода массивным литием
при повышении температуры характеризуется определенной ка-
чественной закономерностью, что подтверждается Данными опыта.
Это изменение легко наблюдать при работе с аппаратурой, рас-
смотренной в разд. II. Реакция начинается при 300—500° и продол-
жается с умеренной скоростью до поглощения нескольких про-
центов водорода от теоретического количества. Затем реакция
замедляется. Вероятно, расплавленный металлический литий на-
сыщается водородом, и скорость после этого определяется медлен-
ной диффузией через слой фазы гидрида на поверхности. Чтобы
заметно увеличить скорость реакции и довести ее до конца, нужно
нагреть систему выше температуры плавления гидрида лития.
15*
232
Глава 8
При температурах 300—350° можно наблюдать небольшое вне-
запное повышение давления водорода, за которым следует сниже-
ние давления, указывающее на начало реакции. Этот эффект обус-
ловлен освобождением водорода в результате реакции
LiOH + LiH = Li2O + Н2.
Примесь гидроокиси лития способна реагировать с гидридом
именно при этой температуре, которая, вероятно, соответствует
эвтектической температуре для смеси ЫН — LiOH. Величина
эффекта соответствует количеству примеси гидроокиси лития и
уменьшается при увеличении чистоты металла. Можно заставить
литий поглощать водород и при более низких температурах. Очень
важное значение при этом имеет чистота поверхности. Реми-Жен-
нет [14] сообщает, что перегнанный литий при комнатной темпе-
ратуре за 24 час поглощает до 9% теоретического количества водо-
рода. Хюттиг и Краевски [15] установили, что литий, растворен-
ный в жидкое аммиаке, реагирует с водородом при комнатной
температуре. Можно заставить литий реагировать с водородом
при низких температурах под давлением, если литий находится в
виде суспензии в инертной жидкости [16, 17], или в присутствии
некоторых катализаторов, например жирных кислот высокого
молекулярного веса или ароматических углеводородов с конден-
сированными ядрами [18].
В литературе имеются лишь скудные данные о скоростях рас-
сматриваемых реакций, но и они сомнительны из-за плохой вос-
производимости результатов вследствие сильного влияния степени
чистоты металла и состояния поверхности. Альберт и Маэ [19]
сообщают, что скорость поглощения водорода при давлении 1 атм
и температуре 680° составляет 1,5 см3/см2-сек. Солиман [20] отме-
чает, что металлический литий при температуре 500° и давлении
0,5 атм поглощает 118 объемов водорода за 30 мин.
Свейн и Хейманн [21 ] изучали скорость реакции при темпе-
ратурах 25—250°. Они обнаружили, что начальная скорость умень-
шения давления водорода пропорциональна давлению и количе-
ству лития: —dPIdT =kmp. В действительности скорость должна
быть пропорциональна р — ре, где ре — равновесное давление
для данного состава и температуры (рис. 1). Но ре для си-
стемы Li — LiH очень мало при этих температурах. Для кон-
станты скорости в начале процесса авторы приводят урав-
нение log&=—1380/7’-|-2,09. Теплота активации равняется
6,3±2,5 ккал1молъ Н2. Изменения величины энергии активации
объясняют влиянием примесей. По мере протекания реакции
константа скорости уменьшается, и процесс, по-видимому, стано-
вится диффузионным.
III. Гидрид лития
233
В. Литература по синтезу гидрида лития
Аппаратура и методика синтеза килограммовых количеств гид-
рида лития описаны Альбертом и Маэ [19]. За 8 час им удалось
получить гидрид лития чистотой 99,6 %. Улучшенный вариант аппа-
ратуры для такого синтеза описан в статье Пономаренко и Миро-
нова [22]. Холдинг и Росс [23] синтезировали дейтерид лития
обычным методом.
Ланда и др. [24] описали синтез гидрида лития под давле-
нием 150—225 ат и температурах 310—345°. Они получали гидрид
из 10 г-атом лития в автоклаве из нержавеющей стали емкостью
2,5 лис добавкой 0,1% WS2 или MoS2 в качестве катализатора.
Продукт был получен с количественным выходом.
Другие указания на источники, касающиеся синтеза гидрида
лития как обычными, так и нестандартными методами, можно
найти в работе Мессера [25].
Г. Методика синтеза гидрида лития
Используют аппаратуру и метод, описанные в разд. 11. Реак-
ционную камеру или бомбу изготавливают из нержавеющей стали,
а лодочку для образца — из нержавеющей стали типа 347 или 316
или из железа Армко. Для получения 0,5 моля или 4 г гидрида
лития пригодна лодочка длиной 89 мм; для более крупных син-
тезов лодочка, бомба, печь и колба для калибровки объема должны
быть соответственно больше.
Для непосредственного использования пригодны литиевые
слитки чистотой 99,96%, если не требуется еще большей чистоты
продукта. В ином случае металл нужно очищать перегонкой в
вакууме [7].
Металлический литий разрезают и очищают от оксидно-нитрид-
ного слоя в сухом боксе, заполненном аргоном. (Можно исполь-
зовать также двуокись углерода, но она менее удобна; азот не
годен для этой цели, так как он реагирует с литием при комнат-
ной температуре с образованием темного нитрида Li3N.)
Для получения продукта наивысшей чистоты лодочку предва-
рительно обрабатывают гидридом лития. С этой целью проводят
предварительный синтез гидрида лития при тех же условиях, что
и основной, за исключением того, что в этом случае можно под-
держивать давление водорода 1 атм в течение всего процесса при
небольшой утечке через затвор; образец выдерживают при 725°
в течение 2 час вместо 8—10 час. После холостого синтеза ло-
дочку извлекают и погружают в чистый метанол, где содержимое
ее растворяется до полной очистки лодочки. Потом лодочку про-
мывают метанолом и безводным эфиром, смоченную эфиром пе-
234
Глава 8
реносят в шлюзовую камеру сухого бокса и возвращают затем в
бокс для загрузки литием. После такой обработки поверхность
лодочки становится более стойкой к действию металлического
лития.
Нужное количество лития (до 4 г) разрезают в сухом боксе на
кусочки, которые легко помещаются в лодочку. Для этой цели
можно пользоваться массивным острым ножом. Лодочку помещают
в реакционную камеру, закрывают камеру, вынимают ее из сухого
бокса, подсоединяют к вакуумной системе и откачивают. После
этого образец лития приблизительно за полчаса нагревают в бом-
бе до 200°, чтобы дегазировать образец и бомбу. Клапан к бомбе
перекрывают, а остальную часть откалиброванной системы за-
полняют водородом. После измерения давления и температуры
водорода клапан открывают и пропускают водород к образцу.
Температуру повышают до 725° и поддерживают ее на этом уровне
до окончания реакции. Каждый раз, когда давление падает при-
близительно до 100лкм рт. ст., клапан реакционной камеры пере-
крывают, измеряют давление, объем и температуру и вводят водо-
род в таком количестве, чтобы давление повысилось до 1 атм.
После этого вновь регистрируют давление, объем и температуру
и открывают клапан бомбы. Эти процедуры продолжают до тех
пор, пока скорость поглощения не станет исчезающе малой. Дав-
ление в конце синтеза должно быть равно или чуть ниже 1 атм.
При этом получается продукт с наивысшим содержанием водо-
рода.
Для получения продукта с содержанием водорода 99,6—99,8%
от теоретического требуется выдержка в печи при температуре 720—
725° в течение 2—10 час с последующим медленным охлаждением.
Некоторое непостоянство состава продукта обусловлено, вероятно,
наличием в металле примесей, так как синтез гидрида из металла
99,96 %-ной чистоты требует меньше времени.
Образец очень медленно охлаждают до 680°, благодаря чему
достигается максимальное поглощение водорода в процессе за-
твердевания. Затем пробу ступенчато (по 100°) охлаждают до 300°
и на каждой стадии охлаждения выдерживают до прекращения
поглощения водорода. Окончательную регистрацию давления,
объема и температуры производят либо при последней повышенной
температуре, либо при комнатной.
Образец выгружают из лодочки в сухом боксе. Если применяют
лодочки из нержавеющей стали, то из небольшой лодочки основ-
ную массу кристаллов можно высыпать, наклонив ее. Из большой
лодочки кристаллы можно удалить постукиванием. Железная
лодочка более хрупкая, и продукт нужно удалять из нее осторож-
но. Если в продукте имеются отличающиеся по цвету от основной
массы кусочки, их нужно отбросить.
IV. Гидрид натрия
235
Д. Анализ
Гидрид лития можно анализировать, используя методы, изло-
женные в разд. II. Для первой стадии гидролиза используют ле-
дяную воду.
Фридман [26] рассматривает методы определения следов не-
которых элементов в гидриде лития и дает обзор литературы по
этому вопросу. Бергстрессер и Уотерберри [27] рассматривают
способ выделения водорода из гидрида лития металлическим свин-
цом при 600°.
IV. ГИДРИД НАТРИЯ
А. Введение
Гидрид натрия нельзя синтезировать из элементов обычным ме-
тодом вследствие высокой температуры плавления (около 860°)
и большой упругости диссоциации. Скорость реакции между мас-
сивным металлом и водородом в той области сравнительно низких
температур, когда гидрид натрия устойчив при давлении 1 атм,
слишком мала для практического осуществления реакции.
Данные по упругости диссоциации и равновесному составу
системы натрий — водород приведены в табл. 2.
Таблица 2
Фазовые равновесия в системе натрий —
гидрид натрия — водород
Температура, °C Горизонтальный участок кривой (плато)а Давление^ над «100%-ным гидридом», мм рт. ст.
NaH в фазе металла, % NaH в фазе гидрида, % Давление, мм рт. ст.
350 — — 74 [28] 220 [29]
422 — — 760 [ 28 , 29] 1 500 [29]
550 30 [29] 80 [29] 16 600 [29] 21 400 [29]
а Плато: log р = -- 6100/Г + 11,66 [28] ; log р = — 5958/Т +
мм мм
+ 11,47 [29].
® «100%-ный гидрид»: log р = — 5070/T + 9,49 [29].
Из таблицы видно, что фаза гидрида натрия не может существо-
вать при давлении 1 атм и температурах выше 422°. В наиболее
чистой почти стехиометрической форме в виде так называемого
236
Глава 8
«100%-ного гидрида» натрия, имеющего более высокую упругость
диссоциации, это соединение не может существовать при темпера-
турах выше 390—400°.
Следует отметить, что, хотя истинно горизонтальные участки
изотерм, согласно Банусу и др. [29], включают состав 30—80%
NaH, пологие участки охватывают состав приблизительно 5—
85% NaH. Эти цифры соответствуют взаимной растворимости
Na и NaH при 500—600°. Вильямс и др. [30] определили раствори-
мость гидрида натрия в металлическом натрии в зависимости от
температуры в области постоянной упругости диссоциации (пла-
то) и установили, что она составляет 0,3 вес. % при 350° и 4,25%
при 445°.
При этих сравнительно низких температурах первые порции
водорода могут растворяться в металлическом натрии и реагиро-
вать с ним весьма быстро, образуя слой гидрида на поверхности
металла. Этот слой гидрида вскоре становится непроницаемым для
водорода, и процесс взаимодействия прекращается. Гибб [2]
предположил, что это частично обусловлено сжатием металла при
образовании гидрида.
В своем раннем исследовании упругости диссоциации гидридов
натрия и калия Кис [31 ] приводит небольшое число полуколи-
чественных данных относительно начальной скорости поглощения
водорода металлом. Скорости поглощения газа у него выражены
как величины понижения давления за определенный интервал
времени. Он отметил, что при температуре 300° скорость реакции
в 16 раз больше, чем при 200°, а при температурах выше 300°
взаимодействие протекает столь быстро, что скорость определить
невозможно.
Взаимодействие между элементами с образованием гидрида
натрия может происходить количественно с практически приемле-
мой скоростью при одном из следующих условий: а) если исполь-
зуется тонко диспергированный металл, б) если добавить очень не-
большие количества поверхностноактивных веществ, которые раз-
рушают поверхность раздела жидкого металла и твердого гидрида,
и в) если взаимодействие происходит в парах металла с конден-
сацией продукта.
Б. Суспензионный метод
Эта методика получения гидрида натрия подробно изложена в
руководстве для лабораторных работ Метсона и Уолей [32].
Суспензию металлического натрия получают в атмосфере азота
при температуре 105—125° перемешиванием натрия с тяжелым
светлым минеральным маслом, содержащим следы олеиновой кис-
лоты. В этом же аппарате суспензию нагревают и вместо азота в
IV. Гидрид натрия
237
систему вводят водород. После окончательной обработки при
280—300° смесь охлаждают и через аппарат снова пропускают ток
азота. Гидрид натрия выделяют из суспензии, промывая ее гекса-
ном и декантируя. Выход продукта количественный.
В. Применение катализатора
или поверхностноактивного вещества
Реакция взаимодействия элементов с образованием гидрида
натрия идет до конца в присутствии некоторых катализаторов или
поверхностноактивных веществ. При этом требуется добавить от
0,1% до нескольких процентов такого вещества. Часто гидрид,
полученный таким образом, достаточно чист и без удаления добав-
ки, но при необходимости примесь можно удалить экстракцией
гексаном или другим способом.
Хансли [18, 33] обнаружил, что добавки жирных кислот с
числом атомов углерода восемь и более, металлических солей
этих кислот, антрацена, флюорена, индена и различных изопро-
пилзамещенных ароматических углеводородов приводят к быст-
рому поглощению водорода при температурах 250—400° и давле-
нии его 1 атм.
Макенфасс [16] синтезировал гидрид натрия, используя сус-
пензию натрия в тетрагидронафталине или белом парафиновом
масле (нуйол"е) при температуре 250° и давлении 35,15 ати в ка-
чающемся автоклаве. Этим методом воспользовались Сайре и
Бивер [34], которые описали метод получения гидрида достаточно
подробно для воспроизведения синтеза.
В качестве автоклава они использовали бомбу из нержавею-
щей стали Аминко 406-22В. Внутрь бомбы был вложен стакан из
стекла пирекс емкостью 600 мл.
