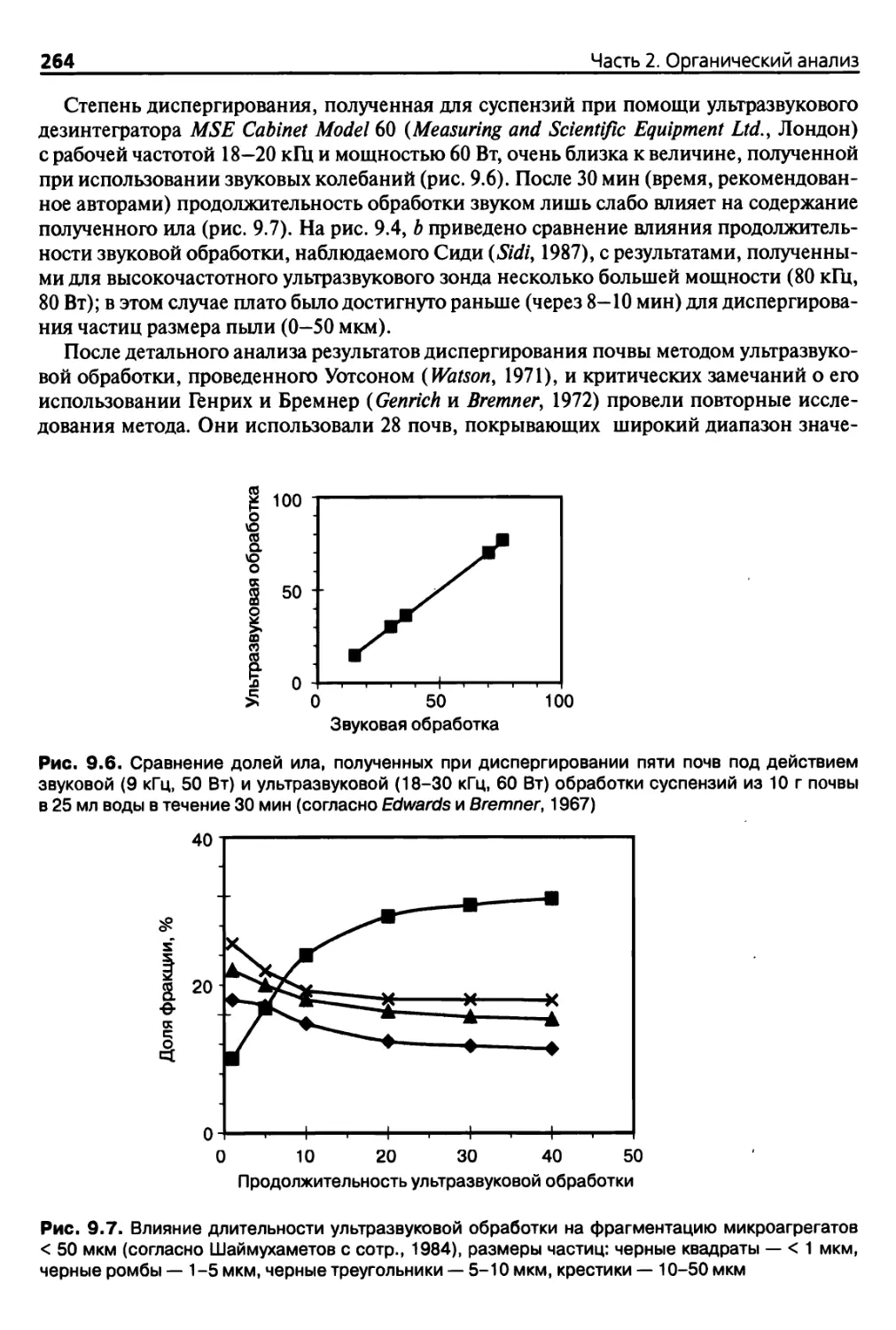
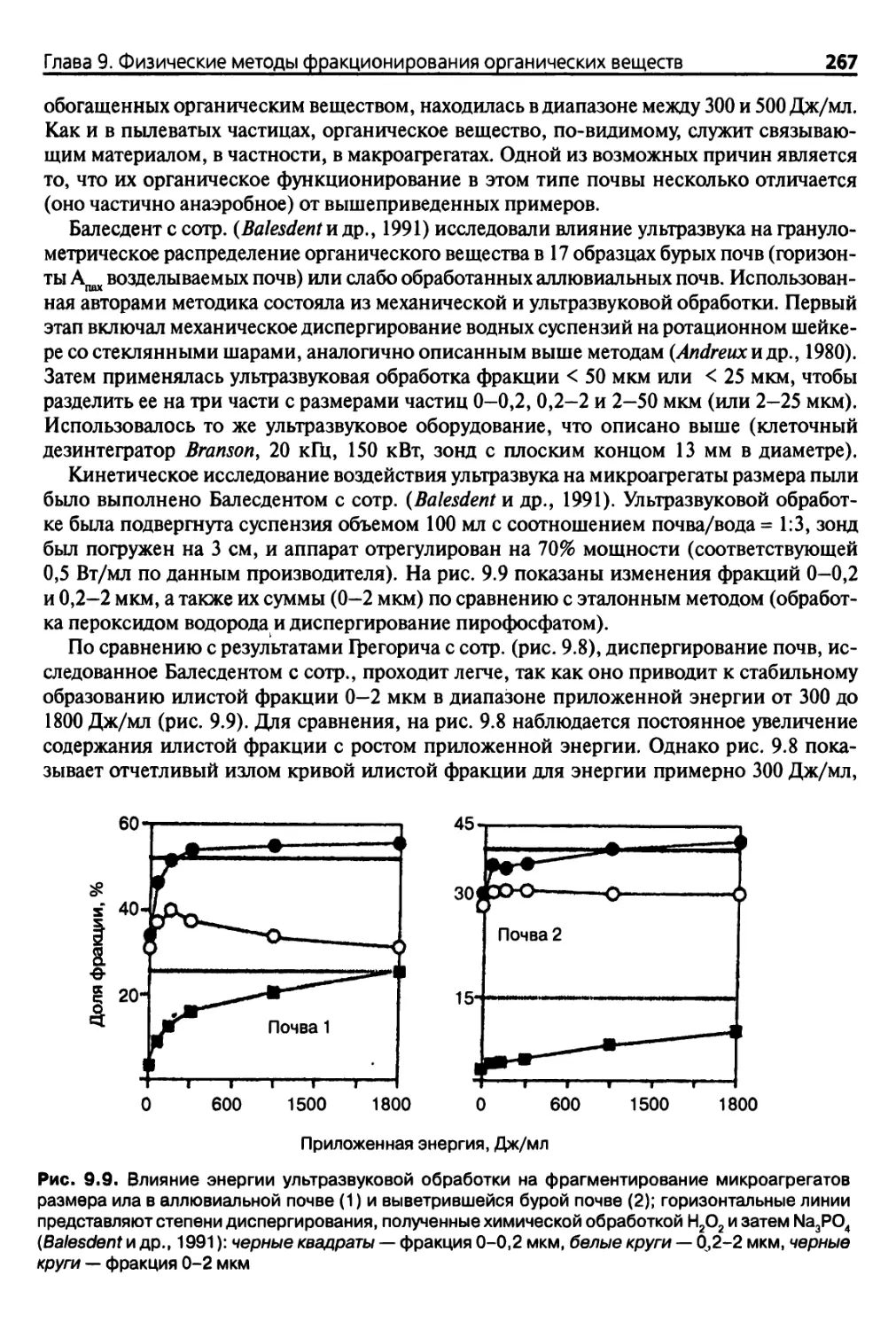
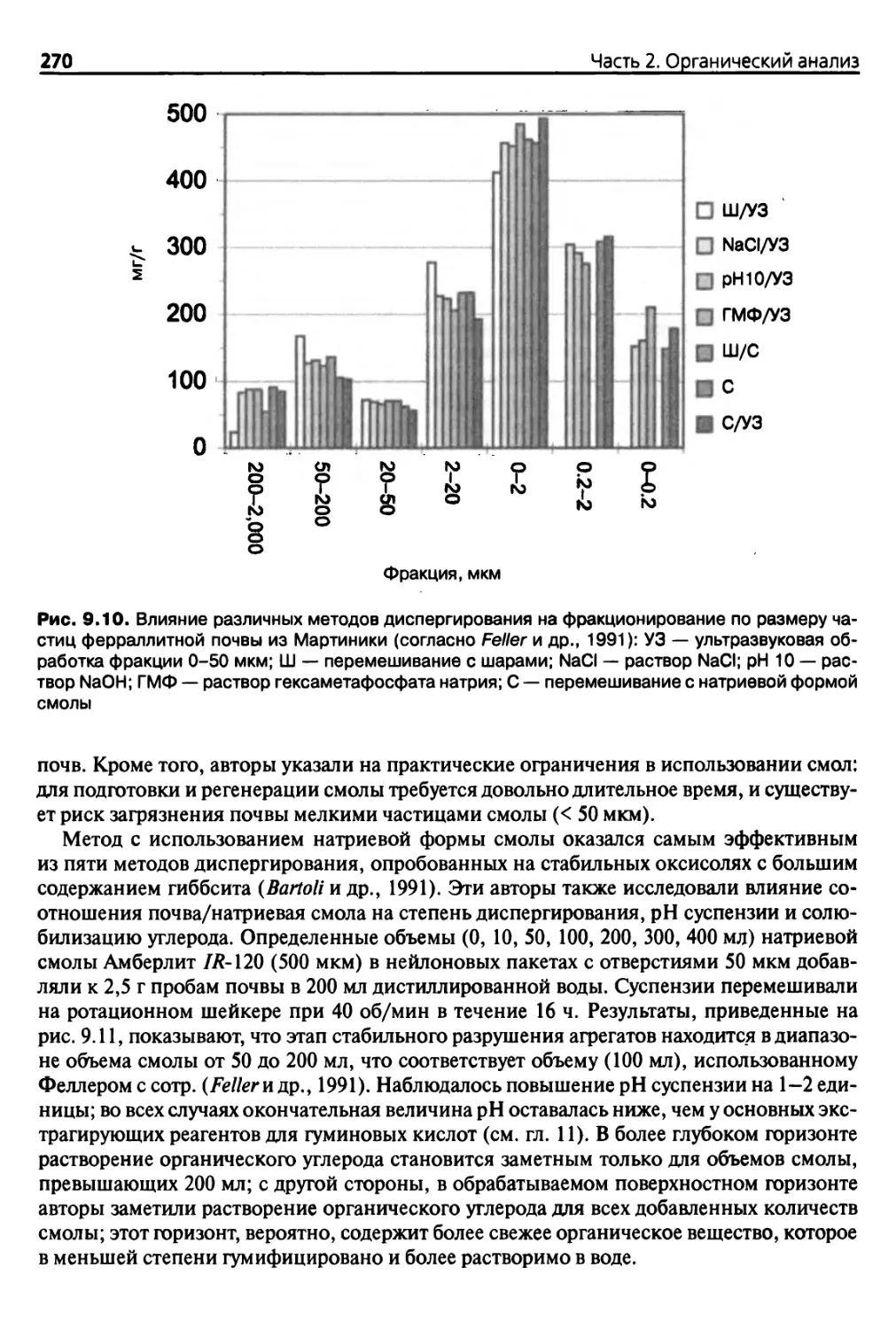
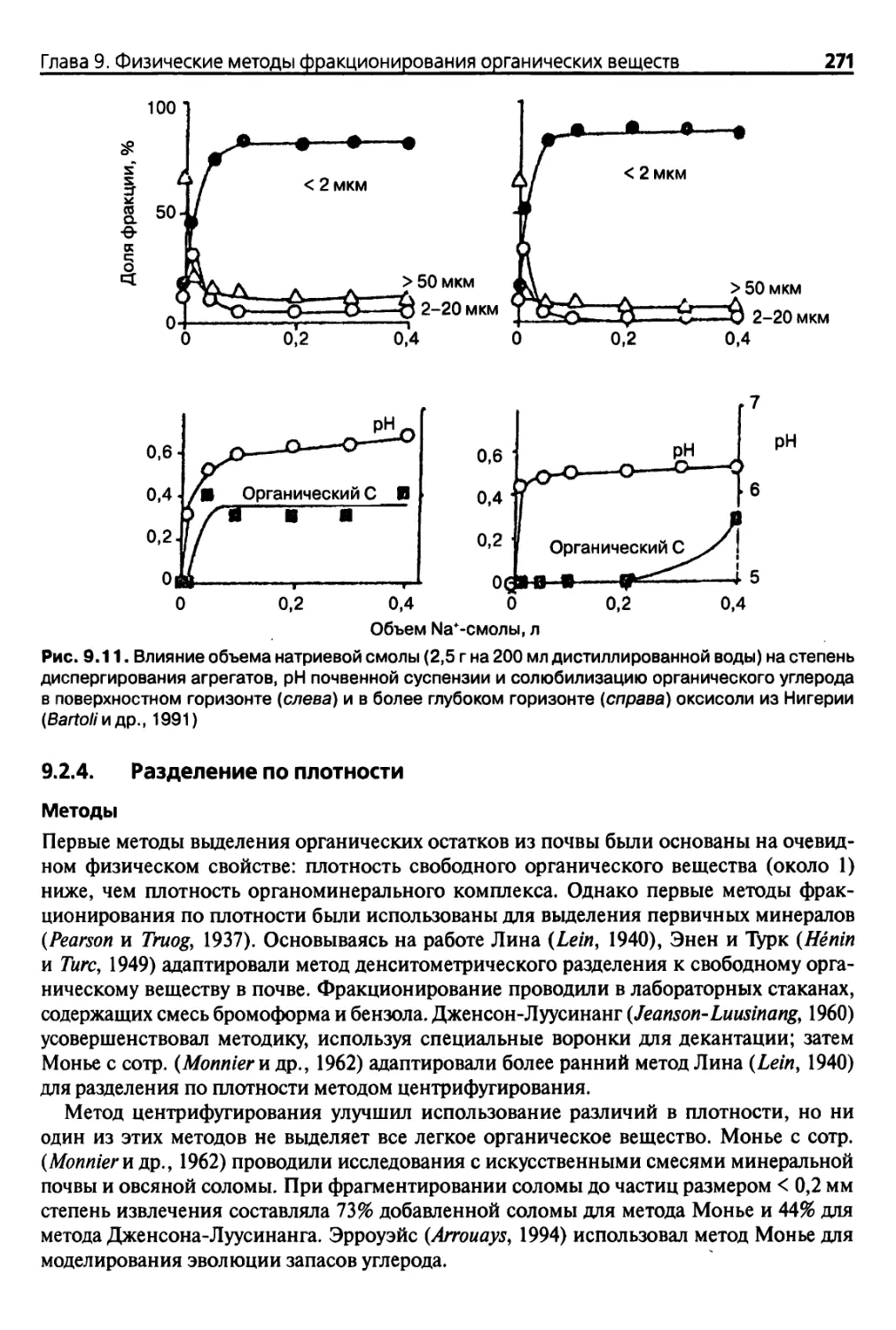
/






































































































































































































































































































































































































































































































































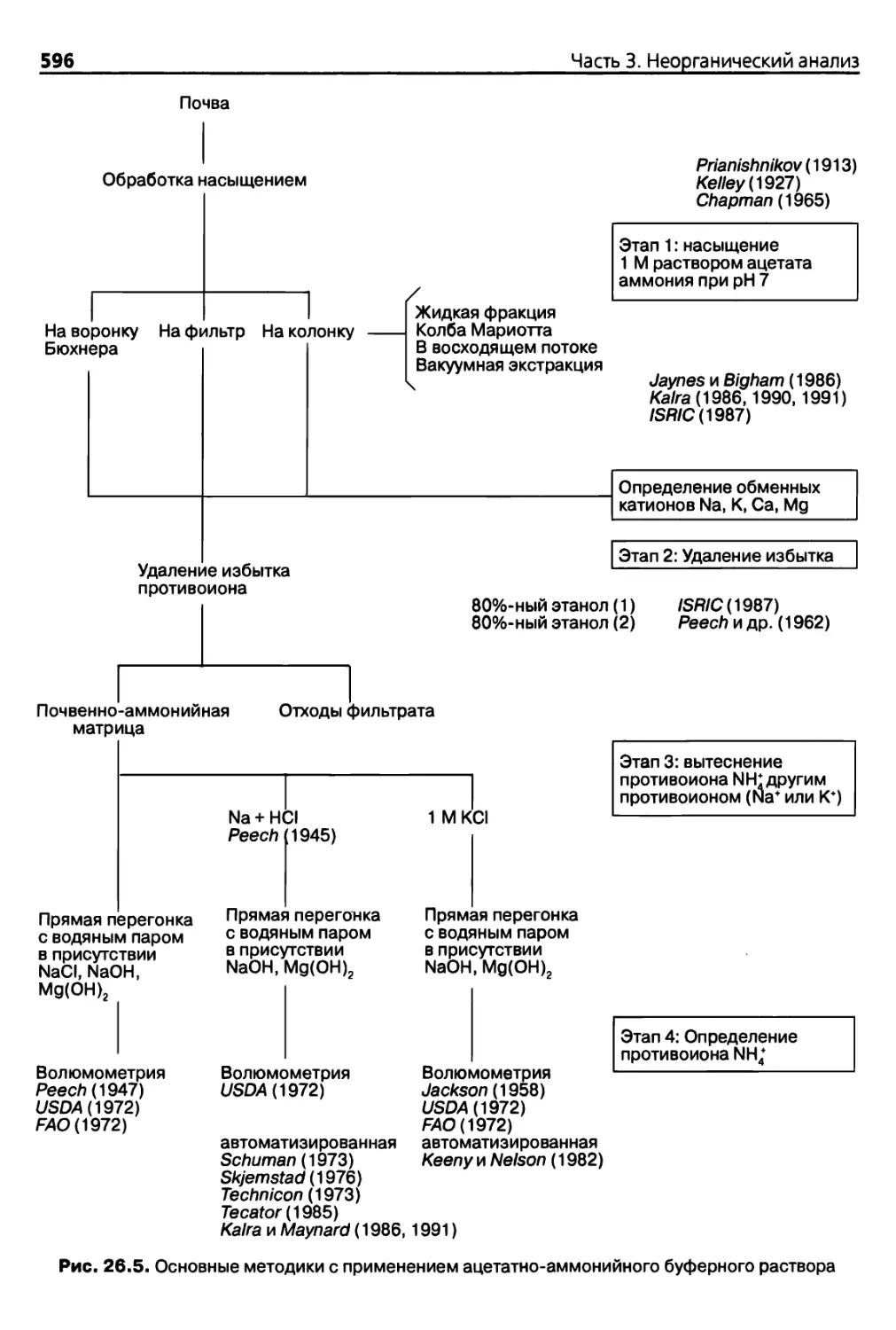









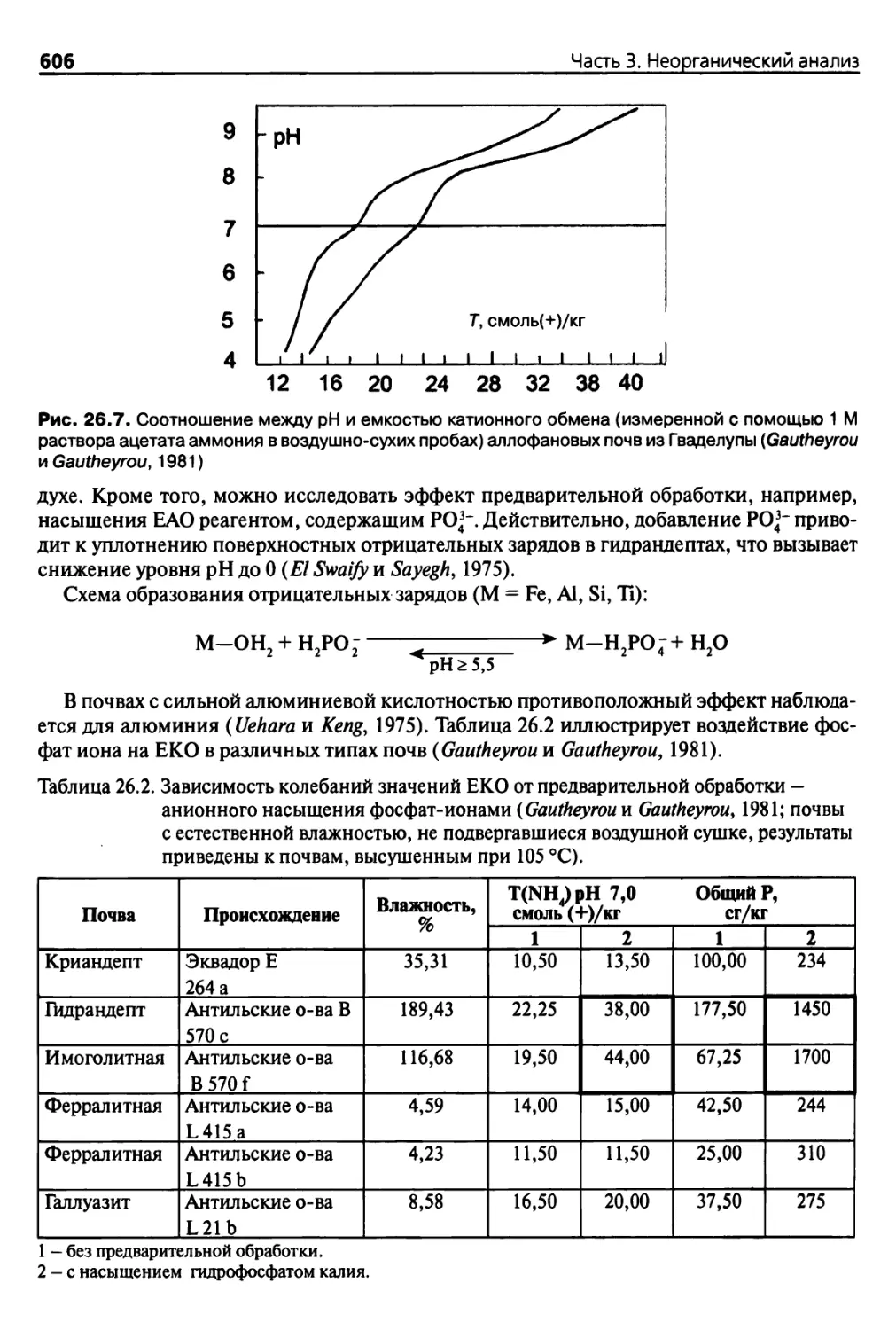







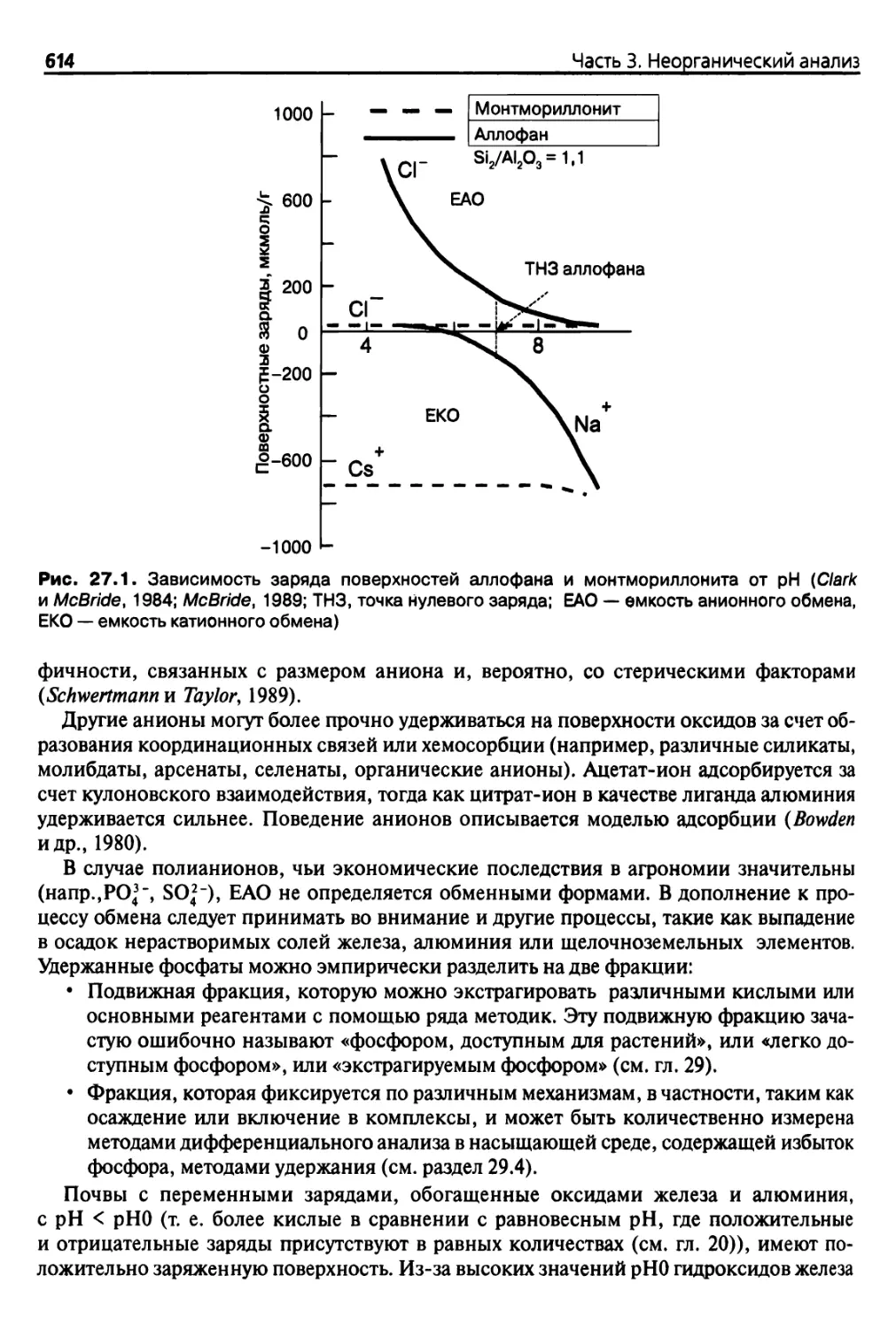










































































































































































Теги: анализ различных веществ аналитический контроль в производстве почвоведение агрономия
ISBN: 978-5-91884-060-3
Год: 2014
Похожие




Текст
М. Пансю, Ж. Готеру
Справочник
Минералогические, органические
и неорганические методы анализа
соон
Marc Pansu
Jacques Gautheyrou
Handbook
of Soil Analysis
Mineralogical, Organic
and Inorganic Methods
with 183 Figures and 84 Tables
Springer
Марк Пансю, Жак Готеру
анализ почвы
Справочник
Минералогические, органические
и неорганические методы анализа
Перевод с английского языка 2-го издания
под редакцией Д. А. Панкратова
и з д О I е (I ». С т и о
Санкт-Петербург
2014
ор
ЦЕНТР
ОВРАЗС
ПРОГРАММ
ПРОФЕССИЯ
ББК40.3
УДК 543.6
П16
М. Пансю, Ж. Готеру
П16 Анализ почвы. Справочник. Минералогические, органические и неорга¬
нические методы анализа : пер. 2-го англ. изд. под ред. Д. А. Панкратова —
СПб.: ЦОП «Профессия», 2014. — 800 с., ил.
ISBN 978-5-91884-060-3
ISBN 978-3-540-31210-9 (англ.)
Приведены минералогические, органические и неорганические методы анализа, и
физические методы обработки образцов. Аналитические методики соответствуют требова¬
ниям стандартов на методы анализа и могут использоваться при контроле состава и качества
почв. Для каждого метода даны принципы, физические и химические основы, преимуще¬
ства, недостатки и область его применения.
Представленные основные методы анализа (ВЭЖХ, ТГА, АЭМ-ИСП, МС-ИСП и др.)
позволят специалистам использовать справочник как при работе в полевых условиях, так и
при проведении фундаментальных и прикладных исследований.
Справочник предназначен для сотрудников профильных лабораторий, служб эколо¬
гического контроля и мониторинга, профильных институтов при проведении анализа окру¬
жающей среды. Он будет полезен агрономам, геологам, климатологам, недропользовате¬
лям, специалистам по гражданскому и промышленному строительству, служб водоканалов,
ЦЛАТИ, исследователям и студентам профильных специальностей.
ББК40.3
УДК 543.6
All rights reserved. Authorized translation from English language edition published by Springer-Verlag Berlin
Heidelberg
Все права защищены. Никакая часть данной книги не может быть воспроизведена в какой бы то ни было
форме без письменного разрешения владельцев авторских прав.
Информация, содержащаяся в данной книге, получена из источников, рассматриваемых издатель¬
ством как надежные. Тем не менее, имея в виду возможные человеческие или технические ошибки, из¬
дательство не может гарантировать абсолютную точность и полноту приводимых сведений и не несет
ответственности за возможные ошибки, связанные с использованием книги.
ISBN 978-3-540-31210-9 (англ.)
ISBN 978-5-91884-060-3
© 2006, Springer-Verlag Berlin Heidelbeig
© ЦОП «Профессия», 2014
© Перевод, оформление: ЦОП «Профессия», 2014
Оглавление
Предисловие к русскому изданию 18
Предисловие 20
Часть 1. Минералогический анализ почв
Пиша 1* Определение влагосодержания почв и потерь при прокаливании 25
1.1. Введение 25
1.2. Определение влагосодержания при 105 °С (Н20~) 26
1.2.1. Основные положения 26
1.2.2. Материалы 28
1.2.3. Образцы 28
1.2.4. Методика 28
1.2.5. Примечания 29
1.3. Определение потерь при прокаливании при 1000 °С (Н20+) 29
1.3.1. Введение 29
1.3.2. Основные положения 32
1.3.3. Оборудование 32
1.3.4. Методика выполнения 32
1.3.5. Обработка результатов 32
1.3.6. Примечания 33
Использованная литература 33
Пшва 2. Анализ гранулометрического состава 35
2.1. Введение 35
2.1.1. Гранулометрический состав в почвоведении 35
2.1.2. Основные положения 37
2.1.3. Закон седиментации 38
2.1.4. Условия применения закона Стокса 46
2.2. Стандартные методы 48
2.2.1. Предварительная обработка образцов 48
2.2.2. Суспендирование и диспергирование частиц 52
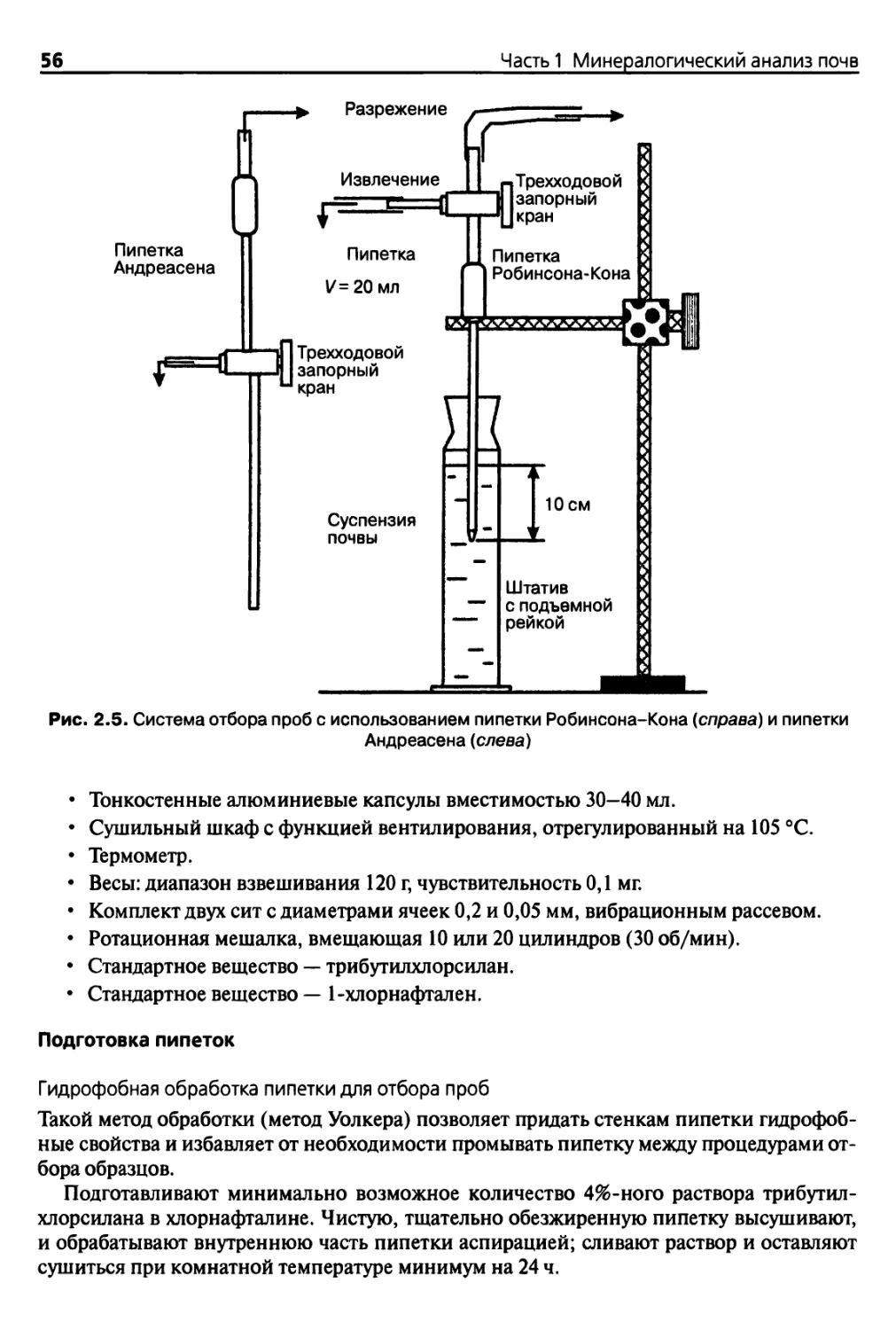
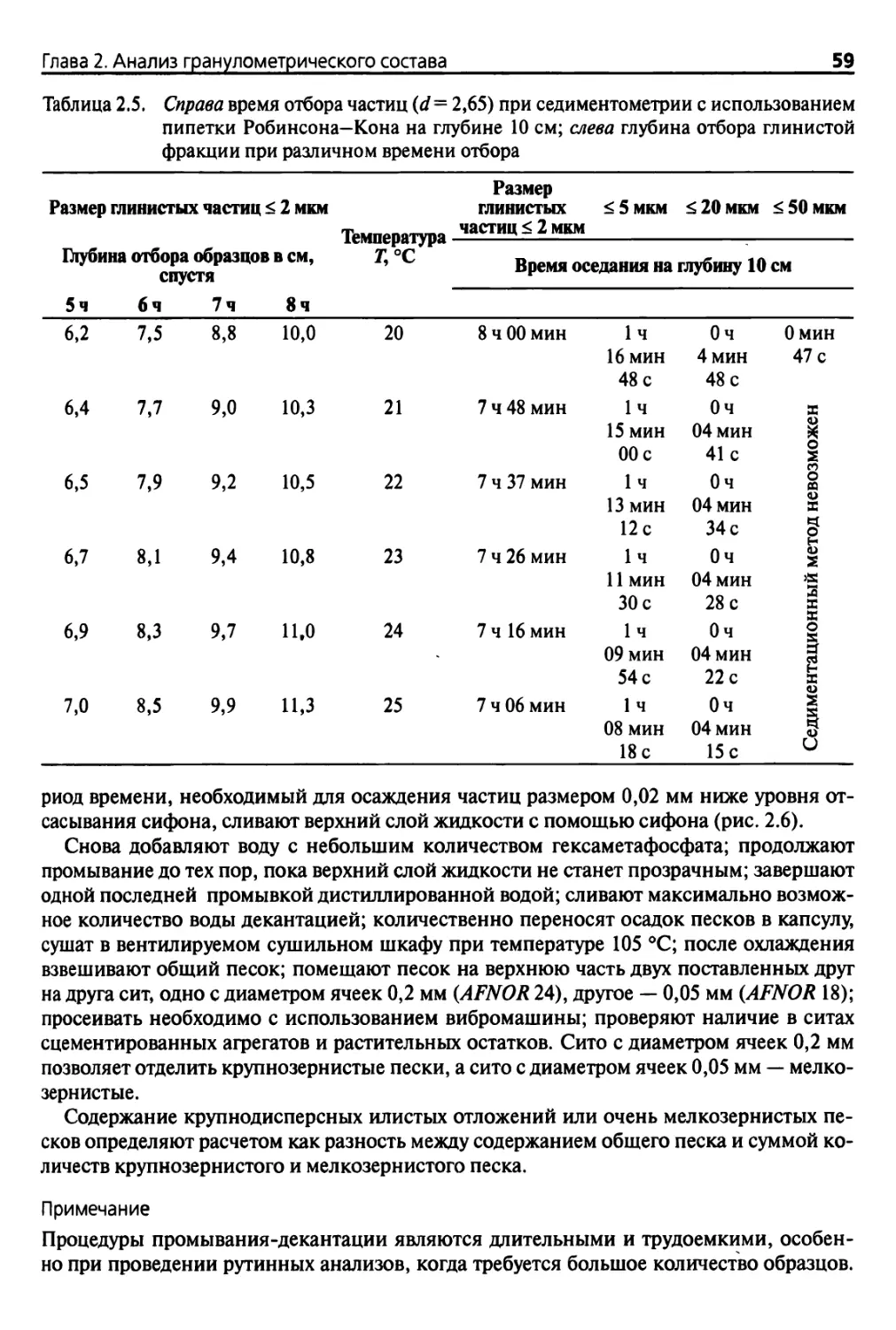
2.2.3. Пипеточный метод согласно Робинсону-Кону
или Андреасену 55
2.2.4. Метод определения плотности при погружении ареометра
на различную глубину 62
2.2.5. Метод определения плотности при погружении ареометра
на фиксированную глубину 66
2.2.6. Анализ гранулометрического состава песка 67
2.3. Автоматизированное оборудование 68
2.3.1. Введение 68
2.3.2. Седиментационный анализ при прямом использовании
гравитационного поля 69
6
Оглавление
2.3.3. Методы, использующие принцип ускоренной
седиментации 71
2.3.4. Методы, использующие рассеяние и дифракцию лазерного
излучения 72
2.3.5. Методы, использующие оптические и электрические свойства... 72
2.3.6. Методы, использующие прямое наблюдение частиц 73
2.3.7. Методы, использующие электропроводные свойства
материалов 74
Использованная литература 74
Дополнительная литература 75
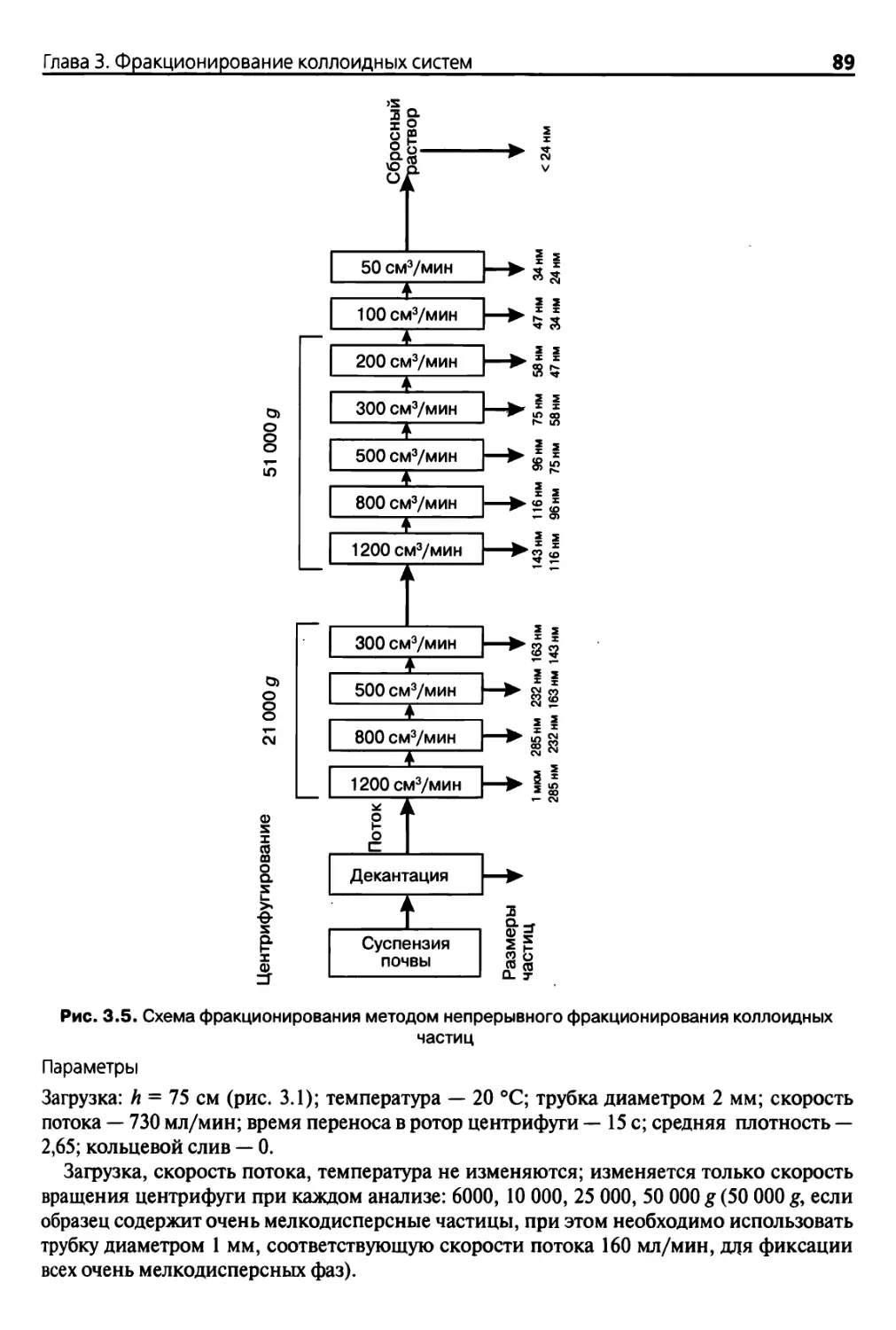
ПгаваЗ. Фракционирование коллоидных систем 79
3.1. Введение 79
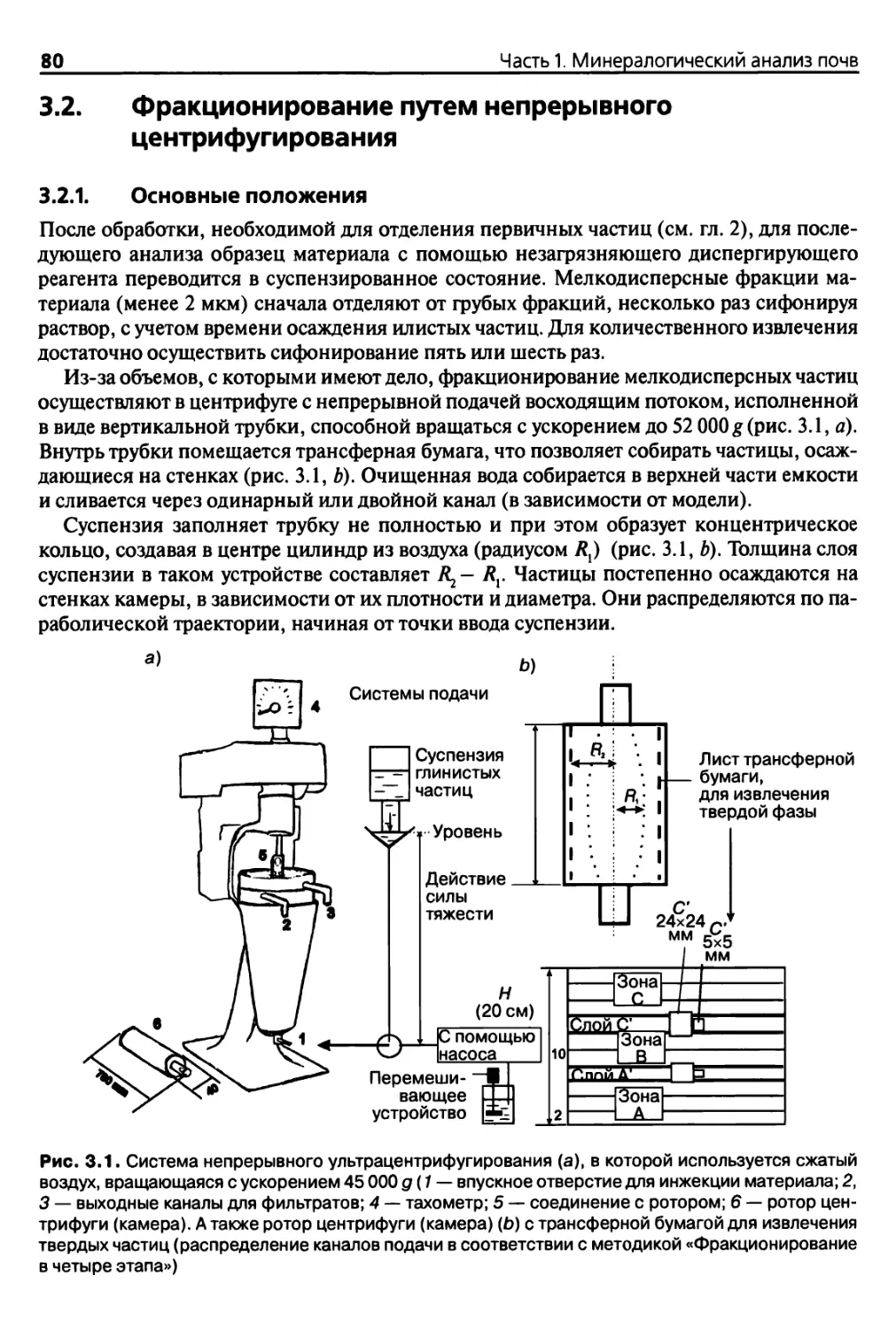
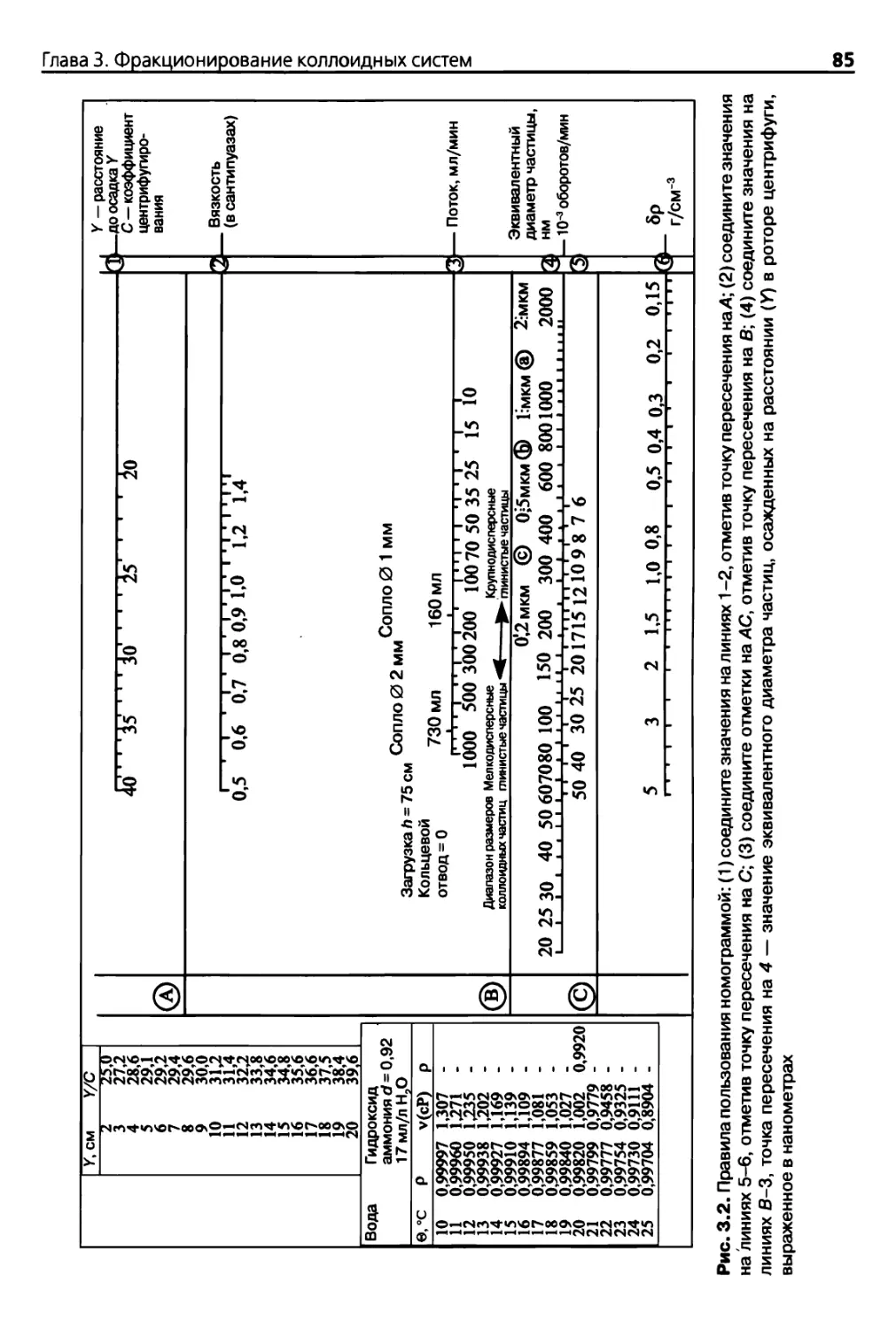
3.2. Фракционирование путем непрерывного центрифугирования 80
3.2.1. Основные положения 80
3.2.2. Теоретические основы 82
3.2.3. Оборудование и реактивы 86
3.2.4. Методика 87
3.3. Предварительная обработка экстрагированных фаз 92
Использованная литература 92
Глава 4. Характеристика минералогического состава методом рентгеновской
дифрактометрии 94
4.1. Введение 94
4.1.1. Рентгеновская дифракция и минералогия 94
4.1.2. Основные положения 96
4.1.3. Оборудование для проведения рентгеновской дифракции 97
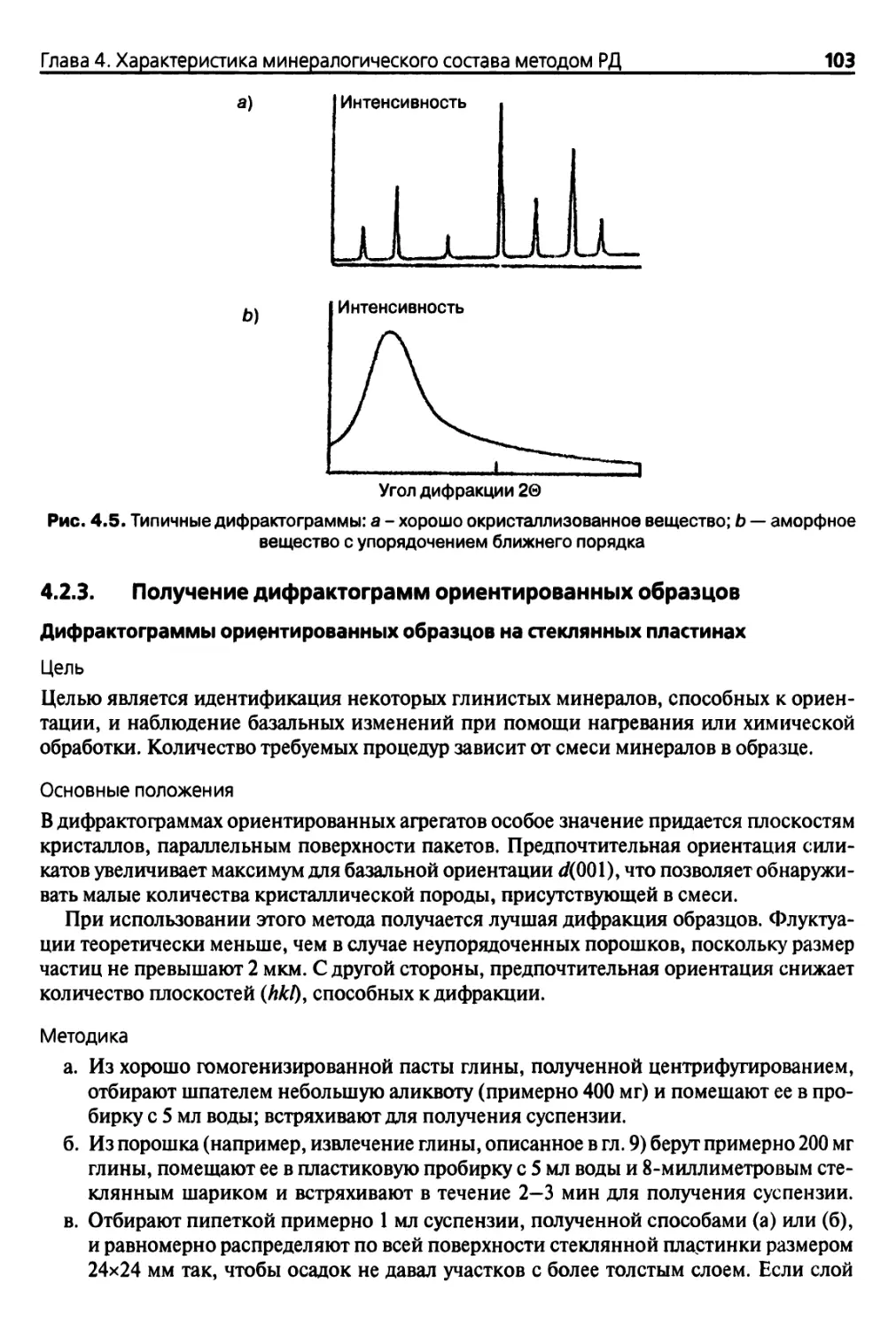
4.2. Качественная дифрактометрия 99
4.2.1. Краткий обзор методов подготовки образцов 99
4.2.2. Получение дифрактограмм порошкообразных материалов 99
4.2.3. Получение дифрактограмм ориентированных образцов 103
4.2.4. Предварительная обработка глин 107
4.2.5. Качественная дифрактометрия 120
4.3. Количественный минералогический анализ 123
4.3.1. Практическая значимость 123
4.3.2. Количественный минералогический анализ методом
рентгеновской дифракции 123
4.3.3. Количественный минералогический анализ
с использованием сложного технического оборудования 127
Использованная литература 129
Дополнительная литература 129
Глава 5. Минералогический анализ методом инфракрасной спектрометрии 133
5.1. Введение 133
5.1.1. Основные положения 133
5.1.2. Оборудование, используемое в методе инфракрасной
спектрометрии 134
5.2. Применение ИК-спектрометрии в минералогии 137
5.2.1. Оборудование и материалы 137
Оглавление
7
5.2.2. Подготовка образцов 138
5.2.3. Краткое руководство по интерпретации спектров 144
5.2.4. Количественный анализ 148
5.3. Другие ИК-методы 151
5.3.1. Ближняя инфракрасная спектрометрия (БИКС) 151
5.3.2. Сочетание термических методов анализа и ИК-спектрометрии
с Фурье-преобразованием при анализе летучих соединений... 153
5.3.3. Инфракрасная микроскопия 153
5.3.4. Спектроскопия комбинационного рассеяния 154
Использованная литература 155
Дополнительная литература в хронологической последовательности 155
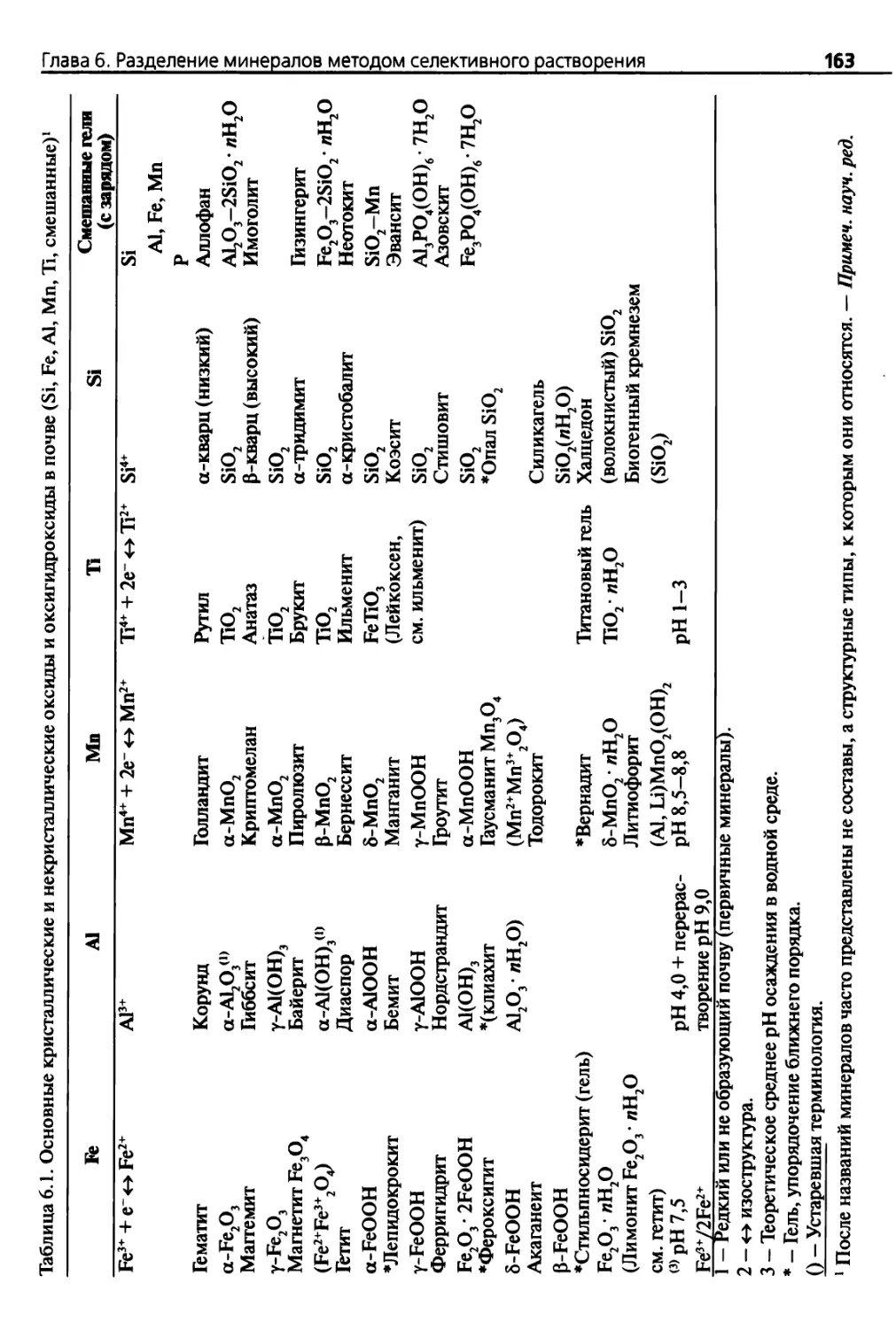
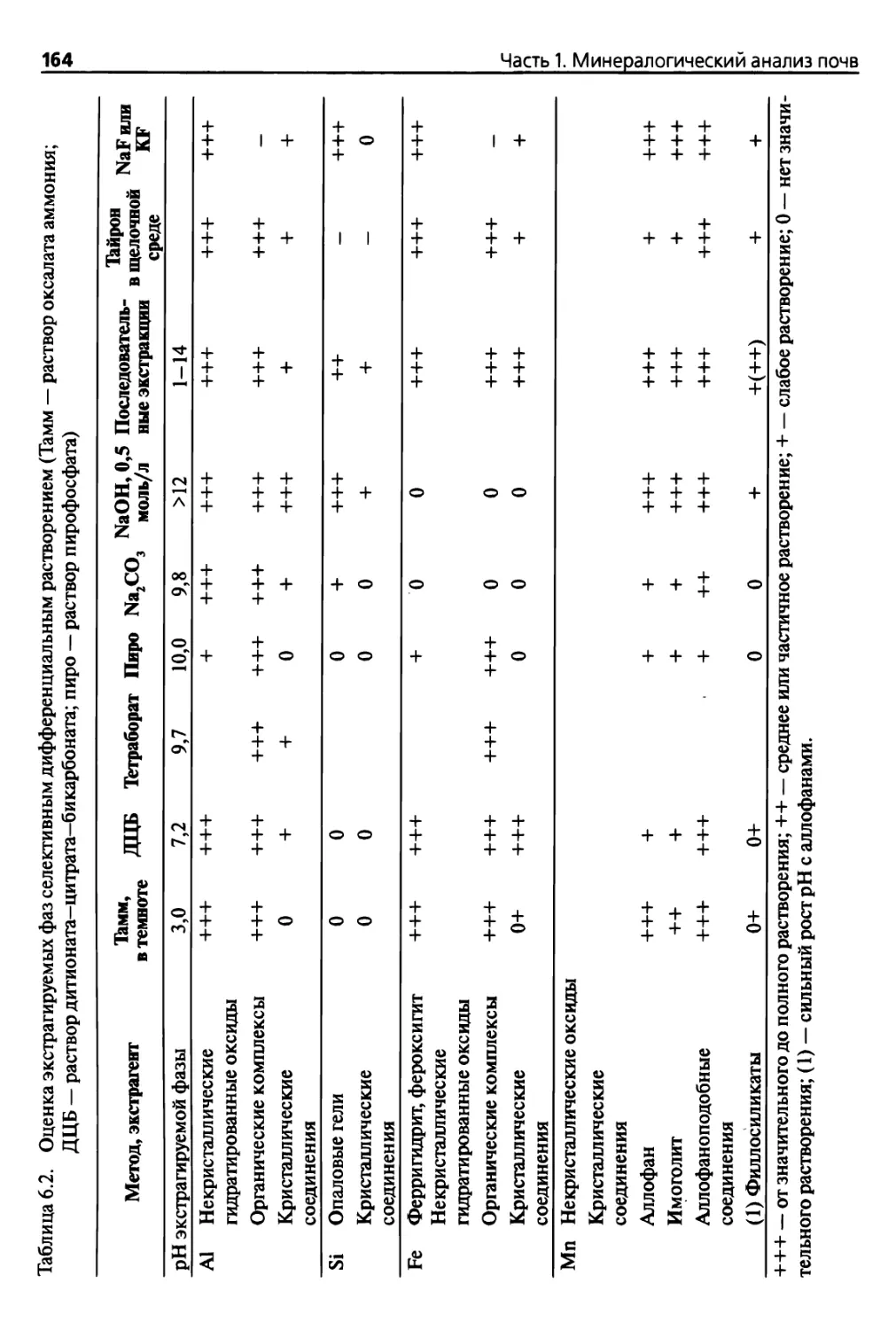
Глава 6. Разделение минералов методом селективного растворения 158
6.1. Введение 158
6.1.1. Кристаллическое состояние глинистых минералов 158
6.1.2. Инструментальные и химические методы анализа 160
6.1.3. Методы селективного растворения 162
6.1.4. Реагенты и искусственные стандарты 166
6.2. Основные методы селективного растворения 169
6.2.1. Метод, основанный на использовании кислого оксалата
в темноте (КОТ) 169
6.2.2. Метод, основанный на использовании
дитионит-цитрат-бикарбоната (ДЦБ) 174
6.2.3. Метод, основанный на использовании
этилендиаминтетрауксусной кислоты (ЭДТА) 179
6.2.4. Пирофосфатный метод 181
6.2.5. Экстракция в сильнощелочной среде 185
6.3. Другие методы исследований, модификации и выбор подходящего
метода 190
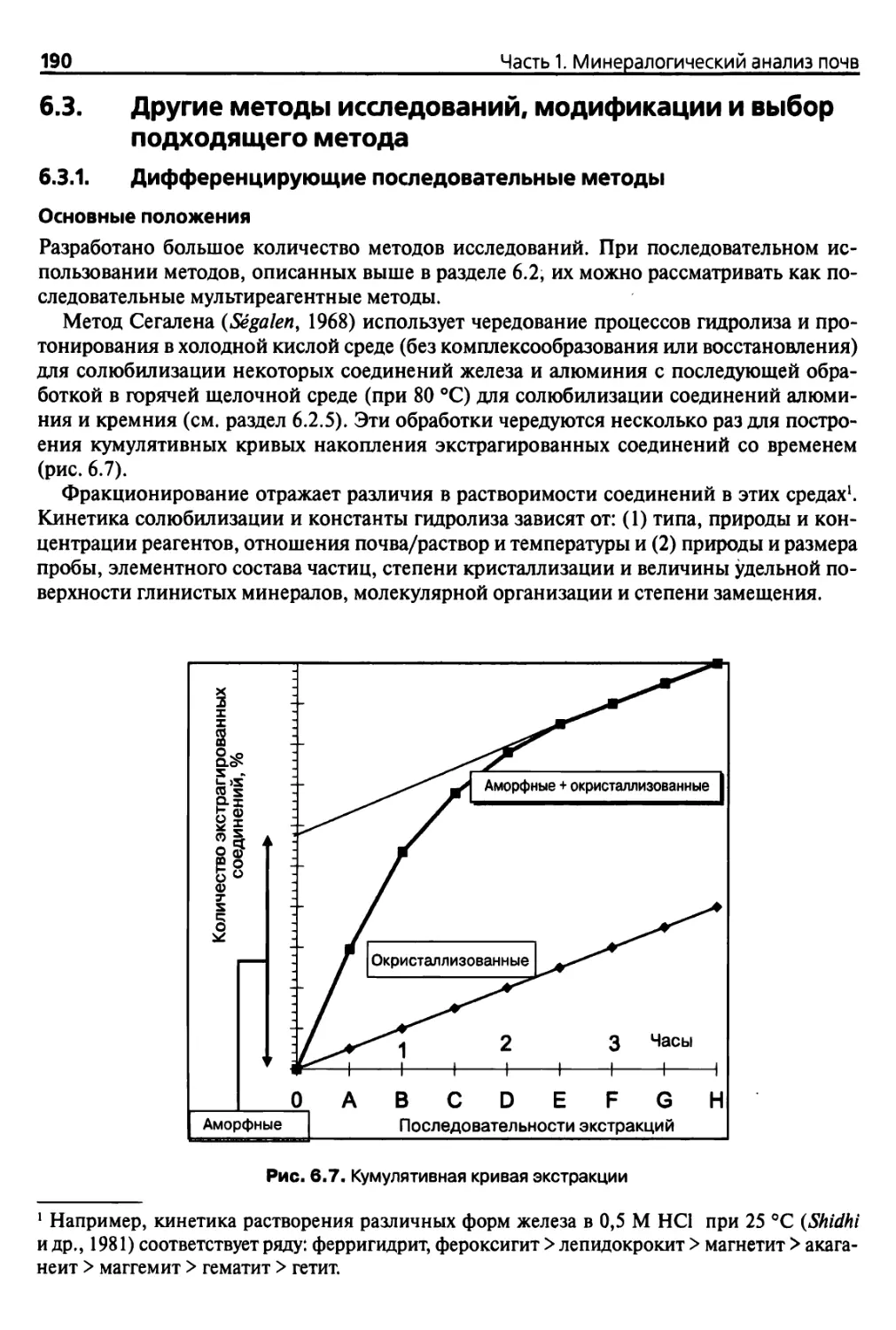
6.3.1. Дифференцирующие последовательные методы 190
6.3.2. Селективные методы анализа аморфных веществ 193
6.3.3. Краткий обзор применения дифференциальных методов
анализа 196
Использованная литература 196
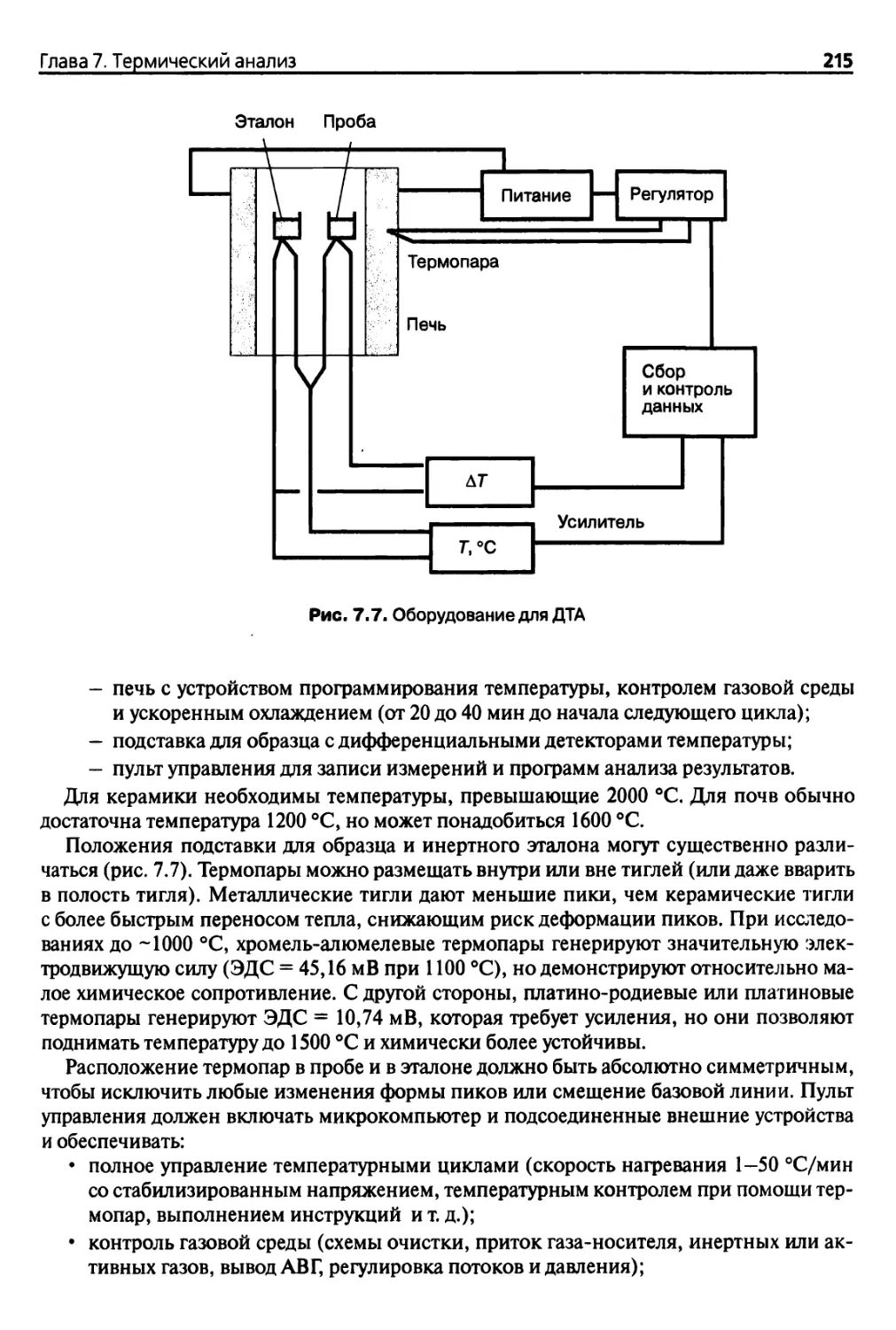
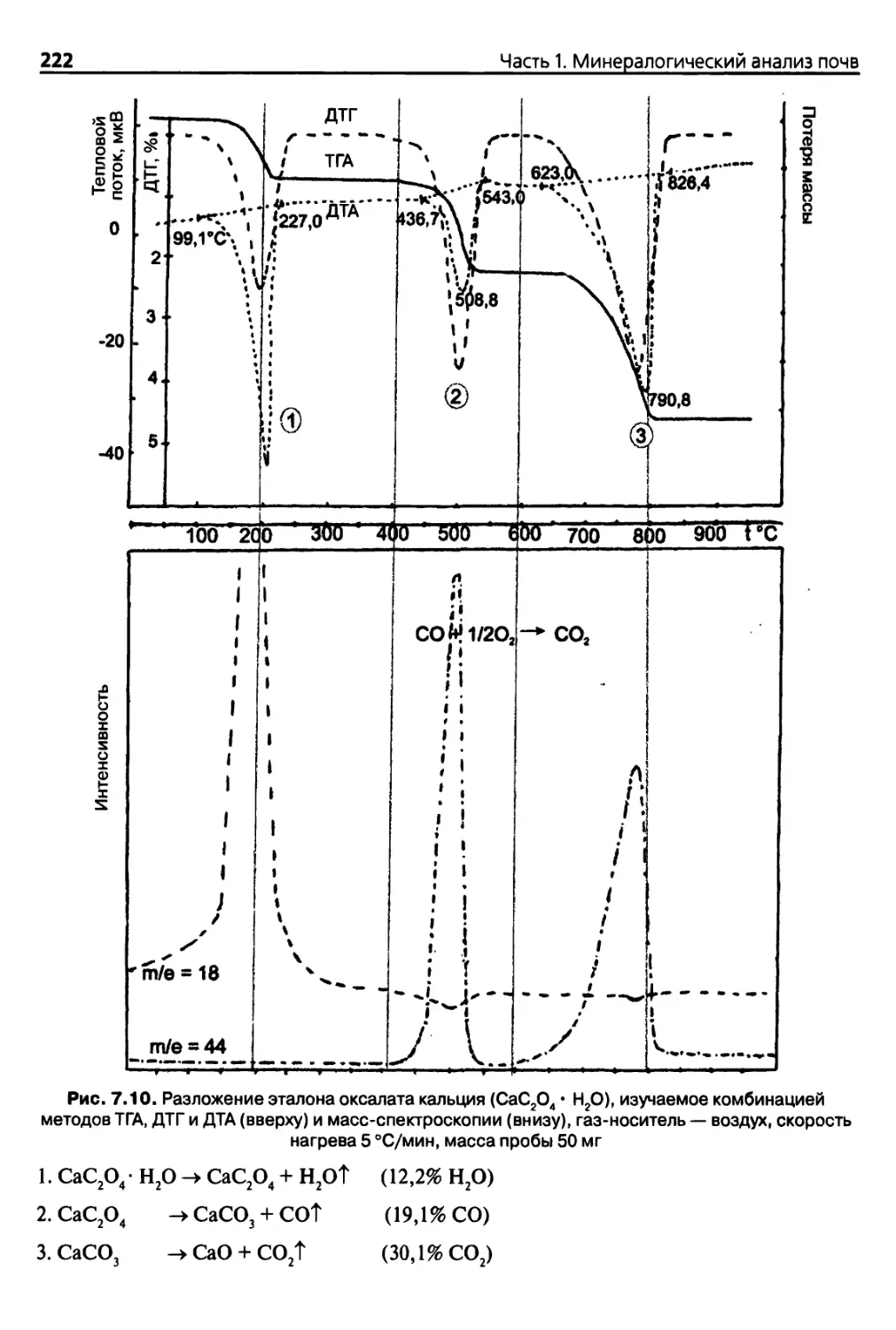
Глава 7. Термический анализ 200
7.1. Введение *. 200
7.1.1. Определения 200
7.1.2. Практическая значимость 201
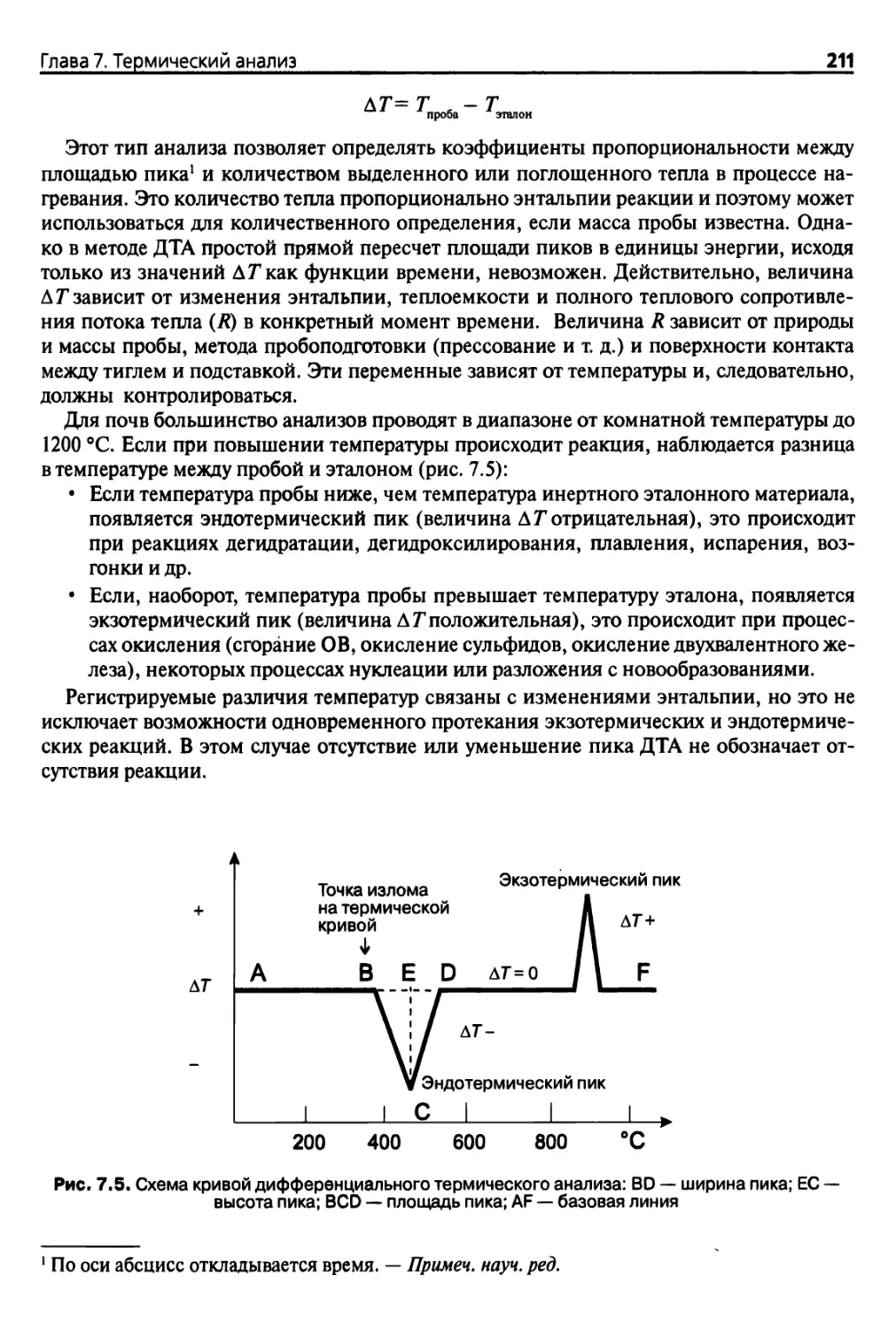
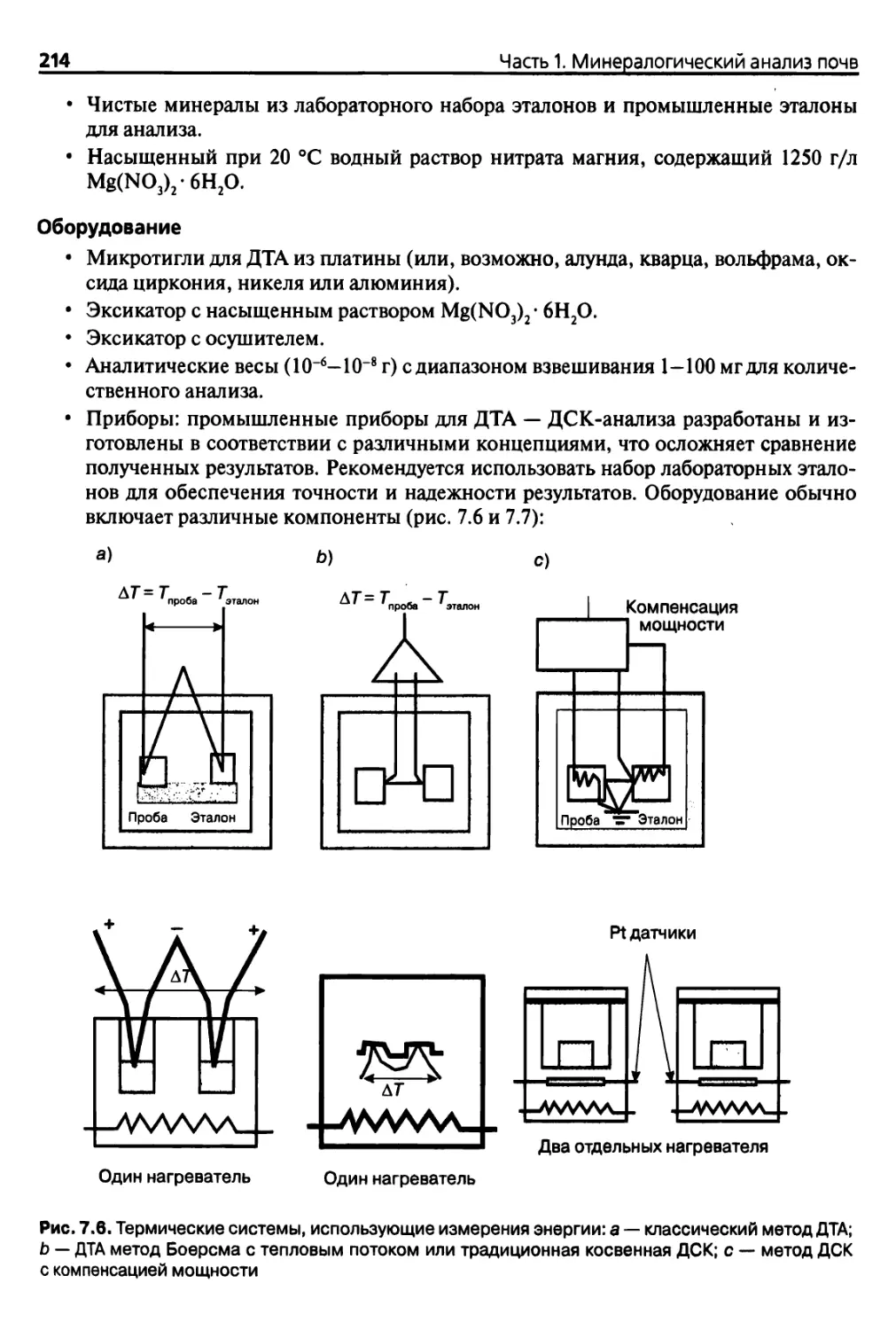
7.2. Классические методы 204
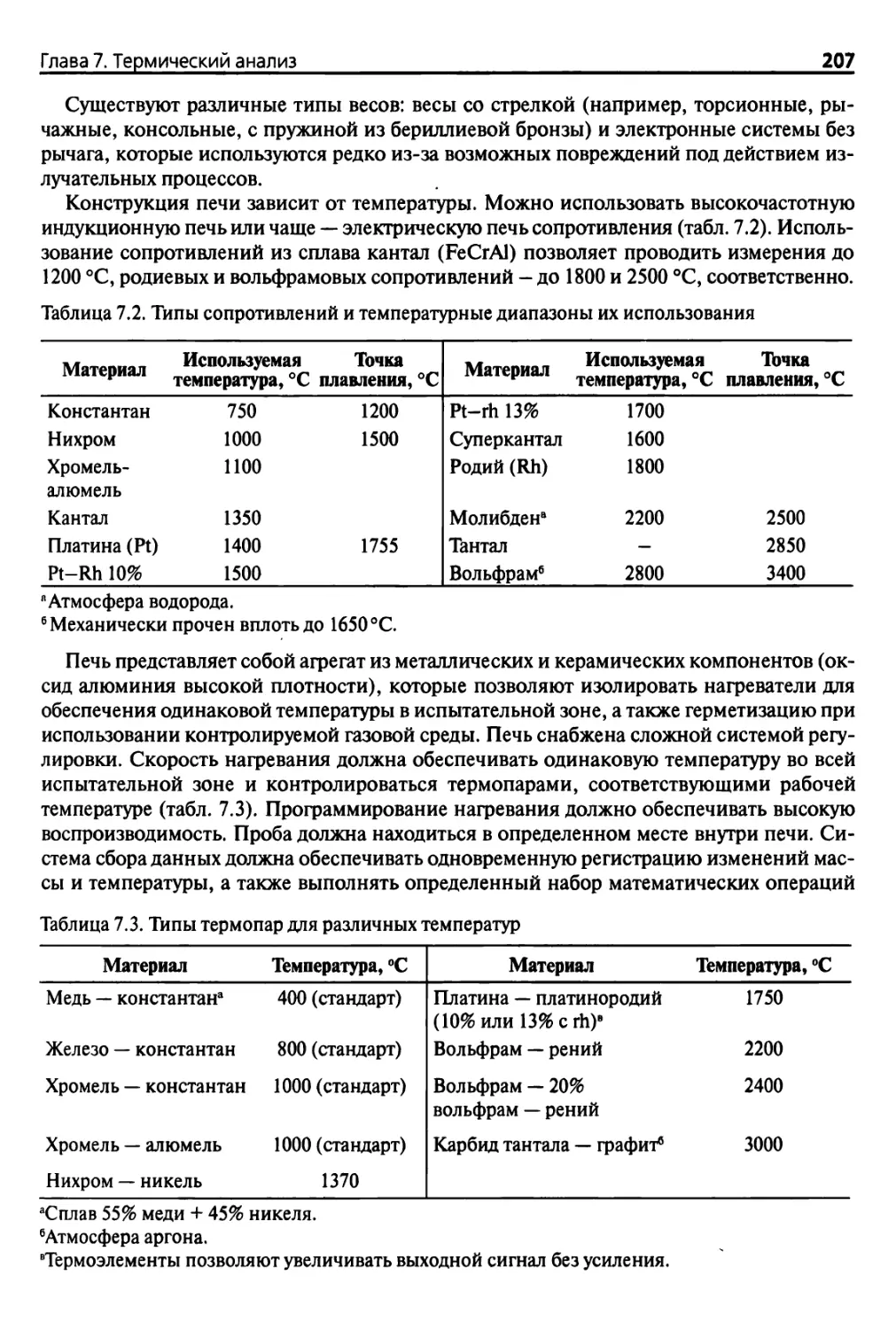
7.2.1. Термогравиметрический анализ 204
7.2.2. Дифференциальный термический анализ
и дифференциальная сканирующая калориметрия 210
7.3. Многокомпонентное оборудование для проведения термического
анализа 220
7.3.1. Основные положения 220
7.3.2. Сочетание термического анализа и анализа выделяемых газов... 220
Использованная литература * 223
Дополнительная литература 223
8
Оглавление
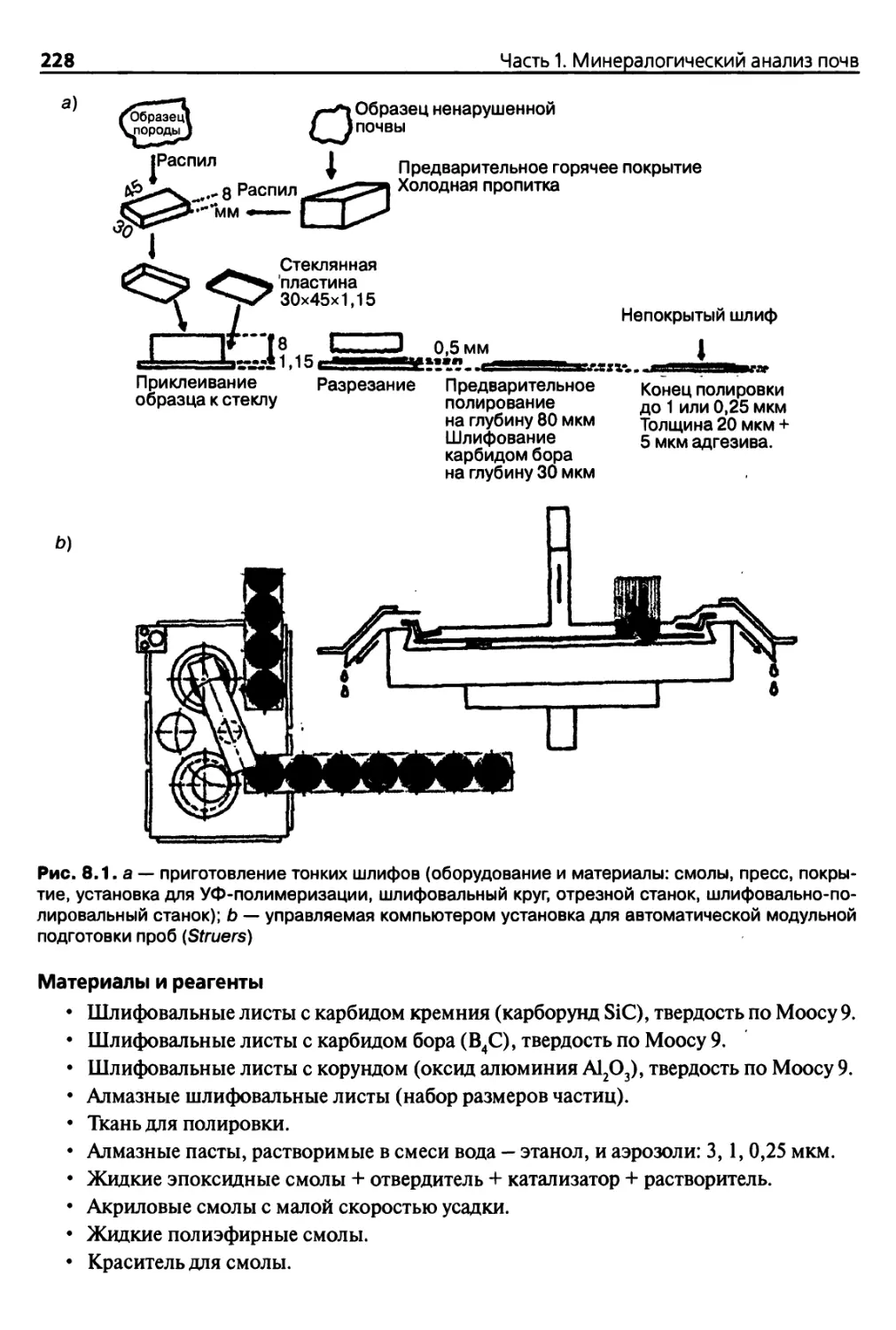
Diana 8. Микроскопический анализ 225
8.1. Введение 225
8.2. Подготовка образцов 226
8.2.1. Способы подготовки образцов 226
8.2.2. Нанесение покрытий и пропитывание тонких пластин 226
8.2.3. Сетки и реплики для просвечивающей электронной
микроскопии 231
8.2.4. Монтаж образцов для проведения сканирующей
электронной микроскопии 233
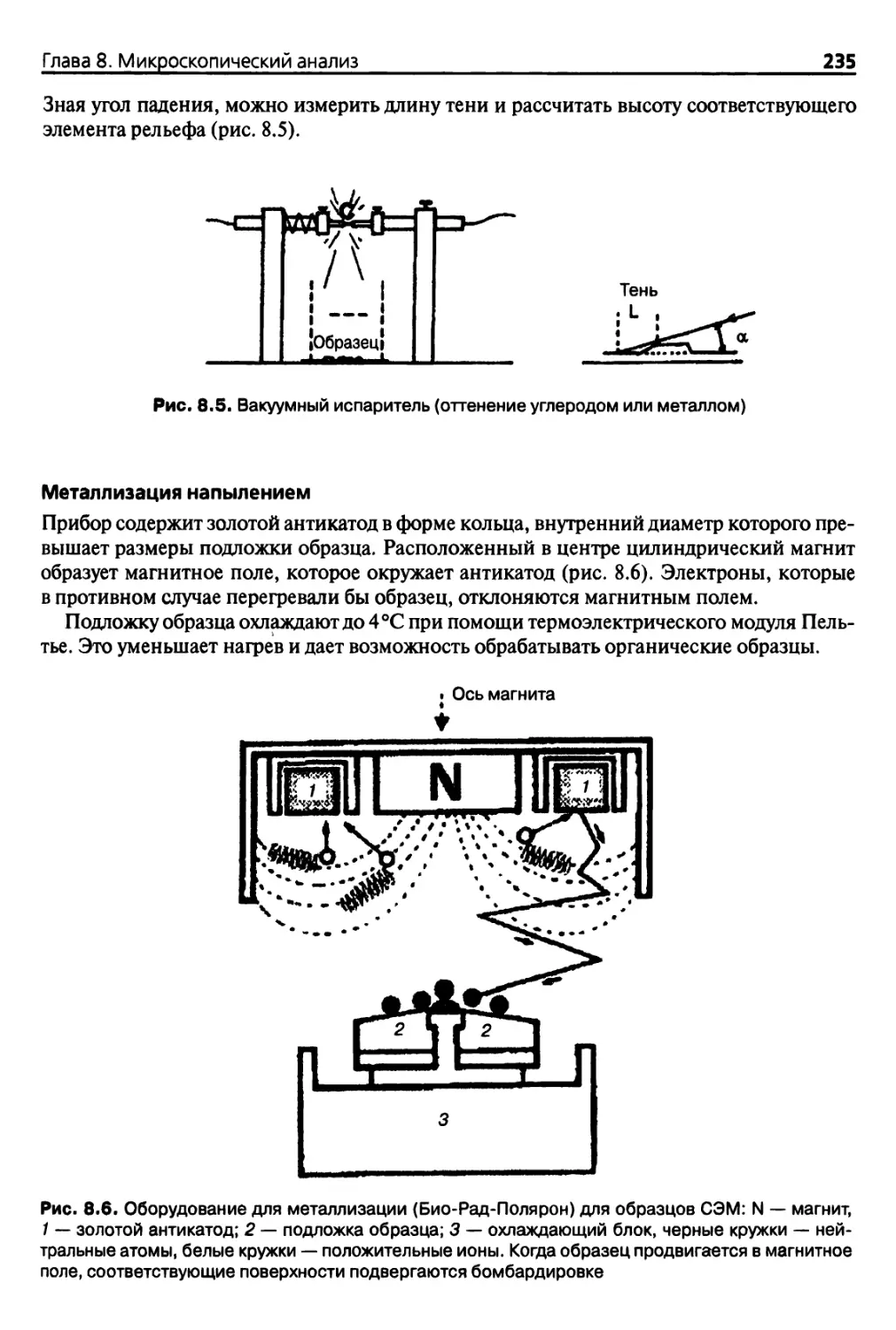
8.2.5. Обработка поверхности (оттенение, покрытие углеродом,
металлизация) 234
8.3. Микроскопические исследования 236
8.3.1. Оптическая микроскопия 236
8.3.2. Электронная микроскопия, общая информация 238
8.3.3. Просвечивающая электронная микроскопия,
микродифракция 240
8.3.4. Сканирующая электронная микроскопия 247
8.3.5. Полный элементный анализ методом рентгеновской
спектрометрии 249
Использованная литература 250
Библиографические ссылки в хронологической последовательности 251
Часть 2. Органический анализ
Глава 9. Физические методы фракционирования органических веществ 255
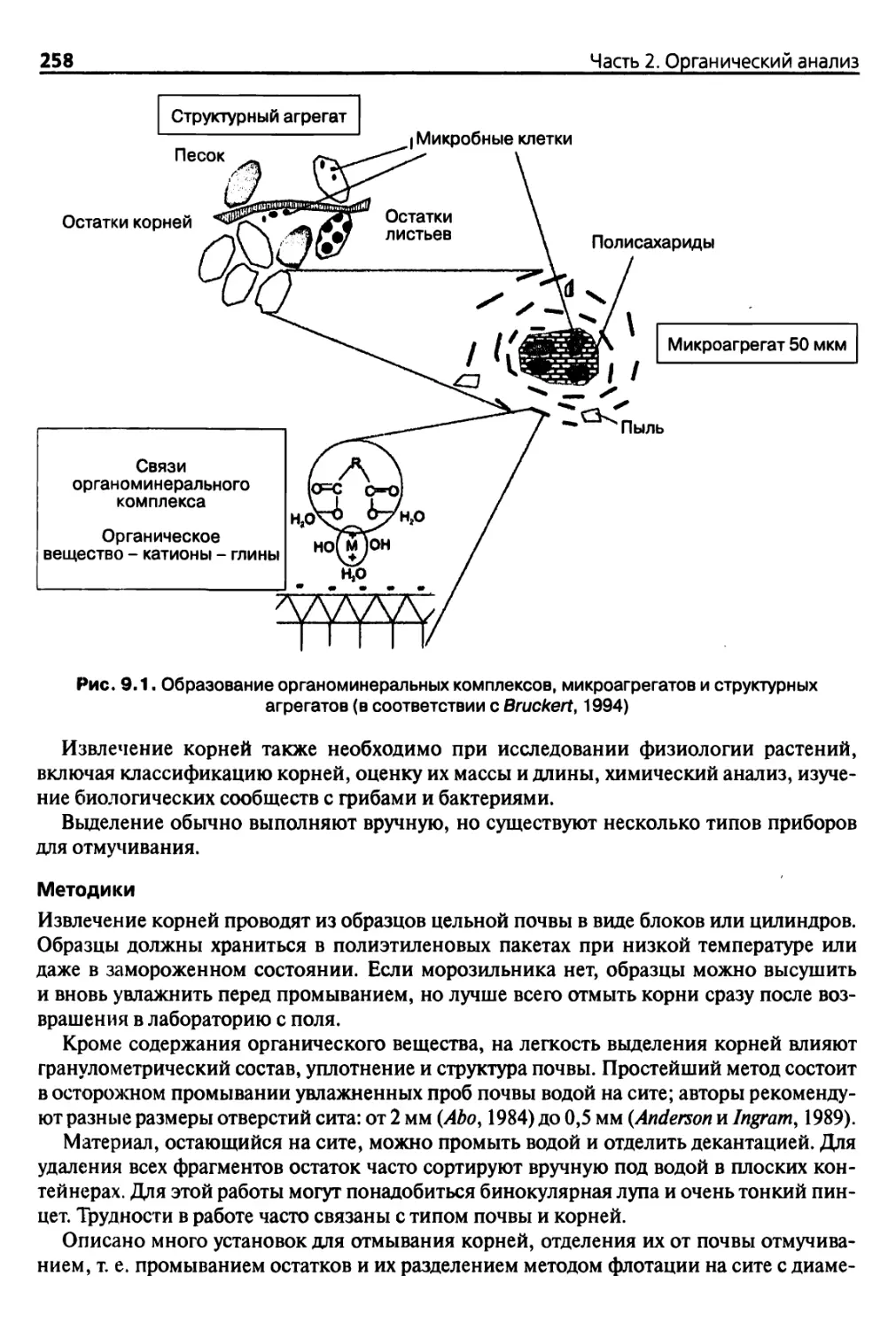
9.1. Принципы и ограничения 255
9.1.1. Формы органического вещества в почве 255
9.1.2. Основные положения 255
9.1.3. Затруднения 256
9.2. Методы фракционирования 257
9.2.1. Ютссификация 257
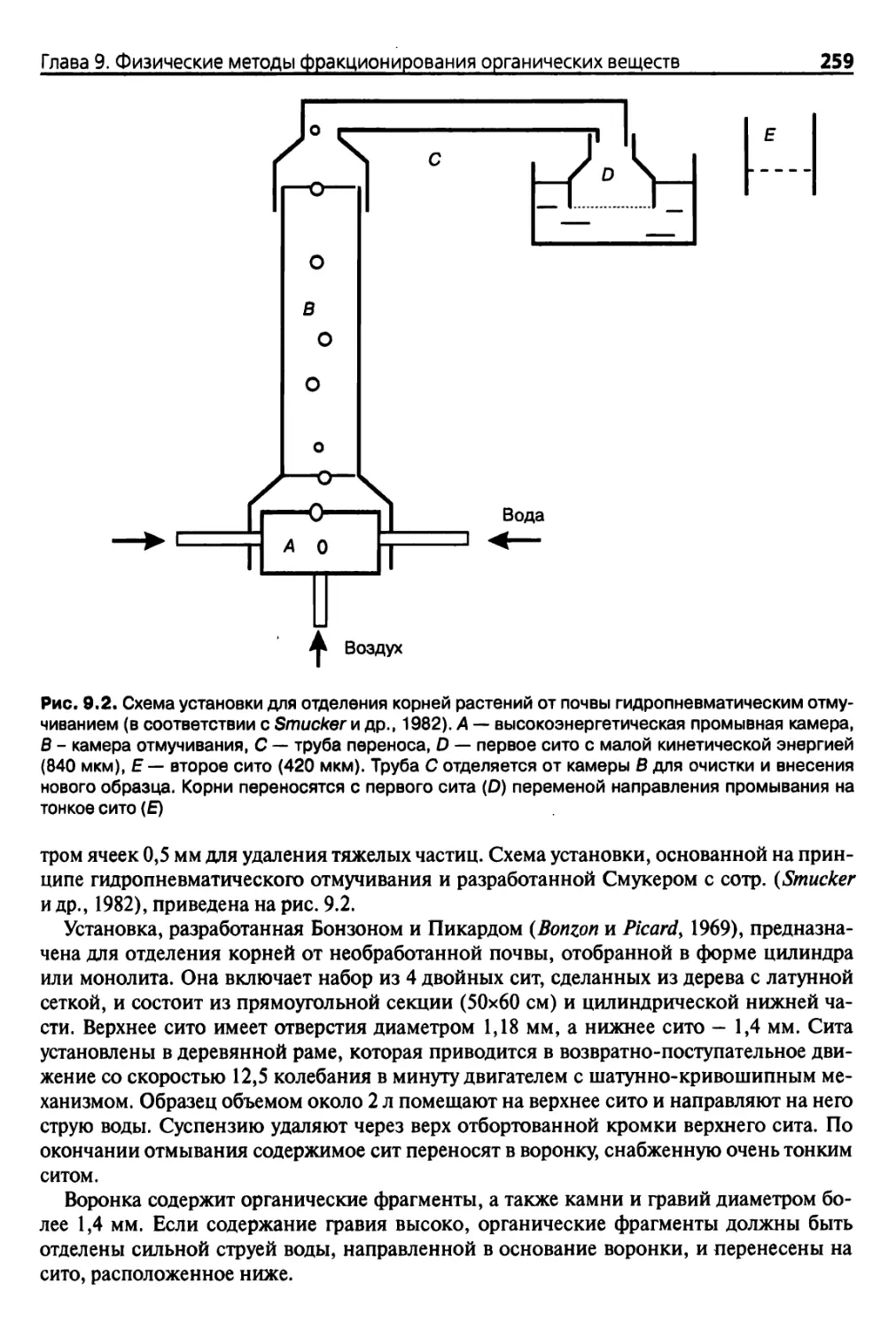
9.2.2. Извлечение корней растений 257
9.2.3. Распределение частиц 260
9.2.4. Разделение по плотности 271
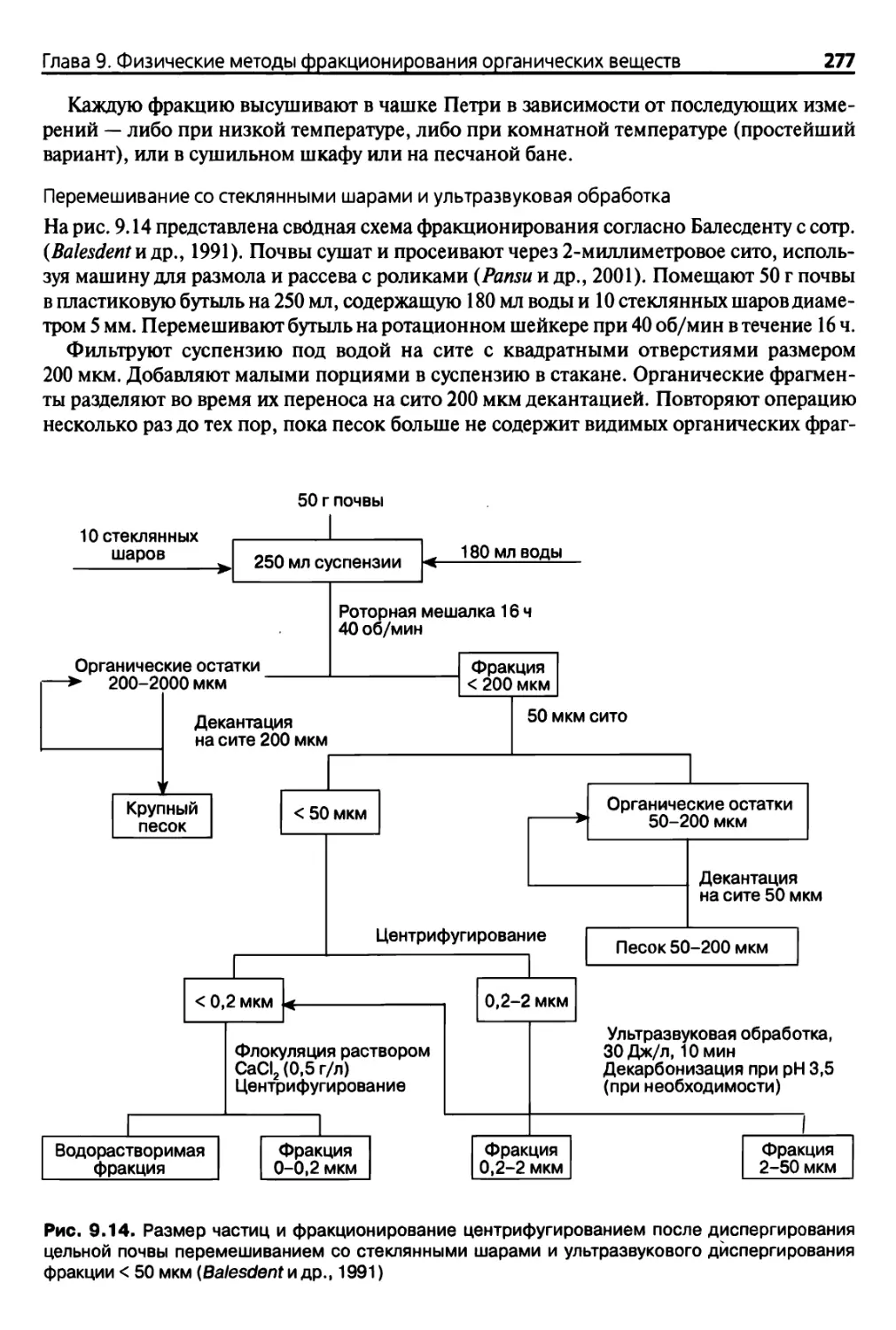
9.2.5. Фракционирование по размерам частиц 275
9.2.6. Точность методов фракционирования 280
9.3. Заключение и перспективы развития методов 281
Использованная литература 282
Глава 10. Определение содержания органического вещества
и общего количества С, N (Н, О, S) 285
10.1. Введение 285
10.1.1. Органическое вещество почвы 285
10.1.2. Отбор и подготовка образцов, аналитическая значимость 287
10.2. Мокрые методы 290
10.2.1. Определение содержания общего углерода: основные
положения 290
10.2.2. Определение содержания органического углерода методом
мокрого окисления при температуре реакции 291
Оглавление
9
10.2.3. Определение содержания органического углерода методом
мокрого окисления при контролируемой температуре 295
10.2.4. Определение содержания органического углерода методом
мокрого окисления с использованием
спектроколориметрии 296
10.2.5. Определение содержания общего азота с использованием
мокрого метода: Введение 297
10.2.6. Определение содержания общего азота методом
Къельдаля с титриметрическим окончанием 298
10.2.7. Определение общего содержания азота методом
Къельдаля со спектроколориметрическим окончанием 302
10.2.8. Определение содержания азота методом Къельдаля
с использованием селективного электрода 304
10.2.9. Механизация и автоматизация метода Къельдаля 306
10.2.10. Модификации методики анализа для определения
содержания ионов NO', N0" и связанного азота 306
10.3. Сухие методы анализа 307
10.3.1. Определение содержания общего углерода методом
простого выпаривания 307
10.3.2. Совместный инструментальный элементный
СН1Ч(08)-анализ методом сухого озоления 308
10.3.3. Определение CHNOS термическими методами анализа 312
10.3.4. Определение содержания углерода и азота неразрушающими
инструментальными методами 313
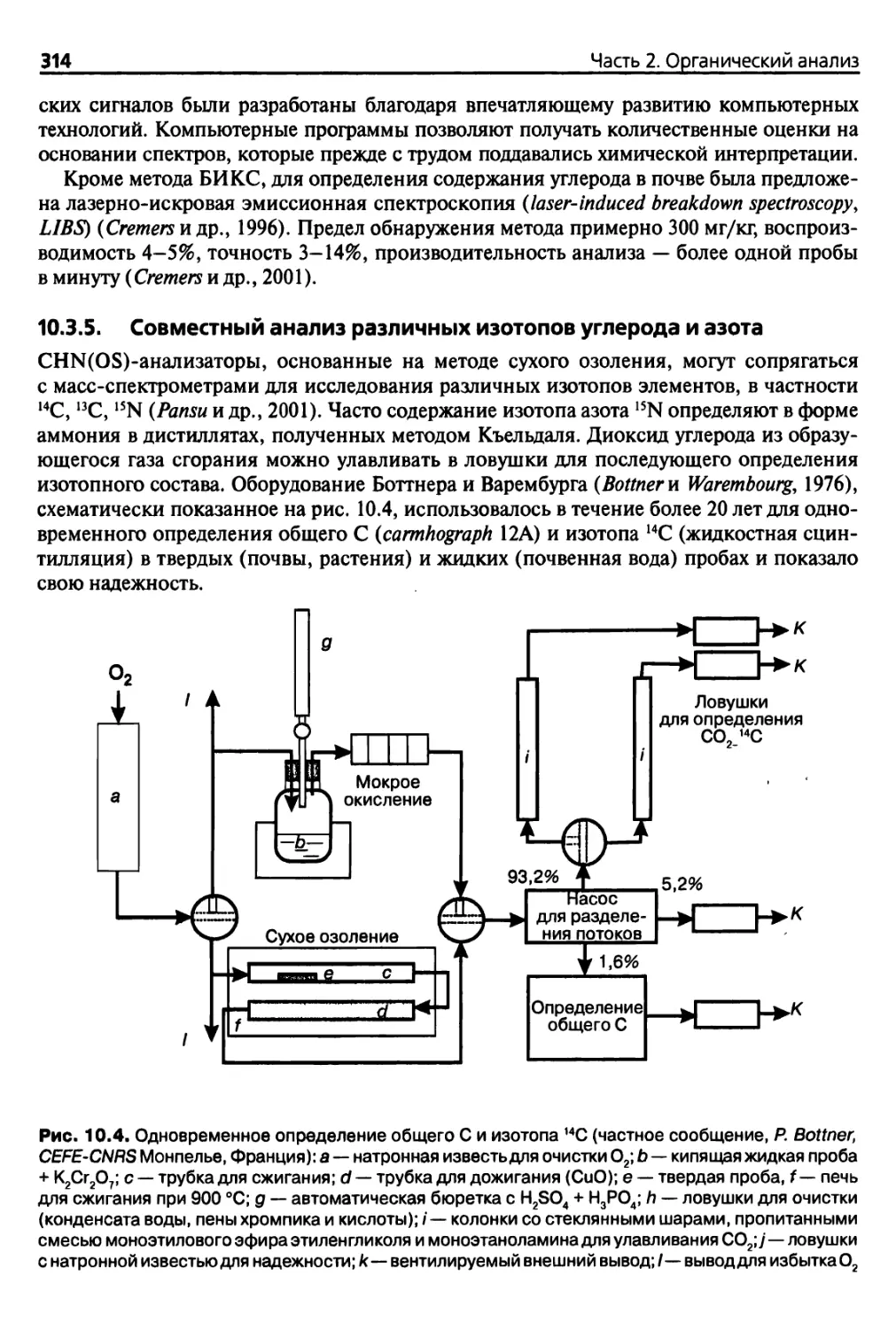
10.3.5. Совместный анализ различных изотопов углерода и азота 314
Использованная литература 315
Дополнительная литература 316
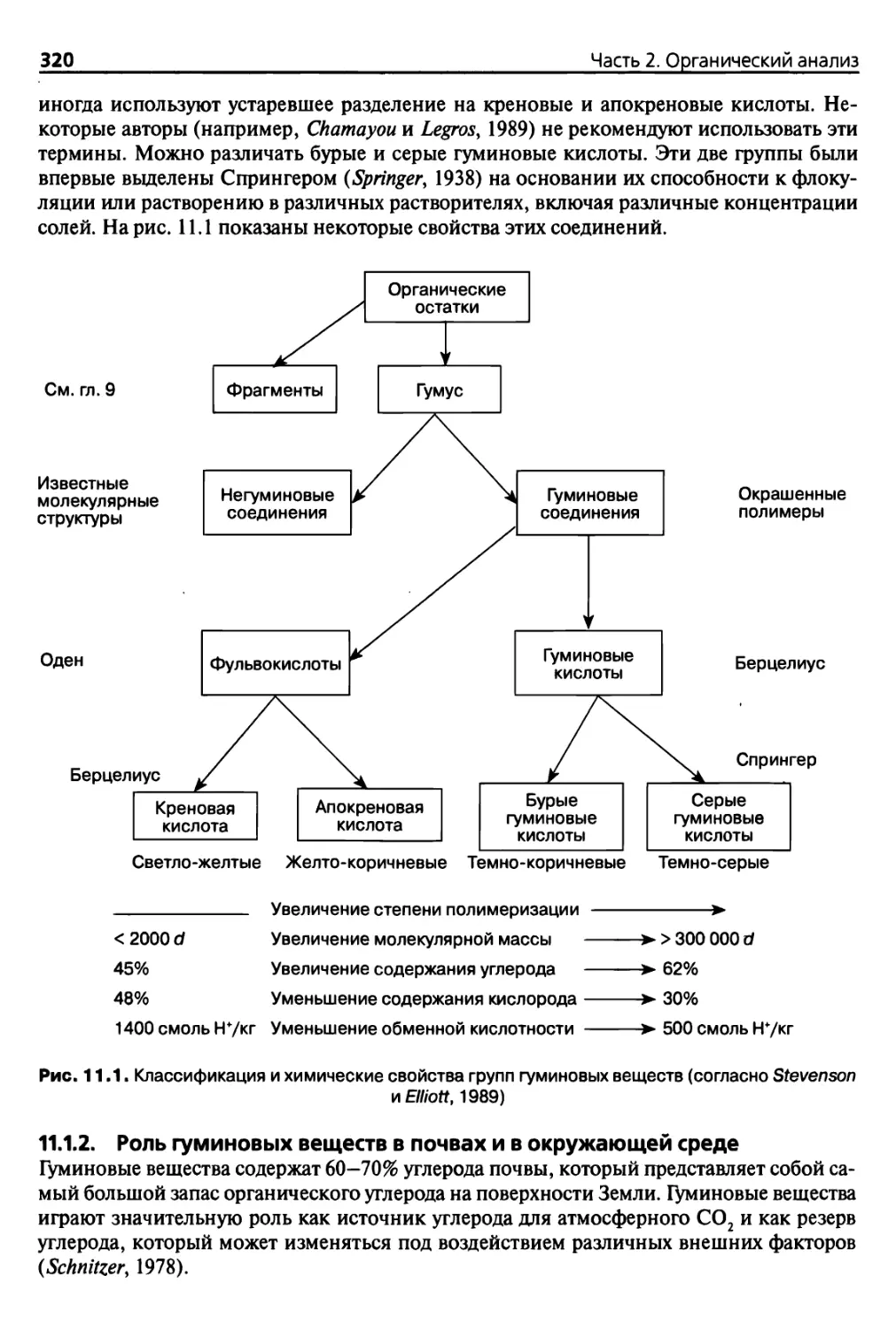
Шва 11. Количественное определение куминовых веществ 319
11.1. Гумус в почвах 319
11.1.1. Определения 319
11.1.2. Роль гуминовых веществ в почвах и в окружающей среде 320
11.1.3. Методы экстракции 321
11.2. Основные методы анализа 322
11.2.1. Экстракция 322
11.2.2. Количественный анализ экстрактов 325
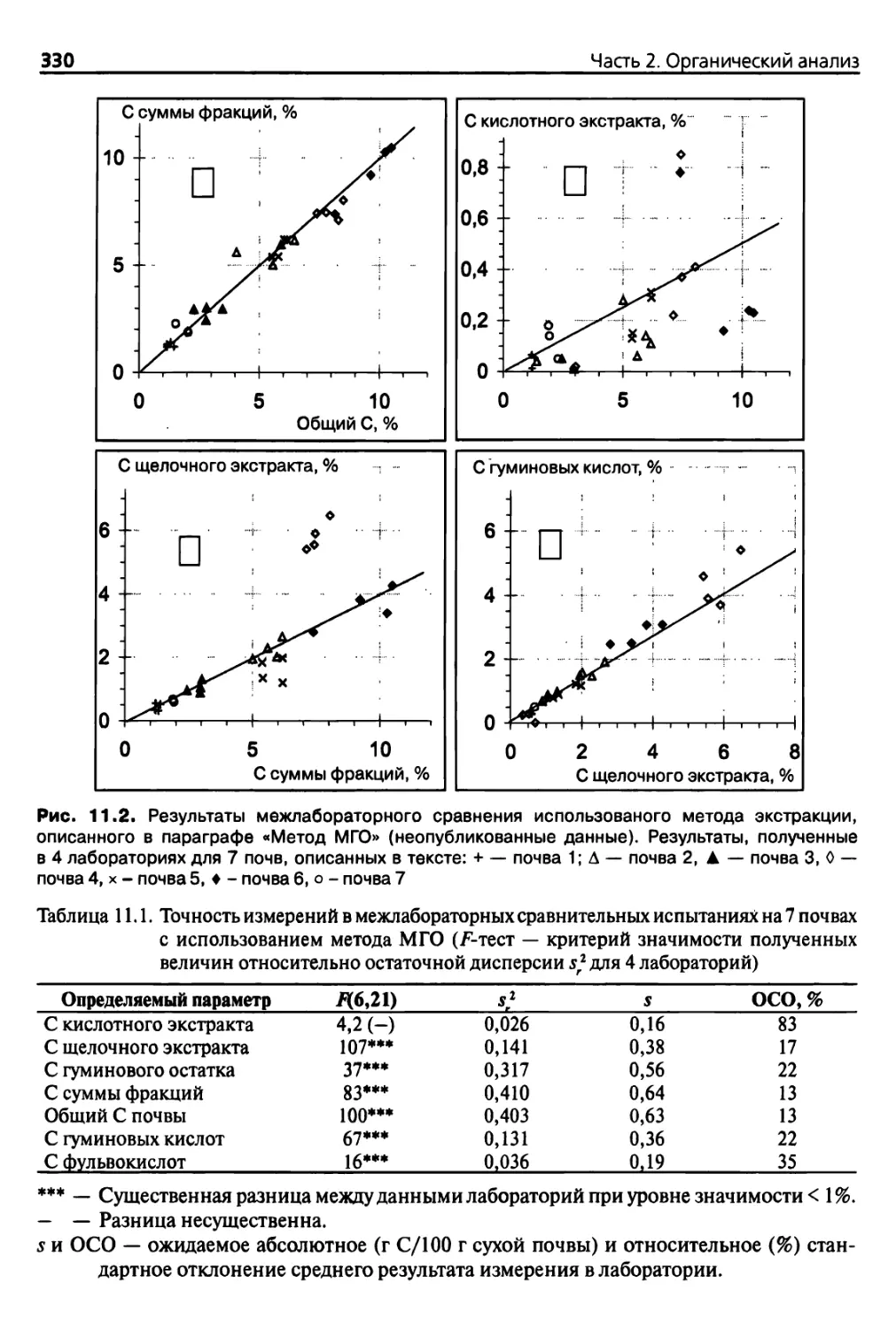
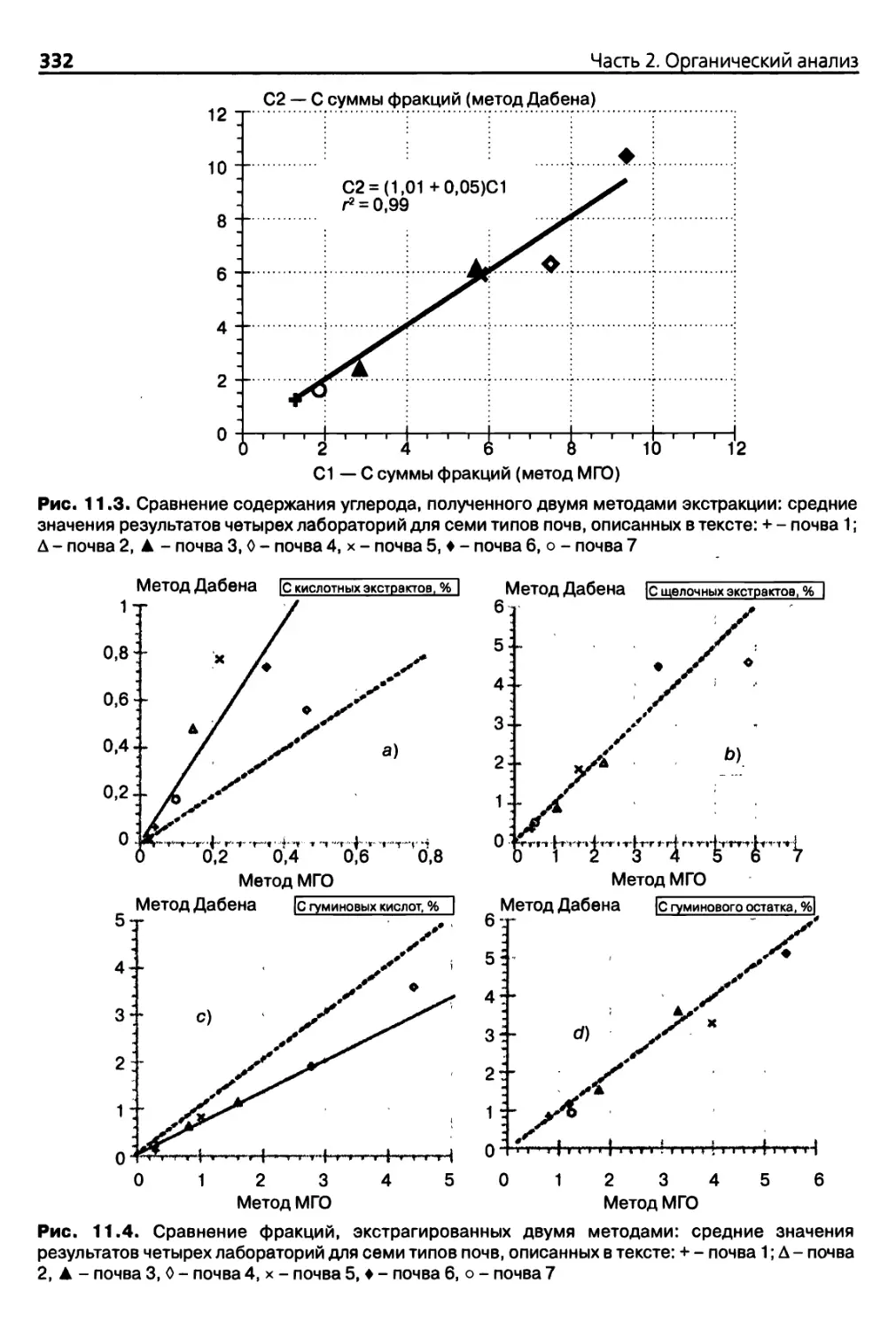
11.2.3. Точность и сопоставимость методов экстракции 328
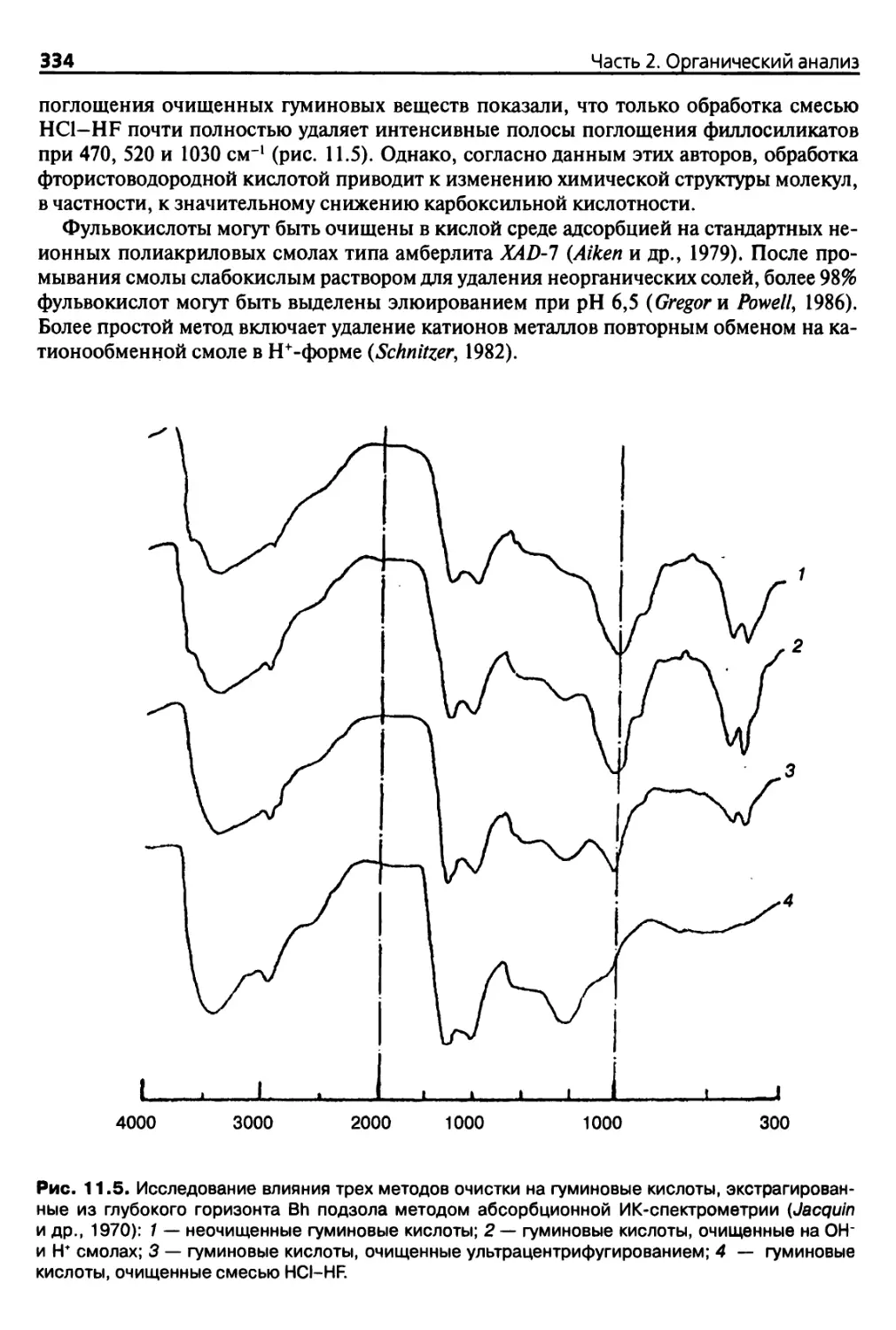
11.2.4. Методы очистки гуминовых веществ 333
11.3. Другие альтернативные и дополнительные методы 335
11.3.1. Альтернативные методы экстракции 335
11.3.2. Фракционирование гуминового остатка 336
Использованная литература 338
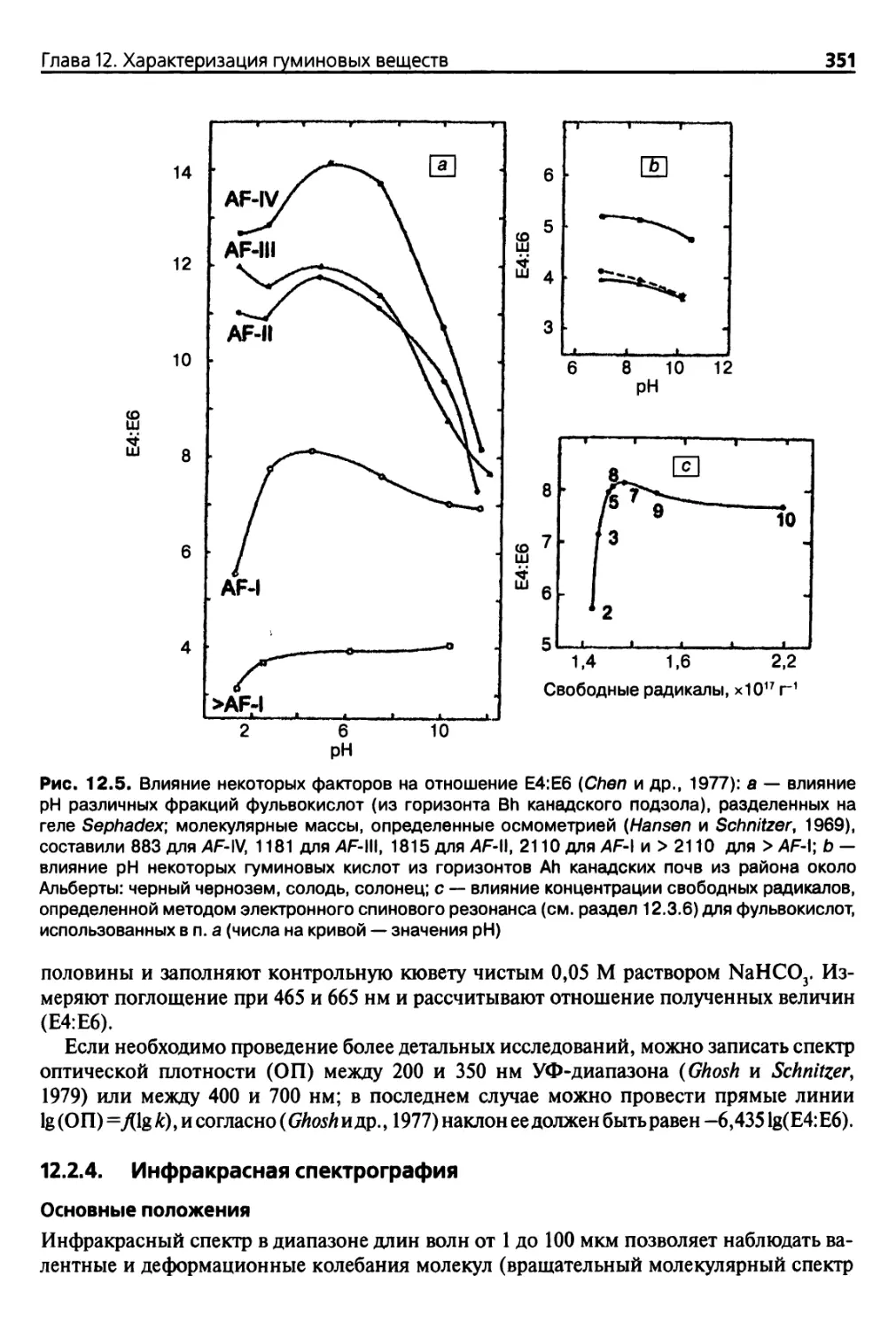
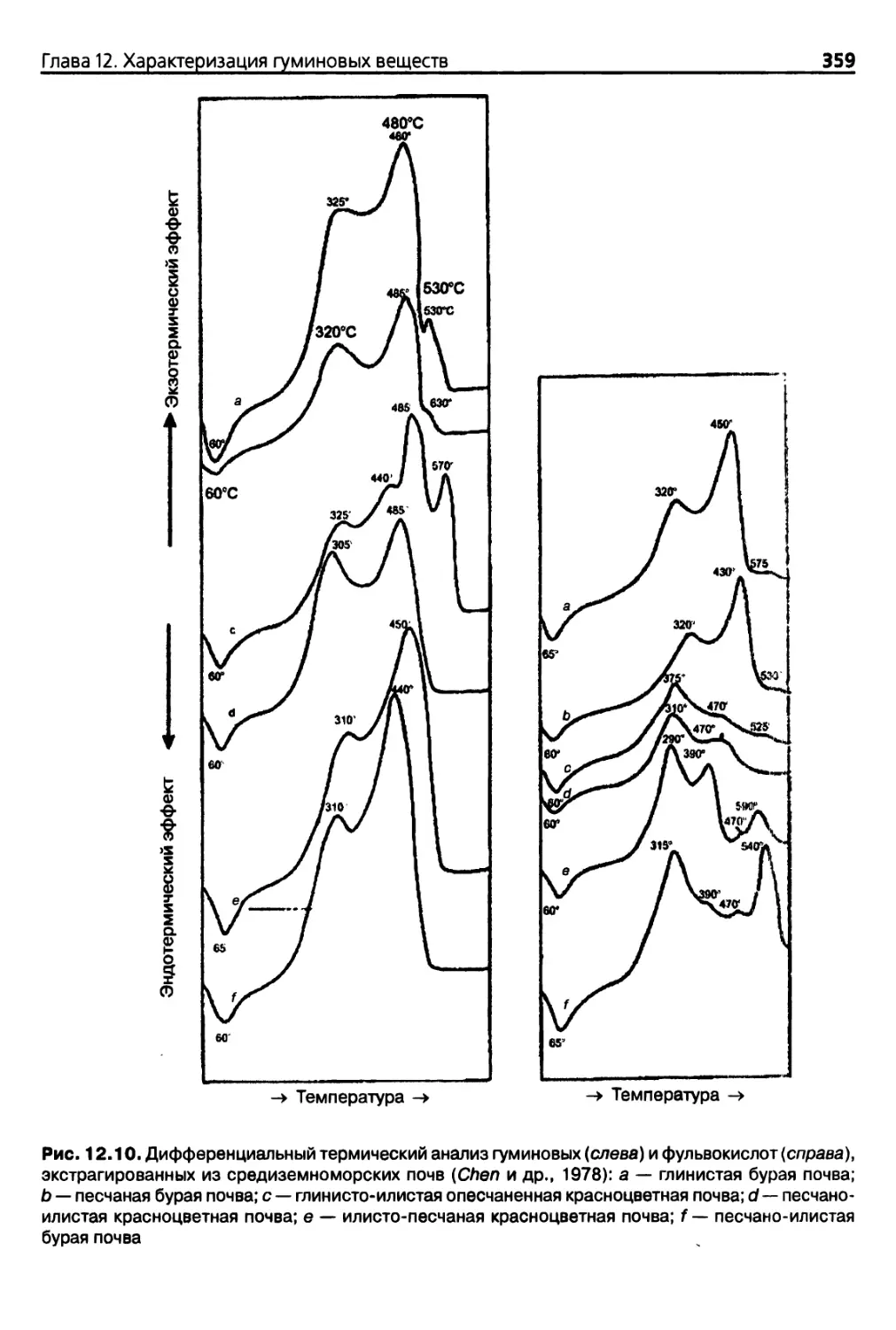
Шва 12. Характеризация гуминовых веществ 341
12.1. Введение 341
12.1.1. Механизмы образования 341
12.1.2. Молекулярная структура 341
12.2. Традиционные методы анализа 342
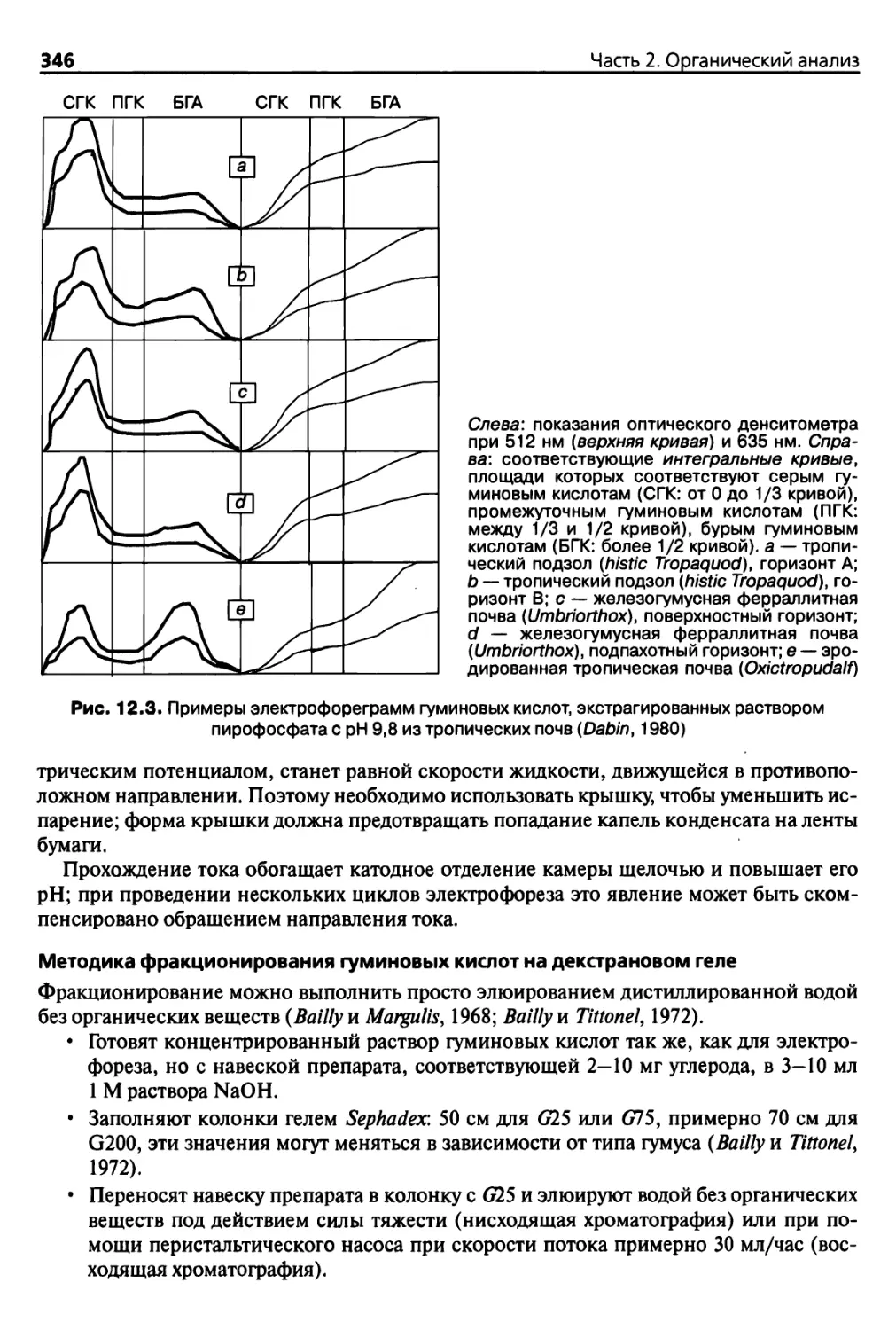
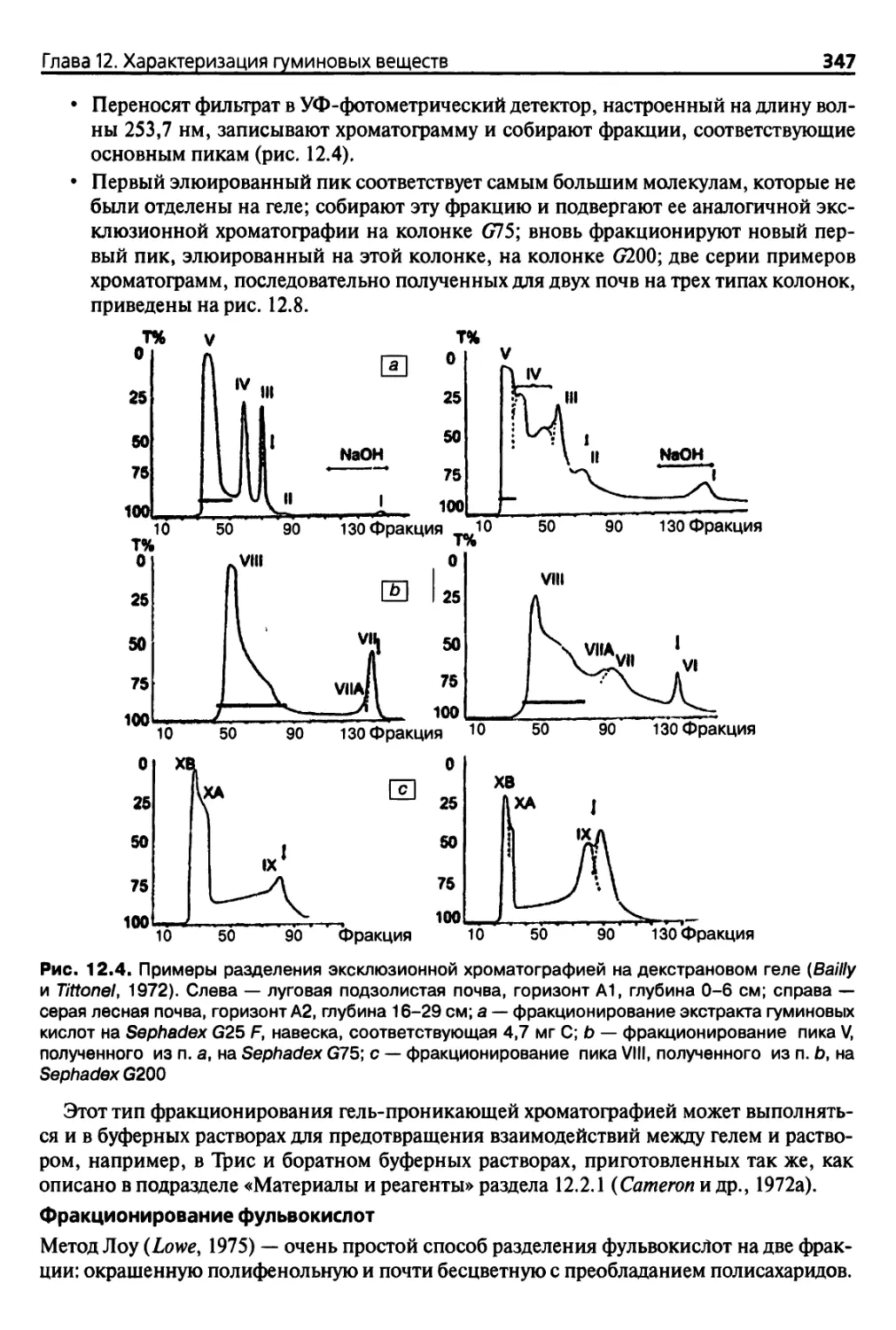
12.2.1. Фракционирование гуминовых соединений 342
12.2.2. Определение основных функциональных групп 348
12.2.3. Спектрометрия УФ-видимого диапазона 349
10
Оглавление
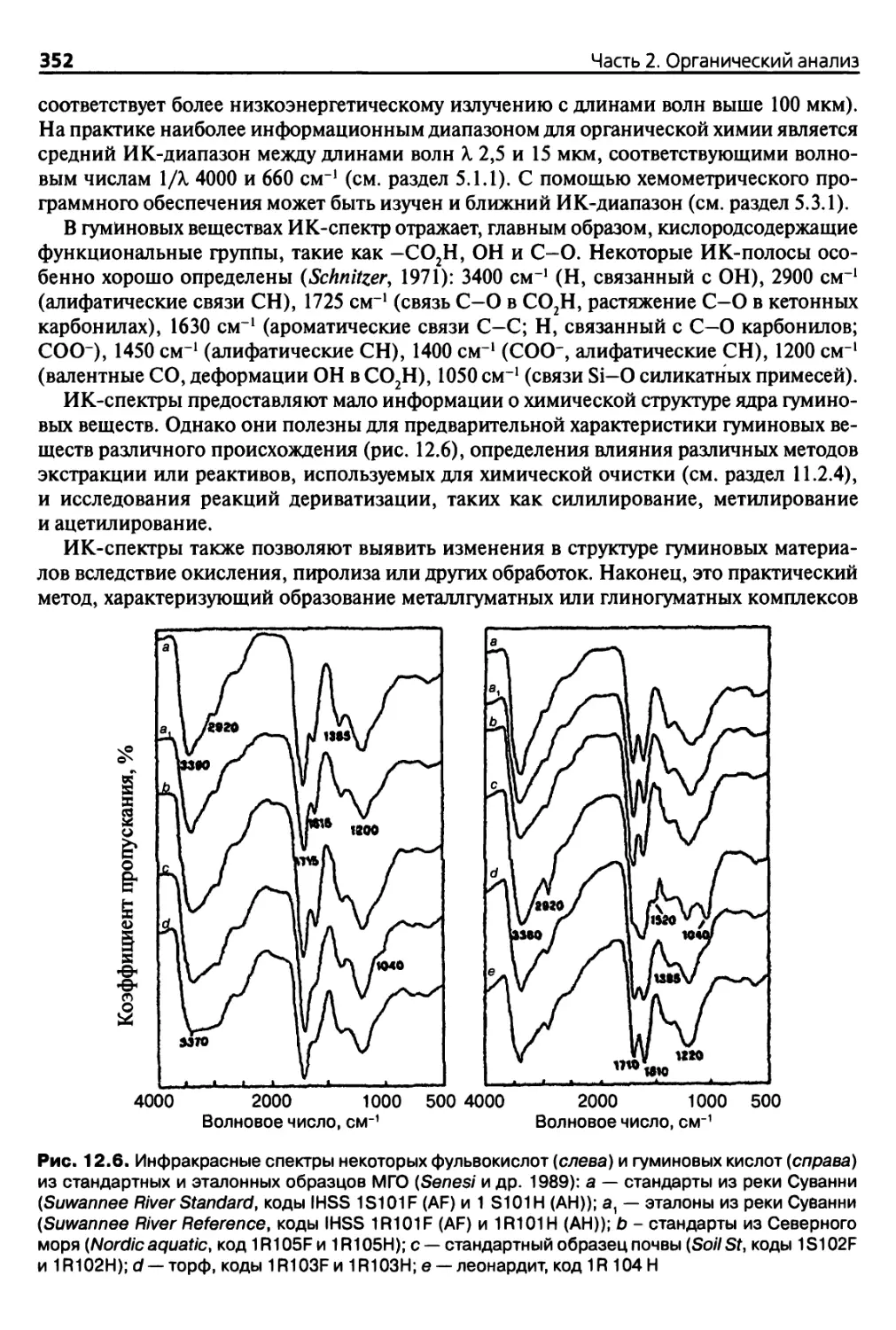
12.2.4. Инфракрасная спектрография 351
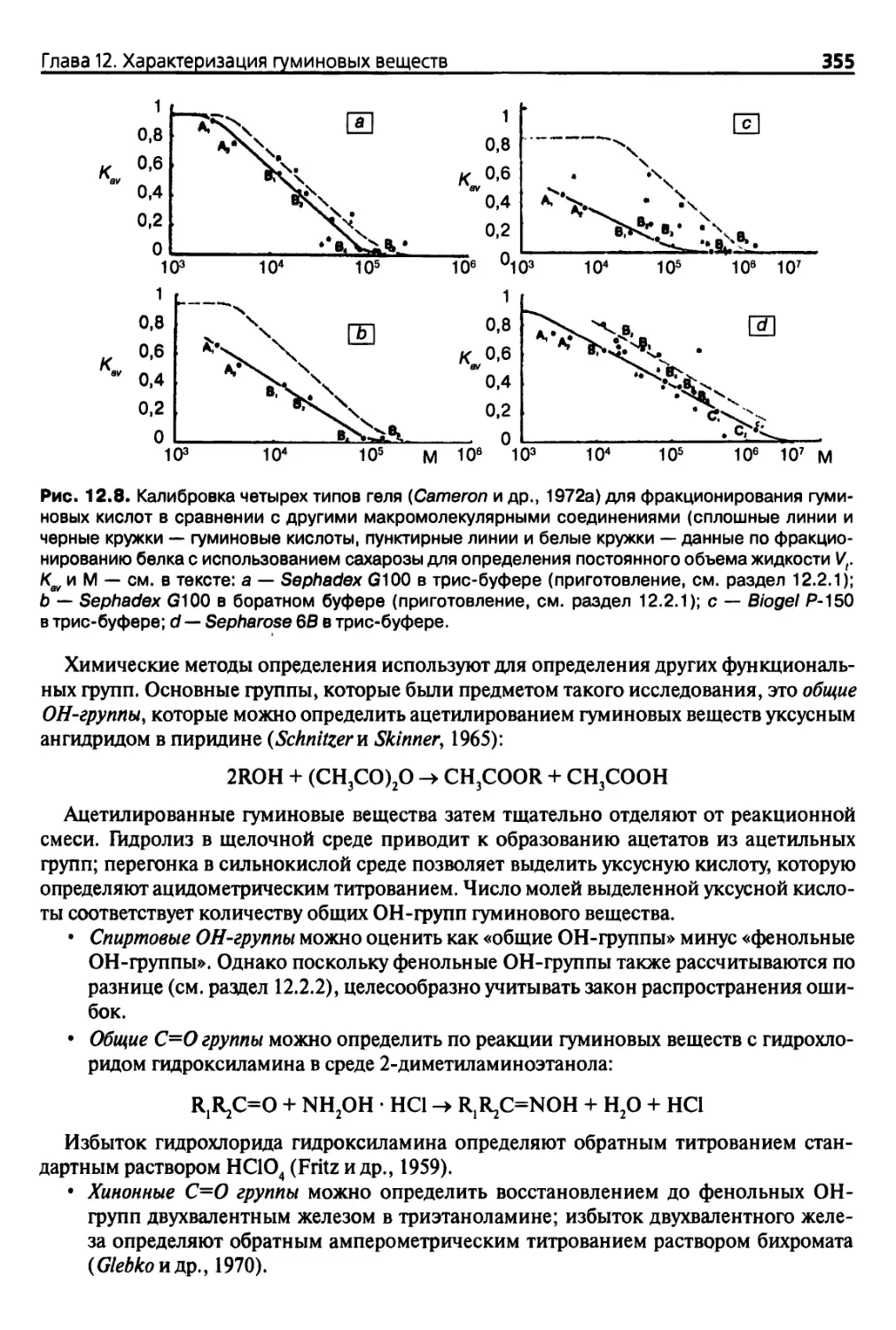
12.3. Дополнительные методы 353
12.3.1. Усовершенствование методов фракционирования 353
12.3.2. Определение функциональных групп 354
12.3.3. Характеристика гуминовых веществ методом фрагментации .. 356
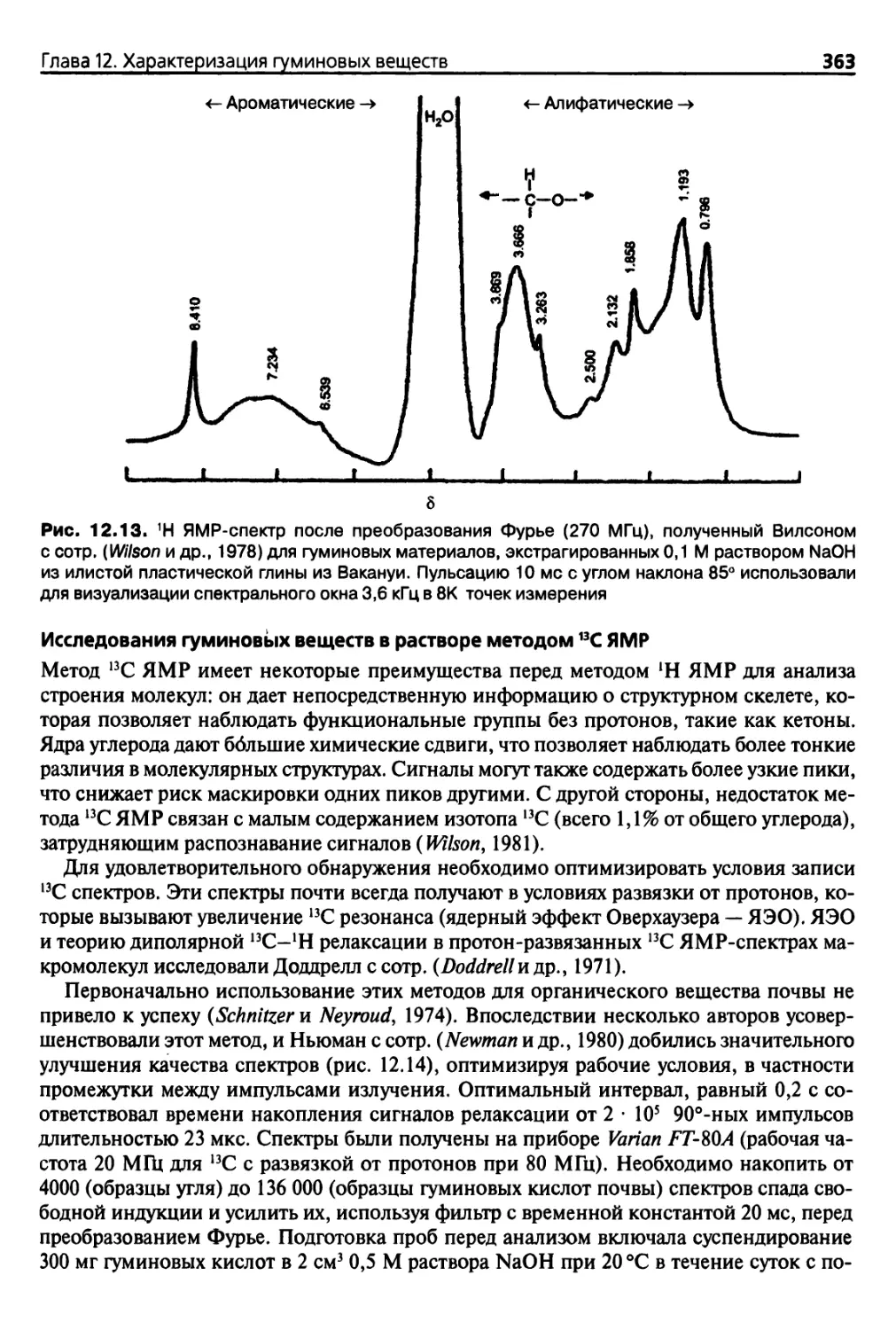
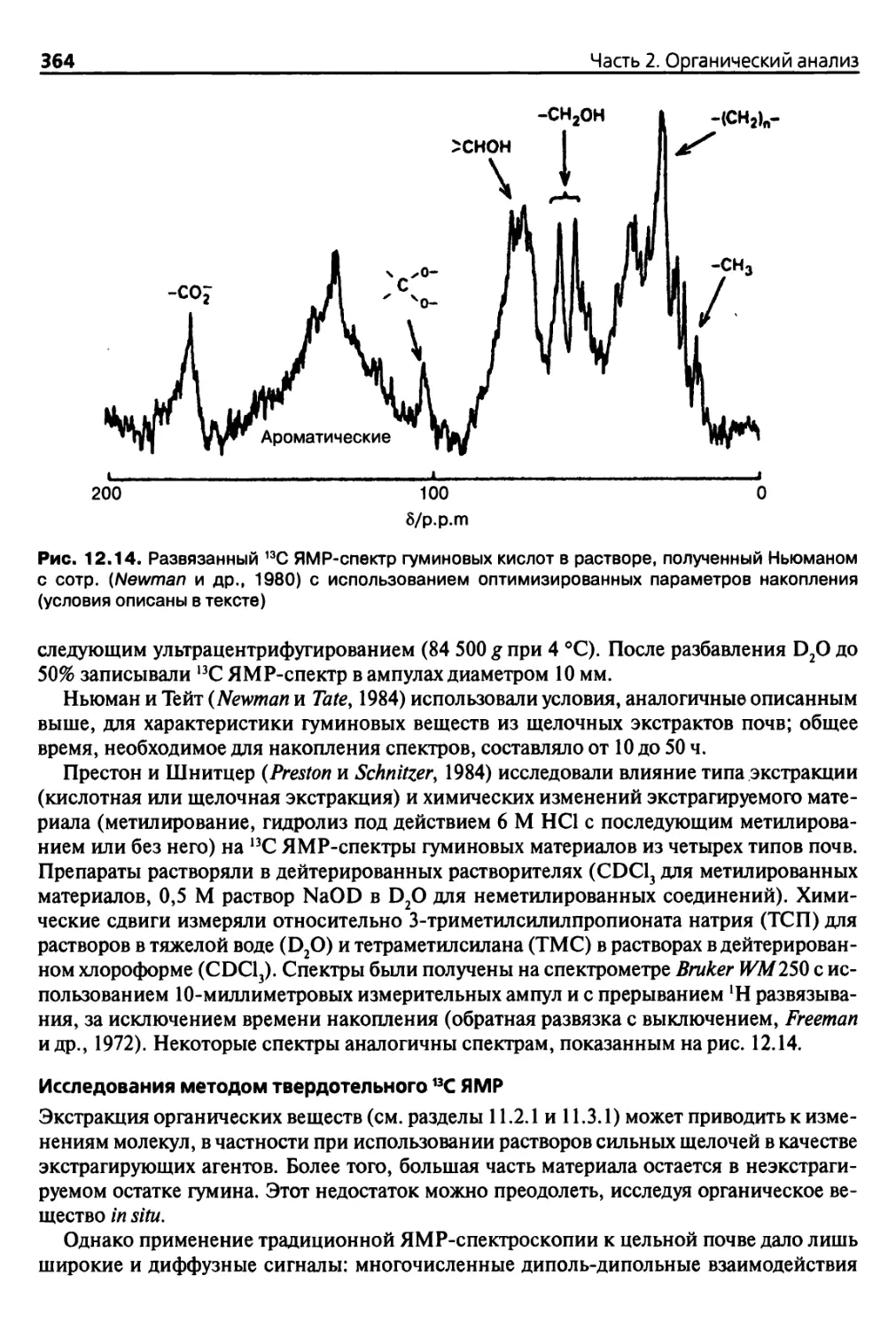
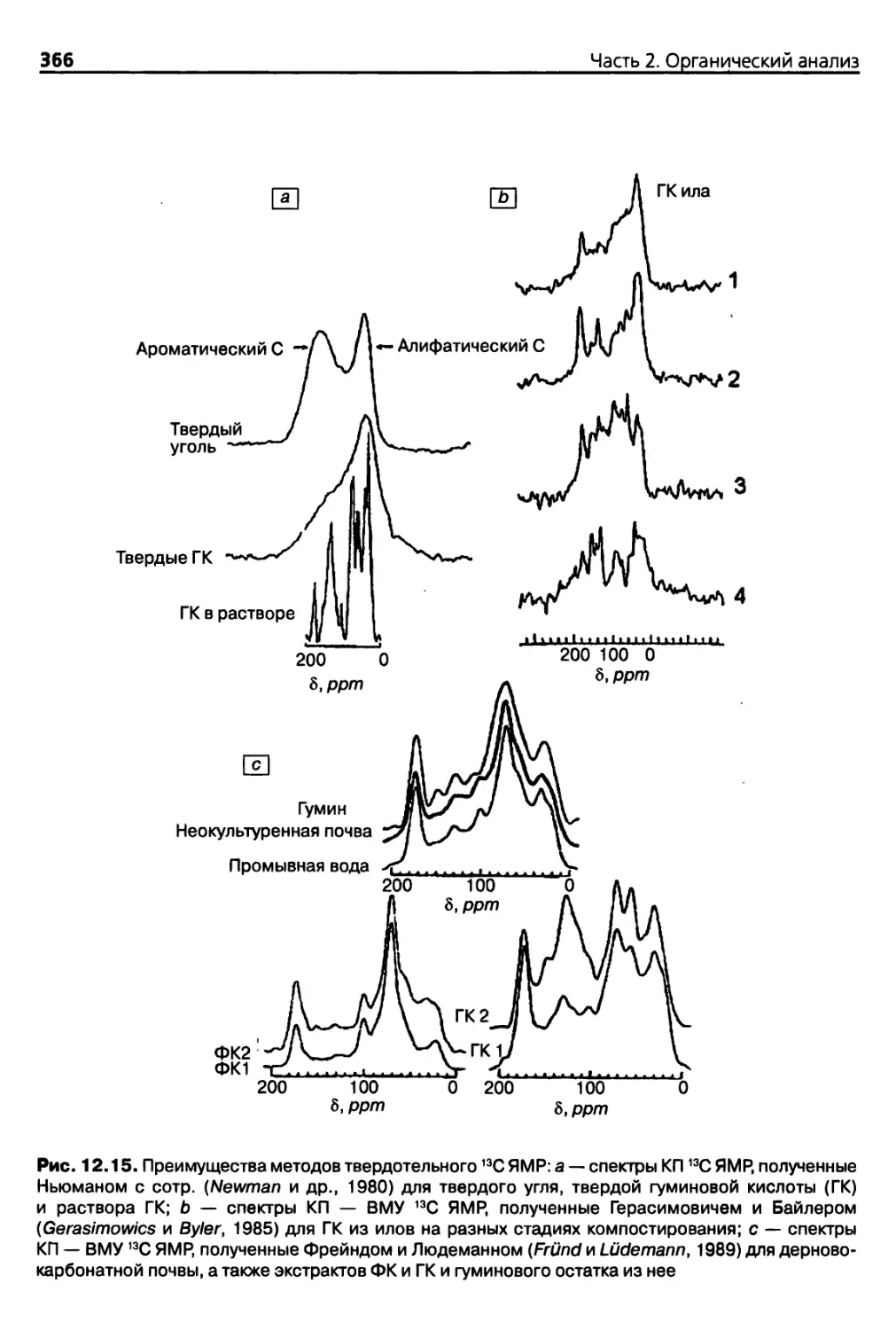
12.3.4. Ядерный магнитный резонанс (ЯМР) 358
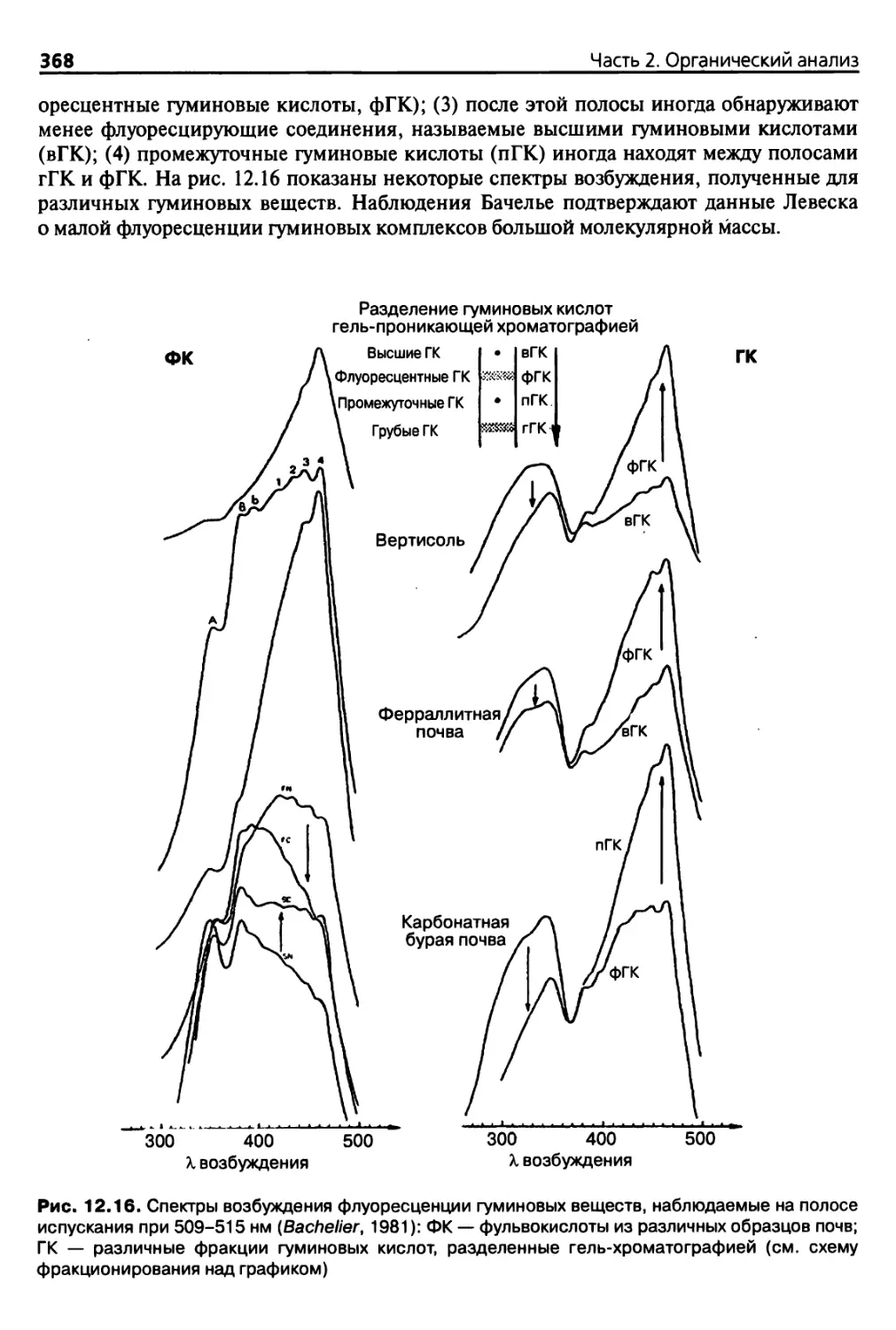
12.3.5. Флуоресцентная спектроскопия 367
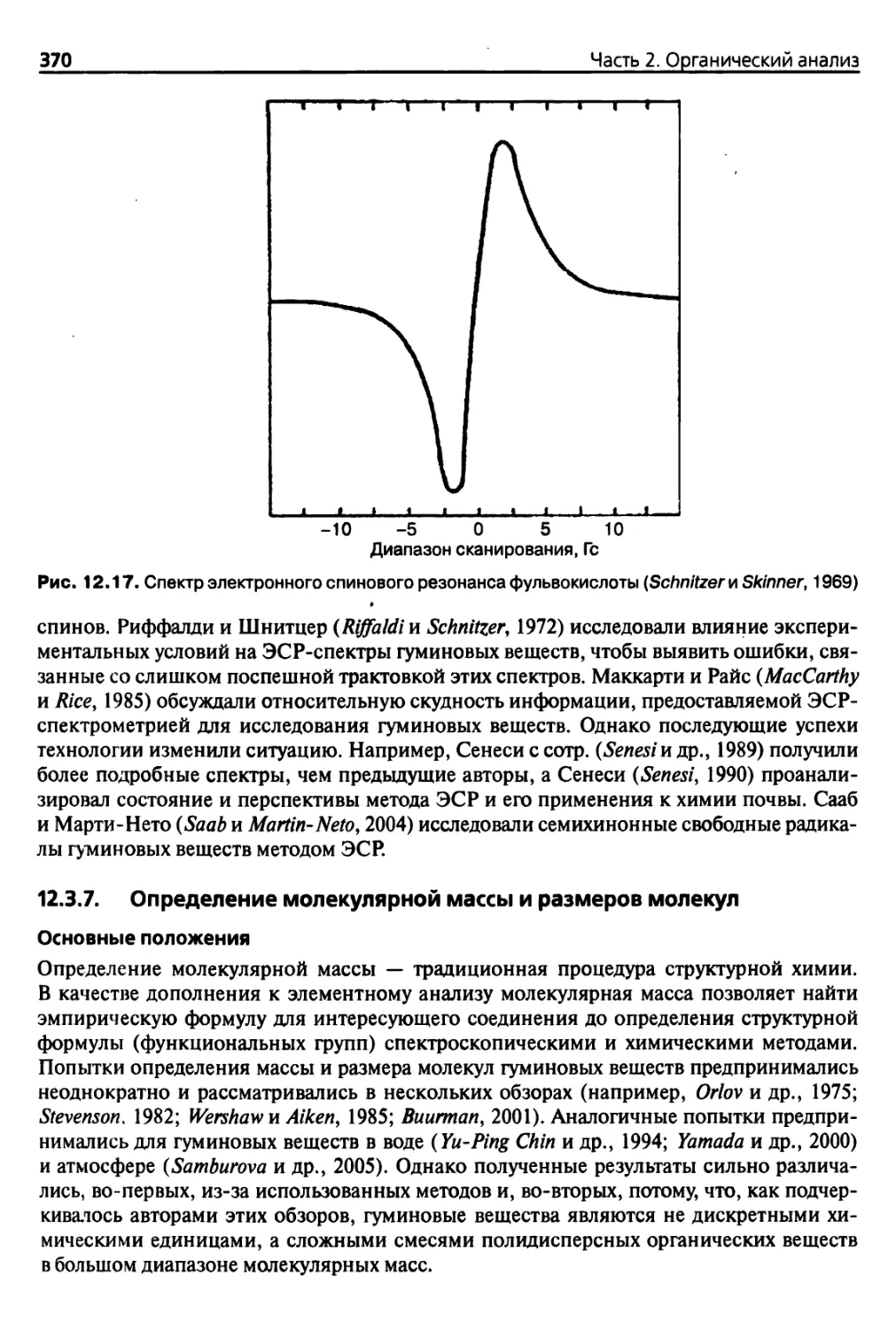
12.3.6. Спектроскопия электронного парамагнитного резонанса
(ЭПР) 369
12.3.7. Определение молекулярной массы и размеров молекул 370
12.3.8. Микроскопические исследования 373
12.3.9. Другие методы анализа 373
Использованная литература 374
Ншва 13. Определение иегуминовых веществ 381
13.1. Введение 381
13.1.1. Негуминовые вещества 381
13.1.2. Углеводороды в почве 381
13.1.3. Липиды в почве 383
13.1.4. Пестициды и поллютанты 383
13.2. Классические методы анализа 384
13.2.1. Кислотный гидролиз полисахаридов 384
13.2.2. Очистка кислотных гидролизатов 388
13.2.3. Колориметрическое определение сахаров 390
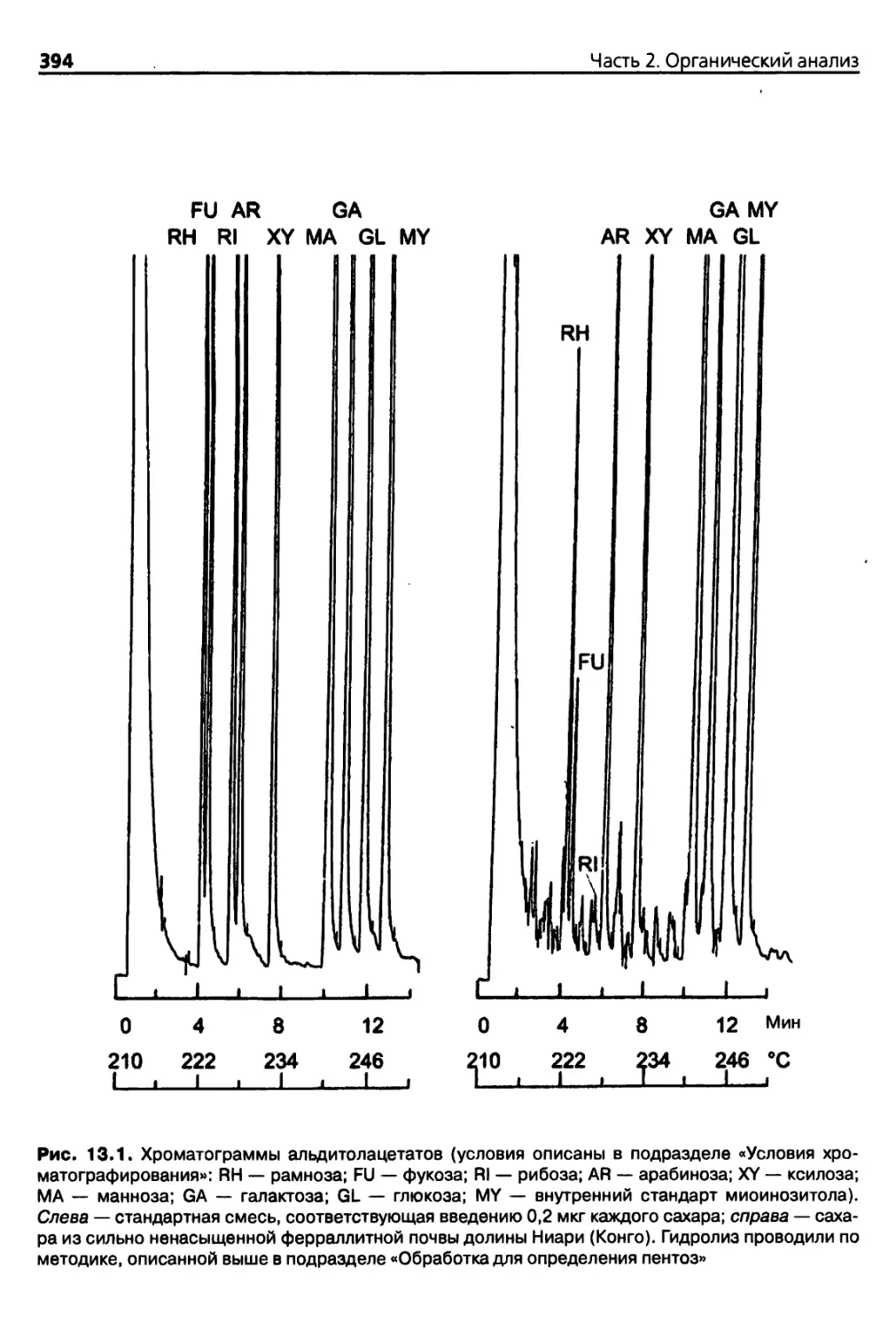
13.2.4. Определение содержания сахаров методом газовой
хроматографии 392
13.2.5. Определение общего содержания липидов 396
13.2.6. Количественное определение содержания
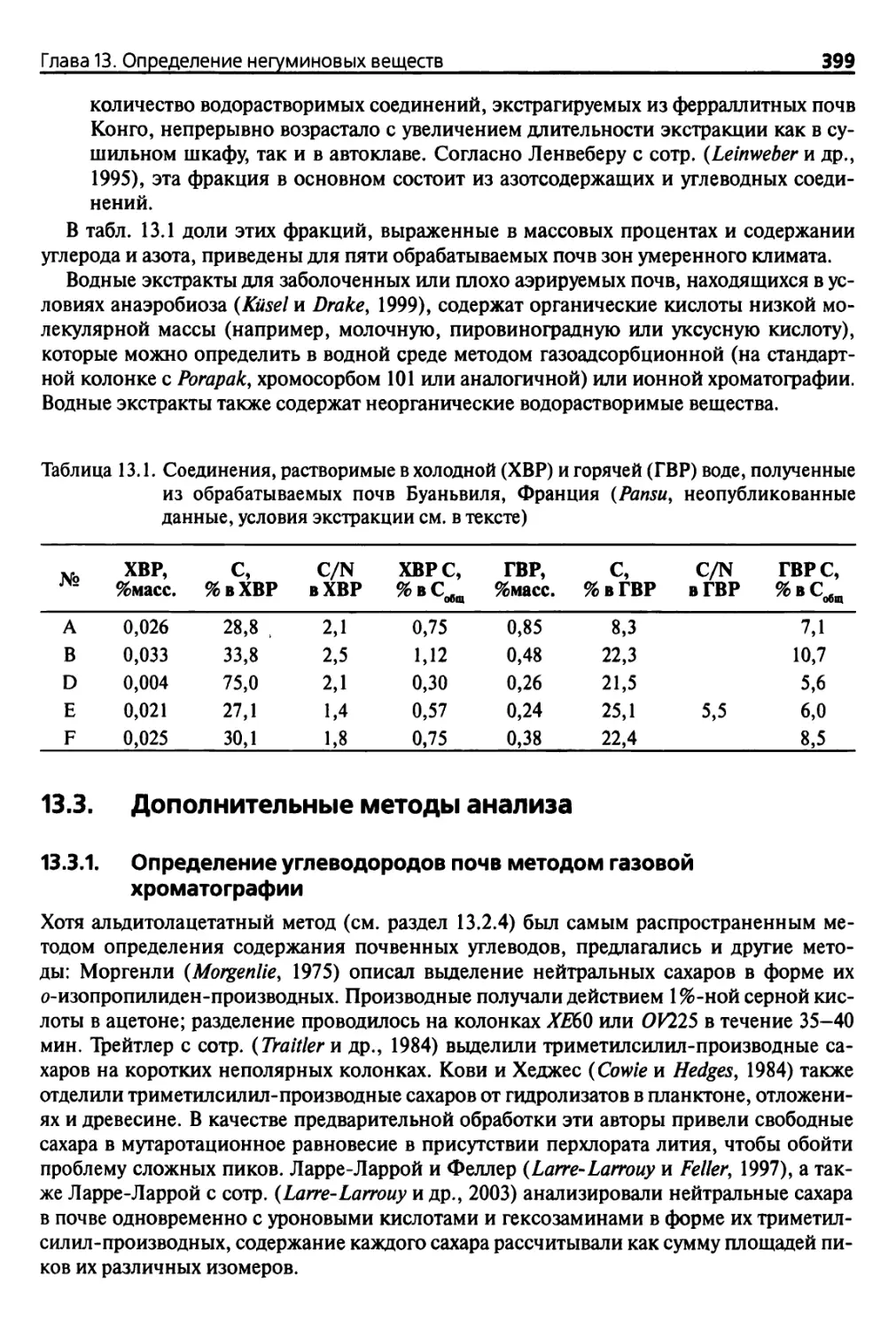
водорастворимых органических соединений 398
13.3. Дополнительные методы анализа 399
13.3.1. Определение углеводородов почв методом газовой
хроматографии 399
13.3.2. Определение углеводородов методом жидкостной
хроматографии 400
13.3.3. Фракционирование и исследование почвенной фракции
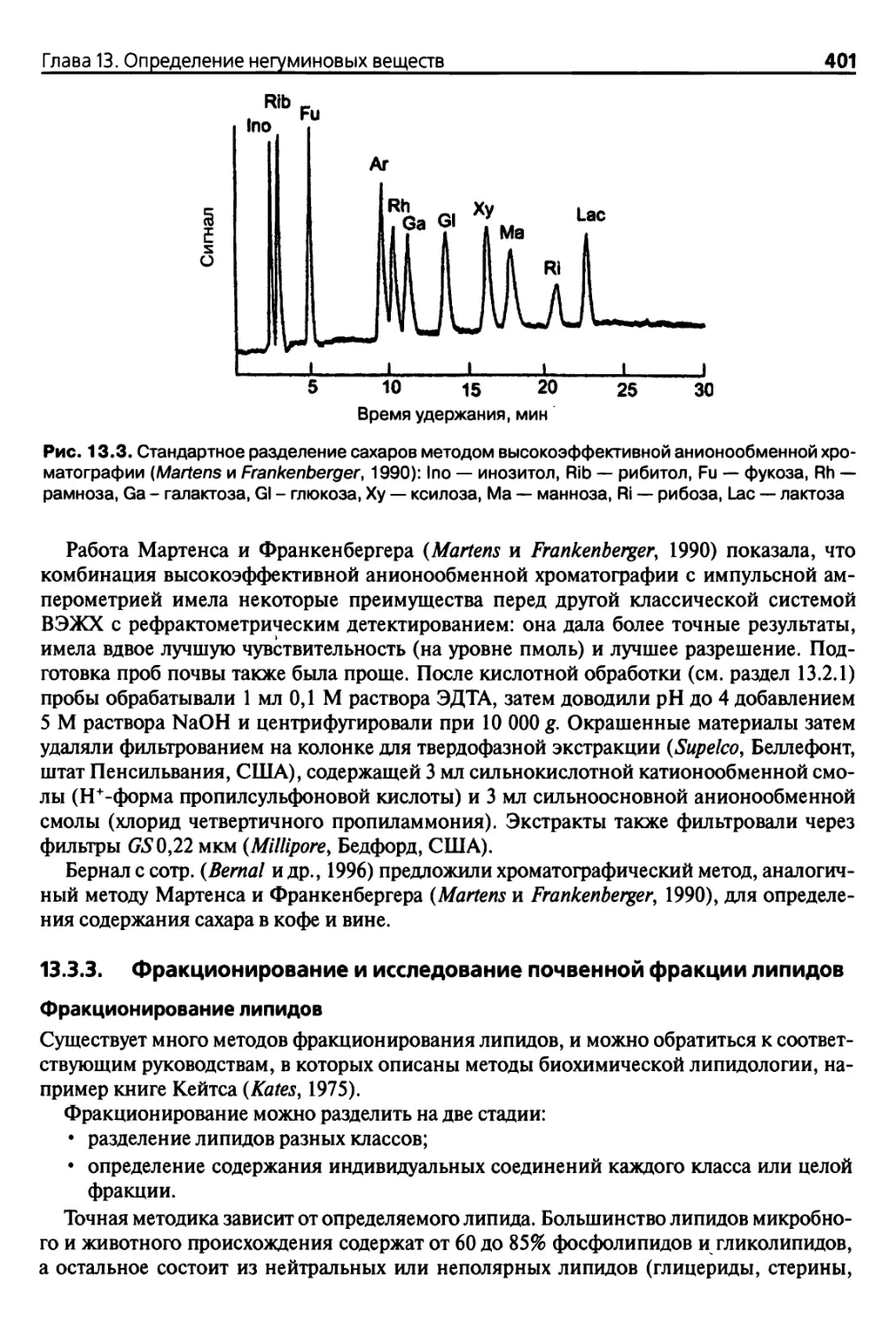
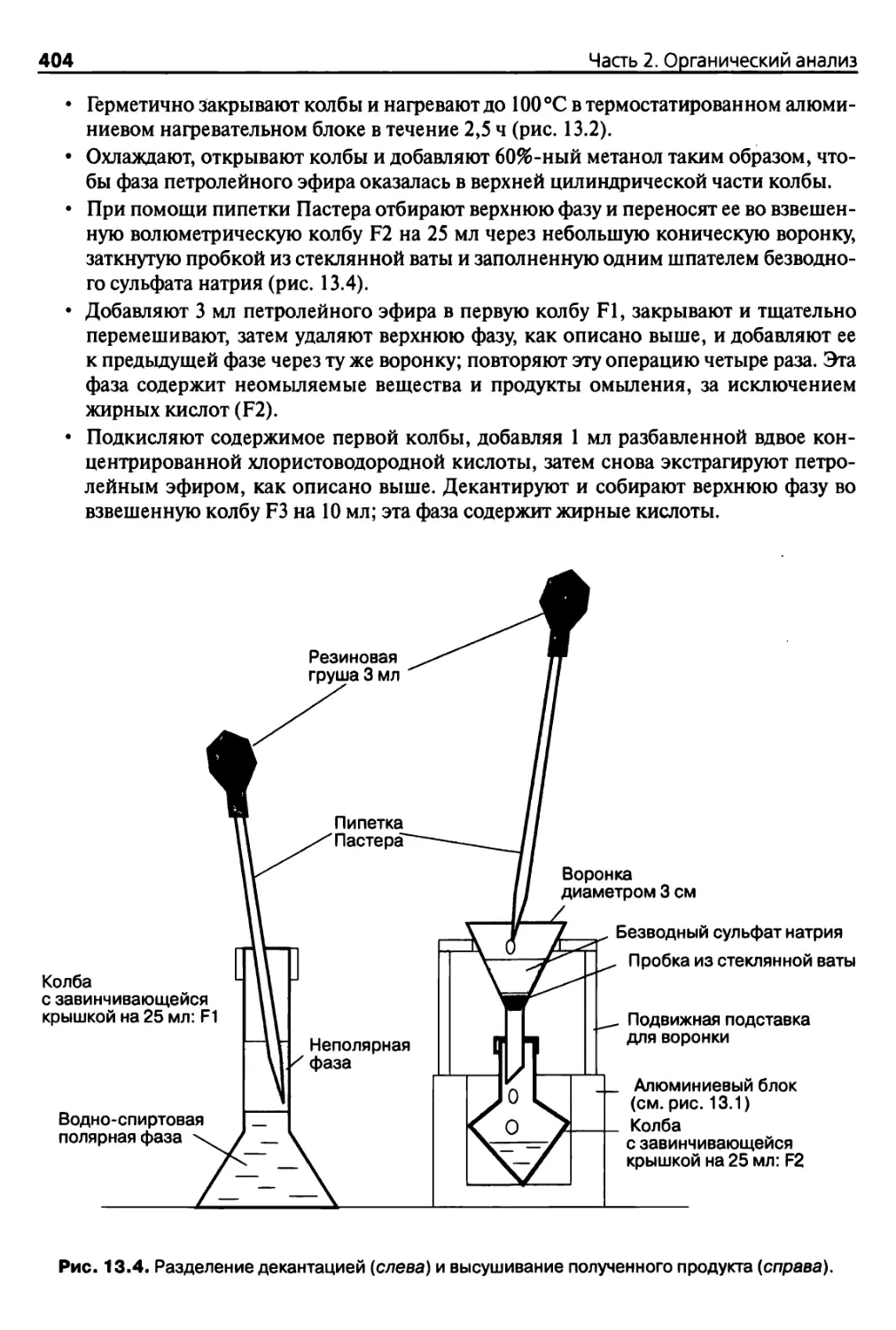
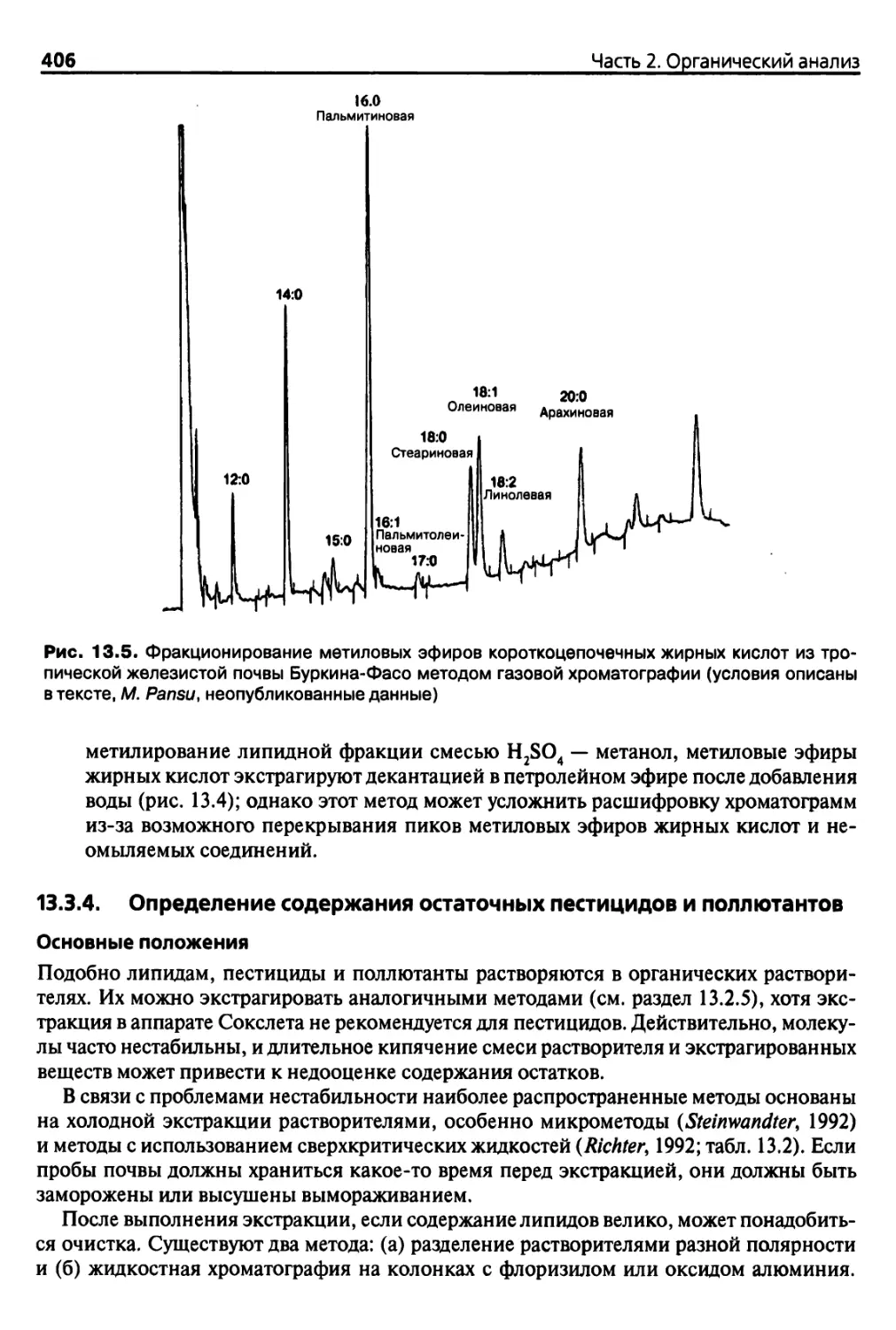
липидов 401
13.3.4. Определение содержания остаточных пестицидов
и поллютантов 406
Использованная литература 413
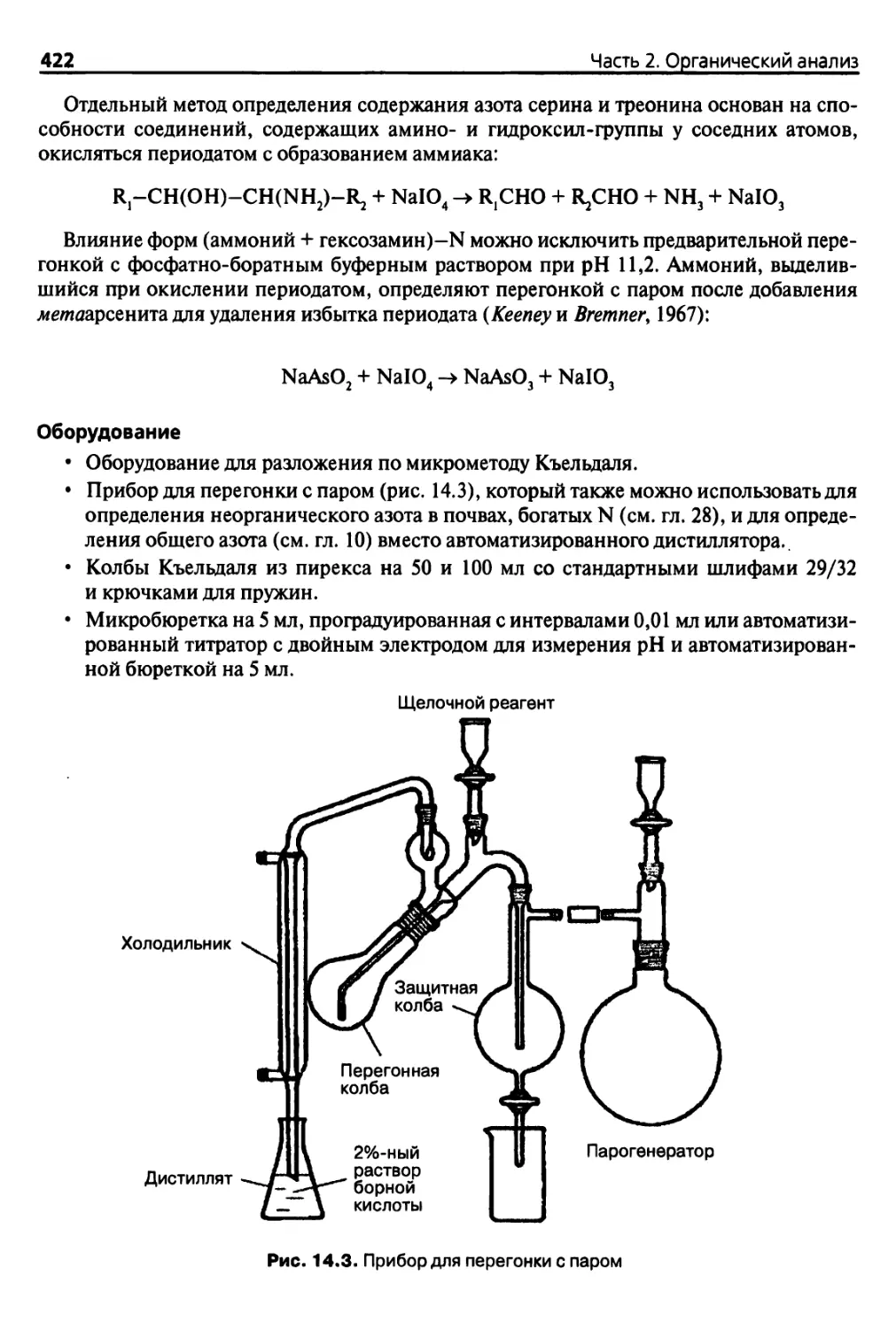
Diaea 14. Органические формы азота, минерализуемый азот (и углерод) 417
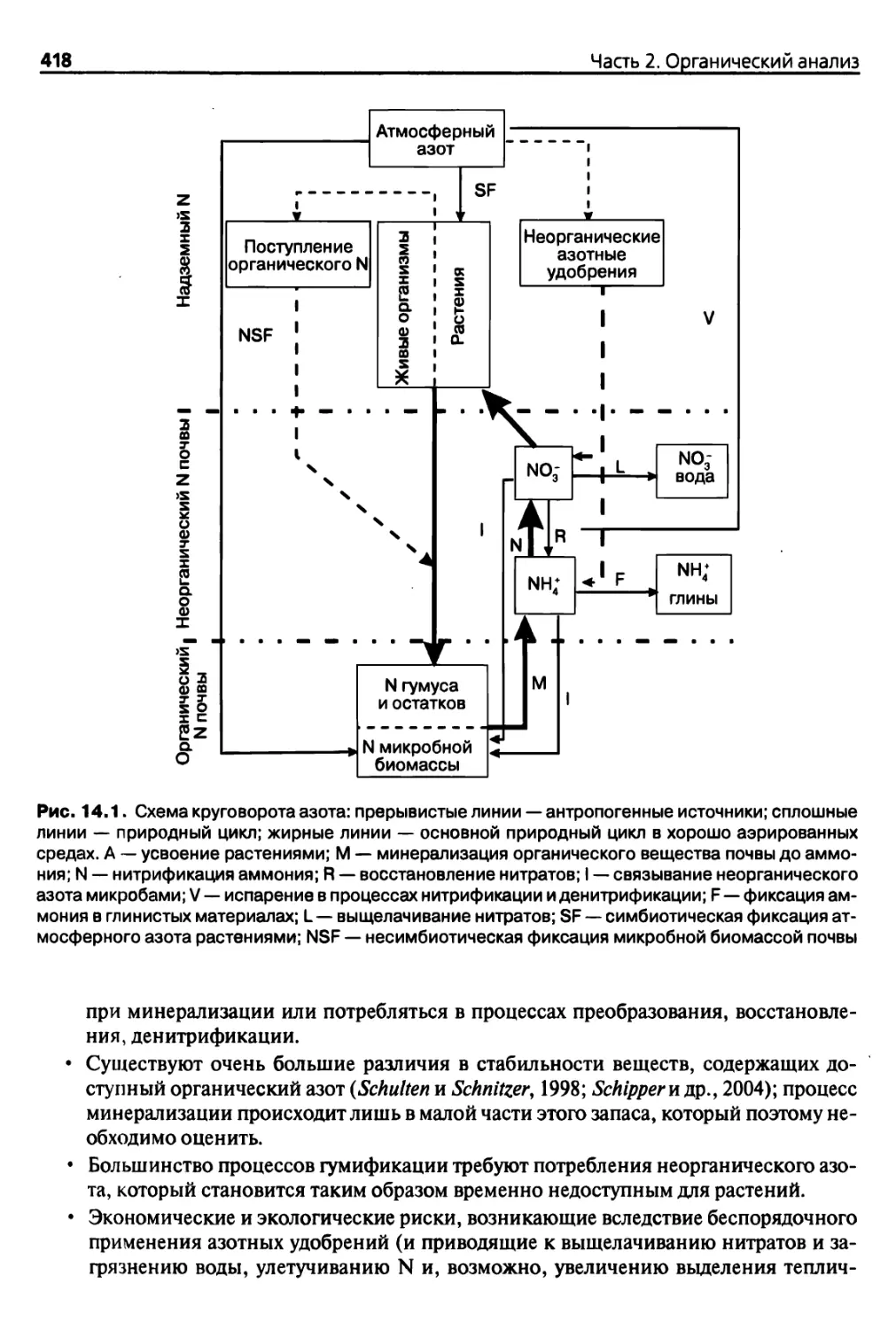
14.1. Введение 417
14.1.1. Круговорот азота 417
14.1.2. Типы методов анализа 419
14.2. Классические методы анализа 419
14.2.1. Формы органического азота, выделяемого при кислотном
гидролизе 419
14.2.2. Определение органических форм азота: упрощенные методы — 426
14.2.3. Определение содержания мочевины 427
Оглавление
11
14.2.4. Определение содержания потенциально доступного азота:
биологические методы 429
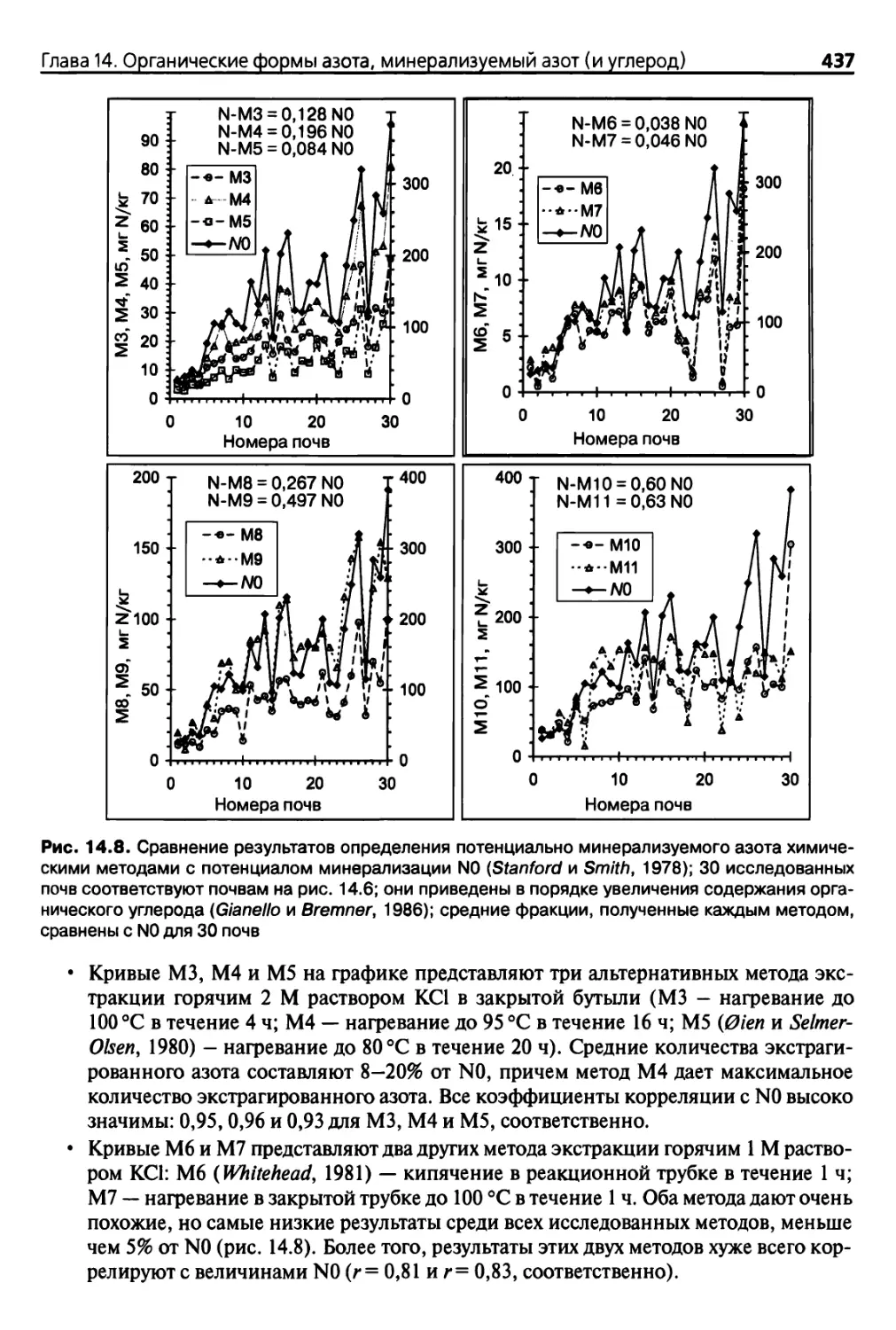
14.2.5. Определение содержания потенциально минерализуемого
азота: химические методы 436
14.2.6. Кинетика процессов минерализации 439
14.3. Дополнительные методы анализа 443
14.3.1. Альтернативные методики кислотного гидролиза 443
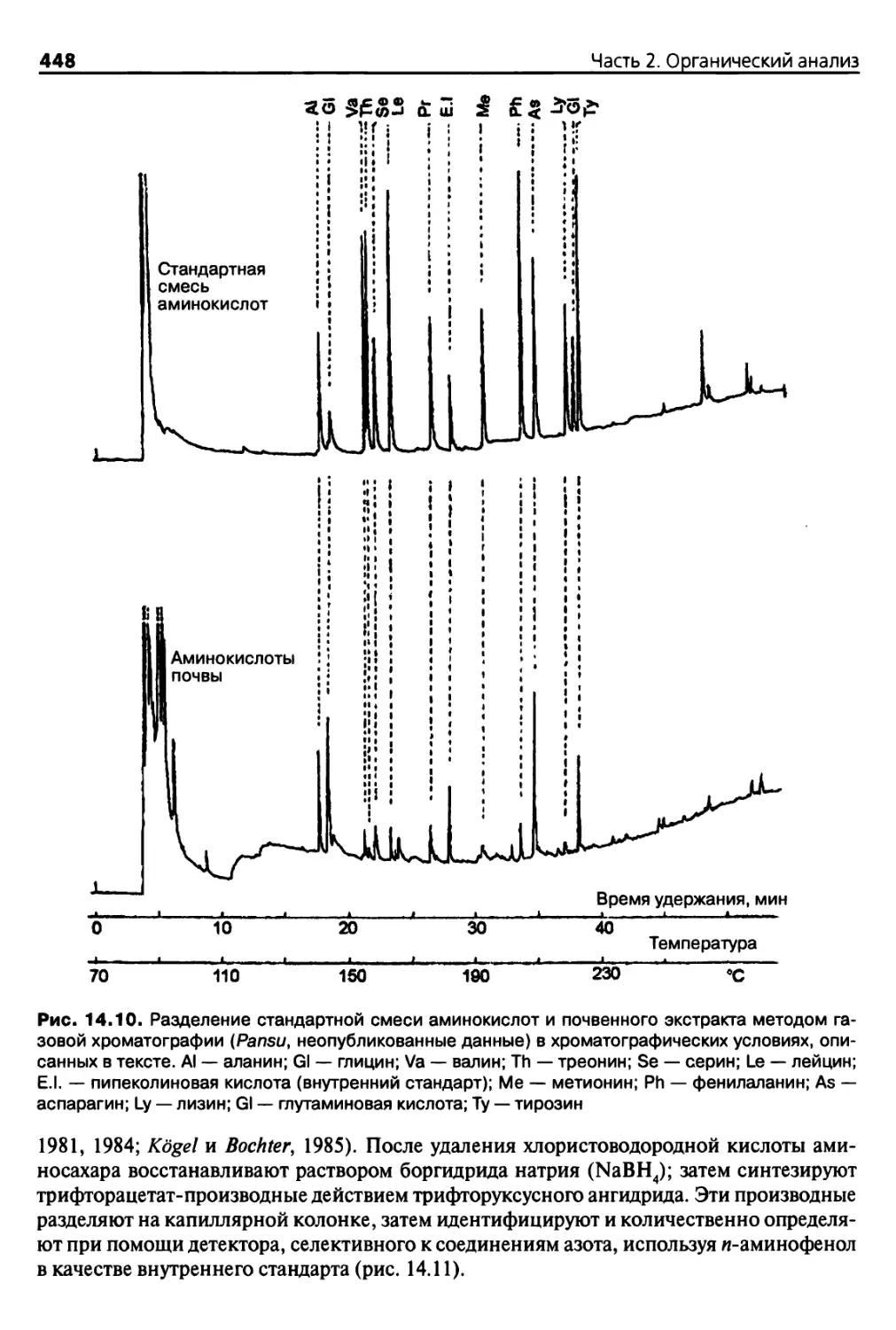
14.3.2. Определение содержания аминокислот 444
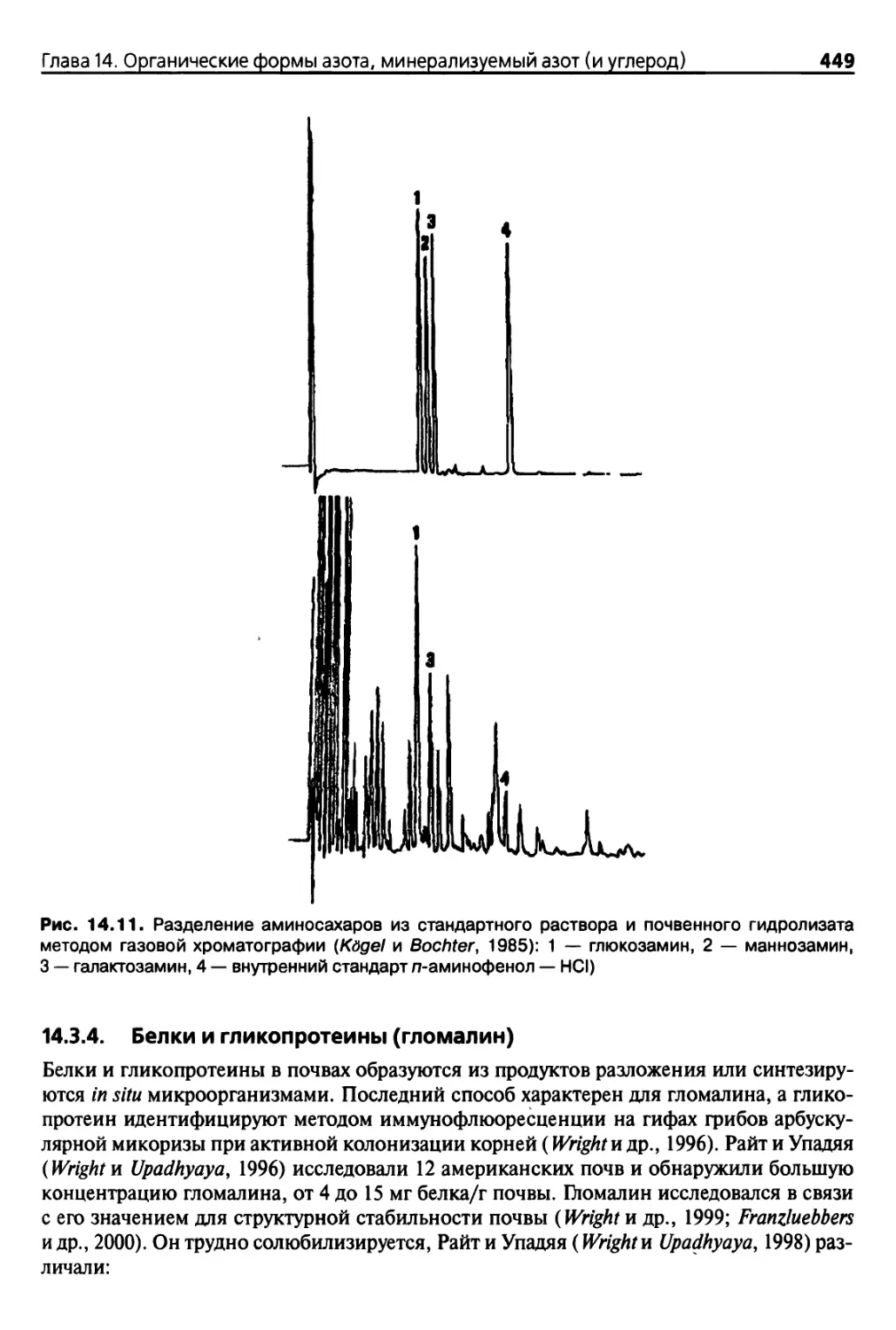
14.3.3. Определение содержания аминосахаров 447
14.3.4. Белки и гликопротеины (гломалин) 449
14.3.5. Определение содержания потенциально минерализуемого
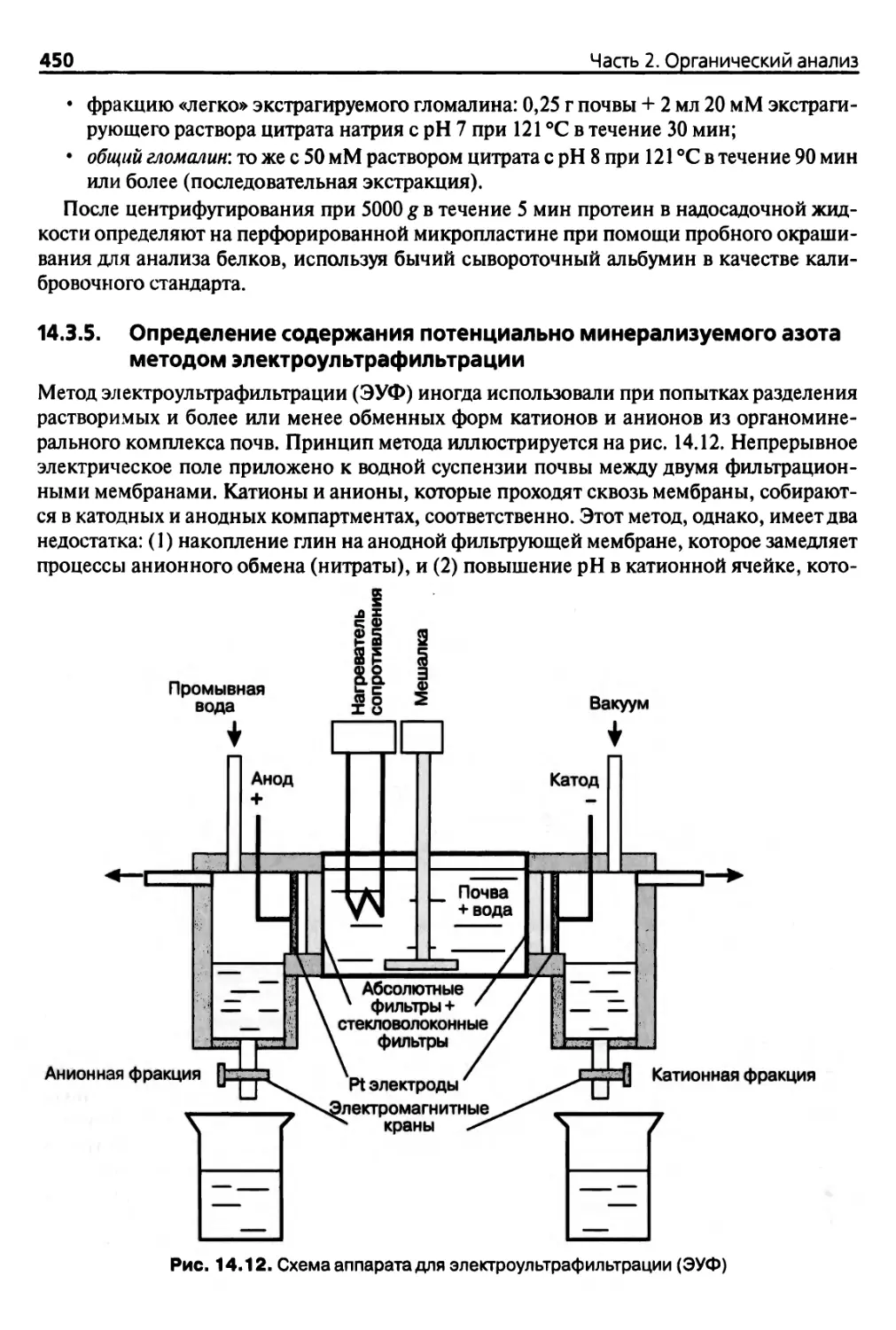
азота методом электроультрафильтрации 450
Использованная литература 451
Часть 3. Неорганический анализ — определение обменных ионов
и полный элементный анализ
Ппава 15. Определение pH среды 459
15.1. Введение 459
15.1.1. pH почвы 459
15.1.2. Ограничения 461
15.1.3. Теоретические аспекты 461
15.2. Классические методы анализа 463
15.2.1. Методы испытаний 463
15.5.2. Колориметрический метод 464
15.2.3. Электрометрический метод 466
15.2.4. Электрометрический контроль и калибровка 470
15.2.5. Исследование водных суспензий почв 470
15.2.6. Определение рНк и рН^ 472
15.2.7. Определение pH «насыщенных паст» 473
15.2.8. Определение pH насыщенных экстрактов 473
15.2.9. Определение pHNaF 474
15.3. Анализ почв в полевых условиях 474
15.3.1. Оборудование 475
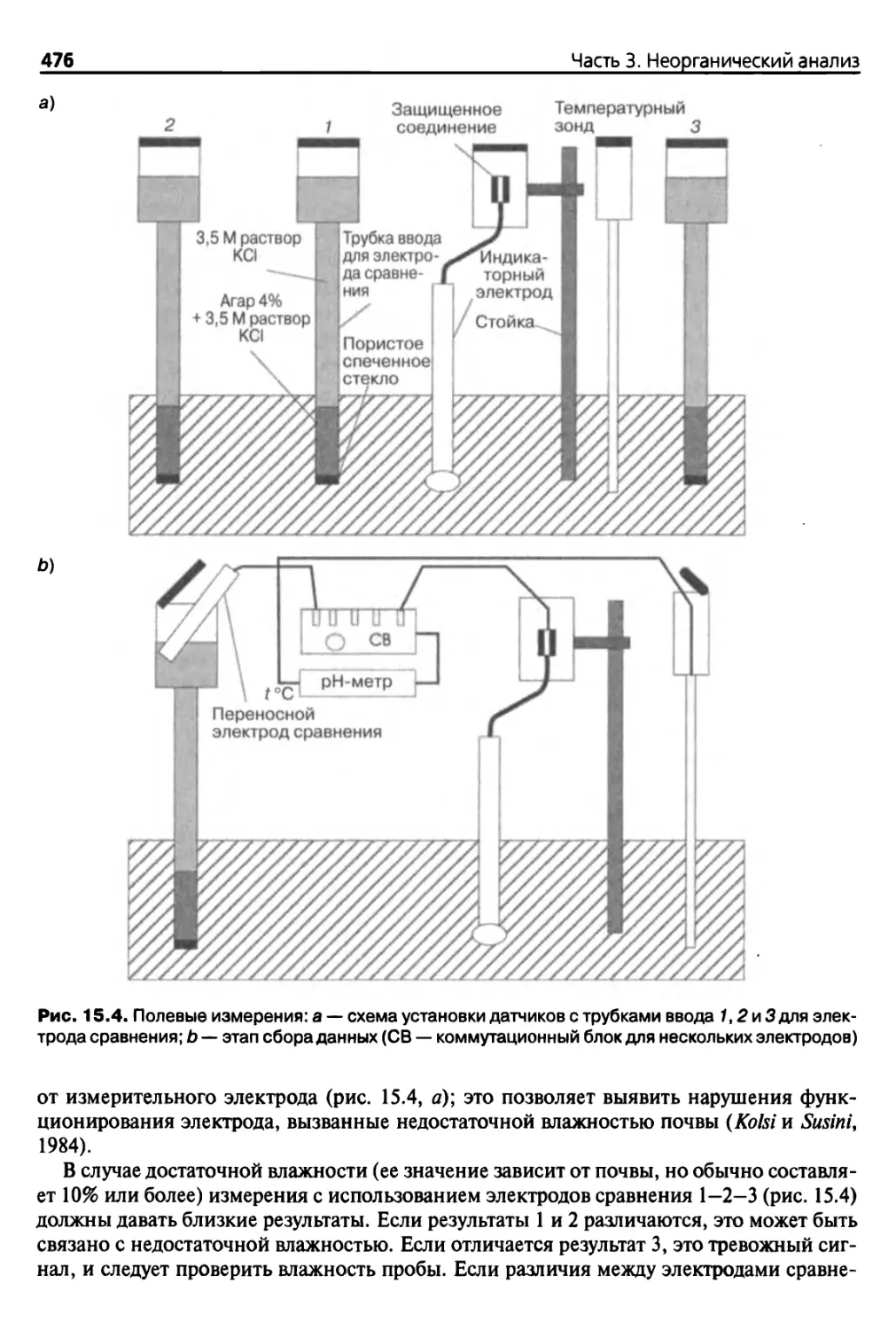
15.3.2. Полевые измерения 475
15.3.3. Измерения на почвенных монолитах 477
Использованная литература 477
Дополнительная литература 478
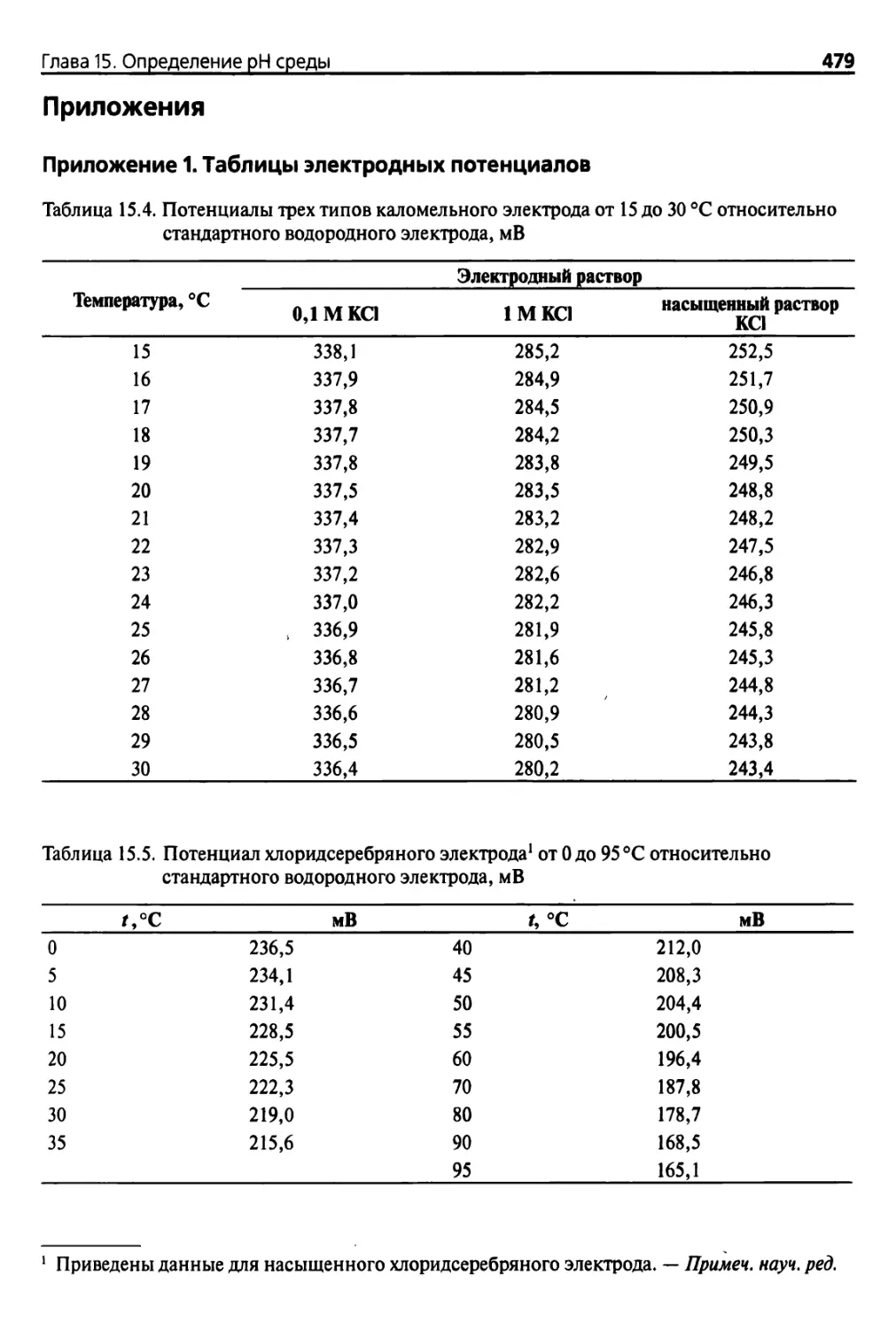
Приложение 1. Таблицы электродных потенциалов 479
Приложение 2. Константы диссоциации для некоторых равновесий .. 480
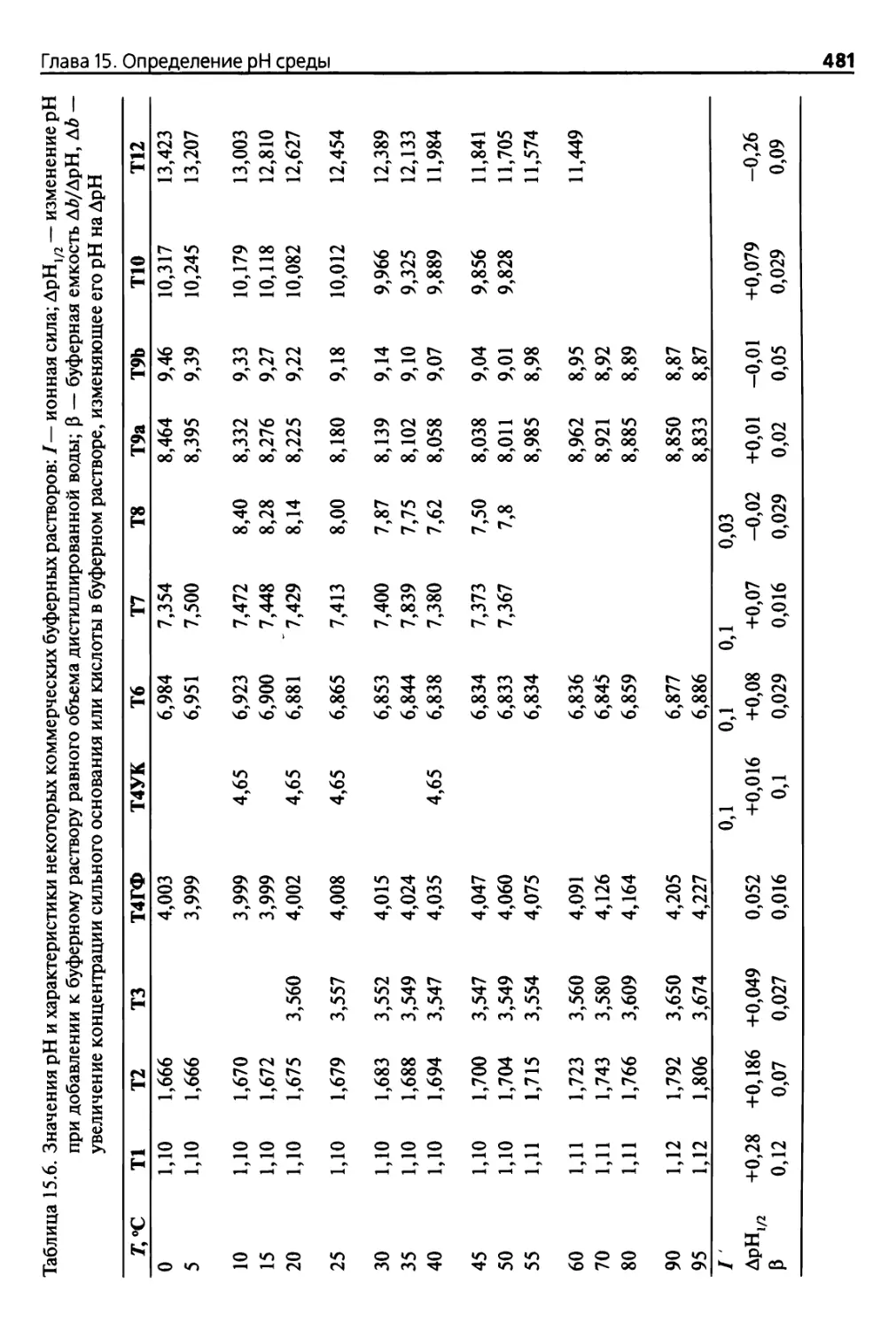
Приложение 3. Буферные растворы 480
Приложение 4. Цветные индикаторы 482
Гпава 16. Окислительно-восстановительный потенциал 483
16.1. Определения и основные положения 483
16.2. Оборудование и реактивы 484
16.2.1. Электроды 484
16.2.2. Соединительный солевой мостик 485
12
Оглавление
16.2.3. Система измерений 485
16.2.4. Калибровочные растворы 486
16.3. Методика 486
16.3.1. Предварительная обработка электрода 486
16.3.2. Анализ образцов почв 487
16.3.3. Анализ почвенных монолитов 487
16.3.4. Исследования в полевых условиях 487
16.3.5. Определение скорости диффузии кислорода 488
16.3.6. Колориметрическое определение Eh 489
Использованная литература 490
Дополнительная литература 490
Diana 17. Карбонаты 492
17.1. Введение 492
17.2. Определение содержания общих карбонатов 493
17.2.1. Введение 493
17.2.2. Волюметрические измерения методом кальциметрии 494
17.2.3. Ацвдиметрия 497
17.3. Определение активных карбонатов 498
17.3.1. Общие положения 498
17.3.2. Порядок выполнения 498
17.3.3. Индекс хлорозного потенциала 500
Использованная литература 500
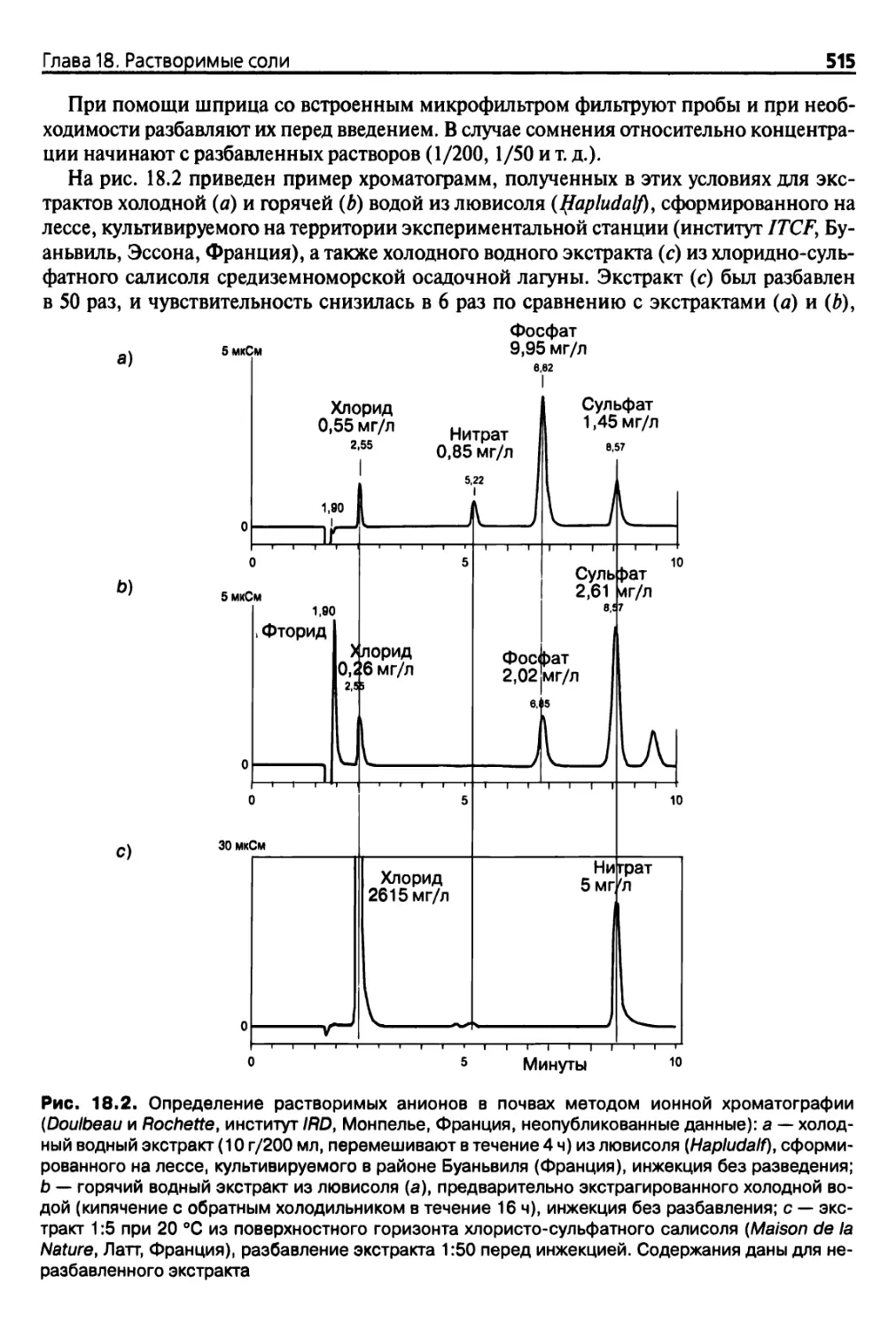
Гпава 18. Растворимые соли 501
18.1. Введение 501
18.2. Экстракция 502
18.2.1. Соотношение почва : раствор 502
18.2.2. Экстракция насыщенных паст 502
18.2.3. Разбавленные экстракты 503
18.2.4. Отбор проб воды из почв 504
18.2.5. Экстракция горячей водой 504
18.3. Анализ и определение содержания солей 505
18.3.1. Электропроводность экстрактов 505
18.3.2. Измерение удельной электропроводности в полевых
условиях 507
18.3.3. Определение общего содержания растворенных веществ 507
18.3.4. Растворимые катионы 508
18.3.5. Экстрагируемые карбонаты и бикарбонаты (щелочность) 509
18.3.6. Экстрагируемые хлориды 510
18.3.7. Экстрагируемые сульфаты, нитраты и фосфаты 511
18.3.8. Экстрагируемый бор 512
18.3.9. Определение экстрагируемых анионов методом
ионообменной хроматографии 513
18.3.10. Представление результатов 516
Использованная литература 517
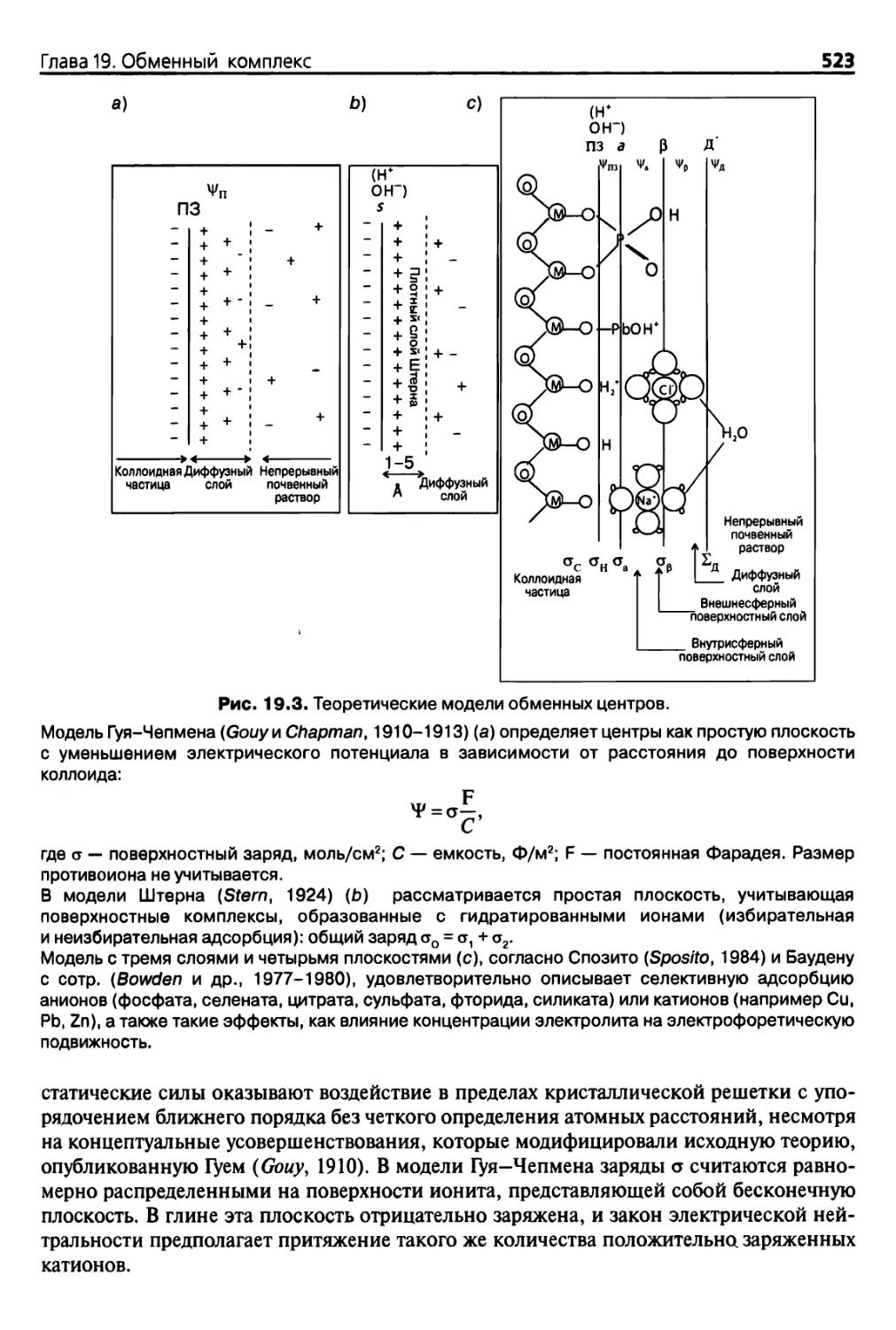
Diana 19. Обменный комплекс 519
19.1. Введение 519
Оглавление
13
19.2. Источники зарядов 519
19.2.1. Ионный обмен 519
19.2.2. Обменный комплекс 520
19.2.3* Теория 522
Использованная литература 524
Дополнительная литература 525
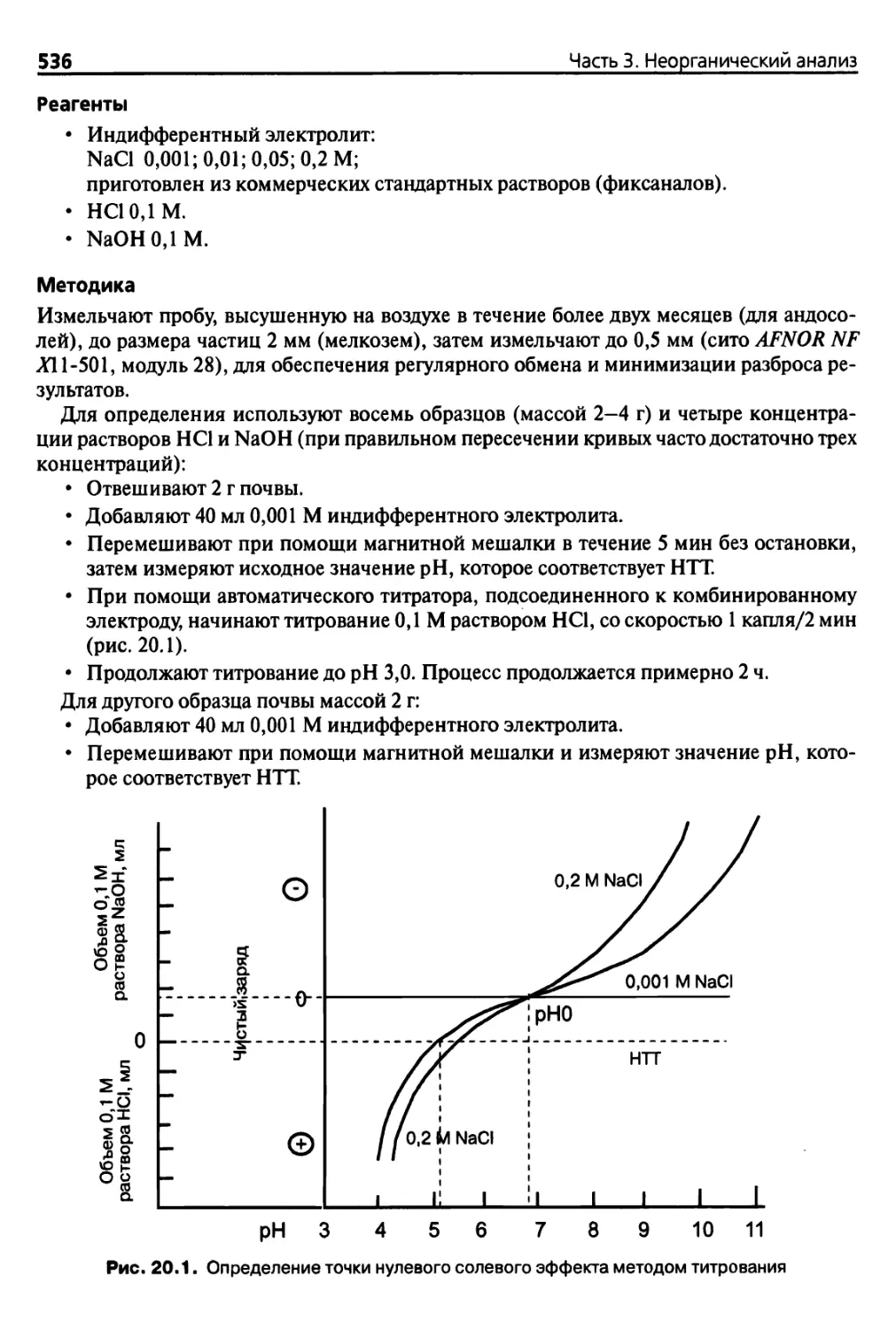
Глава 20. Изоэлектрическая точка и точка нулевого заряда 530
20.1. Введение 530
20.1.1. Заряды коллоидных растворов 530
20.1.2. Определения 532
20.1.3. Условия измерения заряда 533
20.2. Основные методы 534
20.2.1. Определение рНО (ТНСЭ), длительное установление
равновесия 534
20.2.2. Точка нулевого солевого эффекта (ТНСЭ), быстрое
установление равновесия 535
Использованная литература 537
Става 21. Постоянные и переменные заряды 539
21.1. Введение 539
21.2. Основные методы определения 542
21.2.1. Измерение переменных зарядов 542
21.2.2. Определение постоянных зарядов 543
Использованная литература 545
Дополнительная литература 545
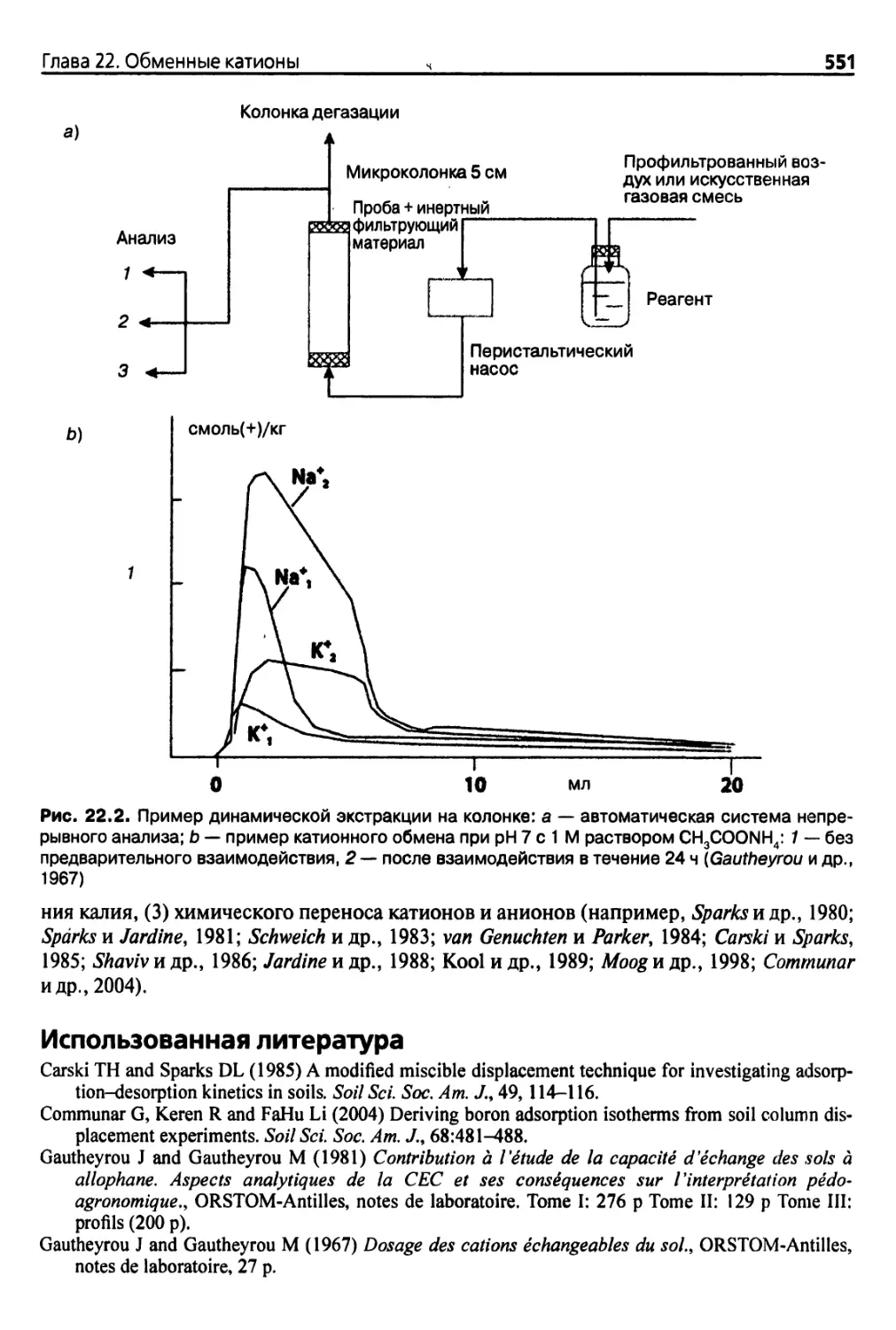
Diaea 22. Обменные катионы 546
22.1. Введение 546
22.1.1. Обменные катионы в почве 546
22.1.2. Экстрагирующие реагенты. ♦ 547
22.1.3. Оборудование 547
22.2. Метод экстракции ацетатно-аммонийным буфером при pH 7 548
22.2.1. Основные положения 548
22.2.2. Методика 549
22.3. Автоматизированный метод непрерывной экстракции 550
Использованная литература 551
Дополнительная литература 552
Става 23. Обменная кислотность 553
23.1. Введение 553
23.1.1. Природа кислотности 553
23.1.2. Цели анализа 554
23.2. Метод определения 555
23.2.1. Основные положения 555
23.2.2. Реагенты 555
23.2.3. Методика 556
23.3. Другие методы анализа 558
Использованная литература '. 558
14
Оглавление
Дополнительная литература 559
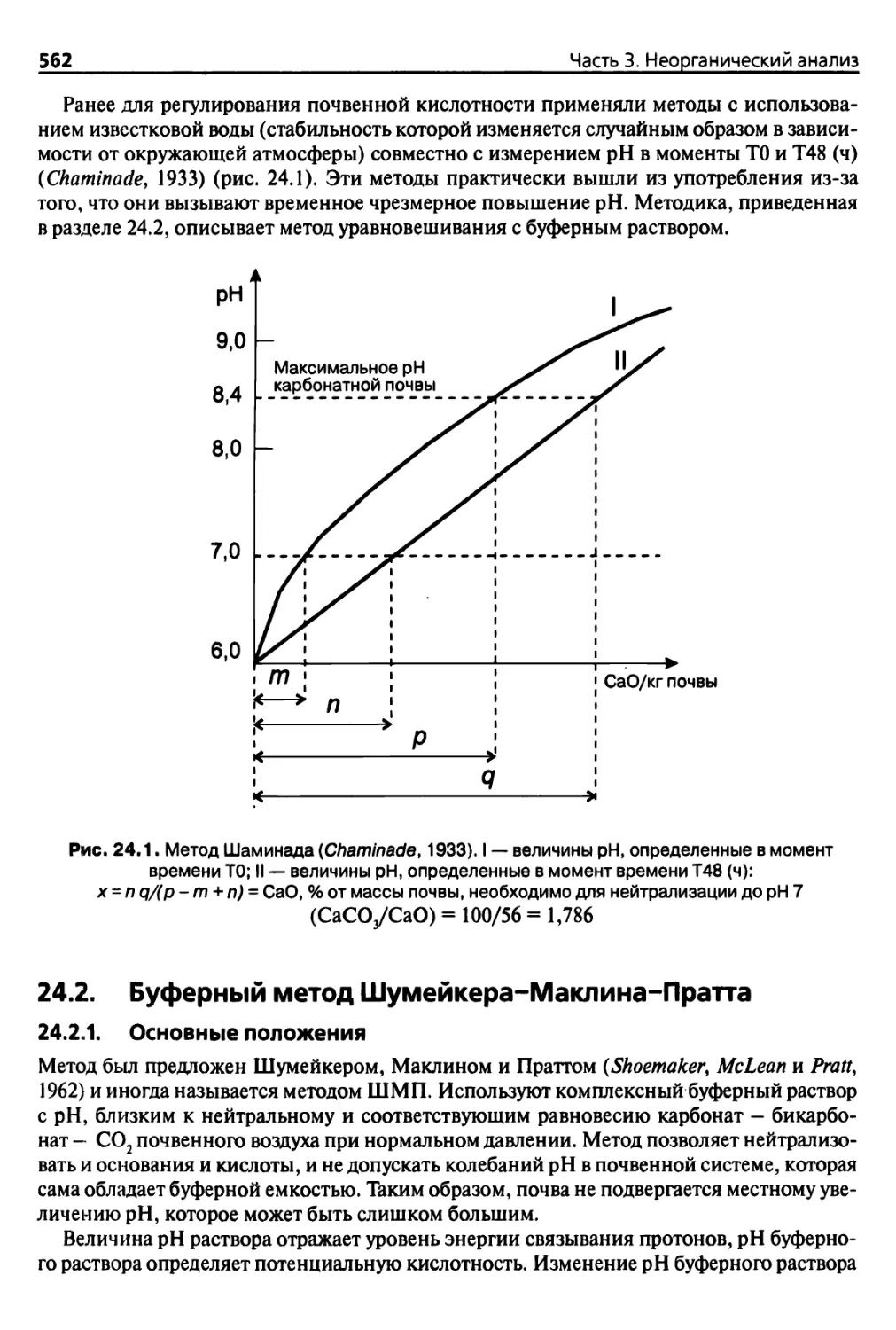
Diana 24. Потребность в известковании почв 560
24.1. Введение 560
24.1.1. Регулирование кислотности почв 560
24.1.2. Расчет необходимого количества извести 561
24.2. Буферный метод Шумейкера-Маклина—Пратта 562
24.2.1. Основные положения 562
24.2.2. Реагенты 563
24.2.3. Методика 563
24.2.4. Примечания 564
Использованная литература 564
Дополнительная литература 565
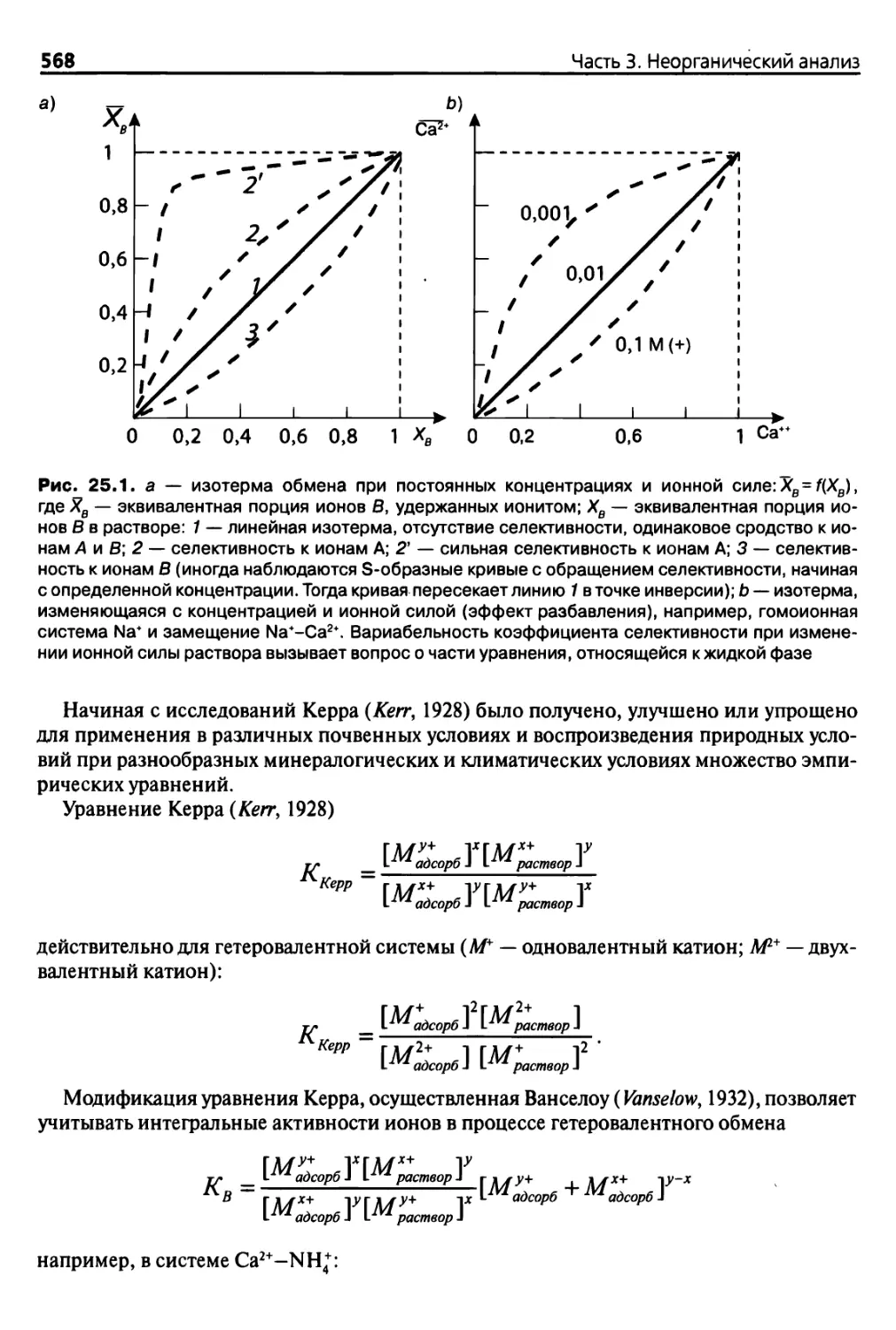
Diana 25. Селективность обмена, изотерма катионного обмена 567
25.1. Введение 567
25.2. Построение изотермы обмена 571
25.2.1. Основные положения 571
25.2.2. Реагенты 571
25.2.3. Методика 571
25.2.4. Примечания 573
Использованная литература 573
Дополнительная литература 574
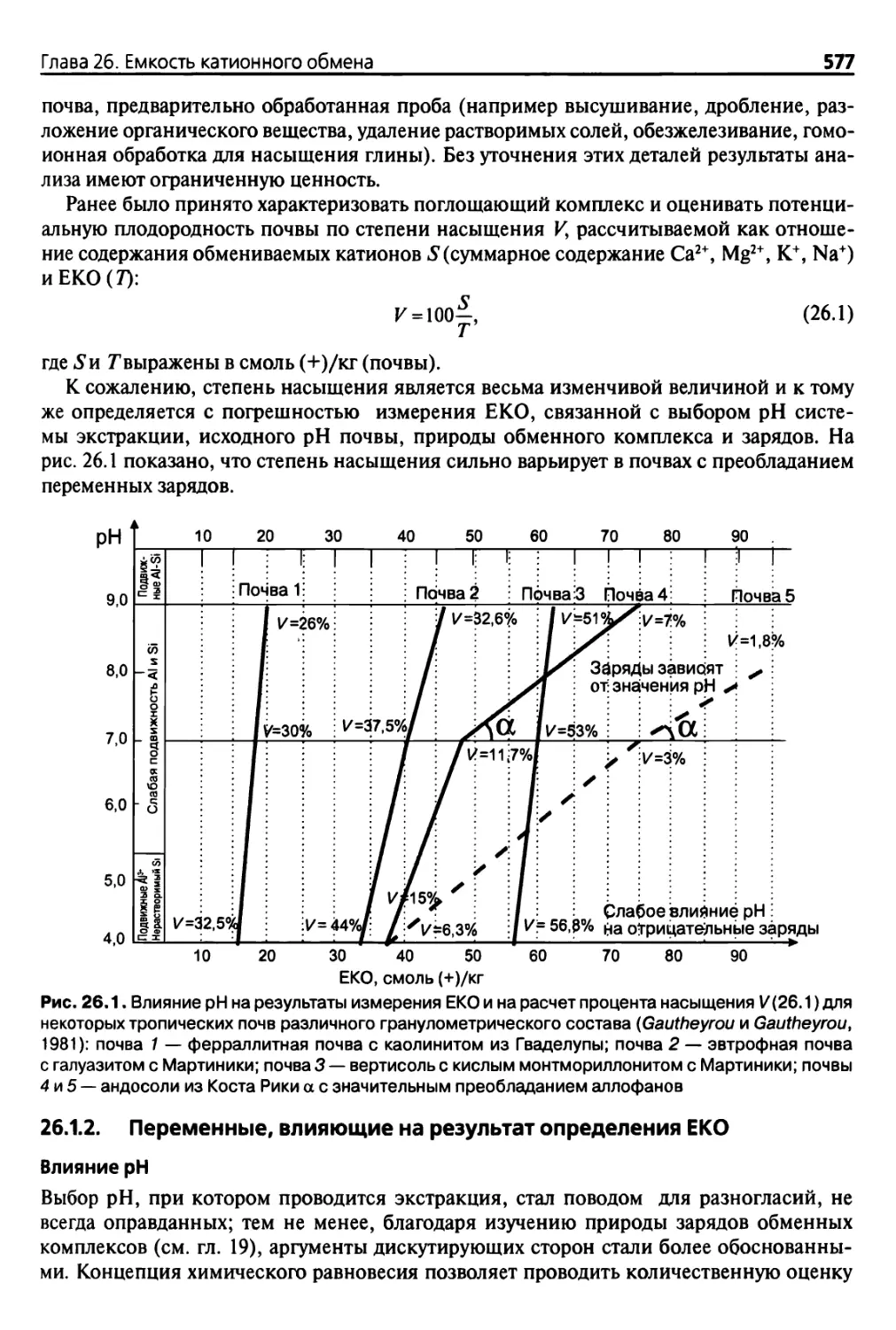
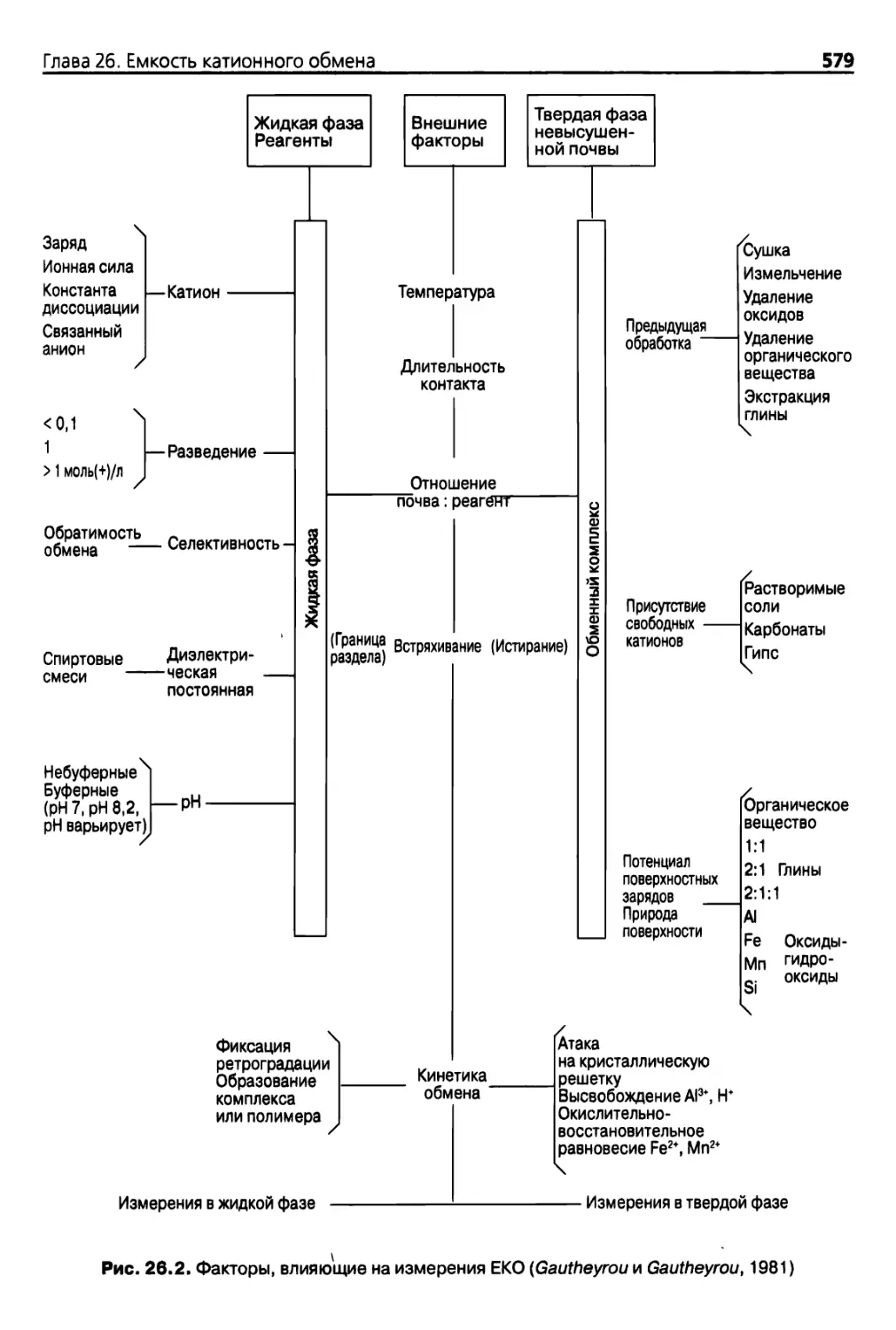
Diana 26. Емкость катионного обмена 576
26.1 Введение 576
26.1.1 Теоретические аспекты 576
26.1.2. Переменные, влияющие на результат определения ЕКО 577
26.2. Определение эффективной ЕКО путем суммирования (ЭЕКО) 583
26.2.1. Основные положения 583
26.2.2. Альтернативные методы 584
26.3. Измерение ЕКО на уровне pH почвы в небуферной среде 584
26.3.1. Основные положения 584
26.3.2. Методики, использующие небуферные растворы солей
металлов 584
26. 3.3. Методики с использованием металлорганических катионов
в небуферной среде 587
26.3.4. Методики с использованием органических катионов
в небуферных растворах 591
26.4. Измерения ЕКО в буферной среде 593
26.4.1. Методики измерения ЕКО в буферных средах —
общая информация 593
26.4.2. Методика с применением ацетата аммония при pH 7,0 594
26.4.3. Методика с применением буферных систем при pH 8,0-8,6... 600
26.4.4. Методики, использующие буферные растворы с различными
pH 605
Использованная литература 607
Дополнительная литература 609
Оглавление
15
Diana 27. Емкость анионного обмена 613
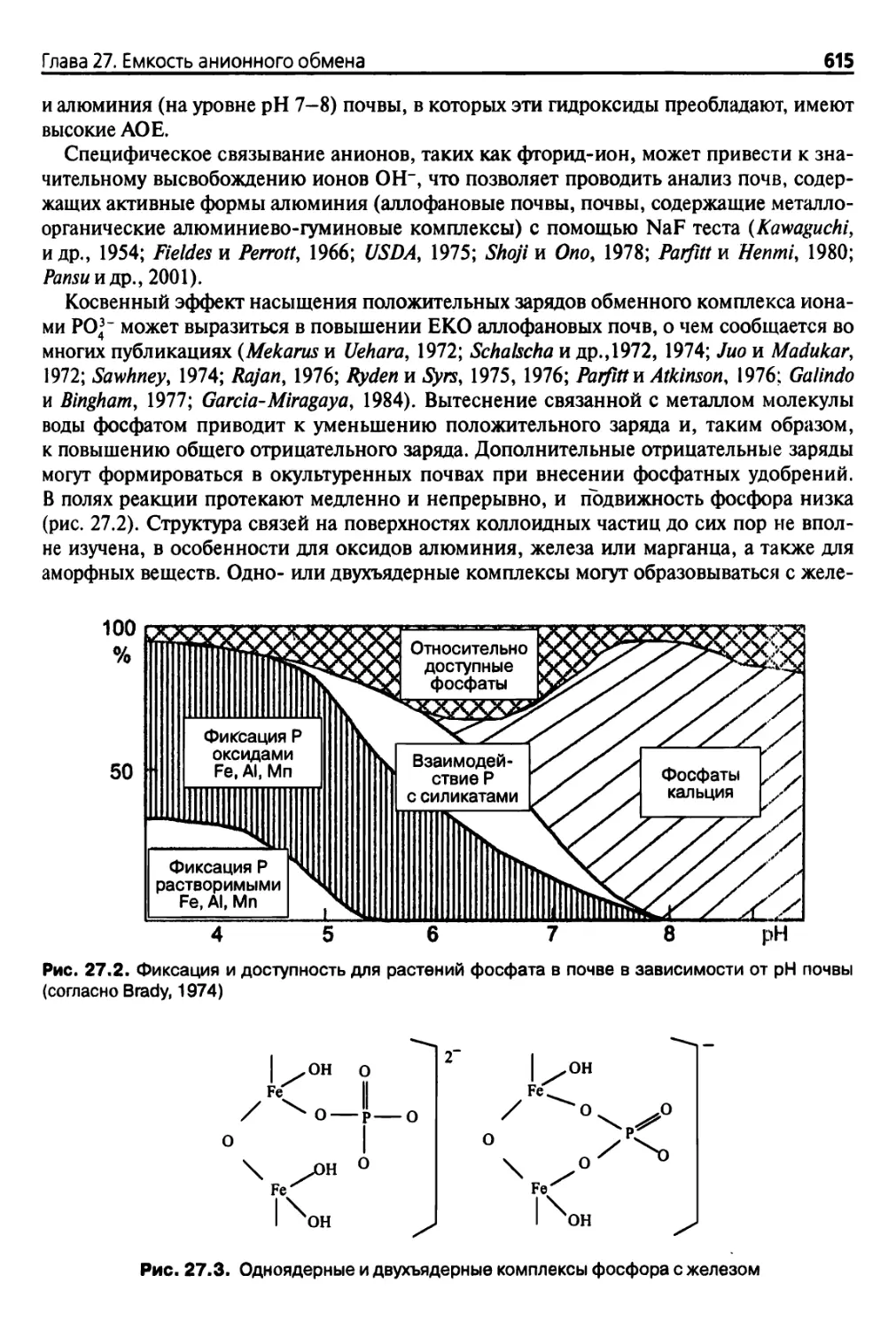
27.1. Теория 613
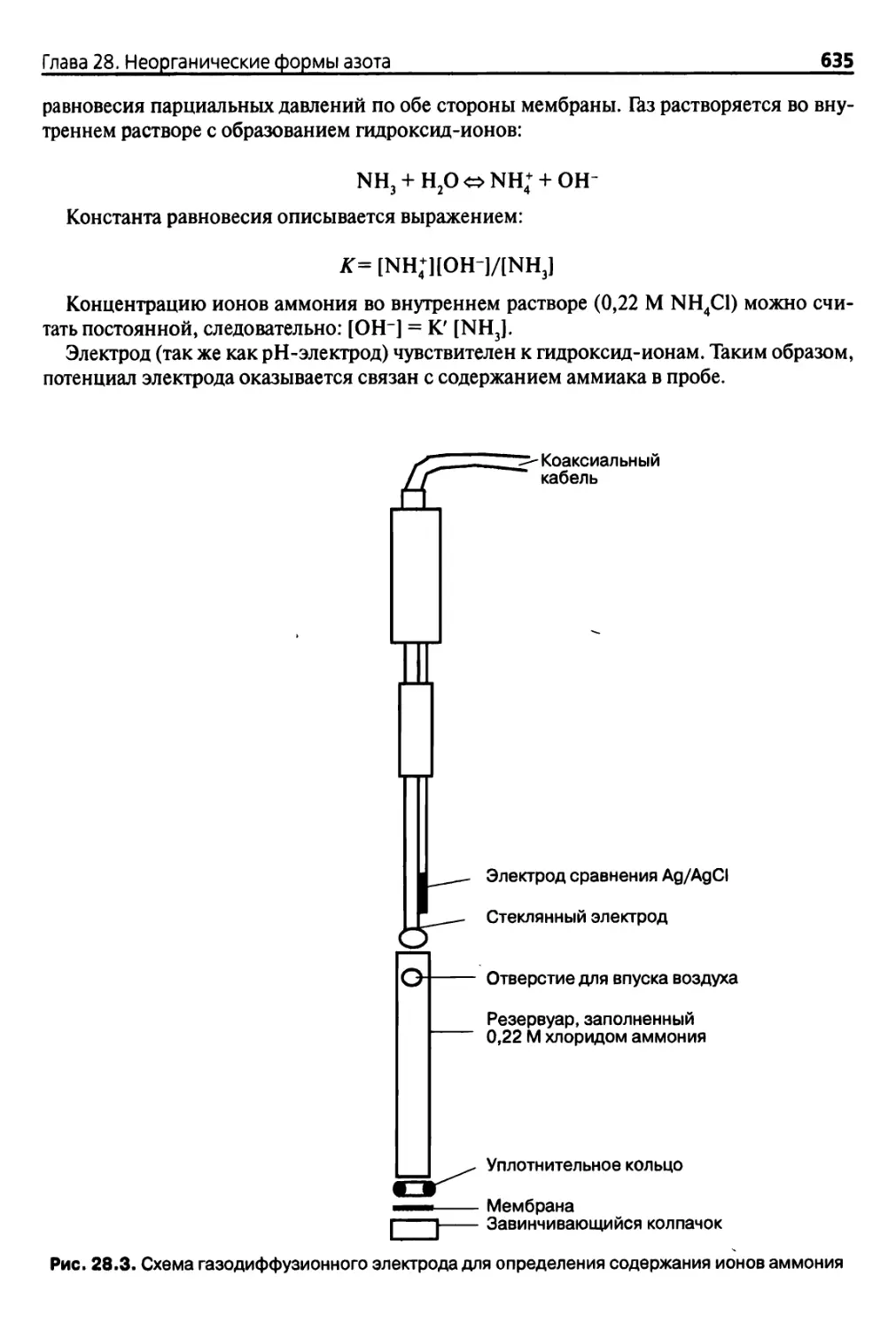
27.2. Измерения 616
27.2.1. Основные положения 616
27.2.2. Методика 617
27.3. Одновременное измерение ЕАО, ОКат, ЕКО и чистой ЕКО 617
27.3.1. Цель 617
27.3.2. Описание методики 618
Использованная литература 620
Diana 28. Неорганические формы азота 622
28.1. Введение 622
28.1.1. Ионы аммония, нитрата и нитрита 622
28.1.2. Проблемы, связанные с отбором проб 622
28.1.3. Аналитические проблемы 623
28.2. Традиционные методики 623
28.2.1. Экстракция обменных форм 623
28.2.2. Разделение микродиффузионным методом 624
28.2.3. Колориметрическое определение аммония 626
28.2.4. Колориметрическое определение нитритов 629
28.2.5. Колориметрическое определение нитратов 631
28.2.6. Экстрагированный органический азот 632
28.3. Другие методы 632
28.3.1. Определение нитратов и нитритов методом УФ
фотометрического поглощения 632
28.3.2. Определение аммония с помощью селективного
электрода 634
28.3.3. Анализ нитратов с помощью ион-селективного электрода 637
28.3.4. Измерения in situ 639
28.3.5. Необменный аммоний * 641
Использованная литература 642
Дополнительная литература 642
Diana 29. Фосфор 643
29.1. Введение 643
29.2. Общее содержание фосфора в почве 644
29.2.1. Введение 644
29.2.2. Минерализация мокрым озолением при общем анализе 645
29.2.3. Сухая минерализация 647
29.3. Разделение различных форм фосфора 648
29.3.1. Введение 648
29.3.2. Последовательные методики 648
29.3.3. Селективная экстракция — индексы доступности 652
29.3.4. Методы изотопного разбавления 659
29.3.5. Анализ органического фосфора 660
29.4. Удержание фосфора 664
29.4.1. Введение 664
29.4.2. Определение параметров удержания Р 664
29.5. Анализ Р в экстрактах 665
16
Оглавление
29.5.1. Введение 665
29.5.2. Анализ ортофосфатного Р спектроколориметрическим
методом 667
29.5.3. Анализ Р методами атомной спектрометрии 671
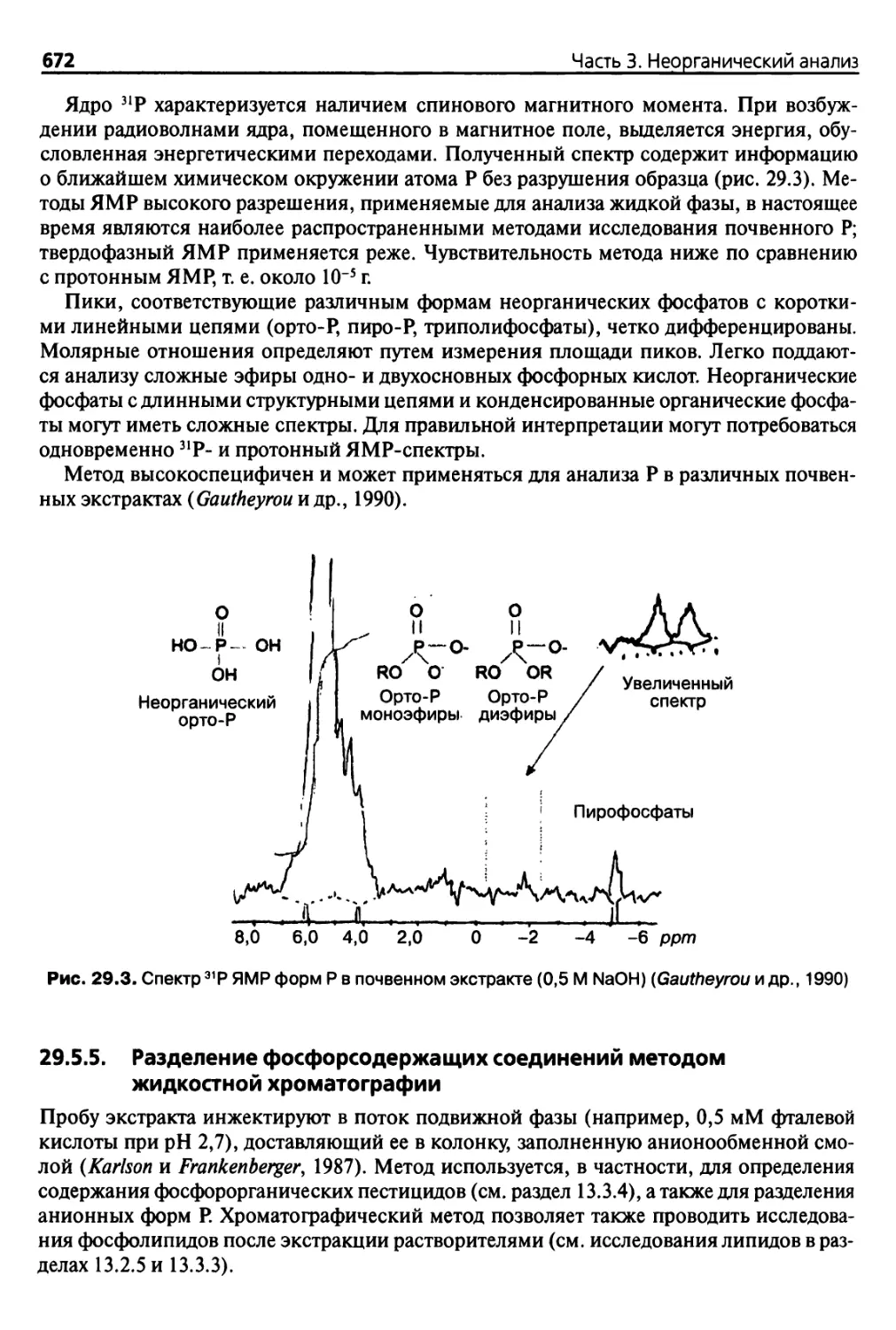
29.5.4. Определение различных форм Р методом 31Р-ЯМР 671
29.5.5. Разделение фосфорсодержащих соединений методом
жидкостной хроматографии 672
29.6. Прямое определение форм Р in situ или в экстрагированных
частицах 673
Использованная литература 673
Дополнительная литература 675
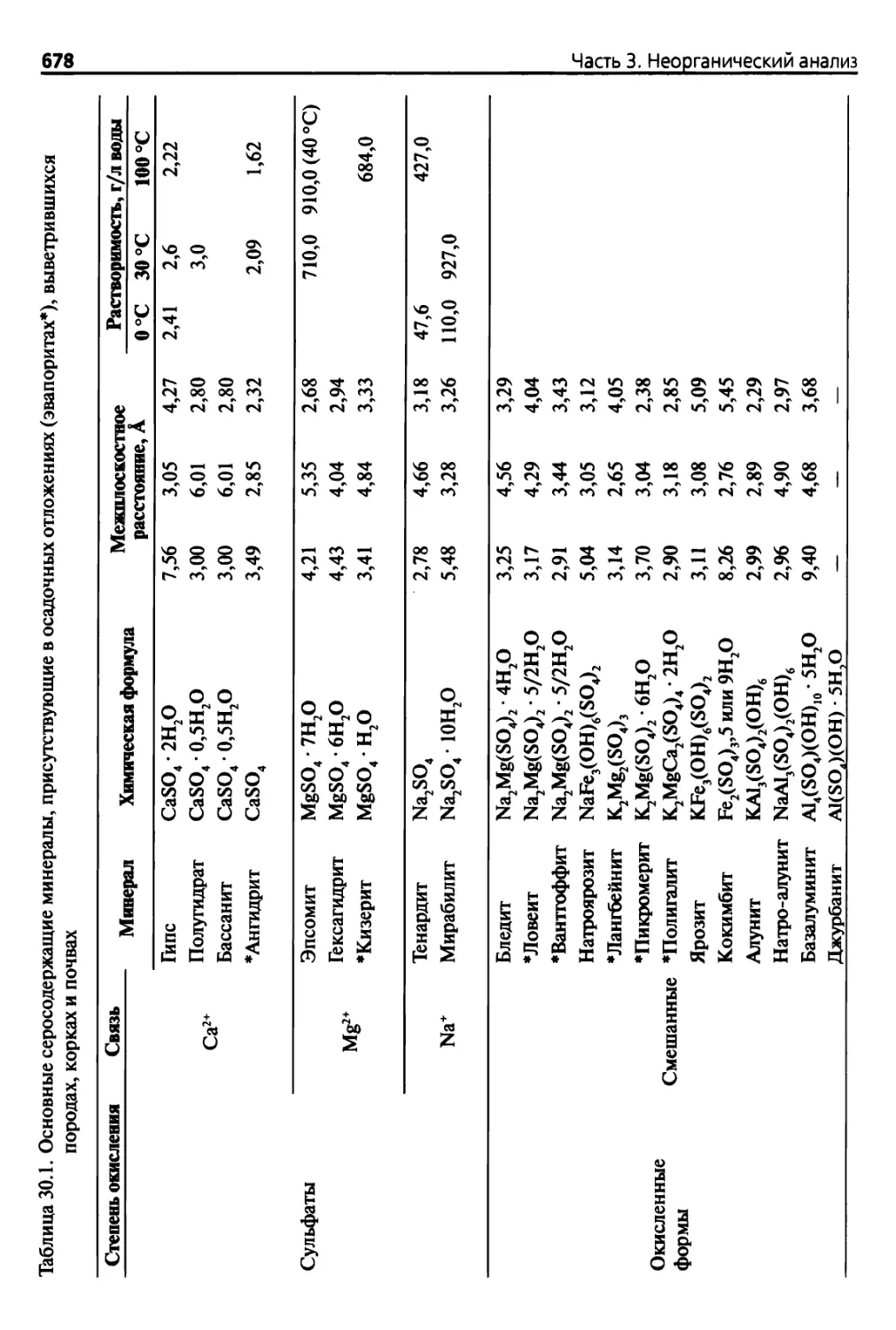
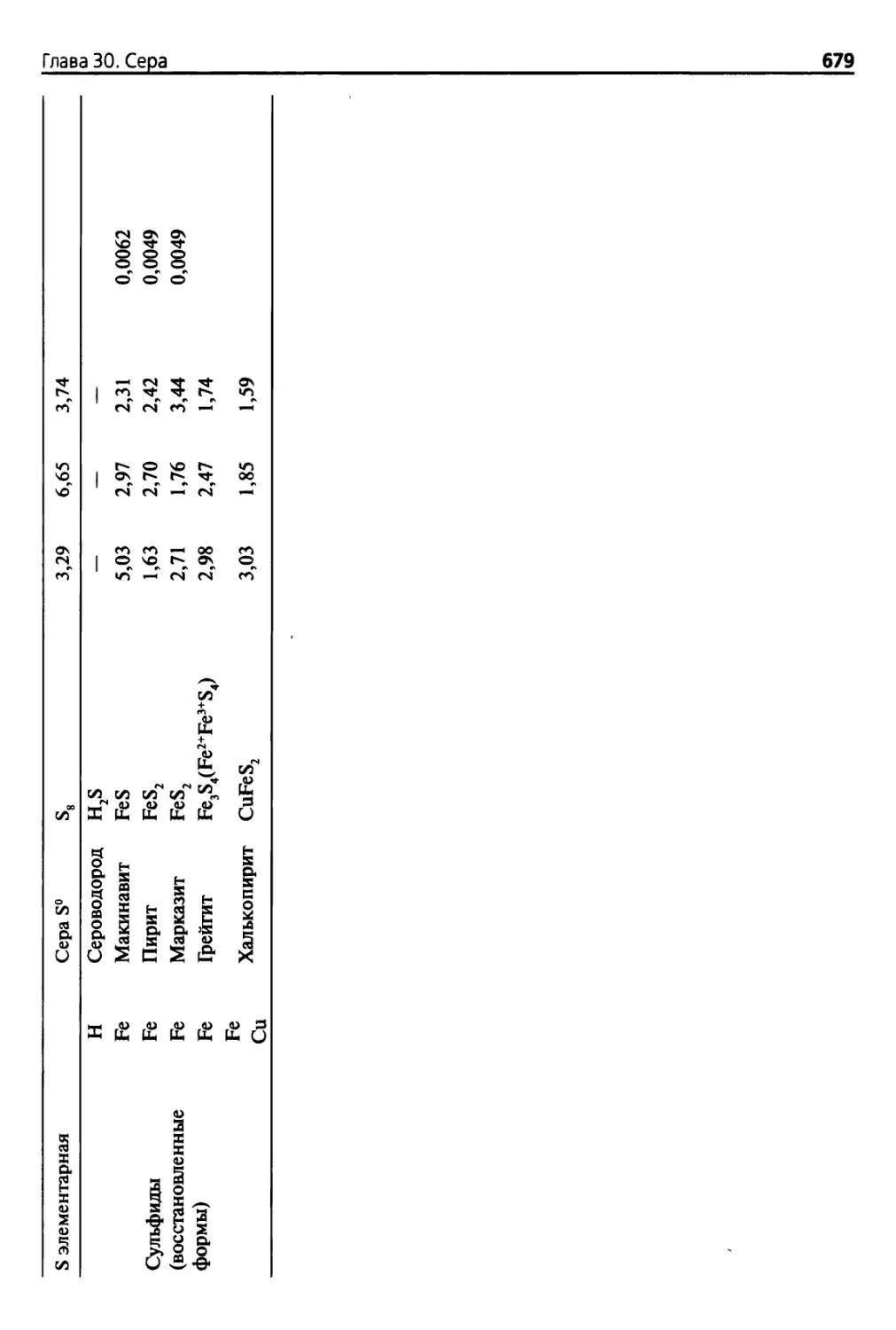
Пиша 30. Сера 677
30.1. Введение 677
30.1.1. Соединения серы 677
30.1.2. Минералогические исследования 677
30.2. Общее содержание серы и соединений серы 681
30.2.1. Характеристики почв, сформировавшихся на морских
и речных наносах (Fluviomarine Soils) 681
30.2.2. Отбор и подготовка проб почвы 681
30.2.3. Анализ растворимых форм серы 682
30.2.4. Анализ общей серы 683
30.2.5. Солюбилизация общей серы методом окислительного
щелочного плавления 683
30.2.6. Полная солюбилизация гипобромитом натрия в щелочной
среде ,... 684
30.2.7. Анализ S колориметрическим методом с использованием
метиленового синего 685
30.2.8. Анализ сульфатов колориметрическим методом
с использованием метилтимолового синего 690
30.2.9. Определение общей серы автоматизированным сухим
CHN(OS) элементным анализом 693
30.2.10. Анализ общего содержания S в форме SO*- методом
ионной хроматографии 694
30.2.11. Анализ общей S методом плазменной эмиссионной
спектрометрии 696
30.2.12. Анализ методом рентгеновской флуоресценции 696
30.2.13. Анализ методом атомно-адсорбционной спектрометрии 696
30.2.14. Аналитическое фракционирование соединений серы 696
30.2.15. Анализ органической S, связанной с С 698
30.2.16. Анализ органической S, не связанной с С 699
30.2.17. Экстракция и анализ содержания растворимых сульфидов 700
30.2.18. Анализ серы в пиритах 702
30.2.19. Анализ элементарной серы 704
30.2.20. Анализ водорастворимых сульфатов 706
30.2.21. Анализ сульфатов, экстрагированных №3-ЭДТА 707
30.2.22. Анализ ярозита 709
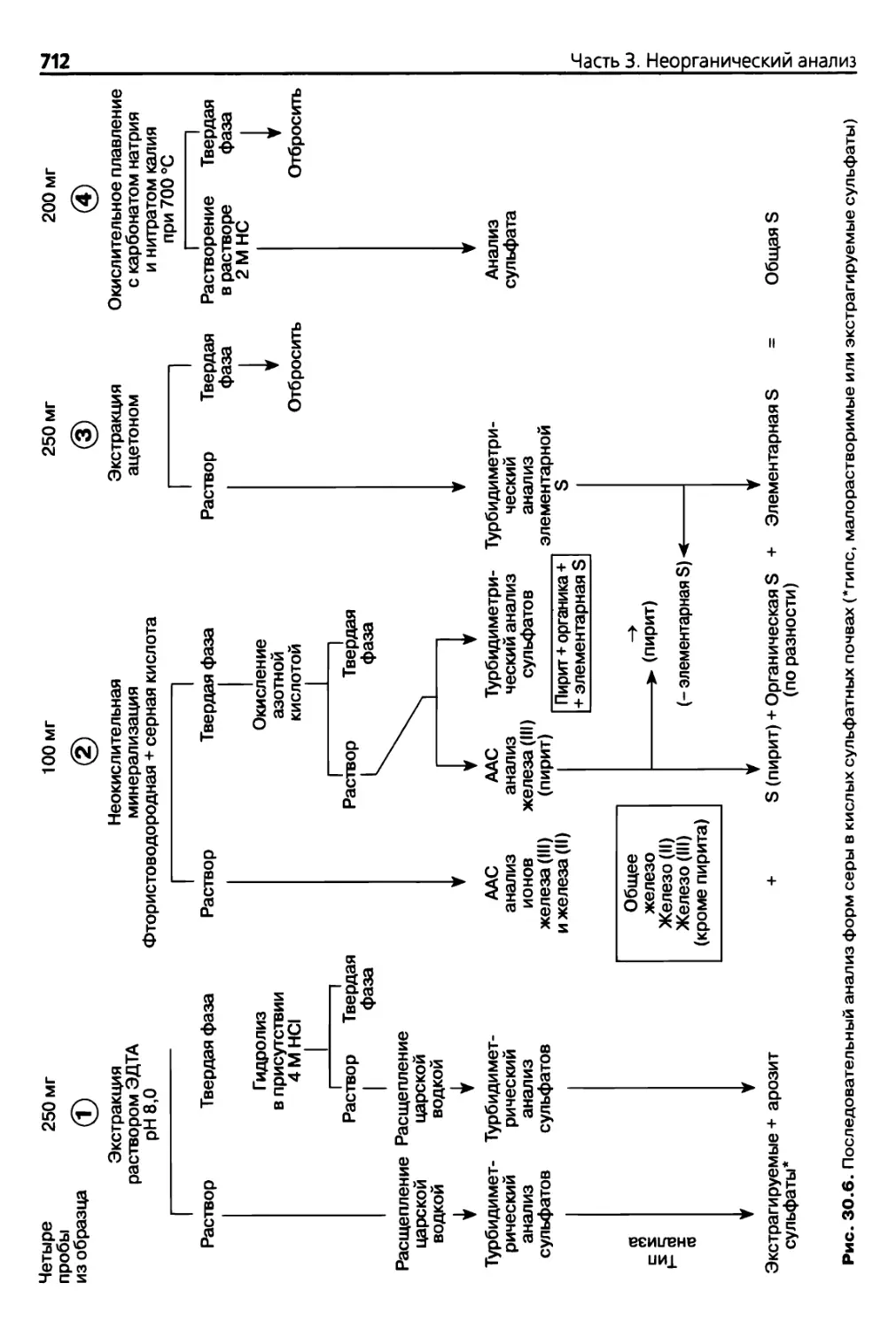
30.2.23. Последовательный анализ форм S 711
Оглавление
17
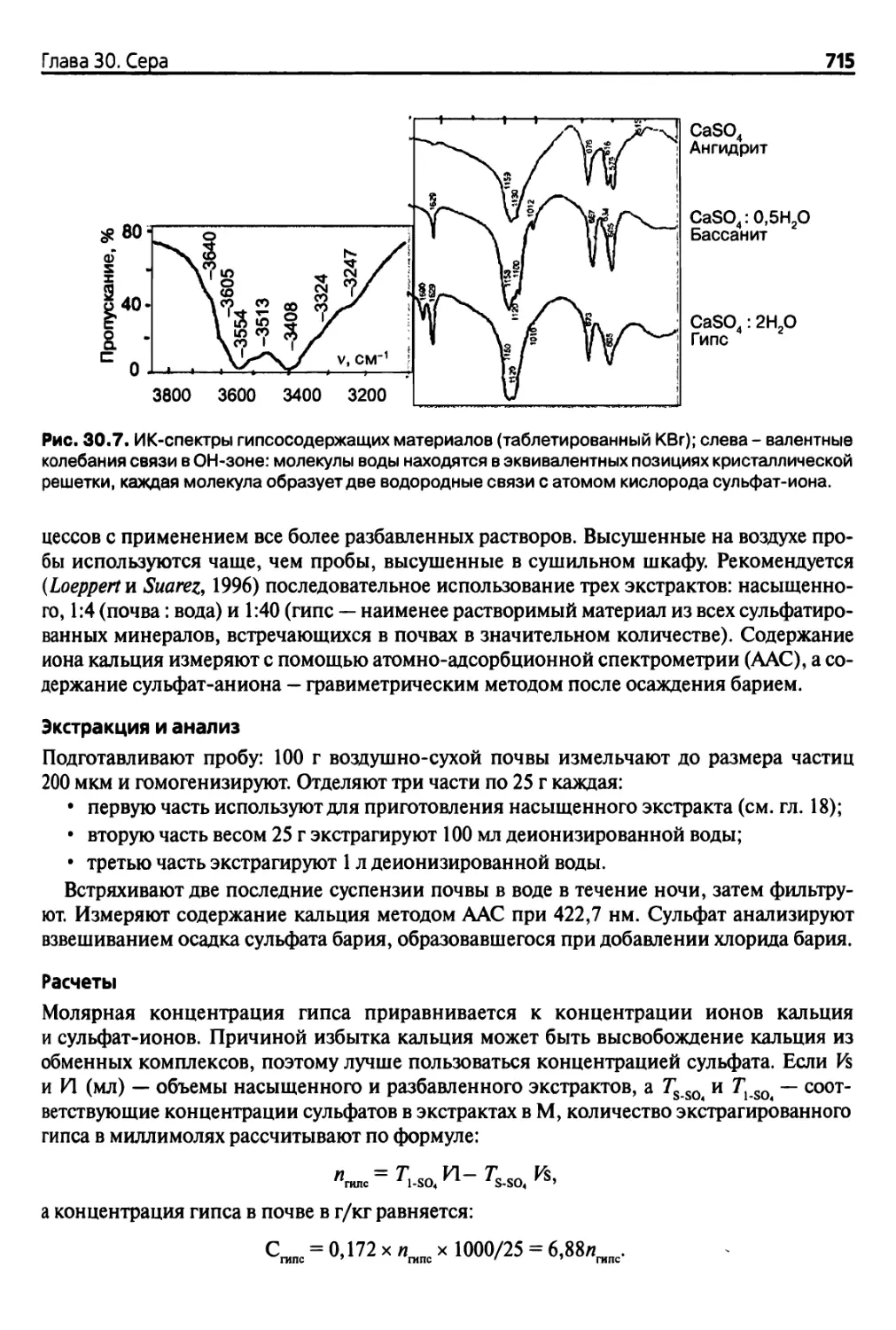
30.3. Сера в гипсоносных почвах 713
30.3.1. Пшсоносные почвы 713
30.3.2. Предварительные анализы 713
30.3.3. Экстракция и количественный анализ при многоступенчатой
экстракции 714
30.3.4. Анализ путем осаждения ацетоном 716
30.4. Потребность почвы в сере и гипсе 717
30.4.1. Введение 717
30.4.2. Потребность растений в сере 718
30.4.3. Потребность в гипсе 719
Используемая литература 720
Дополнительная литература 722
Глава 31. Анализ экстрагируемых элементов и полный элементный анализ 725
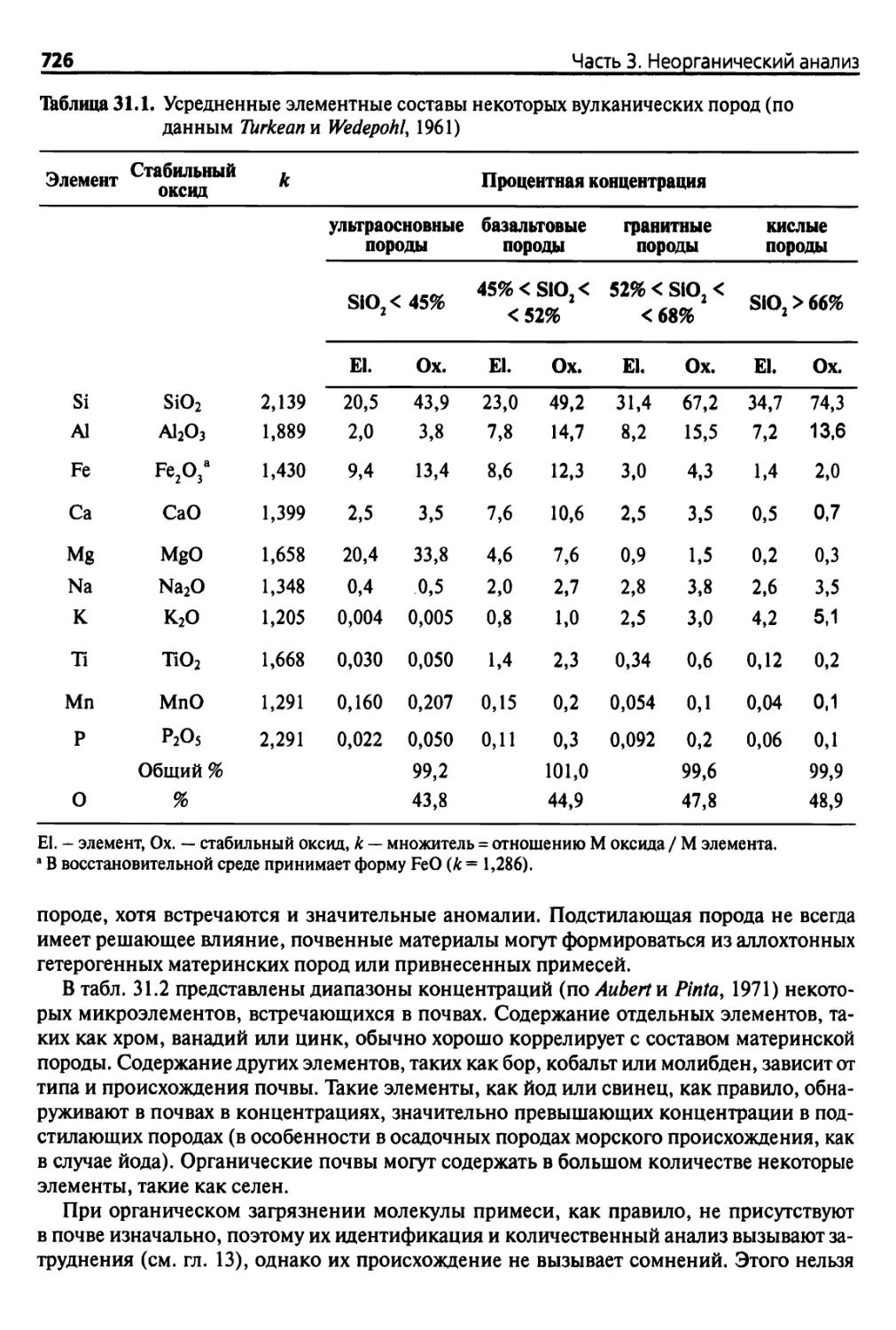
31.1. Элементы почвы 725
31.1.1. Основные элементы 725
31.1.2. Микроэлементы и примеси 725
31.1.3. Биогенные и токсичные элементы 728
31.1.4. Полный элементный анализ 729
31.1.5. Экстрагируемые элементы 729
31.2. Методы, включающие солюбилизацию 730
31.2.1. Методы полной солюбилизации 730
31.2.2. Основные реагенты для полного растворения 731
31.2.3. Кислотное вскрытие пробы в открытом сосуде 734
31.2.4. Кислотное вскрытие в закрытом сосуде 738
31.2.5. Микроволновая минерализация 740
31.2.6. Щелочное сплавление 741
31.2.7. Селективная экстракция .. 746
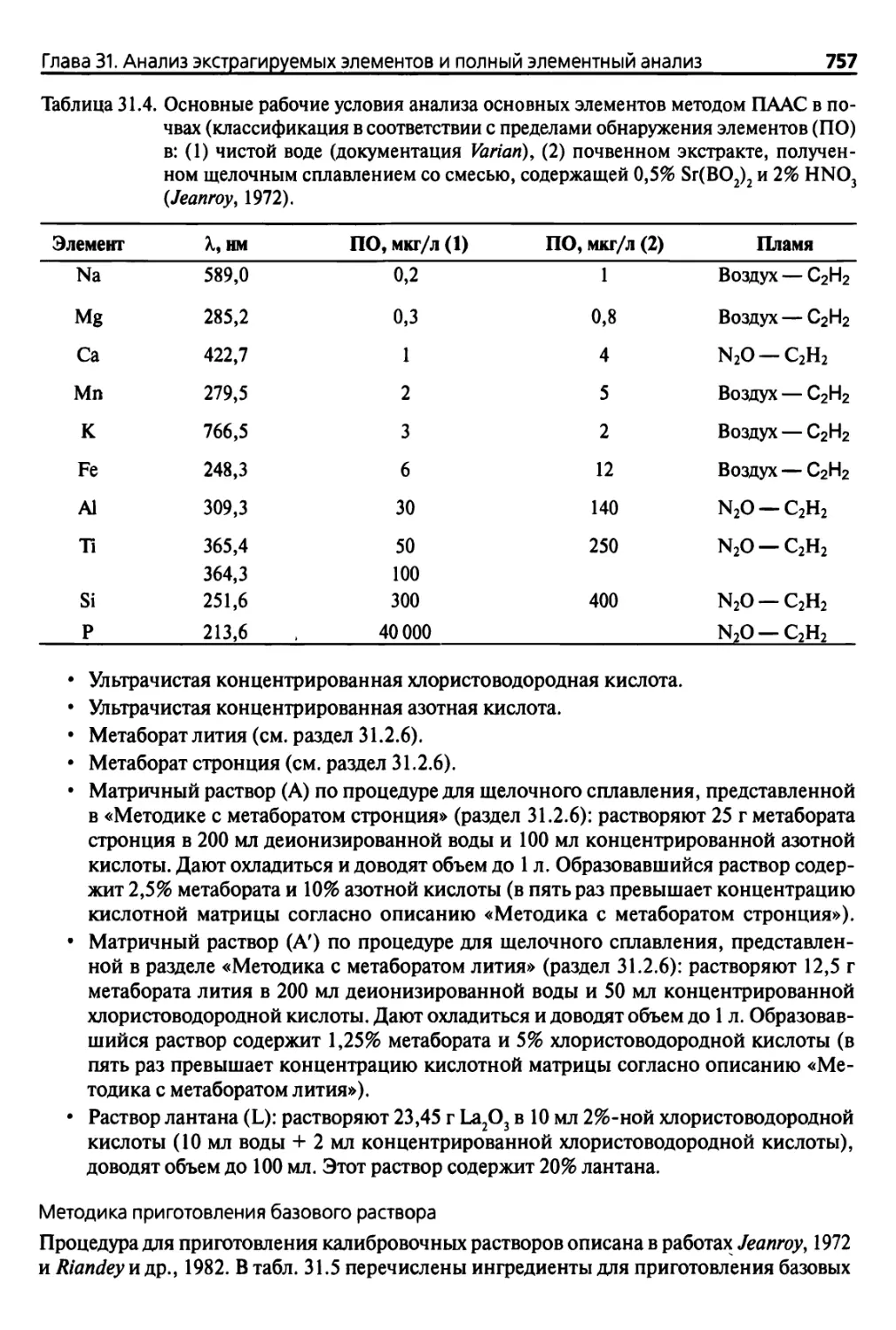
31.2.8. Методы измерений 751
31.2.9. Спектроколориметрический анализ 752
31.2.10. Пламенно-эмиссионная спектрометрия 755
31.2.11. Пламенная атомно-абсорбционная спектрометрия 756
31.2.12. Анализ микроэлементов ААС с генерацией гидридов
и методом холодного пара 760
31.2.13. Анализ микроэлементов электротермической ААС 763
31.2.14. Анализ методом атомно-эмиссионной спектрометрии
с индуктивно связанной плазмой (ИСП-АЭС) 763
31.2.15. Анализ методом масс-спектрометрии с индуктивно
связанной плазмой 767
31.3. Анализ в твердых средах 772
31.3.1. Методы 772
31.3.2. Рентгенофлуоресцентный анализ 773
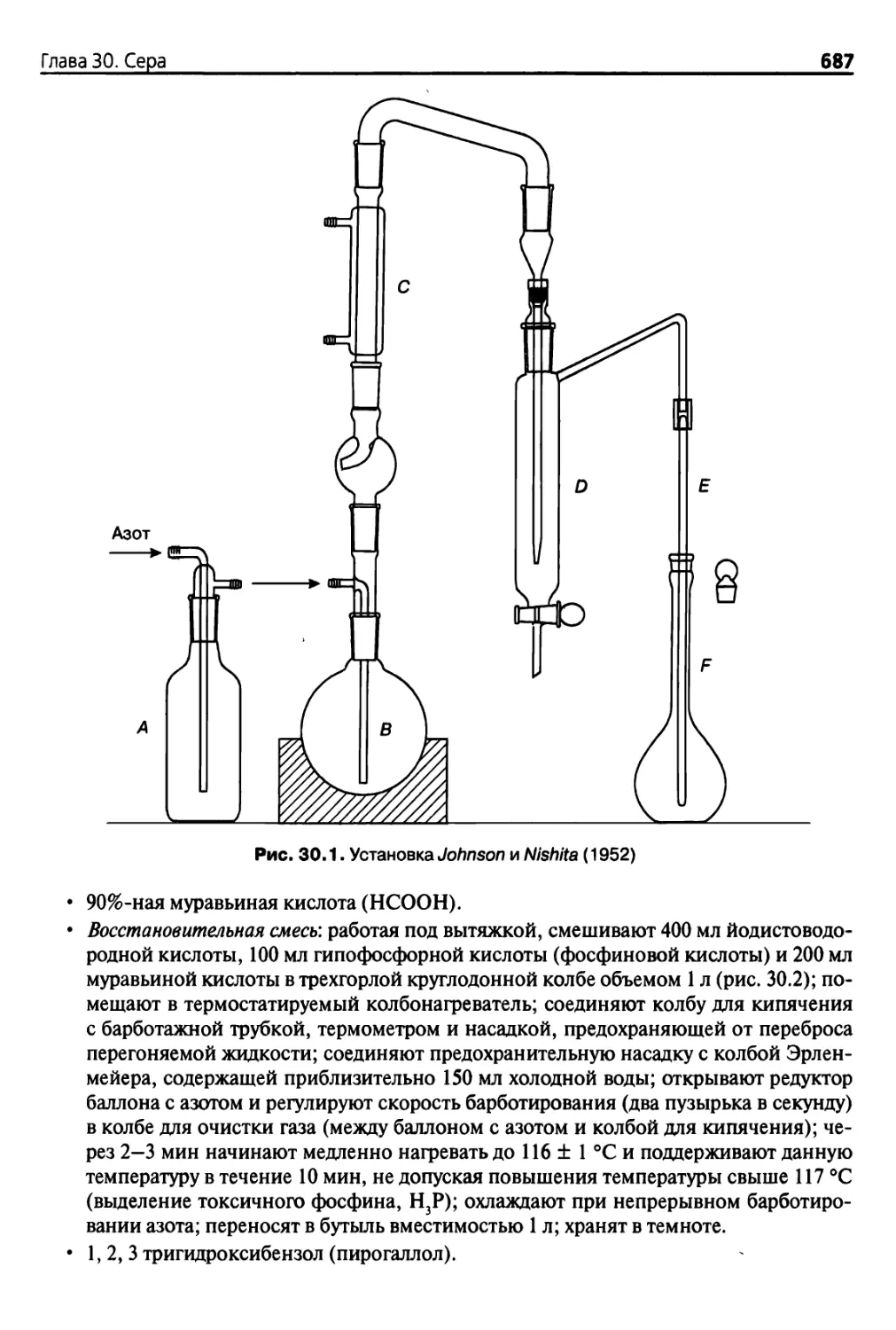
31.3.3. Нейтронно-активационный анализ 780
Используемая литература 786
Предметный указатель 790
Предисловие к русскому изданию
Вы держите в руках перевод уникального справочного руководства, которое уже выдер¬
жало издание как на французском, так и на английском языках. Преимущество этой
книги в том, что в ней собраны подробные сведения как о классических аналитических
методах анализа почвы на содержание различных неорганических и органических ком¬
понентов, так и о современных инструментальных методах, адаптированных для из¬
учения самых разнообразных физико-химических свойств почв. Авторы руководства
делятся с читателем своим огромным практическим опытом применения самых разно¬
образных методов (начиная от весового анализа и заканчивая ядерно-химическими ме¬
тодами) при исследовании реальных образцов почв различных регионов. В справочном
пособии подробно описаны не только многочисленные методы анализа, но и методы
отбора, консервации и хранения проб почв. Материал хорошо структурирован, каждый
метод обязательно предваряется описанием его основ, включая анализ преимуществ и
недостатков различных методик и областей их применения, подробные прописи мето¬
дик с указанием требуемого оборудования и реагентов, примеры обработки и интер¬
претации результатов, таблицы, формулы, примеры спектров, диаграмм и пр. И если
отдельные материалы изложены несколько конспективно, подчас даже сжато, то бла¬
годаря тому, что все материалы пособия сопровождаются подробной библиографией,
охватывающей период с 1789 по 2005 год, читатель при необходимости может уточнить
особенности применения любой из описанных методик в первоисточнике. Обширная и
подробная библиография — еще одна из сильных сторон данного справочника. Слож¬
но найти издания на русском языке, аналогичные по широте и полноте охватываемого
материала.
