/































































































































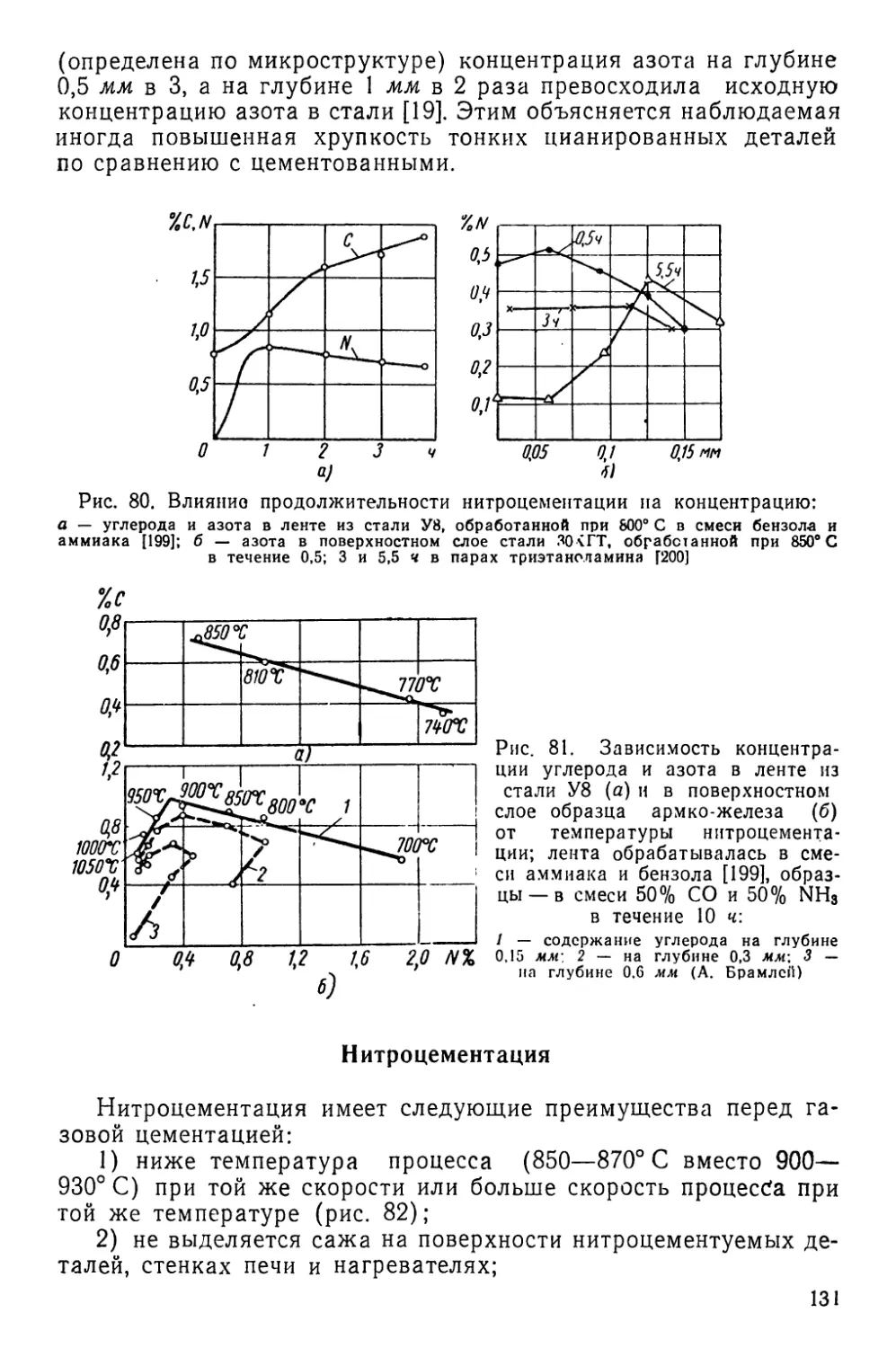





























































































































































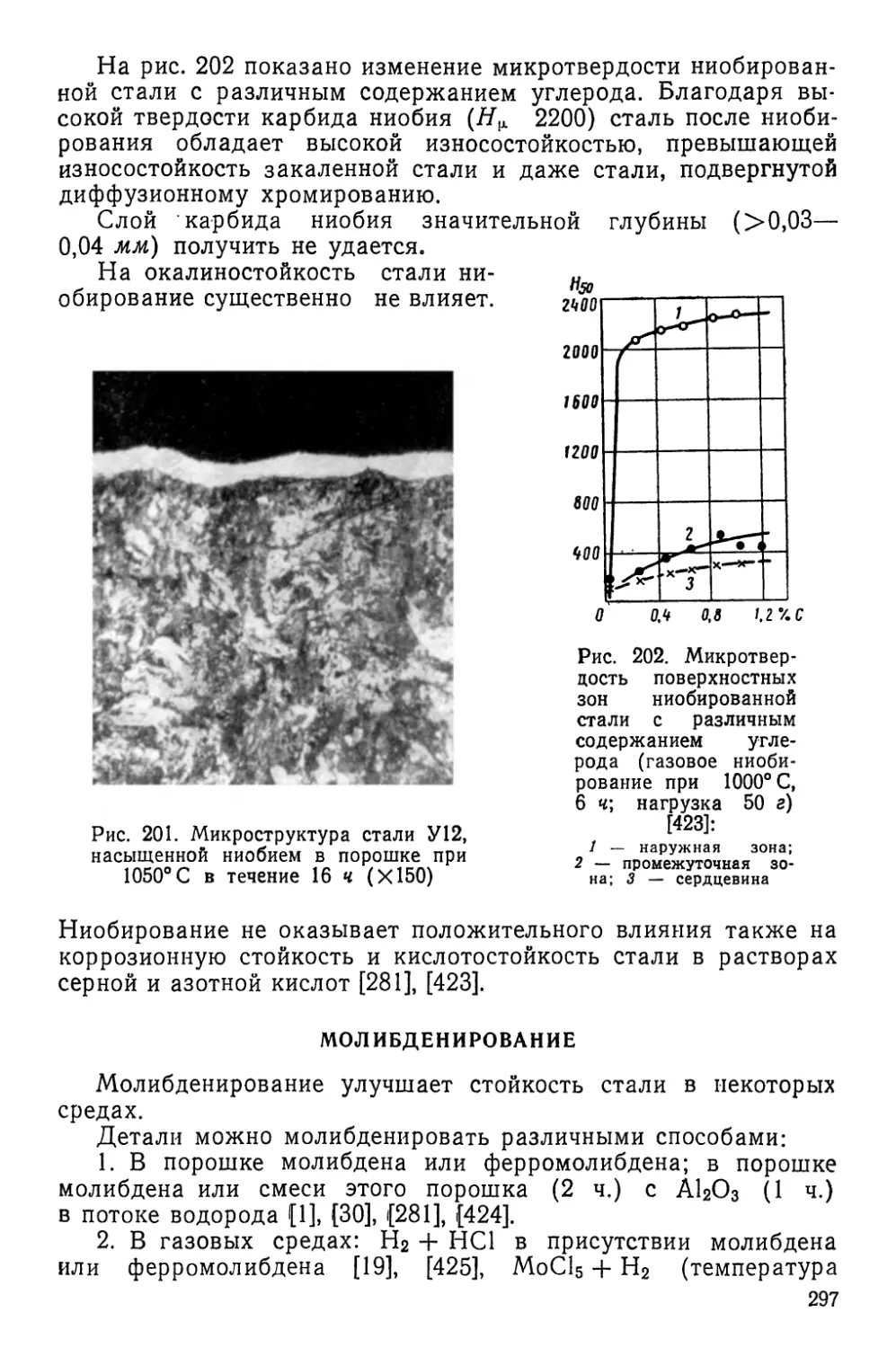










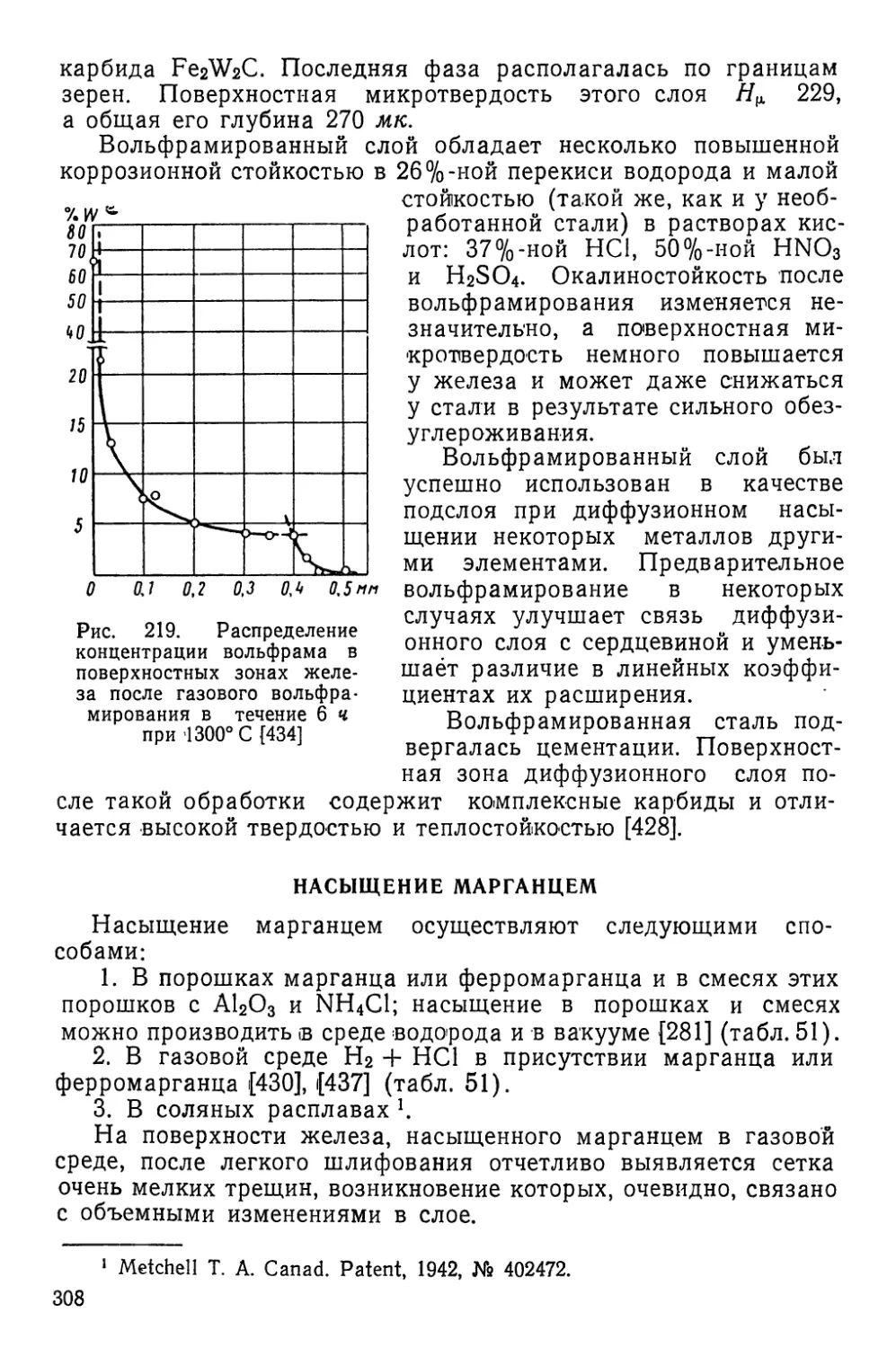










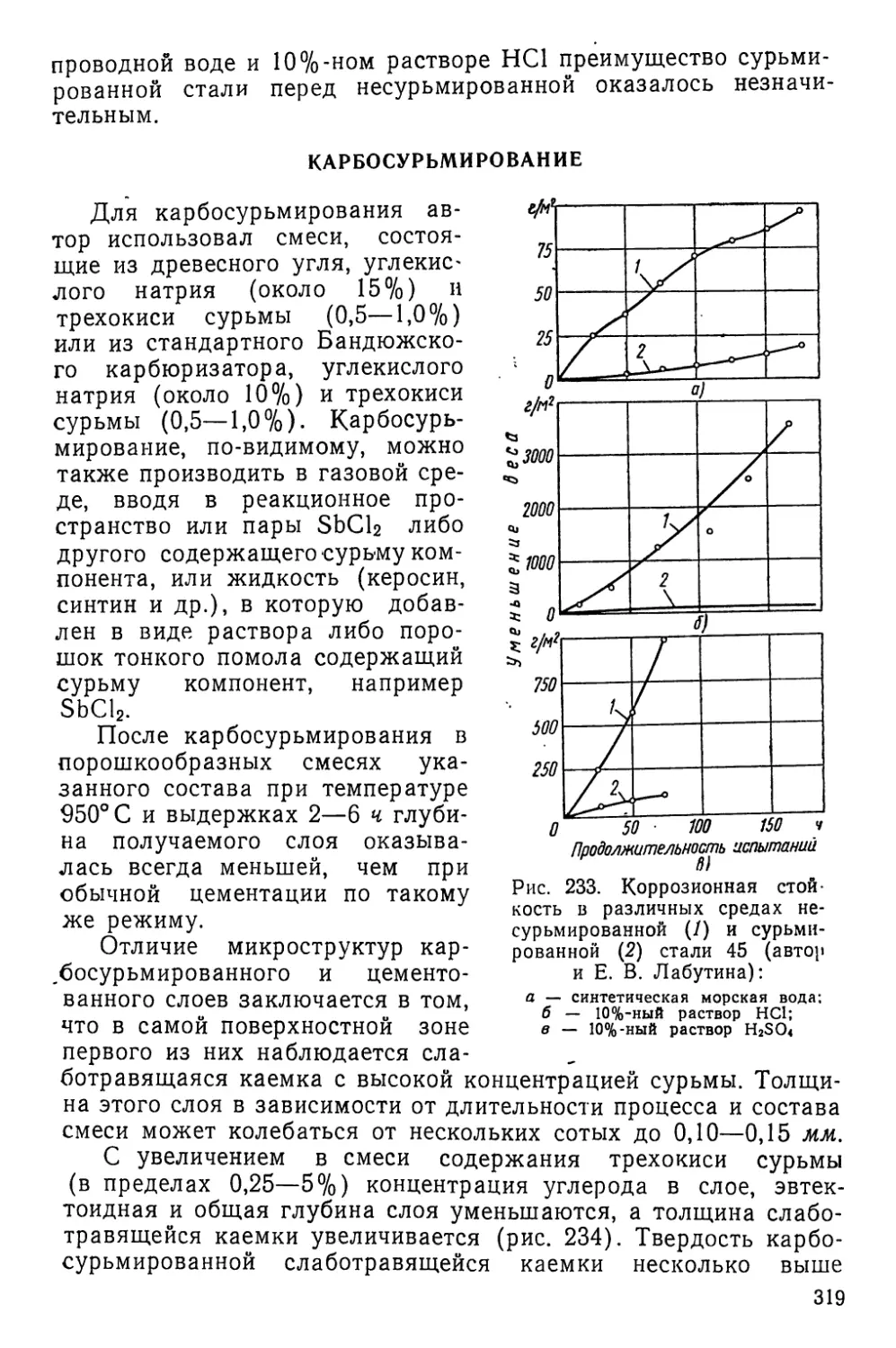


































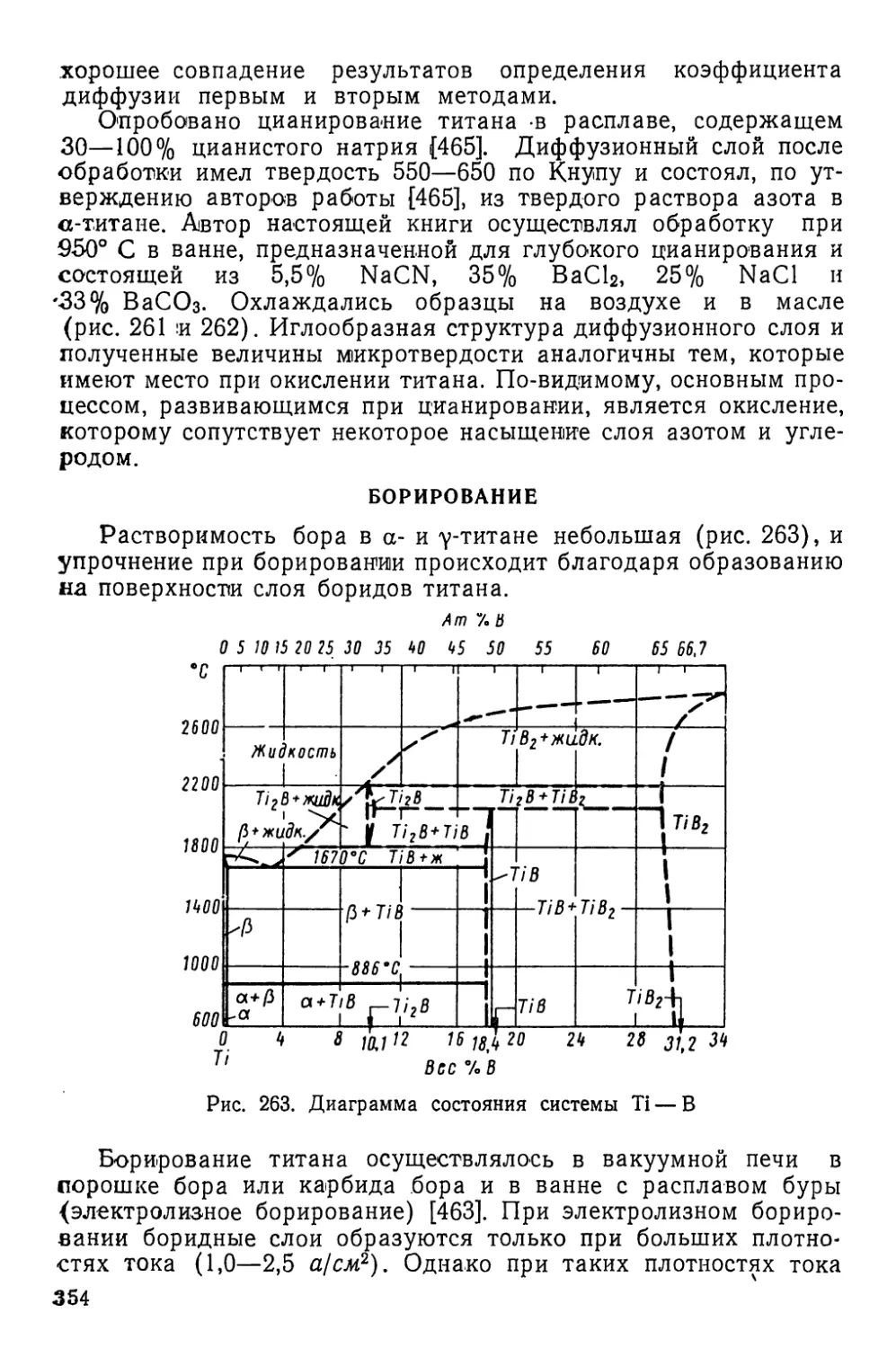















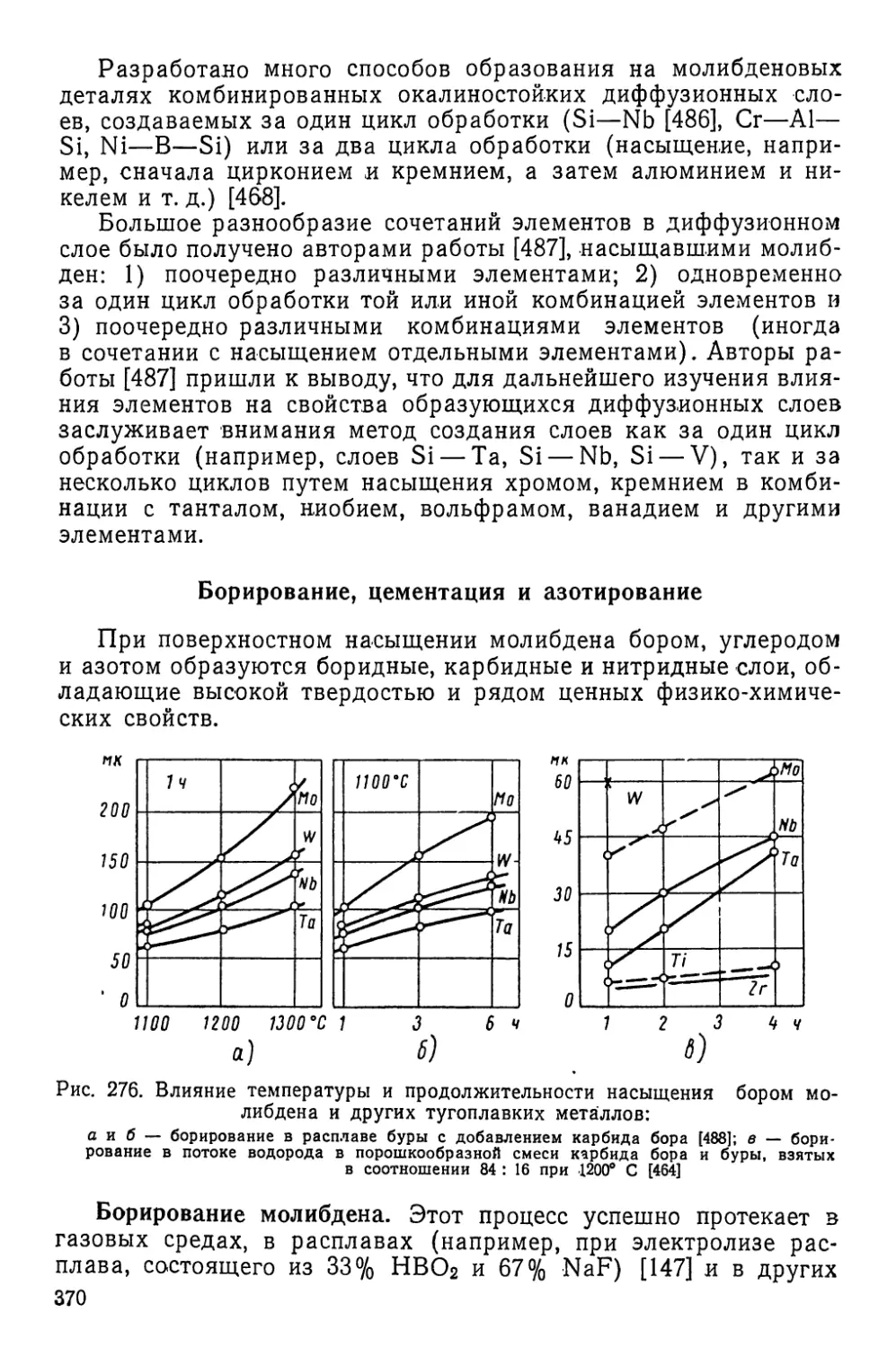






































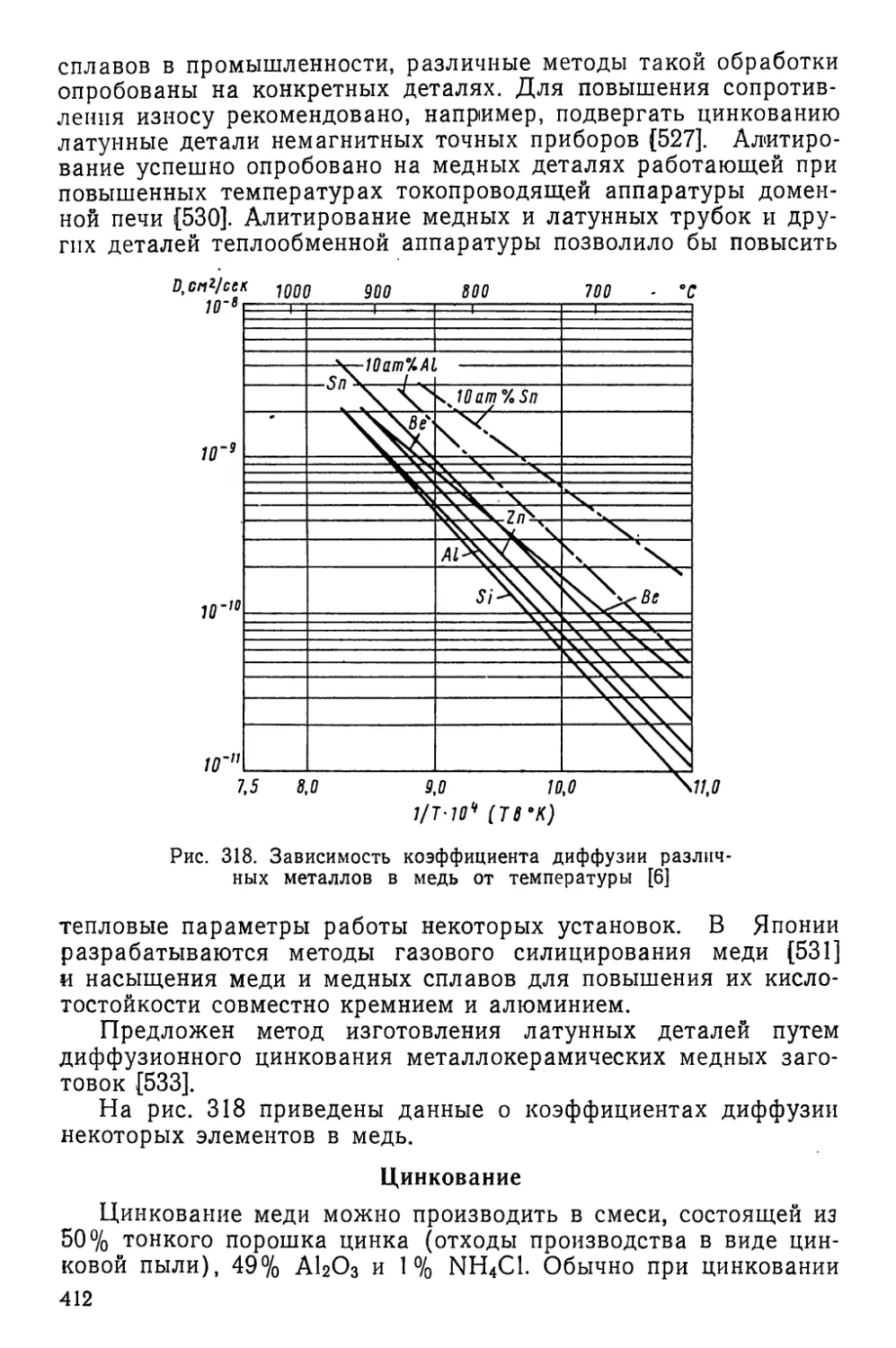








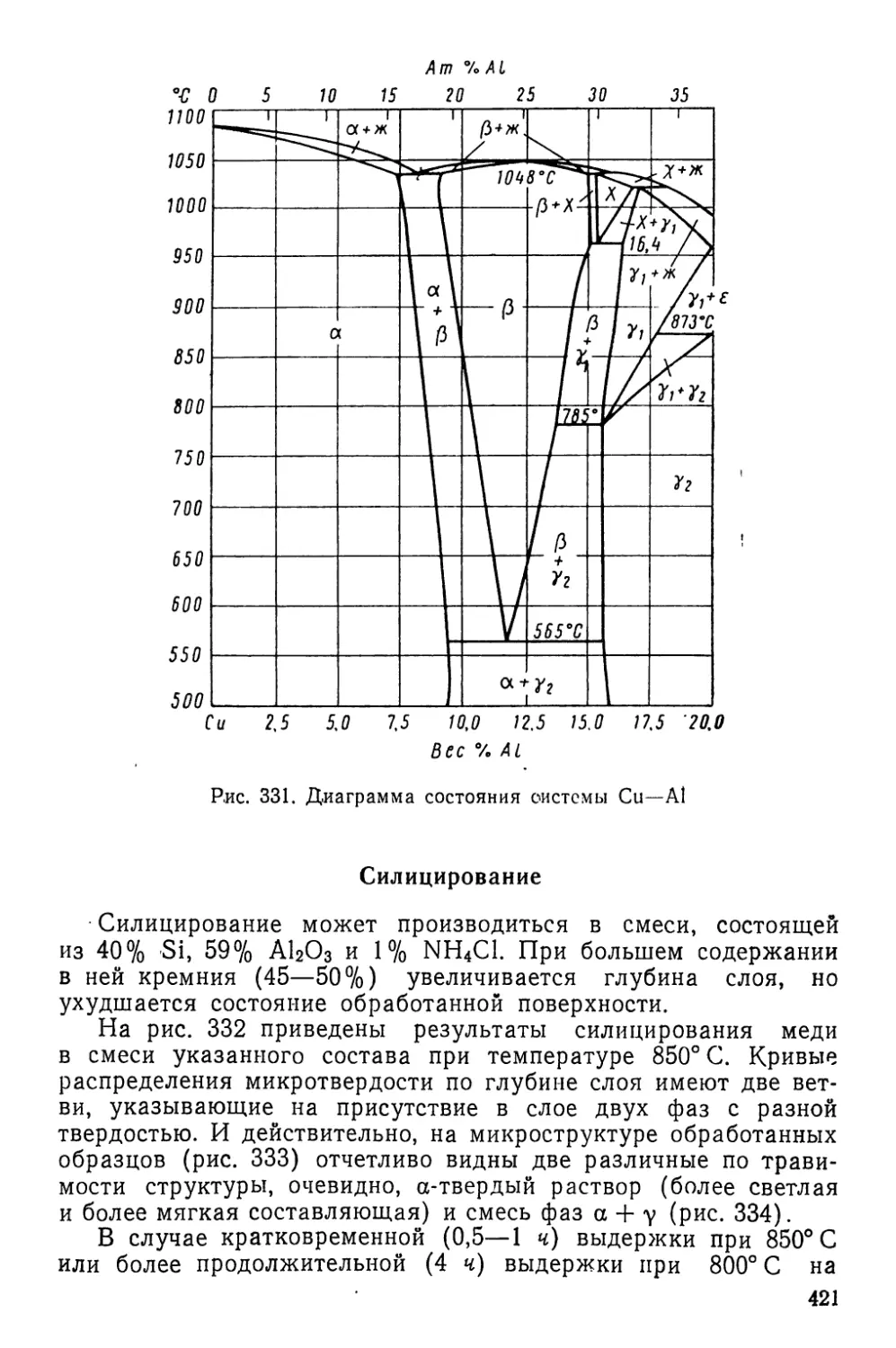




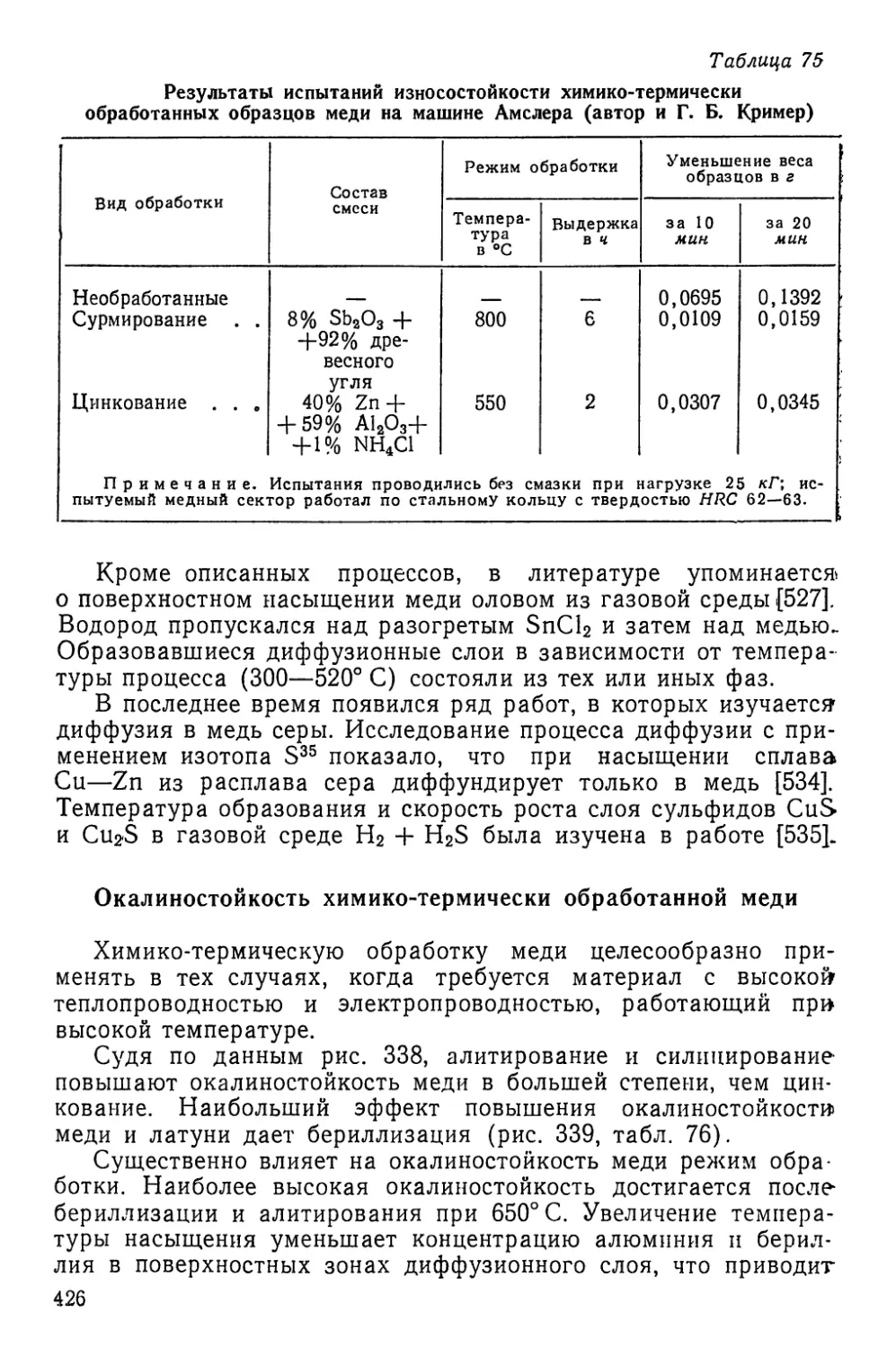



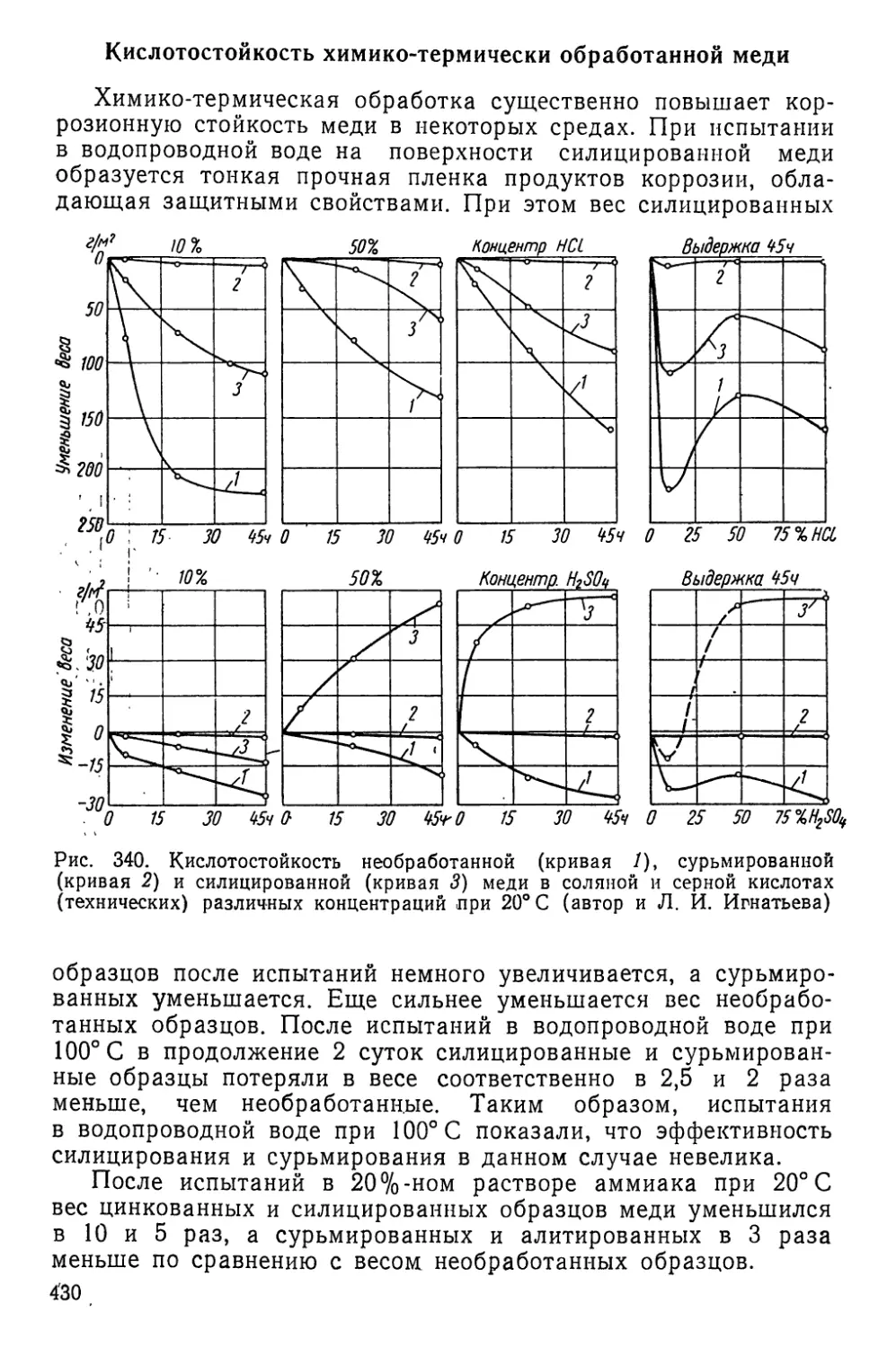





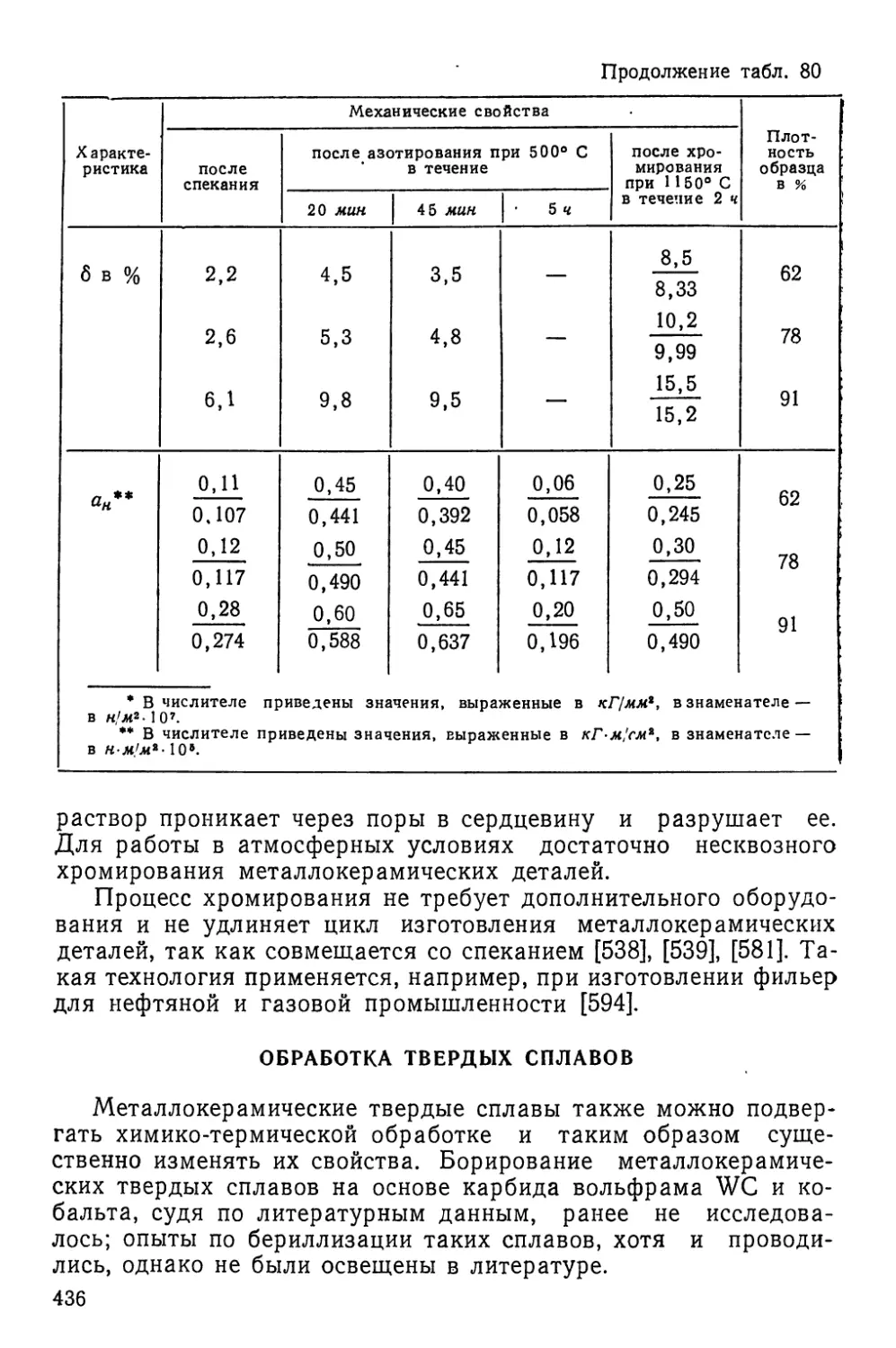






























































Текст
5,2 Ё 78
А.Н.МИНКЕВИЧ
ХИМИКО-
ТЕРМИЧЕСКАЯ
ОБРАБОТКА
МЕТАЛЛОВ
И СПЛАВОВ 7
5 I
s<
л ; rnyHOg ?
УДК 621.78.794
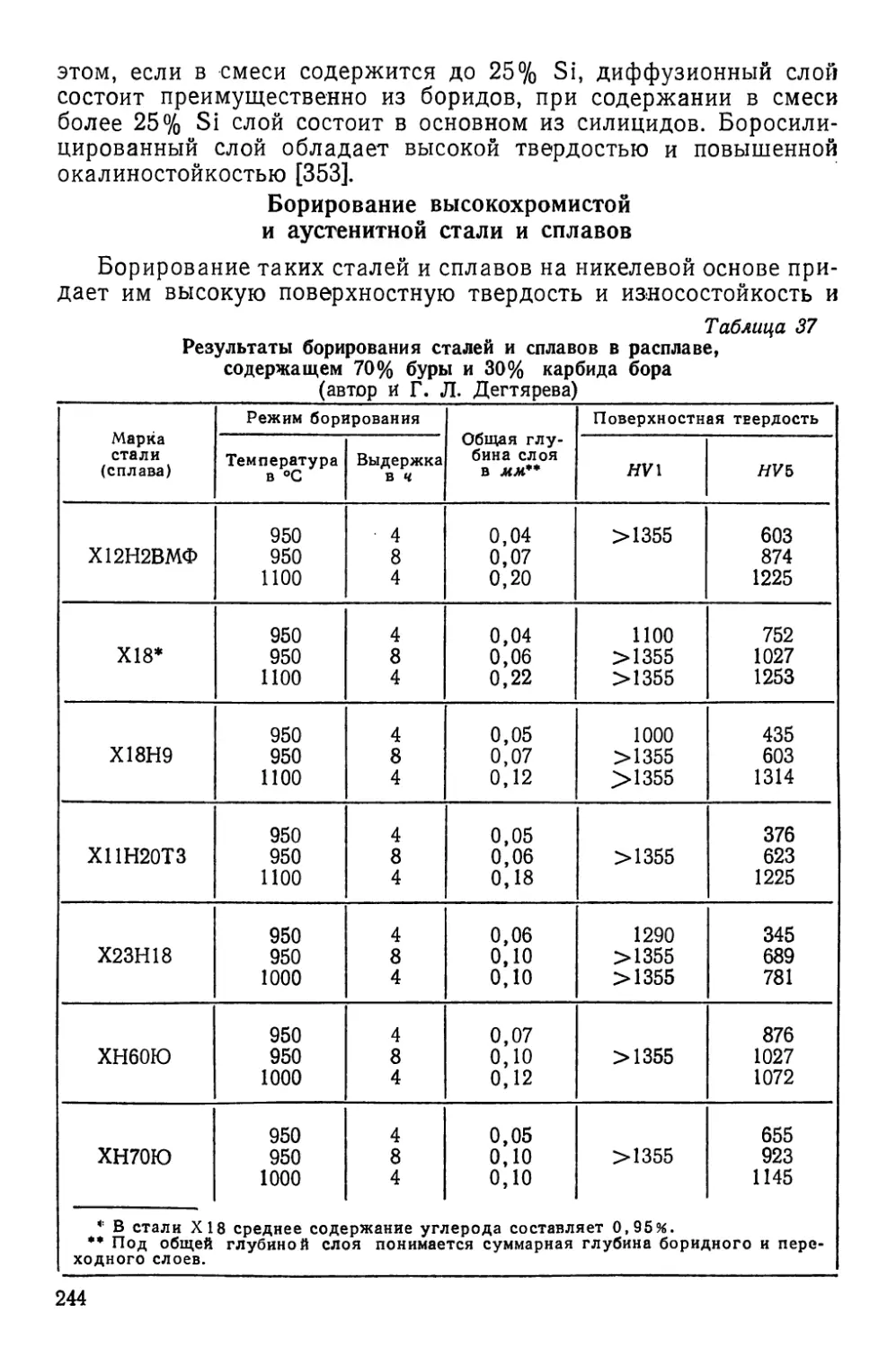
В монографии кратко изложены, общие закономерности
диффузионных процессов и методы получения диффузионных
слоев на металлах.
Рассмотрены основы процессов цементации, нитроцемента-
ции, цианирования, азотирования, алитирования, хромирова-
ния, борирования и цинкования стали, а также процессов
поверхностного насыщенця стали бериллием, кремнием, ти-
таном, ванадием, молибденом, вольфрамом, ниобием, марган-
цем, серой и некоторыми другими элементами. Освещены
процессы химико-термической обработки титана, молибдена,
ниобия, вольфрама, тантала, циркония, никеля, меди, кобаль-
та и сплавов на их основе.Дано краткое описание процессов
химико-термической обработки металлокерамических сплавов,
гальванических покрытий и полупроводниковых материалов,
а также рассмотрены условия осаждения на поверхности ме-
таллов карбидных, боридных, нитридных и силицидных по-
крытий.
Для каждого вида обработки указаны методы насыщения,
описан химизм процесса насыщения и рассмотрены влияние
различных факторов на ход процесса, а также структура и
свойства, приобретаемые металлами и сплавами после обра-
ботки.
Монография предназначена для инженерно-технических и
научных работников, специализирующихся в области химико-
термической обработки, и может быть использована студен-
тами. вузов, изучающими курс термической обработки метал-
лов.
Рецензент д-р техн, наук проф. А. Д. Ассонов
Редактор д-р техн, наук проф. Ю. М. Лахтин
3—12—5
306—65
9
ПРЕДИСЛОВИЕ
В постановлениях Партии и Правительства по вопросам
развития отечественной промышленности неоднократно отмеча-
лось, что одной из важнейших задач машиностроения является
повышение прочности, надежности и долговечности изделий.
Большую роль в решении этой проблемы должно сыграть даль-
нейшее совершенствование и широкое внедрение в производство
различных методов химико-термической обработки.
Химико-термическая обработка широко применяется в ма-
шиностроении. Это определяет большую потребность в литера-
туре, рассматривающей различные методы химико-термической
обработки, как применяемые в производстве, так и находящиеся
в стадии лабораторных исследований. Разрозненность обшир-
ных опубликованных материалов, посвященных вопросам хими-
ко-термической обработки, и то, что часть этих материалов уста-
рела, затрудняет пользование ими.
В предлагаемой вниманию читателей книге автор попытался
обобщить основные материалы, связанные с вопросами химико-
термической обработки стали, различных металлов, а также
сплавов на нежелезной основе, и изложил свои некоторые на-
блюдения и исследования.
По сравнению с первым изданием, вышедшим в 1950 г., кни-
га значительно переработана. В настоящем издании не рас-
сматривается оборудование для химико-термической обработки,
так как оно достаточно полно описано в литературе по обору-
дованию термических цехов. Сокращены главы, посвященные
цементации, цианированию и азотированию стали. За счет
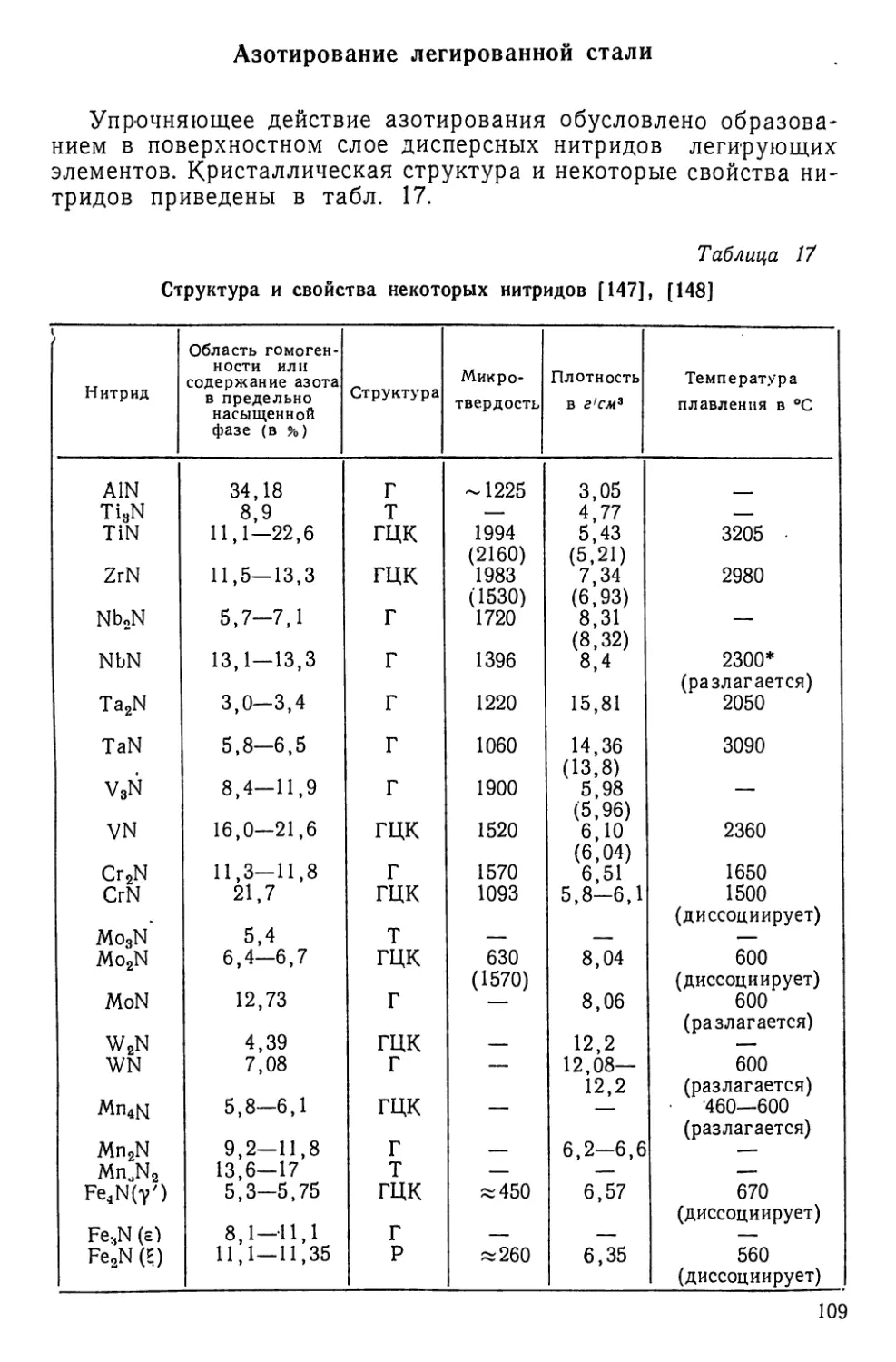
этого расширены разделы по алитированию, хромированию, бо-
рированию и цинкованию стали, т. е. процессам, которые заслу-
живают более широкого применения в промышленности.
В книгу включены новые главы и разделы, посвященные насы-
щению стали титаном, ниобием, марганцем, танталом, сурьмой,
серой, фосфором и некоторыми другими элементами. Вторая
половина книги посвящена химико-термической обработке ти-
тана, молибдена, ниобия, вольфрама, тантала, циркония, ко-
бальта, меди, а также металлокерамических сплавов и гальва-
з
нических покрытий. Эти процессы, а также процесс осаждения
на поверхности металлов карбидных, нитридных, боридных и
силицидных слоев и процессы химико-термической обработки
полупроводниковых материалов, которым посвящены две по-
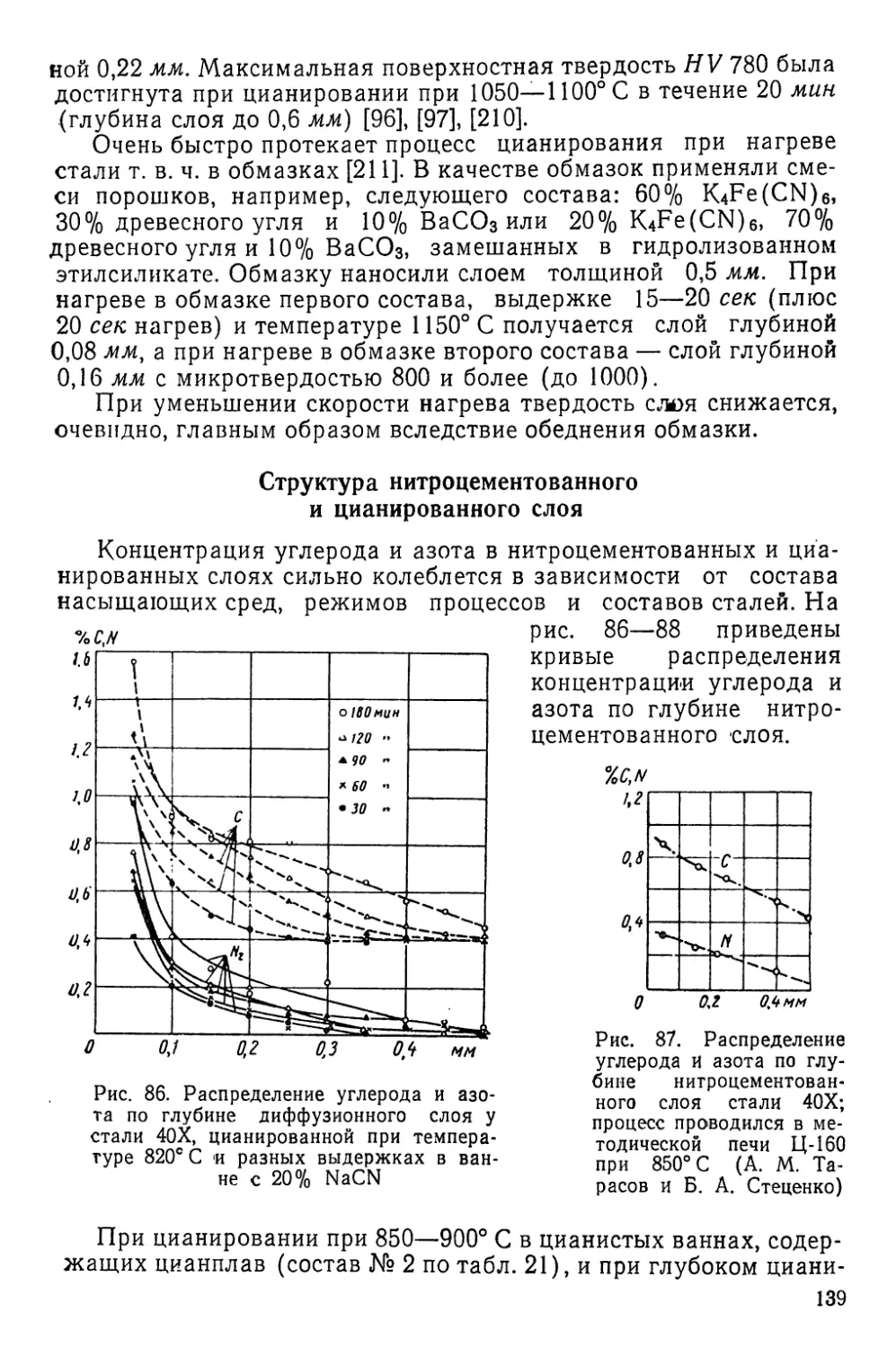
следние главы книги, представляют особый интерес для отрас-
лей новой техники. В книге, помимо процессов, применяемых в
производстве, описаны и новые, еще разрабатываемые пер-
спективные процессы, а также некоторые процессы (насыщение
стали фосфором, мышьяком, церием, медью; насыщение титана
рядом элементов и др.), которые, очевидно, не перспективны
с практической точки зрения, но имеют познавательный интерес.
В каждой главе, посвященной тому или иному процессу хи-
мико-термической обработки, изложены особенности его тех-
нологии, характеризуются микроструктура и фазовый состав
образующихся диффузионных слоев и описаны свойства, при-
обретаемые металлами и сплавами в результате обработки.
В книге лишь очень кратко освещены процессы, протекаю-
щие во внешней насыщающей среде, и взаимодействие этой
среды с поверхностью металла. Закономерности перемещения
элемента в глубь металлов и особенности возникающих при
этом структур подробнее рассмотрены в гл. I.
Приведенные в книге диаграммы состояний двойных систем
заимствованы из общеизвестных справочников (А. Е. Вол
«Строение и свойства двойных металлических систем», т. 1,
1959; т. 2, 1962, Физматиздат и М. Хансен и К. Андерко «Струк-
туры двойных сплавов», том 1 и 2, 1962, Металлургиздат).
Микрофотографии, в подписях к которым не указан автор ис-
следования, выполнены непосредственно автором настоящей
книги или при его участии.
В перечне литературы отсутствует большинство источников,
использованных в первом издании книги; при использовании
материалов этих источников в настоящем издании автор ссы-
лается на первое издание [19].
ВВЕДЕНИЕ
Одним из наиболее эффективных и широко применяемых
iB промышленности методов повышения долговечности многих
ответственных деталей является, их химико-термическая обра-
ботка, которая воздействует, на поверхностные слои металла,
т. е. на те слои, в которых концентрируются максимальные на-
пряжения, возникают трещины, развиваются процессы износа и
коррозии.
Химико-термической обработкой достигаются:
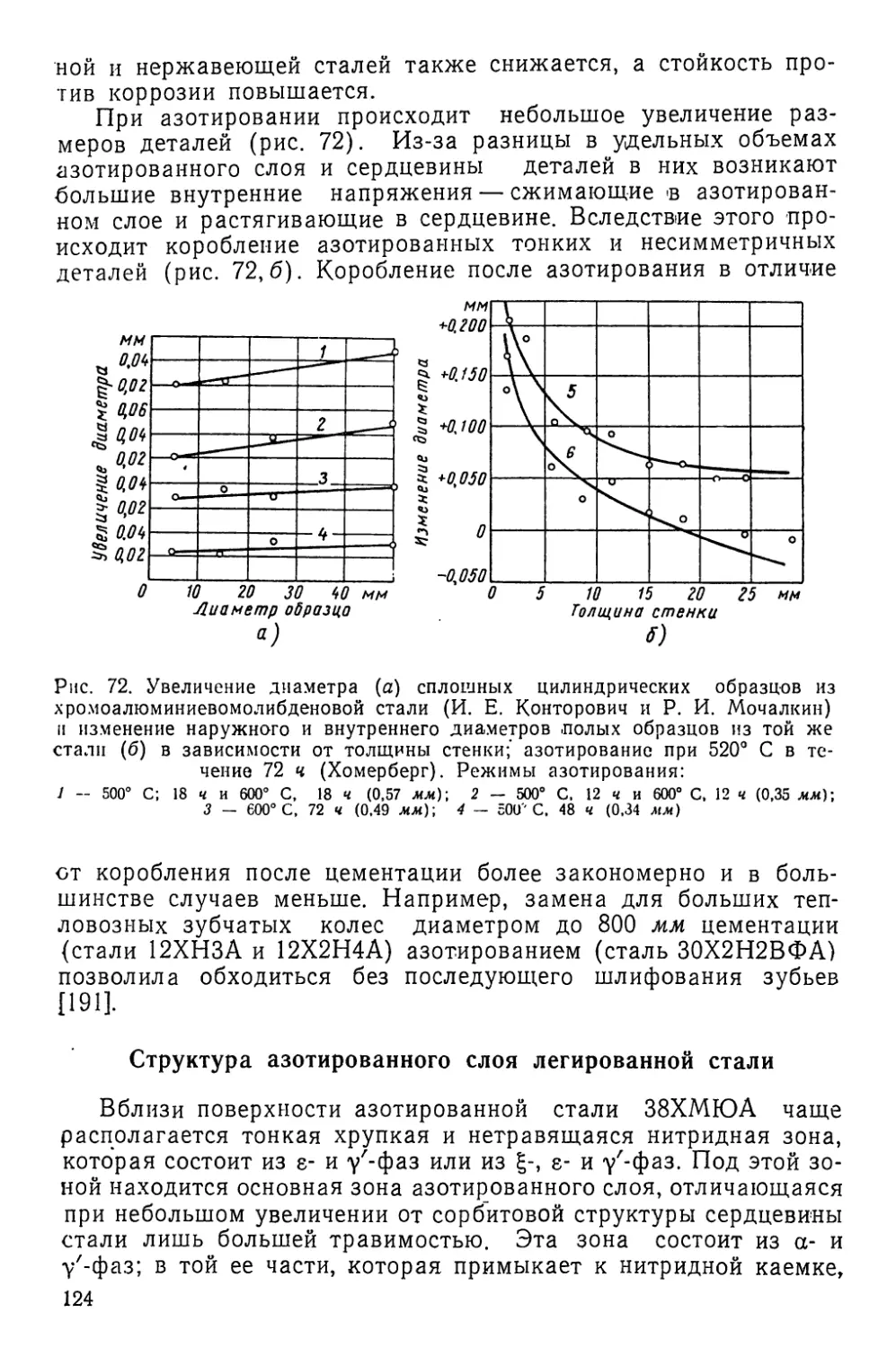
а) поверхностное упрочнение металлов и сплавов (повы-
шаются поверхностная твердость, износостойкость, усталостная
прочность, теплостойкость и т. д.);
б) повышение стойкости металлов и сплавов против воздей-
ствия внешних агрессивных сред при нормальных и повышен-
ных температурах (повышаются стойкость против коррозии, ка-
витационной эрозии, кислотостойкость, окалиностойкость и т. д.).
Химико-термическая обработка металлов и сплавов заклю-
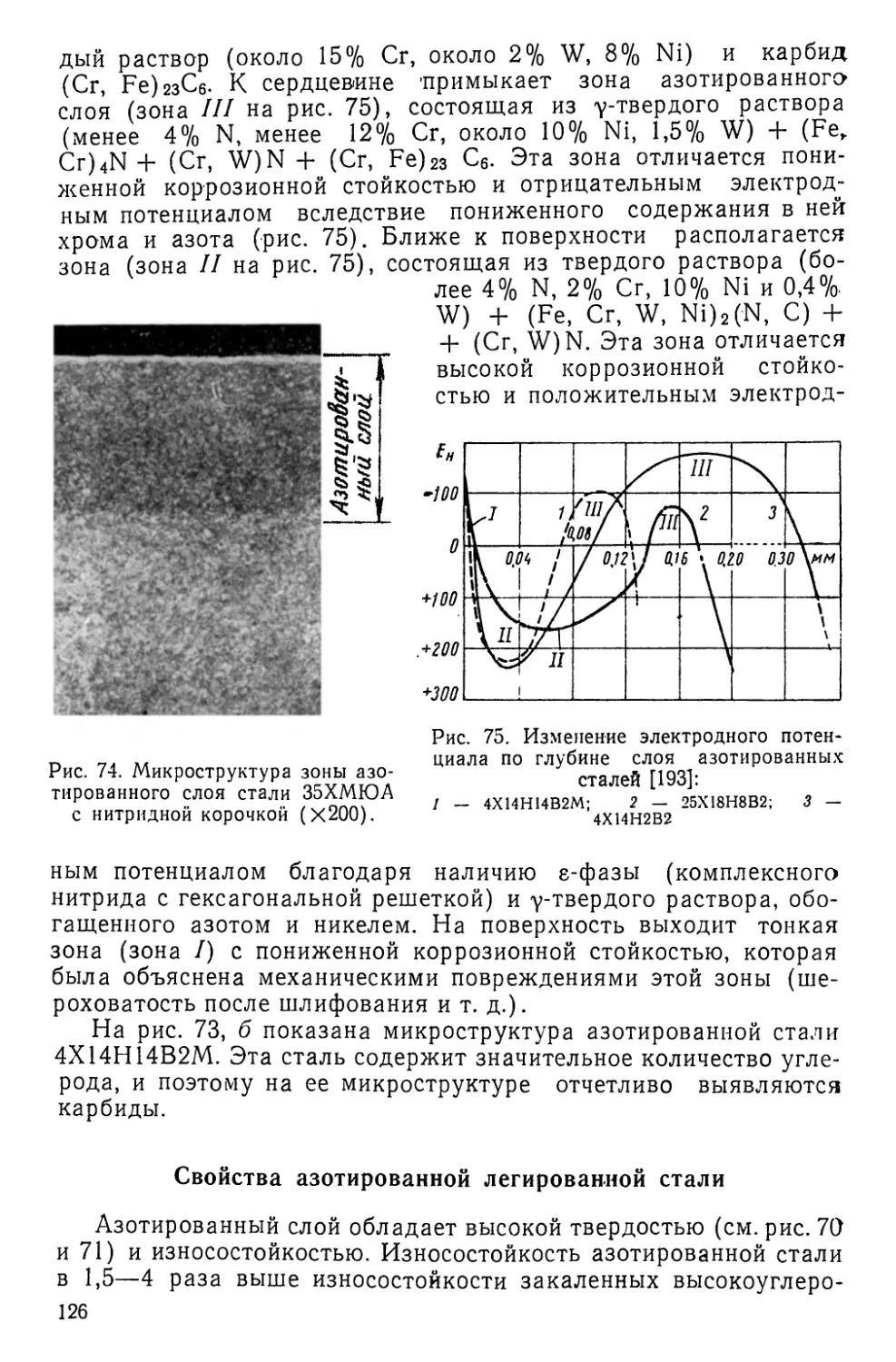
чается в нагреве и выдержке их при высокой температуре в
активных газовых, жидких или твердых средах, в результате
чего изменяются химический состав, структура и свойства по-
верхностных слоев металлов и сплавов. В отличие от термиче-
ской обработки химико-термическая обработка изменяет не
только структуру, но и химический состав поверхностных слоев,
что позволяет в более широких пределах изменять свойства
металлов и сплавов. После некоторых видов химико-термиче-
ской обработки для улучшения свойств сердцевины и поверх-
ностных слоёв проводят термическую обработку. Иногда
термическая обработка предшествует химико-термической
(азотированию, низкотемпературной нитроцементации и низко-
температурному цианированию, цинкованию и т. д.).
До недавнего времени химико-термической обработке под-
вергали лишь стали. При этом наибольшее распространение в
промышленности получили процессы цементации, нитроцемен-
тации, цианирования и азотирования. Реже применяют алити-
рование (алюминирование), хромирование и сульфоцианирова-
ние стали (насыщение одновременно серой, азотом и углеродом).
5
Такие процессы, как борирование, силицирование, цинкование,
бериллизация, получили пока лишь ограниченное применение.
В последние годы химико-термическую обработку начали
использовать для улучшения свойств поверхностных слоев раз-
личных металлов: титана, молибдена, ниобия, тантала, цирко-
ния, кобальта, никеля, меди и др., а также сплавов на их ос-
нове.
Большую роль играет химико-термическая обработка и в
производстве полупроводников.
Методы химико-термической обработки, применяемые в про-
изводстве, основаны на обогащении поверхностных слоев метал-
лов и сплавов тем или иным элементом или комплексом эле-
ментов. Обработка, основанная на обеднении поверхностных
слоев сплавов элементами, в производстве почти не приме-
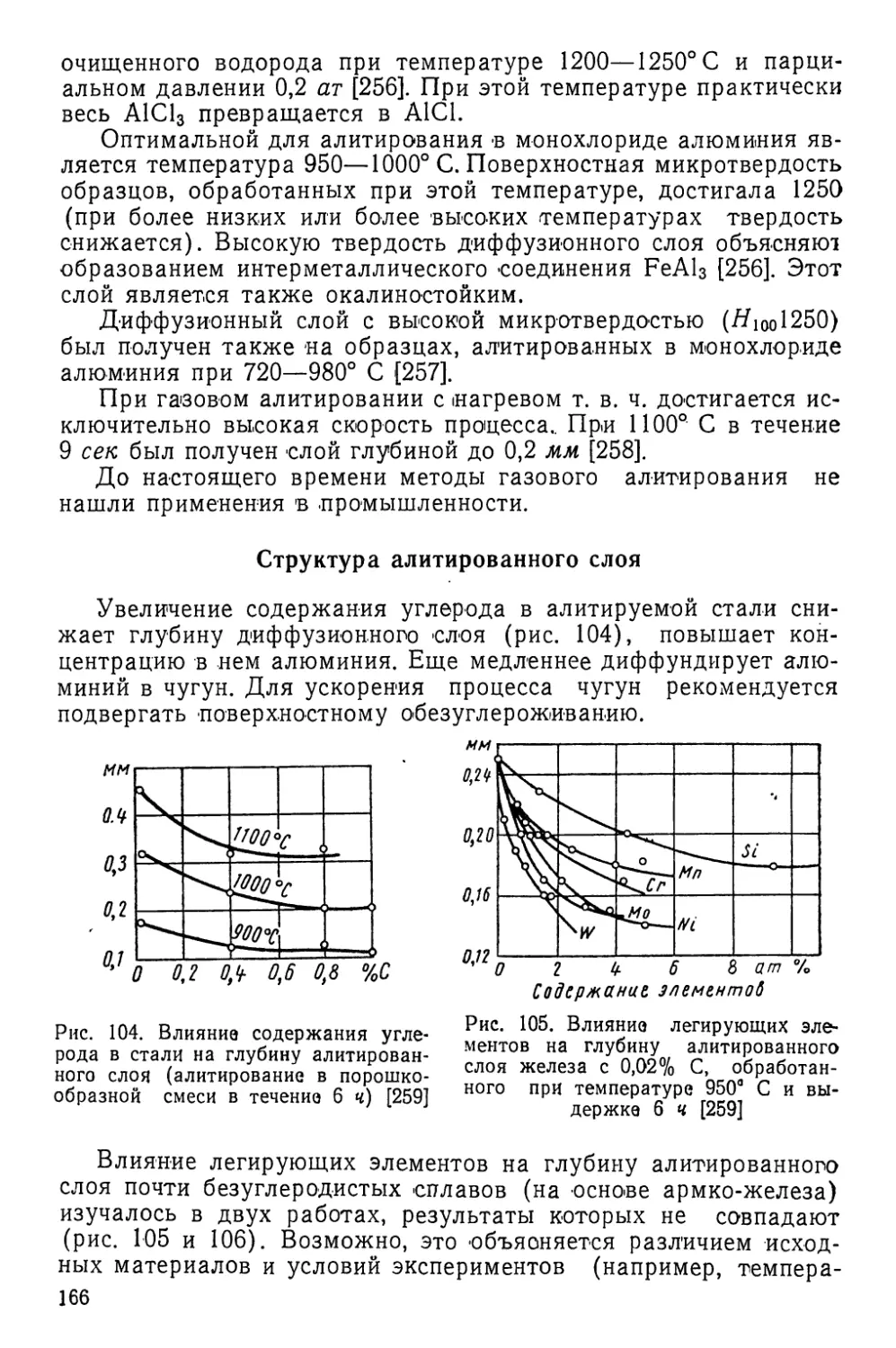
няется, если не считать отжига в водороде трансформаторного
железа, в результате чего достигается сквозное обезуглерожи-
вание листа, и отжига в вакууме титановых сплавов с целью
их сквозного обезводороживания для повышения вязкости.
Однако ряд исследований, в процессе которых изучались
методы химико-термической обработки, основанные на поверх-
ностном обеднении сплавов, показал весьма интересные резуль-
таты. Около 30 лет назад был опробован оригинальный способ
изготовления гетерогенной брони с твердой лицевой и вязкой
тыльной сторонами. Этот способ заключался в одностороннем
обезуглероживании в водороде плит высокомарганцовистой
стали типа Г13 (13% Мп). После обезуглероживания содер-
жание углерода в поверхностных слоях этой стали понижалось
с 1,2% до примерно 0,3%, в результате чего повышалась тем-
пература мартенситного превращения. Это приводило к тому,
что при охлаждении на воздухе происходило мартенситное пре-
вращение и твердость поверхностного слоя повышалась с НВ
220—250 (до обработки — аустенит и карбиды) до НВ 550
(мартенсит и небольшое количество остаточного аустенита).
Из сплава меди и цинка можно изготовлять пористые
фильтры путем нагрева его в вакууме; при этом цинк посте-
пенно диффундирует к поверхности, где испаряется, оставляя
в металле поры.
Удаление легирующей примеси из поверхностных слоев ма-
териала путем испарения дает возможность создать электронно-
дырочные переходы вблизи поверхности германия, т. е. получить
германиевые диоды и триоды (см. гл. IX).
У сплава алюминия с 3% Mg* можно существенно повысить
стойкость против коррозии под напряжением, нагревая сплав
в умеренно окислительной атмосфере, при этом в поверхност-
* Магний добавляется для повышения механических свойств, но при этом
снижается стойкость против коррозии под напряжением.
6
ном слое сплава возникает зона, почти лишенная магния, так
как магний диффундирует к поверхности, где окисляется.
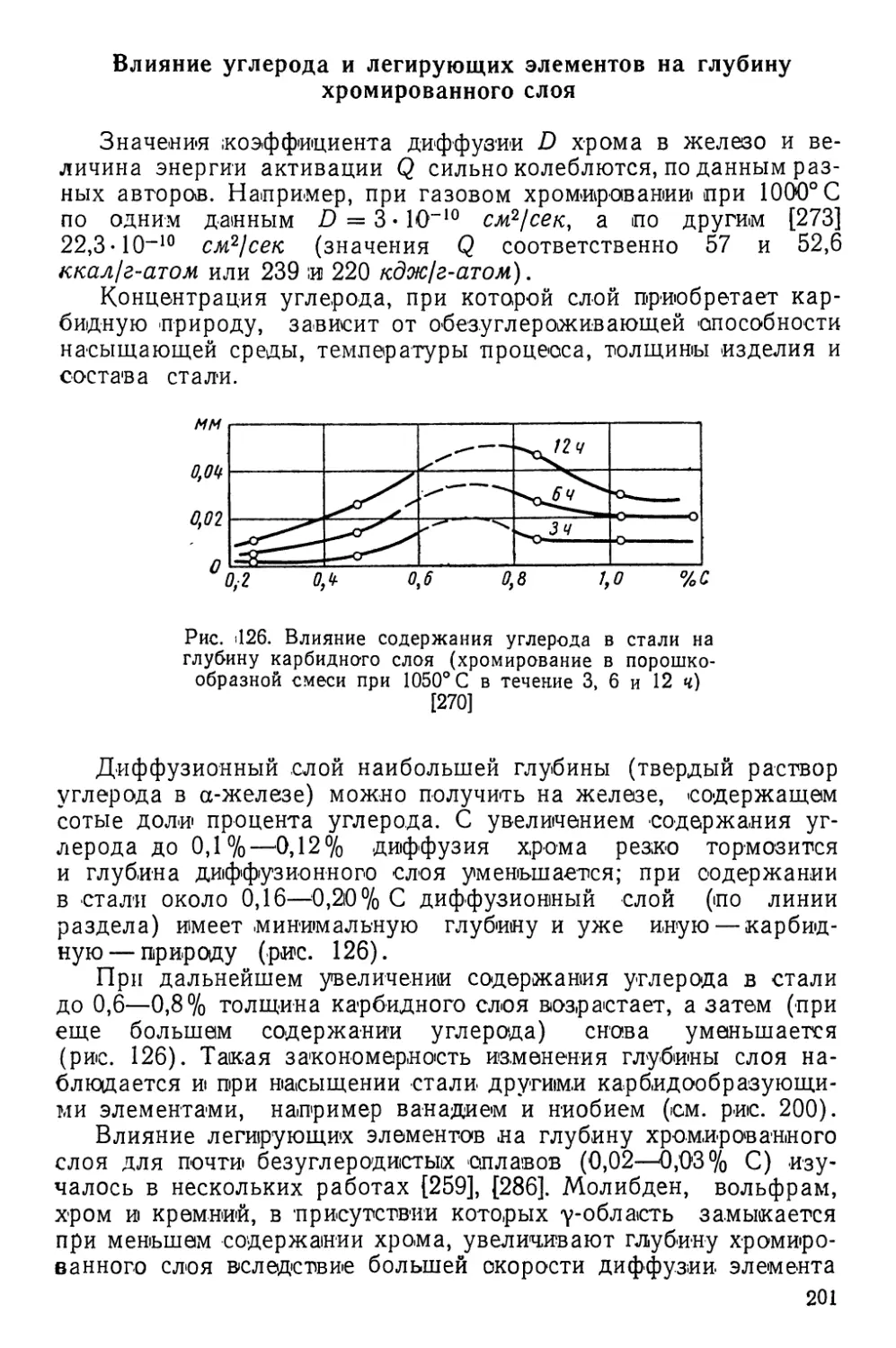
Приведенные примеры показывают, что химико-термическая
обработка, основанная на поверхностном обеднении металлов
и сплавов, также приводит к резкому повышению их твердости,
коррозионной стойкости и других свойств, и несомненно, что
способы обработки, основанные на этом принципе, со временем
найдут должное применение в производстве.
Химико-термическая обработка, основанная на поверхност-
ном обогащении металлов, в большинстве случаев включает в
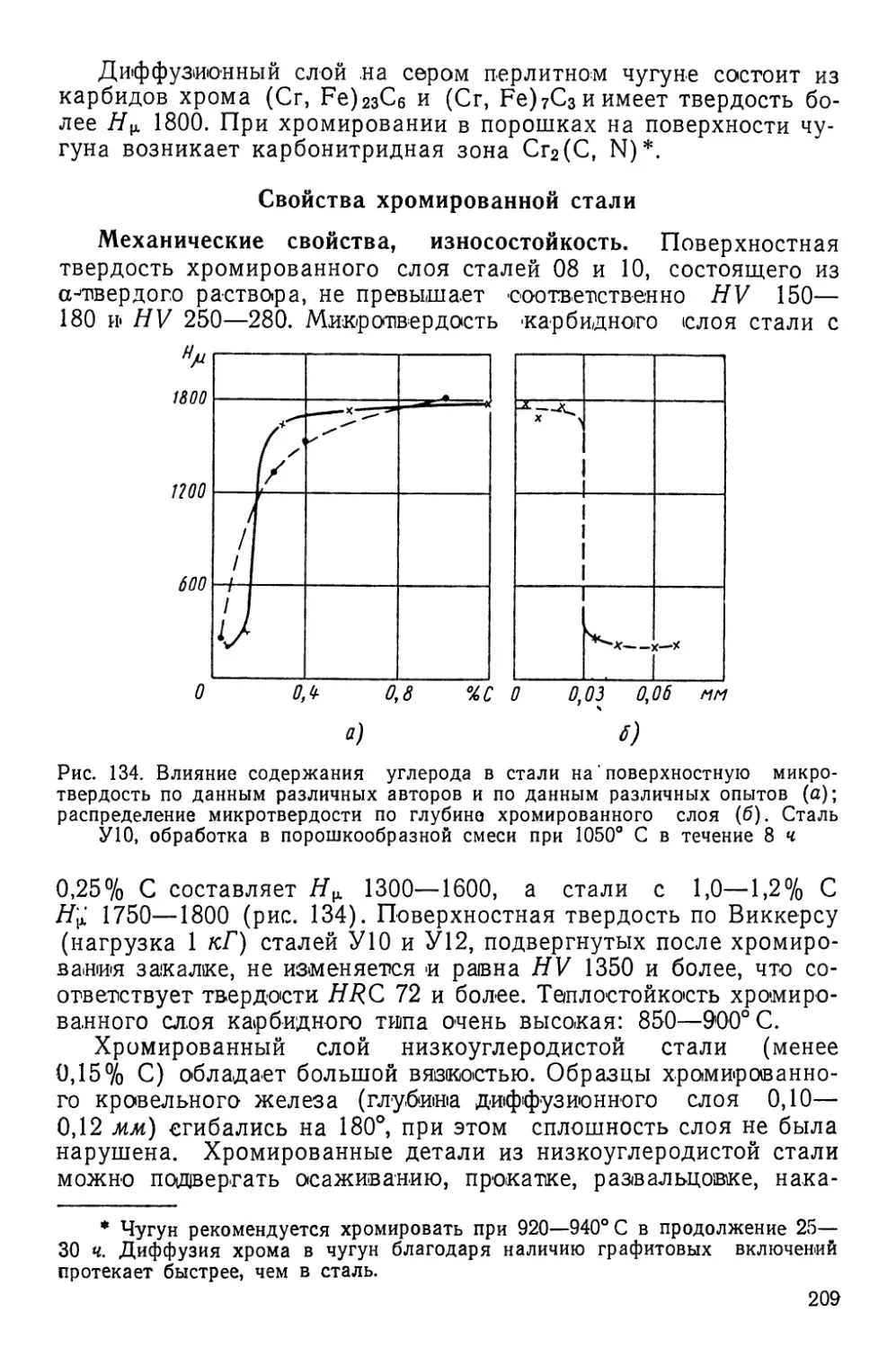
себя три одновременно идущих элементарных процесса:
1) образование во внешней среде диффундирующего эле-
мента в активном атомарном состоянии;
2) контактирование активных атомов диффундирующего
элемента с поверхностью металла, адсорбция атомов и поглоще-
ние части этих атомов с установлением химических связей с ато-
мами металла (абсорбция);
3) диффузия — проникновение в глубь металла абсорбиро-
ванных атомов.
Состав, строение и физико-химические свойства диффузион-
ного слоя зависят главным образом от состава насыщающей
среды, а также температуры и продолжительности процесса.
В качестве насыщающих сред применяются твердые, жид-
кие и газообразные вещества.
Взаимодействие твердого вещества с поверхностью металла
происходит в местах их контакта. Остальные участки поверхно-
сти металла могут насыщаться из паровой фазы, образующейся
вследствие испаряемости твердых тел. Поскольку активность
твердых веществ невелика, чтобы создать условия, необходимые
для ускоренного насыщения в твердых компонентах, к ним
либо добавляют так называемые активизаторы, или ускорители
(NH4C1, NH4J, NH4Br, НО, С12, ВаСОз), которые (или продукты
диссоциации которых) взаимодействуют с твердым компонентом,
в результате чего образуется активная газовая среда, либо осу-
ществляют насыщение в упаковке, состоящей из твердых компо-
нентов, в вакууме — при разрежении 10"2—10-4 мм рт. ст. (1,33—
0,013 н/м2), резко повышающем испаряемость твердых тел. Та-
ким образом, насыщение в твердых компонентах по существу
превращается в насыщение из газовой фазы.
При обработке в жидкой среде насыщение происходит либо
•благодаря газам или элементам в атомарном состоянии, выде-
ляющимся в результате протекающих в расплаве реакций или
электролиза расплава, либо вследствие непосредственного со-
прикосновения расплава металла (сплава) с поверхностью
насыщаемого металла.
Наилучшей средой для химико-термической обработки яв-
ляется газовая среда.
7
Насыщение непосредственно в газовых средах (а также в
газовых средах, образующихся при упаковке в твердые компо-
ненты и при погружении в жидкие среды) в простейших слу-
чаях происходит по реакциям следующих видов:
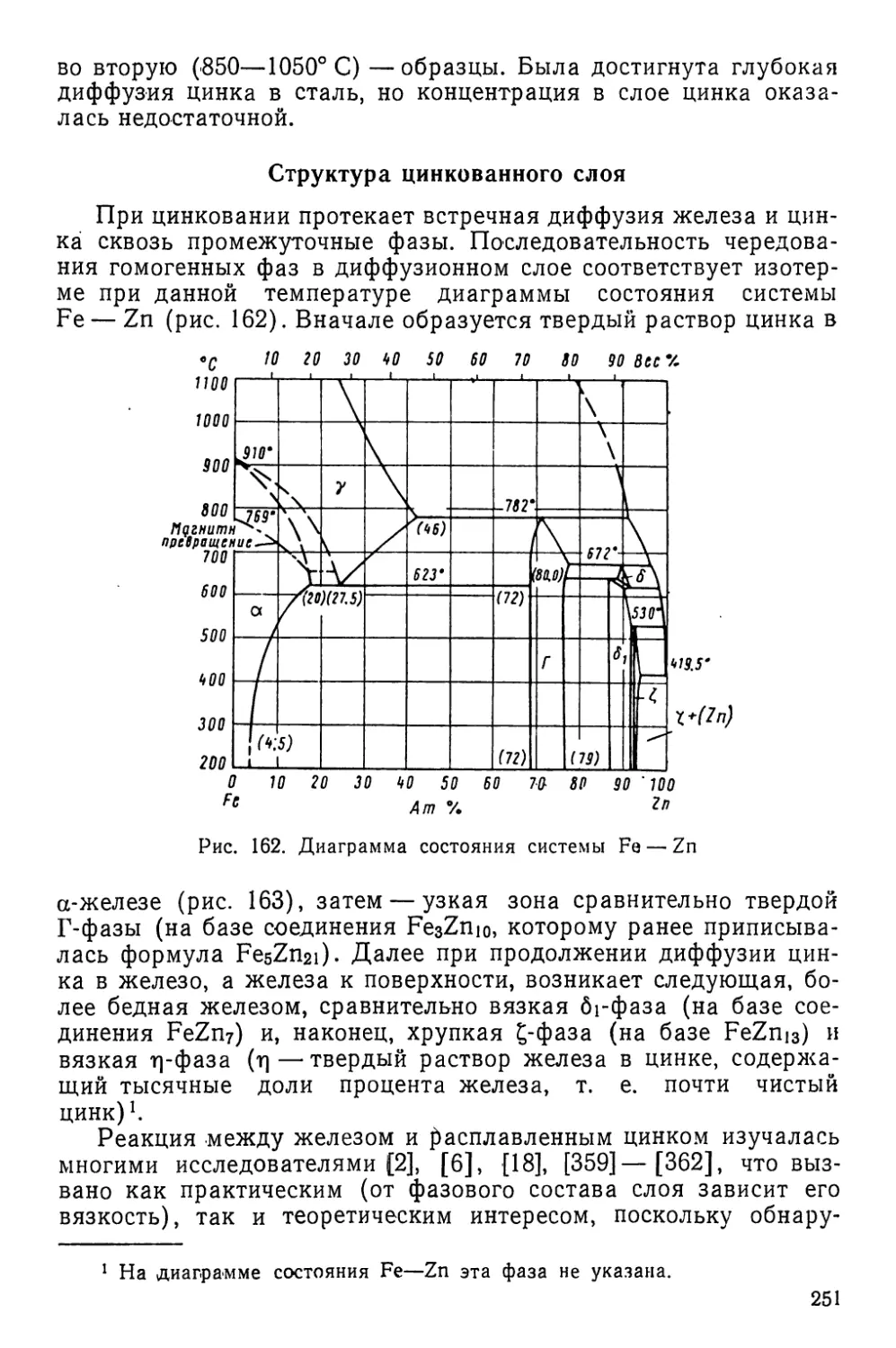
1) обмена
СгС12 + Fe = FeCl2 + Cr,
VC12 + Fe = FeCl2 + V;
2) восстановления
VC12 + H2 = 2HC1 + V;
B2O3 -I- 6Na = 3Na2O -I- 2B
(электролизное борирование);
3) термического разложения:
а) диссоциации
2NHS = 2N + 6Н; СН4 = С + 4Н;
| 1 I
N2 ЗН2 2Н2
б) диспропорционирования
2СО = С + СО2, ЗА1С1 = A1C1S + 2А1; 3TiCl2 = 2TiCl3 + Ti.
Насыщение происходит также благодаря образованию паров
элементов (в вакууме или в среде водорода при высокой темпе-
ратуре).
При данных температуре, давлении, а также составе обраба-
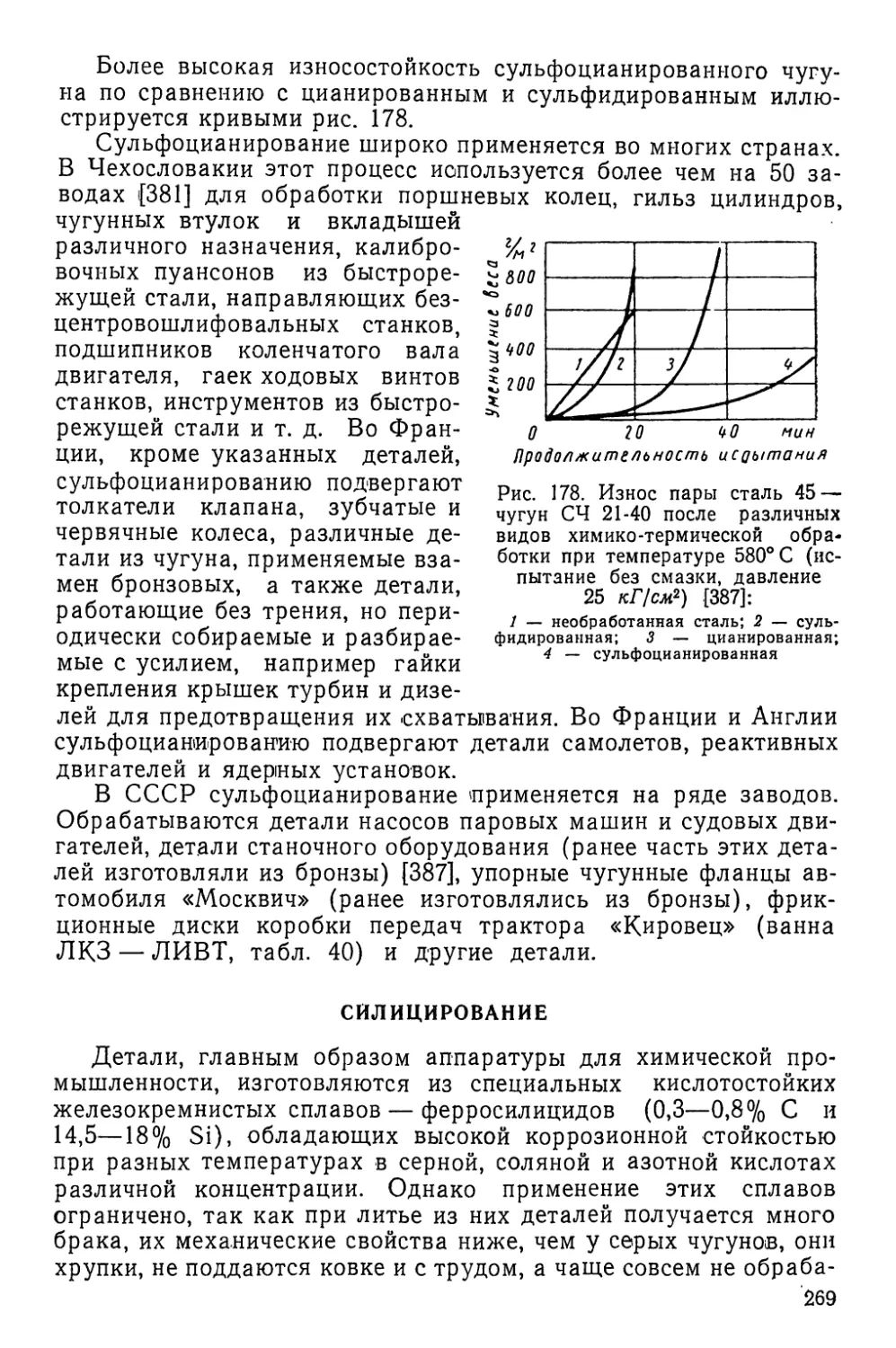
тываемого металла скорость абсорбции элемента из газовой
фазы пропорциональна концентрации активной составляющей,
участвующей в реакциях обмена, восстановления или термиче-
ского разложения.
Температура процесса существенно влияет на абсорбцион-
ную способность: чем выше температура, тем больше подвиж-
ность атомов в окружающей среде и в металле и тем больше
абсорбируется атомов элемента из газовой фазы (если при
этом с повышением температуры не снижается активность
среды).
Влияние давления в газовой среде на скорость абсорбции,
казалось бы, должно быть аналогично влиянию концентрации
активной составляющей, так как число молекул активного газа
в единице объема пропорционально давлению. Однако, говоря
о влиянии давления на абсорбцию, следует учитывать характер
реакций в газовой фазе. Если активная составляющая газовой
фазы разлагается с увеличением (например, СН4 = 2Н2 + С)
или уменьшением (например, 2СО = СО2 + С) объема, то уве-
личение давления в обоих случаях будет приводить к отклоне-
нию в ту или иную сторону от прямолинейной зависимости ско-
рости абсорбции от давления. И, наконец, зависимость скорости
8
абсорбции от давления будет прямолинейной в том случае,
когда активная составляющая реагирует без изменения объема
(например, СгС12 + Fe = FeCl2 + Сг).
В зависимости от обрабатываемого металла (сплава), цели
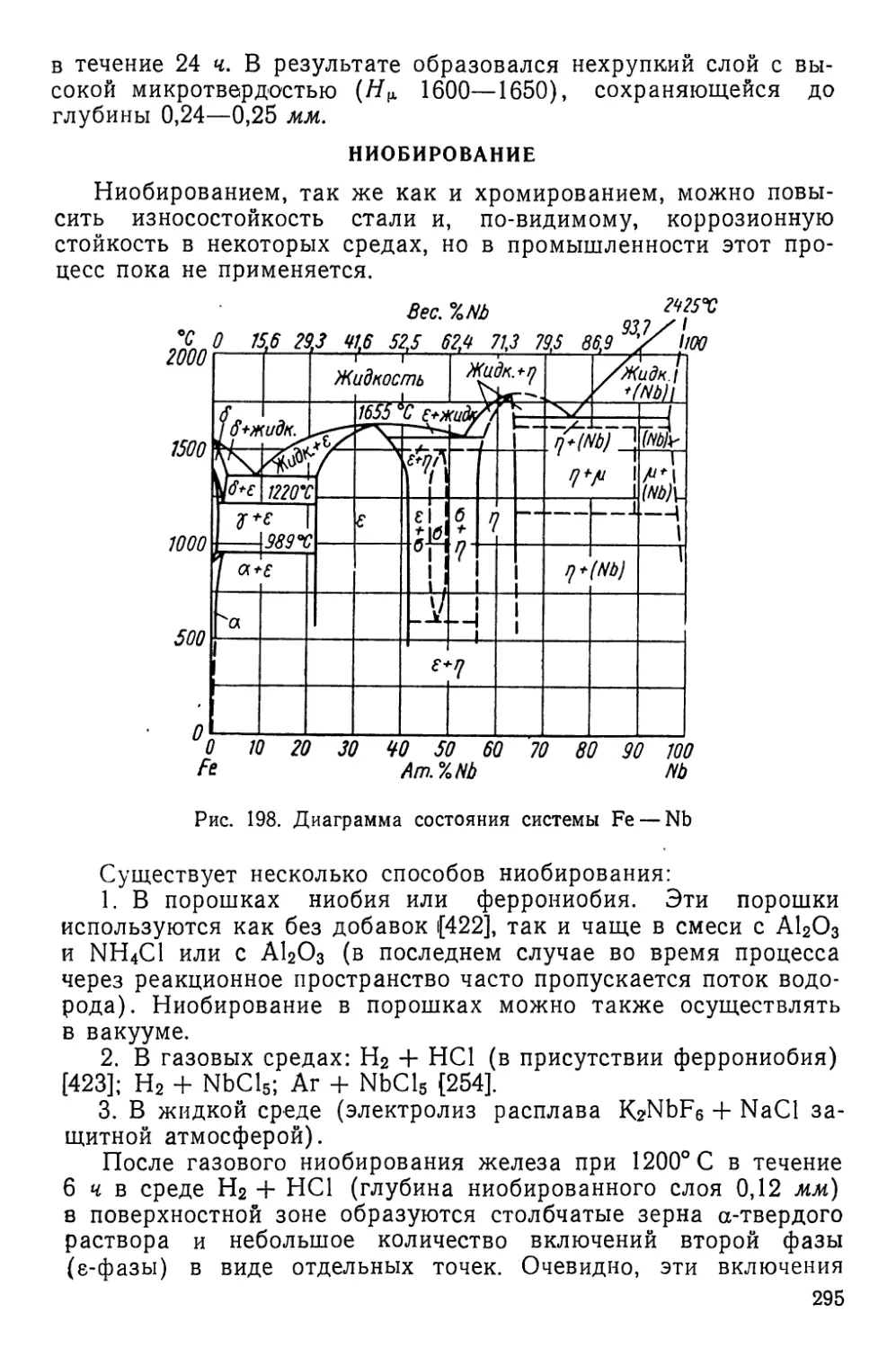
обработки, элемента, используемого для поверхностного насыще-
ния, температура и продолжительность процесса химико-терми-
ческой обработки колеблются в весьма широких пределах: тем-
пература преимущественно от 350 до 1450° С, а продолжитель-
ность от нескольких минут до десятков часов (при нагреве т. в. ч.
до высоких температур продолжительность выдержки может
исчисляться секундами).
От температуры процесса зависят глубина, химический и фа-
зовый состав диффузионного слоя. Для получения более глу-
боких диффузионных слоев температура поддерживается на
верхнем допустимом для данного процесса пределе, превышение
которого отрицательно влияет на структуру сердцевины, фазо-
вый состав и структуру поверхностного слоя. Глубина диффузи-
онного слоя при данной температуре зависит главным образом
от продолжительности процесса.
Один из недостатков почти всех процессов химико-термиче-
ской обработки — их большая продолжительность. Поэтому
многие работы были направлены на изыскание методов уско-
рения этих процессов. В частности, удалось несколько ускорить
их путем установления оптимальных параметров обработки
(температуры, давления, расхода и состава насыщающей сре-
ды), применения катализаторов и т. д.
В последние годы появилось много работ по интенсификации
химико-термических процессов пут.ем применения индукционно-
го нагрева, тлеющего разряда, воздействия на металл или на-
сыщающую среду ультразвука и электромагнитного поля, об-
лучения ультрафиолетовыми лучами и быстрыми нейтро-
нами.
Наибольшее количество исследований по интенсификации
химико-термической обработки посвящено применению для этой
цели нагрева т. в. ч. Были проведены исследования по цемен-
тации стали с нагревом т. в. ч. в газовом, жидком и твердом
карбюризаторах, по нитроцементации, цианированию в распла-
вах и растворах, хромированию и борированию в газовой сре-
де, порошках и обмазках, а также по насыщению стали крем-
нием, алюминием, титаном, марганцем, вольфрамом и другими
элементами. Опробована также химико-термическая обработка
с нагревом т. в. ч. тугоплавких металлов. Большинство исследо-
вателей отмечало резкое ускорение процессов поверхностного
насыщения при нагреве т. в. ч. Достигаемый эффект в ряде слу-
чаев во много раз превосходил ожидаемый только от сокраще-
ния времени нагрева стали до температуры обработки. В связи
с этим о причинах ускорения процессов были выдвинуты раз-
9
личные гипотезы, правильность двух из которых была недавно
доказана (см. стр. 29 и 122).
Вопрос о рациональности использования индукционного на-
грева для цементации и нитроцементации в термических цехах
массового производства пока еще остается не решенным. Одна-
ко удобство и преимущество этого способа нагрева при прове-
дении высокотемпературных процессов поверхностного насыще-
ния стали металлами и при химико-термической обработке ту-
гоплавких металлов дают основание полагать, что нагрев т. в. ч.
будет в значительных масштабах применяться при осуществле-
нии этих процессов.
ГЛАВА I
ОБЩИЕ ЗАКОНОМЕРНОСТИ ДИФФУЗИОННЫХ
ПРОЦЕССОВ
МЕХАНИЗМ ДИФФУЗИИ
Под диффузией понимается изменение местоположения ато-
мов (ионов) в газах, жидкостях и твердых телах. Диффузия
определяется тепловым движением частиц; с повышением тем-
пературы скорость диффузии увеличивается. Во многих случаях
диффузия вызывает выравнивание концентрации, протекающее
во времени. Однако и при отсутствии разницы в концентрации
вследствие энергетических флуктуаций также идет процесс
диффузии; такой процесс, не связанный с изменением концен-
трации, называется самодиффузией. Часто перемещение атомов
происходит в направлении возрастания концентрации (образо-
вания и роста карбидов, других интерметаллических фаз
и т. д.). Этот процесс называется восходящей диффузией. Вос-
ходящая диффузия имеет место также в твердых растворах при
наличии градиента напряжений.
Явление диффузии сначала было обнаружено в жидкостях
и газах. Многие явления, свидетельствующие о подвижности
частиц в твердом теле, долгое время либо оставались незаме-
ченными, либо неправильно трактовались, хотя ими практиче-
ски пользовались очень давно. Например, в течение многих
сотен лет производится «твердая» цементация железа в веще-
ствах, содержащих углерод, с целью получения твердой («це-
ментной») стали. Механизм этого процесса долго оставался
неизвестным.
В XIX в. были поставлены эксперименты, положившие нача-
ло тщательному и систематическому изучению явления диффу-
зии в металлах. В 1820 г. Фарадей и в 1837 г. П. Г. Соболев-
ский наблюдали при спекании порошков образование сплавов
твердых металлов. Выдающийся русский металлург П. П. Ано-
сов в 1837 г. производил газовую цементацию железа в открытом
тигле. Цементация обрезков железа осуществлялась в окиси уг-
лерода, выделяющейся в древесноугольном горне. Затем металл
11
доводился до расплавления, и таким путем получалась литая
сталь.
Основоположник металловедения Д. К- Чернов, судя цо его
работе, вышедшей в 1868 г., уже ясно представлял себе влия-
ние диффузии при структурных превращениях в стали.
Д. К. Чернов писал: «Что же касается нагретого, следователь-
но, размягченного состояния, в особенности при температурах,
соседних с точкой &, то, по всей вероятности, при большей
свободе перемещения частиц стали перемена структуры совер-
шается несколько быстрее».
Количественная сторона диффузии была впервые затронута
в исследовании по диффузии золота в свинце, выполненном в
1896 г. Робертс-Аустеном, который не только наблюдал диф-
фузию в этой системе, но и с большой точностью определил
коэффициент диффузии. В 1909 г. была изучена диффузия*
цинка в медь и латунь, а в 1911 г. диффузия меди в золото.
Большое значение имели работы, выполненные в 1913—1915 гг.
А. П. Чижевским по диффузии азота и бора в железо.
В дальнейшем изучению диффузии были посвящены сотни
экспериментальных и теоретических работ, проведенных в
СССР и за рубежом [1]—[11]. В результате этих и многих других
работ процесс диффузии в металлах получил теоретическое обо-
снование и математическую трактовку.
На первых стадиях взаимодействия металла с активным
насыщающим элементом протекают процессы адсорбции и аб-
сорбции, последний из которых сводится к тому, что внедренные-
атомы, попав в силовое поле кристаллической решетки, образуют
прочные связи с атомами металла. Вследствие теплового движе-
ния абсорбированные атомы не накапливаются в поверхностном
слое, а диффундируют вглубь металла. В начальный период про-
цесса это приводит к образованию в поверхностном слое твер-
дого раствора, а затем, по достижении предельной растворяемо-
сти элемента в этом растворе, происходит перестройка решетки
с образованием новой фазы *. Дальнейшее обогащение поверх-
ности диффундирующим элементом может вновь привести
к перестройке решетки и к последовательному образованию
.слоев новых фаз.
В зависимости от природы образующихся в слое фаз диффу-
зия подразделяется на атомную и реакционную. Под атомной
диффузией понимается диффузия элементов в твердом рас-
творе или химическом соединении переменного состава, изме-
няющая только их концентрацию, под реакционной — диффу-
зия, сопровождающаяся образованием на поверхности новых
фаз (химических соединений .или новых аллотропических моди-
фикаций).
* При некоторых условиях химические соединения могут образоваться^
минуя стадию образования твердого раствора.
12
Атомная диффузия в металлах и сплавах может осуще-
ствляться по различным механизмам.
В твердых растворах внедрения атомы растворенного эле-
мента располагаются в межатомных промежутках кристал-
лической решетки и могут легко мигрировать в решетке из
одного положения в другое.
В твердых растворах замещения атомы обоих компонентов
распределены в узлах решетки. Если принять, . что почти все
узлы решетки заняты атомами, легче всего допустить, что
процесс диффузии должен происходить путем обмена местами
двух соседних атомов. Однако такой элементарный процесс,
предполагающий одновременное перемещение двух атомов,
маловероятен по энергетическим соображениям.
Один из возможных механизмов диффузии — диффузия по
междуузлиям путем вытеснения. При таком механизме атом,
находящийся в иррегулярном положении в междуузлии, вы-
талкивает соседний атом из его правильного (регулярного)
положения в решетке, а сам занимает освободившееся место.
Осуществление такого механизма маловероятно.
Была предложена также схема кольцевого обмена с уча-
стием трех или четырех атомов, которые одновременно диф-
фундируют согласованным циклическим перемещением по
кольцу. Такое перемещение, хотя и не требует много энергии,
все же маловероятно, особенно в плотноупакованной ре-
шетке.
Наиболее вероятным считается механизм диффузии по
вакансиям, согласно которому атом, находящийся в правиль-
ном (регулярном) положении в узле кристаллической решетки
и имеющий достаточно большую энергию, может переме-
ститься в соседний свободный узел кристаллической решетки,
так называемую атомную «дырку» (вакансию). Механизмом
диффузии по вакансиям удается объяснить наблюдаемую в
ряде случаев весьма большую величину коэффициента диф-
фузии в твердых растворах замещения. При движении атомов
по вакансиям на месте атома, вышедшего из узла решетки,
каждый раз создается новая вакансия, которая может быть
наполнена соседним атомом и т. д. Таким образом происходит
непрерывное «перемещение» вакансий и диффузия атомов
элементов.
Теоретическое и практическое значение вакансионного ме-
ханизма диффузии побудило к проведению все ' увеличиваю-
щегося количества работ по определению концентрации вакан-
сий в металлах и сплавах. Разработанные методы определения
концентрации вакансий не отличаются точностью и позволяют
определить только их порядок, однако и это весьма ценно.
Концентрацию вакансий определяли по кривым зависимости
электросопротивления от температуры и по отклонению линей-
13
ного закона изменения коэффициентов линейного расширения
металлов при температурах, близких к температурам их плав-
ления. Кривая электросопротивления при температуре, близкой
к температуре плавления, существенно отклоняется от прямой
линии и показывает более быстрый рост электросопротивления.
При этом добавочное электросопротивление характеризует
насыщение металла вакансиями.
Концентрация вакансий обычно рассчитывается по отноше-
нию числа свободных узлов к числу всех узлов в том же
объеме кристаллической решетки и выражается в атомных
процентах. Для алюминия, меди и серебра концентрация
вакансий при температурах, близких к температурам плавле-
ния, оказалось равной в среднем 0,1—0,2 ат. %.
Концентрация вакансий может быть достаточно точно
определена по данным измерения плотности сплавов после их
закалки, при которой в кристаллической решетке фиксируются
избыточные вакансии, образовавшиеся в процессе нагрева.
Плотность определяется с высокой точностью методом диффе-
ренциального гидростатического взвешивания [587].
Для определения концентрации вакансий в растворах внед-
рения в объемноцентрированной решетке неоднократно приме-
нялся метод изучения внутреннего трения закаленных образцов.
Однако для растворов внедрения диффузия по вакансиям не
должна иметь большого значения.
В ряде случаев атомы в кристаллической решетке сплава
находятся в ионизированном состоянии. Если диффузия в этих
сплавах происходит под воздействием электрического поля, то
имеет место явление электропереноса [12], [13], [19]. (Впервые это
было обнаружено на твердых железоуглеродистых сплавах,
а позднее и других сплавах). При этом углерод перемещается
к катоду. В этом же направлении перемещаются в железоуглеро-
дистых сплавах бор и водород; это показывает, что атомы дан-
ных элементов находятся в ионизированном состоянии и несут
положительный заряд. Азот перемещается в обратном направ-
лении — к аноду.
Скорость диффузии углерода при прохождении постоянного
тока больше скорости, достигаемой при обычной цементации,
в 1,8—2,5 раза [557].
МАТЕМАТИЧЕСКАЯ ТРАКТОВКА ДИФФУЗИИ
При описании диффузии в твердом кристаллическом теле
так же, как при рассмотрении диффузии в жидкостях и газах,
пользуются законами Фика.
Математическое выражение первого закона Фика, описы-
вающее диффузию в твердом кристаллическом теле, предусмат-
ривает диффузию как непрерывный поток атомов (ионов)
14
вещества dm через изотропный слой определенной толщины dx
и поперечного сечения dF в течение определенного времени dx:
de
dtn = — D — dFdx. (1)
dx
Согласно этой формуле количество продиффундировавшего
вещества прямо пропорционально площади поперечного сече-
ния, градиенту концентрации вдоль направления диффузии и
времени. Коэффициент диффузии D соответствует количеству
вещества, продиффундировавшего за 1 сек через поверхность
площадью I см2, при перепаде концентрации — , равном L
dx
Величина D выражается в см2/сутк,и или чаще в см2!сек. Знак
«минус» перед D показывает, что диффузия сопровождается
перемещением атомов (ионов) с участков, более богатых этим'
веществом, к участкам, более бедным.
Первый закон Фика описывает стационарное состояние
диффузионного потока, когда концентрация в любой точке не
изменяется со временем. При нестационарном потоке, когда
концентрация в любой точке изменяется во времени, справед-
лив второй закон Фика, который для случая независимости
коэффициента диффузии D от концентрации имеет вид
de d2c
— = D-----.
dt dx2
(2)
Это уравнение может быть решено для определенных гра-
ничных условий. Если концентрация на поверхности с0 постоян-
ная, то концентрация с(х, t) в любой точке х со временем изме-
няется в соответствии с выражением
с(х, /) = с0 erf с
(3)
которое является решением уравнения (2) и в котором
представляет собой
Функция erf [ ——Xj
\ 2^Dt
Гаусса, значения которого приводятся в
таблицах [9].
интеграл ошибок
соответствующих
Если экспериментально определена концентрационная кри-
вая, т. е. известны с0 и с в любой точке %, можно определить зна-
чение функции erfc |——) для каждой глубины. Зная х и t,
^2/Dt /
можно рассчитать коэффициент диффузии D.
15
Обработка концентрационных кривых может быть прове-
дена и с учетом диффузии по объему и по границам зерен [14].
При изучении диффузии с постоянной концентрацией эле-
мента на поверхности применяется так называемый торцовый
метод, который заключается в следующем. Изготовляются два
цилиндра либо из разных чистых металлов, дающих непрерыв-
ный ряд твердых растворов, либо из чистого металла и из
твердого раствора элемента в этом металле. Торцы этих
цилиндров перед испытанием обычно сваривают путем кратко-
временного нагрева при слабом сдавливании до температуры,
меньшей, чем та, при которой в дальнейшем определяется
Рис. 1. Концентрационные кривые взаимной диффузии
меди и никеля после отжига в течение 40 ч при
1025° С [6]
скорость диффузии. При этом обращают внимание на безуко-
ризненность контакта торцов образцов и на полное отсутствие
окислов между ними. Поэтому сварку и диффузию следует
производить в вакууме или в защитной атмосфере. После
диффузионного отжига тем или иным методом определяют
концентрацию диффундирующих элементов по продольному
сечению цилиндров.
Результаты подобного исследования взаимодиффузии меди
и никеля приведены на рис. 1. Анализ кривых показывает, что
никель диффундирует в медь быстрее, чем медь в никель.
При подсчете коэффициента диффузии по такому методу за
граничную принимается плоскость, перпендикулярная к на-
правлению диффузии, в которой концентрация каждого из эле-
ментов составляет 50%. Концентрация диффундирующего
металла на расстоянии х от граничной плоскости в этом случае
г / % \
сх = — erf ----- .
2
Во многих случаях концентрация элемента на поверхности
непрерывно повышается в течение процесса (например, при
16
(5)
газовой цементации). Поэтому при подсчете D следует учиты-
вать закон изменения концентрации. Примеры подсчета D для
этих случаев и соответствующие графики, которыми необходимо
пользоваться при этих подсчетах, даны в работах [4], [9].
Все приведенные выше решения справедливы, если диффу-
зия протекает в однородной среде. В практике химико-терми-
ческой обработки часто имеют дело с реакционной диффузией,
когда полученный слой состоит из нескольких фаз, располо-
женных последовательно одна за другой. Для этих случаев
также разработаны методы расчета коэффициентов диффу-
зии [6].
Недавно предложен новый метод определения коэффици-
ентов диффузии для случая, когда в поверхностной зоне насы-
щаемого металла образуется несколько последовательно рас-
положенных фаз [15]. По форме концентрационной кривой
можно определить коэффициент диффузии во всех полученных
фазах, в том числе и в твердом растворе. Предполагается, что
коэффициент диффузии в каждой фазе не зависит от концен-
трации и рост фаз подчиняется параболическому закону. Для
расчета коэффициента диффузии в любой фазе достаточно
знать положение границ данной фазы в какой-либо момент
времени (и соответствующее этому моменту отношение инте-
гральных потоков через эти границы). Эти данные можно
получить из экспериментальной концентрационной кривой.
Так, например, если при диффузии элемента в металл вначале
образуется у-твердый раствор, а затем по достижении предела
насыщения этой фазы возникает и растет слой a-фазы, то
коэффициент диффузии в a-фазе может быть рассчитан по
формуле
где Da — коэффициент диффузии в а-фазе;
г/1 — толщина слоя a-фазы в см\
t — время в сея;
Мо — масса диффундировавшего вещества, определяемая
общей площадью, ограниченной кривой и осями
координат;
Л41 — масса продиффундировавшего вещества в части кри-
вой, отвечающей у-фазе (берется соответствующая
площадь);
С\—концентрация элемента в точке перегиба кривой
(граница а- и у-фаз).
Пример расчета по этой формуле коэффициента диффузии
хрома в а- и у-фазах приведен в работах (15] и {270]. Описан-
17
мая методика весьма удобна для расчетов. Температурная зави-
симость коэффициента диффузии выражается уравнением
D = Dae RT , (7)
где Do — предэкспоненциальный множитель (в см21сек или
см2! сутки), определяемый главным образом типом
кристаллической решетки, частотой колебания диф-
фундирующего атома и некоторыми другими фак-
торами;
R — газовая постоянная, равная 1,987 кал]г-атом, или
8,318 дж]г-атом) ;
Q — энергия активации (теплота разрыхления), которую
можно выражать в любых энергетических единицах
(обычно кал/г-атом или дж/г-атом)-, это энергия, за-
трачиваемая на перемещение 1 г-атома диффунди-
рующих частиц из одного положения равновесия в
Другое.
Таблица 1
Параметры диффузии некоторых элементов в различных сплавах
(по данным разных авторов) [7]
Диффунди- рующий элемент Состав сплава Do в см2!сек Q Метод изучения диффузии
ккал!г - -атом дж]г- -агч ом
Fe Si (монокристалл) 6,2-10—3 20,0 83,74
Fe у-железо 0,11—5,8 56,0— 209,34— Химиче-
68,0 284,70
Fe у-железо (0,18% С) 7-10—1 68,0 284,70 ский
Fe у-железо (1,40% С) 7-10-2 58,0 242,83
Fe у-железо (3,4% С) 1 -10“3 45,0 188,41
C у-железо 7-Ю—2 32,0 133,98 Металле-
C у-железо 1,9-10-2 28,2 117,25 графический
В у-железо (0,0038% В) 2-10“3 21,0 87,92
Cr у-железо (99,5% Fe) 3.0-104 82 343,32
W у-железо (99,5% Fe) 3.8-102 70 293,08 Химиче- ский
Al Si (монокристалл) 4,8±1,9 77,4 322,38
Al Си (0,8% А1) 1,75-10-2 37,7 157,96
Zn Си (30% Zn) 0,73 40,7 170,53
Cu Al (0,17% Си) 8,4-10-2 32,6 136,59 Спектраль- ный и рент-
Si Al (0,5% Si) 9-10-1 30,55 128,00
Be Al (2% Be) 8,4-IO-3 32,6 136,59 геноспект- ральный
18
Значения Q и Do для диффузии элементов в железо и раз-
личные сплавы, по данным разных авторов, приведены
в табл. 1.
Уравнение (7) можно представить графически (рис. 2) в
координатах lg D----По наклону прямой к оси абсцисс оп-
ределяется энергия активации:
(8)
q=
4 0,43
С увеличением продол-
жительности химико-терми-
ческой обработки вследствие
постепенного уменьшения
разности концентраций меж-
ду любыми соседними уча-
стками постепенно уменьша-
ется диффузионный поток.
Поэтому с увеличением про-
должительности процесса
интенсивность его постепен-
Рис. 2. Характер изменения коэффици-
ента диффузии в зависимости от темпе-
ратуры:
а — в координатах D — Т\ б — в координа-
тах lg D------------------
Т
но снижается.
Для многих химико-термических процессов зависимость глу-
бины диффузионного слоя от продолжительности процесса выра-
жается параболой, описываемой уравнением
= (9)
где х — глубина диффузионного слоя;
т — продолжительность процесса;
К— константа (параметр параболы), зависящая от
рода диффундирующего вещества и других факто-
ров.
На практике параболический закон зависимости глубины
слоя от продолжительности процесса в ряде случаев не соблю-
дается, одной из причин этого может быть то, что концентрация
диффундирующего элемента во внешней зоне диффузионного
слоя непрерывно изменяется в течение процесса. Она может
уменьшаться, если активность насыщающей среды со временем
снижается (например, длительная обработка в порошкообраз-
ных смесях), или, наоборот, постепенно возрастает по мере
насыщения поверхностных слоев. Если активность насыщающей
среды большая, то параболический закон роста диффузионного
слоя выполняется, так как процесс в данном случае лимити-
руется диффузионной подвижностью атомов. Если же актив-
ность насыщающей среды небольшая, то зависимость роста
слоя линейная или почти линейная [16], [17]. В других случаях
рост диффузионного слоя со временем резко замедляется и
практически почти совсем прекращается.
19
Закон роста определяется активностью насыщающей среды,
коэффициентом диффузии, скоростью химических реакций на
межфазовых границах.
В качестве примера линейной или почти линейной зависи-
мости можно привести рост цементованного слоя при обработке
стали в древесном угле (без активизаторов) [4], а также рост
боридного слоя на никеле при обработке в смеси Н2 и ВС1з [17].
Воем я выдержки
о)
Время выдержки
В)
Рис. 3. Поглощение углерода армко-железом во время нагрева (участок кри-
вой левее О по оси абсцисс) и цементации при 950° С (древесный уголь -Н15%
Na2CO3) —а и изменение глубины диффузионного слоя армко-жеЛеза во вре-
мя нагрева и азотирования при 650° С [20] — б.
В последней работе экспериментально доказано, что лими-
тирующей стадией этого процесса была подача активного бора
к поверхности никеля.
В других случаях линейный закон кинетики роста слоя
объясняется особой «сетчатой» структурой возникающего
диффузионного слоя. По промежуткам этой кристаллической
структуры компонент быстро достигает поверхности металла,
где и происходит реакция.
Таким путем по прямолинейному закону растет, например,
фаза FeZn7 (ранее этой фазе приписывалась формула FeZnioh
образующаяся при цинковании железа в расплаве цинка [2],
[6], [18].
Возможные варианты кинетики роста диффузионных слоев
при реакционной диффузии рассмотрены в монографии
В. И. Архарова [11]. Были предложены различные эмпирические
уравнения, больше отвечающие получаемым экспериментальным
данным [19].
Так, для цементации предложена зависимость х = /Ст0'75
(х — глубина слоя; К. — опытный коэффициент, т — время),
соответствующая промежуточной между параболической и ли-
нейной.
20
При общепринятой методике построения графиков зависи-
мости глубины слоя от времени выдержки обычно не учиты-
вается толщина диффузионного слоя, образующегося в про-
цессе нагрева. В действительности при медленном нагреве
диффузия насыщающего компонента протекает достаточно
интенсивно. Благодаря такой методике построения этой кривой
форма ее во многих случаях отдаляется от действительной и
приближается к форме параболы. Это убедительно было пока-
зано на примерах цементации и
азотирования стали (штриховая
линия на рис. 3) [20].
Между глубиной х диффузи-
онного слоя, получаемой »при
определенной выдержке, и темпе-
ратурой установлена зависи-
мость [19]
_ а
х — Ае т , (10)
Расстояние от места сварки
Рис. 4. Распределение угле-
рода в пластинках с раз-
личным содержанием крем-
ния после 13-дневного от-
жига при 1050° С; началь-
ная концентрация углерода
в пластинках 0,478 и 0,441 %
(по Л. Даркену) [6]
где Т — абсолютная температура;
А и а — опытные коэффициенты.
Однако эта зависимость во
многих случаях нарушается.
Отметим, что движущей силой
процесса диффузии в металлах
следует 'Считать не принимаемый
обычно градиент концентрации, а
химический потенциал. Градиент
химического потенциала определяет направление диффузии.
Этим можно объяснить «восходящую» диффузию в металлах.
Было показано, например, что при диффузионном отжиге
двух сваренных пластинок, содержащих одинаковое количество-
углерода и различное количество кремния, диффузия углерода
происходит из пластинки с большим содержанием кремния в
пластинку с меньшим содержанием кремния (рис. 4). Если
одна из пластинок была легирована хромом или марганцем
(вместо кремния), диффузия углерода протекала в обратном'
направлении [6].
Следовательно, диффузионный -поток углерода зависит не от
градиента концентрации, а от градиента парциальной свобод-
ной энергии (химического потенциала).
Так как химический потенциал-вещества в растворе опреде-
ляет его термодинамическую активность, можно сказать, что
диффузия происходит в направлении уменьшения термодина-
мической активности компонента. В рассмотренном случае
кремний повышает термодинамическую активность углерода,
растворенного в железе, а хром и марганец понижают. Однако
2*
отсутствие достоверных данных для расчета градиентов
химических потенциалов заставляет пользоваться выражениями,
включающими в себя градиенты концентраций.
Для исследования диффузии в металлах и определения кон-
центрационных кривых используют различные методы: металло-
графические, химические и физические (рентгеноспектральный
.локальный анализ, рентгенографический, электронографический,
метод испарения в вакууме, электросопротивления, магнитный,
внутреннего трения, микротвердости и др.). Все большее рас-
пространение получают методы радиоактивных изотопов, отли-
чающиеся универсальностью (существуют радиоактивные изо-
топы почти всех элементов), высокой чувствительностью, и нако-
нец, тем, что создают возможность для изучения самодиф-
'фузии.
Для исследования процессов диффузии с помощью изотопов
{21] применяют методы, связанные с экспериментальным опре-
делением концентрационных кривых, абсорбционные методы и
вариант абсорбционного метода, позволяющий определить пара-
метры диффузии в объеме и по границам зерен, изложенный в
работе (22], и методы авторадиографии (559].
Выявив изоконцентрационный контур диффузионной зоны
методом авторадиографии, можно рассчитать параметры диффу-
зии в объеме и по границам металла.
ВЛИЯНИЕ РАЗЛИЧНЫХ ФАКТОРОВ
НА ПРОЦЕСС ДИФФУЗИИ
Влияние кристаллической структуры растворителя. Экспери-
ментально доказано, что самодиффузия железа, а также диффу-
зия многих элементов (углерода, азота, цинка, хрома, молибде-
на, вольфрама и др.) протекают быстрее в a-железе, чем в у-же-
.лезе. Это, вероятно, связано с меньшей компактностью кубичес-
кой объемноцентрированной решетки по сравнению с кубичес-
кой гранецентрированной.
Влияние природы диффундирующего элемента. Наибольшим
коэффициентом диффузии в железе обладают элемецты с малым
атомным диаметром (водород, азот, углерод, бор), образующие
твердые растворы внедрения !.
В табл. 2 энергии активации диффузии в a-железо водорода,
азота, углерода и бора сопоставлены с разностью величин атом-
ных диаметров этих элементов и железа. Данные таблицы по-
казывают, что с увеличением атомного диаметра энергия акти-
вации диффузии возрастает.
Элементы, образующие с железом твердые растворы замеще-
ния, диффундируют медленнее.
1 В ряде последних работ высказывается мнение, что бор образует раствор
внедрения с у-железом и раствор замещения с а-железом.
22
Таблица 2
Энергия активации диффузии элементов в а-железо [10]
Элемент Атомный диаметр о в А Разность диамет- ров атомов железа и атомов элемента в А Энергия активации диффузии
в кал'г-атом в дж/г-атом
Н 0,56 2,00 3 750 15712
N 1,42 1,14 18 200 76 258
С 1,54 1,02 20 100 84 219
В 1,78 0,78 21 160 88 660
Анализ результатов ряда исследований диффузии элементов,,
образующих с железом твердые растворы замещения, показыва-
ет, что между диффузионными характеристиками, величиной
атомного диаметра элементов и другими их физическими свой-
ствами прямой связи не обнаруживается. Для некоторых слу-
чаев найдена зависимость величин D от положения элементов а
периодической системе Д. И. Менделеева, т. е. от химической
природы веществ. Например, коэффициент диффузии различных
элементов в свинец оказался тем больше, чем сильнее природа
диффундирующего элемента отличается от природы раствори-
теля. Аналогичная зависимость обнаружена для диффузии оло-
ва, кремния, алюминия и цинка в медь {6].
В большинстве случаев коэффициент диффузии в твердых:
растворах замещения при данной температуре тем меньше, чем
выше температура плавления диффундирующего в железо эле-
мента и больше его атомный радиус [3].
Влияние концентрации. В зависимости от концентрации диф-
фундирующего элемента изменяется и коэффициент его диффу-
зии в основном металле, и коэффициент самодиффузии основного»
металла.
Углерод ускоряет самодиффузию железа. Коэффициент само-
диффузии железа в безуглеродистом железе при 960° С в 16 раз-
меньше, чем в сплаве, содержащем 1,1% С. Коэффициент диффу-
зии углерода в аустените при 1127° С и концентрации углерода
до 0,8% изменяется мало, а при той же температуре и 0,8--
1,7% С резко возрастает.
Коэффициент диффузии углерода в аустените в зависимости»
от концентрации (по Р. Мелю)
32 000
D = (0,07 + 0,06% С) е RT •
Зависимость коэффициента диффузии от концентрации диф-
фундирующего элемента установлена также гТри изучении диф-
фузии в сплавах системы медь — никель и диффузии ряда
23:
элементов в медь [6]. При температуре 750° С коэффициент диф-
фузии цинка при концентрации его до 2% равен (2,5—5) X
X 10~5 см2) сутки, а при концентрации 8 и 24% — (3,5—6) • 10~5 и
(30—35) • 10-5 см2/сутки соответственно.
Влияние концентрации на коэффициент диффузии различно.
Например, при диффузии в медь никеля, алюминия, цинка, крем-
ния, бериллия, олова коэффициент диффузии возрастает с уве-
личением концентрации этих элементов (рис. 5), а при диффу-
зии в алюминий кремния и марганца наблюдается обратное яв-
ление.
Рис. 5. Зависимость коэффициентов диффузии различ-
ных элементов при 800°С от их концентрации в сплавах
на основе меди [6]
Влияние третьего компонента. Примесь, растворенная в ос-
новном металле, ускоряет или замедляет подвижность диффун-
дирующего элемента. Это обусловлено изменением энергии меж-
атомных связей в основном металле, возникновением межатом-
ной связи между примесью и диффундирующим элементов,
изменением химической активности диффундирующего эле-
мента.
Диффузию углерода в аустените замедляют молибден, воль-
фрам, хром и большинство других карбидообразующих элемен-
тов. В противоположность этому некарбидообразующие элемен-
ты (никель и кобальт), ослабляя связь углерода в решетке
аустенита, несколько увеличивают коэффициент диффузии-
Вольфрам уменьшает коэффициент диффузии углерода в аусте-
ните в 2 раза сильнее, чем молибден. Кислород замедляет диф-
фузию углерода, но ускоряет диффузию азота; азот ускоряет
диффузию углерода; углерод резко замедляет диффузию в желе-
зо большинства элементов.
24
всю ее поверхность, не проникая
Рис. 6. Пути диффузионных потоков
в кристаллическом теле
Возможно, что в некоторых случаях одной из существенных
причин влияния третьего компонента на скорость диффузии яв-
ляется вызванное этим компонентом изменение величины зерна
твердого раствора.
Диффузия по границам и объему зерен. Диффузия в метал-
лах может проходить по поверхности, объему зерна и по грани-
цам зерен (рис. 6).
Многочисленными исследованиями доказана большая под-
вижность атомов элементов по поверхности металла. Например,
торий, адсорбированный на одной стороне вольфрамовой нити,
при нагреве быстро покрывает
в сколько-нибудь заметном
количестве внутрь металла.
Экспериментально найдена
температурная зависимость
коэффициента диффузии то-
рия на поверхности, в зерне
и по границам зерен вольф-
рама. Коэффициент диффу-
зии имеет наибольшее зна-
чение для диффузии по по-
верхности, среднее —по гра-
ницам зерен и наимень-
шее— по объему зерен. Пре-
имущественная диффузия
многих элементов по границам зерен (по сравнению с объемом
зерна) доказана во многих работах.
Диффузия цинка в поликристаллической латуни при 700° С
протекает в 40 раз быстрее, чем в монокристалле; углерод при-
мерно в 4 раза скорее диффундирует в мелкозернистый вольф-
рам, чем в монокристалл. Скорость диффузии углерода в ниобий
по границам зерен выше, чем по объему зерна [19].
Более интенсивная диффузия хрома в высокоуглеродистой
стали по границам зерен иллюстрируется микроструктурой
(рис. 7, а) образца, хромированного в расплавленной соляной
ванне (содержащей хлорид хрома) с последующим среднезамед-
ленным охлаждением [19].
Более высокая диффузионная подвижность элементов по гра-
ницам зерен может быть связана с тем, что на границах кристал-
лическая решетка искажена и имеется большее количество ва-
кансий; поэтому энергия активации значительно снижается.
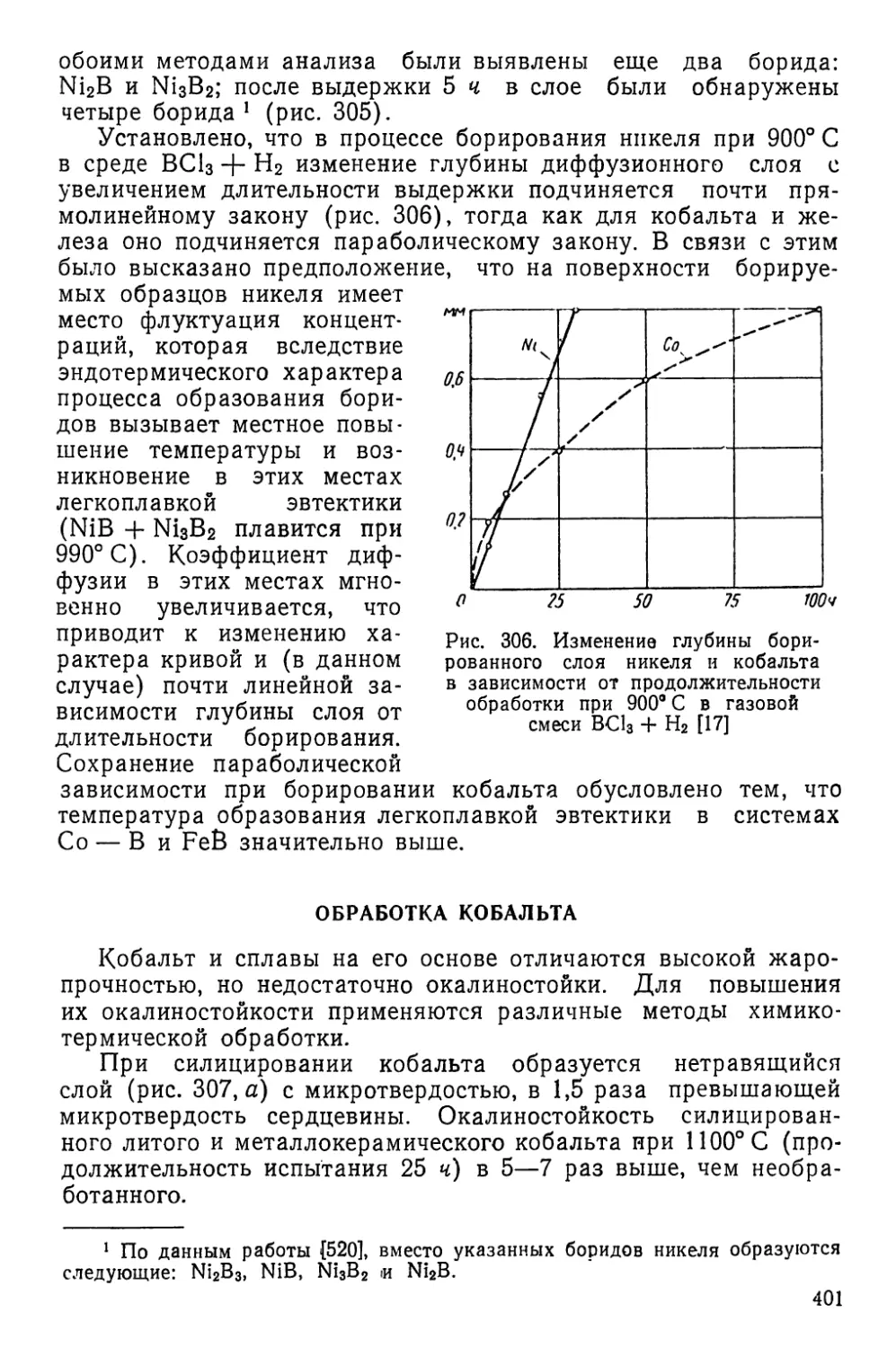
Изучение самодиффузии никеля, свинца, цинка и сереб-
ра по объему и границам зерен подтвердило, что энергия акти-
вации по границам зерен примерно в 2 раза меньше, чем по
объему зерна. Вместе с тем найдено, что скорость межкристал-
лической диффузии цинка в медь лишь незначительно выше ско-
рости внутрикристаллической диффузии.
25
Ряд авторов отмечает, что при диффузии углерода и азота в
-аустените (судя по микроструктуре) отчетливо не обнаружива-
ется преимущественной диффузии их по границам зерен Изу-
чение микроструктуры азотированного при 525° С в течение 24 ч
крупнозернистого железа (1 зерно на 1 жж1 2) и мелкозернистого
железа (200 зерен на 1 жж2) показало, что глубина слоя, судя
Рис. 7. Продвижение:
а — хрома по границам зерен стали У10 (Х1000) [19] и б — азота (е-фазы)
в кремнистом феррите (Х500) [23]
по иглам нитридов, в обоих случаях была равна 1-]- 0,05 жж
[19]. Таким образом, влияние величины зерна на диффузию азо-
та в феррите невелико.