В качестве поверхностноактивного вещества была взята неф-
тяная фракция, кипящая в интервале 167—180°. Авторы считают
очень важным применять именно зту фракцию с указанным темпе-
ратурным интервалом кипения. Растворители, кипящие при более
низких температурах, действуют хуже, вероятно, вследствие
меньшей вязкости. Если пользоваться растворителями, кипящими
выше, потребуется промывка препаратов после синтеза более
летучими растворителями.
Для осуществления синтеза под слоем декана отрезали кусок
натрия (—0,5 моля) и поместили его в автоклав под слой поверх-
ностноактивного растворителя. Натрий гидрировали в качающем-
ся автоклаве при 240° и давлении 28 ати в течение 10 час. Для
опыта использовали около 300 мл растворителя.
Образующуюся пульпу гидрида переносили под слоем этой
жидкости в специальную сухую камеру. В камере имелись стеклян-
238
Глава 8
ные шары, при помощи которых перемешивалось содержимое ка-
меры, в то время как растворитель отгоняли в высоком вакууме.
Благодаря перемешиванию плотная корка из порошка гидрида
разрушалась, и растворитель удалялся полностью.
Данные анализов методами выделения водорода и титрования
щелочи хорошо согласовались между собой и свидетельствовали о
высокой чистоте продукта.
Ланда и др. [24] получали гидрид натрия тем же методом,
что и гидрид лития (разд. III.В). Для этого брали 10 молей на-
трия и обрабатывали их водородом под давлением 130—210 атм
и температуре 220—250° с использованием в качестве катализа-
тора 0,1% WS2.
Михеева и др. [35] очень подробно рассматривают синтез гид-
рида натрия; они приводят несколько методик с диаграммами,
сравнивают различные катализаторы и обсуждают процесс ката-
лиза.
Эти авторы испытали три типа методик синтеза: взаимодействие
натрия и водорода в статических условиях, взаимодействие при
циркуляции водорода над натрием или барботирование газа через
расплавленный натрий и взаимодействие при механическом пере-
мешивании реагентов. Авторы обнаружили, что в статических
условиях при 200—400° образуется очень небольшое количество
гидрида натрия, даже в присутствии пальмитиновой и стеарино-
вой кислот или солей одной из этих кислот. Циркуляция водорода
над натрием лишь незначительно увеличивала образование гид-
рида. Барботирование водорода через взвесь натрия в о-ксилоле
или керосине с 3—6% антрацена увеличивало выход до 76%, но
при этом получался продукт черного цвета.
Однако применение автоклава с мешалкой в виде прямоуголь-
ной перфорированной пластинки, вращающейся со скоростью
300—600 об/мин, увеличивало выход до 96—97 % в лучших опытах,
и при этом получался светло-серый продукт. Давление в этом слу-
чае поддерживали около 0,7 атм, температуру 270°, продолжи-
тельность гидрирования 2,5 час, а количество катализатора 0,25—
0,5%.
Удалось обнаружить, что в этих опытах «масло индустриаль-
ное 30» позволяет получить более высокий выход, чем антрацен,
но еще больший выход дает «бензольная ароматическая фракция»
этого масла.
Авторы высказали интересную гипотезу относительно катали-
тического действия антрацена и других веществ в этой реакции.
Они полагают, что явления, связанные с взаимодействием метал-
лического натрия и антрацена и других полициклических арома-
тических углеводородов, аналогичны явлению, отмеченному при
гидрировании натрия в присутствии этих соединений. В первом
IV. Гидрид натрия
239
случае натрий реагирует с антраценом с образованием 9,10-ди-
натриевой соли 9,10-дигидроантрацена [36]. В последнем случае
предполагают, что антрацен играет роль переносчика натрия, что
он сначала реагирует с металлом с образованием 9,10-соли,
которая потом реагирует с водородом с образованием гидрида
натрия и антрацена. В качестве катализатора реакции, вероят-
но, могут быть использованы и другие ароматические поверхно-
стноактивные вещества, которые реагируют с натрием аналогич-
ным образом.
Подтверждением этого механиза авторы считают исследования
Хугеля [37, 38], который предположил, что гидрид натрия будет
катализировать процесс гидрирования ароматических углеводо-
родов только в том случае, если эти вещества присоединяют натрий,
и что гидрирование происходит только по местам присоединения
натрия.
Г. Реакция в паровой фазе
Гидрид натрия можно получить взаимодействием элементов
в паровой фазе. Газообразный водород пропускают над металлом
или через металл, и гидрид натрия конденсируется в виде мелко-
кристаллической массы на холодных стенках аппаратуры. Этот
метод пригоден только для получения небольших количеств гид-
рида, так как процесс идет медленно, и при синтезе необходимо
принимать меры, предотвращающие осаждение металлического
натрия.
Бардвелл [39] получил гидриды натрия и калия, пропуская
водород через расплавленные металлы в стальной трубке при
400°. На холодных частях аппаратуры конденсировались тонкие
белые иглы. Приведена схема аппаратуры, которая выглядит
очень просто. Количество полученного гидрида не указано, но,
по-видимому, оно составляет около 1 г. Давление паров металли-
ческого натрия при этой температуре чуть ниже 1 мм pm. ст. [40].
Хаген и Сивертс [41 ] нагревали до 420° в течение 4—8 час
технический гидрид натрия в железном тигле, установленном в
стеклянном аппарате, и получили до 30 мг тонких белых игл
NaH.
Автор с сотрудниками [42] использовали этот метод для полу-
чения гидрида натрия в количествах порядка нескольких грам-
мов. Над расплавленным натрием в течение 24—48 час пропускали
водород под давлением 1 атм и температуре 650—750°. Давление
паров натрия составляло приблизительно 100 мм рт. ст. Как пра-
вило, гидрид конденсировался в виде волокнистой массы, хотя в
одном или двух случаях наблюдали игольчатые кристаллы. На-
ряду с белыми препаратами получалось иногда некоторое количе-
240
Глава 8
ство серого вещества. Такое вещество отбрасывали. По-видимому,,
полученные препараты были очень чувствительны к остаточной
влажности в сухом боксе, поэтому их нужно было обрабатывать
очень быстро.
Несмотря на то что этот метод лишь с большим трудом можно
использовать для получения сравнительно больших количеств-
гидрида, он дает возможность получить чистое вещество. Метод,
заслуживает тщательного изучения, и особенно таких факторов,
как температура, скорость пропускания водорода, температурные
градиенты, положение конденсирующей поверхности и т. д.
V. ГИДРИДЫ ДРУГИХ ЩЕЛОЧНЫХ МЕТАЛЛОВ
Гидрид калия очень близок по свойствам к гидриду натрия,
поэтому условия его получения почти те же. Упругость диссоциа-
ции гидрида калия немного ниже, чем гидрида натрия [28, 31].
Скорость образования гидрида калия при сопоставимых условиях
несколько больше, чем гидрида натрия [24].
Михеева и Шкрабкина [43] описали способ получения гидрида
калия в автоклаве при температуре 200—250° и давлении водорода
3—4 атм в присутствии 1 % машинного масла. Выход составил
97-96% КН.
Гидриды рубидия и цезия менее известны. Упругость их дис-
социации выше, чем гидридов натрия или калия [28]. Вероятно,
и у этих гидридов наблюдается увеличение скорости образования
из элементов с увеличением атомного номера.
VI. ГИДРИД КАЛЬЦИЯ
А. Введение
Гидрид кальция с содержанием водорода по меньшей мере до
99% теоретического можно получить обычным методом. Это
белый порошок или мелкокристаллическое твердое вещество с
температурой плавления выше 1000° [44]. В условиях постоянной
упругости диссоциации (в области плато на кривой зависимости
упругости диссоциации от состава; разд. I)гидрид кальция устой-
чивее, чем гидрид лития, но при почти стехиометрическом составе
он менее устойчив. Следовательно, присоединение следовых
количеств водорода происходит с трудом. Взаимная растворимость
металла и гидрида при температурах 500—800° больше, чем
лития и гидрида лития.
Петерсон и Фатторе [44] термическим анализом и исследова-
нием равновесия определили диаграмму состояния для твердых
VI. Гибрид кальция
241
и жидких фаз системы Са — СаН2. Эта система осложнена суще-
ствованием трех аллотропных форм кальция и двух форм гидрида
в различных температурных интервалах.
Изотермы упругости диссоциации, аналогичные приведенным на
рис. 1, и значения упругости диссоциации в области плато были оп-
ределены многими исследователями. Три последних исследования—
работа Джонсона и сотрудников [45], Тредвелла и Стичера [46],
а также работа Куртиса и Чиотти [47 ] — рассмотрены в дан-
ной главе; ссылки на более ранние работы можно найти в указан-
ных источниках.
Значения упругости диссоциации и растворимости по резуль-
татам этих работ приведены в табл. 3. Данные этих авторов нес-
колько различаются, но качественно хорошо согласуются. Упру-
гость диссоциации в области плато при температурах ниже 600°
невелика, а растворимость водорода в кальции мала при темпера-
турах ниже 400°. При температурах 800—900° растворимость
СаН2 в Са составляет приблизительно 20%; фаза гидрида при рав-
новесии содержит около 95% СаН2.
Б. Кинетика образования гидрида кальция
Скорость взаимодействия металлического кальция с водородом
изучали многие исследователи, в результате чего были получены
важные результаты. Однако примеси, остающиеся в металле, по-
видимому, влияют на воспроизводимость величины константы ско-
рости реакции от опыта к опыту.
Джонсон и др. [45] изучали эту реакцию при температурном
интервале 0—200°, используя тонкоизмельченный металл, полу-
ченный испарением раствора металлического кальция в жидком
аммиаке. При температурах выше 100° и давлениях более 0,05 атм
результаты не воспроизводились. При температурах 27—98° и
давлениях ниже 15 мм рт. ст. измерения удовлетворительно согла-
совывались. Результаты указывают на первый порядок реакции
(logpH, линейно изменяется со временем) с теплотой активации
7,7 ±0,5 ккал/молъ.
Тредвелл и Стичер [46 ] нашли, что реакция имеет первый поря-
док при температурах 327—356° и давлениях 8—130 мм рт. ст.
с теплотой активации 7,8 ккал/молъ. В работе они применяли ме-
талл в виде компактного куска.
Шушунов и Шафиев [48] изучали реакцию при температурах
125—500°. Они использовали пленку металла, полученную конден-
сацией паров на стеклянной поверхности. Авторы получили две
серии прямых линий на графике Аррениуса: одну серию с мень-
шим наклоном для высоких температур и небольших давлений,
другую с большим наклоном для низких температур и больших
242
Глава 8
Таблица 3
Равновесия в системе кальций —
гидрид кальция — водород
Горизонтальный участок кривой (плато)а Давление над 99,14%- ным СаН2, мм рт. ст. [46]
Температура, °C СаН2 в фазе металла, мольная доля [44] СаН2 в фазе гидрида, мольная доля [44, 46] Давление р, м т. ст. [47]
400 0,02
500 0,05 — — —
600 0,07 -0,95 0,2 —
700 0,11 -0,95 2,2 10
800 0,20 -0,95 19 70
900 0,22 -0,95 94 550
log р мм = — 10 870/Г + 11,49, 650-900° [45],
log р мм =-- — 9840/7' + 10,7, 711-941° [46],
log р мм log р мм = — 9610/7 + 10,23, = - 8890/Г + 9,54, 578—780° 780-900° [47], [47].
давлений. Теплота активации для первой серии прямых с неболь-
шим наклоном составляет 5,5—7,2 ккал/молъ, для второй 12—15
ккал!моль. Первый порядок реакции наблюдали только при низких
теплотах активации. Авторы пришли к выводу, что более высо-
кие значения энергии диссоциации соответствуют переходу реак-
ции в диффузионную область, когда скорость реакции определи-,
ется диффузией через слой гидрида кальция.
Кавана [49—51] определил экспериментально и рассчитал
скорости поглощения водорода и дейтерия кальцием при темпера-
турах 250—325°. Он рассматривал влияние поверхности и вопросы
образования центров кристаллизации. Им обнаружено, что реак-
ция имеет первый порядок, а теплоты активации равны соответ-
ственно 13,8 и 13,9 ккал!молъ.
Солиман [20] нашел, что: а) максимум скорости реакции
наблюдается при температуре 350° и б) при давлениях выше ат-
мосферного происходит заметное повышение скорости поглощения.
Шушунов и Шафиев [48] определили влияние примесей кисло-
рода и паров воды на скорость реакции. При введении 0,5% кисло-
VI. Гибрид кальция
243
рода скорость реакции снижается до 42% первоначальной; 1%
кислорода и более снижают скорость до 1—3% первоначальной.
Аналогичным образом влияют на скорость реакции пары воды.
Соответствующие снижения значения скорости для паров воды
составляют 15 и 1,5—2% первоначальной скорости.
В. Синтез гидрида кальция
Джонсону и др. [45] удалось приготовить 99—100%-ный
СаН2 при 230—270°, но при 650—900° они не смогли получить пре-
параты чище, чем 91%-ный. Тредвелл и Стичер [46] синтезирова-
ли 99,14%-ный СаН2 при 380°, используя один из вариантов обыч-
ной аппаратуры в статических условиях. Однако они установили,
что без перемешивания нельзя получить продукт с более высоким
содержанием водорода. Ими описан аппарат для синтеза. Авторы
гидрировали в нем металл до 96% при 500° без перемешивания.
Затем поворачивали аппарат так, что он попадал в поле действия
магнитной мешалки. После этого гидрирование продолжали при
500°, и во всех случаях происходило дополнительное поглощение
водорода. Лучшие образцы содержали 99,6—99,9% СаН2.
Все эти исследователи перед гидрированием очищали метал-
лический кальций перегонкой, обычно при остаточном давлении
10-3—10-4 мм рт. ст. и 850—900°. Джонсон [45] и Тредвелл [46]
описывают установку для дистилляции.
Вопрос о необходимости вакуумной перегонки кальция реша-
ется в зависимости от чистоты исходного металла и требуемой
степени чистоты продукта.
Для синтеза можно использовать обычную аппаратуру и мето-
ды, применяемые для получения гидрида лития (разд. III). Реак-
цию следует вести по возможности при температурах 300—350°.