При переводе книги мы старались максимально сохранить исходный стиль изло¬
жения материала и применяемую авторами терминологию, чтобы донести до читате¬
ля авторский взгляд на рассматриваемые вопросы. При этом мы обязаны указать, что
некоторые из применяемых в оригинальном тексте терминов отличаются от принятых
в русскоязычной научной литературе. Так в частности, для обозначения спектроскопи¬
ческих методов исследования авторами преимущественно используются термин «спек¬
трометрия» и аналогичные. В отечественной литературе термины с окончанием «-ме-
трия» включают весьма обширную область наук как об измерении, так и обо всем, что с
ним связано (законы, техника, методы, приложения и т. д.). В контексте данного посо¬
бия в большинстве случаев имеется ввиду, конечно же, спектроскопическое примене¬
ние методов анализа. Другой пример: рассматривая процессы обмена катионов, авторы
широко применяют непринятый в отечественный литературе термин «обменный ком¬
плекс». Аналогом данного термина считается «поглощающий комплекс почвы», но так
как в оригинале данный термин употребляется отдельно, и только в контексте поглоще¬
ния ионов, мы посчитали необходимым сохранить авторскую терминологию, призывая
читателя быть внимательным при ее употреблении. Аналогично, у авторов градуировоч¬
ные растворы и процедуры называются калибровочными.
Кроме того, мы столкнулись с проблемой перевода названий некоторых типов почв.
Действительно, помимо международной, существует несколько национальных почвен¬
ных классификаций: американская, канадская, китайская, французская и т. п. Даже
в нашей стране на сегодняшний момент активно применяются классификации, относя¬
щиеся к трем различным школам. Очевидно, что национальные классификации, прежде
Предисловие к русскому изданию
19
всего, направлены на систематизацию почв, встречающихся в соответствующих, порою
уникальных, местностях. Поэтому при попытке адаптировать терминологию названий
почв в некоторых случаях просто невозможно подобрать не только прямых, но и кос¬
венных аналогов. А внесение искажения при такой «адаптации», в отличие от прямого
перевода, может привести к ошибкам. Поэтому при подготовке перевода мы стреми¬
лись, прежде всего, сохранить аутентичность исходному тексту, исходя из того, что это
справочник прежде всего по методам анализа почвы.
С учетом вышесказанного, мы надеемся, что это объемное издание станет настольной
книгой как для опытных специалистов, ученых, инженеров, так и для начинающих ис¬
следователей в областях почвоведения, агрономии, экологии, геологии, аналитической
химии, градостроительства и других смежных дисциплинах, связанных с почвой. Также
этот справочник будет полезен для преподавателей, студентов, магистрантов и аспиран¬
тов соответствующих специальностей в качестве практического пособия по подготовке
и анализу проб почвы.
Панкратов Денис Александрович,
кандидат химических наук, доцент
Предисловие
В представленном новом справочном руководстве Марка Пансю (Marc Pansu) и Жака
Готеру (Jacques Gautheyrou) приводится краткий обзор аналитических методов, приме¬
няемых при физико-химическом исследовании почв. Они представлены в соответствии
с требованиями стандартов для методов анализа и контроля качества почв. Основное
внимание уделено методам минералогического, органического и неорганического ана¬
лизов, при этом также описываются методики обработки образцов, предваряющие не¬
которые исследования. Настоящее руководство поможет широкому кругу читателей
выбрать наиболее подходящий метод анализа в зависимости от типа исследуемого мате¬
риала и специфических задач, с которыми им приходится сталкиваться. Представленная
работа является результатом практического опыта, накопленного авторами в лаборато¬
риях Института исследований в целях развития (Institute of Research for Development (IRD))
во Франции и в тропических странах, а также включает в себя обширный обзор литера¬
туры. Ссылки на использованную литературу, приведенные в конце каждой главы, пере¬
числяют данные первоисточников, начиная от пионерских работ вплоть до современ¬
ных, например таких как предположения о структурных моделях гуминовых молекул,
и сами по себе представляют собой ценный источник информации.
Почвоведы IRD обобщили данные о почвах средиземноморского региона, а также
о тропических почвах из Западной и Северной Африки, Мадагаскара, Латинской Аме¬
рики и Юго-Восточной Азии. Почвенные материалы в этих регионах достаточно часто
отличаются от почв регионов с умеренным климатом. Поскольку при анализе прояви¬
лись новые проблемы, возникла необходимость в разработке убедительных и точных
физико-химических методов. Ученые из IRD объединили усилия с физиками, химиками
и биологами, чтобы те внесли вклад знаний от своих дисциплин, тем самым значитель¬
но расширяя их сферу применения. Настоящая книга является плодом этих экспери¬
ментов применительно к сложным системам, включающим почвы и окружающую среду.
Методологический материал представлен достаточно широко. В каждой главе описа¬
ны как простые методы анализа, так и методы, которые требуют использования дорого¬
стоящего оборудования и специальных навыков. Это обусловлено тем, что справочник
предназначен как для специалистов, работающих в полевых условиях, так и исследова¬
телей, ведущих фундаментальные и прикладные изыскания. В книге приводятся прин¬
ципы, физические и химические основы каждого метода, соответствующих аналитиче¬
ских процедур, а также недостатки и области использования каждого из них. Описания
основаны на практическом опыте, легко понятны и реализуемы. В итоговых таблицах
представлен краткий обзор данных. Сущность сложных методов анализа подробно опи¬
сана в разделе под названием «Основные положения» и включает конкретные примеры
применения методов: спектральные методы анализа (спектрометрия ближней и дальней
ИК-области, УФ-видимого диапазона, ‘Н-ЯМР, 13С-ЯМР, ЭСР, ИСП-АЭС, ИСП-МС,
рентгенофлуоресцентный анализ, ЭД-РСМА и ВД-РСМА, нейтронно-активационный
анализ), дифракционные методы (РД, электронная микродифракция), термические ме¬
тоды анализа (ДТА, ДТГ, ТГА), хроматографические методы (ГХ, ВЭЖХ, ионная хрома¬
тография, эксклюзионная хроматография), электрофорез, ионообменные, электрохи¬
мические, биологические методы анализа, различные методы физического разделения,
селективного растворения, а также методы получения изображений.
Настоящая книга будет полезна не только ученым, инженерам, техническим специали¬
стам и студентам в области почвоведения, но также агрономам и экологам, и другим спе¬
циалистам в сопряженных дисциплинах, таких как аналитическая и физическая химия,
геология, климатология, гражданское строительство и промышленность, связанных
с почвой. Это основное справочное руководство, предназначенное для помощи в на¬
учном анализе окружающей среды. Описанные в нем методики анализа применимы к
широкому диапазону биоклиматических зон: умеренных, засушливых, субтропических
и тропических. Так же как и более ранние книги, созданные этими авторами (Pansu,
Gautheyrou и Loyer, 1998, Masson, Paris, Milan, Barcelona; Pansu, Gautheyrou и Loyer, 2001,
Balkema, Lisse, Abington, Exton, Tokyo), настоящая книга представляет собой справочное
пособие для лабораторий. Мы уверены, что оригинальность и доступность изложения
обеспечат успех настоящей книги.
Ален Авентьюрир (Alain Aventurier),
руководитель аналитической лаборатории
(Director of Analytical Laboratories of CIRAD1)
Кристиан Феллер (Christian Feller),
руководитель научно-исследовательских работ IRD
(Director of Research at I RIP)
Пьер Боттнер (Pierre Bottner),
руководитель научно-исследовательских работ CNRS
(Director of Research at CNRS?)
1 CIRAD — Центр международного сотрудничества по агрономическим исследованиям в це¬
лях развития (Centre International pour la Recherche Agronomique et le Developpement) (Франция).
2IRD — Французский научно-исследовательский институт для целей развития (Institut de Re-
cherche pour le Developpement) (экс-ORSTOM, Франция).
3CNRS — Национальный центр научных исследований (Centre National de la Recherche Scienti-
fique) (Франция).
Часть 1
МИНЕРАЛОГИЧЕСКИМ АНАЛИЗ ПОЧВ
Глава 1. Определение влагосодержания почв
и потерь при прокаливании
1.1. Введение
В первом приближении можно сказать, что почва состоит из твердой фазы, содержащей
минеральные и органические вещества, а также жидкой и газовой фаз. Физические и хи¬
мические свойства твердых компонентов почв зависят как от содержания влаги в почве,
так и от степени устойчивости почвы к потере влаги.
При проведении любых аналитических исследований почв для получения стабильных
и воспроизводимых результатов аналитик должен располагать данными относительно
содержания твердой фракции в составе почвы. Жидкую фазу необходимо отделить, но
так, чтобы не вызывать существенного изменения твердой матрицы (при удалении мо¬
лекул воды, встроенных в кристаллическую решетку).
Существуют разнообразные определения терминов «влажность» и «сухая почва».
Вода, которая может быть выделена умеренным нагреванием образцов или экстраги¬
рована растворителями, представляет собой лишь одну часть от общей влажности по¬
чвы и носит название «гигроскопическая влага». Она включает (1) адсорбированную
воду, удерживаемую на поверхности почвы за счет сил физической абсорбции (взаимо¬
действия Ван-дер- Ваальса) или хемисорбции, (2) капиллярную и внутрипоровую воду,
а также (3) гигрометрическую воду газовой фракции почвы (отношение эффективного
давления водных паров к максимальному давлению). Разграничения между этими типа¬
ми воды не являются очень строгими.
«Воздушно-сухая» почва, которая используется в качестве эталона при подготовке
образцов почв к проведению лабораторных исследований, содержит различное количе¬
ство влаги, которое в определенной степени зависит как от природы вторичных мине¬
ральных веществ, так и от параметров окружающей среды (температуры, относительной
влажности воздуха). Влажность некоторых андисолей и гистосолей, подвергнутых вы¬
сушиванию на воздухе в течение 6 мес., может на 60% превышать влажность образцов
почв, высушенных при 105 °С. Это может привести к недопустимым погрешностям ре¬
зультатов анализа в том случае, если полученные значения не сопоставлять с более реа¬
листичными справочными данными о содержании влаги в почвах.1 * Анализ засоленных
почв также затруднен, поскольку в их составе содержатся гигроскопичные соли.
Для определения влагосодержания можно применять методы, основанные на ис¬
пользовании сил удерживания влаги; при этом могут быть получены воспроизводимые
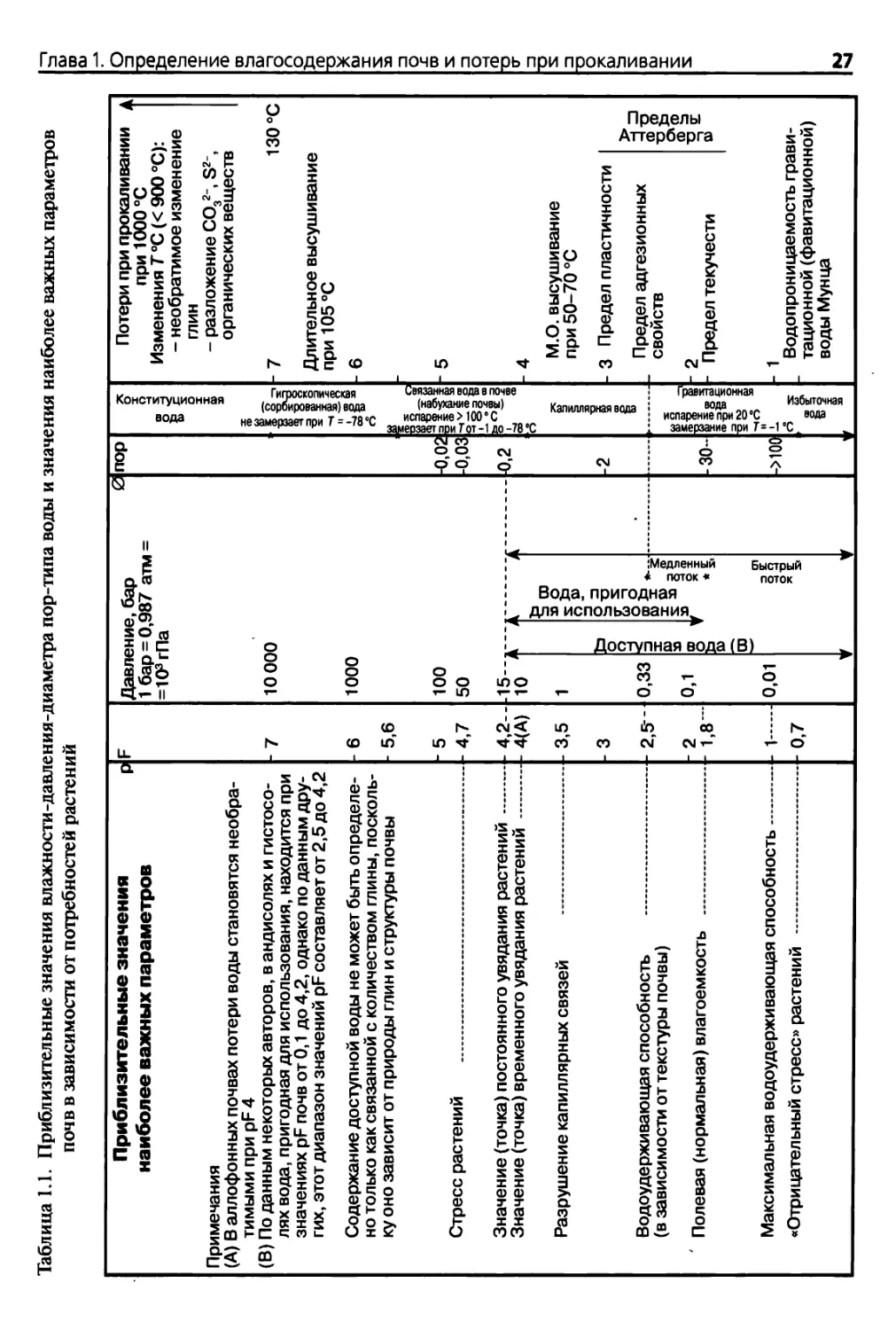
и представительные результаты испытаний (табл. 1.1). Параметры сил удерживания вла¬
ги можно представить в форме капиллярного потенциала (pF), при этом десятичный
логарифм давления, выраженный в миллибарах (табл. 1.1), отражает влагосодержание
образца. Следует отметить, что вследствие действия сил Ван-дер-Ваальса для почвенных
вод могут наблюдаться различия в состоянии, но форма воды остается неизменной, будь
1 Следует отметить, что для почв такого типа погрешности измерений возрастают и вслед¬
ствие увеличения массы образцов (поскольку кажущаяся плотность почвы может достигать
значения 0,3 г/см3), поэтому, по всей вероятности, полученные результаты не могут быть ис¬
пользованы при проведении агрономических исследований.
26
Часть 1. Минералогический анализ почв
это вода, которая испаряется при 20 °С, или вода, не замерзающая при -78 °С. Обычно
влажность определяют в определенных точках (табл. 1.1), например:
• Водоудерживающая способность почвы — влагосодержание почвы, при котором
наибольшее значение в полном потенциале имеют силы давления, а не гравитаци¬
онные силы; она зависит от текстуры и природы минералов почвы и количествен¬
но приближена к понятию полевой влагоемкости почвы, которая, после соответству¬
ющего дренирования, аналогична отсутствию гравитационной влаги.
• Точка капиллярной хрупкости, т. е. такая влажность, при которой непрерывная вод¬
ная пленка становится мономолекулярной и разрывается.
• Значения (точки) временного и постоянного увядания растений, при которых давле¬
ние пленочной (пелликулярной) воды, удерживаемой прочными связями, урав¬
новешивается осмотическим давлением; в этом случае большая часть растений
(за исключением некоторых галофильных растений) не в состоянии поглощать
воду, которая еще может содержаться в почве.
• Гигроскопическая вода, которая не может быть легко выделена из почвы в есте¬
ственных условиях, поскольку это требует значительных энергетических затрат;
гигроскопическая вода испаряется при температурах, превышающих 100 °С, и не
замерзает при температуре -78 °С.
• Конституционная вода, а также вода, гидратированная минеральными веществами,
которые могут быть удалены только под действием очень высокого давления или
высокой температуры, что сопровождается необратимым изменением или разру¬
шением кристаллической решетки.
Содержание рассматриваемых типов воды в составе почв может быть определено ис¬
пользованием различных методов. Сведения о содержании разных типов воды в составе
почвы позволяют получить информацию о динамических характеристиках воды и оце¬
нить особенности почв, обусловленные их механическими свойствами. Такие данные
могут использоваться в агрономических целях и сельскохозяйственном машинострое¬
нии, например:
• для оценки природных ресурсов, пригодных к использованию (ПР), легкодоступ¬
ных природных ресурсов, пригодных к использованию (ЛПР), или природных ре¬
сурсов, которые могут быть легко извлечены из системы почва-вода-растение.
• пороговых значений пластичности, адгезионных свойств, текучести (пределов
Аттерберга и т. д.).
Это краткое изложение дает представление о сложности понятия влажности почвы
и трудностях, стоящих перед аналитиком при поиске научно обоснованного исходного
состояния сухой почвы, в которой соотношение твердой, жидкой и газовой фаз является
неизменным.
1.2. Определение влагосодержания при 105 °С (Н20~)
1.2.1. Основные положения
Принято считать, что термин «влажность» имеет одно, четко определенное значение. Ее
измерение основано на гравиметрическом определении после высушивания при мак¬
симальной температуре в 105 °С. Такую температуру поддерживают в течение заданно¬
го времени, достаточного для того, чтобы удалить «свободные» (несвязанные) формы
воды, но в тоже время не вызывать существенные потери органического вещества и не¬
стабильных солей в газообразном виде. Если строго соблюдаются методики испытаний,
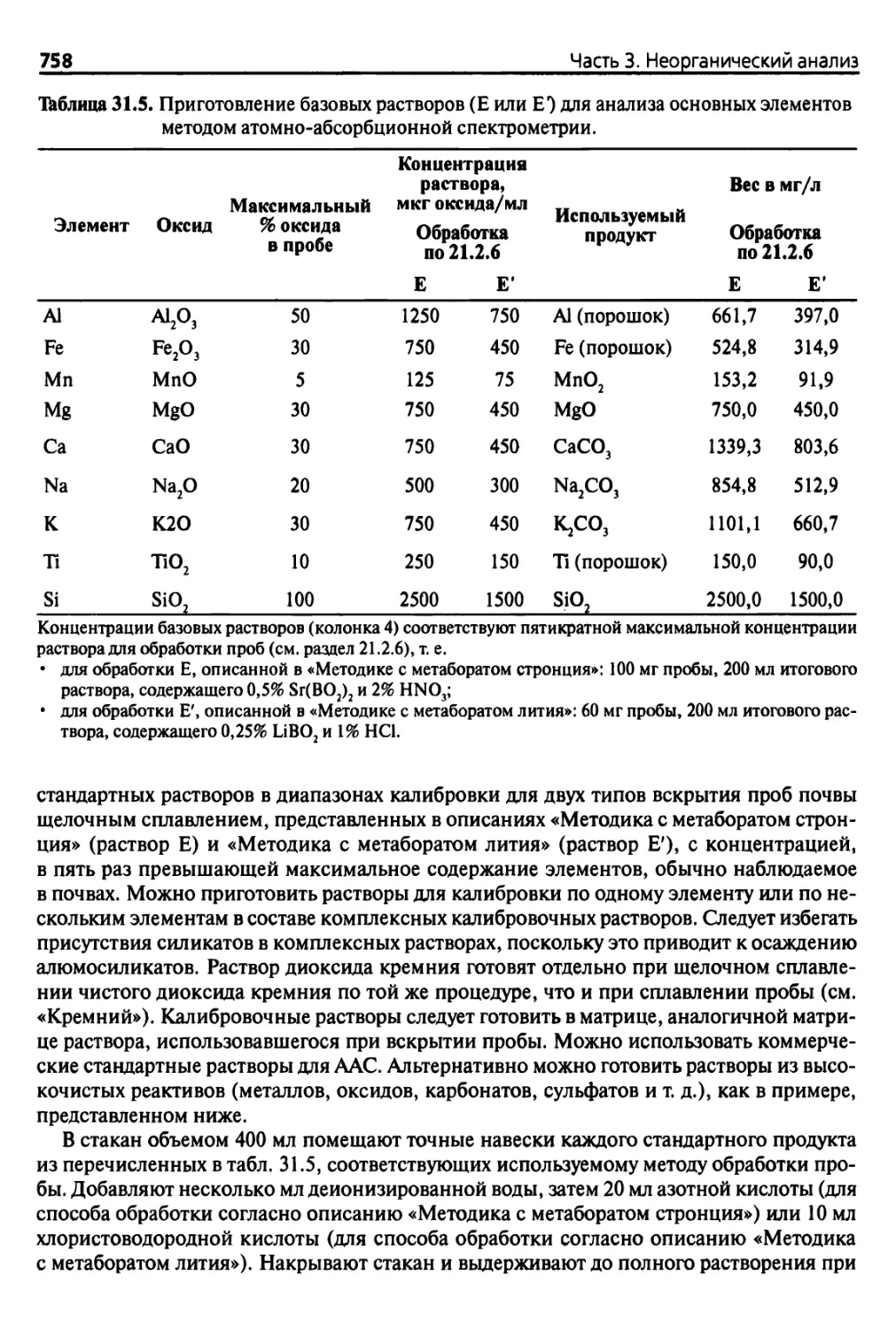
Таблица 1.1. Приблизительные значения влажности-давления-диаметра пор-типа воды и значения наиболее важных параметров
почв в зависимости от потребностей растений
Приблизительные значения
наиболее важных параметров
Давление, бар
1 бар = 0,987 атм =
=103 гПа
Примечания
(A) В аллофонных почвах потери воды становятся необра¬
тимыми при pF 4
(B) По данным некоторых авторов, в андисолях и гистосо¬
лях вода, пригодная для использования, находится при
значениях pF почв от 0,1 до 4,2, однако по данным дру¬
гих, этот диапазон значений pF составляет от 2,5 до 4,2
-7
10000
Содержание доступной воды не может быть определе- - 6
но только как связанной с количеством глины, посколь¬
ку оно зависит от природы глин и структуры почвы - 5,6
1000
Стресс растений
-5 100
- 4,7 50
Значение (точка) постоянного увядания растений
Значение (точка) временного увядания растений
-4,2---15
4(A) 10
Разрушение капиллярных связей
Водоудерживающая способность
(в зависимости от текстуры почвы)
Полевая (нормальная) влагоемкость
3,5
3
2,5-'
2
1,8 -
0,33
0,1
о
=3
X
ш
»
го
5 Ш
8»
го
Максимальная водоудерживающая способность 1—
«Отрицательный стресс» растений
0,7
- 0,01
з Е
33
0-0
* Е
V V
0
пор
о
-2
-30-
з|1
S о
40
->100
Потери при прокаливании А
при 1000 °С
Изменения Т°С (< 900 °С):
- необратимое изменение
глин
- разложение С032~, S2-,
органических веществ
-7 130 °С
Длительное высушивание
при 105°С
-5
-4
М.О. высушивание
при 50-70 °С
- 3 Предел пластичности
Предел адгезионных
свойств
- 2
- Предел текучести
Р
II
0)
Водопроницаемость грави¬
тационной (фавитационной)
воды Мунца
■О
0
2
ь
S
ф
си
1
S
X
41
Глава 1. Определение влагосодержания почв и потерь при
28
Часть 1. Минералогический анализ почв
то повторяемость и воспроизводимость анализа для большинства типов почв удовлет¬
ворительны.
1.2.2. Материалы
• Бюкс низкий (стаканчик для взвешивания) из боросиликатного стекла 50x30 мм*
с плоской притертой крышкой.
• Вакуумный эксикатор диаметром 200 мм, изготовленный из боросиликатного
стекла, с извлекаемой фарфоровой вставкой, заполненный безводным перхлора¬
том магния — Mg(C104)2.
• Сушильный шкаф с термостатом, поддерживающим заданную температуру в пре¬
делах + (0,5—1) °С, оснащенный вентилятором, вращающимся с постоянной ско¬
ростью, для циркуляции воздуха и отводом воздуха в вентиляционное отверстие
в верхней части печи.
• Аналитические весы: точность 0,1 мг, предел взвешивания 100 г.
1.2.3. Образцы
При проведении анализа важно определять влагосодержание для образцов из одной се¬
рии, отобранных одновременно (мелкозем с размером частиц 2 мм или грунт). Следует
отметить, что содержание влаги предварительно подготовленных образцов почв может
изменяться в процессе хранения (колебание влажности и температуры воздуха, окисле¬
ние органического вещества, выделение или связывание летучих веществ и т. д.).
При анализе некоторых типов почв и проведении некоторых исследований настоя¬
щий метод может быть отнесен к группе «разрушающих» методов контроля, поскольку
их физические и химические свойства могут измениться. Образцы, высушенные при
105 °С, не должны использоваться для проведения других исследований.
1.2.4. Методика
• Предварительно высушенные при 105 °С в течение 2 ч бюксы охлаждают в эксика¬
торе, взвешивают бюкс вместе с крышкой, размещенной снизу — т0.
• Приблизительно 5 г воздушно-сухой почвы (мелкозем, просеянный через сито
с диаметром отверстий 2 мм) помещают в бюкс и записывают новый вес — mv
• Емкости для взвешивания (бюксы) и плоские крышки (размещенные снизу) по¬
мещают в сушильный шкаф и выдерживают при 105 °С в течение 4 ч (вентиляци¬
онное отверстие сушильного шкафа должно быть открыто, при этом сушильный
шкаф не должен быть чрезмерно загружен).
• Высушенные образцы охлаждают в эксикаторе и взвешивают (все крышки бюксов,
помещенных в эксикатор, должны быть закрыты для предотвращения увеличения
влажности) — тг
• Открытые бюксы для взвешивания снова помещают в сушильный шкаф, выдер¬
живают в течение 1 ч при температуре 105 °С и взвешивают при тех же условиях;
вес не должен измениться; если не так, продолжают сушить бюксы с образцами до
постоянной массы.
Влагосодержание, % при 105 °С = ЮОх7”1 /”2.
Щ-т0
29
Глава 1. Определение влагосодержания почв и потерь при прокаливании
1.2.5. Примечания
Результаты также могут быть выражены в форме водоудерживающей способности по¬
чвы (ВУС) (характеристика почв):
ВУС =100xw' т\
mj-m0
Значение температуры высушивания (105 °С) до постоянной массы подобрано эмпи¬
рическим путем (рис. 1.1). При температуре 130 °С из образцов почв может быть выделе¬
на практически вся «поровая вода», однако это сопровождается ущербом для стабильно¬
сти органического вещества. Скорость высушивания должна зависеть от температуры,
поверхности диффузии, дисперсности вещества, воздухообмена, давления (вакуум) и др.
Время, ч
Рис. 1.1. Схематическое изображение теоретической кривой, отражающей зависимость выде¬
ления влаги при данной температуре от времени (180 °С.= прекращение потери Н20 аллофанами)
• Первые взвешивания образцов андисолей и гистосолей необходимо систематиче¬
ски проводить каждые 6 ч.
• При анализе засоленых почв, содержащих большие количества растворенных со¬
лей, образцы почв должны быть высушены, при этом растворимые соли переходят
в состав «сухой почвы» либо выводятся из состава почв при предварительной об¬
работке образцов водой.
1.3. Определение потерь при прокаливании при 1000 °С
(Н20+)
1.3.1. Введение
Как уже было отмечено выше, высушивание образцов при температуре 105 °С, выбран-
ной для определения влажности «сухих почв», позволяет установить только содержание
воды в абсолютно гипотетической форме, которую обычно описывают как Н20~.
30
Часть 1, Минералогический анализ почв
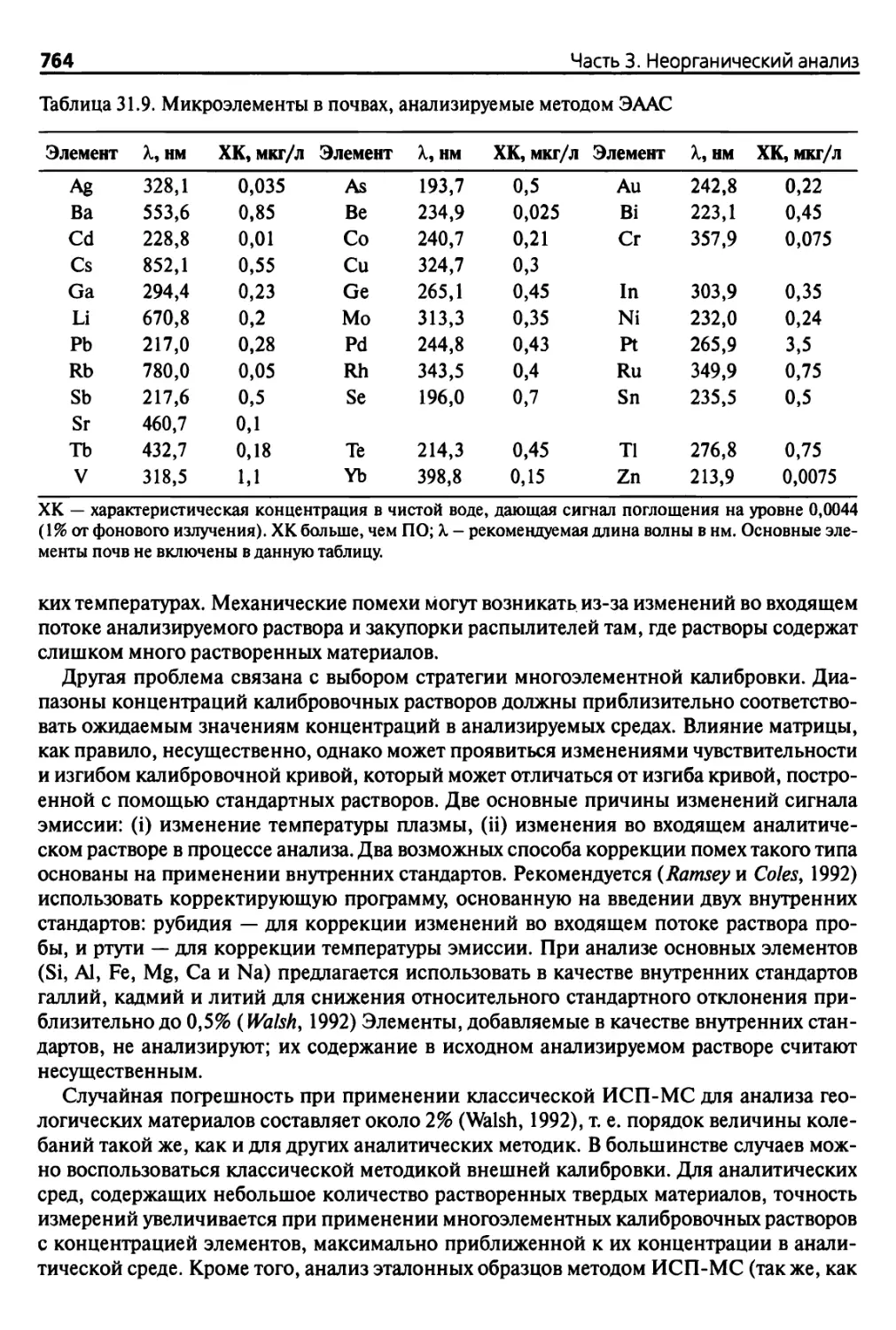
При нагревании образца в контролируемых условиях и непрерывном контроле из¬
менения массы можно получить кривые дегидратации, на которых перегибы характе¬
ризуют потери массы образцов при определенных критических температурах (ТГА)1.
Сравнивая полученную температурную кривую с температурной кривой термически
инертного вещества (рис. 1.2), можно выявить отличия в энергии исследуемого образ-,
ца и стандартного вещества, изменение этой разницы температур может быть изучено
(ДТА, ДСК).
• Если температура образца снижается по сравнению с температурой стандартного
образца, на температурной кривой возникает эндотермический пик, который со¬
ответствует потере Н20 (дегидратация), ОН" (дегидроксилирование), сублимации
или испарению, либо разложению каких-либо веществ и т. д.
• Если температура исследуемого образца возрастает по сравнению с температу¬
рой стандартного образца, на температурной кривой образуется экзотермический
пик, что свидетельствует об изменении кристаллической структуры, окислении
(Fe2+ -> Fe3+) и т. д.
Одновременный анализ выделяемых газов или паров и рентгеновский дифракцион¬
ный анализ (см. гл. 4) изменений структуры позволяют идентифицировать перегибы
кривых либо различные эндо- и экзотермические пики.
Рис. 1.2. Схематичный пример кривых термического анализа ТГА (сплошная линия) и ДТА
(прерывистая линия)
1 ТГА — термогравиметрический анализ; ДТА — дифференциальный термический анализ;
ДСК — дифференциальная сканирующая калориметрия (см. гл. 7).
31
Глава 1. Определение влагосодержания почв и потерь при прокаливании
Из очень упрощенной табл. 1.2 можно отметить, что наиболее распространенные
типы глин подвергаются полному дегидроксилированию при температуре 1000 °С, ок¬
сиды — при 400 или 500 °С, карбонаты, галогениды, сульфаты, сульфиды разрушают¬
ся либо подвергаются дегидрированию при температурах от 300 до 1000 °С, при этом
свободные или связанные органические вещества разрушаются при температурах от 300
до 500 °С. Таким образом, температура 1000 °С может использоваться в качестве ста¬
бильной стандартной температуры д ля определения потерь при прокаливании, при этом
термограммы представляют собой практически ровную линию с эффектами, которые
возникают только при температурах, превышающих 1500 или даже 2500 °С.
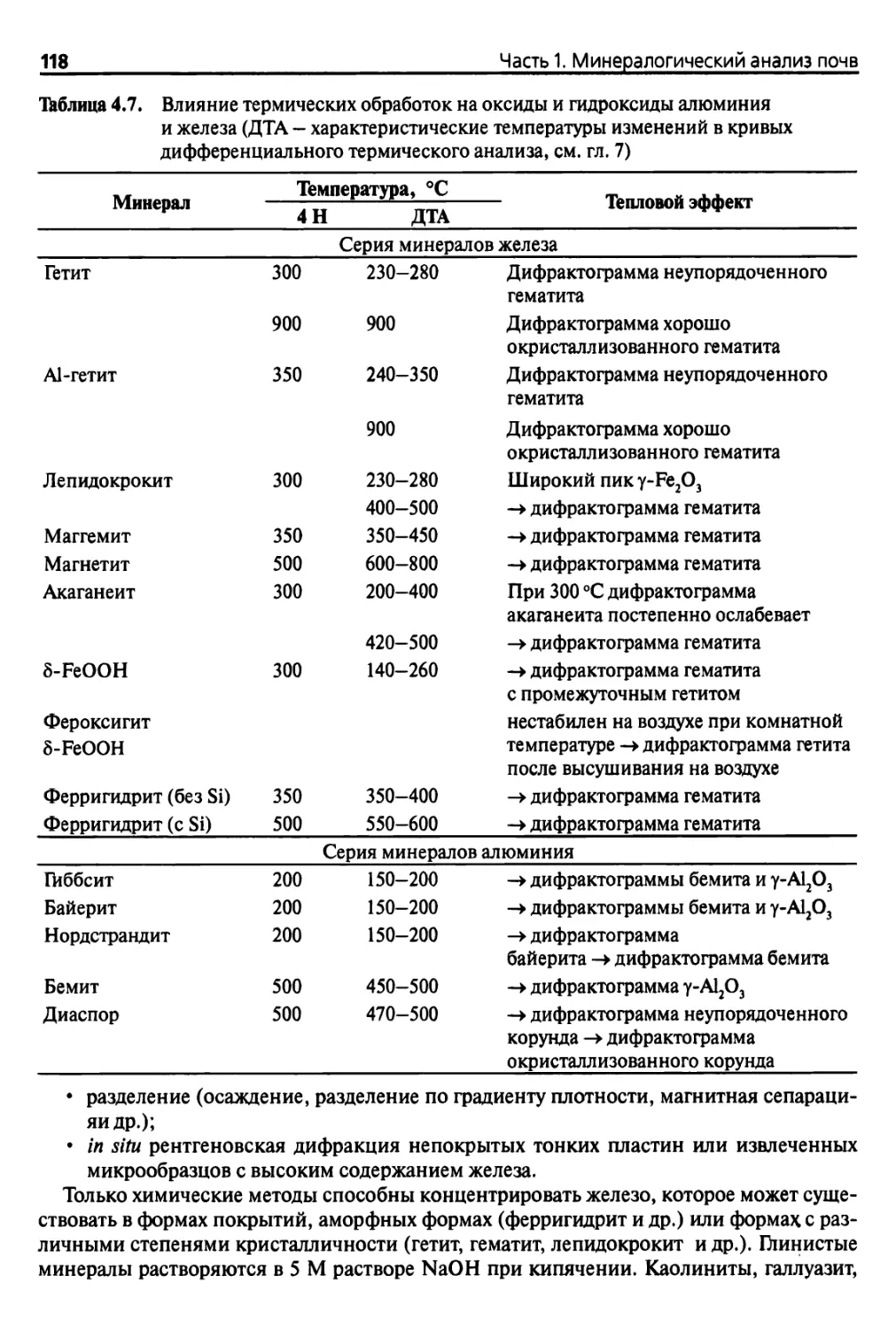
Таблица 1.2. Дегидратация и дегидроксилирование некоторых глин, оксидов и солей
в зависимости от температуры, в °С
Тйп
Наименование
Дегидратация11
*
5
I
1
Глины 1:1
Каолинит-галлоизит
350 •
1000
Глины 2:1
Смектит-монтмориллонит
370
1000
Глины 2:1
Иллит-слюдистые минералы
350-370
1000
Глины 2:1
Вермикулит
700
1000
Глины 2:1:1
Хлорит
600
800
Глины
Сепиолит-палыгорскит
300
800-900
с волокнистым
строением
аллофан
200
900-1000
Оксиды железа
Гематит a-Fe203
(нет эффектов)
1000
Гётит a-FeOOH
100
370
Магнетит Fe304
375
650
Оксиды А1
Гиббсит у-А1(ОН)3
100
350
Карбонаты кальция
Кальцит-арагонит СаС03
—
950-1000
Карбонаты магния
Магнезит MgC03
—
710
Г&логенсодержащие
соединения
Хлорид натрия NaCl
—
800 (плавится)
Сульфаты
Гипс CaS04 * 2Н20
—
300
Сульфиды
Пирит FeS2
—
615
Органические
соединения
Свободные или связанные
органические вещества
—
300-500
а Дегидратация — потеря молекул воды, адсобированных на внешней или внутренней по¬
верхности веществ, сопровождающаяся обратимым или необратимым изменением кристал¬
лической решетки глины в зависимости от типа глины, т. е. в зависимости от того, присут¬
ствует ли вода в виде мономолекулярной пленки на поверхностных атомах кислорода или
около способных к обмену катионов.
ь Дегидроксилирование (+ реакции декарбонизации и десульфуризации) — потеря воды, свя¬
занной кристаллической решеткой (ОН ), необратимые реакции или разрушение структуры
воды, находящейся в порах, кислорода, формирующего основу тетраэдров. -
32
Часть 1. Минералогический анализ почв
1.3.2. Основные положения
Образцы подвергаются постепенному нагреванию в окислительной среде до температу¬
ры 1000 °С и затем выдерживаются при этой температуре в течение 4 ч.
Определение потерь при прокаливании осуществляют гравиметрическим методом.
При этом определяют потери воды, связанной кристаллической решеткой, плюс не¬
много остаточной неструктурной адсорбированной воды, органического вещества, в не¬
которых случаях также летучих растворимых солей (F~, S2-) и карбонатов (СО2-, С02).
Создание окислительной среды при прокаливании имеет большое значение, поскольку
позволяет обеспечить полное сгорание органического вещества и способствует протека¬
нию частичного окисления восстановленных форм железа, что сопровождается увели¬
чением массы для почв, богатых соединениями Fe2+. Полный анализ обычно включает
последовательное определение содержания Н20 и Н20+ в составе одного и того же об¬
разца.
1.3.3. Оборудование
• Платиновый тигель или тигель из инконеля (Ni-Cr-Fe) с крышкой, диаметром
46 мм.
• Аналитические весы (аналогично разделу Н20~).
• Эксикатор (аналогично разделу Н20_).
• Муфельная электрическая печь (диапазон температур 100-1100 °С) с пропорци¬
ональным электронным контроллером, позволяющим осуществлять модуляцию
импульсов с отклонением относительно заданной температуры приблизительно
в 1 °С; встроенная система вентиляции для отведения дыма и паров.
• Термозащитные перчатки.
• Тигельные клещи с наконечником 300 мм.
1.3.4. Методика выполнения
• Взвешивают тигель, нагревают его до 1000 °С и охлаждают в эксикаторе с закрытой
крышкой — mQ.
• Помещают в охлажденный тигель 2-3 г воздушно-сухой почвы, измельченной до
размера частиц 0,1 мм — mv
• Сушат в сушильном шкафу при 105 °С в течение 4 ч.
• Охлаждают в эксикаторе и взвешивают — тг
• Накрывают тигель крышкой приблизительно на 2/3 и помещают его в электриче¬
скую муфельную печь.
• Задают скорость нагревания печи приблизительно на 6 °С в минуту с выдержива¬
нием в течение 20 мин при 300 °С, затем осуществляют быстрое повышение темпе¬
ратуры до 1000 °С и выдерживают в течение 4 ч (дверца печи после полного сгора¬
ния органического вещества обязательно должна быть закрыта).
• Охлаждают тигель в эксикаторе и взвешивают — ту
1.3.5. Обработка результатов
т\ — то = масса воздушно-сухой почвы
т1 - т2 = влагосодержание при 105 °С
т2 “ то = масса почвы, высушенной при 105 °С
Глава 1. Определение влагосодержания почв и потерь при прокаливании
33
т2 — т}= масса потерь при прокаливании
Н2СГ, %=100x^3—ЧЬ,
относительно воздушно-сухой почвы,
Н20\ % = 100х"*г
относительно почвы, высушенной при 105 °С.
1.3.6. Примечания
При известной влажности воздушно-сухой почвы можно рассчитать ее вес, необходи¬
мый для подготовки образца почвы, для определения влагосодержания при 105 °С, что
упростит процесс обработки результатов анализа.
Для отбора образца почвы, эквивалентного 1 г почвы, высушенной при 105 °С, не¬
обходимо отвесить:
100
100-wc’
где ж — влажность воздушно-сухой почвы, %.
Платиновые тигли очень дорогие, и при температуре 1000 °С возможна потеря их
массы, т. е. перед проведением каждого этапа анализа необходимо их взвешивать, в осо¬
бенности при работе в восстановительной среде.
Сжигание органических веществ при недостатке кислорода может привести к образо¬
ванию карбида платины, сульфидов платины, вызвать взаимодействие хлора и платины
ит.д.
Использованная литература
Campbell GS, Anderson RY (1998) Evaluation of simple transmission line oscillators for soil moisture
measurement. Comput. and Electron, Agric, 20, 31-44.
Chin Huat Lim, Jackson ML (1982) Dissolution for total elemental analysis. In Methods of Soil Analysis,
Part 2, Page A.L., Miller R.H., Kenny D.R. ed. Am. Soc. Agronomy, pp. 1-11.
Dixon JB (1977) Minerals in soil environments. Set Soc, Am,
Dubois J, Paindavoine JM (1982) Humidite dans les solides, liquides et gaz. Techniques de Pingenieur.,
(P3760).
Gardner WH (1986) Water content. In Methods of Soil Analysis, Part I, Klute ed. Am. Soc. Agronomy,
Soil Sci. Soc. Am., pp. 493-544.
Henin S (1977) Cours de physique du sol: Peau et le sol tome II, Editest, Paris: 1-164.
Lane PNJ, Mackenzie DH, Nadler AD (2002) Note of clarification about: Field and laboratoiy calibration
and test of TDR and capacitance techniques for indirect measurement of soil water content. Aust. J.
Soil Res,, 40, 555-1386.
Lane PNJ, Mackenzie DH (2001) Field and laboratory calibration and test of TDR and capacitance
techniques for indirect measurement of soil. Aust, J, Soil Res., 39,1371-1386.
NF ISO 11465 (ХЗЫ02) (1994) Determination de la teneur ponderale en matiere seche et en eau. In
Qualite des sols, AFNOR, 1996, 517-524.
34
Часть 1. Минералогический анализ почв
Rankin LK, Smajstrla AG (1997) Evaluation of the carbide method for soilmoisture measurement in
sandy soils. Soil and Crop Science Society of Florida, 56, pp. 136-139.
Skieracha W (2000) Accuracy of soil moisture measurement by TDR technique. Int. Agrophys., 14,417-
426.
Slaughter DC, Pelletier MG, Upadhyaya SK (2001) Sensing soil moisture using NIR spectroscopy. Appl.
Eng. Agric, 17, 241-247 .
Walker JP, Houser PR (2002) Evaluation of the Ohm Mapper instrument for soil moisture measurement.
Soil Sci. Soc. Am. J., 66, 728-734.
X31-505 (1992) Methode de determination du volume, apparent, et du contenu en eau des mottes. In
Qualite des sols, AFNOR, 1996, 373-384.
Yu C, Warrick AW, Conklin MH (1999) Derived functions of time domain reflectometry for soil moisture
measurement. Water Resour. Res., 35, 1789-1796.
Глава 2. Анализ гранулометрического состава
2.1. Введение
2.1.1. Г ранулометрический состав в почвоведении
Определение гранулометрического состава образцов почв принадлежит к числу наибо¬
лее важных видов анализа, применяемых в различных областях почвоведения, агроно¬
мии, седиментологии, а также в других областях, например, дорожной геотехнике.
Гранулометрический состав почвы оказывает чрезвычайно большое влияние на ее
физические и механические характеристики и на все свойства, определяемые содержа¬
нием и перемещением воды (плотность, пластичность, удельное сопротивление почвы,
растрескивание, влагоудерживающая способность, влагоемкость, выраженная в различ¬
ных потенциалах, водопроницаемость, капиллярное движение и т. д.).
Анализу гранулометрического состава образцов почв, который иногда называют
«анализом механического состава», уделяется повышенное внимание (Henin, 1976).
Почва представляет собой структурированную среду, содержащую набор минеральных
и органических частиц, с непрерывным распределением по размеру. Первая сложность
состоит в приведении таких различных частиц в соответствие со стандартной классифи¬
кацией, которая из-за этого в некоторой степени искусственна.
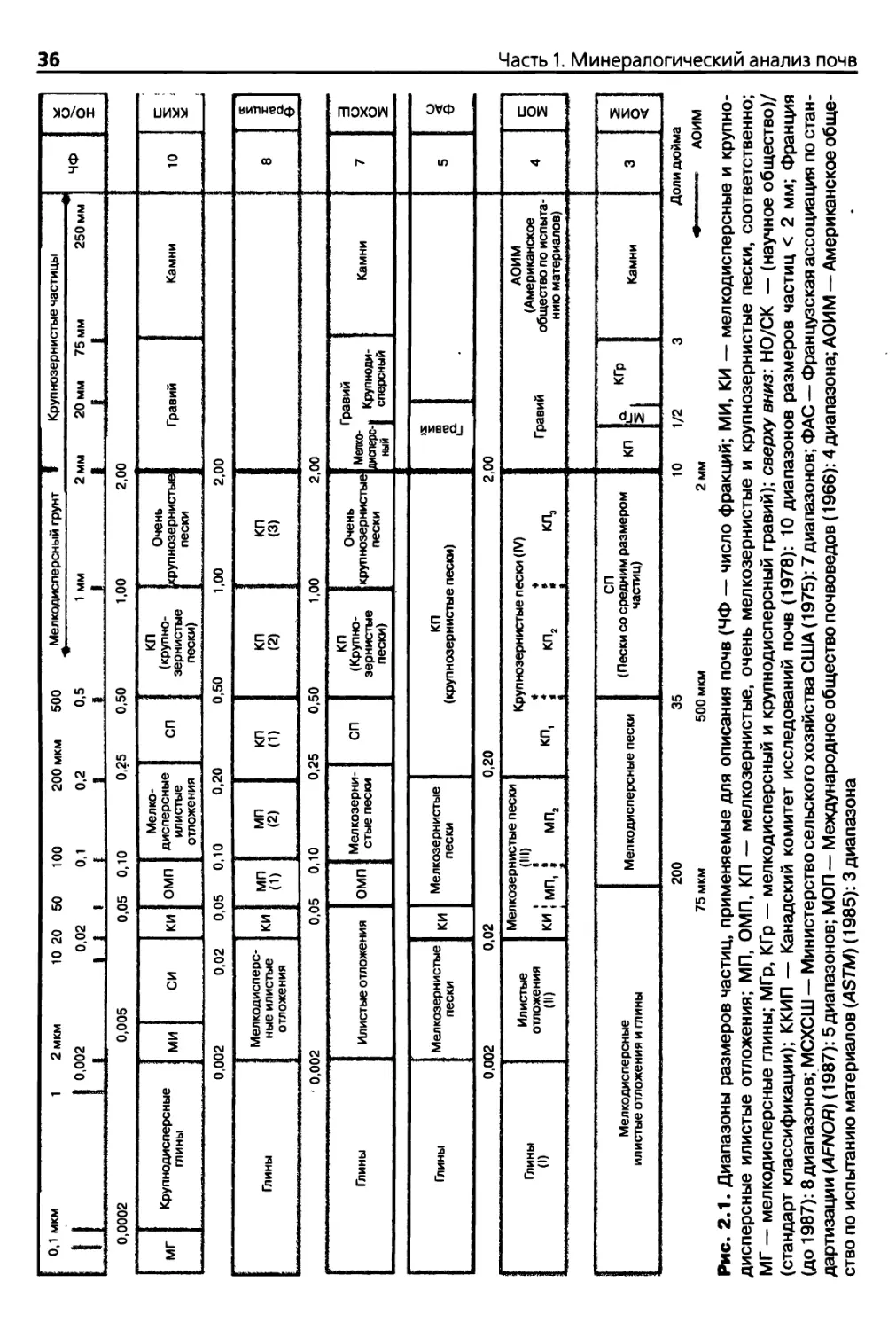
Один из вариантов классификационной шкалы был предложен Аттербергом
(Atterberg, 1912). В настоящее время эта шкала признана на различных национальных,
международных уровнях и включает две основные фракции: мелкодисперсный грунт1
(глина, илистые отложения и пески с частицами диаметром менее 2 мм) и крупнозер¬
нистые частицы (гравий, камни с частицами диаметром свыше 2 мм). При грануломе¬
трическом анализе мелкодисперсного грунта (рис. 2.1) образцы обычно разделяют на
три фракции по размеру частиц (глинистая фракция с размерами частиц менее 0,002 мм,
илистая фракция с размерами частиц от 0,002 до 0,02 мм и песчаная фракция с размера¬
ми частиц от 0,02 до 2 мм). В некоторых странах2 или в целях специфической характери¬
стики состава почв могут использоваться более подробные классификационные шкалы,
например, включающие пять фракций: мелкодисперсные глины, илистые отложения,
крупнозернистые илистые отложения или очень мелкозернистые пески, мелкозерни¬
стые пески или крупнозернистые пески (рис. 2.1).
Следует отметить, что используемая терминология не позволяет получить более по¬
дробную информацию о действительной природе фракций; так, фракция глин, опре¬
деляемая как частицы диаметром равным или меньше 0,002 мм, содержит не только
глины, соответствующие их минералогическому определению, но и полуторные окси¬
ды, очень мелкодисперсные илистые частицы, органическое вещество, карбонаты или
соединения, не обладающие коллоидными свойствами. Пески, которые, как правило,
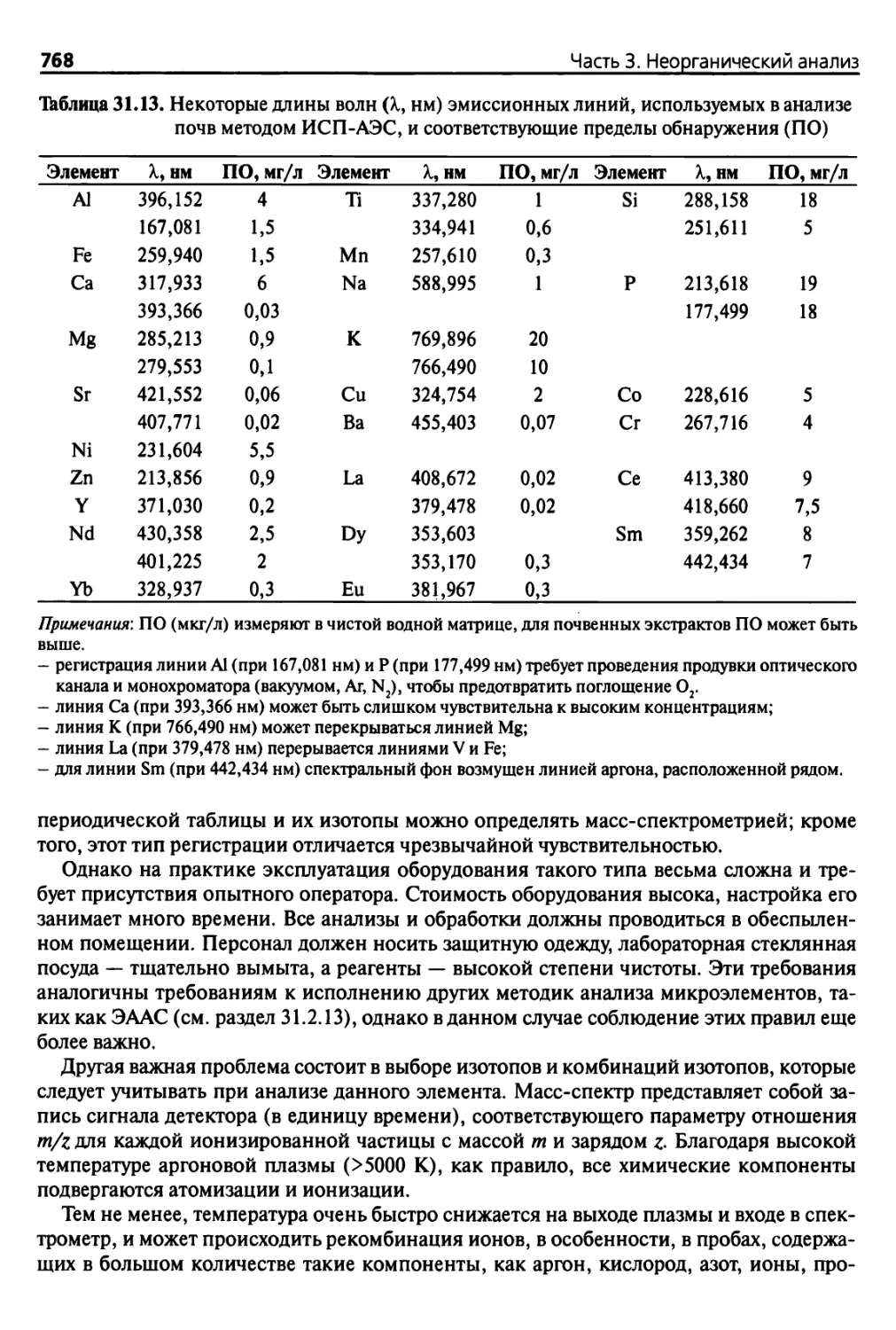
образуются в результате дробления материнской породы, могут включать псевдопески,
небольшие железистые конкреции, крошку известняка или сцементированные друзы,
1 В отечественной литературе к мелкозему относят частицы мельче 1 мм. — Примен. науч,
ред.
2 Рекомендуем ознакомиться с содержанием ГОСТ 25100-2011 1фунты. Классификация. —
Примеч. науч. ред,
°*1мкм 1 2 мкм 10 20 50 100 200 мкм 50
1 I 1 0,002 0,02 0.1 0,2 0,
—1.Л 1 1 -LJ ! _1_ I
0 ^Мелкодисперсный грунт f Крупнозернистые частицы
Чф
НО/СК
,5 1 мм 2 мм 20 мм 75 мм 250 мм
! 1 1 1 !
о,<
)002
0,0
05 0,05 0,10 0.25 0,50 1,00 2,0
>0
МГ
Крупнодисперсные
глины
МИ
си
КИ
ОМП
Мелко¬
дисперсные
илистые
отложения
СП
КП
(крупно¬
зернистые
пески)
Очень
крупнозернистые
пески
Гравий
Камни
10
ККИП
0,0
02 0,02
о,<
)5 0,10 0,20 0
|,50 1,00 2,00
Глины
Мелкодисперс¬
ные илистые
отложения
КИ
МП
(1)
МП
(2)
КП
(D
КП
(2)
КП
(3)
8
Франция
' о.ос
>2 0,05 0,10 0,25 0,50 1,00 2.
00
Глины
Илистые отложения
ОМП
Мелкозерни¬
стые пески
СП
КП
(Крупно¬
зернистые
пески)
Очень
крупнозернистые
пески
Гравий
Мелко- „
оисперс-| Крупноди-
1 слерсный
Камни
7
МСХСШ
Глины
Мелкозернистые
пески
КИ
Мелкозернистые
лески
КП
(крупнозернистые пески)
Гравий
1
5
1
0,002 0,02
0,20 2,00
Глины
(О
Илистые
отложения
(II)
Мелкозернистые пески
(III)
1 в
ки; мп, Л мп.
Крупнозернистые пески (IV)
кп, ; кп2 ; кл,
L. 1
АОИМ
(Американское
Гравий общество по испыта¬
нию материалов)
4
Г
! МОП
1
Мелкодисперсные
илистые отложения и глины
Мелкодисперсные пески
СП
(Пески со средним размером
частиц)
КП
МГр
ж
Камни
3
АОИМ
200 35 10 1/2 3 Доли дюйма
75 мкм
500 мкм
2
АОИМ
Рис. 2.1. Диапазоны размеров частиц, применяемые для описания почв (ЧФ — число фракций; МИ, КИ — мелкодисперсные и крупно¬
дисперсные илистые отложения; МП, ОМП, КП — мелкозернистые, очень мелкозернистые и крупнозернистые пески, соответственно;
МГ — мелкодисперсные глины; МГр, КГр — мелкодисперсный и крупнодисперсный гравий); сверху вниз: НО/СК — (научное общество)/
(стандарт классификации); ККИП — Канадский комитет исследований почв (1978): 10 диапазонов размеров частиц < 2 мм; Франция
(до 1987): 8 диапазонов; МСХСШ — Министерство сельского хозяйства США (1975): 7диапазонов; ФАС — Французская ассоциация по стан¬
дартизации (AFNOR) (1987): 5диапазонов; МОП — Международное общество почвоведов (1966): 4диапазона; АОИМ — Американское обще¬
ство по испытанию материалов (ASTM) (1985): 3 диапазона
и«
о>
Часть 1. Минералогический анализ почв
37
Глава 2. Анализ гранулометрического состава
которые с трудом поддаются разделению. Наличие таких псевдопесков может привести
к необъективным результатам анализа гранулометрического состава.
Другая сложность возникает с фракционированием элементарных частиц при их от¬
делении от исходных скоплений. В этой области тоже разработаны аналитические стан¬
дарты, но следует отметить, что в некоторых случаях разрушение всех когезионных сил
недостаточно (например, в случае отвержденных цементированных почв), в то же время
в других — эти силы чрезмерны.
Наконец, следует подчеркнуть, что анализ гранулометрического состава позволяет
получить информацию о размере частиц, но не о форме частиц или их природе. При
необходимости такие данные могут быть получены с помощью дополнительных морфо¬
логических или минералогических анализов. Результатом анализа гранулометрического
состава почвы является распределение по фракциям, относительные доли которых мож¬
но представить в форме треугольной диаграммы, отображающей гранулометрический
состав образца, горизонта или почвы. В зависимости от научной школы используются
различные типы треугольных диаграмм, которые позволяют отобразить гранулометри¬
ческий состав почв: GEPPA (Groupe d’Etude des Problemes de Pedologie Appliquee — Иссле¬
довательская группа по проблемам прикладного почвоведения, AFES, Цжньон (Grignon),
Франция), включает 17 фракций гранулометрических элементов; USDA (United States
Department of Agriculture — Министерство сельского хозяйства США), включает 12 фрак¬
ций (Gras, 1988). Другие типы в большей либо в меньшей степени упрощены в зависи¬
мости от почвоведческих или агрономических целей исследования. На основании таких
данных делают различные выводы, как правило, с точки зрения почвообразования (на¬
пример, сравнение процентного содержания песка в вертикальном срезе для проверки
однородности анализируемого материала в данном почвенном профиле, расчет различ¬
ных показателей выщелачивания, перенос глин и т. д.). Некоторые интерпретации носят
более прикладной характер (например, определение связи гранулометрического состава
и водообеспеченности для первичного расчета интенсивности и частоты орошения почв
либо для выбора механизмов, необходимых для культивации почв).
2.1.2. Основные положения
Анализ гранулометрического состава является процессом, выполняемым в лаборатор¬
ных условиях, в ходе которого на начальном этапе разлагают материал до элементарных
частиц, что предполагает устранение действия цементирующих веществ для разрушения
агрегатов. Такая операция не должна быть чрезмерно интенсивной, иначе могут обра¬
зовываться частицы, которых в естественных условиях не существует. Таким образом,
процедура диспергирования частиц должна быть достаточно эффективной для разру¬
шения агрегатов на отдельные компоненты, но не слишком сильной, чтобы избежать
образования модифицированных частиц.
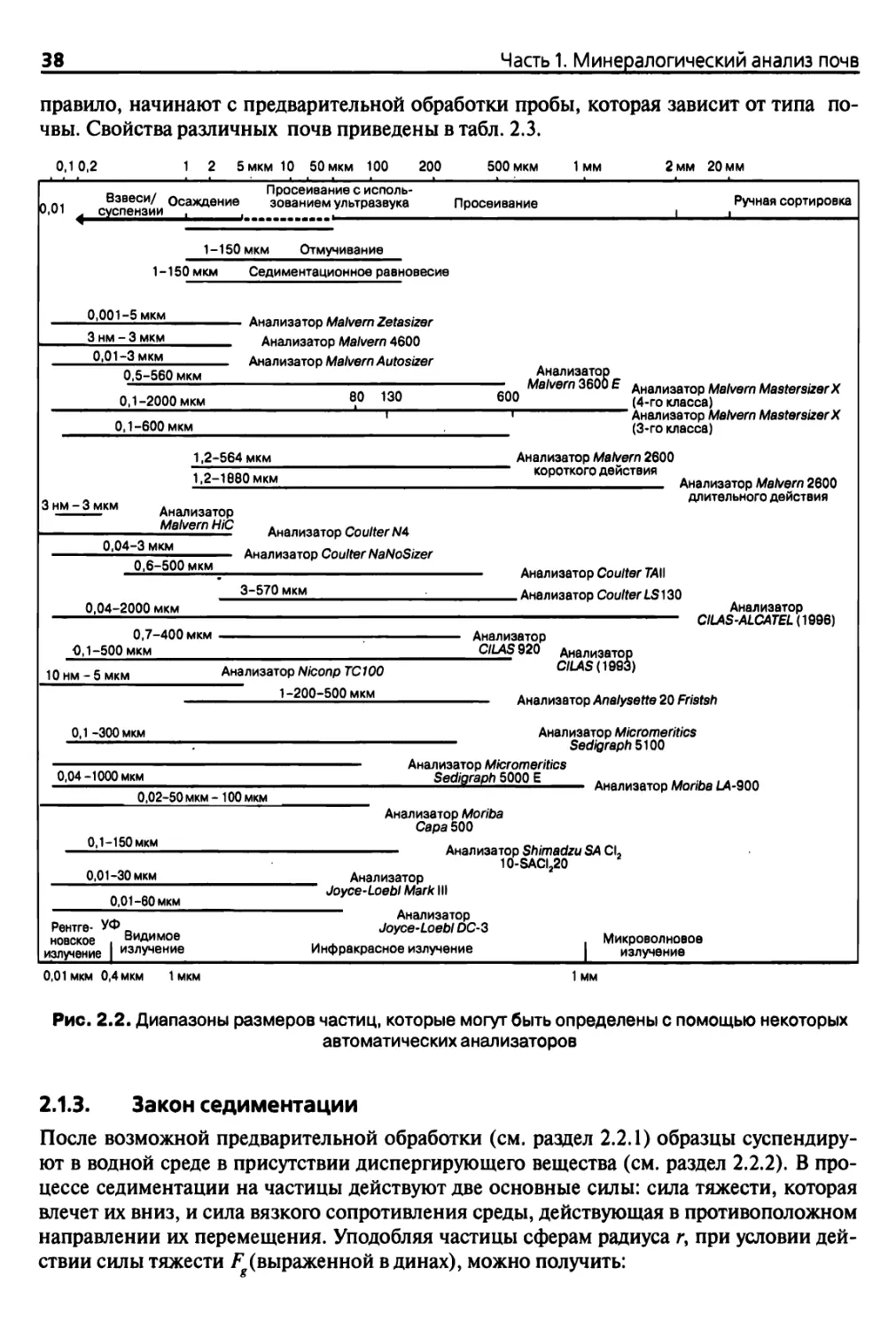
Измерения (табл. 2.1, рис. 2.2) позволяют связать размер частиц с физическими свой¬
ствами суспензии почвы после диспергирования (см. раздел 2.1.3). Однако результаты
таких измерений могут быть искажены вследствие присутствия в почве некоторых со¬
единений: органического вещества, растворимых солей, полуторных оксидов, карбона¬
тов или гипса. Наличие гипса вызывает наибольшие трудности, поскольку может при¬
вести к двум противоположно направленным эффектам (Vieillefon, 1979): флокуляция
вследствие наличия растворимых ионов кальция приводит к относительному снижению
содержания глины, а низкая плотность гипса по сравнению другими минералами —
к увеличению содержания глины. Таким образом, гранулометрический анализ, как
38
Часть 1. Минералогический анализ почв
правило, начинают с предварительной обработки пробы, которая зависит от типа по¬
чвы. Свойства различных почв приведены в табл. 2.3.
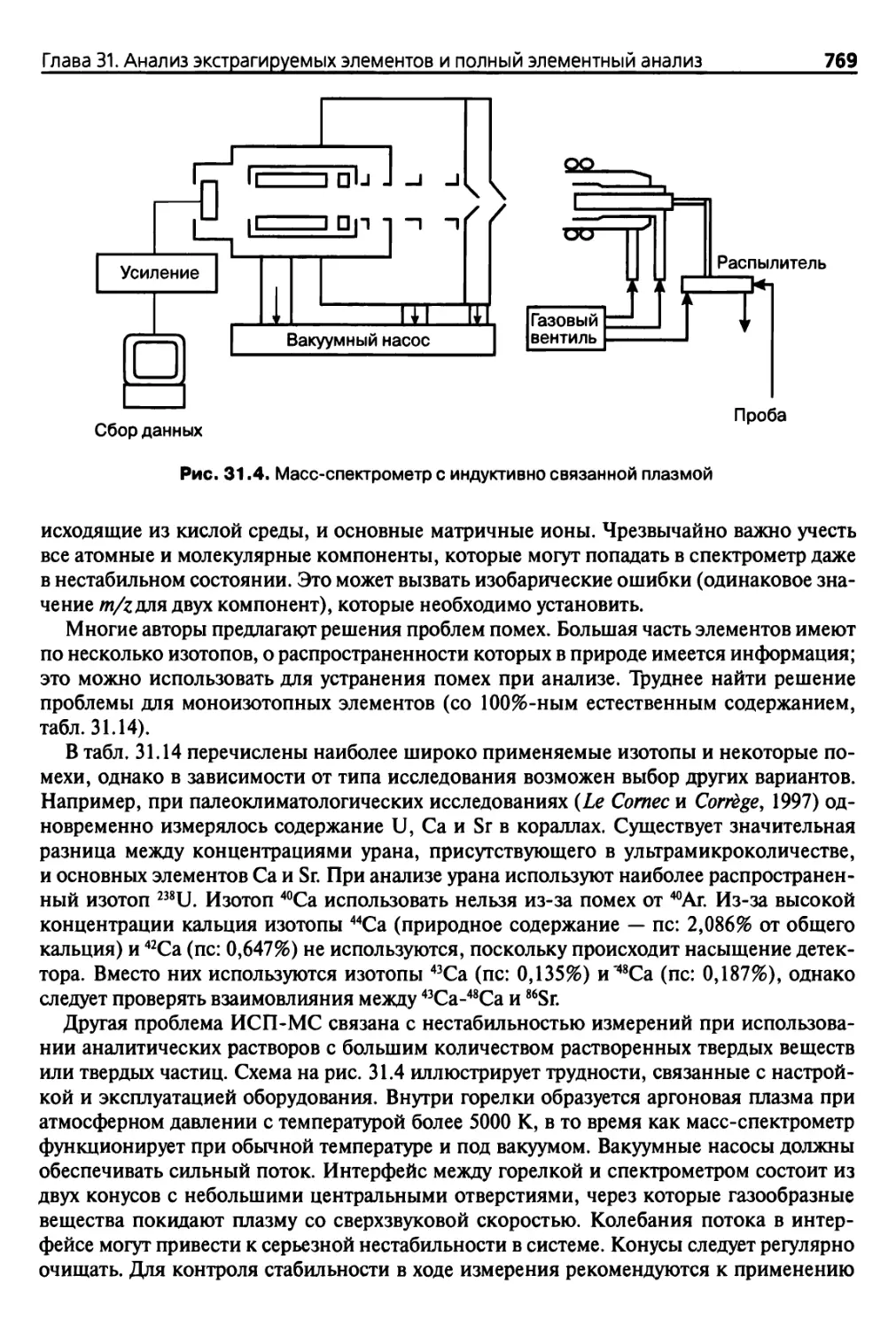
0,10,2 1 2 5 мкм 10 50мкм 100 200 500 мкм 1мм 2 мм 20 мм
Взвеси/ Осаждение
0,01 „ суспензии
Просеивание с исполь¬
зованием ультразвука Просеивание t Ручная сортировка
1-150 мкм Отмучивание
1-150 мкм
Седиментационное равновесие
0,001-5 мкм
Анализатор Malvern Zetasizer
Анализатор Malvern 4600
Анализатор Malvern Autosizer
3 нм - 3 мкм
0,01-3 мкм
0,5-560 мкм
Анализатор
м Aila/t/АГл OfiAn С л . . . . . ..
0,1-2000 мкм
80 130
елл Malvem dbUU fc Анализатор Malvern MastersizerX
600 (4-го класса)
0,1-600 мкм
i
• Анализатор Malvern Ma9tersizerX
(3-го класса)
1,2-564 мкм
Анализатор Malvern 2600
1,2-1080 мкм
короткого действия
Днялыэатпр Mah/arn 9ft0ft
3 нм 3 мкм Анализатор
Malvern НЮ
Анализатор CoulterN4
Анализатор Coulter NaNoSizer
длительного действия
0,04-3 мкм
0,6-500 мкм
Анализатор Coulter TAW
Анализатор Coulter LS130
'
3-570 мкм
0,04-2000 мкм
Анализатор
Q МКМ
UILAo-AL\JAI tL \ 1 bbOj
0,1-500 мкм
CILAS 920 Аналиэатоо
10 нм - 5 мкм Анализатор Niconp ТС 100
CILAS (1933)
1-200-500мкм
Анализатор Analysette 20 Frist9h
0,1 -300 мкм
Анализатор Micromeritics
Sedigraph 5100
0,04-1000 мкм
Sedigraph 5000 E „ . ™
0,02-50 мкм - 100 мкм
0,1-150 мкм
Анализатор Moriba
Capa 500
0,01-30 мкм
Аналиэатоо
a iwu wnimau4M yn wu
10-SACI220
0,01-60 мкм
Joyce-Loebl Mark III
Рентге- УФ -
новское , Видимое
излучение | излучение
Анализатор
Joyce-Loeb1 DC-3
Инфракрасное излучение
. Микроволновое
| излучение
0,01 мкм 0,4 мкм 1мкм
1 MM
Рис. 2.2. Диапазоны размеров частиц, которые могут быть определены с помощью некоторых
автоматических анализаторов
2.1.3. Закон седиментации
После возможной предварительной обработки (см. раздел 2.2.1) образцы суспендиру¬
ют в водной среде в присутствии диспергирующего вещества (см. раздел 2.2.2). В про¬
цессе седиментации на частицы действуют две основные силы: сила тяжести, которая
влечет их вниз, и сила вязкого сопротивления среды, действующая в противоположном
направлении их перемещения. Уподобляя частицы сферам радиуса г, при условии дей¬
ствии силы тяжести /^(выраженной в динах), можно получить:
Глава 2. Анализ гранулометрического состава
39
р*=\™г{р,-р/)я>
где г — эквивалентный радиус сферической частицы, см; g — гравитационная постоян¬
ная, 981 см/с2; ps — плотность частиц (от 2,4 до 2,8 г/см3 для почв); рг — плотность дис¬
персионной жидкости, г/см3.
Сила сопротивления среды Fr (в динах) может быть описана следующим выраже¬
нием:
F = 6nrr\V,;
где V — скорость падения, см/с; г) — вязкость среды, П (г/(см • с)), при температуре
0, °- (табл. 2.2).
Частицы достигают равновесной1 скорости, когда силы F и Fr становятся равными,
откуда скорость их падения может быть оценена помощью закона, изначально установ¬
ленного Стоксом (1851):
^ = 2(р,-р>2 (21)
9г\
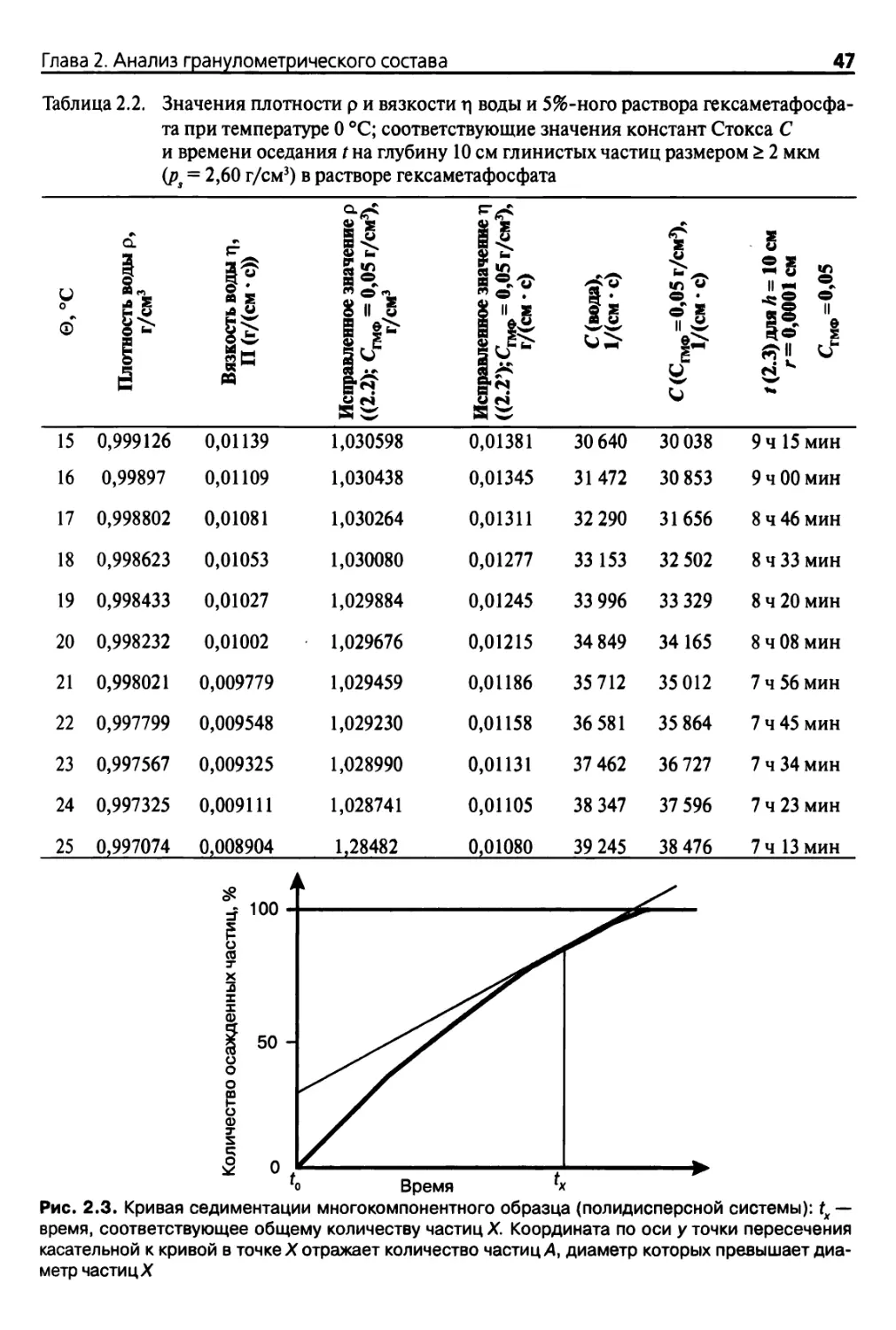
Для проведения вычислений средняя плотность рл частиц почвы в дисперсионных
системах почв часто принимается равной 2,65 или 2,60 г/см3. Разработано эмпирическое
уравнение для расчета р/и rj в водных растворах гексаметафосфата, обычно используе¬
мого для получения распределения частиц почв по размеру (Gee и Bander, 1986):
р=р0(1 +0,630Сшф), (2.2)
Л = л0О+4,25Сшф), (2.2е)
где р0 — плотность воды, г/см3 при рабочей температуре (табл. 2.2); ri0 — вязкость
воды, П при рабочей температуре (табл. 2.2); Сшф — концентрация гексаметафосфата
в г/см3.
Таким образом, константа Стокса для среды может быть определена как:
с = 2( р - р)фх\.
Уравнение (2.1) свидетельствует о том, что скорость оседания пропорциональна ква¬
драту радиуса частицы и не изменяется на протяжении всего процесса седиментации
при строгом соблюдении определенных условий (см. раздел 2.1.4). Скорость также мо¬
жет быть определена как V=АД гДе t—время, с, затраченное частицей радиуса г, см при
падении с высоты А, см. Глубина ее осаждения в течение заданного времени или время,
необходимое для осаждения на данную глубину, могут быть определены как:
t= 9/rn =h erV~2.
2(p ,-pf)gr (23)’
Анализ гранулометрического состава, основанный на явлении седиментации, заклю¬
чается в определении содержания частиц, размер которых меньше или равен заданному
1 Постоянной. — Лримеч. науч. ред.
40
Часть 1. Минералогический анализ почв
Таблица 2.1. Методы определения характеристик частиц, используемые для установления
гранулометрического состава почв
Выделение частиц
Суспендирование
(диспергирование)
— разложение органического вещества (Н202, гипохлорит натрия),
гипобромит....
— разложение цементирующих компонентов (Al, Fe, Si):
— в воде
разбавление (исключение катионов2+)
— предварительные обработки
химические методы
подбор pH
разнообразные ПАВ
обработки ультразвуком
физические методы
механическое перемешивание (диспергирование:
40 возвратно-поступательных движений/мин)
предельно допустимые концентрации
— подбор концентрации
эффекты прилипания к стенке
Методы разделения, методы измерения
Диапазон
размеров частиц
Восста¬
новление
фазы
Измерение размеров
частиц
сухое
2 мм
1. Просеивание:
0,050 (5 мкм)
Да
мокрое
2. Измерение площади (для справки)
< 2 мкм
Нет
Плотность
сухое
1-150 мкм
да
почв — 2,65
3. Отмучивание:
влажное
Сухие или влажные
Радиоактивность (для справки)
образцы
Изменение электропроводности
Нет
(либо фотометрических свойств)
Нет
4. Подсчет
0,5-500 мкм
При микроскопических исследованиях:
Нет
оптических, ПЭМ, РЭМ... и анализ изо¬
бражения
Глава 2. Анализ гранулометрического состава
41
- кислая либо основная среда
- восстановительная либо комплексообразующая среда
кислая среда (некоторые андосоли)
основная среда: NaOH, NH4OH, пирофосфат, гексаметафосфат
Принцип, лежащий в основе метода Достоинства
Недостатки
Компании-
производители
по состоянию
на 1990 г.
В основе метода лежит разделение Простой
частиц на вибрирующих ситах с и без
воздействия ультразвуковых волн,
взвешивание фракций (дискретное)
Недолговечность сит, Saulas Tamisor
дефекты ячеек сит, заку- и т. д.
порка ячеек сит и т. д.;
при использовании
метода влажного
просеивания тонко¬
дисперсные частицы
слипаются в крупные
Измерение массы молекулярного Внутренняя
слоя (азот, моноэтиловый эфир эти- и внешняя
ленгликоля...), расположенного на поверхность
поверхности частиц (предваритель¬
ное разделение фаз)
Разделение, измерение обратной
седиментации: противодействие
силам тяжести действием потока газа
или жидкости (в восходящем потоке)
(периодического)
Для меченых элементов
Калиброванные отверстия, распре- Исключается
деление данных частиц пропорцио- влияние плот-
нально изменениям в вытесненном ности
электролите или пропорционально
экстинкции
Подсчет количества частиц с помо- форма,
щью анализатора изображений или внешний вид
подсчет вручную
Сложность проведения Micromeretics
измерений
Очень медленно;
используется для
изучения осадка
Требуется калибровка
отверстий;
закупорка отверстий
Дорогостоящее
оборудование
Coulter
Quantimatl 20
Micro-
Videomat (Zeiss)
Integramat
(Leitz)
42
Часть 1. Минералогический анализ почв
Таблица 2.1 (продолжение)
Методы разделения, методы измерения
Диапазон
размеров частиц
Восста¬
новление
фазы
Седиментационные весы
1— 150 мкм
Нет
Пипеточный метод (Robinson, Andreaseri)
эталонный метод ФАС (AFNOR)
1— 150 мкм
Да
Простая весовая и фото-седиментация
(турбидиметрия) динамическое рассеяние
света, лазерная дифракция
1-20 мкм-50 мкм
Нет
Только один метод
не может быть ис¬
пользован для всех
фракций частиц
5. Седиментация
Малоугловое рентгеновское и нейтронное
рассеяние
Измерение плотности на различной
глубине1
0,1-100 мкм
1—50 мкм
Нет
Погружение ареометра
на фиксированную глубину2
длительное
0,02—500 мкм
Нет
5'. Центрифугирование
периодическое
Глава 2. Анализ гранулометрического состава
43
Принцип, лежащий в основе метода Достоинства
Гравиметрическое определение мас¬
сы накопленных частиц за опреде¬
ленный промежуток времени
Отбор определенного объема мелко- Определен-
дисперсных частиц, просеивание для ный уровень
отделения ила и песков седиментации
за задан¬
ное время.
Критическая
концентрация
на единицу
объема
Оптический метод: поглощение, Лазер:
диффузия или рассеяние света (бело- высокая ин-
го либо монохроматического света тенсивность
или лазерного излучения) излучения,
малый объем,
низкая кон¬
центрация
Недостатки
Компании-
производители
по состоянию
на 1990 г.
Анализ занимает очень Mettler,
много времени Becker, Cahn,
Sartorius,
Prolabo
Влияние формы, цвета Stanton,
частиц на коэффици- Malvern.
ент экстинкции, за- Cilas
висимость дифракции
от длины волны для
малых частиц
Измерение электромагнитного
излучения: рентгеновского или
у-изл учения
Высокая сте- Концентрации выше, Micromeretics
пень детекти- чем в фотометрии 5100.
рования
Метод Боюкоса (Bouyoucos): измере- Ареометр
ние глубины погружения ареометра (Casagrande,
ASTM)
Влияние плотности,
температуры, вязкости
среды, вертикальность
цилиндров
Метод De Leenher. цепной ареометр
Испарение
с поверхности
Усиление действия гравитационных
сил под действием центробежной
силы. Изменение концентрации на
данной глубине, фотодетектирование
Shimadzu.
Сара 500
Horiba 300
Horiba 700
Horiba
Beckman
44
Часть 1. Минералогический анализ почв
Таблица 2.1 (окончание)
Методы разделения, методы измерения
Диапазон
размеров частиц
Восста¬
новление
фазы
Закон Стокса: скорость расслоения твер- 0,02—2 мкм Да
дых веществ: одна неподвижная жидкая
фаза - одна подвижная твердая фаза,
отсутствие деформации, вещество не
взаимодействует с жидкой фазой, равно¬
мерное рассредоточение частиц. Влияют:
температура (вязкость, конвекционные
потоки), плотность, диаметр частиц.
Электростатический эффект отталкива-
ния частиц
6. Броуновское движение, электрокине- 0,003—2 мкм
тический (дзета-) потенциал
1 Метод ареометра постоянного веса. — Примеч. науч. ред.
2 Метод ареометра постоянного объема. — Примеч. науч. ред.
Глава 2. Анализ гранулометрического состава
45
Принцип, лежащий в основе метода
Достоинства
Недостатки
Компании-
производители
по состоянию
на 1990 г.
- Взвешивание накопленного осадка
или измерение высоты осадка
- Возможное примерное разделение
фаз
- Пипеточный метод анализа
центрифугата
Sharpies
Simcar
Частота смещения обратно
пропорциональна размеру частиц
Подвиж¬
ность частиц,
эффективный
поверхност¬
ный заояд
Только для анализа
мелкодисперсных
частиц
Malvern
46
Часть 1. Минералогический анализ почв
пороговому значению. Известные значения объемов раствора (пипеточный метод), как
правило, используют для выбора глубины и времени седиментации в качестве точки от*
сечки порогового значения. После высушивания пипетированной пробы, взвешивания
и корректировки объема может быть определено содержание частиц, размер которых не
превышает выбранного порогового значения. В примере, представленном в табл. 2.2,
в пипетированной пробе при 20 °С на глубине 10 см спустя 8 ч 08 мин после начала се¬
диментации выявлено значительное содержание фракции глинистых частиц (диаметр
частиц не превышает 2 мкм).
В методе, основанном на определении плотности, соотношение между размерами ча¬
стиц (радиус г) и продолжительностью седиментации / может быть выражено как:
г = 5г1/2, (2.4)
где S — коэффициент седиментации.
С учетом (2.3)
г= Сг'1Щч\ (2.4').
где Нг — глубина погружения весов-плотномера (гидрометра, ареометра), которая от¬
ражает величину эффективной глубины для частиц радиуса г.
2.1.4. Условия применения закона Стокса
Формула закона Стокса теоретически может быть применима только к частицам,
диаметр которых не превышает 0,1 мм, однако, согласно отдельным исследованиям
(Meriaux, 1954), закон может быть применен для частиц до 0,2 и даже 0,5 мм. Для ана¬
лиза частиц, размер которых превышает указанные значения, рекомендуется применять
формулу Осина (Oseen); однако частицы диаметром более 0,1 мм могут быть с большей
точностью определены с помощью просеивания на ситах.
Для проведения гранулометрического анализа используются седиментационные ци¬
линдры, стенки которых снижают скорость оседания частиц из-за действия сил трения.
Так, для сферических частиц кварца диаметром 0,05 мм скорость оседания на рассто¬
янии 0,1 мм от стенок цилиндра замедляется на 12%, тогда как на расстоянии в 1 мм
замедление пренебрежимо мало (0,28%). На практике целесообразно применять седи¬
ментационные колонки (цилиндры) достаточно большого диаметра, как минимум 5 см.
Кроме того, константа Стокса рассчитана для минеральных веществ со средней плот¬
ностью частиц от 2,60 до 2,65 г/см3, при этом в состав почв могут входить илистые ве¬
щества плотностью 2,1-2,7 г/см3, монтморрилонит плотностью 1,7-2,6 и т. д. Однако
основная сложность этого метода заключается в том, что частицы не обладают ни сфе¬
рической, ни гладкой поверхностью, что приводит к необходимости вводить понятие
эквивалентного радиуса.
Анализ гранулометрического состава основан на осаждении частиц различного раз¬
мера. Некоторые частицы оседают более быстро, чем другие, что приводит к изменению
вязкости системы в ходе эксперимента, а также изменению плотности жидкости. Та¬
ким образом, для того чтобы не отклоняться слишком сильно от теоретических условий,
установленных для монодисперсных систем, следует избегать слишком высоких кон¬
центраций почвы в образцах (не выше, чем 1%).
Кривая седиментации гетерогенного образца соответствует кривой седиментации
полидисперсных систем. Такой вид кривой (рис. 2.3) позволяет оценить процентное
содержание частиц диаметром, большим, чем Л, для которых характерно время осаж¬
дения tx.
Глава 2. Анализ гранулометрического состава
47
Таблица 2.2. Значения плотности р и вязкости г\ воды и 5%-ного раствора гексаметафосфа¬
та при температуре О °С; соответствующие значения констант Стокса С
и времени оседания t на глубину 10 см глинистых частиц размером £ 2 мкм
(рз = 2,60 г/см3) в растворе гексаметафосфата
и
О
<£
Плотность воды р,
г/см3
Вязкость ВОДЫ Т|,
П (г/(см • с))
$ ^
§2
w О «Ь,
1" Д
1 р
§°с
И
sS
м
£ ь
15?
1 "||
1&
s S
С (вода),
1/(см • с)
I.
«п О
о •
fl
р
у
3
S3 £
11 "s
Я и о
О,
15
0,999126
0,01139
1,030598
0,01381
30640
30038
9ч 15 мин
16
0,99897
0,01109
1,030438
0,01345
31472
30853
9 ч 00 мин
17
0,998802
0,01081
1,030264
0,01311
32 290
31656
8 ч 46 мин
18
0,998623
0,01053
1,030080
0,01277
33 153
32 502
8 ч 33 мин
19
0,998433
0,01027
1,029884
0,01245
33 996
33 329
8 ч 20 мин
20
0,998232
0,01002
1,029676
0,01215
34 849
34 165
8 ч 08 мин
21
0,998021
0,009779
1,029459
0,01186
35 712
35012
7 ч 56 мин
22
0,997799
0,009548
1,029230
0,01158
36 581
35 864
7 ч 45 мин
23
0,997567
0,009325
1,028990
0,01131
37 462
36727
7 ч 34 мин
24
0,997325
0,009111
1,028741
0,01105
38 347
37 596
7 ч 23 мин
25
0,997074
0,008904
1,28482
0,01080
39 245
38 476
7 ч 13 мин
Рис. 2.3. Кривая седиментации многокомпонентного образца (полидисперсной системы): tx —
время, соответствующее общему количеству частиц X. Координата по оси у точки пересечения
касательной к кривой в точке X отражает количество частиц Д диаметр которых превышает диа¬
метр частиц X
48
Часть 1. Минералогический анализ почв
2.2. Стандартные методы
2.2.1. Предварительная обработка образцов
Особенности обработки образцов в зависимости от типа почвы
Образец высушивают при комнатной температуре, затем просеивают через сито диаме¬
тром ячеек 2 мм, после чего аккуратно разделяют на части помощью ручного пробо-
делителя (Pansu и др., 2001). Масса каждого опытного образца должна составлять 10 г;
если необходимо проанализировать почвы с низким содержанием глин, масса образца
может быть увеличена до 20 г. В табл. 2.3 приводятся методики обработки образцов для
различных типов почв (А-Н).
Карбонатные почвы (В)
Реактивы: 1 М HCI
Методика
• Требования стандарта на метод анализа А" 31-107 (1983) рекомендуют проведение
этой обработки после разложения органического вещества пероксидом водорода
(Н202). Однако окисление органического вещества может привести к промежуточ¬
ному образованию щавелевой кислоты, что, в свою очередь, приводит к новооб¬
разованию оксалата кальция. Таким образом, желательно отделить известняк на
стадии предварительной обработки.
• Помещают образец (эквивалентный 10 г почвы, высушенной, например, при
105 °С), взвешивают с точностью ±1 мг в низком лабораторном стакане марки
Ругех объемом 1000 мл.
• Добавляют 100 мл воды.
• Перемешивают и медленно с помощью бюретки добавляют 1 М раствор хлористо¬
водородной кислоты до полного разложения присутствующего известняка.
• Величина pH не должна быть ниже 3,0.
• После выделения всего углекислого газа осторожно кипятят в течение 5 мин. Про¬
мывают декантацией для удаления кальция и избытков кислоты (тест на хлорид-
ионы).
Примечания
• Следует избегать введения слишком большого количества хлористоводородной
кислоты, которая может вызвать разрушение хлоритов в составе некоторых триок-
таэдрических хлоритов.
• Можно использовать ацетат натрия при pH 5, чтобы предотвратить разрушение ре¬
шетки некоторых глин.
• В присутствии частиц известняка, входящих в гранулометрический состав почвы,
необходимо проводить дополнительные измерения без разложения СаС03 (Baize,
2000).
Разложение органического вещества
Органическое вещество имеет высокую склонность к агрегации. Поэтому для большин¬
ства почв оно должно быть разложено (A—F в табл. 2.3). Для почв тропического климата,
Глава 2. Анализ гранулометрического состава
49
Таблица 2.3. Характеристики анализируемых почв, а также особенности их
предварительной обработки
Органоминеральный комплекс Свойства Обработка
А: комплекс является в большей
или меньшей степени
водонасыщенным
В: наличие карбонатов
С: большое содержание
органического вещества
D: высокое содержание органо¬
минеральных цементирующих
веществ и аморфных или
индивидуализированных
полуторных оксидов
Е: высокое содержание солей
натрия, отсутствие гипса
F: наличие Мп02
G: наличие гипса < 10%
Н; наличие гипса > 10%
Несколько капель
концентрированной НС1
не приводят к выделению
С02, pH Н20 < 7
НС1 вызывает выделение
со2
pH Н20 > 7
Содержание
органических
веществ > 15%, сухого
мелкодисперсного грунта
размером < 2 мм
(Si02 + А1203) > 10%
от тонкодисперсного
грунта — ферраллитные
почвы, железистые
тропические почвы
Электропроводность
1/5 > 0,3 мСм/см;
добавление к 5 мл
водного экстракта 5 мл
ацетона не вызывает
выпадения осадка
Энергичная
реакция Н202;
фиолетовая окраска
перманганат-иона после
окисления
Положительная
реакция ацетоновой
пробой
Определение наличия
гипса путем нагревания
образца до 60 °, затем до
105 °С (см. раздел 2.2.4)
Обработка 1%-ным
раствором Н202, затем 30%-
ным
Необходимо провести два
анализа: 1) не вызывающий
разложения карбонатов,
а затем 2) с разложением
Соответствующая обработка
НА
Растворение минеральных
цементов в НС1,
а органических цементов
вН2°2
Обработка реактивом Тамма
Выделение солей промывкой
и декантацией/фильтрацией
Предобработка
бисульфитом до разложения
органического вещества
Выделение гипса промывкой
перед перемешиванием
Интерпретация результатов
анализа является сложной.
Могут использоваться
различные методы
как правило, используется 30%-ный раствор пероксида водорода (110 объемов) или ста¬
билизированный пероксид водорода (пергидроль или подобное соединение). Некото¬
рые авторы предлагают использовать для этого гипохлорит натрия или бром в щелочной
среде (2 М раствор КОН).
50
Часть 1. Минералогический анализ почв
Реактивы
• Чистый пероксид водорода (110 объемов 30%-ного).
• Раствор гидроксида аммония (20%-ный, d = 0,92).
• Диспергирующее вещество: 5%-ный раствор гексаметафосфата натрия.
• Бисульфит натрия NaHS03.
Почвы, содержащие менее 15% органического С
Помещают анализируемые образцы в химический стакан марки Ругех объемом 1000 мл.
Добавляют 100 мл 1%-ного пероксида водорода. Выдерживают при низкой температуре
в течение 1 ч, при этом необходимо избегать избыточного вспенивания перемешивани¬
ем или применением аэрозоля (например, спирта) для изменения поверхностного на¬
тяжения.
Нагревают до 60 °С и вводят небольшое количество 30%-ного раствора пероксида
водорода для повторной обработки. При этом Н202 вводят небольшими порциями до
прекращения выделения газа и обесцвечивания жидкости над осадком. Кипятят образец
в контролируемых условиях д ля разрушения избыточного Н202, а также для снижения
объема пробы, избегая ее полного высушивания.
Почвы с высоким содержанием органического вещества (гистозоли, андосоли и др.)
При обработке таких почв необходимо поддерживать их естественную влажность, чтобы
избежать необратимых изменений, связанных с сушкой, поскольку эти почвы приобре¬
тают гидрофобные свойства. Для этого необходимо использовать пробу, эквивалентную
почве, высушенной при 105 °С. Воздействие должно быть очень осторожным, посколь¬
ку после начала реакции окисления она протекает очень бурно; возможно внезапное
увеличение температуры и интенсивное пенообразование.
При отборе влажных почв добавляют дистиллированную воду до образования суспен¬
зии. Добавляют 50 мл пероксида водорода, разбавленного до 1%-ного раствора, и остав¬
ляют на холоде.
Каждый стаканчик нагревают до 60 °С для запуска реакции. В случае необходимости
регулируют температуру, добавляя кубик льда из деионизированной воды.
Небольшими порциями вводят пероксид водорода, пока не перестанет образовывать¬
ся пена, затем доводят до кипения. Надосадочная жидкость должна быть прозрачной.
Промывают осадок декантацией и продолжают анализ.
Примечания
В некоторых случаях органическое вещество может быть «защищенным», образуя одно¬
родную смесь с карбонатами, и пероксид водорода на него не действует. Если процедуры
предварительного разложения карбонатов (см. «Карбонатные почвы») недостаточно, то
необходимо обработать органическое вещество гипобромитом (3 мл брома, растворен¬
ного в 100 мл ледяного раствора 2 М КОН).
Для этого смешивают 50 мл такой смеси с образцом и оставляют на 1 ч; кипятят в те¬
чение 30 мин, охлаждают, добавляют 200 мл дистиллированной воды; оставляют на
ночь, помещают на фильтр и промывают (достаточно трех процедур промывки) перед
диспергированием.
Присутствие Mn02 (F)
Наличие солей марганца может привести к быстрому разрушению пероксида водорода.
В этом случае обработку необходимо повторить заново, при этом следует контролиро¬
Глава 2. Анализ гранулометрического состава
51
вать цвет жидкости над осадком. Несвязанный диоксвд марганца растворяется в перок¬
сиде водорода (Jackson, 1969).
Поскольку диоксид марганца вызывает интенсивное разложение пероксида водоро¬
да, необходимо сначала восстановить его бисульфитом натрия (Petard, 1993). До введе¬
ния пероксида водорода к пробе добавляют 1 г бисульфита натрия или 10 мл 37,5%-ного
водного раствора бисульфита. Затем вводят 50 мл деионизированной воды и кипятят
в течение 20 мин для восстановления диоксида марганца до ионов Мп2+. Затем проводят
обработку пероксидом водорода, как описано выше.
Наличие аморфных органоминеральных цементирующих веществ (D)
Реактивы
• Буферный раствор Тамма (Татт, 1922): 10,92 г щавелевой кислоты + 16,11 г окса¬
лата аммония растворяют в 1000 мл воды; устанавливают pH = 3.
• Хлористоводородная кислота, раствор 2 М: растворяют 166 мл концентрированной
НС1 (d = 1,18) в 800 мл воды, перемешивают, охлаждают и доводят до 1 л.
Метод, основанный на использовании реактива Тамма
К пробе массой 20 г добавляют 800 мл реактива Тамма; перемешивают на холоде, выдер¬
живают в темноте в течение 4 ч, центрифугируют и затем фильтруют.
Метод, основанный на использовании реактива НС1/Н202
Пробу обрабатывают 300 мл 2 М раствора НС1, нагревая на песчаной бане в течение 1 ч
при 60 °С. С осадка сливают жидкость и промывают деионизированной водой. Раствор
кислоты и вода от промывания должны быть собраны и высушены при 105 °С. Взве¬
шивают сухой остаток, получая массу Мп фракции растворимых минеральных веществ.
Если т представляет массу исходной пробы почвы, процентное содержание фракции
минеральных растворимых веществ Fm рассчитывается как Fm = 100 MJm. Полученное
значение должно учитываться при расчете баланса различных фракций частиц.
После растворения минеральных цементирующих веществ органические цементи¬
рующие вещества должны быть растворены, как описано выше («Разложение органи¬
ческого вещества»).
Наличие гипса (G и Н)
Метод грубой оценки содержания гипса
Пробу почвы, просеянной через сито диаметром ячеек 2 мм, массой 10 г помещают
в алюминиевую капсулу. Капсулу помещают в вентилируемый сушильный шкаф и вы¬
сушивают при 60 °С в течение 24 ч для удаления гидратационной воды. Охлаждают
в эксикаторе и взвешивают (Р1), затем высушивают в вентилируемом сушильном шкафу
при 105 °С (не менее 3 ч) для удаления конституционной воды; охлаждают в эксикаторе
и взвешивают (Р2).
Приблизительное процентное содержание гипса =100
Р1-Р2 172
10 х 36
*50(/>1-Р2).
Методика анализа в случае содержания гипса < 10% (почва типа G)
При 25 °С растворимость гипса в воде составляет 2 г/л. После разложения органиче¬
ского вещества (см. раздел «Разложение органического вещества») и после разложения
52
Часть 1. Минералогический анализ почв
или неразложения известняка (см. раздел «Карбонатные почвы») помещают пробу (10 г)
в химический стакан объемом 500 мл, добавляют 300 мл дистиллированной воды и пере¬
мешивают магнитной мешалкой. Спустя 1 ч оставляют стакан отстаиваться и декантиру¬
ют прозрачную жидкость, вновь добавляют 300 мл дистиллированной воды и повторяют
эту операцию до тех пор, пока ацетоновая проба не будет отрицательной (как правило,
достаточно еще одного раза).
Для ацетоновой пробы (см. гл. 30) отбирается 5 мл водного экстракта, добавляют 5 мл
ацетона, тщательно перемешивают; при отсутствии гипса выпадения осадка не наблю¬
дается.
Методика анализа в случае содержания гипса > 10% (почва типа Н)
Определение точного размера частиц затруднено и рекомендуется применять специали¬
зированные методики, например, метод Виеллефона (Vieillefon, 1979). Полное раство¬
рение гипса позволяет определить элементарный состав части почвы, не содержащей
гипса, однако не позволяет установить полный состав почвы. Инструментальные ме¬
тоды обладают более высокой точностью, поскольку измерение требует мало времени.
Использование ионообменных смол сопровождают сильным разбавлением, эти методи¬
ки позволяют получить удовлетворительные результаты, до образования хлопьевидного
осадка.
2.2.2. Суспендирование и диспергирование частиц
Введение
Ионы, которые могут вызвать образование осадка глинистых частиц, можно располо¬
жить в порядке убывания:
А13+ > Са2+ > NH/ >К+ > Na+ > Li+.
Для диспергирования почвы, т. е. обеспечения удерживания элементарных частиц
в суспензии, необходимо заместить природные компенсационные катионы, обладаю¬
щие высокой флокулирующей способностью. Стабильность суспензий поддерживается
только за счет взаимодействий между диффузными слоями частиц глины одною знака
заряда и контролем над силами притяжения Ван-дер-Ваальса.
Электростатическое отталкивание зависит от концентрации электролитов и валент¬
ности ионов. В том случае, если силы Ван-дер-Ваальса сильнее сил электростатическо¬
го отталкивания, потенциальная энергия результирующего взаимодействия приводит
к флокуляции. Поэтому в таком случае требуется осуществить химическую модифика¬
цию распределения зарядов и pH.
Например, инверсия краевых зарядов может устранить притяжение между краевыми
узлами кристаллической решетки и вызывать образование противоположно заряжен¬
ных частиц, обладающих слабым потенциалом сил притяжения. Так, хемосорбция гек¬
саметафосфата на октаэдральные катионы, расположенные на боковых поверхностях
кристаллитов, приводит к образованию хорошей дисперсии. Такое диспергирующее
вещество наиболее часто используется в тех случаях, когда не планируются дальнейшие
исследования фракций после определения гранулометрического состава образца.
Желательно установить значение pH раствора, наиболее отдаленное от pH изоэлек-
трической точки (см. гл. 20), при котором наблюдается выпадение осадка. Как правило,
среда должна быть основной (приблизительно 10). При слишком высоком значении pH
может наблюдаться явление солюбилизации.
53
Глава 2. Анализ гранулометрического состава
Для гидроксидов, наподобие Fe(OH)3, которые в диспергированном виде могут иметь
положительный или отрицательный заряд, устанавливают величину pH, учитывая зави¬
симость диспергирования от изоэлектрической точки. Для диспергирования андисолей
чаще всего устанавливают величину pH, соответствующую кислой среде (~3,5), при этом
всегда придерживаются правила удаленности от изоэлектрической точки.
Такие методы обработки не препятствуют объединению узлов кристаллической ре¬
шетки, что характерно для каолинитов с низким отрицательным зарядом или для слюд
(иллиты, мусковиты и т. д.). В этом случае для дополнительного механического воздей¬
ствия необходимо использовать ультразвук1. Оптимальное количество диспергирующего
вещества зависит от эффективности стадии предварительной обработки. В свою оче¬
редь, оптимальная эффективность предварительной обработки достигается при таком
количестве гексаметафосфата натрия, которое превышает величину ЕКО2 в 20—50 раз.
Однако следует избегать избытка этого реагента, поскольку это может привести к фло¬
куляции вследствие его адсорбции на коллоидных частицах. По этой причине, а так¬
же при необходимости проведения дальнейших исследований с фракциями в каче¬
стве диспергирующего вещества предпочтительно использовать ионообменные смолы
в Ыа+-форме, например, амберлитовую смолу Amberlite IR 120 Na (Rouillern др., 1972).
Они обладают определенными преимуществами, поскольку не вызывают какого-либо
дополнительного ионного обмена со средой, что имеет большое значение в случае гори¬
зонтов с низким содержанием глинистых веществ, где солевая среда приводит к значи¬
тельному увеличению массы фракции глинистых веществ.
В число диспергирующих веществ, которые могут быть использованы, входят гидрок¬
сид аммония, гидроксид натрия, карбонат натрия и пирофосфат натрия. Все эти веще¬
ства предотвращают сжатие двойного слоя кристаллитов и препятствуют увеличению
дзета-потенциала, поддерживая, таким образом, силы отталкивания между частицами.
Следует отметить, что при содержании аммиака в количестве 17 мл на 1 л он не вызыва¬
ет завышения определяемой массы, а при использовании гидроксида натрия растворя¬
ется органическое вещество и осаждается железо, тогда как пиро- и гексаметафосфаты
оставляют его в растворе. Алюминий вводится в раствор в форме алюминатов, раство¬
ренных в гидроксиде натрия, но не в аммиачном растворе. Результаты применения та¬
ких диспергирующих веществ всегда немного различаются.
1 Ультразвук: (см. дополнительную литературу по влиянию ультразвуковых волн на почвы).
Колебания ультразвуковых волн создаются с помощью магнитострикционного осциллятора.
При помещении стержня из ферромагнитного материала в магнитное поле его длина изменя¬
ется из-за магнитострикционного эффекта. При воздействии такого поля вдоль оси стержня
наблюдается его колебание с частотой, в два раза превышающей частоту приложенного поля.
Эти колебания через водную среду передаются суспендированным частицам. Явление кави¬
тации при частоте колебаний ультразвуковых волн от 20 до 30 кГц приводит к прекращению
действия когезионных сил агрегатов, но при этом не вызывает существенного повреждения
элементарных частиц, при условии непродолжительного воздействия. Такая обработка при¬
водит к повышению температуры, которое необходимо контролировать. Применяемое обо¬
рудование должно быть сконструировано таким образом, чтобы предотвратить образование
зон резонансных волн, которые оказывают наиболее разрушительное действие. Как правило,
используется оборудование двух типов, оснащенное резервуарами либо зондами с механиче¬
скими лопатками для перемешивания. Магнит для перемешивания не применяют, за исклю¬
чением извлечения, если потребуется, магнитных частиц.
2 ЕКО — емкость катионного обмена (СЕС — Cation-exchange capacity). — Примен. наун. ред.
54
Часть 1. Минералогический анализ почв
При использовании смеси гексаметафосфат-аммиак в качестве диспергирующего ве¬
щества имеем один компонент с плотностью, превышающей плотность воды, и один
компонент с более низкой плотностью, конечная плотность оказывается приближенной
к значению плотности воды.
Оборудование и реактивы
• Седиментационные цилиндры: обычно используют градуированные цилиндры объ¬
емом 1000 мл с притертыми крышками (45/40 мм), длиной 400 мм и диаметром
60 мм, однако эквивалентные результаты могут быть получены и при использова¬
нии прозрачных ПВХ-трубок с внутренним диаметром 71 мм, длиной 30 см, от¬
меткой уровня наполнения на 1000 мл, прямоугольным основанием, закрываемых
резиновыми пробками для перемешивания.
• Диспергирующий раствор: 15%-ный раствор гексаметафосфата натрия (калгон,
(NaP03)6) в деионизированной воде.
• 20%-ный раствор аммиака (d = 0,92).
• Ионообменная смола (Amberlite IRI20 Na или подобная).
Методика
Обработка смешанной диспергирующей добавкой гексаметафосфат-аммиак
После соответствующей предварительной обработки образца почвы его количественно
переносят на развернутый аналитический фильтр и промывают деионизированной во¬
дой, пока не начнется диспергирование образца. Протыкают фильтр и смывают почву
струей воды из промывной бутылки непосредственно в цилиндр (рис. 2.4). Добавляют
10 мл 15%-ного раствора гексаметафосфата натрия и 5 мл 20%-ного раствора аммиака.
Доливают еще приблизительно 500 мл воды, закрывают цилиндр и помещают на рота¬
ционную мешалку на 2 ч (на 4 ч в случае анализа глинистых почв). При анализе почв
андосоль-гистосольного типа сперва обрабатывают образец ультразвуком в течение
15 мин.
После перемешивания образец должен быть хорошо диспергирован, т. е. его элемен¬
тарные частицы (песок, илистые частицы и глинистые частицы) полностью отделены
друг от друга. Спустя несколько минут необходимо удостовериться в том, что осадок не
выпал. Далее доводят объем до 1000 мл деонизированной водой и гомогенизируют рас¬
твор.
Обработка с использованием Ыа+-формы ионообменной смолы //?120 Resin
Перед обработкой смолу необходимо просеять для удаления частиц, размер которых ме¬
нее 200 мкм.
Образцы, подвергнутые соответствующей предварительной обработке, количествен¬
но переносят на фильтр и промывают деионизированной водой до начала дисперги¬
рования. Затем прокалывают фильтр и размещают образец почвы на сите с ячейками
200 мкм в воронку на седиментационном цилиндре для отделения крупнозернистого пе¬
ска. Промывают, высушивают и отфильтровывают полученные пески (см. «Промывка
и определение количества мелко- и крупнодисперсных песков»).
Мелкодисперсные частицы переносят в цилиндр. Затем добавляют 50 мл влажной ио¬
нообменной смолы в 1Ча+-форме, затем добавляют приблизительно 500 мл деонизиро¬
ванной воды и перемешивают с помощью ротационной мешалки в течение 4 ч (5 ч для
почв, содержащих менее 10% гипса).
Глава 2. Анализ гранулометрического состава
55
Рис. 2.4. Перенос образца в седиментационный цилиндр для диспергирования частиц пробы
после предварительной обработки, фильтрования и промывки
После перемешивания смолу отделяют на сите с размером ячеек 200 мкм, размещен¬
ном на воронке над седиментационным цилиндром объемом 1000 мл, затем смолу от¬
дельно промывают и сливают промывочную воду (смола может быть восстановлена для
проведения других анализов).
Теперь объем пробы можно довести точно до 1000 мл деионизированной водой для
приготовления образцов мелкодисперсных частиц, которые могут понадобиться для по¬
следующих анализов. Также можно ввести 10 мл 15%-ного раствора гексаметафосфата
натрия и 5 мл 20%-ного аммиака, согласно методике, описанной в разделе «Обработ¬
ка смешанной диспергирующей добавкой гексаметафосфат-аммиак», плотность и вяз¬
кость практически не изменяются.
2.2.3. Пипеточный метод согласно Робинсону-Кону или Андреасену
Принцип
Пипеточный метод основан на осаждении частиц под действием силы тяжести, соглас¬
но закону Стокса (2.1). Отбор аликвоты на данной глубине в определенный момент вре¬
мени позволяет идентифицировать определенную фракцию частиц, когда все частицы
больше выбранного диаметра исключаются.
Оборудование
• Песчаная баня.
• Пипетка Робинсона-Кона на переносном штативе с зубчатой рейкой (рис. 2.5); от¬
сасывание осуществляется с помощью микронасоса со скоростью потока, отре¬
гулированной на 60 мл/мин. Пипетка должна быть подвергнута предварительной
обработке для снижения смачиваемости и придания ей гидрофобных свойств (см.
ниже «Гидрофобная обработка пипетки для отбора проб»).
56
Часть 1 Минералогический анализ почв
Рис. 2.5. Система отбора проб с использованием пипетки Робинсона-Кона (справа) и пипетки
Андреасена (слева)
• Тонкостенные алюминиевые капсулы вместимостью 30-40 мл.
• Сушильный шкаф с функцией вентилирования, отрегулированный на 105 °С.
• Термометр.
• Весы: диапазон взвешивания 120 г, чувствительность 0,1 мг.
• Комплект двух сит с диаметрами ячеек 0,2 и 0,05 мм, вибрационным рассевом.
• Ротационная мешалка, вмещающая 10 или 20 цилиндров (30 об/мин).
• Стандартное вещество — трибутилхлорсилан.
• Стандартное вещество — 1-хлорнафтален.
Подготовка пипеток
Гидрофобная обработка пипетки для отбора проб
Такой метод обработки (метод Уолкера) позволяет придать стенкам пипетки гидрофоб¬
ные свойства и избавляет от необходимости промывать пипетку между процедурами от¬
бора образцов.
Подготавливают минимально возможное количество 4%-ного раствора трибутил-
хлорсилана в хлорнафталине. Чистую, тщательно обезжиренную пипетку высушивают,
и обрабатывают внутреннюю часть пипетки аспирацией; сливают раствор и оставляют
сушиться при комнатной температуре минимум на 24 ч.
Глава 2. Анализ гранулометрического состава
57
Калибровка пипетки - введение избыточного количества реагента
Калибровку пипетки необходимо проводить регулярно. Для калибровки используется та
же диспергирующая жидкость, что и для анализов, однако необходимо контролировать
температуру, поскольку она влияет на вязкость диспергирующей жидкости. Помещают
пять порций раствора по 20 мл в капсулы и взвешивают с точностью ±0,1 мг. Получен¬
ная средняя масса соответствует величине отобранного объема. Затем высушивают в те¬
чение 5 ч при температуре, не превышающей 105 °С, взвешивают капсулы (±0,1 мг) для
определения избытка объема пробы, вызванного введением реагента.
Методика
Все операции должны выполняться при температуре 20 °С в помещении с кондициони¬
рованным воздухом, при этом оборудование и реактивы также должны храниться при
этой же температуре.
Контроль диспергирования
После перемешивания расставляют седиментационные цилиндры в линию на лабора¬
торном столе и открывают, соблюдая при этом осторожность, чтобы не потерять часть
осадка, отложенного на крышках.
Сначала контролируют состояние суспензии. Тщательно проверяют, не выпал ли
осадок, хотя он мог выпасть частично и остаться незамеченным. При наличии сомнений
прибегают к выполнению следующих операций: измеряют температуру суспензии, рас¬
считывают (уравнение 2.1) время оседания осадка на 5 см и 10 см (для частиц с диаме¬
тром 0,02 мм), вручную встряхивают цилиндры, переворачивая их вверх дном в течение
1 мин; запускают хронометр, опускают пипетку на 5 см. За десять секунд до истечения
времени отбирают 10 мл образца и переносят в капсулу. Затем опускают пипетку на 10 см
и по истечении указанного времени извлекают образец при тех же условиях. Далее вы¬
сушивают капсулы, заполненные образцами, в сушильном шкафу при 105 °С в течение
24 ч, затем взвешивают (с точностью ±1 мг); массы двух образцов должны совпадать,
расхождение масс свидетельствует об образовании осадка и его величине. При наличии
любых сомнений по поводу выпадения осадка рекомендуется провести анализ заново.
Оборудование специальной конструкции позволяет определить оптимальную зону
диспергирования различных глин в зависимости от ориентации их частиц в электриче¬
ском поле.
Первичный отбор образцов (глины + ил)
При необходимости отбора большого количества образцов для выполнения процедуры
может привлекаться еще один человек, функцией которого является тщательная реги¬
страция времени отбора образцов (табл. 2.4). Вручную встряхивают цилиндр объемом
1000 мл с суспензией, переворачивая цилиндр вверх дном и обратно, переводя весь оса¬
док в суспензию, затем помещают его на рабочую поверхность и запускают хронометр
(снимают пробку после удаления всех частиц при перемешивании).
• Закрывают трехходовой запорный кран пипетки; опускают пипетку таким об¬
разом, чтобы наконечник касался поверхности суспензии. Отмечают положение
указателя на шкале. Приблизительно за 30 с до истечения указанного времени
(4 мин 48 с при 20 °С для 10 см, см. табл. 2.5) аккуратно опускают пипетку в первый
цилиндр на выбранную глубину (в данном случае на 10 см).
• Точно за 10 с до установленного времени отбора образца начинают отбирать 20 мл
раствора (скорость притока 1 мл/с), медленно открывая запорный кран. Отбор
58
Часть 1. Минералогический анализ почв
пробы «вокруг» точного времени позволяет получить «усредненную пробу». В тот
момент, когда столбик жидкости поднимется выше уровня запорного крана, от¬
крывают кран и сливают избыточную жидкость через боковой носик.
Таблица 2.4. Методика перемешивания группы седиментационных цилиндров
Перемешивание
Перемешивание
первого цилиндра
1 мин
1 мин
второго цилиндра
1 мин
1 мин
После окончания процедуры
Длительность
Длительность
перемешивания запускают
хронометр: начало указанного
периода времени
выдержки
выдержки
Убирают пипетку, быстро вытирают внешнюю поверхность трубки и переносят ее со¬
держимое в заранее взвешенную колбу объемом 50 мл. Выпаривают содержимое колбы
до сухого остатка и затем высушивают в сушильном шкафу при температуре не более
105 °С в течение 3 ч. Взвешивают сухой остаток с точностью 0,1 мг. Далее корректируют
значение полученной массы с учетом массы реагента.
Примечания
Процесс отбора образцов не должен быть слишком быстрым, чтобы предотвратить за¬
вихрение частиц и избежать отбора частиц, диаметр которых превышает заданные раз¬
меры, и протекать равномерно.
Не следует промывать пипетку между этапами отбора образцов, поскольку она была
подвергнута гидрофобной обработке (см. раздел «Щдрофобная обработка пипетки для
отбора образцов»), при этом любые ошибки, связанные с остатками отбираемой жид¬
кости в пипетке, пренебрежимо малы по сравнению с другими источниками ошибок
измерения.
Холостые пробы должны быть выполнены с использованием того же диспергирую¬
щего вещества (см. раздел «Калибровка пипетки — введение избыточного количества
реагента»). Взвешивают образец и фиксируют массу образца после высушивания его
в сушильном шкафу в течение 24 ч.
Вторичный отбор образцов: глинистые частицы
Повторяют описанную выше процедуру при 20 °С спустя 8 ч после протекания седимен¬
тации (в случае необходимости может использоваться меньшая глубина для отбора образ¬
цов глин в течение одного дня: например, 7 ч на глубине 8,8 см при 20 °С (см. табл. 2.5)).
Взвешивают остаток с точностью ±0,1 мг и корректируют полученное значение мас¬
сы с учетом массы реактива (контрольная проба).
Можно выполнить промежуточный отбор образцов; необходимо помнить, что пер¬
выми отбирают суспензии, содержащие крупнодисперсные фазы, а мелкодисперсные —
последними. Для этого целесообразно использовать пипетку Андреасена.
Промывка и определение крупно- и мелкодисперсных песков
После последнего отбора проб глин сифонируют надосадочную жидкость до 5 см от дна;
переливают осадок в лабораторный стакан объемом 1000 мл. Добавляют деионизиро¬
ванную воду с небольшим количеством гексаметафосфата (приблизительно 700-800
мл); интенсивно перемешивают, чтобы перевести осадок в суспензию; затем спустя пе-
59
Глава 2. Анализ гранулометрического состава
Таблица 2.5. Справа время отбора частиц (d = 2,65) при седиментометрии с использованием
пипетки Робинсона—Кона на глубине 10 см; слева глубина отбора глинистой
фракции при различном времени отбора
Размер глинистых частиц < 2 мкм
Температура
1)1убина отбора образцов в см, Г, °С
спустя
5ч 6ч 7ч 8ч
Размер
глинистых
частиц < 2 мкм
<5 мкм
£20 мкм
<50 мкм
Время оседания на глубину 10 см
6,2
7,5
8,8
10,0
20
8 ч 00 мин
1ч
0ч
0 мин
16 мин
4 мин
47 с
48 с
48 с
6,4
7,7
9,0
10,3
21
7 ч 48 мин
1 ч
0ч
X
15 мин
04 мин
1
00 с
41 с
§
6,5
7,9
9,2
10,5
22
7 ч 37 мин
1ч
0ч
о
со
13 мин
04 мин
X
12 с
34 с
1
6,7
8,1
9,4
10,8
23
7 ч 26 мин
1ч
0ч
(U
2
11 мин
04 мин
«
30 с
28 с
X
X
6,9
8,3
9,7
11,0
24
7 ч 16 мин
1ч
0ч
о
я
09 мин
04 мин
54 с
22 с
X
V
7,0
8,5
9,9
11,3
25
7 ч 06 мин
1 ч
0ч
2
я
08 мин
04 мин
1
18 с
15 с
и
риод времени, необходимый для осаждения частиц размером 0,02 мм ниже уровня от¬
сасывания сифона, сливают верхний слой жидкости с помощью сифона (рис. 2.6).
Снова добавляют воду с небольшим количеством гексаметафосфата; продолжают
промывание до тех пор, пока верхний слой жидкости не станет прозрачным; завершают
одной последней промывкой дистиллированной водой; сливают максимально возмож¬
ное количество воды декантацией; количественно переносят осадок песков в капсулу,
сушат в вентилируемом сушильном шкафу при температуре 105 °С; после охлаждения
взвешивают общий песок; помещают песок на верхнюю часть двух поставленных друг
на друга сит, одно с диаметром ячеек 0,2 мм (AFNOR 24), другое — 0,05 мм (AFNOR 18);
просеивать необходимо с использованием вибромашины; проверяют наличие в ситах
сцементированных агрегатов и растительных остатков. Сито с диаметром ячеек 0,2 мм
позволяет отделить крупнозернистые пески, а сито с диаметром ячеек 0,05 мм — мелко¬
зернистые.
Содержание крупнодисперсных илистых отложений или очень мелкозернистых пе¬
сков определяют расчетом как разность между содержанием общего песка и суммой ко¬
личеств крупнозернистого и мелкозернистого песка.
Примечание
Процедуры промывания-декантации являются длительными и трудоемкими, особен¬
но при проведении рутинных анализов, когда требуется большое количество образцов.
60
Часть 1. Минералогический анализ почв
Рис. 2.6. Промывание песка методом декантации: слева — первое сифонирование, справа — по¬
следующий слив жидкости сифоном до тех пор, пока надосадочная жидкость не станет прозрач¬
ной (4 = высота оседания частиц размером 0,02 мм)
В этих случаях возможно и целесообразно применять автоматизированную систему, по¬
зволяющую промывать пески, например одну из разработанных Сьюзини (Susini, 1978).
Источники ошибок
Необходимо очень тщательно поддерживать стабильность температуры в течение всего
срока осаждения. Этого можно достигнуть погружением цилиндров в термостатируе-
мую баню, однако на практике такое устройство не вполне подходит для проведения
серийных анализов, требуемых во многих лабораториях. Следовательно, цилиндры про¬
сто выставляют в линию на лабораторный стол. В качестве температуры эксперимента
выбирают температуру одного из цилиндров в самом начале. Поскольку жидкая среда
характеризуется хорошей тепловой инерцией, за короткий период изменения не явля¬
ются слишком сильными, т. е. для первого отбора пробы есть приблизительно 4—5 мин.
Наиболее благоприятной температурой является температура от 15 до 25 °С; при темпе¬
ратуре выше 30 °С возможно образование хлопьев; при температуре ниже 15 °С время,
необходимое для седиментации, является слишком большим. По этой причине при от¬
сутствии термостата лучше работать в кондиционируемом помещении.
Однако основными источниками ошибок являются:
• слишком быстрое погружение пипетки в суспензию;
• ошибка в глубине пробоотбора;
• неравномерный или слишком быстрый отбор жидкости; некоторые авторы реко¬
мендуют 20 с на отсасывание 10 мл (такие требования не допускают отбор жидко¬
сти ртом).
Оптимальные условия выполнения процедуры отбора образцов обеспечиваются при
отсутствии различий между способами выполнения операторами серий манипуляций, т.
е. перемещением пипетки, погружением пипетки в суспензию, точной синхронизации
начала пробоотбора, высокой равномерности отбора проб, точном соблюдении отбира¬
емых объемов, осторожном удалении пипетки, сливе образца в капсулу, последователь¬
ности пробоотбора. Идеальным решением для этого является использование автомата-
61
Глава 2. Анализ гранулометрического состава
ческого устройства (Pansu и др. 2001). Однако даже при использовании традиционной
ручной системы желательно отбирать образцы помощью маленького электрического на¬
соса фиксированным потоком: перистальтические насосы успешно справляются с по¬
добной задачей.
Обработка результатов
Полученные результаты
Масса твердого образца почвы (высушенного на воздухе) — /я.
Поправочный коэффициент на нормальную влажность, равный отношению массы
образца после высушивания при 105 °С и массы твердого образца, высушенного на воз¬
духе, — К.
Объем образца — К.
Масса холостой пробы (реагентов без образца) после высушивания при 105 °С — тт.
Масса первого образца (глина + илистые отложения) после высушивания при
105 °С -ту
Масса второго образца (глины) после высушивания при 105 °С — тг
Масса общего количества песка после высушивания при 105 °С — ту
Масса фракции крупнозернистых песков (остаток на сите с размерами ячеек 0,2 мм)
после высушивания при 105 °С — /я4.
Масса фракции мелкозернистого песка (остаток на сите с размерами ячеек 0,05 мм)
после высушивания при 105 °С — т5.
Выражение результатов в % для почвы, высушенной при 105 °С
Глинистые частицы Г = (т2 - тт) х 1000 х 100/( Vnxmx К).
Илистые частицы И = (тх - т2) х 1000 х 100/( Vn х ш х К).
Мелкозернистый песок МП = 100 х т5 /(т х К).
Крупнозернистый песок КП = 100 х тА /(т х К).
Общее содержание песка ОП = 100 х тъ/(т х К).
Содержание крупнодисперсных илистых частиц КИ = ОП - (МП + КП).
При содержании в образце известняка (табл. 2.3, В) и выполнении гранулометриче¬
ского анализа без деструкции карбонатов необходимо по отдельности определить со¬
держание карбонатов во фракциях, что позволит получить данные о распределении из¬
вестняка.
Проверка и корректировка полученных результатов
С учетом величины поправочного коэффициента на нормальную влажность (см. «Об¬
работка результатов» в разделе 2.2.3) сумма: глинистые частицы + илистые частицы +
+ общее количество песка + органическое вещество + если необходимо, карбонаты,
растворимые соли, гипс, должна составлять от 95 до 102%, в идеальном случае от 98 до
102%. Для почв, богатых органическим веществом, результат может получиться слиш¬
ком большим, так как в случае неполного разложения в процессе предварительной об¬
работки органические вещества могут быть посчитаны дважды.
Слишком низкое значение суммы масс обусловлено потерями в процессе предвари¬
тельной обработки. В большинстве случаев невозможно определить точное соотноше¬
ние потерь органического вещества, растворимых солей, карбонатов и гипса. Полная
оценка потерь может быть выполнена следующим образом: в цилиндр, в который отби¬
рали образцы, вводят 10 мл 1 М раствора СаС12 и 1 мл 1 М раствора НС1 для флокуляции
62
Часть 1 Минералогический анализ почв
коллоидных частиц и предотвращения образования карбоната кальция в процессе вы¬
сушивания в сушильном шкафу. После оседания частиц полностью сливают прозрач¬
ный раствор, переносят осадок во взвешенную капсулу, сушат в сушильном шкафу при
105 °С и затем взвешивают. Это позволяет получить значение массы mr, на основании
которой могут быть определены потери в ходе предварительной обработки (органиче¬
ского вещества и т. д.) и исправлено значение массы.
2.2.4. Метод определения плотности при погружении ареометра
на различную глубину
Принцип
Этот тип анализа предпочтительнее, поскольку позволяет построить кривые распреде¬
ления без фракционирования осадка для классификации по размерам. В методе, осно¬
ванном на определении плотности (Bouyoucos, 1927,1935,1962), гетерогенная суспензия
ведет себя таким же образом, что и гомогенная жидкость той же плотности. Касагранд
(Casagrande, 1934) показал, что в таких случаях может быть использован поплавковый
плотномер для измерения средней плотности в суспензионной колонке с поплавком.
Горизонтальное значение средней плотности находится на расстоянии ЯЛот самого вы¬
сокого уровня суспензии. Согласно формулам 2.4 и 2.4' здесь разделяются частицы на¬
чиная от радиуса.
где t — длительность седиментации (время отложения осадка), что позволяет проводить
анализ гранулометрического состава.
Метод, основанный на определении плотности, может быть применен при постоян¬
ном погружении поплавка, что удовлетворяет всем условиям непрерывного измерения,
либо при погружении поплавка на определенное время, что напоминает метод дискрет¬
ного анализа, описанный в главе 1, но не обладает той же точностью. Для массовых из¬
мерений его преимущество в исключении процесса отбора проб и их взвешивания. В то
же время, метод, основанный на определении плотности, требует использования боль¬
шего количества образцов по сравнению с пипеточным методом.
Оборудование и реактивы
• Цилиндры, аналогичные тем, которые описаны в подразделе «Оборудование»
(раздел 2.2.3); специальный конический ареометр, не позволяющий накапливаться
частицам на поверхности; термометр; пикнометры; увеличительное стекло с длин¬
ным эффективным фокусом.
• Диспергирующие вещества, описанные в подразделе «Оборудование и реактивы»;
изоамиловый спирт.
Проверка ареометра
Расчет высоты падения частиц
Отношение Касагранда позволяет рассчитать значение Я, рассматривая эффективную
глубину как высоту падения частиц. Для выполнения расчетов необходимо определить
показания ареометра, как показано на рис. 2.7. Объем ареометра рассчитывают по объ¬
ему вытесненной им жидкости.
63
Глава 2. Анализ гранулометрического состава
Я^ + ОДй-К/З), (2.5)
где V — объем, см3; S — сечение, см2.
Можно представить графически зависимость глубины Я от плотности. Это позволя¬
ет составить таблицу, в которой размеры частиц представлены как функция температу¬
ры, времени и глубины для любого типа ареометра.
Проверка калибровки ареометра
Такая проверка проводится с использованием чистой воды и 2%-ного раствора нитрата
или хлорида бария. Для определения плотности воды используют пикнометр (при этом
регистрируют значение температуры), затем измеряют плотность 2%-ного раствора
(4 значащих цифры после запятой).
Проводят такие же измерения при погружении ареометра в пробирку, последователь¬
но заполненную двумя указанными жидкостями; отмечают значения плотностей с по¬
мощью увеличительного стекла с длинным фокусом (5 или 6 см).
Отмечают расхождения в результатах измерений, полученных с использованием пик¬
нометра и ареометра. В том случае, если относительные значения совпадают, гидрометр
признается годным (поверенным), даже если показания не являются точными, посколь¬
ку в расчетах используются не значения плотностей, а их разница.
Если расхождения между результатами пикнометра и ареометра не являются постоян¬
ными, необходимо соответствующим образом сместить координатную сетку: значения,
считываемые на ареометре/фактические значения (это очень редкий случай).
Этот метод аналогичен методу, описанному в разделе 2.2.3, в котором используется
30 г мелкозема (воздушно-сухая почва, просеянная через сито с диаметром ячеек 2 мм).
Для диспергирования вводят 30 мл раствора гексаметафосфата концентрацией 102 г/л.
Перемешивают в течение 4 ч с помощью ротационного шейкера, переворачивающегося
вверх-вниз, затем переносят в цилиндры и доводят до 1000 мл.
Рис. 2.7. Соотношение между плотностью и высотой падения частиц Нг(2.5) для ареометра
со следующими характеристиками: У=45 см3, S = 28,26 см2, h = 17 см, h, = 15,2 см
64
Часть 1. Минералогический анализ почв
Методика
Предварительная обработка образца
Направляющая
бумага
Рис. 2.8. Центрирование ареометра
Определение содержания фракции глинистых и илистых частиц (0,02 мм)
Таблица 2.6. Размер частиц в зависимости от температуры, времени и глубины:
действительно для стандартного ареометра (лаборатория седиментационных
исследований, институт IRDBondy, Франция, неопубликованные данные)
Размер
и °с
Плотность -►
1,020
1,015
1,010
1,005
1,000
частиц
Время, мин i
8,7 см
11,6 см
14,4 см
17,2 см
20,1 см
4
21 мкм
24,7 мкм
27,4 мкм
30,1 мкм
32,5 мкм
1 с
6
17,4
20,1
22,4
24,5
26,5
15
8
15
17,4
19,4
21
23
9
14,2
16,4
18,2
20
21,6
20 мкм
4
20
23
25,8
28,2
30,3
20
6
16,4
19
21
23
24,5
8
14,2
16,3
18,2
19,8
21,4
9
13,4
15,4
17,2
18,8
20,2
(глинистые
3
21,8
25,2
28,1
30,7
33,2
и илистые
25
4
19,9
21,8
24,3
26,6
28,7
частицы)
6
15,4
17,8
19,8
21,6
23,4
8
13,4
15,4
17,2
18,8
20,3
Размер
Плотность —»
1,020
1,015
1,010
1,005
1,000
частиц
ty °С
Время, ч i
6
2,2 мкм
2,6 мкм
2,9 мкм
3,1 мкм
3,4 мкм
15
8
1,9
2,2
2,5
2,7
2,9
2 мкм
24
1,1
1,3
1,4
1,58
1,7
6
2,1
2,4
2,7
2,9
3,2
20
8
1,8
2,1
2,3
2,5
2,7
(глинистые
24
1
1,2
1,4
1,5
1,6
частицы)
6
2
2,3
2,6
2,8
3
25
8
1,4
2
2,2
2,4
2,6
24
1
М
1,2
1,4
1,5
65
Глава 2. Анализ гранулометрического состава
По возможности измерения проводят в помещении с кондиционированным воздухом
при постоянной температуре в 20 °С. Цилиндры, содержащие суспензии, группируют;
необходимо удостовериться, что их объем доведен до 1000 мл, измеряют температуру,
пользуясь таблицей (наподобие табл. 2.6), в которой приведены значения продолжи¬
тельности седиментации при проведении измерений при выбранной температуре (на¬
пример, 4-6-8-9 мин).
Вручную встряхивают первый цилиндр, переворачивая его вверх дном и обратно в те¬
чение 1 мин, добавляют 1 каплю изоамилового спирта (противовспенивающая добавка),
запускают хронометр, очень аккуратно вводят ареометр таким образом, чтобы избежать
перемешивания суспензии (это является самой сложной частью метода, из-за значи¬
тельного влияния на погрешность), при этом следят, чтобы ареометр находился в сере¬
дине суспензии (при необходимости можно воспользоваться направляющей бумагой,
согласно рис. 2.8), отмечают показания по верхнему уровню мениска в 4-6-8 и 9 мин.
Выполняют аналогичные действия для остальных цилиндров.
Отмечают уровень метки холостой пробы в цилиндре, наполненном только диспер¬
гирующим веществом.
Анализ фракции глинистых частиц (< 0,002 мм)
Отсчет времени начинается с начала перемешивания первого цилиндра и до первой
регистрации результатов (глинистые + илистые частицы). Для установления этого вре¬
мени рассчитывают среднее значение температуры от начала процедуры анализа до
момента регистрации результатов; отмечают показания на ареометре через 6—8—24 ч,
тщательно соблюдают двухКшнутный интервал, предназначенный для первого отбора
образцов; очень осторожно переносят ареометр из одного цилиндра во второй, не до¬
пуская перемешивания.
Примечания
Как следует из табл. 2.6, все выбранные значения времени отбора образцов не в точно¬
сти соотносятся с выбранным размером частиц. Однако предполагают, что изменения
размеров частиц в течение короткого интервала времени протекают непрерывно, что
позволяет построить график вблизи значения глубины погружения (рис. 2.9), на осно¬
вании которой затем можно определить точное время падения частиц данного размера.
Длительность процесса седиментации, мин
Рис. 2.9. Пример координатной сетки: размеры частиц — функция длительности седиментации
для данного ареометра и плотности, составляющей около 1,010 г/см3
66
Часть 1. Минералогический анализ почв
Обработка результатов
Процентное содержание частиц Р, соответствующее данной плотности, определяют из
уравнения:
<2-6)
где ps — плотность твердых частиц (2,65 для почвы); pf — плотность диспергирующе¬
го раствора (в наших условиях при температуре /, °С, можно рассчитать значение pf с
помощью выражения (2.2)); р — плотность, зарегистрированная при температуре /, °С;
6р — поправки + или - на считывание результатов, чтобы привести их к 20 °С (табл. 2.6);
К— объем (1000 мл); т — масса образца почвы (30 г).
Расчет может быть упрощен вычислением К = 100 pjm{ps - р7), на основании кото¬
рого можно вычислить Ру% = А[(р ± 5р) — р^ К, где т = 30 г при 20 °С, вследствие чего
значение К= 5,35.
Определение песков
Используется та же процедура, которая описана в разделе 2.2.3.
2.2.5. Метод определения плотности при погружении ареометра
на фиксированную глубину
Метод определения плотности на переменной глубине (см. 2.2.4) является более бы¬
стрым по сравнению с пипеточным методом, кроме того, при необходимости с помо¬
щью этою метода могут быть проведены измерения в непрерывном режиме. Однако,
поскольку глубина погружения ареометра не является постоянной, точность измерений
является более низкой, а вычисления более длинными.
Цепной ареометр позволяет измерить плотность суспензии на данной постоянной
глубине, составляющей приблизительно 20 см, что, в свою очередь, позволяет прибли¬
зиться к нормам и точности пипеточного метода, избегая при этом отбора проб и взве¬
шивания (De Leenheern Macsy 1952; De Leenheern др., 1955; De Leenheervi van Hove, 1956;
van Ruymbeke и de Leenheer, 1954).
Измерительная установка (рис. 2.10) включает иммерсионное тело, установленное на
конце рычага с указателем уровня жидкости и опорой, к которой может крепиться до¬
полнительный противовес в виде ползунов, а также уравнивающая цепь, позволяющая
очень точно регулировать положение указателя около поверхности жидкости. Глубина
седиментации представляет собой расстояние от острия иглы, расположенной в сере¬
дине ареометра.
За одну минуту до начала измерения необходимо аккуратно поместить ареометр в су¬
спензию; далее следует установить противовесы таким образом, чтобы указатель иглы
располагался на 1-2 см выше уровня жидкости. Во время начала отбора образцов уста¬
навливают иглу таким образом, чтобы она контактировала с жидкостью, быстро регули¬
руя положение цепи с помощью винтового приспособления. Сама цепь может привести
к увеличению массы на 100 мг. Считывание результатов при полной перегрузке позво¬
ляет получить значение массы ареометра. Определив заранее объем ареометра (при по¬
гружении в дистиллированную воду), можно рассчитать:
плотность р = масса ареометра / объем ареометра.
Продолжают расчеты таким же образом, как для расчета плотности в методе с пере¬
менной глубиной (уравнение (2.6)).
Глава 2, Анализ гранулометрического состава
67
2.2.6. Анализ гранулометрического состава песка
В химический стакан объемом 1000 мл, содержащий 100 мл 30%-ного раствора перок¬
сида водорода, переносят 100 г мелкозема (размером 2 мм, подготовленного из тщатель¬
но гомогенизированной пробы); оставляют в холодном месте на ночь для протекания
реакции, затем переносят на умеренно нагретую электрическую плитку; маленькими
порциями вводят пероксид водорода до завершения разложения органического веще¬
ства, после чего выводят избыток пероксида водорода из системы путем кипячения, не
допуская полного высушивания образца.
Переносят остаток в цилиндр, добавляют 500 мл дистиллированной воды и 25 мл рас¬
твора гексаметафосфата натрия с концентрацией 52 г/л. Устанавливают в ротационный
шейкер на 4 ч так же, как при проведении полного анализа гранулометрического со¬
става.
После выполнения описанной операции содержимое цилиндра переносят на
сито с диаметром ячеек 0,05 мм (стандарт Франции AFNOR NF-X-11-504, модуль 18);
промывают осадок под проточной водой.
Хорошо промытый на сите с диаметром ячеек 0,05 мм песок переносят в химиче¬
ский стакан объемом 250 мл. Добавляют 100 мл 6 М раствора НС1, накрывают крышкой
и осторожно кипятят в течение 2 ч для растворения железа.
После охлаждения декантируют и промывают осадок последовательной декантаци¬
ей до полного выведения кислоты; снова переносят на сито с диаметром ячеек 0,05 мм,
промывают и количественно переносят песок в капсулу; высушивают в течение 24 ч
68
Часть 1. Минералогический анализ почв
Таблица 2.7. Две колонки сит, используемых последовательно для анализа
гранулометрического состава песков
мм
Колонка 1
AFNOR
стандарт
Колонка 2
AFNOR
мм
стандарт
2
34
0,315
26
1,6
33
0,25
25
1,25
32
0,20
24
1
31
0,16
23
0,8
30
0,125
22
0,63
29
0,100
21
0,50
28
0,08
20
0,40
27
0,063
19
0,050
18
в сушильном шкафу при 105 °С. Дают остыть и взвешивают для определения общего
содержания песка.
Просеивают высушенный песок последовательно на двух колонках сит (табл. 2.7).
Колонки последовательно помещают в машину с вибрационными ситами (Рати и др.,
2001).
Переносят в колонку 2 фракции, собранные на дне колонки 7. Просеивают в течение
10 мин и взвешивают каждую фракцию (±0,01 г).
Проверка: сумма массы каждой фракции = общей массе песка.
2.3. Автоматизированное оборудование
2.3.1. Введение
Явление растрескивания почв приводит к необходимости наличия точных данных
о гранулометрическом составе фракции почвы с размерами частиц 2-20 мкм; методы
анализа, основанные на явлении седиментации, используются для анализа мелкоди¬
сперсных фаз почвы с размерами частиц 0,1 мкм и менее. В агрономии почвенные го¬
ризонты, включающие образование глинистых частиц, исследуются с использованием
соотношения «крупнодисперсные глинистые частицы < 2 мкм / мелкодисперсная гли¬
на < 0,2 мкм». Поскольку гравитационные методы анализа не способны дать ответы на
все возникающие вопросы, необходимо пользоваться различными методами.
В табл. 2.1 сведены основные методы, применяемые для определения гранулометри¬
ческого состава почв. Некоторые методы успешно используются для повторяющихся
измерений, необходимых при проведении исследований в области почвоведения, агро¬
номии, геологии или седиментологии; другие более пригодны для проведения обстоя¬
тельных и углубленных исследований. Выбор метода зависит от следующих факторов:
• степени требуемой точности, повторяемости и воспроизводимости, а также хоро¬
шей корреляции со стандартным пипеточным методом, несмотря на его недостат¬
ки (см. п. 2.2.3);
• протяженности диапазона частиц по размеру и возможности его расширения до
субмикронных, а также наноразмерных частиц;
69
Глава 2. Анализ гранулометрического состава
• времени, необходимого для получения результатов, гибкости в использовании,
возможности значительного увеличения количества анализов;
• наличия возможности восстановления фракций частиц для последующих иссле¬
дований либо одновременного проведения других исследований (непрерывные
анализы и т. д.);
• стоимости оборудования и затрат на оплату труда персонала, значимости требова¬
ний и доступных площадей;
• непрерывности накопления данных и их представления (мониторинг, вычисления,
гистограммы и интегральные кривые распределения).
Вследствие широкого разброса гранулометрического состава почв часто используют
комбинации индивидуальных методов, которые в отдельности не могут охватить весь
гранулометрический состав почвы. При этом диапазоны методов должны значительно
перекрываться. Пипеточный метод остается стандартным методом для всех сравнитель¬
ных исследований; метод лазерной дифракции позволяет расширить диапазон выявля¬
емых частиц до субмикронного уровня, однако не позволяет идентифицировать или¬
стые частицы. Действительная репрезентативность эквивалентных диаметров в данной
фракции может быть проверена использованием микроскопического анализа и анализа
изображения.
2.3.2. Седиментационный анализ при прямом использовании
гравитационного поля
Большинство типов оборудования, разработанных для автоматизации пипеточного ме¬
тода, не получили широкого распространения, так как в каждой лаборатории уделяется
внимание разработке устройств, соответствующих собственным потребностям. Напри¬
мер, автоматизированное устройство, предназначенное для анализа частиц, разрабо¬
танное компанией Technology Diffusion France (Pansu и др., 2001), основано на явлении
седиментации в термостатируемом шкафу с использованием автоматической пипетки.
Аналитикам, в особенности работающим в промышленном секторе, требуются мето¬
ды, которые могут дать быстрые результаты с хорошей повторяемостью и возможностью
калибровки оборудования (с использованием сферических калиброванных порошков).
Информацию об основных типах имеющегося в продаже автоматизированного обору¬
дования и наименовании производителей можно найти в работе (Pansu и др., 2001).
Седиментационные весы и автоматические рассеивающие устройства для мокрых
методов анализа
Такие весы (Sartorius, Cahn, Mettler и т. д.) позволяют осуществлять непрерывную реги¬
страцию процесса седиментации частиц, с размером от 1 до 150 мкм (рис. 2.11), фрак¬
ции более крупных частиц могут быть проанализированы методами сухого или влаж¬
ного просеивания с использованием специального автоматизированного оборудования
(Micromeritics, Seishin и т. д.).
Процесс седиментации протекает после диспергирования образцов сниженной мас¬
сы, т. е. приблизительно 1-2 г, в термостатируемой камере. Непрерывная автоматиче¬
ская регистрация массы образующихся осадков позволяет получить кумулятивные кри¬
вые зависимости массы образца от времени. В зависимости от степени автоматизации
и метода обработки результатов также могут быть построены графики плотности рас¬
пределения. Измерения являются воспроизводимыми, однако занимают много времени
и к тому же непригодны для проведения серийных анализов.
70
Часть 1, Минералогический анализ почв
Рис. 2.11. Схема определения размера частиц с компенсацией веса постоянным
уравновешиванием
Системы, основанные на прямом использовании гравитационных сил
и рентгеновского излучения
Частицы подготавливают и помещают в суспензию (как описано в разделах, посвящен¬
ных рентгеновскому анализу). Частицы поглощают часть рентгеновского излучения,
пропорциональную их числу. Итоговое значение интенсивности излучения измеряется
сцинтилляционным детектором.
На начальной стадии итоговая интенсивность рентгеновского излучения являет¬
ся минимальной, затем оседающие частицы приводят к увеличению проходящей ин¬
тенсивности. Для сокращения продолжительности измерений ячейку, содержащую
суспензию, постепенно перемещают вниз и регистрируют рентгеновское излучение,
захватывая все более близкие к поверхности части суспензии. Все эти перемещения
управляются компьютером, а положение ячейки представляет собой логарифмическую
функцию от времени, отображаемой на оси Xрегистрирующего устройства, что позво¬
ляет определить диаметр, соответствующий положению ячейки. В зависимости от мо¬
дели оборудования таким методом можно определить частицы размером от 0,1 мкм до
более чем 100—300 мкм.
Проведение непрерывных измерений позволяет представить результаты в форме
интегральных кривых гистограмм массы либо количества поверхностных частиц и т. д.
Программные интерфейсы позволяют сохранить результаты исследований, а устройство
для отбора проб с карусельным механизмом — осуществлять непрерывную обработку
нескольких образцов.
Следует отметить, что рентгеновское излучение длиной волны менее 10 нм подходит
для анализа частиц, которые в диапазоне видимого излучения рассмотреть нельзя (100—
800 нм, т. е. частицы, диаметр которых приблизительно соответствует диаметру мелко¬
дисперсных частиц глины). Повторяемость метода удовлетворительна, а измерения для
частиц до 2 мкм занимают приблизительно 10 мин.
Оборудование этого типа применяли для сравнительных исследований пипеточным
методом анализа почв (например, Delaune и др., 1991). Такой метод удобен для фракций
с размером от 50 до 1 мкм, но требует больше времени для более мелких частиц (для
частиц менее 0,2 мкм требуется 50 мин). Если в состав почв входит более 20% крупноди¬
сперсных илистых частиц, данный метод дает заниженные результаты.
Глава 2. Анализ гранулометрического состава
71
Системы, основанные на прямом использовании гравитационных сил
и поглощения или рассеивания света
Методы, основанные на фотоседиментации (нефелометрия, турбвдиметрия), зависят
от воздействия различных факторов. Их воспроизводимость и повторяемость низки
вследствие использования белого света или монохроматического излучения видимого
спектра.
Такие методы могут быть использованы только для экспресс-анализов однородных
серий.
Методы, основанные на элютриации
В таких методах скорость падения частиц (подвижная твердая фаза, которая легко изме¬
ряется) является более низкой, равной или более высокой по сравнению со скоростью
жидкости (нестационарной подвижной жидкой фазой).
Жидкая фаза циркулирует в обратном направлении по отношению к частицам, что
позволяет осуществить их сортировку; самые мелкие частицы перемещаются вверх либо
оседают вниз под действием гравитационных сил. Различные фракции могут быть вы¬
делены. Такая система подходит для определенных исследований осадков, однако изме¬
рения занимают много времени и мало пригодны для повторных анализов.
2.3.3. Методы, использующие принцип ускоренной седиментации
Основные положения
На практике методы, прямо использующие гравитацию, неприменимы для анализа ча¬
стиц менее 2 мкм, поскольку для их осаждения требуется очень продолжительное вре¬
мя. Трудно обеспечить в седиментационных цилиндрах отсутствие конвекции в течение
длительного периода и удержать частицы от броуновского движения. Однако усиление
действия гравитационных сил за счет центрифугирования позволяет преодолеть ограни¬
чения и снизить влияние броуновского движения.
Методики разделения и выделения гранулометрических фаз с использованием такого
метода подробно описаны в гл. 3.
Оборудование, использующее центрифужные диски
В некоторых типах оборудования последовательно используются непосредственно сила
тяжести применяемая с вертикальным ротором в неподвижном состоянии для анализа
более крупных частиц, и тяготение за счет центрифугирования со скоростью от 1800 до
8000g(Horiba, Shimadzu, Seishin, Union-Giken, Joyce-Loebl- Vickerss и т. д.).
Анализ осуществляется непрерывно, а регистрация результатов позволяет получать
кривые и гистограммы в автоматическом режиме. Таким способом могут быть проана¬
лизированы образцы почв массой 1 г.
В зависимости от производителя измерительные ячейки просвечивают либо фильтро¬
ванным излучением лампы накаливания с измерением поглощения света, либо (в ис¬
ключительных случаях) лазерным или рентгеновским излучением (Brook Haven), Таким
образом могут быть идентифицированы частицы размером от 0,01 мкм до 100 мкм. Дру¬
гие изготовители используют горизонтальные диски и устройства для отбора проб на
определенном расстоянии и в заданное время. Фракции высушивают, взвешивают и, по
возможности, подвергают другим исследованиям (Frltsch, Simcar, Joyce-Loebl и т. д.).
Технические данные оборудования не всегда одинаковы для серийных анализов,
и повторяемость не всегда лежит в пределах, получаемых для пипеточного метода.
72
Часть 1. Минералогический анализ почв
Например, для субмикрометрического анализа один производитель предлагает ис¬
пользовать ультрацентрифугу с титановым диском при 100 000 g в условиях частичного
вакуума, чтобы избежать перегрева оборудования и снизить шум. УФ-сканер (280 нм)
позволяет осуществлять анализ частиц почвы с размерами менее чем 500 нм (Beckmann
Spinco).
Микрометоды, основанные на использовании фракционирования в поперечном си¬
ловом поле (ФПСП), все еще недостаточно надежны для широкого применения в ис¬
следованиях почв.
2.3.4. Методы, использующие рассеяние и дифракцию лазерного
излучения
Лазерные приборы для измерения размеров частиц претерпели впечатляющее разви¬
тие и в настоящее время могут применяться для анализа все возрастающего диапазо¬
на размеров частиц. Некоторое оборудование позволяют охватывать диапазон от 0,1 до
2000 мкм, но в основном оборудование является особенно эффективным для более огра¬
ниченного диапазона. Одно оборудование предназначено для субмикронных или даже
наноразмерных частиц, тогда как другое, более широко распространенное, особенно
полезно для анализа гранулометрического состава, чаще всего требуемого для почвовед¬
ческих лабораторий.
Такие измерения не основаны на явлении седиментации, поэтому требуется кали¬
бровка.
Диспергирующая жидкость, содержащая взвешенные частицы, циркулирует в изме¬
рительной ячейке под пучком монохроматического лазера, коллимированного конденса¬
тором в окне анализатора на определенную поверхность. Излучение лазера рассеивается
на внешней поверхности частиц, а значения углов дифракции обратно пропорциональ¬
ны размеру частиц. Оптическая система накапливает сигналы, которые анализируются с
помощью Фурье-преобразования и различаются на детекторе под предварительно опре¬
деленными углами. Сигнал обрабатывается для получения распределения частиц. Резуль¬
таты могут быть выражены в форме кривых: среднего диаметра (гранулометрический со¬
став), представленного в процентном содержании к общей массе, или в виде гистограмм
по весу, поверхности, количеству частиц, объему и т. д. С помощью такого метода может
быть измерено 32-64 фракции размеров частиц (Malvern, Cilas-Alcatel, Coulter и т. д.).
Некоторые типы оборудования позволяют дозировать либо суспензии, либо сухой
порошок, что может быть полезным при анализе илов. Периодическая дефлокуляция
в процессе работы возможна с помощью ультразвука. Загрузка 40 образцов с помощью
распределительного устройства и использование распределительного устройства для по¬
дачи реагентов позволяют проводить анализ без непрерывного контроля.
Библиотеки анализов допускают применение определенных методов и диспергиру¬
ющих веществ. Методики просты, однако значительно различаются в зависимости от
используемого оборудования, вследствие чего в настоящей книге нельзя привести их
подробное описание.
2.3.5. Методы, использующие оптические и электрические свойства
Анализ распределения частиц субмикронного размера (от 3 нм до 3 мкм) проводится со¬
вместным определением значения pH, температуры, электропроводности и относитель¬
ной вязкости, что позволяет контролировать стабильность суспензии и значение элек¬
73
Глава 2. Анализ гранулометрического состава
трокинетического потенциала (дзета-потенциала, разности потенциалов между слоем
диспергированной поверхности и дисперсной средой).
Ионная сила раствора при этом измеряется методом электрофореза в кварцевом
капилляре с использованием Pt-Мо-электрода на частицах размером от 1 до 1000 мкм
(Malvern, Brookhaven, Coulter, Micromeritics, Mono-Research Lab., Zetameter Inc., Matec
Applied Science и т. д.).
Эффективный поверхностный заряд частиц определяется за счет измерения подвиж¬
ности в системе «жидкость — твердые вещества» при известном значении диэлектриче¬
ской проницаемости жидкости. Это позволяет исследовать явления флокуляции и дис¬
пергирования.
Для исследования и оптимизации процессов диспергирования различных глин в про¬
мышленных условиях разработаны другие типы оборудования. Они позволяют отличать
флокулированные и дефлокулированные частицы. Например, первичные частицы ка¬
олинита представляют собой правильные гексагональные пластинки. Благодаря своей
природе под действием электрического поля такие частицы образуют диполи, которые
ориентируются в соответствии с полем. Новообразованные агрегаты, сформированные
в процессе флокуляции, состоят из кластеров первичных частиц, соединенных в слу¬
чайном порядке, они не выстраиваются в направлении электрического поля. Для из¬
мерения относительного количественного соотношения флокулированных и дисперги¬
рованных частиц на суспензию направляют лазерный луч и анализируют интенсивность
рассеянного света. Результат отказывается дискретным. Измерения, выполненные при
различных условиях (концентрации диспергированного вещества, природе диспергиру¬
ющего вещества, pH и т. д.), позволяют оптимизировать этот метод анализа.
2.3.6. Методы, использующие прямое наблюдение частиц
Оптическая и электронная микроскопия — дозиметр излучения и анализатор
изображения
Такие прямые методы основаны на использовании оптической микроскопии либо, в не¬
которых случаях, электронной микроскопии (см. гл. 8). Среди прочих методов может
применяться фракционирование частиц за счет действия гравитационных сил. В элек¬
тронной микроскопии препараты должны быть высушены и нанесены на сетку или пла¬
стинку (ПЭМ-РЭМ).
Микроскопический анализ позволяет осуществить непосредственное наблюдение
совокупности частиц и их морфологических параметров. Для микрообразцов могут
быть определены форма, длина, ширина, толщина или диаметр частиц, а также оценены
фрактальные свойства. Сравнение частиц из одной и той же фракции позволяет оценить
выбранное разбиение частиц на фракции (тесты с использованием массива диатомей,
оценка правильности, влияние плотности и т. д.). Подсчет количества частиц может
быть осуществлен с использованием сцинтилляционного счетчика либо анализатора
изображений и структуры.
С помощью метода электронной микроскопии с применением ЭД-РСМА можно вы¬
полнить химические исследования; при этом в оптической микроскопии применяется
инфракрасное излучение. Такое оборудование обладает широким диапазоном возмож¬
ностей (Quantachrome, Zeiss, Leitz и т. д.) и позволяет определить процентное содержание
накопленной массы, гранулометрический состав, выраженный в форме числа, массы
и удельной поверхности (0,1-300 мкм).
74
Часть 1. Минералогический анализ почв
2.3.7. Методы, использующие электропроводные свойства
материалов
При прохождении через откалиброванное отверстие частица перемещает некоторый
объем электролита, что вызывает изменение электрического сопротивления раствора
(дифференциальная проводимость). Такие изменения сопротивления являются функ¬
цией объема. Подсчет количества частиц позволяет сгруппировать частицы по фракци¬
ям с использованием амплитудного дискриминатора.
Результаты в виде данных 16-канального накопителя представляются как массовые
проценты или количества частиц определенного размера; результаты могут быть пред¬
ставлены в форме графиков или таблиц.
Оборудование, в основе которого лежит этот принцип, представляет собой счетчи¬
ки, не учитывающие плотность, что может быть полезным при анализе почв, богатых
оксидами железа, имеющих высокую плотность (4—5), т. е. значительно превышающую
значение 2,65, используемое для седиментации при прямом действии силы тяжести.
Форма частиц оказывает большое влияние на точность измерений. Измерения могут
быть выполнены в диапазоне от 1 до 5000 мкм {Coulter), однако оптимальная область
измерения зависит от выбора подходящего отверстия. Для частиц размером менее 1 мкм
часто получают заниженные результаты. Подготовка образцов аналогична используемой
в эталонном методе (см. подразделы 2.2.1 и 2.2.2).
Использованная литература
Atterberg А (1912) Die mechanische Bodenanalyse und die klassifikation der mineral boden sechwedens.
Int. Mitt Bodenk, 2, 312-342.
Baize D (2000) Guide des analyses courantes enpedologie., INRA, France, 257 p.
Bouyoucos GS (1927) The hydrometer as a new method for mechanical analysis of soils. Soil Set, 23,343.
Bouyoucos GS (1935) A hydrometer method for making mechanical analysis of soils. Bull. Am. Ceram.
Soc, 14, 259.
Bouyoucos GS (1962) Hydrometer method improved for making particle size analysis of soils. Agron. J.,
54,464-465.
Casagrande A (1934) Die Araometer-methode zur Bestimmun der komverteilung von Boden und anderen
materialen. Springer J.
De Leenheer L and Van Hove J (1956) Werkwijze voor de mechanische analyse met de kettinghydrome-
ter. Rijksland Bouwhogeschool (Gand), XXI, 249-274.
De Leenheer L and Maes L (1952) Analyse granulon^trique avec l’hydromdtre к chame. Bull. Soc. Beige
de Giologic, 61, 138-164.
De Leenheer L, Van Ruymbeke M and Maes L (1955) L’analyse тёсашяие au moyen de l’hydrom£tre к
chaine. Silicates Indus trie Is., Tome XX, n° 6-7, 1-7.
Delaune M, Reiffsteck M and Feller C (1991) L’analyse granulometrique de sols et sёdiments к l’aide du
microgranulometre sёdigraph 5000 et comparaison avec la ntfthode к la pipette Robinson. Cahiers
ORSTOMser. Pedol, 26, 183-189.
Gee GW and Bauder JW (1986) Particle-size analysis. In Methods of Soil Analysis. Part 1 Physical and
Mineralogical Methods., Klute A. Ed. Chap. 15. American Society of Agronomy. Soil Sci. Soc. Am.,
383-411.
Gras R (1988) Physique du sol pour I'amenagement, Masson, Paris, 587 p.
Нёпт S (1976) Cours de physique du sol, vol. 1. Orstom-Editest, Braxelles, 159 p.
Jackson (1969) Soil Chemical Analysis -Advanced Course, 2nd ed. University of Wisconsin, Madison, WI.
Мёпаих S (1954) Contribution к l’etude de l’analyse granulometrique. Ann. Agro., I, 5-53, II, 149-205.
Pansu M, Gautheyrou J and Loyer JY (2001) Soil Analysis - Sampling, Instrumentation and Quality
Control, Balkema publishers, Lisse, Abington, Exton, Tokyo, 512 p.
Petard J (1993) Les methodes d’analyse. Tl Analyse de sols., Notes techniques laboratoires communs
d’analyse, Orstom, Noumea, Paris.
Глава 2. Анализ гранулометрического состава
75
Rouiller J, Burtin G and Souchier В (1972) La dispersion des sols dans Panalyse granulometrique.
Methode utilisant les resines echangeuses d’ions. Bull. ENSAIA, Nancy, France, 14, 183-204.
Stokes GG (1851) On the effect of the lateral friction of fluids on the motion of pendulums. Trans.
Cambridge Phil Soc, 9, 8-106.
Susini J (1978) Realisation d’un ensemble automatique de lavage des sables de l’analyse granulomS-
trique. Cah. ORSTOMSerie Pedol., 16, 339-344.
Tamm О (1922) Eine Methode zur Bestian on vag der anorganischen komponenten des Gelkomplexes in
Boden. Meddel. Staters Skogsfirsoksanst (Suede), 19, 385-404.
Van Ruymbeke M and De Leenheer L (1954) Etude comparative d’analyses granulometriques par
decantations successives et par Phydromfcre a chaine. Actes et C R. du Verne Congres International
de la Science du Sol (Leopoldville), 11, 322-328.
Vieillefon J (1979) Contribution h Pamelioration de Petude analytique des sols gypseux. Cah. ORSTOM
Sir. Pedol, XVII, 195-223.
X 31-107 (1983) Analyse granulometrique par sedimentation. Methode de la pipette. In Qualite des sols
3°ed., AFNOR, 357-371.
Дополнительная литература
Основные положения
Barth HG and Shao-Tang Sun (1991) Particle size analysis. Anal Chem., 63, 1R-10R.
Chamayou H and Legros JP (1989) Les bases physiques, chimiques et mineralogiques de la Science du
sol, Tech. Vivantes, ACCT Presses. Univ. de France, 593 p.
Guillet В and Rouiller J (1979) La granulometrie. In Pedologie, constituants et proprietes des sols,
Bonneau and Souchier ed., Masson, 317-321.
Johnston, Farina MPW and Lawrence JY (1987) Estimation of soil texture from sample density. Commun.
Soil Set Plant Anal, 18, 1173-1180.
Jones JL, Kay JJ, Park JJ and Bishop CK (1980) The determination of particle size distribution in soil.
A collaborative study. J. Sci. FoodAgric, 31, 724-729.
Loveland PJ and Whalley WR (1991) Particle size analysis. In Soil Analysis: Physical Methods., Smith
KA, Mullins CEJ ed., Dekker, New York 271-328.
Smith RB and Pratt DN (1984) The variability in soil particle size test results by various sub sampling
techniques. J. Soil Sci., 35,23-26.
Syvitski JPM (1991) Principles, methods and applications of particle size analysis. Cambridge Univ.
Press., 366 pages.
Предварительная обработка
Органические вещества
Douglas LA and Fiessinger F (1971) Degradation of clay minerals by H202 treatments to oxidize organic
mater. Clays Clay Miner., 19,67-68.
Fisher WR (1984) The oxidation of sol organic matter by KBrO for particle size determination. Commun.
Soil Sci. Plant Anal, 15, 1281-1284.
Harada Y and Inoko A (1977) The oxidation products formed from soil organic matter by hydrogen
peroxide treatment. Soil Sci. Plant Nutr., 23, 513-521.
Langeveld AD Van, Gaast SJ Van der and Eisma D (1978) A comparison of the effectiveness of eight
methods for the removal of organic matter from clay. Clays Clay Miner., 26, 361-364.
Lavkulich LM and Wiens JH (1970) Comparison of organic matter destruction by hydrogen peroxide
and sodium hypochlorite and its effects on selected mineral constituents. Soil Sci. Soc. Am. Proc, 34,
755-758.
Omueti JAI (1980) Sodium hypochlorite treatment for organic matter destruction in tropical soils of
Nigeria. Soil Sci. Soc. Am. J., 44, 878-880.
Sequi P and Aringhieri R (1977) Destruction of organic matter by hydrogen peroxide in the presence of
pyrophosphate and its effect on soil specific surface area. Soil Sci. Soc. Am. J., 41, 340-342.
76
Часть 1. Минералогический анализ почв
Visser SA and Caillier М (1988) Observations on the dispersion and aggregation of clays by humic
substances. I - Dispersive effects of humic acids. Geoderma, 42,331-337.
Vodyannitskii Yu N, Trakhin VT and Bagina OL (1989) The action of perhydral upon iron oxides in soil.
Dokuchzer soil Sci. Inst. (Moscou), 1,20-21.
Удаление органоминеральных компонентов
Harward ME, Theisen AA and Evans DD (1962) Effect of iron removal and dispersion methods on clay
mineral identification by X-Ray difraction. Soil Sci. Soc. Am. Proc, 26, 535-541.
Mehra OP and Jackson ML (1960) Iron oxide removal from soils and clays by a dithiomite-citrate system
buffered with sodium bicarbonate. In Clays and Clay Minerals. Proc. Seventh Conf. Natl Acad. Sci.
Natl Res. Counc. Pub., 237-317.
Выделение растворимых солей - гипса
Rengasamy Р (1983) Clay dispersion in relation to changes in the electrolyte composition of dialysed red-
brown earth. J. Soil Sci., 34, 723-732.
Rivers ED, Hallmark CT, West LT and Drees LR (1982) A technique for rapid removal of gypsum from
soil samples. Soil Sci. Soc. Am. J., 46, 1338-1340.
Суспендирование - диспергирование - флокуляция
Balli P (1965) Criteres de la qualite de la suspension en vue de Г analyse granulometrique. Science du
sol, 1,15.
Bartoli F, Burtin G and Herbillon AJ (1991) Disaggregation and clay dispersion of oxisols: Na Resin, a
recommended methodology. Geoderma, 49, 301-317.
Brewster GR (1980) Effects of chemical pretreatment on X-Ray powder diffraction characteristics of clay
minerals derived from volcanic ash. Clays Clay Miner., 28, 303-310.
Colmet-Daage F, Gautheyrou J, Gautheyrou M, Kimpe C de (1972) Dispersion et 6tude des fractions fines
des sols к allophane des Antilles et d’Antique latine. lere partie: Techniques de dispersion. Cah.
Orstom, Ser. Pedol, Vol. X(2), 169-191.
Demolon A and Bastisse E (1935) Sur la dispersion des colloi’des aigil eux. Applications a leur extraction.
Annales Agronomiques, 1-15 Dong A, Chesters G and Simsiman GV (1983) Soil dispersibility. Soil
Sci, 136, 208-212.
Egashira К (1981) Floculation of clay suspensions separated from soils of different soil type. Soil Sci.
Plant Nutr, 27, 281-287.
Forsyth P, Marcelja S, Mitchell DJ and Ninham BW (1978) Stability of clay dispersions. In Modidication
of Soil Structure., Emerson, Bond, Dexter Ed. Wiley, New York. 2,17-25.
Goldberg S and Forster HS (1989) Floculation of reference clays and arid soil clays as affected by
electrolyte concentration, exchangeable section percentage, sodium adsorption ratio, pH and clay
mineralogy. Annual Meeting- Clay Minerals Society, 26, 35.
Gupta RK, Bhumbla DK and Abrol IP (1984) Effect of sodicity, pH, organic matter and calcium carbonate
on the dispersion behavior of soils. Soil Sci., 137, 245-251.
Keren R (1991) Adsorbed sodium fraction’s effect on rheology of montmorillinite-kaolinite suspensions.
Soil Sci. Soc. Am. J., 55, 376-379.
Manfredini T, Pellacani GC, Pozzi P and Corradi AB (1990) Monomeric and oligomeric phosphates as
deflocculants of concentrated aqueous clay suspensions. Appl. Clay Sci, 5,193-201.
Miller WP, Frenkel H and Newman KD (1990) Floculation concentration and sodium/calcium exchange
of kaolinitic soil clays. Soil Sci. Soc. Am. J., 54,346-351.
Ohtsubo M and Ibaraki M (1991) Particle-size characterzation of floes and sedimentation volume in
electrolyte clay suspensions. Appl Clay Sci., 6, 181-194.
Oreshkin NG (1979) Device for tating suspension samples for the particle-size analysis of soils. Soviet
Soil Sci, 4, 136-138.
Reddy SR and Fogler HS (1981) Emulsion stability: determination from turbidity. Colloid Interface Sci,
79,101-104.
77
Глава 2. Анализ гранулометрического состава
Reddy SR, Fogler HS (1981) Emulsion stability: delineation of different particle loss mechanisms.
Colloid Interface Sci, 79, 105-113.
Robinson GW (1933) The dispersion of soils in mechanical analysis. Bur. Soil Sci. Tech. Commun., 26,
27-28.
Shaviv A, Ravina I and Zaslavsky P (1988) Floculation of clay suspensions by an anionic soil conditioner.
Appl. Clay Sci., 3, 193-203.
Ультразвуковое диспергирование
Arastamyants YEI (1990) Optimizing the ultrasonic preparation of soils for particle-size analysis.
Pockvovedeniye, 12, 55-68.
Busacca AJ, Aniku JR and Singer MJ (1984) Dispersion of soils by an ultrasonic method that eliminates
probe contact. Soil Sci. Soc. Am. J., 48, 1125-1129.
Edwards AP and Bremner JM (1967) Dispersion of soil particules by sonic vibrations. J. Soil Sci., 18,1.
Feller C, Burtin G and Herbillon A (1991) Utilisation des resines sodiques et des ultra-sons dans le
fractionnement granulometrique de la matiere organique des sols. Interet et limites. Science du sol,
29, 77-93.
Gregorich EG, Kachandski RG and Voroney RP (1988) Ultrasonic dispersion of aggregates: distribution
of organic matter in size fractions. Can. J. Soil Sci., 68, 395-403.
Hinds AA and Lowe LE (1980) Dispersion and dissolution effects during ultrasonic dispersion of gleysolic
soils in water and in electrolytes. Can. J. Soil Sci, 60, 329-335.
Hinds AA and Lowe LE (1980) The use of an ultrasonic probe in soil dispersion and in the bulk isolation
of organo-mineral complexes. Can. J. Soil Set, 60,389-392.
Ilnicki P and Matelska U (1984) Ultrasound application for dispersion of soil samples for particle size
analysis. Roezniki Gleboznaweze, 35, 15-24.
Mikhail EH and Briner GP (1978) Routine particle size analysis of soils using sodium hypochlorite and
ultrasonic dispersion. Aust. J. Soil Res., 16, 241-244.
Minkin MB, Mulyar IA and Mulyar AI (1985) An ultrasonic method of analysing of water extracts from
soils. Pockvovedeniye, 3, 136-140.
