/
































































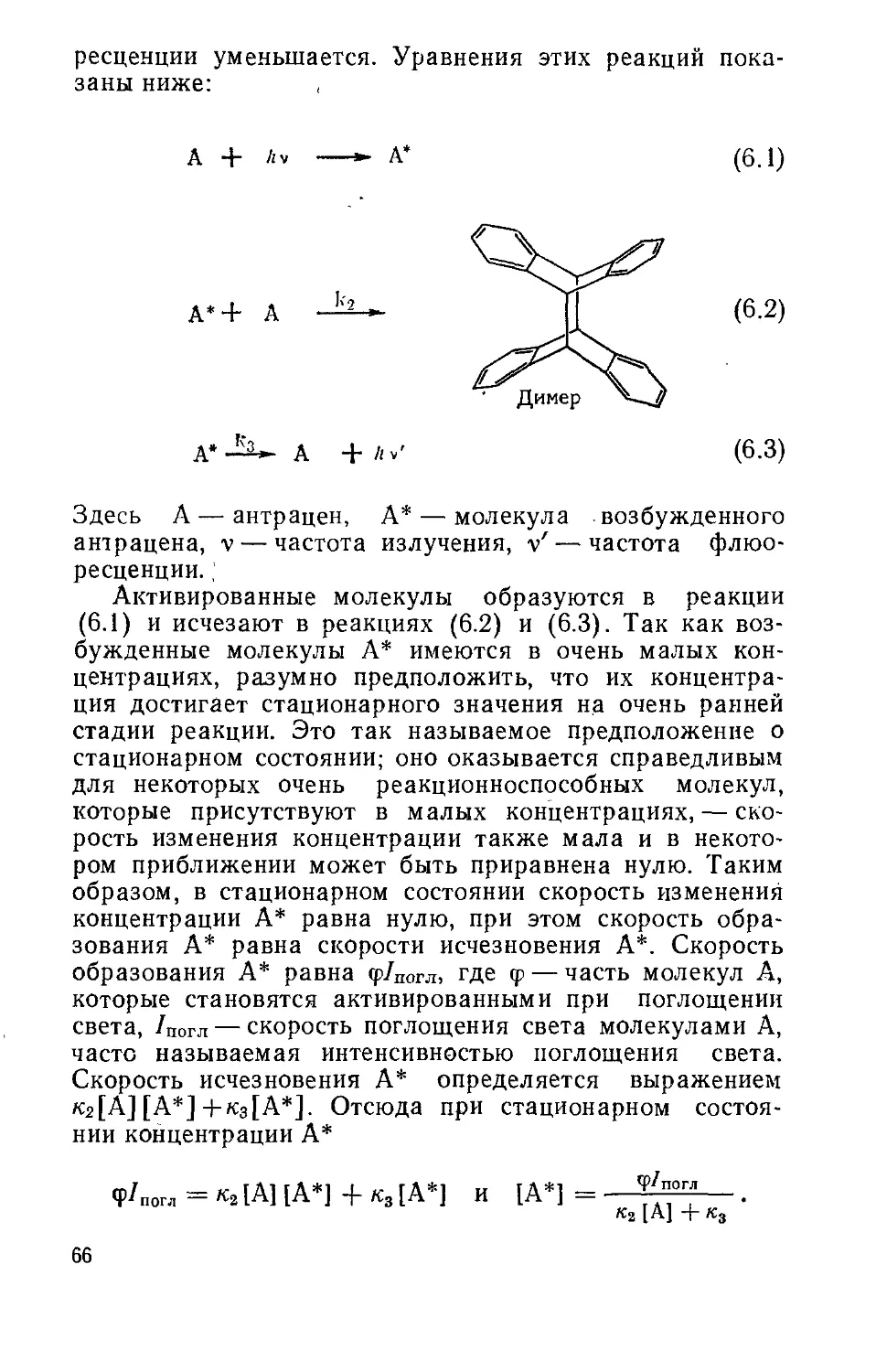




























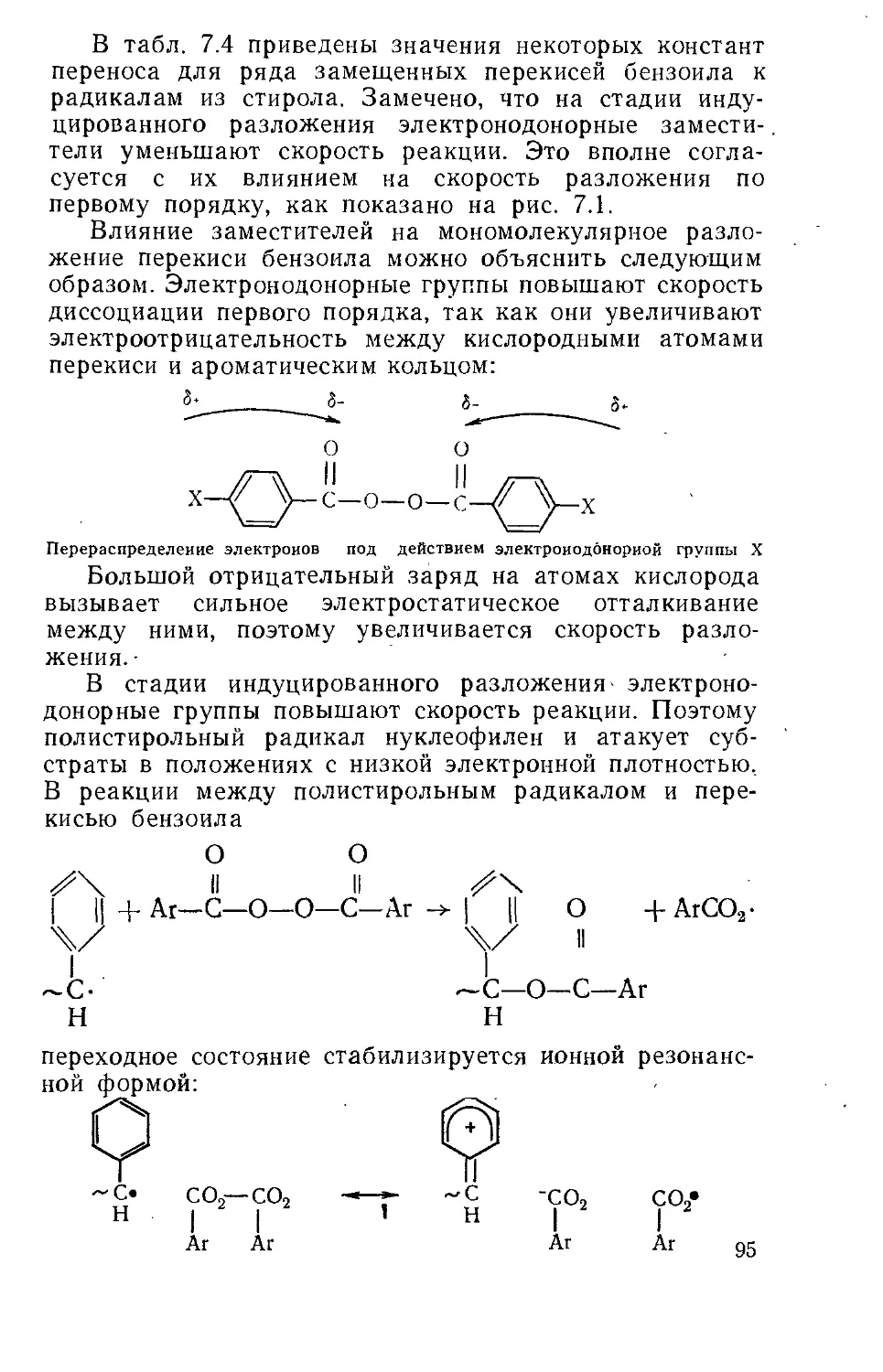






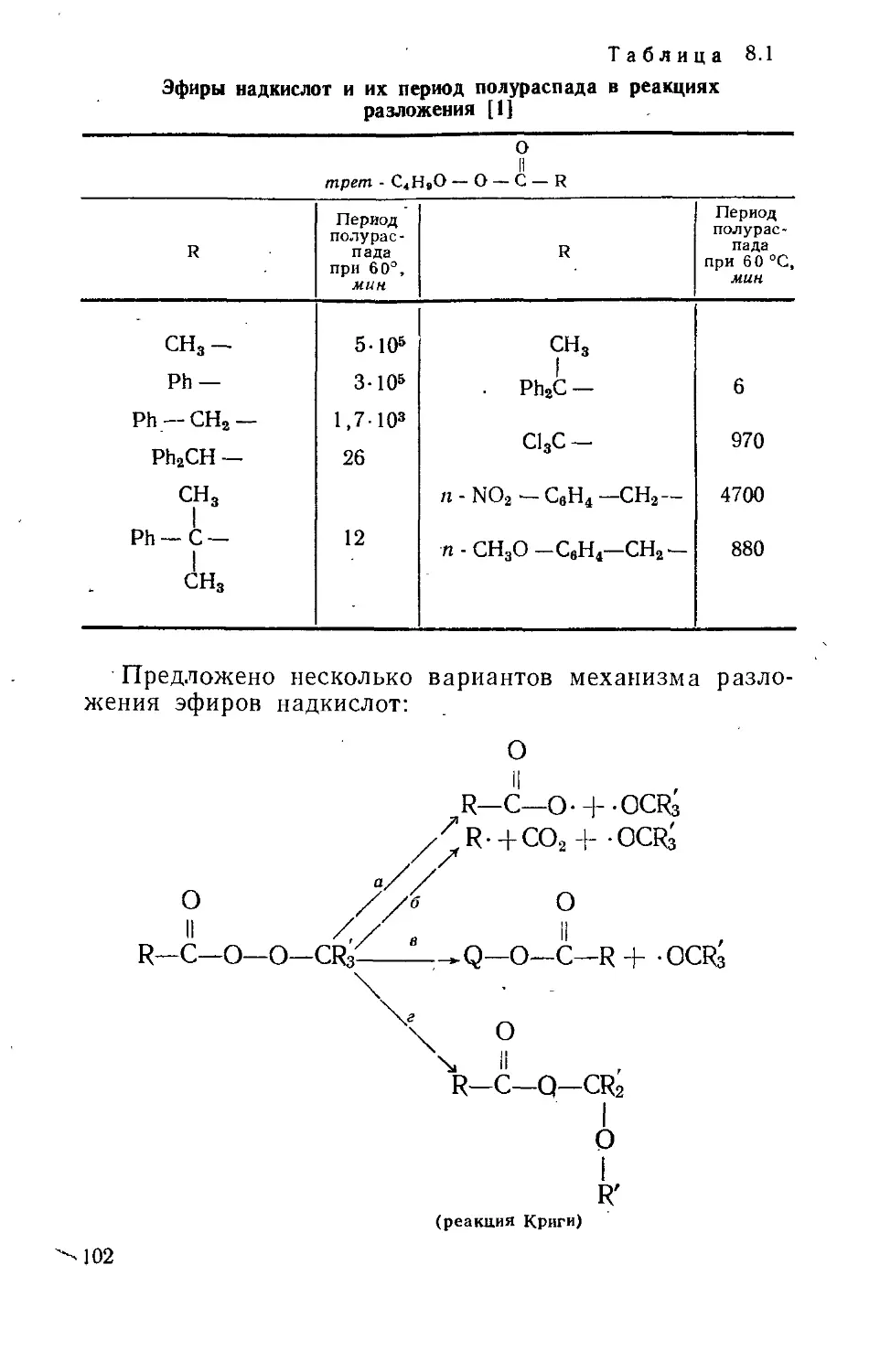


























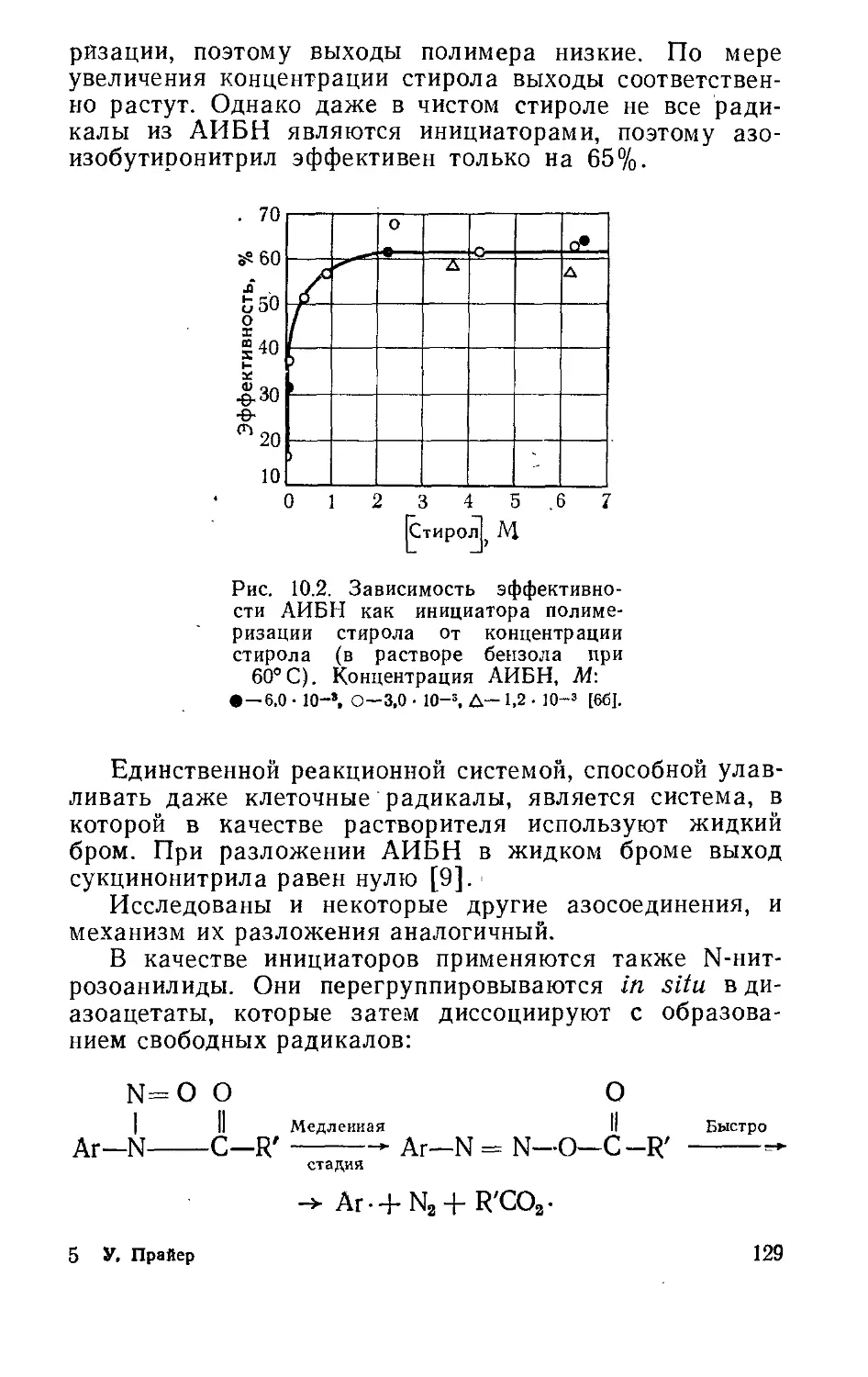






























































































































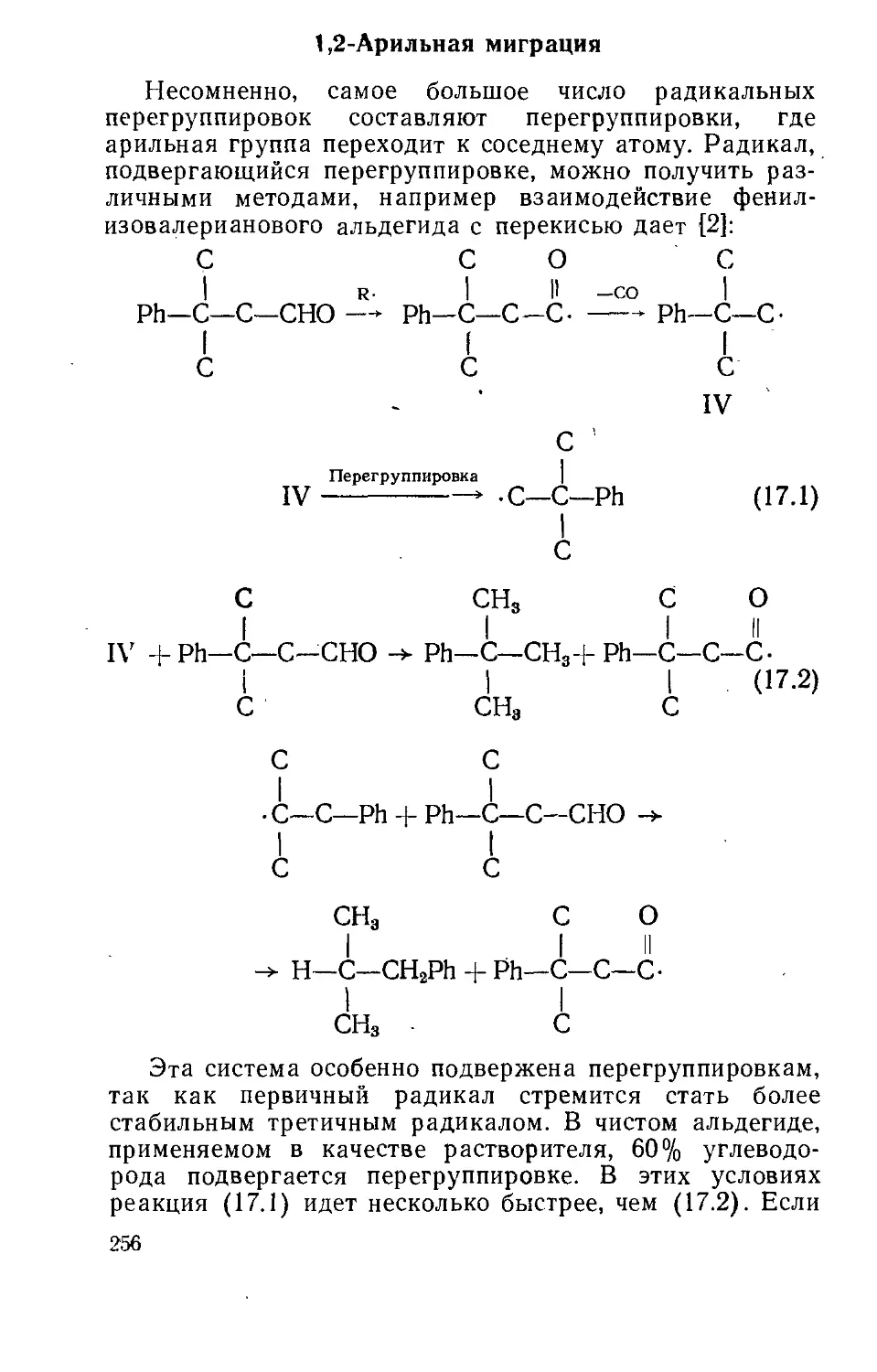



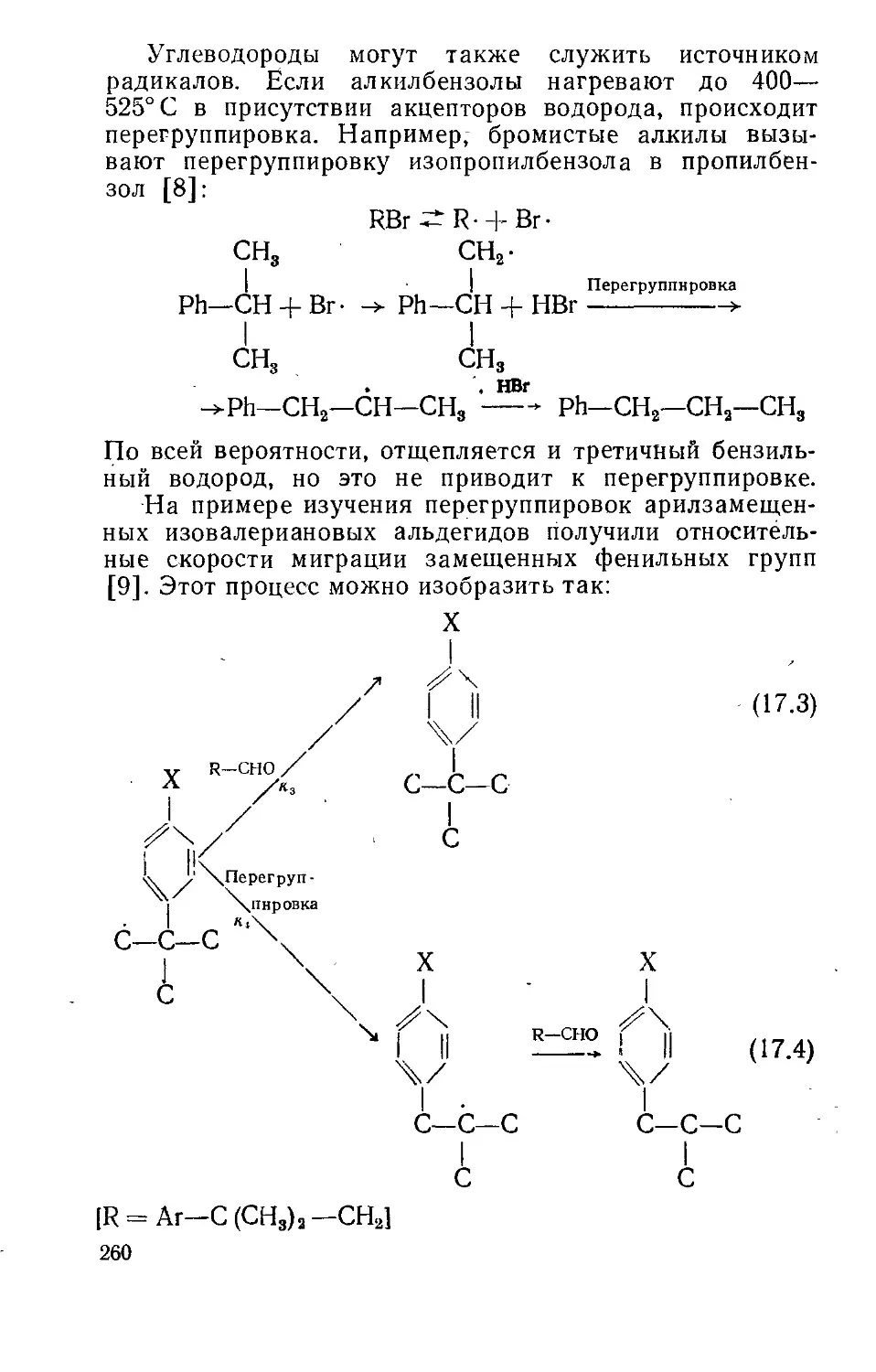



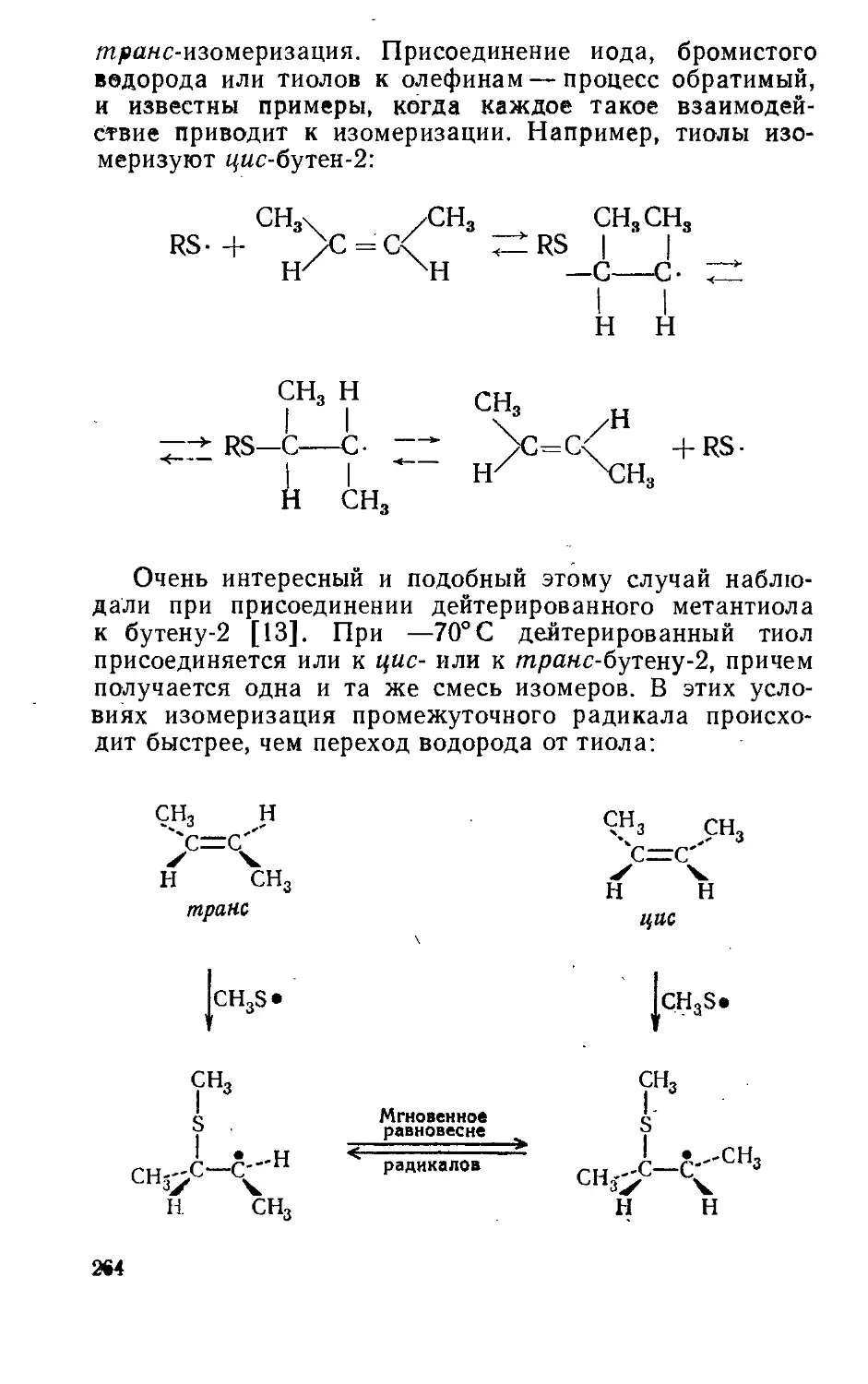






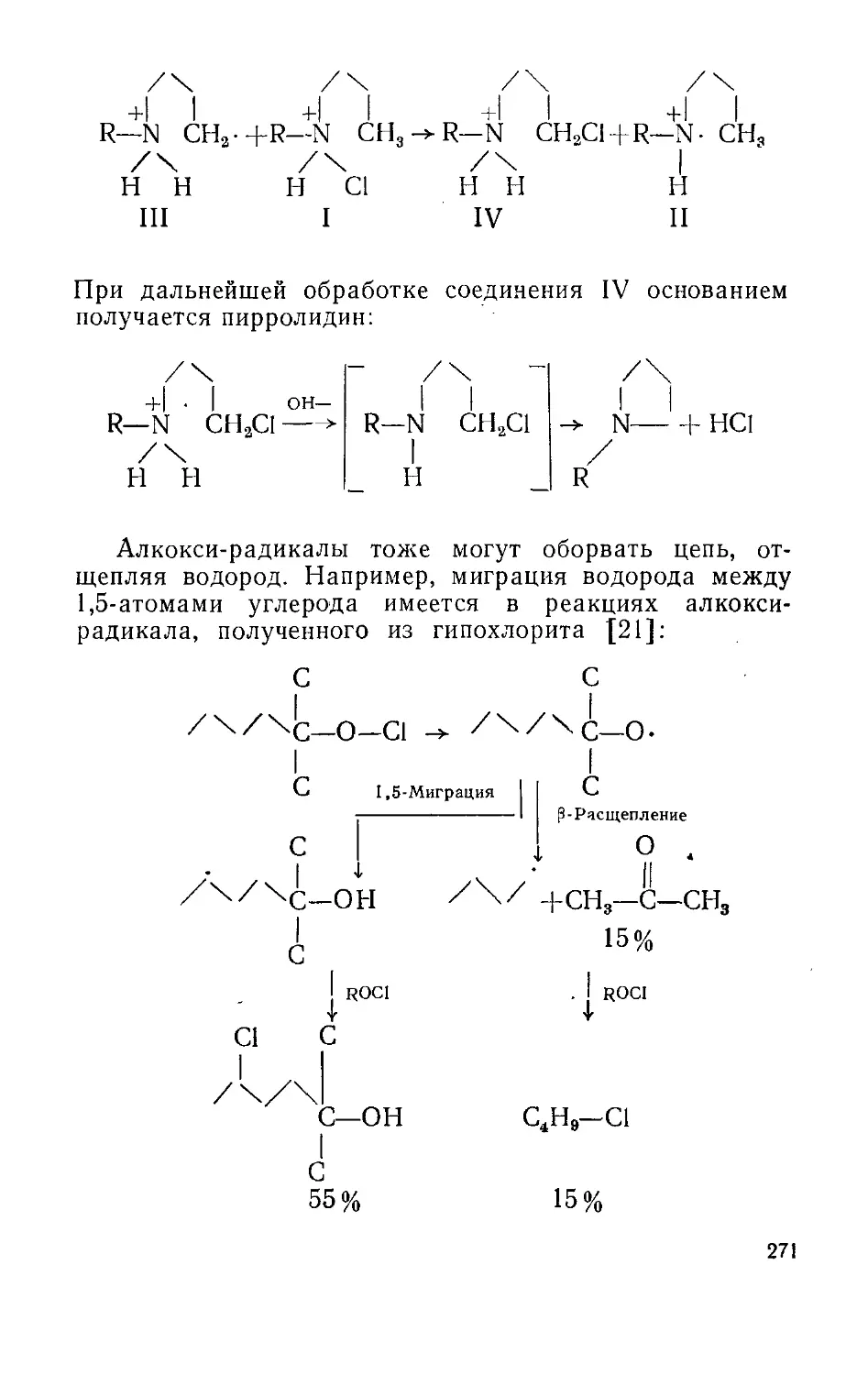

















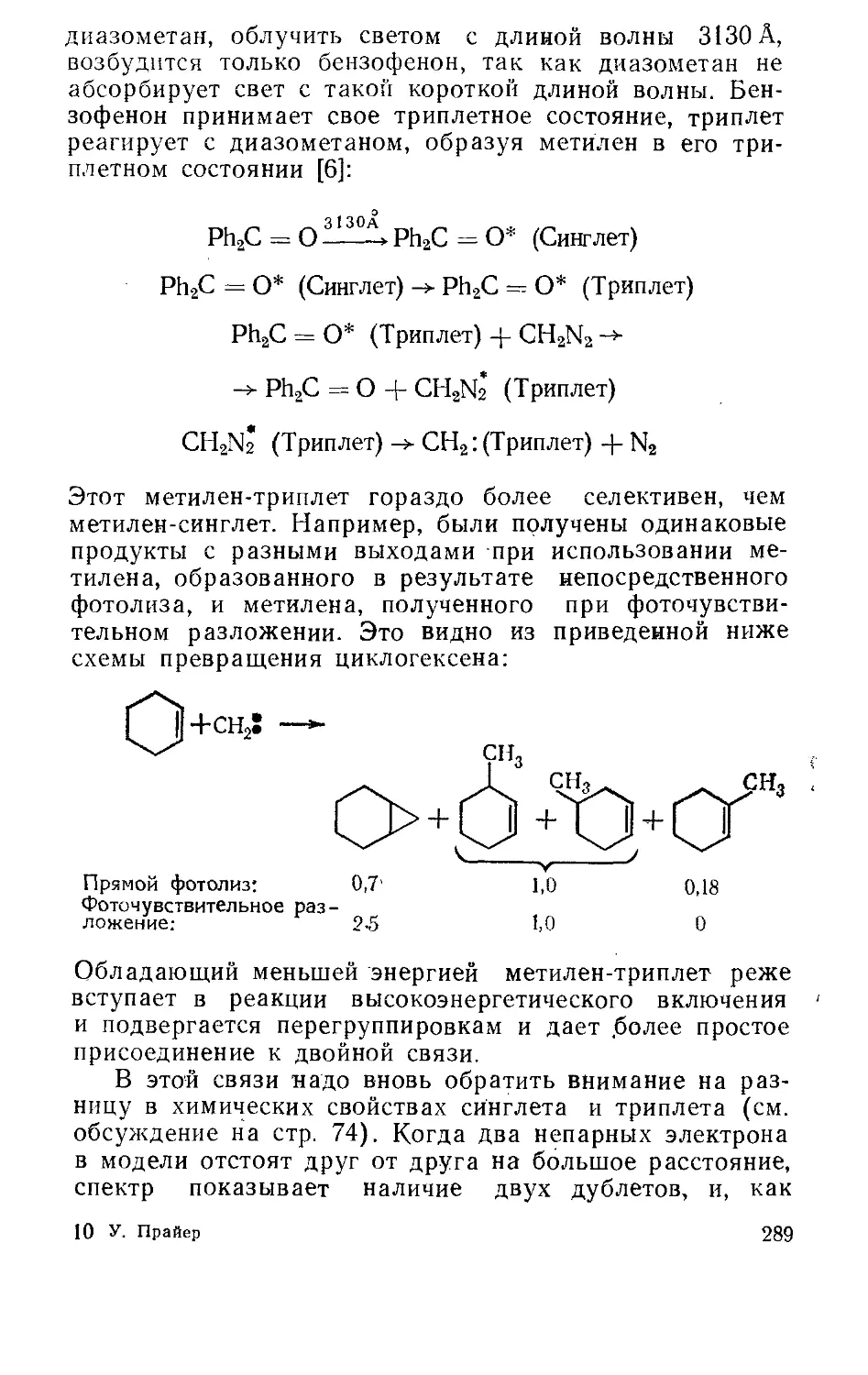





















































Текст
WILLIAM A. PRYOR
FREE RADICALS
MCGRAW-HILL BOOK COMPANY
NEW YORK, ST. LOUIS, SAN FRANCISKO,
TORONTO, LONDON, SYDNEY
У. ПРАЙЕР
СВОБОДНЫЕ
РАДИКАЛЫ
Перевод с английского: А. Ф. Усатого,
Е. В. Дегтерева, Л. Д. Шустова, К. И. Ратмановой
Под редакцией канд. хим. наук
Л. Н. Николенко
АТОМИЗДАТ
Москва 1970
УДК 541.515
П р а й е р У. Свободные радикалы. Перев.
с англ. Атомиздат, 1970 г:, стр. 336
Книга является первой монографией по физи-
ке н химии свободных радикалов — области ис-
следований, бурно развивающейся в последнее
время.
В 1книге рассматриваются природа свободных
радикалов, методы их образования, реакции
с участием свободных радикалов, конечные реак-
ции (реакции присоединения и диспропорциони-
рования, реакции ингибирования) и т. д.
Книга рассчитана на широкий круг научных
работников — физиков, химиков- и биологов. Она
также может служить учебным пособием для
студентов и аспирантов вузов.
Рисунков 28, таблиц 55, библиография —
439 названий.
2-5-2
142—69
ПРЕДИСЛОВИЕ К ПЕРЕВОДУ
Представления о свободных радикалах получили
исключительно широкое распространение в химии, хими-
ческой технологии, химической кинетике, биологии, фи-
зике. Без участия свободных радикалов немыслимы та-
кие процессы, как полимеризация, цепные реакции горе-
ния и медленного окисления, свободнорадикальное
галогенирование, фотохимические и радиационнохими-
ческие реакции. Важную роль играют свободные ра-
дикалы в ферментативных процессах и гетерогенном
катализе.
Особое место занимают стабильные радикалы. По-
мимо их самостоятельной роли они позволяют вести
моделирование и играют роль индикатора процесса при
изучении механизма Химических и биохимических реак-
ций методом электронного парамагнитного резонанса
(ЭПР). Применение стабильных радикалов в сочетании
с методом ЭПР открывает возможности вести самые
различные кинетические исследования и исследования
структуры радикалов.
Книга У. Прайера «Свободные радикалы», как спра-
ведливо указывает сам автор, представляет собой вве-
дение в курс химии радикалов, которая в настоящее
время приобретает полную самостоятельность.
Книга состоит из четырех частей (21 глава). В ней по-
следовательно разбираются природа радикалов, их об-
разование и радикальные реакции. Большое внимание
уделено механизмам реакций и применению свободных
радикалов. Каждая часть и главы имеют внутреннюю
логику в изложении материалов.
Важной частью книги являются упражнения, кото-
рые служат не столько контролем усвоения прочитанно-
го материала, сколько школой активного мышления.
5
Книга вышла из печати в 1966 г. и содержит лите-
ратуру по 1964 г., и только некоторые главы дополнены
материалом до начала 1965 г. За прошедшее со дня
издания книги'время в этой области появилось много
нового. Однако мы не сочли целесообразным изменять
или дополнять настоящую монографию, хотя 'она не ли-
шена недостатков. В книге мало внимания уделено ис-
тории и развитию учения об органических свободных
радикалах и свободнорадикальных процессах в биоло-
гии. Автор практически не затрагивает вопроса о ста-
бильных радикалах. С сожалением должны отметить,
что автор книги отметил весьма мало 'работ русских и
советских ученых. Чтобы в какой-то мере компенсиро-
вать этот недостаток, перевод книги снабжен дополни-
тельным списком литературы, помещенным в конце
книги.
Книга У. Прайера одинаково полезна как для сту-
дентов химических факультетов вузов и техникумов, так
и для аспирантов, научных работников, инженеров и
техников, работающих в области химии, химической
технологии, биоорганической, физической органической
и радиационной химии.
Л. Н. НИКОЛЕНКО
ПРЕДИСЛОВИЕ
Эта книга представляет собой введение в курс хи-
мии радикалов для студентов, прослушавших началь-
ный цикл лекций по органической и физической химии.
Учебник первого года курса органической химии обыч-
но содержит очень мало материала по радикальным
реакциям. С другой стороны, эта область исследований
в настоящее время развивается очень активно. Книга
является попыткой построить мост между многочислен-
ными последними публикациями, относящимися к химии
радикалов, и традиционным начальным курсом органи-
ческой химии.
Хотя история современной химии радикалов имеет
возраст всего около 30 лет, результаты исследований
в этой области уже сейчас имеют огромное зна-
чение.
Радикалы используют в многочисленных реакциях,
имеющих промышленное применение, включая термиче-
ский крекинг нефти и многие процессы получения пла-
стиков. Некоторые биологические процессы, например
дыхание и фотосинтез, включают в себя радикальные
промежуточные продукты.
В этой книге сделана попытка обрисовать целую
область исследований на ознакомительном уровне. Здесь
обсуждаются экспериментальные результаты измере-
ний в газовой и жидкой фазах, результаты изучения
скоростей и продуктов реакций. Рассмотрение ведется
на уровне физической органической химии с акцентом
на механизм и кинетику радикальных реакций. Осве-
щается большое количество проблемных вопросов, а ли-
тературные ссылки на оригинальные работы дают воз-
можность читателю сравнить свой ответ с мнением са-
мих исследователей.
7
Может возникнуть вопрос, соответствует ли приве-
денная литература современному состоянию исследова-
ний. Первый черновик этой монографии был написан
в 1963 г. и был доработан в течение 1964 г., таким об-
разом, большинство литературных ссылок ограничива-
ются сентябрем 1964 г. В частности, в то время не были
опубликованы ни механическая модель бромирования
N-бромсукцинймида (стр. 187), ни представления
о свободных радикалах, образующих галогеновые мос-
тики. Эти вопросы были введены в текст при правке.
С согласия издателя небольшая часть текста была пе-
реписана и добавлены некоторые новые ссылки — на
работы с конца 1964 г. до начала 1965 j.
Очень приятно выразить благодарность моим друзь-
ям и коллегам, которые нашли время прочитать книгу,
сделать критические замечания и помогли отредакти-
ровать некоторые главы. Я особенно обязан профессо-
рам .4. Уоллингу, Р. Дэвнсу, Р. Фаги, К. Райнхарту,
Дж. Трейнхему, Р. Снину, Ф. Скеллу, Дж. Питту,
Г. Расселу, Г. Гриффину, С. Мак-Глину и доктору
Е. Чиуффарин. Майкл Гриффис и доктор Джеймс Шрек
помогали читать гранки. Профессора Д. Де Тар, Е. Ко-
совер, Г. Рассел, П. Скелл и доктор М. Поуцма любезно
позволили прочитать некоторые их книги до публи-
кации.
УИЛЬЯМ А. ПРАИЕР
ЧАСТЬ ПЕРВАЯ
ПРИРОДА РАДИКАЛОВ
Глава 1
ВВЕДЕНИЕ
Двухэлектронная химическая/ связь может разор-
ваться симметрично или несимметрично:
X — Y-+X- + -Y; X —Y->X++:Y-
В первом случае получаются, радикалы, во втором —
ионы. Эта книга посвящена изучению реакций, включа-
ющих радикалы как промежуточные продукты реакции.
Радикальные реакции распространены очень широко
и -имеют чрезвычайно большое значение. Большинство
реакций, инициируемых светом, включая фотосинтез,
являются радикальными. Взрывы, горение, большая
часть процессов галогенирования, многие реакции поли-
меризации и большинство реакций пиролиза также яв-
ляются радикальными. Много реакций с участием
кислорода, в том числе дыхание, имеют радикальный
механизм.
История
В настоящее время термином «радикал» могут обо-
значаться каК различные группы атомов в молекуле,
так и соединение, имеющее нечетное число электронов,
так называемый свободный радикал. Такая двойствен-
ность возникла в прошлом столетии, когда природа и
само существование радикалов были покрыты тайной.
В начале XIX столетия термин «радикал» означал
часть молекулы, и большинство химиков верило, что эти
части способны к независимому, отдельному от моле-
кулы существованию. Однако, когда была развита тео-
рия валентности и выяснилось, что атом углерода нор-
мально четырехвалентен, а методы определения моле-
кулярных весов стали более точными, стало ясно, что
соединения, которые,' как предполагали, были радика-
9
лами, оказались в действительности молекулами. На-
пример, в реакциях, в которых ожидалось получение
стабильного метильного радикала СН3-, в действи-
тельности в качестве продукта был определен этан
СН3—СН3. Противоречие было столь сильным, что при-
близительно до конца прошлого века считалось, что
«свободных» органических радикалов не существует.
Хотя было известно, что атомы , иода, натрия и некото-
рых других элементов существуют в газовой фазе в
виде радикалов, считалось, что органические радикалы
недостаточно стабильны, чтобы их выделить в изолиро-
ванном состоянии. Два исследователя опровергли эту
крайнюю точку зрения.
В 1900 г. М. Гомберг пытался получить гексафенил-
этан, воздействуя серебром или цинком на раствор три-
фенилхлорметана. Вместо ожидаемого стабильного со-
единения он получил раствор желтого цвета, который
быстро обесцвечивался под воздействием воздуха, иода
и некоторых других веществ, которые, как теперь изве-
стно, быстро реагируют с радикалами. Гомберг сделал
правильный вывод, что он имеет дело с растворами
поглощающего свет трифенилметильного (тритильного)
радикала:
PhsC — Cl + Ag -+ Ph3C • -J- AgCl
2Ph3C- ^Ph3C — CPh3
Реакция обесцвечивания, которую наблюдал Гомберг,
является типичной реакцией радикалов со своим акцеп-
тором, например молекула иода реагирует с радикала-
ми и при этом выделяется атом иода
Ph3C • 12 —> Ph3C —I -|- I •
Окрашен Не окрашен
В ,1929 г. Ф. Панет представил тщательно получен-
ные данные, которые недвусмысленно свидетельствовали
о том, что радикалы могут существовать в газовой фа-
зе. На рис. 4.1 показана схема ^использованной Панетом
установки. Азот под давлением 1—2 мм рт. ст. пропус-
кался в объем, где он насыщался парами тетраметил-
свинца. Газовая смесь затем увлекалась в ту часть си-
стемы, которая могла нагреваться до 450 °C передвиж-
ной печью, при этом на стенках вследствие разложения
тетраметилсвинца образовывалось свинцовое зеркало.
Газообразные продукты разложения двигались далее по
10
трубке на расстояние 5—30 см и попадали на предва-
рительно приготовленное металлическое зеркало, нагре-
ваемое до 100° С. Панет наблюдал, как второе зеркало,
состоящее из цинка, сурьмы или свинца, постепенно ис-
чезало. Эти эксперименты свидетельствовали о том, что
Нзот
Передвижная
450°С
Жидкий
Предварительно
нанесенное
зеркало
Ловушка
---*-К насосу
тетраметилсвинец
Рис. 1.1. Схема установки Ф. Панета,
тетраметилсвинец разлагается при ,450° С с образова-
нием метильных радикалов:
(СН3)4РЬ—-4СН3- + РЬ
Далее радикалы реагировали со вторым металлическим
зеркалом с регенерацией соответствующего алкилме-
талла:
4СН3- +РЬ-^(СН3)4РЬ
2СН3 • -|- Zn -> (СН3)2 Zn
После проведения этой изящной работы были обна-
ружены и идентифицированы другие радикальные ре-
акции. В 1937 г. Хей и Уотерс в Англии и Караш, Эн-
гельман и Майо в США опубликовали детальные
механизмы некоторых реакций, которые до этого трудно
было объяснить. Наиболее примечательно, что был
идентифицирован радикальный механизм реакций, не
подчиняющихся правилу Марковникова — присоединение
бромгидрата к олефинам и гомолитическое ароматиче-
ское замещение. В том же году Флори впервые предпо-
ложил наличие теперь уже признанного радикального
механизма для процесса привитой полимеризации. Та-
ким образом, начало современной истории химии ради-
калов может быть датировано 1937 ,г.
Вторая мировая война побудила многие страны раз-
вивать производство синтетических материалов, так как
природного сырья не хватало; многие из этих синтети-
11
ческих материалов стали получать с помощью процес-
сов радикальной полимеризации. В качестве примера
можно привести производство синтетической резины из
стирола и бутадиена. Производство стирола выросло от
отдельных опытов до большой индустрии, которая про-
извела буквально переворот в развитии промышлен-
ности. С открытием неопрена, полиэтилена и других
пластиков химия радикалов достигла индустриального
значения и стала областью огромных вкладов в Иссле-
дования.
> Определения
В настоящее время свободным радикалом называ-
ют атом или группу атомов, обладающих неспаренным
электроном. Бирадикал— это соединение с двумя неспа-
ренными электронами. Исходя из этого определения,
атомы хлора и натрия, окись азота, метил СН3- яв-
ляются радикалами. Метильные радикалы и атомы хло-
ра реагируют с углеводородами одинаково*:
СН3- -MR-H->CH4+R-
Cl- +R- H->HC1+R-
Реакции щелочных металлов с органическими соеди-
нениями могут приводить к образованию радикалов.
Например, кетоны подвергаются одноэлектронному вос-
становлению до ион-радикалов, которые затем димери-
зуются :
О 0“
II I
R—С—R + Na • -> R—С • -|-Na+
i
R
0“ О-О- ОН ОН
1 I I но I I
2R-C- -> R-C - С—R—!2> R—С—С—R
R R R R R
* Условимся в атоме хлора и в метильном радикале неспарен-
ный электрон обозначать в виде точки. Заметим также, что терми-
ны «метил», «метильный радикал» и «метильный свободный ра-
дикал» — все могут быть использованы для обозначения СНз«. Ме-
тилен СНг: имеет два неподеленных электрона, которые могут, быть
спарены или не спарены.
12
Как показано выше, радикалы не обязательно ней-
тральны. Одноэлектронное восстановление кетонов при-
водит к появлению отрицательных ион-радикалов. По-
ложительные ион-радикалы образуются в камерах мас’с-
спектрографов при взаимодействии быстрых электронов
с органическими молекулами — происходит отрыв ва-
лентных электронов:
R:R + e-->R-R++2e-
При реакции нафталина с натрием образующийся про-
дукт реакции представляет собой окрашенный ион-ра-
дикал нафталина:
| II J + Na •-> I || I Na+
Основные типы радикальных реакций
Радикальные реакции можно разделить на три ос-
- новных типа: реакции, в которых радикалы образуются;
реакции, в которых меняется местоположение радикала,
и реакции, в которых радикалы исчезают. Полезно рас-
смотреть некоторые примеры каждого из этих процес-
сов.
Механизмы процессов образования и исчезновения
радикалов очень просты. Радикальные соединения об-
разуются парами при разрыве двухэлектронной связи *,
например при диссоциации перекисей
ROOR->2RO-
фотолизе галогенов
или разложении азосоединений
СН3 — N = N — СН3-> 2СН3- +N2
Радикалы могут исчезать также парами. Этот про-
цесс называют рекомбинацией радикалов или атомов.
* Радикалы могут образовываться индивидуально при неко-
торых электродных процессах.
13
Например, завершающей стадией реакции хлорирования
является соединение двух атомов хлора
2С1 • С12
Метильные радикалы исчезают при взаимном связыва-
нии с образованием этана:
2СН3 • -> СН3 - СН3
Однако высшие алкильные радикалы исчезают в ре-
зультате двух конкурирующих процессов рекомбинации
и диспропорционирования. При диспропорционировании
атом водорода перемещается от одного радикала к дру-
гому. Этильный радикал исчезает, например, по реак-
циям
2СН3— СН2 • СН3 — СНа — СН2 — СН3
Диспропорциями-
2СН3 — сн2 • —сн3 - сн3 + сн2 =. сн2
Для изопропильного радикала конечными реакциями
являются
сн3 сн3 сн3
I I I
2СН3—С - сн3-сн—сн—сн3
н
сн3
2СН3- с - сн3—сн2—сн3 + сн3—сн = сн2
н
Последовательность радикальных реакций услож-
няется тем, что большинство из них являются цепными
процессами. Реакционноспособные радикалы образуют-
ся в реакциях инициирования, и затем по цепной после-
довательности образуются продукты реакции до завер-
шающей стадии — обрыва цепи. Например, хлорирова-
ние углеводородов RH
Инициирование:
С12 — -2C1. (1.1)
14
Рост цепи (развитие):
Cl- +RH->R- + НС1
R. + C12->R-CI + Cl-
Обрыв цепи:
2С1 • -> С12
2R- ->R — R
Cl - +R. ->R — Cl
(1-2)
(1-3)
(1-4)
(1-5)
(1-6)
Молекулы хлора поглощают свет и активируются в до-
статочной степени, чтобы рвалась связь CI—С1. Цепная
реакция развивается в результате двух цепей переноса:
первая заключается в замещении атома хлора на водо-
род с образованием алкильного радикала; вторая — во
взаимодействии алкильного радикала с молекулой хло-
ра. Заметим, что сумма реакций (1.2) и (1.3) есть
R —Н + С12-> R-Cl+ НС1 (1.7)
Реакции (1.2), (1.3) представляют собой обменные
реакции, аналогично ионным обменным реакциям. По-
этому удобно рассматривать эти реакции как гомоли-
тические обменные и использовать для их обозначения
символ 8ц2 (обменная, гомолитическая, бимолекуляр-
ная). Этот символ предполагает аналогию между нук-
леофильной реакцией замещения, которая обычно обо-
значается как SN2 или вальденовским инверсионным
процессом и процессом 5Н2. Процесс Зл-2:
R —X + СГ->R —С1 + Х~ (1.8)
был тщательно исследован. Как известно, механизм это-
го процесса предусматривает атаку нуклеофильным
агентом со стороны, противоположной замещаемой свя-
зи центрального атома углерода («атака с тыла»). Этот
механизм включает также изменение конфигурации цен-
трального атома углерода (вальденовское обращение),
при этом меняется характеристический профиль группы
R [1].
Однако большинство реакций SH2 наблюдается с
одновалентными атомами, и изменение центра, связан-
ного с оптической активностью, нельзя использовать
для изучения стереохимии реакции. Так, в уравнении
(1.2) реакция SH2 предусматривает замещение атома
15
водорода, а реакция (1-3)—замещение хлора. Исходя
из этих соображений, стереохимию реакций SH2 экспе-
риментально наблюдать очень трудно. В настоящее вре-
мя достоверно не известно, связано радикальное заме-
щение у многовалентных атомов с «атакой с тыла» и
вальденовским обращением или с «фронтальной» атакой
и сохранением конфигурации. Все-таки, как. мы увидим
в гл. 12, имеются веские данные, свидетельствующие о
том, что радикальные реакции типа SH2 включают ме-
ханизм «атаки с тыла», подобно тому, как это известно
для ионных реакций SN2.
Природа стадии роста цепи
Радикальные цепные реакции состоят из начальной
стадии — инициирования, в процессе которой образу-
ются радикалы; стадии роста (развития) цепи, в кото-
рой образуются продукты реакции, и завершающей ста-
дии— обрыва цепи. Стадия роста цепи представляет
большой интерес, так как она обычно является ключом
для понимания природы образуемых продуктов. В ре-
акциях развития цепи меняется место локализации
неспаренного электрона, однако число радикальных об-
разований не меняется *.
Реакции развития цепи подразделяются на четыре
основных типа: |1) перенос атомов; 2) присоединение;
3) фрагментацию и 4) перегруппировку.
1. Реакции переноса атома являются обычно реакци-
ями отщепления водорода или галогена. Например,
R- +R'H->RH + R'- R- +HC1->RH + C1.
Cb+RH->HC1+R. R-+R'C1-э-RCl + R'-
r- +cci;^rci+ -ecu
* При некоторых радикальных процессах число радикалов мо-
жет возрастать в стадии роста цепи. Такие реакции называют раз-
ветвленными — они имеют место при горении и взрыве. Например,
при некоторых условиях водородно-кислородное пламя включает
ступень Н--f-Oz-j-HO'-f-O". В этой книге не будут рассматри-
ваться разветвленные цепные реакции; для ознакомления с этим
вопросом читатель может обратиться к специальной литературе
12, 3].
16
2. Реакции присоединения включают в себя присо-
единение радикалов к обычным олефинам:
С1 • + RCH = СН2 RCH - СН2С1
СН3 • + RCH = СН2 -> RCH — СН2 — СН3
Присоединение происходит таким ^образом, чтобы обра-
зовать более стабильный радикал. В приведенных при-
мерах вторичный по отношению к замещаемой связи
радикал образуется более предпочтительно по сравне-
нию с первичным. Для алифатических радикалов ста-
бильность их возрастает в последовательности первич-
ный< вторичный <третичный.
Стадия роста в радикальной полимеризации олефи-
нов является также реакцией присоединения. В этом
случае полимерный радикал, содержащий п олефиновых
мономерных звеньев, присоединяет к себе еще одно зве-
но олефина, увеличивая цепь до п+1 звена. Это присо-
единение может быть представлено * процессом, пока-
занным для олефина CH2 = CHR:
R
R' - (СН2 - d Н)я • + СН2 = CHR
R R
I I
R' — (СН2 — СН)„ — СН2 - СН •
В этом случае направление присоединения также тако-
во, что предпочтительнее образование более стабильного
вторичного радикала, а не менее стабильного первич-
ного. Реакцию, показанную выше, можно представить
в виде
М„- + М->М„+1 •
где М — молекула полимеризуемого олефина (называе-
мая мономером) и Мп-—полимерная цепь, содержа-
щая п мономерных единиц.
В другом написании концевое звено можно показать
отдельно, а остальную полимерную цепь можно обозна-
чить сплошной-волнистой линией:
~CHR + СН2 = CHR -> ~ CHR — СН2 — CHR
* Природу группы R' при этом рассмотрении учитывать не обя-
зательно. Мы вернемся к этой теме на стр. 224.
17
Стадия конденсации в реакциях замещения ароматиче-
ских
ния:
соединений также является реакцией присоедине-
3.
R'
Акцептор
водорода .
Присоединение
R
Реакции фрагментации. Большинство реакций
фрагментации представляют собой p-расщепление, в ко-
тором неспаренный электрон образует связующую пару
с одним из электронов р-связи,
радикальный фрагмент:
при этом выделяется
реакции присоедине-
Эта реакция является обратной реакции присоедине-
ния. Вообще имеется три электронных пары, которые
могут принимать участие в p-расщеплении. Эксперимен-
тально было определено, что происходит преимущест-
венно p-расщепление, что приводит к появлению наи-
более стабильных радикалов. Например, радикал алко-
голят, показанный ниже, претерпевает три различных
p-расщепления, в результате получается трн набора
различных продуктов:
О
<0,5%
О
3%
сн3«
С2Н5.
снх
СН-
СН<
— с — с—
R
О
+
+
С = С
18
95%
Как показано, количество продуктов [4] симбатно со
стабильностью образующихся продуктов: СзН?«>
>С2Н5->СН.3.
4. Реакции перегруппировки. Иногда стадия роста
цепи представляет собой перегруппировку радикалов,
например [5]:
Н
I
CI • +снз—С—СН2С1 -> СН3—С—СН2С1 + HCI
н н
сн3-с-сн2С1^^вк4
н
С1
I .
сн3—с—сн2
I
н
С1 С1
I I
СН3—С—-СН2 • +НС1 -> сн3-с—сн3 + С1 •
н н
Полярные характеристики радикалов
Первое приближение состоит в том, что радикалы
считаются электрически нейтральными и, следовательно,
нр подвержены полярным влияниям, существенным
в случае ионных реакций. С этой точки зрения ради-
кальные реакции должны были мало зависеть или
совсем не зависеть от влияния растворителя и не зави-
сеть от электронной природы реагирующих веществ.
Иногда эта упрощенная точка зрения подтверждается,
но ;часто радикальные реакции заметно подвержены по-
лярным влияниям.
Примером нечувствительности к полярному окруже-
нию в радикальных реакциях может служить радикаль-
ная диссоциация перекиси трет-бутила. Целесообразно
сравнить ее с ионной диссоциацией алкилгалогенидов.
Перекись mpe/n-бутила диссоциирует с одинаковой ско-
19
ростью как в газовой фазе, так и в растворе бензола,
кумола, аминов и многих других растворителей:
ROOR-> [RO . . .OR] -> 2RO-
Переходное состояние
где R = трет-С4Н 9
По-видимому, в переходном состоянии происходит не-
большое разделение зарядов, и растворение в полярных
растворителях только в небольшой степени Способствует
диссоциации. Эта ситуация сильно отличается от ион-
ной диссоциации алкилгалогенидов, происходящей толь-
ко в тех растворителях, которые могут сольватировать
образующиеся ионы:
R — Cl —> [R5+ . . . С15~] -»• R+ (сольват) + С1~ (сольват)
Расчет показал, что диссоциация трет-бутилхлори-
да происходит в 1О100 раз' быстрее в водном растворе,
чем в газовой фазе. Нечувствительность к природе рас-
творителя настолько типична для радикальных реакций,
что часто используется в качестве доказательства того,
явдяется вновь открытая реакция радикальной или
ионной.
Однако многие радикальные реакции подвержены
полярному влиянию другого рода, чем ионные реакции.
Как можно было ожидать, полярное влияние обычно
проявляется в реакциях соединений, обладающих раз-
личной электроотрицательностью. По-видимому, про-
стейшим примером этого положения может служить тот
факт, что наиболее предпочтительно рекомбинируют
два радикала, отличающиеся по типу полярности. Если
два различных радикала присутствуют в одной реакци-
онной системе, возможны три различные комбинации
реакций:
2А- — АА
2В- — ВВ
А- +В- — АВ
Отношение констант
• — 1/ •
(каак1>1>) г
20
Из статистических соображений можно ожидать, что ф
могло бы достигать величины, равной 2 *. В согласии
с этим было определено, что <р = 2 в случае рекомбина-
ции радикалов одинаковой полярности, например, ког-
да один радикал этил, а другой — пропил. Однако при
комбинированных реакциях рекомбинации между двумя
радикалами, отличающимися электроотрицательностью,
часто оказывается, что <р>2. При окислении некоторых
* Тот факт, что это отношение статистически равно 2, наибо-
лее очевиден при рассмотрении случая, когда концентрации А*
и В* равны, хотя это рассмотрение справедливо при любых концен-
трациях. Определим скорости образования продуктов АА, ВВ и
АВ, как показано ниже:
d
[АА]=каа[А-] [А-];
d
Явв = ^-[ВВ1 = «»ЯВ ] [В-];
d
/?дв = & [АВ] = Kat) [А• ] [В•].
Данное количество радикалов А- имеет одинаковую вероятность
встречи как с А-, так и с В-. Если мы предположим, что все соу-
дарения одинаково эффективны, тогда в единицу времени A* pay
дикалов образует одинаковое количество продуктов АА и АВ. Из
тех же соображений, если все соударения одинаково эффективны,
данное количество радикалов В» образует одинаковое количество
продуктов ВВ и АВ. Таким образом, во всей системе продукт АВ
образуется со скоростью, вдвое большей, чем АА или ВВ:
^Ав = 2«АА “ 2^вв>
отсюда 1
(^АА'^Вв)
используя приведенные выше обозначения, получим
^АВ кпЯА]-[В-]
(Каа-Явв)17* {«aaIA-1 [A]KW[B][B.]}*'«
1 / • *
(^аак/>й) *
Если соударения одинаково эффективны и только статистика опре-
деляет отношение количеств, получаемых продуктов, - то <р должно
быть равным 2.
21
соединений молекулярным кислородом в системе при-
сутствуют как радикалы R-, так и ROO-.
Реакции роста цепи представляют собой
R- +OS->ROO-
ROO- +RH^ROOH +R-
а тремя завершающими реакциями являются
2R • Нерадикальные продукты
2ROO • -> Нерадикальные продукты
R- +ROO- -> Нерадикальные продукты
Значение <р>2 часто имеет место в этих окислительных
системах. Наиболее вероятен перекрестный обрыв це-
пи, так как алкильные и перекисные радикалы разли-
чаются электроотрицательностью; переходное состояние
в перекрестной завершающей "реакции стабилизируется
резонансными полярными формами, в которых пере-
кисный радикал несет отрицательный заряд:
[ROO- R-<—ROO:-+R]
Переходное состояние перекрестного обрыва цепи
Перекрестный обрыв цепи часто доминирует над
однородным при радикальной сополимеризации двух
олефинов. При сополимеризации стирола CH2=CHPh
с бутилакрилатом СН2=СН—СО—С4Н9 радикал со сти-
рольным звеном на конце предпочитает радикал с
акрилатной концевой группой *:
II
С-ОС4Н9
•С~
I
н
Действительно, при сополимеризаций стирол—бутил-
акрилат, <р имеет очень большое значение, равное 150.
Предпочтительность перекрестного завершения реакции
* Предполагается, что только природа конечного олефинового
звена определяет реакционную способность полимерного радикала.
22
следует из того, что стирольный радикал является от-
носительно хорошим электронным донором, а акрильный
радикал — электронным акцептором. Переходное состо-
яние перекрестного обрыва цепи стабилизируется резо-
нансными формами, в которых электрон передается от
одного радикала к другому:
О
I
С-ОС4НЭ
Полярные эффекты часто также влияют на ради-
кальные реакции замещения. Приведенные ниже данные
показывают относительные скорости, с которыми атом
хлора отщепляет водород в различных положениях в
бутане и Д-фторбутане:
СН3—СН2—СН2—СН3 CH2F—СН2—СН2—СН3
1,0 3,7 3,7 1,0 0,9 1,7 3,7 1,0
Данные для бутана иллюстрируют известную пред-
почтительность, с которой происходит отщепление атомов
водорода вторичной С—Н-связи по сравнению с первич-
ной. Такая селективность связана с тем фактом, что вто-
ричные связи С—Н слабее первичных, и вследствие этого
атомы водорода вторичной С—Н-связи более лабильны.
В 1-фторбутане эта нормальная селективность в от-
щеплении атомов водорода вторичмой С—Н-связи вид-
на из данных по относительным скоростям воздействия
на атомы водорода, наиболее удаленные от замести-
теля— атома фтора. Однако скорость отщепления
атомов водорода от углерода в 0-положении по отноше-
нию к заместителю изменилась на 1,7/3,7, или на 46%,
против ожидаемого значения, а скорость отщепления
атомов водорода от углерода в a-положении составляет
0,9/1,0, или 90% ожидаемого значения.
Так как атомы водорода и фтора близки по разме-
рам, маловероятно, что такое изменение скорости про-
исходит в результате стерических эффектов. Более при-
емлемое объяснение состоит в том, что электроотрица-
тельный атом хлора обладает электрофильными свой-
ствами (старается присоединить электрон); так что
он имеет тенденцию преимущественно атаковать те
23
атомы водорода, у которых относительно большая плот-
ность электронов. Атом фтора в качестве заместителя
(в 1-фторбутане) с его большим электроноотталкиваю-
щим индуктивным эффектом понижает электронную
плотность вблизи атомов водорода и делает их менее
восприимчивыми для атаки электрофильным атомом
хлора.
Упражнения
1.1. Показать цепную стадию- в фотобромнровании метана.
1.2. Напишите все возможные продукты при 0-расщеплении
радикалов, показанных ниже. Какие продукты можно ожидать
в наибольшем количестве.
а) СН3—СН2—С—СН2—СНа—СН3
СНа-СН (СН3)а
б) СН3—СН2—О-
в) PhCHa—СН2—СН—СН2—СН3
1.3. При окислении углеводородов иногда наблюдают преиму-
щественно перекрестную терминальную реакцию. Например, при
окислении этиллинолеата <р=3 [6]. Обозначив два имеющихся ра-
дикала как R« и ROO-, опишите переходное состояние для трех
терминальных реакций. Объясните предпочтительность перекрест-
ной терминальной реакции.
1.4. Изобразите структуры ; 1-фторбутана, иллюстрирующие
электроноотталкивающие свойства атома фтора. Напишите пере-
ходное состояние реакции атома хлора с атомом водорода при
0-атоме углерода в 1-фторбутане; изобразите электронную струк-
туру переходного состояния, которая объясняет эффект замедле-
ния скорости отщепления при наличии заместителя — атома фтора.
Сопоставьте возможные объяснения тому факту, что скорость за-
мещения атомов водорода при 0-атоме углерода больше, чем для
атомов водорода при а-атоме углерода [7].
ЛИТЕРАТУРА I
1. Streitwieser A. (Jr.) Solvolic Displacement Reactions.
McGraw-Hill Book Company. N. Y., 1962, p. 5.
2. Benson S. W. Foundations of Chemical Kinetics. McGraw-Hill
Book Company, 1960, p. 427.
3. Moore W. J. Physical Chemistry. Preutice-Hall. Inc. Englewood
Cliffs. N. Y„ 3d ed., 1962, p. 289.
4. Green F. D. et al. J. Org. Chem., 28, 55 (1963).
5. В en s on H. L, W i 11 a r d J. E. J. Amer. Chem. Soc., 83, 4672
(1961).
6. ButemanL. Quart. Rev., 8, 152 (1954).
7. G a 1 i b a J., T e d d e r J. M., Watson R. A. J. Chem. Soc.,
1321 (1964).
24
Глава 2
РЕГИСТРАЦИЯ РАДИКАЛОВ
Возникает вопрос: имеются ли доказательства того,
что соединения, обладающие неспаренным электроном,
действительно существуют? В случае некоторых ста-
бильных радикалов, например NO2, 'доказательством
служат обычные химические данные. Для более реак-
ционноспособных радикалов, как это видно из работ
Гомберга и Панета, можно использовать косвенные со-
ображения. Однако, используя магнитные свойства
неспаренного электрона, можно получить прямые дан-
ные о существовании соединений с неспарениым элек-
троном.
Магнитная восприимчивость
Большая часть веществ диамагнитна: если их по-
местить между полюсами магнита, появляется сила,
выталкивающая их из магнитного поля. Эта сила возни-
кает вследствие того, что все спаренные электроны
молекул рриентируются таким образом, чтобы компен-
сировать внешнее магнитное поле. Напротив, вещества,
обладающие неспаренными электронами, парамагнитны,
они втягиваются в магнитное поле. В этих веществах
вклад парамагнетизма неспарениых электронов проти-
востоит вкладу в диамагнетизм всех спаренных элек-
тронов.
Полную магнитную восприимчивость" вещества мож-
но измерить по действующей на него выталкивающей
силе в магнитном поле. Самым первым прибором, спо-
собным измерять эти эффекты, были весы Гюи, создан-
ные в 1889 г. В 1910 г. Паскаль сделал заключение, что
диамагнетизм молекул является простой аддитивной
функцией составляющих их элементов. Поэтому, если
определено, что фактически измеренная магнитная вос-
приимчивость вещества меньше, чем это предполагается
из правила аддитивности Паскаля, то разницу можно
отнести в счет парамагнетизма неспаренных электронов.
Например, этим методом было показано, что гексафе-
нилэтаи на 2% диссоциирован на радикалы при 20°С
в бензоле [1],
25
Фактический расчет представляет собой следующее:
ед. СГС
Молярная восприимчивость 5%-ного раствора
Ph6C2 в бензоле............................... —272-10— 8
Восприимчивость, рассчитанная при использова-
нии констант Паскаля..........................—(—325-10~’ )
Вклад парамагнетизма ........................ —J-53-10—8
Раствор обладает меньшей отрицательной восприимчи-
востью, чем рассчитано, так как радикалы дают поло-
жительный парамагнитный эффект. Известно, что при
20° С 1 моль радикалов имеет восприимчивость, равную
1270-10-6 ед. СГС. Поскольку каждая диссоциирован-
ная молекула образует два радикала, то гексафенилэтан
диссоциирован на 53/(2-1270) =2,1 % при 20° С. Необ-
ходимо заметить, что этому методу присуща малая точ-
ность, так как положительная величина парамагнитного
вклада получена в результате вычитания двух величин,
близких по значению.
Электронный парамагнитный резоиаис (ЭПР)
Начиная с 1945 г. получила развитие эксперимен-
тальная техника, позволяющая непосредственно изме-
рять парамагнетизм неспаренных электронов. Этот
метод называется электронным парамагнитным резо-
нансом или электронным спиновым резонансом. * Изме-
рения методом ЭПР обладают чрезвычайно высокой
чувствительностью регистрации радикалов. В настоящее
время можно измерить такие низкие концентрации, как
IO-8 моль радикалов. Эта методика продолжает улуч-
шаться — повышается чувствительность.
Электронные 'приборы, используемые для регистра-
ции спектров ЭПР, достаточно сложны и не рассматри-
ваются в этой книге. Теория электронного парамагнит-
ного резонанса представляет определенные трудности
для понимания и требует знания квантовой механики,
здесь мы дадим упрощенное описание этого явления.
Сигнал ЭПР появляется вследствие того, что элек-
трон имеет спин, с которым связан соответствующий
магнитный момент. При наложении внешнего магнит-
ного поля электронный магнитный момент может ори-
ентироваться «по» или «против» поля, в соответствии
* Явление ЭПР открыто советским ученым Е. К. Завойским.
26
с этим электрон может находиться на одном из двух
энергетических уровней. На рис. 2.1 показано это изме-
нение, когда одиночный электронный энергетический
уровень расщепляется магнитным полем на два под-
уровня. Линия поглощения в спектре ЭПР связана с
переходом между этими двумя подуровнями. Частота
v, при которой наблюдается эта линия поглощения,
дается выражением
v=^, (2.1)
Л
где g— константа пропорциональности, называемая ги-
ромагнитным отношением; р— постоянная, называемая
магнетоном Бора и связывающая механический момент
Рис. 2.1. Электронный переход в магнитном
поле Н.
электрона с его магнитным моментом; Н — напряжен-
ность внешнего магнитного поля; h — постоянная План-
ка. Для свободного электрона g имеет значение 2,0023;
в органических радикалах g варьирует от 2,002 до 2,006.
Подставив значение констант в уравнение (2.1), полу-
чим
v = 2.8026Я, (2.2)
где Н — напряженность поля, ас; v — частота, МгЦ. Как
видно из уравнений (2.1) и (2.2), при использовании
большей частоты для осуществления перехода необхо-
димо увеличивать напряженность магнитного поля Н.
Практически обычно используют поле около 3000 гс, в
этом случае линия наблюдается при частоте около
9000 Мгц. Эта частота соответствует длине волны око-
ло 3 см.
27
До сих пор мы рассматривали свободный электрон в
магнитном поле и видели, что полученный спектр ЭПР
представляет одиночную линию. Однако почти все сво-
бодные радикалы дают спектры, состоящие более чем
из одной линии. Это явление есть результат так назы-
ваемого сверхтонкого расщепления. Если неспаренный
электрон в радикале локализован на атоме, ядро кото-
рого обладает магнитным моментом, и этот магнитный
момент взаимодействует с электроном, то происходит
дальнейшее расщепление энергетических уровней элек-
трона.
Примерами ядер, обладающих магнитными момен-
тами, являются Н1, F19, N15, С13 и Р31.
Рис. 2.2. Электронные переходы при взаи-
модействии иеспарениого электрона с од-
ним протоном (внешнее магнитное поле Н).
В том случае, когда неспаренный электрон взаимо-
действует с одним протоном, магнитное поле протона
складывается или вычитается из внешнего магнитного
поля (в соответствии с двумя возможными ориентаци-
ями протона во внешнем поле. — Прим. ред.). Отсюда
каждый уровень, показанный на рис. 2.1, при взаимо-
действии. с полем протона расщепляется на два, как
это показано на рис. 2.2. Спин ядра не изменяется при
переходах между электронными уровнями, и поэтому
из- всех возможных осуществляется только половина пе-
реходов, а именно между 'теми уровнями, показанными
на рис. 2.2, которые имеют одинаковые спины
ядер.
Таким образом, происходит два перехода, и элек-
трон, который давал одиночную линию ЭПР в условиях,
иллюстрируемых рис. 2.1, теперь дает две линии.
28
Примерами радикалов, для которых можно предпо-
ложить наличие дублетного спектра, являются Н- и
• СНС12.
Вообще говоря, магнитные моменты ядер расщеп-
ляют линию ЭПР на 2/4-1 линий, где I — ядерный спин.
В случае водорода, так как у протона / = V2, получа-
ются две линии. Если с электроном взаимодействуете
эквивалентных протонов, то спектр состоит из п+1
линий. Например, метильный радикал с тремя протона-
ми должен давать спектр, состоящий из четырех линий.
На рис. 2.3 показаны спектры Н- и СН3-; как и предпо-
лагалось, они состоят из двух и четырех линий соответ-
ственно.
Рис. 2.3. Спектры ЭПР атома водорода (а) и ме-
тильного радикала (б).
Заметим, что величина расщепления больше в спек-
тре Н-, чем в СН3-. В случае водорода электрон непо-
средственно находится в атоме, имеющем ядерный спин.
В метильном радикале электрон расположен на угле-
роде, а атомы с ядерными спинами являются соседними,
что приводит к меньшему взаимодействию и соответст-
венно дает меньшую величину расщепления *.
Семихиноны представляют собой ряд соединений, на
которых можно проиллюстрировать эти явления. Хинон
* Другие примеры спектров ЭПР углеводородных радикалов
см. в работе [2].
29
может быть одноступенчато восстановлен до семихинон-
ного ион-радикала:
Семихиноны сравнительно стабильны и могут быть по-
лучены в больших концентрациях. Их стабильность
связана с наличием резонансных форм, в которых не-
спаренный электрон докализован как на углеродных
атомах кольца, так и на обоих атомах кислорода:
Так как электрон делокализован на атомах углерода
кольца, спектр ЭПР должен быть расщеплен, если с
этими атомами связаны атомы водорода. Такое расщеп-
ление наблюдалось, и число регистрируемых линий, как
и предполагалось, соответствовало правилу п.4-1. На-
пример, тетрахлорсемихинон дает только одну линию.