Если требуется нагреть образец до более высокой температуры,
его нужно выдерживать при 300—350° в процессе охлаждения так
долго, чтобы в последний час или дольше не происходило погло-
щение водорода.
Если образец гидрида кальция потребуется нагреть выше
600°, то следует использовать аппаратуру только из железа или
нержавеющей стали. В любом случае лодочка для синтеза должна
быть из железа или нержавеющей стали.
Гидрид кальция можно приготовить конденсацией из смеси
водорода и паров кальция. Цинтл и Хардер [52] для рентгеногра-
фического исследования гидридов кальция, стронция и бария по-
лучали таким способом кристаллы этих веществ, иногда размером
до 1 мм.
244
Глава 8
VII. ДРУГИЕ СОЛЕОБРАЗНЫЕ ГИДРИДЫ
А. Введение
К солеобразным гидридам относятся также дигидриды бария,
стронция, европия, иттербия и магния. Все они, за исключением
MgH 2, по своим физическим и кристаллографическим свойствам
очень близки к СаН2. Гидриды ЕнН2 и YbH2 имеют такую же ор-
торомбическую структуру [53], как СаН2, SrH2 и ВаН2 [52]. Это
объясняется исключительной устойчивостью этих металлов в окис-
лительном состоянии 2+.
Б. Гидриды бария и стронция
Эти гидриды изучены не так подробно, как гидрид кальция.
Петерсон и Индиг [54] исследовали диаграмму состояния твердых
и жидких фаз системы Ва — ВаН2. Гибб [55] определил упругость
диссоциации для систем Sr — SrH2 и Ba — ВаН2. Он дает следую-
щие эмпирические уравнения, которые относятся предположитель-
но к упругостям диссоциации в области плато изотерм давление —
состав:
Sr — SrH2: log рмм = —10 400/Г + 11,10 (92,3% SrH2),
Ba —BaH2: log pMM = — 6450/Г + 8,20 (93,7% BaH2).
Из фазовой диаграммы следует более высокая взаимная раство-
римость в системе Ва — ВаН2 по сравнению с системой Са — СаН2.
В соответствии с данными по упругостям диссоциации, устойчи-
вости SrH2 и СаН2 в области плато приблизительно одинаковы;
ВаН2 определенно менее устойчив, чем два других гидрида.
В. Гидрид магния
Гидрид магния MgH2 имеет структуру рутила [56] в отличие от
орторомбической структуры СаН2, SrH2 и ВаН2. Он значительно
менее устойчив, чем гидриды кальция, стронция и бария, и упру-
гость диссоциации в области плато достигает 1 атм при 280—
290° [56-58].
Скорость образования MgH2 [56—58] такова, что для синтеза
соединения за приемлемый промежуток времени требуется давле-
ние более 1 атм и температура выше 290°. В пределах давлений
100—1000 мм рт. ст. и температур 250—300° для достижения
равновесия требуется несколько суток [58].
Виберг и др. [59] синтезировали MgH2 из элементов при 570°
и 200 атм с применением в качестве катализатора Mgl2, но выход
Литература
245
составил только 60%. Дымова и др. [60] провели синтез гидрида
магния во вращающемся автоклаве при давлении 100—200 атм
и температурах 310—450° в присутствии СС14 в качестве катализа-
тора. Фауст и др. [61] провели синтез при давлении 5 атм и 175°
с использованием аллилиодида, пропаргилиодида и иода в каче-
стве катализатора. Однако результаты измерения упругости дис-
социации, по-видимому, указывают на то, что при температурах
400° и выше применение катализаторов не обязательно.
Анализы продуктов свидетельствуют о том, что очень трудно
приготовить гидрид магния без значительной примеси окисла и
металла. Стемпфер и др. [57] дают состав продукта: 93,1% MgH2,
3,0% MgO и 3,9% Mg.
Гидрид магния MgH2 можно получить также термическим раз-
ложением диалкилов магния [62, 63].
ЛИТЕРАТУРА
1. Hurd D. Т., Chemistry of the Hydrides, Wiley, New York, 1952.
2. G i b b T. R. P., Jr., «Primary Solid Hydrides», in Progress in Inor-
ganic Chemistry, Vol. Ill, Cotton F. A., ed., Interscience, New York,
1962, pp. 315—509.
3. Pretzel F.E., Rupert G.N., MaderC. L.,StearnsE.K.,
G r i t t о n G. V., Rushing С. C., Phys. Chem. Solids, 16, 10 (1960).
4. Proskurnin M., Kasarnowsky J., Z. anorg. allgem. Chem.,
170, 301 (1928).
5. Z i n t 1 E., Harder A., Z. Physik. Chem. (Leipzig), B14, 265 (1931).
6. G i b b T. R. P., Jr., Anal. Chem., 29, 584 (1957).
7. Douglas T.B., Epstein L.F., Dever J. L., Howland
W. H., J. Am. Chem. Soc., 77, 2144 (1955).
8. M e s s e г C.E., Damon E. B., Maybury P. C., Mellor J.,
Seales R. A., J. Phys. Chem., 62, 220 (1958).
9. Weaver E. R., J. Am. Chem. Soc., 38, 352 (1916).
10. P r e t z e 1 F. E., R u s h i n g С. C., Phys. Chem. Solids, 17, 232 (1961).
11. Moers K., Z. anorg. allgem. Chem., 113, 179 (1920).
12. Hurd С. B., Moore G. A., J. Am. Chem. Soc., 57, 332 (1935).
13. H e u m a n n F. K., Salmon O. N., «The Lithium Hydride, Deute-
ride, and Tritide Systems», KAPL-1667, Dec. 1, 1956; Nucl. Sci. Abstr.,
11, 4811 (1957).
14. Remy-GenneteP. A., Ann. Chim., 19, 263 (1933).
15. H ii t t i g G. F., Krajewski A., Z. anorg. allgem. Chem., 141,
133 (1924).
16. M u c k e n f u s s A. M., пат. США 1958012, May 8, 1934; through Chem.
Abstr., 28, 4185 (1934).
17. The Roessler and Hasslacher Chemical Co., англ. пат. 405017, Jan. 29,
1934; through Chem. Abstr., 28, 4545 (1934).
18. Han.sley V. L., пат. США 2372670 and 2372671, April 3, 1945;
through Chem. Abstr., 39, 3129 (1945).
19. A 1 b e r t P., M a h ё J., Bull. Soc. Chim. France, 1950, Ц65,
20. Soliman A., J. Appl. Chem. (London), 1, 98 (1951).
21. S w a i n E. E., Heimann F. K., «The Reaction berween Lithium
and Hydrogen at Temperatures between 29° C. and 250° С», KAPL-1067,
March 1, 1954; Nucl. Sci. Abstr., 9, 5589 (1955).
246
Глава 8
22. Пономаренко В. А., Миронов В. П., Изв. АН СССР
ОХН, 1954, 497.
23. Н о 1 d i n g A. F. I., Ross W. A., J. Appl. Chem. (London), 8, 321
(1958).
24. Landa S., Petru F., Mostecky J., Vit J., P г о c h a z-
k a V., Collection.Czech. Chem. Commun., 24, 2037 (1959).
25. Messer С. E., NYO-9470, Oct. 27, 1960; Nucl. Sei. Abstr., 15, 12906
(1961).
26. Friedman H. A., Anal. Chem., 32, 137 (1960).
27. Bergstresser K. S., Waterbury G. R., «Determination of
Hydrogen in LiH>, LAMS-1698, May, 1943, deci. Oct. 5, 1956; Nucl. Sci.
Abstr., 13, 1116 (1959).
28. H e т о 1 d A., Compt. Rend., 228, 686 (1949).
29. В a n u s M. D., McSharry J. J., Sullivan E. A., J. Am.
Chem. Soc., 77, 2007 (1955).
30. W i 1 1 i a m s D. D., Grand J. A., Miller R. R., J. Phys.
Chem., 61, 379 (1957).
31. К e у e s F. G., J. Am. Chem. Soc., 34, 779 (1912).
32. Mattson G. W., Whaley T. P., «Sodium Hydride», in Inorga-
nic Syntheses, Vol. 5, Moeller T., ed., McGraw-Hill, New York, 1957,
pp. 10—-13.
33. Hansley V. L., Carlisle P. J., Chem. Eng. News, 23, 1332
(1945).
34. Sayre E. V., Beaver J. J., J. Chem. Phys., 18, 584 (1950).
35. Михеева В. И., Дымова Т. Н., Шкрабкина М. М.,
ЖНХ, 4, 709 (1959).
36. Schlenk W., Appenrodt J., Muchael A., ThalA.,Chem.
Ber., 47, 473 (1914).
37. Hu gel G., Fr i ess J., Bull. Soc. Chim. France, 49, 1042 (1931);
through Chem. Abstr., 26, 363 (1932).
38. H u g e 1 G., G i d a 1 y, Bull. Soc. Chim. France, 51, 639 (1932); through
Chem. Ahstr., 26, 5002 (1932).
39. Bardwell D. C., J. Am. Chem. Soc., 44, 2499 (1922).
40. M a k a n s i M. M., Muendel С. M., Selke W. A., J. Phys.
Chem., 59, 40 (1955).
41. Hagen H., Sieverts A., Z. anorg. allgem. Chem., 185, 239 (1930).
42. M e s s e г С. E., Fasolino L. G., Thalmayer С. E., J. Am.
Chem. Soc., 77, 4524 (1955).
43. Михеева В. И., Шкрабкина М. М., ЖНХ, 7, 463 (1962).
44. Peterson D. Т., Fattore V. G., J. Phys. Chem., 65, 2062 (1961)
45. J о h n s о n W. C., Stubbs M. F., Sidwell A. E., P e c h u-
k a s A., J. Am. Chem. Soc., 61, 318 (1939).
46. T r e a d w e 1 1 W. D., Sticher J., Helv. Chim. Acta, 36, 1820
(1953).
47. C u r t i s R. W., C h i о 11 i P., J. Phys. Chem., 67, 1061 (1963).
48. Шушунов В. А., Шафиев А. И., ЖФХ, 26, 672 (1952).
49. К a wan a Y., Nippon Kagaku Zasshi,71, 494 (1950); through Chem.
Abstr., 45, 6468 (1951).
50. К a w a n a Y., Nippon Kagaku Zasshi, 71, 554 (1950); through Chem.
Abstr., 45, 6468 (1951).
51. H i g u c h i I., К a w a n a Y., Nippon Kagaku Zasshi, 71, 624 (1950);
through Chem. Abstr., 45, 7461 (1951).
52. Zintl E., Harder A., Z. Elektrochem., 41, 33 (1935).
53. War f J. C., Korst W. L., Acta Cryst., 9, 452 (1956).
54. Peterson D. T., In dig M., J. Am. Chem. Soc., 82, 5645
(1960).
Литература
247
55. G i b b T. R. P., Jr., «Hydrides and Metal-Hydrogen Systems. Fibal
Report», NEPA-1841, April 30, 1951, declass. July 18, 1961; Nucl.Sci.
Abstr., 16, 8713 (1962).
56. E 11 i n g e r F. H., Holley С. E., Jr., M с I n t e e г В. В., P a-
vone D., Potter R. M., Staritzky E., Z achariasen
W. H., J. Am. Chem. Soc., 77, 2647 (1955).
57. Stampfer J. F., Jr., Holley С. E., Jr., Suttle J. F., J. Am.
Chem. Soc., 82, 3504 (1960).
58. Kennelley J. A., V a r n i g J. W., Myers H. W., J. Phys.
Chem., 64, 703 (1960).
59. Wiberg E., Goeltzer H., Bauer R., Z. Naturforsch., 6b, 394
(1951).
60. Дымова Т.Н., Стерлядкина 3.K., Сафронов В.Г.,
ЖНХ, 6, 763 (1961).
61. Faust J.P., Whitney E.D., Batha H.D., Heyring
T. L., Fogle С. E., J. Appl. Chem., 10, 187 (1960).
62. W i b e r g E., Bauer R., Chem. Ber., 85, 593 (1952).
63. Freundlich W., Claudel B., Bull. Soc. Chim. France, 1956,
967.
ГЛАВА 9
СОЕДИНЕНИЯ СЕРЫ С АЗОТОМ И ФТОРОМ
Г лемаер
Институт неорганической химии
Гёттингенского университета
1. ВВЕДЕНИЕ
Соединения серы с азотом и фтором были впервые синтезиро-
ваны в 1955 г. [1]. С тех пор получено еще несколько новых
соединений этого типа. Исследование их структуры показало, что
эти соединения можно разделить на две группы: ациклические и
циклические.
Ациклические соединения: тиазилфторид NSF; тиодитпазплди-
фторид S3N2F2; тиазилтрифторид NSF3.
Циклические соединения: тетратиазилтетрафторид N4S4F4; три-
тиазилтрифторид N3S3F3; тиотритиазилфторид S4N3F.
Все эти фториды можно получить из тетратиотетранитрида
S4N4. Химическая связь в тетратиотетранитриде и его производ-
ных, ациклических и циклических соединениях серы с азотом
и фтором, несколько своеобразна и необычна для неорганической
химии. В этих соединениях имеются одинарные, двойные и трой-
ные связи S — N, а также локализованные и делокализованные
тг-связи по кольцу. Структурные формулы соединений показаны
на рис. 1.
При получении этих соединений важное значение имеют побоч-
ные реакции с материалом контейнеров (так же, как и в других
случаях фторирования). Поэтому такие реакции заслуживают осо-
бого внимания. Обычно используют сосуды из кварца, меди, по-
лиэтилена и тефлона.
Все указанные выше фториды, за исключением N4S4F4, и особен-
но NSF3, чрезвычайно чувствительны к влаге, что следует иметь в
виду при их синтезе.
Детальная теория ступенчатого гидролиза этих соединений
уже разработана, но относительно механизма реакций их образо-
вания почти ничего неизвестно. Известно только, что очень
часто характер реакций с участием тетратиотетранитрида застав-
ляет предположить существование промежуточного радикала SN.