Moen DE and Richardson JL (1984) Ultrasonic dispersion of soil aggregates stabilized by polyvinyl
alcohol and T 403-glyoxal polymers. Soil Sci. Soc. Am. J., 48, 628-631.
Morra MJ, Blank RR, Freeborn LL and Shafil В (1991) Size fractionation of soil organo-mineral
complexes using ultrasonic dispersion. Soil Sci, 4, 294-303.
Schulze DG and Dixon JB (1979) High gradient enzymatic separation of iron oxydes and other magnetic
minerals from soils clays. Soil Sci. Soc. Am. J., 43, 793-799.
Пипеточный метод
Andreasen AHM and Andersen J (1930) Etude de rinfluence de la dilution sur les resultats de l’analyse
granulometrique par sedimentation. KolloidZ., 50,217.
Bloom PR, Meter К and Cram JR (1985) Titration method for determination of clay-sized carbonates.
Soil Sci. Soc. Am. J., 49, 1070-1073.
Godse NG and Sannigrahi AK (1988) Comparative study on methods of particle-size analysis for vertisols.
J. Indian Soc. Soil Sci., 36, 780-783.
Indorante SJ, Follmer LR, Hammer RD and Koenig PG (1990) Particle-size analysis by a modified pipette
procedure. Soil Sci. Soc. Am. J., 54, 560-563.
Krumbein WC (1935) A time chart for mechanical analyses by the pipette method. J. Sediment. Petrol,
5, 93-95.
Miller WP and Miller DM (1987) A micro-pipette method for soil mechanical analysis. Commun. Soil Sci.
Plant Anal, 18, 1-15.
Oreshkin NG (1979) Device for taking suspension samples for the particle-size analysis of soils. Soviet
Soil Sci. (Pockvovedeniye), 4, 136-138.
Richter M and Svartz H (1984) Analisis granulometrico de suelos en escala reducida. Ciencia del suelo,
2, 1—8.
Shetron SG and Trettin CC (1984) Influence of mine tailing particle density on pipette procedures. Soil
Sci. Soc. Am. J., 48, 418-420.
78
Часть 1. Минералогический анализ почв
Ареометрический метод
American Society for Testing and Materials (1972) Standard test methode for particle-size analysis of
Soils - D 422-463. Annual Book ofASTM, 1985.
Barthokur NN (1986) Clay fraction determinations with Beta-ray gauge. Commun. Soil Sci. Plant Anal,
17, 533-545.
Fontes LEF (1982) A new cylinder for sedimentation of soil suspension in the determination of the clay
fraction by the hydrometer method. Revista brasileira de Ciencia do Solo, 6, 152-154.
Gee GW and Bauder JW (1979) Particle size analysis by hydrometer, a simplified method for routine
textural analysis and a sensivity test of measurement parameters. Soil ScL Soc. Am. J., 43, 1004-
1007.
Johnson JE, Bowles JA and Knuteson JA (1985) Comparison of pretreatments and dispersants on clay
determination by the hydrometer method. Commun. Soil Sci. Plant Anal 16, 1029-1037.
Sur HS and Kvkal SS (1992) A modified hydrometer procedure for particle size analysis. Soil Sci, 153,
1-4.
Инструментальные методы анализа
Arastamyants YEI (1992) Instrumental methods for determining the particle-size composition of soils.
Scr. Tech., 101-117.
Barth, HG (1984) Modern Methods of Particle Size Analysis., Wiley, New York, 209 pages.
Cooper LR, Haverland RL, Hendricks DM and Knisel WG (1984) Microtrac particle-size analyzer: an
alternative particle-size determination method for sediment and soil. Soil Sci., 132,138-146.
Devyatykh GG, Karpov YU A, Krylov VA and Lazukina OP (1987) Laser-ultra microscopic method of
determining suspended particles in high-parity liquids. Talanta, 34, 133-139.
Hendrix WP and Orr C (1970) Automate sedimentation size analysis instrument. Particle Size Analysis,
133-146.
Hutton JT (1955) A method of particle size analysis of soils (balance de Plummet). CSIRO, Report,
11/55.
Karsten JHM and Kotze WAG (1984) Soil particle analysis with the gamma alternation technique.
Commun. Soil Sci. Plant Anal, 15, 731-739.
Kirkland JJ and Yau WW (1983) Simultaneaous determination of particle size and density by sedimentation
field flow fractionation (FFF). Anal Chem., 55, 2165-2170.
Kirkland JJ, Rementer SW and Yav WW (1981) Time-delayed exponential field-programmed sedimentation
field flow fractionation for particle-size distribution analysis. Anal. Chem., 53, 1730-1736.
Marshall TI (1956) APlummett Balance for measuring the size distribution of soil particles. Aust. J. Appl.
Sci., 7, 142-147.
Me Connel ML (1981) Particle size determination by quasielastic light scattering. Anal. Chem., 53,1007-
1018.
Novich BE and Ring ТА (1984) Colloid stability of clays using photron correlation spectroscopy. Clays
Clay Miner., 32, 400-406.
Oakley DM and Jennings BR (1982) Clay particle sizing by electrically induced birefringence. Clay
Miner., 17, 313-325.
Pennington KL and Lewis GC (1979) A comparison of electronic and pipet methods for mechanical
analysis of soils. Soil Sci, 28, 280-284.
Rybina W (1979) Use of conductimetry for the determination of the particle-size composition of soils.
Pochvovedeniye, 7, 134-138.
Salbu B, Bjomstad HE, Linstrom NS, Lydersen E (1985) Size fractionation techniques in the determination
of elements associated with particulate or colloidal material in natural fresh waters. Talanta, 32,
907-913.
Svarovsky L and Allen T (1970) Performance of a new X-Ray sedimentometer. Particle Size Analysis,
147-157.
Yang КС and Hogg R (1979) Estimation of particle size distributions from turbidimetric measurements.
Anal. Chem., 51, 758-763.
Yonker CR, Jones HK and Robertson DM (1987) Non aqueous sedimentation field flow fractionation.
Anal. Chem., 59, 2574-2579.
Глава 3. Фракционирование коллоидных систем
3.1. Введение
Для объяснения процессов преобразования и трансформации минералов большое зна¬
чение имеет идентификация и количественный анализ наиболее мелкодисперсных
фракций почвы, поскольку подобные фракции оказывают непосредственное влияние
на процесс почвообразования и на возможное плодородие почв.
Природа и свойства подобных частиц вызывают определенный интерес у агрономов
(химия и физика почвы: группа механического состава почвы, плодородие, система пор,
возможность накопления влаги, когезия, стойкость к растрескиванию и т. д.), почво¬
ведов (почвообразование, характеризация и функционирование почвы, литологическая
природа, продукты преобразований и т. д.), геологов кварцевого цикла (седиментология:
происхождение переносимых воздушными потоками отложений в морях, озерах, типо¬
логия вулканического пепла и тяжелых пород и т. д.), минералогов и геохимиков (анализ
изменений в почве, возможные изменения запасов полезных ископаемых, происхожде¬
ние и природа минералов и т. д.).
Большинство методов инструментального анализа, используемых для определения
гранулометрического состава почв (см. гл. 2), не позволяют произвести разделение
определяемых фракций, однако применяют отдельные методы анализа, основанные
на осаждении, предполагающие повторное применение песчаных, иловых и глинистых
фракций, до тех пор, пока они не загрязнятся диспергирующими реагентами. На прак¬
тике обычные методы, основанные на гравитационном разделении, ограничены разме¬
рами фракций приблизительно до 1 мкм. При определенных условиях для получения на-
норазмерной фракции частиц можно использовать методы ультрацентрифугирования.
Фракции размером частиц менее 0,5 мкм практически не содержат кварца или пер¬
вичных минералов. Фракции размером частиц менее 0,2 мкм используются для более
достоверного анализа горизонтов, подвергшихся аргиллизации. Этот порог, предло¬
женный приблизительно в 1931 г., в те времена рассматривался как типичная граница
«коллоидного состояния», поскольку после удаления оксидов и гидроксидов фракция
частиц представляется однородным химическим составом, подобно монодисперсной
системе, т. е. так же обладает обменной емкостью и структурным составом в виде частиц
размером 0,2 мкм, 0,1 мкм и 0,05 мкм.
Если окончательной целью фракционирования частиц является физический или хи¬
мический анализ, то различные обработки для поддержания суспензии очень мелких ча¬
стиц в стабильном состоянии могут существенно отличаться от методов, используемых
для гранулометрического анализа, поскольку вторичные продукты (в частности, глины
и оксиды) не могут быть существенно модифицированы. В некоторых случаях можно
просто перевести мелкие фракции частиц в суспензированное состояние обработкой
ультразвуком, а также использовать ионообменные смолы для снижения степени на¬
сыщения. Для этих целей не следует использовать диспергирующие реагенты гексамета-
фосфатного или пирофосфатного типа (см. гл. 2).
Требования закона Стокса распространяются и на процесс центрифугирования,
включая сложности, связанные с частицами, скорость которых изменяется. После ко¬
личественного разделения всех фракций могут быть уточнены их химические и физи¬
ческие свойства, а также откорректированы кривые распределения для всего множества
частиц.
80
Часть 1. Минералогический анализ почв
3.2. Фракционирование путем непрерывного
центрифугирования
3.2.1. Основные положения
После обработки, необходимой для отделения первичных частиц (см. гл. 2), для после¬
дующего анализа образец материала с помощью незагрязняющего диспергирующего
реагента переводится в суспензированное состояние. Мелкодисперсные фракции ма¬
териала (менее 2 мкм) сначала отделяют от грубых фракций, несколько раз сифонируя
раствор, с учетом времени осаждения илистых частиц. Для количественного извлечения
достаточно осуществить сифонирование пять или шесть раз.
Из-за объемов, с которыми имеют дело, фракционирование мелкодисперсных частиц
осуществляют в центрифуге с непрерывной подачей восходящим потоком, исполненной
в виде вертикальной трубки, способной вращаться с ускорением до 52 000 g (рис. 3.1, а).
Внутрь трубки помещается трансферная бумага, что позволяет собирать частицы, осаж¬
дающиеся на стенках (рис. 3.1, Ь). Очищенная вода собирается в верхней части емкости
и сливается через одинарный или двойной канал (в зависимости от модели).
Суспензия заполняет трубку не полностью и при этом образует концентрическое
кольцо, создавая в центре цилиндр из воздуха (радиусом /?,) (рис. 3.1, Ь). Толщина слоя
суспензии в таком устройстве составляет Rv Частицы постепенно осаждаются на
стенках камеры, в зависимости от их плотности и диаметра. Они распределяются по па¬
раболической траектории, начиная от точки ввода суспензии.
Рис. 3.1. Система непрерывного ультрацентрифугирования (а), в которой используется сжатый
воздух, вращающаяся с ускорением 45 000 д(1 — впускное отверстие для инжекции материала; 2,
3 — выходные каналы для фильтратов; 4 — тахометр; 5 — соединение с ротором; 6 — ротор цен¬
трифуги (камера). А также ротор центрифуги (камера) (Ь) с трансферной бумагой для извлечения
твердых частиц (распределение каналов подачи в соответствии с методикой «Фракционирование
в четыре этапа»)
Глава 3- Фракционирование коллоидных систем
81
Центробежные силы, приложенные в радиальном направлении, сочетаются с вер¬
тикально направленной силой тяжести. Частица определенного диаметра и плотности
будет осаждаться под действием результирующего вектора сил. При осуществлении цен¬
трифугирования можно контролировать вязкость раствора за счет изменения темпера¬
туры, давления, создаваемого высокими скоростями и, возможно, наличием тиксотроп¬
ных материалов. Для почв, содержащих аллофаны, может наблюдаться флокуляция,
если дисперсионная среда является неоднородной в течение повторяющихся циклов
экстракции, или из-за потери части поглощенной воды (воды набухания). Следует от¬
метить, что волокна имоголита, слои слюды, пробы диатомей и т. д. не подчиняются
закону Стокса и разделяются случайным образом.
Распределение компонентов перпендикулярно оси может быть относительно легко
проанализировано с помощью закона Стокса (см. раздел 3.2.2), однако следует отме¬
тить, что при центрифугировании скорость осаждения частичек является непостоян¬
ной, что объясняется изменением величины центробежных сил, которые определяются
диаметром ротора. Вертикальная составляющая должна рассчитываться в зависимости
от скорости потока суспензии, которая, в свою очередь, зависит от скорости подачи су¬
спензии, от диаметра канала для ввода, а также от диаметра кольцевого канала.
На практике для верхней и нижней части камеры нельзя строго применять формулы
расчета для стандартной центрифуги компании Sharpies из-за турбулентности в этих об¬
ластях (канал ввода и дефлектора в нижней части камеры и кольцевой канал слива —
в верхней части). Слишком много осадка также может изменить радиус нижней части
камеры. Однако высота осаждения слоя может быть определена с достаточно высокой
точностью, если следовать строгой процедуре. Предпочтительнее при каждой обработке
собирать относительно небольшое количество осадка (с массой не более 10 г).
При определенных условиях испытаний (число оборотов в минуту, скорость пото¬
ка и т. д.) мельчайшие частицы могут проходить сквозь седиментационный ротор, по¬
тому что время прохождения при данной величине центробежной силы недостаточно
для переноса их к стенке ротора. Однако при изменении хотя бы одного из параметров
испытания, например, при уменьшении скорости потока за счет изменения диаметра
трубки ввода, подобные частички также могут быть отобраны. При низкой скорости по¬
тока разделение отличается большей эффективностью, чем при высокой. Для повыше¬
ния эффективности разделения лучше использовать трубки небольшого диаметра и не
слишком высокую загрузку. Для простого отделения (для очистки методом концентри¬
рования) следует использовать трубку диаметром 2 мм и более высокую загрузку (на¬
пример, 75 см). Слив все равно может содержать мелкие частицы, в зависимости от при¬
меняемой скорости вращения камеры. Система позволяет направлять отфильтрованную
жидкость снова с меньшей скоростью потока, и собирать твердые фракции на новой
трансферной бумаге, размещенной в роторе центрифуги.
Важно подобрать скорость вращения центрифуги и потока, которые позволяют по¬
лучать неравномерные осадки для удовлетворительной классификации частиц. При
расчетах сложно учесть все переменные, и прогноз будет ненадежным, если средняя
плотность отличается от общепринятой для почв величины — 2,65. Действительно, при
использовании смеси минералов различной плотности будут микрочастицы, которые
проявляют индивидуальные свойства в суспензии, и на любом уровне могут находиться
очень различные частицы одного диаметра (табл. 3.1).
В свое время казалось, что вполне точными и быстрыми являются графические ме¬
тоды. Саундерс (Saunders, 1948) изучил номографическое представление для непрерыв¬
ного ультрацентрифугирования (1936-1940), в результате чего ему удалось определить,
82 Часть 1. Минералогический анализ почв
Таблица 3.1.
Осаждение частичек различных минералов при непрерывном
ультрацентрифугировании (скорость потока — 730 мл/мин, ускорение
вращения — 20 000 g, вязкость —
о
Минерал
Плотность
Диаметр частиц, которые осаждаются на
высоте 10 см в роторе центрифуги (мкм)
Опал
2,10
360
Гиббсит
2,40
320
Кварц
2,65 (среднее значение,
используемое для почвы)
280
Гематит
5,26
115
что некоторые параметры в уравнении Хаузера (Hauser) и Линна (Lynn) (см. раздел 3.2.2)
остаются постоянными для данной процедуры, что позволяет, таким образом, упростить
подход и перейти к номографическому представлению с пятью переменными, которые
могут быть расширены до шести переменных при использовании метода Дэвиса (Davis,
1943).
Таким образом, появилась возможность быстро определять высоту осаждения эле¬
ментарных частиц различной плотности, но одинакового диаметра.
3.2.2. Теоретические основы
Метод Хаузера и Линна (Hauser и Lynn, 1940) для расчета размера частиц является одним
из наиболее мощных инструментов, который можно использовать при ультрацентрифу¬
гировании с непрерывной подачей суспензии. Уравнение позволяет выразить расстоя¬
ние по вертикали (К, см) до уровня осаждения частиц данного размера, которое измеря¬
ется начиная от основания ротора центрифуги:
ус \ЩОц
(3.1)
где
(3.0
R2 — расстояние от оси вращения до стенки камеры ротора, см; Rt — расстояние от оси
вращения до поверхности жидкости в роторе, см; Х0 — расстояние от оси вращения, на
котором заданная частица должна начинать осаждаться на стенке камеры ротора, см;
К{ — функция, зависящая от конструкции ротора центрифуги, см-2,
.
(3/4Х +(1/4)1$-/^2-1$1п(Ц//д ’
(3.2)
Q — скорость подачи жидкости (скорость потока суспензии, мл/с); г| — вязкость дис¬
персионной среды, пуаз; D — диаметр сферы, эквивалентной частицам, которые осаж¬
даются на высоте К, см; оз — угловая скорость вращения, рад/с; 5р — разница в плотно¬
сти диспергируемых частиц (табл. 3.2) и дисперсионной среды, г/мл.
Глава 3. Фракционирование коллоидных систем
83
Таблица 3.2. Удельная плотность некоторых минералов (среднее значение плотности,
используемое для почвы, составляет 2,65)
Минералы
Плотность
А1
Бемит АЮ(ОН)
3,07
Диаспор АЮ(ОН)
3,40
Байерит А1(ОН)3
2,53
Гиббсит А1(ОН)3
2,40
Нордстрандит А1(ОН),
2,43
Корунд AljOj
4,10
Акдалаит 4А1203, Н20
3,68
Боксит А1203, 2Н20
2,55
Fe
Акаганеит Fe3+0(0H, Cl)
3,55
Гетит FeO(OH)
4,0-4,4
Лепидокрокит FeO(OH)
3,85
ГЬматит Fe203
5,26
Маггемит Fe203
5,10
Магнетит Fe304 (Fe^Fe^O^
5,17
Ильменит Fe0-T102
4,44-4,90
Пирит FeS2
5,02
Мп
Манганозит MnO .
5,36
Пиролюзит Mn02
5,06
Рамсделит Mn02
4,50
Манганит MnO(OH)
4,33
Файткнехтит MnO(OH)
3,80
Цюутит MnO(OH)
4,15
Пирохлорит Mn(OH)2
3,25
Нсутит Mn+2Mn+402(0H)2
4,50
Г&усманит Мп+2Мп2+304
4,84
Глины
Каолинит
2,60
1&ллуазит
2,10
Диккит
2,60
Монтмориллонит
2-3,00
Нонтронит
2-3,00
Бейделлит
2-3,00
Слюды
Биотит
3,00
Флогопит
2,80
Глауконит
2,60
Мусковит
2,80
Иллит
2,6-2,9
Полевые
Белый полевой шпат Na20, А1203,6Si02
2,61-2,64
шпаты
Андезин CaO, Na20 А1203,4Si02
2,65-2,69
Олигоклаз NaAlSi308 +CaAl2Si208
2,62-2,67
Ортоклаз KAlSi308
' 2,56
84 Часть 1. Минералогический анализ почв
Таблица 3.2 (окончание)
Минералы
Плотность
Si
Коэсит Si02
2,93
Кристобалит Si02
2,33
Кварц Si02
2,65
Тридимит Si02
2,26
Силгидрит 3Si02, Н20
2,14
Опал Si02, пН20
2,10
Са
Кальцит СаСОэ
2,7-2,9
Арагонит СаСОэ
2,9
Тйпс CaS04, 2Н20
2,3
При стандартных условиях (скорости потока и диапазоне размеров частиц) уравне¬
ние Хозера и Линна зависит от значений величин Х0 и D. Для частиц данного диаметра
уравнение зависит только от Х0, что позволяет построить кривую зависимости Сот Y На
основании этого Саундерс (Saunders, 1948) разработал систему монограмм, которая по¬
зволяет с достаточной точностью рассчитать значение эквивалентного диаметра частиц,
осаждающихся в камере.
Определяя постоянную
, 18*
из (3.1) получаем:
Y Ar\Q
С DW 6р‘ (3'3)
Это уравнение может быть преобразовано с использованием «номографического мето¬
да» Дэвиса (Davis, 1943), что позволяет получить соотношение Y/Сддя каждого значения
Y. Если значение т\ выражено в сантипуазах, Q в мл/мин, D в нм, ш в об/мин, 5р в г/мл,
Ув см и С в см2, уравнение (3.3) можно представить в виде:
— = 152х1012
С ’ D2 coV 3‘3
Это выражение может быть сокращено до четырех уравнений тремя переменными
и тремя параметрами а, р и у:
log а =log г| - log( У/С);
log р =log бр - 2 log ю;
logy=loga + logP;
2 log ZMog у + log Q.
Номографический метод позволяет получить решение указанной системы постро¬
ением четырех прямых, которые отображают решение каждого из этих уравнений
(рис. 3.2). Постоянная А вводится в начале исходя из размерности численного решения
уравнения (3.3'). Например, для трубки для центрифугирования, для которой значение
= 2,175 см и *2 = 0,735 см, величина постоянной А составляет 2,44 • 1012. Окончательная
формула для непосредственного расчета примет вид:
D= 1,93 * 1012 (r)Q/(<s>2bp(Y/Qy*
со значением Y/С на высоте Y, и единицах измерения (3,3').
Рис. 3.2. Правила пользования номограммой: (1) соедините значения на линиях 1 -2, отметив точку пересечения на А; (2) соедините значения
на линиях 5-6, отметив точку пересечения на С; (3) соедините отметки на АС, отметив точку пересечения на В; (4) соедините значения на
линиях В-3, точка пересечения на 4 — значение эквивалентного диаметра частиц, осажденных на расстоянии (Y) в роторе центрифуги,
выраженное в нанометрах
оо
1Л
Глава 3. Фракционирование коллоидных систем
86
Часть 1. Минералогический анализ почв
3.2.3. Оборудование и реактивы
Оборудование
• Стандартная ультрацентрифуга Sharpies Т\ с непрерывной циркуляцией и турби¬
ной, оснащенной ротором из нержавеющей стали 8RY диаметром 44 мм,
• Бутылка из пирексного стекла объемом 6 л, с широкой горловиной и пробкой.
• Пробка со стеклянной трубкой внутренним диаметром 10 мм, обрезанная наиско¬
сок (для ввода суспензии).
• Конические щипцы на 12 см с плоскими браншами (для извлечения трансферной
бумаги).
• Маленький пластмассовый шпатель.
• Скальпель (для извлечения трансферной бумаги с рукояткой).
• Полимерная трансферная бумага с шероховатой внутренней поверхностью
(Integraph, Invar 15 мкм, Kodatrace, Chronaflex или Tymek Dupont de Nemours), листы
размером 204x150 мм; толщина такой подложки должна быть равномерной, листы
должны иметь постоянное значение соотношения массы к единице поверхности,
должны быть устойчивыми к действию воды, иметь диффузный спектр рентгенов¬
ской дифракции либо четкие пики, расположенные за пределами областей изме¬
рения образца (рис. 3.3).
• Весы с точностью 0,1 мг.
• Комплект соответствующих небольших контейнеров и микрофлаконов (пластико¬
вых, одноразовых, рис. 3.4).
• Алюминиевая подложка для проведения сканирующей электронной микроско¬
пии.
Реактивы
См. гл. 2.
Рис. 3.3. Дифрактограмма: пунктирная линия — используется только подложка Tymek; сплошная
линия — каолинит на подложке Tymek (достаточно толстослойная для предотвращения загрязне¬
ния)
Глава 3. Фракционирование коллоидных систем
87
Подготовка решеток
для ПЭМ и СПЭМ
Порошок для РД
Таблетирование для
ИК-спектроскопии
СЭМ + микропроба
Ориентированная
РД
Полный анализ
порошкообразных
или хлопьевидных
глинистых частиц
Сухой Влажный
Рис. 3.4. Пример системы обработки мелкодисперсных фракций, разделенных на трансфер*
ной бумаге (рис. 3.1): 1 — перегруппировка на ПЭМ-подложке; 2 — только андисоли-гистозоли;
3 — микроконтейнер, микрофлаконы
3.2.4. Методика
Стандартное непрерывное ультрацентрифугирование
• Выбирают диаметр канала инжектора, диаметр кольцевого отвода и высоту h уста¬
навливают на уровне X(рис. 3.1).
• Помещают в ротор центрифуги взвешенную трансферную бумагу с изображенной
на задней стороне схемой разделения (масса трансферной бумаги Р0 позволяет рас¬
считать значение масс Рор Р02, /^3соответствующих областей А, В, Сна рис. 3.1).
• Подвешивают ротор центрифуги на якорь мотора и под сливным каналом помеща¬
ют контейнер для сбора жидкости.
• Заполняют раструб центрифуги до постоянного уровня Xтем же диспергирующим
веществом, которое вводилось в образец, не допуская попадания воздуха в подво¬
дящую трубку.
• Перемешивают содержимое бутылки, наполненной глинистой суспензией.
• Отбирают аликвоту раствора объемом 2 мл (для решетки ПЭМ).
• Переворачивают бутылку над воронкой; включают центрифугу на заданной ско¬
рости.
• Устанавливают канал ввода под камерой ротора центрифуги и открывают входной
кран.
После того как вся жидкость будет удалена, добавляют 200 мл дисперсионной жид¬
кости, чтобы удалить все мешающие взвешенные частицы в роторе центрифуги (при¬
близительно 150 мл). Устанавливают максимальную скорость вращения центрифуги —
52 000 g на 2—3 мин для укрепления осадка.
88
Часть 1. Минералогический анализ почв
Отсоединяют впускную трубку и выключают центрифугу (собирают жидкий остаток
в трубке в кристаллизатор и выливают ее в том случае, если она является прозрачной).
Аккуратно удаляют трансферную бумагу, держа ротор под наклоном, чтобы предот¬
вратить загрязнение верхней части ротора крупнодисперсными частицами. Распрямля¬
ют бумагу на плоской поверхности. Выделяют любые следовые количества осадка на
дефлекторе, добавляют на нижнюю часть трансферной бумаги.
Высушивают при комнатной температуре (при необходимости, перед высушиванием,
шпателем восстанавливают положение влажной глины в соответствующей зоне транс¬
ферной бумаги).
Взвешивают трансферную бумагу и высушенную глину: Рг Масса глинистого осадка
составляет Рх — Р0.
Обрезают трансферную бумагу, следуя разметке на обороте. Это позволяет отдельно
взвесить зоны А, В, С (рис. 3.1), т. е. определить PiV Plv Р1У
Непрерывное разделение на фракции коллоидных частиц
Сложный и длительный метод Хозера и Линна позволяет выделить из суспензии фрак¬
ции мелкодисперсных частиц (рис. 3.5) и получить кумулятивные кривые распределе¬
ния частиц по их размерам, используя оборудование для автоматического измерения
размеров частиц (см. гл. 2). Такой тип оборудования предусматривает две скорости цен¬
трифугирования и семь различных потоков, т. е. 11 вариантов настроек центрифуги для
группирования частиц размерами от 1 мкм до 24 нм в центрифуге непрерывного дей¬
ствия с турбинным приводом фирмы Sharpies.
Такой метод не используют для рутинных задач, поскольку он занимает слишком
много времени, и для хрупких образцов, которые сложно поддерживать в суспендиро¬
ванном состоянии, как например, почвы с аллофаном.
Однако его успешно применяют в металлогении для идентификации обогащенных
фракций (выделяемых ситами). Суспензии должны быть разбавлены для достижения
фракционирования без грубого воздействия механизмов.
Фракционирование в четыре этапа
Метод Гатеруа и Гатеруа (Gautheyrou и Gautheyrou, 1967) основан на уравнениях Хойзера
и Линна и номографической системе Сондерса (см. раздел 3.2.2). Трансферная бума¬
га, изображенная на рис. 3.1, Ъ, является одним из примеров разметки, отвечающей по¬
требностям минералогического анализа; возможны и альтернативные схемы, если учи¬
тывать, что осаждающиеся в горизонтальных плоскостях частицы одинаковы, а состав
твердой фазы изменяется снизу вверх.
Зоны А, Д С (рис. 3.1 и 3.4) обеспечивают разделение фаз по размеру частиц, значе¬
ния которых зависят от использованной методики (такие фракции используются для
химических и физических исследований). Зоны А9 и С (рис. 3.1 и 3.4) включают фрак¬
ции, которые могут быть выделены для проведения рентгеновской дифракции: как для
порошковых рентгенограмм (после измельчения), так и (без предварительной обработ¬
ки) для изучения ориентированных образцов размером 24x24 мм2 (см. гл. 4). После фик¬
сации частиц углеродным лаком на подходящей подложке площадью 5x5 мм2 (см. гл. 8)
может быть проведен анализ методом электронной сканирующей микроскопии ЭД-РС
с микроанализом.
После разбавления и соответствующей подготовки решеток образец каждой суспен¬
зии объемом 1 мл исследуется методом просвечивающей электронной микроскопии (см.
гл. 8).
Глава 3. Фракционирование коллоидных систем
89
о>
о
8
Ю
сз>
о
0
S
X
2
а
Рис. 3.5. Схема фракционирования методом непрерывного фракционирования коллоидных
частиц
Параметры
Загрузка: h = 75 см (рис. 3.1); температура — 20 °С; трубка диаметром 2 мм; скорость
потока — 730 мл/мин; время переноса в ротор центрифуги — 15 с; средняя плотность —
2,65; кольцевой слив — 0.
Загрузка, скорость потока, температура не изменяются; изменяется только скорость
вращения центрифуги при каждом анализе: 6000, 10 000, 25 000, 50 000 g (50 000 g, если
образец содержит очень мелкодисперсные частицы, при этом необходимо использовать
трубку диаметром 1 мм, соответствующую скорости потока 160 мл/мин, для фиксации
всех очень мелкодисперсных фаз).
90
Часть 1. Минералогический анализ почв
Фракция 2~1 мкм
Эта фракция отделяется при скорости центрифугирования 6000 g и выделяется на первой
трансферной бумаге (ТБ); масса только ТБ — Р01; ТБ + осадок — Рп; масса глинистых
частиц 2-1 мкм Рсх = (Рп- Р01), выражена в % относительно исходной массы почвы.
Фракция 1~0,5 мкм
Выделяется на второй ТБ при скорости центрифугирования 10 000 g: Рс2.
Фракция 0,5-0,2 мкм
На третьей ТБ при скорости центрифугирования 25 000 g: Рсг
Фракция 0,2-0,05 мкм
На четвертой ТБ при скорости центрифугирования 50 000 g и очень низкой скорости по¬
тока в 160 мл/мин, диаметр трубки 1 мм: Рс4.
Глинистые частицы < 0,05 мкм
Такие очень мелкодисперсные глинистые частицы содержатся в последней порции
сливных вод и могут быть выделены флокуляцией. Такая фракция, обычно прозрачного
экстракта, может быть выброшена, поскольку содержит большую часть остатков приме¬
сей реагентов, используемых при первичной обработке глин. Для анализа таких частиц
может быть использован метод электронной микроскопии, однако концентрирование
испарением может привести к гидролизу и преобразованию веществ.
Примечания
Если осадок слишком толстый, в глинах с высоким коэффициентом усадки может про¬
исходить беспорядочное растрескивание, что делает невозможным вырезание образцов
размером 5x5 мм2 для исследования ориентированных частиц методом РД. В этом слу¬
чае квадратные образцы не используются, вместо этого осколки кусочков глины закреп¬
ляют на подложке углеродным лаком.
В некоторых образцах почв практически отсутствуют мелкодисперсные фракции.
В таком случае можно остановиться на втором или третьем центрифугировании.
Если основные различия наблюдаются между самой крупнодисперсной и самой мел¬
кодисперсной фазами, подготовка и анализы частей образца должны проводиться по¬
следовательно.
Некоторые исследования могут потребовать сохранения части свежих образцов гли¬
ны, не подвергшихся высушиванию. В этом случае трансферная бумага должна иметь
дополнительное вертикальное разделение. Свежие образцы глины должны быть ото¬
браны на выходе ротора пластмассовым шпателем и быстро перенесены в маленький
контейнер. Точность количественного определения можно проверить на высушенной
фракции, однако существует риск ошибки из-за преобразования образца в процессе вы¬
сушивания.
Такой метод может быть также использован для:
* разделения и обогащения двух фаз; например, двух фаз с одинаковым значением
минеральной плотности, но различными размерами частиц (микрочастиц слюды
и более крупных частиц кристаллического каолинита);
* разделения двух компонентов с одинаковыми размерами частиц, но с различной
плотностью, например, глиноземного компонента плотностью < 3,0 и железо(Ш)-
или железо(Н)-содержащих компонентов плотностью 4—5;
Глава 3. Фракционирование коллоидных систем
91
• сбора частиц определенного диаметра и плотности в узком диапазоне (металлоге¬
ния — ситовое разделение), и т. д.
Достаточно часто состав крупнодисперсных фракций обогащен хлоритом, вермику¬
литом или каолинитом. Хорошо окристаллизованный каолинит обычно присутствует во
фракциях с размерами частиц > 0,5 мкм. В составе фракций с размерами частиц < 0,5 мкм
практически отсутствует кварц и мусковит, что позволяет «очистить» распределения.
В состав крупнодисперсных фракций, как правило, входит гиббсит, а в мелкодисперс¬
ных фракциях он присутствует в очень малых количествах.
Может также наблюдаться обогащение галлуазитом частиц размером < 0,5 мкм или
0,2 мкм, содержащих материалы с упорядочением ближнего порядка, тогда как хорошо
окристаллизованный каолинит исчезает.
Обезжелезивание увеличивает однородность частиц, препятствуя объединяющему
действию железа.
Изучение различных фракций методом рентгеновской дифракции позволяет оценить
возможную кристаллохимическую неоднородность коллоидной фракции и определить
минеральное происхождение в процессе развития и выветривания. Также могут быть
выявлены минералы, интенсивность дифракционных линий которых чуть выше 5% (на¬
пример, сепиолит).
Фракционирование использованием различных потоков
Согласно этого методу (Биологический центр почвоведения, Нанси, Франция (Biological
Centre of Pedology, Nancy, France)) глинистая суспензия размером частиц > 2 мкм при кон¬
центрации не более 1% сначала подвергается серии циклов центрифугирования с неиз¬
менной скоростью и величиной потока (рис. 3.6).
Повторная серия центрифугирования с более низкой скоростью потока (31 мл/мин)
позволяет отделить более тонкие фракции. Благодаря повторному центрифугированию
и низкой концентрации среды разделение фракций считается количественным.
V = 6000 д D-248 мл/мин
✓ "Ч
Рис. 3.6. Схема фракционирования методом фракционирования в различных потоках
92
Часть 1. Минералогический анализ почв
3.3. Предварительная обработка экстрагированных фаз
Минералогические исследования, выполненные непосредственно для всего образца по¬
чвы, позволяют получить информацию о наиболее преобладающих компонентах, однако
не позволяют обнаружить присутствие компонентов низкой концентрации вследствие
недостаточной чувствительности инструментальных методов анализа и большого коли¬
чества помех. Такие фазы при концентрациях < 5% могут иметь очень большое значение
при объяснении определенных процессов почвообразования, но часто маскируются фо¬
новым шумом, даже когда их концентрация превышает порог обнаружения.
Вследствие этого необходимо устранять мешающие факторы и повысить концентра¬
цию фазы «минералогических компонентов глины».
При использовании соответствующего метода фракционирования глины очищают
и концентрируют, а также переводят в гомоионную форму: NH4+, Са2+ или Н+, в зависи¬
мости от случая. Поскольку минеральные связующие и органоминеральные связи раз¬
рушаются, можно добиться достаточного разделения частиц.
Использование ультразвука позволяет достичь хорошего разделения элементарных
частиц, при этом глины могут быть подвергнуты дополнительным предварительным об¬
работкам, что позволит использовать конкретные инструментальные методы анализа.
Селективное растворение позволяет отделить оксиды железа, наличие которых мо¬
жет затруднить РД (см. гл. 4) из-за флуоресценции при использовании медного анода,
а также ДТА-ТГА (см. гл. 7) из-за окисления Fe2+. Различные предобработки позволяют
проводить анализы использованием методов ЯМР, ЭСР, Мессбауэровской спектроско¬
пии и т. д.
1Ьли и вещества, которые аморфны для рентгеновской дифракции и имеют кристал¬
лические решетки с упорядочением ближнего порядка, могут быть растворены, что по¬
зволяет осуществлять их количественное определение и идентифицировать их методом
дифференциальной РД.
Любой карбонат кальция, который все еще присутствует в составе образца, может
быть удален с образованием комплекса ионов Са2+ со стандартным раствором ЭДТА.
Предварительная обработка также позволяет улучшить ориентацию глин (которая на¬
рушается железом, например во внешних слоях), интенсивность дифракционных реф¬
лексов и ее отношение к фоновому шуму.
Достаточно часто глины необходимо анализировать после гомоионного насыщения
катионами (например, Mg2+, который упорядочивает адсорбцию воды глинами с расши¬
рением межпакетного пространства, либо К+, который ограничивает адсорбцию воды,
а значит и разбухание слоев).
Другие обработки, такие как сольватация полярными растворителями либо образова¬
ние интеркаляционных комплексов, позволяют идентифицировать определенные типы
глин. Специально для разрушения кристаллических решеток или модификации свойств
поверхности глин также используют методы термообработки. Все эти методы подробно
описаны в главах 4-7.
Использованная литература
Davis DS (1943) Empirical Equations and Nomography. Me Graw Hill, New York, 1,104-114.
Gautheyrou J and Gautheyrou M (1967) Mode operatoire pour l’extraction et la purification de la fraction
argileuse < 2 pm. Notes de laboratoire, Orstom-Guadeloupe, mars 1968,1-9, Orstom.
Hauser EA and Lynn JE (1940) Separation and fractionation of colloidal systems. Ind. Eng Chem., 32,
659-662.
Saunders E (1948) Nomograph for particle size determination with the Sharpies supercentrifuge. Anal
Chem., 20, 379-381.
93
Глава 3. Фракционирование коллоидных систем
Дополнительная литература
Atterberg А (1912) Die mechanische bodenanalyse und die klassification der mineralbuden schwedens.
Intern. Mitt. Bodenk, 2, 312-342.
Coca Prados J and Bueno de las Heras J (1977) Dinamica de particulas en suspensions solido-liquido.
I - Sedimentacion de particulas. Ingeniera quimica, 153-162.
Colmet-Daage F, Gautheyrou J, Gautheyrou M, Kimpe de C, Fusil G and Sieffermann G (1972) Disper¬
sion et etude des fractions fines de sols a allophane des Antilles et d’An^rique latine. Нёше partie:
Modifications de la nature et de la composition de la fraction inftrieure a 2 microns selon la taille des
particules. Cahiers Orstom, serie. Pedol., X, 219-241.
Davis JM (1986) General retention theoiy for sedimentation Field-Flow-Fractionation. Anal. Chem., 58,
161-164.
Essigton ME, Mattigod SV and Ervin JO (1985) Particles sedimentation rates in the linear density gradi¬
ent. Soil Sci. Soc. Am. J., 49, 767-771.
Gautheyrou J and Gautheyrou M (1982) Fractionnement des systames collondaux argileux par centrifu¬
gation continue. Notes laboratoire Orstom Bondy, 1-38.
Hauser EA and Reed CE (1936) Studies in thixotropy. I - Development of a new method for measuring
particle-size distribution in colloidal systems. J. Phys. Chem., 40,1169-1182.
Horrocks M (2005) A combined procedure for recovering phytoliths and starch residues from soils, sedi¬
mentary deposits and similar materials. J. Archaeological Sci., 32,1169-1175.
Jackson ML, Whittig LD and Pennington RP (1949) Segregation procedure for the mineralogical analysis
of soils. Soil Sci. Soc. Am. Proc., 14, 77—81.
Jacobsen AE and Sullivan WF (1946) Centrifugal sedimentation method for particle size distribution. Ind.
Eng. Chem., 18, 360-364.
Jaymes WF and Bigham JM (1986) Concentration of iron oxides from soil clays by density gradient cen¬
trifugation. Soil Sci. Soc. Am. J., 50, 1633-1639.
Johnson L (1956) Particle size analysis and centrifugal sedimentation. Trans. Bull. Ceram Soc., 55,267-285.
Kamack HJ (1951) Particle size determination by centrifugal pipet sedimentation. Anal. Chem., 23,844-850.
Kittrick JA and Hure EW (1963) A procedure for the particle-size separation of soils for X-Ray diffraction
analysis. Soil Sci., 96, 5, 319-325.
Koch T and Giddings JC (1986) High-speed separation of large (> 1 pm) particles by steric Field-Flow-
Fractionation. Anal. Chem., 58, 994-997.
Levitz PE (2005) Confined dynamics, forms and transitions in colloidal systems: from clay to DNA.
Magn. Reson. Imaging, 23, 147-152.
Marshal CE (1931) Studies in the degree of dispersion of the clays, I - Notes on the technique and accu¬
racy of mechanical analysis using the centrifuge. J. Soc. Chem. Ind., SDT, 444-450.
Muog E, Taylor JR, Pearson RW, Weeks AE and Simonson RW (1936) Procedure for special type of me¬
chanical and mineralogical soil analysis. Soil Sci. Soc. Am. Proc., 101-112.
Rouiller J, Brethes A, Burtin G and Guillet В (1984) Fractionnement des argiles par ultra-centrifugation
en continu: Evolution des illites en milieu podzolique. Sci. Geol. Bull., 37,319-331.
Schachman HK (1948) Determination of sedimentation constants in the Sharpies supercentrifuge. J. Phys.
Colloid. Chem., 52, 1034-1045.
Tan KH (1996) Soil Sampling, Preparation and Analysis. Dekker, New York, 278-361.
Tanner CB and Jackson ML (1947) Nomographs of sedimentation times for soil particles under gravity or
centrifugal acceleration. Soil Sci. Soc. Am. Proc., 12, 60-65.
Tran-Vinh-Ann and Ndejuru E (1972) Analyse granulom£trique de la fraction argileuse par centrifugation
en flux continu. Mise au point d’une m^thode et application a quelques sols tropicaux. Pedologie,
XXII, 366-382.
Truo E, Taylor JR, Simonson RW and Week ME (1936) Mechanical and mineralogical subdivision of the
clay separate of soils. Soil Sci. Soc. Proc., 175-179.
Tu Y, O’Carroll JB, Kotlyar LS, Sparks BD, Ng S, Chung KH and Cuddy G (2005) Recovery of bitumen
from oilsands: gelation of ultra-fine clay in the primary separation vessel. Fuel, 84, 653-660.
Whittig LD and Allardice WR (1986) X-Ray diffraction techniques - separation of particle - size frac¬
tion. In Klute A (ed.), Method of Soil Analysis Part Physical and Mineralogical Methods, second
edition, American Society or Agronomy, 340-342.
Глава 4. Характеристика минералогического состава
методом рентгеновской дифрактометрии
4.1. Введение
4.1.1. Рентгеновская дифракция и минералогия
Методы, основанные на использовании оптической микроскопии в петрографии, не¬
пригодны для идентификации тонкодисперсных глинистых минералов, кристалли¬
ческие решетки которых изменяются в зависимости от содержания влаги и ионного
окружения, а химический состав часто неизвестен (табл. 4.1,4.2). Среди других методов
одним из наиболее эффективных является рентгеновская дифракция (РД). Когерент¬
ное рассеяние падающего излучения в методе РД позволяет четко идентифицировать
параметры кристаллической решетки и геометрическое распределение атомов в ячейках
решетки.
Метод РД можно комбинировать с геохимическим или изотопным анализом (AAS,
ICP, ICP-MS, EXAFS и др.), термическим анализом (DTA-TGA, DSC, EGD, EGA2 и др.),
методами оценки энергии межатомных или межмолекулярных связей (например, ИК-
Фурье-спектроскопия, рамановская3 и мессбауэровская спектроскопии, ЯМР), транс¬
миссионной электронной микроскопией высокого разрешения (+ электронная микро¬
дифракция) и электронной сканирующей микроскопией. Энергодисперсионные (ЭД)
и волнодисперсионные (ВД) рентгеновские датчики позволяют установить связь хими¬
ческого состава с профилем наблюдаемых частиц. Общий химический состав определя¬
ют полным анализом после минерализации в средах, позволяющих осуществлять солю¬
билизацию всех компонентов. Селективное растворение позволяет разделить образец на
фракции, различающиеся химической устойчивостью. Такое подразделение необходимо
как для количественного определения, так и для очистки проб перед анализом инстру¬
ментальными методами (например, РД-, ИК- или ЯМР-спектроскопией).
Межлабораторные исследования могут проводиться на разных уровнях: определе¬
ние структуры глин (геохимические соотношения, педологическая дифференциация
по профилю и пространственная дифференциация), водных свойств (порозность, про¬
ницаемость, функциональное обводнение, вызванное природой и содержанием глин),
адсорбции комплексов (распределение зарядов, ЕКО и др.). 11AAS — Atomic Absorption Spectrometry (AAC — атомно-абсорбционная спектрометрия); ICP—
Inductively Coupled Plasma (ИСП — индуктивно-связанная плазма); ICP-MS— (ИСП-МС —
масс-спектрометрия с индуктивно-связанной плазмой); EXAFS — Extended X-ray Absorption
Fine Structure (ДТС РСП — дальняя тонкая структура рентгеновских спектров поглоще¬
ния). — Примеч. науч.ред.
2 DTA- TGA — Differential Thermal Analysis и Thermal Gravimetric Analysis (ДТА-ТГА — дифферен¬
циальный термический анализ и термогравиметрический анализ); DSC— Differential Scanning
Calorimetry (ДСК — дифференциально-сканирующая калориметрия); EGD — Evolved Gas
Detection, EGA - Evolved Gas Analysis (ГТА — газовый термический анализ). — Примеч«науч, ред,
3 КР — спектроскопия комбинационного рассеяния. — Примеч. науч. ред.
Глава 4. Характеристика минералогического состава методом РД 95
Таблица 4.1. Классификация глинистых минералов, предложенная Международной
ассоциацией по изучению глинистых минералов (AIPEA1)
Ъш
Ц>уппа(дг-заряд
формульной
единицы)
Подгруппа (и — число катионов
в октаэдрических слоях)
Минерал
1:1
Каолинит -
серпентин
(*=0)
Каолинит (п = 2) серпентин (п = 3)
Каолинит, галлуазит,
хризолит, лизардит,
антигорит
2:1
Пирофиллит —
Пирофиллит (п =2)
Пирофиллит,
тальк (х = 0)
тальк (п = 3)
тальк
Смектиты
Диоктаэдрические смектиты или
Монтмориллонит,
монтмориллонит
монтмориллониты (п = 3)
бейделлит, нонтронит
Сапонит
Триоктаэдрические смектиты или сапонит
Сапонит, гекторит,
(х = 0,25-0,6)
(п = 3)
сауконит
Вермикулит
(х = 0,6—0,9)
Диоктаэдрические вермикулиты (п = 2)
Триоктаэдрические вермикулиты (п = 3)
Диоктаэдрический
вермикулит
Триоктаэдрический
вермикулит
Слюды® (х= 1)
Диоктаэдрические слюды (п = 2)
Мусковит, парагонит
Хрупкие слюды
Триоктаэдрические слюды (п = 3)
Биотит, флогопит
(*=2)
Диоктаэдрические хрупкие слюды (п = 2)
Триоктаэдрические хрупкие слюды (п = 3)
Маргарит
Клинтонит
2:1:1
Хлорит
Диоктаэдрические хлориты (4 < п < 5)
Донбассит
(переменный х)
Ди-триоктаэдрические хлориты
Триоктаэдрические хлориты (5 < п < 6)
Кукеит, судоит
Пеннинит, хлорит,
прохлорит
а Иллиты (или гидрослюда), серицит и др. материалы, относящиеся к иллитам (могут чередоваться).
Таблица 4.2. Словарь по структуре кристаллов (.AIPEA, 1972)
Русский
English
French
(атомная) плоскость
(atomic) plane
plan (atomique)
(тетраэдрическая или
(tetrahedral or octahedral)
couche (tetra^drique ou
октаэдрическая) сетка
sheet
octaSdrique,
(комбинация плоскостей)
(plane combination)
combinaison de plans)
слой (пакет) 1:1 или 2:1
1:1 or 2:1 layer (sheet
feuillet 1:1 ou 2:1 (combinaison
(комбинация сеток)
combination)
de couches)
межслоевое (межпакетное)
пространство (межслой)
interlayer space
espace interfoliaire
пакет = комбинация сеток +
unit structure = combination
assemblage de feuillets + materiel
межслоевых материалов
of layers + interlayer
materials
interfoliaire = unite structurale
решетка
lattice
teseau
1 AIPEA — Association Internationale pour I'Etude des Argiles, GPO Box 2434, Brisbane, Qld 4001
Australia.
96
Часть 1. Минералогический анализ почв
Для детального количественного изучения кроме глинистой фракции обычно необ¬
ходимо исследовать фракцию тонкой пыли, которая может содержать смешанно-слой¬
ные минералы, особенно в случае слюд и хорошо окристаллизованного каолинита. Рас¬
чет баланса учитывает глины и связанные с ними фазы, оксиды, гидроксиды и другие
соединения, что позволяет объяснить кажущееся несоответствие результатов (избыток
содержания Si4+ вследствие присутствия диатомей, очень мелкого кварца, высокой кон¬
центрации К* из калиевых полевых шпатов, слюд и пр.).
4.1.2. Основные положения
Кристалл — это твердое тело, состоящее из атомов, собранных в трехмерную периоди¬
ческую структуру. Расстояния а, Ь, с и углы а, р, у между гранями определяют параметры
ячейки основного блока1 (рис. 4.1).
С
Рис. 4.1. Кристаллическая ячейка
Когда монохроматические рентгеновские лучи определенной длины волны падают на
грань кристалла, они отражаются атомами кристалла. Сигнал усиливается в определен¬
ном направлении, если лучи, отраженные различными плоскостями, совпадают по фазе
(рис. 4.2). Это явление подчиняется закону Брэгга2 *:
2dsinQ = пХ, (4.1)
где d — межплоскостное расстояние или расстояние между плоскостями атомных сеток
в кристалле (d(hkt))\ X — длина волны, 0 — угол между лучом и атомной плоскостью; п —
порядок отражения (целое число).
Все плоскости кристалла дифрагируют рентгеновское излучение, когда кристалл по¬
вернут под углом 6 к падающему лучу с длиной волны X, в соответствии с уравнением
(4.1).
Угол 0 связан с длиной волны X и расстоянием d, выраженными в ангстремах или на¬
нометрах (1 А = 0,1 нм = 10‘10м). Если длина волны известна, измерение угла отражения
позволяет определить межплоскостное расстояние.
1 Элементарной ячейки. — Примеч. науч. ред,
2 В отечественной литературе закон упоминается как уравнение Брэгга—Вульфа. — Примеч.
науч. ред.
Глава 4. Характеристика минералогического состава методом РД
97
Падающий
Рис. 4.2. Дифракция падающего луча плоскостями атомных сеток кристалла. Линии р, p^ и р2
представляют параллельные и равноотстоящие ретикулярные плоскости на расстоянии d друг от
друга. Рентгеновский луч, падающий на верхнюю плоскость, отражается под углом, равным углу
падения 0. Для получения отражения, которое можно измерить, все лучи, отраженные плоскостя¬
ми д р1 и р2 и т. д., должны совпадать по фазе. Для этого сумма GE + ЕН, равная разности хода
между лучами АВС и DEF, должна быть равна целому числу длин волн. Поскольку GE = EH = d sin 0,
это условие соответствует закону Брэгга (4.1)
Примечания
Некоторые минералы могут быть «аморфными» по отношению к рентгеновским лучам,
если они не имеют определенного кристаллического упорядочения (истинные стекла)
или их упорядочение ближнего порядка слишком мало для детектирования длиной вол¬
ны в 1—2 А.
РД — не лучший метод для изучения некристаллических твердых тел, таких как алло¬
фаны, состоящие из кластеров атомов Si (структурных элементов с межпакетными рас¬
стояниями, соответствующими одному или двум межатомным расстояниям).
Атомы кремния в тетраэдрической позиции и атомы алюминия с октаэдрической ко¬
ординацией, но без регулярной симметрии не дают отчетливых пиков, а только широкие
и плохо определяемые, которые проявляются около 0,33 и 0,22 нм.
4.1.3. Оборудование для проведения рентгеновской дифракции
Рентгеновские лучи были открыты Рентгеном в 1895 г., но явление дифракции на кри¬
сталлах было обнаружено только в 1912 г.
Длины волн рентгеновских лучей расположены в диапазоне между длинами волн
УФ- и гамма-излучения, т. е. примерно от 10 до 10_2нм. В РД используется «жесткое»
излучение с длинами волн от 1,5 до 1,9 А, в зависимости от используемого анода. Рент¬
геновское излучение распространяется по прямой линии. Необходимо обеспечить, что¬
бы излучение было максимально монохроматическим, используя для этого набор филь¬
тров, щелей и монохроматор. Оборудование включает следующие элементы:
* Источник питания со стабилизированным высоким напряжением и токами поряд¬
ка микроампер для питания рентгеновской трубки и счетчика.
98
Часть 1. Минералогический анализ почв
• Запаянная рентгеновская трубка, включающая источник электронов, ускоряемых
разностью электрических потенциалов от 20 до 50 кВт, и анод (или антикатод)
(табл. 4.3), сделанный из толстого металла и охлаждаемый водяным контуром; вы¬
сокоскоростная бомбардировка электронами вызывает перенос энергии к атомам
анода-мишени, переводя их на более высокий энергетический уровень, и таким
образом создает вакансии электронов на орбиталях; атомы анода испускают харак¬
теристичные кванты энергии; фотоны рентгеновского излучения выходят из труб¬
ки через бериллиевое окно толщиной 300 мкм, прозрачное для рентгеновских лу¬
чей (пятно должно быть как можно меньше для концентрация энергии электронов
в ограниченной зоне анода и обеспечения высокой интенсивности рентгеновского
излучения); мощность трубки ограничивается количеством тепла, рассеиваемого
анодом, и выражается через максимально допустимую величину в мА для данного
напряжения (1—3 кВ или более, в случае вращающегося анода); характеристиче¬
ское излучение получают только начиная с определенной величины напряжения
возбуждения, поэтому необходима тщательная регулировка напряжения и интен¬
сивности во избежание сдвига длины волны.
• Гониометр, позволяющий вращать образец и счетчик в пределах, определенных
уравнением Брэгга; он образует основание для образца, плоскость которого на¬
строена очень точно, и сфокусированного детектора, который вращается в том же
направлении вокруг той же оси с соответствующей скоростью.
• Линейный луч получают, используя щели Соллера и степень расходимости, которая
ограничивается открытием пучка1; в настоящее время используют регулируемые
щели, апертура которых связана с углом, что делает возможным облучение оди¬
наковой площади поверхности, это особенно удобно при малых углах, поскольку
прямой луч имеет ограниченное применение.
• Приемная щель ограничивает ширину луча в фокальной плоскости; чем уже щель,
тем выше разрешение, хотя и происходит потеря интенсивности; графитовый мо¬
нохроматор ограничивает излучение флуоресценции, некогерентные излучения
и комптоновское рассеяние; качество измерений зависит от точности регулировки
всех элементов.
• Система детектирования (счетчик) позволяет измерять интенсивность проходя¬
щего рентгеновского излучения; число пульсаций в единицу времени пропорцио¬
нально количеству прошедшего излучения; можно использовать линейный или
пропорциональный счетчик, сцинтилляционный или даже полупроводниковый
детектор (последний должен находиться в жидком азоте при -196 °С).
Меры безопасности
Рентгеновское излучение опасно и может вызывать ожоги, генетические изменения,
раковые заболевания и т. д. Следует учитывать и риск, связанный с использованием вы¬
сокого напряжения.
Необходимо тщательно соблюдать меры профилактики и строго выполнять правила
обращения со всем оборудованием, которое должно быть оснащено защитными устрой¬
ствами:
• запаянная трубка должна быть защищена толстыми стенками для устранения ри¬
ска облучения;
1 С помощью коллиматоров. — Примеч. науч. ред.
Глава 4, Характеристика минералогического состава методом РД
99
• гониометр должен быть изолирован свинцовым стеклом или свинцовым пласти¬
ком для защиты всего аппарата и открываться только при выключенной трубке;
9 оператор обязан носить бейдж мониторинга с чувствительной пленкой (дозиметр),
рассчитывающий возможное общее облучение (который должен регулярно прове¬
ряться), а также кольцо для измерения облучения рук;
• оператору необходимо проходить регулярные медицинские осмотры для обнару¬
жения изменений состава крови (количество белых кровяных телец и т. п.);
9 для проверки возможной утечки радиации нужно использовать счетчик Гейгера:
люминесцирующие экраны должны находиться на основаниях, выполненных из
цинка с добавками никеля, — для идентификации зон, пораженных рентгенов¬
ским излучением в процессе регулировки оборудования.
Таблица 4.3. Характеристики некоторых анодов
Анод
Длина
волны
Кос, А
Фильтр
кр
Индуци¬
рованная
флуоресценция
Напряжение
возбуждения
(кВ)
Рабочее
напря¬
жение (кВ)
Примечание
Си
1,542
Ni
Со—Fe
9,0
25-45
Проникающая
способность и среднее
рассеяние, слабое
влияние воздуха
Со
1,791
Fe
Мп-Сг
7,8
25-35
Низкая проникающая
способность, большое
рассеяние, слабое
влияние воздуха
Fe
1,937
Mn
Cr-V
7,1
25-35
Низкая проникающая
способность, большое
рассеяние
4.2. Качественная дифрактометрия
4.2.1. Краткий обзор методов подготовки образцов
Методы подготовки образцов представлены на рис. 4.3.
4.2.2. Получение дифрактограмм порошкообразных материалов
Цель
Этот метод обычно используют для идентификации минералов без преимущественной
ориентации. Он позволяет проводить количественный анализ, поскольку относитель¬
ная интенсивность дифракционных максимумов приблизительно пропорциональна ко¬
личеству кристаллизованного вещества при низком уровне анизотропии.
Основные положения
В методе, основанном на «записи спектра1 неориентированного образца» (Thomson и
др., 1972; Peterson и др., 1986; Decarreau, 1990), кристалл анализируют в форме тонко-
1 Непосредственными результатами порошковой дифрактографии являются дифрактограм-
мы, Понятие спектра предполагает изучение изменения какой-либо величины от энергии, на
дифрактограммах же получают зависимость интенсивности отраженного излучения от угла.
— Примеч. науч. ред>
100
Часть 1. Минералогический анализ почв
Экстракция
глины
I крупная 2-0,5 мкм
< 2 мкм ► средняя 0,5-0,2 мкм
тонкая < 0,2 мкм
1
Перенос Химическая Центрифугирование
Ориентирование Умеренное Ориентированные Ориентированная Пористые Умеренное
на пластиковой
пленке
измельчение
I
Неориентированный
порошок
агрегаты
уг
паста пластины измельчение
1
Неориентированный
порошок
Слайды
Необработанный
образец
Базальное расстояние
Сольватированный Насыщенный Нагретый
образец образец образец
Расширение между слоями Сжатие решетки
Рис. 4.3. Подготовка образцов для рентгеновской дифракции
дисперсного изотропного порошка при облучении монохроматическим рентгеновским
излучением. Случайная ориентация должна статистически представлять все возможные
ориентации различных частиц и обеспечить получение полной дифрактограммы мине¬
ралов, способных к дифракции рентгеновских лучей (глины, оксиды, гидроксиды, ок-
сигидроксиды, невыветренные первичные минералы, различные соли и т. д.).
Каждая частица рассматривается как микрокристалл или агрегат микрокристаллов,
и весь объем порошка может сравниваться с монокристаллом, поворачивающимся не
вокруг одной оси, а вокруг всех возможных осей. Дифрактограмма порошка позволя¬
ет связать относительные интенсивности пиков с данными, внесенными в сборники
JCPDS (Объединенного комитета порошковых дифракционных стандартов)1, и поэтому
должна быть записана перед любой другой операцией.
1 JCPDS - ICDD - Joint Commitee on Powder Diffraction Standards - International Center
for Diffraction Data, Newtown Square Corporate Campus, 12 Campus Bdv, Newtown Square
Pennsylvania 19073-3273 (USA).
101
Глава 4. Характеристика минералогического состава методом РД
После уплотнения в держателе, в полумикромасштабе, порошковый образец содер¬
жит микроагрегаты фрактальных размеров с нагроможденной структурой и открытой
порозностыо (зерна очень слабо связаны), и его характеристики зависят от степени из¬
мельчения, сжатия и гомогенности среды. Общая средняя ориентация в этом масштабе
очень слабая.
С другой стороны, в субмикромасштабе (несколько десятков или сотен нанометров)
смешанные пакеты глинистых кристаллитов являются первичными морфологическими
единицами и, следовательно, могут представлять ориентацию с переменной степенью
беспорядка в зависимости от типа глины. Использование вращающегося держателя об¬
разца повышает уровень рандомизации, но требует относительно большого количества
порошка (примерно 400 мг).
Методика
Порошковые дифрактограммы могут быть получены для самой почвы или ее фракций
(см. гл. 3). Ил, полученный центрифугированием (отцентрифугированную гранулу или
пластичную массу, перенесенные на бумагу), высушивают на воздухе и затем измельча¬
ют в агатовой ступке для получения гомогенного порошка:
• шпателем помещают в полый держатель количество порошка, необходимое для
почти полного заполнения углубления (рис. 4.4);
• осторожно разравнивают порошок при помощи стеклянной пластины;
• постепенно добавляют порошок, чтобы заполнить углубление доверху, и осторож¬
но прессуют так, чтобы совместить поверхность порошка с плоскостью фокуси¬
ровки.
а)
Ь)
с)
Рис. 4.4. Держатели с углублениями для измерения дифракции: а - пластиковый держатель;
b - вращающийся держатель Siemens; с - стеклянный держатель с углублением
Наблюдения
Рандомизация
Порошок должен быть достаточно компактен, чтобы обеспечить сцепление без исполь¬
зования связующего вещества или необходимости полировать его поверхность. Слиш¬
ком сильное давление может вызвать ориентацию образца. Поэтому необходимо регули¬
ровать степень «рандомизации-ориентации», готовя образцы максимально одинаково.
102 Часть 1. Минералогический анализ почв
Поскольку слои глины плоские, они стремятся к ориентации, что может привести к не¬
правильным результатам.
Этот эффект может быть снижен при использовании связующего вещества, инертно¬
го по отношению к рентгеновскому излучению и материалу образца (ацетон, каучук +
спирт, коллодий + бутил- или амилацетат, трагакант и др.). Такая обработка стабилизи¬
рует порошок. Вместе с тем, использование вертикального гониометра предотвращает
осыпание порошка, помещенного под рентгеновское излучение, что делает эту стадию
подготовки образца в большинстве случаев ненужной.
Если количество материала мало, то можно использовать двусторонний скотч, на ко¬
торый наносят образец (избыток материала удаляется легким постукиванием), или еще
лучше поместить образец в кремниевый держатель.
Лиофильная сушка позволяет получать менее ориентированные образцы (хотя су¬
ществует риск остаточной ориентации некоторых пакетов слоев). Вращение образца
обеспечивает максимум возможных отражений, что снижает риск ошибки за счет улуч¬
шения рандомизации и уменьшения флуктуаций интенсивностей из-за недостаточного
количества дифрагирующих частиц. Однако вращающийся держатель требует большего
количества порошка.
Гранулометрия — фокусировка
Для удовлетворительной фокусировки луча необходимо использовать поверхность об¬
разца. Неровность поверхности заметно влияет на относительные интенсивности ли¬
ний. Если поверхность грубая, как в случае крупнозернистых порошков, коэффициент
поглощения будет большим, а интенсивности при малых углах - чрезвычайно малыми.
Размеры частиц в порошке должны быть достаточно малы (около 10 мкм), чтобы из¬
бежать таких флуктуаций поглощения, но и не слишком мелкие, чтобы не допустить
искусственного увеличения доли аморфных минералов:
• при 10 мкм флуктуации не превышают 2%;
• при 50 мкм флуктуации могут достичь 20%.
С другой стороны, при размере частиц менее 0,02 мкм получаются диффузные пики
дифракции и их интенсивность снижается.
Ширина дифракционной линии увеличивается с уменьшением толщины кристалла.
Структура «аморфных» веществ характеризуется отсутствием периодичности или упо¬
рядочением ближнего порядка. В последнем случае они дают только одно статистически
предпочтительное значение для межатомного расстояния, и РД не может привести к удо¬
влетворительным результатам, а показывает только широкие и нечеткие пики (рис. 4.5).
Отдельные случаи использования порошковых дифрактограм
Порошковые дифрактограммы можно использовать для идентификации непреобразо-
ванных первичных минералов (кварц, кальцит и др.) или оксидов и гидроксидов желе¬
за и алюминия и др. Так, степень кристалличности в случае каолинитов и огнеупорной
глины будет распознаваться более четко: поскольку упаковка слоев различна, некоторые
пики, присутствующие в каолините, отсутствуют в огнеупорной глине. В дифрактограм-
ме ориентированного образца это различие может не наблюдаться.
Ди- или триоктаэдрические слои могут быть выделены при использовании линии
060 (заполнение октаэдрических пустот двухвалентными (1,54 А) или трехвалентными
(1,49 А) катионами).
Для полиморфных иллитов могут быть получены данные, характеризующие способ
их образования.
103
Глава 4. Характеристика минералогического состава методом РД
Рис. 4.5. Типичные дифрактограммы: а - хорошо окристаллизованное вещество; Ь — аморфное
вещество с упорядочением ближнего порядка
4.2.3. Получение дифрактограмм ориентированных образцов
Дифрактограммы ориентированных образцов на стеклянных пластинах
Цель
Целью является идентификация некоторых глинистых минералов, способных к ориен¬
тации, и наблюдение базальных изменений при помощи нагревания или химической
обработки. Количество требуемых процедур зависит от смеси минералов в образце.
Основные положения
В дифрактограммах ориентированных агрегатов особое значение придается плоскостям
кристаллов, параллельным поверхности пакетов. Предпочтительная ориентация сили¬
катов увеличивает максимум для базальной ориентации */(001), что позволяет обнаружи¬
вать малые количества кристаллической породы, присутствующей в смеси.
При использовании этого метода получается лучшая дифракция образцов. Флуктуа¬
ции теоретически меньше, чем в случае неупорядоченных порошков, поскольку размер
частиц не превышают 2 мкм. С другой стороны, предпочтительная ориентация снижает
количество плоскостей (hkl), способных к дифракции.
Методика
а. Из хорошо гомогенизированной пасты глины, полученной центрифугированием,
отбирают шпателем небольшую аликвоту (примерно 400 мг) и помешают ее в про¬
бирку с 5 мл воды; встряхивают для получения суспензии.
б. Из порошка (например, извлечение глины, описанное в гл. 9) берут примерно 200 мг
глины, помещают ее в пластиковую пробирку с 5 мл воды и 8-миллиметровым сте¬
клянным шариком и встряхивают в течение 2—3 мин для получения суспензии.
в. Отбирают пипеткой примерно 1 мл суспензии, полученной способами (а) или (б),
и равномерно распределяют по всей поверхности стеклянной пластинки размером
24x24 мм так, чтобы осадок не давал участков с более толстым слоем. Если слой
104
Часть 1. Минералогический анализ почв
осадка слишком тонок, то может сказываться влияние держателя; если он слиш¬
ком толст, то дифракция будет происходить только на илистой глине, а пленка гли¬
ны будет покрываться сетчатым узором. Образец оставляют сохнуть при комнат¬
ной температуре, затем досушивают в эксикаторе. Готовят три препарата:
- первый препарат для исследования исходного образца без обработки;
- второй препарат для исследования образца после обработки глицерином или
гликолем;
- третий препарат для нагревания пробы при 490 °С (если необходимо, этот пре¬
парат может быть использован для нескольких термических обработок при по¬
вышенных температурах).
Наблюдения
После того как суспензия распределена на стеклянной пластине, происходит микро¬
фракционирование по размеру частиц глины, поскольку крупные частицы оседают бы¬
стрее, чем мелкие. Однако при быстром высушивании эта сегрегация не повлияет на
качественную интерпретацию.
В отличие от работы с порошком (см. раздел 4.2.2), слой которого должен быть доста¬
точно толстым для снижения влияния ориентации, ориентированные слайды должны
быть тонкими, чтобы максимальное число базовых блоков было ориентировано долж¬
ным образом. Нанесение черной линии на держатель позволяет оценить качество пре¬
парата: она должна быть слабо видима сквозь почти прозрачную пленку глины.
В сравнительном полуколичественном анализе суспензированный осадок каждого
образца должен содержать примерно одинаковое количество глины. Этого легко до¬
биться при использовании сухой глины, которую взвешивают перед приготовлением
суспензии. Отбор аликвоты образца в процессе перемешивания позволяет получить
препараты, содержащие почти одинаковое количество минерального вещества, рас¬
пределенного по одинаковой площади. После перемешивания и гомогенизации суспен¬
зии быстро отбирают аликвоту такого же объема, сушат и взвешивают одну из проб для
определения концентрации.
Из суспензий влажных материалов извлекают глину на пленку определенной площа¬
ди и взвешивают идентичную аликвоту после высушивания.
Перед получением дифрактограммы необходимо удалить аморфные материалы, ко¬
торые могут маскировать ее часть.
Дифрактограмма ориентированной пасты
Основные положения
Этот метод наиболее подходит для минералов типа 2:1 и четырехводного галлуазита,
влажный образец должен быть высушен при комнатной температуре и храниться при
относительной влажности воздуха около 80%.
Методика
После отделения требуемого количества глины центрифугированием (см. главу 3) от¬
бирают влажный осадок, гомогенизируют маленьким шпателем из нержавеющей стали,
помещают в полость держателя (рис. 4, а), распределяют в углублении и выравнивают
стеклянной пластиной так, чтобы совместить поверхность образца с плоскостью фоку¬
сировки. Медленно высушивают при комнатной температуре, не допуская чрезмерного
высыхания.
105
Глава 4. Характеристика минералогического состава методом РД
Наблюдения
Для крупных глин с размером частиц 2 мкм полученную пасту следует гомогенизировать
после центрифугирования, так как возле поверхности концентрируются волокнистые ча¬
стицы глины, для которых не соблюдается закон Стокса. Смектиты оседают преимуще¬
ственно вблизи поверхности осадка, а хлориты и иллиты осаждаются быстрее и накапли¬
ваются в нижней части отцентрифугированной гранулы. То же происходит с оксидами
железа (плотность 4,5-5), которые тяжелее, чем оксиды алюминия (плотность около 3).
Во время высушивания глины с высоким коэффициентом сокращения могут тре¬
скаться и разделяться. Можно добавить связующее вещество, хотя в этом случае суще¬
ствует риск введения трудно контролируемой переменной (нарушенная ориентация,
флоккуляция, неаморфность для рентгеновских лучей связующего вещества, и т. д.).
Отдельные случаи использования
В некоторых случаях можно работать с влажными образцами. Для глин типа 2:1 насыще¬
ние влажной пасты ионами Na+ или Li+ позволяет наблюдать явление неограниченного
набухания d(001) > 30 А. При высушивании на воздухе глина вновь достигает величин
d(001) = 18,15,14 А.
Агрегаты, ориентированные на пористой керамической пластине
Цель
Фиксация минералов, которые не прилипают к стеклянным пластинам, и/или проведе¬
ние последовательных обработок одного и того же образца: исходный образец, обработ¬
ка катионами, многоатомным спиртом, затем термическая обработка. Такая подготовка
позволяет достигнуть удовлетворительной ориентации глин и чрезвычайно полезна для
количественного анализа дифрактограмм.
Основные положения
Глину фиксируют на пористой пластине, подсасывая вакуумным насосом или центрифу¬
гируя с использованием пористого керамического диска Поретикс и держателя Хетгиха.
Методика
Переносят суспензию глины на пористую пластину размером 24x24 мм, помещенную
на воронку Бюхнера диаметром 40 мм, с дном из спеченного стекла, котором оставлено
проницаемым только окно размером 22x22 мм (рис. 4.6). Наливают гомогенизирован¬
ную жидкость на пластину и подключают воронку к вакуумному насосу. После образо¬
вания тонкого сплошного слоя глины образец высушивают и анализируют дифракцию
рентгеновских лучей.
Наблюдения
Эта методика позволяет получать хорошо ориентированные осадки с пониженным
фрактальным размером поверхности, который сохраняется после высушивания и по¬
этому прекрасно подходит для РД. Слой осадка должен быть однородным по всей пла¬
стине и достаточной толщины для устранения влияния материала держателя, но в то же
время и достаточно тонким, чтобы сохранять определенную степень эластичности и не
допускать растрескивания из-за реологических свойств среды.
Измерения на пористой пластине позволяют избежать излишней сольватации и вы¬
полнить необходимые процедуры: химическое насыщение ионами Mg24" и К+, нагрева-
106
Часть 1. Минералогический анализ почв
Пористая пластина
Основание из
модифицированного
спеченного стекла
Рис. 4.6. Приготовление образца на пористой керамической пластине
ние и др. Микропористые фильтры (0,20 мкм) на стеклянных пластинах можно исполь¬
зовать для измерений (сбор водных суспензий, воздушной пыли и др.).
Очистка пористой пластины — деликатная процедура, которая должна проводиться
после мытья поверхности, бывшей в контакте с глиной, с использованием легких абра¬
зивных средств. Важно сохранить совершенную гладкость пластины и удостовериться
в отсутствии загрязнений.
Ориентированные агрегаты, осажденные ультрацентрифугированием
на полугибкой пленке
Цель
Целью данного метода является получение образцов одинаковой толщины, минерало¬
гического состава и кажущегося размера частиц.
Основные положения
Частицы осаждают с помощью суперцентрифуги Шарплес (Sharpies), оборудованной
цилиндром с полугибкой внутренней пластиковой пленкой (см. гл. 3). Скорость регу¬
лируют так, чтобы собрать частицы известного среднего диаметра (от грубой до тонкой
глины). В случае необходимости пленку можно анализировать на восьми последова¬
тельных уровнях (шириной 24 мм), что позволяет контролировать изменения в приро¬
де глин в зависимости от размера частиц и плотности. Для того чтобы пленка не была
слишком толстой, необходимо знать содержание крупной, средней и мелкой фракции
глины и использовать только зоны, соответствующие оптимальной плотности (проверка
по черной линии на пленке, см. раздел «Дифрактограммы ориентированных образцов
на стеклянных пластинах»).
Методика
Снимают пластиковую пленку с цилиндрической чаши и сушат плашмя в течение при¬
мерно 1 ч. Когда образец еще слегка влажный и не совсем жесткий, его разрезают на ку¬
ски размером 24x24 мм по рисунку, нанесенному на обратную сторону пленки (рис. 4.7).
Каждый уровень должен быть маркирован. Кладут куски полугибкой пленки размером
24x24 мм на стеклянную пластину и помещают ее в держатель перед ограничителем, со¬
ответствующим плоскости фокусировки.
Глава 4. Характеристика минералогического состава методом РД
107
Рис. 4.7. Диаграмма, показывающая, как разрезать пластиковую пленку после проточной
ультрацентрифуги и установить ее для РД
Наблюдения
РД может выполняться непосредственно на ориентированном образце или после обра¬
ботки (см. раздел 4.2.4) многоатомным спиртом; можно также нагреть до 110 °С, в за¬
висимости от природы используемого пластика. Поверхность пластикового держателя
должна быть неполированной и без добавок, так как они могут вызвать фоновый шум,
недопустимый при монотонных измерениях.
4.2.4. Предварительная обработка глин
Влияние предварительной обработки
Для облегчения идентификации глинистых минералов можно использовать различные
виды предварительной обработки, которая вызывает селективные изменения межпло¬
скостных расстояний в слоях глины. Виды обработок и их влияние показаны в табл. 4.4.
Таблица 4.5 показывает максимальные межплоскостные расстояния, наблюдаемые
в глинистых минералах почв.
Насыщение катионами
Основные положения
Насыщение катионами позволяет заполнить существующие катионные вакансии и, за¬
мещая обменные катионы, получить гомоионные образцы с равномерным расширени¬
ем слоев расширяющихся филлосиликатов (количество межпакетной воды зависит от
обменных катионов). Двухвалентные катионы, например, Mg2+, Са2\ Sr2+, образуют ги¬
драты с двумя слоями, относительно стабильные в широком диапазоне относительной
влажности воздуха и мало подверженные влиянию ионов гидроксония при промывке
водой.
Насыщение ионами дает стабильный комплекс с двумя слоями межпакетной
воды, которая доводит межплоскостное расстояние в расширяющихся филлосиликатах
до tf(001) £ 14. Расширение слоев позволяет дифференцировать нерасширяющиеся виды
глин, в которых межпакетное расстояние составляет примерно 10 А (табл. 4.5).
108
Часть 1. Минералогический анализ почв
Насыщение ионами К+ ограничивает адсорбцию воды в межпакетных промежутках
и делает возможной дифференциацию хлоритовых глин типа 2:1:1 и вермикулитовых
глин типа 2:1. Эта обработка не изменяет нерасширяющиеся хлориты, в то время как
вермикулит разрушается при межпакетном расстоянии 10 А. Ту же пробу можно исполь¬
зовать для определения смектитов после нагревания до 550 °С.
Насыщение ионами LF с последующей дегидратацией при -300 °С и сольватацией
глицерином позволяет отличить монтмориллонит от бейделлита при помощи теста
Грин—Келли, основанного на эффекте Гофмана—Клемена. Когда насыщенный литием
смектит подвергается дегидратации при 300 °С, прослойка лития мигрирует к октаэдри¬
ческим слоям, которые испытывают дефицит положительных зарядов после замещения.
Структура становится нерасширяющейся, и дальнейшего расширения образца при об¬
работке глицерином не происходит (9,5 А). Бейделлит (или сапонит), чьи заряды об¬
разуются из-за замещений в тетраэдрических слоях, не изменяется под действием таких
обработок и расширяется под действием глицерина (17,7 А).
Методика насыщения Мд2+
• Берут навеску, содержащую 125—250 мг глины.
• Готовят суспензию и подкисляют разбавленной хлороводородной кислотой до pH
3,5-4,0, чтобы избежать осаждения гидроксида магния (особенно если первона¬
чальное диспергирование проводилось в щелочной среде).
• Добавляют 5 М раствор ацетата магния для получения суспензии, содержащей
-0,5 моля Mg в литре.
• Оставляют смесь на 30 мин.
• Центрифугируют при -3000 g в течение 5 мин и сливают надосадочную жидкость.
• Дважды промывают осадок после центрифугирования 0,5 М раствором
Mg(CH3COO)2 для удаления ионов Н+ из кислой суспензии и затем дважды 0,5 М
раствором MgCl2 (примерно 10 мл).
• Центрифугируют и промывают осадок 50%-ным метанолом (примерно 10 мл), за¬
тем 98%-ным метанолом (примерно 10 мл) и 85%-ным ацетоном. Проба с нитра¬
том серебра на ионы СГ должна быть отрицательной.
• Сушат образец при комнатной температуре для получения порошковой дифракто-
граммы или готовят препараты для непосредственного получения дифрактограмм
ориентированных образцов, добавляя небольшое количество воды.
Методика насыщения К+
• Берут навеску, содержащую 125-250 мг глины.
• Готовят суспензию и добавляют 1 М раствор КС1.
• Центрифугируют флоккулированную глину после контакта в течение 30 мин.
• Сливают надосадочную жидкость.
• Промывают осадок после центрифугирования 1 М раствором КС1.
• Затем промывают его 50%-ным и 95%-ным метанолом и затем 95%-ным ацетоном
до отсутствия СГ ионов в промывном растворе (отсутствие осадка при добавлении
AgN03).
• Оставляют сохнуть при комнатной температуре или сразу готовят препараты для
анализа ориентированных образцов, добавляя дистиллированную воду.
Таблица 4.4. Влияние предварительной обработки образцов на межплоскостные расстояния в основных типах глин: воздушно¬
сухие образцы, насыщенные ионами Mg2+ (М — метагалуазит или галлуазит, 2Н20 (7 А), Н — гидратированный
галлуазит, 4Н20 (10 А)). Обработка разделяет переходные формы хлорит-вермикулита и хлорит-смектита
Группа
iff А)
1:1
(Td-Oh)
7
7-10
2:1
(Td-Oh-Td)
9.4
10
10-12
14-15
2:1:1
(Td-Oh-Td-Oh)
Стабильность
(межслойная)
Семейство
Каолинит
Серпентин
н2о
м
Галлуазит
Н
— Катион
Тальк Слюда
Пирофиллит (иллит)
Волокнистый
Сепиолит
Палыгорскит
Катионы
воды
Смектиты
Вермику¬
литы
Октаэдрический
Хлориты
Заняли х
0
0
1-2
0.2—0.6
0.6—0.9
Пеоеменные
Сольватация
глицерином
Нет
7,1 А
Нет
7,3 А
Нет
М7,2 А
Н10А
Нет
Нет
10,1 А
Нет
S 12,1 А
Р 10.48 А
Расширение
17,7 А
Нет
14-15 А
Нет, кроме
набухания
хлоритов
Насыщение К+
Нет
7,1 А
Нет
7,3 А
Нет
М7,2 А
НЮ А
Нет
Нет
10,1 А
Нет
S 12,1 А
Р 10.48 А
Сжатие
12,4 А
12.8 А
Сжатие
10 А
(с 14 А)
Нет
14 А
Нагрев
до 550 °С
Разрушение
Нет
7,3 А
М коллапс
Н коллапс
Нет
Нет
10 А
10,4-8,2 А
9,2 А
4.7 А
Сжатие
10 А
Сжатие
Нет
14 А
Иигеркаляция
формамцда
Нет
7,1 А
< 4ч
Нет
7,3 А
Расширение
М 10,48 А
ДМСО
Расширение
11.18 А
Нет
7.3 А
м
расширение
Проба Цнш-
Келли, 250 °С,
глицерин
Монтмо¬
риллонит
9,5 А
Бейделлит
17.7 А
Глава 4. Характеристика минералогического состава методом РД 109
110
Часть 1. Минералогический анализ почв
Таблица 4.5. Максимальные межплоскостные расстояния в почвенных минералах,
насыщенных ионами Mg2+ и К+ или сольватированных глицерином
Минерал
d(A)
Плоскость hkl
(а) Образцы, насыщенные Mg2+
Хлорит
13,6-14,7
001
Вермикулит
14,0-15,0
002
Монтмориллонит
9,9-10,1
001
Слюда (иллит)
7,2-7,5
001
Тальк
9,2-9,4
002
Галлуазит
10,1
001
Метагаллуазит
7,2-7,5
001
Каолинит
7,1-7,2
001
Лепидокрокит
6,27
020
Бёмит
9,11
020
Гйббсит
4,85
002
Силикаты
4,4-4,6
110
Гйпс
4,27
121
Гетит
4,18
110
Кристобалит
4,04
101
Ильменит
3,73
102
Кварц
3,34
101
Полевой шпат
3,1-3,25
Кальцит
3,03
100
Гематит
2,69
104
Магнетит
2,53
311
Триоктаэдрические силикаты
1,54
060
Диоктаэдрические силикаты
1,49
(б) Образцы, насыщенные Mg2+ и сольватированные глицерином
Монтмориллонит
17,7
001
Вермикулит
14,4
002
Хлорит
13,6-14,7
001
Галлуазит
10,1-10,7
001
Монтмориллонит (2-й порядок)
9,5
001
Хлорит-вермикулит (2-й порядок)
7,15
002
Минералы с межпакетными промежутками, близкими к (а)
(в) Образцы, насыщенные К* и высушенные на воздухе
Хлорит
13,6-14,7
001
Монтмориллонит
11,0-13,0
001
Вермикулит
10,0-11,0
002
Метагаллуазит
7,2-7,5
001
Хлорит (2-й порядок)
7,15
002
Минералы с межпакетными промежутками, близкими к (а)
(г) Образцы, насыщенные К+ и нагретые до 550 °С в течение 2-3 ч
Хлорит
13,6-14,7
001
Монтмориллонит
9,9-10,1
001
Вермикулит
9,9-10,1
002
Слюда (иллит)
9,9-10,1
001
Хлорит (2-й порядок)
7,15
002
111
Глава 4. Характеристика минералогического состава методом РД
Методика насыщения Li+
• Берут навеску, содержащую 110-250 мг глины.
• Готовят суспензию и добавляют 3 М раствор LiCl.
• Оставляют на 30 мин.
• Центрифугируют и сливают надосадочную жидкость.
• Быстро промывают остаток после центрифугирования 1 М раствором LiCl и затем
небольшим количеством воды.
• Готовят препарат ориентированного образца и высушивают при 250 °С в течение
ночи.
• Проводят обработку глицерином (в течение ночи) и получают дифрактограмму.
Примечания
• Это не полностью селективный тест.
• Приготовление препарата на обычном стекле может привести к ошибкам при на¬
гревании, так как ионы Na+ в стекле могут обмениваться на ионы Li+ в глине, что
приводит к неполной нейтрализации октаэдрических зарядов.
• Обработка глицерином должна проводиться в течение нескольких часов для обе¬
спечения полного расширения слоев.
• Иррациональная последовательность базальных рефлексов указывает на присут¬
ствие смешанно-слойных образований.
Удаление железа
Основные положения
Часто необходимо удалить железо из образца, во-первых, чтобы ослабить его влияние на
результаты РД с использованием Cu-анода рентгеновской трубки (рентгеновская флуо¬
ресценция усиливает фоновый шум) и, во-вторых, чтобы избежать снижения интенсив¬
ности дифракции.
Различные формы железа удаляют разными методами. Эти методы различаются по
жесткости и они не должны изменять присутствующие филлосиликаты, но давать воз¬
можность связывать и восстанавливать аморфное и окристаллизованное железо в сла¬
бокислой и минимально агрессивной среде. Эти методы аналогичны методам, исполь¬
зованным в главах 2 и 6, но растворение соединений железа обычно не контролируется
(потому что РД будет подвергаться конечный остаток образца).
Следует отметить, что аморфные кремний-железные соединения, присутствующие
в некоторых осадках, будут диссоциировать в кислой среде с образованием растворимых
форм железа и выделением аморфного кремнезема.
Оксиды гематита и гетита мало подвержены влиянию обработки раствором кислого
оксалата с pH 3. Восстановительная обработка с образованием комплексов продуктами
растворения (щавелевая кислота + дитионит) — лучший способ для удаления большей
части железа, если необходимо экономить образец глины. Щавелевая кислота раство¬
ряет аморфное железо, а дитионит растворяет оксиды железа. Слабокислая среда дает
возможность экстрагировать «свободное железо», но одновременно экстрагирует часть
железа из кристаллической решетки некоторых глин, например, вермикулитов. При ис¬
пользовании дитионита присутствие серы, осажденной после восстановления, не ме¬
шает РД, за исключением эффекта разбавления образца. Серу следует удалить из осадка
после центрифугирования и высушивания.
112
Часть 1. Минералогический анализ почв
Методика
Основные методы основаны на растворении в кислом или щелочном растворе комплек-
сообразователя и/или восстанавливающем растворе (см. гл. 6).
♦ Метод ДЦБ (использование смеси: дитионит + цитрат + бикарбонат) экстрагирует
железо из большинства аморфных и кристаллических соединений восстановлени¬
ем и удалением без значительной модификации алюмосиликатов или литогенного
гематита.
♦ Метод, основанный на использовании кислого оксалата при pH 3,0 (в темноте или
с ультрафиолетовым фотолизом), экстрагирует некристаллические формы железа.
♦ Метод экстракции пирофосфатом натрия при pH 9—10, ЭДТА при pH 9-10 или
ацетилацетоном извлекает металлоорганические формы.
♦ Тетраборатный метод при pH 8—9 экстрагирует Fe из мономерных комплексов.
Удаление железа сопровождается растворением алюминиевых продуктов, кремнезема
и других соединений, что должно контролироваться химическим анализом экстрактов.
Обработка методом сольватации
Основные положения
Сольватация полярными молекулами, такими как одно- или многоатомные спирты,
простые эфиры, амины, приводит к образованию межпакетных органических комплек¬
сов. Образующаяся структура более стабильна, чем структура дегидратированной глины
типа 2:1. Набухание проходит легче всего, поскольку заряд пакетов меньше или ограни¬
чен октаэдрическим пакетом. Природа катионов межпакетного пространства влияет на
пределы стабильности органического комплекса. Базальное расстояние достигает 17,7 А
для смектитов с двойной прослойкой глицерина и 17,1 А с этиленгликолем. Скорость
гидратации может значительно изменяться. Монтмориллониты расширяются легче, чем
большинство глин.
Пропитывание может проводиться в жидкой или паровой фазе при нагревании до
60 °С. В некоторых случаях обработка должна продолжаться в течение 24 ч, с учетом низ¬
кой скорости расширения межпакетных промежутков. Конденсация пара не вызывает
никаких механических нарушений и позволяет получать дифракционные линии боль¬
шей интенсивности.
Методика обработки глицерином
Эта методика основана на процедуре Модре и Диксона (Modre и Dixon, 1970):
♦ Готовят смесь глицерина и воды 1:10.
♦ На пластину с ориентированным образцом, предварительно высушенным на воз¬
духе, очень мелкими каплями аэрозоля напыляют пленку глицерина, стараясь не
создавать избытка реагента.
♦ Оставляют сохнуть не менее, чем на 1 или 2 ч, и затем выполняют рентгенодифрак¬
ционный анализ.
Предупреждение. Соединение теряет эффективность со временем, поэтому рекомен¬
дуется использовать его не позднее, чем через 20 ч. Использование керамической пла¬
стины для этой обработки позволяет удалить избыток глицерина.
Методика обработки этиленгликолем
Эта методика основана на процедурах Элтантавии-Арнольда (Eltantawy и Arnold, 1974)
и Чассина (Chassin, 1974):
Глава 4 Характеристика минералогического состава методом РД
113
• Помещают образец (обычно ориентированные агрегаты) в эксикатор с этиленгли¬
колем (рис. 4.8).
• Создают частичный вакуум при помощи вакуумного насоса и оставляют образцы
в контакте с паровой фазой по крайней мере на ночь.
• Вынимают образец и выполняют рентгенодифракционный анализ как можно ско¬
рее.
Предупреждение. Соединение теряет эффективность со временем, поэтому рекомен¬
дуется использовать его не позднее, чем через 10 ч.
Меры безопасности. Этиленгликоль (1,2-этилендиол), НОСН2—СН2ОН, точка кипе¬
ния 196 °С; гигроскопичный, токсичный при приеме внутрь, может повредить почки,
легкие и сердце.
Соединения включения
Основные положения
Образование соединений включения (интеркалятов) очень полезно, особенно для вы¬
деления глин типа 1:1 и различения хорошо окристаллизованных форм, разупорядо-
ченных форм и галлуазитов. Поскольку эти формы похожи, их нельзя точно разделить
(каолинит и галлуазит могут находиться в плоской, трубчатой и глобулярной формах).
Общая методика включает образование соединения включения, получение дифракто-
граммы, смешивание соединения с водой и проведение сольватации этиленгликолем
или глицерином. Устойчивость соединений включения переменна, и рентгеновские
дифрактограммы следует записывать безотлагательно.
Обработка гидразингидратом (Wada и Yamada, 1968; Range и др., 1969; Calvert, 1984)
позволяет изменить межпакетное расстояние упорядоченного каолинита с 7,15 до
10,48 А без модифицирования хлорита. Присутствие смешанно-слойных минералов мо¬
жет осложнить измерения.
При обработке диметилсульфоксидом (Gonzalez Garciaand и Sanchez Camazano, 1968;
Olejniknjip., 1968; Antonandn Rouxhet, 1977; Calvert, 1984): (i) каолинит образует соедине¬
ние включения, в котором межпакетное расстояние увеличивается с 7,15 до 11,18 А и за¬
тем остается постоянным после нагревания до 300 °С; (И) галлуазит и диккит ведут себя
похожим образом, за исключением того, что нагревание до 300 °С не приводит к даль¬
нейшему расширению; (Ш) в вермикулитах и смектитах это расстояние увеличивается
с 18 до 19 A; (iv) хлорит не подвергается изменению.
Рис. 4.8. Обработка образцов этиленгликолем
114
Часть 1. Минералогический анализ почв
Метагаллуазит образует соединение включения с формамидом. Дифракционная по¬
лоса быстро достигает 10,4 А и более, тогда как реакции с каолинитом не наблюдается
даже после контакта в течение 4 ч (Churchman и др., 1984).
Методика обработки гидразин гидратом
• Помещают образец глины на стеклянную или пористую керамическую пластину
в сатуратор с гидразингидратом.
• Создают частичный вакуум при помощи вакуумного насоса и оставляют образец
в контакте с паровой фазой при 65 °С по крайней мере на ночь.
• Вынимают образец и незамедлительно проводят рентгенодифракционный ана¬
лиз.
Примечания
Следует отметить, что соединение теряет эффективность со временем, поэтому реко¬
мендуется провести измерения дифрактограмм не позднее, чем через 1 ч.
Последовательная обработка гидразином, водой и глицерином позволяет увеличить
базальное межпакетное расстояние в галлуазите до 11,1 А, тогда как в каолините оно
остается равным 7,15 А.
Меры безопасности
Гидразингидрат (H2N-NH2H20, точка кипения 119 °С) смешивается с водой и этано¬
лом, является сильным основанием, очень агрессивен (разрушает стекло и кожу), высо¬
котоксичный (вызывает раздражение глаз, может иметь канцерогенные свойства).
Методика обработки диметилсульфоксидом
• Помещают навеску глины массой от 100 до 250 мг в 5 мл диметил сульфоксида для
создания суспензии.
• Оставляют на 8 ч в водяной бане при ~40 °С.
• Время от времени перемешивают.
• Центрифугируют, готовят препараты и оставляют сохнуть.
• Сразу после высушивания выполняют рентгенодифракционный анализ.
Меры безопасности
Диметилсульфоксид ((CH3)2OS, точка кипения 189 °С) гигроскопичен, раздражает кожу
(образование волдырей); следует остерегаться попадания в глаза.
Методика обработки формамидом
• Помещают образец глины на стеклянную или предпочтительно пористую керами¬
ческую пластину и оставляют сохнуть.
• Испаряют формамид и замечают время; после удаления избытка формамида (при¬
мерно через 20 мин) выполняют рентгенодифракционный анализ (измерения по¬
вторяют в конце дня и сравнивают два результата).
Меры безопасности
Формамид, HCONH2, ионизирующий растворитель, который может слегка пахнуть ам¬
миаком. Растворяет лигнин. Степень опасности для здоровья неизвестна.
115
Глава 4. Характеристика минералогического состава методом РД
Термические обработки
Основные положения
Гкдратированные минералы подвергаются изменениям под влиянием повышенных тем¬
ператур. Эти изменения происходят при определенных характеристических температу¬
рах. Продолжительность выдержки при определенной температуре также существенна.
Превращения происходят с разной скоростью в зависимости от природы и степени
кристалличности термочувствительных минералов. Обычно требуется нагрев в течение
2-4 ч.
Методика
Помещают образцы на стеклянных пластинах в холодную печь, доводят до требуемой
температуры (110, 350,490, 530 °С и др.) и выдерживают при ней не менее 4 ч. Нагрева¬
ние должно быть постепенным, чтобы избежать растрескивания стеклянных пластин
и образования сетки трещин на пленке глины. Затем печь медленно охлаждают, откры¬
вая дверцу, если необходимо несколько ускорить процесс. Если не произошло необрати¬
мых деформаций, пластины с препаратами можно хранить в эксикаторе до выполнения
рентгенографического анализа. Влияние термических обработок на дифракции глин
показано в табл. 4.6. Данные для оксидов алюминия и гидроксидов железа приведены
в табл. 4.7.
Наблюдения
Потеря межпакетной воды приводит к сокращению ячейки и смещению основных ли¬
ний дифракции.
Нагрев до высокой температуры может привести к разрушению кристаллической ре¬
шетки и рассеиванию характеристических линий рентгеновской дифракции.
Пример
Диссоциация каолинита при нагревании до -490 °С (рис. 4.9) может быть представлена
следующими реакциями:
(1) при 500 °С
Al203*2Si02-2H20 -> Al203*2Si02 (метакаолинит) + 2Н20;
(2) при 925 °С
CAl203‘2Si02 2H20 -> Al203 Si02 (силлиманит) + Si02 ((3-кварц) 4* 2Н20;
(3) при 1100°
Al203*2Si02*2H20 -> а-А1203 + 2Si02 (p-кварц) + 2Н20;
(4) при 1400°
CAl203‘2Si02*2H20 -► 1/3 (3Al203-2Si02) 4- 4/3 Si02(P-KBapn) + 2Н20.
Все эти реакции могут начинаться при 800 К (примерно 527 °С). Легко протекают
превращения: каолинит -» метакаолинит -> силлиманит -> муллиты -» оксиды. По¬
следние являются наиболее стабильными системами, зависящими от температуры. По
термодинамическим причинам первой протекает реакция (1) (рис. 4.9).
116 Часть 1. Минералогический анализ почв
Таблица 4.6.
Влияние термической обработки на дифракцию глин (ДТА — характеристиче¬
ские температуры изменений в кривых дифференциального термического ана-
лиза, см. гл. 7)
Температура, °С
Минерал
4Н
ДТА
■ Тепловой эффект
Глины 1:1
Хорошо
окристаллизованный
500
575-625
Отсутствие дифракции
каолинит
Разупорядоченный
500
550-562
Отсутствие дифракции
каолинит
Дискит
500
665-700
Отсутствие дифракции
Накрит
500
625-680
Отсутствие дифракции
Четырехводный
по
125-150
Удаление воды
галлуазит
500
560-605
Отсутствие дифракции
Двухводный
110
125-150
Удаление воды
метагаллуазит
500
560-590
Отсутствие дифракции
Алюмосилика- Аллофан
110
140-180
удаление воды, отсутствие
ты со структу¬
дифрактограммы
рой ближнего Имоголит
350
Трудная идентификация
порядка
500
500
поРД-»18 А
Исчезновение пика при
18 А, разрушение решетки
Слюды и 2:1
Окристаллизованные
490
700
Дифрактограмма слюды
глины
слюды (мусковит)
350
125-250
до 1000°С
Потеря воды
Иллит-слюдяные
500
350-550
Дифрактограмма слюды
глины
500
700
Дифрактограмма слюды
(001)
Глауконит
500
530-650
Потеря структуры водной
слюды
Селадонит
500
500-600
Структура слюды
Биотит
500
700
Спектр слюды до 700-
1000°С
Вермикулит
300
300
Постепенная потеря воды;
исходный базальный
промежуток (001) зависит
от влажности: изменения
14-13, 8-11,6-9 А
Монтмориллонит
300
300
Исчезновение пика
при 15 -> 9 А
Глава 4. Характеристика минералогического состава методом РД
Таблица 4.6 (окончание)
117
Минерал
Температура, °С
4 Н
ДТА
Тепловой эффект
2:1:1
Хлорит
500
680-800
Отсутствие изменений
глины
Mg-хлорит
500
650
хорошо упорядоченной
структуры
Усиление полосы
при 14 А; полоса при
3,54 А не изменяется
(октаэдрический слой -
820 °С, промежуточный
слой - 640 °С)
Fe-хлорит
500
500
Ослабление полосы при
14 А, которая становится
широкой и диффузной;
октаэдрический слой —
530 °С, Fe2+
промежуточный слой —
430 °С, Fe2+
промежуточный слой —
250 °С, Fe3+
Смешанно-слойный
500
500
(октаэдрический слой —
А1-хлорит
<600
750 °С, промежуточный
слой - 900 °С) эффект
зависит от типа минерала
<200
Дегидратация,
промежуток 12 А становится
>200
слабым и диффузным,
Волокнистые
Сепиолит
350
-►9,8 А;
минералы
350
промежуток 7,6 А —
более интенсивная
800
рекристаллизация
<400
Дегидратация без изменения
300
структуры, пик при 10,5 А
Глины
Палыгорскит
500
440
становится широким
и диффузным, разрушение
800
структуры
Получение оксидов железа для рентгеновской дифракции
Основные положения
Использование рентгеновской дифракции для исследования оксидов железа в почве
требует достаточных концентраций различных железосодержащих фаз. Следовательно,
продукты должны быть концентрированы и очищены без изменения их степени кри¬
сталличности или степени замещения алюминием, а также без химических превраще¬
ний фаз. Для этого могут быть использованы следующие методы:
118 Часть 1. Минералогический анализ почв
Таблица 4.7. Влияние термических обработок на оксиды и гидроксиды алюминия
и железа (ДТА - характеристические температуры изменений в кривых
дифференциального термического анализа, см. гл. 7)
Минерал
Температура, °С
4Н ДТА
Тепловой эффект
Серия минералов железа
Гетит
300
230-280
Дифрактограмма неупорядоченного
гематита
900
900
Дифрактограмма хорошо
окристаллизованного гематита
А1-гетит
350
240-350
Дифрактограмма неупорядоченного
гематита
900
Дифрактограмма хорошо
окристаллизованного гематита
Лепидокрокит
300
230-280
Широкий пику-Ре203
400-500
дифрактограмма гематита
Маггемит
350
350-450
-> дифрактограмма гематита
Магнетит
500
600-800
-> дифрактограмма гематита
Акаганеит
300
200-400
При 300 °С дифрактограмма
акаганеита постепенно ослабевает
420-500
дифрактограмма гематита
8-FeOOH
300
140-260
-> дифрактограмма гематита
с промежуточным гетитом
Фероксигит
5-FeOOH
нестабилен на воздухе при комнатной
температуре -> дифрактограмма гетита
после высушивания на воздухе
Ферригидрит (без Si)
350
350-400
-> дифрактограмма гематита
Ферригидрит (с Si)
500
550-600
дифрактограмма гематита
Серия минералов алюминия
Тйббсит
200
150-200
дифрактограммы бемита и у-А1203
Байерит
200
150-200
-► дифрактограммы бемита и у-А1203
Нордстрандит
200
150-200
-* дифрактограмма
байерита -► дифрактограмма бемита
Бемит
500
450-500
дифрактограмма у-А1203
Диаспор
500
470-500
дифрактограмма неупорядоченного
корунда -> дифрактограмма
окристаллизованного корунда
• разделение (осаждение, разделение по градиенту плотности, магнитная сепараци-
яидр.);
* in situ рентгеновская дифракция непокрытых тонких пластин или извлеченных
микрообразцов с высоким содержанием железа.
Только химические методы способны концентрировать железо, которое может суще¬
ствовать в формах покрытий, аморфных формах (ферригидрит и др.) или формах с раз¬
личными степенями кристалличности (гетит, гематит, лепидокрокит и др.). Глинистые
минералы растворяются в 5 М растворе NaOH при кипячении. Каолиниты, галлуаэит,
Глава 4. Характеристика минералогического состава методом РД
119
гиббсит, аморфные алюмосиликаты и др. разлагаются и растворяются, но глины типа
2:1 более устойчивы и, следовательно, повреждаются лишь частично. Кроме железа,
в остатке находят также смектиты, иллит, кварц, анатаз и рутил. Если количество при¬
сутствующего кремнезема недостаточно, при нагревании с 5 М раствором NaOH фер-
ригидриты способны превращаться в гематит или гетит. Обработка образца раствором
5 М NaOH + 0,2 М диоксида кремния не вызывает значительного изменения скорости
замещения алюминием.
Методика
• Взвешивают в тефлоновых стаканчиках 2-5 г почвы или илистой фракции
(< 2 мкм), в зависимости от общего содержания железа.
• Добавляют 20 мл раствора 5 М NaOH 4* 0,2 М диоксида кремния, накрывают ста¬
канчик тефлоновой пластиной и кипятят в течение 1 часа.
• Охлаждают и декантируют.
• Разбавляют смесь деионизированной водой, затем промывают водой на фильтре до
нейтрального pH.
• Выполняют рентгенодифракционный анализ.
Примечания
• Если почва или глина содержит много филлосиликатов типа 2:1, концентрация
может оказаться слишком низкой; в этом случае полезно записать две дифферен¬
циальные дифрактограммы (дифференциальная рентгеновская дифракция), одну
для исходного продукта, а другую для химически сконцентрированного продукта
или после растворения железа в смеси цитрат - бикарбонат - дитионит (см. гл. 6).
• Следует использовать кобальтовую рентгеновскую трубку (анод) для предотвраще¬
ния флуоресценции железа, которая происходит при использовании медной трубки.
• Политетрафторэтиленовые стаканчики можно использовать .до температуры
250 °С.
120 Часть 1. Минералогический анализ почв
• В некоторых случаях разрушение матрицы силикатов может быть достигнуто об¬
работкой HF в тефлоновом стаканчике.
• Нагревание до 600 °С дает безводные оксиды -> гематит.
4.2.5. Качественная дифрактометрия
Образец почвы, размолотой до размера частиц 0,1 мм, и илистые фракции < 2 мкм (или
субфракции в 0,5 или 0,2 мкм), приготовленные для получения спектров порошковых
неориентированных или ориентированных образцов, анализируют методом рентгенов¬
ской дифракции.
Каждая группа глин имеет собственную пакетную структуру, которая дает базальные
рефлексы, положение, интенсивность и форма которых делает возможным или непо¬
средственную идентификацию или после специальных обработок. В данном разделе
описывается интерпретация стандартных ситуаций, которые позволяют классифици¬
ровать глинистые минералы по шкале групп и подгрупп и очень редко по шкале видов.
Особенности регулировки экспериментальных условий
Экспериментальные условия должны быть определены заранее, включая тип трубки
(Си, Со и т. д.), напряжение нити накала, апертуру щелей, скорость вращения гонио¬
метра, измеритель напряжения, усиление, чувствительность, отношение сигнал/шум,
угловой интервал, скорость сканирования и т. д.
Для образцов, богатых железом, должна использоваться кобальтовая рентгеновская
трубка, потому что она не вызывает флуоресценцию с этими ионами. Как правило, при
соответствующей геометрии медная трубка, соответствующая международному стандар¬
ту, дает удовлетворительные характеристики; и таблицы и программы JCPDS (см. при¬
мечание к разделу 4.2.2) основаны на этом стандарте.
Скорость вращения гониометра и выбор углового интервала сканирования опреде¬
ляют время, необходимое для анализа. Выбор скорости сканирования образца влияет
на относительную точность дифрактограмм. Слишком высокая скорость может вы¬
звать недостаточное разрешение. Для стандартного определения скорость перемещения
1-2 °/мин (угол 20) может быть достаточной, но она не позволяет адекватно разделить
тонкую структуру дублетов каолинита (002) и хлорита (004), для которых требуется бо¬
лее низкая скорость (< 0,5 °/мин). Для быстрой охарактеризации глины (определение
группы и подгруппы) угловой интервал сканирования может быть ограничен зоной
20 = 2-40°, если не требуется определения дифракции tf(060), которое занимает гораздо
больше времени. Сильное отражение при 20 = 60° позволяет оценить параметр b и та¬
ким образом отличить диоктаэдрические минералы (1,48—1,50 А) от триоктаэдрических
минералов (1,53—1,55 А), а иногда и предоставить дополнительные данные для прибли¬
женной оценки величин, приближая эти величины к интенсивности дифракции £/(001),
например, при идентификации некоторых слюд. Порошковые образцы анализируют
при вращении держателя образца в плоскости фокусировки, что требует высокоточно¬
го оборудования. Метод ориентированных образцов позволяет усилить рефлексы (001),
ориентируя частицы по плоскости развития глинистых минералов. Все препараты (или
любая другая форма представления) для каждого образца должны готовиться одновре¬
менно и максимально единообразно (с использованием одной и той же суспензии, гли¬
ны того же качества и т. д.). Обычно требуются три препарата образца:
• контрольный необработанный ориентированный препарат, высушенный и про¬
анализированный при известной относительной влажности;
121
Глава 4. Характеристика минералогического состава методом РД
* сольватированный ориентированный препарат для гидратированных глин типа
2:1;
• ориентированный препарат для термической обработки (500 °С в течение 4 ч.).
Образцы следует анализировать один за другим, и для более точного сравнения спек¬
тров оборудование должно быть отрегулировано абсолютно аналогично для всех об¬
разцов. В более сложных случаях может понадобиться готовить гомоионно (К+, Mg2+)
насыщенные образцы, используя подходящую последовательность обработок и ин-
теркаляций. Операции должны выполняться в одинаковых условиях, с учетом отно¬
сительной влажности воздуха, относительной стабильности сольватов и соединений
включения, времени реакции (обработка формамидом галлуазита проходит быстро,
а для каолинита необходимо 4—5 ч и, следовательно, их обработка должна выполняться
в начале серии экспериментов). Если образец очень мал, можно выполнять обработки
и измерения последовательно на одном и том же образце. В этом случае следует исполь¬
зовать пористую керамическую пластину (или фильтры Миллипор размером 0,20 мкм,
если не предусмотрена тепловая обработка). Однако когда измерения должны прово¬
диться в течение нескольких дней, поддерживать постоянные условия трудно.
Анализ дифрактограмм
Фоновый шум
Фоновый шум вызывается неспецифическими сигналами, свойственными матери¬
алу, процессу, минералам с упорядочением ближнего порядка и, возможно, явлениям
флуоресценции вследствие эмиссии вторичных рентгеновских лучей. Последнее явле¬
ние может быть устранено использованием монохроматора с обратным прохождением
лучей и амплитудной дискриминацией. Фоновый шум можно сгладить и при помощи
соответствующего программного обеспечения. В области малых углов (0 < 20 < -10°)
в зависимости от регулировки щелей счетчик может получать часть прямого пучка или
отражений от краев держателя. Использование регулируемых щелей обычно предотвра¬
щает эти явления.
Широкие и диффузные полосы указывают на присутствие аморфных веществ (алю¬
мосиликаты, различные оксиды и пр.). Программы интерпретации дифрактограмм по¬
зволяют селективно удалить эти области и стабилизировать базовую линию.
Геометрия пиков
Геометрия пиков делает возможным определение степени кристалличности, особенно
для хорошо окристаллизованных минералов, размеры кристаллов которых соответству¬
ют длине волны рентгеновского излучения, а регулярные межплоскостные изменения
дают четкие дифракции и многочисленные гармоники (рис. 4.5, а). Плохо ©кристалли¬
зованные минералы с неупорядоченной структурой и размером кристаллов, не соответ¬
ствующим рентгеновским лучам, часто дают широкие двояковыпуклые пики, площадь
которых не может быть использована для количественного анализа (например, смектит,
гетит, см. рис. 4.5, б). Слишком много таких частиц как диатомеи, кварц или полевые
шпаты, могут ограничить ориентацию глин и затруднить интерпретацию спектров. На¬
ложение пиков, вызванных совместным присутствием нескольких минералов, приво¬
дит к уширению сигналов и аномальному увеличению относительных интенсивностей,
например, для пар хлорит (001) - вермикулит или каолинит (001) - хлорит (002). Эти
образцы требуют различной предварительной обработки для уменьшения неопределен¬
ности и выявления скрытых пиков (см. раздел 4.2.4).
122
Часть 1. Минералогический анализ почв
Если слой образца недостаточно толстый, то пики могут быть широкими и малоин¬
тенсивными даже в случае минералов, для которых они обычно хорошо различимы, та¬
ких как кварц, кальцит или упорядоченный каолинит (001 или 002). Гетиты и смектиты
имеют более диффузные пики, которые могут маскироваться. При некоторых углах ска¬
нирования появляются также рефлексы от подложки.
Материалы с размером частиц около 0,5 мкм используют для сравнительного иссле¬
дования различных илистых фракций и для количественного анализа, когда разделение
проводилось методом ультрацентрифугирования. Должно контролироваться уширение
на полувысоте пиков и других, более крупных или более мелких фракций. Действи¬
тельно, если кристаллиты слишком тонкодисперсные, рефлексы уширяются; если они
слишком крупные, явления поглощения могут исказить интенсивность. Следует отме¬
тить, что смектиты накапливаются в самых мелких фракциях; каолиниты, иллиты, хло¬
риты, оксиды железа — во фракциях 0,2—0,5 мкм; обломочные слюды, хлориты, кварц,
полевые шпаты — во фракциях 0,5-2 мкм. Хорошо окристаллизованный, но не полно¬
стью расслоенный каолинит можно обнаружить в форме мелкой пыли.
Определение межплоскостных расстояний из углов
Значение угла расположения вершин пиков дифракции должно быть преобразовано
в межплоскостное расстояние d (А) в соответствии с уравнением Брэгга (4.1) (с помо¬
щью таблиц или компьютерных программ с учетом типа используемого анода). Каждый
пик нумеруют в порядке регистрации и приводят соответствующее межплоскостное рас¬
стояние в А или нм (международный стандарт), а также относительную интенсивность
пика. Компьютерные программы позволяют проводить визуальное сравнение на экране
со справочными дифрактограммами и автоматический поиск по базе данных JCPDS(см.
примечание к разделу 4.2.2).
Рентгенограмма порошка цельной почвы дает представление обо всех присутствующих
минералах и относительных интенсивностях пиков различных компонентов. Спектр
может быть труден для интерпретации, если он получен без сравнения с другими образ¬
цами той же серии, но можно отобрать соответствующие образцы для более тщательно¬
го исследования. Илистые фракции порошков, очищенные и концентрированные рас¬
сеиванием и сепарацией по размеру частиц, дают более точную информацию по глинам
и ассоциированным минералам. Можно определить основные пики с высокой интен¬
сивностью и комбинации характеристических пиков для межплоскостных расстояний
7—, 10—, 12-, 14—15 А, а также пики кварца, кальцита и др. минералов и, возможно,
пики при 4,40-4,50 А, которые идентифицируются в большинстве глин. Те же ограниче¬
ния существуют для сложных смесей. Пики часто широкие, а их профили асимметрич¬
ные; смектиты дают воспроизводимое межслоевое пространство, только если контроли¬
руется состояние их гидратации и ионного насыщения.
Присутствие смешанно-слойных минералов может вызывать трудности, особенно
для ди- и триоктаэдрических подгрупп из-за помех в зоне (060) около 1,50 А. В зави¬
симости от целей исследования дифрактограмма помогает определить стратегию, на¬
пример, использование коррекции условий эксперимента или определение количества
ориентированных пластин после конкретных обработок и необходимости дополнитель¬
ного использования других методов, таких как термический анализ, ИК-спектроскопия,
электронная микроскопия, селективное растворение, химическая очистка и т. д.
На дифрактограммах ориентированных образцов, полученных после соответствую¬
щих обработок, базальные рефлексы, относительные интенсивности и перегруппировки
диагностических рефлексов при использовании интерпретационных таблиц JCPDS-ICDD
123
Глава 4. Характеристика минералогического состава методом РД
(см. примечание к разделу 4.2.2) могут быть быстро представлены на экране с помощью
соответствующего программного обеспечения. Можно использовать таблицу Бриндли
и Брауна (Brindley и Brown, 1980), в которой учитываются самые интенсивные характери¬
стические линии глин и связанных минералов почв, расположенных в порядке убывания
величины параметра d. Каталог минералов Ханаволта произвольно делит межплоскост¬
ные расстояния от 999,99 до 1,00 А на 40 групп. Первая позиция соответствует линии мак¬
симальной интенсивности. Величина d второй линии соответствует второй по интенсив¬
ности линии и определяет подгруппу. Внутри каждой подгруппы записи расположены по
убыванию интенсивности шести дополнительных линий. Поскольку интенсивность опре¬
делить трудно, несколько записей позволяют объединять экспериментальные варианты:
если дифракция минерала включает 2 или 3 пика большой интенсивности, могут быть 2
или 3 записи (с интенсивностями в диапазоне от 100 до 75). Запись положения восьми
пиков должна сопровождаться названием минерала и номером карточки этого минерала
в таблицах JCPDS. Рассмотрение превращений минералов при разных химических и тер¬
мических обработках облегчает идентификацию глинистых минералов (см. табл. 4.4-4.7).
4.3. Количественный минералогический анализ
4.3.1. Практическая значимость
Количественный минералогический анализ используют для определения факторов,
влияющих на текущие или предыдущие педогенные показатели, а также физические
и химические свойства почв. Этот тип анализа является логическим продолжением ка¬
чественного анализа, однако встречается со многими препятствиями, и его точность за¬
висит от химического состава и сложности структуры субстрата. Для простого субстрата,
состоящего из двух видов минералов, в зависимости от используемого метода, величи¬
на ошибки может составлять около 3%, но она может достигать 5—10% для смеси трех
видов минералов и около 30% для смесей из п видов. Присутствие веществ с упорядо¬
чением ближнего порядка еще более увеличивает степень неопределенности и может
даже помешать использованию некоторых методов, если такие вещества преобладают
в смеси. Используют следующие методы:
* простые инструментальные методы, из которых наиболее распространенным явля¬
ется рентгеновская дифракция, или
• группу методов, позволяющих лучше идентифицировать процентный состав об¬
разца, хотя и за счет усложнения процедуры и удорожания оборудования.
4.3.2. Количественный минералогический анализ методом
рентгеновской дифракции
Основные положения
Преимуществом данного аналитического метода являются его относительная простота
и особенно использование одного прибора. Интенсивности дифракции рентгеновских
лучей, полученные для каждого компонента смеси, прямо коррелируют с его содержа¬
нием в соответствии с уравнением:
г kW.
I — р р
1 р “ — »
Ц
(4.2)
где Wp - вес компонента Р в образце; 1р - интенсивность дифракционной линии чистого
соединения Р; кр - константа, зависящая от природы соединения Р и эксперименталь¬
ных условий; р - массовый коэффициент ослабления смеси.
124
Часть 1. Минералогический анализ почв
Если вещества хорошо закристаллизованы и имеют определенный химический со¬
став, дающий интенсивные неперекрывающиеся рефлексы рентгеновской дифракции,
они легко определяются количественно с приемлемой точностью, особенно при ис¬
пользовании стандарта с одинаковыми химическими характеристиками. К сожалению,
несовершенство почвенных минералов (структурное упорядочение или разупорядоче-
ние), размер кристаллитов, непостоянство химического состава, связанное с замещени¬
ями, и преимущественные ориентации1 приводят к изменениям в интенсивности и даже
угловому смещению пиков (что осложняет выбор эталонных минералов для калибро¬
вочных измерений). По возможности, эталонные минералы должны выбираться из той
же геологической формации и быть подвергнуты педологическому изменению того же
типа с тем, чтобы воспроизвести матрицу с аналогичной степенью беспорядка и хими¬
ческим составом (глины в процессе трансформации).
Образцы для количественного анализа должны готовиться с особой тщательностью
для ограничения уширения полос (чрезмерное измельчение может приводить к разу-
порядочению структуры, а также увеличению количества аморфной фазы), эффектов
ослабления и микропоглощений (кристаллиты и слишком крупные частицы) и ориента¬
ции, из-за взаимных связей между частицами (преимущественная ориентация).
Измерения проводят на:
* образцах порошков, в которых учитываются все виды рефлексов; поскольку отно¬
сительная интенсивность базальных рефлексов мала, может понадобиться концен¬
трация около 10% для количественного определения данного соединения в смеси;
• ориентированном образце, что увеличивает базальные рефлексы глин, однако вле¬
чет риск помех для регулярности ориентации со стороны некоторых компонентов;
этот метод более чувствителен и расширяет пределы определения, но требует при¬
готовления образца не на стеклянной, а на пористой керамической пластине для
устранения влияния седиментации (см. раздел «Агрегаты, ориентированные на по¬
ристой керамической пластине»).
Подготовка образцов может включать насыщение ионами Mg2+, сольватацию глице¬
рином и, наконец, смешивание с внутренним стандартом и гасителем интерференции
матрицы. Гомогенизация образца и внутреннего стандарта должна проводиться в мик¬
сере с агатовым шаром. Если образцы глин разделяются ультрацентрифугированием,
следует удостовериться в отсутствии изменений состава (относительное содержание
каждой глины в смеси может различаться и приводить к сдвигу результатов). Можно
использовать три типа количественных методов рентгеновской дифракции: непосред¬
ственный анализ без стандарта, анализ с использованием внешнего стандарта и анализ
с использованием внутреннего стандарта (добавление стандарта, гасителя интерферен¬
ции матрицы и т. д.).
В прямом анализе без стандарта эталонный минерал выбирается из матрицы образца.
Получают интересующие относительные величины для сравнения образцов из одной
и той же последовательности.
Анализ с использованием внешнего стандарта не требует длительной подготовки, но
выбор стандартных веществ для расчета интенсивностей также сложен, как в других ме¬
тодах. Поэтому точность может быть сомнительной, за исключением соединений, даю¬
щих четкие рефлексы на относительно чистом фоне с малым числом основных компо¬
нентов, например, хорошо окристаллизованный каолинит или кварц.
1 Текстурирование. — Примеч. науч. ред.
125
Глава 4. Характеристика минералогического состава методом РД
Анализ с использованием внутреннего стандарта: такие анализы выполняют в присут¬
ствии стандартного вещества, имеющего (i) низкий коэффициент ослабления, (ii) силь¬
ный, узкий и неперекрывающийся пик рентгеновской дифракции (или пики определя¬
емых минералов) с малым числом гармоник, если возможно, и (ш) плотность, близкую
к плотности минералов в смеси, что улучшает гомогенизацию.
Корунд а-А1203 был принят JCPDS (см. примечание к разделу 4.2.2) в 1976 г. в каче¬
стве эталонного стандарта для количественного исследования минералов (синтетиче¬
ский корунд высокой чистоты, размер частиц 1 мкм, производства Линде, Юнион Кар-
байд или похожие). Имеет четкий базальный рефлекс при 2,085 А (/= 100 для плоскости
ИЗ). Степень кристалличности можно увеличить нагреванием до 800 °С в течение 1 ч.
Преимущественная ориентация отсутствует. Можно также использовать линию MgO
при 2,106 А и линию бемита у-АЮОН при 6,11 А.
Можно использовать различные методики и способы расчета, например, метод стан¬
дартных добавок или гашение дифракции матрицы. Метод стандартных добавок длителен
и требует построения градуировочных кривых. Метод Чанга (Chung, 1974а, Ъ) с гашением
дифракции матрицы требует меньше времени и позволяет, если необходимо, определить
присутствие аморфных минералов с упорядочением ближнего порядка в дифференци¬
альном балансе1. Однако необходимо получить дифрактограммы, основные рефлексы
которых не содержат диффузных линий или зон перекрывания, поскольку эти сигналы
требуют слишком сложной обработки. Этот метод позволяет исключить влияние ма¬
трицы на зависимость интенсивности от концентрации, а все интенсивности получа¬
ют при одном сканировании, что снижает возможные инструментальные погрешности.
Методика
Спектры записывают для порошковых образцов, чистого корунда и смеси «образец —
корунд»:
• Взвешивают корунд (к) и образец (А) в соотношении 1:1.
• Гомогенизируют в горизонтальном миксере с агатовым шаром в течение 20 мин.
• Насыпают порошок в держатель и слегка уплотняют; разравнивают лезвием брит¬
вы для получения гладкой, но неориентированной поверхности; плоскость фоку¬
сировки должна быть безупречной2.
• Помещают в стандартных условиях в дифрактометр с медной трубкой, с перемен¬
ной щелью и графитовым монохроматором с постоянной времени, позволяющей
выполнить не менее 20 000 отсчетов на пик (без фонового шума) при скорости ска¬
нирования 20 = 0,5 °/мин (или даже 20 = 0,25 °/мин); сканируют только зону, со¬
держащую существенные пики.
• Определяют положение наиболее интенсивных пиков дифракции, представитель¬
ных для каждого компонента, определяют их интенсивность и сравнивают с вну¬
тренним стандартом матрицы.
1 Если образец почвы содержит неразложенное органическое вещество или вещества, про¬
зрачные для рентгеновских лучей, эти вещества могут быть определены в дифференциаль¬
ном балансе.
2 Некоторые авторы предпочитают готовить образцы таблетированием, хотя существует риск
определенной ориентации частиц порошка под сильным давлением. Однако останутся по¬
грешности, связанные с качеством поверхности или плотностью участков образца, способ¬
ных к дифракции.
126
Часть 1. Минералогический анализ почв
Расчеты
Основываясь на природе монохроматического рентгеновского излучения с определен¬
ной длиной волны, природе эффектов матрицы (поглощение) и базовом уравнении
Клуга-Александера (Klug и Alexander, 1959), Чанг математически выделил эффекты
ослабления массы:
KiXJQi KiXiIQi
2>д, А
где /. — интенсивность рентгеновского луча, дифрагированного выбранной плоскостью
компонента / (неизвестна); Kt — константа, зависящая от геометрии дифрактометра
и природы компонента /; X. — вес фракции компонента /; Q. — плотность компонента /;
р, — коэффициент массовой абсорбции (или массовый коэффициент ослабления) чи¬
стого компонента /; рг — коэффициент поглощения всего образца, включая компонент
I, внутреннего стандарта и, возможно, эталонного материала.
Две последних переменных характеризуют влияние поглощения, которое часто труд¬
но измерить другими методами. Введение определенной массы гасителя матрицы (ко¬
рунд, похожий на внутренний стандарт) позволяет ввести: Xf — вес гасителя матрицы
(f — выравнивающий агент) и Х0 — вес образца, а также уравнение:
^+*„=^+£*,.=1,
i=i
где п — число компонентов в образце, и
ии^а'/Ф^/х,)^/»,),
где If и If обозначают интенсивности рентгеновского луча, дифрагированного выбран¬
ной плоскостью каждого чистого компонента. Вводя отношение между интенсивностя¬
ми эталона kf = IJIC и другие подстановки, получим уравнение:
Х,= Х,(к,/к) (/,//,), (4.4)
описывающее соотношение между интенсивностью и концентрацией, из которого ис¬
ключено влияние матрицы, и используемое в количественном многокомпонентном ана¬
лизе. При использовании корунда в качестве гасителя дифракции матрицы получаем
простое конечное уравнение (при kf = kc= 1):
Х,=(Хс/к()(1(/1с), (45)
где Xt — вес фракции образца; Хс — вес корунда; I — интенсивность луча, дифрагирован¬
ного образцом; / — интенсивность луча, дифрагированного корундом; к{ = 1(/1с — от¬
ношение интенсивностей эталона (табл. 4.8).
Примечания
Используя компьютеризированное оборудование, можно определить высоту рефлекса
и его ширину на полувысоте1 или площадь рефлексов £/(001), которые программа рас¬
считывает автоматически с учетом стабилизированного фонового шума.
1 Если необходимо сравнить образцы, измеряя интенсивности, следует отметить, что отно¬
шение высот пиков двух образцов можно использовать, только если ширины их пиков на
полувысоте идентичны.
Глава 4. Характеристика минералогического состава методом РД 127
Таблица 4.8. Рекомендуемые отношения /// (уравнение 4.5, Bayliss, 1986)
Минерал
hi
</, А
Минерал
щ
d, А
Аллофан
0,1
3,3
Иллит
0,7
10
Биотит
0,0
10,0
Иллит-монтмориллонит
0,4
12
Бемит
1,0
6,11
Каолинит lMd
М
7,1
Кальцит
3,7
3,03
Каолинит1Т
2,1
7,1
Корунд
1,0
2,09
Мусковит
2,2
10
Диккит
2,9
7,2
Кварц
4,3
3,35
Гкббсит
1,6
4,85
Смектит
3,0
15
Гетит
1,4
4,18
Тальк
1,5
9,3
Вше
2,2
2,87
Гематит
0,9
2,70
Могут понадобиться дополнительные измерения на ориентированных образцах: на¬
сыщенных Mg2+ или сольватированных образцах, а также использование умножающих
коэффициентов, учитывающих структуру минералов (волокнистые глины с псевдослоя¬
ми, не дающие интенсивных отражений, и т. п.).
Можно объединять средние результатов по двум рефлексам, так как это предостав¬
ляет информацию о степени окристаллизованности и т. п. Выбор зависит от профиля
дифрактограммы, природы компонентов и желаемой точности.
Величины к{ для некоторых минералов изменяются в зависимости от геологического
источника и природы педологических изменений, поэтому измерения должны прово¬
диться в одинаковых условиях. Выбор минералов проводят после химического анализа
и рентгеновской дифракции.
Отношение It/Ic зависит от степени кристалличности, оно приближается к 0, если
минерал не кристаллический (аллофан — материал с упорядочением ближнего порядка)
и может достигать 8-9 при оптимальном размере кристаллитов и степени кристаллич¬
ности. Изменение химического состава (например, тяжелые ионы Fe2+ в минералах за¬
мещаются легкими Mg2+) изменяет отношение 1{/1с.
4.3.3. Количественный минералогический анализ с использованием
сложного технического оборудования
Количественные методы, основанные на рентгеновской дифракции, имеют ограничен¬
ную точность, в частности при работе со сложными агрегатами, дающими диффузные
рефлексы или рефлексы, маскируемые из-за наложения, либо в присутствии больших
количеств веществ, аморфных для рентгеновского излучения. Мультиинструменталь¬
ные методы объединяют измерения, основанные на использовании рентгеновской диф¬
ракции и других химических и физических методов, что позволяет характеризовать раз¬
личные компоненты. Измерения обычно проводят на илистых фракциях, но они могут
быть дополнены другими измерениями, например, органического вещества, разрушен¬
ного пероксидом водорода, карбонатов, растворимых солей, оксидов железа и других
веществ, удаленных в процессе выделения илистой фракции < 2 мкм, и, наконец, ана¬
лизами песка и пыли, отделенных рассевом.
Эти методы могут использоваться только в специализированных лабораториях, об¬
ладающих широким ассортиментом инструментальных методов, таких как РД, ИК-
спектроскопия, ТЭМ, ДТА—ТГА, ААС, ИСП и др. Каждый компонент смеси имеет соб¬
ственные химические и физические характеристики, которые могут быть определены.
128
Часть 1. Минералогический анализ почв
Сначала качественно интерпретируют спектры рентгеновской дифракции для иденти¬
фикации глинистых минералов (см. разделы 4.2.4 и 4.2.5).
Робертс и Роберт с сотр. (Roberts, 1974; Robert и др., 1991) количественно определи¬
ли различные компоненты, используя их специфические свойства. Структура, размер
и форма частиц позволяют сделать правильный выбор и подтвердить идентификацию
такими методами, как ПЭМВР, СПЭМ, РСМА (см. гл. 8) для минералов, присутству¬
ющих лишь в малых количествах и не поддающихся определению методом РД. Терми¬
ческий анализ может играть существенную роль в количественном определении као¬
линита, оксигидроксидов и хлорита. Используют потери массы в диапазонах 100—300,
300-600 и 600—950 °С. При этом необходимо вводить поправки на окисление железа
при высоких температурах.
Общий химический анализ глины после обработки смесью HF—НС1 (см. гл. 31) позво¬
ляет определить содержание различных присутствующих элементов, что имеет большое
значение для идентификации структуры минеральных фаз. Содержание общего калия
позволяет определить слюды, исходя из 7,5% К в иллитах и 8,3-10% К в других слю¬
дах. Селективное растворение (см. гл. 6) с использованием соответствующих методик
позволяет проводить преимущественное растворение отдельных фаз без существенного
влияния на другие компоненты. Например, обработка образца (нагретого до 110 °С в те¬
чение 4 ч) 0,5 М раствором NaOH с последующим кипячением в течение 2,5 ч позволяет
выделить аллофан (42,7% Si02,36,3% А12Оэ) и такие некристаллические соединения, как
коллоидный кремнезем (также растворяется немного монтмориллонита и вермикулит).
Сплавление с пирофосфатом или обработка смесью трех кислот (H2S04—HN03—НС1)
позволяет отделить кварц и полевые шпаты в остатке после обработки. Затем остаток
взвешивают. Его вес следует увеличить на 3% для компенсации растворения неболь¬
ших количеств кварца и полевых шпатов. Остаток анализируют методом рентгеновской
дифракции для выявления присутствия полевого шпата и затем проводят химический
анализ после растворения. Растворение в реактиве Тамма (в темноте или в условиях УФ
фотолиза) дает возможность выделить некристаллические формы железа, а метод ЦБД
(см. гл. 6) — кристаллические гидроксиды железа.
Анализы, определяющие активность глин, позволяют проводить разделение некото¬
рых глин типа 2:1 по их емкости катионного обмена, используя различные обработки:
насыщение ионами Na* и замещение ионами Mg2*, насыщение ионами Mg2* и замеще¬
ние ацетатом аммония и т. д. Общую удельную поверхность (внешнюю и/или внутрен¬
нюю) можно определить, используя этиленгликоль моноэтиловый эфир, поглощение
метиленового голубого или БЭТ-метод.
Таблица 4.9. Некоторые свойства минералов, используемые для уточнения результатов
Минерал
ЕКО,
смоль/кг
Сдельная поверхность,
М2/г
Потеря воды при 540-900 °С, %
Слюда
25
175
0,50
Кварц
2
25
0
Полевые шпаты
2
25
0
Аллофан
100
800-1000
0
Каолинит
5-10
45
0
Коллоидный Si02
20
200
2
Монтмориллонит
80-100
800
0,88
Вермикулит
175-200
440
1,83
Хлорит
25
175
12,30
Глава 4. Характеристика минералогического состава методом РД
129
Вся эта количественная информация, введенная в компьютерные программы, позво¬
ляет уточнить содержание каждого компонента в смеси, установить пределы свойств для
различных соединений и выбрать условия для расчетов.
Использованная литература
Anton О and Rouxhet PG (1977) Note on the intercalation of kaolinite, dickitte and halloysite by
dimethyl-sulfoxide. Clays Clay Minerals, 25, 259-263.
Bayliss P (1986) Quantitative analysis of sedimentary minerals by powder X-Ray diffraction. Powder
diffraction, 1,37-39.
Brindley GW and Brown G (1980) Crystal structure of clay minerals and their X-Ray identification.
Mineralo. Soc., 415-438.
Calvert CS (1984) Simplified, complete CsCl-hydrazine-dimethylsulfoxide intercalation of kaolinite.
Clays Clay Miner, 32, 125-130.
Chassin P (1974) Influence de la st^ochimie des diolssur la formation des complexes interfoliaires de la
montmorillonitecalcique. Clay Miner., 11,23-30.
Chung FH (1974a) Quantitative interpretation of X-Ray diffraction pattern of mixtures. I — matrix
flushing method for quantitative multi-component analysis. J. AppL Crystallog., 7, 519-525.
Chung FH (1974b) Quantitative interpretation of X-Ray diffraction patterns of mixtures. II - Adiabatic
principle of X-Ray diffraction analysis of mixtures. J. AppL Crystallog., 7, 526-531.
Churchman GJ, Whitton JS, Claridge GGC and Theng BKG (1984) Intercalation method using formamide
for differentiating halloysite from kaolinite. Clays Clay Miner., 32, 241-248.
Decarreau A (1990) Les poudres: techniques expdrimentalesetinterprdtation des diagrammes -
Facteursd&erminant le mode d’empilement. In Structure, propri£t£set applications. Soci6t6Fran9aise
de Min6ralogieetcristallographie, GroupeFran9ais des Argiles, 209-236.
Eltantawy IM and Arnold PM (1974) Ethylene glycol sorption by homoionicmontmorillonites. J. Soil
Sci., 25, 99-110.
Gonzalez Garcia S. and Sanchez Camazano M. (1968) Differenciation of kaolinite from chlorite by
treatment with dimethylsulfoxyde. Clay Miner. Bull., 7, 447-450.
Klug HP and Alexander LE (1974) X-Ray diffraction procedures. Wiley 2nd edition.
Modre DZ and Dixon JB (1970) Glycerol vapor adsorption on clay minerals and montmorilllonite soil
clays. Soil Sci. Soc. Am. Proc., 34, 816-822.
Olejnik S, Aylmore LAG, Posner AM and Quirk JP (1968) Infra-red spectra of kaolin mineral-dimethyl
sulfoxide complexes. J. Phys. Chem., 72,241-249.
Paterson E, Bunch JL and Duthie DML (1986) Preparation of randomly oriented samples for X-Ray
diffractometry. Clay Miner., 21, 101-106.
Range KJ, Range A and Weiss A (1969) Fireclay type kaolinite or fire-clay mineral? Experimental
classification of kaolinite - halloysite minerals. Proceedings of the International Clays Conference
(Tokyo). Israel Universities Press, 3-13.
Roberts JM Jr (1974) X-Ray diffraction and chemical techniques for quantitative soil clay mineral
analysis. Engineering Thesis, Pennsylvania State University, 78 pages.
Robert M, Hardy M and Elsass F (1991) Crystallochemistry, properties and organization of soil clays
derived from major sedimentary rocks in France. Clay Miner., 26: 409-420.
Thomson A, Duthie DM, Wilson MT (1972) Randomly oriented powders for quantitative determination
of clay minerals. Clay Miner., 9, 345-348.
Wada К and Yamada H (1968) Hydrazine intercalation, intercalation for differentiation of kaolin mineral,
from chlorites. Am. Miner., 53, 334-339.
Дополнительная литература
Основные положения
Alekseeva TV, Alekseev AO, Sokolovska Z, Khainos M, Sokolowska Z and Hajnos M (1999) Relation¬
ship between mineralogical composition and physical properties of soils. Pochvovedenie, 5, 604—
613.
130
Часть 1. Минералогический анализ почв
Brindley GW and Brown E (1980) Crystal structure of clay minerals and their X-Ray identification.,
Mineralogical Society, 495 p.
СаШёге S, H<5nin S and Rautureau M (1982) Mineralogie des argiles, 7, Masson, 184 p.
Cailldre S, H6nin S and Rautureau M (1982) Miniralogie des argiles, 2, Masson, 189 p.
Charley H (1989) Clay Sedimentology, Springer Belin Heidelberg New York, 623 p.
Dixon JB and Weed SB (1989) Minerals in soil environments, Soil Science Society of America (USA),
2e edition, 1244 pp.
Gautheyrou J and Gautheyrou M (1979) Etude des argiles par diffraction X. Synthesebibliographique
pour Гidentification des argiles., Guide pratique.ORSTOM-Antilles, notes de laboratoires, OR-
STOM (26 pages + 2 annexes).
1CDD (JCPDS-ASTM)) Mineral powder diffraction File - PDF-1 DATA BASE (powder diagramms,
interlayer spaces, relative intensity, chemical name, mineralogical formula) — PDF-2 DATA BASE
(powder diagramm, interlayer spaces, relative intensity, chemical name, mineralogical name, Mill¬
er’s indice, unit cell, physical properties, references) - Shorten ICDD ref: eliminate - version papier
(SET 1 a 36, SET 1 a 8 revises, SETS 37,38,39,40,41) - version disque compact (CD-ROM DISC
SETS 1-41 inorganique - organique pour IBM PC, VAX, McIntosh) - SEARCH MANUAL Alpha¬
betical index - inorganic phases. Hanawalt index - inorganic phases (remise a jour annuelle), ICDD
Newton Square PA 19073-3273 (USA).
Inigo AC, Tessier D, Pemes M (2000) Use of X-ray transmission diffractometry for the study of clay-
particle orientation at different water contents. Clays Clay Miner, 48,682-692.
Kovda IV, Morgun EG, Tessier D, Pemes M (2000) Particle orientation in clayey soils according to trans¬
mission diffractometiy data, 8,989-1003.
Manhaes RST, Auler LT, Sthel MS, Alexandre J, Massunaga MSO, Carri6 JG, dos Santos DR, da Silva
EC, Garcia-Quiroz A and Vargas H (2002) Soil characterisation using X-ray diffraction, photoacous¬
tic spectroscopy and electron paramagnetic resonance. Appl. ClaySci., 21, 303-311.
Martins E de S, de S Martins E (2000) Integrated method of mineralogical characterization of deeply
weathered soils.ComunicadoTecnicoEmbrapaCerrados., Brazil, 37, 5 pp.
Millot G (1964) Geologie des argiles. Masson, Paris, 499 pp.
Newman AC (1987) Chemistry of clays and clay minerals, Mineralogical society, monograph no 6,480 p.
Chen PY (1977) Table of key lines in X-Ray powder diffraction patterns of minerals in clays and
associated rocks. Dept, of Natural resources, (Indiana, USA). Geological Survey occasional paper
no 21, 67 p.
Robert M (1975) Principes de determination quantitative des minfrauxargileux&l’aide des rayons X.
Prob1emesparticulierspos£s pour les min£rauxargileux les plus fr£quentsdans les sols des r£gion-
stemperdes. Ann. Agron., 26, 363-399.
Stucki JW (Goodman BA and Schwertmann U (1985) Iron in soils and clay minerals., D. Reidel, 893 p.
Teissier D (1984) Etude experimental de Гorganisation des materiauxargileux.Hydratation, gonflement
et structuration au cours de la dessiccation et de la rehumectation., INRA, These doc. Etat, 361 pp.
Thorez (1975) Phyllosilicates and clay minerals. A laboratory handbook for their X-Ray diffraction
analysis., Lelotte Ed., 579 p.
Wilson MJ (1987) A handbook of determinative methods in clay mineralogy. Blackie - Chapman and
Hall, 308 p.
Получение ориентированных агрегатов на пористой керамической пластинке
Kinter ЕВ and Diamond S (1956) A new method for preparation and treatment of oriented-aggregat speci¬
mens of soil clays for X-Ray diffraction analysis. Soil Sci., 81,111-120.
La Manna Ш and Bowers FH (1985) A suction apparatus for orienting clay minerals into porous ceramic
tile. Soil Sci. Soc. Am. J.9 49,1318—1319.
Rich Cl (1969) Suction apparatus for mounting clay specimens on ceramic tile for X-Ray diffraction. Soil
Sci. Soc. Am. Proc.9 33, 815-816.
Shaw HF (1972) The preparation of oriented clay mineral specimens for X-Ray diffraction analysis by a
suction unto ceramic tile method. Clay Miner., 9, 349-350.
Глава 4. Характеристика минералогического состава методом РД
131
Насыщение глин катионами
Brindley GW and Ertem G (1971) Preparation and solvation properties of some variable charge mont-
morillonite. Clays Clay Miner., 19, 399-404.
Buhmann C, Fey MV and De Villiers JM (1985) Aspects of the X-Ray identification of swelling clay
minerals in soils and sediments. S. Afri. J. Sci., 81, 505-509.
Calvet R and Prost R (1971) Cation migration into empty octahedral sites and surface properties of clays.
Clays Clay Miner., 19, 175-186.
Hofmann U and Klemen R (1980) Verlust der Austanschfahigeit von lithiumionenanbentonitdarcherhit-
zung (perted’6changeabilit6 des ions lithium dans les bentonites aprfcs chauffage). Zeit. Anorganisc
Chem., 262,95-99.
Lim CH and Jackson ML (1986) Expandable phyllosilicate reactions with lithium on heating. Clays Clay
Miner., 34,346-352.
Насыщение, сольватация, образование соединений включения, растворение
Bamhisel RI and Bertsch PM (1989) Chlorites and hydroxy-interlayered vermiculite and smectite. In
Minerals in soil environments Dixon JB and Weed SB ed., Soil Sci. Soc. of Am., 729-740.
Bamhisel RI (1977) Chlorites and hydroxy interlayered vermiculite and smectite, 331-356. In Minerals
in soil environments, Dixon JB and Weed SB ed., Soil Sci. Soc. Am., Monogr., 331-356.
Brindley GW (1966) Ethylene glycol and glycerol complexes of smectites and vermiculites. Clay Miner,
6,237-260.
Brindley GW and Ertem G (1971) Preparation and solvation properties of some variable charge mont-
morillonites. Clays Clay Miner., 19, 399-404.
Churchman GJ (1990) Relevance of different intercalation tests for distinguishing halloysite from kaolin-
ite in soils. Clays Clay Minerals, 38, 591-599.
Follet EAC, McHardy WJ, Mitchell BD and Smith BFL (1965) Chemical dissolution techniques in the
study of clays. Part 1. Clay Miner., 6,23-24.
Novich BE and Martin RT (1983) Solvation methods for expandable layers. Clays Clay Miner., 31,235-238.
Suquet H, Iiyama JT, Kodama H and Pezerat N (1977) Synthesis and swelling properties of saponites
with increasing layer charge. Clay Miner., 25,231-242.
Suquet M, Calle de la C and Pezerat H (1975) Swelling and structural organization of saponite. Clays
Clay Miner., 23, 1-9.
Theng BKG, Churchman GJ, Whitton JS and Claridge CGC (1984) Comparison of intercalation methods
for differentiating halloysite from kaolinite. Clays Clay Miner, 32,249-258.
Walker GF (1958) Reactions of expanding-lattice clay minerals with glycerol and ethylene glycol. Clay
Miner Bull., 302-313.
White JL and Jackson ML (1947) Glycerol solvation of soil clays for X-Ray diffraction analysis. Soil Sci.
Soc. Am. Proc., 11,150-154.
Получение оксидов железа
Brown G and Wood IG (1985) Estimation of iron oxides in soil clays by profile refinement combined with
differential X-Ray diffraction. Clay Minerals, 20,15-27.
Campbell AS and Schwertmann U (1985) Evaluation of selected dissolution extractants in soil chemistry
and mineralogy by differential X-Ray diffraction. Clay Miner., 20, 515-519.
Meunier A and Velde В (1982) X-Ray diffraction of oriented clays in small quantities (0,1 mg). Clay
Miner., 17, 259-262.
Paterson E, Bunch SL and Duthie DML (1986) Preparation of randomly oriented samples for X-Ray dif-
fractometry. Clay Miner., 21,101-106.
Schwertmann U and Taylor RM (1989) Iron oxides. In Minerals in Soil environments, Dixon JB and
Weed SB ed. Soil Sci. Soc. Am., 379-438.
Schwertmann U, Murad E and Schulze DG (1982) Is there Holocene reddening (hematite formation) in
soils of a xeric temperate area. Geoderma, 27,209-223.
Torrent J, Schwertmann U and Schulze DG (1980) Iron oxyde mineralogy of some soils of two river ter¬
race sequences in Spain. Geoderma, 23, 191-208.
132
Часть 1. Минералогический анализ почв
Количественная рентгеновская дифракция
Austin GS and Leininger RK (1976) Effect of heat-treating mixed-layer illitesmectite as related to quan¬
titative clay mineral determinations. J. Sedim. Petrol., 46,206-215.
Brime C (1985) The accuracy of X-Ray diffraction methods for determining mineral mixtures. Miner.
Mag., 49, 531-538.
Carter RJ, Hatcher MT and Di Carlo L (1987) Quantitative analysis of quartz and cristobalite in bentonite
clay based products by X-ray diffraction. Anal. Chem., 59, 513-519.
Cody RD and Thomson GL (1976) Quantitative X-ray powder diffraction analysis of clays using an ori¬
ented internal standard and pressed discs of bulk shale. Clays Clay Miners, 24, 224-231.
Davis BL (1980) Standardless X-Ray diffraction quantitative analysis. Atmospk Environ., 14, 217-220.
Decleer J (1985) Comparaison between mounting techniques for clay minerals as a function of quantita¬
tive estimations by X-ray diffraction. Bull. Soc. BelgeGeol, 94, 275-281.
Gavish E and Friedman GF (1973) Quantitative analysis of calcite and Mg-calcite by X-ray diffraction
effect of grinding on peak height and peak area. Sediment., 20,437-444.
Goehner RP (1982) X-Ray diffraction quantitative analysis using intensity ratios and external-standards.
Adv. in X-Rpy Analy., 25, 309-313.
Heath GR and Pissas NG (1979) A method for the quantitative estimation of clay minerals in North Pa¬
cific deep sea sediments. Clays Clay Miner., 27, 175-184.
Hogson M. and Dudgney ANL (1984) Estimation of clay proportions in mixtures by X-Ray. Diflfaction
and computerized chemical mass balance. Clays and Clay Miner., 32,19-28.
Hooton DH and Giorgetta NE (1977) Quantitative X-Ray diffraction analysis by a direct calculation
method. X-Ray Spectrom., 6, 2-5.
Hubbard CR and Smith DK (1977) Experimental and calculated standards for quantitative analysis by
powder diffraction. Adv. X-Ray Anal, 20, 27-39.
Hubbard CR, Evans EH and Smith DK (1976) The reference intensity ratio ///c for computer simulated
powder patterns. J. Appl. Cryst., 9, 169-174.
Johnson LJ, Chu CH and Hussey GA (1985) Quantitative clay mineral analysis simultaneous linear equa¬
tions. Clays and Clay Miner., 33, 107-117.
Kahle M, Kleber M and Reinhold J (2002) Review of XRD-based quantitative analyses of clay minerals
in soils: the suitability of mineral intensity factors. Geoderma, 109,191-205.
Norrish К and Taylor RM (1962) Quantitative analysis by X-Ray diffraction. Clay Miner. Bull., 5:98-109.
Ouhadi VR and Yong RN (2003) Impact of clay microstructure and mass absorption coefficient on the
quantitative mineral identification by XRD analysis. Appl. Clay Sci., 23, 141-148.
Parrot JF, Verdoni PA and Delaune-Mayere (1985) Analysemodalesemiquantitative d aprfcs 1 &ude des
Rayons X. Analusis, 13, 373-378.
Pawloski GA (1985) Quantitative determination of mineral content of geological samples by X-Ray dif¬
fraction. Am. Mineral., 70, 663-667.
Persoz (1969) Fid^litd de Г analyse quantitative des poudres de roches par diffration X. Bull. Centre Reck
Pau(SNPA), 3, 324-331.
Renault J (1987) Quantitative phase analysis by linear regression of chemistry on X-Ray diffraction in¬
tensity. Powder Diffract., 2, 96-98.
Ruffell A. and Wiltshire P. (2004) Conjunctive use of quantitative and qualitative X-ray diffraction analy¬
sis of soils and rocks for forensic analysis.Forensic Science International, 145, 13-23.
Taylor RM and Norrish К (1972) The measurement of orientation distribution and its application to quan¬
titative determination of clay minerals. Clay Miner., 9, 345-348.
Tomita К and Takahashi H (1985) Curves for the quantification of mica/smectite and chlorite/smectite
interstratifications by X-Ray powder diffraction. Clays Clay Miner., 33, 379-390.
Глава 5. Минералогический анализ методом
инфракрасной спектрометрии
5.1. Введение
5.1.1. Основные положения
Взаимодействие вещества с ИК-излучением позволяет различать энергии колебаний
молекул нескольких типов (рис. 5.1): колебания вдоль оси химической связи (валент¬
ные колебания или сжатие - растяжение, которые редко бывают простыми, кроме как
в двухатомных молекулах) и деформационные, которые перпендикулярны оси связи
(вращение, кручение, сдвиг, качание, либрация, изгиб). Энергетические уровни этих ко¬
лебаний лежат в ИК-диапазоне. Поглощение ИК-излучения происходит, когда частота
излучения совпадает с частотой одного из колебаний.
Ось
• • *
А
Валентные Сдвиг Ножничные Веерные Маятниковые Крутильные
Рис. 5.1. Примеры колебаний простой многоатомной молекулы
Абсорбционная ИК-спектроскопия использует излучения с длинами волн в области,
расположенной между видимым и микроволновым диапазонами. Эту область обычно
разделяют на три энергетических зоны: ближний, средний и дальний ИК-диапазоны
(рис. 5.2). В этих зонах энергии ИК-излучения соответствуют различные молекулярные
колебания.
• Ближний ИК- (БИК-) диапазон, так же как видимый и ультрафиолетовый (УФ),
соответствует высокоэнергетичным электронным спектрам, связанным с атомны¬
ми орбиталями, например, с переходом со связывающей орбитали на свободную
с более высокой энергией. В настоящее время развитие БИК-спектрометрии свя¬
зано с исследованиями органического вещества почвы (см. раздел 5.3.1 и часть 2
книги).
• Средний ИК-диапазон, расположенный между 300 и 5000 см-1, позволяет наблю¬
дать колебания с участием протонов (колебания, которые хорошо коррелируют со
структурой, и переходы, соответствующие слабым изменениям валентных связей
или деформациям валентных углов в молекуле). Элементарные ячейки глин (кри¬
сталлографические единицы) содержат многоатомные ионы или молекулы, вну¬
тренние моды колебаний которых встречаются между 4000 и примерно 400 см-1.
134
Часть 1. Минералогический анализ почв
Эти колебательные состояния являются объектом тщательного изучения в мине¬
ралогии. Полосы поглощения позволяют удовлетворительно характеризовать ак¬
тивные молекулярные группы.
Видимый
свет
Ближний
ИК
Средний И К
Дальний
ИК
Микроволны
0
7 2
5 3 3,5 4 4,5 5 5,5 6 7 8 9 10 12 18 20
30 40
X, МКМ
13
300 4
000 3 000 2 000 1 600 ' 1 000 800 4
1 1 1 1 1 1 1 ■ 1 1
ю 'юо
» !■
и; см'1
2 800 1 430 1 100 823 350
V, СМ'1
Кварц | LiF | CaF2 | NaCI | КВг | РЕ
Диапазоны
оптической
BaF,
Густая суспензия
Фторированное
углеводородное масло
Электронные
переходы
Валентное силовое поле - функциональные группы
Валентные колебательные переходы
gacj
Т
г
JH
IRTRAN
|AgCI |Csl|
Парафиновое1 масло (NUJOL) I
прозрачности
подложек
Спектральная
| характеристика
Изменения углов
- деформация
молекул
-либрация
Спектраль¬
ный
диапазон
Рис. 5.2. Область инфракрасной молекулярной спектрометрии и прозрачность оптических эле¬
ментов приборов. Положение полос по координате х выражается или в длинах волн X (нм или мкм),
или в волновых числах v (см-1) = 1 /X (см) = 10*/\ (мкм). По оси ординат коэффициент пропускания
выражают в процентах излучения, прошедшего через образец (РЕ — полиэтилен, Mylar)
В дальнем ИК-диапазоне на очень низких частотах между 200 и 10 см-1 можно на¬
блюдать колебания в решетке, вызванные перемещением многоатомной группы внутри
элементарной ячейки. Этот малоисследованный диапазон стал в настоящее время до¬
ступен благодаря развитию ИК-спектрометрии. Полосы вращательных переходов, рас¬
полагающиеся недалеко друг от друга, позволяют определять частоту вращения вокруг
оси без участия валентных колебаний или заметных изменений валентных углов, харак¬
теристичных для геометрии кристалла.
Таким образом, ИК-спектрометрия используется в дополнение к РД и химическому
и термическому анализам. РД (см. гл. 4) удовлетворительно определяет периодичность
дальнего порядка, но она неэффективна в случае веществ, аморфных в рентгеновских
лучах, или минералов с упорядочением ближнего порядка, образующихся в серии мета¬
морфических вытеснений.
В ИК-области можно определить спектральные характеристики вещества, а также
природу и направление связей, и углубить знания об атомных структурах и изоморфных
замещениях в тетраэдрических (Si—А1) и октаэдрических (Al-Mg) слоях. Эти данные
необходимы для идентификации некоторых минералов, количественного определения
молекулярной воды и конституционных гидроксилов, а также для обнаружения кри¬
сталлических или некристаллических примесей, влияющих на регулярность структуры
решетки.
В глинах и глинистых минералах только некоторые молекулярные группы способны
колебаться, и поэтому часто имеют менее сложные спектры, чем спектры некоторых
органических веществ.
5.1.2. Оборудование, используемое в методе инфракрасной
спектрометрии
ИК-спектрометр состоит из источника ИК-излучения, оптических элементов, детек¬
тора и блока компьютерной обработки измерений. Оптическое оборудование, пригод¬
1 Вазелиновое. — Примеч, науч. ред.
Глава 5. Минералогический анализ методом инфракрасной спектрометрии
135
ное для всего ИК-спектра от ближнего до дальнего диапазона, найти довольно трудно
(рис. 5.2).
Источник может излучать интенсивное полихроматическое излучение, охватываю¬
щее весь ИК-диапазон. УФ и видимое излучения, которые испускаются одновременно,
удаляются фильтром.
• Источники с вольфрамовой нитью накала охватывают только область ближнего
И К-диапазона.
• Никель-хромовые нити накала позволяют расширить диапазон до 600 см 1.
• Газовые ртутные лампы позволяют проводить изменения в дальнем ИК-диапазоне
между 300 и 10 см-1.
• Пюбары — стержни из карборунда с тугоплавкими оксидами (для стабилизации
температуры источника на уровне примерно 1500 °С часто используют водяное ох¬
лаждение), излучают в диапазоне примерно до 200 см-1.
• Источники из карбида кремния излучают в диапазоне между 6500 и 50 см1.
Оптическое оборудование (линзы, окна, дисперсионные системы) следует выбирать
исходя из его пропускающей способности (рис. 5.2) и устойчивости к воде и растворите¬
лям. Линзы лучше заменить зеркалами (рис. 5.3 и 5.8).
Для фильтрации видимой части спектра обычно применяется покрытие поверхности
германием или черными полиэтиленовыми пленками различной толщины.
Выбор детектора ограничен площадью его поверхности, охватываемой спектральной
областью, чувствительностью и временем отклика оборудования, детектируемыми ча¬
стотами, особенностями обслуживания (работа с жидким азотом или гелием). Детектор
может быть:
• неселективным, как например, термопары или термобатареи (несколько после¬
довательно соединенных термопар); болометры на легированном германии для
дальней ИК-области, которые работают в жидком гелии (4,2 К); ртутно-кадмие¬
во-теллуровые детекторы; детекторы DTGS (на основе обогащенного дейтерием
триглицинсульфата), термостатируемые для работы в среднем ИК-диапазоне; де¬
текторы Голея с газовой ячейкой для дальнего ИК-диапазона (эти детекторы очень
чувствительные, но весьма хрупкие);
• селективным (фотон-электрон-преобразование зависит от длины волны), напри¬
мер, детекторы из сульфида свинца, селенида свинца или антимонида индия, ра¬
ботающие в жидком азоте.
Оптическая система может быть основана на принципе рассеияния (дисперсии) или
интерференции в системе одиночного или двойного луча.
В дисперсионном режиме излучение проходит сквозь образец и частично выходит из
него (пропускание — поглощение), попадает на дифракционную решетку или моно¬
хроматор, который разделяет излучение в зависимости от длины волны (рис. 5.3, а).
Энергия регистрируется, точка за точкой, при вращении решетки. Сначала необходимо
определить нулевую точку прибора, а затем записать базовый спектр без пробы для уче¬
та, в частности, содержания С02 и Н20 в воздухе. Коэффициент пропускания рассчиты¬
вают для каждой длины волны как отношение двух величин сигнал/шум. Разрешения
часто недостаточно и энергия уменьшается с увеличением длины волны, поэтому не¬
обходимо постепенно открывать щель монохроматора и вносить поправку на фоновый
шум. Эти операции могут быть автоматизированы, а сходимость можно улучшить ис¬
пользованием двойного луча и проведением измерений образца и контроля на каждой
длине волны с помощью модулятора. Таким образом, энергию луча поддерживают на
136
Часть 1. Минералогический анализ почв
а)
Закрепленное
Рис. 5.3. Типы ИК-спектрометров: а) дисперсионный спектрометр: 1 — дисперсионная система
(призмы, решетки, монохроматор, непрерывный интерференционный клин высокого разреше¬
ния); 2 — выходная щель для точного выделения области частот; 3 — детектор; 4 — усилитель;
5 — блок сбора данных; /0 и /, /х — интенсивности общего падающего, прошедшего луча и луча на
выбранной длине волны х\ Т = ///0 — коэффициент пропускания, соответствующий поглощению
А = -log 7; Ь) спектрометр с интерферометром и преобразованием Фурье: 1 — светоделитель
из КВг, покрытого пленкой из германия (100-4000 см-1) или Майлара (0-700 см-1); 2 — подвиж¬
ное зеркало (спектральный состав сигнала зависит от положения зеркала, разрешение — от ам¬
плитуды перемещения); 3 — гелиево-неоновый лазер для измерения перемещения подвижного
зеркала и определения направления движения зеркала; 4 — камера для образца под вакуумом
или в контролируемой атмосфере; 5 — отношение интенсивность сигнала/положение зеркала;
6 — интерферограмма; 7, в — система сбора данных в реальном времени
постоянном уровне. Для дальнего ИК-диапазона из-за используемого оборудования
и недостаточной энергии работа в дисперсионном режиме невозможна. Радиометриче¬
ские приборы работают медленно, а модели высшего класса очень дорогие, поскольку
необходимо использовать несколько решеток с высоким разрешением для получения
удовлетворительной линейной дисперсии.
Лучшие характеристики могут быть получены при применении интерферометриче¬
ской системы, использующей преобразование Фурье для обработки данных (рис. 5.3, b).
Интерферометрия основана на быстрых движениях зеркала. Каждая длина волны мо¬
дулируется на характеристической частоте, определяемой скоростью зеркала. Данные
записываются в виде интерферограммы, которую обрабатывают в реальном времени
с использованием преобразования Фурье. Это позволяет получить спектр, где амплиту¬
да сигнала является функцией частоты.
В интерферометре Майкельсона полихроматическое излучение из источника разде¬
ляется на два луча светоделителем, сделанным из КВг или Майлара (покрытым йленкой
из германия для фильтрации излучения в видимой области спектра). Один из двух лучей
посылается на закрепленное зеркало, другой — на подвижное зеркало, передвигаемое
с известной скоростью линейным двигателем. Затем оба луча объединяются светодели¬
телем. Разрешение зависит от максимального хода подвижного зеркала. Регулировка
должна обеспечивать передвижение подвижного зеркала без вибраций и наклона, ко¬
торые могут исказить спектры и привести к ошибочным значениям коэффициента про¬
пускания. Одновременный анализ ИК-излучения на всех длинах волн производится за
одно передвижение зеркала вперед-назад.
137
Глава 5. Минералогический анализ методом инфракрасной спектрометрии
Изменения сигнала отражают различия в модуляции интерферограммы. Результаты
записывают при различных положениях зеркала, которые должны точно определяться
при помощи лазера. Каждый индивидуальный сигнал мультиплексируется и декодиру¬
ется. Полный спектр сканируется последовательно как функция времени менее чем за
одну секунду.
Чтобы ограничить влияние влажности и атмосферного С02, оптические схемы по¬
мещены в герметичный корпус, за исключением отделения с образцом, которое можно
продувать сухим азотом.
Разрешение обычно составляет около 2-4 см-1, но может достигать 0,1 см-1 (при уста¬
новке дополнительной памяти или увеличении производительности системы обработки
данных).
Оборудование для ИК-спектроскопии с преобразованием Фурье обычно основано на
технологии одинарного луча. Иногда (в моделях высшего класса) используется двойной
луч, который поддерживает непрерывное измерение отношения сигнал/шум. Соответ¬
ствующее программное обеспечение позволяет управлять спектрометром, собирать ре¬
зультаты и обрабатывать данные, восстанавливая спектр в режиме реального времени.
Поиск по базам данных ИК-спектров позволяет при необходимости сравнивать полу¬
ченные спектры для идентификации, калибровки, вычитания спектров и других опера¬
ций. Калибровку выполняют с использованием полистироловых пластин, которые дают
узкие интенсивные полосы в диапазоне между 699 и 3104 см-1.
На практике выбор оборудования зависит прежде всего от целей исследования, ко¬
торые определяют основные требования: сканируемый диапазон длин волн, природу
анализируемого материала (твердый, жидкий, газообразный), требуемую чувствитель¬
ность, разрешение, производительность обработки данных, качество программного
обеспечения, возможности расширения базового модуля (объединение с жидкостным
или газовым хроматографом, EGA термическим анализом, рамановской спектроскопи¬
ей, ИК- или рамановской микроскопией). Что касается цены, то два специализирован¬
ных прибора, дополняющих друг друга, могут быть использованы для повторяющихся
лабораторных исследований вместо одного дорогостоящего «универсального» прибора
высшего класса.
Для рутинных почвенных анализов достаточно прибора, охватывающего средний ИК-
диапазон до 220-250 см-1 с оптической системой из КВг (или, еще лучше, Csl) и разре¬
шением около 2 см-1. Для более специализированной лаборатории в настоящее время
необходимо оборудование, позволяющее работать в областях дальнего ИК-диапазона.
5.2. Применение ИК-спектрометрии в минералогии
5.2.1. Оборудование и материалы
• Ультрамикровесы, чувствительность 10_6 г, диапазон 2-5 г.
• ИК или ИК-Фурье-спектрометр с соответствующим диапазоном длин волн.
• ИК-микроскоп.
• Бинокулярная лупа (х 100).
• Пресс для таблетирования (10 т/см2).
• Центрифуга с центрифужными пробирками (см. гл. 4).
• Охлаждаемая влагозащитная вибрационная мельница с агатовым шаром для раз¬
мола в жидкой среде.
• Сушильный шкаф (105 °С) с точной электронной регулировкой.
138
Часть 1. Минералогический анализ почв
• Эксикатор из пирексового стекла.
• Лабораторная посуда.
• Продукты высокого качества для ИК-спектроскопии: КВг, Csl, полиэтилен, Nujol
(парафиновое масло), AgCl, CaF2, IR TRAN 4, растворители, гексахлорбутадиен
(Voltalef oil), Fluorolube (фторированное углеводородное масло), различные мине¬
ральные стандарты.
5.2.2. Подготовка образцов
Типы подготовки образцов
Подготовка образца для анализа методом ИК-спектрометрии чрезвычайно важна: она
определяет спектральные области анализа и их границы, а также, косвенным образом,
чувствительность и избирательность измерений.
Измерения должны проводиться на частицах размером не более 5 мкм, а при использо¬
вании суспензий в инертном масле или при количественном анализе — даже 1 или 2 мкм.
Обычно для анализа используют илистую фракцию с размером частиц < 2 мкм в Н+-,
NH/- или Х+-формах или фракции < 0,5 мкм, очищенные в стандартной ультрацен¬
трифуге Шарплес (Sharpies) (см. гл. 3). Из-за размеров элементарных ячеек глин в их
спектрах наблюдается лишь незначительное искажение полос поглощения. Образцы
измельчают и гомогенизируют в агатовой ступке в присутствии летучей органической
жидкости, не поглощающей в ИК-области (этанол, ацетон и т. д.), избегая модифика¬
ции или разрушения кристаллической структуры. Слишком крупные частицы могут вы¬
зывать искажения спектра, рассеяние побочного излучения и уширение полос поглоще¬
ния (эффект Христиансена).
Часто необходимо предварительное разложение органического вещества для того,
чтобы, во-первых, уменьшить его поглощение, которое может перегрузить спектр, и,
во-вторых, избежать удержания излишнего количества адсорбированной воды, которая
может маскировать отдельные полосы поглощения минералов. Однако в некоторых слу¬
чаях такая обработка может привести к неогенезису минералов.
Для измерений на твердых образцах
• В режиме «пропускание — поглощение» глины можно анализировать в трех основ¬
ных формах:
а) самоподдерживающиеся тонкие пленки или пленки на соответствующей под¬
ложке;
б) диски, приготовленные со связывающими веществами, прозрачными для ИК-
излучения;
в) в смеси со специальными жидкими составами.
• В БИК-области или в режиме диффузного отражения или комбинационного рас¬
сеяния образец просто упаковывают в держатель, стараясь уменьшить преимуще¬
ственную ориентацию.
• В режиме многократного зеркального отражения или нарушенного полного вну¬
треннего отражения (НПВО) проблемы на поверхности контакта с почвами часто
приводят к невоспроизводимости результатов, поэтому эти методы могут быть ис¬
пользованы только на тонких пластинах.
• В ИК-микроскопии образцы можно анализировать на пленке или как микропробы
без подготовки.
139
Глава 5. Минералогический анализ методом инфракрасной спектрометрии
Предварительная обработка может изменить свойства образцов: это может быть исполь¬
зовано для сравнения с образцами без химической обработки, сохраненными в их ис¬
ходном состоянии (аморфные фазы и пр.).
Для измерений на жидких образцах, выделенных из почвы химическими методами,
можно либо (i) использовать растворители, прозрачные для ИК-излучения и не реаги¬
рующие с минералами, или (ii) проводить БИК-спектроскопические измерения лиофи-
лизированных экстрактов.
Для измерений газообразных образцов, полученных контролируемым пиролизом или
разложением минеральных фракций в процессе термического анализа (ГТА и ТГА-ДТА,
см. главу 7), используют порошковые образцы, идентичные используемым для стан¬
дартного термического анализа.
Чтобы улучшить чувствительность анализа, используют специальные ячейки для
того, чтобы удлинить путь луча в газовой среде, поскольку поглощение пропорциональ¬
но длине хода луча. Необходима предварительная очистка воздуха в камере для пробы
для удаления присутствующих Н20 и С02.
Подготовка самоподдерживающихся пленок
Основные положения
Подготовка самоподдерживающихся пленок подходит только для некоторых типов
глин, например, монтмориллонитов, вермикулитов и волокнистых глин, которые мож¬
но приготовить в виде тонких, но прочных пленок.
Метод Фармера и Пальмиери (Farmer и Palmieri, 1975) был слегка модифицирован для
проведения количественных измерений. Преимуществом этого метода является отсут¬
ствие необходимости применения к образцу сильного давления и отсутствие обменных
реакций между образцом и связующим веществом, добавляемым при приготовлении
таблеток. Основные сложности связаны с критической толщиной пленки (не более 4—8
мкм), ее способностью пропускать излучение значительной интенсивности, механиче¬
ской прочностью и трудностью ее удаления, требующей определенных навыков. Сили¬
катные минералы часто осаждаются в преимущественно ориентированном состоянии,
что позволяет идентифицировать колебания, вызываемые осцилляциями диполей, ко¬
торые должны быть перпендикулярными плоскости решетки. Кроме того, при помощи
гониометра можно изучать полихроизм, связанный с плоскостью.
Методика
* Измельчают порции глины известной массы в агатовой ступке с небольшим коли¬
чеством воды для образования текучей пасты и затем готовят суспензию в извест¬
ном объеме дистиллированной воды.
9 Всю суспензию на пластине, покрытой гибкой полиэтиленовой пленкой, пере¬
носят в цитокамеру Хеттиха (Hettich) (рис. 5.4); центрифугируют при умеренной
скорости (примерно 2500 g). Выбранный объем камеры равен 4 мл, что позволяет
получать пленку диаметром 12,4 мм, т. е. с площадью поверхности 120 мм2; таким
образом можно определить количество осадка на единице площади (плотность
должна быть примерно 1—2 мг/см2).
9 Удаляют пипеткой чистую надосадочную жидкость и высушивают на воздухе плен¬
ку, содержащую осадок.
• Снимают пленку, перегибая гибкую подложку через край с острой фаской, и пере¬
носят ее на держатель для ИК-измерения; осадок также можно перенести на тон¬
140
Часть 1. Минералогический анализ почв
кий держатель фильтров с жесткой полимерной сеткой, которая предотвращает
перемещение благодаря ее сплошной структуре.
• Оставляют осадок на 48 ч в эксикаторе с Р205.
Рис. 5.4. Приготовление образца самоподдерживающихся пленок минералов: а) центрифугиро¬
вание при 2500 д в цитокамере Хеттиха; Ь) отделение самоподдерживающейся пленки
Приготовление пленки на держателе, прозрачном для ИК-иэлучения
Основные положения
Эта методика позволяет получать тонкие пленки для ориентированного осадка под дей¬
ствием силы тяжести или центрифугированием, как описано выше.
Следует принимать во внимание следующие факторы: толщину осадка глины, вы¬
бор подходящего растворителя для предотвращения растворения материала подложки,
спектральную область для данного типа подложки, реакционную способность систе¬
мы «глина — растворитель - подложка». Обычно используют пластины КВг (они не¬
дороги, просты в использовании и позволяют проводить сканирование в среднем ИК-
диапазоне примерно до 400 см-1) или Csl (для ИК-диапазона до 220 см-1)- Для дальнего
ИК-диапазона используют полиэтилен. Образец превращают в суспензию без растворе¬
ния. Поверхность можно насыщать микроиспарением с парафиновым маслом для того,
чтобы уменьшить явления отражения на границе раздела фаз «воздух — глина».
Методика
В зависимости от ИК-области готовят таблетки диаметром 13 мм методом прессования
(10 т/см2, AgCl, CaF2, IRTRAN2, IRTRANA, Ge, Si, КВг, Csl, полиэтилен и др.).
Суспензию, приготовленную из образца в соответствующем органическом раствори¬
теле, осаждают аналогично предыдущей методике под действием силы тяжести или цен¬
трифугированием. После высушивания осторожно нагревают диск, покрытый пленкой,
в сушильном шкафу при 100 °С в течение 5 ч для удаления следов воды и затем в эксика¬
торе до проведения анализа.
Приготовление дисков (твердого раствора)
Основные положения
Твердый образец полностью измельчают в агатовой ступке в присутствии органической
жидкости (например, этанола) и высушивают под вакуумом в эксикаторе с фосфорным
141
Глава 5. Минералогический анализ методом инфракрасной спектрометрии
ангидридом. Затем его смешивают с матрицей, что позволяет создать под сильным дав¬
лением самоподцерживающиеся диски; эти диски можно использовать в достаточно
широком диапазоне ИК-излучения.
В случае набухающих смектитов предпочтительнее размолоть образец известной мас¬
сы с небольшим количеством воды для создания густой пасты, затем добавить связы¬
вающее вещество и вновь растереть влажный образец. После полного высыхания гомо¬
генизируют разбавленный образец в микрошаровой мельнице.
Эта методика проста в использовании и находит широкое применение, но обладает
некоторыми недостатками:
• все материалы, используемые в качестве связывающих веществ для дисков, имеют
ограниченные области пропускания в ИК-диапазоне, поэтому при использовании
одной таблетки невозможно наблюдать поглощение глины во всем ИК-диапазоне
от ближнего до дальнего ИК;
• реакционная способность подложки может приводить к обменным реакциям
с глиной.
Например, бромид калия (КВг), который прозрачен до 400 см-1 (рис. 5.2), образует
прекрасные таблетки, дающие хорошие спектры в широком ИК-диапазоне. Но в глинах
типа 2:1, контактирующих с ионами К+, таких как смектиты, обменные явления могут
вызвать искажение спектра поглощения (Nyquist и Kagel, 1971). Поэтому такие диски
непригодны для изучения уменьшения водоудержания ионами калия или для исследо¬
вания поверхностных катионов.
Поскольку КВг гигроскопичен, таблетки следует хранить в эксикаторах под вакуумом
с фосфорным ангидридом для снижения поглощения воды и связанных с ней неопре¬
деленностей в интерпретации полос гидроксилов адсорбированной минералами воды
и подложки. Для дальнего ИК-диапазона следует использовать полиэтилен, политетра¬
фторэтилен (тефлон) или парафин.
Давление может вызывать неуместные превращения за счет усиления химической
реакционной способности, но в то же время его можно использовать для in situ исследо¬
вания влияния очень высокого давления, получаемого в алмазной наковальне, что по¬
зволяет анализировать методом ИК-спектроскопии вызванные им превращения (Weir
идр., 1959; Пии Memagh, 1992).
Методика (качественный анализ)
• Выбирают матрицу и диаметр таблеток.
• Высушивают образец в эксикаторе в течение 48 ч для удаления неструктурной
воды, полоса которой при 3440 см-1 может маскировать полосу структурных ОН-
групп; этот метод высушивания пригоден не для всех минералов (см. гл. 1).
• Высушивают тонкоразмолотое связывающее вещество для ИК-анализа под ваку¬
умом в сушильном шкафу при 100 °С в течение ночи, если позволяет его термиче¬
ская стабильность.
• Для диска толщиной около 1 мм и диаметром 13 мм взвешивают 0,5-3 мг глины
с точностью 0,01 мг.
• Добавляют 300 мг КВг (можно изменить).
• Гомогенизируют в микрогомогенизаторе с пластиковым или агатовым шаром в те¬
чение 2 мин или размалывают в агатовой ступке еще раз для лучшего перемеши¬
вания.
• Переносят в пресс-форму из нержавеющей стали диаметром 13. мм (обозначена
буквой А на рис. 5.5, Ь).
142
Часть 1 Минералогический анализ почв
• Прилагают небольшое давление и дегазируют под вакуумом в течение 5 мин.
• Прилагают давление (рис. 5.5, а) 10 т/см2 в течение 10 мин (при этом давлении КВг
становится пластичным).
• Вынимают диск из формы при помощи отделительного кольца (обозначено буквой
Е на рис. 5.5); он должен выглядеть однородным, гладким и прозрачным (руками
его не трогают).
• Высушивают при 100 °С в течение 2 ч и хранят в эксикаторе над Р205 до использо¬
вания.
Ют
а) Ь) I
Рис. 5.5. Приготовление образца в форме твердого раствора: а) ручной гидравлический пресс
на 12 т; Ь) детали пресс-формы: А — корпус; В — сменное основание; С — поршень диаметром
13 мм; D — полированные цилиндры диаметром 13 мм; Е — отделительное кольцо формы, запол¬
ненный круг представляет уплотнительное кольцо
. На практике лучше приготовить две или даже три таблетки с одним и тем же связы¬
вающим веществом и двумя разными концентрациями:
• таблетка из 3 мг образца и 300 мг КВг для достижения полного поглощения в зонах
около 1000 и 500 см*1 (силикаты);
• таблетка из 0,25—0,5 мг образца глины для получения подробных спектров в райо¬
нах интенсивного поглощения силикатов;
• третья таблетка со связывающим веществом, прозрачным в дальнем ИК-диа-
пазоне, понадобится, если нужны спектры ниже 200 см*1;
• получают спектры в выбранных условиях.
Время записи спектра зависит от типа спектрометра (дисперсионный или интерферо¬
метр), области сканирования, доступной для дисперсионного оборудования, и требуе¬
мого разрешения. После измерения таблетки можно хранить в эксикаторе над Р205.
Чтобы избежать коррозии, пресс-форму необходимо очистить сразу после использо¬
вания без применения абразивных средств.
Приготовление образцов глины в виде густой суспензии (пасты)
Основные положения
В случае если возможны взаимодействия между связывающим веществом и образцом,
если невозможно приготовить самоподдерживающуюся пленку или нежелательно при¬
Глава 5. Минералогический анализ методом инфракрасной спектрометрии
143
менение давления, можно приготовить густую суспензию в нелетучем инертном масле.
Нужол (парафиновое масло), гексахлорбутадиен (Voltalef oil) или Fluorolube (фториро¬
ванное углеводородное масло) смешивают с образцом до образования жидкой пасты,
которую сжимают между двумя окошками. Метод быстрый, но только качественный;
образец получается ориентированным. Нельзя высушивать смесь в сушильном шкафу.
Методика
• Помешают 10 мг сухого образца глины в 50-миллиметровую агатовую ступку; ми¬
кропипеткой или шпателем добавляют известное количество масла для смачива¬
ния порошка.
• Измельчают до получения густой пасты, в которой равномерно распределен об¬
разец (концентрация должна быть около 0,3-0,5%).
• Шпателем намазывают смесь на окне, прозрачном для ИК-излучения, накрывают
другим окном для получения однородной толщины без попадания воздуха внутрь.
• Помещают образец в спектрометр и записывают спектр.
При интерпретации следует учитывать спектральные границы окон и специфиче¬
ские зоны поглощения матрицы масла. Например, нужол сильно поглощает между 3000
и 2800 см-1 (СН), при 1460 и 1375 см-1; Fluorolube не поглощает между 4000 и 1400 см-1.
Оба масла дополняют друг друга, и следует проверить, что их полосы поглощения не
маскируют специфические полосы образца. Трудно достичь и поддерживать однород¬
ность суспензии: подготовленные образцы должны всегда храниться в горизонтальном
положении.
Подготовка образцов для измерения зеркального или диффузного отражения
и нарушенного полного внутреннего отражения (НПВО)
Поскольку интенсивность поглощения зависит от угла падения, поверхность образца
должна быть мелкозернистой, т. е. необходимо гранулометрически однородное измель¬
чение до размера 0,2 мм. Уплотнение выполняют так же, как для рентгеновской дифрак¬
ции (см. гл. 4), избегая чрезмерной ориентации.
Толщина слоя порошка (примерно 1 мм) несущественна, поскольку излучение про¬
никает на глубину лишь нескольких микрон. В отдельных случаях можно использовать
спрессованные таблетки с добавлением связывающих веществ или без них, но в этом
случае ориентация будет сильнее. Следует отметить, что поскольку показатель прелом¬
ления имеет большое значение для измерений отражения, наблюдаются значительные
различия между спектрами поглощения и отражения при высоких длинах волн.
Дейтеризация
В специализированных лабораториях дейтеризация является идеальным методом из¬
учения воды в глинах. В тяжелой воде дейтерий замещает водород. Когда молекула Н20
замещена молекулой D20, дейтерируются группы ОН поровой воды, но не воды в ре¬
шетке (Wada, 1966). Межатомные расстояния не изменяются, масса удваивается, часто¬
ты колебаний уменьшаются. Поэтому можно разделить ретикулярные ОН и адсорбиро¬
ванную воду и исключить неопределенность измерений в исследованиях минеральных
гелей (Ми/идр., 1976).
Примечания
Следует избегать атмосферного загрязнения исходных образцов во время приготовле¬
ния, например:
144
Часть 1. Минералогический анализ почв
• аммоний может поглощать около 3250 и 1400 см*1 (валентные и деформационные
колебания);
• в присутствии кальция обработка органического вещества перекисью водорода мо¬
жет привести к образованию нерастворимого оксалата кальция, который поглоща¬
ет около 1400 см*1; разложение органического вещества гипохлоритом натрия не
дает оксалата и, следовательно, более применимо в данном случае;
• отметим, что окисление органического вещества сопровождается окислением ми¬
неральных компонентов, таких как Fe2+;
• декарбонизация и деферризация в кислой среде может разрушать такие минералы, как
аморфные силикаты, с осаждением кремнезема и удалением Fe, и т. д. (Frohlich, 1980).
5.2.3. Краткое руководство по интерпретации спектров
Основные положения
В ИК-диапазоне переходы между различными энергетическими уровнями подчиняются
правилам отбора, так как поглощение связано с изменениями дипольного момента мо¬
лекул. В многоатомных молекулах нельзя наблюдать все возможные частоты, потому что
энергетические уровни вырождаются, например, из-за симметрии молекулы. Поэтому
очень сложно точно предсказать частоты фундаментальных колебаний в сложных струк¬
турах, таких как глины, хотя в последнее время компьютерные программы позволили
добиться определенного прогресса.
Кремнийсодержащие тетраэдры и октаэдры, содержащие алюминий и магний,
формируют основные единицы глинистых минералов: у тетраэдрической структуры
возможны четыре типа колебаний, у октаэдрической — шесть, но не все они актив¬
ны и могут быть модифицированы либо изоморфными замещениями, либо природой
структурных катионов.
Интерпретация спектра глины может проводиться после его сравнения со спектром
«чистого» вещества близкой природы, чтобы устранить неопределенности, вызываемые
вариациями в химическом составе и степени упорядочения. Потребность в эталонных
спектрах предполагает, что каждая лаборатория должна записывать все результаты ис¬
следований почвенных минералов, которые могут использоваться в качестве эталонов,
в дополнение к имеющимся базам данных.
В качественном анализе необходимо сначала определить интенсивность диагности¬
ческих полос поглощения минералов и затем отнести их к молекулярным группам и,
возможно, типам конкретных колебаний. Для некоторых минералов с интенсивными
полосами (например, каолин, кварц, гиббсит, кальцит) чувствительность достаточна.
Могут быть определены количества порядка 1%.
Например, протонное окружение чистых гидроксидов и оксигидроксидов приводит
к специфичным валентным колебаниям: с другой стороны, в минералах типов 2:1 и 2:1:1
с частыми химическими вариациями и изоморфными замещениями может наблюдаться
смещение полос; в этом случае рекомендуется одновременно использовать РД (см. гл. 4)
и химический анализ селективного растворения (см. гл. 6) для определения второсте¬
пенных компонентов почвы, что позволяет определить отношения (тетраэдрический
кремнезем)/(октаэдрический глинозем), которые удовлетворительно отражают окруже¬
ние гидроксилов.
В каолините типа 1:1 отношение Si02/Al203 = 2 и поверхностные гидроксилы под¬
вергаются колебаниям, приводящим к характеристическим полосам поглощения при
четырех частотах ИК-диапазона (табл. 5.1).
Глава 5. Минералогический анализ методом инфракрасной спектрометрии 145
Таблица 5.1. Полосы поглощения хорошо ©кристаллизованного каолинита А1203 • 2Si02* 2Н20
(таблетка с КВг толщиной < 2 мкм) в ИК-диапазоне
Полоса,
см*
Колебания
Полоса, см*
Колебания
3695
Внешние ОН
690
Mg/Al
3669
Внешние ОН
538
Деформа- SiO
ционные
3653
Валентные
Внешние ОН
470
4- +
3619
Внутренние ОН
430
3410-3450»
Гйдратированные
364
Колебания Октаэдрический
ОН
Al/Mg
1630»
Деформа¬
Гидратированные
345
Смешан¬
ционные
НОН
ные
1106-1112
Валентные
Внеслоевые SiO
275
1036
SiO...
195
1010
SiO...
938
Деформа¬
Внешние ОН
ционные
912
Внутренние ОН
а Гидратированную воду (около 3450 см-1) можно отличить от структурных гидроксилов по
присутствию полосы НОН около 1630 см-1.
В смектитах и слюдах Si02/Al203 равно примерно 3. Поскольку гидроксилы на вну¬
тренних позициях связаны с различными октаэдрическими катионами, полосы погло¬
щения в районе 3600 см*1 неоднородны, и можно наблюдать смещение частот валентных
колебаний гидроксилов. Уровень заполнения октаэдрических (ди- и триоктаэдрических)
позиций можно определить рентгено-дифракционным анализом линии 060, а дополни¬
тельную информацию получить из ИК-анализа.
В случае триоктаэдрических минералов заняты три позиции и ось связи ОН внутрен¬
него гидроксила перпендикулярна ретикулярной плоскости (001) глин, тогда как в ди-
октаэдрических минералах заняты только две позиции; протон внутреннего гидроксила
оттеснен к вакантным октаэдрическим позициям, и, следовательно, спектр деформиру¬
ется.
Методика
Неизвестные спектры вручную разделяют на несколько полей, выбирают полосы мак¬
симальной интенсивности и сравнивают их длины волн со справочными данными. Этот
поиск может быть автоматизирован при работе с электронными базами данных, но на
практике необходимы также данные лабораторных эталонов и постоянные обращения
к литературе.
В твердых минералах интерпретация основана главным образом на частотах коле¬
бания внешних молекулярных групп, поскольку значительные колебания внутренней
части кристалла наблюдаются только в дальнем ИК-диапазоне. ИК-спектрометрия
удовлетворительно определяет молекулярные группы, в которых атомы находятся
в специфическом окружении. Можно выделить молекулярные структуры с характери¬
стическими полосами поглощения.
146
Часть 1. Минералогический анализ почв
ИК-полосы поглощения филлосиликатов
Кажущаяся простота почвенных минералов маскирует сложность полос поглощения ос¬
новных типов колебаний (рис. 5.6). Полосы смещены в зависимости от кристаллическо¬
го окружения, замещений и т. д. Определение частот и отнесение полос требуют очень
четких спектров для выделения небольших изменений фаз, которые часто составляют
около 2 см-1. Например этому порядку величин соответствуют различия между аморф¬
ным кремнеземом, опалом и биогенным кремнеземом на основании колебаний связей
Si—О, Si—О—Si и Si—ОН.
В областях 3700-3400
и 950-600 см-1
- протоны гидроксильных групп, даже в отсутствие молекуляр¬
ной структуры дальнего порядка, которые полезны в случае
«аморфных» веществ
- активные формы колебаний тетраэдров и октаэдров: колеба¬
ния связи ОН, дихроизм позиций ОН в диоктаэдрических ми¬
нералах
— смещение полос поглощения в зависимости от природы октаэ¬
дрических катионов, влияние обмена (например, взаимодей¬
ствие структурной ОН-связи с межслоевыми катионами)
— выделение валентных колебаний несвязанной воды позволяет
лучше различать галлуазиты, структура которых нарушена мо¬
лекулами межпакетной воды
В области 1100-500 см-1
— колебания аниона силиката, которые считаются спектральны¬
ми «отпечатками пальцев» глин, слабо связанных с колебани¬
ями других структур (связи кремний-кислород), валентные
колебания связи Si—О около 1000 см-1, деформационные коле¬
бания связи Si-О около 500 см~1
— изоморфные замещения тетра- и октаэдрических минералов
могут вызывать полосы поглощения в дальнем ИК-диапазоне
В области < 400 см-1
- ионы А13+ октаэдров и кислородные связи соседних слоев
— колебания обменных катионов, уравновешивающих заряды
в межпакетном пространстве глин, и замещение обменных ка¬
тионов
Рассматривая в качестве примера полосы поглощения обычной глины, каолинита
типа 1:1 (табл. 5.1), можно отметить:
• сходную форму полос для колебаний протонов (валентных колебаний внешних
и внутренних гидроксилов) по отношению к степени кристалличности;
• уровень разупорядочения, определяемый по коалесценции дублета при 3669-
3653 см*1;
• гидратированную воду легко отличают дейтерированием и появлением полосы
НОН около 1630 см1;
• полосы при 1110, 1036 и 1010 см_1 соответствуют характеристическим валентным
колебаниям связи SiO.
В случае галлуазитов уширение полос можно наблюдать между 3700 и 3600 см-1 (ва¬
лентные колебания связи ОН) из-за структурного искажения, вызванного переменной
гидратацией. Полосы при 795 и 758 см1 в каолинитах имеют примерно равную интен¬
сивность, тогда как интенсивность полосы при 795 см*1 в галлуазитах очень слаба.
Октаэдрические позиции заняты
cm
4000
3000
АР
ОН(т)
2000
—г——
«
*
!
I
I
•
I
карбоксилаты
1500 | 1
латыт SO+
1000
500
100
НОН
1630*- 1640
МВ- + пиридин
карбонаты рл 4 ^ 1 X*4VJ)
NaC!
SiO (t) SKVAKwb)^ §n»b
SJOAl(mb)
> ^
■OHKrr}
HOH
X'W)—►
'sow1
о SO«g(VI)
(Wrnb)
X^OVH* SO(f)
Fe3
HOH
OH(m)
FeJ*
pH(m)'
HOH
x*av)~*
sto(t)
(f) (mb)
i —11| |
SOSIO-Fe'4VI)
Валентные колебания структурных гидроксилов "валентные колебания
гидратационной НОН
SIO AIO,AIOSi SiO SiOFe
Q.
со
Q.
3
Колебания протона
- ОН-дегидроксилирование — различается доступностью
и обратимостью
- дейтеризация (D20)
- либрация ОН (диоктаэдрического)
Структура
- порядок-беспорядок, соединения, аморфные
для рентгеновской дифракции (отношение Si/AI)
- ОН, связанные с (AI,Fe2+) и (Fe2*,Fe3*)
- преобразование тетраэдрических и октаэдрических
слоев
(Валентные колебания) (Деформационные
октаэдрические
замещения Fe/AI
тетраэдры Si04
октаэдры АЮ6
колебания)
- окисление или восста¬
новление железа
- вытеснение Fe3+ из
октаэдрических слоев
- обработка гидразином
Рис. 5.6. Отнесение полос поглощения в филлосиликатах (Stubican и Roy, 1961). Интенсивности полос: (t) очень большая, (т) средняя, (f)
сильная, (Ь) низкая; изменение интенсивности, -> изменение частот при замещениях (X3* или Х2+) в тетраэдрических (IV) и октаэдрических
(VI) позициях
Глава 5. Минералогический анализ методом инфракрасной спектрометрии
148
Часть 1. Минералогический анализ почв
5.2.4. Количественный анализ
Определение
В минералогии совершенствование количественного анализа с использованием ИК-
спектроскопии сопровождалось совершенствованием ИК-Фурье-оборудования, и ко¬
личественный анализ является важным инструментом для исследования педогенных
хронопоследовательностей, потоков и осадочных серий (например, в палеогидрологии
и палеоклиматологии) или функционирования бассейнов. Также интересно наблюдать
минералы в их первоначальном состоянии без обработки, за исключением измельчения
в неагрессивной жидкости для уменьшения размера частиц до 1—2 мкм и высушивания.
Отношения между интенсивностью поглощения ИК-излучения и концентрацией ми¬
неральных пород подчиняются закону Бера—Ламберта, который применим для твердых
тел (Keller и Picket, 1949; Duyckaerts, 1959). Однако этот закон не всегда применим к по¬
чвенным минералам, для которых характеристические полосы не полностью разделены
и часто недостаточно однородны. Степень упорядоченности влияет на ширину полос,
а размер частиц — на их интенсивность. В этом случае ограничения очевидны и фото¬
метрический отклик редко бывает линейным: поглощение полосы больше не пропор¬
ционально концентрации, и количественное определение не может быть таким точным,
как для газа или органической жидкости.
Если стандартные минералы (со структурой, степенью кристалличности, химическим
и гранулометрическим составом, близкими к параметрам образцов) отсутствуют, то
с помощью этого метода может быть проведен только относительный количественный
анализ; однако он позволяет измерять изменения в составе минералов в профиле или
топографической последовательности почв с приемлемой точностью.
Для отдельных минералов с относительно характеристичными спектрами и хорошо
определенной базовой линией, такими как некоторые кварцы, каолинит и карбонаты,
уровни обнаружения могут достигать 1-2%. Очистка методом седиментации позволяет
концентрировать и упрощать спектры. Степень кристаллической упорядоченности мо¬
жет вызывать значительные изменения, например, в каолинитах при анализе структур¬
ных ОН-связей между 3700 и 3600 см-1.
Подготовка образца является особенно важной для количественного анализа. Тре¬
буется достижение максимального поглощения (без насыщения) и минимальной дис¬
персии. Размер частиц должен быть менее 2 мкм. Гомогенность дисков должна быть со¬
вершенной, компоненты — высушиваться на каждой стадии подготовки или хранения,
и толщина образца должна быть постоянной и менее чем 1 мм.
Строгая стандартизация процедур позволяет оптимизировать измерения компонен¬
тов почвенных минералов вне зависимости от полноты окристаллизованности порош¬
ков, как в РД (см. гл. 4). Таким методом молено количественно определять некоторые
минералы с упорядочением ближнего порядка, которые «аморфны» в рентгеновских
лучах, например, некоторые алюмосиликаты, криптокристаллические соединения (на¬
пример, аллофаны, имоголит) и некоторые формы кремнезема и железа (например,
ферригидриты).
Методика
Общее представление
Могут использоваться методики качественного анализа (см. разделы 5.2.1 и 5.2.2), при¬
чем особое внимание должно уделяться следующему:
Глава 5. Минералогический анализ методом инфракрасной спектрометрии
149
• Образцы и связывающие вещества высушивают при 105 °С в течение 1 ч перед точ¬
ным взвешиванием (до 10_6 г); для термочувствительных веществ следует исполь¬
зовать температуру 40 °С.
• Образцы глины измельчают до размеров менее 1—2 мкм в агатовой ступке или луч¬
ше с тщательно отрегулированным охлаждением в вибрационной мельнице с ага¬
товыми шарами в жидкой среде (в присутствии этанола или ацетона); после вы¬
сушивания все частицы должны проходить сквозь сито с ячейками 0,1 мм; можно
также использовать фракции, количественно отделенные ультрацентрифугирова¬
нием (см. гл. 3).
• Взвешивают количество глины, позволяющее измерять пропускание в диапазо¬
не приблизительно между 20 и 70% для всех областей поглощения спектра (ка¬
чественные тесты позволяют найти точные пропорции в диапазонах от 2 до 5 мг
для образца на 1 г связывающего вещества); смешивают с соответствующим свя¬
зывающим веществом (обычно КВг) и гомогенизируют в вибрационном смесителе
с агатовыми шарами в течение 5 мин.
• Из 1 г полученной смеси готовят три диска диаметром 13 мм каждый, взвешивая
одинаковые количества смеси (300—330 мг) для получения постоянного оптическо¬
го пути, равного 1 мм или меньше; переносят смесь в установку для таблетирова-
ния под вакуумом (рис. 5.5) и прикладывают давление 10 т/см2 в течение 2-3 мин;
диски должны быть прозрачными, гладкими, без дефектов на поверхности, по¬
стоянной толщины и с равномерным распределением частиц глины в связываю¬
щем веществе (должно проверяться под микроскопом); их следует брать пинцетом
и хранить в эксикаторе с фосфорным ангидридом; аналогичным образом готовят
калибровочные диски.
• Измеряют поглощение дисков, приготовленных из связывающего вещества (для
контрольного анализа), диски образцов и калибровочные диски из чистых веществ
или смесей в зависимости от используемых в лаборатории эталонов для проведе¬
ния калибровки с различными пропорциями минералов.
Отложения
Фрелих (Frolich, 1980,1989) рекомендовал проводить измельчение с агатовым шаром до
менее чем 2 мкм в охлажденной инертной жидкой среде. Тонкость помола должна кон¬
тролироваться под микроскопом. Оптимальное разбавление около 0,25%.
• Готовят 1 г смеси: 2,5 мг осадка (с точностью 10-5 г) и 997,5 мг КВг.
• Тщательно гомогенизируют в вибрационной мельнице с агатовым шаром.
• Таблетируют 300 мг смеси в пресс-форме диаметром 13 мм под вакуумом в течение
2 мин.
Толщина диска КВг1 должна составлять 0,83 мм и обеспечивать постоянный оптиче¬
ский путь для всех измерений. Диск должен быть гладким и прозрачным и содержать
0,75 мг образца.
Расчеты
Любое поглощение, вызванное разбавителем (КВг), должно вычитаться из полного
поглощения (образец + КВг). Пропускание (затем пересчитанное в коэффициент по¬
глощения А) измеряют на спектре начиная от базовой линии. Эту линию часто трудно
1 Плотность КВг 2,75.
150
Часть 1. Минералогический анализ почв
определить для сложных смесей (рис. 5.7), и приходится проводить аппроксимирование
(с учетом влияния матрицы, наложения полос и т. д.). Относительная погрешность ча¬
сто составляет около 5% и может быть снижена для некоторых минералов1. Калибровка
с использованием «чистых» минералов или смесей минералов, состав которых близок
к составу образцов, позволяет построить кривые коэффициент поглощения =/(масса
минерала).
Рис. 5.7. Определение величины Г0: а, b — хорошая базовая линия; с — аппроксимация
Использование 1 г смеси дает возможность оценить повторяемость с помощью трех
дисков массой 300 мг.
Примечания
Сильная ориентация минералов в диске может приводить к ошибкам. В глинах типа 2:1
изменения интенсивности полос валентных колебаний гидроксилов могут быть вызва¬
ны триоктаэдрическими компонентами. В слюдах, ориентированных перпендикулярно
лучу, проявляются только колебания, параллельные плоскости b (флогопит). В каолини¬
те, напротив, интенсивность полосы при 3619 см-1 не зависит от ориентации (внутрен¬
ний гидроксил направлен в сторону вакантной октаэдрической позиции).
Если измерения выполняют на традиционной дисперсионной аппаратуре, разреше¬
ние можно улучшить, используя более узкие щели, но энергия при этом уменьшится.
Поскольку ширина щели непостоянна по всему спектру, необходимо следить, чтобы
щели не были слишком широкими, потому что сигналы могут деформироваться, и за¬
кон Бера-Ламберта не будет выполняться.
1 Несмотря на использование эталонных минералов, значительная вариативность полос по¬
глощения в зависимости от химической структуры часто приводит к непреодолимым труд¬
ностям при калибровке минералов типов 2:1 и 2:1:1. В этом случае можно проводить только
полуколичественные измерения для выявления изменений в профиле, используя минераль¬
ный индикатор в качестве образца сравнения на данном уровне.
Глава 5. Минералогический анализ методом инфракрасной спектрометрии 151
5.3. Другие ИК-методы
5.3.1. Ближняя инфракрасная спектрометрия (БИКС)
Основные положения
Для анализа органоминеральных или органических веществ используют колебания лег¬
ких атомов, имеющих прочные молекулярные связи с протонами (N, С или О). В случае
слабых связей или тяжелых элементов выявление и количественное определение коле¬
баний затруднено.
Широкие полосы поглощения легко воспроизводимы, но они подвержены влия¬
нию проникающего излучения и поэтому чувствительны к размеру частиц и влажно¬
сти. Обычно необходим тонкий размол для того, чтобы получить частицы одинаково¬
го размера и снизить фоновый шум до максимально низкого уровня. Но приемлемые
результаты можно получить и для материалов без тонкого помола (частное Сообщение,
Д. Брюне, Исследовательский институт IRD, Монпелье, Франция). Колебания связи
являются причиной отклика, который зависит от количества присутствующих молекул
и их окружения и позволяет проводить количественное определение.
В ближнем ИК-диапазоне полосы расположены на большем расстоянии друг от дру¬
га, чем в среднем и дальнем ИК, что уменьшает их перекрывание. Для повышения точ¬
ности можно использовать первую производную сигнала.
Измерения проводят в режиме пропускания - поглощения или диффузного из¬
лучения - отражения (NIRA-DRIFT)1 на порошках в диапазоне длин волн от 1000 до
2500 нм (волновые числа от 10 000 до 4000 см*1), в ряде случаев, таких как почвенная
подстилка (содержание воды, белка, общего азота, сахаров и др.) или жидкие пробы
с погруженными оптоволоконными датчиками.
Материал
Измерительное оборудование, применяемое для диффузной отражательной ИК-спектро-
метрии, несколько отличается от приборов, работающих в режиме пропускания — по¬
глощения (см. рис. 5.8). Оптические элементы выполняются из кварца. Полный спектр
в ближнем ИК-диапазоне получают в течение нескольких секунд.
Метод
Этот неразрушающий метод требует предварительного измельчения проб, но не требует
ни реагентов, ни определения массы или объема пробы. Измерения проводятся очень
быстро (~30 с) и стоимость одного анализа невелика.
Результаты измерений зависят от физических факторов, влияющих на отражение, т. е.
от размера частиц (отражение, преломление, дифракция), их распределения (неодно¬
родность) и распределения пустот (уплотнение и связанная с ним ориентация).
Современные БИК-системы существенно улучшены в связи с прогрессом, достигну¬
тым в хемометрическом программном обеспечении, которое позволяет проводить не¬
возможные ранее вычисления. Количественный анализ основан на многофакторной ка¬
либровке с использованием всей спектральной информации, а не только на поглощении
на конкретных длинах волн. Калибровку проводят с использованием разнообразных
1 NIRS (near-infrared reflectance spectrometry) — отражательная ближняя инфракрасная спек¬
трометрия; NIRA (near-infrared analysis) — ближний инфракрасный анализ; DRIFT (diffuse
reflectance IR with Fourier transform) — диффузная отражательная инфракрасная спектрометрия
с преобразованием Фурье.
152
Часть 1. Минералогический анализ почв
Диафрагма
щающаяся кювета
(образец)
Рис. 5.8. Схема отражательного ближнего ИК-спектрометра: 1 — поворотное зеркало для ре¬
гулировки луча, падающего на пробу, и улучшения отражения на стены интегрирующей сферы.
Должны быть представлены все возможные углы, начиная с 90°. Полный поток из источника, ко¬
торый возбуждает образец, концентрируется кварцевыми линзами; 2 — интегрирующая сфера,
которая уменьшает влияние различий в размере частиц, собирает наиболее интенсивное излуче¬
ние, но отделяет его зеркальные компоненты
методов расчета, основные из которых: метод главных компонент {principal component
analysis, РСА) ’регрессия на главных компонентах (regression on principal components, RPC),
метод частных наименьших квадратов {partial least square, PLS) и множественная линей¬
ная регрессия {multiple linear regression, MLR). Программы помогают в выборе лучшего
метода калибровки, хотя выбрать не всегда легко {Dardenne и др., 2000).
Метод подходит, главным образом, для анализа органических веществ, но использу¬
ется и во многих других методиках почвенного анализа. Чанг с сотр. {Chang и др., 2001)
использовали калибровку методом регрессии на главных компонентах для определе¬
ния методом ИК-спектрометрии с преобразованием Фурье содержания влаги, общего
углерода, общего азота, ЕКО, песка, пыли, ила, макроагрегатов, потенциально мине¬
рализуемого азота, углерода биомассы, интенсивности полного и базального дыхания
почв. Единственным условием является необходимость составления базы данных для
калибровок по стандартным образцам почв. Для конкретной переменной калибров¬
ка включает: (1) измерение определяемого параметра стандартным методом для всех
эталонных образцов почвы (предпочтительно, содержащих определяемый компонент
в широком диапазоне концентраций), (2) получение БИК-спектров тех же самых образ¬
цов почвы, (3) расчет и построение прямой линии множественной линейной регрессии
на графике с результатами стандартного метода по оси абсцисс и результатами БИК-
анализа по оси ординат. После калибровки измерение неизвестного образца происходит
очень быстро. Реальную концентрацию компонента по оси абсцисс можно вычислить из
спектральных данных, отложенных на оси ординат. Однако кажущаяся универсальность
метода не вполне соответствует действительности. Калибровка возможна всегда, но не
всегда надежна (например, вариабельность калибровочных кривых см. в Chang и др.,
Глава 5. Минералогический анализ методом инфракрасной спектрометрии
153
2001). Какие почвы выбирать для эталонных образцов (все типы или конкретный тип
почв)? Какой метод подготовки почвы использовать?
Список дополнительной литературы в конце этой главы содержит описание областей
применения БИК-спектроскопии для исследования почв и подстилок.
5.3.2. Сочетание термических методов анализа и ИК-спектрометрии
с Фурье-преобразованием при анализе летучих соединений
Измерения, проводимые во время ТГА-ДТА1, позволяют определять природу газовых
продуктов, образующихся при термическом разложении образца (регистрация выделяе¬
мого газа или газовый термический анализ)2. Анализы выполняют методом пролетной
ИК-Фурье-спектрометрии (Фурье-ИКС), измеряя зависимость пропускания или погло¬
щения от температуры и времени нагрева.
Этот динамический метод позволяет осуществлять мониторинг в режиме реального
времени химических или физико-химических превращений, происходящих при быстром
нагревании пробы (можно проводить контролируемый термолиз или пиролиз органиче¬
ских и неорганических материалов). Повышение температуры с умеренной скоростью
дает возможность обнаруживать нестабильные радикалы и молекулярную фрагмента¬
цию, связывая их с изменениями массы и температуры (плавление, экзо- и эндотер¬
мические реакции, разложение минеральных карбонатов, N-, С-, S- и Н-соединений,
окисление, восстановление, перенос протонов и т. д.).
Давление является параметром, который влияет на сублимацию и испарение. Дав¬
ление может изменяться при слишком быстром разложении нестабильного продукта,
но может приводить и к молекулярному синтезу при определенной температуре. При
низком давлении наиболее реакционно-способные газы быстро диффундируют, из¬
бегая возможной рекомбинации. Обычно рекомендуется работать в атмосфере аргона
при низком давлении. Но контролируемая атмосфера может выдвинуть на первый план
окислительно-восстановительные процессы или, наоборот, подавить их.
Дополнительную информацию можно получить, выбирая скорость нагрева в диапа¬
зоне от 20 до 400 °С/мин в зависимости от скорости удаления образовавшегося газа и его
обнаружения или быстрого титрования прежде, чем начнутся другие газовые реакции.
5.3.3. Инфракрасная микроскопия
Анализ методом Фурье-ИКС можно проводить на микропробах размером от 20 до
500 мкм. ИК-микроскоп (см. гл. 8) состоит из линз с зеркалом Кассегрена, сопряжен¬
ных с высокочувствительным KPT-детектором3, охлаждаемым жидким азотом. Он мо¬
жет работать в режиме пропускания или отражения. Разрешение примерно 8 см-1 в за¬
висимости от качества исследуемого материала и количества накопляемых спектров.
Чтобы избежать последствий флуоресценции, возможной в присутствии некоторых
органических материалов, можно использовать рамановскую спектрометрию, в которой
источником возбуждения является монохроматический лазер, излучающий в красной
области спектра. Этот метод позволяет определять количества порядка пико- и даже
фемтограммов.
1ДТА — дифференциальный термический анализ; ТГА — термогравиметрический анализ.
2 EGD (evolvedgas detection) — регистрация выделяемого газа; EGA (evolvedgas analysis) — ана¬
лиз выделяемого газа.
3 KPT — кадмий, ртуть, теллур (МСТ = HgCdTe).
154 Часть 1. Минералогический анализ почв
5.3.4. Спектроскопия комбинационного рассеяния1
Общие сведения
Рамановская спектроскопия не нашла широкого применения в почвоведении, потому
что дисперсионное оборудование очень дорогое, а его эксплуатационные качества недо¬
статочно высокие из-за проблем выбора длин волн с высоким разрешением.
Прогресс в области электроники позволил разработать высокочувствительные детек¬
торы, высокоселективные монохроматоры и мощные монохроматические лазеры и сде¬
лал Фурье-ИК-спектрометры доступными приборами, дополняющими другие методы
ИК-спектрометрии. Компьютерная обработка данных обеспечивает очень быструю
трактовку спектров.
Этот метод позволяет дополнить информацию, полученную методами абсорбцион¬
ной ИК-спектрометрии, так как согласно правилам отбора некоторые колебания актив¬
ны только в одном из двух методов или их интенсивности отличаются. Полосы симме¬
тричных колебаний более интенсивны в рамановской спектроскопии, а асимметричных
колебаний интенсивнее в ИК-спектроскопии.
Использование микропроб позволяет исследовать некоторые трудно дифференцируе¬
мые молекулярные структуры, такие как рутилы, анатазы и брукиты. Можно определить
природу химических связей и ориентацию ОН-групп, а поровая вода, если она присут¬
ствует, не привносит искажений. Это недеструктивный метод, который не требует слож¬
ной подготовки проб. Можно работать с порошками, и даже с влажными порошками,
что невозможно для других ИК-методов.
Основные положения
Рамановская спектроскопия основана на неупругом рассеянии ИК-излучения2 с вто¬
ричным ИК-излучением на частотах биения. Спектры состоят из узких линий, которые
требуют использования приборов с высоким разрешением:
• флуоресценция, вызываемая электронными переходами, может искажать спектры
и маскировать рамановский сигнал, если используется возбуждающий лазер, ко¬
торый излучает в видимом диапазоне (448 нм); возбуждение NdrYAG-лазером, из¬
лучающим на 1064 нм (частоте, соответствующей электронным переходам в мало
заселенной зоне), обычно не вызывает флуоресценции, и его сопряжение с высо¬
кокачественным Фурье-ИК-спектрометром исключает проблемы, связанные с не¬
достаточным разрешением;
• когда монохроматический луч попадает на образец, небольшая часть его переиз-
лучается, демонстрируя изменения частот, которые отражают частоты колебаний
образца. Эту часть излучения и измеряют в рамановской спектрометрии; неизме¬
ненная часть излучения (упругое рассеяние Релея) удаляется с помощью фильтров.
Оборудование
Основным прибором является Фурье-ИК-спектрометр, состоящий из интерферометра,
снабженного программным обеспечением для сбора и обработки данных. Он должен иметь
внешнее окно для прохождения излучения от Nd:YAG-лазера, камеру для порошкообраз¬
ных образцов, позволяющую облучать образцы под углами 90 и 180°, модуль спектральной
1 Рамановская спектроскопия. — Примеч. науч, ред.
2 Рамановская спектроскопия в видимом и УФ-диапазонах может привести к фотодеструк¬
ции образца, а также к термическому повреждению.
155
Глава 5. Минералогический анализ методом инфракрасной спектрометрии
фильтрации и, если необходимо, определенный детектор. К Фурье-ИК-спектрометру
могут быть добавлены рамановский Фурье-микроскоп и вспомогательное оборудова¬
ние для рамановских измерений при очень высоком давлении (алмазные наковальни).
Лазер для возбуждения атомов и молекул должен быть монохроматическим или пере¬
страиваемым на разные длины волн (для лучшей селективности). Он должен обеспечи¬
вать высокую интенсивность (чувствительность измерений), когерентность излучения
(пространственные и временные характеристики) и, наконец, в случае использования
оптоволокон — низкую расходимость.
ИК и рамановскую спектроскопию можно дополнить другими методами для изуче¬
ния структуры (например, ЯМР, EXAFS). Несмотря на развитие этих методов, ИК- и ра-
мановская спектроскопии остаются конкурентоспособными (легкость в обращении,
разумная стоимость оборудования); они дают возможность достаточно детально изучать
структуру (включая вакансии и замещения, природу связей в молекулах) и ее влияние,
например, на реологию или явления педогенезиса, происходящие при выветривании
(например, образование наносов, взаимодействия с поверхностью, адсорбция молекул).
Однако количественное определение фаз часто трудновыполнимо, если вообще воз¬
можно, из-за проблем, связанных с ориентацией глин и несовершенством спектров.
Использованная литература
Chang C.-W., Laird DA, Mausbach M. et Hurburgh CRJr (2001) Near-Infrared Reflectance Spectroscopy-
Principal Components regression analysis of soil properties. Soil Sci. Soc. Am. J., 65,480-490.
Dardenne P, Sinnaev G. et Baeten V. (2000) Multivariate calibration and chemometrics for near infrared
spectroscopy: which method. Journal of Near Infrared Spectroscopy, 8, 229-237.
Duyckaerts G. (1959) The infra red analysis of solid substances. Analyst, 84,201-214.
Farmer VC et Palmieri F (1975) The characterization of soil minerals by Infrared spectroscopy. In: Soil
components - 2 - Inorganic components, Gieseking JE ed., Springer, 573-670.
FrOhlich F (1980) №oformation de silicates ferrifdres amorphes dans la sedimentation peiagique r6cente.
Bull Mindral., 103, 596-599.
Frdhlich F (1989) Les silicates dans l’environnement p£lagique de Госбап indien du c6nozoi‘que. M6-
moire Mus6um National d’Histoire Naturelle, Paris, XLVI, 206 p.
Keller WD et Pickett FE (1949) Absorption of IR radiation by powdered silice minerals. Am. Miner., 34,
855-868.
Liu LG et Memagh TP (1992) Phase transitions and Raman spectra of anatase and rutile at high pressures
and room temperature. Eur. J. Mineral, 4,45-22.
Nail SL, White JL et Hem SL (1976) IR studies of development of order in aluminium hydroxide gels.
J. Pharm. Sci., 65, 231-234.
Nyquist RA et Kagel О (1971) Infrared spectra of inorganic compounds, Academic Press, New York.
Stubican V et Roy R (1961) Infrared spectra of layer silicates. J. Am. Ceram. Soc., 44, 625.
WadaK (1966) Deuterium exchange of hydroxyl groups in allophane. Soil Sci. Plant Nutr., 12,176-182.
Weir CE Lippincott ER Van Valkenbuig A et Bunting EN (1959) Infra-red studies in the 1 and 15 microns
region to 30 000 atmospheres. J. Res. Natl Bur. Stud., 63A, 55.
Дополнительная литература в хронологической
последовательности
Tuddenham WM et Lyon R.P. (1960) Infrared techniques in the identification and measurement of miner¬
als. Anal Chem., 32, 1630-1634.
Mitchell BD Farmer VC et Me Hardy WJ (1964) Amorphous inorganic materials in soils. Academic
Press. Adv. Agron., 16, 327-383.
Hayashi H et Oinuma К (1965) Relationship between infrared absorption spectra in the region of 450-
900 cm-1 and chemical composition of chlorite. Am. Miner., 50,476-483.
156
Часть 1. Минералогический анализ почв
Hayashi Н et Oinuma К (1967) Si-0 absorption band near 1000 cm1 OH absorption bands of chlorite.
Am. Miner., 52, 1206-1210.
Russell JD, McHardy WJ et Fraser AR (1969) Imogolite: a unique aluminosilicate. Clay Miner., 8,87-99.
Wada К et Greenland DJ (1970) Selective dissolution and differential infrared spectroscopy for character¬
ization of amorphous constituents in soil clays. Clay Miner., 8, 241-254.
Conley RT (1972) Infra-red spectroscopy. Allyn-Bacon, 2nd edition.
Fieldes M, Furkert RJ et Wells N (1972) Rapid determination of constituents of whole soils using IR
absorption. N. Z. J. Sci., 15, 615-627.
Miller RGT et Stace BC (1972) Laboratory methods in Infrared spectroscopy., Heyden and Son.
Farmer VC (1974) The Infrared spectra of minerals. Minerals Sci. (London).
Stepanov IS (1974) Interpretation of the IR spectra of soils. Pochvovedenie, 6, 76-88.
Gadsden JA (1975) Infrared spectra of minerals and related inorganic compounds., Butterworth.
Griffiths PR (1975) Chemical infrared fourier transform spectroscopy., Wiley, New York Chemical Anal¬
ysis, 43.
Brame EG, Grasselli JG (1976) Infrared and Raman spectroscopy., Marcel Dekker, 1A.
White JL, Nail SL et Hem SL (1976) Infrared technique for distinguishing between amorphous and crys¬
talline aluminium hydroxide phase. Proceedings. 7th Conference. Clay Mineral Petrology (Czecho¬
slovakia), 51-59.
Marel HW, Van der et Beutelspacher H (1976) Atlas of infrared spectroscopy of clay minerals and their
mixtures., Elsevier Amsterdam.
Proshina NV (1976) Use of infrared spectroscopy for identification of soil samples. Nauch. dokl. Vsshei
Shk., Biol. Naudi, 3, 114-118.
Brame EG, Grasselli JG (1977) Infrared and Raman spectroscopy., Marcel Dekker, IB, 1C.
Hlavay J, Jonas K, Elek S et Inczedy J (1977) Characterization of the particle size and the cristallinity of
certain minerals by infrared spectrophotometry and instrumental methods. I - Investigations on clay
minerals. Clays Clay Miner., 25, 451-456.
Hlavay J, Jonas K, Elek S et Inczedy J (1978) Characterization of the particle size and the crystallinity of
certain minerals by infrared spectrophotometry and other instrumental methods. II — Investigation
on quartz and feldspar. Clays Clay Miner., 26, 139-143.
Ferraro JR et Basile LJ (1978) Fourier transform infrared spectroscopy. Applications to chemical
systems., Academic, New York, vol. 1.
Slonimskaya MV, Besson G, Dainyak LG, Tchoubar C et Drits VA (1978) Interpretation of the IR spectra
of celadonites and glaucomites in the region of OH-streching frequencies. Clay Miner., 21,377-388.
Smith AL (1979) Applied infrared spectroscopy: fundamentals, techniques and analytical problem¬
solving., Wiley, New York, vol. 54 (chemical analysis).
Farmer VC (1979) The role of infrared spectroscopy in a soil research institute: characterization of inor¬
ganic materials. Eur. Spectrosc. News, 25, 25-27.
Ferraro JR et Basile LJ (1979) Fourier transform infrared spectroscopy Applications to chemical systems,
Academic, vol. 2.
Hlavay J et Inczedy J (1979) Sources of error of quantitative determination of the solid crystalline miner¬
als by inrared spectroscopy. Acta Chim., (Budapest), 102,11-18.
Olphen H Van et Fripiat JJ (1979) Data handbook for clay materials and other поп-metallic minerals.,
Pergamon.
Martin AE (1980) Infrared interferometric spectrometers. In Vibrational spectra and structure, Durig J.R.
ed., Elsevier, Amsterdam, vol. 8.
Pouchert CJ (1981) The Aldrich library of infrared spectra., Aldrich Chemical Co, 1850 p.
Shika A, Osipova NN et Sokolova ТА (1982) Feasibility of characterizing the mineralogical composition
of soils by infrared spectrophotometry. Moscow Univer. Soil Sci. Bull., 37, 34-40.
Theng BKG, Russel M, Churchman GJ et Parfitt RL (1982) Surface properties of allophane, halloysite
and imogolite. Clays Clay miner., 30, 143-149.
Ferraro JR et Basile LJ (1983) Fourier transform infrared spectroscopy. Applications to chemical
systems., Academic, New York, vol. 3.
Fysh SA et Fredericks PM (1983) Fourier transform infrared studies of aluminous goethites and hema¬
tites. Clays clay Miner., 31, 377-382.
Глава 5. Минералогический анализ методом инфракрасной спектрометрии 157
Velde В (1983) Infra-red OH-stretch bands in potassic micas, talcs and saponites: influence of electronic
configuration and site of charge compensation. Am. miner., 68,1169-1173.
Gillette PC et Koenig JL (1984) Objective criteria for absorbance subtraction. Appl. Spectrosc., 38,334-337.
Kosmas CS, Curi N, Bryant RB et Franzmeier DP (1984) Characterization of iron oxide minerals by
second-derivative visible spectroscopy. Soil Sci. Soc. Am. J., 48,401-405.
Prost R (1984) Etude par spectroscopie infra-rouge к basse temperature de groupes OH de structure de la
kaolinite, de la dickite et de la nacrite. Agronomie, 4, 403-406.
Kodama H (1985) Infrared spectra of minerals. Reference guide to identification and characterization of
minerals for the study of soils. Res. Branch, Agric. Can. Tech. Bull., IE.
Mulla DJ, Low PF et Roth CB (1985) Measurement of the specific surface area of clays by internal reflec¬
tance spectroscopy. Clays Clay Miner., 33, 391-396.
Keller RJ (1986) The Sigma library of FT-IR spectra., Sigma chemical Co, vols. 1-2,2894 p.
Griffiths et Haseth PR (1986) Fourier transform infrared spectrometry, Chemical Analysis Series, Vol.
83, Wiley New York, 672 p.
Russel JD (1987) Infrared spectroscopy of inorganic compounds. In Laboratory methods in infra-red
spectroscopy, Willis H. ed., Wiley, New York.
Johannsen PG, Krobok MP et Holzapfel WB (1988) High-pressure FT-IR spectrometry., Bruker report,
39-43.
Pouchert CJ (1989) The Aldrich library of FT-IR Spectra., Aldrich Chemical Co, vols. 1-3,4800 p.
Mottana A et Burragato F (1990) Absorption spectroscopy in mineralogy., Elsevier, Amsterdam, Oxford,
New York, Tokyo, 294 p.
Delvigne JE (1998) Atlas of Micromorphology of mineral alteration and weathering. The Canadian Min¬
eralogist, special publication 3, Ottawa et IRD (ex-Orstom), Paris.
Silverstein RM et Webster FX (1998) Spectrometric Identification of organic compounds. Wiley New
York, 482 p.
McHale JL (1999) Molecular spectroscopy., Prentice-Hall, London, Sydney, Toronto, 463 p.
Gillon D, Joffre R et Ibrahima A. (1999) Can litter decomposability be predicted by near infrared reflec¬
tance spectroscopy. Ecology, 80, 175-186.
Confalonieri M, Fomasier F, Ursino A, Boccardi F, Pintus В et Odoardi M (2001) The potential of near
infrared reflectance spectroscopy as a tool for the chemical characterisation of agricultural soils.
J. Near Infrared Spectrosc., 9, 123-131.
Joffre R, Agren GI., Gillon D. et Bosatta E (2001) Organic matter quality in ecological studies: theory
meets experiment. Oikos, 93,451-458.
Feam T (2001) Standardisation and calibration transfer for near infrared instruments: a review. J. Near
Infrared Spectrosc., 9, 229-244.
Ludwig В et Khanna PK (2001) Use of near infrared spectroscopy to determine inorganic and organic
carbon fractions in soil and litter. In Assessment methods for soil carbon, Lai R, Kimble JM, Fol-
let RF et Stewart BA ed., Lewis, UK.
Ozaki Y, Sasic S et Jiang JH (2001) How can we unravel complicated near infrared spectra? - Recent
progress in spectral analysis methods for resolution enhancement and band assignments in the near
infrared region. J. Near Infrared Spectrosc., 9, 63-95.
Reeves JB et McCarty GW (2001) Quantitative analysis of agricultural soils using near infrared reflec¬
tance spectroscopy and a fibre-optic probe. J. Near Infrared Spectrosc., 9, 1, 25-34.
Tso, Ritchie GE, Gehrlein L et Ciurczak EW (2001) A general test method for the development, valida¬
tion and routine use of disposable near infrared spectroscopic libraries. J. Near Infrared Spectrosc.,
9, 165-184.
Fidencio PH, Poppi RJ et de Andrade JC (2002) Determination of organic matter in soils using radial
basis function networks and near infrared spectroscopy. Anal. Chem. Acta., 453, 125-134.
Couteaux MM, Berg В and Rovira P (2003) Near infrared reflectance spectroscopy for determination of
organic matter fractions including microbial biomass in coniferous forest soils. Soil Biol. Biochem.,
35, 1587-1600.
Brown DJ, Shepherd KD, Walsh MG, Dewayne Mays M and Reinsch TG (2005). Global soil characteriza¬
tion with VNIR diffuse reflectance spectroscopy. Geoderma, doi: 10.1016/j.geoderma.2005.04.025.
Глава 6. Разделение минералов методом
селективного растворения
6.1. Введение
6.1.1. Кристаллическое состояние глинистых минералов
Минералогическая характеристика скрытокристаллических минералов или минера¬
лов с ближним порядком расположения атомов (Fe, Al, Si, Mn, 'll, P) имеет большое
значение для понимания геохимических и педохимических процессов, происходящих
при выветривании первичных минералов, а также для объяснения направления разви¬
тия и относительной устойчивости систем, кинетики химических процессов в почве.
Эти вещества могут представлять собой промежуточный этап между кристаллической
материнской породой и вторичными минералами и часто рассматриваются в качестве
индикаторов изменений. Почва — это открытая система, которая может обмениваться
энергией и веществом с окружающей средой. Большинство реакций происходят в не¬
равновесных условиях, и переходные состояния зависят от потоков жидких или газо¬
образных веществ (химические реакции протекают на границах раздела фаз твердое —
жидкость и жидкость — жидкость), времени релаксации (диффузия частиц, перенос
вещества и т. д.) и, разумеется, деятельности микроорганизмов.
Накопление индивидуальных веществ или их осаждение в виде тонкого слоя изме¬
няет активность структурных центров кристаллических материалов и может подавлять
движение ионов, нейтрализовать заряды или вызывать замещения в решетках. 1Ьли, ок¬
сиды или оксигидроксиды и алюмосиликаты могут заряжаться (некоторые из них имеют
амфотерный характер). Высокая реакционная активность, вызываемая состоянием их
поверхности раздела, делает возможной адсорбцию катионов и анионов. Они могут ней¬
трализоваться органическими веществами. Эти реакции придают почве большую устой¬
чивость к выветриванию и действию микроорганизмов. Таким образом, конечной це¬
лью анализа некристаллических продуктов может являться почвенный генезис, а также:
• почвенная классификация (через процессы подзолизации, андосолизации, лате-
ризации и др.);
* почвенная минералогия, минералогические балансы, очистка перед применением
других методов (в частности, методов, требующих удаления парамагнитных эле¬
ментов), например, ЭПР, ДТС РСП, Мессбауэровская спектроскопия, РД, Фурье-
ИК-спектроскопия, СЭМ, ЭД-РСМА, ВД-РСМА, ПЭМВР, СПЭМ1;
1 ЭПР — электронный парамагнитный резонанс (ESR — electron spin resonance); ДТС РСП —
дальняя тонкая структура рентгеновских спектров поглощения (EXAFS — Extended X-ray
Absorption Fine Structure); РД — рентгеновская дифракция (XRD — X-ray diffraction); СЭМ —
сканирующая электронная микроскопия (SEM— scanning electron microscopy); ЭД-РСМА —
энергодисперсионный рентгеноспектральный микроанализ (EDX — energy dispersive X-ray);
ВД-РСМА — волнодисперсионный рентгеноспектральный микроанализ (WDX— wavelength
dispersive X-ray); ПРЭМ — просвечивающая растровая электронная микроскопия (STEM —
scanning transmission electron microscopy).
Глава 6. Разделение минералов методом селективного растворения
159
• исследование физических и химических свойств почвы, почвенного плодородия
(дефицит Fe, фиксация Р, токсичность А13+, перенос тяжелых металлов, разруше¬
ние связывающих частицы веществ, факторы агрегации и др.).
Идентификация некристаллических веществ требует использования нескольких ме¬
тодов: РД не очень удобна для гелей, поскольку дает только широкие полосы, если со¬
держание геля велико или развитие кристаллической структуры дальнего порядка на¬
ходится на ранних стадиях. Методы химического растворения недостаточно селективны
для минералогии, поскольку они основаны на использовании кислотных, основных,
восстанавливающих или комплексообразующих реагентов и включают, например:
• разрыв электростатических (например, реакции обмена, А13+ мостики) или коорди¬
национных (например, Fe3+ мостики) связей;
• ионизацию функциональных групп (органическое вещество).
Растворение должно не только выделять различные фазы, аморфные в рентгеновских
лучах, но также:
• минимизировать химические изменения (относительная нестабильность экстраги¬
руемых компонентов по сравнению с почвенной матрицей) и предотвратить раз¬
рушение решеток глин и первичных минералов;
• ограничивать гидролиз экстрагированных веществ;
• не допускать молекулярных перегруппировок в жидкой фазе (зародышеобразова-
ния);
• сохранять экстрагированные вещества в растворе;
• не допускать образования химических барьеров (нерастворимых осадков под воз¬
действием реагентов) и новообразования твердых веществ.
Все методы экстракции (с использованием одного или нескольких реагентов, одно¬
стадийной или ступенчатой экстракции) зависят от термодинамических ограничений:
• активность ионов, pH, концентрации реагентов, отношение почва/реагент, поря¬
док применения реагентов;
• фактор времени, кинетика экстракции, скорость перемешивания, длительность
контакта, старение геля;
• температура;
• фотолитическая энергия (УФ катализ химических реакций).
Начальная скорость минералогической экстракции обычно довольно высока, но
затем падает; она также зависит от размера кристаллитов и степени кристаллического
совершенства (наличие дефектов, степень разупорядочения) или природы и концен¬
трации элементов в жидкой фазе. Таким образом, согласование с результатами других
исследований, воспроизводимость и надежность метода зависят от используемой мето¬
дики экстракции. За исключением необходимости адаптации к специфическим услови¬
ям, любая модификация методики (соотношения или концентрации реагентов, время
контакта и т. д.) может привести к серьезным ошибкам определения. Требуемую степень
селективности экстракции минералов можно достичь только при сравнении различных
экстрактов, тщательно очищенных ультрацентрифугированием, и проведении химиче¬
ских измерений (1) в жидкой фазе, содержащей экстрагированные вещества (конгру¬
энтные и неконгруэнтные реакции), и (2) в твердой фазе (например, с использованием
дифференциальной РД, СЭМ и ЭД-РСМА). Некоторые из надежных методов включают
автоматические расчеты и интерпретацию, что позволяет проводить количественное
определение каждой фазы и устанавливать точные и воспроизводимые геохимические
балансы.
160
Часть 1. Минералогический анализ почв
6.1.2. Инструментальные и химические методы анализа
Рентгеновская дифракция хорошо работает на атомных решетках с упорядочением даль¬
него порядка, но для веществ с упорядочением ближнего порядка без упорядоченной су¬
перструктуры РД дает малоинтенсивные широкие рефлексы, непригодные для исполь¬
зования (рис. 6.1). Поэтому вещества, дающие дифрактограммы такого типа, называют
аморфными.
50 А
Примеры структур ближнего
порядка
Аллофан Si02, А12Оэ, лН20
структура полой сферы = 35 А
Сфера
аллофана
Микроагрегат
Монослой
воды
S\OJM2Oz = 2
SICVAIA < 1
Дифракто грамма
аллофана
Дифрактограмма
имоголита
/vA/V.
@ о •
ОН О Si
о
AI
Рис. 6.1. Кристаллические и аморфные для рентгеновского излучения вещества
Глава 6. Разделение минералов методом селективного растворения
161
В настоящее время развитие инструментальных методов позволяет более точно опре¬
делить природу фаз и их расположение. Кристаллическое состояние характеризуется
периодическим повторением атомной структуры по трем некомпланарным направле¬
ниям в пространстве (Maziere, 1978). В настоящее время допустимо использовать терми¬
ны «некристаллические» (бесструктурные, аморфные) или криптокристаллические (со
скрытой структурой), но не паракристаллические (почти структурированные) вещества.
Это твердые вещества, структура которых не содержит повторяющихся элементов на
дальних расстояниях (молекулярная область по крайней мере 3 нм в диаметре), но кото¬
рые представляют упорядочение на коротких расстояниях, что придает им особые свой¬
ства. Упорядочение ближнего порядка учитывает взаимное расположение ближайших
соседних атомов в масштабе межатомных расстояний. Эти расстояния не имеют четких
характеристичных наборов рефлексов в рентгеновской дифракции. В другой среде или
в большем масштабе можно наблюдать различные типы некристалличности:
• зоны беспорядочных замещений или структурной дислокации в случае аморфных
веществ с хорошо организованной периодической структурой (структура с дефек¬
тами); фазово-контрастная просвечивающая микроскопия высокого разрешения
и просвечивающая растровая электронная микроскопия (STEM) в режиме микро¬
дифракции способны обнаружить и локализовать этот тип дефектов;
• протяженные непериодические зоны, состоящие из кластеров случайным образом
распределенных частиц с упорядочением ближнего порядка в случае гелей окислов
алюминия, железа, кремния и некоторых алюмосиликатов (опалин, аллофанопо-
добные, протоаллофан, аллофан, протоимогодит; гелеподобный, стеклообразный,
стекловатый кремнезем и др.).
Некоторые вещества обнаруживают начальную стадию организации среднего поряд¬
ка (рис. 6.1), которая приводит к образованию широких линий на рентгеновской диф-
рактограмме (например, имоголит, ферригидрит, фероксигит). Для исследования этих
минералов можно использовать спектроскопические методы (рис. 6.2):
• Метод EXAFS позволяет получить точную информацию о межатомных расстояниях
в ближайшем окружении и об организации первой координационной сферы, но ми¬
нимум информации о связях между полиэдрами (координация среднего порядка).
• ПСВР анализирует сферу, окружающую атом, но требует хорошего знания струк¬
туры.
• ЯМР-спектрометрия является селективным методом для химических структур,
однако не очень чувствительным; исследование сверхтонкого магнитного поля
оксидов железа позволяет измерить степень кристаличности; в силикагелях ис¬
пользование ядер 29Si позволяет различать состояния связей в Si04. Можно также
исследовать различные формы31Р.
• ЭПР- и ДЭЯР1-спектроскопия исследуют сверхтонкие и суперсверхтонкие струк¬
туры методом резонанса спина электрона на атомном уровне.
Все эти инструментальные методы используют излучения, которые отвечают диа¬
пазонам расстояний, соответствующих субмикронным уровням, необходимым для ис¬
следования структуры почвенных материалов. Диапазон излучений простирается от
радиочастот до рентгеновских и гамма-лучей. Высокая стоимость и уровень сложности
оборудования, а также очень специфические аналитические области, где они исполь-
1 ПСВР — припороговая спектроскопия высокого разрешения (XANES — X-ray absorption
near-edge spectroscopy); ДЭЯР — двойной электронно-ядерный резонанс (ENDOR - electron
nuclear double resonance).
162
Часть 1. Минералогический анализ почв
Ралмп* I ИК
частоты Микроволны | р 1 m'In"
V
УФ Рентгеновский^ Гамма-лучи
\
10е ' 10т ' 10‘ ' 10’ ' 10 1 КГ1 1<Н
Х.нм
1 1 1 1 1 1 1111 I 1 Г“
ю' Ю10 10” 10м ю1* ю1' ю”
V, Гц
1 10 100 10' ' 10* ' Ш7 ' 1'0'
Л, СМ'1
Спиновые переходы
)
вращения
Молек
тяс
ны
*
Внешние
лектроны
Внутренние электроны
Ядро
Природа
переходов
(Ядро) (Электроь
Молекулярные
Ьан
с
„ ЯМР (| ( ЭПР
Raman
ИК
Мессбауэр
ц РФА XAFS
Методы
Микроволны
ПСВР ДТС РСП
1<Гв |10-‘|
КГ* | 1
LJ0
о
о
Энергия, эВ
Рис. 6.2. Анализ молекулярных структур спектроскопическими методами (электромагнитные
спектры и зонды окружения); к — длина волны; v — частота; п — волновое число; F — дальний
ИК; М — средний ИК; N — ближний ИК; V — видимый; РФА — рентгено-флюоресцентный анализ;
XAFS — спектроскопия тонкой структуры спектра рентгеновского поглощения; ПСВР — припоро-
говая спектроскопия высокого разрешения; ДТС РСП — дальняя тонкая структура рентгеновских
спектров поглощения
зуются, ограничивают исследования такого рода узкоспециализированными лаборато¬
риями.
Инструментальные методы позволяют проводить наблюдения на субмикронном уров¬
не, но химический анализ позволяет исследовать объекты, которые проявляют среднюю
активность отдельных частиц, связанную с их поверхностью и зарядами. Таким обра¬
зом, материал, образованный в процессе выветривания, может быть удовлетворительно
разделен последовательными экстракциями, что позволяет выделить составы с увели¬
чивающейся степенью кристалличности, которые соответствуют (или нет) различным
химическим соединениям.
6.1.3. Методы селективного растворения
Область применения методов селективного растворения, очевидно, ограничивается раз¬
нообразием почвенных минералов (табл. 6.1) и сложностью растворения только одной
строго определенной фазы (табл. 6.2).
Растворение зависит от различных факторов:
• размера «кристалла» и уровня атомного разупорядочения;
• дефектов в стехиометрии или блокирования активных центров;
• свойств кристаллографических поверхностей (анизотропия);
• порозности систем, плотности дефектов поверхности и т. д.
Используемые реагенты и возможные методы предварительной обработки не должны
приводить к образованию осадков. Они должны обеспечивать сохранение экстрагиро¬
ванных веществ в растворе и, если это возможно, ограничивать рекомбинации в жидкой
фазе (рис. 6.3 и 6.4),
Оксиды, гидроксиды и оксигидроксиды приводят к образованию связанных заря¬
дов в почве, и их структура, связи, поверхность и реакционная способность изменяют¬
ся в зависимости от степени кристалличности и степени разупорядочения их решеток
(табл. 6.1).
Таблица 6.1. Основные кристаллические и некристаллические оксиды и оксигидроксиды в почве (Si, Fe, Al, Mn, Ti, смешанные)1
Fe
А1
Мп
Ti
Si
Смешанные гели
(с зарядом)
Fe2 3* + е<-> Fe2*
А13*
Мп4+ + 2е~ <-> Мп2+
Ti4+ + 2e-<->Ti2+
Si4+
Si
Al, Fe, Mn
n
Гематит
Коруцд
Голландит
Рутил
a-кварц (низкий)
r
Аллофан
a-Fe203
a-AlgOj03
а-Мп02
ТЮ2
Si02
А^О,—2Si02 • лН20
Маггемит
Гиббсит
Криптомелан
Анатаз
p-кварц (высокий)
Имоголит
y-Fe203
у-А1(ОН)э
а-Мп02
ТЮ2
Si02
Магнетит Fe304
Байерит
Пиролюзит
Брукит
a-тридимит
Гизингерит
(Fe2*Fe3*204)
а-А1(ОН)3<1)
Э-Мп02
ТЮ2
Si02
Fe203—2Si02* nHfi
Гетит
Диаспор
Бернесе ит
Ильменит
a-кристобалит
Неотокит
a-FeOOH
а-АЮОН
б-Мп02
FeTiO,
Si02
Si02—Mn
♦Лепидокрокит
Бемит
Манганит
(Лейкоксен,
Коэсит
Эвансит
Y-FeOOH
Y-A100H
Y-MnOOH
см. ильменит)
Si02
A13P04(0H)6 7Н20
Ферригидрит
Нордстрандит
Гроутит
Стишовит
Азовекит
Fe203 * 2FeOOH
♦Фероксигит
5-FeOOH
Акаганеит
(3-FeOOH
♦Стильпносидерит (гель)
Fe203 • лН20
(Лимонит Fe203 • лН20
см. гетит)
(3) pH 7,5
Fe?+/2Fe2+
А1(ОН)3
♦(клиахит
А1203 /|Н20)
pH 4,0 + перерас-
творение pH 9,0
a-MnOOH
Гаусманит Мп304
(Мп2+Мп3+204)
Тодорокит
♦Вернадит
5-Мп02 nHfi
Литиофорит
(Al, 1л)Мп02(0Н)2
pH 8,5-8,8
Титановый гель
ТЮ2 лН20
pH 1-3
Si02
♦Опал Si02
Силикагель
Si02(nH20)
Халцедон
(волокнистый) Si02
Биогенный кремнезем
(Si02)
^езР04(0Н)б4 тр
2 — <-» изоструктура.