К такому же выводу пришли в работе [23] относительно прод-
вижения богатой азотом е-фазы в сплавы железо — никель и же-
лезо— марганец. В сплавах же железо — алюминий, железо —
молибден, железо — вольфрам, железо — хром и железо — крем-
ний наблюдалось усиленное продвижение s-фазы по границам
зерен (рис. 7, б). На основании имеющихся данных можно сде-
лать вывод, что диффузия по границам зерен в большинстве слу-
чаев протекает значительно быстрее, чем внутри зерна. Это раз-
личие уменьшается с повышением температуры; вблизи точки
плавления оно, по-видимому, весьма мало.
1 Однако методом авторадиографии доказана большая ско*рость диффузии
углерода по границам зерна аустенита, чем в объеме зерна.
26
При рассмотрении диффузии внутри зерен возникает вопрос
о ее скорости по различным кристаллографическим направле-
ниям.
Диффузия углерода в аустените всегда отличается равно-
мерным проникновением .углерода в каждое кристаллическое
зерно аустенита. Такая же картина наблюдалась при диффузии
азота в a-железе и кремния в меди. На основании этих и
других аналогичных данных можно считать, что скорость диф-
фузии в металлах с кубической решеткой практически не зави-
сит от кристаллографического направления. Различия в скоро-
сти диффузии по разным кристаллографическим направлениям
для металлов с гексагональной решеткой (например, цинка и
кадмия) существуют, но степень этой анизотропии невелика и с
ростом температуры еще более уменьшается.
Можно предполагать, что эти различия в скорости диффузии
должны быть тем резче, чем менее симметрична кристаллическая
решетка металла. Так, большая анизотропия скорости диффузии
обнаружена при изучении самодиффузии при 265° С в висмуте,
ромбоэдрическая решетка которого отличается весьма низкой
симметрией.
Влияние искажений кристаллической решетки и дислокаций.
Внутренние напряжения и особенно искажения кристаллической
решетки существенно влияют на диффузию. Еще большее влия-
ние на диффузию оказывает деформация. Так, скручивание мо-
нокристалла вольфрама заметно увеличивает скорость диффузии
в него тория. Скорость диффузии никеля в медь возрастает в
1000 раз после деформации последней. Толщина карбидного слоя
при цементации танталовой пластинки увеличивается на 75%
после предварительной холодной прокатки {3]. При некоторых
видах деформации коэффициент диффузии цинка в а-латуни уве-
личивается в 2—5 раз.
Самодиффузия в a-железе при 750—800° С в области малой
упруго-пластической деформации возрастает более чем в 2 раза,
а при больших деформациях изменяется ее порядок.
Ускорение диффузии при деформации наблюдается главным
образом по плоскостям сдвига и границам зерен, т. е. там, где
сохраняется энергия остаточных упругих напряжений. При на-
личии неоднородного напряженного состояния атомы с большим
радиусом стремятся переместиться в растянутые области, а ато-
мы с меньшим радиусом (рис. 8) — в сжатые. Таким образом,
диффузия может приводить к разделению компонентов твердого
раствора (восходящая диффузия).
Реальные технологические процессы поверхностного насы-
щения сплавов происходят обычно при температурах, достаточ-
ных для возврата и даже рекристаллизации деформированного
металла. Это уменьшает, а чаще устраняет влияние деформации
на скорость диффузии.
27
Дислокации, по мнению ряда исследователей, влияют на диф-
фузию: при большой плотности дислокаций наблюдается ускоре-
ние диффузии. Диффузия вдоль краевых дислокаций обладает
более низкой энергией активации вследствие сравнительно
большого объема, в котором
Рис. 8. «Восходящая» диффузия
в упруго-изогнутом блоке
(С. Т. Конабеевский)
может перемещаться атом.
В таком объеме атом стано-
вится как бы «наполовину
внедренным». Диффузия по
краевым дислокациям име-
ет важное значение при
сравнительно низких темпе-
ратурах, когда концентра-
ция вакансий уменьшается.
Некоторые исследователи
считают, что количество ве-
щества, переносимое по дислокациям, не может быть большим.
Влияние дислокаций на диффузию заключается скорее в том,
что повышение их плотности или их
перемещение обусловливает повыше-
ние плотности вакансий. В непосредст-
венной близости от дислокаций воз-
можно непрерывное возникновение ва-
кансий. Это и может приводить к уско-
рению диффузии.
Влияние магнитных превращений и
воздействие излучений. В ряде работ
показано ускорение образования диф-
фузионных слоев в случае насыщения
металлов при температуре, близкой к
точке Кюри. Впервые это было обнару-
жено при азотировании никелевоко-
бальтовых сплавов [558] и позднее —
при цементации железа в твердом кар-
бюризаторе [24], [25]. В последнем слу-
чае повышение температуры с 766 до
768° С (выдержка 8 ч) привело к уве-
личению глубины слоя с 0,18 до
0,36 мм. При цианировании стали в
Рис. 9. Влияние температу-
ры вблизи точки Кюри (К)
на глубину азотированного
слоя железа при обработке
на разных установках в
продолжение 2 ч:
1 — по данным автора и
М. Г. Вейнова; 2 — по данным
автора и В. Д. Демидовой
цианистой ванне аномалия в глубине слоя была обнаружена при
750° С (азот снижает температуру точки Кюри) [26].
В экспериментах, проведенных в Московском институте ста-
лей и сплавов, полностью подтверждены аномалии в глубине
слоя при цементации в твердом карбюризаторе и азотировании
стали при температуре, близкой к точке Кюри (рис. 9). Однако
полученный эффект был менее резким, чем описанный в опубли-
кованных работах. Это объясняется, по-видимому, различными
23
условиями экспериментов и, в частности, различной амплитудой
колебания температуры около точки Кюри. Лишь дальнейшие
эксперименты помогут установить причину ускорения диффузии
при температуре магнитных превращений. Возможно, что вслед-
ствие колебания температуры около точки Кюри на диффузию
оказывают влияние стрикционные эффекты, а также непрерыв-
ные переориентировки элементарных моментов в доменах. В про-
цессе цементации при температуре, близкой к точке Кюри, боль-
ших партий деталей ускорения процесса
не обнаруживалось. Авторы данной рабо-
ты объясняют это незначительной ампли-
тудой колебания температуры стали .при
таких условиях проведения цементации.
Воздействие на сплавы радиоактив-
ных излучений вызывает образование ва-
кансий и внедренных атомов. Поэтому
можно было ожидать, что коэффициент
диффузии у облученных металлов будет
выше. Однако прямое доказательство это-
го получить трудно, так как при высокой
температуре происходит быстрое залечи-
вание этих нарушений строения, а при
низких температурах возникают трудно-
сти с измерением коэффициента диффу-
зии. Поэтому 'считают, что эффект уско-
рения можно наблюдать только в узком
интервале температур, например для
550° С [8].
Рис.. 10. Объем и глуби-
на (высота сектора)
диффузионных слоев, об-
разующихся при насы-
щении равных по площа-
ди вогнутой и выпуклой
поверхностей
меди в пределах 330—
Влияние скорости нагрева. В ряде исследований показано, что
при быстром нагреве стали (т. в. ч. или контактным нагревом)
в активных насыщающих средах и небольшой выдержке (или
без выдержки) насыщающий элемент быстро проникает на глу-
бину, в несколько раз превышающую глубину слоя, образующе-
гося при обычной скорости нагрева до этой же температуры
[560]. Это можно объяснить повышенной активностью процесса’
насыщения аустенита, близкого к зародышевому состоянию [27].
Чем больше скорость нагрева, тем более мелкозернистым оказы-
вается аустенит и меньше величина блоков при достижении за-
данной температуры и выходе на изотерму, а следовательно, тем
-больше протяженность границ зерен и блоков, обеспечивающих
ускоренное продвижение насыщаемого элемента. Обнаруженная
закономерность, по-видимому, может быть распространена на
многие другие металлы и сплавы, зерно которых при нагревании
склонно к сильному росту.
Влияние формы и размера насыщаемой поверхности. На глу-
бину диффузионного слоя оказывают существенное влияние
форма и размер насыщаемой поверхности. Как следует из рис. 10,
29
в зависимости от кривизны насыщаемых поверхностей образу-
ются диффузионные слои разной толщины и объема. Это объяс-
няется различными условиями отвода насыщающегося элемента
в глубь образца по направлениям, расходящимся от центра от-
верстия или сходящимся к оси цилиндра. При насыщении по-
верхностных слоев стенок отверстия в стали отвод элемента по
расходящимся направлениям в слой большого объема приводит
Рис. 11. Зависимость глубины цементации от кривизны
поверхности при различных выдержках и температуре
920° С (сталь 20, карбюризатор — древесный уголь
+ 10% ВаСОз):
1 — общая глубина слоя; 2 — сумма заэвтектоидных и эвтек-
тоидных слоев [4]
к более низкой концентрации элемента в слое и на поверхности,
чем при насыщении плоской поверхности и тем более цилиндри-
ческой. В последнем случае концентрация элемента на поверх-
ности и глубина слоя получаются максимальными.
Математические расчеты и экспериментальное изучение влия-
ния кривизны поверхности на глубину цементованного слоя были
выполнены в работе [4]. На рис. 11 показана зависимость глуби-
ны цементации от кривизны поверхности.
Размер насыщаемого объекта также оказывает существен-
ное влияние на результаты обработки. При сквозном проникно-
вении элемента в металл определенная концентрация его на по-
верхности пластинки или цилиндра небольшого диаметра и за-
данная глубина слоя достигаются быстрее, чем при несквозном
30
проникновении L Эксперименты показали, что, например, цемен-
тованный слой глубиной 1 мм при несквозной цементации стали
в древесном угле образуется за 6 ч при 910° С, а на пластинке
толщиной 2 мм через 3,5 ч наблюдается сквозная цементация [4].
Это связано с тем, что при сквозном проникновении диффузион-
ные потоки смыкаются в центре образца и отвод элемента в глубь
металла замедляется. Последнее приводит к быстрому повыше-
нию содержания в слое насыщающего элемента, в данном случае
углерода, до концентрации, которую учитывают при определении
глубины цементованного слоя.
СТРУКТУРА ДИФФУЗИОННЫХ СЛОЕВ
Образующиеся последовательно на поверхности и в более
глубоких зонах металла диффузионные слои соответствуют од-
нофазным областям диаграммы состояния металл — элемент в
пределах концентраций от нуля
до максимального содержания
элемента на поверхности. При
этом области, представленные на
диаграмме в виде смеси фаз, иск-
лючаются. В результате этого на
кривых распределения элемента
по глубине слоя возникают сту-
пеньки, высота которых соответ-
ствует двухфазным областям, от-
сутствующим в ’ слое (рис. 12).
Появление этих ступеней обуслов-
лено тем, что при взаимодействии
металла с элементом возникают
лишь термодинамически возмож-
ные фазы, тогда как гетерогенные
смеси, расположенные на диа-
грамме состояний между этими
фазами, не могут образовываться.
Смесь фаз в диффузионном
слое при взаимодействии двух
элементов образуется лишь в ре-
зультате распада твердого раст-
вора или соединения при последу-
Расс таяние от поверхности
Рис. 12. Изменение концентрации
внедряемого элемента по глубине
диффузионного слоя:
1 — концентрация ниже предельной в
твердом растворе; 2 — концентрац1я
достигла предела насыщения; 3 — по-
степенное образование первого хими-
ческого соединения переменного со-
става (/); 4 — образование второго
химического соединения (//); 5 — об-
разование третьего химического соеди-
нения или твердого раствора {III)
ющем медленном охлаждении с
температуры насыщения либо вследствие преимущественной диф-
фузии внедряемого элемента вдоль границ зерен1 2. Когда поверх-
1 Глубина диффузионных слоев обычно определяется по микрошлифу, в
она существенно меньше фактической обшей глубины проникновения элемента
в металл.
2 При обработке многокомпонентных сплавов возможно образование мно-
гофазного слоя непосредственно при температуре диффузии.
31
С
Рис. 13. Тройная система А — В — С,
из компонентов, образующих ограничен-
ные твердые растворы и двойное хими-
ческое соединение [28]
и В) не изменялся во всех точках
постному насыщению подвергается двухкомпонентный сила©
или когда чистый металл насыщается одновременно двумя ком-
понентами, пользуются разрезами тройных диаграмм при тем-
пературе диффузии [28].
При насыщении компонентом С сплава А — В, содержащего
эти элементы в количестве, отвечающем точке X (рис. 13), ком-
понент С вначале будет растворяться в исходных а- и р-фазах,
изменяя их состав в направлении 1—2 и 1'—2'. Средний состав
слоя меняется от X др а. Дальнейшее насыщение компонентом С
приводит к скачкообразному
изменению концентрации;
при этом вместо p-фазы по-
является фаза СпВт и слой
состоит уже из СпВт и а-фаз
(Ь — с). По достижении фа-
зой СпВт предельной кон-
центрации С (Я7), а фазой а-
концентрации, отвечающей
точке 3, на поверхности воз-
никает новый двухфазный
слой, состоящий из фаз у и
a (d— 4'), и наконец воз-
никает однофазный слой у с
непрерывно повышающейся
в нем концентрацией компо-
нента С. Следует отметить,
что в рассмотренном случае
для упрощения предполага-
лось, что состав основного
сплава (т. е. соотношение А
слоя, что обычно не соблю-
дается.
С помощью аналогичных рассуждений можно .описать строе-
ние диффузионного слоя при совместной диффузии компонентов
А и В в сплав С. При соотношении в насыщающей среде ком-
понентов, соответствующем лучу ХС, диффузионный слой будет
состоять из следующих зон: а + (3 (поверхностная зона слоя),
сх + СпВт, а + у, у (зона слоя, примыкающая к сердцевине).
Если при этом диффузионная способность компонентов А и В
различна, то луч ХС будет искривляться в ту или иную сторону
(при большей диффузионной способности компонента А сме-
щаться к вершине /1).
Хотя при использовании двойных и тройных диаграмм для
расшифровки фазового состава диффузионных слоев вводится
ряд допущений и не учитываются некоторые явления, примене-
ние таких диаграмм весьма полезно.
32
Некоторые исследователи [29] предлагают интенсифициро-
вать диффузию компонента С в сплав А—В или в тройной сплав
А — В — С путем дости-
жения в поверхностном
слое этих сплавов в про-
цессе диффузии фазового
состояния, соответствую-
щего большему коэффи-
циенту диффузии компо-
нента С. Для этого в по-
верхностном слое изме-
няется соотношение меж-
ду компонентами А и В
путем введения вместе с
С одного из этих компо-
нентов *. На рис. 14, а по-
казана диаграмма трой-
ной системы (при высоких
температурах), типичная
для сплавов Fe—Сг—Ni,
Fe — Мп — Сг и др. Из-
менение состава диффу-
зионного слоя сплава X
при постоянном соотно-
шении компонентов А и В
будет протекать по лучу
ХС, Допустим, что пре-
дельное насыщение ком-
понентом С поверхностно-
го слоя будет соответст-
вовать точке Х2, лежащей
в области ₽-фазы. Допу-
стим, что коэффициент
диффузии компонента С в
Nt. X бес—~-
6)
Рис. 14. Тройные системы А — В — С (а)
и Fe — Ni — Сг (б); в системе Fe —
Ni —Сг точка Xi соответствует исходно-
му составу сплава, точка Х2 — сплаву,
насыщенному хромом, и точка X 2—спла-
ву, насыщенному хромом совместно
с железом [29].
3-фазе меньше, чем в
a-фазе. Тогда смещение
в процессе диффузии точ-
ки Х2 (при неизменном со-
держании компонента С)
из области 3-фазы в об-
ласть a-фазы (точках Х'2)
приведет к ускорению
* Впервые аналогичный метод интенсификации химико-термической обра-
ботки был предложен Г. Н. Дубининым [270], рекомендовавшим при хромиро-
вании сплавов на нежелезной основе подвергать их предварительно диффузи-
онному железнению для уменьшения энергии активации кристаллической ре-
шетки сплава и увеличения глубины насыщения его хромом.
2 А. Н. Минкевич 33
диффузии насыщающего компонента С в сплав. Смещение точ-
ки Х2 в положение Х2 происходит, если при диффузии вместе с
компонентом С в сплав А — В (Л — В — С) вводить определен-
ное количество компонента А. Изменяя таким путем соотноше-
ние компонентов в поверхностном слое, можно интенсифициро-
вать процесс насыщения. Это положение было проверено при
диффузии хрома в сплав Fe — Сг — Ni, содержащий 35% Ni и
15% Сг. Хромирование проводилось в смесях, содержащих:
1) 60% Сг, 1% йодистого аммония, 39% глинозема; 2) 60% Сг,
10% Fe, 1% NH4J и 29% глинозема. На изотермическом раз-
резе диаграммы тройной системы Fe — Сг — Ni при 1100° С
(рис. 14, б) показано положение экспериментальных точек Х2
и Х2 , соответствующих составу первой снятой стружки толщи-
ной 0,025 мм.
Глубина слоя после обработки во второй смеси (точка Х2 )
была в 4—5 раз больше, чем после обработки в первой смеси
(точка Х2).
Иногда в диффузионном слое обнаруживаются фазы с боль-
шей концентрацией диффундирующего элемента, а фазы с мень-
шей концентрацией диффундирующего элемента отсутствуют.
Так, при силицировании молибдена первым обнаруживается слой
соединения MoSi2, тогда как слой с менее богатыми кремнием
соединениями Mo3Si и Mo5Si3 обнаруживается лишь после про-
должительного насыщения. При цинковании меди вначале был
обнаружен слой у-фазы, затем 8-фазы и, наконец, p-фазы. Пос-
ледняя фаза содержала минимальное количество цинка и поэто-
му, казалось бы, должна была возникнуть первой. То же наблю-
дается и при алитировании железа в расплаве алюминия, когда
сразу возникает слой с большой концентрацией алюминия (сое-
динение Fe2Al5).
Одни исследователи считают, что эти аномалии объясняются
первенствующей ролью химического взаимодействия в образова-
нии диффузионных слоев, другие объясняют отклонения природы
образующихся слоев от соответствующих диаграмм состояния
кристаллографическими соображениями. Они считают, что в
диффузионном слое могут образовываться те или иные фазы в
зависимости от структурного соответствия возникающей фазы и
основы и соотношения величин атомных радиусов внедряемого
элемента и основы. При этом в предпочтительно ориентирован-
ной основе возникают текстуры диффузии.
Можно предположить, что первой всегда обнаруживается фа-
за, скорость возникновения которой максимальна. Невозмож-
ность обнаружения в ряде случаев существующими методами бо-
лее бедных фаз не дает, однако, оснований утверждать, что они
отсутствуют в слое. Эти фазы могут иметь малую толщину вслед-
ствие ничтожной скорости их образования. При длительных про-
34
цессах толщина этих фаз, по-видимому, увеличивается до раз-
меров, позволяющих их наблюдать.
Однако в противоположность этому мнению следует "все же
заметить, что диффузионные процессы не могут быть отнесены к
полностью обратимым, изображаемым равновесными диаграм-
мами состояния, поскольку в лучшем случае в них имеет место
стационарный поток вещества. Поэтому нет убедительных осно-
ваний ожидать во всех без исключения случаях точного соответ-
ствия между диаграммой состояния и последовательностью воз-
никающих при диффузии фаз.
Микроструктуры диффузионного слоя в зависимости от типа
бинарной диаграммы состояния системы отличаются друг от дру-
га. Если диффундирующий элемент,
вызывает в процессе насыщения фа-
зовых превращений, то (независимо
от наличия при последующем охла-
ждении перекристаллизации) глуби-
ну диффузии удается приблизитель-
но установить лишь по весьма сла-
бому различию в травимости обыч-
ных по форме равноосных зерен.
Концентрация диффундирующего
элемента в этом случае постепенно
изменяется от сердцевины к поверх-
ности металла (рис. 15, линии /, 3,
4, 6). При этом в большинстве слу-
чаев различие в травимости диффу-
растворяясь в металле, не
% диффундирующего элемента
а) б)
Рис. 15. Левый угол диа-
грамм состояния систем ме-
талл — диффундирующий
элемент
зионного слоя и сердцевины на-
столько невелико, что глубину диффузионного слоя невозможно
определить микроскопическим методом.
Диффузия при температуре, соответствующей линии 2, соп-
ровождается после насыщения a-фазы перестройкой решетки а
в у. Перекристаллизация начинается от поверхности и идет вглубь
по направлению диффузии, вследствие чего образующиеся зерна
у-фазы приобретают вытянутую форму. После охлаждения вы-
тянутость зерен не сохраняется вследствие образования равно-
осных зерен новой фазы (а). Общая глубина слоя так же, как
и в предыдущем случае, может быть определена по слабому раз
личию в травимости сердцевины и внутренней части слоя. Гра-
ница фаз а и у (при температуре диффузии) наблюдается на
шлифе в виде резкой полосы (линия раздела), расстояние кото-
рой от поверхности обычно принимается в исследованиях за ус-
ловную глубину диффузионного слоя. Линия раздела а- и у-фаз
соответствует резкому перепаду концентрации по глубине слоя,
а следовательно, и изменению физико-химических свойств.
Диффузия по изотерме 5 (рис. 15) сопровождается вначале
насыщением у-фазы, а затем перестройкой решетки .у в а. При
35
этом столбчатость зерен вновь образовавшейся на поверхности
a-фазы отчетливо сохраняется при последующем охлаждении, так
как прй этом не происходит перекристаллизации a-фазы. Линия
раздела а- и у-фаз (при температуре диффузии) отчетливо выяв-
ляется на микроструктуре и соответствует перепаду концентра-
Рис. 16. Схема роста зе-
рен при неодновремен-
ном зарождении центров
на поверхности
ции диффундирующего элемента. Об-
щая глубина слоя выявляется неотчет-
ливо, аналогично предыдущим слу-
чаям.
Столбчатые зерна часто проника-
ют на значительную глубину за линию
раздела. Объясняется это тем, что при
перекристаллизации сердцевины во
время охлаждения столбчатые зерна
a-фазы служат, очевидно, базой пере-
кристаллизации для близлежащих уча-
стков у-фазы. Поэтому происходит
развитие столбчатых зерен глубже линии раздела [1].
Образование столбчатых зерен схематически показано на
рис. 16. Перекристаллизация в диффузионном слое в процессе
насыщения начинается с образования зародышей новой фазы,
которые растут в радиальных
направлениях во все стороны, об-
разуя объемы полусфероидаль-
ной формы. Радиальное развитие
новой фазы идет до тех пор, пока
не произойдет столкновения меж-
ду зернами, растущими от раз-
личных зародышей. Зерна, посте-
пенно увеличиваясь, приобретают
столбчатый характер. Рост менее
благоприятно ориентированных
зерен подавляется ростом сосед-
них зерен, ориентация которых
благоприятнее.
Такой механизм образования
столбчатых зерен при возникно-
вении в диффузионном слое твер-
дых растворов может быть пере-
несен и на случай образования
химических соединений. Часто
химические соединения (напри-
мер, слой карбидов, 8-фазы при
Рис. 17. Столбчатые зерна кар-
бонитридной фазы в поверхно-
стной зоне железа, цианирован-
ного при 640° С (X 250)
азотировании железа, карбонитридной фазы) при травлении
обычными травителями выявляются в виде нетравящегося
светлого слоя, однако при соответствующем травлении отчетливо
выявляются столбчатые зерна (рис. 17). Для понимания меха-
36
низма реакционной диффузии важно выявить роль отдельных
компонентов в образовании диффузионного слоя. Если процесс
образования слоя осуществляется встречной диффузией компо-
нентов через решетку химического соединения, то рост слоя бу-
дет происходить одновременно с обеих сторон. При односторон-
ней диффузии компонента из насыщающей фазы внутрь металла
рост слоя будет происходить на внутренней границе слой — ме-
талл, а при преимущественной односторонней диффузии металла
основы фронтом реакции образования слоя будет наружная по-
верхность.
В диффузионном слое иногда возникает пористость, которая
в ряде случаев может быть объяснена так называемым эффек-
том Киркендалла. В опытах, поставленных этим автором [5], ря-
ды тонких молибденовых проволок
помещали вдоль боковых
1
Рис. 18. Схема опытов Киркендалла:
1 — метка из фольги; 2 — проволочная метка
р-латунь
СдВиг
6)
граней прямоугольного образца латуни, содержащей 30% Zn, и
затем с боковых сторон электролитически покрывали образец
медью. После диффузионного отжига ряды молибденовых про-
волок, расположенных у противоположных граней образца, ока-
зались придвинутыми друг к другу на расстояние, пропорцио-
нальное квадратному корню из величины продолжительности от-
жига (рис. 18, а). Последнее указывало на диффузионную при-
роду этого эффекта, впоследствии обнаруженного во многих си-
стемах. Во всех случаях перемещение меток происходило в нап-
равлении части образца, состоящего из вещества с более низкой
температурой плавления (например, к латуни в системе латунь—
медь, к меди в системе медь — никель и т. д.). Сдвиг молибде-
новых проволок наблюдали, когда в одной плоскости с ними по-
мещали молибденовую фольгу (рис. 18,6). Через фольгу диффу-
зия не происходила, и поэтому фольга указывала начальное по-
ложение поверхности.
37
Эффект Киркендалла при изучении системы медь — латунь
позволяет сделать следующий вывод: число атомов цинка, пере-
ходящих из латуни в медь, больше числа атомов меди, перехо-
дящих в противоположном направлении. Вблизи линии раздела
со стороны меди возникают новые атомные плоскости, необходи-
мые для размещения возрастающего числа атомов; если до ка-
кого-то периода латунь сохраняет недефектную решетку, то
вследствие потери атомов она должна сжаться. Таким образом
создается перемещение плоскостей решетки в диффузионной зоне
и проволочных меток — в направлении областей, обогащенных
более быстро диффундирующим элементом. Этот эффект указы-
вает на неравенство потоков двух элементов, диффундирующих в
противоположных направлениях. Он доказывает несостоятель-
ность обменного и кольцевого (циклического) механизмов диф-
фузии, предусматривающих одновременное перемещение раз-
ных элементов с одинаковой скоростью. Эти механизмы,
возможно, могут иметь место только для металлов, в которых не
наблюдается эффект Киркендалла.
Эффект Киркендалла сопровождается образованием пор
вблизи границы раздела со стороны быстро диффундирующего
элемента, возникающих вследствие слияния избыточных вакан-
сий. Наличие пор ускоряет диффузию, которая осуществляется
перемещением атомов по поверхности пэр или через газовую
фазу.
В некоторых работах [5], [10] приводятся примеры проявле-
ния эффекта Киркендалла при диффузии в различных системах.
При этом иногда диффузия может привести к разрушению метал-
ла. Например, если стержень из цинка покрыть медью и подверг-
нуть диффузионному отжигу (при 380° С в течение 22 ч), то в
результате диффузии цинка в медь, образования зоны сплава
медь — цинк и возникновения пористости медная оболочка об-
разца разрывается.
Эффект Киркендалла изучен на системах Fe — Та и Fe — Сг
[10]. Пластины соответствующих металлов сваривались и под-
вергались диффузионному отжигу в вакууме при 1450° С, т. е. в
области существования объемноцентрированного б (а)-железа.
После отжига в обоих случаях граница раздела металлов сме-
щалась в сторону железной пластины и в последней на некото-
ром расстоянии от границы железо — металл возникала пори-
стость вследствие большего диффузионного потока атомов желе-
за в тантал и хром, чем этих элементов в железо. В результате
твердый раствор хрома и тантала в железе пересыщался вакан-
сиями, которые, соединяясь друг с другом, образовывали поры.
Поры, образовавшиеся в твердом растворе тантала в железе,
имели трехугольную, квадратную, пяти-шести- и семиугольные
формы. Большая часть крупных пор имела пяти- и семиугольную
форму. Эти формы могут соответствовать только сечениям
38
плоскостей решетки ромбического додекаэдра, наиболее густо
усеянных атомами. Отсюда вывод, что моры ограничены плоско-
стями решетки с наименьшей поверхностной энергией [10].
Исследование диффузии в системе Fe — Сг при температуре
у-железа показало иные результаты, чем при диффузии при
температуре существования б (а)-железа. В диффузионной паре
хрома с у-железом поток атомов, идущий от хрома в железо,
больше потока, идущего в обратном направлении. В результате
первоначальная поверхность раздела перемещается в сторону
хрома и в нем образуется пористость. Таким образом, поверх-
ность раздела перемещалась, как и в предыдущих опытах, в нап-
Рис. 20. Распределение по глу-
бине диффузионного слоя фос-
фора и углерода после насыще-
ния стали, содержащей 0,4% С,
в смеси 10% РНз+90% Н2 в
течение 80 ч при 1000°С
(А. В. Брамлей и др. [19])
Расстояние от поверхности
a) S)
Рис. 19. Распределение по глубине диффу-
зионного слоя углерода (С) и диффундиру-
ющего элемента (Э) в случаях образова-
ния в слое:
а — зоны с a-фазой (при температуре насыще-
ния); б — карбидной зоны
равлении компонента, обладающего большей подвижностью.
Диффузионная природа пористости убедительно доказывается
тем, что область максимальной пористости перемещается пропор-
ционально времени диффузии, т. е. по тому же закону, по кото-
рому изменяется глубина диффузионного слоя [6], [10]. Все ска-
занное свидетельствует о вакансионном механизме диффузии
как в объемноцентрированном, так и в гранецентрированном же-
лезе.
Рассмотрим поведение углерода при насыщении стали различ-
ными элементами и особенности возникающих в связи с этим
структур диффузионных слоев.
Во многих случаях наблюдается поверхностное обезуглеро-
живание стали под действием газовых, жидких или порошкооб-
разных сред, в которых происходит насыщение. Но даже при
отсутствии такого действия равномерное распределение углерода
39
в поверхностных слоях стали при ее насыщении всегда наруша-
ется. Одни элементы оттесняют углерод от поверхностной зоны
слоя, другие, наоборот, «подтягивают» его к этой зоне. К первым
Расстояние от поверхности
ж} з)
Рис. 21. Изменение микро-т. э. д. с. по глу-
бине диффузионных слоев (автор, Е. В
Панченко, Е. М. Струг, В. Устогал,
А. С. Салтыкова):
а — после цементации железа в твердом кар-
бюризаторе с последующей закалкой; б — после
азотирования стали 38ХМЮА; в — после хро-
мирования железа; г — после хромирования ста-
ли У12; д — после силицирования железа; е —
после цинкования; ж — после алитирования же-
леза; з — после борирования железа
относятся, например, не-
карбидообразующие эле-
менты, суживающие об-
ласть у-фазы,— кремний,
алюминий, фосфор, медь.
В процессе диффузии та-
ких элементов после насы-
щения до предела раство-
римости в у-фазе возника-
ет зона слоя с a-фазой, в
которой углерод имеет
ничтожную раствори-
мость. Поэтому углерод
оттесняется растущей зо-
ной a-фазы к сердцеви-
не, находящейся в аусте-
нитном состоянии (рис.
19, ау и 20). Имеются так-
же данные об оттеснении
углерода никелем, даю-
щим открытую у-область
[30], и бором [31].
При насыщении стали
с повышенным содержа-
нием углерода карбидооб-
разующими элементами
(хромом, ванадием, нио-
бием, титаном, вольфра-
мом, марганцем и т. д.)
на поверхности стали об-
разуется тонкая сплошная
зона карбидов соответст-
вующих элементов.
Содержание углерода
в зоне таких карбидов
повышается до 5—10%, а
в некоторых случаях и бо-
лее. Под карбидным сло-
ем обнаруживается широ-
кая обезуглероженная зо-
на. Иногда (например,
при хромировании стали) между обезуглероженной и карбидной
зонами наблюдается промежуточная темная зона (рис. 19, б) —
подслой с повышенным содержанием углерода (глобули карби-
40
дов на ферритной основе). В работе [9] отрицается возможность
образования такого подслоя, но это мнение не подтверждено
экспериментально.
Глубина проникновения элементов в аустенит с увеличением
содержания в нем углерода всегда уменьшается в той или иной
степени в зависимости от природы диффундирующего элемента.
С увеличением в стали содержания углерода глубина проникно-
вения в аустенит элементов, не образующих карбидов (кремния,
алюминия, бора, фосфора, серы), уменьшается значительно в
меньшей степени, чем карбидообразующих элементов. Влияние
углерода на глубину проникновения в аустенит элементов, спо-
собствующих расширению у-области (никеля, кобальта, меди) и
не склонных к образованию карбидов, незначительно.
Для изучения строения диффузионных слоев можно приме-
нять метод послойного измерения микротермоэлектродвижущей
силы. Результаты измерения микротермоэлектродвижущей силы
различных диффузионных слоев на приборе, разработанном в
МИСиС [32], приведены на рис. 21 *. Этот метод оказывается осо-
бенно полезным в тех случаях, когда образующиеся фазы трудно
дифференцировать вследствие их почти одинаковой травимости
и близкой твердости. Это относится, налоимер, к некоторым бо-
ридам, образующимся в поверхностных слоях металлов при бо-
рировании. Из рис. 21 следует, что величины микротермоэлектро-
движущих сил для боридов Fe2B и FeB существенно отличаются,
что позволяет определить толщину слоев этих боридов.
* Отклонение кривой в поверхностной зоне цементованного слоя, по-види-
мому, связано с поверхностным обезуглероживанием при медленном охлажде-
нии после цементации.
ГЛАВА II
ХИМИКО-ТЕРМИЧЕСКАЯ ОБРАБОТКА СТАЛИ
ЦЕМЕНТАЦИЯ
Цементация — наиболее распространенный в машиностроении
способ химико-термической обработки стальных деталей — при-
меняется для получения высокой поверхностной твердости, из-
носостойкости и усталостной прочности деталей. Эти свойства
достигаются обогащением поверхностного слоя низкоуглероди-
стой, легированной и нелегированной стали углеродом до кон-
центрации эвтектоидной или заэвтектоидной и последующей тер-
мической обработкой, сообщающей поверхностному слою струк-
туру мартенсита с тем или иным количеством остаточного аусте-
нита и карбидов.
Глубина цементованного слоя обычно находится в пределах
0,5—2,0 мм (иногда для мелких деталей в пределах 0,1—0,3 мм,
а для крупных — более 2,0 мм). Цементацию стальных деталей
осуществляют в твердых, газовых и жидких карбюризаторах. За
последние годы все большее развитие получает газовая цемен-
тация.
Диффузия углерода в сталь
По количественной характеристике диффузии углерода в же-
лезо накоплены многочисленные данные.
Коэффициент диффузии углерода в a-железо более чем на
порядок выше, чем в у-железо, имеющее значительно более
плотно упакованную решетку (рис. 22).
Энергия активации Q углерода в a-железе значительно ниже,
чем в у-железе, и по данным работы [33] составляет соответствен-
но 18 100 и 31 350 кал!молъ, или 75 839 и 131 356 дж!молъ
(рис. 22). Такая величина энергии активации диффузии углерода
в a-железе подтверждается и работой [34], согласно которой Q =
= 75 362 дж)моль = 18 000 кал!молъ и Do = 3-10~5 см21сек.
Данные ряда других авторов по диффузии углерода в а-желе-
зо приведены в работах [10], [35], [36].
42
Диффузия углерода в феррите обусловливает возможность
протекания таких низкотемпературных процессов, как коагуля-
ция и сфероидизация карбидов в отожженной стали, карбидооб-
разование при отпуске закаленной стали, графитизация и т. д.
Однако цементация при температурах существования а-железа
не производится ввиду ничтожной растворимости в этой фазе
углерода. Цементация проводится при температурах 920—950° С
и выше, при которых сталь нахо-
дится в аустенитном состоянии.
Концентрационная зависи-
мость коэффициента диффузии
углерода в аустените выражается
уравнением [37]
32 000
£>с = (0,07 + 0,06 • С %)е RT ,
или, по другим данным [38],
_ 31 3S0
£>с= (0,04 + 0,08-С%) е RT .
Из приведенных зависимостей
следует, что коэффициент диффу-
зии углерода в аустените увеличи-
вается с увеличением содержания
в стали углерода. Это, очевидно,
связано с увеличением искажения
кристаллической решетки аусте-
нита и термодинамической актив-
ности углерода [10].
По данным работы [35], пара-
метры диффузии углерода в аус-
тените в интервале 950—1050° С
следующие: Do — 0,1 см{сек, Q —
= 32 400 кал/г-атом
Рис. 22. Зависимость коэффи-
циента диффузии углерода в
феррите и аустените от темпе-
ратуры (Стэнли [33]):
18 100
- 29 • ю-3 • е
с 31 3S0
_ РТ
DC >7 - 0,064 . е
(135 756 дж/г-атом).
Легирующие элементы оказывают существенное влияние на
диффузию углерода в аустените, что связано с искажением
кристаллической решетки, изменением ’ энергии межатомной
связи в твердом растворе и термодинамической активности
углерода.
Результаты изучения влияния легирующих элементов на коэф-
фициент диффузии углерода в аустените при 1100° С приведены
на рис. 23. При других температурах влияние некоторых элемен-
тов на коэффициент диффузии углерода в аустените изменяется.
Автор работы [38] сделал следующее обобщение: карбидообразу-
ющие элементы обычно замедляют, а некарбидообразующие уско-
ряют диффузию углерода. Однако следует заметить, что это обоб-
43
щение требует существенного уточнения. Так, например, кремний
увеличивает коэффициент диффузии углерода в аустените при
низких температурах (ниже 950° С), что согласуется .с представ-
лением о кремнии как о некарбидообразующем элементе, иска-
жающем кристаллическую решетку аустенита и вследствие это-
го ускоряющем диффузию углерода. Однако при более высоких
температурах кремний уменьшает коэффициент диффузии угле-
рода в аустените. Зависимость коэффициента диффузии углерода
в аустените от содержания кобальта имеет сложный характер —
увеличивается [38] или уменьшается в зависимости от содержа-
ния кобальта [10]. Медь ускоряет диффузию углерода, а алюми-
Рис. 23. Влияние содержания легирующих
элементов на коэффициент диффузии угле-
рода в аустените при 1100°С [38]
ний и хром замедляют
ее [10].
При добавлении в
сталь 1 % легирующего
элемента на первом месте
по степени ускорения диф-
фузии углерода в аустени-
те при 950° С стоит крем-
ний, затем никель и на
последнем месте медь.
Добавление одновременно
кремния и кобальта * в
системы с карбидообразу-
ющим элементом понижа-
ет подвижность атомов
углерода, что указывает
на существование взаимо-
действия легирующих элементов в твердом растворе.
Влияние карбидообразующих элементов на снижение коэф-
фициента диффузии углерода в аустените при 840—1100° С в
сплавах, содержащих 1,2% Si и около 1% легирующего элемен-
та, усиливается, по данным работы [39], в такой последователь-
ности: марганец, молибден, ванадий, вольфрам, хром. Хром за-
медляет диффузию углерода в 20 раз сильнее марганца.
Большинство карбидообразующих элементов (хром, молиб-
ден, вольфрам, ванадий, титан и др.) увеличивает содержание
углерода в поверхностной зоне цементованного слоя в связи с
интенсивным карбидообразованием (рис. 24). Наиболее резко
увеличивает содержание углерода в поверхностной зоне слоя
хром.
В ряде случаев с повышением содержания в стали карби-
дообразующих элементов концентрация углерода в слое повы-
шается лишь до определенного предела, после чего снижается
(рис. 25). Это объясняется различиями в составе и количестве
карбидов, образующихся в стали с различным содержанием
легирующих элементов.
44
Рис. 24. Влияние температуры на содержание углерода в поверхностном слое
стали, содержащей 0,20% С и 3% различных легирующих элементов; цемента-
ция в твердом карбюризаторе в течение 10, 30 и 60 ч\ С — нелегированная
сталь (Е. Гудремон и X. Шредер)
О 0/t 0,8 1.2 мм
Глубина слоя
Рис. 25. Влияние хрома на глубину цементованного слоя
и содержание в нем углерода; сталь с 0,15% С, цемента-
ция в твердом карбюризаторе (85% древесного угля
и 15% ВаСОз) при 930° С в течение 7 ч (автор
и Л. В. Федосова).
45
В цементованном слое стали, легированной карбидообразую-
щим элементом, углерод в большом количестве концентрируется
лишь в самой поверхностной зоне слоя на глубине 0,1—0,2 мм.
Здесь обнаруживаются глобули карбидов вместо цементитной
Рис. 26. Влияние легирующих элементов на содержание углерода в цементо-
ванном слое; цементация в твердом карбюризаторе (80% древесного угля
и 20% соды) при 930° С в течение 8 ч [40]: Сп — содержание углевода в це-
Рис. 27. Распределение углерода по глубине цементованного слоя, образовав-
шегося в твердом карбюризаторе при температуре 950° С в течение 8 ч (кри-
вая /) и при 900° С в течение 13 v (кривая 2) [41]:
а — сталь 20; б — сталь 20ХНМ; в — сталь 20ХГР
сетки, наблюдаемой в заэвтектоидной зоне соответствующего
слоя углеродистой стали.
Некарбидообразующие элементы (кремний, никель, кобальт,
медь, сера, фосфор) несколько уменьшают содержание углерода
в слое, что объясняется снижением растворимости углерода в
аустените под их влиянием. Наиболее резко снижает содержание
46
углерода в слое кремний, воздействие которого в несколько раз
сильнее воздействия никеля. Относительно воздействия такого не-
карбидообразующего элемента, как алюминий, имеются противо-
речивые мнения: по одним данным [93] алюминий уменьшает со-
держание углерода в слое, а по другим |[19] — увеличивает
(рис. 26). В полном соответствии со сказанным в цементованном
а) б)
Рис. 28. Влияние температуры на глубину цементованного слоя (цементация
в твердом карбюризаторе) стали, содержащей 0,2% С и 3% различных леги-
рующих элементов (С — нелегированная сталь).
а — выдержка 10 ч; б — выдержка 30 ч
значительно больше углерода, чем в соответствующем слое угле-
родистой стали. При этом с повышением температуры цемента-
ции содержание углерода в поверхностной зоне слоя этих сталей
снижается (рис. 27).
Влияние легирующих элементов на глубину цементованного
слоя обусловлено их воздействием на концентрацию углерода в
поверхностной зоне этого слоя и на коэффициент диффузии угле-
рода в аустените.
Никель увеличивает коэффициент диффузии углерода в аусте-
ните, но несколько снижает глубину цементованного слоя. Это
объясняется тем, что, уменьшая растворимость углерода в аусте-
ните, никель снижает концентрацию углерода в поверхностной
зоне цементованного слоя, что приводит к небольшому уменьше-
нию глубины слоя; аналогичным образом, но более резко, влияют
на глубину слоя кремний и медь [19].
47
Хром, вольфрам, молибден, ванадий и титан, снижая коэффи-
циент диффузии углерода в аустените, повышают концентрацию
углерода в поверхностной зоне слоя, что приводит к небольшому
увеличению глубины цементованного слоя (рис. 28, а).
Сталь для цементации
Цементованные детали после соответствующей термической
обработки должны иметь твердый, прочный поверхностный слой,
стойкий против износа и продавливания, и достаточно прочную и
вязкую сердцевину. В связи с последним требованием для цемен-
тации применяют низкоуглеродистую сталь, содержащую 0,08—
0,25% С.
В последние годы для высоконагруженных зубчатых колес и
других ответственных, в том числе крупных, деталей начали ис-
пользовать цементуемую сталь с более высоким (0,25—0,35%)
содержанием углерода. Поэтому оказалось возможным умень-
шить глубину цементованного слоя, не опасаясь его продавлива-
ния при больших нагрузках, предотвратить преждевременное
разрушение поверхностного слоя из-за пластической деформации
слоев металла, лежащих непосредственно под этим слоем, а так-
же закаливать сердцевину с более низкой температуры без пере-
грева цементованного слоя [42].
Положительное влияние повышения содержания углерода в
цементованной стали отмечалось и в ряде последующих работ.
Показано, что увеличение содержания в некоторых сталях угле-
рода повышает предел их выносливости лишь в случае одновре-
менного некоторого снижения глубины цементованного слоя [19].
Для цементации широко используют низкоуглеродистую ка-
чественную сталь (08, 10, 15 и 20) и автоматную сталь (А12, А15,
А15Г, А20), а для неответственных деталей низкоуглеродистую
сталь обыкновенного или повышенного качества (Ст. 2, Ст. 3, Ст. 4,
Ст. 5 [43], М12, М16, БОЭ, Б16 и др.). Ответственные изделия из-
готовляют из легированной стали.
Основное назначение легирующих элементов в цементуемой
стали — повышение ее прокаливаемости и механических свойств
сердцевины. Большинство легирующих элементов понижает
склонность зерна стали к росту при нагреве, а некоторые из них
улучшают механические свойства цементованного слоя.
В табл. 3 приведены режимы термической обработки и меха-
нические свойства некоторых широко применяемых легированных
цементуемых сталей, а в табл. 4 — механические свойства не-
скольких сталей в зависимости от сечения заготовок, подвергну-
тых термической обработке. На рис. 29 показано изменение пре-
дела текучести при статическом изгибе некоторых сталей. В на-
стоящее время в промышленности применяют марки стали, отли-
чающиеся более высокими свойствами. Для тяжелонагруженных
48
Таблица 3
Режимы термической обработки и механические свойства
цементуемых сталей
Марка стали Закалка* Отпуск в °C Механические свойства (не менее)** ан
Температура в °C Охлаждающая среда ат ав со «о со -э- в кГ-м/см2 I в нм/м2- 10е
I II | zWvTjx я в Х107 н/м2 а? а? С СП о хЗ и %
15Х 880 770—820 Вода или масло 180 50 49 70 68 12 45 7 6,8
20Х- 880 770—820 То же 180 65 63 80 78 12 45 7 6,8
20ХГ • 880 — » 180 75 73 90 88 10 40 6 5,9
15ХФ 880 770—820 » 180 55 54 75 73 13 50 8 7,8
20ХФ 880 770—820 » 180 60 59 80 78 14 50 8 7,8
20ХН 860 760-810 » 180 60 59 80 78 11 50 7 6,8
13Н2ХА 860 770—810 Масло 180 40 39 60 59 15 50 11 10,8
12ХН2 860 760—810 » 180 60 59 80 78 12 50 9 8,8
12ХНЗА 860 760—810 » 180 70 68 95 93 11 55 11 10,8
12Х2Н4 860 780—800 » 180 100 98 110 108 10 50 9 8,8
20Х2Н4А 860 780—800 » 180 110 108 130 127 9 45 8 7,8
18Х2Н4ВА 950 850 Воздух или масло 180 105 103 120 117 12 55 11 10,8
15ХГНВА 860 — Масло 160 65 63 90 88 12 55 14 13,7
18ХГН 860 770—810 » 180 70 68 85 83 12 50 7 6,9
18ХГТ 880 870 » 200 85 83 110 108 9 50 8 7,8
25ХГТ 830— 840 — » 200 ПО 107 130 127 9 45 6 5,9
25ХГМ*** — — » 190 100 107 120 117 10 55 10 9,8
16ХГТА 900 790—860 » 180 80 78 105 103 12 55 10 9,8
15ХГНТА 850 — 180 75 73 95 93 10 55 10 9,8
15Х2ГН2ТА 860 770—810 » 180 90 88 100 98 12 55 11 10,8
25Х2ГНТА 900 860 » 180 135 132 150 147 10 45 7 6,8
15ХР 860 770—810 Масло или вода 180 60 59 80 78 12 45 8 7,8
20ХГР 910 870 Масло 200 80 78 100 98 9 50 8 7,8
20ХГНР 820 — » 200 120 117 130 127 10 50 9 8,8
18ХСНРА 900 — Масло или вода 200 НО 108 130 127 10 45 7 6,9
15Х2ГН2ТРА 850 Масло 170 90 88 105 103 12 55 11 10,8
* В графе «Закалка» для многих сталей приведены температуры I i: !Т зака-
лок по данным ГОСТа и другим источникам. Вместо двойной закалки часто при-
меняют одинарную или непосредственную закалку с подстуживанием.
** Определены на образцах, вырезанных из круглых и квадратных за-
готовок размером 15 мм, за исключением сталей 15ХНГВА и 15Х2ГН2ТРА (заго-
товки размером 25 мм), а также 18ХГТ, 15Х2Г2Т, ЗОХГТ 25Х2ГНТА (образцы).
*** Сталь 25ХГМ применяется только для нитроцементации.
49
о
Таблица 4
Механические свойства сердцевины ложно-цементованных образцов в зависимости от сечения заготовок
(нагрев до 910° С, 9 ч\ закалка с 810° С в масле, отпуск при 200° С) (С. Г. Перельман)
Предел текучести ат (при растяжении) Ударная вязкость ан в кГ-м/см2*
Диаметр заготовки в мм
5 15 20 25 35 50
Марка
стали
Л(2 'М2 м* м* 'м2 м* 10 15 20 25 35
« £ « £ « Sв, » ". « « £
zO z0 г и*/ zO z0 ww zO O', zO ww,
« x « x'Jx^x^x^x
15Х 110 108 55 54 50 49 45 44 35 34 30 29 8,5 5,0 4,5 4,0 7,5
18ХГТ 130 127 80 78 60 59 55 54 - - - - 7,5 5,0 6,5 6,5 —
ЗОХГТ 145 142 95 93 80 78 75 73 70 68 60 59 6,0 7,0 6,5 6,0 6,5
12ХН2А 95 93 65 64 60 58 55 53 — — — — 18,5 16,0 17,0 16,0 —
12Х2Н4А 110 108 100 98 90 88 85 83 — — — — 10,5 10,5 13,5 16,0 —
20ХНЗА 125 122 120 117 110 107 100 98 95 93 80 78 9,5 8,5 8,5 8,5 8,5
18Х2Н4ВА 125 122 120 117 115 112 115 112 110 108 95 93 9,5 8,0 7,5 9,0 10,0
Примечание. Предел текучести у некоторых сталей (например, ЗОХГТ) при диаметре заготовки 15 мм получился понижен-
ным по сравнению с данными ГОСТа (возможно, вследствие заниженной температуры закалки).
* При переводе в единицы системы СИ (нм/м2) указанные в графе* цифры следует помножить на 9,8 -104.
деталей на автозаводе им. Лихачева (ЗИЛ) используют сталь
25ХГТ [44], которая имеет более твердую сердцевину, чем ранее
применявшаяся сталь 18ХГТ. Переход на сталь 25ХГТ позволил
Рис. 29. Условный предел текучести сго.а при статическом изгибе ложно-цемен-
тованных (а) и цементованных по двум граням (б) образцов в зависимости
от сечения заготовок; газовая и ложная цементация при 915° С в течение 9 ч,
последующая закалка с 810°С и отпуск при 200°С (С. Г. Перельман):
1 — сталь 30ХН2МФА; 2 — сталь 18Х2Н4ВА; 3 — ста.^ь ЗОХГТ; 4 — сталь 12Х2Н4А;
5 — сталь 12ХНЗА; 6 — сталь 18ХГТ; 7 — сталь 25Х; 8 — сталь 12ХН2А; 9 — сталь 15Х
уменьшить глубину цементованного слоя с 0,9—1,3 до 0,6—
0,9 мм и, следовательно, сократить продолжительность цемента-
ции (на 20—25%). Сталь 25ХГТ имеет повышенный условный
предел прочности и более высокую усталостную прочность.
На Горьковском автозаводе наряду со сталью 20ХНМ приме-
няются для изготовления тяжелонагруженных зубчатых колес,
кулачков, шарниров и т. д. боросодержащие стали 20ХГР [45] и
27ХГР. На Ярославском моторном заводе взамен стали 12ХНЗА
применяется для крупных зубчатых колес сталь 15ХГНТА.
51
Применяют также ряд новых цементуемых сталей, большая
часть которых для увеличения прокаливаемости содержит бор,
а для измельчения зерна — титан (табл. 3). Из таких сталей сле-
дует особо отметить стали 15Х2ГН2ТА и 15Х2ГН2ТРА [46]. Эти
стали получают все большее применение взамен сталей 12Х2Н4А,
12ХНЗА, 13Н5А, 21Н5А и 18Х2Н4ВА для изготовления различных
ответственных тяжелонагруженных деталей, работающих в ус-
ловиях динамических знакопеременных нагрузок. В цементован-
ном слое этих сталей сохраняется значительно меньше остаточ-
ного аустенита, чем в сталях 12Х2Н4А, 12ХНЗА и 18Х2Н4ВА, и
их можно цементовать без заметного роста зерна при температу-
рах до 1000° С. К недостаткам этих сталей следует отнести их
повышенную поражаемость волосовинами [46].
В некоторые сложнолегированные опытные стали был успешно
введен ванадий (25ХНФР, 20ХЗФ, 25Х2ГН2Д2Ф) [47]. Взамен вы-
соколегированных сталей 18Х2Н4ВА и 20Х2Н4А разработаны
также малоникелевые стали типа 18ХГСН2МВА и 18ХГСН2МА,
а взамен сталей 12ХНЗА и 12Х2Н4А — сталь типа 14ХГСН2МА
[48]. Эти новые стали по механическим свойствам не уступают
соответствующим сталям, содержащим большое количество ни-
келя.
Из сказанного следует, что добавка никеля сохранена в ряде
ответственных сталей (15ХГВА, 15ХГНТА, 15Х2ГН2ТА,
25Х2ГНТА 18ХСНРА, 12ХН2РА, 15Х2ГН2ТРА, 18ХГСН2МВА,
18ХСН2МА, 14ХГСН2МА). Это объясняется тем, что никель бла-
гоприятно воздействует на вязкость цементованного слоя и серд-
цевины и понижает порог хладноломкости цементованной стали.
Испытания ударной вязкости различных сталей, проведен-
ные при комнатных, пониженных и повышенных температурах
на ложно-цементованных и цементованных образцах, отчетливо
показывают преимущество сталей, содержащих никель. Высокие
свойства этих сталей подтверждаются их испытаниями на долго-
вечность путем многократно изгибающих ударов на копре по-
вторного удара (КПУ-2).
Доказана целесообразность введения в цементуемую сталь
кремния. Кремний уменьшает глубину заэвтектоидной зоны и ее
насыщенность карбидами и повышает механические свойства це-
ментованной стали, особенно ударно-усталостную выносливость.
Эффективно легировать кремнием стали с большой и средней
прокаливаемостью и стали, склонные к пересыщению поверхност-
ного слоя углеродом. Предлагается повысить содержание крем-
ния в стали 12ХН2 до 0,9—1,2% (12ХН2С) и в стали 25ХГТ до
1,1—1,3% (25ХГСТ). Сталь типа 12ХН2С может в некоторых
случаях заменить сталь 12ХНЗ [561].
Сравнение контактной выносливости сталей различных сос-
тавов позволило рекомендовать в качестве стали, обладающей
высокой контактной выносливостью, сталь 17СГ2М [49].
52
В связи с расширением выплавки стали из керченских руд
были продолжены работы по исследованию влияния мышьяка на
свойства цементуемых сталей. Содержание мышьяка в сталях,
выплавленных из керченских руд, составляет 0,15—0,18%, а в
сталеплавильном скрапе большинства отечественных заводов не
более 0,02—0,03%. В исследованных сталях, легированных од-
ним элементом (никель, кремний и др.), содержание мышьяка
колебалось в пределах 0,23—0,67%, а в сложнолегированных
(12ХНЗА, ЗОХНЗА, 18Х2Н4ВА) — от 0,15 до 0,31% [562]. С увели-
чением содержания в сталях мышьяка глубина цементованного
слоя несколько снизилась. Кроме того, в сталях, легирован-
ных одним элементом, с увеличением содержания мышьяка
немного снизилась поверхностная твердость, что вызвано
некоторым уменьшением количества углерода в цементованном
слое.
Работа, выполненная в центральном научно-исследователь-
ском институте черной металлургии (ЦНИИЧМ) на сталях типа
20ХГ и 20ХН, содержащих до 0,3% As, показала, что мышьяк
в указанных количествах практически не влияет на механические
свойства стали.
Теллур, введенный в цементуемую сталь (сотые доли процен-
та), снижает содержание углерода в цементованном слое и умень-
шает склонность зерна аустенита к росту, не ухудшая прокали-
ваемое™ стали [563].
При разработке составов машиностроительных цементуемых
сталей до настоящего времени не учитывается склонность сталей
к образованию при цементации дефектной структуры в виде гло-
булей или темной сетки, обнаруживаемой на нетравленых или
слаботравленых шлифах (см. стр. 83). При расположении этой
сетки на глубине более 20 мк резко снижается усталостная проч-
ность стали. Образованию такой сетки способствует хром (более
0,5%) и марганец. Это обстоятельство следует учитывать при
создании новых марок цементуемой стали.
Ряд работ посвящен исследованию цементации нержавеющих
высокохромистых сталей Х13, Х17, Х17Н2, 13Х14НВФРА,
12Х12НВМА и др. Из этих сталей изготовляют детали приборов
и аппаратов, работающих в коррозионной среде на износ [50] —
[55].
Некоторые заводы подвергают цементации пуансоны, матри-
цы и другой штамповый инструмент, изготовленный из стали
ШХ15. При цементации этой стали в поверхностном слое" выде-
ляется много глобулярных карбидов, и содержание углерода
в слое увеличивается до 2,5—3%; в результате после закалки
резко повышаются теплостойкость и износостойкость штампов
[56], [57].
В некоторых случаях целесообразно подвергать цементации
быстрорежущую сталь. Цементация быстрорежущей стали
53
протекает достаточно быстро и приводит к заметному повыше-
нию ее теплостойкости [58]. При цементации аустенитных сталей
(типа 4Х14Н14В2М) содержание углерода в слое увеличивается
до 2,2—2,6%; при этом твердость повышается лишь на HRC 20—
25 и после закалки не изменяется.
Подлежащие цементации детали обычно подают в термиче-
ский цех после предварительной механической обработки с при-
пуском на грубое и окончательное шлифование до 0,25 мм или с
припуском только на окончательное шлифование 0,05—0,15 лш.
Глубину слоя для зубьев колес рекомендуется принимать: при
модулях 1,5—3,0 в пределах 0,3—0,5 мм\ при модулях 3—5 в
пределах 0,7—0,9 мм (при твердости сердцевины зубьев в преде-
лах HRC 35—45).
Для изоляции поверхностей, не подлежащих цементации, при-
меняют гальваническое малопористое меднение (слой толщиной
0,01—0,05 мм, в зависимости от продолжительности и метода це-
ментации) . Иногда используют обмазки (например, 2,5 ч. одно-
хлористой меди, 1 ч. свинцового сурика), замешанные в раство-
ре, состоящем из 1 л этилового спирта или бензина и канифоли
(250 г). Отверстия забивают медными пробками или смесью из
50% кварцевого песка и 50% окалины, а затем асбестом и глиной.
В редких случаях на деталях оставляют припуски, снимаемые
после цементации.
Цементация в твердом карбюризаторе
Цементация в твердом карбюризаторе протекает при нагреве
стали в ящике вместе с древесным углем. В результате взаимо-
действия древесного угля с кислородом воздуха, находящимся
в ящике, образуется окись углерода, при последующем разложе-
нии которой возникает активный углерод (2СО = С + СОг), диф-
фундирующий в сталь. Освободившаяся двуокись углерода взаи-
модействует с древесным углем, и вновь образуется окись углеро-
да и т. д. Таким путем обеспечивается непрерывность процесса.
Для ускорения процесса цементацию ведут в карбюризаторах,
основой которых является древесный уголь с добавкой активи-
заторов, ускорителей процесса — углекислого бария и натрия.
Эти соли разлагаются с выделением соответствующих окислов и
двуокиси углерода (ВаСОз = ВаО + СОг; Ма2СОз = Na2O +
+ СО2). Двуокись углерода, реагируя с древесным углем, дает
дополнительное количество окиси углерода, что согласно преж-
ним представлениям является причиной ускорения процесса це-
ментации. Однако такое объяснение действия углекислых солей
нельзя считать исчерпывающим, так как известно, что почти
аналогичное ускорение цементации достигается добавлением к
древесному углю других соединений: К2СГ2О7, КМпО4, ВаО,
BaSO4, BaS и т. д.
54
Некоторые исследователи полагают, что влияние солей, окис-
лов и других соединений, обусловливается, главным образом
каталитическим воздействием входящих в их состав металлов
на реакцию взаимодействия
двуокиси углерода с углем,
причем наиболее сильное воз-
действие оказывают щелочные
и щелочноземельные металлы.
Предполагается, что эти ме-
таллы, входящие в состав ак-
тивных добавок, внедряются в
решетку графита и разрыхля-
ют ее. Ослабление связей меж-
ду базисными плоскостями
графита приводит к ускорению
взаимодействия СОг с угле-
родом.
Различие в действии от-
дельных активизаторов обус-
ловлено различием в степени
деформирующего воздействия
на решетку графита и, воз-
можно, разницей в теплоте
диссоциации-карбидов, окислов
воздействия хлоридов и других
Рис. 30. Изменение концентрации
СО в газовой фазе, образующейся
в цементационном ящике при це-
ментации в древесном угле с 15%
NaaCO3:
1 — температура в центре ящика;
2 — концентрация СО [20]
и других соединений. Механизм
солей, вероятно, такой же.
Рис. 31. Диаграммы равновесия железа с газовыми фазами
СН4 —Н2 и СО —СО2
50 60 70 S0 30 lOO^CO
50 <>0 30 Z0 10 OKCOi
При цементации в карбюризаторе с углекислым натрием в на-
чальный период в газовой среде цементационного ящика содер-
жится более 50% СО, а затем количество его постепенно умень-
шается (рис. 30). Из рассмотренной диаграммы равновесия СО—
СО2 с железом (рис. 31) следует, что для предельного насыще-
56
ния аустенита углеродом (например, при 950° С) соотношение
СО и СО2 должно быть примерно 99:1. Поэтому в начальный пе-
риод процесса количество СО2 в газовой среде должно составлять
около 0,5%. Остальной объем газовой среды цементационного
ящика в основном занимают азот и небольшое количество водо-
рода.
Глубина цементованного слоя увеличивается с повышением
содержания в карбюризаторе углекислого бария или натрия до
4—5%. При дальнейшем повышении содержания этих солей глу-
бина слоя почти не изменяется, концентрация же углерода в его
поверхностной зоне продолжает немного повышаться. Карбюри-
заторы, содержащие углекислый барий, медленнее истощаются,
и поэтому их более широко используют в производстве. К таким
карбюризаторам относится, например, древесноугольный карбю-
ризатор (ГОСТ 2407—51), содержащий 20—25% ВаСО3, менее
3,5% СаСО3 (добавляется для предотвращения спекания частиц),
менее 0,06% S, менее 0,5% SiO2, менее 5% Н2О, менее 10% лету-
чих, остальное древесный уголь зернистостью 3,5—10 мм (92%).
Выпускаются также карбюризаторы на основе полукокса и тор-
фяного кокса.
Иногда в карбюризатор добавляют мазут (3—5%) для улуч-
шения связи солей с частицами угля. Простейший карбюризатор
составляется из 85—90% древесного угля и 10—15% ВаСО3
или Na2CO3.
В производстве применяется смесь из 20—25% свежего и
75—80% отработанного карбюризатора. Иногда для снижения
содержания углерода в цементованном слое в смесь добавляют
всего лишь 5—10% свежего карбюризатора или уменьшают со-
держание углекислых солей в свежем карбюризаторе.
При повышении содержания в карбюризаторе серы гальвани-
ческое меднение частей деталей, не подлежащих цементации,
становится малоэффективным. Присутствие в карбюризаторе сер-
нистого колчедана (пирита) и сернокислых солей недопустимо,
так как вызывает резъедание поверхности деталей, а повышенное
содержание SiO2 ведет к образованию на поверхности деталей
стекловидных наплывов, под которыми отсутствует цементован-
ный слой.
Предложены новые карбюризаторы, в которых в качестве ак-
тивизатора применен ацетат натрия — CH3COONa или ацетат ба-
рия— Ва(СН3СОО)2 [60], [61]. Ацетат бария при нагреве разла-
гается с образованием углекислого бария и ацетона, который,
в свою очередь, разлагается на атомарный углерод, метан и окись
углерода: 2Ва(СН3СОО)2 = 2ВаСО3 + 2(СН3)2СО; 2(СН3)2СО =
= ЗСН4 + 2СО + С.
Цементация в таком карбюризаторе ускоряется благодаря
радикальному изменению состава газовой фазы в цементацион-
ном ящике (табл. 5).
56
Таблица 5
Содержание метана, окиси углерода и водорода в газовой среде,
образующейся при применении различных карбюризаторов [61]
Состав карбюризатора Состав газовой среды в %
СН4 СО н2
70% древесного угля; 20% ВаСО3; 7% Na2CO3; 3% мазута 0,0 25,7 17,3
90% полукокса; 10% ацетата натрия 11,0 23,3 41,8
Рекомендуется проверить в производственных условиях кар-
бюризаторы следующих составов: 10—15% древесного угля, 78—
80% полукокса, 5—10% ацетата бария или 90—95% полукокса
и 5—10% ацетата бария [61];
За рубежом также исследованы карбюризаторы с ацетатом
бария, состоящие из зерен угля, пропитанных ацетатом бария,
и кокса [62].
Контакт между карбюризатором и деталью необязателен.
Газовая среда в цементационном ящике достаточно активна, что-
бы мог образоваться почти нормальный цементованный слой у
деталей, находящихся на некотором расстоянии от карбюризато-
ра. В связи с этим был предложен способ раздельной укладки в
ящике карбюризатора и деталей. При этом поверхность деталей
после цементации получается чистой, почти блестящей [63]. Одна-
ко данные о большом преимуществе в скорости цементации при
применении этого способа укладки деталей являются, судя по
нашим опытам, сильно преувеличенными.
Температуру цементации в твердом карбюризаторе поддер-
живают в пределах 910—950° С. В последние годы в некоторых
случаях применяют высокотемпературную цементацию при 980—
1000° С. При этом процесс ускоряется в 1,5—2 раза. Получения
крупного зерна после такой цементации йожно избежать, приме-
няя наследственно мелкозернистую сталь или правильно подби-
рая режимы последующей термической обработки. Пересыщение
стали углеродом при высокой температуре процесса также легко
устраняется подбором карбюризатора необходимой активности;
кроме того, надо иметь в виду, что обычно с повышением темпе-
ратуры цементации содержание углерода в слое уменьшается
(рис. 27).
Механические свойства стали, подвергнутой высокотемпера-
турной цементации, после закалки с повторного нагрева обычно
не снижаются (табл. 6). Эти свойства часто не ухудшаются да-
же тогда, когда заметно вырастает зерно стали. Объясняется это
57
тем, что увеличение однородности твердого раствора оказывает
в ряде случаев большее влияние на вязкость стали, чем некрто-
рый рост зерна.
Таблица 6
Механические свойства сталей 20Х, 20ХГР и 12ХНЗА,
подвергнутых цементации при разных температурах
(А. М. Тарасов и М. В. Семенченко)
Марка стали Режим цемен- тации Темпе- ратура в °C Механические свойства образцов
ложно-цементованных цементованных
Температура в °C Продолжи- тельн ость в ч закалки отпуска ав и ю и ан HRC поверхность HRC сердцевина ан
С ХЮ’ н/м2 м 2 1 о X кГ-MjcM2 - Л xi м д к I ХЮ» 1 нм/м*
20Х 910 980 12 7 870 200 136 139 133 136 159 162 155 159 12 12 51 51 5,5 5,9 5,4 5,8 60—63 59—63 42-43 43 0,9 1.3 0,8 1,2
20ХГР 910 980 12 7 870 200 133 130 130 128 152 150 149 147 12 12 54 53 10,0 10,3 9,8 10,0 60—63 61-63 4 2—43 43 0,8 1,1 0,7 1,0
12ХНЗА 880 1000 20 10 800 160 — 103 105 100 103 — 58.8 61,5 13,8 14,1 13,5 13,8 — — — —
Применяя в промышленности высокотемпературную твердую
цементацию, следует учитывать, что при этом усиливается короб-
ление деталей и ускоряется выход из строя цементационных
ящиков и печей.
Зависимость продолжительности цементации в твердом кар-
бюризаторе от требуемой глубины слоя показана на рис. 32.
Ниже приведены данные, характеризующие глубину слоя при
твердой цементации в больших ящиках при температуре 920—
930° С в зависимости от продолжительности процесса:
Глубина слоя Продолжитель-
в мм ность в ч
0,4—0,7 4—5
0,6—0,9 5,5—6,5
0,8—1,2 6,5—10
1,0-1,4 8—11,5
1,4—1,8 11,5—16
2,0—2,4 19—24
Цементация высокохромистых нержавеющих сталей (1X13,
Х17, Х17Н2 и др.) протекает медленнее, чем обычных машино-
строительных сталей. При цементации этих сталей рекомендует-
ся применять более высокие температуры (950—1000° С) и более
активные карбюризаторы [52], чтобы достигнуть необходимого
насыщения поверхности стали углеродом (2,5—3,5%). При ис-
пользовании менее активных карбюризаторов [53] твердость пос-
58
ле закалки не превышает HRC 57—58. Кроме того, рекомендует-
ся защищать поверхность деталей, чтобы предотвратить образо-
вание плотной пленки -окислов хрома, препятствующей
проникновению углерода в сталь и возникающей при медленном
нагреве деталей в карбюризаторе. Для защиты поверхности
предлагается фосфатирование деталей [51] либо их обмазка гра-
фитом или специальной пастой [52].
Цементация в твердом карбюризаторе с нагревом т. в. ч. Цемен-
тация стальных деталей в твердом карбюризаторе протекает
интенсивно при быстром нагреве т. в. ч. и высоких темпера-
турах.
Первая работа по цементации стали с индукционным нагре-
вом была выполнена еще в 1938 г. [64]. Позднее цементация с
нагревом т. в. ч. была освещена в работе [65] (рис. 33).
Рис. 32. Зависимость
глубины цементован-
ного слоя хромонике-
лемолибденовой стали
от продолжительно-
сти и температуры
твердой цементации в
карбюризаторе (65%
древесного угля, 23%
кокса, 12% ВаСОз)
Рис. 33. Зависимость глуби-
ны цементованного слоя
стали от продолжительно-
сти процесса цементации в
твердом карбюризаторе с
нагревом т. в. ч. [65]
Изучена высокотемпературная (1050—1100° С) цементация
стали в твердом карбюризаторе с нагревом т. в. чь в специально
приспособленных для этого индукционных плавильных печах [66].
Существенный недостаток этого метода цементации — раз-
личие в глубине слоя у разных деталей вследствие перепада
температуры по объему тигля.
Цементация в твердом карбюризаторе в плавильных печах
или стаканах с нагревом т. в. ч. едва ли может быть рекомен-
дована.
59
Цементация в пастах. При индивидуальном производстве
иногда применяют цементацию в пастах, позволяющую сокра-
тить продолжительность процесса. Детали покрывают слоем
пасты толщиной 3—4 мм и загружают в ящики, кромки крышек
которых тщательно промазывают огнеупорной глиной. В состав
большинства паст входит газовая сажа, углекислые соли и же-
лезосинеродистый калий, которые разводятся канцелярским
клеем, жидким стеклом, маслом и другими веществами. Соста-
вы некоторых паст приведены в табл. 7. Из-за наличия почти во
всех пастах железосинеродистого калия поверхностный слой
стали немного насыщается азотом.
Таблица 7
Составы паст для цементации
Составляющие паст
Содержание в%
Газовая сажа
Na2CO3
BaCOg
K4Fe (CN)e
27*
3,5
30—60
20—40
Веретенное масло
1,5
68
10—20
50
15
20
15
* При использовании этой пасты за 1 ч при температуре 920—
930° С глубина слоя достигает 1 мм, а твердость после закалки
HRC 56-60 [67].
Цементация в пастах с нагревом т. в. ч. Этот метод цемента-
ции после соответствующей доработки (подбора составов об-
мазок) несомненно будет применен в мелкосерийном и единич-
ном производстве. Метод несложен, позволяет быстро получать
цементованные слои большой глубины и производить местную
цементацию. При использовании пасты, состоящей из порошка
древесного угля, замешанного на гидролизованном этилсили-
кате, в течение 1 мин при температуре 1200° С был получен
цементованный слой глубиной 0,46 мм (причем заэвтектоидная
зона составляла 0,37 мм) с твердостью после закалки HRC 62.
Слой был сильно пересыщен углеродом [68].
Предложено также несколько составов паст для нитроцемен-
тации с нагревом т. в. ч., которые рассмотрены в гл. IV.
Газовая цементация
Возможность цементации стали в газовой среде была пока-
зана еще в работе П. П. Аносова, выполненной в 1837 г. Однако
только почти через 100 лет (в 1935 г.) этот процесс начали
впервые внедрять в производство в высокопроизводительных
муфельных печах непрерывного действия на автозаводе
60
им. Лихачева. При этом в качестве газового карбюризатора была
использована среда, получаемая при пиролизе и крекинге керо-
сина [69], [19].
Для газовой цементации пока еще часто применяют шахт-
ные муфельные печи и печи непрерывного действия с длинными
горизонтальными муфелями из окалиностойкого сплава. Изред-
ка применяют также печи с вращающимися ретортами.
В последние годы начали получать все большее распростране-
ние безмуфельные печи непрерывного действия, нагреваемые
излучающими трубками из стали Х23Н18 или Х18Н25С2. Увели-
чивается выпуск шахтных безмуфельных печей типа ШЦН
с рабочей температурой до 1050° С [564].
Детали загружают в печи на поддонах (в корзинах) или в
различных приспособлениях, на которых они располагаются, на
расстоянии 5—10 мм между цементуемыми поверхностями;
мелкие детали загружают навалом на этажерки, помещаемые в
корзины.
В массовом производстве применяют автоматические агре-
гаты, включающие в себя безмуфельные однорядные или двух-
рядные печи, закалочный бак, моечную машину и отпускную
печь. Производительность таких агрегатов колеблется от 170
до 920 кг/ч .
Для газовой цементации используют различные карбюриза-
торы— газы: природный (92—97% СН4)*; природный, разбав-
ленный для городских нужд (65—90% СН4); светильный (20—
35% СН4, 5—25% СО); нефтяной (50—60% СН4); коксовый
(20—25% СН4, 4—10% СО); сжиженные (пропан С3Н8, бу-
тан С4Н10, пропано-бутановая смесь), а также получаемые при
газификации синтина, осветительного керосина (желательно
грозненского месторождения), бензола, пиробензола, веретен-
ного и трансформаторного масел и т. д. Из жидких карбюриза-
торов рекомендуется применять синтин, не содержащий
сернистых соединений и при разложении выделяющий меньше
сажи по сравнению с другими жидкими карбюризаторами [70].
Жидкие карбюризаторы подают в печи в виде капель (шахтные
печи с вентиляторами) или, чаще, в пылевидном состоянии фор-
сунками, соединенными с двух- или четырехплунжерным на-
сосом.
Сложные углеводороды, которые входят в состав карбюри-
заторов или образуются при их разложении в результате ряда
промежуточных реакций, распадаются в основном до метана.
При крекинге углеводородов, который производится для сни-
жения их активности или для получения эндогаза, образуется
также СО. Таким образом, химизм выделений атомарного
* Использование природного газа, пропана и бутана без предварительной
переработки не оекомендуется.
61
углерода при газовой цементации сводится в основном к распа-
ду метана (СН4 = С + 2Н2) и окиси углерода (2СО = СО2 + С).
Диаграммы, приведенные на рис. 31, показывают, что СН4
является более активным карбюризатором, чем СО. Для наугле-
роживания железа при 900—1000° С в смеси СН4—Н2 доста-
точно наличия всего лишь нескольких процентов СН4, тогда
как для цементации в смеси СО—СО2 необходима концентра-
ция около 95—97% СО.
Рис. 34. Диаграмма Неймана равновесного состояния между цементующими
атмосферами и сталью; аа — граница атмосфер, образующих сажу; по краям
диаграммы — константы равновесия основных уравнений [71]
Сравнение диаграмм равновесия железа с газовыми средами
СО—СО2 и СН4—Н2 показывает, что в первой смеси содержа-
ние углерода в железе, находящемся в равновесии с газовой
фазой постоянного состава, уменьшается с повышением темпе-
ратуры, а во второй смеси увеличивается. Следовательно, с по-
вышением температуры науглероживающая способность смеси
СО—СО2 определенного состава уменьшается, а смеси СН4—Н2
растет.
На рис. 34 дана диаграмма равновесного состояния между
цементующими атмосферами и углеродосодержащими фазами
железа, показывающая концентрацию углерода в аустените
в зависимости от парциального давления в атмосферах Н2—СН4
и СО—СО2, а также границу аа между атмосферами, в которых
выделяется сажистый углерод или в которых углерод находится
в составе газообразных соединений.
62
Рассмотрим примеры применения этой диаграммы [71].
1. При каких температурах газовые смеси Н2 и СН4 с константами равно-
весия 10 и 0,5 не выделяют сажи?
Ответ. Смесь с константой 10 не выделяет сажи до температуры 775° С, а
с константой 0,5 — до температуры 530° С.
2. До какого содержания будет насыщаться углеродом аустенит при 925° С
в смеси Н2 и СН4 с константой равновесия 200?
Ответ. До содержания всего лишь 0,2—0,4% С. Для насыщения аустенита
углеродом при этой температуре, например, до 0,8% константа равновесия сме-
си должна быть 60.
Вопрос о самообразовании при газовой цементации богаты-
ми газами (природный, бутан, пропан, газы, получаемые из
керосина, масел и т, д.) имеет большое практическое значение.
На деталях и на стенках муфелей выделяются сажа и кокс,
что влияет на скорость процесса цементации и ведет к быстрому
прогоранию муфелей и излучающих трубок из-за местных пере-
гревов. Чтобы предотвратить выделение сажи, на некоторых
заводах в печи непрерывного действия дополнительно вводят
аммиак. Добавление аммиака в количестве 2,5—5% несколько
ускоряет процесс цементации. По данным ЗИЛ, рекомендуется
в большие прямоточные безмуфельные печи вводить 80—90%
эндогаза, 5—8% природного газа и 2—3% аммиака [44].
Механические свойства деталей значительно повышаются,
если содержание углерода в поверхностной зоне слоя не пре-
вышает 0,75—0,85%. При этом в слое не образуется избыточных
карбидов, а содержание остаточного аустенита минимально.
Такое содержание углерода в поверхностной зоне слоя может
быть достигнуто лишь при .автоматической регулировке состава
газа и применении газа-разбавителя, например эндогаза. Этот
газ получают в эндотермическом генераторе [72], [73]. Светиль-
ный газ, пропан, бутан или пропано-бутановые смеси и воздух
подаются в определенной пропорции в вертикальную реторту,
нагреваемую до 950—1050° С и наполненную катализатором
ГИАП-3. Верхняя часть реторты охлаждается с помощью водя-
ной рубашки, что необходимо для предотвращения реакции
2СО = СО2 + С и получения газа, содержащего около 20% СО
и менее 1 % СН4.
Эндотермический газ без обогащения углеводородами
является нейтральным (табл. 8). Углеродистая сталь может
находиться в равновесии с этой средой лишь при определенном
и весьма незначительном содержании в ней Н2О и СО2. Так
как при изменении содержания в газовой среде одного из этих
компонентов соответственно изменяется и содержание другого,
контроль среды удобно вести по количеству водяных паров,
т. е. по точке росы. Построены эмпирические кривые равнове-
сия различных сталей со средой, имеющей различную точку
росы [72]—[73]. На основании этих данных градуируют автома-
тические регуляторы концентрации углерода в цементованном
63
слое, которые связаны с дозирующими клапанами, изменяющи-
ми соотношение газа и воздуха, подаваемых в генератор.
Таблица 8
Состав газа, полученного из смеси городского газа (природного)
и воздуха в результате эндотермической реакции,
протекающей при наличии катализатора ГИАП-3 [44]
Газ Состав в %
о, со со2 н2 N, сн4
Г ородской <0,2 <2 <0,4 <10 <3 85—95
Газовоздушная смесь 14,5—16 <1 — <3 54—56 27—30
Эндотермический <0,2 19—23 <0,2 39—44 33—37 <1
Промышленность начала выпускать газоанализаторы, с боль-
шой точностью определяющие содержание в газе СО2. Они
более удобны в работе, чем приборы, контролирующие состав
газа по точке росы.
В шахтных печах насыщение стали углеродом контролирует-
ся прибором, принцип действия которого основан на том, что
при постоянной температуре электросопротивление стали яв-
ляется линейной функцией содержания углерода в аустените.
В качестве датчика применяют тонкую железную проволоку
диаметром 0,1 мм, содержание углерода в которой почти
безынерционно изменяется в соответствии с изменением угле-
родного потенциала атмосферы в печи. Прибор дает команду
устройству, подающему жидкий карбюризатор в печь. Такой
прибор может быть применен только при использовании мало-
сажистых сред. Он был опробован на Челябинском тракторном
заводе и на ряде других заводов.
Давление в печах для газовой цементации обычно не превыша-
ет 50 мм вод. ст. (490 н/м2). Повышая его до 5—8 ат
(490—785 кн/м2), можно ускорить цементацию [75]. Однако для
проведения процесса под давлением необходимы сложные и до-
рогостоящие установки, что пока исключает рациональность
применения такого способа цементации. Количество газа или
жидкого углерода, подаваемое в цементационную печь, зави-
сит от ее размера и конструкции и оказывает влияние на ско-
рость и равномерность цементации, на содержание углерода
в цементованном слое и на выделение на детали и стенках пе-
чи сажи и кокса. Скорость газовой цементации значительно из-
меняется также в зависимости от величины садки и условий
проведения процесса и обычно превышает скорость цементации
в твердом карбюризаторе не более чем на 10—20%.
Примерные расходы жидких и газовых карбюризаторов при
64
цементации в шахтных печах приведены в работе {76]. Напри-
мер, в печи Ц-60 и ШЦН-65А рекомендуется подавать 60—90
капель в минуту синтина, керосина, пиробензола или около
0,9—1,2 м31ч газообразных карбюризаторов, в печи Ц-105 и
ШЦН-95А соответственно 120—150 капель в минуту или 1,2—
1,6 м3/ч. При нагреве и охлаждении садки расходы карбюри-
заторов уменьшаются в 2 раза и более.
Составы отходящих из печи газов колеблются в широких
пределах. Например, в случае применения керосина в шахтной
печи Ц-205 отходящий газ имеет следующий состав: 2—6% СН4;
20-25% СО; 55—70% Н2; 0,1 —1,0% СО2; 0,2—0,8% О2;
8—12% N2.
Чтобы ускорить насыщение поверхностных слоев стали уг-
леродом, в первый период процесса (30—50% общей продол-
жительности) на некоторых заводах в шахтные печи подают
больше жидкого и газового карбюризатора, а во второй период
для снижения концентрации в слое углерода уменьшают рас-
ход. Поступают и иначе: в первый период вместе с газом-разба-
вителем вводят 8—10% природного газа, 3—5% пропана или
2—3% бутана, а во второй период подают лишь газ-разбавитель.
В результате диффузии углерода в глубь стали и взаимодействия
с газом-разбавителем достигается снижение содержания углеро-
да в поверхностной зоне цементованного слоя до требуемой кон-
центрации.
В печах непрерывного действия такой же эффект достигает-
ся подачей жидких карбюризаторов только в загрузочную зону,
вследствие чего к разгрузочной зоне печи приходит уже сильно
обедненный газ. При подаче в такие печи карбюризатора в не-
скольких точках более богатый карбюризатор или большее его
количество вводят у загрузочной зоны печи.
Однако полученные в последнее время данные показывают,
что применение цементации с понижающимся к концу процесса
углеродным потенциалом атмосферы не всегда может быть
рекомендовано (см. стр. 68) [77].
Наиболее быстро газовая цементация протекает в печах
с вращающимися ретортами: При 910° С в течение 2 ч 45 мин
глубина слоя достигает 0,8—0,9 мм, а в течение 3 ч 30 мин 1,0—
1,15 мм. На одном из заводов в шахтной печи Ц-105 с вентиля-
тором при садке 0,8—1,2 т, температуре 930° С и применении
природного газа слой глубиной 1,0—1,2 мм получается в тече-
ние 8—9 ч (нагрев длится 3 ч). На другом заводе, использую-
щем веретенное масло, в эту же печь загружают лишь 0,6 т,
при этом продолжительность выдержки для получения слоя той
же глубины сокращается до 7 ч (нагрев длится 3 ч). Таким
образом, в зависимости от величины садки,, толщины деталей
и состава карбюризатора продолжительность процесса может
изменяться в значительных пределах. ,
65
Высокохромистые нержавеющие стали (1X13, Х17, Х17Н2
и др.) рекомендуется подвергать газовой цементации при 950° С
в шахтных печах с применением, например, пиробензола.
Цементация этих сталей протекает медленнее, чем конструк-
ционных (54].
Продолжительность процесса цементации в больших без-
муфельных прямоточных печах, применяемых на ЗИЛе, приве-
дена в табл. 9.
Таблица 9
Продолжительность газовой цементации при 950°С стали 25ХГТ
в безмуфельной печи ЗИЛа
(состав газа приведен в табл. 8)
Глубина слоя в мм Продолжительность процесса
без добавки аммиака | с добавкой аммиака
0,5—0,7 6 Ч 5 Ч 15 MUH
0,6—0,9 7 > 6 » 35 »
0,9—1,3 9 > 7 » 30 >
1,2—1,6 12 > 9»
Для крупногабаритных деталей, применяемых в тяжелом
машиностроении (кольца и шары подшипников, детали шагаю-
щих экскаваторов и т. д.), продолжительность цементации до-
стигает десятков часов (78]. Крупногабаритные детали обраба-
тываются в больших шахтных печах Ц-250 (садка 2—4 т) при
930° С, например, в газах, получаемых при разложении пиро-
бензола.
Ниже указана продолжительность выдержки для получения
на стали 12Х2Н4А цементованного слоя определенной глубины.
Глубина слоя в мм Выдержка в ч
3—3,4 36
6—6,4 84
7—7,4 108
8,4—9,0 144
(Продолжительность прогрева садки 7—9 ч)
В последнее время благодаря более детальному изучению
структуры и химического состава цементованного слоя в их
связи с механическими свойствами были обнаружены новые
важные закономерности, вынуждающие нас с новых позиций
подходить к разработке режимов цементации, обеспечивающих
получение оптимальных механических свойств у деталей, рабо-
чая поверхность которых после цементации не подвергается
шлифованию. К таким деталям относятся, например, зубчатые
колеса, изготовляемые в больших количествах во многих от-
раслях промышленности.
66
Известно, что оптимальные характеристики прочности цемен-
тованной стали получаются при определенном насыщении угле-
родом поверхностной зоны цементованного слоя. По данным
разных авторов, для различных конструкционных сталей это
содержание углерода колеблется в пределах 0,75—1,1%. Однако
указанное условие является недостаточным для получения
у цементованной стали оптимальных свойств. Важным обстоя-
тельством является технология цементации, при которой в слое
была достигнута оптимальная концентрация углерода.
В промышленности широко применяется газовая цементация,
при которой углеродный потенциал атмосферы вначале поддер-
живается высоким, обеспечивающим получение в поверхностной
зоне слоя, например, концентрации в 1,3—1,5% С, а затем угле-
родный потенциал атмосферы снижается с тем, чтобы получить
в этой зоне оптимальное содержание . углерода, например,
0,8%.
Такое ступенчатое изменение углеродного потенциала атмо-
сферы позволяет сократить продолжительность процесса и по-
этому нашло широкое применение.
При цементации в методических печах непрерывного дей-
ствия перед закалкой часто применяют подстуживание деталей
с 930—950° С до 830—850° С в течение 30—35 мин при понижен-
ном углеродном потенциале атмосферы. После цементации
в шахтных печах обычно детали охлаждают на воздухе, а по-
следующую закалку проводят в печах без защитной атмосферы.
Во всех перечисленных случаях в конце процесса наблюдается
понижение концентрации углерода в слое.
Однако было установлено, что любое понижение концентра-
ции углерода в цементованном слое многих легированных
сталей, в том числе понижение его концентрации до оптималь-
ной, всегда приводит к уменьшению прочности этих сталей.
Ниже приведены данные механических испытаний образцов
сталей 20ХНМ и ЗОХГТ, обработанных по трем режимам [77]:
1) при постоянном углеродном потенциале атмосферы
(0,8% С) в течение всего процесса и при подстуживании с 930
до 820° С;
2) в первый период насыщения углеродный потенциал атмо-
сферы поддерживался равным 1,3—1,4% С, а во второй период
и при подстуживании — 0,8% С;
3) в течение всего периода насыщения углеродный потен-
циал атмосферы равен 1,3—1,4% С, а при подстуживании —
0,8% С.
На рис. 35 приведены результаты определения предела проч-
ности при изгибе и предела выносливости образцов, обработан-
ных по указанным выше режимам и имеющих в поверхностном
слое различную концентрацию углерода. Рассмотрение этих
кривых позволяет сделать следующие выводы:
67
1) подтверждается явление снижения прочности стали с уве-
личением в цементованном слое углерода;
2) при широко применяемой в настоящее время цементации
по третьему режиму (в муфельных или шахтных печах с газо-
вой средой, богатой углеводородами, снижение концентрации
углерода в слое до оптимального значения при подстуживании
под закалку) прочность стали на 25—30% ниже, чем прочность,
достигаемая при оптимальном (первом) режиме.
Рис. 35. Предел прочности при изгибе и предел выносливости стали ЗОХГТ,
цементованной по трем режимам (/, 2 и 5), с различным содержанием углеро-
да в поверхностном слое [77]
При цементации по второму режиму прочность выше, чем
при цементации по третьему режиму, и несколько ниже, чем
при цементации по оптимальному (первому) режиму.
Такое влияние обезуглероживания на механические свойства
объясняется тем, что в сталях, легированных карбидообразую-
щими элементами, снижение концентрации углерода в слое
происходит в основном за счет обеднения углеродом твердого
раствора поверхностных зон слоя (см. стр. 93). Поэтому макси-
мальная концентрация углерода в твердом растворе оказывает-
ся сдвинутой в глубь слоя. Уменьшение концентрации углерода
в твердом растворе поверхностной зоны слоя вызывает сниже-
ние напряжения сжатия на поверхности, а следовательно, пони-
жение предела выносливости стали. В связи с этим во второй
стадии процесса и тем более в конце процесса нельзя допускать
68
обезуглероживания деталей, не подвергаемых после цементации
шлифованию.
За рубежом некоторое распространение получила цемента-
ция в садочных печах с подачей в них двух жидкостей: более
бедной, из которой образуется газ-носитель, и богатой, обеспе-
чивающей заданную науглероживающую активность атмосферы.
В качестве газов-носителей применяют, например, метанол
(СН3ОН) и изопропил (С3Н7ОН). Первая жидкость при кре-
кинге дает газ состава: 32% СО; 65,5% Н2 и 0,68% СН4; а вто-
рая жидкость — газ состава: 19% СО; 78% Н2 и 1% СН4. В ка-
честве богатого газа применяют, например, бензол [79]. Приме-
няются также смеси из 80% изопропилового спирта и 20%
бензола (карбодрип) или из 40% терпинтинового масла, 30%
ацетона и 30% этилового спирта.
Высокотемпературная газовая цементация стали в печах.
Газовую цементацию обычно ведут при 910—930° С, а на ряде
заводов без заметного ущерба для качества изделий при 940—
960° С. В ряде случаев рационально устанавливать еще более
высокую температуру (980—1000° С), но закалку необходимо
проводить как самостоятельную операцию. При этом условии
механические свойства стали после высокотемпературной и
обычной (920° С) газовой цементации практически одинаковы
(табл. 10), а продолжительность процесса во втором случае
Таблица 10
Механические свойства стали после обычной
и высокотемпературной газовой цементации*
(Н. А. Мишин)
Марка стали Испытания на растяжение Испытания на изгиб
о в 8 в % Ф в % а н Стрела прогиба в мм • Газрушающая на- грузка в кГ
а? С 5. о X в * 5 х о X
18ХГТ 146** 143 7,2 48* 6,6 6,4 1,25 2272
141 138 5,6 50 7,3 7,1 1,30 3522
12XH3A 130 127 L3 53 8,1 7,9 1,20 2030
131 128 7,5 53 9,9 9,7 1,25 2720
20Х 128 125 1,7 36 5,6 5,4 1,0 2050
134 131 4,2 44 6,5 6,3 1,5 2980
* Дальнейшая обработка: охлаждение на воздухе, одинарная закалка с 850° С и низкий отпуск. * * В числителе приведены данные для обычной цементации (920° С, 15 ч), в знаменателе — для высокотемпературной (1000° С, 8 ч).
69
значительно меньше: 8 ч вместо 15 ч при глубине цементации
1,0—1,3 мм и 12 ч вместо 20 ч при глубине 1,5—1,8 мм.
Однако при внедрении в производство высокотемпературной
цементации следует учитывать, что этот процесс вызывает боль-
шее (чем обычная цементация) коробление деталей, и поэтому
его нельзя рекомендовать для обработки деталей любой формы
и назначения. В частности, таким способом нельзя обрабаты-
вать ответственные зубчатые колеса сложного профиля, так как
после цементации их не шлифуют. Для уменьшения коробления
можно использовать ступенчатую закалку в горячих средах
(180—200°С). Однако такая закалка способствует сохранению
в слое остаточного аустенита. Чтобы избежать этого, в цемен-
тованном слое должно быть не более 0,8% С.
Для высокотемпературной цементации с непосредственной
закалкой следует применять мелкозернистые стали 18ХГТ,
25ХГТ и др. Зерно этих сталей при нагреве до 1050° С несколь-
ко возрастает, но механические свойства не снижаются, так как
увеличивается однородность твердого раствора; лишь при тем-
пературе минус 60° отмечается небольшое снижение ударной
вязкости (А. Д. Ассонов).
Высокотемпературная газовая цементация — прогрессивный
процесс, применение которого непрерывно расширяется.
Впервые высокотемпературная цементация была внедрена на
отечественных заводах, в последние годы она рекомендуется
также за рубежом [80]—[82], [588].
Цементация с нагревом т. в. ч. Этот способ цементации пред-
ложен на ЗИЛе, где в 1954 г. был создан первый промышлен-
ный агрегат для цементации зубчатых колес с нагревом т. в. ч.
[65], {83]. Ускорение цементации в этом агрегате достигается
благодаря более быстрому нагреву зубчатых колес и высокой
температуре процесса.
Основной частью агрегата является печь, в которой проте-
кает цементация в смеси природного газа и крекированного
газа (или эндогаза). Индуктор, расположенный в центре печи,
представляет собой многовитковую катушку из медной трубки,
питаемую от генератора мощностью 50 кет, дающего ток часто-
той 2000 гц. Внутри индуктора имеется футеровка в виде кера-
мических стаканов, удерживающих деталь на определенном
расстоянии от индуктора и предохраняющих его от теплового
излучения.
Весь процесс работы агрегата автоматизирован. Темпера-
тура деталей определяется фотопирометром. Темп толкания
заготовок 1,5—3 мин. В течение 1 ч цементуется 20—40 зубча-
тых колес (100—120 кг). При 1080° С цементованный слой глуби-
ной 0,8—1,2 мм на деталях из стали 18ХГТ получается за 40—
45 мин, после чего следует подстуживание до 850° С и закалка
в масле. В муфельной печи непрерывного действия для полу-
70
чения слоя указанной глубины процесс при 930° С продолжает-
ся 8—10 ч.
При высокотемпературной цементации деталей из сталей
18ХГТ и ЗОХГТ механические свойства этих сталей не
снижаются.
Если требуется большая производительность, то целесообраз-
нее применять аналогичные двухручьевыё агрегаты производи-
тельностью 200—240 кг/ч (потребляемая мощность ПО—120 кет
и частота тока 2500 гц).
Позднее исследования по газовой цементации с нагревом
г. в. ч. были выполнены на Горьковском автозаводе и в Научно-
исследовательском институте токов высокой частоты [84]. Бы-
ли также сконструированы агрегаты для цементации зуб-
чатых колес трактора, колец подшипников и других деталей
[565].
Вопрос о использовании индукционного нагрева для газовой
цементации в термических цехах массового производства пока
еще не решен. Хотя агрегат, опробованный на ЗИЛе, работал
в течение нескольких лет в основном успешно, его производи-
тельность была меньше производительности прямоточных без-
муфельных печей, а цементованные в нем зубчатые колеса
имели несколько повышенное коробление (однако в пределах
допуска). ЗИЛ сконструировал новый более совершенный
агрегат такого типа.
Комплексные агрегаты с полным циклом обработки, в кото-
рые входят не только печь для газовой цементации с нагревом
т. в. ч. и закалочный бак, но и моечная машина и отпускная
печь, рационально применять в потоках механических цехов
и в автоматических линиях. Возможно, перспективными окажут-
ся установки непрерывного действия, в которых нагрев поверх-
ностных слоев стали осуществляется т. в. ч. (с использованием
всех преимуществ такого нагрева), а температура в зоне изо-
термической выдержки поддерживается электрическим или
газовым нагревом.
Ионная цементация [85], [566]. При газовой цементации с на-
гревом в тлеющем разряде в вакууме ионы газа бомбардируют
и быстро нагревают поверхность стали до высокой температуры.
Поэтому на процесс затрачивается почти столько же времени,
сколько и при нагреве т. в. ч. Этот способ нагрева для цемента-
ции, по-видимому, не является перспективным. Однако возмож-
но, что он будет с успехом использован для некоторых других
процессов (см. стр. 121).
Газовая цементация кислородно-ацетиленовым пламенем.
При этом методе цементации рекомендуется [86], [87] применять
инжекторные горелки, например, типа СУ-48 с диаметром вы-
ходного отверстия 1 мм. При нагреве поверхности до 1100—
1150° С в течение 5 мин (3 мин цементация и 2 мин нагрев
71
в нейтральном газе) получается слой глубиной 0,38 мм; в течение
15 мин — глубиной 0,68 мм и 20—30 мин — глубиной 0,8—1,0 мм.
Этот метод цементации описан и в зарубежной литературе [88].
Цементация в жидкой среде
Цементация стали в соляной ванне состава: 78—85% Ма2СОз;
10—15% NaCl и 6—8% SiC (карборунд) получила небольшое
применение (89]—(90].
В ванне протекает реакция
2Na2CO3 + SiC = Na2SiO3 + Na2O + CO + C
ИЛИ
3Na2CO3 -p SiC — Na2SiO3 -p 2Na2O -p 4CO;
2CO = C + CO2.
Каждые 3 ч ванну освежают, добавляя около 0,5% (от веса
солей) карборунда (зерно № 26—50); кроме того, добавляют
углекислый натрий до требуемого уровня ванны. Один раз
в 1—1,5 месяца ванну полностью заменяют. В такой ванне при
880—900° С в течение 30 мин получается общий слой глубиной
0,15—0,20 мм и эвтектоидный слой глубиной 0,07—0,10 мм,
HRA 72—78. Более продолжительные процессы в такой ванне
вести нерационально. Для получения более глубоких слоев
в Экспериментальном научно-исследовательском институте ме-
таллорежущих станков (ЭНИМС) (91] разработана аналогичная
ванна с добавкой хлористого аммония. В этом случае сталь
насыщается также азотом (см. подробнее стр. 138).
Цементация в ванне с карборундом имеет преимущество
перед цианированием в цианистых ваннах, так как применяемые
в ней соли не ядовиты; существенный недостаток этого вида
цементации — неравномерность глубины слоя. Тем не менее
эти ванны (без добавок к ним хлористого аммония) в течение
ряда лет применяются на заводе Станконормаль, Московском
заводе пишущих машин и др.
Для улучшения технологичности ванны (повышения жидко-
текучести и устранения шлаковой корки на поверхности распла-
ва) предложено заменить в ней Na2CO3 на К2СО3 [567].
Разработан также автоматический агрегат с программным
управлением для жидкостной цементации и закалки деталей
(емкость тигля 200 кг расплава, производительность 3100—
3900 кг в сутки) (91].
Худшие результаты по скорости процесса, чем в описанной
выше ванне, достигаются в расплаве, содержащем 44% NaCl,
43% КС1, 6% NaOH и 7% порошка древесного угля (в ванне
образуется СО). Вместо части древесного угля в ванну можно
добавить карбид кремния. Несколько быстрее протекает цемен-
тация в ванне, содержащей 65% Na2CO3, 15.% NaCl, 9% порош-
ка высокоуглеродистого ферромарганца и 3% порошка древес-
%
ного угля ([92]. Недостатками этих двух последних ванн
являются быстрая их истощаемость, трудность замешивания
древесного угля; кроме того, последняя ванна склонна к за-
густеванию. Все это препятствует распространению этих ванн
в промышленности.
Цементация в соляной ванне с применением ультразвука.
Эффект ускорения «твердой» цементации под воздействием
ультразвуковых колебаний известен еще из прежних работ
[93]—[95]. Влияние ультразвука на цементацию в жидкой среде
(Na2CO3, NaCl, SiC) было изучено в ЭНИИМСе (Б. Н. Бату-
рин). Облучение ванны ультразвуком осуществлялось при
помощи системы магнитострикционных излучателей с волново-
дами мощностью 1,5 кет и частотой 23,7 кгц. Воздействие
ультразвука привело к повышению скорости цементации при
870° С в 1,5—2,0 раза. Это ускорение связано главным образом
с интенсивным перемешиванием расплава солей под действием
ультразвуковых колебаний.
Электролизная цементация. Этот метод цементации [19] за-
ключается в электролизе расплавленных карбонатов щелочных
и щелочно-земельных металлов (Na2CO3, ВаСО3 и др.). На
катоде (детали) выделяется Na или Ва, которые восстанавли-
вают СО2, образующийся в расплаве солей, до СО и С. В ванне,
содержащей 1 часть ВаСО3 и 1 часть ВаС12, в течение 4 ч при
955° С образуется слой глубиной 1,6 мм. Этот метод цементации
не получил пока промышленного применения.
Цементация с нагревом в электролите. Для этого процесса
используют те же установки, что и для обычного нагрева стали
в электролите, с той лишь разницей, что в состав электролита
вводят вещества, диссоциирующие с выделением углерода.
И. 3. Ясногородский опробовал электролит с 15% глицерина
и 10% сахара, обеспечивающий большую скорость цементации.
Активность процесса объясняется выделением углерода в иони-
зированном состоянии и большой концентрацией ионов у по-
верхности стали, а также воздействием на поверхность искрово-
го разряда. Позднее В. С. Ваниным был рекомендован более
дешевый электролит (80% ацетона и 20% воды) [76].
Цементация с нагревом т. в. ч. В 1939 г. М. Г. Лозинский
доказал возможность цементации стали, нагреваемой т. в. ч.,
при погружении ее в керосин. Позднее в Польше [96], [97]
и в ФРГ [98] были проведены опыты по цементации вначале
в керосине, а затем в метиловом спирте, который выделяет
меньше сажи. Газовая рубашка, образующаяся при разложении
метилового спирта вокруг нагретой т. в. ч. детали, имеет
следующий состав: 28% СО; 4,2% СН4; 61,5% Н2; 2,0% О2,
другие составляющие — 4,3%. Скорость цементации в парах
метилового спирта с нагревом т. в. ч. близка к скорости газовой
цементации с нагревом т. в. ч.
73
Большая работа по изучению цементации стали в различных
жидкостях (в том числе в синтине) с нагревом т. в. ч. выпол-
нена в НИИТракторосельхозмаше Г. 3. Словоохотиевым. Были
проведены опыты по цементации в толуоле, этиловом спирте [99],
уайт-спирите и синтине [100]. Авторы последней работы пришли
к выводу, что в ряде случаев цементацию-в жидкой среде с на-
гревом т. в. ч. применять рационально.
При этом способе нет необходимости в герметичных муфелях
и процесс можно проводить независимо от наличия на заводах
газовых магистралей или специальных газовых генераторов.
Недостатками этого процесса в сравнении с газовой цемен-
тацией с нагревом т. в. ч. являются: необходимость энергичного
охлаждения жидкости, непроизводительные потери электроэнер-
гии, расходуемой на нагрев жидкости, а также трудности,
связанные с обработкой мелких деталей и с точным контролем
температуры металла.
Цементация в расплавленном чугуне при 1300° С (568]. Этот
исключительно быстрый метод имеет большой недостаток: чугун
прилипает к цементируемой поверхности. При выдержке 15 сек
образуется общий слой глубиной 0,27 мм (прилипший чугун
0,08 мм и цементованный слой глубиной 0,19 мм), а при вы-
держке 1 мин 20 сек — 0,43 мм (прилипший чугун 0,16 мм
и цементованный слой глубиной 0,27 мм). Этот недостаток
исключает возможность широкого использования этого метода,
который, однако, применим для повышения твердости ломов,
зубил и различного слесарно-монтажного инструмента.
Режимы термической обработки
цементованной стали
Режимы охлаждения после цементации и режимы последую-
щей термической обработки определяются кинетикой распада
аустенита в цементованном слое и сердцевине стали.
При переохлаждении цементованной стали до температуры
перлитной области превращение одновременно начинается на
поверхности слоя (образуется феррито-цементитная смесь:
перлит, троостит) и в сердцевине (образуется избыточный фер-
рит, рис. 36) [101]. По мере увеличения выдержки зона непре-
вращенного аустенита постепенно сужается. Примерно так же
протекают превращения при более низких температурах (500—
450°С): область аустенита, не превращающегося при коротких
изотермических выдержках, становится более узкой, и продукты
распада имеют большую твердость. При изотермической вы-
держке в интервале 350—200° С превращение происходит
в одном направлении — в основном от сердцевины к поверхно-
сти и протекает по мартенситному типу. Цементованный слой
претерпевает мартенситное превращение при более низких
74
благодаря чему увеличиваются
Рис. 36. Диаграмма структурного
состояния цементованного слоя
стали 18ХГМ в зависимости от
времени изотермического превра-
щения при 690° С [101]
температурах, чем сердцевина. С повышением температуры за-
калки точка Мн заметно снижается; положение точки Мк из-
меняется незначительно.
Наиболее распространенные режимы термической обработки
цементованных сталей приведены в табл. 11 (рис. 37).
Во многих случаях детали из легированных сталей после
цементации целесообразно подвергать дробеструйной обработке.
Дробеструйный наклеп повышает предел выносливости стали,
так как под его влиянием остаточный аустенит в цементованном
слое превращается в мартенсит,
сжимающие напряжения. Если
рассмотреть эпюру остаточных
напряжений в цементованном
слое образцов, обработанных
по технологии, принятой на не-
которых заводах, то во многих
случаях она будет характери-
зоваться уменьшением сжима-
ющих напряжений на поверх-
ности слоя. Этот недостаток
устраняется наклепом. По дан-
ным Горьковского автозавода
(рис. 38), дробеструйная обра-
ботка цементованных зубчатых
колес из стали 20ХНМ повы-
шает их долговечность в 2 раза
[104].
Аналогичные результаты
дает обкатка роликом. Так,
после обкатки роликом образцов
закаленных на твердость HV 650—700, их твердость повышается
на 100 единиц по Виккерсу, износостойкость в абразивной сре-
де— на 30%, предел прочности надрезанных образцов — на 40—
50% [105].
Один из наиболее распространенных видов брака цементо-
ванных деталей, особенно тонкостенных, несимметричных
и сложных по форме, а также имеющих неравномерный или
односторонний цементованный слой — коробление. При ‘этом
детали, цементованные в твердом карбюризаторе, в большин-
стве случаев коробятся меньше, чем цементованные в газовом.
Наиболее эффективный путь уменьшения коробления —
сокращение числа высокотемпературных нагревов, уменьшение
температуры и скорости охлаждения при закалке. Однако за-
калка с пониженных температур в ряде случаев нежелательна,
гак как не обеспечивает необходимых свойств сердцевины дета-
ли. Минимальное коробление деталей достигается при ступен-
чатой закалке, которая неприменима для массивных деталей.
из сталей 12Х2НЗМА и 20ХНЗА,
75
Исследования, проведенные НИИТавтопромом на шлицевых
отверстиях зубчатых колес из сталей 18ХГТ и 20Х, показали,
что на деформацию почти не влияют степень осадки металла
при ковке, направление волокна, режим термической обработки
поковок и условия механической обработки. Напротив, сильное
влияние оказывают глубина цементованного слоя, концентрация
Рис. 37. Схема режимов термической обработки цементованной
стали (температуры Ас3 и Аг3 для сердцевины)
в нем углерода, скорость охлаждения после цементации, режим
термической обработки деталей (особенно скорость охлажде-
ния при закалке) и применение местной защиты от цемен-
тации.
Зубчатые колеса и другие детали из сталей 20Х2Н4А
и 18Х2Н4ВА обычно подвергают нормализации (при 880—
900° С) для устранения избыточных карбидов, а затем высокому
отпуску, закалке и низкому отпуску. При нормализации дефор-
мация шестерен превышает их деформацию при последующей
закалке.
76
Рис. 38. Распреде-
ление тангенциаль-
ных остаточных
напряжений по
толщине стенки ко-
лец из стали
20ХНМ после це-
ментации (/) и до-
полнительной об-
работки дробью
(2) [104]
Деформацию при нормализации можно в 2—3 раза умень-
шить, применяя нагрев т. в. ч. Нормализацию можно не
производить, используя карбюризатор пониженной активно-
сти {106].
Минимальная деформация зубчатых колес из сталей
20Х2Н4А и 18Х2Н4ВА обеспечивается термической обработ-
кой со ступенчатой закалкой в горячей среде без предваритель-
ной нормализации, например, по режиму:
1. Цементация при 900—920° С: а) в ма-
лоактивном твердом карбюризаторе (2—
4% Na2CO3 или ВаСОз) с охлаждением в
ящиках на воздухе; б) в газовом карбюри-
заторе с последующим охлаждением на воз-
духе или в охлаждаемом колодце. .
2. Ступенчатая закалка с 790—810°С
в горячем масле (температура 160—180°С)
с выдержкой в течение 5—10 мин и последу-
ющим охлаждением на воздухе.
3. Низкий отпуск при 140—160° С.
Упразднение нормализации, а также
применение ступенчатой закалки зубчатых
колес из сталей 20Х2Н4А и 18Х2Н4ВА вме-
сто обычной практически не влияет на вели-
чину механических свойств этих сталей [106].
На деталях из стали 12Х2Н4А некото-
рых плавок легко образуются трещины при
медленном охлаждении после цементации в
ящиках. Во избежание этого ящики следует
охлаждать на воздухе, детали вынимать из
ящика при 100—150° С и немедленно поме-
щать в печь для высокого отпуска [107].
Рекомендации по устранению возможно-
сти образования трещин в деталях из ста-
лей 20Х2Н4А, 12ХНЗА и 20ХГНР приводят-
ся в работе [108].
Сталь 12Х2Н4А [109] рекомендуется за-
каливать с 830—850° С в горячем масле или
селитре с температурой 230—260° С, нагре-
вать до 500° С с выдержкой 1 ч и затем охлаждать в масле
(этот режим, по существу, аналогичен режиму 10 на рис. 37).
Оптимальный режим термической обработки стали 18Х2Н4ВА,
обеспечивающий получение стабильной твердости не менее
HRC 60, заключается в следующем: охлаждение после цемента-
ции в ящике, отпуск при 650° С, закалка с 780—800° С, охлажде-
ние в масле или на воздухе, обработка холодом при — 70° С
в течение 20—30 мин, отпуск при 150—160° С (НО].
77
Таблица И
Режимы термической обработки цементованных деталей
Вариант об- работки (см. рис. 37) Условия термической обработки1 Влияние термической обработки на структуру и твердость стали Применение обработки
1 Непосредст- венная закалка с температуры цементации (из печи или ящи- ка) Не обеспечивается Измель- чение зерна стали, наблю- дается значительное короб- ление деталей. У легирован- ной стали — пониженная по- верхностная твердость из-за сохранения в слое значи- тельного количества оста- точного аустенита Наиболее дешевый и простой способ, но применяется редко, лишь для наименее ответственных дета- лей, чаще при цемен- тации их в печах с вращающимся муфе- лем
2 Непосредст- венная закалка с подстужива- нием до 800— 850° С (чаще до температуры вы- ше Ас3 сердце- вины стали) Подстуживание способст- вует уменьшению коробле- ния деталей и повышению поверхностной твердости вследствие уменьшения ко- личества остаточного аусте- нита. Зерно стали при такой обработке не измельчается Широко применяет- ся для ответственных деталей, изготовляе- мых из мелкозернис- той стали и подверга- емых газовой цемен- таций
3 Непосредст- венная ступен- чатая закалка с предваритель- ным подстужи- ванием То же, что и при вариан- те 2. Ступенчатая закалка позволяет добиться мини- мального коробления дета- лей, однако способствует по- вышению в слое количества остаточного аустенита Применяется в тех случаях, когда жела- тельно минимальное коробление деталей 2
4 Непосредст- венная закалка с подстужива- нием до 800— 850° С, затем обработка холо- дом до минус 70—75° С 8 Повышается поверхностная твердость вследствие прев- ращения остаточного аусте- нита в мартенсит при обра- ботке холодом Применяется редко, лишь для высоколеги- рованных сталей
5 Охлаждение после цемента- ции с любой скоростью, аза- тем одинарная закалка с 760— 780° С или с температуры вы- ше Ас3 сердце- вины стали При более быстром охлаж- дении после цементации не образуется цементитная сет- ка, но увеличивается короб- ление деталей. Закалкой до- стигается измельчение зерна стали. .При закалке с 760— 780° С сердцевина стали за- каливается неполностью. Для высоконагруженных де- талей применяют закалку с температуры выше Ас3 серд- цевины Применяется часто после твердой цемен- тации и реже после газовой цементации (главным образом пос- ле газовой цементации в шахтных печах)
78
Продолжение табл. 11
Вариант об- работки (см. рис. 37) Условия термической обработки1 Влияние термической обработки на структуру и твердость стали Применение обработки
6 То же, что и при варианте 5, но закалка сту- пенчатая То же, что и при вариан- те 5, но уменьшается короб- ление Применяется редко
7 То же, что и при варианте 5, но перед оди- нарной закалкой высокий отпуск (смягчающий отжиг) при 620—650° С в течение 3—10 ч4 После высокого отпуска облегчается обработка стали резанием. Выделяющиеся карбиды неполностью успе- вают раствориться при наг- реве под закалку, что при- водит к уменьшению оста- точного аустенита в слое Применяется в ос- новном после твердой цементации и реже — после газовой
8 Охлаждение после цемента- ции на воздухе (или вместе с ящиком), затем двойная закал- ка или норма- лизация и за- калка (темпера- туры закалки указаны в табл. 1) При первой закалке (или нормализации) с 880—900° С устраняется цементитная сет- ка и измельчается зерно сердцевины. Нормализация предпочтительнее с точки зрения снижения коробле- ния, однако она не всегда обеспечивает отсутствие це- ментйтной сетки в слое. При второй закалке (с 760— 830°) Измельчается зерно це- ментованного слоя Применяется редко, главным образом пос- ле цементации в твер- дом карбюризаторе от- ветственных деталей. При такой обработке достигаются высокие механические свойст- ва, однако в связи с двойным нагревом уве- личивается коробление деталей и обезуглеро- живание, а также ус- ложняется процесс об- работки
9 Двойная за- калка с проме- жуточным высо- ким отпуском при 650° С Цель двойной закалки та же, что и при варианте 8. Назначение высокого отпус- ка то же, что и при вариан- те 7 То же
10 Режим обра- ботки с раздель- ной закалкой сердцевины и цементованного слоя Такой обработкой дости- гается минимальное короб- ление деталей. После ох- лаждения до 220° С в це- ментованном слое стали 18Х2Н4ВА сохраняется аус- тенит. При охлаждении пос- ле отпуска при 560° С зна- чительная часть аустени- та превращается в мартен- сит. Сердцевина после ох- лаждения до 220° С в основном приобретает струк- Специальный режим обработки, применяе- мый для ответствен- ных деталей из стали 18Х2Н4ВА, подвергае- мых газовой цемента- ции в шахтной печи
79
Продолжение табл. 11
Вариант об- работки (см. рис. 37) Условия термической обработки1 Влияние термической обработки на структуру и твердость стали Применение обработки
И 12. 13 Охлаждение после цемента- ции на воздухе или в ящике и затем закалка поверхностного слоя с нагревом т. в. ч. Газовая це- ментация при 1050° С с на- гревом т. в. ч., подстуживание до 820—850° С и закалка После высокого отпуска нагре- тая деталь сра- зу же перено- сится в печь для дальнейшего на- грева под за- калку, которая производится в горячей среде гуру мартенсита, а после от- пуска при 560° С — сорбита. Обработка холодом умень- шает количество остаточного аустенита в слое Измельчается зерно толь- ко цементованного слоя и подслоя нагретых т. в. ч. вы- ше критических точек Вследствие мелкозернис- тости стали механические свойства ее под влиянием высокой температуры цемен- таций почти не снижаются Достигается минимальное коробление за счет примене- ния ступенчатой закалки и совмещения нагревов Применяется после цементации в твердом или газовом карбюри- заторе Применялась на ЗИЛе для зубчатых колес из сталей 18ХГТ и ЗОХГТ Применяется после твердой и газовой це- ментации для малоле- гированных сталей. Достигается сокраще- ние цикла обработки
1 При всех вариантах обработки заключительной операцией является низкий от- пуск при 160—200° С в течение 1—2 ч для уменьшения напряжений и некоторого повышения вязкости стали. Поверхностная твердость цементованной стали после за- калки и низкого отпуска должна быть не менее HRC 60. 2 Для уменьшения коробления сложных и склонных к короблению деталей (на- пример, зубчатых колес) закалку часто ведут в штампах. Такую закалку применяют и при других вариантах обработки цементованных деталей. 2 Обработку деталей холодом следует выполнять непосредственно после закал- ки, так как предварительный низкий отпуск или вылеживание при комнатной темпе- ратуре стабилизируют аустенит. Обработка холодом может быть включена и в дру- гие варианты обработки (например, в 3-й, 5-й, 6-й, 7-й). Охлаждение до температур, бо- лее низких, чем минус 70—75°, почти не дает никакого эффекта. Когда под наруж- ным мартенситно-карбидным слоем располагается много остаточного аустенита, об- рабатывать холодом не следует, так как при этом могут возникнуть на поверхности растягивающие напряжения, снижающие предел выносливости стали [102] 4 Высокий отпуск сталей 18Х2Н4ВА и 12Х2Н4А по указанному режиму не всегда обеспечивает необходимое снижение их твердости даже тогда, когда продолжитель- ность этой операции достигает 25—40 ч. Чтобы сократить ее, следует устранить пе- ресыщение цементованного слоя углеродом (применяя малоактивный карбюризатор) и проводить двухкратный высокий отпуск при 650° С или лучше при 620—630° С продолжительностью до 8 ч с последующим медленным охлаждением. Целесообраз- но также перед отпуском подвергать стали обработке холодом [103].
80
Нержавеющую сталь 1X13 рекомендуется закаливать
с 900° С (HRC 67) и затем подвергать низкому отпуску при
150—170° С (HRC 63—65) (55]. Такой режим обработки был
предложен для прессформ, работающих в условиях абразивного
износа.
Детали из сталей 2X13, Х17Н2, 13Х12НВМФА и др. рекомен-
дуется закаливать с 1000—1020° С, обрабатывать холодом при
минус 60—70° С и отпускать при 150—170° (HRC 62—64) [51],
[52], [54].
Для обеспечения стабильности размеров деталей обработку
холодом следует вести при более низкой температуре
(—120° С) [111].
Некоторые нержавеющие стали вместо обработки холодом
можно подвергать высокому отпуску, при котором происходит
явление вторичного твердения. Например, у цементованной
стали Х17Н2, подвергнутой закалке с 1020° С, содержание
аустенита в слое доходит до 40—50%, а после отпуска при
500° С снижается до 12—15%, что обеспечивает получение до-
статочно глубоких слоев (0,4—0,5 мм) с твердостью более
HRC 58 [111]. Механические свойства образцов цементованных
нержавеющих сталей приведены в работе [52].
В некоторых случаях детали после цементации нагревают
под закалку т. в. ч. [112]—(114].
В выполненном исследовании [112] образцы стали 20, цемен-
тованные в твердом карбюризаторе, нагревали под закалку со
скоростью 90 град/сек. до 880° С, со скоростью 225 град/сек до
920° С и 450 град/сек до 980° С.
В первых двух случаях слой имел структуру мелкоигольча-
того мартенсита, а в последнем структуру скрыто-кристалличе-
ского мартенсита. При наличии в слое цементитной сетки на-
грев стали рекомендуется вести со скоростью 400—500 град/сек
до 960—1000° С и выше. Структура слоя после такой закалки
состоит из мартенсита (свободного от цементитной сетки),
имеющего заметную игольчатость.
Влияние параметров индукционного нагрева на твердость
и механические свойства цементованных сталей 20 и 20Х по-
казано на рис. 39, 40. Глубина закаленного слоя при нагреве
т. в. ч. должна превосходить глубину цементованного слоя
в 1,5—2 раза с целью создания под слоем упрочненной по-
душки.
Преимуществами индукционного нагрева под закалку цемен-
тованной стали являются уменьшение коробления деталей, по-
вышение пластичности и прочности цементованного слоя (вслед-
ствие измельчения зерна и блоков, неоднородности мартенсита
и т. д.) (115]. Если детали имеют сложную конфигурацию, а их
сердцевина должна обладать высокой прочностью, этот метод
нагрева неприемлем.
81
Технология изготовления цементуемых деталей простой
формы (кольца подшипников, пальцы поршня и др.) может
быть значительно упрощена при совмещении цементации с на-
гревом под горячую деформацию и закалку. Скоростную цемен-
тацию можно вести при 1050—1100° С, после чего осуществлять
Рис. 39. Изменение механических свойств
цементованной стали 20Х (глубина слоя
1,5 мм) в зависимости от температуры и
скорости нагрева под закалку т. в. ч.
(И. В. Кидин):
1 — скорость нагрева 100 град!сек\ 2 —
200 град!сек\ 3 — 400 град!сек
012 Змм
Рис. 40. Изменение твердости
по глубине цементованного
слоя стали 20 (глубина слоя
1,8 мм), подвергнутой закалке
после нагрева т. в. ч. до 960° С
и закалке с разных температур
при скорости нагрева
450 град/сек [112]:
1 — скорость нагрева 90 град!сек;
2 — 225 град!сек; 3 — 450 град!сек
горячую штамповку или раскатку при температуре около 950° С.
Затем осуществляется непосредственно закалка.
Была опробована термо-химико-механическая обработка
цилиндрических цементованных заготовок колец подшипников
из стали 18ХГТ. Заготовки цементовались при 1050° С на глуби-
ну 1,5 мм, после чего нагревались до 930° С, подвергались горя-
чей раскатке и немедленно закаливались в масле. Горячая
82
деформация цементованной стали, заканчивающаяся непосред-
ственной закалкой, способствует уменьшению количества оста-
точного аустенита в закаленном цементованном слое и придает
мелкозернистое строение как этому слою, так и сердцевине, что
обеспечивает высокие механические свойства деталей, изготов-
ленных с применением такой технологии [116], [117].
Структура цементованного слоя
Примерно 20 лет назад обратили внимание, что при большом
увеличении на нетравленых шлифах цементованной стали в по-
верхностной зоне слоя (на глубине нескольких сотых милли-
Рис. 41. Микроструктуры цементованного слоя стали в нетравленом состоянии
(Х1250) [19]:
7 — нелегированная сталь; 2 — сталь, содержащая 0.7% Сг; 3 и 4 — сталь, содержа-
щая 3,5% Ni и 0,25% Мо (обработка в разных карбюризаторах); 5 — сталь, содержащая
0,5% Сг, 0,6% Ni и 0,25% Мо
метра) иногда наблюдается «темная составляющая» в виде
сетки и глобулей (рис. 41). Было высказано предположение,
что «темная составляющая» является окислами, однако экспе-
риментально это не было доказано {19], [118], [119]. О влиянии
указанной дефектной структуры на поверхностную твердость
слоя после закалки и механические свойства стали в то время
сведений не имелось. Позднее было показано [120], что в сталь
как при твердой, так и при газовой цементации диффундирует
83
кислород, концентрация которого в поверхностной зоне слоя
достигает 0,25—0,30% (рис. 42).
Недавно с помощью микрозонда был изучен химический со-
став поверхностной зоны хромоникельмарганцовистой стали,
цементованной в смеси эндогаза и пропана [121]. Было опреде-
лено содержание хрома, марганца и никеля в основном металле
и в «темной составляющей». В последней оказалось до 15% Сг.
до 11,3% Мп и менее 0,1% Ni. Содержание хрома и марганца
в основном металле поверхностной зоны слоя сильно колеба-
лось, снижаясь вблизи «темной составляющей» до незначитель-
ных концентраций, тогда как содержание никеля изменялось
Рис. 42. Содержание кислорода и водорода в цементованном слое стали Ст. 3
[120]:
а — цементация в твердом карбюризаторе при 930° С в течение 15 ч; б — газовая це-
ментация с применением бензола в печи ШЦН-45А прц 1050° С в течение 1,5 ч
мало (рис. 43). Следовательно, основа металла поверхностной
зоны цементованного слоя стали вблизи «темной составляющей»
может сильно обедняться некоторыми легирующими элементами
(хромом, марганцем), уходящими в образующиеся окислы.
Обеднение твердого раствора хромом и марганцем и образова-
ние окислов понижают устойчивость переохлажденного аустени-
та, что снижает закаливаемость стали и приводит к образова-
нию мягких «пятен», отвечающих участкам слоя с троостито-
образной структурой.
Проблеме внутреннего окисления уделяют в последнее время
большое внимание. Это объясняется тем, что при проникновении
сетки окислов на глубину более 0,02 мм прочностные свойства
стали существенно снижаются. По данным И. С. Козловского
и Е. А. Лебедевой, у образцов из стали 15Г при глубине
внутреннего окисления (трооститной сетки) 0,02—0,04 мм пре-
дел выносливости снижался с 65 кГ)мм2 (63-107 м/л2) до
40—45 кГ]мм2 (39-107—44-107 н/м2). У образцов из стали 25ХГТ
при глубине внутреннего окисления 0,025 мм предел выносливо-
сти снизился с 87 до 67 кГ)мм2 (85-107—66-107 н/л2).
84
Во избежание образования «мягких пятен», возникающих
в результате внутреннего окисления, предложено для повыше-
ния устойчивости аустенита поверхностной зоны цементованного
слоя легировать его азотом,
добавляя аммиак к цемен-
тующему газу в течение 6—
10 мин перед окончанием
процесса [77], [121]. Однако
этот способ не устраняет
внутреннего окисления, а
лишь уменьшает его вред-
ное действие.
Внутреннее окисление
можно устранить подбо-
ром соответствующих насы-
щающих сред и составов це-
ментуемых сталей. В связи
с этим предлагают пере-
смотреть составы некоторых
отечественных цементуемых
сталей, предназначенных
для изготовления деталей,
не подвергаемых после це-
ментации шлифованию и со-
держащих в среднем 1 % Сг
и (или) 1 % Мп, а также ти-
тан, которые легко окисля-
ются при цементации. Мож-
но заметить, что за рубежом
для цементации применяют
последнее время стали, со-
держание хрома в которых
почти всегда не превышает
0,6%.
В противоположность это-
му в стали для изготовления
ответственных деталей ра-
ционально вводить никель
и молибден, не участвую-
щие во внутреннем окис-
лении и благоприятно
влияющие на свойства
Рис. 43. Изменение содержания никеля,
марганца и хрома в поверхностной
зоне цементованной стали, содержащей
1,48% Ni, 0,86% Сг и 0,75% Мо (ис-
следование выполнено на микрозонде;
на шлифе видны участки с темной тро-
оститообразной структурой [il21].
стали.
Предметом большого числа исследований, проведенных на
протяжении ряда лет, явился наблюдаемый иногда особый вид
структуры заэвтектоидной зоны цементованного слоя углероди-
стой и легированной стали. Эта структура отличается от
85
нормальной, тем, что в заэвтектоидной зоне цементованного
слоя наряду со структурно свободным цементитом наблюдается
структурно свободный феррит в виде сетки, окружающей сетку
цементита, или в виде отдельных скоплений. Цементитные вы-
деления в подобной структуре бывают более крупными вслед-
ствие уменьшения содержания углерода в соседних участках
структуры. Сталь с такой структурой называется анормальной.
Преимущественно анормальной бывает мелкозернистая сталь.
О причинах анормальности стали имеется ряд противоречи-
вых мнений. В частности, некоторые исследователи видят эту
причину в повышенном содержании кислорода в стали, подвер-
гающейся цементации.
Можно считать доказанным, что чистое от кислорода железо,
цементованное в карбюризаторе с незначительным количеством
кислорода, не дает анормальной структуры. Анормальность
чистого железа и стали с небольшим содержанием кислорода
всегда связана с поглощением кислорода при нагреве перед
цементацией в среде, содержащей кислород, или при цемента-
ции в карбюризаторе, содержащем большое количество кисло-
рода. На рис. 44 показаны микроструктуры склонной к анор-
мальности низкоуглеродистой стали, цементованной в различных
условиях. После длительного нагрева на воздухе и цементации
в древесном угле с углекислым барием сталь имела анормаль-
ную структуру (рис. 44, а). Структура этой же стали после на-
грева на воздухе и цементации в парах бензола с добавкой
водорода была почти нормальной (рис. 44, б), так как кислород,
поглощенный из воздуха, в значительной степени был удален из
стали при цементации в восстановительном газе. Несмотря на
предварительный нагрев в водороде, при последующей цемен-
тации в древесном угле с углекислым барием та же сталь по-
глотила настолько много кислорода, что превратилась в анор-
мальную (рис. 44, в). Сталь, нагретая в водороде и цементован-
ная в парах бензола с добавкой водорода, имела нормальную
структуру, так как не содержала кислорода (рис. 44, г).
Установлено, что точка Агх для анормальной стали лежит
выше, чем для нормальной; выделение феррита у анормальной
стали начинается при более высокой температуре, а стойкость
переохлажденного аустенита у этой стали меньше, чем у нор-
мальной стали [19]. Поэтому условия для коагуляции цементита
в анормальной стали более благоприятные.
Очевидно, анормальная структура образуется вследствие
диффузии углерода по направлению к ранее выделившемуся
заэвтектоидному цементиту, чему способствует более высокая
температура начала перлитного превращения в этой стали.
Анормальность структуры стали отчетливо выявляется
только после цементации в твердом карбюризаторе. При газо-
вой цементации, выполняемой в среде, содержащей углеводо-
56
роды, количество кислорода оказывается меньше, чем при
цементации в твердом карбюризаторе, и, следовательно, воз-
Рис. 44. Микроструктуры склонной к анормальности низкоуглеродистой стали,
нагретой в течение 36 ч при 930° С, а затем цементованной при 930° С в тече-
ние 8 ч (Х200):
а — нагрев в воздушной среде, цементация в древесном угле с углекислым барием;
б — нагрев в воздушной среде, цементация в смеси паров бензола и водорода; в — на-
грев в водороде, цементация в древесном угле с углекислым барием; г — нагрев в во-
дороде, цементация в смеси паров бензола и водорода
можность увеличения содержания кислорода в стали умень-
шается.
Поэтому анормальность- стали при газовой цементации
в газах, содержащих углеводороды, почти не обнаруживается.
87
Некоторые исследователи связывают влияние элементов на
склонность стали к анормальности с их воздействием на величи-
ну поверхностного натяжения на границах аустенит — феррит
и аустенит — цементит [122], другие — с изменением свободной
энергии граничного слоя зерен и степенью обогащения его теми
или иными элементами [123].
Б. С. Натапов считает, что горофильные по отношению
к аустениту элементы (О2, Al, Р, W, Mo, V, Si и др.) понижают
поверхностное натяжение на границе аустенит — феррит и спо-
собствуют образованию анормальной структуры, а горофобные
элементы (Мп, Сг, Ni) повышают его и препятствуют образо-
ванию анормальной структуры.
При изучении причин образования анормальной структуры
ни один из исследователей не попытался установить связь меж-
ду склонностью стали к анормальности и наличием тонкой
темной «окисной» сетки, наблюдаемой на нетравленых шлифах
цементованного слоя (см. стр. 83). Это объясняется, по-види-
мому, тем, что изучением каждого из этих дефектов занимались
разные исследователи. Между тем, появление как темной сетки
(окислов), так и анормальной структуры наблюдается после
твердой цементации и реже после газовой.
Отсюда следует предположить, что условия образования
этих дефектов совпадают.
В наших исследованиях при рассмотрении при большом
увеличении нетравленых микроструктур анормальной стали
в ряде случаев в поверхностной зоне слоя по границам зерна
была видна тонкая темная сетка (сетка окислов), к которой
с обеих сторон (после травления) примыкали полосы цементита
(цементитная сетка), а далее также с обеих сторон наблюда-
лась ферритная сетка. Таким образом, появление «темной со-
ставляющей» может совпадать с анормальностью структуры.
Следовательно, между этими дефектами структуры возможна
тесная связь.
Очевидно, влияние легирующих элементов на склонность
стали к анормальности связано с влиянием их на склонность
к образованию сетки и глобулей окислов по границам зерен.
Эта сетка и глобули отчетливо видны на глубине до 0,05—
0,06 мм; но в дисперсном виде они, несомненно, распространяют-
ся значительно глубже. Вследствие выделения этих включений
уменьшается устойчивость аустенита и повышается темпера-
тура точки Ari, что облегчает диффузию углерода в а-железе
к участкам цементита. Диффузия углерода вблизи границ зерен
ускоряется также вследствие обеднения областей, прилегающих
к границам зерен, элементами, образующими на границах зерен
окислы (см. рис. 84).
Таким образом, несмотря на большое количество работ по
изучению анормальной структуры стали, до сего времени еше
88
не ясен механизм ее образования. Это не дает возможности
объяснить все явления, связанные с образованием такой струк-
туры.
Анормальная сталь при закалке склонна к образованию
«мягких пятен». Для получения равномерной высокой твердости
сталь с анормальной структурой следует закаливать с более
высокой температуры или давать большую выдержку при
Рис. 45. Микроструктуры стали 15:
а — цементованной на небольшую глубину; б — закаленной (Х200)
нормальном нагреве под закалку. Охлаждение стали с анор-
мальной структурой нужно производить в более резких охла-
ждающих средах.
На рис. 45 и 46 показаны характерные микроструктурные
цементованных сталей 15 и 18ХГТ.
Предельная концентрация углерода в цементованном слое
нелегированной стали определяется максимальной раствори-
мостью углерода в аустените при температуре насыщения.
Цементитная сетка, часто наблюдаемая в структуре поверхност-
ной зоны слоя нелегированной стали, цементованной в излишне
активном карбюризаторе, выделяется лишь при медленном
охлаждении после цементации. Такого рода структура встре-
чается также в сталях, легированных 'некарбидообразующими
элементами. После закалки таких сталей слой1 приобретает
89
структуру мелкоигольчатого или скрытокристаллического мар-
тенсита (при условии растворения при нагреве цементитной
сетки, если она имелась).
В сталях, легированных карбидообразующими элементами,
по достижении предельной растворимости углерода в аустените
Рис. 46. Микроструктуры цементо-
ванной и закаленной стали 18ХГТ
(ЗИЛ):
а — нормальная структура; б — недо-
пустимо высокое количество остаточ-
ного аустенита; в — недопустимо
большое количество карбидов (сталь
со структурой б подвергается вторич-
ной термической обработке, а со
структурой в бракуется)
в поверхностной зоне слоя при температуре цементации обра-
зуются карбиды (в виде глобулей). При медленном охлаждении
количество карбидов в слое увеличивается. В некоторых сталях
при определенных условиях охлаждения часть карбидов при-
обретает форму игл или пластин.
При образовании во время цементации карбидов и их росте
в поверхностной зоне слоя происходит непрерывное диффузион-
ное перераспределение карбидообразующего элемента между
90
карбидом и аустенитом. При этом в карбиде уменьшается
содержание углерода и карбидообразующего элемента (на-
пример, хрома), а в аустените уменьшается содержание хрома
и увеличивается содержание углерода (хром снижает раство-
римость углерода в аустените). По мере удаления от поверхно-
сти в глубь слоя количество карбидов уменьшается, а содержа-
ние в них хрома и углерода увеличивается; растет также со-
держание хрома в твердом растворе.