II. Ациклические соединения
249
а. Ациклические соединения
NSF
Кратность связи
S—N порядка 2,5 [2,3]
83МЛ
F—S—N=S=N—SF
F—S—N—S=N—SF
<-> (+>
Структура неизвестна
NSE,
n»s4f„
fStN,]+F“
Кратность связи
S—N порядка 2,7 [2-4]
6. Циклические соединения
F
| Локализованные
/S- двойные связи S=N
N3S3F3 |NX м| неплоские кольца
I II 12,5]
F—Sk IS—F
''N
Вероятно,
делокализованные связи
S—N [В]
S*N» Делокализованные двойные связи е 8-членном кольце [7]
Рис. 1. Структурные формулы соединений серы с азотом и фтором.
С этой гипотезой вполне согласуется реакция фторирования тет-
ратиотетранитрида S4N4 с образованием газообразного мономера
NSF. Однако, как это будет указано в разделе III.А.1, такая ги-
потеза не объясняет образования N4S4F.
11. АЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
А. Тиазилфторид NSF
1» Получение
Тиазилфторид NSF можно получить различными способами.
Так, при пропускании тока аммиака через суспензию серы и ди-
фторида серебра в четыреххлористом углероде образуется тиазил-
фторид [8] (а также тиазилтрифторид NSF3; разд. II.В) по урав-
нению
NHS + S + 4AgF2 —'U NSF + 3HF + 4AgF,
18 Заказ № 547
250
Глава 9
Тиазилфторид можно получить также взаимодействием аммиака
с избытком SF4 [9]
NH3 4- SF4---> NSF + 3HF.
Наиболее удобный метод получения тиазилфторида — фторирова-
ние тетратиотетранитрида S4N4, причем наилучшие результаты
получаются при фторировании суспензии S4N4 в кипящем четырех-
хлористом углероде дифторидом серебра AgF2 [10] или фторидом
ртути HgF2 [11 ]. При использовании AgF2 наряду с NSF образуется
также ряд других соединений, которые трудно отделить от основ-
ного вещества. Напротив, при использовании HgF2 получают от-
носительно чистый сырой продукт. Получение NSF с применением
фторида ртути более удобно и дает лучший выход
ось
S4N4 + 4HgF2----> 4NSF -|- 2Hg2F2.
Методика синтеза. Тиазилфторид NSF [И] получают в ци-
линдрическом кварцевом сосуде с обратным холодильником, к ко-
торому подсоединяют две охлаждаемые ловушки; в кварцевый со-
суд помещают 3 г S4N4 и 100 мл свежеперегнанного безводного че-
тыреххлористого углерода. Из медной пробирки в сосуд переносят
около 80 г HgF2, который приготовляют непосредственно перед
использованием в той же пробирке нагреванием 100 г HgCl2 в
газообразном фторе до 100° с последующим охлаждением в токе
фтора. Сразу же после добавления HgF2 суспензию нагревают до
температуры кипения четыреххлористого углерода при энергич-
ном перемешивании магнитной мешалкой. Летучие продукты
реакции уносятся очень медленным током азота через обрат-
ный холодильник в ловушку, охлаждаемую до —80°. Следую-
щая ловушка, охлаждаемая до —18°, предотвращает попадание в
аппаратуру влаги воздуха. Фторирование заканчивается через
5—6 час.
Сырой продукт очищают фракционной конденсацией в глубоком
вакууме. Для этого ловушку, в которой скапливался сырой про-
дукт, перестают охлаждать и летучие компоненты пропускают че-
рез три холодные ловушки. В первой ловушке (—80°) конденсиру-
ется четыреххлористый углерод, во второй (—115°) собирается
NSF, а в третьей (охлаждаемой жидким воздухом) — сильно
летучие примеси, такие, как SOF2, SF4 и CC13F. Тиазилфторид мож-
но вторично очистить таким же фракционированием.
Выход. Из 3 г S4N4 получается 1,5—2 г NSF, что соответствует
35—47% теоретического выхода.
II. Ациклические соединения
251
2. Свойства NSF
Тиазилфторид — бесцветный газ с резким запахом. Темпера-
тура его кипения (0,4°) определена по кривой давления паров.
Температура замерзания —89°. Химический сдвиг сигнала ядер-
ного магнитного резонанса фтора (при использовании в качестве
эталона насыщенного водного раствора фторида калия) составляет
+358 ч. на млн. [2]. Инфракрасный спектр имеет полосы погло-
щения при основных частотах 1372, 640 и 360 см'1.
Тиазилфторид очень реакционноспособен; под действием водя-
ных паров газ гидролизуется с образованием промежуточного про-
дукта тионилимида HNSO, который легко идентифицируется по
инфракрасному спектру [10]. В результате полного количествен-
ного гидролиза разбавленной соляной кислотой образуются ионы
фторида, сульфита и аммония. Тиазилфторид можно хранить в
медном сосуде при комнатной температуре и давлении около 500 мм
рт. ст. в течение нескольких суток без разложения. В стеклянном
сосуде при аналогичных условиях вещество разлагается. На стек-
лянных стенках образуются тетратиотетранитрид S4N4 и зелено-
вато-желтые кристаллы S3N2F2. При нагревании стеклянного сосу-
да до 110° в течение 3 час весь тиазилфторид разлагается с обра-
зованием S4N4, SiF4, SO2, SOF2 и N2. Тиазилфторид аналогично
NSC1 [12] может полимеризоваться до N3S3F3 (разд. III.Б.1).
Б. Тиодитиазилдифторид S3N2F2 [13]
1. Получение
Если 6-литровую стеклянную колбу наполнить тиазилфторидом
при комнатной температуре и начальном давлении около 600 мм
рт. ст., то через 1 час на внутренних стенках начнет появляться
налет желтого вещества. Позднее начинают расти зеленовато-
желтые кристаллические пластинки, которые через неделю можно
соскрести со стенок, применяя с этой целью иглу, управляемую,
снаружи магнитом. Кристаллы отделяют от образующегося одно-
временно с ними желтого налета и собирают их в многокамерную
трубку для сублимации, подсоединенную ко дну большой колбы.
В результате сублимации в глубоком вакууме при 40° собирают
зеленовато-желтые пластинки, а при 65° — ярко-зеленые кристал-
лы. После растворения обеих фракций в четыреххлористом углеро-
де получаются одинаковые желтовато-зеленые растворы. Оба
раствора дают идентичные ультрафиолетовые спектры поглощения
с широким максимумом около 375 ммк. Обе фракции являются,
вероятно, полиморфными модификациями одного и того же соеди-
18*
252
Глава 9
нения. За 30 опытов было получено около 300 мг этого вещества,
причем израсходовано 300 г S4N4. Выход составляет приблизитель-
но 0,1% теоретического.
2. Свойства
Тио дитиа зилдифторид S3N2F2 плавится при 83°. В интервале
85—97° начинается разложение вещества, а при температурах выше
100° соединение разлагается со взрывом. При контакте с влагой
фторид немедленно чернеет. При щелочном гидролизе азот коли-
чественно выделяется в виде аммиака.
В. Тиазилтрифторид
1. Получение
Как указано в разд. II.A.1, тиазилтрифторид NSF3 (так же,
как и NSF) можно приготовить, пропуская аммиак через суспен-
зию серы и дифторида серебра в четыреххлористом углероде [8].
Относительно механизма реакций образования этого соединения
ничего не известно. Можно только предположить следующее урав-
нение суммарной реакции:
NH3 + S + 6AgF2 —Д- NSF3 + 3HF + 6AgF.
Вероятно, при взаимодействии исходных веществ образуется сна-
чала NSF, который при дальнейшем фторировании превращается в
NSF3. Действительно, один из способов получения NSF3 заклю-
чается в пропускании тока NSF над AgF2 при 100° [14]:
NSF + 2AgF2----> NSF3 + 2AgF.
Во всех случаях, когда в результате фторирования (например,
S4N4 [10]) образуется NSF, при более глубоком фторировании обра-
зуется также NSF3. Реакцию S4N4 с AgF2 в кипящем четыреххло-
ристом углероде с образованием NSF3 можно выразить уравнением
S4N4 + 12AgF2 —'i- 4NSF3 + 12AgF.
Кроме NSF3, образуется также несколько других соединений, из
которых можно выделить или идентифицировать по инфракрасным
спектрам следующие вещества: N4S4F4, SiF4, SF4, SF6, COF2,
SOF2, SO2F2, CC13F, CC12F, SO2, N2 и Cl2. Эти примеси нужно тща-
тельно удалить.
Недавно появилось сообщение [15] о том, что при взаимодей-
ствии S2F10 с недостаточным количеством аммиака образуются
II. Ациклические соединения
253
значительные количества NSFa. Более подробная информация
отсутствует.
Методика синтеза [11]. 5 г S4N4 и около 50—70 г свежепри-
готовленного дифторида серебра смешивают со 100 мл четырех-
хлористого углерода в аппарате, описанном ранее (разд. II.А.1).
При перемешивании смесь быстро нагревают до температуры
кипения четыреххлористого углерода. В дальнейшем поступают
так же как при получении NSF. Реакция заканчивается через
1,5 час. Сырой продукт промывают 50 мл 3 %-ного водного раствора
перманганата калия и очищают пропусканием над тонкоизмель-
ченной двуокисью свинца. Для дальнейшей очистки применяют
метод газовой хроматографии. В качестве неподвижной фазы ис-
пользуют трикрезилфосфат на кизельгуре, а в качестве носителя —
газообразный водород.
Выход. Из 5 г S4N4 получают около 5 г NSFa, что соответ-
ствует 24% теоретического выхода.
2. Свойства
Тиазилтрифторид NSFa — бесцветный газ с резким запахом.
При нормальном давлении он конденсируется при температуре
—27,1±0,1° в бесцветную жидкость с температурой замерзания
—72,6±0,5°. Химический сдвиг сигнала ядерного магнитного ре-
зонанса фтора (при использовании в качестве эталона насыщенного
водного раствора фторида калия) составляет —f-187 ч. на млн.
[2]. Инфракрасный спектр имеет интенсивные полосы поглощения.'
при частотах 1515, 811, 775, 521, 429 и 342 см"1 и при частотах,
соответствующих комбинационным полосам [2].
В противоположность NSF тиазилтрифторид очень устойчив.
В стеклянном сосуде при 200° никакой реакции не происходит.
При 500° начинается быстрое взаимодействие со стеклом с образо-
ванием SiF4, SO2, N2, серы и фторидов металлов. Газообразный
хлористый водород не реагирует с NSF3 при нагревании; даже га-
зообразный аммиак не реагирует с тиазилтрифторидом при ком-
натной температуре и низких давлениях. Металлический натрий
также не реагирует с NSF3 при комнатной температуре; при нагре-
вании приблизительно до 400° происходит взаимодействие с обра-
зованием сульфида натрия, фторида натрия и азота. .В безэлек-
тродном светящемся разряде NSF3 разлагается с образованием
NSF, SF4 и SF6 [И].
Для очистки тиазилтрифторида важное значение имеет его
устойчивость по отношению к разбавленным кислотам. Газ лишь,
медленно реагирует с водой при комнатной температуре. В вод-
ных растворах он разлагается только при нагревании в разбавлен-
ном растворе едкого натра; при разложении образуется натриевая
254
Глава 9
соль амидосерной кислоты, гидролизующаяся в концентрирован-
ной соляной кислоте с образованием ионов аммония и сульфата.
Эту реакцию можно использовать для количественного анализа
NSFa.
111. ЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
А. Тетратиазилтетрафторид N4S4F4
1. Получение
До сих. пор тетратиазилтетрафторид N4S4F4 удавалось получить
только фторированием тетратиотетранитрида S4N4 [1 ]. Попытки
получить N4S4F4 таким же способом, как и N3S3F3 (т. е. полимери-
зацией NSF), оказались безуспешными. Естественно предположить,
что в реакции фторирования S4N4 до N4S4F4 не участвует проме-
жуточный радикал SN, а происходит непосредственное присоеди-
нение фтора к атомам серы в восьмичленном кольце S4N4. В ре-
зультате этой реакции взаимное расположение атомов серы и азо-
та изменяется по сравнению с исходным соединением (S4N4), и
ароматический характер кольца исчезает, как это показано на
рис. 1.
Тетратиотетранитрид S4N4 фторируют дифторидом серебра
AgFa в четыреххлористом углероде.
S4N4 4- 4AgF2 —N4S4F4 -J- 4AgF.
Как оказалось, взаимодействие облегчается, если фторирование
ведут в медном сосуде и к дифториду серебра добавляют некоторое
количество СпЕг [11].
Методика получения [9, И]. В кварцевом сосуде, который
описан в разд. II.A.1, смешивают 5 г S4N4 и 50 г свежеприготов-
ленного дифторида серебра со 100 мл четыреххлористого углерода,
высушенного предварительно хлористым кальцием и перегнанно-
го непосредственно перед употреблением. Раствор медленно, в те-
чение 2 час, нагревают до кипения. В процессе нагревания раствор
зеленеет. Затем смесь, еще горячую, быстро фильтруют и остаток
дважды экстрагируют четыреххлористым углеродом. При охлаж-
дении в течение ночи раствор приобретает красный цвет. Осаж-
дающиеся белые иглы тетратиазилтетрафторида N4S4F4 следует
быстро отфильтровать. Можно получить дополнительное коли-
чество загрязненного тетратиазилтетрафторида, если упарить ма-
точный раствор. Эту порцию вещества можно очистить перекри-
сталлизацией из четыреххлористого углерода.
Выход. Из 5 г S4N4 получают 1 г N4S4F4, что соответствует 14%
теоретического выхода. При использовании медных сосудов мож-
но получить более высокий выход.
III. Циклические соединения
255
2. Свойства
Тетратиазилтетрафторид N4S4F4 образует белые игольчатые
кристаллы, которые начинают разлагаться при 128° и плавятся
при 153°. N4S4F4 можно сублимировать без разложения в глубоком
вакууме при 80°. Растворимость в четыреххлористом углероде при
2° составляет 3,44 г/л\ растворимость увеличивается с ростом тем-
пературы. Плотность при 20° составляет 2,326 г!см3. Для иден-
тификации вещества можно использовать полосы инфракрасного
спектра [2], соответствующие частотам 1117, 786, 760, 709, 645
и 520 слг1. Химический сдвиг сигнала ядерного магнитного резо-
нанса фтора (при использовании в качестве эталона насыщенного
водного раствора фторида калия) составляет -}-155 ч. на млн.;
наблюдается только одна полоса поглощения [2].