3 — Теоретическое среднее pH осаждения в водной среде.
* — Гель, упорядочение ближнего порядка.
() — Устаревшая терминология.
1 После названий минералов часто представлены не составы, а структурные типы, к которым они относятся. — Примеч. науч. ред.
Глава 6. Разделение минералов методом селективного растворения
Таблица 6.2. Оценка экстрагируемых фаз селективным дифференциальным растворением (Тамм — раствор оксалата аммония;
ДЦБ — раствор дитионата—цитрата—бикарбоната; пиро — раствор пирофосфата)
Метод, экстрагент
Тамм,
в темноте
ДЦБ
Тетраборат Пиро Na2CQ3
NaOH, 0,5 Последователь-
моль/л ные экстракции
Тайрон
в щелочной
среде
NaF или
KF
pH экстрагируемой фазы
3,0
7,2
9,7
10,0
9,8
>12
1-14
А1
Некристаллические
гидратированные оксиды
+++
+++
+
+++
+++
+++
+++
+++
Органические комплексы
+++
+ + +
++ +
+++
+++
+++
+++
+++
—
Кристаллические
соединения
0
+
+
0
+
+++
+
+
+
Si
Опаловые гели
0
0
0
+
+++
++
—
+++
Кристаллические
соединения
0
0
0
0
+
+
—
0
Fe
Ферригидрит, фероксигит
Некристаллические
+++
+++
+
0
0
+++
+++
+++
гидратированные оксиды
Органические комплексы
+++
+++
+++
+++
0
0
+++
+++
Кристаллические
соединения
0+
+++
0
0
0
+++
+
+
Mn
Некристаллические оксиды
Кристаллические
соединения
Аллофан
+++
+
+
+
+++
+++
+
+++
Имоголит
++
+
+
+
+++
+++
+
+++
Аллофаноподобные
соединения
+++
+++
+
++
+++
+++
+++
+++
(1) Филлосиликаты
0+
0+
0
0
+
+(++)
+
+
+++ — от значительного до полного растворения; ++ — среднее или частичное растворение; + — слабое растворение; 0 — нет значи¬
тельного растворения; (1) — сильный рост pH с аллофанами.
Часть 1. Минералогический анализ почв
Глава 6. Разделение минералов методом селективного растворения
165
Матрица
пробы
/
Природа, источник
Размер частиц, дисперсность
Окристаллизованность,
активные поверхности
Влияние pH
\
Влияние
комплексообразования
Влияние окислительно¬
восстановительных
условий
Концентрации ионов
Химическое
действие
экстрагента
Физические факторы
Экспериментальные
условия
Термодинамика
Отношение
проба/реагент
/
Температура
Кинетика реакции
Константы
равновесия
■ Хемосорбция
Площадь контакта
функция
дзета-потенциала
Условия
перемешивания
Фотолиз
Остаточная твердая фаза **
Рентгеновская дифракция—
Дифференциальный _
термический анализ
Термогравиметрический
анализ
Магнитное __
фракционирование
Мессбауэровская _
спектроскопия
Баланс выхода
Слабый поток
(или закрытая система)
низкое отношение твердая
фаза/реагент
Нестехиометрическое
растворение
Накопление ионов в растворе
риск осаждения
(комплексообразующий
реагент)
фактор задержки
перенасыщение
подавление растворения
Осаждение экстрагированной
фазы
нуклеация, реакционные
барьеры
рекомбинации фаз
новообразования
и преобразования матрицы
Сильный поток
(или открытая система)
высокое отношение
твердая фаза/реагент
Конгруэнтное,
неконгруэнтное или
селективное растворение
Стехиометрическая
разрядка на реакционной
поверхности раздела фаз
Быстрое растворение
до равновесия
с остаточной матрицей
Рис. 6.3. Факторы, контролирующие селективное растворение
166
Часть 1. Минералогический анализ почв
Нуклеация
ОН Старение Рекомбинация
Рис. 6.4. Превращение гелей гидроксидов (вверху) и растворимость гидроксидов
в зависимости от pH и концентрации (внизу)
6.1.4. Реагенты и искусственные стандарты
Сложные формы железосодержащих и некристаллических веществ часто требуют ис¬
пользования беспримесных синтетических моделей минералов со степенью кристал¬
личности или организацией ближайшего окружения, сходных с теми, что наблюдаются
у почвенных веществ.
Твердые вещества должны быть приготовлены из высокочистых исходных веществ,
потому что гидроксиды склонны адсорбировать примеси из-за их очень большой удель¬
ной поверхности. Флокуляция достигается добавлением ионов Н+ или ОН-. Кипяче¬
ние приводит к дегидратации и приводит к увеличению количества геля (нуклеация).
Временной фактор позволяет состарить гель, т. е. осуществить постепенную медленную
кристаллизацию (рис. 6.4) с переходом от структуры ближнего порядка в структуру с бо¬
лее высокой степенью упорядочения. Например, для ионов алюминия сначала будет на¬
блюдаться осаждение мономера, а затем полимеризация гидроксида:
А13+ + ЗОН->А1(ОН)3.
Процесс идет в несколько стадий:
А1(ОН)2+->А1(ОН)2+-> А12(ОН)24+и т. д.,
и дегидратация сопровождается потерей ионов Н+ в ходе осаждения. Осадки амфотер¬
ных соединений алюминия могут снова растворяться в щелочной среде с образованием
167
Глава 6. Разделение минералов методом селективного растворения
растворимых алюминатов. Все операции должны проводиться в строгом соответствии
с методикой, чтобы получить продукты осаждения, которые соответствуют воспроизво¬
димым стадиям образования.
Эти методики были определены в работах (Henry, 1958), (Towe и Bradley, 1967), (Atkin¬
son и др., 1968), (Schwertmann и Taylor, 1972; 1977), (Murphy и др., 1976), (Jeanroy, 1983),
(Farmern Fraser, 1978), (Pollard, 1992), (Lewis и Schwertmann, 1979).
Приготовление железосодержащих соединений
Гетит
• Растворяют 8,08 г нитрата трехвалентного железа (Fe(N03)3 * 9Н20) в 80 мл воды
в конической колбе (колбе Эрленмейера) на 250 мл.
• Доводят pH до 7,5 добавлением по каплям примерно 20 мл 3 М раствора КОН при
перемешивании магнитной мешалкой.
• Оставляют на 3 ч для образования осадка, удаляют надосадочную жидкость с по¬
мощью сифона и четыре раза промывают осадок водой для полного удаления об¬
разовавшегося в растворе нитрата калия.
• Суспензируют осадок в 100 мл воды, затем добавляют 3 М раствор КОН до концен¬
трации ионов ОН", равной 0,3 М.
• Хранят раствор в полипропиленовой бутыли при 20 °С, время от времени пере¬
мешивая, в течение 2—5 лет в зависимости от требуемой степени кристаллизации.
• Промывают до полного удаления КОН.
• Высушивают в вентилируемом сушильном шкафу при 50 °С.
Акаганеит
• Растирают порцию чистого FeCl2 • 4Н20 в агатовой ступке так, чтобы частицы про¬
ходили сквозь сито с ячейками размером 0,5 мм.
• Распределяют порошок тонким слоем и оставляют для гидролиза в контакте
с влажным воздухом на 6 мес. (вещество со временем приобретает бурую окраску).
• Промывают водой для удаления оставшегося Fe2+ и высушивают при 50 °С.
Лепидокрокит
• Растворяют 0,6 г FeCl2 • 4Н20 в 150 мл 0,2 М раствора NaCl, насыщенного азотом
барботированием с использованием перистальтического насоса при скорости по¬
тока 15 мл/мин.
• Перемешивают в атмосфере азота и доводят pH до 6,0, добавляя по каплям 1 М
раствор NaOH.
• Оставляют до стабилизации pH и замещают азот воздухом путем барботирования,
поддерживая pH во время окисления (2,5 ч).
• Промывают водой и высушивают при 50 °С.
Слабо упорядоченный ферригидрит
• Растворяют 2,02 г Fe(N03)3 • 9Н20 в 500 мл дистиллированной воды и доводят pH
до 7,5, добавляя по каплям 15 мл 1 М NaOH.
• Оставляют при pH 7,5 на 18 ч.
• Промывают водой и высушивают при 50 °С.
168
Часть 1. Минералогический анализ почв
Гематит
• Готовят 1 М раствор хлорида трехвалентного железа (FeCl3 • 6Н20).
• Осаждают добавлением NH4OH при 60 °С.
• Фильтруют на быстром фильтре.
• Промывают до отрицательной реакции на ионы С1“ (проба с AgN03).
• Прокаливают при 500 °С в течение 1 ч.
Маггемит
• Взвешивают 5 г оксалата двухвалентного железа (FeC204 • 2Н20) и помещают
в кварцевый тигель с крышкой.
• Постепенно нагревают в электрической печи до 410-420 °С и выдерживают при
этой температуре в течение 1 ч для удаления конституционной воды.
Полученный продукт близок к маггемиту.
Аморфное органическое железо
• Экстрагируют органическое вещество (ОВ) из образца подзолистой почвы массой
20 г с высоким содержанием гумуса с помошью 900 мл 0,2 М раствора NaOH.
• Центрифугируют экстракт и отфильтровывают (рассчитывают содержание ОВ, вы¬
раженное в содержании С).
• Готовят раствор нитрата трехвалентного железа Fe(N03)3 • 9Н20 (С = 108 г/л).
• К 225 мл этого раствора (3,35 гжелеза) добавляют требуемое количество экстракта ОВ.
• Перемешивают, контролируя pH с помощью pH-метра; окончательное значение
pH должно быть 5,0; осаждается красно-коричневый гель.
• Промывают дистиллированной водой до полного удаления натрия.
• Хранят гель под водой и определяют содержание железа (мг/мл).
Приготовление соединений АР*
Бемит
• Готовят 0,1 М раствор А1С13.
• Нейтрализуют до pH 6, добавляя 0,4 М раствор NaHC03.
• Оставляют в контакте на 1 ч.
• Доводят pH до 8 медленным добавлением 0,15 М раствора NaOH.
• Выдерживают в закрытой пластиковой (ПТФЭ) бутыли при 160 °С в течение 60 ч.
• Охлаждают, промывают до полного удаления бикарбоната и удаляют избыток ги¬
дроксида натрия центрифугированием.
• Хранят в виде суспензии в закрытой полиэтиленовой бутыли.
Приготовление смешанных соединений
Имоголит
• Добавляют 30 ммоль перхлората алюминия в 2,5 л деионизированной воды.
• Добавляют 15 ммоль тетраэтилсиликата, что соответствует примерно 3,3 мл ком¬
мерческого раствора.
• Гомогенизируют и доводят pH до 4,5 раствором соды.
• Смесь становится опалесцирующей; оставляют на ночь, и жидкость осветляется.
Глава 6. Разделение минералов методом селективного растворения
169
• Осторожно кипятят с обратным холодильником в течение 5 сут.
• Оставляют охлаждаться, затем добавляют раствор гидроксида аммония до pH 9,0;
гель осаждается.
• Промывают до полного удаления ионов натрия; хранят в воде и измеряют концен¬
трации А1 и Si.
6.2. Основные методы селективного растворения
6.2.1. Метод, основанный на использовании кислого оксалата в темноте
(КОТ)
Основные положения
Растворяющий реагент называют также реактивом Тамма. Метод позволяет растворять
аллофан и гели, органические комплексы железа и алюминия, гидратированные оксиды
железа и алюминия (ферригидрит, фероксигит). Имоголит при одной обработке полно¬
стью не растворяется. Филлосиликаты разрушаются лишь в очень небольшой степени,
за исключением минералов со значительным уровнем разупорядоченности. Лепидокро-
кит чувствителен к оксалатному реагенту. При использовании нескольких (2—3) обра¬
боток некоторые кристаллические соединения алюминия и железа могут растворяться
в значительной степени.
Буферный раствор (оксалат аммония + щавелевая кислота) вызывает процессы про¬
тонирования, комплексообразования и восстановления. Таким образом, это может вы¬
зывать перенос протонов, электронов и ионов (Stum, 1985; Furrer, 1985—1987; Cornell
и Schindler, 1987; Schwertmann, 1991).
Оксалат-ионы образуют три типа комплексов с ионами трехвалентного железа:
Fe3+ + С2042- о FeC204+ (6.1)
Fe3+ + 2ОД- о Fe(C204)2- (6.2)
Fe3+ + 3C2042-oFe(C204)3- (6.3)
(Двухвалентное железо также образует комплексы с другими константами устойчивости
и растворимости.)
Для осуществления третьей стадии (6.3) необходим избыток оксалатного буфера;
в противном случае ионы С202-, являясь акцепторами ионов Н+ и Fe3+, могут вызвать
конкурентные реакции, особенно если повышение pH уменьшает константы раство¬
римости.
С ионами А13+ происходит комплексообразование по схеме1:
1 Оксалат-анион, выступая в качестве бидентантного лиганда, координируется посредством
атомов кислорода, образуя пятичленный цикл — АЮ2С2. Еще по два атома кислорода оста¬
ются направленными наружу (на рисунке «утеряны» образующие связь атомы кислорода). —
Примеч. науч. ред.
170
Часть 1, Минералогический анализ почв
При pH ниже 3,5 поверхности оксидов насыщаются протонами. Таким образом, бла¬
гоприятная зона имеет pH 3,0, потому что в ней контролируются заряды ниже точки
нулевого заряда (см. главу 20). Что касается стадий реакции, протонирование должно
быть первой стадией растворения соединения, так как эта реакция приводит к лучшей
адсорбции комплексов и одновременному синергетическому действию.
В случае некристаллических соединений железа и марганца преобладает реакция вос¬
становления:
Fe3+ + e-<->Fe2+.
Применение комплексообразующего реагента имеет два последствия:
• Ионы трехвалентного железа становятся менее сильными окислителями, тогда
как ионы двухвалентного железа — более сильными восстановителями; отношение
Ре3+/Ре2+снижается, растворимость увеличивается; реакция автокаталитическая.
• Это влияет на pH (а буферная система поддерживает его на уровне 3,0) и таким
образом влияет на растворимость и устойчивость комплексов, что предотвращает
изменения окислительно-восстановительного потенциала, которые иначе были бы
вызваны изменением pH.
Модификации метода
Тти(Татт, 1922,1931,1934а, Ь) рекомендовал использовать буферный раствор, содер¬
жащий оксалат аммония и щавелевую кислоту, для растворения неорганических гелей,
в том числе оксидов железа, свободного кремнезема и глинозема. Автор предложил ис¬
пользовать pH 3,25.
Беглый обзор составов реагентов, используемых в методе Тамма с 1922 г., выявляет
различные модификации основной методики (например, изменения концентраций ре¬
агентов, pH, отношения почва/реагент, времени контакта, перемешивания, температу¬
ры, фотолиза (рис. 6.5)).
Активное
и окристаллизованное
железо
Tamm, 1922
pH 3,25
отношение 1:50
облучение в течение 2 ч
Тамм, облучение УФ
т
Невоспроизводимые результаты
Тамм, в темноте
Активное некристаллическое
и скрытокристаллическое
железо
DeEndredy, 1963
pH 3,25
отношение = 1/100 0,2 М
облучение УФ в течение 1 ч
Schwertmann, 1964
pH 3,25
отношение = 1/100 0,2 М
в темноте в течение 2 ч
Международные стандарты
Министерство сельского хозяйства США, 1972 (4 ч)
Blakemore, 1981 (4 ч)
Jackson, 1982 (2 ч)
Jeanroy, 1983 (4 ч)
Международный информационно-справочный центр
по почвам, 1987 (4 ч)
Wilson, 1987 (4 ч)
Е
МсКвадив и Day, 1966 .
pH 3
отношение = 1/40 0,2 М
4ч
1
pH 3,5 0,15 М
в темноте
/лоиё, 1981
Parfitt, 1982
Рис. 6.5. Модификации метода Тамма с использованием оксалата аммония и щавелевой
кислоты (основные методики)
Глава 6. Разделение минералов методом селективного растворения
171
Некоторые изменения вносились для адаптации метода к природе образца и его ком¬
понентов. Сравнение со старыми данными иногда затруднено из-за сложности почвен¬
ной матрицы и взаимодействий, и особенно из-за того, что точные условия обработки
неизвестны.
Джанг (Jung, 1934) установил, что метод пригоден для легких, но не карбонатных почв,
и что он не дает воспроизводимых результатов для тяжелых почв из-за разрушения глин.
Авторы варьировали условия обработок: меняли концентрации реагентов и pH (от 3
до 6); заменяли щавелевую кислоту другими органическими кислотами (винной, лимон¬
ной, салициловой, бензойной, фталевой, малоновой и др.); сочетали с восстановлением
сероводородом, водородом или дитионитом (Duchaufourn Souchier, 1966).
Значительными этапами в развитии метода Тамма явились открытие фоточувстви¬
тельности при растворении (Schoefield, 1959), влияния фотосенсибилизации (метод из¬
вестен как УФ-метод Тамма, (DeEndredy, 1963) и особенно разработка Швертманном
стандартной методики (Schwertmann, 1964), также называемой методом Тамма в темноте.
В настоящее время метод используется в качестве международного стандарта.
Временной фактор оказывает различное влияние на реакции; обычно растворение
вначале проходит быстро, но после 4 или 5 ч замедляется. Влияние времени обычно су¬
щественно только в первые 2 ч. Швертманн определил среднее время равным 2 ч на
основании исследования профиля традиционного растворения, демонстрирующего
предпочтительное растворение отдельных фракций (ферригидрит, вещества с упорядо¬
чением ближнего порядка и др.). Маккиг и Дей (McKeague и Day, 1966) показали, что
при их условиях растворение проходило лучше, когда время контакта увеличивалось до
4 ч (в темноте). Хотя последствия обработки такой длительности могут иметь кинетиче¬
ский эффект, основой метода является одностадийная экстракция.
Отношение почва/раствор обычно имеет ограниченное влияние на результаты. Пар-
тифф (Party?\ 1989) показал, что экстракция реагентом с концентрацией 0,15 М при
pH 3,0, при отношении почва/раствор 1:100 и перемешивании в течение 4 ч при 20 °С
(в темноте) давала удовлетворительные результаты для большинства почв, однако ее
нельзя использовать, если содержание алюминия или экстрагируемого железа превы¬
шает 5%. В этом случае необходимо применять реагент с концентрацией 0,20 М и от¬
ношение почва/раствор 1:200.
Экстракция раствором оксалата с концентрацией 0,15 М или реагентами с концен¬
трацией 0,20 М при pH 3,0 дает аналогичные результаты для аллофана, если его концен¬
трация не является лимитирующим фактором.
Определяющее влияние на экстракцию оказывает pH. Он контролирует кинетику
растворения кристаллических и некристаллических соединений. Максимальное рас¬
творение достигается при pH между 2,6 и 3,0, протонирование является синергичным
восстановительному или комплексообразующему действиям оксалатного реагента. Если
pH растет выше 4,0, эффективность буфера существенно снижается, количество экс¬
трагируемого железа уменьшается, и селективность изменяется. Поэтому величина pH
должна поддерживаться на уровне 3,0, чтобы избежать возможных изменений. Повыше¬
ние температуры ускоряет реакцию и изменяет селективность. Стандартная температура
составляет примерно 20 °С.
Приготовление реагентов
Все реагенты должны готовиться с использованием стандартных реактивов, бидистил-
лированной воды или воды, деионизированной на ионообменной колонке, способной
удерживать Si.
172
Часть 1. Минералогический анализ почв
Кислый оксалат аммония: реактив Тамма, содержащий оксалат (С = 0,2 М) при pH 3,0:
• Растворяют 16,15 г оксалата аммония (COONH4)2* H2Ol и 10,90 г щавелевой кисло¬
ты (СООН)2 • 2Н202 в примерно 900 мл воды и доливают до 1000 мл.
• Проверяют pH и доводят его до 3,0, добавляя раствор гидроксида аммония или ща¬
велевой кислоты (С = 0,2 М).
• Готовят еженедельно и хранят в бутыли из темного стекла, защищенной от света.
• 0,2%-ный раствор суперфлока (флоккулянт) в воде (Cyanamid Согр.).
• Корректор матрицы для разбавления перед проведением атомной спектрометрии,
для 10 000 ррт К:
— отвешивают 19 г КС1;
— растворяют в примерно 900 мл воды;
— когда раствор достигнет комнатной температуры, доводят объем до 1000 мл.
Методика
• Измеряют влажность почвы, используя отдельную субпробу для определения по¬
правки на влажность (см. главу 1).
• Взвешивают на лабораторных весах 1 г воздушно-сухой почвы, просеянной через
0,2-миллиметровое сито (избегать излишнего измельчения).
• Помещают почву в колбу на 100 мл.
• Добавляют 50 мл кислого оксалатного реагента; для почв, содержащих хорошо рас¬
творимые в оксалатах соединения (содержание экстрагируемых соединений А1 или
Fe более 2%), добавляют 100 мл оксалатного реагента в колбу на 250 мл.
• Перемешивают в темноте в течение 4 ч.
• Сливают часть надосадочной жидкости в центрифужную пробирку на 50 мл.
• Центрифугируют при 10 000 g в течение 10 мин; если жидкость не совершенно про¬
зрачна, вновь перемешивают, добавляют 3 капли суперфлока и снова центрифуги¬
руют (каждая серия измерений должна включать два холостых опыта в дополнение
к стандартному образцу почвы и два повтора каждого из образцов серии).
После соответствующего разбавления выполняют анализы экстракта, используя:
• Si: ИСП- или ААС-спектрометрию на 251,6 нм, пламя N20-C2H2;
• Fe: ИСП- или ААС-спектрометрию на 248,3 нм, пламя воздух-С2Н2;
• А1: ИСП- или ААС-спектрометрию на 309,3 нм, пламя N20-C2H2;
• Мп: ИСП- или ААС-спектрометрию на 279,5 нм, пламя воздух—С2Н2
и, при необходимости, Т1 и Р,
Если используют спектрофотометрию:
• разлагают оксалатную матрицу кипячением с концентрированной азотной кисло¬
той;
• высушивают и растворяют в 5 М хлористоводородной кислоте;
• испаряют почти досуха и растворяют в воде, затем доливают до требуемого объема.
1 (NH4)2C204 • Н20, молекулярная масса 142,12; может быть непригодным для некоторых
глин (можно заменить оксалатом натрия Na2C204); меры безопасности: относится к ядам,
нельзя принимать внутрь.
2 СООН-СООН • 2Н20, молекулярная масса 126,07; разлагается под УФ-излучением; вы¬
сушивание при 100 °С приводит к потерям за счет сублимации, разлагается при 160 °С; меры
безопасности: относится к ядам, едкая, не принимать внутрь.
Глава 6. Разделение минералов методом селективного растворения
Расчеты
173
Полученные данные:
- Рх - вес влажной или воздушно-сухой пробы для измерения влажности;
- Р2 — вес пробы почвы, высушенной при 105 °С;
- Р- вес пробы почвы для экстракции (мг);
-Ли В- содержание в экстракте и холостой пробе, соответственно (мг/л);
- D — разбавление экстракта;
- VR - объем оксалатного реагента, использованного для экстракции (мл).
Поправочный коэффициент на влажность
Этот коэффициент необходим для того, чтобы привести к обычным условиям результа¬
ты, полученные для почв, высушенных при 105 °С, особенно почв, обогащенных некри¬
сталлическими веществами, таких как аллофановые почвы:
Я = 100^—^, %.
Поправочный коэффициент на влажность/= 100/#.
Расчет содержания элементов (Fe, Al, Si, Mn, Т1, Р и др.):
% элемента = 0MDVJLA - В)]/Р.
Коэффициент пересчета содержания элемента в содержание оксида:
%Fe203 = %Fe х 1,43 %Мп02 = %Мп х 1,58
%А1203 = %А1 х 1,89 %Si02 = %Si х 2,14
Примечания
Экстракция дает достаточно надежные результаты для большинства почв. Идентифи¬
кация некоторых фаз может представлять трудности для неупорядоченных глин. Даже
если экстракты защищены от света, они могут подвергаться изменениям. Поэтому они
должны быть быстро проанализированы, чтобы избежать осаждения из-за нестабиль¬
ности реагента.
При высокой ионной силе реагента и образующихся комплексов добавление супер-
флока обычно не обязательно. Надосадочную жидкость можно отфильтровать сильфо-
нированием с помощью шприца, снабженного фильтром 0,45 нм (после декантации).
Оксалатную экстракцию используют в различных областях.
• Почвоведение и почвообразование, геохимия (дифференциальное растворение
и идентификация некристаллических или мало упорядоченных фаз, переходов
между кристаллическими фазами, химические и методические исследования, вли¬
яние почвенной структуры и др.), предпочтительно с использованием данных по
растворенным фазам.
• Минералогия одновременно использует солюбилизированные фазы и остаточные
твердые фазы для:
— исследования замещений, степени упорядочения, соединений с атомной орга¬
низацией ближнего порядка;
- для приготовления проб (растворение связующих частицы веществ, удаление
оксидов для повышения интенсивности дифракции кристаллических соедине¬
174
Часть 1. Минералогический анализ почв
ний), проведения анализов методом дифференциальной рентгеновской диф¬
ракции (DXRD) и анализов после удаления парамагнитных соединений (Месс-
бауэрская, ЭПР-, ЕЛ^/Ф-спектроскопии), наблюдений пространственного
распределения на шлифах растворимых оксалатных фаз (Агосепа, 1988) и, на¬
конец, моделирования процессов разрушения (образующихся соединений и др.)
• Микробиология и агрономия для анализа биофизической и биохимической ак¬
тивности (влияние на водоудерживание, пластичность, доступность для растений
соединений активного железа, содержания щавелевой кислоты, образовавшейся
в почве (щавелевая кислота биохимической природы вызывает разрушение окси¬
дов железа)).
• В карбонатных почвах оксалат аммония осаждает катионы Са2+ в форме оксалата
кальция, растворимость которого менее 0,006 г/л воды или уксусной кислоты; по¬
этому карбонат должен быть разрушен минимальным количеством уксусной кис¬
лоты перед экстракцией оксалатом аммония и дополнительно измерено содержа¬
ние элементов, солюбилизированных уксусной кислотой.
• В восстановительных условиях (например, гидроморфные почвы, гистосоли, ан-
досоли в условиях влажного климата) оксалатный метод не может предоставить
информацию об исходной степени окисления железа и марганца в почве перед
экстракцией.
• В андичных почвах и андосолях-андисолях оксалатный метод может исполь¬
зоваться аналогично ДЦБ-методу (см. раздел 6.2.2); органические комплексы
железа растворяются; аллофан, который экстрагируется после перемешивания
с оксалатом в течение 2-4 ч, можно определить исходя из содержания экстраги¬
рованного кремния на основании гипотезы о преобладании связей Si-0-Al; со¬
держание ферригидрита можно оценить исходя из количества экстрагированного
железа.
6.2.2. Метод, основанный на использовании дитионит-цитрат-
бикарбоната (ДЦБ)
Основные положения
Этот метод (Mehra-Jackson, 1959—1960) позволяет растворять педогенные оксиды и ги¬
дроксиды:
• кристаллические оксиды железа (гематит, гетит), некристаллические оксиды же¬
леза и органические комплексы железа и алюминия, также как оксиды замеще¬
ния железа и марганца, некоторые некристаллические соединения с отношением
Si02/Al203 менее чем 0,5;
• магнетит и ильменит разрушаются лишь в небольшой степени, так же как гиббсит
и алюмосиликаты типа аллофан-имоголит; однако в некоторых случаях магнетит
может быть растворен в значительной степени (магнетит легко окисляется в магге¬
мит в присутсвии сильного окислителя);
• глины не разрушаются, но железо, присутствующее в кристаллической решетке
вермикулитов и нонтронита, может быть растворено в значительной степени, осо¬
бенно если экстракция проводится при пониженном pH.
Основным процессом метода является восстановление, а дитионит — очень активный
восстановитель при pH ниже 9—10 (Deb, 1950). Элементы, подверженные биологическо¬
му восстановлению, такие как железо и марганец, восстанавливаются и удерживаются
Глава 6. Разделение минералов методом селективного растворения
175
в растворе за счет комплексообразования с лимонной кислотой в системе с pH 7,3, под¬
держиваемым бикарбонатом натрия.
Оптимальное значение pH для реакций восстановления равно 7—8. Ниже pH 6,5
коллоидная сера может осаждаться и образовывать суспензии в экстрактах, что препят¬
ствует измерениям методом абсорбционной спектроскопии. Использование буферных
растворов может уменьшить этот процесс. Поскольку раствор дитионита быстро теря¬
ет свои восстановительные свойства, комплексообразование предотвращает повторное
окисление, так же как осаждение сульфида железа, и способствует сохранению экстра¬
гированных фаз в растворе, если продолжительность экстракции не превысит 15 мин.
Первоначально предлагалось проводить однократное добавление раствора дитионита
при первой экстракции. Впоследствии, под влиянием международных организаций и по
рекомендации основоположника метода, было рекомендовано двухступенчатое добав¬
ление дитионита.
При необходимости можно проводить две или три последовательных экстракции,
чтобы захватить железо из кристаллических соединений относительно большого раз¬
мера. В этом случае экстракты можно либо смешать перед анализом, либо проанали¬
зировать отдельно для измерения изменения кинетики растворимости и суммировать
результаты после каждого измерения.
Температура должна поддерживаться на уровне 75 °С, чтобы ускорить реакцию
и уменьшить количество коллоидной серы и сульфида железа, а также минимизировать
разложение дитионита. Нельзя повышать температуру, и во избежание местного пере¬
грева следует использовать водяную баню.
Содержание железа должно быть не более 0,5 г Fe203, чтобы обеспечить избыток вос¬
становителя и комплексообразователя. Реакцию восстановления железа в слабощелоч¬
ной среде можно представить следующими уравнениями:
S2042- + 40Н- -► 2S02- 4- 2Н20 + 2е"
2Fe3+ + 2e-->2Fe2+
или в растворе нитратного комплексообразователя:
S2042* 4- Fe203 4- 2НОС(СОО)2- + 2Н+ -► 2S02" 4- 2Fen-HOC(COO)-4- Н20
Масса пробы должна составлять от 1 до 5 г без изменения состава реагента (если же¬
лезо не может образовать комплекс из-за недостаточного количества цитрата, возможно
осаждение черного сульфида железа).
Используют буферный раствор для предотвращения изменений pH, влияющих на
окислительно-восстановительный потенциал, поскольку для восстановления каждого
иона Fe3+ требуется два иона ОН".
Некоторыми авторами проверялись восстановительные методики, использующие
дитионат в цитратном растворе при различных значениях pH (Homgren, 1967; Avery
и Bascom, 1982) или концентрации реактива Тамма (Duchaufour и Souchier, 1966; Hetier
и Jeanroy, 1973; Loveland и Bullock, 1976), или в других буферных или комплексообра¬
зующих средах, таких как раствор тартрата-ацетата натрия (Deb, 1950), или в щелочных
средах, таких как раствор пирофосфата (Franmeiervuip., 1965).
Комплексы с лимонной кислотой (тридентантный лиганд) подобны комплексам
с щавелевой кислотой и образуют очень стабильные соединения железа и алюминия.
Константа устойчивости комплекса алюминия с лимонной кислотой = 7,37.
176
Часть 1. Минералогический анализ почв
В природных условиях присутствие цитрата предотвращает или замедляет осаждение
алюминия, поскольку центры координации заняты цитрат-ионом, и его гидролиз про¬
ходит медленно1:
О
В экстрагенте ДЦБ, содержащем высокую концентрацию лимонной кислоты, проис¬
ходят замещение молекул воды и блокирование центров координации. Поэтому гидро¬
лиз становится невозможным (Kwong и Huang, 1979). Некоторые минералы, например,
псевдобемит, проявляют особые свойства {СатЫеги Sposito, 1991).
Было обнаружено, что два реагента (гидроксиламин гидрохлорид и подкисленный
пероксид водорода) более эффективны, чем ДЦБ, для селективного растворения окси¬
дов марганца (Neaman и др., 2004а, Ь).
ДЦБ-обработка может вызывать разупорядочение структуры, которое можно наблю¬
дать с использованием рентгеновской дифракции или электронной микродифракто-
метрии. Адсорбция групп лимонной кислоты на некоторых глинах может существенно
замедлить удаление межслоевой воды. Это следует учитывать в анализе осадков, содер¬
жащих формы соединений, известные как «свободные». Следует отметить, что в таких
условиях протоимоголит не может превращаться в более структурированный имоголит.
Приготовление реагентов
Все реагенты должны готовиться с использованием стандартных реактивов «для анали¬
за», бидистиллированной воды или воды, деионизированной на ионообменной колон¬
ке, способной удерживать кремнезем.
• Дитионит натрия применяют в виде порошка (в зависимости от количества анали¬
зов используют небольшие флаконы продукта, чтобы он был всегда свежим).
• Цитрат-бикарбонатный буферный раствор: растворяют в дистиллированной воде
непосредственно перед использованием 79,40 г трехзамещенного цитрата натрия
(C6H5Na307 • 2Н20) и 9,24 г бикарбоната натрия (NaHC03), проверяют pH, который
должен быть равен примерно 7,3, и доводят объем до 1 л.
• Для флокуляции используют или насыщенный раствор хлорида натрия — 400 г
NaCl в 1 л воды, или насыщенный раствор хлорида калия — 375 г КС1 в 1 л
воды; если затем проводят измерения методом атомной абсорбции или ИСП-
спектрометрии, необходимо использовать хлорид калия, чтобы избежать повы¬
шенных содержаний натрия; или ацетон.
1 Цитрат-ион может координироваться с центральным атомом только тремя карбоксильными
группами. Гидроксильная группа лимонной кислоты в процессе диссоциации и координации
не участвует. — Примеч. науч. ред.
177
Глава 6. Разделение минералов методом селективного растворения
Методика
• В плоскодонную центрифужную пробирку из полипропилена или ПТФЭ на 100 мл
помещают от 1 до 5 г почвы (размер частиц 0,2 мм) в зависимости от предполага¬
емой концентрации оксида железа (карбонаты, органическое вещество и раство¬
римые соли следует предварительно удалить из образца).
• Помещают в пробирку магнитный якорь и добавляют 45 мл цитрат-бикарбонатного
буферного раствора при помощи распределителя, снабженного фторопластовым
шприцем.
• Помещают в водяной бане, отрегулированной на 75 °С, на магнитную мешалку;
когда образец достигнет температуры бани, добавляют 1 г порошка дитионита
и продолжают перемешивание с умеренной скоростью в течение 5 мин.
• Добавляют еще 1 г дитионита и перемешивают в течение 10 мин.
• После 15 мин перемешивания центрифугируют в течение 5 мин при 2500 g для
получения прозрачного раствора (если жидкость еще мутная, взбалтывают ее
и добавляют насыщенный раствор хлорида натрия или калия, чтобы вызвать
флокуляцию, а затем опять центрифугируют при 2500 g; эта обработка делает от-
центрифугированный осадок более компактным и таким образом затрудняет по¬
вторное ресуспендирование для второй стадии обработки; в почвы, происходящие
из вулканического пепла, часто необходимо добавлять 10 мл ацетона перед центри¬
фугированием для достижения удовлетворительной флокуляции).
• Декантируют чистую надосадочную жидкость в мерную колбу на 250 мл.
• Если осадок интенсивного бурого, черного или красного цвета, добавляют 45 мл
буферного раствора и обрабатывают, как описано выше, с двукратным добавлени¬
ем дитионита и нагреванием до 75 °С в течение 15 мин, затем повторно ресуспен-
дируют центрифужный осадок для более однородного разрушения.
• Центрифугируют и декантируют в ту же самую 250-миллилитровую колбу (или
анализируют второй экстракт отдельно).
• Дважды или трижды промывают осадок 10 мл буферного раствора, флокулируют,
центрифугируют промывной раствор и добавляют экстракт к предыдущей порции.
• Добавляют 250 мл дистиллированной воды и гомогенизируют.
• В каждую серию проб добавляют две холостые пробы (только реактивы) и один
стандартный образец. После соответствующего растворения (в течение 2—10 мин)
фильтрат, содержащий свободные оксиды и гидроксиды, должен быть проанализи¬
рован методом атомно-абсорбционной спектрометрии:
- А1 при 309,3 нм в пламени ацетилен — оксид диазота;
- Fe при 309,3 нм в пламени ацетилен - оксид диазота;
- Si при 309,3 нм в пламени ацетилен — оксид диазота;
- Мп при 279,5 нм в пламени ацетилен - воздух;
- Т1, Р, К, Mg также могут определяться при необходимости.
Если используют колориметрию, некоторые методы позволяют работать непосред¬
ственно с экстрактами, но предпочтительно разложить забуференную, хелатирующую
и восстановительную матрицу кипячением с азотной или серной кислотой и пергидро¬
лем. Содержание железа определяют, используя 1,10-ортофенантролин или феррон1,
алюминия — эриохром цианин, кремнезем — молибдат, с учетом присутствия фосфора
(см. гл. 31).
1 Купферон. — Примеч. науч. ред.
178
Часть 1. Минералогический анализ почв
Взвешивают очищенный осадок. Его можно анализировать, используя рентгенов¬
скую дифракцию как инструментальный метод; интенсивность линий увеличивается
после обработки ДЦБ (см. гл. 4); ИК-, ЭПР-, ЯМР-, i^FS-спектрометрии (см. гл. 12);
термический анализ, ДТА-ТГА (см. гл. 7) или химический анализ (общий анализ, см. гл.
31), ЕКО (см. гл. 26), растворение алюмосиликатов и др.
Расчеты
Полученные данные:
• А, В - содержание в экстрактах из анализируемой и холостой пробы, соответствен¬
но, мг/л;
• D - коэффициент разбавления;
• /- поправка на влажность (см. «Расчеты» в разделе 6.2.1);
• Р- масса воздушно-сухой пробы, мг.
Расчет
Процентное содержание оксидов рассчитывают для всех элементов: Fe203, А1203, Si02
и др. Вычитая массу остатка из массы исходной пробы, получаем сумму всех свободных
оксидов и гидроксидов:
Al, Fe, Si...% = 25(А - B)Df/P.
О коэффициентах пересчета содержания см. «Расчеты» в разделе 6.2.1.
Примечания
Воспроизводимость результатов при этом методе вполне приемлема, если соединения
кристаллического железа находятся в дисперсном состоянии, достаточном для того,
чтобы получить необходимую величину поверхности частиц. Было предложено большое
количество различных методик, но в настоящем стандартном методе используются кон¬
центрации реагентов, первоначально предложенных в работе (Mehra и Jackson, 1959):
0,42 г бикарбоната натрия и 3,52 г двухводного цитрата натрия в 45 мл воды. Авторы
внесли следующие основные изменения в методику: двукратное добавление дитионита
к одному и тому же экстракту и приготовление единого буферного и комплексообразую¬
щего реагента, что облегчает выполнение анализа.
По цвету остатка осадка можно судить об эффективности обработки, хотя присут¬
ствие магнетита или ильменита, которые не разрушаются под действием ДЦБ, может
окрасить осадок в черный или серый цвет. Метод ДЦБ можно использовать для облегче¬
ния диспергирования глин, которое может быть затруднено поверхностными педоген-
ными оксидами и гидроксидами.
Иногда вместо метода ДЦБ используют метод Холмгрена (Holmgren, 1967), в котором
реагент состоит из нестабильной смеси 17% цитрата натрия и 1,7% дитионита натрия.
Он считается эквивалентным методу Меры-Джексона, но проще по исполнению и бо¬
лее подходящим для рутинных анализов.
Размол до размера частиц 0,2 мм приводит к лучшему разрушению соединений же¬
леза, находящихся, например, в конкрециях. Следовательно, измельчение обычно по¬
зволяет сравнивать массу остатков после обработки.
Нагревание до температуры выше 80 °С (или местный перегрев) может приводить
к осаждению черного сульфида железа. В этом случае лучше начать анализ сначала, чем
удалять осадок обработкой ацетоном и четыреххлористым углеродом.
179
Глава 6, Разделение минералов методом селективного растворения
Обработка дитионитом изменяет отношение Fe3+/Fe2\
Райан и Пивенд {Ryan и Gschwend, 1991) предложили заменить дитионит титаном (III)
в растворе цитрат-бикарбоната и этилендиаминтетрауксусной кислоты (ЭДТА). Этот
восстановительный метод с очень сильным комплексообразующим реагентом при 80 °С
теоретически позволяет достичь более полного растворения аморфных оксидов железа
и гетита (гематит растворяется в меньшей степени). Экстракция методом li (III) дости¬
гается легче, чем методом ДЦБ, что облегчает выбор методов растворения. Поведение
соединения алюминия отличается. Использование метода Т\ (III) на холоде увеличивает
степень селективности, но содержание титана в почве должно быть низким.
6.2.3. Метод, основанный на использовании
этилендиаминтетрауксусной кислоты (ЭДТА)
Основные положения
Согласно данным Борггаарда (Borggaard, 1976) метод с использованием ЭДТА (натрие¬
вой соли) позволяет проводить экстракцию железа в аморфном или очень мало упоря¬
доченном состоянии, в результате биологического разрушения (неорганического или
органического некристаллического железа) ферригидрит растворяется. Раствор должен
содержать водорастворимые и обменные формы железа.
Этот реагент не пригоден для экстракции аморфных и органических форм алюминия
из-за высоких значений pH экстрагирующего раствора. Силикаты и кристаллические
формы железа и алюминия не растворяются.
Экстракция железа характеризуется хорошей сходимостью и селективностью. Метод
надежен, но имеет один значительный недостаток: медленность экстракции, когда рав¬
новесие достигается лишь после трех месяцев экстракции. Наиболее широко использу¬
емыми процессами являются гидролиз и комплексообразование в щелочной среде при
комнатной температуре (20 °С).
ЭДТА — аминополикарбоновая кислота, содержащая шесть атомов, к которым ка¬
тион металла присоединяется при помощи координационных связей с органическими
радикалами (например, два атома азота и четыре карбоксильных группы (рис. 6.6), с об¬
разованием хелатов (от греческого Khele — клешня краба). Шесть позиций вокруг атома
металла (Fe2+) приводят к большой комплексообразующей способности и малой изби¬
рательности, поскольку большинство двух- и трехвалентных элементов способны об-
HOOC-CH2N^ ^сн-соо-
H-N+-CH,-CH,-N+-H
-оос-сн, хСН2СООН
Рис. 6.6. Молекула ЭДТА (IV) (вверху) и координационный комплекс с железом (внизу)
180
Часть 1. Минералогический анализ почв
разовывать этот тип комплексов. Константы устойчивости, log К при 20 °С, равны 13,8
для Мп2+, 14,3 для Fe2+, 16,1 для А13+ и 25,1 для Fe3+.
В щелочном растворе (pH 10) образуются комплексы типа 1:1 с почвообразующими
элементами. Для приемлемо управляемой скорости растворения необходим избыток
комплексообразующего агента.
В зависимости от pH можно получить формы H4Y, H3Y_, Н^ и HY3_(me Y4- - анион
ЭДТА, см. рис. 6.6).
В среде, где происходит экстракция, экстрагируемые ЭДТА формы железа, по-
видимому, являются наиболее активными формами в почвообразовании, т. е. «свобод¬
ными» наименее окристаллизованными соединениями с развитой поверхностью кон¬
такта и, следовательно, высокой реакционной способностью. Солюбилизируется как
обменное железо, так и его органические хелатные формы, потому что реагент имеет
высокий pH и комплексы с ЭДТА стабильны.
В агрономии доступность железа для растений (питательный фактор или возмож¬
ность хлороза) часто оценивают, используя два других комплексообразователя (Lindsay-
Norvell, 1976): диэтилентриаминопентауксусную кислоту (ДТПК, DTPA) или этилендиа-
минди (0-гидроксифенилуксусную) кислоту (ЭДДК, EDDHA).
Корреляция между ЭДТА- и оксалат-экстрагируемым железом довольно высока: фер-
ригидрит растворяется в ЭДТА или оксалате, но почвы, содержащие гидроферритные
комплексы, могут иначе реагировать с этими реагентами (Jeanroy, 1983). Существенное
значение имеет фактор времени. Экстракция происходит медленно и продолжается
вплоть до 90 сут, когда состояние стабилизируется.
Температура имеет существенное значение, и предпринимаются попытки ускорить
реакции, поднимая температуру до 75 °С. В методе ДЦБ это приводило к недопустимым
изменениям селективности: некоторые кристаллические продукты разрушались или
солюбилизировались.
Влияние pH исследовалось Борггаардом (Borggaard, 1976). Его значение не может
быть ниже 7,5. При pH 10, который также используют в пирофосфатном и тетраборат-
ном методах, можно сравнивать органические фазы, экстрагируемые этими методами,
и исключить влияние pH. Однако для алюминия, который дает растворимые алюмина¬
ты при pH 8—9, селективность неопределенна.
В ЭДТА концентрационный фактор меньше влияет на процесс экстракции, так как
кинетику реакции контролирует гидролиз. Периодическое перемешивание позволяет
обновлять реагент на границе раздела жидкой и твердой фаз и избегать местного перена¬
сыщения. Осветление растворов обычно не вызывает затруднений.
Приготовление реагентов
Исходная методика Борггаарда (Borggaard, 1976) относится к динамическим методам.
Автор исследовал влияние концентрации комплексообразователя в диапазоне от 0,01 до
0,1 М и pH в диапазоне от 7,5 до 10,5. Этот метод чрезвычайно медленен и в его ориги¬
нальной форме не может быть адаптирован к рутинным анализам.
В качестве стандарта используют 0,1 М раствор ЭДТА при pH 10. Это позволяет про¬
водить сравнение с реагентами, экстрагирующими органоминеральные комплексы при
том же pH, т. е. пирофосфатом при pH 9,6-10 и тетраборатом при pH 9,7. ЭДТА часто
продается в виде натриевой соли под различными названиями: Версенат, Секвестрен,
Титриплекс II, Трилон Б и др. Эмпирическая формула C10H16N2O8 соответствует молеку¬
лярной массе 292,25 г.
Раствор А: растворяют примерно 29,225 г ЭДТА в 500 мл воды.
181
Глава 6. Разделение минералов методом селективного растворения
Раствор Б: растворяют 20 г NaOH в 250 мл воды.
Постепенно добавляют раствор Б в раствор А, чтобы довести pH до 10.
Доливают до 1 л дистиллированной водой.
Методика
♦ Отвешивают на аналитических весах точно 2 г почвы, измельченной до размера
частиц 0,2 мм в 100-миллилитровой центрифужной пробирке из полипропилена
или ПТФЭ.
• Добавляют 50 мл 0,1 М раствора ЭДТА.
* Закрывают пробирку и встряхивают на вибраторе Vortex в течение 1 мин.
♦ Помещают пробы на подвижную платформу шейкера и хранят в защищенном от
света месте в течение 90 сут, ежедневно встряхивая в течение 5 мин.
• Через 90 сут центрифугируют при 5000 g в течение 5 мин и фильтруют.
Экстрагированные элементы (главным образом, Fe, Al, Si и Р) анализируют методом
эмиссионной спектрометрии с индуктивно-связанной плазмой или атомно-абсорбци¬
онной спектрометрии. В последнем случае матрица ЭДТА должна быть разрушена (см.
параграф «Методика» в разделе 6.2.1). В каждую серию проб включают две холостые
пробы (только реагенты) и стандартный образец.
Расчеты
Полученные данные:
А, В, D,f, Р— то же, что и в предыдущих методах (см. параграфы «Расчеты» в разделах
6.2.1 и 6.2.2).
Содержание Fe, Al, Mn, Si, Р (%) = 5(А - B)Df/P.
Содержание элементов выражают в процентах оксидов (см. «Расчеты» в разделе
6.2.1).
Примечания
Этот метод является одним из наиболее воспроизводимых и селективных для определе¬
ния аморфного железа; металлоорганические комплексы также могут экстрагироваться,
если pH превышает 9. Содержание экстрагированного железа следует сравнить с содер¬
жанием железа, экстрагированного оксалатным реагентом, с учетом того, что глинозе¬
мистые соединения, растворенные при высоком pH, могут выделять железо, например
вследствие изоморфных замещений.
Полезно сравнивать результаты с экстракцией активного железа, доступного для рас¬
тений (с использованием ДТПК или ЭДДК), чтобы связать результаты с явлением хло¬
роза и перейти от педогенных наблюдений к идентификации агрономических свойств
системы почва - растение - климат. С агрономической точки зрения, экстракция фос¬
фора раствором ЭДТА также помогает идентифицировать долю Р, связанного с Fe или А1
и входящего в состав «доступного Р».
6.2.4. Пирофосфатный метод
Основные положения
Этот метод используют для определения форм железа и алюминия, образующих ком¬
плексы с органическим веществом почвы, в частности для дифференцирования сподо-
вых и подзолистых горизонтов, где может наблюдаться замещение этих комплексов.
182
Часть 1. Минералогический анализ почв
Обычно хорошая корреляция наблюдается между экстрагированными Al, Fe и орга¬
ническим С. Хорошо окристаллизованные оксиды железа, такие как гетит и гематит,
не разрушаются, а слабо упорядоченные оксиды железа только немного солюбилизиру¬
ются.
Исходную методику, рекомендованную Александровой (1960) и доработанную Мак-
кигом (McKeague, 1967), нельзя применять без соответствующей модификации для
осветления экстрактов.
Пирофосфат анион — Р20*~ обладает хелатирующими свойствами и может реагиро¬
вать с поливалентными катионами с образованием нерастворимых соединений и рас¬
творимых комплексов с органическим веществом, например,
R(COO)4Ca2 + Na4P207 -► R(COONa)4 растворимый + Ca2P207!
Однако полный механизм действия пирофосфата не столь ясен, как для предыдущих
методов. Действие пирофосфата на органические соединения железа и алюминия было
поставлено под сомнение: как методики и достоверность результатов, так и механизмы
экстракции и природа экстрагируемых продуктов. Кламт (Klamt, 1985) упоминал «отказ
от экстракции Fe из почв пирофосфатом (крайне недостоверна)», указав, что междуна¬
родный консенсус по этому вопросу не достигнут.
О методиках
Фактор времени обработки, длящейся 16 ч, считается критическим, особенно при срав¬
нении результатов метода с результатами экстракции ЭДТА в течение 90 сут.
Были исследованы различные уровни pH и выбрано значение pH 10 для сравнения
с другими методами экстракции в щелочных растворах.
Пирофосфат обладает хелатирующими свойствами. Первоначально выбор между
пирофосфатом калия или натрия был более или менее случайным. Пирофосфат калия
экстрагирует несколько ббльшие количества, чем пирофосфат натрия, и облегчает про¬
ведение спектрометрических измерений благодаря отсутствию высоких концентраций
ионов Na\ которые всегда мешают измерениям. Однако для некоторых глин использо¬
вание соли калия имеет серьезные недостатки1. Поэтому считается, что пирофосфат Na
дает более воспроизводимые результаты экстракции, и в настоящее время используется
в концентрации 0,1 М (Loveland и Digby, 1984).
При pH 10 пирофосфат обладает пептизирующими свойствами, которые чрезвычай¬
но затрудняют очистку экстрактов. Присутствие взвешенных частиц объясняет низкую
воспроизводимость и недостаток точности метода, так как суспендированный матери¬
ал не может быть химически экстрагирован пирофосфатом. Эффективность (скорость
и продолжительность) центрифугирования была исследована при 100 000 g и сравнена
с параметрами ультрафильтрации. Перед центрифугированием должен быть добав¬
лен флокулирующий агент, обычно сульфат натрия в концентрации между 0,25 и 1 М
(Schuppli и др., 1983) или Суперфлок Цианамид N-100 (либо Floerger Kemflock F20H).
В этом случае решающее значение имеет концентрация (Ballantyne и др., 1980). Она
1 Например, ион К+ специфически адсорбируется вермикулитом или искажает структуру
слюды, потому что его диаметр сравним с размером адсорбционных центров. Селективность
ионов К+ по отношению к ионам Са2+ или Mg2+ увеличивается полимерными отложениями
гидроксидов алюминия на межслоевых поверхностях. В присутствии больших концентра¬
ций Р и при определенных условиях совместное присутствие К и оксидов алюминия может
приводить к образованию таранакита.
183
Глава 6. Разделение минералов методом селективного растворения
определена на уровне 0,2 мл суперфлока на 50 мл экстракта. Центрифугирование при
20 000 g в течение 15 мин дает прозрачные экстракты. Ультрафильтрацию с фильтрами
0,02-мкм можно использовать для удаления всех коллоидных частиц, которые могут
присутствовать в суспензии. В этом случае воспроизводимость составляет 10-15%.
О механизмах солюбилизации
В реакции между пирофосфатом натрия и почвой комплексообразование не может быть
основным механизмом, потому что железо, связанное в формах органических комплек¬
сов, не может раствориться без солюбилизации аморфных форм железа, которые об¬
ладают высокой реакционной способностью, как в случае солюбилизации ЭДТА. По
сравнению с аморфными оксидами железа комплекс пирофосфата железа считается не
очень устойчивым при pH 10.
Брукерт (Bruckert, 1979) считал, что пирофосфат натрия вытесняет органическое
вещество из комплексов с металлическими центрами глин (мостики из ионов железа)
и адсорбируется вместо гуминовых веществ, которые солюбилизируются в щелочной
среде. Соединения металлов с высокими зарядами, такие как аморфные гидроксиды,
могут вести себя аналогичным образом. Все комплексы, экстрагируемые пирофосфа¬
том, входят в состав «неподвижных комплексов».
Микроагрегаты разрушаются, а глинистые и коллоидные связывающие вещества
диспергируются. Комплексы фульвокислот с аморфными гидроксидами железа экстра¬
гируются наряду с органическими молекулами пленок на глинах.
Жанрой (,Jeanroy, 1981,1983) считал, что растворение запускает механизм пептизации
и солюбилизацию. Адсорбция пирофосфата на частицах почвы увеличивает отрицатель¬
ные заряды и повышает их растворимость в воде. В отличие от ЭДТА, которая оказывает
флокулирующее действие, пирофосфат переводит железосодержащие частицы во взве¬
шенное состояние и таким образом рассеивает их в экстракте.
Разделение на миллипоровых фильтрах показывает, что в ЭДТА экстрактах железо
соединяется с малыми молекулами и таким образом проходит через мембрану С дру¬
гой стороны, пирофосфатные экстракты содержат соединения большего молекулярного
объема, которые не проходят через мембрану, так как лишь небольшая часть хелатной
фракции способна на это. Аналогичным образом, ультрацентрифугирование ЭДТА экс¬
тракта не разделяет фазы, показывая, что железо находится в растворимой хелатной
форме, тогда как использование пирофосфата приводит к образованию центрифужного
осадка значительной доли коллоида.
Обобщая, можно сказать, что пептизирующее влияние пирофосфата суспендирует
мелкие железосодержащие частицы, вероятно гидроксидов железа, чьи неоднородность
и низкая степень атомного порядка объясняются присутствием органического вещества,
подавляющего кристаллизацию оксидов железа. «Неподвижные комплексы» Брукерта,
по-видимому, являются в основном гидроксокомплексами железа, указывающими на
преобладающий минерал.
С практической точки зрения пирофосфат эффективен, только если почва содержит
органическое вещество. По мнению Шуппли с сотр. (Schuppli и др., 1983), осаждение
железа в пирофосфатном экстракте может быть связано со старением экстрактов. Дру¬
гим механизмом растворения может быть высвобождение пирофосфатом натрия малых
количеств железа из органических комплексов, так что комплексы приобретают отри¬
цательный заряд и становятся растворимыми в воде. Органическое вещество позволяет
поддерживать большое количество железа в растворе, но при достижении определенно¬
го соотношения происходит осаждение (Petersen, 1976).
184
Часть 1. Минералогический анализ почв
Если данное описание механизма солюбилизации достаточно точное, экстракция
пирофосфатом может использоваться в качестве метода селективного растворения, осо¬
бенно если преодолеть неопределенности, связанные с очисткой экстрактов. С прак¬
тической точки зрения экстракция пирофосфатом позволяет оценивать поведение не¬
которых почв для целей классификации, хотя и без определения точного источника
экстрагируемого железа (однако наиболее вероятными являются органические формы).
Приготовление реагентов
Экстракция
Пирофосфат натрия (С = 0,1 М): растворяют 44,6 г Na4P207- ЮН20 в дистиллированной
воде и доводят объем раствора до 1 л; проверяют, чтобы pH был равен 10,0.
Осветление
Суперфлок Цианамид N-100 (Cyanamid Согр.> Госпорт, Хэмпшир, Великобритания):
растворяют 0,2 г суперфлока в 100 мл воды и перемешивают ПТФЭ якорем магнитной
мешалки в темноте в течение 16 ч, защищая от света в темной бутыли; следует готовить
свежий раствор еженедельно.
Методика
На точных лабораторных весах отвешивают 1 г почвы (размер частиц 0,2 мм) в поли¬
этиленовую пробирку на 250 мл (с завинчивающейся пробкой). Добавляют 100 мл 0,1 М
раствора пирофосфата натрия и перемешивают в течение 16 ч при комнатной темпера¬
туре (20 °С).
Добавляют 0,2 мл суперфлока и гомогенизируют на роторном шейкере в течение
10 мин, затем центрифугируют при 20 000 g. С помощью шприца с миллипоровым филь¬
тром отбирают порции надосадочной жидкости, необходимые для анализа требуемых
элементоэ (главным образом Fe, Al, Si и С). Добавляют две холостых пробы (содержа¬
щие только реагенты) и два стандартных образца в каждую серию.
Измеряют концентрации Al, Fe и Si методом эмиссионной спектрометрии с индук¬
тивно-связанной плазмой или атомно-абсорбционной спектрометрии (см. главу 31),
используя стандарты, разведенные в экстрагирующей матрице. При измерениях мето¬
дом абсорбционной спектроколориметрии предварительно разрушают пирофосфатную
матрицу (см. «Методика» в разделе 6.2.1).
Расчеты
Полученные данные:
А, В, D, Pff — то же, что и в предыдущих методах (см. параграфы «Расчеты» в разделах
6.2.1 и 6.2.2).
Содержание Fe, Al, Si (%) = 10(Л - B)Df/P.
Содержание элементов выражают в процентах оксидов (см. «Расчеты» в разделе
6.2.1).
Примечания
Этот метод хорошо подходит для дифференциации подзолистых горизонтов В. Условия
центрифугирования и осветления должны быть максимально одинаковыми, скорость
Глава 6. Разделение минералов методом селективного растворения
185
и время центрифугирования должны строго выдерживаться. Если серия проб должна
храниться в течение некоторого времени перед проведением спектрометрических из¬
мерений, ее хранят в темноте в холодильнике при 6-8 °С.
Процентное содержание экстрагированного органического углерода обычно изме¬
ряют на автоматизированных CHN-анализаторах (см. гл. 10) одновременно со спек¬
трометрическим измерением содержания Fe, А1 и Si (см. гл. 31). Часто используют
серии экстрагентов с постепенно увеличивающимися значениями pH. Такая диффе¬
ренциальная экстракция позволяет характеризовать формы с возрастающей устойчи¬
востью:
• Предварительная экстракция буферным 0,1 М раствором тетрабората натрия с pH
9,71 разрушает электростатические связи простой реакцией обмена. Экстрагиру¬
ются адсорбированные органические молекулы малой молекулярной массы. Они
содержат комплексы железа и алюминия, включая недавно десолюбилизирован-
ные «подвижные комплексы». Почвенные агрегаты не разрушаются (Bruckert, 1970,
1974,1979).
• Метод с использованием 0,1 М раствора пирофосфата с pH 10 позволяет разрушать
координационные связи с гидроксидами и оксидами, покрывающими глинистые
частицы.
• Наконец, метод с использованием 0,1 М раствора гидроксида натрия при pH > 12
(см. раздел 6.2.5) разрушает органо-минеральные связи, даже в комплексах алло¬
фана с гуминовыми кислотами.
6.2.5. Экстракция в сильнощелочной среде
Основные положения
Методы с использованием карбоната натрия основаны на:
• растворении некоторых соединений кремния, алюминия и алюмосиликатов
в сильнощелочном растворе; при этом образуются растворимые силикаты или
алюминаты в соответствии со следующим упрощенным уравнением:
А14- NaOH + Н20 -> NaA102 4- 3/2 Н2;
• концентрировании в остатке нерастворимых соединений, особенно железа и мар¬
ганца.
Обработка кипящим 0,5 М раствором гидроксида натрия в течение 2,5 ч солюбили¬
зирует органические формы алюминия и кремния, гидратированные некристалличе¬
ские и кристаллические (гиббсит) оксиды алюминия, опаловый кремнезем и диатомеи,
и наконец, аморфные или скрытокристаллические алюмосиликаты, такие как аллофан
и имоголит (соотношение Si02 /А1203 = 1,5-2,3). Некоторые силикаты типа 1:1 разруша¬
ются и частично растворяются. Соединения железа не экстрагируются.
1 Приготовление реактива: 0,1 н. тетраборат натрия с pH = 9,7: растворяют 21 г Na2B407- ЮН20
в примерно 900 мл воды, добавляют 1,8 г гранулированного пероксида натрия, гомогенизи¬
руют, проверяют, чтобы pH был равен 9,7, и доводят объем до 1 л добавлением деионизиро¬
ванной воды.
Экстракция, смесь 1 г почвы, измельченной до размера частиц 0,2 мм, + 100 мл 0,1 н. раство¬
ра тетрабората натрия перемешивают в течение 1 ч, центрифугируют при 20 000 g и продол¬
жают анализ аналогично описанному для пирофосфатных экстрактов (Al, Si, Fe', С и др.).
186
Часть 1. Минералогический анализ почв
Обработка 0,5 М раствором карбоната натрия при 20 °С в течение 16 ч (Follet и др.,
1965) обеспечивает более мягкие условия и позволяет солюбилизировать органические
и некристаллические соединения алюминия, а также определенную долю гиббсита. Вы¬
сокодисперсные кремнийсодержащие соединения и опаловый кремнезем могут частич¬
но растворяться, аморфные алюмосиликаты солюбилизируются, но полной солюбили¬
зации аллофана и имоголита не происходит. Филлосиликаты и соединения железа не
подвергаются разрушению.
В полученном остатке можно исследовать соединения железа, незатронутые преды¬
дущими реагентами. Более жесткий метод с использованием 5 М раствора NaOH и ки¬
пячением в течение 2 ч позволяет растворить большинство присутствующих глин типа
1:1 и глинистых минералов и достичь большей концентрации. Однако даже при исполь¬
зовании данного метода некоторые минералы, такие как кварц, анатаз, рутил, кристоба-
лит и некоторые глины типа 2:1, могут присутствовать в остатке.
Воздействие реагентов главным образом зависит от размера частиц, степени разде¬
ления кремний- и алюминийсодержащих веществ, степени кристалличности и реакци¬
онной способности поверхностей упорядоченных или рентгеноаморфных соединений.
Экстракция 0,5 М раствором NaOH с непродолжительным кипячением позволя¬
ет проводить дифференциальную солюбилизацию соединений алюминия и кремния,
а также органических веществ с высокоактивной поверхностью, тогда как хорошо ©кри¬
сталлизованные соединения сохраняются, поскольку их солюбилизация требует гораздо
более длительного кипячения.
Растворимость гидроксидов алюминия увеличивается под действием первоначаль¬
ного гидролиза мономерных форм алюминия (при малых концентрациях): А10Н2+,
А1(ОН)2, А1(ОН)3, А1(ОН)~, последняя из которых существует только в щелочной среде
в соответствии с реакцией:
А13+ + 4Н20 -► А1(ОН)4- + 4Н+.
Алюминий имеет тетраэдрическую1 координацию. При высоких концентрациях по¬
лимерные формы постепенно приобретают форму структур [А16(ОН)12- (Н20)12]6\
Кроме того, все кислые группы органических макромолекул (гуминовые и фульво-
кислоты) диссоциируют, а полярные и анионные центры легко сольватируются. В таких
условиях разрывается большинство органоминеральных связей, даже очень прочные
связи между аллофановым алюминием и гуминовыми кислотами андосолей. Плохо упо¬
рядоченные соединения алюминия и кремния или их органические и неорганические
производные переходят в раствор в виде алюминатов и силикатов.
Растворение в растворе гидроксида натрия в окислительных условиях может приво¬
дить к разрушению некоторых форм гуминовых кислот в присутствии кислорода, кото¬
рый изменяет отношение Fe2+/ Fe3+ в остатке. Проведение обработки в атмосфере азота
может уменьшить интенсивность этого процесса, а также минимизировать карбониза¬
цию гидроксида натрия атмосферным С02.
Наконец, кроме вышеуказанных процессов, следует учитывать сильное диспергирую¬
щее воздействие гидроксида натрия на филлосиликаты. Элементарные компоненты ста¬
бильных микроагрегатов, удерживаемых на месте пленками оксидов Fe, А1 и Si, сначала
освобождаются в результате процесса растворения и затем диспергируются под усили¬
вающимся действием реагента на поверхности раздела между твердой и жидкой фазами.
Щелочной характер поверхности оксидов снижается в следующем ряду:
*В растворах — как правило, октаэдрическую А1(ОН)4 • (Н20)2. — Примеч. науч. ред.
Глава 6. Разделение минералов методом селективного растворения
187
Амфотерные >у-А10(0Н) >а-А1(ОН)3 >у-А1(ОН)3 >у-А1203
гидратированные бёмит байерит гиббсит
оксиды А1
pH в изоэлек- 9,45 9,40 9,20 8,00
трической
точке
Кроме гидроксида и карбоната натрия и пирофосфатов натрия и калия, для экстрак¬
ции органических веществ и органоминеральных комплексов используются следующие
реактивы: этилендиамин (ЭДА), N,N-диметилформамид, сульфолан, пиридин, диме-
тилсульфоксид идр.
Реагенты
Здесь описаны три наиболее широко применяющихся реагента, так как результаты их
использования хорошо коррелируют с результатами других методов селективной экс¬
тракции и удовлетворительно характеризуют устойчивость к растворению алюминий-
и кремнийсодержащих продуктов. Были также предложены многочисленные альтер¬
нативные реагенты, относительная эффективность (но не селективность) которых
приблизительно классифицирована в табл. 6.3. Иногда вместо гидроксида натрия ис¬
пользуют растворы гидроксида калия.
Таблица 6.3. Сильнощелочные реагенты в порядке снижения эффективности
NaOH,
NaOH,
NaOH,
Na2C03,
Na2C03,
NaOH,
5 М,
> 0,5 М,
> 1,25 М
>0,5 М,
> 0,5 М,
>0,1М
pH £14
рН> 12
pH 10,7
pH 10,7
кипячение
кипячение
80 °С
кипячение
20 °С
20 °С
2ч
2,5 мин
< 20 мин
1 ч (или
16 ч
переменное
время)
обогащение
селективная
удаляет
удаляет вещества,
селективная
дисперги¬
железом
экстракция
гиббсит
связывающие
экстракция
рование
свободных
и свободные частицы,
аллофана,
глин
оксидов,
оксиды
солюбилизирует
имоголита
аллофана,
минералы типа 1:1
имоголита
Norrish
Hashimoto и
Folletvmp.
и Taylor (1961)
Jackson (1960)
0965)
Приготовление
• 5 М раствор NaOH: осторожно растворяют 200 г гранулированного гидроксида на¬
трия (чда) в 1 л дистиллированной воды, предварительно прокипяченной для уда¬
ления С02. Оставляют охлаждаться без доступа воздуха и хранят в полиэтиленовой
бутыли. Свежую порцию готовят еженедельно.
• 0,5 М раствор NaOH: растворяют 20 г гранулированного гидроксида натрия в 1 л
предварительно прокипяченной дистиллированной воды. Хранят в полиэтилено¬
вой бутыли. Этот реактив должен готовиться ежедневно.
♦ 0,5 М раствор Na2C03: растворяют 53 г безводного Na2C03 в 1 л дистиллированной
воды и хранят в полиэтиленовой бутыли.
♦ 0,5 М раствор НС1: берут 42 мл НС1 (d = 1,19) и доводят объем водой до 1 л.
188
Часть 1. Минералогический анализ почв
Методики
Селективное растворение в 0,5 Мрастворе Ш2СОг при 20° С (Follet идр., 1965)
• Взвешивают 100 г почвы, измельченной до 0,2 мм, и помещают в центрифужную
пробирку на 100 мл.
• Добавляют 80 мл 0,5 М раствора Na2C03; закрывают пробирку и встряхивают на
роторном шейкере в течение 16 ч.
• Центрифугируют при 5000 g в течение 10 мин.
• Переносят надосадочную жидкость в мерную колбу на 200 мл.
• Промывают осадок после центрифугирования дистиллированной водой и повтор¬
но центрифугируют.
• Добавляют центрифугат к предыдущему экстракту и доводят объем до 200 мл деио¬
низированной водой.
• Без промедления выполняют спектрометрические измерения экстракта методом
атомно-абсорбционной спектрометрии или эмиссионной спектрометрии с индук¬
тивно-связанной плазмой (Si и А1).
Селективное растворение кипящим 0,5 Мраствором NaOH (Hashimoto и Jackson, 1960)
• Взвешивают 100 мг почвы, измельченной до 0,2 мм (или используют осадок после
экстракции раствором ДЦБ) в никелевом или ПТФЭ тигле на 250 мл.
• Добавляют 100 мл кипящего 0,5 М раствора NaOH и гомогенизируют.
• Продолжают кипячение в течение 2,5 мин.
• Быстро охлаждают и переносят в полиэтиленовую центрифужную пробирку на
250 мл и центрифугируют при 5000 g в течение 10 мин.
• Переносят надосадочную жидкость в мерную колбу на 500 мл.
• Промывают осадок после центрифугирования дистиллированной водой, повторно
центрифугируют и добавляют центрифугат в мерную колбу.
• Доливают дистиллированной водой до метки и незамедлительно измеряют содер¬
жание Si и А1 методом атомно-абсорбционной спектрометрии или эмиссионной
спектрометрии с индуктивно связанной плазмой.
Растворение кипящим 5 Мраствором NaOH (2 ч) (Norris И и Taylor, 1961)
• Взвешивают 100 мг почвы, измельченной до 0,2 мм (или используют осадок после
селективной экстракции) в стаканчике из нержавеющей стали или ПТФЭ.
• Добавляют 100 мл 5 М раствора NaOH.
• Кипятят в течение 2 ч.
• Охлаждают и центрифугируют при 5000 gв течение 10 мин.
• Сливают надосадочную жидкость (которую отбрасывают или анализируют).
• Промывают осадок после центрифугирования небольшим количеством воды, за¬
тем трижды 0,5 М раствором НС1 для растворения образовавшегося содалита и уда¬
ления хлорида натрия, затем снова промывают водой до отрицательной реакции
на хлорид-ионы.
• Высушивают пробу для анализа на оксиды марганца и железа. Проверяют отсут¬
ствие каолинита и содалита методом рентгеновской дифракции.
189
Глава 6. Разделение минералов методом селективного растворения
Расчеты
Все результаты выражают в содержании оксидов (см. «Расчеты» в разделе 6.2.1).
Примечания
Отношение NaOH:Al влияет на образование гидроксидов алюминия (Hsu, 1977). По¬
этому важно стандартизовать методику на стадии осаждения А1. Исследования методом
27А1ЯМР показали, что для данной концентрации и pH природа геля изменяется со вре¬
менем (Hsu, 1984). Такие изменения происходят как в природных условиях, так и в син¬
тетических средах (хотя и в другом масштабе) (Stol и др., 1976). ЯМР-спектрометрия
является мощным инструментом, позволяющим различать взвешенные и растворенные
полиядерные соединения. Иногда наблюдаемые изменения могут быть связаны с вариа¬
циями методов разделения (центрифугирование, ультрафильтрация или диализ). Поэто¬
му предпочтительно выполнять спектрометрические измерения на продуктах, недавно
экстрагированных различными методами.
При использовании 5 М раствора NaOH Кампф и Швертманн (Kampf и Schmrtmann,
1982) рекомендуют проводить экстракцию кипящим раствором и в присутствии гиббси-
та модифицировать реагент, доводя содержание кремнезема до 0,2 М для того, чтобы
минимизировать фазовые изменения и возможное увеличение кристалличности, а так¬
же подавить растворение и перекристаллизацию замещенного алюминия в гетитах.
Ферригидрит, который может превращаться в гематит и/или гетит, остается почти не¬
измененным. Однако увеличение содержания кремнезема приводит к большему осаж¬
дению силиката натрия и алюминия (содалита), который должен быть удален из остатка
неоднократным промыванием 0,5 М раствором НС1.
Использование 5 М раствора NaOH позволяет более четко идентифицировать ок¬
сиды марганца в осадке. Эти оксиды обычно присутствуют в малых концентрациях
и проявляют малую окристаллизованность в почвах. Бернессит и литиофорит встре¬
чаются с оксидами железа. Бернессит может быть растворен гидроксиламинхлоридом,
тогда как гетит и литиофорит не изменяются под действием этого реагента. Заключи¬
тельная обработка раствором ДЦБ позволяет растворить гетит (Shuman, 1982; Tokashiki
идр., 1986).
В методе с кипячением с карбонатом натрия в течение 2,5 ч (см. «Селективное рас¬
творение кипящим 0,5 М раствором NaOH») время обработки играет существенную
роль и должно тщательно выдерживаться для того, чтобы избежать излишней солюби¬
лизации каолинита и галлуазита, которые постепенно переходят в раствор. При этом
хлориты и монтмориллониты, которые предварительно нагревались до 500 °С, почти не
затрагиваются. Спектрометрические измерения должны проводиться вскоре после экс¬
тракции, чтобы избежать старения экстрактов и осаждения алюмосиликата.
Чтобы не допустить насыщения экстрактов алюминием и кремнием, следует ис¬
пользовать большие объемы экстрагента по сравнению с массой пробы почвы. Чтобы
избежать загрязнения, рекомендуется использование лабораторного оборудования из
ПТФЭ, нержавеющей стали или никеля. Пирексовое стекло может вызывать загрязне¬
ние такими элементами, как Si, А1 и Fe.
Растворение гиббсита может быть скорректировано на основании данных ДТА.
Обработка 0,5 М кипящим раствором NaOH не дает возможности проводить очень
точную дифференциацию аморфных и кристаллических продуктов из-за неустойчиво¬
сти плохо упорядоченных глин типа 1:1 и гиббсита. Однако обычно наблюдается хоро¬
шая корреляция с оксалатным методом.
190
Часть 1. Минералогический анализ почв
6.3. Другие методы исследований, модификации и выбор
подходящего метода
6.3.1. Дифференцирующие последовательные методы
Основные положения
Разработано большое количество методов исследований. При последовательном ис¬
пользовании методов, описанных выше в разделе 6.2, их можно рассматривать как по¬
следовательные мультиреагентные методы.
Метод Сегалена (Segalen, 1968) использует чередование процессов гидролиза и про¬
тонирования в холодной кислой среде (без комплексообразования или восстановления)
для солюбилизации некоторых соединений железа и алюминия с последующей обра¬
боткой в горячей щелочной среде (при 80 °С) для солюбилизации соединений алюми¬
ния и кремния (см. раздел 6.2.5). Эти обработки чередуются несколько раз для постро¬
ения кумулятивных кривых накопления экстрагированных соединений со временем
(рис. 6.7).
Фракционирование отражает различия в растворимости соединений в этих средах1.
Кинетика солюбилизации и константы гидролиза зависят от: (1) типа, природы и кон¬
центрации реагентов, отношения почва/раствор и температуры и (2) природы и размера
пробы, элементного состава частиц, степени кристаллизации и величины удельной по¬
верхности глинистых минералов, молекулярной организации и степени замещения.
1 Например, кинетика растворения различных форм железа в 0,5 М НС1 при 25 °С (Shidhi
и др., 1981) соответствует ряду: ферригидрит, фероксигит > лепидокрокит > магнетит > акага-
неит > маггемит > гематит > гетит.
191
Глава 6. Разделение минералов методом селективного растворения
Кумулятивная кривая экстрагированных соединений объединяет эти параметры: (1)
начальный участок кривой представляет собой фрагмент параболы, которая возрастает
в большей или меньшей степени в зависимости от типа почвы и соответствует, главным
образом, быстрому растворению некристаллических веществ с очень большой удельной
поверхностью (рис. 6.7); кинетика растворения показывает, что часть мелкокристалли¬
ческих веществ также может переходить в раствор в это же время; (2) верхняя прямоли¬
нейная часть графика имеет небольшой наклон, указывающий на слабое растворение
хорошо окристаллизованных и химически малоактивных веществ.
Постепенное изменение наклона кривой может указывать на окончание растворения
мелкокристаллических частиц или, наоборот, появление практически неупорядоченных
зон предпочтительного растворения. Экстраполяция верхнего сегмента до пересечения
с осью ординат дает оценку процентного содержания аморфных веществ.
Реагенты
Сегален (Segalen, 1968) рекомендовал использовать растворы хлористоводородной кис¬
лоты (с концентрациями 2,4 и 8 М) для кислотной обработки (чаще применяют послед¬
нюю концентрацию) и кипячение с 0,5 М раствором NaOH для щелочной обработки.
Последовательная экстракция состоит из 8 чередующихся кислотных и щелочных об¬
работок:
• 8 М НС1: в мерную колбу на 1 л помещают 100 мл дистиллированной воды, до¬
бавляют 830 мл химически чистой хлористоводородной кислоты, перемешивают,
оставляют охладиться и доводят объем до 1 л; для приготовления растворов НС1,
содержащих 6, 4, 2 или 0,5 М, в колбу соответственно добавляют 623, 415, 208
и 52 мл хлористоводородной кислоты.
• 0,5 М NaOH: помещают 20 г гранулированного NaOH в мерную колбу на 1 л, рас¬
творяют в дистиллированной воде и доводят объем до метки.
Предложенные модификации метода касаются температуры (от комнатной до кипе¬
ния), продолжительности контакта и, наконец, использования только одного реагента
(2 М НС1) без чередования с NaOH.
Методика
(Segalen, 1968):
• Отвешивают 500 мг почвы (измельченной до 0,2 мм) в полиэтиленовую центри¬
фужную пробирку на 100 мл.
• Добавляют 50 мл 8 М раствора НС1, гомогенизируют и оставляют в контакте на
30 мин при комнатной температуре.
• Центрифугируют при 5000 g в течение 10 мин.
• Переносят надосадочную жидкость в мерную колбу на 100 мл.
• Добавляют 45 мл деионизированной воды и суспендируют осадок после центрифу¬
гирования в смесителе Vortex.
• Центрифугируют при 5000 g и добавляют промывную воду в мерную колбу.
• Доводят объем до 100 мл; это экстракт А (кислый) для определения железа, алю¬
миния и кремния.
• Помещают 50 мл 0,5 М раствора NaOH в центрифужную пробирку и суспендируют.
• Помещают на кипящую водяную баню на 5 мин.
• Центрифугируют при 5000 g в течение 10 мин.
192
Часть 1. Минералогический анализ почв
• Собирают надосадочную жидкость в мерную колбу на 100 мл.
* Промывают осадок 45 мл дистиллированной воды и добавляют промывную воду
к экстракту; доводят объем до 100 мл; это экстракт В (щелочной) для определения
алюминия и кремния.
Повторяют двойную экстракцию 8 раз.
Расчеты
Все результаты, полученные методом эмиссионной спектрометрии с индуктивно-свя¬
занной плазмой или атомно-абсорбционной спектрометрии, выражают в процентах ок¬
сидов (см. «Расчеты» в разделе 6.2.1).
Для алюминия и кремния все результаты, полученные для экстрактов А и В, должны
быть сложены. Для железа используют только кислый раствор. Строят кумулятивные
кривые для железа, алюминия и кремния (рис. 6.7); из касательной к правой части кри¬
вой определяют процентное содержание аморфных соединений.
Примечания
Условия реакции, описанные в методике с использованием вышеуказанных концентра¬
ций (чередование 8 М НС1 и 0,5 М НС1 с кипячением в течение 5 мин), оказались слиш¬
ком жесткими и недостаточно селективными для многих почвенных образцов и многих
минералов, поскольку содержание аморфных веществ часто завышается. Таким об¬
разом, этот метод должен быть адаптирован к типу анализируемой почвы. Исходный
^модифицированный метод растворяет аморфное железо и железо из комплексов, но
если использование агрессивных реагентов приводит к быстрому растворению аморф¬
ных веществ, он также солюбилизирует кристаллические продукты: глины типа 1:1,
слюду, хлорит, биотит, роговую обманку, нонтронит, гиббсит и др. (Colmet-Daage и др.,
1973; Quantin и Lamouroux, 1974; Quantin и 1975; Коя# идр., 1979; Bentley идр., 1978,
1980; Quigley идр., 1980,1985; Torrance vi др., 1986).
Степень селективности может оказаться недостаточной для некоторых почв, а со¬
держание аморфных веществ будет значительно завышено. Сами по себе кинетические
кривые не показывают, какие именно формы солюбилизированы.
Кинетика растворения тесно коррелирует с концентрацией реагентов, временем
и температурой. Изменение этих параметров может существенно изменить кинетику
растворения и селективность. Результаты всегда зависят от точного соблюдения мето¬
дики. Как и во всех других методах, необходим избыток реагента, чтобы избежать на¬
сыщения. Этот метод плохо подходит для рутинных измерений без роботизации из-за
большой длительности последовательных экстракций. На малой серии проб ежедневно
можно проводить шесть двойных экстракций.
Исследование растворения гетита в 0,5 М НС1 при 20 и 60 °С с использованием
просвечивающей электронной микроскопии (ПЭМ) (Cornell и др., 1974, 1975, 1976;
Schmrtmann и др., 1985) показало, что в зонах предпочтительного растворения создают¬
ся новые поверхности, что может привести к образованию о-образных кривых раство¬
рения. Этот механизм растворения связан со структурой молекулярных конфигураций.
Кинетика растворения гетита замедляется при значительном замещении ионами А13\
Эти исследования выдвигают на первый план изменения в представлениях о раство¬
рении и «аморфных» веществах, а также необходимость проверять размер и состояние
поверхности минералов методами СЭМ и рентгеновской фотоэлектронной спектроско¬
пии (РФЭС).
193
Глава 6. Разделение минералов методом селективного растворения
6.3.2. Селективные методы анализа аморфных веществ
Для описания аморфных веществ используют многие другие методы, в частности, вы¬
теснение гидроксилов фторидами и триметилсилирование.
Вытеснение гидроксилов фторидами
Этот метод основан на общей реакции:
(минерал-ОН) + F' -Цминерал-F) + ОН',
которая удовлетворительно учитывает реакционную способность скрытокристалличе¬
ских соединений, алюмосиликатов, подобных аллофану или имоголиту, или аморфных
форм алюминия, поскольку реакции протекают очень быстро.
А1(ОН)3 + 6NaF -► NajAlF6 + 3NaOH
Эта реакция позволяет разработать полевой тест на NaF для андосолей. Общее ко¬
личество выделенных ионов ОН" хорошо коррелирует с аморфными формами А1 и Si,
при слабом влиянии Fe. Количественное определение проводят титрованием при по¬
стоянном pH:
• Смешивают 25 мг почвы, измельченной до 0,2 мм, с 0,85 М раствором NaF при
pH 6,8 и 25 °С.
• Используя титрометр, поддерживают pH 6,8 в течение 30 мин, добавляя титрован¬
ный раствор кислоты. Количество кислоты, необходимой для нейтрализации, дает
оценку содержания некристаллических веществ.
Реакция может быть проведена стехиометрически только в отсутствие органического
вещества и карбонатов. Кристаллические вещества недостаточно реакционноспособны
для того, чтобы прореагировать в такой короткий промежуток времени.
Триметилсилирование
Этот метод используется исключительно для кремниевых соединений; он основан на
реакции кремниевой кислоты с кремнийорганическими соединениями, приводящей
к образованию стабильных и летучих органосилильных производных, которые могут
быть разделены и идентифицированы методом газовой хроматографии. Основная реак¬
ция может быть записана в общем виде:
Si(CH3)3
> (CH3)3SiO—4i—OSi(CH3)3
Si(CH3)3
Мономерная фаза Кремниевая Триметилсилильные
со связями O-Si кислота производные
Эта реакция позволяет проводить определение степени полимеризации оксида крем¬
ния и связать его с различными типами разрушения. Неорганические гели, которые со¬
держат много алюминия, особенно реакционноспособны. Кристаллические вещества
не очень активны.
194
Часть 1. Минералогический анализ почв
Смешивают 50 мг образца почвы с 9 мл гексаметилдисилоксана (ГМДС)1 в 0,2 мл
воды и внутренним стандартом (я-эйкозан, СН3(СН2)18СН3) при 4 °С. После гомогени¬
зации добавляют 2 мл триметилхлорсилана (ТМХС)2 при 4°С. При гидролизе выделяет¬
ся хлористоводородная кислота, которая реагирует с аморфными силикатами почвы по
механизму катионного обмена и протонирования. Оставляют в контакте при 4 °С на 16
ч. Кремниевая кислота образует производные триметилсилила.
Добавляют 2 мл воды для гидролиза избытка ТМХС. Центрифугируют для разделения
жидкой и твердой фаз и переносят органический раствор на 1 г катионообменной смолы
(Амберлайт 15). После контакта в течение 5 ч и фильтрования отделяют экстракт и опре¬
деляют количество триметилсилильных производных методом газовой хроматографии
с использованием азота в качестве газа-носителя.
Таблица 6.4. Примерный баланс минеральных фаз почвы по данным различных методов
экстракции
Формы Fe
Обозначение
Описание
Al, Si, Мпидр.
Обшее железо
Fe(o6iu)
— Важнейший анализ для установления
геохимического и минералогического ба¬
лансов и характеристических отношений.
- Обработка кипящей НС1 недостаточна
для удовлетворительного количествен¬
ного определения. Должна проводиться
щелочная плавка. Такие минералы, как
пириты, марказит и магнетит трудно
минерализуются. Отношения Fe(o6iu)/
глины и Fe(flUB)/Fe(o6iu) являются по¬
казателями выветривания минералов.
Степень развития гидроморфных почв.
Свободное
железо
Fe(ce)=
= Fe(flUB)
Железо, экстрагируемое раствором ДЦБ
(см. раздел 6.2.2): железо, мобилизуемое
в почвообразующих процессах.
Это железо представляет интерес, если
оно не связано со структурой решеток
силикатов (глины, первичные мине¬
ралы), представляющих различные ги-
дроксилированные и гидратированные
формы с разными степенями кристал¬
личности.
Общий свобод¬
ный А1 (обменная
кислотность, об¬
менный А13+, эф¬
фективная ЕКО
и др.), индикатор
разрушения (аци-
долиз, комплексо-
образование)
свободный Мп
Кристалличе¬
ское свобод¬
ное железо
Отношения Ре(св)/глины и Fe(KOT)/
Ре(ДЦБ) указывают на степени выве¬
тривания и кристалличности свобод¬
ных оксидов и конкреций. Отношение
FeCQUB)/Fe(o6iu) является показате¬
лем разрушения и подвижности железа
в профиле.
Ре(ДЦБ)—Fe(KOT)
1 Гексаметилдисилоксан (ГМДС) — (CH3)3Si-0-Si(CH3)3.
2 Триметилхлорсилан (ТМХС) — (CH3)3SiCl.
Глава 6. Разделение минералов методом селективного растворения
Таблица 6.4 (окончание)
195
Формы Fe Обозначение
Описание
A!, Si, Мп и др.
Железо в си-
Fe(cnn)
Ре(общ)-Ре(ДЦБ)
ликатах
Железо в силикатах, происходящих из
первичных минералов и решеток фил¬
литовидных минералов; восстановление
указывает на прогрессирующее разру¬
шение (горизонт камбик)
Плохо окри-
Fe(OA)
Fe(KOT)—Ре(ЭДТА)
сталлизован-
ные оксиды
железа
Хорошо
Fe(OC)
FeCaUB)-Fe(KOT)
окристалли-
Хорошо окристаллизованные оксиды
зованные ок-
железа, идентифицируемые инструмен-
сиды железа
тальными методами (РД, ДТА и др.). Это
унаследованные или новообразованные
оксиды. Присутствие лепидокрокита
и магнетита, чувствительных к обра¬
ботке оксалатом, может повлиять на ре¬
зультаты. Магнетит — унаследованный
минерал, а не образованный в процессе
педогенезиса.
Железо
Fe(impo)
Неподвижный комплекс
Отношение
в аморф¬
А1(тетра)/А1(пиро)
ных формах
уменьшается при
и в органиче¬
старении геля
ских ком¬
(растворимые
плексах
алюминаты
в щелочной среде)
Железо ком¬
Fe(TeTpa)
Подвижный комплекс. Метод использу¬
плексов
ется с реагентами в порядке увеличения
эффективности: тетраборат с pH 9,7 <
пирофосфат с pH 9,8 <NaOH с pH 12.
- Отношение Ре(тетра)/Фе(пиро) харак¬
теризует степень превращения комплек¬
сов железо-органических веществ, если
содержание Fe(TeTpa) уменьшается,
a Fe(nnpo) увеличивается — почвообра¬
зование протекало с сильным биологи¬
ческим разрушающим воздействием
Fe(KOT)
Железо, экстрагируемое оксалатом
в темноте (раздел 6.2.1), если содержа¬
ния Fe(KOYO), Ре(ЭДТА) и Fe(nnpo)
малы, степень разрушения невелика.
196 Часть 1. Минералогический анализ почв
6.3.3. Краткий обзор применения дифференциальных методов анализа
Каждому методу экстракции посвящены многие публикации, так что можно найти
примеры использования всех этих методов по отдельности или в сочетании с другими
(Aomine и Jackson, 1959; Bruckert, 1979; Jeanroy, 1983; Baize, 1988; Krasnodebska-Ostrega
идр., 2001).
Конечной целью может быть просто удаление веществ (например, связывающих ма¬
териалов), которые мешают диспергированию или инструментальному наблюдению
(например, РД или ИК-спектров). В этом случае не обязательно идентифицировать рас¬
творимые продукты; напротив, для понимания механизмов развития минералогических
и почвообразовательных процессов необходимо рассматривать как твердую, так и жид¬
кую фазу. Сравнение результатов производят, выражая их в процентном содержании ок¬
сидов; это практический способ количественного определения минералогических фаз,
однако он не определяет химические формы, реально присутствующие в почве.
Сочетание нескольких методов позволяет установить примерный баланс различных
форм алюминия и железа в почве (табл. 6.4). Отдельные объекты часто недостаточно
специфичны, а полученные результаты до некоторой степени эмпирические, но они
дают достаточно точное представление о явлениях выветривания. В целом, изменения
в количествах экстрагированных элементов (сначала железа, а затем алюминия, крем¬
ния и марганца) в одном и том же образце, сравнивают по результатам общего и затем
селективного анализа (свободные, аморфные и др. формы) одной и той же пробы. Затем
можно сравнить изменения между горизонтами почвенного профиля и, наконец, изме¬
нения между профилями. Могут быть выявлены корреляции между содержанием и по¬
ведением компонентов. Железо — основной элемент, используемый в качестве индика¬
тора разрушения. Оно нерастворимо в трехвалентном состоянии, и его потеря обычно
указывает на наличие восстановительных процессов почвообразования, которые также
идентифицируются по цвету почвы. Процессы биохимической гумификации приводят
к образованию железо-органических комплексов.
В андосолях сначала измеряют содержание А1 и Si, компонентов аллофана и имого-
лита (рис. 6.1). В этих почвах аллофановые материалы иногда можно определить кос¬
венным путем, например, при измерении емкости катионного обмена пробы, из кото¬
рой предварительно удалено органическое вещество. Одну часть пробы обрабатывают
раствором карбоната натрия в течение 60 мин, а другую - кипящим раствором ацетата
натрия в течение 15 мин, получающаяся разница, выраженная в емкости катионного
обмена, хорошо коррелирует с содержанием аллофана (Aomine и Jackson, 1959).
Инструментальный метод дифференциальной дифрактометрии позволяет оценить
влияние растворяющих реагентов и поведения почвенных материалов. Снимают не¬
сколько дифрактограмм пробы до и после обработки селективного растворения. Можно
использовать соответствующую компьютерную программу для расчета разницы между
данными для обработанной и необработанной пробы. Кварц или а-глинозем исполь¬
зуется в качестве внутреннего стандарта для определения коэффициента пересчета при
вычитании дифрактограмм. Это позволяет проводить реконструкцию дифрактограмм
растворенных веществ во время селективной обработки (Campbellи Schmrtmann, 1985).
Использованная литература
Alexsandrova LN (1960) The use or pyrophosphate for isolating free humic substances and their organic-
mineral compounds from the soil. Soviet Soil Sci., 190-197.
Aomine S and Jackson ML (1959) Allophane determination in andosoils by cation exchange delta value.
Soil Sci. Soc. Am. Proc23, 210-214.
197
Глава 6. Разделение минералов методом селективного растворения
Atkinson RR, Posner AM and Quirk J (1968) Crystal nucleation in Fe (III) solutions and hydroxyde gels.
J. Inorg. Nucl. Chem., 30, 2371-2381.
Avery BW and Bascomb CL (1982) Soil Survey Laboratory Methods, Soil Survey of England-Wales
(Harpenden), 6.
Baize D (1988) Guide Des Analyses Courantes En Pedologie: Choix — Expression — Prisentation —
Interpretation., INRA, 172 pages.
Ballantyne AKD, Anderson DW and Stonehouse HB (1980) Problems associated with extracting Fe and A1
from Saskatchewan soils by pyrophosphate and low speed centrifugation. Can. J. Soil Sci., 60,141-143.
Bentley SP, Clark NJ and Smalley IJ (1980) Mineralogy of a Norwegian postglacial clay and some
geotechnical implications. Can. Miner., 18, 535-547.
Borggaard OK (1976) Selective extraction of amorphous iron oxide by EDTA from a mixture of
amorphous iron oxide, goethite and hematite. J. Soil Sci., 27, 478-486.
Bruckert S (1979) Analyse des complexes organo-mindraux des sols. In Pedologie 2, constituants et
proprietes du sol, Bonneau M. and Souchier B. ed. Mason, IX, 187-209.
Cambier P and Sposito G (1991) Adsorption of citric acid by synthetic pseudoboehmite. Clays Clay
Miner., 39, 369-374.
Campbell AS and Schwertmann U (1985) Evaluation of selective dissolution extractants in soil chemistry
and mineralogy by differential X-Ray diffraction.Clay Miner., 20, 515-519.
Colmet-Daage F, Gautheyrou J, Gautheyrou M and De Kimpe C (1973) Etude des sols к allophane diriv&s
de matdriaux volcaniques des Antilles et d’Amdrique latine к Paide de techniques de dissolution
diffdrentielle. I£re partie. Etude des produits solubilises. Cah. ORSTOMserie Pedol., XI, 97-120.
Cornell RA, Posner AM and Quirck J.P (1976) Kinetics and mechanisms of the acid dissolution of
goethite (a-FeOOH). J. Inorg. Nucl. Chem., 38, 563-567.
Cornell RM and Schindler PW (1987) Photochemical dissolution of goethite in acid/oxalate solution.
Clays Clay Miner., 35, 347-352.
Cornell RM, Posner AM and Quirck J.P (1974) Crystal morphology and the dissolution of goethite.
J. Inorg. Nucl. Chem., 36, 1937-1946.
Cornell R.M., Posner A.M and Quirk JP (1975) The complete dissolution of goethite. J. Appl. Chem.
Biotechnol., 25, 701-706.
De Endredy AS (1963) Estimation of free iron oxides in soils and clays by a photolytic method .Clay
Miner. Bull., 29, 209-217.
Deb BC (1950) The estimation of free iron oxides in soils and clays and their removal. J. Soil Sci., 1,
212-220.
Duchaufour Ph and Souchier В (1966) Note sur une m6thode detraction сотЫпёе de l’aluminum et du
fer libres dans les sols. Sci. du Sol, 1, 17-29.
Farmer VC and Fraser AR (1978) Synthetic imogolite, a tubular hydroxy- aluminium silicate. In
International Clay Conference., Elsevier, Amsterdam, 547-553.
Farmer VC, Fraser AR and Tait JM (1979) Characterization of the chemical structures of natural and
synthetic aluminosilicate gels and soils by infrared spectroscopy. Geochim.Cosmochim.Acta, 43,
1417-1420.
Follett EAC, McHardy WJ, Mitchell BD and Smith BFL (1965) Chemical dissolution techniques in the
study of soil clays. Clay Miner., 6, 23-43.
Franzmeier DP, Hajek BF and Simonson CH (1965) Use of amorphous material to identifiyspodic
horizons. Soil Sci. Soc. Am.Proc., 29, 737-743.
Hashimoto I and Jackson ML (1960) Rapid dissolution of allophane and kaolinite and halloysite after
dehydratation. Clays clay Miner., 7,102-113.
Henry S (1958) Synthase de quelques oxydes de fer au laboratoire. C.R. du XXXI Congres Intern, de
Chimie Industrielle (Liege)., Mercurius, 1-3.
Hetier JM and Jeanroy E (1973) Solubilisation diffiSrentielle du fer, de la silice et de l’alumine par le
r^actif oxalate-dithionite et la soude dilute. Pidologie, 23,85-99.
Holmgren GGS (1967) A rapid citrate-dithionite extractable procedure. Soil Sci. Soc. Am. Proc., 31,210-211.
Hsu PH (1977) Aluminum hydroxydes and oxyhydroxydes. In Minerals in Soil Environments, Dixon JB
Weed SB and ed., Soil Sci. Sc. Am., 99-143.
Hsu PH (1984) Aluminum hydroxides and oxyhydroxides in soils: recent developments.,4««tt. Meeting
and Am. Soc. Agron.
198
Часть 1. Минералогический анализ почв
Jeanroy Е and Guillet В (1981) The occurrence of suspended ferruginous particles in pyrophosphate
extracts of some soil horizons. Geoderma, 26,95-105.
Jeanroy E (1983) Diagnostic des formes du fer dans les pedogeneses tempertes. Evaluation par les riactifs
chimiques detraction et apports de la spectrometrie Mossbauer (etudes des formes organiques du
fer amorphe dans les sols)., These Doctorat, Nancy, 109-129.
Kampf N and Schwertmann U (1982) The 5M-NaOH concentration treatment for iron oxides in soils.
Clays clay Miner, 30, 401-408.
Klamt E (1985) Reports of meetings. Iron in soil and clay minerals. Bad Windesheim, West Germany,
July 1-13 1985. Bull. Soc. Int. Sci. du Sol, 2, 9.
Krasnodebska-Ostrega B, Emons H and Golimowski J (2001) Selective leaching of elements associated
with Mn-Fe oxides in forest soil, and comparison of two sequential extraction methods. Fresenius
J. Anal Chem., 371, 385-390.
Kwong KF and Huang PM (1979) The relative influence of low-molecular weight complexing organic
acids on the hydrolysis and precipitation of Aluminum. Soil Sci.9 128, 337-342.
Lewis DG and Schwertmann U (1979) The influence of A1 on iron oxides. Part III — Preparation of A1
goethites in M KOH. Clay Miner., 14, 115-126.
Lewis DG and Schwertmann U (1979) The influence of A1 on the formation of iron oxides. Part IV: The
influence of [Al], [OH] and temperature. Clays Clay Miner., 27,195-200.
Loveland PJ and Bullock P (1976) Chemical and mineralogical properties of brown podzolic soils in
comparison with soils of other groups. J. Soil Sci., 27, 523-540.
Loveland PJ and Digby P (1984) The extraction of Fe and Al by 0.1 M pyrophosphate solutions:
a comparison of some techniques. J. Soil Sci., 35, 243-250.
McKeague JA and Day JH (1966) Dithionite and oxalate-extractable Fe and Ag as aids in differentiating
various classes of soils.Canad. J. Soil Sci., 46, 13-22.
McKeague JA (1967) An evaluation of 0.1 M pyrophosphate and pyrophosphate-dithionite in comparison
with oxalate as extractants of the accumulation products in podzols and some other soils. Can.
J. SoilSci., 47, 95-99.
Neaman A, Moueie F, Trolard F, Bourrie G (2004a) Improved methods for selective dissolution of Mn
oxides: applications for studying trace element associations. Appl. Geochem19,973-979.
Neaman A, Waller В, Moueie F, Trolard F, Bourrie G (2004b) Improved methods for selective dissolution
of manganese oxides from soils and rocks. Eur. J. Soil Sci., 55,47-54.
Norrish К and Taylor RM (1961) Theisomorphous replacement of iron byaluminum in soil goethites.
J. Soil Set, 12, 294-306.
Petersen L (1976) Podzols andpodzolization., Thesis Copenhagen (Danmark).
Pollard RJ, Cardile CM, Lewis DG and Brown LJ (1992) Characterization of FeOOH polymorphs and
ferrihydrite using low-temperature applied-field, Mdsshauer spectroscopy. Clay Miner., 27, 57-71.
Quantin P et Lamouroux M (1974) Adaptation de la methode cinetique de Segalen к la determination des
constituents mindraux de sols varies, Cah. ORSTOM, ser. Pidol., XII, 1, 13-46.
Quigley RM, Haynes JE, Bohdanowicz A and Gwyn QHJ (1985) Geology, geotechnique, mineralogy and
geochemistry of Leda clay from deep Boreholes, Hawkesbury Area. Ontario Geol. Surv., study 29,
128 pages.
Ryan JN and Gschwend PM (1991) Extraction of iron oxides from sediments using reductive dissolution
by titanium (III).Clays Clay Miner., 39, 509-518.
Schuppli PA, Ross GJ and McKeague JA (1983) The effective removal of suspended materials from
pyrophosphate extracts of soil from tropical and temperate regions. Soil Sci. Soc. Am. J„ 47, 1026—
1032.
Schwertmann U (1964) Differenzierung der Eisenoxide des Bodens durch photochemische extraktion mit
saurer Ammoniumoxalate-Losung.Z Pflanzenernahr. Dueng.Bodenk., 105, 194-202.
Schwertmann U (1991) Solubility and dissolution of iron oxides .Plant and Soil, 130, 1-25.
Segalen P (1968) Note sur une methode de determination des produits amorphes dans certains sols к
hydroxy des tropicaux. Cahiers ORSTOM SeriePedol., 6, 106-126.
Shuman LM (1982) Separating soil iron and manganese oxide fractions for microelement analysis. Soil
Sci. Soc. Amer. J., 46, 1099-1102.
Глава 6, Разделение минералов методом селективного растворения
199
Stol RJ, Van Helden AD and De Bruyn PL (1976) Hydrolysis-precipitation studies of aluminum solution.
II—A kinetic study and a model. J. Colloid Interface Sci., 57,115-131.
Stumm W (1985) The effects of complex-forming ligands on the dissolution of oxides and aluminosilicates.
In The chemistry of weathering. ,RQidei\ D,Drever J ed., 55-74.
Tamm О (1922) Einemethode zur Bestimmungder anorqanischen komporentem des Gelkomplexesim
Boden. Meddal. Statens Skogforsdksanst, 19, 385-404.
Tamm О (1931) Monthly letter., Imperial Bureau of Soil Science, 1 October.
Tamm О (1934a) Monthly letter., Imperial Bureau of Soil Science, 34, August.
Tamm О (1934b) Ober die Oxalat-methode in der chemischen Boden analyse. Medd. Skogforsokamsanst,
27, 1-20.
Tokashiki Y, Dixon JB and Golden DC (1986) Manganese oxide analysis in soils by combined X-Ray
diffraction and selective dissolution methods .Soil Sci. Soc. Amer. J., 50,1079-1084.
Torrance JK, Hedges SW and Bowen LH (1986) MOssbauer spectroscopic study of the iron mineralogy
of post-glacial marine clays. Clays clay Miner., 34, 314-322.
Yong R, Sethi AJ and La Rochelle P (1979) Significance of amorphous material relative to sensitivity in
some Champlain clays. Canad. Geotechn. 16, 511-520.
Глава 7. Термический анализ
7.1. Введение
7.1.1. Определения
Большинство методик анализа фазовых превращений используют различные темпера¬
турные измерения. Например, простые гравиметрические измерения после высушива¬
ния при данной температуре (см. гл. 1) или высушивание до постоянного веса после хи¬
мического осаждения. Общий термин «термический анализ» обычно применяют только
к методам, выполняемым в температурных условиях, изменяющихся в соответствии
с динамическим нагревом, контролируемым программой, что позволяет выявить и ко¬
личественно оценить различные физико-химические переходы. Самые распространен¬
ные методы, используемые при анализах почвы, включают регистрацию изменений мас¬
сы, энергии или механических свойств образца (рис. 7.1) в зависимости от температуры.
Измерения изменений массы
Аббревиатура ТГА (TGA) или ТГ (TG) устоялась для термогравиметрического анализа
и ДТГ (DTG) — для дифференциальной термогравиметрии. Потери массы образца про¬
исходят через выделение газов, которые легко определить (регистрация выделяющегося
газа, РВГ) и проанализировать (анализ выделяющегося газа, АВГ).
Переходы в термическом анализе
Изменения
массы
Изменения
энергии
Изменения
механических
Изменения
оптических,
Рис. 7.1. Основные методы термического анализа: ТГА — термогравиметрический анализ (TGA,
thermo gravimetric analysis); ДТГ—дифференциальная термогравиметрия (DTG, differential thermo
gravimetry); ДТА — дифференциальный термический анализ (DTA, differential thermal analysis);
ДСК — дифференциальная сканирующая калориметрия (DSC, differential scanning calorimetry);
TMA — термомеханический анализ (TMA, thermo mechanical analysis); ДМА — динамический ме¬
ханический анализ (DMA, dynamic mechanical analysis); СТА — синхронный термический анализ
(STA, simultaneous thermal analysis)’, АВГ — анализ выделяющегося газа (EGA, evolved gas analysis).
Международная конфедерация термического анализа (ЮТА) использует термин «деривация» для
данных, следующих из математических преобразований (расчеты производных по первоначально
измеренным данным), и «дифференциальный» — для экспериментальных измерений изменений
величин с интересующим шагом задержки
Глава 7. Термический анализ
201
Измерения изменений энергии
Главным образом используются методы дифференциального термического анализа
(ДТА) и дифференциальной сканирующей калориметрии (ДСК). Они позволяют про¬
водить количественное определение экзотермических и эндотермических изменений
энергии при каждой температуре без неизбежного изменения массы образца.
Измерения изменений размеров и физических свойств
Используются методы ТМА — термомеханического анализа (дилатометрия) и ДМА —
динамического механического анализа (вязкоупругость). Хотя эти методы в основном
используются для керамики и пластмасс, они применимы и для исследования физиче¬
ских свойств почв и особенно их усадки и набухания, связанных с агрегированием и во¬
доудерживающими свойствами (Braudeau, 1987,1988; Braudeau и др., 1999,2004).
Возможные изменения различных свойств (оптических, магнитных, электрических
или звуковых) могут быть измерены (например, термолюминесценция широко исполь¬
зуется для датирования, а потеря магнитных свойств определяется по точке Кюри и т. д.).
Оборудование, используемое для этих анализов, достигло высокой степени совер¬
шенства благодаря их промышленному использованию в производстве керамики, стек¬
ла, цемента, гипса, взрывчатых веществ, радиоактивных изотопов, фармацевтических
препаратов и полимеров, где они находят широкое применение в исследованиях и кон¬
троле качества. Простейшее оборудование относительно дешево, но приборы высшей
категории качества (мультипараметрические, высокотемпературные, высокого давле¬
ния и пр.), снабженные дополнительными устройствами и программным обеспечением,
могут стоить очень дорого.
7.1.2. Практическая значимость
Методы термического анализа, используемые в сочетании с РД (см. гл. 4), ИК-
спектроскопией (см. гл. 5) и другими методами химического анализа, являются не¬
оценимым инструментом для минералогии, геологии, почвоведения, химии и физики
почвы. Они позволяют осуществлять качественную идентификацию и количественный
анализ глин (табл. 7.1) и многих минералов, а также идентификацию различных форм
воды в почвах, исследование окисления всех форм органического вещества и неоргани¬
ческих материалов, фазовых переходов и др.
Чувствительность методов ДТА—ДСК позволяет выявить присутствие минералов с та¬
ким же пределом обнаружения, как в РД (например, гетит и гиббсит при содержании ме¬
нее 0,25%, вещества с ближним порядком расположения атомов и др.), в глинах и почве.
Все используемые методы позволяют установить баланс превращений минералов как
результат различных геохимических процессов в выветриваемом профиле и в топогра¬
фической последовательности почв.
Можно проводить достаточно точный количественный анализ для гидроксидов и ок-
сигидроксидов железа и алюминия, а также для глин и частично каолинита 1:1 и гал-
луазита. Континентальные отложения, содержащие органические комплексы, можно
исследовать методом контролированного окислительного или неокислительного пи¬
ролиза, а выделяющиеся газы — ИК-Фурье-спектрометрией. Методом ДСК можно
количественно оценить изменения энтальпии в процессах дегидратации, дегидрокси-
лирования и других форм структурных разложений в интервале температур от —150 до
+725 °С. Анализ выделяющихся газов методами ИК-Фурье-спектрометрии или масс-
спектрометрии в процессе температурного сканирования позволяет точно рассчитать
химические превращения, происходящие в образце.
Таблица 7.1. Превращения глин до температуры 1200 °С (поглощенная и связанная вода)
К
°с
Муллит
t
r-AiA
Лейцит
1373
1100
Метакаолин
Корунд
Энстатит
1273
1000
Конец
дегидро-
ксилирования
Кристобалит
t
1073
800
Вода, связан*
ная с кри¬
сталлической
решеткой
Вода, связан- Связан¬
ная с кри- ная вода
сталл ической
решеткой
Связанная
вода
Связанная
вода
Связанная
вода
Вода, связан- Вода, связан¬
ная с кри- ная с кри¬
сталлической сталлической
решеткой решеткой
Связанная
вода
973
700
-16,0%
-16,0% -5,3%
-4,7%
~5,1%
-5,0%
700°С
500°С
-14,8%
873
600
-4,8% воды
в пустотах
-3,1%
-4,8% воды
в пустотах
-4,7%
673
400
(350 °С)
(350 °С)
(350 °С)
370 °С
Адсорбиро¬
ванная вода
Адсорби¬
рованная
вода
573
300
(250 °С)
Адсорбиро¬
ванная вода
в пустотах
~0,5%
(250 °С)
в пустотах
-3,4%
(250 °С)
(250 °С)
(250 °С)
202 Часть 1. Минералогический анализ почв
473
200
Адсорбиро¬
Адсорбиро¬
Адсорби¬
Адсорби¬
Адсорби¬
(150 °С)
ванная вода
ванная вода
рованная
рованная
рованная
(150 °С)
<1,0%
-13,2%
вода
межслоевая
межслоевая
Адсорби¬
Адсорби¬
-0,5-1,5%
вода
вода
рованная
рованная
-1,0%
-20,1%
межслое¬
межслоевая
вая вода
вода
373
100
-14,0%
-20,0%
Адсорбиро¬
ванная вода
-10,1%
вода
<0,5%
ГЛИНЫ 1:1
ГЛИНЫ 2:1
ГЛИНЫ
2:1:1
Галлуазит
Каолинит 4Н20
Пиро¬
филлит
Слюды
Мусковит
(иллиты)
СМЕКТИТЫ
Са2+— монт- Na+—
мориллонит монтмо¬
риллонит
Са2+
ВЕРМИКУЛИТЫ
Na+
Клино-
хлор
IS)
о
Глава 7. Термический анализ
204
Часть 1. Минералогический анализ почв
Термический анализ особенно важен для исследования генезиса почв, изучения почв,
богатых яддякристаллическими соединениями (например, андосоли, гистосоли), и ха¬
рактеризации превращений соединений с атомной организацией ближнего порядка,
которые не могут быть непосредственно проанализированы методом РД.
В некоторых случаях можно использовать термогравиметрический метод в диапазоне
температур от комнатной до 200 °С для косвенного измерения удельной поверхности
(внешней и внутренней) глин, пропитывая образец мономолекулярным слоем органи¬
ческого материала (например, метод с использованием ЭГМЭ — этиленгликольмоно-
этилового эфира) или газа (например, БЭТ-метод).
Инструментальные термические дилатометрические методы, разработанные специ¬
алистами по керамике, можно использовать вместо ручных измерений процессов усад¬
ки — расширения (коэффициента линейного расширения).
7.2. Классические методы
7.2.1. Термогравиметрический анализ
Основные положения
Изменения массы пробы (уменьшение или увеличение) записывают как функцию от
температуры или времени. Для изучения почв обычно достаточен диапазон от комнат¬
ной температуры до 1100 или 1200 °С, хотя может возникнуть необходимость исследо¬
вать реакции при температурах выше 2000 °С, в частности при определении точек плав¬
ления.
В динамическом анализе пробу массой т нагревают с постоянной скоростью в со¬
ответствии с линейной программой (рис. 7.2). Исходя из данных о диапазоне реакции,
форме кривой и природе выделяемых газов, можно получить информацию о природе
почвенной пробы и ее термической стабильности.
В ТГА записывают первую производную изменений массы пробы как функцию вре¬
мени или температуры. В некоторых случаях используют два «статических» метода, ко¬
торые, однако, требуют длительного времени:
• изотермический метод, в котором пробу подвергают воздействию постоянной тем¬
пературы, и массу пробы записывают в течение некоторого времени до достиже¬
ния равновесного значения;
• изобарный метод, в котором пробу подвергают воздействию постоянного давле¬
ния, и массу пробы записывают как функцию от температуры; для некоторых ма¬
териалов давление может превышать 500 атм.
О 500 1000 f,° С 0 500 1000 t,° С
Рис. 7.2. Данные термогравиметрического анализа т = f(t) (слева) в сравнении
с дифференциальной термогравиметрией dm/dt = /^(справа)
Глава 7. Термический анализ
205
Пшнистые минералы содержат молекулы воды и гидроксид-ионы, связанные с кри¬
сталлической решеткой с различными энергиями связи. При повышении температуры
эта вода постепенно мигрирует и выделяется в форме газа.
В почве при дегидратации обычно сначала выделяется несвязанная промежуточная
вода гидратации или адсорбции. Дегидратация не приводит к разрушению решетки, но
может вызывать изменения в расположении сплошных поли- и мономолекулярных сло¬
ев (внутренние и внешние поверхности глин типа 2:1 и межслоевые обменные ионы),
наряду со сжатием межслоевого пространства. Это обратимый процесс. Вода в пустотах
около оснований тетраэдров связана более прочно. С другой стороны, процесс может
стать необратимым в почвах, содержащих аллофан, и при регидратации достигается
не более 10% первоначального содержания промежуточной воды. Это также проис¬
ходит в случае гидратированного четырехводного галлуазита, который превращается
в двухводный метагаллуазит.
Затем мигрируют гидроксид-ионы, связанные с атомами кислорода в вершинах тетраэ¬
дрических или октаэдрических полиэдров или находящиеся во внешних сплошных слоях
глин типа 1:1 или во внутренних или внешних слоях глин типа 2:1 (или гидратирован¬
ного галлуазита 1:1). Их удаление является необратимым процессом и сопровождается
разрушением структуры (дегидроксилированием). Затем может быть зарегистрировано
появление новых форм при более высокой температуре, например, каолинит превраща¬
ется в метакаолинит и затем в муллит. Однако метод ТГА не может выявить превращения,
происходящие без потери массы. В этом случае следует записать спектры ДТА или ДСК.
Следует отметить, что переход между свободной водой Н20 и гидроксильной ОН_-
группой не всегда ясно виден, поскольку вода, связанная с кристаллической решеткой,
может начать выделяться до полного удаления промежуточной воды. В этом случае
можно использовать термический анализ при контролируемой скорости превращения
(Rouquerol, 1970,1989; Rouquerol и ., 1985,1988)1.
Выполнение
Реагенты
• Реагенты, описанные в главах 2 и 3 для разложения органического вещества или
карбонатов, гомоионного насыщения и т. д.
• Глины, очищенные для стандартизации (пробы для эталонной системы лаборатор¬
ных и/или международных стандартов).
• Насыщенный раствор нитрата магния (Mg(N03)2 • 6Н20, С = 1250 г/л при 20 °С) (для
выравнивания влажности глин - относительная влажность воздуха 56% при 20 °С).
• Высушивающие агенты различной степени эффективности:
а) перхлорат магния (дигидрит), Mg(C104)2, молекулярная масса (Мм) составляет
223,23;
1 Метод анализа при контролируемой скорости превращения позволяет определять исходное
состояние воды с ббльшей точностью. В традиционном анализе программируется линейный
рост температуры со временем. При контролируемой скорости превращения скорость повы¬
шения температуры определяет давление, вызываемое выделением газа. Сопряжение с ква-
друпольным масс-спектрометром позволяет проводить анализ выделяющегося газа. Когда
адсорбированная вода с низкой энергией активации удалена, рост температуры останавли¬
вается до тех пор, пока первоначальное давление не восстановится до заданного уровня. За¬
тем нагревание продолжается. Эта методика облегчает исследование механизмов гидратации,
а возможно и атомных структур ближнего и дальнего порядков.
206
Часть 1. Минералогический анализ почв
б) оксид алюминия, А1203, Мм = 101,94;
в) диоксид кремния (силикагель), Si02, Мм = 60,08;
г) безводный хлорид кальция, СаС12, Мм = 110,99;
д) безводный сульфат кальция (ангидрит/драйерит), CaS04, Мм = 136,04;
е) пентаоксид фосфора (фосфорный ангидрид), Р205, Мм = 141,96.
Оборудование
• Платиновые микротигли;
• Эксикатор с насыщенным раствором нитрата магния.
• Эксикаторы с различными осушающими агентами.
• Термовесы (рис. 7.3). Термовесы состоят из печи, весов и различных приспособле¬
ний для регулировки и сбора данных. Взвешивание производится непрерывно в те¬
чение теплового цикла. Существует два типа оборудования: весы, помещенные над
передвигающейся печью и весы, помещенные под передвигающейся печью.
В весах первого типа платформа держателя пробы подвешена на проволоке; в весах
второго типа проба находится на подставке, установленной на вертикальном штоке.
Каждая система имеет свои недостатки, которые должны быть минимизированы для
оптимизации условий измерений.
Весы должны позволять проводить измерения уменьшения или увеличения массы
пробы в зависимости от температуры или времени при любых экспериментальных ус¬
ловиях. Температура в печи может достигать 2400 °С, так что возможны различные из¬
лучательные и магнитные процессы. Качество измерений должно быть таким же, как
при использовании любых других аналитических микровесов (Рати и др., 2001). Масса
взвешивания может изменяться в зависимости от предполагаемого использования. Для
почв должен быть сделан выбор между (а) относительно большими пробами, от 100 мг
до 1 г, при чувствительности 10-5—10~6 г или (б) микропробами, от 1 до 50 мг (например,
пьезоэлектрические весы с чувствительностью 10-8—10-12 г).
Рис. 7.3. Пример системы для термогравиметрического анализа
Глава 7. Термический анализ
207
Существуют различные типы весов: весы со стрелкой (например, торсионные, ры¬
чажные, консольные, с пружиной из бериллиевой бронзы) и электронные системы без
рычага, которые используются редко из-за возможных повреждений под действием из¬
лучательных процессов.
Конструкция печи зависит от температуры. Можно использовать высокочастотную
индукционную печь или чаще — электрическую печь сопротивления (табл. 7.2). Исполь¬
зование сопротивлений из сплава кантал (FeCrAl) позволяет проводить измерения до
1200 °С, родиевых и вольфрамовых сопротивлений - до 1800 и 2500 °С, соответственно.
Таблица 7.2. Типы сопротивлений и температурные диапазоны их использования
vto^hot. Используемая Точка
Материал температура, °С плавления, °С
Мятепиал Используемая Точка
1 затериал темПература, °с плавления, °С
Константан
750
1200
Pt-rh 13%
1700
Нихром
1000
1500
Суперкантал
1600
Хромель-
1100
Родий (Rh)
1800
алюмель
Кантал
1350
Молибденв
2200
2500
Платина (Pt)
1400
1755
Тантал
-
2850
Pt-Rh 10%
1500
Вольфрам6
2800
3400
"Атмосфера водорода.
6 Механически прочен вплоть до 1650 °С.
Печь представляет собой агрегат из металлических и керамических компонентов (ок¬
сид алюминия высокой плотности), которые позволяют изолировать нагреватели для
обеспечения одинаковой температуры в испытательной зоне, а также герметизацию при
использовании контролируемой газовой среды. Печь снабжена сложной системой регу¬
лировки. Скорость нагревания должна обеспечивать одинаковую температуру во всей
испытательной зоне и контролироваться термопарами, соответствующими рабочей
температуре (табл. 7.3). Программирование нагревания должно обеспечивать высокую
воспроизводимость. Проба должна находиться в определенном месте внутри печи. Си¬
стема сбора данных должна обеспечивать одновременную регистрацию изменений мас¬
сы и температуры, а также выполнять определенный набор математических операций
Таблица 7.3. Типы термопар для различных температур
Материал
Температура, °С
Материал
Температура, °С
Медь — константан3
400 (стандарт)
Платина — платинородий
(10% или 13%crh)B
1750
Железо — константан
800 (стандарт)
Вольфрам — рений
2200
Хромель — константан
1000 (стандарт)
Вольфрам — 20%
вольфрам — рений
2400
Хромель — алюмель
Нихром — никель
1000 (стандарт)
1370
Карбид тантала — графит6
3000
аСплав 55% меди + 45% никеля.
6Атмосфера аргона.
Термоэлементы позволяют увеличивать выходной сигнал без усиления.
208
Часть 1. Минералогический анализ почв
(например, расчет первой производной, площади пиков), контролировать программу
нагрева и, наконец, хранить и распечатывать данные.
Методика
Если необходимо сравнивать результаты различных серий измерений, используемые
методы должны быть стандартизованы. Технические характеристики термовесов предъ¬
являют определенные требования к некоторым показателям. Природа анализируемых
материалов определяет выбор других диагностических параметров. Измельчение об¬
разцов не должно сопровождаться их нагреванием, которое может исказить результаты
термического анализа.
Сначала исходные образцы (из которых удалено органическое вещество) измельча¬
ют методом мокрого помола. Образец должен иметь однородный размер частиц и после
высушивания на воздухе проходить сквозь сито 0,1 мм. Можно использовать образцы
глины, очищенные и насыщенные с применением методов, описанных в гл. 3. Следует
отметить, что слишком тонкий сухой помол может исказить результаты1.
Регулируют влажность образца, поместив его в эксикатор с насыщенным раствором
нитрата магния на 48 ч (относительная влажность должна составлять 56% при 20 °С
и нормальном давлении). Эта обработка гомогенизирует гидратацию слоев,любых меж¬
слоевых катионов, которые могут присутствовать в образце.
Взвешивают пробу (5—20 мг), масса которой соответствует диапазону взвешивания
и чувствительности весов. Насыпают пробу в платиновый тигель максимально ровным
слоем для уменьшения различий в теплопроводности. Помещают пробу в термовесы.
Регулируют положение пробы относительно термопары в печи.
Используя управляющие программы, задают параметры нагревания, в том числе ли¬
нейную скорость нагревания (например, 10 °С/мин), состав газовой среды в печи, ко¬
нечную температуру и др.
Наблюдения
Для глин лучшие количественные данные получены на гомоионно насыщенных пробах
(Na+, К+, Са2+или Mg2+) после удаления органического вещества, растворимых солей,
ионов двухвалентного железа и т. д. Гомоионное насыщение глин позволяет:
• при насыщении ионами Na+ — улучшить разделение адсорбированной воды
и воды, связанной в кристаллической решетке;
• при насыщении ионами Mg2+ — улучшить разделение глин типа 2:1 и глин типа 1:1,
исходя из измерений адсорбированной воды.
Присутствие органического вещества (ОВ) изменяет потерю массы минеральных ве¬
ществ. Следует измерять потерю Н20 + С02 из ОВ. Если она не слишком велика, ее
можно еще уменьшить, используя инертную газовую среду, или потери можно оценить,
анализируя выделяющийся газ (например, методом АВГ — Фурье-ИК-спектроскопии).
Двухвалентное железо при нагревании окисляется и увеличивает массу образца в соот¬
ветствии с реакциями:
Fe2+->Fe3+ + e-
1 Сухой помол может изменить природу базальных граней и, следовательно, их физические
свойства. Например, обменная емкость и некоторые термические свойства каолина могут
измениться (исчезновение пиков) после слишком тонкого помола, при котором возможно
образование трещин, перпендикулярных базальным граням, и последующего разрушения
слоев.
Глава 7. Термический анализ
209
или Fe(OH)2 -> FeO + H2Ot
2FeO + О ->Fe203’
Так как изменения массы при окислении двухвалентного железа определить нелегко,
то предпочтительнее удалить его. Это же верно для марганца и кобальта. Можно также
использовать инертную газовую среду. Удаление растворимых солей позволяет избежать
вторичной рекомбинации.
Выбор материала тигля и его геометрии также может повлиять на результаты. Тигель
не должен реагировать с пробой, компонентами газовой среды или выделяющимися га¬
зами. Некоторые металлы обладают каталитическим действием. Алундовые тигли име¬
ют пористую структуру, серебро можно использовать при средних температурах, а пла¬
тину можно использовать до 1500 °С. Стенки тигля должны быть максимально тонкими
(примерно 0,5 мм) для минимизации отклонений в температуре. Аналогичным образом,
слой пробы должен быть максимально тонким для того, чтобы обеспечить равенство
температуры в центре и по краям пробы. Поскольку некоторые минералы могут расши¬
ряться или выделять газы, иногда следует использовать более глубокие тигли или полу¬
проницаемые крышки, однако это может изменить поток выделяющихся газов.
Скорость повышения температуры влияет на разложение пробы. Для конкретного
интервала температуры лучше использовать медленное разложение, чем быстрое, кото¬
рое может привести к смещению характеристических температур, более крутой кривой
разложения и т. д. Например, Котра с сотр. (Kotra и др., 1982) показали, что сидерит
(FeC03), нагреваемый со скоростью 1 °С/мин, характеризуется диапазоном разложения
от 400 до 500 °С. При скорости нагрева 20 °С/мин этот диапазон сдвигается до 480-
610 °С. Однако малые пробы характеризуются меньшими сдвигами, чем пробы большей
массы. Медленная скорость нагрева также облегчает идентификацию соединений, даю¬
щих лишь слабый изгиб на кривой при высоких скоростях нагрева.
Газовая среда в пени может существенно влиять на результаты с учетом природы про¬
дуктов разложения и типов реакций. Применение вакуума, инертная или реакционно-
способная газовая среда приводят к различным термоаналитическим кривым. Анализ
выделяющихся газов может предоставить дополнительную информацию.
Дифференциальный термогравиметрический анализ (ДТГ)
В этом типе анализа получают пики, площадь которых прямо пропорциональна измене¬
ниям массы пробы1 2 (рис. 7.2 и 7.4), что облегчает проведение количественного анализа.
При нулевых изменениях массы пробы кривая возвращается к базовой линии, для ко¬
торой dm/dt = 0. Кривые имеют точку перегиба в области, где изменение массы пробы
достигает максимума. Использование производных облегчает разделение накладываю¬
щихся процессов.
Обсуждение
Влажность глины можно определить на пробе, находящейся в равновесии с газовой сре¬
дой при относительной влажности -56% (получаемой при использовании насыщенного
водного раствора нитрата магния). Таким образом, можно получить исходную точку от-
1 Так как окислителем выступает кислород воздуха, реакцию следует записывать в виде:
4FeO + 02-> 2Fe203. — Примеч. науч. ред.
2 По оси абсцисс откладывается время. — Примеч. науч. ред.
210
Часть 1. Минералогический анализ почв
Рис. 7.4. Пример кривых ТГА и ДТГ для каолина (Макон, Франция): масса пробы — 25 мг, скорость
нагревания — 10 °С/мин, газовая среда — азот, скорость 30 мл/мин, платиновый тигель
счета с учетом сложных структур минералов и органоминералов и достичь лучшей вос¬
производимости.
Можно измерить содержание воды, которое зависит от присутствия обменных кати¬
онов, оно может быть очень высоким в монтмориллонитовых глинах типа 2:11 и низким
в каолиновых глинах типа 1:1. Однако энергию активации, необходимую для выделения
воды, невозможно измерить методами ТГА и ДТГ Для этой цели необходимо исполь¬
зовать метод ДТА, в котором определяется количество калорий, поглощенное эндотер¬
мическим пиком, а количество выделенной воды определяется методом ДТГ в тех же
условиях. Существующее оборудование позволяет одновременно использовать методы
ТГА - ДТГ и ДТА в одном и том же цикле нагрева одной и той же микропробы до тем¬
пературы 1750 °С и выше.
Прямое количественное определение возможно только в случае, если при конкрет¬
ной температуре происходит одна реакция. При изолировании компонентов можно
определять количество органического вещества в окислительных условиях, а также ма¬
лых количеств карбонатов (например, доломита, арагонита, кальцита, сидерита) и суль¬
фидов (пиритов). В этом случае необходимо комбинировать определение количества
с анализом выделяющихся газов.
7.2.2. Дифференциальный термический анализ и дифференциальная
сканирующая калориметрия
Основные положения ДТА
В ДТА фиксируют разницу меж1у температурами образцов почвы или глины и инертно¬
го эталонного материала как функции времени или температуры при одновременном
нагревании двух веществ с постоянной скоростью:
1 Слои в глинах типа 2:1 адсорбируют не менее двух слоев молекул воды. Они не ориенти¬
рованы относительно друг друга и проявляют тенденцию к образованию комбинаций слоев
(турбостратные структуры из 5 или 6 слоев).
Глава 7. Термический анализ
211
АТ=Т
проба
- т
эталон
Этот тип анализа позволяет определять коэффициенты пропорциональности между
площадью пика1 и количеством выделенного или поглощенного тепла в процессе на¬
гревания. Это количество тепла пропорционально энтальпии реакции и поэтому может
использоваться для количественного определения, если масса пробы известна. Одна¬
ко в методе ДТА простой прямой пересчет площади пиков в единицы энергии, исходя
только из значений А Г как функции времени, невозможен. Действительно, величина
А Г зависит от изменения энтальпии, теплоемкости и полного теплового сопротивле¬
ния потока тепла (R) в конкретный момент времени. Величина R зависит от природы
и массы пробы, метода пробоподготовки (прессование и т. д.) и поверхности контакта
между тиглем и подставкой. Эти переменные зависят от температуры и, следовательно,
должны контролироваться.
Для почв большинство анализов проводят в диапазоне от комнатной температуры до
1200 °С. Если при повышении температуры происходит реакция, наблюдается разница
в температуре между пробой и эталоном (рис. 7.5):
• Если температура пробы ниже, чем температура инертного эталонного материала,
появляется эндотермический пик (величина А Г отрицательная), это происходит
при реакциях дегидратации, дегидроксилирования, плавления, испарения, воз¬
гонки и др.
• Если, наоборот, температура пробы превышает температуру эталона, появляется
экзотермический пик (величина А Г положительная), это происходит при процес¬
сах окисления (сгорание ОВ, окисление сульфидов, окисление двухвалентного же¬
леза), некоторых процессах нуклеации или разложения с новообразованиями.
Регистрируемые различия температур связаны с изменениями энтальпии, но это не
исключает возможности одновременного протекания экзотермических и эндотермиче¬
ских реакций. В этом случае отсутствие или уменьшение пика ДТА не обозначает от¬
сутствия реакции.
Рис. 7.5. Схема кривой дифференциального термического анализа: BD — ширина пика; ЕС —
высота пика; BCD — площадь пика; AF — базовая линия
А
Экзотермический пик
I С I
200 400 600 800
«I
С
1 По оси абсцисс откладывается время. — Примеч. науч. ред.
212
Часть 1. Минералогический анализ почв
В отличие от ТГА, в методе ДТА пики образуются даже при отсутствии потери или
увеличения массы пробы, например, при обратимых переходах второго рода: изменение
удельной теплоемкости, магнитной восприимчивости, точки Кюри или аллотропного1
перехода между а- и р- формами кварца1 2. ТГА и ДТА являются взаимно дополняющими
методами.
Форма, величина и температура пиков зависят от таких инструментальных факторов,
как скорость нагревания, тип держателя образца и термопар.
Малые пробы дают лучшее разрешение пиков и допускают более быстрое нагревание.
Более медленное нагревание может увеличить чувствительность, но в ущерб точности
и разрешению. Динамический состав газовой среды предпочтительней самогенери-
рующейся статической газовой среды. Это дает возможность проводить непрерывное
удаление выделяющихся газов и таким образом уменьшать риск побочных реакций при
повышенных температурах. Анализ выделяющихся газов позволяет определять молеку¬
лярную структуру соединений, выделяющих эти газы.
Широкий ассортимент различных типов оборудования, выпускаемого различными
производителями, позволяет не проводить сложных математических оценок учитывае¬
мых параметров, см., например Duval (1963), Watson и др. (1964), Garrets и Christ (1965),
Allen (1966), Gray (1968), Mackenzie (1970), Brennan (1971), Miller и др. (1973), McNaughton
и Mortimer (1975).
Основные положения дифференциальной сканирующей калориметрии
Другие методы измерения энергии сгруппированы под названием дифференциальной
сканирующей калориметрии (ДСК). Методы ДСК часто плохо определены, так как
в патентах используют один и тот же термин для обозначения различных понятий.
Термин ДСК применяют к приборам, способным измерять удельную теплоемкость
вещества, или теплоемкость образца, и количественно определять энергию реакций
в процессе нагревания. В методе ДСК с компенсацией мощности пробу и эталон не¬
прерывно поддерживают при указанной температуре, используя отдельные нагреватели.
Регистрируемым параметром является количество энергии, потребленное компенсиру¬
ющими сопротивлениями, т. е. величина d(AQ)/dt или dtf/df, выраженная в мкал/с, как
функция температуры (при контролируемом линейном росте температуры).
Когда в пробе протекает реакция, тепловая энергия добавляется или удаляется. Ко¬
личество добавленной или удаленной энергии эквивалентно количеству энергии, выде¬
ленной или поглощенной при данном превращении. Регистрация этого баланса энергии
является калориметрическим измерением энтальпии.
В методе Боерсма (Boersma), также известном под названием ДСК или ДТА с тепло¬
вым потоком, как и в методе ДТА, измеряют величину АГ= Гпроба - 7^^. Сенсоры по¬
мещают под тиглями для уменьшения влияния изменений теплового сопротивления
пробы. Этот тип сборки компонентов оборудования подобен тому, который используют
в другом методе — ДСК теплового потока, регистрирующем величины dAq/dt или, при
использовании термоэлементов, dQnpo6a/df - d£?OTanoH/df = d(AQ)/dt, которые позволяют
проводить количественные измерения энтальпии.
В модулированной ДСК пробу нагревают с линейной скоростью (например,
10 °С/мин), на которую накладывают синусоидальную модуляцию температуры (с пе¬
риодом 30 с и амплитудой 1 °С), что дает циклический профиль нагревания. Таким об¬
1 Полиморфного. — Примеч. науч. ред.
2 Полиморфные переходы относятся к фазовым переходам первого рода. — Примеч. науч. ред.
Глава 7. Термический анализ
213
разом, имеется постоянная скорость, лежащая в основе нагрева, и мгновенная величина
у синусоидальной скорости. Обратное преобразование профиля потока профилем на¬
гревания дает общий тепловой поток, как в традиционном методе ДСК, но к тому же
разделяет поток на два компонента: обратимой теплоемкости и необратимого кинетиче¬
ского тепла. Этот метод позволяет повысить разрешение и чувствительность, потому что
медленная скорость улучшает разрешение, а высокая скорость нагревания способствует
повышению чувствительности.
Рабочая температура приборов ДСК с компенсацией мощности обычно ограниче¬
на 750 °С, что слишком мало для исследования некоторых реакций в образцах почвы.
Классический метод ДСК позволяет достичь температур, близких к используемым в ме¬
тодах ДТА и ТГА.
Материалы
Используют эталонные материалы, измельченные до 0,1 мм в агатовой ступке.
• Вещества с известными температурами плавления для калибровки (табл. 7.4).
• Инертный эталон: окись алюминия (А1203), прокаленная при 1200 °С и измельчен¬
ная до 0,1 мм в агатовой ступке, не проявляет никакого эффекта при нагревании,
за исключением возможных небольших неровностей базовой линии.
• Очищенные глины из набора лабораторных эталонов, гомоионно насыщенные
ионами Mg2+, Са2+ или Na\ Образцы глин из промышленно разрабатываемых оса¬
дочных месторождений должны быть отделены от почвенных глин. 1Таблица 7.4. Калибровочные вещества для термического анализа
Вещество
Формула
Точка плавления, °С
Мм (г/моль)
Нафталин
С10Н8 ОО
80,2
128,16
Фенантрен
ад. сЯз
99,3
178,22
Бензойная кислота
c7h6o2G-C0OH
122,3 (возгонка)
122,12
Хлорид бария
ВаС12- 2Н20
130,0 (—2Н20)
244,31
Сульфат кальция8
CaS04- 2Н20
180,0 (-2Н20)
172,18
Олово
Sn
231,97
118,70
Карбонат кадмия
CdCOj
350,00
172,42
Цинк
Zn
419,4 (возгорание
при контакте с воздухом)
65,38
Бромид лития
LiBr • H20 (расплывается)
553
104,87
Кварц
Si02
573*
60,06
Алюминий
А1
660,2
26,98
Карбонат кальция8
CaC03
850
100,09
(468 кал/г)
(С02 + СаО)
Серебро
Ag
960,8
107,88
"Стандартный материал, используемый для измерения теплоты реакции.
1 Фазовый переход: P-Si02-> a-Si02, — Примеч. науч. ред.
214 Часть 1. Минералогический анализ почв
• Чистые минералы из лабораторного набора эталонов и промышленные эталоны
для анализа.
• Насыщенный при 20 °С водный раствор нитрата магния, содержащий 1250 г/л
Mg(N03)2-6H20.
Оборудование
• Микротигли для ДТА из платины (или, возможно, алунда, кварца, вольфрама, ок¬
сида циркония, никеля или алюминия).
• Эксикатор с насыщенным раствором Mg(N03)2 • 6Н20.
• Эксикатор с осушителем.
• Аналитические весы (10_6—10-8 г) с диапазоном взвешивания 1—100 мг для количе¬
ственного анализа.
• Приборы: промышленные приборы для ДТА — ДСК-анализа разработаны и из¬
готовлены в соответствии с различными концепциями, что осложняет сравнение
полученных результатов. Рекомендуется использовать набор лабораторных этало¬
нов для обеспечения точности и надежности результатов. Оборудование обычно
включает различные компоненты (рис. 7.6 и 7.7):
а)
Ь)
с)
^"проба ^эталон
^проба ^эталон
Сдатчики
__^ww\a
Один нагреватель
Рис. 7.6. Термические системы, использующие измерения энергии: а — классический метод ДТА;
Ь — ДТА метод Боерсма с тепловым потоком или традиционная косвенная ДСК; с — метод ДСК
с компенсацией мощности
Глава 7. Термический анализ
215
Эталон Проба
Рис. 7.7. Оборудование для ДТА
- печь с устройством программирования температуры, контролем газовой среды
и ускоренным охлаждением (от 20 до 40 мин до начала следующего цикла);
- подставка для образца с дифференциальными детекторами температуры;
- пульт управления для записи измерений и программ анализа результатов.
Для керамики необходимы температуры, превышающие 2000 °С. Для почв обычно
достаточна температура 1200 °С, но может понадобиться 1600 °С.
Положения подставки для образца и инертного эталона могут существенно разли¬
чаться (рис. 7.7). Термопары можно размещать внутри или вне тиглей (или даже вварить
в полость тигля). Металлические тигли дают меньшие пики, чем керамические тигли
с более быстрым переносом тепла, снижающим риск деформации пиков. При исследо¬
ваниях до -1000 °С, хромель-алюмелевые термопары генерируют значительную элек¬
тродвижущую силу (ЭДС = 45,16 мВ при 1100 °С), но демонстрируют относительно ма¬
лое химическое сопротивление. С другой стороны, платино-родиевые или платиновые
термопары генерируют ЭДС = 10,74 мВ, которая требует усиления, но они позволяют
поднимать температуру до 1500 °С и химически более устойчивы.
Расположение термопар в пробе и в эталоне должно быть абсолютно симметричным,
чтобы исключить любые изменения формы пиков или смещение базовой линии. Пульт
управления должен включать микрокомпьютер и подсоединенные внешние устройства
и обеспечивать:
• полное управление температурными циклами (скорость нагревания 1-50 °С/мин
со стабилизированным напряжением, температурным контролем при помощи тер¬
мопар, выполнением инструкций и т. д.);
• контроль газовой среды (схемы очистки, приток газа-носителя, инертных или ак¬
тивных газов, вывод АВГ, регулировка потоков и давления);
216
Часть 1. Минералогический анализ почв
• одновременный сбор данных по температуре и переносу тепла с малой инерцией,
термодинамические расчеты и распечатка результатов.
Как и в методе ТГА, необходима стандартизация методик выполнения измерений.
Образец
Используют образец необработанной почвы, измельченной до 0,1 мм (избегать излиш¬
него измельчения). Органическое вещество в перегнойных почвах вступает в сильно
экзотермическую реакцию с максимумом при 300 °С (573 К), которая маскирует эндо¬
термические пики дегидратации при 250—400 °С, если анализ выполняется в воздухе или
окислительной газовой среде. Поэтому лучше разложить ОВ в соответствии с методи¬
ками, описанными в главах 2 и 3 (или экстрагировать его и использовать метод ДТА).
Необработанная почва обычно дает пики малой интенсивности. Кварц может мешать,
но его пик при 573 °С можно использовать в качестве маркера.
Для повышения чувствительности предпочтительно использовать выделенные, очи¬
щенные и обогащенные фракции с определенным размером частиц, гомоионно на¬
сыщенные в случае глин и, после высушивания на воздухе, хранящиеся в присутствии
насыщенного раствора Mg(N03)2 • 6Н20 для поддержания постоянной относительной
влажности (размер и степень кристалличности частиц влияют на температуру, величину
и форму пиков).
Во фракциях песка (2,0-0,05 мм) можно измерить содержание первичных минералов
и конкреций, содержащих много железа и марганца. Обычно наблюдается эндотерми¬
ческий пик кварца при 573 °С.
Фракции пыли (0,05-0,005 мм) часто представляют собой более сложную среду: они
содержат как первичные минералы, так и преобразованные формы и, возможно, формы
глинистых минералов > 2 мкм.
Фракции грубой или тонкой глины (< 2 мкм) часто дают гораздо более интенсивные
и количественно измеряемые пики, чем необработанные почвы. Эти фракции содер¬
жат вещества с организацией ближнего порядка (например, аллофаны, ферригидриты).
Гомоионное насыщение катионом (Na+, К+, Са2+, Mg2+, А13+) позволяет выявлять неко¬
торые свойства, такие как уровни гидратации. Первая эндотермическая реакция испаре¬
ния адсорбированной воды зависит от количества молекул воды, связанной с катионом.
Катионы влияют на величину и форму пиков, обычно без существенного изменения
температуры их появления.
Допускается смешивание глинистой фракции с окисью алюминия, чтобы избежать
спекания, сжатия или растрескивания образца, но это вызывает эффект разбавления.
Две пробы глины из одного и того же профиля можно сравнивать при одинаковых
экспериментальных условиях. В качестве эталонного образца можно использовать одну
из этих проб. Если пробы идентичны, АГможет быть равно 0, и в таком случае получит¬
ся базовая линия без пиков, поскольку одинаковые процессы в одно и то же время будут
происходить в обеих пробах.
Эталон
Эталон должен быть химически инертным в используемом диапазоне температуры. Его
теплопроводность и тепловое рассеяние должны быть близки к соответствующим па¬
раметрам пробы. Размер частиц должен совпадать с размером частиц почвы (измель¬
ченной до 0,1 мм), что обеспечивает тесный контакт со стенками тигля и подходящую
насыпную плотность (одинаковое с измельченной почвой количество пор).
Глава 7. Термический анализ
217
Обычно используют оксид алюминия (А1203, прокаленный при 1200 °С в 1 ч). Такой
эталон можно использовать для нескольких измерений, но после 4 или 5 измерений мо¬
жет наблюдаться некоторая гигроскопичность продукта.
Можно использовать глину, прокаленную при 1200 °С и затем измельченную, но ча¬
сто возникают проблемы с базовой линией. Например, при переходе необработанного
каолина и каолина, прокаленного при 1200 °С до превращения в муллит, теплопровод¬
ность изменится в два раза и могут происходить некоторые обратимые реакции. Иногда
используют в качестве эталонов кварц, окись магния или пластины алюминия.
Методика
Методика аналогична классическому количественному или качественному анализу ме¬
тодом ДТА - ДСК, с учетом вышеупомянутых факторов.
• Взвешивают на лабораторных весах (±0,01 мг) пробу (1-100 мг), параметры ко¬
торой соответствуют характеристикам прибора, и инертный эталон идентичного
качества; для метода ДСК с компенсацией мощности в качестве инертного эталона
можно использовать пустой тигель, аналогичный тому, в котором находится проба.
• Размещают пробу и эталон максимально однородно в платиновых тиглях.
• Находят оптимальную позицию для тиглей (в соответствующих держателях) отно¬
сительно термопар в наиболее гомогенной зоне печи.
• При необходимости накрывают тигель крышкой (может измениться направление
потока выделяемого газа).
• Устанавливают скорость линейного подъема температуры; высокая скорость
может привести к увеличению температуры некоторых пиков, а также повы¬
сить чувствительность (высоту пика); обычно используют скорость в диапазоне
от 5 до 20 °С/мин, но с некоторыми материалами можно работать в диапазоне
0,1—200 °С/мин (а также использовать обратный метод и записывать изменения
при охлаждении пробы).
• Создают газовую среду в печи с учетом возможного анализа газовой фазы при ис¬
пользовании сопряжения с АВГ.
Рабочие условия могут контролироваться соответствующими компьютерными про¬
граммами, включая расчеты и вывод результатов, что позволяет оптимизировать все па¬
раметры.
Интерпретация результатов
Для необработанных образцов и выделенных фракций с различными степенями очист¬
ки используют одну и ту же методику в инертной или активной газовой среде, в зависи¬
мости от природы пробы.
Для построения калибровочных кривых можно использовать «чистые» стандартные
минералы или известные смеси минералов (90:10,80:20 и т. д.), что позволяет проводить
количественное определение образцов (необходим стандартный набор лабораторных
минералов).
Следует различать глинистые минералы осадочных отложений, обычно дающие хо¬
рошо определенные пики, и минералы различных почв после очистки и концентриро¬
вания.
Сначала идентифицируют хорошо определенные пики (например, эндотермический
пик при 500 °С и экзотермический пик около 900 °С для каолинита). Форма пиков ука¬
218
Часть 1. Минералогический анализ почв
зывает на тип теплового процесса: плавление приводит к образованию узкого эндотер¬
мического пика, разложение дает широкий, часто асимметричный, эндотермический
пик, переход второго порядка вызывает только слабый перегиб. Некоторые минералы
дают хорошо определенные пики, используемые в программе термоанализатора в каче¬
стве маркеров характеристических переходов.
Следует учитывать наклон базовой линии и величину пиков. Основные тепловые
переходы, наблюдаемые в глинах, приведены в табл. 7.5. Зона «низких температур»
предоставляет информацию об адсорбированной воде и присутствии некоторых глин
и аморфных веществ. Зона средних температур соответствует пикам, в основном опре¬
деляемым влиянием степени кристалличности. Наконец, зона высоких температур вы¬
являет такие процессы, как нуклеация или перекристаллизация глин.
Отдельные эталоны используют в количественном анализе, например CaS04* 2Н20
используют для расчета в милликалориях на единицу площади (калибровка по соотно¬
шению между площадью пика и известным количеством выделенного или поглощенно¬
го тепла).
Каждый тип структуры можно идентифицировать по характеристической термиче¬
ской кривой (рис. 7.8). Пшны необходимо очищать для уменьшения влияния артефак¬
тов, вызванных примесями, которые могут накладываться в той же зоне температур.
В глинах с тетраэдрическим и октаэдрическим слоями могут быть обнаружены про¬
цессы, связанные с разрушением октаэдрического слоя. Экзотермическое испарение
гигроскопической воды может сопровождаться рекомбинацией элементов тетраэдриче¬
ских и октаэдрических слоев.
Количественный анализ методом ДТА возможен для глин, имеющих эндотермиче¬
ские пики, связанные с потерей воды из кристаллической решетки. Это происходит
в случае глин типа 1:1 (каолинит), но глины типа 2:1 (монтмориллонит, вермикулит или
иллит) трудно определяются этим методом.
Обезжелезивание усиливает экзотермические пики каолинита, а железо подавляет
экзотермические процессы. Следует учитывать присутствие карбоната, органического
вещества и щелочных ионов. Гйдроксиды вначале теряют воду, а затем сжимаются из-за
разрушения кристаллической решетки.
Таблица 7.5. Основные тепловые эффекты почвенных глин
Диапазон температуры, °С
Сигналы
50-250
Эндотермические пики
Потеря адсорбированной воды, межслоевой
воды (например, аллофаны, галлуазит,
смектит)
400-700
Экзотермические и эндотермические пики
Реакции кристаллизации (например, гели
оксидов железа и алюминия)
Диагностические пики кристаллических форм
/
Реакции дегидроксилирования
900-1050
Экзотермические пики
Реакция нуклеации или перекристаллизации
(например, каолинит-хлориты)
Глава 7. Термический анализ
219
«£
ЮЛ
ини
Т (1
1:1)
Диккит(1:1)|
| Накрит (1:1)
Iv'""*
V
S
Бейделлит(2:1)
Г
Монтмориллонит (2:1)
LX 1JLX-1
| Нонтрс
>нит (2
:1)
i ■■
Г-
—
■2
500 1000°Со
Магнезит
МдС03
500
'А
'"ТР
Гипс (CaS04)
Гетит
a-FeOOH
Лепидокрокит
y-FeOOH
1000°С о
Маггемит (y-Fe?On)
500 1000°С
Рис. 7.8. Примеры термических диаграмм ДТА почвенных минералов (Ж. Готейруа, набор стан¬
дартных образцов, Исследовательский институт IRD, Бонди, Франция, неопубликованные данные)
Высоту пиков легче оценить, чем их площадь, и ее можно использовать для прибли¬
женной оценки. Термодинамические расчеты используют всю информацию термограм¬
мы, а также данные о количестве выделившейся воды, измеренном методом ТГА.
В ДСК кривые пропорциональны энтальпии:
Площадь пика - тАН/к,
где ДЯ — теплота перехода; т — реакционная масса; к — калибровочный коэффициент
(не зависящий от температуры при измерении методом ДСК с компенсацией мощно¬
сти).
220
Часть 1. Минералогический анализ почв
7.3. Многокомпонентное оборудование для проведения
термического анализа
7.3.1. Основные положения
Развитие новых приборов было основано на нескольких различных концепциях. В мно¬
гокомпонентном оборудовании несколько специальных приборов подчиняются одному
пульту управления, а измерения компьютеризированы. Этот тип многофункциональ¬
ного оборудования позволяет проводить различные измерения одной и той же пробы,
нагреваемой в условиях контролируемых газовой среды и давления. Использование со¬
ответствующих датчиков позволяет одновременно проводить измерения методами ТГА,
ДТГ, ДТА и ДСК, что сокращает время, необходимое для анализа, увеличивает его точ¬
ность и позволяет более полно охарактеризовать пробу.
Автоматическое введение проб в печь полезно в случае оборудования с охлаждающим
циклом менее 30 мин (возврат к комнатной температуре после нагревало 1700 °С).
Некоторые специальные периферические устройства могут также фиксировать или
измерять количество выделяющихся газов. Такое количественное определение необхо¬
димо для полного анализа кривых ДТА (например, при осуществлении одновременных
экзотермических и эндотермических реакций).
Выбранные методы могут программироваться и применяться последовательно, при
условии надежной системы эталонов, например, для идентификации глин и исследова¬
ния структурных превращений, перекристаллизации, форм воды, термической стабиль¬
ности или термического окисления.
7.3.2. Сочетание термического анализа и анализа выделяемых газов
Анализ выделяющегося и исходящего газа может потребовать совместного использо¬
вания оборудования различной степени сложности. Следует использовать небольшую
печь открытой конфигурации, позволяющей быстрое удаление газов, образующихся при
разложении (рис. 7.9). Циркуляция газа-носителя и возможное присутствие активных
газов приводят к изменению давления и процессов конвекции, связанных с давлени¬
ем и локальными температурами (а также с термомолекулярными силами, способными
создавать неоднородное температурное поле внутри печи), которые должны контроли¬
роваться системой экранов. Ниже приводится краткий обзор наиболее распространен¬
ных систем.
Простая регистрация выделяющегося газа (РВГ): нейтральный газ-носитель уносит
выделяющийся газ в детектор по теплопроводности (катарометр); выходящий сигнал
пропорционален концентрации выделяющегося газа в газе-носителе. Природу выде¬
ляющегося газа не определяют. Можно улавливать тяжелые продукты жидким азотом
и подвергать их дальнейшему анализу, например, контролируемому пиролизу путем тер¬
мического анализа.
Анализ выделяющегося газа (АВГ) можно осуществлять, соединяя оборудование раз¬
личной сложности. Газовый хроматограф, соединенный с оборудованием ТГА-ДТА, по¬
зволяет характеризовать выделяющийся газ при автоматическом непрерывном введении
(можно также улавливать фракции, которые не анализируются в жидком азоте). Инте¬
грирование пиков позволяет количественно определять выделившиеся и разделенные
газы как функции их времен удержания.
Подсоединение ИК-Фурье-спектрометра дает возможность анализировать време¬
на пролета благодаря скорости захвата и чувствительности спектрометра. Поток азота
Глава 7. Термический анализ
221
а)
Весы
Ь)
Весы
с)
Вывод ^ Активный газ
Весы
Нейтральный 4
газ
1
1
1
I 1 | Нейтральный 4
ш газ
1
1
1
I * | Нейтральный 4"
LU газ
1
1
1
Печь ^
й
| Печь |
й
Hh
Ловушка
с жидким
Печь ^
й
А
азотом \
у
Ч*
Связь с гель-
проникающим Вакуум
хроматографом д ◄—v
илиИК-Фурье- Активныи|
спектрометром газ
ИГ
спектрометром
Рис. 7.9. Примеры газоотводящих устройств для методов ТГА и ДТА — ТГА: а — простое вытесне¬
ние нейтрального газа, проходящего весы и защищающего их от коррозии; b — система с двумя
газами (нейтральным и активным) с горизонтальным отводом от пробы; с — система с вакуумной
очисткой с последующим отводом нейтрального газа или смеси нейтрального и активного газа
автоматически переносит выделяющиеся газы в спектрометр. Разрешение прибора -
около 4 см-1. Каждая точка температуры сохраняется в файле с номером, соответствую¬
щем значению выбранной длины волны в этой точке. Воду идентифицируют при 3600
и 1600 см-1, СО и С02 — при 2000 и 2400 см_!.
Сопряжение с масс-спектрометром является мощным методом, но трудным в испол¬
нении, поскольку масс-спектрограф работает в высоком вакууме (~10~5-10~6мбар). Со¬
пряжение с термоанализатором достигается при помощи двустадийных сепараторов.
Квадрупольный масс-спектрометр позволяет проводить быстрое сканирование выде¬
ляющегося газа в массовом диапазоне примерно до 100, включая |2С, 16СН4,28СО, *COv
|8НОН, 1бО, 3202, 34H2S, 64S02, 80SO3. Этот диапазон недостаточен для наблюдения орга¬
нических веществ, образующихся при контролируемом пиролизе (см. гл. 12), так как
молекулярные массы распределены в более широком диапазоне, до 500 и более.
На рис. 7.10 показаны результаты анализов термического разложения оксалата каль¬
ция, который можно использовать в качестве калибровочного стандарта. При исполь¬
зовании сопряженного масс-спектрометра с относительно низкой скоростью захвата
и воздушной атмосферой реакция разложения (2), которая приводит к образованию
карбоната кальция и монооксида углерода, дает только один пик с массой 44 (С02).
В среде азота наблюдается образование смеси СО + С02, что указывает на начало разло¬
жения новообразованного карбоната. Можно проводить одновременную регистрацию
СО и С02 при сопряжении термоанализатора с ИК-Фурье-спектрометром.
Анализ радиоактивных газов, выделяющихся1 из пород в твердом состоянии при раз¬
личных температурах, может быть выполнен сочетанием термического анализа с реги¬
страцией а-частиц. 11 Эманационный метод. — Примеч, науч. ред.
222
Часть 1. Минералогический анализ почв
Рис. 7.10. Разложение эталона оксалата кальция (СаС204 • Н20), изучаемое комбинацией
методов ТГА, ДТГ и ДТА (вверху) и масс-спектроскопии (внизу), газ-носитель — воздух, скорость
нагрева 5 °С/мин, масса пробы 50 мг
1. СаС204 • Н20 -)• СаС204 + H2Ot (12,2% Н20)
2. СаС204 -> СаСОэ + COt (19,1% СО)
3. СаС03 -> CaO + C02t (30,1 % C02)
Потеря массы
223
Глава 7. Термический анализ
Использованная литература
Allen JA (1966) Energy Changes in Chemistry., Allyn-Bacon Newton, MA.
Braudeau E (1987) Mesure automatique de la r&raction dtechantillons de sol non remantes. Science du
Sol, 25,85-93.
Braudeau E (1988) Equation g£n£ralis6e des courbes de retrait dtechantillons de sols structures. Comptes
rendus acad. Sci. Fr., s£rie 2, 307, 1731-1734.
Braudeau E, Costantini Ш, Bellier G and Colleuille H (1999) New device and method for soil shrinkage
curve measurement and characterization. Soil Sci. Soc. Am. J., 63, 525-535.
Braudeau E, Frangi JP and Mothar RH (2004) Characterizing non-rigid dual porosity structured soil me¬
dium using its characteristic shrinkage curve. Soil Sci. Soc. Am. J., 68, 359-370.
Duval C (1963) Inorganic Thermogravimetric Analysis., Elsevier. Amsterdam. 2eme edition.
Garrels RM and Christ CL (1965) Solutions, Minerals and Equilibra., Harper-Row New York.
Gray AP (1968) Symposium Analytical Chlorimetry, Porter R.S. and Johnson JF ed., Proc. Am. Chem.
Soc., Plenum New York, 209.
Kotra RK, Gibson EK and Urbancic MA (1982) Icarus., 51, 593.
Mackensie RC (1970) Differential Thermal Analysis. Academic London. Vol. 1.
McNaughton JL and Mortimer CT (1975) La calorimStriedifferentielleabalayage.Btttteniwf/*, Physical
Chemistry., S6rie 2, 10. 43 pages.
Miller B, Giuham JM, Brennan WP, Nentzer C and Whitwell JC (1973) Thermochim.Acta, 5,257.
Pansu M, Gautheyrou J and Loyer JY (2001) - Soil analysis - sampling, Instrumentation and Quality
Control, Balkema Amsterdam, Lisse,Abington, Exton, Tokyo, 512 p.
Rouquerol J (1970) L’analyse thermique de decomposition construite. J. Therm.Anal., 2, 123-140.
Rouquerol J (1989) Reciprocal thermal analysis.The hidden face of the thermalanalysis. Thermodyn.Acta.
Rouquerol F, Rouquerol J, Thevand G and Triaca M. (1985) Desorption of chemisorbed species: its study
by controlled rate thermal analysis. Surf Sci., 239-244.
Rouquerol J, Rouquerol F, Grillet Y and Ward RJ (1988) A critical assessmentof quasi equilibrium as
adsorption techniques in volumetry gravimetry, calorimetry. In Characterization of Porous Solid,
Kunger KK ed., Elsevier Amsterdam, 67-75.
Watson ES, O’neill MJ, Justin J and Brenner N (1964) DSC. Anal. Chem., 36,1233.
Дополнительная литература
Bang DV and Atanasov I (1984) Thermal characteristics of the clay fraction ofsoils overlying zeolites.
Pochvoznanie i Agrokhimiya, 19, 58-65.
Barshad I (1965) Thermal analysis techniques for mineral identification andmineralogical composition. In
Methods of Soil Analysis, Part 1., Black C.A. ed., A.S.A., S.S.S.A., 9, 699-742.
Bishop JL, Banin A, Mancinelli R and Klovstad MR (2002) Detection of solubleand fixed NH4+ in clay
minerals by DTA and IR reflectancespectroscopy: a potential tool for planetary surface exploration.
Planetary Space Sci., 50, 11-19.
Brennan WP (1974) Application of differential scanning calorimetry for thestudy of phase transitions. In
Analytical Calorimetry, Porter RS and Johnson JF ed., Plenum New York.
Caillere S and Henin S (1963) Mineralogie des argiles., Masson Paris.
Daniels T (1973) Thermal Analysis., Kogan Page.
Dunn JG (1980) L’analyse thermique, une technique de centrale de qualitS dansles industries de l’argile,
des c£ramiques et des verres. Silicateslndustriels, 10, 203.
Duval C (1953) Inorganic Thermogravimetric Analysis., Elsevier Amsterdam. 1st edition.
Earnest CM (1983) Thermal analysis of Hectorite. Part II. Differential thermalanalysis.Thermochim.
Acta., 63, 291-306.
Emmerich WD and Kaisers Berger E (1979) Simultaneous TG-DTA mass-spectrometry to 1550 °C.
J. Therm. Anal., 17, 197-212.
FerencPaulik (1995) Special Trends in Thermal Analysis., Wiley New York, 478 p.
Fordham CJ and Smalley IJ (1983) High resolution derivative thermogravimetryof sensitive clays .Clay
Sci, 6, 73-79.
224
Часть 1. Минералогический анализ почв
Gallagher РК (Ed.) (1998) Handbook of Thermal Analysis and Calorimetry.,Elsevier.
Gam PD (1965) Thermo Analytical Methods of Investigation., Academic London.
Giovannini G and Lucchesi S (1984) Differential thermal analysis and infra-redinvestigations on soil
hydrophobic substances. Soil Sci., 137,457-463.
Hatakeyama T and ZhenhaiLui (Ed.), (1998) Handbook of Thermal Analysis., Wiley New York.
Keyser de WL (1953) Differential thermobalance: a new research tool. Nature, 172, 364.
Khanafer К and Vafai К (2002) Thermal analysis of buried land mines over adiumal cycle. IEEE Trans.
Geosci. Remote Sensing, 40, 461-473.
Lombardi G (1984) Thermal analysis in the investigation of zeolitized andalteradvolcanics of Latium,
Italy. Clay Miner., 19, 789-801.
Mackenzie RC (1963) SCIFAX, Differential Thermal Analysis Data Index.,Cleaver-Hume Press.
Mackenzie RC, Keattoh CJ, Dollimore D, Forester JA, Hodgson AA and Redfem JP (1972) Nomencla¬
ture in thermal analysis II. Talanta, 19, 1079-1081.
Mackenzie RC and Caillere S (1975) The thermal characteristics of soil mineralsand the use of these
characteristics in the qualitative and quantitative determination of clay minerals in soils. In Inorganic
components,Gieseking E. ed. vol. 2, Springer Berlin 529-571.
Madkensie RC and Robertson HS (1961) The quantitative determination of halloysite, goethite and gibb-
site. Acta Univ. Caral. Geol. SuppL, 1, 139.
Maizenberg MC, Karpachevski LO, Markovich MN and Kurakof VN (1991) The use of differential scan¬
ning calorimetry in soil studies. MoscowUniv. Soil Sci. Bull., 46, 31-34.
Muller F, Drits V, Plancon A and Robert JLL (2000) Structural transformationof 2:1 dioctahedral layer
silicates during dehydroxylation-rehydroxylation reactions. Clays Clay Miner., 48, 572-585.
Murphy CB (1962) Differential thermal analysis. A review of fundamental developments in analysis Anal.
Chem., 298-301.
Paulik F, Paulik J and Arnold M (1984) Simultaneous TG, DTG, DTA and EGA technique for the deter¬
mination of carbonate, sulphate, pyrite andorganic materials in minerals, soils and rocks. II - Op¬
eration of thethermo-gas-titrimetric device (TGT) and examination procedure. J.Therm. Anal., 29,
333-344.
Poerschmann J, Gorecki T and Parsi Z (2005) Analytical non-discriminatin gpyrolysis in soil analysis.
LabPlus Int., 19,8-14.
Sarikaya Y, Onal M, Baran В and Alemdaroglu T (2000) The effect of thermal treatment on some of the
physicochemical properties of a bentonite. Clays Clay Miner., 48, 557-562.
Schnitzer M and Kodama H (1972) Differential thermal analysis of metal-fulvicacid salts and complexes.
Geoderma, 1, 93-103.
Schomburg J (1991) Thermal reactions of clay minerals: their significance as“archaeological thermom¬
eters” in ancient potteries. Appl. Clay Sci., 6,215-220.
Schultze D (1969) Differential Thermo-Analysis., Verlag.
Smothers WJ and Chiang YZO (1966) Handbook of Differential ThermalAnalysis., Chem. Publ. Co.
Smykatz-Kloss W and Heide К (1988) Progress of thermal analysis in earth Sciences. J. Therm. Anal,
(GBR) 33, 1253-1257.
Taichev T, Konishev P and Donov D (1984) Application of thermal analysis instudies on the structural
evolution of humic substances.Pochvoznanie i Agrokhimiya, 19, 82-87.
Tan KH and Clark FE (1969) Polysaccharide constituents in fulvic and humicacids extracted from soil.
Geoderma, 2,245-255.
Tan KH, Hajek BF and Barshad I (1986) Thermal analysis techniques. In Methods of soil analysis,
Klute A. ed., A. S.A., S.S.S.A., 9,151-183.
Trofimov SA, Tolpeshta II, Sokolova ТА (1999) A study of plant material and organic soil horizons using
thermal analysis techniques. Pochvovedenie, 54, 3-9.
Van Olphen H and Fripiat JJ (1979) Data Handbook for Clay Minerals and Other Non-Metallic Minerals.,
Pergamon New York, 350 pages.
Wendlandt WW (1974) Thermal Methods of Analysis, Part 1. Wiley New York.
Глава 8. Микроскопический анализ
8.1. Введение
Изучение процессов образования и эрозии почв и характеризация минералов требуют
проведения наблюдений в различных пространственных и временных масштабах. Пе-
доны, горизонты, макро- и микропробы используют как основные объекты для ряда
методов исследования, в которых микроскопические методы позволяют проводить на¬
блюдения поверхностей раздела различных фаз и даже последних стадий образования
молекулярных агрегатов. Дополнительные аналитические методы способствуют опре¬
делению, в частности, химической природы (например, энергодисперсионный (ЭД)
или волнодисперсионный (ВД) рентгеноспектральные методы микроанализа (РСМА))
и структурной организации (электронная микродифракция) этих фаз.
В то время как «мокрый» химический анализ предоставляет информацию об общих
изменениях образца, микроскопический анализ позволяет проводить идентификацию
сверхтонкой химической гетерогенности и способствует пониманию механизмов пре¬
вращений. С помощью микроскопического анализа можно получить качественную
и количественную информацию о строении индивидуальных частиц, например, форме,
присутствии склеивающих материалов, включений, пор.
Детальный морфологический анализ (пространственных соотношений различных
фаз, преимущественной ориентации, и т. д.) выполняют, комбинируя микроскоп с ана¬
лизатором изображения или структуры и анализируя шлифы для выявления картогра¬
фии химической сегрегации, микропрофилей.
Кристаллическую структуру можно установить при изменении масштаба наблюде¬
ния: исследования элементарных ячеек глин, регулярных последовательностей силика¬
тов в пакетах, модификаций базальной прослойки, проявлений апериодичности, дефек¬
тов, микротрещин, ступеней роста, гелей с атомным упорядочением ближнего порядка
вблизи зародышей кристаллов позволяют получить информацию о превращениях од¬
ного минерала в другой на наноструктурном уровне. Метод электронной микродифрак¬
ции, который можно применять на большинстве электронных микроскопов, позволяет
идентифицировать кристаллические структуры. Все эти возможности делают микроско¬
пию чрезвычайно мощным инструментом.
Оптическая и электронная микроскопия с дополнительными внешними устройства¬
ми для определения распределений химических элементов в образце широко исполь¬
зуются в почвоведении, минералогии, кристаллографии, петрографии, металлографии
и геологии глин и предоставляют точную информацию о механических свойствах, лока¬
лизации поверхностных зарядов и обменных свойствах почв.
Однако чрезвычайно важно оценивать надежность наблюдений и приводить масштаб
измерений в соответствие со свойствами конкретного образца. Следует точно опреде¬
лять условия пробоотбора и различных стадий пробоподготовки. Необходимо учиты¬
вать и погодные условия: образцы почв, содержащих много глин типа 2:1, отобранные
в сухой или влажный сезон, обладают различной порозностью. Следует использовать
данные не только по почвообразованию и геостатистике, но также по геоморфологии
(может понадобиться дополнительный пробоотбор для исследования изменений про¬
филя или отбор проб в больших масштабах для оценки гомогенности зоны). При про¬
ведении наблюдений следует помнить о физических и химических процессах, которые
226
Часть 1. Минералогический анализ почв
могут изменить пробу и привести к ошибочным результатам (например, загрязнение,
окисление, новообразование). Также важно полностью использовать все технические
возможности оборудования, чего часто не происходит.
Электронный пучок может повредить поверхность материалов. Проникновение из¬
лучения на глубину может быть ограничено близкими слоями поверхности, т. е. пример¬
но 1-3 нм, или, наоборот, может постепенно эродировать поверхность в соответствии
с концентрационным профилем (например, при плазменной бомбардировке, вторично¬
ионной масс-спектрометрии).
Так же как и для других методов, необходима градуировка оборудования, которая
требует применения эталонного набора наблюдений образцов, что позволяет проводить
сравнение с известными и четко идентифицированными случаями.
8.2. Подготовка образцов
8.2.1. Способы подготовки образцов
Визуальный осмотр с последующим исследованием под увеличительным стеклом не¬
достаточен, так как он дает только приблизительную оценку способов формирования
агрегатов и об их физических свойствах. Поэтому данный тип исследования всегда со¬
провождается тактильным и другими сенсорными и химическими анализами (Рати
идр.,2001).
Наблюдения с помощью оптического или электронного микроскопа проводят на об¬
разцах почвы, подвергнутых следующим обработкам:
• очистка, концентрирование, количественное разделение на классы по размеру ча¬
стиц;
• разделение в тяжелых жидкостях с увеличивающейся плотностью вплоть до d = 4,28
при 20 °С (например, бромоформ, тетрабромметан, дийодметан, раствор Клеричи);
• магнитное разделение в магнитном сепараторе Франца (или подобном).
В этом случае исследуют индивидуальные частицы (форму, природу, относительные
содержания минералов, влияние и природу эрозии). Для получения точных данных об
агрегатах готовят шлифы на стекле, что позволяет изучать превращения в почвах и свя¬
зать полученные данные с результатами полевых наблюдений.
Чаще всего используют петрографические стекла размером 28x48 мм. Для микропе-
дологического и микроморфологического анализа можно применять стекла Маммота
размером 110x76 мм или даже 200x180 мм (однако это очень сложные операции).
8.2.2. Нанесение покрытий и пропитывание тонких пластин
Основные положения
Термин «покрытие» обычно используют для массивных компактных проб с простой гео¬
метрией, таких как куски камня, которые можно заделать в быстро твердеющую смолу,
что ускоряет и облегчает подготовку к анализу.
Пропитка смолой используется для укрепления мягких пористых пород, продуктов
разрушения пород и почв (Delvigne, 1988), для которых необходимо сцепление. Влажные
почвы следует высушить во избежание структурных изменений образца из-за сжатия
или появления усадочных трещин. Одним из способов уменьшения этого явления в по¬
чвах с большим содержанием глин типа 2:1 и низкой порозностыо является насыщение
образца хлоридом натрия перед высушиванием методом лиофилизации.
227
Глава 8. Микроскопический анализ
В почвах, обогащенных аллофанами, с водоудерживающей емкостью до 250% от мас¬
сы сухой почвы вода может быть постепенно замещена ацетоном или диоксаном. Этот
метод применим и к глинистым почвам, но образцы, обработанные ацетоном, более
хрупки, чем образцы, обработанные диоксаном (Tessier, 1985). Использование ацетона
и диоксана можно совмещать с некоторыми смолами, используемыми для пропитки.
Лиофильная сушка обычно сохраняет особенности образца лучше, чем высушивание
на воздухе или в печи. Для малых образцов (примерно 30 см3) прекрасные результаты
дает использование аппаратов для сушки при критической температуре С02. Для объ¬
емных влажных образцов единственным решением является использование водораство¬
римых эпоксидных смол (Moran и др., 1989).
Приготовление шлифов вручную является очень длительным процессом, требующим
значительного мастерства для получения качественных шлифов с требуемой степенью
прозрачности и одинаковой толщиной в диапазоне от 20 до 30 мкм. Процессы нане¬
сения покрытия и пропитки, нарезания и полировки можно автоматизировать, но это
требует больших первоначальных вложений, которые могут окупиться при выполнении
не менее 60 покрытий или 40 пропиток в неделю, т. е. примерно 2000 шлифов размером
28x48 мм в год. Некоторые модульные компьютерные системы приспособлены для ав¬
томатического производства сверхтонких шлифов толщиной 10 мкм при глубине по¬
лировке 0,25 мкм. Стандартные шлифы имеют толщину 30 мкм с глубиной полировки
примерно 1 мкм (рис. 8.1).
Из-за необходимости использования взрывобезопасного электрического оборудова¬
ния, систем для удаления паров растворителей ловушками, содержащими растворители,
а также стоимости технического обслуживания оборудования и нестабильности смол
часто предпочтительнее готовить шлифы в специально оборудованной лаборатории.
Оборудование
• Лаборатория, оборудованная вытяжным шкафом (с пожаробезопасными электри¬
ческими приборами).
• Комплексная система для шлифования и полировки с диском 250 мм в диаметре.
• Отрезной станок с алмазным шлифовальным кругом 350 мм в диаметре.
• Оборудование для холодной пропитки и вакуумного покрытия: эксикатор 300 мм
в диаметре с вакуумным краном и приспособлением для подачи смеси смолы с ка¬
тализатором.
• Вакуумный насос с манометром (0-750 мм ртутного столба).
• Ультразвуковая баня.
• Сушильный шкаф с вентиляцией (нагрев до 100 °С).
• Гравировальный инструмент с наконечником из карбида вольфрама.
• Петрографические стеклянные пластины размером 28x48 мм и 1,2 мм толщиной.
• Стеклянные пластины толщиной 0,13 мм для защиты шлифов.
• Коробки для хранения шлифов.
• Ультрамикротом с терморегулируемой системой перемещения.
• Лиофилизатор.
• Вакуумные эксикаторы с осушителями, например, силикагелем.
• Бинокулярный микроскоп.
• Мелкое лабораторное оборудование (стеклянные пластины, алюминиевые листы,
пинцеты).
• Холодильник (для хранения смол).
228
Часть 1. Минералогический анализ почв
а)
.„8 Распил
•*мм
а Образец ненарушенной
почвы
Предварительное горячее покрытие
Холодная пропитка
Стеклянная
ааг„
Приклеивание
образца к стеклу
0±5 мм
разрезание Предварительное
полирование
на глубину 80 мкм
Шлифование
карбидом бора
на глубину 30 мкм
Непокрытый шлиф
i
Конец полировки
до 1 или 0,25 мкм
Толщина 20 мкм +
5 мкм адгезива.
Ь)
Рис. 8.1. а — приготовление тонких шлифов (оборудование и материалы: смолы, пресс, покры¬
тие, установка для УФ-полимеризации, шлифовальный круг, отрезной станок, шлифовально-по¬
лировальный станок); Ъ — управляемая компьютером установка для автоматической модульной
подготовки проб (Struers)
Материалы и реагенты
• Шлифовальные листы с карбидом кремния (карборунд SiC), твердость по Моосу 9.
• Шлифовальные листы с карбидом бора (В4С), твердость по Моосу 9.
• Шлифовальные листы с корундом (оксид алюминия А1203), твердость по Моосу 9.
• Алмазные шлифовальные листы (набор размеров частиц).
• Ткань для полировки.
• Алмазные пасты, растворимые в смеси вода — этанол, и аэрозоли: 3,1,0,25 мкм.
• Жидкие эпоксидные смолы + отвердитель + катализатор + растворитель.
• Акриловые смолы с малой скоростью усадки.
• Жидкие полиэфирные смолы.
• Краситель для смолы.
229
Глава 8. Микроскопический анализ
• Канадский бальзам (+ ксилол).
• Смазочные материалы и растворители: глицерин НОСН2-СН(ОН)СН2ОН, масло,
этанол, о-ксилол С6Н4(СН3)2, стирол С6Н5СН=СН2,1,4-диоксан, ацетон, 99%-ный
диэтилентриамин H2N-CH2CH2NH-CH2CH2NH2.
• Силиконовая смазка для шлифования.
Выбор смолы зависит от следующих факторов:
• показатель преломления (около 1,54);
• растворимость в органическом растворителе или воде;
• низкая степень сжатия и низкая вязкость;
• условия полимеризации;
• твердость и прочность при требуемой температуре, особенно при наблюдениях
и количественном определении методом электронной микроскопии (СЭМ или
ЭД-РСМА).
Методика
Плотные твердые образцы (породы)
Петрографические методы подобно описаны Хартшорном и Стьюартом (Hartshome
и Stuart, 1970).
Образец ориентируют соответствующим образом и распиливают по одной грани, за¬
тем постепенно полируют и, наконец, приклеивают на петрографическое стекло разме¬
ром 28x48 мм (1 х2 дюйма). После отпиливания параллельного шлифа толщиной 2—3 мм
уменьшают его толщину до 30 мкм. Полируют шлиф на глубину 1 мкм. Шлиф можно
защитить от царапин и окисления, приклеив на тонкое стекло при помощи канадско¬
го бальзама. Шлиф не следует накрывать, если предполагается применение красителей,
химикатов (удаление карбонатов) или образцы будут подвергаться исследованию мето¬
дами СЭМ — ЭД-РСМА.
Хрупкие, пористые или трещиноватые образцы
Образцы, отобранные в поле при естественной влажности методами вдавливания ци¬
линдра или отбора монолита (Рати и др., 2001), часто ориентированы вертикально или
в соответствии с наклоном поверхности земли (рис. 8.2). Они должны храниться в гер¬
метичных коробках и затем использоваться для приготовления стандартных шлифов
размером 28x48 мм или шлифов Маммона.
Всякий раз, когда в процессе подготовки размеры блока уменьшают, необходимо
учитывать направление поверхности шлифа (рис. 8.2). Образцы сушат методом лиофи-
лизации или замещением воды ацетоном или диоксаном. Небольшое сжатие позволит
вынуть образец из формы, если не используется система пробоотбора с двумя полуци¬
линдрами.
Блок следует обрёзать примерно до формы квадрата, чтобы уменьшить его толщину,
и поместить, с учетом выбранной ориентации, в полужесткую пластиковую форму (на
практике дно формы соответствует дну профиля).
Пропитка
Блок почвы помещают вместе с формой в вакуумный эксикатор с притертой крышкой,
смазанной силиконовой смазкой. Откачивают эксикатор примерно до 60 см ртутного
столба (или больше) в зависимости от температуры кипения растворителя и смолы. По¬
чву дегазируют в течение 30 мин.
230
Часть 1. Минералогический анализ почв
Верх
В эластичном пластиковом контейнере быстро смешивают смолу с отвердителем
(в соотношении, рекомендованном производителем) и равным объемом растворителя
для разжижения смеси. Заливают смесь через воронку в крышке эксикатора. Смесь вво¬
дится постепенно, при постоянном контролировании вакуума, входящего потока и ро¬
ста уровня впитываемой жидкости до тех пор, пока образец не покроется полностью
(плюс один сантиметр для последующего удаления смолы). Смесь также должна плавать
вокруг образца, чтобы происходило восходящее впитывание по капиллярам. В зависи¬
мости от порозности и размера образца обычно необходимо 5-8 ч для последующего
приготовления шлифов размером 28x48 мм.
Осторожно снимают вакуум и переносят образец в вытяжной шкаф. Растворитель
испаряется примерно в течение недели. Продукт становится все более вязким и затем
начинает твердеть (этот процесс занимает примерно месяц, в зависимости от смолы
и степени разбавления). Помещают его в вентилируемый сушильный шкаф при 45 °С
на 2-3 сут, пока образец не перестанет липнуть. Вынимают его из формы с учетом ори¬
ентации.
Разрезание
Помещают блок на слой окрашенной быстро твердеющей смолы (толщиной пример¬
но 2 мм) для возможности определить дно профиля почвы. Распиливают затвердевший
пропитанный блок в соответствии с преимущественной ориентацией (на боку блока
можно сделать небольшую зарубку для указания направления уклона поверхности).
Циркулярная пила должна быть смазана растворителем, который не растворяет соли,
соответствует смоле и не будет мешать последующим химическим анализам. По окон¬
чании пропитывания разрезают блок на параллельные пластины толщиной 5-6 мм.
Каждая пластина должна соответствовать формату петрографических шлифов. Быстро
проверяют качество поверхности на такие дефекты впитывания, как трещины или пло¬
скостность. Может потребоваться дополнительная пропитка поверхности после очистки
спиртом и сжатым воздухом. При помощи шпателя покрывают пластину смесью смолы
с катализатором и помещают ее на плоский стеклянный блок, накрывают тонкой алю¬
миниевой фольгой, прижимая ее для исключения воздушных пузырьков. После загусте¬
ния открывают образец, обрезают его при помощи скальпеля и оставляют затвердевать
в вентилируемом сушильном шкафу при 45 °С. После этого шлиф готов для полировки.
231
Глава 8. Микроскопический анализ
При помощи грубого абразивного диска из карбида кремния полируют до почти пол¬
ного истирания повторного слоя пропитки, затем, после очистки струей сжатого возду¬
ха, продолжают полировать алмазными абразивными пастами последовательно увели¬
чивающейся гладкости (6,3 и 1 мкм). Поверхность должна быть абсолютно гладкой, без
царапин и остатков пленки повторной пропитки. Материал должен иметь тусклый вид
на фоне полированной смолы.
Приклеивание образца на предметное стекло размером 48x28 мм
Очищают стекло растворителем и высушивают. Наносят тушью номер. Смешивают
смолу для приклеивания образца с отвердителем (при 60 °С или меньшей, в зависимо¬
сти от природы образца и смолы). При помощи шпателя наносят смолу на одну по¬
верхность подложки и полированную поверхность образца, очень осторожно сжима¬
ют намазанные поверхности, медленно вращая относительно друг друга для удаления
избытка склеивающего вещества и воздушных пузырьков. Оставляют для отвердения
и обрезают края. Через 48 ч шлифуют внешнюю поверхность образца, спиливая его до
толщины примерно 300 мкм и затем полируя набором алмазных порошков с уменьша¬
ющимся размером частиц примерно до 30 мкм (обычная толщина тонких шлифов для
оптических наблюдений вызывает оранжевое двойное лучепреломление в кварце). При
каждой смене размера частиц алмазной пасты тщательно очищают шлиф для удаления
всех крупных частиц, которые могут поцарапать его. Если образец содержит много же¬
лезистых минералов, толщина шлифа в 30 мкм слишком велика для наблюдений, и не¬
обходимо уменьшить ее примерно до 20 мкм, чтобы шлиф стал достаточно прозрачным.
Для проб, предназначенных для сканирующей электронной микроскопии или ЭД-
РСМА, заключительное полирование проводят алмазной пастой с размером частиц
0,25 мкм. Для ПЭМ высокого разрешения стеклянную подложку удаляют и в отдель¬
ных микрозонах (диаметром 3 мм) уменьшают толщину, затем их разделяют и вырезают
скальпелем. Для утоньшения используют аргоновую пушку. Для резки минералов, ори¬
ентированных параллельно плоскости стекла, можно использовать микротом. Некото¬
рую информацию можно получить, наблюдая микродифракцию, но для очень тонких
шлифов интерпретация часто затруднена.
Отделение микрочастиц размером около 50-100 мкм на стеклах возможно проводить
с помощью ультразвукового зонда, оборудованного углеродной иглой на микромани¬
пуляторе; шлиф наблюдают через реверсивный оптический микроскоп. Микрочастицы
располагают на кремниевой пластине и анализируют методом рентгеновской дифрак¬
ции с медленным сканированием. Получающиеся дифрактограммы можно сравнивать
по угловым расстояниям и стандартным интенсивностям.
8.2.3. Сетки и реплики для просвечивающей электронной микроскопии
Основные положения
Если образцы тоньше 1 мкм или собраны на очень тонких проводящих пленках, то их
можно исследовать только методом просвечивающей электронной микроскопии. Объ¬
ект, подвергаемый бомбардировке электронами, должен пересекаться пучком электро¬
нов. Приемлемая толщина образца зависит от энергии этого пучка, т. е. проникновения
примерно на 0,2 мкм при энергии 50 кВ и в 3—4 раза больше — при энергии 100 кВ.
Взаимодействие пучка электронов с веществом нагревает образец-мишень, что может
привести к испарению поровой воды; в этом случае морфология галлуазитов серьезно
нарушается, а также загрязняется электронная пушка микроскопа. В случае элементов
232
Часть 1. Минералогический анализ почв
с большими атомными номерами нагревание может быть очень сильным (и приводить
к плавлению, сдвигу фаз, возгонке, разрушению поддерживающей пленки).
Поддерживающая пленка должна быть прозрачной для электронов, достаточно проч¬
ной для поддержания образца и устойчивой к нагреванию и электростатическим заря¬
дам; она не должна быть толще, чем 100 А.
Реагенты
• Поливинилформаль (формвар, /ij° = 1,50).
• Дихлорэтан, С1-СН2-СН2-С1.
• 0,15%-ный раствор формвара в дихлорэтане.
• Коллодий (нитрат целлюлозы или пироксилен C12H16N4018).
• Амилацетат (СН3—С02—С5НП) или бутилацетат (СН3С02(СН2)3СН3).
• 1 %-ный раствор коллодия в амилацетате.
• 1 %-ный раствор фтористоводородной кислоты в воде.
Оборудование
• Стеклянная лабораторная посуда.
• Медные (или никелевые) сетки диаметром 3,05 мм (рис. 8.3); также существуют
сетки, покрытые формвар-углеродной пленкой или перфорированной формвар-
углеродно-золотой пленкой, позволяющей проводить прямые наблюдения микро¬
частиц.
110-миллиметровые немагнитные стальные пинцеты с ультратонкими кончиками.
Пластиковая пленка для реплик 0,034 мм.
Микросушильный шкаф для плавления реплик при 45 °С.
Петля для отбора проб.
Рис. 8.3. Сетка для просвечивающей электронной микроскопии с диаметром ячеек 3 мм
(322 меш); номера — расположение абсциссы х\ буквы — расположение ординаты у
Методика
Сетки
• Необходимое количество 3-миллиметровых сеток кладут на стеклянную пласти¬
ну, предварительно увлажненную водой. Пластину помещают в пробирную чашку
с водой и очень медленно затопляют ее; сетка не должна плавать на поверхности.
Глава 8. Микроскопический анализ
233
• Добавляют каплю раствора формвара в дихлорэтане на поверхность воды; капля
должна образовать очень тонкую пленку (< 100 А) без складок или дыр, которая
затвердевает на воздухе после испарения растворителя (пленка будет тоньше, если
температура воды близка к 0 °С); медленно удаляют жидкость, чтобы пленка кос¬
нулась пластины и сетки, которую она покроет; дают высохнуть и затем напыля¬
ют на сетку углерод, который армирует пленку и делает ее токопроводящей; когда
сетка становится серой, она готова к использованию; проверяют ее качество под
бинокулярным микроскопом.
• Разбавляют каплю суспензии образца в воде до получения почти прозрачной жид¬
кости; обрабатывают ультразвуком для разделения частиц; наносят одну микро¬
каплю в центр сетки1 (сетка не должна переворачиваться во время операции);
оставляют сохнуть на воздухе при комнатной температуре; и если необходимо, вы¬
сушивают при критической температуре С02.
Реплики
Образцы, которые изменяют форму при высыхании, слишком хрупкие или слишком
плотные для того, чтобы пропускать электроны, могут быть исследованы при использо¬
вании одно- или двухстадийного метода приготовления реплик, который приводит к не¬
которому уменьшению разрешающей способности.
Сначала образец должен быть быстро подвергнут направленному оттенению золотом
или платиной, затем поверхность покрывают вертикально напыленной токопроводящей
и прочной углеродной пленкой.
При двухстадийном методе образец покрывают специальной легкоплавкой пленкой
толщиной 0,034 мм, которая размягчается при 45 °С и сохраняет реплику поверхности
минерала. Растворяют глинистый образец вместе с покрытием в разбавленной фтори¬
стоводородной кислоте. Реплика остается в углублениях образца и может подвергаться
обработке золотом или углеродом (если это еще не сделано). Затем растворяют полисти¬
роловую пленку в этиленхлориде. Собирают углеродную реплику на сетке и исследуют
ее методом ПЭМ. Все операции должны выполняться чрезвычайно тщательно.
8.2.4. Монтаж образцов для проведения сканирующей электронной
микроскопии
Основные положения
Образцы могут быть массивными и необработанными. В зависимости от характери¬
стик образца используют вакуумную камеру и диски 20 см в диаметре и 4 см толщиной
(8-дюймовые подложки), но дегазация больших образцов возможна только для плотных
пород с ограниченным числом трещин. На практике предпочтительнее использовать
малые пробы. Поверхность для наблюдения должна быть чистым разломом образца, по¬
зволяющим исследовать, например, плоскость скола, ориентацию кристаллов, дефекты
кристаллической решетки, наличие окклюдированных примесей.
Поверхностям придают проводящие свойства напылением углерода или металлиза¬
цией, если не проводится микрозондовый анализ. В противном случае, если СЭМ обо¬
рудован микрозондом для ЭД-РСМА или ВД-РСМА, необходимы плоские, тщательно
отполированные (0,25 мкм) поверхности.
1 Сетку следует брать при помощи пинцета с ультратонкими кончиками. Используют микро¬
шприцы Гамильтона на 0,1 мл либо оттянутые на пламени гидрофобные стеклянные трубки,
обработанные Просилом-28.
234
Часть 1. Минералогический анализ почв
Оборудование
• Специальные держатели для СЭМ.
• Коробки для хранения образцов.
• Калибровочные сетки размером 5x5 мм с отверстиями 2 мкм.
• 3-миллиметровые углеродные пластины для установки на держатели СЭМ.
• Маркер для СЭМ (с проводящими чернилами).
• Двусторонняя самоклеящаяся лента с низким содержанием летучих элементов.
Материалы
• Цапоновый лак.
• Проводящий углеродный лак.
• Набор эталонных минералов для количественного анализа.
Малые пробы можно собрать на алюминиевых подложках, наклеивая их при помощи
цапонового или углеродного лака. В некоторых случаях можно использовать углеродные
подложки или пластины толщиной 3 мм, наклеенные на алюминиевые подложки. Угле¬
родный лак обычно предпочтителен для микрозондового анализа — ЭД-РСМА. Очень
важно точно расположить образцы (выровнять, пометить, отметить правильное направ¬
ление на подложке); также важно не допускать пустот под образцом (рис. 8.4), потому
что это нарушает непрерывность и может помешать удалению зарядов, что приведет
к образованию штрихов на изображениях, которые делают фотографии непригодными
к использованию. Лак не должен покрывать образец или заполнять трещины. После
длительного высушивания для удаления растворителя образцы покрывают углеродом
при помощи распылителя с ионной бомбардировкой или оттеняют металлическим по¬
крытием из испарителя.
8.2.5. Обработка поверхности (оттенение, покрытие углеродом,
металлизация)
Вакуумный испаритель
Чтобы усилить пленку формвара, которая поддерживает образцы, а также придать ей
проводящие свойства, часто используют напыление углерода. Углерод следует напылять
вертикально и равномерно. Иногда микропробы слабо поглощают свет и плохо видны
под ПЭМ. В этом случае на пробу можно нанести направленное покрытие углерода, ча¬
стицы которого слабо различимы, или металлизировать под касательным углом плати¬
ной или золотом. Каждая область, защищенная рельефом, будет отображаться тенью.
Методика
Правильно Неправильно
Стекло
Рис. 8.4. Расположение образцов на подложке СЭМ
Глава 8. Микроскопический анализ
235
Зная угол падения, можно измерить длину тени и рассчитать высоту соответствующего
элемента рельефа (рис. 8.5).
Рис. 8.5. Вакуумный испаритель (оттенение углеродом или металлом)
Металлизация напылением
Прибор содержит золотой антикатод в форме кольца, внутренний диаметр которого пре¬
вышает размеры подложки образца. Расположенный в центре цилиндрический магнит
образует магнитное поле, которое окружает антикатод (рис. 8.6). Электроны, которые
в противном случае перегревали бы образец, отклоняются магнитным полем.
Подложку образца охлюкдают до 4°С при помощи термоэлектрического модуля Пель¬
тье. Это уменьшает нагрев и дает возможность обрабатывать органические образцы.
I Ось магнита
t
Рис. 8.6. Оборудование для металлизации (Био-Рад-Полярон) для образцов СЭМ: N — магнит,
/ — золотой антикатод; 2 — подложка образца; 3 — охлаждающий блок, черные кружки — ней¬
тральные атомы, белые кружки — положительные ионы. Когда образец продвигается в магнитное
поле, соответствующие поверхности подвергаются бомбардировке
236
Часть 1. Минералогический анализ почв
Обработку выполняют под вакуумом 10-1 торр в течение 30—180 с. Приложенное на¬
пряжение может достигать 3 кВ. Чистка струей нейтрального сухого газа (аргона) по¬
зволяет удалять остаточные следы воды, углекислого газа, кислорода и, возможно, мас¬
ляных загрязнений от вакуумного насоса. Иногда необходимо дегазировать пористые
образцы в течение нескольких часов. Это снижает загрязнение, однако лучше использо¬
вать аппарат для сушки при критической температуре С02, перед дегазацией в металли-
заторе и последующей металлизацией.
Для органического вещества и очень хрупких образцов часто рекомендуется крио¬
фиксация.
Эта обработка позволяет достичь высокой проводимости поверхности и подчеркнуть
рельеф необработанных образцов оттенением для метода СЭМ.
8.3. Микроскопические исследования
8.3.1. Оптическая микроскопия
Описание
Оптические микроскопы позволяют наблюдать объекты, которые слишком малы для
наблюдения невооруженным глазом или через лупу. Прямые наблюдения выполняются
в диапазоне излучения от ИК до УФ, включая видимый свет, и могут сопровождаться
фотографированием на пленку (черно-белую или цветную) или записью цифровых ви¬
деоизображений.
Увеличение лупы составляет от 2 до 60 раз, а для самых мощных микроскопов до¬
стигает 1500 раз. Объект может быть массивным или слишком тонким (тонкий шлиф
с покрытием или без) для определения свойств почв или почвенных материалов на ос¬
нове поглощения и пропускания света. Покровные стекла защищают поверхность от
окисления. Защищенные шлифы можно использовать для оптических наблюдений при
погружении, но их нельзя затем использовать для электронной микроскопии.
Оптический микроскоп состоит из штатива, который поддерживает механический
держатель, обеспечивающий вертикальное перемещение объектива, и окуляра над пред¬
метным стеклом.
Оптическое увеличение и диаметр поля зрения определяют соотношение между изо¬
бражением и объектом. Яркость имеет отношение к светосиле используемой оптики.
Глубина резко изображаемого пространства и диапазон фокусировки, а также предель¬
ное разрешение определяют зоны, в которых объект можно наблюдать при оптимальных
условиях для данного материала. Система освещения позволяет проводить наблюдения
в режимах отражения или пропускания. Для массивных объектов можно использовать
направленное освещение (низковольтные лампы, оптические волокна). Для очень тон¬
ких объектов освещение может быть сконцентрировано в точку конденсаторами по от¬
ношению к различным фонам: бледный или темный фон, поляризованный свет, фазо¬
вый контраст, УФ-излучение (покрытые или непокрытые стекла).
ИК-микроскопы используют оптические системы с зеркалами, чтобы избежать по¬
глощения ИК-излучения материалами, обычно используемыми при производстве
линз.
Поляризационный микроскоп
В науках о земле поляризационные микроскопы являются основными инструментами
для исследования кристаллов и характеризации их оптических свойств. Интерферен¬
Глава 8. Микроскопический анализ
237
ционные микроскопы или фазово-контрастные микроскопы (для наблюдения про¬
зрачных объектов на бледном фоне) используются редко. Изменения в коэффициенте
пропускания могут выявить структуры, которые невидимы при естественном освеще¬
нии и позволяют идентифицировать явления плеохроизма, изотропии и анизотропии
структуры и ассоциаций минералов (формы, фации, плоскости скалывания, двойнико¬
вые кристаллы), используя скрещенные или несмещенные поляризаторы. Эти микро¬
скопы (рис. 8.7) позволяют проводить наблюдения изменений пропускания по срав¬
нению с направлением поляризации падающего света. Калиброванный компенсатор,
помещенный перед анализатором, дает возможность измерять наблюдаемые изменения.
Выбор объектива (увеличение, иммерсия или нет) влияет на качество наблюдений об¬
разцов почв.
Образец
(шлиф/тонкий срез)
◄ ►
Окуляр
Анализатор
Линзы
тьшж ушшт
Гониометрический
столик
Конденсатор
Диафрагма
Поляризатор
Конденсатор
Диафрагма
Источник света
Рис. 8.7. Схема поляризационного микроскопа
Для исследования структуры и порозности почв размер, форму, ассоциации отдель¬
ных зерен и распределение различных фаз определяют либо на выделенных фазах, либо
на тонких срезах (Jongerius и др., 1972; Bullock и Murphy, 1980).
238
Часть 1. Минералогический анализ почв
Методика
Вращающийся держатель, откалиброванный в градусах, позволяет измерять углы по¬
гасания и идентифицировать еще присутствующие первичные минералы, а также
уточнять масштаб и тип эрозии. Интерпретация требует большого опыта и выполнять
ее способны только специалисты-петрографы. Ортоскопические методы требуют мно¬
го времени, но они обычно точнее, чем коноскопические (Wahlstom, 1969; Hartshorne
и Stuart, 1970).
Это качественный анализ, но количественное определение может быть осуществлено
подсчетом минеральных частиц, происходящих из почвообразующей породы и эроди¬
руемых минералов, во фракциях, предварительно выделенных фракционированием по
размеру частиц, плотности, твердости, магнитным свойствам и др. Форма частиц (за¬
кругленные края, округление более мягких минералов), вид поверхности (сглаженность
поверхности частиц, плоскости спайности, трещины, различные наносы) являются ин¬
дикаторами типа повреждения, эрозии и переноса (химическая эрозия, микрокоррозия,
ледяная и ветровая эрозия).
Природу минералов можно определить на основании цвета, мутности и особенно
коэффициента преломления, а также модификаций, наблюдаемых в поляризованном
свете (например, плеохроизм). Некоторые минералы проявляют более или менее четкое
двойное лучепреломление. Угла погасания можно достичь с различной степенью бы¬
строты; он может быть частичным или полным. Форма некоторых кристаллов является
характеристичным признаком.
Эти наблюдения можно дополнить методом сканирующей электронной микроско¬
пии, быстро закрепив образцы на двусторонних клеящих подложках, на которые напы¬
лен углерод для придания электропроводности (см. раздел 8.2.5).
Использование шлифов позволяет проводить наблюдения, не нарушая природной
структуры образца, ориентации и ассоциации минералов, непрерывности минералоги¬
ческого состава профиля (уменьшение содержания или исчезновение некоторых мине¬
ралов; отношение между минералами, устойчивыми к эрозии, и минералами, подвер¬
женными эрозии, дающее индекс разрушения).
Формирование конкреций, вкраплений, появление склеивания, наличие органиче¬
ского вещества на различных стадиях разложения можно наблюдать и количественно
оценить, последовательно используя методы СЭМ и ЭД-РСМА (легко выявляются не¬
которые артефакты, возникшие в процессе приготовления, такие как заполнение све¬
жих трещин или отверстия, образовавшиеся при полировке оксидом алюминия).
Единицы структурной организации (скелеты, плазма, полости) могут быть подробно
исследованы в различных масштабах, используя выделенные фракции или/и шлифы:
скелетные компоненты соответствуют частицам, которые не были реорганизованы,
плазма соответствует мелким элементам, которые могут перемещаться и реорганизовы¬
ваться (глины, оксиды), пустоты относятся к порозности (расчет содержания воздуха
и раствора в почве). Для исследования порового пространства можно смешать флуорес¬
центные краски с пропитывающей смолой в процессе приготовления шлифов (см. раз¬
дел 8.2.2).
8.3.2. Электронная микроскопия, общая информация
Принцип действия всех электронных микроскопов основан на взаимодействии
электронов с веществом. Энергия электрона, ускоряющегося напряжением V, равна
Е = т\?/2 = еУ(где mfvte — масса, скорость и заряд электрона, соответственно). Высокая
Глава 8. Микроскопический анализ
239
энергия излучения (быстрые электроны) может изменять уровень глубоких электрон¬
ных слоев атомов. Низкая энергия излучения (медленные электроны) влияет только на
внешние электронные слои, которые отражают химическое состояние атомов. Общее
воздействие электронного пучка связано с электронным облаком из Zэлектронов (ег) на
электронных орбиталях вокруг ядер атомов.
При рассмотрении влияния излучения на вещество должны приниматься во внима¬
ние следующие факторы:
♦ интенсивность: интенсивность прошедшего или отраженного излучения меньше,
чем интенсивность падающего излучения, так как происходит его поглощение;
♦ направление: происходит рассеяние с потерей энергии (неэластичное рассеяние,
изменяющее внутреннюю структуру) или без потери энергии (когерентное эла¬
стичное рассеяние, допускающее дифракцию);
♦ энергия: поскольку часть энергии теряется, количество отраженной, прошедшей
или рассеянной энергии меньше, чем исходное количество энергии.
Потери интенсивности и энергии могут сопровождаться превращением вещества под
воздействием излучения:
♦ в случае электронных микроскопов очень высокой энергии (3000 кВ) образец мо¬
жет постепенно разрушаться;
♦ в случае микроскопов с энергией ниже, чем 400 кВ происходит перенос энергии за счет
возбуждения электронов, тепловых колебаний, выброса частиц и эмиссии вторич¬
ных излучений, используемых для количественного определения. Нагревание при¬
водит к генерированию фононов. В некоторых случаях происходят химические про¬
цессы восстановления (приобретение электронов). Химические связи могут рваться.
Когда перенос энергии превышает порог смещения (от 15 до 30 эВ), излучение мо¬
жет вызывать перемещения атомов. Побочное корпускулярное электронное излучение
достаточно высокой энергии может выбить из глубоких слоев орбитальный электрон
с кинетической энергией, соответствующей разнице между потерей энергии падающе¬
го излучения и собственной энергией электронов (вторичные электроны). Поскольку
возбужденное состояние нестабильно, атом возвращается в основное состояние; проис¬
ходит испускание рентгеновских фотонов или Оже-электронов (явления релаксации).
Электромагнитное излучение, такое как падающие или вторичные рентгеновские
лучи, приводит к испусканию фотоэлектронов (от ИК- до УФ-фотонов, катодолюми-
несценция). Электронные микроскопы можно разделить на два класса в зависимости от
геометрии образца:
• массивные образцы, поскольку они очень большой толщины, можно анализиро¬
вать только методом отражения сигналов от их поверхности;
• образцы критической толщины, сквозь которые излучение может проходить (ми¬
крокристаллы, тонкие пленки и др.), можно анализировать методом пропускания.
Однако развитие приборостроения привело к изменениям в этой дихотомии с появ¬
лением комбинированных приборов, способных проводить измерения на тонких образ¬
цах, используя оба процесса. Существуют следующие типы оборудования:
• традиционные просвечивающие электронные микроскопы (ПЭМ) (иногда с до¬
полнительными функциями просвечивающего сканирования);
• просвечивающие растровые электронные микроскопы (ПРЭМ);
• сканирующие отражательные микроскопы (стандартные сканирующие электрон¬
ные микроскопы, СЭМ);
240
Часть 1. Минералогический анализ почв
• сканирующие отражательные микроскопы с дифференциальной откачкой, в ко¬
торых наблюдательная камера находится под частичным вакуумом (сканирующие
электронные микроскопы с режимом естественной среды, ССЭМ).
Каждый тип оборудования позволяет проводить измерения, дополняющие измере¬
ния, проводимые на других типах оборудования. Приборы могут применяться либо для
очень высокого разрешения, либо при сложном наборе оборудования для различных
химических и физико-химических исследований. Получаемые сигналы дополняют друг
друга в энергетических полях.
8.3.3. Просвечивающая электронная микроскопия, микродифракция
Основные положения
Просвечивающие электронные микроскопы используют падающий электронный пучок,
который, проходя через очень тонкие образцы, предоставляет информацию о форме
и структурном распределении элементарных почвенных частиц. Взаимодействие элек¬
тронного пучка с веществом дает изображения и микродифрактограммы (и позволяет
подобрать элементарные химические анализы в дополнение к различным электронным
микрозондам в ПРЭМ).
Геометрию электронного микроскопа можно сравнить с геометрией оптического
микроскопа (рис. 8.8). Вместо источника фотонов используется источник электронов
(высоковольтная электронная пушка). Система облучения с электромагнитными кон¬
денсаторами концентрирует электронный пучок на объекте; электронный объектив об¬
разует промежуточное изображение, которое захватывается проекционными линзами,
образующими окончательное изображение на флуоресцентном экране или фотографи¬
ческом устройстве.
Используются не все излучения, генерируемые падающим электронным пучком
(рис. 8.9), поскольку приборы обычно снабжены только одним-двумя датчиками1. Рент¬
геновское фотонное излучение (7) можно регистрировать детектором ЭД-РСМА(при Si—
Li детектировании, или диодами без входного окна фотоприемника для анализа легких
элементов, таких как азот и углерод) или ВД-РСМА (с кристаллом-монохроматором).
ИК, видимое, УФ фотонное излучение (2) может быть обнаружено по катодолюми-
несценции. Рассеянные электроны (5) можно анализировать спектрометрией характе¬
ристических потерь энергии электронами (СХПЭЭ), легкие элементы можно анализи¬
ровать методом ПРЭМ. Обратно рассеянные электроны (б), вторичные электроны (7)
и Оже-электроны используют для получения изображений СЭМ, а проходящие элек¬
троны (12) для методов ПЭМ и ПРЭМ.
Выбор просвечивающего электронного микроскопа зависит от характера необходи¬
мых наблюдений (увеличение, высокое разрешение, необходимость применения высо¬
кого напряжения для возбуждения или проникновения электронов, возможность ко¬
личественного определения химического состава) и стоимости оборудования, а также
его универсальности и потенциала модернизации, способности охватить весь диапазон
увеличения, включая слабое увеличение, степени автоматизации и легкости использо¬
вания, степени и чистоты вакуума, контраста и эффективности работы на темном фоне,
возможности сопряжения с системами химического анализа и анализаторами изображе-
1 Максимальная конфигурация некоторых высококлассных коммерческих анализаторов
включает до пяти датчиков.
Глава 8. Микроскопический анализ
241
Источник излучения
- Фотоны Электроны -
Конденсатор .
(Линза) (Катушка) L I
Объект
(Шлиф) (Сетка)
Линза тят
.(Линза) (Катушка) □
□
Отклоняющие
катушки
Компенсация
астигматизма
(Атмосферное Вспомогательный
[давление вакуум
В/
2000 А Предел 2 А
разрешения
. Промежуточное
1А’ изображение
► Окуляр КатУшка [Zf
7 н проектора
□
Линза камеры
АЧ
Конечное изображение
Рис. 8.8. Сравнение оптического и просвечивающего электронного микроскопа
ний и т. д. Следует учитывать и стоимость годового технического обслуживания и рас¬
ходных материалов, которая может составлять 3-6% первоначальной цены оборудова¬
ния.
Эмиссия электронов
Источником электронного излучения служит электронная пушка. Обычно излучение
генерируется термоэлектронной эмиссией вольфрамовой нити, нагретой до 2500 °С (или
наконечника гексаборида лантана, нагретого до 1600 °С), образующей катод (рис. 8.10).
Вольфрамовая нить имеет диаметр около 0,1 мм, она имеет V-образную форму и за¬
острена на конце, чтобы фокусировать излучение. Нить нагревают в вакууме до высокой
температуры и подают на нее рабочее напряжение, соответствующее 4,5 эВ. Электроны
проводимости могут преодолеть барьерный потенциал и образовать электронное обла¬
ко. Эти электроны ускоряются потенциалом К0. Образуется электронный пучок, энергия
которого Е0 = eVQ. Испускаемые электроны двигаются в колонну с постоянной скоро-
242
Часть 1. Минералогический анализ почв
1. Рентгеновские фотоны Падающий пучок
6. Обратно рассеянные
3. Сток электроно
2. ИК, видимое,
излучение(ф
с образца
5. Неупруп
электронов (потеря энергии)
4. Проше
рентге
11. Упругое рассеяние
электронов (тормозное
излучение)
10. Ионы, десорбированные
атомы (искусственная
десорбция)
электроны
7. Вторичные электроны
9. Фононы, ультразвук
8. Оже-электроны
12. Прошедшие электроны,
ПЭМ-ПРЭМ (наблюдение
на светлом фоне)
Рис. 8.9. Типы излучений, испускаемых при бомбардировке образца пучком электронов
Рис. 8.10. Термоэмиссионная электронная пушка: 1 — вольфрамовая (слева) или LaB6 (справа)
нить; 2 —• фокусирующий электрод, отрицательно поляризованный относительно нити (электрод
Венельта); 3 — анод; 4 — минимальное поперечное сечение электронного пучка (10-50 мкм).
Электрод Венельта имеет отрицательный потенциал в несколько вольт, чтобы отклонить излучае¬
мые электроны от их пути и сконцентрировать в узкий пучок
стью благодаря высокому электрическому потенциалу между нитью и анодом (напря¬
жение питания анода).
Катод, сделанный из гексабората лантана (LaB6) с рабочим напряжением, соответ¬
ствующим 2,7 эВ, т. е. ниже, чем напряжение вольфрама, можно использовать при более
низкой температуре (примерно 1600 °С); яркость значительно повышается. Однако не¬
обходим вакуум 10~7 торр, и может вызвать затруднения химическая активность LaB6 по
отношению к некоторым металлам. Существуют электронные пушки с автоэлектрон-
ной эмиссией, которые обеспечивают гораздо бблыиую яркость по сравнению с термо¬
электронными пушками и пониженное рассеяние энергии.
3
3
Глава 8. Микроскопический анализ
243
Падающий пучок электронов, испускаемый электронной пушкой (< 1 мм в мини¬
мальном сечении), пересекает колонну микроскопа, следуя оптической оси. Электро¬
магнитные линзы в виде соленоидов генерируют магнитное поле, которое фокусирует
электроны. Внешнее поле предотвращает рассеивания этого магнитного поля. Напря¬
жение ускорения обычно находится в диапазоне от 50 до 1250 кВ, но может достигать
3000 кВ. На практике используют микроскопы, характеризующиеся (1) напряжением
менее 100 кВ, (2) средним напряжением от 200 до 500 кВ, а также (3) электронные ми¬
кроскопы высокого и сверхвысокого напряжения (ЭМВН), 1250 кВ и выше. Приборы
группы (3) очень дорогие, громоздкие и требуют специальных средств защиты.
Наблюдения
В методе ПЭМ электронные микроскопы высокого разрешения (ЭМВР - ПЭМВР)
(200-300 кВ) могут оборудоваться 15 А зондами, которые дают возможность исследовать
морфологию образцов в различных масштабах, проводить прямые наблюдения атомной
структуры кристалла и упаковки атомов (1,5 А при напряжении 400 кВ) на микропро¬
бах, для которых метод РД неэффективен. Таким образом, эти микроскопы можно ис¬
пользовать для исследования проблем фундаментальной кристаллографии, процессов
разрушения (зарождения и роста кристаллов, переходов фаз, аморфных в рентгеновских
лучах, в скрытокристаллические и кристаллические фазы в повторяющихся структурах
глин). Например, перемежающиеся слюды — хлориты и минералы с межпакетными рас¬
стояниями 7-14 А исследовались Амуриком (Amouric, 1987,1990; Amouricи др., 1988); Ан
и Пикор изучали ассоциации слюда — каолинит (Ahn и Реасог, 1987)1.
Используя эти методы, можно обнаружить двумерные дефекты, реликтовые пакеты
и бледные края межпакетных уровней. Приборы высокого разрешения должны исполь¬
зоваться с большой осторожностью, чтобы не допустить радиационных повреждений
электронами больших энергий из-за сильных колебаний электронного вещества.
Микродифракция
В минералогии микродифракцию электронов обычно осуществляют одновременно
с наблюдением изображений, освещенных под нормальным углом падения. Объекты
готовят на микросетках при достаточно низкой плотности для изолирования отдельных
частиц таким же способом, как и для получения изображений.
Микродифракцию можно наблюдать при помощи большинства приборов ПЭМ, про¬
сто регулируя диафрагму. Пучок быстрых электронов с короткой длиной волны и вы¬
сокой энергией (20-60 кэВ и более) проникает сквозь чрезвычайно тонкие микрокри¬
сталлы (< 100 А); когда угол падения на плоскости атомных сеток соответствует закону
Брэгга (см. гл. 4), наблюдаются пятна дифракции (рис. 8.11). На субмикропробах полу¬
чают дифрактограммы, характерные для монокристаллических структур. Такой деталь¬
ный вид кристаллических структур и дефектов не может быть получен традиционным
методом РД (см. гл. 4).
Для микродифракционных исследований в состав микроскопа включают регулиру¬
емую диафрагму, которая расположена в плоскости изображения и ограничивает ак-
1 В этих тщательных работах интересующие зоны выделялись на шлифах методом СЭМ при
увеличении около 10 000—20 000. Эти зоны разделялись на секции и утоньшались ультрами¬
кротомом и аргонной пушкой, чтобы обеспечить их достаточную прозрачность для электро¬
нов в методе ПЭМВР.
244
Часть 1. Минералогический анализ почв
Выбранная зона
тивную поверхность площадью около 1 мкм2. Этот метод позволяет получать четкие
дифрактограммы ориентированных микрокристаллов, но очень короткие длины волн
электронов приводят к малым углам дифракции и соответствующим интенсивностям,
которые отличаются от наблюдаемых в методах традиционной рентгеновской дифрак¬
ции, описанных в гл. 4.
Поскольку образец очень тонкий, полученные пятна дифракции типичны для струк¬
тур монокристаллов. Это метод часто используют в качестве дополнительного к тради¬
ционной РД. На поликристаллических материалах, таких как глины, можно получить
кольцевые дифрактограммы в течение минуты.
Специализированные методы ПЭМ
Визуализация зарядов с использованием коллоидного золота
Основные положения
Поскольку коллоидное золото непроницаемо для электронов, его используют в качестве
метки для обнаружения краевых зарядов или структурных дефектов в кристаллических
структурах (фото 8.1, слева).
Оборудование и реагенты
• Сетки для ПЭМ диаметром 3,05 мм, покрытые пленкой формвара и углеродом.
• Раствор коллоидного золота с размером частиц 5 нм (хранить в холодильнике за¬
щищенным от света).
Методика
• Суспендируют раствор коллоидного золота взбалтыванием.
• Берут 0,5 мл суспензии золота и смешивают в стеклянной пробирке с 0,5 мл образ¬
ца низкой плотности, чтобы получить хорошо разделенные минералы; оставляют
Глава 8, Микроскопический анализ
245
Фото 8.1. Специализированные методы просвечивающей электронной микроскопии: слева —
высвечивание краевых зарядов каолинита коллоидным золотом (см. методику в тексте раздела);
справа — фото (х90 000), напечатанное методом параглифа (методику см. ниже) (Ж. Готейруа, на¬
бор стандартных образцов, Исследовательский институт/ЯО, Бонди, Франция, неопубликованные
данные)
в контакте на несколько минут; перемешивают и помещают микрокаплю на сетку
3 мм.
• Оставляют на воздухе для высыхания и рассматривают под ПЭМ с увеличением
около 60 000. Золото мигрирует предпочтительно к зонам разрыва или кристаллиза¬
ции и таким образом выявляет способ сборки и активные центры некоторых глин.
Печать методом параглифа
Основные положения
Метод основан на получении псевдорельефа наложением с очень маленьким сдвигом
прозрачных негативов и позитивов одного и того же изображения (фото 8.1, справа).
Оборудование и материалы
• Высококонтрастная негативная пленка размером не менее 6,5 х 9 см.
• Прозрачная позитивная пленка.
• Материалы для проявления фотографий (проявитель, закрепитель).
Методика
• Выбирают четкий негатив изображения.
• Контактным методом получают прозрачное позитивное изображение близкой
плотности.
246 Часть 1. Минералогический анализ почв
• Накладывают позитив и негатив друг на друга и находят оптимальный сдвиг между
ними для получения эффекта рельефа.
• Получают изображение копированием или увеличением. Этот тип изображе¬
ния позволяет более четко увидеть покрытия частиц и получить яркий эффект
рельефа.
Затемнение образцов, «прозрачных» для электронов
Обогащенные железом минералы мало прозрачны для электронов и могут вызывать
сложности, если они имеют слишком большую толщину, так как изображения получа¬
ются слишком контрастными и детали не различимы.
С другой стороны, некоторые очень мелкие минералы, такие как аллофаны, прак¬
тически прозрачны для электронов, если присутствуют в малых концентрациях. Такие
препараты можно сделать более контрастными добавлением соли свинца (1%-ный рас¬
твор РЬС12 в воде).
Образец оставляют в контакте с раствором соли свинца в течение часа, затем промы¬
вают, снова суспендируют и разбавляют для приготовления сетки для ПЭМ.
Просвечивающая растровая электронная микроскопия
Электронная пушка и система конденсаторов, используемая для создания электронного
пучка, основаны на том же принципе, что и традиционная ПЭМ, но в ПЭМ сигнал пере¬
дается в плоскость изображения, которое можно наблюдать на флуоресцентном экране
через систему электронных линз, тогда как в просвечивающей растровой электронной
микроскопии (ПРЭМ) сигнал непосредственно получается электронными или рентге¬
новскими детекторами и передается на экран (рис. 8.12).
В ПРЭМ электронная пушка с автоэлектронной эмиссией и диаметром пучка около
5 мм, яркость которого превышает яркость традиционного вольфрамового источника
более чем в 1000 раз, испускает пучок электронов, который пересекает конденсатор, да¬
ющий уменьшенное изображение источника (микрозонда).
Этот зонд сканирует поверхность образца при помощи отклоняющей катушки. Элек¬
троны, прошедшие сквозь образец или дифрагированные им, собираются на детекторе
Автоэмиссионная
электронная пушка
Конденсатор
Образец
Детектор
электронов
Рис. 8.12. Схема просвечивающего растрового электронного микроскопа
Глава 8. Микроскопический анализ
247
с откликом, пропорциональным их интенсивности. После усиления изображение по-
стадийно создается на экране при синхронизации с генератором развертки.
Электронные пушки с автоэлектронной эмиссией очень чувствительны к загрязне¬
нию. Они требуют сверхвысокого «чистого» вакуума (10~10 торр), что запрещает исполь¬
зование масляного диффузионного насоса для вторичного вакуума. Несмотря на ис¬
пользование криоскопических ловушек, пушка может выйти из строя из-за присутствия
следов масла.
В ПРЭМ можно добиться очень высокого пространственного разрешения. Оборудо¬
вание может быть снабжено энергоанализаторами, такими как спектрометры энергети¬
ческих потерь электронов (СЭПЭ). Они способны анализировать поверхности микро¬
проб площадью около одного нанометра на содержание всех элементов периодической
системы (от 3Li до 92U).
Специализированные ПРЭМ не находят очень широкого применения, многие про¬
изводители предпочитают продавать комбинированные ПЭМ, оборудованные допол¬
нительным ПРЭМ, который имеет те же рабочие характеристики при цене в 2—3 раза
меньше.
8.3.4. Сканирующая электронная микроскопия
Отражательные сканирующие микроскопы и микрозонды
Сканирующие электронные микроскопы (СЭМ) и электронные микрозонды (ЭМЗ) ос¬
нованы на дополняющих друг друга принципах; используемые микрозонды менее 1 мкм
оптимизированы для рентгеновского анализа.
На термоионную электронную пушку подается отрицательный потенциал, равный
10-50 кВ. Образец помещают на прецизионный гониометрический держатель (бино¬
кулярная лупа, позволяет осуществлять визуальный выбор точки взаимодействия на
микрозонде).
Электромагнитные конденсаторы формируют изображение зонда, которое проеци¬
руется на образец. Луч перемещается отклонением зонда. Массивный образец может
быть толщиной 2-4 см и диаметром 20 см (8-дюймовая подложка). Для создания ва¬
куума в объемной камере для пробы требуется мощная система с большой произво¬
дительностью (турбомолекулярный насос). Увеличение равно отношению амплитуды
сканирования изображения (постоянная) к амплитуде сканирования объекта (пере¬
менная).
Для аналитических измерений могут использоваться различные взаимодействия
между электроном и веществом (вторичные и обратно рассеянные электроны, рентге¬
новские лучи, Оже-электроны, фотолюминесценция, проходящие электроны).
Изображение создается постадийно (пиксель за пикселем) и может подвергаться
оцифровке и обработке с использованием соответствующей системы обработки данных.
Создание изображений основано на двух методах:
- В методе вторичных электронов падающий пучок первичных электронов теряет
энергию при контакте с веществом; часть энергии возвращается в форме вторичных
электронов, которые пересекают сетку коллектора и затем ускоряются в поле сцинтил¬
лятора; получается пригодный для использования сигнал, который смешивается с об¬
ратно рассеянными электронами, способными пересечь диафрагму детектора.
- В методе обратно рассеянных электронов электроны собираются коллектором сцин-
тилляционного детектора; сигнал относительно слабый, но детектирование улучшается
при использовании полупроводникового детектора в форме диска, с отверстием в цен-
248
Часть 1. Минералогический анализ почв
тре и помещенного над образцом; устройство, установленное в двух или четырех различ¬
ных секторах, позволяет создать топографический контраст.
Химический состав образца иногда меняется случайным образом, потому что глубина
проникновения пучка очень мала. ГЬубина оттенков серого зависит от атомных номеров
наблюдаемых элементов.
Разрешение составляет около 20-100 А в зависимости от наблюдаемого элемента.
Интенсивность электронного пучка и условия сканирования выбирают с учетом обе¬
спечения максимального разрешения и оптимального отношения сигнал/шум для дан¬
ного увеличения. Даже в лучших условиях высокая энергия падающего пучка (прибли¬
зительно 30 кэВ) не позволяет наблюдать очень мелкие детали, но в целом получаются
довольно хорошие результаты. С другой стороны, если материал слабо проводящий и не
может быть покрыт металлом методом напыления, возможно, лучше уменьшить заряд,
используя энергию ниже 5 кэВ.
Необходимо выбрать подходящий размер диафрагмы для получения соответствую¬
щей глубины резкости, а также для регулировки рабочего расстояния, если образец име¬
ет развитый рельеф.
Сканирующая электронная микроскопия с режимом естественной среды
Эти микроскопы позволяют получать изображения высокого разрешения методом от¬
ражения на образцах, сохраняющих естественную влажность, без дегазации или прида¬
ния проводящих свойств поверхности. Некоторые исследования в режиме естественной
среды можно проводить без деформации или превращения образца. Используют две
системы:
• низковакуумные сканирующие электронные микроскопы (НВ-СЭМ) — относи¬
тельно простые приборы, которые могут использоваться для традиционной СЭМ;
они могут давать частичный вакуум около 2—4 торр в камере для образцов и обыч¬
но снабжены детектором обратно рассеянных электронов;
• сканирующие электронные микроскопы с режимом естественной среды (ССЭМ) —
специализированные микроскопы, способные создавать высокий вакуум (10~7 торр)
в электронной пушке и одновременно низкий вакуум (близкий к атмосферному
давлению) в камере наблюдения. Это различие в степени вакуума образуется сту¬
пенчато при последовательном снижении давления.
Расстояние между образцом и выходящим электронным пучком под высоким вакуу¬
мом должно быть как можно меньше, чтобы избежать ухудшения рабочих параметров.
В традиционной СЭМ более 95% электронов не подвергаются рассеиванию. В СЭМ
с режимом естественной среды с образцом, расположенным близко от выходящего пуч¬
ка при давлении в 1 торр, доля нерассеянных электронов может достигать 90%, но она
уменьшается при увеличении количества молекул газа на пути пучка.
Специальный газовый детектор вторичных электронов (ГДВЭ) позволяет улучшить
качество изображения разделением обратно рассеянных электронов и вторичных элек¬
тронов, образующихся при взаимодействиях электронного пучка с атомами образца.
Ложные сигналы от заряда, подобного тому, что имеет место в традиционной СЭМ (на¬
пример, ионизация газа, образование свободных электронов или положительных ионов,
компенсирующих отрицательные заряды) не наблюдаются. Этот детектор не чувствите¬
лен к свету или температуре.
Газовая среда в камере для образцов может контролироваться одновременно с давле¬
нием и температурой, что позволяет проводить наблюдения в газовой среде почти по-
249
Глава 8. Микроскопический анализ
стоянного состава. Интерпретация результатов требует учета таких явлений, как кон¬
денсация на минералах (например, закругление углов), присутствие поровой воды или
загрязнителей и учета баланса газов. Можно проводить количественные измерения ме¬
тодом ЭД-РСМА (см. раздел 8.3.5). В настоящее время СЭМ с режимом естественной
среды может применяться во многих областях почвоведения, включая исследования ор¬
ганического вещества, глинистых материалов и микроорганизмов (Mathieu, 1998; Leroux
и Morin, 1999), например:
• физические проблемы, связанные с расширяющимися минералами, аллофано-
выми почвами с высоким алагоудержанием, структурой, текстурой, порозностыо,
агрегатами, переносом вещества между почвой и окружающей средой, процессами
гидратации и дегидратации, усадкой, сжатием и адгезией почвы;
• влияние нагрева или химической обработки, плавления, возгонки, роста кристал¬
лов, стабилизации структуры, испытаний под давлением;
• динамика разложения органического вещества и микрофауны.
8.3.5. Полный элементный анализ методом рентгеновской
спектрометрии
Энергодисперсионная рентгеновская спектрометрия
Микроопределения обычно проводят методом рентгенофлуоресцентной спектрометрии
(см. раздел 31.3.2) с помощью ЭД-РСМА-спектрометра. Эта система (рис. 8.13) позво¬
ляет строить карты качественного распределения элементов в поверхностном слое тол¬
щиной примерно 1 мкм. Лучше использовать почти плоские поверхности; для точного
количественного анализа поверхности должны быть отполированы на глубину 0,25 мкм,
чтобы уменьшить возможные топографические эффекты.
Падающий пучок электронов
Е0 = 100-3000 КВ
Криостат
с жидким
азотом
Электронная ЭД-РСМА
измерительная
система
Кристалл
Рентгеновские
* лучи
Полевой
транзистору^ детектор
Предусилитель дд q
ВДС Детектор
с линейной
характеристикой
Рис. 8.13. Микрозонды с дисперсией энергии и длины волны: ЭДС (энергодисперсионная спек¬
трометрия), ЭД-РСМА (энергодисперсионный рентгеноспектральный микроанализ); ВДС (волно¬
дисперсионная спектрометрия); ВД-РСМА (волнодисперсионный рентгеноспектральный микро¬
анализ) (круг Роуланда, влияние расфокусировки электронного пучка; 0 — угол Брэгга: Д0 — от¬
клонение угла Брэгга, вызванное расфокусировкой)
250
Часть 1. Минералогический анализ почв
Для менее точного количественного микроанализа, чем анализ с применением спе¬
циальных аналитических зондов, можно использовать фиксированный зонд; программа
ZAFjsjгя корректировки матрицы (Z- атомный номер, А - абсорбция, F- флуоресцен¬
ция) позволяет улучшить результаты. Такой анализ может проводиться только для эле¬
ментов тяжелее, чем uNa. Для легких элементов необходима эмиссия Оже-электронов;
однако сверхвысокий вакуум (Ю-10 торр), требующийся для Оже-спектроскопии, не мо¬
жет быть достигнут в обычных сканирующих микроскопах. Необходим сканирующий
Оже-микроскоп (СОМ), в котором вакуум достигается ионным насосом.
Волнодисперсионная рентгеновская спектрометрия
Источник рентгеновских лучей, излучаемых на область контакта электронного луча с об¬
разцом, помещен на фокусирующий круг, называемый «кругом Роуланда» (рис. 8.13).
Детектирование осуществляется перемещением кристаллического анализатора и вход¬
ной щели детектора (счетчик с линейной характеристикой) по кругу. Детектор должен
находиться на эффективном фокальном пятне (20 относительно падающего пучка).
Для выполнения количественного анализа направление измерения и апертура долж¬
ны быть постоянными. На практике угол отражения не может выходить за диапазон
5—70°. Поэтому может понадобиться использовать четыре анализирующих кристалла
(система «Микроспек», США) для различных межплоскостных расстояний (закон Брэг¬
га) для того, чтобы охватить весь диапазон длин волн, доступных с применением это¬
го подхода (фторид лития UF, пентаэритрит, кислый фталат рубидия, стеарат свинца).
В настоящее время наблюдается тенденция к замене системы ВД-РСМА более быстро¬
действующей системой ЭД-РСМА.
Использованная литература
Ahn JH and Peacor DR (1987) Kaolinization of biotite: ТЕМ data and implications for an alteration
mechanism. Am. Miner., 72,353.
Amouric M (1987) Growth and deformation defects in phylllosilicates as seen by HRTEM. Acta Cryst.,
R43, 57.
Amouric M (1990) Etude de Г interstratification mica-chlorite par microscopie 61ectronique. In
Materiaux argileux, structure, proprietes et applications, Decarreau A ed., Soc. Fr. de Min£ralogie et
Cristallographie, Groupe Fran$ais des Argiles, 283-287.
Amouric M, Bianetto T and Proust D (1988) 7.1 and 14 A mixed layer phyllosilicates structurally studied
by ТЕМ in pelitic rocks. Bull. Miner, 111, 29.
Bullock P and Murphy CP (1980) Towards the quantification of soil structure. Microscopy, 120,317-328.
Delvigne JE (1998) Atlas of micromorphology of mineral alteration and weathering, The Canadian
Mineralogist, special publication 3, Ottawa, IRD, Paris.
Hartshome NM and Stuart A (1970) Crystals and the polarizing microscope., Arnold, 219-251.
Jongerius A, Schoonderbeeek D, Jager A and Kowalinski S (1972) Electrooptical soil porosity by means
of Quantimet В equipment. Geoderma, 7,177-198.
Leroux A and Morin P (1999) Evolution de la microscopie к balayage - un progrds pour les applications
g6o-environnementales. Bull. Lab. Fonts et Chaussees, 222, 85-89.
Mathieu C (1998) Effects of electron-beam/gas interaction on X-ray microanalysis in the variable pressure
SEM, Microchim. Acta., 15,295-300.
Moran C, McBratney AB and Koppi AJ (1989) A rapid analysis method for soil macropore structure. I.
Specimen preparation and digital binary image production. Soil Sci. Soc. Am. J., 53,921-928.
Pansu M, Gautheyrou J and Loyer JY (2001) - Soil analysis - sampling, instrumentation and quality
control, Balkema Publishers, Lisse, Abington, Exton, Tokyo, 512 p.
Tessier D (1985) Validity des techniques de d6shydratation pour P&ude de la micro-organisation des sols.
In Soil Micromorphology, Fedoroff N., Bresson LM and Courty MA ed., AFES.
Wahlstrom EE (1969) Optical crystallography., Wiley New York.
Глава 8. Микроскопический анализ
251
Библиографические ссылки в хронологической
последовательности
Deer WA, Howie RA and Zussman J (1962,1963, 1966) Rock-forming minerals., vols. 1-6, Longmans-
Green London.
Beutelspacher H and van den Marel HW (1968) Atlas of electron microscopy of clay minerals and their
admixtures., Elsevier Amsterdam.
Reid WP (1969) Mineral staining tests. Mineral Ind. Bull., 12, 1-20.
Spry A (1969) Metamorphic textures., Pergamon, Oxford.
Hartshome NM and Stuart A (1970) Crystals and the polarizing microscope., Arnold.
Gard JA (1971) Electron-optical investigation of clays., Mineral Society.
Hutchison CS (1974) Laboratory handbook of petrographic techniques., Wiley New York.
Wells OC (1974) Scanning electron microscopy., McGraw-Hill New York.
Brewer R (1976) Fabric and mineral analysis of soils., Krieger USA.
Goldstein JI and Yakowitz H (1976) Practical scanning electron microscopy., Plenum New York.
Zussman JB (1977) Physical methods in determinative mineralogy., Academic New York.
Tessier D and Berrier J (1979) Utilisation de la microscopie Electronique к balayage dans l’Etude des sols.
Observation des sols humides soumis к difFErents pF. Sci. du Sol., 1, 67-82.
Jogerius A and Bisdum EBA (1981) Porosity measurements using the quantimet 720 on backscattered
electron, scanning images of thin sections of Pub. and Doc., Wageningen, Pays-Bas.
Smart T and Tovey NK (1981) Electron microscopy of soils and sediments, tl et t2. Clarendon Oxford.
Fleischer M, Wilcox RE and Matzko JJ (1984) Microscopic determination of opaque minerals. US Geol.
Survey Bull., 1627.
Low AJ, Low EJ and Douglas LA (1984) A motorized grinder for making soil thin sections. Geoderma,
32, 335-337.
Bullock P, Fedoroff N, Jongerius A, Stoops G, Tursina T (1985) Handbook for Soil Thin Section Descrip¬
tion, Waine Research. Publication. Wolverhampton, England.
Goldstein JI, Newbury DE, Echlin P, Joy DC, Fiori C and Lifshin E (1985) Scanning electron microscopy
and X-ray microanalysis., Plenum New York.
Maurice F, Keny L and Tixier R (1985) Microanalyse et microscopie electronique a balayage., Les Edi¬
tions de Physique.
Murphy CP (1985) Fasten methods of liquid-phase acetone replacement of water from soils and sedi¬
ments prior to resin impregnation. Geoderma, 35, 39-45.
Willaime C (1987) Initiation a la microscopie electronique par transmission en mineralogie, science des
materiaux., Soc. Fr. de MinEralogie et de Cristallographie.
Blackburn M, Caillier M, Bourbeau GA and Richard G (1988) Utilisation d’une solution de chlorure de
sodium pour le remplacement de l’eau dans les Echantillons d’argile lourde avant 1’imprEgnation.
Geoderma, 41, 369-373.
Takeda H (1988) A rapid method for preparing thin sections of soil organic layers. Geoderma, 42, 159-
164.
Chartres CJ, Ringrose-Noase AJ and Raupach M (1989) A comparison between acetone and dioxane and
explanation of their role in water replacement in indisturbed soil samples. J. Soil Sci., 40, 849-863.
Wright D, Stanley D, Chen HC, Shultz AW and Fang JM (1990) A frame based expert system to identify
minerals in thin section. Microcomputer applications in geology II. Pergamon Oxford, 289-299.
Zhurov AV (1990) Preparation of polished sections for the study of soil pores and their differentiation by
size. Pochvovedenie, 8, 144-147.
Sludzian G and Galle P (1992) Cartographie de matEriaux et d’Echantillons biologiques par microscopie
ionique к balayage. La vie des sciences, Compte rendu serie generale, 9, 157-177.
Gribble CD and Hall AJ (1993) Optical mineralogy: principles and practice., Chapman and Hall Lon¬
don.
Fitzpatrick EA (1993) Soil microscopy and micromorphology., Wiley.
Ringrose-Voase AJ and Humphreys GS (1994) Soil micromorphology, studies in management and
genesis: Proceedings of the IX international working on soil micromorphology., Elevier Science Ltd.
252
Часть 1. Минералогический анализ почв
Lavoie DM, Little BJ, Ray RI, Bennett RH, Lambert MW, Asper V and Baerwald (1994) Environmental
scanning electron microscopy of marine aggregates. J. Microscopy, 178, 101-106.
Vempati RL, Hess TR and Cocke DL (1996) X-ray photoelectron spectroscopy. In Methods of soil
analysis, Sparks DL ed., SSAA book series No 5.
Jonhson RA (1996) Environmental Scanning electron microscopy - An introduction of ESEM, Philips
Electron Opt.,-FEU 55 p.
Mathieu С (1996) Principle and application of the variable pressure SEM, Microscopy and analysis, 43,13.
Pichler H, Schmitt-Riegraf C and Hoke L (1997) Rock-forming minerals in thin section., Chapman, Lon¬
don.
Hitachi, (2000) Low temperature microscopy using a cooling stage. Hitachi technical data, 62.
Astley OM, Chanliaud E, Donald AM and Gidley MJ (2001) Structure of Acetobacter cellulose compos¬
ites in the hydrated state. Int. J. Biol. Macromole, 29, 193-202.
Slowko W (2001) Secondary electron detector with a micro-porous plate for environmental SEM,
Vacuum, 63,457—461.
Tai SSW and Tang XM (2001) Manipulating biological samples for environmental scanning electron
microscopy observation. Scanning, 23,267-272.
Часть 2
ОРГАНИЧЕСКИМ АНАЛИЗ
Глава 9. Физические методы фракционирования
органических веществ
9.1. Принципы и ограничения
9.1.1. Формы органического вещества в почве
В почве находится множество различных органических фрагментов, многие из которых
растительного происхождения (живые или отмершие корни, обрывки древесных во¬
локон, куски стеблей и опавшие листья), другие - животного (например, трупы, фе¬
кальные пеллеты, выбросы дождевых червей) от таких представителей макрофауны как
насекомые, паукообразные, многоножки, ракообразные, брюхоногие моллюски или
земляные черви.
Увеличивая масштаб наблюдений, можно обнаружить при помощи увеличительного
стекла органические остатки меньшего размера, например, корневые волоски, частично
разложившиеся остатки животных, нематод, грибов и водорослей.
Для наблюдения микроорганизмов (например, бактерий, актиномицет, простейших)
и остатков животного и растительного происхождения, которые все больше смешивают¬
ся с органоминеральными коллоидами, необходима бблыиая степень увеличения.