Когда хлор замещается водородом, число линий увели-
чивается, как показано ниже:
О
I
ci—а
С1-\ЛС1
О—
1 линия
0_
2 линии
о
н-А-с1
H-UJ-c
I
0_
3 линии
5 линий
30
Подобная серия спектров ЭПР наблюдается при
ферментативном окислении олефинов с помощью комп-
лекса фермент пероксидаза — перекись водорода. Окис-
ление диоксифумаровой кислоты, аскорбиновой кислоты
и 1,2-диоксициклопентенон-(3) кислоты приводит к по-
явлению радикалов соответственно I, II и III:
НО.2с" ^СО2Н
CHjOH-CHOH^^O-
II Hl
Можно предположить, что спектры ЭПР этих трех
радикалов должны состоять из одной, двух и пяти ли-
ний соответственно. На рис. 2.4 представлены эти спек-
тры, состоящие из предсказанного числа линий.
Рис. 2.4. Спектры ЭПР радикалов I,
| II и III.
Величину расщепления в сверхтонкой структуре
спектра, которая наблюдается в том случае, когда
электрон все время локализован на определенном атоме,
можно рассчитать из теоретических соображений. В свя-
зи с этим экспериментально определенное расщепление,
связанное с взаимодействием с данным атомом, может
быть связано с определенной долей электронной плот-
31
ности у этого атома. Экспериментально можно измерить
резонансное распределение неспаренного электрона в
молекуле. Величина расщепления, в гауссах, называется
константой сверхтонкого расщепления ан. [Из уравне-
ния (2.2) следует, что, умножая ан на 2,80, получаем
величину расщепления в частотных единицах — мега-
герцах.]
Ион-радикал бензола может быть получен при вос-
становлении бензола калием
Спектр этого ион-радикала состоит из семи эквидистант-
ных (равноотстоящих) линий с ан = 3,75 гс. Из сообра-
жений симметрии на каждый атом углерода должна
приходиться одна шестая плотности неспаренного элек-
трона, и поэтому можно ожидать, что константа рас-
щепления для фенильного радикала должна равняться
‘/б величины ан для . метила, или 76-23 = 3,8 гс, что
весьма близко к наблюдаемому значению.
Оптические спектры
Во многих случаях для идентификации свободных
радикалов можно использовать оптические спектры по-
глощения. Раствор, содержащий радикал трифенилме-
тил, имеет желтый цвет
Ph3C------CPh3 2Ph3C •
Бесцветный дна- Желтый пара-
магнитный магнитный
Если присутствуют иод или кислород, то желтая окраска
исчезает
Ph3C • 4- О2 -> Ph3C—О—О •
Бесцветный
Дифенилпикрилгидразин может быть окислен до ради-
кала дифенилпикрилгидразила (ДФПГ):
Дифенилпикрилгидразин Дифенил пикрил г и дразил
32
Радикал ДФПГ обладает глубокой фиолетовой окрас-
кой и в твердом состоянии стабилен в течение несколь-
ких лет. Даже раствор концентрации 10-5 М окрашен,
и этот факт можно использовать как индикатор присут-
ствия радикалов, как, например, кислотно-щелочной
индикатор используют при титровании. Иногда, однако,
возникают некоторые трудности: продукты реакции
ДФПГ с радикалами часто не определены. А в том ма-
лом числе случаев, когда они известны, эти продукты
не являются результатом простой реакции присоедине-
ния к атому азота, обладающему неспаренным элек-
троном (см. структуру выше).
Захват
Если известно, что соединение быстро реагирует с
радикалами, влияя на скорость реакции, то можно сде-
лать вывод, что оно содержит радикал. Например,
ДФПГ подавляет полимеризацию мономерных олефи-
нов, таких, как стирол. Следовательно, процесс поли-
меризации включает радикалы в виде промежуточных
продуктов, и они захватываются ДФПГ. В растворе
иод, хиноны, кислород, сера и многие другие вещества
можно использовать как акцепторы радикалов. В га-
зовой фазе можно использовать бутадиен; радикалы
связываются с диеном, образуя продукты, которые
димеризуются. Димеры могут быть выделены и иден-
тифицированы.
ЛИТЕРАТУРА
1. Roy М. Е., Marvel С. S. J. Amer. Chem. Soc., 59, 2622 (1957).
2. Fesenden P. W., Schuler R. H. J. Chem. Phys., 39, 2147
(1963).
Дополнительная литература
Электронный парамагнитный резонанс
Carrington A. Quart. Rev., 17, 67 (1963).
Инграм Д. Электронный парамагнитный резонанс в свободных
радикалах. Перев. с аигл. М., Изд-во иностр, лит., 1961.
Symons М. С. R. Identification of Organic Free Radicals by Elec-
tron Spin Resonance. In V. Gold (ed.) «Advances in Physical
Organic Chemistry». Vol. 1, Academic Press Inc., N. Y., 1963,
p. 284. r •
2 У. Прайер
33
Магнитная восприимчивость
М i с h а е 1 i s L. In: Л. Weissberger (ed). «Technique of Organic
Chemistry». Vol. 1, pt. II. Interscience Publishers. Inc. N. Y.,
Lst. ed., 1946.
Общая
Trotman-Dickenson A. F. Free Radicals. Methuen Co., Ltd.
London, 1959, pp. 1—24.
Walling C. Free Radicals in Solution. John Wiley and Sons. Inc.
N. Y., 1957, pp. 1—24. (Уоллинг Ч. Свободные радикалы в
растворе. Перев. с англ. Под ред. Г. А. Разуваева. М., Изд-во
иностр, лит., 1960).
Waters W. A. Chemistry of Free Radicals. Oxford University
Press. London, 2d. ed., 1948, pp. 1—34. (Уотерс У. Химия
свободных радикалов. Перев. с англ. Под ред. Я. К. Сыркина.
М., Изд-во иностр, лит., 1948.)
Глава 3 I
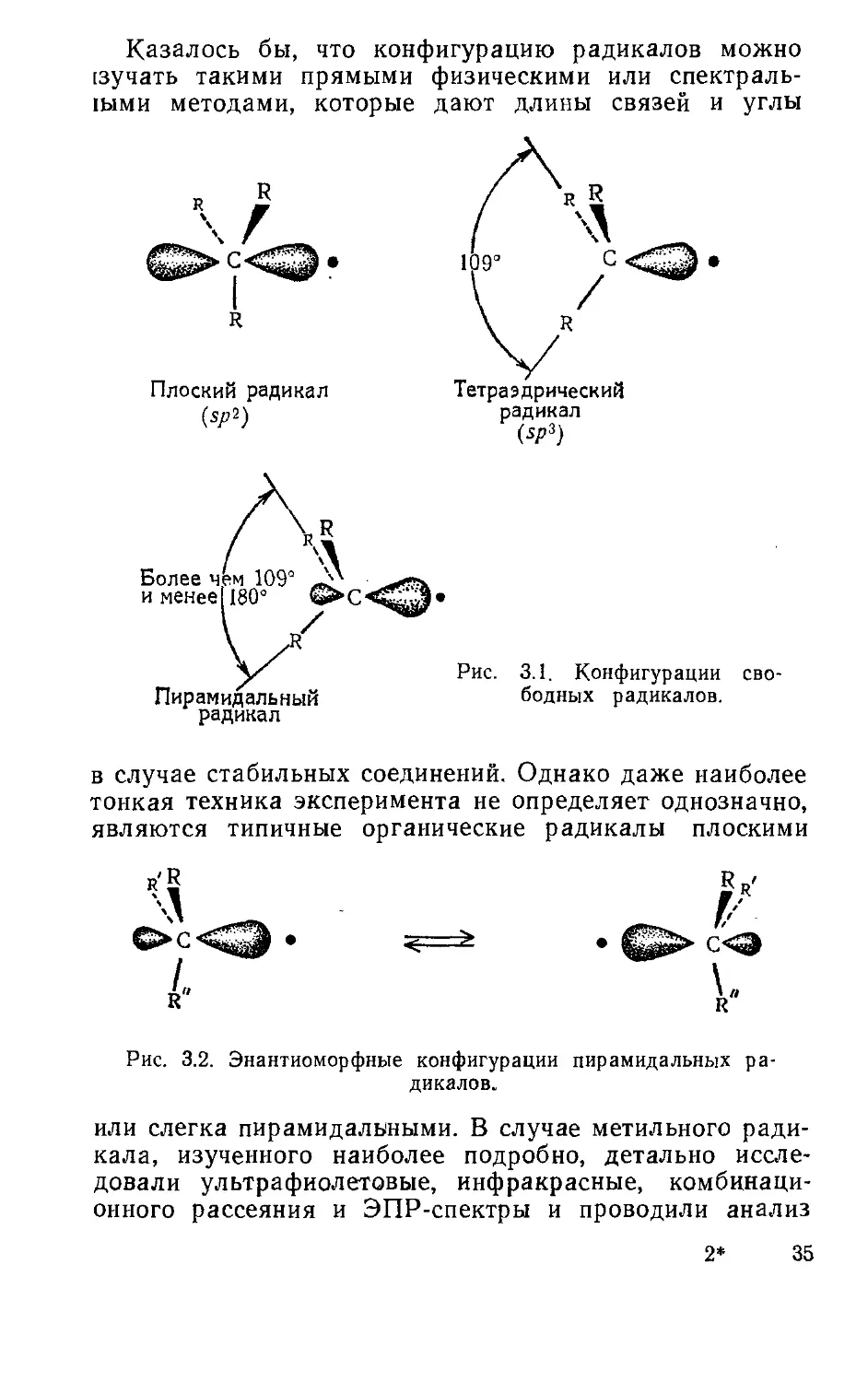
КОНФИГУРАЦИЯ РАДИКАЛОВ
Наиболее стабильная конфигурация свободных ради-
калов может осуществляться в трех случаях: связи ато-
ма углерода, содержащего неспаренный электрон, лежат
в одной плоскости (как в ионе карбония), составляют
тетраэдр или образуют промежуточную конфигура-
цию— плоскопирамидальную. Рис. 3.1 иллюстрирует
эти три возможные конфигурации. Хотя вопрос еще на-
ходится в стадии изучения, представляется наиболее
вероятным, что органические свободные радикалы очень
близки, по-видимому, к плоским, т. е. имеют геометрию
плоской пирамиды. В то же время энергия, необходи-
мая для перевода такой пирамиды в плоскую или тетра-
эдрическую конфигурацию, по-видимому, очень мала.
Если бы радикал был плоским, имела бы место sp2-
гибридизация, как в ионе карбония, а остающаяся
р-орбита содержала бы неспаренмый электрон (см.
рис, 3.1). При пирамидальном строении р-орбита, со-
держащая неспаренный электрон, в некоторой степени
носила бы «-характер, т. е. имелась бы некоторая гибри-
дизация между чистой р и «р3, при этом должна по-
явиться асимметрия в распределении электронной плот-
ности при любом строении радикала. На рис. 3.2 пока-
заны две энантиоморфные конфигурации пирамидально-
го радикала.
34
Казалось бы, что конфигурацию радикалов можно
[зучать такими прямыми физическими или спектраль-
1ыми методами, которые дают длины связей и углы
Плоский радикал
(V2)
3.1. Конфигурации сво-
бодных радикалов.
в случае стабильных соединений. Однако даже наиболее
тонкая техника эксперимента не определяет однозначно,
являются типичные органические радикалы плоскими
С О
Рис. 3.2. Энантиоморфные конфигурации пирамидальных ра-
дикалов.
или слегка пирамидальными. В случае метильного ради-
кала, изученного наиболее подробно, детально иссле-
довали ультрафиолетовые, инфракрасные, комбинаци-
онного рассеяния и ЭПР-спектры и проводили анализ
2*
35
& "гбчкй зрения определения конфигурации радикала [1.
2]. В итоге можно только сделать заключение, что СНз-
имеет или плоскую, или очень близкую к плоской кон-
фигурацию. К сожалению, ни один из этих методов
неприменим в тех случаях, когда отклонения от плос-
кости не превышают 10—15°. Эти эксперименты могут
свидетельствовать о том, что энергетический барьер
между плоской конфигурацией СН3- и плоскопирами-
дальной очень мал.
Определенные трудности при регистрации спектров
радикалов состоят в том, что большинство радикалов
настолько реакционноспособны, что их можно изучать
только в замороженных матрицах при очень низких
температурах. Однако спектры ЭПР некоторых алкиль-
ных и циклоалкильных радикалов регистрировали с
помощью современной экспериментальной техники —
жидкие углеводороды облучались электронами высокой
энергии непосредственно в резонаторе спектрометра
ЭПР и спектры регистрировались во время облучения
[3, 4].
Этим методом при обычной температуре были полу-
чены концентрации радикалов достаточно высокие, что-
бы зарегистрировать спектры ЭПР. К сожалению, как
правило, анализ спектров не дает ответа на вопрос,
является радикальный центр плоским или пирамидаль-
ным. Эти трудности можно проиллюстрировать при
обсуждении результатов для циклогексильного ради-
кала.
Спектр ЭПР циклогексильного радикала как в жид-
кой фазе при 10° С, так и в твердом состоянии при
—10° С показывает, что два атома водорода у (5-атома
углерода, соседнего с радикальным центром, не явля-
ются эквивалентными. Один из них взаимодействует с
неспаренным электроном сильнее, чем другой, и констан-
ты расщепления соответственно равны 41 и 5 гс. Цикло-
гексан имеет конфигурацию «кресла», при этом удобно
подразделить его атомы водорода на аксиальные и эква-
ториальные. Изучение моделей показывает, что аксиаль-
ные и экваториальные атомы водорода не могут быть
эквивалентными в спектрах ЭПР циклогексильного ра-
дикала независимо от того, является радикальный центр
плоским или пирамидальным. На рис. 3.3 показан цик-
логексильный радикал в предположении, что его ради-
кальный центр является плоским. Связь экваториаль-
36
ного атома водорода лежит вблизи узла /7-орбиты,
содержащей неспаренный электрон, и поэтому эквато-
риальный водород должен сравнительно слабо взаимо-
действовать с неспаренным электроном. Аксиальный
атом водорода должен взаимодействовать сильнее.
Н
Экваториальный 0-атом
водорода с С—Н-связью
вблизи узла плоскости ор-
биты, содержащей неспа-
ренный электрон
Рис. 3.3. Циклогексильиый радикал с
плоским радикальным центром.
На рис. 3.4 показан циклогексильиый радикал в
предположении, что радикальный центр пирамидален.
В этом случае, орбита неспаренного электрона может
занимать как аксиальное, так и экваториальное поло-
жение. Когда неспаренный электрон аксиален, он опять
Рис. 3.4. Циклогексильиый радикал с пирамидальным центром:
а — аксиальный неспарениый электрон; б — экваториальный неспаренный
электрон.
же сильно связывается с аксиальным и слабее с эквато-
риальным атомом водорода; если неспаренный электрон
экваториален, то он приблизительно одинаково взаимо-
действует с обоими атомами водорода в 0-положении.
37
Общий вывод состоит в том, что неспаренный элек-
трон должен сильнее взаимодействовать с аксиальным
атомом водорода независимо от того, является ради-
кальный центр плоским или пирамидальным *.
В связи с вопросом о геометрии связей тригональ-
ного атома углерода интересно рассмотреть геометрию
других атомов, содержащих неспаренный электрон.
Особый интерес представляет серия азотсодержащих
соединений:
• •
N
115"
Ион нитрита
•
N
> О
134’ •
Двуокись азоте
O=N = O
. 180’
Ион нитрония
В этом ряду ион нитрита согнут почти до тетрагональ-
ного угла; двуокись азота, которая является стабиль-
ным свободным радикалом, имеет слегка согнутую
конфигурацию, и ион нитрония линеен.
Сравнение этих трех соединений с тремя углерод-
ными соединениями
Р-А. ..
Карбанион
Радикал
позволяет сделать предположение, что радикал пред-
* Циклические и линейные насыщенные углеводородные ради-
калы дают константы расщепления для атомов водорода при
а-атоме углерода, сильно отличающиеся от констант для виниль-
ных и циклопропильных радикалов. Однако из этого не следует,
что обычные углеводородные радикалы точно плоские, а виниль-
ные и циклопропильные радикалы не плоские. Детальное рассмотре-
ние этого вопроса, которое в случае циклогексильного радикала
достаточно сложно, имеется в работе [4].
38
ставляет собой плоскую пирамиду, а карбанион — более
глубокую пирамиду или тетраэдр *.
Если бы радикалы были плоские, они имели бы
плоскость симметрии и не могли бы существовать в оп-
тически активных формах. Поэтому одним из подходов
для определения, является ли радикал плоским, может
служить приготовление радикалов из соединений, кото-
рые в исходном состоянии являются оптически актив-
ными. Одиако ни один эксперимент такого рода не
выявил предпочтительную геометрию радикалов. Напри-
мер, хлорирование оптически активного 1-хлор-2-метил-
бутана дало продукты реакции, не сохранившие ника-
кой оптической активности [6]:
Н
I сь
С2Н5-С-СН2С1->
СН3
Оптически активен
С2Н5—-С—СН2С1
I
сн3
С12
С12
С1
I
C2HS—С—-СН2С1
сн3
Рацемат
Такой результат можно трактовать двояким обра-
зом. Во-первых, потерю оптической активности продук-
тов можно объяснить тем, что радикал плоский и не
может существовать в оптически активной форме:
Н
С2Н5-С-СН2С1
сн3
Оптически активен
,СН2С1
ед-с<сн, -
Рацемат
Плоский радикал ие активен ввиду
плоскости симметрии
Второе объяснение потери оптической активности со-
стоит в том, что инверсия радикалов происходит быст-
рее, чем реакция с молекулами хлора. Рассмотрим,
например, реакцию D-молекул, дающих D-радикалы:
* Краткое обсуждение неорганических оксианионов и предпо-
ложения о том,- что их геометрия может быть непохожей на гео-
метрию водородзамещенных соединений, даны в работе [5].
39
н
I CI.
C2H5-C-CH2C1 -
сн3
D
Очень быстрая
С2НБ—С—СН2С1 -------- С2НБ—С—СН2С1
। инверсия I
сн3 сн3
D L
Рацемическая смесь радикалов
С1
СаН6—С—СНаС1
СН8
Рацемат
Переход D в L-радикалы, показанный выше, является
простым инверсионным процессом
Быстро
«Г
/С2Н5
•®>с;-СНз
СН2С1
Так как известно, что в аминах инверсия происходит
быстро, вполне разумно предположить, что в радикалах,
если они обладают пирамидальной структурой, инвер-
сия также должна осуществляться с очень большой
скоростью
Быстро
Даже в тех случаях, когда получаются оптически
активные продукты реакции, результат исследования не
поддается четкому объяснению. Бромирование оптиче-
ски активного 1-хлор- или 1-бром-2-метилбутана при-
водит к появлению оптически активных продуктов [7].
40
Наиболее убедительное объяснение такого результата
состоит в том, что молекулы брома связываются с оп-
тически активными радикалами до того, как они раце-
мизуются в результате инверсии. Например, £)-молекулы
могут реагировать способом, показанным ниже:
Н
сн3 сн3
D D
(Х=С1 или Вг)
Вг
C2HS—С—СН2Х Очень быстро С2Н8—С—СН2
сн3 сн3
D D
С2Н,-С-СН2Х
сн3
D
С2Н8—С—СН2Х
I
сн3
L
Так как бром является лучшим передающим агентом,
чем хлор, возможно, что бромирование может давать
оптически активные продукты, а хлорирование дает
рацематы.
Однако вероятно, что оптически активные продукты
получаются прямо из этих а-галогенорадикалов, так как
атом галогена способен к замыканию, образуя «замкну-
тый» («мостиковый») радикал, который не инвертирует
и не рацемизуется. Этот факт иллюстрируется схемой
и х
Т /*\
1 Вг* ' • ВГо
С2Н5—С—СН2Х -----*- С2Н5—С=^=^СН2 -*— D-соединение
СН3 СН3
D D
Такую возможность прохождения реакции нельзя
игнорировать, поскольку известно, что атомы как хлора,
так и брома в радикальных процессах могут мигриро-
41
вать к смежным’ атомам, и эта перегруппировка может
включать переходную стадию, в которой галоген частич-
но связан с каждым атомом углерода (см., например,
стр. 19). В реакции, показанной выше, постулировалось,
что исходное соединение образует замкнутый радикал
прямо, без какой-либо предварительной ступени в виде
«открытого» радикала. Если осуществляется описанный
выше случай, тогда сохранение оптической активности
в продуктах бромирования никак не связано с пробле-
мой конформации обычных открытых радикалов.
Существует также и другая возможность: замкнутые
радикалы могут образовываться из открытых радика-
лов:
Н Д
; с2н3—с—СН2Х С2Н5 сн3 сн2х —*с2н—6^сн2
1 СН3 СН3
Оптически активен ’ Оптически активен |вг2
Вг
С2Н5~ С—СН2Х
сн3
Оптически активен
Ясно, что если этот механизм соответствует действи-
тельности, то замкнутый радикал является просто про-
межуточным продуктом в схеме реакции и открытые
радикалы должны существовать в оптически активной
форме *.
Проблема, какие радикалы — замкнутые или откры-
тые—более стабильны, является предметом интенсив-
ных исследований, и окончательный ответ еще не полу-
чен. Однако для определенных систем существуют
строгие доказательства того, что бромзамещенный
мост образует радикалы с меньшей энергией, чем ана-
* Объяснение стереоспецифичности при галогенировании 2-га-
логенобутанов было дано с привлечением конформационных эффек-
тов, а не замкнутых радикалов [8]. Наличие замкнутых радикалов
будет оставаться просто гипотезой до получения более конкретных'
доказательств их существования. Этот спорный вопрос имеет ана-
логию в ионной химии [9].
42
логичные открытые радикалы [10]. В цис-1-бром-4-трет-
бутилциклогексане (I) объемистая mpem-бутильная
группа держит молекулу в конформации, в которой атом
брома аксиален. При бромировании это соединение дает
1,2-транс-дибромид (II) как основной продукт:
Два типа данных подтверждают, что только замкну-
тые радикалы участвуют 'в этой реакции. Во-первых, от-
крытый радикал III, как можно ожидать, реагирует
с образованием основных количеств как соединения II,
так и продукта, в котором два брома находятся в цис-
положении:
С другой стороны, наличие замкнутого радикала типа
IV объяснило бы преимущественное образование транс-
продукта (изомера):
молекулой Вг2
Атаку брома на радикал IV в аксиальном направлении
приходится постулировать, однако известно, что это
43
направление атаки является преимущественным при
ионных реакциях.
Вторая группа данных (кинетические) свидетельст-
вует о том, что замкнутый радикал IV образуется пря-
мо из соединения I. Известно, что галогенирование
обычно замедляется у а-галогензамещенных (см., на-
пример, табл. 13.6). Поэтому образование а-галогени-
рованного продукта II как преимущественного продукта
нельзя объяснить без постулирования каких-то специ-
альных эффектов. Наиболее разумное объяснение пре-
имущественного бромирования соединения I в а-поло-
жении представляет собой «эффект соседства» у а-бром-
замещенного *
При этом объяснении соседство бромных мостиков ста-
билизирует переходное состояние, в котором а-С—Н-
связь растянута. Одно из стерических требований для
такого замыкания заключается в том, чтобы бромзаме-
стители были аксиальны. Такой механизм подтверж-
дается тем фактом, что эпимерное соединение V, кото-
рое имеет экваториальный замещенный атом брома,
бромируется менее чем на ’/is быстрее по сравнению с
соединением I:
* Влияние соседних групп в иодзамещеиных при гомолизе над-
эфирной О—О-связи будет обсуждено в гл. 9.
44
Ясно, что аксиальный бром, соседний с С—Н-связью,
в циклогексане приводит к увеличению эффективности
отрыва водорода в результате влияния «соседней груп-
пы» *. Полная схема реакции представляет собой •
К дальнейшему рассмотрению замкнутых радикалов
вернемся в гл. 14.
Данные о преимущественной конфигурации радика-
лов можно получить при изучении скоростей, с которы-
ми радикалы образуются из соединений с известной кон-
фигурацией. Эти исследования показали, что радикалы
образуются в обычных ненапряженных системах пример-
но с одинаковыми скоростями независимо от того в ка-
кой конфигурации находится система: в плоской или
неплоской конфигурации. Этот факт опять же может
говорить'о том, что разница в энергии между плоским
и неплоским радикалом мала. Соединения с двумя цик-
лами не могут быть плоскими при положении «голова к
голове» из-за стерических ограничений в мостике. На-
пример, преимущественно плоские ионы карбония
* В работе [11] обсуждается влияние соседних групп. Боль-
шинство примеров относится к ионным реакциям, радикальные ре-
акции коротко рассмотрены на стр. 104 цитируемой работы.
45
образуются чрезвычайно медленно в положении «голова
к голове»
Очень
медленно
+ сг
Однако соответствующие радикальные реакции проте-
кают с примерно нормальными скоростями. Например,
аналог перекиси апокамфорила диссоциирует в четырех-
хлористом углероде, давая обычные продукты [12]:
CI + Дру
.гие
продукты
36%
Очевидно, что в этих случаях образуется бициклический
радикал, и для этого не требуется особенно высокая
энергия.
Подобно этому, соли серебра апокамфорной кислоты
подвержены обычному радикальному бромированию в
четыреххлористом углероде [13]:
Вг2
СС14
Другие продукты
реакции с растворителем
Производные триптицила с бициклическими заме-
стителями представляют другой пример того же явле-
ния:
46
.X
Триптицил (T r).-X=
(Tr—X)
Катион триптицила образуется очень медленно, так как
ион не может быть плоским:
_L Очень .
Tr — X------------ Tr+ + X-
медленно
Перекись триптоила, с другой стороны, реагирует, как
показано ниже [14]:
О О
Tr—С—О—О—С—Тг 2^+12 Тг_ !
| 80° 41%
Бензол 'I' 80°
О
Тг — Н + Тг—С—ОТг + СО2 + Другие продукты
45%
Связывание триптицильных радикалов с иодом
Тг—I свидетельствует об их участии в этих реакциях.
Тот факт, что триптицильный радикал отнимает водо-
род у бензола, который является очень неактивным
донором водорода, показывает, что он обладает не-
сколько большей энергией, чем обычный радикал.
Приведем другой пример из работы [15]. Бициклоги-
похлорит подвергается фотолизу, образуя соответствую-
щий алкокси-радикал. Этот радикал подвергается реак-
ции p-расщепления двумя основными путями: отрывом
метильного (3.1) или бициклического радикала (3.2).
Интересное свойство этой реакции состоит в том, что
бициклический радикал отщепляется чаще, чем метиль-
47
ный, и что бициклический радикал остается неповреж-
денным и образует бициклический продукт:
(ROCI)
|CFCI3
Свет Растворитель
упри 0°С
+ СН3«
(3.1)
сн3— с—<н3
22% ,
(3.2)
ROCI
22%
+ RO.
Упражнения
3.1. По реакциям (3.1) и (3.2) получаются самые различные
продукты, если в качестве растворителя использовать циклогексен.
При этом основными продуктами являются диметил-(1-норбориил)
карбинол
С(СН3)2ОН
и 3-хлорциклогексен с 94%-ным выходом. Объясните этот результат.
3.2. Константы скорости образования радикалов из перекисей
ацетила, бензоила, триптоила и апокамфоила
-отличаются на величину, меньшую 10 при 80° С [14].
48
Какие предположения о стабильности бициклических радика-
лов вы должны сделать, исходя из этой информации?
Рассмотрите подробно, какие связи рвутся в переходном со-
стоянии.
3.3. Бромирование оптически активного 1-бром-2-метилбутана
с помощью треот-бутилгипобромида дает некоторое количество
1,2-дибром-2-метилбутана, который оптически активен, плюс другие
продукты. отреот-Бутилгипогалоид дает продукты хлорирования, ко-
торые все неактивны [7]. Объясните эти результаты.
3.4. Ионы карбония преимущественно плоские. Карбанионы
изоэлектронны с аммиаком, который, как известно, пирамидален,
но с энергией активации для инверсии только 6—8 ккал! моль [16].
Можно ли ожидать, что энергия активации для инверсии радикала
будет больше или меньше, чем в карбанионе.
3.5. Изобразите структуры для циклопентильиого • радикала,
предполагай: а) плоское кольцо и плоский радикал; б) плоское кольцо
и неплоский радикал; г) неплоское кольцо и неплоский радикал. От-
метьте структуры, в которых атомы водорода при р-атоме углерода
эквивалентны. Спектр ЭПР циклопентильиого радикала показывает,
что атомы водорода в p-положении эквивалентны при —40° С, но
становятся неэквивалентными при —80° С.
Дает ли этот факт возможность сделать выбор для геометрии
радикала? Объясните.
ЛИТЕРАТУРА I
1. Herzberg G. Proc. Roy. Soc. (Lond.), A262, 291 (1961); Ann.
Rev. Phys. Chem., 9, 327 (1958).
2. Karplus M. J. Chem. Phys., 30, 15 (1959); Karplus M.,
Fraenkel G. K. Ibid., 35, 1312 (1961).
3. Fessenden R. W., S c h u 1 e г R. H. J. Chem. Phys., 39, 2147 .
(1963).
4. Ogawa S„ Fessenden R. W. Ibid., 41, 994 (1964). )
5. Atkins P. W., Keen N., S у m о n s M. C. J. Chem. Sot., 250
(1963).
6. В г о w п H. С., К h a r a s c h M. S., C h а о T. H. J. Amer.
Chem. Soc., 62, 3435 (1940).
7. Skell P. S„ Tuleen D. L, Re a di о P. D. Ibid., 85, 2849
(1963).
8. F r e d r i c k s P. S., Tedder J. M. J. Chem. Soc., 3520
(1961).
9. Cram D. J. J. Amer. Chem. Soc., 86, 3767 (1964);
Brown H. C., Morgen K- J-> Chloupek F. J. Ibid., 87,
2137 (1965).
10. Skell P. S., Readio P. D. Ibid., 86, 3334 (1964).
11. C a p on B. Quart. Rev., 28, 45 (1964).
12. К h a r a s c h M. C., Engel man F., UrryW. H. J. Amer.
Chem. Soc., 65, 2428 (1943).
13. Wilder P„ Winston A. Ibid., 75, 5370 (1953).
14. Bartlett P. D„ Greene F. D. Ibid., 76, 1088 (1954).
15. Greene F. D. et al. J. Organ. Chem., 28, 55 (1963).
16. К i n c a i d J. F., H e n r i q u e s F. C. J. Amer. Chem. Soc., 62,
1474 (1940).
49
Глава 4
ЭНЕРГЕТИКА И СКОРОСТИ РЕАКЦИЙ
Скорости
Радикалы являются реакционноспособными образо-
ваниями, которые обычно реагируют с другими веще-
ствами очень быстро. Однако отдельный изолированный
радикал в полном вакууме, очевидно, может быть ста-
бильным неопределенно долгое время независимо отего
реакционной способности. Скорость, с которой радикалы
исчезают, зависит от собственной стабильности (илн,
наоборот, реакционности) и концентрации, а также от
реакционной способности и концентрации других при-
сутствующих веществ.
С этой точки зрения полезно рассмотреть выраже-
ние для скорости реакции. Скорость реакции
аА ЬВ
дается выражением
_L. =J_..+UB]._K[Ar,
a dt b dt
где квадратные скобки обозначают концентрации
(моль/л), а дроби ’/а и '/& являются статистическими
множителями, зависящими от стехиометрии реакции.
Скорость реакции зависит от концентрации А, возве-
денной в n-ю степень, и говорят, что она n-го порядка.
В гл. 15 будут даны ссылки на работы, в которых при-'
водятся экспериментальные правила определения поряд-
ка реакции.
Количество энергии, которую молекула должна полу-
чить, чтобы прореагировать, называется энергией акти-
вации Е. На рис. 4.1 показана эта энергия, а также
энергия Е', необходимая для обратной реакции. Заме-
тим, что теплота реакции ЛЯ равна разности двух
энергий активации:
\Н--Е — Е’. (4.1)
(Уровни энергии, показанные на рис. 4.1, выше мини-
мума энергии из-за энергии так называемых нулевых
колебаний.)
Знание энергии активации очень важно для пони-
мания движущих сил химических реакций. Количествен-
50
ную оценку энергии активации можно получить при
изучении изменений скорости реакции с температурой.
Наиболее часто использующимся соотношением для вы-
ражения этого изменения является уравнение Арре-
ниуса
_ Еа
к = Ае *г . (4.2)
Большинство реакций подчиняется этому уравнению,
т. е. константу скорости можно определить, если из-
вестны два параметра: аррениусовская энергия актива-
ции.
ции Еа и предэкспоненциальный множитель А, который
связан с вероятностью реакции, идущей при какой-либо
данной температуре. Энергия активации из уравнения
Аррениуса является хорошим приближением к Е.
Теория переходного состояния дает другое выраже-
ние для констант скорости. В этом приближении пред-
полагается, что реагирующие соединения находятся в
равновесии с «активированным комплексом» или «пере-
ходным состоянием», и выражение для константы ско-
рости имеет вид
к’= kj- (4.3)
h
(4.4)
51
где k — постоянная Больцмана; h — постоянная План-
ка; Т — абсолютная температура; AS*— энтропия ак-
тивации и АН* — энтальпия (теплота) активации. Еа и
АН* для реакций в газовой фазе связаны соотношением
АН* = Ea — nRT,
где п — порядок реакции.
Для реакций в растворе
АН*—Еа — RT.
В случае мономолекулярной реакции уравнения (4.2) и
(4.4) могут быть объединены, что дает
_ ekT
h
Величина ekT/h при 25° С есть 1,7 • 1013 сек-1, и если
величина AS * близка к нулю, то в указанном прибли-
жении можно написать
к== 1013е-Еа/«г сек-1. (4.5)
Определенная для мономолекулярных реакций величи-
на предэкспоненциального множителя близка к
1013 сек-1.
Возвратимся теперь к реакционной способности
радикалов и скорости радикальных реакций. Константа
скорости димеризации метильных радикалов измерена
методами, обсуждаемыми в гл. 15. Константа скорости
к оказалась равной 2-Ю10 сек-1. Если этот результат
сравнить с уравнением (4.2), то, можно заметить, что
энергия активации димеризации метильных радикалов
равна нулю, А имеет значение 2 • 1010 сек-1 и константа
скорости не зависит от температуры. Следовательно,
метильные радикалы настолько реакционны, что обра-
зуют этан при каждом соударении *.
Время жизни радикалов в газовой фазе можно по-
лучить в экспериментах типа опытов Панета (см.
стр. 10). В этих опытах генерировались свободные ра-
дикалы, которые затем увлекались носителем по труб-
* Так как реакция образования связи между двумя радикала-
ми весьма экзотермична, то радикалы могут сталкиваться упруго
без образования связи до тех пор, пока постороннее тело или
стенка сосуда не поглотят избыток энергии. Влияние третьего
тела обсуждается в гл. 20.
52
ке и снимали предварительно нанесенное металлическое
зеркало, помещенное на пути потока. Зеркало не исче-
зало, если оно было далеко от места образования ради-
калов, что означало, что радикалы разрушались окон-
чательно прежде, чем достигали зеркала. Если скорость
потока и ' расстояние между местом возникновения
радикалов и зеркалом известны, можно подсчитать вре-
мя жизни радикалов. При скорости потока около
10 м/сек найдено, что метильные радикалы имеются до
расстояний около 0,1 м, т. е. их время жизни около
0,01 сек. Даже очень реакционные радикалы существу-
ют определенное время, если завершающие реакции
протекают медленно.
Энергия диссоциации связей
Энергию связи можно определить различными мето-
дами, детально (Уписанными в учебниках по физической
химии. В химии радикалов успешно применяют два ме-
тода. В одном из них аррениусовская энергия актива-
ции отождествляется с энергией диссоциации связи.
В реакции
АВ->А- + В-
энергия диссоциации связи £)(А—В) равна теплоте ре-
акции: ' ,
О(А —В) = АЯ.
Если энергия активации для обратной реакции (т. е.
рекомбинации радикалов) равна нулю и если радикалы
образуются в своем основном колебательном состоянии,
то из уравнения'(4.1) следует
Яа = АЯ
и
£> (А — В) = = АЯ. (4.6)
Как пример использования этого метода рассмотрим
диссоциацию перекиси трет-бутил а
/пре/п-С4Н9О—OC4H9-mpem —
/пре/п-С4Н9О • + • ОС4Н9-/пре/п.
Энергию диссоциации связи О—О, т. е. D
(/пре/п-С4Н9О—ОС4Н9-/пре/п) можно определить как
энергию активации, найденную для этой реакции кине-
тическим методом, и ее значение 37 ккал/моль.
53
Второй метод определения энергии связи преду-
сматривает использование закона Гесса. Этот закон
гласит, что ДН реакции не зависит от механизма реак-
ции (более детальное описание закона Гесса можно
найти в курсе физической химии — см. литературу к
этой главе). Например, энергию диссоциации при раз-
рыве одной из связей С—Н в метане можно получить
следующим образом: энергии активации прямой и об-
ратной реакций могут быть измерены в реакции
£а=17
СН4 + Вг--?—- СН3- + НВг
Еа=2
Из уравнения (4.1) ДН= 15 ккал/моль. Величина
О(Н—Вг) определена спектроскопическими методами
и равна 87 ккал/моль. Отсюда энергию связи СНз—Ы
можно получить из следующего ряда реакций: 1
«ДН, ккал/моль
СН4 + Вг-^СН3- + НВг 15
НВг^Н-4-Вг. ' 87
Сумма СН4-> Н--|-СН3- 102
Теплота образования
Теплоту образования можно рассчитать из данных
по энергии связи и знаний некоторых теплот образова-
ния, учитывающих предварительно вычисленную энер-
гетику других реакций. Теплота образования АЯ/° оп-
ределяется как изменение энергии системы, когда со-
единение образуется из элементов в их 1 основном
состоянии при 25° С и 1 атм. Пример расчета теплоты
образования для атома водорода из диссоциации моле-
кулы водорода в газовой фазе:
Н2 -> 2Н- ДН = 103,4 ккал/моль.
Теплота реакции равна сумме теплот образования всех
продуктов минус сумма теплот образования всех реа-
гентов:
ДН — ^,ДН/ (продукты) — (реагенты).
Поэтому для водорода
ДН = 103,4 = 2ДЯ° (Н-) — ДН°[ (Н2);
ДН0/ (Н •) = 103,^+ ° ~ 52 ккал/моль.
54
Рассмотрим расчет теплоты образования mpem-бу-
токси-радикала. Теплоту образования mpem-бутилового
спирта можно рассчитать из теплоты сгорания
(—77 ккал/моль). Энергия диссоциации связи О—Н
измерена различными методами и равна 102—
104 ккал/моль. Поэтому
mpem-C4H9O—Н -> m/?em-C4H9O • -f- Н-
\Н = 104 ккал/моль
и затем
АЯ°(Н-) + А/7° (mpem-C4H9O •) — Atfz° (mpe/n-C4H9OH) =
= 104 ккал!моль\
(mpem-C4H9O-) = 104 — 52 + (—77) = —25 ккал/моль.
Рассчитанную теплоту образования игрет-бутокси-ра-
дикала можно использовать для определения энергии
активации реакции:
тре/п-С4Н9О— OC4H9-mpem -> 2 mpem-C4H9O-
Теплоту образования перекиси трет-бутила (ТБП)
можно рассчитать, измерив его теплоту сгорания; най-
дено, что она равна —85 ккал/моль. Поэтому энергия
активации диссоциации перекиси, которая должна быть
равна энергии связи О—О, есть
\Н = Еа- 2АЯ° (mpem-C4H9O-) — ЬН° (ТБП) =
= 2 (—25) — (—85) — 35 ккал/моль.
Заметим, что такое оценочное вычисление дает значе-
ние энергии диссоциации всего на 2 ккал/моль меньше,
чем полученное экспериментально (стр. 53).
Теплоту реакции можно рассчитать из энергии дис-
социации связи. Рассмотрим, например, реакции
А —В->А + В (4.7)
В — С -> В + С (4.8)
Разница А — В + С -> В — С + А (4.9)
Реакция (4.9) получена при вычитании реакции (4.8)
из (4.7). Если теплота этих реакций есть АН7, и
Д/79, из закона Гесса следует
А/79 = АЛ/, — А/78.
55
Если удовлетворены условия, необходимые для выпол-
нения уравнения (4.6), то
АЯ, = D (А — В),
АЯ8 - D (В — С),
и поэтому
АЯ9 = О(А —В) —D(B —С).
Таким образом теплота реакции равна разности
энергий диссоциации рвущихся и образующихся связей.
Например, теплоту реакции атомов хлора с метаном
можно рассчитать следующим образом:
сн4 + сь ->нс1.4- сн3,
\Н =х D (СН3 — Н) — D (Н — С1) = 101 — 102 =
= — 1 ккал/мо ль.
Реакция с АЯ= 1 ккал/моль экзотермична, так как об-
разуется несколько более прочная связь, чем та, кото-
рая рвется. ,
Упражнения 1
4.1. Предположим, что для некоторой мономолекулярной реак-
ции в газовой фазе А —10’3 сек~1. Подсчитайте период полурас-
пада исходного соединения при 100° С, предполагая, что энергия
активации равна (а) 15,0 ккал/моль и (б) 30,0 ккал/моль.
4.2. Используя значения энергии активации, данные в этой
главе, определить ДЯ для реакции
CH3-4-/npe/n-C4HeOH -» CH4-(-mpem-C4HsO-
Дополнительная литература
Кинетика
Benson S. W. Foundations of Chemical Kinetics. McGraw-Hill
Book Company. N. Y., 1960.
Frost A. A., Pearson R. G. Kinetics and Mechanism. N. Y.
John Wiley and Sons, Inc., 1961.
M о о r e W. J. In: Physical Chemistry. N. Y. Printice-Hall, Inc., Eng-
lewood Cliffs, 3d. ed,, 1963, p. 253. —
Энергия диссоциации связей. Теплота образования
С о 11 г е 11 Т. L. Strengths of Chemical Bonds. Academic Press. Inc.
N.Y., 1958.
Gray P., Williams A. Chem. Rev., 59, 239 (1959) (к алкокси-
радикалам).
M о о r e W. J. In: Physical Chemistry. N. Y., Prentice-Hall, Inc.,
Englewood Cliffs, 3d. ed., 1963, p. 50.
Natl. Bur. Standards. Circ. 500, Washington D. C.
56
ЧАСТЬ ВТОРАЯ
ОБРАЗОВАНИЕ РАДИКАЛОВ
Глава 5
ВВЕДЕНИЕ. СПОСОБЫ ПОЛУЧЕНИЯ РАДИКАЛОВ
Радикалы могут быть получены при облучении, тер-
мическом гомолизе и при окислительно-восстановитель-
ных реакциях. В этой главе дается общее рассмотрение
этих процессов, в последующих главах мы вернемся к
описанию каждого из них.
Облучение
В процессе облучения энергию, । необходимую для
образования радикалов, можно использовать в виде
электромагнитного излучения (ультрафиолетовый или
видимый свет, рентгеновское излучение и т. д.) или
корпускулярного излучения (электроны высокой энер-
гии, а-, 0-, у-частицы, нейтроны, протоны и т. д.).
В этой книге мы ограничимся рассмотрением процессов,
связанных с поглощением света и относящихся к раз-
делу радиационной химии, называемому фотохимией.
Процессы, происходящие при поглощении высокоэнерге-
тичных частиц, связанные с ионизацией и последующи-
ми реакциями, в книге не рассматриваются.
Термический гомолиз
Энергия простой связи С—С равна примерно
90 ккал!моль, и теплового возбуждения молекул ока-
зывается достаточно, чтобы порвать 'эти связи при тем-
пературах 350—550° С. Термический крекинг нефти, в
частности, происходит как раз в этом диапазоне темпе-
ратуры. Однако в некоторых соединениях имеются
исключительно слабые связи, которые рвутся и обра-
зуют радикалы при более низких температурах. Эти
соединения могут быть использованы для инициирова-
57
ния радикальных процессов при температурах от 50 до
150° С.
Выражение 1 (4.5) определяет константу скорости мо-
номолекулярной реакции в виде
к == 1013е~£а'да сек-1.
Это выражение предполагает, что молекулы, содержа-
щие связи с энергией диссоциации около 25 ккал/моль,
будут диссоциировать с периодом полураспада 1 ч при
50° С, соединения, содержащие связи с Энергией
32 ккал/моль, должны диссоциировать с периодом полу-
распада 1 ч при 150° С. Таким образом, молекулы с
энергиями связей 25—35 ккал/моль можно использо-
вать как радикальные инициаторы при температурах
50—150° С.
Имеется несколько типов соединений, имеющих связи
указанной прочности, однако чаще всего используются
соединения, содержащие перекисные связи О—О.
В табл. 5.1 перечислены некоторые доступные переки-
си, используемые как инициаторы. В таблице приво-
дится энергия активации реакции диссоциации и тем-
пература, при которой перекиси разлагаются на ради-
Таблица 5.1
Часто используемые радикальные инициаторы
Название Структура ккал/моль Темпера- тура для периода полурас- Диада 1 ч, °C
Перекись трет-бутила трет - CtHfiOCtti^-mpem 37 150
Пербензоат mpem-бутила трет - CiHsOOOCPh 34 125
Перекись бензоила PhCOOOOCPh 30 95*
Перекись ацетила СН3СООООССН3 30—32* 85**
Азоизобути- ронитрил (CH3)2CN=NC(CH3)2 CN CN 30 85
* Зависит от растворителя, данные для инертного растворителя.
••В газовой фазе.
58
калы с периодом полураспада 1 ч. Можно заметить, что
предположения, основывающиеся на уравнении Арре-
ниуса, оправдываются. В табл. 5.1 приведены также
данные для азоизобутиронитрила (АИБН), который
является общепринятым радикальным инициатором, это
соединение диссоциирует вследствие сил напряжения,
возникающих при образовании стабильной молеку-
лы N2:
СН3 сн3 сн3
СН3—С—N = N—С—СН3 -+ 2СН3—С- + N2
CN CN CN
АИБН
Соединения, представленные в табл. 5.1, удобно
использовать из-за их достаточной стабильности при
синтезе при обычной температуре; их можно хранить
длительное время в холодильнике.
Эти соединения можно использовать как инициаторы
при температурах, при которых большинство связей не
разлагается при термическом гомолизе.
Окислительно-восстановительные источники радикалов
Часто окислительно-восстановительные реакции ис-
пользуются как источники образования радикалов. На-
пример, полимеризация стирол-бутадиеновой смеси для
получения синтетической резины дает продукт с луч-
шими физическими свойствами, если полимеризация
идет при 0—10° С, чем при более высоких температурах.
Трудно найти легко синтезирующийся инициатор, кото-
рый разлагался бы при таких низких температурах.
В этом случае можно использовать реакцию железа
(II) с гидроперекисью кумола:
СН3 СН3
I I
Fe2+ + Ph—С—ООН -> Fe3+ + Ph—С—О - + ОН-
СН3 СН3
Энергия активации этой реакции всего 12 ккал1моль.
В гл. 11 мы рассмотрим другие примеры окислительно-
восстановительных реакций ионов металлов, подобные
приведенной выше.
59
Дополнительная литература
Образование и стабилизация свободных радикалов. Под ред.
А. Басса и Г. Бройда. Перев. с англ. М., Изд-во иностр, лит.,
1962.
Химия высоких энергий. Сборник трудов конференции. М., «Наука»,
1965.
Глава 6
ФОТОХИМИЯ
В некоторой данной системе свет может распростра-
няться, преломляться, рассеиваться и поглощаться.
В фотохимии имеется фундаментальное правило —
только поглощенный свет может производить химиче-
ские или физические изменения. Отдельную молекулу
активирует в основном каждый квант поглощенного
света.
Квант света частоты v обладает энергией
E = h»,
где h — постоянная Планка. В фотохимии единицей из-
мерения количества квантов света является эйнштейн *.
Энергия, которую несет 1 эйнштейн света, связана с ча-
стотой света формулой
Е = 6,02- 1023/iv = • = 2’8—-- ккал! эйнштейн.
К Л (А)
Так, свет с длиной волны 7500 А несет 38 кк.ал1эйнштейн,
свет с Л=3130А несет 91 ккал/эйнштейн, т. е. энергию,
достаточную для разрыва связи С—С.
В фотохимических экспериментах необходимо ис-
пользовать источники света большой интенсивности.
Например, обычная 1000-ваттная лампа дает только
0,05 вт своей энергии в видимой области в данном
направлении в полосе 10 А. Для света с X =4000 А
0,05 вт соответствует 1017 к.вант]сек, и, чтобы при такой
интенсивности получить один эйнштейн света, необхо-
* 1 эйнштейн равен числу квантов света определенной частоты,
которое вызывает в системе, способной к фотохимическим реак-
циям, фотохимическое превращение 6.02 • 1023 молекул, или 1 моля,
вещества. (Физический энциклопедический словарь.) Определение
автора характеризует падающий свет. — Прим, перев.
60
Дймо время Ь • 106 сёк, что составляет около 10 недель.
Однако в настоящее время становятся доступными
источники и реакторы со специально разработанной гео-
метрией, которые дают свет в диапазоне 2000—4000 А
с интенсивностью до одного эйнштейна за время 8—
24 ч.
Процессы, протекающие при фотовозбуждении веществ
Когда молекула поглощает квант света и становится
возбужденной, она может потерять энергию возбужде-
ния различными путями:
физические процессы
тепловая закалка (переход в тепло);
флюоресценция;
фосфоресценция;
внутренняя конверсия;;
межсистемный переход; -
химические процессы
радикальные реакции;
нерадикальные реакции.
Нас в основном будут интересовать химические про-
цессы, которые следуют за актом поглощения света.
Однако прежде чем обсудить примеры этих химических
реакций, необходимо рассмотреть физические процессы,
поскольку, как мы увидим, они могут конкурировать с
химическими. Такое рассмотрение полезно также для
лучшего понимания природы фотовозбужденного со-
состояния. II. . . - „
Большинство известных физических процессов, с по-
мощью которых молекулы могут дезактивироваться,
происходят при передаче энергии возбуждения другим
молекулам, присутствующим в системе. Это так назы-
ваемая передача энергии молекулам растворителя, или
тушение. Такие процессы наблюдаются как в газовой
фазе, так и в жидкости — различные молекулы раство-
рителя обладают различной эффективностью тушения.
Например, в газовой фазе атомы ртути могут перехо-
дить в возбужденное состояние Hg*, а различные при-
меси М в газе могут снимать это возбуждение;
Hg + hi -> Hg*
Hg* + M -> Hg + M*
61
Интересно отметить, что метан в качестве тушителя
мало эффективен, более эффективны водород и высшие
углеводороды. Водород и высшие углеводороды явля-
ются особенно эффективными тушителями прежде всего
потому, что они могут терять энергию возбуждения в
процессах диссоциации:
Hg* + H2+ Hg + 2H-
Hg* + RH -> Hg+R-+H-
Тушение наблюдается, также при рекомбинации ато-
мов иода. Когда молекулярный иод подвергается фото-
лизу, образующиеся атомы иода имеют очень большей
избыток энергии, что мешает их рекомбинации. Моле-
кулы растворителя могут снимать избыток энергии и
способствовать реакции рекомбинации. Измерена эф-
фективность различных растворителей как в газе, так
и в жидкости [1]. При этом найдено, что выход сво-
бодных атоМов иода меньше в растворе, чем в газе.
В растворе иод диссоциирует, образуя два возбужден-
ных атома иода, которые удерживаются в непосредст-
венной близости «клеткой» из молекул растворителя
(природу этой клетки более детально обсудим в гл. 7)
Атомы иода могут потерять избыток энергии в клетке
растворителя и затем рекомбинировать. Так как моле-
кулы растворителя присутствуют в растворе в большей
концентрации, то реакции рекомбинации в растворе
имеют значительно большую вероятность, чем в газовой
фазе.
. Другие физические процессы, связанные с дезакти-
вацией молекулы, лучше всего могут быть проиллюстри-
рованы рис. 6.1. На этом рисунке схематически
показаны несколько электронных уровней и часть колеба-
тельных и вращательных уровней типичной органиче-
ской молекулы. Синглетные электронные уровни пока-
заны слева, а триплетные — справа *. Сначала рассмот-
рим только синглетные состояния: вслед за поглоще-
нием может следовать безызлучательный процесс. Он
показан слева на рисунке в виде волнистой стрелки,
* В спектроскопии мультиплетность состояния определяется
членом 2S+1, где S есть общий электронный спин. Если присутст-
вуют - только спаренные электроны, все спины компенсируются
S=0 и система синглетна. Если два электрона имеют параллельные
спины, каждый со значением 1/2, то S=1 и состояние триплетное.
62
соответствуя каскадному переходу по ряду колебатель-
ных уровней и в результате внутренней конверсии до
более низких синглетных электронных уровней; энер-
гетические переходы в этих процессах тушатся другими
молекулами системы. За поглощением может следо-
вать переход, при котором испускается излучение — та-
кие переходы показаны сплошными стрелками. Излуча-
тельные переходы между двумя состояниями одной и
Синглетные состояния
Триплетные состояния
Рис. 6.1. Физические процессы, следующие за фотовозбуждением:
Сплошные прямые стрелки соответствуют излучательным процессам, волни-
стые— безызлучательным процессам, пунктирные стрелки — переходы между
уровнями различной мультиплетности.
той же мультиплетности называются флюоресценцией.
Так как время жизни системы в возбужденном состоя-
нии очень мало, флюоресценция происходит в течение
Ю~9—10-6 сек после поглощения.
Теперь рассмотрим триплетные состояния, показан-
ные на рис. 6.1 справа. В части молекул может произой-
ти переход с возбужденного синглетного состояния в
метастабильное триплетное. Безызлучательные перехо-
ды между состояниями различной мультиплетности
иногда называют межсистемными переходами, эти пере-
ходы на рис. 6.1 показаны пунктирными линиями. Заме-
тим, что межсистемные переходы происходят с одного
63
электронного уровня на колебательный уровень с той
же энергией в другой системе, таким образом, этот
процесс происходит адиабатически. Теоретически пере-
ходы между состояниями различной мультиплетности
запрещены, однако реальные состояния не являются
чисто триплетными или синглетными, поэтому межси-
стемный переход в данном случае происходит, но с
меньшей вероятностью, чем обычная внутренняя кон-
версия. Время жизни в триплетном состоянии может
быть дольше, чем в возбужденном синглетном состоя-
нии, и излучательный переход (высвечивание) с три-
плетного обратно на синглетный уровень может длиться
10~3( сек и даже больше. Такие излучательные переходы
между состояниями различной мультиплетности назы-
вают фосфоресценцией. В органических веществах фос-
форесценция обычно осуществляется, как показано на
рис. 6.1: с самого низкого возбужденного триплета па
колебательный уровень основного электронного со-
стояния. '
Сформулируем определения основных понятий:
1) виутреиняя конверсия: - безызлучательный пере-
ход между состояниями одинаковой мультиплетности;
2) флюоресценция: излучательный переход между
состояниями одинаковой мультиплетности;
3) межсистемный переход: безызлучательный пере-
ход между состояниями разной мультиплетности;
4. фосфоресценция: излучательные переходы между
состояниями разной мультиплетности;
5) тушение: дезактивация возбужденного состояния
другими компонентами системы.
Прежде чем обратиться к примерам различных хи-
-мических реакций, которые могут следовать за актом
поглощения света, представляет интерес вопрос, каким
способом измерить количество квантов света. 1
Актинометрия
Актинометрией называется измерение полного коли-
чества радиации, попавшего на изучаемую систему.
Одним из типов актинометра является термостолбик,
представляющий собой группу термопар, у которых опре-
деленные концы прижаты к черной поверхности. Для из-
мерения излучения, поглощенного системой, термостол-
бик помещается за реакционным сосудом и измеряется
64
разница в интенсивности света при полном и пустом
сосудах. Термостолбик может быть калиброван при
использовании ламп с известными характеристиками
(стандартные лампы можно получить в Палате мер- и
весов).
Для определения интенсивности света можно также
использовать химическую реакцию с известным кванто-
вым выходом. Квантовый выход Ф равен числу молекул
продукта, образованных при поглощении одного кванта
света. Более удобное определение: квантовый выход
равен числу молей продукта на эйнштейн поглощенного
света. Так как поглощенная энергия может теряться в
результате физических процессов, при которых могут
не протекать химические реакции, то квантовый выход
может быть очень маленьким. С другой стороны, про-
дукты реакции могут образоваться в результате реак-
ции, инициированной первичным фотохимическим про-
цессом, и Ф может быть очень большим.
В актинометрии часто используют химическую
реакцию разложения щавелевой кислоты, фотосенсиби-
лизированную солями ураиила. Ион уранила UO2+ по-
глощает свет в области 2500—4500 А. Возбужденный
ион уранила (UO2+)* способен передавать избыток
энергии молекуле щавелевой кислоты, что приводит к ее
разложению: ;
UOf+ + Ь (1Ю2+Г
(UO2+)‘ + НООССООН ис>2+ + со2 + СО + Н2О
Квантовый выход СО и СОг измерен в различных экс-
периментальных условиях, и реакция достаточно чув-
ствительна, чтобы детектировать 7*1013 квантов све-
та [2].
Примеры фотохимических реакций
В некоторых случаях в облученных молекулах про-
исходят как физические, так и химические процессы,
конкурирующие между собой. Например, если антрацен
облучать светом в растворе бензола, он димеризуется
и одновременно флюоресцирует. Если раствор бензола
сделать более концентрированным, квантовый выход ди-
мерных продуктов возрастает, а интенсивность флюо-
3 У. Прайер
65
ресценции уменьшается. Уравнения этих реакций пока-
заны ниже:
А + Av —*- А*
(6.1)
Здесь А — антрацен, А* — молекула возбужденного
антрацена, v — частота излучения, у'— частота флюо-
ресценции. ,
Активированные молекулы образуются в реакции
(6.1) и исчезают в реакциях (6.2) и (6.3). Так как воз-
бужденные молекулы А* имеются в очень малых кон-
центрациях, разумно предположить, что их концентра-
ция достигает стационарного значения на очень ранней
стадии реакции. Это так называемое предположение о
стационарном состоянии; оно оказывается справедливым
для некоторых очень реакционноспособных молекул,
которые присутствуют в малых концентрациях, — ско-
рость изменения концентрации также мала и в некото-
ром приближении может быть приравнена нулю. Таким
образом, в стационарном состоянии скорость изменения
концентрации А* равна нулю, при этом скорость обра-
зования А* равна скорости исчезновения А*. Скорость
образования А* равна <р7Погл, где <р — часть молекул А,
которые становятся активированными при поглощении
света, /погл — скорость поглощения света молекулами А,
часто называемая интенсивностью поглощения света.
Скорость исчезновения А* определяется выражением
к2[А] [А*] Н-к3[А*]. Отсюда при стационарном состоя-
нии концентрации А*
ф/погл = «2 [А] [А*] + к3[А*]
и [А*] = Ф7"°гл
К2 [А] + к3
66
Скорость образования димеров
d [димер] __ % .д, __ [Д] чАюгл _ <р/погл [А]
dt 2 ‘ Л2[А] + к3 [А] + к3/к2
Таким образом, квантовый выход димеров
1 _ d [димер] = ф ср [А]
/погл dt [А] + К3/К2
При малых концентрациях к3/к2>[А'] и ФДИмер=
= ф —[А], выход димеров растет с увеличением кон-
центрации антрацена. При высоких концентрациях
[A] >K3/«2 и ФДимер=Ф, т. е. достигается предельное
значение квантового выхода димеров.
В некоторых случаях облучение приводит к появле-
нию как радикальных, так и нерадикальных продуктов
в конкурирующих процессах. Например, при облучении
бутиральдегида в газовой фазе имеют место две реак-
ции разложения: одна дает пропильные и формильные
радикалы, а другая—этилен и ацетальдегид [3]:
СН3—СН2—СН2—СНО + hv -> (СН3—СН2—СН2—СНО)*
(СН3—СН2—СН2—-СНО)* -> СН3—СН2—сн2- + -сно
(СН3-СН2—СН2-СНО)* -> Н2С = СН2 + СН3-СНО
Реакция хлора и водорода, включающая фотохими-
ческий процесс, изучена очень тщательно:
С12 + Ь -> 2С1 • (6.4)
С1- + Н2-\ НС1 + Н- (6.5)
H-H-CU —6—НС1 + С1- (6.6)
2С1 - С12 (6.7)
Скорость образования атомов хлора равна 2 <р7Погл, а
скорость их исчезновения есть 2к7[С1«]2. В стационар-
ном состоянии
2Ф/погл = 2к, [С1-]2,
[С1-] = ПОГЛ \ 1^2
\ *7 / ’
Скорость образования хлоргидрата
rf[”C11 = Кх [С1 • ] [Н2] + к„ [Н • ] [С12].
3*
67
В стационарном состоянии для атомов водорода реак-
ции (6.5) и (6.6) должны иметь одинаковую скорость.
Поэтому
= 2кб [С1 • ] [Н2] = (ф/погл)*/! [Н2].
dt K-f*
Квантовый выход хлоргидрата Фна в этом процессе
лежит в пределах от 104 до 106. Каждый атом хлора
образует при инициировании [реакция (6.4)] многих
тысяч цепных последовательностей (6.5) и (6.6), преж-
де чем реакция (6.7) выведет активный атом хлора из
цепи *.
Целесообразно провести различие между квантовым
выходом первичного фотохимического процесса ср и кван-
товым выходом конечного продукта Ф. Первичный фото-
химический выход ф определяется как та часть погло-
щающих радиацию молекул, которая прореагировала
при поглощении одного фотона. Величина ф должна ле-
жать между нулем и единицей. Квантовый выход Ф дан-
ного продукта в фотохимической системе есть число
молекул продукта, образованного при поглощении одно-
го фотона. Величина Ф лежит в пределах от нуля до
миллионов и, как мы видели, больше. При фотолизе
хлора, обсужденном выше, квантовый выход первичного
фотохимического процесса [реакция (6.4)] не может
быть больше единицы, так как каждый фотон приводит
к диссоциации только одной молекулы хлора. Кванто-
вый выход образования хлоргидрата Фна может иметь
порядок величины 106, в зависимости, от длины цепи
реакции**.
Карбонильные соединения
Для протекания фотохимических реакций необходи-
мо, чтобы субстрат поглощал свет используемой длины
волны или должен присутствовать фотосенсибилизатор,
который способен поглощать энергию излучения и пере-
* Детальное описание реакции водорода с бромом дано в мо-
нографии Фроста и Пирсона [4].
** Необходимо различать термины «выход» и «квантовый вы-
ход». Под выходом продукта обычно понимают число молей про-
дукта, образованного на моль израсходованных исходных веществ.
Квантовый выход есть число молей продукта, образованного на
одни эйнштейн поглощенного света.
68
давать ее на субстрат. Многие из ранних исследований
[5] фотохимических реакций были проведены па карбо-
нильных соединениях, так как они имеют'полосу погло-
щения вблизи 3200 А и поэтому могут быть возбуждены
при использовании относительно простого оборудования.
а
п электронов (показана
только одна пара\
^^/Несвяэывающая п*-орбита
б OS ^с—о*
п- Электроны кислорода
Рис. 6.2. Два представления п—л*-процесса возбуждения.
Длинноволновая полоса поглощения карбонильных
соединений связана с возбуждением неподеленного (п)
электрона на несвязывающую л-орбиту (обозначаемую
л*). Такой п—л*-процесс возбуждения показан на
рис. 6.2. При рассмотрении п—л*-процесса наглядно
проявляется структура молекулярных орбит (см.
рис. 6.2, а). л*-Связь характеризуется высокой элект-
ронной плотностью между атомами углерода и кисло-
рода: л*-орбита имеет здесь узел и электронная плот-
ность между этими атомами равна нулю. Процесс
п—л*-возбуждения приводит к переходу одного из непо-
деленных электронов кислорода на л*-орбиту. На
рис. 6.2, а этот электрон обозначен как б-—он присут-
ствует частично на атоме как кислорода, так и угле-
рода. Однако л*-орбита имеет центр тяжести, смещен-
ный к углероду, и из этих соображений лепестки элект-
ронной плотности в атоме углерода большего размера,
чем в атоме кислорода.
На рис. 6.2, б представлены две резонансные струк-
туры возбужденного состояния. Из рис. 6.2, б ясно, что
атом углерода имеет большую электронную плотность
в возбужденном состоянии, чем в основном состоянии,
из чего следует чрезвычайная локализованность места
фотохимической реакционности у n-орбиты на атоме
69
кислорода. Кроме того, кислород оказывается лишен-
ным электрона и реагирует подобно электрофильному
алкокси-радикалу. Например п—л*-состояние может
отщеплять атомы водорода:
\ \-------- RH \
J>C = О: — 2е’ =- °- —” /С-—О—Н + R-
Если реагент не взаимодействует с атомом кислорода
за время жизни возбужденного состояния п—л*, моле-
кула возвращается в исходное состояние в результате
излучательного перехода или процесса внутренней кон-
версии.
До сих пор мы не уточняли, является возбужденное
состояние п—л* триплетным или синглетным. Конечно,
нет ничего удивительного в том, что молекулы в три-
плетном состоянии способны проводить реакции, подоб-
ные радикальным, как, например, отщепление водоро-
да. Можно ожидать, что молекула в синглетном воз-
бужденном состоянии обладает такой способностью в
меньшей степени. Как синглетное, так и триплетное
состояния характеризуются определенным разделением
зарядов — в синглете электронные пары остаются с ан-
типараллельными спинами, а в триплетном состоянии
один из спинов переворачивается в результате внутри-
системного перехода. В то же время пространственное
распределение электронной пары может быть абсолютно
подобным в этих двух случаях. Так, в одной и той же
молекуле лишний электрон пары не может сильнее раз-
делиться в триплетном, чем в синглетном состоянии.
Более детальное рассмотрение [6], показывает, на-
сколько важным является учет разницы в энергиях син-
глетного и триплетного состояний. Если разница мала,
оба состояния стремятся иметь одинаковое электронное
распределение, и синглетное состояние также может
подвергаться реакциям, подобным радикальным. Если
оазница в энергиях велика, то два неподеленных элект-
рона дольше разделены в триплете — это образование
ведет себя как бирадикал, в этом случае нельзя ожи-
дать, чтобы синглетное состояние характеризовалось
реакциями, подобными радикальным. Во всех случаях
триплетное состояние парамагнитно, а синглетное со-'
стояние не парамагнитно *.
* Разница в реакционной способности триплетных и синглетных
состояний в настоящее время не полностью ясна. Кинетический
подход при изучении этих состояний показывает, что триплетное
70
Ознакомимся более детально с основными особенно-
стями фотохимических реакций карбонильных соедине-
ний. Когда альдегиды возбуждаются светом, в них
могут происходить четыре различных типа химических
реакций, в зависимости от структуры альдегида и длины
волны света. Ниже показаны четыре реакции типичного
линейного альдегида:
R—сн2—сн2-сно -> r-ch2—сн2+-сно
R-CH2—СН2-СНО -> R—СН2—СН34-СО
R-CH2—СН2-СНО R—СН -= СН2 + нсно
R--CH2-CH2-CHO -> R—СН2 • + сн2—сно
Кетоны также подвергаются целому ряду фотохими-
ческих превращений. При фотолизе ацетона получаются
ацетильный и метильный радикалы. Ацетильные ради-
калы в свою очередь подвергаются p-расщеплению, об-
разуя другой метильный радикал и молекулу окиси
углерода. Конечными продуктами являются этан и окись
углерода:
С3Н6О 4- Ь С3Н6О*
С3Н6О* -> СН3- -Г СН3—С=О
О
' II
сн3—с- -> сн3 + со
2СН3. ->.С2Н6
где СзН6О* — молекула ацетона, поглотившая квант
света и находящаяся в фотовозбужденном состоянии.
Квантовый выход окиси углерода Фео равен 1,0 при
2537 А и 0,7 при 3130 А. Фотолиз ацетона тщательно
состояние, вероятно, способно к типичному отщеплению водорода.
Синглетное возбужденное состояние обычно переходит в основное
за время около 10-8 сек. Отщепление водорода, которое обычно
имеет энергию активации около 7 ккал/моль, как правило, не за-
вершается за время такого быстрого процесса дезактивации.
В тоже время триплетные состояния часто имеют время жизни
10-3 сек и более и таким образом отщепление водорода может
происходить достаточно эффективно по сравнению с процессами
синглет-триплетного перехода или фосфоресценции. Мы рассмотрим
разницу в реакционной способности синглетных и триплетных со-
стояний еще раз в гл. 19, в которой будут рассматриваться бира-
дикалы. 1
71
изучен, и выделение окиси углерода часто используют
в актинометрии. Эту реакцию применяют также как
источник метильных радикалов при различных исследо-
ваниях в газовой фазе.
Кетоны, содержащие одну алкильную группу и по
крайней мере три атома углерода в жирной цепи, раз-
рушаются по внутримолекулярному механизму без уча-
стия свободных радикалов. Например, метилбутидкетон
дает ацетон и пропилен:
hv
сн3-со—сн2—сн2-сн2-сн3 —►
hv
-> сн3 —со -сн3 + СН2 = СН—сн3
Квантовые выходы олефина и.ацетона почти равны
во всех экспериментальных условиях, и отсюда можно
заключить, что они образуются в одном первичном фо-
тохимическом процессе. Наиболее приемлемое объясне-
ние такого разложения предусматривает миграцию ато-
ма водорода при у-атоме углерода с образованием
энольной формы ацетона и олефина:
О
IU
сн—с—с4н9
км
СИ-
ОН
I
—*- сн3—с=сн2 + сн2=сн—сн3
Такой механизм реакции подтверждается следующими
наблюдениями: 1) метиламилкетон дает ацетон и
бутен-1; 2) метилизопропилкетон и диизопропилкетон,
который не имеет атома водорода в у-положении, не
разлагаются внутримолекулярным путем; 3) метил-
втор-бутилкетон дает этилен и метилэтилкетон:
СН3— С^-уСН2
СН
I
СН3
ОН
СНз—С=СН—СН3 + СН2=СН2
В результате такого внутримолекулярного процесса раз-
лагаются и другие кетоны:, дипропилкетон, метилпро-
пилкетон, метиламилкетон, метилгексилкетон и метил-
неопентилкетон.
72
Для получения возбужденных молекул, которые
трудно возбудить прямыми методами, можно использо-
вать явление передачи энергии. Например, при облуче-
нии циклопентадиена выход триплетных состояний мал
из-за низкой вероятности синглет-триплетного перехода.
С другой стороны, триплетное состояние бензофенона
генерируется с высоким квантовым выходом. Если облу-
чать смесь этих двух соединений, образуется триплет-
ное состояние кетона, который затем передает свое воз-
буждение диену. Триплетное состояние циклопентадие-
на реагирует по схеме, показанной ниже [7, 8]:
Синглет-
Ph2c=o (Ph2c-O)‘ T°n\n™iiHPh2c-0)>
Возбужденный Возбужденный
синглет триплет
(Ph2C—о)
Триплет
О
Триплет
33%
Этот результат используется практически, так как пря-
мая термическая димеризация циклопентадиена при
помощи обычного механизма Дильс-Альдера приводит
к 100%-ному выходу эндо-продукта.
Тепло
Дильс-Лдьдер
73
Аналогично тому как большинство радикалов спо-
собны к отщеплению атомов водорода, не удивительно,
что триплеты также могут реагировать таким путем.
Отщепление водорода карбонильными соединениями в
триплетном состоянии, как и можно было ожидать, осо-
бенно облегчено, так как атом кислорода в триплетном
состоянии карбонила является электрононедостаточным
и должен вести себя подобно электрофильному алкокси-
радикалу. Облучение бензохинона в присутствии раз-
личных доноров водорода позволяет определить отно-
сительные скорости отщепления водорода, которые
оказались симбатны скоростям, полученным при ис-
пользовании щрет.-бутокси-радикала. В табл. 6.1 при-
ведены данные, иллюстрирующие это положение.
Таблица 6.1
Скорости реакции отщепления водорода бензофеноном в триплетном
состоянии по сравнению с трет-бугокси-радикалом [9]
Доноры водорода Относительные скорости Доноры водорода Относительные скорости
Ph2C — О- трет - -С4Н9О< Ph2C — О- трет - - С:Н9О-
Толуол Мезитилен м- Ксилол 1 5,5 з.о 1 4,1 2,3 п-Хлортолуол Циклогексан 0,97 2,1 0,71 6,0
Не удивительно, что mpem-бутоксй-радикал и бензо-
хинон в триплетном состоянии отщепляют атомы водо-
рода с одинаковыми относительными скоростями, так
как они оба образуют связь с водородом с одинаковой
энергией связи.. Энергия диссоциации связи О—Н в
mpem-CiHsO—Н равна 104 кк.ал!моль. Из показанного
в табл. 6.2 термодинамического цикла можно опреде-
лить энергию связи О—Н в Ph2C—О—Н, она оказалась
равной приблизительно 102 ккал!моль.
Хотелось бы рассмотреть еще одно свойство фотохи-
мических реакций. Принцип Франка—Кондона утверж-
дает, что поглощение энергии излучения молекулой про-
исходит настолько быстро, что в течение процесса
поглощения атомы не успевают перегруппироваться.
Поэтому структура молекулы в возбужденном состоянии
должна быть такой же, как и в исходной молекуле.
74
Таблица 6.2
Энергия связи
Реакция Д?/, ккал/моль
Ph2CH — ОН- Ph2C = О + Н2 9*
Ph2C = О - Ph2C —О- 69**
Н- + Ph2C — ОН - Ph2CH — ОН —80***
Н2- 2Н- 104*
Сумма: Ph2C— ОН - Ph2C— О- + Н 102
* Из энергии связи.
** Из спектроскопии [10]. -
*** В предположении о равенстве обычной энергии связи С — Н бензола.
Например, при фотолизе ацетона поглощение света с
2, = 3130А происходит в карбонильной группе
О
II
сн3—с-сн3 + Ь ->
/ ° у
11
\СН3—с —сн3/
Однако двойная связь углерод — кислород очень силь-
на, и вслед за поглощением следует перераспределение
избытка энергии на другие колебательные формы моле-
кулы— этот процесс требует 10-12—10-8 сек. Расщепле-
ние углерод-углеродной связи происходит тогда, когда
на ней концентрируется достаточное количество энергии
О
II
СН3—С—сн3
о
II
-> СН3—с-+ сн3-
Хотя расщепление происходит непосредственно в ре-
зультате электронного возбуждения, молекула в воз-
бужденном состоянии находится настолько малое вре-
мя по сравнению с последующими химическими реак-
циями, что часто реакция пишется как одна ступень
схемы фотолиза
О
II h.
СН3—С—сн3 СН3СО- + сн3-
75
Упражнения
6.1. Выход tp первичной фотохимической реакции равен пример-
но 0,05, а квантовый выход продукта Фп около 104. Сколько Пасов
потребуется на получение 1 моля продукта П, если интенсивность
используемого света 1017 квант/сек? Оценить время при Фп = 10.
6.2. В типичном при фотолизе эксперименте ртутная лампа
снабжается фильтром, пропускающим свет %=3130А. С помощью
термостолбика было измерено, что в течение 10 ч поддерживалась
интенсивность излучения 1.0- 10s эрг! сек. Сколько килокалорий на
эйнштейн дает свет 3130 А? Каково общее количество поглощенной
энергии (1 эрг=2,39-10-11 ккал)? Сколько получено эйнштейнов?
Сколько молей продукта получится при квантовом выходе 0,25?
6.3. При облучении бензохинона Ph2C=O в изопропиловом
спирте при температуре 25° С светом с длиной волны 3660 А полу-
чаются Ph2C(OH)—(OH)CPh2 с квантовым выходом 0,93 и ацетон
с квантовым выходом 0,92. При наличии в системе растворенного
кислорода квантовый выход ацетона не меняется, а бензопинакола
падает до 0. Предложите механизм для этой реакции [11]. '
Примечание: Бензохинон переходит в возбужденное со-
стояние, в котором возможно отделение водорода. Заметим, что на
каждые две молекулы использованного бензохинона образуется одна
молекула ацетона. Разумеется, прёдложенный механизм должен
соответствовать этому. Найдено Также, что если вместо изопропи-
лового спирта использовать оптически активный 2-бутанол, то вос-
становленный 2-бутанол не рацемизуется. Соответствует ли это
механизму реакции [16]?