Во влажном воздухе тетратиазилтетрафторид N4S4F4 мед-
ленно гидролизуется; в теплом разбавленном растворе щелочи он
гидролизуется быстро в соответствии с уравнением
N4S4F4 + 8ОН- + 4Н2О---»• 4NH4F + 4SO^“.
В запаянной стеклянной ампуле N4S4F4 разлагается при темпе-
ратуре 100°. Кроме основного продукта — тетратиотетранитрида
S4N4, образуются газообразные соединения SiF4, SOF2 и SOa.
При нагревании N4S4F4 с AgFa в медном сосуде до 100° образуются
NSFa и другие газообразные вещества.
Б. Тритиазилтрифторид N3S3F3
1. Получение
Если тиазилфторид NSF оставить в запаянном стеклянном
сосуде на 3 суток, образуется смесь кристаллов, из которой воз-
гонкой можно выделить летучий тритиазилтрифторид N3S3F3
[И]. При этом, как предполагалось, происходит полимеризация
NSF до N3S 3F3 аналогично полимеризации газообразного мономера
NSC1 до образования N3S3CI3 [12]
3NSF-----> N3S3F3.
Более удобным методом получения N3S3F3 является фториро -
вание N3S3CI3 дифторидом серебра в четыреххлористом углероде,
так как N3S3CI3 легко можно получить из S4N4. Кроме того, с
хлоридом легче работать, чем с тиазилфторидом NSF [16].
Методика синтеза [15]. В небольшую кварцевую колбу с маг-
нитной мешалкой (рис. 2) помещают суспензию 10 г измельчен-
ного N3S3CI3 в 40 мл сухого и свежеперегнанного четыреххлори-
256
Глава 9
стого углерода; туда же добавляют 20 г AgFa. От проникновения
влаги смесь защищают U-образной трубкой, наполненной хлори-
стым кальцием. Реакционную смесь перемешивают при комнат-
ной температуре примерно в течение 18—20 час, пока не исчезнет
желтая окраска и раствор не обесцветится. После этого колбу
подсоединяют к аппарату для фильтрования (рис. 2). Аппарат
предварительно тщательно проду-
вают азотом, а шлиф смазывают
смазкой KEL-F. Бесцветный раст-
вор отделяют от осадка соли се-
ребра фильтрованием через стек-
лянный фильтр 3 (G4). Ток азота
затем отключают и закрывают
краны 4 и 5. После этого колбу
1 подсоединяют к аппарату для
фракционной конденсации в глу-
боком вакууме (рис. 3) и произво-
дят разгонку фильтрата. Для этой
цели колбу 1 охлаждают смесью
твердой углекислоты и ацетона
до —80° и откачивают при откры-
том кране 4. После откачивания
кран 4 закрывают, а ловушки 6, 7
и 9 подогревают в глубоком ва-
кууме. При непрерывном откачи-
вании ловушку 9 охлаждают жид-
ким воздухом до —180°, ловушку
7— смесью твердой углекислоты и
метанола до —40°, а ловушку 8 да
—20°. Кран 4 снова открывают и,
уменьшая уровень холодной ван-
ны из твердой углекислоты с аце-
тоном, дают веществу в колбе 1
медленно нагреваться и испарять-
ся. При этом в ловушках 6 и 7 со-
бираются сильно преломляющие свет бесцветные кристаллы
N3S3F3, а четыреххлористый углерод конденсируется в ловушке
2. После разгонки веществ из колбы 1 чистый тритиазилтри-
фторид перегоняют из ловушек 9 и 7 в ловушку 8. Из ловушки
8 вещество можно перегонять в колбы, для анализа, пробирки для
определения температуры плавления и другие сосуды для соот-
ветствующих исследований.
Выход. Из 10 г N3S3CI3 получается около 7 г N3S3F3, что соот-
ветствует 90% теоретического выхода.
III. Циклические соединения
257
2. Свойства
Тритиазилтрифторид' образует бесцветные кристаллы, обладаю-
щие заметной летучестью даже при комнатной температуре. Тем-
пературы плавления и кипения вещества равны 74,2 и 92,5° соот-
ветственно. Тритиазилтрифторид легко растворяется в четырех-
хлористом углероде и бензоле. Химический сдвиг сигнала ядерного
магнитного резонанса фтора (при использовании в качестве эта-
лона насыщенного водного раствора фторида калия) составляет
4-147 ч. на млн. [2]; наблюдается только одна полоса поглощения.
Для идентификации вещества можно использовать полосы погло-
щения инфракрасного спектра при частотах 1085, 720 и 650 слг1
[2]. Несмотря на то что тритиазилтрифторид устойчив в сухом
воздухе, во влажном воздухе он разлагается и чернеет. NaSaFa
значительно более чувствителен по отношению к влаге, чем
NaSaCla и N4S4F4. В холодном разбавленном растворе едкого
натра тритиазилтрифторид количественно гидролизуется
N3S3F3 + 6ОН- + ЗН2О---3NH4F + 3SO|”
В. Тиотритиазилфторид S4N3F
1. Получение [17]
Все соединения тиотритиазила содержат катионное семичлен-
ное кольцо S4N+. Для получения этих соединений, в том числе
для получения фторида, используют относительно устойчивый
17 Заказ № 547
258
Глава 9
хлорид. Для замещения ионов хлора на ионы фтора S4N3CI обра-
батывают безводным фтористым водородом при 80—100° в поли-
этиленовой или тефлоновой трубке
S4N3C1 + HF----> S4N3F + НС1.
Образуются желтовато-коричневые кристаллы состава S4N3F-
• 1,5HF. Фтористый водород можно удалить только при одновре-
менном разложении соединения.
Методика синтеза. В полиэтиленовую трубку длиной около
30 см, которая введена в холодильник Либиха, помещают 0,5 г
S^NsCi [18]. Вещество затем нагревают до 80—100°, пропуская
через холодильник горячий глицерин. После достижения нужной
температуры через трубку с хлоридом начинают пропускать
безводный фтористый водород. При этом образуется желтая жид-
кость и выделяются пузырьки хлористого водорода. Когда весь
S4N3C1 прореагирует, прекращают нагревание и пропускание
фтористого водорода. Полиэтиленовую трубку отпаивают у од-
ного конца холодильника так, что ее длина уменьшается прибли-
зительно до 20 см. Под защитной атмосферой СОг полиэтиленовую
трубку в вертикальном положении вводят в вакуумную систему.
После откачивания избытка фтористого водорода препарат сушат
еще в течение 3 час при 40° и затем охлаждают, после чего аппарат
заполняют сухой двуокисью углерода.
2. Свойства
Тиотритиазилфторид существует в виде коричневато-желтых
кристаллов, которые во влажном воздухе сразу же становятся зе-
леновато-черными. Тиотритиазилфторид значительно более чув-
ствителен к присутствию влаги по сравнению с хлоридом.
ЛИТЕРАТУРА
1. Glemser О., Schroder Н., Haeseler Н., Naturwissen-
schaften, 42, 44 (1955); Z. anorg. allgem. Chem., 279, 28 (1955).
2. Richert H., Glemser O., Z. anorg. allgem. Chem., 307, 328
(1961).
3. Kirchhoff W. H., Dissertation, Harvard University, USA, 1962.
4. Kirchhoff W. H., Wilson E. B., J. Am. Chem. Soc., 84, 334
(1962).
5. W i e g e r s G. A., Vos A., Acta Cryst. (Copenhagen), 14, 562 (1961);
16, 152 (1963).
6. W e i s s J., Angew. Chem., 74, 216 (1962).
7. С r a i g D. P., Chem. Soc. (London), Spec. Publ., 12, 343 (1958); C r a-
i g D. P., P a d d о c k N. L., Nature, 181, 1052 (1958); Craig D. P.,
J. Chem. Soc., 1959, 997.
8. Glemser O., Wyszomirski E., Meyer H., unpublishde
results.
Литература
259
9. G 1 е m s е г О., Austin S., unpublished results; Austin S.,
theisis, Gottingen, 1961.
10. Glemser O., R i c h e r t H., Z. anorg. allgem. Chem., 307, 313
(1961).
11. Glemser O., Meyer H., Haas A., Ber., in press; Meyer H.,
Dissertation, Gottingen, 1964.
12. G 1 e m s e r O., Perl H., Naturwissenschaften, 48, 620 (1961).
13. Glemser O., Schroder H., Wyszomirski E., Z. anorg.
allgem. Chem., 298, 72 (1959).
14. Glemser O., Schroder H., Z. anorg. allgem. Chem., 284, 97
(1956); Glemser O., Richert H., ibid., 307, 313 (1961).
15. Cohen B., MacDiarmid A. G., Chem. Ind. (London), 1962,
1866.
16. Schroder H., -Glemser O., Z. anorg. allgem. Chem., 298, 78
(1959).
17. Glemser O., Wyszomirski E., Chem. Ber., 94, 1443 (1961).
18. For the preparation SniNsCl see Meuwsen A., Ber. Deut. Chem. Ges.,
65, 1724 (1932).
17*
ГЛАВА 10
ГИПОГАЛОГЕНИТЫ И СОЕДИНЕНИЯ,
СОДЕРЖАЩИЕ ОХ -ГРУППУ
Вилъямсон
Калифорнийский институт, Беркли, Калифорния
I. Введение
Данная глава посвящена в основном методам получения сое-
динений, известных под названием гипогалогенитов. Атом галоге-
на в гипогалогените «одновалентен», и его «валентность» израсхо-
дована на связь с атомом кислорода. В некоторых случаях группа
X—О несет отрицательный заряд (например, С1—О~ в ионных
солях соответствующих кислот). В других случаях «валентность»
кислорода участвует в образовании ковалентной связи с другим
атомом или другой группой. Здесь не будут рассмотрены те соеди-
нения галогенов, в которых имеются «галогенные катионы» и об-
зор которых был недавно составлен Ароцким и Симонсом [1 ], хо-
тя в некоторых из них галоген проявляет валентность 1-]-.
Одна из первых реакций, которая должна быть здесь отмечена,
это реакция галогенов (кроме фтора) с раствором едкого натра, в
результате которой образуются ионы галогенидов и гипогалогени-
тов.
Х2+20Н- Х0- + Х- + Щ0. (1)
Как известно, при взаимодействии фтора с водой или щелочными
растворами не образуется ионных форм, соответствующих ионным
формам других галогенов [21. В результате реакций с участием
фтора не образуется ни фторноватистой кислоты, ни других ион-
ных производных, поэтому реакции фтора с образованием кова-
лентных гипофторитов будут рассмотрены в специальном разделе.
При более подробном изучении оказывается, что окислитель-
ные потенциалы различных кислородсодержащих ионных форм
галогенов в водных растворах не благоприятствуют устойчивому
существованию гипогалогенитов или других промежуточных окис-
лительных состояний. Латимер [3 ] приводит следующие значения
потенциалов для хлорных систем:
Cl- + 2ОН-----» СЮ" + Н2О + 2е', Е« = — 0,88, (2)
II. Ионные гипогалогениты
261
СЮ" + 40Н- ----» CIO3 + 2Н2О + 4е-, £» = — 0,50. (3)
Комбинируя эти уравнения, получим
ЗС10-----> CIO3 + 2С1-, Е<> = 0,38. (4)
Положительное значение стандартного потенциала для реакции
(4) указывает на то, что гипохлорит термодинамически неустойчив
в отношении диспропорционирования. Соответствующая констан-
та равновесия для комнатной температуры, рассчитанная из соот-
ношения E°nF — RT\nK, равна 1025>7. Аналогичный расчет для
ВгО- дает Х = 1014’9 и для Ю~ Х = 1023’6, если воспользоваться
соответствующими потенциалами для брома и иода.
Еще более серьезной проблемой, чем реакции диспропорцио-
нирования, являются реакции гипогалогенитов с галогенидами в
растворах с низкими значениями pH. В кислых растворах галоге-
ниды и галогеноватистые кислоты взаимодействуют по реакции,
обратной реакции образования. В результате такого взаимодей-
ствия образуется вода и выделяется свободный галоген.
Чистые гипогалогениты или их растворы можно получить в
том случае, если удалять из сферы реакции побочный продукт —
ионы галогенида, например, осаждая их серебром. Поэтому жела-
тельно вести процесс галогенизации щелочей так, чтобы примеси,
в частности ионы галогенов и галогенатов, удалялись перед вы-
делением гипогалогенитов. Гипогалогениты, как правило, очень
растворимы, поэтому для их выделения требуется низкотемпера-
турное упаривание, при быстром проведении которого удается
получить гипогалогениты в виде солей.
Изучали также процесс разложения гипогалогенитов в водных
растворах. Для НОС1 [4а], ВгО" [46] и 10" [4в] реакция разло-
жения имеет второй порядок по отношению к гипогалогениту.
Наиболее медленной стадией, определяющей скорость процесса,
является реакция 2ХО_-»ХО“ +Х~, за которой следует быстрая
реакция ХО_г+ХО--> ХО--»Х“. На практике разложение этих
соединений происходит не так быстро, чтобы предотвратить выде-
ление гипогалогенитов, за исключением гипоиодитов.
П. Ионные гипогалогениты
Один из способов получения гипохлорита, который применяют
для получения LiOCl хорошего качества [5], заключается в обра-
ботке хлором смеси раствора едкого натра и едкого лития. Хлорид
удаляется в виде NaCl до кристаллизации более растворимого
LiOCl из растворов после упаривания.
Другой метод, который, по-видимому, найдет более широкое
признание, использовали для получения гипобромита LiOBr-
• 2НгО [6]. Раствор едкого лития смешивают с раствором брома
262
Глава 10
с таким расчетом, чтобы молярное соотношение равнялось 1:1;
после этого к смеси добавляют стехиометрическое количество све-
жей влажной окиси серебра. Выделившийся LiOH удаляют титро-
ванием смолой дауэкс-50УУ-Х8 в водородной форме с размером гра-
нул 40 меш. После этого холодный раствор LiOBr, не содержащий
СОг и посторонних ионов, упаривают при пониженном давлении.