Таким образом, при первом взгляде органическое вещество почвы (ОВП) представ¬
ляется сплошной средой, состоящей из все более мелких остатков, которые могут быть
разделены физически.
9.1.2. Основные положения
Методы фракционирования, описанные в настоящей главе, включают ручную и меха¬
ническую сортировку, а также использование физических методов разделения по плот¬
ности, размеру частиц просеиванием и седиментационный анализ. Эти методы похожи
на методы, используемые для фракционирования минеральных частиц (см. гл. 2).
Ручную сортировку используют главным образом для исследования живых корней
в почве. Сортировку облегчает использование, например, аппарата для отмучивания
(см. раздел 9.2.2).
Фракционирование по плотности основано на различии между плотностью вещества
органического (около 1) и минерального происхождения (около 2,65 для первичных ми¬
нералов). Теоретически это идеальный метод для отделения неразложившихся в почве
растительных остатков. В настоящее время этот вид измерений широко используется
в исследованиях динамики углерода в почвах. Некоторые блоковые модели основаны
на данных, полученных методом разделения по плотности (Рати и Sidi, 1987; Arrouays,
1994). Эти методы описаны в разделе 9.2.4. Однако в зависимости от типа почвы ме¬
тод фракционирования по плотности может быть затруднен из-за прочной связи между
минеральными и органическими частицами. Его можно усовершенствовать, объединив
с методом фракционирования по размеру частиц (Sallih и Рати, 1993) и различными ме¬
тодами измельчения, описанными в разделе 9.2.3.
Целью фракционирования по размеру частиц является полное отделение органиче¬
ских компонентов почвы. В идеальном случае грубые фракции с размером частиц более
256
Часть 2. Органический анализ
50 мкм должны содержать неповрежденные растительные остатки, пылеватые фракции
с размером частиц 50—2 мкм (или 20—2 мкм) — микробные клетки и фрагменты, фрак¬
ции крупного ила (2—0,2 мкм) — органическое вещество органоминерального комплек¬
са, и, наконец, фракции мелкого ила (0,2-0 мкм) — недавно образованные метаболиты.
Некоторые методы, такие как датирование (Anderson и Paul, 1984) и измерение содер¬
жания изотопа 14С (Hassink и Dalenberg, 1996) или 5l3C (Puget и др., 1995), частично под¬
тверждают эту теорию. Другие исследования показали, что микроорганизмы и органи¬
ческие вещества тесно связаны с минеральными коллоидами и, следовательно, «чистое»
фракционирование биологических компонентов почвы невозможно {Ahmed и Oades,
1984). Однако в некоторых обзорных исследованиях (например, Christensen, 1992; Feller,
1994) различают три основных класса органических веществ:
• компартмент растительных остатков (> 20 мкм), не тесно связанных с минераль¬
ными песками с относительно высоким соотношением C/N (15-25) или высоким
соотношением ксилоза/манноза, указывающим на растительное происхождение
этого органического вещества;
• смесь органической пыли, включающей смесь органического вещества почвы
(ОВП) растительного и грибного происхождения, минеральной пыли и очень ста¬
бильных органоминеральных микроагрегатов; соотношения C/N и ксилоза/ман¬
ноза ниже, чем в предыдущем компартменте; происхождение этого органического
материала менее ясно, чем происхождение более крупного вещества;
• глино-органический компартмент (< 2 мкм), обогащенный аморфным ОВП; это
гумифицированный компартмент, который тесно связан с минеральными части¬
цами, с низкими соотношениями C/N (8—11) и ксилоза/манноза, указывающими,
что это органическое вещество может быть микробного происхождения.
9.1.3. Затруднения
Фракционирование по размеру частиц основано на методах рассева (обычно мокрого
рассева) для разделения частиц размером до 50 или 20 мкм. Для отделения самых мелких
частиц необходимо использовать методы седиментации, аналогичные методам, опи¬
санным в главе 2 для фракционирования минеральных частиц. Однако дополнитель¬
ные сложности вызывает учет плотности частиц, влияющий на осаждение, в соответ¬
ствии с законом Стокса (см. гл. 2). Среднее значение плотности, равное 2,65, которое
используется для минеральных частиц, не подходит для органических остатков {Elliott
и Cambardella, 1991). Однако при таком размере частиц органическое вещество часто
прочно связано с минеральными частицами. Так как содержание ОВП относительно
мало по сравнению с содержанием минеральных частиц, можно считать, что плотность
минеральных фракций изменяется незначительно.
Основную трудность метода физического фракционирования по плотности или раз¬
деления по размеру частиц представляет прочная связь между минералами и органиче¬
ским веществом, которая приводит к образованию агрегатов различных типов (рис. 9.1).
Эти агрегаты должны разрушаться, а органические компоненты — выделяться без раз¬
рушения. Различные методы измельчения, а также их ограничения и преимущества до¬
вольно подробно описаны в разделе 9.2.2.
Приготовление и особенно повторное увлажнение образца может приводить к из¬
менению органических компонентов. Образцы обычно высушивают перед хранением
в лаборатории. Высушивание, а также другие методы пробоподготовки {Pansu и др.,
2001) приводят к прекращению органического функционирования почвы, что вызывает
Глава 9. Физические методы фракционирования органических веществ
257
деактивацию или гибель микроорганизмов. При обработке используют жесткие методы,
поэтому предпочтительно снизить активность микроорганизмов хранением в холодном
месте или даже замораживанием свежей почвы.
При использовании любого метода консервации прекращение биологической актив¬
ности необходимо для сохранения состояния органического вещества почвы при пробо-
отборе, поскольку динамика развития некоторых органических компонентов опережает
изменения основных неорганических компонентов.
Однако повторное увлажнение снова запускает биологическую активность. Быстрый
рост численности микроорганизмов связан с их питанием растительными остатками,
которые освобождаются от их защитной глинистой оболочки в процессе пробоподго-
товки (эффект измельчения) и становятся доступными для микроорганизмов, а также
потреблением биомассы микроорганизмов, убитых в ходе этих операций. Часть этих
углеродсодержащих веществ затем минерализуется или трансформируется.
Таким образом, существует риск изменения содержания ОВП при повторном увлаж¬
нении образцов, которое необходимо в большинстве методов физического фракцио¬
нирования, описанных ниже. Однако этот риск уменьшается в присутствии большого
количества воды, так как наиболее активные пищевые цепи являются главным образом
аэробными. Риск можно также уменьшить использованием реагентов, неблагоприятно
влияющих на рост микроорганизмов, и проведением фракционирования как можно бы¬
стрее после увлажнения.
9.2. Методы фракционирования
9.2.1. Классификация
Методы физического фракционирования органических компонентов можно разделить
на три основные группы:
• извлечение корней растений;
• разделение по плотности;
• фракционирование по диапазонам размера частиц.
При использовании этих методов следует учитывать структуру почвенного материала,
т. е. мелкодисперсного гумифицированного органического вещества, связанного с не¬
органическими веществами в органоминеральные комплексы. Эти комплексы содержат
связи между твердыми частицами, приводящие к образованию различных типов агре¬
гатов в почве. Некоторые компоненты, подлежащие разделению, оказываются заклю¬
ченными в этих агрегатах (рис. 9.1). Трудности при фракционировании состоят в рас¬
щеплении этих агрегатов без разрушения компонентов, которые подлежат измерению.
Основные методы расщепления агрегатов описаны и обсуждены в разделе 9.2.3.
9.2.2. Извлечение корней растений
Цель и принцип
Этот тип разделения используется для оценки корневой продуктивности в почве. Дей¬
ствительно, продуцирование и кругооборот корней являются одним из основных источ¬
ников углерода в почве, наряду с экссудацией корней и продуцированием растениями
мертвой наземной биомассы. Углеродный баланс в почве и атмосфере являлся объектом
тщательных экспериментальных и методических исследований, например, Андерсон
и Инграм (Anderson и Ingram, 1989) описали методы оценки поступления органических
веществ в почву.
258
Часть 2. Органический анализ
Рис. 9.1. Образование органоминеральных комплексов, микроагрегатов и структурных
агрегатов (в соответствии с Bruckert, 1994)
Извлечение корней также необходимо при исследовании физиологии растений,
включая классификацию корней, оценку их массы и длины, химический анализ, изуче¬
ние биологических сообществ с грибами и бактериями.
Выделение обычно выполняют вручную, но существуют несколько типов приборов
для отмучивания.
Методики
Извлечение корней проводят из образцов цельной почвы в виде блоков или цилиндров.
Образцы должны храниться в полиэтиленовых пакетах при низкой температуре или
даже в замороженном состоянии. Если морозильника нет, образцы можно высушить
и вновь увлажнить перед промыванием, но лучше всего отмыть корни сразу после воз¬
вращения в лабораторию с поля.
Кроме содержания органического вещества, на легкость выделения корней влияют
гранулометрический состав, уплотнение и структура почвы. Простейший метод состоит
в осторожном промывании увлажненных проб почвы водой на сите; авторы рекоменду¬
ют разные размеры отверстий сита: от 2 мм (Abo, 1984) до 0,5 мм (Anderson и Ingram, 1989).
Материал, остающийся на сите, можно промыть водой и отделить декантацией. Для
удаления всех фрагментов остаток часто сортируют вручную под водой в плоских кон¬
тейнерах. Для этой работы могут понадобиться бинокулярная лупа и очень тонкий пин¬
цет. Трудности в работе часто связаны с типом почвы и корней.
Описано много установок для отмывания корней, отделения их от почвы отмучива-
нием, т. е. промыванием остатков и их разделением методом флотации на сите с диаме-
Глава 9. Физические методы фракционирования органических веществ
259
Воздух
Рис. 9.2. Схема установки для отделения корней растений от почвы гидропневматическим отму-
чиванием (в соответствии с Smucker и др., 1982). А — высокоэнергетическая промывная камера,
В - камера отмучивания, С — труба переноса, D — первое сито с малой кинетической энергией
(840 мкм), Е — второе сито (420 мкм). Труба С отделяется от камеры В для очистки и внесения
нового образца. Корни переносятся с первого сита (D) переменой направления промывания на
тонкое сито (Е)
тром ячеек 0,5 мм д ля удаления тяжелых частиц. Схема установки, основанной на прин¬
ципе гидропневматического отмучивания и разработанной Смукером с сотр. (Smucker
и др., 1982), приведена на рис. 9.2.
Установка, разработанная Бонзоном и Пикардом (Bonzon и Picard, 1969), предназна¬
чена для отделения корней от необработанной почвы, отобранной в форме цилиндра
или монолита. Она включает набор из 4 двойных сит, сделанных из дерева с латунной
сеткой, и состоит из прямоугольной секции (50x60 см) и цилиндрической нижней ча¬
сти. Верхнее сито имеет отверстия диаметром 1,18 мм, а нижнее сито — 1,4 мм. Сита
установлены в деревянной раме, которая приводится в возвратно-поступательное дви¬
жение со скоростью 12,5 колебания в минуту двигателем с шатунно-кривошипным ме¬
ханизмом. Образец объемом около 2 л помещают на верхнее сито и направляют на него
струю воды. Суспензию удаляют через верх отбортованной кромки верхнего сита. По
окончании отмывания содержимое сит переносят в воронку, снабженную очень тонким
ситом.
Воронка содержит органические фрагменты, а также камни и гравий диаметром бо¬
лее 1,4 мм. Если содержание гравия высоко, органические фрагменты должны быть
отделены сильной струей воды, направленной в основание воронки, и перенесены на
сито, расположенное ниже.
260
Часть 2. Органический анализ
После механического фракционирования почвы и корней может понадобиться руч¬
ное отделение корней от других органических остатков, которое может занять несколь¬
ко часов. Следовательно, не существует идеальной установки для исключения всех руч¬
ных операций.
Все методы сепарации приводят к потере мелких корней, поэтому промывные воды
и отстоявшиеся осадки должны периодически контролироваться для оценки этих потерь.
Замачивание на ночь в водном растворе гексаметафосфата натрия ускоряет процесс
отмывки корней из глинистых почв, но химическое воздействие может обесцветить кор¬
ни и разрушить некоторые растительные ткани, тем самым делая последующую иденти¬
фикацию живых корней невозможной. Такая предварительная обработка может также
влиять на последующий химический анализ корней. Кроме того, все эти методы обра¬
ботки могут повредить присутствующие растительные ткани, поэтому предпочтительно
отделять субпробу корней вручную и осторожно промывать корни минимальным коли¬
чеством воды для обеспечения точного химического анализа.
Промытые корни можно хранить в холодильнике в герметичных полиэтиленовых па¬
кетах, но замораживание предпочтительнее. Можно также добавить небольшое количе¬
ство бактерицидного вещества (например, тимола).
Массу сухого вещества и содержание органического углерода и азота (см. гл. 10) мож¬
но определять после высушивания при 70 °С в течение 48 ч. Бонзон и Пикард (Bonzon
и Picard, 1969) измеряли в дополнение к массе сухого вещества удельную поверхность
корней. Прокаливание с постепенным повышением температуры до 550 °С позволяет
определять содержание золы в корнях, а также неорганических элементов после раство¬
рения остатков в растворе кислоты.
9.2.3. Распределение частиц
Структура почвы и органических компонентов
Как указывалось во вводном разделе данной главы, почва всегда содержит переменные
количества крупных частиц неорганического (грубый песок, гравий) или органического
(фрагменты растений) происхождения в дополнение к структурным агрегатам, форма
и стабильность которых изменяются в зависимости от типа почвы.
В активной среде (мулль) процессы гумификации приводят к образованию относи¬
тельно больших количеств трансформированного органического вещества: микробные
метаболиты с быстрым круговоротом (например, многие полисахариды) и очень устой¬
чивые вещества фенольной структуры. Количество веществ обоих типов было оценено
с использованием моделей разложения ОВП (Рати и др., 2004). Оба типа веществ могут
связываться с минеральными веществами, образуя органоминеральные комплексы, та¬
кие как связывающие вещества, присутствующие в микроагрегатах почвы. Эти микро¬
агрегаты содержат и строительные материалы для более крупных агрегатов, включаю¬
щих органические частицы, органические остатки и бактерии (рис. 9.1).
В почвах с низким уровнем активности {модер, мор) возможно образование хорошо
различимого профиля, включающего горизонт стабильного органического вещества
{модер, мор) наряду с горизонтом, в котором накапливается перераспределенное органи¬
ческое вещество, что приводит к образованию органоминеральных комплексов структур
осаждения (Bruckert, 1994).
Таким образом, эффективность фракционирования зависит от различных сил сцеп¬
ления в почвенной структуре. В некоторых случаях простого смачивания достаточно для
разрушения макроагрегатов и рассеивания мелких частиц (гашение). В других случа-
Глава 9. Физические методы фракционирования органических веществ
261
ях необходимо использовать более жесткие дисперсионные методы для освобождения
микроагрегатов и органических остатков, включенных в структурные агрегаты.
Методы диспергирования
Диспергирование представляет собой разрыв некоторых органоминеральных связей
без фрагментирования растительных остатков и, по возможности, без повреждения
микробных клеток или структуры микроагрегатов. Следует иметь в виду, что цель гра¬
нулометрического фракционирования органического вещества отличается от цели гра¬
нулометрического анализа почвы (см. гл. 2). В гранулометрическом анализе использу¬
ются очень жесткие методы для разрушения веществ, связывающих глино-гумусные
комплексы, например, разрушение органического вещества пероксидом водорода или
разрыв органоминеральных связей при помощи реагентов с высокой комплексообразу¬
ющей способностью по отношению к железу и алюминию, таких как тетраборат натрия
или пирофосфат натрия.
В данном случае подобные методы неприменимы, поскольку органические компо¬
ненты должны быть выделены без повреждения или растворения. Наиболее широко ис¬
пользуемые методы можно разделить на три группы:
• диспергирование водой, возможно при механическом перемешивании различной
интенсивности;
• звуковое и ультразвуковое диспергирование;
• химическое диспергирование при помощи диспергирующих реагентов, не слиш¬
ком агрессивных по отношению к органическому веществу.
Механическое диспергирование под действием воды
Брукерт (Bruckert, 1994) рекомендовал механическое диспергирование под действием
воды вместо методов с использованием ультразвука. В этих методах эффективность об¬
работки существенно зависит от типа органоминерального вещества, связывающего
агрегаты; ультразвук может оказаться слишком деструктивным для некоторых соедине¬
ний в почве, так как разрушает хрупкие минералы и повреждает некоторые органиче¬
ские вещества, и, что особенно неприятно, «он приводит к разрушению микробных кле¬
ток, содержимое протоплазмы из которых адсорбируется на глинах» (McGuillи др., 1975).
Метод Брукерта с сотр. (Bruckert и др., 1978) представляет собой щадящую механиче¬
скую обработку при помощи контролируемого перемешивания в присутствии агатовых
шаров (35 г сухой почвы, просеянной через сито с диаметром ячеек 2 мм, и 200 мл воды
помещают в ротационный шейкер с пятью агатовыми шарами и перемешивают на ско¬
рости 50 об/мин в течение 15 ч). Феллер (Feller, 1979) разработал аналогичный метод для
тропических песчаных почв с низким содержанием гумуса. В этом случае рекомендуется
еще более мягкое механическое воздействие: 100 г почвы перемешивают в течение часа
с тремя стеклянными шарами в 300 мл дистиллированной воды.
Андро с сотр. (Andrewс и др., 1980) исследовали стандартную степную почву (тип -
чернозем) с очень устойчивым глино-гумусным комплексом. Сухую почву просеивали
через сито с диаметром ячеек 2 мм, перемешивали при медленном вращении (40 об/мин)
в воде (35 г почвы на 200 мл воды) в течение ночи при 20 °С с различным числом ага¬
товых шаров. Доля тонкой пылевато-илистой фракции (0-50 мкм), полученной этими
авторами, возрастала от 57% от массы почвы при перемешивании без агатовых шаров
до 90% при использовании механической фрагментации. Число шаров, если оно было
больше двух, мало влияло на долю тонкой фракции (рис. 9.3). С другой стороны, доля
262
Часть 2. Органический анализ
Число стеклянных шаров
(перемешивание в течение 15 ч)
Продолжительность перемешивания, ч
(15 стеклянных шаров)
Рис. 9.3. Влияние числа 10-миллиметровых агатовых шаров (а) и продолжительности
перемешивания (Ь) на фрагментацию агрегатов > 50 мкм (в соответствии с Andreux и др., 1980);
черные ромбы — доля фракции 0-50 мкм, масс. %; черные квадраты — содержание углерода во
фракции 0-50 мкм; % от общего С
фракции увеличивалась со временем вплоть до 15 ч перемешивания, не достигая следу¬
ющей стадии, указывая, что продолжается разрушение всех агрегатов крупнее 50 мкм.
Однако после определенного уровня механического воздействия (более трех шаров при
перемешивании в течение 15 мин или пять шаров при перемешивании в течение более
8 ч) наблюдалась частичная солюбилизация соединений углерода, поэтому не рекомен¬
дуется использовать такие жесткие условия.
Сиди (Sidi, 1987) также использовал перемешивание со стеклянными шарами для
фрагментирования карбонатной почвы из Туниса. На рис. 9.4 показано влияние на
распределение частиц по размерам (гранулометрический состав) длительности пере¬
мешивания без шаров или в присутствии трех стеклянных шаров (15 г почвы и 100 мл
воды, встряхивание со скоростью одно возвратно-поступательное движение в секунду).
Основным результатом механической обработки явилось разрушение самых крупных
макроагрегатов (200-2000 мкм), тогда как доля агрегатов промежуточного размера (50-
Рис. 9.4. Диспергирование средиземноморской почвы под действием воды (согласно Сиди, 1987,
15 г почвы/150 мл воды, возвратно-поступательный шейкер, одно возвратно-поступательное дви¬
жение в секунду): а) влияние продолжительности перемешивания с тремя стеклянными шарами
и без них: черные и белые квадраты — агрегаты 200-2000 мкм, с шарами и без, соответственно;
черные и белые треугольники — агрегаты 50-200 мкм, с шарами и без них, соответственно; чер¬
ные и белые круги — агрегаты 0-50 мкм, с шарами и без них, соответственно; Ь) влияние ультра¬
звука (80 Вт, 80 кГц) на перемешивание без шаров в течение 1 ч
Глава 9. Физические методы фракционирования органических веществ
263
200 мкм) оставалась практически одинаковой при перемешивании с шарами и без них.
Форма кривых также указывает, что процесс фрагментации происходит в две стадии: (1)
разделение самых крупных агрегатов (> 200 мкм) с образованием агрегатов промежу¬
точного размера (50-200 мкм) в течение первых 30 мин перемешивания и (2) разделе¬
ние промежуточных агрегатов с образованием микроагрегатов ила и пыли (с размером
частиц 0-50 мкм). В отличие от ситуации, представленной на рис. 9.3, перемешивания
с тремя стеклянными шарами в течение 1 ч достаточно для достижения плато на кривой
диспергирования для почвы этого типа.
Монье с сотр. (Monnier и др., 1962) рекомендовали выполнение диспергирования
перед фракционированием по плотности (см. раздел 9.2.4): сухое просеивание до разме¬
ра 500 мкм или кипячение в воде с последующим однократным промыванием спиртом
и высушиванием в сушильном шкафу.
Звуковое и ультразвуковое диспергирование
Хотя некоторые авторы считают звуковое и ультразвуковое диспергирование слишком
жесткими методами для органического вещества (Bruckert, 1994), их обычно рекоменду¬
ют для физического фракционирования органического вещества почвы.
Эдвардс и Бремнер (Edwards и Вгетпег, 1967) подвергли водную суспензию образца
почвы (10 г почвы на 25 мл воды) воздействию звуковых колебаний (9 кЩ, 50 Вт), ис¬
пользуя вибратор Raytheon S-IQ2A (Raytheon Со., Норвуд, США). Диспергирование мел¬
ких частиц размера ила (< 2 мкм) под действием звуковых колебаний в течение 30 мин
было оценено для 14 сильно различающихся структурой почв при определении грануло¬
метрического состава методом пипетирования (см. гл. 9). На рис. 9.5 видно, что степень
диспергирования была всегда гораздо выше, чем при простом перемешивании в воде,
и сравнима с той, которая была получена при использовании двух химических диспер¬
гирующих реагентов: смеси калгон — пероксид и натриевой смолы.
Рис. 9.5. Доли илистых фракций, полученные ультразвуковой обработкой, и три других метода
диспергирования 14 почв (согласно Edwards и Вгетпег, 1967)
264
Часть 2. Органический анализ
Степень диспергирования, полученная для суспензий при помощи ультразвукового
дезинтегратора MSE Cabinet Model 60 (Measuring and Scientific Equipment Ltd., Лондон)
с рабочей частотой 18—20 кЩ и мощностью 60 Вт, очень близка к величине, полученной
при использовании звуковых колебаний (рис. 9.6). После 30 мин (время, рекомендован¬
ное авторами) продолжительность обработки звуком лишь слабо влияет на содержание
полученного ила (рис. 9.7). На рис. 9.4, Ъ приведено сравнение влияния продолжитель¬
ности звуковой обработки, наблюдаемого Сиди (Sidi, 1987), с результатами, полученны¬
ми для высокочастотного ультразвукового зонда несколько большей мощности (80 кГц,
80 Вт); в этом случае плато было достигнуто раньше (через 8-10 мин) для диспергирова¬
ния частиц размера пыли (0-50 мкм).
После детального анализа результатов диспергирования почвы методом ультразвуко¬
вой обработки, проведенного Уотсоном (Watson, 1971), и критических замечаний о его
использовании Генрих и Бремнер (Genrich и Вгетпег, 1972) провели повторные иссле¬
дования метода. Они использовали 28 почв, покрывающих широкий диапазон значе-
Рис. 9.6. Сравнение долей ила, полученных при диспергировании пяти почв под действием
звуковой (9 кГц, 50 Вт) и ультразвуковой (18-30 кГц, 60 Вт) обработки суспензий из 10 г почвы
в 25 мл воды в течение 30 мин (согласно Edwards и Вгетпег, 1967)
Рис. 9.7. Влияние длительности ультразвуковой обработки на фрагментацию микроагрегатов
< 50 мкм (согласно Шаймухаметов с сотр., 1984), размеры частиц: черные квадраты — < 1 мкм,
черные ромбы — 1-5 мкм, черные треугольники — 5-10 мкм, крестики — 10-50 мкм
Глава 9. Физические методы фракционирования органических веществ
265
ний pH (3,6-8,2), содержания карбонатов (0-34% СаС03), гранулометрического состава
(2-59% песка, 7-72% ила) и органического вещества (0,14-9,4% органического С). Ис¬
следовались два типа приборов {Heat Systems Ultrasonics Inc., Плейнвыо, США): (i) стан¬
дартная модель Branson W-\%5Cc зондом (20 кГц, 80 Вт) и (й) ультразвуковой очиститель
Branson 220 с баком из нержавеющей стали (40 кГЦ, 100 Вт). Для модели с баком ис¬
пользовались различные методики: изменялись соотношения почва/вода в суспензиях,
ультразвуковая обработка проводилась в конической колбе или непосредственно в баке.
При использовании зондовой модели конец зонда (диаметром 1,27 см) погружали на
2 см в суспензию (10 г почвы в 25 мл воды) в стальных пробирках, охлажденных снаружи
ниже 20 °С.
Во всех случаях более высокая степень диспергирования была достигнута при ис¬
пользовании зондовой модели. Однако степень диспергирования существенно зависела
от качества поверхности зонда: продолжительность обработки при использовании зонда
с щербатой поверхностью была в 2—4 раза больше, чем для зонда в хорошем состоянии.
Поэтому следует осторожно полировать конец зонда тонкой абразивной бумагой после
каждых 30 мин использования. Согласно данным Генриха и Бремнера, неудовлетвори¬
тельные результаты, полученные ранее другими авторами, могут объясняться недоста¬
точно хорошим состоянием используемых зондов.
Они также показали, что после обработки ультразвуковым зондом в течение 15 мин
в описанных выше условиях доли ила, полученные для 28 почв, были всегда равны или
больше, чем при использовании пероксида и полифосфата натрия в методе Килмера
и Александера {Kilmer и Alexander, 1949), который в это время был стандартным методом
диспергирования {Soil Survey, 1960). Это исследование ясно продемонстрировало дис¬
пергирующую способность ультразвуковых зондов. Однако авторы осторожно заклю¬
чили, что ни один из методов диспергирования не может считаться универсальным для
всех почв.
Андерсон с сотр. {Anderson и др., 1981) исследовали распределение органического ве¬
щества в гранулометрических фракциях из двух почв типа чернозем. Они провели дис¬
пергирование этих почв ультразвуковой обработкой в течение 8 мин при большей мощ¬
ности, чем ранее (300 Вт, аппарат Bransonic 1510), но для более разбавленных суспензий
(соотношение почва/вода = 1:10). Тиссен и Стюарт {Tiessen и Stewart, 1983) исследовали
влияние культивации на состав органического вещества во фракциях частиц при ис¬
пользовании методики, аналогичной методике Андерсона с сотр. {Anderson и др., 1981).
На почве типа чернозем Шаймухаметов с сотр. (1984), так же как Андерсон с сотр.
{Anderson и др., 1981), наблюдали стадию фрагментации микроагрегатов (< 50 мкм) по¬
сле звуковой обработки в течение 30 мин, тогда как 96,4% более крупных агрегатов были
разрушены уже в течение одной минуты. Этот эксперимент подчеркнул огромную раз¬
ницу между стабильностью микроагрегатов и структурных агрегатов (рис. 9.1). Следует
также подчеркнуть (рис. 9.7), что микроагрегаты трех размеров от 1 до 50 мкм имеют
сходную стабильность, что может объясняться увеличивающимся количеством выде¬
ленных мелких илистых частиц из трех классов микроагрегатов размера пыли, без раз¬
личий между классами. Это отличается от поведения структурных агрегатов, для кото¬
рых наблюдается четкое различие в стабильности между фракциями 50—200 и 0—50 мкм
(рис. 9.4).
Чтобы исследовать органическое вещество почвы аквол, Катру и Шнитцер {Catroux
и Schnitzer, 1987) выполнили ультразвуковое диспергирование почвы в водной суспен¬
зии при соотношении почва/вода, равном 1:5 (это промежуточное значение между со¬
отношениями, использованными Генрихом и Бремнером {Genrich и Вгетпег, 1972) и Ан¬
266
Часть 2. Органический анализ
дерсоном с сотр. (Anderson и др., 1981)). 100 г почвы в 500 мл дистиллированной воды
перемешивали в магнитной мешалке и обрабатывали ультразвуком с помощью генера¬
тора Blackstone SS2 (400 Вт) в течение 15 мин (это более жесткая обработка, чем исполь¬
зованные предыдущими авторами), конец зонда был погружен в жидкость на 2-3 см,
чтобы уменьшить бурление.
1])егорич с сотр. (Gregorich и др., 1988) попытались более точно определить и оценить
воздействие ультразвука. Ультразвуковая вибрация вызывает кавитацию из-за лопания
микроскопических пузырьков, образующихся при местном снижении давления. Когда
пузырьки лопаются в суспензии, они образуют волны давления с механической энерги¬
ей, достаточной для разрыва связей в агрегатах. Эти авторы использовали зонд Branson
(20 кШ), мощность которого может регулироваться в диапазоне от 0 до 150 Вт. Голов¬
ка зонда (диаметром 12 мм) погружалась в суспензию (15 г агрегатов размером 1—2 мм
в 75 мл воды) на глубину от 5 до 10 мм. Выходная мощность зонда оценивалась по из¬
мерению повышения температуры известной массы воды в течение данного периода
времени. 1]эегорич с сотр. считали, что самым важным параметром является количество
приложенной энергии на миллилитр суспензии:
J=PtV\
где J — приложенная энергия, Дж/мл; Р— выходная мощность зонда, Вт; t— время, с;
К- объем суспензии, мл.
На рис. 9.8 представлены результаты, полученные этими авторами для темного глее-
вого гумусового горизонта возделываемой брунисоли. Этот тип почвы содержит очень
устойчивые пылеватые частицы. Для нее ни один из методов ультразвуковой обработки
не вызывает такого полного фракционирования, как перемешивание в присутствии пе¬
роксида водорода. Таким образом, органические связи, по-видимому, являются основ¬
ным типом связи между этими частицами. Эти авторы также наблюдали более прочные
связи между макроагрегатами (или псевдопесками), чем в большинстве работ, цитируе¬
мых выше; энергия, необходимая для диспергирования этих агрегатов, относительно
&
0)
а
8
Приложенная энергия, Дж/мл
Рис. 9.8. Ультразвуковое диспергирование материала из темного глеевого гумусового горизон¬
та возделываемой брунисоли (Gregorich и др., 1988): черные треугольники - фракция > 50 мкм,
черные квадраты - фракция 2-50 мкм, черные ромбы - фракция < 2 мкм; горизонтальные линии
представляют степень диспергирования, полученную после обработки Н202 для разрушения ор¬
ганического вещества
267
Глава 9. Физические методы фракционирования органических веществ
обогащенных органическим веществом, находилась в диапазоне между 300 и 500 Дж/мл.
Как и в пылеватых частицах, органическое вещество, по-видимому, служит связываю¬
щим материалом, в частности, в макроагрегатах. Одной из возможных причин является
то, что их органическое функционирование в этом типе почвы несколько отличается
(оно частично анаэробное) от вышеприведенных примеров.
Балесдент с сотр. (Balesdent и др., 1991) исследовали влияние ультразвука на грануло¬
метрическое распределение органического вещества в 17 образцах бурых почв (горизон¬
ты возделываемых почв) или слабо обработанных аллювиальных почв. Использован¬
ная авторами методика состояла из механической и ультразвуковой обработки. Первый
этап включал механическое диспергирование водных суспензий на ротационном шейке¬
ре со стеклянными шарами, аналогично описанным выше методам {Andreux и др., 1980).
Затем применялась ультразвуковая обработка фракции < 50 мкм или < 25 мкм, чтобы
разделить ее на три части с размерами частиц 0-0,2, 0,2-2 и 2—50 мкм (или 2-25 мкм).
Использовалось то же ультразвуковое оборудование, что описано выше (клеточный
дезинтегратор Branson, 20 кГЦ, 150 кВт, зонд с плоским концом 13 мм в диаметре).
Кинетическое исследование воздействия ультразвука на микроагрегаты размера пыли
было выполнено Балесдентом с сотр. (Balesdent и др., 1991). Ультразвуковой обработ¬
ке была подвергнута суспензия объемом 100 мл с соотношением почва/вода =1:3, зонд
был погружен на 3 см, и аппарат отрегулирован на 70% мощности (соответствующей
0,5 Вт/мл по данным производителя). На рис. 9.9 показаны изменения фракций 0—0,2
и 0,2-2 мкм, а также их суммы (0—2 мкм) по сравнению с эталонным методом (обработ¬
ка пероксидом водорода и диспергирование пирофосфатом).
По сравнению с результатами Г^егорича с сотр. (рис. 9.8), диспергирование почв, ис¬
следованное Балесдентом с сотр., проходит легче, так как оно приводит к стабильному
образованию илистой фракции 0-2 мкм в диапазоне приложенной энергии от 300 до
1800 Дж/мл (рис. 9.9). Для сравнения, на рис. 9.8 наблюдается постоянное увеличение
содержания илистой фракции с ростом приложенной энергии. Однако рис. 9.8 пока¬
зывает отчетливый излом кривой илистой фракции для энергии примерно 300 Дж/мл,
Приложенная энергия, Дж/мл
Рис. 9.9. Влияние энергии ультразвуковой обработки на фрагментирование микроагрегатов
размера ила в аллювиальной почве (1) и выветрившейся бурой почве (2); горизонтальные линии
представляют степени диспергирования, полученные химической обработкой Н202 и затем Na3P04
(Balesdent и др., 1991): черные квадраты — фракция 0-0,2 мкм, белые круги — 6,2-2 мкм, черные
круги — фракция 0-2 мкм
268
Часть 2. Органический анализ
т. е. энергии, необходимой для достижения этапа, показанного на рис. 9.9. Роско с сотр.
(Roscoe и др., 2000) обнаружили, что энергия, равная 260-275 Дж/мл, достаточна для
того, чтобы разрушить неустойчивые агрегаты (2000—100 мкм) и в то же время оставить
стабильные агрегаты (100-2 мкм) неизменными в глинистой латосоли.
Результаты Балесдента с сотр. (Balesdentuwp., 1991) (рис. 9.9) трудно сравнимы с дан¬
ными Шаймухаметова с сотр. (рис. 9.7), где порог измельчения тонких фракций был
равен 1 мкм. Однако продолжительность ультразвуковой обработки для достижения
стадий измельчения 0-2 и 0—1 мкм была близка в обоих случаях. Более детальное ис¬
следование фракционирования фракции 0—2 мкм, проведенное Балесдентом с сотр.,
предоставило интересную дополнительную информацию относительно двух аспектов:
• Даже при наибольшей энергии ультразвуковой обработки стабильная стадия не до¬
стигается для тонкой фракции < 0,2 мкм, а степень диспергирования этой фракции
всегда ниже, чем получаемая при использовании эталонного химического метода.
• Для одной из почв содержание промежуточной фракции (0,2—2 мкм) достига¬
ло максимума примерно через 5 мин ультразвуковой обработки (150 Дж/мл). Это
предполагает, что начальная стадия фрагментирования включала деление агрега¬
тов размера пыли (5-25 мкм) до более мелких элементов (0,2-2 мкм), а не деле¬
ние фракции 0,2—2 мкм. Поведение ассоциатов частиц во фракции размера ила
отличается от того, что наблюдалось во фракции размера пыли, для которой три
исследованные диапазона размеров частиц (рис. 9.7) продемонстрировали одина¬
ковую стабильность. Оно скорее напоминало поведение макроагрегатов (рис. 9.4):
крупные структурные агрегаты (> 200 мкм) менее стабильны, чем промежуточные
макроагрегаты (50-200 мкм).
В заключение, Балесдент с сотр. рекомендовали для исследованных почв ультразву¬
ковую обработку (600 Дж/мл) в течение 10 мин в указанных выше условиях. Дисперги¬
рование пылеватых микроагрегатов (до 2 мкм) можно считать завершенным, тогда как
грубую фракцию ила (0,2—2 мкм) следует считать микроагрегированной.
Балесдент с сотр. также исследовали влияние ультразвука на крупные органические
остатки, отделенные в воде после воздействия стеклянных шаров. Исследование про¬
водили на аллювиальной почве, содержащей 27% ила и 0,9% органического углерода
при pH 7. На этой почве выращивали пшеницу и кукурузу в течение 17 лет, поэтому
крупные остатки принадлежали, главным образом, этим растениям. Ультразвуковую об¬
работку водных суспензий проводили при различных уровнях энергии и соотношении
почва/раствор 1:200 для каждой из трех легких фракций: 200-2000,50-200 и 25-50 мкм.
Суспензии затем просеивали через сито с отверстиями 25 мкм и, если необходимо, 50
и 200 мкм. Суспензия с размером частиц 0-25 мкм была разделена методом седимента¬
ции на фракции с размером частиц 0-5 и 5-25 мкм.
Результаты исследования показали сильное деструктивное воздействие ультразвука
на органические остатки. После обработки в течение 10 мин (рекомендуемая продол¬
жительность обработки для фракционирования илистых частиц) более 60% углерода из
исходной грубой органической фракции попало во фракции частиц меньшего размера,
и это наблюдалось для каждого изученного диапазона размеров частиц. Авторы показа¬
ли, что это фракционирование частично происходит из-за сепарации илистых фракций,
связанных с растительными остатками, однако очистка растительных остатков является
недостаточным объяснением количества органического вещества, перенесенного в бо¬
лее мелкие фракции.
Использование ультразвука в условиях, необходимых для диспергирования ила, при¬
водит к заметному фракционированию крупных растительных остатков. Это важное
Глава 9. Физические методы фракционирования органических веществ
269
наблюдение позволило авторам предложить метод для фракционирования по размеру
частиц с использованием только ультразвука для суспендирования частиц мельче 50 мкм
(см. раздел 9.2.4).
Химическое диспергирование
Методы химического диспергирования находят меньшее применение для фракциони¬
рования органического вещества, чем классический гранулометрический анализ почвы
(см. гл. 2).
Как указано в разделе 9.2.2, только такие изолирующие реагенты, как тетраборат или
гексаметафосфат натрия, можно использовать для диспергирования глинистых почв для
выделения корней или крупных фрагментов растений {Anderson и Ingram, 1989). Но даже
в этом случае существует риск обесцвечивания (что впоследствии затрудняет идентифи¬
кацию живых корней) и модификации органических веществ.
Не рекомендуется использовать для исследования гранулометрического состава ор¬
ганического вещества диспергирующие реагенты, обладающие высокой деструктивной
способностью по отношению к органоминеральным связям. Их слишком высокая экс¬
трагирующая способность, в частности по отношению к гуминовым и фульвокислотам
(см. гл. 11), может искажать результаты таких исследований. Следует использовать ме¬
нее агрессивные экстрагирующие реагенты, такие как нейтральные соли одновалентных
металлов, которые вызывают диспергирование глин через обмен с двух- и трехвалент¬
ными катионами обменного комплекса и последующий разрыв определенных органо¬
минеральных связей.
Ладц с сотр. {Laddи др., 1977), Удино (Oudinot, 1985) и Саллих и Пансу {Sallih и Рати,
1993) использовали раствор бикарбоната натрия в дополнение к механическому пере¬
мешиванию для первоначального диспергирования почв.
Натриевые смолы также использовались для диспергирования почв {Edwards
и Вгетпег, 1967; Rouillern др., 1972). Основанные на результатах Эдвардса и Бремнера
{Edwards и Вгетпег, 1967) данные (рис. 9.5) показывают, что при использовании смол
степень дисперсии немного выше, чем при использовании двух других методов, опро¬
бованных для 14 почв в этом эксперименте.
Феллер с сотр. {Feller ъ др., 1991) сравнили различные методы диспергирования, ис¬
пользующие амберлитовую смолу IRN 77 в натриевой форме. Смолу исследовали при
отдельном использовании (С) или в сочетании с ультразвуком (С/УЗ) при фракциони¬
ровании фракции < 50 мкм. Метод с использованием смолы сравнивали с пятью дру¬
гими методами диспергирования, которые были объединены с тем же ультразвуковым
фракционированием фракций < 50 мкм: метод Ш/УЗ, аналогичный описанному выше
методу Балесдента с сотр. {Balesdent и др., 1991); метод NaCl/УЗ, в котором вода в су¬
спензии замещена на 1 М раствор хлорида натрия ; метод, использующий 1 М раствор
гидроксида натрия для доведения pH суспензии до 10 (pH 10/УЗ) и метод с использова¬
нием раствора гексаметафосфата натрия (3,3 г/л) (ГМФ/УЗ).
На рис. 9.10 показана сравнительная эффективность различных методов на феррал-
литной почве из Мартиники. Вплоть до уровня мелкой пыли, наиболее эффективное
диспергирование было достигнуто при использовании метода С/УЗ (обработка цельной
почвы смолой, затем фракции < 50 мкм ультразвуком). В среднем, солюбилизация ор¬
ганического вещества составила менее 4% общего углерода для 19 исследованных почв.
По результатам этого эксперимента метод С/УЗ представляется предпочтительным ме¬
тоду с использованием стеклянных шаров плюс ультразвук (Ш/УЗ), описанному выше
{Balesdent и др., 1991). Однако эти два метода не проверялись на одних и тех же типах
270
Часть 2. Органический анализ
2
Ш/УЗ
NaCI/УЗ
рНЮ/УЗ
ГМФ/УЗ
Ш/С
С
С/УЗ
Фракция, мкм
Рис. 9.10. Влияние различных методов диспергирования на фракционирование по размеру ча¬
стиц ферраллитной почвы из Мартиники (согласно Feller и др., 1991): УЗ — ультразвуковая об¬
работка фракции 0-50 мкм; Ш — перемешивание с шарами; NaCI — раствор NaCI; pH 10 — рас¬
твор NaOH; ГМФ — раствор гексаметафосфата натрия; С — перемешивание с натриевой формой
смолы
почв. Кроме того, авторы указали на практические ограничения в использовании смол:
для подготовки и регенерации смолы требуется довольно длительное время, и существу¬
ет риск загрязнения почвы мелкими частицами смолы (< 50 мкм).
Метод с использованием натриевой формы смолы оказался самым эффективным
из пяти методов диспергирования, опробованных на стабильных оксисолях с большим
содержанием гиббсита (Bartoli и др., 1991). Эти авторы также исследовали влияние со¬
отношения почва/натриевая смола на степень диспергирования, pH суспензии и солю¬
билизацию углерода. Определенные объемы (0, 10, 50, 100, 200, 300, 400 мл) натриевой
смолы Амберлит IR-120 (500 мкм) в нейлоновых пакетах с отверстиями 50 мкм добав¬
ляли к 2,5 г пробам почвы в 200 мл дистиллированной воды. Суспензии перемешивали
на ротационном шейкере при 40 об/мин в течение 16 ч. Результаты, приведенные на
рис. 9.11, показывают, что этап стабильного разрушения агрегатов находится в диапазо¬
не объема смолы от 50 до 200 мл, что соответствует объему (100 мл), использованному
Феллером с сотр. {Feller и др., 1991). Наблюдалось повышение pH суспензии на 1-2 еди¬
ницы; во всех случаях окончательная величина pH оставалась ниже, чем у основных экс¬
трагирующих реагентов для гуминовых кислот (см. гл. И). В более глубоком горизонте
растворение органического углерода становится заметным только для объемов смолы,
превышающих 200 мл; с другой стороны, в обрабатываемом поверхностном горизонте
авторы заметили растворение органического углерода для всех добавленных количеств
смолы; этот горизонт, вероятно, содержит более свежее органическое вещество, которое
в меньшей степени гумифицировано и более растворимо в воде.
Глава 9. Физические методы фракционирования органических веществ
271
Рис. 9.11. Влияние объема натриевой смолы (2,5 г на 200 мл дистиллированной воды) на степень
диспергирования агрегатов, pH почвенной суспензии и солюбилизацию органического углерода
в поверхностном горизонте (слева) и в более глубоком горизонте (справа) оксисоли из Нигерии
(Bartoli и др., 1991)
9.2.4. Разделение по плотности
Методы
Первые методы выделения органических остатков из почвы были основаны на очевид¬
ном физическом свойстве: плотность свободного органического вещества (около 1)
ниже, чем плотность органоминерального комплекса. Однако первые методы фрак¬
ционирования по плотности были использованы для выделения первичных минералов
(Pearson и Truog, 1937). Основываясь на работе Лина (Lein, 1940), Энен и Турк (Henin
и Тигс, 1949) адаптировали метод денситометрического разделения к свободному орга¬
ническому веществу в почве. Фракционирование проводили в лабораторных стаканах,
содержащих смесь бромоформа и бензола. Дженсон-Луусинанг (Jeanson-Luusinang, 1960)
усовершенствовал методику, используя специальные воронки для декантации; затем
Монье с сотр. (Monnieru др., 1962) адаптировали более ранний метод Лина (Lein, 1940)
для разделения по плотности методом центрифугирования.
Метод центрифугирования улучшил использование различий в плотности, но ни
один из этих методов не выделяет все легкое органическое вещество. Монье с сотр.
(Monnier и др., 1962) проводили исследования с искусственными смесями минеральной
почвы и овсяной соломы. При фрагментировании соломы до частиц размером < 0,2 мм
степень извлечения составляла 73% добавленной соломы для метода Монье и 44% для
метода Дженсона-Луусинанга. Эрроуэйс (Arrouays, 1994) использовал метод Монье для
моделирования эволюции запасов углерода.
272
Часть 2. Органический анализ
Пжнланд и Форд (Greenland и Ford, 1964) использовали ультразвук для диспергирова¬
ния агрегатов перед разделением по плотности (см. раздел 9.2.3). Метод был усовершен¬
ствован Фордом с сотр. (Ford и др., 1969), использовавшими поверхностно-активное
вещество и дибромхлорпропан (ДБХП, плотность 2,06) вместо бромоформа для разде¬
ления по плотности. В то время авторы не знали о возможной токсичности этих соеди¬
нений, которая сегодня широко известна.
Турченек и Оадес (Turchenek и Oades, 1979) исследовали методы фракционирования
органического вещества по плотности, комбинируя их с предварительным фракциони¬
рованием по размеру частиц. Они проводили от 4 до 7 опытов по фракционированию
смесей в декалине (декагидронафталин, d- 0,88), дибромхлорпропане (ДБХП, d= 2,06)
и бромоформе (d =2,88) на большинстве из семи стандартных диапазонов размера ча¬
стиц (крупный песок, мелкий песок, крупная пыль, мелкая пыль, крупный ил, средний
ил, мелкий ил).
Их наблюдения показали, что более 50% легкой фракции (d < 2,06) с частицами раз¬
мера крупного и мелкого песка состоит из органического вещества. Фракция, содержа¬
щая более крупные частицы, состоит главным образом из распознаваемых растительных
остатков с высокими соотношениями C/N и низкой растворимостью. Фракция, состоя¬
щая из более мелких частиц (от мелкого песка до крупного ила) содержит большую долю
распознаваемых остатков микробных клеток и растворимые ароматические гуминовые
соединения.
Фракции легкого ила также обогащены органическими веществами. Все фракции со¬
держат значительные количества окисного железа, алюминия и кремния, что указывает
на широкий диапазон различных взаимодействий между неорганическим и органиче¬
ским веществом.
В настоящее время не используют ни один из методов разделения по плотности с тя¬
желыми хлорированными растворителями из-за их токсичности и требований к безо¬
пасности работ в лаборатории.
Дабин (Dabin, 1976) предложил метод фракционирования органических материалов
(см. гл. 11). Первая часть этого метода включала фракционирование по плотности в 2 М
фосфорной кислоте (d=1,2). Кроме низкой токсичности фосфорной кислоты по сравне¬
нию с использованными жидкостями, преимущество этого метода кислотной обработки
состоит в разрушении карбонатов в известковых почвах и выделении определенного ко¬
личества связанного растительного материала. Сиди (Sidi, 1987) использовал этот метод
для выделения легких фракций из смесей почв и пшеничной соломы, выдержанных в тер¬
мостате в контролируемых лабораторных условиях. Метод был использован для разработ¬
ки описательной модели динамики углерода с тремя компартментами (Pansu и Sidi, 1987).
Метод Ладда с сотр. {Ladd и др., 1977) включает серию операций фракционирова¬
ния по размеру частиц и плотности, которые также применимы к известковым почвам.
Модификация этого метода позволяет проводить на первом этапе фракционирование
легких материалов in vitro в инкубационных экспериментах на смесях почв и пшенич¬
ной соломы, меченой 14С (Cortez, 1989; Sallih и Pansu, 1993). Предполагаемая модифика¬
ция включала использование насыщенного водного раствора сульфата цинка {d = 1,4)
в качестве тяжелой жидкости, тогда как Ладд с сотр. использовали четыреххлористый
углерод (d-1,59). Среди различных насыщенных солевых растворов высокой плотности
наиболее перспективными являются растворы сульфата цинка и сульфата двухвалент¬
ного железа (d = 1,6). Использовался раствор сульфата цинка, чтобы не допустить свя¬
зывания железа в органоминеральные комплексы. Однако использование цинка также
может привести к образованию определенных комплексов. Применяли и другие плот-
273
Глава 9. Физические методы фракционирования органических веществ
ные минеральные жидкости, включая растворы хлорида цинка (Besnard и др., 1996), ме¬
тавольфрамата натрия (Elliott и др., 1991), поливольфрамата натрия (Cambardella и Elliott,
1993; Golchin и др., 1994; Six и др., 1999), лудокс (водную суспензию коллоидных частиц
окиси кремния) (Meijboom и др., 1995).
Андерсон и Инграм (Anderson и Ingram, 1989) рекомендовали использовать методы
фракционирования легких материалов в воде, весьма похожие на методы, описанные
для отделения корней. Легкая фракция определяется как фракция, (1) которая всплыва¬
ет при диспергировании в воде и (2) проходит сквозь 2-миллиметровое сито, но задер¬
живается на 0,25-миллиметровом сите. Однако авторы указывали, что методы отмучи-
вания и рассева выделяют значительно меньшие количества свободного органического
вещества, чем методы разделения по плотности. Тем не менее, методы разделения в воде
имеют смысл, поскольку в воде солюбилизируется меньшее количество органического
вещества, чем в тяжелых жидкостях, которые часто обладают коррозионными свойства¬
ми (Веагеидр., 1994; Pugetnjsp., 1996).
Методики
Здесь описаны только методы Монье с сотр. (Моптеги др., 1962), Дабина (Dabin, 1976)
и с использованием концентрированного раствора ZnS04 (о модификациях см. раздел
2.4.1).
Метод с использованием органической тяжелой жидкости
Плотность жидкости доводится до требуемой величины смешиванием бромоформа с бо¬
лее легким растворителем, предпочтительно спиртом (Моптеги др., 1962). Авторы, ра¬
ботавшие с пылеватыми почвами из района Версаля (Франция), рекомендовали исполь¬
зовать жидкость с плотностью, равной 2. Они указывали, что более полное разделение
может быть достигнуто при последовательном использовании жидкостей с плотностью
1,75,2 и 2,25.
Образец почвы должен быть высушен на воздухе и измельчен до размера частиц 2 мм.
Отвешивают 5—10 г почвы, в зависимости от содержания свободного органического ве¬
щества. Вес пробы может быть уточнен с учетом методов, используемых для количествен¬
ного определения после фракционирования (например, определение содержания угле¬
рода во всей легкой фракции). Помещают пробу в центрифужную пробирку на 100 мл
и заполняют пробирку тяжелой жидкостью. После перемешивания стеклянной палоч¬
кой центрифугируют с ускорением в центре пробирки около 1000 g в течение 5 мин. Со¬
бирают надосадочную жидкость (супернатант) на плоском фильтре и повторяют опера¬
цию, начиная с суспендирования отцентрифугированной гранулы в тяжелой жидкости.
Можно разрушить агрегаты для высвобождения включенного легкого органического
вещества перед фракционированием, прокипятив в воде, промыв спиртом и высушив
в сушильном шкафу, либо просеяв сухую пробу через сито с отверстиями 500 мкм.
Для почв с высоким содержанием свободного органического вещества высок риск
изоляции тяжелых частиц внутри легкого органического материала, поэтому рекомен¬
дуется снова отцентрифугировать легкие материалы после промывания в другой про¬
бирке с тяжелой жидкостью.
Метод разделения по плотности с использованием фосфорной кислоты
Метод Дабина (Dabin, 1976) применяют для почвы, просеянной через сито с ячейками
0,5 мм. Масса пробы может быть от 5 до 40 г, в зависимости от содержания органиче¬
ского вещества. Перемешивают в течение 30 мин в возвратно-поступательном шейке¬
274
Часть 2. Органический анализ
ре (одно возвратно-поступательное движение в секунду) с 200 мл водного 2 М раствора
фосфорной кислоты (136 мл кислоты на 1 л воды). Центрифугируют при 3000 g в те¬
чение 20 мин и переносят супернатант на фильтр. Повторяют эту операцию дважды.
Высушивают и взвешивают растительный материал, собранный на фильтре. Содержа¬
ние общего углерода в этом материале можно измерить сжиганием и определением вы¬
делившегося диоксида углерода; одновременно можно измерить содержание азота на
CHN-анализаторе или отдельно методом Къельдаля (см. гл. 10).
Метод просеивания и тяжелой неорганической жидкости
На рис. 9.12 в обобщенном виде представлена методика выделения свободного органи¬
ческого вещества (СОВ). Просеивают 80 г почвы сквозь 5-миллиметровое сито и готовят
суспензию в 300 мл водного 0,2 М раствора NaHC03. После умеренного перемешивания
на ротационном шейкере в течение 1 ч центрифугируют при 12 000 g в течение 30 мин.
Собирают легкую фракцию фильтрованием супернатанта. Экстракцию можно повто¬
рить дважды.
Суспендируют отцентрифугированный осадок в 500 мл воды. Для диспергирования по¬
мещают на роторную мешалку и перемешивают на максимальной скорости в течение
2 мин. Просеивают сквозь сито с отверстиями 50 мкм в воде. Суспендируют крупную
Продолжение на рис. 9.13
Рис. 9.12. Схема отделения свободного органического вещества по плотности и просеиванием
Глава 9. Физические методы фракционирования органических веществ
275
фракцию (> 50 мкм) в тяжелом водном растворе (насыщенный раствор сульфата цинка
с плотностью 1,4). После перемешивания с умеренной скоростью в течение 30 мин цен¬
трифугируют при 3000 g и фильтруют супернатант через мембрану 3 мкм «Миллипор».
Осторожно промывают легкую фракцию, собранную на этом фильтре, водой четыре
раза, добавляют к предыдущей легкой фракции и сушат в сушильном шкафу при 30 °С.
Если не требуется отделять песок от СОВ, метод можно упростить, исключив разде¬
ление по плотности крупной фракции. В этом случае содержание СОВ оценивают, опре¬
деляя содержание органического углерода во фракции «легкий материал, отделенный
в NaHC03 + фракция >50 мкм». Исследование некоторых типов почв с применением
метки 14С показало, что эта упрощенная оценка значительно точнее, чем предыдущая
(Sallih и Bottner, Cefe-CNRS, Монпелье, Франция, неопубликованные данные).
9.2.5. Фракционирование по размерам частиц
Ограничения методов разделения по плотности
Метод разделения по плотности для исследования органических компонентов иногда
подвергался критике, но не тогда, когда он использовался совместно с фракционирова¬
нием по размеру частиц. Одной из причин была вышеупомянутая токсичность тяжелых
органических жидкостей. Это препятствие может быть преодолено использованием тя¬
желых насыщенных водных растворов минеральных солей. Согласно Брукерту (Bruckert,
1994), использование плотности в качестве единственного критерия также может быть
оспорено по нескольким причинам:
• Идеальная величина плотности используемой жидкости зависит от типа почвы.
Так, при плотности жидкости 1,8 в легкую фракцию может быть выделено 90% ор¬
ганического вещества андосолей, тогда как соответствующая доля в бурых почвах
составляет только 20%.
• Плотность растительных остатков увеличивается при разложении из-за внедрения
минерального вещества, содержание которого можно рассчитать после определе¬
ния золы.
• При использовании тяжелых органических жидкостей органические компоненты
могут фиксироваться на глинах и искажать результаты последующих определений.
Как указано выше, тяжелые неорганические жидкости не имеют этого недостатка,
но они могут изменить органоминеральные комплексы.
Методики фракционирования по размеру частиц
С учетом замечаний о диспергировании агрегатов, приведенных в разделе 9.2.3, трудно
найти единую методику фракционирования по размеру частиц для всех типов почв. Од¬
нако можно выбрать четыре методики для различных типов почв:
• продолжение метода просеивания и тяжелой неорганической жидкости (см. ниже),
адаптированного с учетом данных Ладца с сотр. (Ladd и др., 1977) для известковых
почв;
• перемешивание со стеклянными шарами и ультразвуковая обработка (Balesdent
и др., 1991), используемые для различных возделываемых почв Франции;
• применение смолы, насыщенной ионами Н+, и ультразвуковая обработка (Feller
и др., 1991), используемая для тропических почв различного происхождения
с упрощенной методикой седиментации (Gavinelli и др., 1995);
• специальная методика для песчаных почв (Feller, 1979; Feller и др., 1991).
276
Часть 2. Органический анализ
Продолжение метода просеивания и тяжелой неорганической жидкости (см. раздел 9.2.4)
В дополнение к отделению фракции «свободного органического вещества», описанному
в разделе «Метод просеивания и тяжелой неорганической жидкости» (рис. 9.12), этот
метод предусматривает выделение:
• водорастворимой органоминеральной фракции;
• фракции крупнее 50 мкм (главным образом, неорганические вещества, плот¬
ность > 1,4);
• органоминеральной фракции частиц меньше 50 мкм.
Метод включает разделение последней фракции на частицы размера пыли (2—
50 мкм), крупного ила (0,2—2 мкм) и мелкого ила (0—0,2 мкм). Должна быть использо¬
вана методика разделения, описанная Ладдом с сотр. (Ladd и др., 1977) и схематически
представленная на рис. 9.13: (1) центрифугирование в пробирке на 250 мл при 4000 g
в течение 15 мин позволяет отделить мелкую фракцию (менее 0,2 мкм) в супернатанте;
(2) повторное суспендирование отцентрифугированного осадка в воде и центрифуги¬
рование при низкой скорости (800 g) в течение 5 мин; повторенная дважды, эта опе¬
рация позволяет выделить в отцентрифугированный осадок частицы с размером пыли
(50—2 мкм); (3) получение супернатанта, содержащего частицы размера крупного ила
(0,2-2 мкм).
Две илистых фракции и водорастворимую фракцию концентрируют в вакуумном ро¬
торном испарителе при 40 °С. Таким образом, метод (рис. 9.12 и 9.13) позволяет полу¬
чить шесть фракций: водорастворимая фракция 0—0,2 мкм, фракция 0,2—2 мкм, фрак¬
ция 2—50 мкм, тяжелая крупная фракция (частицы > 50 мкм и плотность > 1,4) и легкая
крупная фракция (частицы > 50 мкм и плотность < 1,4).
См. рис. 9.12
Рис. 9.13. Фракционирование пылевато-илистой фракции центрифугированием (дополнение
к рис. 9.12с учетом данных Ладда с сотр. (Ladd и др., 1977))
277
Глава 9. Физические методы фракционирования органических веществ
Каждую фракцию высушивают в чашке Петри в зависимости от последующих изме¬
рений — либо при низкой температуре, либо при комнатной температуре (простейший
вариант), или в сушильном шкафу или на песчаной бане.
Перемешивание со стеклянными шарами и ультразвуковая обработка
На рис. 9.14 представлена сводная схема фракционирования согласно Балесденту с сотр.
(Balesdent и др., 1991). Почвы сушат и просеивают через 2-миллиметровое сито, исполь¬
зуя машину для размола и рассева с роликами (Pansu и др., 2001). Помещают 50 г почвы
в пластиковую бутыль на 250 мл, содержащую 180 мл воды и 10 стеклянных шаров диаме¬
тром 5 мм. Перемешивают бутыль на ротационном шейкере при 40 об/мин в течение 16 ч.
Фильтруют суспензию под водой на сите с квадратными отверстиями размером
200 мкм. Добавляют малыми порциями в суспензию в стакане. Органические фрагмен¬
ты разделяют во время их переноса на сито 200 мкм декантацией. Повторяют операцию
несколько раз до тех пор, пока песок больше не содержит видимых органических фраг-
50 г почвы
Рис. 9.14. Размер частиц и фракционирование центрифугированием после диспергирования
цельной почвы перемешиванием со стеклянными шарами и ультразвукового диспергирования
фракции < 50 мкм (Balesdent и др., 1991)
278
Часть 2. Органический анализ
ментов. Выполняют ту же самую операцию с фракцией < 200 мкм и ситом 50 мкм для
получения фракций Ф200-2000, М200-2000, Ф50-200, М50-200 (где Ф - органические
фрагменты, М - органоминеральная часть).
Центрифугируют суспензию, содержащую частицы < 50 мкм, чтобы отделить частицы
< 0,2 мкм (см. Продолжение описанного метода); сохраняют супернатант. Суспендируют
отцентрифугированный осадок при массовом соотношении твердая фаза/вода, равном
примерно 1:3. Подвергают суспензию ультразвуковой обработке в течение 10 мин в ус¬
ловиях, описанных выше, т. е. при приложенной энергии около 300 Дж/мл. Для проб,
содержащих известняк, рекомендуется удалять карбонаты из суспензии после ультразву¬
ковой обработки добавлением раствора НС1 до pH 3,5 при контроле pH-метром и про¬
мывать твердый остаток перед последующим разделением.
Фракции 2-50, 0,2-2 и 0-0,2 мкм разделяют методами центрифугирования, анало¬
гичными тем, что описаны ранее. Выбранные условия лишь слегка отличаются от ус¬
ловий, описанных предыдущими авторами: 25 мин при 2900 g для отделения мелкой
фракции < 0,2 мкм декантацией и 3 мин при 800 g для фракции 0,2-2 мкм. Эти условия
должны пересчитываться каждый раз на основании закона Стокса в зависимости от ра¬
бочих условий (см. гл. 2).
Вышеуказанные условия были рассчитаны Балесдентом с сотр. (Balesdent и др., 1991)
для стоксовых диаметров 0,2 или 2 мкм, плотности частиц 2,5 г/см3 и параметров, опре¬
деленных для их оборудования. Использованное значение плотности соответствует ско¬
рее минеральным, чем органическим частицам. Таким образом, указанные фракции не
точно соответствуют размеру органических частиц, но трудно разделить неорганическое
и органическое вещество, связанное во фракциях размера ила. После каждой деканта¬
ции супернатанта 0-0,2 или 0,2-2 мкм суспендируют отцентрифугированный осадок
в воде, перемешивают в течение 30 мин и снова центрифугируют. Авторы советуют
проводить четыре операции седиментации для фракции 0-0,2 мкм, а затем четыре для
0,2-2 мкм. Центрифугируют суспензию 0,2-2 мкм при 2900 g в течение 25 мин и полу¬
чают отцентрифугированный осадок. Смешивают супернатант с предварительно полу¬
ченными фракциями 0-0,2 мкм. Флокулируют суспензию добавлением раствора СаС12
(0,5 г/л); оставляют на ночь и затем центрифугируют. Отцентрифугированный осадок
представляет собой фракцию 0-0,2 мкм, а супернатант - это конечная водорастворимая
органическая фракция.
Фракционирование частиц размера ила может выполняться легче ультрацентрифуги-
рованием в непрерывном потоке (см. гл. 2).
Фракции крупнее 50 мкм должны высушиваться при 60 °С, а фракции мельче 50 мкм
следует гомогенизировать, заморозить и лиофилизировать. Их взвешивают и затем из¬
мельчают до 50 мкм для химического анализа, особенно для измерения содержания
С и N (см. гл. 10).
Фракционирование с использованием Н+-смолы и ультразвука
Метод декантации. В этом методе, описанном Феллером с сотр. (Feller и др., 1991), пер¬
воначальное диспергирование почвы выполняют с использованием катионобменной
смолы (Амберлит IRN11, в Na-форме), тщательно просеянной через сито 500 мкм. Опе¬
рацию просеивания выполняют перед каждым фракционированием. Разделяют смолу
на порции по 100 мл и помещают их в полиамидные пакеты (NytrelTUS) с отверстиями
45 мкм. Помещают эти пакеты в другие пакеты (Nytrel TI60) с отверстиями 60 мкм. Па¬
кеты закрывают резиновой лентой. Двойные пакеты защищают почву от загрязнения
при разрыве одного из них.
279
Глава 9. Физические методы фракционирования органических веществ
Затем выполняют диспергирование перемешиванием 20 г воздушно-сухой почвы
с 300 мл дистиллированной воды в бутыли на 1 л, содержащей пакет со смолой, в те¬
чение 16 ч. Удаляют пакет из суспензии, обильно промывают водой и оставляют для
измерений небольших количеств почвенной фракции 20—50 мкм, которая может быть
захвачена в пакет (взвешивают фракцию, остающуюся на сите 20 мкм после извлечения,
и промывают смолу на сите 50 мкм).
Остальные операции могут выполняться в соответствии с методикой, описанной
в разделе «Перемешивание со стеклянными шарами и ультразвуковая обработка». Од¬
нако хотя методика Феллера с сотр. очень похожа на описанную в предыдущем разде¬
ле, она отличается уровнем ультразвуковой энергии и методом фракционирования ила.
Просеивают суспензию почва - вода через сита 200 мкм и 50 мкм. Промывают материал,
остающийся на сите, и подвергают суспензию 0-50 мкм, полученную в литровой бу¬
тыли, ультразвуковой обработке. Установка (250 ТН, USAnnemasse) использует частоту
20 кЩ и переменную мощность (от 0 до 300 Вт) и оборудована зондом с плоской голов¬
кой 9 мм в диаметре. Эта головка расположена на расстоянии 2,5 см от дна суспензии,
ультразвук подается непрерывно в течение 7 мин при 75% максимальной мощности, т. е.
0,23 Вт/мл, или примерно 100 Дж/мл.
Просеивают суспензию 0-50 мкм, промывают материал, оставшийся на сите, затем
переносят суспензию 0-20 мкм в два седиментационных цилиндра и доливают дистил¬
лированной водой до 1 л. Встряхивают цилиндры, переворачивая их вверх дном и обрат¬
но (30 раз); помещают их на лабораторный стол для седиментации (см. гл. 2) и отбирают
фракцию 0-2 мкм при цомощи пипетки. Повторяют эту операцию до исчерпания (не
менее пяти раз). Осадок на дне цилиндра является фракцией 2—20 мкм. Центрифугиру¬
ют отобранные суспензии 0—2 мкм при 2500# в течение 1 ч для разделения отцентрифу-
гированного осадка (0,2-2 мкм) и супернатанта (0-0,2 мкм). Повторяют эту операцию
дважды. Флокулируют собранные порции супернатанта, добавляя на каждый литр жид¬
кости 2 мл насыщенного раствора SrCl2. Отделяют чистый супернатант от отиентрифу-
гированного осадка (фракция 0-0,2 мкм) центрифугированием.
Таким образом, получают следующие фракции:
• фракции 200-2000, 50-200 и 20-50 мкм мокрым рассевом;
• фракции 2-20,0,2-2 и 0-0,2 мкм седиментацией;
• водорастворимую органическую фракцию.
В зависимости от типа почвы или в случае, когда слишком энергичное диспергиро¬
вание нежелательно, можно применить тот же метод к суспензии 0—50 мкм без ультра¬
звуковой обработки.
Метод с отбором аликвот. Метод был описан йвинелли с сотр. (Gavinelli и др., 1995).
Для измерения пылеватых и илистых фракций он быстрее, чем описанный выше. Алик¬
воты из седиментационных цилиндров отбирают при помощи пипетки Робинсона
(см. гл. 2).
Специальная методика для песчаных почв
Методика Феллера (Feller, 1979) разработана для песчаных почв. Она включает низко¬
энергетическое механическое диспергирование перемешиванием суспензий почва -
вода (100 г почвы и 300 мл воды) с тремя стеклянными шарами в течение 1 ч. Просеивание
под водой и последующее разделение фракций, как описано в разделе «Фракциониро¬
вание с использованием Н+-смолы и ультразвука», позволяет выделить фракции М2000,
Ф2000, М200, Ф200, М50, ОМ, В (где М - органоминеральные фракции, Ф - органи-
280
Часть 2. Органический анализ
ческие фрагменты; числа обозначают нижнюю границу размера частиц во фракции,
ОМ - органоминеральная фракция < 50 мкм, отделенная центрифугированием, В - во¬
дорастворимая фракция).
В методику, описанную в разделе «Фракционирование с использованием Н+-смолы
и ультразвука», вносится одно изменение для песчаных и песчано-глинистых почв. Про¬
должительность перемешивания суспензий почва — вода — смола в пакетах уменьшает¬
ся, чтобы не допустить сильного разрушения растительных остатков частицами песка.
Методика состоит в следующем.
Помещают 40 г воздушно-сухой почвы в бутыль на 1 л, содержащую 300 мл дис¬
тиллированной воды и 100-миллилитровый пакет с катионобменной смолой Амбер-
лит IRN77 в Na-форме (см. раздел «Фракционирование с использованием Н+-смолы
и ультразвука»). Перемешивают бутыль на возвратно-поступательном шейкере при
умеренной скорости в течение 2 ч. Отделяют фракции крупнее 50 мкм просеиванием.
Перемешивают суспензию 0—50 мкм со смолой в течение 14 ч. Остальные операции
идентичны методу, описанному в разделе «Фракционирование с использованием Н+-
смолы и ультразвука».
9.2.6. Точность методов фракционирования
Точность методов физического фракционирования органического вещества существен¬
но изменяется в зависимости от типа почвы и особенно от стадии развития органиче¬
ского вещества. В целом, чем меньше количество фракции, тем выше вариабельность.
Повторяемость увеличивается с размером частиц во фракции. Относительная погреш¬
ность фракционирования изменяется аналогично изменению процентного содержания
по весу или процентному содержанию углерода фракции относительно общего содер¬
жания углерода.
В связи с малым относительным весом крупной и легкой органических фракций по¬
грешность их определения часто является наиболее значительной. Эта погрешность
также может быть связана с используемым методом, так как Монье с сотр. (Monnier
и др., 1962) определили для четырех типов почв интервал отклонений результатов опре¬
делений от +30 до +60% при сравнении метода фильтрования Дженсона-Луусинанга
(Jeanson-Luusinang, 1960), использующего воронку, с методом центрифугирования.
Удино (Oudinot, 1985) обнаружил, что для фракции плотностью менее 1,4 относитель¬
ное стандартное отклонение составляло 28% для известковой бурой почвы и 62% для
ферросиаллитной почвы. Феллер (Feller, 1979) также получил коэффициент вариации,
равный 63%, для 60 повторных измерений содержания крупной органической фракции
Ф2000, отделенной просеиванием через сито 2 мм и всплывающей в воде. Монье с сотр.
{Monnier и др., 1962) получили суммарное относительное стандартное отклонение ~2%
для двух повторных экспериментов на четырех типах почв.
Феллер (Feller, 1979) также измерил погрешность определения процентного содержа¬
ния углерода в каждой фракции (относительно общего углерода), полученной мокрым
рассевом и декантацией (см. раздел «Специальная методика для песчаных почв»). Полу¬
ченные результаты (табл. 9.1) подчеркивают значимость погрешности для количествен¬
ных анализов органического вещества почвы и ее изменение с размером фракции.
Работа Феллера с сотр. {Feller и др., 1991) также содержала оценку точности опреде¬
ления массы фракций и доли углерода в них относительно общего содержания углерода
(табл. 9.2). Показано, что погрешность зависит от типа почвы. Ее величина для слитой
почвы была меньше, чем для ферраллитной почвы.
Глава 9. Физические методы фракционирования органических веществ
281
Таблица 9.1. Погрешность в точности фракционирования по размеру частиц для песчаной
почвы из Сенегала (Feller, 1979): Ф — органические фрагменты; М — органоми¬
неральные фракции; числа — нижняя граница размера частиц, ОМ — органо¬
минеральная фракция < 50 мкм; В — водорастворимая фракция; т — массовая
доля углерода фракции в общем углероде всех фракций, %; ОСО — относитель¬
ное стандартное отклонение для 10 повторных экспериментов
Фракция
т
ОСО
Ф2000
3,0
63
Ф200
21,6
11
Ф50
8,8
25
М50
5,8
28
В
2,0
25
Таблица 9.2. Повторяемость фракционирования по размеру частиц для ферраллитной и сли¬
той почв (РеИегищу., 1991): ОСО% т и ОСО% Ct - относительное стандартное
отклонение массовой доли фракции и массовой доли углерода фракции (об¬
щий углерод фракции/общий углерод) для четырех повторных экспериментов
Тип почвы
Фракция, мкм
ОСО%т
осо% ct
Ферралитная
0-2
10
23
2-20
25
48
20-2000
8
48
Слитая
0-2
4
8
2-20
4
14
20-2000
10
30
9.3. Заключение и перспективы развития методов
Методы физического фракционирования часто используют перед другими исследова¬
ниями органического вещества почвы. По сути они сами представляют собой один из
методов исследования органического вещества.
Нет метода, обеспечивающего полное отделение каждого компонента почвы (корни
растений, растительные остатки, остатки животных, микроорганизмы и метаболиты
органоминеральных комплексов). Методы, описанные в этой главе, представляются
наиболее пригодными для дальнейшего развития. Они были разделены на три группы:
выделение корней растений, выделение «свободного органического вещества», соответ¬
ствующего частично разложенным органическим остаткам, и фракционирование орга¬
нического вещества по диапазонам размера частиц.
Оборудование для отделения корней растений основано на принципах отмучивания
и мокрого рассева. Сложность этих методов и неполнота разделения заставляют некото¬
рых авторов предпочитать ручные методы.
Методы разделения по плотности относительно просты в исполнении. Объединение
разделения по плотности с разделением крупных фракций по размеру частиц, а также
применение не очень агрессивных методов диспергирования структурных агрегатов по¬
вышают надежность определения.
Методы фракционирования по размеру частиц позволяют осуществлять более по¬
дробную классификацию органического вещества, в частности трех основных органи¬
ческих и органоминеральных компартментов, указанных Феллером (Feller, 1994).
282
Часть 2, Органический анализ
Наряду с описанием методов физического фракционирования, в данной главе опи¬
саны основные типы почв, на которых эти методы были испытаны. Возможно, необхо¬
дима адаптация методов для других почв или специфических объектов исследования.
Такой адаптации могут помочь данные наблюдений Кристинсена (Christensen, 2001),
Сикса с сотр. (Six и др., 2002), Ровиры и Валледжо (Rovira и Vallejo, 2003) или Ксю с сотр.
(Хи и др., 2003). Данные, приведенные в этой главе, следует принимать во внимание,
особенно это касается мер предосторожности при использовании ультразвука и диспер¬
гирующих реагентов для более полного разделения органических фрагментов и органо¬
минеральных комплексов при меньшем разрушении органических объектов.
Использованная литература
Abo F (1984) Influence du bore et du manganese sur la nutrition, le developpement et la production de
ble sur sols de regions temperee et aride.9 Thfcse d’6tat, university Paris VII, 390 p.
Ahmed M and Oades Ш (1984) Distribution of organic matter and adenosine triphosphate after
fractionation of soils by physical procedures. Soil Biol. Biochem., 16, 465-470.
Anderson DW and Paul EA (1984) Organo-mineral complexes and their study by radiocarbon dating. Soil
Sci. Soc. Am. J.9 48, 298-301.
Anderson DW, Saggar S, Bettany JR and Stewart JWB (1981) Particle size fractions and their use in
studies of soil organic matter: I. The nature and distribution of forms of carbon, nitrogen and sulfur.
Soil Sci. Soc. Am. J.9 45, 767-772.
Anderson JM and Ingram JSI (1989) Tropical soil biology and fertility (TSBF): a handbook of methods9
C.A.B. International, 171 p.
Andreux F, Bruckert S, Correa A and Souchier В (1980) Sur une m^thode de fractionnement physique et
chimique des agr£gats des sols: origines possibles de la mattere organique des fractions obtenues.
C.R. Acad. Sci. Paris, 291, 381-384.
Arrouays D (1994) Int6ret du fractionnement densimytrique des matiyres organiques en vue de la
construction d’un module bi-compartimental devolution des stocks de carbone du sol. Exemple
apres dyfrichement et monoculture de maTs grain des sols de “touyas”. C.R. Acad. Sci. Paris, 318,
II, 787-793.
Balesdent J, Pytraud JP and Feller C (1991) Effets des ultrasons sur la distribution granulomytrique des
matiyres organiques des sols. Sci. du Sol,9 29, 95-106.
Bartoli F, Burtin G and Herbillon AJ (1991) Disaggregation and clay dispersion of oxisols: Na resin, a
recommended methodology. Geoderma, 49,301-317.
Beare MH, Hendrix PF and Coleman D.C (1994) Water-stable aggregates and organic matter fractions in
conventional-and no-tillage soils. Soil Sci. Soc. Am. J,9 58, 777-786.
Besnard E, Chenu C, Balesdent J, Puget P and Arrouays D (1996) Fate of particulate organic matter in soil
aggregates during cultivation. European Journal of Soil Science, 47,495-503.
Bonzon В and Picard D (1969) Matyriel et mythodes pour l’etude de la croissance et du dyveloppement
en pleine terre des systymes racinaires. Cah. ORSTOMser. Biol.9 9, 3-9.
Bruckert S, Andreux F, Correa A, Ambouta KJM and Souchier В (1978) In Proc. IP Congris A.I.S.S.,
Edmonton, Canada.
Bruckert S (1994) Analyse des complexes organo-ттУгаих des sols. In - Pedologie 2. Constituants et
proprietes du sol Bonneau and Souchier ed., 2nd ed., Masson, Paris, 275-295.
Cambardella CA, Elliott ET (1993) Carbon and nitrogen distribution in aggregates from cultivated and
native grassland soils. Soil Sci. Soc. Am. J.9 57, 1071-1076.
Catroux G and Schnitzer M (1987) Chemical, Spectroscopic and Biological Characteristics of the organic
matter in particle size fractions separated from an aquoll. Soil Sci. Soc. Am. J., 51, 1200-1207.
Christensen ВТ (1992) Physical fractionation of soil and organic matter in primary particle size and density
separates. In Advances in soil science, No. 20, Springer Berlin Heidelberg New York Inc, 1-90.
Christensen ВТ (2001) Physical fractionation of soil and structural and functional complexity in organic
matter turnover. Eur. J. SoilSci.9 52, 345-353.
283
Глава 9. Физические методы фракционирования органических веществ
Dabin В (1976) Methode d’extraction et de fractionnement des matieres humiques du sol. Application
к quelques etudes pedologiques et agronomiques dans les sols tropicaux. Cah Orstom, Ser. Pedol.,
XIV, 287-297.
Edwards AP and Bremner JM (1967) Dispersion of soil particles by sonic vibration. J. Soil Sci., 18,
47-63.
Elliott ET and Cambardella C.A(1991) Physical separation of soil organic matter. Agriculture, ecosystems
and environment. Elsevier Science Amsterdam, 34,407-419.
Elliott ET, Palm CA, Reuss DE and Monz CA (1991) Organic matter contained in soil aggregates from
a tropical chronosequence : correction for sand and light fraction. Agriculture, Ecosystems and
Environment, 34, 443-451.
Feller C (1979) Une methode de fractionnement granulometrique de la matiere organique du sol. Cah
ORSTOM sir. Pedol., XVII, 339-346.
Feller C (1994) La matiere organique dans les sols tropicaux a argile 1:1. Recherche de compartments
organiques fonctionnels. Une approche granulometrique., IRD-Orstom, Paris, theses et documents
microfiches.
Feller C, Burtin G, Gerard В and Balesdent J (1991) Utilisation des resines sodiques et des ultrasons dans le
fractionnement granulometrique de la matiere organique des sols. Interet et limites. Sci. du Sol., 29,77-93.
Ford GW, Greenland DJ and Oades JM (1969) Separation of the light fraction from soils by ultrasonic
dispersion in halogenated hydrocarbons containing a surfactant. J. Soil Sci., 20,291-296.
Gavinelli E, Feller C, Larre-Larrouy MC, Bacye B, Djegui N and Nzila JdD (1995) A routine method to
study soil organic matter by particle-size fractionation, examples for tropical soils. Commun. Soil
Sci. Plant Anal., 26, 1749-1760.
Genrich DA and Bremner JM (1972) A reevaluation of the ultrasonic vibration method of dispersing soils.
Soil Sci. Soc, Amer. Proc., 36,944-947.
Golchin A, Oades JM, Skjemstad JO and Clarke P (1994) Study of free and occluded particulate organic
matter in soils by solid state ,3C CP/MAS NMR spectroscopy and scanning electron microscopy.
Austral. J. Soil Res., 32, 285-309.
Greenland DJ and Ford GW (1964) Separation of partially humified organic materials from soils by
ultrasonic dispersion. Trans. 8th Int. Congr. Soil Sci., 3,137-148.
Gregorich EG, Kachanoski RG and Voroney RP (1988) Ultrasonic dispersion of aggregates: distribution
of organic matter in size fractions. Can. J. Soil. Sci., 68, 395-403.
Hassink J and Dalenberg JW (1996) Decomposition and transfer of plant residue 14C between size and
density fractions in soil. Plant Soil, 179, 159-169.
H6nin S and Turc L (1949) Essai de fractionnement des matures organiques du sol. C.R. Acad. Sci., Paris,
35, 41-43.
Jeanson Luusinang C (1960) Fractionnement par density de la matiere organique des sols. Ann. Agron.,
11, 481-496.
Kilmer VJ and Alexander LT (1949) Methods of making mechanical analyses of soils. Soil Sci., 68,
15-24.
Ladd JN, Parsons JW and Amato M (1977) Studies of nitrogen immobilization and mineralization in
calcareous soils -1, Distribution of immobilized nitrogen amongst soil fractions of different particle
size and density. Soil Biol. Biochem., 9, 309-318.
Lein ZJ (1940) Les formes de liaison de l’humus avec la partie mindrale des sols. Pochvovedeniye.
Me Gill W.B., Shields J.A. and Paul E.A (1975) Relation between carbon and nitrogen turnover in soil
organic fraction of microbial origin. Soil Biol.Biochem., 16, 465-470.
Meijboom FW, Hassink J and van Noordwijk M (1995) Density fractionation of soil macroorganic matter
using silica suspensions. Soil Biol. Biochem., 27, 1109-1 111.
Monnier G, Turc L and Jeanson-Luusinang C (1962) Une methode de fractionnement densimetrique par
centrifugation des matieres organiques du sol. Ann. Agron, 13, 55-63.
Oudinot S (1985) Fractionnement physique de la matidre organique. Distribution du carbone natif et
marqud entre les fractions granulometriques de deux sols miditerraneens incubes avec du matёriel
vigital marque au 14C, DEA, ENSAIA-INP de Lorraine, France, 30 p.
Pansu M and Sidi H (1987) Cindtique damnification et de mineralisation des melanges sols-residus
vegetaux. Sci. du sol, 25, 247-265.
284
Часть 2. Органический анализ
Pansu М, Bottner Р, Sarmiento L and Metsellaar, К (2004) Comparison of five soil organic matter
decomposition models using data from a l4C and 15N labeling field experiment. Global Biogeockem.
Cycles, 18, GB4022, doi: 10.1029/2004GB002230.
Pansu M, Gautheyrou J and Loyer JY (2001) Soil Analysis - Sampling Tokyo, 489 pp.
Pearson RW and Truog E (1937) Procedure for the mineralogical subdivision of soil separates by means
of heavy liquid specific gravity separations. Soil Sci. Soc. Am. Proc.9 2,109-114.
Puget P, Besnard E and Chenu C (1996) Une mdthode de fractionnement des matifcres organiques
particulaires des sols en fonction de leur localisation dans les agrCgats. Comptes Rendus de
l 'Academie des Sciences, Paris, 322, 965-972.
Puget P, Chenu C and Balesdent J (1995) Total and young oiganic matter distributions in aggregates of
silty cultivated soils. Eun J. Soil Sci.9 46, 449-459.
Roscoe R, Buurman P and Velthorst EJ (2000) Disruption of soil aggregates by varied amounts of
ultrasonic energy in fractionation of organic matter of a clay latosol: carbon, nitrogen and 513C
distribution in particle-size fractions. Eur. J. Soil Sci.9 51, 445-454.
Rouiller J, Burtin G and Souchier В (1972) La dispersion des sols dans Г analyse granulomCtrique.
Methode utilisant les resines Cchangeuses d’ions. Bull. ENSAIA Nancy, France, XIV, 193-205.
Rovira P and Vallejo VR (2003) Physical protection and biochemical quality of organic matter in
Mediterranean calcareous forest soils: A density fractionation approach. Soil Biol Biochem., 35,
245-261.
Sallih Z and Pansu M (1993) Modeling of soil carbon forms after organic amendment under controlled
conditions. Soil Biol Biochem., 25, 1755-1762.
Shaymukhametov MS, Titova NA, Travnikova LS and Labenets YM (1984) Use of physical fractionation
methods to characterize soil organic matter. Translated from : Pochvovedeniye, 8,131-141.
Sidi H (1987) Effet de Vapport de matikre organique et de gypse sur la stabilite structurale de sols de
region mediterraneenne, These Docteur ingCnieur, INA Paris Grignon.
Six J,. Callewaert P, Lenders S, De Gryze S, Morris SJ, Gregorich EG, Paul EA and Paustian К (2002)
Measuring and Understanding Carbon Storage in Afforested Soils by Physical Fractionation. Soil
Sci. Soc. Am. J.9 66, 1981-1987.
Six J, Schultz PA, Jastrow JD and Merckx R (1999) Recycling of sodium polytungstate used in soil
organic matter studies. Soil Biol. Biochem., 31,1193—1196.
Smucker AJM, McBumey S and Srivastava AK (1982) Separation of roots from compacted soil profiles
by the hydropneumatic elutriation system. Agron. J.9 74, 500-503.
Soil Survey Staff (1960) Soil classification -A comprehensive system, 7th Approximation. USDA, SCS,
265 p.
Tiessen H and Stewart JWB (1983) Particle-size fractions and their use in studies of soil organic matter:
II. Cultivation effects on organic matter composition in soil fractions. Soil Sci Soc. Am. J.9 47, 509-
514.
Turchenek LW and Oades JM (1979) Fractionation of organo-mineral complexes by sedimentation and
density techniques. Geoderma, 21, 311-343.
Watson JR (1971) Ultrasonic vibration as a method of soil dispersion, Soils and Fertilizers, 34, 127-134
Xu YC, Shen QR and Ran W (2003) Content and distribution of organic N in soil and particle size
fractions after long-term fertilization. Chemosphere9 50, 739-745.
Глава 10. Определение содержания органического
вещества и общего количества Cf N (Н, О, S)
10.1. Введение
10.1.1. Органическое вещество почвы
Органическое вещество играет определяющую роль в почвообразовании и может суще¬
ственно изменить физические, химические и биологические свойства почвы (структу¬
ру, пластичность, цвет, водоудержание, емкость катионного и анионного обмена (ЕКО
и ЕАО, соответственно)).
Основные процессы развития почвы включают минерализацию и иммобилизацию,
особенно углерода и азота. В процессе минерализации органические остатки превра¬
щаются в неорганические вещества в почве, атмосфере и гидросфере и становятся до¬
ступными для флоры и микроорганизмов. Иммобилизация — это превращение органи¬
ческого вещества в более стабильные органические и органоминеральные соединения
с большой молекулярной массой, которые связываются в межпакетных промежутках
глинистых минералов. Эти процессы обобщены следующей схемой:
со21\ СН/Г
NHjt
tN20, N2, NOx
NOj-oNO,-
Этот цикл включает фазы минерализации, гумификации, аммонификации, иммоби¬
лизации, нитрификации и испарения под воздействием специфических микроорганиз¬
мов (Pansu и др., 1998) и зависит от многочисленных факторов, самыми важными из
которых являются:
• климат (температура и влажность и их влияние на активность микробов, микро¬
фауны и микрофлоры);
• рельеф;
• типы растительного покрова и подстилок;
• природа почвообразующих пород (главным образом, гранулометрический и мине¬
ралогический состав и pH);
• время (возраст почвы и состояние равновесия);
кроме того, для возделываемых почв:
• влияние методов обработки, таких как вспашка, орошение, выжигание, добавле¬
ние навоза, удобрений, пестицидов:
• типы сельскохозяйственных культур, вынос питательных веществ с урожаем и ис¬
пользование остатков переработки урожая.
Органическое
вещество
почвы
и некромасса
Минерализация
Иммобилизация
Прирост Потеря
Видимое равновесие
286
Часть 2. Органический анализ
Содержание С и N в почве может существенно варьировать, например, тропическая
оксисоль может содержать менее 2% общего С, тогда как его содержание в андосолях
и гистосолях может превышать 30%.
Динамика превращений С и N очень сложна и трудно поддается точному модели¬
рованию. Кроме измерения содержания общего С и N, необходимо иметь простой
индикатор для выяснения динамики и сравнения почв из различных зон, а также для
установления изменений органического вещества в почвенном профиле или наличия
погребенного органического горизонта А.
Полевые наблюдения гумифицированных подстилок (А00) различной толщины в со¬
ставе органического горизонта \ и элювиальных или иллювиальных горизонтов позво¬
ляют получить важную информацию. Затем эволюцию органического вещества анали¬
зируют в процессе изучения его химической структуры и физико-химических свойств.
Различные формы вещества можно разделить на характеристические объекты различ¬
ной степени полимеризации и количественно определить физическим разделением
(см. гл. 9) по их устойчивости к гидролизу (в кислой и щелочной среде) и их раствори¬
мости в специфических растворителях и реагентах (см. гл. 11 и 12). Лабораторные мето¬
ды, такие как гель-проникающая хроматография, позволяют определять молекулярную
массу гумифицированных веществ после очистки. Поглощение УФ, видимого или ИК-
излучения и другие спектрографические методы дают возможность идентифицировать
молекулярные структуры и определить степень полимеризации. Можно исследовать как
активные функциональные группы, так й глино-гумусные комплексы (см. гл. 12). Эле¬
ментный анализ, описанный в данной главе, позволяет установить валовый химический
состав органических веществ.
Различные факторы, контролирующие гумификацию, особенно климат, материн¬
ская порода и биомасса, накладывают физико-химические ограничения, которые,
в свою очередь, приводят к образованию определенного типа гумуса, например, мор,
модер, лесной и известковый мулли, анмур или торфы. Микроскопическое исследова¬
ние (см. гл. 8) морфологии систем в различных масштабах позволяет характеризовать
границы раздела между органическим и неорганическим веществом и механизмы гуми¬
фикации. Кинетические методы используют для анализа биогеохимической динамики
органических остатков:
• при измерении, например, С02, выделившегося в единицу времени, при помощи
переносного хроматографа или И К-датчиков, установленных на месте (респиро¬
метрия, биологические выделения газа);
• использовании изотопных меток 14С, 13С и 15N для мониторинга превращений ор¬
ганических веществ, добавленных в почву (скорость круговорота органического
вещества почвы);
• изучении изменений содержания стабильных изотопов, таких как 13С, 14С и 15N,
и изотопных отношений в палеоклиматологических и геохронологических иссле¬
дованиях.
Большинство этих методов требуют применения высокоспецифичных и высокочув¬
ствительных датчиков, потому что необходимо измерять чрезвычайно малые изменения.
Это длительные и дорогостоящие измерения, которые недостаточно универсальны для
использования в рутинных анализах. С другой стороны, содержание общего С и N мо¬
жет быть определено простейшими методами, доступными для любых лабораторий. Со¬
вершенствование оборудования для сухого анализа (например, СНЫ(08)-анализаторы)
в настоящее время позволяет стандартизовать анализы и сочетать точность, быстро-
287
Глава 10. Определение содержания органического вещества...
ту и автоматизацию измерений. Некоторые из современных приборов могут работать
с представительными пробами массой более чем 100 мг.
Соотношение C/N в поверхностных горизонтах почвы можно определить только
на основании данных об общем содержании этих элементов (табл. 10.1). Эту величи¬
ну можно затем использовать в качестве индикатора, дающего относительно надежную
информацию о биологической активности и равновесии двух элементов, участвующих
в противоположно направленных процессах минерализации и иммобилизации. В ре¬
гионах умеренного климата соотношение C/N составляет примерно 10—12 для невоз-
делываемых почв и обычно снижается с глубиной почвы. В некоторых почвах N может
быть окклюдирован в глинах, особенно в глубоких горизонтах. В лесных почвах, тор¬
фяных горизонтах или подзолах соотношение C/N может достигать 20—30 или даже
больше из-за образования слабо биоразлагаемых комплексов, обедненных азотом (на¬
пример, сподовые горизонты). При соотношении C/N ниже пороговой величины 20
обычно наблюдается положительная нетто-минерализация N. В возделываемых почвах
пожнивные остатки, повторно используемые в поле, имеют соотношение C/N от 15 до
60 из-за присутствия лигнин-целлюлозных соединений с низкой скоростью разложе¬
ния. В лесах с подкисляющей подстилкой соотношение C/N может достигать 150 или
даже выше.
Таблица 10Л. Типичные соотношения C/N некоторых типов гумуса
Ъш
C/N
pH
Известковый эвтрофный мулль
~10
7,0-8,4
Лесной мулль
12-15
5,5-6,5
Модер
15-15
4,0-5,0
Мор
>20
<4,0
известковые эвтрофные торфы
<30
-
Гйдроморфные
кислые олиготрофные торфы
~40
-
анмур
Переменное
-
10.1.2. Отбор и подготовка образцов, аналитическая значимость
Оборудование
• Решето с круглыми отверстиями 2 мм в диаметре AFNOR 7V/34.
• Режущая мельница, оборудованная ситом с отверстиями 125 мкм (AFNOR NF22)
и водонепроницаемым приемником (для подстилки).
• Мельница с регулируемым молотом, оборудованная ситом AFNOR NF22 и водо¬
непроницаемым приемником (для проб из минеральных и органоминеральных
горизонтов).
• Агатовая ступка и пестик.
• Аналитические весы (±0,1 мг или ±0,01 мг, в зависимости от анализируемых об¬
разцов).
• Сушильный шкаф, устанавливаемый на 105 °С.
Методики и меры предосторожности
Поскольку неоднородность поверхностных горизонтов почвы вблизи подстилки очень
высока, отбор образцов затруднен и должен выполняться с большой осторожностью.
288
Часть 2. Органический анализ
Способ отбора образцов влияет на надежность результатов. Высушивание образцов
должно проводиться в контакте с воздухом в хорошо вентилируемом помещении.
В лаборатории образцы измельчают до 2 мм, чтобы отделить неразложившиеся или
слабо разложившиеся остатки растений. Следует соблюдать меры предосторожности,
чтобы не разрушить органические фрагменты, сохранившие свое исходное строение,
поскольку это может значительно перегрузить образец. Этот этап влияет на достовер¬
ность результата анализа, так же как масса анализируемой пробы (Рати и др., 2001).
Высушивание увеличивает фиксацию аммония, особенно если материнская матри¬
ца содержит глинистые минералы типа 2:1, такие как монтмориллонит и вермикулиты.
Действие микроорганизмов может не прекращаться во время хранения в зависимости от
типа почвенного дыхания, содержания лигнина или остаточной влаги в воздушно-сухом
образце (в андосолях и гистосолях влажность воздушно-сухой почвы через 6 мес. может
составлять 60%).
Измельчение субпробы с размером частиц 2 мм до 125 мкм (сто AFNOR NF22) может
изменить влажность и баланс активных поверхностей. Содержание влаги используется
для корректировки результатов анализа и должно измеряться одновременно с отбором
образцов для анализа. Если продолжительность высушивания при 105 °С не превышает
3 или 4 ч, обычно не происходит существенной потери С или N в газовой форме, но
сушка может несколько увеличить фиксацию N в кристаллической решетке глинистых
минералов. В случае инструментального анализа (СНЫ(08)-анализатор) высушивание
образцов при 105 °С позволяет исключить необходимость в корректировке результатов
и уменьшить загрязнение водных ловушек.
Осторожный размол до однородных по размеру мелких частиц необходим для улуч¬
шения воспроизводимости результатов; навеска порошка пробы почвы может варьи¬
роваться от 5 до 500 мг в зависимости от используемого для сухого анализа прибора
(СН1Ч(08)-анализатор). В случае мокрого анализа измельчение обеспечивает более рав¬
номерное и полное взаимодействие значительно ббльших поверхностей раздела твердая
фаза - жидкость.
Представление результатов
Если результаты анализа необходимы для использования в агрономических целях, следу¬
ет учитывать плотность почвы, особенно для торфов, андосолей и гистосолей, где кажу¬
щаяся плотность может составлять около 0,30. В этом случае концентрация на единицу
массы может существенно отличаться от содержания доступных растениям неорганиче¬
ских веществ в единице объема и должна быть исправлена с учетом плотности.
Для почв с высоким содержанием гравия, камней, фрагментов породы или нераз-
ложившихся остатков растений, которые обнаруживают значительный процент брака
при подготовке проб почвы с размером частиц 2 мм, также предпочтительно исправлять
первичные результаты для получения величин, близких к показателям почв на единицу
объема.
Предварительные испытания
Хорошее знание процессов образования и сельскохозяйственного использования иссле¬
дуемых почв позволяет уменьшить количество необходимых испытаний. Число измере¬
ний зависит и от используемого аналитического метода (табл. 10.2).
Следует отметить, что по определению «общее органическое вещество почвы» соот¬
ветствует преобразованным органическим формам и исключает неизмененные расти¬
тельные и животные остатки. На практике, поскольку отделение легких или неразло-
Глава 10. Определение содержания органического вещества»., 289
Таблица 10.2. Аналитические методы
Метод
Tim
Источник помех
Сухие
Озоление при <360 °С
Гипс, потеря воды выше 150 °С
методы
Озоление при > 360 °С
Разные формы воды (гидратация)
Разложение карбонатов, С02Т
Плавление Na2C03 (загрязнение)
различные формы воды (связанной)
Мокрые
Классический метод
Случайное обнаружение NO^", NOj (не определяются
методы
Къельдаля
без предварительного восстановления)
NH+ фиксированный в кристаллической решетке
глинистых минералов типа 2:1 (случайное обнаружение)
Холодное окисление С
Заниженные результаты (мультипликативный
коэффициент)
Присутствие хлорида, окисляющих
и восстанавливающих агентов
Горячее окисление С
Присутствие хлорида, окисляющих
и восстанавливающих агентов
жившихся фрагментов органического вещества (см. гл. 9) трудновыполнимо в массовых
анализах, считается, что частицы мельче 2 мм составляют существенную часть образца.
Живые микроорганизмы входят в состав образца. Чтобы получить детальные сведения
об аналитическом субстрате, достаточные для выбора соответствующих аналитических
процедур, должны быть выполнены различные типы испытаний.
Исследование при помощи лупы позволяет подтвердить присутствие морских ракушек,
известняковых мелиорантов, углей и т. д. (почвы под сельскохозяйственными культура¬
ми, прибрежные почвы, карбонатные почвы и др.).
Тест с НС1: при pH ниже 7,4-7,0 указывает на присутствие карбонатов и бикарбона¬
тов только в форме отдельных частиц.
Тест на NO~, NO~: быстрый тест с использованием аналитического набора для ин¬
тенсивно возделываемых и заболоченных почв.
Тест на фиксированный NH+: используется только для почв, содержащих глинистые
минералы типа 2:1, для проверки концентрации аммония двумя разными методами
и возможен при разрушении кристаллической решетки (см. раздел 28.3.5).
Предварительное разрушение карбонатов и бикарбонатов
(методы сухого озоления)
Образцы обрабатывают 0,1 М раствором НС1 при комнатной температуре до оконча¬
ния реакции. Если возможно присутствие доломита, сидерита или биогенного кальцита,
время контакта следует увеличить до 2 или 3 ч.
СаС03 + 2Н+ -> Са2+ + С02Т + Н20
Образцы, содержащие сидерит, можно обработать горячей Н3Р04 или СН3СООН
(0,3 Н) в течение 5 ч. Разрушение протекает трудно и не полностью. Тщательно высу¬
шивают.
Общий С - неорганический С = органический С „ (10.1)
290
Часть 2. Органический анализ
Следует принять меры к тому, чтобы не потерять растворимый органический С в фор¬
ме кислот (например, аминокислот или фосфолипидов). Образование гигроскопиче¬
ских солей может помешать взвешиванию. Присутствие диоксида марганца в почве мо¬
жет вызвать выделение С12 из НС1.
В аридных или полуаридных регионах почвы могут содержать растворимые соли (на¬
пример, карбонаты, хлориды или соединения серы), которые могут замедлить окисле¬
ние С из-за их низких точек плавления (например, карбонат натрия) и, следовательно,
помешать определению методом сухого озоления.
10.2. Мокрые методы
10.2.1. Определение содержания общего углерода: основные положения
Строго говоря, «общий углерод» в почве происходит из двух источников (10.1):
• органический углерод (частично преобразованные органические остатки расти¬
тельного и животного происхождения, гумус, древесный уголь, ископаемое орга¬
ническое вещество, микроорганизмы);
• неорганический углерод, который может присутствовать в форме карбонатов и би¬
карбонатов.
В большинстве методов не учитываются газовые фазы, присутствующие в почвенном
воздухе (С02, связанный с биологической активностью; СН4).
Сохраняется некоторая двусмысленность в терминологии и используемых методах.
Измерения, проводимые на необработанных образцах почвы (без предварительного
удаления карбонатов) с использованием оборудования для сухого озоления (CHN(OS)-
анализаторы), определяют органические и неорганические формы «общего углерода».
Окислительно-восстановительные измерения с использованием мокрого озоления
определяют только «органический углерод», соответствующий гумифицированным
формам и органическому веществу остатков, которые содержат еще много неперерабо-
танной целлюлозы, но не включают древесного угля или ископаемого органического
вещества. Хотя мокрые методы при комнатной температуре не полностью разрушают
гумус (необходимо использовать коэффициент пересчета), они используются для опре¬
деления «общего органического углерода».
«Общий неорганический углерод» можно определить методами, описанными в главе
17, но с различающейся точностью в связи с медленным химическим разложением кар¬
боната магния (MgC03) и особенно сидерита (FeC03).
Термин «общее органическое вещество» используется широко. Практически он соот¬
ветствует содержанию «общего органического углерода», определенного методом окис¬
ления - восстановления с поправкой, основанной на предположении, что органическое
вещество состоит в основном из гуминовых кислот, содержащих около 58% углерода
(100/58 = 1,724 - коэффициент ван Беммелена). В действительности это отношение да¬
леко не постоянно даже для верхних горизонтов почвы. Таким образом, коэффициент
не слишком реалистичен, особенно для почв, содержащих слабо гумифицированный
мор или модер, лесных почв и торфов. Для них необходимо использовать коэффициент,
равный 2 или даже 2,5. Таким образом, термин «общее органическое вещество» является
приближенным и не может использоваться в качестве показателя.
На практике некоторые авторы неправильно используют термин «общий углерод»
для обозначения «общего органического углерода», а также для установления баланса
валового анализа и соотношений C/N или сравнения содержания общего С в различных
горизонтах почвенного профиля и распределения органического С с глубиной.
291
Глава 10. Определение содержания органического вещества...
Термин «общий органический углерод» должен включать все органические вещества,
образующиеся при гумификации углеродсодержащих веществ (микробные остатки, гу-
миновые вещества) под воздействием биохимических и химических процессов. Он так¬
же представляет еще не полностью разложенное легкое органическое вещество, которое
не может быть выделено на сите в процессе подготовки образцов (подстилки, грубые ор¬
ганические фрагменты растений или животных размером менее 2 мм). Ископаемое ор¬
ганическое вещество (уголь, нефти, смолы и др.) и древесный уголь в регионах, подвер¬
женных лесным пожарам, или регионах подсечно-огневого земледелия не изменяются
по этому типу, и это означает, что они не подлежат учету при использовании влажных
окислительно-восстановительных методов. Однако они всегда учитываются в методах
сухого озоления, и это вызывает сложности при сравнении результатов, полученных
разными методами.
Таким образом, исследование общего органического углерода в сырье может потребо¬
вать уточнения при помощи:
• микроморфологических наблюдений в различных масштабах для определения от¬
носительных долей неразложившихся и гумифицированных органических веществ
в комбинации с селективной экстракцией в различных средах;
• определения происхождения, природы и скорости минерализации различных
форм в зависимости от pH, природы глин и глино-гумусного комплекса, а также
методов обработки почвы.
В мокрых методах используется относительно недорогое оборудование, т. е. они до¬
ступны для любой лаборатории. Эти методы позволяют работать с большими пробами,
более представительными для объектов окружающей среды. С другой стороны, они дли¬
тельны, требуют использования очень агрессивных реагентов и устранения загрязняю¬
щих веществ (Сг3+, H2S04), что может нанести урон окружающей среде.
10.2.2. Определение содержания органического углерода методом
мокрого окисления при температуре реакции
Введение
Метод определения общего органического углерода окислением бихроматом калия
в сильно кислой среде был впервые предложен Шеленбергером (Schollenberger, 1927)
и затем Уолки и Блэком (Walkley и Black, 1934), именами которых он назван. После этапа
окисления/минерализации при температуре реакции в течение определенного периода
времени избыток невосстановленного бихромата определяется обратным титрованием
двухвалентным железом.
Многие авторы исследовали факторы, влияющие на минерализацию углерод-со-
держащих веществ: концентрация кислоты (H2S04, Н3Р04), концентрация бихромата
калия, температура окисления (от температуры реакции до +210 °С), время реакции,
необходимость конденсации паров воды, чтобы не допустить повышения концентра¬
ции реактивов в растворе и уменьшить расход окислителя из-за перегрева стенок. Выбор
температуры приводит к двум различным типам методов:
• при температуре взаимодействия ~ 120 °С (Walkley и Black, 1934);
• при кипячении в стандартизованных условиях -150 °С (Аппе, 1945; Mebius, 1960).
Предлагались различные методики для обратного титрования избытка бихромата,
такие как разделение смеси почва — раствор центрифугированием и фильтрованием,
однако наиболее широкое применение нашло прямое волюмометричеекое титрование
почвенной суспензии. Индикаторы окислительно-восстановительных реакций (дифени-
292
Часть 2. Органический анализ
ламин, дифениламин сульфонат бария, N-фенилантраниловая кислота, о-фенантролин
железа(Н)) могут сорбироваться на глинах. Добавки (например, NaF, Н3Р04) позволяют
более четко различать образующиеся или растворенные окрашенные продукты (напри¬
мер, Fe3+), которые могут маскировать протекание реакции.
Основные положения
Минерализация
Органические формы С окисляются в присутствии избытка бихромата. Реакция в кон¬
центрированной кислой среде экзотермична (-120 °С). Она быстро протекает в соот¬
ветствии с уравнением:
ЗС + 2Сг2072-+ 16Н+ 1222 ► 4Сг3++ 8Н20 + ЗС02
Считается, что количество восстановленного бихромата связано с содержанием ор¬
ганического С в пробе. Принято, что вероятность восстановления одинакова для раз¬
личных форм органического С, а восстанавливающая способность остается постоянной
в течение минерализации. На практике при температуре реакции без нагревания необ¬
ходимо использовать поправочный коэффициент, потому что окисляются только наи¬
более активные формы, т. е. 60-80% органического вещества. Чтобы учесть различную
реакционную способность органических форм углерода, этот коэффициент принят рав¬
ным 1,30 (100/76), но он может изменяться от 1,10 до 1,45 в зависимости от почв и типов
растительности.
Неорганические формы углерода (карбонаты, бикарбонаты) разлагаются и не игра¬
ют никакой роли, за исключением реакции с кислотой и образования пены. Осаждение
сульфата кальция может вызывать помехи при конечном определении, если использует¬
ся спектро-колориметрический метод.
СаС03 + H2S04 -► CaS04i + Н20 + C02T
Титрование
Волюмометрическое обратное титрование Cr+VI в форме бихромата, не прореагировав¬
шего с органическим С, выполняют восстановлением солью Мора (сульфат двухвалент¬
ного железа) в присутствии индикатора.
Сг2072' + 6Fe2+ + 14Н+ -► 2Сг3+ + 6Fe3+ + 7Н20
Можно добавить фторид натрия или фосфорную кислоту (Н3Р04) для стабилизации
образовавшегося или растворенного трехвалентного железа и лучшего определения точ¬
ки эквивалентности титрования:
Fe203 + 3H2S04 -> Fe2(S04)3 + 3H20
Fe3+ + 6F" -> FeF3- (бесцветный)
Тем не менее, добавление фторида может привести к образованию фтороводородной
кислоты, которая реагирует со стеклом и силикатами.
2NaF + H2S04 -> Na2S04 + 2HF
293
Глава 10. Определение содержания органического вещества...
Si02 + 6HF -► H2SiF6 + 2Н20
Поэтому необходимо мыть стеклянную посуду сразу же после титрования и исполь¬
зовать всегда одну и ту же посуду для этого метода определения С.
Влияющие факторы
В засоленных почвах присутствие хлоридов приводит к завышенным результатам из-за
образования хромилхлорида:
+ 6H2S04 + 4КС1 -> 2Сг02С12 + 6KHS04 + ЗН20
Двухвалентное железо, которое может присутствовать, окисляется бихроматом и та¬
ким образом изменяет количество сульфата железа (II), необходимое для обратного ти¬
трования. Для заболоченных почв, которые иногда содержат много Fe2+, необходимо
вводить поправки.
Метод непригоден для кислых сульфатных почв, обогащенных пиритом (FeS2):
5Cr+VI + FeS2 -► Fe3+ + 2S+VI + 5Cr3+
Большое количество Mn2+ также мешает определению, как и металлическое железо,
которое может попасть из размалывающего оборудования.
Оборудование
• Аналитические весы (±0,1 мг) и весы с верхней загрузкой и пределом взвешивания
500 г (±1 мг).
• Широкогорлые конические колбы из пирекса на 500 мл.
• Изолирующие плиты.
• Тефлоновый дозатор.
• Бюретка для титрования.
• Магнитная мешалка с тефлоновыми магнитными якорями.
Реагенты
Все реагенты должны быть аналитической степени чистоты:
• Дистиллированная или бидистиллированная вода (избегать употребления воды,
деионизованной на ионообменной смоле, так как она может содержать мелкие ча¬
стицы смолы).
• Стандартный 1 н. раствор бихромата калия: растворяют 49,040 г KjCrflj (высушен¬
ного в вакууме или над P2Os в эксикаторе) в 800 мл бидистиллированной воды в 1 л
бутыли и затем доливают до 1 л.
• Концентрированная серная кислота, H2S04, d = 1,84.
• 0,5 н. раствор серной кислоты: наливают 800 мл дистиллированной воды в мерный
стакан из пирекса на 1000 мл, затем медленно добавляют 13,9 мл концентрирован¬
ной серной кислоты и перемешивают. Оставляют до охлаждения и доливают дис¬
тиллированной водой до 1 л.
• 0,5 н. раствор соли Мора (железо(Н)-аммоний сульфат): растворяют 196,05 г
Fe(NH4)2S04 • 6Н20 (высушенного в эксикаторе над Р205) в примерно 800 мл 0,5 н.
раствора H2S04 в мерной колбе на 1000 мл. Доливают до 1000 мл 0,5 н. раствором
H2S04. Жидкость должна быть прозрачной и светло-зеленого цвета.
294
Часть 2. Органический анализ
• Концентрированная фосфорная кислота H3P04, d= 1,71 (85%).
• Фторид натрия NaF в виде порошка.
• Раствор дифениламина Раств°Ряют 0,5 г дифениламина в 100 мл кон¬
центрированной H2S04. Наливают в 20 мл воды и хранят во флаконе из темного
стекла с притертой пробкой с капельной пипеткой.
Можно использовать другие индикаторы:
о-Фенантролин (1,10-фенантролин) , который образует комплекс с Fe2+
(ферроин): растворяют 14,85 г моногидрата о-фенантролина и 6,95 г сульфата двух¬
валентного железа (FeS04 • 7Н20) в 800 мл дистиллированной воды. Доводят объем
раствора до 1000 мл в мерной колбе и хранят в темном стеклянном флаконе.
Дифениламин сульфонат бария Ba(C6H5-NH-C6H4-S03)2. Растворяют в дистил¬
лированной воде.
N-Фенилантралиновая кислота
,соон
Методика
Если содержание общего N было предварительно определено, то определяют примерную
массу почвы, необходимую для получения пробы, содержащей 10-25 мг С (табл. 10.3).
Таблица 10.3. Рекомендуемая масса пробы почвы (Р, г) в зависимости от содержания азота
(для соотношения C/N = 10)
N, г/кг
Проба, Р, г
С, мг
(теоретически)
N, г/кг
Проба, Р, г
С, мг
(теоретически)
о
Т
о
К)
10
10-20
1-2
1
10-20
0,3-0,4
5
15-20
3-4
0,5
15-20
0,5-0,9
2,5
12,5-22,5
5-10
0,2
10-20
Отвешивают пробу почвы (±0,1 мг), переносят в широкогорлую коническую колбу
на 500 мл и добавляют точно 10 мл 1 н. раствора бихромата калия. Осторожно пере¬
мешивают, чтобы не оставлять суспензии на стенках колбы. Быстро добавляют 20 мл
концентрированной серной кислоты при помощи тефлонового дозатора. Перемешива¬
ют вращением в течение 1 мин (температура реакции примерно 120 °С). Помещают на
изолирующую пластину и оставляют для продолжения окисления в течение 30 мин.
Добавляют 200 мл дистиллированной воды, затем 10 мл фосфорной кислоты (или
примерно 5 г фторида натрия при помощи шпателя). Гомогенизируют. Добавляют три
капли дифениламина.
Титруют избыток бихромата 0,5 н. раствором двухвалентного железа (концентрация
реагента должна проверяться титрованием ежедневно). Окончание титрования опреде¬
ляется по изменению цвета раствора с сине-фиолетового на светло-зеленовато-голубой.
Определение конечной точки облегчается добавлением 1-2 капель индикатора, как
только цвет раствора начинает меняться.
Примечание
После окончания взаимодействия с органическим веществом раствор должен быть еще
оранжевым, что указывает на избыток бихромата. Если раствор зеленый, реакцию по¬
вторяют с меньшей навеской почвы.
295
Глава 10. Определение содержания органического вещества...
Представление результатов
Признано, что количество потребленного кислорода пропорционально количеству отти¬
трованного углерода, исходя из теоретического положения, что 1 мл 1 н. раствора бихро¬
мата окисляет 3 мг С, т. е. с учетом поправочного коэффициента (1,3 = 100/76) = 3,9 мг С.
Этот поправочный коэффициент может изменяться в зависимости от формы С и на
основании сравнения с результатами сухого озоления:
• Общий органический С, г/кг почвы, высушенной при 105 °С = 3,9(10 - 0,5 V)/P,
где Р — масса пробы, г; К— объем (мл) 0,5 н. раствора Fe2+ (если концентрация
отличается от 0,5, заменяют точным значением); 10 — количество добавленного
раствора бихромата, мл.
• Общее органическое вещество, г/кг = общий органический С х 1,724.
Контроль
Ежедневно проверяют точную концентрацию раствора Fe2+, выполняя два определения
с 10 мл 1 н. раствора бихромата.
В каждой серии определений выполняют два холостых опыта с кварцем, предвари¬
тельно прокаленным при 1100 °С, и два определения контрольных проб почвы того же
типа, что и анализируемые пробы.
10.2.3. Определение содержания органического углерода методом
мокрого окисления при контролируемой температуре
Введение и принцип определения
В процессе мокрого окисления при температуре реакции процесс минерализаиии/окис-
ления активного органического углерода никогда не проходит до конца. Использование
«стандартизованного» коэффициента корреляции, равного 1,3, вводит трудно контро¬
лируемую переменную, потому что на практике величина коэффициента варьирует от
1,10 до 1,40.
Для смягчения проблемы Анне (Аппе, 1945) и затем Мебиус (Mebius, 1960) предложи¬
ли проводить полную минерализацию, поддерживая пробу при постоянной температуре
в течение всего процесса минерализации, не вызывая термического разрушения бихро¬
мата. Эффективность и воспроизводимость исследовались в диапазоне температуры от
130 до 210 °С при различной продолжительности окисления и различных соотношениях
кислота/бихромат. Выше 150 °С бихромат проявляет тенденцию к все более быстрому
разложению, что указывает на необходимость проведения реакции в относительно ко¬
роткий срок. Четкое соблюдение методики и строгий контроль времени и температуры
реакции позволяют достичь приемлемой точности. В принципе, этот тип измерений
и возможное влияние различных факторов — такие же, как в методе, описанном выше
в разделе 10.2.2.
Оборудование
• Блок минерализации, содержащий от 20 до 40 ячеек, термостатируемых при 150 °С
(Tecator, Scalar, Technicon и др. изготовители); возможна автоматизация в лабора¬
ториях, выполняющих многочисленные рутинные анализы; например, в системе
Скаляр (Skalar) держатель образцов накрывается рамой с 20 обратными холодиль¬
никами. После введения проб и реагентов помещают узел в блок программируе¬
мого нагрева; после окончания минерализации вынимают узел и охлаждают, от¬
296 Часть 2. Органический анализ
деляют раму с холодильниками; держатель проб перемещают по рельсу к узлу
разбавления и возможно к системе титрования.
• Аналитические весы (±0,1 мг).
• Пробирки для разложения проб со шлифованным переходом к холодильнику.
• Бюретка для титрования.
• Точный объемный дозатор (±0,1 мл) со стеклянным шприцом из пирекса и тефло¬
новым плунжером.
• Магнитная мешалка с 15 мм тефлоновыми магнитными якорями.
Реагенты
• См. «Реагенты» в разделе 10.2.2.
• 0,2 М раствор Fe2+: отвешивают 78,5 г соли Мора (высушенной над Р205 в экси¬
каторе) и растворяют в 800 мл 0,5 н. раствора серной кислоты. Доводят объем до
1000 мл добавлением раствора серной кислоты.
Методика
Взвешивают (±0,1 мг) от 200 мг до 2 г почвы (измельченной до размера частиц 125 мкм
и высушенной над Р205) чтобы получить пробу, содержащую примерно 15 мг С
(табл. 10.3).
В пирексовую пробирку (со шлифованным переходом к холодильнику) помещают
10 мл 1 н. раствора бихромата калия и 15 мл концентрированной серной кислоты. Го¬
могенизируют. Помещают пробирку в блок нагревания, отрегулированный на 150 °С,
и устанавливают холодильник. Реакцию следует проводить при постоянной температуре
в течение 30 мин.
Охлаждают реакционную смесь на воздухе и затем переносят в широкогорлую ко¬
ническую колбу на 250 мл; доводят до объема около 100 мл водой смывов из пробирки.
Титруют 0,2 М (или 0,5 М) раствором Fe2+ в присутствии индикатора и пассиватора (см.
«Методика» в разделе 10.2.2). В конечной точке титрования темно-фиолетовый цвет
раствора изменяется на светло-зеленый.
Контроль и расчет
• Титрование раствора Fe2+ (два определения).
• Холостой опыт с прокаленным кварцем при нагревании в тех же условиях для вве¬
дения поправки на термическое разложение реагента и определения оптимальной
концентрации бихромата (два определения).
• Анализ стандартного углевода в тех же условиях для проверки полноты окисления
и отсутствия необходимости в поправочном коэффициенте.
Без поправочного коэффициента общий органический С, г/кг почвы, высушенной
при 105 °С = 3,9(10- 0,5 V)/R
Объяснение символов и чисел — см. «Представление результатов» в разделе 10.2.2.
10.2.4. Определение содержания органического углерода методом
мокрого окисления с использованием спектроколориметрии
Французский стандарт NF АЗ 1-109 (1993), разработанный для определения органиче¬
ского углерода окислением хромовой смесью, позволяет рассчитать содержание органи¬
ческого вещества при помощи пересчетного коэффициента для использования в агро¬
номических исследованиях.
297
Глава 10. Определение содержания органического вещества...
Окисление проводят в термостатируемом блоке при 135 °С. Окисление атома углеро¬
да требует переноса четырех электронов. В качестве стандартного вещества используют
глюкозу, и конечное определение проводят методом абсорбционной спектрометрии на
длине волны 585 нм, используя аликвоты, центрифугированные в течение 10 мин при
2000 g и отфильтрованные для удаления взвешенных частиц.
Метод неприменим в присутствии минеральных восстановителей (например, С1~,
Fe2+) или при загрязнении органическими соединениями. Подробная методика ссыла¬
ется на стандарт NFX31-109 (1993).
Примечания
♦ После центрифугирования титрование органического С в растворе может выпол¬
няться на длине волны 590 или 625 нм (в зависимости от стандарта сахарозы).
• Чтобы не переносить пробу, можно использовать зондовый спектрометр с опти¬
ческим световодом, который погружают непосредственно в очищенный раствор
{Baker, 1976).
10.2.5. Определение содержания общего азота с использованием
мокрого метода: Введение
Азот — один из самых распространенных элементов в живых тканях (после углерода,
водорода и кислорода). Он играет важную роль в сельском хозяйстве, будучи необходи¬
мым элементом для роста растений. В почве органические формы могут составлять до
90% от общего азота.
В количественном отношении содержание общего азота включает не только соедине¬
ния N в органическом веществе почвы и биомассы (см. гл. 14), но также неорганические
соединения азота (см. гл. 28). Все эти соединения составляют как краткосрочные, так
и долгосрочные запасы, т. е. азот, непосредственно или потенциально доступный для
растений и способный повысить урожай.
С другой стороны, содержание общего азота не может количественно определить
величину переноса различных форм азота между живыми организмами и неорганиче¬
скими веществами. Таким образом, содержание общего N не может ни количественно
выразить разнообразие форм азота, которые существенно варьируют в почвах, подвер¬
женных специфическим климатическим условиям и условиям окружающей среды, та¬
ким как типы растительности или различным системам земледелия, ни описать сложные
взаимодействия между микроструктурами, контролирующими цикл азота. Компоненты
белков, углеводов, гемицеллюлоз, целлюлозы и лигнина из живых организмов разлага¬
ются в почве, причем самой стабильной фракцией является лигнин.
Фазы иммобилизации N существуют в форме органического и фиксированного N,
а также окклюдированных или обменных форм N—NH4+. Фазы нитрификации/дени-
трификации приводят к образованию N02~, N03~, N2 и оксидов азота (включая погло¬
щение корнями растений и потерю через сток). Азотные процессы в почве могут быть
изменены симбиотическими ассоциациями между растениями (например, бобовыми)
и бактериями (микориза, актиномицеты), которые включают образование клубеньков,
способствующих фиксации атмосферного азота.
Анализ «общего N» мокрым методом основан на методе, предложенном Къельдалем
(Kjeldahl, 1993). Без длительных дополнительных обработок этот метод не позволяет
полностью определять все формы запасов N. Органические и неорганические соедине¬
ния азота включают (в переменных долях):
298
Часть 2, Органический анализ
♦ Неорганические формы: (1) NH4+-N, который может быть обменным или фикси¬
рованным в минеральных или органоминеральных решетках; (2) N03~-N, кото¬
рый содержится в больших количествах в сильно удобренных интенсивно культи¬
вируемых почвах; (3) N02~-N, содержание которого обычно ничтожно, исключая
заболоченные почвы или присутствие загрязнений; N03~-N и N02~-N хорошо
растворимы и, следовательно, легко выщелачиваются просачивающимися и по¬
верхностными водами.
• Объекты, химическое, физическое и биохимическое поведение которых достаточ¬
но хорошо определено, чтобы сгруппировать их по отдельным пулам: растительные
фракции на различных стадиях разложения; активная микробная биомасса; биоло¬
гические формы (аминокислоты, аминосахара, белки бактериальных клеток); N, ги¬
дролизуемый в кислой среде; негидролизуемый N недостоверных соединений. Часть
N включена в состав гумифицированных комплексных соединений различной ста¬
бильности: относительно нестабильные фульвокислоты, гуминовые кислоты с раз¬
личными степенями поликонденсации, гумины (связанные с глинами и связываю¬
щими Fe—А1 соединениями) и особые формы белков, слабо разрушенные протеазами.
Эволюция органического азота почвы связана с молекулярными формами гумусовых
соединений. Процессы конденсации гумусовых молекул и образования органомине¬
ральных соединений изменяют стабильность различных пулов. Эти пулы можно распо¬
ложить в соответствии с возрастающей стабильностью в следующий ряд: фульвокислоты
< алюминийорганические соединения < железоорганические соединения < гуминовые
кислоты < различные органоминеральные соединения < гумины, связанные с глинами.
Выработка аммония служит индикатором нестабильности.
На физическом уровне органические слои, адсорбированные на поверхности мине¬
ральных и органоминеральных матриц, реагируют легче, чем зафиксированные в решет¬
ках. На химическом уровне короткие азотные цепи гидролизуются быстрее, чем длин¬
ные цепи или соединения N, зафиксированные в глинах.
Добавление воды перед анализом освобождает различные доли фиксированного N,
вызывая набухание решеток некоторых глинистых минералов типа 2:1. Этот фиксиро¬
ванный или окклюдированный N может играть определенную роль в питании расте¬
ний (Mengel и Scherer, 1981; Keerrthisinghe и др., 1984). Классический метод Къельдаля
позволяет количественно определять только часть его; поэтому чтобы контролировать
эту переменную, минеральная матрица должна быть сначала разрушена смесью кислот
HF-HC1 (см. раздел 10.3.5).
Вопрос о возможности полностью описать относительную доступность соединений
N в почве при помощи моделей химических и физических компартментов, различаю¬
щих все активные и пассивные формы элемента, еще не решен. Но при любом ответе на
этот вопрос анализ общего органического и неорганического N является существенным
компонентом математических моделей, основанных на динамическом моделировании
форм N в системах почва - растения - климат.
10.2.6. Определение содержания общего азота методом Къельдаля
с титриметрическим окончанием
Основные положения
Минерализация
Целью стадии минерализации является превращение органических форм N в аммоний¬
ную форму в концентрированной серной кислоте в присутствии катализаторов.
Глава 10. Определение содержания органического вещества...
299
(Rj)N + H2S04 Кяпшоат1& (NH4)2S04 + Н20 + С02
2NH; + H2S04 -» (NH4)2S04 + 2Н+
Весь азот в формах амида, имида, нитро N-N, нитрозо N-0 или других превращает¬
ся в соль аммония. Термическая стабильность и повышение температуры реакционной
среды обеспечиваются добавлением К^04. Нитраты и нитриты, вероятно, не подверга¬
ются этому превращению даже в присутствии ртутного катализатора:
2HN03 + 6Hg + 3H2S04 -► 3Hg2S04 + 4H20 + 2NOT
Нитраты и нитриты могут восстанавливаться салициловой кислотой
посульфитом натрия Na2S203.
<5-
соон
он и ги-
Титрование NH4+
Образовавшийся NH4+—N переносят в щелочной среде перегонкой с паром и затем ти¬
труют волюмометрически в присутствии индикатора.
2NH4OH + H2S04 -> (NH4)2S04 + 2Н20
(NH4)2S04 + 2NaOH -> Na2S04 + 2NH3t + 2H20
После переноса NH3 собирают в борной кислоте (Winkler, 1913) и титруют ацидиме-
трически:
4Н3В03 + 2NH4OH -> (NH4)2B407 + 7Н20
(NH4)2B407 + ЗН20 о 2Н3В03 + 2NH4BOz
2NH4B02 + 4Н20 о 2Н3В03 + 2NH4OH
Можно отложить титрование до окончания перегонки всех образцов.
Оборудование
• Аналитические весы (±0,1 мг).
• Штатив для колб для минерализации с газовым подогревом или нагревательные
блоки, термостатируемые при 360 °С.
• Пирексовые колбы Къельдаля на 350 мл или пирексовые цилиндры для нагрева¬
тельных блоков.
• Дозирующий шпатель для катализатора (-500 мг, 1 г, 2 г).
• Стеклянные шары 6 мм в диаметре.
• Тефлоновый дозатор.
• Пирексовые насадки с шаром (рис. 10.1, а).
• Вытяжной шкаф с выводами для тяжелых паров.
• Прибор для перегонки или перегонки с паром.
• Пирексовая коническая (Эрленмейера) колба на 250 мл.
• Бюретка на 50 мл (±0,1 мл).
• Магнитная мешалка с магнитными якорями в тефлоновой оболочке.
BOO
Часть 2. Органический анализ
• Центрифуга на 5000 g и пирексовые центрифужные пробирки на 100 мл.
• Спектроколориметр.
Реагенты
• Серная кислота H2S04, 1,83.
• Серная кислота, содержащая салициловую кислоту (50 г/л).
• Катализатор: измельчают и просеивают (Afnor NF22) 100 г сульфата калия (K^SO^,
20 г сульфата меди (CuS04* 5Н20), 2 г порошка серого селена (Se); перемешивают
и хранят в широко горлом флаконе.
• 2%-ный раствор борной кислоты Н3В03 в дистиллированной воде.
• Индикатор Таширо (Ма Zuazaga, 1942): смешивают одну часть 0,1%-ного раствора
метилового красного в этаноле с тремя частями 0,1%-ного раствора бромкрезола
зеленого в этаноле; хранят в темном флаконе с капельницей.
• 10 М раствор гидроксида натрия (NaOH): отвешивают 2,5 кг гранулированного
NaOH и осторожно растворяют в 6 л дистиллированной воды; охлаждают в закры¬
той пирексовой колбе (С02 удаляют осаждением Na2C03); сливают жидкость и от¬
брасывают осадок; хранят в герметичном флаконе.
• 1 %-ный раствор фенолфталеина в 30%-ном этаноле.
• Стандартные 0,1 и 0,05 н. растворы серной кислоты.
• Стандартный раствор сульфата аммония: отвешивают 4,714 г (NH4)2S04, высушен¬
ного над Р205; растворяют в деионизированной воде и доливают до 1 л; 1 мл рас¬
твора = 1 мг N.
Методика (макрометод)
Минерализация
Отвешивают 2 г (±0,1 мг) почвы (измельченной до размера частиц в 125 мкм) на не-
проклеенной папиросной бумаге (не содержащей азота). Тщательно закрывают, скрутив
бумагу.
Переносят пробу в пирексовую колбу на 350 мл. Увлажняют почву дистиллирован¬
ной водой из промывалки. Осторожно перемешивают до полной гомогенизации почвы.
Оставляют на ночь.
Осторожно (особенно для карбонатных почв) добавляют 20 мл концентрированной
серной кислоты. Добавляют 2 г катализатора с помощью дозирующего шпателя. До¬
бавляют 3 стеклянных шара (диаметром 6 мм), надевают на колбу насадку и помеща¬
ют колбу в штатив (рис. 10.1, а). Уменьшают газовое пламя и проверяют образование
пены. Колба должна быть наклонена под углом 45-60°, чтобы снизить риск выбросов
и улучшить конденсацию на стенках (рис. 10.1, а). Когда органическое вещество почвы
разложится (цвет пробы бледнеет), увеличивают нагрев и кипятят в течение 2—3 ч, не
допуская высыхания.
Перегонка и ацидиметрическое титрование
Охлаждают реакционную смесь. Споласкивают насадку и стенки колбы примерно 10 мл
дистиллированной воды. Устанавливают колбу в прибор для перегонки (рцс. 10.1, б).
Добавляют 100 мл раствора 10 М гидроксида натрия. Закрывают кран и начинают пере¬
гонку с паром.
Глава 10. Определение содержания органического вещества...
301
Рис. 10.1. Минимальное оборудование для определения общего N по Къелвдалю: а — кислотная
минерализация с устройством для уменьшения потерь кислоты; b — перегонка образовавшегося
аммиака при добавлении гидроксида натрия (менее производительный прибор для перегонки
с паром см. на рис. 14.3)
Собирают дистиллят в пирексовой конической колбе, содержащей 20 мл 2% раство¬
ра борной кислоты и 3 капли индикатора Таширо; следят, чтобы конец выводной труб¬
ки находился под поверхностью жидкости в конической колбе. Для количественного
определения необходимо примерно 80 мл дистиллята.
Титруют дистиллят волюмометрически 0,1 или 0,05 н. раствором серной кислоты,
в зависимости от предполагаемого содержания азота. Конечную точку титрования опре¬
деляют по изменению цвета раствора с зеленого на серо-фиолетовый.
Представление результатов
Полученный результат TN выражают в мг азота — N на кг почвы, высушенной при
105 °С:
1 мл 0,1 н. H2S04 0,1 ммоль NH3 равно 1,4 мг N
Если VA — объем 0,1 н. раствора H2S04, мл; Р— масса анализируемой пробы почвы,
высушенной при 105 °С, г, то содержание N в почве, мг (N)/r (почвы) находят по фор¬
муле:
Гм=1,4Иа/Р.
Контроль
Контроль осуществляют перегонкой раствора сульфата аммония известной концентра¬
ции: (1) однократная перегонка после обработки в тех же условиях, что для анализируе¬
302
Часть 2. Органический анализ
мой пробы, в присутствии кварца, прокаленного при 1000 °С (холостой опыт); (2) пря¬
мая перегонка раствора сульфата аммония. Результат холостого опыта рассчитывают как
разницу между двумя операциями. Два стандартных образца и два определения случай¬
ной пробы из каждой серии должны анализироваться в тот же день.
Примечания
Экспериментальный стандарт X 31111 (1983) и затем международный стандарт NF
ISO 11261 (1995) были опубликованы по определению общего азота почвы перегонкой
после минерализации по Къельдалю.
Обесцвечивание не является признаком окончания минерализации, но это признак
окончания окисления окрашенных гумифицированных органических веществ, указы¬
вающий на прекращение образования пены. Превращение азотистых органических ве¬
ществ в аммоний зависит от других факторов, таких как температура, катализатор, вре¬
мя реакции и природа соединений N.
Добавление сульфата калия позволяет повысить температуру кипения серной кисло¬
ты и уменьшить время реакции. Чтобы избежать потерь, используют менее 0,5 г KjSC^
на 1 мл серной кислоты с селеном в качестве катализатора. Температура кипения H2S04
повышается с 330 до 364 °С после добавления 1 г I^SC^ на 1 мл серной кислоты.
Катализаторы: (1) оксид ртути — очень эффективный катализатор, но загрязняет
окружающую среду; оксид ртути может образовывать аминокомплексы, которые не
выделяются при перегонке до обработки пробы тиосульфатом; (2) селен позволяет ко¬
личественно определять N; температура должна быть не слишком высока (< 367 °С),
и реакция должна быть ограничена во времени (максимум 3 ч), чтобы избежать потерь;
(3) стандарт NFISO11261 (1995) рекомендует заменить Se анатазом ТЮ2, который мень¬
ше загрязняет окружающую среду (катализатор: смесь 200 г К^С^, 6 г CuS04 • 5Н20, 6 г
ТЮ2).
Измельчение проб до размера частиц 125 мкм позволяет использовать микрометоды
без существенного снижения точности (Pamas и Wagner, 1921; Markham, 1942), тем са¬
мым уменьшая себестоимость, загрязнение и объем необходимых работ.
Использование нагревательных блоков уменьшает выбросы реакционной смеси, по¬
скольку тепло распределяется более равномерно. Программирование этапов нагревания
позволяет выполнять минерализацию без постоянного контроля.
10.2.7. Определение общего содержания азота методом Къельдаля
со спектроколориметрическим окончанием
Основные положения
После разложения в среде серной кислоты с селеновым катализатором (без меди или
титана) реакция (Berthelot, 1859; Bolleter и др., 1961) продолжается в щелочной среде
(фосфатно-щелочной буфер).
Ионы NH4 реагируют с гипохлоритом натрия и фенолятом натрия (или салицилатом
натрия) с образованием зелено-голубого комплекса. Реакция катализируется нитро-
пруссидом натрия.
303
Глава 10. Определение содержания органического вещества...
Оборудование
• См. «Оборудование» в разделе 10.2.6.
• Центрифуга.
• Автоматический сегментный проточный анализатор с коллектором NH4.
Реагенты
Все реагенты должны быть аналитической степени чистоты:
• Реагенты для минерализации: см. «Реагенты» в разделе 10.2.6.
• Детергент Brij 35: лауриловый эфир полиоксиэтилена С12Н25(ОСН2СН2)ОН) (для
уменьшения загрязнения аналитического оборудования).
• 20%-ный исходный раствор гидроксида натрия (NaOH).
• Двузамёщенный фосфат натрия Na2HP04 • 2Н20.
• 20%-ный исходный раствор тартрата калия-натрия (NaKC4H406 * 4Н20, сегнетова
соль).
• Исходный буферный раствор: растворяют 89 г Na2HP04 • 2Н20 в 800 мл деионизи¬
рованной воды; охлаждают и добавляют 50 мл 20%-ного раствора NaOH; переме¬
шивают и доливают до 1000 мл.
• Буферный раствор: смешивают 200 мл исходного буферного раствора и 250 мл ис¬
ходного 20%-ного раствора тартрата калия-натрия; добавляют 60 мл 20%-ного ис¬
ходного раствора NaOH и около 300 мл воды; перемешивают и охлаждают; добав¬
ляют 1 мл Brij 35; дрводят объем до 1 л и гомогенизируют.
• Салицилат натрия: растворяют 150 г салицилата натрия и 300 мг нитропруссида на¬
трия в 800 мл воды; доводят объем раствора до 1 л; фильтруют через воронку Бюх¬
нера (фильтр с синей лентой без NH4) и добавляют 1 мл Brij 35; хранят в темной
бутыли в защищенном от света месте.
• Нитропруссид натрия Na2Fe(CN)5NO • 2Н20.
• Гипохлорит натрия NaCIO: добавляют 5 мл NaCIO в 80 мл дистиллированной воды
и доводят объем до 100 мл; добавляют 2 капли Brij 35; ежедневно готовят свежий
раствор.
• Сульфат аммония (NH4)2S04.
• Соляная кислота НС137%.
• Стандарт NH4: добавляют 80 мл 37%-ной НС1 к 800 мл воды; охлаждают и добавляют
воды до 1000 мл; готовят исходный раствор сульфата аммония (100 мг NH4—N/мл);
хранят в холодильнике; свежие рабочие растворы в диапазоне от 0 до 50 мкг/мл
должны готовиться ежедневно из исходного раствора.
Методика
Минерализацию выполняют, как описано в параграфе «Минерализация» («Методика
(макрометод)») раздела 10.2.6, но без сульфата меди в смеси катализаторов. После окон¬
чания реакции пробу охлаждают и после полного охлаждения разбавляют и переносят
в мерную колбу на 250 мл.
После доведения объема раствора до метки и перемешивания центрифугируют каж¬
дую аликвоту при 3000 g в течение 15 мин для получения прозрачного раствора. Отби¬
рают пробы для анализа из надосадочной жидкости и разбавляют, если необходимо,
в соответствующее число раз (например, в пять раз) перед введением в пробы в N—NH4
коллектор. Определение проводится по поглощению на длине волны 660 нм в кол ори-
304
Часть 2. Органический анализ
метрической ячейке толщиной 15 мм. Установка, предложенная Буурмансом с сотр.
(Buurmans и др., 1996), показана на рис. 10.2 без стадии разбавления. В случае сильно
карбонатных почв иногда необходимо кроме тартрата калия-натрия добавлять ЭДТА,
чтобы избежать побочных эффектов, связанных с присутствием кальция, который осаж¬
дается в среде серной кислоты и может образовывать коллоидный осадок, трудно раз¬
личимый невооруженным глазом (Gautheyrou и Gautheyrou, 1965).
Скорость
потока, Ввод
мл/мин
Промывание
^A/yVVSt-AAA*—г
Водяная баня
при 37 °С
Спектрометр
видимой части
спектра
►
t
Отходы
2,00
Вода
0,32
Воздух
|l,00
Буферный раствор
t
0,10
Проба
0,32 Салициловая кислота + нитропруссид
0,16 NaCI
1,02 Вывод кюветы
Кювета 15 мм
Фильтр 660 нм
Рис. 10.2. Определение общего азота методом сегментного проточного анализа после
минерализации (Buurmans и др., 1996)
10.2.8. Определение содержания азота методом Къельдаля
с использованием селективного электрода
Основные положения
Принцип метода аналогичен описанному в разделе 10.3.2 для определения аммонийного
азота, с небольшими изменениями условий применения. Использование газодиффузи¬
онного электрода позволяет непосредственно считывать результаты по калибровочной
кривой. Отклик линеен в диапазоне концентраций азота от 0,5 до 500 мг/л, точность
определения снижается в области высоких концентраций из-за логарифмического ха¬
рактера кривой. Рекомендуемый диапазон концентраций - от 0,5 до 10 мг/л.
Преимуществом этого метода анализа является чрезвычайная простота и широкий
рабочий диапазон концентраций. Если температура остается постоянной, ежедневная
калибровка не требуется. При нормальных рабочих условиях помехи отсутствуют.
Оборудование
• Оборудование для минерализации: см. параграф «Оборудование» в разделе 10.2.6.
• Газовый электрод (модель 95-100, Orion).
• Ионометр (pH-метр, потенциометр) с разрешением 0,1 мВ.
• Термостатируемая баня, отрегулированная на 25 °С.
• Магнитная мешалка.
305
Глава 10. Определение содержания органического вещества...
Реагенты
• Кислота для минерализации: см. параграф «Реагенты» в разделе 10.2.6.
• ЮМ раствор гидроксида натрия.
• Стандартный раствор: растворяют 1,179 г сульфата аммония ((NH4)2S04,
М = 132,12) в дистиллированной воде и доводят объем раствора до 250 мл; этот рас¬
твор содержит 1 мг N/мл; разводят в 10 раз для получения исходного стандартного
раствора (0,1 мг N/мл).
• 0,1 М раствор хлорида аммония для заполнения и обслуживания электрода.
Методика
Минерализацию проводят по методике, описанной в параграфе «Минерализация»
(«Методика (макрометод)») раздела 10.2.6. После окончания реакции пробу охлаждают
и переносят в мерную колбу на 200 мл. После полного охлаждения доводят объем до
200 мл.
Построение стандартной кривой: в 50-миллилитровые стаканы помещают объемы
стандартного раствора (0,1 мг N/мл), приведенные в табл. 10.4; доводят объемы до 20 мл
добавлением холостого реакционного раствора (см. раздел 10.2).
Таблица 10.4. Диапазон определения общего азота методом ионометрии; содержание при¬
ведено для пробы массой 2 г, объем раствора по окончании реакции — 200 мл
(см. параграф «Минерализация» («Методика (макрометод)») раздела 10.2.6);
объем аликвоты для титрования — 20 мл
Объем стандартного Объем реакционного
Объем раствора
Концентрация
Концентрация
раствора
раствора
NaOH
раствора,
MrN/л
азота в почве,
(0,1 мг N/мл), мл
в 20 мл, мл
(10 моль/л), мл
MrN/r
1
19
2
5
0,5
2
18
2
10
1
4
16
2
20
2
6
14
2
30
3
8
12
2
40
4
10
10
2
50
5
Доводят температуру стаканов и холостого раствора до 25 °С, погружают электрод
в холостой раствор, избегая попадания воздуха под мембрану; добавляют 2 мл 10 М рас¬
твора гидроксида натрия; начинают осторожное перемешивание для уменьшения эф¬
фекта Вортекса; через 5 мин устанавливают нуль ионометра.
Повторяют операции для различных точек диапазона калибровки. Записывают вели¬
чину сигнала в мВ и строят калибровочную кривую, откладывая логарифмы концентра¬
ций азота на оси абсцисс и показания прибора в мВ на оси ординат.
Измерения должны проводиться на аликвотах объемом 20 мл из каждого реакцион¬
ного раствора аналогично операциям при калибровке. Важно выполнять определение
сразу после добавления гидроксида натрия, чтобы избежать потери аммиака.
Примечания
Перед началом анализа необходимо проверить pH измеряемых растворов, которое
должно быть от 11 до 13 после добавления гидроксида натрия (табл. 10.4). Если pH ниже,
немного увеличивают содержание 10 М раствора гидроксида натрия.
306
Часть 2. Органический анализ
После использования электрод должен храниться с мерами предосторожности, ука¬
занными производителем. Заполняющий раствор следует периодически обновлять.
10.2.9. Механизация и автоматизация метода Къельдаля
Преимуществами метода Къельдаля являются использование только простого оборудо¬
вания и высокая воспроизводимость метода. Однако ручные методы очень длительны.
Они требуют рабочих мест большой площади и вытяжных шкафов для удаления тяже¬
лых паров. Метод может улучшить автоматизация, которая позволяет исключить непо¬
средственный контакт с опасными реагентами, такими как кипящая серная кислота или
концентрированная щелочь.
Использование программируемых блоков нагревания (например, Technicon, Scalar,
Tecator) позволяет регулировать температуру минерализации и уменьшить случаи вне¬
запного вскипания и образования пены.
Существует частично автоматизированное оборудование, включающее пульты управ¬
ления для стадий минерализации, перегонки и титрования в макро- и микромасштабе
(например, Bicasa, Bucchi, Gerhart, Skalar, Foss- Tecator, Velp). Оборудование фирм Prolabo,
Сет, Questron позволяет ускорять минерализацию СВЧ-нагреванием; реакционные со¬
суды обрабатываются автоматически.
Полностью автоматизированные системы поставляются такими производителями,
как Tecator, Foss, Perstop, или Gerhart. Минерализация автоматизирована, а конечная
точка титрования определяется потенциометрически или спектроколориметрически.
Работа оператора сводится к заливанию реагентов, регулировке программы нагревания
и сохранению результатов в конце рабочего дня. Мониторинг облегчается тем, что в слу¬
чае неполадки раздается сигнал тревоги.
10.2.10. Модификации методики анализа для определения содержания
ионов NO', NO~ и связанного азота
Эти методы редко используют в рутинных анализах из-за неопределенности результа¬
тов, длительности процесса и часто малых концентраций определяемых форм азота.
Нитраты и нитриты
Определение содержания общего N мокрыми методами не учитывает нитратов, по¬
скольку они обычно содержатся в объектах окружающей среды в малых количествах
и поэтому не влияют на результаты, полученные для высушенных проб. Однако в случае
возделываемых почв, удобренных навозом с большим содержанием N, предпочтитель¬
нее быстро проверить присутствие нитратов, используя коммерческие тесты, потому что
нитраты и нитриты могут привнести случайную погрешность.
Нитраты и нитриты могут быть учтены в общем N при помощи окислительно-вос¬
становительных реакций, выполненных перед минерализацией по Къельдалю: (1) под
действием KMnOyFe2* — нитрит окисляется в нитрат раствором перманганата калия,
а нитрат затем восстанавливается в аммоний двухвалентным железом перед операцией
минерализации в традиционном методе (см. параграф «Методика (макрометод)» в раз¬
деле 10.2.6); (2) в системе салициловая кислота/тиосульфат аммония — салициловая
кислота может количественно нитроваться только в отсутствие воды (Fuson, 1962);
нитрованные соединения затем восстанавливаются тиосульфатом аммония Перед вы¬
полнением минерализации по Къельдалю, как описано в параграфе «Методика (ма¬
крометод)» раздела 10.2.6 (Nelson и Sommers, 1980; Du Freeze Bate, 1989). Международ¬
307
Глава 10. Определение содержания органического вещества...
ный стандарт NF ISO 11261 (1995) рекомендует: (1) действие 4 мл смеси салициловой
и серной кислот (25 г салициловой кислоты в 1 л концентрированной серной кисло¬
ты) в течение нескольких часов или ночи, (2) добавление 0,5 г тиосульфата натрия
с умеренным нагреванием до исчезновения пены, (3) охлаждение колбы и добавление
катализатора (диоксид титана вместо селена) и серной кислоты для минерализации
по Къельдалю. Нитраты и нитриты также можно определять отдельно методами, опи¬
санными в гл. 28.
Фиксированный и окклюдированный азот
Определение содержания фиксированного или окклюдированного N — очень тонкая
операция. Действительно, некоторые авторы поднимали вопрос относительно приро¬
ды (только неорганический NH4 —N или органический и неорганический N) и спосо¬
бов фиксации N. Фиксированный N не определяется количественно традиционным
методом, особенно в почвах, содержащих глинистые минералы типа 2:1. Для опре¬
деления фиксированного N используют полиэтиленовый флакон, а решетку глини¬
стых минералов разрушают смесью HF-KC1 или концентрированной КОН (см. раздел
28.3.5). Органически усвоенный N определяют в обычных колбах Къельдаля (см. раз¬
дел 10.2.6).
Примечания
Поскольку стекло колб Къельдаля сильно разрушается, их качество быстро снижается,
и они должны регулярно заменяться. Фиксация N-NH4 в кристаллических решетках
минералов более важна в глубоких горизонтах, обогащенных глинами и содержащих
мало соединений N; увеличение способности к фиксации N в зависимости от природы
глинистых минералов наблюдается в ряду: вермикулиты > иллиты > монтмориллониты
> каолиниты.
В некоторых случаях расширение решеток, которое наблюдается при добавлении
воды, может привести к увеличению содержания общего N (Bal, 1925). Следовательно,
степень извлечения N03-N может уменьшиться (Вгетпег и Mulvaney, 1982).
10.3. Сухие методы анализа
10.3.1. Определение содержания общего углерода методом простого
выпаривания
Эти методы основаны на окислении органического вещества почвы и разложении кар¬
бонатов при нагревании проб до относительно высоких температур в течение заданного
периода времени. Измерение потери массы (см. гл. 1) позволяет оценить газовые по¬
тери, состоящие главным образом из С02 и Н20. Печи сопротивления и индукционные
печи могут работать при температурах 1000-1500 °С.
В простейшем случае проводят реакцию или на открытом воздухе, возможно обога¬
щенном кислородом (низкотемпературное озоление), или в закрытых системах с кон¬
тролем температуры и времени. Можно использовать ловушки для захвата некоторых
соединений, катализаторы ускоряют реакции. Системы для газового разделения раз¬
личной степени совершенства используют в последовательном анализе выделяющегося
газа. Таким образом, С02, выделенный органическим веществом, можно собрать при
температуре ниже 500 °С, а С02 из карбонатов — при температуре выше 500 °С.
Хотя методы прокаливания на открытом воздухе для определения содержания обще¬
го N очень просты в исполнении, они не могут считаться количественными и обычно
308 Часть 2. Органический анализ
должны дополняться методами термического анализа (см. гл. 7). В окислительной среде
гравиметрические измерения определяют:
• органический С, превратившийся в С02;
• неорганический С, начиная примерно с 400 °С;
• различные формы воды (гигроскопическая, поровая, кристаллизационная, ги¬
дроксильных групп), которые выделяются в течение всего времени анализа;
• летучие формы N, S, некоторые металлы и металлоиды (С1~).
Эти превращения зависят от температуры и времени нагревания. Однако и другие
превращения также могут мешать измерениям.
В окислительной атмосфере масса некоторых соединений в почве, чувствительных
к окислительно-восстановительным реакциям, будет увеличиваться из-за перехода эле¬
ментов в более высокие степени окисления, например, Fe2+ -* Fe3+. Соединения серы
в реакциях рекомбинации могут одновременно терять и увеличивать массу в зависимо¬
сти от среды: H2S S02 -> S03 -► SO2-.
Таким образом, результаты случайны и зависят от используемой газовой среды (окис¬
лительная, нейтральная, восстановительная) и влияния матрицы. Термически стабиль¬
ный остаток почвы может быть получен между 1100 и 1600 °С. В большинстве случаев
общий органический и неорганический С не может быть приравнен к С, полученному
простым измерением потерь при прокаливании. Ниже 500 °С потери С при низких тем¬
пературах могут легко характеризовать окисление С, но эту переменную трудно опреде¬
лить количественно.
Однако для некоторых почв получаются результаты с малым пределом погрешности,
в частности для песчаных почв с низким содержанием глинистых минералов типа 1:1, но
не карбонатных почв или почв, обогащенных органическим веществом. Декарбоксили-
рование органического вещества начинается около 180°С; считается, что 70% органиче¬
ского С окисляется при 250 °С и 90% — при 500 °С. Для окисления 99% органического С
необходимы температуры от 800 до 950 °С.
В случае карбонатов разложение начинается примерно при 400 °С: карбонаты каль¬
ция и магния (например, кальцит-арагонит, магнезит, доломиты) обычно демонстри¬
руют тот же тип термического поведения, но биогенный кальцит (оболочки, скелеты,
известковые остатки), карбонат железа (сидерит), ископаемые угли и смолы могут отли¬
чаться по поведению, так же как карбонаты натрия с низкой точкой плавления (851 °С).
10.3.2. Совместный инструментальный элементный СН1Ч(05)-анализ
методом сухого озоления
Основные положения
Благодаря совершенствованию оборудования приобретение полностью автоматизиро¬
ванного СНМ(ОБ)-анализатора (Pansu и др., 2001) очень заманчиво. Эти приборы очень
выгодны в лабораториях, выполняющих массовые анализы, поскольку они одновре¬
менно определяют содержание С, N и Н, а также, при соответствующих операционных
условиях, О и S. Для обеспечения непрерывного функционирования приборов операто¬
ры должны проходить специальное обучение. Приборы должны регулярно проверяться
и обслуживаться; точные условия использования должны быть четко определены. В этом
типе оборудования каждый элемент связан со всеми другими (печь, СиО, катализаторы,
различные ловушки) и окончательное качество анализов определяется самым слабым
звеном той цепи. Метод регулируется международным стандартом NF ISO 10694 (1995)
для анализа органического и общего углерода в почвах.
Глава 10. Определение содержания органического вещества...
309
Герметизируют представительную пробу определенной массы в оловянной (или сере¬
бряной) микрокапсуле и помещают ее в печь с контролируемой газовой средой в при¬
сутствии кислорода и оксида меди при 1100°С. Различные формы органического и не¬
органического С и N окисляются или быстро разлагаются в соответствии с принципом
Либиха (1830)
C + 2CuO C02f + 2Си
Температура 1800 °С достигается при взрывном сгорании пробы в оловянной капсуле.
Карбонаты и бикарбонаты разрушаются одновременно. Для определения органического
С карбонаты и бикарбонаты должны быть химически удалены перед помещением про¬
бы в печь
СаС03 1000 °с> С02Т +СаО (термическое разложение)
СаС03 + 2Н+ -► С02Т +Н20 + Са2+ (химическое разложение)
Если присутствует метан, который должен быть определен, следует использовать
Сг203 для обеспечения полного окисления. Органический азот, окисляемый во время
озоления, дает оксиды азота, которые должны восстанавливаться медью для превра¬
щения всех оксидов азота в форму N2 методом Дюма {Dumas, 1831). Неорганический
N в соединениях (NH4+, N03~, N02~) вычитают из общего органического азота. Можно
использовать различные катализаторы, чтобы ускорить реакции или сделать их количе¬
ственными.
Наконец, газовые продукты идентифицируют в соответствующих детекторах; газо¬
вые соединения могут быть разделены в специальных временных ловушках или методом
газовой хроматографии, в зависимости от использованной системы. В почвах, не со¬
держащих карбонатов или бикарбонатов и не обработанных известьсодержащими ме¬
лиорантами:
• содержание неорганического С = 0;
• общий С = общий органический С.
Оборудование
• Элементный СНМ(08)-анализатор, предпочтительно для проб массой 100 мг или
более для компенсирования неравномерности распределения углерода в почве
(Pansu идр., 2001).
• Микроаналитические весы.
• Остроносые щипцы для закрывания капсул.
• Микропипетка переменной емкости на 0,1 -0,5 мл.
• Керамическая пластина для обработки карбонатных проб.
• Нагревательная плита с контролируемой температурой.
Материалы и реагенты
Все расходные материалы должны соответствовать методике и типу анализируемых ма¬
териалов (степень чистоты - чда):
• топливные газы, газ-носитель: чистота 99,98%;
• оксид меди (СиО);
• медная проволока или стружка (Си);
310
Часть 2, Органический анализ
• перхлорат магния (Mg(C104)2), аскарит (асбест, пропитанный NaOH), Мп02 и др.
поглотители для ловушек;
• оловянные или серебряные микрокапсулы емкостью 100 мг;
• набор катализаторов;
• стандартные образцы для калибровки и контроля: ацетанилид (C8H9ON,
71,09% С, 6,71% Н, 10,36% N, 11,84% О), атропин (C17H2$N03), сульфаниламид
(H2NC6H4S02NH2), пикриновая кислота (2,4,6-тринитрофенол (N02)3C6H20H),
тиомочевина (H2NCSNH2) и др.
Методика
Некарбонатные почвы
Все взвешивания должны производиться на аналитических весах (±0,1 или ±0,01 мг).
Калибровка: отвешивают 5 навесок стандарта возрастающей массы в диапазоне кон¬
центраций, соответствующем техническим характеристикам CHN-анализатора. Точ¬
ность оборудования проверяют сравнением теоретического содержания и полученных
результатов.
Пробы: Измельчают пробы до размера частиц 125 мкм (сито AFNOR NF12) и высуши¬
вают в течение 48 ч в эксикаторе над Р205, чтобы уменьшить насыщение поглотителей
в ловушках. Осторожно отвешивают от 50 до 100 мг пробы (в зависимости от предпола¬
гаемого содержания С и N), высушенной над Р205, в оловянной или серебряной капсуле
и запечатывают ее. Помещают капсулы в распределитель анализатора.
Одновременно отвешивают 1 г той же пробы для измерения остаточной влажности
высушиванием при 105 °С и корректировки результатов (см. гл. 1). Непосредственное
высушивание проб при 105 °С перед CHN-анализом может увеличить фиксацию аммо¬
ния в решетке глинистых минералов типа 2:1, но в большинстве случаев это приемлемо.
Примечание
Андосоли и гистосоли вызывают два типа осложнений: (1) остаточная влажность воз¬
душно-сухой пробы очень велика, до 50-60% после высушивания в течение 6 мес.;
(2) низкий объемный вес (до 0,30-0,25), что не позволяет взвесить 100 мг почвы в капсуле.
Углерод в карбонатных почвах
В случае карбонатных или заизвесткованных почв в отсутствие сидерита (трудно разла¬
гаемый FeC03) обработка 10%-ным раствором хлористоводородной кислоты достаточна
для разрушения карбонатов и бикарбонатов в пробе:
• Отвешивают 100 мг пробы в серебряной капсуле.
• Помещают открытую капсулу на керамическую пластину.
• При помощи микропипетки медленно добавляют 0,1-0,2 мл 10%-ной НС1 в зави¬
симости от предполагаемого содержания карбоната и бикарбоната.
• Оставляют в контакте на 2 ч и затем добавляют 0,1 мл 10%-ного раствора НС1. Удо¬
стоверяются, что выделение газа закончилось.
• Помещают керамическую пластину на нагревательную плиту при 60 °С и сушат.
Охлаждают и запаивают капсулу. Помещают капсулу на CHN-пробоотборник
и определяют содержание общего органического С. Вторую пробу можно взвесить
без разложения карбонатов для измерения содержания общего С. Содержание об¬
щего неорганического С можно рассчитать по разнице.
Глава 10. Определение содержания органического вещества...
311
На некоторых приборах предпочтительно использовать фосфорную, а не хлористо¬
водородную кислоту для уменьшения влияния хлорида на катализаторы и поглотители
в ловушках.
Расчеты
Результаты рассчитываются и распечатываются CHN-анализатором после измерения
площадей пиков С и N. Эти расчеты проводятся с учетом массы пробы.
При определении общего органического азота принимают во внимание результаты,
полученные для общего неорганического азота (см. гл. 28):
Общий N (CHN) = неорганический N + органический N.
Определение водорода, кислорода и серы
Водород определяют вместе с углеродом и азотом по методике, описанной в параграфе
«Методика» раздела 10.3.2, измерением содержания воды, образовавшейся при сжига¬
нии. Однако полученные результаты для большинства почв трудно интерпретировать,
потому что водород, образовавшийся из органических веществ пробы, и водород из
различных форм воды (например, адсорбционная, гидратированная и гидроксилиро-
ванная) не разделены в сигнале Н20. В почвах, содержащих мало глин, и при исполь¬
зовании осторожно высушенных проб сигнал Н20 представляет только водород из ор¬
ганических молекул. Соотношение С/Н выявляет стадии превращения органического
вещества почвы.
Определение кислорода и серы обычно выполняют, используя стандартное CHN-
оборудование, простой модификацией инструментальных параметров. Для определе¬
ния серы используют контролируемое окисление, обычно в присутствии вольфрамового
ангидрида. Для определения кислорода используют пиролиз вместо сжигания (подачу
кислорода отключают) на пробе, специально выбранной для этого определения.
Общий органический кислород• пробу подвергают пиролизу при 1100 °С в присутствии
катализатора (смесь углерода и никеля). Образуется монооксид углерода (СО), который
затем отделяется системой разделения (обычно при помощи ловушки или хроматогра¬
фии) и количественно определяется системой детектирования (например, катароме-
тром). Кислород определяют сравнением со стандартами с известным содержанием О
аналогично тому, как описано для С и N в параграфе «Методика» раздела 10.3.2: одна
молекула СО соответствует одному атому О в пробе.
Общий органический кислород соответствует лишь малой доле общего кислорода
пробы почвы. Неорганический кислород, содержание которого больше, обычно опреде¬
ляют, исходя из валового анализа основных элементов (см. гл. 31), рассчитывая разницу
между суммой содержаний оксидов и суммой содержаний элементов.
Общая сера: пробу окисляют при 1100 °С в присутствии вольфрамового ангидрида
W03 или смеси оксидов вольфрама и ванадия. Образуются оксиды серы (например, три-
оксид серы S03, диоксид серы S02). Стадия восстановления на медной колонке превра¬
щает все оксиды в форму S02. Этот газ затем отделяют и определяют, используя систе¬
му, зависящую от типа используемого оборудования (ловушки, хроматография) и типа
детектора (катарометр, ИК-детектор): одна молекула S02 соответствует одному атому S
в пробе. Возможность (или невозможность) проведения определения серы одновремен¬
но с определением углерода и азота зависит от типа используемого оборудования.
Содержание серы, получаемое инструментальными методами определения CHNS,
обычно считается соответствующим общему содержанию серы в почве: окисление орга-
312
Часть 2. Органический анализ
нической серы и сульфидов, разложение большинства сульфатов (Laurent, 1990). Однако
необходимо учитывать разнообразие природных форм серы. Более подробно определе¬
ние этого элемента рассматривается в главе 30.
Примечания
Щелочные соли (например, карбонат натрия, хлориды, фосфаты) плавятся и замедляют
окисление С в С02, покрывая органические частицы и нарушая цикл анализа.
Соли, которые становятся летучими при 700 °С и выше (например, некоторые хлори¬
ды, бромиды или иодиды), могут загрязнять (или разъедать) линии соединений, ловуш¬
ки, катализаторы.
Перхлорат магния Mg(C104)2, продаваемый под названием ангидрона или дегидрита,
используют для улавливания воды. Это относительно нестабильное соединение, кото¬
рое после использования должно удаляться так же, как другие опасные отходы.
Остатки после прокаливания в печи должны удаляться регулярно, особенно в случае
засоленных карбонатных почв. Смена защитных гондол позволяет: (1) сохранить ка¬
чество фильтров для удаления мелких частиц, которые могут присутствовать в газовой
фазе и (2) уменьшить задержание летучих продуктов сжигания. Ловушки, соединения,
катализаторы и фильтры должны регулярно меняться в соответствии со сроком исполь¬
зования. Большинство ошибок и сомнительных результатов связаны с неправильным
обслуживанием оборудования.
Использование искусственных стандартов может привести к ошибкам, поскольку их
физические и химические свойства могут отличаться от свойств органического веще¬
ства почвы. Однако проведение сжигания в капсулах при температуре 1800 °С позволяет
расщепить все органические соединения. Стандартные образцы почв с аттестованным
содержанием органического С также можно использовать для калибровки. Элементные
СНМ(08)-анализаторы обычно дают точные результаты, но они могут превышать ре¬
зультаты мокрого анализа.
10.3.3. Определение CHNOS термическими методами анализа
В детальных исследованиях важно не упустить возможности, предлагаемые инструмен¬
тальными методами, такими как дифференциальный термический анализ (ДТА) и тер¬
могравиметрический анализ (ТГА), объединенные или нет с анализом выделяемого газа
(АВГ) (см. гл. 7).
Контролируемый пиролиз (с подходящими программами для изменения температуры
со временем) позволяет осуществлять мониторинг окисления органического вещества
и разложения неорганических соединений. Можно идентифицировать газы (например,
С02, СО, СН4, Н20, NH3, H2S, S02), которые соответствуют пикам ДТА и ТГА в зависи¬
мости от температуры, контролировать разложение или превращение соединений, со¬
держащих С и N, разделять экзотермические и эндотермические пики Н20 и С02 и т. д.
Для более полного исследования некоторые фазы улавливают при —180 °С, и затем их
разделяют методом газовой хроматографии или его комбинацией, например, с детекто¬
рами ИК- или масс-спектрометров, что позволяет подробно характеризовать органиче¬
ское вещество почвы.
Сумма различных фаз углерода дает содержание общего С; можно приблизительно
разделить общий органический С и общий неорганический С (рис. 10.3). В органиче¬
ской геохимии молекулярную массу фрагментов, полученных при пиролизе, измеряют
при помощи масс-спектрометра с непрерывным вводом пробы. Можно охарактеризо¬
вать структуру комплексных соединений высокого молекулярного веса.
Глава 10. Определение содержания органического вещества...
313
Окисление ядер гуминовых кислот
Кальцит
а
а
Биогенный 1
кальцит \
Магнезит Арагонит
|а а ■ 1 II
I ■ |1 I U I I I —I 1_1 к I—
0 100 300 500 700 900 7,0С
Преимущественно окисление , Преимущественно разложение
органического С ■ неорганического С
■
Рис. 10.3. Окисление органического и разложение неорганического С
в зависимости от температуры
10.3.4. Определение содержания углерода и азота неразрушающими
инструментальными методами
Анализ методом ближней инфракрасной спектрометрии диффузного отражения
(БИКС, см. гл. 5) выполняют непосредственно на пробе почвы, просеянной через сито
AFNOR NF12 (размер отверстий 125 мкм).
Отбирают пробу массой примерно 1 г после высушивания в сушильном шкафу, тща¬
тельно упаковывают в специальную кювету и помещают в измерительную камеру при¬
бора. Поверхность пробы должна быть абсолютно ровной. Каждый компонент органи¬
ческих комплексов почвы имеет специфическую точку поглощения в диапазоне между
700 и 2500 нм БИК-спектра вследствие валентных и деформационных колебаний связей
между элементами (см. раздел 5.3.1).
Для измерения содержания С и N данные, полученные из БИК-спектров, должны
быть откалиброваны по результатам, полученным другими методами исследования. За¬
тем полученную калибровочную кривую используют для определения содержания С и N
в неизвестных пробах. Хорошую калибровку получали при помощи методов, описанных
выше в разделах 10.3.1—10.3.3 (Krishnan и др., 1980; Dalalvi Henry, 1986; Моггаищ.>., 1991;
Fidencio и др., 2002). Это недеструктивный метод, и пробы могут быть затем использованы
для других анализов. Поскольку каждый прибор имеет индивидуальные характеристики
относительно выбора и оптимизации результатов, необходимо следовать рекомендациям
изготовителя (например, Bran-Luebbe, Bruker, Foss, Leco, Nicolet, Perkin-Elmer и Perstorp).
Экспресс-методы определения содержания С в почве в полевых условиях являются
в настоящее время объектом серьезного изучения в исследовательских проектах, посвя¬
щенных распределению С в почвах, в рамках усилий по снижению выделения парнико¬
вых газов в атмосферу. Хотя эти методы менее точны, чем лабораторные методы мокрого
или сухого озоления, они обладают преимуществом быстрого проведения большого ко¬
личества измерений при низкой стоимости. Методы БИКС для обработки аналитиче-
Потеря воды
из гипса,
аллофана
Потеря воды
из глин, окисление
функциональных
групп гуминовых
кислот .
314
Часть 2. Органический анализ
ских сигналов были разработаны благодаря впечатляющему развитию компьютерных
технологий. Компьютерные программы позволяют получать количественные оценки на
основании спектров, которые прежде с трудом поддавались химической интерпретации.
Кроме метода БИКС, для определения содержания углерода в почве была предложе¬
на лазерно-искровая эмиссионная спектроскопия {laser-induced breakdown spectroscopy,
LIBS) {Cremers и др., 1996). Предел обнаружения метода примерно 300 мг/кг, воспроиз¬
водимость 4-5%, точность 3-14%, производительность анализа — более одной пробы
в минуту {Cremers и др., 2001).
10.3.5. Совместный анализ различных изотопов углерода и азота
СНМ(08)-анализаторы, основанные на методе сухого озоления, могут сопрягаться
с масс-спектрометрами для исследования различных изотопов элементов, в частности
l4C, 13С, 15N {Pansu и др., 2001). Часто содержание изотопа азота 15N определяют в форме
аммония в дистиллятах, полученных методом Къельдаля. Диоксид углерода из образу¬
ющегося газа сгорания можно улавливать в ловушки для последующего определения
изотопного состава. Оборудование Боттнера и Варембурга {Bottner и Warembourg, 1976),
схематически показанное на рис. 10.4, использовалось в течение более 20 лет для одно¬
временного определения общего С {carmhograph 12А) и изотопа 14С (жидкостная сцин¬
тилляция) в твердых (почвы, растения) и жидких (почвенная вода) пробах и показало
свою надежность.
Рис. 10.4. Одновременное определение общего С и изотопа 14С (частное сообщение, Р. Bottner,
CEFE-CNRS Монпелье, Франция): а — натронная известь для очистки 02; Ь—кипящая жидкая проба
+ К2Сг207; с — трубка для сжигания; d — трубка для дожигания (СиО); е — твердая проба, f— печь
для сжигания при 900 °С; д — автоматическая бюретка с H2S04 + H3P04; h — ловушки для очистки
(конденсата воды, пены хромпика и кислоты); / — колонки со стеклянными шарами, пропитанными
смесью моноэтилового эфира этиленгликоля и моноэтаноламина для улавливания С02;у—ловушки
с натронной известью для надежности; к— вентилируемый внешний вывод; /— вывод для избытка 02
315
Глава 10. Определение содержания органического вещества-
Использованная литература
Anne Р (1945) Sur le dosage du carbone organique des sols Ann. Agron., 15,161-172.
Baker KF (1976) The determination of organic carbon in soil using a probe colorimeter. Lab. Practice,
25, 82-83.
Bal DV (1925) The determination of nitrogen in heavy clay soils. J. Agric. Sci., 15,454-459.
Berthelot MP (1859) Violet d’amiline. Repertoire chim. Appl., 1, 284.
Bolleter WT, Bushman, CJ and Tidwell PW (1961) Spectrophotometric determination of ammonia as
indophenol. Anal. Chem., 33, 542-594.
Bottner P and Warembourg F (1976) Method for simultaneous measurement of total and radioactive
carbon in soils, soil extracts and plant materials, Plant Soil, 45, 273-277.
Bremner JM and Mulvaney CS (1982) Nitrogen total. In Methods of soil analysis — part 2. Chemical and
microbiological properties. ASA-SSSA, 9, 595-624.
Buurmans P, van Lager В and Velthorst FJ (1996) Manual for soil and water analysis., Backhuis
Publishers, Leiden, 17-21.
Cremers DA, Ferris MJ and Davies (1996) Transportable Induced Laser Breakdown Spectroscopy (LIBS)
instrument for field-based soil analysis. Proc. Soc. Photo Opt. Inst. Eng., 2835,190-200.
Cremers DA, Ebinger MH, Breashears DD, Uukefer PJ, Kammerdiener SA, Ferris MJ, Catlett KM and
Brown JR (2001) Measuring Total Soil Carbon with Laser-Induced Breakdown Spectroscopy (LIBS).
J Em Qual., 30, 2202-2206.
Dalai RC and Henry RJ (1986) Simultaneous determination of moisture, organic carbon and total nitrogen
by near infrared reflectance spectrometry. Soil Sci. Soc. Am. J., 50, 120-123.
Du Preez DR and Bate GC (1989) A simple method for the quantitative recovery of nitrate N during
Kjeldahl analysis of dry soil and plant samples. Commun. Soil Sci. Plant Anal., 20, 345-357.
Fidencio PH, Poppi RJ and de <Andrade JC (2002) Determination of Organic Matter in Soils using function
networks and near infrared spectroscopy. Anal. Chim. Acta., 453, 125-134.
Fuson RC (1962) Reactions of organic compounds —A text book for the advanced student Wiley, Bristol
1962.
Gautheyrou J and Gautheyrou M (1965) Dosage simultand de l’azote ammoniacal et nitrique dans le sols.
Contribution & l’dtude de la dynamique de l’azote. Cah. Orstom Ser.Pedol., III, 367-391.
Keerrthisinghe GK, Mengel К and De Datta SK (1984) The release of nonexchangeable ammonium (,5N-
labeled) in wetland rice soils. Soil Sci. Soc. Am. J., 48,291-294.
Kjeldahl J (1883) Neue mdthode zur bestimmung des stickstoffs in organischen korpem. Z. Anal. Chem.,
22, 366-382.
Krishnan P, Alexander JD, Butler BJ and Hummel JW (1980) Reflectance techniques for predicting soil
organic matter. Soil Sci. Soc. Am. J., 44, 1282-1285.
Laurent JY (1990) Analyse dldmentaire d’dchantillons de sol et solutions: application de la microanalyse
au dosage simultand de l’azote, du carbone et du soufre. In Actes Journees laboratoire, Orstom,
Bondy, France.
Ma TS and Zuazaga G (1942) Micro — Kjeldahl determination of nitrogen. A new indicator and an
improved rapid method. Ind. Eng. Chem. Anal., 14, 280-282.
Markham R (1942) A steam distillation apparatus suitable for micro - Kjeldahl analysis. Biochem. J., 36,
790-791.
Mebius LJ (1960) A rapid method for the determination of organic carbon in soil. Anal. Chem. Acta., 22,
120-124.
Mengel К and Scherer HW (1981) Release of non exchangeable (fixed) soil ammonium under field
condition during the growing season. Soil Sci., 131,226-232.
Morra MJ, Hall MH and Free Bom LL (1991) Carbon and nitrogen analysis of soil fraction using near
infrared reflectance spectroscopy. Soil. Sci. Soc. Am. J., 55, 281-291.
NF X31-109 (1993) Determination du carbone organique par oxydation sulfochromique. In Qualite des
sols, 1996, AFNOR, Paris, 67-73.
NF ISO 10694 (1995) Dosage du carbone organique et du carbone total aprds combustion sdche (analyse
dldmentaire). In Qualite des sols, 1996, AFNOR, Paris, 189-199.
NF ISO 11261 (1995) Dosage de l’azote total — Mdthode de Kjeldahl modifide. In Qualite des sols,
1996, AFNOR, Paris, 257-260.
316 Часть 2. Органический анализ
X 31-111 (1983) Determination de Pazote total — M6thode par distillation apr£s mineralisation
(Kjeldahl). In Qualite des sols, 1994, AFNOR, Paris, 69-72.
Pansu M, Sallih Z and Bottner P (1998) A process-based model for carbon and nitrogen transfers in soil
organic matter. In Actes 16e congres mondial de science du sol, 20-26 april, Montpellier, France.
Pansu M, Gautheyrou J and Loyer JY (2001) Soil Analysis — Sampling, Instrumentation and Quality
control. Balkema, Lisse, Abington, Exton, Tokyo, 489.
Pamas JK and Wagner R (1921) Uber dieausfuhrung von bestmmungen kleiner stickstoffmengen nach
Kjeldahl. Biochem. Z, 125, 253-256.
Schollenberger CJ (1927) A rapid approximate method for determining soil organic matter. Soil Sci., 24,
65-68.
Walkley A and Black A (1934) An examination of the Degtjareff method for determining soil organic
matter and a proposed modification of the chromic acid titration method. Soil ScL, 37, 29-38.
Winkler LW (1913) Beitrag zur titrimetrishen bestimmung des ammoniaks. ZAngew. Chem., 26, 231—
232.
Дополнительная литература
Allison LE, Bollen WB and Moodie CD (1965) Total carbon. In Black C. A. et A.L. Method of soil an
Analysis part 2. Agronomy Monograph n°9. Am. Soc. Agron., 1346-1366.
Alves В JR, Boddey RM and Urquiaga SS (1993) A rapid and sensitive flow injection technique for the
analysis of ammonium in soil extracts. Commun. Soil ScL Plant Anal., 24, 277-284.
Association of Official Analytical Chemists. AO AC (1975) Official methods of analysis, 924-925.
Bemoux M and Cerri CEP (2005) Soil organic components. In Encyclopedia of Analytical Science, 2nd
Edition, Vol. 4, Elsevier, Amsterdam.
Bowman RA, Guenzin WD and Savory DJ (1991) Spectroscopic method for estimation of soil organic
carbon. Soil Sci. Soc. Am. J., 55, 563-566.
Bremner JM and Tabatabai M.A (1972) Use of an ammonia electrode for determination of ammonium in
Kjeldahl analysis of soils. Commun. Soil Sci. Plant Anal., 3, 159-165.
Burton DL, Gower DA, Rutherford PM and McGill WB (1989) Amino acid interference with ammonium
determination in soil extracts using the automated indophenol method. Commun. In Soil Sci. Plant
Anal., 20, 555-565.
Carr CE (1973) Gravimetric determination of soil carbon using the Leco induction furnace. ,J. ScL Food
Agric., 24, 1091-1095.
Cheng HH and Kimble J (2000) Methods of Analysis for Soil Carbon: An Overview. In Global Climate
Change and Tropical Ecosystems, CRC, Boca Raton, 333-339.
Chichester FW and Chaison RF (1992) Analysis of carbon in calcareous soil using a two temperature dry
combustion infra-red instrumental procedure. Soil Sci., 153, 237-241.
De Bolt DC (1974) A high sample volume procedure for the colorimetric determination of soil organic
matter. Commun. Soil Sci. Plant Anal., 5, 131-137.
Decreider and Meaux R (1973) Utilisation d’une electrode ionique specifique pour le dosage de l’azote
par la methode Kjeldahl. Analusis, 2, 442-445.
Diaz-Zorita M (1999) Soil organic carbon recovery by the Walkley-Black method in a typic.Hapludoll.
Commun. Soil Sci. Plant Anal., 30, 739-745.
Donkin MJ (1991) Loss-on-ignition as an estimator of soil organic carbon in A horizon forestry soils.
Commun. Soil ScL Plant Anal, 22, 233-241.
Froelich PN (1980) Analysis of organic carbon in marine sediments. Limnol.Oceanogr., 25, 564-572.
Gallaher RN, Weldon CO and Bowell FC (1976) A semi-automated procedure for total nitrogen in plant
and soil samplers. Soil. ScL Soc. Am. J., 40, 887-889.
Genty CE and Willis RB (1988) Improved method for automated determination of ammonium in soil
extracts. Commun. In Soil ScL Plant Anal., 19, 721-737.
Graham ER (1948) Determination of soil organic matter by means of a photoelectric colorimeter. Soil
ScL, 65, 181-183.
Guillet В (1979) Etude du renouvellement des matures organiques des sols par les radio isotopes l4C. In
Pedologie—constituants et propriites du sol, Bonneau M and Souchier В ed. Masson, Paris, 210-226.
317
Глава 10. Определение содержания органического вещества...
Heanes DL (1984) Determination of total organic C in soils by an improved chromic acid digestion and
spectrophotometric procedure. Commun. SoilScL Plant Anal., 15,1191-1213.
Henzell EF, Wallis I and Lindquist JE (1968) Automatic colorimetric methods for the determination of
nitrogen in digests and extracts of soils. Int. Cong. Soil Sci. Trans., 9th (Adelaide), 3, 513-520.
Howard PJ A and Howard DM (1990) Use of organic carbon and loss on ignition to estimate soil organic
matter in different soil types and horizons. Biol. Fert. Soils., 9, 306-310.
Kalembasa SI and Jenkinson DS (1973) A comparative study of titrimetric and gravimetric methods for
determination of organic carbon in soils. J. Sci. FoodAgric., 24,1085-1090.
Kalisz PJ and Sainju UM (1991) Determination of carbon in coal “blooms”. Commun. Soil Sci. Plant
Anal., 22, 393-398.
Kempers AJ and Kok CJ (1989) Re-examination of the determination of ammonium as the indophenol
blue complex using salicylate. Anal. Chim. Acta., 221, 147-155.
Lai R, Kimble JM, Follet RF and Stewart BA (2001) Assessment Methods for Soil Carbon, Lewis CRC,
London/Boca Raton, 676.
Lowther JR, Smethurst PJ, Carlyle JC and Nambiar EKS (1990) Methods for determining organic carbon
in podzolic sands. Commun. Soil Sci. Plant Anal., 21, 457-470.
Me Geehan SL and Naylor DV (1988) Automated instrumental analysis of carbon and nitrogen in plant
and soil samples. Commun. Soil Sci. Plant Anal., 19, 493-505.
Merry RH and Spouncer LR (1988) The measurement of carbon in soils using a micro processor -
controlled resistance furnace. Commun. Soil Sci.Plant Anal, 19, 707-720.
Nelson DW and Sommers LE (1975) A rapid and accurate procedure for estimation of organic carbon in
soil. Proc. Indiana Acad. Sci., 84, 456-462.
Nelson DW and Sommers LE (1982) Total carbon, organic carbon and organic matter. In Methods of soil
analysis part 2. Page AL, Miller RH and Keeney DR ed. Agron. Monogr., 9, ASA - SSSA Madison.,
539-579.
Nelson DW and Sommers LE (1996) Total carbon, organic carbon and organic matter. In Methods of soil
analysis, part 3, chemical methods, Sparks DL ed SSSA Book series no 5, 961-1010.
Nelson DW (1983) Determination of ammonium in KC1 extracts of soils by the salicylate method.
Commun. Soil Sci. Plant Anal., 14, 1051-1062.
Nommik H (1971) A modified procedure for determination of organic carbon in soils by wet combustion.
Soil Sci., 111,330-336.
Patton JC and Crouch SR (1977) Spectrophotometric and kinetics investigation of the Berthelot reaction
for the determination of ammonia. Anal. Chem., 3, 464-469.
Pella E (1990) Elemental organic analysis. Part 1, historical development, 116-125; part 2, state of the
art, 28-32, Ann. Lab.
Perrier ER and Kellog M (1960) Colorimetric determination of soil organic matter. Soil Sci., 90,104-106.
Puttanna К and Prakasa Rao EVS (1993) Determination of nitrate in soil by second derivative ultraviolet
spectrometry. Commun. Soil Sci. Plant Anal., 24, 737-743.
Qiu Xing-Chu and Zhu Ying-Quan (1987) Sensitive spectrophotometric determination of ammoniacal
nitrogen analysis. Analusis, 15,254-258.
Quinn JG and Salomon M (1964) Chloride interference in the dichromate oxidation of soil hydrolyzates.
Soil Sci. Soc. Am. Proc., 28,456.
Rowland AP (1983) An automated method for the determination of ammonium-N in ecological materials.
Commun. Soil Sci. Plant Anal., 14, 49-63.
Schepers JS, Francis DD and Thompson MT (1989) Simultaneous determination of total C, total N, and
15N in soil and plant materiel. Commun. Soil Sci. Plant Anal., 20, 949-959.
Schulte EE, Kaufman C and Peter JB (1991) The influence of sample size and heating time on soil weight
loss - on - ignition. Commun. Soil Sci. Plant Anal., 22, 159-168.
Schuman GE, Stanley MA and Knudsen D (1973) Automated total nitrogen analysis of soil and plant
samples. Soil Sci. Soc. Am. Proc., 37, 480-481.
Sheldrick BH (1986). Test of the Leco CHN 600 determination for soil carbon and nitrogen analysis.
Canadian J. of Soil Sci., 66, 543-545.
Sims JJ and Haby VA (1971) Simplified colorimetric determination of soil organic matter Soil Sci., 112,
137-141.
318
Часть 2. Органический анализ
Skjemstad JO, Reeve R (1976) The determination of nitrogen in soils by rapid high temperature Kjeldahl
digestion and auto analysis. Commun. Soil ScL Plant Anal, 7, 229-239.
Soon YK and Aboud S (1991) A comparison of some methods for soil organic carbon determination.
Commun. Soil Sci. Plant Anal, 22, 943-954.
Stewart RA and Porter LK (1963) Inability of Kjeldahl methods to fully measure indogeneous fixed
ammonium in some soil. Soil Sci. Soc. Am. Proc., 27, 41-43.
Szekely E (1991) A rapid colorimetric method for analysis of nitrate nitrogen by reduction to nitrite.
Commun. Soil Sci. Plant Anal, 22, 1295-1302.
Tabatabai MA and Bremner JM (1991) Automated instruments for determination of total carbon, nitrogen
and sulphur in soils by combustion techniques. In Soil analysis — Modern instrumental techniques,
Smith KAed. Marcel Dekker, NY, USA 261-286.
Takesako H (1991) Double-plunger pump system flow injection spectrophotometric determination of
inorganic nitrogen in soil extracts, part 1, flow injection analysis of ammonium nitrogen in soil
extracts, part 2, flow injection analysis of nitrate nitrogen in soil extracts, JpnJ Soil Sci. Plant Nut.,
62, 128-134, 135-140.
Tel DA and Jansen J (1992) Determination of total nitrogen in soil digest using a Traacs 800 Autoanalyser.
Commun. Soil Sci. Plant Anal, 23, 2729-2736.
Tisley J (1950) Determination of organic carbon in soils by dichromate mixtures in Trans. 4th Int. Cong.
Soil. Sci., 1, 161-169. Hoitsemo Brothers, Groningen.
Verardo DJ, Froelich PN and Mcintyre A (1990) Determination of organic carbon and nitrogen in marine
sediments using the Carlo Erba Na. 1500 analyser. Deep Sea Res., 37, 157-165.
Volonteri HJ (1983) Modificacion de un metodo colorimetrico para la determinacion del nitrogeno total
en suelos у pastos. Ciencia del suelo, 1, 98-99.
Walinga J, Kithome M, Novozamsky I, Houba VJG and Vanderlee JJ (1992) Spectrometric determination
of organic carbon in soils. Commun. Soil Sci. Plant Anal., 23, 1935-1944.
Winter JP, Gregorich EG, Voroney RP and Kachanoski RG (1990) Comparison of two samples oxidation
methods for quantitative measurement of 12C and 14C in plant and soil. Canad. J. Soil Sci., 70,
525-529.
Yeumans JC and Bremner JM (1991) Carbon and nitrogen analysis of soils by automated combustion
techniques. Commun. Soil Sci. Plant Anal, 22, 843-850.
Глава 11. Количественное определение гуминовых
веществ
11.1. Гумус в почвах
11.1.1. Определения
Гумус играет фундаментальную роль в экологических процессах как источник углерода
для атмосферы, потребитель углерода из биосферы, потребитель и источник удобрений
для растений и фактор, влияющий на свойства почвы; ему посвящаются регулярные об¬
зоры (Kononova, 1966; Flaig и др., 1975; Schnitzer, 1978; Stevenson, 1982, 1994; Aiken и др.,
1985; Tate, 1992; Carters Stewart, 1995; Piccolo, 1996; Magdoffи др., 1996; Hessen и Tranvik,
1998).
Стивенсон (Stevenson, 1982) определил термин «гумус» (или гумифицированное веще¬
ство) как сумму органических соединений в почве, за исключением живых организмов
в биомассе и неразложившихся или частично разложившихся органических остатков
растительного или животного происхождения (см. гл. 9). Использование термина «ор¬
ганическое вещество почвы» менее определенно: иногда его используют в том же значе¬
нии, что термин «гумус», но в действительности он должен относиться к сумме органи¬
ческих материалов почвы.,
Гумифицированное вещество представляет более половины общего органического
углерода почвы и может быть подразделено на две группы: гуминовые и негуминовые
вещества (Schnitzer, 1978). Физические и химические характеристики негуминовых ве¬
ществ, например, углеводов, белков, пептидов, аминокислот, липидов, восков и органи¬
ческих кислот низкого молекулярного веса, легко определяемы.
С другой стороны, гуминовые вещества не обладают такими заметными физико-хи¬
мическими характеристиками. Они имеют более или менее темный цвет, их молекуляр¬
ная масса изменяется от нескольких сотен до нескольких сотен тысяч дальтон, и они
имеют сложную химическую структуру, гидрофильный характер и кислые свойства.
Однако различие между двумя типами веществ не полностью понятно, потому что
гуминовые вещества всегда содержат негуминовые вещества, которые могут быть вы¬
делены такими химическими методами, как кислотный гидролиз.
Гуминовые вещества обычно разделяют на три основные группы в соответствии с их
растворимостью:
• Гуминовые кислоты, растворимые в разбавленных щелочных растворах и нерас¬
творимые в кислотах; они осаждаются при подкислении щелочных экстрактов из
почвы.
• Фульфокислоты, растворимые в кислых и щелочных средах; эти соединения оста¬
ются в растворе после осаждения гуминовых кислот при подкислении щелочных
экстрактов из почвы.
• Гумин — фракция гумуса, которая не экстрагируется кислотами или разбавленны¬
ми основаниями.
Некоторые гуминовые вещества получили отдельные названия, связанные с их рас¬
творимостью в различных растворителях. Наиболее известны свойства гиматомелано-
вых кислот как растворимой в этаноле фракции гуминовых кислот. Для фульвокислот
320 Часть 2. Органический анализ
иногда используют устаревшее разделение на креновые и апокреновые кислоты. Не¬
которые авторы (например, Chamayou и Legros, 1989) не рекомендуют использовать эти
термины. Можно различать бурые и серые гуминовые кислоты. Эти две группы были
впервые выделены Спрингером (Springer, 1938) на основании их способности к флоку¬
ляции или растворению в различных растворителях, включая различные концентрации
солей. На рис. 11.1 показаны некоторые свойства этих соединений.
Светло-желтые Желто-коричневые Темно-коричневые Темно-серые
< 2000 d
45%
48%
Увеличение степени полимеризации ►
Увеличение молекулярной массы ► > 300 000 d
Увеличение содержания углерода ► 62%
Уменьшение содержания кислорода ► 30%
1400 смоль Н7кг Уменьшение обменной кислотности 500 смоль Н*/кг
Рис. 11.1. Классификация и химические свойства групп гуминовых веществ (согласно Stevenson
и Elliott, 1989)
11.1.2. Роль гуминовых веществ в почвах и в окружающей среде
Гуминовые вещества содержат 60—70% углерода почвы, который представляет собой са¬
мый большой запас органического углерода на поверхности Земли. Гуминовые вещества
играют значительную роль как источник углерода для атмосферного С02 и как резерв
углерода, который может изменяться под воздействием различных внешних факторов
(Schnitzer, 1978).
321
Глава 11. Количественное определение гуминовых веществ
Стивенсон (Stevenson, 1982) привел девять свойств гумуса, оказывающих влияние на
почву:
• темный цвет, который облегчает поглощение солнечного излучения и, следова¬
тельно, нагревает почву;
• водоудерживающая способность: органическое вещество может содержать количе¬
ство воды, превышающее его собственную массу в 20 раз, и поэтому значительно
улучшает водные свойства некоторых (особенно песчаных) почв;
• способность соединяться с глинистыми минералами, склеивая частицы почвы
в структурные единицы, называемые агрегатами, что облегчает газовый обмен
и увеличивает проницаемость;
• хелатообразование с образованием стабильных комплексов со многими полива¬
лентными катионами — влияет на доступность питательных веществ растениям;
• очень малая растворимость в воде и способность образования связей с глинами
и некоторыми поливалентными катионами, что уменьшает потери органического
вещества из-за выщелачивания;
• буферные свойства, которые проявляются в слабокислой, нейтральной и щелоч¬
ной среде;
• емкость катионного обмена: 20—70% от ЕКО многих почв обусловлено наличием
органического вещества;
• минерализация, приводящая к выделению С02, NH+, NO~, РО*~, SO*- и представ¬
ляющая значительный источник питательных веществ для растений;
• сочетание с другими органическими молекулами, которое влияет на биологиче¬
скую активность, стабильность и биоразлагаемость пестицидов.
В данной главе приводится описание основных методик для выделения и количе¬
ственного определения гуминовых веществ почвы. Справочный раздел, приведенный
в конце главы, содержит обширный список методов, используемых для этих целей.
Методы выделения описаны для образцов цельных почв, подготовленных стандарт¬
ными методами. Однако часто предпочтительнее применять эти методы для выделенных
фракций после физического разделения органического вещества (см. гл. 9), в частности
для самых мелких фракций.
11.1.3. Методы экстракции
Органические вещества связаны с поливалентными катионами, гидроксидами и глини¬
стыми минералами с образованием органоминеральных комплексов. Стабильность этих
комплексов значительно меняется в зависимости от типа связей.
Органические вещества могут быть выделены методами экстракции, которые разры¬
вают (по крайней мере, некоторые) органо-минеральные связи. Брукерт (Bruckert, 1979)
различал три типа экстрагирующих растворов:
• Растворы солей, которые могут разрывать электростатические связи через простой
обмен ионов и способствуют растворению органических молекул, ионизируя кис¬
лотные и фенольные функциональные группы; Брукерт предложил использовать
раствор тетрабората натрия с pH 9,7; экстрагированные органоминеральные ве¬
щества относительно малой молекулярной массы были описаны как подвижные
или легкодоступные новообразованные комплексы (Duchaufour, 1977); они харак¬
теризуются относительно малым содержанием металлов; тетраборат не затрагивает
комплексы кальция.
• Комплексообразующие растворы, способные разрывать координационные связи:
самым известным является раствор пирофосфата натрия, обычно используемый
322
Часть 2. Органический анализ
при pH 9,8, он разрывает связи комплексов с металлическим центрами на глини¬
стых минералах; он может солюбилизировать комплексы с высоким содержанием
металлов (аморфные гидроксиды) и растворять гуматы кальция, образуя комплек¬
сы с кальцием, но, как и тетраборат, неэффективен в отношении аллофановых
комплексов; все известные экстрагированные комплексы устойчивы.
• Раствор гидроксида натрия с pH 12 является самым эффективным экстрагентом,
поскольку он способен разорвать большинство органо-минеральных связей и осо¬
бенно связи комплексов гуминовых кислот с аллофаном в андосолях.
Стандартные методы, представленные в разделе 11.2.1, описывают:
• простую экстракцию щелочным раствором после обработки кислотой или без нее;
• двойную экстракцию последовательно растворами пирофосфата и гидроксида на¬
трия (этот метод используется в лабораториях Исследовательского института IRD,
Франция).
В разделе 11.2 сравниваются эффективность и точность этих методов и описываются
основные методы очистки экстрагированных органических веществ.
Некоторые альтернативы этим методам экстракции, включая метод отделения гумина
центрифугированием в гранулу, представлены в разделе 11.3. Методы фракционирова¬
ния и характеризации гуминовых веществ описаны в гл. 12.
11.2. Основные методы анализа
11.2.1. Экстракция
Основные положения
Как указано в разделе 11.1.3, разбавленные растворы гидроксида натрия являются самы¬
ми эффективными экстрагентами для гумифицированного вещества. Однако исполь¬
зование этих экстрагирующих растворов был подвергнуто критике по трем основным
причинам (Bruckert, 1979):
• новообразование растворимых веществ из негумифицированного растительного
материала;
• разрушение гуминовых веществ при гидролизе, окислении или искусственной по¬
лимеризации;
• лизис микробных организмов; гидроксид натрия может разрушать бактерии и осво¬
бождать содержимое цитоплазмы, а стенки клеток затем образуют неэкстрагируе-
мый остаток.
Однако Шнитцер (Schnitzel 1982) не считал, что экстракция разбавленными щело¬
чами в атмосфере азота и при комнатной температуре существенно изменяет структу¬
ру и характеристики экстрагированного органического вещества. Левеск и Шнитцер
(Levesque и Schnitzer, 1966) показали, что 0,1 М раствор гидроксида натрия экстрагирует
больше органического вещества, чем концентрированные растворы. Они также показа¬
ли, что 0,5 М раствор гидроксида натрия экстрагирует органическое вещество с мень¬
шим содержанием золы.
Для максимизации экстракции гуминовых веществ и минимизации их разрушения
были выбраны «метод Шнитцера» (Schnitzer, 1982) и «метод МГО» (Международно¬
го гуминового общества)1. Одноступенчатая экстракция выполняется: (1) в атмосфере
1 1HSS - International Humic Substances Society, Univ. of California, Los Angeles, CA 90024;
Federal Center, mall stop 407, Box 25046, Denver, CO 80225.
Глава 11. Количественное определение гуминовых веществ
323
азота 0,1 М раствором гидроксида натрия, как описано в «методе МГО», или (2) тем
же реагентом или 0,5 М раствором гидроксида натрия или пирофосфата, как описано
в «методе Шнитцера» (Schnitzer, 1982). «Метод Дабена» (Dabin, 1976) разделяет два типа
экстрагированных соединений: органические вещества, экстрагируемые раствором пи¬
рофосфата при pH 9,8, и органические вещества, экстрагируемые затем 0,1 М раствором
гидроксида натрия. Негр с сотр. (Ndgre и др., 1976) наблюдали качественные различия
между двумя экстрактами в частности, в содержании аминокислот; Томанн (Thomann,
1963) обнаружил, что пирофосфат растворяет гуматы кальция за счет образования ком¬
плексов с катионами металла; увеличение pH влияет в основном на диспергирование
агрегатов, а pH 9,8 соответствует стабильной фазе на кривой экстракции гумуса в за¬
висимости от pH.
Другой подход включает предварительную обработку почвы кислотой; такая обра¬
ботка облегчает последующую экстракцию гумифицированного вещества, разрушая
карбонаты и растворяя гидроксиды железа и алюминия; однако количественный эф¬
фект обработки очевиден только для карбонатных почв. «Метод МГО» рекомендует
обязательную предварительную обработку 1 М раствором хлористоводородной кислоты.
В «методе Шнитцера» рекомендуется предварительная обработка 0,05 М раствором хло¬
ристоводородной или серной кислоты только в случае карбонатных почв. «Метод Дабе¬
на» рекомендует обязательную предварительную обработку 2 М фосфорной кислотой.
Эта кислота имеет два преимущества: (1) ее более высокая плотность (примерно 1,2)
более благоприятна для отделения легких органических фрагментов (см. гл. 9), и (2) она
не мешает определению углерода (мокрым озолением) и таким образом позволяет опре¬
делять количество органического вещества, экстрагированного кислотой (несвязанная
фульвокислота).
Оборудование
• Колбы для экстракции и центрифугирования из стекла, полипропилена или поли¬
винила (объем 200 мл и 300-500 мл) с навинчивающимися крышками для исполь¬
зования в качестве центрифужных пробирок, выдерживающие 10 000 g.
• Центрифуга (10 000 g), оборудованная ротором для центрифужных колб.
Реагенты
• Дегазированная вода без органических веществ. Пригодны большинство коммерче¬
ских марок воды. Сначала ее нужно проверить на отсутствие органического веще¬
ства (холостой опыт в условиях последующих испытаний). Для удаления органи¬
ческого вещества из воды либо (1) кипятят воду в присутствии 1% КМп04 и H2S04
в течение 2 ч и затем перегоняют, либо (2) используют деионизированную воду,
очищенную активированным углем (например, фильтром Миллипор), затем дега¬
зируют воду для удаления растворенного кислорода, чтобы исключить окисление
органического вещества во время экстракции (для этого воду кипятят или барбо-
тируют азотом в течение 10 мин).
• 0; 1Мраствор NaOH: растворяют 8 г гранулированного гидроксида натрия в двух¬
литровой мерной колбе в дегазированной воде без органики, доливают до 2 л, пе¬
ремешивают и хранят в тщательно закрытой бутыли.
• 0,5 Мраствор NaOH: растворяют 40 г гидроксида натрия в 2 л воды, как описано
выше.
• 10 Мраствор NaOH: растворяют 400 г гидроксида натрия в 1 л воды, как описано
выше.
324
Часть 2. Органический анализ
• О,1Мраствор NaAP207: растворяют 89,2 г Na4P207 • ЮН20 в дегазированной воде без
органики, доливают до 2 л и хранят в тщательно закрытой бутыли.
• 0,1 Мраствор NaAP207/Na0H: растворяют 89,2 г Na4P207 • ЮН20 и 8 г гранулиро¬
ванного гидроксида натрия в дегазированной воде без органики, доливают до 2 л
и хранят в тщательно закрытой бутыли.
• 2 Мраствор НСк растворяют 166,7 мл концентрированной НС1 (rf = 1,19) в дегази¬
рованной воде без органики, доливают до 1 л.
• 6 Мраствор НСк растворяют 500 мл концентрированной хлористоводородной кис¬
лоты в 1 л дегазированной воды без органики.
• 1Мраствор НСк растворяют 166,7 мл НС1 в 2 л дегазированной воды без органики.
• 0,5 Мраствор НСк растворяют 83,3 мл НС1 в 2 л дегазированной воды без органики.
• 0,05н. раствор H2SOA: растворяют 27,8 мл концентрированной H2S04(d= 1,81) вде-
газированной воде без органики, охлаждают и доливают до 2 л.
• 2 Мраствор НъРОА, растворяют 136 мл концентрированной фосфорной кислоты
(d = 1,71) в дегазированной воде без органики, и доливают до 1 л.
Методики
Метод Шнитцера (1982)
Если почва содержит карбонаты (реакция с разбавленной хлористоводородной кис¬
лотой), ее оставляют в контакте с 0,05 н. раствором хлористоводородной или серной
кислоты при комнатной температуре до окончания выделения газа. Смывают избыток
кислоты водой без органики и сушат почву на пластине при комнатной температуре.
Отвешивают 10 г воздушно-сухой почвы в полипропиленовой колбе на 200 мл. Добав¬
ляют 100 мл выбранного экстрагирующего раствора (0,1 или 0,5 М NaOH, 0,1 М Н3Р04
или смесь 0,1 М Na4P207/Na0H). Вытесняют воздух из колбы потоком азота. Тщательно
закрывают и перемешивают при комнатной температуре в течение 24 ч. Отделяют тем¬
ную надосадочную жидкость от твердой фазы центрифугированием (предпочтительно
в течение 10 мин при 10 000 g), суспендируют остаток в 50 мл дегазированной воды без ор¬
ганики, снова центрифугируют и добавляют промывную воду к предыдущему раствору.
Подкисляют щелочной экстракт до pH 2, добавляя 2 М раствор НС1. Оставляют на
24 ч при комнатной температуре, затем отделяют водорастворимое вещество (фульво-
кислоты) от коагулировавшего вещества (гуминовые кислоты) центрифугированием.
Обе фракции можно высушить лиофизацией или упариванием на роторном испарителе
при 40 °С.
Метод МГО
Смешивают 20 г воздушно-сухой почвы с 1 М раствором хлористоводородной кислоты.
Доводят pH раствора до 1-2 (добавлением 15-20 мл 10 М раствора гидроксида натрия)
так, чтобы конечный объем раствора был равен 200 мл (отношение жидкость : почва =
10 мл : 1 г). Перемешивают в течение 1 ч и отделяют надосадочную жидкость центрифу¬
гированием.
Нейтрализуют отцентрифугированную гранулу до pH 7 добавлением 1 М раствора
гидроксида натрия и добавляют 0,1 М раствор NaOH в атмосфере азота до достижения
отношения раствор : почва = 10 : 1.
Перемешивают смесь не менее 4 ч в атмосфере азота. Оставляют на ночь и центри¬
фугируют.
325
Глава 11. Количественное определение гуминовых веществ
Подкисляют отцентрифугированную жидкость до pH 1 добавлением 6 М раствора
хлористоводородной кислоты при перемешивании. Оставляют стоять на 12-16 ч и цен¬
трифугируют для отделения фульвокислот в растворе от коагулированных гуминовых
кислот.
Метод Дабена (1976)
Помещают 40 г воздушно-сухой почвы, измельченной и просеянной сквозь сито 0,5 мм,
в центрифужную пробирку на 300-500 мл. Добавляют 200 мл 2 М раствора Н3Р04, пере¬
мешивают в течение 30 мин на возвратно-поступательном шейкере и центрифугируют
при 1500 g в течение 5 мин. Отфильтровывают надосадочную жидкость через плоский
фильтр в стеклянную колбу на 1 л. Повторяют экстракцию дважды или трижды в той же
центрифужной пробирке, отфильтровывая через тот же фильтр и собирая кислые экс¬
тракты в ту же колбу. Фильтр содержит легкое органическое вещество (ЛОМ) из негу-
мифицированных растительных и животных остатков (см. гл. 9); кислотный раствор со¬
держит небольшую фракцию органического вещества, названную Дабеном (Dabin, 1976)
свободными фульвокислотами (СФК). Промывают отцентрифугированный осадок
дважды или трижды 200 мл воды без органики в той же пробирке, перемешивая в тече¬
ние 15 мин; центрифугируют и фильтруют промывную воду на использованном фильтре
для сбора легкого вещества, оставшегося в промывной воде, а фильтрат отбрасывают.
Добавляют 200 мл 0,1 М раствора Na4P207 с pH 9,8. Перемешивают в течение 4 ч на
возвратно-поступательном шейкере или оставляют на ночь при периодическом переме¬
шивании. Отделяют надосадочную жидкость центрифугированием при 3000 g в течение
30 мин и переносят ее после фильтрования в мерную колбу на 1 л. Проводят вторую
экстракцию в тех же условиях и объединяют экстракты. Если второй экстракт темного
цвета, выполняют третью экстракцию.
Повторяют аналогичную серию экстракций с отцентрифугированным осадком, ис¬
пользуя 0,1 М раствор NaOH вместо раствора пирофосфата с pH 9,8.
Гуминовые кислоты из пирофосфатных и щелочных экстрактов отделяют от фульво¬
кислот подкислением до pH 1, добавляя 2 М раствор НС1, как описано в параграфе «Ме¬
тод Шнитцера (1982)». В конечном счете, получают следующие фракции: ЛОВ, СФК,
пирофосфатные фульвокислоты (ПФК), пирофосфатные гуминовые кислоты (ПГК),
щелочные фульвокислоты (ЩФК), щелочные гуминовые кислоты (ЩГК) и остаток по¬
сле экстракции, или гумин.
Примечание
В некоторых почвах пирофосфатные или щелочные экстракты могут содержать большие
количества мелких частиц глинистых минералов (менее 0,2 мкм). Эти частицы можно
отделить флокуляцией при добавлении небольшого количества сульфата калия, однако
существует риск одновременной флокуляции некоторых серых гуминовых кислот (см.
раздел 11.2.4 и гл. 12). После центрифугирования флокулированный осадок анализиру¬
ют на содержание углерода индивидуально или после объединения с предыдущим гуми-
новым осадком.
11.2.2. Количественный анализ экстрактов
Основные положения
Количественное определение углерода проводят, измеряя его содержание fe каждом экс¬
тракте (см. раздел 11.2.1). Методы определения содержания углерода в цельной почве
326
Часть 2. Органический анализ
могут применяться и для гуминового осадка (см. гл. 10). Для ЛОВ предпочтителен метод
озоления. Экстракты можно также анализировать озолением остатка после выпарива¬
ния аликвоты досуха. Азот можно определять одновременно с углеродом, водородом и,
возможно, серой и кислородом на CHN-анализаторе. Однако мокрые методы, такие как
окисление бихроматом (см. ниже), часто предпочтительнее для анализа экстрактов. Су¬
ществует также метод с использованием прибора для определения растворенного угле¬
рода. Многие из таких приборов основаны на определении диоксида углерода (обычно
по поглощению в инфракрасной области), полученного при окислении раствора мощ¬
ным окислителем. Приборы для определения растворенного углерода довольно дорогие
и используются для точных исследований объектов окружающей среды; они требуют
точного выполнения инструкции производителя.
В кислой среде бихромат окисляет углерод органического вещества до С02 в соответ¬
ствии с реакцией окисления — восстановления:
Сг2072" + 6е~ + 14Н+ -► 2Сг3+ + 7Н20
С + 2Н20->С02 + 4е+4Н+
В отсутствие автоматизированного оборудования количество выделенного С02 мож¬
но измерить непосредственно. В традиционном окислительно-восстановительном ме¬
тоде используют избыток бихромата, который затем определяют обратным титрованием
раствором двухвалентного железа:
Fe2+ -> Fe3+ + е~
Один моль двухвалентного железа соответствует 1/6 моля KJCtJDj , т. е. 1/4 атома С
или 3 г углерода.
Оборудование
• Лабораторные стаканы из пирекса на 50 и 100 мл.
• Прецизионная бюретка (на 25 или 50 мл).
• Если необходимо, прибор для определения углерода методом сухого озоления, хотя
методы мокрого анализа предпочтительнее.
Реагенты
• 0,1 Мраствор NaAP207 и 0,1 М раствор NaOH (см. параграф «Реагенты» раздела
11.2.1).
• Концентрированная серная кислота ((/=1,81),
• 2 н. раствор серной кислоты: растворяют 56 мл концентрированной серной кислоты
((/=1,81) в 1 л воды без органических веществ.
• 0,1 н. раствор серной кислоты: растворяют 2,8 мл концентрированной H2S04
((/=1,81) в 1 л воды без органических веществ.
• 2%-ный раствор бихромата калия\ растворяют 20 г l^Cr^ в примерно 400 мл воды
без органических веществ, медленно добавляют 500 мл концентрированной серной
кислоты, перемешивают, охлаждают и доводят объем до 1 л добавлением воды без
органических веществ.
• 0,5 н. раствор К2Сг207\ постепенно растворяют 24,52 г бихромата калия в воде
без органических веществ, и доводят объем до 1 л (раствор для титрования соли
Мора).
Глава 11. Количественное определение гуминовых веществ
327
• 0,2 Мраствор соли Мора: растворяют 78,4 г соли Мора (FeS04 • (NH4)2SO; • 6Н20)
в 500 мл воды, добавляют 20 мл концентрированной серной кислоты, доливают
до 1л.
• Фторид натрия в форме порошка.
• Раствор сульфата дифениламина: растворяют 0,5 г порошка дифениламина в 100 мл
концентрированной H2S04, наливают в 20 мл воды, перемешивают и хранят в тем¬
ном флаконе.
Методика
Анализируемые пробы
Общее органическое вещество щелочных и пирофосфатных экстрактов. В низкий стакан
на 100-200 мл помещают точно измеренный объем экстракта, соответствующий 5-8 мг
С. Объем аликвоты рассчитывают, исходя из анализа общего С в пробе (см. гл. 10). Со¬
держание углерода в щелочном экстракте составляет около 40% от общего С, в пиро¬
фосфатном экстракте — около 25%, в щелочном экстракте после экстракции пирофос¬
фатом — около 15% от общего С. Перед определением пробу высушивают в сушильном
шкафу при 70 °С.
Гуминовые кислоты. Берут более 50% экстракта пробы для определения общего орга¬
нического вещества. Осаждают гуминовые кислоты при pH - 1 добавлением 1 М H2S04
(примерно 4-5 мл кислоты на 10 мл общего экстракта, 3 мл на 10 мл пирофосфатного
экстракта, 1,5 мл на 10 мл щелочного экстракта); оставляют для флокуляции не менее
чем на 4 ч, и центрифугируют при 3500 g не менее 5 мин; отделяют надосадочную жид¬
кость (фульвокислоты) и промывают 0,1 н. раствором H2S04. Растворяют отиентрифу-
гированный осадок в 0,1 М растворе NaOH, переносят в стакан и высушивают при 70 °С
перед анализом.
Фосфорнокислый экстракт. Точно отбирают 100 мл аликвоту экстракта, концентриру¬
ют в сушильном шкафу до объема примерно 10 мл (Н3Р04 не упаривается досуха) и ти¬
труют полученный концентрированный раствор.
Фульвокислоты. Фульвокислоты можно определять аналогично общему органиче¬
скому веществу после удаления гуминовых кислот, но обычно их содержание рассчи¬
тывают по разнице между содержанием общего органического вещества и гуминовых
кислот.
Окислительно-восстановительное титрование
После высушивания навесок в стаканах добавляют 10 мл 2%-ного раствора бихромата
калия в серной кислоте. Одновременно выполняют холостой опыт с 10 мл того же рас¬
твора бихромата в стакане. Накрывают стакан крышкой и очень осторожно кипятят на
плитке, отрегулированной на 215-220 °С; кипятят в течение 5 мин, избегая перегрева
или слишком бурного выпаривания.
Охлаждают, споласкивают крышку стакана и добавляют в стакан 100 мл воды без ор¬
ганических веществ, 1,5 г NaF (или 2,5 мл Н3Р04) и три капли раствора дифениламина.
Титруют раствором соли Мора из бюретки до тех пор, пока фиолетовый цвет раствора
не изменится на светло-зеленый. Уи V — объемы раствора двухвалентного железа, не¬
обходимые для титрования холостого раствора и анализируемой пробы, соответственно.
Если Т — молярная концентрация раствора соли Мора; количество Fe2+, экви¬
валентное количеству окислителя, использованному для титрования углерода про¬
бы — Д К— V) ммоль Fe2+, что, согласно уравнениям окисления - восстановления
328
Часть 2. Органический анализ
(см. параграф «Основные положения» раздела 11.2.2), соответствует 3 T(V— V) мг углерода
в стакане, то содержание углерода в образце почвы, выраженное в мг С/г почвы, составит
С = 3 ЦУ-Г)УДАт)9 (11.1)
где Vt — общий объем экстракта гуминовых веществ, мл; А — аликвота гуминового экс¬
тракта, использованная для титрования, мл; mt — масса пробы почвы, использованной
для экстракции, т\Т— величина, определяемая титрованием.
Титрование раствора соли Мора
Помещают в стакан на 250 мл точно 10 мл 0,5 н. раствора YLfi,xfiv 100 мл воды без орга¬
нических веществ и 15 мл концентрированной H2S04.
Охлаждают и добавляют 3,75 г NaF и три капли индикатора дифениламина.
Титруют раствором соли Мора из бюретки. Если Vf— объем раствора соли Мора, мл,
молярная концентрация Тэтого раствора будет равна Т= 5/Vf Тогда содержание углеро¬
да в уравнении (ИЛ) можно выразить в мг С/г почвы:
C=\5(V-\r)VJ{AmtV). (11.1)
Примечания
10 мл 2%-ного раствора бихромата соответствуют 20,4 мл 0,2 М раствора соли Мора.
Объем раствора соли Мора, использованный для титрования пробы, должен быть от 7 до
15 мл; если он меньше, повторяют анализ с пробой большей массы, если больше — берут
пробу меньшей массы.
Количество углерода в фосфорнокислом растворе обычно мало. В этом случае окис¬
ление проводят, используя только 5 мл 2%-ного раствора бихромата и добавив пять ка¬
пель концентрированной H2S04 перед кипячением.
Пирексовые стаканы подвергаются воздействию щелочных растворов и NaF в кис¬
лой среде. Их надо споласкивать сразу после титрования и использовать только для этой
цели.
11.2.3. Точность и сопоставимость методов экстракции
Межлабораторные испытания
Межлабораторные сравнительные испытания, проведенные GEMOS1 (Группой по ис¬
следованию органического вещества почвы Французской ассоциации почвоведов),
включали сравнение результатов определения количества органических веществ, экс¬
трагированных из семи почв различных регионов Франции (Dabin и др., 1983):
1. Илистая почва с плит Буаньвиля.
2. Перегнойно-карбонатная почва из Понтарлье.
3. Горизонт А1 подзола из леса Вилле-Котре.
4. Горизонт Bh того же подзола (п. 3).
5. Феррисиаллитная почва из окрестностей Монпелье.
6. Пгеевая почва с гидромулевым гумусом из Бонво.
7. Рендзина на меловых отложениях из Шалона-на-Марне.
1 GEMOS — Groupe d’Etude des Matieres Organiques des Sols, sub-group of the Association Francaise
d’Etude des Sols (AFES), INRA, 78850 Thiverval-Grignon, France.
Глава 11. Количественное определение гуминовых веществ
329
Каждая почва анализировалась в четырех французских лабораториях:
• лаборатория Центра международного сотрудничества и агрономических исследо¬
ваний в целях развития (C1RADО, г. Монпелье;
• лаборатория почвоведения университета г. Пуатье;
• лаборатория института для целей развития (IRD1) г. Бонди;
• лаборатория почвоведения университета г. Безансон.
В качестве стандартного метода для сравнения результатов был выбран метод, опи¬
санный в параграфе «Метод МГО». Кроме того, лаборатория в Бонди использовала «ме¬
тод Дабена» на тех же пробах.
Результаты экстракции методом МГО
Результаты межлабораторных испытаний приведены на рис. 11.2. Содержание углеро¬
да в г/100 г сухой почвы, полученное для суммы трех фракций (кислотный экстракт +
+ щелочной экстракт + неэкстрагируемый остаток) по сравнению с содержанием обще¬
го углерода, полученного для цельной почвы, показано на рис. 11.2, а. Тот факт, что ре¬
зультаты расположены близко к прямой, проведенной из центра координат под углом
45°, указывает на отсутствие систематического сдвига между двумя методами.
На рис. 11.2, Ъ показано количество углерода, экстрагированного раствором хлори¬
стоводородной кислоты, по сравнению с общим экстрагированным количеством. Оно
оказалось низким, не более 5% общего углерода. Два исключения, для которых полу¬
ченное значение равно 10%, вероятно объясняются присутствием фрагментов легкого
негумифицированного органического вещества. Полученные величины отличались зна¬
чительным разбросом и нё коррелировали с содержанием общего углерода.
На рис. 11.2, с показано содержание углерода в щелочном экстракте по сравнению
с содержанием общего углерода. Можно сделать два замечания:
1. В 6 из 7 случаев щелочной раствор экстрагировал 20-40% общего углерода. Результа¬
ты показывают меньший разброс, чем в случае кислотной экстракции. Распределе¬
ние результатов указывает на существование предела экстракции, равного пример¬
но 40% общего углерода, для шести почв (прямая). Значения, расположенные выше
этого предела, вероятно, связаны с ошибками из-за недостаточной экстракции.
2. Получено аномально высокое значение для почвы 4. Это может быть связано с тем
фактом, что проба почвы была отобрана из глубокого органогенного горизонта Bh
подзола; органическое вещество, выщелоченное до этой глубины, было гораздо
лучше растворимо в щелочном растворе, чем в других шести почвах, и в этом слу¬
чае реагент экстрагировал более ъ/а углерода пробы.
На рис. 11.2, d показано количество углерода в форме гуминовых кислот по сравнению
с углеродом в общем щелочном экстракте. Независимо от типа почвы, углерод гумино¬
вых кислот составлял примерно 2/з углерода щелочных экстрактов, т. е. примерно 27%
общего углерода. Разброс результатов относительно этого значения невелик.
Точность метода МГО
В табл. 11.1 показаны результаты дисперсионного анализа данных, полученных методом
МГО: оценена значимость различий между величинами, полученными для каждой по¬
чвы, по сравнению с экспериментальной погрешностью.
1 CIRAD — Centre International de Recherche Agronomique pour le Developpement, Avenue d’Agropolis,
BP 5035, 34032 Montpellier Cedex, France.
2 IRD — Institute of Research for the Development (ex-Orstom), 32 Avenue Varagnat, 93143 Bondy,
France.
330
Часть 2. Органический анализ
Рис. 11.2. Результаты межлабораторного сравнения использованого метода экстракции,
описанного в параграфе «Метод МГО» (неопубликованные данные). Результаты, полученные
в 4 лабораториях для 7 почв, описанных в тексте: + — почва 1; Д — почва 2, А — почва 3, 0 —
почва 4, х - почва 5, ♦ - почва 6, о - почва 7
Таблица 11.1. Точность измерений в межлабораторных сравнительных испытаниях на 7 почвах
с использованием метода МГО (/’-тест — критерий значимости полученных
величин относительно остаточной дисперсии s* для 4 лабораторий)
Определяемый параметр
Д6.21)
s}
5
осо,%
С кислотного экстракта
4,2 (-)
0,026
0,16
83
С щелочного экстракта
107***
0,141
0,38
17
С гуминового остатка
37***
0,317
0,56
22
С суммы фракций
83***
0,410
0,64
13
Общий С почвы
100***
0,403
0,63
13
С гуминовых кислот
67***
0,131
0,36
22
С фульвокислот
16***
0,036
0,19
35
*** _ Существенная разница между данными лабораторий при уровне значимости < 1%.
- — Разница несущественна.
s и ОСО — ожидаемое абсолютное (г С/100 г сухой почвы) и относительное (%) стан¬
дартное отклонение среднего результата измерения в лаборатории.
331
Глава 11. Количественное определение гуминовых веществ
F-тест — отношение дисперсии между почвами (7 почв, т. е. 6 степеней свободы)
к внутрилабораторной дисперсии (27 измерений, т. е. 20 степеней свободы), представ¬
ленной величиной sr2. Суммарная оценка стандартной погрешности, связанной с из¬
мерениями в данной лаборатории для неопределенной почвы, выражена в абсолютных
значениях — обозначена как s (г С/100 г сухой почвы) и в относительных величинах —
ОСО (относительное стандартное отклонение). Эти величины, представляющие межла¬
бораторную воспроизводимость, являются верхними границами погрешности. Повто¬
ряемость внутри одной лаборатории с квалифицированным персоналом будет лучше.
Содержание углерода измерялось в хлористоводороднокислом экстракте, в щелоч¬
ном экстракте, в гуминовом остатке, в сумме этих трех фракций, и наконец, в гумино¬
вых кислотах и в фульвокислотах.
Данные табл. 11.1 показывают:
• Результаты экстракции хлористоводородной кислотой невозможно контролиро¬
вать, погрешности определения превышают различия между почвами; это под¬
тверждает распределение результатов, приведенное на рис. 11.2, Ь.
• Во всех других случаях различия между почвами были существенны по сравнению
с остаточной ошибкой, представляющей межлабораторную вариабельность.
• В щелочных экстрактах точность измерений гуминовых кислот выше, чем фульво-
кислот, что может быть связано с ббльшим содержанием гуминовых кислот.
• Точность измерения содержания общего углерода почвы, полученная для суммы
первых трех фракций, имеет одинаковый порядок величины с точностью прямого
определения содержания углерода в цельной почве; более того, полученная вели¬
чина согласуется с правилом распространения ошибок (Pansu и др., 2001); действи¬
тельно, для Ссуммы, полученной как:
С = С +с +с
суммы кислотного экстракта щелочного экстракта гумина
В случае нормального распределения:
S — (S2 *4“ 5^ И- )^2,
Ссуммы ' С кислотного экстракта Сшелопного экстракта Сгумина'
Тогда
^Ссуммы = (°>026 + 0,141 + 0,317)1/2 = 0,69.
Эта величина близка значениям 0,64 и 0,63, полученным для углерода (суммы фрак¬
ций и общего углерода, соответственно).
Сравнение методов МГО и Дабена
Данные на рис. 11.3 показывают, что значения содержания общего углерода (кислот¬
ный экстракт + щелочной экстракт -Г гуминовый остаток), полученные двумя методами,
очень близки. Однако более подробное сравнение данных, приведенных на рис. 11.4,
выявляет сходства и различия между методами.
• Экстракция фосфорной кислотой, описанная в методе Дабена, солюбилизирует
от 1,2 до 4 раз больше (в среднем больше в 2 раза) органического вещества, чем
экстракция хлористоводородной кислотой, описанная в методе МГО (рис. 11.4, а);
самые близкие результаты получены двумя методами для глубокого горизонта Bh
подзола.
332
Часть 2, Органический анализ
Рис. 11.3. Сравнение содержания углерода, полученного двумя методами экстракции: средние
значения результатов четырех лабораторий для семи типов почв, описанных в тексте: + - почва 1;
А - почва 2, А - почва 3,0- почва 4, х - почва 5, ♦ - почва 6, о - почва 7
Метод МГО Метод МГО
Рис. 11.4. Сравнение фракций, экстрагированных двумя методами: средние значения
результатов четырех лабораторий для семи типов почв, описанных в тексте: + - почва 1; А - почва
2, А - почва 3,0 - почва 4, х - почва 5, ♦ - почва 6, о - почва 7
Глава 11. Количественное определение гуминовых веществ
333
• Щелочная экстракция дает сравнимые результаты для двух методов (рис. 11.4), если
учитывать сумму пирофосфатного и щелочного экстрактов, описанных в методе
Дабена; сравнимые результаты также получены для углерода гумина, хотя и слегка
заниженные в методе Дабена (рис. 11.4, d).
• С другой стороны, отчетливая разница между двумя методами наблюдается для
качества экстрагированного органического вещества, поскольку «метод Дабена»
определяет значительно меньшее количество гуминовых кислот; на рис. 11.4, с со¬
отношение между количествами С, определенного по методу Дабена, и С по мето¬
ду МГО, составляет примерно 2:3, а поведение пробы 4 (из глубокого горизонта Bh
подзола) несколько отличается; различие может быть связано с влиянием экстра¬
гирующего раствора на экстрагируемые молекулы, т. е. полимеризацией в «методе
МГО» и разрывом макромолекул в «методе Дабена» (см. примечание во введении
к данной главе).
11.2.4. Методы очистки гуминовых веществ
Введение
Существует много методов очистки гуминовых веществ, и трудно выбрать лучший ме¬
тод. Согласно Шнитцеру (Schnitzer, 1982), главной целью очистки является минимиза¬
ция количества золы, а вторая задача — отделить от гуминовых веществ органические
молекулы меньшей молекулярной массы. Однако этого может оказаться недостаточно,
потому что природа связей между гуминовыми, негуминовыми и неорганическими ве¬
ществами в экстрактах очень сложна, и многие методы очистки влияют на структуру
выделенных конечных соединений.
Негр с сотр. (Negre и др., 1976) рекомендовали использовать диализ (диализные меш¬
ки Вискинса с размером пор 24 А) для очистки гуминовых веществ, экстрагированных
раствором пирофосфата. Эти авторы, как и другие, отметили превращение фульвокис-
лот в гуминовые полимеры большей молекулярной массы. Похоже, что «во время диа¬
лиза, который соцровождается последовательным возвращением реакции среды к ней¬
тральным значения^, молекулы, которые были деполимеризованы в процессе щелочной
экстракции, могут полимеризоваться вновь простым восстановлением связей СО—NH,
аналогичным пептидным связям, ведущим к образованию нуклеиновых кислот» (Negre
идр., 1976).
Отедльные простые методы очистки позволяют удалять некоторые минералы из экс¬
тракта простой коагуляцией при добавлении небольшого количества сульфата натрия
и центрифугировании (Kumada и др., 1967) или ультрацентрифугировании (Jacquin и др.,
1970).
Гуминовые кислоты также могут быть очищены растворением в щелочной среде и по¬
следующим осаждением в кислой среде (Lowe, 1980), или повторным циклом экстрак¬
ции — фракционирования экстрактов, предварительно подвергнутых сублимационной
сушке (Schnitzer, 1982), или длительным замораживанием растворов гуминовых кислот,
которое может разделить различные фазы (Bachelier, 1983).
Наиболее эффективным методом очистки, хотя пригодным только для гуминовых
кислот, является химическая реакция минералов с разбавленным раствором смеси кис¬
лот HC1-HF для уменьшения количества золы. Шнитцер (Schnitzer, 1982) отмечал, что
обработка смесью HC1-HF может снизить количество золы до менее 1%. Жакен с сотр.
(Jacquin и др., 1970) получили менее 3% золы в трех экстрактах, очищенных смесью
HC1-HF. Для четырех методов очистки, исследованных этими авторами, ИК-спектры
334
Часть 2. Органический анализ
поглощения очищенных гуминовых веществ показали, что только обработка смесью
HC1-HF почти полностью удаляет интенсивные полосы поглощения филлосиликатов
при 470, 520 и 1030 см-1 (рис. 11.5). Однако, согласно данным этих авторов, обработка
фтористоводородной кислотой приводит к изменению химической структуры молекул,
в частности, к значительному снижению карбоксильной кислотности.
Фульвокислоты могут быть очищены в кислой среде адсорбцией на стандартных не¬
ионных полиакриловых смолах типа амберлита XAD-7 (Aiken и др., 1979). После про¬
мывания смолы слабокислым раствором для удаления неорганических солей, более 98%
фульвокислот могут быть выделены элюированием при pH 6,5 (Gregor и Powell, 1986).
Более простой метод включает удаление катионов металлов повторным обменом на ка¬
тионообменной смоле в Н+-форме (Schnitzer, 1982).
Рис. 11.5. Исследование влияния трех методов очистки на гуминовые кислоты, экстрагирован¬
ные из глубокого горизонта Bh подзола методом абсорбционной ИК-спектрометрии (Jacquin
и др., 1970): 1 — неочищенные гуминовые кислоты; 2 — гуминовые кислоты, очищенные на ОН-
и Н+ смолах; 3 — гуминовые кислоты, очищенные ультрацентрифугированием; 4 — гуминовые
кислоты, очищенные смесью HCI-HF.
335
Глава 11. Количественное определение гуминовых веществ
Оборудование
• Тефлоновые или полипропиленовые пробирки на 150 мл.
• Стеклянные колонки для хроматографии диаметром 1 или 2 см с тефлоновым за¬
порным краном.
• Лиофизатор и морозильная камера до —18 °С (необязательно).
Материалы и реагенты
• Диализные мешки с размером пор 24 А.
• Неионная полиакриловая смола (Amberlite XAD-7 или аналогичная).
• Катионообменная смола Amberlite IR120 или Dowex 50 в Н+-форме.
• Смесь НС1—НР. разбавляют 5 мл концентрированной НС1 и 5 мл 52%-ной фтори¬
стоводородной кислоты в 990 мл воды без органических веществ.
• Экстрагирующие растворы, см. раздел 11.2.1.
Методики
Здесь описаны только методы очистки фульвокислот на катион-обменных смолах или
очистки гуминовых кислот смесью HC1-HF. Однако, как указано во введении, важно
очень тщательно выбирать метод очистки с учетом последующих операций с очищен¬
ными веществами. О других методах очистки — ссылки в последнем абзаце «Введения»
в разделе 11.2.4.
Перемешивают смесь 1 г гуминовых кислот и 100 мл раствора HC1-HF в закрытой
полипропиленовой пробирке в течение 24 ч при комнатной температуре. Фильтруют
и снова суспендируют фильтрат в 100 мл раствора HC1-HF. Повторяют эту обработку
три или четыре раза, тщательно споласкивают остаток водой без органических веществ,
и затем высушивают на воздухе или сублимируют.
Очищают раствор фульвокислот трижды или четырежды на катионообменной смоле
в Н+-форме и затем высушивают сублимированием.
11.3. Другие альтернативные и дополнительные методы
11.3.1. Альтернативные методы экстракции
Хотя вышеописанные методы с использованием разбавленного гидроксида и пирофос¬
фата натрия используются наиболее широко, были предложены и другие методы экс¬
тракции. Например, Кумада с сотр. (Kumada и др., 1967) разработали метод, который
используется в университете Нагойя (Япония); Лоу (Lowe, 1980) использовал еще один
метод в университете Британской Колумбии (Канада); Лоу и Кумада (Lowe и Kumada,
1984) исследовали два метода в сравнительных испытаниях.
Грегор и Пауэлл (Gregor к Powell, 1986) разработали метод экстракции фульвокислот
пирофосфатом в кислой среде; этот метод позволяет избежать двух возможных проблем:
окисления фенольных соединений в щелочной среде и окисления ионами Fe3+ при под¬
кислении для осаждения гуминовых кислот.
Следует также упомянуть некоторые работы по сравнению различных реагентов.
Например, Томанн (Thomann, 1963) сравнивал 3%-ный оксалат аммония, 1%-ный ги¬
дроксид натрия, 1%-ный фторид натрия и пирофосфат натрия; Жакен с сотр. (Jacquin
и др., 1970) сравнивали гидроксид натрия, пирофосфат натрия и ион-обменные смолы;
Хейз с сотр. (Hayes и др., 1975) сравнивали растворы солей, органические хелатообразо-
ватели, биполярные апротонные растворители, пиридин, этилендиамин и раствор ги¬
336
Часть 2. Органический анализ
дроксида натрия. Авторы пришли к выводу, что гидроксид натрия является лучшим из
исследованных реагентов для выделения представительных экстрактов широкого ряда
гуминовых веществ.
11.3.2. Фракционирование гуминового остатка
Основные положения
С учетом того, что от 40 до 80% общего углерода не извлекается щелочными растворите¬
лями, Перро с сотр. (Perraud, 1971; Perraud и др., 1971) предложили метод для фракцио¬
нирования неэкстрагируемого гуминового остатка. Они последовательно выполнили
следующие операции:
• Щелочная экстракция после двукратной обработки горячей H2S04: этот экстракт
назвали «гумином, связанным с железом» (Perraud и др., 1971); возможно, обработ¬
ка серной кислотой также высвобождает сахара в ходе разложения неэкстрагируе-
мых полисахаридов, таких как целлюлоза (см. гл. 13).
• Щелочная экстракция после шестикратной обработки горячей смесью HF-HC1,
если органическое вещество связано с глинистыми минералами.
Однако эти авторы показали, что очень большая фракция еще оставалась неэкстраги-
руемой (37-52% общего углерода в описанном случае), несмотря на полное разложение
глинистых минералов, подтвержденное рентгеновскими исследованиями. От 8 до 23%
этой неэкстрагируемой фракции, солюбилизированной в СН3СОВг, возможно соответ¬
ствовало свежему или слабо преобразованному органическому веществу, которое ранее
не было захвачено минеральной породой. Фракция, которая оставалась нерастворимой
в СН3СОВг, возможно, соответствовала (1) органическому веществу, очень похожему
на лигнин, но преобразованному и ставшему нерастворимым в ацетилбромиде, или (2)
сильно полимеризованным соединениям, в которых восстановление функциональных
групп могло сделать их нерастворимыми в щелочных реагентах.
Методика фракционирования гуминового остатка, разработанная на основе мето¬
да Перро с сотр. (Perraud и др., 1971) и использованная в лабораториях института IRD
(Бонди, Франция), приведена ниже.
Методика
• Отвешивают 10 г остатка после экстракции (раздел 11.2.1), который был высушен,
измельчен и просеян сквозь сито 0,2 мм.
• Добавляют 50 мл 1 н. серной кислоты и нагревают до 70 °С в течение 3 ч.
• Центрифугируют при 3000 g в течение 15 мин.
• Дважды промывают горячей водой и собирают отцентрифугированный осадок.
• Экстрагируют отцентрифугированный осадок 50 мл 0,1 М раствора гидроксида на¬
трия при перемешивании в течение 4 ч и оставляют на ночь.
• Центрифугируют при 3000 g в течение 15 мин. Надосадочная жидкость содержит
гумин, связанный с гидроксидами (ГГ); ее сохраняют для определения фульвокислот
и гуминовых кислот, как описано в разделе 11.2.2; в зависимости от типа почвы
экстракт можно очистить флокуляцией глин солью (например, сульфатом натрия);
добавляют флокулят к отцентрифугированному осадку.
• Отбирают порцию отцентрифугированного осадка для промежуточного определе¬
ния углерода и возможно азота; этот анализ позволяет идентифицировать фрак¬
Глава 11. Количественное определение гуминовых веществ
337
цию, которая растворяется в кислоте (например, полисахариды), рассчитав по раз¬
нице.
• В другой (большей) порции отцентрифугированного осадка разрушают глинистые
частицы:
а) последовательной четырехкратной обработкой 50 мл 1М раствора смеси НС1—HF
при 70 °С в течение 3 ч и центрифугированием при 3000 g в течение 15 мин;
б) обработкой 50 мл 1 М раствора HF при 70 °С в течение 3 ч, центрифугированием
при 3000 g в течение 15 мин, промыванием отцентрифугированного осадка го¬
рячей водой и повторным центрифугированием.
• Экстрагируют отцентрифугированный осадок 50 мл 0,1 М раствором NaOH, пере¬
мешивая в течение 4 ч, и оставляют на ночь.
• Центрифугируют при 3000 g в течение 15 мин. Отделяют гуминовые вещества: гу¬
мин, связанный с силикатами (ГС), от раствора; в этих соединениях определяют
углерод гуминовых и фульвокислот (см. раздел 11.2.2).
• Унаследованный гумин (УГ), который состоит из небольших органических фрагмен¬
тов, напоминающих древесный уголь, может быть выделен из отцентрифугирован¬
ного осадка:
а) добавляют 50 мл Н3Р04 (d =1,4), подвергают ультразвуковой обработке в течение
10 мин и перемешивают в течение 30 мин;
б) центрифугируют при 1500 g в течение 10 мин, затем отфильтровывают надоса-
дочную жидкость через небольшую воронку с тампоном из стеклянной ваты.
Углеродные фрагмент, оставшиеся на этом тампоне, представляют собой нераство¬
римый УГ. Поскольку жидкости от предыдущих кислотных обработок также могут со¬
держать взвешенные частицы, после каждого центрифугирования следует фильтровать
надосадочную жидкость через тот же тампон из стеклянной ваты. После тщательного
промывания водой без органических веществ, высушивают воронку при 50 °С, измель¬
чают унаследованный гумин и стеклянную вату в агатовой ступке и определяют содер¬
жание углерода и азота на CHN-анализаторе.
Последняя часть отцентрифугированного осадка представляет собой остаточный не-
экстрагируемый гумин (ОГ). Дважды промывают его водой без органических веществ
для удаления фосфорной кислоты, высушивают, измельчают и определяют содержание
углерода и азота на CHN-анализаторе.
Расчеты
Полученные данные:
Исходная масса пробы почвы Р0
Масса гумина (отцентрифугированный осадок,
см. «Методики» в разделе 11.2.1) Р1
Масса порции Р1 для обработки серной кислотой Р2
Масса промежуточного осадка (после экстракции ГГ) РЗ
Масса порции РЗ для обработки HF-HC1 Р4
Предположительная масса другой порции РЗ для определения С
(обычно Р4 + Р4’ = РЗ) Р4‘
Масса неэкстрагируемого ОГ Р5
Концентрации экстрактов:
Гумин (исходный отцентрифугированный осадок,
см. «Методики» в разделе 11.2.1) Сг
338
Часть 2. Органический анализ
Гумин, связанный с гидроксидами (мг С в исходном гумине) Сгг
Промежуточный отцентрифугированный осадок (после экстракции ГГ), Сп
мгС/г
Гумин, связанный с силикатами (мг С во всем экстракте) Сгс
Углерод из смеси на стеклянной вате и унаследованный гумин (мг) Суг
Остаточный гумин (мг С/г) Сог
Расчет
В пересчете на исходную пробу почвы получают следующие концентрации углерода (мг
С/г сухой почвы) в гумине перед экстракцией (Г), гумине, связанном с гидроксидами
(ГГ), промежуточном остатке (П), гумине, связанном с силикатами (ГС), унаследован¬
ном гумине (УГ) и остаточном гумине (ОГ):
Г = СгР1/Р0
ГГ = Сгг Р1/(Р2 РО)
П = Сп РЗР1ДР4’ РО)
ГС = Сгс РЗ Р1ДР4 Р2 РО)
УГ = Суг РЗР1ДР4 Р2 РО)
ОГ = Сог Р5 РЗ Р1ДР4 Р2 РО)
Величина Г - (П + ГГ) дает оценку содержания углерода, растворимого в горячей
серной кислоте (полисахариды гумина). Величина П - (ГС + ПГ + ОГ) дает оценку со¬
держания углерода, растворимого смесью HF-HC1.