Описанная закономерность в перераспределении элементов
в слое подтверждена послойным химическим анализом и анали-
Рис. 47. Распределение углерода и хрома по сечению диффузионной зоны на
различных этапах цементации хромистой стали (С££ — среднее содержание
хрома; С^р — среднее содержание углерода) [9]
зом карбидов, полученных при послойном электролитическом
выделении [9]. Таким образом, карбиды, содержащие наиболь-
шее количество углерода и хрома, оказываются не на поверхно-
сти, а на некоторой глубине. Характер изменения распределения
углерода и хрома по сечению диффузионного слоя показан на
рис. 47.
На первом этапе процесса содержание углерода в слое не
превышало предела его растворимости в аустените (рис. 47, а);
на втором этапе (рис. 47, б) в слое возникла зона с карбидами,
которые образуются вначале преимущественно по границам
зерен, а затем внутри зерен; на третьем этапе (рис. 47, в) зона
карбидов расширилась, увеличилась общая глубина слоя и по-
высилась общая концентрация углерода в слое, особенно в зоне
слоя с выделениями карбидов.
Как показывают измерения степени тетрагональности решет-
ки мартенсита и параметров решетки аустенита по глубине
цементованного слоя сталей 20, 18ХГТ, 1X13 и Х17Н2 [28], [124],
9)
встречаются два случая распределения углерода в цементован-
ном слое.
В цементованном слое стали 20 весь углерод при темпера-
туре насыщения находится в твердом растворе и закалкой
фиксируется в мартенсите. Слой с максимальной (1,2%) кон-
центрацией углерода (920° С, 6 ч) находится на расстоянии
0,01—0,02 мм от поверхности. Наличие тонкого слоя с понижен-
ной концентрацией углерода объясняется обезуглероживанием
при переносе детали из цементационного ящика в закалочный
бак.
Максимальная концентрация углерода в мартенсите, а также
максимальное общее содержание углерода в цементованном
Рис. 48. Изменение содержания углерода в мартенсите по глубине цементо-
ванного слоя:
а — стали 20 и 18ХГТ, цементованные при 920° С в течение 6 ч в твердям карбюриза-
торе после непосредственной закалки [28]; б — сталь ЗОХГТ, цементованная в безмуфель-
ном агрегате 477]; 1 — при постоянном углеродном потенциале атмосферы 0,8% С (во
время выдержки при 930° С и подстуживания до 820° С); 2 — углеродный потенциал
атмосферы при выдержке 1,3—1,4% С, при подстуживании 0,8% С
слое сталей 1X13 и Х17Н2, как и в случае, рассмотренном на
рис. 47, находится в поверхностной зоне слоя.
В отличие от этого в цементованном слое стали 18ХГТ мак-
симальная концентрация углерода в мартенсите была лишь на
глубине 0,2 мм (рис. 48), что нельзя объяснить поверхностным
обезуглероживанием, так как производилась непосредственная
закалка стали из цементационного ящика. Наружная часть
слоя, в которой содержание углерода повышается до максиму-
ма, соответствует двухфазной области (мартенсит — карбиды).
Правая ниспадающая ветвь кривой соответствует структуре без
избыточных карбидов. Такое распределение углерода соответ-
ствует лишь содержанию его в твердом растворе. Общее же
содержание углерода, определенное химическим анализом,
уменьшается по мере удаления от поверхности. По-видимому,
в случае низкого содержания хрома в стали при температуре
цементации кинетически быстрее происходит образование кар-
92
бидов, чем насыщение до предела углеродом твердого раствора
•[28], [124].
Слой с максимальной концентрацией углерода в мартенсите
образуется на некотором расстоянии от поверхности и в тех слу-
чаях, когда в процессе цементации применяется более активная
науглероживающая среда, чем в период подстуживания деталей,
или когда охлаждение их до комнатной температуры либо нагрев
под закалку проводят без защитной атмосферы. В этих случаях
снижение общей концентрации углерода в поверхностной зоне
слоя стали, легированной карбидообразующими элементами,
происходит в основном за счет твердого раствора, причем кар-
биды в слое сохраняются. В случае газовой цементации (930°С)
и подстуживания (до 820° С) при постоянном углеродном по-
тенциале атмосферы (0,8% С) слой с максимальной концентра-
цией углерода был лишь немного сдвинут (рис. 48, б) от поверх-
ности стали ЗОХГТ. Однако в случае цементации этой же стали
при углеродном потнциале атмосферы 1,3—1,4% С с последую-
щим снижением при подстуживании (20—40 мин) общей кон-
центрации углерода в слое до 0,8% С (вследствие применения
контролируемой атмосферы с'потенциалом 0,1—0,3% С или под-
стуживания на воздухе) слой с максимальной концентрацией
углерода в мартенсите сдвинулся на расстояние 0,25—0,5 мм от
поверхности стали.
При этом механические свойства стали снижаются на 25—
30% [77]. Поэтому нельзя допускать уменьшения концентрации
углерода (в том числе до оптимального) в периоды подстужива-
ния перед закалкой и охлаждения до комнатной температуры,
или при последующем нагреве под закалку.
В зависимости от состава стали, условий цементации и по-
следующей закалки в цементованном слое легированной стали,
кроме мартенсита и карбидов, сохраняется некоторое количе-
ство остаточного аустенита [125], [126]. При этом в поверхностной
зоне слоя сохраняется меньше аустенита, чем в более глубоких
зонах (рис. 49). В соответствии с количеством остаточного
аустенита в диффузионном слое меняется и твердость послед-
него. В зоне с максимальным количеством аустенита снижаются
твердость [28] и остаточные напряжения сжатия.
Такой характер распределения аустенита по глубине слоя
объясняется тем, что аустенит поверхностной зоны слоя менее
устойчив по сравнению с аустенитом более глубокой зоны из-за
наличия в нем избыточных карбидов и меньшей концентрации
карбидообразующих элементов, а в некоторых случаях и угле-
рода.
Введение в сталь с карбидообразующими элементами не-
карбидообразующих элементов затрудняет образование аусте-
нитно-карбидной зоны. При цементации стали, легированной
только некарбидообразующими элементами, аустенитно-карбид-
93
ная зона не образуется; при этом структура слоя аналогична
структуре, характерной для цементованного слоя нелегирован-
ной стали.
Небольшое количество равномерно распределенного оста-
точного аустенита в слое признается полезным, так как при
этом повышаются проч-
ность, пластичность и осо-
бенно ударная выносли-
вость стали [127]. Наличие
Рис. 49. Распределение остаточного
аустенита в диффузионном слое стали
12Х2Н4, цементованной в твердом
карбюризаторе, на глубину 1 мм
(сплошные линии) и 2 мм (штрихо-
вые линии), закаленной с подстужи-
ванием с 840° С (кривая 1) и обрабо-
танной холодом при минус 72° С (кри-
вая 2) и минус 196° С (кривая 3).
Кривые построены на основании
рентгеноструктурного анализа и маг-
нитотермического изучения сталей с
различным содержанием углерода
[126]
в слое небольшого количест-
ва мелкодисперсных карби-
дов не приносит ущерба ме-
ханическим свойствам и
благоприятно сказывается
на износостойкости стали.
При рентгенографичес-
ком исследовании субструк-
туры цементованного слоя
стали 18Х2Н4ВА обнаруже-
но большое различие в вели-
чине блоков мозаики у- и
a-фаз на различной глуби-
не от поверхности стали
(табл. 12). Для субструкту-
ры у-фазы характерны
большая величина блоков и
меньший наклеп по сравне-
нию с субструктурой а-фазы
(наклеп хара ктер изу ется
высоким значением неоднородной микродеформации и малой ве-
личиной блоков).
Таблица 12
Результаты рентгенографического исследования субструктуры
по глубине цементованного слоя стали 18Х2Н4ВА [128]
Глубина в мм Величина блоков в А Максимальное относительное микроискажение решетки хю-3
7-фазы а-фазы 7-фазы | а-фазы
0,03 0,17 0,29 Прим редственная 270 1100 200 е ч а н и е. Цемент [ закалка, вторая з 210 300 280 ация при 920° С >акалка с 810° С, о 2,4 2,2 1,8 на глубину 1,7— >тпуск при 150° С. 3,0 2,6 2,9 1,9 мм, непос-
94
В поверхностной зоне цементованного слоя нержавеющих
сталей (1X13, 2X13, Х17Н2) общее содержание углерода колеб-
лется в пределах 2,2—3,5%, причем в мартенсите содержится
0,6—0,8% С. Столь высокое содержание углерода в цементован-
ном слое этих сталей следует считать нормальным. При падении
общего содержания углерода в поверхностной зоне слоя ниже
2,2% концентрация углерода в мартенсите уменьшается более
чем до 0,50—0,55%, что приводит к снижению поверхностной
твердости слоя.
При цементации хромистых
сталей (X, ШХ15, Х12Ф) со-
держание углерода в слое так-
же получается высоким (рис.
50). В поверхностной зоне этих,
а также нержавеющих сталей
после цементации имеется
большое количество карбидов,
вкрапленных в перлитную, а
после закалки в мартенситно-
аустенитную основу.
Для контроля глубины це-
ментации по микроструктуре
до настоящего времени нет
единого общепринятого крите-
рия. Обычно глубину слоя оп-
ределяют под микроскопом на
шлифах стали, отожженной
или охлажденной после це-
ментации в песке. При этом
измеряют глубины слоя до
равномерной исходной струк-
туры или до половины пере-
ходной зоны, или до начала
Рис. 50. Микроструктура по-
верхностной зоны слоя стали
ШХ15, цементованной в течение
36 ч при 920° С
выделения феррита. Легче определить глубину слоя до начала
выделения феррита. Однако большее практическое значение
имеют общая глубина слоя и особенно глубина до половины
переходной зоны, близкая по величине к глубине слоя, опреде-
ляемой визуально по излому закаленного образца
Для отдельных марок стали разработаны шкалы для оценки
микроструктуры. Например, на Кировском заводе в Ленинграде
при контроле структуры цементованных сталей 12Х2Н4А
ш 18Х2Н4ВА применяют три пятибалльные шкалы для кон-
троля: 1) избыточных карбидов в цементованном слое после вы-
1 В ряде зарубежных работ предложено за глубину цементованного слоя
принимать расстояние от поверхности до зоны закаленного слоя с твердостью
HRC 50.
95
сокого отпуска; 2) избыточных карбидов после закалки;
3) остаточного аустенита после закалки.
На ЗИЛе для контроля структуры цементованной стали
марки 25ХГТ используются две последние шкалы.
При контроле деталей крупногабаритных подшипников из
стали 20Х2Н4А на Первом государственном подшипниковом
заводе используют четыре шкалы для контроля: 1) карбидной
сетки после цементации; 2) избыточных разрозненных карби-
дов после цементации; 3) микроструктуры после высокого от-
пуска; 4) микроструктуры после закалки. При недопустимой
структуре по первой шкале сталь подвергают закалке с по-
вышенной температуры 940—950° С, по второй шкале — диф-
фузионному отжигу при 940—950° С, по третьей шкале —
вторичному высокому отпуску и по четвертой шкале — вторич-
ному высокому отпуску и вторичной закалке.
Свойства цементованной стали
Оптимальное содержание углерода в поверхностной зоне
цементованного слоя большинства сталей 0,8—0,9%; при таком
его количестве сталь обладает высокой износостойкостью. Даль-
нейшее увеличение содержания углерода уменьшает пределы
выносливости и прочности стали при статических и динамиче-
ских испытаниях (рис. 51). Однако наиболее износостоек
цементованный слой при несколько повышенном содержании
в нем углерода (по некоторым данным до 1,2%). При этом
после термической обработки цементованный слой должен иметь
структуру мелкоигольчатого или скрытокристаллического мар-
тенсита с мелкими глобулями карбидов и небольшим количеством
остаточного аустенита.
Цементация повышает предел выносливости стали. Объ-
ясняется это возникновением в слое остаточных сжимающих на-
пряжений в связи с неодинаковым изменением объема слоя
и сердцевины стали в процессе цементации и закалки (рис. 52).
Наибольшее повышение предела выносливости достигается при
цементации на сравнительно небольшую глубину, когда цемен-
тованный слой приобретает после закалки мартенситную струк-
туру с минимальным количеством остаточного аустенита,
в результате чего в слое возникают максимальные сжимающие
напряжения (рис. 53).
В последнее время проведено много работ по изучению
ударной выносливости цементованной стали. Образцы подвер-
гают многократным изгибающим ударам на специальном копре
(КПУ-2) [135]—([137]. Для образцов диаметром 12 мм глубина
слоя, обеспечивающая оптимальную выносливость, составляет
1,0—1,2 мм (рис. 54) [47]. В ряде работ показано, что никель,
снижающий количество карбидов в цементованном слое, по-
96
вышает ударную выносливость стали. Это подтверждается так-
же данными рис. 54 (кривые 1—<?). Максимальная ударная
выносливость достигается при определенном соотношении в
слое количества остаточного аустенита и количества мар-
тенсита, что зависит от температуры закалки и других
факторов. Так, для сталей 20ХНЗА и 20НЗМА максимальная
ударная выносливость достигается при повторной закалке
с температуры 780°С. Как понижение, так и повышение темпера-
туры закалки резко снижает ударную выносливость [127].
Рис. 51. Влияние содержания углеро-
да в цементованном слое на механи-
ческие свойства стали после закалки
и низкого отпуска; излом всех образ-
цов при испытании на предел вынос-
ливости начинался с поверхности це-
ментованного слоя (И. С. Козловский
и Ю. Ф. Оржеховский):
1 и 4 — сталь 12X2H4A; 2, 3 и 5 — сталь
18ХГМ (глубина слоя 1 мм)
Рис. 52. Распределение остаточных
внутренних напряжений в цементо-
ванном слое двух образцов стали
18ХГТ. Минимум на кривых на глу-
бине 0,2—0,3 мм соответствует зоне
слоя с максимальным количеством
аустенита (К. А. Набатова)
Контактная усталостная прочность, или усталостное выкра-
шивание (число циклов до осповидного разрушения), по одним
данным, возрастает с повышением содержания углерода в по-
верхностной зоне слоя малолегированных сталей по крайней
мере до 1,5% (максимальное содержание углерода в испытан-
ных образцах). По другим данным [129], максимальная контакт-
ная усталостная прочность высоколегированной стали 12ХНЗА
достигается при концентрации в слое 1,2% С.
При содержании углерода в поверхностной зоне слоя более
1,1 —1,2% возникает склонность к образованию шлифовочных
97
трещин, чаще распространяющихся по цементитной сетке. Чрез-
мерное увеличение содержания углерода в поверхностной зоне
цементованного слоя конструкционных сталей в большинстве
случаев нежелательно, так как часто является причиной брака
и преждевременного выхода деталей из строя.
Цементация сталей 1X13 и 1X17 практически не ухудшает
их сопротивления коррозии в пресной и морской воде [53]. Эти
цементованные стали обладают высокой стойкостью к воздей-
Отношение глубины слоя к радиусу образца.
ствию керосина [52]. Испытания
на машине Шкода-Савина показа-
ли, что износостойкость цементо-
5:
3
О
ОЛ
0,8 1,2 мм Рис. 54. Зависимость ударной вязко-
Рис. 53. Влияние глубины цементо-
ванного слоя на предел выносливо-
сти стали, подвергнутой цементации
при 920° С, одинарной закалке и
низкому отпуску; предел выносливо-
сти определяли на образцах:
1 — диаметром 10 мм, сталь 10 (И. В. Куд-
рявцев и В. И. Просвирин); 2 — диамет-
ром 6 мм, сталь 18Х2Н4МА (Б. Г. Гу-
ревич и С. Ф. Юрьев); 3 — диаметром
6 мм, сталь 12ХНЗА; 4 — диаметром
6,3 мм, сталь 18ХГТ; 5 — диаметром
6,3 мм, сталь 12ХНЗ (К. А. Набатова)
сти цементованных образцов диамет-
ром 12 мм от глубины цементован-
ного слоя (работа удара 40 кГ • см,
или 3,92 дж\ цементация в твердом
карбюризаторе при 920° С, первая
закалка из ящика, вторая с 780—
800° С, отпуск при 200° С; стали
25Х2ГН2Д2Ф и 25Х2ГН2Т после про-
межуточного высокого отпуска при
660—680°):
/ — сталь 25Х2ГН2Д2Ф;» 2 — сталь
25Х2ГН2Т; 3 — сталь 25ХНФР; 4 — сталь
18ХТГ; 5 — сталь 25Х2ГТ; 6 — сталь
25ХГР [47].
ванных нержавеющих сталей 1X13, Х17Н2 весьма высока и близ-
ка к износостойкости азотированной стали 38ХМЮА [52]. При-
менение цементованной стали 1X13 вместо цементованной ста-
ли 10 для прессформ, работающих в условиях сильного абразив-
ного износа, позволило повысить стойкость прессформ более чем
в 10 раз [55].
Цементация повышает теплостойкость и износостойкость
сталей ШХ15 и X, в результате чего увеличивается долговеч-
98
ность изготовленных из этих сталей пуансонов и матриц [56],
[57]. Цементацией можно повысить теплостойкость быстрорежу-
щей стали [58].
Рис. 55. Растворимость
азота в железе при пар-
циальном давлении
азота 1 ат (1,013 X
X 105 н/м2).
АЗОТИРОВАНИЕ
Азотированием (азотизацией или нитрированием) стали на-
зывается процесс поверхностного насыщения стали азотом.
Первое систематическое исследование по азотированию
железа и стали и изучению свойств и строения нитридов железа
и специальных элементов было выполнено в 1907—1914 гг.
Н. П. Чижевским [19]. В 1923 г. немец-
ким металлургом Фри была предло-
жена сталь с алюминием, приобрета-
ющая после азотирования высокую
твердость [19]. С 1926 г. азотирование
начали применять в промышленности
[130].
Азотированию, как и цементации,
подвергают детали, работающие на из-
нос и воспринимающие знакоперемен-
ные нагрузки. Азотированные детали
имеют следующие преимущества: вы-
сокую твердость, износостойкость, теп-
лостойкость и коррозионную стой-
кость. Так как азотированию подверга-
ют в основном легированные стали
определенных составов и процесс име-
ет большую продолжительность (30—
60 ч), применение его оказывается
экономически целесообразным лишь
для обработки ответственных инстру-
ментов. и деталей авиамоторов, дизелей, турбин, приборов и т. п.
Небольшое применение получил процесс кратковременного (до
1 ч) антикоррозионного азотирования деталей, изготовленных из
углеродистых сталей и работающих в атмосферных условиях.
Насыщаемость железа молекулярным азотом при атмосфер-
ном давлении и температуре до 1500° С невелика (рис. 55),
однако ее можно увеличить, создав в печи высокое давление
(несколько сот атмосфер). Но этот способ насыщения железа
азотом пока не представляет практического интереса ввиду его
трудоемкости.
Для насыщения целесообразнее использовать атомарный
азот, образующийся в момент разложения соединений, содержа-
щих этот элемент. В качестве такого соединения обычно при-
меняют аммиак, диссоциация которого сопровождается выделе-
нием азота в атомарном активном состоянии, который, однако,
99
вскоре переходит в молекулярное состояние и теряет свою
активность:
2NH3 = 2N + 6Н.
1 Ф
n2 зн2
а) V
Рис. 56. Зависимость количества поглощенного железом азота
от степени диссоциации аммиака:
а — железный порошок (И. El Конторович); б — железные образцы
(А. В Рябченков и В. Д. Яхнина)
ственной близости от азотируемой поверхности. Степень дис-
социаций аммиака зависит от температуры, давления, скорости
1 протекания аммиака через
Рис. 57. Распределение углерода по
глубине диффузионного слоя стали
У8, азотированной по режиму: 510° С,
12 ч + 540° С, 42 ч:
печь и площади поверхности
азотируемых деталей и му-
феля печи (так как сталь
является катализатором, ус-
коряющим эту реакцию).
При данной температуре в
муфеле печи должна под-
держиваться определенная
степень диссоциации аммиа-
ка, от которой зависит ко'
личество поглощаемого
сталью азота (рис. 56). При
малой степени диссоциации
один цикл азотирования; 2 — два
цикла азотирования [131]
аммиака поглощение
уменьшается
недостаточного
азота
вследствие
количества
атомарного азота, контактирующего с поверхностью стали. При
высокой степени диссоциации аммиака в газовой фазе присутст-
вует много водорода, который абсорбируется поверхностью ста-
ли и тормозит поглощение сталью азота. В связи с этим при азо-
100
тировании при 500° С степень диссоциации аммиака стремятся
поддерживать околоЛб—30%, при 550°С — около 35—45%, при
600° С — около 45—60% и т. д.
Водород, образующийся при диссоциации аммиака, вызывает
обезуглероживание стали любых составов (табл. 13, рис. 57)
[131], [132].
Таблица 13
Изменение содержания углерода при азотировании стружки
(режим одного цикла азотирования: 510° С, 12 ч + 540° С, 42 ч) [131]
Содержание углерода в %
Количество циклов азотирования в стали в чугуне
10 УЮ 38ХМЮА 4X13 общее связанно- го
Исходное 0,13 1*,01 0,38 0,38 2,82 0,70
состояние 1 0,08 0,17 0,12 0,10 1,95 0,12
2 0,033 0,18 0,11 0,09 1,61 0,13
3 0,05 — 0,055 0,06 1,37 —
4 0,02 0,04 0,051 0,06 1,40 0,10
5 0,05 0,03 0,07 Следы 0,78 Следы
Диффузия азота в сталь
Данные о параметрах диффузии азота *в а- и у-железо, полу-
ченные разными авторами, сильно колеблются (табл. 14). Боль-
шая 'подвижность азота, как и углерода, объясняется тем, что
его атомы, растворенные в железе, лишены части своих электро-
нов и поэтому имеют уменьшенные размеры.
Коэффициенты диффузии D азота в железо связаны с темпе-
ратурой Т следующими уравнениями:
= 4,67 • 10”4
expPZi^
\ RT I
см2/сек [23];
/ 17 700\
£>“ = 0,0014 ехр (--— см2/сек [135];
\ RT J
Dj, = 3,35 • Ю“3 ехр
/ 34 660 \
k RT /
см2/сек [23].
С увеличением содержания углерода в стали коэффициент
диффузии азота снижается и при температуре 500° С составляет:
5,0 • 10-9 см2/сек при 0,06% С; 1,25 • 10-9 см2/сек при 0,43% С;
0,51 • 10~9 см21сек. при 1,13% С [23]. Большинство легирующих
элементов также снижает коэффициент диффузии азота в желе-
зо, при этом наиболее сильно действуют вольфрам, никель и мо-
либден и в меньшей степени— кремний,, марганец и хром [23].
101
Для объяснения микроструктуры азотированной стали следу-
ет обратиться к диаграмме состояния системы железо — азот
(рис. 58). Следует отметить, что в принятую ранее диаграмму
состояния этой системы в последнее
Рис. 58. Диаграмма со-
стояния системы Fe—N
[136]
время внесены некоторые уточнения,
[136]—(138].
Предел растворимости азота в ни-
триде y/(Fe4N) установлен в диапазо-
не 5,3—5,75% (вместо 5,5—5,95%).
е-Фаза простирается приблизительно
от состава Fe3N до Fe2N, не включая в.
себя последний нитрид £, который при-
сутствует в диффузионном слое при?
концентрации азота, начиная с 11,1—
11,35%. В пределах 11 —11,1% N про-
стирается двухфазовая область & + %.
При диффузии азота в железо на
поверхности последовательно образу-
ются фазы, располагающиеся следую-
щим образом (при температурах диф-
фузии) от края к сердцевине: а) при
температуре ниже эвтектоидной
(591°С) ->а; б) при темпе-
ратуре выше эвтектоидной
у'уа.
Микроструктуры азотированной по различным режимам ма-
лоуглеродистой стали приведены на рис. 59.
Таблица 14
Параметры диффузии азота в а- и у-железо
по данным разных авторов
Кристал- лическая решетка железа Темпера- тура в °C D в см2'сек • в см2 Ice к Q Источник
кал- г-атом в дж1,г-апюм
а 500 600 3,8-IO-® 1,4-Ю-7 0,0066 18 600 77 934 [133]
500 600 800 3,72-10—9 1,4-Ю-8 8,1-10-8 4,67-10—4 17 950 75 210 [23]
У 950 б.б-Ю-8 — — — [133]
955 1,9-10—7 0,019 28 300 118 577 [134]
800 950 2,9-10-’° б,ою-’° 0,335-10-2 34 660 145 225 [23]
102
Фазовый состав
При темпе- ратуре насыщения После медлен- ного охла- ждения
$ i
€ ’
У (звтектоид)
о
g
<§
s
Рис. 59. Микроструктуры стали 10, азотированной при 650° С
в течение 8 ч и затем медленно охлажденной1
1 Примечание: g-фаза в борированном слое рентгеноструктурным ана-
лизом пока не была обнаружена. Однако, судя по химическим анализам, кон«
центоация азота в поверхностной зоне азотированного слоя достигает при не-
которых условиях обработки 11,1—11,3%, что соответствует g-фазе. На микро-
структуре тонкая нетравящаяся зона, расположенная выше эвтектоида (тем-
ная зона) по-видимому отвечает у'-фазе; выше более темная зона — смесь
двух фаз, по-видимому, смесь у'- и 8-фаз. Далее располагаются две нетравя-
щиеся зоны, отделенные друг от друга отчетливой линией раздела. Это, ве-
роятно, 8-фаза и g-фаза.
103
Рис. 60. Изменение концен-
трации азота по глубине
азотированного слоя желе-
за после обработки по раз-
ным режимам (на кривых
указан фазовый состав
слоя при температуре насы-
щения) [23]1
При переходе от поверхност-
ной к более глубоким зонам слоя
имеет место резкий перепад кон-
центраций, устанавливающийся
при температуре диффузии и со-
храняющийся после охлаждения
(рис. 60).
При быстром охлаждении
фиксируется фазовый состав ста-
ли, имеющийся при температуре
насыщения. Лишь у-фаза фикси-
руется частично, остальная ее
часть, менее насыщенная азотом,
претерпевает мартенситное прев-
ращение. При медленном ох-
лаждении от температуры диф-
фузии происходит распад £-, е->
у- и a-фаз и строение азотирован-
ного слоя становится следующим:
а) если температура насыще-
ния азотом была нижё эвтекто-
идной, то
£->£ +е 8 + у' -> у ’ а+ у'-> а;
б) если температура насыще-
ния азотом была выше эвтекто-
идной, то
£ е -> е + у'-*у' -> (а +
+ 7') эвтектоид -> а + у' -> а.
Наименьшим коэффициентом
расширения обладает у-фаза
(0,79-10-5), затем идет а-фаза
(1,33-10~5); наибольший коэф-
фициент расширения у 8-фазы
(2,2- 10-5) [19]. По данным Хэг-
га, удельный вес е-фазы 6,88 г/см1 2,
у'-фазы 7,11 г! см2 и а-фазы
7,88 г/см2 [19].
На рис. 61 приведены микро-
структуры низкоуглеродистой ста-
ли, подвергнутой антикоррозион-
ному азотированию. Внешняя
светлая зона на всех трех микро-
1 Согласно более поздним данным (рис. 58) верхняя точка на кривой, от-
носящаяся к режиму 700° С — 16 ч возможно соответствует g-фазе.
104
структурах представляет собой стойкую против коррозии ни-
тридную зону (£-, е-, у'-фазы), вторая зона слоя после медленного
Рис. 61. Микроструктура стали 10t
подвергнутой антикоррозионному
азотированию при 650° С в течение
45 мин и охлажденной вместе с
печыо (а), на воздухе (б) и в мас-
ле (в) (Х340)
Рис. 62. Микротвердость отдельных состав-
ляющих азотированного слоя железа, на-
сыщенного при 700° С и охлажденного в во-
де (а) и масле (б) [il39]
охлаждения травится сильно, так как имеет эвтектоидную
структуру а + у' (темная зона на рис. 61 и 59). После ускорен-
ного охлаждения (рис. 61, бив) эта зона травится слабо и
105
состоит из азотисто-углеродистого мартенсита и аустенита. Ми-
кротвердость азотистого мартенсита Н 600—700, а азотистого
аустенита около 160 (рис. 62).
Антикоррозионное азотирование стали [140]—[145]
Антикоррозионному азотированию подвергают детали (чаще
из углеродистой стали), работающие в атмосферных условиях
(винты, гайки, мелкие зубчатые колеса приборов паровой арма-
туры, строительно-скобяная
фурнитура и т. д.). Этот
процесс в ряде случаев мо-
Рекомендуемые режимы
| антикоррозионном
азотирования
---------------1-----
Хрупкие
слои
600
700
650
\Слои ч i'
Не
корро- \живош\\& *
зионно^испь1та^30^4,^+~
стойкие* ни е Моде,
слои. \но пористы
-н!-----L—-----1--
30 60 30 мин
550
500L
О
Рис. 64. Изменение глу-
бины залегания 8-фазы в
диффузионном слоз ста-
ли 10 в зависимо-
сти от продолжительно-
сти азотирования [19]
Рис. 63. Диаграмма оптималь*
ных рекомендуемых режимов
(температуры и выдержки) ан-
тикоррозионного азотирования
жет заменить гальваническое цинкование, никелирование, хро-
мирование и др.
Степень подготовки поверхности деталей перед азотировани- •
ем зависит от того, будут ли детали в дальнейшем полироваться
(тогда их до азотирования также полируют *, или они поступят
в эксплуатацию с матовой поверхностью. Детали перед обработ-
кой очищают и затем направляют на азотирование (табл. 15,
рис. 63).
Для снижения хрупкости деталей, подвергаемых антикорро-
зионному азотированию, низкоуглеродистую сталь нужно под-
вергнуть нормализации или закалке, а среднеуглеродистую улуч-
шению [144], [145].
При азотировании стремятся получить беспористый нетравя-
щийся антикоррозионный нитридный слой (£ + 8 + у') толщиной
1 Полирование азотированных деталей — более трудоемкая операция, чеь»
полирование гальванических покрытий.
106
Таблица 15
Режимы антикоррозионного азотирования деталей
из углеродистой стали
Сталь Наименование деталей Темпера- тура процесса в °C Продол- житель- ность процесса в мин Степень диссоциа- ции амми- ака в %
08, 10, 15, 20, 25, Ст.1, Ст. 2, Ст. 3, Ст. 4, Ст. 5, А12, А15, А20, 40, 45, 50, 40Х* Тяги, штыри, болты, венти- ли паропроводов; мелкие де- тали приборов и аппаратов; резаки, сварочные горелки, детали кислородного вентиля, спортивная фурнитура, дета- ли детских велосипедов, строительно-скобяная фурни- тура (скобы, петли, шпинга- леты, задвижки, замочные изделия) и др. 600 60—120* 35—55
650 45—90* 45—65
700 15—30* 55—75
40, 40Х, У7, У8, У10, ШХ15 Детали приборов и аппара- тов (зубчатые колеса, валики, золотники, оси, гайки, винты, штифты и др-.) 770— 850* * 5—10 До 80
600—700 с последу- ющей за- калкой с 770— 850** 15-45 3—10 45—75 До 80
Примечания: 1. Давление азота в муфеле печи обычно колеблется в пределах 5—50 мм вод. ст. (49—490 н/мг). 2. Среднеуглеродистые стали применяют для изготовления деталей, испы- тывающих значительные нагрузки. Процесс азотирования этих улучшенных сталей ведут при температуре не выше 650° С. * Охлаждение после азотирования производят в воде, в масле, на воздухе в муфеле или в муфеле вместе с печью. Для повышения вязкости азотированного слой желательно быстрое охлаждение. ! **‘Температура окончательного нагрева долнсна совпадать с температурой i закалки стали.
0,015—0,030 мм (рис. 64), в зависимости от назначения и формы
детали. Этот нитридный (или ’карбонитридный) слой обладает
повышенной стойкостью во влажной воздушной среде (стоек
в течение нескольких лет), в водопроводной воде (в течение
нескольких месяцев), неочищенном масле, бензине, перегретом
паре, слабых щелочных растворах и других средах. В растворах
кислот и морской воде азотированный слой малостоек против
коррозии.
Коррозионная стойкость азотированной стали определяется
тте только стойкостью нитридного слоя, но и способностью этой
стали пассивироваться с образованием защитной пленки окисно-
107
го характера. Азотированная и неазотированная стали имеют
различные пр величине электрохимические потенциалы (рис. 65).
Азотирование углеродистой стали повышает ее твердость,
пределы прочности и текучести. Пластичность и вязкость тюни-
Продолжительность испытания
Рис. 65. Кривые изменения потен-
циала в водопроводной воде стали
30 до и после азотирования при
разных температурах в течение
30 мин (А. В. Рябченков и
В. Д. Яхнина)
10 ч. NaCl, 25 ч. агар-агара на
слоя синеют).
жаются лишь в случае, если
сталь перед азотированием не
подвергается термической об-
работке [144], [145]. Очень рез-
ко возрастает после азотиро-
вания усталостная прочность
стали (на 25—60%) и особен-
но (примерно на 100%) кор-
розионно-усталостная проч-
ность (табл. 16).
При контроле азотирован-
ных деталей осматривают их
поверхность (она должна быть
матовой), подвергают их ми-
кроанализу (слой 0,015—
0,03 мм), испытывают в тече-
ние суток в водопроводной во-
де или влажной камере (не
должно быть следов ржавчи-
ны) или наносят на детали ре-
актив состава: 6 ч. K4Fe(CN)6,
1 л воды (дефектные участки
Таблица 16
Влияние антикоррозионного азотирования
на коррозионно-усталостную прочность стали 30 [146]
Предел выносливости на базе 107 циклов
Состояние поверхности образцов на воздухе в 0,004%-ном растворе NaCl в 3%-ном растворе NaCl
5= з 5 X о X СЧ * X Л X о X кГ/мм2 5 X о X
Без азотирования После азотирования (600° С, в течение 2 ч) Примечание. В скобках указ носливости по сравнению с неазотиро) 25 35,7 (43) >ано в ванной ( 24,5 35,0 npOUCHI 2талью. 16,7 36,1 (Н6) 'ах уве. 16,4 35,4 пимение 10 19,7 (97) предела 9,8 19,3 1 вы-
108
Азотирование легированной стали
Упрочняющее действие азотирования обусловлено образова-
нием в поверхностном слое дисперсных нитридов легирующих
элементов. Кристаллическая структура и некоторые свойства ни-
тридов приведены в табл. 17.
Таблица 17
Структура и свойства некоторых нитридов [147], [148]
Нитрид Область гомоген- ности или содержание азота в предельно насыщенной фазе (в %) Структура Микро- твердость Плотность в г!см3 Температура плавления в °C
A1N 34,18 Г -1225 3,05
Ti3N 8,9 т — 4,77 —
TiN 11,1—22,6 гцк 1994 (2160) 5,43 (5,21) 3205
ZrN 11,5—13,3 гцк 1983 (1530) 7,34 (6,93) 2980
Nb2N 5,7—7,1 г 1720 8,31 (8,32) 8,4 —
NbN 13,1—13,3 г 1396 2300* (разлагается)
Ta2N 3,0—3,4 г 1220 15,81 2050
TaN V3N 5,8—6,5 г 1060 14,36 (13,8) 3090
8,4—11,9 г 1900 5,98 (5,96) —
VN 16,0-21,6 гцк 1520 6,10 (6,04) 6,51 2360
Cr2N 11,3—11,8 г 1570 1650
CrN 21,7 гцк 1093 5,8—6,1 1500 (диссоциирует)
Mo3N 5,4 т — — —
Mo2N 6,4—6,7 гцк 630 (1570) 8,04 600 (диссоциирует)
MoN 12,73 г — 8,06 600 (разлагается)
w2n 4,39 гцк — 12,2 —
WN 7,08 г — 12,08— 12,2 600 (разлагается)
Мп4ы 5,8—6,1 гцк — — • 460—600 (разлагается)
Mn2N 9,2—11,8 г — 6,2-6,6 —
MnoN2 13,6—17 т — — —
Fe4N(V') 5,3—5,75 гцк ^450 6,57 670 (диссоциирует)
Fe3N (e) 8,1—11,1 г — — —
Fe2N (E) 11,1—11,35 р ^260 6,35 560 (диссоциирует)
109
Наибольшую твердость после азотирования придает стали
алюминий, образующий в ней дисперсные нитриды (рис. 66).
Первоначальное представление о роли частиц нитридов как
«шипов», препятствующих скольжению и повышающих твердость
стали, оказалось недостаточным в связи с развитием дислокаци-
онной теории строения реальных кристаллов. Согласно теории
дислокаций, частицы второй фазы являются препятствиями при
Содержание легирующего элементе
Рис. 66. Влияние легирующих эле-
ментов на поверхностную твер-
дость и глубину азотированного
слоя [23]
передвижении дислокации, так
как последняя не находит про-
должения пригодной для движе-
ния плоскости скольжения в но-
вой решетке второй фазы, имею-
щей другую сингонию. Встречая
на своем пути частицы второй
фазы, дислокации могут либо за-
медлить свое движение, либо
полностью приостановиться. Ес-
ли дислокации все же миновали
(обогнули) Частицу, то вокруг
последней остается петля дисло-
кации и, следовательно, поле
дальнедействующих напряжений.
Приближаясь к таким «окайм-
ленным» частицам, другие ди-
слокации будут испытывать тор-
можение; напряжения, необходи-
мые для преодоления частиц, ок-
руженных петлями, гораздо
больше, чем при отсутствии пе-
тель. Упрочнение сплава будет
тем больше, чем меньше отрезки,
на которые может перемещаться
дислокация, что непосредственно связано с дисперсностью выде-
ляющихся нитридов, т. е. межчастичным расстоянием. Когда
большинство межчастичных объемов будет заполнено петлями
(и будет под влиянием полей напряжений), наступит состояние
максимального упрочнения.
Стали для азотирования. Режимы термической обработки
и механические свойства азотируемых сталей приведены в
табл. 18. Наиболее широко применяется сталь 38ХМЮА. Эта
сталь имеет высокие механические свойства, насквозь прокали-
вается при диаметре до 50 лш, приобретает после закалки твер-
дость НВ 420—480, а после отпуска при 600—625° С твердость
НВ 260—290.
Сталь 38ХМЮА рекомендуется закаливать с 930° С в масле
или воде, причем оптимальная 'прочность достигается при закал-
ке в воде (рис. 67). Минимальная хрупкость азотированного
ПО
Таблица 18
Режим термической обработки и механические свойства азотируемых сталей
Марка стали Термическая обработка Механические свойства (не менее)
Закалка Отпуск °в сэ «о Ф в % ан
Темпера- тура в °C Охлаж- дающая среда Темпера- тура в °C К Д 5 X' о X 3 д <2 д X о X X о X сч * X
38ХМЮА 930 Масло или вода 625—650 85 83 100 98 15 50 9 8,8
35ХЮА 930 Масло 625—650 75 73 90 88 10 45 8 7,8
38ХВФЮА 900—950 600-650 85 83 100 98 12 50 9 8,8
30ХН2ВФА 860 » 540—620 105 102 85 83 12 55 10 9,8
30Х2НВФА 910 » 540 120 117 100 98 10 45 8 7,8
850—870 » 620 100 98 85 83 12 55 12 Н.7
ЗОХЗВА 870—890 » 580—620 85 83 100 98 15 50 10 9,8
18Х2Н4ВА 860—870 » 525—575 105 102 120 117 12 55 12 Н,7
40ХНВА 850 » 620 95 93 по 108 12 50 8 7,8
40НХМА 850 » 620 85 83 по 108 12 55 10 9,8
1X13 1000—1050 Масло или вода 700—790 42 41 60 59 20 60 9 8,8
1Х18Н9Т 1100—1150 Вода — 20 19 55 54 40 55 12 11,7
4Х14Н2В2 1050 Масло 630 95 93 по 108 12 35 2 1,9
20ХЗМВФ 1030—1080 660—700 75 73 90 88 12 40 — —
слоя на стали 38ХМЮА, закаленной с 900° С в масле, достигает-
ся при отпуске на более низкую твердость (HRC 25—27) [150].
По данным работы [149], меньшей хрупкостью обладает азотиро-
ванный слой стали, закаленной с 930° С в воде. Высокочастотный
нагрев этой стали под закалку - обеспечивает получение более
тонкой структуры, способствующей существенному повышению
вязкости азотированного слоя [150].
Наличие -в сталях 38ХМЮА, 35ХЮА значительного количест-
ва алюминия вызывает ряд осложнений в технологии выплавки,
а также в горячей механической и термической обработках.
В связи с этим в ЦНИИЧМ создана и внедрена в производство
сталь 38ХВФЮА, не имеющая в своем составе молибдена и со-
держащая 0,4—0,7% А1 [152]. По сравнению со сталью 38ХМЮА
эта сталь имеет такую же прочность, вязкость, прокаливаемость,
но ее твердость после азотирования заметно ниже, а азотирован-
ный слой менее хрупок.
Для повышения не только износостойкости, но и главным об-
разом предела выносливости все шире подвергают азотированию
детали, работающие в условиях повторно-переменных нагрузок
и изготовляемые из сталей 30Х2Н2ВФА, 18Х2Н4ВА, 30Х2Н2ВА,
40ХНВА, 40ХНМА, ЗОХЗВА, ЗОХНМФ, ЗОХНЗМФ, ЗОХМФ и др.
111
Поверхностная твердость этих сталей после азотирования срав-
нительно невелика 1И колеблется в пределах HV 550—900, нопро-
каливаемость большинста из них выше, чем у стали 38ХМЮА,
а азотированный слой менее тверд и хрупок. Аналогичные стали
азотируют и за рубежом [153].
Иногда азотируют улучшаемые легированные стали, напри-
мер 40Х (рис. 68) [154]. После азотирования эти стали приобре-
тают более низкую твердость, чем после цементации (HRC
50—58 при переводе с HV 10). Однако износостойкость такой
Рис. 67. Влияние температуры закалки на различные механические свойства
азотированных пластинок из стали 38ХМЮА, закаленных в воде (а) [149], и
на предел их прочности при изгибе (б) после закалки в масле (кривые 1 и 2)
[150] и закалки в воде (кривая 3) [149].
стали почти не уступает износостойкости цементованной, а ко-
робление в процессе обработки меньше. Например, на заводе
им. Орджоникидзе азотируют рейки из стали 40Х, а на ГАЗе —
сложного профиля копиры из этой же стали.
Все шире применяется азотирование аустенитных и нержа-
веющих теплостойких сталей [19], [131], [155], [156].
Аустенитная сталь, как известно, имеет низкую износостой-
кость, но в то же время обладает рядом ценных свойств: пара-
магнитностью, высокой жаропрочностью, окалиностойкостью,
коррозионной стойкостью и высокой ударной вязкостью при тем-
пературе ниже 0° С.
Азотирование — наиболее эффективный способ повышения
износостойкости аустенитных нержавеющих сталей.
В ряде зарубежных работ освещены результаты исследова-
ний сталей, содержащих титан. Эти стали азотируются быстрее,
чем хромомолибденоалюминиевая, и отличаются более высокой
поверхностной твердостью и 1красностой1костыо [157], [158].
112
Рекомендуется соотношение между титаном и углеродом в
таких сталях поддерживать более 10 (рис. 69). Такая сталь мо-
жет иметь, например, следующий состав: 0,2—0,3% С; 3,2—
3,8% Ni; 1,0—1,4% Сг; 0,2—0,3% Мо и 2,5—3,0% Ti. Предлага-
ется стареющая сталь
13ХНДФЮТ, температура ста-
рения которой (500—520° С)
совпадает с температурой азо-
тирования. Эта сталь хорошо
обрабатывается резанием, хо-
рошо сваривается и имеет вы-
сокий предел текучести в круп-
ных отливках и поковках [159].
Предлагается и другая ста-
реющая сталь, содержащая
5% Ni; 2% Al; 0,5% Сг; 0,25%
Мо [160]. Разработана сталь,
содержащая 18% Ni, насыще-
ние которой азотом при 425—
455° С в течение 20 ч приводит
к превращению в поверхност-
ном слое феррита в аустенит,
а последний при охлаждении
на воздухе превращается в
мартенсит [161].
Рекомендовано подвергать
азотированию (взамен циани-
рования) инструмент из бы-
строрежущих сталей Р9 и Р18
[162].
Неоднократно предлагалось
детали из углеродистой или
легированной стали сначала
алитировать (насыщать их по-
верхность алюминием), а за-
тем подвергать азотированию,
однако этот процесс не внед-
0 0.0!0,05 0J 0,2 0,3 0,4 0,5 0.6 мм
б)
Рис. 68. Изменение микротвердо-
сти по глубине азотированного
слоя различных сталей [154]:
а — температура 500° С, степень дис-
социации аммиака 20—30%, продолжи-
тельность 48 ч\ б — температура
550° С, степень диссоциации аммиака
35—40%, продолжительность 36 ч
(Е. М. Морозова и А. П. Скузовато-
ва); 1 — сталь 38ХМЮА; 2 — сталь
зОХС; 3 — сталь 20Х; 4 — сталь 18ХГТ;
5 — сталь 40Х; 6 — сталь ШХ15; 7 —
сталь 9ХС
рен ввиду трудоемкости и
малой пригодности для обработки ответственных деталей
[569].
Азотированию подвергают также детали из высокопрочного
магниевого чугуна [163], [164], [165] (в частности, коленчатые валы
тепловоза [166] и детали из специальных чугунов, легированных
алюминием [19]).
Газовое азотирование. Азотирование — завершающая опера-
ция технологического процесса, после которой следуют лишь
шлифование или доводка и протирка деталей.
113
Перед азотированием притупляют острые кромки деталей, а
их поверхность очищают электрическим обезжириванием или
промывкой в бензине. Особенно большое влияние на результаты
газового азотирования оказывает характер подготовки поверх-
ности аустенитных и нержавеющих сталей. Эти стали подвергают
пескоструйной обработке или травят; 'перед травлением места,
не подлежащие азотированию, 'покрывают кислотостойкой крас-
кой. Для травления используют, например, 75%-ный раствор со-
ляной кислоты при 50—60° С (для уменьшения растворения стали
Рис. 69. Распределение микротвердости по глубине
азотированного слоя сталей, содержащих титан в
различных соотношениях с углеродом (550° С, 5 ч)
[157]
30%-ный раствор серной ‘кислоты, или 20%-ный раствор фосфор-
ной кислоты при комнатной температуре).
Для травления нержавеющих сталей 1X13, 2X13 и 3X13 ре-
комендуется ванна, состоящая из 45,5% соляной кислоты, 45,5%
воды и 9% медного купороса. Температура ванны 40—45° С, про-
должительность травления 15—20 мин. При травлении стравли-
вается слой толщиной 0,015—0,02 мм. После травления детали
промывают, сушат и загружают в контейнер для азотирования.
Детали, подвергнутые травлению или пескоструйной обработке,
нельзя брать руками.
Однако и после такой подготовки поверхности к азотирова-
нию не всегда обеспечивается получение на деталях слоя равно-
мерной глубины и твердости.
Наиболее рациональными следует признать два новых спосо-
ба азотирования нержавеющих высокохромистых и аустенитных
сталей. При этих способах окисная пленка на поверхности дета-
лей восстанавливается в печах азотирования.
Первый из этих способов заключается в том, что в муфель
печи помещают плоскую коробку со смесью хлористого аммония
114
(3%) и кварцевого песка (97%) .в количестве 2—3 кг на 1 ж3
объема муфеля [167]. Продукты диссоциации хлористого аммо-
ния восстанавливают окисную пленку. Недостатком этого спосо-
ба является невозможность регулирования действия депассива-
тора и забивание отводящего трубопровода твердым осадком
хлористого аммония. Для проталкивания этого осадка разрабо-
тано шомпольное приспособление.
Второй способ-1 заключается в том, что в течение первых
1,5—2 ч процесса в муфель печи вводятся пары четыреххлори-
стого углерода, полученные путем пропускания аммиака через
жидкий СС14 (расход СС14 составляет 50—75 кг на цикл азоти-
рования в печи ПН-32). В дальнейшем аммиак направляется в
печь по параллельному трубопроводу, минуя сосуд с СС14. Сов-
местное действие хлора и водорода разрушает окисную пленку
[570].
Для подготовки поверхности перлитных и аустенитных сталей
к азотированию часто применяют фосфатирование в ванне с со-
держанием 30—40 а/л препарата Мажеф. Перед фосфати-
рованием детали обезжиривают и декапируют. При такой обра-
ботке окисная пленка, если она есть на поверхности металла,
заменяется пленкой фосфатов, имеющей большую пористость.
Благодаря некоторому растравливанию поверхности и образова-
нию пористой фосфатной пленки активность поверхности насы-
щенного металла возрастает, что ускоряет абсорбцию азота.
Фосфатирование приводит к увеличению твердости азотирован-
ного слоя и его глубины (примерно на 0,05 мм на перлитной
стали).
Для защиты не подлежащих азотированию участков поверх-
ности деталей наиболее часто применяют гальваническое луже-
ние (слой толщиной 12—45 мк) или двухслойное гальваническое
покрытие медью (10—12 мк) и цинком (10—12 мк). После лу-
жения, чтобы олово не растекалось при азотировании, защища-
емую поверхность дополнительно фосфатируют в препарате
Мажеф. Применяют также покрытие жидким стеклом с после-
дующей сушкой при 100—120° С. Однако жидкое стекло несколь-
ко смещается по поверхности, что может нарушить границу меж-
ду азотированной и неазотированной поверхностями.
При азотировании аустенитных и нержавеющих сталей с вве-
дением в муфель печи хлористого аммония или четыреххлористо-
го углерода гальваническое лужение нельзя применять для за-
щиты неазотируемых участков; следует использовать химическое
(толщина слоя 8—10 мк [168] или гальваническое (до 30 мк)
никелирование либо покрытие жидким стеклом.
Для конструкционных, теплостойких и инструментальных
сталей чаще применяют изотермическое азотирование при 500—
1 Способ предложен А. В. Смирновым (Бюллетень изобретений № 16,
1961).
115
520° С [169]. При режимах азотирования, приведенных в табл. 19.
стали 38ХМЮА, 35ХМЮА, 38ХВФЮА и др. приобретают мак-
симальную поверхностную твердость и детали из них минималь-
но коробятся (рис. 70). Для ускорения азотирования сталей
35ХМЮА и 35ХЮА процесс иногда проводят при повышенной
температуре (540° С) или применяют ступенчатый режим: снача-
ла при 500—520° С, а затем при 540—560° С. Для снижения
хрупкости азотированного слоя
Рис. 70. Распределение твердости и микротвердости по
глубине азотированных слоев:
а — сталь 35XMIOA, азотированная при 520° С [169]; б — сталь
2Х18Н8В2, азотированная в течение 48 ч (А. Г. Андреева)
рования (последние 2—3 ч) либо поддерживают высокую сте-
пень диссоциации аммиака, либо полностью прекращают его
поступление в печь и выдерживают детали в среде диссоцииро-
ванного аммиака; назначение этой операции — некоторое де-
азотирование поверхностного слоя.
В некоторых случаях применяют низкотемпературное азоти-
рование при 380—430° С. Снижение температуры азотирования
возможно лишь при введении в печь катализаторов: ферросили-
ция (повышает степень диссоциации аммиака) и обмазки из на-
сыщенного водного раствора хромового ангидрида (повышает
температуру поверхностных слоев стали вследствие реакции
восстановления хрома водородом). При 380°С в течение 60—80 ч
или при 430° С в течение 24 ч на деталях из сталей ЗОХМФ,
ЗОХМА, 35XH3M, 35ХНЗМФ и др. образуется азотированный
слой глубиной 0,20—0,25 мм, имеющий твердость HRC 42—50.
116
При использовании этого режима исключается развитие в серд-
цевине стали отпускной хрупкости второго рода, а азотирован-
ный слой получается нехрупким.
Аустенитные стали азотируют при
550—600° С, и при такой температуре
эти стали азотируются намного мед-
леннее, чем, например, сталь 38ХМЮА
при температуре 500—520° С (рис. 71).
Технология азотирования гильз ци-
линдров дизелей из стали 38ХМЮА,
коленчатых валов из стали 18Х2Н4ВА
и высокопрочного чугуна, пружин из
стали 50ХФА и ряда других деталей
описана в работе [131].
Разработаны некоторые новые спо-
собы азотирования стали.
Азотирование можно проводить пот
давлением с использованием капсул,
наполненных жидким аммиаком (из
расчета 0,5 г на 1 дл*2 поверхности) и
закрытых пробкой из легкоплавкого
металла. Так обрабатывают внутрен-
ние поверхности труб и других дета-
лей, чтобы со-кратить продолжитель-
ность процесса и расход аммиака. При
нагреве пробка плавится и аммиак
испаряется [171].
Азотирование при воздействии уль-
тразвука позволяет несколько увели-
чить глубину и поверхностную твер-
дость азотированного слоя [172].
Азотирование в среде, состоящей
из 1 ч. свежего аммиака, и 3 ч. предва-
рительно диссоциированного аммиака,
повышает вязкость азотированного
слоя и предупреждает образование
сетки и каемки нитридов [173].
На целесообразность азотирования
стали в смеси аммиака с инертными
газами, в том числе азотом, было ука-
зано еще в работе [174]. Позднее в
Венгрии было проведено исследова-
ние, на основании которого для азоти-
рования была рекомендована смесь,
состоящая из 80—90% азота и 10—
20% аммиака [175]. Была отмечена
экономическая выгода применения
Рис. 71. Изменение
глубины и твердости*
азотированного слоя
сталей в зависимости
от температуры азо-
тирования (А. Г. Ан-
дреева) :•
I — перлитный класс
(сталь 38ХМЮА); II —
мартенситный класс
(стали 2X13 и
4X14M2BA); III — ау-
стенитный класс (стали-
25X18Н8; 25X18H8B2 и
4X14H14B2M); I — сталь
Х17Н9Ю; 2 — сталь
38ХМЮА: 3 — сталь
25X18H8B2; 4 — сталь
2X13; 5 — сталь
4X14H2B2; 6 — сталь
40ХНМА; 7 — сталь 45;
8 — железо Армко; 9 —
сталь 25Х18Н8: 10 -
сталь 4X14H14B2M
117
Режимы азотирования сталей различных марок
Таблица 19
Сталь Темпера- тура про- цесса в °C* Степень дис- социации аммиака в % Продол- житель- ность в ч Глубина слоя в мм Получаемая поверхностная твердость HV10 Минимальная поверхностная твердость HV10 по ТУ Наименование обрабатываемых деталей
38ХМЮА— 35ХЮА 510 510 540 1-я 510 20—40 20—40 30—50 ступень 20—40 35 55 35 12 42 0,30—0,35 0,55—0,50 0,45—0,55 0,50—0,70 1000—1150 1000—1150 950—1100 950-1000 950 Гильзы авиамоторов, дизе- лей и насосов, зубчатые ко- леса, коленчатые валы, втулки, кулачки, фиксаторы, детали топливных насосов, шпиндели, пароводная арматура и другие детали
z-я ступень
540 50—60
38ХВФЮА 510 510 20—40 20—40 35 55 0,30—0,35 0,50—0,55 850—950 850—950 750
30Х2Н2ВФА 30Х2Н2ВА 500 500 510 15—30 15—30 20—40 35 55 55 0,25—0,30 0,45—0,50 0,50—0,55 650—750 650—750 600—700 600
ЗОХЗВА 500 500 510 15—20 15—20 20—40 35 55 55 0,25—0,30 0,45—0,50 0,50—0,55 700—850 700—850 650—750 650
18Х2Н4ВА 500 500 510 15—30 15—30 20—40 35 55 55 0,25—0,30 0,45—0,50 0,50—0,55 650—750 650—750 600—700 600
40ХНВА— 40ХНМА, 34ХН4М 500 500 510 15—30 15—30 20—40 35 50 55 0,25—0,30 0,45—0,50 0,50—0,55 550—650 550—650 500—600 500
Продолжение табл. 19
Сталь Темпера- тура про- цесса в °C* Степень дис- социации аммиака в % Продол- житель- ность в ч Глубина слоя в мм Получаемая поверхностная твердость HV10 Минимальная поверхностная твердость HV10 по ТУ Наименование обрабатываемых деталей
20ХЗМВФ 510 20—40 25 0,20—0,30 — — Паровые клапаны, втулки и другие детали турбин
25Х5МА 1-я 530 2-я 550 ступень 25-35 ступень 40—60 36 12 0,35—0,45 820—1000 — Плунжерные пары форсунок насосов
1X13, 2X13, 3X13 510 550 1-я 530 2-я 580 20—40 40—55 ступень ступень 55 55 20 20 0,15—0,25 0,25—0,35 0,25—0,27 950—1100 850-950 800—850 850 Различные детали, испыты- вающие эрозионный износ (ло- патки направляющего аппара- та), работающие на трение в коррозионной среде, детали турбин
15X11МФ, 15Х12ВМФ 1-я 530 2-я 580 ступень 35 ступень 65 10 18 0,32—0,37 HRA 82,5 Лопатки соплового аппарата, штоки, втулки, седла, клапа- ны и другие детали турбин
4Х14Н14В2М 550 570 640 40—55 45—60 45—65 55 55 55 0,08—0,12 0,10—0,15 0,12—0,18 850—1000 800—950 700—800 750 Клапаны двигателей внут- реннего сгорания сферы, ва- лики
120
Продолжение табл. 19
Сталь Темпера- тура про- цесса в °C* Степень дис- социации аммиака в % Продол- житель- ность в ч Глу'ина слоя в мм Получаемая повеохностная твердость НУ\ 0 Минимальная поверхностная твердость HV10 по ТУ Наименование обрабатываемых деталей
25Х18Н8В2 550 570 40—55 45—60 55 55 0,15-0,22 0,18—0,25 850—1000 800—900 800 Клапаны двигателей внут- реннего сгорания, сферы, валики
4Х14Н2В2 550 570 40—55 45—60 55 55 0,18—0,25 0,20—0,30 900—1000 800—950 800 Различные детали, работаю- щие при повышенных темпера- турах в агрессивных средах
Х12Ф 510 510 20—45 20-45 25 55 0,15—0,20 0,20—0,30 900—1000 900—1000 850 Матрицы и пуансоны для горячей штамповки, штампы, прессформы для литья под дав- лением пластмасс, каучука, стекла, валки для горячей прокатки шариков и другой инструмент
4Х8В2, ЗХ2В8 510 20—45 36 0,30—0,35 900—1000 850
4ХВ2С, 7X3 560 560 20—45 20—45 55 55 0,45—0,55 0,45—0,55 700—750 675—750 600
40Х 500 35 48 0,50 450 — Копиры сложного профиля, рейки, прессформы, зубчатые колеса
* Температура обычно поддерживается с точностью ±10° С.
газовой среды и обнаружено некоторое ускорение про-
цесса.
Однако работа, выполненная автором совместно с Ю. В. Со-
рокиным на сталях 38ХМЮА и 4Х14Н14В2М сначала в лабора-
торных, а затем в производственных условиях (применялись га-
зовые смеси, состоящие из 90% Ns, 10% NH3 и 80% N2, 20% NH3)„
не подтвердила, что процесс азотирования в таких средах замет-
но ускоряется по сравнению с азотированием в аммиаке. Было
установлено, что микроструктура стали 38ХМЮА, азотированной
в новой газовой среде, отличается тем, что в ней отсутствуют или
минимально развиты нитридная каемка и прожилки. В полном
соответствии с этим коррозионная стойкость стали 38ХМЮА
оказалась несколько ниже, чем после азотирования в аммиаке.
Внутренние сжимающие напряжения в азотированном слое и
предел выносливости образцов сталей 38ХМЮА и 4Х14Н14В2М
после азотирования в смеси азота и аммиака оказались несколь-
ко выше, чем после азотирования в аммиаке.
Опробовано азотирование магниевого чугуна в смеси, состоя-
щей из 65% N2 и 35% NH3. Поверхностная твердость образцов,
азотированных в этой среде, оказалась несколько большей, чем
после азотирования в аммиаке.
Вопрос о рациональности применения для азотирования ста-
ли и чугуна смеси азота и аммиака остается пока открытым.
Некоторое применение, главным образом в ФРГ [176]—[180],
нашло азотирование в тлеющем разряде. При этом методе дета-
ли подключают в цепь высокого напряжения (800—3500 в) в ка-
честве катода, а анод располагают над ними или вокруг них.
При возбуждении тлеющего разряда ионы газа бомбардируют
поверхность деталей и быстро нагревают ее. В муфеле поддер-
живается низкое давление (3—10 мм рт. ст. или 339—1330 н/ж2).
Такой метод азотирования имеет следующие преимущества: про-
должительность процесса несколько сокращается, на деталях
сложной конфигурации получаются равномерные по глубине
слои, отпадает надобность в печах для нагрева. Поскольку элек-
трический ток расходуется только на нагрев поверхности дета-
лей, коэффициент его использования высок. В тлеющем разряде
азотируют валы, зубчатые колеса, шпиндели и другие детали
машин, изготовляемые из хромоалюминиевомолибденовой и
аустенитной сталей (последняя сталь азотируется без предва-
рительной подготовки поверхности).
При азотировании при 500 и 600° С в тлеющем разряде высо-
копрочного магниевого чугуна глубина слоя получалась пример-
но в 2 раза больше, чем при азотировании с обычным нагревом
[181]. При использовании этого способа азотирования в произ-
водстве детали следует устанавливать в муфеле с учетом тре-
бований, необходимых для возникновения тлеющего разряда
(открытая поверхность деталей, определенное расстояние между
121
деталями и анодом). Это ограничивает применение данного спо-
соба в массовом производстве, но в мелкосерийном производст-
ве он, по-видимому, может быть с успехом использован.
Еще 35 лет назад было обнаружено ускорение процесса азо-
тирования стали при нагреве т. в. ч. Позднее это многократно
подтверждалось. По сравнению с обычным нагревом при нагреве
т. в. ч. при температурах 500—550° С и небольшой выдержке (3—
5 ч) процесс азотирования ускоряется в 2—3 раза [181], [183],
[184].
О причинах ускорения азотирования при индукционном нагреве
высказывались различные предположения. К этим причинам наи-
более часто относили: 1) влияние сжимающих и растягивающих
напряжений, вызываемых явлением магнитострикции ферромаг-
нитного тела под воздействием быстропеременного магнитного
поля, и 2) влияние особых условий диссоциации аммиака, проис-
ходящей только вблизи разогретой поверхности стали.
Экспериментальная проверка последней точки зрения пока-
зала ее правильность [185]. Втулки из стали 40Х нагревали в
индукторе током от лампового генератора (частота 200 кгц),
от машинного генератора (частота 7680 гц), а также от спира-
ли сопротивления, помещенной внутри втулки. Глубина азотиро-
ванного слоя при всех указанных способах нагрева оказалась
одинаковой и несколько большей, чем при нагреве втулок в
печи.
Таким образом, если при нагреве в печи к поверхности дета-
ли подходит смесь аммиака и продуктов его диссоциации, то при
внутреннем нагреве стали к ее поверхности подходит еще не
диссоциированный аммиак. Это создает иные, более благо-
приятные условия для протекания реакций в газовой среде
вблизи насыщаемой поверхности и для абсорбции поверхнос-
тью стали образующегося активного азота, что и является ос-
новной причиной ускорения процесса азотирования.
Следует, однако, отметить малую перспективность азотиро-
вания стали с нагревом т. в. ч., что объясняется невозможнос-
тью обработки одновременно значительного количества дета-
лей и тем, что существенно ускоряются только кратковременные
процессы. При увеличении выдержки влияние газовой фазы на
ускорение процесса уменьшается, и определяющим фактором
становится диффузия азота в глубь стали.
Азотирование в жидкой среде. Азотирование стали в жид-
кой среде можно осуществлять:
1. В цианистых паннах при 550—580° С; при этом получают
азотированный слой, с поверхности которого может возникать
тонкая карбонитридная корочка, глубже которой углерод не
диффундирует. Обработка конструкционных («мягкое азотиро-
вание») и быстрорежущих сталей в таких ваннах описана
в гл. IV.
122
2. В нейтральной соляной ванне (содержащей, например,
31% ВаС12, 48% СаС12 и 21% NaCl), через которую продувается
аммиак. Для электрохимической защиты стали от корродирую-
щего действия расплавленных солей детали подключают в ка-
честве катода в цепь постоянного тока [186]. Этот способ удобен
для антикоррозионного азотирования, заканчиваемого закал-
кой стали. Продолжительность такого процесса при 730° С со-
ставляет 10—15 мин.
3. В водных растворах аммиака с нагревом т. в. ч. (впервые
такой процесс был исследован в Польше). Этот метод азотирова-
ния заключается в том, что вокруг образца, погруженного в вод-
ный раствор аммиака и нагретого т. в. ч., образуется газовая ру-
башка, в состав которой входит аммиак.
В первых работах [96]—[97] рекомендовалось применять на-
сыщенный (25%-ный) водный раствор аммиака, в последую-
щих [187] была показана целесообразность использования ме-
нее насыщенных растворов (12,5%-ных), что уменьшает улету-
чивание аммиака и повышает стабильность состава ванны.
По сравнению с обычным азотированием глубина слоя при таком
способе и глубина азотированного слоя получаются в 2 раза
больше, а твердость почти такой же. •
В широком интервале температур (600—1300° €) было изу-
чено влияние температуры процесса на результаты азотирова-
ния этим методом железа и различных сталей [187]—[190].
У азотированных образцов стали 15 было обнаружено раз-
личие в травимости зоны слоя с s-фазой, образующейся при
насыщении в газовой среде и в водном растворе аммиака. Это
было объяснено различием в составе азотирующих сред, окру-
жающих образцы (наличие кислорода при азотировании в вод-
ном растворе аммиака) [188].
Недостатками метода являются трудности, связанные с из-
мерением температуры стали, бурным выделением аммиака из
ванны во время процесса азотирования и невозможностью об-
работки одновременно больших количеств деталей.
Контроль и механическая обработка азотированных дета-
лей. После азотирования детали подвергают внешнему осмот-
ру, определяют величину их коробления, измеряют поверхност-
ную твердость по Виккерсу (нагрузка 10 или 30 кг) или Супер-
Роквеллу (нагрузка 15 или 30 кг) и определяют глубину слоя
и его хрупкость по виду отпечатка пирамиды прибора Виккер-
са [19].
Азотированные детали подвергают шлифованию или только
полированию и притирке. При обработке деталей из аустенит-
ных сталей снимают припуск менее 0,05 мм, а при обработке
деталей из конструкционных сталей 0,05—0,1 мм. После снятия
припуска хрупкость и коррозионная стойкость азотированного
слоя конструкционных сталей снижаются; хрупкость аустенит-
123
ной и нержавеющей сталей также снижается, а стойкость про-
тив коррозии повышается.
При азотировании происходит небольшое увеличение раз-
меров деталей (рис. 72). Из-за разницы в удельных объемах
азотированного слоя и сердцевины деталей в них возникают
большие внутренние напряжения — сжимающие в азотирован-
ном слое и растягивающие в сердцевине. Вследствие этого про-
исходит коробление азотированных тонких и несимметричных
деталей (рис. 72,6). Коробление после азотирования в отличие
а)
S)
Рис. 72. Увеличение диаметра (а) сплошных цилиндрических образцов из
хромоалюминиевомолибденовой стали (И. Е. Конторович и Р. И. Мочалкин)
п изменение наружного и внутреннего диаметров .полых образцов из той же
стали (б) в зависимости от толщины стенки;’ азотирование при 520° С в те-
чение 72 ч (Хомерберг). Режимы азотирования:
1 - 500° С; 18 ч и 600° С, 18 ч (0,57 мм)\ 2 — 500° С, 12 ч и 600° С, 12 ч (0,35 мм)}
3 — 600° С, 72 ч (0,49 мм)} 4 — 500'С, 48 ч (0,34 мм)
ст коробления после цементации более закономерно и в боль-
шинстве случаев меньше. Например, замена для больших теп-
ловозных зубчатых колес диаметром до 800 мм цементации
(стали 12ХНЗА и 12Х2Н4А) азотированием (сталь 30Х2Н2ВФА)
позволила обходиться без последующего шлифования зубьев
[191].
Структура азотированного слоя легированной стали
Вблизи поверхности азотированной стали 38ХМЮА чаще
располагается тонкая хрупкая и нетравящаяся нитридная зона,
которая состоит из 8- и у'-фаз или из £-, 8- и у'-фаз. Под этой зо-
ной находится основная зона азотированного слоя, отличающаяся
при небольшом увеличении от сорбитовой структуры сердцевины
стали лишь большей травимостью. Эта зона состоит из а- и
у'-фаз; в той ее части, которая примыкает к нитридной каемке,
124
иногда присутствуют нитриды железа в виде тонких прожилок.
Дисперсные нитриды легирующих элементов при обычно приня-
тых увеличениях микроструктуры не видны (рис. 73 и 74).
На основании электронно-микроскопического исследования
пришли к выводу, что в азотированном слое стали 38ХМЮА под
влиянием выделяющихся нитридов легирующих элементов про-
Рис. 73. Микроструктура азотированного слоя стали:
с - сталь 35ХМЮА, 520° С. 48 ч (X150); б — сталь 4X14H14B2M, 560° С, 60 ч (Х100)
исходит дробление блоков. Нитриды имеют размеры около 200—
500 А и располагаются преимущественно по границам этих бло-
ков (192]. Достоверных сведений о величине и твердости нитридов
алюминия, выделяющихся в стали, нет.
При большом содержании в стали нитридообразующих ле-
гирующих элементов нитридные фазы могут быть обнаружены
рентгеновским и микроскопическим методами.
Фазовый состав азотированного слоя стали 15X11МФ
(530° С, 20 ч + 580° С, 20 ч) следующий: на поверхности е + у';
на глубине 0,02 мм у' + а + CrN; на глубине' 0,07—0,28 мм
а + CrN и в сердцевине а + Сг2зС6 [131].
Детально изучен химический состав азотированного слоя
стали 25Х18Н9В2 [193]. Сердцевина представляет собой у-твер-
125
дый раствор (около 15% Сг, около 2% W, 8% Ni) и карбид
(Сг, Ре)2зС6- К сердцевине 'Примыкает зона азотированного
слоя (зона 111 на рис. 75), состоящая из у-твердого раствора
(менее 4% N, менее 12% Сг, около 10% Ni, 1,5% W) + (Fe,
Cr)4N+ (Сг, W)N + (Сг, Fe)23 С6. Эта зона отличается пони-
женной коррозионной стойкостью и отрицательным электрод-
ным потенциалом вследствие пониженного содержания в ней
хрома и азота (рис. 75). Ближе к поверхности располагается
зона (зона 11 на рис. 75), состоящая из твердого раствора (бо-
лее 4% N, 2% Сг, 10% Ni и 0,4%.
W) + (Fe, Сг, W, Ni)2(N, С) +
+ (Сг, W)N. Эта зона отличается
Рис. 74. Микроструктура зоны азо-
тированного слоя стали 35ХМЮА
с нитридной корочкой (Х200).
циала по глубине слоя азотированных
сталей [193]:
1 — 4Х14Н14В2М; 2 — 25Х18Н8В2; 3 —
4Х14Н2В2
ным потенциалом благодаря наличию s-фазы (комплексного
нитрида с гексагональной решеткой) и у-твердого раствора, обо-
гащенного азотом и никелем. На поверхность выходит тонкая
зона (зона /) с пониженной коррозионной стойкостью, которая
была объяснена механическими повреждениями этой зоны (ше-
роховатость после шлифования и т. д.).
На рис. 73, б показана микроструктура азотированной стали
4Х14Н14В2М. Эта сталь содержит значительное количество угле-
рода, и поэтому на ее микроструктуре отчетливо выявляются
карбиды.
Свойства азотированной легированной стали
Азотированный слой обладает высокой твердостью (см. рис. 70
и 71) и износостойкостью. Износостойкость азотированной стали
в 1,5—4 раза выше износостойкости закаленных высокоуглеро-
126
дистых, цементованных, а также цианированных и нитроцемен-
тованных сталей.
Азотирование снижает вязкость стали, повышает ее проч-
ность, ослабляет влияние концентраторов напряжений на сниже-
ние предела выносливости стали (рис. 76) и существенно повы-
шает предел выносливости, особенно тонких деталей и деталей,
работающих в некоторых коррозионных средах [194], {196].
Предел выносливости стали &
марки 38ХМЮА при повторно- 1D7^m2 о/о
переменном изгибе повышается
с 49 кГ/мм2 (48 X 107 н/м2) до
58 лег/мм2 (56,9 X 107 н/м2), ес-
ли глубина слоя 0,30 мм, и до
60 кГ)мм2 (58,8 X 107 н/ж2) при
глубине слоя 0,45 мм.
Правка детали после азотиро-
вания снижает сжимающие внут-
ренние напряжения и, следова-
тельно, предел выносливости ста-
ли [197]. Декремент затухания
колебаний после азотирования
может изменяться по-разному.
У сталей 38ХМЮА и 25Х2МФА
азотирование снижает декремент
колебаний, а у аустенитных ста-
лей Х14Н14В2С2Т и 1Х17Н13М2Б,
наоборот, повышает [131].
Азотирование повышает со-
противление задираемости и на-
липанию металла под нагрузкой
и особенно при повышенных тем-
пературах. В связи с этим азоти-
рованная сталь 38ХМЮА рекомендуется для пароводяной арма-
туры, работающей при температурах до 500—520° С, а сталь
1Х17Н13М2Б — до 600° С.
Азотированная сталь обладает теплостойкостью (красностой-
костью), и ее твердость сохраняется после воздействия высоких
температур. Например, сталь 38ХМЮА сохраняет свою твер-
дость при нагреве до 500—520° С в течение нескольких десятков
часов. Еще большую устойчивость твердости против воздейст-
вия высоких температур (до 600° С) имеет аустенитная
сталь. Однако при длительной эксплуатации в условиях высоких
температур азотированный слой постепенно рассасывается, на
поверхности образуются окислы и происходит глубокая диффу-
зия кислорода по нитридным прожилкам, образующимся как в
процессе азотирования, так и при длительном нагреве во время
эксплуатации.
тированных (кривая /) и неазо-
тированных (кривая 2) образцов
диаметром 40 мм, имеющих раз-
личные дефекты и изготовленных
из стали, содержащей 0,36% С;
0,98% Мп; 1,48% Сг и 0,39% Мо
(С. В. Серенсен и И. Е. Конторе-
вич):
I — гладкий образец;
с буртом, г = 2,2 мм; х
с буртом, г = 2 мм; IV — образец
поперечным отверстием, d
— образец
— образец
Ill
5 мм
с
127
Ниже указаны стали, которые рекомендуются для изготовле-
ния азотируемых деталей, работающих при высоких температу-
рах [131]:
Рабочая тем- пература в °C 400—490 490—510 510—540 540—570 570—590 590—600 Рекомендуемые марки стали 38ХМЮА, 25Х2МФА 25Х2М1Ф, 1X13 15X11МФ 15Х12ВМФ 1Х18Н9Т, 1Х14Н14В2М 25Х18Н8В2. 1Х17Н13М2Б, ЗХ19Н9МВБТ
В результате азотирования коррозионная стойкость конструк-
ционной стали (в среде воздуха, водопроводной воде, перегре-
том паре, слабых щелочных растворах) повышается и, наоборот,
Рис. 77. Влияние режима азотирования на ка-
витационно-эрозионную стойкость стали
38ХМЮА:
1 — азотирование по режиму А(525° С, 35 ч); 2 — азо-
тирование по режиму Б(510° С, 12 ч + 540° С. 40 ч}\
3 — шесть циклов азотирования по режиму Б; 4—один
цикл азотирования по режиму Б с последующей
пескоструйной обработкой; 5 — один цикл азотирова-
ния по режиму Б с последующим шлифованием;
6 — сталь после улучшения J131]
аустенитной хромоникелевой и нержавеющей хромистой стали
некоторых марок понижается. Окалиностойкость последних ста*
лей также понижается. Это объясняется тем, что в азотирован-
ном слое этих сталей из твердого раствора устраняется значи-
тельная часть хрома, входящего в состав образующихся нитри-
дов. В аустенитной стали некоторых составов, например с малым
содержанием никеля, это может сопровождаться даже выпаде-
нием в азотированном слое a-фазы, в результате чего поверхно-
стный слой становится слегка магнитным [198].
Азотированная сталь обладает высокой эрозионной стойкос-
тью в потоках горячей воды и водяного пара.
Так как разрушение при кавитационной эрозии начинается
с у'- и 8-фаз, следует стремиться к получению в слое наимень-
128
шего количества нитридных выделений. В этом случае, по дан-
ным работы [131], азотирование повышает сопротивление стали
38ХМЮА кавитационной эрозии в 17 раз, тогда как цинковое
покрытие повышает его в 8 раз (рис. 77).
НИТРОЦЕМЕНТАЦИЯ И ЦИАНИРОВАНИЕ
Поверхностное насыщение стали одновременно углеродом и
азотом в газовой среде называется нитроцементацией, а в рас-
плавленной цианистой ванне — цианированием.
Нитроцементации и цианированию подвергают при темпера-
туре 820—950° С конструкционную низкоуглеродистую сталь,
среднеуглеродистую (легированную и нелегированную), а также
нержавеющую для повышения их поверхностной твердости, из-
носостойкости и предела выносливости. Улучшенную среднеуг-
леродистую (легированную и нелегированную) сталь обрабаты-
вают при 570—600° С («мягкое азотирование») для повышения
износостойкости и предела выносливости, а быстрорежущую
при 550—560° С для повышения поверхностной твердости, изно-
состойкости и теплостойкости.
Особенности совместной диффузии в сталь углерода
и азота
При повышении температуры процесса содержание азота в
диффузионном слое уменьшается, а содержание углерода может
увеличиваться непрерывно или до определенной температуры, а
затем снижаться (рис. 78). Таким образом, в зависимости от на-
углероживающей способности среды максимальное насыщение
поверхностного слоя стали углеродом достигается при различных
температурах.
Азот, диффундируя в сталь вместе с углеродом, оказывает
существенное влияние на степень насыщения поверхностного
слоя углеродом и на глубину диффузии углерода (рис. 79). По-
нижая температурную область существования у-железа, азот
способствует интенсивному науглероживанию стали при более
низких температурах, чем при цементации. При чрезмерном на-
сыщении стали азотом и образовании в поверхностном слое кар-
бонитридных фаз азот может затруднять диффузию углерода в
сталь.
При нитроцементации процесс насыщения имеет две стадии,
различные по кинетике. На первой стадии (1—3 ч) сталь насы-
щается углеродом и азотом одновременно [199], [200]. На второй
стадии при продолжающемся насыщении углеродом происходит
десорбция азота, т. е. переход части абсорбированных атомов
азота с поверхности металла в газовую среду (рис. 80). Доказа-
но, что десорбция азота является не следствием изменения
129
состава газовой фазы, которая поддерживается постоянной, а
результатом кинетического взаимодействия атомов азота и уг-
лерода в стали.
С повышением температуры нитроцементации увеличивает-
ся содержание углерода и уменьшается содержание азота в тон-
кой ленте и поверхностной зоне диффузионного слоя образ-
цов, и соотношение в них этих эле-
ментов носит линейный характер
(рис. 80).
Однако в более глубоких зонах
диффузионного слоя образцов эта
зависимость нарушается (рис. 81).
Не соблюдается она и при более вы-
соких температурах, что полностью
б) *)
Рис. 78. Влияние температуры на со-
держание углерода и азота в по-
верхностном слое стали глубиной
0,076—0,15 мм (по данным разных
авторов) :
а — нитроцементация в смеси 50% СО и
60% NH3; б — цианирование в цианистой
ванне с 23—27% NaCN; в — цианирование
в ванне с 50% NaCN; г — цианирование
в ванне с 30% NaCN; 8,5% NaCNO; 25%
NaCl и 36.5% Na2CO3
Рис. 79. Изменение концентрации уг-
лерода и твердости по глубине диф-
фузионного слоя хромоникелевой
стали:
/ — предварительно азотированной, а за-
тем цементованной; 2 — цементованной
(В. И. Просвирин); а — содержание угле-
рода; б — твердость после закалки в во-
де 820° С; в — твердость после закалки и
отпуска при 260° С
согласуется с ранее отмеченной закономерностью влияния тем-
пературы нитроцементации и цианирования на содержание уг-
лерода в диффузионном слое.
Таким образом, в определенных условиях механизм взаимо-
действия углерода и азота при совместной диффузии их в сталь
заключается в том, что преобладающее проникновение одного
элемента тормозит проникновение другого.
При цианировании глубина проникновения в сталь азота
больше глубины проникновения углерода, определяемой по мик-
роструктуре. Так, при глубине цианированного слоя 0,15 мм
130
(определена по микроструктуре) концентрация азота на глубине
0,5 мм в 3, а на глубине 1 мм в 2 раза превосходила исходную
концентрацию азота в стали [19]. Этим объясняется наблюдаемая
иногда повышенная хрупкость тонких цианированных деталей
по сравнению с цементованными.
Рис. 80. Влияние продолжительности
нитроцементации па концентрацию:
а — углерода и азота в ленте из стали У8, обработанной при 800° С в смеси бензола и
аммиака [199]; б — азота в поверхностном слое стали 30 <ГТ, обработанной при 850е С
в течение 0,5; 3 и 5,5 ч в парах триэтаноламина [200]
Рис. 81. Зависимость концентра-
ции углерода и азота в ленте из
стали У8 (а) и в поверхностном
слое образца армко-железа (б)
от температуры нитроцемента-
ции; лента обрабатывалась в сме-
си аммиака и бензола [199], образ-
цы — в смеси 50% СО и 50% NH3
в течение 10 ч:
1 — содержание углерода на глубине
0,15 мм: 2 — на глубине 0,3 мм: 3 —
на глубине 0.6 мм (А. Брамлсй)
Нитроцементация
Нитроцементация имеет следующие преимущества перед га-
зовой цементацией:
1) ниже температура процесса (850—870° С вместо 900—
930° С) при той же скорости или больше скорость процесса при
той же температуре (рис. 82);
2) не выделяется сажа на поверхности нитроцементуемых де-
талей, стенках печи и нагревателях;
131
Рис. 82. Влияние температу-
ры на глубину цементован-
ного и цианированного слоя
стали 10 (выдержка 3 ч):
1 — газ пиролиза керосина и
25°/гного аммиака; 2 — газ пи-
ролиза керосина (С. К. Иль-
инский и А. Н. Мясоедов,
Л. А. Бабушкин)
3) выше износостойкость деталей вследствие дополнительно-
го насыщения стали азотом;
4) меньше коробление деталей.
В качестве газовой среды при нитроцементации применяют
смесь, состоящую из 2—10% аммиака и 90—98% науглерожи-
вающих газов, обычно используемых при газовой цементации.
В муфельные печи непрерывного действия науглероживающий
газ и аммиак обычно подают раздельно. Жидкие карбюризато-
ры в такие печи вводят, как и при цементации, под давлением
в тонкораспыленном виде форсункой, соединенной с четырех-
плунжерным насосом. Процесс обычно протекает при 850—
870° С, а затем детали подвергают-
ся непосредственно закалке и низ-
кому отпуску.
На ЗИЛе для нитроцементации
разработаны и внедрены агрегаты
непрерывного действия, включаю-
щие в себя печь с радиационными
трубами, закалочный бак, моечную
машину и отпускную печь. При ни-
троцементации в этих агрегатах де-
талей из сталей 25ХГТ и 25ХГМ
при 840—830° С применяют газовую
смесь, состоящую из 80—90% эн-
догаза, 5—8% природного газа (см.
табл. 8) и 2,5—5% аммиака. При
850° С диффузионный слой глуби-
ной 0,5—0,7 мм образуется в тече-
ние 5 15 мин, а глубиной 0,8—
1,0 мм в течение 9 ч. Детали подсту-
закаливают в масле с температурой
живают до 820—830° С и
170—190° С. Отпуск проводят при 160—180° С. Структура сердце-
вины зуба зубчатого колеса послё обработки при указанном ре-
жиме— сорбит или троостит (HRC 35—45), твердость поверхно-
сти стали более HRC 60; коробление зубчатых колес незначи-
тельное [44].
На ряде заводов нитроцементацию успешно проводят в шахт-
ных муфельных печах, снабженных вентиляторами. На Харьков-
ском тракторном заводе в печи Ц-25 нитроцементации в смеси
паров керосина и аммиака на глубину 0,1—0,8 мм подвергают
различные детали топливной аппаратуры, изготовляемые из
сталей 20, 20Х, 18ХГТ и 18Х2Н4ВА. Температура процесса
850° С. Скорость образования нитроцементованного слоя 0,20—
0,25 мм]ч. Закалку производят непосредственно из печи (твер-
дость HRC 59—62).
Нитроцементацию инструментальной легированной
Нитроцементацию инструментальной легированной стали
(ШХ15, 9Х) рационально вести при 840—850° С в течение 2—4 ч
132
в такой газовой среде, в которой поверхностный слой стали насы-
щается азотом до 0,2—0,3%, а содержание углерода в слое поч-
ти не изменяется [201]. Обработку измерительного инструмента
из сталей 20Х, 12ХНЗА и 18ХГТ ведут в печи Ц-105 при 930° С.
Глубина слоя 0,8—1,3 мм достигается за 4,5 ч.
Нитроцементация сталей обычного качества описана в рабо-
те [202]. Нержавеющую сталь Х17Н2 подвергали нитроцемента-
ции в смеси пиробензола и аммиака [203]. На этой стали слой
глубиной 0,3—0,5 мм, твердостью более HRC 58 можно получить
при 850° С в течение 6 ч. После нитроцементации сталь проходит
закалку с 1020° С, обработ-
ку холодом при —70° С в те-
чение 2 ч и низкий отпуск
при 160° С. Поверхностная
зона нитроцементованного
слоя обладает низкой кор-
розионной стойкостью.
Предложенный НИИТАв-
топромом для нитроцемен-
а)
б)
Рис. 83. Содержание углерода и азо-
та в нитроцементованном слое ста-
лей, обработанных с применением
триэтаноламина:
а — сталь 40, печь Ц-25, нагрев в тече-
ние 40 мин и выдержка 5 ч\ б — сталь
18ХГТ, печь Ц-60, расход триэтаноламина
0,5 л!ч; нагрев до 850е С в течение 1,5 ч
и выдержка 1,5 ч (А. Т. Калинин, Н. М.
Сергеева и А. Я. Новикова)
тации жидкий карбюри-
затор триэтаноламин
(C2H5O)3N (рис. 83) внед-
рен на ряде заводов — Ку-
таисском и Кременчугском
автомобильных заводах,
МЗМА, Черниговском заво-
де запасных частей, Карбю-
раторном и др.
На Кременчугском заво-
де в печь ШПН-65 подают 550 мл/ч триэтаноламина при давле-
нии в печи 18—20 мм вод. ст. (176—196 н)м2). Отходящие газы
проверяют один раз в сутки; состав их должен быть следующим:
до 0,2% СО2; до 0,2% Ь2; 16—20% СО; 30-40% Н2; 8—10%
СН4. При 840° С в течение 3 ч на сталях 10 и 20 получается слой
глубиной 0,35 мм и твердостью более HRC 60. После такой
обработки биение зубчатых колес по начальной окружности
снизилось на 30% по сравнению с биением после цемен-
тации.
Оптимальными для получения качественного нитроцементо-
ванного слоя являются расходы триэтаноламина 300, 500, 500—
600 и 700 см21ч соответственно для печей Ц-35, Ц-60, Ц-75 и
Ц-105 [200].
Расход триэтаноламина определен из условий полной за-
грузки печи деталями. Во избежание закоксования входного от-
верстия триэтаноламин необходимо подавать непосредственно
в горячую зону печи через охлаждаемую водой трубку или фор-
сунку.
133
Чтобы не пересыщать сталь азотом, что может привести к
образованию дефектных слоев, снижающих прочность стали, пе-
риод восстановления состава атмосферы в печи должен быть
меньше времени прогрева деталей и не превышать 1,5—2 ч. Ре-
комендуется для восстановления атмосферы в печи применять
углеводородные жидкости, не содержащие азот. При закалке
следует принимать меры против обезуглероживания, так как оно
Рис. 84. Зависимость пределов вынос-
ливости 0-1 и прочности при изгибе
Qu, а также ударной вязкости ан от
суммарного содержания углерода и
азота в поверхностной зоне слоя ста-
ли ЗОХГТ, нитроцементовапной на
глубину 0,5—0,7 мм [200]
может привести к снижению
прочности [200].
При нитроцементации,
так же как и при цемента-
ции, нужно строго контро-
лировать степень поверхно-
стного насыщения. Лишь
при оптимальном суммар-
ном содержании в нитроце-
ментованном слое углерода
и азота (табл. 20) обеспечи-
вается получение у стали
оптимальных механических
свойств (рис. 84). При пе-
ресыщении азотом в слое
появляется темная состав-
ляющая (видимая только
на нетравленых шлифах),
резко снижающая свойства
стали (см. стр. 140). Опти-
мального содержания угле-
рода и азота в слое недо-
пустимо достигать путем
обезуглероживания и деазо-
тирования в конце процесса или при нагреве под закалку, так
как прочность при этом будет существенно снижаться аналогич-
но тому, как это происходит при цементации.
За рубежом рекомендуются [204], [205] следующие варианты
режимов нитроцементации:
1) в смеси цементующего газа и 2—8% аммиака при 840° С
с охлаждением в соляной ванне с температурой 180° С и обра-
ботке холодом при температуре минус 30—40° С (глубина слоя
0,2—0,4 мм);
2) при температуре выше Асз сердцевины стали (например,
820° С) с последующим подстуживанием до 700° С, закалкой в
соляной ванне и обработкой жидким воздухом (глубина слоя
0,2—0,25 мм, твердость HV 950—980, на глубине от поверхности
0,1 мм содержится 0,9% С и 0,8% N, сердцевина сохраняется
незакаленной);
3) при температуре ниже Act (см. стр. 147—150).
134
Таблица 20
Оптимальное суммарное содержание углерода и азота
в поверхностной зоне нитроцементованного слоя [200]
Марка стали Глубина нитро- цементованного слоя в мм Суммарное содержание С и N в % при толщине стружки для химического анализа в мм
0,925 0,05 0,10
40Х 25ХГТ 25ХГМ 25ХГМТ 0,25—0,35 0,5—0,7 0,5—0,8 0,5—0,65 0,9-1,25 1,05—1,25 1,3—1,65 1,25—1,65 0,85—1,2 1,0—1,15 1,25—1,55 1,2-1,6 0,75-1,0 0,95-1,05 1,15-1,25 1,15—1,5
В ряде работ изучена нитроцементация с нагревом т. в.ч. [84],
[206]. Глубина получаемого слоя оказалась несколько больше,
чем в случае обычного нагрева при той же продолжительности
процесса. Содержание азота в поверхностном слое сталей 30 и
ЗОХГТ, полученном при индукционном нагреве, примерно в 2 ра-
за выше, чем после обработки в тех же условиях с обычным
нагревом [206]. Это дает возможность проводить нитроцемента-
цию при более высокой температуре, сохраняя высокое содержа-
ние азота в слое.
Эксперименты, проведенные совместно работниками ЗИЛа
и МИСиС на автоматическом агрегате для газовой цементации с
нагревом т. в. ч., показали, что при 930—950° С в течение 40 мин
мвжет быть получен нитроцементованный слой глубиной 0,35—
0,5 мм, содержащий 0,10—0,25% N.
Цианирование
Для цианирования на небольшую глубину используют ванны
составов № 1 и 2 (табл. 21). В ванне № 1 с тиглем емкостью
130—150 кг расходуется 1,3—1,5 кг!ч NaCN и 0,8—1,0 кг)ч NaCl.
Углекислый натрий обычно не вводят, так как он образуется в
ходе процесса по реакциям
2NaCN + О2 = 2NaCNO;
2NaCNO + О2 = Na2CO3 + 2N + СО.
Наряду с Na2CO3 в ванне образуются атомарный азот и окись
углерода. Для освежения такой ванны добавляют (обычно один
раз в смену) цианистый натрий, а для сохранения необходимого
уровня — NaCl. Непосредственно после цианирования в высоко-
процентных ваннах при 820—870° С проводят закалку, а затем
низкий отпуск при 160—200° С (рис. 85).
135
Характеристики ванн для цианирования конструкционной стали
Таблица 21
ЛГв состава ванны! Назначение ванны Максимальная глубина слоя, на которую рацио- нально цианиро- вать, в мм Период анализа Состав ванн* в % Температура циа- нирования в °C Продолжитель- ность процесса в ч и мин Глубина слоя в мм
NaCN цианплав ГИПХ NaCl 8 св Z ВаС12 О и св СП СаС1»
1 Ванна с NaCN для циа- нирования на неболь- шую глубину (ГАЗ и ДР-) 0,35 При расплав- лении Рабочий 50 20-25 — 50 25-50 25 -50 — — — 840 840 870 870 0.30 1 .00 0.30 1.00 0,15-0,20 0,20—0,25 0,20-0,25 0,25—0,35
2 Ванна с цианплавом для цианирования на не- большую глубину 0.60 При расплав- лении Рабочий — 9** 36 — — — 55 840 840 870 870 870 0.30 1.00 0.30 1.00 2.00 0,15—0,25 0,25—0,30 0,20-0,30 0,35—0,45 0,55—0,65
3 Активная ванна с NaCN и ВаС12 для цианирова- ния на различную глу- бину (ГАЗ и др.) 1,0 При расплав- лении Рабочий 10 8—12 — 40 30-55 <10 50 >15 (фактиче- ски чаще < 15) 35—50 — 840 900 900 900 1 .20 1 .00 2.00 4.00 0, 25—0.30 0,50—0,60 0,70—0 80 1,0-1,2
4 Ванна для глубокого ци- анирования (1-й ГПЗ, Минский и Киевский мотоциклетные заво- ды, Харьковский вело- сипедный и др.) 2,0 При расплав- лении Рабочий 8 3-8*** 10 <30 Нет 82 >30 <4 0 — 900 900 900 950 950 950 0.25 0.45 1.30 2.00 3.00 5.30 0.20—0,25 0,30—0,50 0,50—0,80 0,8—1,0 1,0—1,2 1 ,4—1,6
* Технический высокопроцентный цианистый натрий: более >85% NaCN (чаще 96% NaCN). Цианплав ГИПХ: 43—48% Ca(CN)>;
2—3% CaCN2; 14 — 16% СаС12; 30—35% NaCl; 4—5% С и 0,8% CaF .
*♦ Содержание группы CN должно составлять 0,4—1,0%.
*** На 1 вес. ч. введенного цианистого натрия вводят 3,5—4,0 ч. ВаС12.
Ванна № 2 более дешевая, чем ванна № 1, но в ней выделяет-
ся значительно больше пены и осадка, которые приходится пе-
риодически удалять. В такой ванне протекают реакции
Са (CN)2 = CaCN2 + С;
CaCN2 + О2 = СаО + СО + 2N;
2Са (СЫ)2 + ЗО2 = 2СаО + 4СО + 4N.
Освежают ванну № 2 через каждые 2,5—4 ч работы, добав-
ляя циапплав и нейтральные соли.
После цианирования непо-
средственно из ванны производит-
ся закалка, а затем детали под-
вергают низкому отпуску.
Глубокое цианирование [207]
осуществляют в ваннах с соста-
вами № 3 и 4, содержащими хло-
ристый барий, взаимодействую-
щий с цианистым натрием по
реакции
ВаС12 + 2NaCN = 2NaCl + Ba (CN)2 ;
Ba (CN)2 = BaCN2 + C;
BaCN2 + O2 - BaO + CO + 2N.
Рис. 85. Зависимость глубины
цианированного слоя от продол-
жительности процесса -при 850° С
в ванне состава 1 (табл. 22).
Данные получены в ванне ем-
костью 150 кг [208]:
1 — сталь 20X; 2 — сталь 20; 3 —
стали 40 и 40X
В процессе эксплуатации ак-
тивность таких ванн постепенно
понижается. Продолжительность
их работы (до значительного
снижения активности ванн, когда необходимо заменить 50—
100% солей) около 60—90 суток.
На ГАЗе применяют активную цианистую ванну с составом
№ 3 (табл. 21) [208], содержащую 8—12% NaCN, до 12% ВаС12,
40—50% ВаСОз, Ю—40% NaCl и 0,5—5% Na2CO3. Эту ванну
используют для цианирования при 830—870° С на сравнительно
небольшую глубину, но ее можно применять и для глубокого
цианирования.
В небольших тигельных ваннах глубокое цианирование при
930—950° С можно осуществлять с использованием цианплава
(65% ВаС12, 25% NaCl и 10% цианплава или 65% СаС12,
25% NaCl и 10% цианплава). В течение 3 ч в ванне с цианпла-
вом на деталях образуется общий слой глубиной 1,1 мм и эвтек-
тоидный слой глубиной 0,55 мм. Глубокое цианирование в таких
ваннах сопровождается выпадением больших осадков и вспени-
ванием ванны при ее освежении, из-за чего работа сильно ос-
ложняется.
Так как глубокое цианирование продолжается в течение не-
скольких часов при высокой температуре (900—950°С), для
137
измельчения зерна стали и снижения количества остаточного
аустенита в слое детали обычно охлаждают на воздухе, а затем
закаливают с нагревом в соляной ванне или печи и подвергают
низкому отпуску.
На некоторых зарубежных заводах детали из цианистой ван-
ны с температурой 940° С после выдержки в течение 3,5—4,0 ч
(глубина слоя 1,25—1,5 мм) переносят в стабилизирующую ван-
ну (например, с температурой 800° С), а затем после выдержки
в течение 5 мин закаливают. При такой обработке уменьшается
коробление и повышается поверхностная твердость, но не устра-
няется перегрев стали, который происходит в процессе цианиро-
вания. Иногда после глубокого цианирования или цианирования
при 780—840° С для повышения твердости и предела выносли-
вости сталь обрабатывают холодом или подвергают дробеструй-
ной обработке.
Для цианирования опробована ванна, не содержащая циа-
нистых солей (70—76% Na2CO3, 9—12% NaCl, 6—9% NH4C1
9—10% SiC) [89]. В такой ванне при 850° С поверхностная зона
слоя обогащается азотом лишь до 0,08—0,15%; цианированпый
слой при выдержке 15—30 мин имеет глубину 0,1—0,25 мм и при
выдержке 1,0—1,5 ч глубину 0,7—0,85 мм. Однако использование
такой ванны сопряжено с рядом трудностей: сильное испарение
хлористого аммония при его загрузке в ванну, осадки в ванне
и неравномерность слоя.
Разработан способ цианирования крутильных колец из ста-
ли 4X13 с нагревом т. в. ч. в расплавленной желтой кровяной со-
ли [90% K4Fe(CN)6 + 10% NaCl] [76]. Аналогичный способ ис-
пользуют на другом заводе для обработки мелких зубчатых ко-
лес из стали 40 и деталей приборов небольших размеров [76].
При цианировании зубчатых колес глубина слоя составляет
0,023 мм при 840° С и выдержке 25 сек и 0,04—0,07 мм при 860° С
и выдержке 70 сек. После непосредственной закалки в масле
твердость сердцевины зуба в обоих случаях HRC 50—52,
а поверхности HRC 59—62 (перевод с показаний прибора Вик-
керса).
Опробовано также цианирование стали с применением пасты
(мелкий порошок желтой кровяной соли, перемешанный с дек-
стриновым клеем до консистенции сметаны) и нагревом т. в. ч.
Показана возможность получения в цианистых ваннах с циа-
нидом кальция тонких твердых поверхностных слоев на сталях
1X13 и Х17Н2 [209].
В настоящее время все чаще переходят от жидкостного циа-
нирования, при котором используют ядовитые цианистые соли,
к нитроцементации.
Предложен метод цианирования с нагревом стали т. в. ч. в сме-
сях спиртов (метилового и этилового) и водного 20%-ного раство-
ра аммиака. При 800° С в течение 20 мин получается слой глуби-
138
ной 0,22 мм. Максимальная поверхностная твердость HV 780 была
достигнута при цианировании при 1050—1100° С в течение 20 мин
(глубина слоя до 0,6 мм) [96], [97], [210].
Очень быстро протекает процесс цианирования при нагреве
стали т. в. ч. в обмазках [211]. В качестве обмазок применяли сме-
си порошков, например, следующего состава: 60% K4Fe(CN)6,
30% древесного угля и 10%ВаСО3или 20% K4Fe(CN)6, 70%
древесного угля и 10% ВаСО3, замешанных в гидролизованном
этилсиликате. Обмазку наносили слоем толщиной 0,5 мм. При
нагреве в обмазке первого состава, выдержке 15—20 сек (плюс
20 сек нагрев) и температуре 1150° С получается слой глубиной
0,08 мм, а при нагреве в обмазке второго состава — слой глубиной
0,16 жж с микротвердостью 800 и более (до 1000).
При уменьшении скорости нагрева твердость слоя снижается,
очевидно, главным образом вследствие обеднения обмазки.
Структура нитроцементованного
и цианированного слоя
Концентрация углерода и азота в нитроцементованных и циа-
нированных слоях сильно колеблется в зависимости от состава
насыщающих сред, режимов процессов и составов сталей. На
рис. 86—88 приведены
кривые распределения
концентрации углерода и
азота по глубине нитро-
цементованного слоя.
Рис. 86. Распределение углерода и азо-
та по глубине диффузионного слоя у
стали 40Х, цианированной при темпера-
туре 820е С и разных выдержках в ван-
не с 20% NaCN
Рис. 87. Распределение
углерода и азота по глу-
бине нитроцементован-
ного слоя стали 40Х;
процесс проводился в ме-
тодической печи Ц-160
при 850° С (А. М. Та-
расов и Б. А. Стеценко)
При цианировании при 850—900° С в цианистых ваннах, содер-
жащих цианплав (состав № 2 по табл. 21), и при глубоком циани-
139
ровании при 900—950° С в низкопроцентных ваннах с цианистым
натрием и хлористым барием (составы № 3 и 4) сталь с поверхно-
сти насыщается углеродом примерно до той же концентрации, что
и при цементации, и лишь немного азотом (0,08—0,4%). При циа-
нировании в ванне состава № 1 сталь
Рис. 88. Распределение уг-
лерода и азота по глубине
нитроцементованного слоя
стали ЗОХГТ, обработанной
в безмуфельном агрегате
ЗИЛа в среде эндотермиче-
ского газа с добавкой при-
родного газа И аммиака при
850 и 870° С [200]
насыщается углеродом несколько
меньше, чем при цементации, а азотом
в поверхностной зоне слоя больше,
чем в других ваннах (до 0,7—1,1%).
Иногда на нетравленых шлифах зака-
ленной нитроцементованной стали на
глубине до 0,05—0,15 мм наблюдается
темная сетка или отдельные мелкие
темные глобули, располагающиеся по
границам зерна стали и напоминаю-
щие по своему внешнему виду анало-
гичный дефект цементованной стали.
Это снижает механические свойства
стали, особенно предел выносли-
вости.
Наиболее сильно снижается предел
выносливости при глубоком залегании
дефектов [200]. Даже при незначитель-
ной глубине залегания дефектов
(0,04—0,05 мм) резко (в 6 раз и более)
снижается контактная выносливость.
Таким образом, этот дефект структуры
является весьма опасным.
Описанию и выяснению 'природы
этого дефекта нитроцементованной и цианированной стали по-
священ ряд отечественных и зарубежных работ [200], [212]—
[216]. Установлено что:
1) при нитроцементации поверхностные дефекты образуются
чаще и распространяются глубже, чем при цементации (напри-
мер, после нитроцементации при 830° С в течение 2 ч глубина рас-
положения их иногда доходит до 0,1 мм);
2) чем больше в газовой смеси’аммиака, тем больше в структу-
ре слоя образуется дефектов; при небольших добавках аммиака
дефекты в слое отсутствуют;
3) дефекты образуются в слое, когда содержание азота в
нем превышает 0,5%;
4) образованию и интенсивному развитию темной состав-
ляющей способствует увеличение продолжительности про-
цесса;
5) при нитроцементации в триэтаноламине темная составляю-
щая возникает лишь при больших расходах жидкости;
140
6) очень важным (на взгляд автора) обстоятельством являет-
ся то, что количество азота в зоне залегания темной составляю-
щей меньше, чем в более глубокой зоне слоя [200]. Это свиде-
тельствует о протекании процесса деазотирования в зоне зале-
гания темной составляющей (рис. 80, б), объясняемого процес-
сом вытеснения азота углеродом при совместном насыщении
стали этими элементами;
7) в дефектной зоне слоя наряду со связанным углеродом име-
ется значительное количество свободного углерода [215].
Последние данные позволили некоторым авторам [212] прийти
к выводу, что темная составляющая в цианированном и нитро-
цианированном слое имеет иную природу, чем аналогичная со-
ставляющая в цементованном слое, и представляет собой поры,
образовавшиеся вследствие диссоциации нитридов и выделения
молекулярного азота, которое, по данным [217], происходит под
большим давлением. Эти поры заполняются свободным углеро-
дом, образующимся в результате протекающей графитизации.
Однако, по мнению автора, вопрос о природе темной составляю-
щей в цианированном и нитроцементованном слое нельзя считать
окончательно решенным. Не исключено, что эта составляющая
при высоком содержании в газовой среде кислорода имеет окис-
ную природу, т. е. ту же, которую имеет темная составляющая
в цементованном слое.
Во избежание образования темной дефектной структуры
в нитроцементованном слое добавка аммиака в газовой смеси
должна быть минимальной и, по данным НИИАвтопрома, не пре-
вышать 7—10%, а при более длительных процессах, когда может
происходить деазотирование, количество аммиака должно сни-
жаться до 1—3%. Следовательно, чем больше глубина нитроце-
ментованного слоя, тем меньше должно содержаться в нем
азота. При нитроцементации, так же как и при цементации,
нельзя на конечных стадиях процесса допускать обезуглерожи-
вание.
Микроструктура нитроцементованного и цианированного слоя
после медленного охлаждения в травленом состоянии не отличает-
ся от микроструктуры цементованного слоя или же отличается
лишь наличием тонкой поверхностной карбонитридной корочки
либо отдельных включений карбонитридов. После закалки и низ-
кого отпуска структура слоя состоит из мартенсита (рис. 89 и
90) с тем или иным количеством остаточного аустенита, который
обнаруживается в микроструктуре при больших количествах.
Иногда в диффузионном слое присутствует троостит. Количество
остаточного аустенита в нитроцементованном и особенно циани-
рованном слое больше, чем в цементованном, вследствие допол
нительного легирования слоя азотом.
Карбонитридная корочка или отдельные карбонитридные
включения соответствуют соединениям Fe2(N, С), Еез(М, С)
141
Рис. 89. Микроструктура стали
20, цианированной при 840°С в
течение 40 мин и закаленной
(Х200)
Рис. 90. Микроструктура стали
25ХГМ, нитродементованной при
870° С в течение 5 ч и закален-
ной (структура соответствует
первому баллу по карбонитридам
и первому баллу по остаточному
аустениту по шкале ЗИЛа) (Х200)
Рис. 91. Схема изотермического разреза системы Fe — С — N при
450° С
142
и Fe4(N, С). При незначительном насыщении азотом светлые вы-
деления в виде глобулей и сетки могут быть построены на базе
цементита Рез (С, N). Образованию карбонитрида на базе це-
ментита способствует не только высокая науглероживающая
способность среды, но и повышение температуры процесса. На
рис. 91 дана схема изотермического разреза системы Fe — С — N
при 450° С [218], построенная на основании химического и рентге-
ноструктурного анализа продуктов взаимодействия нитридов
с окисью углерода и карбида железа с аммиаком. Правильность
этой схемы подтверждена в работе [219]. На схеме, кроме
карбонитридов: £ (гомогенного в пределах примерно от Fe8N4
до Fe8C3N), е, содержащего 25—30 ат. % (N + С) и у' (все эти
нитриды аналогичны соответствующим нитридам в системе
Fe — N), также указана тройная фаза (обозначена %), гранича-
щая с карбидом Fe2oC9 и областью с £-фазой, возникающая при
длительной реакции нитрида железа с окисью углерода при тем-
пературе ниже 500° С.
О возможности образования таких соединений при низкотем-
пературной нитроцементации или цианировании убедительных
данных пока не имеется.
Свойства нитроцементованной и цианированной стали
Нитроцементованная и цианированная конструкционная
сталь благодаря присутствию азота более износостойка, чем це-
ментованная.
Нитроцементация и цианирование существенно повышают
предел выносливости, причем нитроцементация в большей сте-
пени, чем цианирование, а в ряде случаев в большей степени,
чем цементация.
По данным ЗИЛа, предел выносливости стали 25ХГМ после
цементации на глубину 1 мм (950° С, подстуживание до 820° С,
закалка в масле, отпуск при 200° С) составляет 65—75 кГ/мм2
(64 • 107 — 73 • 107 h/jw2), а после нитроцементации на глубину
0,7—0,8 мм (850° С, подстуживание до 830° С, закалка в масле
с температурой 160°С, отпуск при 200°С)—90 кГ)мм2 (88 X
X Ю7 нМ2).
При цианировании невозможно регулировать концентрацию
азота и углерода в слое. Поэтому в цианированием слое количе-
ство остаточного аустенита всегда больше, чем в нитроцементо-
ванном.
В связи с этим сжимающие напряжения создаются в циани-
рованием слое лишь на некотором расстоянии от поверхности,
что приводит к снижению предела выносливости стали. Этим
и объясняется меньшая долговечность цианированных деталей
по сравнению с нитроцементованными.
143
Рис. 92. Распреде-
ление внутренних
остаточных напря-
жений в цианиро-
ванном слое стали
40Х после обра-
ботки при 815° С в
ванне, содержащей
20—25% NaCN, с
последующей за-
калкой и низким от-
пуском при 200°С
(кривая /) и пос-
ле дополнитель-
ного наклепа
дробью (кривая 2)
[Ю4]
После цианирования необходимо производить наклеп деталей
дробью, создающий на поверхности (вследствие превращения
остаточного аустенита в мартенсит) высокие напряжения сжатия
(рис. 92). Усталостные испытания зубьев цианированных зубча-
тых колес (сталь 40Х) коробки передач автомобиля «Волга» на
изгиб с циклической нагрузкой показали, что наклеп дробью по-
вышает предел выносливости с 43 до 72 кГ/мм1 2 * * * *
(42 - 107—71 • 107 нМ2), т. е. на 67%.
Испытания на стенде показали, что после
наклепа дробью стойкость (до разрушения)
цианированных зубчатых колес увеличилась
с 9 до 140 ч [104].
Сталь, подвергнутая нитроцементации и
имеющая на поверхности тонкий нетравящий-
ся карбонитридный слой (что бывает не всег-
да), корродирует медленнее нецианированной
стали. Например, в 3%-ном растворе поварен-
ной соли стойкость такой стали против корро-
зии в 2 раза выше, чем нецианированной. Кор-
розионная стойкость нержавеющих сталей
(Х17Н2, 1X13) после нитроцементации и циа-
нирования снижается.
Низкотемпературная нитроцементация
и цианирование
Низкотемпературной нитроцементации и
цианированию при 560—700° С подвергаются
стали различного назначения для повышения
их поверхностной твердости, износостойкости,
предела выносливости, теплостойкости и про-
тивозадирных свойств. Обычно такая обработ-
ка проводится при 560—580° С, т. е. при темпе-
ратуре, которая немного ниже минимальной
температуры существования у-фазы в системе Fe — N. Поэтому
в процессе обработки при такой температуре на стали образует-
ся, по существу, азотированный слой, а углерод проникает на
глубину лишь нескольких микрон, где может образовываться
тонкая карбонитридная зона L
1 В соответствии с этим в некоторых странах низкотемпературное циани-
рование быстрорежущей стали принято называть азотированием, а низкотем-
пературное цианирование конструкционной стали (не предназначенной для
азотирования) — мягким азотированием (твердость после азотирования по-
лучается сравнительно невысокой). В отечественной литературе эти термины
не приняты, так как они не отражают различия между азотированием в ам-
миаке и процессами, которые протекают в средах, содержащих и активный
углерод, что придает некоторую, специфику кинетике процесса, а также фазо-
вому составу и свойствам диффузионного слоя.
144
Низкотемпературная нитроцементация и цианирование быст-
рорежущей стали [220]. Такой обработке подвергают быстрорежу-
щую сталь для повышения стойкости режущего инструмента.
Процессы проводятся в активных газовых, жидких и порошко-
образных смесях, обеспечивающих насыщение поверхностных
слоев стали в основном азотом на глубину 0,01—0,025 мм. Пред-
варительно режущей инструмент подвергают термической и ме-
ханической обработке (включая шлифование и заточку). Тем-
пература процесса совпадает с температурой отпуска инструмен-
та (560° С).
Нитроцементация проводится в смеси, которая состоит из
65—75% углеродосодержащего газа (природного, нефтяного, све-
тильного, генераторного и т. д.) и 25—35% аммиака; продолжи-
тельность процесса 1—2 ч. На некоторых заводах применяют га-
зовую среду, полученную путем пропускания аммиака через дре-
весный уголь, нагретый до 850—900° С или до 560° С (при нагре-
ве до 560° С уголь помещают в муфеле для нитроцементации).
В первом случае образуется газ, содержащий примерно 35% СО;
2% СН4; 2% СО2; 10% Н2; остальное N2 и немного аммиака, а во
втором—0,17% СО; 1,26% СН4; 0,02% СО2; 16,4% Н2; 7,1% N2;
75% NH3.
Следует отметить, что малоактивные газы (древесноугольный,
генераторный и др.), не пригодные для цементации и высокотем-
пературной нитроцементации, вполне пригодны для обработки
быстрорежущей стали, так как основным элементом, создающим
в данном случае диффузионный слой высокой твердости, являет-
ся азот.
Основное назначение газа, содержащего углерод, — разбав-
ление аммиака, чтобы предотвратить пересыщения поверхност-
ного слоя азотом и устранить образования хрупкой нитридной
корочки.
На некоторых заводах вместо нитроцементации применяют
азотирование быстрорежущей стали, снижая азотирующую спо-
собность аммиака добавлением к нему до 70% N2. Несколько за-
водов используют неразбавленный аммиак (т. е. осуществляют
азотирование) [221]. При этом свойства диффузионного слоя, за
исключением несколько повышенной хрупкости, почти аналогич-
ны получаемым после нитроцементации. Во многих случаях азо-
тирование может заменить нитроцементацию инструментов из
быстрорежущей стали.
Для нитроцементации быстрорежущей стали в последние
годы все чаще применяют триэтаноламин, подаваемый непосред-
ственно в печь [222]. При внутреннем диаметре печи 450 мм и вы-
соте 600 мм триэтаноламин подается со скоростью 50—55 капель
в минуту (продолжительность процесса около 2,5 ч). По-видимо-
му, вначале лучше подвергать триэтаноламин пиролизу в специ-
альной печи, а затем подавать газ в муфель с инструментами
145
аналогично тому, как это предложено для обработки конструкци-
онной стали.
При цианировании применяют цианистые среднепроцентные
ванны следующего состава: 35—50% NaCN, остальное Иа2СОз
и NaCl. Для освежения этих ванн используют высокопроцентный
цианистый натрий. Продолжительность обработки в таких ван-
нах 5—30 мин [19]. Общий расход солей составляет около 2% от
веса обрабатываемых деталей. На один инструмент средних раз-
меров расходуется следующее количество высокопроцентного
цианистого натрия: при обработке сверл и метчиков 1,5 г, развер-
ток и зенкеров — 2 г, фрез — 5 г.
Процессы, протекающие в низкотемпературной цианистой
ванне, можно разбить на три стадии [223]: 1) накопление в яан-
не кислорода и окисление NaCN и KCN до NaCNO и KCNO;
2) разложение этих соединений с образованием карбонатов, оки-
си углерода и азота (на этой стадии протекает интенсивное азо-
тирование и насыщение стали углеродом, что приводит к образо-
ванию с поверхности слоя тонкой, хрупкой, нетравящейся каем-
ки) и 3) дальнейшее разложение этих сединений (однако выде-
ление активного углерода в ванне резко уменьшается и карбо-
нитридная каемка не образуется).
При обработке инструментов в ванне, эксплуатировавшейся
более 12—15 ч, хрупкая карбонитридная каемка не возникает, а
диффузионный слой образуется быстрее. Поэтому цианировать
инструменты в свежеприготовленной ванне не рекомендуется.
При обработке в твердой упаковке применяют стандартный
твердый карбюризатор, к которому добавляют 15—
20% K4Fe(CN)6, а иногда дополнительно 3—5% Na2CO3. Отра-
ботанная смесь освежается добавлением 10—15% свежей смеси.
Продолжительность процесса 1,5—3,5 ч. Такой способ обработки
можно встретить даже на передовых заводах, так как он дает
возможность обрабатывать инструмент любых размеров, напри-
мер длинные протяжки, которые невозможно разместить в тигле
ванны или муфеле печи.
Предлагается ряд видоизмененных режимов обработки инст-
рументов из быстрорежущей стали:
а) высокотемпературное глубокое цианирование при 850° С
(до глубины 0,25 мм)\ б) закалка стали в расплаве солей KCN
(47%) и NaCN (53%) при 500—550° С; в) глубокая нитроцемен-
тация (до глубины 0,09 мм); обработка с применением паст
в среде аммиака [76]. Для снижения хрупкости режущей кромки
инструменты следует подвергать отпуску при 200—300° С или,
по другим данным, при 360—380° С [224].
Микроструктура быстрорежущей стали, подвергнутой низко-
температурной нитроцементации и цианированию, при неболь-
шом увеличении аналогична микроструктуре азотированного слоя
этой стали (рис. 93). Основная зона нитроцементованного слоя,
146
отличающаяся от сердцевины лишь более сильной травимостью,
соответствует смеси фаз а + у', в которую вкраплены дисперс-
ные нитриды легирующих элементов, не видимые под микроско-
пом. На поверхности иногда обнаруживается тонкая светлая
корочка с решеткой нитридов железа или цементита в зависи-
мости от науглероживающей и азотирующей активности при-
меняемой среды. Для быстрорежущей стали карбонитридной
каемке соответствуют соединения (Fe, W, V, Сг)з(С, N);
(Fe, W, V, Cr)2(N, С) и (Fe, W, V, Cr)3(N, С). Общая глубина
слоя обычно составляет 0,015—0,025 мм.
Рис. 93. Микро-
структура циани-
рованием быстро-
режущей стали Р18
(560° С; 60 мин)
(Х800)
Рис. 94. Теплостойкость стали
Р18 после нормальной терми-
ческой обработки (кривая 2) и
дополнительного цианирования
(кривая 1) в ванне с 30%
NaCN в течение 25 мин (кри-
вые построены на основании
измерения твердости после
трехкратного нагрева продол-
жительностью 1 ч каждый)
[19]
Поверхностная твердость быстрорежущей стали после циани-
рования практически достигает HV 1000—1050, т. е. на
HV 150—200 выше, чем до цианирования. Планированный слой
обладает хорошей теплостойкостью (рис. 94); поэтому стойкость
цианированного инструмента из быстрорежущей стали в
1,5—2,5 раза выше, чем нецианированного.
Последнее время в литературе неоднократно упоминалось
об успешном применении низкотемпературной нитроцементации
(580° С; 3,5 ч) прессформ из хромовольфрамовой стали ЗХ2В8,
предназначенных для литья под давлением алюминиевых спла-
вов [225]. Иногда применяется и азотирование таких прессформ
при 540° С в течение 20 ч.
Низкотемпературная нитроцементация и цианирование («мяг-
кое азотирование») стали и чугуна. В некоторых странах, осо-
147
бенно в ФРРГ [226] — [231], [589], .в течение ряда лет широко
применяют так называемое «мягкое азотирование цементируе-
мой стали (типа 20Х, 15ХН и др.), среднеуглеродистой улучшен-
ной стали (типа 35Х, 40Х, 50ХФ и др.), нержавеющей, аустенит-
ной стали и чугуна. Такой обработке подвергают зубчатые коле-
Рис. 95. Изменение микротвердости
по глубине цианированного и азоти-
рованного слоев предварительно
улучшенной стали 40ХГТР:
1 — азотирование в печи при 570° С в те-
чение 6 ч\ 2 и 3 — цианирование в ванне
при 570° С соответственно в течение 2 и
6 ч (автор, В. Ф. Никонов, П. Я. Груздов,
В. В. Каражас)
Рис. 96. Микроструктура стали
40ХГТР, цианированной в ванне при
570° С в течение 6 ч (Х400)
са, шатуны, коленчатые валы, кулачки, толкатели, направляю-
щие втулки, чугунные гильзы цилиндров, поршневые кольца
и т. д.
Мягкое азотирование 1 осуществляют за рубежом в цианистых
ваннах, чаще содержащих 30—45% NaCN или 30—35% KCNO,
при 570° С в течение 1—3 ч. При этом образуется диффузионный
слой с поверхностной микротвердостью около 500—800 и глуби-
ной (судя по микрошл-'ифу и изменению микротвердости) около
0,15 мм. Это повышает противозадирные свойства, усталостную
прочность <и износостойкость стали.
1 Правильнее этот процесс называть низкотемпературным цианированием,
а при использовании газовой среды — низкотемпературной нитроцементацией.
148
После обработки в цианистой ванне усталостная прочность
легированной стали повышается на 20—40%, а нелегированной —
на 30—60%. О высокой износостойкости цианированной стали
свидетельствуют данные, полученные в результате работы, вы-
полненной МИСиС совместно с ЗИЛом на образцах улучшенной
стали 40ХГТР (рис. 95—97).
Образцы испытывались на машине МИ (типа Амслер) при
нагрузке 50 кг. Следует, однако, отметить, что предел выносли-
Рис. 97. Износостойкость, стали 40ХГТР (сум-
ма уменьшения веса верхнего и нижнего роли-
ков, обработанных одинаково):
1 — образцы улучшенной стали; 2 и 6 — цианиро-
ваны при 560° С соответственно в течение 2 и 6 ч;
3 — нитроцементованы при 560е С, 6 ч (среда: ам-
миак, пропущенный через раскаленный уголь); 4 и
5 — азотированы соответственно при 560° С, 6 ч и
при 520° С 36 ч\ 7 — цементованные образцы стали
ЗОХГТ (автор, В. Ф. Никонов, П. Я. Груздов,
В. В. Каражас)
вости цианированной при 560° С стали 40ХГТР оказался замет-
но ниже, чем цементованной стали ЗОХГТ.
При низкотемпературном цианировании размеры деталей из-
меняются незначительно, поэтому в зарубежной практике при-
нято детали из улучшенной стали сначала подвергать оконча-
тельной механической обработке и притирке, а затем мягкому
азотированию.
Применению этого процесса в отечественной промышленно-
сти препятствовала необходимость использовать ядовитые
149
цианистые соли. В связи с этим представляют большой интерес
данные о низкотемпературной нитроцементации стали в продук-
тах пиролиза триэтаноламина [232].
Пиролиз триэтаноламина рекомендуется осуществлять при
900° С. В случае нитроцементации при 600° С отходящий из печи
газ содержит 32,9% СО; 13,1% СН4; 0,9%’ CmHn; 0,5% СО2;
40,4% Н2; 11,6% Н2; 0,6% О2. Рекомендуется проводить нитро-
цементацию при 600° С в течение 6—10 ч. В этом случае общая
глубина слоя достигает 0,15—0,40 мм, а глубина е-фазы
4—10 мк. После выдержки 6 ч поверхностная микротвердость
улучшенной стали 45 Ни. 290, стали 30Х2Н2ВФА Н^ 560 и стали
36ХН1МФА /7|Х 717. Распределение микротвердости по глубине
нитроцементованного слоя и характер микроструктуры послед-
ней стали аналогичны полученным для цианированной стали
40ХГТР (рис. 95 и 96).
После нитроцементации резко уменьшается коэффициент тре-
ния, например у стали 30Х2Н2ВФА с 0,94 (после улучшения)
до 0,22 (после нитроцементации и в течение 10 ч). Снятие слоя
толщиной лишь 0,01 мм, т. е. карбонитридного слоя, повышает
коэффициент трения до 0,63. Это указывает на высокую изно-
состойкость образующейся на поверхности карбонитридной
е-фазы. Рентгеноструктурным анализом на поверхности обнару-
жены фазы Fe2(N, С), Fe3(C, N), Fe4N, a-фаза (последняя фаза
располагается глубже карбонитридных фаз одновременно с из-
быточным нитридом Fe4N). Карбонитридная корочка при боль-
шой толщине отличается хрупкостью. Однако при рекомендуемых
режимах ее толщина колеблется в пределах 4—10 мк.
После нитроцементации предел выносливости улучшенной
стали 45 повышается на 50—60%, а стали 30Х2Н2ВФА—на
30—35%. В поверхностном слое этих сталей сжимающие напря-
жения достигают 40—60 кГ)мм2 (39-107—59-107 н/л^2). Стендо-
вые испытания некоторых деталей на контактную прочность
после низкотемпературной нитроцементации при 600°С в течение
6 ч, проведенные на Кировском заводе в Ленинграде, показали
существенное повышение контактной прочности.
Внедрение низкотемпературной нитроцементации в промыш-
ленность позволит повысить долговечность многих деталей и лик-
видировать некоторое отставание от зарубежной практики, ши-
роко применяющей процесс мягкого азотирования.
Низкотемпературное цианирование стали 35ХМЮА и нержа-
веющей стали в цианистой ванне с продуванием аммиака. Эт!и
стали вместо обычного газового азотирования можно подвергать
цианированию в цианистой ванне. Однако в такой ванне скорость
насыщения стали азотом оказывается значительно ниже. Для
ускорения процесса предложено продувать через расплав ванны
предварительно подогретый аммиак, что увеличивает скорость
образования азотированного слоя более, чем в 2 раза (рис. 98)
150
[233]. Продувание через ванну аммиака изменяет соотношение
между диффундирующими в сталь азотом и углеродом. Коли-
чество диффундирующего азота увеличивается в 2—3 раза (по
сравнению с обычным цианированием) и становится примерно
миака.
Р'ис. 98. Влияние сте-
пени диссоциации ам-
миака, подаваемого в
ванну (трубка для
подачи аммиака име-
ет внешний обогрев),
на глубину и поверх-
ностную твердость по
Виккерсу азотирован-
ного слоя стали
38ХМЮА: 1 — расход
аммиака 45 л/ч; 2—
70 л!ч [233]
Рис. 99. Влияние
плотности постоянно-
го тока на аноде
(кривая /) и катоде
(кривая 2) на глуби-
ну диффузионного
слоя и изменение ве-
са образцов стали
38ХМЮА (состав
ванны: 40% KCN,
40% Na2CO3 и 20%
NaCl; 640° С; 45 мин)
[233]
равным количеству азота, поглощаемого сталью при газовом азо-
тировании, а количество углерода, сосредоточивающегося глав-
ным образом в карбонитридной каемке, уменьшается в 2—4 раза.
При обработке таким методом стали 38ХМЮА по непродол-
жительному процессу (5—15 ч) глубина диффузионного слоя
получается такой же или даже большей, чем при газовом азоти-
ровании, а его твердость почти всегда выше. Так, в случае обра-
ботки стали 35ХМЮА в течение 5 ч при 520° С и продувании
через ванну аммиака со скоростью 200 л/ч (степень диссоциации
35%) глубина цианированного слоя составляет 0,20 мм, а по-
верхностная твердость HV 1050. При азотировании этой стали
в лабораторной печи глубина слоя достигала 0,16—0,18 мм,
151
а твердость HV 850. При длительных процессах, проводимых по
этому методу, получается глубина слоя несколько меньше, чем
при газовом азотировании (поверхностная твердость в обоих
случаях одинакова).
Обычное цианирование стали Р18 при 560° С в течение 1 ч
позволяет получить слой глубиной 0,044 мм, а цианирование
при дополнительном продувании через ванну аммиака — слой
глубиной 0,069 мм.
Хорошие результаты дает описываемый метод при обработке
аустенитных сталей. При азотировании стали 1Х18Н9Т в течение
20 ч при 640° С глубина слоя составляет 0,17 мм, а твердость
HV 1050. В зарубежной литературе неоднократно приводились
примеры применения в производстве азотирования аустенитных
сталей (в частности клапанных) в цианистых ваннах с продува-
нием аммиака. В отечественной практике такие примеры автору
неизвестны.
Таким образом, в мелкосерийном производстве при наличии
цианистой ванны и отсутствии печи для азотирования послед-
нюю в ряде случаев вполне можно заменить цианистой ванной,
через которую нужно продувать аммиак.
Пути ускорения низкотемпературного цианирования стали. Для
ускорения процесса низкотемпературного цианирования различ-
ных сталей можно также применять электролиз расплава и про-
дувание через расплав воздуха. При электролизе расплава на
аноде и катоде выделяются активные азот и углерод, и диффу-
зионный слой образуется примерно в 2 раза быстрее, чем без
применения электролиза (рис. 99). Это объясняется тем, что при
электролизе NaCN разлагается на катион Na, направляющийся к
катоду, и анион CN, идущий к аноду, где последний теряет заряд
и разлагается на азот и углерод. Выделяющийся на катоде нат-
рий восстанавливает накапливающуюся в ванне группу CNO
по реакции
CNO + 2Na = Na2O + С + N.
Уменьшение глубины слоя при повышении плотности тока
связано с увеличением толщины осадка графитизированного
углерода на аноде и железа и натрия на катоде.
В ряде работ отмечается большой эффект, который дает про-
дувание воздуха [234], [235] или кислорода [236] через цианистые
низкотемпературные ванны, предназначенные для мягкого азо-
тирования конструкционных сталей, а также обработки нержа-
веющих (типа 1Х18Н9 и Х13) и других сталей. Благодаря этому
ускоряется окисление NaCN и KCN до NaCNO и KCNO (см.
стр. 135), что и активизирует ванну. Например, в ванну диамет-
ром 600 мм рекомендуется подавать 700 л)ч воздуха под давле-
нием 1,5 ат (147 кн)м2), а охлаждать детали в растворе NaCl.
При этом предел выносливости нелегированных сталей повы-
152
при /ии с. при этой температуре
Рис. 100. Микроструктура железа, нитроце-
мрнтовзнного в течение 6 ч при 700° С и за-
каленного в воде (Х200) [205]
шается на 80—120%, а легированных—на 60—80%. Размеры
деталей при этом увеличиваются на 4—8 мк. Особенно эффек-
тивным оказывается введение в ванну, содержащую KCN -и
KCNO, кислорода; при этом в течение 1,5—2 ч при 540—565° С
глубина упрочненного слоя достигает 0,46 мм [236].
Нитроцементация и цианирование при 700° С. В зарубежной
литературе неоднократно указывалось на возможность нитроце-
ментации и цианирования низко- и среднеуглеродистых сталей
в течение нескольких часов ~
поверхностный слой стали
вследствие насыщения
азотом переходит в аусте-
нит, а после окончания
процесса и закалки в 'во-
де превращается в мар-
тенсит тонкого строения.
Однако в слое сохраняет-
ся некоторое количество
остаточного аустенита,
для устранения которого
сталь обрабатывают хо-
лодом. В поверхностной
зоне слоя имеются нетра-
вящиеся участки карбо-
нитридной фазы. При та-
ком способе обработки в
сердцевине образцов ста-
ли отсутствуют фазовые
превращения, деформа-
ция практически не на-
блюдается. Процесс осуществляется в газовой среде, состоящей
из 30% СО; 43% Н2; 25% Ы2; 1% СН4; 1% NH3 (очевидно, отхо-
дящий из печи газ). На низкоуглеродистой стали в течение 6 ч
при 700° С образуется слой азотистоуглеродистого мартенсита
толщиной 0,075 мм с твердостью HV 900 (рис. 100); на стали,
содержащей 0,35% С, в течение 24 ч при 700° С глубина такого
слоя достигает 0,2 мм, а твердость HV 890. Глубже этого слоя
располагается переходной слой, состоящий из зерен мартенсита
и феррита [205].
Описанным способом можно и цианировать указанные стали
при 700° С. Рекомендована ванна с 20% KCNO, в которой в тече-
ние 90 мин на стали с 0,15% (охлаждение в воде) достигается
поверхностная микротвердость 620—780 [237]. В ваннах не-
сколько иного состава в течение 60 мин получался слой глубиной
0,08 мм с поверхностной микротвердостью 900, а в течение
160 мин—слой глубиной 0,15 мм с той же поверхностной мик-
ротвердостью [238].
153
АЛИТИРОВАНИЕ
Алитирование (алюминирование)— это процесс поверхност-
ного насыщения стали и чугуна алюминием, придающий им по-
вышенную окалиностойкость и сопротивление атмосферной кор-
розии. Алитированию подвергается чаще низкоуглеродистая
сталь и реже среднеуглеродистая сталь и серый чугун. Алитиро-
ванные сталь и чугун применяются в качестве заменителей вы-
соколегированной окалиностойкой стали и сплавов, имеют высо-
кую стойкость при нагреве до 800—900° С, а в некоторых случаях
даже до 950—1000° С. В последнее время алитированию начали
подвергать также некоторые окалиностойкие и жаропрочные ста-
ли и сплавы, например СХ8, Х18Н9Т, 4Х14Н14В2М и др., для
дополнительного повышения их окалиностойкости.
Алитирование производится при 700—1100° С. Глубина алити-
рованного слоя колеблется в пределах 0,02—0,8 мм.
Разработаны следующие методы диффузионного алитирова-
ния: I) в порошкообразных смесях; 2) в ваннах с расплавлен-
ным алюминием; 3) металлизация стали алюминием с после-
дующим диффузионным отжигом; 4) в аэрозолях; 5) электро-
лизное— в ваннах с расплавленными солями алюминия; 6) га-
зовое. Три первых метода применяют в промышленности, а
остальные проверены лишь в лабораторных условиях.
Методы и режимы алитирования
Алитирование в порошках * 1
Первые исследования по разработке технологии алитирова-
ния в порошкообразных смесях были проведены в СССР в 1927—
1930 гг. [19]. Позднее этот метод алитирования получил преиму-
щественное распространение в промышленности.
Порошкообразные смеси для алитирования в ящиках состав-
ляют из следующих компонентов: алюминия или ферроалюми-
ния; хлористого аммония — NH4C1, являющегося ускорителем
процесса (кроме того, продукты разложения NH4C1 способствуют
вытеснению из ящика воздуха); окиси алюминия (глинозема,
каолина), предотвращающей сплавление частиц порошка алю-
миния и спекание ферроалюминия.
При высоких температурах в ящике протекают следующие'
реакции:
NH4C1 = NH3+HC1;
6НС1 + 2А1 = 2А1СЬ + ЗНа.
Ранее предполагали, что затем при контакте газообразного
хлористого алюминия с поверхностью нагретой стали протекает
обменная реакция
Fe + А1С13 = FeCl3 + Al.
1 В зарубежной литературе этот процесс часто называют калоризацией.
154
Однако в последнее время было доказано, что такое представ-
ление о переносе алюминия на поверхность стали не подтвер-
ждается термодинамическими расчетами. Кроме того, согласно
этой реакции на поверхности стали должна происходить замена
одного атома железа на один атом алюминия. Поскольку удель-
ный вес железа и алюминия различен, после алитирования сле-
довало бы ожидать уменьшения веса стали. Однако эксперимен-
ты показывают обратное.
Нужно также учитывать, что хлор имеет большее сродство к
алюминию, чем к железу.
Наиболее вероятными реакциями алитирования являются:
вначале образование субхлорида алюминия, а затем самовосста-
новление алюминия вследствие окисления некоторого количества
соединения алюминия низшей валентности до соединения
высшей валентности [239]. При этом протекают следующие ре-
акции:
а) на поверхности частиц ферроалюминия и алюминия об-
разуется субхлорид алюминия:
6НС1 + 2А1 = 2А1С1 з + ЗН<,
А1С13 + 2А1 = ЗА1С1
ИЛИ
А1С13 = A1G1 + с12;
б) на поверхности стали происходит самовосстановление
алюминия:
ЗА1С1 — AICI3+2А1.
Интересно отметить, что приведенное выше объяснение реак-
ции алитирования с участием субхлорида алюминия, вероятно,
может быть распространено и на реакции силицирования, хро-
мирования и некоторых других процессов диффузионного насы-
щения поверхности стали, протекающих в большинстве случаев
также с участием субхлоридо-в соответствующих элементов. Хи-
мический перенос становится возможным лишь при условии об-
разования соединений низшей валентности. Из предложенного
механизма переноса следует, что хлориды металлов высшей ва-
лентности, принимая участие в реакциях перноса, не расходу-
ются, а полностью возвращаются в сферу реакции.
Достоинство этой схемы в том, что она позволяет обобщить
процессы насыщения некоторыми металлами с процессом цемен-
тации, протекающей за счет разложения субоксида углерода:
2СО = С + СО2.