Таким образом можно получить ярко-желтое твердое вещество
состава LiOBr-2Н2О 95%-ной чистоты. Анализ этого продукта не
представляет трудности, так как анионы гипобромида, бромита и
бромата можно определить при совместном присутствии [7].
При комнатной температуре LiOBr-2НгО обладает только очень
слабым запахом брома. Содержание [ВгО;] с течением времени
практически не увеличивается.
К моменту написания этой главы, по-видимому, никому еще не
удалось выделить кристаллический гипоиодит. Получены только
три гипобромида: ВЮВг-2НгО [6], ЫаОВг-бНгО и КОВг-ЗНгО
[8]. Две последние соли при комнатной температуре заметно
пахнут бромом; их первоначальная желто-оранжевая окраска
быстро исчезает.
Другой способ получения гипохлоритов — взаимодействие
хлорноватистой кислоты с гидроокисями. Хлорноватистую кис-
лоту НОС1 [9] можно получить в концентрациях до 5 М взаимодей-
ствием жидкого С12О с водой. Из хлорноватистой кислоты получают
довольно устойчивое белое вещество Са(ОС1)2 94%-ной чистоты.
Получают также довольно чистую бромноватистую кислоту
[6,10], но она устойчива лишь в сравнительно низких концентра-
циях, что препятствует получению препаративных количеств ги-
побромитов путем взаимодействия ее с гидроокисями.
III. Ковалентные гипогалогениты
Первую группу ковалентных гипогалогенитов составляют ги-
пофториты. Дифторид кислорода OF2 можно рассматривать в ка-
честве простейшего по структуре бифункционального гипофтори-
та. В эту группу включают также диоксидифторид O2F2, триок-
сидифторид O3F2 и тетраоксидифторид O4F2, хотя их структура
точно не известна.
При замещении атома водорода в сильных кислотах атомом
фтора получаются гипофториты, составляющие другую группу.
К таким гипофторитам относятся: нитрат фтора FONO2, перхло-
рат фтора FOCIO3, фторсульфонат фтора FOSO2F, трифтораце-
тилгипофлорит (трифторацетат фтора)
О
II
FOCCF3
IV. Фториды кислорода
263
и пентафторпропионилгипофторит (пентафторпропионат фтора)
О
II
FOCC2F6
Третью группу гипофторитов получают фторированием некоторых
окислов или оксифторидов, для которых не известны соответствую-
щие кислоты. К ним относятся: трифторметилгипофторит CF3OF,
гипофторит пентафторида серы SFsOF и гипофторит пентафторида
селена SeFsOF.
Гипофториты очень реакционноспособны в отношении фтори-
рования других веществ. Они очень летучи по сравнению с другими
соединениями тех же молекулярных весов. С гипофторитами мож-
но работать без затруднений (за исключением взрывчатых гипо-
фторитов) в стеклянной вакуумной аппаратуре. Они не очень чув-
ствительны к присутствию влаги, что оказывается очень благопри-
ятным, так как отпадает необходимость тщательного удаления
паров воды. При гидролизе гипофторитов происходит медленное
выделение кислорода, образование ионов фторида и других анио-
нов, например нитрата, перхлората, фторсульфоната и т. д.
Гипофториты являются сильными окислителями и реагируют с ор-
ганическими веществами со взрывом, хотя в некоторых случаях
реакцию удается замедлить. В результате этого удалось получить
новый класс углеродных соединений [11—13].
IV. Фториды кислорода
Прямое фторирование воды приводит к образованию дифторида
кислорода OF2 вместе с другими соединениями [14—16]. При
пропускании пузырьков фтора через 1-сантиметровый слой 0,5 М
раствора КОН в дифторид превращается приблизительно 50%
фтора. Раствор пропускают в аппарат при температуре возможно
более близкой к 0° с такой скоростью, чтобы концентрация ОН"
поддерживалась постоянной. Большая часть образующегося OF2
выделяется в виде газа и может быть вынесена из аппарата с из-
бытком непрореагировавшего фтора и выделившимся кислородом.
Растворимость OF2 при 0° и нормальном давлении составляет
68 мл/л воды. Дифторид кислорода лучше всего улавливать в стек-
лянной U-образной ловушке, открытой в атмосферу и охлаждае-
мой жидким воздухом (—183°), так как при этой температуре не
конденсируются ни фтор, ни воздух. Если процесс ведут без газа-
носителя, дифторид кислорода очень хорошо улавливается при
температуре —183°, несмотря на то что при этой температуре он
обладает заметной растворимостью. Эту реакцию нужно вести в
хорошем вытяжном шкафу; в аппаратах не должно быть стеклян-
264
Глава 10
ных шлифов, смазанных углеводородными смазками. Другие фто-
риды кислорода получают созданием электрического разряда в '
газовых смесях кислорода и водорода, взятых в различных соот- *
ношениях, при низких давлениях и температурах. Основные спо-
собы получения O2F2 [17], O3F2 [18а] и О4Гг [19] похожи один
на другой. Смесь фтора и кислорода пропускают через разрядную
камеру при общем давлении 5—15 мм рт. ст. Образующийся про-
дукт конденсируется на холодных стенках (~90°К) аппаратуры.
Интересно отметить [19], что для получения О4Гг требуются мень-
шие энергии разряда и более низкие температуры, чем для полу-
чения O2F2 и ОзР2. В работе Киршенбаума и Гроссе [18а, рис. 1]
приведена схема аппарата, который оказался вполне удовлетвори-
тельным. Вполне вероятно, что этот метод синтеза найдет более
широкое применение [20а]. Примером этому может служить ис-
пользование метода для синтеза KrF4 [206].
Дифторид кислорода не разлагается при нагревании до темпе-
ратур примерно 250°, но разложение более сложных окси-
фторидов происходит ступенчато с образованием устойчивого
OF2 при очень низких температурах. Ступенчатое разложение ок- ‘
сифторидов используют для определения состава соединений.
V. Гипофториты сильных кислот
Получение гипофторитов взаимодействием газообразного фтора ]
с сильными кислотами не представляет технической трудности.
Таким способом синтезировано пять гипофторитов. Нитрат фтора !
FONO2 [21] был получен фторированием 3 М HNO3 при темпе-
ратуре около 2° в аппарате, аналогичном по конструкции аппарату »
для получения OF2. Поскольку нитрат фтора взрывоопасен, аппа-
рат изготовляют из никеля или монельметалла, и он должен иметь ।
отверстие для аварийного выброса газов [21, рис. 2 ]. По-видимому, ‘ *
более удобным и эффективным методом является прямое фториро-
вание твердого KNO3 [22]. В этом случае над нитратом калия, по-
мещенном в платиновую или никелевую лодочку, пропускают фтор.
Газообразный FNO3 выносится вместе с избытком F2 и конденси-
руется в твердое вещество в ловушке, охлаждаемой жидким воз-
духом до —183°, в то время как фтор проходит через ловушку.
Рекомендуется получать FNOs непосредственно перед предполагае-
мым использованием, так как он иногда взрывает без видимой при-
чины. При фторировании 60 %-ной хлорной кислоты в стеклянном
аппарате [23, рис. 1 ] образуется взрывчатое газообразнсе вещест- н
во, которое было идентифицировано как FOClOg. Фтор пропускают
противотоком навстречу хлорной кислоте, которая стекает по |
измельченному стеклу. При пропускании неразбавленного фто- i
ра над 72 %-ной хлорной кислотой в платиновой лодочке был полу-
VI. Гипофториты окислов
265
чен 90%-ный выход FOCIO3. Фтористый водород и перхлорат фтора
FOCIO3 достаточно различаются между собой по летучести, и их
можно разделить перегонкой. Реакция F2 и НС1О4, по-видимому,
довольно сложна. Если фторировать 60%-ную хлорную кислоту в
угольной аппаратуре, перхлорат фтора не образуется, но вместо
этого образуются приблизительно равные количества OF2 и О2.
Исследователи не упоминают о другом возможном методе полу-
чения FOCIO3 — реакцией F2 с КС1О4.
Трифторацетилгипофторит CF3CO2F [24] и пентафторпропио-
нилгипофторит C2F5CO2F [25] были получены фторированием
трифторуксусной и пентафторпропионовой кислот в газовой фазе.
Судя по тому, что для взаимодействия веществ требуется присут-
ствие влаги, механизм реакции довольно сложен. Эти два гипо-
фторита очень неустойчивы и сильно взрывают при поджигании их
искрой. При разложении образуются насыщенный фтороуглерод и
двуокись углерода. Эта реакция показывает, что для фторирования
при комнатной температуре не всегда требуется сложная аппара-
тура из устойчивых материалов. Резиновые, полиэтиленовые и са-
рановые части аппарата [24, рис. 1] не вызывают образования
фтороуглеродов в мешающих количествах.
VI. Гипофториты окислов
Методом «каталитического» фторирования [26 ] были получены
три гипофторита. Это трифторметилгипофторит CF3OF [27],
фторсульфонат фтора FOSO2F [28] и гипофторит пентафторида
серы FsSOF[29]. Для получения трифторметилгипофторита можно
использовать метанол, окись углерода или карбонилфторид.
При 170° выход CF3OF при реакции фтора с метанолом составляет
50%, а при реакции с окисью углерода —70%. Несмотря на избы-
ток фтора, например при отношении F2 : СО=5, образуется неко-
торое количество COF2. Метод получения CF3OF фторированием
COF2 неудобен, поскольку СО значительно более доступен. При
использовании окиси углерода нет необходимости в удалении фто-
ристого водорода, который образуется как побочный продукт при
получении CF3OF из метанола. По-видимому, COF2 и CF3OF
нельзя разделить перегонкой, потому что они образуют азеотроп-
ную смесь. Примесь COF2 легко можно удалить из CF3OF обработ-
кой водой. При комнатной температуре COF2 быстро разлагается
водой с образованием СОг и HF, a CF3OF лишь медленно окисляет
воду.
«Каталитический» реактор представляет собой по существу мед-
ную трубку с наружным обогревом, термоизоляцией и термопа-
рами, размещенными внутри или снаружи медной трубки. Размеры
реактора определяются необходимым расходом газа и временем
266
Глава 10
его пребывания в реакторе. Обычно реактор имеет длину около
1 м и свободный объем 1—4 л. В качестве насадки можно исполь-
зовать покрытые серебром медную ленту, стружку или проволоку.
Эффективная однородная насадка получается из медных шаров.
Для ввода и вывода газов используют медные трубки диаметром
0,6 см, припаянные серебром к основной трубе реактора. При на-
личии соответствующего вспомогательного оборудования реактор
может работать как при пониженном, так и нормальном или повы-
шенном давлении.
Каталитическое действие AgF2 при образовании гипофторитов
изучено еще не до конца. Сам по себе AgF2 не способен фторировать
СО или другие окислы до гипофторитов. Кроме AgF2, для фториро-
вания требуется еще элементарный фтор. ।
При 220° серный ангидрид SOs в присутствии AgF2 фторируется
избытком фтора с образованием FOSO2F и выходом 60%. Осталь- 1
ная сера превращается в сульфурилфторид. При температуре
100° и ограниченном количестве фтора из SO3 образуется перок-
сидисульфурилдифторид ЗгОвРг [30]. SO3F2 и ЗгОвРг можно лег-
ко разделить перегонкой. Температуры кипения этих веществ j
равны соответственно —31,3 и +61,4°. От этих веществ легко от-
делить также и SO2F2, который кипит при —562°. Исследователи
не упоминают о другом возможном методе синтеза — реакции
F2 с K2SO3.
При 215° в присутствии катализатора тионилфторид легко
фторируется с образованием гипофторита пентафторида серы
FsSOF. Основными побочными продуктами реакции являются гек-
сафторид серы и сульфурилфторид. Выход гипофторита составляет
50% и более. Он легко отделяется от более летучих примесей, так ’
как имеет температуру кипения —35,1°.
Следует отметить, что SOCI2 не пригоден для получения SFsOF
методом фторирования. Тионилхлорид сначала нужно превратить ,
в тионилфторид [31 ], а если этого не сделать, хлор будет погло-
щаться серебром в реакторе, а, кроме того, в продукт будет попа-
дать некоторое количество хлора, удалить который очень трудно.
Последний гипофторит этой группы — гипофторит пентафто-
рида селена FsSeOF [32 ] — образуется с небольшим выходом при
прямом фторировании двуокиси селена или оксихлорида селена
при температурах 60—90°.
VII. Окислы галогенов (I) I
Как указано выше, при действии хлора или брома на водные
растворы оснований образуются соответствующие гипогалогениты,
в то время как фтор в этих условиях образует F2O — ангидрид L
фторноватистой кислоты. Соответствующие соединения хлора CI2O
VIII. Нитраты галогенов (1)
267
[9] и брома ВггО [33] образуются при взаимодействии галоге-
нов со свежеприготовленной желтой окисью ртути HgO. Окись
хлора С1гО достаточно устойчива, и ее можно хранить в виде крас-
новато-коричневой жидкости при температуре несколько ниже
температуры ее кипения, равной -|-2о. Окись хлора хорошо раство-
рима в четыреххлористом углероде, и ее можно хранить в виде
такого раствора. Коричневую окись брома лучше всего хранить
в виде раствора в четыреххлористом углероде при —30°. Суще-
ствование свободной окиси иода 1гО весьма сомнительно. Относи-
тельная устойчивость моноокисей галогенов — очень хороший при-
мер периодичности свойств соединений.
VIII. Нитраты галогенов (I)
Получение нитрата фтора [21] уже было рассмотрено. Полу-
чены также нитраты всех остальных галогенов: нитрат хлора (I)
CIONO2 [34], нитрат брома (I) ВгОИОг [35] и нитрат иода(1)
IONO2 [35]. Нет сомнения, что молекулярная структура ВтОИОг
не похожа на молекулярную структуру IONO2, и обе они отли-
чаются от структур FONO2 и CIONO2 [36], которые напоминают
структуру азотной кислоты.