Использованная литература
Гуминовые вещества
Aiken GR McKnight DM Wershaw RL and MacCarthy P eds (1985) Humic substances in soil, sediment
and water. I. Geochemistry, Isolation and Characterization. Wiley, New York, NY, 692 p.
Bruckert S (1979) Analyse des complexes organo-mindraux des sols. In: Pedologie 2.Constituants et
proprietes du sol, Bonneau M and Souchier В ed., Masson, Paris, 187-209.
Carter MR and Stewart BA eds. (1995) Structure and Organic Matter Storage in Agricultural Soils (Ad¬
vances in Soil Science), Lewis Publishers, Inc., Boca Raton, FL.
Chamayou H and Legros JP (1989) Les bases physiques, chimiques et mineralogiques de la science du
sol. Presses Universitaire de France (Techniques vivantes), 608 p.
Duchaufour Ph (1977) Pedologie. 1. Pedogdndse et classification. Masson, Paris, 477 p.
Flaig W, Beutelspacher H and Rietz E (1975) Chemical composition and physical properties of humic
substances. In Soil Components, Vol.l: Organic Components, Gieseking JE ed., 1-211.
Hessen DO and Tranvik LJ eds. (1998) Aquatic Humic Substances: Ecology and Biogeochemistry (Eco¬
logical Studies, Vol .133). Springer, Berlin, Heidelberg, New York.
Kononova MM (1966) Soil Organic Matter - Its Nature, Its Role in Soil Formation and in Soil Fertility
(translated from the Russian by Nowakowski PhD and Newman ACD), Pergamon Press, New York.
339
Глава 11. Количественное определение гуминовых веществ
Magdoff F, Tabatabai МА and Hanlon Е (1996) Soil Organic Matter: Analysis and Interpretation (SSSA
Special Publication, No 46), American Society of Agronomy.
Piccolo A Ed. (1996) Humic Substances in Terrestrial Ecosystems. Elsevier, Amsterdam.
Schnitzer M and Kahn SU (1972) Humic Substances in the Environment. Dekker, New York, 327 p.
Schnitzer M (1978) Humic substances: chemistiy and reactions. In: Soil Organic Matter, Schnitzer M and
Kahn SU ed., Elsevier, Amsterdam, 1,58.
Springer U (1938) Der Heutige stand der Humusuntersuchungsmethodik mit besonderer Berucksichti-
gung der Trennung, Bestimmung und Charakterisierung der Huminsauretypen und ihre Anwendung
auf Charakteristische Humusformen. Zeitsc.fPflanzen, 6,312-373.
Stevenson FJ and Elliott ET (1989) Methodologies for assessing the quantity and quality of soil organic
matter. In: Dynamics of Soil Organic Matter in Tropical Ecosystems, Coleman DC, Oades JM, Ue-
hara G ed. University of Hawaii Press, Honolulu Hawaii 96822,173-199.
Stevenson FJ (1982) Humus Chemistry, Wiley, New York, 443 p.
Stevenson FJ (1994) Humus Chemistry: Genesis, Composition Reactions, 2nd edition. Wiley, New York,
496 pages.
Tate RL (1992) Soil Organic Matter. Biological and Ecological Effects. Krieger, Melbourne FL, 304 p.
Экстракция, титрование, очистка и фракционирование гуминовых
материалов
Aiken GR, Thurman ЕМ and Malcolm RL (1979) Comparison of XAD macroporous resins for the con¬
centration of fulvic acid from aqueous solution. Anal. Chem., 51, 1799-1803.
Bachelier, G (1983) Variations p6riodiques dans le degte de condensation des acides humiques: mise en
Evidence par spectrofluorimdtrie, correlation avec la stability structural des sols. Cahiers ORSTOM.
Sdrie Pedologie (FRA), |20(3): 247-254.
Dabin B, Gavinelli E and Pelloux P (1983) Rdsultats de l’enquete analytique sur l’extraction et le dosage
des matteres humiques des sols-R6union GEMOS Montpellier, Mai 1983. Document Orstom-Ge-
mos, 16 p. multigr.
Dabin (1976) Methode d’extraction et de fractionnement des matures humiques du sol, application к
quelques etudes pedologiques et agronomiques dans les sols tropicaux. Cah. ORSTOM ser. Pedol.,
XIV, 287-297.
Gregor JE and Powell HKJ (1986) Acid pyrophosphate extraction of soil fulvic acids. J. Soil Sci., 37,
577-585.
Hayes MHB, Swift RS, Wardle RE and Brown JK (1975) Humic materials from an organic soil: A com¬
parison of extractants and of properties of extracts. Geoderma, 13,231-245.
Jacquin F, Calvez C, Dormaar JF and Metche M (1970) Contribution к Г etude des processus d’extraction
et de caracterisation des composes humiques. Bull. Ass. Fr. Etude du Sol, 4, 27-38.
Kumada K, Sato O, Ohsumi Y and Ohta S (1967) Humus composition of mountain soils in central Japan
with special reference to the distribution of P type humic acid. Soil. Sci. Plant Nutr., 13, 151-158.
Levesque and Schnitzer (1966) Effects of NaOH concentration on the extraction of organic matter and of
major inorganic constituents from a soil. Can. J. Soil Sci., 46, 7-12.
Lowe LE and Kumada К (1984) A comparison of two methods for routine characterization of humus in
pedological studies. Soil Sci. Plant Nutr., 30, 321-331.
Lowe LE (1980) Humus fraction ratios as a means of discriminating between horizon types. Can. J. Soil
Sci., 60,219-229.
N£gre R, Ghiglione Cl, Pugnet T and Giraud M (1976) Influence des ntethodes d’extraction et de purifi¬
cation sur la nature des acides humiques de la cddraie du Petit Lub£ron. Cah. ORSTOM ser. Pedol.,
XIV, 337-350.
Pansu M, Gautheyrou J and Loyer JY (2001) Soil Analysis - Sampling, Instrumentation and Quality con¬
trol. Balkema, Lisse, Abington, Exton, Tokyo, 489 pp.
Perraud A (1971) La matiere organique des sols forestiers de la Cote d ’Ivoire., Tltese Docteur Ss-sciences
naturelles, Univ. Nancy I, 87 p. + annexes.
Perraud A, Nguyen Kha, Jacquin F (1971) Essai de caracterisation des formes de l’humine dans plusieurs
types de sols. C R Acad. Sci. Paris, sdrie D, 272, 1594-1596.
340
Часть 2. Органический анализ
Schnitzer М (1982) Organic Matter Characterization. In: Methods of Soil Analysis, Part 2 Chemical and
Microbiological Properties, 2nd edition, Page AL, Miller RH and Keeney DR ed. Agronomy mono¬
graph №9, Am. Soc. of Agronomy, Madison, Wisconsin USA, 581-593.
Thomann Ch (1963) Quelques observations sur Г extraction de Phumus dans les sols : m6thode au pyro¬
phosphate de sodium. Cah. ORSTOMser. Pidol., 3, 43-72.
Глава 12. Характеризация гуминовых веществ
12.1. Введение
12.1.1. Механизмы образования
Образование гуминовых веществ является объектом обсуждения в течение многих лет.
Андро (Andreux, 1994) рассматривал механизмы растворения (лизис клеточных стенок,
протеолиз, расщепление лигнина, преобразование полифенолов и других органических
компонентов), механизмы дубления и меланизации, которые включают внедрение азота
и кислорода (реакция Майяра: конденсация углеводов в присутствии аминного азота,
конденсация полифенолов и аминокислот в окислительной среде), и внедрения унасле¬
дованных соединений.
Шнитцер (Schnitzer, 1978) описал четыре гипотезы образования гуминовых веществ.
Разложение растительного материала. Определенные фракции растительных тканей,
особенно древесных материалов, подвергаются в почве лишь поверхностному разложе¬
нию с образованием гуминовых веществ; поэтому природа этого «унаследованного гу¬
муса» сильно зависит от природы исходного растительного материала; на первой стадии
гумификации образуются самые тяжелые гуминовые материалы, которые могут затем
разрушаться до более легких веществ и, в конечном счете, до С02 и Н20.
Химическая полимеризация. Растительный материал разрушается до малых молекул,
которые используются микроорганизмами в качестве источника энергии и углерода; эти
микроорганизмы затем синтезируют фенолы и аминокислоты, которые полимеризуют-
ся в гуминовые вещества; в этом случае природа исходного материала не влияет на тип
образовавшихся веществ.
Клеточный автолиз. Фрагменты, образующиеся в ходе автолиза микробных и расти¬
тельных клеток (аминосахара, кислоты, фенолы и другие ароматические соединения),
конденсируются и полимеризуются через свободные радикалы.
Микробный синтез. Микробы используют растительные ткани в качестве источника
углерода и энергии для синтеза высокомолекулярных межклеточных органических ве¬
ществ; после гибели микробов эти вещества выделяются в почву; они представляют со¬
бой результат первой стадии гумификации и могут затем подвергаться межклеточному
микробному разложению до более легких молекул.
12.1.2. Молекулярная структура
В зависимости от их растворимости гуминовые вещества обычно подразделяют на три
категории: гуминовые кислоты, фульвокислоты и гумины (см. гл. 11).
В обзоре литературы, опубликованном Шнитцером и Каном (Schnitzer и Kahn, 1972),
показано, что эти три гуминовых фракции имеют аналогичную структуру. Структуру гу-
миновой молекулы можно схематически описать как ядро, обогащенное гидрокси-хино-
новыми агрегатами, соединенными связями С-С или С-О-С (Andreux, 1994). Содержа¬
ние белковых и полипептидных цепей, связанных с этим ядром, зависит от типа почвы
и предшественников. Молекулярные размеры этих структур (рис. 12.1) зависят как от
размеров ядра, так и от природы фенольных предшественников. Поэтому их сродство
к водным растворам зависит от количества и длины периферических гидрофильных це-
342
Часть 2. Органический анализ
Рис. 12.1. Структура двух гуминовых макромолекул; только ядра представлены в масштабе
молекулярной массы (М): а — М > 50 000 и b — М < 50 000; • — С=0; о — СН; □ — NH;
пептидные связи; ОН карбоксила (Andreux, 1994)
пей, имеющих свободные -СООН группы полипептидов, при значительной кислотно¬
сти матрицы СAndreux, 1994).
Свойства трех гуминовых фракций значительно различаются, особенно молекуляр¬
ной массой, элементным составом и количеством функциональных групп. Фульво-
кислоты имеют самую низкую молекулярную массу, содержат больше кислорода, но
меньше углерода и азота, чем две другие фракции. Содержание кислородсодержащих
функциональных групп (С02Н, ОН, С=0) на единицу массы в фульвокислотах больше,
чем в гуминовых кислотах и гумине (Stevenson, 1982).
Гуминовые кислоты можно экстрагировать из древесного угля (Lawson и Stewart, 1989).
Исследования с помощью 13С ЯМР-спектроскопии (см. раздел 12.3.4) показали, что ста¬
бильный углерод в австралийских почвах относится главным образом к древесному углю
(Skjemstad и др., 1996). Хотя для гуминовых молекул были предложены структурные
модели (например, Schulten, 1995; Schulten и Schnitzer, 1997; Schulten и Leinweber, 2000;
Schulten, 2002; Lodygin и Beznosikov, 2003), большинство концепций молекулярной струк¬
туры не применимы к гуминовым кислотам (McCarthy, 2001). Необходимы дальнейшие
исследования для получения точной информации об этих молекулах, их связях с неор¬
ганическими (Schulten и Leinweber, 2000) и ксенобиотическими (Piccolo и др., 1999) мо¬
лекулами и их распределении в различных типах почв, которая, в свою очередь, способ¬
ствует пониманию их роли в окружающей среде. Для таких исследований необходимы
все современные инструментальные методы {Hatcher и др., 2001, Piccolo и Conte, 2003).
12.2. Традиционные методы анализа
12.2.1. Фракционирование гуминовых соединений
Основные положения
В течение многих лет гуминовые кислоты характеризовались по степени их связи с гли¬
нистыми минералами (Springer, 1938; Tiurirt, 1951; Duchaufour, 1954,1956).
Глава 12. Характеризация гуминовых веществ
343
Бурые, или свободные, гуминовые кислоты считались слабо связанными с глинисты¬
ми минералами, имеющими самую низкую молекулярную массу и относительно мало
подверженными флокулирующему воздействию электролитов; считалось также, что они
образуются при окислении лигнина под действием полифенолоксидазы и характеризу¬
ют кислые почвы, например, лесные почвы влажного климата (Duchaufour, 1956).
Серые гуминовые кислоты имеют более темный цвет, они образуют более тесные свя¬
зи с минеральными коллоидами, имеют более сжатые молекулы, легко флокулируются
в присутствии электролитов; этот тип гумуса характерен для черноземов, но относитель¬
но часто встречается во всех почвах, богатых кальцием.
Таким образом, ранние методы фракционирования гуминовых кислот были основаны
на свойствах этих двух типов соединений: экстракция в присутствии флокулирующего
агента для бурых гуминовых кислот с последующим промыванием остатка водой для се¬
рых гуминовых кислот (Duchaufour, 1956), прямая щелочная экстракция бурых гуминовых
кислот и последующая экстракция двух фракций серых гуминовых кислот в зависимо¬
сти от образования ими связей с кальцием или железом и алюминием (Duchaufour, 1957).
Для облегчения процедуры Дюшофур и Жакен {Duchaufour nJacquin, 1966) предложи¬
ли электрофоретическое фракционирование гуминовых кислот с использованием пиро¬
фосфатных экстрактов (см. подраздел «Метод Дабена» в гл. 11). Дабен и Томанн {Dabin
и 7Ъотапп, 1970) сравнили этот метод с методом Норина (Tiurin, 1951); он широко ис¬
пользовался во Франции, особенно в лабораториях института 1RD1 (Ratsimbazafy, 1973;
Dabin, 1980). Этот первый метод фракционирования описан ниже. Метод фракцио¬
нирования гуминовых кислот эксклюзионной хроматографией (ВаШу и Margulis, 1968;
Bailly и Tittonel, 1972) также описан ниже, вместе с методикой фракционирования фуль-
вокислот. Другие сложные методы фракционирования гуминовых соединений коротко
описаны в разделе 12.3.1.
Оборудование
• Пластиковая камера для электрофореза с тремя отделениями (рис. 12.2).
• Стабилизированный источник постоянного тока с регулировкой от 0 до 600 В.
• Фотоэлектрический денситометр для чтения электрофореграмм.
• Колонки для жидкостной хроматографии диаметром 2,5 см и длиной 80 см.
• Детектор для УФ и видимой области, оборудованный самописцем или системой
сбора данных.
• Коллектор фракций (факультативно).
• Перистальтический насос со скоростью потока 50 мл/час (факультативно).
Материалы и реагенты
• Листы фильтровальной бумаги {Arch 302 или Whatman No. 1, 2) шириной 5 см
и длиной 35 см, разрезанные перпендикулярно направлению волокон (если удер¬
живать квадратный лист бумаги с одной стороны, направление, перпендикулярное
направлению волокон, это то, где бумага изгибается под собственным весом силь¬
нее всего).
• Экстрагирующие растворы (см. раздел 11.2.1).
1IRD — Institute of Research for Development (ex-Orstom), BP 64501, 911 Avenue of Agropolis,
34394 Montpellier Cedex 5, France.
344
Часть 2. Органический анализ
Нагрузка полоса бумаги
Анод
+
• Буферный раствор для электрофореза: в мерную колбу на 2 л помещают 13,6 г мо-
нозамещенного фосфата калия, 2,5 г гранулированного гидроксида натрия и при¬
мерно 1,5 л воды, не содержащей органических веществ (см. раздел 11.2.1); при
необходимости добавляют раствор гидроксида натрия до pH 7,4, хорошо переме¬
шивают и доливают водой до 2 л.
• Стандартный декстрановый гель Sephadex <325 для молекулярных масс < 5000.
• Стандартный декстрановый гель Sephadex G75 для молекулярных масс < 50 000.
• Стандартный декстрановый гель Sephadex <3200 для молекулярных масс < 200 000.
• Поливинилпирролидон.
• Трис-буфер (pH 9, ионная сила 0,5): смешивают с 414 мл 2-амино-2-(гидроксиметил)
пропан-1,3-диола 50 мл 1 М раствора НС1 и доводят водой без органических ве¬
ществ до 1 л.
• Боратный буфер (pH 9,1; ионная сила 0,075): 0,025 М раствор Na2B407.
Методика электрофоретического фракционирования гуминовых кислот
• Отбирают порцию гуминового экстракта (пирофосфатного или щелочного), соот¬
ветствующую 25-50 мг углерода (см. подраздел «Титрование» в разделе 11.2.2).
• Осаждают гуминовые кислоты при pH 1 серной кислотой, центрифугируют и про¬
мывают отцентрифугированный осадок несколько раз 1 н. раствором H2S04.
• Растворяют гуминовые кислоты примерно в 1 мл 1 н. раствора гидроксида натрия,
чтобы получить довольно густой раствор, но без осадка (см. об очистке в разделе
11.2.4) и с равномерным распределением углерода; хранят в плотно закрытой про¬
бирке для гемолиза.
• Наполняют два внешних отделения камеры до одного и того же уровня буферным
раствором для электрофореза; увлажняют полосу бумаги буферным раствором,
сушат ее между двумя листами фильтровальной бумаги и помещают в камеру для
электрофореза, туго натянув между двумя внешними отделениями, как показано
на рис. 12.2.
Глава 12. Характеризация гуминовых веществ
345
• С помощью микропипетки на 100 мм3 наносят примерно 40 мм3 гуминового рас¬
твора на расстоянии 5 см от катода по прямой к центру полосы бумаги, оставляя
по 1 см с каждой стороны бумаги; в случае очень концентрированных растворов
объем может быть уменьшен до 20 мм3.
• Закрывают камеру крышкой и включают электрический ток; регулируют напряже¬
ние на 10 В/см бумаги, или примерно 200 В; сила тока зависит от числа полос бу¬
маги для одновременного электрофореза и электропроводности электролита (при¬
мерно 15 мА для четырех полос). Отрицательно заряженные гуминовые молекулы
движутся к аноду; чем меньше молекулы, тем быстрее они движутся (например,
бурые гуминовые кислоты (БГК)). Вначале скорость движения большая, затем оно
замедляется. Время, необходимое для стандартного электрофореза, составляет 3 ч,
что соответствует перемещению бурых гуминовых кислот на 10—12 см.
• Выключают ток и быстро вынимают полосы бумаги, помещают их на плоскую по¬
верхность и сушат под ИК-лампой или в сушильном шкафу при 60 °С. Фиксируют
значения коэффициента пропускания излучения для каждой полосы бумаги при
помощи фотоэлектрического денситометра.
• Получают электрофореграмму (рис. 12.3), на которой:
1) серые гуминовые кислоты (СГК) часто дают четкий узкий пик на расстоянии
до 1 см от стартовой линии; для черноземов Дюшофур и Жакен (Duchaufour
и Jacquin, 1963) описали второй пик, который может мигрировать до 2 см от
стартовой линии; в тропических почвах СГК часто продвигаются до 3 или 4 см
от стартовой линии в виде одного, двух или более пиков; иногда довольно слож¬
но определить границу СГК, и тогда границу их пика условно отмечают на трети
полной длины диаграммы, т. е. на 3-4 см для ленты длиной 9-12 см;
2) гуминовые соединения между 1/3 и 1/2 длины диаграммы называются промежу¬
точными гуминовыми кислотами (ПГК);
3) бурые гуминовые кислоты (БГК) находятся между серединой и концом диа¬
граммы.
• Определяют площадь поверхности каждой фракции; ПС — площадь СГК, ПП —
площадь ПГК, ПБ — площадь БГК, ПО — общая площадь и ок — общее количе¬
ство гуминовых кислот в %; общее количество каждой фракции выражают форму¬
лами:
СГК % = ок ПС/ПО;
ПГК %= окПП/ПО;
БГК %= окПБ/ПО.
На рис. 12.3 приведены некоторые примеры электрофореграмм, полученных этим
методом.
Примечания
Любая жидкость, двигающаяся по лентам бумаги, может вызвать миграцию гуминовых
кислот. Этого можно избежать, если жидкость у катода и анода будет находиться на од¬
ном и том же уровне (в противном случае возможно сифонирование бумагой) и если не
будет испарения с бумаги. Испарение может заставить жидкость двигаться из-за вса¬
сывания на обеих сторонах ленты бумаги. Таким образом, даже в отсутствие электри¬
ческого тока некоторые гуминовые кислоты могут мигрировать до центра полосы. При
включенном токе движение прекратится, когда скорость движения, вызванного элек-
346
Часть 2. Органический анализ
СГК ПГК БГА СГК ПГК БГА
Слева: показания оптического денситометра
при 512 нм (верхняя кривая) и 635 нм. Спра¬
ва: соответствующие интегральные кривые,
площади которых соответствуют серым гу-
миновым кислотам (СГК: от 0 до 1/3 кривой),
промежуточным гуминовым кислотам (ПГК:
между 1/3 и 1/2 кривой), бурым гуминовым
кислотам (БГК: более 1/2 кривой), а — тропи¬
ческий подзол (histic Tropaquod), горизонт А;
b — тропический подзол (histic Tropaquod), го¬
ризонт В; с — железогумусная ферраллитная
почва (Umbriorthox), поверхностный горизонт;
d — железогумусная ферраллитная почва
(Umbriorthox), подпахотный горизонт; е — эро¬
дированная тропическая почва (Oxictropudalf)
Рис. 12.3. Примеры электрофореграмм гуминовых кислот, экстрагированных раствором
пирофосфата с pH 9,8 из тропических почв (Dabin, 1980)
трическим потенциалом, станет равной скорости жидкости, движущейся в противопо¬
ложном направлении. Поэтому необходимо использовать крышку, чтобы уменьшить ис¬
парение; форма крышки должна предотвращать попадание капель конденсата на ленты
бумаги.
Прохождение тока обогащает катодное отделение камеры щелочью и повышает его
pH; при проведении нескольких циклов электрофореза это явление может быть ском¬
пенсировано обращением направления тока.
Методика фракционирования гуминовых кислот на декстрановом геле
Фракционирование можно выполнить просто элюированием дистиллированной водой
без органических веществ (ВаШуи Margulis, 1968; Bailly и Tittonel, 1972).
• Готовят концентрированный раствор гуминовых кислот так же, как для электро¬
фореза, но с навеской препарата, соответствующей 2-10 мг углерода, в 3-10 мл
1 М раствора NaOH.
• Заполняют колонки гелем Sephadex: 50 см для G25 или G75, примерно 70 см для
G200, эти значения могут меняться в зависимости от типа гумуса (Bailly и Tittonel,
1972).
• Переносят навеску препарата в колонку с G25 и элюируют водой без органических
веществ под действием силы тяжести (нисходящая хроматография) или при по¬
мощи перистальтического насоса при скорости потока примерно 30 мл/час (вос¬
ходящая хроматография).
Глава 12. Характеризация гуминовых веществ
347
• Переносят фильтрат в УФ-фотометрический детектор, настроенный на длину вол¬
ны 253,7 нм, записывают хроматограмму и собирают фракции, соответствующие
основным пикам (рис. 12.4).
• Первый элюированный пик соответствует самым большим молекулам, которые не
были отделены на геле; собирают эту фракцию и подвергают ее аналогичной экс-
клюзионной хроматографии на колонке G75; вновь фракционируют новый пер¬
вый пик, элюированный на этой колонке, на колонке G200; две серии примеров
хроматограмм, последовательно полученных для двух почв на трех типах колонок,
приведены на рис. 12.8.
Рис. 12.4. Примеры разделения эксклюзионной хроматографией на декстрановом геле (ВаШу
и Tittonel, 1972). Слева — луговая подзолистая почва, горизонт А1, глубина 0-6 см; справа —
серая лесная почва, горизонт А2, глубина 16-29 см; а — фракционирование экстракта гуминовых
кислот на Sephadex G25 F, навеска, соответствующая 4,7 мг С; Ь — фракционирование пика V,
полученного из п. а, на Sephadex G75; с — фракционирование пика VIII, полученного из п. Ь, на
Sephadex G200
Этот тип фракционирования гель-проникающей хроматографией может выполнять¬
ся и в буферных растворах для предотвращения взаимодействий между гелем и раство¬
ром, например, в Трис и боратном буферных растворах, приготовленных так же, как
описано в подразделе «Материалы и реагенты» раздела 12.2.1 {Cameron и др., 1972а).
Фракционирование фульвокислот
Метод Лоу (Lowe, 1975) — очень простой способ разделения фульвокисЛот на две фрак¬
ции: окрашенную полифенольную и почти бесцветную с преобладанием полисахаридов.
348
Часть 2. Органический анализ
Поскольку СА, С^. и Cfl — углероды гуминовых кислот, фульвокислот и их окрашенной
полифенольной фракции, соответственно, соотношения Ch:Cf и Со:Сг были связаны
с типами горизонтов, использованных в Канадской системе классификации почв, что
облегчает разделение некоторых из этих типов (Lowe, 1980):
• обрабатывают фракцию экстракта фульвокислот, используя раствор поливинил-
пирролидона с концентрацией 1 г/100 мл;
• периодически перемешивают в течение 30 мин и фильтруют;
• определяют содержание углерода в фильтрате (см. раздел 11.2.2) для определения
содержания фракции Со; количество фульвокислот в объеме должно быть доста¬
точно для определения с приемлемой точностью.
12.2.2, Определение основных функциональных групп
Основные положения
Методики, описанные в настоящей главе, основаны на измерении общей и карбоксиль¬
ной кислотности (Wright и Schnitzer, 1959; Schnitzer и Gupta, 1965).
Для измерения общей кислотности пробу обрабатывают раствором гидроксида бария
в атмосфере N2 в течение 24 ч. Ва(ОН)2, оставшийся в растворе после реакции, опреде¬
ляют обратным титрованием стандартным раствором кислоты.
Для определения карбоксильных групп гуминовые вещества перемешивают в течение
24 ч с избытком раствора ацетата кальция, который приводит к выделению уксусной
кислоты в соответствии с реакцией:
2RCOOH + (СН3СОО)2Са -► (RCOO)2Ca + 2СН3СООН
Выделившуюся уксусную кислоту затем титруют стандартным раствором щелочи.
Долю фенольных групп рассчитывают как разницу между общей кислотностью и кис¬
лотностью карбоксильных групп.
Другие методы определения функциональных групп (фенольных, спиртовых, кетон-
ных, хинольных) кратко описаны в разделе 12.3.
Оборудование
• Конические колбы с навинчивающимися пробками на 125 мл;
• Титрометр, оборудованный комбинированным электродом для измерения pH.
Реактивы
• 0,2 н. раствор гидроксида бария: отвешивают 31,548 г Ва(ОН)2- 8Н20 (содержаще¬
го минимум карбонатов), растворяют в воде, не содержащей С02 и органических
веществ, доводят объем до 1 л и защищают от атмосферного С02 ловушкой с на¬
тронной известью.
• 0,5 М раствор хлористоводородной кислоты: готовят из фиксанала; разбавляют во¬
дой, не содержащей С02 и органических веществ.
• 1 н. раствор ацетата кальция Са(СН3СОО)2: сушат чистый реактив при 105 °С
и взвешивают 79,085 г в безводной атмосфере, растворяют в воде без органических
веществ и доводят объем до 1 л.
• 0,1 М раствор гидроксида натрия: готовят из фиксанала; разбавляют водой без
органических веществ, доводят объем до 1 л; при хранении защищают ловушкой
с натронной известью.
349
Глава 12. Характеризация гуминовых веществ
Методика
Общая кислотность
Для получения максимального числа обменных центров в активной кислотной форме
рекомендуется работать с тщательно очищенными гуминовыми веществами, чтобы сни¬
зить содержание золы в них (см. раздел 11.2.4).
Помещают точно взвешенную навеску лиофилизированного гуминового материала
массой 50—100 мг в коническую колбу на 125 мл с навинчивающейся пробкой и добав¬
ляют точно 20 мл 0,2 М раствора Ва(ОН)2. Выполняют холостой опыт только с 0,2 М
раствором Ва(ОН)2 без пробы. Помещают колбы в атмосферу азота, тщательно закрыва¬
ют и перемешивают при комнатной температуре в течение 24 ч. Фильтруют суспензию,
тщательно смывают остаток дистиллированной водой, не содержащей С02, добавляют
промывные воды к фильтрату и выполняют потенциометрическое титрование получен¬
ного раствора 0,5 М раствором НС1 до pH 8,4. Если Vb и Vs - объемы стандартного рас¬
твора кислоты для титрования холостого опыта и пробы, соответственно, Na — нормаль¬
ность кислоты и Р — масса пробы (мг), то общую кислотность А, (мэкв/г гуминового
материала) находят по формуле:
At=\m{Vb-V)NJP.
Карбоксильные группы
Точную навеску гуминового материала массой 50—100 мг помещают в коническую кол¬
бу с навинчивающейся крышкой; добавляют 10 мл 1 н. раствора Са(СН3СОО)2 и 40 мл
воды, не содержащей С02 и органических веществ. Одновременно выполняют холостой
опыт со всеми реагентами, но без гуминового материала. После встряхивания при ком¬
натной температуре в течение 24 ч фильтруют суспензию, споласкивают остаток дис¬
тиллированной водой, не содержащей С02, объединяют фильтрат и промывную воду
и выполняют потенциометрическое титрование 0,1 М раствором NaOH до pH 9,8. Если
Vsn Vh — объемы титрующего раствора для пробы и холостого опыта, соответственно
(мл), Nh — нормальность стандартного раствора щелочи (моль/л) и Р — масса пробы
(мг), то карбоксильную кислотность Ас (моль СООН/г гуминового материала) находят
по формуле:
Ac=m0(V-VJNb/P
Фенольная кислотность
Фенольную кислотность А можно выражать в (моль фенольных ОН/г гуминового мате¬
риала) по формуле:
А =А-А.
Р * с
12.2.3. Спектрометрия УФ-видимого диапазона
Основные положения
УФ-спектрометрия исследует электронные переходы между энергетическими уровнями
в молекулах, а изменения молекулярной кинетической энергии фиксируются с помо¬
щью низкоэнергетического инфракрасного излучения.
350
Часть 2. Органический анализ
Хотя поглощение в ультрафиолетовой и видимой областях электромагнитного спек¬
тра не дает характеристических полос для гуминовых соединений (Schnitzer и Kahn, 1972;
Schnitzer, 1978), отношение Е4:Е6 между поглощением при 465 (Е4) и 665 нм (Е6) ча¬
сто используют для характеристики гумуса. Отношения ниже 5 типичны для гуминовых
кислот, тогда как фульвокислоты характеризуются более высоким отношением; это от¬
ношение не зависит от концентрации гуминовых веществ, но различается для гумино¬
вых веществ, экстрагированных из различных типов почв.
Кононова (1966) считала, что отношение Е4:Е6 определяется степенью конденсации
сети ароматического углерода; его низкое значение указывает на высокую степень кон¬
денсации ароматических гуминовых компонентов, а высокое значение — на ббльшую
долю алифатических структур.
Чен с сотр. (Chen и др., 1977) подробно изучили данные об отношении Ё4:Е6. Они
показали, что отношение Е4:Е6:
• зависит, главным образом, от молекулярных размеров (или молекулярной массы
или массы частиц, рис. 12.5, а, Ъ);
• сильно зависит от pH (рис. 12.5, а, Ь)\
• коррелирует с концентрацией свободных радикалов (рис. 12.5, с), содержанием О,
С, С02Н и общей кислотностью (эти данные также коррелируют с размером ча¬
стиц);
• не коррелирует напрямую с концентрацией конденсированных ароматических
ядер, что может опровергнуть предположение Кононовой (1966);
• не зависит от концентрации гуминовых или фульвокислот, по крайней мере, в рай¬
оне 100-500 ppm, что подтверждает другое предположение Кононовой.
Эти авторы показали, что наиболее благоприятный диапазон pH для измерения от¬
ношения Е4:Е6 находится между 7 и 8 (что соответствует данным Кононовой (1966)).
Его можно получить растворением гуминовых веществ в 0,1 М растворе NaHC03 с кон¬
центрацией 200-400 мг/кг.
Гош и Шнитцер (Ghosh и Schnitzer, 1979) предложили механизм, связывающий макро-
молекулярные характеристики гуминовых веществ с поглощением в УФ и видимом диа¬
пазонах: оптическая плотность снижается при увеличении концентрации нейтрального
электролита, указывая на уменьшение размера частиц, вероятно из-за сворачивания ма¬
кромолекулы.
Оборудование
• Регулируемый двухлучевой спектрограф УФ и видимой областей с фиксирован¬
ной длиной волны (465 и 665 нм) или предпочтительно переменной длиной волны
между 200 и 700 нм.
• Кварцевые кюветы для УФ-спектроскопии.
Реагенты
0,05 М раствор NaHC03: в мерной колбе на 1 л растворяют в дистиллированной воде, не
содержащей органических веществ, 4,200 г NaHC03 (спектроскопически чистого), до¬
водят объем до 1 л и тщательно закрывают колбу перед хранением.
Методика
Растворяют 2-4 мг гуминового материала в 10 мл 0,05 М раствора NaHC03. После рас¬
творения проверяют pH, который должен быть близок к 8 (pH 0,05 М водного раствора
NaHC03 равен 8,3). Заполняют этим раствором кварцевую измерительную кювету до
Глава 12. Характеризация гуминовых веществ
351
ш
S
14
12
10
8
б
4
pH
Рис. 12.5. Влияние некоторых факторов на отношение Е4:Е6 (Chen и др., 1977): а — влияние
pH различных фракций фульвокислот (из горизонта Bh канадского подзола), разделенных на
геле Sephadex; молекулярные массы, определенные осмометрией (Hansen и Schnitzer, 1969),
составили 883 для AF-IV, 1181 для AF-III, 1815 для AF-II, 2110 для AF-I и > 2110 для > AF-1; Ь —
влияние pH некоторых гуминовых кислот из горизонтов Ah канадских почв из района около
Альберты: черный чернозем, солодь, солонец; с — влияние концентрации свободных радикалов,
определенной методом электронного спинового резонанса (см. раздел 12.3.6) для фульвокислот,
использованных в п. а (числа на кривой — значения pH)
половины и заполняют контрольную кювету чистым 0,05 М раствором NaHC03. Из¬
меряют поглощение при 465 и 665 нм и рассчитывают отношение полученных величин
(Е4.Е6),
Если необходимо проведение более детальных исследований, можно записать спектр
оптической плотности (ОП) между 200 и 350 нм УФ-диапазона (Ghosh и Schnitzer,
1979) или между 400 и 700 нм; в последнем случае можно провести прямые линии
lg (ОП) =Дlg к), и согласно (Ghosh и др., 1977) наклон ее должен быть равен -6,435 lg(E4: Е6).
12.2.4. Инфракрасная спектрография
Основные положения
Инфракрасный спектр в диапазоне длин волн от 1 до 100 мкм позволяет наблюдать ва¬
лентные и деформационные колебания молекул (вращательный молекулярный спектр
352
Часть 2. Органический анализ
соответствует более низкоэнергетическому излучению с длинами волн выше 100 мкм).
На практике наиболее информационным диапазоном для органической химии является
средний ИК-диапазон между длинами волн X 2,5 и 15 мкм, соответствующими волно¬
вым числам \/Х 4000 и 660 см-1 (см. раздел 5.1.1). С помощью хемометрического про¬
граммного обеспечения может быть изучен и ближний ИК-диапазон (см. раздел 5.3.1).
В гуминовых веществах ИК-спектр отражает, главным образом, кислородсодержащие
функциональные группы, такие как —С02Н, ОН и С-О. Некоторые ИК-полосы осо¬
бенно хорошо определены (Schnitzer, 1971): 3400 см-1 (Н, связанный с ОН), 2900 см-1
(алифатические связи СН), 1725 см-1 (связь С—О в С02Н, растяжение С—О в кетонных
карбонилах), 1630 см-1 (ароматические связи С—С; Н, связанный с С—О карбонилов;
СОО), 1450 см-1 (алифатические СН), 1400 см-1 (СОО", алифатические СН), 1200 см-1
(валентные СО, деформации ОН в С02Н), 1050 см-1 (связи Si—О силикатных примесей).
ИК-спектры предоставляют мало информации о химической структуре ядра гумино¬
вых веществ. Однако они полезны для предварительной характеристики гуминовых ве¬
ществ различного происхождения (рис. 12.6), определения влияния различных методов
экстракции или реактивов, используемых для химической очистки (см. раздел 11.2.4),
и исследования реакций дериватизации, таких как силилирование, метилирование
и ацетилирование.
ИК-спектры также позволяют выявить изменения в структуре гуминовых материа¬
лов вследствие окисления, пиролиза или других обработок. Наконец, это практический
метод, характеризующий образование металлгуматных или глиногуматных комплексов
Волновое число, см1 Волновое число, см~1
Рис. 12.6. Инфракрасные спектры некоторых фульвокислот (слева) и гуминовых кислот (справа)
из стандартных и эталонных образцов МГО (Senesi и др. 1989): а — стандарты из реки Суванни
(Suwannee River Standard, коды IHSS 1S101F (AF) и 1 S101H (АН)); а, — эталоны из реки Суванни
(Suwannee River Reference, коды IHSS 1R101F (AF) и 1R101H (AH)); b - стандарты из Северного
моря (Nordic aquatic, код 1R105F и 1R105Н); с — стандартный образец почвы (Soil St, коды 1S102F
и 1R102H); d — торф, коды 1R103F и 1R103H; е — леонардит, код 1R 104 Н
353
Глава 12. Характеризация гуминовых веществ
и выявляющий взаимодействия между гуминовыми веществами и другими органиче¬
скими молекулами, такими как пестициды.
Необходимо тщательно расшифровывать спектры и особенно не путать органические
и минеральные источники полос поглощения (Russel и Anderson, 1977).
Оборудование
• Двухлучевой ИК-абсорбционный спектрограф с интервалом волновых чисел от
300 до 4000 см-1.
• Ручной гидравлический пресс для приготовления таблеток для ИК-спектрографии
(например, стандартный 12-тонный Spex-Carver).
• Вакуумируемая пресс-форма из полированной нержавеющей стали для таблеток
диаметром 13 мм.
Примечание: Можно работать и без пресс-формы или гидравлического пресса; спек¬
тры можно получать, используя суспензии, расположенные между двумя пластинками,
прозрачными для ИК-издучения, либо растворы в растворителях, прозрачных для ИК-
излучения. Метод, описанный ниже, не самый дешевый, но, как было показано, чрез¬
вычайно подходящий для ИК-спектрометрии.
Реагент
Порошок бромида калия для ИК-спектрометрии.
Методика
В агатовой ступке готовят таблетку с КВг, смешивая 1 мг гуминового материала с 400 мг
сухого КВг. Помещают порошок в пресс-форму, вакуумируют и прессуют под давлением
7600 кг/см2 в течение 20 мин (см. подраздел «Приготовление дисков (твердого раство¬
ра)» в гл. 5). Вынимают таблетку из пресс-формы, которая должна быть прозрачной,
и помещают ее в измерительную кювету ИК-спектрометра; кладут таблетку чистого КВг
в эталонную кювету и записывают спектр в диапазоне от 300 до 4000 см-1.
12.3. Дополнительные методы
12.3.1. Усовершенствование методов фракционирования
Описанные ниже методы являются результатами усовершенствования электрофореза
и гель-проникающей хроматографии (см. раздел 12.2.1).
Электрофорез-электрофокусирование
Какко с сотр. (Сассо и др., 1974) предложили усовершенствовать электрофорез гумино¬
вых веществ, используя метод электрофокусирования, описанный Ригетти и Дрисдей-
лом (Righetti и Drysdale, 1971). В этом методе гуминовые вещества движутся от анода
к катоду в полиакриламидном геле в присутствии амфолинов, вызывающих градиент
pH. Движение прекращается, когда каждое соединение достигает своей изоэлектриче-
ской точки. На рис. 12.7 показаны результаты изоэлектрической характеризации (Сассо
и Maggioni, 1976) гуминовых и фульвокислот, экстрагированных пирофосфатом при pH
7 из альпийского подзола. Русина с сотр. (Rusina и др., 1983) предложили систему рас¬
четов молекулярных параметров на основе электрофоретической подвижности в полиа¬
криламидном геле. Методы электрофореза обычно используют для исследования моле¬
кулярных размеров, а также электрических зарядов гуминовых веществ (Duxbury, 1989).
354
Часть 2. Органический анализ
Рис. 12.7. Изоэлектрофоретическая характеризация фульвокислот (глубокий горизонт Bh) и гу¬
миновых кислот (поверхностный А1 и глубокий Bh горизонты), экстрагированных из альпийского
подзола (Сассо и Maggioni, 1976)
Гель-проникающая хроматография
Гель можно откалибровать в молекулярных массах гуминовых кислот, измеренных дру¬
гими методами. На рис. 12.8 показаны калибровочные кривые, полученные Камеро¬
ном с сотр. (Cameron и др., 1972а) для четырех типов геля; молекулярные массы (на оси
абсцисс) были определены методом седиментации, используя ультрацентрифугирова¬
ние (Cameron и др., 1972b). В таких исследованиях гель характеризуют параметром Kav
(Laurent и Killander, 1964):
Km=(v*-vj/(K-V>
где VR - объем удерживания; Vo — объем пор; Vt — общий объем колонки.
Средние значения Kav устанавливают в соответствии со средними значениями молеку-
лярных масс М (рис. 12.8), используя две константы к{ик2в соответствии с формулой:
К =к.1пМ+кг
OV 1 I
В настоящее время фракционирование методом гель-фильтрации можно выполнять
методом высокоэффективной жидкостной хроматографии (ВЭЖХ), и, кроме того, но¬
вые гели более эффективны, чем старые гели Sephadex. Например, силикагель Zorbax
PSM 1000, использованный Моризуром с сотр. (Morizur и др., 1984), позволил осуще¬
ствить более эффективное выделение всех органических веществ, обладающих емко¬
стью ионного обмена примерно в 100 меньшей, чем гель Sephadex.
Беккет с сотр. (Beckett и др., 1987) использовали метод проточного фракционирования
в поперечном силовом поле (проточное ФПСП) для фракционирования гуминовых и фуль¬
вокислот.. который оказался мощным инструментом для получения информации о мо¬
лекулярных массах.
12.3.2. Определение функциональных групп
В разделе 12.2.2 описаны методы определения основных функциональных групп гуми¬
новых соединений: карбоксильных и фенольных групп, причем последние рассчитыва¬
ются по разнице между общей кислотностью и карбоксильной кислотностью.
Глава 12. Характеризация гуминовых веществ
355
Рис. 12.8. Калибровка четырех типов геля (Cameron и др., 1972а) для фракционирования гуми¬
новых кислот в сравнении с другими макромолекулярными соединениями (сплошные линии и
черные кружки — гуминовые кислоты, пунктирные линии и белые кружки — данные по фракцио¬
нированию белка с использованием сахарозы для определения постоянного объема жидкости Vr
Kw и М — см. в тексте: а — Sephadex G100 в трис-буфере (приготовление, см. раздел 12.2.1);
b — Sephadex G100 в боратном буфере (приготовление, см. раздел 12.2.1); с — Biogel Р-150
в трис-буфере; d—Sepharose 6В в трис-буфере.
Химические методы определения используют для определения других функциональ¬
ных групп. Основные группы, которые были предметом такого исследования, это общие
ОН-группы, которые можно определить ацетилированием гуминовых веществ уксусным
ангидридом в пиридине (Schnitzern Skinner, 1965):
2ROH + (CH3C0)20 -► CH3COOR + СН3СООН
Ацетилированные гуминовые вещества затем тщательно отделяют от реакционной
смеси. ГНцролиз в щелочной среде приводит к образованию ацетатов из ацетильных
групп; перегонка в сильнокислой среде позволяет выделить уксусную кислоту, которую
определяют ацидометрическим титрованием. Число молей выделенной уксусной кисло¬
ты соответствует количеству общих ОН-групп гуминового вещества.
• Спиртовые ОН-группы можно оценить как «общие ОН-группы» минус «фенольные
ОН-группы». Однако поскольку фенольные ОН-группы также рассчитываются по
разнице (см. раздел 12.2.2), целесообразно учитывать закон распространения оши¬
бок.
• Общие С=0 группы можно определить по реакции гуминовых веществ с гидрохло¬
ридом гидроксиламина в среде 2-диметиламиноэтанола:
R1R2C=0 + NH2OH • НС1 -► R1R2C=NOH + H20 + HC1
Избыток гидрохлорида гидроксиламина определяют обратным титрованием стан¬
дартным раствором НС104 (Fritz и др., 1959).
• Хинонные С=0 группы можно определить восстановлением до фенольных ОН-
групп двухвалентным железом в триэтаноламине; избыток двухвалентного желе¬
за определяют обратным амперометрическим титрованием раствором бихромата
(Glebko идр., 1970).
356
Часть 2. Органический анализ
• Кетонные С=0 группы можно определить как разницу между общими С=0 группа¬
ми и хинонными С=0 группами.
Кроме того, Шнитцер (Schnitzer, 1978) описывает несколько попыток характериза¬
ции кислотных групп прямым потенциометрическим титрованием. Было сложно чет¬
ко различить этим методом два основных типа кислотности (функциональные группы
ОН и С02Н), даже в неводной среде. Некоторым авторам это удалось, включая Росселя
с сотр. (Rosell и др., 1972), одновременно определившим три типа кислотных групп по¬
тенциометрическим титрованием в 80% водном растворе N-метилацетамида.
Де Нобили с сотр. (De Nobili и др., 1990) описали метод определения карбоксильных
групп, альтернативный методу, описанному в разделе 12.2.2: осаждение гуминовых ве¬
ществ катионным ПАВ-цетилтриметиламмонием.
12.3.3. Характеристика гуминовых веществ методом фрагментации
Один из способов исследования крупных гуминовых макромолекул состоит в их расщеп¬
лении и идентификации полученных фрагментов. Такие методы можно разделить на
четыре основные группы: окислительная фрагментация, восстановительная фрагмента¬
ция, другие методы химического расщепления и термическая фрагментация пиролизом.
Окислительная фрагментация
Эта группа методов может быть подразделена на две подгруппы: (i) окисление перманга¬
натом и (И) другие методы окисления.
Окисление перманганатом нашло широкое применение для гуминовых материалов
из различных типов почв: например, горизонтов Ah солонцов, солодей и черноземов
(Kahn и Schnitzer, 1971а; Kahn и Schnitzer, 1972b), серых лесных почв при различных си¬
стемах земледелия (Kahn и Schnitzer, 1972b), вулканических тропических почв (Griffith
и Schnitzer, 1975), средиземноморских почв (Chen и др., 1978а), негидролизуемых гуми¬
новых остатков (Ogner, 1973) и фракций фульвокислот (Khan и Schnitzer, 1971b).
Методики определения зависят от авторов. Гуминовые вещества можно сначала
фракционировать или ввести в реакцию дериватизации (метилирования) перед окис¬
лением. Кан и Шнитцер (Kahn и Schnitzer, 1972b) окисляли 1 г гуминового материала
кипячением с обратным холодильником в 250 мл 4%-ного водного раствора КМп04 в те¬
чение 8 ч. Избыток перманганата должен быть разрушен контролируемым добавлением
малых объемов метанола, а раствор отделен от нерастворимого Мп02 фильтрованием
и промыванием. Подкисленный фильтрат затем экстрагируют этилацетатом в жидкост¬
ном экстракторе1 в течение 48 ч. Экстракт высушивают в роторном испарителе, рас¬
творяют в малом количестве метанола и метилируют до эфира раствором диазометана.
Продукты реакции затем разделяют хроматографически и идентифицируют обычными
спектрографическими методами, используемыми для молекулярной характеризации ве¬
ществ (УФ-, ИК-, масс- и ЯМР-спектрометрия); наиболее распространенным методом
является газовая хроматография, сопряженная с масс-спектроскопией (ГХ—МС).
Для исследования расщепления органического вещества использовались и другие
окислители. Нейрод и Шнитцер (Neyroud и Schnitzer, 1974) а затем Гриффит и Шнитцер
(Griffith и Schnitzer, 1976) исследовали продукты щелочного окисления гуминовых и фуль¬
вокислот оксидом двухвалентной меди. Окисление 1 г гуминового вещества, смешанного
с 100 мл 2 М раствора NaOH и 5 г СиО, выполняется в автоклаве при 170 °С; окончание
реакции почти такое же, как при окислении перманганатом (см. выше в данном разделе).
1 Делительной колонке. — Примен. наун. ред.
Глава 12. Характеризация гуминовых веществ
3S7
Другие методы включают щелочное окисление нитробензолом (Morrison, 1963), а так¬
же окисление азотной кислотой (Hansen и Schnitzer, 1967), гипогалогенитом (Chakrabartty
и др., 1974) и перуксусной кислотой (Schnitzer и Skinner, 1974). Гриффит и Шнитцер
(Griffith и Schnitzer, 1989) опубликовали обзор методов окислительной деструкции, одно¬
го из аналитических инструментов для исследования гуминовых веществ (Hatcher и др.,
2001).
Восстановительная фрагментация
Самым широко используемым реагентом является амальгама натрия (Mendez^ Stevenson,
1966; Stevenson и Mendez* 1967; Piper и Posner, 1972), но использовались и другие вос¬
становители, такие как порошок цинка (Hansen и Schnitzer, 1969). Стивенсон (Stevenson,
1989) представил обзор методов восстановительной фрагментации.
Другие методы химического расщепления
Кипячение гуминовых кислот в воде высвобождает полисахариды и малые количества
фенольных кислот и альдегидов, полипептидов, алканов и жирных кислот (Neyroud
и Schnitzer, 1975).
Кислотный гидролиз и кипячение (с обратным холодильником) растворяет от 1/3
до 1/2 органического вещества большинства почв (Schnitzer, 1978), но этот метод чаще
всего применялся для изучения органических форм азота (см. раздел 14.2.1); Андерсон
с сотр. (Anderson и др., 1978) исследовали продукты кислотного гидролиза гуминовых
и фульвокислот, растворимые в эфире.
Щелочной гидролиз также использовали, как метод расщепления для изучения гу¬
миновых молекул. Нейрод и Шнитцер (Neyroud и Schnitzer, 1975) подвергали гуминовые
материалы четырем последовательным циклам гидролиза под действием 2 н. раствора
NaOH в автоклаве при 170 °С в течение 3 ч. Выделение и идентификация продуктов ги¬
дролиза выполнялись методами, аналогичными другим методам расщепления (см. под¬
раздел «Окислительная фрагментация»). Парсонс (Parsons, 1989) анализировал основные
механизмы фрагментации кислотного и щелочного гидролиза органических веществ.
Метод, разработанный для деполимеризации углей (Ouchi и Brooks, 1967), был при¬
менен для расщепления гуминовых кислот (Jackson и др., 1972): реакция с фенолом
в присутствии я-толуолсульфоновой кислоты в качестве катализатора. Хейз и О’Калаган
(Hayes и O’Callaghan, 1989) опубликовали обзор методов расщепления под действием
фенола и сульфида натрия. Чешир с сотр. (Cheshire и др., 1968) исследовали влияние на
гуминовые кислоты щелочного сплавления после кипячения с кислотой.
Термическое расщепление
Термогравиметрия (ТГ), дифференциальная термогравиметрия (ДТГ), дифференци¬
альный термический анализ (ДТА) и изотермический (равнотемпературный) нагрев
использовались для исследования механизма термического разложения гуминовых
материалов (Schnitzer, 1978). Шнитцер и Хофманн (Schnitzer к Hoffmann, 1964) иссле¬
довали химическую эволюцию гуминовых и фульвокислот под действием температур
до 540 °С; наблюдаемые ими кривые дифференциальной термогравиметрии показаны
на рис. 12.9.
Кодама и Шнитцер (Kodama и Schnitzer, 1970) использовали дифференциальный тер¬
мический анализ для исследования механизма термического разложения фульвокислот.
Чен с сотр. (Chen и др., 1978b) также применяли этот метод для сравнения физико-хи¬
мических свойств гуминовых и фульвокислот из средиземноморских почв (рис. 12.10).
358
Часть 2, Органический анализ
Рис. 12.9. Кривые дифференциальной термогравиметрии (Schnitzer и Hoffmann, 1964) для
органических материалов из подзола: а — поверхностный горизонт 02; b — глубокий горизонт Bh.
Сочетание пиролиз — газовая хроматография было разработано Кимбером и Сирлом
(Kimbern Searle, 1970) для исследования органического вещества почвы. На рис. 12.11
показана одна из хроматограмм продуктов пиролиза фульвокислот из глубокого гори¬
зонта Bh подзола, полученных Мартином (Martin, 1976).
Комбинация пиролиз — масс-спектрометрия, описанная Мейзелааром с сотр. (Meuze-
laar и др., 1973), применялась некоторыми авторами для гуминовых соединений
(Meuzelaarnwp., 1977; Saiz-Jimenezvuip1979). Пробу гуминовых веществ диспергирова¬
ли в растворе щелочи или в метаноле, накрывали, помещали на ферромагнитную катушку
и разлагали при 510 °С. Пример масс-спектра продуктов пиролиза показан на рис. 12.12.
В обзорах Брейсвела с сотр. (Bracewellи др., 1989) и Шультена (Schulten, 1996) приведены
дополнительные данные о продуктах термического разложения гуминовых материалов.
12.3.4. Ядерный магнитный резонанс (ЯМР)
Основные положения
Большинство атомных ядер вращаются вокруг своей оси и поэтому обладают угловым
моментом, который выражается формулой [И/2ш][1(1+ 1)]1/2, где А - постоянная Планка
и /— спиновое квантовое число. Спиновое квантовое число может принимать значения
0, 1/2, 1, 3/2, 2 и т. д,, в зависимости от природы ядра: для *Н и 13С /= 1/2, а для 12С —
I- 0. Поскольку ядро обладает электрическим зарядом, его вращение создает магнитное
поле. И наоборот, если ядро помещено в магнитное поле Я0, оно может ориентироваться
в одном из (2/+ 1) направлений, связанных с направлением поля. Каждое направление
соответствует определенному энергетическому состоянию, и возможно возникновение
резонанса между энергетическими состояниями при использовании электромагнитного
излучения частоты v, равной:
v = yHJ2n, (12.1)
где гиромагнитное отношение у — константа, зависящая от типа ядра.
Атом водорода (/= 1/2) дает (2/+ 1) = 2 возможных ориентации ядра, и его резонанс
может быть обнаружен. С другой стороны, ориентация большинства ядер не может быть
обнаружена: для 12С (2/+ 1) =1, т. е. существует только одна возможная ориентация ядра
Глава 12. Характеризация гуминовых веществ
359
Рис. 12.10. Дифференциальный термический анализ гуминовых (слева) и фульвокислот (справа),
экстрагированных из средиземноморских почв (Chen и др., 1978): а — глинистая бурая почва;
b — песчаная бурая почва; с — глинисто-илистая опесчаненная красноцветная почва; d— песчано¬
илистая красноцветная почва; е — илисто-песчаная красноцветная почва; 1 — песчано-илистая
бурая почва
360
Часть 2. Органический анализ
0 4 8 12 16 20 24
Время, мин
Рис. 12.11. Хроматограмма продуктов пиролиза фульвокислот, экстрагированных щелочью из
глубокого горизонта Bh подзола, полученных Мартином (Martin, 1976)
Почва до экстракции
vo
О
Рис. 12.12. Масс-спектры продуктов пиролиза, полученные Сейз-Джименезом с сотр. (Saiz-
Jimenez и др., 1979) для пробы коричневой почвы на гранитной породе (typical Xerochrept);
фракция гуминового остатка была получена щелочной экстракцией почвы с последующей
экстракцией гуминовых кислот и полным разложением силикатов смесью HF-HCI
Глава 12. Характеризация гуминовых веществ
361
в магнитном поле и возникновение резонанса невозможно. Только изотоп углерода 13С
доступен для исследования методом ЯМР, но он гораздо менее распространен в приро¬
де, чем изотоп 12С.
Наконец, наиболее важными факторами оценки обнаруживаемости элемента в почве
или почвенном экстракте являются спиновое квантовое число /и гиромагнитное отно¬
шение у, а также распространенность элемента п и относительная распространенность
исследуемого изотопа N. В табл. 12.1 приведены значения обнаруживаемости атомных
ядер в почве, рассчитанные Вилсоном (Wilson, 1981) по формуле yW/(/+ 1 )п. Эта табли¬
ца не представляет общую обнаруживаемость элемента, а только его обнаруживаемость
в почве или материалах органического вещества почвы. Таким образом, даже элемент,
чувствительный к ЯМР, такой как 31Р, трудно выявить из-за его малого содержания.
Таблица 12.1. Относительная обнаруживаемость (00) ядер органического вещества почвы
методом ЯМР (Wilson, 1981)
Ядро
оо
iH.
102
27А1
10'
23Na
10°
'Н6
10°
55Мп
ю-'
29Si
ю-'
,3С
10-3
uN
ю-3
39К
ю-3
170
ю-3
25Mg
ю-3
67Zn
ю-3
з.р
ю-4
43Са
ю-4
57 Fe
ю-5
ISN
1<Н
8 Очень непостоянный, зависит от содержания воды и pH.
6 Только Н. органического вещества почвы.
Традиционный метод ЯМР заключается в радиочастотном сканировании образца
(ЯМР с непрерывной разверткой) в фиксированном магнитном поле (или наоборот)
и в записи резонанса, когда частота излучения соответствует частоте ядерного перехо¬
да согласно уравнению (12.2). ЯМР с преобразованием Фурье часто позволяет быстрее
получить спектры высокого качества. В ЯМР Фурье-спектроскопии1 наблюдается по¬
ведение ядра под воздействием коротких и интенсивных импульсов излучения. Все ядра
резонируют одновременно, и получающийся спектр сигнала как функция от времени
(спад свободной индукции (ССИ\free induction decay, FID)) не очень информативен для
химиков. Необходима обработка сигнала преобразованием Фурье для получения более
легко интерпретируемых спектров, аналогичных тем, что получаются в ЯМР с непре¬
рывной разверткой. Для повышения чувствительности сначала необходимо накопить на
1 Импульсный ЯМР. — Примеч. науч. ред.
362 Часть 2. Органический анализ
компьютере большое количество сигналов ССИ и затем использовать преобразование
Фурье для суммы.
Метод ЯМР предоставляет мало полезной информации по структурной химии,
если определять только спиновые переходы ядер на частотах, соответствующих урав¬
нению (12.1). В самом деле, электронное окружение ядра защищает его от приложен¬
ного магнитного поля (Но), и действительное магнитное поле при постоянной частоте
(или частота при постоянном поле), необходимое для ядерного резонанса, зависит от
эффективности защиты ядра. В органических молекулах функциональные группы име¬
ют различные распределения электронов, и частоты резонанса каждого ядра этих групп
слегка сдвигаются в зависимости от природы функциональной группы. Это делает воз¬
можным идентифицировать эти группы. На практике сдвиг частоты определяется срав¬
нением его со стандартом, обычно тетраметилсиланом (ТМС), и величину химического
сдвига (8) рассчитывают как «химический сдвиг частоты» относительно «частоты стан¬
дарта». Поскольку значения химических сдвигов малы, величины 8 обычно выражают
в млн-1:8 (ppm).
Другим важным параметром является релаксация. После возбуждения ядра проходит
некоторое время, прежде чем оно возвращается в основное состояние энергии. Теоре¬
тически это возвращение происходит двумя путями: взаимодействием с молекулярной
решеткой или обменом спиновой энергии с таким же ядром. Константы времени, соот¬
ветствующие этим двум процессам, называются временем спин-решетчатой релаксации
и временем спин-спиновой релаксации (Т1 и Т2, соответственно). Это явление имеет
большое значение для химии почвы. В ЯМР-исследованиях с использованием изотопа
13С оно может влиять на результаты количественного измерения содержания углерода
в функциональных группах.
Развитие ЯМР-метода позволило использовать его для исследований органического
вещества почвы. Первоначально почвенные экстракты анализировали в растворе мето¬
дом 1Н ЯМР, потом стали применять 13С ЯМР; сначала качественные, а затем попыта¬
лись выполнить количественные наблюдения. Исследуемый материал должен был быть
растворим в растворителе, который не дает резонанса с исследуемым элементом (напри¬
мер, при исследованиях изотопа *Н использовали D20 вместо Н20), но теперь в этом нет
необходимости (Wilson, 1981). В более поздних работах использовали методы ЯМР для
твердой фазы. Вилсон (Wilson, 1981, 1987) опубликовал обзор этих методов и их приме¬
нение для исследования органического вещества почвы. Стилинк с сотр. (Steelink и др.,
1989) получили дополнительные данные по 1Н и 13С ЯМР гуминовых веществ в растворе.
Вилсон (Wilson, 1989) и Тейт (Tate, 1998) опубликовали дополнительную теоретическую
информацию по твердотельному ЯМР и его использованию для исследования гумино¬
вых веществ. Симпсон (Simpson, 2004) применил сочетание ЯМР и методов разделения.
Метод 15N ЯМР использовали для изучения азотного цикла (Thom и Mikita, 2000).
Исследования гуминовых веществ методом1Н ЯМР
Шнитцер и Бартон (Schnitzer и Barton, 1963) первыми наблюдали ЯМР-спектры орга¬
нических экстрактов почв. Они использовали непрерывное сканирование для фракций
метилированных фульвокислот, которое давало относительно мало структурной инфор¬
мации. Впоследствии многие авторы применяли этот вариант метода 1Н ЯМР. Лентц
с сотр. (Lentz и др., 1977) получили спектры лучшего качества, используя преобразова¬
ние Фурье. Вилсон с сотр. (Wilson и др., 1978) использовали методы с преобразованием
Фурье в сильном магнитном поле с очень высокой частотой и тем самым увеличили ко¬
личество информации, получаемой из *Н ЯМР-спектров (рис. 12.13).
Глава 12. Характеризация гуминовых веществ
363
Рис. 12.13. 1Н ЯМР-спектр после преобразования Фурье (270 МГц), полученный Вилсоном
с сотр. (Wilson и др., 1978) для гуминовых материалов, экстрагированных 0,1 М раствором NaOH
из илистой пластической глины из Вакануи. Пульсацию 10 мс с углом наклона 85° использовали
для визуализации спектрального окна 3,6 кГц в 8К точек измерения
Исследования гуминовых веществ в растворе методом 13С ЯМР
Метод 13С ЯМР имеет некоторые преимущества перед методом 1Н ЯМР для анализа
строения молекул: он дает непосредственную информацию о структурном скелете, ко¬
торая позволяет наблюдать функциональные группы без протонов, такие как кетоны.
Ядра углерода дают ббльшие химические сдвиги, что позволяет наблюдать более тонкие
различия в молекулярных структурах. Сигналы могут также содержать более узкие пики,
что снижает риск маскировки одних пиков другими. С другой стороны, недостаток ме¬
тода 13С ЯМР связан с малым содержанием изотопа 13С (всего 1,1% от общего углерода),
затрудняющим распознавание сигналов (Wilson, 1981).
Для удовлетворительного обнаружения необходимо оптимизировать условия записи
13С спектров. Эти спектры почти всегда получают в условиях развязки от протонов, ко¬
торые вызывают увеличение 13С резонанса (ядерный эффект Оверхаузера — ЯЭО). ЯЭО
и теорию диполярной 13С—*Н релаксации в протон-развязанных 13С ЯМР-спектрах ма¬
кромолекул исследовали Доддрелл с сотр. (Doddrellи др., 1971).
Первоначально использование этих методов для органического вещества почвы не
привело к успеху (Schnitzern Neyroud, 1974). Впоследствии несколько авторов усовер¬
шенствовали этот метод, и Ньюман с сотр. (Newman и др., 1980) добились значительного
улучшения качества спектров (рис. 12.14), оптимизируя рабочие условия, в частности
промежутки между импульсами излучения. Оптимальный интервал, равный 0,2 с со¬
ответствовал времени накопления сигналов релаксации от 2 • 105 90°-ных импульсов
длительностью 23 мкс. Спектры были получены на приборе Varian FT- 80/1 (рабочая ча¬
стота 20 МШ для 13С с развязкой от протонов при 80 МГц). Необходимо накопить от
4000 (образцы угля) до 136 000 (образцы гуминовых кислот почвы) спектров спада сво¬
бодной индукции и усилить их, используя фильтр с временной константой 20 мс, перед
преобразованием Фурье. Подготовка проб перед анализом включала суспендирование
300 мг гуминовых кислот в 2 см3 0,5 М раствора NaOH при 20 °С в течение суток с по-
364
Часть 2. Органический анализ
„д-и. j
200 100 0
6/р.р.т
Рис. 12.14. Развязанный 13С ЯМР-спектр гуминовых кислот в растворе, полученный Ньюманом
с сотр. (Newman и др., 1980) с использованием оптимизированных параметров накопления
(условия описаны в тексте)
следующим ультрацентрифугированием (84 500 g при 4 °С). После разбавления D20 до
50% записывали 13С ЯМР-спектр в ампулах диаметром 10 мм.
Ньюман и Тейт (Newman и Tate, 1984) использовали условия, аналогичные описанным
выше, для характеристики гуминовых веществ из щелочных экстрактов почв; общее
время, необходимое для накопления спектров, составляло от 10 до 50 ч.
Престон и Шнитцер (Preston и Schnitzer, 1984) исследовали влияние типа экстракции
(кислотная или щелочная экстракция) и химических изменений экстрагируемого мате¬
риала (метилирование, гидролиз под действием 6 М НС1 с последующим метилирова¬
нием или без него) на 13С ЯМР-спектры гуминовых материалов из четырех типов почв.
Препараты растворяли в дейтерированных растворителях (CDC13 для метилированных
материалов, 0,5 М раствор NaOD в D20 для неметилированных соединений). Хими¬
ческие сдвиги измеряли относительно 3-триметилсилилпропионата натрия (ТСП) для
растворов в тяжелой воде (D20) и тетраметилсилана (ТМС) в растворах в дейтерирован-
ном хлороформе (CDC13). Спектры были получены на спектрометре Bruker WM250 с ис¬
пользованием 10-миллиметровых измерительных ампул и с прерыванием *Н развязыва¬
ния, за исключением времени накопления (обратная развязка с выключением, Freeman
и др., 1972). Некоторые спектры аналогичны спектрам, показанным на рис. 12.14.
Исследования методом твердотельного 13С ЯМР
Экстракция органических веществ (см. разделы 11.2.1 и 11.3.1) может приводить к изме¬
нениям молекул, в частности при использовании растворов сильных щелочей в качестве
экстрагирующих агентов. Более того, большая часть материала остается в неэкстраги-
руемом остатке гумина. Этот недостаток можно преодолеть, исследуя органическое ве¬
щество in situ.
Однако применение традиционной ЯМР-спектроскопии к цельной почве дало лишь
широкие и диффузные сигналы: многочисленные диполь-дипольные взаимодействия
365
Глава 12. Характеризация гуминовых веществ
давали сигналы, содержащие неясную информацию о химических сдвигах; более того,
времена спин-решеточной релаксации (Т1) были слишком велики для накопления сиг¬
налов спада свободной индукции, необходимых для получения количественной инфор¬
мации. Прогресс в теории метода ЯМР {Pines и др., 1973) позволил впоследствии ре¬
шить эту проблему и наметить дальнейшие разработки. В ЯМР с кросс-поляризацией
(КП 13С ЯМР) протоны развязаны от ядер 13С и используются для увеличения скорости
релаксации ядер 13С. Ширину пика сигналов можно уменьшить до такой степени, что
функциональные группы можно будет частично идентифицировать.
Для выявления форм углерода в цельной почве методом КП 13С ЯМР необходимы по¬
чвы с высоким содержанием органического вещества (6% С, согласно Wilson и др., 1981).
Это ограничение было постепенно ослаблено благодаря улучшению качества приборов,
а также предварительному использованию методов физического фракционирования,
таких как описанные в главе 9, однако применение спектрального анализа ограничива¬
ется фракциями с наибольшим содержанием органики {Barron и др., 1980).
Метод ЯМР при вращении образца под магическим углом (ВМУ ЯМР), который был
первоначально описан Лоу {Lowe, 1959) и затем применен для исследования полимеров
Шеффером и Стейскалом {Schaefer и Stejskal, 1976), также может помочь в решении про¬
блем, связанных со спектральным разрешением, и получении спектров непосредствен¬
но для образцов почв; в этом методе образец быстро вращается под углом 54°44г, и это
позволяет уменьшить анизотропные эффекты и выбрать изотропные сдвиги. Этот метод
часто используется совместно с кросс-поляризацией под общим названием КП—ВМУ
13СЯМР.
Для исследования почв многие авторы предпочитают применять методы ЯМР к пред¬
варительно концентрированным гуминовым материалам, обычно гуминовым или фуль-
вокислотам. Работы Ньюмана с сотр. {Newman и др., 1980) четко выявили различие в раз¬
решении, которое еще существует между методами 13С ЯМР в растворах и КП 13С ЯМР
в твердой фазе (рис. 12.15, а).
Благодаря совершенствованию этого метода спектры КП—ВМУ 13С ЯМР, получен¬
ные Герасимовичем и Вайлером {Gerasimowics и Byler, 1985) для гуминовых веществ, по¬
казали лучшее разрешение (рис. 12.15, Ь), чем полученное Ньюманом с сотр. {Newman
и др., 1980). Фрейнд и Людеманн {Freundи Liidemann, 1989) осуществили показательное
сравнение методов 13С ЯМР в растворе и КП-ВМУ13С ЯМР, которое продемонстриро¬
вало, что эти методы характеризуются сравнимым разрешением; кроме того, сравнение
спектров, полученных для дерново-карбонатной почвы (4,6% углерода), и ее экстрак¬
тов гуминовых веществ и гуминового остатка из этой почвы дали удовлетворительные
результаты (рис. 12.15, с). Конт с сотр. {Conte и др., 1997, а) рекомендовали два метода
(жидкофазный ЯМР и твердотельный КП-ВМУ 13С ЯМР) для изучения органическо¬
го вещества почв. Современный КП—ВМУ 13С ЯМР позволяет проводить наблюдения
органических веществ в естественном окружении (без фракционирования), если по¬
чвы содержат достаточно углерода. Это было продемонстрировано Кинчешом с сотр.
{Kinchesh и др., 1995) на почвах Роттамстеда (Великобритания). Другие работы, такие
как исследования Конта с сотр. {Conte и др., 1997, Ъ) на вулканических почвах, исполь¬
зовали КП—ВМУ 13С ЯМР на экстрагированных гуминовых веществах.
Количественный анализ методом ЯМР
Существуют различные методы количественной обработки данных ЯМР по гуминовым
веществам. Самые первые из них относятся к исследованиям угля и углеподобных ма¬
териалов.
366
Часть 2. Органический анализ
Рис. 12.15. Преимущества методов твердотельного 13С ЯМР: а — спектры КП 13С ЯМР, полученные
Ньюманом с сотр. (Newman и др., 1980) для твердого угля, твердой гуминовой кислоты (ГК)
и раствора ГК; b — спектры КП — ВМУ 13С ЯМР, полученные Герасимовичем и Вайлером
(Gerasimowics и Byler, 1985) для ГК из илов на разных стадиях компостирования; с — спектры
КП — ВМУ 13С ЯМР, полученные Фрейндом и Людеманном (Frund и Ludemann, 1989) для дерново¬
карбонатной почвы, а также экстрактов ФК и ГК и гуминового остатка из нее
Глава 12. Характеризация гуминовых веществ
367
Метод Брауна и Ладнера (Brown и Ladner, 1960) позволяет оценивать степень аро¬
матичности этих углеродистых материалов на основе спектра *Н ЯМР. Вилсон (Wilson,
1981) предложил адаптацию этого метода для исследования гуминовых материалов.
Степень ароматичности fa чаще всего оценивают количественной обработкой сиг¬
налов 13С ЯМР. Однако методы для прямого количественного анализа различных спек¬
тральных пиков следует использовать с осторожностью. Атомы 13С различных функ¬
циональных групп обладают различными временами релаксации ядер; ядра с самыми
короткими временами релаксации вносят меньший вклад в общий спектр, чем ядра
с продолжительными временами релаксации. Кроме того, благодаря развязке от прото¬
нов ЯЭО приводит к увеличению сигнала от ядер другой природы (Wilson, 1981). Однако
некоторые исследования показали, что эти два фактора не оказывают существенного
влияния на прямое измерение степени ароматичности (Newman и др., 1980).
Фрейнд и Людеманн (Friind и Liidemann, 1989) усовершенствовали количественный
анализ гуминовых материалов. Разработанный ими метод позволяет проводить одно¬
временные измерения содержания углерода карбоксильных, ароматических, углевод¬
ных и алифатических фрагментов. Смерник и Оадес (Smemik и Oades, 2000а) подчерки¬
вали эффект влияния парамагнитных примесей и степени чистоты HF для обработки
(Smemik и Oades, 2000b). Смерник и Оадес (Smemik и Oades, 2003) и затем Мосер и Ле-
фебвр (Moser и Lefebvre, 2004) исследовали новые способы улучшения количественного
анализа органического вещества почвы методом 13С ЯМ Р. Конт с сотр. (Conte и др., 2004)
опубликовали обзор современного состояния КП—ВМУ 13С ЯМР-спектроскопии при¬
родных гуминовых веществ.
12.3.5. Флуоресцентная спектроскопия
Хотя этот метод находит меньшее применение, чем видимая, УФ- (см. раздел 12.2.3) или
ИК- (см. раздел 12.2.4) абсорбционная спектрометрия, флуоресцентная спектрометрия
была опробована несколькими авторами в качестве дополнительного метода для харак¬
теристики гуминовых веществ.
Левеск (Levesque, 1972) использовал этот метод для изучения гуминовых комплексов
железа и фосфора. Спектр излучения фульвокислоты содержит основной пик, положе¬
ние которого изменяется от 500 к 520 нм, при изменении длины волны возбуждения от
400 до 468 нм. Поскольку спектры излучения обычно дают мало информации, вместо них
использовали спектры возбуждения. Длину волны излучения фиксировали в диапазоне
500-520 нм, а длина волны возбуждения непрерывно изменялась между 250 и 500 нм.
Гош и Шнитцер (Ghosh и Schnitzer, 1980а) наблюдали в спектрах гуминовых веществ
две хорошо различаемых полосы возбуждения, при 465 и 360 нм, а также снижение ин¬
тенсивности флуоресценции при увеличении ионной силы и снижении pH; их данные
были впоследствии использованы для расчета констант диссоциации гуминовых кислот
(Goldberg и др., 1987).
Бачелье (Bachelier, 1981) дополнил эти результаты точным описанием спектров воз¬
буждения для большого количества почв; он отметил наличие семи пиков (включая
два редких), отвечающих за искажение склона большого пика при 465 нм и двух пиков
(включая один редкий) на большом пике при 360 нм.
Бачелье также исследовал флуоресценцию нескольких типов гуминовых кислот, раз¬
деленных на геле Sephadex G25: (1) гуминовые кислоты большой молекулярной массы,
элюированные с головы колонки (грубые гуминовые кислоты, гГК) дают слабый пик
флуоресценции; (2) следующая полоса менее конденсированных желто-коричневых гу¬
миновых кислот дает яркую желтую флуоресценцию при облучении УФ-светом (флу¬
368
Часть 2. Органический анализ
оресцентные гуминовые кислоты, фГК); (3) после этой полосы иногда обнаруживают
менее флуоресцирующие соединения, называемые высшими гуминовыми кислотами
(вГК); (4) промежуточные гуминовые кислоты (пГК) иногда находят между полосами
гГК и фГК. На рис. 12.16 показаны некоторые спектры возбуждения, полученные для
различных гуминовых веществ. Наблюдения Бачелье подтверждают данные Левеска
о малой флуоресценции гуминовых комплексов большой молекулярной массы.
Разделение гуминовых кислот
гель-проникающей хроматографией
X возбуждения X возбуждения
Рис. 12.16. Спектры возбуждения флуоресценции гуминовых веществ, наблюдаемые на полосе
испускания при 509-515 нм (Bachelier, 1981): ФК — фульвокислоты из различных образцов почв;
ГК — различные фракции гуминовых кислот, разделенные гель-хроматографией (см. схему
фракционирования над графиком)
Глава 12. Характеризация гуминовых веществ
369
Бачелье (Bachelier, 1984) использовал флуоресцентную спектрометрию для определе¬
ния степени конденсации гуминовых кислот. Наяк с сотр. (Nayak и др., 1985) исследова¬
ли флуоресценцию гуминовых кислот в различных растворителях. Метод поляризации
флуоресценции позволял получить дополнительную информацию по агрегации и кон¬
формации гуминовых молекул и был использован Лапеном и Зейтцем {Lapen и Seitz,
1982) для этой цели в исследовании фульвокислот.
12.3.6. Спектроскопия электронного парамагнитного резонанса (ЭПР)
Когда молекулы, содержащие неспаренные электроны, попадают в магнитное поле, маг¬
нитные моменты этих электронов взаимодействуют с приложенным полем, что приво¬
дит к расщеплению энергии каждого неспаренного электрона на два дискретных уров¬
ня. Это явление называется электронным спиновым резонансом (ЭСР), которое иногда
еще называют электронным парамагнитным резонансом (ЭПР)1. ЭСР-спектроскопия
использует излучение электромагнитного возбуждения в спектральном диапазоне ми¬
кроволн для исследования соединений, содержащих неспаренные электроны, прежде
всего свободные радикалы.
Энергия неспаренного электрона в магнитном поле определяется уравнением
2T = -gP^tf0,
где g - коэффициент спектрального расщепления, который равен 2,0023 для свободного
электрона; р - магнетон Бора; Af — компонент углового момента спина в направлении
оси z приложенного магнитного поля, который может принимать дискретные значения
+1/2 и “1/2; Н0 - сила магнитного поля. Для данного значения Н0 разница в энергии АЕ
между двумя состояниями спина электрона составляет
А£=*РЯ0.
Явление резонанса происходит, когда эта энергия равна энергии приложенного поля
AE = hv,
где h - постоянная Планка; v - частота.
В своей пионерской работе Рекс {Rex, 1960) использовал ЭСР-спектроскопию для
того, чтобы подчеркнуть радикальную природу гуминовых веществ. Маккарти и Райс
{MacCarthy и Rice, 1985) перечислили некоторые работы по использованию ЭСР-
спектроскопии для исследования гуминовых веществ. Спектры гуминовых веществ
обычно дают простые сигналы, которые идентифицируются по их положению и ширине
(рис. 12.17). Линии сверхтонкой структуры, которые иногда присутствуют в некоторых
молекулах, обычно отсутствуют в спектрах гуминовых веществ.
Шнитцер и Скиннер {Schnitzer и Skinner, 1969) показали для десяти образцов гуми¬
новых материалов, что наиболее изменяемым параметром из тех, которые можно уста¬
новить из этих спектров, является концентрация спинов. Этот вывод был получен при
сравнении с калибровочным стандартом с учетом того, что количество радикалов про¬
порционально площади пика. В гуминовых веществах эта концентрация составляет
примерно 1018 спинов/г. Шнитцер и Скиннер исследовали влияние некоторых факторов
на концентрацию спинов в гуминовых веществах (химические превращения, нагрева¬
ние), а также соотношения между другими параметрами (молекулярная масса, отноше¬
ние Е4:Е6, количество молекул данной массы на свободный радикал) и концентрацией
1 В отечественной литературе чаще применяют этот вариант. — Примен. наун. ред.
370
Часть 2. Органический анализ
Диапазон сканирования, Гс
Рис. 12.17. Спектр электронного спинового резонанса фульвокислоты (Schnitzer и Skinner, 1969)
спинов. Риффалди и Шнитцер (Riffaldi и Schnitzer, 1972) исследовали влияние экспери¬
ментальных условий на ЭСР-спектры гуминовых веществ, чтобы выявить ошибки, свя¬
занные со слишком поспешной трактовкой этих спектров. Маккарти и Райс (MacCarthy
и Rice, 1985) обсуждали относительную скудность информации, предоставляемой ЭСР-
спектрометрией для исследования гуминовых веществ. Однако последующие успехи
технологии изменили ситуацию. Например, Сенеси с сотр. (Senesi и др., 1989) получили
более подробные спектры, чем предыдущие авторы, а Сенеси (Senesi, 1990) проанали¬
зировал состояние и перспективы метода ЭСР и его применения к химии почвы. Сааб
и Марти-Нето (Saab и Martin-Neto, 2004) исследовали семихинонные свободные радика¬
лы гуминовых веществ методом ЭСР.
12.3.7. Определение молекулярной массы и размеров молекул
Основные положения
Определение молекулярной массы — традиционная процедура структурной химии.
В качестве дополнения к элементному анализу молекулярная масса позволяет найти
эмпирическую формулу для интересующего соединения до определения структурной
формулы (функциональных групп) спектроскопическими и химическими методами.
Попытки определения массы и размера молекул гуминовых веществ предпринимались
неоднократно и рассматривались в нескольких обзорах (например, Orlov и др., 1975;
Stevenson. 1982; Wershaw и Aiken, 1985; Buurman, 2001). Аналогичные попытки предпри¬
нимались для гуминовых веществ в воде (Yu-Ping Chin и др., 1994; Yamada и др., 2000)
и атмосфере (Samburova и др., 2005). Однако полученные результаты сильно различа¬
лись, во-первых, из-за использованных методов и, во-вторых, потому, что, как подчер¬
кивалось авторами этих обзоров, гуминовые вещества являются не дискретными хи¬
мическими единицами, а сложными смесями полидисперсных органических веществ
в большом диапазоне молекулярных масс.
371
Глава 12. Характеризация гуминовых веществ
Тем не менее, проблема определения молекулярной массы смесей исследовалась в те¬
чение многих лет. В 1935 г. Лансинг и Креймер (Lansing и Кгаетег, 1935) указывали, что
самые распространенные методы позволяют определить средние молекулярные массы, ко¬
торые зависят от используемого метода измерения, поэтому эти средние массы не всегда
могли быть сравнимы. В настоящее время используют три типа средних молекулярных
масс, соответствующих трем типам методов измерения.
Среднечисловая молекулярная масса, Д
Среднечисловую молекулярную массу определяют методами, измеряющими количе¬
ство молекул (обычно в разбавленном растворе) независимо от их размера. Она выража¬
ется следующей формулой:
Д — Ея, Д/Ея,,
где п( — количество молекул массой Д
Величину Д определяют всеми методами, соответствующими термодинамическому
свойству, связанному с количеством молекул в растворе (коллигативное свойство): сни¬
жение давления насыщенного пара, понижение точки замерзания, повышение точки
кипения (законы Рауля), осмотическое давление.
Средневзвешенная молекулярная масса, Mw
Средневзвешенную молекулярную массу определяют методами, применимыми для
различных материалов, такими как рассеяние света и осаждение ультрацентрифугиро¬
ванием; она выражается формулой:
Mw = Е^Д/Е*, = ЕяД2/ЕяД,
где wt — массовая доля каждого компонента.
Z-средняя молекулярная масса Д
Z-среднюю молекулярную массу также рассчитывают по результатам ультрацентри¬
фугирования и выражают формулой:
Д = ЕиДУЕиД, = ЕиД3/ЕлД2.
В монодисперсной системе Д = Mw — Д, но для полидисперсной системы это вы¬
ражение недействительно; в этом случае наименьшее значение обычно имеет средне¬
числовая молекулярная масса, представляющая, как правило, меньшие молекулярные
массы, тогда как Mw представляет самые тяжелые частицы смеси (Wershaw и Aiken,
1985). Орлов с сотр. (Orlov и др., 1975) предполагали, что величины Mw лучше корре¬
лируют с известными свойствами гуминовых веществ. В гетерогенных системах Д >
Mw > Д; отношение Д; Д обычно используют для расчета степени .полидисперсности
(Stevenson, 1982).
В дополнение к методам определения средней молекулярной массы другую группу
методов (например, гель-фильтация, ультрафильтрация и малоугловое рассеяние рент¬
геновских лучей) используют для определения размеров молекул. В этих методах мо¬
дельные соединения известной молекулярной массы и известного состава используют
для определения молекулярной массы гуминовых веществ, но есличони слишком от¬
личаются от гуминовых веществ, могут возникнуть сложности (Wershaw и Aiken, 1985).
372
Часть 2. Органический анализ
Методы определения размера молекул
Гель-проникающая хроматография
Методы гель-вытеснительной (или гель-проникающей) хроматографии описаны в раз¬
делах 12.2.1 и 12.3.1. Следует отметить, что Рейтер и Пердю (Reuter и Perdue, 1981) обна¬
ружили очень большое различие между ожидаемыми молекулярными массами фракций
гуминовых веществ, выделенных на геле Sephadex, и измеренными среднечисленными
молекулярными массами. Плеханов (Plechanov, 1983) использовал систему «Sephadex
1Я60 — диметилформамид — уксусная кислота» для определения молекулярной мас¬
сы гуминовых веществ. Нобили с сотр. (Nobili и др., 1989) опубликовали обзор методов
гель-хроматографии для определения размеров молекул гуминовых веществ.
Ультрафильтрация
Ультрафильтрация (фильтрация под давлением через мембрану) также используется для
разделения макромолекул в зависимости от их молекулярной массы. Этот метод анало¬
гичен обратному осмосу, за исключением размера частиц, которые могут быть разделе¬
ны. Обратный осмос отделяет частицы, молекулярный размер которых близок к размеру
молекул растворителя, тогда как ультрафильтрация отделяет частицы, размеры которых
примерно в 10 раз больше, чем размеры частиц растворителя, т. е. от 0,5 мкм (Wershaw
и Aiken, 1985). Существует много мембран различных типов, которые производители
классифицируют по порогу разделения, выраженному в молекулярных массах. Тем не
менее, ультрафильтрация — это метод разделения по размеру молекул, а не молекуляр¬
ной массе. Много полезной информации об этом методе можно найти в обзоре Вершау
и Эйкена (Wershaw и Aiken, 1985).
Рассеяние электромагнитного излучения
Принцип этого метода для светорассеяния был описан Керкером и Милтоном (Кегкег
и Milton, 1968), а Гинье и Фурне (Guinier и Fournet, 1955) — для малоуглового рассея¬
ния рентгеновского излучения. Целесообразно познакомиться с этими публикациями
для всестороннего объяснения явления, а также с работами Вершау и Эйкена (Wershaw
и Aiken, 1985) и Вершау (Wershaw, 1989) об использовании этого метода для исследова¬
ния гуминовых веществ.
Методы определения молекулярной массы
Определение среднечисловой молекулярной массы измерением коллигативных свойств
Коллигативное свойство — это термодинамическое свойство, которое зависит от коли¬
чества частиц в растворе и не зависит от их природы. При бесконечном разбавлении
каждое из этих свойств пропорционально количеству молекул растворенного вещества
в растворе. Классическая теория каждого коллигативного свойства представлена во всех
справочниках по химии и физике. Самыми распространенными методами определе¬
ния молекулярной массы гуминовых веществ являются криоскопия и метод измерения
осмотического давления (осмометрия). В криоскопии фиксируют снижение температу¬
ры при замерзании растворителя в присутствии исследуемого растворенного вещества.
При осмометрическом методе измеряется изменение осмотического давления в резуль¬
тате прохождения растворителя сквозь мембрану из разбавленного раствора в более кон¬
центрированный. Описание этих методов и их применения для исследования гумино¬
вых веществ можно найти в публикациях Стивенсона (Stevenson, 1982), Вершау и Эйкена
(Wershaw и Aiken, 1985) и Эйкена и Питлама (Aiken и Gillam, 1989).
373
Глава 12. Характеризация гуминовых веществ
Ультраценрифугирование
Существуют две различных группы методов ультрацентрифугирования, основанных на
кинетике седиментации и равновесном ультрацентрифугировании. Первую группу ме¬
тодов чаще всего используют для гуминовых веществ (Wershaw и Aiken, 1985), иногда
в дополнение к другим методам фракционирования (Cameron и др., 1972b), хотя равно¬
весное ультрацентрифугирование дает обширную информацию, поскольку позволяет
одновременно определять значения Мп, Mw и Mz {Posner и Creeth, 1972). Теория и прак¬
тика первого метода детально описаны Камероном с сотр. {Cameron и др., 1972b), второ¬
го — Поснером и Гбитом {Posner и Creeth, 1972); в обзоре Свифта {Swift, 1989) приведено
подробное описание всех методов ультрацентрифугирования.
Вискозиметрия
Измерения вязкости могут дать важную информацию о размере и форме молекул. Хо¬
рошо известный вискозиметр Освальда фиксирует время прохождения раствора и рас¬
творителя между двумя точками, отмеченными на приборе. Молекулярную массу можно
оценить на основе измерений вязкости, используя уравнение Штаудингера: [г|] = кМа,
которое связывает характеристическую вязкость г| с молекулярной массой Мпри помо¬
щи двух контролируемых параметров {Ghosh и Schnitzer, 1980b). Обзор методов, основан¬
ных на измерении вязкости, представлен Клаппом с сотр. {Clapp и др., 1989).
Некоторые авторы также считают протонное ФПСП (см. раздел 12.3.1) перспективным
методом для определения молекулярной массы гуминовых веществ {Beckett и др., 1987).
12.3.8. Микроскопические исследования
В некоторых публикациях приведены результаты исследования гуминовых веществ
с использованием оптической микроскопии, просвечивающей электронной микро¬
скопии и сканирующей электронной микроскопии. Первая сложность связана с раз¬
личными изменениями образца в процессе подготовки (например, степень разделения
с неорганическими материалами; молекулярные изменения, связанные с ионной силой
и pH), а вторая сложность относится к условиям наблюдения (нагрев образца). Баче-
лье {Bachelier, 1983) исследовал девять различных типов почв с помощью трех методов:
растворы замороженных гуминовых кислот под бинокулярной лупой, высушенные рас¬
творы гуминовых кислот под просвечивающим электронным микроскопом и растворы
гуминовых кислот, лиофизированные на позолоченной алюминиевой пленке, под ска¬
нирующим электронным микроскопом. В последних случаях проводилась металлиза¬
ция непроводящих веществ перед исследованием.
Чен и Шнитцер (Chen и Schnitzer, 1976) исследовали влияние pH на вид гуминовых кис¬
лот под сканирующим электронным микроскопом, а Стивенсон и Шнитцер {Stevenson
и Schnitzer, 1982) исследовали тот же эффект методом просвечивающей электронной
микроскопии. Чен и Шнитцер {Chen и Schnitzer, 1976) использовали просвечивающую
электронную микроскопию для изучения комплексов металлов с фульвокислотами.
Чен с сотр. {Chen и др., 1978) использовали сканирующую электронную микроскопию
для сравнения гуминовых кислот из среднеземноморских почв. Тан {Tan, 1985) опубли¬
ковал подробные методологические данные по подготовке образцов.
12.3.9. Другие методы анализа
В данной главе описаны основные методы, используемые для структурных исследова¬
ний. Однако для углубления наших знаний о структуре гуминовых веществ и их связи
с минеральными материалами находят свое применение и другие методы.
374
Часть 2. Органический анализ
Методы рентгенографии не ограничиваются малоугловым рассеянием рентгеновско¬
го излучения, описанным в подразделе «Рассеяние электромагнитного излучения» раз¬
дела 12.3.7. Шнитцер (Schnitzer, 1978) использовал рентгеновскую дифракцию, которая
также может предоставить полезную информацию, несмотря на некристаллическую
природу гуминовых веществ.
Что касается электрохимических методов, то Шинозука и Хайано (Shinozuka и Науапо,
1987) описали исследование гуминовых веществ методом полярографии.
Использование Фурье ИК-спектроскопии позволяет увеличить разрешение и снизить
фоновый шум ИК-спектров. Однако эти преимущества могут оказаться неочевидны¬
ми при исследовании гуминовых веществ из-за их молекулярной природы (MacCarthy
и Rice, 1985).
Более современные методы, такие как рентгеновская фотоэлектронная спектроско¬
пия (РФЭС) и мессбауэровская спектроскопия, не нашли широкого применения для
исследования гуминовых веществ. РФЭС, которую также называют электронной спек¬
троскопией для химического анализа (ЭСХА), может использоваться только для твер¬
дых материалов, поскольку для нее необходим глубокий вакуум; она основана на анали¬
зе электронов, испускаемых с внутренних электронных оболочек атомов, подвергнутых
бомбардировке рентгеновскими лучами достаточной энергии; Дефосс и Роуксет (Defosse
и Rouxhet, 1980) применили этот метод для анализа почв.
Мессбауэровскую спектроскопию нельзя непосредственно применять для иссле¬
дования структуры соединений металлов, но она может использоваться для изучения
металл-органических связей, в частности комплексов железо-гуминовых соединений
{Goodman и Cheshire, 1979).
Интересно, будет ли будущий прогресс связан с развитием новых технологий или
с обобщающими исследованиями на молекулярных моделях, как в случае идентифика¬
ции двойной спирали ДНК Ватсоном и Криком (Watson и Creek),
Использованная литература
Молекулярные модели
Hatcher PG, Dria KJ, Sunghwan Kim, Frazier SW (2001) Modem analytical studies of humic substances.
Soil Science, 166, 770-794.
Lawson GJ and Stewart D (1989) Coal Humic Acids. In Humic substances II, search of structure, Hayes
HB., MacCarthy P, Malcolm RL and Swift RS ed., Wiley, 641-686.
Lodygin ED and Beznosikov VA (2003) The ,3C NMR study of the molecular structure of humus acids
from podzolic and bog-podzolic soils. Pochvovedenie, 9,1085-1094.
Piccolo A and Conte P (2003) Comments on “Modem analytical studies of humic substances” by Hatcher
et al. Soil Sci., 168, 73-74.
Schulten HR (1995) The three-dimensional structure of humic substances and soil organic matter studied
by computational analytical chemistry. FreseniusJ. Anal. Chem., 351, 62-73.
Schulten HR and Leinweber P (2000) New insights into organic-mineral particles: composition, proper¬
ties and models of molecular structure. Biol Fertil Soils, 30, 399-432.
Schulten HR and Schnitzer M (1997) Chemical model structures for soil organic matter and soils. Soil
ScL, 162, 115-129.
Skjemstad JO, Clarke P, Taylor JA, Oades JM and McClure SG (1996) The chemistry and nature of pro¬
tected carbon in soil. Aust. J. Soil Res., 34, 251-271.
Schulten HR (2002) New approaches to the molecular structure and properties of soil organic matter:
humic-, xenobiotic-, biological-, and mineral-bonds. In 3rd Symposium on Soil Mineral-Organic
Matter-Microorganism Interactions and Ecosystem Health, Naples-Capri, Italy, 22-26 May 2000.
Violante A, Huang PM, Bollag JM and Gianfreda L. ed. Developments in soil science, volume 28A, 351-381.
375
Глава 12. Характеризация гуминовых веществ
McCarthy Р (2001) The principles of humic substances. Soil Sci., 166, 738-751.
Piccolo A, Celano G, Conte P, Zena A and Spacini R (1999) Adsorption of atrazine on humic substances
of different molecular structure and their hydrolyzed products as modified by pH. In Human and
environmental exposure to xenobiotics Del Re AAM, Brown CD, Capri E, Evans SP and Trcvisan M,
ed., Proceedings of the XI Symposium of Pesticide Chemistry, Cremona, Italy, September 12-15
1999,425-431.
Фракционирование, определение молекулярной массы и размеров молекул
Aiken GR and Gillam АН (1989) Determination of molecular weights of humic substances by colliga-
tive property measurements. In: Humic substances //., Hayes MHB., MacCarthy P, Malcolm RL and
Swift RS ed. Wiley New York, 515-544.
Bailly JR and Margulis H (1968) Etude de quelques acides humiques sur gel de dextrane. Plant and soil,
XXIX, 343-361.
Bailly JR and Tittonel E (1972) Etude de quelques acides humiques sur gel de dextrane (II). Plant Soil,
37, 57-80.
Beckett R, Zhang Jue and Giddings JC (1987) Determination of molecular weight distributions of fulvic
and humic acids using flow field-flow fractionation. Environ. Sci. Technol., 21, 289-295.
Buurman P (2001) Understanding humic substances: advanced methods, properties and applications. Soil
Sci., 166, 950-951.
Cacco G and Maggioni A (1976) Isoelectrophoretic characterization of humic and fulvic acids extracted
from an alpine podzol. Agrochimica, 20,20-28.
Cacco G, Maggioni A and Ferrari G (1974) Electrofocusing: a new method for characterization of soil
humic matter. Soil Biol. Biochem., 6,145-148.
Cameron RS, Swift RS, Thornton BK and Posner AM (1972a) Calibration of gel permeation chromatog¬
raphy materials for use with humic acid. J. Soil Sci., 23,343-349.
Cameron RS, Thornton BK, Swift RS and Posner AM (1972b) Molecular weight and shape of humic
acid from sedimentation and diffusion measurements on fractionated extracts. J. Soil Sci., 23, -342.
Clapp CE, Emerson WW and Olness AE (1989) Sizes and shapes of humic MHB, MacCarthy P. Mal¬
colm RL and Swift RS ed. Wiley New York, 497-514.
Dabin В (1980) Les matures organiques dans les sols tropicaux normalement drainSs. Cah. ORSTOMsir.
Pidol, 18,197-215.
Dabin В and Thomann Ch (1970) Etude comparative de deux mdthodes de ffactionnement des composes
humiques (m£thode Tiurin et mSthode 61ectrophor6tique). ORSTOM ser. Initiation-documentation
technique No. 16, 66 p.
Duchaufour Ph and Jacquin F (1963) Recherches d’une mdthode detraction et de fractionnement des
composds humiques controls par 61ectrophor£se. Ann. Agron., 19, 6.
Duchaufour Ph and Jacquin F (1966) Nouvelles recherches sur l’extraction et le fractionnement des com¬
poses humiques. Bull. ENSAN, VIII, 1, 3-24.
Duchaufour Ph (1954) Propri6t£s des complexes humiques dans diffSrents types de sols. Ecole Nationale
eaux et forets, Nancy, 29 p.
Duchaufour Ph (1956) PSdologie: Applications forestifcres et agricoles. Ecole Nationale eaux et forets,
Nancy, 310 p.
Duchaufour Ph (1957) P&iologie: tableaux descriptifs et analytiques des sols. Ecole Nationale eaux et
forets, Nancy, 87 p.
Duxbury JM (1989) Studies of the molecular size and charge of humic substances by electrophoresis. In:
Humic substances II, Hayes MHB, MacCarthy P, Malcolm RL and Swift RS ed. Wiley New York,
593-620.
Ghosh К and Schnitzer M (1980b) Macromolecular structures of humic substances. Soil Sci., 129,266-276
Guinier A and Foumet G (1955) Small angle X-ray scattering, Wiley New York, 268 p.
Kerker and Milton (1968) Light scattering, lnd. Eng. Chem., 60, 31-46.
Lansing WD and Kraemer EO (1935) Molecular weight analysis of mixtures by sedimentation equilib¬
rium in the Svedberg ultracentrifhge. J. Am. Chem. Soc., 57, 1369-1377.
Laurent TC and Killander J (1964) A theory of gel filtration and its experimental verification.
J. Chromatog., 14, 317-330.
376
Часть 2. Органический анализ
Morizur JP, Monegier du Sorbier В, Silly L and Desbene PL (1984) Etude par chromatographie sur gel
avec detection spectromdtrique de Involution de la conformation des acides humiques en fonction de
la force ionique et du pH. C. R. Acad. Sc. Paris, 299, 1269-1272.
Nobili Maria De, Gjessing E and Sequi P (1989) Sizes and shapes of humic substances by gel chroma¬
tography. In: Humic substances II., Hayes MHB, MacCarthy P, Malcolm RL and Swift RS ed. Wiley
New York, 561-591.
Orlov DS, Ammosova YaM, Glebova GI (1975) Molecular parameters of humic acids. Geoderma, 13,
211-229.
Plechanov N (1983) Studies of molecular weight distributions of fulvic and humic acids by gel perme¬
ation chromatography. Examination of the solute molecular composition using RI, UV, Fluorescence
and weight measurement as detection techniques. Org. Geochem., 5, 143-149.
Posner AM and Creeth JM (1972) A study of humic acids by equilibrium ultracentrifugation. J. Soil Sci.,
23,333-341.
Ratsimbazafy CA (1973) Protocole de fractionnement et d’Stude de la mattere organique des sols hydro-
morphes de Madagascar. Cah. ORSTOMser. Pedol, XI, 227-236.
Reuter JH and Perdue EM (1981) Calculation of molecular weights of humic substances from colligative
data: application to aquatic humus and its molecular size fractions. Geochim. Cosmochim. Acta, 45,
2017-2022.
Righetti PG and Drysdale JW (1971) Isoelectric focusing in polyacrilamide gels. Biochem. Biophys.
Acta., 236, 17-28.
Rusina TV, Kasparov SV and Zharikov AV (1983) Method of electrophoretic research of humus sub¬
stances and proteins in soi 1 solutions (texte Russe, r£sum6 Anglais). Pochvovedenie, 1, 38-46.
Samburova V, Zenobi R and Kalberer M (2005) Characterization of high molecular weight compounds in
urban atmospheric particles. Atmos. Chem. Phys., 5, 2163-2170.
Stevenson FJ (1982) Colloidal properties of humic substances. In: Humus chemistry, Stevenson FJ ed.
Wiley and Sons, 285-308.
Swift RS (1989) Molecular weight, shape, and size of humic substances by ultra-centrifugation. In:
Humic substances II., Hayes MHB, MacCarthy P, Malcolm RL and Swift RS ed. Wiley New York,
467-495.
Tiurin (1951) Vers une m&hode d’analyse par T6tude comparative des constituents de l’humus du sol.
Trav. Inst, des Sols Dokutchaev, 38,32 p.
Yamada E, Doi K, Okano К and Fuse Y (2000) Simultaneous determinations of the concentration and
molecular weight of humic substances in environmental water by gel chromatography with a fluores¬
cence detector. Analytical Sci., 16, 125-132.
Yu-Ping Chin, Aiken G, and O’Loughlin E (1994) Molecular weight, polydispersity, and spectroscopic
properties of aquatic humic substances. Environ. Sci. Technol., 28, 1853-1858.
Wershaw RL and Aiken GR (1985) Molecular size and weight measurements of Aiken GR, McK-
night DM, Wershaw RL and MacCarthy P ed. Wiley New York, 477-492.
Wershaw RL (1989) Sizes and shapes of humic substances by scattering techniques. In: Humic
substances II., Hayes MHB, MacCarthy P, Malcolm RL and Swift RS ed. Wiley New York, 545-
559.
Функциональные группы гуминовых веществ
De Nobili M, Contin M and Leita L (1990) Alternative method for carboxyl group determination in humic
substances. Can. J. Soil Sci., 70, 531-536.
Fritz JS, Yamamura SS and Bradford EC (1959) Determination of carbonyl compounds. Anal. Chem., 31,
260-263.
Glebko LI, Ulkina JU and Maximov OB (1970) A semi-micro method for the determination of quinoid
groups in humic acids. Microchim. Acta., 1247-1254.
Rosell RA, Allan AL, Agullo E and Gelos В (1972) Estudio potentiometrico del humus. II. Determinacion
de varios tipos de acidez (o grupo funcionales) de acidos humicos de suelos de la provincia de Bue¬
nos Aires, Argentina. Turrialba, 22,327-332.
Schnitzer M and Gupta UC (1965) Determination of acidity in soil organic matter. Soil Sci. Soc. Am.
Proc., 29, 274-277.
377
Глава 12. Характеризация гуминовых веществ
Schnitzer М and Skinner SIM (1965) Organo-metallic interactions in soils: 4. Carboxyl and hydroxyl
groups in oiganic matter and metal retention. Soil Sci., 99, 278-284.
Wright JR and Schnitzer M (1959) Oxygen-containing functional groups in the organic matter of a Podzol
soil. Nature, London, 184, 1462-1463.
Спектрометрический анализ
Спектрометрии УФ, видимого и ИК-диапазона, флуоресцентная
и ЭПР- спектрометрии
Lapen AJ and Seitz WR (1982) Fluorescence polarization studies of the conformation of soil fulvic acid.
Anal Chim. Acta., 134, 31-38.
Bachelier G (1981) Etude spectrographique de la fluorescence des acides humiques et des acides ful-
viques de divers sols. Cah. ORSTOMser. Pedol, 18, 129-145.
Chen Y, Senesi N and Schnitzer M (1977) Information provided on humic substances by E4:E6 ratios.
Soil Sci. Soc. Am. J., 41, 352-358.
Ghosh К and Schnitzer M (1979) UV and visible absorption spectroscopic investigations in relation to
macromolecular characteristics of humic substances. J. Soil Sci., 30, 735-745.
Ghosh К and Schnitzer M (1980a) Fluorescence excitation spectra of humic substances. Can J. Soil Sci.,
60, 373-379.
Goldberg MC, Cunningham KM and Weiner ER (1987) The use of isosbestic points in the fluorescence ex¬
citation spectrum of humic acid to calculate the dissociation constant. Can. J. Soil Sci., 67,715-717.
Kononova MM (1966) Soil organic matter, its nature, its role in soil formation and in soil fertility, 2nd
English ed. Pergamon Oxford, 544 p.
Levesque M (1972) Fluorescence and gel filtration of humic compounds. Soil Sci., 113, 346-353.
MacCarthy Pet Rice JA (1985) Spectroscopic methods (other than NMR) for determining the function¬
ality in humic substances. In: Humic substances in soil, sediment and water., Aiken GR, McK-
night DM, Wershaw RL and MacCarthy P ed. Wiley New York, 527-559.
Nayak DC, Barman AK, Varadachari C, Ghosh К (1985) Fluorescence excitation spectra of humic acids.
J. Indian Soc. Soil Sci., 33, 785-787.
Rex RW (1960) Electron paramagnetic resonance studies on stable free radicals in lignins and humic
acids. Nature, 188, 1185-1186.
Riffaldi R and Schnitzer M (1972) Effects of divers experimental conditions on ESR spectra of humic
substances. Geoderma, 8, 1-10.
Russel JD and Anderson HA (1977) Comment on “spectroscopie infra-rouge de quelques fractions
d’acides humiques obtenues sur Sephadex” Plant Soil, 48, 547-548.
Schnitzer M and Skinner SIM (1969) Free radicals in soil humic compounds. Soil Sci., 108, 383-390.
Saab SC and Martin-Neto L (2004) Studies of Semiquinone Free Radicals by ESR in the Whole Soil, HA,
FA and humin substances. J. Braz. Chem. Soc., 15, 34-37.
Senesi N (1990) Application of ESR spectroscopy in soil chemistry. Adv. in Soil Sci., 14, 77-129.
Ядерный магнитный резонанс
Barron PF, Wilson MA, Stephens JF, Cornell BA and Tate KR (1980) Cross-polarization 13C NMR spec¬
troscopy of whole soils. Nature, 286, 585-587.
Brown JK and Ladner WR (1960) A study of the hydrogen distribution in coal-like materials by high
resolution NMR spectroscopy (II). Fuel, 39, 87-96.
Conte P, Piccolo A, van Lagen B, Buurman P and de Jager PA (1997a) Quantitative differences by liquid-
and solid-state 13C NMR spectroscopy. Geoderma, 80, 339-352.
Conte P, Piccolo A, van Lagen B, Buurman P and de Jager PA (1997b) Quantitative aspects of solid-state
13C NMR spectra of humic substances from soils of volcanic systems. Geoderma, 80, 327-338.
Conte P, Spaccini R and Piccolo A (2004) State of the art of CPMAS C-13-NMR spectroscopy applied to
natural organic matter. Progress in Nuclear Magnetic Resonance Spectroscopy, 44, 215-223.
Doddrell D, Glushko V and Allerhand A (1972) Theory of the Nuclear Overhauser Enhancement and
,3C 1H dipolar relaxation in protondecoupled carbon-13 NMR spectra of macromolecules. J. Chem.
Physics, 56,3683-3689.
378 Часть 2. Органический анализ
Freeman R, Hill HDW, Kaptein R (1972) Proton-decoupled NMR spectra of carbon-13 with the nuclear
Overhauser effect suppressed. J. Magn. Res., 7, 327-329.
Frtlnd R. and Ltidemann HD (1989) The quantitative analysis of solution and CPMAS-C13 NMR Spectra
of humic material. Sci. Total Environ., 81/82, 157-168.
Gerasimowicz WV and Byler DM (1985) Carbon-13 CPMAS NMR and FTIR spectroscopic studies of
humic acids. Soil Sci., 139, 270-278.
Kinchesh P, Powlson DS and Randall EW (1995) ,3C NMR studies of organic matter in whole: a case
study of some Rothamsted soils. Eur. J. Soil Sci., 46, 139-146.
Lentz H, Ludemann HD and Ziechmann W (1977) Proton resonance spectra of humic acids from the
solum of a podzol. Geoderma, 18, 325-328.
Lowe IJ (1959) Free induction decay of rotating solids. Phys. Rev. Lett., 2, 285-287.
Moser A, Lefebvre В (2004) Identifying Residues in Natural Organic Matter through Spectral Prediction
and Pattern Matching of 2-D NMR datasets. Magn. Resonance Chem., 42, 14-22.
Newman RH and Tate KR (1984) Use of alkaline soil extracts for 13C NMR characterization of humic
substances. J. Soil Sci., 35, 47-54.
Newman RH, Tate KR, Barron PF and Wilson MA (1980) Towards a direct, non-destructive method of
characterizing soil humic substances using 13C-NMR. J. Soil Sci., 31, 623-631.
Pines A, Gibby MG and Waugh JS (1973) Proton enhanced NMR of dilute spins in solids. J. Chem. Phys.,
59, 569-590.
Preston CM and Schnitzer M (1984) Effects of chemical modifications and extractants on the 13C NMR
spectra of humic materials. Soil Sci. Soc. Am. J., 48,305-311.
Schaefer J and Stejskal EO (1976) C-13 NMR of polymers spinning at the magic angle, J. Am. Chem.
Soc., 98, 1031-1032.
Schnitzer M and Barton DHR (1963) A new approach to the humic acid problem. Nature, London 198,
217-219.
Schnitzer M and Neyroud JA (1974) The chemistry of high molecular weight fulvic acid fractions. Can.
J. Chem., 52,4123-4132.
Simpson AJ, Kingery WL, Williams A, Golotvin S, Kvasha M, Kelleher BK, Simpson AJ, Tseng L, Spraul
M, Brauman U, Kingery WL, Kelleher B, Simpson MJ (2004) The application of LC-NMR and LC-
SPE-NMR for the separation of Natural Organic Matter. The Analyst, 129:1216-1222.
Smemik RJ and Oades MJ (2000) The use of spin counting for determining quantitation in solid state I3C
NMR spectra of natural organic matter. 1. Model systems and the effects of paramagnetic impurities.
Geoderma, 96, 101-129.
Smemik RJ and Oades MJ (2000) The use of spin counting for determining quantitation in solid state l3C
NMR spectra of natural organic matter. 1. HF-treated soil fractions. Geoderma, 96,159-171.
Smemik RJ and Oades MJ (2003) Spin accounting and RESTORE, two new methods to improve quan¬
titation in solid-state 13C NMR analysis of soil organic matter, Eur. J. Soil. Sci., 54, doi:10.1046/
j. 1365-2389. 2003.00497.x.
Steelink C, Wershaw RL, Thom KA and Wilson MA (1989) Application of liquid-state NMR spectros¬
copy to humic substances. In: Humic substances II, Hayes MHB, MacCarthy P, Malcolm RL and
Swift RS ed. Wiley New York, 281-338.
Tate RL (1998) Humic substances and organic matter in soil and water environments: characteriza¬
tion, transformations and interactions. Soil Thom KA and Mikita MA (2000) Nitrite fixation by
humic substances: nitrogen-15 nuclear magnetic resonance. Evidence for potential intermediates in
chemodenitrification. Soil Sci. Soc. Am. J., 64, 568-582.
Wilson MA (1981) Applications of nuclear magnetic resonance spectroscopy to the study of the structure
of soil organic matter J. Soil Sci., 32, 167-186.
Wilson MA (1987) Techniques and applications of NMR spectroscopy in geochemistry and soil science.,
Pergamon, Oxford.
Wilson MA (1989) Solid-state NMR spectroscopy of humic substances, basic concepts and techniques.
In: Humic substances II, Hayes MHB, MacCarthy P, Malcolm RL and Swift RS ed. Wiley and Sons,
309-338.
Wilson MA, Jones AJ and Williamson В (1978) NMR spectroscopy of humic materials. Nature, London,
276, 487-489.
Глава 12. Характеризация гуминовых веществ
379
Методы исследования гуминовых веществ, основанные на фрагментации
Anderson НА, Hepburn A and Sim А (1978) Ether-soluble hydrolysis products in humic and fulvic acids.
J. Soil Sci., 29, 84-87.
Bracewell JM, Haider K, barter SR and Schulten HR (1989) Thermal degradation relevant to structural
studies of humic substances. In: Humic substances II, Hayes MHB., MacCarthy P, Malcolm RL and
Swift RS ed., Wiley New York, 181-222.
Chakrabartty SK, Kretschmer HO and Cherwonka S (1974) Hypohalite oxidation of humic acids. Soil
Sci., П7, 6.
Chen Y, Senesi N and Schnitzer M (1978a) Chemical degradation of humic and fulvic acids extracted
from mediterranean soils. J. Soil Sci., 29, 350-359.
Chen Y, Senesi N and Schnitzer M (1978b) Chemical and physical characteristics of humic and fulvic
acids extracted from soils of the mediterranean region. Geoderma, 20, 87-104.
Cheshire MV, Cranwell PA and Haworth RD (1968) Humic acid - III. Tetrahedron, 24, 5155-5167, Per-
gamon UK.
Griffith SM and Schnitzer M (1989) Oxidative degradation of soil humic substances. In: Humic
substances II, Hayes MHB, MacCarthy P, Malcolm RL and Swift RS ed., Wiley New York, 69-98.
Griffith SM and Schnitzer M (1975) Oxidative degradation of humic and fulvic acids extracted from
tropical volcanic soils. Can. J. Soil Sci., 55, 251-267.
Griffith SM and Schnitzer M (1976) The alkaline cupric oxide oxidation of humic and fulvic acids ex¬
tracted from tropical volcanic soils, Soil Sci., 122, 191-201.
Hansen EH and Schnitzer M (1967) Nitric acid oxidation of Danish illuvial organic matter. Soil Sci. Soc.
Am. Proc., 31, 79-85.
Hansen EH and Schnitzer M (1969) Zn-dust distillation and fusion of a soil Humic and fulvic acid. Soil
Sci. Soc. Am. Proceed., 33, 29.
Hatcher PG, Dria KJ, Kim S., Frazier, SW (2001) Modem Analytical Studies of Humic substances. Soil
Sci., 166, 770-794.
Hayes MHB and O’Callaghan MR (1989. Degradations with sodium sulfide and with phenol. In: Humic
substances II, Hayes MHB, MacCarthy P, Malcolm RL and Swift RS ed., Wiley New York, 143-180.
Jackson MP, Swift RS, Posner AM, Knox JR (1972) Phenolic degradation of humic acid. Soil Sci., 114,
75-78.
Kahn SU and Schnitzer M (1971a) The permanganate oxidation of methylated and unmethylated humic
acids extracted from Solonetz, Solod and Chernozem Ah horizons. Israel J. Chem., 9,667-677.
Khan SU and Schnitzer M (1971b) Further investigations on the chemistry of fulvic acid, a soil humic
fraction. Can. J. Chem., 49,2302-2309.
Kahn SU and Schnitzer M (1972a) Permanganate oxidation of humic acids, fulvic acids, and humins
extracted from Ah horizons of a black chernozem, a black solod and a black solonetz soil. Can.
J. Soil Sci., 52, 43-51.
Kahn SU and Schnitzer M (1972b) Permanganate oxidation of humic acids extracted from a GrayGrey
wooded soil under different cropping systems and fertilizer treatments. Geoderma, 1,113-120.
Kimber RWL and Searle PL (1970) Pyrolysis gas chromatography of soil organic matter. 1. Introduction
and methodology, Geoderma, 4,47-55.
Kodama H and Schnitzer M (1970) Kinetics and mechanism of the thermal decomposition of fulvic acid.
Soil Sci., 109, 265-271.
Martin F (1976) Effects of extractants on analytical characteristics and pyrolysis gas chromatography of
podzol fulvic acids. Geoderma, 15, 253-265.
Mendez J and Stevenson FJ (1966) Reductive cleavage of humic acids with sodium amalgam. Soil Sci.,
102, 85.
Meuzelaar HLC, Haider K, Nagar BR and Martin JP (1977) Comparative studies of pyrolysis mass spec¬
tra from melanins of soil fungi, model phenolic polymers and humic acids from soil, peat and com¬
posted straw. Geoderma, 17, 239-252.
Meuzelaar HLC, Posthumus MA, Kistemaker PG and Kistemaker J (1973) Curie point pyrolysis in direct
combination with low voltage electron impact ionization spectrometry. Anal. Chem., 45,1546-1549.
Morrison RI (1963) Products of the alkaline nitrobenzene oxidation of soil organic matter. J. Soil Sci.,
14,2.
380 Часть 2, Органический анализ
Neyroud JA and Schnitzer М (1974) The exhaustive alkaline cupric oxide oxidation of humic and fulvic
acid, Soil Sci. Soc. Am. Proc., 38,907-913.
Neyroud JA and Schnitzer M (1975) The alkaline hydrolysis of humic substances. Geoderma; 13, 171—
188.
Ogner G (1975) Oxidation of nonhydrolyzable humic residue and its relation to lignin, Soil Sci., 116,
93-100.
Ouchi К and Brooks JD (1967) The isolation of certain compounds from depolymerized brown coal.
Fuel, 46, 367-377.
Parsons JW (1989) Hydrolytic degradations of humic substances. In: Humic substances II, Hayes MHB,
MacCarthy P, Malcolm RL and Swift RS ed., Wiley New York, 99-120.
Piper TJ and Posner AM (1972) Sodium amalgam reduction of humic acid (I and II). Soil Biol Biochem.,
4,513-531.
Saiz-Jimenez C, Haider К and Meuzelaar HLC (1979) Comparisons of soil organic matter and its frac¬
tions by pyrolysis mass-spectrometiy. Geoderma, 22, 25-37.
Schnitzer M and Hoffmann I (1964) Pyrolysis of soil organic matter. Soil Sci. Soc. Proced., 520-525.
Schnitzer M and Skinner SIM (1974) The peracetic acid oxidation of humic substances, Soil Sci., 118,
322-331.
Schulten HR (1996) Direct pyrolysis-mass spectrometry of soils: a novel tool in agriculture, ecology,
forestry and soil science. In Mass spectrometry of soils, Boutton TW and Yamasaki S-i ed., Marcel
Dekker New York.
Stevenson FJ and Mendez J (1967) Reductive cleavage products of soil humic acids. Soil Sci., 103, 383.
Stevenson FJ (1989) Reductive cleavage of humic substances. In: Humic substances II, in search of
structure, Hayes MHB, MacCarthy P, Malcolm RL and Swift RS ed., Wiley New York, 122-142.
Другие методы (микроскопия, рентгеновская спектроскопия, электрохимические
методы анализа и т. д.)
Bachelier G (1983) Figures de dessication des acides humiques, Rapport multig. ORSTOM, 14 p.
Chen Y and Schnitzer M (1976) Scanning electron microscopy of a humic acid and of a fulvic acid and its
metal and clay complexes. Soil Sci. Soc. Am. J., 40,682-686.
Chen Y, Senesi N and Schnitzer M (1976) Chemical and physical characteristics of humic and fulvic acids
extracted from soils of the Mediterranean region. Geoderma, 20, 87-104.
Shinozuka N and Hayano S (1987) Polarographic characterization of humic acid. Soil Sci., 143, 157—
161.
Stevenson FJ and Schnitzer M (1982) Transmission electron microscopy of extracted humic and fulvic
acids, Soil Sci., 133, 334-345.
Tan KH (1985) Scanning electron microscopy of humic matter as influenced by methods of preparation.
Soil Sci. Soc. Am. J., 49, 1185-1191.
Defosse C and Rouxhet PG (1980) Introduction to X-ray photoelectron spectroscopy. In: Advanced
chemical methods of soil and clay mineral research., Stucky J.W and Banwart W.L. ed., Reidel,
Dordrecht, 165-204.
Goodman BA and Cheshire MV (1979) A Mussbauer spectroscopic study of the effect of pH on the reac¬
tion between iron and humic acid in aqueous media. J. Soil Sci., 30, 85-91.
Глава 13. Определение негуминовых веществ
13.1. Введение
13.1.1. Негуминовые вещества
Органическое вещество почвы может содержать большую часть биохимических соеди¬
нений, синтезируемых живыми организмами (Stevenson, 1982). Кроме гуминовых мо¬
лекул, количественное определение которых (описано в гл. 11) технически проще, чем
определение структуры молекул (см. гл. 12), в почве присутствует также большое коли¬
чество других молекул известной структуры. Наиболее распространенные из них можно
разделить на три большие группы.
1. Азотсодержащие молекулы, которые часто изучают, исследуя продукты их фраг¬
ментации в результате кислотного гидролиза.
2. Полисахариды, главным образом не содержащие азота, которые также часто ис¬
следуют, анализируя продукты их фрагментации.
3. Липидная фракция, которую иногда называют почвенными битумами (содержит
ряд молекул, экстрагируемых жирорастворяющими растворителями); эта фракция
также содержит много органических поллютантов и остатков ксенобиотиков, ко¬
торые могут загрязнять почву.
Методы определения азотсодержащих молекул (1) описаны в гл. 14. В настоящей гла¬
ве описаны основные методы определения молекул второго и третьего типа.
13.1.2. Углеводороды в почве
Общий состав
Сахара (также неправильно называемые углеводами потому, что их эмпирическая фор¬
мула соответствует Сй(Н20)т) составляют 5-25% органического вещества почвы. Кроме
следов свободных сахаров, которые можно экстрагировать из почвы водой, почвенные
углеводы являются компонентами полисахаридов. Практически невозможно выделить
растворимые фракции полисахаридов для идентификации (Cheshire, 1979). Поэтому
сложно выяснить, входят сахара в гетерогенную смесь полисахаридов или в единый
очень сложный полисахарид.
Восемь нейтральных углеводов можно идентифицировать гидролизом почв. Их мож¬
но разделить на три основные группы: гексозы, дезоксигексозы и пентозы. Также можно
идентифицировать два кислых сахара (уроновые кислоты) и два основных сахара (гек-
созамины).
Гексозные сахара составляют от 4 до 12% органического вещества: это глюкоза, га¬
лактоза и манноза. Уроновые кислоты составляют 1-5% органического вещества почвы
и включают примерно равные части галактуроновой и глюкуроновой кислот. Пентозные
сахара присутствуют в малых количествах и включают в основном арабинозу и ксилозу,
а также следы рибозы. Дезоксигексозы — фукоза и рамноза присутствуют в количествах,
близких к содержанию пентозных сахаров.
Аминосахара (см. гл. 14) присутствуют в почве в еще меньших концентрациях; их ос¬
новные компоненты — гексозамины в форме галактозамина и глюкозамина.
382
Часть 2. Органический анализ
В почве можно найти и следы других сахаров: четыре метилсахара, два спиртосахара
(инозитол и манитол), два гексозных сахара (фруктоза и сорбоза), пентозный сахар (де-
зоксирибоза) и гексозамин (А-ацетилглюкозамин).
Сахара и типы почв
Многие авторы пытались характеризовать почвы методом количественного определе¬
ния сахаров, образующихся при кислотном гидролизе полисахаридов. Фолсом с сотр.
(Folsom и др., 1974) обнаружили почти линейную зависимость между общим содержа¬
нием углеводов и содержанием органического вещества почвы; однако зависимость ста¬
новится нелинейной для горизонтов, содержащих много органических веществ и малые
концентрации сахаров. Эти авторы сообщили, что луговые почвы содержат больше пен-
тоз и меньше гексоз, чем лесные почвы, а также что доля маннозы увеличивается с глу¬
биной залегания почвы, что указывает на бблыную стабильность этого сахара.
Сингал и Шарма (Singhal и Sharma, 1985) сообщили, что содержание общих углеводов
и органического углерода в лесных почвах изменяется сходным образом, независимо от
типа деревьев. Они не обнаружили различия в относительных долях этих сахаров. Мак¬
Грат (MacGrath, 1973) также нашел, что относительный состав сахаров в 38 ирландских
луговых почвах очень стабилен.
Чешир и Андерсон (Cheshire и Anderson, 1975) обнаружили большее содержание об¬
щих сахаров в обрабатываемых почвах, чем в некультивируемых, однако относительные
доли индивидуальных соединений оставались неизменными.
Распределение и происхождение сахаров
Другой аспект проблемы связан с распределением полисахаридов во фракциях органи¬
ческого вещества почвы. Разбавленные щелочные растворы являются лучшим реакти¬
вом для экстракции полисахаридов, хотя большинство последних остаются связанными
с гуминовыми остатками. Подкисление щелочных экстрактов осаждает гуминовые кис¬
лоты, а ббльшая часть полисахаридов остается в кислоторастворимой фракции фульво-
кислот (Cheshire, 1979; Barriuson др., 1985). Багаутдинов с сотр. (Bagautdinov и др., 1984)
отделили от раствора фульвокислот фракцию, содержащую в основном полисахариды
с молекулярной массой 27 000-28 000.
Многие авторы пытались определить, являются почвенные сахара соединениями
микробного или растительного происхождения. Это непростая задача, поскольку ни
один из сахаров не может быть отнесен к веществам исключительно растительного или
микробного происхождения (Cheshire, 1979). Существует больше сходств, чем различий
в сахарах из гуминовых кислот и меланинов грибов (Coelho и др., 1988).
Инкубационные эксперименты с использованием меченой глюкозы приводят к пере¬
ходу метки на все сахара и, в меньшей степени, аминокислоты. Дезоксигексозы были
идентифицированы как самые стабильные синтезированные сахара (Cheshire, 1979).
При увеличении времени инкубации доля меченого углерода в гумине увеличивается
(Gucker и др., 1971). Гексозы являются основными сахарами, синтезируемыми почвен¬
ными микроорганизмами (Oades, 1974).
Франсуа (Franqois, 1988) и Мураяма (Мигауата, 1983, 1984, 1988) сообщали, что они
не смогли определить конкретный источник глюкозы, галактозы и рибозы, но выяс¬
нили, что ксилоза и арабиноза — в основном растительного происхождения, тогда как
рамноза, манноза и фукоза очень часто синтезируются микроорганизмами, хотя также
присутствуют в экссудатах корней различных растений.
383
Глава 13. Определение негуминовых веществ
Франсуа (Frangois, 1988) показал, что общие сахара концентрируются главным об¬
разом в свежих корнях и фракции почвенных частиц размером 5—25 мкм. Заметное
относительное уменьшение содержания ксилозы и увеличение содержания маннозы
и рамнозы наблюдались в более тонких фракциях. Феллер (Feller, 1991) использовал от¬
ношение ксилоза/манноза в качестве индикатора микробного или растительного проис¬
хождения органического вещества. Это отношение имело наименьшее значение (0,5-2)
в органическом веществе глино-органического компартмента < 2 мкм, находилось в ди¬
апазоне между 1 и 3 для фракции 2—20 мкм и от 5 до 10 во фракции > 20 мкм (см. гл. 9).
Принцип определения сахаров
Свободные сахара можно определить в водных экстрактах почв. Определение полисаха¬
ридов включает три основных стадии:
• кислотный гидролиз полисахаридов;
• возможную очистку гидролизата;
• анализ гидролизата.
Для определения свободных или гидролизованных сахаров используют методы двух
типов:
• общий колориметрический метод для восстанавливающихся сахаров;
• хроматографические методы, позволяющие определять каждый сахар индиви¬
дуально.
13.1.3. Липиды в почве
Почвенные липиды представляют собой довольно сложные смеси соединений, одним
из общих признаков которых является растворимость в ряде органических раствори¬
телей или смесях растворителей. Эта фракция содержит свободные жирные кислоты,
углеводы, полярные или неполярные липиды, стероиды, воски и смолы.
Большинство липидов можно отнести к трем основным группам: жиры, воски и смо¬
лы. Смолы являются самыми полярными соединениями и поэтому лучше всего раство¬
римы в метаноле и этаноле; это свойство можно использовать для их выделения.
Липиды присутствуют в почве в меньших количествах, чем соединения азота и поли¬
сахариды. Согласно Стивенсону (Stevenson, 1982), они составляют 2-6% от органическо¬
го вещества; по данным Жамбу с сотр. (Jambu и др., 1978), в некоторых почвах их содер¬
жание может достигать 20%. Концентрации липидов в кислых почвах обычно больше.
Самые плодородные почвы обычно бедны липидами. Присутствие липидов может даже
быть связано со старой концепцией болезни почвы (Stevenson, 1982) и зависит от содер¬
жания гумуса, а также аэрации и текстуры почвы {Jambu и др., 1978).
Экстракция почвенных липидов может осложняться тем, что они образуют различ¬
ные связи с другими органическими или неорганическими компонентами почвы. Боль¬
шинство пестицидов и других органических веществ, загрязняющих почвы, также экс¬
трагируется вместе с липидной фракцией.
13.1.4. Пестициды и поллютанты
В почве можно обнаружить большинство соединений, используемых в пищевой и сель¬
скохозяйственной химии.
Органические поллютанты (загрязнители), образующиеся в результате разложения
продуктов в окружающей среде, могут быть чрезвычайно разнообразными, хотя боль¬
шинство из них принадлежит к следующим основным группам:
384
Часть 2. Органический анализ
• полихлорированные бифенилы (ПХБ) — род хлорзамещенных поллютантов,
включающий более 30 соединений; следы ПХБ можно обнаружить во многих суб¬
стратах;
• полициклические ароматические углеводороды (ПАУ), включающие 15—20 соеди¬
нений (например, нафталин, фенантрен, антрацен или пирен), устойчивых к раз¬
ложению;
• диоксины также являются продуктами разложения, но они присутствуют в мень¬
ших количествах.
Пестициды — это обычно не очень устойчивые соединения, используемые в сельском
хозяйстве, включают широкий спектр продуктов, таких как инсектициды, акарициды,
нематоциды, репелленты, фунгициды, гербициды и яды. Они включают очень большое
число соединений. Хотя наблюдается общая тенденция к использованию соединений,
все менее и менее токсичных для людей и более и более легко разлагаемых, общее коли¬
чество областей их применения растет, что в итоге приводит к генным мутациям и раз¬
витию сопротивляемости вредителей. Кальве с сотр. (Calvet и др., 2005) обобщили со¬
временные данные о пестицидах и агрономических и экологических последствиях их
применения на почвах.
Более тысячи соединений продают в качестве пестицидов, которые трудно классифи¬
цировать. Вот некоторые из основных типов пестицидов:
• хлорорганические соединения, включая первые коммерческие синтетические со¬
единения, такие как ДДТ, дильдрин или линдан1; сегодня эти продукты исполь¬
зуются в меньшей степени из-за их токсичности и малой скорости разложения;
однако они все еще находятся в окружающей среде, поскольку накапливаются
в липидах живых организмов; в настоящее время используют новые галоген-за-
мещенные соединения;
• фосфорорганические соединения, часто используемые вместо первых хлороргани-
ческих соединений;
• триазиновые гербициды;
• кислотные гербициды, такие как хлорфеноксиуксусные кислоты, пиклорам или
дикамба;
• карбаматы2 и тиокарбаматы, часто используемые как системные инсектициды;
• пиретриноиды3 — синтетические соединения, производные природных пиретри-
нов, обычно используемые в качестве инсектицидов потому, что они не вступают
в реакции обмена в крови человека или теплокровных животных.
13.2. Классические методы анализа
13.2.1. Кислотный гидролиз полисахаридов
Принцип и основные проблемы метода
Горячие разбавленные кислоты способны полностью гидролизовать полисахариды, од¬
нако в большинстве почв они гидролизуются приблизительно на 75%.
1 Гексахлоран. — Примеч. науч.ред.
2 Уретаны. — Примеч. науч. ред.
3 Пиретроиды. — Примеч. науч. ред.
Глава 13. Определение негуминовых веществ
385
Для достижения полного гидролиза полисахаридов, например, целлюлозы или по¬
чвенных полисахаридов, требуется предварительная обработка более концентрирован¬
ной кислотой (Cheshire, 1979). Во время гидролиза разбавленными кислотами выделение
гексозы увеличивается при увеличении концентрации кислоты. Использование только
разбавленных кислот обычно не позволяет определять количества гексозных сахаров.
Пентозные сахара выделяются легче, чем гексозные сахара, но они также легче раз¬
лагаются в условиях кислотного гидролиза и в зависимости от типа почвы могут эффек¬
тивнее определяться в более разбавленной среде.
Иварсон и Сауден (Ivarson и Sowden, 1962) первыми рекомендовали применять пред¬
варительную обработку холодной 12 М серной кислотой (72%) с последующим разбав¬
лением кислоты до 0,5 М и кипячением с обратным холодильником. Из образцов под¬
стилки такая обработка выделяла втрое больше гексоз, чем гидролиз без предобработки,
но выделение пентоз было снижено примерно на 20% в случае хвойной подстилки.
ГУпта и Сауден (Gupta и Sowden, 1965) подтвердили для четырех почв сильное положи¬
тельное влияние предварительной обработки на определение гексоз и пентоз, за исклю¬
чением некоторых случаев, когда дезоксигексозы частично разлагались.
Чешир и Мунди (Cheshire и Mundie, 1966) оптимизировали время холодной предо¬
бработки 12 М серной кислотой. Содержание сахаров, определенное орцинольным
колориметрическим методом, достигало максимума через 16 ч и затем снижалось, тог¬
да как содержание гексозов, определенное антроновым колориметрическим методом,
продолжало увеличиваться вплоть до 40 ч. Таким образом, длительность предваритель¬
ной обработки влияла на время реакции при кипячении с обратным холодильником
с 0,5 М раствором серной кислоты, необходимое для выделения максимального коли¬
чества сахара. После предварительной холодной обработки в течение 16 ч обработка
горячей кислотой в течение 5 ч была достаточна для достижения максимального вы¬
хода сахаров, тогда как после 2-часовой предобработки требовалось почти 20 ч горя¬
чей обработки. Наконец, эти авторы заключили, что не существует идеального метода
гидролиза для полного выделения глюкозы без частичного разложения пентоз или де-
зоксигексоз. Была рекомендована 16-часовая обработка 12 М H2S04 при 20 °С с после¬
дующим кипячением с обратным холодильником с 0,5 М H2S04 в течение 5 ч, потому
что в некоторых случаях она приводила к максимальной скорости выделения глюкозы.
Однако это не очень убедительный метод определения источника сахаров в почве, где
глюкоза не является представителем сахаров растительного или микробного происхож¬
дения (см. раздел 13.1.1).
Оадес с сотр. (Oades и др., 1970) исследовали условия гидролиза при хроматографи¬
ческом определении сахаров. Они обнаружили, что лучшие результаты экстракции из
почвы, не содержащей растительных фрагментов, наблюдались при непосредственной
обработке образца 5 н. раствором H2S04 в течение 1 ч при кипячении с обратным холо¬
дильником. Однако такая обработка приводила к выделению меньшего количества глю¬
козы, чем другие методы, аналогичные использованному Чеширом и Мунди (холодная
обработка 13 М раствором H2S04 в течение 16 ч с последующим кипячением с обратным
холодильником с 0,5 М H2S04 в течение 2 ч). Однако метод Оадеса позволял получать
ббльшие количества других сахаров, в частности ксилозы, рамнозы и фукозы. Однако
в песчано-пылеватой почве существует опасность значительного расщепления сахаров,
если продолжительность гидролиза превышает 20 мин для ксилозы, 40 мин для араби-
нозы и около 1 ч для других сахаров (не равная риску в случае метода с предобработкой).
В действительности для общего содержания сахаров результаты метода с кипячением
с обратным холодильником в 5 н. растворе H2S04 в течение 20 мин незначительно отли-
386
Часть 2. Органический анализ
чались от результатов, полученных с холодной предобработкой кислотой, за исключени¬
ем образцов, содержащих большие количества материалов, для которых рекомендуется
использование предобработки. Метод Оадеса дает больше пентоз и дезоксигексоз, но
гораздо меньше глюкозы. Для выделения глюкозы требовалась более длительная обра¬
ботка 5 н. раствором H2S04, однако в этом случае присутствовал риск разложения пенто-
зы и дезоксигексозы. Для одновременного получения максимальных количеств глюко¬
зы и других сахаров Оадес с сотр. рекомендовали кипячение с обратным холодильником
с 5 н. раствором H2S04 в течение 20 мин, фильтрование, вымачивание в холодном 26 н.
растворе H2S04 в течение 16 ч и кипячение с обратным холодильником с 1 н. раство¬
ром H2S04 в течение 5 ч. Первоначальное кипячение с обратным холодильником с 5 н.
раствором H2S04 в течение всего 20 мин может быть достаточно для некоторых почв
с низким содержанием органического вещества, но в этом случае появляется риск недо-
оценки содержания глюкозы.
В большинстве более поздних работ не рассматривается оптимизация условий гидро¬
лиза: Коэльо с сотр. (Coelho и др., 1988), Мураяма (Мигауата, 1987), Чешир и Гриффитс
(Cheshire и Griffiths, 1989) использовали метод Оадеса с сотр. (Oades и др., 1970) с двуста¬
дийным гидролизом. В некоторых случаях разделялись сахара, выделенные при кипяче¬
нии с обратным холодильником с 5 М раствором H2S04 в течение 20 мин, называемые
нецеллюлозными сахарами, и сахара, выделенные последующим гидролизом (кипяче¬
ние с обратным холодильником с 1 н. раствором H2S04 в течение 5 ч после вымачивания
в 26 н. растворе H2S04) и называемые целлюлозными сахарами.
Аршад и Шнитцер (Arschad и Schnitzer, 1987) и Балдок с сотр. (Baldock и др., 1987)
использовали метод, разработанный Спителлером (Spiteller, 1980), который аналогичен
методу Чешира и Мунди (Cheshire и Mundie, 1966) в отношении условий гидролиза: вы¬
мачивание в 24 н. растворе H2S04 в течение 16 ч с последующим кипячением с обратным
холодильником с 1 н. H2S04 в течение 5 ч.
Сингал и Шарма (Singhalи Sharma, 1985) использовали метод ГУпта (Gupta, 1967): экс¬
тракция 72% H2S04 при низкой температуре в течение 2 ч и кипячение с обратным хо¬
лодильником в течение 16 ч после разбавления. Бензинг-Пурди и Никифорук (Benzing-
Purdie и Nikiforuk, 1989) применяли более простой метод гидролиза в 2 н. H2S04 при
105 °С в течение 18 ч и сравнивали его с методом Чешира и Мунди (Cheshire и Mundie,
1966). Гуккерт (Guckert, 1973) также использовал аналогичный метод гидролиза 3 н. рас¬
твором H2S04 при 80 °С в течение 24 ч после экстракции.
Гйдролиз без холодной предобработки в течение 4,5 ч с последующим определени¬
ем содержания сахаров методом колориметрии с антроном и феррицианидом позволял
получить максимальное содержание сахаров при использовании 5 н. раствора кислоты
и температуры 100 °С (в закрытой колбе) и указывал на значительно меньшее влияние
концентрации кислоты, чем температуры (Pansu, 1992). Холодная предобработка увели¬
чивала количество выделенных сахаров, а оптимальная температура для последующей
обработки колебалась от 70 до 90°С в более разбавленной кислоте (1 н. H2S04), что соот¬
ветствовало результатам Чешира и Мунди (Cheshire и Mundie, 1966).
Пансу (Pansu, 1992) также показал хорошую устойчивость к разложению моноса¬
харидов, известные концентрации которых добавляли к реагентам. Коммерческие
препараты кристаллической целлюлозы показали хорошую устойчивость к концен¬
трированным кислотам при 105 °С, не зависящую от концентрации серной и даже хло¬
ристоводородной кислоты. Значительный гидролиз целлюлозы (88%) наблюдался толь¬
ко при использовании метода, включающего холодную предобработку 26 н. раствором
H2S04. Гидролиз коммерческого препарата целлюлозы привел к выделению не только
387
Глава 13. Определение негуминовых веществ
глюкозы (66-86% общего выхода), но также галактозы (10-30%), маннозы (1-2%), кси¬
лозы (примерно 1%) и рибозы (примерно 1%).
Оборудование и реагенты
• Очищенная вода, не содержащая органических веществ: деминерализованная вода,
очищенная активированным углем (стандарт Миллипор), или вода, перегнанная
после кипячения с обратным холодильником с небольшим количеством перман¬
ганата и серной кислоты.
• Шесть круглодонных перегонных колб на 100 мл с притертой пробкой или шесть
колб (100 мл) с завинчивающимися крышками из ПТФЭ.
• Набор из шести обратных холодильников (только для метода с нагреванием с об¬
ратным холодильником).
• Воронки Бюхнера со стандартными фильтрами из стекловолокна GF/A 4 см в диа¬
метре (Whatman No. 1820 042 или аналогичные).
• 13 М раствор серной кислоты: добавляют 278 мл воды без органических веществ
в мерную колбу на 1 л; осторожно при перемешивании и охлаждении в холодной
водяной бане добавляют 722 мл концентрированной серной кислоты высокой сте¬
пени чистоты; доводят объем раствора до 1 л.
• 2,5 М (5 н.) раствор H2S04: готовят, как указано выше, но из 139 мл кислоты, кото¬
рую доводят до 1 л водой без органических веществ.
Методика
В разделе 13.2.1 указано, что на современном уровне знаний трудно найти точную стан¬
дартизованную методику обработки, пригодную для всех почв. Необходимо уточнять
условия гидролиза в соответствии с типом почвы. На основании обзора литературы,
приведенного в разделе 13.2.1, можно рекомендовать три варианта методик, которые
приведены ниже.
Обработка для преимущественного определения пентоз
Помещают 5 г почвы и 40 мл 2,5 М серной кислоты в круглодонную перегонную колбу
на 100 мл. Присоединяют обратный холодильник и кипятят в течение 20 мин (либо на¬
гревают в колбе с завинчивающейся крышкой при 100 °С в течение 20 мин).
Охлаждают реакционную смесь и фильтруют через воронку Бюхнера с фильтром из
стекловолокна или центрифугируют.
Общая обработка
Помещают остаток после предыдущей обработки (с фильтром из стекловолокна) и 2 мл
13 М раствора H2S04 в ту же самую круглодонную колбу на 100 мл.
Закрывают колбу и оставляют при комнатной температуре на 16 ч (на ночь).
При охлаждении аккуратно добавляют 50 мл воды, не содержащей органических ве¬
ществ.
Подсоединяют к колбе обратный холодильник и кипятят реакционную смесь в тече¬
ние 5 ч (либо нагревают до 90 °С в закрытом флаконе).
Фильтруют через фильтр из стекловолокна, как описано выше.
Для определения содержания общих сахаров можно смешать фильтраты, описан¬
ные в подразделах «Обработка для преимущественного определения пентоз» и «Общая
обработка». Анализ каждого гидролизата определяет: (а) легко гидролизуемые сахара
388
Часть 2. Органический анализ
(в основном пентозы) в гидролизате, описанном в подразделе «Обработка для преиму¬
щественного определения пентоз», (б) трудно гидролизуемые сахара, такие как целлю¬
лоза, в гидролизате, описанном в подразделе «Общая обработка».
Упрощенная общая обработка
Помещают в 100-миллилитровую круглодонную колбу 5 г почвы и 2 мл 13 М раствора
H2S04.
Закрывают колбу и оставляют при комнатной температуре на 16 ч (на ночь).
При охлаждении аккуратно добавляют 50 мл воды, не содержащей органических ве¬
ществ.
Подсоединяют к колбе обратный холодильник и кипятят реакционную смесь в тече¬
ние 5 ч (либо нагревают до 90 °С в закрытом флаконе).
Фильтруют через фильтр из стекловолокна, как описано выше.
13.2.2. Очистка кислотных гидролизатов
Принцип и основные проблемы метода
Перед хроматографическим определением гидролизаты должны быть очищены для уда¬
ления значительного количества неорганических соединений, особенно сульфат-ионов,
а также растворенных кислотной обработкой почвы неорганических твердых веществ
(особенно соединений железа и алюминия). Нейтрализация экстрактов включает осаж¬
дение гидроксидов алюминия и железа. Если нейтрализацию выполняют добавлением
карбоната или гидроксида щелочноземельных металлов (Са, Sr или Ва), могут также
осаждаться сульфаты. Риск ошибок связан с возможной адсорбцией осадками нейтраль¬
ных сахаров. При использовании гидроксида или карбоната бария нейтральные сахара
и бариевые соли уроновых кислот могут быть извлечены фильтрованием и промывани¬
ем осадков водой, иногда горячей водой или экстракцией этанолом в аппарате Сокслета
(Cheshire и Mundie, 1966). Для нейтрализации кислотных экстрактов с экстракцией метано¬
лом сульфата натрия после высушивания используется и бикарбонат натрия (Oades, 1967).
Использование карбоната кальция позволяет проводить нейтрализацию удалени¬
ем сульфатов вместе с большинством окрашенных органических веществ (Brink и др.,
1960). Оадес с сотр. (Oades и др., 1970) предпочитали использовать карбонат стронция
для того, чтобы довести pH гидролизата до 7. Авторы наблюдали соосаждение бурого
органического вещества и комплексов железа после хранения растворов в течение ночи.
После фильтрования и упаривания фильтрата досуха в роторном испарителе сахара рас¬
творяли в 4-5 порциях метанола по 2—4 мл и фильтровали.
Экстракт можно также очищать последовательным пропусканием через катионооб¬
менные и анионообменные смолы, которые также разделяют нейтральные сахара и уро-
новые кислоты. В качестве альтернативы можно элюировать сахара 50%-ным водным
раствором этанола с колонки, заполненной смесью угля и цеолита, после вымывания
солей водой (Cheshire, 1979).
В исследовательском институте IRD1 (неопубликованные данные) были проведены
сравнительные испытания семи методов очистки экстрактов:
• элюирование водой на амберлитовой ионообменной смоле МВ1;
• элюирование метанолом на амберлитовой ионообменной смоле МВ1;
1 IRD — Institute of Research for Development (ex-Orstom), BP 64501,911 Avenue of Agropolis, 34394
Montpellier Cedex 5, France.
Глава 13, Определение негуминовых веществ
389
• нейтрализация карбонатом стронция;
• нейтрализация бикарбонатом натрия;
• нейтрализация карбонатом бария;
• нейтрализация сульфатом бария;
• нейтрализация гидроксидом натрия.
Выделение максимального количества сахаров было достигнуто при нейтрализации
карбонатом стронция; этот результат соответствовал данным Оадеса с сотр. (Oades и др.,
1970).
Поправочные коэффициенты
Использование искусственных растворов позволило исследовать влияние нейтрализа¬
ции карбонатом стронция на выделение сахаров (Pansu, 1992). В три раствора добавляли
внутренний стандарт (мио-инозитол) перед нейтрализацией; в три других раствора его
добавляли перед восстановлением боргидридом очищенных концентрированных рас¬
творов. Каждый из конечных растворов дважды вводили в хроматограф. Расчеты абсо¬
лютного содержания показали следующее:
• Погрешность метода подготовки проб не превышала погрешности хроматографи¬
ческого определения (F-тест). Для большинства сахаров погрешность составляла
около 1%.
• Скорость адсорбции внутреннего стандарта осадком была выше, чем адсорбции
других сахаров. Поэтому предпочтительно вводить внутренний стандарт после
очистки экстрактов^ но перед дериватизацией сахаров.
• Средняя доля выхода сахаров составляла 85%, колеблясь от 71% для рибозы до 91%
для рамнозы. Рибоза всегда выделялась в самых малых количествах. На основе этих
величин процентных долей выхода мы предложили использовать следующие по¬
правочные коэффициенты: умножать полученные значения абсолютных содержа¬
ний каждого сахара на 1,21 для рамнозы, 1,08 для фукозы, 1,41 для рибозы, 1,10для
арабинозы и ксилозы, 1,25 для маннозы, 1,21 для галактозы и 1,15 для глюкозы.
Эти величины учитывают коэффициенты отклика каждого сахара по сравнению
с внутренним стандартом.
Оборудование и реагенты:
• Шесть лабораторных стаканов на 100 мл.
• Магнитные мешалки.
• Шесть круглодонных колб на 250 мл для роторного испарителя.
• Карбонат стронция, М = 147,63 г/моль.
Методика
Переносят фильтраты или отцентрифугированные растворы, описанные в разделе 13.2.1,
в лабораторные стаканы на 100 мл, перемешивают на магнитной мешалке. Добавляют
порошок карбоната стронция, количество которого рассчитывают, исходя из количества
серной кислоты, которое необходимо нейтрализовать, плюс 10%, т. е.:
• гидролизА: 1,1 х 40 х 2,5 х 147,63/1000 = 16,2 г;
• гидролиз Б: 1,1 х 2 х 13 х 147,63/1000 = 4,2 г;
• гидролиз В: то же, что гидролиз Б + 2 мл 2,5 М раствора кислоты для пропитывания
остатка, т. е. всего 4,9 г.
390 Часть 2. Органический анализ
SrC03 необходимо добавлять к раствору медленно, при перемешивании, чтобы не до¬
пускать чрезмерного образования пены.
Перемешивают реакционную смесь не менее часа. Проверяют, чтобы pH был ней¬
тральным или слабощелочным (при необходимости добавляют гранулы щелочи). Про¬
веряют осаждение гидроксидов железа, которые окрашивают осадок и обесцвечивают
раствор.
Центрифугируют смесь и собирают надосадочную жидкость в круглодонную колбу
для последующего анализа методом газовой хроматографии (ГХ1), если это необходимо
(см. раздел 13.2.4), и в мерную колбу на 100 мл в ином случае. Споласкивают остаток
25 мл метанола при перемешивании стеклянной палочкой. Снова центрифугируют и до¬
бавляют надосадочную жидкость к предыдущему раствору. Повторяют операцию еще
раз.
13.2.3. Колориметрическое определение сахаров
Методы
Большинство колориметрических методов основано на одном или двух из следующих
свойств сахаров: их восстановительной способности или образовании в сильных кис¬
лотах фурфурольных соединений, которые легко вступают в реакции с образованием
окрашенных производных.
Методы можно разделить на группы в зависимости от определения общего сахара,
гексоз или пентоз.
Колориметрическое определение общего сахара
Были исследованы два метода определения, основанные на восстановительной способ¬
ности углеводов: (а) восстановление щелочных растворов солей двухвалентной меди
(Фелингова жидкость) с образованием комплексов Си+ или (б) восстановление желтых
растворов феррицианидов до бесцветных ферроцианидов. Последний способ считался
предпочтительным для определения сахаров в почве и был автоматизирован Чеширом
и Мунди (Cheshire и Mundie, 1966).
В трех других методах используется свойство, основанное на свойствах фурфуроловых
производных: определение общего сахара с антроном, фенолом или орцинолом. Антрон
реагирует с фурфуроловыми производными в концентрированной серной кислоте с об¬
разованием красивого сине-зеленого окрашивания. Этот реагент обеспечивает лучшее
поглощение комплексов с дезоксигексозными и гексозными сахарами (за исключени¬
ем маннозы). Однако пентозные сахара дают меньший отклик и не могут быть опреде¬
лены, если содержание антрона превышает 0,05%. Кроме того, предполагается, что на
этот метод влияет присутствие других органических веществ, а также железа и нитратов
(|Cheshire, 1979).
Фенол реагирует с фурфуроловыми производными с образованием желтого окраши¬
вания (Dubois и др., 1956). Похожесть этого окрашивания на цвет гидролизатов почв мо¬
жет приводить к завышению содержания общего сахара при реакции с этим реагентом
(McGrath, 1973). Завышенные результаты были получены при прямом фенольном ко-
1 В оригинале авторы использовали термин «газофазовая хроматография» с аббревиату¬
рой GPC, обычно применяемой для обозначения гель-проникающей хроматографии (gel
permeation chromatography). — Примеч. науч.ред.
391
Глава 13. Определение негуминовых веществ
лориметрическом анализе без очистки гидролизатов (Pansu, 1992). Однако Дутр с сотр.
(Doutre и др., 1978) считали, что этот метод дает лучшие результаты, чем антроновый
метод.
Орцинол, или 3,5-дигидрокситолуол, также реагирует с фурфуроловыми произво¬
дными с достаточно близкими откликами всех сахаров. Бачелье (Bachelier, 1966) исполь¬
зовал его для почвенных гидролизатов.
Колориметрическое определение гексозных сахаров
Хотя антрон был первоначально предложен для определения общего сахара, он находит
большее применение для определения гексоз и дезоксигексоз в гидролизатах почв.
Иварсон и Сауден (Ivarson и Sowden, 1962) предложили использовать хромотроповую
кислоту для определения содержания гексоз. Этот реагент малочувствителен к присут¬
ствию пентоз и уроновых кислот и дал большие отклики, чем антрон, на четырех пробах
почв и подстилок.
Колориметрическое определение пентозных сахаров
Чешир и Мунди (Cheshire и Мundie, 1966) использовали смесь орцинола и FeCl3, описан¬
ную Томасом и Линчем (Thomas и Lynch, 1961), для максимального выделения пентоз¬
ных сахаров во время гидролиза. В уксусной кислоте анилин также реагирует с пентоз-
ными сахарами при комнатной температуре, образуя красные соединения, при слабом
влиянии гексозных сахаров и уроновых кислот (Ivarson и Sowden, 1962; Tracey, 1950).
Колориметрическое определение дезоксигексозных сахаров
Желтое окрашивание, образующееся при нагревании экстракта углеводов с цистеином
в серной кислоте, считается специфическим для дезоксигексозных сахаров; Чешир
и Мунди (Cheshire и Mundie, 1966) использовали этот реагент для почвенных гидроли¬
затов.
Определение уроновых кислот и аминосахаров — см. гл.14.
Оборудование и реагенты
• Градуированные стеклянные пробирки 150x25 мм.
• Водяная баня.
• Спектрофотоколориметр для видимого диапазона.
• Пластиковые колориметрические кюветы д линой 1 см.
• Колотый лед.
• Концентрированная (18 М) серная кислота (d = 1,84).
• 5%-ный раствор фенола в воде.
• 0,2%-ный раствор антрона в концентрированной H2S04.
• Стандартные растворы глюкозы для фенольного метода: 0, 5,10, 25, 50 и 100 мг/л.
• Стандартные растворы глюкозы для антронового метода: 0, 5,10,15, 20 и 25 мг/л.
Методика для фенольного метода
Помещают в пробирки 2 мл почвенного гидролизата (см. раздел 13.2.1) или очищенного
почвенного гидролизата (см. раздел 13.2.2), который был предварительно разбавлен до
100 мл в мерной колбе (можно предварительно проверить необходимость очистки мето¬
дом стандартных добавок).
392
Часть 2. Органический анализ
Добавляют 1 мл раствора фенола и затем сразу добавляют 5 мл концентрированной
серной кислоты, не допуская стекания ее по стенкам пробирки (и не допуская образо¬
вания брызг).
Оставляют пробирку стоять на 10 мин, перемешивают и помещают в водяную баню
при 25-30 °С на 20 мин, затем охлаждают под проточной водой.
Измеряют поглощение при 485 нм (490 нм для гексозных сахаров и 480 нм для пен-
тозных сахаров и уроновых кислот). Цвет остается стабильным в течение нескольких
часов. Иногда необходимо гомогенизировать раствор непосредственно перед колори¬
метрическим анализом.
Повторяют операцию для каждой точки калибровочной кривой.
Методика для антронового метода
Доводят объем гидролизованного раствора (см. раздел 13.2.1 или 13.2.2) до 100 мл и тща¬
тельно перемешивают.
Помещают 5 мл этого раствора в градуированной пробирке на лед. Повторяют эту
операцию для каждой точки калибровки.
Медленно добавляют в каждую пробирку 10 мл антронового раствора, позволяя ему
стекать по стенке пробирки; вращают пробирку для перемешивания.
Закрывают пробирки парафильмом и сразу же помещают их в водяную баню при
85 °С на 35 мин.
Охлаждают на ледяной бане, помещают в колориметрическую кювету и измеряют по¬
глощение при 625 нм. Следует использовать одноразовые пластиковые колориметриче¬
ские кюветы (длиной 1 см) для того, чтобы уменьшить количество операций по перено¬
су концентрированных растворов серной кислоты и отмыванию от них.
13.2.4. Определение содержания сахаров методом газовой
хроматографии
Основные положения
Определение содержания сахаров методом газовой хроматографии довольно затрудни¬
тельно по двум причинам:
• сахара являются слишком полярными соединениями для удовлетворительного
разделения непосредственно на хроматографических колонках; поэтому необходи¬
мо получить их производные, чтобы превратить гидроксильные функциональные
группы в менее полярные формы;
• газовая хроматография — более селективный метод, чем жидкостная хроматогра¬
фия; разделение углеводов может привести к образованию многих пиков, пред¬
ставляющих изомеры различных молекулярных конфигураций, и хроматограммы
могут стать слишком сложными для расшифровки.
Большинство авторов использовали методы, аналогичные методу Оадеса с сотр.
(Oades и др., 1970). К нейтрализованным и очищенным кислотным экстрактам добавля¬
ли боргидрид натрия, что переводило изомеры сахаров в их альдитоловые формы. После
удаления борной кислоты, образовавшейся при последовательном выпаривании в среде
уксусной кислоты, альдитолы были ацетилированы уксусным ангидридом.
Затем растворяли альдитолацетаты в дихлорметане для введения в хроматограф. Оа-
дес с сотр. (Oades и др., 1970) использовали колонку длиной 2 м с внутренним диаметром
3,5 мм, заполненную носителем GAS Chrom Q (100—120 меш), пропитанным 5%-ным
393
Глава 13. Определение негуминовых веществ
ECNSS-M. Восемь основных почвенных сахаров были разделены этим методом в тече¬
ние 70 мин, хотя рамноза и фукоза разделялись довольно плохо.
Спителлер (Sp (teller, 1980) усовершенствовал этот метод. Он разделил альдитолацета-
ты на капиллярной колонке длиной 25 м с неполярным носителем OV1 в течение 25 мин
при обнаружении следовых количеств глюкозамина и галактозамина. Чешир с сотр.
(Cheshire и др., 1983) разделили альдитолацетаты на капиллярной колонке размером
50 м х 0,3 мм с SILAR 10 СР в течение 90 мин, используя программируемое изменение
температуры. Дормаар (Dormaar, 1984) провел разделение примерно за то же время,
что Спителлер (Spiteller, 1980), используя стеклянную капиллярную колонку с 572330.
Балдок с сотр. (Baldock и др., 1987) применили методику Спителлера, также как Ар-
шад и Шнитцер (Arshad и Schnitzer, 1987), которые использовали капиллярную колонку
и стационарную фазу с трифторпропилметилом, что увеличивало продолжительность
анализа. Коэльо с сотр. (Coelho и др., 1988) использовали набивную колонку с разде¬
ляющей фазой той же длины, что Оадес с сотр. (Oades и др., 1970). Чешир и Гриффитс
(Cheshire и Griffiths, 1989) и Мураяма (Мигауата, 1988) также разделяли альдитолацетаты
методом хроматографии, но не указали, какие колонки использовали.
Операции восстановления и ацетилирования, которые следуют за очисткой гидро¬
лизатов, довольно продолжительны, поэтому производительность метода относительно
мала. Блакени с сотр. (Blakeney и др., 1983) рекомендовали использовать метод, позво¬
ляющий сократить время подготовки, но исследование, проведенное в лаборатории ин¬
ститута IRD, показало неудовлетворительные результаты (неопубликованные данные).
С другой стороны, время хроматографического анализа (рис. 13.1) было снижено до
12,5 мин при использовании капиллярной колонки с фазой 572330, аналогичной колон¬
ке, использованной Дормааром (Dormaar; 1984), но выполненной из кварцевого стекла
вместо боросиликатного стекла (Pansu, 1992).
Оборудование и реагенты:
• Пробирки с коническим дном на 5 мл и завинчивающимися крышками из ПТФЭ
(рис. 13.2).
• Пипетки пастера на 3 мл, оборудованные дозатором.
• Термостатированный алюминиевый нагревательный блок (рис. 13.2).
• Устройство для продувания азота для выпаривания (рис. 13.2).
• Газовый хроматограф с пламенно-ионизационным детектором.
• Кварцевая капиллярная колонка с 57>2330 (supelco) длиной 15 м и внутренним диа¬
метром 0,25 мм.
• Стандартный раствор углеводов в 85%-ном метаноле, содержащий 1 мг/мл каждо¬
го углевода: рамнозы, фукозы, рибозы, арабинозы, ксилозы, маннозы, галактозы
и глюкозы.
• Стандартный раствор, содержащий 1 мг/мл миоинозитола в 50%-ном метаноле.
• Метанол.
• Карбонат стронция.
• Боргидрид натрия.
• Ледяная уксусная кислота.
• Безводный раствор 10%-ной уксусной кислоты в метаноле (сушат над безводным
Na2S04).
• Уксусный ангидрид.
• Хлороформ.
394
Часть 2. Органический анализ
FU AR GA
RH Rl XY МА GL MY
GA MY
AR XY МА GL
L i » ■ I 1__1 I
0 4 8 12 Мин
210 222 234 246 вС
1 » t 1 1 I I 1
Рис. 13.1. Хроматограммы альдитолацетатов (условия описаны в подразделе «Условия хро¬
матографирования»: RH — рамноза; FU — фукоза; RI — рибоза; AR — арабиноза; XY — ксилоза;
МА — манноза; GA — галактоза; GL — глюкоза; MY — внутренний стандарт миоинозитола).
Слева — стандартная смесь, соответствующая введению 0,2 мкг каждого сахара; справа — саха¬
ра из сильно ненасыщенной ферраллитной почвы долины Ниари (Конго). Гидролиз проводили по
методике, описанной выше в подразделе «Обработка для определения пентоз»
Глава 13. Определение негуминовых веществ
395
Платиновый дапмкк
температуры i
Система регулировки i
температуры i
Завинч ивающаяся крыш ка
П рокладка иааГШШЭ
Граду и рован ная п роб и рка на 5мл i
АЛюмишевыйнагревательныйблокг
Реаггирук&щи© соединения
Нагреватель о регулируемой i
температурой!
Нагреватель сопротивления^
С и стем а охл ажде н и я
Электромагнитный кланам *
Трубке из неокиеляющегося^
Рис. 13.2. Рекомендуемая система для реакций дериватизации перед газохроматографическим
определением. Вверху— реакции в закрытых микропробирках при контролируемой температуре;
внизу— испарение избытка растворителей и реагентов в атмосфере азота
396
Часть 2. Органический анализ
Приготовление альдитолацетатов
а) В каждую колбу, содержащую очищенный гидролизат, добавляют точный объ¬
ем раствора мио-инозитола в качестве внутреннего стандарта, соответствующий
определяемому количеству сахаров (0,5-2 мл).
б) Выпаривают почти досуха на роторном испарителе; споласкивают колбу тремя-
четырьмя набольшими порциями метанола при помощи пипетки Пастера и пере¬
носят промывной раствор в пробирку на 5 мл с завинчивающейся крышкой.
в) Добавляют примерно 10 мг боргидрида натрия и оставляют реагировать на ночь.
г) Добавляют 0,1 мл ледяной уксусной кислоты, выпаривают досуха в токе азота при
70°С (рис. 13.2), добавляют 1 мл 10%-ного раствора уксусного ангидрида в этаноле
и выпаривают аналогичным образом; повторяют эту операцию пять раз.
д) Добавляют 1 мл уксусного ангидрида, закрывают пробирку и нагревают до 135°С
в течение 2 ч.
е) Охлаждают до 70 °С и выпаривают досуха в токе азота.
ж) Охлаждают и растворяют в точном объеме (0,5—2 мл) хлороформа в зависимости от
предполагаемого содержания сахаров.
Растворы можно оставить храниться или непосредственно вводить в хроматограф.
Приготовление стандартов для альдитолацетатов
Добавляют 1 мл стандартного раствора сахаров и 1 мл внутреннего стандарта в пробирку
на 5 мл с завинчивающейся крышкой и продолжают, как описано выше, начиная с пун¬
кта в.
Условия хроматографирования
• капиллярная колонка из кварцевого стекла с Supelco SP2330 (или аналогичная)
длиной 15 м и внутренним диаметром 0,25 мм;
• газ-носитель — гелий (0,7 бар);
• инжектор для ввода пробы с делением потока, скорость потока 100 мл/мин;
• объем инжекции - 1 мкл;
• пламенно-ионизационный детектор;
• температура колонки программируется:
от 210 до 250 °С при скорости 3 °С/мин;
инжектора — 300 °С;
детектора — 250 °С.
На рис. 13.1 показан пример хроматограмм, полученных для стандартного раствора
сахаров и для ферраллитной почвы из Конго.
13.2.5. Определение общего содержания липидов
Основные положения
Содержание общих липидов определяли гравиметрически после экстракции органи¬
ческими растворителями в аппарате Сокслета. Большинство липидных компонентов
почв — малополярные соединения, хотя некоторые из них обладают несколько большей
полярностью (Ambles и др., 1990). Поэтому выбор полярности экстрагента существенен
и требует проведения предварительных испытаний. Фад-Рашид (Fahd-Rachid, 1993) ис¬
пользовал хлороформ. Часто рекомендуют смесь петролейного эфира с этилацетатом
397
Глава 13. Определение негуминовых веществ
(3 : 1 по объему), поскольку она менее токсична, чем хлорсодержащие растворители.
Однако было показано, что эта смесь менее эффективна при экстракции сложных липи¬
дов, чем хлороформ (Jambu и др., 1987).
Растворители экстрагируют не все липиды, потому что некоторые из них связаны
с гуминовыми соединениями, глинами и различными минералами. Кислотная обра¬
ботка позволяет высвобождать связанные липиды, которые становятся доступными для
второй экстракции методом Сокслета. Хлористоводородную кислоту также можно ис¬
пользовать для кислотной обработки, однако согласно методу, рекомендованному Ван¬
гом с сотр. (Wang и др., 1969), лучше использовать смесь хлористоводородной и фтори¬
стоводородной кислот. Хлористоводородная кислота способна высвобождать липиды,
связанные с катионами, а фтористоводородная кислота может выделять липиды, свя¬
занные с органоминеральной матрицей (Fahd-Rachid, 1993).
Оборудование и реагенты
• Экстрактор Сокслета на 200 мл с обратным холодильником и круглодонной пере¬
гонной колбой из боросиликатного стекла на 500 мл с коническими шлифами.
• Электрический полусферический колбонагреватель с регулировкой температуры
для колб на 500 мл.
• Патроны для аппаратов Сокслета 38 мм в диаметре и 150 мм высотой.
• Роторный вакуумный испаритель.
• Пипетки Пастера.
• Волюметрические колбы на 25 мл (градуировка необязательна) с завинчивающей¬
ся крышкой из ПТФЭ.
• Стеклянные воронки диаметром 3 см.
• Пластиковые лабораторные стаканы на 250 мл.
• Пластиковые воронки диаметром около 8 см.
• Фильтровальная бумага для пластиковых воронок.
• Водный раствор HC1-HF (2,5% HF и 2,5% НС1).
• Экстрагирующий раствор - смесь «петролейный эфир: этилацетат» (3 :1 по объе¬
му): смешивают 750 мл петролейного эфира и 250 мл этилацетата.
Методика
Свободные липиды
• Помещают точную навеску Р( 100-150 мг) воздушно-сухой почвы, просеянной че¬
рез сито 2 мм, в экстракционный патрон и вставляют патрон в аппарат Сокслета.
• Помещают 200 мл экстрагирующего раствора и две гранулы пемзы в круглодонную
колбу на 500 мл; подсоединяют колбу к экстрактору Сокслета.
• Устанавливают обратный холодильник на экстрактор.
• Начинают нагревать перегонную колбу и регулируют температуру колбонагревате-
ля так, чтобы обеспечить конденсацию растворителя в холодильнике.
• Продолжают экстракцию в течение 48 ч.
• Охлаждают и устанавливают колбу на роторный испаритель; испаряют раствори¬
тель, пока его объем не уменьшится до нескольких мл.
• Одновременно взвешивают мерную колбу с цилиндрической горловиной на 25 мл
(после высушивания в сушильном шкафу и охлаждения).
398
Часть 2. Органический анализ
• Открывают колбу и помещают под маленькую воронку, заткнутую пробкой из сте¬
клянной ваты и наполовину заполненную безводным сульфатом натрия.
• При помощи пипетки Пастера декантируют остаток от выпаривания в маленькую
воронку и собирают сухой экстракт в колбу на 25 мл.
• Несколько раз споласкивают колбу для выпаривания и воронку экстрагирующим
раствором при помощи пипетки Пастера до получения объема около 20 мл в колбе
на 25 мл.
• Испаряют растворитель из колбы потоком азота (см. рис. 13.2) или под вакуумом
в вакуумном испарителе, используя шлифованные соединения.
• Закрывают колбу, охлаждают и взвешивают; вычитая массу тары, получают массу
свободных липидов Рг
• Рассчитывают процентное содержание свободных липидов = 100Р/Р.
Связанные липиды
• Помещают остаток после экстракции свободных липидов в пластиковый стакан
на 250 мл и добавляют раствор НС1—HF исходя из соотношения 150 мл раствора
на 100 г почвы.
• Оставляют взаимодействовать в течение 48 ч при периодическом перемешивании.
• Фильтруют через бумажный фильтр в пластиковой воронке.
• Промывают остаток водой до pH 5; декантируют в мелкую чашку или стеклянную
крышку и оставляют сушиться на лабораторном столе или в эксикаторе.
• Экстрагируют липиды, как описано выше для свободных липидов (см. «Свобод¬
ные липиды»).
Общие липиды
Можно непосредственно экстрагировать общие липиды обработкой исходной пробы
смесью HC1-HF с последующей экстракцией в аппарате Сокслета. Фад-Рашид (Fahd-
Rachid, 1993) обнаружил прекрасное соответствие между количеством экстрагирован¬
ных общих липидов и их количеством, рассчитанным как сумма свободных и связанных
липидов.
13.2.6. Количественное определение содержания водорастворимых
органических соединений
Для исследования пространственной и временной динамики органического вещества
необходимо учитывать водорастворимые органические фракции почвы. Эти фракции
включают органические кислоты, простые сахара или легкие полисахариды и азотсо¬
держащие соединения. Эти различные соединения оказывают существенное влияние
на структурную стабильность и плодородие почвы. Различают две группы соединений:
• Соединения, растворимые в холодной воде: их получают перемешиванием по¬
чвы с водой, используя различные методики, например, встряхивают 10 г почвы
в 200 мл воды на роторном шейкере в течение 2 ч, оставляют на ночь, встряхивают
еще в течение 2 ч и центрифугируют при 14 000 g.
• Соединения, растворимые в горячей воде: для их выделения можно использовать
различные методики, например, кипятить с обратным холодильником 4 г почвы
с 200 мл воды в присутствии трех стеклянных шаров в течение 16 ч, охладить и от-
центрифугировать при 14 000 g. Куакуа с сотр. (Коиакоиа и др., 1997) показали, что
399
Глава 13. Определение негуминовых веществ
количество водорастворимых соединений, экстрагируемых из ферраллитных почв
Конго, непрерывно возрастало с увеличением длительности экстракции как в су¬
шильном шкафу, так и в автоклаве. Согласно Ленвеберу с сотр. (Leinweber и др.,
1995), эта фракция в основном состоит из азотсодержащих и углеводных соеди¬
нений.
В табл. 13.1 доли этих фракций, выраженные в массовых процентах и содержании
углерода и азота, приведены для пяти обрабатываемых почв зон умеренного климата.
Водные экстракты для заболоченных или плохо аэрируемых почв, находящихся в ус¬
ловиях анаэробиоза (Ktisel и Drake, 1999), содержат органические кислоты низкой мо¬
лекулярной массы (например, молочную, пировиноградную или уксусную кислоту),
которые можно определить в водной среде методом газоадсорбционной (на стандарт¬
ной колонке с Рогарак, хромосорбом 101 или аналогичной) или ионной хроматографии.
Водные экстракты также содержат неорганические водорастворимые вещества.
Таблица 13.1. Соединения, растворимые в холодной (ХВР) и горячей (ГВР) воде, полученные
из обрабатываемых почв Буаньвиля, Франция (Pansu, неопубликованные
данные, условия экстракции см. в тексте)
№
ХВР,
%масс.
С,
%вХВР
C/N
в ХВР
ХВР С,
ГВР,
%масс.
с,
%вГВР
C/N
в ГВР
ГВР с,
А
0,026
28,8 ,
2,1
0,75
0,85
8,3
7,1
В
0,033
33,8
2,5
1,12
0,48
22,3
10,7
D
0,004
75,0
2,1
0,30
0,26
21,5
5,6
Е
0,021
27,1
1,4
0,57
0,24
25,1
5,5
6,0
F
0,025
30,1
1,8
0,75
0,38
22,4
8,5
13.3. Дополнительные методы анализа
13.3.1. Определение углеводородов почв методом газовой
хроматографии
Хотя альдитолацетатный метод (см. раздел 13.2.4) был самым распространенным ме¬
тодом определения содержания почвенных углеводов, предлагались и другие мето¬
ды: Моргенли (Morgenlie, 1975) описал выделение нейтральных сахаров в форме их
о-изопропилиден-производных. Производные получали действием 1%-ной серной кис¬
лоты в ацетоне; разделение проводилось на колонках 2Ж60 или OV225 в течение 35-40
мин. Трейтлер с сотр. {ТУаШеги др., 1984) выделили триметилсилил-производные са¬
харов на коротких неполярных колонках. Кови и Хеджес {Соте и Hedges, 1984) также
отделили триметилсилил-производные сахаров от гидролизатов в планктоне, отложени¬
ях и древесине. В качестве предварительной обработки эти авторы привели свободные
сахара в мутаротационное равновесие в присутствии перхлората лития, чтобы обойти
проблему сложных пиков. Ларре-Ларрой и Феллер {Larre-Larrouy и Feller, 1997), а так¬
же Ларре-Ларрой с сотр. {Larre-Larrouy и др., 2003) анализировали нейтральные сахара
в почве одновременно с уроновыми кислотами и гексозаминами в форме их триметил-
силил-производных, содержание каждого сахара рассчитывали как сумму площадей пи¬
ков их различных изомеров.
400
Часть 2. Органический анализ
13.3.2. Определение углеводородов методом жидкостной хроматографии
В течение многих лет методы жидкостной хроматографии давали неудовлетворитель¬
ные результаты при определении содержания сахаров. Чешир с сотр. (Cheshire и др.,
1969) разделили восемь нейтральных сахаров методом ионообменной хроматографии
в градиенте pH, но разделение продолжалось 14 ч. На выходе из колонки сахара анали¬
зировались колориметрически после реакции с орцином и серной кислотой. Гйдразид
п-гидроксибензойной кислоты является лучшим реагентом для щелочных элюатов; без
подкисления он дает желтое окрашивание с углеводами.
Хамада и Оно (Hamada и Опо, 1984) анализировали сахара в почвах, обогащенных
вулканической золой, методом высокоэффективной жидкостной хроматографии
(ВЭЖХ) на анионной колонке и регистрацией методом флуоресцентной спектроскопии
после взаимодействия с этаноламином. Разделение длилось 70 мин, но арабиноза, фрук¬
тоза и фукоза элюировались единым пиком, так же как рамноза и рибоза. Плюиджмен
(Pluijmen, 1987) анализировал сахара из различных растений методом ВЭЖХ на колонке
SUGAR РАК ТМ(WatersAssociates) с водой (или смесью ацетонитрил - вода) в качестве
подвижной фазы, рефрактометрической регистрацией и анионной и катионной пред-
колонками. Но этот автор сконцентрировал внимание на глюкозе и фруктозе и не пред¬
ставил хроматограмм.
Рейм и Ван Эффен (Reim и Van Effen, 1986) использовали более перспективный метод.
Они предложили новый детектор, более чувствительный, чем рефрактометр, который не
требовал предварительной дериватизации, как спектрометрические методы. Используя
ионообменную колонку, они одновременно разделяли простые сахара и низкомолеку¬
лярные олигомеры, но не представили хроматограмм восьми основных сахаров почвы.
Анжер с сотр. (Angers и др., 1988) разделили сахара из почвенных гидролизатов на ко¬
лонке aminex НРХ-87Р (BIO-RAD labs), но они определили только пять сахаров при до¬
вольно высоком пределе обнаружения.
Мартенс и Франкенбергер (Martens и Frankenberger, 1990) разделили десять почвенных
сахаров методами высокоэффективной анионообменной хроматографии и импульсной
амперометрии (HPAC-PAD, Dionex, Саннивейл, штат Калифорния, США) в следующих
условиях хроматографирования (рис. 13.3):
• петлевой дозатор на 200 мкл;
• предколонка CarboPac РА (25x3 мм);
• хроматографическая колонка (250x4 мм), заполненная пелликулярной анионооб¬
менной смолой (CarboPac РА\)\
• скорость элюента 0,8 мл/мин, комнатная температура;
элюент а: очищенная вода без органических веществ (18 МОм),
элюент б: водный раствор 50 мМ NaOH + 1,5 мМ CH3COONa,
93% элюента а и 7% элюента б в течение 15 мин, градиент до 100% за 25 мин,
та же подвижная фаза должна быть дегазирована для предотвращения поглоще¬
ния С02 и образования карбонатов, которые могут привести к движению ионов
и уменьшать время удержания;
• детектирование методом импульсной амперометрии с тремя последовательными
импульсами на золотом электроде:
Е\: 0,1 V; /1: 300 мс, окисление групп СНОН;
Е\\ 0,6 V; t\: 120 мс, перемещение продуктов реакции;
Е\: -0,8 V; Л: 300 мс, очистка электрода при отрицательном потенциале, время
отклика 1.
401
Глава 13. Определение негуминовых веществ
Рис. 13.3. Стандартное разделение сахаров методом высокоэффективной анионообменной хро¬
матографии (Martens и Frankenberger, 1990): Ino — инозитол, Rib — рибитол, Fu — фукоза, Rh —
рамноза, Ga - галактоза, GI - глюкоза, Ху — ксилоза, Ма — манноза, Ri — рибоза, Lac — лактоза
Работа Мартенса и Франкенбергера (Martens и Frankenberger, 1990) показала, что
комбинация высокоэффективной анионообменной хроматографии с импульсной ам-
перометрией имела некоторые преимущества перед другой классической системой
ВЭЖХ с рефрактометрическим детектированием: она дала более точные результаты,
имела вдвое лучшую чувствительность (на уровне пмоль) и лучшее разрешение. Под¬
готовка проб почвы также была проще. После кислотной обработки (см. раздел 13.2.1)
пробы обрабатывали 1 мл 0,1 М раствора ЭДТА, затем доводили pH до 4 добавлением
5 М раствора NaOH и центрифугировали при 10 000 g. Окрашенные материалы затем
удаляли фильтрованием на колонке для твердофазной экстракции (Supelco, Беллефонт,
штат Пенсильвания, США), содержащей 3 мл сильнокислотной катионообменной смо¬
лы (Н+-форма пропилсульфоновой кислоты) и 3 мл сильноосновной анионообменной
смолы (хлорид четвертичного пропиламмония). Экстракты также фильтровали через
фильтры GS 0,22 мкм (Millipore, Бедфорд, США).
Бернал с сотр. (Bernal и др., 1996) предложили хроматографический метод, аналогич¬
ный методу Мартенса и Франкенбергера (Martens и Frankenberger, 1990), для определе¬
ния содержания сахара в кофе и вине.
13.3.3. Фракционирование и исследование почвенной фракции липидов
Фракционирование липидов
Существует много методов фракционирования липидов, и можно обратиться к соответ¬
ствующим руководствам, в которых описаны методы биохимической липидологии, на¬
пример книге Кейтса (Kates, 1975).
Фракционирование можно разделить на две стадии:
• разделение липидов разных классов;
• определение содержания индивидуальных соединений каждого класса или целой
фракции.
Точная методика зависит от определяемого липида. Большинство липидов микробно¬
го и животного происхождения содержат от 60 до 85% фосфолипидов и гликолипидов,
а остальное состоит из нейтральных или неполярных липидов (глицериды, стерины,
402 Часть 2. Органический анализ
углеводы, пигменты). Липиды растительного происхождения содержат бблыиую долю
нейтральных липидов и меньшую долю фосфолипидов.
Для разделения липидов по классам используют методы двух основных типов:
• фракционирование растворителями и
• жидкофазная хроматография на колонке, бумаге или в тонком слое.
Характеризация и количественное определение компонентов липидов требуют хими¬
ческого расщепления сложных липидов с последующим разделением такими методами,
как газовая хроматография, обычно с пламенно-ионизационным детектированием, воз¬
можно в комбинации с масс-спектрометрией для идентификации молекул.
Фракционирование растворителями
Осаждение ацетоном является простейшим методом разделения фосфолипидов и ней¬
тральных липидов. Он основан на том, что большинство фосфолипидов (особенно
кислых фосфатидов в форме солей) нерастворимо в холодном ацетоне (0°С), тогда как
нейтральные липиды растворимы в нем. Этот метод подходит для липидов животного
и микробного происхождения, но он менее эффективен в присутствии больших коли¬
честв нейтральных липидов, включая большинство липидов растительного происхож¬
дения (Kates, 1975).
Методы разделения несмешиваемых растворителей (полярных и неполярных) также
могут применяться, особенно для липидов с высоким содержанием глицеридов. В каче¬
стве первой стадии фракционирования почвенных липидов Стивенсон (Stevenson, 1982)
рекомендовал разделение хлороформом и водным раствором гидроксида натрия. Затем
водную фазу подкисляют и экстрагируют эфиром для выделения свободных жирных
кислот. Фаза хлороформа содержит другие липиды, которые затем разделяют хромато¬
графией.
Фракционирование жидкофазной хроматографией
Было предложено много методов фракционирования жидкофазной хроматографией.
Для предварительного фракционирования экстракта общих липидов почвы Джем-
бу с сотр. (Jambu и др., 1991, 1993) и Амбле с сотр. (Ambles и др., 1989, 1990) использо¬
вали метод, рекомендованный МакКарти и Дьюти (McCarthy и Duthie, 1962). Липиды
разделяли на колонках калийного кремнезема (кремниевая кислота, обработанная ги¬
дроксидом калия в изопропаноле) на три фракции: нейтральную (первое элюирование
диэтиловым эфиром), кислую (элюирование 2%-ным раствором муравьиной кислоты
в эфире) и полярную. Зеллес и Бей (Zelles и Вах, 1993) использовали аналогичный метод
с твердофазной экстракцией.
Затем было можно отделить нейтральную фракцию липидов на колонках с двуоки¬
сью кремния (Kiesselgel 60, Merck) (Jambu и др., 1993) или флорисилом, обработанным
или не обработанным кислотой (Kates, 1975). Элюирование проводили сначала гексаном
или петролейным эфиром для отделения углеводов, а затем смесями с увеличивающейся
полярностью для отделения других классов липидов. Чаще всего использовали смеси,
содержащие увеличивающиеся доли диэтилового эфира в петролейном эфире, которые
позволяли последовательно разделять сложные эфиры (метиловые эфиры), кетоны,
триглицериды, диглицериды, моноглицериды, свободные спирты и стерины.
Методы газожидкостной хроматографии
Эти методы часто используют для фракционирования, характеристики и количествен¬
ного определения компонентов липидов. Их можно применять непосредственно к опре¬
Глава 13, Определение негуминовых веществ
403
деленным фракциям, таким как углеводы, или после расщепления тяжелых липидных
соединений до жирных кислот и неомыляемых веществ (см. «Фракционирование жир¬
ных кислот и неомыляемых веществ»).
Фракционирование жирных кислот и неомыляемых веществ
Основные положения
Реакция омыления может разрушить тяжелые липидные вещества до жирных кислот
и других продуктов омыления (глицерин и стерины), а также неомыляемых веществ.
Жирные кислоты затем метилируют, разделяют и определяют методом газо-жидкостной
хроматографии (ГЖХ), неомыляемые вещества также определяют методом ГЖХ после
силилирования гидроксильных групп. Метод можно применять к экстрактам общих ли¬
пидов или липидных групп, которые уже были разделены, как описано выше в подраз¬
деле «Фракционирование липидов».
Оборудование и реагенты
• Колбы F1: волюметрические колбы из боросиликатного стекла на 25 мл (мерные
колбы без градуировки, см. подраздел «Оборудование и реагенты» в разделе 13.2.5
и рис. 13.4) с навинчивающимися крышками из ПТФЭ.
• Колбы F2: волюметрические колбы на 25 мл с навинчивающимися крышками из
ПТФЭ.
• Колбы F3: волюметрические колбы из боросиликатного стекла на 10 мл (мерные
колбы без градуировки) с навинчивающимися крышками из ПТФЭ.
• F4: волюметрические колбы из боросиликатного стекла с градуировкой и навин¬
чивающимися крышками из ПТФЭ.
• Воронки диаметром 3 см.
• Пипетки Пастера.
• Весы с точностью 0,1 мг.
• Газовый хроматограф с инжектором для ввода проб с делением/без деления потока
для капиллярной колонки и пламенно-ионизационным детектором.
• Безводный петролейный эфир.
• Безводный метанол (хранят над безводным сульфатом натрия).
• 0,3 М раствор NaOH в метаноле: растворяют 1,2 г гидроксида натрия в 100 мл без¬
водного метанола.
• 3%-ный раствор H2S04 в метаноле: добавляют 3 мл концентрированной серной
кислоты к 70 мл безводного метанола в мерной колбе на 100 мл, перемешивают,
охлаждают, доводят объем до 100 мл и плотно закрывают.
• 60%-ный раствор метанола в воде.
• Безводный сульфат натрия.
Методика
Омыление
• Добавляют 4 мл петролейного эфира и 2 мл метанола в колбы F1 на 25 мл, содер¬
жащие экстракты свободных или связанных липидов (см. подраздел «Методика»
в разделе 13.2.5), и перемешивают в течение 1 ч.
• Добавляют 8 мл 0,3 М раствора NaOH в метаноле.
404
Часть 2. Органический анализ
• Герметично закрывают колбы и нагревают до 100 °С в термостатированном алюми¬
ниевом нагревательном блоке в течение 2,5 ч (рис. 13.2).
• Охлаждают, открывают колбы и добавляют 60%-ный метанол таким образом, что¬
бы фаза петролейного эфира оказалась в верхней цилиндрической части колбы.
• При помощи пипетки Пастера отбирают верхнюю фазу и переносят ее во взвешен¬
ную волюметрическую колбу F2 на 25 мл через небольшую коническую воронку,
заткнутую пробкой из стеклянной ваты и заполненную одним шпателем безводно¬
го сульфата натрия (рис. 13.4).
• Добавляют 3 мл петролейного эфира в первую колбу F1, закрывают и тщательно
перемешивают, затем удаляют верхнюю фазу, как описано выше, и добавляют ее
к предыдущей фазе через ту же воронку; повторяют эту операцию четыре раза. Эта
фаза содержит неомыляемые вещества и продукты омыления, за исключением
жирных кислот (F2).
• Подкисляют содержимое первой колбы, добавляя 1 мл разбавленной вдвое кон¬
центрированной хлористоводородной кислоты, затем снова экстрагируют петро-
лейным эфиром, как описано выше. Декантируют и собирают верхнюю фазу во
взвешенную колбу F3 на 10 мл; эта фаза содержит жирные кислоты.
Рис. 13.4. Разделение декантацией (слева) и высушивание полученного продукта {справа).
Глава 13. Определение негуминовых веществ
405
Не жирнокислотные соединения
• Выпаривают содержимое колбы F2 досуха под током азота или в роторном испа¬
рителе.
• Закрывают и взвешивают колбу, чтобы определить общее содержание веществ.
• Добавляют 100 мкл триметилхлорсилана (ТМХС), 300 мкл гексаметилдисила-
на (ГМДС) и 600 мкл пиридина, закрывают и нагревают до 70 °С в течение 5 ч
(рис. 13.2).
• Вводят 2 мкл смеси в газовый хроматограф в следующих условиях:
а) капиллярная колонка CP-Wax (или аналогичная) длиной 25 м и внутренним
диаметром 0,3 мм, температура 230°С;
б) газ-носитель гелий при 0,5 бар;
в) инжектор для ввода пробы с делением потока, скорость потока 50 мл/мин.
Жирные кислоты
• Высушивают содержимое колб F3 на 10 мл досуха и взвешивают для определения
массы общих жирных кислот.
• Добавляют 3 мл 3%-ного раствора серной кислоты в безводном метаноле; закрыва¬
ют колбы и нагревают до 70°С в течение 5 ч (рис. 13.2).
• Охлаждают и добавляют 2 мл петролейного эфира и примерно 3 мл воды для под¬
нятия эфирной фазы в цилиндрическую часть колбы (рис. 13.4).
• Переносят метиловые эфиры жирных кислот в колбы F4 на 10 мл, как описано
выше для нежирнокислотной липидной фракции, доливают до 10 мл и вводят
2 мкл в ГХ в следующих условиях (короткоцепочные кислоты < С20, рис. 13.5):
а) капиллярная колонка из кварцевого стекла 25 мх0,3 мм с фазой CP-Wax (или
аналогичной),
б) газ-носитель гелий, входное давление 0,6 бар,
в) инжектор для ввода пробы с делением потока, 180 °С, скорость потока
60 мл/мин,
2 °С/мин
г) программа повышения температуры 180°С ►240°С,
д) пламенно-ионизационный детектор, 240 °С.
Примечания
• Существуют реагенты, которые позволяют получить метиловые эфиры жирных
кислот практически моментально при комнатной температуре (например, BF3-
СН3ОН или диазометан), что гораздо быстрее, чем смесью Me0H-H2S04; реагент,
описанный в подразделе «Методика» раздела 13.3.3, также используется, потому
что он недорогой, а нагревательная система позволяет проводить дериватизацию.
• Для почв, содержащих мало жирных кислот, можно использовать волюметриче¬
ские колбы F4 на 10 мл для концентрирования смеси выпариванием растворителя
в токе азота.
• Хроматографическая колонка, используемая для разделения (рис. 13.5), непри¬
годна для фракционирования длинноцепочечных жирных кислот из почвы (С20-
С34); для этого необходима более короткая колонка или другая фаза.
• Методику, описанную далее в подразделе «Оборудование и реагенты», можно
упростить: для определения только жирных кислот используют прямое транс-
406
Часть 2. Органический анализ
16.0
Пальмитиновая
14:0
12:0
15:0
18:1 20:0
Олеиновая драхиновая
16:0
Стеариновая
16:1
Пальмитолеи-
новая
17:0
18:2
Линолевая
к
Рис. 13.5. Фракционирование метиловых эфиров короткоцепочечных жирных кислот из тро¬
пической железистой почвы Буркина-Фасо методом газовой хроматографии (условия описаны
в тексте, М. Pansu, неопубликованные данные)
метилирование липидной фракции смесью H2S04 — метанол, метиловые эфиры
жирных кислот экстрагируют декантацией в петролейном эфире после добавления
воды (рис. 13.4); однако этот метод может усложнить расшифровку хроматограмм
из-за возможного перекрывания пиков метиловых эфиров жирных кислот и не¬
омыляемых соединений.
13.3.4. Определение содержания остаточных пестицидов и поллютантов
Основные положения
Подобно липидам, пестициды и поллютанты растворяются в органических раствори¬
телях. Их можно экстрагировать аналогичными методами (см. раздел 13.2.5), хотя экс¬
тракция в аппарате Сокслета не рекомендуется для пестицидов. Действительно, молеку¬
лы часто нестабильны, и длительное кипячение смеси растворителя и экстрагированных
веществ может привести к недооценке содержания остатков.
В связи с проблемами нестабильности наиболее распространенные методы основаны
на холодной экстракции растворителями, особенно микрометоды (Steinwandter, 1992)
и методы с использованием сверхкритических жидкостей (Richter, 1992; табл. 13.2). Если
пробы почвы должны храниться какое-то время перед экстракцией, они должны быть
заморожены или высушены вымораживанием.
После выполнения экстракции, если содержание липидов велико, может понадобить¬
ся очистка. Существуют два метода: (а) разделение растворителями разной полярности
и (б) жидкостная хроматография на колонках с флоризилом или оксидом алюминия.
Глава 13. Определение негуминовых веществ 407
Таблица 13.2. Некоторые методы с использованием сверхкритических жидкостей для
экстракции остаточных пестицидов из почв (Richter, 1992)
Пестицвд
Условия экстракции
Перметрин, атразин, дельтаметрин,
диэлдрин, карбофуран, диурон, 2,4-Д,
метилпаратион
Метанол, 250“С, 150 бар, 1 мл/мин, 2 ч
дат
С02, 40вС, 100 бар, 0,7 г/с, 10 мин
дат
С02 + 5%-ный метанол или толуол, 40 или 100°С,
0,7 г/сек, 5 мин
Линдан, адцрин, ДДТ
С02,138 бар, 15 мин
Линурон,диурон
С02 + метанол, этанол или ацетонитрил,
75-120 °С, 100-400 бар, 2,5-8,5 мл/мин,
15-180 мин
Сульфонилмочевина
С02 + 2%-ный метанол, 40 °С, 223 бар, 6 мл/мин,
2-15 мин
Симазин, атразин, пропазин,
тербутилазин, цианазин
С02, до 48 °С, 230 бар, 1,7 мл/мин, 30 мин
Хлор- и фосфорорганические
100%-ный С02 и С02 + 10%-ный метанол,
соединения
50-70 °С, 150 или 300 бар, 25-60 мин
ДЦТ, ДЦЭ, дда, линдан, алдрин
СО, + 5%-ный ацетон, 75 °С, 400 бар, 60 мин
Обычно необходимо концентрирование почвенных экстрактов для снижения предела
обнаружения. Конечную стадию концентрирования полезно проводить, пропуская ток
азота над поверхностью экстракта в мерных остродонных колбах с цилиндрической гор¬
ловиной (см. раздел 13.3.3).
Рекомендуемым методом для анализа экстракта является газовая хроматография
из-за (а) высокой селективности, особенно на капиллярных колонках, и (б) большого
числа существующих селективных и очень чувствительных детекторов. Для большин¬
ства нелетучих пестицидов рекомендуется использовать инжектор со стеклянной иглой,
который обеспечивает максимально возможную концентрацию растворенных веществ
во время инжекции. Можно также использовать инжектор для ввода проб с делением/
без деления потока. Другие методики, основанные на ВЭЖХ, обычно совмещают с УФ-
детектором.
Разнообразие пестицидов и поллютантов, присутствующих в почве (см. раздел
13.1.4), привело к развитию очень большого количества специальных аналитических
методик, которые не могут быть подробно описаны здесь. На основе литературных дан¬
ных и лабораторной практики был обеспечен выбор методик для экстракции, очистки
и хроматографического анализа основных классов продуктов. В будущем эти методы
изменятся в результате дальнейшего развития аналитической химии, и некоторые из
рекомендаций потребуют уточнения. Например, Дженнаро с сотр. (Gennaro и др., 1996)
описали одновременное разделение остаточных гербицидов феноксиуксусной кислоты,
триазинов и фенилмочевин в почвах методом ВЭЖХ. Сели с сотр. (CW/идр., 1993) пред¬
ложили способ определения содержания феноксапропа и фенаксопроп-П-этила в ряде
почв. Анон (Апоп, 1993) предложил метод для одновременного определения 27 герби¬
цидов в почве методом ВЭЖХ. Кианг и Пюб (Kiang и Grob, 1986) предложили метод для
одновременного определения содержания 49 поллютантов методом капиллярной газо¬
вой хроматографии.
408
Часть 2. Органический анализ
Оборудование и реагенты
• Возвратно-поступательный шейкер.
• Делительные воронки из боросиликатного стекла со притертыми тефлоновыми
пробками на 50,125, 250 мл.
• Колонки для жидкостной хроматографии диаметром 1 см и длиной 50 см с проб¬
кой из ПТФЭ и резервуаром для растворителя.
• Мерные колбы на 10 мл.
• Роторный испаритель с колбами на 50,125,250 мл.
• Регулируемый алюминиевый нагревательный блок и система для выпаривания под
током азота (рис. 13.2). Или вакуумный центрифужный испаритель типа SpeedVac.
• Газовый хроматограф с несколькими селективными детекторами (например, детек¬
тор электронного захвата для галогенированных соединений, термоионный детек¬
тор для фосфорорганических соединений) и неселективными (например, пламен¬
но-ионизационным или масс-спектрометрическим). Рекомендуется использовать
инжектор со стеклянной иглой; при его отсутствии можно использовать инжектор
для ввода проб с делением/без деления потока или инжектор для прямого ввода
проб в колонку (Pansu и др., 2001).
• ВЭЖХ с УФ-детектором, если необходимо.
• Аттестованные растворители, не содержащие пестицидов и поллютантов. Каждый
растворитель должен быть проверен введением в хроматограф после концентриро¬
вания выпариванием в роторном испарителе (получая 1 мл из примерно 200 мл).
Наиболее широко распространенные растворители — ацетон, петролейный эфир,
гексан, диэтиловый эфир, метанол, ацетонитрил, дихлорметан, этилацетат.
• Различные стандарты пестицидов.
• Активированный флоризил (размер частиц 60-80 меш, 3% воды).
• Безводный сульфат натрия.
• Хлорид натрия.
Экстракция
Кислые гербициды
Йенсен и Diacc (Jensen и Glass, 1990) проверили несколько методик перед тем, как реко¬
мендовать холодную экстракцию диэтиловым эфиром в кислой среде. При энергичном
перемешивании в течение довольно длительного времени этот метод эффективен даже
для остаточных количеств, внесенных в почву год назад. Необходимо добавить достаточ¬
ный объем воды и кислоты для снижения вязкости пробы почвы и обеспечения хороше¬
го контакта с эфирной фазой. Авторы рекомендовали проводить три последовательных
экстракции эфиром в течение часа каждая.
Триазиновые гербициды
Можно использовать несколько различных растворителей. Для экстракции из водной
среды может подойти смесь равных объемов диэтилового эфира и петролейного эфи¬
ра. Для почв предпочтительна смесь дихлорметана и этилацетата в соотношении 1:5
по объему. Рекомендуется проводить три последовательные экстракции в возвратно¬
поступательном шейкере, добавляя последовательно 100, 50 и 50 мл смеси растворите¬
лей к пробе массой 20 г.
409
Глава 13. Определение негуминовых веществ
Хлорорганические соединения
Вилер и Томпсон (Weeler и Tompson, 1990) рассмотрели большое число методов, ис¬
пользуемых для экстракции соединений этого класса. Методы, применяемые для почв,
обычно основаны на методах анализа пищевых продуктов. Простой способ включает
перемешивание пробы почвы (50 г) с ацетоном (100 мл) или смесью «ацетон — петро-
лейный эфир» на возвратно-поступательном шейкере. К влажным пробам добавляют
безводный сульфат магния в качестве осушителя.
Фосфорорганические соединения
Для консервации проб почвы обычно рекомендуется использовать лиофилизацию.
Использовались несколько растворителей или смесей растворителей для стандартной
экстракции фосфорорганических соединений из почвы: ацетон, ацетон - вода, дихлор-
метан, этил ацетат, ацетон - гексан, ацетон - дихлорметан, метанол - вода (Barcelo
и Lawrence, 1992). Эти соединения могут экстрагироваться совместно с хлорорганиче-
скими соединениями (ацетон, ацетон — гексан), пиретриноидами (ацетон — гексан)
и карбаматами (ацетон — дихлорметан).
Карбаматы
Карбаматы можно определять, используя довольно сложную методику многоостаточ¬
ного анализа (Seiber, 1990). Пансу с сотр. (Рати и др., 1981а) исследовали следующий
метод со средним выходом экстрагируемых веществ 72±8%. Готовят пробы почвы лио-
филизацией, просеивают через сито с отверстиями 2 мм и делят на навески массой 10 г.
Встряхивают навеску с 50 мл метанола и 50 мл воды в возвратно-поступательном шей¬
кере в течение 6 ч, добавляют 100 мкл 1 М раствора НС1 и фильтруют под вакуумом.
Промывают остаток экстрагирующим раствором и добавляют промывные растворы
к фильтрату. Трижды экстрагируют фильтрат в делительной воронке на 200 мл последо¬
вательными порциями хлороформа 30, 20 и 20 мл.
Пиретриноиды
В биологических субстратах пиретриноиды выделяют двумя последовательными экс¬
тракциями гексаном в присутствии безводного сульфата натрия (Pansu и др., 198lb).
Для почв более эффективна смесь гексана с 3% ацетона.
Приготовление экстрактов для хроматографии
Основные положения
Большинство экстрактов должны очищаться перед хроматографированием для исклю¬
чения влияния других липидных компонентов почв. Использование детекторов, селек¬
тивных по отношению к большинству классов соединений (таких как хлор- и фосфор¬
органические соединения), позволяет уменьшить количество повторных операций
очистки. Однако некоторые из этих детекторов (например, детектор электронного за¬
хвата) очень чувствительны к загрязнению, и экстракты должны подвергаться очистке,
чтобы не снижать чувствительность детектора.
Экстракты следует максимально возможно концентрировать, чтобы улучшить выяв¬
ление ультраследовых количеств остатков. Экстракты обычно концентрируют в вакуум¬
ных роторных испарителях на первой стадии с использованием микрометодов, таких
как поток азота (рис. 13.2), или центрифужный испаритель SpeedVac на конечной ста¬
дии. Если для экстракции используют высокополярные растворители (трудно вымыва-
410
Часть 2. Органический анализ
емые из хроматографических колонок) или если они несовместимы с системой детекти¬
рования, экстракт можно перевести в другой растворитель перед инжекцией. Например,
если используют детектор электронного захвата, хлорсодержащие растворители должны
быть полностью удалены перед инжекцией. За исключением случая летучих пестици¬
дов, эта операция выполняется многократной сушкой экстракта после его растворения
в растворителе, не содержащем галогенов.
Наконец, для некоторых полярных соединений необходимо проводить реакции дери-
ватизации перед введением в хроматограф.
Кислые гербициды
Очистку можно проводить разделением между растворителями (Jensen и Glass, 1990).
Эфирные экстракты (см. подразделы «Кислые гербициды» и «Экстракция» в разделе
13.3.4) перемешивают с водным раствором бикарбоната натрия. В водном растворе кис¬
лые гербициды превращаются в соли, тогда как большинство окрашенных органических
соединений остается в органической фазе. Эту фазу удаляют, и кислые гербициды ре-
экстрагируют эфиром после подкисления водной фазы и насыщения хлоридом натрия.
Если анализ проводят методом газовой хроматографии, экстракты обычно необхо¬
димо метилировать перед инжекцией, поскольку кислые гербициды обычно высокопо¬
лярные соединения. Можно использовать различные реагенты в методах, аналогичных
метилированию жирных кислот (см. выше подраздел «Методика» в разделе 13.3.3). Осо¬
бенно эффективным метилирующим агентом является диазометан.
Триазины
Основные липиды можно удалить разделением на границе раздела ацетонитрил - гек¬
сан. Концентрируют дихлорметан-этилацетатные экстракты до 2 мл (см. подраздел
«Триазиновые гербициды»). Добавляют 20 мл ацетонитрила, насыщенного петролей-
ным эфиром (АН). Добавляют 10 мл петролейного эфира, насыщенного ацетонитрилом
(ПЭ), и перемешивают. Отделяют фазу АН. Реэкстрагируют ПЭ 10 мл АН. ПЭ отбра¬
сывают. Переносят все фазы АН в делительную воронку, добавляют 120 мл воды и 10 мл
насыщенного водного раствора NaCl. Дважды экстрагируют триазины 50 мл смеси «ди-
этиловый эфир - петролейный эфир» в равных объемах. Смешивают эфирные фазы,
высушивают над сульфатом натрия и концентрируют точно до требуемого объема (на¬
пример, 2 мл) перед инжекцией.
Хлорорганические соединения
Сначала ацетоновый экстракт необходимо перевести в гексановую фазу. Помещают аце¬
тоновый экстракт в делительную воронку (см. выше: параграф «Хлорорганические со¬
единения» в подразделе «Экстракция» раздела 13.3.4) с четырехкратным объемом воды
и 25 мл гексана. Осторожно перемешивают переворачиванием и декантируют органиче¬
скую фазу. Снова экстрагируют водную фазу 25 мл гексана, смешивают гексановые экс¬
тракты, высушивают над безводным сульфатом натрия и концентрируют до 5 или 10 мл.
Экстракты очищают на колонке с активированным флоризилом (3% воды). Взвеши¬
вают 5 г флоризила и помещают его в колонку диаметром 1 см, закрытую пробкой из
стеклянной ваты и частично наполненную петролейным эфиром. Добавляют 2—3 см
безводного сульфата натрия, споласкивают 50 мл гексана, наливая жидкость до верха
твердой фазы сульфата.
Помещают гексановый экстракт в колонку при помощи пипетки Пастера. Регулиру¬
ют поток до скорости 15-20 капель/мин при помощи крана из ПТФЭ. Когда уровень
411
Глава 13. Определение негуминовых веществ
жидкости достигает верха сульфата натрия, добавляют немного гексана из пипетки, по¬
вторяют эту операцию еще раз. Элюируют точным объемом гексана, необходимым для
выделения полихлорированных бифенилов (ПХБ), т. е. примерно 20-30 мл. Этот элюат
Э1 может также содержать гексахлорбензол (ГХБ) и дихлордифенилэтан (ДДЭ), один
из продуктов разложения ДДТ. Точное количество растворителя для элюата Э1 сначала
определяют с использованием стандартных смесей в аналогичных условиях. Для ПХБ
эти стандарты состоят из набора веществ, дающего типичные хроматограммы (напри¬
мер, смеси dp5 и dp6).
После отбора элюата Э1 продолжают элюирование 10%-ным раствором дихлорметана
в петролейном эфире (или гексане) до получения достаточного объема для сбора других
хлорорганических соединений (элюат Э2). Сначала определяют точный объем Э2 при
помощи одного или более стандартных соединений, например, диэлдрина.
Элюат Э2 необходимо отделить от дихлорметана. Высушивают почти досуха в ротор¬
ном испарителе. Добавляют немного петролейного эфира и снова высушивают почти
досуха. Повторяют эту операцию четыре или пять раз. Доводят элюаты Э1 и Э2 до точ¬
ною объема гексаном для хроматографического определения.
Фосфорорганические соединения
Сначала превращают ацетоновый экстракт в гексановую фазу. Помещают экстракт
в делительную воронку (см. выше: параграф «Фосфорорганические соединения» в под¬
разделе «Экстракция» раздела 13.3.4) с четырехкратным объемом воды и 25 мл дихлор¬
метана. Хорошо перемешивают и сливают органическую фазу. Снова экстрагируют,
используя 25 мл дихлорметана, смешивают экстракты, высушивают их над безводным
сульфатом натрия и выпаривают почти досуха в роторном испарителе. Добавляют не¬
много петролейного эфира и снова высушивают почти досуха. Повторяют эту опера¬
цию трижды. Несколько раз промывают гексаном, содержащим 10% (по объему) ацето¬
на (общий объем 10 мл), смешивают промывные растворы, закрывают и оставляют для
хроматографического определения.
Карбаматы
Хлороформные экстракты (см. выше: параграф «Карбаматы» в подразделе «Экстрак¬
ция» раздела 13.3.4) можно очистить следующим образом. Выпаривают досуха, добав¬
ляют в смесь 10 мл метанола и 10 мл 0,1 М раствора НС1, охлаждают в течение 15 мин,
отфильтровывают взвешенные вещества и промывают фильтр смесью равных объемов
воды и 0,1 М раствора НС1. Трижды экстрагируют фильтрат 25 мл хлороформа, высуши¬
вают экстракты над безводным сульфатом натрия и переносят в круглодонную перегон¬
ную колбу на 100 мл. Для удаления хлороформа выпаривают почти досуха в роторном
испарителе, добавляют немного метанола и снова выпаривают почти досуха; повторя¬
ют эту операцию трижды. Затем переносят остаток в мерную колбу с завинчивающейся
крышкой, содержащую точно 10 мл метанола, промывая несколько раз круглодонную
колбу несколькими миллилитрами метанола. Если нельзя немедленно ввести пробу
в хроматограф, экстракт хранят при -20 °С. Чтобы понизить предел обнаружения, экс¬
тракт можно концентрировать в пробирках под током азота при 40 °С (рис. 13.2).
Примечание: Растения могут превращать некоторые карбаматы в гликозиды. В ходе
экстракции (см. «Карбаматы» в подразделе «Экстракция» раздела 13.3.4) эти формы не
попадают в хлороформную фракцию, а остаются в водно-спиртовой фракции. Для их
определения необходимо разорвать гликозидные связи омылением при кипячении с об¬
ратным холодильником и реэкстрагировать хлороформом (Pansu и др., 1981а).
412
Часть 2. Органический анализ
Пиретриноиды
Экстракты, полученные в соответствии с параграфом «Пиретриноиды» в подразделе
«Экстракция» раздела 13.3.4, следует очистить перед введением в хроматограф, оборудо¬
ванный детектором электронного захвата. Очистку выполняют на хроматографических
колонках (диаметром 1 см), содержащих 5 г флоризила с 3% воды и 2 г безводного суль¬
фата натрия.
Применяют два различных метода для экстрактов, содержащих жирные вещества,
и экстрактов, не содержащих таких веществ (Pansu и др., 1981b). В отсутствие липидов
пиретриноиды выделяют элюированием 70 мл раствора 10%-ного диэтилового эфира
в петролейном эфире. В присутствии липидов предварительное элюирование пример¬
но 110 мл гексана позволяет удалить липиды перед элюированием пиретриноидов, как
описано выше. Точные объемы элюирующих растворов предварительно определяют
в экспериментах со стандартными пиретриноидами (декаметрин) и взвешиванием элю¬
ированных жирных веществ. Элюат, содержащий пиретриноиды, высушивают почти
досуха и снова растворяют точно в 10 мл гексана в мерной колбе с завинчивающейся
крышкой из ПТФЭ для хроматографического определения. Чтобы снизить порог обна¬
ружения, экстракты можно предварительно сконцентрировать в пробирках.
Хроматографическое определение
Существует широкий ассортимент хроматографических колонок и условий для опреде¬
ления пестицидов и поллютантов, наиболее интересные из них приведены ниже.
Кислые гербициды
Для определения содержания кислых гербицидов методом ГХ необходимо предвари¬
тельное метилирование кислотных функциональных групп (см. выше подраздел «Фрак¬
ционирование жирных кислот и неомыляемых веществ»). Для разделения метиловых
эфиров 2,4-Д и 2,4,5-Т используют колонку длиной 1,5 м и внутренним диаметром 4 мм,
содержащую 5% OV225 на хромосорбе Wс размером частиц 100-120 меш, с детектором
электронного захвата, в изотермических условиях при 220 °С. Кислые гербициды можно
также определять жидкостной хроматографией.
Триазины
Атразин и симазин могут быть определены методом ГХ в условиях, аналогичных услови¬
ям, описанным выше для кислых гербицидов, однако с использованием термоионного
детектора. Для одновременного определения всех азотсодержащих гербицидов предпо¬
чтительно использовать капиллярную колонку, такую как РТЕ-5 (Supelco), длиной 30 м
и внутренним диаметром 0,25 мм с программированием температуры от 40 °С (5 мин) до
100°С (30°С/мин) и затем до 275 °С (5°С/мин).
Хлорорганические соединения
Лучшая чувствительность при определении остаточных хлорорганических пестицидов
была достигнута методом ГХ с детектором электронного захвата. Можно использовать
различные типы неподвижных фаз для колонок, включая SE-30, OV-1 или OV-225.
Для разделения многочисленных ПХБ пиков рекомендуется использовать капилляр¬
ную колонку из кварцевого стекла типа SPB-Octyl (Supelco) длиной 60 или 30 м в зави¬
симости от программы нагрева с внутренним диаметром 0,25 мм и пленкой 0,25 мкм
SPB-Octyl
Глава 13. Определение негуминовых веществ
413
Для хлорированных пестицидов используют капиллярную колонку типа SPB-5 дли¬
ной 15 м и внутренним диаметром 0,20 мм с 0,20 мкм пленкой (Supelco) и температур¬
ной программой в диапазоне 120-290 °С.
Фосфорорганические пестициды
Фосфорорганические пестициды обычно определяют методом ГХ с термоионным или,
возможно, масс-спектрометрическим детектором. Для хлорорганических соединений
были рекомендованы различные колонки. Капиллярная колонка типа РТЕ-5 длиной
30 м и внутренним диаметром 0,25 мм с пленкой (Supelco) 0,25 мкм и программирова¬
нием температуры в диапазоне 50-300 °С рекомендуется для фракционирования смеси
девяти фосфорорганических пестицидов.
Карбаматы и пестициды — производные мочевины
Пестициды классов карбаматов и производных мочевины могут быть разделены мето¬
дом высокоэффективной жидкостной хроматографии с колонкой Supelcosil LC-8 длиной
15 см и внутренним диаметром 4,6 мм, частицы размером 5 мкм (Supelco), с градиентом
ацетонитрил - вода 18-65% в течение 9 мин, скорость потока 2 мл/мин, температура
40 °С, УФ-детектор при 240 нм.
Другой метод позволяет проводить определение всех ^-метилкарбаматов методом
ГХ, получая только один пик (Рати и др., 1981а). Введенную смесь трансметилируют
in situ в верхней части колонки и измеряют пик образующихся О-метил TV-метил карба¬
матов на выходе из колонки. Используют следующие условия: термоионный детектор,
колонка из пирекса длиной 1,4 м и внутренним диаметром 2 мм наполнена хромосорбом
101 (80-100 меш) на 1,3 м и VolaspherAl (80-100 меш, катализатор трансметилирования)
на последних 10 см около ввода, газ-носитель N2, скорость 30 мл/мин. Вводят 2 мкл
смеси из 950 мкл метанольного экстракта (см. подраздел «Карбаматы») и 50 мкл 0,2 М
метанольного раствора NaOH, приготовленного непосредственно перед введением.
Пиретриноиды
Эти довольно тяжелые и нестабильные соединения, тем не менее, могут определяться
с использованием ГЖХ с детектором электронного захвата, короткими колонками и
большими скоростями газа-носителя, чтобы уменьшить разложение (Рати и др., 198lb).
Для одновременного определения перметрина и декаметрина используют колонку дли¬
ной 80 см и внутренним диаметром 4 мм, наполненную 3% SE-30 на хромосорбе Ж80-
100 меш, при температуре колонки 215 °С, газ-носитель азот, скорость 85 мл/мин.
Использованная литература
Углеводороды почв
Angers DA, Nadeau Р and Mehuys GR (1988) Determination of carbohydrate composition of soil
hydrolysates by high-performance liquid chromatography. J. Chromatogr., 454,444-449.
Arshad and Schnitzer (1987) Characteristics of the organic matter in a slightly and in a severely crusted
soil. Pftanzenernahr Bodenk., 150, 412-416.
Bachelier G (1966) Les sucres dans les sols et leur dosage global. Cah. ORSTOM Sen PedoL, 4,9-22.
Bagautdinov FY, Khaziyev FK and Shcherbukhin VD (1984) Polysaccharide fraction of humic substances
from a typical chernozem and a gray forest soil. Soviet Soil Sci., 16, 37-42.
Baldock JA, Kay BD and Schnitzer M (1987) Influence of cropping treatments on the monosaccharide
content of the hydrolysates of a soil and its aggregate fractions. Can. J. Soil Sci., 67, 489-499.
Barriuso E, Andreux F and Portal JM (1985) Etude de la repartition des glucides associes aux constituents
humiques dans un sol humiffcre de montagne. C.R. Acad. Sci. Paris, 300, II, 16, 827-830.
414 Часть 2. Органический анализ
Benzing-Purdie LM and Nikiforuk JH (1989) Carbohydrate composition of hay and maize soils and their
possible importance in soil structure. J. Soil Sci., 40,125-130.
Bernal JL, Del Nozal MJ, Toribio L and Del Alamo M (1996) HPLC analysis of carbohydrates in wines
and instant coffees using anion exchange chromatography coupled to pulsed amperometric detection.
J. Agric. Food Chem., 44, 507-511.
Blakeney AB, Harris PJ, Henry RJ and Stone BA (1983) A simple and rapid preparation of alditol acetates
monosaccharide analysis. Carbohydrate Res., 113, 291-299.
Brink RH, Dubach P and Lynch DL (1960) Measurement of carbohydrates in soil hydrolysates with
anthrone. Soil Sci., 89, 157-166.
Cheshire MV and Anderson G (1975) Soil polysaccharides and carbohydrate phosphates. Soil Sci., 119,
356-362.
Cheshire MV and Griffiths BS (1989) The influence of earthworms and cranely larvae on the
decomposition of uniformly 14C labelled plant material in soil. J. Soil Sci., 40, 117-124.
Cheshire MV, Mundie CM, and Shepherd H (1969) Transformation of 14C glucose and starch in soil. Soil
Biol. Biochem. 1: 117-130.
Cheshire MV and Mundie CM (1966) The hydrolytic extraction of carbohydrates from soil by sulfuric
acid. J. Soil Sci., 17, 372-381.
Cheshire, MV, Mundie CM, Bracewell JM, Robertson GW, Russell JD and Fraser AR (1983) The
extraction and characterization of soil polysaccharide by whole soil methylation. J. Soil Sci.,
34:539-554.
Cheshire MV (1979) Nature and origin of carbohydrates in soils, Academic London, 216 p.
Coelho RRR, Linhares LF and Martin JP (1988) Sugars in hydrolysates of fungal melanins and soil humic
acids. Plant Soils, 106, 127-133.
Cowie GL and Hedges JI (1984) Determination of neutral sugars in plankton, sediments and wood by
capillary gas chromatography of equilibrated isomeric mixtures. Anal. Chem., 56,497-504.
Dormaar JF (1984) Monosaccharides in hydrolysates of water-stable aggregates after 67 years of cropping
to spring wheat as determined by capillary gas chromatography. Can. J. Soil Sci., 64, 647-656.
Doutre DA, Hay GW, Hood A and van Loon GW (1978) Spectrophotometric methods to determine
carbohydrates in soil. Soil Biol. Biochem., 10, 457-462.
Dubois M., Gilles K.A., Hamilton J.K., Rebers P.A., and Smith F. (1956) Colorimetric Method for
Determination of Sugars and Related Substances. Analytical Chemistry 28:350-356.
Folsom BL, Wagner GH and Scrivner CL (1974) Comparison of soil carbohydrate in several prairie and
forest soils by gas-liquid chromatography. Soil Sci. Soc. Amer. Proc., 38, 305-309.
Feller C., Francois C., Villemin G., Portal J.M., Toutain F., and Morel J.L. (1991) Nature des matures
organiquesassocteesaux fractions argileusesd’un sol ferrallitique. C.R. Acad. Sci. Paris312:1491-1497.
Francis C (1988) Les sucres neutres in “Devenir a court terme de differentes formes de Vazote dans un
ferrisoF, Doctorat de Tuniversit6 de Nancy I, 67-75.
Guckert A, Cure В and Jacquin F (1971) Comparative evolution of the polysaccharides of the humin after
incubation of glucose 14C and straw 14C. Trans. Intern. Symposium “humus etplante F”-Prague, 155-160.
Guckert (1973) Contribution a Vitude des polysaccharides dans les sols et de leur гф1е dans les
mecanismes d’agregation., ThSse doct. d’&at, Univ. Nancy 1,124 p.
Gupta UC and Sowden FJ (1965) Studies on methods for the determination of sugars and uronic acids in
soils. Can. J. Soil Sci., 45, 237-240.
Gupta UC (1967) Carbohydrates. In Soil Biochemistry McLaren et Peterson ed., Marcel Dekker, New
York, 91-118.
Hamada R and Ono A (1984) Determination of carbohydrates in hydrolysates of volcanic ash soil by
liquid chromatography with fluorescence spectroscopy, Soil Sci. Plant Nutr., 30, 145-150.
Ivarson КС and Sowden FJ (1962) Methods for the analysis of carbohydrate material in soil, Soil Sci.,
94, 245-250.
Larre-Larrouy MC and Feller C (1997) Determination of carbohydrates in two ferrallitic soils: analysis by
capillary gas chromatography after derivatization by silylation. Soil Biol. Biochem., 29,1585-1589.
Larr6-Larrouy M.C., Albrecht A., Blanchart E., Chevallier T, and Feller C. (2003) Carbon and
monosaccharides of a tropical Vertisol under pasture and market-gardening: distribution in primary
organomineral separates. Geoderma 117,63-79.
Глава 13, Определение негуминовых веществ
415
Martens DA and Frankenberger WT (1990) Determination of saccharides by high performance anion
exchange chromatography with pulsed amperometric detection. Chromatographia, 29, 7-12.
McGrath D (1973) Sugars and uronic acids in irish soils. Geoderma, 10, 227-235.
Morgenlie S (1975) Analysis of mixtures of the common aldoses by gas chromatography-mass
spectrometry of their O-isopropylidene derivatives. Carbohydrate Research, 41,285-289.
Murayama S (1983) Changes in the monosaccharide composition during the decomposition of straws
under field conditions. Soil Sci. Plant Nutr., 30,367-381.
Murayama S (1984) Decomposition kinetics of straw saccharides and synthesis of microbial saccharides
under field conditions. J. Soil Sci,9 35, 231-242.
Murayama S (1988) Microbial synthesis of saccharides in soils incubated with 13C labelled glucose. Soil
Biol. Biochem., 20, 193-199.
Oades JM (1967) Gas-liquid chromatography of alditol acetates and its application to the analysis of
sugars in complex hydrolysates. J. Chromatogr., 28, 246-252.
Oades JM (1974) Synthesis of polysaccharides in soil by microorganisms. Trans. Intern. Congr. Soil Sci.
10th- Moscow, 93-100.
Oades JM, Kirkman MA and Wagner GH (1970) The use of gas-liquid chromatography for the
determination of sugars extracted from soils by sulfuric acid. Soil Sci. Soc. Am. Proc., 34, 230-235.
Pansu M (1992) Les sucres neutres dans les sols: opportunity et tentative d’amyiioration. de leur
determination. Document IRD (ex-Orstom) Montpellier, 24 p.
Pluijmen MHM (1987) Sugar analysis with the Shaffer-Somogyi micro-analysis, High Performance
Liquid Chromatography and enzymatic analysis in crop samples. Commun. Soil Sci. Plant Anal., 18,
1049-1059.
Reim RE and van Effen RM (1986) Determination of carbohydrates by liquid chromatography with
oxidation at a nickel(III) oxide electrode. Anal. Chem., 58, 3203-3207.
Singhal and Sharma (1985) Status of carbohydrates in the acid hydrolysates of soils and humic acids of
Dehra dun forests (Uttar Pradesh). Proc. Indian ntn. Sci. Acad., B51, 3, 348-352.
Spiteller M (1980) Kapillargaschromatographische bestimmung von zuckem unterschiedlicher boden.
Z Pflanzenernaehr. Bodenkd., 143, 720-729.
Stevenson FJ (1982) Soil carbohydrates. In Humus chemistry, Wiley New York, 147-171.
Thomas and Lynch, (1961) A method for the quantitative estimation of pentoses in soil. Soil Sci., (1961),
91,312-316.
Tracey MV (1950) A colorimetric method for the determination of pentoses in the presence of hexoses
and uronic acids. Biochem. J., 47,433^136.
Traitler H, Del Vedovo S and Schweizer TF (1984) Gas chromatographic separation of sugars by on-
column injection on glass capillary columns. J. High Resol. Chromatog. Chromatogr. Commun., 7,
558-562.
Липиды почв
Wang TSC, Yu-Cheng Liang and Wey-Chiang Shen (1969) Method of extraction and analysis of higher
fatty acids and triglycerides in soils. Soil Sci., 107, 181-187.
Fahd-Rachid A (1993) Effet a long terme d'apports continus de dechets urbains sur les caracteristiques
du sol. Consequences sur les proprietes de la matiere organique en relation avec sa teneur en lipides.,
Th£se Doctorat Sciences agronomiques, INRA-Bordeaux, 151 p.
Ambles A, Jambu P and Ntsikoussalabongui В (1989) Evolution des lipids naturels d’un podzol forestier
induite par l’apport d’engrais mindraux: hydrocarbures, clones, alcools. Science du Sol., 27, 201—
214.
Ambles A, Jambu P and Ntsikoussalabongui В (1990) Evolution des acides gras d’un podzol forestier
induite par l’apport d’engrais mindraux. Science du Sol., 28, 27-42.
Jambu P, Fustec E and Jacquesy R (1978) Les lipides des sols: nature, origine, У volution, propriytys.
Science du Sol., 4, 229-240.
Jambu P, Bilong P, Ambles A, Ntsikoussalabongui В and Fustec E (1987) Influence d’apports ттУгаих
sur Г У volution des lipides naturels des sols acides. Science du sol., 25,161-172.
416
Часть 2. Органический анализ
Jambu Р., Ambles A., Dinel Н., and Secouet В. (1991). Incorporation of natural hydrocarbons from plant
residues into an hydromorphic humic podzol following afforestation and fertilization. Journal of Soil
Science 42: 629-636.
Jambu P., Ambles A., Jacquesy J.C., Secouet B., and Parlanti E. (1993) Incorporation of natural alcohols
from plant residues into a hydromorphic forest-podzol. Journal of Soil Science 44: 135-146.
McCarthy RD and Duthie AH (1962) A rapid quantitative method for the separation of free fatty acids
from other lipids. J. Lipid Res., 3, 117-119.
Stevenson FJ (1966) Lipids in soils. J. Am. Oil Chemists Soc., 43, 203-210.
Stevenson FJ (1982) Soil lipids. In Humus Chemistry., Wiley, 172-194.
Kates M (1975) Techniques of lipidology. In Laboratory Techniques in Biochemistry and Molecular
Biology, Work TS and Work E ed. Elsevier Amsterdam, 269-610.
Zelles L., and Bai Q.Y. (1993) Fractionation of fatty acids derived from soil lipids by solid phase
extraction and their quantitative analysis by GC-MS. Soil Biology Biochemistry 25: 495-507.
Водный экстракт
Leinweber P, Schulten HR and Kurschens M (1995) Hot water extracted organic matter: chemical
composition and temporal variations in a long-term field experiment. Biol Fertil. Soils, 20,17-23.
Kouakoua E, Sala GH, Barthes B, Larre-Larrouy MC, Albrecht A and Feller С (1997) La mattere orgahique
soluble a l’eau chaude et la stability de l’agrdgation. Aspects m6thodologiques et application a des
sols ferrallitiques du Congo. Eur. J. Soil ScL, 48, 239-247.
Kiisel К and Drake HL (1999) Microbial turnover of low molecular weight organic acids during leaf litter
decomposition. Soil Biol. Biochem., 31, 107-118.
Пестициды и загрязняющие вещества
Anon (1993) Determination of herbicides in soils by HPLC with UV-Detection. Agrobiological Res., 46,
155-174.
Barcelo D and Lawrence JF (1992) Residue analysis of organophosphorus pesticides. In Emerging
strategies for pesticide analysis, Cairns T and Sherma J ed. CRC, 127-149.
Calvet R, Barriuso E, Bedos C, Benoit P, Chamay MP and Coquet Y (2005) Les pesticides dans le sol -
Consequences agronomiques et environnementales. Edition France Agricole, Paris, 637 p.
Celi L, N6gre M and Gennari M (1993) HPLC determination of fenoxatrop and fenoxatrop-ethy in
different soils. Pestic. Sci., 38, 43-47.
Gennaro MC, Giacosa D, Baglieto C, Gennari M and Negre M (1996) Simultaneous separation of
phenylurea-, triazine-, and phenoxyacid herbicides by reverse phase ion-interaction HPLC.
Application to soil analysis. J. Liq. Chrom. Rel. TechnoL, 196, 911-924.
Jensen DJ and Glass RD (1990) Analysis for residues of acidic herbicides. In Analysis of Pesticide
Residues, Moye HA ed. Krieger, 223-261.
Kiang PH and Grob RL (1986) Developpement of a screening method for the determination of 49 priority
pollutants in soil. J. Environ. Sci. Health, A21, 15-53.
Pansu M, Gautheyrou J and Loyer JY (2001) Soil Analysis - Sampling, Instrumentation and Quality
Control., Balkema, Lisse, Abington, Exton, Tokyo, 489 pp.
Pansu M, A1 Salti MN, Aubert H and Gry J (1981a) Contribution a l*6tude de Pactivit6 syst6mique du
carbofuran au moyen de la chromatographie en phase gazeuse. Phytiatrie-Phytopharmacie, 30,203-214.
Pansu M, Dhouibi MH and Pinta M (1981b) Determination des traces de pyr6thrinondes (bioperm&hrine
et d6cam6thrine) dans les substrats biologiques par chromatographie en phase gazeuse. Analusis, 9,
55-59.
Richter BE (1992) Supercritical fluid extraction methods. In Emerging Strategies for Pesticide Analysis,
Cairns T and Sherma J ed. CRC, 51-68.
Seiber JN (1990) Carbamate insecticide residue analysis by gas-liquid chromatography. In Analysis of
Pesticide Residues, Moye HA ed. Krieger, 333-378.
Steinwandter H (1992) Development of microextraction methods in residue analysis. In Emerging
Strategies for Pesticide Analysis, Cairns T and Sherma J ed. CRC, 3-38.
Wheeler WB and Thompson NP (1990) Analysis of chlorinated hydrocarbons. In Analysis of Pesticide
Residues, Moye HA ed. Krieger, 199-222.
Глава 14. Органические формы азота,
минерализуемый азот (и углерод)
14.1. Введение
14.1.1. Круговорот азота
Продолжая собственные работы, а также исследования Кавендиша об «мефитическом
воздухе» или атмосферной «мофетте», Лавуазье (Lavoisier, 1789) открыл газ, который он
назвал «азотом», что означает «безжизненный». В соответствии с относительной хими¬
ческой стабильностью молекулы N2, это название вполне приемлемо с геохимической
точки зрения (табл. 14.1), поскольку количество азота, представленного в биосфере,
очень мало по сравнению с атмосферным азотом и составляет ничтожную часть от об¬
щего азота планеты.
Таблица 14.1. Геохимическое распределение азота (согласно Stevenson, 1982с)
Среда
Масса N, т
Литосфера
1,636 ю14
Атмосфера
38,6-1014
Биосфера
0,0028 1014
органическое вещество почвы
0,0022 1014
NH/—N глин
0,0002 1014
Однако для физиолога или агронома азот, наряду с углеродом, водородом и кислоро¬
дом, является одним из основных источников жизни; жизнь появилась на Земле в ре¬
зультате синтеза первых аминокислот.
В начале цикла азот в живых организмах связан, главным образом, с усвоением кор¬
нями растений неорганических форм N (см. гл. 28), прежде всего N-N03, в хорошо
аэрированной почве. Поскольку нитраты составляют лишь малую часть азота, а более
90% азота почвы находится в органических формах (Stevenson, 1982b), очевиден интерес
к изучению азотного цикла. На рис. 14.1 показаны источники нитратов: внешние ис¬
точники вносят некоторый вклад, но основным источником является органический N
почвы. Независимо от источника (1 — неразрушившиеся органические фрагменты (см.
гл. 9), 2 — молекулы гумифицированных веществ (см. гл. 11 и 12) или 3 — живые орга¬
низмы почвы), часть этого азота минерализуется до аммония. Аммоний затем связыва¬
ется микроорганизмами или превращается в нитриты под действием других микроорга¬
низмов, окисляется до нитратов или теряется в форме N20 или других газовых формах.
Таким образом, точное знание резервов органического азота почвы помогает иссле¬
дованию доступности аммонийного и нитратного азота, а также роста растений. Агро¬
номы определяют содержание общего органического азота (см. гл. 10) или запасы не¬
органического азота при выращивании сельскохозяйственных культур (см. гл. 28) для
оценки необходимого количества удобрений (Dahnke и Johnson, 1990). Однако эти два
показателя не отражают реальный цикл азота, а дают лишь приблизительные оценки:
• В данный момент времени количество нитрата или аммония, присутствующего
в почве, не включает неорганические формы N, которые могут вырабатываться
418
Часть 2, Органический анализ
Рис. 14.1. Схема круговорота азота: прерывистые линии — антропогенные источники; сплошные
линии — природный цикл; жирные линии — основной природный цикл в хорошо аэрированных
средах. А — усвоение растениями; М — минерализация органического вещества почвы до аммо¬
ния; N — нитрификация аммония; R — восстановление нитратов; I — связывание неорганического
азота микробами; V — испарение в процессах нитрификации и денитрификации; F—фиксация ам¬
мония в глинистых материалах; L— выщелачивание нитратов; SF — симбиотическая фиксация ат¬
мосферного азота растениями; NSF — несимбиотическая фиксация микробной биомассой почвы
при минерализации или потребляться в процессах преобразования, восстановле¬
ния, денитрификации.
• Существуют очень большие различия в стабильности веществ, содержащих до¬
ступный органический азот (Schulten и Schnitzer, 1998; Schippernnp., 2004); процесс
минерализации происходит лишь в малой части этого запаса, который поэтому не¬
обходимо оценить.
• Большинство процессов гумификации требуют потребления неорганического азо¬
та, который становится таким образом временно недоступным для растений.
• Экономические и экологические риски, возникающие вследствие беспорядочного
применения азотных удобрений (и приводящие к выщелачиванию нитратов и за¬
грязнению воды, улетучиванию N и, возможно, увеличению выделения теплич-
Глава 14. Органические формы азота, минерализуемый азот (и углерод)
419
ных газов, а также фиксации аммония в глинах), являются убедительными аргу¬
ментами в пользу анализов, которые позволят углубить знания об азотном цикле
и, в частности, о формах органического азота, которые представляют собой самый
большой потенциальный резерв в почвах для потребления растениями (Matsumoto
идр., 2002).
14.1.2. Типы методов анализа
Вышеперечисленные проблемы привели к появлению многочисленных работ, посвя¬
щенных исследованию органического азота почвы, которые можно разделить на две
большие группы:
• Исследования форм органического азота, например, работы (Вгетпег, 1965),
(Jocteur Monrozier и Andreux, 1981), (Jocteur Monrozier, 1984), (Kelley и Stevenson,
1995), (Stevenson, 1982a, b, c, 1996).
• Исследования с непосредственной агрономической целью изучить азот, постав¬
ляемый почвой, т. е. потенциально доступный азот, (например, работы (Keeney
и Вгетпег, 1966), (Comforthn Walmsley, 1971), (Juma и Paul, 1984), (Gianello и Вгетпег,
1986), (Catrouxnjxp., 1987), (GirouxnSen Tran, 1987),(СаЬгегаиKissel, 1988)),атак-
же кинетику минерализации С и N.
Первая группа работ обычно исследует формы азота в растворе после гидролиза, с по¬
мощью которого разрушают связи больших органических молекул. Самые последние
результаты исследований этих форм азота представлены в данной главе, наряду с анали¬
зом мочевины как специфической формы азота.
Вторая группа исследований основана на двух подходах к определению параметров
доступности почвенного азота:
• биологический подход к оценке неорганического азота, образующегося при инку¬
бации в контролируемых условиях;
9 методы химической экстракции.
Методы, описанные в настоящей главе, включают экстракцию электроультрафиль¬
трацией (ЭУФ) и методы характеризации форм азота в кислотных гидролизатах. Изо¬
топные методы с использованием метки 15N также являются мощными инструментами
для изучения переноса азота между органическими и неорганическими компартмента-
ми (Guiraud, 1984; Chotte, 1986; Pansu идр., 1998). Методы, описанные ниже, могут ком¬
бинироваться с методами определения изотопа 15N.
14.2. Классические методы анализа
14.2.1. Формы органического азота, выделяемого при кислотном
гидролизе
Основные положения
Арбитражный метод был разработан Бремнером (Вгетпег, 1965) и затем рекомендован
Стивенсоном (Stevenson, 1982а, 1996). Он является адаптацией гораздо более старых ме¬
тодов характеризации белков путем их гидролиза (Van Slyke, 1911-1912).
Согласно Бремнеру, 60-80% общего азота с поверхности почвы солюбилизирует¬
ся обработкой, используемой для кислотного гидролиза белков. Стивенсон (Stevenson,
1982b) утверждал, что 25-35% азота не солюбилизируется под воздействием кислоты.
Согласно Стивенсону, этот азот является структурным компонентом гуминовых ве¬
420
Часть 2. Органический анализ
ществ, а не образуется в известной реакции конденсации между аминокислотами и вос¬
станавливающими сахарами. Дополнительную фракцию можно экстрагировать разбав¬
ленными основаниями и затем солюбилизировать кислотным гидролизом (Griffith и др.,
1976). Предобработка фтористоводородной кислотой (см. раздел 14.3.1) также улучшает
солюбилизацию азота.
Белковый характер органического азота почвы согласуется с фактом, что наиболее
растворимый азот находится в форме аминокислот: 20-40% по Бремнеру (Вгетпег, 1965)
или 30-45% по Стивенсону (Stevenson, 1982b).
Эта обработка также дает 20-35% общего азота почвы (Stevenson, 1982b) в форме ам¬
миака (в количестве, близком количеству аминокислот). Этот аммиак происходит из
различных источников: разложение амидных форм белков, полная или частичная де¬
струкция некоторых аминокислот (таких как триптофан, серин, треонин), частичная
деструкция гексозаминов и выделение аммония, фиксированного на глинах.
Азот в аминосахарах составляет от 5 до 10% азота почвы. Наконец, 10-20% азота по¬
чвы существует в неизвестных формах, т. е. он не включен ни в одну из перечисленных
форм (Stevenson, 1982b).
Ниже приведены условия гидролиза, рекомендованные Бремнером (Вгетпег, 1965)
и затем Стивенсоном (Stevenson, 1982а). Другие возможные варианты рассматриваются
в разделе 14.3.1.
Метод, разработанный Бремнером для оценки различных форм азота в гидролиза¬
тах, включает фракционирование перегонкой с паром свободного аммония, гексоза¬
минов и аминокислот (табл. 14.2). Метод перегонки с паром, возможно, менее точен
и селективен, чем спектрометрические и хроматографические методы (см. раздел
14.3.2), но он прост. Одно и то же перегонное оборудование (рис. 14.3) можно исполь¬
зовать как для определения всех фракций, так и для определения общего азота (см.
гл. 10) и неорганических форм азота в случае почв, достаточно обогащенных азотом
(см. гл. 28). Кроме того, преимуществом этого метода является то, что каждая фрак¬
ция определяется в форме аммиачного дистиллята, пригодного для всех исследований
с использованием азота, меченого изотопом l5N, поскольку содержание этого изотопа
можно определять в молекулярном азоте, образующемся при окислении аммонийной
формы азота.
Определение фракции (NH3 + аминосахара)-Ы в гидролизатах основано на том, что
(1) глюкозамин и галактозамин быстро разлагаются в щелочной среде с образованием
аммония и (2) гексозамины можно определить по количеству аммония, выделившегося
при перегонке с паром гексозаминов в фосфатно-боратном буферном растворе с pH 11,2
(Tracey, 1952).