6.4. В экспериментах, проводящихся для изучения первичного
фотохимического воздействия на один из альдегидов — ацетальде-
гид, последний облучалси в газовой фазе при 60° С светом с длиной
волны 3130 А при различных давлениях паров иода. Иод представ-
ляет собой акцептор радикалов, и в его присутствии квантовый
выход продукта падает. Квантовый выход окиси углерода умень-
шается от 0,34 в отсутствие иода до 0,21 при давлении паров иода
1—3 мм рт. ст. Соответственно квантовый выход метаиа падает от
0,32 до 0,013. Если давление иода увеличивать, то основным про-
дуктом становится метилиод, квантовый выход которого при дав-
лении паров иода 1—3 мм рт. ст. достигает значения 0,20. Учитывая
приведенные результаты, выберите одну из предлагаемых схем
первичного фотохимического акта:
СН3—СНО + ftv -» СН4 + СО
СН3—СНО-[-Ь-^СН3- +НСО
Дайте объяснение тому, что Фс0 в присутствии иода стано-
вится равным Фснаг 112].
6.5. Альдегиды подвергаютси быстрому цепному декарбонилн-
рованию
R +R-CHO —> RH + R—СО
R—СО — R- +СО
Тиолы увеличивают длину цепи этой реакции в результате ряда
ступеней передачи атома водорода. Было показано, что бензохинон
76
Может фотосенсибилизировать декарбонилирование альдегидов, при
этом наилучший результат получается, если присутствуют очень
маленькие концентрации бензохинона и тиола. Покажите, каким
мозйет быть механизм декарбонилирования в этих условиях [13].
6;6. Облучение 5,9-диметилдеканона-2 (оптически активный в
пятому положении) ртутной лампой приводит в образованию 1,2-ДИ-
метил-2-(4-метилфенил) циклобутанола-1, который остается по край-
ней мере на 16°/о оптически активным:
Н О
I II
(СН3)2С—С—С—С—С*—С—С—С—С -<СН3),С—С-С—С‘2------------он
Объясните этот результат, предполагая, что первой ступенью в ре-
зультате воздействия света является образование триплетного
п—л*-состояния [14].
6.7. При фотолизе ацетона в газовой фазе отношение выхода
метана, деленного на -выход этана, пропорционально давлению аце-
тона. Предложите механизм, объясняющий этот результат.
6.8. Облучение 5 г 5,5-диметилгексен-3-деканона-2 в 400 мл
эфира приводит к медленному образованию изомера кетона, кото-
рый достигает максимума выхода 38% через 46 ч. Спектр ЯМР про-
дукта свидетельствует о наличии трех атомов водорода вблизи
нуля ^химического сдвига (характерно для водорода в циклопропа-
не). Предложите механизм реакции и, представьте структуру обра-
зующегося продукта [15]:
С О
I II hi
С—С—С = с—с—с ?
I,
с
ЛИТЕРАТУРА
1. Porter G., Smith J. A. Proc. Roy. Soc., Ser. A621, 28
(1961); Lampe F. W., Noyes R. M. J. Amer. Chem. Soc., 76,
2140 (1954).
2. Volman D. H., Seed J. R. J. Amer. Chem. Soc., 86, 5095 -
(1964).
3. Bl acet F. E., Calvert J. G. Ibid., 73, 661 (1951).
4. Frost A. A., Pearson R. G. Kinetics and Mechanism. John
Wiley and Sons. Inc., N. Y„ 2d. ed„ 1953, p.^236.
5. Pitts J. N. J. Chem. Educ., 34, 112 (1957); Masson C. R.,
В ос k e 1 b e i d e V., N о у e s W. A. In: Technique of Organic-
Chemistry. A. Wessberger (ed.). Vol. 2. Interscience Publishers.
Inc., N. Y., 2d. ed., 1956, p. 307.
6. McGlynn S. P., Smith F. J., Ci lent о G. In; Photoche-
mistry and Photobiology. Vol. 3. Pergamon Press. N.Y., 1964,
p. 269.
W a g п e r P. J., Hammond G. S. J. Amer. Chem. Soc., 87,
4010 (1965).
7. Turro N. J., H a m m о n d G. S. J. Amer. Chem. Soc., 84, 2841
(1962).
77
8. L e e r m а г к е г s Р. А., V е s 1 е у G. F. J. Chem. Educ., 41, 535
(1964). /
9. W а 11 i n g C., Gibia n M. J. J. Amer, Chem. Soc., 86, 3002
(1964).
10. H e г к s t г о c t e г W. G., La mote A. A., Hammond G. S.
Ibid., 86, 4537 (1964). /
11. Pitts J. N. et al. Ibid., 81, 1068 (1959).
12. Bl acet F. E„ H e 1 d m a n J. D. Ibid., 64, 889 (1942).
13. В e r m a n J. D. et al. Ibid., 85, 4010 (1963).
14. Orban J., Schaffner K-, Jeger O. Ibid., 85, 3033 (1963).
15. Jorgenson M. J., Yang N. C. Ibid., 85, 1698 (1963).
16. Moore W. M. et al. Ibid., 83, 2789 (1961); Moore W. M„
К e t c h u m M. D. J. Phys. Chem., 68, 214 (1964).
Дополнительная литература
К a s h a M. In: M. Burton, J. S. К i r b у - S m i t h, J. L. M a -
gee (eds). Comparative Effects of Radiation. Chap. 5. John Wi-
ley and Sons. Inc., N. Y., 1960.
McGlynn S. P„ Smith F. J., Ci lento G. Some Aspects of
the Triplet State. In: Photochemistry and Photobiology. Vol. 3.
Pergamon Press. N. Y., 1964.
Noyes W. A., Hammond G. S., Pitts J. N. (eds). Advances
in Photochemistry. Vol. 1. Interscience Publishers, Inc. N. Y.,
1963.
Hammond G. S., Turro N. J. Science, 142, 1541—53 (1963).
Leer makers P. A., Vesley G. F. J. Chem. Educ., 41, 535
(1964).
Walling C. Free Radicals in Solution. John Wiley and Sons,
Inc. N. Y., 1957. (Уоллинг Ч. Свободные радикалы в растворе.
Перев. с англ. М., Изд-во иностр. лит., 1960.)
Глава 7 i
ТЕРМИЧЕСКИЙ ГОМОЛИЗ. ПЕРЕКИСИ
Рассмотрим реакции получения радикалов из неко-
торых общего типа инициаторов. Эта глава посвящается
химии перекисей. В последующих главах рассмотрим
надкислоты и эфиры надкислот, перекись водорода, гид-
роперекиси и химию азосоединений.
Рассмотрение химии перекисных соединений, обра-
зующих свободные радикалы, целесообразно начать с
номенклатуры. Названия основных перекисей и образу
ющихся из них радикалов даны в табл. 7.1.
Перекиси разлагаются, по крайней мере, согласно
трем механизмам. По одному из них идет мономолеку-
лярное гомолитическое расщепление перекисной связи:
RO—OR 2RO- _
78
Таблица 7.1
Номенклатура перекисных соединений
Основные названия Структура
Соединения
Перекись алкила ROOR
Перекись ацила О О 1! II R __С — О—О — С—- R
Гидроперекись ROOH
Надкислота или R — С — О — ОН
надохсикислота II
Эфиры надкислот или О О II R — С — О — OR'
надоксикислот. О II R — O — О — С — О - O-R
Надкарбонат или над-
оксикарбонат О О
Надоксалат или над- II II R —О —О —С —С —О — О —R
оксиоксалат
Радикалы
Алкокси RO-
Аткилнадокси ROO-
Ацил О II R-C-
Ацилоксн О II R —С — О-
Ацилнадокси О II R —С —О —О-
Специфические радикалы
Метокси СН3О-
Ацетил О II сн3-с-
Ацетокси или ацетат О II СН3 —С—О-
Продолжение табл. 7.1
Основные названия
Надацета' ил i гцетилнад-
о.:си
Бензоат или бензоилокси
Надбензоат или бензоил-
надохси
Структура
О
II
СН3 —С —00-
о
II
Ph —С —О-
О
II
Ph —С —ОО-
Свободные радикалы, получающиеся в ( результате
такой диссоциации, могут разлагаться дальше (напри-
мер, p-расщепление) либо атаковать в той же системе
другие молекулы, инициируя последующие радикальные
процессы.
Вторым механизмом разложения перекисей можно
представить бимолекулярную реакцию, называемую ин-
дуцированным разложением. В этом процессе радикалы
атакуют перекиси и вызывают их разложение. Сами
атакующие радикалы могут возникать непосредственно
из перекисей или в результате какой-либо последующей
реакции. Механизм индуцированного разложения зави-
сит от структуры перекиси. Для простейших алкилпере-
кисей он заключается в отрыве атома водорода от а-
атома углерода
R- -4-R2CH — 00— CHR2 -> RH + RiC-— ОО-CHR^
В случае перекиси бензоила имеет место 5я2-атака
на О—О-связь:
О О О
II II II
R- -4- Ph—С — 00 — С—Ph -> PhCO2- + R—0-С—Ph
Реакция индуцированного разложения подавляется
инициатором, так как он разрушает молекулу перекиси
без увеличения числа радикалов.
Третьим механизмом разложения перекисей является
малоизученный процесс детонации, который делает все
перекисные соединения опасными в обращении. Переки-
80
си различаются по своей чувствительности к взрыву.
Особенно опасны перекиси с низким молекулярным ве-
сом\ Из перекисей алкилов чрезвычайно легко детони-
руетХперекись метила, в то время как перекись трет-бу-
тила 'необычно стабильна и является продажным про-
дуктов. Среди перекисей ацилов весьма опасна
перекись ацетила, тогда как перекись бензоила чувстви-
тельна взрыву в гораздо меньшей степени. Однако
любые йерекиси потенциально взрывоопасны, поэтому
их нельзя растирать, встряхивать или нагревать без
особых предосторожностей. (
Перекиси алкилов
Все перекиси алкилов* подвергаются мономолеку-
лярным реакциям разложения с одинаковыми скоростя-
ми. Значения энергии активации для ряда: перекись
этила — перекись /прет-бутила — в пределах от 34 до
37 ккал/моль соответственно [1]:
RO-OR -> 2RO-
В большинстве случаев продукты последующих реакций
алкокси-радикалов неизвестны. Однако известно, что
при разложении перекиси этила образуются этанол и
ацетальдегид, которые могут получаться диспропорцио-
нированием этокси-радикалов:
2СН3—СН2О- -> СН3—СН2—ОН + сн3—сно
Они могут также возникнуть в процессе отщепления
этокси-радикалом водородного атома от а-атома угле-
рода в молекуле перекиси, которая далее разрушается
по реакции р-расщепления:
СН3СН2ООСН2СН3 + СН3СН2О-
-> СН3СНООСН2СН3 4- СН3СН2ОН
. (Э-Расщепление
СН3СНООСН2СН3-------------- сн3сно + СН3СН2О •
Перекись mpem-бутила благодаря ее исключительной
стабильности изучали более детально, чем другие пе-
*В названиях симметричных диалкилперекисей приставку «ди»
можно опустить. Например, СН3ООСН3 допустимо называть пере-
кисью метила.
81
рекиси алкилов. Константу скорости ее разложения
представляют величиной
к = 101ве 37 000/ЛГ сек~\
Период полураспада (полуразложения) т>/2 = 11 годам
при 60° и 35 сек при 180° С. Скорости расщепленйя этой
перекиси в концентрированном растворе и в/газовой
фазе почти одинаковы. Из этого можно заключить, что
большинство перекисей разлагается по мономблекуляр-
ному механизму без развития цепей. Если большинство
перекисей реагировало бы по механизму, предусматри-
вающему развитие цепи большой длины, то, по-видимо-
му, скорость такого процесса в растворе была бы боль-
ше, чем в газовой фазе, так как концентрация радика-
лов в более концентрированной фазе, т. е. в растворе,
выше. Механизм разложения в газовой фазе показан
схемами (7.1) — (7.3); метильные радикалы димери-
зуются почти количественно, и основными продуктами
разложения перекиси трет-бутила являются ацетон и
этан:
mpem-C4H9O—OC4H9-mpem -> 2mpem-C4H9O- (7.1)
СН3 О
! р-Расщепление II
СН3—С-О--------------СН3-+ СН3-С-СН3 (7.2)
I
сн3
2СН3- -> С2Н6 (7.3)
Если перекись используют в качестве растворителя,
то образуются следующие продукты:
mpem-C4H9O—OC4H9-mpem
О + О
/\ II
Н2с—С—СН3 + mpem-C4H9OH + СН3—С—СН3 +
I 0,50 0,66
сн3
от 1,0 до 0,7
СН4 -|- С2Н6
0,63 0,02
где числа под соединениями обозначают количество
молей продукта на моль перекиси.
82
Образование этих продуктов можно объяснить сле-
дующими реакциями. Ацетон и этан образуются соглас-
но реакциям (7.2) и (7.3). Метан получается в резуль-
тате 'отрыва атома водорода от перекиси после атаки
метильным радикалом:
СН3
I
СН3- 4- преш-С4Н9О—О—С—СН3 ->
I
сн3
•сн2
-^-СН4 4-m/?em-C4H9O—О—С—СН3
СН3
I
Аналогично mpem-бутанол образуется в результате от-
рыва водорода от перекиси трет-бутокси-радикалом:
СН3
I
трет-С^О + трещ-С4Н9О—О—С—СН3 ->
сн3
•сн2
->щрет-С4НвОН 4- трет-С4Н9О—О—С—СН3
СН3
I
Образование окиси изобутилена лучше всего объяс-
няется внутримолекулярной реакцией 5н2-расщепления
О—О-связи в радикале (I):
/•СН2 — сн2
Л рг I / I
трет-С^О—о— С—CH3-*mpem-C4H9O»+ О — С—СН3 (7.4)
! СИ3 СН3
Тщательное исследование продуктов разложения в
газовой фазе показало, что в таких же условиях раз-
83
бавления образуется около 3% окиси изобутилена [21.
Следовательно, даже в газовой фазе развивается не/о-
горая цепь по механизму:
fmnXw С Н О I + трет С^НвО—ОС4Н9-третг
{II1д\_/ * )
_> | ЬГ14 I 4- П)
(mpe/n-C4H9OHj >
(I) ->• mpem-C4H9O-+ окись изобутилена
mpem-C^Q- -> СН3 СО СН3 + СН3 •
Если перекись mpe/n-бутила разлагается .в присутствии
активного донора водорода RH; то метильный и трет-
бутильный радикалы могут реагировать, отрывая водо-
род от RH. В этом случае выход метана и трет-бута-
нола значительно повышается:
mpem-C4H9O • -> СН3 СО СН3 -|- СН3 • (7.5)
mpem-C4HeO- + RH -> mpem-C4H9OH -f- R- (7.6)
CH3- + RH-> CH4 + R- (7.7)
2CH3- -> C2H6 (7.8)
Эти реакции уже рассматривались при сравнении
способности различных растворителей отдавать водо-
род. Если выходы mpem-бутанола и ацетона близки, то
можно определить отношение скоростей реакции (7.5)
и (7.6). Аналогично сопоставление выходов метана и
этана дает отношение скоростей реакций (7.7) и (7.8).
Чем активнее растворитель RH как донор водорода,
тем больше отношения спирт/ацетон и метан/этан.
1 Перекись ацетила
Разложение перекиси ацетила можно представить в
виде реакций:
О О 2СН3-4-2СО2 (7.9а);
II II 7
СН3—С— О—О—С—СН3
[СН, -СО2- О2С—СН3] (7.96)
Клетка растворителя
84
[2СН3—С02 ] -> 2СН3—С02-
СН3-СО2- -> СН3 +СО2
CH3-+RH -> CH4 + R-
2СН3- -> С2Н6
О
[2СН2—СО2 ] -> СН3—С—о—сн3 + со2
[2СН3—СО2.] -> С2Н6 + 2СО2
(7-10)
(7.11)
(7.12)
(7.13)
(7.14)
(7.15)
где RH — донор водорода; радикалы, написанные в
скобках, — клеточные свободные радикалы (см. ниже),
другие радикалы существуют независимо друг от дру-
га. В газовой фазе перекись ацетила быстро диссоци-
ирует на метильные радикалы и двуокись углерода
[реакция (7.9 а)]. Однако в растворе она диссоциирует
на два ацетокси-радикала, которые находятся в непо-
средственной близости в пределах клетки растворителя
[реакция (7.9 6)]. Термин «клетка растворителя» впер-
вые предложили Франк и Рабинович в 1934 г. Дальней-
шее распространение этот термин получил в работах
Нейса [3], который предложил следующую картину
диссоциации. В газовой фазе возбужденные молекулы
фрагментируют на частицы. Вероятность рекомбинации
двух таких первичных частиц чрезвычайно мала. В ра-
створе, однако, молекулы растворителя уменьшают
диффузию первичной пары, и два партнера сближаются
лишь в течение 10”10 сек. За это время они успевают
прореагировать (реакция сдваивания). Тем не менее
раз уж такая пара диффундирует отдельно, то образую-
щие ее радикалы могут рекомбинировать с наименьшей
вероятностью'. Существует две особенности клеточных
реакций:
1) они не протекают в газовой фазе;
2) реакции сдваивания не замедляются и не зату-
хают при наличии радикалов-ловушек (которые могут
ловить только свободные радикалы)*.
* Теория клеточных реакций более сложная, чем рассмотренные
здесь положения. Рекомбинации сдваивания бывают двух типов.
Первичные рекомбинации возникают между радикалами, не успе-
вающими переменить свои позиции, и протекают в течение времени,
необходимого для разрыва связи. Вторичные рекомбинации проис-
85
В табл. 7.2 приведены выходы продуктов разлоя^с-
ния перекиси ацетила в жидкой и газовой фазе в срав-
нимых условиях. Этана образуется больше в газовой
фазе, чем в жидкой. Как и ожидалось, он образуется
Таблица 7.2
Разложение перекиси ацетила в газовой фазе и в растворе
толуола при 65° С [7]
Условия Газ • раствор
Реакционный объем, мл 5,000 10
Перекись, моль 3.8-10-5 19-10—5
Толуол, моль • 0,018 0,094
Перекись/толуол, мольное отноше- 2,010“ 3 2,0-10“3
ние Выход продуктов моль на моль разложившейся перекиси СН4 0,95 0,48
с2н6 0,23 0,05
О II сн3 — с — осн3 <0,005 0,15
со2 1,90 0,72
в результате рекомбинации свободных внеклеточных
метильных радикалов. Однако в присутствии паров
иода, которые служат ловушками свободных метильных
радикалов, в газовой фазе этан почти не обнаружива-
ется. С другой стороны, подавляющая часть этана в ра-
створе образуется в результате клеточной рекомбина-
ходят с радикальными парами, которые диффундируют неразде-
ленными, а затем разделяются за счет одной или более молеку-
лярных перегруппировок. Практически имеют место оба типа ре-
комбинаций. Например, при облучении иода светом с длиной волны
5780 А в гексане при 25° С около 35% атомов иода полностью поки-
дают клетку, около 30% разделяются на расстояние одного моле-
кулярного диаметра, но затем рекомбинируют за счет диффузии,
наконец, около 35% либо не диссоциируют совсем, либо рекомби-
нируют без диффузии из клетки растворителя, в которой они обра-
зовались [3, 4]. Клеточные рекомбинации также могут протекать
в газовой фазе с высокой плотностью паров, в то время как обыч-
ные давления не вызывают в газовой фазе клеточных рекомбина-
ций [5]. Детальные теоретические выкладки, касающиеся клеточ-
ного эффекта, приведены Нейсом [6].
86
ции. Это объясняется тем, что толуол, который служит
растворителем в этой реакции, фактически является ло-
вушкой свободных метильных радикалов. Как иод по-
давляет образование этана во внеклеточных реакциях в
газовой фазе, так и толуол действует аналогичным об-
разом в растворе. Следовательно, образующийся в ра-
створе этан должен возникать в результате клеточной
реакции (7.15). В газовой фазе метилацетат практиче-
ски не обнаруживается. Следовательно, его образование
в растворе является следствием либо клеточных реак-
ций, либо нерадикальных процессов. (Протекание нера-
дикальной реакции можно объяснить образованием на
какой-то промежуточной стадии шестичленного цикла.)
Изучение разложения перекиси ацетила в растворе
изооктана подтверждает эти выводы. Выход двуокиси
углерода на моль разложившейся перекиси составляет
1,81 для 1,3-10-3 М раствора перекиси. Это отношение
не меняется при добавке таких радикальных ловушек,
как иод, хинон или стирол. Очевидно, это отношение
будет равно 2,0, если имеет место только реакция
(7.9 а). Недостаточное количество образующейся дву-
окиси углерода означает, что выход метилацетата со-
ставляет 20%; так как его выход не зависит от наличия
радикальных ловушек, снова можно заключить, что он
всецело образуется в результате клеточного или ради-
кального процесса.
При разложении перекиси ацетила выход метана до-
вольно высокий (0,82 моль на моль образующегося
СО2), но снижается почти до нуля при добавлении
7 моль стирола. Таким образом, метан получается в
основном из радикалов, способных взаимодействовать с
ловушкой вне клетки, и должен появляться в результате
реакции свободных метильных радикалов с растворите-
лем [реакция (7.12)]. Выход этана невелик (0,02 моль
на моль выделяющейся СО2), но он не уменьшается
при добавлении стирола. Следовательно, этан в основ-
ном должен образовываться в результате клеточной ре-
акции (7.15), что подтверждается данными табл. 7.2.
Теоретически возможно сдваивание ацетатных ради-
калов в клетке, что снова может привести к образова-
нию перекиси ацетила. Эту реакцию можно проследить,
используя меченную по карбонильному кислороду пе-
рекись ацетила. Меченая перекись частично разлагается
в растворе, после чего неразложившуюся перекись вы-
87
деляют. Так как последняя не содержит молекул, в ко-
торых О18 мигрирует между карбонильным и перекис-
ным ’ кислородными атомами, становится очевидным,
что рекомбинация ацетатных радикалов не происходит
[7]. Эти реакции представлены ниже:
О* О
11 II г . 1
СНЭ-С—О-О-С—СН3 -> [СНэ-со2- -О2С—СНЭ]-М
Меченая перекись Радикалы с мигрирующим О18
о7*- о
II V II
_/^ СНЭ-С—О /l —О—С—снэ
Перекись с мигрирующим О18 не обнаружена
Метильные радикалы, образующиеся после разло-
жения перекиси трет-бутила, перекиси ацетила или азо-
метана, могут реагировать с дейтерированным этилбен-
золом при том же соотношении CH3D/CH4 независимо
от источника метильных радикалов [8]:
ff?pe/7?-C4H9O—OC^lig-трет -> 2СНЭСОСНЭ + 2СНЭ •
О О
II II
СНЭ—С—О—О—С—СНЭ -> 2СО2 + 2CHS-
СНЭ—N = N—СНЭ -> N2 + 2СНз-
CH3D + Ph—СН—СНЭ
7
СН3- + Ph—CHD-CHg
ЧСН4 + Ph-CD— СНЭ
Поскольку азометан дает только метильные радика-
лы, то он может служить хорошим реактивом на сво-
бодные метильные радикалы, образуемые перекисью
ацетила. Эти радикалы не могут давать комплекс с мо-
лекулой двуокиси углерода или частично разлагать
ацетатный радикал.
Скорость разложения перекиси ацетила зависит от
растворителя. Так, в первичных или вторичных спиртах
она больше, чем в углеводородных. При использовании
меченного дейтерием изопропанола ызо-СзНуОО было
88
замечено, что дейтерий в метане не содержится [9].
Следовательно, водород отрывался от углеродного ато-
ма, а не от окси-группы. Повышенная скорость разло-
жения перекиси ацетила во вторичных спиртах, веро-
ятно, является результатом индуцированного разложе-
ния перекиси под действием этих радикалов, образовав-
шихся из растворителя *.
Перекись бензоила
Перекись бензоила является предметом многих ис-
следований. Период полураспада ее при 100° С ра-
вен 30 мин, энергия активации 30 ккал/моль. В отдель-
ных инертных растворителях скорость разложения пере-
киси должна быть первого порядка, но в концентри-
рованных растворах константа скорости выше, чем в
разбавленных [11]. Объяснение этому находят в ин-
дуцированном разложении, которое, как и ожидалось,
проявляется в большой степени в концентрированных
растворах, где концентрация радикалов выше.
Разложение перекиси бензоила является сложным,
при этом могут образовываться как фенильные, так и
бензоилокси-радикалы:
О О
II II
Ph—С—О—О—С—Ph 2Ph—СО2.
Ph—СО2- -> Ph- + CO2
Бензоилокси-радикалы, как известно, являются первыми
промежуточными продуктами, так как во влажном
четыреххлористом углероде, содержащем иод, обра-
зуется бензойная кислота [12]:
Ph-CO2- + I2 ->
° 1
II
Ph—С—OI
HtO
---- Ph--CO2H
Гидролиз
В чистой перекиси продуктами разложения являются
двуокись углерода и бифенил, а также небольшое коли-
чество фенилбензоата и бензола [13]. В сильно разбав-
ленном бензоле основными продуктами остаются дву-
* Детально изучен механизм разложения перекиси трет-бути-
ла во вторичных спиртах как растворителях [10].
89
окись углерода и бифенил, однако в результате атаки
фенильными радикалами молекул растворителя были
найдены, другие, вещества [14]. Например, разложение
0,025 М раствора перекиси бензоила в бензоле при
80° С дает такие соединения:
О О
II II 80°С
Ph—С—О—О—С—Ph + Ph—Н----------► СО2 + Ph—СО2Н +
Сильно разбавленная Растворитель
Н _ Ph
+ Ph—Ph + \/ +
Н = Н
Ph Н Ph
=/н/Х= н
Реакции, в которых органическая группа замещается
на ароматическое кольцо, называются гомолитическими
реакциями замещения- в ароматическом ряду. Мы вер-
немся к их подробному рассмотрению в гл. 16.
Механизм разложения перекиси бензоила можно
представить в следующем виде:
Р -- 2R- (7.16)
К2
R - + S —- S- ф- Нерадикальные продукты (7.17)
«3
R- + Р —- R- + Нерадикальные продукты (7.18)
«4
S-+ Р —► R- + Нерадикальные продукты (7.19)
Таблица 7.3
Кинетически вычисленный порядок индуцированного
разложения перекиси бензоила
Реакция обрыва цепи Индуцированное разложение Реакция обрыва цепи Индуцированное разложение
Реакция (7.18) Реакция (7.19) Реакция (7.18) Реакция (7.19)
Йсо СО +++ 1,5 2,0 2,0 0,5 1,0 1,5 R- -(-Ингибитор S • + Ингибитор 2,0 2,0 1,0 2,0
90
где Р — перекись; S — молекула растворителя; R«-
фенильный или бензоилокси-радикал и S*—любой ра-
дикал, образованный растворителем [15]. Реакции (7.18)
и (7.19) представляют собой индуцированное разложе-
ние, вероятный механизм которого представлен на
стр. 87, 88. Кинетический порядок индуцированного
разложения зависит от источника радикалов и от типа
реакции обрыва цепи. В табл. 7.3 приведены значения
кинетического порядка реакций (7.18) и (7.19), которые
проводили в различных условиях. Например, без уча-
стия молекул растворителя механизм процесса будет
следующим:
P--2R- (7.16)
«5
2R- —- Нерадикальные продукты
R- + Р —> R- + Нерадикальные продукты (7.18)
Скорость исчезновения перекиси можно записать как
~t[P1 = «1IPJ + MR • 1 IP] • (7-20)
at
Гак как радикальные продукты весьма реакционно-
способны, вполне можно допустить, что их концентрация
быстро достигает постоянной величины. Скорость обра-
зования радикалов
/?об₽ = 2к1[Р1
и скорость их исчезновения
RHC4 = 2k5[R-]2.
Коэффициент 2 означает, что в каждом процессе участ-
вует два радикала.
В устойчивом состоянии обе скорости равны, т. е.
^Обр ~ RhC4I
2MP1 = 2MRF.
Следовательно,
[R-] = f—У/г [Pjl/1! . (7.21)
\ Ч J
Подставив уравнение (7.21) в уравнение (7.20), получим
Щ к'/г
91
Второй член в правой части этого уравнения относится
к реакции (7.18), которая является процессом индуци-
рованного разложения, порядок которого относительно
перекиси, как видно из табл. 7.3, равен 1,5.
Так как может протекать и мономолекулярное, и ин-
дуцированное разложение одновременно, то наблюдае-
мую скорость реакции следует считать суммой обоих
этих процессов. Наиболее легко обнаружить мономоле-
кулярное разложение, если наблюдать разложение пере-
киси в присутствии ингибиторов, которые подавляют
индуцированный процесс. В инертных растворителях пе-
рекись бензоила разлагается со скоростью реакции пер-
вого порядка *. Однако в растворителях, дающих ради-
калы, которые Могут атаковать‘перекись, скорость раз-
ложения и порядок его часто больше 1,0. Разложение
в диоксане, например, протекает наполовину за 23 мин
при 80° С и не является реакцией первого порядка. Меж-
ду тем если к диоксану добавить иод, стирол, тринитро-
бензол или любой другой ингибитор, то реакция разло-
жения перекиси станет первого порядка, а 'период
полуразложения перекиси будет равен 270 мин. Отсюда
следует, что в диоксане индуцированное разложение по-
давляется эффективными ингибиторами. Скорости раз-
ложения ряда замещенных по кольцу перекисей бензои-
ла в диоксане, содержащем 3,4-дихлорстирол, оказались
различными. В присутствии этого ингибитора влияние
заместителей на мономолекулярное разложение можно
изучить независимо от процесса индуцированного разло-
жения. Было найдено, что скорости мономолеКулярных
реакций подчиняются уравнению Гаммета**:
Jg— = op,
к»
где Ко — собственная константа разложения перекиси
бензоила; к—константа скорости разложения замещен-
ной перекиси бензоила; а — эмпирический коэффициент
* Из табл. 7.3 видно, что порядок индуцированной стадии мо-
жет быть равен 1,0; следовательно, общий кинетический порядок
сам по себе не может определять индуцированное разложение.
** Это уравнение, предложенное Гамметом в 1937 г., очень
четко описывает влияние заместителей на органические реакции
[16, 17]. Интересно, что это уравнение, впервые примененное в об-
ласти ионных реакций, также оказалось пригодным для корреляции
скоростей радикальных процессов [18].
92
для каждого заместителя; р — характеристика реакции.
В этом случае было найдено, что р = —0,4.
Это означает, что электронодонорные заместители
повышают скорость разложения перекиси. Кроме того',
если заместитель находится в каждом ядре, то кон-
станта используется в виде суммы этих о-констант для
Рис. 7.1. График уравнения Гаммета
для диссоциации перекиси бензоила
в присутствии 3,4-дихлорстирола
как ингибитора индуцированного
разложения [15].
каждого заместителя*. На рис. 7.1 представлен график
с данными из уравнения Гаммета: kXy— константа ско-
рости разложения перекиси бензоила, содержащей за-
местители X и Y, а ох и оу — константы этих замести-
телей. Поскольку скорость присоединения свободных
радикалов к олефинам велика, то скорость иницииро-
* В действительности эти данные соответствуют уравнению
Гаммета в случае, если, используются сг-*--величины вместо обыч-
ных а-констант. Однако для наших целей их различие не имеет
большого значения. Мы коснемся применения а+-величии при рас-
смотрении радикальных реакций на стр. 165.
93
вания полимеризации стирола под действием бензоил-
окси-радикалов должна соответствовать скорости разло-
жения перекиси *:
Медленно Стирол Обрыв цепи
Перекись---------Радикалы------ М„----------- Полимер
Быстро
В связи с этим влияние заместителей на иницииро-
вание полимеризации стирола также подчиняется урав-
нению Гаммета. При 70° С р имеет то же значение
(—0,4), что и для диссоциации перекиси бензоила [19].
При полимеризации стирола в присутствии перекиси
бензоила растущий полистирольный радикал взаимодей-
ствует с нею, вызывая индуцированное разложение.
Скорость индуцированного разложения можно изме-
рить в процессе полимеризации (см. гл. 15), и ее удобно
выражать в виде константы переноса С, которая пред-
ставляет собой отношение константы скорости атаки на
перекись к константе скорости атаки на мономер
М„- + М-п- м„+1-
о о
II II “пер
M„- + Ph-C—О—О—С—Ph------------
О
II
-> М„— О—С—Ph-f-Ph—СО2-
С = кпер/кп .
Таблица 7.4
Влияние заместителей на констаиты переноса симметрично
замещенных перекисей бензоила [19]
Заместители (в дру- гом ядре — тот же заместитель) с = ^- *П Заместители (в дру- гом ядре — тот же заместитель) кпер С = — кп
п-СН3О 0,07 М-Р 0,25
Н 0,07 м-1 0,26
n-F 0,22 .и-Вт 0,46
п-С1 0,22 n-CN 0,80
п-1 0,29
* Как будет отмечено на стр. 230, скорость полимеризации про-
порциональна квадратному корню из величины скорости разложе-
ния перекиси.
94
В табл. 7.4 приведены значения некоторых констант
переноса для ряда замещенных перекисей бензоила к
радикалам из стирола. Замечено, что на стадии инду-
цированного разложения электронодонорные замести-
тели уменьшают скорость реакции. Это вполне согла-
суется с их влиянием на скорость разложения по
первому порядку, как показано на рис. 7.1.
Влияние заместителей на мономолекулярное разло-
жение перекиси бензоила можно объяснить следующим
образом. Электронодонорные группы повышают скорость
диссоциации первого порядка, так как они увеличивают
электроотрицательность между кислородными атомами
перекиси и ароматическим кольцом:
О о
Перераспределение электронов под действием электроиодонориой группы X
Большой отрицательный заряд на атомах кислорода
вызывает сильное электростатическое отталкивание
между ними, поэтому увеличивается скорость разло-
жения.-
В стадии индуцированного разложения- электроно-
донорные группы повышают скорость реакции. Поэтому
полистирольный радикал нуклеофилен и атакует суб-
страты в положениях с низкой электронной плотностью,
В реакции между полистирольным радикалом и пере-
кисью бензоила
переходное состояние стабилизируется ионной резонанс-
ной формой:
с. со,-со, ~ -с -со, со,.
Аг Аг Аг Аг 95
Наличие такого гибридного резонанса приводит к пред-
положению, что электронодонорный заместитель в ядре
перекиси будет ускорять индуцированное разложение в
соответствии с данными табл. 7.4 *.
Разложение перекиси бензоила ускоряется аминами.
Третичные амины ускоряют образование свободных
радикалов и часто используются в качестве добавок к
катализаторам в процессах полимеризации. Такое явле-
ние как раз служит примером облегчения образования
радикалов веществами, которыми могли бы быть обыч-
ные ионные катализаторы. Особенно показательна в
этом плане система: перекись бензоила — третичный
амин. Так, например, 0,01 М раствор перекиси бензоила
при 20° С имеет период полуразложения 60000 ч. Добав-
ление 0,01 М раствора диметиланилина понижает период
полуразложения до 13 мин. Продуктами реакции пере-
киси бензоила с диметиланилином (после гидролиза)
являются бензойная кислота, метиланилин, формальде-
гид и следы м-бензоилоксидиметиланилина. Механизм
этой реакции сложен и еще до кснца не выяснен.
Возможный его вариант представлен реакциями
(7.22) —(7.24) [21]:
О О
II II
Ph—N(CHS)2 -f-Ph—С—О—О—С—Ph
СН3 О
I II
Ph—N+—О—С—Ph + Ph—СОГ
СН3 I
(Ph-N (CHS)2]+ + Ph-CO2-
II
СН3
Ph—N+=CH2 + PhCO2H
III '
(7.22)
(7.23)
(7-24)
* Или же механизм складывается из атаки полистирольным ра-
дикалом ароматического кольца перекиси с одновременным рас-
щеплением перекисной связи [20]. Кроме того, известно, что элек-
тронодонорные группы в перекисном цикле увеличивают скорость
атаки. ।
96
Образование ионов по реакции (7.22) подтверж-
дается тем, что электронодонорные группы в аромати-
ческом кольце анилина и электронодонорные замести-
тели в перекиси бензоила увеличивают скорость реакции.
Показано, что соединение I реагирует по двум конку-
рирующим направлениям, одно из которых приводит
к образованию свободных радикалов, другое — нет. Это
определяется тем, что эффективность образования ра-
дикалов снижена (25% и менее) и зависит от природы
применяемого растворителя. После гидролиза катион III,
как правило, всегда превращается в формальдегид и
метиланилин, что и наблюдалось в действительности.
Очевидно, по реакции (7.23) должен выделяться только
бензоат-радикал, который может инициировать полиме-
ризацию, поскольку в полимерах, полученных в таких
системах, не было найдено азота. Радикал II, скорее
всего, реагирует дальше и разрушается прежде, чем
стать свободным.
1 Другие перекиси ацилов
При разложении перекисей алкилов ROOR природа
R-группы очень слабо влияет на скорость диссоциации.
Перекиси с R-группой: С2Н5, я-СзН7, изо-С3Н7 или
mpem-CtHg — разлагаются практически с одинаковыми
или близкими скоростями. Однако в перекисях ацилов
RCO — О — О — COR характер R-группы оказывает
большое влияние на скорость диссоциации. В табл. 7.5
приведены относительные скорости разложения ряда
перекисей ацилов. Эти данные показывают, что стабиль-
Таблица 7.5
Относительные скорости разложения перекисей ацилоз
R Относительная константа скорости Литература
60° с 70° С
Метил 1 ~5 [22, 25]
Этил 1 — |[22]
Пропил 1 — [22]
Изопропил 100 — [22]
Фенил — 1 [11, 23]
Беизил — Очень быстро [24
Циклопропил — 0,4 [23
Циклопентил — 32 [23
Циклогексил '— 60 [23]
4 У. Прайер
97
ность R-группы как свободного радикала зависит от
скорости распада перекиси ацила. Это означает, что в
переходном состоянии происходит растягивание связи
С—СО, а также связи О—О:
R—С—О—О—С—R —> R . . . С-0 . . . О-С . . . R
II II II II
О О о о
Переходное состояние
Таким образом, легкость, с которой в R-группе по-
является неспаренный электрон, зависит от скорости
расщепления связи О — О. К этому вопросу мы вер-
немся, когда будем рассматривать эфиры надкислот.
Упражнения
7.1. Перекись /npe/n-бутила может разлагаться в растворах то-
луола, этилбензола и изопропилбензола. Как бы изменялось отно-
шение mpe/n-бутаиол/ацетои в этих трех растворителях?
7.2. В табл. 7.3 показано, что индуцированное разложение яв-
ляется реакцией первого порядка, если имеется ингибитор,
который ловит свободные радикалы R •, и если эти радикалы,
влияющие на индуцированное разложение, образовались из раство-
рителя. Докажите это, используя следующий механизм: '
«д
Р —— 2R.
хД
R- + I----- Продукты
чг
R- -|—S--->- Продукты + S-
S- + Р--->- R- -|- Продукты
К примеру: напишите допущения для стационарного состояния ра-
дикалов R- и S- и затем скомбинируйте так, чтобы получить соот-
ношение, включающее S- только как разновидность радикала.
7.3. Индуцированное разложение перекиси бензоила может про-
текать по одному из двух механизмов. Согласно первому, присоеди-
нение к карбонильному кислороду мбжет предшествовать расщеп-
лению:
О О
II II
R. +Ph—С—О—О—С—Ph -> [Ph—С—О—О—С—Ph] —
I II
OR О
О
II
— Ph—С-О—R + PhCO2 •
98
По другому механизму может происходить прямая атака радикала
на перекисный кислородный атом:
0 0 О
II II II
R • +Ph—С—О—О—С—Ph — R—О—С—Ph + PhCOa •
Разработайте метод для различения этих двух возможных процес-
сов. К примеру, как отличаются эти два типа кислородных атомов
в двух представленных механизмах? Можно ли воспользоваться
метками [26]?
7.4. Энергия активации индуцированного разложения ROOR по-
листирольным радикалом равна 22—24 ккал/моль, где R—СгН5,
Н-С3Н7, U30-C3H7, Н-С4Н9 или mpem-CtHs. На основе этого ответьте,
происходит индуцированное разложение за счет отрыва водородно-
го атома или в результате атаки на О—0-связь? [1].
7.5. Перекись ацетила разлагается с образованием, помимо
других продуктов, метилацетата и двуокиси углерода. Какой необ-
ходим эксперимент, чтобы отличить метилацетат, образующийся из
клеточных радикалов, от метилацетата, образующегося из свободных
ацетатных радикалов? Как различить клеточные радикалы, полу-
чающиеся в результате молекулярного, нерадикального процесса
образования метилацетата [12j? Найдите различия между неради-
кальным механизмом и радикальным процессом, происходящим
исключительно в радикальной клетке (см., например, [27]).
7.6. Найдите среднее время протекания следующих процессов:
а) вибрации связи; б) диффузии из клетки; в) периода полуразло-
жения радикального соединения с нулевой энергией активации и
предэкспоненциальным множителем [28].
7.7. В толуоле при 80° С перекись 6-гептеиоила
(СН2=СН—(СН2)4—С02—)2 разлагается с образованием 1-гекеена,
1,5-гексадиеиа, 1,11-додекадиена, циклогексана, метилциклопентана
и дибензила. Первые три продукта образуются с почти одинаковы-
ми выходами в присутствии или без гальвиноксила — рекомбинато-
ра кислородсодержащих свободных радикалов. Дибензил выде-
ляется полностью, но выходы двух циклических соединений значи-
тельно снижаются в присутствии радикала-ловушки. Представьте
механизм разложения этой перекиси, объясняющий эти факты [29].
ЛИТЕРАТУРА !
1. Pryor W. A. et al. J. Amer. Chem. Soc., 86, 4237 (1964).
2. R a 1 e у J. H., Rust F. F., Vaughan W. E. Ibid., 70, 88,
1336, 2767 (1948); Batt L.( Benson S. W. J. Chem. Phys.,
36, 895 (1962).
3. Noyes R. M. Ibid., 18, 999 (1950); 22, 1349 (1954); 65, 763
(1961).
4. Watts H. P„ Hammond G. S. J. Amer. Chem. Soc., 86, 1911
(1964).
5. Lyon R. K. Ibid., 86, 1907 (1964).
6. Noyes R. M. In: Progress in Reaction Kinetics. Vol. I. N. Y.,
Pergamon Press, 1961, p. 131.
7. H e г k L„ Field M., S z w а г с M. J. Amer. Chem. Soc., 83,
2998 (1961).
8. H e г k L„ Szwarc M. Ibid., 82, 3558 (1960).
4*
99
9. Kharasch M. S., Rowe J. L., Urry W. H. J. Organ.
Chem., 16, 905 (1951).
10. H a у e s e r E. S., BredewegC. J. J. Amer. Chem. Soc., 86,
2401 (1964).
11. Nozak i K-, В a r 11 e 11 P. D. Ibid., 68, 1686 (1946).
12. Hammond G. S„ Soffer L. M. Ibid., 72, 4711 (1951);
Shine H. J., Waters J. A., Hoffman D. M. Ibid., 85,
3613 (1963).
13. Erlenmeyer H., Schoenaur W. Helv. chim. acta, 19, 338
(1936).
14. D e T a r D. F., Long R. A. J. J. Amer. Chem. Soc., 80, 4742
(1958); Hey D. H., P e r k i n s M. J., W i 1 1 i a m s G. H.
J. Chem. Soc., 3412 (1964); Gill G. B., W i 111 a m s G. H.
Ibid., 995 (1965).
15. S w a i n C. G., S t о c k m a у e r W. H., С 1 a r k e J. T. J. Amer.
Chem. Soc., 72, 5426 (1950).
16. Hammett L. P. Physical Organic Chemistry. Chap. 7.
McGraw-Hill Book Company, 1940.
17. Leffler J. E., Grunwald E. Rates and Equilibria of Orga-
nic Reactions. John Wiley and Sons. Inc., N.Y., 1963, p. 171.
18. Howard J. A., Ingold K- U. Canad. J. Chem., 41, 1744
(1963).
19. Cooper W. J. Chem. Soc., 3106 (1951); 2408 (1952).
20. W a 11 i n g C„ Savas E. S. J. Amer. Chem. Soc., 82, 1738
(1960).
21. Horner L. J. Polimer Sci., 18, 438 (1955); W a 11 i n g C., In-
dicator N. J. Amer. Chem. Soc., 80, 5814 (1958); Wal-
ling C. In; Free Radicals in Solution. John Wiley and Sons.
Inc. N. Y., 1957, p. 590.
22. S m i d J., S z w a г с M. J. Chem. Phys., 29, 432 (1958).
23. Hart H., Wyman D. P. J. Amer. Chem. Soc., 81 4891
(1959).
24. Bartlett P. D„ Leffler J. E. Ibid., 72, 3030 (1950).
25. Levy M., Steinberg M., Szwarc M. Ibid., 76, 5978
(1954).
26. P г у о r W. A. Mechanisms of Sulfur Reactions. McGraw-Hill
Book Company. N. Y., 1962, p. 48.
27. D e Tar D. F., W e i s C. J. Amer. Chem. Soc., 79, 3045 (1957);
Lamb R. C., Pacifici J. G. Ibid., 86, 914 (1964).
28. Benson S. W. Foundations of Chemical Kinetics. McGraw-Hill
Book Company. N.Y., 1960, p. 496; Ibid., p. 543.
29. L a m b R . C., Ayers P. W., T о n e у M. K- J- Amer.-Chem.
Soc., 85, 3483 (1963).
Дополнительная литература
Davies A. G. Organic Peroxides. Butterworth Scientific Publica-
tions. Lond., 1961.
Edwards J. O. (ed.). Peroxide Reaction Mechanisms. Interscience
Publishers, Inc. N. Y., 1962.
Hawkins E. G. E. Organic Peroxides. D. Van Nostrand Compa-
ny, Inc., Princeton. N.Y., 1961.
Tobolsky A. V., Mesrobi an R. B. Organic Peroxides. Inter-
science Publishers, Inc. N. Y., 1954.
.100
Глава 8
ТЕРМИЧЕСКИЙ ГОМОЛИЗ. НАДКИСЛОТЫ И ИХ ЭФИРЫ
Надэфиры и надкислоты взаимосвязаны таким же
образом, как и обычные эфиры и кислоты:
О О
II II
R—С—О—ОН R—С—О—OR'
Надкислота Эфир надкислоты
Надкислоты образуются в результате реакции кис-
лот, хлорангидридов кислот или ангидридов с пере-
кисью водорода. Например, надуксусную кислоту можно
получить взаимодействием эквимолярных количеств ук-
сусной кислоты и 98%-ной перекиси водорода в при-
сутствии 1%-ной H2SO4. Через 15 ч при 15°С дости-
гается равновесие, при котором образуется 50%-ный
раствор надуксусной кислоты:
О
сн3- со2н + Н2О2 -> СН3—С—О—он + Н2О
Известно, что надкислоты получаются в качестве
промежуточных продуктов при окислении альдегидов,
и это используют для их синтеза. Например, надуксус-
ная кислота в промышленности получается окислением
ацетальдегида:
О О
R И O2 II
сн3—сно —« сн3—с- —« СН3—С -О-О-
о
II
СН3-СНО4 сн3—с—о—о -->
о о
II II
->сн3-с+ сн3—с—о—он
Эфиры надкислот образуются из хлорангидридов и
гидроперекисей:
О О
II II
R—С—Cl + R'—ООН -> R—С—О—OR' + НС1
Особенно удобно получать их эфиры, применяя легко-
доступную гидроперекись трет-бутила. Этим методом
получают целый ряд других эфиров надкислот
(табл. 8.1).
101
Таблица 8.1
Эфиры надкислот и их период полураспада в реакциях
разложения [ 1 ]
_________________—
II
трет - С4Н9О — О — С — R
R Период полурас- пада при 60°, мин R Период полурас- пада при 60 °C, мин
сн3- 5-Ю5 сн3
Ph — 3-105 Ph2C — 6
Ph — СН2 — 1,7-103
Ph2CH — С13С — 970
26
сн3 n-NO2-CeH4—СН2— 4700
Ph —С — 12 п - СН3О -свн4—сн2 — 880
сн3
Предложено несколько вариантов механизма разло-
жения эфиров надкислот:
О
II
R—С—O- + -OCR3
R • + СО2 4- -OCRs
О /у/ О
11 Л/ в И
R—С—О—О—CR3------.^Q—О—С—R+ OCRs
О
r-c-o-cr;
О
R'
(реакция Криги)
102
Механизм а представляет собой обычный разрыв
связи О — О, аналогично образованию радикалов в пе-
рекисях.
Механизм б — одновременный разрыв связей О — О
и С — С с выделением двуокиси углерода, как это имеет
место при выделении азота из азосоединений.
Механизм в является индуцированным разложением
под действием радикала Q-. Кроме указанных трех
механизмов радикального разложения для некоторых
эфиров надкислот предложен механизм ионного разло-
жения г. Одним из примеров такого механизма может
служит разложение 9-декалилнадбензоата [2]:
Скорость этой реакции в значительной степени за-
висит от ионизирующей способности растворителя, и,
следовательно, процесс должен протекать с образова-
нием заряженных промежуточных продуктов. Разложе-
ние mpem-бутилнадтозилата является другим примером,
в котором надэфир реагирует исключительно по меха-
низму Криги; в метаноле он дает толуолсульфокислоту
и диметилкеталь ацетона [3]:
О сн3
II | Метанол
Ar—S—О—О—С—СН3---------->
II I
О сн3
юз
Метанол
о сн3
II I
Аг— S-О" +О—С—СН3
II I
О сн3
Перегруппировка
сн3 сн3
о о о
II I Метаиолиз I
->Ar—S—О—С—СН3-----------------►Ar—SO3H+CH3O—С—СН3
II I I
О СН3 СН3
(Ar = п-СН3—СвН4)
щрет-Бутилнадтрифторацетат и щрет-бутилнадхлор-
формиат реагируют по механизму а, б или г, в зави-
симости от ионизирующей способности растворителя [4].
Периоды полураспада, приведенные в табл. 8.1,
показывают интересную особенность реакций разложе-
ния надэфиров. Эти эфиры надкислот разлагаются с
различной скоростью в зависимости от природы груп-
пы R, образуя при этом более стабильный радикал.
Например, при переходе R от СН3 к PhCH2 и до Ph2CH
скорость диссоциации возрастает медленно. Отсюда сле-
дует, что стабильность группы, как свободного радикала,
влияет на скорость разрыва связи О — О.
Механизм, объясняющий эти наблюдения, сводится
к тому, что эфиры надкислот, которые не содержат
достаточно стабильных групп, подвергаются обычному
разрыву связи О — О. В то же время надэфиры, кото-
рые могут образовать лишь относительно устойчивые
оксиацильные радикалы, одновременно разлагаются по
двум связям с выделением молекулы СО2.
В этом случае в промежуточном оксиацильном
радикале на группе R локализуется неспаренный элек-
трон. Например, из данных табл. 8.1 видно, что
mpem-бутилнадацетат и надбензоат разлагаются с почти
одной и той же скоростью и эти надэфиры из всех
приведенных в таблице соединений являются наиболее
стабильными. Группой R в этих надэфирах служат
метил и фенил соответственно, но ни тот, ни другой не
|04
образуют достаточно стабильного радикала. Поэтому
оба надэфира разлагаются обычно по связи О — О:
mpem-C4H9O—О—СО—СН3 ->
- > mpem-C4H9O- + СН3—СО—О-
mpem-C4HeO—О—СО—Ph -> mpem-C4H9O• + Ph—СО—О-
Однако mpem-бутилфенилнадацетат разлагается в
102 раз быстрее, чем надацетат или надбензоат. В этом
случае механизм разложения представляется как раз-
рыв двух связей с образованием свободного бензильного
радикала:
mpem-C4H9O—О—СО—CH2Ph ->
- > [mpem-C4H9O- • -СО2- • -CH2Ph] ->
Переходное состояние
- > mpem-C4H9O • + СО2 + PhCH2-
Локализация неспаренного электрона на бензильном
атоме углерода в промежуточной стадии процесса при-
водит к ярко выраженному повышению скорости реак-
ции. Такой случай разложения на первый взгляд может
показаться необычным, так как перекиси чаще подвер-
гаются простому разрыву связи О — О. В то же время
эта реакция встречается не реже реакции образования
азота при разложении азосоединения. Можно сравнить,
например, реакции разложения азосоединения [5] и
надэфира [1];
Ph—N = N— CHPh2 -> Ph • + N2 + • CHPh2
mpem-C4H9O—О—CO—CHPh2 ->
трет-C4H9O • + CO2 + • CHPh2
При 60° С азосоединение имеет период полуразложения
около 60 мин, а надэфир — 26 мин.
В табл. 8.2 приведены значения энтальпии активации
и энтропии диссоциации для трех соединений, взятых
из табл. 8.1. Можно заметить, что с возрастанием кон-
станты скорости уменьшаются значения \Н* и AS*.
Хотя менее положительному значению AS* соответст-
вует малая скорость реакции, при переходе от трет-бу-
тилнадацетата к mpem-бутилнаддифенилацетату наблю-
дается увеличение скорости за счет уменьшения вели-
105
Таблица 8.2
Активационные параметры разложения эфиров надкислот [1]
Эфир надкислоты АН*, ккал/моль AS*, кал /град к, сек 1 (60 °C)
о II трет - С4Н8О — О — С — СН3 38 ' 17 2-IO"8
О II трет - С4Н8О — О — С — CH2Ph 29 3,9 6,8-10-8
О II трет - С4Н8О — О — С — CHPh2 24 -1,0 4,4-10-8
чины Д//*. Причина уменьшения энтальпии активации
обсуждалась выше. Это происходит потому, что трет-
бутилнаддифенилацетат образует более стабильные ра-
дикалы, и для разрыва связи требуется меньшее количе-
ство энергии. Уменьшение энтропии с усложнением R
усматривается в геометрии переходного состояния, в
котором две связи подвергаются одновременному раз-
рыву. Например, для того чтобы фенильная группа в
надфен ил ацетате^, делокализовала неспаренный элек-
трон, необходима компланарность с СОг в переходном
состоянии:
трет-
-С4НЭО—О—СО—СН2— Ph >-
Переходное
состояние
Это ограничение уменьшает возможные повороты моле-
кулы в пространстве в переходном состоянии и снижает
их вероятность и энтропию.
Соединения в табл. 8.3 дают представление о воз-
можности протекания клеточных реакций в эфирах над-
кислот. Из каждого соединения в верхней части таблицы
образуется надэфир, стоящий непосредственно под ним,
если оно разлагается с потерей СО2 и подвергается
последующей клеточной рекомбинации полученного
фрагмента. Например, первое соединение mpem-бутил-
106
Таблица 8.3
Надэфиры и возможность клеточного разложения [6]
Надэфир Относитель- ная константа скорости при 100 °C Клеточйая реакция, %
О О II II трет - С4Н9О—О—С—С—О—ОС4Н9-треот 107 0
О II трет - С4Н9О — О — С — ОС4Н9 - трет ю12 0
трет - С4Н9О — ОС4Н9 - трет 1 —
надоксалат может превратиться во второе — трет-бу-
тилнадмонокарбонат:
О О О
II II II
RO—О—С—С-О—OR -> RO—С—О—OR + СО2
(R—mpem-C4He)
Аналогично с потерей молекулы СО2 и клеточной ре-
комбинацией второй надэфир может давать третье со-
единение: _
О
II
RO—С—O-OR -> RO-OR + СО2
Кроме того, в каждом из этих случаев образующийся
надэфир обычно разлагается медленнее, чем первона-
чальное соединение. Следовательно, если такие клеточ-
ные продукты образуются, то их можно было бы выде-
лить. В действительности, после распада надкарбоната
перекись трет-бутила обнаружена не была, равно как
и два остальных соединения не были выделены после
разложения надоксалата. Из этого следует, что при
разложении надкарбоната или надоксалата. клеточные
реакции не происходят (или, точнее, протекают менее
чем на 1 %).
Результаты, рассмотренные на стр. 88, показывают,
что ацетатные радикалы не подвергаются клеточной
рекомбинации при разложении перекиси ацетила. Таким
107
образом, любые надэфиры или перекиси далеко не
всегда могут участвовать в такого рода процессах.
В то же время трудно объяснить образование связей
С — О, С — N и N — N. Например, mpem-бутилнадмоно-
оксалат [7] разлагается до тцет-бутилкарбоната за
счет присоединения алкокси-радикала к радикалу ROC<
О
О О о
II II 25-с II
RO—O-C-C-OR----------« СО2 7-RO-C-OR->
Бензол
1,5 моль* 0,5 моль
о
г ~ ' II
Гальвиноксил __ „
-----------СО2 + RO-C-OR
25-С, бензол ! моль 0 4 ЯОЛ6
(R=трет-бутил)
Карбонат медленно образуется в результате клеточ-
ной реакции, потому что, как показано выше, его выход
лишь незначительно уменьшается в присутствии галь-
виноксила, который является очень эффективным ра-
дикалом-ловушкой для кислородных радикалов [8]:
•О——СН / = О (R=трет-бутил)
Гальвиноксил
Упражнения
8.1. Опишите продукты, которые получились бы после разло-
жения трет-бутилнадтрифторацетата согласно - схемам а, б и в,
приведенным на стр. 102.
8.2. Перекись п-нитро-п'-метоксибензоила разлагается по ради-
кальному механизму в полярных растворителях и по ионному в не-
полярных.
а) Какие продукты ожидаются после радикального разложения
в присутствии донора водорода?
б) Напишите схему механизма ионного разложения этой пере-
киси.
После ее гидролиза были найдены следующие продукты: п-нит-
робензойная кислота, n-метоксифенол и угольная кислота. Объясни-
те механизм такого ионного разложения, иллюстрируя объяснение
резонансными структурами с дробным распределением зарядов в
переходном состоянии радикальных реакций [9].
108
8.3. При разложении перекиси mpem-бутила в толуоле, содер-
жащем иод как ловушку радикалов, обнаружено, что радикалы об-
разуются точно с такой же скоростью, как и в газовой фазе. Объ-
ясни гр •р'рт факт в свзте данных табл. 8.3 [10].
8.4. Объясните активационные параметры для следующих трех
надэфиров:
AS*, АД*,
R в т/Ж/п-С4Н,О—О—СО—R кал/град ккал!моль
СН3.......... 17 38
PhCH2.......... 4 29
Ph—СН=СН—СН2 . —6 23
При этом следует учесть, что ряд связей разрывается в пере-
ходном состоянии [1].'
ЛИТЕРАТУРА
1. Bartlett Р. D., Hiatt R. R. J. Amer Chem. Soc., 80, 1398
(1958).
2. Criegee R., Kaspar R. Ann. Chem., 560, 127 (1948).
3. Bartlett P. D„ Traylor T. G. J. Amer. Chem. Soc., 83, 856
(1961).
4. В a r 11 e 11 P. D. In: J. O. Edwards (ed.). Peroxide Reaction
Mechanisms. Interscience Publishers, Inc., N. Y., 1962, p. 1.
5. С о h e n S. G., W a n g С. H. J. Amer. Chem. Soc., 77, 3628
(1955).
6. Bartlett P. D., Sakurai H. Ibid., 84, 3269 (1962).
7. Bartlett P. D., Gantarev B. A, Sakurai H. Ibid., 84,
3101 (1962).
8. Bartlett P. D., TunahashiT. Ibid., 84, 2596 (1962).
9. L e f f 1 e r J. E. Ibid., 72, 67 (1956).
10. Bartlett P. D., Riichardt C. Ibid., 82, 1756 (I960).
Дополнительная литература
S w e r n D. Chem. Rev., 45, 1 (1945).
Глава 9
ТЕРМИЧЕСКИЙ ГОМОЛИЗ. ПЕРЕКИСЬ ВОДОРОДА,
ГИДРОПЕРЕКИСИ И МОЛЕКУЛЯРНО-ИНДУЦИРОВАННЫЙ
ГОМОЛИЗ
Перекись водорода существует в водных растворах
различных концентраций. Эта неорганическая перекись
служит исходным веществом для синтеза большинства
органических перекисей, за исключением тех, которые
можно получить прямым окислением с помощью кисло-
рода. Перекись водорода взрывоопасна, и потому при
работе с ней, как и с любой другой перекисью, необ-
109
ходимо соблюдать меры предосторожности. Растворы
перекиси водорода в воде 95—100%-ной концентрации
детонируют, однако более разбавленные растворы без-
опасны. Если перекись водорода загрязнена органиче-
скими примесями, то образуется чрезвычайно взрыво-
опасная смесь даже для низких концентраций.
Чувствительность соединения к взрыву зависит от
содержащегося в нем количества кислорода. Существует
правило, согласно которому вещества с молекулами,
содержащими уже 5% активного кислорода и лабиль-
ную связь, являются потенциально взрывоопасными.
Данные о взрывоопасности некоторых перекисей приве-
дены в табл. 9.1. Из табл. 9.1 видно, что большинства
перекисей относится к классу взрывоопасных. Однака
Таблица 9.1
Перекиси и их взрывоопасность
Содержание Содержа- ние кис- лорода , % Опасность
Перекись водорода 94 Взрывается только, когда за- грязнена или ее концентрация превышает 90%.
Перекись ацетила 13 В твердом виде очень опасна. Нормальной обработке поддают- ся только разбавленные раство- ры (около 30%)
Перекись бензоила 7 В дверном виде взрывается; ра- створы безопасны
Перекись лауроила 4 Не взрывается
Надуксусная кислота 21 Чистый продукт может дето- нировать при 110 °C. Растворы концентрации более 69% взрыва- ются при ударе. Менее опасна, чем перекись ацетила и надму- равьнная кислота
Надмуравьиная кислота 26 Растворы концевтрацйи белее 47% могут детонировать при по- падании органических примесей
Перекись трет-бутила 25 Безопасна
Перекись этила 35 В твердом виде детонирует при растирании, взрывается при высоких температурах (около 200 °C) в газовой фазе (особенно в присутствии кислорода), раст- воры при умеренных температу- рах относительно безопасны
НО
их не используют как взрывчатые вещества, так как
они недостаточно стабильны в обращении.
Перекись водорода при нагревании или облучении
дает гидроксильные радикалы:
Свет
НА------------► 2НО.
или нагрев
Процесс разложения перекиси водорода можно ката-
лизировать железом. На этом основано широкое исполь-
зование в качестве окислителя и источника гидроксиль-
ных радикалов смеси солей железа (II), перекиси водо-
рода и кислоты (реактив Фентона):
НА + Fe2+ + Н+ НО - + Н2О + Fe3+
Подробнее эти реакции см. в гл. И. Органические гид-
роперекиси разлагаются аналогично перекиси водорода
с образованием гидроксильных радикалов. Их разложе-
ние также катализируется ионом железа:
Свет
ROOH----------> RO - + НО-
или нагрев
ROOH + Fe2+ -> RO - + ОН" + Fe3+
Гидроперекись /пре/n-бутила
Третичные гидроперекиси легко получают алкилиро-
ванием перекиси водорода:
ROH (или RC1) + НА ROOH + Н2О
(R=третичный радикал)
По этой причине, а также потому, что третичные гидро-
перекиси более стабильны и менее опасны в обращении,
чем первичные или вторичные, они нашли широкое при-
менение, особенно гидроперекись трет-бутила. Разло-
жение ее в свободном виде либо в инертном растворителе
типа хлорбензола приводит к образованию кислорода и
mpem-бутилового спирта [1]. Механизм этого процесса
следующий:
Инициирование:
mpem-C4H9OOH -> mpem-C4H9O- -(- НО-
Развитие цепи:
mpem-C4H9O- +mpem-CiH9OO'ti ->
->mpem-C4HeOH + mpem-C4H9OO-
2wpem-C4H9OO- -> [mpem-C4H9OO—OOC4H9-mpe.77] ->
-> 2mpe/n-C4H9O • + O2
111
Как можно видеть из этого механизма, растворители,
легко образующие радикалы, увеличивают скорость
разложения гидроперекиси. Например, гидроперекись
/тгрет-бутила разлагается с повышенной скоростью в
спиртах, эфирах и аминах [2]. Все эти растворители
легко дают свободные радикалы, и увеличение ско-
рости разложения гидроперекиси можно объяснить
образованием радикалов из растворителя, принимаю-
щего участие в индуцированном разложении. Такое раз-
ложение имеет следующий механизм (SH — молекула
растворителя с лабильным атомом водорода):
Развитие цепи:
mpem-C4H9O.- + SH -> mpsm-C4H9OH + S •
S • +mpem-C4H9OOH -> mpem-C4H9OO- + SH
2 mpem-CJi9OO • -> 2 mpem-C4H9O-+ Oa
Радикалы, образованные гидроперекисью mpetn-бу-
гила, способны инициировать полимеризацию мономе-
ров, таких, как стирол:
ROOH -> RO- + -OH
пМ
RO- + М -> RO-M- —- RO—М„- (9.1)
(Стирол)
Однако растущий полистирольный радикал может
атаковать гидроперекись, поэтому за счет индуцирован-
ного разложения увеличивается скорость исчезновения
гидроперекиси:
RO—М„ • + ROOH RO—М„—Н + ROO• (9.2)
Так как инициирование гидроперекисью [реакция
(9.1)] включает реакцию первого порядка разложения
перекиси и индуцированное разложение [реакция (9.2)]
по второму порядку, оба этих процесса можно раз-
личать с помощью кинетического анализа. Вычитая
количество перекиси, которая разлагается по реакции
высшего порядка, можно найти скорость мономолеку-
лярного разложения гидроперекиси в растворе стирола
и сравнить со скоростью разложения той же гидропере-
киси в инертных растворителях [3]. Такое действие
приводит к неожиданному результату: после поправки
на бимолекулярное индуцированное разложение ско-
112
рость инициирования гидроперекисью оказалась больше,
чем величина, вычисленная для скорости мономолеку-
лярного разложения гидроперекиси в инертных раство-
рителях. Очевидно, разложение гидроперекиси трет-бу-
тила со столь неожиданно большой скоростью вызывает
стирол. Это явление было названо молекулярно-инду-
цированным гомолизом и будет рассмотрено ниже.
Гидроперекись кумола
Другой детально изученной третичной гидроперекисью
является гидроперекись кумола. Она подвергается
Р-расщеплению с образованием ацетофенона и метана.
При 140°С в декане образуются продукты [4]:
СН3 СН3
I I
Ph—С—О—ОН Ph—С—О-ф НО-
I I
сн3 сн3
сн3 сн3
Ph—С—О- Ph—О = О + СН3-
I 20/,
сн3
сн3 сн3
I I
CH3- + Ph—С-О-ОН СЩ + Р11—С-О—О-
| 30% I
сн3 сн3
СН3 СНз
I I
Ph-С—О - + Ph—С—О—ОН ->
I I
СНз СНз
СН3 СНз
Ph—С—ОН + Ph—С—О—О- (9.3)
I I
СНз СН3
54 %
113
Если в качестве растворителя используют кумол, то
образуется до 90% кумилового спирта, механизм обра-
зования которого можно объяснить схемой:
СН3 СН3 СН3 СН3
I I I I
Ph—С-О- + Ph—С—Н -> Ph—С—ОН + Ph—С-
I I I I
сн3 сн3 сн3 сн3
сн3 сн3
Ph—C-+Ph—С—О—О- ->
I I.
сн3 сн3
сн3 СН3 ~
Ph—С—О—О—С—Ph
СН3 _
_ СН3
-> 2Ph—С—О-
СН3
и далее — по реакций (9.3).
Гидроперекись кумола- также подвергается кислотно-
каталитическому разложению, что приводит к образо-
ванию ацетона и фенола. Разложение гидроперекиси
кумола в условиях гомолитической реакции обычно дает
очень немного фенола как побочного продукта; напри-
мер, в декалине образуется менее 1 % фенола. Однако
в присутствии кислот основными продуктами реакции
являются фенол и ацетон. Эту кислотно-каталитическую
реакцию используют в промышленном синтезе фенола и
ацетона, причем сырьем для кумола служит нефть:
сн3 сн3
Ph—С—О—ОН + Н+ -> Ph-С—0+ + НаО ->
СН3 СН3
СН3
Ионна 1 |
перегруппировка _____q___q_j_
сн3
PhOH + СН3СОСН3
114
Инициирование гидроперекисью кумола полимери-
зации стирола или метилметакрилата служит харак-
терным примером молекулярно-индуцированного разло-
жения.
Истинная скорость полимеризации стирола при
60° С в 100 раз больше вычисленной на основании из-
вестной скорости разложения гидроперекиси кумола в
бензоле. Интересно, что гидроперекись реагирует с обра-
зованием радикалов с еще большей скоростью в смеси
стирола с пиридином или бензиловым спиртом по
сравнению с реакцией в чистом стироле или стирольно-
бензольных смесях [5].
Как мы уже показали, молекулярно-индуцированный
гомолиз имеет место, когда гидроперекись трет-бутила
или кумола разлагается в различных олефиновых рас-
творителях.
В следующем разделе мы коснемся в основных чер-
тах этого интересного явления, происходящего в неко-
торых системах.
\
Молекулярно-индуцироваиный гомолиз
Когда при взаимодействии нерадикальных соедине-
ний с аномально большой скоростью возникают свобод-
ные радикалы, то такой процесс называют молекулярно-
индуцированным гомолизом. Большая часть примеров
этого явления включает взаимодействие олефиновых
растворителей с инициаторами, хотя известны также и
другие случаи. Мы еще остановимся на том, как моле-
кулярно-индуцированный гомолиз часто сопровождается
конкурирующим процессом ионного разложения ини-
циатора, который иногда затрудняет его распознавание,
т. е. не всегда ясно, подвергается инициатор обычному
гомолитическому разложению с повышенной скоростью
или нет.
В системе стирол — гидроперекись mpem-бутила, рас-
смотренной выше, быстрая ионная реакция обусловли-
вает разложение большей части инициатора. Однако в
этом случае не исключена возможность протекания
индуцированного гомолиза, так как свободные радикалы
образуются с аномально большой скоростью [6]. Мы
показали один из простых примеров молекулярно-инду-
цированного гомолиза, который хорошо изучен. В то же
время детально изучить такую систему трудно, ибо ини-
115
циатор разлагается, по крайней мере, четырьмя раз-
личными путями:
мономолекулярным разложением;
молекулярно-индуцированным гомолизом, который
является реакцией первого порядка относительно гидро-
перекиси и стирола;
атакой полистирольного радикала на гидроперекись *
и полярной реакцией, которая приводит к образованию
окиси стирола и mpem-бутилового спирта.
Молекулярно-индуцированный гомолиз можно на-
блюдать в хорошо изученной системе: перекись м, ж'-ди-
бромбензоила — п, п'-диметоксистильбен [7]. В растворе
бензола при 45° С эта перекись и олефин реагируют по
двум различным механизмам. Первую реакцию легко
изучить. Она заключается в нерадикальном процессе
образования аддукта 1:1:
О О
II II
Аг—С—О-О—С—Ar-bRHC =CHR^
О О
II II
-> Ar—С—О—CH ? —CHR—О—С—Аг
(Аг = ж-BrPh)
Однако вторая реакция, как известно, протекает одно-
временно с первой, так как гальвиноксил, как радикаль-
ная ловушка, исчезает быстрее, когда в системе при-
сутствуют и инициатор, и олефин, чем если бы в ней
находились исключительно либо олефин, либо перекись.
В результате радикального процесса около 10% пере-
киси превращается в радикалы, которые затем зарож-
дают короткую цепь. Такой гомолиз должен ускоряться
присутствием олефина, ибо в его отсутствие образование
свободных радикалов из перекиси протекает медленно.
При взаимодействии иода и стирола наблюдается
молекулярно-индуцированный гомолиз [8]. Молекуляр-
* Следует различать молекулярно- и радикально-индуцирован-
ное разложение инициаторов. В первом случае происходит взаимо-
действие нерадикалов с образованием • радикалов. Второй процесс
является реакцией замещения SH2 (см. стр. 15). Радикально-ин-
дуцированное разложение иногда называют просто индуцирован-
ным разложением (примеры см. на стр. 80, 91).
116
ный иод и стирол реагируют при 25° С с образованием
свободных радикалов в 108 раз быстрее, чем можно было
бы ожидать при гомолизе иода или при термическом
инициировании стиролом. Это взаимодействие является
реакцией второго порядка относительно стирола и пер-
вого порядка относительно иода и может быть пред-
ставлено схемой:
2PhCH—СН2 + I2 2PhCH—СН21
Однако система усложняется тем, что одновременно с
этой реакцией протекает ионная реакция, которая также
дает дииодстирол.
По-видимому, примерами наиболее «опасного» мо-
лекулярно-индуцированного гомолиза, все же открытого
исследователями, могут служить взаимодействия трет-
бутилгипохлорита или гипобромита с ацетиленовыми
или олефиновыми растворителями [9]. Образование из
смесей бутена-2 и mpem-бутилгипохлорита протекает,
как правило, настолько экзотермически, что запаянные
трубки взрываются при температурах выше температу-
ры кипения жидкого азота.
Реакции стирола изучены достаточно подробно. Сти-
рол реагирует с mpem-бутилгипохлоритом или гипобро-
митом в темноте при 0°С самопроизвольно с выделе-
нием тепла. Радикальная природа реакции с трет-бу-
тилгипохлоритом доказывается ингибированием ее
кислородом или mpem-бутилпирокатехином (феноль-
ный ингибитор) с образованием продукта при-
соединения вопреки правилу Марковникова:
PhCHCl—CH2OC4H9-mpem, а также тем фактом, что
происходит хлорирование добавленного в систему цик-
Таблица 9.2
Продукты взаимодействия /прет-бутилгипохлорита
со стиролом при 40 °C в темноте и на свету [9]
Продукт Выход, %
На свету В темноте
СН3СОСН3 6 4
(СН3)3 СОН 14 11
РЬСНС1СН2ОС4Н9 - трет 69 63
РЬСНС1СН2С1 2 1
117
логексана, хотя mpem-бутилгипохлорит и циклогексан
не взаимодействуют между собой в темноте при 0°С.
Отношение реакционной способности циклогексана к
реакционной способности стирола в самопроизвольной
реакции в темноте равно 1:1,1 ив обычном процессе,
инициируемом светом, 1 : 1,7. Как показано в табл. 9.2,
продукты этой реакции подобны, независимо от того,
осуществлялся процесс в темноте или на свету.
Х»+У«
Мономолекуляр-
ный гомолиз
XX--S)*4(Y--S>
" Образование
• комплекса
\_(SX)»+(Y—S)»_
Образование
связи
Координата реакции
Рис. 9.1. Образование связи в результате гомолиза
инициатора X—У.
Мономолекулярный гомолиз соединения X —Y можно
представить так:
X—Y -> Х- +Y-
Энергия активации этой реакции равна энергии
диссоциации связи X — Y. Если растворитель S, в кото-
ром протекает гомолиз, может взаимодействовать с
радикалами, стабилизируя их, тогда общая энергия,
необходимая для гомолиза, будет меньше. Это можно
показать схемой:
X—Y + nS[(S- -X)- (Y- -S)]->
Переходное состояние
-> (S--.X).+ (S Y).
где (S«««X)« — комплекс растворителя с радикалом Х-.
Влияние этого комплексообразования на энергию
гомолиза показано на рис. 9.1.
В отдельном случае свободный радикал X- может
связываться с молекулой растворителя:
X-Y + nS -> [(X --S)- .(Y--.S)]->
Переходное , состояние
XS- + -(Y- -S)
118
В этом случае энтальпия реакции, вероятно, умень-
шается, если учитывать энергию связи S —X (см. об-
суждение в гл. 4). Если D(S — X) больше, чем Z)(X —Y),
то реакция в целом будет экзотермической. Это также
показано на рис. 9.1.
Молекулярно-индуцированный гомолиз чаще всего
наблюдается в системах, где растворитель имеет поля-
ризованные электроны, а радикалы относительно элек-
троотрицательны. В этих случаях перенос электронов
-от растворителя к радикалу в момент его выделения
представляется энергетически выгодным. Например, так
протекают реакции олефинов с молекулами галогенов и
диметоксистильбена с перекисью дибромбензоила.
Заметное влияние диметиланилина на разложение
перекиси бензоила обсуждалось в гл. 7. Эта реакция
заключается в переносе электрона основным нуклеофи-
лом к перекисной связи О — О. Электронодонорные
растворители могут вести себя как слабые нуклеофилы,
и их склонность к индуцированному гомолизу можно
рассматривать как вклад в действие сильных нуклео-
филов, таких, как диметиланилин, с одной стороны, и
очень слабых нуклеофилов, например стирола, — с дру-
гой*. Это можно увидеть, написав резонансную струк-
туру с разделением заряда для переходного состояния
при молекулярно-индуцированном гомолизе, вызванном
растворителем S:
X—Y + nS [SX- -YS > SX;- +YS]
* Переходное состояние
Если структура с разделением заряда более ста-
бильна для ионов, чем для радикалов, то эта реакция
является 5м2-замещением связи X — Y растворите-
лем S. Не считается неожиданностью, что ионные про-
цессы так часто сопровождают и Конкурируют с моле-
кулярно-индуцированным гомолизом.