Данный механизм переноса не исключает полностью ранее
принятого механизма, а дополняет его. Например, перенос меди,
никеля, олова и других металлов, которые могут восстанавли-
155
ваться железом, может быть объяснен реакциями восстановле-
ния металла из его хлоридов. Из сказанного следует, что хими-
ческий перенос металла в процессах химико-термической обра-
ботки может идти различными путями.
Для алитирования используются разнообразные смеси. Смесь,
состоящая из 49,5% порошка алюминия, 49,5% окиси алюминия
(каолина) и 1% хлористого аммония, давно применяется на за-
водах. Однако при алитировании в этой смеси состояние алитиро-
ванной поверхности часто -бывает хуже (может происходить
небольшое налипание смеси), чем при использовании смеси, со-
держащей 99% ферроалюминия и 1% хлористого аммония.
Вместо порошка ферроалюминия в этой смеси можно приме-
нять порошок ферроалюминиевомедного сплава, содержащего
2—4% Си и десятые доли процента магния [240], [241]. Присут-
ствие этих добавок в ферроалюминии не оказывает существен-
ного влияния на глубину и качество алитированного слоя. Для
выплавки этого сплава используются отходы дуралюмина.
В смеси, содержащей ферроалюминий, отсутствуют окись
алюминия и каолин, так как порошок ферроалюминия мало под-
вержен спеканию. Однако, если к алитированной поверхности
предъявляются высокие требования или необходимо уменьшить
хрупкость слоя, целесообразно к смеси добавлять до 20—60%
нейтрального порошка (глинозем, каолин и т. д.) или отработан-
ной смеси.
Колебания в содержании хлористого аммония (0,5—2%) в
смесях, применяемых на заводах, незначительно сказываются
на глубине алитированного слоя. Чаще применяются смеси, со-
держащие 1 % хлористого аммония.
Ферроалюминий должен содержать 35—50% железа, осталь-
ное — алюминий с неизбежными небольшими примесями меди,
кремния, марганца и углерода. При выплавке сплава сначала
расплавляют обрезки низкоуглеродистой стали, а затем добав-
ляют заранее сплавленные в слитки отходы алюминия или дур-
алюмина в требуемом по расчету количестве с добавкой на угар
(10—12%). Сплав заливают в ящики, футерованные огнеупор-
ным кирпичом. Застывший сплав легко измельчается в шаровых
мельницах (до величины зерен около 0,5 мм). К порошку сплава
незадолго до упаковки деталей добавляют хлористый аммоний,
после чего смесь тщательно перемешивают. Перед алитирова-
нием поверхность деталей очищают с помощью пескоструйных
или дробеструйных аппаратов или травления в растворах кислот.
Алитирование осуществляется в железных или нихромовых
ящиках. Детали укладывают в них так же, как и при цемента-
ции. Полые и трубчатые детали размещают так, чтобы газы в
процессе нагрева имели свободный выход вверх из полости дета-
ли. После засыпки каждого слоя порошка его слегка утрамбо-
вывают. Между деталями кладут контрольный образец. Для
156
создания лучшей герметичности применяют ящики -с двойными
крышками, между которыми засыпают чугунную стружку, песок
или отработанную смесь. Во внешней крышке делают отверстие
(диаметром 1—2 мм) для предотвращения разры-ва ящика под
действием газов.
Степень обеднения смеси алюминием после алитирования
зависит от герметичности ящиков, состава смеси, продолжитель-
ности и температуры процесса. Порошок ферроалюминия окис-
ляется несколько меньше, чем порошок алюминия. При алити-
ровании в смеси из 98% ферроалюминия и 2% NH4C1 при 1050° С
в течение 24 ч обеднение смеси • алюминием составляет около
8,5—9% А1, следовательно, для алитирования каждой последую-
щей партии деталей к использованной смеси необходимо добав-
лять около 20% свежей смеси. После алитирования в смеси того
же состава при 950—1000° С в течение 6—10 ч к использованной
смеси достаточно добавлять 10—15% свежей. При низкой тем-
пературе (860° С) в течение 10 ч обеднение алюминием смеси,
состоящей из 99% ферроалюминиевого сплав-а и 1% хлористого
аммония, составляет всего 0,5% А1. Поэтому при алитировании
по такому режиму смесь можно применять многократно без осве-
жения (с добавкой только 0,5—-1,0% NH4C1).
При составлении новых смесей или освежении старых их не-
обходимо тщательно перемешивать в механических барабанных
смесителях. Когда содержание алюминия в смеси после много-
кратного употребления уменьшается ниже допустимых пределов
(<20%), смесь переплавляют.
Температуру алитирования обычно поддерживают в пределах
950—1050° С. При -более низкой температуре скорость алитиро-
вания сильно замедляется. Алитирование при более высокой тем-
пературе вызывает сильный рост зерна стали и связано с рядом
технических затруднений. При алитировании очень тонких дета-
лей (толщиной около 1,5 мм) температуру процесса снижают до
850—860° С.
На рис. 101 приведены результаты алитирования при раз-
личных температурах и выдержках в смеси, состоящей из 99,5%
ферроалюминия и 0,5% NH4C1. В смеси, состоящей из 49,5% по-
рошка алюминия, 49,5% окиси алюминия и 1% NH4C1, алитиро-
вание идет медленнее, чем в смесях, указанных выше.
На ряде заводов применяют смесь, состоящую из 48% ферро-
алюминия, 48% песка и 4% NH4C1. Для освежения такой смеси
к ней добавляют 20% свежего сплава и 4% NH4C1. При 1000°С
в течение 12 ч на сталях Ст. 1, Ст. 2 и Ст. 3 получают алитиро-
ванный слой глубиной 0,6 мм, а в течение 24 ч — глубиной 1,1 мм
(в больших ящиках).
Иногда насыщение алюминием производят без контакта дета-
лей с порошком в газовой среде, образующейся в ящике. Это ред-
ко уменьшает расход смеси, но замедляет процесс. Например,
157
на Луганском тепловозостроительном заводе таким образом
алитируют стаканы, компенсаторы, патрубки, тройники, глуши-
тели и т. д. При этом используются ящики, имеющие двойные
крышки с песочными затворами; смесь насыпается толстым сло-
ем (не менее 10% объема ящика) на дно, и на нее помещаются
детали. Смесь состоит из 90% отработанной смеси и 10% све-
жей, содержащей 98% ферроалюминия и 2% хлористого аммо-
ния. В ящиках размером 800 X 650 X 650 мм диффузионный слой
глубиной 0,15—0,25 мм образуется в течение 20 ч при 900° С»
а глубиной 1 мм — в течение 12 ч при 1100° С.
Иногда алитированию подвергают готовые детали, изменение
размеров и состояния поверхности которых не допускается.
В этих случаях процесс осу-
ществляется при 760—850° С в
смеси, состоящей из 20% свежего
ферроалюминия, 79% отработан-
ной смеси и 1 % хлористого алю-
миния, глубина слоя 0,02—
0,04 мм.
При алитировании серого чу-
гуна глубина слоя примерно в
2 раза меньше, чем при алитиро-
вании стали 10 в тех же услови-
ях. При 975° С в течение 3 ч об-
разуется слой глубиной 0,08—
0,10 мм, а при 1050° С в течение
3 ч — глубиной 0,15—0,20 мм.
После алитирования детали
волосяными щетками и сжатым
Рис. 101. Глубина алитированного
слоя стали 10 в зависимости от
продолжительности и температуры
алитирования в смеси 99,5% желе-
зоалюминиевого сплава и 0,5%
NH4C1 [19]
очищают от ферроалюминия
воздухом. Не разрешается долгое время хранить неочищенные
детали с тонким алитированным слоем (до 0,1 мм), так как на-
личие на их поверхности ферроалюминия и следов хлористого
алюминия вызывает коррозию, особенно во влажной атмосфере.
Для снижения содержания алюминия в слое и уменьшения
его хрупкости алитированные детали иногда подвергают допол-
нительному отжигу (нормализации) при 900—1050° С в течение
4—5 ч. Глубина слоя в этом случае увеличивается на 20—40% и
более. Ответственные детали, работающие при сравнительно не-
высоких температурах, после алитирования или длительного от-
жига при высокой температуре подвергают для измельчения зер-
на их сердцевины нормализации при 860—890° С.
Алитирование в ваннах с расплавленным алюминием
Этот процесс был впервые изучен еще в 1932 г. [19]. Тогда же
рекомендовали вводить в алюминиевую ванну (нагретую до
680—800° С) 8—12% Fe во избежание интенсивного растворения
158
стальных деталей в расплавленном алюминии. Результаты али-
тирования в такой ванне приведены на рис. 102.
Для уменьшения хрупкости слоя после алитирования в ван-
нах детали подвергают диффузионному отжигу при 900—1100° С.
Преимуществом алитирования в ваннах является кратковре-
менность процесса и простота выполнения операции. Однако
этому методу присущи и недостатки: стальные тигли, в которых
находится расплав, имеют малую стойкость, трудно полностью
устранить растворение в ванне деталей, расплав обильно нали-
пает на поверхность деталей. В связи с этим алитирование в рас-
плаве в прежние годы получило сравни-
тельно небольшое применение в промыш-
ленности и использовалось главным об-
разом для обработки различных труб,
чехлов термопар, инструментов, приме-
няемых при литье цветных металлов
и т. д.
Литейные инструменты алитировали
в ванне при 750° С в течение 45лшн, полу-
Рис. 102. Изменение
глубины алитирован-
ного слоя стали 10 в
зависимости от про-
должительности и
температуры алитиро-
вания в ванне со
сплавом, содержа-
щим 88% А1 и 12%
Fe (В. Ф. Никонов)
чая алитированный слой глубиной до
0,25 мм. Затем инструменты подвергали
диффузионному отжигу в воздушной ат-
мосфере при 1000° С в течение 5 ч. Глу-
бина диффузионного слоя после отжига
увеличивалась до 0,45 мм.
Трубы рекомендовалось алитировать
при 670—680° С в течение примерно 1 ч.
За это время глубина алитирования до-
стигала 0,08—0,15 мм [19].
В последние годы процесс алитирования в расплавленном
сплаве алюминия и железа был усовершенствован в целях полу-
чения более равномерных по глубине слоев и уменьшения нали-
пания на поверхность стали расплава.
На поверхность расплава наводят слой флюса, который вы-
полняет следующие функции:
1. Защищает расплав от окисления.
2. «Протравливает» (слегка растворяет) поверхность метал-
ла, способствуя удалению загрязнений и тонких окисных пле-
нок, что улучшает смачивание обрабатываемой поверхности
алюминием и способствует образованию однородного по глубине
слоя,
3. Облегчает очистку поверхности металла от налипшего
расплава (путем встряхивания деталей в слое флюса или под
действием центробежных сил).
4. Прогрев изделий в слое флюса уменьшает время контакта
деталей с расплавом, что способствует уменьшению разъедания
поверхности стали. Прогрев деталей в слое флюса, кроме того,
159
позволяет поддерживать температуру -сплава на уровне, лишь
немного превышающем температуру ликвидуса, что уменьшает
растворение стали в алюминии.
5. Слей флюса позволяет применять электродный нагрев.
Составы флюсов, применяемых при алитировании, приведены
в табл. 22. О преимуществах того или иного состава нельзя су-
дить без проверки флюсов в одинаковых условиях эксплуатации.
Таблица 22
Составы флюсов, применяемых при алитировании
№ состава Состав в % Источни- ки
NaCl KCI Na8AlFe A1F, ZnCl2
1 40 40 10 10
2 35 47 12 6 — [242]
3 35 35 10 — 20 [243]
4 44 56 — — — [244]
При длительном использовании флюса в нем накапливаются
нерастворимые оки-слы железа, алюминия и другие загрязнения.
Поэтому флюс нужно обновлять или регенерировать в специаль-
ном тигле.
Вместо расплавленного флюса для подготовки деталей к али-
тированию применяют водные флюсы, состоящие из фтористого
калия и соляной кислоты [245]. В этом растворе детали обраба-
тывают при 60—80° С, затем сушат, подогревают в печи до 400° С
и погружают в расплав алюминия и железа. Созданное таким
образом на поверхности стальной детали покрытие предохраняет
ее от окисления при подогреве и обеспечивает получение после
обработки чистой поверхности.
Для устранения налипания расплава на поверхность стали
применяют различные способы: а) обдувают поверхность выну-
той из ванны детали сильной струей воздуха; б) встряхивают
или подвергают вибрации детали в слое флюса; в) вращают де-
тали в слое флюса [244].
При вращении деталей в слое флюса их закрепляют на валу,
приводимом во вращение электродвигателем. Первый раз
электродвигатель рекомендуется включать на короткое время
после погружения деталей из слоя флюса в расплав; при этом
улучшается смачивание сплавом поверхности детали и удаляют-
ся с нее окислы, которые могут налипнуть в слое флюса. Вто-
рично электродвигатель включают на 30—60 сек по окончании
процесса, когда детали переносятся из расплава в слой флюса.
При этом под действием центробежных сил с деталей сбрасывает-
160
ся максимальное количество налипшего расплава. Скорость вра-
щения деталей не менее 100 обIмин (рис. 103).
Тигли для алитирования в расплавленном алюминии изготов-
ляют из магнезита или шамота (для лабораторных эксперимен-
тов из фарфора, кварца, графита). Сначала в тигле расплавляют
флюс -и прогревают его до 900—950° С для удаления влаги, за-
тем плавят алюминий и в зону алюминия вводят железную
стружку из расчета получения расплава, содержащего 10—12%
Fe.
Меньшее разъедание поверх-
ности стали наблюдается при
алитировании в никельалюминие-
вом расплаве, содержащем 20—
22% Ni [244].
Подогрев и флюсование мож-
но производить также в отдель-
ной ванне, а затем переносить де-
тали в ванну с расплавом. После
извлечения деталей из ванны из-
быток расплава удаляется вибра-
цией или сжатым воздухом. Вы-
держка во флюсе при 700—730° С
длится 8—15 мин, а в расплаве,
имеющем ту же температуру, 1 —
3 мин [246]. Глубина алитирован-
Рис. 103. Влияние скорости вра-
щения образцов во флюсе на
сброс с них расплава алюми-
ний — никель при 820° С
(В. А. Илларионов)
ного слоя достигает примерно
0,05 мм. Для повышения вязкости
и улучшения качества, внешнего
вида поверхности деталей в рас-
плав иногда вводят немного
кремния и бериллия или кремния,
цинка и титана (1—5% Si; 2% Zn; 0,18% Ti; 0,2—0,6% Be) [242].
Некоторые зарубежные фирмы алитируют, автомобильные
клапаны [242]. Клапаны устанавливают в приспособлении голов-
ками вниз и опускают на 3 мин в слой расплавленного флюса,
нагретого до 730° С (состав № 1 в табл. 22). Затем головки кла-
пана опускают в расплав алюминия, выдерживают в нем 10 сек
(730° С), вынимают из ванны и обдувают сильной струей возду-
ха. При этом глубина образующегося слоя составляет 0,025—
0,035 мм.
Поддоны для обжига пеносиликатного кирпича алитирова-
лись с применением флюса № 3 при 700° С в течение 3 мин
с последующим встряхиванием в слое флюса. Глубина диффу-
зионного слоя достигала 0,02—0,04 мм, сверху оставался, слой
налипшего алюминия толщиной 0,1—0,33 мм. Стальные листы й
проволоку алитируют в поточных установках, аналогичных тем,
которые применяются при цинковании [246].
161
Алитирование методом металлизации с последующим отжигом
Этот процесс состоит из четырех операций [247]:
1. Подготовка поверхности пескоструйной или дробеструй-
ной обработкой (или подогревом до 300° С). Очистка поверхно-
сти, создание шероховатости и подогрев повышают прочность
сцепления напыленного алюминия со сталью.
2. Нанесение слоя алюминия толщиной 0,7—1,2 мм. Для ме-
таллизации применяют электрические аппараты ЭМЗА, ЭМ6 и
ЭМС1-57 либо газовые аппараты ГИМ-2 и МГИ1-57, работаю-
щие на ацетилене, пропано-бутановой смеои, .метане или смешан-
ном газе. Режим металлизации с применением аппаратов типа
ЭМ следующий:
ЭМЗА ЭМ6 ЭМС1-57
Расстояние от поверхности в мм . . . . 75—150 100—200 100—250
Напряжение дуги в в . . 25—30 35—40 35-45
Сила тока в а . . 60—80 100—120 100—120
Давление воздуха в ат . . 5—6 5—6 5—6
» > » кн/м2 . . 490—588 490—588 490—588
3. Обмазка металлизированной поверхности для предохране-
ния алюминия от окисления во время диффузионного отжига.
Применяются обмазки следующих составов (в %):
Состав 1 Состав 2
Серебристый графит..............50 Серебристый графит.............50
Огнеупорная глина...............20 Огнеупорная глина..............20
Кварцевый песок.................30 Кварцевый песок................30
Жидкое стекло...................10 Жидкое стекло.................. 8
Хлористый аммоний.............. 2
Примечание. Жидкое стекло и хлористый алюминий добавляются к
приготовленной смеси остальных компонентов.
Подогретую до 80—100° С обмазку наносят на металлизи-
рованную поверхность детали слоем толщиной 0,8—1,5 мм
кистью, путем окунания или пневматическим распылителем. Про-
сушивается обмазка на воздухе, затем в печи при 80—100° С.
4. Диффузионный отжиг: посадка изделий в печь при 500—
600° С, повышение температуры до 900—950° С, выдержка 2—4 ч,
охлаждение вместе с печью до 600° С. Иногда производят отжиг
и при более высоких температурах. При таком режиме обработ-
ки глубина алитированного слоя достигает 0,20—0,40 мм.
Описанная технология алитирования путем металлизации с
последующим диффузионным отжигом обеспечивает получение
достаточно качественных диффузионных слоев. Этот метод во
многих случаях вполне может конкурировать с другими метода-
ми, его стоимости меньше стоимости алитирования в порошко-
образных смесях.
16?
На Волгоградском и Харьковском тракторных заводах алити-
рованию путем металлизации с последующим отжигом подверга-
лись детали тракторных газогенераторных установок, на Ногин-
ском заводе топливной аппаратуры — опоки, на ряде других за-
водов— муфели 'и горелки для отжига и другие детали. В оте-
чественной и зарубежной промышленности внедрены в производ-
ство автоматические линии для алитирования таким методом
клапанов автомобиля [248], [249]. По технологии, принятой на Че-
лябинском автомеханическом заводе, алюминий наносят на не-
нагретые клапаны, затем нагревают их т. в. ч. для диффузии
нанесенного алюминия и производят опрессовку фаски нагрето-
го клапана в оправке, имеющей профиль посадочного гнезда.
Это позволяет получить поверхность высокой чистоты, не тре-
бующую доводки и притирки.
Алитирование клапанов из сталей Х9С2 и Х10С2М на глуби-
ну около 0,02 мм увеличивает их стойкость в Г,5—2 раза и дает
возможность работать на высокооктановом бензине, а следова-
тельно, увеличить мощность и быстроходность автомобилей.
По другой технологии [249] клапаны в вертикальном положе-
нии движутся на цепном конвейере и одновременно вращаются
вокруг оси со скоростью 550 об!мин. Сначала они проходят зо-
ну индукционного подогрева, затем подвергаются металлизации
с помощью пневматического распылителя и поступают во вторую
индукционную установку, где осуществляется диффузионный от-
жиг при 780° С. Глубина диффузионного слоя не достигает
0,02—0,025 мм.
Алитирование в аэрозолях
Алитирование в аэрозолях (как и насыщение стали хромом,
кремнием и другими элементами) предложено в работе [250].
Этот процесс осуществляется в газообразных и парообразных
фазах с наличием мельчайших взвешенных частиц твердой фа-
зы в аппаратуре, близкой по конструкции к аппаратуре, приме-
няемой для диффузионных газовых процессов.
Над печью расположен дозатор с порошкообразной смесью.
Смесь посредством конусного затвора, приводимого в движение
электромагнитом, соединенным с реле времени, подается не-
большими дозами в реакционную реторту, в верхней части ко-
торой установлен вентилятор.
Выход для газов находится в нижней части реторты. Образ-
цы, помещенные в металлическую сетку, быстро вводятся в ра-
зогретую печь, при этом немедленно включается дозатор, на-
строенный на подачу двойного или тройного расхода смеси, что
делается в течение 10—15 мин во избежание окисления деталей.
Дозы порошкообразной смеси подаются в реторту через каждые
1—2 мин. Для реторты диаметром 120 и высотой 350 мм расход
смеси, которая состоит из Al, NaCl и NH4C1, взятых в соотноше-
163
ниях 8:2:1 или 4:2:1 (обе смеси близки по активности), со-
ставляет примерно 65 а/^. Замена в смеси хлористого аммония
иодистым ускоряет процесс алитирования: при 1050° С в течение
2 ч на образцах стали 35X12H12B3K9MTNb глубина слоя увели-
чилась с 0,096 до 0,12 мм.
Насыщение стали в аэрозолях алюминием и другими элемен-
тами позволяет несколько сократить продолжительность про-
цесса, однако в производстве пока не применяется.
Электролизное алитирование
Электролизное алитирование осуществляют в солевом рас-
плаве, состоящем, например, из 25 мол. % безводного хлористого
алюминия и 75 мол. % хлористого натрия (251]. Соли расплавля-
ют в стальном тигле, выложенном внутри шамотом. Детали
включаются в качестве катода в цепь постоянного тока. Анодом
служит помещаемый на дно тигля расплавленный алюминий,
к которому подводится ток по железной проволоке, изолируемой
фарфоровой трубкой. Чтобы уменьшить испарение и окисление
солей, зеркало ванны покрывается слоем графита. При плотно-
сти тока на поверхности алитируемой детали 0,5 а/дм2 и темпера-
туре 800° С глубина алитированного слоя в течение 4 ч достига-
ет 1,5 мм. Для уменьшения хрупкости слоя дополнительно про-
водится диффузионный отжиг.
Электролитическое алитирование в соляных расплавах за-
служивает большего внимания; но для того чтобы внедрить этот
метод в производство, необходимо усовершенствовать процесс —
автоматизировать его и найти способы борьбы с разрушением
тиглей и летучестью солей.
Значительно позднее, чем у нас, ванна аналогичного состава
(80% А1С13 и 20% NaCl) была (рекомендована за рубежом для
промышленного применения при температуре 940° С и плотно-
сти тока 46 а/дм2. При меньших плотностях тока ухудшается
сцепление алитированного слоя с поверхностью стали. Переме-
шивание электролита ускоряет процесс образования диффузион-
ного слоя.
В последнее время предложен ряд ванн, в состав которых
входит криолит (Na3AlF6). В ванне, содержащей 100% криоли-
та, рекомендуется проводить процесс при 980° С и плотности то-
ка 10 а/дм2 [252]. В такой ванне особенно хорошо протекает али-
тирование меди. Так, при 900° С и плотности тока 0,5 а/см2 в те-
чение 10 мин глубина диффузионного слоя достигает 0,2 мм, а
при 950° С за 10 мин — 0,42 мм. Преимуществом криолита яв-
ляется его недефицитность, но при температурах порядка 900° С
он сильно испаряется [253].
Рекомендуется ванна, составляемая из смеси КС1, NaCl и
NasAlFe, в которой при 700° С и плотности тока 7 а/дм2 за 5 мин
164
глубина слоя достигала 6 мк. Может быть применена также
ванна, составленная из 0,5—1,3% A1F3; 8—20% Na3AlF4; 25—
45% КС1 и 37—57% NaCl. Процесс в этой ванне ведут при
680—870° С и плотности тока 1,5—4,5 а/дм2.
Электролизное осаждение алюминия на поверхность стали
можно осуществлять также при 300° С в ваннах следующих со-
ставов: 1) 75% А1С13; 23% КС1; 2% KJ и 2) 80% А1С13; 18,5%
NaCl и 1,5% NaF [252]. После осаждения может проводиться
диффузионный отжиг с предварительной обмазкой поверхности
специальной пастой.
Электролизное алитирование не -получило распространения в
промышленности ввиду необходимости индивидуальной подвес-
ки деталей при обработке, неравномерности слоя на деталях
сложной формы и большой испаряемости расплавов.
Газовое алитирование
Уже давно был предложен способ газового алитирования в
горизонтальных ретортах. Одна половина реторты с порошкооб-
разной смесью, состоящей из 45% алюминия, 45% А12О3 и 10%
NH4C1, нагревается до 600° С; другая половина с обрабатывае-
мыми образцами — до 900—1000° С. Через реторту непрерывно
пропускается водород, который увлекает за собой образующийся
в первой половине реторты хлористый алюминий.
Алитирование может осуществляться также с применением
хлора или хлористого водорода, подаваемого в печь [241]. Во
время нагрева через трубчатую печь с кусками ферроалюминия
и образцами пропускают аргон, хлор же или хлористый водород
пропускают лишь при высокой температуре. При газовом али-
тировании в потоке хлористого водорода или хлора скорость
процесса была меньше, чем при алитировании в порошкообраз-
ных смесях, очевидно, вследствие недостаточного количества
омывающих поверхность стали хлоридов алюминия. Кроме того,
глубина насыщения образцов с разных сторон неравномерная.
Лишь засыпка образцов порошком ферроалюминия позволяет
получить глубокие диффузионные слои.
Газовое алитирование можно проводить в смеси А1С1з + Н2
и А1Вг3 + Н2 при 1000° С (температура испарения А1С13 125—
135° С, а А1Вг3 115—130° С) [254].
При алитировании в парах алюминия в вакууме не удалось
получить хороших результатов по глубине слоя и концентрации
в нем алюминия [255].
Представляют интерес работы по газовому алитированию в
монохлоридах и монофторидах алюминия. При обработке образ-
цов в этих средах получаются весьма твердые алитированные
слои.
Монохлорид алюминия может быть получен в результате воз-
действия паров А1С13 на расплавленный алюминий в среде
165
очищенного водорода при температуре 1200—1250° С и парци-
альном давлении 0,2 ат [256]. При этой температуре практически
весь А1С13 превращается в А1С1.
Оптимальной для алитирования в монохлориде алюминия яв-
ляется температура 950—1000° С. Поверхностная микротвердость
образцов, обработанных при этой температуре, достигала 1250
(при более низких или более высо-ких температурах твердость
снижается). Высокую твердость диффузионного слоя объясняют
образованием интерметаллического ’Соединения FeAl3 [256]. Этот
слой является также окалиностойким.
Диффузионный слой с высокой микротвердостью (Яюо125О>
был получен также на образцах, алитированных в монохлориде
алюминия при 720—980° С [257].
При газовом алитировании с (Нагревом т. в. ч. достигается ис-
ключительно высокая скорость процесса.. При 1100° С в течение
9 сек был получен слой глубиной до 0,2 мм [258].
До настоящего времени методы газового алитирования не
нашли применения в -промышленности.
Структура алитированного слоя
Увеличение содержания углерода в алитируемой стали сни-
жает глубину диффузионного слоя (рис. 104), повышает кон-
центрацию в нем алюминия. Еще медленнее диффундирует алю-
миний в чугун. Для ускорения процесса чугун рекомендуется
подвергать поверхностному обезуглероживанию.
Рис. 104. Влияние содержания угле-
рода в стали на глубину алитирован-
ного слоя (алитирование в порошко-
образной смеси в течение 6 ч) [259]
Рис. 105. Влияние легирующих эле-
ментов на глубину алитированного
слоя железа с 0,02% С, обработан-
ного при температуре 950° С и вы-
держке 6 ч [259]
Влияние легирующих элементов на глубину алитированного
слоя почти безуглеродистых сплавов (на основе армко-железа)
изучалось в двух работах, результаты которых не совпадают
(рис. 105 и 106). Возможно, это объясняется различием исход-
ных материалов и условий экспериментов (например, темпера-
166
тура алитирования отличалась на 76° С, что, несомненно, ока-
зывало существенное влияние на результаты).
Влияние легирующих элементов на глубину алитированного
слоя в безуглеродистых сплавах зависит главным образом от
их влияния на замыкание у-области (чем уже у-область, тем
быстрее возникает a-фаза, скорость диффузии алюминия в кото-
рой значительно выше) и влияния на скорость диффузии алю-
миния в у- и сс-фазах. Повышение содержания углеродов в леги-
рованных сплавах должно
также оказывать существен-
ное влияние на глубину али-
тированного слоя, однако
влияние углерода изучалось
лишь на нелегированной
стали (рис. 104).
Легирующие элементы
влияют и на микротвер-
дость алитированного слоя
[260]. Повышают микро-
тв ер д ость >п р е и»м ущ еств енн о
те элементы, которые повы-
шают твердость феррита.
При этом кремний и марга-
нец в большей степени по-
вышают микротвердость зо-
ны слоя, примыкающей к
сердцевине. Хром сравни-
тельно равномерно повыша-
Рис. 106. Влияние легирующих эле-
ментов на глубину алитированного
слоя, образовавшегося при 1025° С
и выдержке 5 ч (сплавы выплавля-
ет МИКротвердость всех зон лись на основе армко-железа) [260]
слоя. При содержании в
стали 12% Сг это повышение равно примерно 150—200. Мо-
либден, вольфрам и кобальт влияют на микротвердость диффу-
зионного слоя незначительно.
Структура и фазовый состав алитированного слоя зависят от
метода алитирования. После алитирования в порошкообразных
смесях или газовой среде и травления обычными реактивами
алитированный слой выявляется в виде нетравящейся зоны
(рис. 107). Длительным травлением иногда удается выявить
столбчатое строение зерен слоя, отделенных от сердцевины ли-
нией раздела, и наличие игольчатых включений и сетки по гра-
ницам зерен примерно в средней части слоя L Зону диффузион-
ного слоя с несколько повышенным содержанием алюминия,
1 Выявляются только после травления в 5%-ном кипящем растворе
NaOH или в травителе, составленном из 8% HF, 13% НС1 и 79% Н2О при
комнатной температуре.
167
Рис. 107. Микроструктура диффузионно-
го слоя стали 15, алитированной в по-
рошкообразной смеси при 1050° С в
течение 6 ч (Х200)
лежащую глубже линии раздела, на микрошлифах выявить не
удается.
Строение и природа составляющих алитированного слоя изу-
чены еще совершенно недостаточно. Предполагается следующее
чередование 'фаз в алитированном слое. К линии раздела с -серд-
цевиной примыкает почти нетравящий-ся a-твердый раствор
(рис. 108). Далее пример-
но в середине слоя наблю-
даются сетка и иглы
(рис. 109), которые пред-
положительно относятся к
соединению ЕезА1. Еще
ближе к поверхности дол-
жны располагаться фазы
FeAl, FeAl2 и, возможно»
Fe2Als. Эти фазы должны
присутствовать в слое, так
как содержание алюми-
ния в поверхностных зо-
нах слоя достигает 50% и
более (рис. 190). Одной из
этих фаз принадлежат
шестигранные зерна, вы-
ходящие на поверхность
образца (рис. 110).
В случае примене-
ния железоалюминиево-
медного сплава в сталь
диффундирует также
медь, содержание которой
в поверхностных зонах
диффузионного слоя до-
ходит до 0,6—0,8%; это,
однако, не сказывается на
строении слоя.
При алитировании ме-
тодом металлизации с по-
содержание алюминия в
следующим диффузионным отжигом
слое, по данным послойного анализа, может доходить до 75—
79% [247].
На м-икрошлифах алитированного слоя углеродистых сталей
к линии раздела, со стороны сердцевины, часто примыкает зона,
обогащенная углеродом -и возникающая -вследствие оттеснения
углерода в аустените по мере роста столбчатых зерен сс-фазы
при температуре насыщения (рис. 111). Однако оттеснение угле-
рода в глубь стали не всегда наблюдается на микрошлифах
глубже линии раздела. Применение рентгеноструктурного ана-
168
лиза о авторадиографии позволило раскрыть причины отсутст-
вия зоны, обогащенной углеродом на микрошлифах алитирован-
Рис. 108. Диаграмма состояния системы
Fe — Al
алюминия по глубине слоя
при различных режимах
алитирования в порошкооб-
разной смеси:
1 — при 900е С, 6 ч; 2 — при
1000° С, 6 ч\ 3 — при 1000е С,
12 ч [241]
Рис. 110. Микроструктура алитированного слоя двух образцов, обработан-
ных в порошкообразной смеси (снимки сделаны под углом 90° к поверхности
образцов):
а — поверхностная зона алитированного слоя; б — средняя зона алитированного слоя;
в и г — соответственно те же зоны второго образца, алитированного по другому ре-
жиму (241]
ных при 930—950° С сталей, содержащих более 0,7% С [261].
В алитированном слое сталей У7 и У12 были обнаружены вклю-
чения тройной фазы Fe — Al — С, имеющей решетку гранецент-
рированного куба (3,73—3,75 А) и выявляющейся обычно в сред-
ней и прилегающей к линии раздела зонах слоя. Эта фаза, по-
169
видимому, является 8-фазой, существование которой было уста-
новлено в литых сплавах тройной системы Fe — Al — С (сплав
с 15% А1 и 3,6% С состоит преимущественно из е-фазы). Кроме
того, в самой поверхностной зоне алитированного слоя этих ста-
лей были обнаружены столбчатые иглообразные включения,
тип решетки и параметры которых такие же, как и у карби-
да А14С3.
Чтобы изучить характер распределения углерода в алитиро-
ванном слое,- образцы технического железа цементовались в
Расстояние от поверхности
Рис. 111. Изменение концентра-
ции алюминия и углерода в
алитированном слое стали с
0,4% С (фазовый состав дан
для температуры диффузии)
[259]
твердом карбюризаторе, часть
которого содержала радиоактив-
ный изотоп С14. Затем цементован-
ные образцы алитировались. Ав-
торадиограммы подтвердили, что
углерод концентрируется во вклю-
чениях, располагающихся в на-
ружной части слоя, и в предпола-
гаемой 8-фазе, включения которой
находятся между вытянутыми
зернами a-твердого раствора.
Впервые карбид алюминия
AI4C3 был обнаружен в алитиро-
ванном слое в исследованиях, ко-
торые проводились в связи с на-
блюдающимися в практике слу-
чаями частичного разрушения алитированного слоя чугуна и вы-
сокоуглеродистой стали после длительного пребывания в среде
влажного воздуха. Исследование проводилось на железоуглеро-
дистых сплавах пяти различных составов. Оказалось, что на
образцах сплавов, содержавших 0,02 и 0,6% С, алитированный
слой во влажном воздухе не разрушается, а на образцах спла-
вов, содержащих 1,6; 2,6 и 3,6% С, разрушается уже через 3 дня
(в среде сухого воздуха он остается неизменным длительное
время) [262]. На поверхности диффузионного слоя высокоуглеро-
дистого и высокоалюминиевого сплава с помощью рентгено-
структурного анализа и был обнаружен карбид алюминия А14С3,
наблюдавшийся под микроскопом в виде столбчатых кристалли-
ков. Карбид алюминия, как известно, легко разлагается во влаж-
ном воздухе.
Микроструктура диффузионного слоя образцов низкоуглеро-
дистой стали, алитированной в расплаве алюминия и железа,
имеет на границе слоя и сердцевины извилистый иглообразный
характер (рис. 112).
Диффузионные слои такого вида образуются намного быст-
рее, чем -слои, характер которых был рассмотрен ранее.
Иглообразный характер алитированных слоев указывает на
то, что при их образовании происходит 'быстрый ро-ст фаз в на-
170
правлении, перпендикулярном к поверхности образца, тогда как
вдоль поверхности развитие этих фаз протекает слабо. Многие
авторы связывали такой характер алитированного слоя с пре-
имущественной диффузией алюминия по границам зерен 'Стали.
Однако тщательное рассмотрение микроструктуры алитирован-
ных слоев опровергает это мнение. Направление роста игл .почти
Рис. 112. Микроструктура стали 10, подвергнутой алитиро-
ванию при 770° С в ванне, составленной из 90% А1 и 10% Fe.
(Х500)
всегда не совпадает с границами зерен. Иглообразные диффу-
зионные слои аналогичного вида встречаются и в других слу-
чаях, например при поверхностном насыщении бором железа,
кобальта и никеля (см. рис. 150).
Показано [263], что при обработке железа в расплаве алюми-
ния диффузия алюминия в железо протекает быстрее, а фазо-
вый состав образующегося слоя получается иным, чем при об-
работке железа в других средах, -содержащих алюминий. Реак-
ция между расплавленным алюминием и железом начинается
уже при 590—600° С (для чистых исходных материалов). После
холодной деформации железа со -степенью деформации 50% эта
температура снижается до 560° С, а при большей степени дефор-
мации еще ниже — вплоть, до 400° С [263]. Коэффициент диффу-
зии алюминия в недеформиро-ванное железо (из расплава алю-
миний— железо), определенный при температуре 715—980° С
(судя по скорости роста фазы РегАЬ), составляет Dai =
13000
= 0,0323 е RT см2!сек.
171
Рентгеноструктурный анализ показал, что иглообразный ха-
рактер -структуры алитированного слоя железа, обработанного в
расплаве алюминий — железо, связан с особой природой кри-
сталлической решетки, возникающей при этом в слое -фазы
Fe2Als (обращает на себя внимание тот факт, что именно эта
фаза не обнаруживается в диффузионных слоях, образующихся
в порошкообразных смесях). Из-за этих особенностей решетки
фазы Fe2A15 другие фазы при таких условиях алитирования не
Рис. 113. Модель трех поставленных друг на
друга элементарных ячеек фазы Fe2Ai5 [263]
образуются в диффузионном слое, очевидно, потому, что ско-
рость диффузии в них алюминия несравненно меньше, чем че-
рез фазу Fe2Als [263].
Фаза Fe2Als принадлежит к ромбической системе, три ячейки
которой показаны на рис. ИЗ «поставленными друг на друга с
направленной вверх осью С. Часть атомов алюминия расположе-
на вдоль оси С на ребрах ячейки; остальные атомы алюминия и
все атомы железа лежат на гранях элементарной ячейки или
внутри нее, располагаясь кольцеобразно вокруг основных цепо-
чек атомов алюминия на расстоянии -- от них. В связи с такой,
особой, природой структуры фазы Fe2Als с относительно густо
следующими друг за другом атомами алюминия 2,1
становится возможным предпочтительное перемещение алюми-
ния в направлении оси С (для атомов основной цепочки).
Повышенной подвижности атомов алюминия в решетке спо-
собствует еще и то, что, судя .по интенсивности рентгеновских
172
интерференций, основные цепочки заполнены алюминием лишь
на 70% [263]. Такая ненормально высокая концентрация вакан-
сий на параллельно направленных рядах узлов решетки и объ-
ясняет предпочтительную подвижность атомов алюминия в опре-
деленном направлении.
Рентгенограммы боковых сторон игольчатой фазы и поверх-
ности образца подтвердили, что плоскость базиса ромбической
решетки всегда лежит в основании игольчатых кристаллов, а
ось С решетки всегда совпадает с осью игл, т. е. с направлением
диффузии алюминия.
Таким образом, игольчатые кристаллы фазы БегА^ возника-
ют вследствие того, что зародыши этой фазы благоприятно ори-
ентируются к поверхности образца и кристаллографические на-
правления -с несовершенствами решетки оказываются ориенти-
рованными нормально к фронту диффузии; это и вызывает ани-
зотропию скорости диффузии и быстрый преимущественный рост
фазы в направлении к сердцевине.
Быстрый рост интерметаллических соединений при поверх-
ностном насыщении чаще, чем предполагали ранее, происходит
вследствие особенностей их структуры. Эти особенности заклю-
чаются в существовании таких кристаллографических направле-
ний, перемещение активного элемента в которых облегчено. Ин-
тересно, что после алитирования в расплаве алюминий — железо
при температурах выше 1000° С диффузионный слой уже не име-
ет иглообразного строения [263].
Поверхностная зона алитированного слоя, образовавшегося
при любой температуре в расплаве аллюминий — железо, пред-
ставляет собой механически приставший к поверхности сплав,
состав которого соответствует сплаву ванны (на микроструктуре
рис. 112 граница между этим слоем и диффузионным слоем от-
четливо не различается).
Свойства алитированной стали
Механические свойства. Поверхностная зона алитированного’
слоя имеет повышенную твердость (до HV 500, рис. 114). Не-
смотря на это, алитированный слой обладает низкой износо-
стойкостью.
При высоком содержании алюминия алитированный слой от-
личается хрупкостью. Для уменьшения хрупкости диффузион-
ного слоя применяют более бедные алюминием порошкооб-
разные и газовые смеаи, а также производят диффузионный
отжиг. Когда в поверхностной зоне алитированного слоя при-
сутствует не более 20—30% алюминия, слой обнаруживает
удовлетворительную вязкость и не разрушается, если деформа-
ции невелики (при удлинении не более 5% на стандартном раз-
рывном образце).
173
Испытания пластинок толщиной 1,2 мм из стали 20, подвер-
гнутых алитированию на глубину 0,1—0,15 мм, показали, что
предел прочности огв стали после алитирования повышается на
5—8 кГ/мм2 (4,9-107 — 7,8-107 н/м2), удлинение 6 и вязкость по
Эриксену снижаются соответственно на 5—8% и на 1,5—2 мм.
Тонкие алитированные слои (0,05—0,07 мм), полученные при
850° С в течение 3—5 ч в порошкообразной -смеси, хорошо де-
формируются в холодном и горячем состоянии, сохраняя после
этого высокую окалиностойкость и коррозионную стойкость.
При навивке проволоки с таким алити-
Рис. 114. Распределе-
ние микротвердости
по глубине диффузи-
онного слоя стали 10,
алитированной по
разным режимам
[241]:
/ — при 1000° С. 12 ч;
2 — при 1000° С, 6 ч\ 3 -
при 900° С, 6 ч
рованным слоем его сплошность сохра-
няется.
Антикоррозионные свойства. Алитиро-
ванный слой обладает повышенными ан-
тикоррозионными свойствами в некото-
рых средах, в том числе в воздушной сре-
де. Особенно высокой стойкостью в этой
среде обладает сталь, алитированная в
расплаве алюминия и железа.
Ранее детали подвергали алитирова-
нию лишь с целью повышения их окали-
ностойкости. В последние годы алитиро-
вание в расплаве алюминия и железа все
шире используют для защиты проволоки,
ленты, предметов домашнего обихода,
строительных деталей и т. д. от атмос-
ферной коррозии.
Алитированная сталь оказывается бо-
лее стойкой против атмосферной корро-
зии и коррозии в морской воде [245], чем
сталь, оцинкованная в расплавленном
цинке.
Алитированная сталь стойка в условиях тропиков [264]. При
испытании коррозионной устойчивости в тумане, содержащем
пары соли (температура 36° С, продолжительность 120 ч), оцин-
кованная сталь (толщина слоя 0,05 мм) потеряла в весе 7\2г/дм2,
а алитированная (слой 0,05—0,07 мм) лишь 49—174 г]дм2 [245].
При весьма длительных атмосферных испытаниях алитирован-
ная сталь оказалась почти в 3 раза более стойкой, чем оцинко-
ванная (стойкость соответственно 19 и 7 лет).
Процесс алитирования не сложнее процесса цинкования и
требует аналогичного оборудования [246}. Стоимость же алити-
рования примерно такая же, как и цинкования [245].
Для обеспечения высокой коррозионной стойкости стали не-
обходим алитированный слой глубиной 0,05—0,06 мм. Такой
слой образуется в расплаве при 700° С в течение нескольких ми-
нут,
174
Алитирование -стали в газовой среде или порошкообразных
смесях применяется лишь для повышения ее окалиностойкости,
и сравнительных данных о коррозионной стойкости стали, али-
тированной этими методами и в расплаве, нет.
Антикоррозионная стойкость деталей понижается, если по
окончании процесса алитирования с них не смыты следы хлори-
стых солей; поэтому детали, подлежащие длительному хране-
нию, необходимо промывать и смазывать маслом. Нормализация
или диффузионный отжиг могут повысить антикоррозионную
стойкость тонкого алитированного слоя вследствие улетучива-
ния следов хлористого алю-
миния, а также увеличения
глубины и уменьшения по-
ристости слоя.
Алитированный слой вы-
сокоуглеродистой стали и чу-
гуна после очень длительно-
го пребывания во влажной
среде неустойчив и подвер-
гается частичному разруше-
нию (см. стр. 170), однако
это не исключает примене-
ния алитирования для обра-
ботки деталей из этих мате-
риалов.
Окалиностойкость. Мини-
мальное содержание алюми-
ния в железоалюминиевом
Продолжительность испытания
а) 6)
Рис. 115. Окалиностойкость при
1000° С алитированных (кривая 1) и
неалитированных (кривая 2) сталей:
а — сталь 10; б — сталь 1X13 [19]
сплаве, обеспечивающее его высокую окалиностойкость, состав-
ляет около 8%. При алитировании в поверхностных слоях стали
достигается значительно более высокое содержание алюминия.
Нагрев алитированной стали в окислительной среде приводит
к образованию на поверхности стали тонкой, но прочной пленки
окиси алюминия А12О3, предохраняющей сталь от окисления. На
рис. 115 показано влияние алитирования на окалиностойкость
сталей 10 и 1X13.
Глубина алитированного слоя и концентрация в нем алюми-
ния предопределяют стойкость деталей в рабочих условиях. Али-
тированные тонкие детали, например спирали электросопротив-
лений, обладают несколько более высокой окалиностойкостью,
чем крупные детали. В процессе эксплуатации тонких деталей
при высокой температуре алюминий успевает продиффундиро-
вать до их -сердцевины, и поэтому дальнейшее .понижение кон-
центрации алюминия в поверхностном слое стали замедляется,
что и способствует сохранению окалиностойкости.
Окалиностойкость сталей 10 и 45 после алитирования при-
мерно одинакова, а сталей У8, У10 и чугуна заметно ниже.
175
Рис. 116. Изменение
глубины распростра-
нения а- и у-фаз в
алитированном слое
стали 4Х14Н14В2М
при температуре на-
сыщения в зависимо-
сти от температуры и
продолжитель н о с т и
процесса [259]
Влияние температуры на окалиностойкость образцов алити-
рованной и неалитированной стали и чугуна показано в табл. 23.
После кратковременных испытаний при 850° С увеличение веса
алитированных образцов чугуна в 14 раз, а при 1000° С в 7 раз
меньше, чем неалитированных; увеличе-
ние веса алитированных образцов стали
после испытаний при 1050 и 1200° С соот-
ветственно в 52 и 20 раз меньше, чем не-
алитированных.
Однако по результатам кратковремен-
ных испытаний (рис. 116 и табл. 23) нель-
зя судить об окалиностойкости стали в
условиях длительной эксплуатации. При
длительных испытаниях окалиностойкость
понижается в результате снижения кон-
центрации алюминия в поверхностном
слое вследствие его диффузии в глубь
стали.
Наибольший эффект дает алитирова-
ние деталей, длительное время работаю-
щих при температуре до 850° С; хорошие
результаты получены при температуре
до 900—950° С; при более высоких тем-
пературах эффект от алитирования сни-
жается, однако целесообразность приме-
нения алитирования деталей, предназна-
ченных для работы и при таких темпе-
ратурах, не теряется. Сравнительно про-
должительные испытания алитированных
образцов низкоуглеродистой стали показали, что при 850° С их
окалиностойкость выше, чем у неалитированных, примерно в
15—20 раз; при 900° С — в 10—15 раз; при 1000° С — в 5—6 раз;
при 1100° С — в 3—4 раза и при 1200° С — в 2—3 раза.
Таблица 23
Влияние алитирования окалиностойкости стали и чугуна [241]
Материал образца Состояние образца Увеличение веса образца после 5 ч нагрева при различных температурах в г!м2ч
О о иэ со 950° С о о о о о Э oOSOl 1125° С 1200° С
Сталь 10 Неалитированный — — — 210 287 334
Алитированный — — — 3—5 5—7 10—25
Сдпитй uvrvu Неалитированный 56 64 154 — — —
syiyri Алитированный 3—5 8—10 18-25 — — —
176
Эффект от применения алитированных деталей при высокой
температуре увеличивается, если они работают в среде сероводо-
рода, который способствует быстрому выходу из строя стальных
деталей, в то-м числе из окалино-стойких сталей. Особенно повы-
шается окалиностойкость деталей, -работающих в среде сероводо-
рода при невысоких температурах.
По данным табл. 24, увеличение веса образцов неалитирован-
ной стали в сероводороде в несколько раз больше, чем в окисли-
тельной среде, хотя продолжительность испытания в сероводоро-
де в 42 раза меньше; стойкость алитированных образцов в серо-
водороде намного больше, чем неалитированных.
Таблица 24
Результаты испытаний окалиностойкости образцов из разных сталей
при 650° С в окислительной среде
(1000 ч) и в сероводороде (24 ч) [19]
Состав стали в % Увеличение веса образцов в мг’см*
в окислительной среде в сероводороде
С Сг Ni Мо W неали- тирован- ных алити- рован- ных нсалити- рован- ных алити- рован- ных
0,08—0,18 -X- 173,5 0,6
0,08—0,18 1,25 — 0,5 — — — 101,3 0,7
0,08—0,18 5,0 — 0,5 — 36,6 0,8 172,8 1,3
0,08—0,18 5,0 — — 1,0 17,5 1,0 175,6 0,5
0,08-0,18 18,0 8,0 — — 1,1 0,6 36,5 0,1
Алитированию в целях повышения окалиностойкости реко-
мендуется подвергать самые различные детали и изделия: муфе-
ли и ящики для термообработки (стойкость повышается более
чем в 4 раза), ящики для цементации, чехлы для термопар, тиг-
ли для солей и свинца (алитируется только наружная поверх-
ность), оболочки электротиглей для разливки силумина, ковши
для разливки алюминия и дуралюмина, газоотводные трубы дви-
гателей, трубы воздухопроводов и па-ронагревателей, трубы уста-
новок для крекирования, печную арматуру, чугунные колосники
и решетки котлов, камеры газификации, топливники и колосники
газогенераторных тракторов и автомобилей, ящики для отжига
литья по выплавляемым моделям (стойкость увеличивается в 8—
9 раз), изложницы для взятия проб (в 5—6 раз), сквозные круп-
ные изложницы (стойкость изложниц весом 6,7 т после алитиро-
вания методом металлизации повышается с 45 до 115—-127 пла-
вок) [241], [247], [265]. Алитированию можно подвергать изготов-
ленные из перлитных сталей цилиндры высокого давления
177
турбин, работающие при 560—585° С [266], лопатки газовых тур-
бин [267], направляющие дисков газовых турбин и валы газо-
турбинных установок из аустенитных жаропрочных сталей. Все
чаще встречаются случаи алитирования весьма сложных по со-
ставу сплавов и различных металлов (молибдена, ниобия, тита-
на, меди, кобальта и др.)1.
Неоднократно предлагалось вместо азотируемых деталей из
специальных сталей, легированных алюминцем, применять де-
тали из углеродистой или малолегированной стали (например,
35ХМА), подвергая их алитированию, диффузионному отжигу,
шлифованию, а затем азотированию (образуется диффузионный
•слой глубиной 0,4—0,6 мм с поверхностной твердостью
HV 1100—1150). Однако такой метод не нашел применения ввиду
его трудоемкости и малой пригодности для обработки ответст-
венных нагруженных деталей.
Алитирование аустенитной стали
Алитирование повышает окалиностойкость и эрозионную стой-
кость многих окалиностойких и жаропрочных аустенитных ста-
лей. Однако применение алитирования для жаропрочных сталей
ограничивается тем, что в результате такой обработки у многих
из них понижаются предел ползучести и длительная прочность.
Аустенитные стали алитируются тем медленнее, чем сильнее
они легированы хромом, никелем и другими элементами. В ре-
зультате алитирования при 1050° С в течение 8 ч в порошке фер-
роалюминия -с добавкой 1% NH4C1 на стали X16H36BT3 обра-
зуется диффузионный слой глубиной 0,08—0,10 мм, а на стали
Х16Н25М6—слой глубиной 0,18—0,21 мм [268].
Скорость образования алитированного слоя на менее легиро-
ванной хромом и никелем стали 4Х14Н14В2М заметно увеличи-
вается (см. рис. 116).
После алитирования жаропрочные стали подвергаются закал-
ке и старению.
Чаще применяются следующие два режима алитирования и
термической обработки жаропрочных сталей:
1) алитирование, а затем термическая обработка, нагрев до
1100—1200° С с последующим охлаждением на воздухе или в
масле и старение в течение 16—24 ч при 750—850° С;
2) алитирование совмещается со старением предварительно
закаленной стали.
По первому режиму алитирование можно проводить при тем-
пературах порядка 1050—4100° С в течение 6—40 ч. Из несколь-
ких марок аустенитных сталей были изготовлены модели лопаток
газовых турбин, которые после обработки по первому режиму
1 Подробнее об этом см. в соответствующих главах.
178
проходили старение при 750—-800° С. Испытания при 600—620° С
показали, что алитирование в 2—3 раза уменьшило эрозионный
износ моделей. Лучшие результаты (износ уменьшался в 3—4 ра-
за) показали хромоалити-рованные лопатки, поэтому для повы-
шения эрозионной стойкости лопаточного аппарата турбин, рабо-
тающих на твердом топливе, было рекомендовано хромоалитиро-
вание по этому режиму [268].
При обработке по второму режиму температура алитирова-
ния, совпадающая с температурой старения, колеблется в пре-
делах 750—850° С [269]. По этому режиму алитируются готовые
детали, не подвергающиеся в даль-
нейшем механической обработке.
Поэтому для повышения вязкости
диффузионного слоя и улучшения
чистоты поверхности рекомендуется
применять менее активную алити-
рующую смесь, например, состоя-
Рис. 418. Влияние алитирова-
ния на окалиностойкость стали
4Х14Н14В2М; продолжитель-
ность испытаний 400 ч (Ю. М.
Лахтин и П. И. Георгиевский):
1 — неалитированная сталь;
2 — алитированная
Продолжительность испытания
Рис. 117. Окисление алитированной
(кривая /) и неалитированной (кри-
вая 2) стали 4Х12Н12КЮМ2ФТМЬ
на воздухе при 800° С (образцы ис-
пытывались после предварительного
окисления в течение 1370 ч) [269]
щую из 20% ферроалюминия, 79% отработанной смеси и 1% хло-
ристого аммония. В такой смеси при 800° С в течение 20 ч глу-
бина слоя на деталях из стали 35Х12Н12ВЗК9ФМТЫЬ дости-
гает 0,02—0,04 мм и сохраняется 6-й класс чистоты поверхности.
В процессе эксплуатации деталей в течение нескольких тысяч
часов при 800° С глубина слоя увеличивается до 0,10—
0,12 мм [269].
Заготовка вала опытного ротора турбины из указанной аусте-
нитной стали подвергалась закалке с 1220° С, первому стабили-
зирующему отпуску (старению) при 700° С, механической обра-
ботке и, наконец, алитированию при 800° С в течение 20 ч, сов-
мещенному со вторым стабилизирующим отпуском (старением).
Длительная прочность алитированного образца после испытаний
в течение 9000 ч при 750° С не изменилась. Окалиностойкость в
179
воздушной среде, испытанная при 800° С в течение 11 000 ч. после
алитирования возросла в 6,5 раз; состояние поверхности образ-
цов после .испытаний почти не изменилось (рис. 117).
На рис. 118 приведены данные о влиянии алитирования в по-
рошкообразной смеси на окалиностойкость стали 4Х14Н14В2М,
а на рис. 119 — данные об окалиностойкости стали Х23Н16, али-
тированной методом металлиза-
ции с последующим диффузион-
ным отжигом.
Сталь Х16Н25М6 подвергалась
алитированию при 860° С в тече-
ние 15 ч в порошкообразной сме-
си (99% ферроалюминия и 1 %
NH4C1) [19]. Глубина алитирован-
ного слоя составляла 0,04—
0,07 мм. Результаты испытания
образцов этой стали на окалино-
стойкость приведены на рис. 120.
Рис. 120. Влияние алитирования
на окалиностойкость стали
Х16Н25М6 при температурах
(увеличение веса неалитированных
образцов при 1000° С за 24 ч со-
ставило 6760 г • 1 /м2\ [19]:
а — 800° С; б — 900° С; и в —
1000° С; сплошные линии — неалитиро-
ванные образцы; штриховые —• али-
тированные
Продолжительность
испытания
Рис. 1.19. Влияние алитиро-
вания методом металлиза-
ции на окалиностойкость
стали Х23Н16 при 4150° С
[247]:
1 — алитированная сталь;
2 — неалитированная
После испытания при 800° С разница между алитированными
и неалитированными образцами была ничтожной, после испыта-
ния при 900° С в течение 96 ч стойкость алитированных образцов
оказалась в 3,5 раза выше, чем неалитированных; при 1000° С
и том же времени испытаний неалитированный образец совсем
разрушился, а алитированный увеличился в весе лишь на
15 г/л<2.
Таким образом, с точки зрения повышения окалиностойкости
аустенитных сталей эффект алитирования сказывается тем силь-
нее, чем выше температура испытаний. И наоборот, для углеро-
180
дистих и малолегированных сталей ферритного и перлитного
классов повышение температуры снижает эффект алитирования.
Это объясняется следующими причинами:
1. У ферритных и перлитных сталей с повышением темпера-
туры интенсивно развивается диффузия алюминия в сердцевину
и снижается концентрация алюминия в поверхностном слое, что
приводит к снижению окалиностойкости этих сталей.
2. Аустенитные стали до определенных температур окалино-
стойки вообще, поэтому эффект алитирования почти не заметен.
При более высоких температурах окалиностойкость этих сталей
снижается и эффект алитирования проявляется все сильнее.
Для торможения диффузии алюминия из алитированного
слоя в сердцевину и повышения окалиностойкости было предло-
жено (Н. В. Масютиным) гальваническим способом предвари-
тельно наносить на поверхность стали 1Х18Н9 никелевый под-
слой толщиной 0,04—0,05 мм. Диффузия алюминия в никеле про-
текает значительно медленнее, чем в железе, и поэтому падение
концентрации алюминия в никелевом слое за счет его диффузии
к сердцевине стали сильно тормозится. Таким путем удалось
поднять окалиностойкость стали 1Х18Н9 до 1200° С, а стали 10 —
до 1050° С.
Микроструктура алитированного слоя аустенитной стали
имеет две или три ярко выраженные зоны. При двухзонном строе-
нии слоя, наблюдаемом, например, на микроструктуре стали
4Х14Н14В2М, внешняя зона слабо травится и представляет со-
бой безуглеродистый сложный a-твердый раствор, содержащий
20—40% алюминия [259], [268]. Вторая, более глубоко располо-
женная зона, при температуре насыщения является у-фазой,
имеющей повышенное содержание углерода, вытесненного из
внешней зоны a-фазы. После охлаждения эта зона состоит из
фаз а, у и карбидов.
ХРОМИРОВАНИЕ
Насыщение низкоуглеродистой стали хромом, когда образует-
ся слой твердого раствора хрома в a-железе глубиной 0,05—
0,15 мм *, производят для повышения ее коррозионной стойкости
и, реже, окалиностойкости. Хромирование стали с содержанием
углерода более 0,4% в результате чего образуется тонкий
твердый карбидный слой глубиной 0,01—0,03 мм. применяют
главным образом для повышения ее поверхностной твердости и
износостойкости. Хромирование как метод повышения окалино-
стойкости, эрозионной и коррозионной стойкости иногда
применяется также для обработки аустенитных сталей,
* Особый случай хромирования на значительно большую глубину описан
на стр. 192.
181
хромоникелевых и кобальтовых сплавов, ниобия, молибдена и
других металлов и сплавов.
Первые сведения о процессе хромирования стали были опуб-
ликованы еще в 1928 г. В настоящее время хромирование широ-
ко применяется в промышленности многих -стран. На отечествен-
ных заводах процесс ’пока используется недостаточно полно.
Методы и режимы хромирования
Хромирование в порошках и обмазках
Хромирование в порошкообразных смесях с активизаторами.
Благодаря своей простоте этот метод наиболее широко приме-
няется как в отечественной, так и в зарубежной промышленно-
сти. Хромирование осуществляют в смеси порошков, содержащей
около 50% феррохрома (или хрома), 48—49% окиси алюминия
и 1—2% (чаще 1%) хлористого аммония.
В контейнере (ящике) с обрабатываемыми деталями и хро-
мирующей смесью протекают различные реакции, основные из
которых приведены ниже.
Хлористый водород, образующийся при разложении хлори-
стого аммония:
NH4C1 = NH3 + НС1,
вступает при высокой температуре во взаимодействие с хромом
по реакции
2НС1 + Сг = СгС12 + Н2.
Далее при контакте газообразного хлорида хрома с поверх-
ностью нагретой стали происходят обменные реакции между
хлоридами хрома и железом:
СгС12 Fe = FeCl2 Сг.
При избытке водорода может идти реакция вытеснения
хрома
СгС12 + Н2 - 2НС1 + Сг.
Окись алюминия, вводимая в состав смеси для предотвра-
щения спекания частиц хро-ма или феррохрома и их приварива-
ния к поверхности -стали, считалась ранее «инертной добавкой».
Однако в работе [270] показано, что в хромированном слое содер-
жится 1—5% алюминия. Из-за наличия в смеси для хромирова-
ния хлористого аммония в сталь диффундирует также азот и на
хромируемой поверхности может образовываться очень тонкая
(до 5 мк) нитридная — Сг2Ы — или кар-бонитридная —
Cr2(N, С)—корочка [270].
Соотношение составляющих порошкообразной смеси сущест-
венно -влияет на результаты хромирования. Увеличение в смеси
182
хрома (феррохрома) приводит к увеличению глубины диффузи-
онного слоя железа и стали при одновременном повышении
концентрации хрома в поверхностном слое железа (рио. 121).
При хромировании в смесях, содержащих одинаковое количе-
ство либо хрома, либо феррохрома, глубина диффузионного слоя
у железа и стали больше при использовании феррохрома, а не
хрома, как можно было бы предполагать. Концентрация хрома в
поверхностном слое стали больше при использовании хрома, а в
поверхностном слое железа — при использовании феррохрома.
С увеличением 'содержания “в'смеси NH4C1 до 5% концентра-
ция хрома в поверхностном слое железа сначала уменьшается,
затем увеличивается, а в поверхностном слое стали существенно
не изменяется (рис. 121).
Обнаруженные закономерности трудно поддаются объясне-
нию в связи с недостаточной изученностью тех реакций, проте-
кающих при диффузионном насыщении, которые должны отра-
жать механизм не только поверхностного насыщения стали хро-
мом, но и алюминием, азотом, водородом, а также механизмы
обеднения поверхностных слоев углеродом и перераспределения
легирующих элементов в слое.
В хромирующей смеси (см. стр. 182) следует применять низ-
коуглеродистый феррохром (не более 1% С), марок ХрОООО,
ХрООО, ХрОО, ХрО, XpOl, Хр1. Феррохром «1ЛИ1 хром размалывают-
ся в шаровых мельницах до получения зерен величиной менее
0,15—0,35 мм, а окись алюминия и хлористый аммоний (предва-
рительно просушенный) растираются и просеиваются. Смесь при-
готовляется непосредственно перед употреблением и может быть
повторно попользована 4—5 раз.
Смесь рекомендуется прокаливать в герметичном ящике. Это
предотвращает налипание частиц хрома на поверхность стали.
При повторном использовании смеси введение в нее хлористого
аммония не обязательно, так как частицы хрома (феррохрома)
и окиси алюминия абсорбируют хлорид хрома, что сохраняет
активность смеси. После 4—5-кратного употребления смесь сле-
дует обогащать свежим феррохромом и хлористым аммонием
(добавляется до 20% свежей смеси). Для хромирования 1 м2 по-
верхности стали расходуется в среднем около 400 г феррохрома.
Окись алюминия в смеси может быть заменена кварцевым
песком, каолином, окисью хрома или металлургическим магне-
зитом.
Детали, подлежащие хромированию, должны иметь чистую,
обезжиренную поверхность (они кипятятся в щелочном растворе,
промываются водой, декапируются раствором азотной кислоты
и опяты промываются).
Упаковываются детали в нихромовые или железные контей-
неры (ящики) с двойными крышками либо трубы, так же как
при алитировании.
183
Армко -железо
^~А1г03°/а
Сталь У8
Рис. 121. (Влияние состава хромирующих смесей на глубину диффузионного
слоя и на концентрацию хрома и алюминия на поверхности армко-железа и
стали У8 (хромирование при 1100° С и выдержке 3 ч) [270]: сплошная ли-
ния — смесь с хромом; штриховая — с FeCr
184
Иногда для повышения герметичности используются контей-
неры с плавкими затворами. За рубежом их изготовляют из по-
рошка (кусочков) силикатного происхождения, плавящихся при
500—600° С. В отечественной промышленности рекомендовано
[271] использовать для этой цели натросиликатное и боросиликат-
ное стекло, плавящееся при 750—800° С и обладающее малой
летучестью при температуре 1050° С. Предпочтительнее приме-
нять дешевое натросиликатное стекло, являющееся полуфабри-
катом для получения жидкого стекла.
Натросиликатное стекло (по ГОСТу 917—41—силикатная
глыба) содержит: 72% SiO2, 27% Na2O и 1% СаОЗЮз и
Ре20з + А120з. При нагреве воздух из контейнера вытесняется
продуктами разложения хлористого аммония через затвор
с еще не расплавленным силикатом. Затем силикат расплавляет-
ся и герметизирует ящик. После охлаждения контейнера за-
стывший силикат разбивают.
Измерение давления при хромировании в контейнере с плав-
ким затвором (смесь состава: 70% Сг, 29% А12О3, 1% NH4C1)
показало, что, начиная с температуры 750° С, в контейнере соз-
дается разрежение, достигающее при 900—1150° С величины
160—ПО мм рт. ст. (15—21 кн)м2) [271]. Причина образования
в контейнере вакуума пока не объяснена, ню самый факт его
образования указывает на то, что при недостаточной герметиза-
ции контейнера в него должно проникать большое количество
воздуха. Это подтверждено экспериментально. При хромирова-
нии в контейнере с двумя крышками, между которыми находит-
ся чугунная стружка, в верхней части контейнера протекали
реакции окисления хром^,
Недостаточная герметизация контейнеров ускоряет истоще-
ние смеси, ослабляет процесс насыщения, ухудшает стабиль-
ность результатов хромирования и может вызвать спекание
смеси. Поэтому при внедрении хромирования в производство
следует ориентироваться на контейнеры с герметичными затво-
рами.
На рис. 122 приведены данные, характеризующие влияние
температуры, продолжительности процесса и содержания угле-
рода в стали на глубину хромированного слоя.
Наиболее часто процесс хромирования осуществляется при
температурах 1000—1050° С и выдержках 6—12 ч. При таком
режиме, например, на низкоуглеродистой стали 08 образуется
слой типа твердого раствора глубиной 0,08—0,15 мм, а на высо-
коуглеродистых сталях — карбидный слой глубиной 0,01 —
0,03 мм.
Вместо хлористого аммония в порошкообразные смеси иног-
да вводят 5% соляной кислоты [272]. Для этого порошок ферро-
хрома или хрома с величиной зерна <0,35 мм порциями загру-
жают в керамическую или фарфоровую посуду, заливают под
185
вытяжным шкафом соляной кислотой и перемешивают, а затем
в течение суток сушат при комнатной температуре. Затем к сме-
си» добавляют глинозем и каолин, предварительно прокаленные
и размолотые до величины частиц 0,25—0,35 мм, и смесь еще
раз пропускают через шаровую мельницу. Хранить смесь следует
в сухом месте. Смесь можно использовать не освежая 3—4 раза,
после чего ее нужно вновь обрабатывать соляной кислотой.
В заграничной практике вместо хлористого аммония в порош-
кообразные смеси чаще вводят галогенидные соли аммония
Рис. 122. Влияние температуры (а) и продолжительности (6) хромирования
на глубину диффузионного слоя (хромирование в порошкообразной смеси в
ящиках с двойными крышками) [270]:
/ — армкп-железо; 2 — сталь У8; 3 — сталь 40; 4 — сталь 20
NH4J, NH4F и NH4Br [273], [274]. Эти соединения применяют при
хромировании как обычным методом в ящиках, так и в кон-
тейнерах с,рудными или силикатными затворами. Обрабатывае-
мые детали находятся в средней части контейнера, а смесь хро-
ма (30—60%), окиси алюминия (40—60%) и галогенидных солей
аммония — на дне контейнера. Над деталями вблизи крышки
контейнера помещают на сетке куски феррохрома. В контейнер
небольшими -порциями непрерывно подается водород.
Внутри контейнера протекают следующие реакции:
NH4F = NH3+HF;
2NH3 - N2 + ЗЫ2;
Сг + 2HF = CrF2 + Н2
(важно, что CrF2 негигроскопичен);
CrF2 + Fe = FeF2 Сг;
CrF2 + Н2 = 2HF + Сг; FeF2 + Н2 = 2HF + Fe.
186
Образующаяся плавиковая кислота взаимодействует с хро-
мом или феррохромом, и таким образом возобновляется при-
ток CrF2.
Такой способ хромирования (применяемый во Франции под
названием ONERA-процесс) правильнее было бы отнести к газо-
вому методу.
О глубине хромирования стали с 0,1 % Св смеси, содержащей
фтористый аммоний, можно судить по следующим данным, полу-
ченным при выдержке 4 ч;
Температура в °C............... 900 1000 1100 1200
Глубина слоя в мм.............. 0,02 0,12 0,20 0,30
При хромировании в смеси, содержащей иодистый аммоний
(такой способ применяется в Англии под названием DAL-npo-
цесс), протекают аналогичные реакции [273]:
NH4J = NH3 + HF; 2HJ + Сг = CrJ2 + Н2;
CrJ2 + Fe = FeJ2 + Сг;
CrJ2 + H2 = 2HJ + Cr; FeJ2 + H2 = 2HJ + Fe.
Смеси с йодистым аммонием применяют, например, фирма
Chrom-Alloying Со. Смесь состоит из 1 ч. хрома и 1 ч. каолина
или 2 ч. хрома и 1 ч. каолина, к которым добавляется всего
0,1% NH4J.
В качестве защитной атмосферы вместо водорода можно при-
менять аммиак [274]. В ящик засыпается гранулированный хром
с примесью нескольких процентов фтористых солей хрома. При
нагреве фтористый хром разлагается с выделением хрома, а
фтористый водород реагирует с гранулированным хромом, в ре-
зультате чего вновь образуются пары фтористого хрома. При
1100° С в течение 1,5 ч глубина слоя достигает 0,1 мм. В по-
верхностном слое углеродистых сталей образуются карбонитри-
ды; слой получается матовый, блеск ему придают электрополи-
рованием.
При обработке сталей ХВГ, ШХ15 и У8 обычным способом
(с упаковкой в порошкообразную смесь) не было установлено
существенного влияния природы активной добавки (NH4C1,
NH4J, NH4F или NH4Br) на результаты хромирования [270].
Иная картина наблюдается при хромировании железа. Оказа-
лось, что с увеличением атомного номера галогена *, введенного
в хромированную смесь, количество хрома, поглощенного желе-
зом, возрастает, поскольку чем больше атомный номер галогена,
тем выше содержание данного галогенида хрома в газовой
среде [275].
1 В периодической системе элементов фтор, хлор, бром и йод соответст-
венно имеют следующие порядковые (атомные) номера: 9, 17, 35, 53.
187
Хромирование в порошкообразной смеси хрома и глинозема.
Этот способ, разработанный в США [19], предусматривает хро-
мирование деталей, упакованных в ящиках или трубах, в смеси,
состоящей из 55% глинозема и 45% порошкообразного хрома
при 1250—1380° С. Ящик помещают в печь, через .которую про-
пускается водород, предварительно очищенный от (влаги и кис-
лорода. В хромирующей смеси нет веществ, которые могут
вызвать образование газообразных соединений хрома, и насы-
щение поверхности стали хромом протекает в основном из паро-
вой фазы, возникающей в результате испарения хрома при вы-
сокой температуре и в меньшей степени благодаря контакту ча-
стиц хрома с обрабатываемой поверхностью. На образцах
стали 10, хромировавшихся таким способом в течение 3 ч, при
1300° С, образовывался диффузионный слой глубиной 0,15 лш,
а при 1350°С— глубиной 0,45 мм.
Недостатками метода являются необходимость применения
чрезмерно высоких температур и сильный перегрев стали. Све-
дений о применении метода в промышленности нет.
Хромирование в обмазке. Обмазка, предназначенная для
хромирования деталей в закрытых муфелях1, содержит 4 ч. по>
рошка феррохрома (65% Сг) с величиной зерен <0,12 мм, 1 ч.
порошкообразного каолина или глинозема. К 6 ч. такой смеси
добавляется 1 ч. ГО % иного водного раствора (NH4)2HF2; при
этом происходит бурная реакция с выделением газов и тепла и
образованием тестообразной массы, в которой во взвешенном
состоянии находятся железохромистые соединения и NH3. Для
предотвращения спекания к массе добавляется глинозем, а для
разжижения — вода. Обмазка наносится на детали слоем толщи-
ной 0,25 мм. Хромирование осуществляется в муфеле при 900—
1050° С. По окончании процесса обмазка легко удаляется с де-
талей.
Хромирование при контактном нагреве. Контактный нагрев
с использованием сварочных трансформаторов позволяет резко
ускорить процесс хромирования. В порошкообразной смеси, со-
держащей 65% феррохрома, 30% А12О3 и 5% NH4C1, при 1050° С
в течение 50 мин на стали Ст.З образуется диффузионный слой
глубиной 0,08 мм, а на стали У10 — глубиной 0,043 мм [276].
При опробовании хромирования с контактным нагревом в
обмазке образцы вначале покрывались тонким слоем жидкого
стекла, на которое затем наносилась смесь порошков феррохро-
ма (75—80%) и древесного угля (20—25%) [277].
Хромирование в порошкообразной смеси и в обмазке с на-
гревом т. в. ч. При хромировании в порошкообразной смеси
с нагревом т. в. ч. длительность процесса можно сократить до
1 Возможно, эту обмазку можно использовать и при хромировании с на-
гревом т. в. ч.
188
10—15 мин (вместо 6—8 ч при обычных способах нагрева) [270].
На железных образцах, хромированных этим способом в не-
большом цилиндрическом контейнере, в течение указанного вре-
мени при 1050° С образовывался диффузионный слой глубиной
до 0,2 м, а на стальных образцах — глубиной до 0,08—0,1 мм.
Удобно хромировать в порошкообразной .смеси с нагревом т. в. ч.
внутренние поверхности цилиндрических деталей.
Английская фирма Сготе Alloying Со применяет этот способ
для хромирования различных мелких деталей. Детали и хроми-
рующая порошкообразная смесь помещаются в небольшие ци-
Рис. 123. Распределение микротвердости по глубине диффузионного слоя ста-
ли 45, хромированной с нагревом т в. ч. при 4050 и 1200° С в течение /, 2
и 3 мин (соответственно кривые Л 2 и 3) [автор и О. И. Батенина]
линдрические патроны. Режим хромирования: температура
1000° С, выдержка 100 сек.
Перспективным является, по-видимому, использование при
хромировании с нагревом т. в. ч. обмазок [277], [278]. Опробова-
на обмазка, состоящая из 75% порошка хрома (величина зерна
0,05 мм) и 25% криолита, замешанных в гидролизованном этил-
силикате до консистенции сметаны. Обмазка слоем толщиной
0,5—1 мм наносится на образец путем окунания, после чего по-
крытие подвергается сушке при 100° С. Для нагрева использова-
лась установка Л Г-60 с частотой 350 кгц. Температура контроли-
ровалась фотоэлектрическим пирометром на участке поверхно-
сти образца, не покрытом обмазкой. Изменение глубины и
твердости полученного при этом хромированного слоя показано
на рис. 123.
В табл. 25 приведены полученные автором совместно с
О. И. Ботениной данные, которые характеризуют изменение
189
глубины и микротвердости диффузионного слоя образцов ста-
лей 15 и 45 в зависимости от режима хромирования при нагреве
т. в. ч. (скорость нагрева 150 град!сек).
Таблица 25
Влияние режима хромирования на глубину и микротвердость
хромированного слоя
Темпера- тура в °C Продол- житель- ность процесса в мин Сталь 15 Сталь 45
Глубина слоя в мм Микротвер- дость 7/юо на поверх- ности Глубина слоя в мм Микротвердость Н100
на поверх- ности на глубине 0,015— 0,02 мм
0* 0,01 187 0,01 243
1050 1 0,07 298 0,05 398 574
2 о,н 307 0,09 598 642
3 0,14 340 0,12 703 742
0 0,018 212 0,015 287
1200 1 0,10 345 0,08 513 642
2 0,14 420 0,12 587 724
3 0,17 487 0,15 710 842
* Нагрев в течение 7 сек до дением на воздухе. 1050° С и 8 сек до 1200° С с последующим охлаж-
При хромировании этим методом сталей 45 ю У10 диффузион-
ных слоев карбидного типа не образуется и микротвердость
слоя достигает лишь 725—930 вместо 1700—1800 после хроми-
рования с обычным нагревом.
Это связано главным образом с быстрой диффузией хрома
в сталь при скоростном нагреве 1 до высоких температур. На
поверхности стали в этих условиях не успевают создаваться вы-
сокие концентрации хрома и углерода, необходимые для обра-
зования сплошного карбидного слоя. Поэтому хромирование
высокоуглероди'стых сталей с нагревом т. в. ч. следует произво-
дить при относительно медленном нагреве (.по крайней мере при
скорости нагрева менее 30 град!сек).
Хромирование стали в обмазках с нагревом т. в. ч. почти в
100 р$з и более сокращает продолжительность процесса. Этот
способ хромирования целесообразно применять для обра-
1 Влияние скоростного нагрева рассматривается на стр. 29.
190
ботки многих деталей, в том числе длинных труб. Трубы при не-
прерывном поступательном движении вначале должны механи-
чески покрываться обмазкой, затем .подвергаться сушке и, на-
конец, хромироваться с нагревом т. в. ч.
Однако для внедрения этого метода в производство необхо-
димо подобрать оптимальные составы обмазок, которые позво-
ляли бы получать стабильные результаты в отношении качест-
ва хромированной поверхности.
Хромирование в вакууме
При хромировании в вакууме насыщение поверхности дета-'
лей хромом происходит в основном благодаря образованию па-
ровой фазы, содержащей хром, и в незначительной степени
вследствие контакта деталей с порошком хрома.
Детали засыпаются порошком хрома в металлическом или
керамическом тигле и помещаются в вакуумную камеру [2S0]—
[283]. Хромирование проводится при разрежении 10“2 —
10-3 мм рт. ст. (1,33—0,13 н/м2). Такой вакуум может быть
создан одним форвакуумным ротационным масляным насосом,
что упрощает проведение процесса.
Хром при высокой температуре хорошо испаряется в вакууме.
Температура испарения хрома, при которой давление его паров
достигает 10-2 мм рт. ст. (1,33 h/jw2), равна 917° С, а железа со-
ответственно 1157° С. Тем не менее для ускорения процесса и
получения более равномерного по глубине слоя рекомендуется
располагать детали на минимальном расстоянии от частиц воз-
гоняемого хрома (-размер частиц 1—3 мм). Поэтому изделия ча-
ще засыпаются хромом.
Полученные автором результаты вакуумного хромирования
образцов армко-железа и стали приведены в табл. 26.
Таблица 26
Влияние температуры и продолжительности хромирования
в вакууме на глубину диффузионного слоя
Хромируемый материал Глубина слоя в мм
950° С; 6 ч 1050° С; 1 ч 1050° С; 3 ч 1050° С; 6 ч
Армко-железо 0,08 0,03 0,08 0,15
Сталь У12 0,015 0,008 0,015 0,03
По данным работ [280], [281], хромирование в вакууме проис-
ходит быстрее. Однако результаты других работ по хромирова-
нию в вакууме больше согласуются с данными, приведенными
191
в табл. 26. Эти разногласия можно объяснить тем, что авторы
работ [280], [281], по-видимому, принимали за глубину хромиро-
ванного слоя не расстояние от поверхности до линии раздела,
а расстояние от поверхности до конца распространения темного
подслоя.
Хромирование в вакууме углеродистых сталей, сталей 1X13,
1Х18Н9Т, X15H36BT3 и др. было изучено в работе [283].
Предложен водородно-вакуумный способ хромирования [280].
Нагрев производится в среде водорода, выдержка — в вакууме,
а охлаждение — снова в среде водорода. Присутствие водорода
позволяет ускорить прогрев садки, однако создает взрывоопас-
ность.
Осуществляя вакуумное хромирование с нагревом т. в. ч.,
Л. К. Гущин и Г. В. Земсков показали, что добавление к по-
рошку хрома хлористого аммония интенсифицирует процесс;
при этом в реакционном пространстве наблюдается свечение
(по-видимому, происходит ионизация). Процесс еще более ин*
тенсифицируется, если насос, создающий вакуум, не работает
непрерывно. Так, в течение 30 мин при 1000° С на образцах ста-
ли 10 при давлении 10-3 мм рт. ст. (0,13 н!м2) глубина диффу-
зионного слоя достигает 35 л<лс, при давлении 1 —10 мм рт. ст.
(133,3—1333 н/м2) —120 мк и при давлении 150 мм рт. ст.
(19935 н/м2) —150—170 тик.
Большой интерес представляет опробованное в нашей про-
мышленности глубокое хромирование (на глубину до 1,5—8 мм)
в вакууме при весьма высокой температуре слябов, сутунок, за-
готовок труб и проката из низкоуглеродистой стали. В качестве
насыщающего материала используются кусочки феррохрома,
переходящего в указанных условиях в сублимированную фазу.
Последствия сильного перегрева стали устраняются по оконча-
нии хромирования нагревом и горячей пластической деформаци-
ей, необходимыми для получения готовых хромированных ли-
листов, труб и прутков [571].
Хромирование в газовой среде
Хромирование в среде Н2 + HCI. Впервые хромирование га-
зовым методом было осуществлено именно в этой среде [19],
[284]—[286].
В одном конце реторты помещаются феррохром или хром,
а в середине и другом конце — детали. Водород, просушенный
и очищенный от кислорода, подается по трубке в реторту во вре-
мя ее нагрева. По достижении требуемой температуры водород
до поступления в реторту предварительно пропускается через
сосуд с дымящейся НС1. Смесь Н2 + HCI поступает в рето-рту
с той стороны, где находится хром или феррохром; при этом про-
исходит образование хлоридов хрома. Хлорид хрома движется
192
по реторте вместе с потоком -водорода и омывает хромируемую
поверхность. При этом, очевидно, протекает главным образом
реакция
СгС12 + Н2 = 2НС1 + Сг.
На образцах сталей 10 и У10 было исследовано влияние ряда
факторов на результаты хромирования этим способом [285].
Повышение расхода газовой смеси Н2 + НС1, проходящей
над кусками хрома (92,9% Сг и 0,02% С), ведет к увеличению
глубины хромированного слоя, очевидно, потому, что при не-
большом расходе газа образуется, по-видимому, недостаточное
количество хлоридов хрома. Применение феррохрома вместо
металлического хрома приводит к уменьшению глубины хроми-
рованного слоя. Например, при температуре 980° С, выдержке 4 ч
и расходе водорода 40—50 л!ч (диаметр трубки 55 мм, высота
750 мм) глубина диффузионного слоя на образцах низкоугле-
родистой стали достигала в случае применения феррохрома
0,05 мм, а в случае применения хрома 0,08 мм, при выдержке
6 ч— соответственно 0,085 мм и 0,105 мм.
При хромировании стали в среде Н2 + НС1 всегда происхо-
дит в той или иной степени поверхностное обезуглероживание
стали. Со стороны .поступления газов в печь образцы обезугле-
роживаются быстрее и после хромирования становятся иногда
блестящими и имеют пониженную твердость. С противополож-
ной стороны образцы имеют нормальный матовый цвет и более
высокую твердость, которая, однако, ниже, чем при хромирова-
нии в порошкообразной смеси, где обезуглероживание значи-
тельно меньше.
Неравномерные твердость и глубина хромированного слоя
наблюдаются далеко не всегда и связаны главным образом с по-
вышенным расходом водорода и нагревом стали в среде одного
водорода (в начале процесса) до слишком высокой температу-
ры. Засыпка образцов феррохромом или хромом способствует
получению более равномерной твердости (табл. 27); поэтому,
если хромирование производится с целью получить высокую по-
верхностную твердость, детали рекомендуется засыпать кусоч-
ками феррохрома или хрома.
Первые большие промышленные печи для газового хромиро-
вания длинных труб, построенные за границей, имели горизон-
тальные длинные реторты и поэтому не оправдали себя, так как
глубина хромированного слоя на обработанных в них трубах
оказывалась неравномерной. Это объясняется значительной
разницей в молекулярных весах СгС12, НС1 и Н2, вызывавшей
расслоение газов. Поэтому печи с горизонтальными ретортами
могут применяться лишь при условии непосредственной засыпки
деталей феррохромом или хромом и вращения реторт для уве-
личения равномерности хромирования.
193
Таблица 27
Результаты газового хромирования стали в смеси Н2-[-НС1 [19]
Расположение кусков хрома относительно образцов Режим хромирования Марка стали Вид поверхности Поверхност- ная твердость по Виккерсу
температура в °C выдержка в ч
На расстоянии 150 мм 980 4 У8 Блестящая сторона. Противоположная — ма- товая сторона 330 876—953
1150 5 У8 Блестящая сторона. Противоположная — ма- товая сторона 251 1103-1219
Образцы засы- паны кусками 980 2 45 У8 Все стороны матовые То же 850—1000 1051—1219
хрома 1100 3 45 У8 » 1003 1219—1355
По данным заграничной практики при хромировании в вер-
тикальных печах 'Слой глубиной 0,10—0,15 мм образуется при
1000° С в течение 5 ч. Расход хрома при пересчете из ферро-
хрома составляет при этом около 200 г)м2 хромированной по-
верхности.
Недостатками хромирования в среде Н2 + НС1 являются
взрывоопасность газовой смеси и непостоянство состава среды
вследствие постепенного снижения концентрации кислоты при
непрерывной продувке через нее водорода.
Хромирование в среде НС1 или С12. Хромирование можно
осуществлять в муфеле, в который детали загружаются вместе
с кусочками феррохрома (величина зерна 3—10 мм). Муфель
продувается хлористым водородом, получаемым путем воздей-
ствия серной кислоты на соляную [287]. Во время .нагрева хло-
ристый водород не пропускается через печь; по достижении
температуры 950° С его подача возобновляется. Процесс прово-
дится при 1000—1050° С в течение 6—8 ч\ при этом глубина хро-
мированного слоя на низко-, средне- и высокоуглеродистых
сталях достигает соответственно 0,1; 0,03—0,05 и 0,02 мм.
Феррохром используется многократно и каждый раз просу-
шивается при 200° С перед употреблением. При хромировании
низкоуглеродистой стали на глубину 0,1 мм требуется 150 г хро-
ма или 300 г феррохрома (с 50% Сг) на 1 м2 поверхности из-
делий..
’ В работе [280] предложен почти аналогичный способ, но с ис-
пользованием хлора, который продувается только через холод-
194
ный муфель; затем его подача .прекращается, а на выходной
трубке устанавливается клапан Бунзена. Однако при таком спо-
собе не ’исключается порча хромируемой поверхности.
Описанные способы более трудоемки по сравнению с газовым
методам, поэтому они в настоящее время не могут считаться
перспективными.
Хромирование с использованием хлороводородной установки.
В работе [270] приведены результаты исследования газового
хромирования с использованием специальной хлороводородной
установки, позволяющей изменять состав и количество активной
газовой смеби, в реакционном пространстве, а следовательно,
управлять .процессом хромирования. Способ оонован на исполь-
зовании прямого синтеза для получения хлористого водорода из
хлора и водорода.
Установка имеет хлорную и водородную линии, питаемые из
баллонов. Смесь газов поступает в реактор и зажигается за-
пальной свечой. Получение в реакторе нужной газовой смеси
(Н2 + НС1 или С12 + НО) достигается регулированием подачи
водорода и хлора. В муфеле печи, со стороны входа газов, по-
мещается феррохром (хром), а во второй зоне печи — обрабаты-
ваемая сталь.
Позднее в Бельгии газовое хромирование аналогичным спо-
собом было применено в производственных условиях [288]. Хло-
ристый водород получается синтезом хлора и водорода, а водо-
род в крекинг-аппарате—из аммиака.
Описанные способы хромирования имеют несомненные до-
стоинства, однако их осуществление связано со значительными
трудностями и взрывоопасностью.
В литературе сообщалось о применении в производстве га-
зового метода хромирования деталей в шахтных печах и печах
с вращающимися ретортами (процесс «альфатайзинг»), но тех-
нология процесса детально не изложена [289], [290].
Хромирование с использованием галогенидов хрома. Газо-
вое хромирование может производиться с использованием соли
СгС1з, помещаемой в муфеле печи со стороны впуска водорода.
Водород восстанавливает СгС13 до СгС12, перенося его в зону,
где находятся детали. Недостатком этого метода является необ-
ходимость использовать взрывоопасный водород.
При использовании соли CrJ2 процесс хромирования может
осуществляться в вакууме или в потоке нейтрального несущего
газа, так как йодистый хром претерпевает термическую диссо-
циацию с выделением хрома.
По мнению автора, наиболее перспективно газовое хромиро-
вание с использованием соли СгС12, так как в настоящее время
организуется ее промышленное производство, что во многом об-
легчит внедрение этого способа на отечественных заводах.
195
г. 7Г
7
Рис. 124. Схема лабораторной установки для поверхностного насыщения ста-
ли и различных металлов из газовой фазы с нагревом т. в. ч.
/ _ колба с КМпОч и HCI; 2 и 3 — сосуды с водой и H2SO4 д/я очистки и сушки
хлора; 4 — печь для получения хлоридов металлов (хрома) в потоке хлора; 5 — «ло-
дочки» с металлом (хромом); 6 — хромируемый образец; 7 — индуктор; 8 — сосуд с
NaOH для поглощения хлора и создания постоянного давления газа [258]
Рис. 125. Установка для насыщения изделий различными металлами (хро-
мом) из газовой фазы с нагревом т. в. ч.:
1 — перфорированная трубка для подачи газов; 2 — перфорированная труба (контейнер);
3 — порошкообразный металл (хром); 4 — цилиндр; 5 — огнеупорная крышка; 6 — ог-
неупорная пробка с резиновым уплотнением; 7 — электронагреватель печи; 8 — труб-
ка-контейнер из кварца; 9 — индуктор; 10 — обрабатываемое изделие; 11 — подъемная
платформа; 12 — вакуумный патрубок; 13 — коническая часть с индивидуальным элек-
тронагревом (Ф. Л. Самюэль)
196
Хромирование с использованием СгС12 может производить-
ся в ретортах, а также ящиках с двойными крышками или плав-
кими затворами. Определенное количество хлористого хрома
помещается при этом на дно контейнера. Лучшие результаты да-
ст хромирование в реторте при небольшом разрежении, напри-
мер 20 мм рт. ст. (2666 н/м2). Для газового хромирования в
присутствии СгС12, кроме водорода, могут быть использованы
нейтральные газы, с помощью которых можно осуществлять пе-
ренос с необходимой скоростью хлорида хрома к деталям, что
облегчает регулирование процесса.
Хромирование в аэрозолях [250]. Сущность метода поверх-
ностного насыщения стали в аэрозолях была описана на
стр. 163. Для хромирования в аэрозолях рекомендуется порош-
кообразная смесь, состоящая из 1 ч. хрома (величина зерен
0,55 мм), 0,25 ч. NH4C1 и 0,5 ч. NaCl.
Хромирование с нагревом т. в. ч. Газовая смесь для хроми-
рования получается путем пропускания хлора над кусками хро-
ма (рис. 124) [258]. Хлориды хрома направляются далее к об-
разцам, помещенным в зоне, окруженной индуктором. При ис-
пользовании индукционного нагрева в течение 8—9 сек при
1000—1100° С глубина хромированного слоя на армко-железе
достигает 0,2 мм.
В Англии предложена1 установка (рис. 125) с нагревом
т. в. ч. для газового насыщения металлов и сплавов различными
элементами, в том числе хромом. Контейнер 2 заполняется по-
рошкообразным хромом 3 (размер частиц 0,08—-0,4 мм) и закры-
вается крышкой 5, после чего вставляется в цилиндр 4 больше-
го диаметра для образования активного газа.
Через трубку 1 в рабочее пространство генератора, в котором
находится хром, нагретый до 1050° С, подается смесь водорода,
насыщенного при 20° С парами брома и сухого аргона. Образую-
щиеся газообразные бромиды хрома потоком газа-носителя вы-
тесняются через перфорацию контейнера 2 в коническую часть 13
установки, после чего поступают в зону индукционного нагрева,
где взаимодействуют с обрабатываемой поверхностью. Скорость
подачи смеси паров брома с водородом 100 см21мин, аргона
20 см2!мин. Хромирование производится при 1100—1200° С в те-
чение 5 мин, время нагрева 50 сек. При таком режиме глубина
хромированного слоя составляет 50 мк, а концентрация хрома на
поверхности превышает 17,5%. Нагрев т. в.ч. по сравнению с
.обычным нагревом ускоряет процесс хромирования в 10 раз.
Опробован способ хромирования с нагревом т. в. ч. в газовой
среде, содержащей карбонил хрома [291]. Хром может быть
осажден как на железе, так ина железных сплавах. Применение
индукционного нагрева удешевляет процесс и уменьшает рас-
1 Samuel R. Brit. pat. № 897559, 1962.
197
ход карбонила (inо сравнению с аналогичным процессом, осу-
ществляемым в муфеле с внешним обогревом).
Хромирование в керамических материалах
Этот способ, называемый за рубежом BDS-inpoueccoM, требу-
ет предварительного насыщения газообразными хлоридами
хрома пористых керамических материалов (пористого фарфора,
пеношамотного кирпича, глин и т. д.). Во время хромирования
хлориды выделяются из пор. Кусочки керамического материала-
размером 10—15 мм вместе с феррохромом или хромом загру-
жаются в ящик, из которого водородом вытесняется воздух.
При нагреве ящика до температуры выдержки (1000—
1100° С) -в него непрерывно подается водород, при выдержке —
сначала смесь Н2 + НС1 (непродолжительное время), а затем1
снова водород (до окончания процесса, длящегося 6—8 ^). При
поступлении в рабочее пространство смеси Н2 + НС1 протекает
реакция
(Fe, Сг) + 4НС1 = FeCl2 + СгС12 + 2Н2.
Охлаждаются ящики также в потоке водорода.
После хромирования поверхность деталей получается всегда»
чистой. Процесс -применяется, например, на арматурном заводе
в г. Магдебурге (ГДР).
Хромирование в жидкой среде
Хромирование в жидкой среде осуществляется в ванне с рас-
плавленными солями (ВаС12, MgCl2, СаС12 и др.). В расплав,
вводится СгС12 в количестве 10—20%; над зеркалом ванны про-
пускается водород. В результате соответствующих реакций в
ванне образуется хлористый хром СгС1з. При соотношении
СгС1я 3 rim
в ванне - =— процесс протекает интенсивно [19].
Автором была предпринята попытка осуществить хромиро-
вание стали в такой ванне с добавкой разных количеств просу-
шенного СгС13, выпускаемого промышленностью [19]. Однако
хромирования при этом не происходило. Хромирование проте-
кало только при введении в ванну СгС12. Это позволило заклю-
чить, что хромирование в ванне происходит, возможно, благо-
даря реакции
ЗСгС12 = 2СгС13 + Сг.
В такой -расплавленной соляной ванне сплошной тонкий хро-
мированный слой получался за весьма короткое время. Выдер-
жанный в ванне 10 мин при 980° С образец стали У10 после ох -
198
лаждения на воздухе хорошо резал стекло и в течение несколь-
ких суток не растворялся в 30%-ном растворе азотной кисло-
ты [19].
В соляную ванну, состоявшую ив 70% ВаС12 и 30% NaCl,
вводили 20—25% хрома или феррохрома (от веса солей), обра-
ботанного соляной кислотой (.способ такой обработки описан
выше) [272].
Есть сведения о применении в полупроизводственных услови-
ях жидкого хромирования в ванне, содержащей 30% СаС12,
49% ВаС12 и 21% NaCl. На дно ванны помещаются куски хро-
ма. Количество вводимого в ванну хлорида хрома не указывает-
ся. При 1200° С глубина хромированного слоя на образцах же-
леза достигает за 3 ч 0,2 мм. а на образцах никеля, молибдена
и вольфрама за 6 ч соответственно 0,05; 0,03 и 0,005 мм.
Недавно предложен новый состав ванны для хромирования:
75% хрома (величина частиц менее 30 мк) и 25% криолита.
Рабочая температура 950—1200° С [292], [573].
Недостаток хромирования в ваннах — довольно быстрое на-
капливание в них продуктов реакции и понижение в связи с
этим их активности.
Контроль качества хромирования.
Термическаяобработкахромированныхдеталей
Детали после хромирования очищаются металлической щет-
кой или промываются горячей водой для удаления следов хло-
ридов хрома или солей.
Поверхность деталей после хромирования бывает блестящей
серебристой или матовой светло-серой. Участки поверхности, не
соприкасавшиеся с порошкообразной смесью, становятся бле-
стящими, а соприкасавшиеся — матовыми. При равных условиях
насыщения поверхность низкоуглеродистой стали чаще бывает
блестящей, а высокоуглеродистой — матовой. Повышение тем-
пературы способствует получению блестящей поверхности.
После газового хромирования повышается класс чистоты по-
верхности, по-видимому, благодаря сглаживанию микрорелье-
фов поверхности (данные М. П. Кольяновой).
Качество диффузионного слоя проверяется путем погружения
деталей или контрольных образцов в 30%-ный водный раствор
азотной кислоты на 20—40 мин. Выделение пузырьков водоро-
да, помутнение раствора и разъедание поверхности указывают
на нарушение сплошности диффузионного слоя. Иногда после
такого испытания образцы промывают в воде и дополнительно
погружают в 15%-ный раствор медного купороса. Отсутствие
осадка меди на поверхности образцов подтверждает хорошее
качество диффузионного слоя. Для контроля применяют также
раствор, состоящий из 10 г/л K4Fe(CN)6, 15 г/л NaCl и 20 г/л
199
желатина. В местах, где сплошность хромированного слоя на-
рушена, появляются пятна турбулентной сини. Контроль хроми-
рованного слоя, помимо перечисленных способов, периодически
осуществляется путем микроанализа.
При хромировании рекомендуется получать:
а) на деталях, работающих в средах, вызывающих сильную
коррозию при /высоких температурах, слой со структурой а-
твердого раствора глубиной 0,10—0,15 мм;
б) на деталях, работающих в средах, вызывающих незначи-
тельную коррозию (например, в атмосферных условиях), слой
со структурой a-твердого .раствора, глубиной 0,06—0,08 мм;
в) на деталях, работающих в условиях сильного износа и
коррозии, карбидный слой глубиной 0,025—0,03 мм;
г) на деталях малого сечения с острыми углами, работаю-
щих на износ, карбидный слой глубиной 0,01—0,015 мм;
д) на деталях из чугуна карбидный слой глубиной 0,02—
0,04 мм.
Поверхностная твердость стали' с карбидным слоем, контро-
лируемая по Виккерсу с нагрузкой 0,5 и 1 кГ, должна быть бо-
лее HV 1200, а микротвердость такой стали и чугуна — более
Я1Х 1500.
Для повышения твердости сердцевины стали и улучшения ее
механических свойств хромированные детали часто подвергают-
ся термической обработке (нормализации, закалке, отпуску)»
после которой их поверхность становится темной.
Термическая обработка почти не оказывает влияния на стой-
кость хромированных деталей в мало агрессивных средах, но в
сильно агрессивных средах (кислотах) эта стойкость иногда
снижается, очевидно, вследствие образования в диффузионном
слое микротрещин [19]. На поверхностную твердость хромирован-
ной стали закалка и отпуск заметно не влияют.
При хромировании происходит изменение размеров деталей.
Для сплошных деталей увеличение размеров чаще составляет
0,01—0,02 мм на сторону и не превышает 0,03—0,04 мм (по од-
ним данным примерно 20% от глубины слоя, а по другим
33%). Величина изменения размеров зависит от метода хроми-
рования. Изменение размеров, вызываемое структурными из-
менениями в основном металле, иногда может быть большим»
особенно у длинных деталей. В процессе хромирования (так же
как и азотирования) цилиндров и колец при некоторой опреде-
ленной толщине стенок этих деталей может произойти увеличе-
ние их наружного и уменьшение внутреннего диаметров. Напри-
мер, при хромировании в порошкообразной смеси при 1050° С в
течение 15 ч (HV 750—1000) у партии колец, изготовленных из
сталей 45 и 40Х, наружный диаметр (46 мм) увеличился в
среднем на 0,01—0,02 мм, а внутренний (28 мм) уменьшился на
0,02—0,04 мм [272].
200
Влияние углерода и легирующих элементов на глубину
хромированного слоя
Значения коэффициента диффузии D хрома в железо и ве-
личина энергии активации Q сильно колеблются, по данным раз-
ных авторов. Например, при газовом хромировании при 1000° С
по одним данным D = 3 • 1О“10 см21сек, а по другим [273]
22,3-10“10 см2] сек (значения Q соответственно 57 и 52,6
ккал!г-атом или 239 ;и 220 кдж!г-атом).
Концентрация углерода, при которой слой приобретает кар-
бидную природу, зависит от обезуглероживающей способности
насыщающей среды, температуры процесса, толщины изделия и
состава стали.
Рис. 426. Влияние содержания углерода в стали на
глубину карбидного слоя (хромирование в порошко-
образной смеси при 1050° С в течение 3, 6 и 12 ч)
[270]
Диффузионный слой наибольшей глубины (твердый раствор
углерода в a-железе) можно получить на железе, содержащем
сотые доли процента углерода. С увеличением содержания уг-
лерода до 0,1%—0,12% диффузия хрома резко тормозится
и глубина диффузионного слоя уменьшается; при содержании
в стали около 0,16—0,20% С диффузионный слой (по линии
раздела) имеет минимальную глубину и уже иную — карбид-
ную— природу (рис. 126).
При дальнейшем увеличении содержания углерода в стали
до 0,6—0,8% толщина карбидного слоя возрастает, а затем (при
еще большем содержании углерода) снова уменьшается
(рис. 126). Такая закономерность изменения глубины слоя на-
блюдается и при насыщении стали другими карбидообразующи-
ми элементами, например ванадием и ниобием (см. рис. 200).
Влияние легирующих элементов на глубину хромированного
слоя для почти безуглеродистых сплавов (0,02—0,03% С) изу-
чалось в нескольких работах [259], [286]. Молибден, вольфрам,
хром и кремний, в присутствии которых у-область замыкается
при меньшем содержании хрома, увеличивают глубину хромиро-
ванного слоя вследствие большей скорости диффузии элемента
201
в a-фазе, чем. в; у-фазе. Элементы, расширяющие у-область
(марганец, никель), действуют в противоположном направле-
нии (рис. 127). Данные работы [259] подтверждают это, кроме
влияния на глубину слоя хрома, который, судя по р.ис. 128, не
увеличивает глубину диффузионного слоя.
Приведенные данные о влиянии легирующих элементов на
глубину, хромирования почти безуглеродистых сплавов трудно'
попользовать практически, так как достаточно повышения содер-
жания в стали углерода более 0,10—0,15%,.чтобы скорость фор-
мирования диффузионного слоя опре-
делялась не только влиянием легирую-
щих элементов на скорость диффузии
хрома, но и их влиянием на скорость
диффузии углерода в аустените из
сердцевины стали навстречу хрому.
Карбидообразующие элементы (титан,
ванадий, хром, молибден, марганец),
тормозят диффузию углерода, умень-
Рис. 127. Влияние содержания легирующих
элементов на глубину хромированного слоя
железа (950° С, 3 ч) [286]
в
Рис. 128. Влияние содержания
легирующих элементов на
бину хромированного
стали (хромирование
рошкообразной смеси
.1100° С) [259]
глу-
слоя
по-
при
шают его скопление .в диффузионном слое и этим облегчают диф-
фузию хрома в сталь.
Подвергать хромированию можно стали большинства марок.
Слабонагруженные детали и изделия (резервуары, патрубки,
химическая посуда, м-алоответствеиные гайки, болты), подле-
жащие хромированию для повышения их антикоррозионных
и кислотоупорных свойств, изготовляют из' сталей 08, 10, 15;.
средненагруженные (штоки, тяги, болты, фланцы, детали паро-
водяной арматуры) — из сталей 40, 15Х, ЗОХ, 15ХФ, 20ХН и др.;
высоконагружеиные— из сталей ЗОХГСА, 25ХГТ и др. Хромиро-
ванию можно подвергать также детали из чугуна-[277].
• Если требуется получить вязкий диффузионный слой, кото-
рый должен выдерживать -вытяжку, чеканку, осадку, и другие
подобные’операции, сталь нужно выбирать, с .учетом размера
детали. Чем толще деталь,'тем меньше углерода должна содер-
жать сталь, из'Которой она изготовлена, или в стали должны
202
присутствовать элементы, тормозящие диффузию углерода из
сердцевины к поверхности (титан, ванадий, хром, молибден,
марганец).
В Германии были разработаны специальные стали для хро-
мирования (IK-стали), содержащие: 0,1% С; 0,45% Ti (IK-1);
3% Мп (IK-2); 0,9.% Si; 2,5% Сг и 0,2% Си (IK-25) и 0,8% Si;
7,5% Сг; 0,5% V и 1,2% Си (IK-85).
Если хромирование применяют для повышения износостой-
кости деталей, то в стали должно содержаться не менее 0,30—
0,35% С.
В ряде случаев оказывается более рациональным применение
легированных сталей, позволяющих сократить длительность хро-
мирования для получения диффузионного слоя заданной глу-
бины.
Для облегчения доводки хромированных изделий содержание
углерода в стали не должно превышать 0,6%. Стали У10 и У12
можно применять только для изготовления напильников или
деталей, которые не доводятся.
Иногда для ускорения процесса хромирования и максималь-
ного смягчения диффузионного слоя применяется предваритель-
ное обезуглероживание низкоуглеродистой стали в среде влаж^
ного водорода.
Структура хромированного слоя
После травления в 2—4 %-ном растворе азотной кислоты
микроструктура основной части хромированного слоя низкоуг-
леродистой стали выявляется в виде нетравящегося светлого
слоя (рис. 129, а). После длительного травления в слое обнару-
живаются слабые контуры столбчатых зерен, ориентированных
перпендикулярно поверхности стали и представляющих собой
твердый раствор хрома в а-железе.
Более отчетливо строение основной части слоя выявляется
только после травления царской водкой или раствором хлорного
железа в серной кислоте.
При больших увеличениях в слое -столбчатых зерен стали,
содержащей около 0,1% С, удается рассмотреть вторую фазу,
располагающуюся По границам столбчатых зерен (в виде тон-
ких прожилок) и внутри зерен (в виде мелких включений) и
представляющую собой железохромистые карбиды. Структура
зоны слоя, примыкающей с внутренней стороны к столбчатым
зернам (глубже линии раздела), почти не отличается от струю
туры сердцевины стали вследствие сравнительно невысокого со-
держания хрома; .поэтому за глубину хромированного слоя ус-
ловно принимают лишь глубину поверхностного нетравящегося
светлого слоя с высоким содержанием хрома. Содержание хрома
в поверхностной зоне хромированного слоя . 'низкоуглеродистой
203
стали достигает 30—60% (рис. 130). Резкое падение концентра-
ции хрома (начиная примерно с 13% Сг) соответствует линии
раздела, разграничивающей фазы а и у при температуре насы-
щения.
Рис. 129. Микроструктура
хромированной стали:
а —• низкоуглеродистая сталь
(Х100); б — сталь 40 (Х100);
в — сталь У8, подвергнутая
после хромирования закалке и
низкому отпуску (Х500)
Согласно электрографическим исследованиям, тончайшая по-
верхностная зона хром иров а иного слоя может при -некоторых
условиях хромирования даже полностью состоять из ориентиро-
ванных кристаллов хрома [19].
Как уже отмечалось, при хромировании с использованием
хлористого аммония (и других солей аммония) в сталь диффун-
дируют, кроме хрома, азот и алюминий. Присутствие этих эле-
ментов в хромированном слое приводит к образованию более
204
сложной структуры и фазового состава слоя. В поверхностной
зо-не хромированного слоя на образце железа, обработанного
в порошках с хлористым аммонием, «спектральный анализ пока-
зывает присутствие более 0,2% N. При этом послойный рентге-
ноанализ 'позволил установить, что на поверхности хромирован-
ного слоя, представляющего собой твердый раствор хрома в
a-железе, располагается тонкая (5—10 мк) корочка нитрида хро-
ма Cr2N, содержащая (судя по пара-
Рис. 130. Распределе-
ние храма в поверх-
ностных зонах стали
после газового хро-
мирования при 1000°
С в течение 1 ч [270]:
1 —• сталь с 1,03% С;
2 — сталь с 0,03% С
метрам решетки) около 10% N [270].
Светлый диффузионный слой на стали
с повышенным содержанием углерода
Расстояние от поверхности
Рис. 131. Схема распреде-
ления углерода по глубине
хромированного слоя угле-
родистой стали:
1 — промежуточная зона;
2 — зона обезуглероживания
(примерно более 0,2% С) не травится обычными травителями
и имеет карбидную природу.
Химический анализ хромированного слоя образцов сталей
с различным содержанием углерода показал, что чем больше это
содержание, тем выше концентрация хрома и углерода в кар-
бидном слое. Содержание хрома в карбидном слое достигает
75—95%, а углерода 6—8% (,рис. 130).
Карбидный слой состоит из двух зон карбидов: СггзСб (на
поверхности) и СГ7С3 (ближе к сердцевине). Оба эти карбида
на структуре представляют собой сплошную светлую полосу.
Разделить карбидный слой на две карбидные зоны возможно
только путем травления шлифа реактивом Гросбека, который вы-
травливает границу между двумя карбидами [270]. В табл. 28
205
Таблица 28
Фазовый состав и распределение микротвердости по глубине'
хромированного слоя (от поверхности до линии раздела)
в зависимости от содержания в стали углерода и способа хромирования [270]
Содержание углерода в стали в % Хромирование в порошкообраз- ной смеси Газовое хромирование
Фазовый состав Фазовый состав
0,03 Cr2N a-твердый раствор >1500 >300 а-твердый состав 250
0,25 Cr2 (N. С) а 1514 725 СГ2зСд а 4- Сг23Сз 1645 500
0,47 Cr2 (N, С) Сг2зСв Сг7С3 >1550 1780 Сг23Сб а Сг23Сб 1650 500
0,65 — — Cr2dC(j а 4~ Сг23С6 1750 580
0,85 Cr2(N. С) Сг23С6 Сг7С.> 1600 1800 Сг23Сб Сг7С3 1780 2000
1,03 — — Сг23Сб Сг7С3 1800
* Существует значительная разница между значениями микротвердости карбидов, приведенными в таблице и указываемыми в различных литературных источниках, согласно которым микротвердость Сг88С8, Сг7С8 и Сг8С8 составляет соответственно 1650. 1336 и 1350. Микротвердость нитрида CrsN по литературным данным 1570 [148], что согласуется с приведенными данными.
приведен послойный фазовый состав хромированного слоя в за-
висимости от способа хромирования и содержания в стали угле-
рода.
Образование карбидов в диффузионном слое средне.- и высо-
коуглеродистых сталей происходит благодаря диффузии угле-
рода из сердцевины к слою. В сталям, содержащих 0,2—0,8% С,
это приводит к образованию отчетливо видимой на микрострук-
туре обезуглероженной зоны. Между обезуглероженной, ц кар-
бидной зонами образуется промежуточная сильно травящаяся
зона, имеющая повышенное содержание, углерода по сравнению
с более глубоко расположенной обезуглероженной ,. додой
(рис. 129, б, и 131). По мнению некоторых исследователей, тем-;
ный слой по составу близок к эвтектоиду, образованному, хроми-
стым ферритом и карбидом (Сг,Ее)7,Сз. , , :.
206
Толщина промежуточной темной зоны возрастает с увеличе-
нием температуры процесса, ню мало изменяется с увеличением
выдержки (рис. 132). Толщина обезуглероженной зоны увеличи-
вается с повышением температуры и выдержки, но не превыша-
ет 0,1 мм.
В эвтектоидной и заэвтектоидной стали микроанализ чаще
не позволяет обнаружить темный промежуточный слой и зону
с подслойным обезуглероживанием. В этом случае после мед-
ленного охлаждения карбидный слой примыкает к перлитной
структуре сердцевины, а после быстрого охлаждения — к мар-
тенситной структуре сердцевины (рис. 129, в). При определен-
Рис. 132. Влияние температуры (а) и продолжительности (б) хромирования в
порошкообразной смеси на глубину промежуточной зоны (штриховые линии)
и зоны обезуглероживания (сплошные линии):
1 — сталь 20; 2 — сталь 40 [2701
ных условиях охлаждения и у этой стали удается все же зафик-
сировать переходную зону, однако несколько иного характера.
Как уже отмечалось, при хромировании с нагревом т. в. ч.
на средне- и высокоуглеродистых сталях не образуются диффу-
зионные слой карбидного типа. Это связано с весьма быстрой
диффузией хрома в сталь вследствие большой скорости нагре-
ва 1 до высокой температуры, в результате чего на поверхности
не успевают создаваться высокие концентрац'иигхро)ма и углеро-
да, «необходимые для образования карбидного слоя. В отноше-
нии хрома это подтверждается данными спектрального анализа
поверхностного слоя образцов стали-' У10 — глубина слоя
0,01 мм i(глубина' проникновения искры),—среднее содержание
хрома в котором составляло в проведенных автором опытах
1 1 Влияние скоростного нагрева на'глубийу диффузионных Слоев’рассмо-
трено на стр. 29.
207
33%, тогда как после хромирования в порошках с обычным на-
гревом в такой поверхностной воне содержится 92—95% Сг. На
образцах стали 15, хромированных с нагревом т. в. ч. и обычным
нагревом, различие в концентрации хрома было также большим
(соответственно 25% и 60—7-0% Сг).
Микроструктуры сталей 45 и У10, хромированных с нагре-
вом т. в. ч., иные, чем после обычного хромирования; они весь-
ма похожи на микроструктуры хромированной низ коугл ер о ди-
етой стали. Отличие первых от последних заключается в том, что
Рис. 133. Микроструктура стали У8 после хромирования
при 1150° С в течение 2 мин с нагревом т. в. ч.
после перетравливания видно, что слой не представляет собой
гомогенный твердый раствор (микротвердость это также под-
тверждает). Нетравящийся слой разделен темным скелетом на
участки, многие из которых имеют удлиненную форму в направ-
лении, перпендикулярном к поверхности шлифа (рис. 133). Су-
дя по микротвердости (Яр. 550—850), слой представляет собой
а-гвердый раствор с вкраплениями карбидов.
Микроструктуры сталей 10 и 15, хромированных с нагревом
т. в. ч. и обычным нагревом, не отличаются.
208
Диффузионный слой на сером перлитном чугуне состоит из
карбидов хрома (Сг, Ре)2зСб и (Сг, Ре^Сзиимеет твердость бо-
лее Hfi. 1800. При хромировании в порошках на поверхности чу-
гуна возникает карбонитридная зона Сгг(С, N)*.
Свойства хромированной стали
Механические свойства, износостойкость. Поверхностная
твердость хромированного слоя сталей 08 и 10, состоящего из
атвердого раствора, не превышает соответственно HV 150—
180 № HV 250—280. Микротвердость карбидного слоя стали с
а) б)
Рис. 134. Влияние содержания углерода в стали на ‘ поверхностную микро-
твердость по данным различных авторов и по данным различных опытов (а);
распределение микротвердости по глубине хромированного слоя (б). Сталь
У10, обработка в порошкообразной смеси при 1050° С в течение 8 ч
0,25% С составляет 1300—1600, а стали с 1,0—1,2% С
H\L 1750—1800 (рис. 134). Поверхностная твердость по Виккерсу
(нагрузка 1 кГ) сталей У10 и У12, подвергнутых после хромиро-
вания закалке, не изменяется и равна HV 1350 и более, что со-
ответствует твердости HRC 72 и более. Теплостойкость хромиро-
ванного слоя карбидного типа очень высокая: 850—900° С.
Хромированный слой низкоуглеродистой стали (менее
0,15% С) обладает большой вязкостью. Образцы хромированно-
го кровельного железа (глубина диффузионного слоя 0,10—
0,12 мм) сгибались на 180°, при этом сплошность слоя не была
нарушена. Хромированные детали из низкоуглеродистой стали
можно подвергать осаживанию, прокатке, развальцовке, нака-
* Чугун рекомендуется хромировать при 920—940° С в продолжение 25—
30 ч. Диффузия хрома в чугун благодаря наличию графитовых включений
протекает быстрее, чем в сталь.
209
тыван1ию резьбы, шлифованию, полированию и» другим операци-
ям, причем диффузионный слой не будет -растрескиваться [19].
Карбидный слой высокоуглеродистой стали хорошо связан
с сердцевиной. Сжатие образцов стали У12 на 5—10% не вызы-
вает «шелушения» диффузионного слоя, наблюдается лишь его
растрескивание. Хром и ров а иная сталь обладает высокой терми-
ческой усталостью.
Статическое растяжение образцов стали 40, хромирован-
ных при НОЮ0С, а затем подвергнутых отжигу или улучшенных,
показался что с увеличением глубины диффузионного слоя незна-
чительно1 снижается предел .прочности сталй, при этом пределы
Рис. 135. Кривые выносливости стали 40 при 20° и 500° С до и после хроми-
рования [270]:
1 — хромированная; 2 — нехромированная
пропорциональности и текучести, а также пластичность заметно
не изменяются. Хромирование повышает усталостную выносли-
вость стали при комнатной и особенно повышенной температуре
(рис. 135), что связано с возникновением в хромированном слое
сжимающих напряжений. После закалки напряжения сжатия в
слое снижаются вследствие увеличения объема сердцевины
(рис. 136).
Хромирование повышает длительную прочность стали 40 при
500° С и ©плаве Х20Н80Т при 800° С [270]. Если термически обра-
ботанная сталь 40 при 500° С и нагрузке 24 кГ1мм2 (23,5 X
X Ю7 нДи2) разрушается за 17 ч, то после газового'хромирова-
ния в течение 15 мин цри 1100° С и последующей термообработ-
ке — за 22 ч, после хромирования в течение 6 ч — за 100 ч и по-
сле хромирования в течение 8 ч — за 147 ч. Хромирование повы-
шает также предел ползучести и эрозионную стойкость сталей
и сплавов.
Хромированная сталь с карбидным слоем отличается высокой
и зн осостойкостыо.
Испытания, на, трение, проведенные г(а мащине Х4-Б при аб-
разивном износе (карборундовая шкурка),. показали, что если
210
износ образцов оловянно-свинцов.истого баббита - принять за
единицу, то износостойкость образцов закаленной стали ШХ15
выше в 54,6 раза,, а образцов хромированной, стали,- содержащих
0,65% С,— в 227,5 раза. Таким образом, абразивная износостой-
кость хромированной высокоуглеродистой стали в несколько раз
выше, чем стали ШХ15 [270].
На одном из заводов хромированию были подвергнуты бар-
хатные и личные напильники. Испытания показали, что их стой-
Рис. 136. Послойное изменение остаточных на-
пряжений сталей 10, 40 и У10 после хромиро-
вания и последующей термической обработки
(остаточные напряжения определялись на
оптико-механической установке) [270]:
./ — после хромирования, сталь 10; 2 — после
хромирования, сталь 40; 3 — после хромирования с
последующей термической обработкой, сталь У10;
.4 — после хромирования, сталь У10
кость при обработке стали с твердостью HRC 38, в 2,5—3 раза
выше, чем обычных напильников, Бархатными хромированными,
напильникамц легко обрабатывались закаленные калибры тверг
достью HRC 50—62, в то время как обычно для, этого- применя-
лись дорогостоящие .алундовьш-бруски. На .другом < заводе .хро-
мированными'; напильниками- обрабатывались шаблоны и калибр
ры, которые из-за высокой твердости обычно обрабатываются
абразивами с доследующей доводкой, , J •
-Был иопщтан ряд хромированных- деталей, применяемых в
станкостроении. Хорошие результаты показали ^хромированные,
малонагруженные шпиндели (сталь 35ХМЮА) быстроходных
21.1
точных станков-автоматов, применяемых в часовой (Промышлен-
ности. При круглосуточной эксплуатации станков в течение не-
скольких лет хромированные шпиндели продолжали успешно ра-
ботать и станки не потеряли необходимой точности. Длитель-
ность работы этих шпинделей значительно превзошла нормы,
испытания
Рис. 137. Уменьшение
веса хромированных и
нехромированных об-
разцов из стали 10
при испытании в 25 % -
предусмотренные для азотированных
шпинделей из той же стали.
Испытания на лесозаготовках хроми-
рованных пильных цепей из стали 85ХФ
показали, что их износостойкость по
сравнению с нехромированными’выше бо-
лее чем в 2—3 раза [270], [293].
В табл. 29 приведены данные о повы-
шении хромирования стойкости матриц
для холодной высадки.
Коррозионные свойства. Хромирован-
ный слой долгое время хорошо сохраняет-
ся во влажном воздухе, в воде, щелочах,
перегретом паре, растворах азотной кис-
лоты и в ряде других сред (табл. 30,
1рис. 137).
Продолжительные испытания показа-
ли, что сталь, подвергнутая диффузион-
ному хромированию, обладает при ком-
натной температуре более высокой стой-
ном растворе азотной
кислоты [19]:
/ —• нехромированная;
2 — хромированная
костью против разрушения, чем электро-
литическая хромированная сталь и даже
высокохромистая сталь XI7. Стойкость
хромированного слоя превысила 135—
280 суток (затем испытания были прекра-
щены). Стойкость хромированной стали в кипящей азотной кис-
лоте во много раз превосходила стойкость нехромированной ста-
ли, однако время, в течение которого продолжалось защитное
действие хромированного слоя, оказалось сравнительно неболь-
шим и колебалось в зависимости от концентрации кислоты
в пределах 11—20 ч [294].
Уменьшение веса (на образование окалины) в среде паров
кипящей азотной кислоты составило при 780° С в течение 4,5 ч
166,6 г) мм2 у нехромированных образцов и 1,8 г/ж2 у хромиро-
ванные (сталь ЗОХГСА) [294].
Коррозионная стойкость хромированной стали после термиче-
ской обработки (нормализации, закалки и отпуска) понижается
в разной степени у различных .сталей.
Так^хромированные образцы стали 18Х2Н4МА показали в
25 %-ной азотной кислоте высокую стойкость, а в концентриро-
ванной значительно меньшую (стойкость разных образцов силь-
но колебалась, возникающие дефекты имели характер отдельных
212
Таблица 29
Износостойкость хромированных матриц из стали У10 [270]
Наименование детали Стойкость матрицы в штуках высаженных деталей
нехромирован- ной хромированной
Стержень велосипедного руля Колпак бензонасоса Сепаратор Крышка сальника Корпус бензоотстойника Стакан реактивной тяги 750—1200 560 2000 900 700 2000—3000 7 000—10 000 20 000 18 000 16 000 7 900 8 000—12 000
Таблица 30
Коррозионная стойкость хромированной стали с различным содержанием
углерода
Среда Коррозионная стойкость
чистого железа стали чугуна
ДО 0,1596 С л 95 до 25% С о,’б% С 0,6- 1,2% с ковкого серого
Воздух Хорошая Ограни- ченная
Щелочь Хорошая
Пресная вода Ограни- ченная
Соленая вода Хорошая Ограниченная Плохая
Раствор HNO3 Хорошая Ограни- ченная :
Раствор НС1 Плохая Очень плохая Плохая Очень плохая Очень плохая
Перегре- тый пар При Alloying Хорошая м е ча н и е. Таблица заимствована из каталога Interchrome фирм! Со, выпущенного в Англии в 1961 г. Ограни- ченная л Chrome
больших и глубоких трещин и углублений). Сталь ЗОХГСА,.
наоборот, показала меньшую стойкость в разбавленной азотной
кислоте и очень высокую в концентрированной. Это объясняется
21S
различаем свойств диффузионных слоев этих сталей (типа твер-
дого раствора и карбидного типа) в отношении характера мик-
родефектов, зарождающихся при термической обработке.
В растворе серной (рис. 138), уксусной и фосфорной кислот
стойкость хромированной стали заметно выше, чем нехромиро-
ванной, однако различие в стойкости менее резко, чем в азотной
' кислоте.
Продолжительность испытания
Рис. 138. Уменьшение веса
при испытаниях в 35 Уо-
ном растворе серной кислс-
В растворах соляной кислоты
стойкость низкоуглеродистой стали
после хромирования мала, даже не-
сколько меньше, чем нехромирован-
ной стали (уменьшение веса за 5 су-
ток в 20 %-ном растворе НС1
18,6 а/ти2 вместо 14,2 г/мм2). С по-
вышением содержания в стали угле-
рода стойкость хромированной ста-
ли в среде НС1 повышается (для
сталей 45, У7 и У12 уменьшение веса
составляет соответственно 7,8; 4,4
и 2,5 г/м2) [294].
Хромированная сталь обладает
высокой стойкостью в дистиллиро-
ванной воде с примесью перекиси,
водорода (0,02%), в морской воде,,
во влажном воздухе с примесью се-!
роводорода (10%) или сернокисло-
го газа (15%) и в ряде других сред.1
Окалиностойкость. Окалиностой-
ты образцов стали диамет-
ром 15 и высотой 5 мм пос-
ле газового хромирования, и
хромирования в твердой
смеси [19]:
кость хромированной низкоуглеро-;
диетой стали, хотя и достаточно вы-;
сока, однако ниже, чем алитирован-i
ной. При 960° С в течение 15 ч уве-
1 — нехромированные; 2 —
хромированные в твердой сме-
си при 980° С в течение 4 ч\
3 — хромирование в твердой
смеси при 1100° С в течение 4 ч;
4, 5 И 6 — газовое '.хромирова-
ние при 980° С в течение соот-'
ветстденно 2, 4 и 6 ч
личение веса образцов стали 15 со-
ставляло: алитированной 1,2—•
1,8 г/м2-ч, хромированной 3,5—।
4,2 г/м2-ч и. необработанной 50—;
60 г/м2-ч [19]. Продолжительные ис-1
пытания показали, что высокой ока-
линостойкостью хромиройайная;
^таль обладает при температурах до 800°С, при более высоких'
Температурах хорошие результаты получены лишь при испыта I
ниях продолжительностью в несколько Десятков часов. При низ-;
кой температуре окалиностойкость хромированной стали высокая
Из табл. 31 следует, что при 700° G: и выдержке 1000 « ока-,
лино’етойкость. образцов хромированной низкоуглеродистой..ста-
ли может быть почти в 1000'раз выше, чем нехромироваадюй.
214
Таблица 31
Окалиностойкость хромированной и алитированной низкоуглеродистой
стдли при 700°С
Метод обработки Увеличение веса в мг^м?
в течение 100 ч в течение '1000 ч
Необработанная Алитирование путем металлизации с 40,4 . 147,7
последующим отжигом То же с дополйительной термической 6,68 ' 13,8
обработкой (глубокий слой) Горячее алитирование в расплаве алюми- 4,06 9,33
НИЯ 0,73 2,2
Хромирование 0,04 0,15
Примечание. К сожалению, в таблице не приведены данные о стойкости
стали, алитированной методом, обеспечивающим получение наиболее качественно-
го алитированного слоя, получаемого при обработке в порошкообразной смеси.
Окалиностойкость хромированной стали с диффузионным
слоем карбидного типа по крайней мере при кратковременных
испытаниях выше, чем стали с диффузионным слоем типа твер-
дого раствора. На-пример, при 1000° С в течение 6 ч увеличение
веса хромированных образцов железа 'составило 8 мг/см2 и ста-
ли (0,6% С) 0,3 мг)см2, а при 1Г00°С соответственно 19 и
0,5 мг)см2 (В. И. Архаров). Это различие в окалиностойкости
объясняется, в частности, тем, что при высоких температурах
«рассасывание» диффузионного елся карбидного типа происхо-
дит менее интенсивно, чем слоя типа твердого раствора, [270].
Хромирование обеспечивает длительную защиту низкоуглеро-
дистой стали от коррозии,при 420—450° С в смеси водорода и
сероводорода. При 500—550° С в атмосфере сернистых газов-
образцы нехромированной низкоуглеродистой стали увеличились
в весе за 45 ч на 258,2 г/м2, а хромированной лишь на 14,4 г/м2.
' Хромирование существенно повышает окалиностойкость при
1050—1100° С окалиностойких и жаропрочных сталей и сплавов,
однако лишь при наличии глубоких диффузионных слоев,
(табл. 32).
Хромирование стоит примерно на 3-м месте по степени повы-
шения окалиностойкости нелегированных или малолегированных
сталей . (после берилл из ации и алитирования); на 4*м месте сто-,
ит силицирование. Поверхностное насыщение стали большинст-
вом других элементов не приводит к повышению окалиностой-
кости стали (рис. 13,9)»
; Хромированная сталь обладает высокой, стойкостью также
в расплавах цинка, свинца и олова [295].
215'
Высокие антикоррозионные свойства, повышенные кислото-
стойкость и акалиностойкость, а также высокая твердость и из-
носостойкость хромированной стали позволяют применять хро-
мирование для обработки большой номенклатуры деталей. В оте-
чественной (Промышленности успешно опробовано хромирование
матриц для холодной штамповки, пильных цепей (стали 85ХФ
и У10, глубина хромированного слоя 0,01 мм) [293], колец пря-
дильных машин, штоков и втулок узлов уплотнителей стопорных
регулирующих клапанов паровых турбин (стали X15H36BT3)
J296], плоских пружин лабиринтных уплотнителей (сталь
ЭИ723) [297], [298], деталей шарниров втулочно-роликовых цепей,
Рис. 139. Окалиностойкость при 1000° С низ-
коуглеродистой стали, насыщенной различны-
ми элементами [270]
комбайнов [299], нитепр сводников для текстильной промышлен-
ности и многих других деталей.
Хромируют различные крупногабаритные изделия (900X
X 200 X 200 мм и более) из низко- и высокоуглеродистых сталей
при 940° С в течение 25 ч в порошке, содержащем 65% ферро-
хрома, 33% речного песка и 2% хлористого аммония [300].
Таблица 32
Окалиностойкость при 1100° С и продолжительности испытания 50 ч [270]
Материал Увеличение веса на единицу поверхности образца в %
нехромированных хромированных
Армко-железо (0,03% С) Сталь 15 » Х18Н11В » Х20Н80ТЗ » Х23Н18 Примечания: 1. Газовое хромирс пение 6 ч. 2. Увеличение веса образца стали X23 819,3 669,5 368,2 57,2 100 1вание осуществляло^ Н18 условно принято 81,0 8,1 10,4 18,3 6,3 ь при 1200° С в те- за 100%.
216
Наибольший экономический эффект достигается при хроми-
ровании толстостенных деталей. Так, если для изготовления од-
ной муфты весом 1,5 кг из высокохромиютой стали (30% хрома)
расходуется около 0,5 кг хрома, то при изготовлении ее из низ-
коуглеродистой стали и последующем газовом хромировании
требуется всего лишь около 10 г хрома [19].
Доказана целесооб|разность хромирования прецизионных де-
талей топливной аппаратуры (плунжерных пар, игл распылите-
лей из сталей ХВГ, ШХ15 и У10), поршневых чугунных колец,
фрезерных цепей и строгальных ножей для деревообрабатываю-
щей промышленности.
Согласно данным иностранной литературы, хромирование при-
меняют для сопловых устройств, клапанов, сальников, фильтров
нефтяных скважин, стержней соленоидных клапанов (повышается
коррозионная стойкость), глушителей автомобилей, стержней
паяльников, радиаторов, ножей машин, штампов для автомобиль-
ных панелей, металлокерамических железных деталей, электро-
дов запальных свечей, паронапревателей, паровой арматуры,
труб теплообменников, вентилей, сердечников электромагнитов
(повышаются магнитные свойства), деталей тракторного мотора,
форм для стекольного производства, калибров, деталей пище-
вой и химической аппаратуры, призм и подушек весов, магнито-
мягких материалов, различных матриц и пуансонов, в текстиль-
ном машиностроении, судостроении, авиационной промышлен-
ности, ракетостроении и в других отраслях [301]—[305], [572].
Насыщение стали хромом совместно с другими элементами
Хромоалитирование. Хромоалитированный слой имеет тем
большую глубину, чем больше в насыщающей среде алюминия
и меньше хрома. Алюминий диффундирует в сталь быстрее хро-
ма (при 1150° С коэффициент диффузии алюминия в у-железе
D = 170-10"5 см2/сутки, а хрома £> = 5,9-10"5 см2/сутки) [305].
Алюминий оттесняет углерод в глубь стали и способствует
перестройке решетки у в а, чем облегчается диффузия хрома
в сталь.
По сравнению с алитированным слоем хромоалитированный
может быть менее или более окалиностойким, в зависимости от
соотношения в нем хрома и алюминия. Это было показано в опы-
тах, проведенных как в порошкообразных смесях, так и в газовой
среде (НС1). В последнем случае смесь порошков ферросплавов
находилась с обеих сторон от образцов на расстоянии 30—50 мм,
для получения равномерной глубины слоя направление потока
хлористого водорода изменяли каждые 30 мин. Поскольку с уве-
личением в смеси содержания феррохрома глубина диффузион-
ного слоя резко уменьшалась, продолжительность процессов при
980° С соответственно увеличивали с 2 до 10 ч, с тем чтобы при
217
разных соотношениях в смеси ферросплавов получать диффузи-
онные слои примерно одинаковой глубины (0,13—0,16 мм).
Как видно из рис. 140, с увеличением содержания в смеси
феррохрома окалиностойкость образцов вначале немного повы-
шается, а затем начинает резко снижаться1.
После хромоалитирования, осу-
ществленного при 1000° С в смеси из
75% феррохрома, песка и НО, к кото-
рым добавили 2,5% алюминиевой пуд-
ры, окалиностойкость образцов ста-
ли 45, испытанных при 800° С в течение
100 ч, оказалась в несколько раз выше,
чем хромированных (по данным Науч-
но-исследовательского института хими-
ческого машиностроения).
Преимуществом хромоалитирован*
ного слоя перед алитированным яв-
ляется его большая вязкость.
Кроме того, по данным Е. В. Масю-
тина, хромоалитированный слой отли-
чается большим сопротивлением тер-
мическому удару (производились на-
гревы до 950° С), чем алитированный,
что является весьма ценным свойством
такого слоя.
Рис. 140. Окалиностой-
кость стали 10 при 950° С
и выдержке 15 ч после
алитирования и хромо-
алитирования в газовой
среде (кривая /) и по-
рошкообразных смесях с
добавкой 1% NH4C1
(кривая 2) или 1%
NH4CI и 8% огнеупорной
глины (кривая 5). Вес
образцов, не подвергну-
тых обработке, увеличил-
ся в среднем на 960 г/м2
(автор)
При хромоалитировании стали 20
ее механические свойства сохраняются
лучше, чем при хромировании, когда
происходит сильное обезуглерожива-
ние сердцевины образцов в результате
диффузии углерода к поверхности.
Хромоалитирование жаропрочных
сталей описано в работе [270].
Следует отметить, что хромоалити-
рованию стали посвящено много работ
и в зарубежной литературе [273],
[306].
Хромосилицирование. Хромосили-
цированный слой низкоуглеродистой
стали в некоторых средах отличается
большей окалиностойкостью и кислотоупорностью, чем хромиро-
ванный, и большей вязкостью, чем силицированный.
1 Спектральный анализ поверхностной зоны диффузионного слоя, полу-
ченного с использованием смеси, содержащей 50% феррохрома и 50% ферро-
алюминия, показал наличие в этой зоне 29% алюминия и 4,5% хрома. Таким
образом, в этом случае в сталь диффундировал в основном алюминий.
218
Рис. 141. Глубина хромоси-
лицированного слоя в зави-
симости от содержания
ферросилиция в смеси
FeSi + FeCr (процесс про-
текал в среде хлора при
980° С в течение 2 ч\ об-
разцы располагались от-
дельно от ферросплавов)
[автор]:
1 — смесь занимает все сече-
ние трубки со стороны ввода
хлора; 2 — смесь занимает !/з
сечения трубки
Кремний диффундирует в сталь бьистрее, чем хром: при
1150° С коэффициент диффузии кремния в у-железе D —
= 125•10-5 см2/сутки, а хрома D = 5,9-Ю"5 см2/сутки [305]. Крем-
ний оттесняет углерод в глубь стали и способствует перестройке
решетки у в а, что облегчает диффузию хрома в сталь.