Определение содержания аммачных форм азота NH3-N основано на наблюдениях
Бремнера (Вгетпег, 1960): влияние гексозаминов и других не очень стабильных форм
на определение содержания аммония перегонкой в щелочной среде может быть исклю¬
чено, если выполнять перегонку с паром с малым количеством MgO в течение очень
короткого времени дистилляции.
Метод определения содержания форм а-аминокислот основан на методе определе¬
ния аммиака, выделенного при окислении нингидрином (рис. 14.2). Он был разрабо¬
тан после открытия (Вгетпег, 1960), что реакция конденсации В (рис. 14.2), приводящая
к захвату выделенного аммиака (реакция А) в форме окрашенного соединения, не про¬
исходит при pH 2,5. Таким образом, определение а-аминокислот может осуществляться
обработкой нингидрином при pH 2,5 с последующей перегонкой с паром продуктов ре¬
акции в присутствии фосфатно-боратного буферного раствора при pH 11,2.
Глава 14, Органические формы азота, минерализуемый азот (и углерод) 421
Таблица 14.2. Методы перегонки с паром для определения содержания различных форм азота
в почвенных гидролизатах и процентное содержание каждой формы в зависи¬
мости от общего азота почвы (Вгетпег, 1965; Keeney и Вгетпег, 1967; Stevenson,
1982а, Ь)
Форма N
Метод
%otN
почвы
Аммиачный N
Перегонка с паром с MgO
20-35
N аминосахаров
Перегонка с паром с фосфатно-боратным буферным
5-10
N аминокислот
раствором при pH 11,2 и вычитание NH4 - N
Перегонка с паром с фосфатно-боратным буферным
30-45
(серин + треонин)-Ы
раствором после обработки NaOH при 100 °С
для разложения гексозаминов и удаления аммония и затем
с нингидрином (pH 2,5; 100 °С) для перевода а-амино-N
в NH3-N
Перегонка с паром с фосфатно-боратным буферным
(аммиак +
раствором после удаления форм (аммоний+
гексозамин)-Ы перегонкой с паром с тем же буферным
раствором и обработок перйодатом для превращения
(серин + треонин)-Ы в NH3—N и с метаарсентом
для восстановления избытка перйодата
Перегонка с паром с фосфатно-боратным буферным
+ аминосахара +
раствором после обработки нингидрином (pH 2,5; 100 °С)
+ аминокислотьО-N
для перевода а-амино-N в NH3-N
Общий
Перегонка с паром с NaOH после разложения методом
65-80
гидролизуемый N
Къельдаля со смесью H2S04+ K2S04 — катализатор
Неизвестный
Общий гидролизуемый азот - (NH3-N + амино-N)
гидролизуемый N
(сахаров + а-аминокислот)^ + азот (серин + TpeoHHH)-N
10-20
Рис. 14.2. Реакция а-аминокислотс нингидрином. При pH 2,51МН3 является стабильным продуктом
реакции (реакция А); когда реакционная смесь приводится к pH 5, аммиак, выделившийся
в реакции А, соединяется с восстановленной и окисленной формами нингидрина (реакция
В) с образованием соединения, окрашенного в синий цвет (Вгетпег, 1965; Stevenson, 1982а)
422
Часть 2. Органический анализ
Отдельный метод определения содержания азота серина и треонина основан на спо¬
собности соединений, содержащих амино- и гидроксил-группы у соседних атомов,
окисляться перйодатом с образованием аммиака:
R-CHCOHJ-CHCNHj)-^ + NaI04 -► R,CHO + ЩСНО + NH3 + NaI03
Влияние форм (аммоний + гексозамин)-Ы можно исключить предварительной пере¬
гонкой с фосфатно-боратным буферным раствором при pH 11,2. Аммоний, выделив¬
шийся при окислении перйодатом, определяют перегонкой с паром после добавления
и«е/и0арсенита для удаления избытка перйодата (Keeney и Вгетпег, 1967):
NaAs02 + NaI04 -> NaAs03 + NaI03
Оборудование
• Оборудование для разложения по микрометоду Къельдаля.
• Прибор для перегонки с паром (рис. 14.3), который также можно использовать для
определения неорганического азота в почвах, богатых N (см. гл. 28), и для опреде¬
ления общего азота (см. гл. 10) вместо автоматизированного дистиллятора..
• Колбы Къельдаля из пирекса на 50 и 100 мл со стандартными шлифами 29/32
и крючками для пружин.
• Микробюретка на 5 мл, проградуированная с интервалами 0,01 мл или автоматизи¬
рованный титратор с двойным электродом для измерения pH и автоматизирован¬
ной бюреткой на 5 мл.
Щелочной реагент
Рис. 14.3. Прибор для перегонки с паром
423
Глава 14. Органические формы азота, минерализуемый азот (и углерод)
Реагенты
1. 6 М хлористоводородная кислота: добавляют 513 мл концентрированной НС1
(d = 1,19) в примерно 500 мл воды, охлаждают и доводят объем до 1 л в мерной
колбе.
2. я-октиловый спирт.
3. Смесь катализаторов с сульфатом калия для разложения по Къельдалю (см. при¬
готовление в подразделе «Реагенты» раздела 10.2.6).
4. 10 Мраствор гидроксида натрия (см. приготовление в подразделе «Реагенты» раз¬
дела 10.2.6).
5. 5 Мраствор гидроксида натрия: разбавляют 500 мл 10 М раствора гидроксида на¬
трия до 1 л и хранят в плотно закрытой бутыли.
6. 0,5 Мраствор гидроксида натрия: разбавляют 50 мл 10 М раствора гидроксида на¬
трия до 1 л и хранят в плотно закрытой бутыли.
7. Раствор борной кислоты с индикатором: растворяют 100 г борной кислоты в 4 л
деионизированной воды, добавляют 100 мл смешанного индикатора (0,495 г бром-
крезола зеленого и 0,33 г метила красного в 500 мл этанола), доводят pH до 4,8-5,0
(красновато-фиолетовый цвет) добавлением небольшого количества разбавленно¬
го гидроксида натрия или хлористоводородной кислоты и доводят объем раствора
до 5 л.
8. 0,005 н. раствор серной кислоты: используют коммерческий стандартный раствор
(фиксанал).
9. Безводный оксид магния. При необходимости прокаливают в муфельной лечи при
600-700 °С в течение 2 ч.
10. Нингидрин (С9Н403 • Н20), реактив на аминокислоты.
11. Фосфатно-боратный буферный раствор с pH 11,2. помещают 100 г фосфата натрия
(Na3P04* 12Н20), 25 г буры (Na2B407- ЮН20) и примерно 900 мл воды в мерную
колбу на 1 л, перемешивают до растворения, доливают до 1 л и хранят в плотно
закрытой колбе.
12. Лимонная кислота (С6Н807* Н20).
13. Цитратный буферный раствор с pH 2,6: смешивают 2,06 г двухводного цитрата на¬
трия (Na3C6H507* 2Н20) и 19,15 г лимонной кислоты (С6Н807* 2Н20), тщательно
размалывают в ступке и хранят в небольшом закрытом флаконе.
14. 0,2 Мраствор периодной кислоты: растворяют 4,6 г НЮ4- 2Н20 в 100 мл воды и хра¬
нят в стеклянном флаконе с притертой пробкой.
15. 1Мраствор метаарсенита натрия: растворяют 13 г NaAs02 в 100 мл воды и хранят
в плотной закрытой колбе.
16. Стандартный раствор A (NH* + аминосахар + аминокислота)-N: растворяют 0,189 г
(NH4)2S04, 0,308 г гидрохлорида глюкозамина и 0,254 г аланина в воде; разбавля¬
ют до 2 л в мерной колбе при энергичном перемешивании; этот раствор содержит
20 мкг NH4 -N, 10 мкг аминосахара-N и 20 мкг а-аминокислоты-N в 1 мл; хранят
в холодильнике при 4°С.
17. Стандартный раствор В (серин + треонин)-N: растворяют 150 мг серина и 170 мг
треонина в воде, разбавляют раствор до объема 2 л в мерной колбе и тщательно
перемешивают; этот раствор, приготовленный из сухих чистых веществ, содержит
10 мкг серина-N и 10 мгтреонина-N в 1 мл; хранят в холодильнике при 4°С.
424
Часть 2, Органический анализ
Методика
Кислотный гидролиз
Взвешивают тонко измельченную (< 100 меш) воздушно-сухую пробу почвы, содержа¬
щую примерно 10 мг общего азота (см. гл. 10), и помещают ее в круглодонную перегон¬
ную колбу из пирекса на 125 мл со стандартной притертой пробкой.
Добавляют две капли октанола и 20 мл 6 М раствора НС1. Тщательно перемешивают,
подсоединяют к перегонной колбе холодильник Либиха и кипятят в течение 12 ч.
Охлаждают реакционную смесь и фильтруют под вакуумом через воронку Бюхнера
со стандартной фильтровальной бумагой Whatman А°50. Споласкивают остаток малыми
порциями до тех пор, пока не набирается 50 мл общего фильтрата. Охлаждают фильтрат
на ледяной бане и нейтрализуют до pH 6,5 + 0,1 добавлением гидроксида натрия, из¬
меряя pH при помощи pH-метра. При перемешивании осторожно добавляют гидрок¬
сид натрия (раствор не должен становиться щелочным), используя сначала 5 М раствор
примерно до pH 5, а затем 0,5 М раствор до нейтрализации. Переносят раствор в мер¬
ную колбу, доводят опять до комнатной температуры и доливают до 100 мл, осторожно
споласкивая использованные колбы и электроды дистиллированной водой; осторожно
закрывают и тщательно перемешивают.
Разделение перегонкой, меры предосторожности
В каждом из следующих методов для определения азота помещают 5 или 10 мл гидро¬
лизата в колбу для перегонки на 50 или 100 мл (см. подраздел «Кислотный гидролиз»
раздела 14.2.1). После соответствующей обработки подсоединяют колбу к аппарату для
перегонки с паром. Определяемая форма азота соответствует количеству аммиака, пере¬
гнанному в течение 2-4 мин.
Аппарат следует промывать перед каждым использованием для удаления всех следов
аммиака. Скорость перегонки должна быть отрегулирована для сбора 7-8 мл дистиллята
в минуту. Поток охлаждающей воды в холодильнике следует отрегулировать так, чтобы
температура дистиллята при данной скорости перегонки не превышала 22 °С.
Для получения представительной субпробы гидролизата очень важно перемешивать
содержимое мерной колбы перед каждым отбором. Кончик пипетки не должен быть
слишком мал, чтобы можно было быстро перенести пробу в колбу для перегонки. Ис¬
пользование пипеток с очень маленькими отверстиями может приводить к ошибкам
пробоотбора из-за удержания отдельных компонентов суспензий или закупоривания
отверстия. Если определение проводят не сразу после перегонки с паром, то предпо¬
чтительнее хранить гидролизат в кислой среде и нейтрализовать его непосредственно
перед определением.
Определение общего азота в гидролизате
Помещают 5 мл нейтрализованного гидролизата в колбу для перегонки на 50 мл, до¬
бавляют 0,5 г смеси катализатора с K2S04 и 2 мл концентрированной серной кислоты.
Осторожно нагревают на плите микроаппарата Къельдаля до удаления воды и прекра¬
щения выделения белого дыма. Увеличивают нагрев до осветления смеси и завершают
разложение спокойным кипячением в течение 1 ч.
Охлаждают реакционную смесь и осторожно добавляют 10 мл дистиллированной
воды при перемешивании. Охлаждают водой и в ледяной бане.
Добавляют 5 мл раствора борной кислоты с индикатором в коническую колбу на
50 мл с отметкой, соответствующей объему 35 мл, и помещают колбу под холодильник
аппарата для перегонки (рис. 14.3).
425
Глава 14. Органические формы азота, минерализуемый азот (и углерод)
Устанавливают колбу для перегонки в аппарат, помещают 10 мл 10 М раствора NaOH
в капельную воронку и медленно приливают в перегонную колбу. Когда почти вся
щелочь добавлена, споласкивают воронку примерно 5 мл воды, затем добавляют еще
примерно 3 мл воды перед тем, как закрыть кран. Начинают перегонку, закрывая кран
для отвода пара, и останавливают ее, открывая этот кран, когда дистиллят достигнет
отметки 35 мл (после примерно 4 мин перегонки). Ополаскивают конец холодильника
и определяют содержание NH3-N, соответствующее содержанию общего N в гидроли¬
зате, титрованием 5 мн. раствором серной кислоты. В конечной точке титрования цвет
раствора изменяется с зеленого на розовый.
Если Vx — объем титрующего раствора в мл, количество азота в субпробе равно
70 • Vx мкг. Если Р — вес образца почвы в г, количество гидролизованного азота равно
70 * Kj • 100/(5 • Р) мкг/г почвы, т. е. 1.4 • К/Рмг/г почвы.
Определение (NH3 + аминосахараЬЫ
Помещают ровно 10 мл гидролизата в колбу для перегонки на 100 мл, добавляют 10 мл
фосфатно-боратного буферного раствора и продолжают операции, как описано выше,
до получения примерно 35 мл дистиллята (примерно 4 мин). Определяют выделив¬
шийся аммоний, как описано выше. Если V2 — объем 5 мн. раствора H2S04, количество
(NH3 + аминосахара)—N будет равно 0JV2/Pmt/t почвы.
Определение NH+-N
Помещают 10 мл гидролизата в колбу для перегонки на 50 или 100 мл, добавляют
0,07 ± 0,01 г MgO и продолжают операции, как описано выше, до получения пример¬
но 20 мл дистиллята (примерно 2 мин перегонки). Титруют дистиллят, как описано
выше. Если К3 — объем использованного 5 мн. раствора H2S04, количество NH| -N,
образовавшегося при гидролизе почвы, будет равно 0JV3/P мт/г почвы. Количество
аминосахаров-N определяют по разнице: 0,7(К2 - V3)/P мг/г почвы.
Определение (NH + + аминосахара + а-аминокислотьО-N
Помещают 5 мл гидролизата в колбу для перегонки на 50 мл, добавляют 100 мг цитрат-
ного буферного раствора и 100 мг нингидрина, помещают колбу в кипящую водяную
баню на 10 мин; через одну минуту перемешивают. Охлаждают колбу, добавляют 10 мл
фосфатно-боратного буферного раствора, перегоняют до получения 35 мл дистиллята
(примерно в течение 4 мин) и титруют дистиллят, как описано выше. Если V4 — объем
5 мн. раствора H2S04 в конечной точке титрования, количество (NH4 + аминосахара +
+ а-аминокислотьО-N будет равно 1,41^/Рмг/г почвы. Количество (а-аминокислот)-Ы
можнооценить по разнице: 0,7(2^— У2)/Рмг/г почвы. Можно также определить количество
(а-аминокислоты)-N, предварительно удалив (NH4 + аминосахара)-М, как описано ниже.
Определение (а-аминокислоты)-Ы
Помещают 5 мл гидролизата в колбу для перегонки на 50 мл, добавляют 1 мл 0,5 М рас¬
твора NaOH и нагревают колбу на кипящей водяной бане до тех пор, пока объем ре¬
акционной смеси не уменьшится до примерно 2—3 мл (примерно 20 мин). Охлаждают,
добавляют 500 мг лимонной кислоты и 100 мг нингидрина, помещают колбу в кипящую
водяную баню на 10 мин; через 10 мин перемешивают. Охлаждают колбу, добавляют
10 мл фосфатно-боратного буферного раствора и 1 мл 5 М раствора NaOH. Перегоняют
до объема дистиллята 35 мл (примерно в течение 4 мин) и титруют дистиллят, как описа¬
но выше. Если V5 — объем 5 мн. раствора H2S04, количество (а-аминокислоты)^ будет
равно 1,4У5/Рмг/г почвы.
426
Часть 2. Органический анализ
Определение(серин + треонин)-М
Выполняют операции до определения (NH|+ аминосахара)-1Ч, как описано выше в под¬
разделе «Определение (NH4 + аминосахара)-1Ч». Затем удаляют колбу для перегонки,
споласкивают трубку для ввода пара и охлаждают под холодной водой. Добавляют 2 мл
раствора периодной кислоты. После перемешивания в течение 30 с, добавляют 2 мл рас¬
твора арсенита натрия. Подсоединяют к аппарату для перегонки и перегоняют до по¬
лучения примерно 35 мл дистиллята (4 мин). Титруют дистиллят, как описано выше.
Если V6 — объем 5 мн. раствора H2S04, количество (серин + TpeoHHH)-N будет равно
0,7 VJP мг/г почвы.
Калибровка метода
Эти методики дистилляции можно проверять, используя стандартные растворы А и В;
рекомендуемые объемы аликвот — 5 мл раствора А для NH4-N, (а-аминокислоты)-Ы
или (NH4+ + гексозамины + аминокислоты) - N, 10 мл раствора А для N аминосахаров
и 10 мл раствора В для (серин + треонин)-М При хранении в холодильнике оба стан¬
дартных раствора остаются стабильными в течение нескольких месяцев. Другие меры
предосторожности, необходимые для этих методов, можно найти в исходной публика¬
ции (Вгетпег, 1965).
14.2.2. Определение органических форм азота: упрощенные методы
Основные положения
В части кислотной обработки методика аналогична методике, описанной в разделе
14.1.1. Методика перегонки упрощена, в гидролизате определяют только две фракции:
• nrN: фракция, перегоняемая в щелочной среде, которая включает, прежде всего,
формы NH+-N и аминосахара-N, а также некоторые амидные и аминофенольные
формы азота (Egouminides и др., 1987).
• HnrN: неперегоняемая фракция, которая содержит в основном аминокислоты
и неидентифицированый азот. Эту фракцию определяют по разнице между rN (об¬
щий гидролизуемый азот, определяемый по методу Къельдала) и nrN (перегоняе¬
мый гидролизуемый азот).
Негидролизуемый азот HrN можно определить, как описано выше, либо по разни¬
це между общим азотом, определенным по Къельдалю, и общим гидролизуемым азо¬
том или методом минерализации по Къельдалю остатка после гидролиза (см. раздел
14.2.1). Этот упрощенный метод был разработан и использован Эгуминидесом с сотр.
(Egouminides и др., 1987) для оценки потенциального плодородия тропических почв,
которые обычно содержат гораздо меньше азота, чем почвы умеренных климатических
зон.
Оборудование и реагенты:
• То же оборудование, что и в подразделе «Оборудование» раздела 14.2.1.
• Реагенты 1, 3, 4, 7 и стандартные растворы 16, 17 подраздела «Реагенты» раздела
14.2.1.
• 0,02 н. раствор серной кислоты: готовят из коммерческого стандарта (фиксанала).
Методика
Кислотный гидролиз
Анализируемый образец должен быть подготовлен в соответствии с содержанием азота,
как описано в разделе 14.2.1. На практике масса воздушно-сухих проб почвы составляет
427
Глава 14, Органические формы азота, минерализуемый азот (и углерод)
5 г для андисоли, 10-20 гдля ферраллитной почвы, 20-50 г для железистой тропической
почвы.
Помещают пробу с круглодонную перегонную колбу на 250 мл со стандартной при¬
тертой пробкой и добавляют 100 мл 6 М раствора хлористоводородной кислоты. Соеди¬
няют колбу с холодильником Либиха и кипятят в течение 16 ч (т. е. с 5 ч вечера до 9 ч
следующего утра).
Прекращают нагревание, охлаждают и переносят реакционную смесь в центрифуж¬
ные колбы на 250 мл, ополаскивают холодильник и реакционную колбу дистиллирован¬
ной водой. Центрифугируют при 4000 g в течение 15 мин и собирают надосадочную жид¬
кость в мерные колбы на 200,250 или 500 мл, в зависимости от количества промываний.
Дважды или трижды промывают отцентрифугированный осадок порциями по 20-40 мл
дистиллированной воды, каждый раз центрифугируя в тех же условиях и добавляя про¬
мывные воды в мерную колбу. Доводят раствор до метки при тщательном перемешива¬
нии. Гйдролизат в кислой среде можно хранить в мерной колбе до проведения анализа.
Выделяют отцентрифугированный осадок, высушивают при 40 °С и точно взвешивают.
Определение перегоняемого гидролизуемого азота nrN
Помещают аликвоту гидролизата в колбу для перегонки на 100 мл. Устанавливают ко¬
ническую колбу на 100 мл, содержащую 10 мл раствора борной кислоты с индикатором,
под холодильник аппарата для перегонки с паром (рис. 14.3). Соединяют колбу для
перегонки с аппаратом и добавляют через воронку (рис. 14.3) 10 М раствор гидрокси¬
да натрия объемом, необходимым для нейтрализации, плюс небольшой избыток (для
гидролизата, содержащего 3 моль/л, добавляют объем 10 М раствора NaOH, равный 1/3
объема аликвоты). Ведут перегонку до получения 50-80 мл дистиллята. Титруют содер¬
жимое конической колбы 0,02 М раствором серной кислоты. Если Р, V0, Vol и V{ — масса
пробы почвы (г), общий гидролизуемый объем, объем субпробы гидролизата и объем
титрующего раствора (мл), соответственно, тогда nrN = 0,28 V0 • VJ( V0l • Р) мг перегоняе¬
мого гидролизуемого азота на грамм почвы.
Определение общего гидролизуемого азота rN
Проводят минерализацию аликвоты гидролизата по Къельдалю (см. раздел 14.2.1): если
У02 — объем субпробы гидролизата (мл), a V2 — объем титрующего раствора (мл), тогда
rN = 0,28 V0 - V2/(V02 • Р)- Неперегоняемый гидролизуемый азот определяют по разнице:
HnrN = rN — nrN.
Определение негидролизуемого азота нгЫ
Содержание негидролизуемого азота определяют минерализацией по Къельдалю остат¬
ка после гидролиза, высушенного при 40 °С (см. раздел 11.2.1). Если масса остатка мень¬
ше, чем 10 г, используют весь остаток, в противном случае отвешивают точную порцию
остатка. Если Р0, Р01 и V3 — масса остатка, масса порции остатка и объем титрующего
раствора, соответственно, тогда HrN = 0,28 Р0 • К3/(Р01 * Р).
14.2.3. Определение содержания мочевины
Основные положения
Мочевину используют в качестве азотного удобрения на сельскохозяйственных почвах
по всему миру (Beaton, 1978). Соответственно, необходимы надежные методы для опре¬
деления остаточных количеств мочевины в почве. Можно отметить две основные груп¬
пы методов для ее определения:
428
Часть 2. Органический анализ
• колориметрические методы, основанные на цветной реакции мочевины с диа-
цетилмонооксимом (ДАМ) (Fearon, 1939) или с л-диметиламинобензальдегидом
{Wattvi Crisp, 1954) в кислой среде;
• ферментативные методы, позволяющие определить аммоний, образующийся
в процессе гидролиза мочевины; в этом методе после инкубации проводят перегон¬
ку с паром, используя оборудование, описанное выше (рис. 14.3 и разделы 14.2.1
и 14.2.2).
Реакция мочевины с ДАМ приводит к образованию желтого соединения. В при¬
сутствии тиосемикарбазида (ТСК) образуется красное соединение. Дуглас и Бремнер
{Douglas и Вгетпег, 1970) разработали метод с использованием последней реакции, ко¬
торый позволяет проводить точные измерения при анализе почвенных растворов, со¬
держащих менее 20 мг/л мочевины. Мочевина почвы экстрагируется раствором КС1
в присутствии ингибитора уреазы. Экстракты затем анализируют методом спектроко-
лориметрии. Исходный метод Дугласа и Бремнера был слегка модифицирован Мулвани
и Бремнером {Mulvaney и Вгетпег, 1979). Последний метод описан ниже. Растворы после
экстракции также можно анализировать на автоматическом колориметре {Douglas и др.,
1977). Более подробно методы определения мочевины описаны Бремнером (Вгетпег,
1982).
Реагенты
• Раствор фенилртути ацетата (ФРА): растворяют 50 мг ФРА в 1 л очищенной
воды.
• Хлорид калия и раствор ФРА (2 М КС1-ФРА): растворяют 1500 г КС1 в 9 л воды
и добавляют 1 л раствора ФРА.
• Раствор ДАМ: растворяют 2,5 г ДАМ в 100 мл воды.
• Раствор ТСК: растворяют 0,25 г ТСК в 100 мл воды.
• Кислотный реактив: добавляют 40 мл концентрированной серной кислоты к 1 л
раствора фосфорной кислоты (85 %масс.), разбавляют смесь водой до 2 л (добавляя
кислоту к воде) при перемешивании.
• Окрашивающий реактив: смешивают 50 мл раствора ДАМ с 30 мл раствора ТСК
и разбавляют смесь кислым реактивом до 1 л; этот реактив не может храниться
и должен готовиться непосредственно перед использованием.
• Стандартный раствор моневины-N: растворяют 0,4288 г чистой сухой мочевины
в растворе КС1—ФРА, доливают тем же раствором до 2 л и тщательно перемешива¬
ют. Этот раствор содержит 100 мкг мочевины-N в 1 мл. Хранится в холодильнике.
Методика
Помещают 5 г почвы во колбу на 100 мл и добавляют 50 мл 2 М раствора КС1-ФРА.
Закрывают колбу, перемешивают механической мешалкой в течение 1 ч и фильтруют
(фильтровальная бумага Whatman А°42).
Отбирают точную аликвоту экстракта (1—10 мл), содержащую от 10 до 100 мкг
мочевины-N, и помещают ее в мерную колбу на 50 мл. Доливают 2 М раствором КО¬
ФРА до 10 мл и добавляют 30 мл окрашивающего реактива. Быстро встряхивают кол¬
бу для перемешивания и помещают ее на термостатируемую баню при 85 + 0,5 °С на
30 мин. Охлаждают холодной водой (12—15 °С) в течение 10 мин, доливают водой до
50 мл и тщательно перемешивают. Измеряют поглощение колориметром на длине вол¬
ны красного света — 527 нм. Если колориметр не имеет монохроматора, можно исполь¬
429
Глава 14. Органические формы азота, минерализуемый азот (и углерод)
зовать зеленый фильтр. Рассчитывают концентрацию по калибровочной кривой, по¬
строенной по точкам, соответствующим 0,10, 50 и 100 мкг мочевины-N.
Для построения калибровочной кривой разбавляют 20 мл стандартного раствора
мочевины-N раствором КС1—ФРА в мерной колбе на 200 мл и тщательно перемешивают.
При помощи пипетки помещают аликвоты раствора объемами 0,1, 5 и 10 мл в мерные
колбы на 50 мл. Доливают 2 М раствором КС1-ФРА до 10 мл и продолжают операции,
как для почвенных экстрактов. Строят калибровочную кривую для каждой серии ана¬
лизов.
14.2.4. Определение содержания потенциально доступного азота:
биологические методы
Основные положения
Эти методы используют для анализа способности почвы снабжать растения неорганиче¬
ским азотом. Определяемые показатели обозначают как показатели доступности азота,
потенциально минерализуемый азот, нетто-минерализация и потенциал минерализации.
Биологические методы стремятся моделировать in vitro эволюцию почвы в естествен¬
ных условиях. Они измеряют количество неорганического азота, произведенного в про¬
бе почвы после инкубации в контролируемых условиях в течение данного периода вре¬
мени.
Эти методы кажутся простыми для выполнения, но на практике очень трудно выбрать
подходящую методику из-за существования многочисленных вариантов. Кини (Keeney,
1982) привел 29 методик инкубации, которые были протестированы относительно изме¬
рения потребления азота различными типами растений. Фад-Рашид (Fahd-Rachid, 1990)
перечислил 11 методов, три из которых не были упомянуты Кини.
Из этих методов чаще всего упоминается метод, первоначально предложенный Стэн¬
фордом и Смитом (Stanford и Smith, 1972). Он основан на концепции разложения пула
доступного азота в соответствии с кинетикой первого порядка (рис. 14.4). Изменение
содержания органического азота d[No] в течение данного интервала времени dt может
быть выражено через константу скорости минерализации1 к следующим образом:
d[No]/df=-fc[No] (14.1)
Рис. 14.4. Схема минерализации азота по Стэнфорду и Смиту (Stanford и Smith, 1972): Not —
общий органический азот почвы; No - минерализуемый органический азот почвы; N0 — потен¬
циально минерализуемый органический азот; Nт - минерализуемый азот (NH^+ NO~ + NO~);
к — константа скорости, равная доле органического азота, минерализованного в течение интер¬
вала времени d t
1 Авторы применяют аппарат формальной кинетики для описания множества параллельных
многостадийных процессов превращения ряда исходных веществ в ряд минерализованных
продуктов. Поэтому под «константой скорости минерализации» следует понимать «эффек¬
тивную константу скорости процессов минерализации». — Примеч. науч. ред.
430
Часть 2. Органический анализ
Поскольку N0 — количество минерализуемого органического азота почвы в момент
времени 0 (потенциально минерализуемый азот), интегрирование (14.1) дает:
[N0] = NOe-*'. (14.2)
Кроме того, если рассчитать количество исходного неорганического азота в почве,
баланс азота будет:
[No] + [N/w] = 7V0. (14.3)
В соответствии с (14.2) и (14.3), эволюция N/w неорганического азота будет выражена
уравнением:
[N/w] = N0(1 — е-*0. (14.4)
Эта методика заключается в измерении количества неорганического азота, образо¬
вавшегося в пределах инкубационного времени, и нанесении на график накопленных
величин как функции от времени. Преобразовав данные в соответствии с (14.4), можно
одновременно оценить количество потенциально минерализуемого азота М) и констан¬
ту скорости минерализации к. Были предложены несколько методов преобразований.
Стэнфорд и Смит (Stanford и Smith, 1972) сначала рассчитали линейную корреляцию
между накопленным неорганическим азотом и временем инкубации методом наимень¬
ших квадратов. Для расчетов потенциально минерализуемого азота и скорости минера¬
лизации они использовали линейную регрессию. После логарифмического преобразо¬
вания (14.4) можно записать следующим образом:
lg(N0 - N/w) = IgNO - kt (14.5)
Задав величину N0 в левой части уравнения, из линейного соотношения между пере¬
менными lg(N0 — N/w) и / можно определить значение к. Кривые, полученные в полуло¬
гарифмических координатах для различных выбранных значений N0, имеют выпуклую
форму, если расчетные N0 < истинных N0, вогнутую — если расчетные N0 > истинных
N0, и линейны, если расчетные N0 = истинным N0. Итерационная процедура позволяет
оценить оптимальное значение N0. Используя этот метод, Стэнфорд и Смит получили
значения N0 в диапазоне между 5 и 40% общего азота для 39 сильно различающихся почв.
С другой стороны, константы скорости к для различных почв отличаются несуществен¬
но, и авторы оценили ее как 0,054 ± 0,009 недель-1. Этот метод преобразований был впо¬
следствии подвергнут критике, особенно потому, что логарифмическое преобразование
данных приводит к одновременному преобразованию экспериментальной погрешности
(Campbell, 1978; Smith и др., 1980; Reynolds и Beauchamp, 1984). Метод сглаживает значе¬
ния, расположенные далеко от начала координат, и этим эффектом можно объяснить
низкую вариабельность к, наблюдаемую Стэнфордом и Смитом (Магу и Remy, 1979).
В настоящее время большинство авторов предпочитает нелинейные методы преобразо¬
вания. Бенедетти и Себастиани (Benedetti и Sebastiani, 1996) сравнили три метода оцен¬
ки: максимального правдоподобия, линейного преобразования по (14.5) и нелинейного
преобразования. Согласно их результатам, последний метод кажется предпочтитель¬
нее. На рис. 14.5 приведены два примера нелинейной интерполяции, полученные Фад-
Рашидом (Fahd-Rachid, 1990) в соответствии с алгоритмом Маркардта (Marquardt, 1963).
Варианты
Недостатком этого метода является требуемое количество измерений и длительность
эксперимента, хотя авторы и предложили снизить ее с 210 дней (аэробная инкубация,
Stanford и Smith, 1972) до 26 дней (Stanford и др., 1974).
Глава 14. Органические формы азота, минерализуемый азот (и углерод)
431
Время, недели
Рис. 14.5. Наблюдаемые и рассчитанные величины (Fahd-Rachid, 1990) накопленного неоргани¬
ческого азота как функции от времени инкубации для метода Стэнфорда и Смита (Stanford и Smith,
1972) с нелинейной трансформацией (14.4): с — нейтральная коллювиальная почва; а —аллюви¬
альная карбонатная почва, холодные пробы, хранившиеся 50 дней перед инкубацией
Были предложены более быстрые, хотя и менее точные, биологические методы. Джи-
анелло и Бремнер (Gianello и Вгетпег, 1986) сравнили различные биологические и хими¬
ческие варианты, проведя свои тесты на 30 типах почв с широким диапазоном содержа¬
ния органического вещества (0,3—9% углерода).
Сравнивались следующие биологические методики:
• ш13 — метод Воринга и Бремнера (Waring и Вгетпег, 1964), модифицированный
Кини и Бремнером (Keeney и Вгетпег, 1966); состоял в определении аммония, об¬
разующегося при анаэробной инкубации 5 г почвы в насыщенной водной среде
при 40 °С в течение 7 сут.
• т14 — метод Кини и Бремнера (Keeney и Вгетпег, 1967); состоял в определении
(NH+ + NO3- + NO~)-N, образующегося при аэробной инкубации 10 г почвы, сме¬
шанной с 30 г кварцевого песка (размер частиц 30-60 меш.) и 6 мл дистиллирован¬
ной воды, при 30 °С в течение 14 сут.
• ш15 — метод Стэнфорда и Смита (см. подраздел «Методика» раздела 11.2.4) для
времен инкубации 2,4, 6, 8 и 12 недель, накопленный результат за 12 недель.
• ш16 —результат метода m 15 для 2 недель.
• ш17 — расчет N0 по Стэнфорду и Смиту (14.5) с использованием всех данных
ш15.
На рис. 14.6 представлены результаты, опубликованные этими авторами в соответ¬
ствии с номером почвы в порядке увеличения содержания углерода (рис. 14.6, о, с).
С одной стороны, для сравнения величин N0 с содержанием общего азота в по¬
чве и, с другой стороны, для сравнения методов т14, ш15, ш16 с методом N0 (т17).
432
Часть 2. Органический анализ
Рис. 14.6. Сравнение результатов определения для 30 проб различных типов почв (Gianello
и Вгетпег, 1986): а и b - потенциал минерализации (N0 по Stanford и Smith, 1972) и общий
азот (общий N); с и d - N0 и четыре других биологических методики измерения потенциально
доступного азота
Сравнение с содержанием общего азота показало аналогичные колебания, иллюстри¬
рующие корреляцию, наблюдаемую Джианелло и Бремнером (г— 0,86; Р< 0,1%). Одна¬
ко в некоторых случаях значительная корреляция между N0 и общим N может привести
к существенным погрешностям: на рис. 14.6, Ъ показаны различия между преобразо¬
ванными и действительными величинами, которые могут достигать 100 мг/кг; это со¬
ответствует приблизительно 300 кг неорганического N/ra и не позволяет точно оценить
внесение азотных удобрений в поле. Рассчитанный коэффициент пропорциональности
дает среднюю оценку потенциально минерализуемого азота, примерно равную 6,5%
общего азота в почве.
Сравнение метода N0 и других тестов с использованием биологических методов (рис.
14.6, с) показывает корреляции (значимые при Р< 0,1%) между результатами Джианелло
и Бремнера: г= 0,96 между N0 и т13 или т15; г= 0,90 между N0 и ш16; г-0,81 между
N0 и ш14. Коэффициенты пропорциональности на рис. 14.6, Смогут использоваться для
сравнения методов с меньшей неопределенностью, чем предыдущее сравнение с общим
433
Глава 14. Органические формы азота, минерализуемый азот (и углерод)
азотом (рис. 14.6, Ь). Таким образом, можно оценить N0 на основе более простых экс¬
периментов, чем эксперименты Стэнфорда и Смита (Stanford и Smith, 1972):
• анаэробная инкубация типа М13 позволяет примерно оценить
N0 = NH4+-N/0,44;
• аэробная инкубация типов М14 и М16 позволяет примерно оценить
N0 = (NH; + N03- + NO-) - N/0,27.
Два метода ш14 и ш16, использующие аэробную инкубацию в течение 2 недель, дают
одинаковые значения коэффициента пропорциональности. Однако различия, наблю¬
даемые между преобразованными и действительными значениями (рис. 14.6 ,d), более
значительны, чем для других двух методов. Метод m 13 анаэробной инкубации в течение
1 недели кажется более надежным, по крайней мере, по сравнению с методом N0. Срав¬
нение с методом ш15 не представляет интереса, поскольку величина N0 была рассчита¬
на изданных ш 15.
Поскольку предложено большое количество методов инкубации, единственный ме¬
тод, описанный в настоящей главе, является адаптацией метода Стэнфорда и Смита
(Stanford и Smith, 1972), который используется в центре IRD, Монпелье (J.C. Talineau,
частное сообщение; Fahd-Rachid, 1990).
Оборудование
• 10-30 полипропиленовых воронок Бюхнера (в зависимости от размеров проб се¬
рии) диаметром 5,5 или 7 см для тропических почв с низким содержанием азота;
• стандартные фильтры из стекловолокна типа Whatman GF/A, соответствующие раз¬
меру воронок Бюхнера;
• воздухопроницаемая водонепроницаемая пленка (например, парафильм Rhone-
Poulenc, Франция) для того, чтобы накрывать стаканы;
• сушильный шкаф (для инкубации) с точной регулировкой температуры между 20
и 50 °С (пропорциональная регулировка);
• 10-30 лабораторных стаканов на 150 мл;
• 10-30 колб Бунзена на 250 мл;
• манометр в диапазоне 0-1 атм;
• микрокомпьютер со статистическими программами, позволяющими рассчиты¬
вать линейную и нелинейную регрессии, или полулогарифмическая масштабно¬
координатная бумага.
Реагенты и материалы
• Чистый песок, не содержащий органических веществ, просеянный до размера ча¬
стиц между 0,5 и 2 мм.
• 0,01 М раствор СаС12: растворяют 7,35 г СаС12 • 2Н20 в дистиллированной воде, до¬
ливают до 5 л и тщательно перемешивают.
• Раствор для насыщения ионами, не содержащий азота, 0,002 М CaS04, 0,002 М
MgS04, 0,005 М СаНР04, 0,0025 М KjSO^ растворяют 0,689 г CaS04* 2Н20, 0,482 г
MgS04, 1,720 г СаНР04- 2Н20 и 0,871 г KjSC^ в дистиллированной воде, доливают
до 2 л и тщательно перемешивают.
434
Часть 2. Органический анализ
Методика
а) Для непесчаных почв смешивают 30 г воздушно-сухой почвы, просеянной через
сито 5 мм, с 10 г крупного просеянного песка и помещают в воронку Бюхнера (диа¬
метром 55 мм, емкостью 80 мл) с фильтром из стекловолокна. Для песчаных почв
с низким содержанием N помещают 50 г почвы (без добавления песка) в воронку
диаметром 70 мм. Некоторые авторы предлагают использовать свежую почву для
уменьшения первоначального всплеска минерализации после добавления воды
к высушенной почве (Fahd-Rachid, 1990). Прикрывают воронку слоем стеклянной
ваты для защиты поверхности материала от размывания при смачивании.
б) Смачивают пробу, поливая 100 мл 0,01 М раствора СаС12, затем 25 мл раствора для
насыщения ионами, собирая весь фильтрат в стакан на 150 мл.
в) Регулируют влажность проб, снизив давление на 0,03 МПа по сравнению с атмо¬
сферным (до абсолютного давления 0,0713 МПа). Согласно некоторым авторам,
это снижение давления соответствует оптимальной влажности минерализации для
многих почв (Talineau, IRD, Монпелье, частное сообщение). Оборудование, пока¬
занное на рис, 14.7, позволяет выполнять эту операцию очень просто при помо¬
щи стеклянного водоструйного насоса и зажима для шлангов. Раствор, собранный
в колбе для фильтрования, добавляют к предыдущим перколяционным растворам;
его можно временно хранить охлажденным перед анализом.
г) Накрывают воронку парафильмом и инкубируют в инкубаторе при 28 °С. Помеща¬
ют плоский контейнер с водой в инкубатор для поддержания влажной атмосферы.
Для поддержки воронок Бюхнера можно использовать широкую полиуретановую
пластину толщиной 5 см с просверленными отверстиями для ножек воронок.
д) Убирают воронки из инкубатора и начинают снова с пункта б для выбранных перио¬
дов инкубации, например, 2,4,6,8,10,12 и 16 недель (Fahd-Rachid, 1990; рис. 14.5).
е) С помощью перколяционных растворов определяют различные формы неоргани¬
ческого азота (аммоний, нитриты или нитраты), перегоняя аммиак в присутствии
восстанавливающего реактива (сплав Деварда, железо) при помощи аппарата для
перегонки с паром (рис. 14.3) или используя любой другой метод, пригодный для
определения неорганического азота (см. гл. 28). Перколяционные растворы в мо¬
мент времени 0 можно отбросить или использовать для определения исходного не¬
органического азота почвы. Эти исходные значения не учитывают при определе¬
нии потенциально минерализуемого азота.
ж) Строят график накопленных содержаний неорганического азота как функцию вре-
мени инкубации (рис. 14.5) и оценивают параметры преобразования (см. подраз¬
дел «Основные положения» раздела 14.2.4).
Обсуждение
Результаты в значительной степени зависят от хранения и подготовки проб почвы. Эти
операции всегда приводят к дополнительной минерализации в начальный период инку¬
бации из-за выделения органического вещества, заблокированного в почвенных агрега¬
тах, но особенно из-за частичной гибели микробной биомассы. Мертвая биомасса и вы¬
деленные вещества поглощаются быстрее в начальный период инкубации, что может
привести к завышенной оценке содержания минерализуемого азота. Однако сравнивая
результаты, полученные для образцов воздушно-сухой почвы и тех же образцов почвы,
хранившихся в свежем виде при пониженной температуре, Фад-Рашид (Fahd-Rachid,
1990) сделал два интересных наблюдения:
Глава 14. Органические формы азота, минерализуемый азот (и углерод)
435
Рис. 14.7. Оборудование для регулирования влажности пробы и ионного равновесия перед
инкубацией
• Различия между субпробами особенно влияют на величину к, которая для свежей
почвы была примерно в четыре раза меньше, чем для воздушно-сухой почвы; ве¬
личины N0 изменялись примерно на 20%.
• В случае сухой почвы, вычитая два первых значения накопленного неорганическо¬
го азота (1 и 2 недели инкубации) из других величин, получают кривую и параме¬
тры, близкие к величинам, наблюдаемым для свежей почвы.
Модель Стэнфорда и Смита, соответствующая уравнению (14.5), была не единствен¬
ной, но самой простой из исследованных моделей. Мари и Реми {Магу и Вету9 1979)
предложили использовать произведение Ш0 для грубой оценки минерализационно-
го потенциала азота почвы. Некоторые авторы исследовали несколько более сложные
модели эволюции: Nт = N0(1 — егк\1) + k2t {Chaussod и др., 1986; Seyfried и Rao, 1988),
N t= N01(1—+N02(1 - егк*) (Nuske и Richter, 1981; Deansvmp., 1986) и даже модель,
состоящую из п пулов азота с п кинетиками минерализации {Richter и др., 1982). Ис¬
пользование этих различных моделей не меняет методики инкубации, описанной ранее.
Часто содержание минерализованного азота просто оценить, исходя из содержания
нитратов, аммония и нитритов, являющихся трансформированными формами, присут¬
ствующими в малых количествах в хорошо аэрируемой среде.
Стандартная методика Стэнфорда и Смита является скорее приблизительным моде¬
лированием процессов, происходящих в естественных условиях. Ее можно критиковать
по двум основным причинам:
• Методика не моделирует цикл азота (рис. 14.1), так как регулярное выщелачивание
неорганического азота способствует процессам минерализации и нитрификации,
а не иммобилизации и восстановления. Она полезна для определения содержания
потенциально минерализуемого азота (которое может быть завышено), но не мо¬
жет оценить содержание неорганического азота, реально доступного для растений
в данный период времени.
436
Часть 2. Органический анализ
• Выщелачивание 0,01 М раствором СаС12 удаляет растворимый органический азот,
который может быть минерализован позднее (Robertson и др., 1988), приводя к за¬
нижению количества минерализуемого азота.
Более того, если моделирование динамического аспекта образования неорганическо¬
го азота обладает неоспоримым преимуществом надежности, то длительность экспери¬
ментов может явиться препятствием для их осуществления. Впоследствии было пред¬
ложено много методов для более быстрого определения потенциально доступного азота:
(1) более быстрые биологические методы (см. подраздел «Варианты») или (2) химиче¬
ские методы экстракции (см. 14.2.5).
14.2.5. Определение содержания потенциально минерализуемого азота:
химические методы
Основные положения
Поскольку потенциально минерализуемый азот составляет только часть (от 5 до 10%)
общего азота (в среднем 6,5%, согласно результатам сравнения, приведенным на
рис. 14.6, й), жесткие методы кислотной обработки, используемые для определения
форм азота (см. разделы 14.2.1 и 14.2.2), в этом случае непригодны.
Многие авторы разрабатывали методы с использованием менее агрессивных экстра¬
гентов, которые часто испытывались по сравнению с потребностями растений в кон¬
кретных ситуациях. Эти методы основаны на действии таких реагентов, как (1) ней¬
тральные соли (КС1, СаС12) в холодных, а чаще горячих, растворах и (2) более или менее
щелочные горячие или слабо окислительные растворы солей. Затем измеряется количе¬
ство аммониевого азота, экстрагированного этими растворами.
Велли с сотр. (Velly и др., 1980) наблюдали очень тесную корреляцию между по¬
требностью растений в N и общим экстрагированным азотом, используя метод Жиро
и Фардо (Giraud и Fardeau, 1977) для экстрактов холодным раствором КС1. Большин¬
ство авторов (например, 0ien и Selmer-Olsen, 1980; Whitehead, 1981; Gianello и Bremner,
1986) исследовали методы с использованием экстрактов горячим раствором КС1. Уайт¬
хед (Whitehead^ 1981) отмечал, что разработанный им метод экстракции при кипячении
с 1 М раствором КС1 в течение 1 ч дал хорошую оценку количества азота, поглощенного
пастбищами. Фокс и Пикелек (Fox и Piekielek, 1978) исследовали экстракцию 0,01 н. рас¬
творами NaHC03 в соответствии с методом Маклейна (Mac Lean, 1964) с последующим
измерением поглощения нитратов и нитритов в УФ-диапазоне на длине волны 205 нм.
Кини (Keeney, 1982) рекомендовал использовать раствор СаС12 в автоклаве. Джианелло
и Бремнер (Gianello и Bremner, 1986, 1988) рекомендовали определять аммоний пере¬
гонкой в присутствии фосфатно-боратного буферного раствора при pH 11,2. Стэнфорд
и Смит (Stanford и Smith, 1978) предложили определять аммоний, выделившийся под
действием КМп04 в кислой среде. Стэнфорд (Stanford, 1978) предложил проводить экс¬
тракцию раствором перманганата в щелочной среде.
Исследования 30 почв с различным содержанием органического вещества, проведен¬
ные Джианелло и Бремнером (Gianello и Bremner, 1986), позволили сравнить относитель¬
ную эффективность различных методов.
На рис. 14.8 сгруппированы все их результаты для образцов, расположенных в со¬
ответствии с увеличивающимся содержанием органического вещества в пробах. Ана¬
логично данным на рис. 14.6, для сравнения использовалась величина N0, полученная
Стэнфордом и Смитом (см. раздел 14.2.4). Сравниваемые методы были сгруппированы
на четырех графиках, представляющих различные уровни экстрагированного азота.
Глава 14. Органические формы азота, минерализуемый азот (и углерод)
437
Рис. 14.8. Сравнение результатов определения потенциально минерализуемого азота химиче¬
скими методами с потенциалом минерализации N0 (Stanford и Smith, 1978); 30 исследованных
почв соответствуют почвам на рис. 14.6; они приведены в порядке увеличения содержания орга¬
нического углерода (Gianello и Вгетпвг, 1986); средние фракции, полученные каждым методом,
сравнены с N0 для 30 почв
• Кривые М3, М4 и М5 на графике представляют три альтернативных метода экс¬
тракции горячим 2 М раствором КС1 в закрытой бутыли (М3 — нагревание до
100 °С в течение 4 ч; М4 — нагревание до 95 °С в течение 16 ч; М5 (0ien и Selmer-
Olsen, 1980) - нагревание до 80 °С в течение 20 ч). Средние количества экстраги¬
рованного азота составляют 8-20% от N0, причем метод М4 дает максимальное
количество экстрагированного азота. Все коэффициенты корреляции с N0 высоко
значимы: 0,95,0,96 и 0,93 для М3, М4 и М5, соответственно.
• Кривые Мб и М7 представляют два других метода экстракции горячим 1 М раство¬
ром КС1: Мб (Whitehead, 1981) — кипячение в реакционной трубке в течение 1 ч;
М7 — нагревание в закрытой трубке до 100 °С в течение 1 ч. Оба метода дают очень
похожие, но самые низкие результаты среди всех исследованных методов, меньше
чем 5% от N0 (рис. 14.8). Более того, результаты этих двух методов хуже всего кор¬
релируют с величинами N0 (г = 0,81 и г = 0,83, соответственно).
438
Часть 2. Органический анализ
• М8 и М9 представляют два метода: «перегонка с фосфатно-боратным буферным
раствором» (М8, Gianello и Вгетпег, 1986, 1988) и «экстракция СаС12 в автоклаве»
(М9, Keeney, 1982). Количество экстрагированного азота значительно больше, чем
во всех методах с КС1; средние значения составляют 27% от N0 для М8 и 50% от
N0 для М9. Таким образом, метод М8 с фосфатно-боратным буферным раствором
экстрагирует почти вдвое меньше азота, чем автоклавный метод М9. Однако его
результаты лучше коррелируют с величинами N0 (г = 0,95 для М8 и г = 0,92 для М9,
рис. 14.8).
• Ml0 и Ml 1 представляют два метода контролируемого окисления перманганатом
в кислой (М10, Stanford и Smith, 1978) и щелочной (Ml 1, Stanford, 1978) среде. Эти
методы экстрагируют самые большие количества азота. Средние значения очень
близки: 60-65% от N0. Однако обнаружена очень слабая корреляция между их ре¬
зультатами. Корреляции же с N0 самые низкие среди всех исследованных методов,
особенно для щелочного окисления (г = 0,85 для М10 и г = 0,48 для Ml 1).
Ниже приведено описание только методики М8 перегонки аммония в присутствии
фосфатно-боратного буферного раствора (Gianello и Вгетпег, 1988). Эта методика по¬
казала лучшую корреляцию с величинами N0 (Stanford и Smith, 1978) (рис. 14.8). Ее реа¬
лизация относительно проста и использует тот же тип оборудования для перегонки с па¬
ром, как при определении форм азота (см. рис. 14.3, разделы 14.2.1 и 14.2.2). Согласно
Джианелло и Бремнеру, высушивание и хранение проб почти не влияет на результаты.
Тем не менее, эта методика редко применялась для сравнения с реальной потреб¬
ностью растений в азоте, поэтому не следует отбрасывать другие методы экстракции
нейтральными солями (Campbell и др., 1997). Кроме того, окисление перманганатом
в кислой среде может дать результаты, более близкие к величинам N0 по абсолютному
значению. Более подробно о других методиках можно прочитать в оригинальных пу¬
бликациях, упомянутых выше. Коэффициенты, приведенные на рис. 14.8, позволяют
оценить соответствие между результатами различных методов.
Оборудование и реагенты
Оборудование
• Аппарат для перегонки с паром (см. рис. 14.3 и разделы 14.2.1 и 14.2.2).
• Колбы Къельдаля для перегонки на 200 мл со стандартными шлифами 29/32
и крючками для пружин.
• Тйтриметрическое оборудование для ацидиметрии или точная ручная бюретка на
5 мл, проградуированная с интервалами 0,01 мл.
Реагенты
• Фосфатно-боратный буферный раствор с pH 11,2 (см. раствор 11 в подразделе «Ре¬
агенты» раздела 28.2.1): помещают 200 г Na3P04 • 12Н20,50 г Na2B407 • ЮН20 и при¬
мерно 1800 мл воды в мерную колбу на 2 л. Перемешивают до полного растворе¬
ния. Доводят pH раствора до 11,2 добавлением Н3Р04, затем доводят объем до 2 л.
Хранят в плотно закрытой бутыли.
• Раствор борной кислоты с индикатором (см. раствор 5 в подразделе «Реагенты»
раздела 28.2.1).
• 0,005 н. раствор серной кислоты (см. раствор 6 в подразделе «Реагенты» раздела
28.2.1).
439
Глава 14. Органические формы азота, минерализуемый азот (и углерод)
• Порошок MgO (см, реагент 9 в подразделе «Реагенты» раздела 28.2.1).
• 2 М раствор КС1: растворяют 149,1 г КО в дистиллированной воде. Доливают до
1 л, гомогенизируют и хранят в полно закрытой бутыли.
Методика
• Наливают 5 мл раствора борной кислоты с индикатором в коническую колбу на
100 мл с отметкой объема 50 мл и помещают под холодильник аппарата для пере¬
гонки с паром (рис. 14.3).
• Помещают 4 г почвы, просеянной через сито 2 мм, в колбу для перегонки на 200 мл.
Добавляют 40 мл фосфатно-боратного буферного раствора с pH 11,2 и подсоединя¬
ют колбу к аппарату для перегонки.
• Сразу начинают перегонку, открыв паровой кран, и прекращают, когда дистиллят
достигнет отметки 50 мл.
• Титруют аммоний 0,005 н. раствором серной кислоты вручную или автоматически.
Если V{ мл — объем титрующего раствора, тогда количество общего аммония (ис¬
ходный + минерализованный) в пробе будет равно oNH3-N = 17,5 V} мкг/r почвы.
• Если обменный аммоний NH3-N в пробе был определен ранее (см. гл. 28), со¬
держание минерализуемого азота можно рассчитать по разнице между oNH3-N
и NH3-N. Если нет, то исходное содержание NH3—N можно определить, исполь¬
зуя то же оборудование для перегонки с паром и метод Кини и Бремнера (Keeney
и Вгетпег, 1966): перегонка 4 г почвы в присутствии 20 мл 2 М раствора КС1 и 0,2 мг
MgO в течение 3,3 Мин. Если К2 — объем 0,005 н. раствора серной кислоты, не¬
обходимой для титрования перегнанного аммиака, количество минерализуемого
NH3-N из органического азота будет равно Y!,5(VX - V2) мкг/г почвы.
14.2.6. Кинетика процессов минерализации
Основные положения
Методы определения потенциально минерализуемого азота (разделы 14.2.4 и 14.2.5)
определяют максимальный потенциал азотного плодородия, но они далеки от опенки
реального цикла азота, который тесно связан с углеродным циклом (Pansu и др., 1998).
Кинетические исследования включают одновременное измерение минерализации
и трансформации углерода и азота (1) в естественных условиях с использованием ме¬
ченых 14С или 13С и 15N (Bottner и др., 2000) или немеченых веществ или (2) в контроли¬
руемых лабораторных условиях: измерение количеств С02, NH+и NO~, произведенных
почвами и смесями почв с мечеными или немечеными веществами. Адаптированный
лабораторный метод, используемый в институте IRD, Монпелье (Thurids и др., 2000),
описан ниже. Методика отличается в части, относящейся к длительности инкубации.
Для исследования кинетики превращения нестабильных компартментов часто проводят
эксперименты в течение одного месяца или даже меньше. Для исследования превраще¬
ний более стабильных компартментов период инкубации должен превышать 100 дней
(Blet-Charaudeau и др., 1990). Часто он длится 6 мес. Дополнительная работа, связанная
с большой длительностью эксперимента, не является большим недостатком, поскольку
планируется логарифмическая схема пробоотбора в зависимости от времени инкубации.
Таким образом, на начальных стадиях эксперимента необходимо большое количество
проб, а на последних стадиях оно очень невелико.
440
Часть 2. Органический анализ
Оборудование
• Большой бактериологический инкубатор с контролем температуры ±0,3 °С, в дан¬
ном случае 28 °С.
• Защитные стеклянные сосуды на 1,2 л с резиновым уплотнением.
• Держатель пробы из ПВХ трубки диаметром 8 см и высотой 12 см с просверлен¬
ными отверстиями и двумя перекрещенными проволоками на одной стороне для
поддержки коробок с пробами (рис. 14.9).
• Цилиндрические полипропиленовые сосуды диаметром 5 см на 50 мл.
• Банки (Nalgene) на 60 мл с завинчивающейся крышкой.
• Эксикатор с окисью кальция или бария (для поглощения С02).
• Титриметрическое оборудование с ячейкой для титрования в токе азота.
• Точный дозатор объема (для 20 мл стандартного раствора гидроксида натрия), за¬
щищенный ловушкой для С02 (с гранулированным NaOH) на крышке бутыли.
• Реактивы для определения неорганического азота (см. гл. 28).
г I
. \ . ■ Г . :
• J' ' ,) ’ V '
> с с
Герметичная чаша
|- Резиновое уплотнение
— Защитный стеклянный сосуд на 1,2 л
Полипропиленовый сосуд на 25 мл
Проба
Трубка из ПВХ с отверстиями
Банка Nalgene с завинчивающейся крышкой
20 мл 0,25 М раствора NaOH
Рис. 14.9. Инкубационный сосуд для изучения дыхания почвы и кинетики минерализации С и N
Реагенты
• Коммерческий стандартный 0,25 М раствор NaOH: количественно переносят со¬
держимое пластиковой ампулы с коммерческим стандартным 0,5 М раствором
NaOH (фиксаналом) в мерную колбу на 2 л, доливают до метки и гомогенизируют.
Хранят в бутыли дозатора объемов с ловушкой в крышке, наполненной гранулиро¬
ванным гидроксидом натрия.
• Коммерческий стандартный 0,25 М раствор НС1: разбавляют требуемое количе¬
ство коммерческого стандартного 0,5 М раствора НС1 в мерной колбе на 2 л, до¬
ливают до метки и гомогенизируют.
• 20%-ный раствор хлорида бария: растворяют 200 г ВаС12 в 800 мл воды, перемеши¬
вают и доливают до 1 л; если раствор мутный, фильтруют через тонкую фильтро¬
вальную бумагу.
441
Глава 14. Органические формы азота, минерализуемый азот (и углерод)
• Реактивы для экстракции (1 М раствор КС1 или KjSO,) и определения неорганиче¬
ских форм азота (см. гл. 28).
Методика инкубации
Определяют максимальную водоудерживающую способность ВУС (г/кг) и фактическую
влажность В (г/кг) анализируемой почвы.
Рассчитывают массу воды МВ, которую надо добавить к массе исследуемой пробы
почвы МП (г, в данном случае 25 г):
МВ = 0,75(ВУС - В)МП/1000.
Выбирают схему пробоотбора (табл. 14.3) для исследования минерализации С и N как
функции от времени t (дни).
Таблица 14.3. Схема эксперимента в зависимости от времени инкубации (г, дни) для
исследования кинетики минерализации С и N (х - отбор пробы)
т
0
1
2
3 5
7 10
14 21
28 41
61 90
100 120 130 152 180
с
X
X
X
X X
X X
X X
X X
X X
X X X X X
N
X
X
X
X
X
X
X
X
X
Отвешивают в полипропиленовые коробки требуемое количество порций проб почвы
по 25 г, с учетом того, что определение азота является деструктивным, а определение
углерода — недеструктивным методами. Планируют три серии измерений.
На квадратиках алюминиевой фольги взвешивают порции органического вещества
(добавленное органическое вещество, ДОВ), которые добавляют к пробам почвы в слу¬
чае исследования кинетики минерализации ДОВ. ДОВ высушивают и готовят в соответ¬
ствии с выбранным размером частиц, массу рассчитывают в соответствии с отношением
C:N в ДОВ (табл. 14.4). Осторожно сворачивают алюминиевую фольгу с порциями ДОВ,
наносят фломастером номера порций и хранят в холодильнике. В эксперименты по
кинетике минерализации ДОВ всегда включают пробы почвы без добавок (холостые).
В эксперименты по измерению дыхания почвы включают холостые пробы без почвы.
Таблица 14.4. Рекомендуемые добавки для исследования кинетики минерализации
добавленного органического вещества (ДОВ) в почве
ДОВ
Отношение C:N в ДОВ
Масса ДОВ
Органоминеральное удобрение, органическое
удобрение
<3
75-125 мг
Органическое удобрение
3-15
250 мг
Органические добавки, пожнивные остатки
с низким содержанием N
> 15
500 мг или
более
В день начала эксперимента увлажняют пробы почвы на весах, добавляя массу воды
< МВ, добавляют ДОВ, тщательно перемешивают при помощи шпателя и заканчивают
увлажнение на весах, добавляя массу воды точно до МВ. Регистрируют общую массу ОМ
увлажненной пробы. Заканчивают гомогенизацию.
Помещают пробы в бактериологический инкубатор, отрегулированный на выбран¬
ную температуру (28 °С в институте IRD, Монпелье). Большинство из проб почвы долж¬
ны быть помещены в пластиковые сосуды, покрытые пластиковой пленкой с отверсти¬
ями, проделанными булавкой (проницаемой для воздуха, но непроницаемой для воды).
442
Часть 2. Органический анализ
Только пробы, предназначенные для отбора в момент времени t = 1 (табл. 14.3), должны
быть помещены в инкубационные сосуды (рис. 14.9). Помещают в каждый сосуд банку
(Nalgene), содержащую точно 20 мл 0,25 М водного раствора NaOH. Все тщательно за¬
крывают и помещают в инкубатор.
В момент времени t = 1 открывают стеклянный сосуд, вынимают полипропилено¬
вую коробку, содержащую пробу, и помещают в холодильник для определения неорга¬
нического азота, вынимают банку (Nalgene), содержащую гидроксид натрия, закрывают
и хранят в эксикаторе над окисью кальция или бария. Перезагружают каждый сосуд сле¬
дующей пробой и новой банкой (Nalgene), содержащей 20 мл 0,25 М водного раствора
NaOH, и повторяют ту же операцию при каждом отборе пробы на N (табл. 14.2). В дни,
когда отбирают пробы только для анализа С, меняют только раствор гидроксида натрия
и помещают обратно для инкубирования с той же самой пробой.
Раз в 5 дней влажность каждой пробы регулируют на весах, доводя ее до значения
ОМ, записанного ранее.
Титрование
Для минерализованного азота экстракцию выполняют с 1 М водными растворами КС1
или KjSO^ а титрование проводят в соответствии с инструкциями, приведенными в гл. 28.
Для минерализованного углерода:
• Калибруют титратор, используя буферные растворы с pH 4 и 7; выбирают програм¬
му титрования: без исходной добавки, детектирование в фиксированной конечной
точке с pH 8,6, низкая скорость добавления.
• Быстро переносят содержимое банки (Nalgene) в ячейку для титрования с малень¬
ким магнитным якорем, ополаскивая ее струей воды из промывного флакона, за¬
тем добавляют 5 мл раствора ВаС12.
• Начинают пропускать азот в раствор с умеренной скоростью; вводят рН-электрод
и вводную трубку титрующего раствора кислоты.
• Начинают титрование и продолжают его до автоматической остановки; определя¬
ют добавленный объем титрующего раствора кислоты, мл: sVдля пробы, сУцдя
контроля (только почва для кинетики минерализации ДОВ) и bVдля холостого
опыта (без пробы почвы для измерения дыхания почвы).
Расчеты
Микробное дыхание в инкубационных горшках вызывает карбонизацию щелочи в со¬
ответствии с реакцией:
2NaOH + С02 -► Na2C03 + Н20
Образующийся карбонат осаждается хлоридом бария по реакции:
Na2C03 + ВаС12 -» BaC03! +2NaCl
Избыток щелочи нейтрализуют хлористоводородной кислотой:
NaOH + НС1 -> NaCl + Н20
Если вначале было п моль гидроксида натрия, которые затем провзаимодействова-
ли с х моль С02, в пробе останутся п — 2х моль гидроксида. Титрование холостого (из¬
мерение дыхания почвы) или контрольного (кинетика ДОВ) раствора связывает п моль
гидроксида натрия; следовательно, выдыхаемый С02 будет составлять п — (п — 2х) = 2х,
443
Глава 14. Органические формы азота, минерализуемый азот (и углерод)
и если Г— концентрация кислоты (М), то содержание С02-С в пробе можно выразить
следующим образом (мМ):
bV-cV
х= — аТ для дыхания почвы,
2
cV-sV
х = аТ для кинетики ДОВ.
2
За более полной информацией о представлении этих результатов и подробностями
моделирования кинетики минерализации следует обратиться к работам Турье (Thuries
идр., 2001,2002), ПансуиТурье(Pansuи Thuries,2003) иПансуссотр. (Лшиидр., 2003).
Примечания
• Для кинетики ДОВ разница между неорганическим азотом в пробе и неорганиче¬
ским азотом в контроле может быть положительной (нетго-минерализация) или
отрицательной (иммобилизация исходно присутствующего неорганического азо¬
та). Нетто-минерализация происходит, если ДОВ содержит относительно много N;
иммобилизация — если мало. Реку с сотр. (Recous и др., 1995), а также Хенриксен
и Бреланд (Henriksen и Breland, 1999), наблюдали, что когда содержание азота из
почвы + ДОВ меньше, чем 0,012 (сухое вещество ДОВ), рост микробной биомассы,
а также минерализация С, значительно снижаются.
• Описанная выше методика пригодна для исследований неорганических форм
С и N с использованием изотопов 14С, 13С и 15N: в растворах гидроксида натрия для
изотопов Сив экстрактах КС1 или KjSC^ для изотопов N.
• Кроме титриметрии, можно использовать и другие методы для определения С02
непосредственно в атмосфере инкубационных сосудов: газовую хроматографию
(газоадсорбционная хроматография, детектор — катарометр) или инфракрасную
спектрометрию.
• При расчете исходной массы проб почвы следует учитывать риск недостатка кис¬
лорода в инкубационных сосудах. Чтобы не допустить ослабления микробного ды¬
хания, содержание кислорода в газовой среде сосуда не должно опускаться ниже,
чем на треть его первоначального уровня.
• Расположение инкубационных сосудов (рис. 14.9) основана на том, что С02 тя¬
желее воздуха. Поэтому логично поместить коробку с почвой над коробкой со
щелочью.
14.3. Дополнительные методы анализа
14.3.1. Альтернативные методики кислотного гидролиза
Периодический кислотный гидролиз
Джанел с сотр. (Janeln др., 1979) критиковали использование методов гидролиза для ис¬
следования таких сложных сред, как почвы (см. разделы 14.2.1 и 14.2.2), поскольку этот
метод может приводить к «вторичным реакциям между соединениями азота и другими
продуктами разложения, сахарами и прочими восстанавливающими веществами, уча¬
ствующими в процессах инсолюбилизации и дезаминирования».
Эти авторы сравнили два метода гидролиза кипячением с обратным холодильником
с 3 М растворами хлористоводородной кислоты для букового опада. Гидролиз проводили
в течение 40 ч одновременно методами (1) непрерывного гидролиза и (2) периодическо¬
444
Часть 2. Органический анализ
го гидролиза с декантированием гидролизата через определенные интервалы времени
и заменой его свежими порциями экстрагирующего раствора. Изменения в содержании
общего, аминного и аммонийного азота контролировались для каждой серии растворов.
Некоторая схожесть результатов между двумя методами гидролиза наблюдалась, но
изменения содержания различных форм азота в случае периодического гидролиза на¬
блюдались гораздо чаще: содержание аминного азота увеличивалось при увеличении со¬
держания общего гидролизуемого азота; скорость выделения NH3-N была гораздо ниже
и почти постоянна, а скорость выделения негидролизуемого азота была ниже на 10%,
т. е. гораздо ниже, чем описанная Бремнером (Вгетпег, 1965) и Стивенсоном (Stevenson,
1982а, Ь) (табл. 14.4). С другой стороны, содержание фракции так называемого объеди¬
ненного N, представляющего собой гидролизованный азот неизвестного происхожде¬
ния, оставалось практически постоянным и равным примерно 35% азота в каждый
момент гидролиза в обоих методах. Это могли быть неустойчивые продукты, легко под¬
вергающиеся превращениям с самого начала процесса гидролиза.
Таким образом, периодический гидролиз оказался более надежным и был рекомендо¬
ван для использования вместо методов, описанных выше в разделах 14.2.1 и 14.2.2. Од¬
нако этот метод использует другую концентрацию кислоты и не испытывался на боль¬
шом количестве проб. Кроме того, он значительно сложнее для выполнения. Вопрос
о предпочтительном типе гидролиза все еще остается открытым. Например, Эгуменидес
с сотр. (Egoumenides и др,, 1987) использовали классическую методику, тогда как Бар-
риусо с сотр. (Barriuso и др., 1990) предпочли адаптировать метод Джанела с сотр. (Janel
идр., 1979).
Кислотный гидролиз с предварительной обработкой фтористоводородной
кислотой
Некоторые наблюдения показали низкую степень выделения аминокислот из глинистых
отложений с низким содержанием азота. Поэтому иногда рекомендуют предваритель¬
ную обработку фтористоводородной кислотой (например, Cheng и др., 1975; Stevenson
и Cheng, 1970). Стивенсон (Stevenson, 1982а) также рекомендовал предварительную обра¬
ботку в случае последующего колориметрического определения аминокислот (см. раз¬
дел 14.3.2):
• Помещают 50-125 мг тонкоизмельченной почвы в центрифужную пробирку из
полипропилена на 50 мл, добавляют 2,5 мл раствора 5 М HF - 0,1 М НС1 и пере¬
мешивают на возвратно-поступательном шейкере в течение 24 ч (для карбонатных
проб подкисляют 6 М раствором НС1 перед добавлением раствора НС1—HF).
• Добавляют 5 мл дистиллированной воды и лиофилизируют для удаления фтори¬
стоводородной кислоты.
• Добавляют 10 мл 6 М раствора НС1 и нагревают на масляной бане при 110°С с вну¬
тренним охлаждающим пальцем, чтобы не допускать испарения.
• Охлаждают и фильтруют через фильтровальную бумагу Whatman №42; промывают
остаток 10-15 мл дистиллированной воды.
14.3.2. Определение содержания аминокислот
Основные положения
Метод перегонки в присутствии нингидрина, описанный в разделе 14.2.1, хорошо под¬
ходит для анализа почв и грунтов, содержащих относительно много аминокислот. Для
Глава 14. Органические формы азота, минерализуемый азот (и углерод)
445
субстратов с низким содержанием N следует использовать более чувствительные мето¬
ды, например, колориметрические или хроматографические.
Для полного и относительно быстрого определения чаще всего применяют колори¬
метрический метод (например, Moore и Stein, 1948; Stevenson, 1965,1982а, b, с), при кото¬
ром измеряют поглощение синего комплекса, образующегося с нингидрином при pH 5
(рис. 14.2). Влияние катионов металлов из гидролизатов можно исключить, проводя
реакцию в присутствии комплексообразователя. Аммоний и другие соединения азота,
которые нестабильны в щелочной среде (аминосахара), удаляют щелочной предобра¬
боткой перед проведением реакции с нингидрином.
Хроматографические методы более трудны для реализации. Однако они более на¬
дежны и менее чувствительны к помехам, чем колориметрические методы. Кроме того,
они предоставляют примерно в 20 раз больше информации при определении каждой
индивидуальной аминокислоты. Тем не менее, полученную информацию трудно интер¬
претировать в таких сложных средах, как почвы; этому аспекту посвящены многочис¬
ленные работы.
Свободные аминокислоты в почвенных растворах можно разделить методом дву¬
мерной тонкослойной хроматографии (ТСХ) (Monreal и McGill, 1985). Однако наиболее
надежным методом является жидкостная ионообменная хроматография; существуют
специальные аппараты для определения содержания аминокислот методом двойного
ионного обмена. ВЭЖХ с обращенной фазой и дериватизацией ортофгалевым альде¬
гидом на предколонке также успешно используется в химии почвы (например, Warman
и Bishop, 1985,1987). Может быть применена и газовая хроматография (например, Jocteur
Monrozier, 1984; Barriuso и др., 1990; Рати, неопубликованные данные). Недостатком
этих методов является необходимость двойной дериватизации для блокирования кис¬
лотных и аминных функций перед введением в хроматограф, а их преимуществами —
хорошее разрешение и высокая чувствительность.
Колориметрия с нингидрином
Методика была предложена Стивенсоном (Stevenson, 1965, 1982а, Ь, с).
• Проводят гидролиз 6 М раствором НС1 после предобработки фтористоводородной
кислотой (см. раздел 14.3.1).
• Собирают фильтрат и промывные воды после гидролиза во вторую полипропиле¬
новую центрифужную пробирку на 50 мл и лиофилизируют для удаления НС1.
• Чтобы удалить аммоний и аминосахара, растворяют остаток в 5 мл дистиллирован¬
ной воды, добавляют две-три капли фенолфталеина (0,1 %-ный этанольный раствор)
и титруют 5 М раствором NaOH до розового окрашивания (примерно при pH 11).
Помещают пробирку на масляную баню при 100 °С; через 10 мин обдувают поверх¬
ность легким током воздуха, чтобы уменьшить объем раствора примерно до 2 мл.
• По каплям добавляют 6 М раствор НС1 до растворения гидроксидов металлов (рас¬
твор становится светло-желтым) и разбавляют водой, не содержащей аммония.
• Колориметрия. Помещают 0,5 мл раствора в пробирку и добавляют 0,5 мл 0,2 М
раствора цитрата натрия (177,6 мг безводного цитрата натрия в 1 л воды). Тщатель¬
но встряхивают, затем добавляют 2 мл раствора, приготовленного следующим об¬
разом: 25 мл ацетатного буферного раствора с pH 5 (500 г CH3COONa • ЗН20 +
+ 100 мл СН3СООН в 1 л воды); 50 мл нингидринного реагента (4%-ный раствор
в метилцеллозольве, Kodak), хранящегося в темноте под азотом в присутствии смо¬
лы Dowex-50 в Н-форме; 25 мл воды и 80 мг SnCl2- 2Н20. Накрывают алюминиевой
446
Часть 2. Органический анализ
крышкой и помещают пробирку на кипящую водяную баню при 100 °С на 30 мин.
Охлаждают холодной водой, добавляют 5 мл 50%-ного этанола и измеряют по¬
глощение при 570 нм. Разбавляют слишком концентрированные пробы 50%-ным
этанолом. Сравнивают со стандартным набором, приготовленным из стандартного
раствора лейцина, содержащего 28 мг амино-Ы/л: 0,262 г лейцина + 100 мл 0,1 М
раствора НС1 в 1 л воды.
Газожидкостная хроматография
Основные принципы
Методика, разработанная Занеттой и Винсендоном (Zanetta и Vincendon, 1973) для
определения аминокислот протеинов, была адаптирована для анализа почв. Газожид¬
костная хроматография выполняется на N(О)-гептафторбутират-производных изоами-
ловых эфиров аминокислот. Метод аналогичен ранее описанному методу разделения
N-трифторацетатов бутиловых эфиров аминокислот (Gehrke и др., 1971), который ис¬
пользуется в анализе почв (Jocteur Monrozier, 1984; Barriuso и др., 1990). Согласно Занет-
те и Винсендону, >1(0)-гептафторбутиратный метод может иметь два преимущества по
сравнению с N-трифторацетатным методом: (1) все аминокислоты легче разделяются на
широко применяемых колонках и (2) производные менее летучи, а продукты ацилиро¬
вания могут быть удалены перед инжекцией без риска потери. Степень разделения, до¬
стигнутая Занеттой и Винсендоном, была улучшена в лабораториях института IRD при
использовании капиллярной колонки (рис. 14.10).
Подготовка проб
• Помещают 5 мл почвенного экстракта или гидролизата и 100 мкл раствора
10 мкМ/мл пипеколиновой кислоты (внутренний стандарт) в коническую колбу из
пирекса с цилиндрической горловиной и завинчивающейся крышкой из ПТФЭ.
• Высушивают лиофилизацией, добавляют 400 мкл 1,25 М безводного метанольного
раствора НС1 (приготовленного растворением в безводном метаноле паров НС1,
полученных действием H2S04 на NaCl, и высушенных барботированием через чи¬
стую H2S04). Оставляют охлаждаться на 30 мин или 1 ч, затем высушивают, про¬
пуская ток азота над поверхностью при 50 °С.
• Добавляют 400 мкл 1,25 М раствора НС1 в изоамиловом спирте (приготовленного
так же, как метанольный раствор НС1) и нагревают герметически закрытую реак¬
ционную колбу до 110°С в течение 2,5 ч. Охлаждают и высушивают током азота
при 80 °С, как описано выше.
• Растворяют изоамиловые эфиры в 400 мкл ацетонитрила и добавляют 60 мкл геп-
тафторбутирового ангидрида. Нагревают герметически закрытую реакционную
колбу до 150°С в течение 10 мин. Охлаждают и высушивают под током азота, как
описано выше.
• Растворяют М(0)-гептафторбутират-производные изоамиловых эфиров амино¬
кислот в 0,5 мл этилацетата и вводят в хроматограф или хранят в холодильнике до
введения.
• Одновременно аналогичным образом готовят стандартную смесь из (1) 1 мл рас¬
твора, содержащего 1 мкМ каждой аминокислоты (коммерческий стандарт), и (2)
100 мкл 10 мкМ раствора пипеколиновой кислоты (внутренний стандарт). Этот
раствор используют для определения коэффициентов отклика каждой аминокис¬
лоты относительно пипеколиновой кислоты.
447
Глава 14. Органические формы азота, минерализуемый азот (и углерод)
Примечание
Если растворы содержат много неорганических веществ, рекомендуется отделить их пе¬
ред определением аминокислот. Простейший способ состоит в фильтровании смеси или
непосредственно перед инжекцией, или на первом этапе (метанол - НС1) при помощи
шприцевого фильтра. Можно также использовать предварительное фракционирование
методом ионного обмена (Gehrke и др., 1971).
Условия хроматографирования
• Капиллярная колонка с неполярной фазой, например, SE30, внутренним диаме¬
тром 0,3 мм и длиной 50 м.
• Газ-носитель — гелий, давление на входе 1,1 бар.
• Программирование печи: 70—270 °С при скорости нагрева 4 °С/мин.
• Пламенно-ионизационный детектор, 250 °С.
• Инжектор с делением пробы потоком газа, скорость 50 мл/мин.
На рис. 14.10 показана хроматограмма, полученная в этих условиях.
Примечание
Хотя предел обнаружения для почв обычно достаточен, его можно значительно улуч¬
шить, (1) используя инжектор с удалением растворителя (стеклянная игла или инжектор
для ввода проб с делением/без деления потоком) и (2) используя детектор электронного
захвата (чувствительный к фторированным производным), который позволяет опреде¬
лять ультраследовые количества аминокислот.
14.3.3. Определение содержания аминосахаров
Колориметрическое определение
Колориметрические методики для определения содержания аминосахаров в почве осно¬
ваны на методе, описанном Элсоном и Морганом (Elson и Morgan, 1933). В щелочной
среде аминосахара реагируют с ацетилацетоном с образованием пирроловых производ¬
ных. В кислой среде эти производные дают красный продукт конденсации с реактивом
Эрлиха (и-диметиламинобензальдегид в смеси этанол - НС1).
Этот метод мало селективен; многие вещества, такие как железо, смеси аминоса-
хар - аминокислота, а также темноокрашенные продукты гумификации, могут мешать
определению. Кроме того, методика, предложенная Стивенсоном (Stevenson, 1982а), до¬
вольно сложна. Перед колориметрированием необходимо провести двойную очистку
экстракта, сначала на анионообменной, потом на катионообменной смоле.
Позднее Шейдт и Зех (Scheidt и Zech, 1990) разработали упрощенный метод для почв,
основываясь на работе Бурцевой с сотр. (Burtseva и др., 1985). Он также основан на ме¬
тоде Элсона и Моргана (Elson и Morgan, 1933), но не требует предварительной очистки.
После колориметрирования типичный хромофор аминосахаров отделяют алкализаци¬
ей и экстрагируют диэтиловым эфиром для колориметрического определения. Метод
представляется перспективным, но используется не часто.
Хроматография
Хотя, так же как для аминокислот, наиболее распространенные методы основаны на ио¬
нообменной жидкостной хроматографии, для определения содержания аминосахаров
в почвах был разработан специальный метод газовой хроматографии (Benzing-Purdie,
Рис. 14.10. Разделение стандартной смеси аминокислот и почвенного экстракта методом га¬
зовой хроматографии (Pansu, неопубликованные данные) в хроматографических условиях, опи¬
санных в тексте. AI — аланин; GI — глицин; Va — валин; Th — треонин; Se — серин; Le — лейцин;
E.I. — пипеколиновая кислота (внутренний стандарт); Me — метионин; Ph — фенилаланин; As —
аспарагин; Ly — лизин; GI — глутаминовая кислота; Ту — тирозин
1981, 1984; Kogel и Bochter, 1985). После удаления хлористоводородной кислоты ами-
носахара восстанавливают раствором боргидрида натрия (NaBH4); затем синтезируют
трифторацетат-производные действием трифторуксусного ангидрида. Эти производные
разделяют на капиллярной колонке, затем идентифицируют и количественно определя¬
ют при помощи детектора, селективного к соединениям азота, используя и-аминофенол
в качестве внутреннего стандарта (рис. 14.11).
Глава 14. Органические формы азота, минерализуемый азот (и углерод)
449
Рис. 14.11. Разделение аминосахаров из стандартного раствора и почвенного гидролизата
методом газовой хроматографии (Kdgel и Bochter, 1985): 1 — глюкозамин, 2 — маннозамин,
3 — галактозамин, 4 — внутренний стандарт л-аминофенол — HCI)
14.3.4. Белки и гликопротеины (гломалин)
Белки и гликопротеины в почвах образуются из продуктов разложения или синтезиру¬
ются in situ микроорганизмами. Последний способ характерен для гломалина, а глико¬
протеин идентифицируют методом иммунофлюоресценции на гифах грибов арбуску-
лярной микоризы при активной колонизации корней (Wright и др., 1996). Райт и Упадяя
(Wright и Upadhyaya, 1996) исследовали 12 американских почв и обнаружили большую
концентрацию гломалина, от 4 до 15 мг белка/г почвы. Пюмалин исследовался в связи
с его значением для структурной стабильности почвы (Wright и др., 1999; Franzluebbers
и др., 2000). Он трудно солюбилизируется, Райт и Упадяя (Wright и Upadhyaya, 1998) раз¬
личали:
450
Часть 2. Органический анализ
• фракцию «легко» экстрагируемого гломалина: 0,25 г почвы + 2 мл 20 мМ экстраги¬
рующего раствора цитрата натрия с pH 7 при 121 °С в течение 30 мин;
• общий гломалин: то же с 50 мМ раствором цитрата с pH 8 при 121 °С в течение 90 мин
или более (последовательная экстракция).
После центрифугирования при 5000 g в течение 5 мин протеин в надосадочной жид¬
кости определяют на перфорированной микропластине при помощи пробного окраши¬
вания для анализа белков, используя бычий сывороточный альбумин в качестве кали¬
бровочного стандарта.
14.3.5. Определение содержания потенциально минерализуемого азота
методом электроультрафильтрации
Метод электроультрафильтрации (ЭУФ) иногда использовали при попытках разделения
растворимых и более или менее обменных форм катионов и анионов из органомине¬
рального комплекса почв. Принцип метода иллюстрируется на рис. 14.12. Непрерывное
электрическое поле приложено к водной суспензии почвы между двумя фильтрацион¬
ными мембранами. Катионы и анионы, которые проходят сквозь мембраны, собирают¬
ся в катодных и анодных компартментах, соответственно. Этот метод, однако, имеет два
недостатка: (1) накопление глин на анодной фильтрующей мембране, которое замедляет
процессы анионного обмена (нитраты), и (2) повышение pH в катионной ячейке, кото-
8.S. э
Промывная t с £
вода х 8 2 Вакуум
Рис. 14.12. Схема аппарата для электроультрафильтрации (ЭУФ)
Глава 14. Органические формы азота, минерализуемый азот (и углерод)
451
рое может приводить к потере аммония из-за испарения (Nemeth и др., 1988). Вторую
проблему можно решить, добавляя раствор хлористоводородной кислоты в катодный
компартмент.
Этот же метод был предложен для исследования различных форм азота (Nemeth и др.,
1979,1985). Использовались две ступени экстракции:
• в течение 0—35 мин при 20 °С и 200 В; это соответствует экстракции фактического
неорганического азота (аммоний и нитрат);
• 35-40 мин при 80 °С и 400 В; это характеризует минерализуемый органический
азот, доступный для растений в период их роста.
Методы ЭУФ были подвергнуты некоторой противоречивой критике: воспроизводи¬
мость считалась приемлемой (Sheehan, 1985), но иногда хуже, чем при более распростра¬
ненных методах экстракции, которые также легче в исполнении (Houba и др., 1986).
Фад-Рашид (Fahd-Rachid, 1990) нашел, что воспроизводимость обычно приемлема для
экстрагируемого нитратного азота, однако обычно неприемлема для ЭУФ минерализу¬
емого органического азота. Менгель (Mengel, 1996) использовал ЭУФ для исследования
круговорота органического азота почвы и его доступности для сельскохозяйственных
растений, Диез и Валеджо (Diezn Vallejo, 2004) сравнивали ЭУФ с другими методами при
определении потенциально доступного органического N.
Использованная литература1
Органические формы азота: общие работы
Chotte JL (1986) Evolution d’une biomasse racinaire doublement marqu6e (l4C, 15N) dans un syst&ne
sol-plante : £tude sur un cycle annuel d’une culture de mais. Thdse Doctor. Univ. Nancy I, 116
p.+annexes.
Guiraud G (1984) Contribution du marquage isotopique a devaluation des transferts d*azote entre les
compartiments organiques etmindrauxdans lessystkmessol-plante., Th£se Doct. es Sciences, Paris6,335 p.
Jocteur Monrozier L and Andreux F (1981) L’azote organique des sols, exemples de quantification des
formes prot£iques et des combinaisons complexes. Science du sol., 3, 219-242.
Jocteur Monrozier L (1984) Nature et evolution de l ’azote organique dans les sols et les sediments marins
recents., Th£se Doct. Etat, Univ. Nancy 1,176 p.
Kelley KR and Stevenson FJ (1995) Forms and nature of organic N in soil. Fert. Res., 42, 1-11.
Lavoisier AL (1789) Trait6 £16mentaire de chimie, Paris.
Matsumoto S, Yamagata M, Koga N and Ae N (2001) Identification of organic forms of nitrogen in soils
and possible direct uptake by plants. Dev. Plant Soil Sci., 92,208-209.
Schipper LA, Percival HJ and Sparling GP (2004) An approach for estimating when soils will reach
maximum nitrogen storage. Soil Use Manage., 20,281-286.
Schulten HR and Schnitzer M (1998) The chemistry of soil organic nitrogen: a review. Biol. Fertil Soils,
26,1-15.
Stevenson FJ (1982a) Nitrogen-organic forms. In Methods of Soil Analysis, Page AL, Miller RH and
Keeney DR ed. ASA-SSSA№9 part 2, 2nd edition, 625-641.
Stevenson FJ (1982b) Organic forms of soil nitrogen. In Humus Chemistry, Wiley, New York, 55-119.
Stevenson FJ (1982c) Origin and distribution of nitrogen in soil. In Nitrogen in Agricultural Soils,
Stevenson FJ ed. American Society of Agronomy, 1-42.
Stevenson FJ (1996) Nitrogen-organic forms. In Methods of Soil Analysis, Bigham JM and Bartels JM ed.
ASA-SSSA №5 part 3, 3rd edition, 1185-1200.
1 Авторы, упомянутые в тексте несколько раз, указаны только в той работе, в которой их вклад
является самым значительным.
452
Часть 2. Органический анализ
Формы азота» выделяемого методами кислотного гидролиза
и дистилляции
Bremner (1960) Forms of nitrogen in soils and plants. Rothamsted Exp. Stat. Rep., for 1959, p 59.
Bremner (1965) Organic forms of soil nitrogen. In Methods of Soil Analysis, Black CA et al. ed. American
Society of Agronomy, USA 9, part 2, 1238-1255.
Egoumenides C, Risterucci A and Melebou KE (1987) Appreciation de la fertility azot6e des sols
tropicaux: etude des fractions organiques de l’azote. L 'agronomie tropicale, 42, 85-93.
Keeney DR and Bemner JM (1967) A simple steam distillation method of estimating P-hydroxy-a-amino
acids. Anal. Biochem., 18, 274-285.
Tracey MV (1952) The determination of glucosamine by alkaline decomposition. Biochem. J., 52, 265-
267.
Van Slyke (1911-1912) The analysis of proteins by determination of the chemical groups characteristic of
the different amino-acids. J. Biol. Chem., 10,15-55.
Усовершенствование методов кислотного гидролиза
Barriuso Е, Andreux F and Portal JM (1990) Caract^risation par hydrolyse acide de l’azote des fractions
organiques et organo-min6rales d’un sol humiffcre. Science du sol., 1990,28, 223-236.
Cheng CN, Shufeldt RC and Stevenson FJ (1975) Amino acid analysis of soils and sediments: extraction
and desalting. Soil Biol. Biochem., 7, 143-151.
Griffith SM, Sowden FJ and Schnitzer M (1976) The alkaline hydrolysis of acid-resistant soil and humic
acid residues. Soil Biol. Biochem., 8, 529.
Janel Ph, Jocteur Monrozier L and Toutain F (1979) Caract6risation de l’azote des litidres et des sols par
hydrolyse acide, Soil Biol. Biochem., 11, 141-146.
Определение содержания аминокислот
Gehrke CW, Zumwalt RW and Kuo К (1971) Quantitative amino acid analysis by gas-liquid
chromatography. J. Agric. Food Chem., 19, 605-618.
Monreal CM and McGill WB (1985) Centrifugal extraction and determination of free amino acids in soil
solutions by TLC using tritiated 1 -fluoro-2,4-dinitrobenz£ne. Soil Biol. Biochem., 17, 533-539.
Moore S and Stein WH (1948) Photometric ninhydrin method for use in the chromatography of amino
acids. J. Biol. Chem., 176, 367-388.
Stevenson FJ and Cheng CN (1970) Amino acids in sediments: recovery by acid hydrolysis and
quantitative estimation by a colorimetric procedure. Geochim. Cosmochim. Acta., 34, 77-88.
Stevenson (1965) Amino acids. In Methods of Soil Analysis, Black C.A. et al. ed. American Society of
Agronomy 9, part 2,1437-1451.
Warman PR and Bishop C (1985) The use of reverse-phase HPLC for soil amino-N analysis, J. Liquid
Chromat., 8, 2595-2606.
Warman PR and Bishop C (1987) Free and HF-HCl-extractable amino acids determined by high
performance liquid chromatography in a loamy sand soil. Biol Fertil. Soils, 5, 215-218.
Zanetta JP and Vincendon G (1973) Gas-liquid chromatography of the N(0)-heptafluorobutyrates of the
isoamyl esters of amino acids. I. Separation and quantitative determination of the constituent amino
acids of proteins. J. Chromat., 76, 91-99.
Определение содержания аминосахаров
Benzing-Purdie L (1981) Glucosamine and galactosamine distribution in a soil as determined by gas-
liquid chromatography on soil hydrolysates: effect of acid strength and cations. Soil Sci. Soc. Am. J.,
45, 66-70.
453
Глава 14. Органические формы азота, минерализуемый азот (и углерод)
Benzing-Purdie L (1984) Amino sugar distribution in four soils as determined by high resolution gas
chromatography. Soil Sci. Soc. Am. J., 48,219-222.
Burtseva TI, Cherkasova SA and Ovodov YuS (1985) Quantitative determination of amino sugars in
bacterial lipopolysaccharides. Khimiya Prirodnykh Soedinenii, 6, 739-743.
Elson LA and Morgan WTJ (1933) A colorimetric method for the determination of glucosamine and
chondrosamine. Biochem. J., 27,1824-1828.
Kugel I and Bochter R (1985) Amino sugar determination in organic soils by capillary gas chromatography
using a nitrogen-selective detector. Z Pflanzenernaehr. Bodenk, 148,260-267.
Scheidt M and Zech W (1990) A simplified procedure for the photometric determination of amino sugars
in soil. Z Pflanzenerndhr. Bodenk, 153, 207-208.
Гломалин
Franzluebbers AJ, Wright SF and Stuedemann JA (2000) Soil aggregation and glomalin under pastures in
the southern piedmont USA. Soil Sci. Soc. Am. J., 64, 1018-1026.
Wright SF, Franke-Snyder M, Morton JB and Upadhyaya A (1996) Time-course study and partial
characterization of a protein on hyphae of arbuscular mycorrhizal fungi during active colonization of
roots. Plant Soil, 181, 193-203.
Wright SF and Upadhyaya A (1996) Extraction of an abundant and unusual protein from soil and
comparison with hyphal protein of arbuscular mycorrhizal fungi. Soil Sci., 161, 575-586.
Wright SF and Upadhyaya A (1998) A survey of soils for aggregate stability and glomalin, a glycoprotein
produced by hyphae of arbuscular mycorrhizal fungi. Plant Soil, 198,97-107.
Wright SF, Starr JL and Paltineanu IC (1999) Changes in aggregate stability and concentration of
Glomalin during tillage management transition. Soil Sci. Soc. Am. J., 63,1825-1829.
Определение содержания мочевины
Beaton JD (1978) Urea - its popularity grows as a dry source of nitrogen. Crops Soils, 30, 11-14.
Bremner JM (1982) Nitrogen-Urea. In Methods of Soil Analysis, Part 2, Page AL et al. ed. ASA-SSSA
№9 part 2,2rd edition, 699-709.
Douglas LA and Bremner JM (1970) Extraction and colorimetric determination of urea in soils. Soil Sci.
Soc. Am. Proc., 34, 859-862.
Douglas LA, Sochtig H, and Flaig W (1977) Colorimetric determination of urea in soil extracts using an
automated system. Soil Sci. Soc. Am. J., 42,291-292.
Fearon WR (1939) The carbamido diacetyl reaction: a test for citrullin. Biochem. J., 33,902-907.
Mulvaney RL and Bremner JM (1979) A modified diacetyl monoxime method for colorimetric
determination of urea in soil extracts. Commun. Soil Sci. Plant Anal., 10,1163-1170.
Watt GW and Chrisp JD (1954) Spectrophotometric method for determination of urea. Anal. Chem., 26,
452-453.
Определение содержания потенциально минерализуемого азота:
общие работы
Catroux G, Chaussod R and Nicolardot В (1987) Appreciation de la foumiture d’azote par le sol. C. R.
Acad. Agric. Fr., 73, 71-79.
Comforth IS and Walmsley D (1971) Methods of measuring available nutrients in west indian soils. 1.
Nitrogen. Plant Soil, 35, 389-399.
Dahnke WC and Johnson GV (1990) Testing soils for available nitrogen. In Soil Testing and Plant
Analysis, 3rd. ed. SSSAbook series, n°3, 127-139.
454
Часть 2. Органический анализ
Fahd-Rachid (1990) Mise аи point methodologique sur Гestimation de l ’azote organique potentiellement
mineralisable dans le sol., DEAINP-ENSAT Toulouse, Document ORSTOM-Montpellier №1, 60
p. multig.
Gianello C and Bremner JM (1986) Comparison of chemical methods of assessing potentially available
organic nitrogen in soil, Commun. Soil Sci. Plant Anal, 17,215-236.
Giroux M and Sen Tran T (1987) Comparaison de dififerentes mdthodes d’analyse de l’azote du sol en
relation avec sa disponibilite pour les plantes. Can. J. Soil Sci., 67, 521-531.
Juma NG and Paul EA (1984) Mineralizable soil nitrogen: Amounts and extractability ratios, Soil Sci.
Soc. Am. J., 48, 76-80.
Keeney DR and Bremner JM (1966) Comparison and evaluation of laboratory methods of obtaining an
index of soil nitrogen availability. Agron. J., 58, 498-503.
Keeney DR (1982) Nitrogen-Availability indices. In Methods of Soil Analysis, Part 2, Page AL et al. ed.
ASA-SSSA №9 part 2, 2nd edition, 711-733.
Определение содержания потенциально минерализуемого азота:
биологические методы
Benedetti A, Sebastiani G (1996) Determination of potentially mineralizable nitrogen in agricultural soils.
Biology and Fertility of Soils 21,114-120.
Cabrera ML and Kissel DE (1988) Evaluation of a method to predict nitrogen mineralized from soil
organic matter under field conditions. Soil Sci. Soc. Am. J., 52, 1027-1031.
Campbell CA (1978) Soil organic carbon, Nitrogen and fertility. In Soil Organic Matter, Schnitzer and
Khan ed. Elsevier, Amsterdam, 224-225 and 254.
Chaussod R, Nicolardot B, Soulas G and Joannes H (1986) Mesure de la biomasse microbienne dans les
sols cultiv^s. Il-Cin&ique de mineralisation de mattere organique microbienne marqude au C14. Rev.
Ecol Biol. Sol., 23,183-196.
Deans JR, Molina JAE and Clapp СЕ (1986) Models for predicting potentially mineralizable nitrogen and
decomposition rate constants. Soil Sci. Soc. Am. J., 50, 323-326.
Keeney DR and Bremner JM (1967) Determination and isotope ratio analysis of different forms of
nitrogen in soil: 6. Mineralizable nitrogen. Soil Sci. Soc. Am. Proc., 31, 34.
Marquardt DW (1963) An algorithm for least-squares estimations of nonlinear parameters. J. Soc. Ind.
Appl. Math., 11, 431-441.
Mary В and R6my JC (1979) Essai d’appreciation de la capacity de mineralisation de Pazote des sols de
grande culture. I. Signification des cinetiques de mineralisation de la matiere organique humifiee.
Ann. Agron., 30, 513-527.
Nuske A and Richter J (1981) N-mineralization in lOss-parabrownearthes: incubation experiments. Plant
Soil, 59, 237-247.
Reynolds WD and Beauchamp EG (1984) Comments on “Potential errors in the first-order model for
estimating soil nitrogen mineralization potentials”, Soil Sci. Soc. Am. J., 48, 698.
Richter J, Nuske A, Habenicht and W Bauer J (1982) Optimized N-mineralization parameters of loess
soils from incubation experiment. Plant Soil, 68, 379-388.
Robertson K, Schnbrer J, Clarholm M, Bonde ТА and Rosswall T (1988) Microbial biomass in relation to
C and N mineralization during laboratory incubations. Soil Biol. Biochem., 20,281-286.
Seyfried MS and Rao PSC (1988) Kinetic of nitrogen mineralization in Costa Rican soils: model
evaluation and pretreatment effects. Plant Soil, 106,159-169.
Smith JL, Schnabel RR, McNeal BL and Campbell GS (1980) Potential errors in the first-order model for
estimating soil nitrogen mineralization potentials. Soil Sci. Soc. Am. J., 44, 996-1000.
Stanford G and Smith SJ (1972) Nitrogen mineralization potentials of soils. Soil Sci. Soc. Am. Proc., 36,
465-472.
Stanford G, Carter JN, and Smith SJ (1974) Estimates of potentially mineralizable soil nitrogen based on
short-term incubations. Soil Sci. Soc. Am. Proc., 38, 99-102.
455
Глава 14. Органические формы азота, минерализуемый азот (и углерод)
Waring SA and Bremner JM (1964) Ammonium production in soil under waterlogged conditions as an
index of nitrogen availability. Nature (London), 201, 951.
Определение содержания потенциально минерализуемого азота:
химические методы
Campbell СА, Jame YW, Jalil A and Schoenau J (1997) Use of hot KC1-NH4-N to estimate fertilizer N
requirements. Can. J. Soil Sci., 77,161-166.
Fox RH and Piekielek WP (1978) A rapid method for estimating the nitrogen-supplying capability of a
soil. Soil Set Soc. Am. J., 42, 751-753.
Gianello C and Bremner JM (1988) A rapid steam distillation method of assessing potentially available
organic nitrogen in soil, Commun. Soil Sci. Plant Anal, 19, 1551-1568.
Guiraud G and Fardeau JC (1977) Dosage par la m&hode Kjeldahl des nitrates contenus dans les sols et
les v^taux. Ann. Agron., 28, 329-333.
Mac Lean OA (1964) Measurement of nitrogen supplying power of soils by extraction with sodium
bicarbonate. Nature, 203, 1307-1308.
0ien A and Selmer-Olsen AR (1980) A laboratory method for evaluation of available nitrogen in soil.
ActaAgric. Scand., 30, 149.
Stanford G and Smith SJ (1978) Oxidative release of potentially mineralizable soil nitrogen by acid
permanganate extraction, Soil Sci., 126, 210.
Stanford G (1978) Evaluation of ammonium release by alcaline permanganate extraction as an index of
soil nitrogen availability. Soil Sci., 126, 244.
Velly J, Egoumenides C, Pichot J and Marger JL (1980) L’azote extractible par une solution de KC1 et la
foumiture d’azote & la plante dans 40 sols tropicaux. Agronomie tropicale, 35, 374-380.
Whitehead DC (1981) An improved chemical extraction method for predicting the supply of available
soil nitrogen. J. Sci. FoodAgric., 32, 359-365.
Определение содержания потенциально минерализуемого азота методом
ЭУФ
Diez JA and Vallejo А (2004) Comparison of two methods for nitrogen extraction of irrigated Spanish
soils and related nitrogen balance calibrations. Commun. in Soil Sci. Plant Anal., 35, 2227-2242.
Houba VJG, Novozamsky I, Huybregts AWM and Van der Lee JJ (1986) Comparison of soil extractions
by 0.01M CaCl2, by EUF and by some conventional extraction procedures. Plant Soil, 96, 433-437.
Mengel К (1996) Turnover of organic nitrogen in soils and its availability to crops. Plant soil, 181,
83-93.
Nemeth К (1985) Recent advances in EUF research (1980-1983). Plant and Soil, 83,1-19.
Nemeth K, Bartels H, Vogel H and Mengel К (1988) Organic nitrogen compounds extracted from arable
and forest soils by EUF and recovery rats of amino acids. Biol. Fertil. Soils, 5,271-275.
Nemeth K, Makhdum IQ, Koch К and Beringer H (1979) Determination of categories of soil nitrogen by
electroultrafiltration (EUF). Plant Soil, 53,445-453.
Sheehan MP (1985) Experiments on the reproducibility of results from EUF soil extracts with possible
improvements resulting from these experiments. Plant Soil, 83, 85-92.
Кинетика процесса минерализации
Blet-Charaudeau C, Muller J and Laudelout H (1990) Kinetics of carbon dioxide evolution in relation to
microbial biomass and temperature. Soil Sci. Soc. of Am. J., 54,1324-1328.
Bottner P, CoQteaux MM, Anderson JM, Berg B, Billfcs G, Bolger T, Casabianca H, Romanya J and
Rovira P (2000) Decomposition of ,3C labelled plant material in a European 65-40° latitudinal
transect of coniferous forest soils: simulation of climate change by translocation of soils. Soil Biol.
& Biochem., 32, 527-543.
456
Часть 2. Органический анализ
Henriksen, ТМ and Breland, ТА (1999) Nitrogen availability effects on carbon mineralization, fungal and
bacterial growth, and enzyme activities during decomposition of wheat straw in soil. Soil Biol. &
Biochem., 31, 1121-1134.
Pansu M, Sallih Z and Bottner P (1998) Modelling of soil nitrogen forms after organic amendments under
controlled conditions. Soil Biol. & Biochem., 30, 19-29.
Pansu M and Thurtes L (2003). Kinetics of C and N mineralization, N immobilization and N volatilization
of organic inputs in soil. Soil Biol & Biochem., 35, 37-48.
Pansu M, Thurtes L, Larrd-Larrouy MC and Bottner P (2003) Predicting N transformations from organic
inputs in soil in relation to incubation time and biochemical composition. Soil Biol. & Biochem., 35,
353-363.
Recous S, Robin D, Darwis D and Mary В (1995) Soil inorganic N availability: effect on maize residue
decomposition. Soil Biol. & Biochem., 27, 1529-1538.
Thurifcs L, Larr6-Larrouy MC and Pansu M (2000) Evaluation of three incubation designs for
mineralization kinetics of organic materials in soil. Commun. Soil Sci. Plant Analy., 31,289-304.
Thurtes L, Pansu M, Feller C, Hermann P, and R6my JC (2001) Kinetics of added organic matter
decomposition in a mediterranean sandy soil. Soil Biol. & Biochem., 33,997-1010.
Thurtes L, Pansu M, Larre-Larrouy MC and Feller C (2002) Biochemical composition and mineralization
kinetics of organic inputs in a sandy soil. Soil Biol. & Biochem., 34, 239-250.
Часть 3
НЕОРГАНИЧЕСКИЙ АНАЛИЗ -
ОПРЕДЕЛЕНИЕ ОБМЕННЫХ ИОНОВ
И ПОЛНЫЙ ЭЛЕМЕНТНЫЙ АНАЛИЗ
Глава 15. Определение pH среды
15.1. Введение
15.1.1. pH почвы
Среди методов, используемых для характеризации почвы в данный момент времени,
одним из наиболее распространенных является измерение величины pH (показателя
концентрации ионов Н+). Простота и скорость измерения этого показателя позволяют
использовать измерение pH почвы в массовых лабораторных процедурах.
В почвоведении практически используемый диапазон pH сокращен с 0-14 до 1-12.
Почвы, характеризующиеся крайними значениями pH, содержат значительные количе¬
ства солей: от сильнокислых сульфатных до сильнощелочных карбонатных почв. Цели
определения pH зависят от потребителя.
В лабораторной практике предварительное определение pH позволяет выбрать соот¬
ветствующие методы экстракции и измерения для кислых, нейтральных или щелочных
почв в зависимости от их pH (например, для определения обменных катионов или эле¬
ментов, доступных для растений, таких как фосфор). Однако следует иметь в виду, что «pH
почвы» представляет собой только pH раствора, находящегося в равновесии с почвой.
Измерение pH пробы является общепринятым методом, используемым для характе¬
ризации почвы.
В базе данных французского Института агрономических исследований (INRA, 1995)
приводится следующая классификация диапазонов pH почв:
pH < 3,5 - очень сильнокислые почвы
3.5 < pH < 5,0 - сильнокислые почвы
5,0 < pH < 6,5 - кислые почвы
6.5 < pH < 7,5 — нейтральные почвы
7.5 < pH < 8,7 — щелочные почвы
pH > 8,7 — сильнощелочные почвы.
Величина pH характеризует физико-химическое окружение почвы на данном участке
и является результатом мгновенных равновесий, контролируемых различными компо¬
нентами среды, например:
• смешанные сульфаты с очень сильнокислой реакцией, образующиеся при окис¬
лении сернистых соединений почв мангровых болот (кислые сульфатные почвы);
• ряд органических или неорганических кислот и таких элементов, как алюминий
или железо, которые способны подкислять почвенный раствор из-за кислотного
гидролиза компонентов, начиная с минеральных веществ и до обменных комплек¬
сов1;
• нейтральные соли сильных кислот и сильных оснований или почвы, насыщенные
обменными комплексами, но с низким содержанием карбоната кальция, pH кото¬
рых близок к нейтральному;
• в присутствии карбоната кальция в открытой системе при парциальном давлении
атмосферного С02 равновесие в почвенной суспензии устанавливается при pH
1 Очевидно, имеется в виду «комплекс почвенный поглощающий». — Примеч. науч. ред.
460
Часть 3. Неорганический анализ
около 8,4; давление С02 может достигать больших значений, особенно в глубоких
горизонтах почвы, и может существенно влиять на равновесное pH;
• в некоторых почвах карбонат магния образуется при высоких значениях pH (около
9). Присутствие карбонатов натрия приводит к повышению pH до величин, кото¬
рые могут превышать 10.
Настоящий обзор диапазона pH в почвах подчеркивает со статической точки зрения
влияние факторов двух типов на это сложное равновесие:
• на концах диапазона pH — влияние больших количеств легкорастворимых кислых
или основных солей и, в меньшей степени, влияние органических и неорганиче¬
ских кислот и всех соединений, способных вызывать кислотный гидролиз;
• в почвах, система обычно в большей степени зависит от давления С02 и содержа¬
ния элементов в обменном комплексе почвы (ионы Н+ и ионы металлов), которые
компенсируют возможные колебания pH за счет постоянного обмена между по¬
чвой и почвенным раствором.
Однако очень важно рассмотреть величину pH с динамической точки зрения, посколь¬
ку многие типы равновесий могут устанавливаться в течение различных промежутков
времени, которые зависят от различных внутренних и внешних факторов. Самое боль¬
шое влияние на физико-химическое окружение, несомненно, оказывает переувлаж¬
нение почвы. Сезонные колебания влажности и особенно ритм этих колебаний могут
значительно изменять концентрацию почвенного раствора вследствие гидролиза и вы¬
деления протонов1 или катионов, растворения и выщелачивания или, наоборот, концен¬
трирования и осаждения. Эти общие замечания, касающиеся экологических аспектов,
позволяют выделить различные концепции, связанные с pH почвы:
• актуальная кислотность или щелочность, выраженная через концентрацию диссо¬
циированных протонов в почвенном растворе;
• обменная кислотность, обеспечиваемая протонами, закрепленными в обменном ком¬
плексе почв и способными к миграции после обмена с нейтральными солями (КС1);
• потенциальная кислотность, выраженная через кислотность (измеренную в усло¬
виях насыщения) сульфидов (в кислых сульфатных почвах) или чаще после заме¬
щения всех кислотных функциональных групп почвы в процессе гидролиза;
• буферная емкость, которая уменьшает колебания pH через постоянный обмен
между почвой и почвенным раствором, причем наиболее важными определяю¬
щими факторами являются степень разбавления и качество обменного комплекса
(тип глинистых минералов и степень насыщения).
С агрономической точки зрения pH является, прежде всего, показателем состояния
почвенного плодородия. Он дает информацию о возможной химической деградации по¬
чвы из-за снижения степени насыщения, возможном присутствии определенных ток¬
сичных солей и микробной активности, а также степени усвояемости элементов рас¬
тениями, причем оптимальный диапазон pH для растворимости элементов расположен
между 5,5 и 6,5. При pH ниже 5,5 некоторые элементы (например, свободный алюми¬
ний, марганец) могут быть токсичными; другие элементы (например, фосфор) могут су¬
ществовать в недоступных формах или удерживаться твердой фазой. При pH выше 6,5
другие элементы (например, микроэлементы) могут оказаться недоступными.
Знание pH почвы позволяет также выбирать сельскохозяйственные культуры для
выращивания, например, ацидофильные растения, такие как чай или кофе, или расте-
1 Здесь и далее: ионов гидроксония — Н30\ — Примеч. науч. ред;
461
Глава 15. Определение pH среды
ния с нейтрофильными клетками. Наконец, pH предоставляет полезную информацию,
позволяющую сделать правильный выбор исправляющего воздействия: (1) удобрения
с подкисляющим (например, соли аммония), нейтрализующим или подщелачивающим
(например, селитра или аммонийные соли) эффектом и (2) удобрения или подкормки
для увеличения или уменьшения pH и повышения плодородия культивируемых почв
(например, известкование). Однако знание только величины pH недостаточно, напри¬
мер, для точной оценки потребности в извести (см. гл. 24) или обменной кислотности
и токсичности алюминия (см. гл. 23).
15.1.2. Ограничения
Определение pH — простая операция, но эта простота может дезориентировать, по¬
скольку определенная величина pH может оказаться неправильной, если не уделять
внимания деталям (таким, как состояние контактов электродов и стабилизации напря¬
жения питания). Необходимо точно следовать методике измерения и выбирать соответ¬
ствующее оборудование. Следует учитывать и некоторые другие аспекты при интерпре¬
тации результатов:
• Измерения pH чаще всего проводят в стандартизованных условиях с использова¬
нием проб с нарушенной структурой и почвенных суспензий, но это означает, что
они не отражают реальных условий.
• Проблема пространственной вариабельности измерений связана с неоднородными
условиями почвенных горизонтов, агрегатов и организованных микробиотопов.
• Природные или искусственные временные колебания, связанные с влиянием та¬
ких внешних факторов, как влажность или система возделывания культур.
Все это говорит в пользу in situ измерения pH. При непрерывном измерении в иде¬
альных технических условиях результаты определения pH in situ (см. раздел 15.3) будут
ближе всего к реальным условиям — в поле и, следовательно, способны предоставить
информацию о сложных равновесиях между почвой и почвенным раствором, а также о
многих внешних влияющих факторах. Тем не менее стандартизованные лабораторные
измерения из-за их простоты остаются наиболее часто используемыми.
15.1.3. Теоретические аспекты
Согласно Брёнстеду, кислоты при диссоциации выделяют протоны по реакции:
Кислота <-» основание + Н+ (15.1)
Эта реакция происходит в присутствии акцептора электронов. Сильная кислота — это
кислота, которая выделяет протоны наиболее легко. Для нейтрализации растворов двух
кислот (хлористоводородной и уксусной) с равными молекулярными концентрациями
требуются одинаковые количества гидроксида натрия. Но эффект повышения темпера¬
туры в процессе нейтрализации более выражен для раствора хлористоводородной кис¬
лоты: таким образом, можно сказать, что хлористоводородная кислота «сильнее» уксус¬
ной кислоты. Эту концепцию «силы» кислот (остававшуюся неясной в течение долгого
времени) можно количественно оценить при измерении pH. В случае сильной кислоты
равновесие реакции (15.1) сильно сдвинуто вправо. Можно количественно определить
силу кислоты, определяя количество ионов Н+, выделенных при диссоциации кислоты.
Диссоциацию кислоты (НА) в растворителе (S) можно описать уравнением:
НА + S <-» А- + SH
462
Часть 3. Неорганический анализ
Чаще всего кислоту применяют в виде водного раствора. Вода как растворитель мо¬
жет играть роль либо кислоты:
Н20 о он- + Н+
либо основания:
Н20 + Н+ Н30+
Применив закон действующих масс, получаем:
[0Н-][Н30Ч/(Н20)2 = *,
где К — константа диссоциации, определенная при данной температуре.
Так как диссоциированная часть остается чрезвычайно малой по сравнению с общим
числом молекул воды, знаменатель может считаться постоянной величиной, тогда
[Н30+][ОН-] = *Н20, (15.2)
где Kw = 10’14 при температуре 25 °С.
В чистой воде количества ионизированных форм Н30+ и ОН-равны: [Н30+] = [ОН-] =
= 10-7. Таким образом, в водном растворе добавление кислоты увеличит количество ио¬
нов гидроксония Н30+: Н30+ > 10-7. В нейтральной среде Н30+ = 10-7 и в щелочной среде
Н30+ < 10~7. Для удобства используют показатель Сёренсена (Sorensen, 1909), называемый
pH:
pH = —lg[H30+] (15.3)
Величина pH отражает активность ионов гидроксония. В случае водного раствора
сильной кислоты эта активность сравнима с концентрацией кислоты. В случае сильного
основания ее можно рассчитать из уравнения (15.2). Величины pH изменяются между 0
(очень кислая) и 14 (очень щелочная). Десятикратное разбавление изменяет pH на одну
единицу (в случае сильных кислот или оснований). На практике если концентрацию
ионов Н+ умножить на 2, величина pH уменьшается примерно на 0,3. Согласно формуле
(15.1) и закону действующих масс:
[основание] [Н30+]/[кислота] = КА (15.4)
Величина КА определяет константу кислотности пары кислота/основание (в актив¬
ностях). Удобнее использовать показатель:
РЛГА = -1вЛГА (15.5)
Более сильная кислота имеет меньшее значение рКк Из уравнений (15.3)—(15.5) мож¬
но вывести общую формулу pH для раствора кислоты в равновесии с ее сопряженным
основанием:
pH = р КА + 1§([основание]/[кислота]) (15.6)
Уравнение (15.6) показывает, что отклонения pH раствора уменьшаются с такими
изменениями концентраций, когда кислотная форма находится в равновесии с анало¬
гичным количеством соответствующей основной формы. В этом случае pH приближа¬
ется к рКА. Такой раствор имеет стабильное значение pH и мало чувствителен к добав¬
лению кислоты или основания. Этот стабилизирующий эффект называется буферным
эффектом. Он наблюдается при смешении кислоты и соответствующего основания или
кислоты и одной из ее ионизируемых солей. Буферные растворы используют в качестве
Глава 15. Определение pH среды
463
стандартов pH или для поддержания определенного уровня pH в некоторых реакциях.
Обменный комплекс почв также оказывает буферное влияние на равновесный почвен¬
ный раствор, присоединяя или выделяя протоны.
Уравнение (15.6) неприменимо для растворов, содержащих только кислоты или осно¬
вания (в этом случае pH стремилось бы к —а> или +оо, соответственно). Однако pH всегда
легко рассчитать по формулам (15.2)—(15.5), используя следующие приближения.
В водном растворе слабой кислоты равновесная реакция диссоциации
ан + н2о<->н3о+ + а-
показывает, что концентрация основной формы А- равна концентрации ионов Н30+,
образованных в процессе диссоциации. Кроме того, если СА — общая концентрация
кислоты, концентрация кислотной формы АН становится равной СА — [Н30+]. В случае
слабой кислоты концентрацию ионов гидроксония можно считать малой по сравнению
с СА. Тогда уравнение (15.4) приобретает вид
*а = [Н,0+]УСа;
применяя уравнения (15.3) и (15.5) с величинами СА, выраженными в единицах нор¬
мальности, получаем:
pH = (pAA-lgCA)/2 (15.7)
В водном растворе слабого основания равновесная реакция имеет вид:
в + н2о<->вн + он-
Концентрация кислотной формы ВН равна концентрации ионов ОН-, образованных
при диссоциации. Если Св — общая концентрация основания, концентрация основной
формы В равна Св — [ОН-]. В случае слабого основания концентрация ионов ОН- оста¬
ется малой по сравнению с Св, и уравнение (15.4), относящееся к равновесию кислоты
ВН, можно записать в следующем виде:
А:а=Св[Н30+]/[0Н-];
используя выражения (15.2), (15.3) и (15.5), получаем:
pH = 7 + 1/2(рКд + lgCB) (15.8)
Льюис применил кислотно-основную концепцию Брёнстеда к непротонным системам
и показал, что она применима ко всем растворителям, если соединение, принимающее
электроны, рассматривать как кислоту, а соединение, отдающее электроны — как ос¬
нование. Объединение пары электронов основания со свободной орбиталью кислоты
приводит к нейтрализации. Ешнистые минералы с центрами, способными выделять
электроны, можно рассматривать как основания Льюиса, тогда как гидроксильные
и карбоксильные группы органического вещества почвы, которые могут принимать
электроны, соответствуют кислотам Льюиса.
15.2. Классические методы анализа
15.2.1. Методы испытаний
Как указано в разделе 15.1.3, pH определяет активность ионов гидроксония. Активность
этих ионов сравнима с концентрацией ионов только в случае достаточно разбавленных
растворов. Концентрация кислоты соответствует ее титруемой кислотности (общей кис-
464
Часть 3. Неорганический анализ
лотности). Активность определяет количество «свободных» (т. е. диссоциированных)
ионов Н+, определяемых при измерении pH. Для этой цели используют два метода: (1)
колориметрический метод, недорогой и быстрый, но не очень точный, особенно в мут¬
ных или окрашенных средах, и (2) электрометрический метод, который используется
гораздо чаще.
15.5.2. Колориметрический метод
Основные положения
Это недорогой экспрессный метод, который можно использовать для быстрого исследо¬
вания почвы. Основную трудность представляет визуальное сравнение с цветным стан¬
дартом, особенно в случае окрашенных или мутных растворов. Можно использовать
оборудование для фотоэлектрического сравнения, чтобы избежать ошибок, возникаю¬
щих при наблюдениях невооруженным глазом.
Известно, что окрашенные вещества способны изменять цвет в зависимости от кис¬
лой или основной природы раствора (табл. 15.1). Механизм действия цветных индика-
Таблица 15.1. Основные цветные индикаторы, используемые в диапазоне pH почв
Тривиальное
название
Химическое название
Кислотная
форма
pH перехода
Основная
форма
Тимол синий
Тймолсульфонфталеин
Красный
1,9
оранжевый
Желтый
Динйтрофенол
2,6- или 2,4-динитрофенол
Бесцветный
3,1
Желтый
Метилоранж
Диметиламиноазобензол-
сульфонат натрия
Красный
3,7 желтый
Оранжевый
Бромфеноловый
Тетрабромфенолсульфон-
Желтый
4,0
Фиолетовый
синий
фталеин
пурпурный
Бромкрезоловый
Тетрабром-и<-крезолсуль-
Желтый
4,6 зеленый
Синий
зеленый
фонфталеин
Хлорфеноловый
Дихлорофенолсульфон-
Желтый
5,6 розово¬
Пурпурный
красный
фталеин
оранжевый
«-нитрофенол
«-нитрофенол
Бесцветный
5,2
Желтый
Метиловый
Диметиламиноазобензол-
Красный
5,7
Желтый
красный
о-карбоновая кислота
оранжевый
Бромкрезоловый
Дибром-о-крезолсуль-
Желтый
6,2
Фиолетовый
пурпурный
фонфталеин
пурпурный
Бромтимоловый
Дибромтимолсульфонфта-
Желтый
6,9 зеленый
Синий
синий
леин
Феноловый
Фенолсульфонфталеин
Желтый
7,3 красно¬
Фиолетовый
красный
оранжевый
jw-крезоловый
л<-крезолсульфонфталеин
Желтый
8,3
Красный
пурпурный
оранжевый
Фенолфталеин
Фенолфталеин
Бесцветный
8,3 розовый
Розовый
Тимол синий
Тймолсульфонфталеин
Желтый
8,9
Сине¬
пурпурный
фиолетовый
Ализариновый
Нитробензолазосалицило-
Желтый
10,3
Красный
желтый
вая кислота
оранжевый
Глава 15. Определение pH среды
465
торов хорошо описывается теорией электролитической диссоциации. Цветной инди¬
катор можно рассматривать как слабый электролит, один из ионов которого окрашен,
тогда как конденсированная форма индикатора бесцветна или имеет другой цвет в соот¬
ветствии с равновесием диссоциации:
Индикатор-Н (бесцветный) <-> индикатор (окрашенный) + Н\
Индикаторы pH часто используют для пропитки полосок бумаги, которые можно
приводить в непосредственный контакт с большинством почв. Это очень быстрый ме¬
тод, но не очень точный (подвержен влиянию ионных мицеллярных сред, солей, зави¬
сит от времени установления равновесия и т. д.). Его используют для предварительных
тестов в полевых или лабораторных условиях (Рати и др., 2001).
Проведение измерений
Изменение цвета наблюдается только в переходной зоне, которая для большинства ин¬
дикаторов соответствует изменению pH на единицу. Существующие индикаторы покры¬
вают диапазон pH от 1 до 12 (табл. 15.1 и 15.2). Определение включает сравнение цвета
индикатора в буферном растворе определенного pH (табл. 15.3) с цветом, полученным
при добавлении индикатора к неизвестному раствору. Используя хризоидин, Ки и Жу
(Qui и Zhu, 1986) одновременно определили величину pH, буферную емкость и потреб¬
ность почв в известковании.
Таблица 15.2. Приготовление индикаторов для почв в диапазоне pH от 2,8 до 11
Ицдикатор
pH перехода
Переход цвета
Концентрация
раствора
рКпри 18 °С
а-динитрофенол
2,4-4,4
Бесцветный в желтый
0,1 г в 200 мл воды
4,06
у-динитрофенол
4-5,4
Бесцветный в желтый
0,1 г в 440 мл воды
5,15
«-нитрофенол
5,2-7,6
Бесцветный в желтый
0,1 г в 100 мл воды
7,18
ж-нитрофенол
6,6-8,8
Бесцветный в желтый
0,3 г в 100 мл воды
8,33
Фенолфталеин
8,5-10,5
Бесцветный в красный
0,04 г в 100 мл
30%-ного спирта
9,73
Ализариновый
желтый
10-12
Бесцветный в желтый
0,05 г в 100 мл
50%-ного спирта
11,16
Неизвестный раствор помещают в пробирку такой же формы, как пробирка, содер¬
жащая стандартные буферные растворы, и добавляют к раствору заранее определенное
количество капель индикатора. Как указано выше, визуальное сравнение часто за¬
труднительно, поскольку неизвестные растворы могут быть сами слегка окрашенными
или мутными. Использование стандартного компаратора может улучшить результаты
(рис. 15.1).
Методика
Для приготовления шкал сравнения наливают 6 мл буферного раствора (табл. 15.3) в гра¬
дуированную пробирку, пригодную для использования в стандартном компараторе Уол¬
пола (рис. 15.1). Добавляют 1 мл раствора индикатора, подходящего для интересующего
диапазона pH (табл. 15.2). Повторяют эти операции с почвенным раствором.
Целью является получение одинаковых цветов (1) в пробирке, содержащей неизвест¬
ный раствор (позиция 2 на рис. 15.1) и (2) в стандартных пробирках, представляющих
466
Часть 3. Неорганический анализ
Таблица 15.3. Приготовление стандартных растворов pH с использованием универсального
буферного раствора Бриттона—Робинсона
V— объем (мл) раствора 2, добавляемый к 100 мл раствора 1 для получения
соответствующего значения pH.
Раствор 1 Фосфорная кислота Н3Р04, М = 98, d= 1,70 (85%) 2,71 мл
Уксусная кислота СН3СООН, М = 60, d= 1,05 (100%) 2,28 мл
Борная кислота Н3В03, М = 61,8,99% 2,5 г
Растворяют и доводят объем до 1000 мл.
Раствор 2 0,2 М водный раствор NaOH (не содержащий карбонатов).
а-динитрофенол у-динитрофенол л-нитрофенол ж-нитрофенол Фенолфталеин
V
pH
V
pH
V
pH
V
pH
V
PH
17,5
2,87
25
4,10
37,5
5,3
50
6,8
65
8,69
20
3,29
27,5
4,35
40
5,72
52,5
7
67,5
8,95
22,5
3,78
30
4,56
42,5
6,09
55
7,24
70
9,15
25
4,10
32,5
4,78
45
6,37
57,5
7,54
72,5
9,37
27,5
4,35
35
5,02
47,5
6,59
60
7,96
75
9,62
37,5
5,33
50
6,80
62,5
8,36
77,5
9,91
52,5
7
65
8,69
80
10,38
постепенные изменения pH (и, следовательно, цвета) в позициях 1 и 3. Позиции б и 4
заняты неизвестным раствором без индикатора для исключения влияния исходного цве¬
та раствора. Это быстрый метод, позволяющий достичь точности 0,2 единицы pH. Если
раствор мутный, рекомендуется отфильтровать его через фильтр Millipore.
Глаз
<
1 6
о о
2 5
О О
о3 4о
<—
Свет
<
<
Рис. 15.1. Наблюдения с компаратором Уолпола.
15.2.3. Электрометрический метод1
Основные положения
Электрометрический метод чаще всего используется для анализа почв, поскольку он
точнее колориметрического метода. Он позволяет анализировать мутные и окрашенные
среды, а также проводить непрерывные измерения.
1 Электрохимический метод. — Примеч. науч. ред.
Глава 15. Определение pH среды
467
Для измерения истинного значения pH необходимо получить тонкий слой водорода,
насыщая платиновый электрод, покрытый платиновой чернью, водородом в момент вы¬
деления. Погружая этот электрод в раствор, измеряют разницу потенциалов Е (в воль¬
тах) как функцию концентрации ионов Н+, которую можно оценить из уравнения Нерн-
ста (в стандартных условиях для парциального давления водорода):
Е= (RT\n[W])/nF= 0,0001983 74g(H+) = 0,058 lg(H+) (при 20°С), (15.9)
где R — газовая постоянная; Т — абсолютная температура; п - заряд иона; F— постоян¬
ная Фарадея (96 500).
С водородным электродом трудно работать. Вместо него используют другие электро¬
ды сравнения с известным потенциалом относительно водородного электрода. Вели¬
чину pH определяют из значения электродвижущей силы (ЭДС), измеренной между
электродом сравнения и индикаторным электродом.
рН-метры
Электронное оборудование для измерения pH можно разделить на две группы: (1) по¬
тенциометрические системы и (2) системы прямого (аналогового или цифрового) отсче¬
та. Потенциометрический метод с измерениями при нулевом токе является самым точ¬
ным, но длительным и поэтому больше не используется для массовых анализов. Чаще
всего используются модели прямого отсчета, в частности цифровые модели. Выбор рН-
метра зависит от требуемого качества измерений:
• для массовых анализов прибор должен обладать входным сопротивлением около
1010 Ом для проведения измерений с точностью 0,1 единиц pH, ручной настройкой
температуры, масштаба шкалы и угла наклона шкалы;
• для более точных измерений входное сопротивление должно быть не менее 1012 Ом
и разрешение ±0,002 единицы pH, соответствующее 0,1 мВ, с автокоррекцией мас¬
штаба шкалы, ее угла наклона и температуры. Если выходной сигнал измеряется
в милливольтах, значит прибор может быть снабжен адаптором сопротивления
и подсоединен к устройству записи или компьютеризированной системе сбора
данных.
Для некоторых специфических типов электродов необходимы модели, настройка
и калибровка которых программируются микропроцессором (см. раздел 15.3).
Индикаторный электрод
Индикаторный электрод чувствителен к ионам Н+, для достоверных измерений он дол¬
жен использоваться вместе с электродом сравнения.
Стеклянный электрод является наиболее распространенным pH-электродом. Нернст
первым описал принцип его работы, но идея использовать стеклянный электрод была
впервые высказана Хабером {Haber) и Клеменсивицем (Klemensiewicz) в 1909 г. Если два
раствора различной кислотности разделены очень тонкой стеклянной мембраной (тол¬
щиной в несколько микрон), между ними устанавливается разница потенциалов, кото¬
рая зависит от разницы в концентрациях ионов Н\
На практике электрод состоит из шарика, выдутого из стекла на конце трубки. Не
все сорта стекла пригодны для изготовления электродов. Обычно используют «мягкое»
стекло, содержащее много натрия (тип Корнинг 015). Затем электрод наполняют рас¬
твором НС1 или буферным раствором, содержащимся в Ag/AgCl электроде. Две системы
отсчета применяют для этих внутренних растворов, одна дает нулевую ЭДС при pH 7 от¬
468
Часть 3. Неорганический анализ
носительно насыщенного каломельного электрода в качестве электрода сравнения; дру¬
гая дает ЭДС = 0 при pH = 0. Первый вариант является наиболее часто используемым.
Время отклика стеклянного электрода на изменение pH очень мало. Он нечувстви¬
телен к окислительным или восстановительным средам. Образующийся потенциал
стабильно изменяется на ~58 мВ/единицу pH при 25 °С, шкала измеряемой разницы
потенциалов простирается на ±500 мВ. Диапазон внутреннего сопротивления очень ве¬
лик (100—1000 МОм), что требует применения измерительных приборов с очень большим
входным сопротивлением, не менее 1010 Ом.
Могут наблюдаться небольшие различия в отклике между электродами. Этот сдвиг
связан с асимметрией потенциала из-за небольших различий в конструкции. Стеклян¬
ный электрод особенно чувствителен к обезвоживанию. Он должен всегда храниться
в дистиллированной воде. Перед первым использованием электрода его следует выдер¬
жать в дистиллированной воде в течение 24 ч.
Стеклянные электроды часто продаются в форме единого зонда, включающего ин¬
дикаторный электрод и электрод сравнения. Эта модель не рекомендуется для анализа
почвенных суспензий, так как пористое спеченное стекло в месте контакта электрода
сравнения легко блокируется в присутствии глинистых минералов, что приводит к боль¬
шой нестабильности сигнала во времени. Однако комбинированные электроды посте¬
пенно улучшаются и становятся более надежными, чем модели из пористого спеченного
стекла (тип Hack-one)1.
Электроды, состоящие из стеклянной мембраны на плоской поверхности, так¬
же были рекомендованы для измерения pH почвы, особенно при низкой влажности
(см. раздел 15.3, in situ измерения). Используя эти электроды, Брельтембек и Бремнер
(Breltembeck и Вгетпег, 1984) нашли, что величины pH были стабильными, воспроизво¬
димыми и коррелировали с другими классическими измерениями для 15 почв с водным
потенциалом от -15 до -0,3 бар.
Прочный и простой в использовании в труднодоступных средах сурьмяный элек¬
трод также использовался для определения pH почв, илов или шламов, хотя он менее
точен, чем стеклянные электроды. Сурьмяный стержень приобретает потенциал, кото¬
рый изменяется линейно с pH в диапазоне pH от 1 до 10. Конклинг и Бланчар (Conkling
и Blanchar, 1988) опубликовали данные по pH почвы, измеренным с использовани¬
ем стеклянного электрода, которые хорошо коррелировали с данными, полученными
с сурьмяным электродом, хотя последний немного завышал значения в некоторых слу¬
чаях.
С исторической точки зрения, следует упомянуть и платино/хингидронный электрод.
Первоначально его применяли как аналог водородного электрода, поскольку хингидрон
диссоциирует в растворе, выделяя атомарный водород. Он был рекомендован Вторым
международным конгрессом почвоведов в 1927 г., но уже давно не используется в химии
почвы.
Электроды сравнения
Электроды сравнения содержат в себе вторую половину элемента с известным электриче¬
ским потенциалом (который должен быть максимально стабильным), необходимого для
сравнения с индикаторным электродом. Наиболее распространенным и изученным яв¬
ляется каломельный электрод, но существуют и другие модели, которые могут использо¬
ваться в конкретных случаях, в частности хлоридсеребряный электрод или амальгамный
таллиевый электрод, который позволяет проводить измерения при температурах до 135 °С.
1 Hack Europe SA, LP 51, Namur, Belgium.
469
Глава 15. Определение pH среды
Каломельный электрод
Каломельный электрод состоит из ртутьсодержащей основы, покрытой слоем каломе¬
ли (хлорида одновалентной ртути) в растворе хлорида калия. Его параметры описаны
в Приложении 1 в конце данной главы. Хотя существующие pH-метры проводят изме¬
рения при нулевом токе, на поверку малые отрицательные или положительные электри¬
ческие потоки не влияют на потенциал электрода. Потенциал зависит от концентрации
КС1 в растворе, заполняющем электрод. Используют три концентрации КС1:0,1 М, 1 М
и насыщенный раствор. 0,1 М раствор рекомендуют использовать для точных измере¬
ний; он не очень чувствителен к температуре, но плохо хранится. Обычно используют
электрод с насыщенным раствором, который прост в обращении, но имеет довольно
высокий температурный коэффициент (см. Приложение 1). При температуре 25 °С
его потенциал равен +245,8 мВ относительно водородного электрода сравнения. Одна
из возможных проблем использования электрода сравнения связана с электрическим
контактом между заполняющим раствором КС1 и анализируемым раствором. Обычно
используют электрод из пористого спеченного стеклянного электрода со скоростью
утечки около 0,2 мл/ч. Для точных измерений можно использовать аппаратуру со шли¬
фованными соединениями, задается та степень утечки, которая обеспечивает желаемую
степень герметизации. Можно также использовать гелевый электролит для решения
проблемы контакта без риска диффузии, но он может храниться только в течение огра¬
ниченного времени.
Важно обратить особое внимание на проблемы, которые могут возникнуть при ис¬
пользовании пористых диафрагм при анализе почвенных суспензий. Диафрагмы легко
блокируются, что приводит к ухудшению воспроизводимости результатов. Их надо всег¬
да хорошо очищать при помощи щетки и обдува. Большинство проблем при измерени¬
ях связаны с использованием электрода сравнения, особенно в мутных средах, таких
как почвенные экстракты. Устройства для обеспечения контакта с раствором хлорида
калия быстро засоряются, что искажает результаты. Было рекомендовано использовать
твердотельный электрод сравнения (TBI— Recomat SA). Он выполнен из деревянных ко¬
лец, которые пропитаны насыщенным раствором хлорида калия, и прекрасно работает
в сильно загрязненных средах, таких как на станциях очистки воды.
Если присутствие ионов хлорида или калия нежелательно в процессе измерений, сле¬
дует использовать промежуточный мостик с двумя диафрагмами. В этом случае кали¬
бровку проводят с трис-буферными растворами (см. Приложение 3).
Хлоридсеребряный электрод
Этот электрод применяют реже, но его прочность делает его достойным использова¬
ния. Его простота также позволяет изготавливать миниатюрные установки. Его часто
используют в сочетании со стеклянным электродом или ионоселективными электро¬
дами.
Этот электрод дает возможность работать при температурах в диапазоне от -30 до
+135 °С. При 25 °С, когда электрод наполнен насыщенным раствором хлорида калия,
его потенциал равен +200 мВ относительно стандартного водородного электрода (см.
Приложение 1). Относительно каломельного электрода при 25 °С его потенциал ра¬
вен —45 мВ. Рекомендуется контролировать стабильность этого электрода, поскольку
измерительный ток может вызвать превращение серебра при микроэлектролизе (элек¬
трод можно улучшить, покрывая его тонкой тефлоновой пленкой).
470
Часть 3. Неорганический анализ
15.2.4. Электрометрический контроль и калибровка
Перед измерениями pH-метр должен быть откалиброван. Выбранный аппарат должен
иметь возможность коррекции угла наклона шкалы и температуры. В большинстве слу¬
чаев достаточна точность измерений ±0,1 единицы pH, потому что другие источники
ошибок оказывают большее влияние (например, неоднородность пробы). Для измере¬
ния pH суспензий предпочтительно выбирать устройство с отдельным электродом срав¬
нения, поскольку риск ошибок, вызванных загрязнением пористой диафрагмы, умень¬
шается, кроме того при возникновении проблем оно более доступно.
Аппарат должен быть включен за некоторое время до начала измерений (часто суще¬
ствует режим ожидания, в котором система поддерживается в равновесии), а электроды
помещены в соответствующий буферный раствор. Рекомендуется начинать с исполь¬
зования Т4УК-буфера (см. Приложение 3 в конце данной главы), который отличает¬
ся большой устойчивостью к изменениям температуры. Корректируют показание рН-
метра до соответствующей величины, затем переходят к другому буферному раствору,
который должен иметь pH, близкий к pH измеряемых растворов. Прибор должен затем
показать значение для нового буферного раствора (с поправкой на температуру, см. При¬
ложение 3). Вводят компенсацию на любые небольшие изменения с помощью коррек¬
тировки угла наклона шкалы. Если корректировка невозможна, комбинация электродов
может быть неудовлетворительной, в частности из-за стеклянного электрода, который
не дает строго линейного отклика между двумя выбранными значениями pH. В этом слу¬
чае выбирают более узкий диапазон pH, например, 7-9 или 6-8 вместо 4-9. Чем ближе
pH стандартного буферного раствора к измеряемому значению, тем точнее измерение.
Не рекомендуется перемешивать растворы во время измерения pH . В случае проблем
с линейностью тщательно очищают пористую диафрагму электрода при помощи жест¬
кой щетки. Также можно очистить ее, продувая при помощи насоса для фильтрации.
Измерения pH почв можно разделить на четыре основных группы:
1. Измерения в водных суспензиях почв.
2. Измерения в насыщенных пастах.
3. Измерения в насыщенных экстрактах.
4. In situ измерения.
15.2.5. Исследование водных суспензий почв
Технические требования изменяются с изменением соотношения почва: раствор из-за
разнообразия почвенных материалов. Во Франции экспериментальный стандарт AFNOR
А/’ЛГ-ЗЫОЗ (1998) рекомендует использовать соотношение почва : вода, равное 1:2,5.
Международный стандарт NF ISO 10390 (1994) рекомендует соотношение почва : вода,
равное 1:5.
Методика
• Взвешивают 10 г почвы, высушенной при комнатной температуре и просеянной
через сито 2 мм (в стандарте ISO используют анализируемую пробу объемом не ме¬
нее 5 мл), добавляют 25 мл (или пятикратный объем анализируемой пробы в стан¬
дарте ISO) кипяченой (для удаления С02) дистиллированной воды, встряхивают
в течение 1 ч на качающемся столе (энергичное перемешивание в течение 5 мин,
согласно стандарту ISO).
• Дают отстояться раствору в течение 30 мин (по стандарту ISO - в течение не ме¬
нее 2, но не более 24 ч), погружают электроды в раствор так, чтобы пористая часть
Глава 15. Определение pH среды
471
электрода сравнения была погружена в светлую часть суспензии (рис. 15.2). Запи¬
сывают значение pH после стабилизации показаний. Отмечают температуру и про¬
веряют температурный корректор рН-метра.
Примечания
Поскольку величина pH уменьшается при повышении температуры, обычно все зна¬
чения приводят к температуре 25 °С. Для точных измерений следует использовать тер¬
мостатированную баню; почвенные суспензии часто имеют буферный эффект, который
нельзя скорректировать расчетным методом.
Измерения должны выполняться без перемешивания. Для некоторых почв показания
pH-метра могут оказаться нестабильными и показывать постоянное смещение. В таких
случаях рекомендуется записывать показания спустя некоторое время, например, 3 мин,
и использовать тот же самый промежуток времени для других измерений, не забывая
упомянуть об этом при записи результатов.
Измерения почвенных суспензий и, что еще более важно, насыщенных паст вызыва¬
ют явление, называемое «эффектом пасты» или «эффектом суспензии», который может
исказить результаты на ±1 единицу pH (рис. 15.3). Этот эффект проявляется особенно
Индикаторный Электрод
электрод сравнения
Надосадочная
жидкость
+300 мВ
+100 мВ
О
0 мВ
■ы
+200 мВ
Рис. 15.3. Возможные ошибки, связанные с влиянием суспензии для почвы в дистиллированной
воде (Bates, 1973)
472
Часть 3. Неорганический анализ
сильно, когда электроды (и особенно электрод сравнения) находятся в контакте с осад¬
ком. Этот эффект может быть связан с различием в подвижности ионов К+ и С1_, диф¬
фундирующих из раствора электрода сравнения в присутствии коллоидов (заряженных
частиц высокой емкости катионного обмена). Пэюлинг и Пич (Grewling и Peech, 1960)
сообщали, что это явление можно минимизировать, используя 0,01 М раствор СаС12
(PHJ.
Хотя соотношение почва: раствор влияет на результаты определения pH в воде, оно
оказывает слабое влияние на растворы солей, таких как 0,01 М раствор СаС12 (Conyers
и Davey, 1988). Нилссон с сотр. (Nilsson и др., 1995) рекомендовали использовать
объемно-весовые соотношения вода: почва больше, чем 10 (или в случае объемных от¬
ношений больше, чем 2) для получения надежных результатов при определении водного
рНн о в органических почвах.
Время контакта воды с почвой также влияет на величину pH. Кониерс и Дави (Conyers
и Davey, 1988) не рекомендовали перемешивать в течение нескольких часов, поскольку
это приводит к колебаниям результатов и не позволяет получить стабильную величину.
Ки и Жу (Qui и Zhu, 1986) сообщили о стабилизации результатов их измерений после
перемешивания более 30 мин (но до 1 ч).
15.2.6. Определение рНк и рНСа
При измерении в воде величина pH не учитывает общую кислотность, в частности про¬
тоны и формы алюминия, зафиксированные в обменных комплексах почвы, которые
представляют потенциальную кислотность. Следовательно, кроме первого измерения
в воде, необходимо выполнить измерение в 1 М водном растворе КС1 с тем же соотно¬
шением почва : раствор (1:2,5 для методики, описанной в параграфе «Методика» под¬
раздела 15.2.5) и используя те же технические средства. Это дает величину рНк.
Полученные величины рНк обычно меньше1, чем величины рНно. Разница (АрН)
может достигать 1 единицы pH. Величина АрН > 0 указывает, что емкость катионного
обмена превышает емкость анионного обмена. Существует значимая корреляция между
положительным значением АрН и обменной кислотностью. Однако, это может быть
верно для одного семейства почв, но неверно в общем случае.
Эти измерения были объектом экспериментального исследования стандарта AFNOR
Л^-Л31-104 (1988) и стали частью международного стандарта NFISO 10390 (1994).
Измерения pH также проводят в 0,01 М водном растворе СаС12, потому что ионы Са
вызывают флокуляцию раствора и минимизируют влияние пасты и разбавления. Поэто¬
му значение pH становится более стабильным, и легче сравнивать значения pH для раз¬
личных почв, особенно засоленных. Определение рН^ приводит к меньшим значениям
АрН, чем определение рНк. Исследования Кониерса и Дави (Conyers и Davey, 1988) по¬
казали, что величины рНводн, рНк и рНСа тесно связаны для почв в широком диапазоне
pH. Для несоленых почв с отрицательным суммарным зарядом они нашли следующее
соотношение:
рНСа=1,05рНюян-0,9.
Методика, описанная в параграфе «Методика» подраздела 15.2.5, применима во всех
случаях, как для водных, так и для солевых растворов.
1 В некоторых андосолях с высоким содержанием аллофана, pH КС1 может превышать pH
Н20 (амфотерная среда).
473
Глава 15. Определение pH среды
15.2.7. Определение pH «насыщенных паст»
Определение pH насыщенных паст проводят в целях возможно более полного воспро¬
изведения условий природной среды. Этот метод очень сложен для выполнения. Для
начала следует приготовить «насыщенную пасту»:
• Отвешивают 200 г воздушно-сухой почвы, просеянной сквозь сито 2 мм, поме¬
щают ее в цилиндрический контейнер объемом около 500 мл с широким носиком
и плотно закрывающейся крышкой.
• Добавляют 50—70 мл кипяченой дистиллированной воды в зависимости от механи¬
ческого состава почвы (т. е. от содержания глины); количество воды должно быть
достаточным для того, чтобы только увлажнить почву.
• Отмечают объем добавленной воды в мл.
• Закрывают сосуд и оставляют стоять на 30 мин.
• При помощи лабораторной бюретки при перемешивании добавляют дистилли¬
рованную воду малыми порциями для получения однородной пасты. Основная
сложность — это понять, когда необходимо прекратить добавление воды, т. е. когда
паста насыщается водой. Отмечают общий объем добавленной воды, закрывают
сосуд и оставляют стоять на 30 мин.
При насыщении паста должна быть блестящей и достаточно текучей, чтобы отлипать
от шпателя. Если в центре пасты сделать лунку, вода не должна собираться на ее дне,
в противном случае существует избыток воды. Если вода появляется, необходимо доба¬
вить небольшое количество почвы и учесть его массу в окончательных расчетах. Несмотря
на субъективность оценки, на практике повторные определения для одной пробы дают
очень близкие результаты. Почвы, содержащие мало глины, требуют добавления очень
небольшого количества воды. Почвенную пасту характеризуют степенью насыщения:
% насыщения = масса добавленной воды х 100 / масса почвы.
Электрод следует погружать чрезвычайно осторожно. Электрод сравнения может
иметь двойную диафрагму, чтобы исключить загрязнение пористой диафрагмы. Пред¬
почтительно использовать электрод, наполненный насыщенным раствором КС1 или
гелевым раствором агар-агара для снижения диффузии жидкости. Что касается индика¬
торного электрода, то разработано несколько моделей для пенетрации, менее хрупких,
чем модели со стеклянным шариком, хотя и менее чувствительных.
Показания pH-метра должны записываться после их стабилизации, которая может
занимать несколько минут. Этот метод в большей степени, чем другие, зависит от явле¬
ния, называемого «эффектом пасты», который может исказить результаты измерения на
1 единицу pH. Полученные результаты будут выше, чем результаты, полученные для со¬
отношения почва: водная суспензия, равного 1:2,5 (см. параграф «Методика» подразде¬
ла 15.2.5), но сравнимы с результатами для насыщенных экстрактов, описанными ниже.
15.2.8. Определение pH насыщенных экстрактов
Столкнувшись с трудностями при получении водного экстракта, максимально представ¬
ляющего действительный почвенный раствор, in situ отвечающий за водоудерживающую
способность почвы, Ричардс (Richards, 1954) предложил использование насыщенного
экстракта в качестве промежуточного между полевым и лабораторным измерениями.
В самых распространенных методах определения степени засоления почвы использу¬
ют насыщенный экстракт, pH которого можно измерять при соблюдении мер предосто¬
рожности и методик, описанных в разделе 15.2.5.
474
Часть 3, Неорганический анализ
Сначала готовят насыщенную пасту (см. подраздел 15.2.7). Хранят ее в течение 24 ч,
предохраняя от высыхания. Затем переносят ее на фильтровальную бумагу на ворон¬
ке Бюхнера под вакуумом. Собирают фильтрат, что может занять довольно много вре¬
мени, иногда более 2 ч. Если экстракт мутный, фильтруют его еще раз. Измеряют pH.
Если измерение не может быть выполнено немедленно после экстракции, хранят рас¬
твор в плотно закрытом флаконе в холодильнике. Раствор также можно использовать
для определения других параметров, например, проводимости и содержания катионов
и анионов (см. гл. 18).
15.2.9. Определение pHNaF
Основные положения
Этот анализ дополняет in situ пробу с NaF на фильтре (Рати и др., 2001). Его использу¬
ют для обнаружения веществ с упорядочением ближнего порядка в почвах, в частности
алюминия в активной форме А1(ОН)3. Это определение основано на реакции:
А1(ОН)3 + 6NaF -► NayMFg + 3NaOH
Выделение ионов ОН~ вызывает увеличение pH, которое указывает на присутствие
довольно больших количеств веществ с упорядочением ближнего порядка. Недостатком
этого метода является то, что присутствующие фториды быстро повреждают стеклян¬
ный электрод, и поэтому рекомендуется использовать один из электродов только для
этой цели.
Методика
• Готовят 1 л насыщенного раствора фторида натрия из 45 г NaF. Тщательно переме¬
шивают в полиэтиленовой бутыли и доводят объем до 1000 мл дистиллированной
водой. Оставляют стоять на 2 дня при периодическом встряхивании бутыли. Если
происходит частичная кристаллизация NaF, используют только прозрачную часть
надосадочной жидкости.
• Взвешивают 1 г тонкодисперсной почвы, просеянной через сито 2 мм, воздушно¬
сухой или предпочтительно с исходной влажностью. Измеряют влажность, ис¬
пользуя другую пробу, чтобы затем пересчитать результаты на почву, высушенную
при 110°С. Помещают пробу в полиэтиленовый стакан на 100 мл. Добавляют 50 мл
раствора NaF, перемешивают и погружают электроды. Измеряют исходное значе¬
ние pH в момент времени t = 0 и затем при t = 2, 10 и 30 мин. Наносят результаты
на график pH = ДО*
15.3. Анализ почв в полевых условиях
Полевые измерения в природных условиях обеспечивают получение самых точных дан¬
ных для исследования почвенных процессов. Это особенно справедливо для измерения
pH, учитывая, как много физико-химических реакций могут влиять на величину pH.
Кроме измерения pH, используют другие электрохимические сенсоры (Pansu и др.,
2001) для измерения проводимости, окислительно-восстановительного потенциала,
а также ионометрию с электродами, селективными к другим ионам: Na+, К+, Са2+ и NH4+
(см. гл. 16 и 18). Более того, использование измерительной станции позволяет получать
временные серии данных для характеризации изменений процессов во времени, кото¬
рая более информативна, чем одномоментная характеризация в конкретный момент
Глава 15- Определение pH среды
475
времени. Электрометрические измерения могут также предоставлять информацию об
активности ионных форм, которая более полезна для термодинамических расчетов, чем
данные о концентрациях (Le Brusq и др., 1987). Этот тип данных неоценим для исследо¬
вания механизмов переноса в почве.
15.3.1. Оборудование
pH-метр должен иметь входное сопротивление не менее 109 Ом или лучше 1011 Ом,
и кроме того — возможность корректировки нуля, угла наклона и температуры. Коррек¬
тировка должна быть автоматической, если измерения проводятся в течение длитель¬
ного промежутка времени. Должна быть возможность регулярно проверять калибровку
путем программирования этих операций с помощью микропроцессора, что также спо¬
собствует обеспечению хорошей воспроизводимости.
Чувствительность, равная 0,1 единицы pH, достаточна с учетом неоднородности сре¬
ды. Прибор должен быть способен функционировать независимо (снабжен батареями).
Выходной сигнал должен быть выражен в мВ. Желательно использовать электронную
систему сбора и хранения данных для последующей повторной обработки.
pH-метр должен иметь возможность экранированного соединения с датчиками, трех¬
проводным коаксиальным кабелем для исключения помех, вызванных длиной кабелей.
Более простое решение состоит в использовании электродов со встроенным усилите¬
лем. Это позволяет избежать проблем, связанных с длиной кабеля (до 200 м) и проник¬
новением влаги через нарушенную изоляцию. Предпочтительнее использовать модель
с отдельными индикаторными электродами и электродами сравнения (рис. 15.4). Для
контроля нескольких электродов коммутационный блок обычно включает пять рабочих
каналов с возможностью потенциометрической подстройки каждого канала, корректи¬
рующей небольшие различия между нулевыми точками электродов.
15.3.2. Полевые измерения
Ручные измерения
После выбора места проводят первоначальную характеризацию почвы. Отбор проб вы¬
полняют с глубин, на которые будут погружены электроды. Минимальный набор изме¬
ряемых параметров включает влажность, электропроводность, ионный баланс и обмен¬
ные катионы. Устанавливают метеорологический пункт, включающий, как минимум,
дождемер и термометр. Температурный зонд помещают в почву вблизи других электро¬
дов. Индикаторный электрод очень осторожно погружают в отверстие, предваритель¬
но проделанное в почве при помощи пробоотборника. Отверстие вокруг электрода на¬
полняют почвой, которая была вынута ранее. Следует принять меры для обеспечения
хорошего контакта с шариком электрода. Можно использовать электроды с плоской
стеклянной мембраной (Breltenbeck и Вгетпег, 1984). Электрический кабель около метра
длиной с коаксиальным разъемом на конце ведет в пластиковую камеру с крышкой для
защиты соединения в периоды между измерениями (рис. 15.4). Ввод электрода сравне¬
ния состоит из стеклянной трубки диаметром 3—4 мм и длиной 30 см, с контейнером
50 мл с крышкой на одном конце. Нижняя секция погруженной части сделана из по¬
ристого спеченного стекла (рис. 15.4). Наполняют трубку до 3 или 4 см выше пористой
секции 3,5 М водным раствором КС1, загущенным добавлением 4% агара, содержащего
кристалл тимола для защиты от плесени. Продолжают заполнение 3,5 М водным раство¬
ром КС1 до заполнения половины контейнера. Гель над пористой секцией предотвращает
излишнюю диффузию раствора КС1. Вводы расположены на расстояниях 10-50-100 см
476
Часть 3. Неорганический анализ
а)
Агар 4%
3,5 М раствор
KCI
Температурный
зонд 3
3,5 М раствор
KCI
ния
Защищенное
соединение
Трубка ввода
для электро-^^Индика
да сравне- \ торный
электрод
Стойка.
Пористое
спеченное
Ь)
Рис. 15.4. Полевые измерения: а — схема установки датчиков с трубками ввода 1,2 и 3 для элек¬
трода сравнения; Ь — этап сбора данных (СВ — коммутационный блок для нескольких электродов)
от измерительного электрода (рис. 15.4, а); это позволяет выявить нарушения функ¬
ционирования электрода, вызванные недостаточной влажностью почвы (Kolsi и Susini,
1984).
В случае достаточной влажности (ее значение зависит от почвы, но обычно составля¬
ет 10% или более) измерения с использованием электродов сравнения 1-2-3 (рис. 15.4)
должны давать близкие результаты. Если результаты 1 и 2 различаются, это может быть
связано с недостаточной влажностью. Если отличается результат 3, это тревожный сиг¬
нал, и следует проверить влажность пробы. Если различия между электродами сравне-
477
Глава 15. Определение pH среды
ния велики, эти результаты отбрасывают. Обычно измерения проводят, используя элек¬
трод сравнения 1 после проверки электродов других позиций.
Все измерительное оборудование, включая коммутационный блок с четырьмя входа¬
ми, подключенный к pH-метру с автономным источником питания, должно упаковы¬
ваться в контейнер для легкой транспортировки в поле.
Коммутационный блок регулируют с помощью буферного раствора для каждого
электрода, чтобы привести все потенциалы сдвига к одной и той же величине (отмечают
величины на потенциометрах).
Измерения
Открывают защитные крышки датчиков и помещают насыщенный каломельный
электрод в трубку ввода 1. Подключают индикаторный электрод и электрод сравнения
к коммутационному блоку. Считывают значение pH, помещают переносной электрод
сравнения в трубку ввода 2 и считывают новое значение. Повторяют эти операции
с трубкой ввода 3 и другими, если имеется несколько измерительных электродов, ис¬
пользуя коммутационный блок. Для введения поправки на температуру термометр дол¬
жен быть подсоединен к pH-метру (рис. 15.4, Ь).
15.3.3. Измерения на почвенных монолитах
Поскольку измерения на почвенных монолитах похожи на полевые измерения, они по¬
зволяют получить большое количество ценной информации. Даже если результаты из¬
мерений не идентичны результатам полевых измерений, их легко регистрировать и по¬
лучать в контролируемых климатических условиях.
Монолит, использованный Сусини и Лойером (Susini и Loyer, 1967), представлял со¬
бой почвенную колонку, заключенную в ПВХ параллелепипед квадратного поперечно¬
го сечения размером 30 см, длиной 1 м, три стороны которого были непрозрачными,
а четвертая - прозрачной. Почвенный монолит был вырезан в поле по размерам камеры
минус несколько сантиметров в нижней части для дренирующей гравийной подушки.
Весь объект был изолирован при помощи цемента Рабсона. В прозрачном окне были
проделаны 3x3 отверстий на трех различных уровнях для индикаторного pH электрода,
электрода сравнения и, при необходимости, платинового электрода для измерений Eh
(см. гл. 16). Три отверстия 40 мм в диаметре были проделаны с левой стороны монолита
для отбора проб и закрыты резиновыми пробками. С другой стороны было проделано
по отверстию на каждом уровне для введения влагомеров Буйукоса (Bouyoucos). Только
один электрод сравнения использовался для всех трех уровней с помощью гибких труб-
кок для транспорта раствора хлорида калия.
Использованная литература
Bates R (1973) Determination du pH, Wiley, New York.
Breltenbeck CA and Bremner JM (1984) Use of a flat-surface combination pH electrode for measurement
of soil pH. Commun. Soil Set Plant Anal., 15, 87-98.
Conkling BL and Blanchar RW (1988) A comparison of pH measurements using the antimony microelec¬
trode and glass electrode. Agron. J., 80, 275-278.
Conyers MK and Davey BG (1988) Observation on some routine methods for soil pH determination. Soil
ScL, 145,29-36.
Grewling, T and Peech (1960) Chemical soil tests. Cornell Univ, Agric. Exp. Sta. Bull., 960 p.
INRA (1995) R6f6rentiel p6dologique. Association Fran9aise d’6tude des sols, INRA, 332 p.
Kolsi and Susini J (1984) Publications ES 209 - Direction des Sols, ORSTOM, Tufiis.
478
Часть 3. Неорганический анализ
Le Brusq JY, Zante P and Peraudeau M (1987) La mesure in situ des paramiitres physico-chimiques (pH
et Eh) dans un sol sulfate acide de Casamance (Senegal). Cah. Orstom Sen Pedol, XXIII, 55-66.
NF ISO-10390 (1994) Determination du pH. In Qualite des sols, AFNOR, 1996.
NF X31-103 (1988) Determination du pH dans l’eau — Methode eiectrometrique. In Qualite des sols,
AFNOR, 1994.
NF X31-104 (1988) Determination du pH dans une solution de KC1 - Methode eiectrometrique. In
Qualite des sols, AFNOR, 1994.
Nilsson T, Kranz-Eliasson В and Bjurman M (1995) Measurement of pH in soil samples from a cutover
peatland in Sweden: the effect of electrolyte and solution/soil ratio. Commun. Soil Sci. Plant Anal,
26, 371-374.
Pansu M, Gautheyrou J and Loyer JY (2001) Soil Analysis-Sampling, Inorganic Analysis. Instrumentation
and Quality Control, Balkema, Lisse, Abington, Exton Tokyo, 489 p.
Qiu Xing-Chu and Zhu Ying-Quan (1986) Spectrophotometric determinations of pH value, Buffer capac¬
ity and rate of lime need in acidic soil, using chrysoidine as a chromogenic agent. Soil Sci., 142,
275-278.
Richards LA (1954) Saline and alkali soil - United States Agriculture - Handbook no. 60: 84.
Susini J and Loyer JY (1967) Utilisation d'un ensemble automatique pour la, mesure en continu du pH.,
DRES-ORSTOM-Tunis.
Дополнительная литература
Beaumann EW (1973) Determination du pH dans les solutions salines concentrees. Anal Chim. Acta.,
64, 284-288.
Billmann E (1927) L’eiectrode & quinhydrone et ses applications. Bull Soc. Chem. Fr, 213,41-42.
Bower (1961) Studies on the suspension effect with a sodium electrode. Soil Sci. Amer. Proc., 25, 18-21.
Cheng KI (1989) pH glass electrode and its mechanisms. In Am. Chem. Soc. Symp. Ser., Stock JT and
OmaMV ed., 390, 286-302.
Clark JS (1964) An examination of the pH of calcareous soils. Soil Sci., 9, 145-151.
Clark J.S (1996) The pH values of soils suspended in dilute salt solutions. Soil. Sci. Soc. Am. Proc., 30,
11-14.
Colin C, Collings K, Drummond P, and Lund E. (2004) A Mobile Sensor Platform for Measurement
of Soil pH and Buffering., American Society of Agricultural and Biological Engineers, St. Joseph,
Michigan, USA.
Peech M, Olsen RA and Bolt GH (1953) The significance of potentiometric measurements involving
liquid junction in clay and soil suspensions. Soil Sci. Soc. Proc., 214-218.
Perley, GA (1939) Sur l’eiectrode antimoine. Ind. Eng. Chem. Anal, 11,316-319.
Peverill KI, Sparrow LA and Reuter DJ (2001) Soil Analysis -An interpretation manual, CSIRO Publish¬
ing, Australia.
Raupach (1954) The essor involved in pH determinations in soils. J. Agric. Res., 5, 716-729.
Schofield RK and Taylor AN (1955) The measurement of soil pH. Soil Sci. Soc. Am. Proc., 19,164-167.
Sistverson DL and Wurfelt BE (1984) Methods for reliable pH measurements of precipitation samples.
Inter J. Environ. Anal. Chem., 18, 143-160.
Soil and Plant Analysis Council Inc (1999) Soil Analysis - Handbook of reference methods, CRC, Boca
Raton.
Stevens G, Dunn D and Phipps В (2001) How to diagnose soil acidity and alkalinity problems in crops: A
comparison of soil pH test kits. J. Extension, 39, 4.
Thomas GW (1996) Soil pH and soil acidity. In Methods of Soil Analysis, Part 3, Chemical Methods,
Bigham JM and Bartels JM ed., ASA-SSSA, Madison, Etats-Unis, 475-490.
Thunjai T, Boyd CE, Dube К (2001) Pond soil pH measurement. J. WorldAquacult. Soc., 32,141-152.
Yang SX, Cheng KL, Kurtz LT and Peck TR (1989) Suspension effects in potentiometry. Part. Sci.
Technol, 7, 131-152.
Глава 15. Определение pH среды 479
Приложения
Приложение 1. Таблицы электродных потенциалов
Таблица 15.4. Потенциалы трех типов каломельного электрода от 15 до 30 °С относительно
стандартного водородного электрода, мВ
Электродный раствор
Температура, °С
0,1 М КС!
1М KCI
насыщенный раствор
KCI
15
338,1
285,2
252,5
16
337,9
284,9
251,7
17
337,8
284,5
250,9
18
337,7
284,2
250,3
19
337,8
283,8
249,5
20
337,5
283,5
248,8
21
337,4
283,2
248,2
22
337,3
282,9
247,5
23
337,2
282,6
246,8
24
337,0
282,2
246,3
25
, 336,9
281,9
245,8
26
336,8
281,6
245,3
27
336,7
281,2
244,8
28
336,6
280,9
244,3
29
336,5
280,5
243,8
30
336,4
280,2
243,4
Таблица 15.5. Потенциал хлоридсеребряного электрода1 от 0 до 95 °С относительно
стандартного водородного электрода, мВ
t,° С мВ
t, °С
мВ
0
236,5
40
212,0
5
234,1
45
208,3
10
231,4
50
204,4
15
228,5
55
200,5
20
225,5
60
196,4
25
222,3
70
187,8
30
219,0
80
178,7
35
215,6
90
168,5
95
165,1
1 Приведены данные для насыщенного хлоридсеребряного электрода. — Примен. науч. ред.
480 Часть 3. Неорганический анализ
Приложение 2. Константы диссоциации для некоторых равновесий
Соединение
Формула
К в воде при 18 °С рК при 18 °С
Уксусная кислота
Аммония ион
Борная кислота
Ортофосфорная кислота 1
Ортофосфорная кислота 2
Ортофосфорная кислота 3
Лимонная кислота 1
Лимонная кислота 2
Лимонная кислота 3
Кислый сульфат-ион
СН2С(0Н)-(С02Н),
СН2С(0Н)-(С02Н)2С02
СН2С(0Н)-СО2Н(СО2)22-
HSO;
CHjCOOH
1.8 • 10-5 4,75
3,2 • 10-10 9,5
5,25 • 10-10 9,26
7,6 • 10-3 2,12
7.5 • 10-* 7,12
3.5 • 10-13 12,45
8,4 • 10-4 3,08
1,77 • 10-5 4,75
3.9 • Ю-4 5,41
1,27 • 10-3 2,89
Приложение 3. Буферные растворы
Буферный раствор — это химическая система, которая стремится поддерживать посто¬
янную концентрацию ионов Н+ и, следовательно, pH, несмотря на разбавление или до¬
бавление ограниченных количеств некоторых кислот или оснований. Обычно буферные
растворы являются смесями кислоты и одной из ее ионизированных солей. Показатель
буферной емкостсти р определяют из уравнения р = Дй/ДрН или Да/ДрН, которое опи¬
сывает изменение pH, вызванное добавлением кислоты (Да) или основания (АЬ). Значе¬
ние pH буферного раствора (табл. 15.6) находят из уравнения:
Т1 = 0,1 М раствор НС1;
Т2 = 0,05 М раствор тетраоксалата калия КН3С408 • 2Н20;
ТЗ = насыщенный раствор гидротартрата калия КН5С406;
Т4ГФ = 0,05 М раствор гидрофгалата калия КН5С804;
Т4УК = 0,1 М CHjCOOH + 0,1 М CHjCOONa (особенно рекомендуется из-за стабиль¬
ности pH при изменении температуры);
Тб = 0,025 М КН2Р04 + 0,025 М Na2HP04;
Т7 = 0,087 М КН2Р04 + 0,0304 М Na2HP04;
Т8 = 0,05 М трис(гидроксиметил)аминометан + 0,0292 М НС1 (особенно рекомендуется
в биологии и ионометрии);
Т9а = 0,01 М раствор Na2B407- ЮН20;
Т9Ь = 0,05 М раствор Na2B407- ЮН20;
Т10 = насыщенный раствор Са(ОН)2.
pH буфера = рК+ ШСС0П1/Сакяат).
КИСЛОТЫ3
Таблица 15.6. Значения pH и характеристики некоторых коммерческих буферных растворов: I— ионная сила; ДрН1/2 — изменение pH
при добавлении к буферному раствору равного объема дистиллированной воды; р — буферная емкость АЬ/АpH, АЪ —
увеличение концентрации сильного основания или кислоты в буферном растворе, изменяющее его pH на АрН
г,°с
Т1
Т2
тз
Т4ГФ
Т4УК
Тб
17
Т8
Т9а
Т9Ъ
тю
Т12
0
1,10
1,666
4,003
6,984
7,354
8,464
9,46
10,317
13,423
5
1,10
1,666
3,999
6,951
7,500
8,395
9,39
10,245
13,207
10
1,10
1,670
3,999
4,65
6,923
7,472
8,40
8,332
9,33
10,179
13,003
15
1,10
1,672
3,999
6,900
7,448
8,28
8,276
9,27
10,118
12,810
20
1,10
1,675
3,560
4,002
4,65
6,881
~ 7,429
8,14
8,225
9,22
10,082
12,627
25
1,10
1,679
3,557
4,008
4,65
6,865
7,413
8,00
8,180
9,18
10,012
12,454
30
1,10
1,683
3,552
4,015
6,853
7,400
7,87
8,139
9,14
9,966
12,389
35
1,10
1,688
3,549
4,024
6,844
7,839
7,75
8,102
9,10
9,325
12,133
40
1,10
1,694
3,547
4,035
4,65
6,838
7,380
7,62
8,058
9,07
9,889
11,984
45
1,10
1,700
3,547
4,047
6,834
7,373
7,50
8,038
9,04
9,856
11,841
50
1,10
1,704
3,549
4,060
6,833
7,367
7,8
8,011
9,01
9,828
11,705
55
1,11
1,715
3,554
4,075
6,834
8,985
8,98
11,574
60
1,11
1,723
3,560
4,091
6,836
8,962
8,95
11,449
70
1,11
1,743
3,580
4,126
6,845
8,921
8,92
80
1,11
1,766
3,609
4,164
6,859
8,885
8,89
90
1,12
1,792
3,650
4,205
6,877
8,850
8,87
95
U2
1,806
3,674
4,227
6,886
8,833
8,87
I
0,1
0,1
0,1
0,03
ДрН1/2
+0,28
+0,186
+0,049
0,052
+0,016
+0,08
+0,07
-0,02
+0,01
-0,01
+0,079
-0,26
р
0,12
0,07
0,027
0,016
0,1
0,029
0,016
0,029
0,02
0,05
0,029
0,09
00
Глава 15. Определение pH среды
482
Часть 3. Неорганический анализ
Приложение 4. Цветные индикаторы
Состав растворов
Ли В
Отношение
Цвет
рн
перехода
А: В
(по объему)
кислотная
форма
основная
форма
Примечание
А: метилоранж
(0,1%-ныйвводе)
В: индигокармин
(0,25%-ный вводе)
1:1
фиолетовый
зеленый
4,1
очень
полезен для
титрования при
искусственном
освещении
А: бромкрезол синий
(0,2%-ный в воде)
В: метиловый красный
(0,2% в воде)
3:1
красный
зеленый
5,1
быстрое
изменение цвета
в точке перехода
А: нейтральный
красный
(0,1%-ныйвводе)
В: метиленовый синий
(0,1%-ныйвводе)
1:1
сине¬
фиолетовый
зеленый
7,0
хранят в темном
флаконе
А: фенолфталеин
(0,1%м в 50%-ном
спирте)
В: нафтолфталеин
(0,1%-ный в 50%-ном
спирте)
3:1
светло-
розовый
фиолетовый
8,9
светло-зеленый
при pH 8,6
А: тимоловый синий
(0,1%-ный в 30%-ном
спирте)
В: фенолфталеин
(0,1%-ный в 50%-ном
спирте)
1:3
желтый
фиолетовый
9,0
зеленый при pH
9,0
Глава 16. Окислительно-восстановительный
потенциал
16.1. Определения и основные положения
Анализы, описанные в данной главе, позволяют характеризовать переменные, главным
образом связанные с диффузией воздуха в почве:
• Eh — окислительно-восстановительный потенциал;
• СДК — скорость диффузии кислорода.
Сначала дадим некоторые определения: окисление характеризуется потерей электро¬
нов, восстановление характеризуется приобретением электронов. Таким образом, в ре¬
акции окисления — восстановления окислитель восстанавливается, а восстановитель
окисляется с обменом п электронами (е~):
окислитель + пег восстановитель.
Измерение окислительно-восстановительного потенциала Eh позволяет количе¬
ственно оценить силу и тенденцию развития системы. Его можно определять по разнице
между потенциалом стандартного водородного электрода (или, что легче, каломельного
электрода) и потенциалом платинового электрода, помещенного в среду (рис. 16.1). Он
выражается формулой:
t 0,058
п
lg
[OxY
[Redf
где [Ох] и [Red] — активности окислителя и восстановителя, соответственно; а и
b — количества их эквивалентов в реакции; п — число обмениваемых электронов;
Е0 — стандартный окислительно-восстановительный потенциал, характеризующий пару
окислитель — восстановитель и свойственный этой паре (например, для Fe3+ + е_ Fe2+,
Е0 = 770 мВ).
В почвах Eh изменяется в диапазоне от 900 до —300 мВ. Потенциал может изменяться
под влиянием pH или в присутствии ионов-комплексообразователей. Таким образом,
можно определить кажущийся усредненный потенциал. Полезно контролировать изме-
Рис. 16.1. Измерение потенциала Eh
484
Часть 3. Неорганический анализ
нения Eh в зависимости от pH (Е0 = ДрН)). Следует отметить, что Eh уменьшается при¬
мерно до 100 мВ, когда pH увеличивается на одну единицу.
Другие факторы, такие как влажность и содержание органического и неорганическо¬
го вещества, также влияют на величину Eh, поэтому оценить состояние почвы можно
только качественно. Часто более информативно рассмотрение изменений Eh, которые
указывают направление развивающихся процессов. В табл. 16.1 обобщены основные
биохимические процессы для различных переувлажненных почв в умеренных клима¬
тических зонах.
Таблица 16.1. Некоторые из основных биогеохимических процессов при различных
величинах Eh
Стадия
Процесс
Eh, мВ
Микробный
метаболизм
Растворимое
органическое
вещество
Стадия I
Исчезновение 02
Исчезновение NOf
От 600 до 300
От 500 до 300
Аэробиоз
Биологическое
разрушение
Стадия II
Восстановление Мп4+
Восстановление Fe3+
От 400 до 200
От 300 до 100
Факультативный Промежуточное
анаэробиоз накопление
Стадия III
Восстановление SOJ"
Образование Н2 и СН4
От 0 до—150
От-150 до-220
Полный
анаэробиоз
Активное
накопление
Биологическое
разрушение
анаэробиозом
Кроме окислительно-восстановительного потенциала, полезно измерить скорость
диффузии кислорода (СДК). Она представляет потенциальный запас кислорода и опи¬
сывает диффузию в газовой фазе с растворением и переносом в жидкой фазе. Рост
растений зависит от кислорода в этой растворенной форме. Удовлетворительный рост
наблюдается при СДК выше 20; оптимальный рост достигается при СДК около 40. Ве¬
личина СДК может снизиться до 5 в переувлажненной почве и до 0 в восстановительных
грунтовых водах.
Используют также обозначение rH = -lg рН2 (рИ2 - давление молекулярного водоро¬
да). Эта величина, которая используется реже, чем Eh, связывает Eh с pH:
„и _ £A+0,06pH
гН 0,03 •
На шкале гН окислительно-восстановительный потенциал среды нейтрален при
гН = 27,7 при температуре 20 °С и pH 7. Меньшие величины соответствуют восстано¬
вительным растворам. Ббльшие величины (от 27 до 40) соответствуют окислительным
растворам.
16.2. Оборудование и реактивы
16.2.1. Электроды
Платиновый электрод изготовлен из платиновой проволоки диаметром 1 мм, нижние
2 см которой имеют форму штопора. Эта форма позволяет ввинчивать ее в почву и та¬
ким образом обеспечивать хороший контакт (рис. 16.2). Корпус электрода представляет
собой стеклянную или ПВХ трубку диаметром 8 мм и длиной 20 см.
Глава 16. Окислительно-восстановительный потенциал
485
В качестве электрода сравнения должен использоваться насыщенный каломельный
электрод (см. гл. 15).
Рис. 16.2. Платиновый электрод: 1 — платиновая проволока в форме штопора; 2 — смола, напри¬
мер аралдит; 3 — соединение, погруженное в ртуть для обеспечения контакта; 4 — стеклянная или
ПВХ трубка; 5 — электрический провод
16.2.2. Соединительный солевой мостик
Солевой мостик выполнен из непрозрачной ПВХ трубки диаметром 15 мм, длина ко¬
торой зависит от выбранной глубины. Она обеспечивает постоянный контакт с почвой.
Окно (рис. 16.3) размером в несколько миллиметров вблизи дна трубки обеспечивает
электрическое соединение с почвой. Оно заклеено куском фильтровальной бумаги.
Верхний конец мостика выступает из почвы; он немного больше по размеру для под¬
держания электрода сравнения в процессе измерений. Этот блок наполняют нагретой
(до текучести) смесью 350 г/л КС1 + 3% агара и несколькими крупинками фенола для
консервации и затем закрывают до затвердения геля. Описанный солевой мостик похож
на тот, что был предложен Венеманом и Пикерингом (Veneman и Pickering, 1983).
16.2.3. Система измерений
Можно использовать pH-метр (см. гл. 15) со шкалой, проградуированной в мВ, и нуле¬
вой отметкой в центре (дающей возможность для измерения отрицательных величин)
или просто милливольтметр. Однако одна характеристика — определяющая: входное
сопротивление должно быть высоким, чтобы позволять проводить измерения очень
малых токов во избежание поляризации платинового электрода, которая может приве¬
сти к ошибочным результатам. Для этого к входу контроллера может быть подсоединен
адаптер импеданса, установленный на усилителе в режиме измерения потенциала.
486
Часть 3. Неорганический анализ
Рис. 16.3. Соединительный солевой мостик: 1 — пробки, ПВХ держатель электрода; 3 — со¬
единение из смолы (аралдита); 4 — проводящий гель; 5 — ПВХ трубка диаметром 15 мм и пере:
менной длины; 6 — окно, закрытое фильтровальной бумагой
16.2.4. Калибровочные растворы
• Раствор Зобелла (Zobell): смешивают 1,26 г ферроцианида калия, 0,99 г феррици-
анида калия и 7,50 г хлорида калия; растворяют и доводят объем до 1000 мл. При
температуре 25 °С Eh этого раствора равен 429 ± 2,4 мВ относительно стандартного
водородного электрода.
• Раствор Лайта (Light, 1972): смешивают 39,21 г сульфата железа(Н) аммония
(б^О)1, 48,22 г сульфата железа(Ш)2 аммония (12Н20) и 56 мл серной кислоты
(id = 1,84); доводят объем до 1000 мл. При температуре 25 °С Eh этого раствора ра¬
вен 675 мВ относительно стандартного водородного электрода.
Для измерений с использованием электрода сравнения, отличного от стандартного
водородного электрода, используют следующее соотношение:
Eh(Н2) = измеренный Eh + Ег электрода сравнения.
Величины Ег при 25 °С: насыщенный каломельный электрод сравнения 244,4 мВ;
ртутно-сульфатный электрод сравнения 636,0 мВ; хлоридсеребряный электрод сравне¬
ния 198,7 мВ.
16.3. Методика
16.3.1. Предварительная обработка электрода
Перед использованием обрабатывают поверхность платинового электрода, удаляя все
следы предыдущих операций:
• Вымачивают в пергидроле в течение 15 мин.
• Ополаскивают дистиллированной водой.
1 Соль Мора. — Примеч. науч. ред.
2 Железоаммонийные квасцы. — Примеч. науч. ред
Глава 16. Окислительно-восстановительный потенциал
487
• Вымачивают в чистой азотной кислоте (d = 1,37) в течение 15 мин.
• Ополаскивают чистой хлористоводородной кислотой (d — 1,19) в течение 15 мин.
• Промывают проточной водой в течение 2 ч.
16.3.2. Анализ образцов почв
Измерения этого типа используют относительно редко. Полученные данные зависят от
концентрации окисленных или восстановленных веществ, но не указывают на степень
аэрации почвы, которая зависит от влажности и порозности. Однако при соблюдении
некоторых условий измерения можно проводить на насыщенной пасте. Пасту готовят
с использованием того же оборудования, что и для экстракции растворимых солей (см.
подраздел 16.2.2), с соблюдением определенных мер предосторожности: дистиллиро¬
ванная вода должна быть полностью дегазирован^ кипячением в вакууме; установка
(рис. 16.4) должна позволять проводить измерения в вакууме. Результаты записывают,
если разница между двумя показаниями не превышает 2 мВ.
Лю и Ю (Liu и Yu, 1984) предложили усовершенствовать эти измерения, используя
поляризацию платинового электрода с последующим построением кривых деполяриза¬
ции как функций времени. Пересечение двух кривых, полученных после анодной и ка¬
тодной поляризации, дает величину Eh.
Рис. 16.4. Установка для измерения Eh почвенной суспензии или насыщенной пасты
16.3.3. Анализ почвенных монолитов
Измерения, проводимые в контролируемых лабораторных условиях, являются альтер¬
нативой полевым измерениям. Почвенную колонку помещают в непрозрачную ПВХ
трубу диаметром от 30 до 40 см и длиной 1 м (0,12-0,15 м3). Солевой мостик аналогичен
описанному в разделе 16.2.2, с окном для электрического контакта на уровне каждого из
платиновых электродов, расположенных на разных высотах.
При отборе почвы в поле следует тщательно следить за точным сохранением поле¬
вого профиля почвы в монолите. Окна доступа для проведения измерений снабжаются
пробками и позволяют при необходимости проводить отбор малых проб, например, для
контроля влажности почвы.
16.3.4. Исследования в полевых условиях
Измерения этого типа наиболее информативны. После выбора участка начинают бу¬
рение отверстия с диаметром, примерно равным диаметру электрода (8 мм), при помо-
488
Часть 3, Неорганический анализ
щи бура, стараясь как можно меньше нарушить структуру почвы. Вставляют электрод
и ввинчивают платиновую проволоку в почву. Это обеспечивает хороший электриче¬
ский контакт между почвой и платиной.
Уплотняют почву вокруг электрода, чтобы исключить просачивание, поскольку
электрод остается в почве в промежутках между измерениями, и необходимо защи¬
тить электрические контакты. Размещают солевой мостик на расстоянии около 6 см
от электрода (рис. 16.5). Верхнее отверстие должно быть закрыто между измерениями.
Целесообразно поместить второй мостик на расстоянии 20 см от первого для проверки
правильности измерений. Для этого измерения проводят непосредственно после под¬
соединения первого мостика. Полученные два значения должны быть идентичными,
если почвенные условия (в частности, влажность) не менялись. Неточные результаты
часто связаны с плохими электрическими контактами. Поэтому желательно поддержи¬
вать в исправности постоянные контакты, используя коммутатор с многопроводными
соединениями, управляемыми поворотным переключателем.
Рис. 16.5. Полевые измерения
16.3.5. Определение скорости диффузии кислорода
Это определение основано на принципе полярографических измерений. В кислород¬
содержащей среде, когда потенциал поляризуемого электрода достигает —200 мВ, на¬
чинается восстановление кислорода и возникает электрический ток. Восстановление
кислорода возрастает с ростом потенциала. В зоне, где сила тока не зависит от напряже¬
ния, концентрация растворенного кислорода и скорость его диффузии к катоду являют¬
ся единственными лимитирующими факторами. Этот метод был предложен Лемоном
и Эриксоном (Lemon и Erickson, 1955) для измерения диффузии кислорода к корням рас¬
тений. Корень похож на цилиндр, окруженный преимущественно жидкой контактной
зоной, которая изменяется в зависимости от степени насыщения среды. Атмосферный
кислород растворяется и диффундирует из жидкой фазы к корням. Устройство для из¬
мерения Eh и СДК должно быть собрано, как показано на рис. 16.6.
Глава 16. Окислительно-восстановительный потенциал
489
4,5 В
Рис. 16.6. Объединенное устройство для измерения (а) скорости диффузии кислорода (СДК)
и (Ь) Eh (Vilain и Ruelle, 1974)
Солевой мостик (рис. 16.3) вставляют в почву до глубины около 15 см для подсо¬
единения насыщенного каломельного электрода сравнения. Индикаторный электрод
(платиновая проволока диаметром 1 мм и длиной 6,5 мм, см. раздел 16.2.1) проверяют,
используя калибровочные растворы (см. раздел 16.2.4) и затем располагают на той же
глубине (15 см).
Измерение Eh предшествует измерению СДК. Последнее требует поляризации пла¬
тинового электрода с использованием рекомендуемого потенциала —650 мВ относитель¬
но электрода сравнения. Показания равновесного тока записывают через 3 мин после
включения цикла поляризации. Интегрирование уравнения диффузии показывает, что
СДК примерно пропорциональна равновесному току в соответствии с уравнением:
СДК =(0,5/А)Г,
где СДК — скорость диффузии кислорода, 10~8 г/(см2 • мин); А — площадь поверхности
платинового электрода, см2; I— сила тока, мкА.
Если влияние температуры на результаты измерения Eh незначительно, то при из¬
мерении СДК ситуация другая. Вилен и Рюэль-Дрюэль (Vilain и Ruelle Druelle, 1974)
рекомендовали использовать поправочный коэффициент, равный в среднем 2%/°С.
Повторяемость результатов измерений может быть около 10% через 2-3 месяца после
установки устройства в поле.
16.3.6. Колориметрическое определение Eh
Окислительно-восстановительный потенциал можно определять с использованием
цветных индикаторов.
Метод включает наблюдение перехода окрашенной формы индикатора в бесцветную
при характеристическом для используемого реагента значении Eh (табл. 16.2). Приме¬
нение этого метода встречает много трудностей при анализе почв, но все же он может
использоваться для их предварительной оценки.
490 Часть 3. Неорганический анализ
Таблица 16.2. Окрашенные окислительно-восстановительные индикаторы, бесцветные
в восстановительных условиях, при pH 7 и 20 °С (согласно Voiret и Froquet, 1950)
Цветной индикатор
Д мВ
гН
Нейтральный красный
-340
10
Сафранин Т
-290
Индиго-дисульфонат калия
-125
Метиленовый синий
+11
23
Хингидрон
+50
24,4
Чистая вода
+ 145
27,7
Индофенол
+248
31,2
Феррицианид калия
+430
37,5
Использованная литература
Lemon ER and Erickson АЕ (1955) Principe of the platinum micro electrode as a method of character¬
izing soil aeration. Soil Sci., 79, 383-392.
Liu ZG and Yu TR (1984) Depolarisation of platinum electrode in soil its utilisation for the measurement
of redox potential. J. Soil Sci., 35, 469-479.
Veneman PLM and Pickering W (1983) Salt bridge for field redox potential measurements. Commun. Soil
Sci. Plant Anal., 14,669-677.
Vilain M and Ruelle JP (1974) Appreciation d’un 6tat deration par l’utilisation de techniques dlectro-
chimiques, principes et observations. Ann. Agron., 25,1-23.
Voiret EG and Froquet L (1950) Le rH : la pratique de sa mesure. Applications. Chimie et industrie, 64,
439.
Дополнительная литература
Bohn HL (1968) Electromotive force of inert electrodes in soil suspensions. Soil Science Society American
Proceedings, 32, 211-215.
Eshel G and Banin A (2002) Feasibility study of long-term continuous field measurement of soil redox
potential. Communications in Soil Science and Plant Analysis, 33, 695-709.
Fisterer UP and Gribbohm S (1989) Constructing platinum electrodes for redox measuring. Zeitschrift fur
Pflanzenernahrung und Bodenkund, 152, 455-456.
Gao S, Tanji KK, Scardaci SC and Chow AT (2002) Comparison of redox indicators in a paddy soil dur¬
ing rice-growing season. Soil Science Society of America Journal, 66, 805-817.
Grundl TJ and Macalady DL (1989) Electrode measurement of redox potential in anaerobic ferric/ferrous
chloride systems. Journal of Contaminant Hydrology, 5, 97-117.
Holm TR and Curtiss CD (1989) A comparison of oxidation-reduction potentials calculated from the
As(V)/As(III) and Fe(III)/Fe(II) couples with measured platinum-electrode potentials in groundwa¬
ter. Journal of Contaminant Hydrology, 5,67-81.
Hunter JD, Scoggins DK, Hawk RM and Sims RA (1986) Development of a microcomputer controlled
multi-probe instrument for automated time-dependent measurement of redox potential and oxygen
diffusion rate. Analytical Instrumentation, 15, 51-62.
ISO 11271 (2002) A field method for the determination of soil redox potential (Eh). International Orga¬
nization for Standardization.
Komada M (1990) Redox potential measurements in a flooded paddy field using a compact computer
system. In Transactions Nth International Congress of Soil Science, Kyoto, Japan, 44-49.
Kukec A, Berovic M, Celan S and Wondra M (2002) The role of on-line redox potential measurement in
Sauvignon blanc fermentation. Food Technology and Biothechnology, 40,49-55.
Le Brusq JY, Zante P and Pёraudeau M (1987) La mesure in situ de param&res physico-chimiques (pH-
Eh) dans les sols sulfates de Casamance. Cah. ORSTOM Ser. Pedol., XXIII, 55-66.
Глава 16, Окислительно-восстановительный потенциал
491
Mueller SC, Stolzy LH and Fick GW (1985) Constructing and screening platinum microelectrodes for
measuring soil redox potential. Soil Science, 139, 558-560.
Patrick WH, Gambrell RP and Faulkner SP (1996) Redox measurements of soils. In Methods of Soil
Analysis, Part 3 Chemical Methods, Bigham JM and Bartels JM ed. SSSA-ASA, Madison, Wiscon¬
sin, Etats-Unis.
Susini J and Loyer JY (1977) Realisation d’un ensemble automatique pour la mesure en continu et in situ
du pH, du Eh, du pNa du sol,, ORSTOM-DRES, Tunis, 17 p.
van Bochove E, Beauchemin S and Theriault G (2002) Continuo.us multiple measurement of soil redox
potential using platinum microelectrodes. Soil Science Society of America Journal, 66,1813-1820.
Vizier JF (1971) Etude de F6tat d’oxydo-r6duction du sol et ses consequences sur la dynamique du fer
dans les sols hydromorphes. Cah. ORSTOMSer. Pedol., IX, 376-380.
Vizier JF (1989) Etude du fonctionnement des milieux satur£s d’eau. Cah. ORSTOM ser. Pedol., XXV,
431-442.
Глава 17. Карбонаты
17.1. Введение
Карбонаты широко распространены в биосфере земли; они составляют группу минера¬
лов из более чем 130 видов, их основные формы включают:
• Са2+ (СаС03, кальцит и некоторые более редкие полиморфные формы, арагонит,
ватерит, икаит);
• Mg2+ (MgC03, магнезит, лансфордит и замещенные формы CaMg(C03)2, доломит,
гантит);
• Na+ (Na2C03, например, натрон, термонатрит);
• Fe2+ (FeC03, например, сидерит и такие замещенные формы, как анкерит (Fe-Ca-
Mg-Mn)).
Многие группы содержат промежуточные виды с двумя или тремя элементами метал¬
лов и различными замещениями. Карбонаты образовывались в различные геологические
эпохи под воздействием физических, химических и биохимических факторов (Chamayou
и Legros, 1990), в частности во время трансгрессии юрского и мелового периодов, в соот¬
ветствии с реакциями, описывающими упрощенный баланс:
Са2+ + 2НСОг ► СаСО,4- +Н,0 + CO,t
3 /, °С, давление, С02 5 i г
Карбонаты осаждались в морских или озерных средах и могли образовываться из
продуктов осаждения, отличавшихся по растворимости (Garrets и Christ, 1967): около
0,014 г/л для кальцита, 0,106 г/л для магнезита, 71 г/л для натрона и 0,067 г/л для сиде¬
рита (для получения двухвалентного железа сидерита необходимы восстановительные
условия).
Карбонаты могут также происходить из органических веществ, образовавшихся из
живых организмов (биогенные известняки из полипов, водорослей, различных форами-
ниферов, ракушек и т. д.) или продуктов эрозии известняковых пород.
Педогенные и биогеохимические процессы отвечают за другой тип дифференциации:
малорастворимые карбонаты кальция и магния приводят к образованию различных
кальциевых и магниевых почв, различающихся в зависимости от климатических усло¬
вий, рельефа, происхождения материнской породы, pH, типа растительности и уровня
биохимической активности. Магний более растворим и обычно более мобилен.
В пустынных и полупустынных климатических условиях могут наблюдаться затвер¬
девшие иллювиальные горизонты с высоким содержанием кальция (петрокальциевый
горизонт) и возможным осаждением гипса.
Хорошо растворимый карбонат натрия обычно очень быстро вымывается, но в сухом
климате он может накапливаться и приводить к образованию содовых солонцовых почв.
Его геохимическое образование порождается тремя основными реакциями:
СаХ + 2NaCl Na2X + СаС12 (выщелачивание);
Na2X + 2Н20 -► Н2Х + 2NaOH (гидролиз);
2NaOH + С02 -> Na2C03 + Н20 (карбонизация).
Глава 17. Карбонаты
493
Этот карбонат определяется вместе с растворимыми солями (см. гл. 18). Дезагрега¬
ция карбонатной материнской породы ведет к нарастающему удалению Са2+, но также
вызывает образование сильно дифференцированных частиц, которые изменяются при
образовании карбонатов в почвах: пески, илы, конкреции, оолиты, корки, пленочные
покрытия или межчастичные скрепляющие вещества.
Мел и известковый мергель, которые содержат большое количество пор размером по¬
рядка микрона, обогащают мелкие пески и илы С02, растворенным в контактирующей
с ними воде. Менее порозные и более плотные известняки обычно образуют более круп¬
ные частицы гравия и песка.
Дезагрегация растениями и микроорганизмами также может приводить к одновре¬
менным механическим и биохимическим процессам под совместным воздействием кор¬
ней и органических кислот. Концентрирование и осаждение кальцита может наблюдать¬
ся в близости от корней и даже внутри корней в не слишком влажных климатических
условиях (Jaillard, 1984). Если карбонат кальция присутствует в большом количестве,
особенно в виде мелкодисперсных частиц, его большая активная поверхность приводит
к чрезвычайно высокой реакционной способности. Карбонат кальция может объеди¬
няться с органической фазой и влиять на процессы разложения. Почвы, образующиеся
из мела или известкового мергеля, могут вызвать насыщение почвенного раствора ио¬
нами Са2+ согласно реакции:
давление С02
СаС03 + С02 + Н20 < рН Са(НС03)2 растворимый1
В этом случае для агрономов содержание общих карбонатов не слишком важно. Две
почвы с одинаковыми концентрациями общих карбонатов могут иметь различную спо¬
собность вызывать хлороз у растений.
Для определения пороговых концентраций, вызывающих кальциевый и железный
хлорозы, были проведены исследования, основанные на анализе активного карбоната
кальция.
Анализ «общих карбонатов» позволяет определять градиенты профильного распре¬
деления карбонатов, вклад материнской породы (унаследованных карбонатов), удобре¬
ний, коллювиального осаждения и т. д. Но этот анализ не может различить педогенный
Са и унаследованный Са. Содержание общих карбонатов может колебаться от 2 до 50%
и даже больше.
Определение «активных карбонатов», важность которых подчеркивали Калло и Дю¬
пюи (Callot и Dupuis, 1980), связано с содержанием известковых илов и долей экстраги¬
руемого железа (,Juste и Pouget, 1972). Наличие корреляции между активными и общими
карбонатами может быть обнаружено при использовании фракции менее 20 мкм.
17.2. Определение содержания общих карбонатов
17.2.1. Введение
Значения pH в диапазоне от 7,5 до 8,5, получаемые в полевых условиях, указывают на
присутствие карбонатов. Более высокие значения pH (до 10) могут указывать на при¬
сутствие карбоната натрия.
Экспресс-тест с 10%-ным раствором хлористоводородной кислоты приводит к бур¬
ному вспениванию пробы. Интенсивность вспенивания указывает на наличие карбо-
1 Более подробное описание уравнений см.: Garrel и Christ, 1967; Chamayou и Legros, 1990.
494
Часть 3. Неорганический анализ
ната, но не объясняет его происхождение (Са, Mg, Na, Fe и т. д.). Карбонаты Са2+ и Na+
мгновенно реагируют с хлористоводородной кислотой, тогда как карбонаты Mg2+ и Fe2+
выделяют С02 медленно. Распределение карбонатов может значительно варьировать
в зависимости от природы почвенных частиц, и одним из способов увидеть различия
является обнаружение вспенивания вокруг изолированных мелких частиц, таких как
конкреции или оолиты.
Важно определить природу минеральных компонентов для того, чтобы выбрать луч¬
шую методику: РД (гл. 4), СЭМ + ЭД-РСМА, или ИК-Фурье-микроскопию (гл. 8), или
термический анализ (гл. 7).
Методы для определения общих карбонатов обычно основаны на выделении двуоки¬
си углерода под действием кислоты. Варианты методов определения:
1) измерение объема С02, образованного в тщательно контролируемых условиях
анализа (в частности, температуры и давления);
2) исследование выделенного С02 методом инфракрасной абсорбционной спек¬
трометрии;
3) исследование выделенного С02 методом газовой хроматографии после отделе¬
ния от других газов;
4) термогравиметрический анализ совместно с анализом выделяющегося газа
(АВГ, гл. 7);
5) разложение карбонатов раствором кислоты с последующим обратным титрова¬
нием избытка кислоты в растворе.
Ни один из этих методов не является совершенным. В методе 1 можно использо¬
вать представительные пробы, поскольку этот метод допускает изменение массы про¬
бы и тонкий размол пробы до частиц одинакового размера, что позволяет регулировать
время реакции. Это простой и экспрессный метод, который дает удовлетворительную
воспроизводимость, если значения давления и температуры отвечают соответствующим
требованиям. Его используют для почв с содержанием карбонатов более 2-5%. Методы
2, 3 и 4 могут использоваться только для микропроб, и их результаты не могут считаться
представительными; кроме того, они требуют применения относительно сложного обо¬
рудования, которое доступно не для всех лабораторий. Из-за неоднородности почв эти
методы используют для тщательных минералогических исследований, например, для
дифференцирования кальцита и магнезита или сидерита. Метод 5 прост в исполнении,
но часто недостаточно точен при малых содержаниях карбонатов.
17.2.2. Волюметрические измерения методом кальциметрии
Основные положения
Карбонаты разлагаются хлористоводородной кислотой, и объем выделенного диоксида
углерода измеряется при контролируемых температуре и давлении:
СаС03 + 2НС1 -> СаС12 + Н20 + С02 (быстрая реакция)
или MgC03 + 2НС1 -> MgCl2 + 2Н20 + С02 (медленная реакция).
Этот метод является частью международного стандарта (NF ISO 10693 1995). Балац
с сотр. (Balazs Horvatha и др. 2005) предложили усовершенствовать этот метод, используя
кальциметр. Изменение давления, вызванное реакцией между НС1 и пробой почвы, из¬
меряют цифровым врезным манометром, через силиконовую прокладку в завинчиваю¬
щейся крышке пробирки.
495
Глава 17 Карбонаты
Оборудование
• Аналитические весы (±0,1 мг).
• Кальциметр Бернарда с двухходовым запорным краном и конической колбой на
250 мл с отводом (рис. 17.1).
• Большой деревянный захват.
• Барометр.
Реактивы
• Деионизированная кипяченая вода.
• Порошок карбоната кальция (СаС03), высушенный в эксикаторе.
• 18 Мраствор хлористоводородной кислоты (НС1): смешивают один объем деиони¬
зированной кипяченой воды с равным объемом концентрированной хлористово¬
дородной кислоты (d=1,19).
• Заполняющий раствор для кальциметра: насыщенный раствор хлорида натрия, со¬
держащий цветной реагент.
Методика
• Работу ведут в комнате с кондиционированием воздуха при 20 °С с пробой, измель¬
ченной до размера частиц 0,1 мм и высушенной в эксикаторе.
• Взвешивают от 1 до 10 г почвы для получения объема С02 около 60-80 мл.
• Насыпают почву на непроклеенную папиросную бумагу и тщательно сворачивают,
не допуская потерь пробы.
• При помощи пипетки на 10 мл вводят хлористоводородную кислоту в отвод кони¬
ческой колбы, не допуская попадания кислоты на дно колбы (рис. 17.1).
• Добавляют пробу почвы.
496 Часть 3. Неорганический анализ
• Увлажняют почву, добавляя 2 мл деионизированной воды для освобождения почвы
от бумаги.
• Избегая нагрева деталей кальциметра собственными руками, открывают кран а
и закрывают коническую колбу
• При помощи подвижного резервуара с устанавливают уровень жидкости в градуи¬
рованной колонке на 0 и затем закрывают кран а.
• При помощи деревянного захвата встряхивают колбу для смешивания кислоты
с почвой.
• Перемешивают и постепенно опускают резервуар с (рис. 17.1), контролируя пони¬
жение уровня жидкости в градуированной трубке.
• Когда уровень стабилизируется, уравнивают уровень жидкости в резервуаре с уров¬
нем в градуированной колонке и фиксируют объем Умл С02.
• Отмечают температуру и атмосферное давление.
Калибровка аппарата
Взвешивают 0,3 г карбоната кальция, высушенного в эксикаторе, и помещают его в не-
проклеенную папиросную бумагу. Продолжают, как описано выше, и отмечают полу¬
ченный объем. Повторяют операцию с 0,2 г и 0,1 г карбоната кальция.
Расчет
Результат обычно выражают как процентное содержание известняка:
КхМх|2ЫЮ
3 1000 хР
где V— объем выделенного С02 из пробы, мл; М— масса 1 л С02при давлении и темпера¬
туре определения, г (табл. 17.1); Р— масса пробы, г; 2,28 = СаС03/С02 = 100,09/44,01.
Таблица 17.1. Масса 1 л сухого С02, г, при различных значениях температуры и давления
t,° с
миллибары
MMHg
986,66
740
1000,00
750
1013,33
760
1026,8
770
20
1,7423
1,7665
1,7906
1,8147
21
1,7338
1,7530
1,7818
1,8059
22
1,7251
1,7443
1,7730
1,7970
23
1,7164
1,7355
1,7641
1,7880
24
1,7075
1,7265
1,7551
1,7789
Примечания
• Колонку заполняют окрашенным насыщенным раствором хлорида натрия, чтобы
уменьшить растворение диоксида углерода и облегчить считывание показаний.
• Измеряемая температура является температурой окружающей среды; поскольку
реакция НС1 + СаС03 экзотермическая, действительная температура может быть
несколько выше комнатной; поэтому значение объема записывают через минуту,
подождав, пока она стабилизируется.
• Кальциметр можно откалибровать, приготовив соответствующие шкалы между 5
и 50% СаС03.
Глава 17 Карбонаты
497
• Результаты измерений могут быть искажены в присутствии избытка сульфидов;
в конце анализа проверяют наличие запаха сероводорода в выделенном диоксиде
углерода.
• Сидерит не разлагается полностью, что приводит к занижению содержания кар¬
боната.
• В присутствии доломита [CaMg(C03)2] снимают два показания: первое через 1 мин
и второе через 3 мин, поскольку доломит реагирует медленнее, чем известняк.
17.2.3. Ацидиметрия
Основные положения
Этот экспрессный метод основан на нейтрализации пробы раствором кислоты извест¬
ной концентрации. Обратное титрование основанием позволяет определять эквива¬
лентное количество карбоната кальция. Поскольку растворение кальцита не селектив¬
но, магнезит, доломит и частично сидерит также растворяются.
Оборудование
• Стеклянная лабораторная посуда.
• Центрифуга с центрифужными пробирками на 250 мл с завинчивающимися крыш¬
ками.
• Мешалка.
Реагенты
• Кипяченая деионизированная вода.
• Хлористоводородная кислота, d= 1,19.
• 1Мраствор хлористоводородной кислоты: добавляют 42,5 мл концентрированной
хлористоводородной кислоты в примерно 400 мл деионизированной воды при пе¬
ремешивании; охлаждают и доводят объем до 500 мл.
• Стандартный коммерческий раствор (фиксанал) 0,5 М хлористоводородной кисло¬
ты.
• 0,5 М раствор гидроксида натрия. растворяют 20 г гранулированного гидроксида
натрия в 1000 мл деионизированной кипяченой воды в перегонной колбе на 1 л;
охлаждают, защищая от проникновения воздуха; гомогенизируют и немедленно
титруют 0,5 М раствором НС1. Этот раствор не хранится и должен готовиться каж¬
дые два дня и титроваться перед каждым использованием.
• 0,1 %-ный спиртовой раствор индикатора фенолфталеина.
Методика
• Взвешивают 5 г почвы, измельченной до 0,1 мм (или 2,5 г для почв с высоким со¬
держанием известняка).
• Добавляют 100 мл 1 М раствора НС1 и перемешивают в центрифужной пробирке
на 250 мл.
• Оставляют смесь на ночь.
• Закрывают пробирку и перемешивают в течение 2 ч.
• Центрифугируют при 2000 g и отбирают пипеткой 10 мл надосадочной жидкости
в коническую колбу на 100 мл.
498
Часть 3. Неорганический анализ
• Добавляют 25 мл воды и две капли фенолфталеина и титруют 0,5 М раствором
NaOH.
• В тех же самых условиях обрабатывают два холостых и один контрольный образец
карбоната кальция массой 500 мг.
Расчеты
Результат обычно выражают как процентное содержание известняка (Ml/2CuCOj = 50 г):
СаСО„%=50 N-,
J
где а — объем NaOH, использованный для холостых определений, мл; Ъ - объем NaOH,
использованный для титрования пробы почвы; S- масса пробы, высушенной в эксика¬
торе, г9 N — концентрация раствора гидроксида натрия, М; (вводят поправку на влаж¬
ность после высушивания отдельной пробы при 105 °С).
Примечания
• Содержание СаС03 может быть завышено, если НС1 реагирует с некарбонатными
веществами почвы.
• Доломит и магнезит растворяются полностью, а сидерит только частично.
• Определение содержания Са и Mg в растворах позволяет различить СаС03
и MgC03.
17.3. Определение активных карбонатов
Метод определения активных карбонатов, также называемый методом Друино-Ruie
(Drouineau-Galet), стандартизован в международном стандарте NF X 31-106 (1982)
и предложен Леппертом и Суаресом (Loeppert и Suarez, 1996).
17.3.1. Общие положения
Пробу почвы вводят в контакт с оксалатом аммония на время, обычное для взаимодей¬
ствия и перемешивания. Оксалат аммония, не прореагировавший с карбонатами, опре¬
деляют обратным титрованием перманганатометрически:
СаС03 + (NH4)2Cj04 -> + (NH4)2CO,
5(NH4)2C204 + 2KMn04 +8H2S04 -> 2MnS04 + KjS04 + 5(NH4)2S04 + 8H20 +10CO2
Метод не пригоден для почв, содержащих много органического вещества или гипс.
Следует избегать присутствия восстанавливающих веществ.
17.3.2. Порядок выполнения
Оборудование
• Аналитические весы.
• Реверсивная мешалка.
• Набор стеклянной лабораторной посуды.
Глава 17. Карбонаты
499
Реагенты
• Деионизированная кипяченая вода.
• Дистиллированная вода для окисляющих растворов,
• Оксалат аммония (NHJ2C20A, М = 142,11 г.
• 0,2 М экстрагирующий раствор (NHJ2 С2 0А:
а) взвешивают 14,2110 г оксалата аммония (высушенного в эксикаторе в течение
48 ч);
б) растворяют в деионизированной воде при 80 °С и после охлаждения доводят
объем до 1 л;
в) титруют 0,1 н. раствором КМп04.
• Серная кислота (d = 1,84).
• Разбавленный раствор серной кислоты: постепенно влить 50 мл H2S04 в 200 мл деио¬
низованной воды, дать остыть.
• Перманганат калия, КМпОА, М = 158,03.
• 0,1 н, стандартный раствор КМпОА: используют коммерческий стандартный рас¬
твор (фиксанал) или взвешивают 3,1606 г перманганата калия; растворяют в 800 мл
дистиллированной воды в мерной колбе на 1 л; перемешивают до полного раство¬
рения перманганата калия; доводят объем до 1000 мл (проверяют титр, используя
0,1 н. стандартный раствор Н2С204).
Методика
Измельчают образец воздушно-сухой почвы с частицами размером 2 мм до 1 мм. Изме¬
ряют остаточную влажность почвы, измельченной до 1 мм, для пересчета результатов на
почву, высушенную при 105 °С.
• Отвешивают 2,5 г почвы и помещают в коническую колбу на 500 мл.
• Добавляют 250 мл 0,2 н. раствора (NH4)2C204 и перемешивают в ротационной ме¬
шалке в течение 2 ч.
• Фильтруют через фильтр с синей лентой, отбрасывают первые фракции, если они
мутные.
• Отбирают 10 мл прозрачного фильтрата и наливают в коническую колбу на
250 мл.
• Добавляют примерно 75 мл дистиллированной воды.
• Добавляют 25 мл разбавленной серной кислоты.
• Нагревают до 60 °С на плите магнитной мешалки и титруют 0,1 н. раствором пер¬
манганата калия до получения стабильной розовой окраски — (Амл). Аналогичным
образом титруют 10 мл раствора оксалата — (Умл). Разница между результатами
двух титрований соответствует количеству карбоната кальция, прореагировавшему
с оксалатом аммония.
Расчет
Процентное содержание активного известняка в пробе определяют по формуле:
Активный СаСО„ % =50х0,1(Г-ЛГ)^^^=50с(К-Л,
где У и X— объемы раствора перманганата для титрования холостой и анализируемой
пробы, соответственно, мл; с — действительная концентрация этого раствора, Н.
500
Часть 3. Неорганический анализ
Примечания
Органическое вещество может быть частично растворимо и потребить некоторое коли¬
чество перманганата. Следует отметить, что разработано много других методов анализа
для восстановительных условий при pH 4,8 (для экстрагируемых Fe, Al, Si), или в при¬
сутствии комплексообразователей: ЭДТА, ДТПА, ДЦТА и ЭДФА1 (см. гл. 31).
17.3.3. Индекс хлорозного потенциала
В присутствии избыточной концентрации активного карбоната кальция развитие каль-
цефобных растений может быть угнетено, и растения проявят симптомы патологии. Ак¬
тивный карбонат кальция также является антагонистом железа и может вызывать желез¬
ный хлороз. Железо, которое необходимо для хлорофильных растений, иммобилизуется,
что приводит к дефицитарному развитию растений. Торн и Уоллес (Thome и Wallace,
1944) рекомендовали определять «свободное» железо, экстрагируемое 0,5%-ным раство¬
ром щавелевой кислоты, для оценки дефицита железа. Жуст и Пуге (Juste и Pouget, 1972)
определили «индекс хлорозного потенциала» (ИХП), который связывает активный кар¬
бонат кальция и экстрагируемое железо, титруемое в той же среде оксалата аммония (Fe
можно определить атомной абсорбционной спектроскопией на длине волны 243,3 нм
в воздушно-ацетиленовом пламени).
активный карбонат кальция, % ,
ИПХ = 2 *—104.
экстрагируемое железо, ррп
Метод был предложен в качестве стандарта FDX31-146 (1996), но воспроизводимость
межлабораторных испытаний оказалась неудовлетворительной.
Использованная литература
Bal&zs Н, Opara-Nadib О and Beesea F (2005) A simple method for measuring the carbonate content of
soil. Soil Set Soc. Am. J., 69, 1066-1068, DOI: 10.2136/sssaj2004.0010.
Callot G and Dupuis M (1980) Le calcaire actif des sols et sa signification. Sci. du Sol., 1,17-26.
Chamayou H and Legros JP (1990) Les bases physiques, chimiques et mineralogiques de la science du
sol. Presses Univ. France, 593 pages.
FD X31-146 (1996) Determination de l’indice de pouvoir chlorosant (IPC) selon Juste et Pouget. In
Qualite des sols, AFNOR, 117-125.
Garrels MA and Christ GL (1967) Equilibre des mineraux et de leurs solutions aqueuses. Gauthier-Vil-
lard, Paris, France.
ISO 10693 (1995) Soil quality — Determination of carbonate content — Volumetric method. Interna¬
tional Organisation for Standardization.
Jaillard В (1984) Mise en evidence de la n6ogen£se des sables calcaires sous l’influence des racines: inci¬
dence de la granulom6trie du sol. Agronomies 4, 91-100.
Juste C and Pouget R (1972) Appreciation du pouvoir chlorosant des sols par un nouvel indice faisant
intervenir le calcaire actif et le fer facilement extractible. C.R. Acad. Agric. de Fr., 58, 352-357.
Loeppert RH and Suarez DL (1996) Carbonate and gypsum. In Methods of Soil Analysis, Part 3, Chemical
Methods, Bigham JM and Bartels JM ed. SSSA, ASA, Madison, Wisconsin, Etats-Unis, 437-474.
NF ISO 10693 (1995) Determination de la teneur en carbonate — Methode volumetrique. In Qualite des
sols, AFNOR, 177-186.
NF X31-106 (1982) Determination du calcaire actif. In Qualite des sols, AFNOR, 55-58.
Thome DW and Wallace A (1944) Some factors affecting chlorosis on high-line soils. I — Ferrous and
Ferric iron. Soil Sci., 57, 299-312.
1ЭДТА (EDTA) — этилендиаминтетрауксусная кислота; ДТПА (DTPA) — диэтнлентриамин-
пентауксусная кислота; ДЦТА (CDTA) — /ирдяс-1,2-диаминоциклогексан-Ы^,К,К — те-
трауксусная кислота; ЭДФА (EDDHA) — этилендиаминдигидроксифенилуксусная кислота.
Глава 18. Растворимые соли
18.1. Введение
К растворимым солям обычно относят ряд анионов и катионов, присутствующих в по¬
чве либо в твердой кристаллической форме (например, высолы, корки, кластеры микро¬
кристаллов), либо в почвенном растворе в диссоциированной форме (исключая раство¬
римое органическое вещество) (см. гл. 13). Растворимые соли отличаются от катионов
обменного комплекса почвы (см. гл. 19), адсорбированных на поверхности глинистых
минералов (см. гл. 22), с которыми они находятся в равновесии.
Растворимые соли почвы часто сравнивают с комбинацией основных элементов,
включая катионы Na\ К+, Са2+ и Mg2+ и анионы С1_, НС03" СО2- и SO2-. Присутствуя
в почве в достаточных количествах, эти солевые системы входят в группу засоленных
почв, называемых галоморфными, солончаками, салисолями или засоленными почвами
(Halomorphes, Solontchaks, Salisols или Salic soils), в зависимости от используемой системы
классификации (INRA, 1995; FAO, 1998). Эти почвы распространены во всех сухих реги¬
онах мира и вблизи морей (первичное засоление). Орошаемые земли также часто про¬
являют свойства засоления (вторичное или антропогенное засоление). Это может быть
связано с плохим качеством используемой воды, внесением удобрений или неудовлет¬
ворительной агротехникой, в частности, отсутствием дренажа (Bouteyre и Loyer, 1995;
Qadirnjip., 2001).
Эти соли могут происходить из разнообразных морских, петрографических или вул¬
канических источников. В осадочных толщах они находятся, главным образом, в форме
хлоридов и сульфатов, тогда как в кристаллических средах преобладают карбонаты и би¬
карбонаты. Кроме этих основных анионов в некоторых системах присутствуют нитраты,
бораты или даже арсенаты, иногда в относительно больших количествах.
Самыми растворимыми и распространенными солями в почвах являются галит или
хлорид натрия (NaCl), сильвинит или хлорид калия (КС1), сульфаты натрия (тенардит
и мирабилит), сульфаты калия и магния (эпсомит и гексагидрит), сульфаты натрия
и магния (блёдит), многие смешанные сульфаты алюминия (типа квасцов), карбонат
натрия (натрон). Высокая растворимость в воде, превышающая растворимость гипса
(2,6 г/л), является основной характеристикой, позволяющей отличать засоленные по¬
чвы, например, от чисто гипсовых почв (Loyer, 1991). Растворимость обычно возраста¬
ет с температурой; растворимость гипса увеличивается до 2,653 г/л при 40 °С и затем
уменьшается до 2,049 г/л при 100 °С. Растворимость некоторых растворимых солей при
20 °С приведена в табл. 18.1.
Среди факторов, влияющих на растворимость каждой соли, нельзя пренебрегать
присутствием других солей. Например, растворимость гипса значительно увеличивает¬
ся в присутствии хлорида натрия, но уменьшается в присутствии сульфата натрия из-за
влияния общих ионов (Pouget, 1968; Harvie и Weare, 1980; Harvienjsp., 1984).
Лабораторное определение растворимых солей в почве включает три этапа:
• экстракция водой при различных соотношениях почва: раствор (насыщенные экс¬
тракты, водные экстракты 1:1,1:5,1:10);
• измерение общей концентрации солей в экстракте (электропроводность, раство¬
ренного твердого вещества);
• определение различных анионов и катионов, присутствующих в экстракте.
502 Часть 3. Неорганический анализ
Таблица 18.1. Растворимость некоторых солей, присутствующих в почвах (А—
растворимости)
произведение
Соль
Минерал
г/л при 20 °С
lg*
NaCl
Галит
360
+ 1,55
Na2S04
Тенардит
209
-0,86
Na2C03 • Н20
Термонатрит
215
+0,1
NaN03
Чилийская селитра
880
KC1
Сильвинит
350
+0,80
w
Арканит
109
MgCl2
Хлоромагнезит
543
+22
CaCl,
Гидрофилит
427
+11,5
18.2. Экстракция
18.2.1. Соотношение почва: раствор
Выбор соотношения почва : раствор для экстракции растворимых солей зависит от раз¬
личных критериев и в частности от цели и скорости исполнения. Длительный метод экс¬
тракции насыщенной пасты используют для возможно более точного воспроизведения
природного почвенного раствора, из которого растения поглощают питательные эле¬
менты. Более быстродействующие методы с использованием разбавленных экстрактов
дают возможность контролировать вертикальные и объемные изменения засоленности,
а также ее изменения во времени из-за влияния внешних и внутренних факторов. От¬
ношения, связывающие различные экстракты, не являются одними и теми же повсюду
(см. раздел 18.23), но зависят от типа солей в соответствующем географическом районе
(Le Brusq и Loyer, 1982).
18.2.2. Экстракция насыщенных паст
Основные положения
Это международный арбитражный метод, рекомендованный лабораторией засоления
в г. Риверсайд (США) (Richards, 1954). В связи с трудностью получения экстракта, со¬
ответствующего почвенному раствору в диапазоне между полевой влажностью почвы
и точкой завядания в данный момент времени, этот стандартизованный метод включает
доведение пробы до насыщения, т. е. почти до границы текучести (Servant 1975; Baize
1988).
Методика
* В капсуле с крышкой взвешивают от 250 до 500 г почвы, просеянной через сито
2 мм и высушенной на воздухе до известной влажности.
* Готовят пасту добавлением известного количества дистиллированной воды при
перемешивании шпателем до насыщения. Периодически уплотняют смесь, встря¬
хивая контейнер на лабораторном столе.
* За исключением почв с высоким содержанием глины, после насыщения почвенная
паста будет блестеть на свету, слегка перемещаться при наклоне капсулы и легко
скользить вдоль капсулы.
Глава 18. Растворимые соли
503
• Закрывают капсулу крышкой и оставляют стоять на 1 ч или более перед проверкой
насыщения. Если необходимо, добавляют известное количество почвы или воды.
• Взвешивают капсулу и рассчитывают процентное содержание воды при насыще¬
нии (см. параграф «Содержание воды в насыщенной пасте» подраздела 18.2.2)
с учетом количества добавленной воды и исходной влажности почвы (или после
высушивания порции насыщенной пасты при 105 °С).
• Фильтруют через воронку Бюхнера и собирают раствор в колбе Бунзена; если не¬
обходимо определять бор, избегают использовать пирексовое стекло.
• Если фильтрат мутный, фильтруют его еще раз через мембрану или центрифуги¬
руют. Если необходимо определять карбонаты или бикарбонаты, то добавление
капли 0,1%-ного раствора гексаметафосфата натрия (0,1 г гексаметафосфата на¬
трия, разбавленного в 100 мл деионизированной воды) на 25 мл экстракта предот¬
вращает осаждение СаС03. Хранят экстракт в закрытом флаконе для последующих
измерений.
Примечание
Для быстрого определения засоления почвы можно проводить фильтрование через не¬
сколько минут после приготовления пасты. В других случаях предпочтительно подо¬
ждать несколько часов. Для гипсосодержащих почв оставляют пасту стоять на несколь¬
ко часов (4-16 ч) перед экстракцией.
Содержание воды в насыщенной пасте
Если Wn — масса пробы почвы, высушенной при 105 °С, a WB — масса добавленной
воды (включая воду, исходно присутствующую в пробе почвы), то влагосодержание
в процентах при насыщении ВН можно рассчитать из уравнения
ВН = 100 0у Жв. (18.1)
18.2.3. Разбавленные экстракты
Сравнительная значимость
В этом более быстром методе данную массу почвы Wn вводят в контакт с переменной
массой воды WB для получения соотношения WB: Wn = п (вместо ВН/100 как в (18.1)).
Одним из следствий разведения является то, что общая концентрация ионов в более или
менее разбавленных водных экстрактах (Ся) теоретически меньше, чем концентрация
в насыщенном экстракте (Снэ), вследствие только эффекта разбавления:
С = Снэ-ВН/(100я). (18.2)
Если удельная электропроводность УЭП пропорциональна соответствующей концен¬
трации водного раствора, то можно написать:
УЭПЯ= УЭПнэ • ВН /(100 • п). (18.3)
Можно было ожидать, что множитель, необходимый для перевода от одного к другому
измерению, обратно пропорционален разбавлению. Однако значения электропровод¬
ности, определенные в разбавленных экстрактах, часто превышают величины, рассчи¬
танные из уравнения (18.3) (Le Brusq и Loyer, 1982). В действительности использование
некоторых экстрактов, особенно экстракта 1:10, приводит к значительному дополни-
504
Часть 3. Неорганический анализ
тельному растворению солей по сравнению с насыщенным экстрактом. Этот факт ука¬
зывает, что отношение между электропроводностью различных экстрактов не только
прямо пропорционально объему воды, но на практике изменяется в зависимости от раз¬
личных факторов, таких как механический состав почвы, засоленность и ионный состав
растворенных веществ.
Разбавленные экстракты особенно полезны в случае низких концентраций солей
и относительно низкой доли гипса. С практической точки зрения в случае массовых ана¬
лизов больших массивов проб сначала измеряют электропроводность экстрактов 1:2. Те
из них, для которых расчет дает Снэ > 10 дСм/м, можно анализировать снова с исполь¬
зованием (1) насыщенной пасты и (2) сильно разбавленного экстракта для определения
содержания общего гипса. Для проб, для которых Снэ < 10 дСм/м, достаточно исполь¬
зовать экстракт 1:2. В экстрактах 1:2 катионно-анионный баланс изменяется очень мало
или не изменяется совсем. В экстрактах 1:5 и 1:10 следует учитывать гидролиз натрия
и обмен с кальцием экстрагирующего раствора, поскольку эти процессы могут привести
к изменениям в балансе между солями.
Методика
• Взвешивают пробу воздушно-сухой почвы, просеянной через сито 2 мм, и помеща¬
ют ее во флакон, объем которого достаточен для получения экстракта; добавляют
количество воды, необходимое для получения желаемого отношения почва : рас¬
твор (1:1, 1:2, 1:5, 1:10).
• Вводят поправку на исходную влажность почвы, особенно при использовании ма¬
лых отношений почва : раствор (например, для экстракта 1:1 с исходной влажно¬
стью 2% добавляют 98 мл воды к 102 г почвы) или при высоких требованиях к точ¬
ности.
• Механически перемешивают в течение 1 ч (или энергично встряхивают вручную
по 4 мин с 30-минутными интервалами) и фильтруют.
• Добавляют одну каплю 0,1 %-ного раствора гексаметафосфата натрия на 25 мл экс¬
тракта и плотно закрывают контейнер до проведения анализа.
18.2.4. Отбор проб воды из почв
При определении влажности почвы в поле более точно учитывается термодинамическое
равновесие ионов, контактирующих с корнями растений. Можно определить различные
типы почвенной воды (например, промежуточная вода, связанная вода и др., см. гл. 1),
соответствующие различным методам пробоотбора (Pansunjsp., 2001), в частности мето¬
ду пористых трубок, исследованному (Cheverry, 1983) и описанному Роадесом и Остером
(Rhoades и Oster, 1986) для исследования засоленных почв.
18.2.5. Экстракция горячей водой
Горячая вода способна растворить гораздо большее количество вещества, чем холод¬
ная (в 10-50 раз и более из лювисолевых почв, см. гл. 13). Поэтому экстракция горячей
водой может служить индикатором содержания экстрагируемых элементов и соедине¬
ний и рекомендована для определения содержания бора (NF Xil-122 1993). Процеду¬
ры определения довольно сложны для стандартизации, потому что в случае отдельных
почв полное растворение не достигается в течение указанного времени взаимодействия
(Коиакоиа и др., 1997). Селективная экстракция бора (см. подраздел 31.2.7) не требует
жесткого воздействия. Более жесткие условия используются для количественного опре-
Глава 18. Растворимые соли
505
деления водорастворимой органики (кипячение с обратным холодильником в течение
16 ч, см. подраздел 3.2.6). В этих условиях экстрагируется значительно большее количе¬
ство анионов, чем при экстракции холодной водой (см. рис. 18.2).
18.3. Анализ и определение содержания солей
18.3.1. Электропроводность экстрактов
Преимущества метода
В противоположность идеально чистой воде солевые растворы обладают свойством про¬
водить электрический ток. Удельная электропроводность (УЭП) растворов пропорцио¬
нальна количеству растворенных в них солей и зависит от их природы. Таким образом,
существует общая взаимосвязь между удельной электропроводностью (в дСм/м) и об¬
щей концентрацией (Со) анионов или катионов (мг-экв/л или мг/л):
Со (мг-экв/л) = (10-12) х УЭП (18.4)
Со (мг/л) = (600-650) х УЭП (18.4’)
Однако переменное значение коэффициента пропорциональности показывает, что
это отношение не является универсальным. Оно зависит от ионного состава почвенных
растворов {Job, 1985) и поэтому не всегда применимо для любых географических реги¬
онов. Целесообразно установить отношение УЭП =/(Со), применимое к конкретным
условиям засоления, а не использовать среднюю пропорцию, полученную Ричардсом
(Richards, 1954) для почв западных районов США.
Измерения удельной электропроводности чаще всего используются для оценки за¬
соления почв {Rhoades и Miyamoto, 1990; Shirokova и др., 2000). Их обычно выполняют
в насыщенных или разбавленных экстрактах, а иногда непосредственно в насыщенной
пасте. Результаты измерений сильно зависят от температуры (возрастают почти на 2% на
градус) и, следовательно, должны приводиться к базовой температуре 25 °С после опре¬
деления температуры раствора. При одновременном определении pH измерение удель¬
ной электропроводности служит хорошим индикатором качества почвы {Smith и Doran,
1996). Определение удельной электропроводности экстракта 1:5 введено в международ¬
ный стандарт NF ISO 11265 (1995).
Основные положения
Определение УЭП основано на измерении электрического сопротивления между парал¬
лельными электродами, погруженными в водный экстракт, полученный, как описано
в разделе 18.2.
Традиционный кондуктометр основан на принципе мостика Уитстона. Он включает
ячейку, состоящую из двух платиновых электродов определенной площади, расположен¬
ных на определенном расстоянии, обычно покрытых платиновой чернью для снижения
сопротивления на поверхности раздела фаз электрод — раствор. Между электродами
прикладывают переменный ток высокого напряжения для измерения сопротивления.
Удельное сопротивление Ry раствора определяется сопротивлением куба со сторонами,
равными 1 см (поверхность электрода равна 1 см2, расстояние между электродами 1 см).
На практике объем раствора между электродами не точно равен 1 см3 и, следовательно,
сопротивление R (в Ом) не точно равно удельному сопротивлению. Таким образом, по¬
стоянную ячейки можно определить следующим образом:
506
Часть 3. Неорганический анализ
K = R/Ry.
Величина, обратная R, выражает электропроводность (в См), которая для данного
типа электрода дает удельную электропроводность УЭП:
УЭП = 7/Ду = K/R. (18.5)
Выражается она в системе СИ в См/м или их долях в соответствии с выбранной кали¬
бровкой (1 См/м = 10 дСм/м = 10 мСм/см = 1000 мСм/м = 10 000 мкСм/см).
Постоянную ячейки, которая обычно предоставляется производителем, следует про¬
верять с использованием стандартных растворов хлорида калия, удельная электропро¬
водность которого (табл. 18.2) находится в ожидаемом диапазоне измеряемых величин.
Таблица 18.2. Величины УЭП водных растворов КС1 при 25 °С
КС1,М ОДНИ т М2 М5 (U М М I
УЭП,дСм/м 0,147 1,413 2,767 6,668 12,900 24,820 58,640 111,900 * •Большинство кондуктометров имеют логарифмическую шкалу и снабжены пере¬
ключателем, расширяющим диапазон шкалы для измерений с коэффициентом увели¬
чения 10.
Реактивы
• 0,05 М исходный раствор КС1: растворяют 3,728 г сухого КС1 в 1 л деионизирован¬
ной воды при 25 °С.
• 0,01 М раствор КС1: 50 мл исходного раствора в 250 мл деионизированной воды.
• 0,005 М раствор КС1: 25 мл исходного раствора в 250 мл деионизированной воды.
• Очищающий раствор для электрода: смешивают равные объемы изопропанола,
диэтилового эфира и хлористоводородной кислоты.
• Раствор для покрытия платины: растворяют 1 г гексахлороплатиновой кислоты
(H2PtCl6 • 6Н20) и 12 мг ацетата свинца в 100 мл чистой воды.
Приготовление платиновых электродов
В случае нестабильных результатов измерений или результатов, сильно отличающихся
от теоретических значений для стандартных растворов (см. параграф «Контроль по¬
стоянной ячейки» ниже), электроды можно готовить или регенерировать, как описано
ниже.
Очищают электроды хромовой смесью, погружают их в раствор соли платины и под¬
соединяют разъемы двух электродов к отрицательному полюсу источника тока на 1,5 В.
Подсоединяют положительный полюс к платиновой проволоке, помещенной в рас¬
твор, и проводят электролиз при слабом токе (во избежание выделения газа) до тех пор,
пока электроды не покроются платиновой чернью. Осторожно споласкивают электроды
и хранят их в дистиллированной воде.
Контроль постоянной ячейки
Три или четыре раза споласкивают ячейку соответствующим стандартным раствором
КС1 (обычно 0,01 М) перед измерением удельной электропроводности и температуры
(или только электропроводности, если работы проводятся в термостатируемой среде
при 25 °С). Проверяют стабильность результатов измерений, используя новую аликво¬
ту стандартного раствора. Если УЭПТ — теоретическая удельная электропроводность
507
Глава 18. Растворимые соли
стандартного раствора при 25 °С (табл. 18.2); УЭПИ — измеренное значение удельной
электропроводности; 0 — температура ,°С; /—возможный множитель шкалы удельной
электропроводности, то постоянную ячейки А'можно рассчитать по уравнению:
УЭПг
/УЭПИ[1 + 0,019(25 — О)]
(18.6)
Удельная электропроводность пробы
Измерения проводят, как описано в параграфе «Контроль постоянной ячейки» и под¬
разделе 18.3.1, с заменой стандартного раствора раствором пробы. Используя обозначе¬
ния, приведенные в параграфе «Контроль постоянной ячейки» подраздела 18.3.1, удель¬
ную электропроводность при 25 °С (УЭП25 в дС/м) определяют по уравнению:
УЭП25 = КУЭПИ [1 + 0,019(25 - 8)].
(18.7)
18.3.2. Измерение удельной электропроводности в полевых условиях
Прямое измерение удельной электропроводности почвы менее точно, чем методы, опи¬
санные в подразделе 18.3.1, но оно полезно для быстрого картирования распределения
солей в поле или контроля динамики засоления (Simon и Garcia, 1999). Можно исполь¬
зовать различные методы. Например, для измерения удельной электропроводности
почвенного раствора (с дополнительным измерением температуры) в почву вводят по¬
ристые матричные датчики с платиновым электродом. Можно применять более совер¬
шенные инструментальные методы: квадрупольные зонды, электромагнитную кондук¬
тометрию (Boivin и др., 1989; Job и др., 1997), датчики динамической рефлектометрии.
Описание этих методов можно найти, например, в работах Роадеса и Остера (Rhoades
и Oster, 1986) или Корвина и Леша (Corwin и Lesch, 2004).
Другой метод полевых измерений включает измерения удельной электропроводно¬
сти почвенного раствора после отбора проб в поле (см. подраздел 18.2.4).
Существует метод измерения удельной электропроводности насыщенных паст, по¬
лученных непосредственно в поле в соответствующих сосудах (Roades, 1996).
18.3.3. Определение общего содержания растворенных веществ
Основные положения
Общее количество растворенных веществ (ОРВ) в водных экстрактах (сухой остаток экс¬
тракта) определяют взвешиванием остатка после упаривания экстракта, предварительно
отфильтрованного через мембрану 0,45 мкм. Эта величина обычно тесно связана с вели¬
чиной удельной электропроводности (уравнение (18.4)), суммой определенных экстра¬
гируемых катионов и анионов (см. подразделы 18.3.4 и 18.3.5) и, возможно, содержанием
экстрагируемого органического вещества (см. гл. 13). Поэтому при наличии надежных
значений удельной электропроводности необязательно систематически определять со¬
держание ОРВ. Определение ОРВ полезно для точной градуировки согласно уравнению
(18.4*), позволяющей проводить измерения удельной электропроводности на участке.
Оборудование
• Система для фильтрования через мембрану 0,45 мкм (Pansu и др., 2001).
• Чашки Петри из пирексового стекла для упаривания емкостью около 200 мл.
• Вентилируемый сушильный шкаф, работающий при 180 °С.
508
Часть 3. Неорганический анализ
Методика
• Высушивают чашки Петри в сушильном шкафу при 180 °С в течение 2 ч, затем
охлаждают в эксикаторе и взвешивают с точностью до 0,1 мг: Р0.
• Отбирают достаточный объем вводного экстракта (см. раздел 18.2) для получения
50—100 мг остатка, фильтруют через мембрану 0,45 мкм и помещают фильтрат (весь
или часть) в чашки Петри для упаривания.
• Упаривают фильтрат в вентилируемом сушильном шкафу при 105 °С, при необхо¬
димости добавляя остаток фильтрата после уменьшения объема.
• Нагревают сухой остаток при 180 °С в течение 1 ч для удаления воды, оставшейся
в микропорах, охлаждают в эксикаторе и взвешивают: PL
Содержание ОРВ (мг/л) находят по уравнению:
ОРВ = 1000(/>7 - P0)/V (18.8)
18.3.4. Растворимые катионы
Методы
Пламенно-эмиссионная и атомно-абсорбционная спектрометрии являются наиболее
предпочтительными лабораторными методами. Можно также использовать эмисси¬
онную спектрометрию с индуктивно-связанной плазмой. Эти методы обычно дают хо¬
рошие оценки общего содержания элементов в растворе, разрушая некоторые формы,
связанные с органическим веществом, а также некоторые минеральные формы, способ¬
ные осаждаться в водных экстрактах (например, карбонаты). Применение этих методов
описано в главе 31. Единственным параметром, требующим адаптации, является диа¬
пазон калибровки. Влияние матрицы обычно меньше, чем в общих анализах (см. гл. 31),
потому что используют менее концентрированные экстрагирующие растворы.
Ионные формы элементов можно определять, используя электрохимические методы,
в частности ионометрию с селективными электродами. Этот метод рекомендуется и для
мониторинга in situ изменений состава элементов в почвенном растворе, как дополне¬
ния к мониторингу изменений общего засоления электрохимическими, кондуктометри¬
ческими или другими методами (Pansu и др., 2001). Например, натриевый электрод, раз¬
работанный Сусини с сотр. (Sucini и др., 1972) для исследования засоленных почв, был
использован для сбора данных in situ по pH, pNa и Eh (Ьоуеги Susini, 1978).
Ионная хроматография также может использоваться для определения катионов, но
этот метод лучше использовать для определения анионов в связи с удовлетворительным
определением катионов спектрометрическими методами.
Натриевый характер водных экстрактов
Этот широко используемый параметр основан на анализе основных катионов Са, Mg
и Na (в мМ) в водных экстрактах и определении доли адсорбированного натрия относи¬
тельно кальция и магния (ДАН {sodium adsorption ratio, SAR), Richards и др., 1954):
Д^ССа ?Mg)-2- <18'9>
Этот параметр может использоваться для определенных типов водных экстрактов. Он
изменяется в зависимости от количества воды, использованной для экстракции, в част¬
ности из-за возможного растворения кальциевых минералов, таких как кальцит и гипс.
509
Глава 18. Растворимые соли
18.3.5. Экстрагируемые карбонаты и бикарбонаты (щелочность)
Основные положения
Щелочную среду экстрактов определяют карбонат-анионы, хотя другие ионы, такие как
гидроксиды, бораты, фосфаты и силикаты, также могут играть небольшую роль. Бикар¬
бонаты являются ожидаемым компонентом водных экстрактов засоленных почв, тогда
как менее распространенные карбонаты часто присутствуют в континентальных щелоч¬
ных почвах (Cheverry, 1974). Эти анионы не определяются ионной хроматографией с по¬
мощью оборудования, описанного в подразделе 18.3.6, потому что они входят в состав
элюента. Рекомендуемые методы, описанные ниже, основаны на титровании разбавлен¬
ными кислотами методом автоматической титриметрии (Roades, 1982) или с использо¬
ванием двух индикаторов (Bower и Wilcox, 1965). Существуют и другие автоматизирован¬
ные методы (Sa и др., 2002).
Аппаратура и реагенты
• Автоматический титратор со стеклянным электродом для измерения pH и электро¬
дом сравнения (или комбинированным электродом) либо, если это невозможно,
с бюреткой для титрования на 10 мл.
• 0,01 или 0,02 н. раствор серной или хлористоводородной кислоты (с точно опреде¬
ленной концентрацией).
• Стандартные буферное растворы с pH 4 и 7.
• Индикатор фенолфталеин: растворяют 0,25 г индикатора в 100 мл 50%-ного вод¬
ного спирта.
• Индикатор метилоранж: растворяют 0,1 г индикатора в 100 мл воды.
Метод
Автоматическое титрование
• Калибруют титратор буферными растворами с pH 4 и 7; осторожно споласкивают
pH-электроды и помещают их в аликвоту раствора для титрования (1-20 мл). Пе¬
ремешивают магнитной мешалкой. Отмечают исходное значение pH, характерное
для пробы почвы (см. гл. 15).
• Начинают титрование, записывая кривую титрования. Отмечают объем кисло¬
ты И, необходимый для достижения первой точки титрования С032_ при pH 8,3.
Отмечают общий объем кислоты V0, необходимый для достижения второй точки
титрования, которая сопровождается выделением С02 вследствие разложения би¬
карбонатов при pH 4,5.
Использование двух цветных индикаторов
• Отбирают пипеткой аликвоту раствора для титрования в стакан для титрования.
• Добавляют две капли индикатора — фенолфталеина. Если раствор остается бес¬
цветным, он не содержит карбонатов. Если это не так, титруют карбонаты до
обесцвечивания окраски (объем VY).
• Добавляют две капли индикатора — метилового оранжевого. Титруют, бикарбонаты
до изменения окраски. Записывают значение объема: VQ.
510
Часть 3. Неорганический анализ
Расчеты
Если И) - объем аликвоты, мл; Т— концентрация кислоты, н., тогда:
содержание карбонатов, мМ СО2-:
1000
V0
xVlxT;
содержание бикарбонатов, мМ НСО":
1000
V0
х(Г0-П)х7\
Примечания
Некоторые водные экстракты из щелочных почв могут иметь очень темный цвет, что
затрудняет использование метода, описанного в подпараграфе «Использование двух
цветных индикаторов» параграфа «Методика» подраздела 18.3.5. В этом случае следует
использовать титриметрический метод, описаный в подпараграфе «Автоматическое ти¬
трование» параграфа «Методика» подраздела 18.3.5.