Интересно подсчитать' термохимическую эффектив-
ность молекулярно-индуцированного гомолиза, напри-
мер, в случае системы' стирол — трет-бутилгипохлорит.
Энергия связи ГЦтрет-С^дО — С1) равна приблизи-
* Интересно, что даже хлористый и бромистый литий, по-види-
мому, вызывают молекулярно-индуцированный гомолиз перекиси
бензоила [10].
119
тельно 40 ккал/моль, а энергия присоединения атома
хлора к стиролу равна~49 ккал/моль. Следовательно,
теплота реакции
\Н,
ккал/моль
трет-С^НдО—Cl -> mpem-C4H9O-+С1 • 40
mpem-C4H9O— Cl ф PhCH = CH2 -> —9
-> mpem-C4H9O- + C1CH2—CHPh
Отсюда ясно, что для протекания экзотермического
молекулярно-индуцированного гомолиза потребуется го-
раздо меньшая энергия активации, чем для обычного
мономолекулярного разложения.
В некоторых системах олефины могут реагировать
между собой, давая свободные радикалы. Например,
1,1-дихлор-2,2-дифторэтилен взаимодействует с бута-
диеном в темноте при 80° Сив присутствии ингибитора
полимеризации дает циклобутан [11]:
СС12 = CF2 + С = С—С = С -> С — С-С = С
I I
cf2— СС12
Эта реакция подтверждается образованием стабилизи-
рованного бирадикала, который ее инициирует,
СС12—CF2—С—С—С = С
Другим примером молекулярно-индуцированного го-
молиза олефина является термическая полимеризация
стирола. Молекулы его взаимодействуют между собой
в темноте, давая свободные радикалы, инициирующие
собственную полимеризацию, причем скорость ее значи-
тельна даже при 60° С. Относительно стирола эта реак-
ция третьего порядка [12]. В присутствии ингибиторов
полимеризации можно выделить 1-фенилнафталин и
1-фенилтетралин [13].
Эти факты подтверждают механизм, рассмотренный
ниже, согласно которому первой стадией является
обратимая конденсация Дильса-Альдера. Было пока-
зано, что величина скорости замещения дейтерием в
120
a, p-положениях или в кольце согласуется с этим меха-
низмом [14]:
Н Н Ph
I + PhCH -= СН2
Медленно
-------->
+ PhCH—СН3
По-видимому, ясно, что поляризуемость растворителя
играет роль в определении его эффективности в процессе
индуцированного гомолиза. Во всех примерах, рассмот-
ренных выше, растворителями были ненасыщенные со-
единения. Однако в таком насыщенном, но полярном
растворителе, как иодистый метил, разложение перекиси
о-нитробензоила протекает в 300 раз быстрее, чем в
хлороформе [15]. Это объясняется повышенной ско-
ростью разрыва связи О — О в надэфире [16]:
О—О—C4He-mpem
О
Относительные скорости разложения этого надэфира
при 60° С следующие:
X К X К
н трет - С4Н, I 1 4 54 CH3S PhS 1 • 10* 210*
121
Очевидно, сильно поляризованные соседние атомы в'
бензольном кольце в значительной мере способствуют
разрыву связи О — О, причем этот процесс можно рас-
сматривать как внутримолекулярный молекулярно-инду-
цированный гомолиз.
Упражнения
9.1. Пинан можно окислять кислородом воздуха до гидропере-
киси, пинана. При 110° С эта гидроперекись разлагается с образо-
ванием производного циклобутана, которое дает качественную реак-
цию с йодоформом, указывающую, что это — метилкетон. Напишите
механизм его образования. («Ключ» к этой реакции — р-расщепле-
ние [17].)
,ООН
Г идроперекись
пинана
9.2. Напишите резонансную структуру для переходного состоя-
ния гомолиза надэфира, описанного на стр. 121, для случаев, где
а) Х = 1 и б) X=PhS. Можно ли приписать стерическому эффекту
увеличение скорости реакций?
9.3. Перекись бензоила подвергается разложению в иодистом
метиле с обычной скоростью, в то времи как перекись о-нитробен-
зоила распадается неожиданно быстро в этом растворителе. Объяс-
ните это.
9.4. При разложении перекиси ацетила в циклогексане, взятом
в качестве растворителя, образуются циклогексенилацетат (I) и
циклогексилацетат (II):
О—С—СН3
о—с—сн3
Постулировано, что соединение I образуется в результате
Sf/ 2-атаки циклогексенильных радикалов на перекисную связь
О—О. Взаимодействие циклогексенильных радикалов с ацетатными
с образованием соединения I представляется неправдоподобным,
так как, по-видимому, ацетатные радикалы разлагаются прежде,
чем могут встретиться с другим радикалом:
Очень
СН3—СО3------► сн3+со2
быстро
122
В связи с Этим объясните механизм образования соединения Н.
Этот механизм должен объяснить тот факт, что когда перекись
ацетила мечена О18 по карбонильной группе’ образующийся продукт
II имеет распределение О18 частично, но не полностью, между дву-
мя кислородными атомами II [18].
9.5. Определите и проиллюстрируйте примерами: индуцирован-
ное разложение инициатора, S# 2-реакцию инициатора и неинициа-
тора и их молекулярно-индуцированный гомолиз. Укажите разли-
чия в этих процессах.
9.6. Обсудите эти факты относительно образования дииодстиро-
ла в растворе четыреххлорнстого углерода при 25° С: скорость раз-
ложения 0,01 М иода в инертном растворителе при 25° С, вычислен-
ная для газовой фазы, равна 2 • 10~15 моль л~г сек-1; скорость
образования радикалов при термическом инициировании 0,2 М
стирола в инертном растворителе равна также 2 • 10~15 моль • л~‘ X
Хсек~‘; смесь 0,2 М стирола и 0,01 М иода в четыреххлористом
углероде разрушает дифенилпикрилгидразил (ДФПГ) со скоростью
1 • 10~8 моль л~‘сек~1; иод в этом растворе, содержащем ДФПГ,
исчезает в 15 раз быстрее, чем только в ДФПГ; кинетически ие инги-
бированное образование дииодстирола является реакцией первого
порядка относительно иода и 3/2-порядка относительно стирола.
Используйте диаграмму с координатами; энергия — реакция, чтобы
показать термохимическую последовательность молекулярно-инду-
цированного гомолиза. Наконец, укажите единственную возмож-
ность существования переходного состояния, которое включает одну
молекулу иода и две молекулы стирола [8].
ЛИТЕРАТУРА
1. Bell Е. R. et al. Disc. Faraday Soc.; 10, 242 (1951).
2. StannettV., Mesrobian R. B. J. Amer. Chem. Soc., 72,
4125 (1950).
3. Johnson D. H., Tobolsky A. V. Ibid., 74, 938 (1952).
4. Kharasch M. S., Fono A., Nude nb erg W. J. Organ.
Chem., 16, 113 (1951).
5. T о b о 1 s k у A. V., Matlack L. R. J Polimer Sci., 55, 49
(1961).
6. W a 11 i n g С., C h a n g Y. W. J. Amer. Chem. Soc., 76, 4878
(1954); Walling C„ Heaton L. Ibid., 87, 38 (1965);
В r i 11 W. F., Indictor N. J._Organ. Chem., 29, 710 (1964).
7. G г еел e F. D., Adam W„ С a n t r i 11 J. E. J. Amer. Chem.
Soc., 83, 3461 (1961).
8. F r a e n k e 1 G„ Bartlett P. D. Ibid., 81, 5582 (1959).
9. Walling C„ Heaton L, Tanner D. D. Ibid., 87,1715 (1965).
10. Kochi J. K., Graybill В. M., Kurz M. Ibid., 86,5257 (1964).
11. Bartlett P. D, Montgomery L. K., Seidel B. Ibid., 86,
616 (1964).
12. Russell К. E„ Tobolsky A. V. Ibid., 75, 5052 (1953);
Mayo F. R. Ibid., 75, 6133 (1953); H i a 11 R. R., Bart-
lett P. D. Ibid., 81, 1149 (1959).
13. Mayo F. R. Preprints Div. Polymer Chem. Am. Chem. Soc.,
Chicago Meeting, September 1961. Vol. 2, No. 2, p. 55.
14. Pryor W. A. et al. American Chemical Society Meeting Abst-
racts of Papers, Atlantic City, September 1965.
123
15. Leffler J. E., Faulkner R. D., Petropoulous С. C.
J. Amer. Chem. Soc., 80, 5435 (1958).
16. Bentrude W. G., Martin J. C. Ibid., 84, [561 (1962).
17. Schmidt G. A., Fisher G. S. Ibid., 76, 5426 (1954).
18. Martin J. C„ Drew E. H. Ibid., 83, 1232 (1961); S h i-
пе H. J., Slagle J. R. Ibid., 81, 6309 (1959); Shine H. J.,
WatersJ. A., Hoffman D. M. Ibid., 85, 3613 (1963).
Дополнительная литература
Wallace J. G. Hydrogen Peroxide in Organic Chemistry. E. I. du
Pont de Nemours and Company, Electrochemicals Dept., 1962.
Zavgorodnii S. V. Russ. Chem. Rev. (English Transl.), 30,
133 (1961).
Глава 10.
ТЕРМИЧЕСКИЙ ГОМОЛИЗ. АЗОСОЕДИНЕНИЯ
Азосоединения широко используют в качестве ини-
циаторов. Эти соединения оказывают значительное
влияние на стабильность получаемых свободных радика-
лов и на скорость разложения вещества. Энергия акти-
вации разложения азометана СНз — N = N — СНз равна
51 ккал/моль. Он подвергается термическому гомолизу
с обычной скоростью только при температуре, близкой
Таблица 10.1
Энергии активации для азоеоедииеиий
R —N =N — R' ккал/моль Растворитель Лите- ратура
R R'
СН3 сн3 51 Газовая фаза гп
трет - С4Н9 трет - С4Н9 43 Газовая фаза Г2|
Ph2CH Ph 2СН 27 Толуол |31
Ph Р h3C 27 Бензол 14]
CN CN
CH3 — С — СН3 сн3 — С — сн3 31 » [5]
к 400°С*. В табл. 10.1 приведены значения энергии
активации для других азосоединений. Они дают стабиль-
ные радикалы, которые могут разлагаться с повышен-
* Азометан, подобно другим азосоединениям, может подвер-
гаться фотолизу уже при комнатной температуре.
124
ними скоростями; например, фенилазотрифенилметан
Ph—N = N—СРЬз разлагается с энергией активации,
ккал/моль, при оптимальной температуре
Связи С—N и N,= N обладают достаточной проч-
ностью (их энергии равны 70 и 100 ккал/моль соот-
ветственно). Уже одно это, казалось бы, должно свиде-
тельствовать о невозможности азосоединениям служить
инициаторами. Однако образование молекулы азота при
разложении азосоединений обеспечивается достаточным
для разложения количеством энергии. Теплота образо-
вания молекулы N2 равна 225 ккал/моль. Чтобы ста-
бильность молекулы азота могла влиять на скорость
разложения азосоединения, ее образование должно про-
текать через стадию переходного состояния:
R—N = N—R -> [r-•-N = N-•-И]-* 2R-+N2
Переходное состояние
В этом переходном состоянии на R-группах должна
создаваться повышенная электронная плотность, отсюда
ясно, почему способность R-групп нести неспаренный
электрон влияет на скорость разложения азосоединений.
Наиболее распространенным инициатором, содержа-
щим азогруппу, является азоизобутиронитрил (АИБН).
Впервые он был получен в 1949 г. и до сих пор широко
используется в химии. Период полуразложения его
равен 17 ч при 60° С и 1,3 ч при 80° С. Это азосоеди-
нение разлагается примерно с одинаковыми скоростями
в бензоле, толуоле, ксилоле, уксусной кислоте, ани-
лине, нитробензоле, додецилмеркаптане и изобутаноле.
В этом заключается его существенное отличие, напри-
мер, от перекиси бензоила, скорость разложения которой
сильно зависит от природы растворителя. Более того,
скорость разложения АИБН в таких растворителях,
как толуол, практически не изменяется под действием
ингибиторов, например хлоранила, иода или дифенил-
пикрилгидразила (ДФПГ) (см. структуру на стр. 32).
Следовательно, радикалы не атакуют азосоединение по
механизму индуцированного разложения; в противном
случае скорость исчезновения азосоединения снижалась
бы в присутствии веществ, ингибирующих радикальное
цепное разложение. Индуцированное разложение боль-
шинства азосоединений в растворе практически ничтож-
125
но, и этим обстоятельством вместо применения инициа-
тора иногда пользуются при изучении радикальных
реакций.
Тщательные исследования реакций разложения азо-
соединений показали существование заметного клеточ-
ного эффекта, детали которого основательно выявлены
в работах с. АИБН (обсуждение клеточного эффекта
см. на стр. 85).
Эффективность этого инициатора при получении сво-
бодных радикалов менее 100% [6], т. е. вместо обра-
зования двух радикалов на моль азосоединения, как
было предсказано для простого разложения,
R—N = N—R -> 2R- + N2 (10.1)
наблюдается меньший выход радикалов.
Это объясняется тем, что заметная часть радикалов
рекомбинирует в клетке растворителя, прежде чем ра-
дикалы смогли бы диффундировать отдельно друг от
друга. Распад АИБН усложняется также и тем, что
цианпропильные радикалы рекомбинируют с образова-
нием двух конечных продуктов: динитрила тетраметил-
янтарной кислоты (сукцинонитрила R — R) и кетен-
имина (R — R'):
2СН3—С—CN
I
СНз
CN CN Z 4 CN СН3
II I I
СН3—С—С—СН3 СН3—С—N = С = С
II I I
СН3СН3 сн3 сн3
(R-R) (R—R')
Более того, они соединяются с образованием назван-
ных продуктов и в клетке растворителя, и в растворе.
Следовательно, гомолиз АИБН можно представить сле-
дующими реакциями:
R—n=N—R
2R»
R—R
R—R1
126
В этой схеме R • — цианпропильный клеточный радикал
(клеточные радикалы заключены в квадратных скоб-
ках). Замечено, что и клеточные, и свободные циан-
пропильные радикалы дают два продукта. В отличие от
кетенимина R — R', сукцинонитрил R — R является ста-
бильным продуктом. Фактически кетенимин можно счи-
тать инициатором, который имеет константу скорости
разложения, близкую для АИБН. Таким образом, можно
представить следующую схему разложения АИБН [7]:
АИБН разлагается с образованием клеточных ради-
калов. Далее, эти клеточные радикалы могут либо выде-
ляться из клетки, становясь свободными (а), либо ре-
комбинировать с образованием кетенимина (б) или
сукцинонитрила (в). Свободные радикалы могут также
взаимодействовать, давая либо сукцинонитрил (г), ли-
бо кетенимин (д). Интересно, что оба конечных про-
дукта R—R и R—R' образуются примерно в одном и
том же отношении из клеточных и свободных радика-
лов. Это вполне очевидно, так как рекомбинация в
клетке является обычной радикальной реакцией, а не
каким-то особым нерадикальным процессом. Образую-
щийся по реакции (б) или (д) кетенимин разлагается
также с образованием клеточных радикалов (реак-
ция е). Эти клеточные радикалы могут далее претерпе-
вать различные превращения по трем механизмам: пре-
вращаться в свободные радикалы (з), рекомбинировать,
давая сукцинонитрил (ы) или кетенимин (ж).
На схеме (10.2) показаны два пути образования ди-
нитрила тетраметилянтарной кислоты R—R: один —
из клеточных радикалов, другой — из свободных ради-
калов. Отсюда можно заключить, что радикальные ло-
вушки ловят все свободные радикалы, и это должно
снижать выход R—R до определенной величины, пред-
127
ставляющей количество R—R, образующегося непо-
средственно в клетке растворителя:
Пот ря N2
R—N = N—R---------> [2R ] -> R—R
При -утетвие
радикальной
ловушки
Отсутствие
----------- R—R
радикальной
ловушки
Различные продукты, отличные от R—R
Это явление наблюдалось для большого набора ра-
дикальных ловушек. Тиолы являются весьма эффектив-
Рис. 10.1. Зависимость процента выхода
тетраметилсукцинонитрила от молярности
бутантиола в ССЦ. Начальная концентра-
ция АИБН равна 0,2 М [8].
калами. На рис. 10.1 показано влияние бутантиола на
выход сукцинонитрила R—R. Чем больше в системе
тиола, тем быстрее снижается выход сукцинонитрила до
предела около 20%. Это означает, что АИБН подвер-
гается клеточной рекомбинации на 20%, а на свобод-
ные радикалы приходится 80%.
На рис. 10.2 показаны результаты опыта со стиро-
лом в качестве радикальной ловушки. При малых кон-
центрациях стирола к нему присоединяется очень мало
свободных радикалов, чтобы вызвать начало полиме-
128
рйзации, поэтому выходы полимера низкие. По мере
увеличения концентрации стирола выходы соответствен-
но растут. Однако даже в чистом стироле не все ради-
калы из АИБН являются инициаторами, поэтому азо-
изобутиронитрил эффективен только на 65%.
Рис. 10.2. Зависимость эффективно-
сти АИБН как инициатора полиме-
ризации стирола от концентрации
стирола (в растворе бензола при
60°С). Концентрация АИБН, М:
• — 6,0 • 10-’, 0—3,0 • 10—3, Д— 1,2 • 10—3 [66].
Единственной реакционной системой, способной улав-
ливать даже клеточные радикалы, является система, в
которой в качестве растворителя используют жидкий
бром. При разложении АИБН в жидком броме выход
сукцинонитрила равен нулю [9].
Исследованы и некоторые другие азосоединения, и
механизм их разложения аналогичный,
В качестве инициаторов применяются также N-нит-
розоанилиды. Они перегруппировываются in situ в ди-
азоацетаты, которые затем диссоциируют с образова-
нием свободных радикалов:
N=O О
I II
Ar—N----С—R'
Медленная
стадия
о
II
Ar—N = N—О—С—R'
Быстро
-* Аг • 4* N2 -|- R'COa •
5 У. Прайер
129
Упражнения
10.1. Как было найдено, константа скорости выделения азота из
АИБН в бензольном растворе больше, чем константа скорости ини-
циирования стирольной полимеризации с помощью АИБН. Объяс-
ните это наблюдение на основании схемы (10.2).
10.2. При разложении в растворе нзооктана смеси СНз—N=
= N—СНз и CD3—N=N—CD3 образуются только СзНз и C2De.
Однако в газовой фазе выходы продуктов подчиниются выражению
[СН3 —СР8]
{[C2He] [C2De]}l/»
Объясните эти результаты [10].
ЛИТЕРАТУРА
1. Steel С., Trotman-Dickenson A. F. J. Chem. Soc.,
975 (1959).
2. В 1 а с k h a m A. U., Е a t о u g h N. L. J. Amer. Chem, Soc., 84,
2922 (1962)
3. CohenS. G., W a n g С. H. Ibid., 77, 2457 (1955).
4. Alder M. G., Leffler J. E. Ibid., 76, 1425 (1954).
5. В a w n С. E. H., M e 11 i s h S. F. Trans. Faraday Soc., 47,
1216 (1951); Van Hook J. P„ T о b о 1 s k у A. V. J. Amer.
Chem. Soc., 80, 779 (1958).
6. Bevington J. C. J. Chem. Soc., 3707 (1954); Beving-
ton J. C. Trans. Faraday Soc., 51, 1392 (1955).
7. H о m m a n d G. S. et al. J. Amer. Chem. Soc., 82, 5394 (1960).
8. H a m m о n d G. S., S e n J. N., Boozer С. E. Ibid., 77, 3244
(1955).
9. Trapp O. D„ Hammond G. S. Ibid., 81, 4876 (1959).
10. LyonR. K., LevyD. H. Ibid., 83, 4290 (1961).
Глава 11
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ РЕАКЦИИ,
ОБРАЗУЮЩИЕ СВОБОДНЫЕ РАДИКАЛЫ
Для получения свободных радикалов часто исполь-
зуют окислительно-восстановительные реакции с пере-
носом одного электрона. Так как в них не требуется
наличие нестабильного инициатора, то их можно при-
менять для получения радикалов при низких темпера-
турах. Образование свободных радикалов может быть
результатом реакции окисления либо восстановления.
Так, при электролизе солей органических кислот по
Кольбе на аноде анионы окисляются до ацилокси-ради-
130
калов. Последние, потеряв СО2, превращаются в сво-
бодные радикалы, которые затем рекомбинируют:
R—СО? R—СО2- -+ R-+COa
2R- R-R
Эта реакция нашла некоторое применение в органи-
ческих синтезах.
Большинство важнейших окислительно-восстанови-
тельных процессов протекает с участием иона металла,
являющегося переносчиком одного электрона. Из этих
реакций наиболее хорошо изучено взаимодействие пере-
киси водорода с ионом Fe2+. Реакцию открыл Фентон
в 1894 г., а в 1932 г. Габер и Вайсс предложили меха-
низм, который лишь с небольшими изменениями принят
в настоящее время [1]. Перекись водорода реагирует с
ионами железа по следующим схемам:
Fe2+ + Н2О2 -> Fe3+ + НО- + НО • (11.1а)
Fe’++ Н2О2Fe2++ НО2 + Н+ (11.16)
Fe2++ НО • -> Fe3++ ОН- (11.1в)
Fe3+ -f- НОГ Fe2+ + Н+ +ОГ- (П.1г)
Fe2+ + НО2- ->Fe2++ НО? (11.1д)'
НО- + Н2О2 -> Н2О + НО2- . (11.1е)
ОГ-+ Н2О2^ О2 + ОН~-|-НО- (11.ж)
Эти реакции' объясняют каталитическое действие
железа (II) на разложение перекиси водорода:
Соли
2Н2О2 ="♦ 2Н2О 4~ Оз
железа
(И.2)
Реагент Фентона можно применять для окисления
органических веществ. Иногда продуктами окисления
бывают димеры, а иногда и продукты замещения гидро-
ксильными радикалами. Например, бензол превра-
щается в смесь бифенила и фенола с большим отноше-
нием фенол/бифенил, если концентрация ионов железа
поддерживается достаточно высокой. Образование двух
5*
131
продуктов при участии ионов железа объясняется меха-
низмом, представленным ниже [2]:
I + Fe3+ —- PhOH + Н + 4-Fe2*
Н-акцептор
В результате димеризации и диспропорционирования
продукта I могут образоваться также следующие про-
дукты: i
Ph—Ph + 2Н2О
Реакция (11.1 а) справедлива для ионов других ме-
таллов и гидроперекисей, перекисей эфиров надкислот,
а также для перекиси водорода.
В общем виде реакцию можно представить как
Me1 + ROOR -> Me" + RO- + RO“ ,
где Me1 — переходный металл в одновалентном состоя-
нии; Me11 — тот же металл ъ другом (окисленном) со-
132
стоянии. Из других примеров этих реакций можно при-
вести следующие:
Fe2 + , Cr2+, V2+ или Ti3+
н2о2--------------------- но- + НСГ
Fe2+, Со2+ или Си+
ROOH------------------>RO- + HCT
О
II Cu+
RO—О—С—R'--------► RO - + R'COr
Fe2+ о
“O3S—О—O—SO?----------к so4 + so?-
Наиболее важным промышленным применением та-
ких окислительно-восстановительных реакций является
низкотемпературная эмульсионная полимеризация смеси
стирол — бутадиен при получении каучука в присутствии
гидроперекиси кумола и ионов железа в качестве ката-
лизатора. Органические мономеры полимеризуются,
превращаясь в маслообразные капли в водной эмуль-
сии, которая стабилизируется добавлением мыла и ще-
лочей. Типовой промышленный рецепт приведен в
табл. 11.1. Как видно, смесь эта сложная, и в деталях
неизвестно назначение каждого ее ингредиента. Из них
представляют интерес гидроперекись, ион железа, пиро-
фосфат Na4P2O7- 10Н2О (который необходим для рас-
творения железа), и тиол (его добавляют в качестве пе-
реносчика цепи для уменьшения выхода продуктов
с низким молекулярным весом и чтобы обеспечить полу-
чение полимера, легко поддающегося обработке).
Таблица 11.1
Типовой рецепт для эмульсионной полимеризации
стирольно-бутадиеиовых смесей
Реагент Содержа- ние, вес. ч. Реагент Содержа- ние, вес. ч.
Бутадиен Стирол Вода Тиол Едкий натр 75 25 180 0,5 0,061 Г идроперекись кумола Сульфат закисного железа Пирофосфат Фруктоза 0,17 0,017 1,5 0,5
133
Окислительно-восстановительные инициаторы играют
важную роль, поскольку их можно использовать при
гораздо более низких температурах, чем перекиси.
Скорость образования радикалов можно измерить, из-
меняя концентрации окислителя (перекиси) и катали-
затора — иона металла, а также температуру. Кроме
того, известно, что окислительно-восстановительные
системы можно использовать как источники радикалов
в водных растворах.
Другим примером окислительно-восстановительной
реакции, которая разработана для синтетических целей,
является взаимодействие перекисей с веществами, со-
держащими активные атомы водорода. В отсутствие
ионов металлов обычно получается смесь продуктов.
Однако металлические ионы, особенно ионы меди, ката-
лизируют ту реакцию, при которой активный атом водо-
рода замещается —О—СО—R-группой. Эту реакцию
открыли Караш и сотр., и она получила широкое рас-
пространение. В качестве примера [3] может служить
реакция mpem-бутилнадацетата с циклогексеном, ката-
лизируемая бромистой медью:
| || + СН3—С—О—О—C4He-mpem
CuBr
О—с-сн3
(11.3)
Этот процесс является лучшим синтезом циклогексе-
нилацетата. Механизм его [4], по-видимому, заклю-
чается в образовании mpem-бутокси-радикала из эфира
надкислоты по типу разложения по Габеру — Вайссу:
О
II
СН3—С—О—О—C4He-/npem+ CuBr ->
-+ [СН3—С02 • CuBr] + mpem-C4He0 •
(U.4)
134
Далее mpem-бутокси-радикал вытесняет аллильный
водород из циклогексена:
mpem-C4H9O • 4- I || -> mpem-C4H9OH 4- I || (11.5)
Конечные продукты могут получаться в результате
либо радикального процесса, либо ионного. В радикаль-
ной реакций циклогексенильный радикал можно пред-
ставить реагирующим с комплексом, который образо-
вался по реакции (11.4):
О—С—СН3
I II
<\О
|| 4- (СН3СО2 • CuBr) -> I || 4- СиВг (11.6)
Реакцию (11.6) называют лигандным переносом. Для
ионного процесса можно представить комплекс реакции
(11.4) как комплекс Си11 с карбокси-анионом:
СН3СО2 • CuBr
$ _
CH3CO2:CuBr+
-> СН3СОГ + CuBr+ (11.7)
Ионы карбония можно рассматривать как продукты
окисления циклогексенильного радикала ионами меди:
+
|| 4- СиВг+ -> | || 4- CuBr
Продуктами ионной реакции могут быть
О—С—сн3
(П.8)
(И.9)
Реакция (11.8) —ключевая ступень этого ионного про-
цесса — сопровождается переносом электрона.
135
Очевидно, ионы карбония действительно являются
промежуточными продуктами в этих реакциях, исходя
из того, что типичный ион карбония претерпевает пере-
группировки. Например, 1,2-миграция атома водорода и
алкила иногда сопровождается каталитическим дейст-
вием меди; такой тип перегруппировки трудно себе
представить для радикальных промежуточных продук-
тов [4, 5]. Кроме того, продукты реакции алкенов-1 по-
добны тем, которые получаются скорее из аллильного
карбониевого иона, чем из аллильного радикала. Так,
октен-1 реагирует с mpem-бутилнадацетатом, давая в
основном 3-эфир:
CSHU—СН2-СН = сн2
| mpem-C4H9O-
[CSHU—СН—CH = CH2 C8HU-CH = CH-CH2]
I Cu2+
[CsHn-CH-CH=CH2 CSHU-CH = CH-CH2]
I CH3—CO7
С5НЦ-СН-СН=СН2 + CSHU-CH=CH-CH2
о о
j I
CO—CH3 CO—CH3
86: 14
(Общий выход 89 %)
Известны также радикальные реакции алкенов-1, ко-
торые в основном дают 1-замещенные продукты. Напри-
мер, хлорирование пентена-1 по радикальному механиз-
му mpem-бутилгипохлоритом дает 1-хлорид с большим
выходом [6]:
С2Н5—СН2—СН = СН2
40°С mpem-C4H9OCl
С2НВ—СН—СН = СН2 +C2HS—СН = СН—СН2
I I
С1 С1
40% 60%
136
Хотя образование этих продуктов могло произойти в
результате реакций иона карбония, стадия выделения
водорода (11.5) является радикальным процессом. От-
сюда следует, что относительную реакционную способ-
ность различных веществ можно предсказать на осно-
вании реакции, в которой /пое/п-бутокси-радикал вытес-
няет водородный атом. Например, относительная реак-
ционная способность ряда этилбензол : кумол : гексен-2
равна 1,0 : 0,4 : 1,4 в реакции, катализируемой медью, и
в их реакции с трет-бутоксильными радикалами, полу-
чаемыми из трет-бутилгипохлорита [4].
Механизм этих реакций можно представить в об-
щем виде, как указано ниже, где RH — какое-либо со-
единение с подвижным атомом водорода.
Инициирование:
О
II
СН3—С—О—О—С4Н9-/ирет + Си1 -+
-э- (СН3СОГСии) 4- mpem-C4H9O-
трет-С4Н9О- 4- RH -> m/?em-C4H9OH 4- R-
Стадия образования продуктов:
R- 4- (СН3СОГСип) ROOCCH3 4- Cu1
(Перенос лиганда)
или, более вероятно,
r. + Си11 + R+ 4- Си1
(Перенос электрона)
о
II
R+ 4- СН3СОГ -> RO—С—СНз
Эти катализируемые ионом меди реакции нашли ши-
рокое применение в синтезах. Некоторые примеры пред-
ставлены в табл. 11.2.
Упражнения
11.1. Представьте схемы механизмов, объясняющих образова-
ние следующих продуктов, получающихся в реакциях, катализируе-
мых ионами меди:
а) гидроперекись mpem-бутила в изопропилбензоле при 100° С
дает 90%-ный выход Ph—С(СНз)2ООС4Н9-игрепг;
137
Таблица 11.2
Реакции активных доноров водорода с иадэфирами, катализируемые ионами меди
Соединение с активным атомом водорода Перекисное соедннеине Продукт Выход, % Литература
с6нп—сн3—сн=сн2 О ' II О II трет- С4Н9О—О—С—СН3 О II С6Нц—СН=СН-СН2 СН3—СО—О С6Нц—СН—сн=сн2 О ' 1 СО—СН3 О II 12 77 [4]
PhCH2—О—С—СН3 О II PhCH2—О—С—Ph mpem-C4H9O—О—С—Ph О II трет- С4Н9О—О—С—СНа О Ph—СН-О—С—СН3 О—СО—Ph 35 [71
То же 18 [7]
PhO—СН2—СН=СН2 mpem-C4H9O—О— С—Ph PhO—СН—СН=СН2 О—СО—Ph 50 [71
0
Ph—СНО II mpem-C4H9O—0—C—Ph
^\/\ 1 1 1 Х/\/ 0 II mpem-C4H9O—0—C—Ph 0
CgH?—S—C3H7 II mpem-C4H9O—0—C—Ph
Ph—С—0—C—Ph II II 0 0 70 17]
0-С—Ph 1 II У\/\О 15 [7]
CsH7S—CH—QHj 69 [81
0—CO—Ph
б) гидроперекись mpe/n-бутила в диоксане при 70° С дает
50%-ный выход
\оос4н „•трет
в) гидроперекись mpe/n-бутила в диметиланилине при 25° С дает
90%-ный выход Ph—N(CH3)2CH2—00—С4Нв-пгрепг;
г) перекись mpem-бутила реагирует с бензальдегидом с образо-
ванием пгрепг-бутилбензоата, выход 83%;
д) перекись бензоила в изопропилбензоле в отсутствие солей
меди дает Ph—С(СН3)2—С(СН3)2—Ph, В присутствии их при 85° С
получается 30%-ный выход Ph—С(СН3)2—СО—OPh [9].
11.2. а. Перекись водорода, реагируя с циклогексаноном, дает
аддукт, который можно представить как
НОЧ/ООН
Это вещество разлагается солями железа с образованием 1,12-доде-
кандикарбоновой кислоты. Опишите механизм этой реакции.
б. Предскажите продукт разложения в присутствии иона желе-
за гидроперекиси 1-фенилциклогексила.
в. Иои железа катализирует разложение гидроперекиси 1-метйл-
циклопентила в присутствии бутадиена, что дает с выходом 30%
8,12-эйкозадиен-2,19-дион. Напишите механизм [10].
ЛИТЕРАТУРА
1. Uri N. Chem. Rev., 50, 375 (1952).
2. Smith J. R. L„ Norman R. О. C. J. Chem. Soc., 2897 (1963).
3. Kharasch M. C., Sosnovsky G., Y a n g N. C. J. Amer.
Chem. Soc., 81, 5819 (1959).
4. Walling C„ Zavitsas A. A. Ibid., 85, 2084 (1963);
Kochi J. K. Tetrahedron, 18, 483 (1962); Kochi J. K-
J. Amer. Chem. Soc., 85, 1958 (1963).
5. Story P. R. Tetrahedron Letters, 414 (1962).
6. Walling C„ Thaler W. J. Amer. Chem. Soc., 83, 3877 (1961).
7. Sosnovsky G., Yang N. C. J. Organ. Chem., 25,899 (I960).
8. Sosnovsky G. Tetrahedron, 18, 15 (1962).
9. К h a r a s c h M. S., Fono A. J. Organ. Chem., 23, 324 (1958);
24, 72 (1959).
10. Lawess on S. O., Sosnovsky G Svensk Kem. Tidskr., 75.
343 (1963).
Дополнительная литература
Weedon В. C. L. Advances in Organic Chemistry. Vol. I. Inter-
science Publishers, Inc., N. Y., 1960, p. 1.
Lawesson S. O., Sosnovsky G. Svensk Kem. Tidskr., 75,
568 (1963).
Walling C. Free Radicals in Solution. John Willey and Sons, Inc.,
N. Y, 1957, p. 564.
ЧАСТЬ ТРЕТЬЯ
РЕАКЦИИ РАДИКАЛОВ
Глава 12
РЕАКЦИИ ОТРЫВА ВОДОРОДА
Общие особенности реакций отрыва водорода
В первой части книги радикальные процессы были
подразделены на три основные стадии: зарождение
(инициирование), рост (развитие) и обрыв цепи. Во
второй части мы рассмотрели способы получения ради-
калов, т. е. стадию зарождения цепи, а теперь обра-
тимся к реакциям, в результате которых происходит
рост цепи.
Обычно реакция отрыва водорода является процес-
сом, при котором радикалы не претерпевают изменений.
В общем виде это можно записать так:
R-+SH RH + S-
где SH — любой донор водорода.
Перенос водорода наблюдается либо как основная
реакция, либо как неизбежная побочная реакция почти
в каждой радикальной системе.
Обычно энергия, требующаяся для разрыва связи
С—Н настолько велика, что реакция не может проис-
ходить без дополнительной затраты энергии; такое ос-
лабление связи достигается в переходном состоянии
R- +SH [R H:S > R:H-S] -> R—H + S-
Переходное состояние
В этом бимолекулярном процессе радикал отрывает
атом водорода от другой молекулы, и энергия, требую-
щаяся для разрыва связи, частично поступает за счет
образования новой связи. Известны, однако, некоторые
реакции, при которых происходит отщепление атомов
водорода, например пиролиз или фотолиз этилена, ини-
циированный облучением ртутной лампой. В последнем
случае происходит главным образом отщепление моле-
141
кул водорода, но наряду с этим около 4% (от общего
разложения) приходится на отщепление (отрыв) ато-
мов водорода [1]:
ftv
Hg-----Hg*
Hg* + CHa = CHa Hg + (СНа = СН,)*
НС=СН + на
Главным /
образом у/
(СНа=СНа)* <
Частнч- \
н0 \
сна=сн + н.
На основе теории переходного состояния была пред-
принята попытка предсказать скорости реакций отрыва
водорода. Так как эта проблема трудная, изучали глав-
ным образом простейшую реакцию отрыва водорода, а
именно реакцию атома водорода с молекулой водо-
рода [2, 3]
н-н-н-н -> н—н + н-
Скорость этого процесса можно измерить, используя
меченые атомы, т. е. •
H- + D—D -> H-D + D-
а также изучая конверсию орто- И пара-форм водоро-
да *. Вычисления показали, что переходное состояние
* Водород существует в двух формах: орто-, в которой ядра
имеют параллельные спнны, и пара-, где спины антипараллельны.
При температуре, близкой к абсолютному нулю, молекулы . нахо-.
дитси главным образом в пара-форме, в состоянии наиболее низ-
кого энергетическогро уровня. При комнатной температуре около
75% водорода существует в орто-форме. (Две формы отличаются
друг от друга различными физическими свойствами.) Так какядер-
ный спин исчезает в пара-форме водорода, эта форма не прояв-
ляет магнитных свойств газа. Парамагнитные соединении (напри-
мер, О2, редкоземельные элементы и радикалы) способствуют кон-
версии. Механизм конверсии, катализируемой атомами водорода,
можно представить в виде
Н • +пара-Нг—*-орто-Н2+Н-
142
для этой реакции, возможно, представляет собой ком-
бинацию трех атомов водорода, расположенных на
прямой линии:
Н- + На -> [Н - Н - Н]
Переходное состояние
Если это верно, то данный механизм реакции бимо-
лекулярного гомолитического замещения 8Н2 аналоги-
чен механизму ионной реакции Sn2, при котором про-
исходит так называемая «атака с тыла», т. е. атака с
противоположной к отщепляющемуся заместителю сто-
роны, и вальденовское обращение. Неизвестно, все ли
реакции 8^2 протекают по этому механизму (см. стр. 15).
Даже в очень простых реакциях атомов водорода с
молекулами водорода вычисления так трудны, что за-
ключение о линейности переходного состояния может
быть ошибочным.
Для ответа на этот вопрос необходимо установить,
имеет место сохранение конфигурации либо происходит
инверсия при замещении радикалом отщепляющейся
группы в оптически активной молекуле. Однако еще не
описывался пример реакции, в котором было бы точно
доказано, что радикал атакует асимметрический атом
углерода; всегда происходит преимущественный отрыв
водорода или галогена по сравнению с атакой на угле-
род. Например, оптически активный вторичный йоди-
стый бутил, реагируя с изотопом иода, образует продукт
рацемизации, содержащий радиоактивный иод. Меха-
низм этого процесса заключается в атаке радиоактив-
ным иодом атома иода йодистого бутила:
сн3 сн8
| Главным |
I*. + н—С—I------------ Н—С- + I-I*
। образом ।
С2Н4 СаН4
Оптически Быстро
активен рацемизуется
I*
СН3-СН-С2Н5 +1’2 -> СНз—CH-CaHs + I*.
Продукт рацемизации,
содержащий радиоактивный
иод
143
Непосредственно 5н2-замещение у углерода, кото-
рое также должно было бы способствовать рацемизации
и обмену, почти не встречается, если вообще имеет ме-
сто [4, 5]:
СН3 СН3
I Медленно |
!•* + Н-С—I-------------- I*—C-H+L
। или совсем I
I не идет |
c2Hs C2Hg
Продукт
инверсии
Прямым доказательством того, что в реакциях ра-
дикального замещения происходит атака с тыла, может
быть открытие радикального замещения по асимметри-
ческому атому углерода. Однако косвенные доказатель-
ства этого механизма можно -получить, используя кри-
терии скорости реакции. Скорость реакции чрезвычайно
чувствительна к размеру R-группы, так как, атакуя,
Y- должен оказывать давление на R, для того чтобы
приблизиться к связи С—X с противоположной сто-
роны: I
У~ + RCH2—X —*-
—RCH2V +
Переходное состояние
X'
Так как значения типичных скоростей реакций, проте-
кающих по Sjy2-MexaHH3My, подобны значениям скоро-
стей аналогичных ионных реакций дисульфидов, счи-
тают, что они протекают по механизму атаки с тыла
R
RSSR -> lY - S-.-SR.
-> YSR + RS-
Переходное состояние
Также была изучена реакция 5^2-замещения свобод-
ными радикалами в дисульфидах:
R
Y- + RSSR -> 1Y- - S --SR
Переходное состояние
-> YSR + RS-
144
Эти две реакции замещения по атому серы, ионная и
радикальная, имеют близкие по своим значениям ско-
рости [6, 7]. Очевидно, механизм реакций 5Л-2 и Зн2
заключается в атаке двухвалентной серы с тыла.
Рассмотрим теперь факторы, которые влияют на ско-
рости реакций отрыва водорода. Они могут действовать
в противоположных направлениях, либо ускоряя, либо
замедляя реакции. Однако, если отобрать группы тесно
связанных реакций, можно выделить и исследовать влия-
ние одного или двух факторов.
Одним из факторов, влияющих на скорости реакццй
отрыва водорода, является термодинамическая проч-
ность вновь образующихся и разрывающихся связе^.
Прочность связей R—Н и S—Н оказывает влияние iia
прочность частично образующихся и разрывающихся
связей в переходном состоянии:
R- + SH -> [R - H --S] RH-J-S-
Переходное состояние
Так как- образование связи R—Н обеспечивает энер-
гией разрыв связи S—Н, радикалы R«, которые обра-
зуют прочную связь и соединяются в переходном состоя-
нии со слабой связью S—Н, должны особенно легко
осуществлять реакции переноса водорода [8, 9].
В каждом изучавшемся до настоящего времени ра-
дикальном процессе значения скоростей реакций отрыва
водорода от алифатических углеводородов увеличива-
ются в ряду: первичный<вторичный<третичный. Этот
порядок не зависит от природы атакующего радикала
Таблица 12.1
Относительная активность алифатических атомов водорода
к различным радикалам
X-+RH-*XH+R-
Тип С—Н«связи СНз- (182° С) [Ю] Ph- (60° С) [11] (40° С) [12] сь (25° С) [13]
Первичная 1 1 1 1
Вторичная 7 9,3 8 3,6
Третичная 50 . 44 44 4,2
В циклогексане —— 8,3 15 2,5
Примечание. Все значения даны по атому водорода. Активности метиль-
ного радикала приведены для газофазных реакций, остальные значения для реак-
ций в растворе.
145
и легко объясняется прочностью разрывающихся свя-
зей С—Н. Таким образом, третичные связи С—Н явля-
ются наиболее слабыми, и поэтому скорости реакций
отрыва атомов водорода третичных С—Н-связей имеют
высокие значения. В табл. 12.1, которая иллюстрирует
эту точку зрения, приведены значения относительной
активности атомов водорода к четырем различным ра-
дикалам. Как видно из таблицы, отношение скоростей
реакций отрыва атомов водорода при первичной и тре-
тичной С—Н-связях для метильных радикалов рав-
няется 1 :50, и только — 1:4 для атомов хлора, что го-
ворит о влиянии на селективность типа радикалов и
других факторов, которые будут подробно рассмотрены.
Влияние прочности вновь образующихся связей на
скорости реакций отрыва водорода показано в
табл. 12.2. В родственных группах радикалов более
прочные вновь образующиеся связи ведут к увеличению
скорости реакции. Например, в табл. 12.2 показано, что
Таблица 12.2
Влияние прочности вновь образующейся связи
на энергию активации в реакциях отрыва
водорода [14—16]
V I I СН4 МХ4. ( СН*'
х + | с,н, нх+ I ст-
X- D (Н-Х), ккал/моль Еа, ккал/моль
СН4 С.на
F 136 .0,2 0,2
С1 103 i 3,9 Л 1,0
CFS 103 10,3 7,5
Н 103 9
сн3 102 14,0 П,2
Вт 87 18,3 13,9
атомы галогена отрывают атом водорода в ряду
F>Cl>Br и что в этих реакциях существует подобный
ряд прочности Н—Х-связей. Таким образом, фтор от-
рывает водород при наиболее высоких скоростях (т. е.
с наиболее низкой энергией активации) и образует
прочную связь с водородом. Радикалы хлора, водорода,
трифторметильный и метильный радикалы отрывают
атомы водорода с различной энергией активации, не-
146
смотря на то что все они образуют связи с водородом
приблизительно одинаковой прочности. Это явление
можно объяснить более высокой скоростью реакции од-
ноатомных радикалов по сравнению с многоатомными.
Возможно, в переходном состоянии реакций много-
атомных радикалов происходит изменение конфигура-
ции реагентов. Например, метильный радикал, имеющий
Координаты реакции
Рис. 12.1. Селективность и экзотермичность реакций.
плоскую конфигурацию, вынужден занимать более пи-
рамидальную геометрию в переходном состоянии. Это
изменение конфигурации требует затраты энергии:
Н
I
С. + Н—R
Л
Радикал
(очень плоская
пирамида)
н н
С H——R —С—H-+-R*
н'н НН
Переходное состо-
яние (более выра-
женная пирамида)
Метан
(тетраэдр)
Из табл. 12.1 видно, что различные радикалы отли-
чаются по селективности. Понятия «селективность» и
«активность» радикалов взаимосвязаны. Для реакций
CH3-+RH -> CH4 + R.
Cl- +RH HC14-R-
значения из табл. 12.1 и 12.2 показывают, что метильные
радикалы более селективны в своих реакциях с тре-
мя типами С—Н-связи по сравнению с атомами хлора,
147
но обладают меньшей активностью и требуют большей
энергии активации для того, чтобы оторвать определен-
ный атом водорода. Эту обратную взаимосвязь селек-
тивности и активности радикалов можно легко объяс-
нить особенностями термодинамического отношения
между энергией активации и теплотой реакции [17—19].
На рис. 12.1 показаны энергетические уровни для эк-
зотермических и эндотермических реакций. В экзотер-
мической реакции исходные соединения имеют относи-
тельно высокую энергию по сравнению с энергией про-
дуктов реакции, а значения энергии переходного
состояния вдоль оси координат реакции находятся в
непосредственной близости к значениям энергии исход-
ных соединений. Поэтому структура переходного состоя-
ния должна быть подобна структуре исходных соеди-
нений. Под этим подразумевается, что длина разрыва-
ющейся связи в переходном состоянии мало отличается
от длины связи исходных соединений и что скорость
реакции не чувствительна к природе и прочности разры-
вающейся связи.
В эндотермической реакции наблюдается обратная
картина. Исходные соединения обладают низкой энер-
гией, а значение энергии переходного состояния нахо-
дится в непосредственной близости к значениям энергии
продуктов реакции. В этой реакции длина разрываю-
щейся связи в переходном состоянии неравноценна с
длиной связи исходных соединений. Отсюда скорость
реакции очень чувствительна к природе и прочности
разрывающейся связи.
Нужно подчеркнуть, что взаимосвязь между тепло-
той реакции и ее скоростью не является термодинами-
Таблица 12.3
Продукты хлорирования 2,3-диметилбутана [13]
СН, СН3 СН, СН3 II С’2 II сн3—с—с—сн3 —« С1—сн2—с—с—с II л* II НН НН I сн, сн, 1 1 н,+ сн.-с—с—сн,
С! I А
Растворитель I. % п, %
Четыреххлористый углерод 60 40
Бензол 10 90
Сероуглерод 5 95
148
ческой, и на практике обычно происходит таким обра-
зом, что очень активные соединения вступают в экзо-
термические реакции. Так как длина разрывающейся
связи в переходном состоянии мала, эти реакции обыч-
но не чувствительны к ее природе.
На скорость реакций отрыва водорода, кроме того,
влияют полярный эффект заместителя, который был
кратко обсужден в первой части и будет подробно рас-
смотрен позднее, и эффект растворителя. Как было от-
мечено на стр. 20, эффект растворителя оказывает зна-
чительно меньшее влияние в радикальных реакциях,
чем в ионных. Это обычно используют для идентифика-
ции радикальных процессов, однако в некоторых ради-
кальных процессах он значителен. Одним из заслужи-
вающих внимания примеров влияния растворителя на
течение реакции является хлорирование 2,3-диметилбу-
тана. Значения табл. 12.3 показывают различное распре-
деление продуктов хлорирования в зависимости от рас-
творителя *. Механизм реакции следующий:
Cl- +RH -> R- +НС1
R- + С12 -> RC1 + Cl-
Таким образом, положение хлора в продуктах реак-
ции зависит от того, какой атом водорода замещается
хлором. Третичные С—Н-связи гораздо слабее, чем
первичные, и поэтому должны разрываться легче.
2,3-Диметилбутан имеет 12 первичных и 2 третичные
С—Н-связи. Атом хлора в четыреххлористом углероде
как растворителе обладает очень высокой реакционной
способностью и незначительной селективностью. Поэто-
му наличие первичных С—Н-связей в 2,3-диметилбута-
не ведет к высокому выходу первичного хлористого ал-
кила. Бензол и сероуглерод стабилизируют атом хлора,
образуя с ним комплекс, и тем самым повышают его
селективность в этих реакциях:
Cl- + CSa^(CS2-CI)
(CS2-C1) + RH -> HCI 4- R- +CS2
Надо отметить, что даже в четыреххлористом угле-
роде атомы водорода третичной С—Н-связи более ак-
тивны, чем первичной. В табл. 12.3 показано, что выход
* Распределение продуктов реакции зависит не от диэлектри-
ческой постоянной, что наблюдается в ионных реакциях, а от спо-
собности раствооителей образовывать комплекс с атомом хлора [13J.
149
продуктов хлорирования первичной С—Н-связи состав-
ляет 60%, однако в молекуле 2,3-днметилбутана атомов
водорода первичной С—Н-связи в 6 раз больше, чем
третичной. Поэтому относительная селективность по во-
дороду для третичных С—Н-связей по сравнению с
первичными составляет 40/1 к 60/6 или 4 : 1. Таким об-
разом, третичные С—Н-связи в 4 раза более активны,
чем первичные, даже в четыреххлористом углероде.
Скорость реакции бимолекулярного отрыва водоро-
да всегда пропорциональна концентрации молекул и
концентрации радикалов. Например, скорость реакции
R- + SH->RH+S-
равняется k[R-][SH]
Обычно концентрацию радикалов в кинетической
системе невозможно измерить прямыми методами *. По-
этому константы скорости радикальных реакций выво-
дят таким образом, чтобы исключить концентрации ра-
дикалов. Например, скорость отрыва водорода метиль-
ными радикалами относительно скорости их димериза-
ции можно получить без непосредственного измерения
концентрации метильных радикалов. Для реакции
CH3- + RH -- CH4 + R- (12.1)
Ki
2СН3- —*C2He (12.2)
скорость выражается
[СН3] [RH];
а/
d [Ca-H|,L = к2 [СН3]а, (12.3)
dt
и поэтому
К1 [RH] _ d [CH4]/df
КЧ, (dfCaHeJ/d/)1/» ’
* Как обсуждалось в гл. 2, с помощью электронного парамаг-
нитного резонанса (ЭПР) можно непосредственно измерить кон-
центрацию радикалов. Однако современные приборы ЭПР еще не
могут обнаружить радикалы в концентрации менее 10~8 М и обычно
недостаточно чувствительны, чтобы обнаружить в реакциях очень
низкие равновесные концентрации радикалов. С усовершенствова-
нием приборов непосредственные измерения абсолютных значений
в конце концов найдут широкое применение. В исключительных слу-
чаях, когда радикалы достаточно стабильны, чтобы существовать
в обычных концентрациях, реакции могут контролироваться распро-
страненными способами. Пример этого обсуждается на стр. 307.
150
После преобразования
Ki _ 1 [CH4]/d/ (12 4)
[RH] (d [QjH.l/d/)’7’ '
Таким образом, можно получить константу скорости
реакции (12.1) относительно константы скорости реак-
ции димеризации метильных радикалов (12.2), при этом
абсолютная концентрация метильных радикалов не тре-
буется.
Только в одном случае, когда происходит обмен ато-
мов водорода с его молекулами в газовой фазе, кон-
станту скорости получили непосредственным измере-
нием концентраций:
Н-+ Н2* Н2 + Н- (12.5)
В этой реакции концентрацию, атомов водорода в газо-
вой фазе можно получить измерением скорости диффу-
зии или с помощью масс-спектрометрии. Константу ско-
рости вычислили по методике Тротмана-Дикенсона [20]:
кй = 4- ю1ое~7500/ЛГ л-моль~1-сек~'.
Другой способ измерения скорости этой обменной реак-
ции обсуждался на стр. 142.
Реакции отрыва водорода метильными радикалами
Источником метильных радикалов могут быть мно-
гие соединения, из которых чаще всего применяются:
азометан, перекись mpem-бутила, перекись ацетила, ди-
метилртуть и ацетон;
СН3—N = N—СН3 2СН3 + N2
0 0
II II
СН3—С-0—О—С—СНз -> 2СН3- + 2СО2
СН3 СН3 СН3
СН3—С—О—О—С—СН3-> 2СН3—С—О- ->
сн3 сн3 сн3
-> 2СН3СОСН3 4- 2СН3 •
151
Свет
СН3СОСН3 —► сн3- + сн3со-
СН3—Hg—СН3 -> 2СН3- 4- Hg
Метильные радикалы отрывают атом водорода от
любого донора водорода, присутствующего в системе.
Реакция отрыва водорода конкурирует с обычной реак-
цией обрыва цепи [см. реакции (12.1) и (12.2)].
Как обсуждалось выше, отношение скорости образо-
вания метана к квадратному корню скорости образова-
ния этана дает отношение констант скоростей (с по-
правкой на концентрацию) [см. формулу (12.4)]. Абсо-
лютное значение константы скорости измеряли ме-
тодом вращающихся секторов* [21, 22]:
к2 = 2-1010 л-моль""1 сек~\ (12.6)
Подставив это значение к2 в уравнение (12.4), полу-
чим константу скорости отрыва атомов водорода от RH
метильными радикалами:
«! = —-------<ИСН4]/щ (2-1010)1/s л-моль-1-сек-1. (12.7)
[RH] (d[C2HeW)/2 ' ’
Полученные этим методом константы скорости мно-
гих реакций отрыва водорода метильными радикалами
Тротман-Дикенсон свел в таблицу (табл. 12.4).
Из табл. 12.4 видно, что константы скорости реакции
зависят исключительно от источника радикалов. Сле-
дует отметить также, что скорости реакций отрыва водо-
рода увеличиваются в ряду алканов по мере разветвле-
ния их цепи, что объясняется низкими скоростями от-
рыва атомов водорода первичных С—Н-связей. Кон-
станты скорости отрыва водорода от метилового спирта
и диметилового эфира радикалами, полученными из
одного и того же источника, имеют близкие значения.
Это означает, что в том и другом соединении происхо-
дит отрыв атомов водорода от метильных групп. Ацет-
альдегид обладает большей активностью, чем метанол
и метиловый эфир, благодаря высокой лабильности аль-
дегидного атома водорода:
СН3- + СН3—СНО -> СН4 + сн3—со-
* Этот метод заключается в измерении скорости образования
этана по отношению к скорости образования метана при условии
непрерывности и чередования освещения. Этот метод подробнее
будет рассмотрен в гл. 15.
152
Таблица 12.4
Константы скорости реакции отрыва атомов водорода метильными
радикалами [23]
к
CIV+RH----► CH.+R-
RH Источник метильных радикалов «,102 л-моль *Х хсе«—1 (182° С)* £а- ккал}моль
Ацетон Ацетон 120 9,7
Ацетон Перекись /пре/п-бутила 100 9,5
Гексан Ацетон 170 8,1
2-Метилпропан Ацетон 220 7,6
2-Метилпропан Азометан 100 6,6
2-Метилпропан Диметилртуть 160 7,4
2,З-Диметилбутан Ацетон 350 6,9
2,3 - Д иметнлбу тан Диметилртуть 250 6,8
Метанол Ацетон 60 8,2
Диметиловый эфир Ацетон 90 9,5
Ацетальдегид Перекись трет-бутила 2200 7,5
Перекись трет-бутила Перекись znpe/n-бутила 60 И,7
Азометан Азометан 200 7,6
Хлористый водород Ацетон 3-105 2,3
* Коистаиты скорости преобразованы из значений, выраженных в
с М9'МОЛЪ
Из табл. 12.4 также видно, что метильные радикалы
атакуют соединения, являющиеся в то же время их
источником, например перекись трет-бутила и азоме-
тан. Обычно перекись mpem-бутила не рассматривают
как особенно активное соединение по отношению к ра-
дикалам, а азосоединения, как полагают, совершенно
не активны к ним. Однако, как показывают константы
скорости реакций отрыва водорода, метильные ради-
калы с заметной скоростью атакуют эти соединения в
газовой фазе.
На различных соединениях изучали влияние заме-
щения водорода дейтерием на скорость реакции отрыва
водорода. Так как теория изотопного эффекта является
сложной, подробно с ней можно ознакомиться в обзоре
статей [24—26]. Кратко принцип теории изотопного эф-
фекта можно изложить так: замещение водорода на
дейтерий приводит к изменению скорости реакции, так
как для дейтерированного соединения, чтобы достигнуть
переходного состояния, требуется более высокая энергия
153
активации, чем для соединения с обычным водородом.
В реакциях типа
R-4-SH [R-- H- -S] -+RH4-S-
Переходное состояние
R-+SD -> [R-.-D - S] ->RD + S-
Переходное состояние
дейтерий присоединяется к радикалу медленней, чем
водород. Этот эффект называется первичным изотопным
эффектом и обычно выражается как отношение кон-
стант скоростей реакций отрыва водорода и дейтерия
Kh/kd- Значения Кн/kd для реакций водородного отрыва
колеблются в пределах от 1 до 8. Значения эти особен-
но высоки в реакциях отрыва водорода метильными
радикалами [14, 27]:
СН3- + RH (D) -> СН3-Н (D) + R-
Это указывает на то, что связь R—Н значительно растя-
нута в переходном состоянии.
Между величиной изотопного эффекта и энергией
активации наблюдается корреляция. Не удивительно,
что реакции, имеющие высокое значение энергии акти-
вации, часто показывают высокую селективность между
изотопными заместителями (см. обсуждение избира-
тельности селективности и активности на стр. 147).
Примером этого является реакция N-бромсукцинимида
с толуолом, этилбензолом и изопропилбензолом. В этом
ряду соединений энергия активации уменьшается, при
этом значения Кн/kd (при 77° С) равны соответственно
4,8; 2,7; 1,8 [26].
Значения, обсуждавшиеся до сих пор, относились к
реакциям метильных радикалов в газовой фазе. Относи-
тельные скорости реакций метильных радикалов с ря-
дом водородных доноров в растворе того же порядка,
что и в газовой фазе. Они могут быть получены следую-
щим способом. При взаимодействии метильных радика-
лов с различными донорами водорода в четыреххлори-
стом углероде протекают две конкурирующие реакции:
CH3.+ RH--*CH4 + R.
СН3- + СС14-СН3С1 + СС13-
(12.8)
(12.9)
154
Скорости этих реакций
Я. = К8[СНа-] [RH];
at
(12.10)
djCHsCl] =К9[СНз.][СС11Ь
' dt
Разделив одну на другую, имеем
d[CH4] _ кя [RH]
d [СН3С1] к, ’ [СС14] '
Это уравнение является примером способа решения-
для отношения констант скоростей, при этом концентра-
ции радикалов сокращаются в процессе преобразова-
ний. Если количество метильных радикалов невелико,
то концентрации RH и ССЦ остаются неизменными в
течение реакции и их можно принять равными перво-
начальным. Кроме того, так как [СН4] и [СН3С1] в на-
чале реакции равны нулю, отношение Д[СН4]/Д[СН3С1]
можно принять равным отношению количества метана
к хлорметану в продуктах реакции. Тогда уравнение
(12.10) приобретает вид
«8 _ [СС14] (СН4)
к, [RH] ’ (СН3С1)
(круглые скобки обозначают выход реакции).
Из этого следует, что относительное значение к&
можно получить как отношение количества метана к
хлорметану, которые образуются в результате реакции,
с поправкой на отношение концентраций исходных со-
единений. В табл. 12.5 представлены значения скорости
Таблица 12.5
Скорости реакции отрыва водорода метильными радикалами
относительно скорости реакции отрыва хлора от четыреххлористого
углерода при 100° С [28]
(12.11)
Субстрат «8/«, Субстрат «8/«,
Бензол Метилбензоат Ацетон Толуол Четыреххлорнстый углерод 0,039 0,062 0,40 0,75 (1,00) Октен-1 Циклогексан Хлороформ Метилацетат 3,2 4,8 И,1 21
155
отрыва водорода метильными радикалами относительно
скорости отрыва хлора от четыреххлористого углерода,
вычисленные на основании уравнения (12.11). Перекись
ацетила являлась источником метильных радикалов.
Значения таблицы показывают, что бензол относительно
инертен к отрыву водорода, ацетон и толуол реагируют
медленней, чем четыреххлористый углерод. Циклогек-
сан обладает необычной активностью. Октен, который
имеет аллильные атомы водорода, реагирует быстрее,
чем насыщенные алканы.
В табл. 12.6 сравниваются относительные константы
скоростей реакций, протекающих в растворе и в газовой
фазе (по данным Тротман-Дикенсона).' Значения, при-
веденные в таблице, показывают подобие относитель-
ных скоростей реакций отрыва водорода метильными
Таблица 12.6
Относительные скорости реакций отрыва водорода метильными
радикалами в газовой фазе и в растворе при 100° С [28]*
К»
CH3-!-RH--► CH,+R-
к»
СН3-+СС14---> СН3С1-Ь-СС13
Субстрат (раствор) *8 (газовая фаза) к8 (газ, ф) «3 (раств.)
Бензол 0,04 0,091 2,3
Ацетон 0,40 1,0 2,5
Толуол 0,75 1,9 2,5
Октен-1 3,2 7,7 2,4
Циклогексан 4,8 3,0 0,6
* Значения, полученные в газовой фазе, являются абсолютными значениями
коистаит скоростей (х 10 3), экстраполированных к 100° С с использованием
значений, приведенных в табл. 12.4 [29].
радикалами в газовой фазе и в растворе. Из этого мож-
но заключить, что применение той или иной среды для
реакций водородного отрыва не влияет на их скорости.
Реакции отрыва водорода алкоксильными радикалами
Алкоксильные радикалы можно получить различ-
ными способами, такими, как разложение алкилнитри-
тов, перекисей алкилов и гипохлорита натрия. Эти ра-
156
ДикаЛы участвуют главным образом в реакциях отрыва
водорода. Например, перекись mpem-бутилметила и ме-
тилнитрит образуют метокси-радикалы, которые отры-
вают водород от различных водородных доноров:
200° С
mpem-C4H9O—ОСН3----------” СН3О--|~mpem-C4H9O-
120° С
СН3О—NO ——- СН3О- + NO
CH3O- + RH -> CH3OH + R-
Если RH представляет собой активный донор, такой,
например, как циклогексан, то в результате реакции с
почти количественным выходом образуется метанол.
Алкоксильные радикалы с разветвленной цепью бо-
лее активны. mpem-Бутокси-радикал, например, может
легко отрывать водород от водородного донора либо
подвергаться p-расщеплению с образованием ацетона:
«12
mpew-C4H9O-4- RH------► mpem-C4H9OH 4-R- (12.12)
«13
z77pe/n-C4H9O •-- CH3 • 4-CH3—CO—CH3 (12.13)
Используя уравнение (12.11), можно получить
«12 _ (//гре/п-С4Н6ОН) 1 Н2 14)
«13 (СН3-СО-СНз) ' [RH] ’ '
Это уравнение дает представление об относительной
реакционной способности ряда водородных доноров в
реакциях с перекисью mpem-бутила как источника
mpem-бутоксильных радикалов. С помощью уравнения
(12.14) и полученного при анализе реакционной смеси
количественного отношения mpem-бутанола и ацетона
можно получить относительную константу скорости к12.
В табл. 12.7 приведены значения относительных ско-
ростей реакций отрыва водорода mpem-бутоксильными
радикалами, источником которых был mpem-бутилгипо-
хлорит. В присутствии двух водородных доноров RH и
SH протекают следующие реакции:
RH 4- mpe/n-C4H9O------- R- 4- mpew-C4H9OH (12.15)
R- 4- mpem-C4H9OCl -> RC1 4- mpem-C4H9O •
SH 4- mpem-C4H9O------► S- -|-mpem-C4H9OH (12.16)
S- 4- wpe/n-C4H9OCl -> SCI 4- mpem-C4H9O-
157
Таблица 12.7
Относительные скорости реакций отрыва водорода
mpe/n-бутоксильными радикалами, полученными из перекиси
/npe/n-бутила или /npezn-бутилгипохлорита
«и
RH+mpem-C4HeO------► R-|-mpem-C4H,OH
RH Относительные значение (по водороду)*
Перекись трет- бутила (135° С) [30] трет -Бутилгипохло- рит (40° С) [31]
Толуол 1,0 1,0
Этилбензол 3,2 3,2
Изопропилбензол 5,1; 6,4 [32] 6,8
.м-Ксилол I,2 1,2
п-Ксилол 1,5 1,5
тр«я-Бутилбензол 0,1 о,з
Циклогексан 2,0 1,5
Дифенилметан 4,2 [32] 4,7
Тетралин 7,6 —.
Трифенилметаи — 9,6
• В ароматических субстратах учитываются только участвующие в реакции атомы водорода беизильиого производного.
Используя уравнение (12.11), можно получить
d[RH] d [SH} _ К1в [RH] «1» ’ [SH] (12.17)
После преобразования и интегрирования оно
тает вид
приобре-
тав)
к.» __ d[RH]/[RH] 1g {[RH]/[RH]0)
«1. d[SH]/[SH] lg{[SH]/[SH]0) ’
и [SH] — концентрации двух водородных до-
времени, a [RH]0 Л [SH]0 —их первоначаль-
где [RH]
норов во
ные концентрации.
Используя газовую хроматографию как метод ана-
лиза реакционной смеси, легко получить молярное
отношение двух водородных доноров RH и SH. Исполь-
зуя аналитические приборы с высокой разрешающей
способностью и уравнение (12.18), можно вывести от-
ношение ряда констант скоростей для реакций отрыва
водорода от различных водородных доноров к констан-
те скорости реакции отрыва водорода от некоторого
стандартного водородного донора SH. В табл. 12.7 при-
158
ведены значения, полученные этим способом при 40° С
с циклогексаном в качестве стандартного водородного
донора и /пре/п-бутилгипохлоритом в качестве источника
радикалов.
В табл. 12.7 приведены также относительные кон-
станты скоростей, полученные с перекисью /пре/п-бутила
в качестве источника радикалов, при этом значения для
двух различных источников /npe/n-бутоксильных ради-
калов хорошо согласуются. Замечено, что обычный по-
рядок реакционной способности атомов водорода С—Н-
связей, а именно: первичная<вторичная<третичная от-
вечает ряду толуол — этилбензол — изопропилбензол и
ряду толуол — дифенилметан — трифенилметан.
Реакции отрыва водорода фенильными радикалами
Используя фенилазотрифенилметан в качестве источ-
ника радикалов, можно получить относительную актив-
ность ряда водородных доноров к фенильному радикалу.
Это азосоединение подвержено быстрому термическому
гомолитическому разложению при 60° С. В смеси до-
нора водородов RH и четыреххлористого углерода это
разложение протекает, как показано ниже:
Ph—N = N—CPh3 -> Ph • 4- N2 + Ph3C •
Ph+RH--------►PhH + R. (12.19)
Ph• + CC14 —- PhCl + • CC1S (12.20)
Используя уравнение (12.11), имеем
K18 _ [CC14] (РЪН) z j 2 2 j \
k20 [RH] ‘ (PhCl) * V ' -
Отношение выходов бензола и хлорбензола можно
легко определить, используя газовую хроматографию
как метод анализа реакционной смеси. С помощью этого
соотношения и уравнения (12.21) можно рассчитать
относительное значение Kig. В табл. 12.8 приведены
значения этих реакций. Из данных, полученных для
толуола, этилбензола, изопропилбензола и /прет-бутил-
бензола, можно вычислить реакционную способность
атомов водорода С—Н-связей в порядке: первичная —
вторичная — третичная.
159
Таблица 12.8
Атомы водорода С—Н-связей боковой цепи ароматических соединений
Соединение Число и тип атомов водорода
а-положеиие 3-положение
Толуол 0,27 |3 первичных Нет
Этилбензол 0,84 2 вторичных 3 первичных
Изопропилбензол 0,93 1 третичный 6 первичных
трет- Бути лбензол 0,11 Нет 9 первичных
Как видно из табл. 12.8, активность каждого типа
атома водорода можно получить из уравнений одного и
того же вида. Активность атома водорода первичной
С—Н-связи в a-положении, полученной для толуола,
равняется 0,27/3=0,09. Подобно этому атом водорода
первичной С—Н-связи p-положения имеет активность,
равную 0,11/9 = 0,01 и полученную из значений для
mpem-бутилбензола. Активность атома водорода вторич-
ной С—Н-связи в a-положении можно вычислить из зна-
чений для этилбензола, основываясь на активности
атома водорода первичной С—Н-связи в р-положении.
Активность атома водорода вторичной С—Н-связи в
a-положении равна 1/2 [0,84—3(0,01)] =0,41.
Подобно этому из значения для изопропилбензола:
активность атома водорода третичной С—Н-связи в
a-положении равна 0,93—6(0,01) =0,87. И, наконец,
активность ряда 1°:2°:3° представляет собой отноше-
ние, равное 0,09 : 0,40 : 0,86 или 1 : 4,6 : 9,7. (Символами
1°, 2° и 3° в дальнейшем мы будем обозначать соответ-
ственно атомы водорода первичной, вторичной и третич-
ной С—Н-связей.)
Аналогично для метил-, этил-, изопропил- и трет-
бутилдисульфида получены относительные активности
атомов водорода при а-атоме углерода относительно
связи S—S, равные 1:3,2: 9,1. Таким образом, атомы
водорода, примыкающие к атому серы, обладают актив-
ностью, подобной активности атомов водорода боковой
цепи в бензильных производных. Значения табл. 12.1
показывают избирательность фенильных радикалов к
алифатическим атомам водорода первичной, вторичной
и третичной С—Н-связей, которая выражается отноше-
нием 1 :9,3 : 44. Эти значения можно объединить со значе-
160
ниями табл. 12.8, в результате чего получается ряд от-
носительных значений К19 (по атому водорода), которые
показаны ниже.
Атомы водорода
алифатической цепи боковой цепи алкильных производных бензола
1° 2°’ 3° 0,12 1,0 4,8 1° 2° 3° 1,0 4,6 9,7
Таким образом, атом водорода первичной С—Н-связи
в боковой цепи бензильного производного (как, напри-
мер, в толуоле) обладает такой же активностью к фе-
нильному радикалу, как и атом водорода вторичной
С—Н-связи в алифатических производных. Атомы водо-
рода первичной С—Н-связи в алифатических соедине-
ниях обладают гораздо меньшей активностью.
Для реакций фенильного радикала с толуолом и
ацетоном была измерена величина кинетического изо-
топного эффекта Kh/«d, которая для Ph—СНз и Ph—CD3
равна 4,5, а для СН3—СО—СН3 и CD3—СО—CD3 равна
4,2. Эти значения показывают, что в переходном состоя-
Таблица 12.9
Значения относительной активности водородных доноров
к фенильным радикалам [11,7]
Донор водорода Донор водорода Кц/Кго
Гексан 0,96 Трифенилметан 3,5
2,3-Днметилбутан 1,19 Хлороформ 3,2
Циклогексан 1,08 Хлорметан 0,07
Циклогексен 4,4 Метиловый эфир 0,28
Толуол 0,27 Ацетон 0,17
п -Фенокснтолуол 0,26 Уксусная кислота 0,09
л-Ннтротолуол 0,22 Метилацетат 0,09
Этилбензол 0,84 Метнлднсульфид [7] 0,57
л-Метилтолуол 0,79 Этил дисульфид [7] 1,5
Изопропилбензол 0,93 Изопропнлдисульфнд [7] 1,9
трет-Бутл бензол 0,11 лгрелг-Бутилдисульфид [7] 0,25
Дифенилметаи 1,4
6 V. Прайер
161
нии преобладает значительное количество разрываю-
щихся связей (см. обсуждение на стр. 153).
О влиянии полярного эффекта заместителя на реак-
ции фенильных радикалов с толуолом и производными
толуола, замещенными по бензольному кольцу, можно
судить по различиям в скорости этих реакций
(табл. 12.9):
Ph- + CH3—'V-X —
PhH + -СН2— X
Электроноакцепторная группа X уменьшает значения
Кх, а электронодонорный заместитель увеличивает ее.
Этот вывод подчиняется уравнению Гаммета с низким
значением р, равным —0,1, показывающим, что фениль-
ный радикал нечувствителен к полярным эффектам за-
местителей.
Относительные скорости реакций
отрыва водорода
различными радикалами
В табл. 12.10 приведены вычисленные описанным
способом относительные активности некоторых радика-
лов к различным донорам водорода. Из таблицы видно,
Таблица 12.10
Значения относительной активности (по водороду)
доноров водорода к различным радикалам
Донор водорода Радикал
Метил (65° С) [33] Фенил (60° С) [11] трет- Бутокси (40° С) [12] Атомы хлора (40° С) [И, 13] Атомы брома (40° С) [1 1, 34]
Циклогексан 1,0 1,5 2,0 0,004
Толуол* (I) (1) (1) (1) (1)
Этилбензол* 4,1 4,6 3,2 2,5 17,2
Изопропилбензол* 12,9 9,7 6,9 5,5 37,0
Учитываются только лабильные атомы водорода боковой цепи.
162
что метильный, фенильный и /npe/n-бутильный радикалы
имеют близкие по значениям активности. Более актив-
ный атом хлора менее избирателен, а менее активный
атом брома значительно более селективен.
Полярный эффект в реакциях отрыва водорода
На радикальные реакции отрыва водорода влияет
полярность заместителя. В табл. 12.11 сравниваются ак-
тивности метильных радикалов и атомов хлора к ато-
мам водорода в пропионовой кислоте. Скорость атаки
Таблица 12.11
Относительная селективность С1 • и СН3 к атомам
водорода в пропионовой кислоте [36]
Относительная селективность сн3—сн2—соон
сн3- 1 7,8
сь 30 1
метильными радикалами атомов водорода при а-атоме
углерода в 7,8 раза выше, чем при р-атоме углерода,
в то время как скорость атаки атомами хлора выше
только В 1/30 раза. Это легко объяснить полярным эф-
фектом, в результате которого карбоксильная группа
кислоты оттягивает на себя электроны. Благодаря своей
относительной электроотрицательности хлор предпочи-
тает атаковать самые удаленные от электроотрицатель-
ной карбоксильной группы атомы водорода. Электро-
положительные метильные радикалы действуют наобо-
рот. Используя терминологию ионных реакций, атом
хлора можно назвать относительно электрофильным ра-
дикалом, а метильный радикал — относительно нуклео-
фильным.
Другой пример полярного эффекта показан в
табл. 12.10, откуда видно, что хлор и трет-бутоксиль-
ный радикал отрывают атом водорода от циклогексана
быстрее, чем от толуола, в то время как для брома на-
блюдается обратный эффект.
Чтобы объяснить это, следует рассмотреть индуктив-
ный электроноакцепторный эффект фенильной группы
6*
163
и резонансную устойчивость бензильного радикала [35].
Значения скоростей отрыва водорода в табл. 12.10, при-
нимая во внимание эти факторы, можно объяснить сле-
дующим образом: атом хлора и mpem-бутоксильный ра-
дикал являются электрофильными радикалами, поэтому
индуктивный эффект электронного смещения фенильны-
ми заместителями действует так, чтобы замедлить их
атаку на соседние атомы водорода. Кроме того, эти
радикалы настолько активны, что в переходном состоя-
нии образуется относительно небольшое количество раз-
рывающихся связей С—Н, а более слабые связи боковой
цепи бензильных производных разрываются не быстрее,
чем обычные алифатические связи С—Н. Менее элек-
троотрицательный и более селективный, чем атом хлора,
атом брома, наоборот, отталкивается в меньшей степени
в результате индуктивного эффекта фенильного замести-
теля. Кроме того, в реакциях атомов брома в переход-
ном состоянии образуется значительное количество сья-
зей С—Н. Это приводит к увеличению добавочной
электронной плотности в боковой цепи бензильного про-
изводного в переходном состоянии и значительной резо-
нансной. стабилизации благодаря фенильной группе.