Хро'мосилицирование стали 30 осуществляется при 980° С в.
продолжение 8 ч: 1} ® среде водорода с использованием смеси
порошков феррохрома (60%) и ферросилиция (40%); 2) в среде
НС1 + Н2 и 3) в среде хлора. В двух
последних случаях образцы помещали
отдельно от порошков ферросплавов.
При обработке в среде хлора диффу-
зионный слой достигал глубины 0,4—
0,5 мм, в среде Н2 + НС1 0,20—0,25 мм
и в среде водорода 0,1—0,14 мм *.
Влияние соотношения порошков фер-
росплавов на глубину слоя приведено
на рис. 141.
При 1000° С в течение 2 ч был полу-
чен хромосилицированный слой глуби-
ной 0,25 мм, содержавший в среднем
20% Сг и 5% Si [305].
Хромосилицирование в порошкооб-
разной смеси при нагреве т. в. ч. про-
текает весьма быстро [307]. Например,
при обработке стали 10 в смеси, со-
стоящей из 80% феррохрома, 20%
ферросилиция, к которым добавляли
20% шамота и 4% хлористого аммо-
ния, за 20 мин при 1050° С образовы-
вался слой глубиной 0,3 мм (вместо
0,04—0,05 мм при обычном нагреве).
Фазовый состав хромосилицированнюго слоя приведен в ра-
боте [574].
Азотирование и цементация хромированной стали. Азотиро-
вание хромированной низкоуглеродистой стали позволяет полу-
чать твердые и глубокие диффузионные слои. После такой двой-
ной обработки был получен слой глубиной 0,25 мм с микротвер-
достью на глубине 0,03 мм от поверхности 1770, на глубине
0,21 мм 1550 и на глубине 0,28 мм Н[к 185 [305].
В качестве метода повышения химической стойкости хроми-
рованной стали может быть применена цементация. При твердой
цементации в поверхностной зоне диффузионного слоя образует-
ся тонкая карбидная корочка и повышается сплошность слоя
* Минкевич А. Н. и Б о р з д ы к а А. М. Химико-термические методы
повышения жаростойкости и кислотостойкости стали. ИТЭИН. М. 1944.
21а
вследствие увеличения объема при образовании карбидов и сжа-
тия основной части хромированного слоя. Кроме того, при це-
ментации хромированной стали должна увеличиваться концент-
рация хрома в поверхностной зоне диффузионного слоя благода-
ря встречной диффузии хрома к фронту диффузии углерода [308].
Хромоалитирование и хромоазотирование аустенитной стали
Хромоалитирование и хромоазотирование аустенитной стали
имеет значение, например, для деталей лопаточного аппарата
стационарных газовых турбин, работающих на тяжелом жидком
и особенно пылевидном топливе. Эти детали должны изготовлять-
ся из материалов, обладающих не только высокими механиче-
скими свойствами при рабочих температурах, но и повышенной
эрозионной и коррозионной стойкостью.
Хромоалитированию подвергались образцы аустенитных ста-
лей X16H36BT3, Х18Н25М6, Х23Н18 и др. [309]. Глубина диффу-
зионного слоя при 1050° С в течение 8 ч в порошкообразных
смесях (49,5% ферроалюминия; 49,5% хрома и 1% NH4C1) до-
стигала на образцах сталей X16H36BT3 и Х16Н25М6 соответст-
венно 0,12—0,16 и 0,27—0,35 мм.
Таблица 33
Механические и эрозионные свойства хромоалитированной стали
X16H36BT3 при 650° С [309]
Состояние образца Статический разрыв а* * В К.Г/ММ уст Длительная прочность при нагрузке Уменьшение веса при эрози- онных испыта- ниях в г**
3 д » «л. л to k to « 5 в % Ф в % 28 кПмм2 27 кГ/мм* 28 к Г/мм2 27 кГ/ мм2
в ч в ч в % в %
Без поверхност- ного насыще- ния1 63,745,3 9,8 17,426 132 525 8,4 5,6 0,87
Хромоалитирова- ние1 53,832,4 15,720,721,5 177 794 5,5 4,9 0,21
Примечание. Значения нагрузок, выраженные в кПмм2, при переводе в
систему СИ, где они выражаются в «/лс2, нужно умножить на 10°.
1 Режим термической обработки образцов: нагрев до 1080° С, выдержка
30 мин, охлаждение на воздухе, старение при 800° С в течение 10 ч.
* На базе 1 О8 циклов.
** Эрозионные испытания моделей лопаток турбин проводились на специаль-
ной установке в продолжение 75 ч в тяжелых условиях —в скоростном потоке
горячих продуктов сгорания дизельного топлива (температура 600—610° С), сме-
шанных с золой. Алитированные модели лопаток в этих условиях уменьшились в
весе на 0,33 г.
220
0,06—0,07 мм, а на образцах
Сг
—
-----800 °C--------650 X----------Концентрацион-
ные лучи
Рис. 142. Изотермический разрез диа-
граммы состояния системы Fe—Сг—Ni
(при 800° С); на разрезе нанесены кон-
центрационные лучи, показывающие из-
менение состава хромированного слоя
различных сталей [310]
После хромоалитирования прочностные свойства стали
X16H36BT3 при кратковременно горячем разрыве (850° С) и пре-
дел выносливости, немного снижаются (табл. 33), а длительная
прочность повышается. Эрозионная -стойкость моделей лопаток
турбины, испытанных при 600—610° С, после хромоалитирования
увеличилась в 4 раза.
Диффузионное хромирование сталей Х17Н14М2 и X16H36BT.3
осуществлялось при 1050° С в течение 10 ч в порошкообразных
смесях [310]. Глубина хромированного слоя на образцах стали
Х16Н14М2 составляла в с
стали X16H36BT3 0,03—
0,035 мм *, концентрация
хрома в поверхностной зо-
не диффузионного слоя
(0,01 мм) была соответ-
ственно равна 47 и 31%.
твердость
образцов обеих сталей
составляла HV 1000—
1100.
Установлено, что хро-
мирование снижает пла-
стичность, длительную
прочность и предел уста-
лости этих сталей при вы-
соких температурах, что
объясняется образовани-
ем в хромированном слое
хрупкой а-фазы. Большое
количество а-фазы было
обнаружено рентгеноана-
лизом в осадках, выделен-
ных из поверхностного
слоя стали Х16Н14М2, хромированной как в порошкообразной
смеси, так и вакуумным методом. В поверхностном слое стали
X16H36BT3 a-фаза была обнаружена в меньшем количестве
и только после хромирования в вакууме. Очевидно, при хромиро-
вании в порошкообразной смеси происходит дополнительное ле-
гирование слоя азотом (из-за присутствия NH4C1), что повышает
устойчивость аустенита, расширяет область у-фазы и уменьшает
возможность образования а-фазы. Это различие в склонности
к образованию а-фазы у сталей Х16Н14М2 и X16H36BT3 в ос-
новном полностью согласуется с диаграммой состояния систе-
мы Fe — Сг — Ni (рис. 142). Диаграмма состояния позволяет
1 Способ интенсификации диффузионного насыщения тройных сплавов (в
том числе насыщения хромоникелевожелезных сплавов хромом) описан на
«стр. 33 [270] [29].
221
проследить за изменениями структуры хромированного слоя,,
имеющего: по .глубине переменную концентрацию хрома. Так как,
соотношение между никелем и железом в хромированном слое-
примерно сохраняется, химический состав слоя должен изме-
няться прямолинейно — по лучам, сходящимся к вершине, при-
надлежащей хрому.
На рис. 1’42'линии Лги А2 пересекают фазовые области соот-
ветственно для сталей Х16Н14М2 и X16H36BT3. Ход концентра-
ционных прямых хромированного слон объясняет количественное
Рис. 143. Изменение веса карбидного осадка и содержа-
ние в нем хрома и железа по глубине диффузионного
слоя стали X16H14M2N6N .после хромирования (а) и
после хромирования и азотирования (б) [310]
Дополнительное азотирование хромированной аустенитной
стали вносит коренное изменение в состав и свойства диффузи-
онного слоя. Изменяются количество и природа вторичных фаз.
Азот связывает часть хрома в нитриды и исключает образование
хрупкой о-фазы. Если до азотирования количество выделивше-
гося осадка и количество о-фазы оказывались максимальными,
то после азотирования о-фаза в слое отсутствовала (табл. 34),
а количество осадка по глубине слоя непрерывно снижалось
(рис. 143,).
Хромирование с последующим азотированием стали
Х16Н14М2 повышает (по сравнению с обычной термической обра-
боткой) предел длительной прочности при 600° С с 17 до
19 кГ1мм2 (с 16,6-107 до 18,6-107 н/л2), предел выносливости при
600° С с 20,8 до 29 кГ1мм2 (с 20,4-107 до 28,4:107 к/ж2) и предал
выносливости на надрезанных образцах с 14 до 20 кГ/мм?
(с 13,72-107 до 19,6-107 нМ2), т. е. на 43%. Одновременно увели-
чивается эрозионная стойкость стали в запыленном газовом по-
токе. ПосЛе хромирования и азотирования характеристики ока-
222
Таблица 34
Результаты послойного фазового рентгеноструктурного анализа
хромированного слоя стали X16H36BT3
Расстояние от поверх- ности в мм Фазовый состав после хромирования Расстояние от по- верхности в мм Фазовый состав после хромирования и после- дующего азотирования
0,07 0,022 0,042 0,075 0,112 Приме ченпе 10 ч, Ме2иСб, а-фаза, Сг2О3 a-фаза, Ме23Св, следы TiC TiC, a-фаза, Ме25С0 TiC, следы а-фазы TiC ч а н и е. Хромирование с :табилизация — при 750° С 0,07 0,017 0,057 0,090 0,140 эсуществлялось в вак> в течение 10 ч. TiN, Cr2N, Cr2O3 TiN, TiC, следы Cr2N TiN, TiC TiN, TiC TiN, TiC уме при 1050° С в те-
линостойкости стали X16H36BT3 при 650° С сохраняются такими
же, каки .после обычной термообработки, а эрозионная стойкость
в запыленном газовом .потоке увеличивается.
Процесс хро1моазотировани1я .рекомендован для защиты от
эрозии лопаток газовых турбин из сталей Х16Н14М2 и
X16H36BT3 [310]. Сталь X16H36BT3 следует после хромирования
азотировать при 1100° С <в течение 2 ч ib азоте, а затем «подвергать
старению при 850° С в течение 10 ч или при 700° С в течение 25 ч
(.поверхностная микротвердость Н[к 1400—1500, глубина слоя
0,03—0,1 мм).
Для защиты от эрозионного износа лопаток газовых турбин,
работающих на пылевидном топливе и изготовляемых из аусте-
нитных жаропрочных сталей, разработан метод получения твер-
дых (//[1 1500—1700) диффузионных слоев, содержащих нитриды
хрома типа Cr2N и CrN. Насыщение поверхности стали хромом,
азотом, а '.в некоторых случаях дополнительно и углеродом мож-
но осуществлять в твердых порошках, состоящих, например, из
хрома, обработанного соляной кислотой, хлористого аммония и
угля в количестве не более 2—3 вес. %. Диффузионная обработ-
ка осуществляется в течение 8—10 ч при температуре пе ниже
1050°С. Содержание азота в наружном слое достигает 2—3%,
общая глубина диффузионного слоя 0,1—0,2 мм, толщина нит-
ридной зоны слоя 40—60 мк. Эрозионный износ при 650° С в этом
случае уменьшается в 10—20 раз [311].
БОРИРОВАНИЕ
Насыщение стали (чаще среднеуглеродистой) бором произ-
водится для повышения ее поверхностной твердости, износостой-
кости (главным образом абразивной) и теплостойкости. Глубина
223
борированного слоя обычно не превышает 0,15 мм, его поверх-
ностная твердость доходит до HV 1400—1550, а микротвердость
до //|л 1800—2000. Борирование можно осуществлять в газовых
и жидких средах, а также в порошкообразных смесях.
Процесс борирования известен сравнительно давно. В своей
работе, опубликованной еще в 1915 г., Н. П. Чижевский отметил
чрезвычайно высокую твердость образующихся при борировании
слоев, что дало ему повод предполагать возможность техническо-
го применения процесса «цементации» бором L Н. П. Чижевский
писал: ... «где происходит порча от истирания, бор может оказать
услугу» [312]. Первые опыты по электролизному борированию в
расплаве буры были выполнены в 1934 г. [313]. Этот метод пока
является единственным, внедренным несколько лет назад в про-
изводство. Газовое борирование изучено лишь в лабораторных
условиях и опробовано на полупроизводственной установке.
Методы и режимы борирования
Газовое борирование
Поскольку газовые среды наиболее активны, насыщение ста-
ли бором протекает в них интенсивнее, чем в других средах, и
при более низких температурах. Поэтому газовое борирование со-
вершеннее других методов насыщения бором, но его недостатка-
ми являются токсичность и взрывоопасность применяемых газов.
Борирование в смеси диборана и водорода [314] — [315]. При
температурах выше 500° С диборан (В2Н6) почти полностью раз-
лагается на активный бор и водород. После борирования в чи-
стом диборане на поверхности стали образуется осадок бора, не
успевшего продиффундировать в металл, и процесс насыщения
замедляется^ Поэтому применяется смесь диборана и водорода,
взятых в соотношении 1:25-5- 75. Расход смеси зависит от типа
и размера установки [314]. При рабочем объеме реактора
1000 см3 и нагреве образцов т. в. ч. рекомендуется подавать 75—
,100 л/ч смеси.
Выходящий из печи газ следует пропускать через воду, где
неразложившийся диборан претерпевает гидролиз:
В2Нб + 6Н2О = 2В (ОН)3 + 6Н2.
На рис. 144 приведены результаты борирования в смеси
В2Нб + Н2 при 850° С. При температуре выше 850° С уменьшает-
ся сплошность слоя. Во многих случаях рекомендуется или огра-
ничивать продолжительность борирования 3—4 ч, или после вы-
держки 2—3 ч подвергать детали диффузионному отжигу в про-
должение 2—3 ч без подачи в реактор борирующей смеси (для
улучшения сплошности слоя).
1 Процессы осуществлялись в порошках аморфного бора и ферробора.
224
Борирование в смеси треххлористого бора и водорода [316],
[317]. В начальный момент процесса хлористый водород, по-ви-
димому, активизирует обрабатываемую ‘поверхность, удаляя
окисную пленку, но дальнейшее его воздействие приводит к за-
метному коррозионному разрушению обрабатываемой поверхно-
сти. Диффузионные слои в этом случае
получаются рыхлыми и пористыми. По-
этому рекомендуется [317] применять
смесь треххлористого бора (ВС1з) и водо-
рода в отношении 5:100 и осуществлять
процесс при температуре 850° С и вы-
держке не более 3—6 ч. При этом глубина
борированного слоя на железе достигает
0,11—0,20 мм, а на стали, содержащей
0,4% С, 0Д8—0,16 мм. Химизм реак-
ции борирования в смеси ВС13 и Н2 не
выяснен. Предположение, что в результа-
те взаимодействия треххлористого бора
и водорода при высоких температурах
образуется активный бор, не подтверж-
дено экспериментально. Специально по-
ставленные эксперименты [17] показали,
что при борировании может происходить
замещение бора в галоидном соединении
металлом основы. Это дает основание
Рис. 144. Зависимость
глубины борирован-
ного слоя от продол-
жительности процесса
при 850° С (В2Нв:
: Ня = 1 : 25) [31]
предполагать, что треххлористый бор не
принимает непосредственного участия в
процессе борирования, а является лишь
исходным продуктом для образования
химического соединения (диборана или какого-либо иного), вы-
деляющего активный бор.
Газовое борирование можно также осуществлять с примене-
нием ВВгз + Н2 [314].
Борирование в жидкой среде
Электролизное борирование в расплаве буры [318] — [326]. При
этом методе стальную деталь подключают в качестве катода в
цепь постоянного тока, анодом служит предварительно пропитан-
ный бурой графитовый стержень или цилиндр,
При расплавлении бура частично диссоциирует термически:
Na2B4O7 = Na2O 2В20з
и электролитически:
Na2B4O7 = 2Na + В4О7
выделяет- выделяет-
ся на ка- ся на апо-
тоде де
225
На аноде происходит разряжение аниона В4О7 и образуется
борный ангидрид по реакции
2В4О7 = 4В2О3 ©2-
Натрий, выделяющийся на детали (катоде), частично всплы-
вает на поверхность и сгорает, а частично идет на восстановле-
ние окисла бора до активного бора (6Na + В2О3 = 3Na2O + 2В),
благодаря которому и происходит борирование.
Оптимальный режим борирования по этому методу следую-
щий: плотность тока на катоде 0,15—0,20 а/см2-, напряжение 2—
14 в; температура 930—950° С; выдержка 2—6 ч. При таком ре-
Рис. 145. Влияние плотности тока и температуры борирования на глубину
борированного слоя (Г. И. Юкин):
а — температура борирования 950° С, выдержка 3 ч\ б — плотность тока 0,15 а/см2, вы-
держка 3 ч\ / — сталь 45; 2 — сталь 20; 3 — сталь 20X; 4 — сталь 38ХМЮА;
5 — сталь У10
жиме глубина борированного слоя достигает 0,15—0,35 мм. По-
вышение температуры выше 980° С и увеличение продолжитель-
ности процесса выше 6—8 ч, а плотности тока более 0,25 а/см2
приводит к возрастанию хрупкости слоя при небольшом увели-
чении глубины (рис. 145 и 146).
Недостатком процесса является быстрый выход из строя тиг-
лей. Тигли следует изготовлять из сплавов Х23Н18, Х23Н15, Х28
и предварительно подвергать их борированию (плотность тока
не менее 0,01 а/см2, продолжительность процесса 20—24 */). Не-
обходимо создавать электрозащиту тиглей, подключая их в каче-
стве катода (плотность тока 0,005—0,015 а/см2), но и при этом
стойкость тиглей оказывается низкой.
Пр.и борировании расходуется много буры, большая часть ко-
торой уносится на деталях при их выемке из ванны и затем рас-
творяется в промывочной горячей воде. Путем регенерации уда-
ется возвратить большую часть растворенной в воде буры для
дальнейшего использования.
226
Содержание железа в рабочей ванне не должно превышать
0,5%. По мере накопления в ванне растворенного железа (при
неправильной технологии процесса — отсутствии катодной за-
щиты) скорость борирования па-
дает. В целях рафинирования со-
ли Гипронефтемашем предложен
метод осаждения железа на спе-
циально введенном в ванну же-
лезном стержне, подсоединяемом
в качестве катода (плотность то-
ка 0,15—0,25 а]см2}. После рафи-
нирования соль вместо темного
цвета приобретает светло-дымча-
тый.
Опыт работы Гипронефтема-
ша показывает, что при непра-
вильном ведении процесса элек-
тролизного борирования могут
Рис. 146. Зависимость тол-
щины борированного слоя
на образцах различных ста-
лей от температуры при
выдержке 4 ч (плотность
тока 0,25 а]см2} [320]:
1 — сталь ЗОХГСА; 2 — сталь
12ХНЗА; 3 — сталь 12ХН2А;
4 — сталь 55С2А; 5 — сталь 35;
6 — сталь 40Х
возникнуть следующие дефекты
борированной поверхности:
1. Местное оплавление. Основ-
ная причина — расстояние между
анодом и катодом меньше допу-
стимого при данной плотности
тока. В этом случае возникает
дуги, и бура разогревается до вы-
сокой температуры.
2 . Разъедание поверхности. При отсутствии контакта (деталь
не является катодом) наблюдается осповидное разъедание, при
загрязнении буры железом (более 1% Fe)—пятнистое разъеда-
ние.
3. Волосные трещины в борированном слое. Трещины обра-
зуются при повышенной плотности тока и неправильном режиме
охлаждения.
В целях повышения активности ванны для электролизного бо-
рирования в одной из работ рекомендовалось периодически до-
бавлять в нее В2О3 с таким расчетом, чтобы поддерживать соот-
ношение —В-2^3 * * * *- (где М — Na или К) равным 2,5 и более вместо
М2О
обычного 0,5—2,0. Однако в другой работе рекомендуется для
борирования смесь, содержащая лишь 55—65% В2О3 и 35—45%
Na2B4O7 [325]. Такая ванна несколько дешевле ванны со 100%
буры и дает лучшие результаты (табл. 35).
С целью экономии дорогостоящей буры Гипронефтемаш ре-
комендует вводить в ванну хлористый натрий в количестве до
75% об. (в расплавленном состоянии). При этом глубина слоя
и его твердость остаются неизменными.
227
Таблица 35
Влияние состава ванны на глубину борированного слоя [326]
Состав ванны Глубина борированного слоя в ллс
Na8B4O7 BsO8 Сталь с 0,11 % С Сталь с 0,50% С Сталь с 1,03 % С
85 15 0,28 0,25 0,24
60 40 0,30 0,28 0,27
40 60 0,34 0,32 0,31
30 70 0,33 0,30 0,29
Приме ч а ния. 1. Реж им обработки: те мпература 950° С, выдержка 4 ч,
плотность тока 0,2—0,25 а см*.
2. Поверхностная твердость колебалась в пределах HV5 — 1415 ч- 1580.
Электролизное борирование в смеси метаборной кислоты
(НВО2) и фтористого натрия (NaF). В этом случае рекомендуется
следующий режим обработки: состав ванны 33% НВО2 и 67%
NaF; плотность тока на катоде 9,6 а/см2-, напряжение 11 в [147],
. . । । 1 । । 1 процесса
100 60 60 0-0 20 80 60 ЬО 20 О в)
% бурь) % бурь»
а) б)
Рис. 147. Влияние на глубину борированного слоя:
а — содержания в ванне карбида бора (1000е С, 5 «); б — содержания в ванне хлори-
стых солей (1 ч. ВаС12 и 1 ч. NaCl); 1000° С, 5 ч, 20% карбида бора; в — продолжитель-
ности борирования в ванне: 60% буры и 40% карбида бора {328]
[327]. Такая ванна применялась для борирования молибдена, но
может быть использована и для борирования стали.
Борирование в расплавленной буре с карбидом бора [328]. Ак-
тивный атомарный бор в такой ванне образуется путем восста-
новления окислов бора углеродом карбида бора. К расплавлен-
328
ной буре рекомендуется добавлять 30—40% карбида бора, со-
держащего 76—78% В и 18—20% С (рис. 147). Такая ванна поз-
воляет обрабатывать детали любой формы, что невозможно при
электролизном борировании. Недостатком этого метода является
малая жидкотекучесть ванны, а также быстрое истощение и
уменьшение ее активности. Повышение жидкотекучести за счет
некоторого снижения активности ванны достигается добавлением
б нее нейтральных солей (например, NaCl) или уменьшением ко-
личества карбида бора. Результаты борирования в такой ванне
приведены на рис. 147.
Борирование можно осуществлять в ванне, содержащей
78% ВаС1 и 22% NaCl, к которой после расплавления добавляет-
ся 10% карбида бора или 20% ферробора [272], [329], но такой
метод борирования менее эффективен, чем предыдущий.
Была опробована также ванна, составленная из 80% NaCl,
5% карбида бора и 15% NaBF*. При обработке стали в этой
ванне получается примерно такая же глубина слоя, как и при
электролизном борировании, но не достигается равномерности
глубины [330].
Борирование в порошках и обмазках
Борирование в порошках применяется редко. Опробовано бо-
рирование в порошках аморфного бора, ферробора, ферробора-
ла, карбида бора (в герметичных контейнерах), в среде водоро-
да, в вакууме [272], [280], [312], [331]—[333]. В ряде работ при
проведении процессов в вакууме применялась смесь, состоящая
из 84% карбида бора и 16% буры (слой глубиной 0,14 мм на
стали 45 образовывался за 6 ч при 1000°С).
Удовлетворительные результаты получаются при борирова-
нии в порошке карбида бора с добавлением 0,5—1% NH4CI в
герметичном контейнере (при 950° С за 6 ч образуется слой глу-
биной 0,12—0,14) или в смеси порошка ферробора, кварцевого
песка и 0,5—1% NH4CI.
В работе, выполненной в Московском институте стали и спла-
вов [279], исследовалось борирование с применением быстрого
нагрева т. в. ч. стальных образцов, покрытых обмазкой, состоя-
щей из 50% тонкого порошка карбида бора и 50% криолита
(связующее — гидролизованный этилсиликат). После нагрева
до 1150—1165° С на стали 45 в течение 2—3 мин образовывался
борированный слой глубиной около 0,1 мм с поверхностной твер-
достью около HV 1000 и микротвердостью не более 1150.
Причина пониженной твердости слоя и большой скорости про-
цесса та же, что и при хромировании стали в обмазках с нагре-
вом т. в. ч. (см. стр. 207).
229
Интенсификация процесса с помощью ультразвука
Борирование осуществлялось при 950° С в ванне, состоявшей
из 60% расплава буры и 40% карбида бора. Ультразвуковые ко-
лебания передавались образцу от магнитострикционного вибра-
тора непосредственно через концентратор. Мощность ультразву-
кового генератора 3 /сет, частота 20—25 кгц. В результате воз-
действия ультразвука глубина борированного слоя увеличилась
в 2—2,5 раза по сравнению с достигаемой при обычной обработ-
ке в такой ванне. Прй использовании нагрева т. в. ч. (образец
предварительно покрывался слоем указанного состава) приме-
нение ультразвука способствовало увеличению глубины бориро-
ванного слоя в 1,5 раза.
Защита от борирования
В связи с перспективами более широкого внедрения бориро-
вания в производство возникает вопрос о местной защите изде-
лий от борирования. При электролизном борировании для мест-
ной защиты рекомендуется гальваническое меднение с осажде-
нием слоя толщиной не менее 0,15 мм [334].
Термообработка борированной стали
В большинстве случаев борированная сталь подвергается за-
калке и отпуску, так как наличие под тонким, твердым слоем
вязкой сердцевины может привести к продавливанию и выкра-
Рис. 148. Величины остаточных напряжений в образцах стали, содержащей
0,9% С, толщиной 1.5 мм на различном расстоянии от поверхности. Бориро-
вание в расплаве буры с карбидом бора с последующим охлаждением на
воздухе (кривая /) и дополнительной закалкой в воде (кривая 2)
шиванию слоя. После закалки в слое часто возникают трещины.
Это можно объяснить тем, что при закалке увеличивается объем
сердцевины деталей, тогда как в борированном слое, состоящем
из боридов, существенных объемных изменений не происходит;,
в результате в слое должны возникнуть растягивающие напря-
жения.
230
Для подтверждения этого автор поставил опыты, во время
которых пластинки размером 1,5 X 10 X 50 мм из стали У9 под-
вергались борированию в расплаве буры с карбидом бора при
950° С в продолжение 3 ч (глубина борированного слоя 0,13 мм,
поверхностная микротвердость Яр. 1800). Часть образцов после
борирования подвергалась закалке с 850° С в воде и низкому
отпуску при 180° С. Деформация пластинок определялась на опти-
ко-механической установке Московского авиационного институ-
та, в которой непрерывное снятие слоя с одной стороны пластин-
ки осуществлялось в электролите, состоящем из метилового
спирта, серной кислоты и глицерина, при плотности тока 10 а!дм2
[335]. Напряжения рассчитывались по формуле Саверина. Ре-
зультаты послойного определения напряжений приведены на
рис. 148.
Знак внутренних напряжений в борированном слое после за-
калки изменяется, и вместо сжимающих напряжений в поверх-
ностном слое возникают растягивающие напряжения, величина
которых зависит от размера детали и других факторов. Для сни-
жения этих напряжений рекомендуется борированную сталь под-
вергать поверхностной закалке с нагревом т. в. ч. или изотерми-
ческой закалке. При нагреве стали под закалку следует приме-
нять предварительный подогрев.
Структура борированного слоя
Бор относится к элементам с небольшим атомным радиусом
(0,91 А), что благоприятствует диффузии бора в сталь. Бор стоит
на границе между имеющими минимальные атомные радиусы
элементами, диффундирующими в железо путем внедрения
(водород, азот, углерод), и элементами, диффундирующими
медленнее — путем замещения. До сих пор не было единого
мнения о том, какой твердый раствор — замещения или внедре-
ния — образует бор с железом. В последнее время высказыва-
ются предположения, что бор образует раствор замещения с
a-железом и раствор внедрения с у-железом [336].
Растворимость бора в а- и у-железе невелика. Например,
в у-железе при 1000° С она не превышает 0,003—0,008% [336].
После насыщения железа бором под микроскопом обнаружи-
вается только зона слоя реакционной диффузии (зона боридов).
В системе В — Fe реакционная диффузия осуществляется преи-
мущественно путем диффузии бора через боридный слой к основ-
ному фронту реакции, расположенному на межфазовых границах
железо — борид РегВ и борид БегВ — борид FeB (рис. 149).
Очевидно, бор диффундирует через решетку боридо-в в виде по-
ложительных ионов. Нет оснований предполагать значительную
диффузию железа через борид к границе борид — окружающая
среда.
231
i Микроструктура борированного слоя железа (рис. 150) пред-
ставляет собой иглы боридов, ориентированные перпендикулярно
/поверхности образца и вклинивающиеся в ферритные зерна
‘ (твердый раствор бора в феррите). При формировании диффу-
зионного слоя на поверхности металла по достижении предель-
ного насыщения бором твердого раствора (а или у) вначале воз-
никают зародыши, а затем иглы тетрагонального борида Fe2B
(а = 5,109 А, с = 4,249 А и —= 0,832 А), содержащего
а
8,84% В- Эти иглы растут, постепенно смыкаясь в сплошной слой
боридов ЕегВ, Затем на поверхности этого боридного слоя воз-
никают отдельные разобщенные иглы, а потом и второй сплош-
о О
ной слой ромбического борида FeB (а = 5,506 А, b = 4,061 А п
с = 2,952 А), содержащего 16,25% В [337]. При травлении рас-
твором азотной или пикриновой кислоты слой второго борида
выявляется слабо, и для его выявления чаще используется пик-
рат натрия, а также тепловое (500° С, 5 лшн) или цветное трав-
ление в реактиве следующего состава: 0,5 г пикриновой кислоты,
12,5 г едкого натра и 25 см3 воды. Травление производят в тече-
ние 30 сек при температуре 35—40° С.
Иглообразное строение боридных слоев указывает на то,
что при их образовании происходит быстрый рост боридных фаз
в направлении, перпендикулярном к поверхности образца, в то
время как вдоль поверхности развитие этих фаз протекает сла-
бо. Многие авторы связывают иглообразный характер борид-
ного слоя с преимущественной диффузией бора по границам зе-
рен. Однако рассмотрение микроструктур борированных слоев
не подтверждает этого, так как направление роста игл не совпа-
дает с границами зерен.
Иглообразные диффузионные слои могут возникать при по-
верхностном насыщении железа и некоторыми другими элемен-
тами, например при алитировании железа в расплавленном алю-
минии (см. рис. 112). Рентгеноструктурный анализ показал, что
иглообразный характер структуры при алитировании может
быть объяснен особенностью кристаллографической решетки
возникающей фазы Fe2Als.
Авторы работы [263] пришли к выводу, что при поверхност-
ном насыщении быстрый рост интерметаллических соединений
часто происходит вследствие особенности структуры этих фаз,
допускающей быстрое перемещение атомов активного элемента
в определенном структурно обусловленном направлении (см.
подробнее на стр. 172—173.
Это объяснение, очевидно, может быть распространено и на
механизм образования игольчатых структур в борированных
слоях железа, а также никеля и кобальта (см. рис. 303 и 311).
232
Рис. 149.
Диаграмма состояния системы
Fe —В
Рис. 150. Микроструктура железа (а) и стали 35 (6), подвергнутых электро-
лизному борированию (950° С, 6 ч\ 0,15 а/сж2); более темные иглы борида
FeB едва видны на фоне игл борида FegB (Х300)
233
Наибольшей микротвердостыо обладает борид FeB (7/
1800—2000) и несколько меньшей Fe2B 1600—1800). В свя-
зи с различием в твердости этих боридов на кривых изменения
микротвердости по глубине борированного слоя удается выде-
лить две ступени, соответствующие твердости различных бори-
дов (рис. 151). Кривые изменения микротермоэлектродвижущей
силы полностью воспроизводят изменение фазового состава по
глубине борированного слоя. Так как бориды FeB и Fe2B имеют
различную величину микротермоэлектродвижущей силы, этим
Рис. 151. Изменение фазового со-
става микротвердости (средние
значения из нескольких показа-
ний) и микро- т. э. д. с. (средние
значения) по глубине борирован-
ного слоя железа (автор, Е. В.
Панченко, В. Устагал)
методом можно определять
глубину слоя борида FeB, ко-
торый после обычного травле-
ния выявляется очень плохо.
С повышением температуры
процесса кривая 1 (рис. 152),
характеризующая общую глу-
бину слоя (зона Fe2B + FeB),
имеет перелом, отвечающий
температуре фазового превра-
щения а->-у. Известно, что
во всех случаях скорость диф-
фузии элементов в а-фазу
больше, чем в более компакт-
ную у-фазу. Этим и объясня-
ется торможение диффузии
бора после фазового превра-
щения а->у.
Изменение глубины сплош-
ного слоя боридов имеет дру-
гую закономерность: вначале
глубина слоя возрастает с.по-
вышением температуры, а за-
тем уменьшается (кривая 2,
рис. 152).
С увеличением содержания углерода в стали глубина бори-
рованного слоя постепенно снижается (рис. 153). Влияние угле-
рода на глубину диффузионного слоя при борировании менее
значительно, чем, например, при хромировании, ванадировании
и ниобирова1нии. С увеличением в стали содержания углерода
уменьшается протяженность зоны борида FeB [317]. При бори-
ровании углерод вытесняется из борированного слоя, под кото-
рым образуется зона, обогащенная углеродом, глубина которой
часто в несколько раз больше, чем глубина зоны боридов.
В сердцевине тонких борированных образцов наблюдаются вы-
деления графита [338].
В поверхностной зоне слоя стали 40, подвергнутой электро-
лизному борированию (960°С — 5 ч, плотность тока 0,2 а/см2).
234
рентгеноструктурный анализ обнаружил карбид бора В4С [339].
Однако во всех других исследованиях фазового состава бориро-
ванных -слоев, образующихся на железе и стали при электролиз-
ном и газовом борировании, карбид бора в слое не был обна-
ружен.
При формировании борированного слоя наблюдается пере-
распределение легирующих элементов между слоем и подслоем.
Например, по данным Л. С. Ляховича и Л. Г. Ворошнина, полу-
ченным спектральным анализом, в слое боридов значительно
снижается (в 2 раза и более) содержание хрома, молибдена и
вольфрама и увеличивается (в 1,5—2 раза) содержание никеля,
кремния и марганца.
Влияние легирующих элемен-
тов на глубину борированного
слоя сплавов на железной основе
приведено на рис. 154. Молибден и
Рис. 153. Влияние содержания
углерода в стали на глубину
борированного слоя (процесс
проводился в расплавленной
буро с карбидом бора в тече-
ние 5 ч) [31]
Рис. 152. Влияние температуры бо-
рирования железа в газовой смеси
В2Нб: Н2 = 1 : 25 на глубину диффу-
зионного слоя (продолжительность
2 ч):
1 — полная глубина слоя; 2 — глубина
сплошного слоя боридов (31]
вольфрам сильно уменьшают глубину слоя. Хром, кремний и
алюминий менее резко уменьшают глубину слоя. Кобальт, мар-
ганец и никель практически незначительно уменьшают глубину
слоя. К элементам, сильно уменьшающим глубину борирован-
ного слоя на безуглеродистых сплавах, следует отнести также
титан и ванадий [331], а к элементам, уменьшающим ее незна-
чительно,— медь.
Наличие в сплаве углерода в ряде случаев нарушает уста-
новленную для безуглеродистых сплавов закономерность влия-
ния легирующих элементов. Например, никель, не изменяющий
глубины слоя на безуглеродистых сплавах, резко уменьшает ее
на среднеуглеродистой стали. В случае борирования при темпе-
ратуре 950° С и выдержке 6 ч в смеси диборана и водорода глу-
бина борированного слоя на образцах среднеуглеродистых ста-
лей с 0,41; 2 и 4% Ni соответственно достигала 0,25; 0,14 и
0,08 мм [315]. В противоположность этому на образцах стали,
235
содержащей 0,32% С и 15,5% Сг, никель способствует резкому
увеличению глубины слоя. Хром на образцах стали с 0,38% С,
так же как и безуглеродистых сплавов, резко уменьшает -глуби-
ну слоя. Что касается различных по составу конструкционных
сталей, то после обработки они мало отличаются глубиной бори-
рованного слоя. У сталей 18ХГТ, 37ХС, 50ХФА, 38ХМЮА глуби-
на слоя несколько меньше, чем у сталей 20, 20Х, 45, 45Х. У высо-
колегированных инструментальных сталей ЗХ2В8. Х12 глубина
диффузионного слоя примерно в два раза меньше, чем у конст-
рукционных [340].
Кроме описанных ранее структур диффузионных слоев (две
Рис. 154. Влияние легирующих
элементов на глубину диффузи-
онного слоя железа, борирован-
ного в расплавленной буре с кар-
бидом бора (1100° С, 5 ч) [323]
зоны иглообразных боридов),
при борировании образуются
слои и других типов.
Слой второго типа образу-
ется при обработке стали, на-
пример, в расплаве буры при
температуре немного выше
линии солидуса и имеет эвтек-
тическую или заэвтектическую
структуру (рис. 155,а). Бори-
ды в этом слое выделяются в
виде равномерно распределен-
ных зерен, разделенных вяз-
кой основой [341]. Такой слой
имеет определенные преиму-
щества: заданная глубина на-
сыщения достигается за более
короткое время, чем у слоя
первого типа, кроме того, этот слой отличается меньшей хруп-
костью и в то же время высокой твердостью.
Слой третьего типа (рис. 155, б и в) получен при борирова-
нии стали в течение 2—3 мин в специальной обмазке с нагревом
т. в.ч. до температуры вблизи линии солидуса. Этот слой
отличается хорошей вязкостью, но имеет пониженную микро-
твердость (Яр. 1100—1300) [279], что объясняется особым фазо-
вым составом и структурой полученного борированного слоя.
Особенности фазового состава и структуры связаны, с одной сто-
роны ,с большой скоростью диффузии бора в глубь стали благо-
даря быстрому нагреву до весьма высоких температур (см.
стр. 29), а с другой стороны, с меньшей активностью применяв-
шейся насыщающей боросодержащей среды. В результате этого
в поверхностных слоях стали не успевает создаваться концент-
рация бора, необходимая для образования зоны боридов I Диф-
1 Аналогичная особенность образования диффузионных слоев была обна-
ружена автором при хромировании средне- и высокоуглеродистой сталей с на-
гревом т. в. ч. (см. стр. 207).
236
фузия бора при быстром нагреве т. в.ч. до 1150—1165° С проте-
кает стремительно в первую очередь по границам зерен и вбли-
зи их, и поэтому середина зерен сохраняет свою прежнюю струк-
туру и остается почти не охваченной диффузионным потоком
Рис. 155. Микроструктура
стали с 1% С, борирован-
ной при температуре, близ-
кой к линии солидуса:
а — в расплаве буры [341]; б и
в — в специальной обмазке с
нагревом т. в. ч. (279]
бора (на микроструктурах имеет вид островков с сохранившейся
прежней структурой и микротвердостью, рис. 155, б).
Для создания на поверхности стали твердой боридной зоны
индукционный нагрев следует производить медленнее, а процесс
осуществлять при более низких температурах и в более актив-
ных обмазках.
Слой четвертого типа получается иногда при обработке ста-
ли в малоактивной насыщающей среде — в порошках
237
Рис. 156. Микроструктура разных сторон одного и того же образца стали
с 1% С, борированного в ферроборе в вакуумной печи (1050° С, 8 ч)
238
низкопроцентного ферробора или аморфного алюмотермического
бора (рис. 156).
В этих порошках осуществляется комплексное насыщение
стали бором, кремнием и алюминием, так как два последних
элемента в значительных количествах содержатся в качестве
примесей в ферроборе и аморфном алюмотермическом боре1,
а растворимость их в а- и у-железе во много раз больше, чем бо-
ра. Спектральный анализ обнаружил в поверхностном слое об-
разцов более 1% Si, тогда как в их сердцевине содержалось
всего 0,25% Si [342]. Основой внешней зоны слоя является, по-
видимому, сложный a-твердый раствор, очевидно, с дисперсны-
ми выделениями (микротвердость около Я|А 300—500). Глубже
этой зоны обнаруживаются светлые вытянутые включения.
Можно было предполагать, что эти включения являются цемен-
титом, образовавшимся при температуре насыщения в результа-
те оттеснения углерода кремнием и алюминием, способствующи-
ми возникновению зоны a-твердого раствора. Однако эти
включения имеют в зависимости от режима и условий процесса
различную, иногда высокую микротвердость (Н1к 1000—1300).
В работе [30] на сталях с 1% С и 0,05% С был получен ана-
логичный по структуре борированный слой. При этом на кривой
распределения бора по глубине слоя в зоне вытянутых включе-
ний был обнаружен максимум в концентрации бора (до 4%)2.
Учитывая данные работы [30], а также часто обнаруживае-
мую высокую твердость светлых включений и их своеобразную
вытянутую форму (рис. 156), можно допустить, что эти включе-
ния могут являться карбоборидами. О возможности образования
карбоборидов в сплавах системы Fe — В — С указывается в ли-
тературе. Установлено, что в цементите до 80% углерода может
замещаться бором, при этом образуется соединение Fe3Bo,8Co,2
[343], [344]. В другой работе [345] был обнаружен карбоборид
примерного состава Ре2з(С, В)6. Однако окончательное заключе-
ние о природе этих включений можно сделать лишь после допол-
нительных исследований, тем более что ряд авторов ставит воз-
можность образования карбоборидов в диффузионном слое под
сомнение.
Слой этого типа, хотя и интересен для уточнения механизма
его образования и изучения структуры, но, очевидно, не пред-
ставляет какого-либо практического интереса.
При борировании инструментальных, а также различных ле-
гированных конструкционных сталей получаются и несколько
иные структуры борированных слоев, расшифровать которые
пока не всегда удается [340].
1 Алюмотермический бор может содержать до 3—5% А1, а ферробор
1,5—5% AI и 1,5—2% Si.
2 Борирование проводилось на глубину 1 мм в вакууме с использовани-
ем порошка аморфного бора при 1100° С в течение 250 ч.
239
Свойства борированной стали
Микротвердость внешнего борида FeB колеблется в пределах
Яр. 1800—2000, а борида FezB — в пределах Яр. 1660—1800. По-
верхностная твердость при отсутствии продавливания слоя со-
ставляет HV 1350—1550. В одних работах было замечено сниже-
ние твердости боридов при увеличении содержания в стали угле-
рода [981], в других, наоборот, незначительное увеличение твер-
дости. Судя по данным табл. 36, содержание углерода в стали
практически почти не сказывается на поверхностной микротвер-
дости зон FeB и РегВ.
Таблица 36
Микротвердость различных зон боридов (В. Устагал и автор)
Зона измерения микротвердости Материал Микротвердость Hi.oo
FeB Железо Армко Сталь 20 > 45 » У10 1870; 2050; 1980; 1870 1870; 1870; 1870 1870; 1980; 1800 1880; 1870; 1870
Fe2B Примечани рений микротвердо) 2. Электролизнс Железо Армко Сталь 20 > 45 » У10 я: 1. В таблице приведены "ср сти одного и того же образца >е борирование при температур 1600; 1650; 1650; 1600 1650; 1600; 1650 1650; 1650; 1650J 1650; 1650; 1650 едние значения нескольких изме- с разных сторон. »е 950° С и выдержке 6 ч.
Легирующие элементы существенно влияют на микротвер-
дость борированного слоя. По данным работ [31], [331], никель
снижает микротвердость слоя. Например, сталь с 0,41% Ni име-
ла после борирования микротвердость около Я1Х 2250, а сталь
с таким же содержанием углерода и 12% Ni — лишь Яр 1200.
Борированные в порошкообразной смеси при 1000° С в течение
20 ч стали с 12% Ni (0,2% С) имели твердость HV 847, а желе-
за HV 1430 [315]. Хром, молибден, вольфрам и марганец повы-
шают твердость борированного слоя. Ниже приведены значения
поверхностной микротвердости борированных слоев различных
сталей [341].
Материал образца
Железо.........
Сталь ХВГ . . .
> 12ХМ . .
> Р9 . . . .
> Р18 . . .
Микротвердость
1800—2290
2450—2630
2630—2830
2630—3045
2630—3435
240
Борирование стали долгое время не находило практического
применения главным образом из-за большой хрупкости бориро-
ванного слоя. Однако при надлежащем подборе деталей (отсут-
ствие острых углов, абразивный характер износа, удаление
в процессе эксплуатации продуктов износа с трущихся поверхно-
стей и т. д.) борирование в ряде случаев оказывается весьма эф-
фективным.
Понижение температуры борирования с 1000—1100° С до
920—950° С уменьшает хрупкость слоя, что благоприятно сказы-
вается и на его износостойкости. Поэтому детали обычно бори-
руют при 920—950° С.
Испытания, имитировавшие износ беговой дорожки цапфы,
показали, что через 60 мин уменьшение веса цементованных об-
разцов стали ЗОХГСА составляло 29 мг, а стали 12ХН2А 31,4 мг,
борированные же образцы этих сталей уменьшались в весе со-
ответственно на 17,8 и 16,0 мг, т. е. на 40—50% меньше, чем це-
ментованные [320].
Автор подверг испытаниям на износ (на машине Амслера при
трении скольжения без смазки и нагрузке 50 кГ) цементованные
секторы, работавшие в паре с кольцами из стали У12 [346]. Ис-
пытания показали, что потеря веса борированных секторов из
стали У12 примерно в 2 раза меньше, чем цементованных секто-
ров из стали ЗОХГТ. Если учесть, что на кромках борированных
образцов имелись мелкие отколы, различие в весе было еще бо-
лее значительным.
Хорошие результаты были получены при испытании бориро-
ванных вставок вводных пропусков последних клетей высоко-
производительных непрерывных проволочных станов [347].
Обычно такие вставки изготовляются из серого чугуна, так как
на стальные вставки налипает металл. Были испытаны вставки
из стали 40, гальванические хромированные (толщина покрытия
0,06—0,07 мм) и подвергнутые электролизному борированию на
глубину 0,15—0,20 мм при температуре 950° С, выдержке 6 ч,
плотности 20 а/дм2. Гальванически хромированные вставки пока-
зали стойкость в среднем 57 ч, а борированные 106 ч, что превы-
шает стойкость вставок из серого чугуна соответственно в 7 и
13,5 раз. При этом на хромированные вставки в местах износа
покрытия налипал металл, а рабочая поверхность борированных
вставок оставалась чистой.
В лабораторных условиях на специальной установке испыта-
на износостойкость борированных втулок грязевых насосов [348]
(рис. 157). Для испытаний были использованы модели втулок и
работающих с ними в паре резиновых поршней, уменьшенные в
33 раза по сравнению с натуральными. В качестве рабочей жид-
кости применялся глинистый раствор с кварцевым песком, пода-
вавшийся под давлением 15 ат (14,7• 10s н/м2). Кроме
241
борированных втулок из стали 40 (температура процесса 950° С,
выдержка 4 ч, плотность тока 0,5—0,9а/сл<2, глубина борированно-
го слоя 0,2 лш, микротвердость Нр. 1300—1600), испытанию под-
верглись втулки, изготовленные из других материалов: модифи-
цированного чугуна, закаленного и отпущенного (HV 400—460),
стали 40, улучшенной (HV 400—460), сталей ШХ15 и У10, под-
вергнутых закалке с нагревом т. в. ч., стали Ст. 3, армированной
бористым чугуном (HV 950), азотированной стали 38ХМЮА
(HV 940—1200), стали
40, гальванически хро-
. мированной (толщина
покрытия 135 мк).
Промышленные ис-
пытания втулок (диа-
метр 150 мм,
660 мм) в различных
районах нефтедобычи
показали, что бориро-
ванные втулки по из-
носостойкости в 3—3,5
раза превосходят се-
рийные втулки из ста-
ли 40, подвергаемые
Продолжительность испытаний
Рис. 157. Уменьшение веса моделей втулок,
работающих в паре с резиновым порош-
высота
ком в глиняном растворе:
/ — сталь 40 борированная; 2 — сталь 40, галь-
ванически хромированная; 3 — сталь 38ХМЮА,
азотированная; 4 — сталь, армированная бори-
стым чугуном; 5 и 6 — стали ШХ15 и У10, зака-
ленные с нагревом т. в. ч.; 7 — сталь 40, зака-
ленная; 8 — модифицированный чугун, закален-
ный и отпущенный [350]
закалке с нагревом
т. в. ч. Борирование
втулок буровых грязе-
вых насосов внедрено в
производство [349]. Хо-
рошие результаты бы-
ли получены также при
испытаниях сопел
струйных мельниц тон-
кого помола, звеньев
цепей пил для распиловки древесины и ряда других деталей
[350].
Борированный слой обладает высокой теплостойкостью; после
нагрева до 900—950° С его твердость не снижается. Борирование
немного увеличивает окалиностойкость (в 1,5—2 раза) и значи-
тельно повышает кислотостойкость стали.
На Ленинградском карбюраторном заводе создана полупро-
изводственная установка для газового борирования в среде
диборана и водорода внутренних поверхностей различных трубо-
проводов, а также наружных поверхностей вставок и плит пресс-
форм для литья под давлением. Борированный слой хорошо за-
щищает сталь от разрушения расплавом цинка. Удалось осуще-
ствить подачу расплава цинка по трубопроводам с борированной
внутренней поверхностью.
242
Насыщение стали бором совместно с другими элементами
Бороалитирование и боросилицирование позволяют получать
на деталях окалино- и кислотостойкие диффузионные слои с по-
вышенной твердостью. В некоторых случаях это может пред-
ставлять технический интерес.
Для одновременного насыщения стали 40 алюминием и бо-
ром (бороалитирование) применяли порошок алюмотермическо-
го ферробора (14,7% В и 15,2% А1) [351]. Образцы упаковывали
в порошок и через реакционное пространство пропускали НС1.
В течение 3,5 ч при 1000° С глубина бороалитированного слоя
достигала 0,30 мм, а поверхностная твердость HV 724 вместо
HV 392 после алитирования. Окалиностойкость боросилициро-
ванного слоя была высокой. Как показали испытания . про-
должительностью 100 ч, увеличение веса бороалитирован-
ных образцов при 900 и 1000° С составляло соответственно
2 и 5,1 г)м2-ч, а необработанных 80 и 221 г/м^-ч. В опытах, опи-
санных в работе [332], образцы стали Ст. 3 и ШХ15 последова-
тельно насыщались сначала бором, а затем алюминием, и наобо-
рот, а также одновременно этими элементами. Лучшую
окалиностойкость показали образцы, подвергнутые сначала бо-
рированию, а затем алитированию, и образцы, одновременно
насыщенные бором и алюминием. Более высокая микротвердость
(выше Ян 3600—3700) получена после борирования с последую-
щим алитированием.
Электролизное боросилицирование стальных образцов при
плотности тока 0,7 а/см2, температуре 1040° С и продолжитель-
ности процесса 30 мин осуществляли в расплаве Na2B4O7 и
Na2SiO3 [352]. При соотношении в этих соединениях > 0,86
происходило только борирование стали (в слое присутствовали
g
FeB и Fe2B), при—= 0,5 4-0,86— одновременное борирова-
О1
ние и силицирование (в слое присутствовали FeB, Fe2B и FeSi),
а при -“-<0,4— только силицирование (в слое присутствовали
FeSi и FeSi2). В такие расплавы рекомендуется вводить NaF, что
повышает жидкотекучесть расплава и предупреждает выброс
соли из ванны.
Процесс боросилицирования успешно протекает при 1000—
1050° С в порошкообразной смеси, состоящей из карбида бора
(84%) и буры (16%), к которым добавляется смесь кристалли-
ческого кремния (95%) и хлористого аммония (5%) [353]. По
мере увеличения содержания в смеси кремния глубина диффузи-
онного слоя уменьшается. По данным рентгеноструктурного ана-
лиза, диффузионный слой, полученный при боросилицировании,
состоит из боридов FeB и Fe2B и силицидов FeSi и Fe2Si. При
243
этом, если в смеси содержится до 25% Si, диффузионный слой
состоит преимущественно из боридов, при содержании в смеси
более 25% Si слой состоит в основном из силицидов. Боросили-
цированный слой обладает высокой твердостью и повышенной
окалиностойкостью [353].
Борирование высокохромистой
и аустенитной стали и сплавов
Борирование таких сталей и сплавов на никелевой основе при-
дает им высокую поверхностную твердость и износостойкость и
Таблица 37
Результаты борирования сталей и сплавов в расплаве,
содержащем 70% буры и 30% карбида бора
_____________________(автор и Г. Л. Дегтярева)_________________
Марка стали (сплава) Режим борирования Общая глу- бина слоя в лл*** Поверхностная твердость
Температура в °C Выдержка в ч ЯУ1 НУЪ
950 4 0,04 >1355 603
Х12Н2ВМФ 950 8 0,07 874
1100 4 0,20 1225
950 4 0,04 1100 752
Х18* 950 8 0,06 >1355 1027
1100 4 0,22 >1355 1253
950 4 0,05 1000 435
Х18Н9 950 8 0,07 >1355 603
1100 4 0,12 >1355 1314
950 4 0,05 376
Х11Н20ТЗ 950 8 0,06 >1355 623
1100 4 0,18 1225
950 4 0,06 1290 345
Х23Н18 950 8 0,10 >1355 689
1000 4 0,10 >1355 781
950 4 0,07 876
ХН60Ю 950 8 0,10 >1355 1027
1000 4 0,12 1072
950 4 0,05 655
ХН70Ю 950 8 0,10 >1355 923
1000 4 0,10 1145
* В стали Х18 среднее содержание углерода составляет 0,95%.
** Под общей глубиной слоя понимается суммарная глубина боридного и пере-
ходного слоев.
244
резко повышает коррозионную стойкость в некоторых агрессив-
ных средах. Борированный слой на высокохромистой и аустенит-
ной сталях имеет меньшую толщину и большую твердость и
хрупкость по сравнению с азотированным слоем. Однако повы-
шенная хрупкость не исключает возможности применения в тех-
нике борирования деталей из этих сталей.
В табл. 37 приведены результаты борирования различных
хромистых и хромоникелевых нержавеющих и аустенитных ста-
лей и жаропрочных сплавов. Борирование производилось в ра-
сплаве, состоявшем из 70% буры и 30% карбида бора. Скорость
борирования разных по составу сталей и сплавов на железной и
никелевой основах различна. Например, при борировании в тече-
ние 4 ч при 950° С глубина слоя колебалась от 0,04 до 0,07 мм,
а при 1100° С — от 0,12 до 0,22 мм.
Поверхностная твердость по Виккерсу при измерении с наг-
рузкой 5 кГ в большинстве случаев оказывалась заниженной в
результате частичного продавливания тонкого слоя, покоящего-
ся на мягкой сердцевине, а при измерении с нагрузкой 1 кГ
почти во всех случаях превышала HV 1355. Микротвердость, ко-
лебавшаяся в пределах Яр. 1700
нетравящегося боридного слоя
и резко падала в переходной
зоне (рис. 158).
Испытания борированных
образцов на осадку показали,
что хрупкость диффузионного
слоя после обработки при
950° С и выдержке 4 и 8 ч ни-
же, чем после обработки при
1000 и 1100° С и выдержке 2 и
4 ч.
Испытания износостойко-
сти борированных образцов на
машине Шкода-Савина (на-
грузка 5 кГ, скорость враще-
ния диска 675 об/мин) прово-
дились до углубления диска в
сталь на 0,02 мм. Различие в
необходимых для этого числах
оборотов диска при испыта-
ниях борированных и небори-
рованных образцов оказалось
при этом разительным: для
стали Х18 соответственно
15147 и 216 оборотов (в 70
раз), для стали Х12Н2ВМФ —
и для стали Х11Н80ТЗ— 15444
•2200, мало изменялась в зоне
Рис. il58. Распределение микро-
твердости по глубине борирован-
ного слоя (1000е С, 4 ч) стали
Х23Н18’ (штриховая линия) и
сплава ХН70Ю (сплошная ли-
ния) (автор и Г. Л. Дегтярева)
22 257 и 90 оборотов (в 247 раз)
и 81 оборотов (в 190 раз).
245
После борирования снижается окалиностойкость большин-
ства испытанных сталей и сплавов и повышается коррозионная
стойкость, в том числе в соляной кислоте при комнатной темпе-
ратуре и парах этой кислоты при 85° С (рис. 159).
Кинетика образования борированного слоя на высокохроми-
стых сталях и хромоникелевых аустенитных сталях отличается
от кинетики образования слоя на железе. После достижения пре-
дела насыщения на поверхности таких сталей наряду с насыщен-
Рис. 159. Влияние продолжительности испытаний на кислотостойкость в кон-
центрированной соляной кислоте при комнатной температуре (а) и в парах
соляной кислоты при 85° С (6) сталей и сплавов (автор и Г. Л. Дегтярева):
1 — борированные; 2 — неборированные. Обозначены: штриховой линией — сталь
Х23Н18; сплошной — сплав ХН70Ю; штрихпунктирной — сплав ХН60Ю.
и в объеме зерен. Аустенитно-боридная зона распространяется
на некоторую глубину, при этом на поверхности постепенно об-
разуется сплошная светлая зона боридов (рис. 160). Травимость
этой поверхностной зоны по глубине различна, что сог-
ласуется с данными рентгеноструктурного анализа [352], соглас-
но которым на хромистой стали с поверхности располагается бо-
рид (Fe, Сг)В и глубже (Fe, Сг)2В, а на аустенитной хромонике-
левой стали — с поверхности борид (Fe, Ni, Сг)В и глубже
(Fe, Ni, Сг)2В. На поверхности хромоникелевых сплавов, напри-
мер типа ХН80, образуются бориды (Сг, NO4B3 и (Сг, Ni)sB*.
Темная переходная зона слоя имеет различную протяженность
в зависимости от состава сплава и на аустенитной стали состоит
из смеси боридов и аустенита.
* По данным Н. И. Коспева, Н. Ф. Блок, Н. Ф. Лашко и К. П. Сорока.
246
Борирование аустенитной стали изучалось также в работе
[314], результаты которой в основном совпадают с результатами,
полученными автором. В этом исследовании обработке подвер-
гались стали Х18Н12МЗТ, X15H37BT3, Х18Н20С2, Х25Н20С2,
Рис. 160. Микроструктура борированных сталей и сплавов: (автор и
Г. Л. Дегтярева):
а — Х12Н2ВМФ (1100° С, 2 ч}\ б - Х18 (950° С, 8 ч); в — Х18Н9 (950° С, 8 ч); г —
Х11Н20ТЗ (950° С, 8 ч); д — ЗН70Ю (95CF С, 8 ч); е - ХН70Ю (1000° С, 4 ч) (Х400)
Х25Н25Т, а также стали 1Х18Н9 и Х23Н18, борирование кото-
рых изучалось и автором. В работе [315] на двух последних ста-
лях был получен борированный слой несколько большей глуби-
ны, что объясняется применением более активной газовой сре-
ды — смеси В2Н6 и Н2.
247
Поверхностная микротвердость всех исследованных в работе
[315] сталей колебалась в пределах Ну. 1890—2290.
ЦИНКОВАНИЕ
Цинкование применяется для повышения коррозионной стой-
кости стали в атмосфере и горячих газах (300—550°С), содер-
жащих сероводород.
К основным методам цинкования относятся: 1) горячее цин-
кование (погружение в расплавленный цинк); 2) цинкование в
порошке цинка; 3) цинкование в парах цинка; 4) металлизация;
5) электролитическое цинкование. Первые три метода цинкова-
ния являются химико-термическими, так как предусматривают
нагрев деталей, в результате которого образуется диффузион-
ный слой, представляющий собой сплав железа и цинка. Эти ме-
тоды обеспечивают более прочную связь поверхностного слоя
со сталью, чем два последних метода.
Методы и режимы цинкования
Горячее цинкование. Протравленные и промытые изделия по-
гружают через слой флюса в расплавленный цинк, нагретый до
430—460° С; продолжительность цинкования (от 10—20 сек до
нескольких минут) зависит главным образом от размеров дета-
лей; глубина цинкованного слоя 0,02—0,03 мм.
На Одесском канатно-проволочном заводе по предложению
инженера Золотухиной Н. С. в агрегате патентирования канат-
ной проволоки свинцовая изотермическая ванна была заменена
цинковой. В результате этого удалось не только увеличить ско-
рость прохождения проволоки через агрегат, но и отменить цин-
кование как самостоятельный дополнительный процесс. Продол-
жительность пребывания проволоки в изотермической ванне с
расплавом цинка при температуре 440°С 10—20 сек.
Цинкование в порошке цинка '. Этот метод позволяет получать
на деталях сложного профиля с глубокими сквозными отвер-
стиями, нарезкой, выточками и т. д. диффузионные слои хоро-
шего качества, равномерные по глубине и не имеющие следов
налипшего цинка. Слой, образующийся при обработке в порош-
ке цинка, имеет иной состав и строение, а следовательно, иные
свойства, чем слой, получаемый при «горячем цинковании».
Цинкование в порошке цинка выполняется в железных ящи-
ках и в печах с вращающейся ретортой [19], [354] — [357]. Пер-
вый из этих способов более прост, но менее производителен. Ис-
пользуемый для цинкования порошок (75—89% Zn, остальное —
окись цинка и примеси) является отходом производства, получа-
емым при огневом методе извлечения цинка из руд при дистил-
ляции цинка.
1 Этот процесс в зарубежной литературе часто называется шерардизацией.
248
Для цинкования в ящиках можно применять 100%-ный цин-
ковый порошок, но лучше использовать различные смеси.
П. П. Давыдов предложил применять порошок цинка, обрабо-
танный соляной кислотой. Для этого к порошку цинка добавляют
1,0% соляной кислоты, затем состав просушивают и дробят об-
разовавшиеся куски. Такой обработкой порошка достигается об-
разование некоторого количества хлористого цинка, ускоряюще-
го процесс цинкования. Кроме того, устраняется окисление дета-
лей и порошка при нагреве ящика. Рекомендуется использовать
порошок с размером частиц 0,1—0,25 мм. После каждого процес-
са цинкования порошок вновь обрабатывается тем же количе-
ством соляной кислоты, а после 10-кратного употребления до-
бавляют к нему 10% свежего порошка.
Предназначенные для цинкования детали, очищенные от
ржавчины и окалины дробе- или пескоструйной обработкой, ук-
ладывают в ящики, применяемые при алитировании или хроми-
ровании. Время выдержки при 380° С ограничивается 3—4 ч.
Глубина цинкованного слоя достигает при этом 0,03—0,04 мм.
Влияние продолжительности цинкования в порошке цинка
при 380° С на глубину цинкованного слоя приведено ниже (по
данным П. П. Давыдова):
Продолжительность цинкования вч1 2 3 5 8 10
Глубина слоя в мм ........... 0,020 0,024 0,032 0,043 0,050 0,070
Детали получаются серебристо-серого цвета с незначитель-
ной шероховатостью.
Иногда для цинкования применяют порошкообразную смесь,
состоящую из 75% цинка и 25% кварцевого песка, предотвра-
щающего спекание частиц цинка. Для уменьшения окисления
порошка цинка при нагреве рекомендуется также добавлять к
нему 2% порошка древесного угля.
По окончании цинкования детали промываются в воде и под-
вергаются пассивированию в ванне, содержащей 50 г СгОз и 4 г
H2SO4 или 150 г СгОз и 50 г NaCl. После пассивирования детали
промываются в холодной и горячей воде. Для повышения кор-
розионной стойкости оцинкованные детали рекомендуется под-
вергать фосфатированию или бесщелочному оксидированию.
Цинкование в порошке цинка во вращающихся ретортах, ши-
роко применяемое в США, как это видно из рис. 161, протекает
быстрее, чем в ящиках [354]. Это объясняется, очевидно, тем,
что к поверхности стали непрерывно поступает свежий порошок.
Процесс осуществляют при 370—440° С. Для определения дли-
тельности выдержки при 380—390° С принимают, что за 1 ч глу-
бина диффузионного слоя увеличивается на 0,015—0,020 мм.
Скорость вращения реторты рекомендуется поддерживать в пре-
делах 1—15 об/мин, причем каждая установка имеет свою опти-
мальную скорость вращения, при которой глубина слоя получа-
249
ется максимальной. Расход цинкового порошка на 1 м2 цинкуе-
мой поверхности составляет, по разным данным, 160—300 г.
Стоимость цинкования стали в порошке цинка в 2—2,5 раза ни-
же стоимости гальванического цинкования [354].
Цинкование в порошке цинка можно осуществлять в атмос-
фере водорода или аммиака. В случае использования аммиака
глубина цинкованного слоя за 4 ч при 400° С достигает 0,1 мм [281].
Процесс цинкования в смеси порошков цинка и окиси алюминия
ускоряется в атмосфере
Рис. 161. Влияние про-
должительности цинко-
вания при разных тем-
пературах на глубину
диффузионного слоя
низкоуглеродистой стали:
1 — цинкование во вра-
щающейся реторте; 2 —
цинкование в ящике [355]
хлористого водорода.
При содержании в смеси 50% оки-
си алюминия цинкование можно про-
изводить при температурах до 850° С.
При этом достигается большая глуби-
на диффузионного слоя и высокая кон-
центрация в нем цинка; в результате
повышается коррозионная стойкость
стали.
Цинкование в парах цинка. Этот
метод цинкования осуществляется в
вакууме или в восстановительных
средах.
Цинкование в вакууме при разре-
жении 10“2—10“3 мм рт. ст. (1,33—
0,13 нГм2) обеспечивает, по данным
работы [281], большую скорость про-
цесса и позволяет снизить температу-
ру обработки.
Цинкование в парах цинка при вы-
соких температурах было освоено на
заводе Лейна в ГДР [358], выпускаю-
щем длинные трубы (16 м) из низко-
углеродистой стали для регенераторов, необходимых в производ-
стве синтетического бензина. Трубы после пескоструйной обра-
ботки заполняются смесью, содержащей 78% цинковой пыли и
22% глинозема, и вставляются в каркас, вмещающий 200 труб,
на котором укрепляют корыта со смесью, состоящей из 60% цин-
ковой пыли и 40% глинозема. Каркас помещают в муфель печи
(длина 16,8 .м). Воздух из муфеля вытесняется азотом, который
при нагреве до 200—500° С постепенно заменяется водородом.
Процесс продолжается 48 ч при температуре 870° С и давлении
80 мм вод. ст. (785 н]м2). При такой температуре цинковый по-
рошок полностью испаряется. Глубина цинкованного слоя дости-
гает 0,15' мм. Менее ответственные детали цинкуются при
720—730° С.
Газовое цинкование осуществляли в потоке пропускаемого
через печь водорода. В первую зону печи (350—380° С) помеща-
ли фарфоровые лодочки с расплавленным хлористым цинком, а
250
во вторую (850—1050° С) —образцы. Была достигнута глубокая
диффузия цинка в сталь, но концентрация в слое цинка оказа-
лась недостаточной.
Структура цинкованного слоя
При цинковании протекает встречная диффузия железа и цин-
ка сквозь промежуточные фазы. Последовательность чередова-
ния гомогенных фаз в диффузионном слое соответствует изотер-
ме при данной температуре диаграммы состояния системы
Fe — Zn (рис. 162). Вначале образуется твердый раствор цинка в
a-железе (рис. 163), затем — узкая зона сравнительно твердой
Г-фазы (на базе соединения FesZnio, которому ранее приписыва-
лась формула Fe5Zn2i). Далее при продолжении диффузии цин-
ка в железо, а железа к поверхности, возникает следующая, бо-
лее бедная железом, сравнительно вязкая бгфаза (на базе сое-
динения FeZn7) и, наконец, хрупкая £-фаза (на базе FeZni3) и
вязкая т]-фаза (т] — твердый раствор железа в цинке, содержа-
щий тысячные доли процента железа, т. е. почти чистый
цинк)1.
Реакция между железом и расплавленным цинком изучалась
многими исследователями [2], [6], [18], [359]—[362], что выз-
вано как практическим (от фазового состава слоя зависит его
вязкость), так и теоретическим интересом, поскольку обнару-
1 На диаграмме состояния Fe—Zn эта фаза не указана.
251
жилось, что температурная зависимость скорости роста интерме-
таллических фаз, образующихся при цинковании железа в ра-
сплавленном цинке, имеет весьма своеобразную закономерность.
Скорость роста отдельных фаз и конечные структуры диффузи-
онного слоя определяются соотношением скорости процесса рас-
творения железа в жидком цинке и скорости встречной диффу-
зии железа и цинка сквозь промежуточные фазы. Установлено,
что ниже и выше интервала температур 490—520° С рост интер-
металлических фаз во времени происходит по параболическому
закону, а в указанном интервале температур диффузионный слой
растет с большой скоростью по
прямолинейному закону (рис. 164
и 165).
Рис. Л63. Изменение микро-
гвердости по глубине цин-
кового покрытия, образую-
щегося в расплавленном
цинке [18]
Рис. 164. Зависимость
глубины диффузионных сло-
ев от продолжительности
цинкования при разных
температурах [2]
Таким образом, при взаимодействии железа с жидким
цинком протекают реакции двух типов. Реакции первого типа
соответствуют росту фаз по параболическому закону, реакции
второго типа — росту фаз по прямолинейному закону. При ре-
акциях второго типа (490—520° С) зона слоя с Г-фазой не об-
разуется, а при реакциях первого типа образуется (фиг. 164,
165). По-видимому, аномалия в глубине слоя при температурах
около 500° С связана с тем, что скорость растворения железа
в цинке при этих температурах выше скорости образования
Г-фазы, в результате чего возникают лишь более богатые цин-
ком фазы [2], [18], [359].
Характерной чертой второго типа реакции железа с цинком
является образование на железе сетчатой структуры фазы
б' — FeZn7 (в более ранних работах б'-фазе приписывалась фор-
мула FeZnio). По промежуткам этой кристаллической структуры
жидкий цинк быстро достигает поверхности железа, где и про-
исходит реакция. Поэтому б'-фаза растет линейно, причем ско-
рость роста велика [18].
252
Наиболее хрупкой фазой, резко снижающей пластичность
диффузионного слоя, является g-фаза. Поэтому стремятся к то-
му, чтобы толщина g-фазы была невелика и слой состоял в ос-
новном из б'- и т]-фаз. Г-фаза более хрупка и тверда, чем б'-фа-
за, но ее толщина не превышает 0,004 мм. Г-фаза образуется
в самом начале процесса (быстрее, чем за 1 мин) и в дальней-
шем ее толщина практически не меняется (рис. 166, б).
Цинкование в расплаве цинка чаще осуществляется при
430—460° С. Увеличение выдержки и особенно повышение тем-
Рис. 165. Зависимость постоянных роста фаз и Г от температуры цинкова-
ния железа [6], [36]:
1 — убыль железа а • 108; 2 — линейная зависимость роста фаз от времени; 3 — содержа-
ние железа в брфазе а' • 108; 4 — содержание железа в Г-фазе а" • 10’°; 5 — Г-фаза от-
сутствует
пературы приводит к развитию зоны хрупкой g-фазы и уменьше-
нию толщины б'-фазы. С увеличением длительности цинкования
g-фаза вначале растет быстро, но затем скорость ее роста от-
стает от скорости роста б'-фазы. Это указывает на большую
скорость диффузии железа сквозь Г-7и б'-фазы, чем цинка сквозь
g-фазу.
Повышение в стали содержания углерода, фосфора и крем-
ния способствует развитию g-фазы, что ухудшает пластические
свойства диффузионного слоя. Для цинкования рекомендуется
кипящая бескремнистая сталь, так как присутствие даже 0,15—
0,20% Si (в спокойной раскисленной кремнистой стали) вызыва-
ет получение хрупких слоев [18]. Изменение характера взаимо-
действия железа с расплавленным цинком в зависимости от со-
держания в железе кремния показано на рис. 167. Кремний
253
расширяет температурную зону, в пределах которой растворение
железа протекает по прямолинейному закону. Однако при высо-
ком содержании в железе кремния эта зона полностью замыка-
ется.
При добавлении в цинковую ванну 0,05% А1 уменьшается
угар цинка, и поверхность покрытия становится более светлой.
При добавлении 0,15—0,20% А1 уменьшается толщина £- и
б'-фаз/и покрытие состоит в основном из rj-фазы (твердого рас-
Продолжительность
Рис. 166. Зависимость кинетики роста
фаз Г £ при кратковременной вы-
держке от температуры цинкования
(а) и продолжительности процесса
при 460° С (6) [18]
Рис. 167. Влияние содержания крем-
ния в расплаве цинка на характер
зависимости глубины диффузионно-
го слоя от продолжительности обра-
ботки [362]; обозначения:
# — параболическая зависимость;
+ — прямолинейная зависимость
твора железа в цинке), отделенной от железа тонкой промежу-
точной зоной (смесью фаз £ + т])- Это повышает вязкость диф-
фузионного слоя и исключает его отслаивание.
Механизм влияния алюминия на изменение строения слоя
окончательно не выяснен. Предполагается, что на поверх-
ности железа возникает какая-то тонкая пленка (возможно,
FeAl3), препятствующая диффузии железа в цинк и, следова-
тельно, тормозящая развитие 6'- и Г-фаз.
Термическая обработка цинкового покрытия почти не приме-
няется, однако она весьма увеличивает вязкость и толщину по-
крытия, что удлиняет срок службы оцинкованных деталей. Не-
достатком последующей термической обработки является ухуд-
шение вида поверхности (она становится тускло-матовой), но
254
такая поверхность очень хорошо воспринимает краску. Термиче-
скую обработку рекомендуется производить при 490—570° С с
Рис. 168. Схема изменения строения покрытия пос-
ле нагрева до 490° С в зависимости от времени вы-
держки [18]:
а — без отжига; 6—10 сек, в — 30 сек\ г — 180 сек
Рис. 169. Микроструктура стали 08 после пребывания в те-
чение 17 сек в расплавленном цинке при 480° С (цинкование
совмещено с охлаждением предварительно нагретой до 880° С
проволоки в изотермической среде при патентировании). Косой
шлиф (Х400) [Золотухина Н. С.]
выдержкой 30—10 сек; при этом возникает широкая зона б'-фа-
зы за счет рассасывания трфазы и хрупкой £-фазы (рис. 168).
255
Микроструктура цинкованного слоя проволоки, полученного
методом погружения в расплавленный цинк, совмещенным с про-
цессом патентирования, показана на рис. 169. Слой состоит из
железоцинковых сплавов с различным содержанием цинка и
разной микротвердостью. Самая поверхностная, наиболее мяг-
кая зона слоя (большие расплывчатые отпечатки пирамиды), со-
ответствует составу расплава. В середине слоя отчетливо видна
характерная эвтектическая структура; микротвердость этой зо-
ны несколько выше, чем
соседних. К сердцевине
стали примыкает узкая
почти не травящаяся зона,
наиболее бедная цинком,
наиболее твердая, имею-
Рис. 171. Микроструктура стали 08, об-
работанной в порошке цинка при тем-
пературе 380° С и выдержке 3 ч
(Х400)
Рис. 170. Изменение
микротвердости диффу-
зионного слоя в зависи-
мости от температуры
цинкования (продолжи-
тельность процесса 1 ч)
[354]:
/ — нагрузка 100 Г; 2 —
нагрузка 250 Г
щая столбчатое строение; ее можно предположительно отнести
к Г-фазе.
По данным П. П. Давыдова, при цинковании в порошке цин-
ка среднее содержание железа в поверхностном слое, получен-
ном при 380° С, равно 8—11%, а поверхностная твердость слоя
составляет HV 250—260. По другим данным [354], содержание
железа в цинкованном слое 8—10%, а иногда и ниже (до 6%).
При повышении температуры содержание железа в диффузион-
ном слое повышается до 18—28%, в результате чего коррозион-
ная стойкость слоя снижается. С повышением температуры нес-
колько снижается также и твердость слоя (рис. 170). Твердость
слоя, полученного в порошке цинка, в несколько раз выше, чем
слоя, полученного при горячем или гальваническом цинковании.
25 6
Потенциал поверхностного слоя стали, оцинкованной в порошке,
колеблется в пределах от —0,20 до —0,25 в, а потенциал цинка
равен —0,76 в [354].
Рентгеноструктурный анализ показал [354], что поверхност-
ная зона слоя представляет собой бгфазу с гексагональной ре-
шеткой. Эта фаза гомогенна в интервале 88,5—93% Zn. Зона,
примыкающая к сердцевине, является Г-фазой, которая гомо-
генна в интервале 75—78% Zn (или, по другим данным, 73—
80% Zn). Микроструктура слоя, полученного в порошке цинка,
приведена на рис. 171. Столбчатые зерна являются, очевидно,
61-фазой.
Данные о химическом и фазовом составе диффузионного слоя,
образующегося при высокой температуре в парах цинка, приве-
дены в работе (576].
Свойства цинкованной стали
Цинкование — один из эффективных методов защиты черных
металлов от коррозии. Цинковое покрытие защищает металл от
коррозии не только механически (как это имеет место при галь-
ваническом никелировании, лужении и т. д.), но и электрохими-
чески. Цинк является электроположительным по отношению к
железу, т. е. более активным металлом; поэтому, если под дейст-
вием коррозии или в результате механических повреждений по-
верхность оцинкованного железа обнажится, то в первую оче-
редь будет окисляться цинк, предохраняя железо от коррозии до
тех пор, пока не обнажится большая часть поверхности железа.
Цинковые покрытия имеют высокую коррозионную стойкость
в воздухе, в воде и в некоторых органических средах (бензине,
масле), но нестойки в кислотах и щелочах. Горячему цинкова-
нию (погружение в расплав цинка), помимо листов, труб, прово-
локи, различной посуды, подвергаются детали аппаратуры для
получения питьевой воды, спиртов, детали опреснительных уста-
новок, холодильников, газовых компрессоров и т. д.
Диффузионный слой, образовавшийся при цинковании в по-
рошке, в некоторых средах оказывается менее стойким против
коррозии, чем покрытие, полученное методом погружения; в дру-
гих средах соотношение в стойкости покрытий может быть об-
ратным. В частности, слой, полученный в порошке цинка, пока-
зывает при атмосферных испытаниях почти такие же хорошие
результаты, как и кадмированное покрытие (табл. 38). Сталь,
подвергнутая цинкованию в порошке, показывает также более
высокую коррозионную стойкость в морской воде, чем сталь,
обработанная горячим цинкованием. Диффузионное цинкование
хорошо защищает элементы конструкций нефтепромысловых
сооружений от коррозии в морской воде, во влажном воздухе и
грунтовых водах, содержащих сероводород [363], [365], [366].
257
Например, сваи нефтепромысловых сооружений в течение 4 лет
эксплуатации сохранили 80% цинкованного слоя.
Таблица 38
Коррозионная стойкость образцов стали 25 с различными покрытиями
(П. П. Давыдов)
Условия испытания Продол- житель- ность ис- пытания в ч Изменение веса образцов в г!м,*‘Ч*
без покрытия цинко- ванных в порошке цинка фосфати- рованных кадмиро- ванных
Камера водяного тумана 720 —0,0300 —0,0073 —0,0135 +0,0126
Воздушная среда . . . Переменное погружение в 3%-ный раствор хло- 2160 —0,0860 —0,0005 —0,0380 — 0,0006
ристого натрия . . . 720 —0,2033 —0,0300 — —0,0172
* После испытаний состояние поверхности цинкованных образцов было лучше, чем кадмированных и фосфатированных.
Диффузионное цинкование повышает коррозионно-усталост-
ную прочность стали. Так, коррозионно-усталостная прочность
нормализованной и затем цинкованной стали 35 в 3%-ном рас-
творе хлористого натрия такая же, как у незащищенной на воз-
духе; коррозионно-усталостная прочность стали 20ХН в грунто-
вой воде после цинкования в 2 раза выше, чем до цинкования.
Диффузионное цинкование в несколько раз повышает стой-
кость насосных штанг, работающих в скважинах в коррозионной
среде. Оно применяется для обработки деталей машин, прибо-
ров, электрооборудования, болтов и гаек нефтяного оборудова-
ния [363] — [366]. Большим преимуществом этого метода перед
горячим цинкованием является чистота поверхности и отсутствие
налипшего цинка.
Цинкованная любым способом сталь обладает повышенной
стойкостью в горячих газах, содержащих сероводород. Как из-
вестно, в такой среде происходит интенсивная коррозия стали,
и на ее поверхности образуется пористый рыхлый слой серни-
стого железа, обладающий большим объемом и малой тепло-
проводностью, в результате чего происходит «разбухание» по-
верхности, и детали быстро выходят из строя. В этих средах
стойкость цинковых покрытий, полученных разными способами,
ограничивается следующими предельными температурами:
300° С для горячего и гальванического цинкования; 400—500° С
для цинкования в порошке цинка (глубина слоя 0,05—0,06 мм);
550° С для цинкования в парах цинка (глубина слоя 0,15 мм).
Для повышения стойкости в серосодержащих газах целесо-
образно подвергать цинкованию не только углеродистые, но и
теплостойкие и высокохромистые окалиностойкие стали.
258
Диффузионное цинкование уменьшает склонность к выпаде-
нию сажи на поверхности стали при 400—450° С из углеводоро-
дов и других органических соединений.
Диффузионный слой, полученный в парах цинка на трубах
аппаратуры для синтеза бензина, не разрушается в течение 1 го-
да при работе в газовом потоке с температурой 535° С, содер-
жащем 10% сероводорода. Оцинкованные таким способом тру-
бы и другие детали широко применяются в химической про-
мышленности [358], например в регенераторах для производства
аммиака, метанола, капролактама и т. д.
Аустенитные хромоникелевые стали цинковать не следует,
так как цинк с никелем образуют хрупкие соединения.
СУЛЬФИДИРОВАНИЕ И СУЛЬФОЦИАНИРОВАНИЕ
Сульфоцианирование заключается в поверхностном насыще-
нии чугуна и стали одновременно азотом, серой и углеродом
обычно при температуре 560—580° С. Ранее этот процесс назы-
вали сульфидированием, но это название не соответствует его
сущности. Сульфоцианирование предложено во Франции в
1947 г. и с 1952 г. внедряется в промышленность многих стран
[367].
Для обработки деталей, отпускаемых при 160—200° С (ин-
струментов, цементованных и поверхностно закаленных деталей),
разработан другой процесс, заключающийся в том, что на по-
верхности деталей при температуре отпуска создается пленка
сульфида железа толщиной в несколько микрон. Этот процесс
следует называть сульфидированием.
Стальные и чугунные детали подвергаются сульфоцианиро-
ванию и сульфидированию для предотвращения схватывания и
наволакивания металла, улучшения противозадирных свойств,
ускорения приработки и повышения вследствие этого износостой-
кости. Сопротивление износу повышается не вследствие увели-
чения поверхностной твердости [368], а благодаря созданию на
поверхности пленки сульфида (с малым сопротивлением сдвигу),
предотвращающей непосредственный контакт между металличе-
скими поверхностями и играющей роль постоянной смазки, что
исключает задир и наволакивание металла. Пленка сульфида
железа, кроме того, способна адсорбировать кислород и другие
поверхностно-активные вещества, что благоприятно влияет на
понижение коэффициента трения.
Сульфоцианированными стальными деталями часто удается
заменить бронзовые детали, работающие на трение (подшипни-
ки и др.).
Интересно отметить, что сера также является хорошим про-
тивозадирным средством и поэтому часто добавляется в смазоч-
ные масла. Повышение серой противозадирных свойств объясня-
259
ется тем, что она имеет низкую температуру плавления (НО—
112° С) и аномальную зависимость вязкости от температуры.
Вязкость расплава серы резко возрастает с увеличением темпе-
ратуры до 187° С и лишь затем начинает медленно падать [369].
Поэтому при повышении давления и температуры сера не выдав-
ливается с поверхности трения, как, например, масло, вязкость
которого падает с увеличением температуры. Кроме того, при
трении расплавленная сера образует пленку сульфида железа.
Сульфоцианирование и сульфидирование дают эффект повы-
шения износостойкости лишь при определенных условиях — в
тех случаях, когда детали работают при трении со средними
нагрузками или с недостаточной смазкой, т. е. когда могут воз-
никнуть схватывание и задир металла.
Важным свойством стали после сульфоцианирования являет-
ся также повышение предела выносливости, что связано глав-
ным образом с поверхностным насыщением стали азотом.
Сульфидирование
Сульфидирование при высоких температурах. Первая работа
по насыщению железа серой была осуществлена в 1936 г.
(А. В. Брамлеем и др.). Для насыщения использовалась газо-
Рис. 172. Влияние температуры при выдержке 10 ч (а) и продолжительности
при 950° С (б) процесса на глубину слоя, насыщенного серой, у армко-желе-
за (кривая 1) и стали 45 (кривая 2), обработанных в порошкообразной сме-
си (40% S; 59% А12О3; 1% NH4C1) [автор и Б. Г. Гольдштейн]
образная смесь, состоявшая из 1% H2S и 99% Н2. При выдерж-
ке 40 ч, и температуре 1000° С на образцах железа образовался
обогащенный серой слой толщиной 0,3 мм, в поверхностной зо-
не которого содержалось 0,6% S.
В исследованиях, проведенных автором, для сульфидирова-
ния железа использовались порошкообразные смеси двух соста-
вов: 1) 40% S, 59% А12О3, 1% NH4C1; 2) 75% FeS, 20% А12О3,
5% NH4CI. Скорость процесса в обеих смесях была примерно
одинаковой (рис. 172), но состояние поверхности образцов ока-
260
зывалось лучше после обработки во второй смеси (гладкая, свет-
ло-матовая поверхность).
После обработки в этих смесях при 560—900° С концентра-
ция серы в диффузионном слое была незначительной, что не поз-
воляло выявить путем трав-
ления микрошлифа границу
слоя с сердцевиной. После
обработки во второй смеси
при 950° С диффузионный
Рис. 173. Микроструктура армко-желе-
за, сульфидированного при 950° С
в течение 10 ч (травление в смеси
растворов FeCl3 + HCI) Х315
[автор и Б. Г. Гольдштейн]
Рис. 174. Распределение
концентрации серы по глу-
бине слоя армко-железа,
сульфидированного в тече-
ние 10 ч при 950°С (автор
и Б. Г. Гольдштейн)
Рис. 175. Диаграмма состояния системы Fe — S
слой в результате травления шлифов смесью растворов FeCl и
НС1 выявлялся в виде столбчатых зерен (рис. 173). При этом
в слое обнаруживались круглые и продолговатые выделения
261
желто-коричневого цвета, располагающиеся преимущественно по
границам зерен. Эти включения, судя по их форме, хрупкости
и высокому содержанию серы в зоне столбчатых зерен, являются
сульфидами железа (рис. 174, 175).
Испытания износостойкости сульфидированных образцов
на машине Амслера >и испытания на задир на машине
ЦНИИТМАШа показали, что при ппимененных методиках испы-
таний сульфидирование снижает износостойкость и в то же время
резко повышает сопротивление задиру (табл. 39\.
Таблица 39
Результаты испытания сульфидированных образцов на износ
при трении скольжения без смазки на машине Амслера1
и на задир на машине ЦНИИТМАШа
(автор и Б. Г. Гольдштейн)
Материал образца Режим обработки Уменьше- ние веса сектора в г Нагрузка, при которой происходит задир, в кГ
Сталь А12 Несульфидированная Сульфидирование (750° С. 10 ч) Сульфидирование (850° С, 10 ч) Сульфидирование (950° С, 10 ч) 0,098 0,121 0,145 0,168 12 25 75 (задира нет)
Сталь 40Х Закалка с 850° С, отпуск 560° С Сульфидирование (850° С, 10 ч), закал- ка и отпуск при 560° С Сульфидирование (950° С, 10 ч), закал- ка и отпуск при 560° С 0,0437 0,0923 0,01976 15 25 75 (задира нет)
Серый чугун 1 Нагрузке Несульфидированный Сульфидирование (950° С, 10 ч) 25 кГ, время испытания 1 ч. — 18 75 (задира нет)
Сульфидирование в вакууме и расплаве серы. Для создания
на поверхности стали пленок из сульфидов разработаны два
способа: 1) насыщение из паров серы при разрежении
10-5 мм рт. ст. (0,0013 н]м2) и температуре 150—500° С [370] и
2) сульфидирование в расплаве серы при 120—140° С [371].
Последняя среда практически используется при обработке
железокерамических изделий, главным образом подшипников
скольжения для предотвращения наволакивания и задира ме-
талла (подшипники стартера автомобиля и т. д.). Образование
сульфидов железа происходит не в расплаве серы, а при допол-
нительном диффузионном отжиге.
Сначала железокерамические детали 'пропитываются в рас-
плаве серы при 130° С в течение 10—15 мин (детали толщиной
15—20 мм пропитываются при этом насквозь). Затем следуют
нагрев и выдержка при 600° С в воздушной среде, во время кото-
рых образуются сульфиды железа и выгорает избыток серы.
Заключительной операцией является пропитка деталей мас-
лом.
Сульфидирование инструмента и деталей при 180—200° С.
Для сульфидирования инструментов, цементованных и поверхно-
стно закаленных деталей Научно-исследовательский институт
технологии автомобильной промышленности (НИИТАвтопром)
рекомендует ванну, состоящую из роданистых солей калия и ам-
мония (90% KGNS и 10%
NH4CNS) [372], [373]; Научно-ис-
следовательский институт химиче-
ского машиностроения (НИИ-
ХИММАШ) — из 75% KCNS и
25% ИагЭгОз и Ростовский завод
сельскохозяйственных машин
(Ростсельмаш) — из 2 ч. KCNS
и 1 ч. Иа2$О4. Продолжитель-
ность обработки деталей в ван-
нах 45—60 мин при температуре
180—200° С. После такой обра-
ботки сера сосредоточивается в
поверхностном слое толщиной в
Рис. 176. Распределение серы по
глубине диффузионного слоя се-
рого чугуна (кривая /) и стали 45
(кривая 2), сульфидированных
при 180—220° С в течение 5 ч [374]
несколько микрон.
После сульфидирования при 180—200° С в течение 5ч в ван-
не, предложенной Ростсельмашем, содержание серы в поверхно-
стном слое толщиной 5—8 мк, определенное рентгеноспектраль-
ным методом, достигает 35—50% (рис. 176). Фазовый рентгено-
структурный анализ обнаружил на поверхности исследованных
образцов соединение FeS [374].
Несмотря на свою ничтожную толщину, сульфидная пленка
при определенных условиях работы увеличивает срок службы
деталей. Это, очевидно, объясняется регенерацией сульфидиро-
ванного слоя, а возможно, и тем, что отделяющиеся продукты
износа не сразу уносятся с рабочей поверхности.
НИИТАвтопром рекомендует подвергать низкотемпературно-
му сульфидированию режущий инструмент (метчики, сверла) из
сталей ХВГ и 9ХС, что исключает налипание на него цветных и
мягких черных металлов и повышает при определенных услови-
ях работы его стойкость до уровня стойкости инструмента из
стали Р9. Низкотемпературному сульфидированию при 180—
200° С в ванне НИИТАвтопрома иногда подвергают цементован-
ные зубчатые колеса, например, из сталей 12ХНЗА и 12Х2Н4А.
Эту операцию осуществляют вместо низкого отпуска или в
263
дополнение к нему. Сульфидирование облегчает приработку зуб-
чатых колес.
Низкотемпературное сульфидирование можно весьма эффек-
тивно осуществлять при нагреве в электролите (водный раствор
гипосульфита и нашатырного спирта) с небольшой (3—4. мин)
выдержкой по методу И. 3. Ясногородского. Испытания обрабо-
танных в электролите поршневых 'пальцев из стали 12ХНЗА и
образцов стали 20Х показали хорошие результаты.
Сульфидирование в ванне, состоящей из 3 ч. гипосульфита
и 2 ч. KCNS, при 220° С в течение 5 ч улучшает прирабатывае-
мость и повышает стойкость деталей и инструментов в условиях
больших давлений и полужидкостного трения, когда трению
скольжения сопутствует трение качения. При трении скольже-
ния, абразивном трении и резании, когда при износе отделяется
серосодержащий слой, сульфидирование не благоприятствует
повышению износостойкости трущихся пар.
Сульфидирование, так же как и сульфоцианирование, и фос-
фатирование, относится к группе процессов, при которых на
поверхности металла создаются слои химических соединений ме-
талла, играющих при трении роль постоянной смазки. К этой
группе процессов можно также отнести хлорирование стали (из-
вестно, что хлор, как и серу, часто вводят в смазочные масла).
Хлорирование стальных образцов осуществлялось при 200° С
в струе хлора, при этом на их поверхности образовывался
очень тонкий слой соединения FeCl2 [375]. Наличие на поверхно-
сти стали пленки хлорида обеспечивает повышение ее противо-
задирных свойств, улучшает приработку и в определенных ус-
ловиях повышает износостойкость трущихся пар. Однако процесс
хлорирования еще недостаточно изучен, и это не позволяет ре-
комендовать его для внедрения в производство.
Была сделана попытка доказать, что селенирование стали
(селен — аналог серы) дает примерно такой же эффект, что и
сульфоцианирование. Однако опубликованные данные не убе-
дительны, так как процесс осуществлялся в ванне неудачного со-
става, где сталь могла насыщаться не только селеном, но и (в
небольшом количестве) углеродом и азотом [375].
Сульфоцианирование
Процесс сульфоцианирования проводят при температуре
560—580° С в жидкой и газовой средах. В производстве применя-
ются лишь жидкие среды. За рубежом этот процесс осуществля-
ется в ваннах, в состав которых входят ядовитые цианистые со-
ли. При освоении процесса сульфоцианирования на предприятиях
отечественной промышленности вначале также шли по этому
пути.
В ванны для сульфоцианирования вводятся следующие соли:
264
1) NaCN, NaCNO, KON, KCNO, K4Fe(CN)6, обеспечивающие
цианирование; 2) NaCNS, KCNS, Na2S2O3, tNaSO4, Na2S, обеспе-
чивающие цианирование и одновременно насыщение серой или
только насыщение серой; 3) «нейтральные соли».
Соли, содержащие серу, не только насыщают металл серой,
но также ускоряют образование в ванне цианата и способствуют
увеличению глубины диффузионного слоя [376]—[380], [590].
Таблица 40
Составы ванн для сульфо цианирования (в %)
НИИТвто- НИИТАвто- ЛКЗ—ЛИВТ ЛИВТ-5 Чехо- слова- Фран- цуз-
пром. прома—МЗМА цкие ские
Компоненты
NaCN
(NH2)2CO
Na2S2O3
Na2SO3
Na2SO4
K2S, Na2S
K2CO3
KC1
K4Fe(CN)e
NaOH
S"
CN'
CNO'
CO3
so;
0,2—2
15—45
Oct.
50
5
25
20
2 12
75
13
7—13
20—25
Oct.
5
55
3
s
95
5
* ЛИВТ — Ленинградский институт водного транспорта.
♦♦ ЛКЗ — Ленинградский Кировский завод.
В табл. 40 приведены составы некоторых свежих и рабочих
ванн для сульфоцианирования. В рабочей ванне, содержащей
продукты реакций, количество цианистых и серосодержащих
солей значительно меньше. Интерес представляет ванна
НИИТАвтопрома [372], [373], в составе которой нет ядовитых
цианистых солей. В состав этой ванны входят К2СО3 и CON2H4,
смесь которых в несколько приемов вносится в железный тигель,
нагретый до 300—380° С. Сплавление сопровождается бурной
реакцией с образованием цианата калия, аммиака и двуокиси
углерода:
К2СО3 + 2CON2H4 = 2KCNO + 2NH3 + СО2 + Н2О.
Полученный расплав является высокопроцентным (95%) циа-
натом калия. Вследствие выделения во время реакции газооб-
265
разных продуктов количество полученного в ванне цианата калия
вдвое меньше количества исходных* веществ. Цианат калия не
ядовит и является активным источником насыщения стали азо-
том и углеродом:
2KCNO + О2 = СО + 2N + К2СО3.
В качестве серосодержащей соли в расплав цианата калия
при температуре 500—570° С вводится сульфид калия, в резуль-
тате чего в ванне образуется роданистый калий, обеспечивающий
процесс насыщения стали серой:
KCNO + K2S = KCNS + К2О;
KCNS + Fe + -у O2 = FeS 4- KCNO.
Сернистый натрий вводится через каждые 6—7 ч в количест-
ве 2% от общего веса соли. Для пополнения ванны цианатом ка-
лия в нее вводят только CON2H4, так как К2СО3 непрерывно об-
разуется и накапливается в ванне. Вводить CON2H4 в ванну нуж-
но при пониженной температуре (350—380° С) в количестве, в
два раза превышающем количество содержащегося в ванне
К2СО3. Однако целесообразно приготовлять цианат калия зара-
нее и добавлять в виде кусков, не снижая температуры ванны.
Оптимальный состав рабочей ванны; 30—90% KCNO и 0,2—
2,0% S.
Перед сульфоцианированием в ванне такого состава детали
обезжириваются в 5%-ном растворе NaOH или КОН при 70—
80° С в течение 2—3 мин, промываются в горячей воде и сушатся.
Процесс сульфоцианирования осуществляется при 560—580° С в
течение 1,5—2 ч; наблюдаемая под микроскопом глубина слоя
достигает при этом 0,05—0,10 мм. По окончании процесса детали
охлаждаются на воздухе, промываются в горячей воде, сушатся
и промасливаются.
Для сульфоазотирования опробована газовая смесь NH3 +
+ H2S [380], [382], а для сульфоцианирования — смеси NH3 +CS2
и NH3 + коллоидный раствор серы в масле [383].
В Китае для обработки чугуна и стали при температурах
700° С и выше рекомендовалась порошкообразная смесь следую-
щего состава: 75% сернокислого железа, 5% NH4CI и 20% AI2O3.
На некоторых отечественных заводах были опробованы порош-
кообразные смеси: например, 35% FeS, 55% графита, 10% K-iFe
(CN)6 [384], которые вряд ли следует рекомендовать.
Опробовано одновременное серонасыщение и цементация ста-
ли при 920° С в стандартном карбюризаторе, к которому добав-
лялись различные соли, содержащие серу. После такой обработ-
ки содержание серы в поверхностной зоне слоя достигало 0,1 —
1,0%, глубина цементованного слоя немного уменьшалась, а про-
тивозадирные свойства стали повышались [385].
266
Рис. 177. Изменение содер-
жания серы и азота по
глубине диффузионного
слоя стали, сульфоцианиро-
ванной при 570° С в тече-
ние 2 ч [376]
Структура сульфоцианированного слоя. Сульфоцианирован-
ный слой обогащен азотом, серой и углеродом. После сульфо-
цианирования при температуре 570° С и выдержке 2 ч содержа-
ние серы в поверхностном слое толщиной 0,001 мм выше 5%,
а азота и углерода в слое толщиной 0,005 мм соответственно
более 2,0 и более 2,2%. Азот может проникать на глубину
0,6 мм, а сера на глубину не более 0,15—0,16 мм (рис. 177).
Сульфиды железа обнаруживаются рентгеноструктурным
анализом на глубине не более 0,03—0,04 мм от поверхности.
Углерод сосредоточивается в самой поверхностной зоне слоя
(очевидно, только в зоне карбонит-
ридов, как и при низкотемператур-
ном цианировании).
После обработки чугуна при
570° С в течение 3 ч в ванне НИИТ-
Автопрома содержание азота в по-
верхностном слое толщиной 0,05 мм
достигало в некоторых случаях
2,0—3,8%, а серы—1,5—2,9%.
, Микроструктура сульфоцианиро-
ванного слоя стали почти аналогич-
на микроструктуре слоя, полученно-
ного в результате цианирования в
цианистых солях при 560—590° С.
На поверхности стали наблюдается
тонкая (5—10 мк) слабо травящая-
ся каемка, в состав которой, однако,
входят не только карбонитриды, но
и сульфиды. По данным ряда авто-
ров [379], в этой зоне очень тонкий слой сульфидов находится
над карбонитридным слоем. Кроме того, сульфиды в виде мел-
ких включений вкраплены непосредственно в карбонитридный
слой. Глубже этой зоны расположена зона толщиной 50—100'мк,
состоящая из тех же составляющих, что и обычный цианирован-
ный слой — из эвтектоидной смеси а + у' (в железе). Еще глуб-
же залегает зона, немного обогащенная азотом (иглы у'-фазы на
фоне a-твердого раствора). В случае обработки при пониженной
температуре, как и при обычном цианировании, зона эвтектоида
отсутствует. •
Характер структуры и фазовый состав поверхностной зоны
сульфоцианированного слоя может сильно изменяться в зависи-
мости от состава ванны [385]—[387]. Исследования показали,
что рекомендуемые и применяемые на отечественных заводах,
а также за рубежом ванны можно разделить на следующие
группы:
1. Ванны, в основном сульфидирующие сталь,— в поверхно-
стной зоне слоя обнаруживаются FeS, или FeS и a-Fe, или FeS
и Fe3O4.
267
2. Ванны, обеспечивающие одновременное сульфидирова-
ние и цианирование (ванны, составы которых приведены в
табл. 40). Обработка в таких ваннах дает максимальный
эффект.
3. Ванны, в основном азотирующие сталь,— в слое содержат-
ся Fe2(N,C), Fe4(N, С).
4. Ванны, в основном интенсивно науглероживающие сталь,—
в слое образуется Рез (С, N).
После обработки образцов стали Ст. 3 в ванне состава 75%
K4Fe(CN)6, 13% NaOH и 12% Na2S2O3 при 580° С в течение 1 ч
в поверхностной зоне слоя было обнаружено одновременно шесть
фаз: FeS, Fe3C, Fe2N, Fe3N, Fe4N и a-Fe [387].
Сказанное выше показывает, что некоторые из применяемых
и рекомендуемых ванн для сульфоцианирования .не обеспечива-
ют получения диффузионного слоя с необходимым фазовым со-
ставом, а следовательно, и необходимыми свойствами.
Свойства и применение сульфоцианированной стали. Микро-
твердость различных зон диффузионного слоя сульфоцианирован-
ной низкоуглеродистой стали полностью соответствует их струк-
туре: микротвердость карбонитридной зоны около 550—600,
а зоны феррита с иглами нитридов около 450 [387].
Сульфоцианирование несколько повышает пределы прочно-
сти и текучести стали, значительно повышает предел ее выносли-
вости и немного снижает относительное удлинение и сужение.
Предел выносливости стали Ст. 3 повышается в результате суль-
фоцианирования на 37% [387].
По данным зарубежной литературы, предел выносливости
коленчатых валов из улучшаемой хромоникелевой стали повы-
шается более чем на 50%.
За рубежом широко применяется обработка стали в обычных
цианистых ваннах при 560—580° С в течение 1—2 ч, известная
под названием мягкого азотирования (см. стр. 147). После мяг-
кого азотирования, как и после сульфоцианирования, повышают-
ся износостойкость и предел выносливости, однако сопротивле-
ние схватыванию, наволакиванию металла, противозадирные
свойства повышаются в значительно меньшей степени, чем после
сульфоцианирования. В этом и заключается различие между
сульфоцианированием и мягким азотированием.
Основные свойства, которые приобретает сталь после суль-
фоцианирования, сохраняются даже тогда, когда величина из-
носа превышает глубину сульфоцианированного слоя. При тре-
нии с большими нагрузками в микрообъемах металла создаются
большие давления и происходит нагрев до высокой температу-
ры поверхностного тонкого слоя, что, вероятно, способствует
диффузии серы и, следовательно, непрерывному перемещению
сульфоцианированного слоя в глубь стали. Это было многократ-
но доказано, в том числе с помощью радиоактивной серы [368].
268
Более высокая износостойкость сульфоцианированного чугу-
на по сравнению с цианированным и сульфидированным иллю-
стрируется кривыми рис. 178.
Сульфоцианирование широко применяется во многих странах.
В Чехословакии этот процесс используется более чем на 50 за-
водах [381] для обработки поршневых колец, гильз цилиндров,
чугунных втулок и вкладышей
различного назначения, калибро-
вочных пуансонов из быстроре-
жущей стали, направляющих без-
центровошлифовальных станков,
подшипников коленчатого вала
двигателя, гаек ходовых винтов
станков, инструментов из быстро-
режущей стали и т. д. Во Фран-
ции, кроме указанных деталей,
сульфоцианированию подвергают
толкатели клапана, зубчатые и
червячные колеса, различные де-
тали из чугуна, применяемые вза-
мен бронзовых, а также детали,
работающие без трения, но пери-
одически собираемые и разбирае-
мые с усилием, например гайки
крепления крышек турбин и дизе-
Продолжительность исоытания
Рис. 178. Износ пары сталь 45 —
чугун СЧ 21-40 после различных
видов химико-термической обра-
ботки при температуре 580° С (ис-
пытание без смазки, давление
25 кГ/см2} [387]:
1 — необработанная сталь; 2 — еулъ-
филированная; 3 — цианированная;
4 — сульфоцианированная
лей для предотвращения их схватывания. Во Франции и Англии
сульфоцианированию подвергают детали самолетов, реактивных
двигателей и ядерных установок.
В СССР сульфоцианирование применяется на ряде заводов.
Обрабатываются детали насосов паровых машин и судовых дви-
гателей, детали станочного оборудования (ранее часть этих дета-
лей изготовляли из бронзы) [387], упорные чугунные фланцы ав-
томобиля «Москвич» (ранее изготовлялись из бронзы), фрик-
ционные диски коробки передач трактора «Кировец» (ванна
ЛКЗ— ЛИВТ, табл. 40) и другие детали.
СИЛИЦИРОВАНИЕ
Детали, главным образом аппаратуры для химической про-
мышленности, изготовляются из специальных кислотостойких
железокремнистых сплавов — ферросилицидов (0,3—0,8% С и
14,5—18% Si), обладающих высокой коррозионной стойкостью
при разных температурах в серной, соляной и азотной кислотах
различной концентрации. Однако применение этих сплавов
ограничено, так как при литье из них деталей получается много
брака, их механические свойства ниже, чем у серых чугунов, они
хрупки, не поддаются ковке и с трудом, а чаще совсем не обраба-
269
тываются режущим инструментом. Кроме того, эти сплавы склон-
ны к трещинообразованию при колебаниях температуры.
В связи с этим в последние годы некоторое распространение
получило силицирование (насыщение кремнием). При этом де-
талям из низкоуглеродистой стали предварительно придается лю-
бая форма, после чего они силицируются. Силицированные дета-
ли из низкоуглеродистой стали обладают достаточно высокой
кислотостойкостью. Детали из срёднеуглеродистой стали после
силицирования можно подвергать термической обработке, при
этом в диффузионном слое не образуются трещины, а кислото-
стойкость стали заметно не изменяется. Иногда силицируют от-
ливки из ковкого чугуна и высокопрочного чугуна с шаровидным
графитом [388].
Методы и режимы силицирования
Разработаны методы силицирования стали и чугуна в порош-
кообразных смесях, в жидкой и газовой средах. Силицирование
в порошкообразных смесях и жидкой среде применяется редко,
газовое силицирование — чаще.
Процесс силицирования осуществляют при температуре
950—1000° С. Глубина силицированного слоя обычно лежит в
пределах 0,3—1,0 мм.
Силицирование в порошкообразных смесях. Результаты си-
лицирования в порошкообразных смесях, по данным разных ав-
торов, приведены в табл. 41.
Таблица 41
Глубина силицированного слоя на образцах стали 10
в зависимости от режима силицирования [19]
Состав порошка Режим силицирования Глубина слоя
Темпера- тура в °C Время в ч в мм
Ферросилиций (около 60% Si) 1100 1100 9 18 0,08 0,16
Ферросилиций (80%), шамот (20%) , . . 1100 1100 1200 1200 2 12 2 12 0,02—0,05 0,20—0,23 0,33—0,50 0,60—0,80
Ферросилиций (75%), шамот (20%) и 5% NH4C1 1200 10 0,88—0,90
Ферросилиций (80%), шамот (15%) и 5% | 1050 1 6 1 0,09 NH4C1 [389J 1 1100 1 6 • 1 0,15 Примечания: 1. Вместо шамота в смесь может вводиться А12О8. 2. При силицировании сплавов на нежелезной основе ферросилиций заме- няется порошком кремния.
270
Силицирование в жидких средах. Силицирование этим спосо-
бом может осуществляться в расплаве, состоящем из 50% ВаС12
и 50% NaCl, в который вводится 15—20% ферросилиция (70—
90% Si) с величиной частиц 0,3—0,6 мм. В такой ванне при
1000° С в течение 2 ч глубина слоя на образцах стали 10 дости-
гает 0,35 мм [390]. Примерно в такой же по составу ванне сили-
цировали железные листы при температуре 1000—1050° С и вы-
держке 3,5 ч. По окончании процесса листы охлаждались в воде
для очистки поверхности от соли.
Для электролизного силицирования опробована ванна, состоя-
щая из смеси Na2SiO3 и NaF. Применялась ванна, составленная
из 3 ч. Na2SiO3 и 4,7 ч. NaF, в которой процесс протекал при
температуре 1000—1150° С и плотности тока 0,3—0,6 ajcM2. Изме-
нение глубины слоя в зависимости от температуры и выдержки
подчинялось при этом зависимости [391], [392]
170 000
г/2 = 7,5 • 10~4е RT -г,
где у — глубина слоя; х — продолжительность процесса.
В ванну, составленную из 2 ч. Na2SiO3 и 3 ч. NaF, вводили до-
полнительно Na2B4O7 [353]. В зависимости от соотношения этих
компонентов в расплаве (при температуре около 1000° С и плот-
ности тока 0,7 а) см2) происходило или совместное насыщение
в
стали кремнием и бором (0,5 <— < 0,86), или насыщение толь-
01
ко кремнием либо бором (—>0,86^.
Газовое силицирование. Опыты по газовому силицированию
в СССР были начаты еще в 1931 г. [19]. Через несколько лет пос-
ле этого появились первые упоминания о применении газового
силицирования в зарубежной промышленности [352].
Газовое силицирование осуществляется в различных средах
и различными способами:
I. При непрерывном пропускании хлора через реакционное
пространство с деталями и ферросилицием (или карбидом крем-
ния) либо только при заполнении этого пространства хлором
(непрерывный поток отсутствует) [393].
2. С использованием вместо хлора хлористого водорода [19],
[394].
3. В смеси Н* + SiCl4 или N2 + Н2 + SiCl4 [395].
4. В смеси Аг + SiCl4 или N2 + SiCl4 (в присутствии Si) [396],
[397].
5. В смеси Н2 + SiH4 (моносилан), NH3 + SiH4, Ar + SiH4.
* Водород или другой несущий газ пропускается через сосуд с жид
ким SiCl4.
271
’ В производстве чаще применяется силицирование в потоке
хлора.
Газовое силицирование с применением хлора производится в
герметически закрытых муфелях или, чаще, в печах с вращаю-
щимися ретортами, аналогичных печам, применяемым для газо-
вой цементации. В реторту печи загружают детали и карбид
кремния или ферросилиций (либо смесь того и другого). Можно
также применять более дешевое вещество — аморфный карбид
кремния (силок-сикон) —побочный продукт, получаемый при
производстве кристаллического карбида кремния и содержащий
«) 6)
Рис. 179. Зависимость глубины диффузионного слоя от продолжительности (а)
и температуры (б) газового силицирования в среде хлора, пропускаемого над
порошком ферросилиция (М. Г, Буцик)
около 73% SiC, 13% SiOj, 7% Си другие примеси. Вес загружа-
емых в реторту карбида кремния, ферросилиция или силоксико-
на составляет примерно ’/ю веса обрабатываемых деталей. Ре-
торта при загрузке заполняется на 75—80% объема.
Поковки или отливки могут поступать на силицирование без
предварительной очистки, так как тонкая окалина не оказывает
существенного влияния на глубину силицирования.
Садка нагревается до температуры 950—1050° С (чаще до
980° С), после чего в реторту по трубке подают из баллона хлор
со скоростью, зависящей от размера реторты. Например, при си-
лицировании в реторте диаметром 250 мм и длиной зоны нагре-
ва 1,5 м расходуется 0,9 кг/ч жидкого хлора. Давление в реторте
поддерживают в пределах 10—50 мм вод. ст. (98—490 н/м'2).
Для того чтобы глубина силицированного слоя на всех деталях
была равномерной, по истечении половины времени выдержки
направление потока хлора в реторте изменяют. Через каждые
30 мин отверстие для отвода газов очищают прутком от конден-
сата хлоридов железа и кремния.
Охлаждают детали вместе с печью до 100—200° С в потоке
272
хлора, что предотвращает окисление деталей, ферросилиция и
карбида кремния. Затем детали кипятятся в воде для удаления с
поверхности следов хлорида кремния. Вместо хлора можно при-
менять хлористый водород (НС1), но экономически это менее
целесообразно.
Содержание кремния в ферросилиции существенно влияет на
глубину диффузии кремния. Следует применять высококремни-
стый ферросилиций (марок Си 75 и Си 90 по ГОСТу 1415—49).
Влияние температуры и времени выдержки на глубину силици-
рованного слоя приведено на рис. 179. Образцы засыпались 60%-
ным ферросилицием, через печь пропускался хлор. Из сравнения
данных рис. 179 и табл. 41 видно, что газовое силицирование про-
текает быстрее, чем силицирование в порошкообразных
смесях.
Засыпка деталей ферросилицием или карбидом кремния не
является обязательной при газовом силицировании. Он может
помещаться в невращающейся реторте со стороны ввода хлора,
а детали — в середине реторты и со стороны выхода из нее газа.
При этом глубина силицированного слоя получается меньше, не-
равномерность диффузионного слоя на разных деталях больше,
но зато поверхность деталей чище, чем после засыпки деталей
порошком ферросилиция или карбида кремния.
Таблица 42
Глубина силицированного слоя при газовом силицировании
в зависимости от условий процесса [19]
Материал для силицирования Газ, подаваемый в муфель Глубина слоя в мм
Образцы только омываются газом Образцы засыпаны порошком
Ферросилиций (60% Si) Хлор 0,53 1,38
Карбид кремния 0,51 1,07
Ферросилиций (60% Si) Хлористый 0,59 1,12
Карбид кремния водород 0,62 0,95
Примечание. Силицирование ос уществлялось в трубчатой печи с внут-
ренним диаметром 80 мм и длиной 750 мм-, режим силицирования: температура 980° С, выдержка 2 ч.
В табл. 42 приведены данные о глубине силицированного слоя
в зависимости от условий силицирования. Лучшие результаты с
точки зрения глубины силицирования были достигнуты при за-
сыпке образцов ферросилицием. Поверхность стали при силици-
ровании с карбидом кремния получаемся более чистой и ровной,
поэтому иногда его предпочитают более дешевому ферроси-
лицию.
273
При силицировании следует всегда опасаться разъедания по-
верхности стали избытком хлора. Избытка хлора в рабочем про-
странстве следует избегать даже при силицировании без непре-
рывной подачи хлора [393]. Хорошие результаты получаются
лишь при концентрации хлора 1—2 мл на 1 см2 обрабатываемой
поверхности. При большей концентрации хлора наблюдается
разрушение поверхности деталей. При таком методе силициро-
вания после нагрева в рабочем пространстве-реторты возникает
разрежение, образующееся вследствие протекающих там ре-
акций.
В последние годы появился ряд работ, в которых обсуждает-
ся химизм процесса газового силицирования стали [396]—[399].
При большинстве способов газового силицирования в качест-
ве активного кремнесодержащего газа используется SiCl4, кото-
рый или образуется при взаимодействии хлора с ферросилицием
либо карбидом кремния, или переносится в рабочее пространство
несущим газом (Н2, Аг), пропускаемым через сосуд с жидким
SiCl4.
В последних работах отвергается прежнее представление о
химизме процесса силицирования, согласно которому этот про-
цесс протекает только благодаря обменной реакции между хло-
ристым кремнием и железом:
2Fe + SiCl4 = 2FeCl2 + Si.
Возражения против такого представления о химизме процес-
са базируются как на термодинамических расчетах, так, в част-
ности, и на следующих соображениях:
1. Восстановительные свойства железа по отношению к
SiCl4 слабы.
2. При температурах около 1000° С SiCl4 энергично взаимо-
действует с кремнием, образуя субхлорид кремния SiCl2.
3. При силицировании в смеси N2 + SiCl4 или Аг + SiCl4 на-
сыщение поверхности протекает с очень небольшой скоростью, а
вес образцов резко уменьшается, очевидно, вследствие реакции
SiCl4 + Fe = FeCl2 + SiCl2.
4. При пропускании указанных смесей через зону печи с рас-
каленным Si (Fe — Si) силицированный слой образуется, а по-
тери веса образцов уменьшаются.
5. Силицирование в среде Н2 + SiCl4 позволяет получать бо-
лее глубокие диффузионные слои, вес образцов при этом уве-
личивается.
На основании изложенного, а также результатов некоторых
экспериментов химизм процесса газового силицирования следует
представлять себе, очевидно, следующим образом.
274
При силицировании с использованием хлора или в смеси
Ar + SiCl4 (при наличии кремния в рабочем пространстве) вна-
чале протекает реакция
SiCl4 -I- Si = 2SiCl2-
Далее SiCl2 и непрореагировавший остаток SiCl4 могут участ-
вовать в следующих реакциях:
SiCl2 + Fe = Si + FeCl2;
2SiCl2 = Si 4- SiCl4;
SiCl4+ Fe = SiCl2 + FeCl2.
В зависимости от температуры и других факторов развивает-
ся в большей степени та или иная из этих реакций. При этом
первая и третья реакции, несомненно, имеют место, так как толь-
ко ими и можно объяснить уменьшение веса образцов при сили-
цировании в средах N2 + SiCl4 и Аг + SiCl4 (при наличии крем-
ния). Если кремний отсутствует, в этих средах, очевидно, проте-
кает в основном только реакция
SiCl4 + Fe = FeCl2 + SiCl2;
при этом сталь почти не насыщается кремнием и уменьшается
вес образцов.
При силицировании в смеси Н2 + SiCl4 протекают реакции
SiCl4 + 2Н2 = Si + 4НС1;
SiCl4 + Н2 = SiCl2 + 2НС1
и реакция
2SiCl2 = Si + SiCl4;
при этом вес образцов увеличивается.
По мнению авторов работы [399], при обработке в тетрахло-
риде кремния (при отсутствии кремния в рабочем пространстве)
некоторое количество кремния может поступать к образцу как
вследствие реакции обмена .
SiCl4 + 2Fe = Si + 2FeCl2,
так и непосредственно за счет восстановления кремния атомар-
ным водородом, растворенным в железе. Наибольшее количество
кремния поглощается сталью при силицировании в среде S1C14 +
+ Н2 при наличии в рабочем объеме кремния, меньшее — в сре-
де SiCl4 + Н2 и наименьшее — в среде SiCl4 + Аг при наличии
в рабочем объеме кремния.
Силицирование в среде моносилана (SiH4) предложено
Ю. М. Лахтиным и Т. М. Иващенко. Моносилан получают в спе-
циальной установке путем диспропорционирования триэтаксиси-
лана [SiH(OC2H5)3] в присутствии металлического натрия.
18* 275
Образующийся моносилан смешивают с водородом и подают
в печь для силицирования, а затем во вторую печь для оконча-
тельного разложения остатков моносилана (SiH4 = Si + 2Н2).
Силицирование сопровождается заметным изменением веса
и размеров стальных образцов. В зависимости от условий про-
цесса — ферросилиций или карбид кремния, поток хлора или
несущий газ (Н2, Аг или N2) в смеси с SiCl4— образуется раз-
личное количество хлористого железа, что влияет на вес и раз-
меры образцов. Из-за большого удельного веса силицированного
слоя размеры образцов всегда увеличиваются; вес образцов,
как уже отмечалось, может и увеличиваться и уменьшаться
(-абл. 43).
Таблица 43
Изменение веса и размеров образцов стали 10
после силицирования на глубину около 1,2 мм [19]
Материал для силицирования Изменение
веса в г диаметра в мм длины в мм
Ферросилиций Карбид кремния +0,68 —0,64 + 1,10 +0,60 +2,50 +0,50
Примечание. Исходный вес образцов около метр 10 мм. Образцы засыпались карбидом кремния печь пропускался хлор. 10,5 г, длина или ферросилг 20 мм, диа- щием, через
Структура и свойства силицированной стали
В поверхностной зоне силицированного слоя содержится до
13—15% Si, при этом концентрация кремния по глубине слоя
вначале изменяется мало, а затем резко снижается (рис. 180).
Углерод тормозит диффузию кремния в сталь (рис. 181), в ре-
зультате чего концентрация кремния в силицированном слое и
твердость слоя повышаются [389], [400].
Были проведены эксперименты, в процессе которых выявля-
лось влияние хрома на характеристики силицированного слоя
сталей 20, 20Х, Х6 и XI3. Оказалось, что хром несколько сни-
жает глубину силицированного слоя, но одновременно увеличи-
вает его плотность, что положительно сказывается на коррози-
онной стойкости стали [401].
Силицированный слой обычно не травится и представляет со-
бой раствор кремния в a-железе (рис. 182—183). Иногда удает-
ся выявить столбчатую структуру слоя, что объясняется превра-
щением у->а при температуре диффузии. Иногда слой состоит
из двух фаз (рис. 184). Снаружи образуется слой, соответствую-
щий упорядоченному соединению Fe3Si (а7)» а Далее — твердый
раствор кремния в а-железе [398].
276
При диффузии в сталь кремний оттесняет углерод от поверх-
ности, что объясняется превращением у—>а, протекающим в
слое при температуре на-
сыщения. В связи с этим под
силицированным слоем от-
четливо наблюдается зона,
обогащенная углеродом
(рис. 182). На границах
столбчатых зерен твердого
раствора кремния в железе и
на границах зерен с сердце-
виной обнаружены тонкие
прожилки цементита [396],
[397]. При сквозном силици-
ровании тонких образцов
низкоуглеродистой стали со-
Рис. 180. Изменение содержания
кремния и микротвердости (нагруз-
ка 20 Г) по глубине силицированно-
го слоя армко-железа [389]:
I — силицирование при 1000° С, 2 ч;
II — при 1050° С, 6ч;/ — микротвер-
дость; 2 — содержание кремния по дан-
ным химического анализа; 3 — расчетное
содержание кремния
держание углерода в серд-
цевине резко увеличивается,
что приводит к образованию
крупных выделений цемен-
тита.
Силицированный слой
часто бывает пористым, что
зависит от условий силици-
рования и состава стали. Ранее пористость слоя пытались
связывать с возможностью протекания в нем процесса графити-
Рис. 181. Влияние содержания углерода в стали на глубину силицированного
слоя:
а — обработка в порошкообразной смеси (80% FeSi, 15°/о шамота и 5% NH4C1):
/ — при 1050° С, 6 ч; 2 — при 1100° С, 6 ч; 3 — при 1100° С, 12 ч [389]; б — обработка
в газовой среде (образцы расположены отдельно от ферросилиция): / — в потоке НС1
при 1000° С, 2 ч; 2 — в среде С1г при 1000° С, 2 ч [393]
зации, но это не подтвердилось. Предполагали также, что хлори-
ды кремния проникают по границам зерен и микроканалам через
слой Fe3Si к границе сердцевина — слой и вызывают образова-
277
ние хлоридов железа, что и приводит к возникновению пор и ка-
налов в структуре. В более поздних работах [402] было сделано
предположение, что пористость образуется в результате различ-
ной скорости диффузии движущихся в противоположных направ-
лениях атомов кремния (к сердцевине) и железа (к поверхно-
сти). Многим исследова-
14,02% SL
13,94% SL
13,54% St
11,67% Si
5,40% Si
0,33%, St
0,55%, C
Сердцевина
0,14% St
0^12% C
Рис. 182. Микроструктура силицирован-
ного слоя стали 10 (газовое силициро-
вание в среде хлора при 980° С в тече-
ние 4 ч; глубина слоя около 1 мм; трав-
ление в 2 %-ном растворе азотной кис-
лоты); справа указано среднее содержа-
ние кремния в различных зонах слоя
телям удавалось получать
плотные, беспористые
слои. Например, такой
слой толщиной 0,3 мм на
образцах стали с 0,4% С
удалось получить при
кратковременном (20
мин) газовом силициро-
вании при температурах
не менее 1150° С [403].
Увеличение в стали со-
держания углерода об-
легчает получение беспо-
ристых диффузионных
слоев [404].
Твердость поверхности
силицированного слоя
стали 10 колеблется в
пределах ЯУЮ — 190 ч-
4-250 и несколько возра-
стает с повышением в
стали содержания углеро-
да, а микротвердость до-
стигает Я р. 700—850 [389].
Силицирование пони-
жает предел прочности
стали и- особенно относи-
тельное удлинение и удар-
ную вязкость. Силициро-
ванный слой хрупок. Не-
смотря на низкую твер-
дость силицированного
слоя, он с трудом обраба-
тывается резанием и хорошо сопротивляется истиранию после
предварительной проварки при 170—200° С в масле, которое
впитывается в поры слоя.
В Исследованиях Я. П. Алешина при испытании на машине
Амслера силицированная сталь показала более высокую износо-
стойкость, чем цементованная. Вследствие высокой стойкости
силицированной стали против коррозии она имеет одинаковый
предел усталости при испытании на воздухе и в воде. Силициро-
278
ванне повышает сопротивление стали окислению при нагреве,
однако в меньшей степени, чем алитирование и хромирование.
Силицированные стальные образцы могут длительное время на-
ходиться в окислительной среде при температурах до 700—750° С,
не покрываясь Окалиной.
Как уже отмечалось, силицированный слой обладает высокой
стойкостью против коррозии в морской воде, растворах азотной,
серной и соляной кислот и в других средах при комнатной и по-
вышенной температуре (табл. 44).
Таблица 44
. Коррозионная стойкость несилицированных и силицированных
образцов железа в разных средах [393]
Продолжи- тельность испытаний Уменьшение веса образцов в мг/см*
несилици- рованных силици- рованных несилици- рованных силици- рованных несилици- рованных силици- рованных
в сутках в 10%-ной соляной в 10%-ной серной в 10%-ной фосфор-
кислоте кислоте ной кислоте
1 4,7 0 12,2 0,06 0,73 0,07
3 13,6 0 34,8 0,16 2,22 0,21
6 26,8 0 67,3 0,32 4,08 0,35
10 61,37 0,08 103,1 0,36 7,02 0,41
в 3%-ном хлорис- в 5 %-ном хлорис- в 5%-ном серно-
том натрии том кальции кислом натрии
1 о,3 0,08 0,20 0,01 0,18
3 0,5 0,25 0,47 0,03 0,71 0,04
6 0,8 0,43 0,93 0,05 1,27 0,12
10 1,4 0,48 1,72 0,06 2,15 0,12
В кипящих кислотах силицированный слой разрушается; в
слабых кипящих растворах кислот разрушение происходит мед-
ленно (рис. 185). Например, в 10%-ном кипящем растворе сер-
ной кислоты железный образец диаметром 12 и длиной 50 мм,
силицированный на глубину 0,85 мм, за первые сутки испытаний
уменьшился в весе на 3,1%, за вторые — на 0,5%, за пятые—на
0,4%, за седьмые — на 0,7% и за десятые — на 0,3%. Несилици-
рованный образец железа в тех же условиях полностью раство-
рился в течение 18 ч, а образец стали, содержащей 18% Сг и
8% Ni,—за 30 ч [19].
Силицирование можно применять для обработки деталей хи-
мической, бумажной, нефтяной и маслоочистительной промыш-
ленности (гнезд клапанов, вкладышей, валиков насосов, роторов
водяного насоса, рубашек цилиндров, трубопроводов, различной
арматуры, гаек, болтов и т. д.), для труб судовых двигателей,
279
Рис. 183. Диаграмма состояния си-
стемы Fe — Si
Рис. 184. Распределение микротвер-
дости и фазового состава по глуби-
не диффузионного слоя железа, сили-
цированного в смесях SiCl4 с водоро-
дом (а), аргоном (б) и аргоном
в присутствии кремния (в) [398]
Рис. 185. Внешний вид образца низ-
коуглеродистой стали, разрезанной
на две части. Одна часть подверга-
лась „ кипячению в разбавленной
азотной кислоте до полного раство-
рения сердцевины, другая слегка
протравлена для выявления глубины
силицированного слоя
Рис. 186. Силицированные трубы
двигателя, охлаждаемого морской
водой
280
подводящих и отводящих морскую воду (рис. 186), для патруб-
ков водяных насосов больших промышленных двигателей внут-
реннего сгорания и других стальных и чугунных деталей [405].
Этот процесс, нашедший применение за рубежом, в отечествен-
ной промышленности почти не используется.
БЕРИЛЛИЗАЦИЯ
Бериллизация, как и алитирование (а иногда в большей сте-
пени, чем алитирование), повышает окалиностойкость металлов
и сплавов Г
Методы и режимы обработки. Первые опыты по бериллиза-
ции стали, выполненные еще в 1935 г., и ряд последующих работ
проводили с использованием порошков бериллия и ферроберил-
лия, иногда с добавлением к ним окиси бериллия (для уменьше-
ния спекания порошка). Во всех случаях для предотвращения
окисления порошков либо нагрев производили в атмосфере арго-
на, водорода или в вакууме, либо реакционный стакан помещали
в контейнер с древесным углем.
Позднее для бериллизации начали применять порошкообраз-
ные смеси с хлористым аммонием. Эти смеси чаще содержа!
60—70% бериллия или ферробериллия, 29—30% А12О3 и около
1 % NH4C1. Бериллизация осуществляется при температуре 950—
1100° С и выдержке 4—12 ч. В смеси указанного состава на об-
разцах стали 10 при 1000° С в течение 4 ч образуется слой глу-
биной около 0,20 мм, а в течение 10 ч — около 0,38 мм.
Для бериллизации можно использовать смесь, содержащую
50% окиси бериллия. Последняя полностью восстанавливается
до бериллия металлическим магнием, вводимым в смесь в коли-
честве 48%. Кроме того, в смесь добавляется около 2,0% хлори-
стого аммония. Скорость процесса в этой смеси примерно такая
же, как и в первой [406].
Для интенсификации процесса бериллизации через реакцион-
ное пространство, заполненное обычной порошкообразной
смесью, пропускается хлористый водород. Образцы упаковыва-
ются в порошкообразную смесь в асбестовом стакане, помещен-
ном в трубку, через которую во время процесса пропускается
хлористый водород. Глубина слоя в этом случае заметно боль-
ше, чем при обработке в той же смеси в обычном железном ста-
кане без использования хлористого водорода (табл. 45).
Насыщение стали бериллием осуществлялось также в среде
хлористого водорода без соприкосновения образцов с ферробе-
риллием [19]. Хлористый водород впускался в реакционное про-
1 Приоритет в применении бериллизации для повышения окалиностойко-
сти принадлежит отечественным ученым И. Е. Конторовичу и М. Я. Львов-
скому (Бюллетень изобретений № 13, 1960, заявка 8362/312403 от 5/V 1941 г.
на тему «Способ повышения жаростойкости стальных изделий»).
281
Результаты насыщения стали бериллием
________(автор и В. М. Бирин)________
Таблица 45
Способ насыщения Марка стали Глубина слоя в мм Твердость
HV1 HV10
В обычной смеси1 То же в потоке НС1 10 10 0,19 0,26 986 916 880
В обычной смеси1 То же в потоке НС1 Примечание. Режим обработки: * Состав смеси: 70% FeBe 4- 27% А12 45 45 температу Оа4- 3% NI 0,18 0,27 ра 1050° С, 4.С1. 1219 1219 выдержка 792 880 4 ч.
странство со стороны расположения ферробериллия и, вступая
с ним в реакцию, образовывал летучие хлориды бериллия, кото-
рые далее омывали образцы стали. При этом способе насыщения
Рис. 187. Диаграмма состояния системы Fe — Be
глубина диффузии бериллия в сталь 10 была меньше, чем при
бериллизации в обычной порошкообразной смеси. В диффузион-
ном слое высокоуглеродистой стали под микроскопом не была
обнаружена зона карбида бериллия, что говорит о малой актив-
ности этой насыщающей среды.
Активной газовой средой для бериллизации может служить
смесь Н2 + BeJ2 (температура испарения BeJ2 равна 400° С)
или пары BeJ2, образующиеся в вакуумной печи [254].
282
Бериллизацию стали, а также никеля, меди и алюминия мож-
но осуществлять путем электролиза соляного расплава, состоя-
щего, например, из 50% фторида натрия и 50% оксифторида
бериллия (2ВеО • 5BeF2). Сталь и никель рекомендуется обраба-
тывать в такой ванне при 1100° С, медь — при 900° С, алюми-
ний — при 550° С. Можно применять также ванны, составленные
из смеси ВеС12 и NaCl. Осаждение бериллия в такой ванне про-
изводится при 300—350° С, затем следует диффузионный отжиг.
Рис. 188. Микроструктуры стали, бериллизованной в порошкообразной смеси
(60% Be, 39% А12О3 и 1% NH4C1):
а — армко-железо (1050° С, 4 ч); б - сталь 40 (1050° С, 6 ч); в — сталь У10 (1050° С,
10 ч, охлаждение, повторный нагрев до 1100° С и закалка) (Х150)
Структура бериллизованного слоя. По достижении в процес-
се насыщения предела растворимости бериллия в у-железе про-
исходит фазовое превращение, © результате которого возникают
и растут столбчатые зерна a-фазы (рис. 187, 188). При дальней-
шем повышении концентрации бериллия на поверхности стали
образуется тонкий очень хрупкий слой бериллида FeBe2
(0-фаза). При охлаждении из а-раствора выделяются в виде
игл избыточные бериллиды (рис. 188,а и б). При комнатной
температуре а-ра'створ содержит немного менее 2% Be. На мик-
роструктурах можно наблюдать линию раздела между у- и а-
фазами при температуре насыщения (рис. 188,6)
1 Внешнюю нетравящуюся светлую зону (РеВег) сохранить не удалось,
так как она хрупка и легко откалывается при изготовлении шлифов.
283
При насыщении бериллием высокоуглеродистой стали сна-
ружи вместо светлой корочки бериллида железа обнаруживает-
ся зона легко окисляющихся серых кристаллов, сохраняющихся
при изготовлении шлифа (рис. 188, в). По данным рентгеновско-
го анализа, эти кристаллы являются карбидом бериллия (Ве2С).
После повторного нагрева с последующей закалкой карбид бе-
риллия полностью сохраняется, тогда как избыточные бериллиды
в лежащей глубже зоне слоя переходят в твердый раствор
(рис. 188, в). Зона карбида бериллия, плотная и без микропор,
прочно прилегает к последующей зоне диффузионного слоя. Об-
разование и рост зоны /карбида бериллия с увеличением выдерж-
ки, очевидно, объясняются интенсивным поступлением углерода
из сердцевины к поверхности образца через зону а-фазы. Хотя
растворимость углерода в a-фазе и ничтожна, но скорость его
диффузии в этой фазе во много десятков раз больше, чем в
у-фазе (по некоторым данным, примерно в 200 раз).
С увеличением содержания в стали углерода глубина зоны
карбида бериллия увеличивается, а глубина лежащей ниже двух-
фазной зоны а + FeBe2 уменьшается (табл. 46).
Таблица 46
Глубина различных зон слоя, насыщенного бериллием,
в зависимости от содержания в стали углерода [407]
Зоны слоя Глубина зон в мм на образцах стали
40 У8 У12 У14
Зона карбида бериллия — 0,090 0,095 0,120
Двухфазная зона (бериллиды железа на фоне твердого раствора) . . . Примечание. Обработка образ при температуре 1000° С и выдержке 1 0,220 цов произв» 0 ч. 0,195 одилась в 0,150 порошке б 0,090 ериллия
Структура насыщенного бериллием чугуна принципиально не
отличается от структуры бериллизованной высокоуглеродистой
стали.
Судя по травимости, поверхностная зона слоя бериллизован-
ной среднеуглеродистой стали (0,45% С) состоит примерно из
50% карбида бериллия и 50% бериллида железа [408].
Таким образом, в зависимости от содержания в стали углеро-
да поверхностная зона насыщенного бериллием слоя состоит из
карбида железа, бериллида железа или из смеси этих фаз.
В табл. 47 приведены данные о глубине разных зон берилли-
зованного слоя на стали с различным содержанием углерода.