Нитрат хлора образуется при взаимодействии СКО почти с
любым окислом азота. Изучены реакции СЬО и N2O5 или N2O4.
При реакции с N2O4 образуется смесь CIONO2 и CINO2. Нитрат
хлора представляет собой светло-желтую жидкость с температурой
кипения 18°. «Положительный» характер хлора подтверждается
реакцией с NaCl, в результате которой образуются С1г и NaNOe.
Изучали также реакции CIONO2 с ненасыщенными углеводорода-
ми при низких температурах [37], в результате которых С1 и NO3
присоединяются непосредственно по двойной связи с образованием
2-хлоралкилнитратов. Аналогичные реакции наблюдаются для
некоторых гипофторитов [11—13]. При взаимодействии ВгС1
с CIONO2 при температуре —70° образуется ВгОИОг и С12. Жел-
тый ВгОИОг плавится при температуре —42°, а при температуре
около 0° начинает разлагаться на Вгг, NO2 и Ог. В результате
реакции нитрата хлора с 1С1з образуется 1(НОз)з, а добавление
избытка I2 в растворителе CFCI3 при температурах ниже 0° приво-
дит к образованию твердого IONO2, который разлагается при тем-
пературах около 0°. Нитраты брома(Ш) и иода(Ш) значительно
более устойчивы, чем нитраты брома(1) и иода(1).
IX. ФторсуЛьфонаты галогенов (I)
Получение фторсульфоната фтора прямым синтезом [28]
из F2 и ЭОз уже было рассмотрено. Это соединение можно получить
и другим способом [38], путем фторирования ЭгОвРг [30] при 250°.
268
Глава 10
Такой способ пригоден для получения всей серии фторсульфонатов
галогенов (I) при условиях, соответствующих каждому синтезу.
В оригинальной работе 138] было обнаружено, что при дейст-
вии избытка Вт2 или I® на жидкий БгОеРг при комнатной темпера-
туре в стеклянной колбе образуются BrOSOaF и, вероятно,
lOSOaF. При действии на Вгг или 1гизбытка SaOeFa количествен-
но образуются фторсульфонаты Вг(Ш) и 1(111), из которых можно
получить соединения галогенов(1), если ввести дополнительные ко-
личества галогенов. Фторсульфонат брома BrOSOaF представляет
собой красновато-черную вязкую жидкость с температурой кипе-
ния 120,5°. Соединение устойчиво к разложению, но очень энер-
гично взаимодействует с водой. Фторсульфонат иода(Ш) I(SO3F)3—
светло-желтая жидкость с температурой замерзания 32,2°.
При нагревании до 114° и давлении 30 мм рт. ст. образуются
SaOeFa и нелетучая зеленая жидкость. Такая же зеленая жид-
кость образуется при взаимодействии ограниченного количества
ЗгОвРг с 1г при комнатной температуре. По-видимому, соединения
иода(1) при комнатной температуре очень неустойчивы, если они
вообще образуются при реакциях, для которых наблюдается пере-
менная стехиометрия.
Из фторсульфонатов галогенов труднее всего получить соеди-
нение хлора CIOSO2F [11 ]. Бесцветная жидкость БгОвР® испаряет-
ся, и пары при 125° имеют темно-коричневую окраску. Цвет паров,
вероятно, обусловлен наличием радикала SO3F. При умеренном
давлении SO3F не взаимодействует с С1г, но если С1г и ЗгОвРг
перегнать в цилиндрический сосуд из монельметалла так, чтобы
соотношение в смеси равнялось 1,5 : 1, и, закрыв кран, нагревать
смесь в жидком состоянии при температуре 125° в течение 5 суток,
то образуется почти чистый CIOSO2F. Это очень реакционноспособ-
ная желтая жидкость, которая при стоянии в стеклянном сосуде
при комнатной температуре темнеет и приобретает красный цвет.
X. Перхлораты галогенов(Т)
В одной из недавних работ с неводными растворителями [39]
показано, что в этаноле при —85° иод и перхлорат серебра реаги-
руют с образованием Agl и перхлората иода(1) — ЮСЮз. В одной
из ранних работ [40] исследовали взаимодействие этих веществ
главным образом при комнатной температуре в эфире, но взаимо-
действие осложнилось иодированием растворителя. Результатом
работы было ошибочное заключение об образовании С1О4. В этой
же работе [40] исследовали также взаимодействие брома и пер-
хлората серебра в ароматических растворителях и наблюдалось
бромирование растворителей. В работе не делается вывод, что
ВгОС1О3 может быть активным реагентом. Поскольку ЮСЮз
XI. Алкоксиды галогенов (I)
269
не выделен из спиртового растворителя, перхлорат фтора FOClOs
[23 ] остается единственным выделенным в чистом виде перхлора-
том одновалентного галогена.
XI. Алкоксиды галогенов(1)
Первые литературные сведения о гипогалогенитах органиче-
ских соединений появились в 1885 г. В работе [41] рассматрива-
лось получение метил- и этилгипохлоритов путем прямого хлори-
рования спиртов в присутствии раствора едкого натра. Общая
методика получения маслянистых желтоватых гипохлоритов вклю-
чает хлорирование холодного раствора 2 молей NaOH на 1 моль
спирта. Образующийся слой нерастворимого в воде ROC1 отде-
ляют, промывают и перегоняют.
Первичные и вторичные алкилгипохлориты очень неустойчивы
(теряют НС1 с образованием карбонильных соединений). В чистом
состоянии они настолько неустойчивы, что воспламеняются на све-
ту, иногда взрываются. По-видимому, чистые вещества можно
получить только в темноте и на холоду.
щрет-Бутилгипохлорит, как и другие третичные алкилгипо-
хлориты, довольно устойчив. Желтую жидкость можно хранить
при обычной температуре в течение нескольких месяцев без разло-
жения, если на нее не попадает яркий свет. Жидкость можно
перегонять при нормальном давлении без изменения (79,6° при
750 мм рт. ст.).
Другой метод получения алкилгипохлоритов [43] основан на
взаимодействии хлорноватистой кислоты и спирта. Для этого
смешивают и встряхивают смесь хлорноватистой кислоты и спирта
в инертном нерастворимом вводе веществе, например в четырех-
хлористом углероде. Первичные и вторичные гипохлориты более
устойчивы в растворе, чем в чистом виде. Спирты реагируют также
с раствором С1гО в четыреххлористом углероде с образованием
растворов гипохлоритов.
При кинетическом исследовании процесса образования трет-
BuOCl [44] обнаружено, что это соединение образуется не из
ОС1~. Только при pH<*10, когда концентрация НОС1 становится
заметной, начинается образование ROC1. Ранее отмечалось, что
при хлорировании щелочных растворов спиртов гипохлорит не
образуется до тех пор, пока большая часть щелочи не будет раз-
рушена хлором.
Имеется хороший обзор [45], в котором подробно рассмотрены
получение и свойства органических гипогалогенитов и указаны
другие работы. Недавно появилось предупреждение относительно
взрывчатости mpem-BuOCl [46].
270
Глава 10
шреш-Бутилгипобромит был получен [10] взаимодействием
водного раствора НОВг (не содержащего Вг~) с раствором трет-
бутилового спирта в трихлорфторметане. Выход составил 43%.
Прямым бромированием щелочного раствора щреш-бутилового
спирта не удалось получить шреш-бутилгипобромита.
Красновато-оранжевый жидкий mpm-BuOBr был выделен из
растворителя фреон-11 вакуумной дистилляцией; температура
кипения 44—45° при давлении 85 мм рт. ст. На холоду и в тем-
ноте гипобромит устойчив в течение длительного времени. Он
разлагается либо на ярком свету при температуре выше 85°, либо
в присутствии оснований.
В литературе, по-видимому, нет сообщений о получении и вы-
делении органических гипоиодитов.
XII. Карбоксилаты галогенов(1)
Ацилгипогалогениты изучены и описаны довольно широко, за
исключением упомянутых выше ацил гипофторитов [24, 25]. В
большинстве работ об ацилгипогалогенитах упоминают как о
промежуточных соединениях при реакциях галогенов с металли-
ческими солями карбоновых кислот. В нескольких недавних об-
зорах рассмотрены реакции галогенирования серебряных солей
карбоновых кислот, так называемые реакции Хансдикера [47].
При этих реакциях в результате взаимодействия галогенов и
ВСОгА§, смешанных в эквимолярных количествах, в отсутствие
воды и в присутствии инертного органического растворителя
(например, СС14 или СвНзМОг) образуются промежуточные ацил-
гипогалогениты, которые декарбоксилируются с образованием
соответствующих алкилгалогенидов, двуокиси углерода и пер-
воначально образующегося галогенида серебра. В результате
спектрофотометрического изучения разбавленных растворов аце-
тилгипохлорита и ацетилгипобромита в четыреххлористом угле-
роде твердо установлено наличие связи О — X в этих соедине-
ниях. Эти растворы были приготовлены встряхиванием раствора
хлора в четыреххлористом углероде с ацетатами ртути или серебра
или добавлением избытка уксусной кислоты к соответствующей
моноокиси галогена.
Серебряные соли перфторкарбоновых кислот дают с галоге-
нами более устойчивые гипогалогениты, чем производные обычных
кислот. Это согласуется с тем, что декарбоксилирование СРзСОгА§
при температурах ниже 100° в заметной степени не идет [49], в
то время как соли водородных карбоновых кислот теряют СОа
часто уже при комнатной температуре. Гипогалогениты перфтор-
карбоновых кислот вместе с некоторыми гипофторитами составляют
полные ряды, включающие соединения всех галогенов. Гипогало-
Литература
271
гениты водородных карбоновых менее устойчивы, а гипофториты
этих кислот не образуются.
В данный обзор включены только те работы, которые имеют
общий характер или посвящены синтезу соединений.
Дополнительные сведения читатель может найти в литературе,
цитируемой авторами работ, указанных в списке к этой главе.
ЛИТЕРАТУРА
1. Arotsky J., Symons М. С. R., Quart. Revs., 16, 282 (1962).
2. Cady G. H., J. Am. Chem. Soc., 56, 1647 (1934).
3. Л а т и м e p В. M., Окислительные состояния элементов и их потен-
циалы в водных растворах, ИЛ, Москва, 1954.
4а. Lister М. W., Can. J. Chem., 30, 879(1952); б. Engel Р., О р 1 a t-
ka А., Р е г 1 m u 11 е г - Н а у m а п В., J. Am. Chem. Soc., 76, 2010
(1954); в. L i С. H., W h i t e C. F., J. Am. Chem. Soc., 65, 335 (1943).
5. Cady G. H„ пат. США 2356820; through Chem. Ahstr., 39, 163 (1945)
6. Williamson S. M., «An Investigation into the Preparation of Crys-
talline Hypohromites», Technical Report, Physical Research Laboratory,
The Dow Chemical Company, Midland, Michigan, 1960.
7. Shapin R. M., J. Am. Chem. Soc., 56,2211 (1934).
8a. S c h о 1 d e г V. R., Krauss K., Z. anorg. allgem. Chem., 268,279
(1952); 6. Nouveau Traite de Chimie Minerale, Vol. XVI, Pascal P., ed.,
Masson et Cie, Paris, 1960, p. 430.
9. C a dy G. H., in Inorganic Syntheses, Vol. 5, Moeller T., ed., McGraw-
Hill, New York, 1957, p. 156.
10. W a 11 i n g С., P adwa A., J. Org. Chem., 27, 2976 (1962).
11. G i 1 b r e a t h W. P., Cady G. H., Inorg. Chem., 2, 496 (1963).
12. W i 1 1 i a m s о n S. M., Cady G. H., Inorg. Chem., 1, 673 (1962).
13. A 1 1 i s о n J. А. С., C a d у G. H., J. Am. Chem. Soc., 81, 1089 (1959).
14. L e h e a u P., Damiens A., Compt. Rend., 185, 652 (1927).
15. C a d у G. H., J. Am. Chem. Soc., 57, 246 (1934).
16. И о с т, в книге «Неорганические синтезы», сб. I, ИЛ, Москва, 1951.
17. Ruff О., Menzel W., Z. anorg. allgem. Chem., 211, 204 (1933).
18a. Kirshenbaum A. D., Grosse A. V., J. Am. Chem. Soc., 81,
1277 (1959); б. А о у a m a S., Sakuraba S., J. Chem. Soc. (Japan),
59, 1321 (1938); в. ibid., 62, 208 (1941).
19. Grosse A. V., Streng A. G., Kirshenbaum A. D.,J.
Am. Chem. Soc., 83, 1004 (1961).
20a. Massey A. G., J. Chem. Ed., 40 , 311 (1963); 6. Grosse A. V.,
Kirshenbaum A. D., Streng A. G., Streng L. V.,
Science, 139, 1047 (1963).
21. Cady G. H., J. Am. Chem. Soc., 56, 2635 (1934).
22. Yost D. M., Beer bower A., J. Am. Chem. Soc., 57, 782 (1935).
23. R о h r b a c k G. H., Cady G. H., J. Am. Chem. Soc., 69, 677 (1947).
24. C a d у G. H., Kellogg К. B., J. Am. Chem. Soc., 75, 2501 (1953).
25. Menefee A., Cady G. H., J. Am. Chem. Soc., 76, 2020 (1954).
26. C a d у G. H., Grosse A. V., В a r h e r E. J., В u r g e r L. L.,
Sheldon Z. D., Ind. Eng. Chem., 39, 290 (1947).
27. К e 1 1 о g g К. В., C a d у G. H., J. Am. Chem. Soc., 70, 3986 (1948).
28. D u d 1 e у F. В., Cady G. H., Eggers D. F., Jr., J. Am. Chem.
Soc., 78, 290 (1956).
29. D u d 1 e у F. В., Cady G. H., Eggers D. F., Jr., J. Am. Chem.
Soc., 78, 1553 (1956).
272
Глава 10
30. D u d 1 е у F. В., C a d у G. H., J. Am. Chem. Soc., 79, 513 (1957).
31a. В о о t h H. S., M e r i с о 1 a F. C., J. Am. Chem. Soc., 62, 640 (1940);
б. T u 1 1 о с к C. W.,Coffman D. D., J. Org. Chem., 25, 2016 (I960).