Обобщая сказанное, можно представить переходное
состояние для реакции электроотрицательного и несе-
лективного атома хлора с толуолом следующим образом
(mpem-бутоксильный радикал должен действовать
также):
6+ 8—
ГЧ . Н___СНл_____Ph г Индуктивный электроноакцепторный эффект фе-
V>1 11 JT 11-«Г- гчлчггтгтч оомьппаат п^тплАипт..
Очень мало разры-
вающихся связей
нильной группы замедляет атаку электрофиль-
ного атома хлора.
В переходном состоянии в реакциях атомов брома
существует гораздо большее количество разрывающихся
связей, чем в тех же реакциях хлора. Его можно пред-
ставить двумя резонансными структурами:
Делокализация
/добавочных
Связь, в значитель -
цой степени склон-
ная к разрыву
164
Как обсуждалось на стр. 93, корреляция скоростей
радикальных реакций в соответствии с уравнением Гам-
мета показывает влияние полярного эффекта в гомоли-
тических реакциях. Реакции атомов брома и хлора с
толуолом, имеющим заместителей в бензольном кольце,
хорошо коррелируются согласно уравнению Гаммета.
Однако в обоих случаях скорости лучше коррелируются
со значениями о+, чем по обычным значениям о. Кон-
станты о+ были введены для ионных реакций, в которых
положительный заряд, способный к резонансному взаи-
модействию с бензольным кольцом, возникает в пере;
ходном состоянии [37].
Несмотря на то что константа о+, как это можно
ожидать, взаимосвязана только со скоростью тех реак-
ций, которые обладают значительным ионным характе-
ром, было обнаружено, что многие гомолитические
реакции, в которых участвуют электрофильные ра-
дикалы, лучше коррелируются с о+, чем с о*. Это на-
блюдается, например, в реакциях отрыва водорода от
АгСН3 бутоксильными радикалами, атомами хлора и
брома. Эти реакции являются наиболее ярким доказа-
тельством полярного эффекта, который может возникать
в гомолитических реакциях.
Взаимодействие атомов хлора с замещенными толуо-
лами показано в реакции [40, 41]
* В настоящее время показано, что реакции некоторых ради-
калов лучше коррелируются с а, чем с а+. Действительно реакции
атомов хлора хорошо коррелируются и с а, и с а+, в то время как
реакции атомов брома коррелируются лишь с а+. Эта область ши-
роко изучается, и критерии, по которым можно провести различия
между реакциями с точки зрения корреляции либо с а, либо с <т+,
до сих пор неизвестны. Однако для наших целей эти различия
несущественны. Две точки зрения, представленные здесь, можно
сформулировать следующим (образом: на реакции атомов хлора
и брома влияют полярные эффекты заместителей, и в переход-
ном состоянии в реакциях атомов брома образуется большое коли-
чество разрывающихся связей. Обсуждение корреляции значений
радикальных реакций в уравнении Гаммета и о различии между
о и о+ см. в работах [38, 39].
165
На рис. 12.2, представляющем собой диаграмму Гам-
мета, показаны значения для реакции (12.22), согласно
уравнению
lg— = ст+ Р,
КО
где «о — скорость реакции с толуолом; к — скорость
реакции с X—С6Н4—СН3; ст+ — константа Х-заместителя.
Рис. 12.2. Относительная активность различных производ-
ных толуола, замещенного по бензольному кольцу, к СЬ
в значениях константы о+:
О — значения Рассела и Вильямсона при 40° С [41]; • ~ значения
Валлинга и Миллера при 70° С [40].
По наклону прямой определяют значение р, равное
—0,66. Отрицательное значение р показывает, что элек-
тронодонорный заместитель увеличивает скорость реак-
ции (12.22), в согласии с предыдущими значениями, по-
казывающими, что атом хлора является электрофиль-
ным радикалом. Переходное состояние в этой реакции
отрыва водорода можно представить в виде двух резо-
нансных форм:
CIS
166
По этим резонансным формам можно предсказать, что
электронодонорная группа X должна стабилизировать
переходное состояние и ускорять реакцию.
Наряду с этим изучались реакции атомов брома с
замещенными толуолами:
Как видно, в переходном состоянии в реакциях атома
брома существует большее количество разрывающихся
связей, чем в тех же ре-
акциях атома хлора. По-
этому в реакциях отрыва
водорода, в которых уча-
ствуют атомы брома, от
различных производных
толуола, замещенного по
бензольному кольцу, за-
местители будут более
эффективно влиять на от-
носительную стабилиза-
цию переходного состоя-
ния (а также на скорость
реакции).
На рис. 12.3 показаны
значения для реакции
(12.23), которые согласу-
ются с уравнением Гам-
мета. Как было предска-
зано, значение р для ре-
акций атома брома при
80° С равно —1,36, что
значительно больше, чем
значение р для реакций а
Рис. 12.3. Относительная актив-
ность производных толуола к Вт-
при 80° С в значениях констан-
ты о+ [42].
хлора (—0,66). Несмот-
ря на то что резонансная структура в реакциях атомов
брома аналогична структуре в реакциях атомов хлора,
значительно большее распределение зарядов и более по-
167
ложительный заряд на бензильном углероде в реакциях
атомов брома приобретает особое значение:
На рис. 12.4 показаны энергии активации и теплоты
реакции атомов хлора и брома с толуолом. Эти диа-
гоаммы показывают гораздо большую экзотермичность
Переходное состояние
PhCH3+ Вг*
10 ккал
PhCH2« + НВг
1L_
Переходное состояние
PhCH,+ CI«
9
25 ккал
PhCH2« + HCI
Рис. 12.4. Сравнительные данные по энергиям активации и экзо-
термичности реакций Вг • и С1- с толуолом.
реакций и низкую энергию активации для реакций ато-
мов хлора по сравнению с реакциями атомов броэда.
Исходя из принципов, которые рассматривались на
стр. 147—148 для реакций атомов хлора, в переходном
состоянии происходит сохранение связей субстрата.
Поэтому можно ожидать, что хлорирование менее се-
лективно, чем бромирование. Низкое значение р, наблю-
даемое при хлорировании производных толуола, нахо-
дится в соответствии с предсказанным.
Это свидетельствует о большой энергии активации и
большей селективности в реакциях атомов брома с про-
изводными толуола, чем в тех же самых реакциях ато-
мов хлора. Это дает возможность предположить, что
при замещении водорода на дейтерий кинетический
изотопный эффект должен быть гораздо большим в
реакциях атомов брома (см. стр. 154). В соответствии
168
с предсказанным Кн/кц=4,6 для реакции PhCH2D
с атомами брома и только 1,3 для реакции PhCH2D
с атомами хлора (обе реакции проводились в четырех-
хлористом углероде при 77° С) (43, 18, 40].
Упражнения
12.1. Вывести уравнение (12.17). Объяснить, каким образом
можно сравнивать в одном эксперименте активности трех или более
водородных доноров, используя метод газовой хроматографии
и уравнения, подобные уравнению (12.17).
12.2. Вывести уравнение (12.21).
12.3. Используя значения табл. 12.9, показать, что отношение
активностей атомов водорода при а-атоме углерода относительно
дисульфидной связи равно 1:3,2: 9,1 для 1° : 2°: 3°. Вычислить от-
носительную активность для 1° атомов водорода при а-атоме угле-
рода относительно дисульфидной связи по сравнению с 1° атомами
водорода при р-атоме углерода.
12.4. Используя значения табл. 12.9, сравнить активность (по
атому водорода) атомов водорода при а-атоме углерода, примыка-
ющих к эфирной связи, к дисульфидной связи, к карбонильной свя-
зи С=О и к сложноэфирной связи.
12.5. Используя рис. 12.4, рассмотреть структуру переходного
состояния по отношению к исходным соединениям для реакций хло-
рирования и бромирования толуола.
В каких реакциях больше всего разрывающихся связей?
Каким должно быть значение р в соответствии с уравнением
Гаммета?
12.6. Предсказать знак и приближенное значение величины р в
уравнении Гаммета для реакций бензоилокси-радикалов (из пере-
киси бензоила) с бензиловым эфиром при 90° С [44].
X
СН2—О—CHjPh
12.7. При реакции PhCH2D с метильными радикалами, получен-
ными из перекиси ацетила, при 125° С, образуются дибензил, метан
и СО2. Значения kh/«d могут быть вычислены, исходя либо из 'того,
что дейтерий гГереходит в структуру метана, либо из того, что он
остается в структуре дибензила. Значения Кн/kd, вычисленные та-
ким образом, равны 9 и 7 соответственно.
Представить механизм, который объясняет это противоречие,
если метильные радикалы, отрывают атом водорода бензольного
ядра с такой же скоростью, как и бензильные атомы водорода.
Если это объяснение правильное, какое из двух приведенных значе-
ний кн/кц верно? [45]
12.8. При обсуждении молекулярно-индуцироваиного гомолиза в
гл. 9 было указано, что образование комплекса атомов галогена
может привести к увеличению скорости диссоциации галогена.
В этой главе также констатируется, что образование комплекса
атомов хлора увеличивает их селективность и, по-видимому, умень-
шает их активность. Какое утверждение противоречиво? Доказать
вашу точку зрения, используя диаграмму энергий реакции.
169
ЛИТЕРАТУРА
1. К е b а г 1 е Р. J. Phys. Chem., 67, 716 (4963).
2. Семенов Н. Н. О некоторых проблемах химической кинетики
и реакционной способности. Изд. 2. М., Изд-во АН СССР, 1958.
3. Glasstone S., Laidler К- J, Е у ring Н. Theory of Rate
Processes. McGraw-Hill Book Company. N. Y, 1941, pp. 88, 91,
110, 222.
4. В u j a k e J. E., Pratt M. W. T, Noyes R. M. J. Amer. Chem.
Soc., 83, 1547 (1961).
5. BensonS. W. J. Chem. Phys., 38, 1945 (1963).
6. P г у о r W. A., Pickering T. L. J. Amer. Chem. Soc., 84,
2705 (1962).
7. Pryor W. A., Guard H. Ibid., 86, 1150 (1964).
8. Tedder J. M. Quart. Rev. (Lond.), 14, 344 (1960).
9. Szwarc M. The Transition State, Chem. Soc. (London), Spec.
Publ, 16, 1962, p. 95.
10. Trotman-Dickenson A. F. Gas Kinetics. Butterworth
Scientific Publications. London, 1955, p. 225.
11. Bridger R. F., Russell G. A. J. Amer. Chem. Soc., 85,
3754 (1963).
12. W a 11 in g C, Th a ler W. Ibid, 83, 3877 (1961).
13. R u s s e 11 G. A. Ibid, 80, 4987, 4997 (1958).
14. Szwarc M. Chem. Soc. (London), Spec. Publ, 16, 1962, p. 94
15. T e d d e r J. M. Quart. Rev, 14, 338 (1960).
16. Trotman-Dickenson A. F. Gas Kinetics. Butterworth Sci-
entific Publications. London, 1954, pp. 179, 191.
17. Hammond G. S. J. Amer. Chem. Soc, 77, 334 (1955).
18. Wiberg К- B, S laugh L. H. Ibid, 80, 3033 (1958).
19. Leffer J. E, Grunwald E. Rates and Equilibria of Organic
Reactions. John Wiley Sons. Inc, N. Y, 1963, pp. 162—168.
20. Trotman-Dickenson A. F. Free Radicals. Methuen Co,
Ltd, London, 1959, p. 63.
21. Gomer R, Kistiakowsky G. B. J. Chem. Phys, 19,
85 (1951).
22. SheppA. Ibid, 24, 939 (1956).
23. Trotman-Dickenson A. F. Gas Kinetics. Butterworth
Scientific Publications. London, 1955.
24. W i b e r g К- B. Chem. Rev, 55, 713 (1955).
25. WestheimerF. H. Ibid, 61, 265 (1961).
26. Wiberg К. B, Mote 11 E. L. Tetrahedron, 19, 2009 (1963).
27. Jackson W. M, McNesby J. R, D a r w e n t B. de B. J. Chem.
Phys, 37, 1610 (1962).
28. E d w a r d s F. G, Mayo F. R. J. Amer. Chem. Soc, 72,
1265 (1950).
29. Trotman-Dickenson A. F. Free Radicals. Methuen Co.
Ltd, London, 1959, p. 70.
30. W i 11 i a m s A. L, Oberright E. A, Brooks J. B. J. Amer.
Chem. Soc, 78, 1190 (1956).
31. Walling C, Jacknow В. B. Ibid. 82, 6108 (1960).
32. Brooks J. H. T. Trans. Faraday Soc, 53, 327 (1957).
33. Meyer J. A, Stannett V, Szwarc M. J. Amer. Chem.
Soc, 83, 25 (1961).
34. Russell G. A, DeBoer C. Ibid, 85, 3136 (1963).
35. R u s s e 11 G. A, Brown H. C. Ibid, 77, 4578 (1955).
170
36. Tedder J. M. Quart. Rev. (bond.), 14, 340 (1960).
37. Brown H. C., Okamoto I. J. Amer. Chem. Soc., 80,
4979 (1958).
38. R u s s e 11 G. A. J. Organ. Chem., 23, 1407 (1958).
39. Howard J. A, Ingold K. U. Can. J. Chem., 41, 1744 (1963).
40. W a 11 i n g C., Miller B. J. Amer. Chem. Soc., 79,4181 (1957).
41. Russell G. A., Williamson R. C. Ibid., 86, 2357 (1964).
42. Pearson R. E., Martin J. C. Ibid., 85, 3142 (1963).
43. R u s s e 11 G. A., Brown H. C. Ibid., 74, 3995 (1952).
44. Huang R. L„ Lee H. H., Ong S. H. J. Chem. Soc., 3336 (1962).
45. Wilen S. H., Eliel E. L. J. Amer. Chem. Soc., 80,3309 (1958).
Дополнительная литература
Lewis E. S., Symons M. C. R. Quart. Rev., 12, 230 (1958).
Mayo F. R., Walling C. Chem. Rev. (Lond.), 46, 191 (1950).
Szwarc M. The Transition State, Chem. Soc. (London), Spec.
Publ., 16, 1962, p. 91—'117.
Tedder J. M. Quart. Rev. (Lond.), 14, 336 (1960).
Trotman-Dickenson A. F. Free Radicals. Methuen Co., Ltd.,
London, 1959, p. 61—91.
Глава 13 '
ГАЛОГЕНИРОВАНИЕ
Мы уже видели различные примеры радикальных
цепных реакций галогенов с органическими веществами:
X- + RH -> R- + HX
R-+ Х2RX J-X-
В настоящей главе эти процессы будут подробно рас-
смотрены. В качестве исходных соединений можно ис-
пользовать кроме галогенов их переносчики. Чаще всего
используют N-бромсукцинимид и гипохлориты, что
ведет к увеличению селективности реакций галогени-
рования.
1 Фторирование
Фторорганические соединения обычно получают кос-
венными методами из-за высокой реакционной способ-
ности фтора. Однако при изучении процессов фториро-
вания можно использовать разбавление фтора азотом
или другими инертными газами. Продукты реакции
представляют собой все возможные полифторпроизвод-
ные, при этом обе стадии в цепи последовательных ре-
171
акций чрезвычайно экзотермичны. Например, в реакции
метана с атомом фтора теплота реакции
ЛЯ,
ккал/моль
F-+CH4 -> HF + CH3-
—32
CH3- + F2 CH3F + F-
—70
Фторирование протекает даже в темноте при —80° С.
Стадия инициирования, показанная ниже, по-види-
мому, невероятна из-за большой величины теплоты эндо-
термической реакции:
F2 -> 2F-
ЛЯ = 37 ккал
Поэтому для этой стадии можно предположить бимоле-
кулярный процесс. Например, окислительно-восстанови-
тельный процесс между фтором и метаном
F2 + CH4 -> HF + F- +СН3-
должен иметь теплоту, равную 5 ккал!моль-.
&H = D (F-F) + D (СН3—Н)—D (H-F) =
= 37+ 102—134^5.
Для реакции фтора с олефинами можно предсказать
низкую экзотермичностЬ. Например, для реакции фтора
с этиленом вычисленная теплота реакции приблизитель-
но равна —2 ккал!моль-.
СН2 = СН2 + F2 -> FCH2-CH2 + F •
ЛЯ = — 2 ккал/моль.
Радикальные процессы, такие, как взаимодействие
между фтором и олефинами или алканами, являются
примерами молекулярно-индуцированного гомолиза (см.
стр. 115). Очевидно, фтор особенно активен к взаимо-
действиям, в результате которых образуются радикалы,
как показано на основании следующих фактов.
1. Небольшие количества фтора могут действовать
как инициаторы в реакциях хлорирования или окисле-
ния олефинов в темноте при температуре около 0°С.
172
2. Смесь С12С = СС1г с фтором образует атомы фтора
даже при —78° С, несмотря на то что процесс гомолиза
протекает крайне медленно при этой температуре [1].
Хлорирование
Радикальное хлорирование широко распространено
и является предметом обширных исследований. Если
смесь углеводородов и хлора нагреть до 200° С или под-
вергнуть облучению светом с длиной волны не более
4875 А, наблюдаются следующие реакции:
А Я, ккал/моль
С12 -> 2С1-
С1+СН4 -> НС1 +сн3-
СН3- + С12 -> СН3С1 +С1-
58
— 1
—23
Теплоту реакции, как показано выше, можно рассчи-
тать, исходя из прочности связей Z)(CH3—Cl) =81;
Z) (СН3—Н) = 102; D(H—С1) = 103 и D (Cl—С1) =58.
Атом хлора гораздо менее активен, чем атом фтора,
и только одна из стадий процесса хлорирования более
экзотермична. Однако ни одна из реакций не обладает
значительной энергией активации, а длина реакционной
цепи достигает значения 106.
Широко изучались скорости реакций хлорирования
обычных алканов в газовой фазе [2, 3]. При взаимодей-
ствии водорода с хлором в результате радикальных
реакций образуется хлористый водород:
сы- Н2 НС1 + Н- (13.1)
Н- + С12 -> НС1 +Cl-
Анализ этой системы реакций приводит к константе
скорости Ki, которая, как было обнаружено, равна
8-1010e~5500/RT л-моль-1 -сек-‘. Для реакции хлора со
смесью водорода и метана можно получить относитель-
ные константы скорости реакций
С1- + Н2 -> НС1 + Н-
С1- + СН4 -> НС1 +сн3-
173
Подобным образом можно получить относительные
константы скорости реакции хлора со смесью метана
с другими органическими соединениями.
Таким же способом можно получить относительные
константы скорости реакций атомов хлора с любым
соединением. Например, при взаимодействии атомов
хлора со смесью двух углеводородов RH и SH отноше-
ние констант скоростей krhMsh этих реакций приобре-
тает вид
krh
C1- + RH —HC1+R-
C1- + SH —HC1 + S-
~~1R~ = *rh [RH] [Cl-]
at
jf- = Ksh [SH] [Cl • ]
at
KRH = d [RH]/[RH] = 1g {[RH]0/[RH]}
ksh d[SH]/[SH] 1g {ISH]0/[SHJ}
Подобно этому можно получить относительные кон-
станты скорости реакций двух соединений и в конце
концов константу скорости реакций хлора с водородом.
Вычисленные этим методом значения приведены в
табл. 13.1. При сравнении реакций атомов хлора с водо-
родом и метаном обнаружено, что реакция с метаном
требует энергии активации почти на 2 ккал/моль
меньше, чем с водородом, но предэкспонента А благо-
приятствует реакции с водородом. Если предэкспо-
нента А предполагает вероятность реакции, то можно
взять меньшее значение А для реакций метана, учиты-
вая, что в переходном состоянии происходит некоторое
изменение в геометрии атома углерода, а это уменьшает
вероятность реакции. В табл. 13.1 также показано, что
атомы водорода у третичных С—Н-связей реаги-
руют быстрее, чем атомы водорода у вторичных и
первичных.
В результате исследований природы продуктов реак-
ций хлорирования органических соединений в 1936 г.
174
Таблица 13.1
Параметры реакции газофазного хлорирования соединений*
С1 • 4- RH —> НС! +R • (к = A&~Ea/RT)
RH Второе вещество, использовавшееся для сравнения 1g А Еа- ккал/моль
н2 — 10,9 5,5
сн4 Н2 10,4 3,9
с2нв сн4 П,1 1,0
СН3 — СН2 — СН3 с2нв 10,3 0,7
(СНз)зСН СгНв 11,4 0,7
(СН3)4С с2нв 11,3 0,9
СН3С1 сн4 10,7 3,4
С2Н5С1 с2нв 10,7 1,5
* Скорости выражены в сек \ а концентрации — в моль/л [5,6].
Хасс и др. сформулировали следующие правила [4]:
1) углеродный скелет не претерпевает изменений в
процессе хлорирования;
2) в результате хлорирования всегда образуется
монохлорид;
3) атомы водорода замещаются в ряду 3°>2°>1°;
4) при увеличении температуры отношение скоро-
стей замещения водородов приближается к 1:1:1;
5) скорости жидкофазного хлорирования выше, чем
скорости парофазного хлорирования при любой задан-
ной температуре;
6) влажность, поверхность и освещение не влияют
на отношение активностей.
В табл. 13.2 приведены некоторые относительные
активности* атомов водорода, полученные при анализе
* Относительная активность атомов водорода при любом поло-
жении их в молекуле определяется как отношение скоростей реак-
ций. Например, хлорирование хлорангидрида масляной кислоты
дает 30% 4-хлор-, 65% 3-хлор- и 5% 2-хлорпроизводных.
175
Таблица 13.2
Селективность реакций хлорирования в газовой фазе* [7, 8, 9]
С—С—С-С
1 3,6 3,6 1
С— С —С —СН2С1
1 3,7 2,1 0,8
с — с — с — ch2f
1 3,7 1,7 0,9
о
II
С — С-С — С — С1
1 3,2 0,25
с — с — с — со2 -- сн3
1 з о
* Селективность приведена по атому водорода и преобразована таким обра-
зом, что для конечных атомов водорода в каждой молекуле приведены единицы
активности.
продуктов хлорирования различных субстратов в газо-
вой фазе. Значения показывают, что электроотрицатель-
ные заместители снижают скорость хлорирования сосед-
них атомов углерода. Например, из сравнения двух пер-
вых строк в табл. 13.2 видно, что атомы хлора как
заместители не изменяют относительной активности 3-го
и 4-го атомов углерода. Однако эти заместители значи-
тельно уменьшают относительную активность 1-го и 2-го
атомов углерода. Это проявление полярного эффекта
заместителя. В предыдущей главе при обсуждении
этого эффекта мы видели, что электроноакцепторные
заместители в толуоле снижают скорость хлорирования.
В рассматриваемых алифатических соединениях нали-
чие хлора как электроноакцепторного заместителя так-
же снижает скорость процесса хлорирования [8].
В дальнейшем мы рассмотрим газофазное хлориро-
вание, а сейчас возвратимся к в такой же степени инте-
ресным реакциям в жидкой фазе. Взаимодействие хлора
с некоторыми олефиновыми растворителями приводит
к молекулярно-индуцированному гомолизу связи С1—С1
(см. обсуждение на стр. 115, 116 и 119). Например,
в темноте и при отсутствии кислорода в циклогексане
при '25° С хлор быстро реагирует с образованием про-
дуктов:
Селективность атомов водорода при 4-положении по отношению
к 3-положению и к 2-положению выражается как 30/3 : 65/2 : 5/2,
или 10 : 32 : 2,5 соответственно. Если активность атомов водорода
при 4-положении принять за 1, отношение преобразуется в
1 : 3,2 : 0,25, как показано в табл. 13.2.
176
Молярное отношение этих трех продуктов в чистом
циклогексене как растворителе равно 2:1:0,6. Если
циклогексен разбавлен растворителем не олефинового
типа или если присутствует кислород, то количество про-
дукта III снижается до нуля. Наиболее вероятным
объяснением этого является то, что продукты I, II и
III образуются благодаря радикальному хлорированию
циклогексена, а I й II — в результате конкурирующих
реакций ионного процесса
Известно, что продукт III является результатом реак-
ций, протекающих по радикальному механизму. По-
этому, по-видимому, в результате взаимодействия цик-
логексена и хлора при 25° С в темноте с высокой ско-
ростью образуются радикалы. Циклогексан, если он
присутствует в реакционной смеси, также хлорируется.
При этом образуются атомы хлора, т. е. смесь циклогек-
сана и циклогексена приводит к образованию наряду
с продуктами I, II и III также и продукта:
С1
Г
IV
177
Так как циклогексан инертен к процессу ионного
хлорирования, это убедительно доказывает, что при
взаимодействии хлора и циклогексена должны образо-
ваться атомы хлора, которые, являясь относительно не-
селективными, отрывают затем атом водорода как от
циклогексана, так и от циклогексена. На стр. 116 об-
суждались некоторые молекулярно-индуцированные
реакции гомолиза, которые имели порядок, больший 1.
Поэтому представляет интерес тот факт, что и в данной
радикалообразующей реакции в циклогексене порядок
также больше 1. Это показано разбавлением циклогек-
сена растворителями неолефинового типа, присутствие
которых уменьшает выход радикальных продуктов и
благоприятствует образованию' преимущественно ион-
ных продуктов I и II. Поэтому радикальный процесс
имеет более высокий кинетический порядок, чем ионный
процесс. Если ионное хлорирование циклогексена яв-
ляется по крайней мере реакцией первого порядка, то
радикальный процесс должен иметь порядок, больший!.
Эти реакции показаны ниже:
Ионная реакция
Радикальная реакция, кото-
рая конкурирует
только при высокой концен-
трации олефина н в отсутст-
вие кислорода
С1
CI
+ 1 II
CI
С1
I. + 11 +
Высокая концентрация
олефина-f некоторое
количество циклогексана
.1+И+Ш-Н I
178
Исследование влияния структуры олефина на моле-
кулярно-индуцированный гомолиз хлора показывает,
что различные олефины неодинаково влияют на этот
процесс. В частности, изобутилен не вызывает радикаль-
ного процесса, хотя другие изомеры бутена способны
его вызвать [10].
Как обсуждалось в предыдущей главе, жидкофаз-
ное хлорирование показывает резко выраженный эф-
фект растворителя, а изомерное распределение продук-
тов хлорирования любого субстрата зависит от ис-
пользуемого растворителя. При хлорировании
жирноароматических углеводородов может образовы-
ваться комплекс с атомом хлора; поэтому можно ожи-
дать, что концентрация субстрата наряду с природой
любого используемого растворителя влияет на распре-
деление изомеров (табл. 13.3). Отмечено, что, как только
Таблица 13.3
Относительные константы скоростей* реакций хлорирования
бутилбензола в жидкой фазе при 40° С [11]
а 3 у 3
Ph — СН2 — СН2 — СН2 — СН3
Концентрация бутилбензола М Растворитель «р К1 «8
6,3 13,9 7,3 9,2 1,0
1 ,0 с6н6 20,0 10 13 1,0
3,0 ссц 8,5 4,7 6,0 1,0
1,0 ссц 6,6 3,8 5,0 1,0
0** СС14 5,9 2,7 4,0 1 ,0
* По атому водорода.
** Экстраполировано к нулевой концентрации субстрата бутилбензола.
к бутилбензолу добавляется четыреххлористый углерод
(растворитель, который не дает комплекса с атомами
хлора), селективность хлорирования уменьшается. Как
и ожидалось, использование бензола в качестве раство-
рителя, приводит к очень высокой селективности. Экстра-
полируя значения, полученные при использовании четы-
реххлористого углерода в качестве растворителя, к бес-
конечному разбавлению была получена активность,
показанная в последней строке табл. 13.3. Отмечено, что
атомы водорода в a-положении являются наиболее ла-
бильными, в то время как атомы водорода, находящиеся
179
в p-положении, имеют пониженную активность. Это
уменьшение активности атомов водорода в р-положении
можно объяснить электроноакцепторным индуктивным
эффектом фенильной группы.
Таблица 13.4
Активности углерод-водородных связей по отношению к атомам
хлора при 40° С (активности жидкофазных реакций были
экстраполированы к бесконечному разбавлению) [11]
RH Растворитель Относитель- ная активность (по атому) водорода
Метан В парах* 0,0044
Этан » - 1,05
Пропан (1°) 1,25
(2°) 4,42
Бутан (Г) 1,47
(2°) 5,15
Изобутан (1°) » 1,29
(3°) » 6,25
2,3-Диметилбутан (1 ) Реагенты 1,0
(3°) 3,9
Циклогексан Циклогексан 2,7
Толуол 1,4
Этилбензол (а) » 3,5
(₽) » 1,8
Изопропилбензол (а) 7,8
(Р) » 2,2
трет-Бутил бензол Четыреххлористый углерод 0,63
Дифенилметан Циклогексан 2,7
Трифеннлметан Нитробензол 10
Хлористый метил В парах 0,066
Хлористый метилен Реагенты 0,011
Хлороформ 0,0051
* Значения реакций в паровой фазе взяты из работ [3, 12].
В табл. 13.4 приведены значения жидкофазных реак-
ций, экстраполированные к бесконечному разбавлению,
и значения газофазных реакций. Относительные кон-
станты скорости реакций по атому водорода даны для
большого числа углеводородов. (Результаты реакций,
протекающих в жидких и газообразных фазах, сведены
в таблице вместе, исходя из целесообразного предполо-
жения, что скорость реакции циклопентана одинакова
в обеих фазах [И].) Значения показывают, что заме-
180
щение алканов либо фенильной, либо алкильной груп-
пой активирует атомы водорода. .Таким образом, ряд
СН4, Ph—СНз, Ph2—СН2 и РЬзСН имеет относительные
активности 1 : 320 : 615 : 2300, а ряд СН4, СН3—СН3,
(СН3)2СН2 и (СНз)зСН имеет относительные актив-
ности 1 :240 : 1000 : 1400.
Эти результаты согласуются с выводами, сделан-
ными в предыдущей главе, а именно в реакциях атомов
хлора в переходном состоянии возникает мало лабиль-
ных связей С—Н и реакция относительно нечувст-
вительна к резонансной стабилизации из-за присутствия
а-фенильных групп.
Бромирование
Атом брома значительно более стабилен, чем атом
хлора. Вторая стадия в цепи последовательных реакций
бромирования является эндотермичной:
\Н,
ккал/моль
СН3- + Вг2 -> СН3—Вг + Вг- —21
Вг • 4-СН4-> СН3 • + НВг 4-15
Бромирование поэтому требует повышенной темпера-
туры для любых, но в большинстве своем активирован-
ных связей С—Н.
В реакциях атомов брома влияние растворителя не-
значительно в отличие от тех же реакций атомов хлора.
Поэтому результаты жидкофазных и газофазных реак-
ций можно свести в табл. 13.5. Как отмечалось ранее,
Таблица 13.5
Относительные активности углерод-водородиых связей к атомам
брома при 40° С [13]
RH Фаза Относи - тельная скорость (по атому водорода) RH Фаза Относи- тельная скорость (по атому водорода)
Метан [14] В парах 0,0007 Этилбензол (а) СС14 1,0-10»
Этан Пропан (2°) То же 1,0 220 ' Изопропилбен- зол (а) Дифенилметан СС14 СС14 2,3-Ю8 0,610е
Изобутан (3°) Толуол (а) » СС14 19-103 64-Ю3 (а) Трнфенилметаи (а) СС14 1,1 10»
181
атомы брома более селективны, чем атомы хлора. Поря-
док относительных скоростей в реакциях бромирования
от метана к трифенилметану увеличивается в 109,
в реакциях хлорирования только в 103 (см. табл. 13.4).
По данным табл. 13.6 можно сравнить селективность
атомов фтора, хлора и брома к атомам водорода четы-
Т а б л и ц а 13.6
Относительная селективность галогенов в реакции
с 1-фторбутаном [15]
Галоген Относительная селективность
ch2f сн2 сн2 сн3
F2 при 20° С <0,3 0,8 1,0 1
С12 при 78° С 0,9 1,7 3,7 1
Вг2 при 146° С 10 9 88 1
рех типов С—Н-связей в 1-фторбутане. Бром наиболее
селективен, хлор занимает промежуточное положение,
а фтор неселективен.
Иодирование
Атом иода очень стабилен, поэтому длина реакцион-
ной цепи в случае иодирования невелика. Ниже приве-
дены теплоты реакций для двух стадий иодирования
метана:
А/7,
ккал/моль
12 + СН3- » CH3I + I.
1-4-сн4 -у hi + сн3-
-18
+33
Реакция отрыва водорода атомами иода является
эндотермической, поэтому атомы иода не реагируют с
алканами при умеренных температурах. Однако атомы
иода взаимодействуют с иодистым алкилом с отрывом
иода. О реакции оптически активного вторичного йоди-
стого бутила см. обсуждение на стр. 143. В табл. 13.7
приведены относительные скорости реакции отрыва
атомов иода от ряда иодистых алкилов атомами иода
и метильными радикалами. В этих реакциях наблю-
дается подобие скоростей. Эти данные легко объяснить
атакой атома иода по иоду йодистого алкила:
I- + RI -> R- + 12
182
Таблица 13.7
Относительные скорости реакции I- и СН3-
с иодистыми алкилами [19, 20]
сна +RI -»CH3I + R-
I- +RI I— I + R-
R в RI Относительные скорости
СН3- I-
сн3 1 1
С2н5 4 1,7
цзо-С3Н7 19 29
изо-С4Нв , 37
иео-С6Нц — 2,4
Если происходит атака по иоду, то- можно ожидать,
что иодистый изопропил будет реагировать быстрее,
чем иодистый метил, так как вторичная связь С—I бо-
лее лабильна, чем первичная. Однако при атаке по
углероду в процессе SH2
<^з СН3
!• + н^С-1 ---- ' I-cCH + I.
СН, СНд
изопропильная группа должна реагировать гораздо мед-
леннее, чем иодистый метил, из-за стерических препят-
ствий [16, 17]. Это заключение согласуется с результа-
тами, обсуждавшимися на стр. 143.
При высоких температурах в газовой фазе иод отры-
вает атомы водорода от углеводородов с образованием
непредельных соединений. Например, при 685° С этан,
реагируя с иодом, образует 72% этилена и 10% ацети-
лена. Такие же результаты получены с пропаном, бута-
ном и пентаном [18].
При взаимодействии гексана с иодом в качестве ос-
новного продукта образуется бензол, а побочного — гек-
сен [21]. Во всех этих случаях иод превращается в иоди-
стый водород:
R—СН2—СН3 + 12 R—СН = СН2 + 2HI
Эти реакции, надо полагать, являются радикаль-
ными процессами, так как они протекают в газовой
фазе при высокой температуре, а также ввиду их подо-
бия с другими процессами радикального галогенирова-
ния [16].
183
Гипохлорирование
При хлорировании часто используют гипохлориты.
mpem-Бутилгипохлорит является легко синтезирующейся
жидкостью, с которой необходимо обращаться осторож-
но, так как повышение температуры выше 20° С в про-
цессе получения приводит к взрыву [22]. Он реагирует
с углеводородами с образованием хлористых алкилов.
Механизм этой цепной реакции следующий:
mpem-C4H9O- + RH mpem-C4H9OH + R-
R-+ mpem-C4HeOCl RC1mpem-C4H9O•
Была изучена активность различных углеводородов
к mpem-бутилгипохлориту, которая подобна активности
этих соединений к .перекиси трет-бутила, что указывает
на образование в результате отрыва водорода в том
и другом случае mpem-бутоксильного радикала (см. об-
суждение алкоксильных радикалов на стр. 156—159).
В результате реакций mpem-бутилгипохлорита с некото-
рыми соединениями образуются продукты, отличные от
продуктов подобных реакций молекулярного хлора. На-
пример, в результате взаимодействия mpem-бутилгипо-
хлорита с циклопропаном образуется хлорциклопропан:
СН2
СН2-СН2
трет-С4Н9ОС1
Свет
сн2
сн2—сн
I
С1
Реакция с молекулярным хлором приводит к обра-
зованию главным образом 1,3-дихлорпропана [23]:
С1- +
сн2
С12
сн2—сн —►
сн/
сн2—сн2
сн2
сн/сн
Cl
I • СЬ
Cl—СН2—СН2—сн2 —•
С12
С1—СН2—СН2—СН2С1
184
Нижняя из двух приведенных выше реакций интересна
в том отношении, что она может служить примером
5н2-реакции по атому углерода
С1. +
С помощью гипохлорита можно довольно легко по-
лучить алкоксильные радикалы, а также подробно изу-
чить их реакции. Например, алкоксильные радикалы
участвуют в последовательных реакциях
НОС! Свет
ROH------- ROC1-----> RO- + C1-
Наиболее подробно изучены реакции гипохлоритов тре-
тичных спиртов, которые приводят к 0-расщеплению и
отрыву водорода:
сн3 сн3
I Кпр I
R—С—О- + SH------*R-C—OH + S-
I \кл I
сн3 \ сн3
4R-+ CH3COCH3
В несимметричных алкокси-радикалах может отщеп-
ляться более чем одна алкильная группа. Обычно про-
дукты, образующиеся с большим выходом, являются
продуктами реакций отщепления главным образом ста-
бильных радикалов (некоторые подобные значения см.
на стр. 18). Было обнаружено, что от алкоксильных
радикалов алкильные группы отщепляются в виде сво-
бодных радикалов в ряду тре/п-бутил>изопропил>
> бензил ~ этил > хлорметил > метил [24].
При изучении реакций подобного типа наблюдается
неожиданная аномалия: олефины, используемые в каче-
стве растворителей, ведут к очень резко выраженному
увеличению значений кд относительно Кпр. Таким обра-
зом, в отсутствие олефинов гипохлорит, показанный
185
ниже, реагирует с отрывом атома водорода от цикло-
гексана и отщеплением бензильного радикала:
сн3 сн3
I Инициирование |
Ph—СН2—С—ОС1------------- Ph—СН2—С—О> +С1-
I I
сн3 сн3
сн3
I
Ph—СН2—С—ОН + CeHu-
СН3 с6н12/ I
i сн3
Ph—СН2—С—О • ' I
СН3 кд\
Ph—СН2- + СН3СОСН3
В первом случае продуктом реакции является хлори-
стый циклогексил, во втором — хлористый бензил:
СН3 СН3
I I
C6Hir + Ph—СН2—С—ОС1 -> CeHuCl +Ph—СН2—С—О-
i I
сн3 сн3
сн3
PhCH2- + Ph—СН2-С—ОС1 ->
I
сн3
сн3
-> Ph—СН2С1 + Ph—СН2—С—О-
СН3
Если в качестве растворителя использовать чистый
циклогексан, то кд//Спр = 2. Эта величина указывает на
то, что расщепление алкокси-радикала происходит в
2 раза быстрее, чем он отрывает водород от циклогек-
сана. В циклогексене происходит только реакция раз-
ложения с образованием только хлористого бензила и
186
ацетона. То, что спирт I не образуется, кажется удиви-
тельным, так как циклогексен, имеющий аллильные
атомы водорода, как можно было ожидать, является
более активным водородным донором, чем циклогексан.
Еще более неожиданно, что добавлением к циклогек-
сану уже 4% циклогексена приводит к сложным изме-
нениям в реакции, в результате которой образуются
только продукты разложения, а циклогексен и цикло-
гексан могут быть регенерированы. Это является одним
из примеров огромного влияния непредельных соедине-
ний на скорости некоторых радикальных реакций [25]
(см., например, стр. 115—121).
Бромирование N-бромсукцинимидом
N-Бромсукцинимид бромирует органические со-
единения. Эти реакции инициируются перекисью бензои-
ла или другими типичными источниками радикалов, а
также светом. Эти реакции стали широко исследовать
после того, как в 1944 г. обнаружили их радикальный
характер [26, 27].
Чрезвычайная селективность — характерное свойство
N-бромсукцинимида. Аллильное и бензильное бромиро-
вание протекает с превосходным выходом, например:
N-Бромсукциннмид
Эфир, 25° С
Вг
50—80%
СН2 = СН—СН3 -> СН2 = СН—СН2Вг
29%
PhCH = СН — СН3 -> PhCH = СН— СН2Вг
75%
PhCH3 -> PhCH2Br
На первый взгляд кажется, что эти реакции проте-
кают с образованием радикала сукцинимида как пере-
носчика галогена почти так же, как mpem-бутоксильный
радикал, который участвует в реакции mpem-бутилгипо-
187
хлорита. Однако исследования показали, что это не так.
Были исследованы реакции бензильного бромирования
замещенных толуолов. Полученные данные показывают,
что, возможно, все реакции бромирования с помощью
N-бромсукцинимида протекают по механизму:
Br-|-RH -> HBr + R-
R • + Br2 -> R—Br + Br •
/C0\ ZC0\
HBr + | N—Br _> Br2 + | N—H
XCOZ ‘ XCOZ
В данном случае низкие концентрации брома дейст-
вуют как активные агенты бромирования; N-бромсукци-
нимид действует просто как источник брома. Этот меха-
низм впервые предложил в 1953 г. Гольдфингер, чтобы
объяснить хлорирование N-хлорсукцинимидом. Приме-
нение этого механизма к бромированию с помощью
N-бромсукцинимида теперь широко подтверждено
[28—32]. С этой точки зрения аллильное бромирование
молекулярным бромом протекает предпочтительно с
присоединением по двойной связи. Эти конкурирующие
реакции с циклогексеном показаны ниже:
/ \ Отрыв / \
Вг-+| ||------------| 11+НВг
/J,1 водорода \ /
| Вг,
Вг
Ml+Br-
Вг- +
Присоединение
188
Так как реакция присоединения атомов брома по
двойной связи обратима, низкая концентрация брома
способствует стадии отрыва водорода. Это подтверж-
дается бромированием в результате медленного фото-
лиза молекулярного брома, который также способст-
вует аллильному бромированию [28]. Стадия реакции
отрыва водорода, как было показано, также обратима:
Br-4-RH R- + HBr
Однако в результате реакции N-бромсукцинимида с
бромистым водородом поддерживается низкая концент-
рация брома и минимальная скорость обратной реак-
ции. Таким образом, N-бромсукцинимид является соеди-
нением, которое поддерживает низкую концентрацию
брома в результате взаимодействия его с бромистым
водородом.
Как было обнаружено, в конкурирующих реакциях
бромирования замещенных толуолов молекулярный
бром и N-бромсукцинимид показывают подобные отно-
сительные активности*. Молекулярный бром и N-бром-
сукцинимид реагируют с замещенными толуолами при
скоростях, которые подчиняются уравнению Гаммета,
используя о+ вместо обычного значения о (см. стр. 92,
Таблица 13.8
Относительные активности углеводородов к трем агентам
галогенирования [31]
У глеводород Хлор (40° С) трет-Бутил- гипохлорит (40° С) N - Бромсукцнни- мнд (80° С)
Циклогексен 36 129
Толуол (1) (1) (1)
Этилбензол 3,2 24
Изопропилбензол 4,0 6,8 50
Циклогексан 2,3 1,5 0,003
2,3-Диметилбутаи 3,4 4,2 0,1
165), а р для обеих реакций равно —1,39 [29]. Совпа-
дение значений р указывает на возможность участия
молекулярного брома в обеих реакциях в качестве ак-
тивного начала.
*М-бромтетрафторсукцинимид, N-бромтетраметилсукцинимид,
N-бромсукцинимид и бром имеют подобные активности [29], а
N-бромацетамид и различные N-бромгидантоины обладают еще
большим подобием активностей [33].
189
Из табл. 13.8 видно, что N-бромсукцинимид более
селективен, чем хлор или третичный бутилгипохлорит.
Отношение скоростей реакций циклогексена и циклогек-
сана с N-бромсукцинимидом равно 4-Ю4 и только 24
для реакции с третичным бутилгипохлоритом. Эти зна-
чения указывают на предпочтительность N-бромсукци-
нимида при селективном галогенировании сложных
молекул.
Упражнения
13.1. Используя энергию диссоциации, данную на стр. 172, вы-
числить теплоту реакции:
F+CH4—HF + CH8.
13.2. Вывести уравнение для отношения скоростей реакции ато-
мов хлора с молекулярным водородом и метаном. Уравнение не
должно включать в себя концентрации радикалов.
13.3. Дать объяснение увеличению энергии активации в реак-
циях атомов хлора с RH в ряду СН4>СН3С1>С2Н6С1>С2Н6.
13.4. Объяснить, почему отношение константы скорости реакции
галогенирования трифенилметана к константе скорости реакции га-
логенирования метана равно 109 при бромировании и только 103 при
хлорировании. Каким должно быть это отношение при фториро-
вании?
13.5. Представить относительные выходы кетонов, которые
должны образоваться при разложении следующих гипохлоритов
(см., например, на стр. 15) [34]:
13.6. Как обсуждалось на стр. 186, при разложении
РЬСНзСДСНзДОО в циклогексане образуются хлористый бензил и
хлористый циклогексил, а в циклогексене — только хлористый бен-
зил. Каков механизм реакции в циклогексане? Является ли цикло-
гексен более активным донором водорода, чем циклогексан? Ссы-
лайтесь на полученные значения, чтобы подтвердить ваш вывод.
Объяснить результаты реакций в циклогексене.
13.7. В верхнем горизонтальном ряду табл. 13.3 приведены от-
носительные константы скорости реакции хлорирования четырех
алифатических углеродных атомов бутилбензола в отсутствие раст-
ворителя; в нижней строке представлены эти константы скорости,
экстраполированные к нулевой концентрации бутилбензола в ССК.
Объяснить различия этих значений.
190
13.8. Электроноакцепторные группы в равной степени влияют
на реакции хлорирования и бромирования. Хлорирование протекает
с более низкой скоростью в а-, чем в p-положение, при бромирова-
нии происходит обратное. Обсудить значение газофазного хлориро-
вания и бромирования [8].
Хлорирование (75° C) Бромирование (160° C)
c—c—c—c 1 3,6 3,6 1 c 1 — c-c — 80 80 c 1
c ~ c - c — ch2f 1 3,7 1,7 0,9 c 1 — c-c- 90 7 ch2f 9
C — C — C — CHaCl 1 3,7 2,1 0,8 c 1 _ c — c — 80 32 CH2C1 34
С — С — С — C — CF3 1 4,3 1,2 0,04 c 1 — c — c — 90 7 c - CF3 1
C — C — C — C — COCI 1 3,9 2,1 0,2 c 1 — c — c- 77 32 c — coci 28
С — С — С — C — CO2Me 1 3,6 2,4 0,4 c 1 — c — c — 77 35 C — CO2Me 41
(Числа обозначают относительные активности по атому водорода.)
Дать объяснение наблюдаемым тенденциям.
13.9. Для каждой приведенной ниже реакции вычислить тепло-
ту при X2=F2; С12; Вг2; 12
С = С-[-Х2 —> X—С—С—(“X
2С =С + Х2 2Х—С—С—
RH + X2-»R- + НХ + Х-
Используя вычисленные теплоты, объяснить вероятность каждой ре-
акции как возможного источника радикалов при 0° С для каждого
из четырех галогенов. Объяснить также кинетику реакций, исполь-
зуя ваши вычисления и различия в свойствах галогенов [35, 36].
ЛИТЕРАТУРА
1. Miller W. Т., Koch S. D„ М с L a f f е г t у F. W. J. Amer.
Chem. Soc., 78, 4992 (1956).
2. Trotman-Dickenson A. F. Gas Kinetics. Butterworth
Scientific Publications. London, 1955, pp. 181—188.
3. Pritchard H. O., Pyke J. B., Trotman-Dicken-
son A. F. J. Amer. Chem. Soc., 77, 2629 (1955).
191
4. Walling C. Free Radicals in Solution. Jonn Wiley and Sons.,
Inc, N.Y, 1957, p. 357.
5. Trotman-Dickenson A. F. Gas Kineties Butterworth
Scientific Publication, London, 1955, p. 188.
6. Trotman-Dickenson A. F. Free Radicals. Methuen Co.;
Ltd., London, 1959, p. 65.
7. T e d d e r J. M. Quart. Rev., 14, 342 (1960).
8. S i n g h H., T e d d e r J. M. J. Chem. Soc., 4737 (1964).
9. W a 11 i n g C. Free Radicals in Solution. John Wiley and Sons,
Inc., N.Y, 1957, p. 364.
10. Pouts ma M. L. J. Amer. Chem. Soc., 87, 2161, 2172 (1965).
11. Russell G. A., I to A, H e n d г у D. G. Ibid., 85, 2976 (1963).
12. Knox J. H„ Nelson R. L. Trans. Faraday Soc., 55, 937 (1959).
13. R u s s e 11 G. A., DeBoer C. J. Amer. Chem. Soc., 85,
3136 (1963).
14. Fettis G. C., Knox J. H., T г о t m a n - D i c ken-
s о n A. F. J. Chem. Soc., 4177 (1960).
15. Tedder J. M. Quart. Rev. (Lond), 14, 343 (1960).
16. BensonS. W. J. Chem. Phys, 38, 1945 (1963).
17. Pryor W. A, P i с к e r i n g T. L. J. Amer. Chem. Soc, 84,
2705 (1962).
18. Raley J. H, Mullineaux R. D, Bittner C. W. Ibid,
85, 3174 (1963).
19. Buj ake J. E, Pratt M. W. T, N о у e s R. M. Ibid, 83,
1547 (1961).
20. E v a n s F. W, F о x R. J, Szwarc M. Ibid, 82, 6414 (1960).
21. Mullineaux R. D, Raley J. H. Ibid, 85, 3178 '(1963).
22. Bradshaw С. P. C, Nechvata] A. Proc. Chem. Soc,
213 (1963).
23. W a 11 i n g C, Fredericks P. S. J. Amer. Chem. Soc, 84,
3326 (1962).
24. W a 11 i n g С, P a d w a A. Ibid, 85, 1593 (1963).
25. Walling C, Wagner P. J. Ibid, 86, 3368 (1964).
26. D j e r a s s i C. Chem. Rev, 43, 271 (1948).
27. W a 11 i n g C. Free Radikals in Solution. John Wiley and Sons,
Inc, New York, 1957, pp. 381—386.
28. McGrath В. P, Tedder J. M. Proc. Chem. Soc, 1511 (1961).
29. P e a r s о n R. E, Martin J. C. J. Amer. Chem. Soc, 85,
354, 3142 (1963).-
30. R a s s e 11 G. A, DeBoer C, Desmond К- M. Ibid, 85,
365 (1963).
31. Walling C, Rieger A. L, Tanner D. D. Ibid, 85, 3129
(1963).
32. Rassell G. A, Desmond К. M. Ibid, 85, 3139 (1963).
33. Wai ling C, Rieger A. L. Ibid, 85, 3139 (1963).
34. Greene F. D. et al. J. Organ. Chem, 28, 55 (1963).
35. M i 11 e r W. T, Dittman A. L. J. Amer. Chem. Soc, 78,
2793 (1956).
36. P о u t s m a M. L. Ibid, 87, 2161 1(1965).
Дополнительная литература
Bratolybov A. S. Russ. Chem. Rev. (English Transl.), 30,
602 (1961).
Chiltz G, Goldfinger P, Huybrechts G, Martens G,
192
VerbekeG. Chem. Rev., 63, 355 (1963).
Tedder J. M. Quart. Rev. (Lond.), 14, 336 (1960).
Walling C. Free Radicals in Solution, John Wiley and Sons, Inc.,
N. Y., 1957, chap. 8.
Глава 14
РЕАКЦИИ ПРИСОЕДИНЕНИЯ
Ряд веществ легко присоединяется в форме радика-
лов по углерод-углеродной двойной связи в цепных ра-
дикальных процессах:
АВ+ С = С
Инициатор
или свет
1 I
-е-C-
A. В
Механизм этих процессов представлен в реакциях
(14-1)
в - +^с = cZ -> В—С—С-
(14.2)
(14.3)
Радикал В«, полученный любым доступным методом,
может затем присоединяться к олефину по реакции
7 V. Прайер
193
(14.1). Ниже показаны возможные реакции, протекаю-
щие с продуктом реакции (14.1).
1. Так как реакция (14.1) обратима, то при более
высокой скорости обратной реакции образуются исход-
ные соединения, но такое присоединение и отщепление
радикала В* может приводить к изменению их струк-
туры. Например, quc-олефины могут изомеризоваться в
транс-соединения. В результате присоединения ра-
дикала В*
R R R R
в-+ Хс = с/ ->в—с—с-/
н7 Хн ’ Н \l
образуется радикал, который претерпевает быстрое об-
ращение вокруг простой связи С—С
R R R Н
. в—с—сХ ч* в—с—с7
н Хн Н \
Обе конформации радикала затем отщепляют В« с.об-
разованием цис- и транс-олефинов
R R R R
В—С—С- -> С = С +в-
н Хн нХ цис Хн
R H R Н
в—с—с7 -> Хс = с/ +В-
Н R Н транс R
Примером этого процесса может служить изомеризация
г{ыс-стильбена в транс-форму при взаимодействии с
бромом или индуцировании светом [1].
2. Если АВ является хорошим переносчиком, то кон-
станта скорости реакции переноса (14.2) будет иметь
большое значение. В этом случае происходит образова-
194
ние главным образом аддукта «голова к хвосту»
\ / *пр I I
В-+ С = С--------—С—С— (14.1)
/ \ I •
В
I I «пер ! I
—С—С—+ АВ------► —С—С— 4- В- (14.2)
В В А
Как показано выше, это цепной процесс. Если АВ яв-
ляется хорошим переносчиком, длина цепи может до-
стигать большой величины, т. е. большое количество
молей аддукта может образовываться на каждый моль
В-, выделившийся в первоначальной стадии. Чтобы по-,
лучить высокие выходы аддуктов, требуется достаточ-
но высокая концентрация АВ, и тогда реакция (14.2)
будет протекать быстро. Однако если АВ является
хорошим переносчиком, кпер и кпр будут иметь высокие
значения и хороший выход будет обеспечен даже при
умеренном отношении АВ/олефин. Примером этого про-
цесса служит реакция присоединения бромистого водо-
рода к бутену-1 с 95—100%-ным выходом, иницииро-
ванная перекисью или светом.
3._Есл'и АВ обладает свойством лишь среднего по
активности переносчика и происходит радикальная по-
лимеризация олефина, то реакции (14.3) и (14.4) будут
иметь более высокие константы скорости, чем реак-
ция (14.2). В этом случае в результате полимеризации
образуется полимер с низким молекулярным весом:
В-4^С= —С-С— (14.1)
/ Х В
(14.4)
(14.5)
195
Низкомолекулярные полимеры, такие, как II, назы-
ваются теломерами. Отменено, что полимер II имеет па-
ру агентов теломеризации АВ на каждом конце цепи.
Эти теломеры, которые часто образуются в результате
реакций, являются нежелательными побочными продук-
тами, но в некоторых случаях они находят применение
как полезные синтетические продукты. Примером может
служить реакция [2]
Азоизобутиронитрил
СН2 - СН2 + СС14 -----;----->
z ' 4 9 0° с
-> С13С—(СН2—СН2)„—С1
При 20-кратном молярном избытке этилена образует-
ся продукт, из которого дробным фракционированием
можно выделить следующие теломеры:,
9%; л = 2 44%; лСр=19
12%; п= 3
24%; л = 4—7 11%; лср=40.
Некоторые из этих фракций находят промышленное при-
менение. Например, при гидролизе средней фракции по-
лучают карбоновые кислоты, используемые в мылова-
ренной промышленности.
4. Если АВ обладает слабым по активности свойст-
вом переносчика, то при избытке олефина М (т. е. моно-
мера), протекают реакции
В-+М—Р->В- М- (14.1)
кп
В-М„ • + м —- В-М„+1 • (14.4)
В—М„- + АВ —L В—М„—А + В- (14.5)
В этом случае константа скорости реакции присоедине-
ния (14.4) равна кп, т. е. произведению констант скоро-
стей реакций полимеризации мономера. Константа пере-
носа
кпер
При значении С, равном ~0,1 или меньше, и при избыт-
ке АВ происходит полимеризация М. В этом случае АВ
используется главным образом в качестве растворителя.
Величина л в А — Мп — В, называемая степенью по-
лимеризации, может принимать значения от 103 до зна-
196
чительно больших. В этих случаях Одна молекула АВ
присоединяется к каждой полимерной молекуле.
Примером этого процесса является полимеризация
стирола в четыреххлористом углероде как растворите-
ле (3].
Константа реакции переноса четыреххлористого угле-
рода к стиролу равна 0,01. В результате полимеризации
стирола в эквимолярной смеси с четыреххлористым уг-
леродом образуется полимер со средней степенью поли-
меризации 70 (как было определено из содержания хло-
ра в полимере) ,
ссц
Ph-CH - СН2 СС13—(СН2—СН)70—С1
При более высоком значении молярного отношения сти-
рол/CCU степень полимеризации достигает 10 000.
Итак, мы видели, что активные переносчики (напри-
мер, бромистый водород, бром и меркаптаны) могут при-
соединяться к олефинам с превосходным выходом ад-
дукта «голова к хвосту». Менее активные переносчики
(например, четыреххлористый углерод) могут присоеди-
няться с образованием теломеров. Использование менее
активных переносчиков или избытка олефина, который
участвует в полимеризации, ведет к образованию поли-
мерной молекулы, включающей в себя молекулу пере-
носчика.
В этой главе мы рассмотрели реакции присоединения
с образованием «голова к хвосту» аддуктов и теломе-
ров. Реакции переноса полимерных радикалов будут об-
суждены в гл. 15.
Реакции присоединения галогеноводородов
и галогенов
Бромистый водород является черзвычайно активным
водородным донором, который присоединяется к олефи-
нам и дает превосходные выходы «голова к хвосту»
аддуктов. Реакция присоединения бромистого водорода
к олефинам представляет \ интерес как одна из первых
открытых реакций, протекающих либо по ионному, либо
по радикальному механизму [4].
В течение 1920 г. появились работы, посвященные
изучению реакции присоединения бромистого водорода
197
к аллилбромиду. Одни авторы сообщали, что основным
продуктом реакции является 1,2-дибромпропан, другие—
1,3-изомер. Образование 1,2-дибромзамещенного может
быть предсказано на основании правила Марковникова,
1,3-дибромпроизводное рассматривалось как продукт
аномальной реакции. Эту реакцию детально изучил
Караш с сотрудниками, а в 1937 г. Караш в Америке и
Хей и Уотерс в Англии независимо друг от друга пред-
ложили цепной радикальный механизм реакции присо-
единения бромистого водорода к олефинам, протекаю-
щий не. по правилу Марковникова [5, 6, 7].
Тщательное изучение Карашем этих реакций пока-
зало, что в темноте и в отсутствие каких-либо инициа-
торов возникновения радикалов бромистый водород мед-
ленно присоединяется к аллилбромиду по ионному меха-
низму с образованием ожидаемого по правилу Марков-
никова 1,2-дибромпропана. В присутстрии воздуха, при
индуцировании светом или инициировании перекисями
происходит образование 1,3-дибромида в результате бы-
строго радикального присоединения
СН3—СНВг—СН2Вг
Темнота/’ Ионпый продукт
/ Медленно
СН2 = СН—СНгВг^
к Свет, воздух или
- перекись
Быстро
ВгСН2—СН2—СН2Вг
Радикальный продукт
Позже Караш с сотрудниками показали, что заме-
щенные олефины в реакциях радикального присоедине-
ния обычно образуют продукты не по правилу Марков-
никова. Этот процесс радикального присоединения был
оригинально назван анормальным присоединением, или
перекисным эффектом, или (еще раньше) присоединени-
ем КаРаша- Все эти названия соответствуют истине, од-
нако, используя их, не просто отличить радикальные
процессы присоединения от ионных.
Стабильность радикалов й ионов карбония увеличи-
вается в порядке 1°<2°<3°. Из схематических уравне-
ний, показанных ниже, становится ясно, что увеличение
стабильности радикалов влияет на образование различ-
198
ных продуктов по двум механизмам реакций присоедине-
ния. В случае ионного процесса первым присоединяется
протон и образует более стабильный вторичный ион кар-
бония. В радикальном процессе первым присоединяется
атом брома с образованием более стабильного вторично-
го радикала.
Ионное присоединение
RCH = СН2 + Н+ -> RCH—СН3 — - R—СН—СН3
Вг
Радикальное присоединение,
НВг
RCH = СН2 + Вг- -> RCH— СН2Вг------>
-> R—СН2—СН2Вг + Вг-
При изучении стереохимии реакций радикального
присоединения обнаружено, что происходит преимущест-
венно транс-присоединение. Например, при температуре
от —60 до —78° С бромистый дейтерий егереоспецифич-
но присоединяется к двум изомерам бутена-2; цис-бу-
тен-2 дает трео-З-дейтеро-2-бромбутан, а транс-олефин
образует эратро-продукт [8]
СН3. ЛЗН3
н^с“с^н
РВг
транс-Присо-
единенне
цис
СНзч.^—
Н< ^СН3
РВг
транс-Прнсо-
единение
транс
Реакция присоединения бромистого водорода к
2-бромбутену приводит к образованию либо стереоспе-
цифичного, либо нестереоспецифичного продукта [9]. При
низкой температуре и большом избытке бромистого во-
дорода образуются стереоспецифичные продукты:
199
ch.sJ
снг-г ^сн3
Вг<
Избыток НВг
-806С
/я/»а«с-Прнсоединение
ЦИС
сн3^г
СНг'-С—С'''Н
Вг*' ^сн
транс
мезо
Избыток НВг
-80°С ”
3 траяс-Прнсоединение
сн3^г
dl
н сн3
При более высоких температурах и низких молярных
отношениях бромистого водорода к олефину вероятность
стереохимического преимущественного /пранс-присоеди-
нения уменьшается, пока при 25° С не образуется та же
самая смесь продуктов из любого стереоизомера олефи-
на. Это дает возможность предположить, что механизм
реакций при достаточно низкой концентрации бромисто-
го водорода включает две различные конформации про-
межуточных радикалов, которые уравновешиваются
прежде, чем происходит реакция переноса:
Вг-< „ „ ..СН
сн<
цис
транс
Вг»
СН3 Н
Быстрое достижение
равновесия при 25°С
|НВГ
Смесь мезо -и <П-2,3-дибромбутана
из любого стереоизомера
При высоких концентрациях бромистого водорода
оба промежуточных радикала улавливаются, в результа-
200
те чего из двух стереоизомеров олефина образуются раз-
личные стереосоединения:
СН^Г-.Г--'СН3
ВгГС~С>Н
Вг»
Вг<
сн3 н
с—-сГ
I
Вг
Медленное взаимо-
превращение
изомеров при 80°С
НВт
атакует транс-
-положение
НВг
Быстро
атакует транс-
-положение
Быстро
транс-Присоединение объясняется тем, что промежуточ-
ные радикалы отрывают водород от бромистого водоро-
да с противоположной к брому стороны.,
Можно привести взаимоисключащие объяснения этих
фактов, используя структуру циклических (соединенных
мостиком) радикалов. По этой гипотезе атомы брома
присоединяются к олефину через мостиковую структуру:
' ” ' Вгч 'СН,
СН/ >н
СНзХг_гл'СН3
Вг**'
цис транс
CH3v. ,,СН,
'с.—et
Вг< \ • / >Н
Вг
1
В1Х ,-сн,
СН/
Вг
II
201
Если постулировать, что эти циклические радикалы ата-
куются переносчиком в транс-положение по отношению
к бромному мостику, то можно объяснить стереоспеци-
фичность транс-присоединения. Например, радикал I,
образующийся в результате реакции цнс-олефина, дол-
жен давать .мезо-продукт:
Можно утверждать, что при высоких температурах и
низких концентрациях бромистого водорода возникаю-
щие циклические радикалы I и II взаимопревращаются
по механизму, в который входят радикалы с открытой
цепью [10—15].
Стерические факторы ведут к преобладанию г^нс-при-
соединения бромистого водорода к олефинам. Присоеди-
нение к бициклическому олефину, показанному ниже,
происходит в основном с энзо-стороны, наименее препят-
ствующей реакции [16]:
Вг» а«зо-Лтака
В1
н
202
НВг
зндо-
-Перенос
транс экзо-цис
Продуктом реакции является главным образом транс-
дибромид. Это указывает на то, что атака атомов брома
и бромистого водорода происходит преимущественно с
экзо-стороны.
Как мы видим, радикальное присоединение бромисто-
го водорода к большинству олефинов протекает с боль-
шой скоростью. В противоположность этому, радикаль-
ное присоединение хлористого водорода может быть до-
стигнуто только в специальных условиях, а иодистый
водород вообще не присоединяется к олефинам в ра-
дикальных процессах. В табл. 14.1 приведены теплоты
Таблица 14.1
Теплоты реакций присоединения галогеноводородов к этилену [17]
X-4-СН2 = СН2^Х—СН2 —СН2- ' (14.1)
X —СН2 —СН2- +НХ^Х —СН2 —СН34-Х- (14.2)
нх Д/f, ккал!моль
Реакция присое- динения (14.1) Реакция пере- носа (14.2)
НВг —5 —11
НС1 —26 5
HI 7 —27
реакций присоединения галогеноводородов к этилену.
Несмотря на то что иодистый водород является превос-
ходным переносчиком водорода и снабжает им радикал
с выделением тепла, стабильный атом иода не присоеди-
няется к олефинам с выделением энергии.
203
Поэтому для йодистого водорода реакция присоедине-
ния затруднена.
В случае хлористого водорода ситуация обратная:
атом хлора очень активен и присоединяется к олефинам
с освобождением энергии, а реакция переноса водорода
не экзотермична. Таким образом, можно предсказать
присоединение только бромистого водорода к олефинам
в реакциях с длинной цепью. Создав благоприятные ус-
ловия для реакций радикального присоединения хлори-
стого водорода, можно ее ускорить. Однако в результате
обратимости реакций могут произойти радикальные вза-
имопревращения, которые в процессе радикального при-
соединения хлористого водорода могут привести к про-
дуктам, образующимся по правилу Марковникова. На-
пример, радикальное присоединение хлористого водоро-
да к пропилену может протекать по следующим стади-
ям [18]:
С1 Быстро
С—с = с —С—С—С с—с—с
[Медленно I
НС1 |
СН2—СН2—СН8
С1
С1 11
Быстро |
на 4
сн3—сн-сн3
С1
Как показано выше, несмотря на то что равновесие
между радикалами I и II сдвигается в сторону образо-
вания радикала I, реакция переноса водорода к радика-
лу II происходит быстрее, чем к I, в результате чего об-
разуется вторичный хлорид. Доказательством изомери-
зации радикала I в II служит превращение пропилхло-
рида в изопропилхлорид при у-облучении в присутствии
хлористого водорода. Эта 1,2-миграция галогена извест-
на, и наблюдается также в броманалогах I. Однако в
присутствии бромистого водорода реакция переноса про-
текает так быстро, что первоначально образующийся
радикал моментально вступает в реакцию, что исключает
образование его изомера. Поэтому реакция присоедине-
ния бромистого водорода к пропилену в радикальных
условиях протекает против правила Марковникова с об-
разованием соответствующего продукта:
204
Br-
C = C—С-----► C—C—c
Br
I HBr
Очень быстро у
сн2—сн2—сн3
Вг
Основной продукт
----► С—с—с
Очень ।
мед- 1
ленно В
I HBr
сн3—сн—сн3
I
Вг
Не образуется
В заключение можно кратко охарактеризовать реак-
ции присоединения галогенов к олефинам. В табл. 14.2
приведены теплоты реакций присоединения галогенов к
этилену. Можно предсказать, что реакционная цепь при-
соединения хлора и брома к олефинам достигает боль-
шой длины. Иод присоединяется только в особых усло-
виях и предпочитает реагировать с атомом галогена
алкилгалогенида, как обсуждалось на стр. 183.
Таблица 14.2
. Теплоты реакций присоединения галогенов к этилену [17]
X-+ СН2 = СН2-^Х—СН2 —СН2- (14.1)
X —СН2 —СН2- +Х2-Х — СН2 — СН2 — х + х- (14.2)
Х2 Д Н, ккал/моль
Реакция присоеди- нения (14.1) Реакция переноса (14.2)
Вг2 —5 — 17
С12 —26 —19
Is 7 — 13
Реакции присоединения галогенопроизводных метана
На реакции присоединения галогенопроизводных ме-
тана к олефинам влияют различные факторы, которые
контролируют относительные выходы «голова к хвосту»
аддуктов и теломеров. В смеси СХ4 и мономера радика-
лы могут реагировать либо с СХ4, либо с мономерным
олефином М:
Х3С-М„- + М Х3С-М„+1. (14.4)
205
Х3С-М„ • + СХ4 Х3С-М„-Х + СХ, (14.5)
При п=\ протекают конкурирующие реакции (14.4) и
(14.5) СХ4 и М с Х3С—М«, в результате чего образует-
ся либо аддукт «голова .к хвосту», либо теломер: ;
Х3С—М- + СХ„ 21е₽-. Х3С-М-Х + -СХ3 (14.2)
Дальнейшая
кп реакция
Х3С—М- + М-* X3G—М2- ---------* (14.3)
ведет к тело-
меру
Очевидно, что относительные выводы аддукта «голова
к хвосту» и теломера зависят от активности СХ4 как
переносчика и от способности к полимеризации моно-
мера М.
Рассмотрим активность СХ4 как переносчика. Можно
ожидать, что мономерный радикал Х3С—М* и полимер-
ный радикал Х3С—Мп • будут мало отличаться в своих
реакциях. Относительные скорости реакции Х3С—Мп- с
галогенопроизводными метана определяются значениями
констант реакции переноса С=кпер/кп. Значения кон-
стант переноса уменьшаются в ряду СВг4>СВгС13>
>СС14>СН2С12>СН3С1, т. е. полимерные радикалы реа-
гируют быстрее с СВг4 и,наиболее медленно с СН3С1.
Можно ожидать, что отношение констант скорости реак-
ций (14.2) и (14.3) уменьшается в таком же порядке.
Это означает, что СВг4 должен давать наиболее высокие
выходы аддукта «голова к хвосту», а СН3С1 — наимень-
ший выход.
При рассмотрении активности мономера становится
ясным, что он наиболее активен к полимеризации с об-
разованием теломера, отсюда скорость реакции (14.3)
выше скорости реакции (14.2), а выход аддукта «голо-
, в а к хвосту» уменьшается.
В табл. 14.3 приведены примеры, подтверждающие
это. В реакциях присоединения к октену-1 выход аддук-
та «голова к хвосту» уменьшается, так как активность
переносчика-мала. Однако октен-1 не образует теломер.
Хороший выход аддукта «голова к хвосту» получен
даже с таким неактивным переносчиком, как четырех-
хлористый углерод. При взаимодействии галогенопроиз-
водных метана со стиролом образуется теломер даже с
таким активным переносчиком, как СВгС13.
206
Таблица 14.3
Присоединение галогенопроизводиых метана к октену-1 и стиролу
[19, 20, 21]
Галогенопро- изводное метана Инициатор Продукт Выход, %
Прнсс единение к октену-1 Вг
СВг4 С, А Вг3С— сн2—сн—свн13 Вг 96
CBrCls С, А С13С—СН2—^Н-СвН13 С1 88
СС14 А С13С-СН2—СН—СвН13 85
СНС13 Б . Прнс С13С—СН2—СН2—CeHis оедннеиие к стиролу Вг 22
СВг4 С Вг3С—СНа-СН—Ph Вг 96
СВгОз А С13С—СНа—СН—Ph Теломер 78 10
СС14 А Теломер 90
Примечание. С — бензоила. свет, А — перекись ацетила, Б — перекись
В табл. 14.4 приведены примеры продуктов реакций
присоединения производных галогена к олефинам.
Таблица 14.4
Примеры реакций радикального присоединения ]19]
Ав Олефин Продукт Выход, %
CC13CN Октен-1 NCCClaCH2CHClCeH13 66
СС13СОС1 Октен-1 С1СОСС12СНаСНС1СвН13 81
CF3 = CFI Этилен CFa= CF —СНа—CH2I 67
CF3I Этилен CF3 — CH2 — CH2I 82 (-]- теломер)
207
Реакции присоединения тиолов и меркаптанов
Тиолы и меркаптаны могут присоединяться к олефи-
нам либо по ионному, либо по радикальному механизму,
причем в одном случае присоединение происходит по
правилу Марковникова, а в другом — против правила
Марковникова. Возможность получать в результате этих
реакций любые продукты, так же как и в реакции при-
соединения бромистого водорода, находит определенное
применение [22].
Меркаптан обычно присоединяется по ионному меха-
низму, однако интенсивный источник света, например
облучение ртутной лампой, инициирует радикальное при-
соединение. Реакции присоединенйя тиолов протекают
по радикальному механизму. Чтобы присоединение про-
шло по донному механизму, необходимы чистые свеже-
полученные исходные соединения, полное отсутствие све-
та и кислорода. Элементарная сера является ингибито-
ром радикального присоединения. В ее присутствии
тиолы и меркаптаны присоединяются к олефинам по ион-
ному механизму, образуя продукты по правилу Марков-
никова
С6Н13-СН-сн3
180° с/ I
/ SR
/Сера
RSH + с6н13—сн - сн2
'\перекись
leo^cx*
CeH13—СН2—СН2—SR
Исследования стереохимии реакций присоединения
меркаптанов к циклическим олефинам показали, что
иногда стереоселективность достигается при высокой
концентрации меркаптана. При этом скорость реакции
переноса высокая; низкие концентрации меркаптанов
требуют времени для взаимопревращений двух промежу-
точных радикалов. Например, при взаимодействии
1-хлорциклогексена с меркаптаном, имеющим высокую
концентрацию, образуется главным образом г^мс-про-
дукт [23]. Как показано ниже, цис-аддукты являются
результатом реакции транс-присоединения H2S к оле-
фину. В этом процессе стадия присоединения и стадия
переноса должны протекать либо по вертикальному на-
208
правлению, дибо по горйаднтальному. Реакции в данном
случае, как показано, протекают цо вертикальному на-
правлению. \
Важной особенностью для\настоящего обсуждения
является способность меркаптанох при их высокой кон-
центрации акцептировать промежуточные радикалы не-
зависимо от их конформации, преж^ чем они начнут
инверсироваться
209
В табл. 14.5 приведены отубсительные скорости ре-
акций присоединения радикала додекантиола к различ-
ным олефинам. Данные рголицы показывают влияние
полярного эффекта заме/гителя и предполагают элек-
трофильность радикале тиола. Олефины с электроно-
акцепторными заместителями реагируют быстрее, чем
олефины, имеющир/'электронодонорные группы. Эти зна-
чения объясняю/ртруктуру переходного состояния, в
котором радикал тиола приобретает частичный отрица-
тельный Заряд.
Таблица 14.5
Реакция присоединения радикала додекантнола к олефинам
при 60° С [24]
кпр
C12H25S • + RCH = СН2 ------- C12H26S — СН2 — CHR
Олефнн Относительное значение *пр
PhCH = СН2 17
С4Н9О —СН = СН2 • 4
СвН13-СН = СН2 (1,00)
0 II сн3—с —о —сн = сн2 0,8
CICHj —сн = сн2 0,7
Циклогексан 0,2
Например, переходное состояние в реакции присоедине-
ния радикала тиола к винилбутиловому эфиру можно
изобразить следующим образом:
RS~CH2—СН—ОС4Н» ч—> RS? сн8—СН = ОС4Н9
Следует также упомянуть о некоторых особенностях
реакций радикального присоединения соединений серы.
К олефину присоединяются многие соединения серы, в
число которых можно включить: иоц сернистой кислоты,
сернистый ангидрид, сульфенилгалогенид', сульфогало-
генид, сульфурилгалогенид, хлорпентафторйды серы и
тиокислоты. Некоторые из этих реагентов нашли приме-
нение в препаративных синтезах. Например, иногда при-
210
меняют тиоуксусную кислоту для того, чтобы получить
продукты эквивалентного хрисоединения меркаптана к
олефину, протекающего против правила Марковникова,
исключая таким образом неудобный в работе газооб-
разный меркаптан:
RCH СН2
о О
11 и
СН„—С—SH II
--------R—СН2-СН2—S—С-СН3
Перекись ч
I Гидролиз
R—СН2—СН2—SH
Менее изучены реакции присоединения продуктов серы
к соединениям, содержащим ацетиленовую связь. I
Реакции присоединения других соединений к олефинам
с образованием новых углерод-углеродных связей
Особого внимания в синтезе заслуживают реакции
присоединения с образованием новой углерод-углерод-
ной связи. Большинство работ, которые обсуждались
выше, посвящены реакциям присоединения различных
галогенопроизводных метана. В этом разделе будут рас-
смотрены реакции присоединения различных органиче-
ских производных к олефинам с образованием новых
С—С-связей [25]. Альдегиды, являясь превосходными до-
норами водорода, быстро присоединяются к олефинам с
образованием кетонов. Фактически избыток любого аль-
дегида может присоединяться к любому олефину с обра-
зованием аддукта «голова к хвосту» с хорошим выхо-
дом, за исключением этилена, образующего значитель-
ные количества теломера. Например, масляный альдегид
присоединяется к октену-1
Перекись
С3Н7—СНО + СвН13—сн = сн2 ——-
о
II
С3Н7—С—(СН2)7—сн3
57%.