284
Таблица 47
Глубина различных зон слоя, насыщенного бериллием [408]
Зоны слоя Глубина зон в мм на образцах
железа стали 40 стали У10
Зона бериллидов железа или карбидов бериллия 0,07 0,11 0,12
Зона a-твердого раствора и бериллидов . . 0,25 0,22 0,20
Зона a-твердого раствора 0,06 0,10 0,08
Общая глубина слоя 0,38 0,43 0,40
Зона полного обезуглероживания — 0,25 0,25
Примечание. Режим обработки: температура 1000° С, выдержка 12 ч.
Новые данные о фазовом составе бериллизованного слоя бы-
ли получены в работе [409]. Спектральным анализом в поверх-
ностной зоне слоя образцов стали 20 (обработанных в смеси
50% Be, 49% А12О3 и 1% NH4C1 при 1050° С) обнаружено 40—
45% Be, что соответствует на диаграмме состояния системы
Fe — Be областям, содержащим бериллид FeBe5 (е-фазу). Нали-
чие этого бериллида в поверхностной зоне слоя подтверждено
рентгеноанализом. Кроме бериллия FeBes, в этой зоне слоя был
обнаружен карбид бериллия Ве2С. Фазовый состав бериллизо-
ванного слоя зависит от режима обработки и активности насы-
щающей среды. Поэтому обнаружение в этом слое бериллида
FeBes не противоречит прежним данным о фазовом составе слоя.
Свойства бериллизованной стали. Чем больше содержание в
стали углерода, тем выше ее твердость -после бериллизации. Это
связано, очевидно, с различием в твердости бериллидов железа и
карбида бериллия. Исключительно высокую поверхностную твер-
дость можно получить при насыщении бериллием чугуна. Ниже
приведены значения твердости железа, различных сталей и чу-
гуна, насыщенных бериллием при температуре 1000° С и выдерж-
ке 10 ч [407]:
Стали
Материал............Железо 40 У8 У12 У14 У16 Чугун
Твердость //VI0 .... 1003 1033 1148 1455 1533 1710 1850
На рис. 189 приведены результаты измерения твердости по
глубине диффузионного слоя на образцах железа и стали, насы-
щенных бериллием. Наблюдается разное падение твердости в
месте перехода от зоны химического соединения (карбид берил-
лия или бериллид железа) к зоне, состоящей из двух фаз — твер-
дого раствора и бериллида. В последней зоне твердость снижа-
ется постепенно. Путем двойной термической обработки — за-
285
Рис. 189. Распределение твердости
(по Виккерсу) по глубине поверх-
ностного слоя армко-железа (а) и
сталей 40 (б) и У10 (в) после
насыщения бериллием при 1100° С
в течение 10 ч [408]:
1 — твердость НУГ, 2 — твердость
//V10.
калки с температуры диффузии и длительного отпуска при
500° С —можно повысить твердость бериллизованной низкоуг-
леродистой стали. При закалке бериллий удерживается в твер-
дом растворе, а при отпуске происходит его распад с выделе-
нием мелкодисперсных бериллидов, что сопровождается повы-
шением твердости. Твердость поверхностного слоя, состоящего
из FeBe2, при этом не изменяется [410].
Карбид бериллия, образую-
щийся на поверхности высо-
коуглеродистой стали и чугуна,
химически неустойчив и разру-
шается при длительном нахож-
дении в воздушной среде (в те-
чение 6 месяцев), распадаясь
под действием влаги воздуха
на окись бериллия (тонкий се-
роватый порошок) и метан. Во
влажной среде этот процесс
развивается еще быстрее. Сле-
дует отметить, что на берилли-
зованных образцах железа
процесс разрушения в таких
условиях не наблюдается. По-
сле бериллизации стали и дру-
гие сплавы обладают весьма
высокой окалиностойкостью,
превосходящей во многих слу-
чаях окалиностойкость после
алитирования, хромирования и
силицирования ’. Это объясня-
ется образованием 'при окисле-
нии на бериллизованной по-
верхности прочного, плотного,
тугоплавкого и стойкого против тепловых ударов окисла ВеО.
По данным М. Я. Львовского, использовавшего метод микро-
химического анализа, количества этого окисла в тончайшем на-
лете, образующемся на поверхности окисленной бериллизован-
ной жаростойкой аустенитной стали, составляет около 90%.
Для нелегированных сталей эффект от бериллизации умень-
шается (рис. 190), а для легированных увеличивается с повыше-
нием температуры испытания (то же происходит и при испыта-
ниях алитированной стали). Степень повышения окалиностойко-
сти различных металлов после бериллизации различна. Мини-
мально повышается окалиностойкость хрома (рис. 191) и никеля,
1 Исключение составляет силицированный молибден, однако и в этом слу-
чае .различие в эффекте от силицирования и бериллизации невелико.
286
обладающих высокой окалиностойкостью и в исходном (необра-
ботанном) состоянии.
Рис. 190. Окалиностойкость бериллизованной малоуг-
леродистой стали; испытания при 850° С (а) и 1200° С
Продолжительность испытания
а) д) д) г)
Рис. 191. Окалиностойкость при 1100 и 850° С различных металлов до (кри-
вая /) и после (кривая 2) бериллизации в порошкообразной смеси (автор
и Т. Т. Гребенникова):
а — вольфрам, 1100° С (после- выдержки 26 ч необработанный образец увеличился
в весе на 5600 г/м2)\ б — хром, 1100° С; в — тантал, 1100° С (после выдержки в течение
14 ч необработанный образец увеличился в весе на 4050 г/м2)', г — медь, 850° С
Применяя бериллизацию как метод повышения окалиностой-
кости стали, нужно учитывать следующее.
287
1. Бериллий, необходимый для обработки металлов и сплавов
на нежелезной основе, весьма дорог; ферробериллий, применяе-
мый для обработки стали, хотя и значительно дешевле, но также
относительно дефицитен; предложенный способ использования
окиси бериллия [406] еще недостаточно проверен.
2. Бериллий очень токсичен. Он раздражает дыхательные пу-
ти, вызывает легочную недостаточность и образования на кож-
ных покровах язв и незлокачественных опухолей.
Учитывая последнее, при работе с бериллием нужно строго
соблюдать правила техники безопасности. Так, например, необ-
ходимо процесс приготовления смесей для бериллизации и испы-
тания обработанных образцов на окалиностойкость проводить
под вытяжными зонтами. Микрошлифы также следует изготов-
лять под вытяжными зонтами, используя специально выделен-
ную для этого, отдельно хранящуюся бумагу и особые шлифо-
вальные круги. После соприкосновения с бериллием и берилизо-
ванными образцами нужно мыть руки. Соответствующие меры
следует принимать и при эксплуатации бериллизованных деталей.
По имеющимся сведениям, несколько иностранных фирм, на-
пример фирма Berillium Со (США), применяют в производствен-
ных условиях бериллизацию ответственных деталей.
ТИТАНИРОВАНИЕ
Титанирование (применяется для повышения коррозионной
стойкости и кислотостойкое™ стали.
Первые эксперименты по титанированию, проведенные в
1927 г., и ряд последующих исследований выполнялись с исполь
зованием порошков титана (ферротитана) в вакууме или в среде
водорода [281], [411]—[413]. При титанировании в порошке тита-
на в вакууме при 1100° С в течение 10 ч глубина титанированного
слоя достигает 0,30 мм.
Больший практический интерес представляют разработанные
позднее методы титанирования стали в газовой и жидкой средах.
Сообщают, что газовое титанирование железных листов в сме-
си хлоридов титана и водорода и при 900—1000° С успешно оп-
робовано в Японии. Титанированные листы обладают высокой
стойкостью против коррозии и хорошо свариваются. Титаниро-
вание этим методом позволило получать материал, используе-
мый при производстве анодов термоэлектронных и других ваку-
умных ламп.
Для газового титанирования могут быть использованы [254]:
1) смесь тетробромида титана и водорода (Н2 пропускают
над TiBr4, испаряющимся при 50—100° С, восстановление TiBr4
и титанирование происходят при 1100—1400° С);
2) смесь тетройодида титана и водорода (Н2 пропускают над
TiJ4, испаряющимся при 160—250° С, разложение TiJ4 и титани-
рование происходят при 1200—1500° С).
288
Электролизное титанирование предусматривает использова-
ние растворимого титанового анода или электролиз самого рас-
плава. В первом случае температура расплава, состоящего из
85% KJ и 15% KF, около 750° С, плотность тока 0,4—0,7 а/см2,
температура титанового анода 900—925° С (анод нагревается
т. в. ч.). Во втором случае применяется расплав, состоящий из
16% KaTiFe и 84% NaCl; анод изготовляется из графита, над
зеркалом ванны создается среда аргона. Процесс протекает при
температуре 850—900° С, плотности тока 95 а/дм2, напряжении
3—6 в. По другим данным, в аналогичную ванну дополнительно
вводится 10—20% титановой губки. Достаточным является тита-
нированный слой глубиной 0,020—0,075 мм [414], [415].
При электролизном титанировании образуется диффузионный
слой, состоящий из промежуточной зоны (сплава титана с желе-
зом) и зоны почти чистого титана. В зависимости от температу-
ры процесса в слое преобладает та или иная зона.
Разработан способ титанирования стали и меди в расплав-
ленной соляной ванне [416]—[418], во многом схожий с хорошо
известными способами диффузионного хромирования и силици-
рования стали в ваннах с расплавами хлористых солей, к кото-
рым добавляются порошки хрома, феррохрома или ферроси-
лиция.
Титанирование производится следующим образом. В соляной
расплав (NaCl или NaCl + КС1) вводится порошок сплава ти-
тана, содержащий около 10 ат. % кислорода, или порошок тита-
на, загрязненный кислородом примерно до указанной выше кон-
центрации, так как порошок чистого титана дает плохие резуль-
таты. Над поверхностью ванны пропускается нейтральный газ
для защиты ее от соприкосновения с воздухом при расплавлении
и эксплуатации. Обработанные в такой ванне болты, гайки, не-
большие трубки и другие мелкие детали показывают высокую
коррозионную стойкость.
Автор в своих экспериментах [419] использовал в качестве
защитной среды аргон, очищенный от влаги и кислорода. Тита-
нирование осуществлялось в ванне, рекомендованной в ра-
боте [420] и составленной из 80—90% NaCl и 10—20% порошка
титанового сплава, содержащего около 10 ат. % кислорода (да-
лее обозначается ТЮЖ). Для приготовления сплава порошки маг-
ниетермического титана и двуокиси титана ТЮ2 перемешивались,
брикетировались, плавились в вакуумной печи в среде аргона и
дробились до величины зерен менее 0,15 мм.
Смесь хлористых солей и порошка ТЮЖ загружали в тигель,
из которого аргоном вытеснялся воздух, затем под защитой ар-
гона соль расплавлялась. При увеличенном потоке аргона через
трубку в крышке тигля образцы вводили в ванну, нагретую до
заданной температуры. По окончании выдержки образцы выни-
мали и охлаждали на воздухе.
289
мм
0,5
0,3
Ot2
0,1*
*
Ц4
950 1000 1050 °C
Сравнение активности ванн и удобства работы с ними в за-
висимости от содержания в них порошка титанового сплава по-
казало, что ванна с 10% TiOx отличается большей жидкотеку-
честью, а глубина образующихся в ней диффузионных слоев
практически не уступает глубине слоев, образующихся в ванне
с 20% TiOx. Поэтому, очевидно, лучше
пользоваться ванной, содержащей 10%
TiOx. Однако окончательный вывод мо-
жно будет сделать лишь после того, как
будет решен вопрос о влиянии содержа-
ния в ванне ТЮа на концентрацию тита-
на в поверхностных зонах диффузионного
слоя, от которой зависит коррозионная
стойкость титанированной стали.
Для повышения жидкотекучести и
снижения температуры плавления соли
можно вместо NaCl использовать смесь
NaCl и КС1. Однако при температуре
950—1050° С эта смесь быстро испаряется
и уровень ванны понижается. Такую ван-
ну, очевидно, следует применять лишь
для титанирования меди, так как этот
процесс протекает при 830—850° С.
Автор, не освежая ванну, осуществлял
в ней подряд 8 процессов обработки про-
должительностью 2 ч каждый; результа-
ты титанирования не ухудшались. Более
длительная проверка истощаемости ван-
ны не проводилась.
Значительное количество хлоридов ти-
тана образуется в ванне через определен-
ное время [419], [420]. Однако и свежая
ванна достаточно активна. Поэтому счи-
тают, что титанирование стали в расплав-
ленной соляной ванне протекает, по-ви-
димому, благодаря непосредственному
контакту поверхности стали с мелкими
частицами порошка титанового сплава,
около 10 ат. % кислорода взвешенными в расплав-
Благодаря непрерывной циркуляции расплава
Рис. 192. Изменение глу-
бины титанированного
слоя стали 08 в зависи-
мости от температуры
процесса при выдержке
2 ч (кривая 2) и 6 ч
(кривая /); состав ван-
ны: 80% NaCl и 20%
TiOx (сплошная ли-
ния — глубина слоя до
линии раздела с серд-
цевиной; штриховая ли-
ния — глубина поверх-
ностной нетравящейся
зоны слоя с
концентрацией
[419]
высокой
титана)
содержащего
ленной соли.
вследствие конвекции к поверхности стали постоянно поступают
свежие частицы порошка.
В расплавленной соли, помимо хлоридов титана, обнаружено
[418] соединение NaTiCl4, очевидно, образующееся по реакции
NaCl + TiCl3 = NaTiCl4.
Роль этого соединения, а также роль кислорода в сплаве
TiOx пока не выяснены.
290
На рис. 192 приведена температурная зависимость общей глу-
бины титанированного слоя (до линии раздела) и глубины внеш-
ней тонкой нетравящейся зоны слоя с высокой концентрацией ти-
тана. Глубина внешней зоны с увеличением температуры изме-
няется значительно меньше, чем общая глубина слоя. При по-
вышении температуры с 950 до 1100° С поверхностная твердость
изменилась с HV 250 до HV 480, а микротвердость соответствен-
но с 720 до 1500.
Рис. 193. Микроструктуры титанированного слоя стали 08. Режим обработ-
ки: 1000° С, 4 ч (а) и 1100° С, 0,5 ч (б); состав ванны: 90% NaCl и 10% ТЮЖ
(Х200) [419]
Микроструктура титанированного слоя приведена на рис. 193.
Микротвердость поверхностной зоны с высокой концентрацией
титана в 3—4 раза выше, чем микротвердость расположенной
глубже широкой зоны слоя со столбчатыми зернами. На некото-
рых микрошлифах в зоне столбчатых зерен, вблизи внешней
нетравящейся тонкой зоны, иногда наблюдаются выделения
избыточной фазы (выпадающие при медленном охлаждении),
вероятно, титаниды (рис. 193,6). Глубже слоя со столбчатыми
зернами, непосредственно за линией раздела, замечено неболь-
шое снижение твердости по сравнению с твердостью сердцевины
(рис. 194). Это, по-видимому, объясняется диффузией углерода
и зоны, расположенной глубже линии раздела, в диффузионный
слой, в связи с чем глубже него образуется зона с пониженным
содержанием углерода. Данные об изменении глубины твердо-
сти титанированного слоя у стали 08 в зависимости от темпера-
туры и продолжительности процесса приведены в табл. 48.
291
10*
Таблица 48
Зависимость глубины и твердости титанированного слоя у стали
08 от продолжительности и температуры процесса [419]
Характеристика слоя Температура в °C
950 I 1000
Продолжит ельность в ч
2 4 6 2 4 6
Глубина в мм: внешней тонкой зоны .... 0,008 0,010 0,015 0,01 0,012 0,024
зоны столбчатых зерен . . . 0,050 0,065 0,10 0,054 0,08 0,136
Микротвердость 720 630 600 900 950 1000
Твердость Н VI 200 250 246 270 330 360
На рентгенограммах поверхности образцов стали 08, титани-
рованных при 1000° С в продолжение 2 и 4 ч, обнаружены линии
гексагональной решетки. Периоды этой решетки были близки к
теоретическим для TiFe2 (а = 4,77 кХ и с = 7,75 яХ), но все же
несколько превышали их (рис. 195). На глубине 0,06 лш от по-
верхности (зона столбчатых зерен) были обнаружены линии
о
a-железа с расширенными параметрами (а = 3,16 А вместо
а = 2,861 А), это подтверждает, что зона слоя со столбчатыми
зернами является твердым раствором титана в а-железе.
На фиг. 196 показано изменение концентрации титана по глубине
диффузионного слоя *. Содержание титана в поверхностной зоне
слоя достигает примерно 30%, что соответствует соединению
TiFe2 и вполне согласуется с данными рентгеновского анализа.
Характер распределения титана по глубине диффузионного слоя
согласуется также с микроструктурой слоя и характером кривых
распределения микротвердости.
По данным работы [411], в поверхностной зоне слоя стали,
титанированной аналогичным способом, содержится значитель-
но больше титана (до 70—80%). Однако авторы этой работы
осуществляли процесс при более низкой температуре (900° С),
что могло способствовать повышению концентрации титана в са-
мой поверхностной зоне слоя вследствие меньшей скорости диф-
фузии титана.
1 Кривые построены путем послойного спектрального анализа, выполнен-
ного под руководством В. Г. Корицкого. Для послойного спектрального ана-
лиза. изготовляли специальные эталоны сплавов титана с железом, содержа-
щие 0,1—37% Ti, и строили графики интенсивности спектральных линий в за-
висимости от концентрации титана. Спектральный анализ производили с по-
мощью микрофотометра МФ-2.
292
293
Рис. 194 Изменение микро-
твердости по глубине тита-
нированного слоя стали 08,
обработанной при 1000°С в
ванне, содержащей 90%
NaCl и 10% TiOx в течение
2 и 6 ч;
а и b — общая глубина слоя
наблюдаемая под микроскопом
Г419]
Рис. 196. Изменение кон-
центрации титана по глуби-
не диффузионного слоя ста-
ли 08 (состав ванны: 80%
NaCl и 20% TiOx); режимы
титанирования:
/ — при 1000° С, 4 ч\ 2 — при
•СЕО° С. 4 ч (419]
На поверхности титанированной стали 50 был обнаружен
тонкий (0,005—0,007 мм) нетравящийся слой карбида титана
TiC *, непосредственно примыкающий к перлито-ферритовой
сердцевине и имеющий весьма высокую микротвердость
(до Ярс 2900).
На рис. 197 приведены результаты испытания нетитанирован-
ных и титанированных образцов в 4 %-ном растворе азотной кис-
лоты. Для подтверждения высокой кислотостойкости свойств
титанированной стали с торцов нескольких образцов был снят
титанированный слой. После выдержки этих образцов в течение
Продолжительность
испытания
примерно 100 ч в 4%-ном растворе
HNO3 их сердцевины растворились,
и остались лишь тонкие «стенки»,
образованные сохранившимся тита-
нированным слоем.
Титанированные образцы * 1 испы-
тывали в продолжение 50 ч при 20,
60 и 100° С в 15%-ном растворе азот-
ной кислоты, к которой было добав-
лено 5% двухромовокислого калия.
Уменьшение веса образцов при этом
соответственно составляло 0,038;
0,057; 0,231 г/л2-ч. Необработанные
Рис. 197. Уменьшение веса
титанированных (кривая /)
и нетитанированных (кри-
вая 2) образцов стали 08 в
4 %-ном растворе азотной
кислоты [419]
образцы при всех температурах рас-
твора быстро и полностью раство-
рялись.
При титанировании железа в по-
рошках титана (в вакууме или в
среде водорода) зона слоя с высо-
кой концентрацией титана образу-
ется гораздо медленнее, чем при титанировании в расплавах,
когда эта зона достигает значительной толщины менее чем за 1 ч
при 900—1000° С и менее чем за 20 мин при 1100° С. Очевидно,
это связано со своеобразным механизмом титанировании стали
в соляной ванне.
Опробовано азотирование титанированной стали Ст. 3 [421J.
Образцы сначала подвергались титаниро>ванию при температуре
1050° С и выдержке 3 ч в порошкообразной смеси, содержавшей
75% Ti, 24% А120з и 1% NH4C1, а затем отжигу в среде водоро-
да при 1100° С. После отжига глубина титанированного слоя до-
стигала 0,28—0,30 мм. Далее образцы азотировались при 520° С
* Гранецентрированная кубическая решетка с а =4,305 кХ (теоретиче-
ский период решетки 4,320 кХ).
1 Образцы титанировались при температуре 1000° С и выдержке 2 ч в
среде водорода в смеси порошков титана и окиси титана.
294
в течение 24 ч. В результате образовался нехрупкий слой с вы-
сокой микротвердостью 1600—1650), сохраняющейся до
глубины 0,24—0,25 мм.
НИОБИРОВАНИЕ
Ниобированием, так же как и хромированием, можно повы-
сить износостойкость стали и, по-видимому, коррозионную
стойкость в некоторых средах, но в промышленности этот про-
цесс пока не применяется.
Рис. 198. Диаграмма состояния системы Fe — Nb
Существует несколько способов ниобирования:
1. В порошках ниобия или феррониобия. Эти порошки
используются как без добавок (422], так и чаще в смеси с А12О3
и NH4C1 или с А12О3 (в последнем случае во время процесса
через реакционное пространство часто пропускается поток водо-
рода). Ниобирование в порошках можно также осуществлять
в вакууме.
2. В газовых средах: Н2 + НС1 (в присутствии феррониобия)
[423]; Н2 + NbCl5; Ar + NbCl5 [254].
3. В жидкой среде (электролиз расплава K?NbF6 + NaCl за-
щитной атмосферой).
После газового ниобирования железа при 1200° С в течение
6 ч в среде Н2 + НС1 (глубина ниобированного слоя 0,12 мм)
в поверхностной зоне образуются столбчатые зерна а-твердого
раствора и небольшое количество включений второй фазы
(е-фазы) в виде отдельных точек. Очевидно, эти включения
295
выделяются при охлаждении (рис. 198)*. Содержание ниобия
в тонкой поверхностной зоне слоя достигает 65—68% (рис. 199),
т. е. почти достигает состава.
На поверхности стали, содержащей 0,25% и более углерода,
образуется тонкий слой карбида NbC, под которым располагает-
ся темная зона с повышенной твердостью. По данным рентгено-
анализа, она состоит из a-твердого раствора и карбида ниобия.
Глубже этой зоны лежит зона, обедненная углеродом, анало-
гично тому, как это наблюдается при хромировании. Как и при
хромировании, максимальная
глубина карбидного слоя соот-
ветствует содержанию в стали
0,4—0,5% С (рис. 199).
После газового ниобирования
при 1200° С в течение 6 ч глубина
карбидного слоя на стали, содер-
жащей 1,18% С, достигает
Рис. 200. Послойное распределение
ниобия в поверхностных зонах желе-
за и стали после газового ниобиро-
вания при 1200° С в течение 6 ч [423]
0,04 мм. а концентрация в нем
Рис. 199. Влияние содержания угле-
рода в стали на толщину карбидной
зоны (ниобирование при 1000° С, 6 ч)
[423]
Параметр решетки карбида ниобия (а = 4,38 А), образовав-
шегося на стали, содержащей 1,18% С, оказался меньше пара-
метра карбида ниобия состава NbC (а = 4,457 А). Это расхож-
дение объясняется тем, что соединение NbC кристаллизуется
с дефицитом по углероду, т. е. представляет собой твердый
раствор вычитания.
При ниобировании стали У12 в порошке (50% Nb, 49% А12О3,
1% NH4C1) при 1050° С в течение 16 ч толщина карбидного
слоя составляет 0,02 мм (рис. 201).
♦ s-Фаза, приведенная на рис. 198, по одним данным является твердым
раствором на основе соединения Fe3Nb2, а по другим (более поздним данным)
е-фаза гомогенна в пределах 22—42 ат % и включает в себя два соедине-
ния: Fe3Nb2 и Fe2Nb; трфаза— твердый раствор на основе соединения Fe2Nb3.
296
На рис. 202 показано изменение микротвердости ниобирован-
ной стали с различным содержанием углерода. Благодаря вы-
сокой твердости карбида ниобия 2200) сталь после ниоби-
рования обладает высокой износостойкостью, превышающей
износостойкость закаленной стали и даже стали, подвергнутой
диффузионному хромированию.
Слой карбида ниобия значительной глубины (>0,03—
0,04 мм) получить не удается.
На окалиностойкость стали ни-
обирование существенно не влияет.
^50
Рис. 202. Микротвер-
дость поверхностных
зон ниобированной
стали с различным
содержанием угле-
рода (газовое ниоби-
рование при 1000° С,
6 ч; нагрузка 50 г)
[423]:
1 — наружная зона;
2 — промежуточная зо-
на; 3 — сердцевина
Рис. 201. Микроструктура стали У12,
насыщенной ниобием в порошке при
1050° С в течение 16 ч (Х150)
Ниобирование не оказывает положительного влияния также на
коррозионную стойкость и кислотостойкость стали в растворах
серной и азотной кислот [281], [423].
МОЛИБДЕНИРОВАНИЕ
Молибденирование улучшает стойкость стали в некоторых
средах.
Детали можно молибденировать различными способами:
1. В порошке молибдена или ферромолибдена; в порошке
молибдена или смеси этого порошка (2 ч.) с А12О3 (1 ч.)
в потоке водорода [1], [30], [281], [424].
2. В газовых средах: Н2 + НС1 в присутствии молибдена
или ферромолибдена [19], [425], МоС15 + Н2 (температура
297
испарения МоС15 300° С) [254], [426], [427], МоВг5 + Н2 [254],
Мо(СО)6 + Н2 при 800° С.
3. В жидкой среде: электролиз в ванне с расплавом молиб-
дата (МагМоО4) (исследование Н. Н. Мокина), восстановле-
ние расплавленного молибдата до металла путем продувания
через ванну водорода или предварительно диссоциированного
аммиака [428]:
Рис.
дена
Na2MoO4 + ЗН2 = Мо + 2NaOH + 2Н2О.
На рис. 203 приведены резулы
таты измерения глубины молиб-
денированного слоя при обработ-
ке в порошке в потоке водорода.
Изменение концентрации
и вольфрама по глубине
Рис. 204.
молибдена
слоя при обработке железа в рас-
плавленных молибдатах и воль-
203. Глубина диффузии молиб-
в железо в зависимости от вре-
мени выдержки при различных тем-
пературах в порошке ферромолибде-
на в среде водорода [424]:
I — глубина до равноосных зерен сердце-
вины; 2 — линия раздела; 3 — граница
молибденидов
фрамитах, через которые продувался
водород [428]:
/ — вольфрамирование при 1100° С,
10 ч\ 2 — молибденирование при 1150° С.
20 ч; 3 — молибденирование при 980° С»
6 ч и 10 ч
При газовом молибденированпи в среде Н2 + НС! (образцы
с ферромолибденом не соприкасались) как в экспериментах
автора [19], так и по данным работы [425] скорость процесса
насыщения оказалась не больше, чем при молибденировании
в порошке (рис. 203).
При насыщении в расплаве молибдата [428] скорость про-
цесса снижается с уменьшением расхода Н2 или диссоциирован-
ного NH3, так как перемешивание расплава происходит менее
энергично. Применение недиссоциированного аммиака дает
худшие результаты, чем применение диссоциированного. При
298
молибденировании в расплаве (температура 1150° С, выдержка
20 ч) глубина слоя достигает 0,30 мм (рис. 204).
Концентрация молибдена по глубине диффузионного слоя
повышается с увеличением температуры (рис. 204 и 205). Так,
при обработке при 1300° С в газовой среде содержание молиб-
дена в тончайшей поверхностной зоне слоя достигает 24%
(рис. 206).
С увеличением в стали содержания углерода диффузионная
подвижность молибдена (рис. 207) уменьшается, однако в мень-
Рис. 206. Распределение концентра-
ции молибдена в поверхностных зо-
нах железа после газового молибде-
нирования при 1300° С в течение 6 ч
Рис. 205. Распределение молибдена
по глубине диффузионного слоя же-
леза в зависимости от режима молиб-
денирования в порошке молибдена
[424]:
1 — при 1300° С, 24 ч\ 2 — при 1200° С,
24 ч\ 3 — при 1100° С, 36 ч*, 4 — при
1000° С, 72 ч
шей степени, чем у вольфрама, титана и ванадия. Это объ-
ясняется тем, что при молибденировании <в порошках проис-
ходит сильное обезуглероживание стали.
В случае насыщения железа молибденом при низких темпе-
ратурах (ниже 900° С) в диффузионном слое образуются столб-
чатые зерна, ориентированные в направлении диффузии и имею-
щие извилистую внутреннюю границу. При 900° С и выше
столбчатые зерна перерезаны линией раздела, разделяющей а-
и у-фазы при температуре насыщения (рис. 208). Части зерен,
расположенных между поверхностью железа и линией раздела,
травятся слабо (рис. 209, а); части зерен, расположенные
глубже-линии раздела, травятся сильнее, так же как и сердце-
вина.
В случае молибденирования при 1150—1300° С и последую-
щего медленного охлаждения из зоны a-твердого раствора, при-
мыкающей к поверхности, выделяются в виде иголочек е-молиб-
денида Fe7Mo6 (рис. 209). После быстрого охлаждения с темпе-
ратуры насыщения молибдениды не обнаруживаются. Глубина
» 299
Рис. 207. Распределение вольфрама и молибдена по глубине диффузионного
слоя железа (0,05% С) и стали (1,33% С) после насыщения в порошках в
вакууме в течение 200 ч при 1150° С [30]:
1 — кривые, построенные по экспериментальным данным для железа; 2 — предпола-
гаемый характер кривой; 3 — линия раздела, определенная микроскопическим" спосо-
бом; 4 — кривые для стали
Рис. 208. Диаграмма состояния системы
Fe — Мо
300
залегания линии раздела при температуре насыщения выше
900° С сначала падает, а затем начинает возрастать (рис. 210).
Такой своеобразный ход кривой линии раздела объясняется
следующим. При 900° С перекристаллизация железа у-»-а про-
текает при невысокой концентрации молибдена. При темпера-
туре немного выше 900° С минимальная концентрация молибде-
на в a-фазе резко возрастает. Следовательно, чтобы вызвать
перекристаллизацию, требуется поступление значительно боль-
шего количества молибдена. В то же время небольшое повыше-
Рис. 209. Микроструктура железа, насыщенного молибденом
(1200° С, 3 ч) (Х100) [424]
ние температуры насыщения не может существенно изменить
концентрацию молибдена в слое. Поэтому после обработки
в некотором интервале температур немного выше 900° С види-
мый слой отсутствует.
Твердость железа, насыщенного молибденом, возрастает
с увеличением выдержки и повышением температуры диффузии,
т. е. с увеличением в слое концентрации молибдена. Твердость
диффузионного слоя получается примерно равной твердости
обычных железомолибденовых сплавов, имеющих такой же
состав, как и диффузионный слой (рис. 211).
Диффузионный слой, полученный при 1150—1300° С, может
быть подвергнут дисперсионному твердению. Так, по данным
работы [424], на образцах железа, насыщенных в порошке
молибдена при температуре 1300° С и выдержке 6 ч, образовал-
ся слой глубиной 0,60 мм (до линии раздела), глубина зоны
с молибденидами составляла 0,28 мм, а поверхностная твер-
дость HV 10—280. После закалки с 1250° С в воде и отпуска при
650° С поверхностная твердость повысилась до HV 10—580.
301
Вследствие недостаточной концентрации молибдена в слое
и главным образом вследствие поверхностного обезуглерожива-
ния стали при обработке в порошках не удавалось обнаружить
(как по характеру микроструктуры, так и по измерениям
твердости) на поверхности молибденированной стали выделений
карбида молибдена, а тем более сплошной зоны карбидов.
Лишь позднее удалось установить, что, несмотря на поверхност-
ное обезуглероживание, в диффузионном слое образуются кар-
биды. Например, у стали, содержащей 0,47% С, диффузионный
Рис. 210. Влияние температуры и
продолжительности процесса на глу-
бину диффузионного слоя железа, на-
сыщенного молибденом [424] (круж-
ки — линия раздела; квадраты — гра-
ница молибденидов)
Рис. 211. Изменение твердости по
глубине диффузионного слоя железа,
насыщенного молибденом (при 1200° С,
3 ч), (ХЮО) [424] (кружки — линия
раздела; квадраты — граница молиб-
денидов) :
1 — при 1300° С, 6 ч\ 2 — при 1275° С,
6 ч\ 3 — при 1200° С, 6 ч
слой состоит из a-твердого раствора и карбида РегМо2С *.
После обработки в расплаве молибдата (температура 980° С,
выдержка 14 ч) на сталях, содержащих 0,8% С и 0,4% С, были
обнаружены сплошные поверхности зоны комплексных карбидов
толщиной соответственно 0,015 мм и 0,006 мм с микротвердо-
стыо Hv. 1225. Чем ниже температура, тем больше было кар-
бидной фазы в диффузионном слое.
Окалиностойкость железа и стали после молибденирования
изменяется мало. Молибденированные железо и особенно сталь
обладают повышенной коррозионной стойкостью в 50 %-ной
азотной кислоте.
Путем азотирования молибденированного железа можно
значительно повысить его твердость. Так, после азотирования
* Молибденирование производилось при 1200° С в течение 6 ч (среда
Нг + HCI) в присутствии ферромолибдена; глубина слоя 0,5 мм, микротвер-
дость Ну. 218 [425].
302
по режиму 510° С 4 ч + 540° С, 25 ч твердость слоя достигла
HV 1300 и мало изменялась до температуры 500° С. Были пред-
приняты также попытки цементации молибденированной
стали (428].
ВАНАДИРОВАНИЕ
Ванадирование придает стали высокую износостойкость
и повышенную коррозионную стойкость в некоторых средах, но
на практике не применяется.
Ванадирование осуществляется следующими способами:
1. В порошках ванадия или
феррованадия. Эти порошки мо-
гут использоваться как без до-
бавок (рис. 212), так и в смеси с
NH4C1 или НС1 [19]. Ванадиро-
вание можно производить в сре-
де водорода и в вакууме.
2. В газовых средах: Н2 +
+ НС1 в присутствии ванадия
или феррованадия [19] (429];
VC14 + Н2 [254]; VJ2 + Н2 [254].
После обработки в смеси, со-
стоящей из 60% феррованадия
(38,3% V), 37% каолина и 3%
NH4CI, при 1150° С в течение
2,5 ч на образцах образовался
Рис. 212. Влияние продолжитель-
ности процесса на глубину диф-
фузионного слоя железа, обрабо-
танного в порошке феррованадия
при температуре 1100°С:
1 — столбчатые кристаллы; 2 — ли-
ния раздела
слой глубиной 0,11—0,13 мм, а после обработки в порошке фер-
рованадия— глубиной 0,04—0,05 мм [19]. При обработке по то-
му же режиму в смеси порошка феррованадия с 5% NH4CI в сре-
де водорода был получен слой глубиной 0,093 мм [281].
Таблица 49
Результаты насыщения стали ванадием в порошке
и газовой среде [19]
Насыщение в порошке Насыщение в газовой среде
Марка стали Глубина слоя в мм Н VI Глубина слоя в мм Я VI HV10
10 45 У10 7X3 0,10—0,11 0,01—0,02 0,01—0,015 138 639 713—937 0,12 0,02 0,02 125 893 793 113 413 485
Прим 2. Сост 3. При от кусочно е ч а н и я: 1.1 ав порошка: 6С обработке в г в феррованади? эежим обработг )% феррованади азовой среде 1. <и: температур! 1Я, 37% каолин. Н8+ НС1 обра: а 1150° С, выдс а и 3% ЫН4С1. зны помещали! аржка 2,5 ч. :ь отдельно
303
Обработка в газовой среде Н2 + НС1 при принятых условиях
экспериментов заметно не ускорила процесс ванадирования
(табл. 49).
Рис. 213. Диаграмма состояния системы Fe — V
О 60 120 130 пк
Рис. 214. Распределе-
ние концентрации ва-
надия в поверхност-
ных зонах железа и
стали после газового
ванадирования при
1100°С в течение 6 ч
(429]
Диффузионный слой на ванадированном железе представля-
ет собой столбчатые зерна твердого раствора ванадия в а-желе-
зе (рис. 213). Содержание ванадия в слое
не превышает 50% (рис. 214). В поверх-
ностной зоне диффузионного слоя высоко-
, углеродистой стали содержится более
70% ванадия, и на поверхности образует-
ся тонкий нетравящийся слой карбида
ванадия VC (табл. 50). Микротвердость
ванадированного слоя у железа Нр. 290,
а у сталей, содержащих 0,47—1,18% С,
/7.x 1850—1920 [429].
При одном и том же режиме обработ-
ки глубина карбидного слоя у ванадиро-
ванной стали меньше, чем у хромирован-
ной, а у ванадированного железа, наобо
рот, больше, чем у хромированного желе-
за [429], [430]. Карбидный слой на ванади-
рованной стали обладает хорошей связью
с сердцевиной. При осадке ванадирован-
ной стали на 60% отслаивания диффузи-
онного слоя не происходит, несмотря на
образование в нем мелких трещин.
Благодаря большой твердости карбидного слоя ванадиро-
вапная сталь обладает высокой износостойкостью, во много раз
304
большей, чем закаленная сталь и даже сталь, подвергнутая
диффузионному хромированию. Однако получить значительного
по глубине слоя карбида ванадия на поверхности стали не
удается. На окалиностойкость стали ванадирование существен-
ного влияния не оказывает. Ванадированные железо и сталь
отличаются повышенной коррозионной стойкостью в 50%-ном
растворе азотной кислоты, сталь — также в серной кислоте
(98%), а железо — в 10%-ном растворе NaCl.
Таблица 50
Фазовый и химический состав поверхностных зон
ванадированной стали [429]
Содержание углерода в стали в % Глубина слоя в мм Содержание ванадия в по- верхностной зоне по данным спектрального анализа в % Фазовый состав
по данным рентгеноструктур- ного анализа по данным электронно- графического анализа
0,03 0,09 49,5 а-твердый раствор
0,25 0,004 85,0 VC + a* VC
0,47 0,012 91,3 VC + a* VC
0,65 0,10 80,0 VC —
0,85 0,009 88,7 VC —
1,03 0,007 92,5 VC —
1,18 0,007 83,0 VC + a* VC
Прим е ч а н и е. Реж им обработки: те мпература 1000° C, выдержка 6 ч;
среда Н2 4- НС1.
* Слабые линии a-твердого раствора.
ВОЛЬФРАМИРОВАНИЕ
Вольфрамирование можно производить следующими спо-
собами:
1. В порошках вольфрама или ферровольфрама и в смесях
этих порошков с А12О3 и NH4C1. Обработку можно осуществлять
в среде водорода (431] и в вакууме (281].
2. В газовых средах: Н2 + НС1 (19] (432]—(434], Н2 + WC16
(427], Н2 + WF6 [435] [577], в среде НС1 с нагревом т. в. ч. (258],
в вакууме путем разложения бромида вольфрама [436].
3. В расплавах вольфрамата (Na2WO4) путем электролиза
(работа Н. Н. Мокина) и путем восстановления вольфрамата
до металла при продувании через ванну водорода или аммиа-
ка [428]
Na2WO4 + ЗН2 = W + 2NaOH + 2Н2О.
При обработке в порошке диффузия вольфрама в железо
протекает медленнее (рис. 215), чем молибдена (см. рис. 203),
305
что согласуется с данными о величинах коэффициентов диффу-
зии этих элементов в железо. При газовом вольфрамировании
процесс протекает быстрее. В опытах, проведенных автором,
при температуре 1150° С и выдержке 6 ч в среде Н2 + НС1
(образцы железа не соприкасались с порошком вольфрама)
глубина диффузионного слоя достигала 0,14—0,18 мм [19].
С большой скоростью протекает процесс также в газовой
среде НС1. При нагреве образцов т. в. ч. вольфрамированный
слой глубиной 0,2 мм был получен при 1100° С за 15 сек [258].
Рис. 215. Глубина диффузионного
слоя железа в зависимости от
времени выдержки при разных
температурах в порошке вольфра-
ма в атмосфере водорода [1]:
1 — расстояние до равноосных зерен
сердцевины; 2 — линия раздела
Рис. 216. Микроструктура же-
леза, насыщавшегося вольфра-
мом в течение 3 ч при темпера-
туре 1200° С (ХЮ0) [1]
Диффузионный слой, полученный в результате насыщения
железа вольфрамом при 900° С, представляет собой столбчатые
зерна a-твердого раствора. После насыщения при более высокой
температуре в слое появляется линия раздела (рис. 216, 217),
что согласуется с характером диаграммы состояния системы
железо — вольфрам (рис. 218).
После вольфрамирования железа в порошке при 1150—
1200° С в течение 12 ч вольфрамиды в диффузионном слое не
обнаруживаются, что можно объяснить или недостаточным на-
сыщением железа вольфрамом, или большой дисперсностью
вольфрамидов, или ускоренным охлаждением. Если процесс
протекает при более высоких температурах или в более актив-
ных средах — газовых и жидких, вольфрамиды в слое обнару-
живаются. После насыщения железа в порошке вольфрама при
306
1280° С [431] диффузионный слой состоял из тонкой зоны
(0,05 мм) Fe2W (62,2% W) и зоны a-твердого раствора воль-
фрама в железе (13% W). В более позднем исследовании [433]
(опыты проводились в газовой среде при температуре 1300° С
и выдержке 6 ч) зона Fe2W в вольфрамированном слое железа
обнаружена не была. Диффузионный слой после охлаждения
состоял из трех зон: внешней тон-
кой зоны вольфрамида Fe7W6
(7 мк), двухфазной зоны а +
+ Fe7W6 (45 мк) и зоны а-твердо-
го раствора вольфрама в железе
(348 мк). Среднее содержание
вольфрама в этих зонах было со-
ответственно равно 65,3; 12 и
4,5% (рис. 219), а микротвер-
дость 500, 165 и 141. В этих
опытах, несмотря на медленное
охлаждение, была обнаружена
Ат % IV
Вес % IV
Рис. 218. Диаграмма состоя-
ния системы Fe — W
Рис. 217. Глубина расположения ли-
нии раздела в зависимости от темпе-
ратуры и выдержки при насыщении
железа вольфрамом в порошке воль-
фрама [1]
двухфазная структура (a + Fe7We), устойчивая только в обла-
сти высоких температур.
Углерод сильнее замедляет диффузию вольфрама, чем
молибдена, что, вероятно, связано с различной степенью по-
верхностного обезуглероживания стали в процессе насыщения.
Вследствие поверхностного обезуглероживания, наблюдаемого
при всех опробованных методах вольфрамирования, и недостат-
ка вольфрама* в слое обычно сплошная карбидная зона отсут-
ствует. Например, при обработке образца толщиной 3 мм
(сталь с 1,03% С) в течение 6 ч при 1300° С в газовой среде
Н2 + НС1 в присутствий ферровольфрама содержание углерода
в сердцевине снизилось до 0,3—0,4%. Рентгеноструктурный
анализ диффузионного слоя показал наличие в нем трех зон:
a-твердого раствора, фазы Fe7We и небольшого количества
307
карбида Fe2W2C. Последняя фаза располагалась по границам
зерен. Поверхностная микротвердость этого слоя 229,
а общая его глубина 270 мк.
Вольфрамированный слой обладает несколько повышенной
коррозионной стойкостью в 26%-ной перекиси водорода и малой
Рис. 219. Распределение
концентрации вольфрама в
поверхностных зонах желе-
за после газового вольфра-
мирования в течение 6 ч
при 1300° С [434]
стойкостью (такой же, как и у необ-
работанной стали) в растворах кис-
лот: 37%-ной НС1, 50%-ной HNO3
и H2SO4. Окалиностойкость после
вольфрамирования изменяется не-
значительно, а поверхностная ми-
кротвердость немного повышается
у железа и может даже снижаться
у стали в результате сильного обез-
углероживания.
Вольфрамированный слой был
успешно использован в качестве
подслоя при диффузионном насы-
щении некоторых металлов други-
ми элементами. Предварительное
вольфрамирование в некоторых
случаях улучшает связь диффузи-
онного слоя с сердцевиной и умень-
шает различие в линейных коэффи-
циентах их расширения.
Вольфрамированная сталь под-
вергалась цементации. Поверхност-
ная зона диффузионного слоя по-
сле такой обработки содержит комплексные карбиды и отли-
чается высокой твердостью и теплостойкостью [428].
НАСЫЩЕНИЕ МАРГАНЦЕМ
Насыщение марганцем осуществляют следующими спо-
собами:
1. В порошках марганца или ферромарганца и в смесях этих
порошков с А12О3 и NH4CI; насыщение в порошках и смесях
можно производить в среде водорода и в вакууме [281] (табл. 51).
2. В газовой среде Н2 + НС1 в присутствии марганца или
ферромарганца [430], [437] (табл. 51).
3. В соляных расплавах \
На поверхности железа, насыщенного марганцем в газовой
среде, после легкого шлифования отчетливо выявляется сетка
очень мелких трещин, возникновение которых, очевидно, связано
с объемными изменениями в слое.
1 Metchell Т. A. Canad. Patent, 1942, № 402472.
308
Микроструктура железа, насыщенного марганцем в газовой
среде при температуре 1100° С и выдержке 3 ч, состоит из двух
зон. Наружная зона (0,09 мм), содержащая 80—100% Мп,
согласно данным рентгеноструктурного анализа представляет
Рис. 220. Диаграмма состояния системы Fe — Мп
собой у-марганец или твердый раствор железа в у-марганце
(рис. 220 и 221). Вторая, нетравящаяся зона (0,14 мм) являет-
ся твердым раствором марганца у-железе (после обработки по
данному режиму фаза а-Мп не обнаружена) [437]. На поверхно-
сти образцов стали У10, обработанных по аналогичному режи-
му, обнаружена светлая тонкая (0,015 мм) нетравящаяся
корочка карбида (Мп, Fe)3C с микротвердостью Н^. 1145.
309
В отличие от этого в опытах, проведенных автором на образ-
цах стали У12, при температуре 1150° С, выдержке 16 ч в по-
рошкообразной смеси (50% Мп, 49% А12О3, 1% NH4C1) поверх
Рис. 222. Микроструктура стали
У12, насыщенной марганцем в по-
рошке при 1150° С в течение 16 ч
(Х150)
Рис. 221. Распределение
концентрации марганца в
поверхностных зонах желе-
за (0,03% С) и стали
(1,03% С) после насыще-
ния в газовой среде:
1 — железо, 1100° С, 3 ч\ 2 —
сталь, 800° С, 6 ч [437]
испытания
Рис. 223. Уменьшение веса образцов
железа (0,03% С) в 10%-ном раство-
ре NaCl:
1 — необработанного; 2 — насыщенного
марганцем в газовой среде при 1100° С в
течение 3 ч [430]
карбидного нетравящегося слоя толщиной 0,022—0,033 мм
(микротвердость 1145) был обнаружен более тонкий
310
Таблица 51
Зависимость глубины диффузионного слоя от режима насыщения
Метод обработки Обрабатываемый металл Режим обработки Глубина слоя в мм
Температура в °C Выдержка в ч
В порошке мар- 950 2 0,17
ганца в вакууме 950 4 0,24
[281] Армко-железо 1000 2 0,32
1000 4 0,42
В газовой сре- Армко-железо, 1100 3 0,23
де [430]. [437] Сталь с 1,03% С 800 6 0,016
(0,016—0,020 мм) темный слой, фазовый состав которого уста-
новить не удалось (рис. 222). По внешнему виду этот слой на-
поминает у-марганец (или твердый раствор железа в у-марган-
це), обнаруженный в работе (437] при насыщении марганцем
железа.
Насыщение железа и стали марганцем не влияет на их
окалиностойкость, не обеспечивает коррозионную стойкость
в растворах азотной, соляной и серной кислот, но резко повы-
шает ее в 10%-ном растворе NaCl. В этой среде сталь и особен-
но железо, насыщенные марганцем, весьма стойки (рис. 223).
НАСЫЩЕНИЕ ФОСФОРОМ
Насыщение стали фосфором в газовой среде, состоящей из
10% РН3 и 90% Н2, изучалось еще в работе, выполненной
в 1935 г. А. Брамлеем.
При насыщении железа (стали) фосфором протекает у->-
-> a-превращение, что вызывает оттеснение углерода из поверх-
ностной зоны диффузионного слоя в глубь стали (см. рис. 20)'.
Опыты, проводившиеся при температуре 950° С и выдержке
300 ч, показали, что увеличение в стали содержания углерода
с 0,05 до 1,3% приводит к уменьшению толщины зоны столбча-
тых зерен a-твердого раствора в 10 раз, а глубины проникнове-
ния фосфора (до концентрации 0,05% Р) в 3 раза [30].
Автор проводил исследования по насыщению железа и ста-
ли фосфором при температуре 1150° С и выдержке 8 ч в порош-
кообразной смеси: 20% Р (красного), 79% А12О3 и 1% NH4C1.
Насыщенные образцы имели серебристо-золотистую блестящую
1 Коэффициенты диффузии фосфора в железе определены в работе Гру-
зина П. Л. и Мураля В. В. Изучение диффузии фосфора в железе и его спла-
вах радиометрическим методом. Проблемы металловедения и физики металлов.
Металлургиздат, 1964.
311
Рис. 224. Диаграмма <
системы Fe — Р
поверхность. Диффузионный слой выявлялся в виде нетравя-
щихся столбчатых зерен твердого раствора фосфора в а-железе
(рис. 224 и 225). Толщина зоны столбчатых зерен составляла
0,3 мм. При охлаждении в поверхностной зоне столбчатых зерен
на глубине до 0,05 мм из a-твердого раствора выделялись в виде
мелких темных включений ча-
стицы фосфида железа Fe3P.
После насыщения - фосфо-
ром армко-железа твердость
повышается не только в зоне
столбчатых зерен, но и в зна-
чительно более глубоких зо-
нах (рис. 226), в которых на-
блюдается повышенное со-
держание углерода и фосфо-
ра. Наибольшей твердостью
обладает поверхностная зона
столбчатых зерен с включе-
ниями фосфида. Другие свой-
ства диффузионного слоя, об-
разующегося при насыщении
железа фосфором, изучены не-
достаточно.
Обнаружено, что при насы-
щении легированных низкоуг-
леродистых сталей фосфором
в порошке феррофосфора
(23,5% Р) марганец, хром и
никель диффундируют из на-
сыщенного фосфором слоя к
поверхности и переходят в по-
рошок феррофосфора (рис.
227). Лишь в стали, легиро-
ванной 3,66% Мп (рис. 227,а),
наблюдалось небольшое от-
теснение марганца в более
глубокие слои. Обращает на
себя внимание тот факт, что
глубина поверхностного слоя стали, обедненного хромом, мар-
ганцем и никелем, совпадает с глубиной слоя, насыщенного фос-
фором. В противоположность этому поверхностное насыщение
фосфором стали, легированной молибденом, не вызвало обед-
нения поверхностных слоев стали этим элементом.
Глубина диффузии фосфора оказалась наибольшей у стали,
легированной молибденом и хромом (элементами, увеличиваю-
щими „устойчивость феррита), несколько меньшей — у нелегиро-
ванной стали и наименьшей — у стали, легированной марганцем
312
и никелем (элементами, увеличивающими устойчивость аусте-
нита).
Таким образом, фосфор способствует обеднению а-твердого
раствора элементами, благоприятствующими отпускной хрупко-
сти (Мп, Сг, Ni). Возможно, это происходит потому, что фосфор
Рис. 225. Микроструктура армко-железа, насыщенного
фосфором в порошке (20% Р, 79% А12О3 и 1% NH4C1)
при 1150° С в течение 8 ч (Х150)
Рис. 226. Распределение
микротвердости (нагруз-
ка 50 Г) по глубине слоя
армко-железа, насыщен-
ного фосфором (микро-
структура показана на
рис. 225)
увеличивает подвижность этих элементов или вследствие того,
что они стремятся к диффузионному перераспределению в на-
правлении обогащения растворов, наиболее богатых фосфором
313
рующих элементов в поверхностном слое
стали после насыщения фосфором в ва-
кууме при 1000° С в течение 192 ч. Ис-
следование проводилось на низкоуглеро-
дистых сталях (0,05—0,<10% С): деле-
гированной (А), легированных марган-
цем (Б, В), никелем (Г), хромом (Д)
и молибденом (Е ,и Ж) (438]
(порошок феррофосфо-
ра). В противополож-
ность этому фосфор не
оказывает такого воздей-
ствия на молибден, не-
большого количества ко-
торого достаточно - для
устранения отпускной
хрупкости стали.
Полученные результа-
ты могут оказаться по-
лезными при изучении
сущности причин отпуск-
ной хрупкости стали. В
монографии [439] резуль-
таты данной работы не
обсуждаются, и ее автор
не пытался как-либо увя-
зать их с предложенной
им гипотезой о причинах
отпускной хрупкости ста-
ли. Процесс насыщения
стали фосфором не при-
меняется, не используют-
ся в технике и фосфиды
железа. Однако фосфиды
некоторых других ме-
таллов !, например тита-
на и хрома, обладающие
химической стойкостью и
особыми электрическими
и магнитными свойства-
ми, применяются при из-
готовлении полупровод-
никовых приборов, в тех-
нике пайки и т. д.
Недавно1 2 предложе-
но в целях повышения из-
носостойкости подвергать
аустенитные стали по-
верхностному насыщению
фосфором (950° С, 10 ч
в порошке, содержащем
1 Фосфиды металлов можно получать осаждением из газовой фазы, из
легколетучих галогенных соединений и фосфида, электролизом из расплавлен-
ной среды, воздействием фосфида на нагретый металл и другими методами.
2 Реферативный журнал «Металлургия», 1965, № 1, стр. 111.
314
древесный уголь, Р2О5 и ВаСОз), закалке с 1100° С для получе-
ния твердого раствора и старению при 700° С. В результате полу-
чается упрочненный слой глубиной около 1 мм с поверхностной
твердостью HV 550.
СУРЬМИРОВАНИЕ
Сурьма применяется в качестве добавки к цветным сплавам
(латуни, бронзе, баббиту) для повышения их антикоррозионных
свойств и износостойкости.
Сведения о влиянии сурьмы на свойства стали весьма скуд-
ны, сурьму относят к числу элементов, отрицательно влияющих
на свойства стали [59].
При введении сурьмы в ваграночный чугун оказалось, что
содержание в нем 0,30—0,65% Sb резко снижает его прочност-
ные свойства, но существенно повышает износостойкость и анти-
коррозионные свойства (в морской воде, в растворах щелочей
и некоторых кислот) [440].
Первые исследователи осуществляли сурьмирование стали
в смеси окиси сурьмы и древесного угля [440], [441]. Режимы
процессов и результаты изучения свойств сурьмированной стали
в этих работах не приведены. Указано лишь, что можно полу-
чать сурьмированные слои глубиной до 1 мм с содержанием
до 4—6% и более сурьмы.
Предлагалось сурьмировать сталь в парах сурьмы в вакууме
(2-10-2—5-10-2 мм рт. ст., или 2,66—6,66 н/м2) [442]. При 550° С
за 3 ч на образцах образовывался слой глубиной 15 мк со сред-
ней концентрацией сурьмы 68%, что соответствует соединению
FeSb. Коррозионная стойкость сурьмированных по этому режи-
му образцов технического железа и сталей 20, 45, У8 и У12
повысилась в среднем в 3 раза при испытании в 1%-ном рас-
творе HNO3 и в 2 раза при испытании в 10 %-ном растворе
NaCl.
В экспериментах, поставленных автором, насыщение проис-
ходило за счет сурьмы, восстановленной из трехокиси сурьмы
углеродом древесного угля:
2Sb2Os + ЗС = 4Sb + ЗСО2
или
Sb2O3 + ЗСО = 2Sb + ЗСО2*.
Результаты изучения влияния различных факторов на глу-
бину сурьмированного слоя, образующегося на углеродистых
* Сурьмирование можно также осуществлять в газовых средах:
SbCl3 + Н2(НС1) (температура испарения SbCl3 80—100° С) и SbH3 4-Н2
[254].
315
сталях, приведены на рис. 228. Поверхность образцов после
сурьмирования получалась матовой с желтым оттенком. Коли-
чество БЬгОз в смеси должно быть не более 7—8%. Повышение
Рис. 228. Влияние продолжительности процесса (а), тем-
пературы (б), содержания в смеси трехокиси сурьмы
(в) и углерода в стали (г) на глубину сурьмированного
слоя сталей 10, 45 и У12 (автор и Е. В. Гальперина):
а — 950° С; 30»/о Sb2O3; б — 4 ч\ 30% Sb2O3, в — 95(F С, 6 ч;
г — 950° С; 6 ч, 30% Sb2O3
содержания БЬгОз в смеси способствует увеличению глубины
сурьмированного слоя, но ухудшает чистоту поверхности, на
которой образуются наплывы сурьмы. С увеличением выдержки
Таблица 52
Результаты испытаний износостойкости сурьмированной стали 45
на машине Амслера
№ сек- тора Состояние сектора Работа Момент в кГс м Коэффи- циент трения Уменьшение веса сектора в г
в кГ-м в н-м
1 2 Несурьмиро- ванный 253 260 2479 2549 18,7 19,7 0,48 0,48 0,0394 0,0306
3 4 Г груз: цеме щень Сурьмирован- ный Тримечание. Ис ке 25 кГ без смазки; нтованной стали ЗОХ л при 540° С. 137 143 пытания п] пройденнь ГТ (НРС 6: 1342 1401 РОИЗВОДИЛИ1 лй путь 1,2 2—64); ист 11,6 11,2 сь на трен км; ролик ытуемые cei 0,29 0,28 ие скольже машины из стора закал! 0,0206 0,0105 ния при на- готовлен ИЗ ены и отпу-
316
наплывы уменьшаются и исчезают вследствие диффузии сурьмы
в сталь.
Сурьмированный слой на углеродистых сталях выявляется
в виде нетравящейся светлой зоны (рис. 229). Под диффузион-
ным слоем находится зона с несколько повышенным содержа-
нием углерода (по сравнению с сердцевиной). Очевидно, вместе
с сурьмой в сталь диффундирует небольшое количество углерода
из присутствующего в газовой фазе СО. Возможно также допу-
стить оттеснение углерода к сердцевине за счет образования
твердого раствора сурьмы a-железе при температуре насы-
щения:
При сурьмировании стали Х18
(0,9—1,0% С, 17—19% Сг) на
углах образцов, где концентра-
ция сурьмы была высокой,
иногда наблюдалось (по-видимо-
му, в результате перехода линии
солидуса) возникновение гекса-
гональных включений сурьми-
дов со спиралями роста (рис.
231). Базисные грани сурьмидов,
имеющих форму призм, распола-
гались преимущественно парал-
лельно плоскости шлифа и пер-
пендикулярно сурьмированной
поверхности.
Если до закалки сурьмиро-
ванный слой стали 45 обладал
большей твердостью, чем сердце-
вина, то после закалки, наоборот,
несколько меньшей (рис. 232).
Сурьмированная и затем улуч-
шенная сталь 45 имеет более вы-
Рис. 229. Микроструктура стали
10 после сурьмировании
сокую износостойкость по сравнению с несурьмированной, что
обнаружилось при испытаниях секторов, изготовленных из этой
стали, на машине Амслера (табл. 52). Испытания на разрыв
сурьмированных и затем улучшенных образцов стали 45 пока-
зали, что предел прочности и текучести после сурьмировании
практически не изменяется, а относительные удлинение и суже-
ние площади поперечного сечения немного уменьшаются.
Коррозионные испытания образцов стали 45 показали незна-
чительное повышение после сурьмировании их стойкости в водо-
проводной воде и более существенное — в синтетической мор-
ской воде и в 10%-ных водных растворах НС1 и H2SO4
(рис. 233). Коррозионные испытания стали 10 показали несколь-
ко иные результаты. В синтетической морской воде эффект
сурьмирования был такой же, как и для стали 45, но в водо-
317
Рис. 231. Микроструктура сурьмиро-
ванной стали Х18 (1050° С, 4 ч),
снятая с угла образца
(Ковалев Л. И. и автор)
Рис. 232. Распределение
микротвердости по глубине
сурьмированных слоев ста-
лей 20 и 45 после охлажде-
ния стаканчиков на возду-
хе и после последующей
закалки стали 45 с 850°.
(автор и Е. В. Гальпери-
на)
проводной воде и 10%-ном растворе НС1 преимущество сурьми-
рованной стали перед несурьмированной оказалось незначи-
тельным.
КАРБОСУРЬМИРОВАНИЕ
Для карбосурьмирования ав-
тор использовал смеси, состоя-
щие из древесного угля, углекиС'
лого натрия (около 15%) и
трехокиси сурьмы (0,5—1,0%)
или из стандартного Бандюжско-
го карбюризатора, углекислого
натрия (около 10%) и трехокиси
сурьмы (0,5—1,0%). Карбосурь-
мирование, по-видимому, можно
также производить в газовой сре-
де, вводя в реакционное про-
странство или пары SbCh либо
другого содержащего сурьму ком-
понента, или жидкость (керосин,
синтин и др.), в которую добав-
лен в виде раствора либо поро-
шок тонкого помола содержащий
сурьму компонент, например
SbCl2.
После карбосурьмирования в
порошкообразных смесях ука-
занного состава при температуре
950° С и выдержках 2—6 ч глуби-
на получаемого слоя оказыва-
лась всегда меньшей, чем при
обычной цементации по такому
же режиму.
Отличие микроструктур кар-
.босурьмированного и цементо-
Рис. 233. Коррозионная стой-
кость в различных средах не-
сурьмированной (/) и сурьми-
рованной (2) стали 45 (автор
и Е. В. Лабутина):
ванного слоев заключается в том,
что в самой поверхностной зоне
а — синтетическая морская вода:
б — 10%-ный раствор НС1;
в — 10%-ный раствор H2SO4
первого из них наблюдается сла-
ботравящаяся каемка с высокой концентрацией сурьмы. Толщи-
на этого слоя в зависимости от длительности процесса и состава
смеси может колебаться от нескольких сотых до 0,10—0,15 мм.
С увеличением в смеси содержания трехокиси сурьмы
(в пределах 0,25—5%) концентрация углерода в слое, эвтек-
тоидная и общая глубина слоя уменьшаются, а толщина слабо-
травящейся каемки увеличивается (рис. 234). Твердость карбо-
сурьмированной слаботравящейся каемки несколько выше
319
твердости соседних, более глубоких зон слоя. После закалки
твердость этой каемки несколько ниже твердости зоны, располо-
женной глубже.
Слаботравящаяся каемка, богатая сурьмой, является двух-
слойной. Внешняя, более толстая часть этой каемки двухфазная.
Возможно, в этой части при охлаждении с температуры насы-
щения происходит распад a-твердого раствора с образованием
Рис. 234. Микроструктура по-
верхностной зоны карбосурьми-
рованной стали марки 15
(950° С — 2 ч\ смесь: древесный
уголь, 15% Na2CO3 и 5%
Sb2O3) (Х84)
е-фазы, обнаруживаемой под микроскопом в виде темных игл.
Далее расположен нетравящийся
слой а-фазы, не распадающийся при
охлаждении.
Рентгеноспектральный локаль-
ный анализ диффузионного слоя на
микрозонде (выполненный под ру-
ководством С. Б. Масленкова) и
рентгеноструктурный анализ (вы-
полненный под руководством Л. Н.
Расторгуева) показали, что концен-
трация сурьмы в очень тонкой по-
верхностной зоне высокая (более
60%). Расчет дебаеграмм, снятых
при кобальтовом и хромовом излу-
чениях, показал наличие в этой по-
верхностной зоне слоя соединений
типа FeSb и FeSb2 *.
Коррозионная стойкость карбо-
сурьмированной стали в течение
150 г в морской воде (5 г KJ, 10 г
КВг, 30 г NaCl на 1 л воды) и в
10%-ном растворе НС1 оказалась в
среднем в 2 раза выше, чем цемен-
тованной, а в 10%-ном растворе
H2SO4 — в 2,5 раза выше.
Износостойкость карбосурьми-
рованной стали ЗОХГС испытыва-
лась на машине Амслера при тре-
нии качения с проскальзыванием. Оказалось, что в зависимости
от условий насыщения и методики испытаний износостойкость
карбосурьмированной стали колеблется в пределах от износо-
стойкости цементованной стали до более высокой (примерно в
* Объем кристаллической решетки равновесного сурьмида FeSb(B8) 74,
779кХ, а обнаруженного карбосурьмида FeSb (С) 63, 746кХ, соответственно
для FeSb2(C18) 120, 990кХ и для FeSb2(C) 113, 45кХ. Это сокращение объема
решетки, очевидно, связано с дефицитом атомов железа в образовавшихся
соединениях. Соединение FeSb2 возникает благодаря присутствию на поверх-
ности стали в процессе насыщения очень тонкой пленки расплавленной сурь-
мы (см. рис. 230).
320
Таблица 53
Результаты испытаний свойств высокоуглеродистой стали (1,1% С) с различным содержанием сурьмы1
(автор и М. Ю. Круминьш)
Содержание в стали сурьмы в % Твердость °пч при изгибе Износостойкость Коррозионная стой- кость (уменьшение веса в кг/м2) Окалино- стойкость (900° С. 12 ч) (увеличение веса в г'м2)
в отож- женном состоянии HRB после закалки и отпуска при 180° С HRC в кГ'мм2 в н/м2-107 На машине Амслера2 уменьшение веса в г На машине Х2М* в морской воде (147 ч) в 10%-ном растворе H2SO4 (107 ч)
Обт ем углубле- ния X 10s мм3 Относи- тельный изное
0 88 63,5 234 229 0,0418 30,1 0,93 21,0 34085 312,6
0,37 92 63,5 180 176 0,0221 35,2 0,80 15,5 11210 359,3
0,64 93 64 151 148 0,0200 38,2 0,73 — — 427,0
0,99 95 64 150 147 Отколы на углах колец 39,7 0,70 — — 600,5
1,62 97 63,5 105 103 То же 42,4 0,66 — — —
Влияние сурьмы на свойства стали Немного повышает Не оказы- 4 вает влияния Резко снижает Повышает при трении качения Понижает абра- зивную износо- стойкость в кор- розионной среде Повышает Снижает
1 Испытания проводились на образцах, закаленных и отпущенных при
2 Износ после 50 000 оборотов.
* Испытания на машине Х2М проводились в Институте машиноведения
180° С (HRC 63—64).
__________ ___ . АН СССР канд. техн, наук М. А. Бабичевым. Эта машина
имеет диск из твердого сплава, охлаждаемый 0,5%-ным раствором К2Сг2О7 в дистиллированной воде. За эталон принят образец
стали ШХ15 (относительный износ 1,0); замеры объема лунки износа производили после 2000 оборотов диска. Пониженная изно-
состойкость стали, содержащей сурьму, при таком виде испытания является закономерной в связи с тем, что на поверхности та-
йкой. стали в коррозионной среде (0,5%-ный раствор К2Сг2О7) непрерывно образуются коррозионно-стойкие, пассивные, хрупкие,
. рыстроизнашивающиеся пленки. Коррозионно-стойкие стали при таком методе испытаний вс тда показывают пониженную износо-
стойкость по сравнению с обычными сталями, имеющими такую же твердость (44 3].
2 раза) износостойкости (например, после обработки в смеси,
содержащей 0,75% Sb2O3, и при испытании на трение карбосурь-
мированного кольца по цементованному сектору). Карбосурь-
мированная сталь обладает несколько более высоким сопротив-
лением задиру.
Из механических свойств карбосурьмированной стали опре-
делялась лишь ударная вязкость. У карбосурьмированных
образцов стали ЗОХГТ ударная вязкость (после термообработ-
ки) оказалась выше, чем у цементованных, и составила 1 —
1,7 кГ-м/см2 (0,98-105—1,6-105 н-м!м2) вместо 0,80—0,85 кГ-м/см2
(0,78 • 105—0,83 - 105 н-м/м2). Это можно объяснить тем, что
в карбосурьмированной стали поверхностная слаботравящаяся
каемка имеет несколько пониженную твердость.
Прочностные свойства и особенно предел выносливости карбо-
сурьмированной стали должны быть несколько ниже, чем цемен-
тованной и нитроцементованной, так как сурьма снижает меха-
нические свойства стали (табл. 53), а поверхностный слой карбо-
сурьмированной стали имеет пониженную твердость.
НАСЫЩЕНИЕ ТАНТАЛОМ, ЦЕРИЕМ, МЕДЬЮ, ОЛОВОМ
И МЫШЬЯКОМ
Насыщение танталом {281]. Насыщение низкоуглеродистой
стали танталом осуществлялось в порошкообразной смеси (4 ч.
тантала и 1 ч. А12О3) в среде водорода. После обработки при
температуре 1100° С и выдержках 1 и 2 ч глубина диффузион-
ного слоя достигала соответственно 0,06 и 0,12 мм, а при
1200° С — соответственно 0,24 и 0,36 мм.
Диффузионный слой состоял из a-твердого раствора и со-
единения Fe2Ta.
При насыщении танталом средне- и высокоуглеродистой ста-
ли образуется тонкий диффузионный слой, богатый карбидом
тантала, с микротвердостью около 1500*.
Практическое применение за рубежом нашло осаждение на
поверхности стали слоя почти чистого тантала из газовых
и жидких сред для повышения коррозионной стойкости (см.
гл. VIII).
Насыщение церием [444]. Насыщение церием производилось
при 1000—1100° С в парах церия, образующихся в разреженной
среде. В реторту раздельно помещались ферроцерий и образцы
стали. Глубина слоя после насыщения при 1100° С в течение
8 ч достигала 0,30 мм (сталь с 0,47% С).
Микротвердость железа и стали после длительных (100 ч)
процессов насыщения церием при 1000° С повышается до
Яр. 285—415. Окалиностойкость железа и стали после церирова-
ния почти не изменяется.
* По литературным данным [332], микротвердость ТаС Нр. 1590.
322
Рентгеноструктурный анализ показал, что в диффузионном
слое, помимо a-твердого раствора, присутствует церид РезСег.
Насыщение медью. Насыщение производилось в порошко-
образной смеси (50% Си и 50% AI2O3) в среде водорода.
В течение 1; 2 и 4 ч глубина слоя при 1200° С достигала 70, 87
и 118 мк, а при 1250° С — соответственно 74, 93 и 124 мк. Слой,
судя по травимости, представляет собой, очевидно, эвтектоид
а + е (растворимость меди в a-фазе и е-фазе — твердом раство-
ре железа в меди ничтожна). На поверхности и по границам
столбчатых зерен диффузионного слоя, образовавшегося при
температуре 1200° С и выше, обнаружены выделения свободной
меди [281], [455].
Насыщение медью железа повышает его коррозионную стой-
кость в йекоторых средах. При испытаниях в 10%-ных растворах
соляной кислбты медненые образцы оказались почти в 20 раз
более стойкими, чем немедненые (длительность испытания
10 суток). Хорошие результаты были получены при атмосфер-
ных испытаниях: в течение 6 месяцев внешний вид образцов не
изменился.
При испытаниях в 3%-ном растворе хлористого натрия и
5 %-ном растворе сернокислого натрия насыщение медью эффек-
та не дает.
Насыщать сталь медью можно и в газовой среде CU3CI2 +
+ Нг(НС1). Температура испарения CU3CI2 500—700° С [251].
Насыщение оловом. Сталь насыщалась оловом в порошко-
образной смеси (50% олова и 50% кварцевого песка) в ваку-
уме [442]. Диффузионный слой, полученный при 1000—1150° С,
представлял собой столбчатые зерна, являющиеся твердым
раствором олова в a-железе. После обработки при 1150° С
в продолжение 2 ч концентрация олова по глубине столбчатых
зерен снижалась с 3,2 до 2,1% и затем на глубине 0,20 мм —
на линии раздела — резко падала. После обработки при 1050° С
в течение 2 ч толщина слоя столбчатых зерен снижалась до
0,05 мм, а концентрация олова в поверхностной зоне этого слоя
составляла 2,5%. Судя по диаграмме состояния системы Fe—
Sn, предел растворимости олова в у-железе при 1150° С состав-
ляет примерно 1,9%, а в a-фазе примерно 16%. При 300°С
растворимость олова в a-фазе падает до 9%. Таким образом,
концентрация олова в столбчатых зернах значительно ниже
предельной растворимости олова в (i-фазе. Увеличение в стали
содержания углерода с 0,04 до 1,27% приводит к уменьшению
глубины слоя в 3 раза, а количества поглощенного сталью олова
примерно в 2 раза.
После насыщения оловом коррозионная стойкость стали
в 1%-ных растворах соляной и серной кислот увеличилась
в 3—5 раз, в 0,5 %-ном растворе хлористого натрия не измени-
лась. Насыщение стали оловом можно проводить также в газо-
323
вой среде SnCl2 + Н2 (температура испарения SnCl2 500—
550° С) [254].
Насыщение мышьяком. Сталь насыщалась мышьяком в ваку-
уме (10~2—10-3 мм рт. ст., 'или 1,33—1,135 н/м2) и в порошко-
образной смеси (2—4 ч. мышьяка и 1 ч. А120з). После насыще-
ния в течение 2; 4 и 6 ч глубина диффузионного слоя при 750° С
составляла соответственно 62, 156 и 208 мк, а при 800° С — со-
ответственно 140, 210 и 240 мк. Диффузионный слой выявлялся
в виде слаботравящейся светлой зоны — очевидно, твердого рас-
твора мышьяка в a-железе. Содержание мышьяка в слое не
определялось.
Насыщение железа мышьяком повышает его коррозионную
стойкость, например, в 10%-ных растворах серной и соляной
кислот в 10—15 раз (длительность испытаний 10 суток) [281].
Насыщать сталь мышьяком можно также в газовой среде
AsCl3 + Н2(НС1) [254].
ГЛАВА III
ХИМИКО-ТЕРМИЧЕСКАЯ ОБРАБОТКА ТИТАНА
Титан и сплавы на его основе, имея малый удельный вес, вы-
сокие механические свойства и удельную прочность, очень высо-
кую коррозионную стойкость и т. д., обладают низкой износо-
стойкостью и склонностью к налипанию и образованию задиров
при работе в узлах трения [446], [447]. Это ограничивает -приме-
нение титановых сплавов для изготовления деталей, работаю-
щих на трение. Легирование титана и термическая обработка
титановых сплавов существенно не изменяют их износостойко-
сти. Не дает эффекта и применение жидких смазок, так как их
молекулы не удерживаются на поверхности титана.
Износостойкость титана и его сплавов может быть повышена
химико-термической обработкой и в некоторых случаях гальва-
ническим хромированием и химическим никелированием. Хими-
ко-термическая обработка титана и его сплавов затруднена
большим сродством титана к кислороду, водороду, азоту, хлору»
поэтому активные среды, обычно используемые для насыщения
стали, в большинстве случаев не пригодны для обработки тита-
на. Из опробованных методов химико-термической обработки
титана для промышленного применения наиболее подходят азо-
тирование в азоте, окисление и цементация. Основной недоста-
ток этих методов заключается в том, что для получения упроч-
ненных слоев требуемой глубины обработку приходится произво-
дить при высоких температурах и больших выдержках, что
вызывает рост зерна титана ^и ухудшение1 его механических
свойств.
Кроме перечисленных методов обработки титана, в особых
случаях могут применяться и другие, с помощью которых может
достигаться поверхностное упрочнение, повышение окалино-
стойкости или коррозионной стойкости титана.
АЗОТИРОВАНИЕ В АЗОТЕ
Азотированию титана в азоте посвящено значительное коли-
чество работ как в СССР [448]—[452], так и за рубежом [453]—
[460].
325
Для азотирования чаще применяется технический азот, очи-
щенный от кислорода и влаги. Осушка азота производится пу-
тем его пропускания через сосуды с силикагелем, алюмогелем,
фосфорным ангидридом. В качестве индикатора для определения
у
f)
Рис. 235. Схемы установок для азотирования титановых сплавов в азоте:
а — схема Е. Н. Новиковой (/ — баллон с азотом; 2 — кислородный редуктор; 3 —
реометр; 4 — силикагель; 5 — фосфорный ангидрид; 6 — печи для очистки азота;
7 — геттер, дегазированная стружка титана; 8 — горизонтальная печь для азотирования;
9 — вертикальная печь для азотирования; 10 — детали и контрольные образцы; И —
термопары; 12 — контрольная колба (с водой); б — схема, предусматривающая про-
ведение после азотирования отжига <в аргоне f450] (/ — баллоны; 2 — реометры; 3 — си-
ликагель; 4 — печь с медной стружкой; 5 — холодильник; 6 — алюмогель; 7 — фильтр;
8 — фосфорный ангидрид; 9 — печь с титановой стружкой; 10 — печь для азотирования)
степени влажности газа можно использовать силикагель, про-
питанный 5%-ным раствором хлористого кобальта. Для очистки
от кислорода азот следует пропускать через печь, наполненную
дегазированной титановой стружкой или губкой, нагретыми до
850—900° С (рис. 235). Применяются и более сложные очисти-
тельные установки.
326
Результаты азотирования практически мало зависят от рас-
хода азота в изученных пределах (табл. 54). Незначительное
влияние на результаты азотирования расхода азота, варьируе-
мого в более широких пределах, было подтверждено в работе
[450]. Азотирование осуществлялось при 950° С в продолжение
3 ч, различие между минимальным и максимальным расходом
азота достигало 50 раз. В этой работе было 'показано, что улуч-
Рис. 236. Зависимость глубины
азотированного слоя литого
сплава титана от продолжи-
тельности азотирования при 950
и 1050° С (автор и Л. Я. Кон-
тер)
Рис. 237. Распределение микротвердо-
сти по глубине диффузионного слоя
литого сплава титана, азотированного
при температуре 950° С и различных
выдержках (автор и Л. Я. Контер)
шение очистки азота от кислорода (печь с медной стружкой
была заменена печью с титановой стружкой) приводит к замет-
ному увеличению глубины азотированного слоя.
Таблица 54
Влияние расхода азота на результаты азотирования
литого сплава титана (Л) и металлокерамического сплава (М)
(Автор и Л. Я. Контер)
Расход азота в см31 мин Сплав М Сплав Л
Увеличение веса образцов в мг[см* Глубина слоя в мм* 1 Увеличение веса образцов в мг/см* Глубина слоя в мм
5 1,20 0,060 1,12 0,040
32 1,27 0,055 1,17 0,042
75 1,25 0,058 1,15 0,043
♦ Азотирование производилось в трубке диаметром 25 и длиной 200 мм по
режиму: температура 1050° С, выдержка 4 ч. За глубину слоя принималось рас-
стояние от поверхности до конца залегания слаботравящейся зоны.
327
Рис. 238. Влияние продолжительности
азотирования при 950° С на микро-
твердость азотированного слоя раз-
личных сплавов титана, измеренную
на расстоянии 0,015—0,02 мм от по-
Азотирование титана при 850° С протекает (Медленно, и при
выдержке 24 ч образуется диффузионный слой глубиной в не-
сколько сотых миллиметра. Поверхностная твердость повы-
шается при этом с HV 350 до HV 670, а микротвердость дости-
гает Яр. 800. При 900° С и особенно при 950° С и более высокой
значительно быстрее. На
рис. 236—238 приведены ре-
зультаты определения мик-
ротвердости и глубины слоя
на образцах металлокера-
мического и литого титана,
обработанных при 950 и
1050° С. Поверхностная
твердость после азотирова-
ния при температуре 900—
1050° С и выдержке 8—32 ч
лежит в пределах HV 700—
860, а микротвердость до-
стигает Я(Х 1200—1350.
В результате даже крат-
ковременного азотирования
верхности:
/ — литой сплав с 9% вольфрама; 2 —
металлокерамический титан; 3 — литой
титан (автор и Л. Я. Контер)
заметно понижается проч-
ность и особенно пла-
стичность сплава титана
(табл. 55).
При более высоких температурах -и больших выдержках азо-
тирования механические свойства сплава снижаются сильнее
вследствие роста зерна и увеличения глубины проникновения в
титан азота \ Поэтому не следует рекомендовать азотирование
при температурах выше 930—950° С.
Таблица 55
Механические свойства литого сплава титана до и после
азотирования
Состояние образца ав 5 в % Ф в %
в кГ/мм* в н!м*Х 1 О’ в кг;мм* в н/м2Х 1 0’
До азотирования . . После азотирования 88 76 86 74 77 64 75 63 17 3 40 5
Примечания: 1. Режим азотирования: температура 950° С, выдержка 4 ч. 2. Изломы азотированных образцов крупнозернисты.
1 При глубине азотированного слоя 0,06—0,08 мм (определена микро-
анализом) общая глубина слоя с повышенным содержанием азота составляет
1,5 мм [448].
328
В табл. 56 приведены результаты испытания .на износостой-
кость азотированных и неазотированных секторов из литого
сплава титана. Износ азотированных секторов (судя по умень-
шению веса) в десятки раз меньше, чем неазотированных. Один
из секторов после испытания в течение 37 мин был подвергнут
дополнительному испытанию в продолжение 4 ч, но даже при
этом его весовые потери оказались в 2 раза меньше, чем у неазо-
тированных секторов, испытанных в течение 37 мин. Уменьшение
веса секторов из закаленной стали 45 и цементованной стали 20
в несколько раз больше, чем азотированных секторов из сплава
титана (табл. 56), что указывает на высокую износостойкость
поверхностных зон азотированного слоя титана.
Таблица 56
Результаты испытания износостойкости азотированных секторов
из литого сплава титана на машине Амслера (автор и Л. Я. Контер)
Материал сектора Температура азотирования в °C* Уменьшение веса сектора в г Уменьшение веса сек- тора после азотирования (число раз)
Неазотирован- ный 0,1986 0,1910 —
850 850 0,0020 0,0015 97,5 130
Сплав титана 900 900 0,0010 0,0014 195 139
950 950 0,0020 0,0054 97,5 36
1050 1050 0,0018 0,0034 108 57
1050 0,0934** —
Сталь 45 (HRC 59) — 0,0130 0,0104 —
Сталь 20, цемен- тованная при 925° С; 10 ч (HRC 62—64) Пр имечани кой 25 кГ в продо дость HRC 62—64) * Выдержка 24 *♦ Продолжител е. Испытания проводил лжение 37 мин. Диски С ч. ьность испытания 4 ч 37 0,0094 0,0064 ись при трении ско. >ыли изготовлены из мин. льжения с нагруз- стали У12 (твер-
329
По данным .работы [448] <при испытании на машине Амслера
азотированных роликов из технического титана в паре с азоти-
Рис. 239. Микроструктура металлокерамического титана, азотированного в те-
чение 16 ч при 950°С (а), и литого сплава, азотированного в течение 4 ч-
при 1050°С (б). Х400 (автор и Л. Я Контер)
рованными роликами из стали 38ХМЮА в среде керосина первые
не имели заметного износа, тогда как износ вторых оказался
ощутимым L
Таблица 57
Циклическая прочность при кручении азотированных образцов
технического титана [461]
Режим азотирования Предел усталости при кручении
Температура в °C Выдержка в ч в кГ{ммг в «Си1 2х 107 в %
Неазотир 800 800 800 900 900 П р и м е ч а глубина азотир личением темпе юванный 1 5 10 5 36 н и е. Поверхнос' ованного слоя ко/ ‘ратуры и продола 23 24 22 19 18 17 гная твердость oi гебалась от 0,006 кительности проце 22,5 23,5 21,6 18,6 17,6 16,7 бразцов достигала до 0,08 мм (возра сса). 100 104,5 95,5 82,5 78,5 74 800, а стала с уве-
1 Коэффициент трения азотированной поверхности титана по азотирован-
ной поверхности стали равен 0,14.
330
Рис. 240. Диаграмма состояния си-
стемы Ti—N
Азотирование сущестйвенно «снижает 'циклическую прочность
технического титана (табл. 57).
Микроструктура азотированного слоя титановых сплавов из-
меняется в зависимости от состава сплава и режима обработки.
На рис. 239 приведена микроструктура азотированного слоя,
полученного на разных «сплавах при разной температуре. По-
верхностная ветравящаяся хрупкая корочка (рис. 239, а), тол-
щиной в несколько микрон,
как показал рентгеноструктур-
ный анализ, является нитри-
дом титана (рис. 240) с гране-
центрированной кубической
решеткой. Нитрид титана име-
ет широкую область гомоген-
ности, начинающуюся при со-
держании 10,9% N (соответ-
ствует TiNo42) и доходящую
до 22,6% N (TiN). Микро-
твердость TiN, по литератур-
ным данным, около Нр. 1900
[332]. Корочка нитрида титана
начинает возникать еще при
нагреве титана до температу-
ры азотирования, при этом
поверхность титана приобрета-
ет золотистый цвет. Микро-
твердость этой корочки составляет обычно Нр. 1200—1600, а тол-
щина ее колеблется в пределах 4—20 мк.
Глубже располагается основной слой — широкая «светлая бо-
лее мягкая зона, основой которой является твердый раствор азо-
та в а-титане с гексагональной решеткой.
В верхней части зоны, соответствующей a-фазе, иногда на-
блюдается темная полоса, структура которой, очевидно, соответ-
ствует смесям фаз а + е* и 8 + TiN. Такая структура /может
образовываться при охлаждении с температуры азотирования.
В слоях, расположенных глубже светлой зоны (ее часто назы-
вают альфированной зоной), содержание азота невелико
(рис. 239).
В некоторых сплавах основной «слой, располагающийся глуб-
же нитридной корочки, представляет собой светлые включения
<х-титана, насыщенного азотом, в структуре основы оплава (фа-
зы а + р).
Скорость диффузии азота в титан исследована в интервале
900—1570° С [454], в котором она подчиняется параболической
е-Фаза, вероятно, соответствует соединению Ti3N (8,9% N).
331
зависимости!. Однако в самый начальный /период процесса было
обнаружено отклонение от этого закона — наблюдалась линей-
ная скорость диффузии. Диффузия азота в*р-титан, имеющий
решетку ОЦК, протекает быстрее, чем -в плотно упакованной
решетке а-титана (рис. 241). При этом полученные экоперимен-
Рис. 241. Температурная зависимость
коэффициента диффузии азота в
а- и р-титан [464]
Рис. 242. Температурная
зависимость коэффициен-
та диффузии азота в нит-
рид титана TiN [464]
тально формулы, связывающие коэффициент диффузии азота с
температурой в интервале 900—1570° С, имеют следующий
вид:
Dp -3,5
10 2 ехр
38 000 + 1400'
RT
Da = 1,2 • 10 2 ехр
45 250 ± 2250
RT
По другим данным энергия активации реакции азота с тита-
ном при температурах 800° С равна 45 400 кал! моль [447]. Диф-
фузия азота в нитриде TiN протекает значительно медленнее,
чем в а-титане (рис. 242). Экспериментальная формула, опреде-
ляющая Dtin, имеет следующий вид:
DT1N =5,4-10 3 ехр
52 000 ± 3500
RT
Легирующие элементы заметно влияют на общую глубину
азотированнного слоя и толщину нитридной зоны. Кремний
332
и алюминий (при содержании в сплаве до 2% Si и 4% А1) спо-
собствуют увеличению глубины азотированного -слоя, а хром,
железо и марганец—ее уменьшению. Глубину нитридной зо-
ны слоя увеличивают алюминий (при содержании его в сплаве
более 3%), хром, -кремний и железо [448].
Следует отметить, что образование на поверхности диффу-
зионного слоя тонкой зоны нитрида TiN нежелательно по двум
причинам:
1. Нитридная корочка очень хрупкая. Кроме того, она и при-
мыкающая к ней тонкая зона слоя имеют недостаточно плотное
строение, в результате чего при доводке поверхности азотирован-
ных деталей часто не удается получать чистоту обработки вы-
сокого класса.
2. Нитридная корочка замедляет диффузию азота в титан,
и скорость процесса азотирования фактически определяется ско-
ростью диффузии азота сквозь нитрид TiN.
Уменьшить толщину нитридной корочки можно путем азоти-
рования титана в чистом азоте при пониженном давлении [449]
или в смеси аргона и азота [450]. При азотировании в смеси
довольно трудно с большой точностью подавать в реакционное
пространство малые количества азота (0,1—0,05%), добавляе-
мые -к аргону. Кроме того, технический аргон часто содержит до
0,1% N2, что дополнительно усложняет точное регулирование со-
става газовой среды.
Азотирование сплава ВТ1-Д осуществлялось при 950° С в про-
должение 8 и 24 ч в чистом азоте и смесях азота и аргона, со-
державших 10%; 4,5% и 0,2% N2 [450]. После азотирования при
всех указанных соотношениях газов были получены образцы с
золотистой корочкой нитрида TiN. Увеличение веса и поверхно*
стная твердость образцов, а также общая глубина слоя и тол-
щина нитридной корочки оставались в тех же пределах, что и
при азотировании в чистом азоте. Однако в других условиях
экспериментов [452] даже при содержании в газовой смеси
0,5% N2 поверхность образцов сохранялась светлой, как и до
азотирования, т. е., судя по внешнему виду, нитридная корочка
не образовывалась. Данные рентгеноструктурного анализа под-
твердили, что при уменьшении количества азота в смеси не ни-
же 0,5% снаружи образуется тонкая зона, состоящая из а-твер-
дого раствора титана с включениями TiN, которая постепенно
переходит в зону твердого раствора азота в а-титане. Поверх-
ностный слой образцов, азотированных в смеси, содержащей бо-
лее 0,5% азота, состоял уже из смеси TiN + TiN*.
С увеличением содержания в смеси азота общая глубина
диффузионного слоя заметно уменьшается (рис. 243), хотя вес
образцов при этом увеличивается. Последнее, очевидно, связано
с образованием и постепенным утолщением нитридной каемки.
Увеличение продолжительности азотирования -свыше 24 ч (при
850° С) в смеси с '0,5% N уже вызывает -появление на 'поверхно-
сти титана нитридной корочки. Во 'Ивбежание этого при более
длительных выдержках рекомендовалось или применять перио-
дическую подачу в печь азота, или еще уменьшить его содержа-
ние в смеси [452].
Для рассасывания азота в слое, уменьшения толщины ни-
тридной корочки и снижения хрупкости слоя иногда проводят
дополнительный диффузионный отжиг в вакууме или среде ар-
гона. Рекомендуется 'Следующий оптимальный режим азотиро-
вания и отжига сплавов ВТ1-Д и ВТ5 (450]: азотирование при
температуре 950° С в течение 24 ч; диффузионный отжиг в среде
Рис. 243. Влияние со-
держания азота в сме-
си с аргоном на глу-
бину азотированного
слоя у иодидного ти-
тана (850° С, 4 ч)
[452]
аргона при той же температуре в тече-
ние 8 ч. Отжиг осуществляется в той же
печи по окончании азотирования. После
отжига уменьшаются хрупкость слоя,
толщина нитридной зоны, общая глуби-
на слоя (примерно на 0,01 мм) и немно-
го снижается микротвердость поверхно-
стного слоя (с Др. 1200—1290 до Ну.
1100—1260 у сплава ВТ1-Д) [450]. Ре-
зультаты обработки по такому режиму
приведены в табл. 58.
После обработки по указанным ре-
жимам сплав ВТ1-Д (сердцевина образ-
ца) имеет следующие механические
свойства: ов = 60 кГ!мм2 (59 * 107 h/ju2);
6 = 20%; Ф = 23% и ак — 1,4 кГ-м!см2
(1,37-105 н-м]м2), а сплав ВТ5 (температура процесса 950°С)
соответственно: 92 кГ]мм2 (90-107 н!м2)\ 14%; 28% и 6 кГ-м1см2
(5,9-105 н-м!м2).
На основании вышеизложенного авторы работы рекомендуют
титановые сплавы азотировать при температурах 900—950° С с
выдержкой 24—30 ч при пониженном содержании азота в газо-
Таблица 58
Глубина и микротвердость диффузионных слоев на образцах
сплавов ВТ1-Д и ВТ5 после азотирования и диффузионного отжига [450]
Сплав Температура азотирования и отжига в °C Глубина слоя в мм Микротвердость Hso
общая нитридной каемки поверх- ности на глубине от по- верхности
0,015 мм 0,045 мм
ВТ1-Д 950 0,095 0,008 1155 905 680
ВТ5 950 0,11 0,011 1460 980 645
ВТ5 980 0,13 0,015 1420 870 680
334
вой среде (в смеси Аг + N2) или в чистом азоте с последующим
отжигом в аргоне.
Глубина азотированного слоя на образцах разных сплавов
достигает при этом 0,08—0,13 мм, а поверхностная микротвер-
дость Яр. 1000—1400.
До азотирования и окончательной отделки поверхности де-
тали рекомендуется подвергать кратковременному отжигу в ва-
кууме (800° С, 2 ч) для снятия напряжений и уменьшения ко-
робления, происходящего при азотировании. После азотирова-
ния независимо от метода его осуществления рекомендуется про-
изводить дополнительный отжиг деталей в вакууме при разре-
жении 3-10_4 мм рт. ст. (0,04 н/м2) в течение 2 ч при 800° С.
После отжига уменьшается хрупкость диффузионного слоя и на
10—15% повышаются характеристики пластичности спла-
вов [448].
Для предотвращения скалывания нитридной корочки, обес-
печения необходимой чистоты поверхности и сохранения высо-
кой поверхностной твердости деталей рекомендуется [450]:
1. До азотирования притуплять у деталей острые углы и
оставлять на рабочих поверхностях соответствующий припуск
(с учетам изменения размеров деталей в процессе азотирова-
ния), за счет которого удаляется нитридная корочка путем со-
шлифовывания слоя толщиной 0,01—0,03 мм. На нерабочих по-
верхностях некоторых деталей целесообразно оставлять при-
пуск до 0,5 мм на сторону, который после азотирования уда-
ляется точением (инструментом из твердого сплава) и шлифова-
нием.
2. Притирку производить чугунными притирами с использо-
ванием шаржировочной пасты М28, окончательную доводку —
обкаткой на станке между чугунными дисками с использовани-
ем паст ГОИ (карбид кремния), ЭМ28, М7 и др. Снятие при-
пуска на нерабочих поверхностях производить инструментом из
твердых сплавов. Шероховатость азотированных рабочих по-
верхностей может быть доведена при этом до 10-го класса чи-
стоты.
В некоторых случаях применяемые в приборостроении зуб-
чатые колеса с малым модулем зацепления рекомендуется изго-
товлять из сплава ВТ14 (около 4,3% А1; 3,1% Мо и 1,5% V)
и подвергать азотированию и термической обработке (Е. Н. Но-
викова, Л. М. Никитина). Азотирование следует осуществлять в
среде азота при температуре 900—920° С и выдержке 24—30 ч.
При этом глубина азотированного слоя (до микротвердости
/7р. 500) достигает 0,1—0,12 леи; средняя величина микротвердо-
сти на глубине 0,01—0,02 мм составляет 1100—1150. После
термической обработки (закалка с 860° С в воде и старение
при 500° С в течение 16 ч) глубина азотированного слоя увели-
чивается на 0,03—0,04 мм, а микротвердость на глубине 0,01—
335
0,02 мм немного уменьшается (иногда на 100—200). Предел
прочности и относительное удлинение сплава ВТ14 (После азоти-
рования: Опч = 85-4-95 кГ/мм1 2 * * (83 • 107—93 • 107 н/м2) и 6 =
= 74-11%, а после последующей термической обработки:
апч= 100 4- 115 кГ/мм2 (98 • 107—113 • 107 н/м2} и. 6 = 4 4-6%.
На зубчатых колесах небольшого диаметра рекомендуется
оставлять припуск, так как после азотирования увеличение их
размера составляет 0,02—0,05 мм на диаметр, чего вполне до-
статочно для припуска на доводку1.
Одним из методов ускорения (Процесса азотирования титано-
вых сплавов является обработка при пониженном давлении.
Снижение давления с 760 мм рт. ст. (101 кн!м2) до 1 —
0,1 мм рт. ст. (133—13,3 н/м2) позволяет увеличить глубину азо-
тированного слоя при температуре 950° С и выдержке 8 ч на
0,024—0,063 мм (в 1,5—2,5 раза). При этом толщина нитридной
зоны, в которой коэффициент диффузии азота значительно ниже,
чем в а- и р-титане, уменьшается в несколько раз. Наибольший
эффект дает азотирование при пониженном давлении сплава
ВТ5: глубина азотированного слоя возрастала в 2—2,5 раза,
приращение веса образцов увеличивалось в 3 раза, толщина ни-
тридного слоя уменьшалась с 4—7 мк до 1—2 мк.
Следует отметить, что дальнейшее снижение давления до
3 • 10“2 мм рт. ст. (4 н!м2) приводит к резкому уменьшению глу-
бины азотированного слоя (для всех сплавов).
Были проведены эксперименты по азотированию титановых
сплавов с нагревом т. в. ч. [578]. Благодаря небольшим выдерж-
кам (5—15 мин) сердцевина деталей нагревается до высокой
температуры (950—1050° С) на очень короткое время, и поэто
му механические свойства сплава почти не изменяются.
АЗОТИРОВАНИЕ В АММИАКЕ2
Азотирование титана в аммиаке начали изучать раньше, чем
азотирование в азоте, но этот процесс оказался мало перспек-
тивным, так как сопровождается глубоким проникновением в ти-
тан водорода, что приводит к сильному охрупчиванию титана
Тем не менее процесс азотирования в аммиаке представляет
определенный интерес, особенно в связи с обнаруженными ан<у
малиями в кинетике образования азотированного слоя.
Применяемый для азотирования титана аммиак должен быть
тщательно очищен от влаги и кислорода. Для каждой темпера-
1 Доводку азотированных и термически обработанных зубчатых колес
следует производить на зубошевинговальных станках. При доводке исполь-
зуется паста ГОИ с добавкой 5 вес. % зерен синтетического алмаза (величи-
на зерен 6 мк). Притиры изготовляют из перлитного чугуна.
2 В данном случае термин «азотирование» применен условно, так как при
обработке в аммиаке титан насыщается не только азотом, но и водородом.
336
туры и выдержки должен быть установлен (пю глубине слоя
увеличению веса) оптимальный расход аммиака. Так, для ус
тановки с рабочим диамет-
ром 40 мм и длиной 200 мм "г/с”г\ у.—u. q -------------
оптимальный расход амми-
ака при температурах 850,
950 и 1050° С составляет со-
ответственно 15—20, 50 и
100 см21мин. Для другой
установки, несколько боль-
шего размера, оптимальный
расход аммиака при 950° С
составляет 75 см31мин
(рис. 244). Степень дис-
10 30 50 70 90 110 см3/мйн
Расход аммиака
Рис. 244. Влияние расхода аммиака
на увеличение веса образцов металло-
керамического (/) титана и литого
сплава титана с 5% хрома (2). Азо-
тирование проводилось при 950° в
продолжение 3 ч, (автор и Д. С. Вар-
кая)
социации аммиака при оп-
тимальных расходах была
весьма высокой (около
100%), что исключало воз-
можность ее измерения с
помощью обычного диссо-
циометра.
Влияние продолжительности
рования п-ри 850, 950 и 1050° С
процесса на результаты азоти-
•и установленных оптимальных
а) fi) 0)
Рис. 245. Влияние продолжительности азотирования при разных температу-
рах и оптимальном расходе аммиака на глубину азотированного слоя метал-
локерамического (/), литого (2) титана, сплава с 5% хрома (5) и 9% воль-
фрама (4) [462]:
а — при 850° С, расход аммиака 15—20 см*1мин-, б — при 950° С, расход аммиака
50 см?!мин\ в — при 1050° С. расход аммиака 100 см?!мин
расходах аммиака показано на рис. 245. Глубина азотированного
слоя в зависимости от продолжительности процесса изменяется
337
по кривой с максимумом. Оптимальная глубина видимой части
слоя достигается три определенной критической .продолжитель-
Рис. 246. Распределение микротвердости по
глубине азотированного слоя металлокера-
мического (/), литого (2) титана и сплава
с 5% Сг (5), обработанных при 950° С
в течение 3 ч (автор и Д. С. Баркая)
ности процесса, умень-
шающейся с повыше-
нием температуры азо-
тирования (рис. 245).
Максимум был обна-
ружен и на кривой,
показывающей изме-
нение глубины азоти-
рованного слоя в зави-
симости от темпера-
туры процесса. На-
пример, при выдержке
8 ч глубина слоя
на металлокерамиче-
ском сплаве увеличи-
лась с 0,01 мм при
850° С до 0,05 мм при
950° С, а затем умень-
шилась до 0,03 мм при
1050° С (рис. 245).
Данные, получен-
ные при микрострук-
турном анализе, подтверждены измерением микротвердости об-
разцов на разной глубине от поверхности. На рис. 246 приведен-
Рис. 247. Зависимость микротвердости металлокерами-
ческого (а) и литого (б) титана на расстоянии 0,7 мм
от поверхности от продолжительности азотирования (4621
ные типичные кривые распределения микротвердости по глубине
азотированного слоя для трех сплавов титана. Измерена была
также микротвердость образцов на глубине 0,7 мм. Хотя эта
глубина в несколько раз превышает глубину слоя, наблюдае-
338
мую под микроскопом, тем не менее микротвердость на таком
расстоянии от поверхности непрерывно повышалась с увеличе-
нием выдержки и температуры процесса, что указывает на глу-
бокое проникновение в сплав азота и водорода (рис. 247). •
Почти одновременно с работой, выполненной в СССР, ана-
логичное исследование было проведено в США [455], [460].
В этом исследовании также был -обнаружен максимум на кри-
вой изменения глубины -слоя в зависимости от .продолжительно-
сти и температуры (процесса, а также расхода аммиака. Для
большого интервала температур установлены оптимальные вы-
держки, обеспечивающие получение азотированных слоев мак-
симальной глубины [453]:
Температура в °C . . . 760 788 816 843 871 899 927
Выдержка в ч....... 150—250 60—90 40—60 20—30 10—20 2—6 2
Сравнение этих данных и данных рис. 245 с данными рис. 248
показывает, что найденные закономерности совпадают, хотя
абсолютные значения оптималь-
ной продолжительности процес-
сов различаются.
Причиной обнаруженных ано-
малий в изменении глубины азо-
тированного слоя (альфирован-
ной зоны) является, очевидно,
влияние водорода, который, в
противоположность азоту, резко
Рис. 249. Микроструктура тита-
на, азотированного при 850° С
в течение 24 ч. Х383
Рис. 248. Влияние продолжительности
азотирования при 843°С на глубину
слоя {460]
расширяет (3-область. Поэтому по мере увеличения в твердом рас-
творе содержания водорода возрастает предельное содержание
азота в p-фазе, превышение которого приводит к образова-
нию а-фазы. С увеличением выдержки процесса повышается кон-
центрация в азотированном слое водорода. В связи с этим в
339
прилегающих к сердцевине зонах слоя a-фаза должна превра-
щаться в p-фазу; следовательно, толщина альфированной зоны
будет уменьшаться.
Легирование титана хромом (рию. 244, 246) и вольфрамом
уменьшает глубину азотированного слоя. Оба эти элемента спо-
собствуют повышению твердости поверхностной зоны азотиро-
ванного слоя (рис. 238 и 246).
На рис. 249 показана микроструктура азотированного тита-
на. Поверхностная тонкая корочка имеет кристаллическую ре-
шетку типа NaCl (а = 4,225 А), что соответствует нитриду
TiN, в котором, несомненно, растворен также водород. Основная
светлая зона слоя — твердый раствор азота и водорода в а-ти-
тане с гексагональной решеткой, имеющей увеличенные пара-
метры по сравнению с ненасыщенным а-титаном. Наличие в
слое е-фазы ни на микроструктуре, ни рентгеноструктурным ана-
лизом в нашем исследовании обнаружено не было.
Для оценки влияния азотирования на износостойкость тита-
новых сплавов были проведены испытания образцов на машине
Амслера (табл. 59).
Таблица 59
Результаты испытания износостойкости азотированных секторов
из сплава МХ5* на машине Амслера при трении скольжения [462]
Режим азотирования Уменьшение веса в г Повышение износостой- кости (судя по уменьшению веса после азотирования (число раз)
Температура в °C Выдержка в ч Расход аммиака в см3!мин испытуемого сектора диска из стали v 1 2 (HRC 62-64)
Неазотированные 0,3132 0,2220 0,0263 0,0197 —
850 850 24 24 15 15 0,0040 0,0038 0,0278 0,0210 67 70
1050 1050 П р и м ( скорость вр ♦ Метал. 1 1 гчание. Усл •ащения диска локерамически) 100 100 овия испытани! 200 оо'мин. 5 сплав титана 0,0016 0,0064 й: продолжите. с 5?б Сг. 0,0084 0,0114 льность 1 ч, наг 167 42 рузка 15 кГ,
Механические свойства сплавов титана после азотирования
в аммиаке ниже, чем после азотирования в азоте из-за насы-
щения титана водородом. Однако в тех случаях, когда механи-
ческим свойствам сердцевины сплава титана придается меньшее
значение, азотирование в аммиаке может применяться.
340
По данным -работы [453] нитридная корочка, образующаяся
при азотировании в аммиаке и азоте, повышает коррозионную
стойкость титана в кипящих 10%-ных растворах H2SO4 и НС1
(в 6 раз при азотировании в аммиаке) и «существенно увеличива-
ет ее в растворах NaOH и NH4C1. Кроме того, азотирование по-
вышает сопротивление ползучести! титана при 535—630° С.
ОКИСЛЕНИЕ
Предел растворимости кислорода в а-титане около 14,5%
(рис. 250). При содержании кислорода в а-титане, близком к
максимальному, твердость а-раствора достигает Яр. 950—1100;
/ Рис. 250. Диаграмма состояния системы Ti — О
при дальнейшем увеличении! содержания кислорода в сплаве ти-
тан— кислород твердость его падает, а в области у-фазы (сое-
динение на основе TiO) вновь возрастает (рис. 251).
При окислении титана на воздухе1 образуется зона твердого
раствора кислорода в а-титане, поверх которой возникает
тонкая пленка окислов. Окисел титана TiO, имеющий бронзовый
цвет (почти аналогичный цвету нитрида TiN), появляется
1 Хотя воздух и содержит 77% азота, который имеет большое сродство
к титану, тем не менее основную роль при взаимодействии раскаленного ти-
тана с воздухом играет кислород, скорость диффузии которого в титане зна-
чительно больше, чем азота, а образующиеся окислы отличаются большой хи-
мической прочностью.
341
только при кратковременном нагреве. Обычно образуются все
три окисла: TiO, Ti2O3 и TiO2. Первые два окисла, прилегающие
к сердцевине, имеют ничтожную толщину, поэтому зона окислов
практически состоит из TiO2.
Коэффициент диффузии кислорода в р-титан определялся в
интервале температур 950—1100° С (рис. 252). Между скоростя-
ми диффузии кислорода в а- и р-титан нет существенного отли-
чия, имеющего место для скоростей диффузии азота в этих фа-
зах. При температуре 700—800° С это величины одного порядка:
10-10—ю-9 см2!сек [454].
Рис. 251. Влияние содержания кисло-
рода на твердость сплавов титана с
кислородом (Э. Бампс, Г. Кесслер
и М. Хансен):
1 — сплавы после отливки; 2 — сплавы
после отжига и закалки в воде
Рис. 252. Температурная зависимость
коэффициента диффузии кислорода в
Р-титан [454]
Окисление титана рекомендуется производить на воздухе при
температурах 725—825° С и выдержках, уменьшающихся при-
мерно с 5 до 1 ч с повышением температуры. Образующийся
после обработки по таким режимам слой a-твердого раствора
имеет толщину около 0,02—0,05 мм, снаружи возникает тонкий
(1—2 мк) слой окисла TiOj. Микротвердость титана ВТ1-Д при
этом повышается в среднем с Н1Д. 200 до 700.
Достоинствами метода являются: 1) простота выполнения;
2) низкая температура процесса и 3) небольшие выдержки,
благодаря чему свойства титана и его сплавов почти не изменя-
ются.
Недостатки метода: 1) малая толщина упрочненного слоя;
2)' недостаточное повышение износостойкости; 3) невозмож-
ность получения высокой чистоты поверхности (так как диффу-
зионный слой тонок, детали нельзя шлифовать).
Толщину слоя a-твердого раствора можно увеличить, повы-
шая температуру нагрева на воздухе и удлиняя выдержку. Одна-
342
ко это сопровождается увеличением толщины зоны хрупких
окислов титана. Поэтому окисление при температурах выше
Рис. 253. Распределение микротвердости по глубине диффузионного слоя
сплава титана с 0,5% W в зависимости от продолжительности и температуры
обработки в расплаве буры с применением электрозащиты [463]:
вверху — продолжительность процесса 1 ч (штриховые линии) и 3 ч (сплош-
ные линии); внизу — соответственно 6 ч и 12 ч
850° С не может быть рекомендовано. Исследовано поведение-
титана при нагреве в слабо окисляющих средах, в которых
343-
Рис. 254. Изменение по-
верхностной твердости по
Виккерсу сплава титана
с 0,5% W в зависимости
от продолжительности
процесса при различных
температурах (463]:
образуется лишь твердый раствор кислорода в а-титане, а зона
окислов не возникает. Процесс осуществлялся в вакуумной печи,
в которой создавалось небольшое разрежение, и в расплаве
буры.
При температуре 800—950° С и вакууме (1-4-5) • 10-5 мм рт. ст.
(0,00133—0,00666 н/м2) твердость титана не изменяется. При
снижении степени разрежения до 3 • 10-4 мм рт. ст. (0,04 н/м2)
микротвердость титана немного возрастает, а при снижении до
3- 10-3 мм рт. ст. (0,4 н/м2) и особен-
но до 3 • 10-2 мм рт. ст. (4 н/м2) воз-
растает значительно. После нагрева
при разрежениях 3 • 10~4 и 3- 10~3 мм
рт. ст. (0,04 и 0,4 м/л«2) поверхность
титана сохраняется блестящей, а при
разрежении 3 • 10-2 мм рт. ст. (4 н/м2)
чаще тускнеет. После выдержки 3 ч
при 800° С и разрежении 3 • 10-3 мм
рт. ст. (0,4 н/м2) под микроскопом на
сплаве ВТ1 обнаруживается диффу-
зионный слой глубиной 0,015 мм, а
при разрежении 3- 10"2 мм рт. ст.
(4 н/м2) — глубиной 0,03 мм (судя по
распределению микротвердости, глу-
бина слоев значительно больше); по-
верхностная микротвердость слоев со-
ответственно Ну. 540 и 670 (микро-
твердость сердцевины 240). При
температуре 950°С глубина и микро-
твердость диффузионных слоев (на
расстоянии 0,02 мм от поверхности)
примерно такие же, как и в случае
окисления титана на воздухе. Преиму-
гсления титана в вакуумной печи при
небольшом разрежении по сравнению с окислением на воздухе —
хорошее состояние поверхности после обработки.
При обработке в расплаве буры для защиты поверхности ти-
тана от растворения и разъедания бурой была применена элек-
трозащита. Образцы титана подсоединялись в цепь постоянного
тока в качестве катода (плотность тока 0,1 a/см2, напряжение
12—15 в), а в качестве анода использовался графитовый стер-
жень [463]. По окончании процесса и удалении налипшей буры
(механическим способом или растворением) поверхность образ-
цов была светло-серой.
Результаты измерения микротвердости и глубины диффузи-
онных слоев, образующихся на титане при обработке в расплаве
буры, приведены на рис. 253 и 254. С повышением температуры
и продолжительности процесса глубина слоя и микротвердость
344
щество ограниченного
увеличиваются. Глубина слоя, определенная по изменению мик-
ротвердости, в 2—2,5 раза больше глубины, наблюдаемой под
микроскопом (как и при окислении титана на воздухе).
Сравнение результатов обработки титана при температуре
950° С и выдержке 3 ч в расплаве буры (плотность тока 0,15—
0,30 а!см2) и окисления на воздухе при такой же температуре и
выдержке показало, что микроструктура, поверхностная твер-
дость и микротвердость по глубине слоя в обоих случаях прак-
тически одинаковы. Это указывает на то, что и сущность обоих
процессов одинакова. Различно лишь состояние поверхности об-
разцов.
Таблица 60
Результаты испытаний образцов сплава титана
с 0,5% W (0,5В) на машине Амслера [483]
Материал образца Способ обработки Твердость Уменьше- ние веса образцов в г
Сплав 0,5В Необработанный HV 290 0,1948 0,1910
Сплав 0,5В Окисление при 950° С, Зч HV 660 на поверхности /7 805 на глубине 0,02 мм 0,0030 0,0064 0,0040 0,0050
Сталь 45 Закалка, отпуск при 16О°С HRC 60 на поверхности 0,0130 0,0104
Сталь 20 Цементация и закалка, отпуск при 160° С HRC 63 на поверхности 0,0099 0,0064
П м е ч а н и е. Продолжительность испытания 37 мин, нагрузка 25 кГ, ско- рость вращения 200 об!мин.
Механические свойства после окисления в расплаве буры
сплава титана с алюминием при 950° С в течение 3 ч на глубину
0,3 мм (определена по распределению микротвердости по глу-
бине слоя) снизились: ов с 88 до 76 кг/мм (86,2 • 107 н/м2—
74,5-107 н!м2\ ат с 75 до 65 кг!мм (73,5-107 н!м2—63,7-107 н/м2),
б с 17,3 до 8% и г|) с 40 до 20%.
Испытание на машине Шкода-Савина показало, что объем
углублений, образующихся (после 100 оборотов диска) на неокис-
ленных образцах титана, в 12 и 37 раз больше, чем на окисленных
при выдержке 3 и 6 ч (температура 930° С). При испытании на
машине Амслера уменьшение веса работающих по стальным
345
Рис. 255. Микроструктура сплава титана с 0,5% W,
t, 2, 3 и 4 — при 1000° С, соответственно 1, 3, 6 и 12 ч; 5 и 6 —при 1000° С.
кольцам неокисленных секторов было больше, чем окисленных, в
среднем в 45 раз (табл. 60). Вес окисленных титановых секторов
уменьшался примерно на столько же, на сколько уменьшался вес
секторов из цементованной стали [463].
346
обработанного по разным режимам:
соответственно 1 и 3 ч\ 7 и 8 — 950° С, соответственно 6 и 12 ч [463]. Х200
На микроструктуре окисленного в расплаве буры слоя обна-
руживаются светлые иглы, ориентированные соответственно кри-
сталлографическому строению зерен титана (рис. 255—257).
Иглы а-фазы возникают при температуре насыщения. Рентгено-
347
граммы зон, расположенных на расстоянии 0,01; 0,06 мм от по-
верхности, и сердцевины образца сплава 0,5В, окисленного при
950° С в продолжение 3 ч, показали уменьшение периода решет-
ки (а = 2,96; 2,95 и 2,94 А, с = 4,85; 4,84 и 4,77 А). Это согласу-
ется с влиянием кислорода на период решетки титана.
Рис. 256. Микроструктура
сплава титана с 0,5% W
(обработанного при 1000° С
в течение 6 ч), снятая с ко-
сого шлифа:
а — поверхностная зона
слоя; бив — зоны все более
и более удаленные от поверх-
ности. Х200 (автор и Ю. Н.
Шульга).
В расплаве буры с применением электрозищаты происходит
в основном окисление титана. Количество бора 1 в поверхностном
слое менее 0,1 %.
1 Определено спектральным анализом.
348
Рис. 257. Микроструктура сплава титана с 0,5% W (обработанного при 1000° С
в течение 12 ч. Х86):
а — угол образца; б — плоская поверхность [463]
Рис. 258. Влияние продолжительности процессов на по-
верхностную твердость сплавов ВТ1 (кривая 1) и ВТ2
(кривая 2), обработанных в расплавленной буре с до-
бавлением карбида бора
349
Окисление титана можно осуществлять в расплаве буры с
добавкой 25—40% карбида бора. Эта ванна является активной
борирующей средой для железа, никеля, кобальта и других ме-
таллов. Однако, поскольку сродство титана к кислороду больше,
чем к бору, в этой ванне протекает окисление титана. Глубина
наблюдаемого под микроскопом окисленного слоя, полученного
в этой ванне на образцах сплава BTI при 800° С, составляла
0,05 мм при выдержке 3 ч и 0,07—0,08 мм при выдержке 6 ч.
Изменение поверхностной твердости показано на рис. 258. Сле-
дует отметить, что образцы, обработанные в такой ванне, имеют
чистую ровную слегка блестящую поверхность, не требующую
дополнительной отделки.
ЦЕМЕНТАЦИЯ
Растворимость углерода в а- и р-титане невысокая (рис. 259);
поэтому поверхностное упрочнение титана углеродом возможно
лишь путем создания слоя карбида титана.
В системе Ti — С может образовы-
ваться только карбид TiC, имеющий ши-
рокую область гомогенности (5,2—20%
Ti). Поэтому в зависимости от состава
цементующей среды и температуры про-
цесса образуется слой карбида титана с
тем или иным содержанием титана, а
следовательно, с различной микротвер-
достью (от 1100 до 3200). На-
иболее высокой микротвердости соответ-
ствует карбид титана TiC стехиометри-
ческого состава (20% Ti).
При температуре 950° С за 4 ч в сме-
си порошков угля и графита был полу-
чен карбидный слой толщиной 0,02 мм
с микротвердостью 1290 [451]. Це-
ментация титана в саже в потоке водо-
рода в течение 1; 2 и 4 ч позволила по-
лучить при 900° С слой карбида TiC тол-
щиной соответственно 18; 35 и 66 мк, а
при 1000° С толщиной 50; 60 и 85 мк [464].
Микротвердость слоев, полученных при 900—1000,1200 и 1300° С,,
составляла соответственно 1140—1300, 1820—2000 и 2600—
2780.
Опробована цементация титана в аргоне с добавлением
0,5—5% окиси углерода, пропана или метана [465]. При добав-
лении 5% СО снаружи образовывался слой TiC толщиной 10 мк,.
а глубже располагалась зона твердого раствора кислорода в а-
титане. В смеси аргона с 2,5% пропана при 955° С за 8 ч обра-
350
зовывался слой карбида толщиной 2,5—5 мк. При обработке в
смеси аргона с пропаном или метаном резко повышается содер-
жание в титане водорода, что ухудшает механические свойства
титана. Карбидные слои толще 7,5 мк оказывались во всех слу-
чаях очень хрупкими. Карбидные слои толщиной до 7,5 мк (с
микротвердостью 1500) оказались весьма износостойкими
[465].
При цементации титана в порошке древесного угля образу-
ются несколько более глубокие диффузионные слои, чем при азо-
тировании (рис. 260). Однако при этом титан, кроме углерода,
Рис. 260. Изменение микротвердости по глубине диффузионного слоя титана,
обработанного в порошке древесного угля ори 850° С (а) и 900° С (б) [456]
насыщается кислородом (а также, возможно, азотом и водоро-
дом), что значительно повышает хрупкость сердцевины и диф-
фузионного слоя. В табл. 61 сравнены механические свойства
тйТана, азотированного в азоте и в аммиаке, и цементованного
в порошке древесного угля.
Прочностные свойства титана, цементованного в порошке
древесного угля, примерно такие же, как у титана, азотирован-
ного в азоте или аммиаке, а пластичность и особенно ударная
вязкость значительно ниже, чем у азотированного в азоте, но
выше, чем у азотированного в аммиаке. При цементации в по-
рошке древесного угля поверхностная микротвердость во многих
случаях повышалась до 3200. Износостойкость цементован-
ного титана весьма высокая.
Представляют интерес результаты цементации титана с наг-
ревом т. в. ч. (466]. Цементуемые образцы покрываются пастой,
состоящей из серебристого графита и связующего вещества, и
нагреваются в .среде гелия до 850—1100° С (прерывистый
351
g> Влияние азотирования (в азоте и аммиаке) и цементации (в порошке древесного угля) Таблица 61
ьо __________________________на глубину диффузионного слоя и механические свойства технического титана [456]_____________________
Режим об- работки Насыщающая среда Глубина слоя(до микротвзрдости Н 600) в мм Р' Микротвердость Нг5 ав т 0 а ан
Темпера- тура в °C Выдерж- ка в ч на поверх- ности на глуби- не 0,1 мм 3 * 0 X Ч а X s 0 X ь 0 х 5 с Ч а X
Без обработки - - 36 35,3 22 21 ,G 53 64 27,0 26.5
800 8 48 Азот » 0,013 0,044 1003 1426 205 360 36.7 37 . 4 36,0 36,4 26,0 26,0 25,5 25,5 43 39 66 62 25,1 23,5 24,6 23,0
850 8 Аммиак Древесный уголь 0,05 0,05 0,106 1500 св. 1500 1500 375 335 640 39,1 48,3 46,4 38,3 47 3 45,4 27,5 45,5 38,0 27,0 44,6 37,2 52 30,8 55,6 53,8 17,5 0,3 7,1 17. 1 0,29 6,9
850 16 Азот Аммиак Древесный уголь 0,035 0,040 и, 086 св. 1500 св. 1500 около 3200 370 370 550 38,8 43,5 40,3 38,0 42,6 39,5 28,5 34,0 27,9 33,3 45 20 55,5 45.2 17,7 0,3 10,0 17,3 0,29 9,8
850 48 Азот Аммиак Древесный уголь 0,065 0,077 0, 140 1332 св. 1500 около 3200 505 525 705 35,1 30,5 46,4 34,4 29,9 45,5 31 ,0 43,5 30,4 42,7 20, 0 10,8 42,5 12 5,8 1 ,7 1,3 5,7 1,6 1,2
900 8 Азот Древесный уголь 0,053 0,183 св. 1500 св. 1500 395 880 41 .7 44,4 40,9 43,5 36,0 19,5 35,3 19,2 36,5 19,5 51 17, 1 2,0 16,7 1,9
900 16 Азот Древесный уголь 0,061 0,143 св. 1500 св. 1500 470 720 46,7 41,3 45,8 40.5 22,5 22,0 25 6,0 38,5 11,8 3,0 1 1,6 2,9
950 8 Азот Аммиак Древесный уголь 0,059 0,080 0,087 св. 1500 св. 1500 около 3200 445 490 585 48,1 34,0 45,8 47, 1 33,3 45,9 43,5 43,0 42,6 42,1 43,5 6,0 28,0 12,0 0,5 0,5 11,7 0.49 0,49
950 16 Азот Древесный уголь 0,079 0,088 св. 1500 около 3200 570 530 51,2 40.7 50,2 39,9 44,5 37,0 43,6 36,3 — 16,0 8,9 0,5 8,8 0,49
нагрев). При выдержке 15 мин образуется диффузионный слои
толщиной 0,25 мм с микротвердостью поверхности Нр. 1780. 1
Опыты, выполненные А. В. Щербединской и автором,, позво-
лили определить коэффициент диффузии углерода в карбид ти-
тана. Цементация осуществлялась при 900—1200°С в вакууме в
смеси активированного угля и ВаСОз, содержащего радиоактив-
ный углерод. Распределение концентрации углерода по глубине
Рис. 261. Микроструктура титано-
вого сплава, цианированного при
950° С в течение 3 ч и затем ох-
лажденного на воздухе (автор
и В. Ф. Никонов)
карбидного слоя определялось
методом послойного радиометри-
ческого анализа, коэффициент
Рис. 262. Распределение микротвер-
дости по глубине диффузионного
слоя титанового сплава, цианирован-
ного при 950° С в течение 3 ч и за-
тем охлажденного в масле (кривая 1)
и на воздухе (кривая 2) [автор и
В. Ф. Никонов]
диффузии — по концентрационным кривым с использованием
методики, описанной в работе [15]:
D = 1,3 • 103ехр
/ 83 000\
V RT )
см2/сек.
Была построена температурная зависимость коэффициента
диффузии и по углу наклона прямой линии определена энергия
активации, оказавшаяся равной 83 000 кал/г-атом [591].
Коэффициенты диффузии углерода в карбиде титана опреде-
лялись также и по другой методике — на предварительно выра-
щенном слое карбида титана, полученном с использованием
ВаСО3, не содержащего радиоактивный углерод. В этом случае
продолжительность насыщения выбирали с таким расчетом, что-
бы зона проникновения радиоактивного углерода была намного
меньше толщины карбидного слоя. При расчете коэффициента
диффузии углерода в предварительно выращенных карбидах
было использовано решение уравнения Фика для случая диффу-
зии в однородной среде из постоянного источника, Получено
12 А. Н. Минкевич ,333
хорошее совпадение результатов определения коэффициента
диффузии первым и вторым методами.
Опробовано цианирование титана -в расплаве, содержащем
30—100% цианистого натрия [465]. Диффузионный слой после
обработки имел твердость 550—650 по Кнупу и состоял, по ут-
верждению авторов работы [465], из твердого раствора азота в
«-титане. Автор настоящей книги осуществлял обработку при
950° С в ванне, предназначенной для глубокого цианирования и
состоящей из 5,5% NaCN, 35% BaCU, 25% NaCl и
*33% ВаСОз. Охлаждались образцы на воздухе и в масле
(рис. 261 и 262). Иглообразная структура диффузионного слоя и
полученные величины микротвердости аналогичны тем, которые
имеют место при окислении титана. По-видимому, основным про-
цессом, развивающимся при цианировании, является окисление,
которому сопутствует некоторое насыщение слоя азотом и угле-
родом.
БОРИРОВАНИЕ
Растворимость бора в а- и у-титане небольшая (рис. 263), и
упрочнение при борировании происходит благодаря образованию
на поверхности слоя боридов титана.
Ат 7.В
Рис. 263. Диаграмма состояния системы Ti — В
Борирование титана осуществлялось в вакуумной печи в
порошке бора или карбида бора и в ванне с расплавом буры
(электролизное борирование) [463]. При электролизном бориро-
вании боридные слои образуются только при больших плотно-
стях тока (1,0—2,5 а!см2). Однако при таких плотностях тока
354
происходит сильное растворение титана и поверхность образцов
бывает чаще неровной, а боридный слой неравномерным по тол-
щине (рис. 264, а) .Иногда все же удается получить равномерные
слои глубиной до 0,025—0,03 мм с микротвердостью более
2500 (микротвердость TiB2 3370 [19], микротвердость TiB-
Рис. 264. Микрострук-
тура боридных слоев-
титановых сплавов:
а — электролизное
борирование сплава ти-
тана с 0,5% W при тем-
пературе 1050° С, вы-
держке 3 ч и плотности
тока 2,0 afcM?. х 300;
б, в, г — борирование
в вакуумной печи в кар-
биде бора: сплава с
5% Сг при 1050° С, 3 ч,
X 300 (б), сплава с
5% Сг при 1150° С, 3 ч
Х300 (в), сплава BTf
при 1050° С, 3 ч. X 300
(г); д — борирование в
вакуумной печи в
аморфном боре сплава
ВТ1 при 1000° С, 3 ч.
X 600
и Ti2Bg в литературе не приводится). Спектральным анализом в
поверхностных зонах слоя, полученного при 1050° С за 3 ч, было
обнаружено до 20% В, что немного превышает содержание бора
в бориде TiB (18,4%).
Результаты борирования титана в порошках аморфного бора
или карбида бора в вакуумной печи (10-3—10-4 мм рт. ст., ила
1,33—0,133 н/м2) приведены в табл. 62.
Микроструктура борированных слоев зависит от условий бо-
рирования и состава обрабатываемого сплава. В ряде случае»!
удавалось обнаружить двухслойные боридные каемки,
(рис. 264, б). При этом внешнюю зону боридов предположитель-
но можно отнести к бориду TiB2, а внутреннюю — к бориду TiB.
355-
Таблица 62
Изменение твердости и глубины борированного слоя
в зависимости от условий борирования в вакуумной печи (автор)
Насыщающая среда Темпера- тура про- цесса1 в °C Твердость поверх- ности Глубина слоя в мм
HV5 н
Аморфный бор 1000 904 1600 0,Ol- О.018
Карбид бора 1000 728 1200 0,01 — 0,015
Аморфный бор 1150 1160 2100 0,013— 0,02
То же 1200 1800 2800 —
Карбид бора 1 Выдержка 6 ч. 1200 1600 2400
Между слоем боридов и сердцевиной обнаруживается темная
промежуточная зона той или иной ширины. Например, на спла-
вах титана с 5% хрома возникает широкая промежуточная зона,
в 3—5 раз превосходящая по толщине боридную зону. Возмож-
но, что промежуточной зоной является смесь фаз а + TiB, обра-
зовавшаяся при охлаждении из ₽- и a-твердых растворов вслед-
ствие уменьшения растворимости бора с понижением темпера-
туры.
При борировании титана в смеси порошков карбида бора и
буры (взятых в соотношении 84: 16) в потоке водорода при тем-
пературах 1100 и 1200° С и выдержках соответственно 8 и 4 ч бы-
ли получены слои только с одним боридом TiBj толщиной
8—9 мк и микротвердостью 2950—3100 [464].
Износостойкость титана, борированного в вакуумной печи в
порошке аморфного бора, испытывалась на машине Шкода-Са-
вина при нагрузке 5 кГ и скорости вращения диска 840 об/мин.
Образцы испытывали при различном числе оборотов до образо-
вания углублений примерно одинакового объема (табл. 63).
НАСЫЩЕНИЕ МОЛИБДЕНОМ, АЛЮМИНИЕМ, КРЕМНИЕМ
И НЕКОТОРЫМИ ДРУГИМИ ЭЛЕМЕНТАМИ
Опробовано молибденирование титана методом испарения
молибдена из его карбонильных соединений [Мо(СО)6] в среде
водорода. При температуре 930° С и давлении 0,5 мм рт. ст.
(66,5 н]м2) получен вязкий диффузионный слой с микротвердо-
стью около Яр. 800; за 1 ч выдержки глубина слоя возрастала
на 0,025 мм. На микроструктуре был заметен рост зерен титана.
356
При более низкой температуре (470—750° С) и давлении
0,1 мм рт. ст. (13,3 н!м2) глубина слоя уменьшается (за 1 ч вы-
держки возрастает на 0,0025 мм), а твердость повышается до
Нр. 1000. При обработке дуплекс-процессом (выдержка 3 мин
при 930° С и 20 мин при 540° С) образовывался достаточно вяз-
кий слой глубиной всего 0,0025 мм. Рентгеноструктурный анализ
показал присутствие в нем карбида молибдена. Износостойкость
молибденированного титана намного превосходит износостой-
кость необработанного [467].
Рис. 265. Микроструктура диффузионного слоя сплавов ВТ5 (а) и ВТ2 (б),
насыщенных марганцем при 1000° С в течение 3 ч (в порошке марганца в ва-
куумной печи). X 200 (автор и Э. Н. Островская)
Автор осуществлял молибденирование образцов в порошке
молибдена в вакуумной печи при 1000° С в продолжение 10 ч.
Глубина диффузионного слоя (по микротвердости) достигала
0,4 мм, твердость поверхности HV 572, а микротвердость 640
(микротвердость сердцевины Н{>. 260).
Таблица 63
Износостойкость борированного сплава титана
с 5% хрома, испытанная на машине Шкода-Савина
(автор и Д. С. Варкая)
Режим обработки Число обо- ротов диска Объем углуб- ления в мк3
Необработанные Борированные (1000° С, 6 ч} То же (1150°С, 6 ч) 65 1500 2000 1266,6 1248,3 1280,0
357
В табл. 64 приведены результаты насыщения титана некото-
рыми элементами. Наиболее твердые, но тонкие диффузионные
слои образуются при силицировании и бериллизации, наиболее
глубокие (больше 1 мм), но имеющие небольшую твердость —
при насыщении титана марганцем (основной частью слоя явля-
ется, очевидно, у-фаза — TiMn (рис. 265).
Силицирование, алитирование, бериллизация и борирование
повышают окалиностойкость титана, причем эффект обработки
во многом зависит от глубины диффузионных слоев и условий
насыщения (рис. 266).
Продолжительность испытания
Рис. 266. Окалиностойкость титана ВТ1-Д при 930° С, необработанного
и подвергнутого поверхностному насыщению различными элементами (автор,
Э. Н. Островская, Ю. А. Березкин)
В зарубежной литературе для повышения окалиностойкости
титана чаще рекомендуется алитирование. Наиболее простой
метод алитирования заключается в погружении титана в жидкий
алюминий (760° С, 30 сек), покрытый слоем специального флю-
са с последующим удалением избытка алюминия тем или иным
способом (см. стр. 159—161).
Для повышения окалиностойкости титана рекомендуется так-
же гальваническое никелирование с последующим диффузионным
хромированием в смеси порошков: 50% хрома и 50% каолина, к
которым добавляется 0,2% йодистого аммония.
Химико-термическая обработка титана существенно изменя-
ет его коррозионную стойкость. Азотирование титана в аммиа-
ке и азоте увеличивает стойкость титана в кипящих 10%-ных
растворах серной и соляной кислот, а также в растворах NaOH
и NH4C1. Интересно рассмотреть кислотостойкость химико-тер-
мически обработанного титана в серной кислоте. В этой кислотеч
коррозионная активность титана имеет два максимума: первый
при концентрации 35—45% H2SO4 и второй, более высокий, при
358
Поверхностная твердость и глубина диффузионных слоев, образующаяся на титане при нагреве Таблица 64
в соответствующих порошках в вакуумной печи (автор, Ю. А. Березкин и Э. Н. Островская)___________
Вид обработки Насыщающая среда Режим обработки Обрабатывае- мый сплав Глубина слоя в мм Твердость поверхности Некоторые свойства слоя
температура в °C выдер- жка в ч HV5 Н ц
Алитирование 5096 А14-50% А12О8 (в ва- кууме) 850—1000 10 ВТ1 — 459 580 Повышенная окалиностой- кость, низкая износостой- кость
Силицирование 50% Si + 50 96 А12Оа (в ва- кууме) 950 1000 10 10 ВТ1 0,01 0,03 570 1145 1270 Повышенная окалиностойкость и стойкость в растворах НС1 и H2SO4
Бериллизация 50% Be 4-50% А12Оа (в ва- кууме) 825 24 ВТ1 ВТ 2 ВТ5 — 250 360 410 326 650 1140 Повышенная окалиностой- кость
1000 10 ВТ1 ВТ2 ВТ5 — 300 406 535 785 820 980
Хромирование 25% Сг-|- 75% А12Оа (в ва- кууме) 1000 1000 5 10 ВТ1 ВТ1 0,038 400 550 510 845 Повышенная стойкость в ра- створах H2SO4
Никелирование 2596 Ni -4- 75% А12Оа (в ва- кууме) 800 10 ВТ1 ВТЗ 0,056 0,046 445 452 717 800 —
860 10 ВТ1 ВТЗ — 330 446 512 645
Насыщение мар* ганцем 67% Мп 4-33% А12Оа (в ва- кууме) 950 1000 10 3 ВТ1 ВТ1 ВТ2 ВТЗ ВТ5 0,32 0,4 1,2 1,6 0,8 450 405 650 800 740 520 570 Повышенная стойкость в ра- створах H2SO<
870 24 ВТ2 0,15 365 650
Наводороживанне Водород 1000 2 ВТ1 — — — Высокая стойкость в HaSO4 (благодаря образованию очень тонкого слоя гидрида титана)
концентрации 72—83% H2SO4. Первый максимум активности ти-
тана полностью снимается (испытания продолжительностью
120 ч) хромированием (табл. 65). В то же время насыщение ти-
тана марганцем повышает этот максимум. Второй максимум ак-
тивности титана снимается или резко уменьшается борировани-
ем и силицированием.
Таблица 65
Коррозионная стойкость в серной кислоте сплава ВТ1 - Д
после различных видов химико-термической обработки
(автор, Э. Н. Островская и А. Ю. Березкин)
Вид обработки образцов Уменьшение веса образца в г/мР-ч при концентрации H2SO4 в %
10 22,б| 34| 40 | 52 | 62 64 | 72 78 | 82 86 | 89
Необработан- ные Борирование в порошке бо- ра Насыщение марганцем Молибдениро- вание Хромирование Силицирова- ние П р и м е ч а 0,023 0,3 1,6 1,36 0,40 0,69 0,81 7,92 8,23 6,77 — 0,1 — — — 1,8 0,006 0,004 0,009 0,006 0,088 1,15 1,32 — — — —4,38 — — 0,07 — 0,34 — — — — — —0,147 — — 0,18 2,72 — — 0,76 — 0,027 — —0,032 — — 0,09 — 4,01 — 0,31 — — — — — — — 0,043 — 0,056 — — — н и е. Продолжительность испытания 120 ч.
При испытаниях в течение 120 ч в 5—25%-ных растворах со-
ляной кислоты стойкость сплава ВТ1-Д после силицирования
повысилась более чем в 10 раз, а после борирования в еще боль-
шей степени. Насыщение титана молибденом и марганцем почти
не изменяет кислотостойкость титана в растворах соляной кис-
лоты.