32. M i t r a G., C a d у G. H., J. Am. Chem. Soc., 81, 2646 (1959).
33. Nouveau Traite de Chimie Mineral. Vol. XVI, Pascal P., ed., Masson et
Cie, Paris, 1960, p. 423.
34a. Martin H., Kohnlein E., Z. Physik. Chem. (Frankfurt), 17,
375 (1958); 6. Schmeisser M., Brandie K., Angew. Chem., 73,
388 (1961); в. Fink W., Angew. Chem., 73, 466 (1961).
35. Schmeisser M., Taglinger L., Ber., 94, 1533 (1961).
36a. S к i e n s W. E., Cady G. H., J. Am. Chem. Soc., 80, 5640 (1958);
6. Brandie K.., Schmeisser M., Luttke W., Ber., 93,
2300 (1960).
37a. Fink W., Angen. Chem., 73, 532 (1961); 6. also see, ref 32c.
38. R о b e r t s J. E„ Cady G. H., J. Am. Chem. Soc., 81,4166 (1959).
39. A 1 с о с к N. W., Waddington T. C., J. Chem. Soc., 1962, 2510.
40. Gomberg M., J. Am. Chem. Soc., 45, 398 (1923).
41. S a n d m e у e r T., Chem. Ber., 18, 1767 (1885).
42a. Chattaway F. D., Backeberg O. G., J. Chem. Soc., 1923,
2999; б. T e e t e r H. H., Bell E. W., in Organic Syntheses, Vol. 32,
Arnold R. T., ed., Wiley, New York, 1952, p. 20.
43. T а у 1 о r M. C., MacMullin R. B., G a m m a 1 C. A., J. Am.
Chem. Soc., 47, 395 (1925).
44. Anbar M., Dostrovsky I., J. Chem. Soc., 1954, 1094.
45. Anbar M-, Ginsburg D., Chem. Rev., 54, 925 (1954).
46. Bradshaw С. P. C., Nechuatal A., Proc. Chem. Soc., 1963,
213.
47. В и л ь с о н Ч. В., в сб. «Органические реакции», сб. 9, ИЛ, Москва,
1959.
48. Anbar М., Dostrovsky I., J. Chem. Soc., 1954, 1105.
49. Н е n n е A. L., Zimmer W. F., J. Am. Chem. Soc., 73, 1362 (1951)
СОДЕРЖАНИЕ
Предисловие
5
Предисловие редактора американского издания.....................7
Глава 1. Координационные полимеры. Бейлар, мл...............9
I. Введение.............................................9
А. Циано-полимеры ...................................9
Б. Гидроксо-полимеры ................................11
В. Галогено-полимеры ................................14
Г. Тиооксамидо-полимеры..............................15
II. Свойства координационных полимеров .................16
III. Основные структурные закономерности.................17
IV. Методы синтеза .....................................20
А. Полимеры на основе 1,3-дикетонов.................20
Б. Полимеры, содержащие 8-оксихинолины...............24
В. Фосфинатные полимеры ............................27
Г. Органические полимеры в качестве лигандов.........29
Д. Синтез полимеров из карбонилов металлов...........32
Е. Фталоцианиновые полимеры..........................33
Литература ..........................................35
Глава 2. Оптически активные координационные соединения. Киршнер 38
I. Введение ...........................................38
II. Оптическая активность, поляриметрия и спектрополяриметрия 40
А. Оптическая активность............................40
Б. Поляриметрия .....................................41
В. Спектрополяриметрия ......................42
III. Методы получения оптически активных координационных
соединений. 44
А. Методы получения путем разделения.................44
Б. Методы получения без разделения...................54
IV. Оптическая чистота .................................57
А. Определения......................................57
Б. Критерии оптической чистоты ......................57
В. Определение оптической чистоты..................57
V. Препаративные методики ......................59
VI. Заключение .........................................63
Литература ...........................................63
274
Содержание
Глава 3. Соединения металлов с кетиминами и альдиминами. Д. Ф. Мар-
тин ..............................................70
I. Введение............................................70
А. Номенклатура.....................................70
Б. Побочные реакции..................................71
В. Получение лигандов...............................72
II. Методы получения соединений металлов с альдиминами и кети-
минами .................................................73
А. Непосредственное взаимодействие..................73
Б. Соединение компонентов ...........................75
В. Аминный обмен ...................................76
Г. Хелатный обмен...................................78
Д. Обмен металла....................................80
Е. Темплатный синтез................................80
Ж. Другие методы ....................................82
III. Экспериментальные методы ...........................83
А. Непосредственное взаимодействие..................83
Б. Соединение компонентов ...........................83
В. Аминный обмен ...................................84
Г. Хелатный обмен...................................85
Д. Обмен металла ...................................85
Е. Темплатный синтез................................85
Литература ..........................................86
Глава. 4. Карбонилы металлов. Хилем ан......................88
I. Введение ...........................................88
А. Известные карбонилы металлов.....................88
Б. Структура карбонилов металлов.....................91
В. Предсказанные, но не синтезированные карбонилы ме-
таллов ..............................................93
II. Оборудование........................................96
А. Высокое давление .................................96
Б. Вспомогательное оборудование для синтеза карбонилов
металлов ........................................102
III. Методы получения карбонилов металлов...............106
А. Литература по синтезу карбонилов металлов.......106
Б. Получение карбонилов железа......................106
В. Получение карбонилов кобальта...................112
Г. Получение карбонила технеция ...................114
Д. Получение карбонила марганца....................116
Е. Получение карбонила ванадия.....................120
IV. Идентификация карбонилов металлов..................121
А. Инфракрасные спектры ...........................121
Б. Рентгеноструктурный анализ ......................125
В. Различные методы анализа .......................127
Литература .........................................129
Глава 5. Галогенидные и оксигалогенидные комплексы элементов
подгрупп титана, ванадия и хрома. Фаулс..................134
I. Галогенидные и оксигалогенидные комплексы элементов под-
группы титана ........................................134
А. Для четырехвалентного состояния..................134
Содержание
275
Б. Для трех- и двухвалентных состояний...................136
II. Галогенидные и оксигалогенидные комплексы элементов
подгруппы ванадия ...........................137
А. Для пятивалентного состояния .........................137
Б. Для низших валентностей ..............................138
III. Галогенидные и оксигалогенидные комплексы элементов
подгруппы хрома ........................................140
А. Для состояний окисления 6+ и 5+.......................140
Б. Для состояний окисления 44-> 3-|- и 2-]-..............141
IV. Методы получения галогенидных и оксигалогенидных ком-
плексных солей .........................................144
А. Титан и цирконий ....................................146
Б. Ванадий, ниобий и тантал..............................148
В. Хром, молибден и вольфрам............................149
V. Методики синтеза .......................................151
А. Подгруппа титана.....................................151
Б. Подгруппа ванадия.....................................152
В. Подгруппа хрома .....................................152
Литература ..............................................154
Глава 6. Безводные нитраты металлов. Эдисон и Логан .... 156
I. Введение................................................156
II. Реакции с использованием четырехокиси азота.............158
А. Четырехокись азота без растворителя..................159
Б. Четырехокись азота в инертном разбавителе.............162
В. Четырехокись азота с донорными растворителями . . . 165
III. Реакции с использованием пятиокиси азота................173
А. Пятиокись азота без добавок ..........................173
Б. Пятиокись азота в чистой азотной кислоте..............175
IV. Сравнительная оценка четырехокиси и пятиокиси азота . .175
А. Соединения хрома .....................................176
Б. Соединения ванадия ...................................178
V. Нитраты галогенов.......................................178
Литература ..............................................180
Глава 7. Галогено-и галогеноид-производные силанов. Мак-Дайармид 182
I. Введение................................................182
II. Общие методы синтеза....................................184
А. Из кремния или силицидов ............................184
Б. Из соединений со связью кремний—кремний . . . 185
В. Из соединений со связью кремний— водород .... 185
Г. Из соединений со связью кремний — галоген . . . 187
Д. Из соединений со связью кремний — кислород. . . . 189
Е. Из соединений со связью кремний—азот..192
Ж. Из соединений со связью кремний — фенил .... 193
3. Через реакции перераспределения......................195
III. Производные силана .....................................196
А. Фторосиланы .........................................196
Б. Хлоросиланы ..........................................198
В. Бромосиланы..........................................200
Г. Иодосиланы ...........................................201
Д. Тетрагалогеносиланы ..................................203
Е. Галогеноидные производные............................206
276
Содержание
IV. Производные дисилана...............................211
А. Фтородисиланы ..................................211
Б. Хлородисиланы ...................................211
В. Бромодисиланы ................................. 212
Г. Иододисиланы.....................................214
Д. Галогеноидные производные........................215
V. Производные высших силанов ........................215
А. Фторосиланы ....................................216
Б. Хлоросиланы .....................................216
В. Бромосиланы ....................................217
Г. Иодосиланы ......................................217
Литература .........................................217
Глава 8. Солеобразные гидриды. Мессер......................222
I. Введение ..........................................222
А. Общие замечания .................................222
Б. Упругость диссоциации ...........................222
II. Аппаратура и методика синтезов ....................224
А. Общие замечания ................................224
Б. Реакционная камера ............................. 224
В. Очистка водорода ...............................227
Г. Материалы и их обработка ........................227
Д. Определение количества водорода в образце .... 228
Е. Анализ .........................................229
III. Гидрид лития ......................................230
А. Введение .......................................230
Б. Кинетика образования гидрида лития ..............231
В. Литература по синтезу гидрида лития.............233
Г. Методика синтеза гидрида лития ..................233
Д. Анализ ..........................................235
IV. Гидрид натрия .....................................235
А. Введение .......................................235
Б. Суспензионный метод .............................236
В. Применение катализатора или поверхностноактивного ве-
щества.............................................237
Г. Реакция в паровой фазе...........................239
V. Гидриды других щелочных металлов...................240
VI. Гидрид кальция.....................................240
А. Введение .......................................240
Б. Кинетика образования гидрида кальция.............241
В. Синтез гидрида кальция .........................243
VII. Другие солеобразные гидриды.........................244
А. Введение .......................................244
Б. Гидриды бария и стронция ........................244
В. Гидрид магния ..................................244
Литература .........................................245
Глава 9. Соединения серы с азотом и фтором. Глемзер.........248
I. Введение ..........................................248
II. Ациклические соединения ...........................249
А. Тиазилфторид NSF................................249
Б. Тиодитиазилдифторид S3N2F2 ......................251
В. Тиазилтрифторид.................................252
Содержание 277
III. Циклические соединения ..........................254
А. Тетратиазилтетрафторид N4S4F4..................254
Б. Тритиазилтрифторид N3S3F3......................255
В. Тиотритиазилфторид S4N3F ......................257
Литература........................................258
Глава 10. Гипогалогениты и соединения, содержащие ОХ_-группу.
Вильямсон ..........................................260
I. Введение .........................................260
II. Ионные гипогалогениты .............................261
III. Ковалентные гипогалогениты.........................262
IV. Фториды кислорода..................................263
V. Гипофториты сильных кислот ........................264
VI. Гипофториты окислов ...............................265
VII. Окислы галогенов (I)...............................266
VIII. Нитраты галогенов (I) 267
IX. Фторсульфонаты галогенов (I).......................267
X. Перхлораты галогенов (I) 268
XI. Алкоксиды галогенов (I) 269
XII. Карбоксилаты галогенов (I) 270
Литература .........................................271
СИНТЕЗЫ НЕОРГАНИЧЕСКИХ
СОЕДИНЕНИЙ ТОМ I
Редактор Г. М. Мануйлова
Художник И. А. Литвишко
Художественный редактор Е. И. Бескова
Технический редактор М. И. Грибова
Корректор Л. П. Големинова
Сдано в производство 4/VII 1966 г.
Подписано к печати 27/Х 1966 г.
Бумага 60x90’/ns8,75 бум. л.
17,5 усл. печ. л.
Уч.-изд. л. 17,08. Изд М 3/3523
Цена 1 р. 43 к. Зак. 547.
*
ИЗДАТЕЛЬСТВО «МИР»
Москва, 1-й Рижский пер., 2
*
Ярославский полиграфкомбинат Главполи-
графпрома Комитета по печати при Совете
Министров СССР. Ярославль,
ул. Свободы, 97.
ИЗДАТЕЛЬСТВО «МИР»
выпускает в свет
в 1967 году
книги по химии
Синтезы неорганических соединений, том 2, пе-
ревод с английского, 23 изд. л цена 1 р. 90 к.
Книга представляет собой очередной, 2-й том
серии , .Синтезы неорганических соединений' ‘,
посвященной теоретическим основам синтеза наи-
более актуальных классов неорганических соеди-
нений. Большинство изучаемых классов соедине-
ний в настоящее время приобретает важное про-
мышленное значение.
В книге рассматриваются фосфазеновые сое-
динения, соединения кремния с азотом, ортофос-
форные кислоты и ортофосфаты, алкоксиды метал-
лов, бинарные фториды, соединения платиновых
металлов с фтором, соединения ксенона и др.
Книга предназначена для химиков-неоргани-
ков—работников научно-исследовательских инсти-
тутов и промышленных предприятий.
ИЗДАТЕЛЬСТВО «МИР»
выпускает, в свет, в 1967 году
книги по химии
Айлетт Б., Смит Б. Задачи и упражнения
по неорганической химии, перевод с англий-
ского, 10 изд. л., цена 90 коп.
Книга представляет исключительный интерес
для преподавателей и студентов, специализиру-
ющихся в области физической и неорганической
химии, поскольку материал, приведенный в этом
задачнике по химии, может быть непосредственно
использован в лекционных курсах и на семинар-
ских занятиях.
Авторы рассматривают неорганическую хи-
мию не как отдельную область химии, а как
совокупность сведений, полученных в результа-
те применения общих, физических и физико-хи-
мических методов исследования к определенным
классам соединений, специфичными для которых
являются лишь методы синтеза.
Книга предназначена для химиков-неоргани-
ков и физикохимиков — преподавателей, аспиран-
тов и студентов химических вузов.