В результате присоединения энантового альдегида к
октену-1 образуется гексилоктилкетон с 75%-ным выхо-
дом. Стадии присоединения следующие:
211
о о
II \ / II ! I
R—С- + С = (/ -> R—С—С-С-
/ \ II
ООО о
II I I II II I I II
R—С—С—С- +R—С—Н -> R—С—С—C-H + R—С-
II II
Первичные и вторичные спирты также присоединяют-
ся к олефинам в цепной реакции:
R?C—ОН +^С = -> R2C—С—С-
/ он1 1
R2C—С-С- + R2CH—ОН -> R2C—с—С—Н + R2C—он
III ill
он он
Третичные спирты не присоединяются к олефинам, так
как у них нет ;атомов водорода при ia-атоме углерода.
Значения констант реакций переноса для спиртов не-
велики, поэтому эти реакции протекают при большом из-
бытке спирта. Свет, перекиси или азосоединения иници-
ируют реакции присоединения спиртов к олефинам. На-
пример [26], изопропиловый спирт присоединяется к
октену-1 (при молярном соотношении 24:1) с об-
разованием ,46% аддукта «голова к хвосту» и теломе-
н3с—сн—сн3
свн13-сн = сн2
Перекись
трет- бутила
0,25 моль
СН3
I
-> С8Н17—С—ОН 4- Димер + Теломеры с большим
| молекулярным весом
СН3
0,11 моль 25% 29%
(46%)
212
Образование теломера ^реакциях присоединения спир-
тов к олефинам уменьшатся от первичного спирта к
вторичному. С повышением\;емпературы выход теломе-
ра уменьшается. При высоком^ значении молярного от-
ношения спирт/олефин и достаточно высокой температу-
ре реакции вторичные спирты в результате присоедине-
ния к олефинам образуют аддукт «голова к хвосту» с
хорошим выходом. 4
Амины по а-углерод-водородной связи также присо-
единяются к олефинам. Инициатором этих реакций мо-
жет быть свет или перекись mpem-бутила. Перекись бен-
зоила неэффективна, так как амины катализируют ее
разложение. Амины имеют небольшие значения констан-
ты реакции переноса (около 0,1), поэтому образуются
главным образом теломеры. Однако удовлетворитель-
ные выходы аддуктов «голова к ' хвосту» можно полу-
чить, используя большой избыток амина, например [27]
c4h9-nh2
5 моль
СНа = СН-СвН13
0,25 моль
)
125° С
Перекись
трет- бутила
-+ С3Н,—СН—СН2—СН2—qeH13 + Димер
| 0,09 моль (18%)
NH2 (36%)
+ Тример
(45%)
N
Н
сн2=сн-свн13
0,36 моль
СН-СН2-С6Н13
0,25 моль
(70%)
120° с
Перекись
трет -бутила
+ Димер 4- Тример и тетрамер
(16%) (14%)
213
Типичное раднкал-нннцннрованное присоединение к октену-1 [28 , 29]
Таблица 14.6
реагент Эквива- ленты на один эквива- лент октена-1 Инициатор (эквивален- ты) Г, °C Продукт Выход, %
СН3СО2Н (300) Перекись трет-бутила (0,15) 105 69
СН3СОСН3 (22) Диизопропиловый эфир надугольной кислоты (0,24) 56 н-С8Н1,СНаСОСН3 32
СНаС1СОаС2Н6 (И) Перекись трет-бутила (0,Ю) 155 н-С8Н1,СНС1СОаСаН6 73
CH3COSCHaCH3 (22) Перекись трет-бутила 155 н-С8Н17СН (СН3) SCOCHg 8
(0,10) h-C8Hi7CH2CH2SCOCH3 1
H-C8Hi7CHaCOSCHaCH3 1
СН3СОаСН3 (ЮО) Диизопропиловый эфир иадугольной кислоты (0,24) 57 н-С8Н17СНаСООСН3 н-С8Н17СНаОСОСН3 8 1
Циклогексан (60) । Перекись бензоила (0,50) 82 Октилциклогексаи 42
Эфиры атакуются {Радикалами по а-углерод-водород-
ной связи, в результате \до образуется радикал, кото-
рый может п рисоедин я ться^к олефинам. Например, ди-
этилмалонат радикально присоединяется к октену-1 с
образованием днэтилоктилмалЬцата. Подобно этой ре-
акции, некоторые эфиры, ацетали», амиды и даже неко-
торые углеводороды присоединяются, к конечным олефи-
нам. Значения табл. 14.6 иллюстрируют реакции этих
соединений {28, 25].
Относительные скорости реакций присоединения
Вернемся к факторам, которые влияют на скорости
реакций присоединения. На скорости реакций присоеди-
нения радикала X- к олефинам влияют: стабильность
радикала X-; строение олефина и радикала, образующе-
гося в результате присоединения, стерические препятст-
вия на стадии присоединения и полярный фактор заме-
стителя. В табл. 14.7 представлены значения констант
Таблица 14.7
Константы скорости реакций присоединения и переноса
при взаимодействии бутантиола с олефинами [30, 31]
\ /кт II II лпер I I
RS-+ C=C->RS—С—С- RS—С—C-+RSH—RS—C-CH+RS-
\ I I II II
Олефин Константы скорости, л-моль 1-сеп 1 (25°С)
кпр кпер
Стирол 8-Ю8 1,2-103
Пентен-1 7-106 1,4-10»
скоростей присоединения и переноса реакций тиолов с
олефинами. Радикал бутантиола присоединяется к сти-
ролу быстрее, чем к пентену, так как в результате при-
соединения к стиролу образуется бензильный радикал,
который стабилизируется за счет сопряжения иеспарен-
ного электрона с системой л-электроиов ароматического
ядра
RS- + СН2 = CHR' -> RS—СН2—CHR'
RS- +СН2 = CHPh -> RS—СН2—CHPh
215
Эта резонансная стабилизация з01едляет отрыв водоро-
да от тиола. /
Увеличение скорости реакций радикального присо-
единения конечных олефинбв указывает на влияние сте-
рических факторов. Возможно, что сравнительно низкая
стабильность вторичных радикалов по сравнению с пер-
вичными также является результатом влияния стериче-
ского эффекта. Влияние полярных факторов на ради-
кальные реакции присоединения можно видеть из дан-
ных табл. 14.5.
Наиболее полные значения были опубликованы для
реакций присоединения метильных радикалов к различ-
ным олефинам, ацетиленам и ароматическим соедине-
ниям £32]. Эти значения получены' разложением источни-
ка метильного радикала в смеси изооктана и изучаемых
ненасыщенных соединений. В качестве источника ме-
тильного радикала можно использовать не только чаще
всего применяемую перекись ацетила, но и с тем же
успехом азометан и перекись mpem-бутила. Метильные
радикалы отрывают водород от изооктана (SH) с обра-
зованием метана:
«в
СН3-+ SH — CH4 + S-
(14-6)
Однако в присутствии ненасыщенного соединения А не-
которые метильные радикалы присоединяются к ним:
к,
СНз- + А— СН3А-
(14.7)
Отношение можно вычислить по уменьшению вы-
хода образующегося метана в присутствии ненасыщен-
ного соединения А по сравнению с контрольным опытом
(в отсутствие А).
В табл. 14.8 приведены значения, полученные этим
способом. Значения табл. 14.8 иллюстрируют влияние
различных факторов на скорости процессов присоедине-
ния. Реакция присоединения протекает гораздо быстрее
с конечными олефинами. Замещенные олефины реаги-
руют медленно из-за стерических препятствий, даже если
они могут образовывать стабильные радикалы в реак-
циях присоединения. Например, тетрафенилэтилен реа-
гирует гораздо медленней, чем ди- или тризамещенные
этилены. Стирол, однако, реагирует гораздо быстрее, чем
пропилен. В этом случае фенильный заместитель стаби-
216
Таблица 14.8
Константы скорости реакции присоединения метильных радикалов
по двойной связи (при 65° С) относительно реакции отрыва водорода
от изооктана [32]
Соединение Kj/Ks Соединение
Этилен Этоксиэтилен Пропилеи Метилпропилен 2-Метилбутеи-2 цис-Бутеи-2 транс-Бутен-2 Стирол 1,1-Дифенилэтилен цис-1,2-Дифенилэтилен трансЛ ,2-Дифенилэтилен 26 6 22 36 6 3 7 796 1500 29 104 1,1,2-Трифеиилэтилен Т ет рафенилэтилен Этилмалеат Этилфумарат Бутадиен 2,3- Бутадиен 2,4- Гексадиен 2,5 -Диметилгекса диен-2,4 1,4-Дифенилбутадиен 1,1,4,4-Тетрафенилбута- диен 48 8 333 2000 2015 2200 180 20 380 60
лизирует радикал, который беспрепятственно возникает
на стадии присоединения
СН3- + СН2 = CHPh - СН3—СН2 -CHPh
СНз- + СН2 = СН—СН3 СН3—СН2—СН—СН3
Таким же образом протекают реакции присоедине-
ния метилкных радикалов к ароматическим углеводоро-
дам, относительные скорости которых сведены в
табл. 14.9.
Таблица 14.9
Относительные скорости реакций присоединения метильных радикалов
к ароматическим соединеяним [32]
Субстрат Относи- тельная скорость Субстрат Относи- тельная скорость
(Этилен) (85) Бензантрацен 515
Бензол 1 Антрацен 820
Нафталин 22 Нафтацен 9250
Фенантрен 27 Пиридин 3
217
Например, бензол реагирует следующим образом:
СН3
(—н«).
Акцептор
водорода
Й + сн3*
. (14.8)
И + Другие
продукты
Рис. 14.1. Зависимость относи-
тельных скоростей присоединения
метильных радикалов к аромати-
ческим соединениям от энергии
локализации этих соединений [32].
Относительные скорости метилирования ароматиче-
ских соединений можно связать с энергией локализации,
которую можно количественно вычислить, используя
метод молекулярных орбит [33]. Энергия локализации
определяется как энергия
л-связи, необходимая для
того, чтобы изолировать
один электрон от резонанс-
ной системы и локализо-
вать его на данном атоме.
Каждый акт локализации
образует новую резонанс-
ную систему, содержащую
на один атом и один элек-
трон меньше. Локализо-
ванная система имеет ту
же самую структуру л-
связей, как и переходное
состояние в реакции ме-
тилирования. Локализа-
ция энергии в бензоле,
например, приводит кпен-
тадиенильному радикалу.
См. уравнение ; реакции
(14.8). Энергия л-связи,
которая может быть оп-
ределена по методу моле-
кулярных орбит, вычис-
ляется затем для исход-
ных соединений и локализованной системы, взятая как
приближение для переходного состояния. Так, например,
вычисляется энергия л-связи бензола и пентадиениль-
ного радикала. Разница в энергии должна быть пропор-
циональна энергии, необходимой для образования толу-
ола, и, следовательно, энергии активации (логарифм
константы скорости) реакции (14.8). На рис. 14.1 пока-
зана логарифмическая зависимость относительных ско-
218
Рис. 14.2. Зависимость относи-
тельных скоростей присоедине-
ния фенил-, этил-, пропил- и
трихлорметильных радикалов
к ароматическим соединениям
от относительных скоростей
присоединения метильных ра-
дикалов к этим же соедине-
ниям [34J.
ростей реакций присоединения метильных радикалов от
энергии локализации. По значениям энергии локализа-
ции, вычисленным методом молекулярных орбит, легко
предсказать скорости реакций присоединения.
Если скорости присо-
единения метильных ра-
дикалов подтверждают
основные принципы регу-
лирования радикального
присоединения, изложен-
ные выше, то и другие
радикалы должны пока-
зывать ^подобные резуль-
таты. На рис. 14.2 по-
казана логарифмическая
зависимость скоростей ме-
тильного присоединения
относительно скорости
присоединения этил-, про-
пил-, фенил- и трихлор-
метильных радикалов к
подобным ароматическим
системам. При этом на-
блюдается превосходная'
корреляция значений.
Наклон прямых пока-
зывает относительную се-
лективность следующих
радикалов:
Этил............. 1,0
Пропил............ 1,0
Фенил ............ 1,0
ТрихлОрметил .... 2,0
Таким образом, алкильные и фенильные радикалы оди-
наково избирательно присоединяются к ароматическим
соединениям. Трихлорметильный радикал приблизитель-
но вдвое более селективен, чем метильный радикал.
Аналогичная обработка значений для полистирольных
радикалов показывает, что они также вдвое более селек-
тивны, чем метильные радикалы.
Упражнения
14.1. Написать механизм реакции изомеризации, инициирован-
ной светом и бромом, цис-стильбена в транс-стильбен (PhCH =
=CHPh).
219
14.2. Написать механизм реакции присоединения, индуцирован-
ной светом, бромистого водорода к бутену-1 с образованием 1-бром-
бутана. Почему в этом случае образуется аддукт «голова к хвосту»
и невероятна реакция (14.1)?
14.3. Написать полный механизм реакция образования тример-
ных продуктов в результате инициированного азоизобутиронитрилом
присоединения четыреххлористого углерода к этилену.
14.4. Показать, что в уравнениях
В-+М Л ВМ-
ВМ-+АВ ВМА + В
ВМ-+М вма-
кп
ВМ„-+М------► ВМл+1-
Лпер
ВМЛ-+АВ-----► ВМ„А + В-
где М — мономерный олефин, отношение выхода аддукта «голова к
хвосту» к общему выходу теломеров (димеров и выше) равно
(АВ)/«Пр (М).
Если активность не зависит от длины цепи, можно приравнять кон-
станты скорости этих реакций. Используя это упрощение, перепи-
сать указанное выражение для выходов продуктов.
14.5. На стр. 196 аддукт теломеризации стирола в CCU показан
как С1зС—(СН2—СНРЬ)Л—С1. Почему эта структура является наи-
более вероятной, а не С1зС—(CHPh—СН2)Л—С1? Почему при из-
бытке стирола образуется теломер с-большим молекулярным весом?
14.6. Г1очему название «перекисный эффект» является не совсем
подходящим для реакции присоединения бромистого водорода к ко-
нечному олефину с образованием продуктов, не подчиняющихся
правилу Марковникова?
14.7. Использовать значения табл. 14.1 для объяснения, почему
иодистый водород и хлористый водород не присоединяются к оле-
финам в цепном процессе.
14.8. Написать возможную структуру димерного продукта, об-
разующегося в результате присоединения изопропилового спирта к
октену-1.
14.9. В реакции присоединения четыреххлористого углерода к
этилену отношение константы скорости отрыва хлора от CCU к кон-
станте скорости присоединения к этилену было измерено для ради-
калов различных молекулярных весов. Константа скорости переноса
для н-мера определяется как
№пер, п
Уравнения реакций, константы скоростей которых входят в указан-
ное выражение, следующие:
к
пер, п
ВМЛ-+АВ-------►ВМЛА + В-
ВМЛ +М ВМ„+1-
220
При 27бС Ci= 0,07, С2= 3,0, С3= 9,7, 16 и для очень большого
значения п Сл = 31. Используя влияние полярности в переходном
состоянии, объяснить это увеличение значений констант реакций
переноса [35].
14.10. В реакции присоединения хлороформа к этилену при
28° С С1=0,2, С2= 1,3, С3= 1,5 (см. упражнение 14.9). Объяснить
эту тенденцию к увеличению значений С [36].
14.11. В реакции присоединения четыреххлористого углерода к
стиролу при 60° С С1= 6- 10-4, С2= 25- 10-4, С3= 69- Ю 4, Cl и все
более высокие константы переноса равны 115-10~4 (см. упражне-
ние 14.9). Объяснить эту тенденцию. Почему так велики значения
констант реакций переноса четыреххлористого углерода к этилену
по сравнению с теми же реакциями переноса к стиролу (см. упраж-
нение 14.9) [3]?
14.12. Привести правильное объяснение значений констант реак-
ций переноса для следующих переносчиков:
СВг4 ................. 2,2
СС14 ................1,1-Ю-2
СНС13..................5 10—5[37]
14.13. Объяснить значения к?/Кб в табл. 14.8 для этилена,
2,3-бутадиена и 1,4-дифеиилбутадиена.
14.14. В результате реакции, инициированной перекисью трет-
бутила, либо цис-, либо транс-4-трет-бутилциклогексанола с окте-
ном-1, при 150° С образуется 1-октил-4-/пре/п-бутил-1-циклогексанол
с выходом 40%.
Н^^ОН
1трет - С4 Н„
цис или транс
|С6Н13—СН=СН2
Как видно из уравнения реакции, независимо от применения исход-
ного изомера образуется смесь изомеров продуктов реакции с выхо-
дом 65% и 35%. Обсудить механизм присоединения спиртов к оле-
финам. Объяснить стереохимию указанной реакции присоединения,
если радикал циклогексила имеет плоскую конфигурацию. Объяс-
нить стереохимию этой реакции, 'допуская, что скорость инверсии
221
радикала циклогексила, имеющего пирамидальную конфигурацию,
превышает скорости реакций присоединения их к октену-1 [38].
14.15. При бромировании оптически активного 1-хлор-2-метилбу-
тана образуется 1-хлор-2-бром-2-метилбутан.
а) Объяснить эту реакцию, допуская, что циклические (мости-
ковые) радикалы более стабильны, чем радикалы с открытой
цепью.
б) Объяснить эту реакцию, допуская, что циклические
(мостиковые) радикалы не образуются при этом либо что они
менее стабильны, чем радикалы с открытой цепью, и образуются
только из радикалов с открытой цепью. Какое объяснение
более предпочтительно (см. стр. 40—46 и 201)?
14.16. Смесь 5 -И.ИОЛ6 этилового эфира 2-циангептен-6-овой кис-
лоты и 2,5 ммоль перекиси бензоила кипятят с обратным холодиль-
ником в течение 24 ч в 126 г циклогексана. Получен продукт
(62% выхода), который гидролизуется с образованием 1,1-циклогек-
сандикарбоновой кислоты. Написать механизм реакции и идентифи-
цировать продукт [29].
14.17. (Активности различных олефинов в реакции присоединения
бромистого водорода были измерены относительно активности эти-
лена в той же реакции, как стандартного соединения. Значения для
этих олефинов следующие:
Этилен..................... (1)
Пропилен..................... 15
Бутен-1...................... 20
Бутен-2...................... 69
Изобутилен................... 286
Винилхлорид ................. 0,3
Дать рациональное, объяснение этим значениям и сравнить реакции
этих олефинов с атомами брома и метильными радикалами [39].
ЛИТЕРАТУРА
1. Walling С. Free Radicals in Solution. John Wiley and Sons.,
Inc., New York, 1957, p. 302—308.
2. Nesmeyanov A. N., Freidlina R. Kh., Zakhar-
k i n L. I. Quart. Rev. (London), 10, 360 (1956).
3. M а у о F. R. J. Amer. Chem. Soc., 70, 3689 (1948).
4. Mayo F. R. In: W. A. Waters (ed.). Vistas in Free Radical
Chemistry. Pergamon Press. New York, 1959, p. 139—142.
5. Kharasch M. S., Engelmann FL, Mayo F. R. J. Organ.
Chem., 2, 288 (1937).
6. M а у о F. R., Walling C. Chem. Rev., 27, 351 (1940).
7. Hey D. H., Waters W. A. Ibid., 21, 169 (1937).
8. Skell P. S., Allen R. G. J. Amer. Chem. Soc., 81, 5383 (1959).
9. Goering H. L., Larsen D. W. Ibid., 5937 (1959).
10. G о e r i n g H. L., Ab e 11 P. I., Aycock B. F. Ibid., 74, 3588
(1952).
11. Skell P. S., Wood worth R. C., McNamara J. H. Ibid.,
79, 1253 (1957).
12. Skell P. S„ Tuleen D. L„ Readio P. D. Ibid., 85, 2849
(1963).
222
13. Abell P. I., Piette L. H. Ibid., 84, 916 (1962).
14. Neureiter N. P., В or dwell F. G. Ibid., 82, 5354 (1960).
15. Bohm B. A., Abell P. I. Chem. Rev., 62, 599 (1962).
16. L e В e 1 N. A. J. Amer. Chem. Soc., 82, 623 (1960).
17. Walling C. Free Radicals in Solution. John Wiley and Sons,
Inc., N.Y, 1957, p. 241.
18. M а у о F. R. J. Amer. Chem. Soc., 84, 3964 (1962).
19. Walling C. Free Radicals in Solution, John Wiley and Sons.
Inc., New York, 1957, p. 252.
20. Kharasch M. S, Jensen E. V, Urry W. H. J. Amer. Chem.
Soc., 69, 1100 (1947).
21. Khar asch M. S, Re in mu th O., U г г у W. H. Ibid., 69,
1105 (1947).
22. Pryor W. A. Mechanisms of Sulfur Reactions. McGraw-Hill
Book Company. N. Y, 1962, pp. 75—93, 162—164.
23. G о e r i n g H. L., Relye a D. I., Larsen D. W. J. Amer.
Chem. Soc., 78, 348 (1956).
24. Walling C, Helmreich W. Ibid., 81, 1144 (1959).
25. W a 11 i n g C, Huyser E. S. In: A. C. Cope (ed.). Organic
Reaction^ Vol. 13. John Wiley and Sons, Inc., N.Y, 1963.
p. 91—149.
26. U г г у W. H. et al. J. Amer. Chem. Soc., 76, 450 (1954).
27. Ur г у W..H, Ju vela nd O.O. Ibid., 80, 3322 (1958).
28. A11 e n J. C., Cadogan J. I. G„ Hey D. H. J. Chem. Soc.,
1918 (1965).
29 Cadogan J. I. G, Hey D: H„ Ong S. H. Ibid, 1939 (1965).
30. Onyszchuk M, Si vert z C. Can. J. Chem., 33, 1034 (1955).
31. S i v e r t z C. J. Phys. Chem, 63, 34 (1959).
32. Szwarc M, В i и к s J. H. Theoretical Organic Chemistry. Ke-
kule Symposium, 1958. Butterworth Scientific Publications, Lon-
don, 1958.
33. S t r e i t w i e s e r A. Molecular Orbital Theory for Organic Che-
mists. John Wilev and Sons, Inc, N.Y, 1960, pp. 335, 400.
34. Trotman-Dickenson A. F. Free Radicals. Methuen
Co. London, 1959, p. 109.
35. D a v i d C, Gosselain P. A. Tetrahedron, 18, 639 (1962).
36. M e 11 о w s F. W, Burton M. J. Phys. Chem, 66, 2164 (1962).
37. H e n r i c i - О 1 i v e G, Olive S. Fortschr. Hochpolymer-
Forsch, 2, 496 (1961).
38. G r i 11 e r R. J, Albers R. J. J. Organ. Chem, 29, 728
(1964).
39. A b e 11 P. I. Trans. Faraday Soc., 60, 2214 (1964).
Дополнительная литература
Bohm В. A, Abell P. I. Chem. Rev, 62, 599 (1962).
C a d о g a n J. I. G, Hey D. H. Quart. Rev. (London), 8, 308 (1954).
Stacey F. W, Harris J. F. In: A. C. Cope (ed.). Organic Reac-
tions. Vol. 13. John Wiley and Sons, Inc, N.Y, 1963,
pp. 150—376.
Walling C, Huyser E. S. In: A. C. Cope (ed,). Organic Reac-
tions. Vol. 13. John Wiley and ISons, Inc, N.Y, 1963, pp. 91—149.
223
Глава 15
радикальные полимеризации
Полимер — это большая молекула, либо встречаю-
щаяся в природе, либо полученная искусственным путем;
мы ограничимся рассмотрением синтетических полиме-
ров. Принято считать, что полимерная цепь образуется
из многократно повторяющейся элементарной ячейки,
так называемого мономера. Молекулы, полученные в ре-
зультате радикальных процессов, имеют от 103 до 105
повторяющихся ячеек и молекулярный вес от 105 до !107.
В химии полимеров начиная с 30-х годов, а особен-
но за последние годы, достигнуты значительные успехи.
Полимерные материалы прочно вошли в нашу жизнь:
искусственные ткани, строительные материалы, покры-
тия для столов, посуда и масса других вещей. А ведь
совсем недавно все это требовало дерева, металла или
других природных материалов.
Синтетические полимеры можно получить ионным
или радикальным способом. Для некоторых продажных
пластических масс пригодны оба метода, но большинст-
во все же получают каким-либо одним. Обсуждение
ионной полимеризации выходит за пределы этой моно-
графии, а детальное рассмотрение радикальной полиме-
ризации дано в специальных учебниках * *. Но поскольку
изучение полимерных радикалов расширяет наши по-
знания в области свободных радикалов, рассмотреть
здесь этот вопрос необходимо. Для начала ознакомимся
с некоторыми распространенными пластическими масса-
ми, получаемыми с помощью радикальной полимериза-
ции.
Примеры наиболее часто применяемых пластмасс
Превращение мономера в полимер может протекать
двояко. Чаще всего это полимеризация свободных ра-
дикалов, т. е. взаимодействие их с образованием поли-
мера без выделения каких-либо третьих молекул. При-
мером этого может служить образование полиэтилена**
пСН2 =СН2 -> (-СН2-СН2-)„
* Список литературы дан в конце этой главы.
** Отмечено, что природа концевой группы полимерной цепи
не специфична. Полимер имеет очень большой молекулярный вес и,
следовательно, очень незначительное количество конечных групп на
224
Некоторые ионные полимеризации также представ-
ляют собой рекомбинацию, но большинство их относится
к так называемой поликонденсации *. В такой реакции
при взаимодействии мономеров отщепляется небольшая
молекула. Примером может служить получение найлона
из адипиновой кислоты и гексаметилендиамина:
пСО2Н—(СН2)4—СО2Н + nNH2-(CH3)e—NH2 ->
ECO—(CH2)4—CO—NH—(CH2)e—NH3 „ + 2nH2O
катализ
Простейшим и наиболее часто применяемым полиме-
ром является полиэтилен. Его получают двумя метода-
ми. Полиэтилен «высокого давления» с точкой плавле-
ния около 110° С синтезируют более старым радикаль-
ным методом. Новый ионный метод дает продукт с точ-
кой плавления около 140°С; это полиэтилен «низкого
давления» или «высокой плотности». Вторым способом
получают более высокоплавкий неэластичный материал.
Радикальный метод применяют для получения прозрач-
ного полиэтилена. По этому методу этилен нагревают до
200° при 1000 атм в присутствии небольшого, строго
определенного количества воздуха или перекиси, а по-
лиэтилен непрерывно отводят из реакционной смеси.
Воздух или перекись при взаимодействии с этиленом
дают радикалы [реакция (15.1)]. Затем первичные ра-
дикалы присоединяются к мономеру, инициируя поли-
меризацию [реакция (15.2)]. Полученные таким образом
радикалы, каждый из которых содержит одну мономер-
ную ячейку, соединяются между собой, образуя димер,
тример и т. д.
грамм вещества. Экспериментально определить, какие это группы,
довольно трудно, но в зависимости от способа получения полимера
можно предвидеть, какова будет природа этих групп. Далее в этой
Главе будут рассмотрены способы получения полимеров.
* Разделение полимеров в зависимости от типа полимериза-
ции первоначально предложил Кароферс. Недавно Флори предло-
жил новое деление в зависимости от того, какой характер носит
полимеризация: цепной или ступенчатый. По первому способу обра-
зовавшийся активный центр растет цепным., методом до момента
обрыва. По второму варианту новые связи образуются с по-
мощью ступенчатых реакций любых имеющихся функциональных
групп. Все радикальные полимеризации являются цепными процес-
сами; большая часть ионных полимеризаций (но не все)’ — ступен-
чатые реакции.
8 У. Прайер
225
Воздух
Инициирование:
СН2 = СН2----------* Радикалы (R-) (15.1)
или перекись
R- + СН2 = СН2 -> R—СН2—СН2- (15.2)
Рост цепи:
R-(CH2-CH2)„- + СН2 = СН2
-> R-(CH2—СН2)„-СН2—СН2- (15.3)
Обрыв цепи:
Рекомбинация
2RCH2—СН2- —
R-(CH2)4-R: (15.4)
Диспропор-
ционирование
RCH=СН2+RCH2—СН3
(15.5)
Наращивание прекращается, когда две цепи соеди-
нятся (15.4) или подвергнутся диспропорционированию
(15.5). В промышленном масштабе полиэтилен получают
в США с 1943 г. В настоящее время его используют в
производстве бумаги и пленки, эластичной тары, в каче-
стве покрытия и изоляции для электрических проводов
и во многих других случаях.
Замещая в полиэтилене один или более водородных
атомов различными группами, получают мономеры, на
основе которых синтезируют другие распространенные
полимеры. В табл. 15.1 представлены некоторые наибо-
лее часто применяемые пластики.
Кинетика полимеризации
Механизм полимеризации мономера в общем виде
можно представить уравнениями
Инициирование Закон скорости
И -> 2R- 7?д = Кд[И]
R.-|-M^M- = КиГК1 (MJ
Цепная реакция
м- + м -> м2-
В общем случае
М„-+М + Мя+1-
226
Яобр = «обр [М-1 [М]
Таблица 15.1
Примеры пластических масс, полученных полимеризацией
рекомбинации с использованием радикальных инициаторов [1]
Полимер (и общепри- нятое наиме- нование) Мономер Свойства полимера Использование
Полиэтилен сн2 = сн2 Эластичный, полу- прозрачный, инерт- ный к большинстру химических реа- гентов Тара, пленка, тру- бы, электроизо- ляция
Полистирол СН2 = CHPh Светлый, твердый, хрупкий, воздушные пузырьки могут быть объединены, давая пенопласт различ- ной плотности Контейнеры; пено- пласты, как изоля- ция и строймате- риал
Поливинил- хлорид (Геон, Коро- сеал, Тигон) СН2 = СНС1 Твердый; органиче- ские материалы, такие, как эфиры, добавляют иногда в полимер, чтобы пластифицировать его с целью полу- чить прозрачный эластичный пластик Трубы, изоляция, пластификатор, используется для искусственных кож, шлангов
Поливинил- иденхлорид СН2 = СС12 Полупрозрачный, твердый Обычно полимери- зуется в смеси с винилхлоридом, давая сопо- лимеры
Полихлор- трифтор- этилен (фторо- пласт-3) CF2 = CFC1 Химически инерт- ный Низкомолекулярные полимеры использу- ются в качестве вы- сокотемпературных масел; высокомоле- кулярные полимеры используются как химически инертные прокладки
Политетра- фторэтилен (тефлон, фто- ропласт-4) CF2 = cf2 Необычно высокая точка плавления (327°), химически инертен и эластичен Прокладки, хими- чески устойчивые детали, электро- изоляция
Полиметил- метакрилат (плексиглас, луцит) сн3 сн2 = (!: — — СО2СН3 Прозрачный полу- эластичный Часовые линзы, контактные лиизы, авиационные стекла -
8*
227
Продолжение табл. 15.1
Полимер (и общепри- нятое наиме- нование) Мономер Свойства полимера Использование
Полиакрил- нитрил (ор- лон, акрилан) Стирол-бу- тадиеновый сополимер С г N СН2 = (ЗН СН2 = CHPh, СН2 = СН — — СН — СН2 Кристаллический, очень блестящий Каучукообразный, обычно полимеризу- ется в виде эмуль- сии в воде, стаби- лизированной мыла- ми и другими присадками Искусственные волокна Материал, содержа- щий около 25% сти- рола и 75% бута- диена, является основным синтети- ческим каучуком, который применяет- ся для шин, около 80% стирола в со- полимере дает более твердый полимер, используемый в машиностроении
Rp, к —,Kpt рек [М - ]а
ftp, д = кр, д |М-]2
Обрыв цепи
Мл • > Mrt
Мл- + Мт- -> M„ + Mm
В общем случае
М„ • + Мт • -> Полимер
/?р = кр [М-р
где И — инициатор; R-—радикал с низким молекуляр-
ным весом; М — молекула мономера; Мп • и Ь\т— по-
лимерные радикалы, в цепи которых пит ячеек моно-
мера.
Известно, что существуют две реакции обрыва цепи,
соответствующие одному кинетическому закону, и мож-
но написать общее уравнение. В общем виде для просто-
ты предполагают, что константа скорости любой реакции
не зависит от длины цепи полимерного радикала, при-
нимавшего участие в реакции. Например, предположим,
что во время реакции роста цепи радикалы М200 и М261
присоединяются к какой-либо мономерной ячейке с оди-
наковой скоростью. Если этого предположения не сде-
лать, то для цепи длиной в 1000 и более мономеров
кинетическое уравнение давало бы 1000 различных зна-
чений для констант скоростей реакции роста цепи, что
228
затрудняло бы расчеты. Приняв такое предположение,
радикалы можно обозначить М* без индекса (как пока-
зано в уравнениях скоростей на стр. 226—228).
Из приведенных уравнений можно вывести ряд про-
стых выражений. Принимается во внимание, что динами-
ческое равновесие концентраций будет достигнуто при
равенстве скоростей образования и разрушения радика-
лов, т. е. для динамического равновесия необходимо,
чтобы скорость образования радикалов КОбР была равна
скорости разрушения радикалов /?р. Скорость образо-
вания радикалов выражена уравнением 2Кд/[И], где f —
часть первичных радикалов, возникающих в результате
клеточной рекомбинации; они могут инициировать поли-
меризацию. Коэффициент 2 означает, что каждая моле-
кула-инициатор образует два радикала. Скорость разру-
шения радикалов представлена выражением 2кр[М*]2,
где коэффициент 2 соответствует исчезновению двух ра-
дикалов. Следовательно,
2кд/[И] = 2кр [М • ]2
и
- ( ^-У'”. (1Ь6)
\ *л-р / \ К-D /
Уравнение (15.6) выражает концентрации радикалов,
находящихся в реакционной системе.
Скорость образования полимера представлена урав-
нением
- Кобр = ки [R-1 [М] кобр [М - ] [Мй
В цепных реакциях можно пренебречь молекулами
мономеров, используемых при инициировании. Скорость
исчезновения мономера выражают с некоторым прибли-
жением следующим образом:
Кобр = КобР[М-] [М]. (15.7)
Подставив выражение (15.6) в (15.7), получим
Кобр = (М] (kJ [И])*7* (15.8а)
или в общем виде <
Кобр = [М] (Кобр/г)7’ . (15.86)
к/*
229
Из уравнения (15.8 а) следует, что скорость образо-
ния полимера пропорциональна квадратному корню из
значения концентрации инициатора; это подтверждается
для большинства инициаторов. Из этого же уравнения
делают еще один вывод: скорость должна быть пропор-
циональна концентрации мономера, это тоже часто под-
тверждалось.
Отношение констант скоростей кОбр/Кр! можно полу-
чить из уравнения '(15.8а), вычислив скорость образова-
ния полимера при использовании инициатора, скорость
разложения которого известна.
Отношение кОбр/Кр/г характеризует степень полимери-
зации мономера. Увеличение значения этого соотноше-
ния означает, что одно и то же количество инициатора
дает больше полимера за данный промежуток времени.
В табл. 15.2 представлены эти соотношения для несколь-
ких наиболее часто используемых мономеров. В таблице
Таблица 15.2
Константы скоростей (л-моль~~ '-сек~1) для некоторых
распространенных мономеров при 25° С [2]
Мономер , 1/2 кобР/кР \)бр Кр.1О-7
п-Метоксистирол 0,00284 2,9 0,10
Стирол 0,014 40 0,79
Метилметакрилат 0,055 . 275 2,5
Винилацетат 0,18 1000 3,0
Метилакрилат 0,20 1500 5,5
приводятся также абсолютные значения констант ско-
ростей реакции роста и обрыва цепи (их вычисление
см. ниже). Отмечено, что степень полимеризации моно-
мера определяется не значением константы скорости ро-
ста цепи .кобр, а соотношением кобр/кР/2.
Как видно из таблицы, винилацетат и метилакрилат
имеют приблизительно одну и ту же степень полимери-
зации, но последний имеет большую константу скорости
реакции роста цепи.
Кинетическая длина цепи v есть ряд мономерных мо-
лекул, используемых каждым первичным радикалом. Это
можно выразить уравнением
V = . (15.9)
Яобр
230
Например, если рост цепи происходит в 100 раз быстрее
инициирования, то каждый полимерный радикал увели-
чится в среднем до Мюо. Действительный размер поли-
мерной молекулы зависит от механизма реакции обрыва
цепи. Число мономерных ячеек в средней полимерной
молекул_е называется степенью полимеризации и обозна-
чается Р. Если цепь обрывается в результате диспропор-
ционирования, средний полимер имеет степень полиме-
ризации, равную кинетической длине цепи и P=v. Если
рекомбинация оборвала цепь, число мономерных ячеек
в среднем полимере достигает удвоенной кинетической
длины цепи и P = 2v ।
М„-Мл
Рекомби- Z4 р~— 2v
нация,/
2М„-<
Дис-\
пропор-\
ционнрование
М„+Мл
Не принимая в расчет механизм реакции обрыва цепи,
уравнение (15.9) можно записать
у _ Робр _ кобр[М-][М] _ Кобр [М] 10)
“R/ 2кр [М-р 2кр [М-1 ’ k ‘ ’
используя уравнение (15.7), это уравнение записываем
так:
4р [ME
(15.11)
Как видно из уравнения (15.9), величина длины цепи
обратно пропорциональна скорости инициирования; уве-
личение скорости инициирования приводит к образова-
нию полимеров с более низким молекулярным весом.
Приведенные расчеты полностью подтверждаются.
Из уравнения (15.11) можно видеть также, что измене-
ние длины цепи при любой данной скорости полимери-
зации зависит от природы мономера, а не инициатора.
Так как кОбрЛр/г — эта степень полимеризации мономера,
231
не удивительно, что это отношение в квадрате оказалось
в выражении для длины цепи. Поэтому возникла мысль
о другом методе для вычисления кОбр/Кр!- Если можно
измерить длину цепи v, уравнение ,(15.11) используют
для подсчета кОбР/к'Р‘. Степень полимеризации Р опреде-
ляется теми же методами, что и молекулярные веса вы-
сокомолекулярных веществ * [3]. Если известен механизм
обрыва цепи, степень полимеризации Р можно выразить
через кинетическую длину цепи v, и из уравнения
(15.11) находят значение для Кобр/Кр.
Существует несколько методов, применяемых для
определения механизма обрыва цепи при полимериза-
ции. Один из них — использование радиоактивного ини-
циатора и определение числа его молекул, соединенных
с каждым полимером. Если обрыв цепи происходит в
результате рекомбинации, к каждому полимеру присо-
единяется 1 моль инициатора; в случае диспропорцио-
нирования — 1/2 моль инициатора.
Например,
R-R -> 2R-
R. -» пМ -> R—М„-
R-M„-M„-R
Рекомби- Z4
нация
2R-M„-^
Дис-\
Пропор-4* •
циоиироваииеК—-f- R—М„
Подобные опыты были проделаны для большинства
распространенных мономеров. По большей части обрыв
цепи является результатом рекомбинации, но иногда все
же наблюдается диспропорционирование.
Приведенный метод применим для определения
Ковр/Кр. Для этого степень полимеризации Р находим на
, * Эти методы содержат чувствительные аналитические опреде-
ления числа конечных групп в данном количестве полимера с ис-
пользованием осмотического давления, ультрацентрифугирования
или рассеянного света.
232
основе молекулярного веса полимера. Далее, если обрыв
цепи осуществлен рекомбинацией, можно записать
P=2v, * (15.12)
и значение КобР/кр подсчитывают из уравнения (15.11).
Определение значений констант скоростей
До сих пор константы для реакции роста и обрыва
цепи были получены только в виде отношений. То же по-
ложение мы встречаем для мономерных радикалов; из-за
трудности измерения концентраций радикалов отноше-
ние констант скоростей получить легче, чем сами кон-
станты. Но все же такие абсолютные значения могут
быть определены, хотя методы определения довольно
трудны. Детально их рассматривать не будем. Вкратце
можно сказать, 'что измерение скорости и степени поли-
меризации при динамическом равновесии не может дать
абсолютных значений кОбр или кр. Так можно получить
лишь дополнительный параметр, а именно время жизни
радикала, т. е. весь отрезок времени от начала образо-
вания радикальной цепи до ее деструкции. Продолжи-
тельность жизни растущей цепи равна концентрации
радикалов, деленной на скорость деструкции:
т =----[MJs— =--------1----- (J 5.! 3)
2Кр[М.]2 2Кр[М.]с
и из уравнения (15.7)
= (15.14)
2/Ср[/?обр1с
где индекс с относится к концентрациям при стандарт-
ных условиях. Если тс измеряют при состоянии равно-
весия, то отношение £ОбР/кР можно получить из уравне-
ния (15.14). Это отношение в сочетании с известным
значением Кобр/Кр позволяет подсчитать кОбр и кр.
Чтобы определить тс, необходимо произвести измере-
ния и при неравиовесиом состоянии. В этом случае ини-
циирование полимеризации осуществляется фотоактиви-
рованием инициатора. Известно, что при использовании
равномерного освещения быстро достигается равновес-
ное состояние. Если же применять прерывистый свет,
скорость полимеризации колеблется в некоторых преде-
233
лах. Прерывистое освещение получают, вращая твер-
дый светонепроницаемый диск с клинообразной щелью,
помещенный между источником света и реакционным ра-
створом (рис. 15.1). Отношение периодов освещенности
к периодам, когда освещение отсутствует, равно отноше-
нию площади клинообразной щели к размеру стального
диска. Во время мгновенных оборотов изменяется кон-
центрация радикалов [М-] и, следовательно, скорость
полимеризации 7?Обр-
Свет
Реакционная
система
Вращающийся
диск
Рис. 15.1. Схема прерывистого освещения
реакционного раствора.
Рассмотрим два экстремальных положения: когда из-
меняющаяся скорость близка к значению тс и когда эта
скорость очень мала.
При медленном вращении диска скорость полимери-
зации во время освещения 7?Обр с является величиной по-
стоянной, а в темноте равна нулю. Средняя скорость по-
лимеризации равна среднему арифметическому этих
двух величин. Если освещение длится четверть всего вре-
мени, то
р _ ^обрс
^обрср — 4 •
4»
Если скорость вращения диска очень близка к тс, ради-
калы, полученные во время освещения, продолжают су-
ществовать и в темноте, и полимеризация в этот период
не прекращается. Средняя скорость полимеризации как
раз равна скорости реакции, которая получается под
воздействием света, проходящего через реакционный ра-
створ. Допустим, что освещение длится одну четвертую
часть всего времени. Тогда через раствор проходит чет-
вертая часть того света, который может пройти при по-
стоянном освещении, и одновременно расходуется чет-
234
вертая часть общего количества инициатора. С другой
стороны, скорость полимеризации изменяется пропорци-
онально корню квадратному из значения скорости обра-
зования радикалов [см. уравнение (15.8 6)], и, следова-
тельно, средняя скорость полимеризации
_ ^обрс _ ^Обрс
обРср - - 2 ’
(15.15)
Итак, когда скорость вращения диска принимает
значение тс, скорость полимеризации изменяется. Если
эту скорость нанести на график в виде функции от про-
должительности освещения, получают значение тс.
Ниже приведены данные для винилацетата, получен-
ные описанными методами. Период полураспада расту-
щей цепи составляет около 4 сек, и за это время цепь
вырастает в длину до 104 мономерных единиц. Период
полураспада завершается реакцией с бимолекулярной
константой скорости, большей чем ДО7 л • моль~1 • сек~}.
Цепи растут, несмотря на очень большое значение (ко-
нечной константы скорости, так как концентрация ра-
дикалов чрезвычайно низка (около ДО-8 М).
Абсолютные значения констант скоростей при поли-
меризации чистого винилацетата при 25° С [4] сле-
дующие:
Ru .................... 1 • 10~’ моль-л 1-сек 1
/?обрс................. 0,5-10—4 моль-л~1 • сек—1
кобр1кр ............... 3-10-2 л-моль~1 -сек~~1
.................... 4 сек
кобр/кр ............... 3-Ю—»
Кобр................... 1000 л-моль—1 -сек 1
кр .................... 3-107 л-моль~1-сек~1
[М-] . ..............0,4-10“8 моль! л
Сополимеризация
Многие используемые пластмассы часто получают
сополимеризацией двух различных мономеров. Растущий
полимерный радикал можно присоединить к какому-ли-
бо олефину. (При этом легко подсчитать относительную
реакционную способность двух компонентов. Однако не-
обходим анализ системы, чтобы объяснить существова-
ние двух различных радикалов. При сополимеризации
цвух мономеров имеют место следующие реакции:
235
Mr + Mj -> Mr
Mj • -J- Mg -*• М2 •
Мз-4-М! -> Mr
M2- + M2 -> M2-
Скорость
«н [Mr] [MJ
K12 [Mt • ] [M2]
^21 [M2 • ] [MJ
^22 [М2 * ] [М2]
где Mi — первый мономер; M2 — второй, Мх- и Ма — по-
лимерные радикалы, имеющие первый или второй мо-
номер иа конце цепи. Предположим, что (реакционная
способность полимерного радикала зависит только от
конечного мономера, а не от строения всей кщпи. Пред-
положим также, что в радикалах Мх- и М2- существует
динамическое равновесие, тогда
К21[М2-][М1] = к12‘[Мг][М2]. (15.16)
Скорость исчезновения двух мономеров выражена
уравнениями
[Мг ] [MJ + к21 [Mr ] [MJ; (15.17)
at
Iм! • 1 + «22 [М2 • J [M2]. - (15.18)
at
Отсюда можно определить отношения скоростей
, _ К11 . , к22
' 1 — , /2-------------.
«12 «21
Разделив уравнение (15.17) на (15.18), получим <
d [Mi] [Mj] Г «ц [Mi • ] «21 [Мг - ] ~1 .
d[M2] ~ [М2] I «1-2[М1-] + «22[М2-] J ’
Это можно написать в таком виде*
rf [MJ = [MJ г г, [MJ + [М2] 1
d [М2] ' (М2] I [MJ + г2 [М2] ] ’
Из полученного уравнения определяют относительные
скорости образования полимера из двух мономеров в
виде функции от их концентрации Г\ и г2.
Отношение скоростей ri й г2 называется коэффици-
ентом активности мономера. Когда ri> 1, полимерный
радикал, на конце которого мономер 1, реагирует пред-
почтительно с такой же молекулой 1, а йе с мономе-
* Надо умножить на [M2]/«2i[M2-] и подставить выражение
(15.16) в полученное уравнение.
236
ром 2; когда rt< 1, полимерный радикал с концевым мо-
номером 1 реагирует в основном с .мономером 2. В осо-
бом случае, .когда Г1=Г2=1, полимерный радикал реаги-
рует с любым радикалом, независимо от того, какой
мономер стоит на конце полимера. Получают полимер с
двумя мономерами, беспорядочно распределенными
вдоль цепи, причем {количество их зависит только от со-
отношения концентраций 'мономеров в реакционной сме-
си. Если /[ и г2 очень малы, полимер {будет состоять из
чередующихся мономеров 1 и 2.
В табл. 15.3 представлены значения некоторых ти-
пичных коэффициентов активности. Например, в систе-
ме стирол — метилметакрилат оба значения г} и г2
равны 0,5; следовательно, каждый полимерный радикал
реагирует с другой мономерной ячейкой почти в 2 -раза
быстрее, чем со своей. Это можно объяснить полярными
эффектами.
Таблица 15.3
Коэффициенты активности сополимеров при 60° С [5]
м, м2 Г1 Г2
Стирол Метилметакрилат' 0,5 0,5
Стирол п- Нитростирол 0,2 1,1
Стирол п-Хлорстирол 0,7 1,0
Стирол п-Метоксистирол 1,2 0,8
Стирол Акрилонитрил 0,4 0,04
Стирол Винилацетат - 55 0,01
Стирол Этилвиниловый эфир 90 0
Стирол Бутадиен 0,78 1,4
Стирол Винилхлорид 17 0,02
Винилацетат Винилхлорид 0,2 1,7
Винилацетат Метилметак рилат 0,01 20
Рост цепи при полимеризации
В реакциях роста цепи полимерный радикал {взаимо-
действует с веществом, которое оканчивает одну цепь и
начинает другую. Примером служит реакция с участием
донора водорода RH:
M„- + RH — M„—H-J-R-
кпр
R-J-M----------- М- (15.19)
237
Для простоты будем считать, что константа скорости
присоединения R- к мономеру очень велика, и полиме-
ризация не замедляется. В таком случае молекулярный
вес полимера *, образовавшегося в присутствии RH,
уменьшается, а скорость образования полимера остается
неизменной.
Рост цепи, следовательно, ,есть Процесс, при котором
молекулярный вес изменяется без изменения скорости
полимеризации. Количественная теория реакции роста
цепи требует анализа факторов, влияющих на молеку-
лярный вес. Одной из оценок молекулярного веса яв-
ляется степень полимеризации, которую обозначают
следующим образом:
Р = (15.20)
где А — количество использованных мономерных еди-
ниц; В — образованные полимерные молекулы.
Ограничимся рассмотрением обычного положения,
когда рост цепи происходит без замедления и когда по-
лимеризация оканчивается присоединением. Примером
такого случая является взаимодействие стирола с ак-
тивным донором водорода. Для схемы простой полиме-
ризации
кобр
М- + М — М-
лр
2М •-----* Полимер
степень полимеризации может быть получена из уравне-
ния (15.20)
__к-обр [М • ] [М] __ Кобр [М]
к0[М-]2 кр[М.]
Однако представим, что реакция присоединения
(15.19)—это другой путь образования полимерных мо-
лекул. Тогда степень полимеризации
р _ кобР[М.] [М]__________
Kp[M'l2 + «Pocr[M.][RH]
* Если реинициирование медленное или если реакции обрыва
цепи, включающие первичные радикалы, становятся заметными,
тогда, рост цепи замедляет реакцию образования полимера, при
этом также уменьшается конечный молекулярный вес радикала.
238
Используя уравнение (15.7) и не учитывая радикальных
концентраций, превращаем это уравнение в
1 _ mm-j . ЯрОСТ JRH] _ Кр #0бр с [RH]
р Кобр [М] + Кобр [М] - к02бр [М]2 + RH [М] ’
где константа роста цепи С есть отношение Крост/Добр для
обычного агента роста цепи RH. Действительно, все со-
ставные части, присутствующие в реакционной смеси.
Рис. 15.2. Рост цепи полистирольно-
го радикала в присутствии углеводо-
родных растворителей при 60° С [6].
как мономеры, так и полимеры, могут выполнять роль
агентов роста цепи. При исследовании роста цепи не-
значительным увеличением полимера можно прене-
бречь. Но рост мономера очень велик. Следовательно,
уравнение роста цепи можно записать
1 Кр °обр ,
— = —J----------1- Сумма всех реакции роста цепи =
Р кобр [М]2
= +См И' , (15.21)
Кобр [М]8 Iм!
239
где С’м — константа роста цепи мономера; Са— констан-
та .роста цепи для любого растворителя или агента, в
присутствии которого происходит присоединение, и
[И] — молярность агента роста цепи.
Если агент роста не инициатор, его присутствие не
изменяет скорости полимеризации и первые два члена
уравнения (15.21) являются константами. В этом случае
уравнение роста цепи принимает вид
1 ± п [И]
— — const 4- Си L .
Р ГМ]
(15.22)
На графике изменение степени полимеризации как
функция отношения молярных концентраций агента ро-
ста и мономера выражено прямой 'линией, угол наклона
которой соответствует константе роста. Рис. 15.2 дает
данные для ряда углеводородов, используемых в качест-
ве растворителей при полимеризации стирола.
Таблица 15.4
Константы роста цели при 50° С [7]
Агент роста Константы роста радикалов из
стирола винилацетата
Бензол 0,6-10-6
Толуол 1,6-10-5 —
Изопропилбензол 18-10-5 -—
Циклогексан 0,4-10-5 70-10-5
Дифен илметан 28-10-5 - -—
Трифен илметан 65-10-5 —
Изопропиловый спирт 8-10-5 -—
Пропионовая кислота 0,5-10-5 —
Масляный альдегид 57-Ю-6 6500- Ю-s
Ацетон 5-10-5 120-10-5
Анилин 20-10-5 . —
Бутилхлорид 0,4-10-5 —.
Бутилиодид 18-Ю-6 -—
Метилдисульфид 940-Ю-5 -—
Хлороформ 5-10-5 1,3-10-2
Четырехбромистый углерод 2,2 ~40
Четыреххлористый углерод 1,1-10-2 —
Пеитафенилэтан 2,0 —
Бутантиол 22 48
Сероводород ~5 —
Стирол 8-Ю-6 —
Винилацетат — 22-10-5
240
Характер роста цепи, показанный на рис. 15.2, мо-
жет быть предсказан. Изопропилбензол является луч-
шим агентом роста, чем этилбензол, который, в свою
очередь, лучше, чем толуол. Скорость отрыва водорода
полистирольным радикалом при росте цепи: 3°>2°>1°.
В табл. 15.4 'представлены константы роста цепи. Боль-
шая часть углеводородов является плохими агентами
роста. Из данных, приведенных в таблице, видно, ,что
тиолы, четырехбромистый углерод и пентафенилэтан —
очень активные агенты роста цепи.
Упражнения
15.1. Используя данные табл. 15.3, рассмотрите строение
а) сополимера стирол-акрилонитрил,
б) сополимера стирол-винилацетат.
15.2. Изобразите резонансную структуру для переходного состо-
яния, которая объясняет чередование мономеров в стирол-метилме-
такрилатном сополимере.
15.3. Таблица 15.4 дает константы роста для ряда веществ. На-
пишите _уравнение химического процесса, представляющего наибо-
лее подходящий механизм для роста цепи с помощью толуола, изо-
пропилового спирта, бутантиола, СВг4 и метилдисульфида.
15.4. При использовании радиоактивного АИБН в качестве ини-
циатора образец стирола полимеризовался в среднем до степени
полимеризации 1,0-Ю4. Активность АИБН равна 1,0-Ю8 им-
пульс/(мин-моль) по жидкостному сцинтилляционному счетчику.
Принимая, Что 1 г полистирола имеет активность 100 импульс/мин,
покажите, что обрыв цепи носит характер присоединения.
15.5. Используя уравнение иа стр. 236, которое описывает
сополимеризацию, подсчитайте состав полимера, образованного, ког-
да полимеризация прошла на 1%, ,если концентрации Mi и М2 рав-
ны: а) Г1=г2=1; б) Г1=0,01, г2=10; в) г1=г2=0,01; г) ri =
= г2 = 10.
15.6. Полистирол, полученный нз чистого стирола при 60°С тер-
мической самоинициированной 'полимеризацией (т. е. без введения
инициатора), имеет степень полимеризации 1,0-Ю4 и скорость поли-
меризации 2 • 10-6 моль • л~1 сек~'. Найдите скорость и степень по-
лимеризации для 50%-ного (по объему) раствора стирола в толуоле
(435 г толуола и 450 г стирола на 1 л). Предположите небольшую
степень превращения в полимер. Повторите расчет для 0,1%-иого
(по объему) раствора СВг4 в стироле.
Используйте плотность 3,42 г/мл для СВг4 и 0,90 г/мл для сти-
рола и сделайте предположение, что объем при смешении не изме-
нится.
15.7. Полимеризация стирола может быть инициирована пере-
кисью mpem-бутила при 60° С. Если используется раствор 1,0 М
перекиси и 7,0 М стирола, скорость полимеризации равна
1,05 10-5 моль • л-1 • сек.-' и степень полимеризации 3,23 • 103. Под-
считайте константу роста цепи для трет-бутиловой перекиси. Ис-
пользуйте значение для См, данное в работе [8].
241
ЛИТЕРАТУРА
1. Stille J. К., Introduction to Polymer Chemistry. John-Wiley
and Sons, Inc., N. Y., 1962, pp. 149—180.
2. В u r n e 11 G. M. Mechanism of Polymer Reactions. Interscience
Publishers, Inc., N. Y., 1954, pp. 223, 234.
3. M о о r e W. J. Physical Chemistry. Prentice-Hall, Inc., Engle-
wood Cliffs, N.Y., 3d. ed., 1963, chap. 19.
4. К wart H., Broadbent H. S„ В a r 11 e 11 P. D. J. Amer.
Chem. Soc., 72, 1060 (1950); Bengough W. 1., Melvil-
le H. W. Proc. Roy. Soc. (bond.), Ser. A230, 429 (1955).
5. Young L. J. J. Polymer Sci., 54, 411 (1961).
6. Gregg R. A., M а у о F. R. Disc. Faraday Soc., 2, 328 (1947).
7. Henrici-Olive G., Olive S. Fortschr. Hochpolymer.
Forsch., 2, 496 (1961).
8. P г у о r W. A., Lee A., Witt С. E. J. Amer Chem., Soc. 86,
4229 (1964).
Дополнительная литература
Bamford С. H. et al. Kinetics of Vinyl Polymerization by Radical
Mechanisms. Butterworth Scientific Publications, London, 1958.
Bevington J. C. Radical Polymerization. Academic Press Inc.,
N.Y., 1961.
В i 11 m e у e r F. W. Jr. Textbook of Polymer Science. Interscience
Publishers, John Wiley and Sons, Inc., N. Y., 1962.
Burnett G. M. Mechanism of Polymer Reactions. Interscience Pub-
lishers, inc.j N. Y„ 1954.
D’Al el io G. F. Fundamental Principles of Polymerization. John
Wiley and Sons, Inc., N. Y., 1952.
Flory P. J. Principles of Polymer Chemistry. Cornell University
Press. Ithaca, N. Y., 1953.
Stille J. K. introduction to Polymer Chemistry. John Wiley and
Sons., Inc , N. Y., 1962.
' Глава 16
ЗАМЕЩЕНИЕ В АРОМАТИЧЕСКОМ ЯДРЕ
Замещения в ароматическом ядре могут протекать
по Двум разным механизмам: радикальному и ионному.
Причем в зависимости от механизма получают разные
продукты. В случае ионного замещения исходное веще-
ство может иметь или электронодонорные Труппы, та-
кие, как алкил или алкокси, которые ориентируют за-
мещение в орто- и пара-положения и увеличивают ско-
рость замещения, или электроноакцепторные группы,
такие, как нитро, которые ориентируют замещение в
жеша-положение и дезактивируют ядро. В радикальных
процессах этих правил нет. Например, гомолитическое
242
замещение фенильными радикалами, полученными из
перекиси бензоила протекает быстрее в нитробензоле и
толуоле, чем в самом бензоле. Как нитробензол, так и
толуол замещаются главным образом в орто-положе-
ние. Разница в двух механизмах замещения показана
ниже на примере хлорбензола:
Нитрование Ыоф
из HNO3— H2SO4
31%
Cl
no2
69%
Cl Cl Cl Cl
I | Ph | |
Фенилирование
I II---------------•-> I II I II I II
Ч/ из перекиси \/ \/\ \/
бензоила; . ।
Ph
50% 32% 18%
Нитрование хлорбензола — это типичная реакция
ионного замещения. Ион NO^ воздействует на заме-
щенный бензол с образованием главным образом пара-
изомера. Галоген является дезактивирующим заместите-
лем (хотя ориентирует в пара- и орто-положения), и
поэтому хлорбензол реагирует в 33 раза медленнее бен-
зола. При фенилировании хлорбензола образуется глав-
ным образом орто-изомер, и хлорбензол реагирует
почти в 1,4 раза быстрее бензола.
Предположение о том, что имеются два возможных
механизма замещения в ароматическом ядре, сделал
впервые Гей в 1934 г., а в 1937 г. Гей и Ватере детально
описали ионный и радикальный механизмы, а также от-
метили—-наибольшую вероятность радикального пути.
Несмотря на проведенную огромную исследовательскую
работу с тех пор, механизм гомолитического замещения
в ароматическом ядре все еще не совсем ясен; труд-
ность заключается в том, что монозамещенные продук-
243
ты обычно получаются вместе с труднообрабатываемой
маслянистой массой. Лишь недавно появились 'физиче-
ские методы, позволяющие анализировать реакционную
смесь. В более 'ранних работах вещества Идентифициро-
вали после выделения. Обычно пара-изомер, наименее
растворимый из всех продуктов реакции, кристаллизо-
вался лучше всего. Поэтому в прежних Исследованиях
выделяли главным образом именно этот изомер. Совре-
менные методы анализа |(спектроскопия или газовая
хроматография) показали, что наряду с 'пара-изомером
получаются и другие изомеры.
Введение фенильного остатка
Из гомолитических реакций замещения наиболее хо-
рошо изучено фенилирование. Фенильный радикал мож-
но получить несколькими .методами:
О О
II II
Ph—С—О—О—С—Ph -> 2PhCO2- -> 2Ph- + 2СО2 (16.1а)
N=O
Ph—N—СО—СН3 -> Ph • + N2 + CH3CO2 • (16.16)
Ph—N = N—CPh3 -> Ph • + N2 + Ph3C - (16.1b)
PhN^СГ + NaOH -> Ph- + N2 + NaCl + OH- (16.lr)
Ph-
Ar—H — Ar—Ph (16.2)
Казалось быг использование как самих свободных фе-
нильных радикалов, так и любых их источников должно
привести к одному и тому же эффекту р отношении
полученных изомеров и скорости процесса. Это пред-
положение еще недостаточно изучено, и однозначного
решения вопроса пока нет. |
Рёакция (16.1 а) представляет собой общеизвестное
разложение перекиси бензоила. Когда такое разложе-
ние происходит в ароматических растворителях, наблю-
дается также и замещение в ядре молекулы раствори-
теля. Таким образом, фенильный радикал воздействует
244
на растворитель. Кроме того, до некоторой степени мож-
но себе представить образование двух других возмож-
ных продуктов. Например, для растворителя Аг — Н
можно написать
Ph-
Аг—Н-------- Аг—Ph + Ph—Ph-j-Аг—Ar
В основном Много меньше
Реакция (16.1 б) была открыта Геем и его учениками.
N-Нитрозоацетанилид (ацилфенилнитрозоамин), полу-
ченный нитрозированием ацилфениламина хлористым
ннтрозилом, перегруппировывается in situ в диазоэфир:
N=O О
| Перегруппировка II
Ph—N—СО—СН3----------------> Ph—N = N—О—С—СН3
N- Ннтрозоацетанилнд Фенилдназоацетат
Эта перегруппировка является стадией, определяющей <.
скорость процесса. Далее диазоэфир подвергается быст-
рому гомолизу с образованием фенильных радикалов:
О
II
Ph—N =N—О—С—СН3 -> Ph + N3 + СН3—СО2-
Реакцию (16.1 в) также впервые исследовал Гей. Фе-
нилазотрифенилметан быстро разлагается при 60° С и
дает фенильные радикалы; более стабильный тритиль-
ный радикал не участвует в реакции фенилирования.
В реакции (16.1 г), так называемой реакции Гомбер-
га, используют в качестве- источника радикалов соли
диазония, полученные диазотированием анилина азоти-
стой кислотой. Сильное основание превращает соль ди-
азония (обычно хлорид) в ковалентный диазогидрат,
который при деструкции дает фенильные радикалы. Вся
система гетерогенна: соль диазония превращается в ги-
дроокись в родной фазе, а ковалентная диазогидроокись
диффундирует в органический слой, разрушается, и все
это приводит к фенилированию органического субстрата.
Даже, если выходы составляют от 10 до 40% и реак-
ционная масса осмоляется, реакция представляет инте-
рес из-за доступности исходных материалов.
В табл. 16.1 представлены данные по фенилированию
некоторых субстратов с использованием трех источников
фенильных радикалов. Отмечено, что нитробензол, хлор-
245
Таблица 16.1
Относительные скорости фенилирования
замещенных бензола [1]
X в X — Ph Источник фенила
Перекись бензоила N=O Ph—N—СО—CHS Ph3Bi (фотолиз)
70° С 20° С 80° С
no2 4,0 3,1
н (1,0) (1,0) (1,0)
Cl 1,4 1,5
Br 1,7 1,8
СН3 1,7* 1,8 1,7
* Наблюдается отщепление водорода из метильной
группы почти до 15%.
бензол, бромбензол и толуол реагируют быстрее, чем бен-
зол. В реакции ионного электрофильного замещения элек-
трофильные группы, такие, как NO^, воздействуют на аро-
матическое ядро. Причем электронодонорные заместители
ускоряют реакцию, а электроноакцепторные тормозят
ее. Однако в 'гомолитических замещениях, где речь идет
о нейтральных радикалах, можно ожидать, что 'любые
полярные влияния должны быть много меньше, и как
электроноакцепторные, так и электронодонорные груп-
пы могут стабилизировать стадию роста цепи.
На примере диссоциации замещенных в ядро гекса-
фенилэтанов было доказано, что электронодонорные и
электроноакцепторные группы стабилизируют систему,
содержащую неспаренный электрон:
^равн
Аг3С—САг3 — 2Аг3С- (16.3)
Например [2], константа равновесия Аравн диссоциа-
ции гексафенилэтана при 25°С в бензоле равна 2-Ю-3.
При введении четырех n-метильных групп константа
увеличивается в 7 раз, шести о-метоксигрупп — в
600 раз, а соединение, содержащее шесть п-нитрогрупп,
диссоциировано на 100%.
Из табл. 16.1 видно также, что фенильные радикалы,
полученные из 'перекиси бензоила, нитрозоацетанилида
или фотолизом трифенилвисмута, дают приблизительно
одно и то же отношение скоростей при фенилировании
246
толуола и бензола. (Данные приблизительные, так как
измерения проведены при различных температурах, в то
время как отношения скоростей зависят от Температу-
ры.) Кроме того, в случае толуола наблюдается частич-
ное отщепление водорода из боковой цепи.
В дабл. 16.2 показано распределение изомеров в
реакционной смеси при фенилировании перекисью бен-
зоила. Из таблицы видно, что такая смесь отличается
по своему составу от смеси, полученной методом ионно-
Таблица 16.2
Распределение изомеров при фенилировании С6Н— X с применением
перекиси бензоила при 80° С [3]
X в Изомеры в , %
Свн6 - X
орто мета пара
NO3 62 10 28
Cl 50 32 18
Вг 49 33 18
СН3 67 19 14
го замещения. ,Присутствие как электронодонорных, так
и электроноакцепторных заместителей не отражается на
распределении изомеров.
Механизм фенилировании перекисью бензоила
Между фенильным радикалом и ароматическим суб-
стратом возможны три вида взаимодействия:
РЬ* + АгН
Ph—Аг + Н* (16.4а)
PhH + Аг* (16.46)
(PhArH). (16.4в)
Реакция (16.4а)—это прямой обмен Н* на Ph«;
(16.46) — потеря водорода; (16.4в)—образование ком-
плекса присоединения в результате введения фенильного
остатка в ароматическое кольцо. Из этих трех возмож-
247
ных вариантов реакция (16.46) побочная; она приводит
к образованию в незначительных количествах продукта
Аг — Аг.
Биарильный продукт Аг — Ph можно получить или
в результате прямой обменной реакции (16.4а), или
последовательным двухступенчатым синтезом через ком-
плекс присоединения (16.4в). Разберем подробно двух-
ступенчатый механизм для случая, когда Аг — Н —
бензол:
Ph* + PhH
Ph н
(16.5)
В этой схеме R*—любой радикал, находящийся в
системе, кроме Ph«. Установлено, что бифенил обра-
зуется при взаимодействии перекиси бензоила с бен-
золом. Это двухступенчатый синтез, представленный
уравнениями (16.5) и (16.6). Подсчеты энергий связи
показали, что реакция (16.4а) эндотермична, в то время
как сумма реакций (16.5) и (16.6). экзотермична. Однако
наиболее убедительное доказательство, подтверждающее
двухступенчатый механизм, получено после тщательного
исследования продуктов реакции с помощью современ-
ных аналитических методов.
Было доказано, что при взаимодействии перекиси
бензоила с бензолом (проходит в разбавленном рас-
творе, чтобы уменьшить образование продуктов индуци-
рованного разложения) получается смесь [4J:
О
ph-C-o-o-У-рь-Кнпячение-
0,01 м
о
Phx^H
в беНзол'еСО2+Р11СО2Н+ [I +
1,77 0,08
0,18
Н
Ph — Ph +
0,36
Ph
Ч-Ph—С—О—Ph+
0,03 н
Н =
Н
Ph Н
0,22
РЬ
Н
Н
Н
0.27
Ph
О
248
Цифры обозначают количество молей данного вещества
на моль перекиси. Материальный баланс превосходный,
количество СОг показывает, что (1,77+ 0,08+ 0,03)/2,0,
или 94% перекиси разложилось. Фенильные продукты
получены за счет 82% фенильных групп перекиси, т. е.
I моль перекиси дает 1,63 моль фенильных продуктов.
Характер этих продуктов явно подразумевает обра-
зование в ходе реакции промежуточного комплекса при-
соединения. Комплекс подвергается диспропорциониро-
ванию, присоединению и теряет водород:
Подробное изучение выходов продуктов этих реак-
ций помогает установить механизм процесса [5]. На
рис. 16.1 показана разница в выходах (моль/моль пере-
киси) бифенила, дигидробифенила, бензойной кислоты
и метилбензоата в зависимости от первоначальной кон-
центрации перекиси бензоила. Экстраполируя при бес-
конечном разбавлении, когда разложения быть не мо-
жет, нашли, что выход бензойной кислоты равен нулю,
а выходы бифенила и дигидробифенила равны между
собой. Это позволяет предположить, что при низких
концентрациях перекиси бифенил получается в резуль-
тате диспропорционирования (16.7а). При бесконечном
разбавлении выход бифенила составляет около
249
0,24 моль/моль перекиси; большая часть оставшихся
радикалов превращается в тетрагидрокватерфенилы по
реакции (16.76).
Рис. 16.1. Выход бифенила, дигидробифенила,
бензойной кислоты и фенилбеизоата при реакции
перекиси бензоила с бензолом при 80° С [5].
С увеличением первоначальной концентрации пере-
киси индуцированное разложение становится основным
способом получения главного продукта. Индуцированное
разложение, по-видимому, представляет собой разло-
жение комплекса присоединения:
+ PhCO2H+ PhCO2«
(16.7г)
Если бифенил получали при высоких концентрациях
в результате двух реакций (16.7а) и (16.7г), то выход
его должен быть равен сумме выходов дигидробифенила
и бензойной кислоты. На рис. 16.1 эта сумма представ-
лена в виде пунктирной линии, и она приблизительно
равна выходу бифенила. Реакция (16.7г) представляет
собой передачу водорода от радикалов к перекиси.
Установлено, что именно этот процесс увеличивает ско-
рость разложения перекиси mpem-бутила в некоторых
250
спиртах и аминах [6]. Механизм, по которому проходит
реакция в бутаноле-2, описан ниже:
mpem-C4HBO • + С2Н8—СН (ОН)—СН3 -*•
->mpem-C4HBOH + С2Н8—QOH)—СН3
ОН
С2Н8—С—СН3 + mpem-C4HBOOC4HB-mpem ->
О
II
->С2Н5—С—СН3 + mpem-C4HBOH + mpem-C4HBO •
Реакция комплекса присоединения с бензоатными
радикалами может быть осуществлена также при более
высоких концентрациях перекиси:
Ph
+ PhCO2H
Однако кинетический анализ и другие данные показали,
что за счет этой реакции можно отнести только до 5%
суммы всех продуктов, полученных при взаимодействии
перекиси бензоила с бензолом [5], тогда как в реак-
циях фенилирования замещенных бензола отрыв водо-
рода бензоатными радикалами может стать основным
путем получения бифенила.
На рис. 16.1 видно, что выход фенилбензоата остается
постоянным, в то время как концентрация перекиси
изменяется. Вероятно, в некоторых реакциях образуется
эфир, но ход этих реакций изменяется неодинаково
относительно концентрации перекиси и поэтому их сумма
остается почти постоянной. Можно привести три реак-
ции, в которых возможно образование эфира:
Ph- + PhCO2—CO2Ph + PhCO2Ph + PhCO2- (16.8a)
PhCO2-CO2Ph -+ PhCO2Ph + CO2 (16.86)
R-
PhCO2-+ PhH-> (PhCO2PhH)- —> PhCO2Ph + RH (16.8b)
251
Реакция (16.8а) представляет собой индуцированное
разложение, (16.86)—клеточное разложение и (16.8в) —
двухступенчатый механизм с применением бензоатных
радикалов вместо фенильных. В концентрированных
растворах, несомненно, идет реакция (16.8а) и, веро-
ятно, какое-то количество эфира также образуется в
процессе клеточной реакции (16.86). При обычных кон-
центрациях, однако, эфир образуется в основном по
реакции (16.8в). Это подтверждается тем, что перекись
n-хлорбензоила дает лишь фенил-п-хлорбензоат и не
образует п-хлорфенил-п-хлорбензоата [4, 7]:
0 0 0 0
II II Бензол II II
АгС—О—О—САг----------» PhO—САг + АгО—САг
Образуется Не образуется
(Аг=n-х лорфени л)
Отмечено, что при двухступенчатом получении би-
арила понижается выход бифенила относительно общего
количества комплекса присоединения, которое могло бы
быть получено, так как некоторая его часть превра-
щается в дигидробифенил или в тетрагидрокватерфенил.
Это свидетельствует о трудностях, с которыми прихо-
дится сталкиваться при изучении таких реакций.
При рассмотрении фенилирования замещенных бен-
зола необходимо сделать предположение, что между
относительным выходом ле/па-замещенного бифенила и
относительной скоростью воздействия на мета-положе-
ние имеется некоторая связь. Возможно также, что во
время атаки по мета-положению предпочтительно обра-
зуется продукт димеризации, а не биарил. Например,
на стр. 217 представлены результаты присоединения
метильного радикала к ароматическим соединениям;
подчеркивается, что экспериментальные данные хорошо
совпадают с теоретически вычисленной активностью.
К этим данным надо относиться с осторожностью, так
как образуются и другие продукты помимо тех, кото-
рые удается выделить, и это может исказить опреде-
ление относительной скорости воздействия на любое
положение в ароматическом ядре [8].
В табл. 16.3 представлены относительные скорости
для реакций ряда замещенных фенильных радикалов
с нитробензолом, бензолом и толуолом. п-Нитрофениль-
ный радикал воздействует на эти субстраты в порядке
Ph—CH3>Ph—H>Ph—N02. Следовательно, n-нитрофе-
252
Таблица 16.3
Относительные скорости введения арила в нитробензол и толуол
с помощью различных арильных радикалов при 80° С [1]*
Субстрат
Радикал
Нитробензол 0.9 1,5 4,0 5,1
Бензол (1) (1) ' (1) (1)
Толуол 2,6 1,3 1,7 1,0
* Поправка вводится из-за того, что бензол имеет шесть реакционных поло-
жений, а толуол н нитробензол — только пять.
нильный радикал является электрофильным агентом,
который взаимодействует с фенильным ядром быстрее
при наличии в последнем электронодонорных групп.
Незамещенный фенильный радикал реагирует с толу-
олом и нитробензолом быстрее, чем с бензолом. С самой
большой относительной скоростью п-метилфенильный
радикал атакует нитробензол.
Упражнения
16.1. Фенильный радикал перекиси бензоила реагирует с бензо-
лом с образованием циклогексадиенильного радикала. Этот радикал
должен, как предполагается, подвергаться диспропорционированию,
димеризации, присоединять и обменивать водород. Привести урав-
нение для каждого из этих процессов.
16.2. Обращаясь к рис. 16.1, напишите уравнения, которые пред-
ставляют собой самую сущность взаимодействия перекиси при низ-
ких концентрациях с образованием: бифенила, дигидробифенила,
бензойной кислоты н метилбензоата. Напишите такие же уравнения
и для высоких концентраций перекиси.
16.3. Объясните тот факт, что л-нитрофенильиый радикал реаги-
рует с толуолом быстрее, чем с I нитробензолом, в то время как
/г-метилфенильный радикал быстрее реагирует с нитробензолом.
16.4. В работе [9] проверили предположительный механизм
(16.7), изучили разложение перекиси бензоила в CeH6D. Используя
0,4 Af. раствор перекиси, нашли следующее содержание изомеров:
25% С6Н5—С6Н5; 74% С]2Н9О; 1% С12Н3О2. Объясните образовав
ние каждого продукта.
16.5. Представьте реакцию арилирования вещества Ph—X пере-
кисью бензоила. Могут ли относительные выходы о-, м- и «-заме-
щенных бифенилов не соответствовать относительной скорости, при
253
которой радикалы бифенила воздействуют на о-, м- и /г-положения.