ГЛАВА IV
ХИМИКО-ТЕРМИЧЕСКАЯ ОБРАБОТКА ТУГОПЛАВКИХ
МЕТАЛЛОВ
ОБРАБОТКА МОЛИБДЕНА
Обработка с целью повышения окалиностойкости
Молибден и сплавы на его основе относятся к числу наибо-
лее жаропрочных материалов; их жаропрочность намного пре-
вышает жаропрочность широко применяемых в технике сплавов
на никелевой, кобальтовой и хромоникелевой основах. Молибден
и молибденовые сплавы перспективны и уже начинают находить
применение для изготовления деталей, работающих при темпера-
турах до 1650° С. По данным зарубежной печати, молибден и его
сплавы используются, в частности, в ракетостроении, в ядерной
технике, в космонавтике.
Самая сложная проблема, без решения которой было невоз-
можно воспользоваться высокими свойствами жаропрочности
молибденовых сплавов,— изыскание эффективных способов по-
вышения их окалиностойкости, так как при высоких темпера-
турах молибден и его сплавы катастрофически окисляются.
Полностью эта проблема не решена до сих пор.
При окислении большинства металлов непосредственное со-
прикосновение металла с газами происходит лишь в первой
стадии окисления, а затем развитие процесса затрудняется диф-
фузией атомов металла и газа через слой продуктов реакции.
Иная картина наблюдается при окислении молибдена. Трех-
окись молибдена при температуре выше 495° С имеет исключи-
тельно высокую упругость паров и плавится уже при 795° С.
Поэтому при температуре выше 795° С начинается особо сильное
окисление молибдена, причем продукты реакции окисления не-
прерывно удаляются и поверхность металла постоянно обнажа-
ется.
Над защитой молибдена от окисления работают более 10 лет
многие отечественные и зарубежные исследователи, и уже раз-
работано несколько видов покрытий, достаточно надежно
361
защищающих молибден от окисления в течение сравнительно
длительного времени.
К основным методам защиты молибдена от окисления отно-
сятся [467}: 1) гальванический; 2) осаждение из газовой фазы
окислов и интерметаллидов; 3) погружение в расплав; 4) диф-
фузионный; 5) плакирование (чаще хромоникелевыми сплава-
ми); 6) газопламенное или плазменное напыление и наплавле-
ние, например, покрытий Si — Al, Сг — Si, Al — Сг — Si,
Ni — В — Si; 7) нанесение жаростойких эмалей. Ниже кратко
излагается существо первых четырех методов защиты молибде-
на от окисления.
Гальванический метод [469], [470]. Удовлетворительные ре-
зультаты дает поочередное нанесение на поверхность молибдена
сначала хрома, а затем никеля, причем с утолщением нанесен-
ных слоев окалиностойкость молибдена повышается. При тол-
щине слоя хрома 100 мк и никеля 170 мк стойкость покрытия
колеблется от 400 до 920 ч при 1100° С. Разработано также трех-
слойное покрытие хром (25 мк) —никель (175 мк) —алюминий
(50—75 мк). Алюминий наносится путем электролиза расплава
криолита (Na3AlF6) при 1000° С. Такое покрытие устойчиво со-
храняется в течение 400 ч при 1100° С. Эффективно защищает
молибден от окисления также покрытие никель (нижний
слой)—алюминий. Алюминирование производится при 1050° С
методом электролиза электролита (75% криолита и 25% фтори-
стого натрия).
Осаждение из газовой фазы окислов и интерметаллидов. Этот
метод основан на тепловом разложении летучих металлов, окис-
лами которых должен быть покрыт молибден. Процессы чаще
осуществляются в смеси хлорида металла и углекислого газа
с использованием водорода в качестве несущего газа. При этом
протекают, например, следующие реакции:
ZrCl4 + 2Н2 + 2СО2 = ZrO2 + 4НС1 + 2СО;
2 А1С13 + ЗН2 + ЗСО2 = A12Os + 6НС1 + ЗСО.
Таким методом на поверхность молибдена можно нанести
А12О3, SiO2, ZrO2, Al2O3-SiO2, Al2O3-ZrO2 и т. п., а также туго-
плавкие металлы, карбиды, нитриды, бориды (сущность этих
процессов изложена в гл. VIII).
Погружение в расплав. Метод заключается в погружении мо-
либдена в расплав, состоящий из одного или нескольких метал-
лов, склонных после затвердевания и последующего нагрева
к образованию тугоплавких окислов. Например, покрытие соста-
ва Al2O3-SiO2 создается путем погружения молибдена на
10—60 сек в расплав (75% А1 и 25% Si), нагретый до 1000—
1300° С, после чего следует окислительная обработка покрытия.
Изменяя соотношение алюминия и кремния в ванне, можно по-
лучить покрытие состава ЗА12О3 • 2SiO2. Аналогично создаются
362
покрытия 2MgO-SiO2, MgO-Al2O3 и др. Улучшение смачивания
молибдена сплавом достигается введением в ванну небольшого
количества «активизаторов» (кобальта, титана, железа). Недо-
статок таких покрытий — их хрупкость и высокая чувствитель-
ность к тепловым ударам.
Этим же методом на молибден могут быть нанесены окали-
ностойкие сплавы, например Ni — Al.
Разработан также
метод хромирования
молибдена в расплаве
металлов с образова-
нием диффузионных
слоев хрома [470]. Для
осуществления этого
процесса необходимо,
чтобы металл, являю-
щийся основой рас-
плава, легко растворял
хром, но не растворял
молибден и не обра-
зовывал с ним интер-
металлических соеди-
нений. Подходящими
с этой точки зрения
расплавами являются
Мо + Сг, Си + Сг,
Sn + Сг. Жидкие медь
и олово хорошо рас-
творяют хром, но поч-
ти не растворяют мо-
либден. Хромирование
Рис. 267. Окалиностойкость молибденовой
проволоки, насыщенной различными эле-
ментами из порошкообразных смесей [474]
таким методом произ-
водилось в молибдено-
вых тиглях под защи-
той водорода. Обра-
зующиеся в такой ванне хромированные слои толщиной 25—
100 мк не могут должным образом защищать молибден от оки-
сления, но являются хорошим подслоем для нанесения после-
дующих покрытий.
По такому же принципу выбирают расплавы для нанесения
на молибден при 850—900° С силицированных слоев: Au +
+ 2,5% Si [472], Си + Si [473] и Si + Sn.
Диффузионное насыщение. Окалиностойкость молибдена по-
вышается после силицирования, бериллизации, алитирования,
хромирования, никелирования и в еще большей степени после
одновременного насыщения молибдена несколькими из этих
элементов или в сочетании их с другими элементами. Молибде-
363
Рис. 268. Зависимость глу-
бины силицированного слоя
молибдена, обработанного
при разных температурах, от
продолжительности насы-
щения циркуляционным ме-
тодом [477] (сплошные ли-
нии — среда SiCl4 + Н2;
штриховые линии — НС1 в
присутствии кремния)
новая проволока нагревалась в среде Н2 + НС1 в смеси порош-
ков, состоявшей из 20% окиси алюминия и 80% наносимого ме-
талла или сплава [474]. Испытания на воздухе при 900—1300° С
показали (рис. 267), что наибольшей окалиностойкостыо обла-
дает силицированная проволока. В работе [474], кроме того, ука-
зывалось на исключительно высокую стойкость проволоки, на-
сыщенной цирконием, но позднее это не подтвердилось [468].
Силицирование молибдена можно осуществлять при 1100—
1200°С в порошкообразных смесях (60% кремния, 40% шамота
с добавкой к ним 2% хлористого
аммония) [475], в вакууме в порош-
ке кремния [579] или в газовых сре-
дах [476]. В последнем случае в
печь подается смесь SiCl4 и Н2 в
соотношении, например, 1 : 3 или
1 : 2. Разработан также циркуляци-
онный способ силицирования мо-
либдена в смеси SiCl4 + Н2:
Мо + 2SiC14 + 4Н2 = MoSi2 + 8НС1
или с применением НС1 в присут-
ствии кремния:
8НС1 + 2Si = 2SiCl4 + 4На.
Этот способ отличается от из-
вестных тем, что процесс протекает
в замкнутом объеме с многократ-
ным использованием газовой среды
при непрерывной ее циркуляции
[477], [478]. Преимущества этого
способа силицирования: большая
скорость процесса, уменьшение расхода газов и улучшение са-
нитарных условий работы. На рис. 268 показаны кривые изме-
нения глубины силицированного слоя в зависимости от продол-
жительности циркуляционного способа обработки молибдена.
При насыщении в порошке (Si + 3% NH4C1 в потоке Н2) по
аналогичным режимам глубина диффузионного слоя существен-
но меньше [327], [479].
Микроструктура силицированного слоя может быть различ-
ной в зависимости от условий обработки. Автор работы [480] при-
шел к выводу, что реакция между молибденом и кремнием про-
исходит с образованием дисилицида молибдена MoSi2
(рис. 269) и лишь после продолжительной выдержки на границе
молибдена и дисилицида образуются еще два, более бедных
кремнием силицида: Mo3Si2 и Mo3Si. Микроструктуры (рис. 270),
на первый взгляд, подтверждают это мнение. После кратковре-
менной обработки (рис. 270, а, б) видна только одна фаза, кото-
364
рая согласно рентгеноструктурному анализу является дисилици-
дом молибдена. После продолжительной обработки и диффузи-
онного отжига обнаруживается промежуточная зона
(рис. 270, в, г), которую, очевидно, можно отнести к силициду
Mo3Si3, так как толщина зоны силицида Mo3Si бывает всегда
ничтожной. Однако не исключено, что силициды MosSi3 и Mo3Si
возникают в слое уже при кратковременных выдержках, но их
Рис. 269. Диаграмма состояния системы
Мо —Si
толщина бывает ничтожна, и первой всегда удается обнаружить
ту фазу, которая растет с наибольшей скоростью — в данном
случае дисилицид MoSi2 (см. стр. 34—35).
Микротвердость поверхности силицированного слоя, состоя-
щего из MoSi2, колеблется, по разным данным, в пределах
Яр, 1250—1410. Дисилицид молибдена почти нерастворим в ми-
неральных кислотах, включая царскую водку и плавиковую кис-
лоту. Высокая окалиностойкость MoSi2 и других сплавов молиб-
дена с кремнием объясняется образованием газонепроницаемой,
прочно удерживающейся на поверхности стеклообразной пленки
SiO2. Покрытие из дисилицида молибдена глубиной 0,1 мм, по-
лученное из газовой среды, обеспечивает защиту в течение
4000 ч при 1000° С, 30 ч при 1700° С, несколько часов при 1750° С
и до 1 ч при 1800° С [480]. Интенсивное окисление молибдена, си-
лицированного в порошкообразной смеси, начинается в случае
превышения следующих продолжительностей нагрева [475]:
Температура в ®С ......... 1300 1400 1500 1600 1700
Продолжительность нагрева в ч 100 80 70 60 50
Большим недостатком дисилицида молибдена является его
хрупкость и недостаточная стойкость против термических ударов.
365
Рис. 270. Микроструктуры молибдена:
а — насыщенного кремнием при 1200° С в течение 30 мин циркуляционным методом;
< — силицированного прямоточным методом в течение 18 ч при 1100° С и затем под-
вергнутого диффузионному отжигу при 1400° С в течение 50 ч. X 450 (Б. Н. Арзамасов);
в — силицированного в порошкообразной смеси при 1100° С в течение 10 ч; г — сили-
цированного в порошкообразной смеси при 1100° в течение 10 ч и подвергнутого диф-
фузионному отжигу при 1200° С в течение 2 ч. X 200 (О. К. Якшина); [микроструктуры
а и б не травлены; в и г — травлены в смеси HF и HNO3]
366
При длительных выдержках с переменным нагревом и охлаж-
дением на поверхности силицированного молибдена образуются
микротрещины. Они быстро развиваются, доходя до основного
металла, который начинает бурно окисляться. Трещины образу-
ются вследствие различия термических коэффициентов расши-
рения молибдена и диффузионного слоя [475]. Для уменьшения
коэффициента расширения диффузионного слоя было удачно
применено комплексное на-
сыщение молибдена крем-
нием совместно с другими
элементами.
Рис. 272. Микроструктура молибде-
на, бериллизованного в порошкооб-
разной смеси при 1100° С в течение
10 ч. Х260 (М. И. Захарова)
ротвердости по глубине берил-
пизованного слоя молибдена,
образовавшегося в порошко-
образной смеси при 1100° С
(М. И. Захарова)
Обстоятельный обзор последних работ по силицированию мо-
либдена и других металлов, а также созданию на различных ме-
таллах карбидных, боридных и нитридных слоев дан в недавно
изданной работе [481].
Бериллизация осуществляется в порошкообразных смесях
(например, 80% Be, 18% ВеО и 2% NH4CI) в вакууме и газовых
средах. После обработки в порошкообразной смеси указанного
состава при 1100° С общая глубина бериллизованного слоя, об-
разующегося в течение 2; 6 и 15 ч, достигает соответственно
0,12; 0,15 и 0,175 мм. Диффузионный слой, возникающий при
этой температуре, состоит из трех зон с различной микротвердо-
стью (рис. 271). По другим данным [489], микротвердость слоя
367
несколько ниже: внешней зоны 1200, средней 1350 и внут-
ренней Яр. 1450. Внешняя зона слоя состоит из МоВе28, средняя
(немного более темная) — из MoBej2 и зона, примыкающая
к сердцевине, — из МоВе2 (рис. 272). В результате бериллизации
при более низких температурах (ниже 1050° С) в слое образу-
ются только две зоны: MoBei2 и МоВе2 [482].
С увеличением глубины диффузионного слоя и концентрации
в нем бериллия окалиностойкость слоя увеличивается. При ис-
пытаниях при 1000 и 1100° С на поверхности бериллизованного
Рис. 273. Окалиностойкость
при 1100° С бериллизованного
и силицированного молибдена
(образец необработанного мо-
либдена весом 8,5 г пол-
ностью превратился в лету-
чий трехокисел молибдена за
4 ч) [автор и Т. Т. Гребенни-
кова]
молибдена возникает плотный
достаточно толстый коричневый
слой окисла ВеО, и вес образ-
цов при окислении увеличивает-
ся в большей степени, чем сили-
цированных (рис. 273). Резуль-
татами продолжительных ис-
пытаний бериллизованного мо-
либдена автор не располагает.
Хромирование производится
в различных средах. После хро-
мирования молибдена в порош-
кообразной смеси (49% хрома,
49% шамота и 2% NH4CI) при
1100 и 1200° С в течение 5 ч диф-
фузионный слой имеет соответ-
ственно глубину 0,08 и 0,30 мм,
а в течение 15 ч — глубину 0,12
и 0,45 мм. Судя по измерениям
микротвердости по глубине слоя и характеру микроструктуры
(рис. 274), диффузионный слой состоит из одной зоны, имеющей
микротвердость около Яр. 1400.
После хромирования молибдена в вакуумной печи при тем-
пературе 1200° С и выдержке 25 ч получен диффузионный слой
с двумя зонами (толщина внешней 70 мк, внутренней 40 мк)
[483]. Максимальная микротвердость, зафиксированная на гра-
нице внешней и внутренней зон слоя, составила около Яр. 920
(более высокая микротвердость слоя в предыдущем случае, воз-
можно, объясняется насыщением слоя азотом за счет хлористого
аммония). Рентгеноспектральный локальный химический анализ
и рентгеноструктурный анализ показали, что. диффузионный
слой состоит из двух твердых растворов: внешний твердый рас-
твор имеет гексагональную решетку с а = 2,757 кХ и с = 4,384 кХ
(на глубине 35 мк), а внутренний — объемноцентрированную ку-
бическую решетку [483].
Окалиностойкость хромированного слоя на молибдене суще-
ственно ниже, чем силицированного и бериллизованного, однако
хромированный слой отличается большей устойчивостью против
368
тепловых ударов. Так, при хромировании по специальной техно-
логии удается получить покрытие, выдерживающее 400 ч при
1100° С или 18 циклов нагревов до этой температуры и охлаж-
дений и 100 ч при 1200° С или 5 циклов теплосмен. Хромирова-
нию могут быть подвергнуты такие детали из молибдена, как
лопатки газовых турбин, турбинные колеса, стержни ядерных
реакторов и т. д. [484].
Хорошую окалиностойкость показывает молибден после ком-
плексного насыщения хромом, кремнием и железом [485].
Алитирование молибдена осу-
Рис. 275. Микроструктура мо-
либдена, насыщенного алюми-
нием циркуляционным мето-
дом при 1100° С в течение
90 мин. X 450 [478]
Рис. 274. Микроструктура мо-
либдена, хромированного в
порошкообразной смеси при
1200° С в течение 15 ч. X 200
(М. И. Захарова)
средах, а также в порошкообразных смесях. При циркуляцион-
ном методе насыщения [478] при 1100° С за 5 ч образуется не-
травящийся слой (рис. 275) с микротвердостью 1200 — оче-
видно, соединение Мо3А1.
Алитирование молибдена можно успешно осуществлять в
ванне с расплавленным криолитом [252]. Детали из молибдена
предварительно протравливаются при 230—270° С в ванне, со-
стоящей из 90% NaOH и 10% NaN'O2, и промываются в слегка
подкисленной горячей воде. Затем деталь (катод) помещается
в графитовый тигель (анод) с расплавленным криолитом, содер-
жащим около 70% A1F3. При плотности тока 10 а)дм2 и темпера-
туре 980° С алитирование длится 10—15 мин. После этого деталь
погружается в 20%-ный раствор НС1, а затем подвергается пес-
коструйной очистке.
369
Разработано много способов образования на молибденовых
деталях комбинированных окалиностойких диффузионных сло-
ев, создаваемых за один цикл обработки (Si—Nb [486], Сг—Al—
Si, Ni—В—Si) или за два цикла обработки (насыщение, напри-
мер, сначала цирконием и кремнием, а затем алюминием и ни-
келем и т. д.) [468].
Большое разнообразие сочетаний элементов в диффузионном
слое было получено авторами работы [487], насыщавшими молиб-
ден: 1) поочередно различными элементами; 2) одновременно
за один цикл обработки той или иной комбинацией элементов и
3) поочередно различными комбинациями элементов (иногда
в сочетании с насыщением отдельными элементами). Авторы ра-
боты [487] пришли к выводу, что для дальнейшего изучения влия-
ния элементов на свойства образующихся диффузионных слоев
заслуживает внимания метод создания слоев как за один цикл
обработки (например, слоев Si — Та, Si — Nb, Si — V), так и за
несколько циклов путем насыщения хромом, кремнием в комби-
нации с танталом, ниобием, вольфрамом, ванадием и другими
элементами.
Борирование, цементация и азотирование
При поверхностном насыщении молибдена бором, углеродом
и азотом образуются боридные, карбидные и нитридные слои, об-
ладающие высокой твердостью и рядом ценных физико-химиче-
ских свойств.
Рис. 276. Влияние температуры и продолжительности насыщения бором мо-
либдена и других тугоплавких металлов:
а и б — борирование в расплаве буры с добавлением карбида бора [488]; в — бори-
рование в потоке водорода в порошкообразной смеси карбида бора и буры, взятых
в соотношении 84 : 16 при 1200° С [464]
Борирование молибдена. Этот процесс успешно протекает в
газовых средах, в расплавах (например, при электролизе рас-
плава, состоящего из 33% НВОг и 67% NaF) [147] и в других
370
средах (рис. 276—279) [488]—[490]. Как следует из рис. 276, в, в
расплаве буры с добавлением карбида бора борирование проте-
кает интенсивно, однако при температуре 1200°С и особенно
1300°С проводить процессы не
рекомендуется из-за сильного ис-
парения соли. Расхождения в ско-
ростях борирования вольфрама и
молибдена, обнаруженные в ра-
ботах [464], [488], очевидно, связа-
ны с различиями в применяемых
материалах. Микроструктуры бо-
рированных слоев тугоплавких
металлов показаны на рис. 279.
Борированный слой, образую-
щийся на молибдене, отличается
пористостью, увеличивающейся с
повышением температуры обра-
ботки.
Рентгеноструктурное исследо-
вание, выполненное в Московском
институте стали и сплавов по-
казало, что в поверхностной зоне
борированного слоя в зависимо-
сти от температуры и продолжи-
тельности насыщения в вакуумной
печи в порошке карбида бора при
1300—1500° С образуются либо
две фазы (Mo2Bs и М02В), либо
три фазы (Мо2В5, а-МоВ и Мо2В),
либо четыре фазы (Мо2В5, МоВ2—
слабо, а-МоВ и М02В). Ни по ха-
ми
HV
2000L—1_______
2 k 6 ч1Ш 1500°С
а) б)
Рис. 277. Изменение глубины, по-
верхностной твердости по Виккер-
су (нагрузка 5 кГ) и микротвер-
дости борированных слоев на Мо,
W, Nb, Та, Zr и Re в зависимости
от продолжительности процесса
при 1400° С (а) и температуры
процесса при выдержке 2 ч (б)
[488]
рактеру микроструктуры, ни по
значениям микротвердости не
удавалось обнаружить в бориро-
ванных слоях (вследствие их по-
ристости и малой глубины) ука-
занных зон боридов. Но на косых
шлифах (рис. 280) в поверхност-
ных зонах слоя было замечено
снижение микротвердости с И (Х
2550 до Я1Х 1750—1800 (рис.281).
В исследовании [464] после
борирования молибдена при
1100—1400° С в смеси карбида бора и буры, взятых в соотноше-
нии 84: 16, во внешней зоне диффузионного слоя были обнару-
жены фазы М02В5 и МоВ2, а во внутренней зоне — фаза М02В.
* Рентгеноструктурный анализ проводился Л. Н. Расторгуевым.
371
Энергия активации при диффузии бора в молибден Q =
= 45 000 ± 58 000 кал/г-атом = 188 ± 24 кдж/г-атом, а уравне-
ние температурной зависимости коэффициента диффузии D =
=4,48-10-2 ехр— [464].
По литературным данным, микротвердости боридов Mo2Bs,
а-МоВ и Мо2В близки и колеблются в пределах Н 2350—
2500, а микротвердость борида МоВ2 1200. При электролиз-
ном борировании, по данным работы [322], микротвердость внеш-
Рис. 278. Изменение микротвердости по глубине борированного слоя молибде-
на, вольфрама, ниобия, тантала, циркония и рения (обработка в порошке кар-
бида бора в вакууме при 1400° С (в течение 2, 4 и 6 ч) [488]
него слоя //р. 2650 и внутреннего 1686, а по данным работы
[464], соответственно 1120—1250 и Н,,_ 2450—2580. Борирован-
ный слой, образующийся на молибдене, обладает весьма высокой
износостойкостью.
Твердость борированного слоя на молибдене в горячем со-
стоянии лежит в тех же пределах, что и у широко распространен-
ных металлокерамических твердых сплавов (рис. 282), но его
окалиностойкость существенно выше окалиностойкости метал-
локерамических твердых сплавов типа Со — WC или Со — WC —
TiC. Борирование в 15 раз повышает стойкость молибдена в сме-
си соляной и азотной кислот при 20° С (при кратковременных
испытаниях).
Цементация молибдена. Процесс осуществляется в газовых уг-
леводородных средах или в саже в вакууме либо в потоке водо-
рода. Влияние продолжительности цементации в саже при раз-
372
ных температурах на глубину карбидных слоев, образующихся
на молибдене и некоторых других металлах, показано на
рис. 283. Был получен однофазный кабридный слой, соответст-
вующий, по данным рентгеноструктурного анализа, М02С с мик-
Рис. 279. Микроструктуры борированных
слоев (обработка в вакуумной печи в по-
рошке карбида бора при 1400° С в течение
1 ч). X 135 [488]:
а — молибден; б — вольфрам; в — ниобий;
г — тантал
ротвердостью Н 1420—1500. Как следует из рис. 284, в этих
опытах карбид молибдена имел минимальную микротвердость
по сравнению со всеми другими карбидами и боридами.
В опытах Щербединской А. В. и автора цементацию молибде-
на при 900—1500° С проводили в смеси активированного угля и
373
HV
Рис. 280. Микроструктура
снятой с косого шлифа
средней зоны борированно-
го слоя молибдена (ваку-
умная печь, 1450° С, 5 ч)
(автор и В. А. Тельдеков).
X 150
— / / / / / f s < >ЧО——1 к । \ \
\
\ \ \
\ \ \ \ \ \ \
Рис. 282. Изменение твердости бо-
ридных слоев ниобия, молибдена,
металлокерамических твердых спла-
вов и быстрорежущей стали при на-
греве в вакууме (данные для твер-
дых сплавов и стали Р18 заимство-
ваны из работы [491], (488])
Ци
2500
2000
1500
1000
500
0 0,05 0,10 0,15 0,20 0,25 мм
Рис. 281. Распределение микротвердости на
косом шлифе борированного слоя молиодена:
1 — при 1400° С —5 ч. в вакуумной печи; 2 — при
1450° С — 5 ч (автор и В. А. Тельдеков)
ВаСО3 в вакууме. При температуре 1300° С и выдержке 4 ч на
образцах образовывался слой карбида молибдена Мо2С (что
подтверждено рентгеноструктурным анализом) толщиной 25 мк
(рис. 285), а при температуре 1500° С и выдержке 1 ^ — толщи-
ной 75 мк. Микротвердость слоя по глубине практически не из-
менялась, колеблясь в пределах Ну- 2000—2500 [592]. По одним
литературным данным микротвердость Мо2С Ну. 2000, а по дру-
гим [327], [481], Ну. 1490 и Ну. 1420—1500.
Рис. 283. Влияние продолжительно-
сти цементации на глубину карбид-
ных слоев молибдена, вольфрама,
циркония и тантала (цементация
проводилась в саже в потоке водо-
рода при температурах, указанных
на кривых) [464]
Рис. 284. Зависимость по-
верхностной микротвердо-
сти карбидов и боридов от
температуры насыщения
при выдержке 2 ч (бориро-
вание осуществлялось в
патроне в аморфном боре
с добавкой 3% хлористого
аммония, а цементация —
в саже в потоке водоро-
да) [492]
Для определения параметров диффузии в этом карбиде на
образцах молибдена предварительно создавался слой неактив-
ного карбида, а затем изучалось проникновение в этот слой ра-
диоактивного углерода из смеси активизированного угля и
ВаСО3, содержащего радиоактивный изотоп углерода С14. Рас-
пределение радиоактивного углерода в карбиде молибдена оп-
ределялось методом послойного радиометрического анализа.
Время диффузионного насыщения выбиралось с таким расчетом,
чтобы зона проникновения радиоактивного углерода была на-
много меньше толщины карбидного слоя. Такая постановка экс-
перимента позволила использовать для расчета коэффициента
диффузии решение уравнения Фика для случая диффузии в од-
нородной среде из постоянного источника.
Рассчитанная энергия активации Q = 67 ± 5,4 ккал!г-атом =
= 280 ± 22,6 кдж/г-атом] DQ = 0,3 см21сек\ D = 0,3 X
375
X ехр(—см21сек- При температурах 1500°С (выдержка
15лш«), 1300°С (выдержка 2 ч) и 1100°С (выдержка 24 ч)
коэффициент диффузии соответственно равен 1,92-10-9; 2,03 X
X IO"10 и 1,26-10-" см?/сек. [492].
Важным свойством карбида молибдена, как и многих других
карбидов, является их очень высокая износостойкость, особенно
абразивная, что подтверждено опытами, проведенными в Инсти-
туте машиноведения АН СССР.
Рис. 285. Микроструктура молибдена, це-
ментованного в пропане при 1300° С в тече-
ние 5 ч. X 400 (А. В. Щербединская и ав- •
тор).
Карбид молибдена, как и некоторые другие карбиды переход-
ных металлов IV—VIA групп, характеризуется высокими темпе-
ратурами плавления, стабильностью структуры и состава при на-
греве до высоких температур, хорошей устойчивостью в вакууме
при высоких температурах, а также стойкостью в условиях ион-
ной бомбардировки. Поэтому металлокерамический карбид мо-
либдена начали применять для изготовления катодов электрон-
ных устройств.
А. В. Щербединская и автор изучили эмиссионную способ-
ность и работу выхода электронов карбида молибдена, образо-
ванного диффузионным способом. Полученное значение работы
376
выхода (фо = 3,4-?-3,6 эв) хорошо совпадало с найденным для
металлокерамического карбида молибдена. Эти данные интересны
в связи с тем, что изготовлять катоды электронных устройств,
имеющие весьма тонкое сечение и сложную конфигурацию,
методом порошковой металлургии не представляется воз-
можным; изготовление же их из листового молибдена с после-
дующей цементацией не вызывает каких-либо технологических
Рис. 286. Изменение поверхностной микротвердости ме-
таллов в зависимости от температуры (а) и продол-
жительности (б) азотирования в аммиаке (сплошные
пинии — при 1200° С, штриховые — при 1100° С) [автор
и Л. П. Балабанова]
трудностей. В зарубежной литературе также отмечается практи-
ческое значение цементации молибдена для электронной техни-
ки [486].
Азотирование молибдена. Процесс можно осуществлять в сре-
де азота, тщательно очищенного от влаги и кислорода, или амми-
ака. На рис. 286 показаны кривые, характеризующие изменение
поверхностной микротвердости молибдена и некоторых других
металлов в зависимости от температуры азотирования в аммиа-
ке. При азотировании при 1100° С в течение 6 и 12 ч микротвер-
дость поверхности азотированного молибдена составляла
Ну, 2420. С повышением температуры азотирования микротвер-
дость молибдена резко падала. В литературе отсутствуют дан-
ные о микротвердости нитридов молибдена Mo3N и MoN, а нит-
рид MosN имеет по данным [544] микротвердость 630. На
377
рентгенограммах поверхности молибдена, азотированного при
1100° С в течение 12 ч (рис. 287), были обнаружены линии
кубической гранецентрированной решетки с а = 6,8 ± 0,3 яХ,
что не соответствует параметрам известных нитридов молибдена.
Из трех нитридов только нитрид M02N имеет гранецентрирован-
ную кубическую решетку, но с а = 4,155 ч- 4,160 кХ. Это указы-
Рис. 287. Микроструктура молибдена,
азотированного в аммиаке при
1100° С в течение 12 ч. X 250 (автор
и Л. П. Балабанова)
вает на то, что при азотирова-
нии молибдена в аммиаке об-
разуется фаза, содержащая не
только молибден и азот, но и,
очевидно, водород.
При нитроцементации мо-
либдена в угле в графитовом
тигле в потоке Н2 + NH3 за
1 ч при 1250—1450° С образу-
ются диффузионные слои глу-
биной 0,075—0,12 мм с поверх-
ностной твердостью в среднем
HV 1465 [486].
ОБРАБОТКА НИОБИЯ
В последнее время ниобию
уделяется большое внимание
как возможной основе для соз-
дания весьма жаропрочных
сплавов. Однако ниобий, как и
молибден, отличается очень
низкой окалиностойкостью.
Для защиты ниобия от окис-
ления опробованы те же мето-
ды нанесения покрытий и ис-
пользованы в основном те же
комбинации элементов, что и
для защиты молибдена [493], [494]. Гальванически осаждались
покрытия типа нихром (Ni — Сг — Ni — Сг) и более сложных
составов, подвергавшиеся отжигу. Напылялись различные спла-
вы. Удовлетворительные результаты были получены при напы-
лении слоя сплава А1 — Fe — Si (20 : 40 : 40) толщиной 0,4 мм.
После диффузионного отжига этого слоя при 1200° С в продол-
жение 1 ч образуется диффузионный слой глубиной 0,13 мм, за-
щищающий ниобий от окисления при 1000° С в течение 200 ч.
Производились кратковременное погружение (1—13 мин) молиб-
дена в расплав алюминия с кремнием с последующим диффузи-
онным отжигом при 1040° С в вакууме. При этом в поверхност-
ной зоне слоя образовывалась зона соединения NbAl3. или, если
в ванну вводили еще титан, (NbTi)Al3 [486].
378
Пластичное при высокой температуре покрытие, обладающее
высокой окалиностойкостью до температур 870—1100° С и свой-
ством «самозалечивания», было получено при насыщении ниобия
цинком. При этом в слое образуется соединение NbZna. Такое по-
крытие создается или погружением образцов ниобия в расплав
(температура 550° С), или гальваническим осаждением на них
цинка, после чего производится диффузионный отжиг [482]. Сле-
дует сказать, что, по мнению ряда авторов, в защите ниобия от
Рис. 288. Микроструктуры ниобия (а) и тантала$ (б), силицированных цирку-
ляционным методом при 1100° С в течение 30 мин. X 450 (477]
окисления достигнуты пока меньшие успехи, чем в защите мо-
либдена.
Силицирование ниобия. Этот процесс успешно протекает в га-
зовой среде SiCl4 + Н2 при использовании прямоточного [254]
и циркуляционного [477] способов обработки.
Циркуляционный способ позволяет в течение 30 мин при
1100° С получить слой силицидов ниобия глубиной 0,03— 0,04 мм.
Рентгеноструктурный анализ показал, что основную часть этого
слоя занимает силицид NbSi2, глубже зоны которого расположе-
на тонкая промежуточная зона, примыкающая к сердцевине
(рис. 288).
379
видно из рис.
Si
%
во
20
10
г
0
100 120 МОпк
Рис. 289. Рас-
пределение
концентрации
кремния по
глубине сили-
цированного
слоя ниобия,
насыщенного
из твердой фа-
зы (по данным
рентгеноспек-
трального ло-
кального хи-
мического ана-
лиза) {495]
Более подробно структура силицированного слоя, образующе-
гося на ниобии, изучалась в работе |[495]. Процесс протекал в
твердой фазе при 1100—1200° С в продолжение 0,5—15 ч. Как
289, при общей толщине слоя около 135 мк при-
мерно 106 мк занимает внешняя зона, содержа-
щая 37% Si и представляющая собой, по данным
рентгеноструктурного анализа, силицид NbSi2 с
гексагональной решеткой (а = 4,788кХ, с =
= 6,58 /сХ) микротвердостью 1050. Вторая
фаза — тонкий слаботравящийся слой, содержа-
щий 15% Si, с микротвердостью 700, который
представляет собой соединение Nb3Si3 трех моди-
фикаций: а, у и i|3. Третья, очень тонкая зона со-
держит около 7% Si и соответствует соединению
Nb4Si, кристаллическая структура которого не
установлена.
На ниобиевых проволоках диаметром 0,7 мм,
насыщенных в порошкообразной смеси (20%
кремния, «нейтральный» окисел, хлористый ам-
моний), обнаружен силицированный слой лишь с
одной зоной — дисилицидом ниобия NbSi2
(табл. 66). Аналогичная картина наблюдается
при силицировании молибдена, когда чаще об-
наруживается слой, состоящий только из дисили-
цидов MoSi2, а более бедные кремнием фазы от-
сутствуют.
Таким образом, при силицировании не только
молибдена, но и ниобия отмечается очень слабое
развитие, а в некоторых случаях практически
полное отсутствие в диффузионном слое силици-
дов, бедных кремнием.
Параметры диффузии кремния в ниобии для
фазы NbSi2 определены в работе [471]. Для тем-
пературы 1100, 1200 и 1300° С коэффициент диф-
фузии соответственно равен 0,51 -10“8; 4,50-10“8 и
9,52-10-8 см2/сек (при Q = 29,300 кал/моль — 122,474 дж/моль
и £% = 6-10~4 см2/сек).
Силицированный слой на ниобии обладает высокой окалино-
стойкостью, но легко разрушается под напряжениями при вы-
сокой температуре.
Газовое силицирование ниобия осуществлялось в среде
SiCl4 + Н2 при температуре 1450° С и выдержке 20 мин, в тече-
ние которой образовывался слой силицидов толщиной 0,05 мм
[496]. Лучшей окалиностойкостью обладали образцы ниобия, за-
щищенные двумя слоями силицидов. Сначала эти образцы тра-
вились в специальном реактиве, а затем на них наносился слой
МоО3 погружением в жидкий МоО3 с температурой 800° С.
380
Таблица 66
Результаты рентгеноструктурного анализа силицированного слоя
на ниобиевой проволоке
(Е. Л. Зарубина, Н. Ф. Лашко, А. Ф. Платонова, А. Н. Соколова)
Режим обработки Расстояние от по- верхности в мм* Фазовый состав слоя
Температура в °C Выдержка в ч
1100 3 0 0,013 0,028 0,048 0,093 NbSi2 NbSi2 NbSi2 NbSi2 NbSi2 + Nb
1150 3 0 0,016 0,031 0,044 0,097 NbSi2 NbSi2 NbSi2 NbSi2 NbSi2 + Nb
1100 * Стравливание ра соляной кислоты 5 производилось В эле! в абсолютном этилов 0 0,022 0,045 0,072 0,098 стролите, состоявшем ом спирте. NbSi2 NbSi2 NbSi2 NbSi2 NbSi2 + Nb из 1%-ного раство-
Далее в водороде МоО3 восстанавливался до молибдена, после
чего образцы подвергались силицированию в смеси SiCU + Н2
при 1450° С в течение 20 мин. В результате образовывался внеш-
ний слой силицида MoSi2 толщиной 0,05—0,07 мм и внутренний
слой силицидов ниобия толщиной около 0,05 мм.
При насыщении ниобия из твердой фазы одновременно крем-
нием и титаном при 900—1100° С образовывался один слой с
микротвердостью Я». 1200 [497], [498]. В результате насыщения
при 1200—1300° С образовывалось два слоя, причем второй слой
был чрезвычайно тонким (5—6 мк). Установлено, что основным
поверхностным диффузионным слоем во всех случаях является
дисилицид ниобия NbSi2 с растворенным в нем титаном. Ниобий,
насыщенный одновременно кремнием и титаном, окисляется при
1200° С в 1,5 раза медленнее, чем ниобий, насыщенный только
кремнием. При силицировании сплава ниобия с молибденом диф-
фузионный слой состоял из двух зон — зоны дисилицида ниобия
(15 ;ик) и второй, двухфазной, зоны (45 мк). Окалиностойкость
такого слоя оказалась сравнительно высокой: 100 ч при 1050° С.
Бериллизация ниобия. Насыщение бериллием существенно
повышает окалиностойкость ниобия. В бериллизованном слое,
381
образующемся при обработке в твердой фазе при температуре
900—1300° С и выдержке 6 ч, рентгеноспектральным локальным
химическим анализом 'было установлено наличие четырех зон
бериллидов ниобия (рис. 290) [499].
Рис. 290. Распределение концентрации бериллия по глубине бериллизованно-
го слоя ниобия, насыщенного из твердой фазы (по данным рентгеноспект-
рального локального химического анализа) [499]
Рис. 291. Микроструктура ниобия, алитированного в
порошкообразной смеси при 1100° С в течение 8 ч
(автор и Г. П. Радионова). X 200
Поверхностная толстая зона представляет собой фазу NbBei2
с объемноцентрированной тетрагональной решеткой (а =
= 7,376 кХ, с = 4,280 /сХ), вторая зона — фазу NbBe8 с гексаго-
нальной решеткой (а = 7,56 кХ, с = 10,73 кХ), третья зона —
382
фазу NbBe2 также с гексагональной решеткой, но имеющей дру-
гие параметры (а = 4,516 кХ и с = 7,387 кХ). Кристаллическая
структура четвертой зоны — фазы NbBes не установлена.
Микротвердость поверхностной зоны слоя — бериллида
NbBei2 1200. Коэффициент диффузии бериллия в ниобий,
подсчитанный для NbBei2, равен 6,6 - 10—9 см2/сек при 1100° С и
15,8 • 10-9 см2/сек. при 1200° С.
Хромирование ниобия. Этот способ обработки также повыша-
ет окалиностойкость ниобия. Хромированный ниобий выдержи-
вает испытания при 980° С продолжительностью 320 ч при
13 циклах повторных нагревов и охлаждений.
Рис. 292. Распределение микротвердости по глубине алитированного (а), хро-
мированного (б) и сицилированного (в) слоев ниобия, полученных в порош-
кообразных смесях при 1000 и 1100° С в течение 8 ч (промежуточные зоны
между основным слоем и сердцевиной ничтожны по толщине и при измере-
нии микротвердости не улавливаются) (автор и Г. П. Радионова).
Алитирование ниобия. После насыщения ниобия алюминием
из твердой среды при 1300° С в продолжение 3 ч в поверхност-
ной зоне слоя обнаружена фаза NbAls с тетрагональной (а =
= 3,846 кХ и с = 8,714 кХ) и кубической (а = 3,745 кХ) решет-
ками. Вторая обнаруженная фаза ничтожна по толщине (2—
3 мк). На рис. 291 показана микроструктура ниобия,
обработанного при температуре 1100° С и выдержке 8 ч в смеси,
составленной из 70% алюминиевой пудры, 30% шамота и 3%
хлористого аммония. Микротвердость соединения МЬА1з невысо-
кая— порядка Яр, 650 (рис. 292). Коэффициент диффузии алю-
миния в ниобии для этого соединения при 1100 и 1200° С равен
соответственно 6,3 -10—9 и 7,2* 10-9 см2/сек [499].
Алитирование заметно повышает окалиностойкость ниобия,
особенно при низких температурах. Например, по данным авто-
ра, за 10 ч при 800° С незащищенные образцы металлокерамиче-
ского ниобия увеличивались в весе на 4075 г/м2, а алитирован-
ные— на 59,5 г/м2.
383
Борирование ниобия. Борированный ниобий отличается
высокой поверхностной твердостью (до HV 1890) и микротвер-
достью (до Нр. 2970) [табл. 67]. Важно отметить, что борирован-
ный слой ниобия заметно менее хрупок, чем боридные слои, обра-
зующиеся на вольфраме, молибдене, тантале и цирконии.
Характер кривых (рис. 279) указывает на наличие в слое не-
скольких зон боридов, которые невозможно различить по микро-
структуре (рис. 280).
Таблица 67
Влияние режима борирования ниобия на глубину
и твердость боридного слоя (автор и В. И. Андрюшечкин)
Насыщающая среда Режим обработки Глубина слоя в мм Твердость поверх- ности*
Темпера- тура в °C Выдер- жка в ч HV5
Расплав (70% Na2B4O7 + 1000 3 0,075 1240 1540
+ 30% карбида бора) 1200 3 0,090 1372 2320
Порошок карбида бора 1400 2 0,14 1400 2575
(вакуум 5-10~5 рт- 1400 4 0,22 1839 2620
ст., или 0,00666 н/л2) 1400 6 0,32 1892 2970
1500 2 0,19 1826 2470
* До обработки 250; HV5 — 30 0.
На рентгенограммах образцов ниобия, борированных в ваку-
умной печи при 1400° С в течение 2 ч, обнаружены слабые линии,
принадлежащие боридам NbB2 и Nb3B4 *.
В других условиях обработки образовывался однофазный
(NbB2) борированный слой [488].
Борирование повышает кислотостойкость и окалиностойкость
ниобия. Вес борированных образцов ниобия не изменился после
500 ч испытаний в 50%-ных растворах серной, соляной и азотной
кислот [500]. Борирование резко повышает окалиностойкость ни-
обия, однако только при сравнительно низких температурах. По
данным автора, после испытаний на окалиностойкость при 800° С
в течение 10 ч увеличение веса необработанных образцов метал-
локерамического ниобия составляло 4075 г/м2, а борированных
84 г/м2.
Твердость борированного ниобия в горячем состоянии нахо-
дится на том же уровне, что и у широко распространенных ме-
* Рентгеноструктурный анализ выполнен Л. Н. Расторгуевым.
384
таллокерамических твердых сплавов (см. рис. 282), но его ока-
линостойкость при 800—850° С заметно выше.
Цементация ниобия. Процесс насыщения ниобия углеродом
осуществляется в условиях, аналогичных условиям цементации
молибдена. Данные о микротвердости образующихся при этом
диффузионных слоев приведены на рис. 284. Микротвердость
карбида NbC, по одним данным, Ну. 2055, а по другим — Ну. 1960,
что близко к поверхностной микротвердости слоя после цемен-
тации при температурах 1800—2000° С (см. рис. 284). Сведений
о микротвердости карбида Nb2C в литературе нет.
А. В. Щербединская и автор рассчитали коэффициент диф-
фузии углерода в карбид ниобия по концентрационной кривой,
построенной по результатам гетерофазной диффузии, с исполь-
зованием формул, приведенных в работе [15]. Цементация осу-
ществлялась в смеси активированного угля и ВаСО3, содер-
жащего радиоактивный углерод. Температурная зависимость
коэффициента диффузии углерода в карбид ниобия NbC описы-
вается соотношением [591]
D -= 3,61 • 10“2 ехр
64 500Х
RT /•
Азотирование ниобия. При азотировании ниобия в аммиаке
при 1100—1300° С образуются тонкие и очень хрупкие диффузи-
онные слои. За 6 ч при 1300° С возникает слой глубиной всего
0,02 мм, имеющий микротвердость Ну 2440. Слой с аналогичной
микротвердостыо был получен при 1200° С за 12 ч; после азоти-
рования при этой температуре в течение 24 ч микротвердость
слоя снизилась до Ну 1650 (см. рис. 286, б). В литературе ука-
зывается, что микротвердость нитрида NbN Ну 1400, микротвер-
дость нитрида Nb2N не указана. В работе [147] сообщается об
образовании на азотированной в аммиаке ниобиевой проволоке
зон нитридов, причем внешняя зона соответствует нитриду NbN,
внутренняя — нитриду Nb2N.
ОБРАБОТКА ВОЛЬФРАМА
Кроме хорошо известных областей применения вольфрама,
представляет интерес его использование в ряде новых областей
техники, в том числе для изготовления деталей, работающих
в потоке газов с очень высокими температурой и скоростью исте-
чения. Однако при высоких температурах вольфрам, так же как
молибден и ниобий, быстро окисляется; поэтому очень важно
разработать методы повышения его окалиностойкости. Следует
отметить, что в этом направлении для вольфрама, как и для ни-
обия, сделано значительно меньше, чем для молибдена.
385
Как силицирование, так и бериллизации в несколько раз по-
вышают окалиностойкость вольфрама.
Силицирование вольфрама осуществлялось в порошкообраз-
ных смесях (10% NaF, 5% NH4CI, остальное кремний) [486],
[501] циркуляционным способом (рис. 293) [477] и в вакууме
[502]. В лоследнем случае при более низких температурах и крат-
ковременных выдержках в слое образовывались низшие сили-
циды W5Si3 и W3Si, и при более высоких температурах и продол-
Рис. 293. Микроструктура вольфрама, силицированного циркуляционным
методом при 1000° С (а) и 1200° С (б) в течение 30 мин [478]
жительных выдержках (1240° С, 5 ч) в поверхностной зоне слоя
образовывался дисилицид WSi2.
По данным работы [503], после бериллизации вольфрама
в порошке бериллия при 1100° С диффузионный слой состоит из
трех расположенных друг за другом бериллидов (от поверхности
к сердцевине): WBe24 с тетрагональной решеткой (а = 7,849 кХ„
с = 12,730 кХ); WBei2 с объемноцентрированной решеткой (а =
= 7,29 кХ, с = 4,28 кХ) и WBe2 с гексагональной решеткой (а =
= 4,446 кХ, с = 7,347 кХ), имеющих соответственно следующие
значения микротвердости: 1080, 1145 и 2188. Данные, харак-
теризующие кинетику роста зон слоя с этими бериллидами, при-
ведены на рис. 294. Температурная зависимость коэффициентов;
386
£\уве24 — 1 ’ 175 • Ю 3 exp
^WBei2 — 2,36 • 10 3 exp
£*WBe2 — 1,10-10 4 exp
порошке бериллия:
Л 2, 3 — соответственно
толщина зон бериллидов
WBe24, WBel2, WBe2 [503]
диффузии бериллия в каждой из фаз выражается следующим
образом:
33 800 .
RT ’*
66 950
RT ;
33 115
RT ’
Борирование вольфрама проводилось в вакуумной течи в по-
рошке карбида бора и в расплаве буры с добавкой карбида бора
[488]. Рентгеноструктурным анали-
зом обнаружены в борированном
слое бориды a-WB и W2B (обработ-
ка в вакууме при 1400° С в течение
1 ч). Данные по глубине и твердо-
сти борированных слоев, образую-
щихся на вольфраме, и микрострук-
туры этих слоев приведены на
рис. 276—280.
В среде паров бензина с водо-
родом реакция углерода с вольфра-
мом начинает протекать заметно
уже при 1000° С и подчиняется па-
раболическому закону во всем ис-
следованном интервале температур
(до 1200° С) [504]. Диффузионный
слой состоит из двух зон: внешней
WC (тонкой) и внутренней W2C
(толстой). Микротвердость зоны W2C 1650—1780 [464]. Зави-
симость глубины образующегося на вольфраме карбидного слоя
от продолжительности цементации показана на рис. 283.
Указывается [486], что цементация вольфрама находит прак-
тическое применение. Например, фокус анода рентгеновских
трубок с вращающимся анодом цементуется в гексане при дав-
лении 10—15 мм рт. ст. (1333—1999 н/м2) и температуре 1400° С
до образования W2C. Благодаря образованию карбида повыша-
ются эмиссионные свойства поверхности анода и упрощается его
охлаждение.
Азотирование вольфрама в аммиаке успешно протекает при
температурах 1000 и 1100° С с образованием слоев, состоящих из
внешней (WN) и внутренней (W2N) зон. После азотирования
при 1200 °C цвет и вес образцов не изменяются. Вес образцов,
азотированных при 1000—1100° С и имевших нитридный слой,
после дополнительной выдержки в аммиаке при 1200° С умень-
шался и становился равным весу образцов до азотирования.
Рентгеноструктурный анализ показал, что нитриды при этом
13* 387
исчезли. Таким образом, по приведенным данным, температура
устойчивости нитридов вольфрама в указанной среде ниже
1200° С.
Однако автор получил данные, которые не подтверждают
этот вывод. После азотирования в аммиаке при температуре
1100—1150° С и выдержке 6 ч вес образцов вольфрама не увели-
чивался, а микротвердость поверхности не изменялась, но при
1200° С и той же продолжительности процесса образовался слой
диффузионный глубиной немного менее 0,01 мм, микротвердость
которого составляла около 1540.
При совместном насыщении вольфрама углеродом и азотом
в смеси паров бензина и аммиака в слое образовывались две
зоны с кристаллической решеткой: WC и W2C, что говорит
о большем сродстве к вольфраму углерода, чем азота [504]. Ав-
торы работы [504] считают, что реакционная диффузия в этих
системах осуществляется преимущественно путем диффузии
углерода и азота через карбиды и нитриды в глубь вольфрама.
ОБРАБОТКА ТАНТАЛА
Силицирование тантала осуществлялось в порошке (97% Si
и 3% NH4C1) в атмосфере водорода или аргона [479]. При 900°С
образовывался двухзонный слой (TaSi2 и TaSi), а при 1000,
1100 и 1200° С—однофазный (TaSi2). Глубина однофазного
слоя при 1000° С в течение 4 ч достигала 0,12 мм, а при
1200° С — 0,37 мм. После силицирования в газовой среде
(SiCl4 + Н2) циркуляционным способом при 1000° С за 30 мин
образовывался слой, имевший, судя по микроструктуре, одно-
фазное строение (рис. 288, б). Рентгеноструктурный анализ об-
наружил в этом слое только фазу TaSi2 [478].
После силицирования образцов в вакууме при 1240° С рент-
геноструктурный анализ диффузионного слоя показывал нали-
чие либо двух фаз TaSi2 и TasSi3 (выдержка 1 ч), либо одной
фазы TaSi2 (-выдержка 5 ч). Было показано, что на первой ста-
дии силицирования тантала (так же как ниобия и молибдена)
вначале всегда обнаруживаются только высшие силициды [502].
Микротвердость силицида TaSi2, по разным данным, колеб-
лется в пределах 1250—1650.
Тантал, отличающийся чрезвычайно низкой окалиностой-
костью, после бериллизации очень хорошо противостоит окис-
лению (рис. 295). В работе [486] подтверждается, что особый
интерес представляет создание на поверхности тантала слоя из
очень стабильных бериллидов тантала путем насыщения в ва-
кууме в парах бериллия при температурах 980—1200° С и вы-
держках 2—25 ч. В этой же работе указывается, что в резуль-
тате алитирования тантала при 1040° С в порошкообразной
смеси образуется диффузионный защитный слой, состоящий из
388
ТаА13. Однако в экспериментах, проведенных автором книги,
эффект такой обработки был невелик.
Более высокую, чем после силицирования, окалиностойкость
можно придать танталу, применив следующий способ обработ-
ки: на поверхность образца при 800° С наносится слой Мо03,
затем МоОз при той же температуре восстанавливается в водо-
роде до молибдена и далее силицированием в газовой среде
SiCl4 + Н2 при 1450° С молибденовое
покрытие превращается в дисилицид
молибдена.
Некоторые данные о поверхностной
микротвердости и глубине диффузион-
ных слоев, образующихся на тантале
при борировании, цементации и азоти-
ровании в аммиаке, приведены на
рис. 284 и 276—280.
Боридные слои на тантале имеют
по сравнению с боридными слоями на
других металлах максимальные мик-
ротвердость и твердость по Виккерсу1.
В зависимости от активности боро-
содержащей среды и режима насыще-
Продолжцте льиостъ
испытания
Рис. 295. Окалиностой-
кость при 1100° С небе-
риллизованного (кривая
/) и бериллизованного
(кривая 2) тантала (об-
разцы небериллизован-
ного тантала после ис-
пытания в течение 14 ч
увеличились в весе на
4050 гМ2 [76]
ния в диффузионном слое обнаружи-
ваются или Та2В и ТаВ2, или только
Та2В [488]. По данным работы [464],
в слое обнаружен только борид ТаВ2.
Борирование повышает окалино-
стойкость тантала при температуре
950° С и продолжительности испытания
24 ч в 30 раз, т. е. значительно в мень-
шей степени, чем бериллизация и си-
лидирование [488].
В исследовании, выполненном автором, рентгеноструктурный
анализ насквозь цементованной (температура 1300° С, выдержка
3 ч. среда — пропан) пластинки тантала обнаружил карбиды
ТаС и Та2С. Поверхностная микротвердость этой пластинки со-
ставляла Я[х 1800 (по литературным данным, микротвердость
ТаС 1590, сведений о микротвердости Та2С нет).
После азотирования в аммиаке на ленте из тантала толщи-
ной 0,3 мм за 2 ч при 1100° С образовался нитридный слой глу-
биной 0,07 мм. Увеличение выдержки до 4 ч или повышение
температуры на 200° С приводило к сквозному образованию
1 По литературным данным [544], бориды тантала Та3В2, ТаВ, Та3В4, ТаВ2
имеют соответственно М1икротвердость Нц2770, Нр.3130, Нц3350 и Нр,2500.
Микротвердость боридного слоя на рис. 277 более близка к микротвердости
бопидов ТаВ и Та3В4.
13* А. Н. Минкевич 339
нитридов тантала. Микротвердость тантала, азотированного в
аммиаке при 1300° С в течение 6 ч, достигает 2400 (см.
рис. 286). По литературным данным, микротвердость нитрида
TaN Ну. 1060, значения микротвердости Ta2N и TaNo.eo—TaNo.go
не приводятся.
Неоднократно предлагалось изготовлять некоторые детали
из тантала и цементовать их для получения очень высокой изно-
состойкости (например, фильеры различного назначения, в том
числе для прядильного производства).
ОБРАБОТКА ЦИРКОНИЯ
Окалиностойкость циркония может быть существенно повы-
шена силицированием и бериллизацией.
После бериллизации в порошкообразной смеси (70% Be,
27% шамота и 3% NH4C1) при температуре 1100° С и выдержке
8 ч на образцах циркония образовывался диффузионный слой
глубиной 0,17 мм с поверхностной микротвердостыо Ну. 700.'
Рентгеноструктурный анализ 1 этого слоя показал, что в нем
образуется фаза ZrBeu с кубической гранецентрированной ре-
шеткой типа D23 (а = 10,63 А) и очень небольшое количество
фазы ZrBe2 с гексагональной решеткой (а = 3,46 кХ и с =
= 3,16 кХ). Вследствие малого количества фазы ZrBe2 наличие
ее в слое по кривым 'Микротвердости не обнаруживалось.
Некоторые данные о поверхностной твердости и глубине сло-
ев, образующихся при борировании и цементации циркония, при-
ведены на рис. 278, 279, 283, 284.
При борировании циркония образуется слой, состоящий из
ZrB2 или Z'rB и ZrB2 [339], а по данным работы [464], из ZrB2.
Борирование в 20 раз повышает окалиностойкость циркония при
950° С и продолжительности испытания 24 ч. Кислотостойкость
циркония в смеси соляной и азотной кислот при 20° С повышает-
ся после борирования в 12 раз [488].
При цементации циркония образуется карбид Z'rC, отличаю-
щийся высокой микротвердостью, резко увеличивающейся с по-
вышением температуры цементации (см. рис. 284). По данным
работы [464], после обработки при 1200° С микротвердость це-
ментованного слоя (карбид Z'rC) Ну. 1670—1800, а после обра-
ботки при 1300° С Ну. 2520—2650. Это объясняется широкой
областью гомогенности фазы ZrC (3,3—11,6% Zr).
Азотирование порошка циркония осуществлялось в среде
азота при 2000° С или в среде аммиака при 800—900° С. Азоти-
рование придает цирконию стойкость в азотной кислоте и смесях
соляной и серной кислот. Нитрид циркония ZrN отличается вы-
1 Выполнен Л. Н. Расторгуевым на установке УРС-50М с хромовым излу-
чением (съемки под углами 35’ и 90°).
390
сокой термической устойчивостью, по крайней мере до темпе-
ратуры 1200° С, т. е. он более прочен, чем большинство других
нитридов.
ОБРАБОТКА РЕНИЯ
В .последнее время много внимания было уделено изучению
физико-химических свойств рения и его сплавов [506]. Однако
вопросы химико-термической обработки этого металла не ис-
следовались, и первые сведения об особенностях и результатах
такой обработки могут представлять интерес [507].
Борирование рения. Процесс насыщения рения бором осу-
ществлялся в ванне с расплавом буры и порошком карбида бо-
ра (30%) при 1000 и 1200°С, а также в порошке карбида бора
в вакуумной печи ТВВ-2 (вакуум 5-Ю-5 мм рт. ст., или
0,00666 н/м2) при 1400° С.
Таблица 68
Влияние режима обработки рения на глубину
и твердость борированного слоя [507]
Насыщающая среда Режим обработки Глубина слоя в мм Твердость поверх- ности1
Темпера- тура в °C Выдерж- ка в ч HV5 н м-
Расплав (70?4 Na2B4O7, 1000 3 0,06 443 1890
30% карбида бора) 1200 3 0,07 473 2164
Порошок карбида бора 1400 2 0,08 711 2575
в вакууме 1400 4 0,11 1200 2775
1400 6 0,14 1200 2900
1500 2 0,12 825 2900
1 До обработки Н 550, . HV5 - 400.
Результаты борирования рения 1 приведены в табл. 68 и на
рис. 296, а, б. После борирования в вакуумной печи при 1400°С
в течение 2 ч и в расплаве буры при 1200° С в течение 3 ч глуби-
на диффузионного слоя почти одинакова и его микротвердость
в первом случае заметно выше, очевидно, вследствие образова-
ния иных боридов. Максимально полученная микротвердость
(табл. 68) борированного рения 2900 '(данных о микротвер-
дости боридов рения в литературе не имеется).
1 Образцы рения отрезались от прутка, полученного из металлокера-
мического штабика холодной ковкой с промежуточными отжигами.
391
На микроструктурах (рис. 297) боридный слой выявляется
в виде более темной, чем сердцевина, пористой зоны, травящей-
ся по глубине слоя неравномерно, вероятно, потому, что она со-
стоит из различных боридов.
Рис. 296. Распределение микротвердости по
глубине диффузионных слоев рения после [507]:
а — борирования в расплаве буры с порошком
карбида бора; б — борирования в вакуумной печи:
в — алитирования; г — силицирования [507]
Рентгеноструктурный фазовый анализ диффузионного слоя
(образовавшегося на рении после обработки в вакуумной печи
при 1400° С в течение 2 ч), проводившийся на шлифе при медном
излучении в стандартной камере Дебая, показывает в соответст-
вии с микроструктурой наличие двух зон боридов. На поверхно-
сти расположен тонкий слой гексагонального борида РеВз, а под
ним — толстый текстурированный слой гексагонального борида
РеуВз, кристаллизующегося изоморфно карбиду хрома СГ7В3.
Периоды решетки РеуВз, вычисленные по линиям 330 и 004 в со-
ответствии со структурой, предложенной Аронсоном [508], со-
ставляют: а = 7,5036 кХ; с = 4,8512 кХ, что свидетельствует
392
о некоторой растянутости решетки по оси с, наблюдавшейся и
Аронсоном. Периоды решетки ReB3 вычислить с достаточной
точностью не представляется возможным, так как эта фаза при-
Рис. 297. Микроструктуры диффузионных слоев рения после [507]:
а — борирования (1400° С, 2 ч). Х200; б — хромирования (1100° С, 8 ч). Х135;
в — алитирования (1200. С, 8 ч). X135; г — силицирования (1100° С, 8 ч). Х135.
сутствует в незначительном количестве и представлена линиями,
располагающимися под малыми углами.
Алитирование, хромирование и силицирование рения. Эти
виды химико-термической обработки рения опробованы в по-
рошкообразных смесях, состоявших из 40% порошка шамота и
60% порошков соответствующего элемента (алюминия, хрома,
кремния). К этим смесям добавлялось 3% хлористого аммония.
393
В табл. 69 и на рис. 296 приведены данные о глубине и твер-
дости диффузионных слоев, образующихся на рении при алити-
ровании, силицировании и хромировании.
Таблица 69
Влияние температуры насыщения на глубину и поверхностную
твердость диффузионного слоя, образующегося при алитировании,
силицировании и хромировании рения [507]
Вид обработки Температура1 в °C Глубина слоя в мм Твердость поверхности
HV5
Алитирование 1000 0,09 732 765
1100 0,15 810 864
1200 0,44 1030 1125
Силицирование 1000 1100 0,12 0,14 927 1400 950 1670
Хромирование 1000 0,06 334 362
1100 0,07 349 362
1200 0,08 367 550
1 Продолжительность процессов 8 ч.
Характер микроструктуры (рис. 297, б) и кривых изменения
микротвердости по глубине слоя (рис. 296, в и г) указывает на
фазовую перекристаллизацию, протекавшую в процессе насыще-
ния рения алюминием.
Рентгеноструктурный фазовый анализ алитированного слоя
не проводился.
По данным диаграммы состояния системы Re — Al [509], об-
разуются четыре соединения:
1) Al2Re3 (a-фаза), образуется при содержании в слое
90% Re, микротвердость 700—800;
2) AlxRey (AIRe), образуется -при 82,5% Re, характер струк-
туры и микротвердость не определены;
3) Al2Re, образуется при 77,5% Re, структура не расшифро-
вана, микротвердость около Яц 1000;
4) Ali2Re, структура кубическая, типа A1i2W, микротвердость
Яи 360.
Судя по кривым изменения микротвердости по глубине слоя
(рис. 296, в), после алитирования при 1100°С образуется соеди-
нение Al2Re3, имеющее микротвердость 700—800. После али-
тирования при 1200° С глубина слоя и, очевидно, концентрация
в нем алюминия увеличиваются, так как микротвердость слоя
возрастает до Яи 1125, что близко микротвердости соединения
Al2Re.
394
Рие. 298. Микротвердость
сплавов системы
Re —Сг [289]
После силицирования при 1000° С, судя по микроструктуре
(рис. 297, г) и изменению микротвердости (рис. 296, г), в слое
образуется одно химическое соединение, а при 1100° С — не менее
двух химических соединений с различной микротвердостыо. По
микроструктуре эти соединения различить не удается.
На рентгенограммах поверхности образца, силицированного
при 1100° С в течение 8 ч, обнаружены две системы1 линий:
1) тетрагонального дисилицида ReSis с периодами а = 3,12 кХ
и с = 7,66 кХ и 2) кубического силицида ReSi с периодом а =
= 4,66 кХ. Сопоставляя эти данные с
табличными значениями для силици-
дов рения (ReSig: а = 3,131 кХ, с =
= 7,676 кХ; ReSi: а = 4,774 «X) [510],
можно заключить, что дисилицид ReSis
сохраняет равновесный состав, а сили-
цид ReSi имеет значительный дефицит
по кремнию.
Поверхностная твердость рения пос-
ле хромирования в большинстве слу-
чаев немного снижается или же сохра-
няется на прежнем уровне. Внешний,
нетравящийся, слой отделен от серд-
цевины толстой темной зоной.
Однофазными областями на диаграмме состояния системы
Re — Сг являются p-фаза, представляющая собой твердый рас-
твор хрома в рении с ft = 550 (растворимость хрома в рении
ничтожна), a-фаза (твердый раствор рения в хроме), и o'-фаза
(Re3Crs) [511]. Последняя фаза имеет микротвердость Нц 1560—
1580 (рис. 298). Судя по низкой микротвердости нетравящегося
слоя (Нц 334—550), он, очевидно, состоит из а-фазы, диапазон
изменения микротвердости которой соответствует полученному
на хромированном слое.
Таблица 7Q
Результаты испытаний окалиностойкости необработанного,
хромированного и борированного рения при 1000° С [507]
Продолжительность испытаний в ч Изменение веса образцов в г/м*
необработанных | хромированных борированных
0,5 —8 900 + 129,2 +21,3
2,5 —14 100 +9,4 -28,0
6,5 — —91,0 —136,0
10,0 — —540,0 —298,0
1 Рентгеноструктурный фазовый анализ силицированного слоя рения про-
изводился со шлифа при хромовом излучении в камере Дебая диаметром
57,3 мм Л. Н. Расторгуевым.
395
При нагреве на воздухе до температуры выше 600° С рений
сильно окисляется, выделяя белые и желтоватые пары семиоки-
си рения (Re2O7). При 1000° С уже в течение первого получаса
происходит бурное окисление необработанных образцов рения
(табл. 70), которые через 10 ч разрушаются (остаются только
красно-бурые пленки). Такую же низкую стойкость при 1000°С
показали алитированные и силицированные образцы, после раз-
рушения которых остается сероватый «пепел». Цвет хромиро-
ванных образцов изменяется с серебристого (до испытания) на
темно-матовый; борированных — с серебристо-голубоватого на
темно-серый. Борированные образцы покрываются очень тонкой
Рис. 299. Окалиностойкость рения при 800° С [507]:
1 — необработанного (образцы после испытания в продолжение 6,5 ч потеряли в весе
13 100 г/м2}\ 2 — силицированного; 3 — алитированного; 4 — борированного; 5 — хроми-
рованного
стекловидной пленкой, прочно удерживающейся на металле.
Вес хромированных и борированных образцов при нагреве за-
метно уменьшается, но их форма почти не изменяется. Испыта-
ния при 800° С показали еще больший эффект хромирования и
борирования (рис. 299). Хромирование может предохранять
рений от окисления при этой температуре в течение значитель-
ного времени, тогда как необработанный рений даже при кратко-
временных испытаниях катастрофически окисляется.
ГЛАВА V
ХИМИКО-ТЕРМИЧЕСКАЯ ОБРАБОТКА НИКЕЛЯ,
КОБАЛЬТА И МЕДИ
В последние годы много внимания уделяется исследованию
процессов химико-термической обработки никеля, кобальта, меди
и сплавов на их основе. В ряде случаев такая обработка значи-
тельно улучшает свойства этих металлов и сплавов.
ОБРАБОТКА НИКЕЛЯ
Силицирование никеля в газовой среде SiCh + SiCl2 + Н2
при 1000° С в течение 90 мин позволяет получить слой силицида
никеля Ni2Si глубиной более 50 мк и микротвердостью около
Яр. 820. Свойства этого силицида изучены еще недостаточно
(рис. 300).
В результате бериллизации никеля при 1100° С в течение 8 ч
в порошкообразной смеси (70% Be и 30% шамота + 3% NH4C1)
образуется диффузионный слой, состоящий из двух зон. Рентге-
ноструктурный анализ 1 обнаружил на поверхности слоя фазу,
имеющую деформированную решетку типа у-латуни с а =
= 7,56 А, что совпадает с данными работы [513] для фазы
Ni5Be2i (NisBe2i имеет деформированную решетку типа у-латуни
с а = 7,58 А); на глубине 0,25 мм от поверхности обнаружены
линии решетки В2 с а = 2,60 А, что совпадает с кристалличе-
ской решеткой и периодом фазы NiBe. Характер распределения
микротвердости по глубине бериллизованного слоя показан на
рис. 301.
После бериллизации в смеси порошка бериллия с 2—10%
NH4F или CaF2 при 800° С в течение 1 ч на поверхности образо-
вался слой соединения NiBe4 (по новым данным, это соответст-
вует Ni5Be2i) толщиной 25 мк, а под ним — слой соединения NiBe
толщиной 2 мк [514]. В этой же работе указывается, что на
1 Анализ выполнен на стандартной установке УРС-50М с кобальтовым
излучением под руководством Л. Н. Расторгуева.
397
нимонике 75 поверхностный слой NiBe4 толщиной 25 мк в течение
1 ч при 1000° С переходит в фазу NiBe, окалиностойкую на
воздухе и в продуктах сгорания до температуры 850° С.
Алитирование никеля также 'повышает его окалиностойкость
[580]. Из четырех соединений никеля А1з№ (Р), Al3Ni2 (у) г
AlNi (6) и AlNi (е) наиболее окалиностойкой и наименее хруп-
кой является д-фаза [514], которую можно получить при 950° С
в порошкообразной смеси, состоящей из 10% А1, 90% AhNia и
добавки A1F3, в потоке водорода. При выдержке 90 мин образу-
Расстояние от
фазовой границы
Рис. 300. Изменение микротвердости
по глубине диффузионного слоя ни-
келя, силицированного при 1000° С
в течение 90 мин в газовой смеси
SiCl4 + SiCl2 + На [512]
Рис. 301. Изменение микротвер-
дости по глубине диффузион-
ного слоя никеля, бериллизо-
ванного в порошкообразной
смеси при 1100° С в течение 8 ч
ется слой 5-фазы толщиной 70 мк с подслоем 8-фазы толщиной
4 мк. Аналогичный слой можно образовать в парах AIF3*. При
алитировании нимоника 75 был получен слой 5-фазы толщиной
180 мк и подслой е-фазы.
На воздухе при 1150° С в течение 25 ч слой 5-фазы толщи-
ной 150 мк рассосался (перейдя в е-фазу), а на поверхности
образца образовалась белая пленка AI2O3, после чего вид об-
разца уже не изменялся до конца испытаний длительностью
350 ч [514].
В экспериментах автора окалиностойкость никеля после али-
тирования при 1000° С в течение 10 ч в порошкообразной смеси
возросла при температуре 1100° С (продолжительность испыта-
ния 30 ч) примерно в 4,5 раза.
♦ После алитирования в расплаве (70% А1 и 30% Ni) при 900° С 6-фазу
получить не удалось.
398
Первые опыты по хромированию никеля в порошке хрома
были проведены еще в 1927 г. Тогда было отмечено, что при
температурах процесса 'выше 1250° С диффузия начинает про-
текать в обратном направлении, т. е. наблюдается лишь насыще-
ние хрома никелем.
Образующийся при хромировании никеля диффузионный
слой отличается высокой стойкостью в азотной кислоте, сопро-
Рис. 302. Микроструктура сплава ни-
келя с 5% Сг и 1% Si, насыщенного
хромом. Х600
Рис. 303. Микроструктура никеля, бо-
рированного в расплаве буры с до-
бавлением карбида бора при 950° С
в течение 6 ч. Х200 (автор
и В. Устогал)
гивлением против окисления в парах серы и окалиностойкостью
на воздухе при температурах до 950° С.
Примером успешного практического применения хромирова-
ния сплава на основе никеля может служить обработка
центрального электрода свечей зажигания автомобильных
двигателей
На заводах ряда зарубежных фирм, в том числе английской
фирмы «Лодж» (Lodge Plugs), эта деталь, изготовляемая из
сплава, содержащего 95% Ni и 5% Мп, подвергается диффузи-
онному хромированию. Хромированный слой (рис. 302) резко
повышает сопротивление электродов свечей окислению в присут-
ствии серы и компонентов бензина.
399
При насыщении никеля молибденом или одновременно хро-
мом и молибденом (соотношение хрома и молибдена в насыща-
ющей среде должно быть 1 : 4) возникают диффузионные слои,
отличающиеся высокой стойкостью в некоторых кислотах.
Вольфрамирование никеля повышает его стойкость в соля-
ной кислоте [515].
Насыщение никеля титаном осуществлялось в смеси TiCl4 +
+ TiCl3 + Н2 (при этом возникал слой никеля, легированный
титаном, с соединениями Ni3Ti и NiTi [512]) и в расплаве хло-
ристых солей, в которые вводился титан [516].
Борированию никель подвер-
Рис. 304. Изменение микро-
твердости и микро-т. э. д. с. по
глубине борированного слоя
никеля (автор, Е. В. Панченко,
В. Устогал)
гался в порошкообразных смесях
[431], в расплаве буры с добавле-
нием карбида бора при темпера-
туре 950° С [488] —[490] (рис. 303)
и в газовой среде [17], [517], [518].
Скорость образования борирован-
ных слоев на никеле лишь немно-
го меньше, чем на железе и ко-
бальте [488]. В работе [488] на
Рис. 305. Изменение концентрации ни-
келя в боридном слое (начало отсчета
по оси абсцисс — чистый никель) [5171
кривых изменения микротвердости по глубине слоя были полу-
чены три ступени, соответствующие микротвердости боридов NiB,
ЬИзВ2 и Ni2B. Образование в слое трех боридов было подтверж-
дено также измерением микро-т. э.д. с. по глубине слоя (рис. 304).
Поверхностная микротвердость борированного никеля равна
Ну. 1700 (NiB), что несколько ниже, чем у боридов кобальта
(СоВ) — Ну. 1800 и железа (FeB) — Ну. 1800—2000.
Послойное изучение борированного слоя никеля (обработка
в среде ВС1з + Н2 при 900° С) проводилось с помощью рентге-
ноструктурного анализа и локального химического анализа на
рентгеноспектральной установке «Микрозонд» [517]. На рентге-
нограммах образцов, обработанных в течение 30 мин и 1 ч, бы-
ли обнаружены только линии борида NieB; после выдержки 2 ч
400
обоими методами анализа были выявлены еще два борида:
Ni2B и Ni3B2; после выдержки 5 ч в слое были обнаружены
четыре борида1 (рис. 305).
Установлено, что в процессе борирования никеля при 900° С
в среде ВС13 + Н2 изменение глубины диффузионного слоя с
увеличением длительности выдержки подчиняется почти пря-
молинейному закону (рис. 306), тогда как для кобальта и же-
леза оно подчиняется параболическому закону. В связи с этим
было высказано предположение, что на поверхности борируе-
мых образцов никеля имеет
место флуктуация концент-
раций, которая вследствие
эндотермического характера
процесса образования бори-
дов вызывает местное повы-
шение температуры и воз-
никновение в этих местах
легкоплавкой эвтектики
(NiB + Ni3B2 плавится при
990° С). Коэффициент диф-
фузии в этих местах мгно-
венно увеличивается, что
Рис. 306. Изменение глубины бори-
рованного слоя никеля и кобальта
в зависимости от продолжительности
обработки при 900° С в газовой
смеси ВС13 -I- Н2 [17]
приводит к изменению ха-
рактера кривой и (в данном
случае) почти линейной за-
висимости глубины слоя от
длительности борирования.
Сохранение параболической
зависимости при борировании кобальта обусловлено тем, что
температура образования легкоплавкой эвтектики в системах
Со — Ви FeB значительно выше.
ОБРАБОТКА КОБАЛЬТА
Кобальт и сплавы на его основе отличаются высокой жаро-
прочностью, но недостаточно окалиностойки. Для повышения
их окалиностойкости применяются различные методы химико-
термической обработки.
При силицировании кобальта образуется нетравящийся
слой (рис. 307, а) с микротвердостью, в 1,5 раза превышающей
микротвердость сердцевины. Окалиностойкость силицирован-
ного литого и металлокерамического кобальта при 1100° С (про-
должительность испытания 25 ч) в 5—7 раз выше, чем необра-
ботанного.
1 По данным работы (520], вместо указанных боридов никеля образуются
следующие: Ni2B3, NiB, Ni3B2 и Ni2B.
401
При алитировании металлокерамического кобальта обра-
зуются темные слои (рис. 307, б) с микротвердостью в поверх-
ностной зоне Нр 550. Окалиностойкость литого кобальта повы-
шается после алитирования лишь в 1,5—2 раза, а металлокера-
мического пористого кобальта значительно в большей степени
(рис. 308).
Опробованы титанирование кобальта в расплаве солей
[516] и хромирование в порошкообразных смесях [521]. Хроми-
Рис. 307. Микроструктура металлокерамического кобальта, силицированного:
а — при 1100° С в течение 15 ч в порошкообразной смеси (автор и Т. Т. Гребеннико-
ва). Х200; б — алитированного при 900° С в течение 10 ч в порошкообразной смеси
(автор и М. П. Шелкова). Х86
рование уменьшает окисляемость кобальта и его сплавов при
высокой температуре (980—1090° С) в среде газов, содержащих
сернистые и бррмистые соединения. В связи с этим за рубежом
применяется хромирование изготовляемых из кобальтовых
сплавов деталей реактивных двигателей, в частности сопловые
лопатки газовых турбин.
Предлагается также подвергать кобальтовые сплавы галь-
ваническому никелированию с последующим электролизным
алитированием при 700° С [522].
Борирование кобальта осуществлялось в расплаве буры
(электролиз), в расплаве буры с добавлением карбида бора
[488], [523], а также в газовой среде ВС13 + Н2 [17]. Сопо-
ставление результатов обработки по первому и второму спосо-
бам показывает, что во втором случае борированный слой
402
Рис. 308. Окалиностойкость металлокерамического пористого кобальта при
1100° С (автор и М. П. Шелкова):
1 — неалитированного; 2 — алитированного при 1000° С, 3 ч; 3 — при 900° С, 10 ч;
4 — при 1000° С, 10 ч\ 5 - при 1100° С, 10 ч
а1 S}
Рис. 309. Распределение микротвердости в поверхностной зоне кобальта,
борированного при 950° С в течение 3 ч (а) и 6 ч (б) [523]:
1 — электролизное борирование; 2 — борирование в расплавленной буре с карбидом
бора
403
образуется примерно в два раза медленнее, чем в первом, но со-
стояние поверхности образцов бывает при этом всегда очень
хорошим (после промывки в горячей воде поверхность совер-
шенно чистая, светло-матовая). Судя по данным рис. 309, при
борировании кобальта образуются два борида с различной
микротвердостью: 1800—1850 и 1500—1550. В случае
электролизного борирования оба борида образуются за 3 ч, а
в случае борирования в расплаве с добавкой карбида бора оба
Рис. 310. Влияние продолжительности борирования в расплавленной буре
с карбидом бора при разных температурах на глубину и твердость диффу-
зионного слоя кобальта (за максимальную глубину слоя принимается рас-
стояние от поверхности до конца наиболее длинных игл, за минимальную —
толщина сплошного боридного слоя)
борида образуются лишь через 6 ч (рис. 309). Скорость обра-
зования диффузионных боридных слоев на кобальте примерно
такая же, как на железе (рис. 310).
Микроструктура борированного кобальта напоминает микро-
структуру борированного железа. Диффузионный слой состоит
из двух зон игл боридов, ориентированных перпендикулярно
поверхности образцов (рис. 311). Внешняя зона слоя — борид
СоВ (15,5% В), изоморфный бориду железа FeB (16,25% В),
имеет ромбическую решетку; внутренняя зона — борид C02R
(8,41% В), изоморфный бориду Fe2B (8,84% В),— тетрагональ-
ную решетку. Иглообразный характер боридов кобальта ука-
зывает на то, что их решетка текстурирована, это позволяет
бору перемещаться в определенном структурно обусловленном
направлении (см. стр. 172—173). На границе зоны борида Со2В
и сердцевины иногда можно наблюдать тонкую прослойку про-
межуточной фазы. По-видимому, эта прослойка является бо-
ридом Со3В, о существовании которого до последнего времени
404
имелись противоречивые сведения. Борид Со3В изоморфен бо-
риду Ni3B и имеет ромбическую решетку.
Микротвердость боридов кобальта немного ниже микро-
твердости боридов железа (рис. 309), но хрупкость боридных
слоев у кобальта значительно меньше.
На рис. 312 показан характер распределения по глубине
борированного слоя микротвердости и величины микро-
Рис. 311. Микроструктура кобальта, под-
вергнутого электролизному борированию
в расплаве буры при 950° С в течение
6 ч [523]
г. э. д. с. Кривые микро-
т. э. д. с. полностью воспроиз-
водят изменение фазового
состава по глубине диффузи-
онного слоя.
Рис. 312. Изменение фазо-
вого состава, микротвердо-
сти и микро-т. э. д. с.
по глубине борированного
кобальта (автор, Е. В. Пан-
ченко, В. Устогал)
При изучении оптимальных условий электролизного бориро-
вания кобальта было обнаружено, что с увеличением плотности
тока глубина слоя и его поверхностная твердость по Виккерсу
вначале возрастают, а при дальнейшем увеличении плотности
тока уменьшаются (рис. 313). После электролизного борирова-
ния в эксплуатировавшейся длительное время ванне при не-
большой плотности тока и продолжительности процесса более
6—15 ч нередко можно наблюдать резкое уменьшение поверх-
ностной микротвердости, которая снижается до твердости не-
борированного кобальта и даже несколько ниже. Это сопро-
вождается изменением микроструктуры и уменьшением глубины
борированного слоя. На микроструктуре поверхностной зоны
борированного слоя можно наблюдать при этом слаботравя-
405
щуюся зону плотного металлического осадка значительной тол-
щины и низкой микротвердости (рис. 314).
Спектральный анализ показал, что осадок состоит в основ-
ном из никеля и хрома, которые могли перейти в расплсв из
материала тигля — нихрома (80% Ni и 20% Сг) и нихромовой
проволоки, на которой подвешивались образцы.
Между зоной осад-
ка и зоной борида СоВ
иногда образуется
(вследствие дебориро-
вания) промежуточная
зона борида С02В
(рис. 314, а), которая
имеет меньшую микро-
твердость, чем борид
СоВ.
Следовательно, при
электролизном бориро-
вании кобальта следует
учитывать возможность
растворения в распла-
ве буры металла тигля,
что вызывает анома-
лию в результатах об-
работки. Для уменьше-
ния растворения тигля
в расплаве следует осу-
ществлять его электро-
защиту (см. стр. 226).
На рис. 315 приве-
дены результаты бори-
рования сплавов на ос-
нове кобальта в ванне,
состоящей из расплав-
ленной буры и карби--
да бора. Борирование
сплавов К40Х20 (40% Со, 20% Сг, ост. Fe) и 36НХТЮ (13% Сг,
36% Ni, 1% Al, 3% Ti, ост. Fe) протекает несколько быстрее,
чем сплава К40НХМ (0,08—0,12% С, 36—41% Со, 18—20% Сг,
15—17% Ni, 6—7% Мо, ост. Fe).
Диффузионный слой, образующийся на сплавах, состоит из
нескольких зон (рис. 316). Поверхностная слаботравящаяся зо-
на является зоной боридов. Внешняя часть этой зоны травится
несколько сильнее (отчетливо это видно только на рис. 316, а) и,
по данным рентгеноструктурного анализа ', представляет собой
Плотность тока
Время
Рис. 313. Влияние плотности тока (выдерж-
ка 2 ч) и продолжительности (плотность
тока 1,2 а)см2) электролизного борирования
при 950° С на глубину и твердость диффу-
зионного слоя кобальта (за глубину слоя
принималось расстояние от поверхности до
конца наиболее длинных игл) [523]
1 Выполнен под руководством Л. Н. Расторгуева.
406
ромбический борид типа СоВ и FeB. Внутренняя часть этой
зоны, почти совсем не травящаяся, представляет собой тетраго-
нальный борид типа С02В и Fe2B. Рентгеноструктурный анализ
не показал наличия свободных боридов хрома, никеля и молиб-
дена; очевидно, часть атомов кобальта и железа в кристалли-
Рис. 314. Микроструктуры ко-
бальта, подвергнутого электро-
лизному борированию в дли-
тельно проработавшей ванне
при 950° С в течение 6 ч (а, б)
и 12 ч (в) [523]
ческой решетке боридов замещена хромом, никелем и молиб-
деном.
Микротвердость боридов (Со, Fe, Сг, Ni, Мо)В и (Со, Fe,
Сг)В, расположенных на самой поверхности, практически оди-
накова у обоих исследованных сплавов (рис. 316) и равна при-
мерно Нр. 2300. Это же относится к более глубоко расположен-
ным боридам (Со, Fe, Сг, Ni, Мо)2В и (Со, Fe, Сг)2В, микро-
407
твердость которых у обоих сплавов равна примерно Ям. 1850
(рис. 317).
Переходная зона слоя (твердый раствор + борид) имеет не-
высокую твердость, сильно травится, особенно у сплава
Рис. 315. Изменение глубины и поверхност-
ной твердости по Виккерсу борированного
слоя сплавов К40НХМ (а и б), К40Х20 (в)
и 36НХТЮ (г) в зависимости от продол-
жительности борирования при различных
температурах [523]
К40НХМ (рис. 316, а, б, в). Микроструктура этой зоны у разных
сплавов различна. Легирующие элементы сплавов сильно тор-
мозят диффузию бора. В связи с этим усиливается преимуще-
ственная диффузия бора по границам зерен. Это отчетливо
видно на микроструктурах рис. 316, а, б, на которых глубже
переходной зоны хорошо вытравляются обогащенные бором гра-
ницы зерен.
408
a
Рис. 316. Микроструктуры борированного
при 950° С, 1 ч\ б — при 1000° С, 3 ч; в — при
сплава К40НХМ [523]:
1000° С, 6 ч; г - при 1000° С, 1 *
40$
С увеличением продолжительности борирования и, следова-
тельно, глубины слоя резко возрастает магнитная восприимчи-
вость сплавов (табл. 71), которая у образующихся боридных
слоев, очевидно, больше, чем у металлической основы. Бориро-
Рис. 317. Распределение
микротвердости по глу-
бине борированного слоя
сплавов К40НХМ (кри-
вая /) и К40Х20 (кри-
вая 2), борированных
при 1000° С в течение 6 ч
вание резко увеличивает абразивную износостойкость кобальта
и его сплавов. После борирования при 1000° С в продолжение
6 ч износостойкость кобальта увеличилась в 48 раз, сплава
Таблица 71
Влияние продолжительности борирования при 1000 °C
на магнитную восприимчивость сплавов К40НХМ и К40Х20 [523]
Сплав Длительность борирования в ч Масса в г Сила тока в а Магнитная восприимчивость
К40НХМ 0 1 3 6 2,2601 1,9975 1,9649 2,1311 о,и 0,258 0,34 0,44 1,0 2,66 3,55 4,25
К40Х20 0 1 3 6 4,2433 3,4748 3,4370 3,6211 0,386 0,52 0,686 1,01 1,873 3,08 4,12 5,70
Примечание. Исследования проводились на установке Московского Института стали и сплавов, предназначенной для измерения восприимчивости слабомагнитных тел.
410
К40НХМ в 70 раз и сплава К40Х20 более чем в 100 раз 1 и пре-
восходила износостойкость стали У12, закаленной до твердо-
сти HRC 64.
Борирование несколько снижает коррозионную стойкость
кобальта и его сплавов в условиях тропического климата, но
она все же остается более высокой, чем у закаленной стали У12
(табл. 72).
Таблица 72
Влияние борирования на коррозионную стойкость
кобальта и сплавов К40НХМ и К.40Х20 в условиях
тропического климата [523]
Материал образца Состояние образца Среднее уменьшение веса в г!м* Материал образца Состояние образца Среднее Уменьшение веса в а/л*
Сталь У12 Закаленный 0,393 Сплав К40НХМ Необра- ботанный Бориро- ванный 0,062 0,172
Кобальт Прим шести цикл влажности Необрабо- танный Бориро- ванный е ч а н и е. Исп ов. Режимы ци 100%, 8 ч при 0,087 0,199 ытания прово; клов: 8 ч при 20° С и влажн Сплав К40Х20 лились в тече! 60° С и влажн ости 20%. Необра- ботанный Бориро- ванный 4ие 144 ч и с ости 9Б%, 8 ч 0,049* 0,157 остоялп из при 60° С и
ОБРАБОТКА МЕДИ
Химико-термическая обработка меди и ее сплавов повышает
поверхностную твердость и износостойкость (сурьмирование,
цинкование, бериллизация, силицирование), окалиностойкость
(алитирование, бериллизация, силицирование) и кислотостой-
кость (силицирование, сурьмирование). Однако в отечественной
промышленности химико-термическая обработка меди до сих
пор не применяется, а за рубежом применяется лишь в единич-
ных случаях.
Первые опыты по бериллизация и силицированию меди были
выполнены в 1929 г. Позднее было изучено влияние закалки
и старения на поверхностную твердость предварительно берил-
лизованной меди, алюминиевой бронзы и томпака [524].
В последние годы интерес к химико-термической обработке
меди возрос [525]—[532], [593]. Появились первые рекомендацшг
по использованию химико-термической обработки меди и медных
1 Испытание на износ было проведено на машине Шкода-Савина.
411
сплавов в промышленности, различные методы такой обработки
опробованы на конкретных деталях. Для повышения сопротив-
ления износу рекомендовано, например, подвергать цинкованию
латунные детали немагнитных точных приборов (527]. Алитиро-
вание успешно опробовано на медных деталях работающей при
повышенных температурах токопроводящей аппаратуры домен-
ной печи (530]. Алитирование медных и латунных трубок и дру-
гих деталей теплообменной аппаратуры позволило бы повысить
Рис. 318. Зависимость коэффициента диффузии различ-
ных металлов в медь от температуры [6]
тепловые параметры работы некоторых установок. В Японии
разрабатываются методы газового силицирования меди (531]
и насыщения меди и медных сплавов для повышения их кисло-
тостойкости совместно кремнием и алюминием.
Предложен метод изготовления латунных деталей путем
диффузионного цинкования металлокерамических медных заго-
товок [533].
На рис. 318 приведены данные о коэффициентах диффузии
некоторых элементов в медь.
Цинкование
Цинкование меди можно производить в смеси, состоящей из
50% тонкого порошка цинка (отходы производства в виде цин-
ковой пыли), 49% AI2O3 и 1% NH4CI. Обычно при цинковании
412
EH
Рис. 319. Влияние продолжительности и температуры
процесса на глубину и поверхностную твердость по Вик-
керсу (нагрузка 1 кГ) цинкованного слоя меди (штри-
ховые линии) и латуни ЛС59-1 [253] (сплошные линии):
/ — твердость до обработки.
Рис. 320. Диаграмма состояния системы Си — Zn
Рис. 321. Микроструктура меди, подвергнутой цинкованию при 440° С в тече-
ние 3 ч (а). Х200 и 500°С в течение 3 ч (б). Х200, и латуни ЛС59-1, цинко-
ванной при 500° С в течение 3 ч (в). Х100 [253]
414
к 90% бывшей в употреблении смеси добавляют 10% свежей.
После обработки образцы имеют чистую гладкую поверхность
матово-серебристого цвета.
Результаты цинкования образцов в смеси указанного соста-
ва приведены на рис. 319. Как можно заметить, скорость обра-
зования видимого под микроскопом слоя
на латуни в 2—3 раза больше, чем на
меди. Это различие объясняется разной
подвижностью атомов цинка в меди и ла-
туни в связи со значительной разницей
в температурах их плавления.
В результате цинкования поверхност-
ная твердость меди может быть повыше-
на в 5 раз: с HV 80 до HV 430, а латуни
ЛС59-1 в 3 раза: с HV 150 до HV 460
(рис. 319 и 322). Поверхностная микро-
твердость повышается в еще большей сте-
пени. Повышение твердости объясняется
образованием в слое твердых фаз р' и у
(рис. 320). Внешняя зона слоя (рис. 321),
вероятно, является у-фазой, а внутренняя
Р'-фазой ’.
Цинкование при температуре 395° С и
большой продолжительности процесса
Рис. 322. Распределе-
ние твердости сердце-
вины по Виккерсу (на-
грузка 1 кГ) по глу-
бине диффузионного
слоя латуни, подверг-
нутой цинкованию
в течение 3 ч при тем-
пературе 500° С [253]
позволяет получить глубокие (1,0—1,5 мм) трехфазные слои, со-
стоящие из е-фазы (внешняя зона), у-фазы и тонкой зоны р'-фа-
зы, примыкающей к линии раздела с сердцевиной.
Бериллизация
Бериллизацию меди можно осуществлять в порошкообраз-
ной смеси (40% Be, 59% AI2O3 и 1% NH4C1). Рабочую смесь
составляют из 80% бывшей в употреблении и 20% свежей.
Результаты бериллизации меди и латуни в указанной смеси
приведены на рис. 323 и 324.
Видимый под микроскопом диффузионный слой, как и при
цинковании, на латуни образуется быстрее, чем на меди. По-
верхностная твердость меди после бериллизации повышается
в меньшей степени, чем после цинкования (рис. 319 и 323), за
исключением случая бериллизации при 850° С. Однако диффу-
зионный слой, образовавшийся при этой температуре, имел по-
вышенную хрупкость, а к поверхности образцов прилипали
1 Травление меди производилось в 25%-ном водном растворе NH«OH,
а латуни — в смеси 25%-ного водного раствора NH4OH и 2%-ного водного
раствора Н2О2, взятых в соотношении 1: 1.
415
Рис. 324. Распределение твердости
по Виккерсу (нагрузка 1 кГ)
по глубине диффузионного слоя
латуни ЛС59-1, бериллизованной
по разным режимам [253]:
1 _ при 750° С, 2 ч\ 2 — при 700° С;
2 ч; 3 — при 650°, 2 ч
процесса и температуры на глубину и по-
верхностную твердость по Виккерсу (на-
грузка 1 кГ): бериллизованного слоя
меди (штриховые линии) и латуни
ЛС59-1 (сплошные линии) [253]:
1 — твердость сердцевины
а) б)
Рис. 325. Микроструктура меди, бериллизованной в течение 3 ч при 650° С (а)
и 850° С (б). Х86 [253]
416
частицы порошка (после обработки при более низких темпера-
турах поверхность образцов была гладкой).
Твердость бериллизованного слоя меди и латуни может быть
повышена дополнительной закалкой и старением. У меди,
бериллизованной в продолжение 10 ч при 800° С в среде водо-
рода в порошке бериллия, твердость после закалки с 800° С
составляла HV 230, а после старения при 300° С повысилась до
HV 485 [524]. На рис. 325, б при-
ведены микроструктуры берилли-
зованного слоя. Судя по диаграм-
ме состояния системы Си — Be
(рис. 326), с внешней стороны к
линии раздела примыкает двух-
фазная область (вероятно, а +
+ эвтектоид (а + у), далее рас-
положена темная структура, на-
поминающая эвтектоид (а + у).
Ближе к поверхности появляются
отдельные участки светлой со-
ставляющей, а самая верхняя
слаботравящаяся зона слоя со
столбчатым строением, обладаю-
щая повышенной хрупкостью и
максимальной твердостью, пред-
ставляет собой, очевидно, у-фазу
(СиВе).
ЛИТИрОВЯНИе Рис. Диаграмма состояния
Алитирование можно произво- системы Си —Be
дить в смеси, состоящей из 50%
алюминиевой пудры, 49% А12О3
и 1% NH4C1. Рабочую смесь составляют из 80% бывшей в упо-
треблении и 20% свежей. Поверхность алитированных образцов
после обработки в указанной смеси золотистая. Результаты али-
тирования приведены на рис. 327 и 328. При алитировании на-
блюдается большее, чем при цинковании и бериллизации, разли-
чие в скорости образования диффузионного слоя у меди и лату-
ни. Поверхностная твердость после алитирования повышается
в меньшей степени, чем после цинкования и бериллизации L
После алитирования алюминиевой бронзы Бр.АЮ при 750° С
в течение 1; 2 и 4 ч глубина слоя достигала соответственно
0,78; 0,96 и 1,28 мм. Микротвердость бронзы на глубине 0,05 мм
1 Полученная поверхностная твердость меди и латуни, видимо, не яв-
ляется предельной, так как в отдельных экспериментах, выполненных автором
ранее, была достигнута более высокая поверхностная твердость алитирован-
ной меди (HV 300—390).
417
Рис. 327. Влияние продолжительности и тем-
пературы процесса на глубину и поверхност-
ную твердость по Виккерсу (нагрузка 1 кГ)
алитированной меди (штриховая линия) и ла-
туни ЛС59-1 (сплошные линии) [253]:
сердцевины
Рис. 328. Распределение
твердости по Виккерсу
(нагрузка 1 кГ) по глу-
бине алитированного
слоя латуни ЛС59-1, об-
работанной при 650° С
в течение 3 и 4 ч [253]
418
оказалась выше (Я1Х 320), чем у алитированной латуни
Послойный химический анализ стружки, снятой с образца
меди диаметром 20 мм, алитированного при 750° С в течение
4 ч, показал в первой стружке толщиной 0,05 мм концентрацию
алюминия 18%; на глубине 0,25; 0,45 и 0,68 мм концентрация
Рис. 329. Микроструктура меди, подвергнутой алитированию при 800° С в те-
чение 6 ч (а) и при 850°С в течение 4 ч (б). Х86 [253]
алюминия была соответственно 7,8%; 2,3% и 0,1% (по микро-
шлифу глубина двухфазной зоны слоя около 0,40 мм).
Таблица 73
'Изменение механических свойств латунных трубок после
алитирования [253]
Режим обработки ° в S в %
в кг! мм* в н!мг-1 О7
Неалитированные 39,6 41,7 38,8 40,9 48,8 46,8
Алитированные при 650° С в тече- ние 1,5 ч 50,2 47,4 49,0 46,5 18,0 16,0
Алитированные при 650° С в тече- ние 3 ч 53,1 51,3 52,0 50,3 15,5 14,5
Примечание. Толщина стенок трубки 1 мм, внешний диаметр 10 мм.
419
Алитирование повышает предел прочности и снижает от-
носительное удлинение меди и латуни (табл. 73).
На рис. 329 и 330 приведены микроструктуры алитирован-
ных слоев меди и латуни ЛС59-1.
Сопоставление структуры с диаграммой состояния Си—А1
(рис. 331) и послойный химический анализ указывают, что при
Рис. 330. Микроструктура латуни
ЛС59-1, алитированной при 600° С
в течение 1 ч. Х100 [253]
алитировании меди к линии раз-
дела примыкает зона, состоящая
из светлых (a-фаза) и темных
(эвтектоид а + уг) полей. Ближе
к поверхности расположена зона
эвтектоида, а у самой поверхно-
сти —• эвтектоид и уз-фаза (свет-
лая составляющая); на рис. 329
этой зоны нет.
Хорошие результаты показало
электролизное алитирование меди
в расплаве криолита (На3А1Рб);
анодом служил графитовый ти-
гель, а катодом образец. При про-
пускании тока через расплав
криолита происходят следующие
реакции:
Na3AlF6->3Na++A1F|“;
XlFg— -> Al + 6F.
Результаты обработки при
плотности тока 0,5 а! см2 приве-
дены в табл. 74.
Преимущества электролизного алитирования меди в распла-
ве криолита — большая скорость процессов и чистота обрабо-
танной поверхности; недостатки — сильное испарение криолита,
необходимость контакта между подвеской и изделием и быстрый
выход из строя графитовых тиглей. Этот метод, очевидно, может
быть использован и для алитирования стали.
Таблица 74
Влияние температуры и продолжительности электролизного алитирования
в расплаве криолита на глубину слоя [253]
Режим обработки Глубина слоя в мм Микротвердость на глубине 0,05 мм
Температура в °C Выдержка в мин
900 10 0,20 295
900 20 0,60 —
950 10 0,42 245
420
Р.ис. 331. Диаграмма состояния системы Си—А1
Силицирование
Силицирование может производиться в смеси, состоящей
из 40% Si, 59% AI2O3 и 1% NH4CI. При большем содержании
в ней кремния (45—50%) увеличивается глубина слоя, но
ухудшается состояние обработанной поверхности.
На рис. 332 приведены результаты силицирования меди
в смеси указанного состава при температуре 850° С. Кривые
распределения микротвердости по глубине слоя имеют две вет-
ви, указывающие на присутствие в слое двух фаз с разной
твердостью. И действительно, на микроструктуре обработанных
образцов (рис. 333) отчетливо видны две различные по трави-
мости структуры, очевидно, a-твердый раствор (более светлая
и более мягкая составляющая) и смесь фаз а + у (рис. 334).
В случае кратковременной (0,5—1 ч) выдержки при 850° С
или более продолжительной (4 ч) выдержки при 800° С на
421
образцах образуется однофазный диффузионный слой. При тем-
пературе 900° С возникает двухфазный слой, однако количество
второй структурной составляющей (эвтектоид) настолько
Рис. 332. Влияние продолжительности и температуры силицирования на глу-
бину слоя (а, б) и распределение микротвердости по глубине слоя (в, г)
(автор, Г. Б‘. Кример и Э. И. Нахамкина)
Рис. 333. Микроструктура меди, силицированной
при 850° С в течение 2 ч. Х86
ничтожно, что на кривой распределения микротвердости нет
второй ветви. По-видимому, в процессе силицирования при
900° С благодаря большей, чем при 800° С, скорости диффузии
422
кремния в медь концентрация кремния в слое снижается. Сни-
жение концентрации кремния в слое с повышением температуры
подтверждается и тем, что в процессе силицирования при
850° С иногда происходило оплавление образцов, а при более
высоких температурах (900—950° С) таких случаев не было.
Предложено газовое силицирование меди в смеси SiCU +
+ SiCl2 + Н2 [512].
В этом случае при температуре около 1000°С неизменно
происходило расплавление меди, так как в результате большой
Рис. 334. Диаграмма состояния системы Си—Si
активности газовой среды концентрация кремния в поверхност-
ной зоне диффузионного слоя быстро возрастает до величины,
соответствующей линии солидуса на диаграмме состояния
системы Си—Si (рис. 334).
Опробовано силицирование в парах SiCU с использованием
азота в качестве несущего газа [531]. В этом случае процесс
протекал только в присутствии порошка кремния вблизи по-
верхности меди.
Сурьмирование
Сурьму добавляют в некоторые сплавы, например в типо-
графские сплавы — гарт (12—23% Sb), баббиты (7—16% Sb)
и др. для повышения их износостойкости и улучщения антикор-
розионных свойств. В связи с этим определенный интерес пред-
ставляет изучение процесса диффузионного насыщения меди
и ее сплавов сурьмой.
$3
Рис. 335. Влияние содержания в порошкообразной смеси окиси сурьмы на глу-
бину диффузионного слоя меди при выдержке 6 ч и распределение микро-
твердости по глубине сурьмированного слоя (800° С, 6 ч, 15% БЬгОз)
(автор и Г. Б. Кример)
Рис. 336. Диаграмма состояния системы Си — Sb
424
Сурьмирование меди осуществлялось автором при 800—
900° С в смеси, состоящей из древесного угля и 4% Sb2O3. При
высокой температуре протекает реакция восстановления сурь-
мы (см. стр. 315). Восстановленная сурьма диффундирует в медь.
С дальнейшим увеличением содержания в смеси ЭЬгОз глубина
слоя возрастает (рис. 335), но поверхность иногда частично
оплавляется.
Рис. 337. Микроструктура меди, сурьмирован-
ной при 800’ С в течение 6 ч (травление рас-
твором хлорного железа в НС1). Х86
Так как после сурьмирования в указанной смеси при 800—
900° С на образцах не было следов оплавления, то, следователь-
но, концентрация сурьмы в диффузионном слое не превосходила
предела растворимости сурьмы в a-твердом растворе, т. е. была
ниже примерно 4% при 900° С и 7% при 800° С (рис. 336).
Диффузионный слой под микроскопом обнаруживается
в виде нетравящейся зоны, глубже которой расположена про-
межуточная сильнотравящаяся зона (рис. 337). Фазовый состав
слоя пока не исследовался.
Поверхностная микротвердость меди после сурьмирования
повышается примерно в 2 раза (с //и 60 до Hv. 130).
Сурьмирование, как это видно из табл. 75, значительно по-
вышает износостойкость меди — в большей степени, чем цинко-
вание (вероятно, вследствие большой хрупкости цинкованного
слоя).
14 А. Н. Минкевич 425
Таблица 75
Результаты испытаний износостойкости химико-термически
обработанных образцов меди на машине Амслера (автор и Г. Б. Кример)
Вид обработки Состав Режим обработки Уменьшение веса образцов в г
смеси Темпера- тура в °C Выдержка в ч за 10 мин за 20 мин
Необработанные Сурмирование . . 8% Sb2O3 + +92% дре- весного 800 6 0,0695 0,0109 0,1392 0,0159
Цинкование . . „ угля 40% Zn + + 59о/о А120з+ +1% NH4C1 550 2 0,0307 0,0345
Примечание. Испытания проводились без смазки при нагрузке 25 кГ', ис- пытуемый медный сектор работал по стальному кольцу с твердостью HRC 62—63.
Кроме описанных процессов, в литературе упоминается
о поверхностном насыщении меди оловом из газовой среды [527].
Водород пропускался над разогретым SnCl2 и затем над медью.
Образовавшиеся диффузионные слои в зависимости от темпера-
туры процесса (300—520° С) состояли из тех или иных фаз.
В последнее время появился ряд работ, в которых изучается
диффузия в медь серы. Исследование процесса диффузии с при-
менением изотопа S35 показало, что при насыщении сплава
Си—Zn из расплава сера диффундирует только в медь [534].
Температура образования и скорость роста слоя сульфидов CuS
и Cu?S в газовой среде Н2 + H2S была изучена в работе [535].
Окалиностойкость химико-термически обработанной меди
Химико-термическую обработку меди целесообразно при-
менять в тех случаях, когда требуется материал с высокой
теплопроводностью и электропроводностью, работающий пр»
высокой температуре.
Судя по данным рис. 338, алитирование и силицирование
повышают окалиностойкость меди в большей степени, чем цин-
кование. Наибольший эффект повышения окалиностойкост»
меди и латуни дает бериллизация (рис. 339, табл. 76).
Существенно влияет на окалиностойкость меди режим обра-
ботки. Наиболее высокая окалиностойкость достигается после
бериллизации и алитирования при 650° С. Увеличение темпера-
туры насыщения уменьшает концентрацию алюминия и берил-
лия в поверхностных зонах диффузионного слоя, что приводит
426
к ухудшению окалиностойкости при кратковременных испыта-
ниях. Однако, поскольку при высоких температурах обработки
Рис. 338. Окалиностойкость меди:
1 — необработанной; 2 — алитированной; 3 — силицированной;
4 — цинкованной при разных температурах в зависимости от продол-
жительности испытания (автор, Э. И. Нахамкина и Л. И. Игнатьева) •
образуются более глубокие диффузионные слои, возможно, что
более длительные испытания покажут иную зависимость окалй-
ностойкости от температуры насыщения. Алитирование при
600° С протекает медленно; образующийся при этом диффузион-
ный слой имеет малую глубину (около 0,05 мм) и не обеспечи-
вает защиту меди от окисления (рис. 339).
В табл. 76 проведены сравнительные данные об окалино-
стойкости меди, латуни и литой бронзы Бр. А10, предназначенной
для работы при повышенной температуре. Окалиностойкость
бронзы Бр. А10 при 800° С примерно равна окалиностойкости
бериллизованной латуни и немного выше окалиностойкости
Рис. 339. Окалиностойкость при 800° С необработан-
ной (/), алитированной (а) и бериллизованной (б)
меди {253]. Температуры алитирования и берилли-
зации указаны на кривых. Образцы необработанной
меди после испытания в течение 15 ч увеличились
в весе на 300 г/м2
алитированной латуни. В табл. 77 приведены данные, показы-
вающие влияние алитирования на окалиностойкость при 800° С
медных и латунных трубок.
После обработки аналогичных трубок длиной 60 мм, из-
готовленных другим заводом, окалиностойкость повысилась еще
больше: за 25 ч испытаний при 850° С вес неалитированных
трубок увеличивался па 361 г/л*2, а алитированных — на
14,5 г!м2, за 50 ч соответственно 553 и 18,1 г!м2.
В Московском институте стали и сплавов проведены испыта-
ния алитированных сердечников паяльников. К боковым сторо-
нам таких сердечников не пристает полуда, при замене они
легко вынимаются из паяльника. Для получения надежных
данных о влиянии алитирования на долговечность сердечников
должны быть проведены дополнительные, более широкие испы-
тания.
428
Таблица 76
Окалиностойкость медных и латунных образцов при 800° С
Материал образца Состояние Режим обработки Увеличение веса образца в г1мг в зависимости от времени испытания в ч
образца Темпера- тура в °C Выдерж- ка в ч 2 5 10 15
Медь Необрабо- танный » Алитированный » Бериллизован- ный 650 750 650 650 3 3 3 3 91,0 107 1,92 4,31 1,07 0,87 189,4 172 4,01 5,75 1,8. 1,2 269 230 6,24 8,94 1,90 1,30 322,8 287 8,62 И,7 1,90 1,30
Латунь ЛС 59-1 Необрабо- танный Алитированный » Берйллизован- ный » 600 650 700 650 1 1 2 2 | | О О ND СП bo 4b. О) сл о 12,4 16,1 1,25 1,25 0,8 0 15,21 18,0 2,5 1,99 1,0 0 21,45 21,1 2,8 1,99 1,2 0,4
Бронза Бр. А10 Необрабо- танный — — — 0,05 0,07 1,05 1,30 1,3 1,55
Для защиты меди от окисления можно исхользовать также
диффузионное хромирование, показывающее хорошие резуль-
таты [535].
Таблица 77
Окалиностойкость при 800° С медных и латунных
алитированных трубок [253]
Материал трубки Состояние трубки Увеличение веса в г
Медь Необработанная Алитированная 1,2083 0,0723 0,1043
Латунь Необработанная Алитированная » 0,1015 0,0066 0,0054
Примечания: 1. Режимы обработки трубок: медных — температура 800° С, выдержка 4 ч\ латунных —температура 650° С, выдержка 4 ч. 2. Размеры трубок: толщина стенки 1 мм, внешний диаметр 10 мм, длина 300 мм. 3. Продолжитель- ность испытания 23 ч.
429
Кислотостойкость химико-термически обработанной меди
Химико-термическая обработка существенно повышает кор-
розионную стойкость меди в некоторых средах. При испытании
в водопроводной воде на поверхности силицированной меди
образуется тонкая прочная пленка продуктов коррозии, обла-
дающая защитными свойствами. При этом вес силицированных
Рис. 340. Кислотостойкость необработанной (кривая /), сурьмированной
(кривая 2) и силицированной (кривая 3) меди в соляной и серной кислотах
(технических) различных концентраций при 20° С (автор и Л. И. Игнатьева)
образцов после испытаний немного увеличивается, а сурьмиро-
ванных уменьшается. Еще сильнее уменьшается вес необрабо-
танных образцов. После испытаний в водопроводной воде при
100° С в продолжение 2 суток силицированные и сурьмирован-
ные образцы потеряли в весе соответственно в 2,5 и 2 раза
меньше, чем необработанные. Таким образом, испытания
в водопроводной воде при 100° С показали, что эффективность
силицирования и сурьмирования в данном случае невелика.
После испытаний в 20%-ном растворе аммиака при 20° С
вес цинкованных и силицированных образцов меди уменьшился
в 10 и 5 раз, а сурьмированных и алитированных в 3 раза
меньше по сравнению с весом необработанных образцов.
4'30
Испытания при 20° С в соляной кислоте различных концен-
траций показали, что стойкость сурьмированных образцов
значительно выше, чем силицированных (рис. 340). При 20° С
в растворах серной кислоты уменьшение веса сурьмированных
образцов было ничтожным, а вес силицированных образцов
Умёньш ен и ь о ёсо
Рис. 341. Кислотостойкость необработанной (кривая /), сурьмированной (кри-
вая 2) и силицированной (кривая 3) меди в соляной и серной кислотах
(технических) различных концентраций при 100° С (автор и Л. И. Игнатьева)
существенно увеличивался в результате образования на их по-
верхности пленки продуктов реакций. Следует отметить, что ско-
рость коррозии сурьмированной меди при 20° С практически не
зависит от концентрации серной кислоты (рис. 340).
При испытаниях при 100° С в 10%- и 50%-ных растворах
соляной кислоты стойкость химико-термически обработанной
меди была весьма высокой (рис. 341). В остальных растворах,
а т^кже в концентрированных соляной и серной кислотах ее
стойкость невелика, хотя в несколько раз и превосходит стой-
кость необработанной меди. Следует отметить, что испытаниям,
431
результаты которых приведены на рис. 340, подвергались об-
разцы меди со сравнительно неглубокими диффузионными
слоями (силицированный слой — 0,3 мм, сурьмированный —
0,25 мм).
В другом исследовании автора образцы силицировались на
глубину 0,3—0,4 мм и 0,8—1,0 мм и испытывались в 10 %-ном
растворе серной кислоты при 250° С (табл. 78).
Таблица 78
Результаты испытания1 кислотостойкости образцов меди Ml
Продолжительность испытаний в ч Глубина силицирования в мм Изменение веса образцов в г!м* ч
24 Не обработаны 0,3—0,4 0,8-1,0 —1,4 +0,4 +0,4
200 0,3—0,4 0,8—1,0 —6,8 +0,01
1 Испытания проводились М. Веденеевой в лаборатории коррозии Московско- го института стали и сплавов.
При испытаниях на силицированной поверхности образовы-
валась пленка продуктов реакции, обладающая хорошими за-
щитными свойствами: даже после испытаний в течение 200 ч
вес образцов с глубоким силицированным слоем не умень-
шался.
Измерения, проведенные на паре медь необработанная —
медь силицированная, показали, что силицированная медь имеет
более отрицательный потенциал. Следовательно, в системе
медь — силицированный слой последний будет являться анодом
и усиленно растворяться. Это подтверждается внешним видом
испытанных в течение 200 ч образцов с тонким силицированным
слоем. Раствор постепенно проникал сквозь слой к основному
металлу, и начиналось бурное разрушение слоя.
Предохранить медь от воздействия азотной и соляной кислот
(испытания длились 1—2 месяца) можно путем осаждения на
меди слоя тантала толщиной 0,01—0,06 мм [535]. Процесс
осуществляется в смеси Н2 + ТаС15 при 800—1000° С. Несмотря
на высокую температуру процесса, диффузионного проникнове-
ния тантала в медь не происходит. Это объясняется весьма
незначительной растворимостью тантала в меди и тем, что
они не образуют друг с другом интерметаллических соеди-
нений.
Для повышения кислотостойкости меди рекомендуется диф-
фузионное титанирование при 850° С в продолжение 7—9 ч
432
в ванне из расплава хлористого калия, в которую добавлено
10% порошка сплава TiOK. Зеркало ванны защищается от воз-
духа аргоном или гелием [516]. Этот процесс применительно
к обработке стали описан более подробно на стр. 289.
В Японии для повышения кислотостойкости меди рекомен-
дована порошкообразная смесь, содержащая 60% сплава
Si—Си (25% Si); 6% сплава Fe—Al (25% Al); 0,5% С;
1,5% Fe2O3; 0,8% NH4C1, остальное — порошок, бывший в упот-
реблении. Обработку в такой смеси производят при 860—930°С
в течение 15—18 ч.
ГЛАВА VI
ХИМИКО-ТЕРМИЧЕСКАЯ ОБРАБОТКА
МЕТАЛЛОКЕРАМИЧЕСКИХ СПЛАВОВ
обработка железных сплавов
Металлокерамические сплавы отличаются от компактных
тем, что у них очень развита внутренняя поверхность пор,
а контакт между частицами может быть неполным. В связи
с этим диффузионные процессы в металлокерамических спла-
вах протекают быстрее, чем в компактных, причем скорость их
тем больше, чем меньше плотность металлокерамики. С увели-
чением пористости количество поглощенного элемента увеличи-
вается. Изменяются также механические свойства.
Примером влияния плотности металлокерамических желез-
ных образцов на состав диффузионного слоя и их механические
свойства могут служить данные, приведенные в табл. 79.
После азотирования при 500° С в течение 15 ч общее коли-
чество азота (судя по увеличению веса), поглощенного желез-
ными металлокерамическими образцами с пористостью 91%
и 55%, соответственно в 90 и 190 раз больше, чем компактными
образцами. После хромирования при 950° С глубина слоя на
металлокерамических железных образцах с плотностью 91% —
в 3 раза, а с плотностью в 78% —в 4 раза больше, чем на
компактных железных образцах. После хромирования при
1050° С это различие составляет соответственно 7 и 14 раз [538].
Судя по данным табл. 80, кратковременное азотирование
(20 и 45 мин) при 500° С и хромирование при 1150° С (2 ч)
повышают прочность и пластичность образцов металлокерамики.
Азотирование, кроме того, повышает их износостойкость в;
30 раз, а коррозионную стойкость в 0,5%-ном растворе серной
кислоты в 6—8 раз, хромирование — соответственно в 3 раза
и 8—10 раз.
Высокая коррозионная стойкость хромированнных металло-
керамических железных образцов в 10%-ном растворе азотной
кислоты достигается лишь при сквозном хромировании, иначе
434
Таблица 79
Содержание углерода и азота в слое и механические свойства1
металлокерамических железных образцов, нитроцементованных
______________________при 870° С [537]_________________________
Характеристики Образцы с плотностью 6.75 г!смл Образцы с плотностью 7,5 г/см’
необрабо- танные нитроце- ментованные необрабо- танные нитроце- ментованные
Содержание углерода в поверхностной зоне слоя в % — 0,90 — 0,82
Содержание азота в поверхностной зоне слоя в % — 0,22 — 0,12
в кГ /мм2 в н/м2-107 17,4 17,0 71,0* 67,5 27,5 26,9 41,7 40,8
в кГ/мм2 в ф2-107 9,5 9,3 62* 60,7 16,9 16,5 30,3 29,6
6 В % 13 1,5 33,5 2
Твердость HRA 1 Механические свойс низкого отпуска. ♦ Высокая прочность в 2 раза глубину слоя у тва определи обусловлена б >бразцов с пло' лись во всех ольшой глуби гностью 7,5 г/с 23 случаях после ной слоя, пре м’. 73 закалки и восходящей
Таблица 80
Механические свойства азотированных и хромированных
металлокерамических железных образцов различной плотности [538]
Характе- ристика . Механические свойства Плот- ность образца в %
после спекания после азотирования при 500° С в течение после хро- мирования при 1150° С в течение 2 ч
20 мин । | 45 мин 5 ч
7,3 11,0 9,7 3,6 11,0 62
7,15 10,78 9,5 13,5 10,78
б/ 11,3 17,0 15,0 6,3 20,5 78
11,07 16,67 14,7 6,1 20,0
17,8 24,2 22,0 16,5 25,8 91
17,49 23,6 21,5 16,1 25,2
435
Продолжение табл. 80
Характе- ристика Механические свойства Плот- ность образца в %
после спекания после азотирования при 500° С в течение после хро- мирования при 1150° С в течение 2 ч
20 мин | 4 5 мин | • 5 ч
6 В % 2,2 4,5 3,5 — 8,5 8,33 62
2,6 5,3 4,8 — 10,2 9,99 78
6,1 9,8 9,5 — 15,5 15,2 91
а ♦* 0,11 0,45 0,40 0,06 0,25 62
0.107 0,441 0,392 0,058 0,245
0,12 0,50 0,45 0,12 0,30
0,117 0,490 0,441 0,117 0,294 1 о
0,28 0,60 0,65 0,20 0,50 91
0,274 0,588 0,637 0,196 0,490
* В числителе приведены значения, выраженные в кГ/мм*, в знаменателе — в н/л<2.1 О’.
♦♦ в в «-л</л<2 числителе приведены значения, выраженные в кГ- м'см2, в знаменателе —
раствор проникает через поры в сердцевину и разрушает ее.
Для работы в атмосферных условиях достаточно несквозного
хромирования металлокерамических деталей.
Процесс хромирования не требует дополнительного оборудо-
вания и не удлиняет цикл изготовления металлокерамических
деталей, так как совмещается со спеканием [538], [539], [581]. Та-
кая технология применяется, например, при изготовлении фильер
для нефтяной и газовой промышленности [594].
ОБРАБОТКА ТВЕРДЫХ СПЛАВОВ
Металлокерамические твердые сплавы также можно подвер-
гать химико-термической обработке и таким образом суще-
ственно изменять их свойства. Борирование металлокерамиче-
ских твердых сплавов на основе карбида вольфрама WC и ко-
бальта, судя по литературным данным, ранее не исследова-
лось; опыты по бериллизации таких сплавов, хотя и проводи-
лись, однако не были освещены в литературе.
436
В исследованиях1, результаты которых приводятся ниже,
опыты проводились не только на стандартном сплаве ВК15,
содержащем 15% Со, но и на специально приготовленных для
этого сплавах с 50 и 70% кобальта (ВК50 и ВК70), металло-
керамическом кобальте и карбиде вольфрама (табл. 81). Это
было необходимо для того, чтобы установить, какова в отдель-
ности роль кобальта и карбида вольфрама в упрочнении твер-
дых сплавов при химико-термической обработке.
Таблица 81
Состав и свойства сплавов
Материал Состав в %* Плотность г! см3 Предел проч- ности при изгибе а Твердость HV5 Средний раз- мер зерна в мк Пористость в %
С (общий) С (несвя- занный) Со Fe о,
в кГ!мм* ХЮ? «/ж2
Карбид WC 6,09 0,03 0,18 15,02 40,5 39,7 1145** 4,8 1
Сплав ВК15 5,П 0,05 14,7 0,07 0,36 14,04 189 185 1145 2,5 0,2
Сплав ВК50 3,17 0,05 49,5 0,20 0,41 10,12 157 154 520 2,2 0,2
Сплав ВК70 1,89 0,07 68,2 0,21 0,44 9,98 — — 375 2,1 —
Кобальт — — 99,66 — 0,34 8,59 — — 230 — —
♦ Остальное вольфрам. ** Размер зерна и пористость карбида WC были больше, чем у сплава ВК15; оче-
видно, поэтому его твердость оказалась такой же, как и твердость сплава ВК15.
Борирование сплавов (образцы размером 5 X 5 X 35 мм)
осуществлялось в расплаве буры с добавкой 30—35% карбида
бора при 1000° С в продолжение 4 ч, а бериллизация — в ваку-
умной печи (разрежение 3-105— 5-Ю5 мм рт. ст., или 0,004—
0,007 н!м2) в порошке бериллия при 1000° С в продолжение 2 ч
(за исключением оговоренных случаев). Изломы образцов об-
рабатывались на наждачном камне, шлифовались на чугунных
дисках сначала карбидом бора, а затем алмазной пылью и
полировались на деревянном круге суспензией с алмазной пы-
лью. Полученные шлифы подвергались травлению в концентри-
рованном Feds (для выявления кобальтовой составляющей) и
в реактиве из 20%-ного щелочного раствора и 20%-ного раство-
ра КзРе(СМ)6, взятых в соотношении 1 : 1 (для выявления кар-
бидной фазы).
До борирования микротвердость и твердость по Виккерсу
поверхности сплавов плавно снижались с увеличением содер-
жания в них кобальта; исключение составляли лишь сплав
ВК15 и WC (табл. 81), имевшие одинаковую твердость.
После борирования поверхностная твердость карбида WC
1 Автор, В. В. Функе, Л. Н. Расторгуев, Т. А. Новикова, В. 3. Забржиц-
кий.
437
изменилась незначительно (в пределах ошибки измерений)
(рис. 342)У остальных сплавов-различие в твердости до и
после борирования увеличивалось с увеличением содержания
в них кобальта. Если для кобальта это различие достигло
750 единиц по Виккерсу и 1230 единиц микротвердости, то для
сплава ВК15 — соответственно 150—350 и 400—500.
Судя по данным рис. 343, микро-
твердость сплава ВК15 не изменя-
ется начиная с расстояния 0,01—
0,02 мм от поверхности и глубже.
Рис. 342. Изменение поверхностной твер-
дости по Виккерсу (нагрузка 5 кГ)
и микротвердости (нагрузка 100 Г) спла-
вов в зависимости от содержания в них
кобальта:
1 — до борирования; 2 — после борирования
верхностных слоев бориро-
ванных сплавов; на оси
ординат отложены значения
микротвердости, замерен-
ной на поверхности
образцов:
1 — WC; 2 — ВК15; 3 —
ВК50; 4 — ВК70; 5 — Со
Диффузионные слои остальных сплавов имеют значительно
большую глубину, поэтому удалось измерить их микротвердость.
Микротвердость, измеренная непосредственно на поверхности
(рис. 343), во всех случаях больше микротвердости, измеренной
на микрошлифе среза металла на глубине 0,01 мм.
Из табл. 82 следует, что в большинстве случаев твердость
сплава ВКД5 повышается после борирования в несколько боль-
шей степени, чем сплава Т15К6; очевидно, это объясняется
меньшим содержанием кобальта в сплаве Т15К6. Судя по дан-
ным измерений микротвердости, глубина борированного слоя на
1 Кривые на рис. 342—344 построены на основании усредненных данных,
полученных путем определения твердости в 6—7 точках каждого образца
438
Таблица 82
Поверхностная твердость ИV 5 образцов сплавов разных партий
до и после борирования
Сплав ВК 15 Сплав TI 5К 6
ДО борирования после борирования увеличение твердости До борирования после борирования увеличение твердости
1197 1346 149 1740 1980 240
1171 1486 315 1648 1918 270
1283 1413 130 1605 1880 275
1197 1449 252 1892 2030 138
1253 1600 347 1740 2080 246
1314 1789 375 1740 1839 99
1279 1548 270 1839 1980 141
1314 1648 334 1892 1980 88
образцах сплавов с различным содержанием кобальта дости-
гала:
Сплав
Глубина слоя в мм . .
ВК15 ВК50 ВК70 Кобальт
0,02 0,06 0,08 0,12
По микроструктуре отчетливый диффузионный слой удалось
выявить только на кобальте.
На рентгенограммах 1 * поверхности борированных образцов
сплавов BKI5, ВК50 и ВК70 обнаружены линии, относящиеся
к трем фазам: С02В, СоВ и WC. На рентгенограммах, снятых,
с WC, линии боридов вольфрама обнаружены не были. Для
того чтобы выяснить возможность замещения в карбиде WC
углерода бором, определили объемы элементарных ячеек WC до
и после борирования как чистого карбида WC, так и его компо-
зиций с кобальтом (табл. 83).
Таблица 83
Объемы элементарных ячеек WC в карбиде и сплавах
до и после борирования
Сплав Объем V элементарной ячейки в кХ3
ДО борирования после борирования
Карбид WC • 20,57 20,59
ВК15 20,59 20,61
ВК50 20,59 20,60
ВК70 20,59 20,57
1 Рентгеноструктурный анализ шлифов выполнен на аппарате УРС-70-1К
с использованием стандартной камеры Дебая при углах съемки 35" и 90°.
При анализе необработанных образцов применялся анод с железным фо-
кусом, а борированных и бериллизованных — с фокусом из хрома.
439
Судя по данным табл. 82, объемы ячеек изменяются на-
столько мало, что трудно предполагать возможность замещения
углерода бором. Во всяком случае, по данным В. Пирсона,
объем цементита при замещении углерода бором должен изме-
няться на 3 кХ3 [513]. Таким образом, структура карбида воль-
фрама после борирования сохраняется неизменной.
Борирование, повышая твердость и хрупкость поверхностно-
го слоя, резко снижает предел прочности сплавов при изгибе.
Например, для сплава
ВК15 до обработки Он =
= 189 кГ/мм2 = 185-107
н)м2\ после борирования с
четырех сторон <Тн = 1144-
ч- 120 кГ1м,м2 = (112 4-
4- 118) • 107 н/л<2, а при на-
личии борированного слоя
с двух сторон Он = 151 4-
4- 162 кГ1мм2 = (148 4-
4- 159) • 107 н/м2 (с двух
противоположных сторон
слой был снят).
После бериллизации
твердость и особенно ми-
кротвердость карбида WC
резко возрастает (рис.
343). На рис. 345 показа-
ны микроструктуры бе-
риллизованных сплавов
ВК15 и кобальта.
Глубина бериллизо-
ванного слоя (по резуль-
татам послойного изме-
рения микротвердости)
Рис. 344. Изменение поверхностной твердо-
сти по Виккерсу (нагрузка 5 кГ) и микро-
твердости (нагрузка 100 Г) сплавов в за-
висимости от содержания в них кобальта:
1 — до бериллизации; 2 — после бериллизации
на образцах различных сплавов достигала:
Сплав......................... WC
Глубина слоя в мм........... 0,06
ВК15 ВК50 ВК70 Кобальт
0,18 0,20 9,17 0,16
Бериллизованный слой отличается от борированного боль-
шой хрупкостью (на образцах всех сплавов). При изготовлении
шлифов слой иногда частично откалывается, а на поверхности
некоторых образцов, в том числе и образцов сплава ВК.15, под
микроскопом при малом увеличении и даже невооруженным
глазом можно было увидеть сетку тонких трещин.
При снижении температуры и продолжительности процесса
хрупкость бериллизованного слоя уменьшается. Так, после бе-
440
риллизации при выдержке 30 мин при температуре 950° С тре-
щины на поверхности образцов не обнаруживаются.
В табл. 84 приведены данные, характеризующие влияние
продолжительности и температуры бериллизации сплава ВК15
на твердость и глубину диффузионного слоя.
Рис. 345. Микроструктура сплава ВК15, бериллизованного при 1000° С в тече-
ние 2 ч в нетравленом состоянии (а) и Со после травления в FeCh (б). Х200
В бериллизованном слое карбида WC рентгеноструктурный
фазовый анализ обнаружил две фазы — Be2W и WC, а в берил-
лизованном слое кобальта одну фазу—Be2iCos. В бериллизо-
Таблица 84
Влияние режимов бериллизации
на твердость и глубину диффузионного слоя сплава ВК15
Режим бериллизации Глубина слоя в мм Твердость HV5 Микротвердость
Д° | после ДО | после
Температура в °C Выдержка в ч бериллизации бериллизации
в кГ/мм* в кГ1ммг в к.Пмм* в кГ/мм*
900 950 1000 2 0,04 0,14 0,18 1165 1086 1120 1388 1550 1595 1506 1506 1506 2300 2300 2450
950 0,5 1 2 0,04 0,08 0,14 1145 1095 1095 1450 1515 1550 1506 1506 1506 2250 2300
441
Фазовый состав поверхности бериллизованных сплавов
(Режим обработки: температура 950® С, выдержка 2 ч)
ЯГ
Кобальт Ве21Со5 (а=7,64 кХ)
Сплав о CQ X о . <v ixj* “ ii Be2W (V= 121,61 кХ3) 1
ВК50 ' Be2W (V= 125,23 жХ») Be21Co5 (a=7,654 кХ)
ВК15 Be2W (lz=125,15 кХ3) Be2iCo3 (a=7,641 «X)
Карбид WC Be2W (V’= 124,97 кХ3) WC (17=20,57 кХ3)
Количество фазы (по ин- тенсивности линий) Много (большая) Мало (меньшая)
ванных слоях сплавов обнару-
живалось по две фазы — Be2W
и Ве21Со5. Результаты фазово-
го рентгеноструктурного ана-
лиза бериллизованных сплавов
приведены в табл. 85. Судя по
объемам V кристаллических
решеток фаза Be2W насыщена
бериллием лишь в образцах
карбида WC и сплавов ВКД5,
ВК50; фаза Be2iCo5 более ста-
бильна в чистом кобальте и
сплаве ВК70.
Исследовано влияние хими-
ко-термической обработки на
кислотостойкость сплава ВК15
в 10 %-ном растворе HNO3 при
длительности испытания 30 ч.
Бериллизация ухудшает стой-
кость сплава в этом растворе
примерно в 2 раза, а борирова-
ние повышает ее примерно
в 3 раза.
Вольфрамокобальтовые спла-
вы, имеющие низкую ока-
линостойкость, после бориро-
вания значительно лучше про-
тивостоят окислению. При
800° С (рис. 346) борирован-
ные образцы хорошо сохраня-
лись в течение около 3 ч. И хо-
тя после этого начиналось ин-
тенсивное окисление образцов,
их вес за 9 ч испытаний увели-
чился меньше, чем образцов
необработанного сплава за
20 мин. Причиной внезапного
снижения окалиностойкости бо-
рированных образцов, вероят-
но, является быстрое избира-
тельное окисление частиц WC
или низкая стойкость диффузи-
онного слоя к тепловому удару
(резкое снижение окалиностой-
кости произошло после шести-
кратного нагрева и охлажде-
ния на воздухе), в результате
442
чего нарушается сплошность слоя. После испытаний в течение
1,5 ч при 900° С вес борированного образца сплава ВК15 увели-
чился в 8 раз меньше (90 г/м2),-чем вес необработанного образ-
Рис. 346. Окалиностойкость сплава ВК15 при 800° С (а) и 900° С (б):
1 — необработанный сплав; 2 — борированный; 3 — бериллизованный в течение 0,5 ч:
4 — бериллизованный в течение 2 ч
Рис. 347. Внешний вид образца
сплава ВК15 после испытания на
окалиностойкость при 900° С
в течение 1 ч
Рис. 348. Окалиностойкость при
1250° С металлокерамического
сплава TiC — NiAl:
1 — необработанный сплав; 2 —
сплав, бериллизованный в течение
30 мин при 1500° С в порошкообраз-
ной смеси [540]
ца в течение 5 мин (760 г/м2). Однако влияние борирования на
повышение окалиностойкости сплава ВКД5 кажется ничтожно
малым по сравнению с влиянием бериллизации. Увеличение ве-
са образцов этого сплава, бериллизованных при температуре
443
950° С и выдержке 2 ч, составило всего 13 г]м2 за 75 ч испытаний
при 900° С. Необработанные же образцы .при такой температуре
увеличиваются в весе на 760 г/м2 за 5 мин испытаний и приобре-
тают крестовидную форму (рис. 347).
Однако практическое использование эффекта такой обра-
ботки для ответственных изделий исключается в связи с очень
большой хрупкостью диффузионного слоя и наличием на его по-
верхности сетки трещин.
Бериллизация резко повышает окалиностойкость многих
сложных по составу металлокерамических сплавов. Например,
испытания при 1250° С продолжительностью 40 ч показали, что
окалиностойкость сплава TiC — NiAl* повышается после берил-
лизации в 20 раз (рис. 348).
Бериллизация сплава производилась в смеси порошков бе-
риллия (50%) и двуокиси циркония (50%), к которым добав-
лялся 1 % хлористого аммония, в тигле из окиси циркония по
режиму: температура 1500° С, выдержка 30 мин.
* В сплаве содержится 18,5% NiAl (60% Ni и 40% Al).
ГЛАВА VII
ХИМИКО-ТЕРМИЧЕСКАЯ ОБРАБОТКА
ГАЛЬВАНИЧЕСКИХ ПОКРЫТИЙ
Химико-термическая обработка гальванических покрытий —
новый метод создания на поверхности стали диффузионных
слоев, обладающих теми или иными свойствами. Число метал-
лов и сплавов, которые можно осаждать на поверхности стали
гальваническим или химическим методом, все увеличивается.
Последующая химико-термическая обработка позволяет превра-
щать многие из осажденных покрытий в твердые, износостой-
кие, кислотостойкие и окалиностойкие соединения: карбиды,
бориды, нитриды, силициды и др. В ряде случаев такая обра-
ботка открывает перспективы для получения диффузионных
слоев с особыми фазовым составом и свойствами, достигающих
глубины, не доступной при обычных способах химико-термиче-
ской обработки.
К настоящему времени наиболее подробно изучена цемента-
ция гальванически хромированной стали. Этот процесс позволя-
ет создавать карбидные слои, глубина которых в несколько раз
больше глубины диффузионных слоев, образующихся при хроми-
ровании средне- и высокоуглеродистых сталей (см, стр. 201,
рис. 126).
Цементацию гальванически хромированной стали можно
осуществлять в твердом карбюризаторе или в газовой среде
[541]—[543].
Первые эксперименты, проведенные в обычном твердом
карбюризаторе (древесный уголь и углекислые соли), показа-
ли, что после обработки структура поверхностных зон
хромированного слоя изменяется мало. Поэтому был предло-
жен карбюризатор специального состава: 50% древесного угля,
20% углекислого натрия и 30% железа [543].
После обработки в твердом или газовом карбюризаторе высо-
коуглеродистой стали в хромированном слое образуются с двух
сторон карбидные зоны: с внешней стороны — за счет диффузии
углерода из карбюризатора; с внутренней — за счет диффузии
углерода из сердцевины (рис. 349). Рентгеноструктурный анализ
445
Рис. 349. Микроструктура гальванически
осажденного хромового покрытия после га-
зовой цементации при 1050° С в течение 4 ч,
Х340 (автор и Г. Гулей)
Рис. 350. Зависимость величины износа гальванически
хромированной нецементованной и цементованной ста-
ли при трении по чугуну и бронзе в зависимости от
скорости скольжения (путь 4500 л, удельное давление
4,5 кГ}см2, или 4,3- 107 н!м2)'.
1 — хром по бронзе; 2 — хром по чугуну; 3 — карбид хрома
по бронзе; 4 — карбид хрома по чугуну {546]
показал, что внешняя зона карбидного слоя представляет собой
карбиды Сг7С3 и глубже Сг2зС6.
После цементации при 950 и 1050° С в продолжение 3 и 8 ч
низкоуглеродистой стали с хромовым покрытием толщиной
45—50 мк в активном газовом карбюризаторе — смеси паров
бензола и водорода — образуется диффузионный слой [542], со-
стоящий, как показывает рентгеноструктурный анализ, из трех
последовательно расположенных карбидных зон: 1) внешней тон-
кой зоны — карбида Сг3С2 с ромбической решеткой; 2) средней
толстой зоны, пронизанной прожилками, идущими от поверхно-
сти и образующими сетку — карбида Сг7Сз с гексагональной
решеткой; 3) слаботравящейся зоны — по-видимому, карбида
т/дм2
Рис. 351. Окалиностойкость при
1000° С на воздухе гальванически
хромированной стали, цементованной
в газовой среде по различным
режимам [542]:
1 — нецементованная; 2 — при 950° С,
3 ч\ 3 — при 950° С, 8 ч; 4 — при 1050° С,
3 ч\ 5 — при 1050° С, 8 ч
Рис. 352. Кислотостойкость в 25%-ных
растворах азотной, серной и соляной
кислот нецементованной и дополни-
тельно цементованной гальванически
хромированной стали [543]:
1,2 — хромированная сталь; 1 — в НС1,
2 — в H2SO<; 3, 4, 5 — хромированная и
дополнительно цементованная сталь; 3 —
в HNO3; 4 — в HCI и 5 — в H2SO<
Сг2зС6 с кубической решеткой. Глубже карбидных зон располо-
жен слой нецементованного крупнозернистого хрома, а еще глуб-
же— промежуточный слой стали, в который продиффундировал
хром.
Цементация существенно повышает физико-химические и
механические свойства гальванически хромированной стали.
Микротвердость поверхности увеличивается с Яр. 950—1100 до
Яр. 1500—1800, красностойкость поверхностного слоя возра-
стает до 950—1050° С.
Испытания износостойкости на машине специальной кон-
струкции, в которой образцы истирались при трении о вкладыш
447
из закаленной стали или антифрикционного чугуна, показали,
что цементация в несколько раз повышает износостойкость
хромированной стали [541]|. Испытания на машине КЕ-2 (испы-
туемые образцы терлись о торец эталонного диска) также по-
казали весьма хорошую износостойкость гальванически хроми-
рованной стали после цементации (рис. 350).
Большой эффект повышения окалиностойкости гальваниче-
ски хромированной стали после цементации иллюстрируется
кривыми, показанными на рис. 351 и 352.
ГЛАВА VIII
ОСАЖДЕНИЕ НА ПОВЕРХНОСТИ МЕТАЛЛОВ
КАРБИДНЫХ, НИТРИДНЫХ, БОРИДНЫХ, СИЛИЦИДНЫХ
И МЕТАЛЛИЧЕСКИХ ПОКРЫТИЙ
Один из новых прогрессивных методов придания деталям
повышенной износостойкости, окалиностойкости и коррозион-
ной стойкости — осаждение на их поверхности карбидных, нит-
ридных, боридных, силицидных и металлических покрытий.
Такие покрытия можно осаждать на стали, различных метал-
лах и сплавах, а также графите и других неметаллических ма-
териалах.
Многообразные свойства указанных выше соединений подроб-
но описаны в литературе [147], [322], [544]. Зарубежные авторы
рекомендуют наносить эти покрытия, например, на лопатки газо-
вых турбин, клапаны, цилиндры двигателей внутреннего сго-
рания, ролики для горячей прокатки, фильеры, матрицы, раз-
личные насадки (инжекторов твердого топлива, дробеструйных
и пескоструйных аппаратов) и многие другие детали, рабо-
тающие в условиях интенсивного окисления, коррозии и износа
(особенно абразивного).
Ниже кратко описаны методы осаждения различных покры-
тий из газовых сред путем восстановления или разложения со-
ответствующих летучих соединений.
Карбидные покрытия. Покрытия такого типа осаждаются
на нагретой поверхности из газовой смеси летучих хлоридов тех
или иных элементов, водорода и углеводорода (или окиси уг-
лерода) [2541 [545]. Прямое осаждение карбидов может быть
также результатом пиролиза карбонильных соединений метал-
лов, однако этот вопрос нами не рассматривается.
Карбид титана можно осаждать, например, из газовой
смеси, полученной путем насыщения водорода сначала толуо-
лом при —15° С, а затем парами четыреххлористого титана при
20° С. Температура осаждения 1300—1700° С [254]. На вольфра-
мовой нити карбид титана осаждали при 1500° С из смеси Н2 и
СН4 (взятых в соотношении 15:1), насыщенной парами тетра-
хлорида титана при 20° С.
449
Для осаждения карбида титана применялась также смесь,
получаемая пропусканием водорода через четыреххлористый
титан при 20° С, к которой добавлялось некоторое количество
метана [546]. С увеличением парциального давления метана в
смеси скорость образования карбида титана сначала возрастает
по линейному закону. Это свидетельствует о том, что при малых
парциальных давлениях метана лимитирующим звеном процес-
са является скорость доставки метана к реакционной поверх-
ности. Последующее незначительное изменение скорости оса-
ждения позволяет предположить, что при высоком парциальном
давлении метана лимитирующим звеном является скорость
выделения титана или реакция его взаимодействия с углеро-
дом.
Наиболее вероятным авторы [546], [547] считают следующий
химизм реакций, протекающих в процессе осаждения карбида
титана. При восстановлении тетрахлорида титана водородом
образуется некоторое количество дихлорида титана:
TiCl4 + -у Н2 = TiCls + НС1; TiCl3 + -у Н2 = TiCl2 + НС1.
Из дихлорида титана по реакции диспропорционирования
выделяется титан:
3TiCl2 = Ti + 2TiCls,
который взаимодействует с углеродом, образующимся вследствие
термического распада метана.
Результаты осаждения карбида титана на сталях типа
ЗХ2В8, Х12 и ШХ15 приведены в работах [548], [549]. Водород
проходит через разогретую трубку с древесным углем, обога-
щаясь углеродом, а затем через сосуд с TiCI4, в результате чего
в печи протекает следующая суммарная реакция:
TiCl4 + 2Н2 + С = TiC + 4НС1.
Микротвердость полученного при этом карбидного покрытия
И?. 3900—4150*, а подкладки карбидного слоя — закаленной
стали И?. 860—1020. Отмечается хорошее скольжение железа
по полированному карбиду титана: коэффициент трения оса-
жденного покрытия 0,08—0,1. Шероховатость поверхности после
осаждения карбида титана увеличивается. Рекомендуется оса-
ждать на деталях слой толщиной 5—20 мк, в зависимости от
условий работы деталей. Процесс опробован на фильерах для
протяжки труб и может быть, по-видимому, использован для
упрочнения матриц, штампов и многих других изделий [5481,
[549], [595], [596].
* Нагрузка при измерении 21 Г.
450
Рис. 353. Слой карбида титана,
осажденный из смеси Нг с пара-
ми TiCl4 и бензола при 1100° С
в течение 30 мин на поверхности
стали Х12; микротвердость слоя
Н{) 3500. Х440 (Г. А. Семенова
и автор)
Г. А. Семенова и автор производили осаждение карбида
титана из смеси тетрахлорида титана, бензола и водорода при
температурах 950—1200° С. В качестве основы применялись
различные стали и металлы. В зависимости от материала ос-
новы, величины и формы образцов и скорости охлаждения по
окончании процесса покрытие или растрескивалось, или полно-
стью сохранялось в виде сплошного плотного слоя.
Покрытие не растрескивалось и не отслаивалось при оса-
ждении на металлах, коэффициент расширения которых близок
к коэффициенту расширения карбида титана (а = 7,74 X
ХЮ-6 1/град). К таким металлам
относятся, например, ниобий
(а = 7,2 4- 7,5-10"6 1/град). На
стали удается получить покры-
тие без трещин только в случае
медленного охлаждения и ма-
лых размеров образцов. Получе-
нию нерастрескивающихся покры-
тий на более массивных образ-
цах способствует предваритель-
ное создание на них промежу-
точных слоев металлов и спла-
вов.
Микротвердость осажденных
покрытий из карбида титана
Нр. 2500—4200, в зависимости от
режима осаждения.
На рис. 353 показана микро-
структура стали с покрытием из
карбида титана. Осажденный
слой после травления в специ-
альных реактивах имеет вид кри-
сталлов, вытянутых в направле-
нии, перпендикулярном поверхности образца. Параметр ре-
шетки поверхностной зоны карбидного слоя, определенный на
нескольких образцах, обработанных по разным режимам, коле-
бался в пределах 'а = 4,30 4- 4,32 /сХ и был близок параметру
карбида титана стехеометрического состава.
Образцы стали У8, покрытые карбидом титана, испытыва-
лись на абразивный износ в Институте машиноведения
АН СССР М. А. Бабичевым [443]. Относительная износостой-
кость покрытия достигала е = 353,1, что в два раза выше изно-
состойкости металлокерамического карбида титана.
На графите карбид титана образуется при 1200—1300° С из
смеси Н2 + TiCl4, науглероживание покрытия заканчивается
в атмосфере, содержащей 38% паров пентана.
45.-1
Карбид кремния осаждали из триметанхлорсилана (СН3)3
SiCl в потоке аргона. Предложена следующая схема этого про-
цесса:
(СН8)з SiCl = ЗСН8 + SiCl; 2SiCl = Si + SiCl2;
CHS = C + 3H; C-bSi = SiC,
t. e. вначале предполагается образование кремния по реакции
диспропорционирования субхлорида, а затем — соединение
кремния и углерода с образованием карбида кремния.
Таблица 86
Режимы получения карбидных покрытий [254]
Карбид Температура осаждения в °C Газовая смесь Соединение Температура испарения соединения в °C
Титана 1000—1700 TiCl4+CeH6CH3+H2 TiCl4+CH4+H2 Т1С14+С+Н2* CeH6CH3 T1C14 — 15 20
Циркония 1700 ZrCl4+CeH6CH3+H2 ZrCl4+CO+H2 C,H8CH8 ZrCl4 —15 300—350
Ниобия 1000—1300 NbCl4+CH4+H2 — —
Тантала — TaCl5+C,H8+H2 TaCl6 250
Молибдена 1100—1600 300—800 300—800 MoCl6+CH4+H2 MoCl5+CO+H2 Mo(CO)e+H2 Mo(CO)e+CeHe — —
Вольфрама 1100-1600 300—800 WCle+CH4+H2 WC1,+C8H12+H2 WCle+C,He+(3N2+lH2) W(COe)+H2 WCle W(CO)« 250 30
Ванадия 1500 VC14+C7H8+H2 C7H8; VC14 —15; 50
Кремния 1300 SiCl4+C7H8+H2 C7H8; SiCl, —15; 20
Карбид бора на графите 1800 BC13+H2 (1:1) — —
Примечание. При использовании карбонилов давление находилось в преде- лах 0,1—10 мм рт. ст. (13,3—1330 н!м*), а в остальных случаях было близко к 1 ат (98 кн!м*).
♦ Водород пропускался под раскаленным углем.
452
Карбиды циркония осаждались на вольфрамовых нитях
или из смеси, полученной путем насыщения водорода парами
толуола при —15° С и тетрахлорида циркония при 300—350° С,
или из смеси Н2 + ZrCU + СО.
В табл. 86 приведены некоторые режимы получения карбид-
ных покрытий осаждением из газовой фазы и разложением
карбонильных соединений металлов.
Карбидные покрытия могут быть также получены: 1) це-
ментацией соответствующих металлов или металлических по-
крытий, осажденных на ту или иную основу (см. гл. VII);
2) разложением паров галоидных соединений металлов на гра-
фите; 3) поверхностным насыщением высокоуглеродистой стали
карбидообразующим элементом; углерод диффундирует из
сердцевины к поверхности стали, образуя с насыщающим эле-
ментом карбидное соединение.
Нитридные покрытия. Такие покрытия осаждаются на на-
гретой поверхности из смеси летучих хлоридов тех или иных
элементов и азота или азота и водорода. Примесь Н2 в несу-
щем газе полезна, так как позволяет проводить процесс при
более низкой температуре, чем в случае использования
только азота. Покрытия максимальной плотности наиболее ча-
сто- образуются при пониженном давлении (1—30 мм рт. ст.,
или 133—3999 н/м2).
В табл. 87 указаны режимы осаждения нитридных слоев
и необходимые для осуществления этого процесса газо-
вые среды.
Таблица 87
Режимы осаждения нитридных покрытий из газовых сред [254]
Нитрид Температура осаждения в °C Хлорид Температура испаре- ния хлорида в °C Несущий газ
BN* 1200—1500 ВС13 От —30 ДО 0 3N2+1H2
TiN 1100—1700 TiCl4 20 3N2+ih2
ZrN 2000—2500 ZrCl4 300—350 3N2+1H2
ZrN 2500—2700 ZrCl4 300—350 N2
HfN 2500—2700 HfCl4 300—350
VN 1100—1300 VC14 20 n2
TaN 2100—2300 TaCl4 250—350 n2+h2
Be8N2 1200—2000 BeCl2 280—340 n2+h2
AIN 1200—1600 A1C13 100-130 n2+h2
Si3N2** 1000—1600 SiCl4 От —40 до 0 3N2+H2
♦ Покрытие состоит из двух фаз: BN-J-B.
♦♦ Покрытие, очевидно, состоит из двух фаз:
Получение покрытий из сложных карбидов, смесей карби-
дов или смесей карбидов и нитридов весьма затруднительно.
453
Смесь карбидов или сложные карбиды могут быть получены
лишь в тех случаях, когда реакции осаждения одновременно
разных карбидов возможны с точки зрения термодинамики и
кинетики процесса. Например, в смеси ZrCl4 + TiCl4 +
+ СбН5СНз + Н2 образуется только карбид ZrC, а в смеси
TaCU + ZrCl4 + С6Н5СН3 + Н2 осаждается только свободный
тантал. Это полностью относится и к случаям совместного оса-
ждения карбидов и нитридов. Например, в смеси TiCl4 + N2 +
+ СбН5СНз + Н2 образуется только TiN; в смеси ZrCl4 + N2 +
+ С6Н5СНз образуется карбид, а при добавлении к ней водо-
рода — нитрид.
Боридные покрытия. Осаждение боридных покрытий наибо-
лее легко осуществляется путем восстановления водородом ле-
тучих хлоридов соответствующего металла и хлоридов или бро-'
мидов бора. В некоторых работах отмечается, что предпочти-
тельнее применять хлориды бора.
В табл. 88 указаны основные режимы осаждения боридных
слоев некоторых металлов и приведены необходимые для этого
газовые среды.
Таблица 88
Режимы осаждения боридных покрытий из газовых сред1 [254]
Борид Температура осаждения в °C Хлорид Температура испарения хлорида в °C Соединение бора Температура испарения соединения бора в °C
Алюминия 1000 А1С13 155 ВС13 —22
Кремния 1000—1300 • SiCl4 0 ВС13 —22
Титана 1100-1300 TiCl4 20 ВВг3 20
» 1100—1300 TiCl4 50 ВС13 —22
Циркония 1700—2500 ZrCl4 300—350 ВВг3 20
Гафния 1900-2700 НГС14 300-350 ВВг3 20
Ванадия 900-1300 VC14 20 ВВг3 20
» 900—1300 VC14 75 ВС13 —22
Тантала 1300—1700 TaCl4 170-190 ВВгз 20
Вольфрама 1300—1700 WC16 250—350 ВВг3 20
» 800—1200 WC1„ 235 ВС13 —22
1 Процессы проводятся в потоке водорода.
454
Силицидные покрытия. Силицидные покрытия осаждаются
из газовой среды, состоящей из водорода, хлорида металла и
галогенида кремния (обычно SiCl4).
Таким методом получены покрытия из силицидов титана,
циркония, ниобия, тантала, хрома, молибдена и вольфрама.
Значительное внимание было уделено осаждению силицидов
молибдена, обладающих исключительно высокой окалиностой-
костью. Температура осаждения силицидов колеблется в пре-
делах 1000—1800° С.
Силициды можно также получать электролизом расплава.
Например, в расплаве фторосиликата калия или натрия и
TiO2, ZrO2 или фторида хрома образуются силициды титана, цир-
кония или хрома, а в расплаве смесей силикатов с окислами
тугоплавких и щелочных металлов — дисилициды тантала, цир-
кония и хрома.
Металлические покрытия [254], (582], [583], [597]. Осаждение
на поверхности металла слоя другого металла позволяет прида-
вать деталям удачные сочетания различных полезных свойств и
дает немалый экономический эффект благодаря возможности
осаждать дефицитные металлы на стальную или иную основу
вместо того, чтобы изготовлять детали целиком из этих метал-
лов. Покрытия такого типа образуются в тех случаях, когда
скорость осаждения металла велика, а скорость диффузии ме-
талла основы в осажденный слой и осажденного металла в
основу мала.
Одна из установок, используемая как для обычной химико-
термической обработки с целью получения диффузионных слоев,
так и для процессов осаждения, показана на рис. 354. В этой
установке обрабатываемая деталь находится под колпаком
внутри металлического экрана, нагреваемого т. в. ч. Водород
захватывает пары нагретого галогенного соединения, и смесь
газов направляется к обрабатываемой детали.
Рассмотрим в качестве примера процесс осаждения на по-
верхность металлов слоев тантала и рения. Для осаждения
тантала чаще используется газовая смесь Н2 + TaCls и реже
Н2 + ТаВг5 или Н2 + TaJ5 [537]—[540], [254], [598]. Очищенный
водород пропускается над порошком или брикетами пентахлори-
да тантала (TaCls), нагретыми до температуры, достаточной для
получения необходимого парциального давления паров хлори-
да. Осаждение тантала обычно протекает в две стадии: 1) об-
разование тонкого слоя (около 0,0001 мм) в результате реакции
замещения между покрываемым металлом и парами галоге-
нида тантала; 2) образование основного слоя за счет восста-
новления пентахлорида тантала водородом. Осаждение тантала
на сталь производится при температурах 800—1200° С, скорость
осаждения около 0,0008—0,06 мм]мин, осаждение на медь, ни-
кель и молибден — соответственно при температурах 800—
455
1000, 1400 и 1500° С. Повышение температуры и уменьшение
давления приводят к образованию более крупнозернистых по-
крытий. Чем больше в смеси концентрация хлоридов, тем бы-
стрее протекает процесс. С увеличением скорости газового по-
тока увеличивается гладкость и однородность осажденного слоя.
Рис. 354. Схема установки с нагревом т. в. ч. для осаждения и газового
поверхностного насыщения [254]:
1 — внутренний нагреватель; 2 — сосуд для конденсирования паров; 3 — ловушка
с минеральным маслом; 4 — колокол; 5 — деталь; 6 — индуктор; 7 — излучающий
экран; 8 — держатель; 9 — основание колокола; 10 — резиновые прокладки; 11 — ас-
бестовая прокладка; 12 — электросопротивление; 13 — термопара; 14 — галоид металла;.
15 — внутренний нагреватель; 16 — трубки с водой
Для предохранения металлов от коррозии в агрессивных средах
необходимы плотные покрытия толщиной не менее 0,01—0,06 мм,.
Уменьшение пористости покрытий достигается поочередным на-
несением двух-трех и более тонких слоев, каждый из которых
перед нанесением следующего слоя полируется с целью закры-
тия пор. Таким способом были получены покрытия, предохра-
нявшие в течение 2—3 месяцев стальные и медные детали от
456
воздействия азотной и соляной кислот. Поверхностная твердость
осажденного покрытия HV 180—220 (твердость отожженного
тантала HV 60—80). Осажденное покрытие хорошо связано с
сердцевиной и пластично. Например, покрытие толщиной 0,04—
0,06 мм на стальной проволоке диаметром 3 мм после несколь-
ких сгибаний под углом 30° и выпрямлений проволоки не рас-
трескивается.
Слой, осажденный в среде TaClg + Н2, состоит почти из
чистого тантала, насыщенного водородом, в результате чего
параметры его кристаллической решетки больше, чем у чисто-
го тантала. Тантал обладает очень высокой коррозионной стой-
костью, а также достаточной прочностью, пластичностью и
теплопроводностью. Покрытые им детали служат продолжитель-
ное время во многих химически агрессивных средах. В литера-
туре указывается, что процесс восстановления тантала водоро-
дом успешно опробован для покрытия труб, сопел ракет, тиг-
лей, листов, прутков, проволоки и т. д., изготовленных из стали,
никеля, меди, молибдена, и других металлов и сплавов.
Покрытия из рения можно осаждать на деталях различными
способами, в том числе термической диссоциацией летучих сое-
динений этого элемента — карбонильных [Re(CO)sll2 и галоген-
ных (ReCU, ReOCl4). В случае использования карбонилов ре-
ния покрытия осаждаются при температуре 500—700° С
в вакууме в присутствии водорода; полученные покрытия под-
вергаются диффузионному отжигу [551J, [552].
При осаждении покрытия способом термической диссоциа-
ции галогенида ReCis (пентахлорида рения) тщательно очищен-
ный аргон переносит пары ReCl5 из испарителя установки в ра-
бочую камеру, где находятся нагретые детали. Для получения
качественных покрытий на молибдене, вольфраме и других
металлах рекомендованы следующие оптимальные параметры
процесса: температура покрываемого металла 1200—1400° С,
концентрация ReCl5 в газовой среде порядка 0,05 г/л, что со-
ответствует температуре испарения хлорида 200° С и скорости
потока аргона 10 л/ч для данной установки; осаждение рения
происходит при этом со скоростью 0,14 г/ч [553]. Недостаток
этого способа — малая скорость осаждения рения.
Осаждение покрытия термической диссоциацией моноокси-
тетрахлорида ReOCl4 предложено авторами работы [554]. В ка-
меру с нагретыми до 1200—1500° С деталями вводится смесь
тщательно очищенного аргона с парами ReOCl4, получаемая пу-
тем пропускания аргона над нагретым до 150—228° С ReOCl4
(температура кипения ReOCl4 228°С). Пары ReOCl4 переносятся
аргоном в рабочую камеру; на поверхности детали происходит
диссоциация ReOCl4 по реакции
3ReOCl4 = 2Re + ReO3 + 6С12.
457
На поверхности площадью 15 см2 (молибденовая лента) ско-
рость осаждения равна 150—200 мг/ч или 10—15 мк/ч при коли-
честве испаренного ReOCl4 1 г (при 1200° С). С увеличением
количества испаренного рения толщина слоя возрастает соглас-
но указанной выше зависимости.
Снаружи на металле осаждается чистый рений, между ним
и сердцевиной постепенно образуется промежуточный диффу-
зионный слой, глубина которого зависит от металла основы,
а также от температуры и продолжительности процесса. После
осаждения рения на молибден при 1500° С в продолжение 1 ч
глубина диффузионного слоя всего 1 —15,5 мк при общей тол-
щине покрытия 10 мк.
Дефицитность рения и его соединений ограничивает приме-
нение процесса ренирования. Однако в небольших масштабах
этот процесс уже используется для обработки деталей электро-
вакуумных приборов, вольфрамовой проволоки и ряда других
деталей с целью удлинения срока их службы и улучшения элек-
трических характеристик. Преимуществами рения, например, пе-
ред вольфрамом являются большая прочность с сохранением
пластичности в рекристаллизованном состоянии, более высокое
электросопротивление, меньшая склонность к испарению в при-
сутствии следов влаги и к взаимодействию при высокой темпе-
ратуре с окисью алюминия.
ГЛАВА IX
ХИМИКО-ТЕРМИЧЕСКАЯ ОБРАБОТКА
ПОЛУПРОВОДНИКОВ
Основной частью полупроводниковых приборов является
монокристаллический полупроводниковый материал (кремний,
германий), легированный с поверхности специальными приме-
сями в контролируемых количествах. Одним из основных мето-
дов введения примесей в поверхностные слои полупроводников
является их диффузионное насыщение из газообразной, жидкой
или твердой фазы. Примеси, вводимые в поверхностные слои
полупроводника, подразделяются на акцепторные и донорные
[555].
Атомы акцепторной примеси создают в полупроводнике ак-
цепторные уровни, способные принимать валентные электроны,
что приводит к появлению в полупроводнике дырочной проводи-
мости. У таких полупроводников (дырочных полупроводников,
полупроводников p-типа) основными носителями заряда явля-
ются дырки \
Атомы донорной примеси обладают избыточными электрона-
ми, способными переходить в зону проводимости, что приводит
к появлению электронной проводимости в полупроводнике (элек-
тронные полупроводники или полупроводники п-типа). Зона
диффузионного слоя, на которой концентрация продиффундиро-
вавших атомов примеси (например, акцепторов) равняется
концентрации атомов, легирующих исходный полупроводник
(в данном случае доноров), является электронно-дырочным
п-р-переходом полупроводника, т. е. переходом от материала
п-типа к материалу p-типа. Здесь на границе двух полупровод-
ников разного типа проводимости происходит выпрямление тока.
В одних случаях требуется п — р-переход (диоды, солнечные
батареи), в других р — п — р-переход или п — р — п-переход
(транзисторы, кремниевые триоды). Последние переходы полу-
1 Под дыркой понимается дефектная связь в решетке (возникновение по-
ложительного заряда), вызванная уходом электрона и появлением «вакант-
ного» места.
459
чают, в частности, насыщением полупроводника совместно двумя
примесями (акцептором и донором), из которых одна диффунди-
рует глубже другой. Акцепторы (бор, алюминий, галлий, индий),
относящиеся к III группе периодической системы Д. И. Менделе-
ева, диффундируют в кремний быстрее, чем доноры (мышьяк,
сурьма, висмут) *, относящиеся к V группе элементов (рис. 355).
На рис. 356 показано распределение донорной и акцепторной
примесей в поверхностных опоях кремния при создании двойно-
го «перехода. У поверхно-
сти кремниевой пластин-
ки концентрация донора
значительно превышает
концентрацию акцепто-
ров, и образуется тонкий
слой с электронной про-
водимостью 1 2. Глубже
преобладает концентра-
ция акцептора, и.тип про-
водимости изменяется.
При еще большем удале-
нии от поверхности кон-
центрация акцепторной
примеси, введенной диф-
фузией, будет мала по
сравнению с донорной
примесью, содержащейся
в обрабатываемом крем-
нии, и знак проводимости
снова изменится.
Ряд элементов (цинк,
медь, литий, марганец,
железо), вводимых в «по-
лупроводники, относится
1400 1300 1200 1100 1000 900°С
Рис. 355. Зависимость коэффициента диф- к быстро диффундирую-
фузии в кремний элементов III и V групп щим элементам, коэффи-
от температуры [556] циенты диффузии кото-
рых на несколько поряд-
ков больше, чем у элементов III и V групп. Предполагается, что
эти примеси перемещаются по междуузлиям в отличие от при-
месей III и V групп, диффундирующих по вакантным узлам
решетки.
Химико-термическая обработка полупроводниковых материа-
лов, имея много общего с химико-термической обработкой ме-
1 К донорным примесям относится также фосфор, который, однако, диф-
фундирует быстро (приблизительно с той же скоростью, что и бор).
2 При диффузии в германий имеет место обратное соотношение приме-
сей в слое.
460
таллов и сплавов, одновременно отличается многими специфич-
ними особенностями:
1) требуются диффузионные слои очень небольшой глубины
(0,1—10 мк)]
2) концентрация диффундирующего элемента (примеси) в
этих слоях должна быть незначительной (1014—1021 ат/см3)]
3) в связи с этим применяются особые методы контроля глу*
бины слоя и концентрации в них
примесей;
4) особые требования предъяв-
ляются к чистоте используемой ап-
паратуры и исходных материалов.
Рассмотрим основные способы
создания диффузионных слоев в
кремнии [556]. Пластины кремния
можно насыщать рядом элементов
в замкнутом пространстве. Пласти-
ны вместе с насыщающими элемен-
тами (примесями) помещаются в
кварцевую ампулу, в которой соз-
дается разрежение 10“4—10“5 мм
рт. ст. (0,13—0,013 н/м2). Затем ам-
пула заваривается или наполняет-
Расстояние от поверхности
Рис. 356. Кривые распределе-
ния (а) и разности концен-
траций (б) донорной и акцеп-
торной примесей в поверхно-
стном слое кремния:
D\ — концентрация донорной при-
меси в кремнии; D2 — концентра-
ция донорной примеси в диффузи-
онном слое; А — концентрация
акцепторной примеси в диффузи-
онном слое [555|
ся чистым инертным газом.
При введении некоторых при-
месей элементы помещаются в спе-
циальный отросток, температура
которого регулируется независимо
от температуры нагрева кремния.
Например, при насыщении кремния
фосфором в такой отросток поме-
щается желтый фосфор, изменение
температуры которого в интервале
от —45° до +34° С позволяет в значительных пределах изменять
поверхностную концентрацию фосфора в кремнии 3-1015 —
5-1018 ar/см3), нагреваемом в процессе насыщения до 1200°С.
Легирование поверхности кремния примесями можно осуще-
ствлять также и при непрерывной откачке; при этом наблюдает-
ся некоторое испарение кремния, протекающее с постоянной ско-
ростью.
При легировании кремния в вакууме некоторыми элементами
(алюминием, галлием и др.) поверхность пластин покрывается
многочисленными углублениями, хорошо различимыми невоору-
женным глазом. Причиной эрозии является образование на по-
верхности кремния участков расплава кремний — примесь. Од-
нако, если в качестве источника примеси используются не эле-
менты, а их окислы (Р2О5, В2О3, ЗЬ20з, As2O3 и т. д.), то эрозия
15 А. Н. Минкевич
461
резко уменьшается или совсем устраняется. Предполагают, что
окислы реагируют с кремнием по схеме
2/?О + Si = 2R + SiO2,
где RO — окисел примеси.
Атомы примеси, восстановленные из окислов, диффундируют
в кремний, а пленка SiO2 предохраняет его от эрозии. Слой окис-
ла является при этом постоянным поверхностным источником
примеси.
Легирование кремния может производиться в контейнерах с
конечной величиной утечки (кварцевый контейнер с пришлифо-
ванной пробкой). В качестве источников примеси используются
окислы элементов, растворенные в SiO2. Например, при насыще-
нии бором или фосфором источником примеси служит SiO2 +
+ В2О3 или SiO2 + Р2О5. Утечка приводит к постепенному обед-
нению источника и уменьшению давления паров смеси. Однако
величина обеднения может быть малой.
Кремний можно также легировать в трубчатых печах в пото-
ке инертного или окисляющего газа, несущего пары примеси.
В первой зоне печи образуются пары примеси, во второй поме-
щены пластинки кремния. При необходимости подаваемые
в печь газы можно увлажнять, пропуская через сосуд с во-
дой, нагретой до 30—80° С. В качестве несущего газа можно
также употреблять смесь азота с кислородом или углекислый
газ.
Для создания триодных структур применяются трубчатые пе-
чи с тремя температурными зонами. В качестве источников при-
меси при этом используются как чистые элементы, так и их сое-
динения.
Для управления процессом легирования часто используется
зависимость скорости испарения легирующего элемента и его
соединений от состава парогазовой смеси. Например, скорость
испарения Ga2O3 максимальна во влажном водороде и ничтожно
мала в азоте, гелии, аргоне (чистых или содержащих пары воды
либо кислорода). Скорость испарения чистого галлия больше во
влажном водороде и меньше в сухом, а сурьмы и Sb2O4 значи-
тельна в азоте, водороде, гелии и существенно не зависит от со-
держания в них влаги.
Зависимость кинетики процесса поверхностного легирования
кремния от состава парогазовой смеси позволяет использовать
для изменения глубины и состава слоев сочетание различных га-
зовых сред. Так, диффузия галлия при 1350° С в кремний п-типа
из Ga2Os, нагретого до температуры 950° С, проводится сначала
во влажном водороде (температура воды 80° С), а затем во
влажном азоте (температура воды 30° С). Это позволяет, изме-
няя соотношения продолжительности обработки в этих средах,
462
регулировать поверхностную концентрацию и глубину проник-
новения примеси.
Одновременное легирование кремния n-типа двумя элемен-
тами, например галлием и мышьяком, можно проводить вначале
во влажном азоте, а затем во влажном водороде (температура
воды в обоих случаях 30° С, температура СагОз и AS2O3 соответ-
ственно 900 и 220°С, температура насыщаемого кремния 1200—
1350° С). Изменением соотношения продолжительности выдер-
жек в каждой из этих сред удается получать структуры с двумя
диффузионными слоями определенной глубины с заданной кон-
центрацией примесей.
При легировании кремния бором и фосфором в непрерывном
газовом потоке содержание кислорода в смеси азот — кислород
обычно колеблется в пределах 1—2%. После легирования в этой
среде кремния бором при 1300° С в продолжение 60 мин (источ-
ник примеси содержал 0,01% В2О3 и 0,0015% В) глубина полу-
ченного перехода достигала 8 мк, а поверхностная концентрация
1,5-1018 ат!см\ после легирования фосфором при таких же тем-
пературах и выдержке — соответственно 9 мк и 240 • 1018 ат.1см3
(источник примеси содержал 11,5% Р2О5 и 5% Р).
Разработан двухстадийный способ введения примесей в крем-
ний: вначале при сравнительно низкой температуре в кварцевой
ампуле (в вакууме или в среде неокисляющих газов — аргона, во-
дорода, азота) создается тонкий диффузионный слой, а затем
(после очистки поверхности травлением или другим способом)
при высокой температуре в окислительной среде производится
вторая падия обработки. Образующаяся при этом окисная плен-
ка препятствует испарению примесей с поверхности. Таким спо-
собом удобно образовывать на кремнии диффузионные слои с
пониженной поверхностной концентрацией примеси. Примером
двухстадийной обработки может служить следующий процесс:
1) предварительное насыщение в сухой атмосфере азота в те-
чение 30 мин (источник примеси AS2O3, нагретый до 235° С);
2) диффузионный отжиг во влажной атмосфере азота при
1300° С в течение 30 мин.
Дырочный германий можно легировать сурьмой (например,
в ампуле) при 800° С в течение 30 мин\ при этом глубина слоя
(толщина перехода) достигает 3 мк (коэффициент диффузии
5‘10“п см21сек) при поверхностной концентрации сурьмы при-
мерно 1018—1019 ат!см2.
Применяются способы, при которых в кремний вплавляют
материал, легированный примесью, создающей тип проводимо-
сти, противоположный основной проводимости. При наличии в
электродном материале (вплавляемом) и донорных, и акцептор-
ных примесей могут быть созданы триодные структуры, но толь-
ко п — р — п-типа, так как акцепторы диффундируют в кремний
быстрее доноров.
463
Неравномерного распределения примесей в полупроводнике
можно добиться путем испарения примеси из образца («обратная
диффузия»), нагреваемого в вакууме порядка 10~5 мм рт. ст.
(0,00133 н1м2). Если полупроводник легирован двумя примесями,
создающими противоположные типы проводимости, то вслед-
ствие различных коэффициентов диффузии этих примесей способ
«обратной диффузии» позволяет создать различные электронно-
дырочные переходы вблизи поверхности полупроводника.
Для создания переходов в полупроводниках применяется
также способ эпитаксиальных слоев 1 [585], [586], заключающий-
ся в наращивании на полупроводниковой монокристаллической
основе одноименного (Si на Si) или разноименного (Ge на Si)
полупроводника с кристаллической решеткой, ориентированной
строго определенным образом относительно кристаллической ре-
шетки основы [557]. Процесс ориентированного нарастания мож-
но производить из паровой фазы в вакууме, или из газовой фазы
(SiCU + Н2) при восстановлении галогенидов полупроводников
водородом. Введение различных контролируемых примесей в вы-
ращиваемый эпитаксиальный слой и возможность строго сле-
дить за их концентрацией позволяют создавать области полупро-
водника с наперед заданными свойствами. Так, например, водо-
род захватывает пары SiCU и в трубчатой печи при температуре
1150—1250° С восстанавливает SiCU до кремния, осаждающего-
ся на кремниевую подложку. Добавляя к этой газовой смеси не-
большие количества паров ВВг3 или AsCI3, удается вводить в
осаждаемый кремний то или иное количество бора или мышья-
ка. Таким образом осуществляется контролируемое легирова-
ние эпитаксиального слоя. В настоящее время ведутся работы
по легированию осаждающегося кремния сурьмой, фосфором,
алюминием и рядом других элементов.
Аналогичным образом можно осаждать на германии эпитак-
сиальные слои германия, легированного бором, мышьяком и
другими элементами, используя в качестве основной газовой сме-
си Н2 + GeCU и в качестве подложки германий.
1 Эпитаксия — ориентированное нарастание.
ЛИТЕРАТУРА
1. Прокошин Д. А. Диффузия элементов в твердое тело. Сб. трудов
Московского института стали. Редакция литературы по черной металлургии.
М.—Л., 1938.
2. Бугаков В. С. Диффузия в металлах и сплавах. М., Гостехтеорет-
издат, 1949.
3. Уманский Я. С., Финкельштейн Б. Н., Блантер М. Е. и др.
Физическое металловедение (глава «Диффузия в металлах»). М., Металлург-
издат, 1949 и 1955.
4. 3 а м я т н и н М. М. Кинетика процессов химико-термической обработ-
ки стали. М., Металлургиздат, 1951.
5. Л е - К л е р А. Д. Диффузия в металлах. «Успехи физических наук»,
1956, т. 1.
6. 3 а й т В. Диффузия в металлах. М., И.Л. 1958.
7. Герцрикен С. Д. и Дехтяр И. Я. Диффузия в металлах и спла-
вах в твердой фазе. М., Физматгиз, 1960.
8. Жуховицкий А. А. Теория диффузии. Справочник «Металловеде-
ние и термическая обработка», Т. 1. М., Металлургиздат, 1961, стр. 582.
9. П о п о в А. А. Теоретические основы химико-термической обработки.
Свердловск, Металлургиздат, 1962.
10. К р и ш т а л М. А. Диффузионные процессы в железных сплавах. М.,
Металлургиздат, 1963.
11. Архаров В. И. Окисление металлов при высоких температурах.
Свердловск, Металлургиздат, 1945.
12. Просвирин В. И. К вопросу об ионной диффузии в металлах.—
«Вестник металлопромышленности», 1937, № 12.
13. Францевич Н. Н. и Калинович Д. Ф. Явление электропере-
носа в твердых металлических растворах. Вопросы порошковой металлургии
и прочности металлов, 1956, вып. 3.
14. Борисов В. Т., Голиков В. М. и Щ е б е р д и н с к и й Г. В. Об
определении коэффициента диффузии в поликристалле по концентрационным
кривым. Физические методы исследования. — «Заводская лаборатория»,
1959, № 9.
15. Борисов В. Т„ Голиков В. М. и Дубинин Г. Н. Определе-
ние коэффициента диффузии в сплавах при наличии нескольких фаз. Изве-
стия АН СССР, сер. «Металлургия и горное дело», 1964, № 4.
16. Б ы х о в с к и й А. И. Об одной особенности кинетики диффузион-
ных процессов. «Физика металлов и металловедение», 1963, вып. 1, т. 15.
17. Рудковский М. Л., Ануфриева Н. А., Копьева О, М. и
др. Кинетика газового борирования никеля. «Физика металлов . и металлове-
дение», 1961, т. 12, вып. 2.
• 18. С м и р н о в А. В. Горячее цинкование. М., Металлургиздат, 1953.
19. Минкевич А. Н. Химико-термическая обработка стали. М., Маш-
гиз, 1950.
20. Афонский И. Ф. Сборник статей по вопросу теории химико-тер-
мической обработки изделий. Сибирский лесотехнический институт. Красно-
.ярск, 1957.
465
21. Б ок штейн С. 3.» Жуховицкий А. А. Методы радиоактивных
изотопов (глава в справочнике «Металловедение и термическая обработка
металлов»), т. 1, Металлургиздат, 1961.
22. Борисов В. Т., Голиков В. М., Любов В. Я. и др. Абсорбци-
онный метод изучения диффузии по границам и объему зерен металла. Сб.
«Металлургия и металловедение», Труды Всесоюзной конференции по при-
менению радиоактивных изотопов. АН СССР, 1957.
23. Лахтин Ю. М. Физические основы процесса азотирования. М.,
Машгиз, 1948.
24. Hanke Е., S е h г е с к К. «Wiessenschaftliche Zeitschrift Hochschule
Electrochem». Hmenau, 1957, № 3—4.
25. Hanke E., Schreck K. «Harterei — Techn. Meteilungen», 1963, Bd.
18, № 3, S. 143—149.
26. Kopietz K. «Harterei Techn. u Warmebehandlung», 1958, №6.
27. Кидин И. H. Гипотеза о повышенной активности процесса насыще-
ния аустенита в зародышном состоянии, «Известия высших учебных заведе-
ний», сер. «Черная металлургия», № 3, 1964.
28. Гуляев А. П. Термическая обработка стали. М. Машгиз, 1960.
29. Грибоедов Ю. Н. Закономерности структурообразования хроми-
рованного слоя железохромоникелевых сплавов. — «Металловедение и тер-
мическая обработка металлов», 1964, № 3.
30. Cornelius Н., Bollenratch F. «Arch f. d. Eisenhutenwessen»,
1941, Sept., Kg 3, S. 145.
31. Пчелкина M. А. и Лахтин Ю. M. Газовое борирование. «Науч-
ные доклады Высшей школы», сер. «Металлургия», 1960, № 6.
32. Струг Е. М. и Панченко Е. В. Металлографическое исследова-
ние сплавов методом микро-т. э. д. с.— «Научные доклады Высшей школы»,
сер. «Металлургия», 1959, № 2.
33. S t a n 1 е у I. К. «J. of Metals», 1949, № 10, р. 752.
34. Barducci I. «Nrava Cinonfo», 1955, v. 1, № 2, p. 122.
35. Грузин П. Л., К о с т о н о г о в В. Г., Платонов Н. А. О при-
менении искусственного радиоактивного изотопа С14 для изучения диффузии
углерода в стали. Проблемы металловедения и физики металлов, 1955, вып. 4,
Металлургиздат.
36. Грузин П. Л., Бабикова 10. Ф., Борисов Е. В. и др. Пробле-
мы металловедения и физики металлов. Вып. 5, Металлургиздат, 1958, ст.р. 327
37. Wells С., Mehl R., «Metal Technology AIME, Techn. Public», 1940,
Aug. p. 1180.
38. Бланте p M. E. Диффузия углерода в аустените.— ЖЭТФ, 1947,
№ 17, с. 1331.
39. Thomas W., Lean G. М. «Nature», 1955, v. 176, № 4470, p. 29.
40. Гаев И. С. Физико-химические основы влияния легирующих эле-
ментов на структуру и некоторые свойства стали. Труды ВНИТОМ, т. VI,
М„ Металлургиздат, 1955.
41. Тарасов А. М. и Семенченко М. Р. Интенсификация процесса
цементации в твердом карбюризаторе для автомобильных деталей. — «Ме-
талловедение и обработка металлов», 1955, № 6.
42. Мороз Л. С. и Шураков С. С. Проблемы прочности цементо-
ванной стали. Л. Изд. ЦНИИ, 1947.
43. Котов О. К. Поверхностное упрочнение углеродистых сталей газо-
вой цементацией. — «Металловедение и обработка металлов», 1957, № 10.
44. Шубин Р. П. Внедрение прогрессивных технологических процессов
и оборудования в термическом цехе. — «Металловедение и термическая об-
работка металлов», 1963, № 6.
45. Т а р а с о в А. М. Опыт применения бористой цементуемой стали
20ХГР для автомобильных деталей. — «Сталь», 1961, № 1.
46.. А л е к с е е н к о М. Ф.. и О р е х о в Г. Н. Бористая сталь
15Х2ГН2ТРА — заменитель сталей I12XH4A и 18Х2Н4ВА.— «Сталь». I960, №6.
466
47. Нехендзи Ю. А., Вязников Н. В., Ермаков С. С. Новые
составы цементуемой стали. Научные доклады высшей школы, сер. «Метал-
лургия», 1958, № 4.
48. Алексеенко М. Ф., В а ч и л е н к о Б. С., Натапов Б. С. и др.
Цементуемые и улучшаемые стали.— «Сталь», 1964, № 7.
49. Пятакова Л. Л., Исхаков С. С., Шитов А. П., Мирош-
никова К. Е. О механизме влияния некоторых элементов на свойства це-
ментуемой стали. Сб. «Новое в металловедении и технологии термической
обработки стали». Челябинск, 1962.
50. Вишняков Д. Я., С о в а л о в а А. А. Цементация нержавеющей
стали. Научные доклады высшей школы, сер. «Металлургия», 1958, № 1.
51. Сумароков Н. В. ;и Таек а ев И. П. Цементация хромистых
нержавеющих сталей в твердом карбюризаторе. Ученые записки Пермского
госуниверситета, 1960, т. XVI, вып. 4.
52. Вишняков Д. Я., С о в а л о в а А. А. и Ч у д а р е в а Л. П. Цемен-
тация нержавеющей стали. Труды Московского авиационного технологичес-
кого института, вып. 50, Оборонгиз, М., 1961.
53. Вязников Н. В., Ермаков С. С., Солдатов Н. Н. Цемента-
ция хромистой нержавеющей стали. — «Металловедение и термическая обра-
ботка металлов», 1960, № 3.
54. А н д р е ев а А. Г. и Гуляев А. П. Цементация нержавеющей
стали. — «Металловедение и обработка металлов», 1960, № 3.
55. Шипилов А. Д., Михеев В. Г. Цементация хромистой нержа-
веющей стали. — «Металловедение и термическая обработка металлов»,
1962, № 6.
56. Рустем С. Л. и Зельбет Б. М. Сб. «Термическая обработка де-
талей и инструментов из стали ШХ15». М., Машгиз, 1951.
57. Лаптев Н. Р. и Васильева А. Цементация инструмента, изго-
товленного из стали ШХ15. — «Подшипник», 1952, № 11.
58. Нестеров Н. А. и Грдина Ю. В. О повышении теплостойкости
быстрорежущей стали — «Известия высшей школы», 1962, № 10.
59. Гуд|ремон Э. Специальные стали, т. 1, М., Металлургиздат, 1959.
60. Лукша Л. К. Способ повышения активности твердых карбюрато-
ров. — «Металловедение и обработка металлов», 1956, № 6.
61. Лукша Л. К. Влияние ацетата бария на активность твердых кар-
бюризаторов. — «Металловедение и обработка металлов», 1958, № 7.
62. «Usane Beige», 1957, v. 34, № 1551; Реф. журн. «Металлургия»,
1958, № 8, с. 197.
63. Шашкин П. И. Способ газовой цементации с использованием
твердого карбюризатора. — «Металловедение и обработка металлов», 1956,
№ 6.
64. Гардин А. И. Поверхностная закалка током высокой частоты.—
«Металлург», 1938, № 3.
65. Ассоно в А. Д., Шепелековский К. 3. и Ланкин П. А.
Скоростная цементация с нагревом т. в. ч. — «Вестник машиностроения»,
1954, № 6.
66. Ю р ь е в С. Ф. Гинзбург С. К. Исследование высокотемператур^-
ной цементации в твердом карбюризаторе при нагреве токами высокой
частоты. — «Металловедение и термическая обработка металлов», 1959, № 8.
67. М о р о з о в В. Т. Высокопроизводительный способ цементации ста-
лей специальной пастой. — «Металловедение и термическая обработка ме-
таллов», 1963, № 4.
68. Андрюшечкин В. И., Максименко Г. У. и Беляцкий Л. Я-
Химико-термическая обработка стали с нагревом т. в. ч. в обмазках. «Пере-
довой научно-технической и производственный опыт», 1960, тема 5, № М-60-
212/10.
69. МинкевичН. А., Ильинский С. К. и Просвирин В. И.
Новый способ цементации стали газами, полученными при пиролизе нефте-
продуктов. — «Вестник металлопромышленности», 1936, № 5 и 6.
467
70. Калинин А. И., К у н я в с к и й М. Н., Зайцева А. Я. Синти-
новый рациональный карбюризатор для газовой цементации. — «Металло-
ведение и обработка металлов», 1956, № И.
71. Корецкий Я. Н. Цементация стали, Судпромиздат, 1962.
72. И вен ска я Н. Д. Новые методы химико-термической обработки
стали. Сер. «Зарубежная литература», 1956, вып. 28, изд. ВПТИ,
73. К а л и н и н А. Т. Типовые контролируемые атмосферы для безо-
кислительного нагрева и химико-термической обработки стали. «Технология
автомобилестроения», 1958, № 5.
74. Б р у н з е л ь Ю. М., Р е г и р е р 3. Л., С в е т Б. И. и Пучков В. А-
Автоматическое регулирование потенциала углерода при газовой цемента-
ции.— «Металловедение и термическая обработка металлов», 1962, № 11.
75. Сорокина О. П. и Брон Д. И. Газовая цементация под давле-
нием. — «Металловедение и обработка металлов», 1955, № 7.
76. Минкевич А. Н. Химико-термическая обработка. Справочник «Ме-
талловедение и термическая обработка». М., Металлургиздат, т. II, 1962.
77. Козловский И. С., Лебедева Е. А. и Калинин А. Т. Проч-
ность цементованной стали при различных режимах термической обработ-
ки.— «Металловедение и термическая обработка металлов», 1964, № 3.
78. Гуляев А. П. и Трусова И. И. Термическая обработка глубоко
цементируемых деталей из хромоникелевой стали. М., ВИНИТИ, 1958, № 5-
58-377/13.
79. Wyss Н. Harterei Technische Mitteilungen 1962, Bd. 17, № 3; Metal
Progress», 1963, v. 83, № 1.
80. Unterweisen P. M. «Iron Age», 1958, v. 181, № 21, p. 123.
81. Hus e b у R. A., «SAE Journal», 1960, v. 68, № 1, p. 127.
82 .Ip sen H. N. «Metallurgia», 1959, v. 59, № 354, p. 176; 1960, v. 62,
№ 373.
83. Ассонов А. Д., Шепелековский К. 3, Ланкин П. А. Газо-
вая цементация с индукционным нагревом. М., Машгиз, 1958.
84. Головин Г. Ф. и Замятнин М. М. Высокочастотная термичес-
кая обработка. Машгиз, Л., 1959.
85. Ванин В. С. Ионная цементация стали. Известия АН СССР, ОТН,
Сер. «Металлургия и топливо», 1960, № 3; «Металловедение и термическая
обработка металлов», 1961, № 8.
86. П о д о й н и ц ы н В. Г. Цементация кислородно-ацетиленовым пламе-
нем.— «Металловедение и термическая обработка», 1961, № 3.
87. П о д о й н и ц ы н В. Г. Цементующая способность кислородно-пропа-
но-бутанового пламени. — «Металловедение и термическая обработка», 1962,
№ 6.
88. «Werkst att und Betrieb», 1959, Bd. 92, № 7, S. 459—461.
89. Морозова E. M. и Флоренсова Ф. P. Глубокая цементация в
жидкостной ванне.— «Станки и инструмент», 1949, № 9.
90. Морозова Е. М. и Флоренсова Ф. Р. Применение жидкостной
цементации для деталей станков. — «Станки и инструмент», 1957, № 2.
91. Рафалович В. И. Агрегат для жидкостной цементации и закалки
стальных изделий. ГОСИНТИ, № 7-63-622/11, 1964.
92. Минкевич А. Н. Цементация стали в жидкой среде, «Металлург»,
1940, № 11—12.
93. Schenk Н. Schmidtmann. «Arch. f. d. Eisenhuttenwes», 1954, Bd. 25,
S. 579.
94. Погодин-Алексеев Г. И. Влияние ультразвука на диффузион-
ные процессы в сталях и сплавах при повышенных температурах. — «Металло-
ведение и обработка металлов», 1958, № 6.
95. R oz a nski W. «Arch. Hutnictwa», 1958, Rok. 3, Z. 2, S. 126—149.
96. M о s z e z у n s.k i A., M a t у j a H. «Prace instytutow mechanica»,
1956. Rok. VL Zer. 18; «Przegled mechaniczny», 1956, Rok. XV, Zer. 7.
97. Moszezynski A. «Harterei-Techn. Mitteilungen», 1960, Bd. 15, H. 1,
S. 60.
468
98. Schnorr W., Schuhlmann W. «Harterei Techn. und Warmebehand-
lung», 1959, № 8.
99. Ванин В. С. и T и т о в В. К. Цементация стали в жидких органи-
ческих средах. — «Металловедение и термическая обработка», 1960, № 4.
100. Регирер 3. Л., Свет В. И., Львовский А. Я. и др. Цемента-
ция в жидкой среде с нагревом т. в. ч., «Передовой научно-технический опыт»,
ГОСИНТИ, 1962, № М-62-267/11.
101. Малинкина Е. И. Образование трещин при термической обработ-
ке стали. М., Машгиз, 1958.
102. Вышковский Ю. Г. и Юргенсон А. А. Влияние обработки
холодом на некоторые механические свойства высоколегированных сталей. —
«Металловедение и обработка металлов», 1957, № 10.
103. Вышковский Ю. Г. Смягчающая обработка высоколегированных
цементованных сталей. — «Металловедение и обработка металлов», 1958, № 8.
104. Свешников Д. А. Повышение усталостной прочности цементо-
ванных и цианированных деталей наклепом дробью. —- «Металловедение и
термическая обработка», 1964, № 4.
105. Эпштейн Г. Н. и Паисов И. В. Повышение стойкости цементо-
ванных деталей обкаткой роликом. Передовой научно-технический и произ-
водственный опыт, 1962, М-62-191/5.
106. Регирер 3. Л. и Малинкович А. Н. Информация о научно-ис-
следовательских работах, 1956, Тема 5, № И-57-17.
107. Нахимов А. М. Методы устранения трещин, образующихся при
цементации деталей из хромоникелевой стали. — «Вестник машиностроения»,
1955, № 3.
108. Балтер М. А. и Гринберг Н. М. Причины образования трещин
•при охлаждении деталей после цементации. — «Металловедение и термическая
обработка металлов», 1962, № 6.
109. Натапов Б. С. Термическая обработка цементуемых сталей
18ХНВА и 12Х24ВА. — «Металловедение и термическая обработка металлов»,
1962, № 3.
ПО. Алексеев П. Е. и Смирнова Н. И. Рациональные режимы тер-
мической обработки деталей из стали 18ХНВА. — «Металловедение и терми-
ческая обработка», 1964, № 6.
111. Егоров В. С., Андреева А. Г., Фоменко Г. Д. Газовое циа-
нирование и цементация нержавеющей стали Х17Н2.— «Металловедение и
термическая обработка металлов», 1964, № 3.
112. Кидин И. Н. Термическая обработка стали при индукционном наг-
реве. Металлуогиздат, 1950.
113. Бодяго М. Н. Структура и механические свойства цементованной
стали 18ХГТ, закаленной с нагрева т. в. ч.. Известия АН БССР, 1954, № 5.
114. Бодяго М. Н. Высокочастотная закалка цементованной стали
20ХН. Известия АН БССР, 1954, № 6.
115. Кидин И. Н. Термическая обработка стали при индукционном на-
греве. Справочник «Металловедение и термическая обработка», т. II, М. Ме-
таллургиздат, 1962.
116. Чириков В. Т. Вопросы совмещения технологии процессов термо-
обработки и обработки металлов давлением. — «Вестник машиностроения»,
1959, № 12.
117. Чириков В. Т. Схема совмещения в автоматизированном потоке
операций горячей деформации и химико-термической обработки. Автоматиза-
ция процессов в машиностроении, т. II, сер. «Горячая обработка металлов»,
Изд. АН СССР, 1962.
118. Scheil Е. О. «Harterei — Techn. Mitteilungen», 1954, Bd. 9, H2.
119. S c h e i b 1 W., W u s t B. «Harterei Techn. und Warmebehanlung»,
1959, Okt.
120. Котов О. К. О диффузии кислорода и водорода в сталь при це-
ментации.— «Металловедение и термическая обработка металлов», 1959, № 8.
469
121. Gunnarson S. «Jernkontorets Annall», 1962, Bd. 146, № 5, S. 383—
400; «Harterei— Techn. Mitteilungen», 1963, Bd. 18, № 1.
122. Натапов Б. С. Анормальность структуры цементованной стали. —
«Металловедение и термическая обработка», 1962, № 6.
123. Меськин В. С. Основы легирования стали. Металлургиздат,
1959.
124. Гуляев А. П. и Зеленова В. Д. Распределение углерода в це-
ментованном слое легированных сталей. — «Металловедение .и термическая
обработка металлов», 1960, № 3.
125. Миркин И. Л. и Лейтес А. В. Превращение остаточного аусте-
нита в цементованной легированной стали. Сб. № 2 трудов Московского ме-
ханического института. М., 1951.
126. Максимова О. П. и Никанорова А. И. Проблемы металло-
ведения и физики металлов. Металлургиздат, 1952.
127. Ермаков С. С. и Кочнев В. И. Влияние температуры второй
закалки цементуемых сталей на ударную выносливость. — «Металловедение и
термическая обработка», 1959, № 8.
128. Бабакин Г. Н. и Любченко А. П. Субструктура цементованно-
го слоя.— «Физика металлов и металловедение», 1961, т. 11, вып. 2.
129. Фоменко Г. Д., Егоров В. С. и Андреева А. Г. Исследова-
ние контактной прочности цементованной стали 12ХНЗА. — «Металловедение-
и термическая обработка металлов», 1963, № 1.
130. Ко нто р о в ич И. Е. Азотирование стали и свойства азотирован-
ного слоя. ОНТИ, 1938.
131. Юргенсон А. А. Азотирование в энергостроении. Свердловск^
Машгиз, 1962.
132. Юргенсон А. А. Роль водорода при азотировании стали. — «Фи-
зика металлов и металловедение», 1959, т. VII, вып. 1.
133. Fast J. D., Verrig М. В. «J. Iron a Steel Inst.», 1954, v. 176.
№ 24.
134. Darken L. S., Smith R. P., Filler E. W. «Journ. of Metals»,.
1951, v. 3, № 1174.
135. Wert C. A., Zenner C. «Phys. Review», 1949, v. 76, p. 1196.
136. «Металловедение и термическая обработка», Справочник, т. II, Ме-
таллургиздат, 1961.
137. Maratray F. «Sim la Dacumentation metallyrgu», 1953, t. 16, Oct.,.
Nov., Dec.
138. К о n т о p о в и ч И. E. и С о в а л о в а А. А. Фазовые превращения а
системе железо — азот. Труды МАТИ. Оборонгиз, 4948, № 4, с. 51—67
139. Георгиевский П. И. и Лахтин Ю. М. Структура и свойства
закаленного азотированного слоя. — «Вестник инженеров и техников», 1951,.
№ 1.
140. Просвирин В. И. Азотирование хирургических игол как способ
защиты от коррозии. — «Металлург», 1938, № 2.
141. Минкевич А. Н., Писарев Н. Н. и Солодихин А. Г. Азоти-
рование стали, как метод защиты стали от коррозии. М., ИТЭИН, 1940.
142. Рябченков А. В., Яхни на В. Д. Антикоррозионное азотирова-
ние. Сб. трудов ЦНИИМАШ. Машгиз, 1948, № 1.
143. Конторович И. Е. и Совалова А. А. Коррозионная устойчи-
вость азотированных железных сплавов. Труды МАТИ, Оборонгиз, 1948,.
вып. 4.
144. Соловейчик Е. И. и Финкельштейн М. Л. Охрупчивание-
стали при антикоррозионном азотировании. — «Металловедение и термическая
обработка металлов», 1960, № 7.
145. Могилевский Е. П. Охрупчивание стали при азотировании.—
«Металловедение и термическая обработка металлов», 1960, № 6.
146. Рябченков А. В. Коррозионноустойчивая прочность стали. Маш-
гиз, 1953.
470
147. Самсонов Г. В. и Уманский Я. С. Твердые соединения туго-
плавких металлов. Металлургиздат, 1957.
148. Самсонов Г. В. Тугоплавкие соединения. Справочник, М., Метал-
лургиздат, 1963.
149. Юрген с он А. А. и Погреб ецкая Т. М. О понижении хрупко-
сти азотированного слоя стали 38ХМЮА. — «Металловедение и обработка
металлов», 1959, № 4.
150. Фертик Н. А. Влияние предварительной термообработки на хруп-
кую прочность азотированного слоя стали 38ХМЮА. — «Металловедение и
обработка металлов», 1955, № 1.
151. Фертик Н. А. Повышенная хрупкость азотированного слоя стали
38ХМЮА. — «Металловедение и термическая обработка металлов», 1960, № 3
1152. Безмолибденовая азотиоуемая сталь 38ХВФЮА.— «Технология тран-
спортного машиностроения», 1955, № 2.
153. Simon W. «Iron Age», 1960, v. 185, № 5.
154. Морозова E. M. и Скузоватова А. П. Азотирование кон-
струкционных сталей. — «Станки и инструмент», 1956, № 9.
155. Андреева А. Г. и Алексеенко М. Ф. Новая аустенитная сталь
для азотирования (25Х18Н8В2). — «Сталь», 1959, № 1.
156. Войнич Я. Л. Азотирование лопаток сопловых аппаратов. «Метал-
ловедение и термическая обработка металлов», 1964, № 4.
157. Konkolv Т. «Wissenschftliche Zeitschrift Hachschule Schwerma-
schinenbau», 1959, Bd. 3, № 2—3, «Harterei—Techn. Mitteilungen», 1959, Bd. 13,
H. 2, 1960, Bd. 15, H. 2.
158. «Japan Inst. Metals», 1962, v. 26, № 2, 6.
159. Жиронкин A. H. Новые конструкционные свариваемые стали.
ВПТИ. М. 1958.
160. Mounce W. S., Miller A. I. «Metal Progress», 1960, v. 77, № 2,
p. 91—94.
161. Seabrook I. «Metal Progress», 1963, v. 84, № 1.
162. Бухиник E. H. и Богомолов В. А. Низкотемпературное газо-
вое азотирование режущего инструмента из стали Р9 и Р18. — «Металловеде-
ние и термическая обработка металлов», 1964, № 6.
163. Я х н и н a iB. Д. Азотирование магниевого чугуна. «Литейное произ-
водство», 1955, № 4.
164. Лахтин Ю. М. и Пинчук Д. С. Азотирование высокопрочного
магниевого чугуна. — «Металловедение и обработка металлов», 1958, № 7.
165. Бобро Ю. Г. и Коваленко В. С. Азотирование чугуна с шаро-
видным графитом, Труды ХПИ им. Ленина. Серия металлургическая, Харь-
ков, 1957, т. IX, вып. 1.
166. Ротенберг М. И. и Запольская А. В. Азотирование тепловоз-
ных коленчатых валов из чугуна. — «Литейное производство», 1959, № 10.
167. Устинов Т. И. Активизация процесса азотирования нержавеющих
теплоустойчивых и жаропрочных сталей. Передовой научно-технический и
производственный опыт, 1958, тема 3, № М-58-306/30.
168. Педан Г. П. Местная защита стали 38ХМЮА от азотирования ме-
тодом химического никелирования. — «Металловедение и термическая обра-
ботка», 1962, № 12.
169. Юрьев С. Ф. Азотирование сталей и его применение в автотракто-
ростроении. Л., ОНТИ, 1938.
170. Мишкевич Р. И., Солнцев П. И .и Смирнов А. В. Низко-
температурное азотирование конструкционной стали. — «Металловедение и об-
работка металлов», 1957, № 2, стр. 49—54.
171. «Engineer», 1956, v. 201, № 5235; «Petrol Eng.», 1954, v. 26, № 3.
172. Земсков Г. В., Гущин Л. К-, Домбровская Е. В., Парфе-
нов А. К-, Яркина В. Т. Азотирование стали при воздействии ультразву-
ка.— «Металловедение и термическая обработка металлов», 1961, № 3.
173. Pavesic Е. J. «Canad. Mach, a Manufact. News», 1955, v. 66. № 9.
471
174. Смирнов А. В. и Белоручев Л. В. Контролируемые атмосфе-
ры и их применение для термической и химико-термической обработки метал-
лов, ЛДНТП, 1960.
175. Tomory Tilorne. «Kohaszati latok», 1956, № 5.
176. Knuppel H., Brotzman К., Oberhard F. «Stahl u. Eisen»,
1958, Bd. 78, H. 26.
177. Unterweisen A. «Iron Age», 1958, v. 179, № 18.
178. Mitchell. «Metal Treatment a Drop. Forg.», 1958, v. 25, № 158.
179. Beiwenger H. «Harteren—Techn. Mitteilungen», 1958, № 5.
180. Hornberg H. «Harterei — Techn. Mitteilungen», 1962, Bd. 17, № 2.
181. Лахтин Ю. M., Крымский Ю. H. и Семенов P. А. Азотиро-
вание высокопрочного чугуна в тлеющем разряде. «Металловедение и терми-
ческая обработка металлов, 1964, № 3.
182. Морозова Е. М. и Флоренсова Ф. Р. Азотирование стали с
нагревом т. в. ч. — «Станки и инструмент», 1958, № 6.
183. Просвирин В. И. и Тарасов Б. Г. Азотирование с применени-
ем т. в. ч., Сб. «Превращение в сплавах и взаимодействие фаз». Изд. Рига
АН Латв. ССР, 1961.
184. Флорисова Ф. Р. Азотирование стали с нагревом т. в. ч. Сб. «Ме-
талловедение и термическая обработка», Металлургиздат, М., 1962.
185. Калашникова М. И. и Зотьева А. С. Ускорение азотирова-
ния при нагреве т. в. ч. — «Металловедение и термическая обработка метал-
лов», 1964, № 4.
186. Просвирин В. И. и Рябченков А. В. Новый процесс азотиро-
вания стали в расплавленных слоях, ИТЭИН, 1945, № 13.
187. Mczynski A., Tacikowski J. «Binletjh wzresien —- pardzieznik»,
1960, Rok VI, № 21/22.
188. Palme R., Trock W. «Harterei — Techn. Mitteilungen», 1961, Bd..
16, № 1.
189. Tacikowsi J. «Harterei — Techn. Mitteilungen», 1960, Bd. 15,
№ 1, S. 21—24.
190. S c h a r r W., Stulmann N. «Harterei — Techn. u. Warmebehand-'
lung», 1959, Bd. 5, № 8, S. 57.
191. Борисов А. А. Азотирование шестерен без последующей шлифов-
ки. — «Металловедение и термическая обработка металлов», 1964, № 3.
192. Косолапов Г. Ф. и Сидунова О. И. Электронно-микроскопи-
ческие исследования азотированного слоя. Филиал ВИНИТИ, ПНТПО, 1957.
193. Андреева Г. Г. и Гуревич Л. Я. Влияние азотирования на
коррозионную стойкость нержавеющих сталей. — «Металловедение и терми-
ческая обработка металлов», 1959, № 4.
194. Балашов Б. Ф. Азотирование, как метод повышения прочности
деталей. Тезисы доклада на Московской конференции по методам упрочнения
деталей машин (апрель, 1951), изд. ВНИТОМАШ, 1951.
195. Jun ger A. «Harterei Techn. u Warmebehandlung», 1959, № 5.
196. Гуревич Б. Г. и Юрьев С. Ф. О роли остаточных напряжений в
повышении выносливости стали при химико-термической обработке. Сб. «По-
вышение усталостной прочности деталей машин поверхностной обоаботкой»,
М., Машгиз, 1952.
197. Кудрявцев И. В., Меерович И. Б., Саввина Н. М. и
Ф а ф т В. И. Усталостная прочность валов после азотирования и правки.—
«Металловедение и термическая обработка», 1963, № 10.
198. Беленкова М.М., Костенко А. В., Михеев А. Н. и др.
Влияние термической обработки и азотирования на магнитные свойства аусте-
нитной стали. — «Металловедение и термическая обработка металлов»,.
1960, № 11.
199. Грибоедов Ю. Н. и Просвирин В. И. Кинетика и механизм,
совместного насыщения стали азотом и углеродом. Труды ЦНИИТМАШ, 1954„
№ 64, Машгиз.
472
200. Новикова А. Я., Калинин А. Т., Феофанова А. И. и др.
Рекомендации по -рациональному составу и структуре нитроцементованного
слоя. — «Технология автомобилестроения», 1963, № 10, стр. 3—19.
201. Просвирин В. И. и Грибоедов Ю. Н. Высокотемпературное
газовое цианирование инструментальных сталей. Труды ЦНИИТМАШ, Маш-
гиз, 1954, № 64.
202. Котов О. К. Поверхностное упрочнение углеродистых сталей высо-
котемпературной газовой нитроцементации. — «Металловедение и термическая
обработка металлов», 1958, № 5.
203. Е г о р о в В. С., А н д р е е в а А. Г. и Ф ом'ен ко Г. Д. Газовое циа-
нирование и цементация нержавеющей стали Х17Н2. — «Металловедение и
термическая обработка металлов», 1964, № 3.
204. V i а 11 е J. М. «Rev. Techn. Europ.», 1962, v. 21, № 726.
205. Porne A. «Harterei — Techn Mitteilugen», 1963, Bd. 18, № 3.
206. К и д и н И. H. .и Андреев Ю. Г. Нитроцементация стали с нагре-
вом т. в. ч. — «Известия высших учебных заведений». Сер. «Черная металлур-
гия», 1961, № 5.
207. Минкевич А. Н. и Калинин А. Т. Развитие процесса жидкост-
ной цементации (цианирования) стали. Сб. «Современные методы термичес-
кой обработки». М., Машгиз, 1954.
208. Косов К. В. Применение цианирования в массовом производстве. —
«Вестник машиностроения», 1954, № 2.
209. Сумароков Н. В. Цианирование нержавеющей стали. «Ученые
записки Пермского госуниверситета», 1960, т. XVI, вып. 4.
210. М о s z с z у n s k i A. Tacikiwski J. «Harterei-Techn. Mitteilungen»,
1960, Bd. 15, № 60.
211. Просвирин В. И. и Евтихов Г. В. Скоростное высокотемпера-
турное цианирование на стали с нагревом т. в. ч. — «Металловедение и тер-
мическая обработка металлов», 1964, № 3.
212. Калинин А. Т. и Новикова А. Я. Дефектная (темная) состав-
ляющая в структуре поверхностного слоя стали, возникающая при химико-
термической обработке. — «Технология автомобилестроения», 1963, № 10.
213. Albrecht. «Harterei-Techn. Mitteilungen», 1954, № 2.
214. Pronosil В. «Strojirenstvi», 1956, № 5; «Hutnicke», 1957, № 7.
215. Новикова А. Я. и Л евита нск а я Н. М. Дефекты цианирован-
ного слоя и факторы, способствующие их образованию. — «Автомобильная
промышленность», 1961, № 3.
216. W а 1 е nt i и К. «Metal Progress», 1953, v. 63, № 6.
217. Смирнов А. В. Азотирование технически чистого железа до высо-
кой твердости. ЖТФ, 1953, т. XXIII, вып. 8.
218. Jack К. Н. «Proceeding of the Royal Soc.», 1948, v. A194, № 1090,
p. 41.
219. Bridelle R., Michel A. «Rev. de Metallurgie», 1955, t. 52, № 5,
p. 397.
220. Прокошкин Д. А. и Дубовой В. Я. Цианирование режущего
инструмента, ЦДТ НКСМ, 1941.
221. Бухиник Е. Н. и Богомолов В. А. Низкотемпературное газо-
вое азотирование режущего инструмента из стали Р9 и Р18. — «Металловеде-
ние и термическая обработка металлов», 1964, № 4.
222. Бондаренко Э. В. Газовое цианирование с применением триэтано-
ламина.— «Технология подшипникостроения», 1957, № 12.
223. Немнонов С. А. Старение цианистых ванн и его влияние на при-
роду цианированного слоя. Труды института физики металлов, УФАН СССР,
Сб. работ лаборатории диффузии, 1950, вып. 11.
224. Topp Р. «Industrieblatt», 1957, № 12.
225. Свищова С. Б. и БорилоМ. С Низкотемпературное цианирова-
ние прессформ для литья под давлением из стали ЗХ2В8.— «Вестник машино-
строения», 1958, № 8.
473
226. Miiller I. «Industrieblatt», 1960, № 4; «Maschinenbau», 1961, № 12.
227. Niemann J., RettigH. «VDI.», 1960, Bd. 102, № 21.
228. Miiller I., Krzyminski A. «Technischc Zbl. Prakt. Metalbearbei-
tung», 1961, № 10; 1963, № 3.
229. Eysell F. W. «Feintechnik», 1960, v. 64, № 2.
230. Mitchell E. «Metal Treatment a. Drop Forging», 1964, v. 31,
№ 220—222.
231. Неустроев Г. H. Мягкое азотирование конструкционной стали.—
«Металловедение и термическая обработка металлов», 1962, № 6.
232. Лахтин Ю. М. и Неустроев Г. Н. Низкотемпературное газовое
цианирование конструкционных сталей.— «Металловедение и термическая об-
работка металлов», 1964, № 3.
233. Минкевич А. Н. Цианирование стали при температуре ниже точ-
ки Aci. Сб. трудов Московского института стали, 1940, т. XVII.
234. В en dedio. «Techn. Rundschau», 1961, Bd. 53. № 41.
235. Doliwa Heinz Urlich. «Ind. Anz.», 1963, Bd. 85, № 32.
236. «Metal Progress», 1961, v. 80, № 1, p. 77—82.
237. Eysell F. W. «Harterei Techn. u Warmebehandlung», 1958, № 3.
238. Kopietz К. H. «Harterei Techn. u Warmebehandlung», 1958, № 3.
239. Илларионов В. А. О реакциях диффузионного насыщения стали
металлами. — «Металловедение и обработка металлов», 1957, № 3; Сб. трудов
РВИАВУ. Рига, 1957, вып. 34.
240. Минкевич Н. А. и Зудин И. Ф. Изыскание новых смесей и ре-
жимов алитирования. — «Вестник металлопромышленности», 1939, № 8.
241. Просвирин В. И. и Зудин И. Ф. Повышение жаропрочности
железоуглеродистых сплавов алитированием. М., Машгиз, 1944.
242. D о n a d у Р. «Aluminium Suisse», 1956, Bd. 6, № 2, S. 65.
243. Флоринов К. А. Повышение окалиностойкости и ростоустойчиво-
сти чугунов методом жидкостного алитирования. Труды института химичес-
кого машиностроения, 1957, № 12.
244. Илларионов В. А. Жароупорность стали, алитированной по ни-
келевому подслою. Сб. трудов РВИАВУ. Рига, 1957, вып. 34.
245. Arora S. М., Gurte Р. К., Nyhawan В. R. «Тг. of the Indian Inst, of
Metals», 1958, v. 11, p. 57—72; ВИНИТИ Экспресс-информ., сер. «Металлове-
дение», декабрь, 1958, вып. 45, МТ-178-179.
246. Cobeirn К. G. «J. Metals», 1959, v. 11, № 1, р. 38—39.
247. Г о р о д н о в П. Т. Повышение жаростойкости черных металлов
алитированием. — «Металловедение и термическая обработка металлов»,
1961, № 2; Повышение жаростойкости стальных изделий методом алитирова-
ния, М., Машгиз, 1962.
248. Платонов В. М. и К'рештакова Г. Г. Алитирование клапанов
автомобильных двигателей. «Металловедение и термическая обработка метал-
лов», 1963, № 5.
249. Patton W. G. «Iron Age», 1956, v. 177, № 2.
250. Просвирин В. И., Мясоедов А. Н. и Зудин И. Ф. Алити-
рование в аэрозолях. — «Металловедение и термическая обработка металлов»,
1959, № 6.
251. Плотников В. А., Грацианский Н. Н. и Маховец К- П.
Электролитическое алитирование железа в расплавленной смеси солей. —
«Вестник металлопромышленности», 1934, № 3.
252. «Aluminium», 1957, v. 33, № I.
253. Минкевич А. Н. и Котов А. Н. Химико-термическая обработка
меди и латуни. Труды НТО Машпром, вып. 2, Сб. «Металловедение и терми-
ческая обработка металлов», М., Машгиз, 1960.
254. Powell G. F., Campbell I. Е., Gon sei В. W. Vapor-plating.
N.-I. L., 1955.
255. Титов В. К. и Макаров Е. Ф. Алитирование железа и стали
под вакуумом. ЖПХ, 1962, № 8.
474
256. Семенков и ч С. А. и Смирнов А. В. Алитирование железа па-
рами монохлорида алюминия. «Металловедение и обработка металлов»,
1958, № 5.
257. Schneider A., Schmidt N. «Metallkunde», 1951, № 42.
258. ГрдИ|НаЮ. В. и Гордеева Л. Т. Диффузионные покрытия
стали в газовой среде с нагревом током высокой частоты. «Известия высших
учебных заведений». Сер. «Черная металлургия», 1959, № 7.
259. Лахтин Ю. М. и Георгиевский П. И. К вопросу о диффузии
алюминия и хрома в сталь. — «Вестник машиностроения», 1949, № 9.
260. Б р ж а н В. С. Влияние легирующих элементов на процесс алитиро-
вания железа. — «Записки Воронежского сельскохозяйственного института»,
1954, т. XXV, вып. 1; 1959, т. XXVIII, вып. 2.
261. Красюк Н. С. Распределение углерода в алитированном слое
стали.— «Металловедение и термическая обработка металлов», 1959, № 6.
262. Baukloh W., Boke W. «Arch. f. d. Eizenhuttcnwesen», 1939, Bd.
12, № 7, S. 345—347; Реф. журн. «Металлург», 1939, № 4—5.
263. Heumann T., Dittrich S. «Zeitschrift fur. Metallkunde», 1959,
Bd. 50, № 10.
264. KapporA. H., ChatterjcaA. B., Nijhawan B. R. Scient
and Industry Res. Nat. Metallurg. Lab. 1957 (Индия).
265. Николайчик H. П. и Николайчик Е. Н. Алитирование чу-
гунных изложниц. — «Сталь», 1957, № 6.
266. «VDI — Zeitschrift», 1953, Bd. 95, № 24, S. 818—822; 1957, Bd. 99,
№ 26, S. 1275—1283, № 27, S. 1335—1342.
267. «Mater and Processes Mag.», 1964, № 4% p. 5—9.
268. Просвирин В. И., Федосов А. И. и Мякишев Ю. С. Алити-
рование лопаток газовых турбин из- аустенитной стали. — «Металловедение и
обработка металлов», 1956, № 4.
269. Грибоедов Ю. Н., Иванов Е. В. и Мясоедов А. Н. Термо-
обработка и алитирование валов ротора экспериментальной газотурбинной ус-
тановки. Труды ЦНИИТМАШ, 1960, № 15.
270. Дубинин Г. Н. Диффузионное хромирование сплавов. М., Изд-во
«Машиностроение», 1964.
271. Грибоедов Ю. Н. и Мясоедов А. Н. Диффузионное хромиро-
вание стали методом порошков в контейнерах с плавким затвором. Труды
ЦНИИТМАШ, 1960, № 15, стр. 66.
272. Морозова Е. М. и Спивак Э. Д. Термическая обработка в
станкостроении. М., Машгиз, 1949.
273. Samuel R. L., Lockington N. A. «Metal Treatmant and Drop
Forging», 1955, v. 22, № 117, 119.
274. Galmiche P. «Ingrs. et techniciens», 1959, t. 41, № 121, p. 53—55.
275. Тито в В. К. и Макаров E. Ф. Влияние выбора галлоида на хро-
мируемость железа. ЖПХ, 1963, № 36, вып. 4.
276. Б е и ь к о в с к и й Д. Д. и Елин Л. В. Повышение износоустойчи-
вости деталей машин термохромированием. Трение и износ в машинах. Инсти-
тут машиноведения АН СССР, 1950, № 5.
277. Вирченко И. П. Хромирование поршневых колец.— «Мехашза-
ция сктьского господарства», 1957, № 4.
278. Lockington М. A. «Metal Industry», 1956, v. 83, № 2; «Metal
Treatment and Drop Forging», 1957, v. 24, № 136, p. 28.
279. МинкевичА. H. и Ул ы бин Г. Н. Хромирование и борирование
стали при нагреве т. в. ч. — «Металловедение и термическая обработка метал-
лов», 1959, № 4.
280. Горбунов Н. Св Вакуумный метод хромирования. М., Изд. АН
СССР, 1955.
281. Горбунов Н. С. Диффузионные покрытия на железе и стали. М.,
Изд. АН СССР, 1958.
282. Принц Франтишек, Навара Вацлав, Диффузионное хромирование в
вакууме. «Чехословацкая тяжелая промышленность», 1959, № 7.
475
283. Могилевский Ю. П Диффузионное хромирование стали в ва-
кууме. Труды ЦНИИТМАШ, № 35, ОНТИ ЦНИИТМАШ, 1963.
284. Изгарышев Н. А. и Саркисов Э. С. Взаимное вытеснение ме-
таллов из паров их солей и использование этого процесса для борьбы с кор-
розией. ЖОХ, 1938, т. VIII, вып. 9.
285. Минкевич А, Н. Газовое хромирование стали.— «Сталь», 1943,
№ 5—6.
286. Гуляев А. П. и Дубинин Г. Н. Хромирование стали в газовой
среде.— «Вестник машиностроения», 1945, № 5
287. AtpxapOB В. И. Газовое хромирование, Труды ИММ. УФ АН СССР,
1945. вып. 4.
288. Dure Р., «Revue Universelle des Mines», 1954, v. 10, № 7, p. 531—541.
289. «Industry Heating», 1963, v. 30, № 9, p. 1654.
290. «Iron Age», 1960, v. 185, № 6, p. 193; 1961, v. 188, № 16, p. 133—135.
291. Hoar T. P., Groom E. A., «J. Iron Steel Inst.», 1951, v. 169, № 2,
p. 101—107.
292. Menzies J. A., Mortimer D. «Tr. Inst. Metal Finish», 1964, v. 41, № 1,
p. 15-22.
293. Полищук А. П. Диффузионное хромирование дереворежущего ин-
струмента.— «Станки и инструмент», 1957, № 4.
294. Архаров В. И. К вопросу о хромировании стали и о возможных
областях применения газового хромирования, Изв. АН СССР, ОТН, 1943, № 8.
295. Глускин Д. Я. Влияние предварительной химико-термической об-
работки железа и стали на взаимодействие с жидким цинком.— «Металлове-
дение и термическая обработка», 1961, № 3.
296. Грибоедов Ю. Н. и Юнц Б. Н. Технология диффузионного хро-
мирования способом порошков деталей энергостроения. Сб. «Новое в техноло-
гии термической и химико-термической обработки». Труды ЦНИИТМАШ,
1963, № 35.
297. Волкова Т. А., и Грибоедов Ю. Н. Защита пружин из стали
ЭИ723 от высокотемпературного окисления.— «Металловедение и обработка
металлов», 1958, № 5.
298. Войнич Я. Л. Хромирование пружин из стали ЭИ723, совмещенное
с термообработкой.— «Энергостроение», 1962, № 5.
299. Грибоедов Ю. Н. Повышение сопротивления абразивному износу
посредством диффузионного хромирования. Сб. «Новое в технологии термиче-
ской и химико-термической обработки». Труды ЦНИИТМАШ, 1963, № 35.
300. В у л ь ф о в и ч А. Б. Диффузионное хромирование сложных и круп-
ногабаритных деталей в массовом производстве. Сб. «Термическая и химико-
термическая обработка металлов», ЦИТЭИН, 1960, № 1.
301. Seeling R. Р., «SAE Journal», 1958, v. 66, № 5.
302. Samuel R. L. «Metal Finish J.», 1959, v. 5, № 49, p. 5—11.
303. Becker M., Dorn er H. «Harterei — Techn. Mitteilungen», 1962,
Bd. 11, №2, S. 229—231.
304. Bildi A. L. «Metal Progress», 1964, v. 83, № 4, p. 104—114.
305. G a 1 m i c h e P. «Revue de Metallurgie», 1950, t. 47, № 3, p. 192.
306. Metal Ind., 1959, 95, № 5, p. 98.
307. Земсков Г. В. и Косинский И. В. Хромосилицирование при на
греве т. в. ч. Научные записки Одесского политехнического института
1960, т. 24.
308. Вязников Н. Ф. и ПопандопулоА. Н. Повышение химической
стойкости диффузионно-хромированного слоя. — «Металловедение и обработка
металлов», 1958, № 3.
309. Просвирин В. И., Федосов А. И. и Мякишев Ю. С. Алити-
рование лопаток газовых турбин из аустенитной стали.— «Металловедение и
обработка металлов», 1956, № 4.
310. Грибоедов Ю. Н. и Мясоедов А. Н. Химико-термическая об-
работка аустенитных сталей ЭИ405 и ЭИ612 для повышения их эрозионной
476
стойкости применительно к лопаткам газовых турбин. Сб. «Некоторые вопросы
технологии тяжелого машиностроения», 1960, часть 1. М., Машгиз.
311. Просвирин В И. и Федосов А. И. Эрозионный износ и защита
от износа аустенитовых сталей для лопаток газовых турбин. «Инженерно-фи-
зический журнал», 1959, т. II, № 1.
312. Чижевский Н. П. Цементация железа бором.— «Журнал русского
металлургического общества», 1915, № 5, ч. 19, стр. 645—647.
313. Мок ин Н. Н. Цементация железа бором.— «Известия Сибирского
института металлов», Томск, 1934, т. 1, вып. 4.
314. Ж и г а ч А. Ф., Антонов И. С., Пчелкина М. А. и др. Поверхно-
стное насыщение стали бором из газовой среды.— «Металловедение и терми-
ческая обработка металлов», 1959, № 4.
315. Лахтин Ю. М. и Пчелкина М. А. Борирование высоколегиро-
ванных сталей.— «Металловедение и термическая обработка металлов»,
1961, № 3.
316. Eipeltauer Е. Borierung von Eisenaus der Gasphase, Metalkun-
dishe Berichte. Berlin, 1951, Bd. 12.
317. Пчелкина M. А. и Лахтин Ю. M. Газовое борирование в среде
треххлористого бора.— «Металловедение и термическая обработка металлов»,
1960, № 7.
318. Говоров А. А. Борирование стали.— «Вестник машиностроения»,
1944, № 7, 8.
319. Штрымов А. И., Добродеев А. С. и Яковенко В. А. Про-
мышленное применение электролизного борирования деталей машин. ИТЭИН,
тема 24, 1953.
320. Таран В. Д. и Скугорева Л. П. Борирование как способ упроч-
нения шарошечных долот.— «Вестник машиностроения», 1957, № 5.
321. Таран В. Д. и Скугорева Л. П. Поверхностное борирование
низколегированных сталей.— «Металловедение и обработка металлов»,
1957, № 6.
322. Таран В. Д. и Скугорева Л. П. Упрочнение опоры буровых до-
лот методом борирования. Труды Московского нефтяного института, 1956,
вып. 16.
323. Schaaber О. «Harterei-Techn. Mitteilungen», 1960, № 2.
324. Ка w a kumi М., К о m и г о I. «J. of the Japan Inst, of Metals»,
1949, v. 13, Aug., p. 15—19.
325. Metram. Электролизное борирование. Реферативный журнал «Метал-
лургия», 1959, № 5, стр. 281 (11453П).
326. Ногтев Н. Н. и Рагозин Ю. М. Электролизное борирование в
борном ангидриде.— «Металловедение и термическая обработка металлов»,
1962, № 12.
327. В ес k W. «Metal Ind.», 1955, № 86, р. 43—44.
328. Блантер М. Е. и Беседин А. П. Кинетика образования бориро-
ванного слоя в сплавах железа.—«Металловедение и обработка металлов»,
1955, № 6.
329. Морозова Е. М. и Флоренсова Ф. Р. Борирование стали, Сб.
статей ЭНИМС «Поверхностное упрочнение стали». М., Машгиз, 1949.
330. Ornig Н., Schaaber О. «Harterei — Techn. Mitteilungen», 1962,
Bd. 17, № 3.
331. Конторович И. С. и Львовский М. Я. Поверхностное насы-
щение железа и его сплавов бором.— «Металлург», М., 1939, №-10—41.
332. Самсонов Г. В. и Портной К. И. Сплавы на основе тугоплав-
ких соединений. М., Оборонгиз, 1961.
333. Земсков Г. В., Кайдаш Н. Г. и Правенская Л. Л. Бориро-
вание железа и стали в вакууме.— «Металловедение и термическая обработ-
ка металлов», 1964, № 3.
334. Таран В. Д. и Скугорева Л. П. Борирование стали с гальва-
ническими покрытиями.— «Металловедение и термическая обработка метал-
лов», 1960, № 1.
477
335. Грибовски Л. Установка оптико-механического действия для оп-
ределения остаточных напряжений. Передовой научно-технический опыт.
ЦИТЭИН, 1962, вып. 21, тема 32, № П-61-126/21.
336 Me. Bride С. С., Speth a k I. W., Speiser R. «Тг. Am. Eos. Me-
tals», 1954, № 46, р. 499.
337. К i е s s 1 i n g R. «Acta chem. scand», 1950, v. 4, № 2, p. 209.
338. Лахтин Ю. M. и Пчелкина М. А. Газовое борирование стали.
Сб. «Металловедение и термическая обработка металлов». Труды НТО Маш-
про.м, Сб. Секция металловедения и термической обработки, вып. II 1960, М.,
Машгиз.
339. ОрловА. В., Сандлер Н. И., Кукол В. В. и др. Исследование
борированного слоя среднеуглеродистой стали. Сб. трудов Украинского науч-
но-исследовательского института металлов, вып. 7, 1961.
340. Балтер М. А„ ДукаревичИ. С. и Гольдштейн Л. Я.
Фазовый состав борированного слоя сталей.— «Металловедение и термиче-
ская обработка металлов», 1964, № 12.
341. Пантелеев Ю. Новое в борировании стали. «Народное хозяйство
Советской Латвии», 1958, № 1.
342. МинкевичА. Н. Структура боридных слоев. «Известия высших
учебных заведений», Сер. «Черная металлургия», 1963, № 5.
343. Rundquist S. «Nature», 1958, Bd. 181, S. 259.
344. Самсонов Г. В. Марковский Л. Я., Ж и г ач А. Ф. и др. Бор,
его соединения и сплавы. Киев, Изд. АН УССР, 1960.
345. С а г г о 11 К. G., Darken L. S., Filer Е. W., Z w е 11 L. «Nature»,
1954, Bd. 174, S. 978—979.
346. Минкевич A. H., Расторгуев Л. Н. и Андрюше ч-
к и н В. И. Диффузионные боридные слои на металлах. «Известия Высшей
школы». Сер. «Металлургия», 1960, № 7.
347. Тришевский И. С., Клепандо В. В. и Орлов В. В. Высо-
костойкие вставки для пропусков проволочных станов.— «Сталь», 1959, № 4.
348. Мыльников Г. В. Исследование износоустойчивости цилиндровых
втулок и поршней буровых насосов.— «Нефтяное хозяйство», 1957, № 1.
349. Шрейбер Г. К. и Мархатин Э. Л. Поверхностное упрочнение
деталей оборудования и инструмента нефтяной и газовой промышленности,
Гостоптехиздат, 1959.
350. Маурах А. А. Новый способ повышения износостойкости гусениц
с открытым шарниром.— «Вестник машиностроения», 1956, № 12.
351. Славин Д. О. иМосквин Н. И. Термодиффузионное бороалити-
рованме стали. Сб. статей НИИХИММАШ, 1960, № 6, М., Машгиз.
352. Blum Р., Andrienx I. L. «Revie de Metallurgie», 1954, № 10,
p. 679—682.
353. Земсков Г. В. и Кайдаш Н. Р. Боросилицирование железа и
стали.— «Металловедение и термическая обработка металлов», 1964, № 3.
354. Г а к м а н Э. Л. Диффузионное цинкование деталей машин.—
«Технология транспортного машиностроения», 1956, № 6.
355. «Draht», 1958, 9, № 4.
356. БуздаковА. П. О механизме термодиффузионного цинкования же-
леза. Труды АзНИИ по добыче нефти. Баку 1957, вып. 6.
357. Зильберфарб М. Н. и Приходько Л. Н. Диффузионное цин-
кование. Сб. трудов НИИХИММАШ, 1959, № 28, М., Машгиз.
358. Н а й д и ч И. М. Диффузионное цинкование. Сб. трудов
НИИХИММАШ, 1950, № 4, М., Машгиз.
359. Глузкин Д. Я. О реактивной диффузии в металлах. ЖТФ, 1950,
вып. 2.
360. S с h е i I Е., W u г s t Н. «Zs. MetalIkunde», 1937, № 29, S. 224.
361. Harstmann D. «Stahl und Eiesen», 1953, № 73, S. 659.
362. Harstmann D. «Arch. f. d. Eisenhiittenwesen», 1954, № 11, 12.
478
363. Б уз да ко в А. П. Применение диффузионного цинкования в борь-
бе с коррозией.— «Нефтепромысловое дело», 1959, № 3.
364. Wiliam. «Prod. Finish», 1960, v. 13, № 3.
365. Буздаков А. П. Термодиффузионное цинкование. Технический
бюллетень Совнархоза Азербайджанской ССР, 1959, № 9; Труды научной
межвузовской конференции по вопросам борьбы с коррозией, Гостоптех-
издат, 1962, , ।
366. Знайченко С. Г. Термодиффузионное цинкование в борьбе с мор-
ской коррозией свай. Труды научной межвузовской конференции по вопросам
борьбы с коррозией, Гостоптехиздат, 1962.
367. Dupny G. «Machine Outil Francaise», 1953, № 74—76.
368. Тынный A. H. Влияние сульфидирования на износ металлов.—
«Металловедение и термическая обработка металлов», 1961, № 8.
369. Vi 11 emeu г Y. «Metaux Corr. Ind.», 1962. v. 37, Nov, p. 447.
370. Wil man H., Piggott M. «Acta crystallogr», 1958, v. 11,
№ 12, p. 13.
371. Ефимов M. Г. «Обработка металлокерамических деталей серой».
Автомобильная промышленность, 1959, № 10, с. 40.
372. Калинин А. Т., Иванюк Е. Я. и Иванесян С. В. Сульфиди-
рование в ваннах без применения цианистых солей.— «Металловедение и тер-
мическая обработка металлов», 1961, № 8.
373. Веселов Б. П. Рациональные составы ванн для среднетемператур-
ного процесса сульфидирования.— «Технология машиностроения», 1960, № 7.
374. Блохин М. А., Нестеренко П. С. и Шуваев А. ,Т. Рентгено-
спектральное и структурное исследование сульфидированных образцов. Сб.
«Повышение стойкости деталей машин», М., Машгиз, 1959.
375. Виноградов Ю. М. Хлорирование, сульфидирование и сульфоциа-
нирование, как средства повышения износоустойчивости металлов. Сб. «Кор-
розия и износ металлов», № 27, М., НИИХИММАШ, 1959.
376. Waterfol 1 F. D. «Engineering», 4959, v. 187, 4846, р. 23.
377. Barbas F. «Machine Moderne», 1958, №• 52 (586), p. 21—24.
378. Hosil F., Nainar J., Stepanek K. «Novatarsky Listok», 1955,
№ 163, Listopad, (Praha).
379. Has Zdislaw. «Przeglad mechaniczny», 1963, Rok. XXII, Lipka 10,
c. 411—414.
380. Поспешил А. и Гасил Ф. «Чехословацкая тяжелая промышлен-
ность», 1959, № 7, стр. 38—42.
381. Вайнштейн В. Э. и Виноградов Ю. М. Исследование изна-
шивания сульфидированных металлических поверхностей с помощью радиоак-
тивных изотопов. Сб. «Повышение стойкости деталей машин». М., Маш-
гиз, 1959.
382. Jamadsaki Taketomo, «J. Japan Foundry mens Sos.», 1961, v. 33, № 8,
p. 560—569.
383. ГильманТ. П. Газовое сульфидирование железоуглеродистых
сплавов. Сб. «Повышение стойкости деталей машин». М., Машгиз, 1959.
384. П е т у х о в Е. П., 3 а х а р о в П. А., Ш п и г у н о в а Н. А. и др.
О сульфидировании хромистых нержавеющих сталей.— «Металловедение и об-
работка металлов», 1956, № 5.
385. Зиновии Н. С. Исследование процесса сульфидирования. Сб. «По-
вышение стойкости деталей машин». М., Машгиз, 1959.
386. Виноградов Ю. М., Зеленова В. Д. и Шишокина К. В.
Сб. «Коррозия и износ металлов», НИИХИММАШ, 1959, № 27.
387. Криулин А. В. Сульфидирование и свойства сульфидированного
металла.— «Металловедение и термическая обработка металлов», 1960, № 7.
388. Dumitrescu Т. A. «'Rev. roumaine metallurgie», 1960, т. 5, № 1,
с. 5—23.
389. О р д и н а 3. Г. Диффузионное силицирование железа и стали. Труды
Ленинградского технологического института пищевой промышленности,
1955, т. XII.
479
390. Морозова Е. М. и Флоренсова Ф. Р. Сб. «Поверхностное
упрочнение стали» (Методы химико-термической обработки»), М., ГНТИ, 1949.
391. D о d е г о М., Blum Р. «Rev. de Metallurgie», 1950, № 7, р. 544—546.
392. Ro see v D., «Acad. RPR», 1961, v. 6, № 2, p. 161—170.
393. Горбунов H. С. и Акопджанян А. С. Диффузионное сили-
цирование. ЖПХ, 1956, т. XXIX, вып. 5.
394. Zeilinski W. «Przegl mech.», 1958, № 2.
395. D о v e у D. M., J e n Kins I., R a n d e с К. C. «Sympos. Inst. Metal»,
1952, hon ton 19th, Nov.; «Inst. Metals», 1953, p. 213—236.
396. Fitzen E. «Metallkundische Berichte», 1951, Bd. 22; «Verlag Tech-
nik», Berlin, 1951.
397. Fit zer E. «Arch. f. d. Eisenhiittenwesen», 1954, № 11, 12. S. 601—
612.
398. Schneider A., Siegfiedt I. P. «Galvano», 1960, v. 29, № 278,
p. 154—158; «Metal!», 1959, № 6, p. 98.
399. Ту нин a M. И. и Ключников H. Г. О взаимодействии железа
и четыреххлористого кремния, Журн. неорг. хим., 1962, т. VII, вып. 4.
400. Горбунов Н. С., Акопджанян А. С. и Изгарышев Н. А.
Микротвердосгь диффузионных кремниевых покрытий. Доклады АН СССР,
1952, т. XCI, № 2.
401. Орд ин а 3. Г. Диффузия кремния в хромистую сталь.— «Металло-
ведение и термическая обработка металлов», 1960, № 6.
402. Stahlabs A. «Arch f. d. Eisenhuttenwesen», 1954, № 9, 10,
S. 455—463.
403. Metani Ohisy, Chihou Kundzoki. «J. Japan Inst. Metals», 1959, v. 23,
№ 5, p. 273—276.
404. Metani Chirojsu, Onisi Masatu, Chihon Kundzoki. <J. Japan Inst. Me-
tals», 1960, v. 26, № 3, p. 161.
405. Metteoli L. «Metallurgia Italiana», 1961, № 6; «Atti noture», 1961,
№ 5,6.
406. Парфенов В. А. Применение окиси бериллия для бериллизации
стали.— «Металловедение и обработка металлов», 1956, № 3.
407. Конто рович И. Е. и Львовский М. Я. Насыщение железоуг-
леродистых сплавов бериллием.— «Вестник металлопромышленности»,
1939, № 12.
408. Прокошкин Д. А. и Усов А. Ф. Диффузия бериллия в железо
и сталь. Сб. трудов Московского института стали, 1939, т. XIII.
409. Бернштейн М. Л. и Кишеневский В. Б. Строение диффузи-
онного слоя бериллизовэнной стали. Известия АН СССР, ОТН, 1957, № 2.
410. Конторович И. Е. и Львовский М. Я. Дисперсионное тверде-
ние сталей после насыщения с поверхности бериллием.— «Техника воздушного
флота», 1938, № 7.
411. Chapin Е. J., Hayward С. R. «Тг. Am. Soc. of Metals», 1947,
v. 38, р. 909.
412. Yamaquchi Т. Y., Hikoyama К. Takei T. «Journ. of the Scien-
tific Research Inst.», 1956, v. 50; 1957, v. 51 (Toky).
413. «Metall Finisch Soc. Japan», 1958, v. 19, p. 469.
414. Fortin В. I., Wurm I. O., Crevel L., Portin R. A. «Electro-
chem Soc.», 1959, v. 106, № 5, p. 428.
415. Sibert M. E., Steinberg M. A. «Mater. Design. Eng.», 1957,
v. 46, № 6.
416. Ogawa, Husanats, Kawamura, Tatsume. «Japan Inst. Metals», 1957,
v. 21, ^s 10, p. 599.
417. Schlehten M. W., S t r a u m a n i s M. E., Gill С. B. «Electrochem.
Soc.», 1955, v. 102, № 7.
418. Shil S. T. «Electrochem. Sos.», 1956, v. 103, № 7.
480
419. Минкевич А. Н. и Гвоздев А. Г. Титанирование стали в рас-
плавленной соляной ванне.— «Известия высших учебных заведений». Сер. «Чер-
ная металлургия», 1960, № 2.
420. Straumanis М. Е., Huang V. Р. «Metallwissenschaft», 1958».
Н. 6.
421. ГрдинаЮ. В. иСофрощенко А. Ф. Комбинированная химико-
термическая обработка стали. Известия высших учебных заведений. Сер. «Чер-
ная металлургия», 1962, № 2.
422. Волобуев И. В. Ниобирование стали. Труды Харьковского поли-
технического института, 1951, № 21.
423. Дубинин А. С. Диффузия ниобия в сталь из газовой фазй. ЖТФ„
т. XXII, 1952.
424. Прокошкин Д. А. Диффузия.молибдена в железо.— «Металлург»»
1937, №7. _
425. Дубинин Г. Н. Насыщение поверхности стали молибденом из газо-
вой среды. ДАН СССР, 1952, т. LXXXIV, вып. 5.
426. «Steel», 1957, v. 140, № 20, р. 161—164.
427. Schulze Н. W. «Metal. Progress», 1959, v. 76, № 3, p. 74—80.
428. F i t z e r E. «Arch f. d. Eisenhuttenwesen», 1952, № 9, 10.
429. Дубинин А. Г. Насыщение поверхности стали ванадием из газо-
вой фазы. ДАН СССР, 1952, т. LXXXIV, вып. 2.
430. Дубинин А. Г., Корицкий В. Г., Веселовская И. М. Диф-
фузия хрома, ванадия и марганца в поверхностные слои стали из газовой
среды. ЖТФ, 1953 т. XXIII, вып. 10.
431. Окнов М. Г. и Мороз Л. С. О механизме диффузии различных
элементов в железо и никель, ЖТФ. 1941, т. XI, вып. 7 и 8.
432. Дубинин Г. Н. Вольфрамирование и хромовольфрамирование ста-
ли в газовой среде.— «Вестник машиностроения», 1946, № 9, 10.
433. Дубинин Г. Н. иБогоряцкийЮ. А. О природе фаз, возникаю-
щих на поверхности стали при диффузии вольфрама. ЖТФ, 1952, т. XXII»
вып. 11.
434. Дубинин Г. Н. Насыщение поверхности стали вольфрамом из га-
зовой среды ДАН СССР, 1952, т. LXXXIV, № 4.
435. Chandler R. Н. «Industr. Finish», 1962, v. 14, № 164, p. 77—78.
436. Coves R. M. «Tr. Metallurg. Sos. А1МЕ». 1962, v. 224, № 2„
p. 267—272.
437. Дубинин Г. H. Насыщение поверхностных слоев стали марганцем
из газовой среды. ДАН СССР, 1952, т. LXXXIV, вып. 6.
438. Cornelius Н. «Arch. f. d. Eisenhuttenwesen», 1942, № 4, Oct.»
S. 147—153.
439. Утевский Л. M. Отпускная хрупкость стали. M., Металлург-
издат, 1961.
440. Смирнов А. И., Челпанов Б. В. и Бровкин С. П. Сурьмя-
нистый чугун как подшипниковый материал. Одесское обл правление НТО
Машпром и Одесский дом техники, 1958.
441. Смирнов А. И., Чепанов Б. В. Сурьмирование чугуна и стали.
Научные записки Одесского политехнического института, 1958, т. XVII.
442. Титов В. Н. и Макаров Е. Ф. Диффузионное насыщение железа
и стали оловом и сурьмой.— «Металловедение и термическая обработка метал-
лов», 1961, № 8.
443. Хрущев М. М. и Бабичев М. А. Исследование изнашивания ме-
таллов. Изд. АН СССР, 1959.
444. Г у д ц о в Н. Т. и Д у б и н и н Г. Н. Насыщение поверхностных сло-
ев стали церием. Известия АН СССР, ОТН, 1951, № 4.
445. Горбунов Н. С. и Латухова А. Г. Диффузионное меднение.
ЖПХ, 1957, т. 30, вып. 81.
446. Макквиллэн А. Д. и Макквиллэн М. К. Титан. Металлург-
издат, М., 1958.
447. Еременко В. Н. Титан и его сплавы. Изд. АН УССР, 1955.
481
448. Новикова Е. Н. Азотирование титановых сплавов в чистом азоте.
Сб. «Титан и его сплавы», 1960, вып. 3, Изд. АН СССР.
449. Новикова Е. Н. Азотирование титановых сплавов при понижен-
ном давлении. Металловедение титана. Труды пятого совещания по металлур-
гии, металловедению и применению титана и его сплавов, Изд-во
«Наука», 1964.
450. Сидорин И. И., Рыски на Е. В., Пащенко С. В. и др. Упроч-
нение поверхности деталей из сплавов на основе титана методом азотирова-
ния. Научные доклады высшей школы. Сер «Машиностроение и приборо-
строение», 1959, № 2.
451. ° Смирнов А. В. и Начинков А. Д Поверхностное упрочнение
титана методами химико-термической обработки.— «Металловедение и терми-
ческая обработка металлов», 1960, № 3.
452. Смирнов А. В. и Начинков А. Д. Азотирование титана при
пониженном парциальном давлении азота.— «Металловедение и термическая
обработка металлов», 1960, т. 7.
453. Wyatt I. L., Grant N. I. «Iron Age», 1954, v. 173, №.l, 2, 4.
454. Wa si lewski R. I., Kehl G. L. «Jorn. of the Inst, of Metals»,
1954, Nov.
455. Hansel R. W. «Metal Progress», 1954, v. 65, March.
456. Bungardt K., Rudinger K. «Zf. f. Metallkunde», 1956, Bd. 7,
№ 8.
457. Takenchi Y. «J. Japan Inst. Metall», 1959, v. 23, № 7, p. 410—413.
458. Cuthill I. R., Hayes W. D., Seebold R. E. «J. Res. Nat. Rur.
Standarts», 1960, v. A64, № 1, p. 119—125.
459. «Metal Industri», 1960, v. 77, № 5, p. 87—88.
460. Wyatt J. L., At hers H., «Tr. Am. Sos. of Metals», 1954, v. 46.
461. Ципкина E. Д. Усталостная прочность титана. Сб. докладов «Со-
временные сплавы и их термическая обработка». МДНТП, М., Машгиз, 1958.
462. Минкевич А. Н., Теймер А. Д. и Зотьев Ю. А. Азотирова-
ние титана в аммиаке.— «Металловедение и обработка металлов», 1956, № 7.
463. Минкевич А. Н. ,и Шульга Ю. Н. Поверхностное упрочнение
титана обработкой в расплавленной буре.— «Металловедение и обработка ме-
таллов», 1957, № 12, стр. 53—61.
464. Самсонов Г. В. и Эпик А. П. Исследование условий диффузи-
онного поверхностного насыщения титана, циркония, ниобия, тантала, молиб-
дена и вольфрама углеродом и бором. Сб. докладов по теории жаропроч-
ности металлов и сплавов, М., Институт металлургии им. Байкова, 1963.
465. Grist A., Moorhead Р. Е., Frost Р. D., Jackson J. Н. «Тг.
Am. Soc. of Metals», 1954, v. 46, p. 95.
466. Гр ди н a to. В., Гордеева Л. T. и Тимошина Л. Д. Цемента-
ция титана при индукционном нагреве т. в. ч.— «Известия высшей школы».
Сер. «Черная металлургия», 1963, № 4.
467. Tour S., S t u k a A., Fischer G. «J. of the Metals, Transaction se-
ction», 1958, v. 7, № 2.
468. Борисенко А. И. Защита молибдена от высокотемпературной га-
зовой коррозии. Изд-во АН СССР, 1960.
469. Энциклопедический справочник «Конструкционные материалы», т. 1,
М., Оборонгиз, 1964.
470. Ф ауст Ч. Л. Защитные металлические покрытия. Сб. «Жаропрочные
сплавы в условиях полетов со сверхзвуковыми скоростями», Металлург-
издат, 1962.
471. Wladek S. Т., Wilff I. «Тг. Metallurg. Soc. AIME», 1960, v. 218,
№ 4, p. 714—722.
472. О x x G. D., Coffin L. F. «Tr. Metallurg. Soc. AIME», 1960, v. 218,
№ 3, p. 541—545.
473. Sedlatschek K., Stadler H. I. «Plansceber Pulvermetallurgie»,
1961, v. 9, № 1, 2 p. 39.
482
474. Leadbeater G. I., Rech ar ds D. T. «Met. Treatment», 1954, v. 21 r
№ 106, p. 309.
475. Гуляев А. П., Якшина О. К. и Першина Н. Ф. Силицирова-
ние молибдена. «Специальные стали и сплавы», Сб. трудов ЦНИИЧМ, 1963,
№ 35, М., Металлургиздат.
476. Fitzer Е. «Berd und Huttenmannische Monats», 1952, Bd. 97, H. 5.
477. Прокошкин Д. А. и Арзамасов Б. H. Циркуляционный ме-
тод насыщения молибдена некоторыми элементами. Сб. «Исследования по жа-
ропрочным сплавам», 1962, т. IX, Изд-во АН СССР.
478. Арзамасов Б. Н. и Прокошкин Д. А. Новый циркуляционный
метод насыщения тугоплавких металлов некоторыми элементами. ЦИТЭИН,
Передовой научно-технический и производственный опыт, № 7-63-143/4, 1963.
479. Самсонов Г. В., Ковальченко М. С. и Верхогляд о-
в а Т. С. Диффузия кремния в титан, тантал, молибден и железо. Доклады
АН УССР, 1959, № 1.
480. Jones В. J. «Alloy Metals Rev.», 1953, v. 8, № 70.
481. Самсонов Г. В. и Эпик А. П. Покрытия из тугоплавких соеди-
нений. М., Изд-во «Металлургия», 1964.
482. А р ж а н ы й П. М. Исследование фазового состава диффузионного
слоя. Сб. Исследования по жаропрочным сплавам». Изд-во АН СССР,
1958, т. 8.
483. АржаныйП. М. иВолкова Р. М. Исследование системы хром —
молибден диффузионным методом. ЖНХ, 1963, т. VIII, вып. 3.
484. «Mater. Design. Eng», 1958, v. 47, № 2, p. 157.
485. Дубинин Г. H. Защитные покрытия сплавов на основе молибдена
и ниобия. Сб. «Исследования структуры и свойств жаропрочных сплавов»,
М., Изд-во «Машиностроение», 1964.
486. Braun Н. «Harterei-Techn. Mitteilungen», 1962, Bd. 17, № 4.
487. Чао, Прист, Майерс. Последние достижения в области защит-
ных покрытий на молибдене. Сб. «Защита от окисления тугоплавких метал-
лов». ИИЛ, 1962.
488. Минкевич А. Н. Диффузионные боридные слои на металлах. «Ме-
талловедение и термическая обработка металлов», 1961, № 8.
489. Minkevitch A. N. «iRev. de Metallurgie», 1963, Sept.
490. Minke vic A. N. «Harterei-Techn. Mitteilungen», 1962, Bd. 17, № 3.
491. Лозинский M. Г. Высокотемпературная металлография. M., Маш-
гиз, 1959.
492. Самсонов Г. В. и Латышева В. П. Исследование диффузии
бора и углерода в некоторые металлы переходных групп. «Физика металлов
и металловедение», 1956, т. II, вып. 2.
493. Blumenthal Н., «Planseeber f. Pulwermetallurg», 1961, № 1,
2, -S. 44—46.
494. Миллер К. Защита от окисления некоторых тугоплавких металлов.
Сб. «Защита от окисления тугоплавких металлов». М., ИИЛ., 1962.
495. Аржаный П. М., Волкова Р. М. и Прокошкин Д. А.
О структуре и фазовом составе диффузионного покрытия ниобия кремнием.
«Известия АН СССР». Сер. «Металлургия и топливо», 1959, № 6.
496. Lorenz R. Н., Michael А. В. «J. Electrochem Sos.», 1961, v. 108,
№ 9, р. 885.
497. Аржаный П. М., Волкова Р. М. и Прокошкин Д. А. Диф-
фузия кремния и титана в ниобии и кинетика окисления их сплавов. Изве-
стия АН СССР, Сер. «Металлургия и топливо», 1960, № 5.
498. Аржаный П. М., Волкова Р. М. и Прокошкин Д. А. Иссле-
дование диффузии кремния и титана в ниобий. Труды Института металлургии
им. Байкова, 1962, т. XI.
499. Аржаный П. М., Волкова Р. М., Прокошкин Д. А. О диф-
фузии алюминия и бериллия в ниобий. Известия АН СССР. Сер. «Металлур-
гия и топливо», 1961, № 2.
483
500. Леонова А. П., Герасимов А. Ф., Конев В. И. Иссследова-
ние реакционной диффузии в системах Me — сложный газ, III система Nb —
(В + N). «Физика металлов и металловедение», 1960, т. IX, вып. 5.
501. Goetzel С. G., Landler Р. «Planseeber Pulvermetallurg.», 1961,
Bd. 9, № 1, 2, S. 36.
502. В e p x о p о б и н Л. Ф., Иванов В. Е., М ат ю шенко Н. Н. и др.
О реакционной диффузии в системах Мо—-Si и Та—Si. «Физика металлов и
металловедение», 1962, т. 13, вып. 1.
503. Аржаный П. М., Волков Р. М. и Прокошкин Д. А. Термо-
диффузия в системе вольфрам — бериллий.— «Известия АН СССР». Сер. «Ме-
таллургия и топливо», 1962, № 6.
504. Герасимов А. Ф., Конев В. Н. иТимофеева Н. Ф. Исследо-
вание реакционной диффузии в системах металл — сложный газ. «Физика ме-
таллов и металловедение», 1961, т. 11, вып. 4.
505 Могг, Selikson, «J. Chem. Phys.», 1951, № 19, р. 1539.
506. Тылкина М. А. и Савицкий Е. М. Физико-химические свойства
в области применения рения и его сплавов. Сб. «Рений», М., Изд-во
АН СССР, 1961.
507. Минкевич А. Н., Тылкина М. А., Расторгуев Л. Н. и др.
Химико-термическая обработка рения. Сб. «Рений». М., Изд-во «Наука», 1964.
508. Aronsson В. «Acta Chemica Scandinavica», 1960, р. 733.
509. Савицкий Е. М., Тылкина М. А. и Поворова К. Б. ЖТФ,
т. VI, вып. 8.
510. Knapton. Plansee Seminar. 3, Wieh, 1959, p. 412.
511. Савицкий E. M., Тылкина M. А. и Поворова К. Б. Диаграм-
ма состояния системы хром — рений. ЖНХ, 1959, т. IV, вып. 8.
512. Schneider A., Siegfriedt, «Metall», 1959, № 6, р. 546—552.
513. Pearson W. В. Handbook of lattice spacings and structure of metals
and alloys. 1958.
514. Buckle H., Moisson J. «Rech. Aeronaut.», 1958, № 64, p. 27—33.
515. Samuel R. L., Lockington N. A. «Galvano», 1954, № 213, Okt.
516. Schlechten A. W., Straumanis M. E., Gill С. B. «J. of the
Electrochemical. Sos.», 1955, v. 102, № 2.
517. Масленков С. Б. и Жаркова Д. H. Фазовый состав диффузи-
онного слоя борированного никеля. «Физика металлов и металловедение»,
1962, т. 14, вып. 4.
518. Лахтин Ю. М. и Пчелкина М. А. Механизм образования и
•свойства борированного слоя на сплавах железа и никеля. Сб. трудов НТО
Машпром, 1964, № 3, М., Изд-во «Машиностроение».
519. Хансен М. и Андерко К. Структура двойных сплавов, т. 1, М.,
Металлургиздат, 1962.
520. Вол А. Е. Строение и свойства двойных металлических систем, т. 1,
М, Физматгиз, 1959.
521. «Galvano», 1957, v. 26, № 240.
522. Fabion R., «Mater. Design Eng.», 1961, v. 54, № 6.
523. Минкевич A. H., Шур H. Ф. и Tpекало А. С. Борирование
484
кобальта и сплавов на его основе. Сб. трудов НТО Машпром, 1964, № 3, М.»
Машгиз.
524. Фалин Н. А. и Штильман М. Л. Диффузия бериллия в медь
и ее сплавы.— «Металлург», 1938, № 12.
525. Venturello G., Coen V., «Metallurgio itals, 1955, v. 47, № 10»
p. 453—460.
526. Buckle H., «Rev. de Metallurgies, 1957, v. 54, № 1.
527. «Metal Industries, 1957, v. 90, № 13.
528. Buckle H., Moisson R. «Galvanos, 1957, v. 26, № 240, p. 27.
529. «Mater Products, 1962, v. 1, № 6, p. 76.
530. G о b a 1 s k i S. T. «Prace Inst, mech.s, 1959, № 26.
531. Nagasaki Kyny a, Kaneko Masami, Hukon Kungzoky.
«J. Japan Inst. Metalss, 1959, v. 23, № 2, p. 113.
532. «Anticorros. Mater, and Processes Mags, 1964, № 4, p. 4—9.
533. Терлецкий В. E. Изготовление порошка латуни и латунных изде-
лий методом диффузионного насыщения из газовой фазы.— «Порошковая ме-
таллургия», 1961, № 3.
534. Mikulski I., М rawer St., Strenski Ig., Werber T. «Roczno
cheni.», 1959, № 6.
535. Evans J. W., Fea meh a ugh H., «J. Appl. Chem.s, 1959, v. № 6,
p. 307—310.
536. Пауэл К., Кемпбел И. иГонсерБ. Осаждение ниобия и тан-
тала из их летучих галоидных соединений. Сб. «Ниобий и тантал», М.,
ИИЛ, 1954.
537. Trobery L. «Iron Age», 1957, v. 179, № 2.
538. Горчаков А. В. Диффузионное хромирование и азотирование ме-
таллокерамических изделий. Изд. НИИТАВТОПРОМ, 1957.
539. Москвин Н. И. Коррозионностойкие металлокерамические матери-
алы на основе железа.— «Вестник машиностроения», 1952, № 3.
540. Елютин В. П., Мосжухин Е. И., Шулеп о в В. И. Влияние
химико-термической обработки на жаропрочность сплавов. Сб. Московского
института стали. 1958, № 38, М., Металлургиздат.
541. Архаров В. И. и Немнонов С. А. Карбидизация электролити-
ческих осадков хрома. Известия АН СССР, 1943, № 9—10.
542. Архаров В. И. и Конев В. Н. Повышение износоустойчивости
хромированных покрытий карбидизацией. — «Вестник машиностроения»,
1955, № 11.
543. П л и ш к о Д. С. Повышение износоустойчивости, жаростойкости и
кислотостойкости электролитических хромовых покрытий карбидизацией. «Из-
вестия высших учебных заведений». Сер. «Черная металлургия», 1960, № 8.
544. Самсонов Г. В. Тугоплавкие соединения, М., Металлург-
издат, 1963.
545 Oxley I. Н., Campbell I. Е. «J. of Metals», 1959, Febr.,
р. 135—137.
546. Елютин В. П., П е п е к и н Г. И. и «Лысов Б. С. Исследование
процесса образования карбида титана, осаждаемого из газовой фазы. «Изве-
стия высшей школы», Сер. «Черная металлургия», 1964, № 3.
485
547. Елютин В. П., П е п е к и н Г. И. .и Лысов Б. С. Термодинамиче-
ские расчеты некоторых реакций, происходящих при осаждении карбида ти-
тана из газовой фазы. «Известия высшей школы», Сер. «Черная металлургия»,
1963, № И.
548. Ruppert W. «Metallobreflache», 1960, Н. 7, S. 193—198.
549. Wiegand Н., Ruppert W. «Metalloberflache», 1960, Н. 8,
S. 220—235.
550. Самсонов Г. В. и Константинов В. И. Тантал и ниобий.
М., Металлургиздат, 1959.
551. Белозерский Н; А. и Гинзбург А. А. Нанесение рениевых по-
крытий из газовой фазы на вольфрамовую нить. «Вопросы радиоэлектроники»,
1960. Сер. 1 «Электроника», вып. 3.
552. Гинзбург А. А. Карбонил рения и способ нанесения рениевых по-
крытий путем разложения карбонила рения на нагретой вольфрамовой про-
волоке. Труды института Гипроникель. Л. 1962, № 12.
553. 3 е л и к м а н А. Н. и Барышников Н. В. Изучение условий по-
лучения рениевых покрытий из газовой фазы. «Вопросы электроники». Сер. 1
«Электроника», 1962, № 8.
554. Зеликман А. Н. и Барышников Н. В. «Известия Академии
наук СССР», Сер. «Топливо и металлургия», 1963, № 5.
555. Федотов Я. А. Основы физики полупроводниковых материалов,
М., Изд-во «Советское радио», 1963.
556. Зеликман Г. А., М а з а л ь Е. 3., Пресс Ф. П. и др. Полупровод-
никовые кремниевые диоды и триоды. М.— Л., Изд-во «Энергия», 1964.
557. К а л и н о в и ч Д. Ф., К о в е н с к и й И. И., С м о л и ч М. Д. Фран-
цевич И. Н. Влияние постоянного тока на скорость цементации стали. «Ме-
талловедение и термическая обработка металлов», 1964, № 12.
558. Pierre, Chevenard. «Journal Academie Siens», 1953, № 13.
559. Б о к ш т e й н C. 3., Гудков T. И., Кишкин С. T. и др. Изучение
однородности сплавов и подвижности по границам зерен с помощью радиоак-
тивных «изотопов.— «Заводская лаборатория», 1955, № 4.
560. К и д ин И. Н. и Андрюшечкин В. И. Диффузия хрома из галь-
ванического слоя в железо и сталь при быстром электронагреве.— «Металло-
ведение и термическая обработка металлов», 1964, № 3.
561. Кащенко К. А. Влияние кремния на свойства цементованной ста-
ли. Научные доклады высшей школы. Сер. «Металлургия», 1958, № 3.
562. Добрускина Ш. Р. Влияние мышьяка на химико-термическую
обработку легированных сталей. Доклады на совещании по Керченской ме-
таллургии, Крымиздат, 1958.
563. Гольдштейн Я. Е. Теллур в стали.— «Сталь», 1959, № 2.
564. Рустем С. Л. Оборудование и проектирование термических цехов,
М., Машгиз, 1962.
565. Минкевич А. Н. Современное состояние и задачи в области хими-
ко-термической обработки. Сб. «Современные направления в области техно-
логии машиностроения». М., Машгиз, 1957.
566. Ванин В. С. и Пермяков В. Т. Ускорение высокотемпературной
486
цементации стали.— «Известия АН СССР. Сер. «Металлургия и топливо»,
1962, вып. 5.
567. Андрачников Ш. Л. Жидкостная цементация и нитроцементация
в бесцианистых солях.— «Металловедение и термическая обработка металлов»,
1965, № 2.
568. Розанов А. Н. Цементация стали в расплавленном чугуне.— «Ве-
стник машиностроения», 1954, № 11.
569. Конторович И. Е. Об азотировании стали для поверхностного
упрочнения.— Сб. «Современные методы термической обработки стали», М.,
Машгиз, 1954.
570. Смирнов А. В., Белоручев Л. В., Каплан Р. И. и др. Азо-
тирование пассивирующихся сталей с применением четыреххлористого угле-
рода. Передовой научно-технический опыт. Сер. «Металловедение и термиче-
ская обработка металлов», Л., 1964, ЛДНТП.
571. Бородулин Г. М., Кравченко В. А. и Плышевский А. И.
Исследование толстых термодиффузиопныч покрытий. Исследование сталей и
сплавов. М., Изд-во «Наука», 1964.
572. Подборский Л. К. Твердое диффузионное хромирование полотен
и напильников в пастах. Сб. «Процессы упрочнения деталей машин». М.„
«Наука», 1964.
573. В i 1 d i A. L. «Metal Progress» 1964, v. 85, № 4, p. 109—114.
574. Земсков Г. В., Косинский И. В. и Правенская Л. Л. Хро-
мосилицирование стали.— «Металловедение и термическая обработка метал-
лов», 1964, № 9.
575. Ермаков С. С. и Нестеренкова Ю. П. Борирование стали в
твердых металлизаторах. Труды Ленинградского политехнического института,
1964, № 234, с. 75—82.
576. Гончаревский М. С., Щесно Л. П., Хитрик И. С. Струк-
тура и прочностные свойства цинковых парадиффузионных покрытий в зави-
симости от условий цинкования. Сб. «Производство труб», вып. 12, М., Метал-
лургиздат, 1964.
577. «Nat. Bur. Standards Techn. Navs. Bull», 1960, v. 44, № 2, p. 32—33.
578. Грдина Ю. В., Гардеева Л. Т. и Тимошина Л. Г. Азотиро-
вание титана при индукционном нагреве, т. в. ч. —«Известия высшей школы».
Сер. «Черная металлургия», 1962, № 2.
579. Иванов В. Е., Сомов А. И. и Нечепуренко Е. П. Вакуумное
силицирование металлов. Сб. «Применение вакуума в металлургии», Изд-во
АН СССР, 1963.
580. Becker G., Spyra W., «Metalloberflache», 1963, Bd. 17, № 9,
S. 271.
581. Mitchell E., Dawes C. «Harterei — Techn. Mitteilungen», 1965,
Bd. 19, № 4, S. 224—230.
582. Методы получения чистых металлов. Сб. переводов. М., ИИЛ, 1957.
583. Schultze Н. W. «Metal Progress», 1959, v. 76, № 3.
584. Charles E., Mac Neill. «Metal Finishing», 1963, Juli.
585. Па латник Л. С. и Папиров И. И. Ориентированная кристал-
лизация, М., Изд-во «Металлургия», 1964.
487
586. Чистяков Ю. Д. и Лайнер Б. Д. Ориентированное нарастание
кристаллического вещества (эпитаксия). Сб. «Рост кристаллов», Академиздат,
т. IV, 1964.
587. Авраамов Ю. С., Епанчицев О. Г., Бирюкова Н. А. Уста-
новка для прецизионного определения изменения плотности твердых образцов
дифференциальным методом гидростатического взвешивания. «Передовой на-
учно-технический и производственный опыт», ГОСИНТИ, № 2-64-1156/25.
588. Gaczol J. «Harterei—Techn.» Mitteilungen, 1964, Bd. 19, H. 3.
589. Muller I. «Draht», 1964, 15, № 9, p. 630.
590. Криулин А. В. «Сульфидирование стали и чугуна», М.-Л., Маши-
ностроение, 1965.
591. Щербединская А. В., Минкевич А. Н. Диффузионное на-
сыщение ниобия и титана углеродом, «Известия высшей школы», Сер. Цвет-
ная металлургия, 1965, № 4.
592. Щербединская А. В., Минкевич А. Н. Диффузионное насы-
щение молибдена углеродом, «Известия высшей школы». Сер. «Черная метал-
лургия», 1964, № 11.
593. «Anticorros. Mater, u. Processing Mag.», 1964, № 4, p. 5—9.
594. Андриевский А. И., Шибряев Б. Ф., Юкин Г. И. Металло-
керамические фильтры, применяемые в нефтяной и газовой промышленности»,
ЦНИИТЭНефть. Сер. «Нефтезаводское оборудование», 1963.
595. «Iron Age», 1964, v. 194, № 12, р. 82—83.
596. «American Mashinist», 1964, v. 108, № 18, 57.
597. Owen L. W. «Electroplat and Metal Finish», 1964, v. 17, № 9,
266—304.
598. Поляков Я. M., Нисельсон Л. А., Крестовников А. Н.
Исследование процесса получения тантала и ниобия восстановлением их пен-
тахлорида водородом, «ЖПХ», гл. XXXIV, вып. 1, 1963.
ОГЛАВЛЕНИЕ
Предисловие .........-.................... 3
Введение ........................... ..°..... 5
Г лава I. Общие закономерности диффузионных процессов .......... 11
Механизм диффузии ........-................................. П
Математическая трактовка диффузии ......«................... 14
Влияние различных факторов на процесс диффузии ............. 22
Структура диффузионных слоев ............................. 31
Глава II. Химико-термическая обработка стали ................. 42
Цементация ................................................ 42
Диффузия углерода в сталь.............................-... 42
Сталь для цементации ................«..................— 48
Цементация в твердом карбюризаторе............-....-...... 54
Газовая цементация ....................................... 60
Цементация в жидкой среде .............................. 72
Режимы термической обработки цементованной стали ......... 74
Структура цементованного слоя .......••................... 83
Свойства цементованной стали ........................... 96
Азотирование ............................. -................ 99
Диффузия азота в сталь ......................... «...... 101
Антикоррозионное азотирование стали ..................... 106
Азотирование легированной стали ......«.................. 109
Структура азотированного слоя легированной стали ........ 124
Свойства азотированной легированной стали ............... 126
Нитроцементация и цианирование ............................. 129
Особенности совместной диффузии в сталь углерода и азота... 129
Нитроцементация ......................-................ 131
Цианирование ............................................ 135
Структура нитроцементованного и цианированного слоя ..... 139
Свойства нитроцементованной и цианированной стали ....... 143
Низкотемпературная нитроцементация и цианирование ....... 144
Алитирование ............................................. 154
Методы и режимы алитирования ............................ 154
Структура алитированного слоя .......................... 166
Свойства алитированной стали ............................ 173
Алитирование аустенитной стали ........................ 178
Хромирование ......................................... 181
Методы и режимы хромирования ...........«.........-...... I82
489
Влияние углерода и легирующих элементов на глубину хроми-
рованного слоя ..................-.......................
Структура хромированного слоя ...........................
Свойства хромированной стали ............................
Насыщение стали хромом совместно с другими элементами....
Хромолитирование и хромоазотирование аустенитной стали ..
Борирование ..............-..................................
Методы и режимы борирования .............................
Структура борированного слоя ............................
Свойства борированной стали .............................
Насыщение стали бором совместно с другими элементами-----
Борирование высокохромистой и аустенитной сталей и сплавов
Цинкование ..................................................
Методы и режимы цинкования ...................«..*.......
Структура цинкового слоя ................................
Свойства цинкованной стали ..................... .......
Сульфидирование и сульфоцианирование ........................
Сульфидирование .........................................
Сульфоцианирование ......................................
Силицирование ......«........................................
Методы и режимы силицирования ...........................
Структура и свойства силицированной стали ...............
Бериллизации .......<.......................................
Титанирование ................................. ~..........
Ниобирование ...............................................
Молибденирование ...........................................
Ванадирование
Вольфрамирование ............................................
Насыщение марганцем ........................................
Насыщение фосфором ......~..................................
Сурьмирование.........................................~......
Карбосурьмирование ..........................................
Насыщение танталом, церием, медью, оловом и мышьяком ........
Глава III. Химико-термическая обработка титана ................. 325
Азотирование в азоте......................................... 325
Азотирование в аммиаке ................. ................... 336
Окисление ................................................... 341
Цементация......-............................... «.......... 356
Борирование ............................................... 354
Насыщение молибденом, алюминием, кремнием и некоторыми дру-
гими элементами ........................................... 356
201
203
209
217
220
223
224
231
240
243
244
248
243
251
257
259
260
264
269
270’
276
281
288
295
297
303
305
308
311
315
319
322
Глава IV. Химико-термическая обработка тугоплавких металлов ............. 361
Обработка молибдена ................................................ 361
Обработка с целью повышения окалиностойкости ................... 361
Борирование, цементация и азотирование ..................... 370’
Обработка ниобия ................................................ 378
Обработка вольфрама ................................................ 385-
Обработка тантала ........................ ............-.......... 388
Обработка циркония .................................................. 390
Обработка рения ................................. .................. 391
Г лава V. Химико-термическая обработка никеля, кобальта и меди . 397
Обработка никеля ................ и....»................... 397
Обработка кобальта ....................................... 401
Обработка меди ............................................. 411
490
Цинкование ................................................ 412
Бериллизация .............................................. 415
Алитирование............................................ 417
Силицирование ............................................. 421
Сурьмирование ....................................... 423
Окалиностойкость химико-термически обработанной меди. 426
Кислотостойкость химико-термически обработанной меди .... 430
Г лава VI. Химико-термическая обработка металлокерамических сплавов 434
Обработка железных сплавов .................................. 434
Обработка твердых сплавов ..................... ~.............. 436
Г лава VII. Химико-термическая обработка гальванических покрытий 445
Г лава VIII. Осаждение на поверхности металлов карбидных, нитридных,
боридных, силицидных и металлических покрытий ................. 449
Глава IX. Химико-термическая обработка полупроводников ............ 459
Литература ........................................................ 465
Редактор В. А, Семенченко
Технический редактор В. И. Модель
Корректор И. Г. Петрова
Переплет художника Л. С. Вендрова
Сдано в производство 29/IV 1965 г.
Подписано к печати 5/XI 1965 г.
Т-14266 Тираж 6500 экз. Печ. л. 30,75
Бум. л. 15,38 Уч.-изд л. 31,5
Темплан 1965 г. № 306 Формат 60 х 9О’Д»
Цена 1 р. 73 к. Зак. № 1278
Издательство «МАШИНОСТРОЕНИЕ»,
Москва, Б-66, 1-й Басманный пер., 3
Экспериментальная типография ВНИИПП
Государственного комитета Совета
Министров СССР по печати.
Москва И-51, Цветной бульвар, 30
ЗАМЕЧЕННЫЕ ОПЕЧАТКИ
Стра- ница Строка Напечатано Должно быть
52 23-я сверху 15ХГВА 15ХГНВА
62 8'Я сверху 95 — 97% 98 — 97%
69 11-я сверху богатого газа источника богатого газа
82 Подпись под рис. 39 И. В. Кидин И. Н. Кидин
97 6-я сверху 20НЗМА 20ХНЗМА
98 Подпись под рис. 54 Зависимость ударной вязкости Зависимость ударной выносливости
100 Подпись под рис. 57 [131] [132]
101 В заголовке табл. 13 [131] [132]
102 27-я сверху на рис. 59 на рис. 59 и 61
103 Рис. 59, 8-я строка правой колонки У У'
103 Подпись под рис. 59, 9-я снизу в борированном слое рентгеноструктурным анализом пока не была обнаружена. в азотированном слое рентгеноструктурным анализом обнаружена пока лишь в некоторых исследованиях.
127 Подпись под рис. 76 III — образец с буртом, г — 2 мм III — образец с буртом, г = 1,1 м м
148 Подпись под рис. 95 соответственно в тече- ние 2 и 6 ч соответственно в тече- ние 6 и 2 ч
222 13-я снизу оказывались максималь- ными имели максимум на некотором расстоянии от поверхности
240 3-я снизу 12ХМ Х12М
247 Подпись под рис. 160 ЗН70Ю ХН70Ю
253 Подпись под рис. 165 [6]. [36] [6], [361]
378 3-я сверху а — 6,8 Зг 0,3 кХ а = 4,0 0,3 кХ
437 2-я снизу В. В. Функе В. Ф. Функе
481 10-я сверху 1951 1959
А. Н. Минкевич. Зак. 1278
A. H. МИНКЕВИЧ
ХИМИКО-ТЕРМИЧЕСКАЯ
ОБРАБОТКА МЕТАЛЛОВ
И СПЛАВОВ
ВТОРОЕ ПЕРЕРАБОТАННОЕ ИЗДАНИЕ
ИЗДАТЕЛЬСТВО «МАШИНОСТРОЕНИЕ»
Москва 1965