Напяшите реакции, где укажите хотя бы один вариант, по которо-
му фенильные радикалы могут атаковать n-положение быстрее, чем
.«-положение, но с большим выходом будет выделен .и-изомер.
16.6. При арилировании бензола N-нитрозоацетанилидом выход
бифенила много больше, чем выход СОг. Это подтверждает, 1 что
ацетатные радикалы не участвуют в реакции, так как они должны
при этом разлагаться с образованием СО2. Предложите механизм,
включающий индуцированное разложение, по которому получают
бифенил без участия ацетатных радикалов [10].
ЛИТЕРАТУРА
1. Wi 11 i a m s G. H. Homolytic Aromatic Substitution. Pergamon
Press. N. Y., 1960, p. 57; Huisgen R., G r a s h e у R. Ann.
Chem., 607, 46, (1957).
2. W h e 1 a n d G. W. Advanced Organic Chemistry. John Wiley
and Sons, Inc., N. Y., 3d. ed., 1960, p. 774.
3. W i 11 i a m s G. H. Homolytic Aromatic Substitution. Pergamon
Press, N. Y., 1960, p. 68.
4, De T a r D. F. 17th Organic Symposium, June 1961.
5. H e у D. H„ P e r k i n s M. J., W i 11 i a m s G. H. J. Chem. Soc.,
3412 (1964).
6. H u у s e r E. S., В r e d e w e g C. J. J. Amer. Chem. Soc., 86,
2401 (1964); Huyser E. S„ Bredeweg C. J.,
Van Scoy R. M. Ibid., 86, 4148 (1964).
7. L у п c h В. M., Moore R. B. Can. J. Chem., 40, 1461 (1962).
8. Beckwith A. L. J., Waters W. A. J. Chem. Soc., 1665
(1957); В e c k w i t h A. L. J. Ibid., 2248 (1962).
9. Eliel E. L. et al. J. Amer. Chem. Soc., 82, 2936 (1960).
10. Denney D. B., Gershman N. E., Appelbaum A.
Ibid., 86, 3180 (1964).
Дополнительная литература
Augood D. R., Williams G. H. Chem. Rev., 57, 123 (1957).
D e r m e г О. С., E d m i s о п M. T. Ibid., 57, 77 (1957).
Williams G. H. Homolvtic Aromatic Substitution. Pergamon
Press. N. Y„ 1960.
Глава 17
ПЕРЕГРУППИРОВКА
Перегруппировка свободных радикалов (например,
миграция арильного остатки, водорода, галогена или
алкила) редко, но все же встречается в некоторых
системах. По большей части это побочный процесс,
усложняющий состав реакционной смеси. Иногда,
однако, радикальными п'ерегруппировками пользуются
для синтеза нового вещества. Перегруппировка впер-
вые была описана в 1944 г. Ури и Карашем, но лишь
в последнее десятилетие это явление было изучено.
254
Перегруппировки происходят очень часто в системах
с карбониевым попом, особенно при наличии свободных
радикалов, и очень редко в системах, где имеется карб-
анион. Это можно объяснить обычными правилами тео-
рии молекулярных орбит [1]. При перегруппировке
вещества I в вещество III промежуточным звеном
должна быть конфигурация II:
R
I
а—С—С—х —-
I I
б у
I
Энергия активации перегруппировки зависит от раз-
ницы энергий соединения I и промежуточного положе-
ния II. Трехатомная структура II имеет три орбиты,
одна из которых связывающая, а две других вырож-
денные, антисвязывающие. Электроны антисвязывающих
а Карбониевый ион (2 электрона) б Радикал (3 электрона) в Карбанион (4 электрона) 1_1_
энергия || Ц н
С С - .С.
Рис. 17.1. Энергетические уровни для треугольной системы
трех орбит.
орбит дестабилизируют систему и увеличивают ее энер-
гию. В случае карбониевого иона имеется только два
электрона, и они могут быть спарены и перенесены на
самую низшую связывающую орбиту (рис. 17.1, а).
В радикале имеется еще один электрон, и он должен
идти на одну из антисвязывающих орбит (см.
рис. 17.1,6). В случае карбаниона два электрона могут
перейти на эти орбиты, и, очевидно, такая система
наименее стабильна из всех трех (см. рис. 17.1, в). (Как
показано на рис. 17.1, в, переходным состоянием для
карбаниона является триплет.)
255
1,2-Арильная миграция
Несомненно, самое большое число радикальных
перегруппировок составляют перегруппировки, где
арильная группа переходит к соседнему атому. Радикал,
подвергающийся перегруппировке, можно получить раз-
личными методами, например взаимодействие фенил-
изовалерианового альдегида с перекисью дает [2]:
С С О С
I R- I И -со I
Ph—С—С—СНО —> Ph—С—С-С- —- Ph—С—С-
С С С
IV
С ’
Перегруппировка |
IV-----------* -С—С—Ph (17.1)
I
С
С сн3 с о
I I I II
IV -|-Ph—С—С—СНО -> Ph—С—СН3+Ph—С—С—С-
i I 1 (17.2)
С СН3 С
С с
•С—С—Ph + Ph—С—С—СНО ->
С С
сн3 с о
I I II
-> Н—С—CH2Ph + Ph—С—С—С-
СН3 - С
Эта система особенно подвержена перегруппировкам,
так как первичный радикал стремится стать более
стабильным третичным радикалом. В чистом альдегиде,
применяемом в качестве растворителя, 60% углеводо-
рода подвергается перегруппировке. В этих условиях
реакция (17.1) идет несколько быстрее, чем (17.2). Если
256
этот механизм правилен, степень перегруппировки за-
висит от концентрации доноров водорода в системе.
В чистом альдегиде наиболее активно отдает водород
сам альдегид. Поэтому с уменьшением- концентрации
альдегида скорость реакции (17.1) возрастает по сравне-
нию со скоростью реакции (17.2), и одновременно растет
перегруппировка. По этой же причине степень пере-
группировки возрастает от 60 до 80%, когда альдегид
разбавляют хлорбензолом до 1 М раствора. При введе-
нии такого активного_донора водорода, как тиол, про-
цент неперегруппированного продукта должен возрасти
за счет уменьшения перегруппировок. Это подтверди-
лось в соответствующих системах.
Следовательно, реакции (17.1) и (17.2) нельзя объ-
единить в одну стадию, где фенил вытеснял бы окись
углерода. Это подтверждается и тем, что для такого ги-
потетического одностадийного процесса не удалось обна-
ружить предполагаемое переходное состояние радикала:
’ ’’Ph’
Не наблюдается
Перегруппировка радикалов наблюдается также при
декарбонилировании альдегидов [3]:
Ph Н Ph
I Перегруппировка I I
Ph—С—СН—СНО----------------> Ph—С-С—Н
Ph R - Ph R
(R=H, CH3)
Циклические альдегиды тоже могут подвергаться
декарбонилированию в процессе перегруппировки [4]:
9 У. Прайер
257
В представленном циклопентановом соединении степень
перегруппировки увеличивается с 63 до 92%, когда
альдегид разбавляется хлорбензолом, чтобы получить
1 М раствор. Перегруппировка уменьшается менее чем
на 2%, если прибавить бензилмеркаптан; этот очень
активный донор водорода образует радикал V прежде,
чем он успевает перегруппироваться.
В приведенных случаях первичный или вторичный
радикал перегруппировывается в более стабильный тре-
тичный. Иногда можно наблюдать перегруппировку
одного первичного радикала в другой первичный. На-
пример, 2-С14-3-фенилпропаналь перегруппировывается
на 4% при 170° С. Добавление тиофенола тормозит этот
процесс, что свидетельствует. о промежуточном образо-
вании радикала [5]:
Иннциа- Перегруп-
* торы * пнровка *
Ph—С—С—СНО----------- Ph—С—С-----------> -С—С—Ph
• ради- । ।
калов I RCHO I RCHO
ф ф
Ph—СН3—СН3 Ph—СН3—СН3
96% 4%
Во всех рассмотренных случаях перегруппировке
подвергались радикалы, образованные в результате
декарбонилирования альдегидов. Однако радикалй, по-
лученные другими способами, тоже могут перегруппи-
ровываться. Например, при обработке хлористого нео-
фила реактивом Гриньяра в присутствии бромистого
кобальта получается смесь, содержащая mpem-бутил-
бензол, изобутилбензол, 2-метил-3-фенилпропен-1, 2,3-ди-
метилстирол, а также димеры. Предложенный ниже
механизм объясняет образование этих веществ [6]:
RMgBr + СоВг3 -> RCoBr + MgBr3
RCoBr -> R- + СоВг
С С
Ph—С—СН3С1 + СоВг -> Ph—С—СН2- + CoBrCl
С С
С С
I Перегруп- |
Ph—С—СН3- ----------> -С—CH3Ph
। пнровка ।
258 С С
с
I
Ph—С—СН2-
I
С
с
с—СН Ph -> ДимеРы и продукты
г 2 диспропорционирования
с
Можно привести пример и ионной перегруппировки.
В результате окислительно-восстановительного процесса
получаются карбкатионы:
R- + CoBr2 -> R+ + СоВг + Вг~
(см. обсуждение окислительно-восстановительных про-
цессов в гл. 11). Приведенная перегруппировка идет
через образование карбониевых ионов, а не радикалов.
Однако интересно отметить, что степень перегруппи-
ровки при этом приблизительно близка к степени ради-
кальной перегруппировки соответствующего альдегида.
Азосоединения тоже могут образовывать ради-
калы [7]:
С Н. НС
II I I - S
С С с с
(Дифениловый эфир
в качестве растворителя)
с н
-> Аг—С—С- -> Продукты
С С
I Перегруппировка
I
С н
I I
•С—С—Аг -> Продукты
С С
Как перегруппированные, так и неперегруппированные
радикалы образуют различные соединения, подвергаясь
реакциям диспропорционирования. Так получают, на-
пример, перегруппированные и неперегруппированные
алканы и алкены. В случае, когда Аг = фенил, 23% ве-
щества перегруппировывается.
9
259
Углеводороды могут также служить источником
радикалов. Если алкилбензолы нагревают до 400—
525° С в присутствии акцепторов водорода, происходит
перегруппировка. Например, бромистые алкилы вызы-
вают перегруппировку изопропилбензола в пропилбен-
зол [8]:
RBr^R-4-Br-
СН3 СН2-
I | Перегруппировка
Ph—СН + Вт- -> Ph—СН + НВг------------->
I I
сн3 сн3
. НВг
->Ph—СН,—СН—СН3 --------* Ph—СН,—СН2—СН3
По всей вероятности, отщепляется и третичный бензиль-
ный водород, но это не приводит к перегруппировке.
На примере изучения перегруппировок арилзамещен-
ных изовалериановых альдегидов получили относитель-
ные скорости миграции замещенных фенильных групп
[9]. Этот процесс можно изобразить так:
[R = Аг—С (СН3)2 -СН2]
260
Основываясь на кинетических данных, можно вывести
следующее уравнение для данного процесса:
[RCHO], (17.5)
(В)
где (Л) и (В) — выходы соответственно перегруппиро-
ванного и неперегруппированного продуктов, %. Пред-
положили, что кз — постоянная величина, независимая
от X, и сравнивали значения «4 для различных замести-
телей. Получены следующие результаты:
Относительная
X 1 константа к,
С1...................... 1,8
Н....................... 1,0
СНа .................... 0,6
СНаО ................... 0,35
Эти константы скорости связаны уравнением Гаммета
при р«1. Следовательно, переходное состояние можно
изобразить с помощью некоторой резонансной струк-
туры
Было высказано предположение, что электроноакцептор-
ные Х-группы стабилизируют распределение заряда и
увеличивают степень перегруппировки.
В приведенных примерах была рассмотрена мигра-
ция радикалов к атомам углерода. Но радикалы могут
мигрировать и к другим атомам. Например, арильные
группы мигрируют к атому кислорода в некоторых
алкокси-радикалах. Ниже показано, как перегруппиро-
вывается гидроперекись [10]:
Ph Ph Ph
| | Перегруппи- I
Ar—С—О—OH -> Ar—C—O---------------> -C-O-Ar
। । ровна ।
Ph Ph Ph
I
Ph
H—C—OAr
(Ar = п-нитрофенил) Ph
261
Замечено, что /г-нитрофенильная группа мигрирует
преимущественно к фенилу. В перегруппировках с уча-
стием карбониевого иона фенил мигрирует более легко,
чем п-нитрофенил.
1,2-Алкильная миграция и миграция водорода
Строгого доказательства миграции 1,2-водорода или
алкильной группы не существует. В тех немногих систе-
мах, где постулировали такую миграцию, образование
полученных продуктов можно объяснить и другим путем.
Совершенно очевидно, что миграция 1,2-арила более
распространена в радикальны.х реакциях, чем миграция
1,2-алкила или водорода; в перегруппировках с карбо-
ниевым ионом эти группы мигрируют в порядке
арил>алкил или водород.
Миграция 1, 2-галогенов
Атомы галогена могут мигрировать; этот вид пере-
группировки был ранее рассмотрен в связи с присоеди-
нением галогеноводородов к олефинам, например при-
соединение бромистого водорода к галогенсодержащим
олефинам. Приведем примеры, когда продукт претерпе-
вает полную перегруппировку [11]:
Вг • + С = С-СС13 -> Вг—С-С—СС13
Вг-С-С—СС13 Перегр--^Вг—С-С-СС12
пнровка
С1
Вг—С—С—СС12 + НВг-> ВгСН2-СН-СНС12 + Вг-
С1
С1
Основываясь на этом механизме, можно предска-
зать, что любой радикал в определенных условиях мо-
жет вызвать перегруппировку. Так, радикальное присо-
единение тиолов или брома также приводит к продуктам
перегруппировки:
262
С = С—CC13^>RS—С—С—СС13 22^1.
пировка
->RS—С—С—СС12 RS—СН2—СН—СНС12
I I
С1 С1
С = С—СС13 Вг—С—С—СС13 Перег^
ровка
-> Вг—С—С -СС12 ВгСН2—СН—СС12
Вг
Инициированное светом хлорирование mpem-бутило-
вым гипохлоритом изопропилового бромида и пропило-
вого бромида приводит в обоих случаях к одному
продукту, а именно к 1-бром-2-хлорпропану [12]:
С—С—С------» С—С—С + Другие продукты '
| mpcm-C4H9OCl | |
Вг С1 Вг
40%
С1
__ 70 Ор |
с-с—с—:-----------.с—с—ср с—с—с
| тр£/п-С4Н9ОС1 | | |
Вг С1 Вг Вг
15% 60%
Реакция бромистого изопропила должна проходить че-
рез стадию перегруппировки:
с-с-с с-с-с Перегруп-п-и->
I j ровка
Вг Вг
трет-
С—С—С----------->
I С4Н9ОС1
Вг
С—С—С 4- трет -С4Н9О •
С1 Вг
цис-транс- Перегруппировки
При рассмотрении присоединения НХ к олефинам
было отмечено, что если присоединение представляет
собой обратимый процесс, может наблюдаться цис-
263
т/шнс-изомеризация. Присоединение иода, бромистого
водорода или тиолов к олефинам — процесс обратимый,
и известны примеры, когда каждое такое взаимодей-
ствие приводит к изомеризации. Например, тиолы изо-
меризуют цыс-бутен-2:
снз\ /СНз сн3сн3
RS- + /С = С< „ RS | ’ |
Hz ХН —С—с-
I I
н н
I I \ /Н
—-t RS—С С- —- /С=с\ + RS-
I I Hz хсн3
н сн,
Очень интересный и подобный этому случай наблю-
дали при присоединении дейтерированного метантиола
к бутену-2 [13]. При —70° С дейтерированный тиол
присоединяется или к цис- или к транс-бутену-2, причем
получается одна и та же смесь изомеров. В этих усло-
виях изомеризация промежуточного радикала происхо-
дит быстрее, чем переход водорода от тиола:
СН3 Н
'уС~СС.
Н СН3
транс
CH3S*
сн3 сн3
'С—С''
н н
цис
|CH3S.
СНз
S .
с— с-'Н
CH3/C CV
Н СН3
Мгновенное
равновесие
радикалов
S
I • ,-сн;
СНз>С Ч
н н
284
ICH3SD
CHr-C—C—H
I I
H CH3
Равновесная смесь
трео-и эритро-изомеров
Однако при прибавлении бромистого дейте,рия два изо-
мерных олефина давали разные продукты:
СН3 Н
>=<
н сн3
транс
СН3 СН3
X
н н
цис
CH3S« CH3S»
CH,
]
S
' Л.-Н
сн3->с-с^
н сн,
и
Очень медленно
во взаимо -
превращающиеся
изомеры
сн3
Быстро DBr
Быстро DBr
965
Итак, когда в системе имеется довольно активный
агент переноса, например DBr, два изомерных проме-
жуточных радикала выделяют раньше, чем они могут
изомеризоваться. Интересно, что DBr не присоединяется;
присоединяется лишь DSCH3. Реакция
Вг • + CHsSD -+ CHgS • + DBr
происходит быстрее, чем взаимодействие Вг« с оле-
финами.
Реакции разрыва и замыкания кольца
Присоединение четыреххлористого углерода к 0-пи-
нену, инициированное перекисью, дает перегруппирован-
ный хлорид с высоким выходом:
•СС13
Тиоуксусная кислота, наоборот, присоединяется с
образованием главным образом неперегруппированного
продукта. Этот более, активный агент переноса замед-
ляет появление первого радикала. Перегруппировка
является реакцией p-расщепления, но в циклической
системе можно получить перегруппированный продукт.
При хлорировании спиропентана наблюдается свое-
образный разрыв кольца [14]:
CH2CJ
Дихлорное производное может получиться в результате
реакции
С1* + сн2—сн2
CI
CI—СН2 СН2. —CI—сн2 СН2—CI
26&
Эта реакция подобна описанной на стр. 184, 185.
Известны и другие системы, которые участвуют в
реакциях p-расщепления, образуя продукты с разомкну-
тыми кольцами. Например, в замещенном пиране про-
исходит разрыв кольца при 125°С в момент реакции,
инициированной перекисью [15]:
^-Расщепление
осн3
• сн2 с—осн3
сн3 СО2СН3
Замыкание кольца — процесс, обратный по отноше-
нию к реакциям p-расщепления. Одним из примеров
замыкания кольца является образование трицикличе-
ского продукта в результате присоединения /г-тиокрезола
к норборнадиену [16]:
Было установлено, что степень замыкания кольца умень-
шается пропорционально увеличению количества при-
бавленного тиокрезола; это свидетельствует о том, что
появление первого радикала можно приостановить при
высокой концентрации агента переноса, пока не про-
изойдет замыкание кольца.
Интересным примером реакции, где есть одновремен-
но и размыкание и замыкание кольца, является разло-
жение азокамфана [17]. Пиролиз при 300° С дает смесь
продуктов, куда относятся некоторые перегруппирован-
ные вещества, приведенные ниже:
267
Почти та же смесь продуктов получается при пиро-
лизе азосоединения
В результате ступенчатого синтеза оно дает второй
радикал, как показано выше.
Межмолекулярный перенос водорода
Концевые радикалы сами могут оборвать цепь и
отщепить водород от четвертого или пятого атомов
углерода от конца цепи:
/Н
—С-С< ,СН2-> — С—С\ /СН3
ХС-С/ ХС—(У
СН3
Движущей силой этой реакции является энергия пре-
вращения первичного радикала во вторичный, а пятн-
или шестичленное квазикольцевое переходное состояние
имеет низший энергетический уровень.
Как будет видно ниже, перенос водорода между
1,4- и 1,5-атомами углерода подтверждает это наилуч-
шим образом. Было постулировано даже стерически ме-
нее выгодное перемещение водорода между 1,3-атомами
углерода во время реакций бутильных радикалов, источ-
ником которых является пентанал [18]. Как можно было
268
ожидать в этом случае, при фотолизе должны быть
получены бутен-1, бутан и октан:
С4Н9-СНО С4Н9-+СО
I
Димеризация и дис-
пропорционирование
Однако на самом деле получили большое количество
пропена. Это можно объяснить 1,3-перегруппировкой:
С—С<
Н .
zCH2^C—С—С—сн3
* ^«Расщепление
с—с=с+-сн3
Промежуточным продуктом является напряженное че-
тырехчленное кольцо, или, по другому механизму, совер-
шается менее вероятный 1,2-водородный переход:
Н
C-C-i—СН2-> С—С—С-СН3->--СН3+С=С-СН3
н н
Миграция водорода 1,4-атомами углерода наблю-
дается в реакциях метильных радикалов с этиленом
[19]. Главным продуктом реакции является пропен.
Предполагаемый механизм реакции-следующий:
. СН3+С=С->СН3—С-С -.2.СН3—с-с-с-с •
сн3—с
сн2 ->сн3—с
ЧС—с
сн3 +
сн3—с =с + -С2Н5
В реакционной смеси содержится также и бутен-1,
полученный в результате 1,5-перегруппировки первичных
гептильных радикалов:
С—С-С-С—С • -Л с—С—С—С—С—С—С • ->
->с—с—с—с—с-с—с->с—с—с=с+с—с—с
269
Приведенный механизм реакции был доказан введением
D3C радикалов. При 300° С пропен содержит 30% СзН6,
20% C3H5D, 30% C3H4D2 и~20% C3H3D3. Это наводит
на мысль об обменных реакциях, при которых выде-
ляются атомы дейтерия. Один из возможных механиз-
мов, включающий ряд быстрых обменов водорода между
1,4- и 1,5-атомами углерода, выглядит так:
CD2—D
СН2 СН2
\н2-сн2
I
cd2
5-Миграция
водорода сн2
1,5-Миграция
водорода
CD2H—СН2—СН2—СН2—CHD
СН2-СН2
1,4-Миграция
водорода]
1 ,4-Миграция
водорода
CD2H—СН2-СН2—сн—ch2d
3-Расщепление
cd2h—сн—сн2—сн2—ch2d
3-Расщепление C2H3D2+
сн2=сн—ch2d
cd2h—сн = сн2 +c2h4d
I M'M1”1CD3-CH—сн2-сн2-сн3
водорода
-> CD3—CH = СН2 +C2HS
Примером использования перехода водорода между
1,5-атомами углерода с синтетическими целями служит
реакция Гофманн-Лоффлера. В результате этой реакции
N-хлорамин превращается в пирролидин при освещении
его кислого раствора с последующей обработкой осно-
ванием [20]:
_ +1 । , Свет или +1 I _ 1,5-Миграция I I
R-N СН3------------>R—N- СН3-----------—-R — N СН2-
/ перекись | водорода /
Н С1 Н НН
I II III
270
+11 +11 +11 +11
R—N CHa-+R—N CH3->R—N CHaCl+R—N- CH3
H^H H^Cl ^'h h
III I IV II
При дальнейшей обработке соединения IV основанием
получается пирролидин:
+1 • I он-
R— N СН2С1----->
н н
I
СН2С1
N----h HCI
Алкокси-радикалы тоже могут оборвать цепь, от-
щепляя водород. Например, миграция водорода между
1,5-атомами углерода имеется в реакциях алкокси-
радикала, полученного из гипохлорита {21]:
I ROC1
V
С1 С
С
р-Расщепление
О 4
II
+сн3—с—сн3
15%
ROCI
С4Н9-С1
55% 15%
271
Упражнения
17.1. Напишите, какие продукты получатся при разложении
двух молекул гипохлорита, изображенных ниже, в CCU пря 80° С.
В обоих случаях происходит перегруппировка [21]:
17.2. Изобразите резонансную структуру, объясняющую, почему
n-нитрофенил будет мигрировать преимущественно к фенилу в пере-
группировке ArCPh2—О«.
17.3. Если mpem-бутилбензол кипятить с mpem-бутиловой пере-
кисью, образуется изобутилбензол. Дайте полный механизм реак-
ции. Какие еще продукты можно было бы ожидать? Какое влияние
окажет добавление бензилового меркаптана [22]?
17.4. Перекись (Ph — СН(СН3)—СН2—СО — О—)2 кипятили с
обратным холодильником в бензоле 5 ч и получили следующую
смесь продуктов (лоль/100 моль перекиси): СО2—158;
Ph — СН (СН3) — СН2— СО2Н — 27; PhCH = СН — СН3— 4;
PhCH2CH2CH3— 8; PhCH(CH3)2—19 и Ci8H22—23. Напишите ме-
ханизм реакции, объясняющий образование этих продуктов [23].
17.5. Выведите уравнение (17.5).
ЛИТЕРАТУРА
1. Streitwieser A. Molecular Orbital Theory for Organic Che-
mists. John Wiley and Sons, Inc., N. Y., 1961, pp. 380, 381; Ro-
berts J. D. Molecular Orbital Calculations. Benjamin, W. A.,
Inc., N.Y., 1962, pp. 118—121; Zimmerman H. E.,
Z w e i g A. J. Amer. Chem. Soc., 83, 1196 (1961).
2. WinsteinS., Seubold F. H. Ibid., 69, 2916 (1947); Seu-
b о 1 d F. H. Ibid., 75, 2532 (1953).
3. C u rt i n D. J., Kauer J. C. J. Organ. Chem., 25, 880 (1960).
4. Wilt J. W„ Philip H. Ibid., 25, 891 (1960).
5. S laugh L. H. J. Amer. Chem., Soc., 81, 2262 (1969).
6. Urry W. H-, Kharasch M. S. Ibid., 66, 1438 (1944).
7. Overberger C. G„ Gainer H. Ibid., 80, 4561 (1958).
8. S laugh L. H., R a 1 e у J. J4. Ibid., 82, 1259 (1960).
9. Riichardt C. Chem. Ber., 94, 2609 (1961); Riichardt C.,
EichlerS. Ibid., 95, 1921 (1962).
10. В artlett P. D. Cotman J. D. J. Amer. Chem. Soc., 72, 3095
(1950).
11. Фрейдл ина P. К., Кост В. H., X о p л и н а М. Я. «Усп.
химии», 31, 7 (1962).
272
12. S к е 11 Р. S., Allen R. G., G i 1 m о u r N. D. J. Amer. Chem.
Soc., 83, 504 (1961).
13. Skell P. S„ Allen R. G. Ibid., 82, 1511 (1960).
14. Ар p 1 e q u i s t D. E., Fanta G. E, Henrikson B. W.
Ibid., 82, 2368 (1960).
15. Huyser E. S. J. Organ. Chem., 25, 1820 (1960).
16. Crist о 1 S.' J., В rind ell G. D., Reeder J. A. J. Amer.
Chem. Soc., 80, 635 (1958).
17. Berson I. A., Olson С. I., Walia I. S. Ibid., 82, 5000
(1960).
18. Kerr J. A., Trotman-DickensonA. F. J. Chem. Soc.,
1602 (1960); Gordon A. S„ McNesby J. R. J. Chem. Phys.,
33, 1882 (I960); Quinn С. P. Trans. Faraday Soc., 59, 2543
(1963).
19. Gordon A. S„ McNesby J. R. J. Cherf. Phys., 31, 853
(1959); 33, 1882 (1960).
20. Wawzonek S„ T h e 1 e n P. Y. J. Amer. Chem. Soc., 72, 2118
(1950); Corey E. J., Hertler W. R. Ibid., 82, 1657 (1960).
21. Greene F. D. et al. J. Organ. Chem., 28, 55 (1963).
22. Pines H., Pillai C. N.J. Amer. Chem. Soc., 82,2921 (1960).
23. Rjckatson W„ S t e v e n s T. S. J. Chem. Soc., 3960 (1963).
Дополнительная литература
Fish A. Quart. Rev. (bond.), 18, 243 (1964).
Ф p e й д л и н a P. K-, К о с т В. H., X op л и н a M. Я- «Усп.
химия», 31, 1 1(1962).
Heusler К., KalvodaJ. Angew Chem. (Intern. Ed, Engl.), 3,
525 (1964).
Wa 11 i n g C. In: P. de Mayo (ed.). Molecular Rearrangements.
Vol. 1. Interscience Publishers, Inc., N. Y., 1963.
Глава 18
САМООКИСЛЕНИЕ
Самоокисление (автоокисление)—это медленное
окисление органического вещества кислородом. Опре-
деление «медленное» означает, что окисление не сопро-
вождается пламенем. Эти процессы повсеместно рас-
пространены и очень важны. К ним относятся: высы-
хание лаков и красок при сушке на воздухе, старение
резин и пластических масс, медленное сгорание орга-
нического топлива и многие промышленные окислитель-
ные процессы, где кислород используют в качестве
окислителя. Кислород представляет собой бирадикал и
не удивительно, что процессы самоокисления имеют
радикальный характер.
273
Наиболее распространенной формой самоокисления
является образование гидроперекисей из веществ с
лабильными водородными атомами:
RH + О2 -> ROOH
Примерами самоокисления может служить окисление
изопропилбензола
СН3 СН3
Ph—С—Н + О2 —Я Ph—С—ООН
сн3 сн3
89 %
тетралина
ООН
и циклогексена
ООН
|П + о2->|/Х||
20%
Эти окислительные цепные реакции включают ста-
дию роста цепи
R-4*O2->ROO- (18.1)
ROO-+ RH->ROOH + R- (18.2)
Иногда при самоокислении получается не гидропере-
кись ROOH, а вещество, отвечающее общей формуле
ROH или ROOR. Например, при окислении бензальде-
гида получают бензойную кислоту
О О
I о, II
Ph—С—Н -Ph-C—ОН
274
В этом случае сначала образуется надкислота, которая
в свою очередь взаимодействует с молекулой бензаль-
дегида
О О
II II
Ph—С—Н + О2 -> Ph—С—ООН
О 0 0
II II II
Ph—С—ООН + Ph—С—Н -> 2Ph-C—ОН
Примером реакции такого типа является окисление
10-фенилксантена, которое в общем виде можно запи-
сать так:
RH - ROOR
Этот процесс многостадийный. Сначала идет ини-
циирование
Инициирование, рост и обрыв цепи
Самоокисление может быть вызвано перекисями,
гидроперекисями, азосоединениями или любыми дру-
гими источниками радикалов. Часто инициирование
275
происходит спонтанно. Наиболее вероятно, что причиной
самопроизвольного инициирования является вынужден-
ный молекулярный гомолиз [1]:
rh + o2->r-+-о—он
Цепные реакции (18.1) и (18.2) могут привести к
трем различным результатам:
2ROO • ->Нерадикальные продукты (18.4)
ROO + R • -> Нерадикальные продукты (18.5)
2R-->Нерадикальные продукты (18.6)
Как правило, реакция (18.1) идет очень быстро и
обрыв цепи наступает главным образом тогда, когда
образуются два радикала ROO» [реакция (18.4)].
Однако взаимодействия, описанные реакциями (18.5)
и (18.6), иногда тоже могут оборвать цепь. Например,
реакция (18.3) является разновидностью процесса (18.5).
Рис. 18.1. Относительное участие трех реакций
обрыва роста цепи при самоокислении этилового
эфира линолиевой кислоты при 45° С [2]:
/—.реакция (18.6); 2 — реакция (18.4); 3 — реакция (18.5).
Поскольку отношение R* к ROO« зависит от концентра-
ции кислорода в системе, то механизм обрыва цепи
(один из трех возможных) будет зависеть от изменения
парциального давления кислорода. На рис. 18.1 приве-
дены данные по окислению этилового эфира линолиевой
кислоты. При очень низком давлении кислорода, когда
процесс (18.1) идет медленно и получаются в основном
радикалы типа R«, реакцию обрыва цепи можно пред-
ставить в виде (18.6). При больших значениях давления
276
кислорода обрыв цепи изображают с помощью меха-
низма обрыва цепи [реакция (18.5)]. Однако, когда
давление кислорода увеличивается до 100 мм, про-
цесс (18.1) идет так быстро, что большая часть радика-
лов представляет собой перекиси и основной реакцией
обрыва цепи является реакция (18.4).
RH
в|
R*
ROOH+R*
RO*+*OH
RH
ROOR
*wl
2RO«
RH
2RO*+O2
или
roor+o2
roh+r* roh+r*
Рис. 18.2. Схема реакций, происходящих при само-
окислении.
На диаграмме рис. 18.2 изображены возможные
продукты самоокисления. Реакция- а — процесс иници-
ирования. Реакция б — самоокисление алкильного ра-
дикала. В дальнейшем радикалы ROO- могут превра-
щаться несколькими способами: в, г и д.
По способу в получают вещества, описанные на
стр. 274. Этот способ входит как составная часть в сту-
пенчатый последовательный синтез (18.1) и (18.2).
Реакция (18.3) служит примером стадии обрыва цепи г.
Обе стадии виг дают перекиси, которые могут под-
вергаться гомолизу при некоторых условиях. Такие воз-
можности представлены процессами е и ж. При гомолизе
образуются алкокси-радикалы, а также спирты.
277
Стадия д протекает при условии, что RH — изопро-
пилбензол. Изучая реакцию д, использовали смесь ме-
ченых кислородных молекул, состоящих из О18—О18
и О18—О18. На основании этой реакции можно предска-
зать, что во время окисления образуется некоторое
количество О16—О18:
R- + O—O-^ROO-
R- _|_O18—O18-^RO18O18-
ROO- + RO18O18- [ROOO18O18R] -*
_>RO •О—O18RO18 • (18.7)
Было обнаружено, что О16—О18 получается во время
самоокисления изопропилбензола. Причем реакция
(18.7) имела место несколько раз (от 1,7 до 2,6 раза)
на каждой стадии обрыва цепи. Большие значения полу-
чены при окислении более длинных цепей. Все данные
приводят к выводу о том, что взаимодействие двух
радикалов ROO- иногда ведет к обрыву цепи, а иногда
к продолжению наращивания ее [3].
Было изучено влияние химической природы R-rpynn
на скорость окисления RH в момент самоокисления при
высоком давлении кислорода. Окисление происходит по
схеме
Катализатор, R,
RH -------—> R •
Константы скоростей реакций (18.1), (18.2) и (18.4)
равны Ki, к2 и кр соответственно. Состояния равновесия
можно выразить так:
= 2кр [ROO-]2,
и, следовательно, при состоянии равновесия
~^/Н] = 'ч [RH] [ROO ] = —LIRH] [7?и]‘/г, (18.8)
dt (2кр) /2
где /?и — скорость инициирования; кр — константа ско-
рости реакции обрыва цепи. Давление кислорода очень
велико, так что обрыв цепи происходит исключительно
за счет реакции (18.4). Если сравнивать скорости исчез-
новения различных субстратов RH при постоянной ско-
278
рости инициирования, то можно вывести соответствую-
щие значения для к21крг из уравнения (18.8). Это отно-
шение выражает окисляемость субстрата, а отношение
Кобр/к*7'характеризует поляризуемость мономера в мо-
мент радикальной полимеризации (см. стр. 229).
В табл. 18.1 даны значения отношения для различных
углеводородов.
Таблица 18.1
Окисляемость углеводородов [3]
RH Отношение кг/Крг RH Отношение «s/Кр !
Тетралин Бензиловый эфир Толуол п-Нитротолуол 3,0 7,0 0,015 0,0063 п-Ксилол Этилбензол Кумол (изопропилбензол) 0,052 0,18 1,0
Отмечено, что при окислении толуола электроно-
акцепторные заместители, такие, как п-нитрогруппа,
замедляют окислецие, а электронодонорные —убыстряют
его. Имеется значительно больше данных, чем представ-
лено здесь. Например, производные кумолов подчи-
няются уравнению Гаммета при использовании <т+ и
р=—0,4 [5]. Перекись представляет собой электро-
фильный радикал и атакует углеводороды в местах
относительно большей электронной плотности. Можно
написать резонансные структуры промежуточных ве-
ществ реакции (18.2):
ROO • Н: R<—->ROO:— H+R
Можно ожидать, что электронодонорные группы в
R-заместителях стабилизируют переходное состояние и
скорость реакции.
Из табл. 18.1 видно, что скорость окисления умень-
шается в порядке: кумол >этилбензол>толуол. Следо-
вательно, отрыв водорода перекисным радикалом в ре-
акции (18.2) обычно происходит в таком порядке:
3°>2°>1Х
Абсолютные константы скоростей для стадий можно
получить с помощью прибора с вращающимся сектором
и другими подобными методами. Константа скорости
для реакции (18.1) — величина порядка 10е, а энергия
279
активации меньше 2 ккал!моль. Энергия активации
реакции (18.2) , значительно больше, порядка 8—
12 ккал!моль. Например, для тетралина при 25° С кон-
станты скоростей реакций (18.1), (18.2) и (18.4)
(стр. 274, 278) имеют значения, л • моль~1 • сект1 [6]:
х. К1 = 6,7-107
к2 — 13
v кр = 2-107
Интересные продукты можно получить иногда со-
окислением двух веществ. Особенно наглядным приме-
ром процесса такого рода является система: тиоуксус-
ная кислота — инден [7]. В отсутствие кислорода при-
соединение тиоуксусной кислоты к индену происходит
медленнот"Однако когда на раствор тиоуксусной кис-
лоты и индена действуют окислителем, окисление про-
ходит довольно быстро даже при комнатной темпера-
туре:
Упражнения
18.1. При самоокислении изопропилбензола (кумола) получают
повышенные выходы гидроперекисей, если такое окисление происхо-
дит в присутствии основания. Почему? (Иметь в виду, что гидро-
перекись подвергается автокаталитическому, ионному взаимодейст-
вию в присутствии кислоты.)
18.2. Изобутан стабилен в присутствии кислорода при 163° С.
Однако кислород, содержащий около 8% бромистого водорода,
280
быстро окисляет изобутан. При этом получается 70% mpem-бутило-
вой гидроперекиси и небольшое количество mpem-бутилового спирта
и mpem-бутиловой перекиси. Дайте механизм этой реакции.
ЛИТЕРАТУРА I
1. Tipper С. F. Н. Quart. Rev. (Lond.), 11, 318 (1957);
S е a k i n s M., Hinshelwood C. Proc. Royal. Soc. (Lon-
don), Ser. A276, 324 (1963).
2. Bateman L. Quart: Rev. (Lond.), 8, 147 (1954).
3. В a r 11 e 11 P. D., T г а у 1 о r T. G. J. Amer. Chem. Soc., 85,
2407 (1963).
4. R u s s e 11 G. A. Ibid., 78, 1047 (1956).
5. Russell G. A., Williamson R. C. Ibid., 86, 2357 (1964);
Howard J. A., Ingold K. U. Can. J. Chem., 41, 1750 (1963).
6. В a mJ ord С. H., Dewar M. J. S. Proc. Roy. Soc. (Lond.),
Ser. Al98, 252 (1949).
7. О s w a 1 d A. A., GriesbaumK-, N a e g e 1 e W. J. Amer.
Chem. Soc., 86, 3791 (1964).
Дополнительная литература
В о 11 a n d J. L. Quart Rev. (Lond.), 3, ] (1949).
Fr ank С. E. Chem. Rev., 46, 155 (1950).
Russell G. A. J. Chem. Educ., 36, 111 (1959).
Tipper C. F. H. Quart. Rev, (Lond.), 11, 313 (1957).
Walling C. Free Radicals in Solution. John Wiley and Sons..
Inc., N. Y„ 1957, chap. 9.
i Глава 19
БИРАДИКАЛЫ
Бирадикалы — это соединения, имеющие два неспа-
ренных электрона. Как и можно ожидать, такие, до
некоторой степени экзотические, соединения дают уни-
кальные реакции. Часто полученные из бирадикалов
продукты нелегко синтезировать другими методами.
В этой главе будут рассмотрены реакции соединений
типа -СНг — СН2— СН2— СН2— СН2-, где два неспа-
ренных электрона размещаются на большом расстоянии
друг от друга. Такие вещества являются бирадикалами,
и они должны иметь химические свойства, подобные
свойствам монорадикалов. Их спектр содержит два дуб-
лета, каждый из двух неспаренных электронов имеет
спин 3=1/2 и мультиплетность 23+1=2. Будут рас-
смотрены также реакции соединений, в которых два
неспаренных электрона расположёны довольно близко
друг к другу, так что электронные спины сопряжены.
Как было отмечено ранее (см. стр. 63), подобные сое-
281
динения находятся в триплетном состоянии («трипле-
ты»). Вопрос о том, до какой степени свойства три-
плетов будут похожи на свойства бирадикалов, довольно
сложен. Разберем его на примере реакций метилена.
Самым простым примером триплета является моле-
кула кислорода О2. Кислород парамагнитен и имеет
пару электронов с параллельными спинами:
(t) (t).
:6—О:
Он является триплетом, потому что имеет внешнюю
вырожденную электронную орбиту (т. е. имеет два
уровня одинаковой энергии). По правилу о максимуме
мультиплетности в таком состоянии электроны зани-
мают разные уровни и имеют параллельные спины.
Подобная ситуация наблюдается в метилене* СН2:
Структуру, при которой обособленная пара электро-
нов имеет антипараллельные спины, будем называть
«синглетом». Синглет обладает большей энергйей, чем
триплет, и имеет следующее геометрическое строение:
Заполненная
р- орбита
/Н.
103’Г
Sp’-СвЯЗки
(1,1 ЗА)
ч5/>2-Ррб4га, содер-
жащая 2 слррен-
ных электрона
Сингле’
В основе строения метилена лежит триплет, геомет-
рическая конфигурация которого следующая:
Триплет
2 р-Орбиты,
взаимо пер-
пендикуляр-
ные и содер-
Н----q".---н жащие один
х электрон
sp-Связи
(1.03Л)
* Производные метилена часто называют карбенами.
282
Общий метод получения метилена — фотолиз диазо-
метана в газовой фазе:
CHaN2-~CH2: + N2
Синглет
При этом метилен содержит избыток энергии, погло-
щенной при абсорбции света, и молекула находится не
в основном своем состоянии. Первоначальный метиле-
новый синглет разрушается и дает более стабильный
триплет. Это сопровождается довольно значительным
повышением давления присутствующего инертного ох-
лаждающего газа:
СН3: (синглет) 4- М (инертный газ) -►
-* • СН2 • (Триплет) -j-M
(19-1)
Синтез метилена и его производных
Образование метилена — чрезвычайно эндотермич-
ный процесс, так как должны разрушиться две связи.
Тем не менее все же известны способы получения ме-
тилена. Дигалогенметилены можно синтезировать, ис-
пользуя ионное взаимодействие основания В с галогено-
формом:;
В + Х3СН -> ВН+ + Х3С~ -> X- + :СХ2
В зависимости от того, является X хлором, бромом или
иодом, удаление протона и потеря X происходят или
синхронно, или в две стадии, как изображено выше.
Полученный таким образом дигалогенметилен является
синглетом.
Метилен получают фотолизом кетена или диазоме-
тана:
сн2 = С = О -> СН2 : + СО
CH2 = N = N->CH2 : +N2
Эти реакции, проходящие под воздействием света, —
наиболее распространенный метод получения метилена
и его замещенных в газовой фазе.
Полиметиленовые соединения могут быть получены
фотолизом циклических кетонов:
283
о о
II II
с с-
С^/С-^С^/С-->сн2 сн2+со
с" Сл (СЩ„
или взаимодействием дибромидов с парами натрия
ВгСН2— (СН2)„—CH2Br+2Na->2NaBr+ -СН2—(СН2)„-СН2-
Пиролиз n-ксилола дает п-ксилилол [1]. Если реак-
ционную массу после пиролиза* прикапывать в раство-
ритель, где поддерживается температура —78° С, мо^кно
получить растворы п-ксилилола:
СН3
0,1 сек
сн3
Около 1 000 °C
сн2
Расчеты показывают, что в этом случае синглетное со-
стояние на 12 ккал/моль более стабильно, чем триплет-
ное, и что при комнатной температуре такое вещество
имеет хиноидную структуру, изображенную выше. Ме-
ханизм пиролиза, однако, включает образование га-ме-
тилбензильных радикалов в качестве промежуточных
соединений:
Используя специальные методы исследования, эти мо-
норадикальные промежуточные соединения можно вы-
делить.
284
Реакции метилена
Вероятно, самой распространенной реакцией метилена
является взаимодействие с металлами, что приводит к
исчезновению металлических зеркал. Особенно это ха-
рактерно для теллура, селена, сурьмы и мышьяка. Но
есть металлы, не подверженные этому.
Метилен может отщеплять водород
СН2: +Н2->СН3-+ Н-
Метилен может отщеплять и галоген. Интересным,
но сложным примером такого рода является его взаимо-
действие с четыреххлорйстым углеродом [2]. При облу-
чении диазометана в четыреххлористом углероде полу-
чают 60%-ный выход симметричного тетрахлорида
тетраметнлметана (I). Предполагается, что механизм
этой реакции выглядит следующим образом:
CH2N2-^ :CH2 + N2
: СН2 + СС14 -> • СН2С1 + • СС]3
• СС13 + CH2N2 -> С13С — сн2 • + n2
СН2
С13с—сн2-Перегруппи; ci2c—сн2с1 —Дсс12— ch2ci
ровка
сн2 сн2ск сн2а
| Перегруппировка I CH2N2 z-.rj z^ z^jj z-ч
CC12—CH2C1----------CCI—CH2C1 • <-H2—с—CH2CI
С1
СН2С1 СН2С1
I Перегруппи- |
• СН2—С- СН2С1--------> С1СН2—С—СН2С1 ->
। ровка
С1
сн2а сн2а
CH,N, | ССЦ |
----->С1СН2—С—СН2С1 —> С1СН2—С—СН2С1
СН2- СН2С1
I
285
Механизм реакции предполагает три различных мигра-
ции галогена между 1,2-атомами углерода, как описано
на стр. 262, 263. Хотя этот механизм несколько необы-
чен, трудно представить себе какой-либо другой путь,
объясняющий образование таких продуктов.
Считается, что в 'газовой фазе метилен взаимодей-
ствует с хлористым метилом, главным образом отщепляя
хлор:
СН2: + СН3С1 -> -СНаС1 + СН3-
Между этими двумя радикалами возможны три типа
присоединения, в результате чего получаются три про-
дукта: CH2CI — CH2CI, СНз — СНз и СНз — CH2CI в мо-
лярном отношении приблизительно 1:1:2. Одновре-
менно наблюдается некоторое отщепление водорода [3],
Метилен присоединяется по месту двойных связей.
Если метилен находится в возбужденном синглетном
состоянии, происходит прямое присоединение. Напри-
мер, метилен, полученный при фотолизе диазометана,
присоединяется к цис- и транс-бутену-2 [4]; при этом
образуются два стереоспецифических вещества:
Так как присоединение является стереоспецифичным,
весьма вероятно, что оно одноступенчатое и промежу-
точный бирадикал не образуется. Однако в газовой
фазе при высоких концентрациях инертных газов пер-
воначально образованный метиленовый синглет превра-
щается в более устойчивый триплет перед присоедине-
нием. Затем метилен-триплет присоединяется к олефи-
нам, причем эта реакция нестереоспецифична. Напри-
мер, цис-бутен-2 дает два изомерных диметилциклопро-
пана:
286
СН2»(синглет)-|- At
’СН «(триплет) + М
(19.1)
СН2«(триплет)
СНт—С—СН, г- CHj-C—сн,
2 / \ J Быстро “ / \ 3
г н / Н
•с—СН3 «G-Н
Н Триплеты СН3
(19.2)
Межсистемный
переход
Межсисгемный
переход
CHj-C—сн3 сн—с—сн3
/ н / Н
•С—CHj «с—н
\ \
I1 Синглеты СН3
Быстро! (Быстро
В переходном состоянии в момент присоединения
метиленового синглета к электронной паре в двойной
связи электроны с противоположными спинами могут
соединяться в пару. Это видно из уравнения, где на-
правление электронных спинов показано стрелками:
СН2! <♦♦)
287
Реакция присоединения метилена в триплетном со-
стоянии должна протекать с образованием промежу-
точного продукта — бирадикала:
(1 )-CH2-(t)
Этот радикал необходимо превратить в синглет прежде,
чем произойдет разрыв кольца, и время, требующееся
для такого, изменения мультиплетности, вполне доста-
точно для того, чтобы система из двух стереоизомерных
форм уравновесилась, как показано в реакции (19.2).
Самая характерная реакция метилена — реакция
включения. В этой реакции СНг-группа просто внедря-
ется в С—Н-связь. Метилен, полученный при фотолизе
диазометана, находится в возбужденном состоянии и
внедряется почти произвольно в С—Н-связи. Например,
фотолиз диазометаиа в жидком пентаие при 15° С дает
следующие продукты [5]:
С-С-С—С-С—С С . 49%
С-С-С—С—С-|-СН2: С—С—С—С—С 34%,
С
. С-С—С-С-С 17%
Статистический выход этих продуктов 6:4:2 или
50 : 33 : 17, что очень близко к действительному выходу.
В газовой фазе при высоких концентрациях инертных
растворителей перед реакцией может иметь место дезак-
тивация при соударении. Так достигается несколько
большая селективность.
Хотя прямой фотолиз диазометана в жидкой фазе
приводит к случайным реакциям, большую селектив-
ность можно получить косвенно: фоточувствительным
разложением. Если раствор, содержащий бензофенон и
288
диазометан, облучить светом с длиной волны 3130 А,
возбудится только бензофенон, так как диазометан не
абсорбирует свет с такой короткой длиной волны. Бен-
зофенон принимает свое триплетное состояние, триплет
реагирует с диазометаном, образуя метилен в его три-
плетном состоянии [6]:
Ph2C = О 31-3°^» Ph2C = О* (Синглет)
Ph2C = О* (Синглет) -> Ph2C = О* (Триплет)
Ph2C = О* (Триплет) -|- CH2N2 ->
-> Ph2C = О + CH2Nz (Триплет)
CH2N* (Триплет) -> СН2: (Триплет) + N2
Этот метилен-триплет гораздо более селективен, чем
метилен-синглет. Например, были получены одинаковые
продукты с разными выходами при использовании ме-
тилена, образованного в результате непосредственного
фотолиза, и метилена, полученного при фоточувстви-
тельном разложении. Это видно из приведенной ниже
схемы превращения циклогексена:
Прямой фотолиз: 0,7' 1,0 0,18
Фоточувствительное раз-
ложение: 2-5 1,0 0
Обладающий меньшей энергией метилен-триплет реже
вступает в реакции высокоэнергетического включения
и подвергается перегруппировкам и дает .более простое
присоединение к двойной связи.
В этой связи надо вновь обратить внимание на раз-
ницу в химических свойствах синглета и триплета (см.
обсуждение на стр. 74). Когда два непарных электрона
в модели отстоят друг от друга на большое расстояние,
спектр показывает наличие двух дублетов, и, как
10 У. Прайер
289
можно было бы ожидать, такие модели реагируют по-
добно свободному радикалу. Когда же два непарных
электрона расположены близко друг к другу, картина
менее ясная. Для метилена оба состояния электронов
хорошо охарактеризованы: одно состояние — это три-
плет с преобладанием бирадикального строения, а
другое — синглет, который не является бирадикалом.
В этом случае хотелось бы радикальноподобные реак-
ции (например, нестереоспецифичное присоединение к
олефинам и отщепление водорода) приписать исключи-
тельно триплетному состоянию, а нерадикальные про-
цессы (например, реакция включения) — синглетному
.состоянию. Однако еще не доказано, так ли это. Не-
стереоспецифичное присоединение к олефинам, по всей
вероятности, действительно можно приписать триплет-
ному состоянию, так как образование описанных про-
дуктов идет через бирадикальные промежуточные. Од-
нако метилен-синглет может присоединяться и стерео-
специфично. Кроме того, некоторые возбужденные
синглеты могут взаимодействовать, отщепляя атомы
водорода. Этот вопрос сейчас исследуется [7].
Упражнения
19.1. Отметьте различие между метиленом в его синглетном и
триплетном состоянии по следующим пунктам:
а) энергия; 6) гибридизация несвязанной электронной пары;
в) взаимодействие с двойными связями; а) взаимодействие с С—Н-
связями.
19.2. Фотолиз кетена в газовой фазе в присутствии других газов
изучали как функцию от используемой длины волны света. Исполь-
зуя стереоспецифическое присоединение к цис-бутену-2 в качестве
опытного образца, измерили фракцию а, которая получается из син-
глета в отличие от триплета. При 2650 А в присутствии инертных
газов величина а = 0,98; при длине волны от 3460 до 3820 А величи-
на а<0,5. Предложите механизм фотолиза кетена при длинных и
коротких волнах, который объяснил бы эту разницу. Обратите осо-
бое внимание на соединения, подверженные межсистемному перехо-
ду и реакциям, где получается непосредственно метилеи-триплет [8].
ЛИТЕРАТУРА
1. Szwarc М. Nature, 160, 403 (1947); ErredeL. A., Land-
rum В. F. J. Amer. Chem. Soc., 79, 4952 (1957).
2, Urry W. H., Eiszner J. R. Ibid., 74, 5822 (1952);
Urry W. H„ Bilow N. Ibid., 86, 1815 (1964).
3. Setser D. W., L i 11 r e 11 R., Ha ssler J. C. Ibid., 87, 2062
(1965).
290
4. SkellP. S„ Wood worth R. C. Ibid., 81, 3383 (1959);
A n e t F. A. L., В a d e г R. F. W., Van der Au wer a A. M.
Ibid., 82, 3217 (1960); Frey H.-M. Ibid., 82, 5847 (1960).
5. Doe ring W. E. et al. Ibid., 78, 3224 (1956).
6. Kopecky K. R-, H a m m о n d G. S., Leer makers P A.
Ibid., 84, 1015 (1962).
7. D e More W. В, В e n s о n S. W. Advances in Photochemistry.
Vol. 2. Interscience Publishers, Inc., N. Y., 1964, pp. 219—261;
McGlynn'S. P, Smith F. J., Ci 1 ent о G. Photochemist-
ry and Photobiology. Vol. 3. Pergamon Press. N. Y., 1964,
pp. 269—294.
8. Ho S., Unger I., Noyes W. A., Jr. J. Amer. Chem. Soc.,
87, 2297 (1965).
Дополнительная литература
De Mo re W. В., В e n s о n S. W. Preparation, Properties and
Reactivity of Methylene. In: W. A. Noyes, G. S. Hammond,
J. N. Pitts (eds). Advances in Photochemistry. Vol. 2. Interscien-
ce Publishers, Inc., N. Y., 1964, pp. 219—261.
Hine J. Divalent Carbon. The Ronald. Press Company. N. Y., 1964.
Hine J. Physical Organic Chemistry. McGraw-Hill Book Compa-
ny, N. Y., 2d ed., 1962, pp. 484—504.
Kirmse W. Carbene Chemistry. Academic Press Inc., N. Y., 1964.
Parham W. E., S c h w e i z e r E. E. In A. C. Cope (ed.), Organic
Reactions. Vol. 13, John Wiley and Sons, Inc., N. Y., 1963,
pp. 55—90.
10*
ЧАСТЬ ЧЕТВЕРТАЯ
РЕАКЦИИ ОБРЫВА ЦЕПИ
Глава 20
РЕАКЦИИ РЕКОМБИНАЦИИ И ДИСПРОПОРЦИОНИРОВАНИЯ
Введение
Реакции обрыва цепи происходят в любой радикаль-
ной системе. Одной из важнейших и характерных осо-
бенностей радикальных соединений является то, что они
способны реагировать между собой попарно; причем в
такую реакцию спаривания не вступают ни карбокатио-
ны, ни карбанионы.
Алкильные радикалы обрывают радикальную цепь
двояко: либо в результате рекомбинации, либо диспро-
порционирования. Реакция рекомбинации в простом
случае является обратной реакции диссоциации:
2R->R— R
При диспропорционировании происходит отрыв ато-
ма водорода из p-положения одного из радикальных
центров:
Н
R • + R2C-CR2 -> R —Н + R2C = CR2
При рассмотрении механизма реакций диспропорцио-
нирования имеется некоторая неопределенность в пред-
ставлении структуры переходного состояния. Известно,
что мигрирует атом водорода в p-положении, поскольку
диспропорционируют два радикала СН3 — CD2«:
D D
2СН3—С- -> СН2 = CD2 + CHg-C—Н
D D
292
Следовательно, надо
ние включает известное
«голова к хвосту»:
D
сн,—с«
I
D
думать, диспропорционирова- .
переходное состояние по типу
D
С»
D
Однако, если это верно, то следует ожидать, что
предэкспоненциальиый фактор Аррениуса А должен
быть одинаковым для диспропорционирования и для
реакции отрыва водорода от молекул алканов. В дей-
ствительности в реакциях диспропорционирования
Л~1010 сек-1, в то время как в обычных реакциях с
выделением водорода величина предэкспоненциального
фактора А гораздо ниже, примерно 108 сек-1. Таким
образом, переходное состояние в реакции диспропор-
ционирования имеет меньшую энтропию, чем в обычной
реакции с выделением водорода. Факторы Аррениуса
имеют одно и то же значение в реакциях как диспро-
порционирования, так и рекомбинации. И оба этих типа
реакций обрыва цепи могут протекать через одно и то
же переходное состояние *.
Так как процесс образования связи экзотермичен,
рекомбинация двй^к радикалов дает молекулу с полной
энергией новой ковалентной связи. Чтобы такая моле-
кула была устойчивой, необходимо отвести избыток
энергии за счет колебательного движения или столкно-
вения со стенкой реакционного сосуда или другой мо-
лекулой.
Рассмотрим реакцию рекомбинации двух радикалов
с образованием первоначально возбужденного соедине-
ния XY *:
X-+Y-^>XY* (20.1)
«2
XY* + M^XY фМ (20.2)
* Одна группа исследователей утверждает, что переходные со-
стояния в реакции рекомбинации и диспропорционирования долж-
ны быть очень похожи и могут быть представлены как четырех-
центровая реакция [1, 2]. Существует и противоположное мнение:
два переходных состояния не тождественны, но обладают одной
общей особенностью ионного характера [3].
293
где М — третий компонент, не вступающий в реакцию,
который дезактивирует XY* при столкновении. В мо-
мент устойчивого состояния соединения XY* скорость
образования стабильной молекулы XY можно описать
выражением
d[XY] k1K2[X.][Y][M]
dt к% + Кз [М]
Здесь могут быть два случая: при малых концент-
рациях М К2»/с3[М] и скорость образования XY равна
Ык2) [X.][Y.][M]; при больших концентрациях
М К2<Скз и скорость уменьшается до K|[X*][Y-] и ста-
новится независимой от концентрации третьего вещества.
Экспериментальные данные подтверждают, что веще-
ство М взаимодействует с метильными, этильными ра-
дикалами, а также с атомами иода. Радикалы же боль-
шой величины всегда рекомбинируют со скоростью вто-
рого порядка. Примером этого является рекомбинация
двух этильных радикалов, которая протекает со скоро-
стью третьего порядка только при очень малых давле-
ниях, но выше 1 мм рт. ст. она становится скоростью
второго порядка. Скорость реакций рекомбинации треть-
его порядка зависит от концентрации третьего вещества
и от его структуры. Например, способность различных
молекул ускорять рекомбинацию двух атомов иода, как
было найдено, возрастает с величиной и усложнением
строения вещества М. ,
Как видно, реакции обрыва цепи являются обычно
бимолекулярными. Кинетическим следствием этого яв-
ляется зависимость скорости процесса в целом от квад-
ратного корня из величины скорости инициирования,
потому что концентрация свободных радикалов в пере-
ходном состоянии равна квадрату их концентрации,
помноженному на величину константы скорости иниции-
рования. Так, для стадий инициирования и обрыва цепи
можно записать
Ли
Инициатор —> 2R •
хи
2R- —> Нерадикальные продукты
Гипотеза устойчивого состояния дает
7?и = 2/ср [R-]2,
L 2кр J
294
Обычно наблюдаемая скорость всего процесса про-
порциональна концентрации свободных радикалов, и,
следовательно, она меняется с изменением величины
корня квадратного из скорости инициирования. Напри-
мер, скорость полимеризации, данная в уравнении
(15.8), зависит от величины квадратного корня из кон-
центрации инициатора.
Абсолютные значения констант скоростей
Измерение скорости рекомбинации метильных ради-
калов обсуждалось на стр. 52. Общепринятой констан-
той скорости является величина к=2-1010 л-моль~1Х,
Хсек-1.
Энергия активации равна нулю при допущении
±0,70 ккал!моль. Эту константу впервые получили Го-
мер и Кистяковский в 1951 г. Они использовали метод
«вращающегося сектора», фотолиз ацетона, дающего
свободные метильные радикалы, и пиролиз диметил-
ртути.
Позднее другие авторы помимо метода «вращающе-
гося сектора» использовали масс-спектрографию и чув-
ствительные манометрические измерения. Все s полу-
ченные результаты оказались удовлетворительными.
Скорость рекомбинации этильных радикалов изме-
рена с меньшей точностью. Наиболее вероятная вели-
чина константы скорости
к = 1,6-1010е(_2000±1000)ЛГ л-моль-'-се/с-' .
Было найдено, что величины энергии активации,
измеренные для рекомбинаций радикалов 2СРз«,
2СС13-, 2NC>2’ и НО« + СНз«, равны нулю.
Рекомбинация разных радикалов
При наличии разных радикалов возможны три слу-
чая рекомбинации:
2А- -Л А А
2В- -IBB
А- +В- -^АВ
295
Как подчеркивалось на стр. 21, статистическая веро-
ятность равна *:
ф,_2.
(каа' Kbb) 2
Величина Ф измерена для большей части простых
алифатических радикалов в газовой фазе методом газо-
вой хроматографии, и было найдено, что она всегда
очень близка к 2. Однако для полимерных радикалов
различных типов зарядов величина Ф часто увеличи-
вается в 100 раз и более. Величины Ф, сильно отличные
от 2, были также найдены для самоокислительных си-
стем, в которых рекомбинации между R« и ROO» ча-
сто предпочтительнее, чем любые другие гоморекомби-
нации. Эти различия в величинах объясняют поляр-
ным эффектом. Для некоторых радикалов величины Ф
приведены в табл. 20.1.
Таблица 20.1
Величины Ф = каЬЦкаак)ьь'Ч2 для реакций рекомбинации
разных радикалов [5]
Радикалы
Радикалы
А В
А В
Метил
Метил
Метил
Метил
Этил
Метокси
Ацетил
Пропил
2,0
1,9
1,7
2,1
Этил
Полистирил
Полистирил
Полистирил
Пропил
Поли(п-метоксистирил)
Поли(метилметакрилил)
Поли(бутилахрилил)
1 ,9
1,0
14
150
Сопоставление реакций рекомбинации
и диспропорционирования
Реакции рекомбинации и диспропорционирования
можно сопоставить - на основании анализа продуктов
этих реакций. Обычно алифатические радикалы изуча-
ют в газовой фазе с применением газовой хроматогра-
* Иногда используют альтернативное определение Ф, в кото-
ром Ф = кОй/2 {каа'кььУ!г- При этом статистически ожидается, что
Ф равно скорее 1, а не 2 [4]. Однако все авторы работ, ссылка на
которые приведены в табл. 20.1, придерживались именно этого оп-
ределения. Читатель должен осторожно подходить к тому, какое
Ф было использовано при сравнении величин, полученных раз-
личными авторами.
296
фин. В табл. 20.2 представлены типичные данные для
сравнения реакций рекомбинации и диспропорциониро-
вания. Неразветвленные радикалы предпочтительней
Таблица 20.2
Соотношения между реакциями рекомбинации и
диспропорционирования [5г, 5д, 6]
Радикалы t, °с кд/хрек
СН3- С2Н5- 25—240 0,04
сн3- СН3СН2СН2- 118—144 0,03
2С,Н5- 25—350 0,1
2СН3СН2СН2- 25—150 0,1
2СН3СНСН, 20—200 0,5
2 (СН3)2СН — СН2- 100 0,4
2СН3 — СН2 — СН — СН3 100 2,3
2(СН3)3С- 100 • 4,6
вступают в реакции рекомбинации, однако чем разветв-
леннее радикал, тем характернее для него реакции дис-
пропорционирования. В основном радикалы с большим
числом атомов водорода в 0-положении имеют большее
отношение кд/крек, где кд—константа скорости диспро-
порционирования, а «рек — константа скорости рекомби-
нации.
Если предположить, что все радикалы, представлен-
ные в табл. 20.2, имеют одни и те же величины крек, тог-
да отношения Кд/Крек представляют собой относитель-
ные скорости диспропорционирования этих радикалов.
Если это предположение правильно, то для разветвлен-
ных радикалов в большей степени должны быть харак-
терны реакции диспропорционирования, чем это вытекает
из статистических соображений. Например, изопропиль-
ный радикал имеет в два раза больше атомов водорода
в 0-положении, чем этильный (шесть против трех), но
соотношение кд/крск для него в 5 раз больше, чем для
этильного радикала. Аналогично трет-бутильный ради-
кал имеет девять атомов водорода, этильный — три;
следовательно, статистически mpem-бутильный радикал
должен иметь в 3 раза большее соотношение кд/кРек-
В действительности оно больше в 46 раз.
Расхождение в каждом из этих случаев можно
объяснить, предположив, что абсолютная константа
скорости рекомбинации становится наименьшей, так
как радикал обладает стерически затрудненной струк-
турой. Введем величину напряжения F (напряжение,
возникающее за счет сжатия, необходимого для обра-
зования связи) для реакции рекомбинации. К сожале-
нию, отсутствуют экспериментальные данные, которые
подтвердили бы статистический расчет, указывающий,
что расхождение в величинах Кд/Крек происходит благо-
даря либо изменению кд, либо крек. Однако, как было
указано выше, метильные радикалы рекомбинируют с
нулевой энергией активации, в .то время как для реком-
бинации этильных радикалов требуется энергия около
2 ккал/моль. Таким образом, величины крек могут воз-
растать по мере усложнения структуры радикала.
Большинство полимерных радикалов преимущест-
венно или полностью рекомбинируют, и соотношение
Кд/крек очень мало. Полимерные радикалы, образованные
из метилметакрилата, однако, вступают в реакцию обо-
их типов. Энергия активации реакции диспропорциони-
рования на 5 ккал/моль больше, чем энергия активации
реакции рекомбинации, и, следовательно, доля диспро-
порционирования в процессе обрыва цепи возрастает
с увеличением температуры.’ При 60°С Кд/крек равно
примерно 1,5 [7].
Константы скорости реакции рекомбинации настолько
велики, что скорость обрыва цепи в жидкой фазе под-
дается диффузионному контролю, т. е. диффузия двух
радикалов в растворе при встрече с каждым другим
свободным радикалом является медленным процессом,
и образование связи происходит, как только встретятся
два радикала. В этих условиях константы скорости об-
рыва цепи зависят от природы растворителя и, особен-
но, от его вязкости. Одним очень нежелательным приме-
ром этого является так называемый эффект Тромс-
дорфа; скорость полимеризации некоторых мономеров
возрастает с увеличением выхода реакции. По мере воз-
растания концентрации полимера в реакционном раст-
воре увеличивается его вязкость, и скорость обрыва
цепи снижается. Следовательно, скорость полимериза-
ции, которая пропорциональна ко^/крг , возрастает.
Абсолютные величины констант скоростей полиме-
ризации винилацетата измерены в зависимости от сте-
298
Таблица 20.3
Абсолютные константы скорости реакции полимеризации
винилацетата при 25° С [8]
Полимеризация, 0/ /0 -z, сек кобр Kp.lO' . “обрЛр^
23 -2,5 1 290 126 0,37
46 -10 1 280 90 0,42
57 -15 555 6,7 0,67
65 -23 87 1,1 0,26
пени полимеризации; эти данные приведены в табл. 20.3.
По мере возрастания степени полимеризации реакцион-
ный раствор становится более вязким, и время жизни т
растущего полимерного радикала увеличивается. При
умеренных скоростях /ср уменьшается, а «Обр остается
постоянной величиной. В таких растворах мономер мо-
жет диффундировать к полимерному радикалу со скоро-
стью, близкой к нормальной. При этом начинает подда-
ваться диффузионному контролю рекомбинация двух
полимерных радикалов. При больших скоростях поли-
меризации уменьшается даже /сОбр- В этом случае спо-
собность мономеров к полимеризации, представляемая
величиной /Собр/кр2, проходит через максимум.
Упражнения
20.1. Для реакции между этильным и пропильным радикалами
можно измерить две величины Кд/Крек, так как в результате диспро-
порционирования может получиться как этилен, так и пропилеи:
CaHs'-f-CgH,---» С5Н12
с2н6-н-с3н,- н2с = сн2 + С3Н8
Кобр
С2Н5 • +Сзн, • —► н2с = сн—сн3 + С2Нв .
Найдено, что ковр/крек=0,081. Сравните эту величину с другими из
табл. 20.2. Приемлемо ли это? Предскажите приблизительную вели-
чину К^/Крек [5 г].
ЛИТЕРАТУРА
1. Bradley J. N., RabinovitchB. S. J. Chem. Phys.,. 36,
3498 (1962).
2. Kerr J. A., Trotman-Dickenson A. F. Progress in Reac-
tion Kinetics. Vol. 1, Pergamon Press, N. Y., 1961, p. 113.
299
3. Benson S. W. Advances in Photochemistry. Vol. 2. John Wiley
and Sons, Inc., N. Y.. 1964, pp. 1 —15.
4. Walling C. Free Radicals in Solution. John Wiley and Sons,
Inc., N.Y., 1957, pp. 145, 244, 419.
5. a. Trotman-Dickenson A. F. Ann. Rept. Chem. Soc., 55,
41 (1958).
б. В u r n e 11 G. M. Mechanism of Polymer Reactions. Inter-
science Publishers. Inc., N. Y„ 1954, p. 292.
в. Kerr J. A., T г о t m a n - D i с к e n s о n A. F. Progr. Reac-
tion Kinetics, 1, 108 (1961).
r. G г о t e w о 1 d J., Kerr J. A. J. Chem. Soc., 4337 (1963).
Д. Grotewold J., Kerr J. A. Ibid., 4342 (1963).
«Trotman-Dickenson A. F. Free Radicals. Methuen and
Co., Ltd., London, 1959, p. 53.
7. В e v i n g t о n J. C. Radical Polymerisation. Academic Press
Inc., N. Y„ 1961, p. 136.
8. Ben go ugh W. L, M e 1 v i 1 1 e H. W. Proc. Roy. Soc. (Lon-
don), Ser. A230, 429 (1955).
Дополнительная литература
Benson S. W. Advances in Photochemistry. Vol. 2. John Wiley
Sons, Inc., N. Y., 1964, pp. 1—24.
Lapporte S. J. Angew. Chem., 72, 759 (1960).
Trotman-Dickenson A. F. Ann. Rept. Chem. Soc., 55, 36
(1958).
Глава 21
ИНГИБИРОВАНИЕ
В последней главе уместно разобрать случаи, в ко-
торых радикальные процессы могут ингибироваться,
замедляться и прекращаться. Большинство радикаль-
ных реакций являются цепными процессами, и введение
в систему относительно небольшого числа свободных
радикалов вызывает образование множества различных
продуктов. По той же самой причине связывание ради-
калов, которые способны зарождать цепи, может пол-
ностью устранить образование этих продуктов. Таким
образом, ингибирование играет существенную роль в
радикальных процессах и порой является нежелатель-
ным последствием в цепных реакциях.
Контрольное использование ингибиторов находит
широкое практическое применение. Например, мономе-
ры, предназначенные для полимеризации, продаются с
добавкой ингибитора, чтобы уменьшить количество по-
лимера, который может образоваться в процессе хране-
ния; этот ингибитор обычно удаляют перед использова-
нием мономера. Примером ингибирования является
300
надежное хранение эфира в бутылях с железной прово-
локой. Эфиры самоокисляются с образованием гидро-
перекисей. Они взрывоопасны, их образование протека-
ет автокаталитически, однако в присутствии железа
количество перекиси уменьшается, а их концентрация
поддается контролю.
Смазочные масла и газолины ингибируются во из-
бежание образования «кислого гудрона» (т. е. нежела-
тельных полимеров). Пластиковые оконные панели со-
держат ингибиторы для предотвращения вредного воз-
действия солнечного света (в результате деструктивного
фотолиза). В литературе каждый год описываются
сотни новых соединений, опробованных в процессе поис-
ков лучших ингибиторов для промышленно важных
продуктов или процессов.
Процессы инициирования и ингибирования связаны
между собой. В большинстве радикальных цепных про-
цессов субстрат S превращается в радикал S-, который
принимает участие в развитии цепи. Инициирование
может начаться с распада инициатора на радикальные
фрагменты:
Инициатор-> 2R • (21.1)
R-+S->S- (21.2)
При малых концентрациях инициаторов образуется
мало радикалов, и эти радикалы R» в основном и за-
рождают цепи [реакция (21.2)]. Однако если концент-
рация свободных радикалов велика и если R- относи-
тельно стабилен, тогда реакции обрыва цепей с уча-
стием R- могут протекать помимо обычной реакции об-
рыва цепи между двумя радикалами S<
R. -р s* — -*-Нёрадикальные.
продукты
2S* —1
Как уже отмечалось в предыдущей главе, полярные
эффекты часто делают перекрестный обрыв цепи (21.4)
особенно предпочтительным. Следовательно, умеренно
высокие концентрации свободных радикалов R- могут
обеспечивать другой путь реакции обрыва цепи, поми-
мо реакции (21.5), и уменьшать скорость всего процесса
в целом.
Эти явления хорошо известны в радикальной поли-
меризации. При высоких концентрациях инициатора
(21.3)
(21.4)
301
образуемые им свободные радикалы (так' называемые
первичные радикалы) обрывают растущие цепи. Эта
реакция носит название первичной радикальной реак-
ции обрыва цепи и приводит к уменьшению эффективно-
сти инициаторов и образованию полимера с более низ-
ким молекулярным весом, чем если бы произошла ре-
комбинация двух полимерных радикалов [1а, б].
Ингибиторами могут быть не обязательно свобод-
ные радикалы. Ими могут служить вещества, реагирую-
щие с радикалами, давая другой радикал, который ста-
билен на стадии начала развития кинетической цепи,
либо является реакционноспособным в какой-либо дру-
гой, новой реакции. Ингибиторами могут быть фенолы,
амины и подобные им соединения с активными атомами
водорода:
S-+ I—Н-> SH + I-
I •-> Новая реакция (21.6)
Хиноны также являются ингибиторами, и можно
представить четыре различные реакции-для объяснения
их действия:
(21.7а)
(21.76)
(21.7в)
(21.7г)
Согласно реакции (21.7а) происходит отщепление
водорода от хинона радикалом R-. Реакция (21.76)
описывает отрыв атома водорода от радикала хино-
ном Q:
Н
/ С—СН2 • + Q ->/С=СН2+(QH) •
По схеме (21.7в) происходит присоединение R- к
углерод-углеродной двойной связи в хиноне, и по реак-
ции (21.7г) —присоединение к атому кислорода хинона.
В качестве ингибиторов могут служить также неко-
торые неорганические соединения окислительно-восста-
новительного характера. Например, хлорное железо ин-
гибирует полимеризацию стирола:
М„- + FeCl3 -> Мл—Cl + FeCl2
Это пример переноса лиганда (см. гл. 11).
Механизмы действия ингибитора достаточно слож-
ны и не совсем понятны. Часто многие факты сами по
себе кажутся удивительными. Например, бензохинон
ингибирует полимеризацию винилацетата и обрывает
одну кинетическую цепь на молекулу; он также ингиби-
рует стирольную полимеризацию, но обрывает (в сте-
хиометрическом отношении) две кинетические цепи на
молекулу. С другой стороны, хлоранил ингибирует по-
лимеризацию винилацетата, но является сополимером
стирола. Все эти процессы относятся к инициированным
реакциям полимеризации. В процессе термической поли-
меризации стирола как хлоранил, так и бензохинон яв-
ляются ингибиторами, но их ингибирующее действие
прекращается при аномально быстрых скоростях.
Иногда такая ситуация является следствием того, что
вначале радикальные системы сложны и ингибиторы мо-
гут вызвать различные дополнительные реакции и об-
разование некоторых новых продуктов. Можно предста-
вить три варианта механизма ингибирования:
1. Ингибирование радикалами. Дополнительный сво-
бодный радикал будет ингибировать процесс, описан-
ный реакциями (21.1) и (21.2), в том случае, если он
ловит S«, но не реагирует с S, превращая его в S«.
2. Перенос атома. Перенос атома сопровождается
ингибированием, если агент-переносчик образует ста-
303
бильный радикал, который не вызывает реинициирова-
ние. В реакциях радикальной полимеризации такой
процесс называется деградативным переносом. В этом
случае агент-переносчик можно считать ингибитором.
Примером может служить перенос дифенилпикрилгид-
разином (ДФПГ), который образует весьма стабильный
радикал ДФПГ на стадии переноса:
Мл • + ДФПГ—И -X Мл—И + ДФПГ •
ДФПГ- + М^Хм„.
Так как кПр меньше кОбр, то происходит ингибирование.
3. Присоединение. Некоторые вещества присоединя-
ют свободные радикалы с образованием1 стабильных
соединений, которые препятствуют распространению
кинетической цепи. Примером является ингибирование
кислородом радикальной полимеризации:
мл- + о2->млоо-
Кинетика
Наиболее общей схемой реакций ингибирования ин-
гибитором I является следующая:
«и
ИнициаторS (21.8)
S- + S--.S- (21.9)
S-+ I—XSL(или S + !•) (21.10)
21-| (21.11)
I • + S • I -э-Нерадикальные продукты (21.12)
2SJ (21.13)
Если реакция (21.11) является основном механиз-
мом обрыва цепи, то обычная квадратичная зависи-
мость на стадии инициирования больше не соблюдает-
ся; вернее, скорость процесса распространения цепи за-
висит от первой степени величины скорости инициирова-
304
ния. Если реакция обрыва цепи описывается уравнени-
ем (21.12), тогда каждая молекула ингибитора преры-
вает две растущие цепи; если обрыв цепи представляется
реакцией (21.11) или (21.13), то каждая молекула ин-
гибитора останавливает развитие одной пепи. Обычно
разные процессы конкурируют одновременно, и между
ними найдены стехиометрические соотношения.
Отношение Кю/к0бр определяется как константа инги-
бирования Спит, она аналогична константе переноса
[см. уравнение (15.21)]
р ______
'-'ИНГ - •
/сп
Величины констант ингибирования известны для
различных процессов и ингибиторов. Например, при
полимеризации винилацетата лурохинон имеет кон-
станту ингибирования 90 при 45° С и прерывает одну
цепь на молекулу [2], т. е. растущий полимерный ради-
кал винилацетата реагирует с ингибитором в 90 раз
быстрее, чем присоединяется к винилацетату другой
его мономер. Константа ингибирования дифенилпикрил-
гидцазила в полимеризации метилметакрилата равна
2100 при 45°С [3].
Константы ингибирования хлорного железа, в диме-
тилформамиде как растворителе были измерены в про-
цессе полимеризации ряда мономеров [4]. Константа
имеет величину 500 для стирола и равна 3 для акрило-
нитрила, обе измерены при 60° С. В стадии переноса
FeCl2------------Cl-----------Mn
наиболее важные структуры ионного резонанса пока-
заны следующим образом:
FeCl2 • СГ: +М„ ч—> FeCl?: Cl +М„
Такая резонансная структура с разделенными заряда-
ми может привести к предположению, что стирол дол-
жен иметь константу переноса, большую, чем акрило-
нитрил, что и наблюдается в действительности.
Фенолы и амины являются особенно эффективными
ингибиторами самоокислительных реакций. При инги-
бировании самоокисления стирола функцией фенолов
является перенос водорода к стирилнадоксирадикалу:
ROO • + ArOH ROOH + АгО •
Ц У. Прайер
305
Как было найдено, константы скорости этой реакции
подчиняются уравнению Гаммета при величинах <т+ и
величине р, равной —1,58. Отрицательное значение р
согласуется с электрофильной природой надоксиради-
кала и объясняет резонансные структуры переходного
состояния:
X
о;н
•OOR
Н OOR
H’OOR
Электронодонорные группы X, по-видимому, стабили-
зируют это переходное состояние и ускоряют реакцию,
что и наблюдается в действительности [5].
Реакции с участием стабильных свободных радикалов
Прежде чем закончить обсуждение ингибиторов, сле-
дует рассмотреть реакции с участием необычно ста-
бильных радикалов. Некоторые свободные радикалы
достаточно стабильны, чтобы можно было приготовить
их относительно концентрированные растворы. Напри-
мер, ДФПГ является стабильным радикалом, и его кон-
центрация в растворе может быть измерена благодаря
способности поглощать свет в области 350—520 нм.
Если инициатор разлагается в присутствии ДФПГ, то
окраска раствора последнего исчезает. Этот процесс
можно описать следующими реакциями, где И — ини-
циатор:
H-’-^R- (21.14)
«ч
R- 4- ДФПГ —> Нерадикальные продукты (21.15)
.Скорость реакции (21.15)
= Xis [R • ] [ДФПГ]
Однако константу скорости «15 нельзя вычислить, если
неизвестна концентрация R», но системы этого типа ис-
пользуют для вычисления константы скорости реакции
(21.14) [6]. Для составления равновесия можно напц,-
сать
2«д4/[И] = к1^-ДДФПГД
306
тде /— доля радикалов в инициаторе, которые стано-
вятся свободными. Следовательно,
~^ПГ] - *15 [R• 1 [ДФПГ] = 2Ktif [I]
at
Таким образом, скорость исчезновения окраски
ДФПГ связана с величиной кц, однако собственно кон- _
станта скорости радикальной реакции «15 остается неоп-
ределимой.
Тем не менее уже известны некоторые свободные ра-
дикалы, которые достаточно стабильны, чтобы можно
было получить их в концентрированных растворах,
и их реакционная способность достаточна, чтобы от-
щеплять галоген в обычных реакциях. Ниже показана
реакция восстановления цинком подпетого пиридиния в
высоком вакууме, которая дает раствор пиридиний-ра-
дикала темного изумрудно-серого цвета [7]:
Zn о’с
CH.-jCN
Этот радикал можно извлечь гексаном, затем гексан
удалить и перегнать радикал в высоком вакууме. (Он
перегоняется при 40° С и 10~6 мм рт. ст.) Пиридиний-
радикал мгновенно реагирует с кислородом или четы-
реххлористым углеродом даже при —200° С. Однако
скорость его реакции с менее реакционноспособными
галогеноуглеводородами поддается измерению при на-
блюдении исчезновения окраски раствора радикала.
Например, изучали реакцию:
СН2Вг2 +
К16
CH3CN25°C
СН2Вг
(21.16
Нерадикальные
продукты
11* 307
В этом процессе с участием необычно стабильного пирй-
диний-радикала константу скорости «i6 можно вычис-
лить непосредственно, так как известны концентрации
обоих реагирующих веществ [8]. Скорость реакции
(21.16) можно записать следующим образом:
—-[zPy'] = [СН2Вг2] [Ру-],
at
где [Ру-]—концентрация пиридиний-радикала.
В растворе ацетонитрила при концентрациях 0,012М
пиридиний-радикала и 0,84 М дибромметана Ki8 =
= 2,0-10-4 л-моль~1 -сек~1. Как и ожидалось, скорость
этой реакции первого порядка относительно концентра-
ций каждого реагента. Примечательна одна особен-
ность: константы скорости почти одинаковы в раство-
рах ацетонитрила, изопропанола и дихлорметана. Ока-
залось также неожиданным, что константа скорости
рассматриваемой реакции не зависит от природы раст-
ворителя, по-видимому, потому, что резонансная струк-
тура с разделенными зарядами, которую можно запи-
сать для переходного состояния, является энергетиче-
ски выгодной:
Вг ! СН2Вг *
Переходное состояние
Вг ;сн2вг
Упражнение
21.1. Выведите закономерность для скорости процесса (—dS/dt)
с участием реакций (21.8) — (21.11). Представьте систему, которая
может служить примером зависимости первого порядка от ини-
циатора.
ЛИТЕРАТУРА i
1. Bamford С. Н., Jenkins A. D., Johnston R. Trans. Fa-
raday Soc., 55, 1451 (1959); Henrici-Olive G., Olive S.
Makromol. Chem., 37, 71 (1960).
2. Bartlett P. D„ Kwart H. J. Amer. Chem. Soc., 72, 1051
(1950).
3. Kice J. Ibid., 76, 6724 (1954).
308
4. Bamford С. H., Jenkins A. D„ Johnston R. Proc.
Roy. Soc. (London), Ser. A, 239, 214 (1957).
5. Howard J. A., Ingold K. U. Can. J. Chem., 41, 1744 (1963).
6. Osugi J., Sato M., Sasaki M. Rev. Phys. Chem. Japan.,
33, 53 (1963); Lamb R. С., P a c i f i c i J. G. J.. Amer. Chfem.
Soc., 86, 914 (1964).
7. Kosower E. M., Poziomek E. J. Ibid., 85, 2035 (1963).
8. Kosower E. M., Schwager J. Ibid., 86, 4493 (1964).
Дополнительная литература
Bevington J. C. Radical Polymerisation. Academic Press, Inc.,
N.Y., 1961, chap. 7.
I n g о 1 d K. U. Chem. Rev., 61, 563 (1961).
Walling C. Free Radicals in Solution. John Wiley and Sons, Inc.,
N. Y., 1957, pp. 162—178, 430—436 (Уоллинг С. Свободные ра-
дикалы в растворе. Перев. с англ. Под ред. Г. А. Разуваева.
М., Изд-во иностр, лит., 1960).
Дополнительная литература к переводу
Багдасарьян X. С. Теория радикальной полимеризации. М,
Изд-во АН СССР, 1959.
Бемфорд К. и др. Кинетика радикальной полимеризации вини-
ловых соединений. Перев. с англ. М., Изд-во иностр, лит.,
1961.
Блюменфельд Л. А., Воеводский В. В., Семенов А. Г.
Применение ЭПР в химии. Новосибирск. Изд-во АН СССР.
1962.
Бучаченко А. Л. Стабильные радикалы. М., Изд-во АН СССР,
1963.
Веденеев В. И. и др. Энергии разрыва химических связей. По-
тенциалы ионизации и сродство к электрону. Справочник. М.,
Изд-во АН СССР, 1962.
Глестон С., Лейдлер К., Эйринг Г. Теория абсолютных
скоростей реакций. Перев. с англ. М., 1948.
Гладышев Г. П. Полимеризация виниловых мономеров. Алма-
Ата, Изд-во АН КазССР, 1964.
Жижина Г. П., Кругликова К- Е., Эмануэль . Н. М.
В «Сборнике трудов конференции по химии перекисных соеди-
нений». М., «Наука», 1966.
Ингрэм Д. ЭПР в свободных радикалах. Перев. с англ, под ред.
Л. А. Блюменфельда. М., Изд-во иностр, лит., 1961.
Кондратьев В. Н. Свободные радикалы—активная форма ве-
щества. М., Изд-во АН СССР, 1960.
Кондратьев В. Н. Кинетика химических газовых реакций. М.,
Изд-во АН СССР, 1958.
Кошкин Л. В., Му с а бе ко в Ю. С. Возникновение й развитие
представлений об органических свободных радикалах. М., «На-
ука», 1967.
Минков Г. Замороженные свободные радикалы. Перев. с англ.
М., Изд-во иностр, лит., 1962.
Образование и стабилизация свободных радикалов. Сб. Под
ред. А. Басе и Г. Бройда. Перев. с англ., М., Изд-во иностр,
лит., 1962.
Рюденберг К. Физическая природа химической связи. М., 1964.
Свободные радикалы в биологических системах. Сб. статей. Перев.
с англ. М„ 1963.
309
Сыркин Я. К., Д я т К и н а М. Ё. Химическая связь и строений
молекул. М.—Л., 1946.
Терентьев А. П., Потапов В. М. Основы стереохимии. М.—
Л„ «Химия», 1964.
Элементарные фотопроцессы в молекулах. Сб. статей. М.—Л.,
«Наука», 1966.
Рид С. Возбужденные электронные состояния в химии и биоло-
гии. Перев. с англ. М., Изд-во иностр, лит., 1960.
Уоллинг Ч. Свободные радикалы в растворе. Перев. с англ.
Под ред. д-ра хим. наук Г. А. Разуваева. М., Изд-во иностр,
лит., 1960.
Уотерс У. Механизмы окисления органических соединений. Пе-
рев. с англ. М., «Мир», 1966.
Уотерс У. Химия свободных радикалов. Перев. с англ, под ред.
Я- К. Сыркина. М., Изд-во иностр, лит., 1948.
Химическая кинетика и цепные реакцви. Сб. под ред.
В. Н. Кондратьева. М„ «Наука», 1966.
Эмануэль Н. М., Денисов Е. Т., М а й з у с 3. К. Цепные
рёакции окисления углеводородов в жидкой фазе. М., «Наука»,
. 1965.
Хавкине Э. Органические перекиси. М.—Л., «Химия», 1964.
Веденеев В. И. Химические радикалы. М., «Знание», 1963. Ad-
vances in free-radical chemistry. Ed. by G. H. Williams. Vol. 1 —
2. London, Logos., press-Acad. press 1965—1967.
Бреслер С. E., Казбеков Э. H. Применение ЭПР в полимер-
ной химии. «Усп. химии», 36, вып. 4, 720 (1967).
Бучаченко А. Л., Суханова О. П. Водородные связи и
Л-комплексы в радикальных жидкофазных реакциях. «Усп. хи-
мии», 36, вып. 3, 475 (1967).
Бучаченко А. Л., Сысоева Н. А. Ядериый магнитный ре-
зонанс в радикалах и радикальных реакциях. Усп. химии», 37,
вып. 10, 1952 (1968).
Д ь и ч к о в с к и й П. С., Шилов А. Е. Образование радикалов в
реакциях между валентно-насыщенными молекулами. «Усп. хи-
мии», 35, вып. 4, 699 (1066).
Иванов В. С. Синтез высокомолекулярных соединений методом
радиационной полимеризации. «Усп. химии», 35, вып. 1, 93
(1966).
Кронгауз В. А. Межмолекулярный перенос энергии при фото-
химических реакциях в растворах органических веществ. «Усп.
химии», 35, вып. 9, 1638 (1966).
Лебедев Я. С. Применение ЭПР в химической кинетике. «Усп
химии», 37, вып. 5, 934 (1968).
Мак-Клеланд В. И. Свободные аниои-радикалы. «Усп. хи-
мни», 35, вып. 3, 508 (1966).
Морозова И. Д., Д я т к и н а М. Е. Новые данные о бирадика-
лах. «Усп. химии», 37, вып. 5, 863 (1968).
Налбандян А. В. Изучение кинетики и механизма цепных реак-
ций в газовой фазе. «Усп. химии», 35, вып. 4, 587 (1966).
П и к а е в А. К., Е р ш о в Б. Г. Первичные продукты радиолиза
воды и их реакционная способность. «Усп. химии», 36, вып. 8,
1427 (1967).
Походенко В. Д„ Хижный В. А„ Бидзиля В. А. Ста-
бильные феноксильные радикалы. «Усп. химии», 37, вып. 6, 998
(1968).
310
Рез в ин А. Ф. Абсолютные скорости элементарных стадий ради-
кальной полимеризации. «Усп. химии», 35, вып. 1, 173 (1966).
Рюхардт X. Некатализированиое разложение перэфиров. «Усп.
химии», 37, вып. 8, 1402 (1968).
Семенов Н. Н. Самовоспламенение и цепные реакции. «Усп. хи-
мии», 36, вып. 1, 3 (1967).
Сергеев Г. Б. Химические реакции при температурах, близких
к 77° К. «Усп. химии», 35, вып. 4, 747 (1966).
С к а р ч е н к о В. К. Окислительное дегидрирование углеводородов.
«Усп. химии», 37, вып. 1, 3 (1968).
Степухович А. Д., Улицкий В. А. Кинетика реакций реком-
бинации и диспропорционирования алкильных радикалов. «Усп.
химии», 35, вып. 3, 487 (1966).
Стригун Л. М., Вартанян Л. С., Эмануэль Н. М. Окис-
ление пространственно-затрудненных фенолов. «Усп. химии», 37,
вып. 6, 969 (1968).
Т е р т е р и н Р. А., Б р а у д о Е. Е., Д и н ц е с А. И. Свободно-
радикальная сополимеризация этилена. «Усп. химии», 34,
вып. 4,666 (1965).
Фре.йдлииа Р. X. Перегруппировка радикалов в растворе.
В сб. «Развитие органической химии в СССР». М., «Наука»,
208—215 (1967).
Холмогоров В. Е. Метод ЭПР в фотохимии. «Усп. химии», 37,
вып. 8, 1492 (1968).
Швед чиков А. П. Химия горячих атомов водорода с энергией
1—3v и возбужденных радикалов. «Усп. химии», 36, вып. 3, 495
(1967).
Шляпинтох В. Я. Свободные радикалы и хемилюминесценция.
«Усп. химии», 35, вып. 4, 684 (1966).
Эмануэль Н. М., Гагарина А. Б. Критические явления в
цепных вырожденно-разветвленных реакциях. «Усп. химии», 35,
вып. 4, 619 (1966).
Эмануэль Н. М. Советская физика за 50 лет. «Усп. химии», 36,
вып. 11, 1959 (1967).
До по лниге льна я литература
I. В а 11 е s t е г М. Inert carbon free radicals. Pure and Appl. Chem.,
15, No. 1, 123—1.51 11967).
2. Bentrude Wesley G. Physical organic chemistry: free ra-
dicals. Annual Rev. Phys. Chem., 18, 283—324 (1967).
3. Heusler K. Bildung und Reactionen von Oxyradikalen. Chimie.
21, No. 12, 557 (1967).
4. Htinig S. Stable radical ions. Pure and Appl. Chem., 15, No. 1,
109—122 (1967). t
5. P г у о r W. A. Organic free radicals. Chem. and Engng News,
46, No. 3, 70 (1968).
6. J u 1 i a M. Free radical cyclizations. Pure and Appl. Chem., 15,
No. 1, 167—183 (1967).
7. W a 11 i n g C. A., Pearson M. S. Radical reactions of orga-
nophosphorus compounds. Topics Phosphorus Chem. Vol. 3.
N.Y. — London—Sydney. Interscience, 1966, p. 1—56.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
А, предэкспоиенциальиый мно-
житель Аррениуса 51, 52,293
определение 32; см. Элек-
тронный парамагнитный ре-
зонанс и Сверхтонкого рас-
щепления константа
Абсолютная константа скоро-
сти, см. Константа скорости
Агент роста цепи при полиме-
ризации 240
Адипиновая кислота 225
Азоизобутироиитрил (АИБН)
58, 125, 278
период полураспада 59
— формула 59
Азокамфаи 267
Азометан 124, 151, 216
— как источник метильных
радикалов 88
— реакция с метильными ра-
дикалами 151
Азометаи-Ds 130
Азосоединения 124
— перегруппировка радика-
лов 259
Азот, скорость выделения из
АИБН 129
Акрилонитрил, реакция с хло-
ристым железом 305
— сополимеризация 237
Активации энергия, определе-
ние 50
— реакции обрыва цепи 295
Активность атомов водорода
при первичной, вторичной и
третичной С-—Н-связи, метод
вычисления 160
— взаимосвязь с селективно-
стью 147
Актинометрия 64
— для ацетона 72
312
Алкил, 1,2-миграция 136, 262
Алкилбромиды, как источники
радикалов при высоких тем-
пературах 260
Алкилдибромиды, реакция с
натрием 284
Алкилиодиды, реакция с изото-
пом иода 143
Алкилперекиси, индуцирован-
ное разложение 99
Алкокси-радикалы в реакциях
отрыва водорода 74 ,
—миграция арила 261
— 1,5-миграция родорода 270
— номенклатура 79
Альдегиды, присоединение к
олефинам 211
— фотохимия 71
Амины, влияние перекиси бен-
зоила 96
— присоединение к олефи-
нам 213
Анилин, константа реакции ро-
ста цепи при полимеризации
240
— реакция с АИБН 125
Анормальное - присоединение
198
Антрацен, присоединение ме-
тильных радикалов 217
— фотохимия 64
Апокамфоила перекись 46 ,
Апокамфорная кислота 46
1,2-Арила перегруппировка 256
Арилизовалериановые альдеги-
ды 260
Ароматическое замещение 242
— взаимодействие между
продуктами реакции, меха-
цнзм 252
— метальными радикалами
218
— механизм 218 ;
— пример реакции присоеди-
нения 18
— стабильность промежу-
точных продуктов в реак-
циях присоединения 218 ।
Аррениуса уравнение 51 ,
Атома реакции переноса, опре-
деление 16
— при ингибировании 303
Ацетила перекись 84 ,
— влияние растворителя 89
— изотопный эффект 170
— клеточная реакция 84, 99
— образование метильных
радикалов 84
— предосторожность при хра-
нении |110
— разложение в циклогекса-
не 122
— индуцированное разло-
жение 88
--- механизм 84
----период полураспада 59
---сравнение в газовой и
жидкой фазах 86
----сравнение < с другими
перекисями ацилов 97 ,
Ацетальдегид, окисление до
надуксусной кислоты 101
— реакция с метильными ра-
дикалами |153
Ацетатный радикал 122, 138
— из перекиси ацетила 138
Ацетилен, , молекулярно-ннду-
цнрованный гомолнз 117
— прн фотолизе этилена 141
Ацетон, из гидроперекиси ку-
мола 113
— как источник метильных
радикалов 151
— константа реакции роста
цепи при полимеризации 240
—-присоединение к октену-1
214
— реакция с метильными ра-
дикалами 153, 155 ;
— реакция с фенильными ра-
дикалами ,161
—фотолиз 71, 75
Ацетофенон, из гидроперекиси
кумола 113
Ацила перекиси, влияние
R-группы в RCO—ОО—OCR
на скорость разложения 95
Бензальдегид 140
— самоокисление 275 >
Бензантрацен, присоединение
метильного радикала 217
Бензнлмеркаптан, в перегруп-
пировках 258
Бензиловый эфир, донор водо-
рода ,169
— самоокисление 279 ,
Бензонлацетат 138 , I
Бензоила перекись 89, 214, 244,
246
— влияние растворителя 89,
125
— ндуцнрованное разложе-
ние 80, 89, 98
---возможный порядок ре-
акции 90
---механизм 80
— механизм фенилнровання
247
— период полураспада 58,
82
— предосторожности при
хранении ПО
— разложение в метнлнодн-
де Д 22
— реакция, । катализируемая
медью 137, 140
— реакция с аминами 119,
213
Бензон'локсн-раднкал 89, 138
Бензойная кислота, из бензола
и перекиси бензоила 248
Бензол, аннон-раднкал 32
— константа роста цепи при
полимеризации 240
— растворитель при хлориро-
вании 148, 179
— реакция с АИБН 125 I
— реакция с метильными ра-
дикалами 155, 217
— феннлнрованне перекисью
бензоила 247
Бензофенон, водорода отщеп-
ление 74
— триплетное состояние 73
— фотосенснбнлнзацня 76,
289
—фотохимия 76
— энергия связей 74
313
Бензохинон 303
Бирадикалы 281
— как промежуточные про-
дукты при циклизации ради-
калов 120
— мультиплетность 63, 281
— определение 12, 281
Бифенил 131, 248
Бициклические радикалы 46
Бром 181
— акцептор радикалов 129
— катализ изомеризации
стильбена 194
— присоединение к олефи-
нам с перегруппировкой 262
Брома атом 162
— влияние полярности на ре-
акции 163
— реакция отрыва водорода
146
— цис—транс-изомеризация
263'
Бромбеизол, фенилирование
246
Бромбутен-2, присоединение
бромгидрата 199
Бромирование 181
— бициклических соединений
46
— влияние соседних групп 44
— Гаммета соотношение 165
— экзотермичиость 168
1-Бром-2-метилбутан," броми-
рование 40
N-Бромсукцинимид, галогени-
рование 187
— изотопный эффект 154
1-Бром-2-хлорпропан 263
Бутадиен 133, 140
— присоединение метильных
радикалов 217
— реакция с 1,1-дихлор-2,2-
дифторэтилеиом с образова-
нием радикалов 120
— сополимеризация 237
Бутан 23
— реакция с атомами хлора
180
— хлорирование 176
Бутаиол-2, переносчик водоро-
да 76
— растворитель для переки-
сей 251
Бутантиол, акцептор радика-
лов 128
— константа реакции роста
цепи при полимеризации 240
----с олефинами 215
— скорости реакций присо-
единения и переноса 215
Бутен-1, присоединение броми-
стого водорода 206, 222
Бутен-2, присоединение посред-
ством метилена 286
— стереоспецифическое при-
соединение 263, 290
Бутиламии, присоединение к
октеиу-1 213
— mpem-бутила гидропере-
кись 111
,—влияние растворителя 112
— из изобутана 280
— индуцированное разложе-
ние 111
— использование при приго-
товлении перекисей 101
— приготовление 111
— разложение .111
— реакция, катализируемая
медью '137, 140 .
mpem-Бутила перекись 105, 151,
213
— атака метилом 153
— донор водорода 89
— индуцированное разложе-
ние 82, 152
----в бутаноле-2 251
— источник игреиг-бутоксиль-
ных радикалов 157
— отсутствие влияния рас-
творителя на процесс разло-
жения 19, 82
— период полураспада 58,
82
— предосторожности при
хранении 110
— продукты разложения 82
— разложение в жидкой и
газовой фазах 108
— реакция, катализируемая
медью 137, 140
— реакция с метильными ра-
дикалами 153
— рост цепи при полимери-
зации полистирольиых ради-
калов 240
— энергия диссоциации свя-
зи 53
н-Бутилбензол, хлорирование
314
mpem-Бутилбензол как про-
дукт перегруппировки 258
— реакция с атомами хлора
180
----с mpem-бутоксильными
радикалами 157
----с фенильными радика-
лами 160
трет-Бутилгипобром ит 49 ;
— молекулярио-иидуцироваи-
ный гомолиз 117
трет-Бутилгипохлорит 49, 263
— молекулярно - индуциро-
ванный гомолиз 117 ,
— присоединение радикала к
стиролу 117
— реакция отрыва водорода
158 '
----с пентеном-1 136 1
mpem-Бутилдисульфид, , реак-
ция с фенильными радикала-
ми 160
трет-Бутилдифенилиадацетат
102
втор-Бутилиодид, оптическая
активность, реакция с иодом
143
— рост целя, константа ре-
акции 240
трет-Бутилиадацетат 102, 106,
138
— в окислительио-носстано-
вительиых реакциях 134
— в реакции с октеиом-1 136
mpem-Бутилнадбензоат । 102,
107, 138
— период полураспада 58
mpem-Бутилнадоксалат 102,
138
mpem-Бутилнадтозилат, разло-
жение 103
трет-Бутилиадфторацетат 104
трет-Бутилиадхлорформиат
104
трет-Бутилпирокатехин 117
трет-Бутил-4-фенилбутират-З
109
трет-Бутилфеиилнадацетат
102
mpem-Бутилциклогексанол, при-
соединение к октену-I 221
Бутилхлорид, коистаита реак-
ции роста цепи 240
вгор-Бутнльный радикал 297
«-Бутильный радикал, из пен-
танола 268
— реакции 268
mpem-Бутильный радикал 297
mpem-Бутоксильиый радикал
74, 157, 162
— влияние полярности иа
реакции 163
— из перекиси трет-бутила
82
— реакция отрыва водорода
137, 145
— теплота образования 146
Вальдеиовское обращение, в
реакции атомов водорода с
молекулой водорода 143
— определение 15
Винилацетат 303
— в реакции сополимериза-
ции 237
— значения индивидуальных
констант скоростей 235
— константа реакции роста
цепи 240
— константы скорости поли-
меризации 230
— присоединение к радика-
лам тиола 210
Вииилбутиловый эфир 210
— присоединение радикалов
тиола 210
Винилхлорид, в реакции сопо-
лимеризации .237
— присоединение бромистого
водорода 222
Включения реакции метилена
288
Влияние поляризуемых сосед-
них групп на гомолиз
О—О-связи 121
Внутримолекулярный гомолиз,
перекисей 78, 80
Водород, орто-, пара-конверсия
142
— реакция с хлором 175
— с атомами водорода 150
Водорода атом, отрыв водоро-
да 1141, 146
— реакция с молекулярным
водородом 150
— спектр ЭПР. 29
Водород бромистый, в реакци-
ях присоединения 197
315
— как катализатор самоокис-
ления 280
— как переносчик 266
— относительные скорости
присоединения к олефинам
222
— присоединение к олефинам
с перегруппировкой 262
Водород иодистый, присоеди-
нение к олефинам 203 ' j
Водорода, 1,2-миграция 136,
262
— 1,3-миграция 268
i — 1,4-миграция 270
' —1,5-миграция 270 I
— отрыв, переходное состоя-
ние 141
— — иодом 182 [
----в механизме переноса
237
----метиленами <285 1 1
----полярные эффекты 103
----триплетное состояние
68, 285
----факторы, влияющие на
скорость 144
—перекись 109, 130
----предосторожности Ьри
хранении ПО
----реакции с циклогекса-
ном 140
— перенос — см. Водорода
отрыв
Водород хлористый, присоеди-
нение к олефинам 203
Возбужденное состояние п=л*
69
Время жизни, радикала 233
— триплетного состояния 64
Выход АИБН 125
----в присутствии ловушки
радикалов 128
---- измерения с использо-
ванием дифенилпикрилгид-
разила 125, 306 |
— квантовый, СО из ацето-
на 71
----определение 68
— клеточного продукта . в
присутствии ловушки ради-
калов 128
Вязкость, влияние на скорость
реакции обрыва цепи 298
Габера—Вайсса механизм ре-
акции перекиси водорода и
железа 131
Галогена отщепление метиле-
нами 285
Гальвиноксил 108, 116
— как ловушка радикалов 99
— строение 108
Гаммета уравнение, в ради-
кальных реакциях 92
— гомолиз перекиси бензои-
ла 92
— ингибирования самоокис-
ления -фенолами 306
—индуцированное разложе-
ние перекиси бензоила 93
— использование 92, 165, 279,
306
— определение 92, 165
— отщепление водорода фе-
нильными радикалами от
производных толуола 279
—- перегруппировка арил-
изовалериановых альдегидов
260
— перекись бензоила в каче-
стве ингибитора полимери-
зации 92
Гексаарилэтан, константа рав-
новесия диссоциации 246
Гекса диен-2,4, присоединение
метильных радикалов 217
Гексаметилендиа мин 225
Гексан, реакция с метильными
радикалами 153
— с фенильными радикалами
161
Гексафенилэтан 246
— диссоциация и магнитная
восприимчивость 25
Гексен-2 137
Гептен-6-оила перекись 99
Гептильные радикалы 269
Гесса закон 54
Гея реакция 245
Гибридизация, метилена 282
— радикалов 34
Гидроксильный радикал 130
— в ароматическом замеще-
нии 132
— из перекиси водорода 111,
131
Гидроперекиси 111
— в реакциях самоокисления
273
— перегруппировки 261
316
Гидроперекись триарилметила
261
Гипохлориты 156, 184, 272
Гомберг, опыты с трифенилме-
тилхлоридом 10 1
Гомберга реакция 245
Гомолиз и гетеролиз 10
Гомолитическое замещение в
ароматическом ряду — см.
Ароматическое замещение
— фенильными радикалами
242
Гофмаин-Лоффлера реакция
270
Гриньяра реагент, образование
радикалов 258
Гюи весы 25
Дегидрирование иодом 183
Дейтерий
— как метка, с перекисью
ацетила 88
— — со стиролом 120
— отрыв метильным радика-
лом 154
— переход атома 270, 292 1
Дейтерия бромид 199, 265 (см.
также Водород бромистый)
9-Декалилнадбензоат 103
Декан как растворитель 113
Диазометан, фотолиз 283, 289
Диазониевый реагент в реак-
ции Гомберга 245
Диалкилов перекиси — см. Ал-
килперекиси
2,3-Дибромбутан 200
Ди-трет-бутила перекись —
см. mpem-Бутила перекись
Дигидробифенил, из бензола и
перекиси бензоила 249, 253
Дииодид стирола 117, 123
Дильса-Альдера реакция 73 !
— двух молекул стирола
120
Диметиланилин, реакция с пе-
рекисью бензоила 95, 119
2,3-Диметилбутадиен, присо-
единение метильных радика-
лов 221
2,3-Диметилбутан
— реакция с атомарным хло-
ром 180
----с метильными радика-
лами 153
----с фенильными радика-
лами 161
— хлорирование 148
2,5-Диметилгексадиен-2,4, при-
соединение к метильным ра-
дикалам 217
5,5-Диметил-3-гексен-2-он 77
5,9-Диметилдекан-2-он 76
Диметиловый эфир, реакция с
фенильными радикалами
16
2,2-Диметилпропан, реакция с
хлором 175
Диметилртуть 151
2,2-Днметилстирол, продукт
перегруппировки 258
Диметилциклопропан 286
Диметилцинк 11
Диоксан, в качестве раствори-
теля 92
Диспропорционирование 231,
249, 292
— алкильными радикалами
14
— фактор Аррениуса 293 :
Дисульфиды, реакция с ради-
калами 144
1,4-Дифенилбутадиен, присо-
единение метильных радика-
лов 217, 221
Дифенилметан
— реакция с атомами брома
181
----с атомами хлора 180
----с mpem-бутоксильными
радикалами 158
----с фенильными радика-
лами 161
— константа реакции роста
цепи 240
Дифениловый эфир, раствори-
тель при перегруппировках
259
Д ифенилпикр илгидр азил
(ДФПГ) 32
— в системе иод — стирол 123
— как ингибитор 304
— как индикатор в радикаль-
ных реакциях 306
— константа ингибирования
305
— получение 32 ' 1
— присадка к топливу с азо-
изобутиронитрилом 125
— спектр 32
317
— строение 32
1,1-Дифенилэтилен, присоеди-
нение метильных радикалов
217
транс- 1,2-Дифенилэтилен, при-
соединение метильных ради-
калов 217
цис- 1,2-Дифенилэтилен, присо-
единение метильных радика-
лов 217
Диффузия 99
— в клеточных реакциях 85
— в реакциях обрыва цепи
298
1,1 - Дихлор-2,2-дифторэтилен,
реакция с бутадиеном с об-
разованием радикалов 120
Дихлорметан, реакция с ато-
мами хлора 180 !
3,4-Дихлорстирол, в качестве
присадки к топливу 92
Диэтиловый эфир малоновой
кислоты, присоединение к ок-
тену- 1 215
Диэтилоктилмалрнат 215
Длинноволновый свет 60 i
Додекантиол, присоединение к
олефинам 210
Дублеты, отличие от трипле-
тов 290
Дурохинон 305
Железа ион 140
— реакция с перекисью водо-
рода 131
Железа хлорид 303, 305 '
Замещение в ароматическом
ряду 242
о-Замещениые трет-бутилбен-
зоаты 121
Излучение 57
Изобутан — см. 2-Метилпропан
Изобутилен, присоединение
бромистого водорода 222 _
—метильных радикалов 217
— хлорир ова ние 179
Изобутилена окись, из переки-
си mpem-бутила 83
Изомеризация олефинов под
действием радикалов 194
Изооктан 87, 216
Изопропанол, как донор водо-
рода 88
318
—константа реакции роста-
цепи 240
— присоединение к октену-1'
212
Изопропилбензол — см. Кумол
Изопропилбензол, как агент
роста полимерной цепи 240
— константа реакции роста
цепи 240
— реакция с фенильными ра-
дикалами 161
Изопропилбромид, хлорирова-
ние с перегруппировкой 263
Изопропилдисульфид, реакция
с фенильными радикалами
161
}4зопропил-радикал 297
Изотопный эффект 153
— и азометане 130
— в перекиси ацетила 170
— в реакции атома водорода;
с молекулой водорода 142
— в фенильных радикалах;
161
— в этилбензоле 88, 154
— при галогенировании то-
луола 167
— при термической полиме-
ризации стирола 120
— при фенилировании 253
— связь с энергией актива-
ции 154
Ингибирование 300
— кислородом 117
Ингибирования константа, оп-
ределение 305
Инден, соокисление 280
Индуцированное разложение
80, 91, 250
— возможный кинетический,
порядок 91
—в растворе стирола 112
— гидроперекиси трет-бути-
ла 112
— как реакция первого по-
рядка 92
— конкуренция с мономоле-
кулярным гомолизом 92
— определение 78, 116
— перекисей алкилов 78, 82,
99
— перекиси ацетила 89
— перекиси бензоила 78, 89,
99, 119, 122, 213
Инициаторы, коммерчески до-
ступные, н их период полу-
распада 58
Инициирование, полимеризации
226
— радноактивным инициато-
ром 231
— самоокисления 277
— скорость 231
— фотоактивным инициато-
ром 234
Иод, присоединение к олефи-
нам 264
Ион титана 133
Ионная перегруппировка 259
Ионная полимеризация 224
Ионное хлорирование цикло-
гексена 176
Ионный механизм замещения
в ароматическом ряду 242
Караша присоединение 198
— надэфиров 134
Карбен — см. Метилен
Карбониевый ион, из реаген-
та Гриньяра 258
— образование при окислении
радикалов ионом меди 135
— перегруппировка, сравне-
ние с радикальной перегруп-
пировкой 255
Карбонильная группа, п—л*-
процесс возбуждения 69
Карбонильные соединения, фо-
тохимия 69
Кинетика полимеризации 226
Кинетическая цепь, длина 230
Кислород, в молекулярно-инду-
цированном гомолизе 276
— в присоединении тиоуксус-
ной кислоты к иидену 280
— строение 282
Кислород-18, в перекиси аце-
тила 87
— при самоокислении 278
Клеточная реакция, с АИБН
125
— введение 62
— временная шкала 99
— в газовой фазе, прн высо-
ком давлении 85
— доказательство радикаль-
ного хнрактера реакции 127
— история 85
— обнаружение 85
— рекомбинации, первичные
85
----вторичные 85
— связывание двух атомов
кислорода 107
_—с-надэфира ми 107
— эфира образование прн
ароматическом замещении
251
Кобальт нон 133
— реакция с реактивом
Гриньяра 258
Кольбе, электролиз 130
Комплекс между растворителя-
ми и радикалами при моле-
кулярно-индуцированном го-
молизе 118
Комплекс присоединения 246
Концевые группы в полимери-
зации 224
Константа заместителя а, ис-
пользование в уравнении
Гаммета 92, 165, 279, 306
Константа переноса
— аминов 213
— галогенопроизводных ме-
тана 206
— определение 196, 239
— различных заместителей
240
— спиртов 212
Константа скорости реакции,
атома хлора с водородом
173
— влияние на длину цепи
221, 229
— димеризации метила 152
— индивидуальная оценка
233
— обрыва цепи 295
— отрыва водорода метиль-
ными радикалами 152
— полимеризации различных
мономеров 230
----винилацетатов 299
— присоединения радикалов
тнола 215
' — самоокисления тетрамнна
208
Константа перекрестного за-
вершения реакции 20, 296
— влияние полярного эффек-
та 20
— первичный квантовый вы-
ход 68
319
Конфигурация радикалов 147
Конформация, возможности
для радикалов 34
— циклогексильных радика-
лов 221
Концентрации радикалов 150
Криги реакция надэфиров 103
л-Ксилол, реакция с трет-бу-
токсильными радикалами 158
n-Ксилол, реакция с трет-бу-
токсильиыми радикалами 158
Кумола гидроперекись 113
— из кумилового спирта 114
— окислительно-восстанови-
тельные реакции с ионом
железа 59
— при сополимеризации 133
— разложение 114
— — катализируемое кисло-
той 114
Кумилоксильный радикал 114
— Р-расщепление 114
Кумол 137, 140
— константа реакции роста
цепи 240
— перегруппировка 260
— реакция с атомами брома
181
----с mpem-бутоксильными
радикалами 158
----с атомами хлора 180
----с N-бромсукцинимидом
154
•---с фенильными радика-
лами 160
----с различными радика-
лами 162
Лауроила перекись, взрыво-
опасность ПО
Лиганда перенос 135, 137 ,
Локализации энергия, корреля-
ция с радикальным присо-
единением 218
Магнитная восприимчивость 25
Марковникова правило 208
Масляной кислоты, хлор ангид-
рид, хлорирование 175 i
Масляиый альдегид 211
— константа роста цепи, при
полимеризации 240
— фотолиз 67
Меди ион 133, 137
Медь одиовалентиая, реакция
320
с надэфиром 134
Металлические зеркала,
— удаление бирадикалами
285
------радикалами 11
Метан, как донор водорода
146 ’
— реакция с атомами брома
181
----с атомами хлора 180
----с хлором 175 !
Метанол, реакция с метильны-
ми радикалами 153
Метантиол, дейтерированный,
присоединение к бутеиу-2
265
Метилацетат, из перекиси аце-
тила 87
—присоединение к октену-1
214
— реакция с метильными ра-
дикалами 155
----с фенильными радика-
лами 161
п-Метилбензильные радикалы
284
Метилбензоат, реакция с ме-
тильными радикалами 155
Метилбутилкетон 72
2-Метилбутен-2, присоедине-
ние метильных радикалов
217
Метилгексилкетон, фотолиз 72
Метилдиизопропилкетон, фото-
лиз 72
Метилдисульфид, константа ре-
акции роста цепи 240
•—реакция с фенильными ра-
дикалами 161
Метилен 12
— включение 288
— триплетное и синглетное
состояния 282
— хлористый, реакция с ато-
мами хлора 180
Метилиодид, как растворитель
для разложения перекисей
122
Метилизопропилкетон, фото-
лиз 72
Метилметакрилат, в реакциях
сополимеризации 237
— константы скорости поли-
меризации 230
. — полимеризация с гидропе-
рекисью кумола 115
Метилнеопентилкетон, фотолиз
72 .
Метилнитрит 157
Метиловый эфир, реакция с
фенильными радикалами 161
Метилпентилкетон, фотолиз 72
2-Метилпропан
— реакция с атомами брома
181
----с атомами хлора 180
----с метильными радика-
лами 153
----с хлором 175
— самоокисление 280 ,
2-Метилпропен — см. Изобути-
лен
n-Метилтолуол, реакция с фе-
нильными радикалами 161
2-Метил-3-фенилпропен-1, про-
дукт перегруппировки 258
n-Метилфенильный радикал 253
Метилхлорид 175, 180
1-Метилциклопентила гидро-
перекись 140
Метильный радикал 10, 36, 146,
151, 162, 175
— ароматическое замещение
217 1
— в газовой фазе и в рас-
творе 154
— величины Ф в реакциях
обрыва цепи 296
— изотопный эффект в реак-
ции с толуолом 170
— из гидроперекиси кумола
113
— из перекиси ацетила 89
— из перекиси mpem-бутила
82
— клеточные рекомбинации
87 j
— константа скорости диме-
ризации 52
— конформация 36
— отрыв водорода 146, 151,
154, 155, 162
— полярный эффект замести-
телей 163
— присоединение к олефи-
нам, ацетиленам и аромати-
ческим соединениям 216, 217,
219
— реакции обрыва цепи 294
— реакция с этиленом 269
— спектры ЭПР 36
—сравнение констант скоро-
стей реакций 155
----реакционной способно-
сти в различных условиях 87
— улавливание 86
— фотолиз ацетона 71
Метод вращающегося сектора
150, 234, 279
2-Метоксипиран 267
Метоксильный радикал 157 ,
— Ф, величина 296
n-Метоксистирол, в реакциях
сополимеризации 237
— константы скоростей поли-
меризации 230
Молекулярно-индуцированный
гомолиз 115, 122
— mpem-бутилгипобромита и
олефинов ,117
— mpem-бутилгипохлорита и
бутена-2 117
----и стирола 117
— в реакциях самоокисления
276
— в хлорировании 176
— гидроперекиси кумола и
метилметакрилата 115
----и стирола 115
— гидроперекиси трет-бутн-
ла и стирола 113, 115
— иода и стирола 126
— конкуренция с ионными
реакциями 115
— механизм 116, 120
— олефинов 120
— определение 115
— отличие от радикально-ин-
дуцированного гомолиза 115
— перекиси .ил'-дибромбен-
зоила и диметоксистильбена
116
— с участием кислорода 276
— фтора 173
Мономеров реакционное отно-
шение, коэффициент актив-
ности 236
Мостиковые радикалы 41, 201,
222
— бромный мостик 42
— доказательство существо-
вания 41
— объяснение стереоселек-
тивности присоединения 201
321
— радикалы с галогефвыми
мостиками 41
Мультиплетности 62
Мышьяк 285
Надбензойиая кислота 274
Надкислоты синтез 101
Надмуравьиная кислота, взры-
воопасность ПО
Надокси-радикалы, присоеди-
нение к алкильным радика-
лам ,114
— электрофильность 306
Надуксусная кислота 101
— взрывоопасность ПО
Надэфиры, механизм разложе-
ния 105
— влияние R-группы на ско-
рость распада трет-
C4H9OCO2R 107
— период полураспада 106
— синтез 101
Нафталин, присоединение ме-
тильных радикалов 217
Нафтацен, присоединение ме-
тильных радикалов 217
Нейлон 225
Нейтральные радикалы, фени-
лирование 246
Неофенилхлорид, перегруппи-
ровка 258
Нерадикальные реакции, как
альтернатива клеточной ра-
дикальной рекомбинации 87
о-Нитробензоила перекись, мо-
лекулярно-индуцированный
гомолиз 121
Нитробензол, ароматическое
замещение 253
N-Нитрозоацетанилид 129, 244,
254
п-Нитро-п'-метокснбензоила пе-
рекись 108
n-Нитростирол, в реакциях со-
полимеризации 237
n-Нитротолуол, реакция с фе-
нильными радикалами 161
— самоокисление 279
и-Нитрофенильиый радикал
253
Норборнадиен, присоединение,
перегруппировка 267
Нуклеофильные заместители
119
322
Облучение 57
Обрыв цепи 292
— алкокси-раднкалов 276
— в АИБН 126
— изменения в механизме
276
— при полимеризации, меха-
низм 231
Окисление — см. Самоокисле-
ние, Окислительно-восстано-
вительная реакция
Окислительно-восстановитель-
ная реакция, как источник
радикалов 59
— реактив Гриньяра с ионом
_ кобальта 258
О’кисляемость различных угле-
водородов 279
Окись стирола, из гидропере-
киси mpem-бутила и стиро-
ла 116
Октен-1 138
— присоединение альдегидов
211
----бутиламина 213
----диэтилмалоната 215
— — изопропанола 212
— — пиперидина 213
----тиольных радикалов
208
— реакции присоединения
207
— реакция с mpem-бутил-
иадацетатом 136
----метильными радикалами
155
— типичные реакции присо-
единения 215
Открытое кольцо и закрытое
кольцо 266
Относительная актианость, оп-
ределение 176
Отношение констант скоростей
в радикальных реакциях, ис-
пользование 150
Отношения активностей при
сополимеризации 236
Панет, опыты с тетраметил-
свинцом 10
Парамагнитные соединения,
орто—пара-конверсия водо-
рода 142
Паскаля, правила аддитивно-
сти магнитной восприимчи-
вости 25
Пентан, реакция с метиленом
288
Пентанол, декарбоксилирова-
ние 268
Пентафенилэтан, константа ро-
ста цепи при полимеризации
240
Пентен-1, присоединение ти-
ольных радикалов 215
— реакция в /пре/п-бутилги-
похлоритом 136
Первичный фотохимический
процесс, определение 68
Перегруппировка, атомов хло-
ра 19, 204, 285
— азосоединений 259
— 1,2-алкильная миграция
262
— алкоксильных радикалов
201
— альдегида фенилизовале-
риаиовой кислоты 256
— альдегидов арилизовале-
риановой кислоты 260
— 1,2-арильная миграция 256
— влияние переносчиков 257
— внутренняя 61, 64
— 1,2-водорода миграции 261
— 1,3-водорода миграции 268
— 1,4-водорода миграции 268
— 1,5-водорода миграции 268
— 1,2-галогенов миграция
.262
— изопропилбензола 260
— как пример реакции роста
цепи 19
— при цис-транс-нзомернза-
ции 263
— реактивов Гриньяра 258
— реакции разрыва н замы-
кания кольца 266
— способность арильных
групп к миграции неофил-
хлорида 258
— трифенилпропаналя 257
— З-фенилпропаналя-2-С14
258
— циклических альдегидов
257
— этил-2-циангептен-6-оата
222
Перегруппировки реакция 254
— замена углерода хлором
184, 266
----радикалами 14, 143
— замена циклогексильных
радикалов перекисью ацети-
ла 122
— в дисульфидах 144
— в качестве возможного
механизма при фенилирова-
нии 247
—в реакциях атомов водо-
рода с молекулами водоро-
да 142
— определение 5н2-механиз-
ма 14, 143
Передача заряда, при молеку-
лярно-индуцированном гомо-
лизе 119
Перекиси, в реакциях само-
окисления 273
— взрывоопасность 81
— детонация 80
— индуцированное разложе-
ние 80
— • мономолекулярный гомо-
лиз 80
— номенклатура 79
Перекисный эффект 198
Перекись бензоила, как источ-
ник фенильных радикалов
242
— /пре/п-бутилметила 157
— триптоила 47
Перекрестного обрыва меха-
низм реакции самоокисления
276
Переходное состояние, конку-
ренция вновь образующейся
связи с разрывающейся 141
— при перегруппировке ка-
тионов, радикалов и анио-
нов 255
— при типичных экзотерми-
ческих и эндотермических
реакциях 148
Переходные металлы, в окис-
лительно-восстановительных
реакциях 132
Пинан, гидроперекись 122
— окисление 122
fJ-Пинен, присоединение с пе-
регруппировкой 266
Пиперидин, присоединение к
октену-1 213
323
Пиридин, присоединение ме-
тильных радикалов 217
Пирролидины 270
Полиакрилнитрил 228
Поливинилиденхлорид 227
Поливинилхлорид 227
Полимер, определение 224
Полимеризация 224 i
— винилацетата 237
— кинетика 226
— степень роста цепи, опре-
деляющие символы 17, 233
Полимеризация—конденсация,
определение 225
Полимеризация присоединения
225
Полимеризуемость 229
— метод измерения на осно-
вании длины цепи 232
— мономеров в реакциях те-
ломеризации 206
Полимерные радикалы, в ре-
зультате обрыва цепи 298 !
Полимеры, свойства 227
Полиметилметакрилат, величи-
на Ф 296
Полистирол 227
Полистирольный радикал, ве-
личина Ф 296
Политетрафторэтилен 227 '
Полихлортрифторэтилен 227
Полиэтилен 227
Поляризуемость, мономера 279
— растворителя, влияние на
молекулярно-индуцирован-
ный гомолиз 121
Полярные характеристики ра-
дикалов 19
Полярный эффект 148 1
— в значении Ф в реакциях
обрыва цепи 21, 296
— в реакциях аминов с пе-
рекисью бензоила 96, 119
— в реакциях комбинации
радикалов 20, 295
— в реакциях обрыва цепи
20
— в реакциях отрыва водо-
рода 163
— в реакциях перекисных'
радикалов 305
— в реакциях пиридиние-
вых радикалов 307
— в реакциях присоединения
216
— в реакциях фенильных ра-
дикалов 162
— при индуцированном раз-
ложении перекиси бензоила
95
— при перегруппировке аль-
дегида аллилизовалернано-
вой кислоты 261
— при самоокислении 279
— при хлорировании 23
Присоединение 292
— брома с перегруппировкой
262
— бромистого водорода к
2-бром-2-бутану 199
•---перегруппировка 262
----стереохимия 199
— влияние заместителя в
олефинах 216
— ионное и радикальное 198
— к ацетиленовым производ-
ным 216
— к бициклическим соедине-
ниям 202'
— к ингибиторам 304
— к октену-1 208
— к олефинам, спиртов 212
---альдегидов 212
----аминов 213
---- галогенов 196
— — — энергетика 205
----галогеноводородов 197
----галогенопроизводных
метана 206
----гидробромида, история
197
—-----относительная ско-
рость 222
----йодистого водорода 203
----метилена, роль 286
----тиолов 208
---- хлористого водорода
203
----эфиров 215
— к,стиролу 207 |
— — /пре/п-бутилгипохлори-
та 117
----четыреххлористого уг-
лерода 196
— к хлористому водороду,
присоединение по радикаль-
ному механизу по правилу
Марковникова 204
— механизм 193
324
— образование новых С—С-
связей 211
— примеры различных реак-
ций 17
— радикалов 20, 292
---алкильных 17
----метильных 216
----константа скорости-
295
—селективность 197
— скорость и механизмы 215
— тиолов с перегруппиров-
кой 263
— ти’оуксусной кислоты, к
индену в присутствии кисло-
рода 280
Чис-Присоединение к бицикли-
ческим олефинам 202
Присоединение — диспропор-
ционирование, значения от-
ношений констант скоростей
296;
Пропан, реакция с атомами
брома 181
— атомами хлора 180 :
— с хлором 175
Пропен 269
— присоединение бромистого
водорода 222
----метильных радикалов
217
Пропилбензол 260
Пропилкетон, фотолиз 72
Пропил-радикал, присоедине-
ние к ароматическим соеди-
нениям, Ф значение 296
Пропил сульфид 139
Пропионовая кислота, как до-
нор водорода 163
— константа роста, цепи при
полимеризации 240
Пропорционироваиие 230
Прочность связи, влияние иа
скорость отрыва водорода
145
Радикал-ион, при восстановле-
нии кетонов 12
Радикал пиридина 307
— полиметилметакрильный, в
реакции обрыва цепи 298
— полистирильный 94
----атака на перекись
mpem-бутила 241
---- в реакции с бутантио-
лом 215
—-----с перекисью алкила
99
--значение величины. Ф
296
----при индуцированном
разложении перекиси бен-
зоила 95
----присоединение к арома-
тическому кольцу 95
Радикалы
— гибель 14
— гибридизация 34
— конформация 34, 175, 199,
221 '
— образование 14
— определение 9, 11
----с помощью ЭПР 150
— оптически активные 36
— основные типы образова-
ния 57
— полярный характер — см.
Полярный эффект
— стабильность 19
Радикально - индуцированное
разложение 116
Радикальные реакции
— определение основных ти-
пов 13
— полимеризации 224
— триплетного состояния 289
Разветвленные цепные реакции
16
Растворители, . значение при
индуцированном разложении
91
•— как ловушки фоточувстви-
тельных молекул 61
Растворитель
— влияние на разложение
эфирных перекисей 103
---- на реакции метильных
радикалов 157
— действие на гидроперекись
mpem-бутила 112
----при гомолизе 115
— — при хлорировании 148,
179
— диэлектрическая постоян-
ная, влияние на радикаль-
ные реакции 149
— комплексообразование с
радикалами 118
325
— метилиодид — для переки-
сей 121
— недостаток при разложе-
нии АИБН 125
— нечувствительность ради-
кальных реакций 19
— отличие в действии в ион-
ных и радикальных реакциях
20
0-Расщеплеиие
— алкоксильиыми радикала-
ми ,18, 122 _
— кумилоксильным радика-
лом 113
— определение 18
— приводящее к перегруппи-
ровке 267
— радикала, образованного
из гидроперекиси пинана 122
Рацемизация 39
Реакция
— переноса
----бромом 41
— — DBr и DSCH3 в срав-
нении 266
----полистирольиыми ради-
калами 239
----хлором 41
— присоединения
----в результате цис-транс-
изомеризации 263
----к олефинам 208, 262
— — при декарбоксилирова-
нии 76
—• — стерическиё факторы
202
----тиолов к норборнадиеиу
267
— роста цепи
----влияние иа молекуляр-
ный вес 238
----в полимеризации 237
— — определение
----четыре главных типа
237
— сдваивания — см. Клеточ-
ная реакция
Резина 133
Самоокисление 273
Сверхтонкого расщепления
константа, определение 31
(см. также ЭПР)
Свободный радикал — см., на-
пример, Хлор атом, Метиль-
326
йый радикал
Связей разрыв, изотопный эф-
фект реакции 154
— при галогенировании 165
— при хлорировании 180
— размеры в переходном со-
стоянии 147
Связи, метод измерения 53
— энергия диссоциации 53
Селективность, взаимосвязь с
активностью 147
Селен 285
Семихиионы, восстановление в
радикалы и спектр ЭПР 29
Сера, как ингибитор 208
— как соседняя группа 121
Сероводород, константа роста
цепи при полимеризации 240
— присоединение 208
Сероуглерод, растворитель при
хлорировании 148
Синглетное состояние, опреде-
ление 63
— отличие от триплетного со-
стояния 69, 283, 289
Синглет-триплетиый переход
63, 287
Скорость образования радика^
лов 228
— разрушения радикалов 228
Соедяиеиия серы, в радикаль-
ных реакциях 200
Соли уранила, как фотоэле-
менты 65
Соокнсленне 280
Сополимер стирола и бутадие-
на 227
Сополимеризация 235
— уравнение 236
Соседняя группа, влияние бро-
ма 44
— в о-замещенных трет-
бутилиадбензоатах 121
— при галогенировании 44
Спектр, оптический спектр ка$
способ индентификации ра-
дикалов 32
Спиропеитаи 266
Спирты, как продукты само-
окисления 277
— переноса кристаита 213
— присоединение к олефи-
нам 212
Сравнение SH2 с SN2-реак-
циями 15, 143
Стабильность, Иона карбония
и радикалов 199
— радикалов 18
Стабильные радикалы из солей
пиридина 307
Стадия реакции, определение
226
Степень полимеризации 231
— определение 196, 231
— связь с кинетической дли-
ной цепи 231
Стереоспецифичность, как ме-
тод определения триплета
метилена 286
Стереоселективность в реакци-
ях присоединения 199, 209,
276
Стереохимия, присоединения
меркаптанов 209
— присоединения радикалов
тиола 276
Стерические факторы—см. Ре-
акция присоединения
Стильбен, изомеризация цис- в
транс- 194
Стирол, константа скорости по-
лимеризации 230 -
— константы реакции роста
цепи при полимеризации с
разными агентами роста
240
Структура, ингибитора влияние
на течение реакции стирола
303
— как ловушка радикалов
86, 92, 128 х
— константы реакции роста
цепи при полимеризации 240
----с хлороформом 221
— переноса с четырехброми-
стым углеродом 221
-----с четыреххлористым уг-
леродом 221
— при молекулярно-индуци-
рованном гомолизе 115
----атома хлора 120
----метильных радикалов
217
----радикалов тиола 215
— при сополимеризации 237
— реакции, с гидроперекисью
mpem-бутила 112
----с гидроперекисью изо-
пропилбензойной кислоты
114
---с иодом 117
---с хлоридом железа 303,
305
— сополимера с бутадиеном
133
— теломеризации 206
— теломеризации с СС14 197,
207
— термической полимериза-
ции 120
Сурьма 285
Теллур 285
Теломер, из аминов 213
— из олефинов и спиртов 213
— определение 206
Теломеризации 206
Теория переходного состояния
50 1
— молекулярных орбит при
перегруппировках 255
— применение к реакциям
атомов водорода с молеку-
лами водорода 142
Теплота образования 54
— радикалов 55
— реакций 50
------ отношение к скорости
148
---подсчет на основании
энергий связей 55
Тепловая закалка 61
Термический гомолиз, опреде-
ление 57
Тетрагидрокватерфенилы, из
бензола и перекиси бензоила
249
Тетралилацетат 139
Тетралин 139
— реакции с mpem-буто-
ксильными радикалами 158
— самоокисление 279
Тетраметилсвинец 11
Тетраметилсукциионитрил, из
АИБН 126
1,1,4,4-Тетрафеиилбутадиеи,
присоединение метильных ра-
дикалов 217
Тетрафенилэтилен 217
— присоединение метильных
радикалов 217
Тиильные радикалы, присоеди-
327
йение к стйроЛу и пентену-i
215
— электрофильность 210
п-Тиокрезол 267
Тиоуксусная кислота 266
— присоединение к олефинам
210
— соокнсление при перегруп-
пировке 280
Тиофенол 258
n-Толильный радикал 253
Толуол, ароматические заме-
стители 253
— изотопный эффект при га-
логенировании 169
— как донор водорода 87
— константа реакции роста
цепи при полимеризации 240
— присоединение к трипле-
там и трe/n-бутоксильным ра-
дикалам 74
— реакции
----с атомами брома 181
-----с атомами хлора 180
----с N -бромсукцинимйдом
154
-----с метильными радика-
лами 155
----с радикалами 162
-----с /пре/п-бутоксильными
радикалами 158
----с фенильными радика-
лами 161
— самоокисление 279
— сравнение с циклогекса-
ном как донором водорода
162
— фенилирование 253
Тринитробензол, как ловушка
радикалов 92
Триплетное состояние, бензо-
фенона 73
— время жизни 64, 70
— определение 63
— отличие от синглета 289
— циклопентадиена 73
— энергия передачи 73
Триптицильные радикалы 47
Тритильный (трифени'лметиль-
иый) радикал 10, 244
Трифенилвисмут, как источник
фенильных радикалов 246
Трифенилметаи, как агент ро-
ста цепи при полимеризации
240
328
— константа реакции роста
цепи 240
— реакция с атомами брома
181
----с атомами хлора 180
----с /пре/п-бутоксильны-
ми радикалами 158
----с фенильными радика-
лами 161
Трифенилпропаналь, перегруп-
пировка 257
1,1,2-Трифенилэтилен, присо-
единение метильных радика-
лов 217
Трифториодметан 208
Трифториодэтилен 208
Трифторметильный радикал в
результате реакции отрыва
водорода 146
Трихлорметильный радикал,
присоединение к ароматиче-
ским соединениям 219
3,3,3-Трихлорпропен 262
Углерод четырехбромистый,
константа реакции роста це-
пи при полимеризации 240
— четыреххлористый, агент
переноса 221
— — использование в каче-
стве стандартного раствори-
теля 154
------константа реакции ро-
ста цепи при полимеризации
240
— присоединение к 6-пинену
266
—-растворитель при хлориро-
вании 148, 179
— реакция с метильными ра-
дикалами 155
— — с метиленом 285
Уксусная кислота, присоедине-
ние к октену-1 214
— реакция с фенильными ра-
дикалами 161
Уравнение реакции роста цепи
при полимеризации 239
Уравнение скорости в реакциях
отрыва водорода 150
Ф — квантовый выход реакции
67, 68
Фенантрен, присоединение ме-
тильных радикалов 217
Фенилазотрифенилметан 105,
125, 159, 244
Фенилизовалериановый альде-
гид, декарбонилирование и
перегруппировка 256
Фенилаллиловый эфир 138
Феннлбензоат, из бензола и
перекиси бензоила 248
Феиилбутанола-3 перекись 272
Фенилдиазоацетат 245
Фенилирование, ароматических
соединений, источники фе-
нильных радикалов 244
— механизм присоединения —
отщепления (элиминирова-
ния) 247
— распределение изомеров
247
— соотношение между выхо-
дами бифенила и механизм
250
— толуола 253
Фенилксантен-10 275
1-Феннлнафталин, из стирола
120
З-Фенилпропаналь-2-С14 258
1-Фенилтетралин, из стирола
120
Фенил-п-хлорбензоат 252
1-Фенилциклогексила ’ гидропе-
рекись 140
Фенильная группа, миграция
256
Фенильный радикал 162, 253
— из перекиси бензоила 89,
243, 244
— отрыв водорода 145, 159
— присоединение к аромати-
ческим соединениям 219
ПуФенокситолуол, реакция с
фенильными радикалами 161
Фенол, из гидроперекиси кумо-
ла 114
— как ингибитор 302, 305
Фентона реагент 111, 131
Флюоресценция 61 ।
— антрацена 66
— определение 64
Фосфоресценция 61, 64 ’
— время жизни 64
— определение 64
Фотолиз при облучении ртут-
ной лампой 61
— этилена 141
Фотосенсибилизация 288 1
Фотохимические реакции 60,
283
— кетенов 283
Фотохимия
— ацетона 71
— димеризация антрацена 65
— интенсивность света 60
— карбонильных соединений
68
— кетонов 283
— масляного альдегида 67
— определение 57
— реакция разрушения кето-
нов без участия свободными
радикалами 72
Фотохлорирование — см. Хло-
рирование
Фрагментирования реакция 18
Франк—Кондона принцип 74
Фтор 172
Фтора атом, отрыв водорода
146
1-Фторбутан 23, 182
— бромирование 181
— хлорирование 176, 181
—фторирование 181
Хинон, как ингибитор 302
— как ловушка радикалов
87
Хлор 173
— 1,2-миграция 285
— N-Хлор а мин 270
Хлоранил 125, 303
Хлора атом 162
— вытеснение водорода 145
— комплексы с бензолом и
другими растворителями 149
— поляоный эффект в реак-
циях 163, 182
— присоединение к стиролу-.
120
— селективность 148
n-Хлор бензоила перекись 252
Хлорбензол, нитрование 243
— фенилирование 243
1-Хлорбутан, хлорирование 176
Хлорирование 171
— влияние растворителя 148
— Гаммета соотношение 165
— молекулярно-индуцирован-
ный гомолиз 176
— оптически активного
1-хлор-2-метилбутана 39
329
— относительная селектив-
ность 176
— относительные скорости
176
— 1-фторбутаиа, влияние по-
лярности 23, 24
— цепной механизм 14
— экзотермичиость 168
Хлорметаи — см. Метилхлорид
1-Хлор-2-метилбутаи 39
— бромирование 41, 222
— оптическая активность 222
— хлорирование 39
Хлороформ, константы реакции
роста цепи 240
— реакция
— с атомами хлора 180
— с метильными радикалами
155
— с фенильными радикалами
160
З-Хлорпропеи, присоединение к
тиильиым радикалам 210
n-Хлорстирол, в реакциях со-
полимеризации 237
n-Хлорфеиильиый радикал 253
Хром, ион 133
Цепи длина, определение 230
Цепная реакция, в родикаль-
иой полимеризации 225
Циаиопропильиые радикалы,
из АИБН 125
Циаиотрихлорметаи 208
Циклическая реакция 120
Циклические кетоиы, фотолиз
284
Циклогексадиеиильиый ра-
дикал, как промежуточный
продукт в ароматическом
замещении 218, 248
Циклогексан 177
— как донор водорода 145,
162
—константы реакции роста
цепи 240
— присоединение к октео-
иу-1 214
— реакция, с атомами хло-
ра 180
----с /пре/п-бутоксильными
радикалами 158
----с метильными радика-
лами 155
----с различными радика-
лами 162
-----с фенильными радика-
лами 161
— хлорирование mpem-бу-
тилгипохлоритом 117
Циклогексанон, гидроперекись
140
Циклогексен, присоединение
радикалов тнола 210
— взаимодействие с хлором
177
— как донор водорода 134
— реакция с метиленом 289
----с феиильиым радика-
лом 161
— самоокисление 274
Циклогексеиовой кислоты аце-
тат 134
— из перекиси ацетила 122
Циклогексилацетат, из переки-
си ацетила 122
Циклогексильиый радикал
257
— конформация 221
— спектр ЭПР 36
Циклопентадиен, фотохимии
73
Циклопентильный радикал 257
Эйкозадиен-8,12-диои-2, 19 140
Экзотермическая реакция, ре-
акционная способность и се-
лективность 147
Экзотермичиость 168—см. Хло-
рирование и Бромирование
Электроиодонориые и электро-
ноакцепториые группы, влия-
ние иа радикальные процес-
сы 245
Электронный парамагнитный
резонанс (ЭПР) 26
— алкильных радикалов 35
— сверхтонкое расщепление
28
— свободного электрона 28
— спектр
----атома водорода 29 -
----- метильного радикала 29
---- циклогексильного ради-
кала 36
— чувствительность 150
Электрона перенос 135
Эмульсионная полимеризация
с образованием резины 133
Эндотермическая реакция, ре-
330
акциОйная способность и се-
лективность 147, 148
Энергетика присоединения
— галогенов к олефинам 205
— галогеноводородов к оле-
финам 203
Энергии передача
— в фотохимии 73
— метилена 282
Энзиматическое окисление, об-
разование радикалов и
ЭПР-спектры 31
Энтальпия активации 52
Энтропии изменения, когда две
связи в надэфнрах рвутся
одновременно 106
Энтропия активации 51
Этан, из клеточного соедине-
ния метильных радикалов 86
— как донор водорода 146
— реакция с атомарным бро-
мом 181
----с атомами хлора 180
----с хлором 175
Этантиолацетат, присоединение
к октену-1 214
Этила перекись 81
— взрывоопасность 110
Этилбензол, окисляемость 279
— из N-бромсукцинимида 154
------агент роста цепи 239
— реакция с атомами брома
181
------с атомами хлора 180
------с znpezn-бутоксильными
радикалами 158
------с различными радика-
лами 162
------с фенильными радика-
лами 161
Этилбензол, как донор водо-
рода 88, 154
Этилвиниловый эфир в сопо-
лимеризации 237
Этилен, взаимодействие с воз-
духом и перекисью 225
— присоединение к броми-
стому водороду 222
------метильных радикалов
217
------к хлороформу 221
------к четыреххлористому
углероду 221
— реакция с четыреххлори-
стым углеродом 196, 197
— фотолиз 142
Этилдисульфнд, реакция с фе-
нильными радикалами 161
Этиловый эфир линолевой кис-
лоты, самоокисление 276
Этиловый эфир малоновой
кислоты, присоединение ме-
тильных радикалов 217
Этиловый эфир фумаровой
кислоты, присоединение ме-
тильных радикалов 217
Этилхлорацетат, присоедине-
ние к октеиу-1 214
Этилхлорид, реакция с хлором
175
Этил-2-циангептеи-6-оат 222
Этильный радикал, значение
величины Ф 296
— присоединение к аромати-
ческим соединениям 219
— реакции обрыва цепи 294
Этоксиэтилен, присоединение
метильных радикалов 217
Эффект Тромсдорфа 298
оглавление
Предисловие к переводу..............................- . 5
Предисловие..............................................7
ЧАСТЬ ПЕРВАЯ. ПРИРОДА РАДИКАЛОВ
Глава 1. Введение
История...............................................9
Определения . 12
Основные типы радикальных реакций.....................13
Природа стадии роста цепи.............................16
Полярные характеристики радикалов.....................19
Упражнения............................................24
Литература............................................24
Глава 2. Регистрация радикалов
Магнитная восприимчивость............................25
Электронный парамагнитный резонанс....................26
Оптические спектры..................................32
Захват................................................33
Литература............................................33
Дополнительная литература.............................33
Глава 3. Конфигурация радикалов
Упражнения.......................................... 48
Литература..........................................49
Глава 4. Энергетика и скорости реакций
Скорости . 50
Энергия диссоциации связей.........................53
Теплота образования ........................... 54
Упражнения..........................................56
Дополнительная литература...........................56
ЧАСТЬ ВТОРАЯ. ОБРАЗОВАНИЕ РАДИКАЛОВ
Глава 5. Введение. Способы получения радикалов
Облучение . 57
Термический гомолиз.................................57
Окислительно-восстановительные источники радикалов 59
Дополнительная литература...........................60
Глава 6. Фотохимия.
Процессы, протекающие при фотовозбуждеиии веществ 61
Актинометрия . 64
Примеры фотохимических реакций .....................65
332
Карбонильные Соединения...........................68
Упражнения...........................................76
Литература...........................................77
Дополнительная литература..................' . . .78
Глава 7. Термический гомолиз. Перекиси
Перекиси алкилов.....................................81
Перекись ацетила.....................................84
Перекись бензоила....................................89
Другие перекиси ацилов...............................97
Упражнения...........................................98
Литература .г........................................99
Дополнительная литература...........................100
Глава 8. Термический гомолиз. Надкислоты и их эфиры
Упражнения..........................................108
Литература .........................................109
Дополнительная литература...........................109
Г лава 9. Термический гомолиз. Перекись водорода, гидропе-
рекиси и молекулирио-иидуцироваииый гомолиз
Гидроперекись трет-бутила.............................Ш
Гидроперекись кумола.................................ИЗ
Молекулярно-индуцированный гомолиз..................115
Упражнения..........................................122
Литература . 123
Дополнительная литература...........................124
Глава 10. Термический гомолиз. Дзосоедииеиия
Упражнения......................................... 130
Литература..........................................130
Глава 11. Окислительно-восстановительные реакции, образу-
ющие свободные радикалы
Упражнения..........................................137
Литература..........................................140
Дополнительная литература.........................140
ЧАСТЬ ТРЕТЬЯ. РЕАКЦИИ РАДИКАЛОВ.
Глава 12. Реакции отрыва водорода.
Общие особенности реакций отрыва водорода . . .141
Реакции отрыва водорода метильными радикалами . . 151
Реакции отрыва водорода алкоксильньгми радикалами . 156
Реакции отрыва водорода фенильными радикалами . .159
Относительные скорости реакций отрыва водорода раз-
личными радикалами................................162
Полярный эффект в реакциях отрыва водорода . . . 163
Упражнения...........................................169
Литература..........................................170
Дополнительная литература ............................. 171
Глава 13. Галогенирование
Фторирование . ................................171
Хлорирование . .......................... . 173
Бромирование ..................................... 181
Иодирование . . . .........................182
Гипохлорирование ................................. 184
Бромирование N-бромсукцинимидом.....................187
Упражнения..........................................190
Литература.........................................191
Дополнительная литература..........................192
333
Глава 14. Реакции присоединения
Реакции присоединения галогеноводородов й галогенов 197
Реакции присоединения галогенопроизводных метана . 205
Реакции присоединения тиолов и меркаптанов . . . 208
Реакции присоединения других соединений к олефинам
с образованием новых углерод-углеродиых связей . .211
Относительные скорости реакций присоединения . . . 215
Упражнения.........................................219
Литература.........................................222
Дополнительная литература .......................... 223
Глава 15. Радикальные полимеризации
Примеры наиболее часто применяемых пластмасс . . 224
Кинетика полимеризации.............................226
Определение значений констант скоростей .... 233
Сополимеризация .................................... 235
Рост цепи при полимеризации .........................237
Упражнения....................’......................241
Литература.............................' . 242
Дополнительная литература .......................... 242
Глава 16. Замещение в ароматическом ядре
Введение фенильного остатка......................... 244
Механизм фенилирования перекисью бензоила . . 247
Упражнения...........................................253
Литература...........................................254
Дополнительная литература .......................... 254
Глава 17. Перегруппировка
1,2-Арильная миграция..............................256
1,2-Алкильная миграция н миграция водорода . . . 262
Миграция 1,2-галогенов...............................262
Чис-транс-Перегруппировки............................263
Реакции разрыва и замыкания кольца...................266
Межмолекулярный перенос водорода . . . . . .|268
Упражнения...........................................272
Литература , 272
Дополнительная литература ’ ........................ 273
Глава 18. Самоокисление
Инициирование, рост и обрыв цепи.....................275
Упражнения...........................................280
Литература...........................................281
Дополнительная литература ....................... - • 281
Глава 19. Бирадикалы
Синтез метилена и его производных..................283
Реакции метилена................................... 285
Упражнения...........................................290
Литература...........................................290
Дополнительная литература .......................... 291
ЧАСТЬ ЧЕТВЕРТАЯ. РЕАКЦИИ ОБРЫВА ЦЕПИ
Глава 20. Реакции рекомбинации и диспропорционирования
Введение...........................................292
Абсолютные значения констант скоростей...............295
Рекомбинация разных радикалов . . . . . . • 295
Сопоставление реакций рекомбинации и диспропорцио-
нирования ..................................... • • 296
Упражнения •; >....................................299
334
Литература......................................... . 299
Дополнительная литература..........................300
Глава 21. Ингибирование
Кинетика...........................................304
Реакции с участием стабильных свободных радикалов . 306
Упражнение.........................................308
Литература.........................................308
Дополнительная литература к переводу .......... 309
Дополнительная литература..........................311
Предметный указатель...............................312
У. Прайер
СВОБОДНЫЕ РАДИКАЛЫ
Редактор А. А. Алексеева
Художник А. С, Александров
Технический редактор Н. А. Власова
Корректор Г. Л. Кокосова
Сдано в набор 14/V111 1969 г.
Подписано к печати 3/IV 1970 г.
Формат 84Х1081/зз Бумага тип. № 2
Усл. печ. л. 17,64 Уч.-изд. л. 18,43
Тираж 2350 экз. Цена 2 р. 02 к.
Зак. изд. 68131 Зак. тип. 454
Атомиздат, Москва, К-31, ул. Жданова, 5/7.
Московская типография № 6
Главполиграфпрома
Комитета по печати при Совете
Министров СССР
Москва, Ж-88, 1-й Южно-портовый пр., 17.
Список опечаток
Стра- ница . Строка Напечатано Следует читать
133 - 1 4 сверху Fe2+, CO2+f или Cu+ ROOH— Fe2+, Со8+ ИЛИ Си+ ROOH —-= -RO- 4-НО-
172 3 снизу Наибольшие Небольшие
206 12 сверху иономерный мономерный
13 сверху полимерии полимерный
303 2 снизу ют свободные радикалы с образованием стабильных 2. Перенос атома. Перенос атома - сопро- вождается
330 18 снизу циклогексадианнльиый циклогексадиенильный OQOK УКПЛ
Опечатки, отмеченные в этой таблице, исправлены в тексте
Отсканировал Семенюченко Владимир
chem_vova@mail.univ.kiev.ua; vova2002@mail.ru
У. ПРАЙЕР
СВОБОДНЫЕ
РАДИКАЛЫ