Щелочность (ионы С032 * и НСО") желательно определять непосредственно после
экстракции, чтобы исключить возможные превращения (осаждение, обмен с атмосфер¬
ным диоксидом углерода), хотя эти изменения можно замедлить добавлением гексаме¬
тафосфата. Если необходимо выполнить несколько определений, сначала определяют
щелочность экстрактов.
18.3.6. Экстрагируемые хлориды
Цель определения
Хлориды часто являются основными анионами в засоленных почвах, причем хлориды
Mg, Са, Na и К очень хорошо растворимы (табл. 18.1). Хлорид — традиционный ион,
который используют в качестве метки для мониторинга изменений концентрации вод¬
ных растворов. Определять содержание хлоридов важно и в связи с токсичностью этих
анионов для некоторых растений.
Методы
Из-за специфических химических свойств хлорид-анионы довольно легко и точно
определяются многими методами. Различные волюмометрические, титриметрические
и колориметрические методы используют свойство хлорид-анионов количественно
осаждать катионы серебра. Эти методы можно применять непосредственно к исходным
экстрактам или после определения карбонатов и бикарбонатов (см. подраздел 18.3.5)
в той же аликвоте после повышения pH от 4,2 до 8,3, добавляя небольшое количество
бикарбоната натрия.
Волюмометрия: титрование стандартным 0,025 М раствором нитрата серебра с опре¬
делением окончания реакции с помощью I^Cr^ (образование красно-бурого осадка
хромата серебра).
Титриметрия: автоматическое титрование (см. подпараграф «Автоматическое титро¬
вание» параграфа «Метод» подраздела 18.3.5), основанное на том же принципе, что и во¬
люмометрия, с определением окончания реакции при помощи серебряного электрода
(резкое изменение потенциала в конце осаждения ионов Ag+).
Глава 18. Растворимые соли
511
Кулонометрия: образование ионов Ag+ из серебряного электрода в процессе электро¬
лиза, причем осаждение хлорида серебра перед появлением свободных ионов Ag+ ис¬
пользуется для определения окончания реакции методом амперометрии с двумя другими
индикаторными электродами. Концентрацию хлорид-ионов рассчитывают непосред¬
ственно из количества электричества, необходимого для образования ионов Ag+. Этот
кулонометрический метод очень удобен, потому что он не требует реагентов, а оборудо¬
вание легко транспортировать.
Селективные электроды также являются дешевым оборудованием для мониторинга
активности хлорид-ионов в почвенном растворе в полевых условиях, если необходимо
проводить и другие электрохимические измерения (Susini и Nedhir, 1980).
Колориметрический метод с тиоцианатом ртути (см. параграф «Колориметрическое
определение хлоридов» в подразделе 18.3.6) используется очень часто из-за высокой
чувствительности и хорошей точности.
Колориметрическое определение хлоридов
Основные положения
Хлорид-ионы реагируют с тиоцианатом ртути, образуя хлорид ртути и тиоцианат-
ионы. В присутствии железоаммониевых квасцов тиоцианат-ионы образуют красно¬
оранжевый комплекс: тиоцианат трехвалентного железа. Интенсивность окраски мож¬
но измерить на длине волны 480 нм.
Оборудование и реагенты
• Колориметр с кварцевыми или пластиковыми кюветами или установка для автома¬
тической колориметрии с непрерывно-проточным анализом.
• Железоаммониевые квасцы FeNH4(S04)2* 12Н20: 60 г в 1 л 6 М раствора азотной
кислоты HN03.
• Тиоцианат ртути: 3,5 г в 1 л воды.
• 50 мМ исходный раствор хлорид-ионов: отвешивают 3,728 г сухого КС1 и перено¬
сят в 1 л воды сверхвысокой чистоты.
• Калибровочные стандарты: разбавление исходного раствора до 0-5 мг/л;
разбавление до 0-0,5 мг/л для меньших концентраций.
Метод
На рис. 18.1 показано устройство для определения хлорид-ионов методом автоматиче¬
ской колориметрии, модифицированной Пайченгом (Paycheng, 1980). В методе ручной
колориметрии используют такие же пропорции реагентов, как автоматическом методе.
18.3.7. Экстрагируемые сульфаты, нитраты и фосфаты
Области применения
Наряду с карбонатами, бикарбонатами и хлоридами, сульфаты являются основными
анионами в засоленных почвах. Нитраты являются опасными загрязнителями в связи
с их большой растворимостью. Содержание нитратов должно контролироваться, осо¬
бенно в орошаемых и загрязненных зонах (Cheverry, 1998). Содержание сульфатов мож¬
но приблизительно оценить в миллиэквивалентах из разницы между суммой катионов
(Na, К, Са, Mg) и суммой анионов С032*, НСО", С1“, N0". Фосфаты не типичны для за¬
соленных почв, но они часто присутствуют в водных экстрактах культивированных почв.
512
Часть 3. Неорганический анализ
Потоки, мл/мин
0,8
X = 480 нм
Проба
Вода
Воздух
Тиоцианат
Желеэоаммониевые квасцы
Перекачивание
Промывание
Рис. 18.1. Определение хлорид-ионов методом автоматической колориметрии с непрерывно¬
проточным анализом в сегментированном потоке. Для малых содержаний (0-0,5 мг/л) меняют
местами трубки для пробы и воды
Определение
Методы, описанные далее в гл. 28, можно использовать для определения нитратов. Са¬
мым рекомендуемым методом для обследования засоленных почв является использо¬
вание нитрат-селективного электрода в диапазоне 0-0,01-0,1-1-10-100 мМ N0" при
определении в водных экстрактах или в почвенных растворах in situ.
Определение сульфата описано в гл. 30. Обычно используют два метода для вод¬
ных экстрактов из засоленных почв. В концентрированных пробах можно опреде¬
лить мутность, вызванную осаждением сульфата бария. В более разбавленных пробах
(<5 мн. SO2-) сульфат можно определить автоматическим титрованием 0,002 мМ
раствором перхлората свинца с потенциометрической регистрацией при помощи
РЬ2+-селективного электрода.
Определение фосфатов описано в гл. 29.
18.3.8. Экстрагируемый бор
Цели
Хотя бор присутствует в гораздо меньших количествах, чем основные элементы водных
экстрактов (см. подразделы 18.3.4—18.3.6), в засоленных почвах он стремится накапли¬
ваться вместе с другими солями. Определение этого элемента иногда необходимо, осо¬
бенно из-за его влияния на рост растений. Его порог дефицита очень близок к порогу
токсичности.
Методы
Бор можно определять в водных экстрактах эмиссионной спектрометрией с индуктивно¬
связанной плазмой (см. гл. 31) на длине волны 249,773 нм с пределом обнаружения
около 10 мкг/л. Железо может мешать определению на длине волны 249,782 нм, и «го
концентрация в экстрактах должна быть не слишком большой, т. е. не превышать кон¬
центрацию бора более, чем в 1000 раз (Рати и др., 2001).
Бор в экстрактах можно также определять, используя электрод, селективный для
ионов BF". Этот метод требует довольно длительного превращения в тетрафтороборат-
ионы {Keren, 1996), и электроды нельзя использовать в полевых условиях.
Глава 18. Растворимые соли
513
Также предлагался набор колориметрических методов для определения бора с ис¬
пользованием кармина, куркумина и азометина-Н. Последний реагент был выбран Роа-
десом (Roades, 1982) для засоленных почв и для стандарта NFXi 1-122 (1993).
Колориметрия с азометином-Н
Реагенты
• Буферный раствор для маскировки влияющих веществ; 400 мл деионизированной
воды, 250 г ацетата аммония, 125 мл ледяной уксусной кислоты, 15 г этилендиа-
минтетрауксусной кислоты (ЭДТУ).
• Цветной реагент азометин-Н: 0,5 г азометина-Н и 1 г L-аскорбиновой кислоты
в 100 мл деионизированной воды (готовят перед каждым определением).
• Исходный стандартный раствор 100 мг/л (В): 0,572 г борной кислоты в 1000 мл
воды.
• Стандартные растворы для калибровки в диапазоне от 0 до 4 мг/л (В): готовят раз¬
бавлением исходного стандартного раствора.
Методика
• В пластиковую пробирку добавляют:
- 1 мл водного экстракта, стандартного раствора или воды (холостой опыт);
- 2 мл буферного раствора (для маскировки); тщательно перемешивают;
- 2 мл азометино^ого реагента.
• Тщательно перемешивают, оставляют стоять в течение 1 ч в холодильнике и изме¬
ряют поглощение на длине волны 420 нм.
Примечание
Из-за опасности загрязнения стеклянная посуда не должна использоваться для опреде¬
ления бора. Вместо нее рекомендуется использовать полипропиленовую посуду.
18.3.9. Определение экстрагируемых анионов методом ионообменной
хроматографии
Основные положения
Наиболее эффективным методом анализа водных растворов является ионная хромато¬
графия. В зависимости от колонки и условий определения ее можно использовать для
одновременного определения катионов или анионов. Метод можно использовать для
определения катионов в отсутствие атомного спектрометра. В водных экстрактах одно¬
временный анализ анионов занимает меньше времени, чем последовательное опреде¬
ление каждого аниона (см. подразделы 18.3.5-18.3.7). Однако самые распространенные
методы не позволяют определять карбонаты и бикарбонаты одновременно с другими
анионами, потому что карбонаты и бикарбонаты входят в состав элюента. Метод можно
адаптировать, но мы рекомендуем использовать ацидиметрические методы для опреде¬
ления щелочности, описанные в подразделе 18.3.5. Другая проблема может быть связана
с маскировкой определенных пиков из-за слишком большой концентрации одного из
элементов, например, хлоридов в некоторых засоленных почвах. Одним из возможных
решений является использование рабочих условий, описанных ниже, при которых пик
хлорида четко отделен от остальных (рис. 18.2).
514
Часть 3. Неорганический анализ
Основное оборудование
• Ионный хроматограф с постоянным потоком, без программирования градиента,
рабочее давление 150 бар, снабженный:
а) петлевым дозатором на 10, 25 или 50 мкл;
б) предколонкой OmniPac PAX-100 (Dionex, Саннивейл, США) или аналогичной;
в) аналитической колонкой OmniPac PAX-100 {Dionex) или аналогичной;
г) анионным мембранным подавителем;
д) кондуктометрическим детектором.
• Система фильтрации (0,2 мкм), подходящая для больших объемов (элюента)
и шприц на 1 мл, снабженный встроенным мембранным фильтром (0,2 мкм) {Рати
идр., 2001).
• Микропузырьковый ввод гелия сверхвысокой чистоты.
Реагенты
• Вода сверхвысокой чистоты (удельное сопротивление >18 МОм), профильтрован¬
ная (0,2 мкм) и дегазированная в атмосфере гелия.
• Элюент: 3,9 мМ NaHC03, 3,1 мМ Na2C03, 5% СН3ОН: в мерной колбе на 5 л сме¬
шивают 1,638 г чистого сухого NaHC03 и 1,643 г чистого сухого Na2C03, добавляют
250 мл чистого метанола, растворяют и доводят объем раствора до 5 л водой сверх¬
высокой чистоты.
• Регенерирующий 12,5 мМ раствор серной кислоты: в мерной колбе на 5 л смеши¬
вают 1 л воды сверхвысокой чистоты и 3,472 мл 18 М серной кислоты сверхвысо¬
кой чистоты, перемешивают и доводят объем до 5 л водой сверхвысокой чистоты.
• Стандарты для мультианионной калибровки:
С1-
0,1-15 мг/л
NO,
0,02-1 мг/л
so*-
0,05-10 мг/л
ро t
0,1—5 мг/л
F-
0,1-5 мг/л.
Методика
• Соединяют или проверяют соединения компонентов хроматографической систе¬
мы: петлевого дозатора, предколонки, колонки, подавителя, кондуктометрическо¬
го детектора.
• Проверяют подсоединение регенерирующей линии подавителя.
• Дегазируют элюент барботированием гелия.
• Запускают насос высокого давления, устанавливают скорость потока элюента
1 мл/мин (входное давление 100-130 бар).
• Устанавливают скорость регенерирующего раствора 7 мл/мин под давлением азота.
• Запускают детектор и стабилизируют систему. Базовая линия хроматограммы
должна сохранять стабильность при изменении электропроводности фонового
шума примерно до 20 мСм.
• Вводят стандартную смесь при помощи шприца на 1 мл со встроенным микро¬
фильтром. Ожидают примерно 10 мин до полного выхода хроматограммы, прежде
чем ввести следующую порцию раствора для калибровки или измерения.
515
Глава 18, Растворимые соли
При помощи шприца со встроенным микрофильтром фильтруют пробы и при необ¬
ходимости разбавляют их перед введением. В случае сомнения относительно концентра¬
ции начинают с разбавленных растворов (1/200,1/50 и т. д.).
На рис. 18.2 приведен пример хроматограмм, полученных в этих условиях для экс¬
трактов холодной (а) и горячей (ft) водой из лювисоля (IJapludalj), сформированного на
лессе, культивируемого на территории экспериментальной станции (институт ITCF\ Бу-
аньвиль, Эссона, Франция), а также холодного водного экстракта (с) из хлоридно-суль-
фатного салисоля средиземноморской осадочной лагуны. Экстракт (с) был разбавлен
в 50 раз, и чувствительность снизилась в 6 раз по сравнению с экстрактами (а) и (ft),
Фосфат
Рис. 18.2. Определение растворимых анионов в почвах методом ионной хроматографии
(Doulbeau и Rochette, институт IRD, Монпелье, Франция, неопубликованные данные): а — холод¬
ный водный экстракт (10 г/200 мл, перемешивают в течение 4 ч) из лювисоля (Hapludalf), сформи¬
рованного на лессе, культивируемого в районе Буаньвиля (Франция), инжекция без разведения;
Ь — горячий водный экстракт из лювисоля (а), предварительно экстрагированного холодной во¬
дой (кипячение с обратным холодильником в течение 16 ч), инжекция без разбавления; с — экс¬
тракт 1:5 при 20 °С из поверхностного горизонта хлористо-сульфатного салисоля (Maison de la
Nature, Латг, Франция), разбавление экстракта 1:50 перед инжекцией. Содержания даны для не¬
разбавленного экстракта
516
Часть 3. Неорганический анализ
введенными без разбавления. Эти условия не предотвращали определение нитратов экс¬
тракта (с), несмотря на то что содержание нитратов было в 8 раз меньше (разбавление
1:50), чем в экстракте (а) и в присутствии 5000-кратного избытка хлорида. Этот метод
можно использовать в очень широком диапазоне концентраций для различных типов
почв и исследований.
18.3.10. Представление результатов
Представление данных в миллиэквивалентах на литр экстракта позволяет проводить
немедленную проверку согласованности результатов, особенно при использовании
различных методов измерений (Ludwig п др., 1999). Сумма катионов (2+) должна быть
равна сумме анионов (2-); также следует проверить уравнение (18.4), т. е. соблюдение
двойного равенства:
(2+) = (!-) = (10-12) УЭП.
(18.10)
Результаты также можно выражать в пересчете на почву, п - отношение объема
воды экстракта (мл) к массе анализируемой пробы почвы (г) (см. параграф «Сравни¬
тельная значимость» подраздела 18.2.3). Содержания в почве (7h в ммоль(+)/кг или
ммоль(-)/кг) выражают относительно содержаний в экстракте (ТЬ в ммоль(+)/л или
ммоль(-)/л) согласно простому отношению:
Та =п Тз. (18.11)
Так как М — эквивалентная молярная масса каждого иона, результаты также можно
выразить в мг/л (экстракт) из отношения ТЬ (мл/л) = М ft (ммоль(+ или -)/л). Таким
образом, следует проверить баланс с ОРВ и уравнение (18.4’), если определяют содер¬
жание ОРВ (см. подраздел 18.3.3). Уравнение 18.11 остается справедливым для расчета
экстрагируемых масс почвы, выражая 7hs в мг/кг и Тэ в мл/л.
Результаты, соответствующие примеру ионного баланса (рис. 18.2, с) для экстрактов
1:5 из хлоридно-сульфатной почвы из Латта (Франция), приведены в табл. 18.3.
Таблица 18.3. Пример ионного баланса (рис. 18.2, с) для экстракта из 1:5 хлоридно-суль-
фатного салисоля (Szwarc, CIRAD, Монпелье, Франция, неопубликованные
данные)
pH
УЭП,
Катионы, ммоль(+)/л (экстракт)
Анионы, ммоль(—)/л (экстракт)
дСм/м
Са
Mg К Na NH,
Cl SO, N03 HCOj
7,63
8,18
9,80
19,18 1,04 60,30 0,06
73,66 13,66 0,08 1,48
(4)
I+ = 90,38 = 11,05 УЭП
Е- = 88,88= 10,89 УЭП
катионы, мг/л (экстракт)
анионы, мг/л (экстракт)
(41)
196
233 41 1,387 1
2,615 656 5 90
Общее содержание растворенных веществ = 5,224 мг/л = 639 УЭП
катионы, ммоль(+)/л (почва)
анионы, ммоль(-)/л (почва)
49,0
95,9 5,2 301,5 0,3
368,3 68,3 0,4 7,4
катионы, мг/кг (почва)
анионы, мг/кг (почва)
980
1,165 205 6,935 5
13,075 3,280 25 450
517
Глава 18. Растворимые соли
Использованная литература
Baize D (1988) Guide des analyses courantes en Pydologie. INRA, Paris, 172 p.
Boivin P, Hachicha M, Job JO and Loyer JY (1989) Une ntethode de cartographie de la salinity des sols:
conductivity yiectromagnytique et interpolation par krigeage. Sci. du sol., 27, 69-72.
Bouteyre G and Loyer JY (1995) Sodisols et Salsodisols. In Encyclopaedia Universalis, 235-236.
Bower CA and Wilcox LV (1965) Soluble salts. In Methods of Soil Analysis, Part 2, Chemical and
Microbiological Properties, Black CA ed. ASA, SSSA, Madison, USA.
Cheverry C (1974) Contribution a Vetude pedologique des polders du lac Tchad. Dynamique des sels
en milieu continental sub-aride dans des sddiments argileux et organiques., These University Stras¬
bourg, France, 275 p.
Cheverry C (1983) L’extraction de la solution du sol par le biais de bougies poreuses: synthase bib-
liographique des probtemes ntethodologiques pos6s par ces dispositifs. Bull Groupe Frangais
d’Humidimetrie Neutronique, 14, 47-71.
Cheverry C (1998) Agriculture intensive et quality des eaux. INRA, Paris, 297 p.
Corwin DL and Lesch SM (2004) Characterizing soil spatial variability with apparent electrical
conductivity. Computers and Electronics in Agriculture, doi:10.1016/j.compag.2004.11.002.
FAO, ISSS, ISRIC (1998) World Reference Base for Soil Resources. World Soil Resources Reports, FAO,
Rome no 84, 88 p.
Harvie CE and Weare JH (1980) The prediction of mineral solubilities in natural waters: the Na-K-Mg-
Ca-Cl-S04-H20 system from zero to high concentration at 25°C. Geochim. Cosmochim. Acta., 44,
981-997.
Harvie CE, Moller NE and Weare JH (1984) The prediction in mineral solubilities in natural waters: the
Na-K-Mg-Ca-H-Cl-S04-0H-HC03-C03-C02-H20 system to high ionic strengths at 25°C, Geochim.
Cosmochim. Acta., 48, 723-751.
INRA-AFES, (1995) Referentiel Pedologique., INRA, Paris, 332 p.
Job JO (1985) Essais de correlation entre la conductivite dlectrique et la composition ionique des
solutions du sol., ENSAM-USTL, Montpellier, France, 86 p.
Job JO Gonzalez Barrios JL and Rivera Gonzalez M (1997) Dytermination prycise de la salinity des sols
par conductivimytrie yiectromagnytique. In Giophysique des sols et des formations superficielles.,
Orstom, Paris, 143-145.
Keren R (1996) Boron. In Methods of Soil Analysis, Part 3, Chemical Methods., SSSA book series 5,
603-626.
Kouakoua E, Sala GH, Barthys B, Larre-Larrouy MC, Albrecht A and Feller C (1997) La matiere orga-
nique soluble h l’eau chaude et la stability de Tagrygation. Aspects mythodologiques et application &
des sols ferrallitiques du Congo, Eur. J. Soil Sci., 48,239-247.
Le Brusq JY and Loyer JY (1982) Relations entre les mesures de conductivitys sur des extraits de sols
de rapports sol/solution variables, dans la valiye du fleuve Synygal. Cah. Orstom, ser. Pedol., 3,
293-301.
Loyer JY and Susini J (1978) Ryalisation et utilisation d’un ensemble automatique pour la mesure en
continu et in situ du pH, du Eh et du pNa du sol. Cah. ORSTOM. Ser. Pidologie, 16,425-437.
Loyer JY (1991) Classification des sols sates: les sols Salic. Cah. Orstom Ser. Pedol, XXVI, 51-61.
Ludwig B, Meiwes KJ, Gehlen R, Fortmann H, Hildebrand EE and Khanna P (1999) Comparison of dif¬
ferent laboratory methods with lysimetry for soil solution composition — experimental and model
results. Zeitschrift fur Pflanzenernahrung und Bodenkunde, 162, 343-351.
NF X 31-122, (1993) Extraction du bore soluble к l’eau bouillante. In Qualite des sols, 3e ed., AFNOR,
Paris, 91-95.
NF ISO 11265, (1995) Dytermination de la conductivity yiectrique spycifique. In Qualite des sols, 3e ed.,
AFNOR, Paris, 279-282.
Pansu M, Gautheyrou J and Loyer JY (2001) Soil Analysis — Sampling, Instrumentation and Quality
control. Balkema, Lisse, Abington, Exton, Tokyo, 489 p.
Paycheng C (1980) Mithodes d’analyses utilisees au laboratoire commun de Dakar, IRD-Orstom, Dakar,
Paris, 104 p.
Pouget M (1968) Contribution У ltetude des croutes et encroutements gypseux de nappe dans le Sud Tu-
nisien. Cah. Orstom, ser. pedol., 3-4, 309-366.
518
Часть 3. Неорганический анализ
Qadir М, Schubert S and Ghafoor Aet Murtaza G (2001) Amelioration strategies for sodic soils: A review.
Land degradation and development, 12, 357-386.
Rhoades JD and Miyamoto S (1990) Testing soils for salinity and sodicity. In Soil Testing and Plant
Analysis, 3rded., SSSA book series 3,299-336.
Rhoades JD and Oster JD (1986) Solute content. In Methods of soil analysis, part l, Physical and
Mineralogical Methods, 3rd ed, ASA-SSSA, Agronomy monograph 9, 985-1005.
Rhoades JD (1982) Soluble salts. In Methods of soil analysis, Part 2, Chemical and Microbiological
Properties, 2nd ed, ASA-SSSA, Agronomy monograph 9, 167-179.
Rhoades JD (1996) Salinity: electrical conductivity and total dissolved solids. In Methods of Soil Analysis,
Part 3, Chemical Methods., SSSA book series 5, 417-433.
Richards LA ed (1954) Diagnosis and Improvement of Saline and Alkali Soils, US Salinity Laboratory
Staff. Agricultural Department, Handbook no 60.
S6 SMO, Sartini RP, Oliveira CC and Zagatto EAG (2002) A flow-injection system with a quartz crys¬
tal microbalance for the determination of dissolved inorganic carbon in mineral waters. J. Flow
Injection Anal, 19, 25-28.
Servant J (1975) Contribution а ГEtude des Terrains Halomorphes., Inra, Ensa Montpellier, 194 p. et
annexes.
Shirokova Y, Forkutsa I and Sharafutdinova N (2000) Use of electrical conductivity instead of soluble
salts for soil salinity monitoring in Central Asia. Irrigation-and-Drainage-Systems, 14, 199-205.
Simon M and Garcia I (1999) Physico-chemical properties of the soil-saturation extracts: estimation from
electrical conductivity Geoderma, 90. 99-109.
Smith JL and Doran JW (1996) Measurement and use of pH and electrical conductivity for soil quality
analysis. In Methods for Assessing Soil Quality, SSSA special publication. 49,169-185.
Susini J and Nedhir M (1980) Utilisation d 'electrodes sensibles aux ions pour la mesure en continu, avec
enregistrement du pH, pNa, pCl, pCa + Mg dans Гetude des eaux d'irrigation et de drainage et
essais dans les sols: lere partie. Realisation et description de l ’ensemble de mesure., DRES, Orstom,
Tunis (TN), 17 p.
Susini J, Rouault, M and Kerkeb A (1972) Essais d’utilisation en analyse des sols sates d’une Electrode
sensible aux ions sodium, Сак ORSTOM.Ser. Pedologie, 10, 309-318.
Глава 19. Обменный комплекс1
19.1. Введение
Почва является динамической системой, состоящей из твердой, жидкой и газовой фаз.
В ходе ее эволюции под воздействием геологических, биологических, климатических
и гидрологических факторов почва приобретает электрические и электромагнитные за¬
ряды, которые придают ей специфические физико-химические и термодинамические
свойства. Эти заряды различаются по происхождению и локализации, например, колло¬
идные, молекулярные и атомные. Некоторые их них возникают в процессе разрушения
и образования вторичных веществ, таких как глины, алюмосиликаты, оксиды и гумифи¬
цированное органическое вещество (педогенез).
Таким образом, почва предстает в виде неоднородного континуума. Это открытая
система в состоянии случайного и развивающегося равновесия, регулируемая циклами
иммобилизации и высвобождения ионных форм, которые изменяются в зависимости
от климатических условий. Ее свойства могут быть более или менее произвольно объ¬
единены в аналитические пулы из-за сходства (с учетом соответствующих поправок)
явлений в биосфере. Среди этих явлений объектом исключительной важности является
обменный (или поглощающий) комплекс в связи с его непосредственным влиянием на
плодородие и химические процессы в почве. Во влажных тропических условиях интен¬
сивная биохимическая активность (которая иногда сочетается с интенсивной сельско¬
хозяйственной деятельностью) значительно ускоряет процессы эрозии.
Для химиков обширная аналитическая область, представленная взаимодействиями
между зарядами, включает определение емкостей катионного и анионного обмена, со¬
держания обменных катионов, изотерм ионного обмена, обменной кислотности и ее
агрономической корректировки (известкования), обменного натрия и доли адсорби¬
рованного натрия (ДАН), обмена таких анионов, как фосфаты, определения изоэлек-
трической точки или точки нулевого заряда (ТНЗ), постоянных и переменных зарядов,
энтропий и свободных энтальпий, рассеяния, флокуляции, электрофоретической под¬
вижности, дзета-потенциала, броуновского движения, а также взаимосвязи pH и Eh.
19.2. Источники зарядов
19.2.1. Ионный обмен
Открытие ионного обмена относится к 1848-1850 гг. Томсон (Thomson, 1850) обнаружил,
что промывание почвенной колонки раствором сульфата аммония ведет к появлению
в фильтрате сульфата кальция. Вей (Way, 1850-1852), химик, член английского Коро¬
левского сельскохозяйственного общества, опубликовал первую работу по катионному
обмену:
Са2+ (почва) + 2NH+ (жидкая фаза) <-> 2NH+ (почва) + Са2+ (жидкая фаза).
Из теоретических исследований следует отметить работы Болта (Bolt, 1982), Габи и Ла-
гаша (Gabisw Lagache, 1982), Спаркса (Sparks, 1986), Спозито (Sposito, 1981-1984-1989),
Шамаю и Легро (Chamayou и Legros, 1989), Фентона и Хелиера (Fenton и Helyar, 2000).
1 Поглощающий комплекс почвы. — Примен. наун. ред.
520
Часть 3. Неорганический анализ
В процессе эрозии некоторые первичные и вторичные компоненты переходят в по¬
чвенный раствор, создавая вакантные центры и/или новые поверхности, что приводит
к нарушению локальных равновесий. Реакции между компонентами жидкой фазы или
жидкой и твердой фаз происходят не всегда мгновенно, они зависят от природы хими¬
ческих связей, свободной поверхностной энергии, критического поверхностного натя¬
жения и т. д.
В этом ионном резервуаре можно наблюдать разнообразные явления, такие как аб¬
сорбция - десорбция, образование хелатов с органическими и неорганическими лиган¬
дами, растворение или осаждение на поверхности раздела жидкой и твердой фаз.
Объединение и упорядочение атомов приводят к созданию атомных решеток и струк¬
турных единиц, например, филлосиликатов, с четко определенным зарядом (табл. 19.1).
Однако, поскольку реакции не всегда проходят строго стехиометрически, появляются
дефекты и происходят замещения во внутренней структуре минералов, изменяя рас¬
пределение зарядов и природу компонентов. Кроме того, поверхности минералов и ор¬
ганические функциональные группы могут ионизоваться и создавать новые заряды.
Например, негидратированные слюды, которые теоретически имеют нулевую емкость
катионного обмена (ЕКО), могут обнажаться в процессе эрозии (рис. 19.1) и давать сна¬
чала гидратированные формы с повышенной ЕКО и затем вермикулиты с высокой ем¬
костью катионного обмена, в соответствии с моделями Доусона с сотр. (Dawson и др.,
1974) и Джексона и Джуо {Jackson и Juo, 1986).
Таблица 19.1. Классификация глин, основанная на теоретических отрицательных зарядах
их пакетов (Международное агенство по изучению глин — Agence Internationale
pour Г tude des argiles AIPEA)
Глина
Октаэдрический слой
Отрицательные заряды
1:1
Каолинит
Диоктаэдрический
0
Серпентины
Триоктаэдрический
0
2:1
Пирофиллит
Диоктаэдрический
0
Тальк
Триоктаэдрический
0
Гидратированные слюды
Диоктаэдрический
2
Триоктаэдрический
2
Смектиты
Диоктаэдрический
7
о
Триоктаэдрический
0,5-1,2
Вермикулиты
Диоктаэдрический
Триоктаэдрический
1.2- 1,9
1.2- 1,9
2:1:1
Хлориты
Диоктаэдрический
Переменные
Ди-триоктаэдрический
Переменные
Триоктаэдрический
Переменные
Волокнистые глины
Переменные
Аллофан-имоголит
Переменные
19.2.2. Обменный комплекс
Все твердые, жидкие и газообразные материалы состоят из атомов, состоящих из эле¬
ментарных частиц, набор которых, с точки зрения химиков, может быть ограничен
протонами, нейтронами и электронами. Элемент электрически нейтрален, но если
электрон покидает атом (перенос электрона с внешнего уровня), это вызывает электри-
Глава 19. Обменный комплекс
521
а)
Ь)
‘"'СУ'ТЗ
т-
I I I
0(,ХХ)0
0(,ХХ,)0
Q_._Q.__Q
! Слюда •'
1 необменный К 1
ЕКО ~ О
-К
+К
Гидратированная слюда Вермикулит
«иллит» ЕКО ~ 150 смол(+)/кг
ЕКО ~ 20-50 смол(+)/кг
Рис. 19.1. Модели разрушения слюд: а — Dawson и др., 1974; b — Jackson и Juo, 1986
ческий дисбаланс, ядро приобретает заряд, численно равный, но противоположный за¬
ряду электрона и становится положительно заряженным ионом (катионом). И наоборот,
электрон, связываясь с атомом (заполнение внешнего уровня), приносит дополнитель¬
ный отрицательный заряд, который характеризует заряд частицы (аниона).
Поскольку атомы связаны электростатическими силами, они объединяются, обра¬
зуя ионные структуры, которые легко идентифицируются благодаря их кристалличе¬
ским свойствам или их упорядочению ближнего порядка и являются результатом атом¬
ных связей, соответствующих равновесию между силами притяжения и отталкивания
(рис. 19.2). Взаимопроникновение электронных оболочек с обобществлением электро¬
нов приводит к образованию ковалентных связей, число которых ограничено числом
доступных электронов на внешнем уровне.
Процессы эрозии в почве вызывают переход из ионных состояний в мезоморфные
коллоидные состояния (которые до настоящего времени еще точно не определены),
а затем в состояния, невидимые для рентгеновских лучей, когда соединения приобре¬
тают упорядочение ближнего порядка, после этого начинается вновь создание упорядо¬
чения дальнего порядка и, в конечном счете, соединения переходят в кристаллическое
состояние. Временной масштаб каждого из этих состояний зависит от климатических,
геологических и биологических параметров и может изменяться от нескольких дней до
нескольких тысячелетий.
Электрически заряженные поверхности материалов имеют избыточное или недо¬
статочное количество электронов. Независимо от источника поверхностных зарядов их
522
Часть 3. Неорганический анализ
Рис. 19.2. Основные единицы глин
нейтрализация требует введения добавочного количества положительных или отрица¬
тельных зарядов в жидкую фазу, т. е. в почвенный раствор.
Теория обмена не может объяснить все возможные случаи. Из-за сложности и разно¬
образия обменных явлений приходится использовать несколько различных уравнений.
Для целей моделирования после последовательных приближений оставляют наиболее
влияющие параметры. Можно использовать термодинамический подход для объедине¬
ния законов действующих масс, отбора констант равновесия или отдать предпочтение
рассмотрению некоторых механизмов адсорбции на границе раздела твердой и жидкой
фаз, количественному определению переноса ионов, взаимодействию анионов и т. д.
Важно принимать во внимание исходные работы, в которых впервые сформулирова¬
ны современные теории. Эти теории касаются твердой обменной матрицы, почвенного
раствора или подвижной фазы (выделяющие реагенты: поверхностные заряды, стабиль¬
ность коллоидов, реологические свойства, ионные свойства, селективность, концентра¬
ция, валентность, pH и их взаимоотношения).
Ученые-первооткрыватели, такие как Гельмгольц (Helmotz, 1835), де Кинк (de Quincke,
1861), Гйббс {Gibbs, 1906), Гуй (Gouy, 1910), Чепмен (Chapman, 1913), Фрейндлих (Freundl-
ich, 1916), Штерн {Stem, 1924), Мэттсон {Mattson, 1926), Дженни {Jenny, 1926), Полинг
{Pauling, 1927), Керр {Kerr, 1928), Вагелер (Vageler, 1932), Ванселоу {Vanselow, 1932), Та¬
лон (Gapon, 1933), Ленгмюр (Langmuir, 1938), дали свои имена основным уравнениям,
приведенным в последующих главах.
Явление обмена представляет собой перенос ионов между твердым ионитом и рас¬
сматриваемым ионом в жидкой фазе или химическое явление, связанное с применени¬
ем закона действия масс.
19.2.3. Теория
Несмотря на некоторые недостатки, теория двойного слоя (рис. 19.3) Гуя {Gouy, 1910),
Чепмена (Chapman, 1913) и Штерна {Stem, 1924) в большей степени соответствует дей¬
ствительным зарядам почв, чем равновесие Доннана {Donnan).
Теория двойного слоя применяется к ионному обмену и связывает заряд катиона
в почве, концентрацию электролита и электрический потенциал. Внешние электро-
Глава 19. Обменный комплекс
523
Рис. 19.3. Теоретические модели обменных центров.
Модель Гуя-Чепмена (Gouyu Chapman, 1910-1913) (а) определяет центры как простую плоскость
с уменьшением электрического потенциала в зависимости от расстояния до поверхности
коллоида:
где ст — поверхностный заряд, моль/см2; С — емкость, Ф/м2; F — постоянная Фарадея. Размер
противоиона не учитывается.
В модели Штерна (Stern, 1924) (Ь) рассматривается простая плоскость, учитывающая
поверхностные комплексы, образованные с гидратированными ионами (избирательная
и неизбирательная адсорбция): общий заряд ст0 = + ст2.
Модель с тремя слоями и четырьмя плоскостями (с), согласно Спозито (Sposito, 1984) и Баудену
с сотр. (Bowden и др., 1977-1980), удовлетворительно описывает селективную адсорбцию
анионов (фосфата, селената, цитрата, сульфата, фторида, силиката) или катионов (например Си,
Pb, Zn), а также такие эффекты, как влияние концентрации электролита на электрофоретическую
подвижность.
статические силы оказывают воздействие в пределах кристаллической решетки с упо¬
рядочением ближнего порядка без четкого определения атомных расстояний, несмотря
на концептуальные усовершенствования, которые модифицировали исходную теорию,
опубликованную Гуем (Gouy, 1910). В модели Гуя-Чепмена заряды а считаются равно¬
мерно распределенными на поверхности ионита, представляющей собой бесконечную
плоскость. В глине эта плоскость отрицательно заряжена, и закон электрической ней¬
тральности предполагает притяжение такого же количества положительна заряженных
катионов.
524
Часть 3. Неорганический анализ
Цэуппа ионов, доставленных подвижной жидкой фазой, нейтрализует электрическое
силовое поле. Однако силы диффузии стремятся перераспределить ионы: содержание
катионов уменьшается по экспоненциальному закону, начиная от поверхности, и, сле¬
довательно, существует дефицит анионов вблизи этой поверхности. Заряженная поверх¬
ность и явление перераспределения зарядов составляют основу теории двойного слоя.
Ее количественное применение для почв с переменным зарядом вызывает сомнения,
поскольку ионы рассматриваются как концентрированные заряды, без учета противо¬
ионов. Теория предсказывает завышенные значения.
Теория Штерна (Stern, 1924) исправляет эти завышения, предполагая, что ионы, спо¬
собные входить в контакт с поверхностью, с учетом их объема, могут делать это только
с расстояния в несколько ангстремов (рис. 19.3).
Ионы, адсорбированные на поверхности, образуют плотный слой, толщина которого
равна ионному диаметру, после которого следует диффузный слой Гуя и Чепмена. Эта
модель позволяет отличить сильное сродство при селективной адсорбции, которая от¬
носится к явлениям хемисорбции (координации), и слабое сродство адсорбции, которая
относится к слою Штерна. Этот подход обычно достаточен для почв с постоянными за¬
рядами.
Для почв с переменным зарядом модель тройных слоев с четырьмя плоскостями
(Bowden и др., 1977,1980; Sposito, 1984) позволяет уточнить расчеты и расширить их при¬
менение на реакции образования поверхностных комплексов (рис. 19.3):
• протоны и гидроксилы образуют внутрисферные поверхностные комплексы; эти
комплексы связаны ионными и/или ковалентными связями; между поверхностью
функциональной группы и соседней молекулой нет молекул растворителя;
• другие органические и неорганические катионы и анионы образуют менее ста¬
бильные внешнесферные комплексы, связанные электростатическими связями,
с молекулой растворителя между функциональной группой и связанной молеку¬
лой.
Знание явлений сорбции необходимо в почвоведении и агрономии для понимания
кратко- и среднесрочной эволюции почвы в данных климатических условиях и, возмож¬
но, уменьшения некоторых видов деградации.
Использованная литература
Bolt GH and Bruggenwert MGM (1982) Soil chemistry A - Basic elements., Elsevier developments in
soil science, 5A.
Bowden JW, Posner AM and Quirk JP (1977) Ionic adsorption on variable charge mineral surfaces theo¬
retical charge development and titration curves. Austn J. Soil Res., 15,121-136.
Bowden JW, Nagarajah S, Barow NJ, Posner AM and Quirck JP (1980) Describing the adsorption of
phosphate, citrate and selenite on a variable-charge mineral surface. Austn J. Soil Res., 18,49-60.
Chamayou M and Legros JR (1989) Les bases physiques, chimiques et mineralogiques de la science du
sol., Presses Universitaires de France.
Chapman DL (1913) A contribution to the theory of electrocapillarity. Philos. Mag., 6,475-481.
Dawson M, Foth MD, Page AL and McLean EO (1974) Cation exchange properties of soils. A slide show.
Div. S-2. Soil Chemistry., Soil Science. Society of America, 8 p + 45 diapositives couleur.
Fenton G and Helyar KR (2000) Soil acidification, in PEV Charman and BW Murphy (eds), Soils: Their
Properties and Management, Oxford University Press, Melbourne.
Freundlich H (1926) Colloid and capillary chemistry., Metwuen London.
Gabis V and Lagache M (1982) Les surfaces des solides ттёгаих; adsorption: 109-179; l’£change d’ions.
297-353. Soc. Fr. de Mineralogie et cristallographie, 1-2.
Глава 19. Обменный комплекс
525
Gouy G (1910) Sur la constitution de la charge Electrique a la surface d’un Electrolyte. J. Ann. Phys., 457,
457-468.
Jackson ML and Juo JX (1986) Potassium-release mechanism on drying soils: iron exchangeable to ex¬
changeable potassium by protonation of micas. Soil Sci., 141, 225-229.
Sparks DL (1986) Soil physical chemistry. CRC Boca Raton, FL 308 p.
Sposito G (1981) The thermodynamics of soils solutions., Clarendon Oxford, 223 pages.
Sposito G (1984) The surface chemistry of soils., Clarendon Oxford, 234 p.
Sposito G (1989) The chemistry of soils. Oxford University. Press, Oxford 277 p.
Stem О (1924) Zur theorie der elektrischen doppelschischt. Z. Elektrochem, 30, 508-516.
Thomson HS (1850) On the absorbent power of soils. J. Royal Agric. Soc. Engl., 11, 68-74.
Way JT (1850) On the power of soils to absorb manure. J. Royal Agric. Soc. Eng., 11, 311-379.
Way JT (1852) On the power of soils to absorb manure. J. Royal Agric. Soc. Eng., 13, 123-143.
Дополнительная литература
Johnson SW (1859) On some points of agricultural science. Am. J. Sci. Arts, 2, 71-85.
Freundlich H (1926) Colloid and capillary chemistry., Methuen, London.
Page JB and Bauer LB (1940) Ionic size in relation to fixation of cations by coloidal clay. Soil Sci. Soc.
Am. Proc., 4, 150-155.
Hendricks SB., Nelson RA and Alexander LI (1940) Hydratation mechanism of the clay mineral mont-
morillonite saturated with various cations. J. Am. Chem. Soc., 62,1457-1464.
Barshad I (1948) Vermiculite and its relation to biotite as revealed by base exchange reaction, X-Ray
analysis, differential thermal curses and water content. Am. Miner., 33, 655-678.
Mehrlich A (1948) Determination of cation and anion-exchange properties of soils. Soil Sci., 66, 429-
445.
Standford G (1948) Fixation of potassium in soils under moist conditions and on claying in relation to
type of clay mineral. Soil Sci. Soc. Am. Proc., 12, 167-171.
Schofield RK (1949) Effect of pH on electric charges carried by clay particles. J. Soil Sci., 1,1-8.
Wear JL and White JL (1951) Potassium fixation in clay minerals as related to crystal structure. Soil Sci.,
71, 1-14.
Bolt GH (1952) The significance of the measurement of the zeta potential and the membrane potential in
soil and clay suspensions., Master thesis Cornell University.
Overbeek J Th (1952) Electrochemistry of the double-layer. Colloid Sci., 1, 115.
Pratt PF and Holowaychuck NA (1954) A comparison of ammonium acetate, barium acetate and buff¬
ered barium chloride methods of determining cation-exchange capacity. Soil Sci. Soc. Am. Proc., 18,
365-368.
Schofield RK and Samson HR (1954) Flocculation of kaolinite due to the attraction of oppositely-charged
crystal faces. Disc. Faraday Soc., 18, 138-145.
Bolt GH (1955) Analysis of the validity of the Gouy-Chapman theory of the electric double layer.
J. Colloid Sci., 10, 206-218.
Schuffellen AC and Van der Marel HW (1955) Potassium fixation in soils. Potass. Symp., 2, 157 (180
references).
Diamond S and Kinter EB (1956) Surface areas of clay minerals as derived from measurements of glyc¬
erol retention. Clays Clay Miner., 5,334.
De Mumbrum LE and Jackson ML (1957) Formation of basic cations of copper, zinc, iron and alumini¬
um. Soil Sci. Soc. Am. Proc., 21, 662.
White ML (1957) The occurrence of zinc in soil. Econ. Geol., 52, 645.
Garrett WG and Walker GF (1959) The cation exchange capacity of hydrated halloysite and the formation
of halloysite-salt complexes. Clay Miner. Bull., 4, 75-80.
Johansen RT and Dunning HN (1959) Water-vapor adsorption on clay. Clays Clay Miner., 6, 249.
Van der Marel HW (1959) Potassium fixation, a beneficial soil characteristic for crop production.
Z Pflanzenerdhrung dungung, Bodenkunde, 84, 51-62.
Giles CH, Maewan TH, Nakhwa SN and Smith D (1960) Studies in adsorption XI. A study of classifica¬
tion of solution adsorption isotherms and its use in diagnosis of adsorption mechanisms and in mea¬
surement of specific surface area of solids. J. Chem. Soc., 3973.
526 Часть 3. Неорганический анализ
Hsu PH and Rich Cl (1960) Aluminium fixation in a synthetic cation exchanger. Soil Sci. Soc. Am. Proc.,
24,21-25.
Rich Cl (1960) Aluminium in interlayers of vermiculite. Soil Sci. Soc. Am. Proc., 24, 26-32.
Toujan S (1960) Essai de distribution analytique entre sels solubles et cations 6changeables en sols alcal-
ins sates. Cah. ORSTOM Sen Pedol., X, 25.
Pratt PF (1961) Effect on pH on the cation exchange capacity of surface soils. Soil Sci. Soc. Am. Proc.,
21,96.
Dixon JR and Jackson ML (1962) Properties of intergradient chlorite-expansible layer silicates of soils.
Soil Sci. Soc. Am. Proc., 26, 358-362.
Shen MJ and Rich Cl (1962) Aluminium fixation in montmorillonite. Soil Sci. Soc. Am. Proc., 86,
33-36.
Babcok KL (1963) Theory of the chemical properties of soil colloidal systems at equilibrium. Hilgardia,
V, 11.
Bower CA (1963) Adsorption of o. phenanthroline by clay minerals and soils. Soil Sci., 95, 192.
Coleman NT, Craig D and Lewis RJ (1963) Ion exchange reactions of cesium. Soil Sci. Soc. Am. Proc.,
21, 287-289.
Coleman NT, Lewis RJ and Craig D (1963) Sorption of cesium by soils and its deplacement by salt solu¬
tion. Soil Sci. Soc. Am. Proc., 27, 290-294.
Bear FE (1984) Chemistry of the soil, Reinhold.
Bingham FT, Page AL and Sims JK (1964) Retention of Cu and Zn by H-Montmorillonite. Soil Sci. Soc.
Am. Proc., 28, 351-354.
Coleman NT and Thomas GW (1964) Buffer curves of acid clays as affected by the presence of ferric iron
and aluminum. Soil Sci. Soc. Am. Proc., 28, 187-190.
Greenland DJ and Quirk JP (1964) Determination of total specific surface areas of soils by absorption of
cetyl pyridinium bromide. J. Soil Sci., 15, 178.
Helling CJ, Chesters G and Corey RB (1964) Contribution of organic matter and clay to soil cation
exchange capacity as affected by the pH of the saturating solution. Soil Sci. Soc. Am. Proc., 28, 517.
Marshall CE (1964) The physical chemistry and mineralogy of soils., Wiley New York.
McLean EO, Hourigan WR, Shoemaker HE and Bhumbla DR (1964) Aluminum in soils: V Form of alu¬
minum as a cause of soil acidity and a complication in its measurements. Soil Sci., 97,119-126.
Sawhney BL (1964) Sorption and fixation of micro-quantities of Cs by clay minerals. Effect of saturating
cations. Soil Sci. Soc. Am. Proc., 28, 183-186.
Schnitzer M and Gupta VC (1964) Chemical characteristics of the organic matters extracted from the О
and B2 horizons of a gray wooded soil. Soil Sci. Soc. Am. Proc., 28, 374.
Bhumbla DR and McLean EQ (1965) Aluminium in soils. VI - Changes in pH dependent acidity, cation
ion exchange capacity and extractable aluminium with addition of lime to surface soils. Soil Sci. Soc.
Am. Proc., 29, 370.
Bolt GH and Page AL (1965) Ion-exchange equations based on double layer theory. Soil Science, 99,
357-361.
Carter DL, Heilman MD and Gonzalez CL (1965) Ethylene glycol monoethyl ether for determining sur¬
face area of silicate minerals. Soil Sci., 100, 356.
Follet EAC (1965) The retention of amorphous colloidal “ferric hydroxide” by kaolinites. J. Soil Sci., 16,
334-341.
Fripiat J.J., Cloos P and Poncelet A (1965) Comparaison entre les proprtetes dtechange de la montmoril¬
lonite et d’une tesine vis-a-vis des cations alcalins et alcalino-terreux. I - Reversibilit£ des processus.
Bull. Soc. Chim. de France, 208-214.
Marshall CE and McDowell LL (1965) The surface reactivity of micas. Soil Sci., 99,115-131.
Parks GA (1965) The isoelectric points of solids oxides, solid hydroxides and aqueous hydroxo complex
systems. Chem. Rev., 65, 177-193.
Kittrick JA (1966) Forces involved in ion fixation by vermiculite. Soil Sci. Soc. Am. Proc., 30, 801-803.
Shainberg I and Kemper WD (1966) Hydration status of adsorbed cations. Soil Sci. Soc. Am. Proc., 30,
707-713.
Stanton DA and Burger R de T (1966) Zinc in orange free state soils. I - An assessment of the zinc status
of surface soils. S. Afr. J. Agr. Sci., 601.
Глава 19. Обменный комплекс
527
Stanton DA and Burger R de T (1966) Zinc in orange free state soils. II — Distribution of zinc in selected
soils profiles and in particle size fractions. S. Afr. J. Agr. Sci., 809.
De Villiers JM and Jackson ML (1967) Cation exchange capacity variations with pH in soil clay. Soil Sci.
Soc. Am. Proc., 31, 473.
Parks GA (1967) Aqueous surface chemistry of oxides and couplage oxide minerals in Stumm, W.: Equi¬
librium concepts in natural water systems. Adv. Chem. Series, 67, 121-160.
Parks GM (1967) Isoelectric point and zero point charge. Adv. in Chem., 67, 121-160.
Ruellan A and Deletang J (1967) Les phenomenes d'echange de cations et d'anions dans les sols. IRD
(ex-Orstom), Doc. Techno., no. 5, Paris.
Grim RE (1968) Clay Mineralogy. McGraw-Hill, New York 185-233.
Murray DJ, Healy TW and Fuersteneau DW (1968) The adsorption of aqueous metal and colloidal hy¬
drous manganese oxide. Adv. Chem. Ser., 79, 74-81.
Rich Cl (1968) Mineralogy of soil potassium. In Kilmer VJ, Younts S.E., Brady NC. The role of potassium
in agriculture. Am. Soc. Agron., US, 79-108.
Flegman AW, Goodwin JW and Ottewill RA (1969) Rheological studies on kaolinite suspensions. Proc.
Brit. Ceram. Soc., 13, 31-45.
Gautheyrou J and Gautheyrou M (1969) Index bibliographique “Echange” 1960-1967. ORSTOM-Antil-
les, Notes de laboratoire, no. 10, 56 p.
Kalb GW and Curry RB (1969) Determination of surface area by surfactant adsorption in aqueous sus¬
pension. I - Dodecylamine hydrochloride. Clays Clay Miner., 17,47.
Hingston FJ (1970) Specific adsorption of anions on goethite and gibbsite., Ph. D diss., University West¬
ern Australia Perth.
Sawhney BL, Frinck CR and Hill DE (1970) Components of pH dependent cation exchange capacity. Soil
Sci., 109, 272.
Sawhney BL (1972) Selective sorption and fixation of cations by clay minerals: a review. Clays clay
Miner., 20, 93-100. 1
Van Raij В and Peech M (1972) Electro chemical properties of some oxisols and alfisols of the tropics.
Soil Sci. Soc. Am. Proc., 36, 587-593.
Brace R and Matijevic E (1973) Aluminum hydrous oxide sols. I - Spherical particles of narrow size
distribution. J. Inorg. Nucl. Chem., 35, 3691-3705.
McLaren RG and Crawford DV (1973) Soil copper. I - Fractionation of copper in soils. J. Soil Sci., 24,
172.
McBride MB and Mortland MM (1974) Copper (II) interactions with montmorillonite: evidence from
physical methods. Soil Sci. Soc. Am. Proc., 38,408-415.
Espinoza W, Gast RG and Adams RS Jr (1975) Charge characteristics and nitrate by two andepts from
south-central Chile. Soil Sci. Soc. Am. Proc., 39, 842-846.
Ferris AP and Jepson WB (1975) The exchange capacities of kaolinite and the proportion of homo-ionic
clays. J. Colloid Interface Sci., 51, 245-259.
Gupta SK and Chen KY (1975) Partitioning of trace metals in selective chemical fractions of nearshore
sediments. Environ. Lett., 10, 129.
Baes CF and Mesmer RE (1976) The hydrolysis of cations. Wiley, New York.
Bolland MDA, Posner AM and Quirk JP (1976) Surface charge on kaolinites in aqueous suspensions.
Aust. J. Soil Res., 14, 197-216.
Bolt GHM, Bruggenwert GM and Kamphorst A (1976) Adsorption of cations by Gallez A, Juo ASR and
Herbillon AJ (1976) Surface and charge characteristics of selected soils in the tropics. Soil Sci. Soc.
Am. Proc., 40, 601-608.
Gillman GP and Bell LC (1976) Surface charge characteristics of six weathered soils from tropical N.
Queensland. Austr. J. Soil Res., 14, 351-360.
Herbillon AJ, Mestadgh MM, Vielvoye L and Derouane EG (1976) Iron in kaolinite with special refer¬
ence to kaolinite from tropical soils. Clay Miner., 11,201-220.
Gast RC (1977) Surface and colloid chemistry. In Dixon JB, Weed SB, Kittrick JA, Milford MM, White
JL. Minerals in soils environment., Soil Science Society of America: 27-73.
Laverdidre MM and Weaver RM (1977) Charge characteristics of spodic horizons. Soil Sci. Soc. Am. J.,
41,505-510.
528 Часть 3. Неорганический анализ
Sposito G and Mattigod SV (1977) On the chemical foundation of the sodium adsorption ratio. Soil Sci.
Soc. Am. J., 41, 323-329.
Thomas GW (1977) Historical developments in soil chemistry: ion exchange. Soil Sci. Soc. Am. J., 41,
230-238.
Carr RM, Chaikum N and Paterson N (1978) Intercalation of salts in halloysite. Clays Clay Miner., 26,
144—152.
Gessa C, Melis P, Bellu G and Testin C (1978) Inactivation of clay pH-dependent charge in organo-
mineral complexes. J. Soil Sci., 28, 58.
Gillman GP and Bell LC (1978) Soil solution studies on weathered soils from tropical North Queensland.
Austr. J. Soil Res., 16, 66-77.
Hendershot WH (1978) Measurement technique effects of the value of zero point of charge and its dis¬
placement from zero point of titration. Can. J. Sol Sci., 58, 439.
McBride MB (1978) Copper (II) interactions with kaolinite factors controlling adsorption. Clays Clay
Miner., 26, 101-106.
Parfitt RL (1978) Anion adsorption by soils and soil materials. Adv. Agronomy, 30,1-80.
Warm SS and Uehara G (1978) Surface charge manipulation in constant surface potential soil colloids.
I - Relation to adsorbed phosphorus. Soil Sci. Soc. Am. J., 42, 565-570.
Warm SS and Uehara G (1978) II - Effect on solute transport. Soil Sci. Soc. Am. J., 42,886-888.
Keng JCW (1979) Surface chemistry of some constant potential soil colloids., Thesis University Hawaii.
Lindsay WL (1979) Chemical equilibrium in soils., Wiley New York.
Shuman LM (1979) Zinc, manganese and copper in soil fractions. Soil Sci., 127,10.
Yoon RH, Salman T and Donnay G (1979) Predicting points of zero charge of oxides and hydroxides.
J. Colloid Interface Sci., 70, 483.
Bowden JW, Posner AM, Quirk JP (1980) Adsorption and charging phenomena in variable charge soils.
In Theng BKG soils with variable charge. NZ Soc. Soil Sci., 147.
Bowden JW, Nagarajah S, Barrow NJ, Posner AM and Quirk JP (1980) Describing the adsorption of
phosphate, citrate and selenite on a variable-charge mineral surface. Austr. J. Soil Research, 18,
49-60.
Gillman GP and Uehara G (1980) Charge characteristics of soils with variable and permanent charge
minerals. II — experimental. Soil Sci. Soc. Am. J.t 44, 252-255.
Lim CH, Jackson ML, Koons RD and Helmke PA (1980) kaolins: sources of differences in cation ex¬
change capacities and cesium retention. Clays Clay Miner., 28, 223-229.
Ninham BW (1980) Long-range vs short-range forces. The present state of play. J. Phys. Chem., 84,
1423-1430.
Sposito G (1980) Cation exchange in soils: an historical and theoretical perspective. In: Baker DE
Chemistry in the soil environment., ASA, 40, 13.
Stoops W (1980) Ion adsorption mechanisms in oxidic soils: implications for ZPC determinations. Geod.,
23, 303-314.
Uehara G and Gillman GP (1980) Charge characteristics of soils with variable and permanent charge
minerals. I - Theory. Soil Sci. Soc. Am. J., 44,250-252.
Westhall J and Hohl H (1980) A comparison of electrostatic models for the oxide/solution interface. Adv.
in colloid and interface science, 12, 265-290.
Maksimovic Z, White JL and Logar M (1981) Chromium-bearing dickite and chromium-bearing kaolinite
from Teslic (Yougoslavia). Clays clay mineral, 29, 213-218.
Morel FMM, Westall JC and Yeasted JG (1981) Adsorption models: a mathematical analysis in the frame
work of general equilibrium calculations. In: Anderson MA, Rubin AJ.: Adsorption of inorganics at
solid-liquid interfaces., Ann. Arbor. Sci. Pub.
Tazaki К (1981) Analytical electron microscopic studies of halloysite formation processus-morphology
and composition of halloysite. Proc. Int. Clay Conf. Elsevier, Amsterdam 573-584.
El-Swaify SA (1982) Soil physical chemistry., Hawai Univ., booklet n° 640.
Wada SI and Mizota C (1982) Iron-rich halloysite (10 E) with crumpled lamellar morphology from Hok¬
kaido - Japan. Clays Clay Miner., 30, 315-317.
Gillman GP, Bruce RC, Davey BG, Kimble JM, Searle PL and Skjemstad JO (1983) A comparison of
methods used for determination of cation exchange capacity. Commun. Soil Sci. Plant Anal., 14,
1005-1014.
Глава 19. Обменный комплекс
529
Hodges SC and Zelazny (1983) Interactions of dilute hydrolysed aluminum solutions with clays, peat and
resin. Soil Set Soc. Am. J., 47, 206-212.
Kleijn WB and Oster JD (1983) Effects of permanent charge on the electrical double-layer properties of
clays and oxides. Soil Sci. Soc. Am. J., 47, 821-827.
Bleam W and McBride MB (1984) Cluster formation versus isolated-site adsorption. A study of Mn(II)
and Mg(II) adsorption on boehmite and goethite. J. Colloid Interface Sci., 103: 124-132.
Barrow NJ (1985) Reactions of anions and cations with variable-charge soils. Adr. Agron.9 38,183-230.
Fallavier P (1985) DensitS de charge variable et point de charge nulle dans les sols tropicaux. Definition,
mesure et utilisation. Agronomie tropicale, 40, 239-245.
Gillman GP (1987) Modification of the compulsive exchange method for cation exchange capacity deter¬
mination. Soil Sci. Soc. Am. J.9 51, 840-841.
Lambert К (1987) Cation exchange in Indonesian peat soils., Thesis Gent Univ., 68 p.
Matjue N and Wada К (1987) Comments on “modification of the compulsive exchange method for cation
exchange capacity determination”. Soil Sci. Soc. Am. J.9 51, 841.
Marcano-Martinez E and McBride MB (1989) Comparison of titration and ion adsorption methods for
surface charge measurement in oxisol. Soil Sci. Soc. Am. J.9 53, 1040-1045.
Bolt GH, De Boodt MF, Hayes MHB and McBride MB (1991) Interactions at the soil colloid-soil solution
interface. Kluwer Academic Publishers -NatoAsi Series. Series E: Applied Sciences, vol. 190,603 p.
Song КС and Ishiguro M (1992) Effects of solution pH on ion transport in allophanic andisol. Soil Sci.
Plant Nutr., 38, 477-484.
Sposito G (1994) Chemical Equilibria and Kinetics in Soils., Oxford University Press, Oxford.
Sposito G (ed)itor (1996) The environmental chemistry of aluminum., Lewis UK 464 p.
Comans RNJ, Hilton J, Voitsekhovitch O, Laptev G, Popov V, Madruga MJ, Bulgakov A, Smith JT,
Movchan Net Konoplev A (1998) A comparative study of radiocesium mobility measurements in
soils and sediments from the catchment of a small upland oligotrophic lake (Devoke Water, U.K.).
Water Res. Oxford, 32, 2846-2855.
Vogeler I (2001) Copper and calcium transport through an unsaturated soil column. Journal of
environmental quality, 30, 927-933.
Cervini-Silva Jet Sposito G (2002) Steady-state dissolution kinetics of aluminium-goethite in the pres¬
ence of deferrioxamine-B and oxalate ligands. Environ. Sci. Technol., 36, 337-342.
Глава 20. Изоэлектрическая точка и точка нулевого заряда
20.1. Введение
20.1.1. Заряды коллоидных растворов
Таблица 20.1. Сокращения и обозначения
Сокращение
Обозначение
TH3(PZCimhZPC)
Точка нулевого заряда
ТНЧЗ (PZNC или ZPNC)
Точка нулевого чистого заряда
ТНЧПЗ (PZNPC)
Точка нулевого чистого протонного заряда
ТНСЭ (PZSE)
Точка нулевого солевого эффекта
HTT(ZPT)
Нулевая точка титрования
ИТНЗ (PPZC)
Исходная точка нулевого заряда (при ТНЗ = ИЭТ)
ИЭТ (IEP)
Изоэлектрическая точка
рНО
Определяет знак чистого поверхностного заряда
В последние 50 лет определение точки нулевого заряда (Parks и de Вгиуп, 1962) позво¬
лило развить концептуальный подход к явлениям почвенных зарядов, в частности в тро¬
пических почвах, физико-химические характеристики которых тесно связаны с при¬
сутствием переменных зарядов. Используемая терминология, которая еще изменяется
(табл. 20.1), иногда приводит к путанице, и определенные значения параметров могут
быть ошибочными.
Точка нулевого заряда (ТНЗ) определяет величины pH, связанные с особыми усло¬
виями, примененными к одному или более значению поверхностной плотности заряда,
точнее говоря, величины pH, при которых один или более поверхностных зарядов ком¬
пенсируют друг друга (Sposito, 1984). Величина ТНЗ может изменяться в зависимости от
того, какие слои учитываются при определении твердой поверхности (см. гл. 19).
Исследования Уехара и Гкллмана (Uehara и Gillman, 1981), Спозито (Sposito, 1981,
1984, 1989), Спаркса (Sparks, 1986), Барроу (Barrow, 1987), Гкнгайя и Моррисона
(Gangaiya и Morrison, 1987), Круз-Уэрты и Кинтца (Cruz-Huerta и Kientz, 2000), ГУстафс-
сона (Gustafsson, 2001) показали, что на практике можно использовать более простые ра¬
бочие определения, основанные на результатах анализа (такие как, рНО или ТНЧЗ, см.
подраздел 20.1.2). Возможное расхождение между характеристическими величинами pH
подчеркивает неоднородность молекулярного окружения поверхностей раздела, слож¬
ность почвенной системы и связанных понятий. Поскольку почва одновременно содер¬
жит компоненты, имеющие постоянные или переменные заряды, все заряды системы с0
разделяются на следующие компоненты:
ас — постоянные структурные заряды, возникающие в кристаллических решетках си¬
ликатов и в результате изоморфных замещений ионами различной валентности. В боль¬
шинстве случаев ас имеет отрицательное значение. Величина этих зарядов значительна
в глинах типа 2:1, но обычно мала в глинах типа 1:1 и гидратированных оксидах.
Глава 20. Изоэлектрическая точка и точка нулевого заряда
531
ап (или ан) — переменные заряды или чистые протонные заряды, возникающие в ок¬
сидах железа или алюминия, алюмосиликатах, органическом веществе, а также крае¬
вые заряды и заряды поверхностных функциональных групп. Они преобладают в силь¬
но эродированных почвах, обогащенных гидроксильными группами. Эти заряды могут
быть нулевыми, положительными или отрицательными. Протоны диффузного слоя не
учитываются в соотношении: ап = qH - qOH.
аа (или аетс) — заряды, образующиеся благодаря комплексам, способным создавать
очень прочные ковалентные или ионные связи (кроме связей Н+ и ОН-) неэлектро¬
статической природы, без молекул воды между поверхностью функциональных групп
и ионными комплексами (внутрисферные поверхностные комплексы, Sposito, 1981). За¬
ряды могут быть нулевыми, положительными или отрицательными и могут быть связа¬
ны с избирательной адсорбцией.
ор (или авшс) — заряды электростатической природы из внешнесферных комплексных
ионов (кроме Н+ и ОН-), которые считаются неизбирательными. Между функциональ¬
ной группой и связанным ионом находится молекула воды. Эти ионы слабо адсорбиро¬
ваны.
ад — ионные заряды диффузного слоя, которые компенсируют полные вакантные за¬
ряды и обеспечивают нейтральность. Ионы этого слоя удерживаются слабыми электро¬
статическими силами, но могут диффундировать в почвенный раствор.
Эти заряды или плотности заряда можно выражать в моль/м2 или Кл/м2 и
моль(заряда)/кг. Электрическая нейтральность предполагает использование следующе¬
го уравнения:
ап = а + о„ + о + ол + о = 0.
О с л в р д
Понятие поверхностного заряда частицы апз должно быть определено (в модели двой¬
ного слоя), так же как различных слоев, участвующих в этом явлении. Например, если:
ап, = а + о„ + а + а„,
пэ с п а р7
в этом случае ТНЗ - это точка, где апз = 0, или апэ = -(ад).
Если поверхностный заряд рассматривать как опз = ос + оп + аа, тогда ТНЗ — это точ¬
ка, где опз = 0, или апэ = -(ар + од).
При потенциометрическом титровании поверхностный заряд можно определить как
функцию pH и концентраций электролитов, а величина рНО выводится. Кривые титро¬
вания пересекаются в точке, где поверхностный заряд не зависит от концентраций ин¬
дифферентных электролитов.
Если pH = рНО, величина pH не изменяется при изменении концентрации электро¬
лита, только в этом случае
ап = °
и
однако в ТНЗ: апэ = ос + оп + аа = 0.
В этом типе анализа, если нет значительного постоянного заряда, ТНЗ и рНО могут
быть перепутаны. Однако необходимо учитывать тот факт, что рНО может несколько из¬
меняться, если имеет место адсорбция, которая соответствует избирательному сродству
и связана с образованием более отрицательно или более положительно заряженной по¬
верхности.
532
Часть 3. Неорганический анализ
20.1.2. Определения
Точка нулевого заряда (ТНЗ)
Это величина pH, при котором общий чистый заряд а0 скомпенсирован (а0 = 0). По¬
скольку силы взаимодействия между частицами инактивированы, частицы флокулиру¬
ют и не движутся при приложении электрического поля (электрофоретическая подвиж¬
ность равна нулю). Это свойство имеет большое значение для образования почвенных
агрегатов и удержания ионов. Когда pH почвы превышает величину ТНЗ, заряд а ста¬
новится отрицательным и емкость катионного обмена увеличивается. И наоборот, когда
pH почвы становится меньше величины ТНЗ, появляется положительный заряд, созда¬
ющий емкость анионного обмена.
Оценка ТНЗ позволяет предсказать отклик почвы на изменения условий окружа¬
ющей среды (например, культивация, внесение удобрений). Для почвы в равновесии
с раствором электролита, который содержит катионы и анионы, образующие только
внешнесферные комплексы (см. гл. 19), ТНЗ = ТНЧЗ = ТНСЭ.
Точка нулевого чистого протонного заряда (ТНЧПЗ)
Это значение pH, при котором переменный заряд оп скомпенсирован (ап = 0). Эта точка
обычно снижается с ростом pH. Если в кристаллической решетке нет постоянного
заряда или других потенциал-определяющих ионов (Н+ и ОН-), ТНЧПЗ = ТНЗ = ИЭТ
(изоэлектрическая точка, см. подраздел 20.1.1).
Точка нулевого чистого заряда (ТНЧЗ)
Это величина pH, при которой чистый заряд адсорбированного иона, отличающийся от
ап, например, оа + ар + ад, скомпенсирован {Parker и др., 1979; Sposito, 1984). Эта величи¬
на зависит от концентраций электролитов и также может немного изменяться с приро¬
дой используемого иона, хотя и не очень значительно, в присутствии индифферентных
электролитов (Na+, Li+, Cl-, NO-). Измерения проводят в насыщенном растворе, напри¬
мер, NaCl, при изменяющемся pH и постоянной ионной силе.
Эта величина полезна для понимания явления удержания ионов в почве. В этой точке
эквивалентные количества катионов и анионов адсорбируются в присутствии индиффе¬
рентного электролита, и емкость катионного обмена (ЕКО) равна емкости анионного
обмена (ЕАО), т. е. ЕКО - ЕАО = 0.
Если система содержит заметные количества постоянных или переменных зарядов,
рНО (см. ниже) и ТНЧЗ не соответствуют одинаковым величинам pH.
Если pH (ТНЧЗ) < рНО -> признак постоянного отрицательного заряда.
Если pH (ТНЧЗ) < рНО -> признак постоянного положительного заряда.
ТНЧЗ = ТНЗ, если общий поверхностный заряд комплексов компенсирован.
Точка нулевого солевого эффекта (ТНСЭ)
Величину ап часто определяют титрованием в индифферентном электролите при раз¬
личных значениях ионной силы. Величина pH, при которой кривые пересекаются, на¬
зывается точкой нулевого солевого эффекта (ТНСЭ), это частный случай, в котором ап
постоянная величина. В этой точке концентрация соли больше не влияет на pH, но она
не обязательно равна ТНЗ {Parker и др., 1979), кроме случая, когда плотности зарядов оа
и ор не изменяются при изменении ионной силы электролита. Если постоянный заряд
отсутствует или избирательная адсорбция не изменяется при изменении ионной силы
раствора (индифферентные электролиты), то ТНСЭ = ТНЧЗ.
533
Глава 20. Изоэлектрическая точка и точка нулевого заряда
Нулевая точка титрования (НТТ)
Это точка, используемая при анализе, которая соответствует равновесному pH до до¬
бавления кислоты (Н+) или основания (ОН-). Она характеризует не сами заряды, а pH
начальной точки титрования в данной среде с определенной ионной силой.
Изоэлектрическая точка (ИЭТ)
Изоэлектрическая точка — это pH, при котором зета-потенциал равен нулю, или, с точ¬
ки зрения теории диффузного двойного слоя, pH, при котором заряд плоскости, разде¬
ляющей слой Штерна и диффузный слой 1Уя, равен нулю (Breeuwsma, 1973; Sparks 1986):
ап = 0 и ас = аа = стр = 0. В индифферентном электролите, который не вызывает избира¬
тельной адсорбции в слое Штерна, ИЭТ почти равна рНО в системе с преобладающими
переменными зарядами. Термин ИЭТ используют при определении электрофоретиче¬
ской подвижности.
Точка рНО
Это точка, в которой общий переменный заряд ag равен нулю (Uehara-Gillman, 1981).
При рНО адсорбция ионов Н+ и ОН" одинакова. Эта точка может соответствовать ТНЗ
и ИЭТ, но может и отличаться от ТНЗ; она может быть несколько неточной, так как
ионный обмен между протонами, добавленными при титровании, и катионами, ад¬
сорбированными на центрах с постоянными зарядами, может потреблять протоны, не
влияя на переменный заряд. Спаркс (Sparks, 1986) предложил следующее определение:
рНО — это pH гидроксидированной поверхности, общий поверхностный заряд которой
равен нулю. Это важный показатель в почвах с переменными зарядами, потому что он
определяет положительный или отрицательный знак общего поверхностного заряда.
Для тропических почв с переменными зарядами, в которых постоянные заряды малы,
величины ТНЗ, ТНЧПЗ, ТНСЭ и ТНЧЗ близки, что исключает необходимость исполь¬
зовать очень прецизионные и трудоемкие методы, требующие больших количеств по¬
чвы.
20.1.3. Условия измерения заряда
Можно использовать почвы или илистые фракции, возможно после обработок, ко¬
торые изменяют распределения переменных зарядов в зависимости от pH и ионной
силы растворов. Удаление органического вещества маскирует корреляцию между
общим С и ТНЗ, обработка КС1 позволяет экстрагировать обменный А13+, методы
с использованием кислого оксалата и ДЦБ (см. гл. 6) позволяют удалять слой оксидов
и гидроксидов и выявлять постоянные отрицательные заряды. На эти методы влияют
и другие факторы, такие как:
• валентность Z противоиона;
• диэлектрическая проницаемость е (использование неионных растворителей, таких
как этанол, для удаления избытка реагентов может нарушить поверхностные за¬
ряды и привести к серьезным ошибкам);
• абсолютная температура Т,
• концентрации электролитов.
Добавление электролита к почве с переменным зарядом понижает поверхностный
потенциал, но увеличивает поверхностный заряд. На понижение поверхностного потен¬
циала указывает изменение pH раствора. При отрицательном поверхностном заряде pH
снижается с концентрацией электролита, и наоборот, pH возрастает с положительным
534 Часть 3, Неорганический анализ
поверхностным зарядом. Если не наблюдается изменения pH, полный поверхностный
заряд считается равным нулю.
Поскольку pH почвы определяет величину общего заряда, переменные заряды можно
приблизительно оценить при измерении рНно и рНКС1, рассчитав АрН = рНш - рНно
(см. гл. 15). Если -0,5 < АрН < +0,5, в почвах преобладают компоненты с переменными
зарядами, и рНО можно оценить (Uehara и Gillman, 1981) как сумму рНш + АрН или
PH0 = 2pHKC1-pHHiO.
Эти измерения следует считать предварительным тестом перед анализом. Так как
рНН20 зависит от концентраций электролитов в почвенном растворе, трудно оценить,
связаны наблюдаемые различия с концентрациями электролитов или с природой по¬
верхностных зарядов. Результаты измерения рНКС1 более надежны, так как влияние при¬
сутствия электролита в растворе маскируется, и положение с экстракцией А13+ улучшает¬
ся. Схематично, аналитические операции, необходимые для измерения заряда, просты,
но их реализация может оказаться длительным и сложным процессом, они должны вы¬
полняться при строгом соблюдении рабочих условий, включая:
• контакт между почвой и растворами с постоянными составом и концентрацией,
при контролируемых давлении и температуре и в течение определенного периода
времени для достижения реакционных равновесий; этот контакт может достигать¬
ся в суспензии или в колонке с контролируемой газовой средой, если фазы неста¬
бильны в воздухе (окислительно-восстановительные процессы, равновесие С02);
* возможное гравитационное разделение фаз, фильтрация или центрифугирование;
* химический и физико-химический анализ остатков почвы или жидкой фазы после
экстракции;
• расчеты и графическая интерпретация.
Компьютерные программы позволяют описывать и моделировать реакции ионов с по¬
чвой (например, Barrow, 1987), значительно упрощая обработку данных. Можно выпол¬
нять массовые анализы для контроля изменений зарядов глин до достижения равно¬
весия.
20.2. Основные методы
20.2.1. Определение рНО (ТНСЭ), длительное установление равновесия
Упрощенный метод Уехары и Питмана (Uehara и Gillman, 1981).
Реагенты
хлорид натрия
NaCl
М = 58,44 0,1 М
хлорид кальция
СаС12
М = 110,99 (высушен над
0,1 М
хлорид натрия
2 М
хлорид кальция
2 М
хлористоводородная кислота
НС1
0,1 М
гидроксид натрия
NaOH
0,1 М
Сначала должна определяться остаточная влажность проб, чтобы привести результа¬
ты к массе почвы, высушенной при 105 °С. Таким образом, все результаты приводятся
для массы почвы, высушенной при 105 °С и имеющей одну и ту же относительную по¬
верхность при анализе пробы любой влажности.
535
Глава 20. Изоэлектрическая точка и точка нулевого заряда
Методика
• Отвешивают 4 г почвы, просеянной через сито 0,5 мм (или эквивалентную массу
почвы, высушенной при 105 °С) 15 раз и помещают в стаканчики на 50 мл, прону¬
мерованные от 1 до 15 (табл. 20.2).
• Добавляют 0,5 мл 0,1 М раствора индифферентного электролита (NaCl для почв
с преимущественно одновалентными обменными катионами или СаС12 для почв
с преимущественно двухвалентными обменными катионами Са2+, Mg2+).
• В стаканчики 1-7 добавляют возрастающие количества 0,1 М раствора НС1, а в
стаканчики 9-15 - возрастающие количества 0,1 М раствора NaOH.
• Доводят объем до 20 мл, добавляя дистиллированную воду.
• Оставляют стоять на 4 сут. при периодическом перемешивании.
• Измеряют pH-> pH 0,002 М.
• Добавляют 0,5 мл 2 М раствора электролита и перемешивают в течение 3 ч.
• Измеряют pH -> pH 0,05 М.
• Для каждого стаканчика рассчитывают АрН = [pH 0,05 М - pH 0,002 М] и наносят
на график величины АрН относительно pH 0,002 М, чтобы определить точку, где
АрН = 0, которая соответствует рНО, т. е. величине pH, не зависящей от концен¬
трации соли.
Таблица 20.2. Экспериментальная методика определения рНО (ТНСЭ)
4 г почвы
> 1 2
3
4
5
6
7
8
вода, мл
10 10
10
10
10
10
10
10
0,1 М электролит, мл
0,5 0,5
0,5
0,5
0,5
0,5
0,5
0,5
0,1МНС1
НС1 или NaOH, мл
0,25 0,50
1.0
1.5
2,0
2,5
3,0
/
вода, мл
9,25 9,00
8,5
8,0
7,5
7,0
6,5
9,5
всего, мл
20 20
20
20
20
20
20
20
4 г почвы
9
10
и
12
13
14
15
вода, мл
10
10
10
10
10
L0
10
0,1 М электролит, мл
0,5
0,5
0.5
0,5
0,5
0,5
0,5
0,1 М NaOH
НС1 или NaOH, мл
0,25
0,50
1,0
1,5
2,0
2,5
3,0
вода, мл
9,25
9,00
8,5
8,0
7,5
7,0
6,5
всего, мл
20
20
20
20
20
20
20
20.2.2. Точка нулевого солевого эффекта (ТНСЭ), быстрое установление
равновесия
Основные положения
Метод (Block и de Вгиуп, 1970; Hendershot и Laukulich 1979, 1983) основан на косвенном
измерении ТНСЭ методом потенциометрического титрования общей адсорбции ионов
Н+ и ОН" при различных значениях pH и различной ионной силе.
Из-за амфотерного характера некоторых коллоидов кривые пересекаются при опре¬
деленном значении pH, когда адсорбция протонов не зависит от ионной силы. Это точ¬
ка нулевого солевого эффекта (Parker и др., 1979).
536
Часть 3. Неорганический анализ
Реагенты
• Индифферентный электролит:
NaCl 0,001; 0,01; 0,05; 0,2 М;
приготовлен из коммерческих стандартных растворов (фиксаналов).
• НСЮДМ.
• NaOH 0,1 М.
Методика
Измельчают пробу, высушенную на воздухе в течение более двух месяцев (для андосо-
лей), до размера частиц 2 мм (мелкозем), затем измельчают до 0,5 мм (сито AFNOR NF
А11-501, модуль 28), для обеспечения регулярного обмена и минимизации разброса ре¬
зультатов.
Для определения используют восемь образцов (массой 2-4 г) и четыре концентра¬
ции растворов НС1 и NaOH (при правильном пересечении кривых часто достаточно трех
концентраций):
• Отвешивают 2 г почвы.
• Добавляют 40 мл 0,001 М индифферентного электролита.
• Перемешивают при помощи магнитной мешалки в течение 5 мин без остановки,
затем измеряют исходное значение pH, которое соответствует НТТ.
• При помощи автоматического титратора, подсоединенного к комбинированному
электроду, начинают титрование 0,1 М раствором НС1, со скоростью 1 капля/2 мин
(рис. 20.1).
• Продолжают титрование до pH 3,0. Процесс продолжается примерно 2 ч.
Для другого образца почвы массой 2 г:
• Добавляют 40 мл 0,001 М индифферентного электролита.
• Перемешивают при помощи магнитной мешалки и измеряют значение pH, кото¬
рое соответствует НТТ.
Рис. 20.1. Определение точки нулевого солевого эффекта методом титрования
537
Глава 20. Изоэлектрическая точка и точка нулевого заряда
• Выполняют титрование 0,1 М раствором NaOH со скоростью 1 капля/2 мин до pH
9,5-10,0 (из-за опасности растворения углекислого газа предпочтительно рабо¬
тать в контролируемой атмосфере азота для исключения контакта с атмосферным
С02).
Заканчивают титрование, добавляя 0,01, 0,05 и 0,2 М растворы индифферентного
электролита в тех же условиях, а также выполняют холостой опыт (с реагентами без по¬
чвы) для корректировки результатов, если необходимо.
Расчеты
Результаты рассчитывают в ммоль/кг адсорбированной кислоты или основания; вели¬
чина НТТ-ТНСЭ и заряды 2. приводятся на записывающем с постоянной скоростью.
устройстве (рис. 20.1).
Примечания
Величина рНО соответствует точке максимальной химической стабильности. Этот ана¬
лиз можно выполнять на исходной необработанной почве, а также на подвергнутых
предварительной обработке пробах. Например:
• насыщению 1 М раствором NaCl (Hendershot и Lavkulich, 1979; Gautheyrou и др.,
1981);
• разложению органического вещества гипохлоритом натрия;
• удалению оксидов и гидроксидов (методами с использованием ДЦБ, кислого окса¬
лата аммония, пирофосфата, Segalen и др., 1983).
Для необработанной почвы:
• если pH, определенная в воде, выше, чем определенная в КС1 (рННг0 > рНСаС,2 >
рНш), то ТНЗ расположена ниже, чем НТТ, и величина 2. отрицательна;
• если pH, определенная в воде, ниже, чем рНКС1 (рНн о < рНСаСЬ < рНш), то ТНЗ рас¬
положена выше, чем НТТ, и величина 2, положительна.
Чем больше содержание оксидов и гидроксидов, тем выше ТНЗ.
Для почвы, насыщенной NaCl, колебания ТНЗ и величин 2, отражают баланс между
количествами катионов (А13+) и анионов (РО3-), обменянных на Na+ и С1~. Если содер¬
жание глины относительно мало, колебания будут небольшими.
Сравнение проб, насыщенных ионами Na+, с пробами после более или менее полного
удаления органического вещества под действием гипохлорита показывает подавляющий
эффект органического вещества на ТНЗ.
Оксиды железа и алюминия характеризуются относительно высокими значениями
рНО в диапазоне pH от 7 до 9, в зависимости от их природы и степени их кристаллич¬
ности.
Присутствие кремнезема и органического вещества приводит к относительно низ¬
ким значениям рНО, которые увеличивают ЕКО андосолей и позволяют удерживать
катионы.
Использованная литература
Barrow NJ (1987) Reactions with variable-charge soils. In Martinus Nijhoff- Developments in plant and
soil science, eiden 31, 191 p.
Blok L and de Bruyn PL (1970) The ionic double layer et ZnO/solution surface. I - The experimental
point of zero charge. J. Colloid Interface Sci.9 32, 518-525.
538
Часть 3. Неорганический анализ
Breeuwsma А (1913) Adsorption of ion on hematite (a-Fe203). Ph.D. diss. Wageningen (Hollande)
Cruz-Huerta L and Kientz DG (2000) Electric charge of andosols of Cofre de Perote’, Veracruz, Mexico.
Terra, 18, 115-124.
Gangaiya P and Morrison RJ (1987) A review of the problems associated with applying the terms surface
and zero point of chaise to soils. Commun. in Soil Science Plant Analysis, 18, 1431-1451.
Gautheyrou J and Gautheyrou M (1981) Comparison of electric charges in soils formed in a tropical cli¬
mate. Fourth Intern. Soil classification workshop Rwanda, 7 p.
Gautheyrou J and Gautheyrou M (1981) Point de charge zero des sols a allophane, a imogolite, vertisols
et oxisols de Guadeloupe et Martinique (Antilles framaises). ORSTOM-Antilles, notes laboratoire,
29 p.
Gustafsson JP (2001) The surface chemistry of imogolite. Clays and Clay Minerals, 49, 73-80.
Hendershot WH and Lavkulich LM (1979) The effect of sodium chloride saturation and organic matter
removal on the value of zero point of charge. Soil Sci., 128, 136-141.
Hendershot WH and Lavkulich LM (1983) Effect of sesquioxyde coatings on surface charge of standard
mineral and soil samples. Soil Sci. Soc. Am. J., 47, 1252-1260.
Parker JG, Zelazny LW, Sampath S and Harris WG (1979) A critical evaluation of the extension of zero
point of charge (ZPC) theory to soil systems. Soil Sci. Soc. Am. J., 43:668-673.
Parks GA and De Bruyn PL (1962) The zero point of charge of oxides. J. Phys. Chem., 66, 967-973.
Segalen P, Gautheyrou M, Guenin H, Caracho E, Bosch D and Cardenas A (1983) Etude d’un sol derive
de p6ridotite dans l’ouest de Cuba. Aspects physiques et chimiques (1). Cahiers ORSTOM, serie
Ptdologie, XX, 239-245.
Sparks DL (1986) Soil physical chemistry., CRC, Boca Raton, 308 p.
Sposito G (1981) The operational definition of the zero point charge in soils. Soil Sci. Soc. Am. J., 45,
292-297.
Sparks DL (1986) Soil physical chemistry., CRC, Boca Raton, 308 p.
Sposito G (1984) The surface chemistry of soils., Clarendon Oxford, 234 p.
Sposito G (1989) The chemistry of soils., Oxford University Press, 277 p.
Uehara G and Gillman G (1981) The mineralogy, chemistry and physics of tropical soils with variable
charge clays., Westview Tropical Agriculture Series, 4, 170 p.
Глава 21. Постоянные и переменные заряды
21.1. Введение
Термины «постоянные и переменные заряды» были введены Колеманом с сотр. (Coleman
и др., 1959) для характеристики зарядов, связанных с тремя основными группами ком¬
понентов почвы: глинами, оксигидроксидами и органическим веществом. Полученные
с тех пор результаты исследований больше не позволяют давать такое строгое определе¬
ние природе и происхождению этих зарядов. Например, глины способны нести заряды
обоих типов одновременно. Переменные заряды доминируют в некоторых типах тропи¬
ческих почв, таких как андисоли, тогда как в почвах умеренных регионов преобладают
постоянные заряды.
Хорошо окристаллизованные глинистые минералы имеют нулевой или отрицательный
теоретический заряд, который расположен в кристаллической решетке структуры, и пе¬
ременный заряд, расположенный на краевых группах. Отрицательный заряд зависит от
природы глины и уровня замещения катионов в тетраэдрических и октаэдрических сло¬
ях. Например А13+ может замещать Si4+ в тетраэдрическом слое (например бейделита,
нонтронита и сапонита), или Mg2+ может замещать А13+ в октаэдрическом слое (напри¬
мер монтмориллонита или иллита). Эти изоморфные замещения приводят к дефициту
положительных зарядов и, следовательно, избытку отрицательных зарядов, который
будет скомпенсирован внешними или внутрислоевыми катионами. Эти заряды непо¬
средственно связаны с постоянными зарядами и не зависят от pH или ионной силы.
В 1:1 глинистых минералах типа каолинита, которые содержат тетраэдрический слой
и слегка деформированный октаэдрический слой, структура электрически нейтраль¬
на. Изоморфные замещения А13+ на Fe3+ в октаэдрическом слое редки. Таким образом,
каолиниты имеют незначительный постоянный заряд, который означает, что при от¬
сутствии структурных разупорядочений результаты определения емкости катионного
обмена (ЕКО) не будут сильно искажены колебаниями pH.
Разрушение атомной решетки выявляет нарушения целостности, которые не ском¬
пенсированы на гидроксилированных поверхностях и способны подвергаться иониза¬
ции (рис. 21.1). Эти краевые заряды зависят от pH и площади удельной поверхности.
Атомы А13+ в тетраэдрическом окружении и группы Si-OH могут диссоциировать при
pH около 7.
Рис. 21.1. Алюминольные и силанольные группы в каолините (Sposito, 1984)
540
Часть 3. Неорганический анализ
В четырехводном галлуазите, который содержит слои межпакетной воды, значение
ЕКО выше и может быть изменено за счет катионов с низкой энергией гидратации.
В глинистых минералах типа 2:1 некоторые филлосиликаты, такие как пирофиллит
или тальк, содержат октаэдрический слой (А13+ или Mg2+), заключенный между двумя
тетраэдрическими слоями Si4+, образуя электрически нейтральную единицу. С другой
стороны, смектиты, которые образуются аналогичным образом после замещения иона
Mg2+ ионами А13+ в октаэдрическом слое, имеют дефицит положительных зарядов (или
избыток отрицательных зарядов), который должен быть компенсирован межслоевы¬
ми катионами. Расположение атомов кислорода на поверхности глинистых минералов
типа 2:1, называемых «силоксаном», описывается деформированной гексагональной
симметрией. Они образуют полость размером около 2,6 А, ограниченную шестью орби¬
талями гексагональной симметрии, и ее реакционная способность связана с зарядами,
распределенными в структуре слоев. При отсутствии изоморфных замещений и, сле¬
довательно, дефицита положительных зарядов, силоксановая полость служит донором
электронов, способным образовывать нестабильные комплексы. Если же, напротив,
существует изоморфное замещение А13+ на Mg2+ или Fe2+ в октаэдрическом слое, из¬
быточный заряд расположен близко к поверхностным атомам кислорода, и комплексы
стабильны.
В вермикулитах, образованных из триоктаэдрического талька, Mg2+ замещен на А13+
в тетраэдрическом слое Si4+, создавая отрицательные структурные заряды, которые ком¬
пенсируются межслоевыми катионами й т. д.
Все эти изъяны проявляются в пределах структурной единицы, и отсутствие балан¬
са зарядов может рассматриваться как обычное явление. Концентрации электролитов,
заряд противоиона и потенциал двойного слоя не изменяют поверхностный заряд. На
практике этот постоянный заряд может быть связан с обменной емкостью, измеряемой
при pH почвы в почвах умеренных регионов, поскольку переменные заряды обычно
малы.
Постоянные заряды 1С и переменные заряды 1п составляют характерный поверхност¬
ный заряд, который отражает степень выветривания почвы и присутствия органическо¬
го вещества и оксидов:
Ес + Еп = характерный поверхностный заряд почвы.
Оксиды, гидроксиды и оксигидроксиды в свободном состоянии или на поверхности
кристаллических решеток, а также алюмосиликаты с упорядочением ближнего порядка
имеют гидроксилированную поверхность, способную адсорбировать протоны Н+ или
гидроксиды ОН-. Кислотная среда приводит к избыточной адсорбции ионов Н+, а ще¬
лочная — ионов ОН- (рис. 21.2). Эти ионы также называют потенциал-определяющими
ионами (ПОИ).
При данном значении pH поверхностный потенциал постоянен, но поверхностный
заряд зависит от многих факторов, включая: (1) pH почвенного раствора, (2) природу
электролита, валентность составляющих его частиц и концентрацию, (3) диэлектриче¬
скую проницаемость среды, (4) температуру и условия измерений по сравнению с при¬
родными условиями. Точка нулевого чистого заряда соответствует pH, при котором
плотность положительных зарядов равна плотности отрицательных зарядов.
Переменный заряд обычно развивается строго при pH, равном или превышающем
7,0, но эта величина pH — эмпирическая. Переменные заряды являются поверхност¬
ным явлением и поэтому связаны с поверхностью коллоидов, а не с их абсолютной
концентрацией. В условиях выветривания переменные заряды могут развиваться при
Глава 21. Постоянные и переменные заряды
541
а)
ОН,
\)Н2
+ н
м-
/
он,
+ ОН'
м
/
он
\
он
\
он
Ь)
Твердое
вещество
лО
'/I \Ьон
Г\1/°н>1
лк
А ОН2
1
3+зн!> /
^ /
Протони- /
' 6 он
/|\ж
^Ой-
■ \|/°'
/к,
' О °
^ 1 -
/ |\>н2
рование
Гидроксилированная
F^
/,\>
. 1
поверхность
1
Поверхность раздела
твердой фазы
и раствора
3-
Кислая область pH
Щелочная область pH
Рис. 21.2. Поверхностный заряд гидроксилированных материалов: а — диаграмма Паркса-де
Брайна (Parks и de Вгиуп, 1962); Ь — гематит, оксид железа (Uehara и Gillman, 1981)
неконгруэнтном растворении кристалла или, наоборот, во время перехода из аморфного
состояния в кристаллическое.
В алюмосиликатах аллофанового типа емкость катионного обмена в щелочной сре¬
де может достигать больших значений. Постоянный заряд связан с Al(IV)1, тогда как
за образование переменного заряда отвечает гексакоординированный А1 (Fieldes, 1962;
Fripia, 1964).
Положительные заряды уменьшаются с ростом pH и могут быть измерены на осно¬
вании анализа с использованием ДЦБ (подраздел 6.2.2 в гл. 6), а также, возможно, диф¬
ференцированы с использованием соотношений Fe:Ti и Fe:Mn, если положительные за¬
ряды образуются при изоморфном замещении Ti4+ или Мп4+ в структурах оксида Fe3+.
В органическом веществе присутствие ионизуемых функциональных групп может
приводить к значительным общим отрицательным зарядам на этих группах. Карбок¬
сильные НООС- и фенольные ОН-группы, которые являются самыми активными, об¬
разуют переменные отрицательные заряды, и емкость катионного обмена может дости¬
гать величины 4 моль(+)/кг. Таким образом, органическое вещество играет главную роль
в поверхностных горизонтах и участвует в буферном эффекте почвы (регулирование ве¬
личины pH и концентраций обменных катионов в почвенном растворе).
ГУмусЦ* + почвенный раствор-Са2+ -> гумус-Са2+ + 2Н+ (почвенный раствор).
Неионные органические структуры также могут реагировать за счет сил Ван дер Ва-
альса, которые приводят к образованию только слабых связей, например, посредством
молекул, находящихся на очень близких расстояниях (около одного ангстрема).
Однако очень большие неполярные полимерные гуминовые молекулы не могут при¬
тягивать молекулы воды, что объясняет гидрофобность органического вещества. Таким
образом, в некоторых почвах необходимо использовать пробы естественной влажности.
‘В тетраэдрической позиции. — Примеч. науч. ред.
542 Часть 3. Неорганический анализ
Гумифицированное органическое вещество реагирует с почвенными минералами и об¬
разует агрегаты.
(В+) + (М+=) -► В+= + М+,
где В+ — органическая молекулярная единица в водном растворе; М+= — одновалент¬
ный обменный катион, связанный с почвенными коллоидами; В+= — органическая мо¬
лекулярная единица, связанная с глинистым минералом; М+ — одновалентный катион
в водном растворе.
Органические молекулярные единицы, участвующие в обмене катионов и протонов,
содержат функциональные группы, такие как карбоксильные, карбонильные и амино,
ароматические или гетероциклические азотсодержащие группы. Обмен анионов также
может происходить с карбоксильными группами как лигандами за счет образования бо¬
лее прочных связей.
21.2. Основные методы определения
21.2.1. Измерение переменных зарядов
Основные положения
При рНО, т. е. в точке пересечения кривых титрования в электролите с переменной кон¬
центрацией, емкость катионного обмена, эквивалентную содержанию переменных за¬
рядов, определяют по графику, показывающему увеличение плотности отрицательных
зарядов в диапазоне между pH почвы и рНО (Uehara и Gillman, 1981) (рис. 21.3).
Рис. 21.3. Определение переменных зарядов оп
Реагенты
Хлористоводородная кислота НС1 0,1 М
Гидроксид натрия NaOH 0,1 М
Хлорид калия (М =74,56) 2 М.
543
Глава 21. Постоянные и переменные заряды
Методика
• Помещают 4 г почвы (эквивалент количества почвы, высушенной при 105 °С),
просеянной через сито 0,5 мм, в каждую из 11 центрифужных пробирок на 50 мл
(табл. 21.1).
• Добавляют 10 мл дистиллированной воды и гомогенизируют.
• Добавляют возрастающие количества 0,1 М раствора НС1 в пробирки 1—5 (табл.
21.1) и такие же количества 0,1 М раствора NaOH в пробирки 7-10.
• Пробирку 6 используют для контроля.
• Доводят объемы до 20 мл, добавляя воду.
• Закрывают пробирки и оставляют стоять на 4 сут при периодическом перемеши¬
вании.
• Измеряют pH в каждой пробирке: рН1 (водн).
• Добавляют 1 мл 2 М раствора КС1 в каждую пробирку.
• Перемешивают в течение 3 ч и измеряют pH в каждой пробирке: рН2 (КС1).
• Строят кривые титрования для рН1 и рН2 с поправкой на холостой опыт, если не¬
обходимо.
Таблица 21.1. Экспериментальное определение переменного заряда
Холостые
Номера пробирок
XI Х2
1
2
3
4
5
6
контроль
7
8
9
10
11
Вода, мл
10
10
10
10
10
10
10
10
10
10
10
0,1 МНС1
0,1 М NaOH
НС1 или
NaOH, мл
0,5
1.0
2,0
3,0
4,0
0,5
1,0
2,0
3,0
4,0
Вода, мл
9,5
9,0
8,0
7,0
6,0
10
9,5
9,0
8,0
7,0
6,0
Общий объем 20 мл
Пересечение двух кривых дает значение рНО. Переменная емкость катионного
обмена определяется плотностью зарядов между рНО и рНводн почвы (рис. 21.3).
21.2.2. Определение постоянных зарядов
Основные положения
Определение постоянного заряда ас в почве с переменными и постоянными заряда¬
ми основано на адсорбции ионов при рНО. В этой точке существует равная адсорбция
катионов и анионов на поверхностях с переменными зарядами, при этом плотность
заряда равна нулю (Uehara и Gillman, 1981).
Любая избыточная адсорбция катионов или анионов при рНО представляет меру по¬
стоянного отрицательного или постоянного положительного заряда, соответственно.
Предварительная обработка удаляет селективно адсорбированные ионы и делает среду
гомоионной.
Реагенты
• 1 М раствор хлорида калия (КС1, М = 74,56);
• 0,2 М раствор хлорида калия;
544
Часть 3, Неорганический анализ
• 0,01 М раствор хлорида калия;
• 0,002 М раствор хлорида калия;
• 0,1 М раствор хлористоводородной кислоты (НС1);
• 0,1 М раствор гидроксида калия (КОН);
• 0,1 М раствор гидроксида натрия (NaOH);
• 0,5 М раствор нитрата аммония (NH4N03, М = 80,04).
Методика
Предварительная обработка почвы
• Эта обработка удаляет селективно адсорбированные ионы, такие как SO*-.
• Отвешивают 100 г воздушно-сухой почвы (эквивалент массы почвы, высушенной
при 105 °С), просеянной через сито 0,5 мм.
• Добавляют 500 мл 1 М раствора КС1 и доводят pH до 7,5, добавляя 0,1 М раствор
КОН. Оставляют стоять на 1 ч и сливают надосадочную жидкость.
• Снова добавляют 500 мл 1 М раствора КС1 и повторяют обработку дважды.
• Промывают остаток дистиллированной водой до тех пор, пока проводимость жид¬
кой фазы не станет равна проводимости 0,002 М стандартного раствора КС1.
• Высушивают остаток на воздухе и просеивают сквозь сито 0,5 мм.
Измерение 1с
• Отвешивают 4 г порции предварительно обработанной почвы (эквивалентные мас¬
се почвы, высушенной при 105 °С) и помещают их в стаканчики на 50 мл.
• Измеряют рНО так же, как при определении переменных зарядов (см. подраздел
21.2.1); насыщение ионами К+ и удаление селективно адсорбированных ионов мо¬
гут изменить величину рНО.
• Вынимают остаток почвы. Используют пробирку с pH, самым близким к рНО, для
определения постоянных зарядов.
• Промывают остаток 20 мл 0,2 М раствора КС1 и переносят в центрифужную про¬
бирку на 50 мл.
• Перемешивают в течение 1 ч, центрифугируют и отбрасывают надосадочную жид¬
кость.
• Добавляют 20 мл 0,01 М раствора КС1 и доводят pH до величины, равной получен¬
ному рНО, добавляя 0,1 М раствор НС1 или NaOH.
• Оставляют взаимодействовать в течение 1 ч.
• Центрифугируют и сливают надосадочную жидкость, в которой определяют содер¬
жание ионов К+ и С1“: величины К\ и CI7 (ммоль/мл).
• Взвешивают пробирку, содержащую отцентрифугированный остаток для опреде¬
ления удержанного объема 0,01 М раствора КС1 (V, мл).
• Переводят в раствор ионы К+ и С1“, адсорбированные на отцентрифугированном
остатке, промывая его пять раз 20 мл 0,5 М раствора NH4N03.
• Смешивают пять фильтратов и определяют содержание ионов К+ и С1“: величины
К+2 и С1 ■ (ммоль/мл).
Расчет
Адсорбированный К+ (ммоль(+)/кг): = 25(100 К+2- К+ V).
Глава 21. Постоянные и переменные заряды
545
Адсорбированный С1“ (ммоль(-)/кг): = 25(100 СГ2- C1+V).
Постоянный заряд = адсорбированный К+ - адсорбированный С\~.
Примечания
• Ионы К+ и С1“ обычно определяют методами пламенной эмиссионной спектроме¬
трии и адсорбционной колориметрии, соответственно (Рати и др., 2001).
• Сумма постоянных и переменных ионов примерно соответствует емкости катион¬
ного обмена, определенной с использованием аЦетатно-аммонийного буфера с pH
7,0 (см. гл. 22).
Использованная литература
Coleman NT, Weed SB and McCracken RJ (1959) Cation exchange capacity and exchangeable cations in
Piedmont soils of North Carolina. Soil Sci. Soc. Am. Proc., 23, 146-149.
Fripia JJ (1964) Surface properties of Alumino-silicates. Clays Clay Miner., 12, 327.
Pansu M Gautheyrou J and Loyer JY (2001) Soil Analysis - Sampling, Instrumentation and Quality con¬
trol. Balkema, Lisse, Abington, Exton, Tokyo, 489 pp.
Parks GA and De Bruyn PL (1962) The zero point of charge of oxides. J. Phys. Chem., 66, 967-973.
Sposito G (1984) The surface chemistry of soils., Oxford-Clarendon Press, Oxford, 274 p.
Uehara G and Gillman G (1981) The mineralogy, chemistry and physics of tropical soils with variable
charges clays. West view trop. Agric. Sen, 4, 170 p.
Дополнительная литература
Bortoluzzi EC, Tessier D, Rheinheimer DS and Julien JL (2005) The cation exchange capacity of a sandy
soil in southern Brazil: an estimation of permanent and pH-dependent charges. Eur. J. Soil Sci.,
doi: 10.1111/j. 13652389.00746.x.
Coleman NT, Weed SB and McCracken RJ (1959) Cation exchange capacity and exchangeable cations in
Piedmont soils of North Carolina. Soil Sci. Soc. Am. Proc., 23,146-149.
Conyers MK, Helyar KR and Poile GJ (2000) pH buffering: the chemical response of acidic soils to
added alkali. Soil Sci., 165, 560-566.
Zelazny LW, Liming He and An Vanwormhoudt (1996) - Charge analysis of soils and anion exchange.
In Methods of soil analysis, part 3, Chemical methods, Bigham JM and Bartels JM ed. SSSA-ASA,
Madison, WI Etats-Unis, 1231-1253.
Julien JL and Turpin A (1999) Reactive surfaces and the reasoning of a few chemical characteristics of
acid soils. Comptes-Rendus-de-l Academied Agriculture-de-France, 85, 25-35.
Van-Ranst E, Utami SR and Shamshuddin J (2002) Andisols on volcanic ash from Java Island, Indonesia:
Physico-chemical properties and classification. Soil Sci., 167, 68-79.
Глава 22. Обменные катионы
22.1. Введение
22.1.1. Обменные катионы в почве
Этот тип анализа также широко известен под названием «обменные основания», кото¬
рое было дано ему тогда, когда аналитическая формула выражалась в форме оксидов
(основные оксиды, такие как Кр, Na20; Брей и Уиллхайт (Bray и Willhite, 1929) выпол¬
няли выщелачивание почвы нейтральным раствором ацетата аммония с последующим
прокаливанием для испарения ацетата и превращения элементов в карбонаты и окси¬
ды), а также, возможно, для противопоставления с обменной кислотностью. Прежнее
название может показаться устаревшим, но оно еще часто используется.
Адсорбция обменных катионов в течение процессов старения представляет собой на¬
копление веществ на границе раздела твердой фазы и почвенного раствора, но не под¬
разумевает создания трехмерной молекулярной структуры, как в случае осаждения. Эти
ионы могут переноситься раствором электролита известного состава приданном pH.
В агрономии адсорбция обменных катионов и ЕКО имеют большое значение для
определения собственного плодородия почвы, ее способности удерживать удобрения,
элементов для питания растений и т. д. Адсорбированные катионы доступны для рас¬
тений, которые выделяют ионы Н+ на уровне их малых корней, контактирующих с по¬
чвенным раствором.
Удобрения, но также и поллютанты окружающей среды (такие как пестициды и ток¬
сичные катионы), удерживаются зарядами коллоидных поверхностей, что предотвраща¬
ет выщелачивание. Таким образом, поглощающий комплекс выполняет функции хра¬
нилища элементов и регулятора для ионов (неорганических и органических катионов
и анионов). Определение обменных катионов, присутствующих в почве в естественных
условиях, дает их сумму S(< ЕКО). Вместе с ЕКО (7), отношение S:T( степень насыще¬
ния почвы удобряющими элементами) можно оценить в данный момент времени, обыч¬
но в конце сельскохозяйственного сезона в зонах земледелия. На определение S влияют
многие факторы: содержание ионов Са2+, Mg2+, К+, Na+ может быстро изменяться под
воздействием удобрений, мелиорантов и растительного покрова; орошение может вы¬
зывать изменение содержания обменного натрия.
Другие ионы, вытесняемые более активными реагентами (например, Н+, А13+), и об¬
менные микроэлементы (например, Mn, Zn, Си, Ni, Со, Ti, U, Cs, Sr, Pb, V) не учитыва¬
ются в большинстве работ, за исключением исследований удобрений, токсичности или
загрязнения окружающей среды (например, удаление токсичных или радиоактивных
отходов, эвтрофикация грунтовых вод и рек).
Определение обменного кальция в известняковых или гипсоносных почвах не имеет
смысла из-за насыщения обменного комплекса и ошибок, связанных с растворимостью
карбоната кальция и гипса. Однако определение отношений Ca:Mg и Ca:Na и активного
кальция полезно (см. гл. 17).
В засоленных почвах обменный комплекс насыщен ионами натрия, и присутствие
растворимых солей делает невозможным определение содержания обменных катионов,
требуя использования других показателей, таких как доли адсорбированного натрия
(ДАН) и калия (ДАК) относительно кальция и магния (см. гл. 18). В почвах, характе¬
Глава 22. Обменные катионы
547
ризующихся перенасыщением ионами, сумма экстрагируемых катионов (S) стремится
к ЕКО (7).
Анализ обменных катионов, удерживаемых отрицательными зарядами глин, важен
для оценки скорости переноса ионных соединений по профилю почвы (выщелачива¬
ние) и позволяет исследовать кинетику обмена во времени и пространстве. Для массо¬
вых анализов времена обмена малы (несколько минут), но чтобы осуществить регидра¬
тацию набухающих глин (таких как монтмориллонит) с учетом медленных межслоевых
обменов (как в вермикулите), пробу часто оставляют взаимодействовать с реагентом на
ночь перед промыванием.
В случае иллитов ионы К+ легко попадают в гексагональные полости тетраэдрических
слоев и с трудом извлекаются оттуда. Вследствие этого, обмен больше не будет количе¬
ственным.
22.1.2. Экстрагирующие реагенты
Замещение обменных катионов зависит от природы замещающих катионов. Многова¬
лентные ионы теоретически более эффективны, чем одновалентные. Реагент должен
вытеснить обменные катионы как можно селективнее, но не быть слишком агрессивным
и не растворять вещества, не участвующие в процессах адсорбции-десорбции.
На практике сумму основных обменных катионов (Са2+, Mg2+, К+, Na+), экстрагируе¬
мых нормальным раствором ацетата аммония при pH 7,0, еще используют во многих ла¬
бораториях в качестве контроля для систем классификации и картографии. Катион NH*
представляет интерес, потому что он обычно не присутствует в значимых количествах
в обменном комплексе. Этот метод входит в стандарт AFA"31-108 (1992). Экстракцию
обменных катионов можно также сочетать с другими методами определения ЕКО (см.
гл. 26), как в международном стандарте N/^31-130 (1993).
В оксидных почвах с преобладанием двухвалентных катионов использование таких
катионов, как Ва2+, позволяет выполнять более полную экстракцию. Экстракция затруд¬
нена из-за адсорбции в слое Штерна, и, если эти заряды обменных катионов исполь¬
зуются, необходимо знать, существует ли корреляция с их доступностью для растений.
Для определения эффективной ЕКО (NFISO11260,1994) обменные катионы могут экс¬
трагироваться 0,1 М раствором ВаС12 при pH почвы, а при определении потенциальной
ЕКО (NFISO 13536,1995) — 1 М раствором ВаС12 в буферном растворе при pH 8,1.
Многие авторы, в том числе Оказаки с сотр. (Okazaki и др., 1962) и Гкллман с сотр.
(Gillman и др., 1982), сравнивали различные методы для экстракции обменных катионов.
В большинстве почв, даже почв с переменными зарядами, результаты в основном не за¬
висят от используемого метода экстракции, если pH не превышает 7,0. Однако следует
отметить, что содержание обменного К+ может оказаться несколько меньше, если ис¬
пользуют противоион Ва2+ вместо иона NH4+ (но не в случае ЕКО, см. гл. 26).
22.1.3. Оборудование
Для экстракции обменных катионов можно использовать мануальные или полуавтома¬
тические лабораторные системы (рис. 22.1):
• простое перемешивание со всеми экстрагирующими реагентами с последующим
разделением (декантацией, центрифугированием или фильтрацией) и определени¬
ем катионов в жидкой фазе;
• смешивание с многократным добавлением и отделением реагентов, что позволяет
осуществлять постоянное обновление реагентов:
548
Часть 3. Неорганический анализ
а)
Верхний цилиндр
“на 60 мл
. Экстрагирующий
реагент
6) Многократное добавление
экстрагирующего реагента
С)
^^^вр^^я-Подвижная рама
— Промежуточный цилиндр
Экстрагирующий
реагент
~ Проба почвы
Фильтр
зг
Нижний цилиндр
— для сбора
экстракта
Подвижная рама .
Неподвижная рама I
Ось вращения
Экстрагирующий
реагент
Рис. 22.1. Устройства для смешивания и разделения твердой и жидкой фаз: а — автоматическая
экстракция по Калра и Мейнарду (Ка/га и Maynard, 1991); Ь — фильтрование через фильтр: 1 — во-
ронка Бюхнера для фильтрования в вакууме, 2 — простая система фильтрования под действием
силы тяжести; с — взаимодействие и разделение центрифугированием; d — фильтрация через
колонку: 1 — закрытая колба, 2 — открытая колба, 3 — восходящая схема
• фильтрация на колонке с инертными фильтрующими добавками;
• прямое действие силы тяжести в системе с постоянной подачей реагентов типа
колбы Мариотта;
• система с восходящим потоком, позволяющая удалять пузырьки воздуха, способ¬
ные нарушить контакт между твердой и жидкой фазами;
• низковакуумная система, использующая оборудование с несколькими шприцами
для автоматически программируемой равномерной фильтрации.
При использовании колонок следует избегать локальных изменений скоростей (пред¬
почтительных или ламинарных потоков) и радиального градиента проницаемости. Ка¬
чество наполнения имеет очень большое значение; следовательно, количество образца
почвы должно быть ограничено для обеспечения удовлетворительной проницаемости,
но также для исключения чрезмерной концентрации фронта просачивания.
22.2. Метод экстракции ацетатно-аммонийным буфером
при pH 7
22.2.1. Основные положения
Почва насыщается катионами NH + в буферной (или не буферной) среде при pH 7,0.
Обменные катионы перемещаются и переходят в жидкую фазу, где они определяются
методом атомной абсорбции или спектрометрией с индуктивно связанной плазмой.
Глава 22. Обменные катионы
549
4
+ j^02 Раствор
Са2*
Этот метод дает надежные результаты для большинства почв, за исключением почв
с присутствием больших количеств извести, гипра, растворимых солей (солюбилизация)
и гидратированного вермикулита или слюд (ретроградация).
В небуферной среде экстрагируются обменные катионы при pH, близком к pH по¬
чвы. Метод используют для определения эффективной емкости катионного обмена (см.
раздел 26.2 гл. 26). В почвах с преимущественно переменными зарядами метод с исполь¬
зованием буферного раствора при pH 7,0 дает более высокие результаты, чем в небуфер¬
ном растворе. Обменный марганец можно определять в этой среде для проверки токсич¬
ности марганца.
В почвах, способных фиксировать калий, ионы NH4 могут частично замещать фик¬
сированные ионы К+, давая завышенные результаты для обменного К+.
22.2.2. Методика
Реагенты
• 1М буферный раствор ацетата аммония с pH 7
- Растворяют 77,08 г CH3COONH4 (марки ХЧ) в примерно 950 мл деионизирован¬
ной воды.
- Доводят pH раствора до 7,0, добавляя разбавленный раствор гидроксида аммо¬
ния или уксусной кислоты.
- Доводят объем раствора до 1000 мл деионизированной водой.
Или:
- Добавляют 58 мл ледяной уксусной кислоты (СН3СООН).
- Добавляют примерно 300 мл деионизированной воды, затем 71 мл раствора ги¬
дроксида аммония (марки ХЧ) плотностью 0,90.
- Охлаждают.
- Доводят pH раствора до 7,0, добавляя раствор гидроксида аммония или уксус¬
ной кислоты.
- Доводят объем раствора до 1000 мл деионизированной водой.
• 1Мраствор хлорида аммония
- Растворяют 53,5 г NH4C1 (марки ХЧ) в деионизированной воде и доводят объем
до 1000 мл (pH около 4,5-5,0).
Мануальный метод —традиционная методика
• Отвешивают 10 г (эквивалент массы почвы, высушенной при 105 °С) почвы, про¬
сеянной через сито 2 мм.
• Добавляют почву к 25 мл экстрагирующего реактива (ацетат или хлорид аммония)
в стаканчике на 100 мл.
• Гомогенизируют и оставляют взаимодействовать на ночь.
• Перемешивают и оставляют отстояться, затем фильтруют; повторяют эту операцию
трижды, оставляя взаимодействовать в течение 15 мин перед каждой экстракцией;
смешивают экстрагированные порции и доводят объем до 100 мл; гомогенизируют.
550
Часть 3, Неорганический анализ
• Определяют содержание ионов Са2+, Mg2+, К+, Na+ в фильтрате методом пламен¬
ной эмиссионной или атомно-абсорбционной спектрометрии или спектрометрии
с индуктивно связанной плазмой (Pansu и др., 2001). Определяют содержание тех
же элементов в холостом опыте (только с экстрагентом) и рассчитывают содержа¬
ние элементов.
Результаты выражают в смоль/кг почвы, высушенной при 105 °С.
В каждой серии определений проводят два контрольных опыта: стандартный кон¬
троль воспроизводимости во времени и повторное измерение случайно выбранной про¬
бы почвы для оценки воспроизводимости внутри серии.
Примечания
Международный стандарт NF X 31-108 (1992) рекомендует использовать только одно¬
кратное перемешивание и взаимодействие в течение 1 ч для проб и объемов, приведен¬
ных в табл. 22.1.
Таблица 22.1. Пробы почвы и объемы экстрагента, рекомендуемые в стандарте NFX31-108
(1992)
Проба, г
Объем экстрагента, мл
Объем сосуда, мл
5
100
125-150
10
200
250-300
В случае засоленных почв, если электропроводность превышает 0,5 мСм, почву про¬
мывают для удаления растворимых солей перед определением содержания обменных
катионов. Так как обменный комплекс насыщен Na+, то удаление растворимых солей
изменяет распределение обменных катионов.
В случае карбонатных и/или гипсосодержащих почв ацетат аммония может вызвать
растворение карбонатов или сульфатов кальция и магния.
При большом содержании известняка обменный комплекс может считаться насы¬
щенным ионами Са2+. Метод экстракции при pH 8,1 может уменьшить влияние раство¬
рения карбоната.
Другой подход использует двойное фильтрование: для обменных и солюбилизиро¬
ванных элементов на первом этапе и солюбилизированных элементов на втором этапе.
Обменные элементы определяют, рассчитывая разницу между двумя результатами.
Методику с использованием нормального раствора ацетата аммония при pH 7 можно
объединить с определением ЕКО вместе с NH + (см. гл. 26), для которого она является
начальным этапом насыщения.
22.3. Автоматизированный метод непрерывной экстракции
Использование методов динамического замещения позволяет анализировать явления
адсорбции и десорбции, используя колонки с почвой с фильтрацией восходящей под¬
вижной жидкой фазы соответствующей концентрации (рис. 22.2). Поток контролиру¬
ется и поддерживается постоянным при помощи перистальтического насоса, и прово¬
дится непрерывный анализ элюата (Gautheyrou и др., 1967-1981). Непрерывный поток
выбирают так, чтобы обеспечить быстрое достижение обменного равновесия.
Анализ данных позволяет исследовать законы равновесия обменных катионов. Коло¬
ночные методы были использованы для контроля кинетики обмена с учетом, например,
(1) минералогического состава глин и явлений выщелачивания, (2) емкости поглоще-
Глава 22. Обменные катионы
551
а)
Колонка дегазации
п
Анализ
1
2 <«-
3
Микроколонка 5 см
Проба + инертный
refecj фильтрующий
материал
Профильтрованный воз¬
дух или искусственная
газовая смесь
Реагент
Перистальтический
насос
Рис. 22.2. Пример динамической экстракции на колонке: а — автоматическая система непре¬
рывного анализа; Ь — пример катионного обмена при pH 7 с 1 М раствором СН3С001ЧН4: 1 — без
предварительного взаимодействия, 2 — после взаимодействия в течение 24 ч (Gautheyrou и др.,
1967)
ния калия, (3) химического переноса катионов и анионов (например, Sparks и др., 1980;
Sparks и Jardine, 1981; Schweich и др., 1983; van Genuchten и Parker, 1984; Carski и Sparks,
1985; ЗУнш’уидр., 1986; Jardine и др., 1988; Коо1идр., 1989; А/оо#идр., 1998; Соттипаг
идр., 2004).
Использованная литература
Carski ТН and Sparks DL (1985) A modified miscible displacement technique for investigating adsorp¬
tion-desorption kinetics in soils. Soil Sci. Soc. Am. J., 49,114-116.
Communar G, Keren R and FaHu Li (2004) Deriving boron adsorption isotherms from soil column dis¬
placement experiments. Soil Sci. Soc. Am. J.9 68:481-488.
Gautheyrou J and Gautheyrou M (1981) Contribution a l 'itude de la capacite dfechange des sols a
allophane. Aspects analytiques de la CEC et ses consiquences sur l ’interprdtation pedo-
agronomique., ORSTOM-Antilles, notes de laboratoire. Tome I: 276 p Tome II: 129 p Tome III:
profils (200 p).
Gautheyrou J and Gautheyrou M (1967) Dosage des cations echangeables du sol.9 ORSTOM-Antilles,
notes de laboratoire, 27 p.
552
Часть 3. Неорганический анализ
Gillman GP, Skjemstad JO and Bruce RC (1982) A comparison of methods used in Queensland for deter¬
mining cation exchange properties. CSIROAust. Div. Soils Tech. Pap., 44, 1-18.
Jardine PM, Wilson GV and Luxmoore RJ (1988) Modeling the transport of inorganic ions through undis¬
turbed soil columns from two contrasting watersheds. SoilSci. Soc. Am. J., 52: 1252-1259.
Kalra YP and Maynard DG (1991) Methods manual for forest soil and plant analysis., Forestry Canada,
Northern Forestry Center, Information report NORX-319, 15, 84-94.
Kool JB, Parker JC and Zelazny LW (1989) On the estimation of cation exchange parameters from col¬
umn displacement experiments. Soil Sci. Soc. Am. J., 53 1347-1355.
Moog HC, Streck T, Cammenga HK (1998) Modeling Ca/K exchange kinetics by montmorillonite and
vermiculite. Soil Sci., 163, 382—39.
NF X 31-108 (1992) D6termnation des cations Ca, Mg, K, Na extractibles par Г acetate d’ammonium. In
Qualite des sols, AFNOR, Paris, 1996, 59-65.
NF X 31-130 (1993) Determination de la capacity d’Schange cationique et des cations extractibles. In
Qualite des sols, AFNOR, Paris, 1996, 103—116.
NF ISO 11260 (1994) Determination de la capacite d’echange cationique effective et du taux de satura¬
tion en bases Echangeables к Г aide d’une solution de chlorure de baryum. In Qualite des sols, AF¬
NOR, Paris, 1996, 243-256.
NF ISO 13536 (1995) Determination de la capacite d’echange cationique potentielle et de la teneur en
cations echangeables en utilisant une solution tampon de chlorure de baryum к pH = 8.1. In Qualite
des sols, AFNOR, Paris, 1996, 293-303.
Okazaki R, Smith HW and Moodie CD (1962) Development of a cation-exchange capacity procedure
with few inherent errors. Soil Sci., 93, 343-349.
Pansu M, Gautheyrou J and Loyer JY (2001) Soil Analysis - Sampling, Instrumentation and Quality con¬
trol. Balkema, Lisse, Abington, Exton, Tokyo, 489 p.
Schweich D, Sardin N and Gaudet JP (1983) Measurement of a cation exchange isotherm from elution
curves obtained in a soil column: preliminary results. Soil Sci. Soc. Am. J., 47, 32-37.
Shaviv A, Jury WA and Pratt PF (1986) Exchange, fixation and precipitation of cations during leaching of
soils amended with manure: 1/ column experiments. Soil Sci., 141,237-243.
Sparks DL, Zelazny LW and Martens DC (1980) Kinetics of potassium desorption in soil using miscible
displacement. Soil Sci. Soc. Am. J., 44, 1205-1208.
Sparks DL and Jardine PM (1981) Thermodynamics of potassium exchange in soil using a kinetics ap¬
proach. Soil Sci. Soc. Am. J., 45, 1094-1099.
Van Genuchten MTh and Parker JC (1984) Boundary conditions for displacement experiments through
short laboratory columns. Soil Sci. Soc. Am. J., 48, 703-708.
Дополнительная литература
Hao XY and Chang C (2002) Effect of 25 annual cattle manure applications on soluble and exchangeable
cations in soil. Soil Sci., 167,126-134.
Holmgren GGS, Juve RL and Geschwender RC (1977) A mechanically controlled variable rate leaching
device. SoilSci. Soc. Am. J., 41, 1207-1208.
Kukier U, Sumner ME and Miller WP (2001) Distribution of exchangeable cations and trace elements in
the profiles of soils amended with coal combustion. Soil Sci., 166, 585-597.
Liu CL, Wang MK and Yang CC (2001) Determination of cation exchange capacity by one-step soil
leaching column method. Communications in Soil Science and Plant Analysis, 32, 2359-2372.
Luer В and Bohmer A (2000) Comparison between percolation and extraction with 1M NH4C1 solution
to determine the effective cation exchange capacity (CECeff) of soils. J. Plant Nutr. Soil Sci., 163,
555-557.
Ogwada RA and Sparks DL (1986) Kinetics of ion exchange on clay minerals and soil: evaluation of
methods. Soil Sci. Soc. Am. J., 50, 1158-1162 and 1162-1166.
Van Reuwijk LP (1987) Procedures for soil analysis., ISRIC, 9-1 к 9-11.
Глава 23. Обменная кислотность
23.1. Введение
23.1.1. Природа кислотности
Обменную кислотность, которая появляется в процессах генезиса почвы (например,
подзолизации), можно рассматривать как повреждение поверхностей обмена {Pedro,
1987). Глины могут подвергаться гидролизу (ацидолизу, ацидокомплексолизу), который
вызывает дестабилизацию кристаллических решеток типа 2:1, приводящую к переходу
некоторых катионов АР из октаэдрического слоя в обменные позиции. Таким образом,
содержание поверхностных обменных катионов снижается, и количество алюминия по¬
степенно превышает содержание отрицательных зарядов; pH почвы падает до 4,0, и не¬
конгруэнтное растворение становится возможным.
Кислотные дожди, которые сопровождают выбросы от сжигания горючих полезных
ископаемых, и неоднократное применение подкисляющих удобрений в севооборотах
могут ускорить ухудшение качества почвы. Например, при использовании аммонийных
удобрений в процессе нитрификации образуются протоны:
, NH 4+ + 3/202 -> N02- + Н20 +2Н+
NO-+ 1/202^N03-
Суммарное уравнение:
NH ; + 202 -> N0" + н20 + 2Н+
Если пробу обрабатывают небуферным раствором электролита, например, раствором
хлорида калия, то ионы А13+ вступают в реакцию обмена и переходят в раствор, где они
могут гидролизоваться с выделением протонов. Это сложные реакции. Согласно теории
Льюиса, ион А13+ являясь акцептором электронов, способен к гидратации:
А13+ +6(:ОН2) о [А1(:ОН2)6]3+
Затем происходит ступенчатая диссоциация в водной среде.
[А1(:ОН2)6]3+ + Н20 *+ Н30+ + [А1(Н20)50Н]2+
[А1(Н20)50Н]2+ + Н20 +» Н30+ + [А1(Н20)40Н2]+
Кроме того, А13+ может реагировать с анионами в зависимости от pH.
А13+ + ЗОН- А1(ОН)3
А1(ОН)3 + ОН- -н- [А1(ОН)4]-
[Al(OH)J- + ОН- о [А1(ОН)5]2-
[А1(ОН)5]2- + ОН* о [А1(ОН)6]3-
554
Часть 3. Неорганический анализ
Максимальное содержание мономерных форм наблюдается при низких значениях pH
(до pH 4,0), тогда как полимерные формы становятся преобладающими при pH около
4,5-4,7.
Органические соединения могут выделять протоны, и в кислых почвах, содержащих
восстановленные формы серы (мангровые органогенные почвы, подверженные пере¬
увлажнению), кислотность может быть результатом реакций окисления при контакте
с воздухом. Например, для пирита:
FeS2 + 7/202 + Н20 о Fe2+ + 2S02- + 2Н+
Fe2+ + V402 +5/2Н20 о Fe(OH)3 + 2Н+
При pH ниже 4,5 микробная активность Thiobacillus ferroxidans высока и степень под¬
кисления увеличивается по реакции:
FeS2 + 14 Fe3+ + 8Н20 о 15 Fe2+ + 2 SO2- + 16Н+
В восстановительной среде при pH ниже 5,5 ионы Мп2+ могут также обладать фито¬
токсичностью, которая добавляется к токсичности А13+.
Обменная кислотность отличается от общей кислотности, связанной с концентраци¬
ей ионов Н+ в почвенном растворе. Обменная кислотность представляет собой часть
потенциальной кислотности, которая включает более или менее ионизированные кис¬
лотные функции, слабые органические кислоты и легко обмениваемые катионы.
Потенциальную кислотность можно определить волюмометрически, нейтрализуя
заряды сильным основанием. Потенциальная кислотность является одним из основных
компонентов буферного эффекта почвы. Кислотность, рассматриваемую как «экстраги¬
руемая», определяют при pH 8,2 с раствором смеси ВаС12 и триэтаноламина, и это опре¬
деление можно объединять с измерением зарядов, которые зависят от pH.
23.1.2. Цели анализа
Для целей классификации почв считается, что 1 М КС1 экстракт содержит только об¬
менные формы А13+, А10Н2+, А1(ОН)+ и Н+, возможно без учета необменных солюбили¬
зированных продуктов (например, аморфный гель, гидроксиполимеры, гиббсит, фосфат
алюминия, Fe2+, Мп2+).
Актуальная кислотность почвы измеряется в молях протонов и может выражаться на
единицу массы (эту обменную кислотность иногда называют кислотностью, замещен¬
ной солями). На практике существенных количеств обменного алюминия не обнаружи¬
вается при pH выше 5,2, так как ионы А1 осаждаются при этом pH.
Обменный и необменный алюминий можно однозначно разделить выщелачиванием
сильными электролитами и построением кривых кумулятивной растворимости (Skeen
и Sumner, 1965). Поскольку количество растворенного необменного алюминия в про¬
цессе экстракции постоянно, сумму этих вкладов затем вычитают.
Обменную кислотность по КС1 используют для измерения эффективной емкости ка¬
тионного обмена, суммируя с обменными катионами, экстрагированными при pH по¬
чвы (Са2+, Mg2+, К+, Na+). Для учета влияния диффузного слоя необходима относитель¬
но высокая концентрация (1 М) экстрагирующего раствора.
Определение обменной кислотности полезно в агрономии для определения алюми¬
ниевой фитотоксичности, которая тесно коррелирует с долей обменного алюминия. Од¬
нако результат не определяет индекса токсичности: действительно, формы мономерного
и полимерного алюминия и ионная сила почвенного раствора влияют на биологическую
Глава 23. Обменная кислотность
555
активность алюминия, также как и присутствие фосфора и кальция в окружении корней
растений. Следует также учитывать генотип растений, так как некоторые растения бо¬
лее устойчивы, чем другие. В некоторых тропических почвах КС1, экстрагируемый А13+
не всегда коррелирует с рНводн. Некоторые почвы с pH около 4 могут содержать меньше
обменного алюминия, чем почвы с pH около 5. Корреляция с рНКС| более существен¬
на. Величина ДрН = рНКС1 - рНводн, если она существенна, может служить индикатором
минералогической нестабильности (с другой стороны, если она мала, pH почвы близок
к рНО (см. гл. 20)).
Отношение обменный А1:ЕКО (ЕКО, измеренная при pH почвы) позволяет оценить
риск фитотоксичности А1 или резистентность к А1 для данной культуры. Эспио и Пей-
роннель (Espiau и Peyronnel, 1976, 1977) предложили использовать долю обменной кис¬
лотности:
эффективная Т ’
где А — обменная кислотность, определенная при экстракции 1 М раствором КС1,
равная сумме А13+ + Н+ (смоль/кг); эффективная Т = эффективная ЕКО = ЭЕКО =
= А + l(K+, Na+, Са2+, Mg2+) (смоль/кг).
Степень ненасыщения (%) выражается формулой
100
T-S
т
где Г— емкость катионного обмена; S— сумма обменных катионов.
Во многих случаях достаточно измерить Т — *Упри pH почвы.
23.2. Метод определения
23.2.1. Основные положения
Пробу промывают небуферным 1 М раствором КС1, что позволяет экстрагировать
ионы, образующие обменную кислотность (Н+ и А13+). Определение выполняют
методом волюмометрии. Алюминий определяют волюмометрически или атомной
абсорбционной спектрометрией.
23.2.2. Реагенты
• 1 М раствор хлорида калия: отвешивают 74,56 г КС1; растворяют в примерно 950 мл
деионизированной воды; оставляют стоять для стабилизации температуры и дово¬
дят объем до 1000 мл.
• 0,025 М раствор гидроксида натрия: растворяют 4 г гранулированного NaOH в де¬
ионизированной воде и, после охлаждения, доводят объем до 1000 мл; титруют
стандартным раствором НС1; хранят в пластиковых бутылях, защищенных от С02
воздуха; раствор готовят не реже одного раза в неделю; можно использовать ти¬
трованные коммерческие растворы (фиксаналы), но их концентрация должна быть
проверена титрованием.
• 0,025 М раствор хлористоводородной кислоты: готовят из 0,1 М титрованного ком¬
мерческого раствора.
• Фенолфталеин: растворяют 100 мг фенолфталеина в 100 мл 15%-ного этанола.
• 1 М раствор фторида калия (или 40 г/л раствор менее растворимого фторида на¬
трия): растворяют 58,10 г KF в примерно 950 мл воды и доводят объем до 1000 мл.
556
Часть 3. Неорганический анализ
23.2.3. Методика
Влажность воздушно-сухой почвы определяют на отдельной пробе для пересчета ре¬
зультатов на почву, высушенную при 105 °С.
Экстракция
• Помещают 10 г почвы, просеянной через сито 2 мм, в стаканчик на 100 мл.
• Добавляют 20 мл 1 М раствора КС1 и оставляют взаимодействовать в течение 15 мин
при периодическом перемешивании.
• Фильтруют через бумажный фильтр с синей лентой.
• Собирают фильтрат в мерной колбе на 100 мл.
• Добавляют порциями примерно по 10 мл раствора КС1 и оставляют взаимодейство¬
вать в течение 15 мин после каждого добавления.
• После добавления последней порции доводят объем до 100 мл и гомогенизируют.
• Общая продолжительность экстракции должна быть стандартизована на уровне
120-150 мин.
Определение обменной кислотности
[Н+ + ОН->Н20]
[А13+ + ЗОН- -> А1(ОН)3]
• Помещают 25 мл аликвоту экстракта, описанного в подразделе 2.3.1, в коническую
колбу на 250 мл; добавляют три капли раствора фенолфталеина.
• Титруют 0,025 М раствором NaOH до светло-розового окрашивания смеси.
• Добавляют одну каплю фенолфталеина и ждут 1 мин; окраска должна оставаться
стабильной; не титруют до темно-розовой окраски, чтобы уменьшить осаждение
гидроксида алюминия.
• Проводят два холостых определения. Сохраняют экстракты для определения алю¬
миния титрованием.
Расчет
m(x-y)MAf
Обменная кислотность, смоль(Н+)/кг почвы = >
где w — масса воздушно-сухой почвы; х — объем (мл) раствора NaOH, использованный
для титрования; у — объем (мл) раствора NaOH, использованный для холостого опыта;
М — молярность раствора NaOH; Л — коэффициент аликвоты (=4);/— поправочный
коэффициент для выражения результатов на почву, высушенную при 105 °С.
Примечание
Полученный результат можно использовать для определения эффективной емкости ка¬
тионного обмена (ЭЕКО, см. гл. 26).
ЭЕКО = обменная кислотность, смоль(+)/кг, + обменные катионы (Са2+, Mg2+, К+, Na+).
Определение обменного алюминия
В зависимости от имеющегося лабораторного оборудования содержание обменно¬
го алюминия можно определять методом атомно-абсорбционной спектрометрии или
Глава 23. Обменная кислотность
557
спектрометрии с индуктивно связанной плазмой, либо титриметрией или автоматиче¬
ской колориметрией с непрерывным проточным анализом (Pansu и др., 2001).
Атомно-абсорбционная спектрометрия
• Готовят калибровочные растворы, содержащие: 0, 10, 20, 30, 40, 50 мг Al/л в 1 М
растворе КС1, из исходного раствора, содержащего 500 мг А1/л.
• Измеряют поглощение калибровочных и холостых растворов при 309,3 нм в пла¬
мени C2H2/N20 непосредственно после экстракции.
• Выражают результаты в смоль(-Г) алюминия/кг и вычитают из обменной кислот¬
ности, чтобы получить содержание обменного Н+:
обменная кислотность = Н+ + А13+ (смоль/кг).
Этот результат можно использовать для расчета ЭЕКО (см. гл. 26), различая А13+
и Н+.
Титриметрия
• К 25 мл раствора, использованного для определения обменной кислотности (см.
параграф «Определение обменной кислотности»), добавляют микрокаплю 0,025 М
раствора НС1, чтобы вернуться в состояние непосредственно перед концом титро¬
вания и разрушить розовую окраску.
• Добавляют 10 мл 1 М раствора фторида калия, чтобы связать алюминий в ком¬
плекс. В присутствий алюминия раствор опять станет розовым после реакции под¬
щелачивания:
А1(ОН)3 + 6KF-> I^AIF, + ЗКОН.
(Если раствор не стал розовым, нет необходимости продолжать, и можно считать,
что обменный алюминий отсутствует.)
• Титруют 0,025 М раствором НС1 до обесцвечивания. Ждут 1-2 мин и добавляют
одну каплю фенолфталеина для того, чтобы проверить устойчивость обесцвечива¬
ния. Количество добавленной кислоты соответствует количеству обменного алю¬
миния. Аналогичные операции проводят с холостыми растворами. Они не должны
поглощать НС1. Разница между обменной кислотностью и обменным алюминием
дает содержание обменных протонов.
Расчет
100 VMAf
Обменный алюминий, смоль (1/ЗА13+)/кг почвы = —“—>
где w — масса воздушно-сухой почвы; V— объем, мл раствора НС1, использованный для
титрования; М — молярность раствора НС1; А — коэффициент аликвоты (= 4);/— по¬
правочный коэффициент для приведения результатов к почве, высушенной при 105 °С.
Обменный Н+ (смоль/кг) = обменная кислотность (смоль/кг) - обменный А13+
(смоль( 1 /ЗА13+)/кг).
Наблюдения
• Приведение результатов к почве, высушенной при 105 °С, особенно необходимо
для почв, содержащих большую долю оксидов и гидроксидов или алюмосиликатов
аллофанового типа, из-за очень непостоянной, но высокой остаточной влажно¬
558 Часть 3. Неорганический анализ
сти воздушно-сухих почв. Эта форма представления позволяет сравнивать данные
с результатами других анализов.
• Присутствие оксида железа может привести к ошибкам: если экстракт окрашен
железом, может понадобиться определить содержание железа одновременно со
спектрометрическим определением алюминия.
Присутствие растворимого органического вещества может помешать волюмометри-
ческому определению.
23.3. Другие методы анализа
Дефицит заряда может изменяться в зависимости от экстрагирующего раствора (при¬
роды и концентрации электролита, pH и др.). Лучшим методом была бы экстракция при
pH почвы в поле с использованием жидкой фазы, идентичной почвенному раствору.
Были исследованы многие солевые растворы для селективного определения обмен¬
ного алюминия: небуферные растворы различных концентраций, реагирующие при pH,
близком к pH почвы, или буферные растворы при различных pH.
К небуферным солям относятся: КС1 (Yuan, 1959), NaCl и ВаС12 (McLean и др., 1959),
NH4C1, MgCl2, СаС12 {Skeen и Sumner, 1967), LaCl3 {Bloom, 1979), CuCl2 {Juo и Kamprath,
1979). К буферным солям относятся ацетаты К, Na, La, Си с концентрациями от 0,2 до
1 или 2 М.
Все эти реагенты позволяют экстрагировать различные формы А13+, называемые, на¬
пример, обменным А1, А1, связанным с органическим веществом, межслоевым А1 или
необменным полимерным гидрокси-А1.
Метод с использованием КС1, описанный в разделе 23.2, прост в исполнении и, по-
видимому, имеет лучшие метрологические характеристики, но метод с солью бария
может использоваться для более разнообразных исследований {Pratt и Bair, 1961; Skeen
и Sumner, 1965; Vermeulen и др., 1975; Espiau и Peyronnel, 1976; Gillman, 1979; Espiau
и Pedro, 1980). Поэтому определение обменной кислотности в экстракте хлорида бария
было предложено для международного стандарта NFISO14254 (1997) как метод, позво¬
ляющий также определять эффективную емкость катионного обмена и содержание об¬
менных катионов {NFISO11260 1994).
Использованная литература
Bloom PR (1979) Titration behavior of aluminum organic matter. Soil Sci. Soc. Am. J., 43, 815-817.
Espiau P and Pedro G (1980) Caracterisation du complexe d’echange des sols acides. Le taux d’acidite
d’echange et sa signification p6dog6n6tique sous climat temper. Ann. Agron., 31,363-383.
Espiau P and Peyronel A (1976) L’acidity d’echange dans les sols. Methode de determination de
l’aluminium ^changeable et des protons 6changeables. Sci. du Sol., 3, 161-175.
Gillman GP (1979) A proposed method for the measurement of exchange properties of highly weathered
soils. Austr. J. Soil Res., 17, 129-139.
Juo ASR and Kamprat EJ (1979) Copper chloride as an extractant for estimating the potentially reactive
A1 pool in acid soils. Soil Sci. Soc. Am. J., 43, 35-38.
NF ISO 14254 (1997) Determination de l’acidite echangeable dans un extrait au chlorure de baryum,
AFNOR, Paris, X31^122.
NF ISO 11260 (1994) Determination de la capacite d’echange cationique effective et du taux de saturation
en bases echangeables а Г aide d’une solution de chlorure de baryum. In Qualite des sols, AFNOR,
Paris, 1996.
Pansu M, Gautheyrou J and Loyer JY (2001) Soil analysis - sampling, instrumentation and quality
control. Balkema, Lisse, Abingdon, Exton, Tokyo, 489 p.
Глава 23. Обменная кислотность
559
P&iro G (1987) G6ochimie, mineralogie et organisation des sols. Aspects coordom^s des probl6mes
p£dog6n£tiques. Cah. ORSTOMSer. Pedol., XXIII, 169-186.
Pratt PR and Bair FL (1961) A comparison of three reagents aluminium for the extraction of Aluminium.
Soil Sci., 91, 355-357.
Skeen JB and Sumner ME (1965) Measurement of exchangeable Aluminium in acid soils. Nature, 208,
712.
Дополнительная литература
Hissink DJ (1925) Base exchange in soils. Trans. Far. Soc., 551-617.
Jackson ML (1963) Aluminium bonding in soils: a unifying principle in soil science. Soil Sci. Soc. Am.
Proc., 27, 1-10.
Little I (1964) The determination of exchangeable aluminium in soils. Austr. J. Soil Res., 2, 76-82.
Rich Cl (1970) Conductometric and potentiometric titration of exchangeable aluminium. Soil Sci. Soc.
Am. Proc., 34, 31-38.
Sivasubramaniam S and Talibudeen О (1972) Potassium-aluminium exchange in acid soils. I - Kinetics.
J. Soil Sci., 23, 163-176.
Herbillon AJ (1974) Modifications des propri6t6s de charge provoqu£es par l’alt6ration chimique. Pedol.,
24, 100-118.
Rouiller J, Guillet В and Bruckert S (1980) Cations acides 6changeables et acidtes de surface. Approche
analytique et incidences pёdogёnёtiques. Sci. du Sol., 2, 161-175.
Herbillon AJ (1981) Degree of weathering and surface properties of clays. In Characterisation of soils
in relation to their classification and management, Greenland DJ ed. Oxford University Press, 5,
80-97.
Aleksandova AM, Krupskiy NK and Daragan YuV (1983) The nature of soil acidity. Pochvovedeniye, 3,
34—43.
Gillman GP and Sumpter EA (1985) KCl-extractable aluminium in highly weathered soils. Is it
exchangeable? Commun. Soil Sci. Plant Anal., 16, 561-568.
Logan Kab, Floate MJS and Ironside AD (1985) Determination of exchangeable acidity and exchangeable
aluminium in hill soils. Part I - Exchangeable acidity. Soil Sci. Plant Anal., 16, 301-308.
Wagatsuma T and Ezoe Y (1985) Effect of pH on ionic species of aluminium in medium and on aluminum
toxicity under solution culture. Soil Sci. Plant Nutr., 31, 547-561.
Manrique LA (1986) The relationship of soil pH to aluminum saturation and exchangeable aluminum in
ultisols and oxisols. Commun. Soil Sci. Plant Anal., 17,439-455.
Willoughby EJ (1986) A comparison of methods for measuring aluminium in KC1 extracts of soils.
Commun. Soil Sci. Plant Anal., 17, 667-677.
Wagatsuma T and Kaneko M (1987) High toxicity of hydroxy-aluminum polymer ions to plant roots. Soil
Sci. Plant Nutr., 33, 57-67.
Pansu M, Gavinelli R and Espiau P (1990) Etude de p^cision des mesures de l’acidit6 d^change par KC1
N dans les sols. In Actes Journees laboratoires, IRD (ex-Orstom), Paris, 114-126.
Bertsch PM and Bloom PR (1996) Aluminium. In Methods of soil analysis, part 3 Chemical methods,
Bigham JM and Bartels JM ed., SSSA, ASA, Madison WI, Etats-Unis, 517-550.
Coscione AR, Andrade JC de, Raij В van (1998) Revisiting titration procedures for the determination of
exchangeable acidity and exchangeable aluminum in soils. Communications-in-Soil-Science-and-
Plant-Analysis, 29, 1973-1982.
Derome J, Lindroos AJ (1998) Effects of heavy metal contamination on macronutrient availability and
acidification parameters in forest soil in the vicinity of the Harjavalta Cu-Ni smelter, SW Finland,
Environmental-Pollution., 99, 225-232.
Filep G and Filep T (1999) Characterization of forms of potential soil acidity. A potencialis talajsavanyusag
formainak jellemzese, Agrok Talajtan., 48, 33-48.
Dai KH and Richter DD (2000) A re-examination of exchangeable acidity as extracted by potassium
chloride and potassium fluoride. Commun. Soil. Sci. Plant Anal, 31, 115-139.
Глава 24. Потребность в известковании почв
24.1. Введение
24.1.1. Регулирование кислотности почв
Английский физик Дэви первым объяснил влияние известкования «нейтрализацией
почвенной кислотности» в 1813 г. Потребность в извести (особенно важна при земле¬
пользовании очень кислых почв) можно определить как часть зарядов, зависящих от
pH в диапазоне между естественным pH почвы и pH, необходимым для данной культу¬
ры. Если общая кислотность почвы превышает 15% емкости катионного обмена, могут
возникнуть проблемы фитотоксичности, связанные с:
• самой кислотностью для некоторых растений и микроорганизмов;
• алюминиевой (и марганцевой) токсичностью из-за угнетения роста корней;
• дефицитом основных элементов (вызванным например, выщелачиванием Са или
Mg) или микроэлементов.
Это явление играет большую роль в почвах с постоянными зарядами глин типа 2:1.
Его влияние можно уменьшить, добавляя мелиорирующие вещества, которые снижают
кислотность и увеличивают pH до уровня, необходимого для лучшего использования
земли.
С одной стороны, рост pH увеличивает скорости минерализации, а с другой — умень¬
шает содержание растворимого и обменного А13+ в формах полимеров гидроксиалюми-
ния (см. гл. 23). Эти полимеры повышают доступность фосфора для растений.
В зависимости от культуры оптимальное значение pH получают с учетом: (1) эконо¬
мических ограничений (минимальные дозы внесения и, следовательно, минимальная
стоимость для наилучшего результата), (2) устойчивости растений к А1-Мп, кислотно¬
сти и фитотоксичности, (3) минералогического состава глин и различных коллоидов,
(4) количества органического вещества, (5) климатических ограничений и сезонных ко¬
лебаний pH, (6) орошения, (7) выщелачивания и внесения протонов вместе с подкисля¬
ющими удобрениями.
Осаждение А13+ в виде А1(ОН)3 или A1(0H)S04 * 5Н20 (джурбанит) может быть вы¬
звано внесением карбоната кальция (известняка) или сульфата кальция (гипса). В этом
случае глина постепенно насыщается ионами Са2+, т. е. удобрение должно быть сбалан¬
сированным или, как говорится в старой французской пословице, «известкование без
унавоживания приносит неожиданное разорение».1
В почвах с переменными зарядами, в которых минералы стабильны при относительно
кислых значениях pH, при рНО наблюдается слабый буферный эффект (см. гл. 20), но
он возрастает с обеих сторон рНО. При известковании pH в этих почвах растет медлен¬
но, и исследования в диапазоне от pH 4 до pH 6 (который очень важен для агрономии)
будут достаточны, так же как достижение критического pH для осаждения А1(ОН)3, т. е.
5,2—5,5. Многие крупные хозяйства повысили выход продукции, повышая pH почвы до
этого уровня, поскольку тогда облегчается усвоение N, Р, К, Са, Mg, S, В и Mo. С другой
стороны, превышение pH 7,5 может привести к дефициту Р, В, Zn, Fe, но уменьшить
токсичность Мп.
1 По-французски: «0м/ chaulesansfumer, se mine sansypenser».
561
Глава 24. Потребность в известковании почв
Рост pH первоначально вызывает нейтрализацию центров Н+, а затем останавливает
образование обменного алюминия и, в конечном счете, вызывает образование полимеров
А1(ОН)2+ и А1(ОН)2, которые покрывают активные поверхности коллоидов и уменьшают
обменную кислотность. Кроме того, поглощающий комплекс насыщается ионами Са2*:
2СаС03 + 2Н20 -> Са(ОН)2 + Са(НС03)2
Глина(Н)4 + Са(ОН)2 + Са(НС03)2 -► Г)тина(Са2+)2 + 2Н20 + 2Н2С03
2Н2С03 -> 2С02 + 2Н20
Если pH почвы снижается на уровне корней растений (кислотная экссудация), то ка¬
тионы обменного Са2+опять переходят в раствор. В почвах, содержащих глинистые ми¬
нералы типа 2:1, кислотность обеспечивается присутствием алюминия, фосфор осажда¬
ется в форме фосфата алюминия. Известкование изменяет фиксацию фосфора, удаляя
растворимый А13+.
Если обменный А1 нейтрализован (полимерные, органические соединения А1), то
создаются центры, доступные для катионного обмена, и ЕКО несколько увеличивается.
24.1.2. Расчет необходимого количества извести
В лабораторных условиях потребность в извести выражают в миллиэквивалентах СаС03
на кг почвы, а в агрономии — в тоннах СаС03 на гектар. Первоначально, из-за недоста¬
точного знания сложности процессов обмена в различных почвах, агрономы системати¬
чески стремились достичь нейтральной реакции при корректировке pH почвы. В этом
случае потребность в извести можно было оценить как Т — S (где Т — полная емкость
катионного обмена в буферном растворе при pH 7, и 5 — сумма обменных катионов).
Мелих (Mehlich, 1939) считал, что потребность в извести находится в диапазоне между
обменной кислотностью, экстрагированной небуферным солевым раствором, и общей
кислотностью, нейтрализованной буферным раствором с pH 8,1. Оценка обменной
кислотности величиной Г - S (где Т измерена при pH почвы) также позволяет рассчи¬
тать количество кальция, необходимого для корректировки кислотности (Duchaufour
и Souchier, 1980).
Потребность в извести можно также оценить методами, аналогичными тем, что ис¬
пользуются для определения обменной кислотности (см. гл. 23) или измерения заряда
при известном рНО (см. гл. 20 и 21). Однако установление равновесия в почвах требует
длительного времени, и большая часть кислотности не определяется мгновенной реак¬
цией с основанием. На результаты аналитического определения влияют степень выве¬
тривания, содержания глин и органического вещества, формы кислотности и исходный
pH почвы. Таким образом, количество измельченного известняка, определенное в по¬
чвах из Конго в лабораторных условиях, не приведет к точно такому же увеличению pH
в естественных условиях (Djondo, 1995). На практике используют три следующих подхода:
• Анализ методом инкубации с известняком; это длительный метод, в котором лабо¬
раторные условия (влажность, температура, микробная активность) близки к по¬
левым условиям.
• Инкубация с непрерывно увеличивающимися количествами гидроксида кальция
(известковой воды) и анализ после взаимодействия в течение нескольких часов.
• Уравновешивание системы почва - буферный раствор, которое позволяет нейтра¬
лизовать кислотность без повышения pH почвы выше желаемого уровня, с учетом
почвенной матрицы и выращиваемых культур.
562
Часть 3. Неорганический анализ
Ранее для регулирования почвенной кислотности применяли методы с использова¬
нием известковой воды (стабильность которой изменяется случайным образом в зависи¬
мости от окружающей атмосферы) совместно с измерением pH в моменты ТО и Т48 (ч)
(Chaminade, 1933) (рис. 24.1). Эти методы практически вышли из употребления из-за
того, что они вызывают временное чрезмерное повышение pH. Методика, приведенная
в разделе 24.2, описывает метод уравновешивания с буферным раствором.
Рис. 24.1. Метод Шаминада (Chaminade, 1933). I — величины pH, определенные в момент
времени ТО; II — величины pH, определенные в момент времени Т48 (ч):
х = п q/(p -т + п) = СаО, % от массы почвы, необходимо для нейтрализации до pH 7
(СаССуСаО) = 100/56 = 1,786
24.2. Буферный метод Шумейкера-Маклина-Пратта
24.2.1. Основные положения
Метод был предложен Шумейкером, Маклином и Праттом (Shoemaker, McLean и Pratt,
1962) и иногда называется методом ШМП. Используют комплексный буферный раствор
с pH, близким к нейтральному и соответствующим равновесию карбонат - бикарбо¬
нат — С02 почвенного воздуха при нормальном давлении. Метод позволяет нейтрализо¬
вать и основания и кислоты, и не допускать колебаний pH в почвенной системе, которая
сама обладает буферной емкостью. Таким образом, почва не подвергается местному уве¬
личению pH, которое может быть слишком большим.
Величина pH раствора отражает уровень энергии связывания протонов, pH буферно¬
го раствора определяет потенциальную кислотность. Изменение pH буферного раствора
Глава 24. Потребность в известковании почв
563
позволяет количественно оценить потребность в известковании для почв с исходным
значением pH ниже 6,0 и высокой алюминиевой токсичностью.
г водн 7
Метод, описанный ниже — метод двойного буферного раствора (McLean, 1982; Sims,
1996), отчасти основанный на исходном методе ШМП (Shoemaker, McLean и Pratt, 1962)
уравновешивания в буферном растворе и отчасти — на дополнении к этой методике
(McLean и др., 1978).
24.2.2. Реагенты
Буферный раствор ШМП с pH 7,5
• Отвешивают 3,24 г я-нитрофенола C6H5N03 (М = 139,11).
• Отвешивают 5,40 г хромата калия К2СЮ4 (М = 194,20).
• Отвешивают 95,58 г дигидрата хлорида кальция СаС12 * 2Н20 (М = 147,03).
• Помещают реагенты в колбу из пирекса на 2 л (с градуировочной меткой на
1800 мл), содержащую около 900 мл воды. Перемешивают, переворачивая бутыль
вверх дном и обратно в течение 5 мин для предотвращения слеживания -> рас¬
твор А.
• Отвешивают 3,60 г ацетата кальция Са(СН3СОО)2 * Н20 (М = 176,18) и растворяют
в 500 мл деионизированной воды -> раствор В.
• Смешивают раствор А с раствором В -> раствор С; встряхивают на роторном шей¬
кере в течение 3 ч.
• Добавляют 4,5 мл триэтаноламина N(CH2—СН2ОН)3 (М = 149,19) и встряхивают на
роторном шейкере в тёчение 8 ч -> раствор D.
• Доливают до 1800 мл и доводят pH до 7,5 с помощью 0,2 М раствора NaOH при
контроле рН-метром.
• Фильтруют и защищают от атмосферного углекислого газа ловушкой, сделанной
из трубки, наполненной аскаритом (асбест, пропитанный гидроксидом натрия)
между двумя трубками с Drierite или ангидритом (безводный сульфат кальция); ис¬
пользуют вскоре после приготовления.
24.2.3. Методика
В герметически закрывающейся центрифужной пробирке объемом 50 мл:
• Отвешивают 5 г (эквивалентный массе почвы, высушенной при 105 °С) воздушно¬
сухой почвы, просеянной через сито 0,5 мм.
• Добавляют 5 мл воды.
• Перемешивают и оставляют взаимодействовать в течение 1 ч.
• С помощью pH-метра, проградуированного в диапазоне pH от 7,0 до 4,0, опреде¬
ляют pH почвенной суспензии, перемешивая электродом жидкую пасту до полу¬
чения стабильных показаний (см. гл. 15) -> рН^.
• Добавляют 10 мл буферного раствора ШМП к суспензии и перемешивают в тече¬
ние 15 мин; оставляют стоять на 15 мин, затем перемешивают до стабилизации
показаний и измеряют pH суспензии pHt смеси почва - буферный раствор; это
pH суспензии перед добавлением какой-либо кислоты.
• Добавляют к суспензии аликвоту раствора НС1, необходимую для изменения pH
10 мл буферного раствора с 7,5 до 6,0 (1 мл 0,206 М НС1 = 0,206 ммоль).
• Перемешивают в течение 10 мин, оставляют стоять на 30 мин и измеряют pH су¬
спензии при перемешивании -> рН2 смеси почва - буферный раствор; это конеч¬
564
Часть 3. Неорганический анализ
ный pH смеси почва — буферная суспензия после добавления хлористоводородной
кислоты.
Кислотность «А», выраженную в ммоль Н+ для 5 г почвы, находят по формуле:
А = ДрН2
ДрН,
ч
А4°
ДрН?
-ЛрН2
д4° ¥ 6,5-рН; v
дрН? А РН, -РН2 J
где ДрН, = 7,5 - pH,; ДрНг = 6,0 - рН2;
Ad,
i
ДрН?
изменение кислотности при изменении
pH на единицу в 10 мл буферного раствора с pH 7,5, полученное при титровании стан-
ДЛ°
дартным раствором НС1, т. е. примерно 0,137 ммоль на единицу pH;
ДрН®
изменение
кислотности при изменении pH на единицу в 10 мл буферного раствора с pH 6, т. е. при¬
мерно 0,129 ммоль на единицу pH; 6,5 — желаемое значение pH почвы после известко¬
вания (можно выбрать и другое значение pH).
Потребность в известковании (ПИ), определенную в лабораторных условиях, можно
выразить следующим образом:
ПИ, смоль(+)/кг = 1,69 (20А) - 0,86 = 33,8А - 0,86;
а в полевых условиях, на глубине 20 см:
ПИ, т/га = 45,5/4- 1,16.
24.2.4. Примечания
Этот метод определяет общее содержание ионов Н+ с учетом растворимого и обменного
алюминия. Он может применяться для почв с pH ниже 5,8-6,0, которые требуют внесе¬
ния извести в количествах, превышающих 1 т/га (на глубине 20 см).
Содержание органического вещества не должно быть более, чем 15%. Обычно нет
необходимости проводить эти измерения на почвах с pH выше 6,0, потому что в этом
случае почва требует не известкования, а, возможно, кальцийсодержащего удобрения.
Почвы с большим содержанием органического вещества значительно улучшаются при
внесении извести, только если их pH ниже 5,3.
Метод Адамса и Эванса (Adams и Evans, 1962) также является экспрессным методом
уравновешивания в буферном растворе (pH 8,0), его часто используют для песчаных
почв или почв с низким содержанием органического вещества.
Использованная литература
Adams F and Evans СЕ (1962) A rapid method for measuring lime requirement of red-yellow podzolic
soils. Soil Sci. Soc. Am. Proc., 26, 355-357.
Chaminade R (1933) Mode d’action de la chaux sur les sols et correction de leur activity. Ann. Agron.,
453-477.
Davy H (1813) Elements of agricultural chemistry., Longman 6d.
Djondo MY (1995) Proprietis d'echange ionique des sols ferrallitiques argileux de la vallee du Niari
et sableux du plateau Mbe-Bateke au Congo — application a la correction de leur aciditi., Th£se,
Document IRD (ex-Orstom) Montpellier, France, 5.
Mclean EO, Trieweiler JF and Eckert DJ (1977) Improved SMP buffer method for determining lime
requirement in acid soils. Commun. Soil Sc. Plant Anal., 8,667-675.
McLean EO, Eckert DJ, Reddy GY and Trierweiler JF (1978) An improved SMP soil lime requirement
method incorporating double-buffer and quick-test features. Soil Sci. Soc. Am. J., 42, 311-316.
Глава 24. Потребность в известковании почв
565
McLean ЕО (1982) Soil pH and lime requirement. In Methods of soil analysis. Part 2. Chemical and
microbiological properties 2nd edition), Page AL ed., Am. Soc. Agronomy., Soil Sci. Soc. Am., 199—
224.
Shoemaker HE, McLean, EO and Pratt PF (1962) Buffer methods, for determination of lime requirement
of soils with appreciable amount of exchangeable aluminium. Soil Sci. Soc. Am. Proc., 25,274-277.
Sims JT (1996) Lime requirement. In Methods of soil analysis. Part 3. Chemical methods, Bigham JM
and Bartels JM, Soil Sci. Soc. Agronomy, Madison, WI, 491-515.
Mehlich A (1939) Use of triethanolanine acetate-baryum hydroxide buffer for the determination of some
base-exchange properties and lime requirement of soil. Soil Sci. Soc. Am. Proc., 3,162-166.
Дополнительная литература
Davy H (1813) Elements of agricultural chemistry., Longman 6d.
Clark JS and Nichol WE (1966) The lime potential, percent base saturation relations of acid surface
horizons of mineral and organic soils. Can. J. Soil Sci., 46, 281-315.
Collins JB, Whiteside EP and Cress CE (1970) Seasonal variability of pH and lime requirements in several
southern Michigan soils when measured in different ways. Soil Sci. Soc. Am. Proc., 34, 56-61.
Kamprath EJ (1970) Exchangeable aluminium as a criterion for liming leached mineral soils. Soil Sci.
Soc. Am. Proc., 34, 252-254.
McLean EO (1970) Lime requirements of soils. In active toxic substances or favorable pH range? Soil
Sci. Soc. Am. Proc., 34, 363-364.
CAB (1971) Annotated bibliography no 1529. Liming of tropical soils, 1971-1959. CAB: 25 pages, 101
references.
Yuan TL (1974) A double buffer method for the determination of lime requirement of acid soils. Soil Sci.
Soc. Am. J., 38, 437-440.
Almeida de AM and Bomemisza E (1977) Efecto del encalado sobre las carqas electricas у otros
propriedades quimicas de tres inceptisoles de Costa-Rica. Turrialba, 27, 333-342.
McLean EO (1978) Principles underlying the practice of determining lime requirements of acid soils by
use of buffer methods. Commun. Soil Sci. Plant Anal., 9,699-715.
Cochrane TT, Salinas JG and Sanchez RA (1980) An equation for liming acid mineral soils to compensate
crop aluminium tolerance. Trop. Agric., 57,133-139.
Duchaufour P and Souchier В (1980) pH et besoins en chaux. C. R. Acad. Agric. (Fr.), 66, 391-399.
Totev TP, Palaveyev TD and Kolarov V (1982) Advantages of a KC1 extracts for determining soil acidity
and liming requirements. Pochvovedeniye, 3,117-120.
Nommik H (1983) A modified procedure for rapid determination of titrable acidity and lime requirement
in soils. Acta Agric. Scand., 33, 337-348.
Oates KM and Kamprath EJ (1983) Soil acidity and liming. I: effect of the extracting solution cation and
pH on the removal of aluminium from acid soils. Soil Sci. Soc. Am. J., 47, 686-689.
Oates KM and Kamprath EJ (1983) Soil acidity and liming. II - Evaluation of using aluminum extracted
by various chloride salts for determining lime requirements. Soil Sci. Soc. Am. J., 47, 690-692.
Pavan MA, Bingham FT and Pratt PF (1984) Redistribution of exchangeable calcium, magnesium, and
aluminum following lime or gypsum applications to a Brazilian oxisol. Soil Sci. Soc. Am. J., 48,
33-38.
Haile A, Pieri C and Egoumenides C (1985) Effet des amendements тшёгаих sur les propri6t6s d’dchange
de sols acides tropicaux. Agron. Trop., 40, 98-106.
Alva AK, Edwards DG, Asher CJ and Blarney FPC (1986) Effects of phosphorus/aluminum molar ratio
and calcium concentration on plant response to aluminum toxicity. Soil Sci. Soc. Am. J., 50, 113-
137.
Harvey КС and Dollhoph D (1986) Acid mine-soil reclamation advancements in the Northen plains.,
Montana State Univ. Res. Publ. (01).
Meng Ci-Fu, Luu Yong-Jin, Kong Fan-Gen and Shui jian-Guo (1986) Effects of limestone on soil acidity
and cropyields on a red earth. Soil Science Society of China. Current progress in soil research in
people s republic of China, 377-383.
Borges AL, Braga JM, Defelipo BV, Ribeiro AC and Thiebaut JTL (1987) Evaluation of analytical
methods for estimating soil liming requirement Revista Ceris, 34, 17-32.
566
Часть 3. Неорганический анализ
Nobrega de МТ (1988) Contribuigao ао estudo da estabiliza3ao de solos tropicals com adi3ao de cal
para fins rodoviarios. Aspectos mineralogicos e morfologicos de alguns solos das regioes sul e
sudeste do Brasil., Disserta^ao de maetrado University Sao Paulo, 189 p.
Bailey JS, Stevens RJ and Kilpatrick DJ (1989) A rapid method for predicting the lime requirement of
acidic temperate soils with widely varying organic matter contents: I - development of the lime
requirement model J. Soil Sci., 40, 807-820.
Naidu R, Syers JK, Tillman RW and Kirkman JH (1990) Effect of liming and added phosphate on charge
characteristics of acid soils. J. Soil Sci., 41,157-164.
Rossi PL, Ildefonse P, Nobrega de AT and Chauvel A (1990) Transformations mineralogiques et structurales
d’argiles latfritiques br£siliennes provoqu6es par l’addition de chaux. In ORSTOM Seminaire
«Organisation et fonctionnement des alterites et des sols., (5 f6v.) Thfcme 5 (multigraphi<5).
Aitken RL, Moody PW, Dickson T, Date RA (ed.), Grundon NJ (ed.); Rayment G.E. (ed.), Probert ME
(1995). In Plant-soil interactions at low pH: principles and management. Proceedings of the Third
International Symposium, Brisbane., Kluwer; Dordrecht, 479-484.
Tsakelidou R (1995) Comparison of lime requirement methods on acid soils of northern Greece. Commun.
Soil Sci. Plant Anal., 26, 541-551.
Owusu-Bennoah E, Acquaye DK, Mahamah T (1995) Comparative study of selected lime requirement
methods for some acid Ghanaian soils. Commun. Soil Sci. Plant Anal., 26,937-950.
Coutinho J (1997) Calibration of the single- and double- buffer SMP lime requirement methods by root
elongation bioassay, Commun. Soil Sci. Plant Anal., 28,1127-1139.
Pottker D and Ben JR (1998) Lime for a crop rotation under direct planting. Calagem para uma rotacao de
culturas no sistema plantio direto. Revista Brasileira de Ciencia do Solo, 22,675-684.
Quigley MN, Wallace A (ed.) and Terry RE (1998) Testing soils for lime requirement. In Handbook of soil
conditioners: substances that enhance the physical properties of soil, Dekker, New York, 293-308.
Rossel RAV and McBraney AB (1998) A response-surface calibration model for rapid and versatile site-
specific lime-requirement predictions in southeastern Australia, Aust. J. Soil Res., 2001, 39, 185—
201.
Gustafsson К and Stafford JV (1999) Models for precision application of lime. In Precision agriculture,
Papers-presented at the 2nd European Conference on Precision Agriculture, Odense, Denmark, 11-
15 July 1999, Sheffield Academic, Sheffield, UK.
Pintro JC and Tescaro MD (1999) Correction of an acid soil using the base saturation method and
influence on chemical parameters. Acta Scientiarum, 21,479-482.
Rajkhowa KM and Talukdar MC (1999) Lime requirement of soils as influenced by soil test methods,
J. Agri. Sci. Soc. North East India, 12, 9-12.
Rossel RAV, McBratney AB and Stafford JV (1999) Calibration of a lime requirement buffer for site-
specific lime applications in South-Eastern Australia. In Precision agriculture, Papers-presented at
the 2nd European Conference on Precision Agriculture, Odense, Denmark, 11-15 July 1999, Sheffield
Academic, Sheffield, UK.
Martins E de S, Linhares NW, Giustina C and de S Martins E (2000) Rock analysis reference method for
correcting soil acidity. Metodo de referencia para caracterizacao de rochas utilizadas сото corretivos
de acidez do solo. Comunicado-Tecnico -Embrapa-Cerrados., No.38,3 p.
Gilmour JT and Anderson P (2001) A new approach to lime recommendations in Arkansas. Res. Ser.
Arkansas Agri. Exp. Station, 480, 39-41.
Ozenc DB and Mehlenbacher SA (2001) Methods of determining lime requirements of soils in the Eastern
Black Sea hazelnut growing region. Acta Horticulture, 556,335-341.
Глава 25. Селективность обмена, изотерма
катионного обмена
25.1. Введение
Чтобы лучше понять обменные процессы при выветривании почвы, исследование ад-
сорбционно-десорбционных свойств обменного комплекса с помощью измерения со¬
держания обменных катионов (см. гл. 22) можно дополнить измерениями селективно¬
сти обмена в почве.
Когда почва подвергается циклам выщелачивания-удержания, состояние обменно¬
го комплекса можно определить, оценивая удержание обменных катионов. Эта оценка
лишь косвенно отражает возможную селективность системы по отношению к данному
элементу. Действительно, если бы на обменные свойства почвы влияли только концен¬
трация и заряд катионов, соотношения катионов, удержанных в комплексе, были бы
идентичны их соотношениям в растворе. Растворимые элементы материнской породы
были бы зафиксированы при соответствующих концентрациях. Однако на способность
к удержанию двух катионов (или анионов) Ли Вс одинаковыми зарядами влияют и дру¬
гие факторы:
А в растворе ^ Л адсорбированный
В в растворе В адсорбированный ’
где К — коэффициент селективности.
Он выражает неравенство отношений активностей ионов А и В в растворе и в адсор¬
бированном состоянии.
При определении изменений в составе адсорбированной фазы и жидкой фазы, со¬
держащей ионы, добавленные в определенных пропорциях, можно построить изотерму
обмена. Термин «изотерма», который относится к термодинамике, не должен уменьшать
значимость других существенных переменных системы, таких как валентность и кон¬
центрация ионов, степень гидратации, плотность зарядов коллоидов. Изотерма — это
диаграмма распределения, на основании которой можно построить кривые равнове¬
сий ионных концентраций между ионсодержащей жидкой фазой и твердой обменной
матрицей при постоянной ионной силе. Можно также изменять ионную силу жидкой
фазы или температуру для определения термодинамических параметров.
Как видно из рис. 25.1, а и Ь, можно сравнивать только графики, построенные при
одинаковых условиях — температуре, концентрации и ионной силе, чтобы избежать
ошибочной интерпретации. Определение концентрации ионов, мигрирующих в гомо-
ионной системе в процессе адсорбции, позволяет проверить постоянство емкости кати¬
онного обмена.
Компоненты почвы образуют неорганическую и органическую комплексную обмен¬
ную матрицу, часто включающую очень разные реакции. Этот анализ можно проводить
на цельной почве или ее фракциях, выделенных гранулометрическими и/или химиче¬
скими методами.
Все уравнения для селективности обменных процессов учитывают соотношения
ионных активностей в растворе. Закон обменных отношений Шофилда (Schofield, 1967)
уточняет условия селективности одно-, двух- и трехвалентных ионов.
568
Часть 3. Неорганический анализ
Рис. 25.1. а — изотерма обмена при постоянных концентрациях и ионной силе:Хв=^(ХБ),
где Хв — эквивалентная порция ионов В, удержанных ионитом; Хв — эквивалентная порция ио¬
нов В в растворе: 1 — линейная изотерма, отсутствие селективности, одинаковое сродство к ио¬
нам А и В; 2 — селективность к ионам А; 2’ — сильная селективность к ионам А; 3 — селектив¬
ность к ионам В (иногда наблюдаются S-обраэные кривые с обращением селективности, начиная
с определенной концентрации. Тогда кривая пересекает линию 1 в точке инверсии); Ь — изотерма,
изменяющаяся с концентрацией и ионной силой (эффект разбавления), например, гомоионная
система Na+ и замещение Na+-Ca2+. Вариабельность коэффициента селективности при измене¬
нии ионной силы раствора вызывает вопрос о части уравнения, относящейся к жидкой фазе
Начиная с исследований Керра (Kerr, 1928) было получено, улучшено или упрощено
для применения в различных почвенных условиях и воспроизведения природных усло¬
вий при разнообразных минералогических и климатических условиях множество эмпи¬
рических уравнений.
Уравнение Керра (Kerr, 1928)
К Керр ~
раствор
\МХ* У\МУ*
L адсорб J L-1 раствор
Y
Г
действительно для гетеровалентной системы (А/+ — одновалентный катион; АР+ — двух¬
валентный катион):
[^адсорб ] [^раствор ]
Керр Г А/2+ 1 Г Л/+ I2
L адсорб J L раствор J
Модификация уравнения Керра, осуществленная Ванселоу (Vanselow, 1932), позволяет
учитывать интегральные активности ионов в процессе гетеровалентного обмена
Кв =
WZcope
у\мх+ У
J I раствор л
W,
*+ V\My+ Iх
адсорб J L раствор J
адсорб
+ МХ* Vх
' 1V1 адсорб J
например, в системе Ca2+-NH|:
Глава 25. Селективность обмена, изотерма катионного обмена
569
[С&адсорб 1 Р^Н4раот1вор
]2 [Са
*адсорб
Аадсорб J
На основании статистических исследований Кришнамурти и Оверстрит (Krishna-
moorthy и Overstreet, 1949) вновь модифицировали уравнение, приписав переменные
коэффициенты ионам в соответствии с их валентностью, что привело к общему
уравнению:
к,
[ЛС^ПЛС^У Г,1УЯ.
Шх+ ЛУШУ+ V
-ГаМ:
j-лАЛУ*
где а = 1 для одновалентных катионов, 1,5 — для двухвалентных катионов и 2,0 — для
трехвалентных катионов.
В системе двух- и одновалентных ионов:
^К-0 ~
[М
2+
раствор
[К
раствор
_J
Для корректировки влияния растворителя в формуле Керра Гейнз и Томас (Gaines
и Thomas, 1983) ввели коэффициент, учитывающий долю центров, занятых катионом А,
и молярные количества адсорбированных катионов.
КГ_Т —
l*MZv.+yMZM,r' =
адсорб J
у ^Керр адсорб
В бинарной гетеровалентной системе:
[^раствор 1 адсоиб ]
+ УМадсорб J
КГ_Т -
[КасшорПКдсорб] Шдсорб^Кдсорб]
Все эти уравнения выражены через молярные концентрации.
Часто использовали коэффициент Гйпона (Gapon, 1933), но поскольку он основан на
расчетах эквивалентов, то его применение подвергалось критике (Sposito, 1977):
„
rmm [ACда.](M,*)l,,,
В одно-двухвалентной системе:
„ [АС»*] (М1*)112
л- [<««*,] (Ю '
Установление уравнений селективности для некоторых почв сопровождалось специ¬
фическими проблемами, например, для засоленных (солонцовых-солончаковых) почв,
570
Часть 3. Неорганический анализ
содержащих системы ионов Na\ Са2+, Mg2+ (Richards, 1954; см. гл. 18). Для доли адсор¬
бированного натрия (ДАН) относительно кальция и магния используют уравнение:
Обычно величину ДАН определяют одновременно с долей обменного натрия (ДОН)
В системах ионов К+, Са2+, Mg2+ часто наблюдают необратимый или нелинейный об¬
мен, обычно с низким содержанием ионов К+ по сравнению с ионами Са2+и Mg2+. Доля
адсорбированного калия (ДАК) может помочь объяснить влияние известкования (см.
гл. 24) на отношения активностей и высвобождение ионов К+. Ряд сродства, часто на¬
зываемый лиотропным рядом, может изменяться в зависимости от минеральной матри¬
цы, например, смектиты типа 2:1 адсорбируют прежде всего наименее гидратированные
катионы, способные фиксироваться в межслоевых полостях.
На практике обменный комплекс антропогенных почв может быть изменен при до¬
бавлении селективно адсорбированных анионов (например, фосфатных удобрений),
удалении органического вещества при пожарах или вырубках леса, изменениях содер¬
жания оксидов и т. д. Это может привести к различным скоростям уравновешивания,
которые необходимо измерять в агрономических исследованиях и изучении почво¬
образования. Изменения относительных концентраций катионов в почвенном растворе
также влияют на трофическую способность в единицу времени. Эта селективность
связана с реакциями, контролируемыми законами химической термодинамики.
Например, селективность смектитов типа 2:1 по отношению к одновалентным ионам
отсутствует, если механизмы адсорбции используют только внешнесферные поверх¬
ностные комплексы (например, гексагидратированный ион кальция в монтмориллони¬
те, рис. 25.2). С другой стороны, если образуются внутрисферные комплексы (например,
Рис. 25.2. Адсорбция ионов Ю и Са2+ (Sposito, 1984): внутрисферный поверхностный комплекс
калия в вермикулите (слева) и внешнесферный поверхностный комплекс кальция в монтморил¬
лоните (справа)
[Na+]
([Mg2+]+[Cau])]n
смоль/кг EKO-Nao6M
Глава 25, Селективность обмена, изотерма катионного обмена
571
включение иона калия в вермикулит), обязательно существует обмен между молекулами
воды, сольватирующими катион, и гидроксил-ионами поверхностных функциональных
групп.
Более стабильные внутрисферные поверхностные комплексы образуют с однова¬
лентными катионами основания Льюиса, которые являются скорее слабыми кислотами
из-за их сольватации молекулами воды. Энергия ионизации и ионный радиус однова¬
лентных катионов образуют ряд устойчивости внутрисферных поверхностных комплек¬
сов, образованных в силоксановых полостях: Cs+ > Rb+ > К+ ~ NH4+ > Na+ > Li+.
Для двух- или многовалентных катионов ситуация более сложная, катионы могут
иметь разную валентность в зависимости от окислительно-восстановительных условий
среды, и в растворимой фазе может вновь образоваться лигандный комплекс (конгру¬
энтные и неконгруэнтные изотермы). Ионы Са2+ и Mg2+ могут образовывать комплексы
СаСГ и MgCl+, которые имеют большее сродство, чем свободные Са2+ и Mg2+ катионы.
Селективность Са и Mg может быть связана с большей термодинамической стабильно¬
стью СаСГ (Sposito и др., 1983).
25.2. Построение изотермы обмена
25.2.1. Основные положения
Различные точки для построения изотермы получают с использованием небуферной
жидкой фазы, содержащей два типа ионов с постоянной общей эквивалентной кон¬
центрацией (Sondag и др., 1990). В этой бинарной смеси каждый ион может реагировать
индивидуально или в зависимости от других ионов. Соотношение двух катионов в каж¬
дой точке различно. После насыщения пробы ее оставляют до достижения равновесия
(по окончании адсорбции катионов), затем десорбируют два катиона действием про¬
тивоиона. Определяют оба иона методом атомно-абсорбционной спектрометрии или
спектрометрии с индуктивно связанной плазмой и строят изотерму, используя модель
с двумя центрами (Duffey и Delvaux, 1989), которая позволяет использовать реалистич¬
ный подход к функционированию почвы под действием изменчивых водных и химиче¬
ских условий.
25.2.2. Реагенты
Первая серия равновесных-насыщающих растворов — (равновесие К+-Са2+). Из хло¬
рида калия (М = 74,56) готовят 1 М раствор КС1, а из безводного хлорида кальция
(М = 110,99) — 1 М раствор СаС12. Готовят исходные 1 М растворы (Н-),^, соответству¬
ющие соотношениями К/л/Са, приведенным в табл. 25.1.
Вторая серия равновесных растворов К-Са, Готовят, как описано выше, но исходя из
0,01 Н (+) растворов (табл. 25.1).
Десорбирующий раствор: 1 М раствор хлорида аммония готовят из NH4C1 (М = 53,49).
Стандартные растворы в диапазонах
0-6 мг/л для ионов К+;
0-6 мг/л для ионов Са2+;
0-1 мг/л для ионов А13*.
25.2.3. Методика
• Взвешивают серию из восьми центрифужных пробирок на 50 мл. ч
• Отвешивают по 3 г воздушно-сухой почвы в каждую пробирку.
572 Часть 3. Неорганический анализ
Таблица 25.1. Состав равновесных растворов (общая нормальность 0,01 н. (+), Нк и —
нормальности К и Са)
K/Caw,
К, мМ
Са, мМ
Нк, мг-экв(+)/л
НСа, мг-экв(+)/л
0,2
0,44
4,78
0,44
9,56
0,4
0,86
4,57
0,86
9,14
0,8
1,64
4,18
1,64
8,36
1,6
2,99
3,50
2,99
7,01
2,4
4,12
2,94
4,12
5,88
3,2
5,04
2,48
5,04
4,96
6,6
7,45
1,27
7,45
2,55
29,6
9,78
0,11
9,78
0,22
Первое равновесие
• В каждую пробирку добавляют 25 мл 1 н. (+) раствора ионов К + Са с соотношени¬
ем K/VCa, соответствующим определяемой изотерме.
• Перемешивают в течение 1 ч, центрифугируют и сливают надосадочную жид¬
кость.
• Быстро промывают деионизированной водой для перевода центрифужного осадка
в суспензию.
• Центрифугируют, повторяют обработку дважды.
Построение изотермы
• Готовят шесть последовательных равновесных смесей с 25 мл 0,01 н. (+) раствора
ионов К-Са с соотношениями К/>/Са, соответствующими определяемой изотерме
(без промежуточного промывания водой).
• Каждую смесь выдерживают в течение 30 мин для достижения равновесия.
• Центрифугируют при 5000 g.
• После приготовления и обработки шестой смеси определяют содержание К и/или
Са и продолжают процедуру достижения равновесий, если полученное значение
отличается от значения соответствующего обменного раствора (достаточно про¬
стой проверки равенства величин оптической плотности).
• По достижении равновесия определяют точные концентрации К и Са (которые
всегда несколько отличаются от исходных концентраций), а также алюминия, что¬
бы проверить, что он не участвует в обмене (концентрация <10-5-10-7 М).
Десорбция
• После последнего центрифугирования взвешивают центрифужную пробирку, что¬
бы определить количество раствора, удержанного центрифужным осадком.
• Добавляют 20 мл 1 М раствора NH4C1 и перемешивают в течение 2 ч.
• Центрифугируют и сливают надосадочную жидкость в мерную колбу на 100 мл.
• Повторяют десорбцию дважды, каждый раз оставляя суспензию для взаимодей¬
ствия на 1 ч.
• Доводят объем объединенного экстракта до 100 мл, добавляя деионизированную
воду.
573
Глава 25. Селективность обмена, изотерма катионного обмена
Определение
• Определяют содержание К, Са и А1 и вычитают концентрацию удержанного рас¬
твора, чтобы определить точку К/(К + Са) на изотерме.
• К и Са определяют десять раз методом атомно-адсорбционной спектрометрии
в диапазоне разбавления от 0 до 6 мг/л при добавлении лантана; А1 определяют ме¬
тодом спектрометрии с индуктивно связанной плазмой или спектроколориметрии
{Рати идр., 2001).
25.2.4. Примечания
Обменный комплекс почвы является сложной системой, которая не обладает однород¬
ными поверхностями с однородными плотностями заряда.
Влияние органического вещества на поверхностные свойства можно исследовать на
пробах до и после разложения органического вещества. Каждая гранулометрическая
фракция (песок, пыль, крупный ил, ил < 2 мкм) может обладать сильно отличающейся
селективностью (Sondag идр., 1990).
В связи с экономической важностью кальций-калиевого обмена во многих работах
предпринимались попытки найти более удовлетворительное объяснение адсорбции
К в присутствии Са {Beckett и Nafady, 1967; Goulding и Talibudeen, 1980; Escudey и Galindo,
1988; /Ушей Mamell, 1988; Dufeyn Delvaux, 1989; Sondag идр., 1990).
Попытки одновременно оценить емкости катионного обмена и коэффициентов се¬
лективности обмена на почвенных колонках с нарушенной или ненарушенной струк¬
турой предпринимались с использованием различных одно- и двухвалентных бинарных
систем (например, Scheichnjsp., 1983; Рагкегищр., 1984; Koolидр., 1989).
В настоящее время разработка моделей для обработки данных позволяет исследовать
перенос ионных частиц в почво-ионных обменах. Эти методы можно использовать для
ионов, которые обычно учитывают при определении обменных катионов и емкости
катионного обмена: в этом случае достаточно простейшей модели с двумя катионами
{Duffey и Delvaux, 1989). В экологических исследованиях может возникнуть необходи¬
мость использовать более совершенные многочастичные модели, например, при изуче¬
нии миграции тяжелых металлов в почве в природных условиях, поскольку это может
привести к загрязнению грунтовых вод {Mansellи др., 1986).
Использованная литература
Beckett PHT and Nafady МНМ (1967) Potassium-calcium exchange equilibria in soils: the location of
non specific gapon and specific exchange sites. J. Soil ScL, 18, 263-281.
Delvaux В (1988) Constituants et proprietes de surface des sols derives de pyroclastes basaltiques du
Cameroun occidental Approche genetique de leur fertilite., Th£se UCL Fac. Sc. Agron., 335 p.
Duffey JE and Delvaux В (1989) Modeling potassium-calcium exchange isotherms in soils. Soil ScL Soc.
A. J., 53, 1297-1299.
Escudey M and Galindo G (1988) Potassium-calcium exchange on inorganic clay fractions of Chilean
andepts. Geoderma, 41, 275-285.
Gapon EN (1933) Theory of exchange adsorption in soils. 1 Gen. Chem., USSR, 3, 144.
Goulding KWT and Talibudeen О (1980) Heterogeneicity of cation exchange sites for Са-Mg exchange
in alumino-silicates. J. Colloid Interface Sci., 78, 15-24.
Kerr HW (1928) The identification and composition of the soil aluminosilicate active in base exchange
and soil acidity. Soil ScL, 26, 385.
Kool JB, Parker JC and Zelazny LW (1989) On the estimation of cation exchange parameters from col¬
umn displacement experiments. Soil ScL Soc. Am. J., 53, 1347-1355.
574
Часть 3. Неорганический анализ
Krishna Moorthy С and Overstreet R (1949) Theory of ion-exchange relationships. Soil Sci., 68, 307.
Mansell RS, Bloom SA, Rhue RD and Selim HM (1986) Multispecies cation leaching during continuous
displacement of electrolyte solutions through soil columns. Geoderma, 38, 61-75.
Pansu M, Gautheyrou J and Loyer JY (2001) Soil analysis - sampling, instrumentation and quality
control., Balkema, Lisse, Abington, Exton, Tokyo, 489 pp.
Parker JC and Genuchten MTh van (1984) Determining transport parameters from laboratory and field
tracer experiments. Virg. Univ. Agric. Exp. Station, 84, 3.
Rhue RD and Mansell RS (1988) The effect of pH on sodium-calcium and potassium-calcium exchange
selectivity for Cecil soil. Soil Sci. Soc. Am. J., 52, 641-647.
Richards LA (1954) Diagnosis and improvement of saline and alkali soils., USDA, Agriculture Hand¬
book, 60,160 p.
Schweich D, Sardin M and Gaudet JP (1983) Measurement of a cation exchange isotherm from solution
curves obtained in a soil column - preliminary results. Soil Sci. Soc. Am. J., 47, 32-37.
Sondag F, Feller C and Delcambre L (1990) Etude de la selectivity d’echange K-Ca dans divers sols
tropicaux. Effet de la mature organique. In ORSTOM - Journees Laboratoires (Bondy, France),
127-138.
Sposito G (1977) The Gapon and Vanselow selectivity coefficients. Soil Sci. Soc. Am. J., 41,1205.
Sposito G, Hotzclaw KM, Jouany C and Charlet L (1983) Cation selectivity of sodium-calcium, so¬
dium-magnesium and calcium-magnesium exchange on Wyoming bentonite at 298 K. Soil Sci. Soc.
Am. J., 47,917-921.
Sposito G (1984) The surface chemistry of soils. Oxford University Press, 234 p.
Vanselow AP (1932) Equilibria of the base-exchange reactions of bentonites, permutites, soil colloids and
zeolites. Soil Sci., 33, 95.
Дополнительная литература
Schofield RK (1947) A ratio law governing the equilibrium of cations in the soil solution. Proc. 11th Int.
Congr. Pure Appl. Chem., 3, 257.
Gaines GL Jr and Thomas HC (1953) Adsorption studies on clay minerals. II, A formulation of the ther¬
modynamics of exchange adsorption. J. Chem. Phys., 21, 714.
Assa A (1976) РЬУпотёпе de selectivity d’echange cationique dans certains ттёгаих argileux. I - La
selectivity du potassium dans un systdme potassium-calcium. Cahiers ORSTOM - Ser. Pedol., XIV,
219-226.
Delvaux В (1988) Constituants et proprietes de surface des sols dirives de pyroclastes basaltiques du
Cameroun occidental. Approche genetique de leur fertilite. These Un. Cat. Louvain Fac. Sc. Agron.,
335 p.
Fontaine S, Delvaux B, Dufey JE and Herbillon AJ (1989) Potassium exchange behaviour in Carribean
volcanic ash soil under banana cultivation. Plant Soil, 120,283-290.
Bond WJ and Phillips JR (1990) Cation exchange isotherms obtained with batch and miscible displace¬
ment techniques. Soil Sci. Soc. Am. J., 54, 722-728.
Phillips IR and Black AS (1991) Predicting exchangeable cation distributions in soil by using exchange
coefficients and solution activity ratios. Austr. J. Soil Res., 29, 403-414.
Ishiguro M (1992) Ion transport in soil with ion exchange reaction: effect of distribution ratio. Soil Sci.
Soc. of Am. J., 56, 1738-1743.
Wada SI, Matsuura T and Seki H (1993) Prediction of cation exchange isotherms at different total cationic
concentrations. Soil Sci. and Plant Nutri., 39, 183-187.
Baruah TC, Raj Pal, Poonia SR and Siyag RS (1995) Calcium-potassium, ammonium-potassium and
calcium-ammonium exchange equilibria in soils of semi-arid region of Haryana and humid region of
Assam. J. of Potass. Res., 11, 277-290.
Bond WJ (1995) On the Rothmund-Komfeld description of cation exchange. Soil Sci. Soc. Am. J., 59,
436-443.
Siantar DP and Fripiat JJ (1995) Lead retention and complexation in a magnesium smectite (hectorite).
J. Colloid Interface Sci., 169,400-407.
575
Глава 25, Селективность обмена, изотерма катионного обмена
Borah N, Baruah ТС, Patgiri DK and Thakur AC (1996) Exchange equilibria of calcium versus alu¬
minium, potassium and ammonium in Alfisols of Assam. In Proceedings of the Seminar on Problems
and Prospects of Agricultural Research and Development in North-East India., Assam Agricultural
University, Jorhat, India, 27-28 November 1995,204-212.
Butcher B, Hinz C, Gfeller M and Fluhler H (1996) Cadmium transport in an unsaturated stony subsoil
monolith. Soil Sci. Soc. Am. J., 60, 716-721.
Mukhopadhyay SS (1996) Calcium-potassium exchange and thermodynamics in micaceous soils.
J. Potassium Res., 12, 1-13.
Sumner ME and Miller WP (1996) Cation exchange capacity and exchange coefficients. In Methods of
soil analysis, part 3, chemical methods, Bigham JM and Bartels JM ed., SSSA-ASA, Madison, WI
Etats-Unis, 1201-1229.
Shen SiYan, Tu Shu I and Kemper WD (1997) Equilibrium and kinetic study of ammonium adsorption
and fixation in sodium-treated vermiculite. Soil Sci. Soc. Am. J., 61, 1611-1618.
Moog HC, Streck T and Cammenga HK (1998) Modeling Ca/K exchange kinetics on montmorillonite
and vermiculite. Soil Sci., 163, 382-393.
Endo T, Yamamoto S, Honna T, Eneji AE (2002) Sodium-calcium exchange selectivity as influenced by
clay minerals and composition. Soil Science, 167,117-125.
Saeki K, Wada SI, Shibata M (2004) Ca2+-Fe2+ and Ca2+-Mn2+ exchange selectivity of kaolinite, mont¬
morillonite and illite. Soil Science, 169, 125-132.
Глава 26. Емкость катионного обмена
26.1 Введение
26.1.1 Теоретические аспекты
Емкость катионного обмена (ЕКО) так же, как величина pH или содержание обменных
катионов (см. гл. 22), является важным показателем, используемым в агрономии и по¬
чвоведении для оценки физико-химического состояния почвы. По этому показателю
классифицируют некоторые типы почв, таких как оксисоли, альфисоли и ультисоли.
При точно определенных условиях (например, фиксированном pH) данный параметр
отражает потенциальное количество катионов, способных нейтрализовать отрицатель¬
ный заряд почвы. Преобразование этой величины в единицы смоль (+)/кг позволяет
сравнивать агрономическую ценность почв и обменных комплексов почв, формирую¬
щихся в различных климатических условиях и на различных неорганических и органи¬
ческих матрицах.
Поскольку коллоидная фракция почвы обладает очень большой поверхностью, не¬
сущей электрический заряд, она может удерживать катионы и анионы (источники
питательных веществ для растений), что позволяет до некоторой степени избегать за¬
грязнения подземных вод и воздуха. Сложные взаимодействия между постоянными
и переменными поверхностными зарядами ограничивают изменения pH почвы под
воздействием химических или биологических факторов, в результате чего почва при¬
обретает буферную способность. По мере снижения концентрации почвенного рас¬
твора, удерживаемого в микропорах, из обменного комплекса высвобождаются ионы,
поддерживая равновесие в системе. Общей характеристикой уравнений, описывающих
процесс обмена, является постоянство отношения между адсорбированными катиона¬
ми и свободными катионами в растворе (закон действия масс, теория Гапона (Gapon),
равновесие Доннана (Donnan) и т. п.).
Отрицательные заряды в почве образуются в результате изоморфных замещений
в филлосиликатных структурах, некомпенсированных связей на краях кристаллических
плоскостей или диссоциации функциональных органических групп (см. гл. 19). Эти за¬
ряды можно схематически разделить на (а) постоянные или фиксированные заряды, не
зависящие от pH, валентности и природы противоиона, отношения почва — раствор,
и (Ь) переменные заряды, зависящие от pH и от всех параметров, характеризующих жид¬
кие, твердые среды и межфазные поверхности, таких как:
• концентрация, природа, валентность, ионная сила, константа диссоциации реа¬
гента, природа анионов, связанных с указанным ионом, температура, длитель¬
ность контакта, кинетика обмена, соотношение почва — раствор, а также полиме¬
ры и комплексы, которые могут образоваться;
• диэлектрическая постоянная среды, предварительное высушивание пробы, из¬
мельчение, поверхностный потенциал адсорбента, свойства поверхности и приро¬
да зарядов, преимущественные или селективные поглощения.
Таким образом, выбор метода измерения ЕКО зависит от условий измерения, и огра¬
ничения здесь более очевидны, чем при анализе катионов, способных к обмену. Публи¬
куемая в реферативном бюллетене методика должна быть обоснована, и в тексте должно
быть указано исходное состояние обменной матрицы, т. е. абсолютно необработанная
Глава 26. Емкость катионного обмена
577
почва, предварительно обработанная проба (например высушивание, дробление, раз¬
ложение органического вещества, удаление растворимых солей, обезжелезивание, гомо-
ионная обработка для насыщения глины). Без уточнения этих деталей результаты ана¬
лиза имеют ограниченную ценность.
Ранее было принято характеризовать поглощающий комплекс и оценивать потенци¬
альную плодородность почвы по степени насыщения К, рассчитываемой как отноше¬
ние содержания обмениваемых катионов S(суммарное содержание Са2+, Mg2+, К+, Na+)
и ЕКО (7):
К = 100—, (26.1)
т
где Sn Г выражены в смоль (+)/кг (почвы).
К сожалению, степень насыщения является весьма изменчивой величиной и к тому
же определяется с погрешностью измерения ЕКО, связанной с выбором pH систе¬
мы экстракции, исходного pH почвы, природы обменного комплекса и зарядов. На
рис. 26.1 показано, что степень насыщения сильно варьирует в почвах с преобладанием
переменных зарядов.
ЕКО, смоль (+)/кг
Рис. 26.1. Влияние pH на результаты измерения ЕКО и на расчет процента насыщения У (26.1) для
некоторых тропических почв различного гранулометрического состава (Gautheyrou и Gautheyrou,
1981): почва 1 — ферраллитная почва с каолинитом из Гваделупы; почва 2 — эвтрофная почва
с галуазитом с Мартиники; почва 3 — вертисоль с кислым монтмориллонитом с Мартиники; почвы
4 и 5 — андосоли из Коста Рики а с значительным преобладанием аллофанов
26.1.2. Переменные, влияющие на результат определения ЕКО
Влияние pH
Выбор pH, при котором проводится экстракция, стал поводом для разногласий, не
всегда оправданных; тем не менее, благодаря изучению природы зарядов обменных
комплексов (см. гл. 19), аргументы дискутирующих сторон стали более обоснованны¬
ми. Концепция химического равновесия позволяет проводить количественную оценку
578 Часть 3. Неорганический анализ
параметров, определяющих связи в кристаллической решетке. Применяемые методы
можно разделить на основные категории:
1. Измерение ЕКО при естественном значении pH почвы -> эффективная ЕКО;
2. Измерение ЕКО при заданном буферным раствором pH;
3. Измерение ЕКО при pH, для которого заряд равен нулю (точка нулевого заря¬
да — ТНЗ) или рНО (см. гл. 20).
Измерение ЕКО при естественном pH почвы
Это измерение позволяет определить действительную ЕКО и изучить явления, проис¬
ходящие в условиях, приближеных к реальным. Этим методом можно изучать как эво¬
люцию почв, подверженных энтропийным ограничениям (влияние анионной фиксации
фосфора или силиката кальция, вызывающее снижение pH до 0 и повышение ЕКО, вли¬
яние известкования, повышающего общий заряд, и т. д.), так и обменных комплексов
с точки зрения их влияния на питательную ценность почвы. Метод позволяет рассчиты¬
вать значение эффективной ЕКО путем суммирования, что является хорошим индика¬
тором действительных отрицательных зарядов в полевых условиях.
Небуферные реагенты (например, КС1, NH4C1, органо-металлические катионы) при¬
меняют для количественного анализа, во-первых, обменных катионов (Са2+, Mg2+, К+,
Na+), и, во-вторых, катионов, ответственных за обменную кислотность почвы (таких
как Н+, Al3+, Fe2+, Мп2+).
Описанный метод приемлем для анализа кислых почв с переменными зарядами
и большим количеством межслоевого полимеризованного гидроксида алюминия, кото¬
рый частично нейтрализует отрицательные заряды.
Измерения в буферной среде
Буферные растворы используются для устранения влияния колебаний pH на результаты
измерений и для приведения всех результатов к единой основе, т. е. к выбранному pH.
Методы стандартизированы, и два наиболее часто используемых значения pH составля¬
ют 7,0 и 8,1-8,2.
Методики анализа при pH 7,0 выбраны на международном уровне в целях получения
однородных результатов анализов, используемых для классификации почв. pH 7,0 соот¬
ветствует нейтральной среде. Это значение близко к равновесному pH буферной систе¬
мы почва — НС03 — С02 при нормальном атмосферном давлении.
К этой категории относится методика, использующая молярный раствор ацетата
аммония при pH 7. Методика используется уже около 60 лет в качестве международ¬
ного эталона, поскольку она легка в исполнении, завершается количественным опре¬
делением методом простой дистилляции с последующим титриметрическим анализом,
и ее можно использовать в полевых условиях. Кроме того, поскольку для перемещения
обменных катионов в насыщенную фазу обменного комплекса используется аммоний,
можно объединить две методики и измерять коэффициент насыщения в том же экс¬
тракте. Методика измерения ЕКО с помощью ацетата аммония признана точной, по
меньшей мере, для агрономических целей, что объясняет применение этой методики до
настоящего времени.
Однако это не означает, что методика не подвергалась критике, в особенности при
анализе в присутствии иллитов, смектитов и вермикулитов, где наблюдается селектив¬
ная адсорбция NH4. Для решения этих проблем и адаптации методики к особым ситуа¬
циям были предприняты попытки использования некоторых других солей.
Глава 26. Емкость катионного обмена
579
Жидкая фаза
Внешние
Твердая фаза
Реагенты
факторы
невысушен-
ной почвы
Заряд
Ионная сила
Константа —Катион ■
диссоциации
Связанный
анион
/
<0,1
1
> 1 моль(+)/л
\
—Разведение —
/
Обратимость
обмена —
. Селективность-
Спиртовые Диэлектри-
смеси ческая
постоянная
\
Небуферные
Буферные
(pH 7, pH 8,2,
pH варьирует^
— рН-
Температура
Длительность
контакта
Отношение
почва: реагент-
(Граница встряхивание (Истирание)
Фиксация
ретроградации
Образование
комплекса
или полимера
\
Предыдущая
обработка
Присутствие
свободных -
катионов
'Сушка
Измельчение
Удаление
оксидов
Удаление
органического
вещества
Экстракция
глины
\
'Растворимые
соли
Карбонаты
Гипс
\
Потенциал
поверхностных
зарядов
Природа
поверхности
Органическое
вещество
1:1
2:1
Глины
2:1:
1
AI
Fe
Оксиды-
Мп
гидро¬
Si
оксиды
\
Кинетика
обмена “
Измерения в жидкой фазе
/
Атака
на кристаллическую
решетку
Высвобождение А13\ Н+
Окислительно¬
восстановительное
равновесие Fe2*, Мп2+
\
- Измерения в твердой фазе
Рис. 26.2. Факторы, влияющие на измерения ЕКО (Gautheyrou и Gautheyrou, 1981)
580
Часть 3. Неорганический анализ
Эти методики пригодны для анализа почв с преобладающими постоянными зарядами
или для почв со значениями pH, близкими к нейтральным. Для кислых почв оценка ЕКО
часто оказывается завышенной, в особенности для почв со значительными переменны¬
ми зарядами; в этом случае результат измерения степени насыщения будет занижен.
Методики анализа при pH 8,1—8,2 были разработаны для почв с высокими значениями
pH и обменными комплексами, в которых преобладают двухвалентные катионы. При
25 °С pH 8,2—8,4 соответствует равновесию по С02 при нормальном давлении в системе,
где происходит превращение бикарбоната в карбонат (верхняя точка титрования); кро¬
ме того, оно приблизительно соответствует полной нейтрализации гидроксосоединений
алюминия. Измеренные величины зарядов близки к значениям, реально наблюдаемым
в карбонатных почвах.
Эти методики включают измерения общей обменной кислотности, ЕКО для пере¬
менных зарядов. Применение хорошо забуференной среды при данном pH позволяет
получить результаты, «не зависящие» от исходного pH почвы. В качестве противоиона,
как правило, выбирают катион, такой как двухвалентный барий, поскольку (а) он прак¬
тически отсутствует в большинстве почв и (Ь) валентность у него такая же, как у кальция
и магния, преобладающих в поглощающем комплексе. Однако эта методика непригодна
для исследований процессов почвообразования в кислой среде.
Катионообменная экстракция и количественный анализ при нескольких pH в буферных
средах позволяет проводить оценку изменений заряда при определенном pH (рис. 26.1).
При pH меньше 5,2 влияние Al3+, Fe2+, Fe3+усиливается и становится более выражен¬
ным, что приводит к снижению ЕКО.
В диапазоне pH от 5,2 до 8,5-9,0 происходит осаждение. Это не оказывает существен¬
ного влияния на результаты измерений, поскольку А1 и Fe практически электрически
нейтральны. При pH выше 9,0 А1(ОН)3 превращается в алюминат А10~, добавляющий
глине отрицательный заряд, повышая ЕКО, при этом краевые ионы Al, А1—ОН (алюми-
нолы) трансформируются в анионную форму. Таким образом, диапазоны pH от 4,0 до
9,0 позволяют избежать зон, где высокий уровень растворимости ионов может нарушить
изучаемые обменные процессы.
Измерения при pH, соответствующем ТНЗ-рНО
Эти измерения используются для подробных исследований, однако они непригодны для
повторяющихся анализов из-за длительности и высокой стоимости методики, которую
трудно автоматизировать.
В ТНЗ (см. гл. 20) емкости катионного (ЕКО) и анионного (ЕАО) обмена почвы оди¬
накова. Если pH в системе экстракции выше, чем в ТНЗ, то заряд Iv отрицателен, и зна¬
чение ЕКО оказывается завышенным.
Если pH меньше, чем в ТНЗ, возрастает положительный заряд и завышенной оказы¬
вается анионообменная емкость (ЕАО). Эти методики позволяют проводить исследова¬
ния природы ионной среды и энергетических уровней равновесных состояний в гидро¬
геохимических циклах.
Влияние природы и концентрации катионов
В аналитических методиках наиболее часто используются одновалентные и двухвалент¬
ные ионы. Щдратированные ионы, полностью диссоциированные в разбавленной вод¬
ной среде, обычно легко вступают в обменные взаимодействия. Соответствующие им
лиотропные ряды для концентраций (+) ниже 0,1 М представлены в табл. 26.1.
Глава 26. Емкость катионного обмена
581
Таблица 26.1. Обменные
одновалентные и двухвалентные катионы.
Одновалентные
Cs+
Rb+
к+, nh;
Na*
Li+
Радиус иона (А)
1,67
1,52
1,38
1,02
0,76
Двухвалентные
Ва2+
Sr2+
Са2*
Mg2+
Радиус иона (А)
1,35
1,18
1,0
0,72
Если исключить случаи селективного поглощения, обменная способность изменяет¬
ся в соответствии с валентностью:
М+ < М2+ < М3+
соответственно Na+ < К+ < Mg2+ < Са2+ <А13\
Однако при концентрациях, превышающих 0,1 моль (+)/л, на обменную способность
одновалентных катионов могут влиять различия в селективности поглощения.
В смектитах наименее гидратированные ионы легче достигают межслоевых проме¬
жутков (К+, NH4+), однако при уплотнении 2:1 глин эти ионы могут потерять подвиж¬
ность и стать необменными (фиксированными или возвращаемыми) ионами. Размеры
базального пространства могут доходить до 9,8-10,8 А (см. гл. 4).
С другой стороны, такие ионы, как Са2+ и Mg2+ с высокой энергией гидратации, пол¬
ностью сольватированные, увеличивают межслоевые промежутки до 15-20 А, что делает
эти катионы более подвижными; при этом процесс обмена становится обратимым без
выраженной обменной селективности (см. гл. 25).
Это явление наблюдается в слюдах и иллитах, вермикулитах и минералах с сильным
межслоевым зарядом. Иногда катионы замещаются органоминеральными соединения¬
ми. Существуют стерические затруднения для достижения некоторых обменных пози¬
ций крупными молекулами (например, ионный радиус гексааминокобальта(Ш)-иона
составляет 7 А). В некоторых глинах наблюдают аномально низкие обменные емкости
при развитых межслоевых промежутках, а также в цеолитах, выполняющих функцию
молекулярных сит.
Влияние длительности контакта, перемешивания и уравновешивания
Методики сильно различаются по длительности контакта, что может сильно повлиять
на результат анализа:
• кратковременный контакт (менее 1 ч) необходим при высоких скоростях растворе¬
ния обменной матрицы, в том числе повышенном фоновом шуме (для коррекции
результатов можно применить двойное процеживание); в этом случае диффузия
жидкости в глинистых и коллоидных структурах должна быть достаточно быстрой
для того, чтобы стабильное значение параметров обмена было достигнуто в мак¬
симально короткий срок; осторожное воздействие ультразвука зачастую позволяет
достичь удовлетворительного соотношения обмен : растворение;
• длительность контакта, не превышающая 24 ч, обычно применяется в методиках,
использующих реагенты, позволяющие достичь полного обменного равновесия
в системе реагент — матрица без значительного растворения;
• при длительности контакта, превышающей 24 ч, требуется применение реагентов
с ограниченной растворяющей способностью.
Необходимо проверить содюбилизацию матрицы, наличие возможных новообразова¬
ний в кристаллической или аморфной фазе и стехиометрию обменных процессов.
582
Часть 3. Неорганический анализ
Влияние режимов экстракции: отношение почва — раствор
ЕКО измеряют в двухфазной системе. Анализ проводится для каждой фазы (стационар¬
ной твердой, представленной обменной матрицей почвы и подвижной жидкой, содер¬
жащей противоион (ион-индекс)) (рис. 26.2).
Равновесие между двумя фазами определяет параметры обмена (соотношения ад¬
сорбция - сорбция) и общий заряд твердой фазы. Обмен более полон при высоком со¬
отношении жидкости и твердой фазы (слабо концентрированная жидкая фаза).
Ионная сила противоиона должна быть достаточной для перемещения ионов, фик¬
сированных на обменной матрице, однако при этом не должны переходить в раствор
другие материалы. Условия проведения аналитических измерений далеки от реальных
условий. ЕКО можно измерить в жидкой фазе, обогащенной элементами, высвобожда¬
емыми из почвенной матрицы, в которой снижено содержание противоионов. Избы¬
точное количество противоионов измеряют до и после обмена, а ЕКО рассчитывают по
разности результатов двух измерений.
ЕКО можно измерить и на материале твердой фазы, которая становится гомоионной
после насыщения противоионом и удаления избытка противоионов. Таким образом,
происходит замещение данного иона, и далее возможен его прямой анализ из твердой
фазы при условии, что из этой фазы не высвобождаются другие ионы аналогичной при¬
роды (например, прямая дистилляция почвы с последующим титрованием фиксирован¬
ного NH +). В этом случае ЕКО равна суммарному заряду фиксированных ионов.
Кроме того, ЕКО можно измерить с помощью комбинированной методики, объеди¬
няющей насыщение противоионом и удаление избытка с последующим вытеснением
противоиона другим противоионом; при этом предполагается, что в обоих случаях про¬
исходит стехиометрический обмен.
Раствор, содержащий избыток противоиона, удаляют с помощью сильно разбавлен¬
ного раствора того же иона для того, чтобы избежать диспергирования или избыточного
гидролиза, после чего взвешиванием измеряют количество оставшегося раствора, чтобы
скорректировать результаты. Удаление раствора можно проводить с помощью смешива¬
емых растворителей (метанол, этанол, изопропанол), модифицирующих проницаемость
среды. ISRIC (Международный почвенный справочно-информационный центр)1 (1987)
рекомендует проводить промывание смесью этанол — вода (80% этанола).
Влияние свободных ионов, растворимых солей, известняка, гипса
Присутствие этих веществ в почве мешает измерениям, поскольку немедленно обнару¬
живаются аномальные соотношения между суммой экстрагированных катионов £и об¬
менной емкостью Т. S > 71, в то время как в большинстве других случаев S < Г, или, по
меньшей мере, S= Т. Измеренное значение ЕКО оказывается ошибочным, так же как
и содержание обменных катионов. В зависимости от конкретной ситуации обменный
комплекс насыщается ионами Са2+, Mg2+, Na+.
Влияние предварительной обработки проб почвы
Высушивание
В андосолях с постоянным содержанием влаги высушивание приводит к значительно¬
му снижению значения ЕКО. Поэтому измерения следует производить в пробах, храня¬
щихся при полевой влажности, а вода, содержащаяся в пробах, должна учитываться при
1 ISRIC, International Soil Reference and Information Centre, P.O. Box 353,6700 AJ Wageningen, the
Netherlands. Tel. +31-317-471711; Fax: +31-317-471700. E-mail: soiUsric@wur.nl.
Глава 26. Емкость катионного обмена
583
расчетах. Удельный объем почвы, соответствующий эквивалентному весу почвы, высу¬
шенной при 105 °С, используется для получения результатов, соответствующих одина¬
ковым обменным поверхностям, независимо от исходного содержания влаги в пробах
(между 10 и 300%). Результаты выражают в смоль (+)/ кг почвы, высушенной при 105 °С.
Дробление
В глинах со слабым зарядом, таких как глины 1:1, эффект краевых зарядов усиливает¬
ся при чрезмерном измельчении из-за разрушения структуры, вызванного обработкой.
Аналогичный эффект отмечается при выветривании при присутствии плохо кристалли¬
зированных форм (групп силанол-алюминол, см. гл. 21).
В глинах 2:1 с высоким зарядом краевые заряды пренебрежимо малы в сравнении
с общим зарядом. Следовательно, эффект от умеренного дробления пробы будет менее
значительным, чем в случае глин 1:1.
Дробление аморфных веществ и гелей с очень высокими зарядами практически не
меняет их характеристик, в то же время дробление кристаллических форм, несущих сла¬
бые заряды, может привести к увеличению ЕКО.
Предварительная химическая обработка
ЕКО может существенно измениться при предварительном насыщении пробы андосоли
реагентами, содержащими анионы, склонные к селективному поглощению. Среди этих
анионов особенно активны анионы следующего ряда: F" > РО*_ > SO*-.
Увеличить ЕКО аллофановых почв в три-четыре раза можно, просто добавив фосфат.
Иногда при деструкции органического вещества пергидролем присутствие оксалата
может исказить результаты измерений. Добавление концентрированного гидроксида
аммония для удаления избытка пергидроля может привести к насыщению комплекса
NH+ и стать причиной ошибок при анализе глин 2:1.
Удаление аморфных веществ методом ДЦБ включает восстановление иона железа
(III) до иона железа (II). Если эта реакция происходит в минералах 2:1 на уровне ок¬
таэдральных слоев, отрицательный заряд поверхности повышается. При промывании
почвы ионы железа (II) могут оседать в виде ионов железа (III) и поглощаться ионами
кремния и алюминия матрицы, что приводит к блокировке центров обмена; при этом
измерения ЕКО будут давать ошибочные результаты (Stucki и др., 1984). В этом случае
необходимо оценить возможный эффект подобной обработки в минералах, структура
которых включает железо, таких как нонтронит.
26.2. Определение эффективной ЕКО путем суммирования
(ЭЕКО)
26.2.1. Основные положения
Обменные катионы (Са2+, Mg2+, К+, Na+) (см. гл. 22) добавляют к катионам, экстраги¬
рованным по методу определения обменной кислотности (Н+, Al3+, Fe2+, Fe3+, Мп2' ) (см.
гл. 23); (Yuan, 1959):
ЭЕКО (смоль (+)/кг = [Са2+ + Mg2+ +чК+ + Na+]+(H+ + А13+ + (Fe2+ + Fe3+ + Mn2+)]
Этот анализ (Coleman и Thomas, 1967; Kamprath, 1970) признан репрезентативным для
ЕКО, измеренной в условиях pH почвы и состояния поверхностных зарядов, идентич¬
ного состоянию, существующему в естественных условиях (Juo и др., 1976). Методика
584
Часть 3. Неорганический анализ
пригодна для анализа кислых и нейтральных почв. Однако ионная сила экстрагирующе¬
го раствора выше ионной силы почвенного раствора. Это является преимуществом, так
как позволяет использовать результаты двух различных анализов:
• анализа обменных катионов, оценивающего соотношение и природу доступных
элементов в почве;
• анализа обменной кислотности, определяющего природу кислотности почвы
(напр., Н+, А13+).
Степень насыщения К, измеренная с помощью этих двух анализов, близка к степени
насыщения в полевых условиях, однако является более репрезентативной для кислых
солей с переменным зарядом.
26.2.2. Альтернативные методы
Для экстракции обменных катионов можно использовать методику с применением
0,1 М раствора ацетата аммония при pH 7 или методику с применением небуферного
1 М раствора хлорида аммония.
Анализ обменного алюминия, присутствующего во множественных моно- и поли¬
мерных формах, может дать сильно заниженные результаты, поскольку не все эти фор¬
мы являются обменными.
Для определения ЭЕКО предложены другие методики. Все они предусматривают
использование небуферных реагентов, способных участвовать в обменных процессах,
обменных катионов, или же катионов, определяющих обменную кислотность: Ag-
тиомочевина (PleysiemJuo, 1980), SrCl2 (Edmeadesидр., 1981), NH4-N03 (Stuanesnдр.,
1984), BaCl2 (Hendershot и Duquette, 1986), NH4C1 (Gangaiya и Morrison, 1987).
26.3. Измерение ЕКО на уровне pH почвы в небуферной среде
26.3.1. Основные положения
Почву насыщают противоионами в небуферной среде. Обменный процесс происходит
при pH, близком к уровню pH почвы. После удаления избытка противокатиона с по¬
мощью разбавленного раствора того же иона этот катион вытесняется другим противо-
катионом. Титрование вытесненного катиона позволяет определить ЕКО.
Следует использовать разбавленные растворы, ионная сила которых приближена
к ионной силе почвенного раствора при влажности, соответствующей приблизительно
pF 2 (см. табл. 1.1 в гл. 1), для того чтобы избежать серьезного повреждения поверхности
коллоидов (Gillman, 1979; Gillman и др., 1983; Rhoades, 1982). Эти методики пригодны
исключительно для кислых почв с переменными зарядами, поскольку при измерениях
ЕКО при pH 7,0 получают сильно завышенные результаты. Кислые или нейтральные по¬
чвы не должны содержать растворимых солей или гипса. Кроме того, возможно раство¬
рение органического вещества, приводящее к получению заниженных значений ЕКО.
26.3.2. Методики, использующие небуферные растворы солей металлов
Методика с применением хлорида бария и сульфата магния
Основные положения
Согласно этой методике, подробно описанной в литературе (Gillman, 1979; Rhoades,
1982; NF ISO 11260 1994), обменные катионы экстрагируют небуферным 0,1 М раство¬
ром ВаС12. Содержание обменных катионов (таких как Са2+, Mg2+, К+, Na+) можно опре¬
Глава 26. Емкость катионного обмена
585
делять в экстракте. Остаток почвы обрабатывают 0,02 М раствором MgS04. Ионы Mg2+
вытесняют обменные ионы Ва2+, которые в виде BaS04 оседают в среде, ионная сила
которой поддерживается на уровне, близком к ионной силе почвенного раствора. Раз¬
ность между количеством добавленного Mg2+ и Mg2+, остающегося в растворе, соответ¬
ствует величине ЕКО. Содержание магния определяют методом атомно-адсорбционной
спектрометрии (ААС).
Реагенты
• Раствор для насыщения противоионами — 0,1 М раствор хлорида бария: растворить
23,43 г ВаС12 • 2Н20 (М = 244,3) и довести объем до 1 л деионизированной водой.
• Равновесный раствор — 0,0025 М раствор хлорида бария: взять 25 мл 0,1 М раствора
ВаС12, довести объем до 1 л деионизированной водой;
• Раствор противоиона — 0,02 М раствор сульфата магния: растворить 4,9296 г
MgS04 • 7Н20 (М = 246,50) в 1 л деионизированной воды.
• Реагенты для анализа методами атомно-абсорбционной и эмиссионной спектроме¬
трии:
a) нитрат лантана: к навеске 15,7 г La(N03)3 • 6Н20 (М = 433,02) добавить 42 мл
концентрированной НС1 и довести объем до 500 мл деионизированной водой;
b) хлорид цезия: к навеске 10 г CsCl (М = 168,36) добавить 83 мл концентрирован¬
ной НС1 и довести объем до 1 л деионизированной водой;
c) стандартные растворы Mg. диапазон концентраций - 0, 0,01, 0,02, 0,03, 0,04
и 0,05 мМ (ЕКО);
d) стандартные растворы Na: диапазон концентраций - 0, 4, 8, 12, 16, 20 мг/л
(ЕКО);
e) стандартные растворы К: диапазон концентраций - 0, 10, 20, 30, 40, 50 мг/л
(ЕКО);
f) стандартные растворы Са и Mg. смешанный раствор (ЕКО), содержащий:
Mg: 0, 0,1, 0,2, 0,3,0,4, 0,5 мг/л,
Са: 0, 1, 2, 3, 4, 5 мг/л.
Процедура
Замещение
При необходимости измерить почвенную влажность в другой пробе для корректировки
результатов, полученных при анализе пробы, высушенной при 105 °С (см. гл. 1).
• Поместить 2,5 г почвы < 2 мм в пробирку для центрифугирования емкостью 50 мл
и взвесить пробирку + почву + завинчивающуюся крышку: mv
• Добавить 30 мл 0,1 М раствора ВаС12 и перемешивать в течение 1 ч.
• Центрифугировать при 5000 g и перенести надосадочный раствор в мерную колбу
объемом 100 мл.
• Повторить экстракцию дважды и смешать экстракты в колбе емкостью 100 мл.
• Довести объем до 100 мл; этот раствор содержит обменные катионы: раствор S1.
• Добавить к осадку после центрифугирования 30 мл 0,0025 М раствора ВаС12
и встряхивать в течение ночи.
• Центрифугировать, удалить надосадочный раствор и взвесить пробирку + осадок
после центрифугирования + остаток 0,0025 М раствора ВаС12: т2. .
• Добавить 30 мл 0,02 М раствора MgS02 и перемешивать в течение 2 ч.
586 Часть 3. Неорганический анализ
• Центрифугировать и отфильтровать надосадочную жидкость для количественного
определения ЕКО: раствор 52.
Измерение ЕКО
• Поместить 0,2 мл фильтрата 52 в мерную колбу объемом 100 мл.
• Поместить 0,2 мл раствора противоиона (MgS04) в другую мерную колбу объемом
100 мл.
• Добавить в каждую колбу по 10 мл раствора, содержащего 10 г/л La, и довести объ¬
ем до 100 мл.
• Измерить концентрации Mg методом ААС при 285,2 нм: С0 (концентрация разбав¬
ленного раствора противоиона) и Сх (концентрация разбавленного раствора 52).
Расчет ЕКО
В значение концентрации Сх внести поправку на объем жидкости, оставшейся в пробе
после обработки:
^ С1(30+т2-т1)
“ <,л »
где С2 — скорректированная концентрация Mg, мМ; Сх — концентрация Mg в мМ по
данным ААС; тх — вес пробирки + почвы; т2 — вес пробирки + почвы + удержанной
жидкости.
ЕКО, смоль (+)/кг
3000(С2-С0)
>
т
где т — вес пробы, г (2,5 г); С0 — концентрация разбавленного раствора противоиона.
Если ЕКО превышает 40 смоль (+)/кг, повторить анализ, уменьшив навеску почвы.
Определение содержания обменных катионов
• Пипеткой перенести в мерные колбы объемом 10 мл по 2 мл раствора 51, добавить
по 1 мл раствора CsCl и довести объем до 10 мл деионизированной водой.
• Измерить содержание Na и К методом пламенно-эмиссионной спектрометрии.
• Вновь пипеткой перенести в мерные колбы объемом 10 мл по 1 мл экстракта 51,
добавить по 1 мл раствора нитрата лантана и довести объем до 10 мл деионизиро¬
ванной водой.
• Провести анализ Са и Mg методом ААС.
Расчет содержания обменных катионов
С-Сх 10v
Г = -х ,
°бм т М
где СЛ — концентрация обменных катионов Na, К, Са или Mg, смоль(+)/кг; С и С —
концентрации в экстрактах и холостой пробе, соответственно, мг/л; т — вес, г; v — заряд
катиона (1 для Na и К, 2 для Са и Mg); М— атомная масса катиона, г/моль.
Примечания
Данная методика относительно проста и позволяет проводить измерения постоянных
и переменных зарядов в сильно выветренных тропических почвах, а также обменных
катионов и, возможно, анализ обменной кислотности (NFISO14254 1997).
Глава 26. Емкость катионного обмена
587
Использование MgS04 при анализе андосолей проблематично из-за селективно¬
го связывания сульфат-иона в этих почвах по причине преобладания в них аллофана
и других веществ с упорядочением ближнего порядка. Было предложено (Matsue и Wada,
1985) использовать для насыщения комплекса 0,01 М раствор SrCl2, с последующей де¬
сорбцией обменного Sr 0,5 М раствором НС1.
Согласно другому подходу MgS04 заменяют на MgCl2, поскольку С1~ не подвержен
селективному удерживанию почвами, содержащими аллофан (Henderson и Duquette,
1986) . Аналогичный метод был использован для торфяников: ВаС12 - MgCl2 (Lambert
идр., 1988).
Ва2+, Sr2+, Mg2+ не вызывают усадки глин 2:1, возможно, присутствующих в почве.
Сравнение поведения Sr2+ и Ва2+ в небуферной среде с умеренной ионной силой
(С = 0,01-0,1 М) обнаруживает, что результаты измерений аналогичны результатам, по¬
лученным по аммонийной методике в небуферной среде.
Кроме того, возможно определение алюминия, марганца и железа в экстрактах; эти
элементы в больших количествах могут присутствовать в подзолистых почвах.
26.3.3. Методики с использованием металлорганических катионов
в небуферной среде
Метод хлорида гексааминокобальта(Ш)
Основные положения
Для некоторых металлорганических катионов стерические затруднения позволя¬
ют избежать проникновения в слои глины. Измерение ЕКО с помощью хлорида
гексааминокобальта(Ш) (Esquevin, 1954; Morel, 1957; Amavis, 1959) дает результаты, в об¬
щем, заниженные, однако пригодные для рутинных анализов кислых почв с перемен¬
ными зарядами.
Почву насыщают катионами гексааминокобальта(Ш) [Co(NH3)6]3+ в количестве, пре¬
вышающем ожидаемую ЕКО 3-7 раз (Лету и Orsisni, 1976). Катионы Са, Mg, К, Na и А1
определяют прямо в данном растворе.
Значение ЕКО рассчитывают по разности между количеством добавленного реагента
и количеством реагента, оставшегося в растворе. Рассчитанное содержание поглощен¬
ного Со3+ соответствует ЕКО почвы в небуферной среде при pH, близком к pH почвы.
Можно проводить анализ цельной почвы или ее глинистых фракций; допустимо ис¬
пользование методик макро- и микроанализа.
Реагент
1/20 М (0,05 моль(+)/л) раствор хлорида гексааминокобальта(Ш) (Co(NH3)6Cl3 (М =
= 267,50): отвесить 4,4583 г Co(NH3)6Cl3 и растворить в 1 л воды (1 мл = 0,05 миллиэкви¬
валента ЕКО); готовить свежий раствор каждую неделю, хранить в светонепроницаемой
коричневой бутыли.
Процедура
• Поместить навеску 4 г почвы в пробирку для центрифугирования объемом 150 мл.
* Добавить 100 мл н./20 раствора хлорида гексааминокобальта(Ш).
Следует подбирать концентрацию в зависимости от ЕКО так, чтобы она была при¬
годна для анализа в диапазоне от двух до семи раз от теоретически ожидаемых значений
ЕКО. В противном случае можно изменить вес пробы почвы или глины. Например, при
588
Часть 3. Неорганический анализ
концентрации 0,05 моль (+)/л 100 мл реагента соответствуют 5 ммоль (+) ЕКО; в этом
случае можно с адекватной прецизионностью измерять ЕКО в пробе монтмориллонита
весом 2 г, где ЕКО составляет приблизительно 100 смоль (+)/кг.
• Перемешать и взбалтывать с помощью ротационной мешалки в течение 2 ч.
• Центрифугировать при 5000 g в течение 5 мин, отделить надосадочную жидкость.
• Определить содержание Са2+, Mg2+, К+, Na+ (обменные катионы), А13+ (обменная
кислотность), Со3+ (ЕКО) методами атомно-адсорбционной или эмиссионной
с индуктивно связанной плазмой спектрометрии (Рати и др., 2001). Альтернатив¬
но можно определять ЕКО прямой дистилляцией аммония из экстракта, хлорид
гексааминокобальта(Ш) разрушается при значениях pH > 10 в присутствии ги¬
дроксида натрия по реакции:
Co(NH3)6Cl3 + 3 NaOH -> 6 NH3 + Со(ОН)3 (осадок) + 3 NaCl.
Затем аммиак определяют ацидиметрическим методом, что делает возможным при¬
менение данной методики даже в неопределенных аналитических условиях (Esquevin,
1954; Gautheyrou и Gautheyrou, 1958). Орсини и Реми (Orsini и Remy, 1976) предложили
применять для количественного анализа аммония автоматическую спектроколориме-
трию.
Результаты измерений выражают в смоль(+)/кг, учитывая, что 1/3 моля
Со3+ ► 2 NH3.
Примечания
В растворе хлорида гексааминокобальта(Ш) растворяется лишь очень малая доля карбо¬
натов, что позволяет проводить количественный анализ обменного кальция в умеренно
карбонатных почвах.
При определенных условиях хлорид гексааминокобальта(Ш) может разрушаться, что
приводит к завышению результатов измерения ЕКО (Cornell nAksoyoglu, 1991).
Подробное исследование этих реакций и сравнение методик представлено в работах
(Mantin и Glaeser, 1960). (Jonason, 1961), (Fripiat и Helsen, 1966), (Oliver, 1984), (Fallavier
идр., 1985), (Keita и VanDerPol, 1987).
Хлорид гексааминокобальта(Ш) представляет собой крупный металлоорганический1
катион, образующийся при окислении аммиачного раствора хлорида кобальта, содержа¬
щего хлорид аммония в присутствии активированного угля как катализатора:
2 СоС12 + 2 NH4C1 + 10 NH3 + Н202 о 2 Co(NH3)6Cl3+ 2 Н20.
Координационное число атома кобальта равно шести (рис. 26.3), и каждый
атом N группы NH3 расположен на вершинах правильного октаэдра. Радиус гекса-
аминокобальта(Ш)-иона составляет приблизительно 3,25 А, что может объяснить про¬
странственные ограничения для межслоевых промежутков в глинах и небольшую ион¬
ную силу.
1 В отечественной литературе подобные соединения принято относить к неорганическим
комплексным соединениям. К металлоорганическим соединениям относят органические
соединения, в молекулах которых существует связь атома металла, по крайней мере, с одним
атомом углерода. — Примеч. науч. ред.
Глава 26. Емкость катионного обмена
589
+ 3 сг
Рис. 26.3. Хлорид гексааминокобальта(Ш)
Метод, использующий комплекс Ад — тиомочевина
Основные положения
Почву насыщают одновалентным металлорганическим катионом (серебро — тио¬
мочевина), обладающим сильным сродством к отрицательным зарядам коллоидных
структур.
Обменные катионы переходят в раствор, в котором их можно определить. Анализ со¬
держания иона Ag+, остающегося в растворе, позволяет провести количественное опре¬
деление фиксированных ионов Ag+ по разности результатов и, соответственно, опреде¬
лить ЕКО. Среда не забуферена, экстракция проводится при pH почвы (Pleyssieru Juo,
1980; Searle, 1986; ISRIC, 1987).
Ионная сила раствора реагента меньше ионной силы 1 М раствора КС1 (/« 0,01).
Реагенты
• 0,02 М раствор нитрата серебра: растворить 3,4 г AgN03 (М = 169,89) в 500 мл воды;
хранить раствор в светонепроницаемой коричневой бутыли.
• 0,2 М раствор тиомочевины: растворить 15,25 г тиомочевины (H2N-CS-NH2,
М = 76,12) в 900 мл воды и довести объем до 1000 мл. Выдержать раствор в течение
ночи и профильтровать при наличии осадка.
• Токсичность: тиомочевина слабо токсична при вдыхании и контакте с кожей.
• Раствор для определения ЕКО при изначальном pH почвы: непосредственно перед ис¬
пользованием смешать 1 л 0,2 М раствора тиомочевины с 500 мл деионизированной
воды и перемешать; при перемешивании медленно добавить 500 мл 0,02 М раствора
нитрата серебра; серебро образует с тиомочевиной стабильный комплекс (Bolt, 1982).
• Стандарты для определения серебра методом ААС^ъ мерную колбу объемом 200 мл
поместить 50 мл исходного раствора Ag+ (1,000 мг/л) и довести объем до 200 мл де¬
ионизированной водой: раствор А (концентрация 250 мг/л). В шесть мерных колб
объемом 250 мл поместить по 2,5 мл 0,2 М раствора тиомочевины и 5 мл 1 М рас¬
твора HN03. Точно отмерить объемы 0-5-10-15-20-25 мл раствора А и добавить
в каждую колбу при перемешивании. Довести объем до метки деионизированной
водой и перемешать; в результате получены растворы стандартов в диапазоне кон¬
центраций Ag — 0-5-10-15—20-25 мг/л.
Процедура (/5Я/С1987)
Экстракция
• Взять навеску 1 г почвы (размер частиц 0,5 мм), поместить ее в пробирку для цен¬
трифугирования, добавить 40 мг раствора комплекса серебро — тиомочевина и за¬
590 Часть 3. Неорганический анализ
крыть пробирку. Встряхивать в течение 4 ч, центрифугировать при 5000 g и отде¬
лить надосадочный раствор.
Обменные катионы
Провести количественный анализ Са2+, Mg2+, К+, Na+ и Ag+ методом ААС с помощью
стандартов, приготовленных с добавлением одинакового количества тиомочевины; про¬
анализировать две холостые пробы (те же реагенты без пробы почвы), эталонный обра¬
зец и контрольные пробы серий, выбранные методом случайной выборки.
ЕКО
К 2 мл экстракта добавить 5 мл раствора 1 М HN03, довести объем раствора до 100 мл
водой. Гомогенизировать и измерить содержание Ag+ методом ААС при 328,1 нм с по¬
мощью стандартных растворов с диапазоном концентрации 0-25 мг/л.
Расчет
Ag+ = 107,87
ЕКО, смоль(+)/кг
(6-fl)x50xl00/) 1,85(b-a)f
25x107,87w w
где а — концентрация Ag+ (мг/л) в экстракте при 50-кратном разбавлении; Ъ — концен¬
трация Ag+ (мг/л) в холостой пробе при 50-кратном разбавлении; w — вес воздушно-су¬
хой пробы почвы;/— поправка на влажность.
Примечания
Методика позволяет быстро провести анализ: для количественного определения об¬
менных катионов (Са, Mg, К, Na) и ЕКО требуется только одна экстракция (Pleysier
и Сгетег, 1975; Chabra и др., 1976). Методика позволяет проводить измерения ЕКО на
уровне до 20 смоль (+)/кг почвы. Если ЕКО превышает этот уровень, пробу можно экс¬
трагировать в 80 мл реагента, содержащего тиомочевинный комплекс серебра, или же
использовать пробу почвы меньшего веса; подобные изменения в методике следует учи¬
тывать при расчетах.
Раствор тиомочевинного комплекса серебра может разрушить электроды рН-метров.
Не следует допускать длительного контакта электродов с раствором; непосредственно
после работы необходимо ополаскивать электроды разбавленной азотной кислотой, за¬
тем погружать в деионизированную воду (Siegfridи др., 1986). Чтобы избежать осаждения
нерастворимого соединения (нитрата тиомочевины), не следует использовать HN03,
концентрация которой превышает 1 М.
Методику можно использовать при фиксированном pH при введении в систему
ацетатно-аммонийного или натрий-ацетатного буфера с концентрацией 0,1 мг/л.
При pH до 9,0 методику можно использовать для анализа карбонатных или загип¬
сованных почв (Van Rosmalen, 1980), засоленных почв и, возможно, почв, содержащих
органическое вещество, и почв с переменными зарядами, таких как гистосоли, подзолы
и андисоли (Pleyser и Juo, 1980; Bolt, 1982; Searle, 1986). Стабильность реагента ослабева¬
ет в щелочной среде при pH > 8. Разложение комплекса серебро — тиомочевина приво¬
дит к ухудшению качества экстрагирующего раствора.
Обменная активность велика для глинистых минералов, таких как монтмориллонит,
иллит, каолинит и даже вермикулит, где комплекс серебро — тиомочевина, вероятно,
проникает в межпакетное пространство (Pleysier и Сгетег, 1973, 1975). Селективность
Глава 26. Емкость катионного обмена
591
методики несколько отличается от селективности ацетатно-аммонийной экстракции
при pH 7. Содержание экстрагированного К+ часто оказывается выше при измерении по
методике с применением комплекса серебро — тиомочевина. С другой стороны, содер¬
жание экстрагированных Са2+ и Mg2+, как правило, оказывается заниженным и зависит
от количества органического вещества и pH почвы (Pleysier и др., 1986). Однако неболь¬
шая растворимость кальцита приводит к завышению результата измерений содержания
обменных катионов.
С помощью данной методики можно измерить эффективную ЕКО (см. раздел 26.2).
Результаты анализа несколько занижены в сравнении с результатами, полученными при
использовании методов с применением ацетата аммония и хлорида калия. Тем не менее,
экстракция марганца комплексом серебро — тиомочевина может быть выше, чем для
хлорида калия (Searle, 1986). Влияние на экстракцию железа следует проверить допол¬
нительно, поскольку тиомочевина является нейтральным лигандом, обладающим свой¬
ствами восстановителя.
Небуферный этилендиамин + неорганические катионы
Глину насыщают катионом (например, Со, Си), получая глину в гомоионной модифи¬
кации. Этилендиамин, добавленный в избытке, количественно связывается с добавлен¬
ным катионом. Если образуется растворимый комплекс катиона с этилендиамином, то
произойдет насыщение центров обмена (Fripiat и Helsen, 1996; Mantin, 1969). Эти ком¬
плексы в пределах глины очень устойчивы (Peigneur, 1976; Mas и др., 1978). Величина
ЕКО соответствует концентрации комплекса, удерживаемого матрицей, в результате
происходит снижение концентрации этилендиамина (CornellnAksoyooglu, 1991).
Эти методики имеют те же недостатки, что и методика анализа по разности. Следу¬
ет отметить, что используемый реагент может вызывать раздражение кожи и слизистых
оболочек, вызывать аллергический дерматоз и астму; риск можно уменьшить, работая
в вытяжном шкафу.
Исследователи, изучающие процессы почвообразования и выветривания, использу¬
ют и методики, включающие обмен иона алкиламмония, позволяющие измерить плот¬
ность межслоевых катионов, если известен молекулярный вес базовой обменной еди¬
ницы. Следует использовать только очищенные фракции. В глинах 2:1, в зависимости
от степени кристалличности глины в общей ЕКО, как правило, около 80% приходится
на межслоевую ЕКО и около 20% — на краевую ЕКО. Например, можно вести монито¬
ринг процессов трансформации слюд (иллита в смектит) или трансформации бентонита
всмектиты (Lagaly, 1981).
Кроме того, используются комплексы этилендиамина (NH2-CH2— СН2- NH2) с та¬
кими катионами, как Al, Со, Си, Fe, Mn, Ni, Zn; однако эти методики чаще используют¬
ся при детальных исследованиях глин.
26.3.4. Методики с использованием органических катионов
в небуферных растворах
Интерес к описанным методам возрос с появлением коммерчески доступных органиче¬
ских соединений с большими молекулами (рис. 26.4), применяемых в качестве пести¬
цидов, и осознании вреда, наносимого окружающей среде подобными молекулами (на¬
пример, насыщение поглощающих комплексов, биодеградация, загрязнение подземных
вод паракватом и дикватом).
По возможности эти методики следует комбинировать с методами определения
удельной поверхности глин и макрометодами определения ЕКО.
592
Часть 3. Неорганический анализ
(СНэ)2Н
о
N(CH3)2
Малахитовый зеленый (М = 364,9)
Кристаллический фиолетовый
Рис. 26.4. Органические молекулы, используемые для измерений емкости катионного обмена
Изучение механизмов фиксации требует знаний о поведении одно- или двухвалент¬
ных органических катионов, чтобы различать процессы обмена и физической адсорб¬
ции.
Количество неорганических катионов, замещенных органическими катионами,
должно быть пропорционально или равно количеству поглощенного обменного иона.
Из-за размера органических ионов следует избегать методик, включающих центрифу¬
гирование на высоких скоростях, при которых возможно разделение обмениваемых мо¬
лекул. Обменные емкости определяют по началу плато на обменной изотерме (Margulies
и др., 1988). В основном, используются такие красители, как метиленовый синий, фла¬
вины, малахитовый зеленый и кристаллический фиолетовый (рис. 26.4).
Применяемые методики основаны на выдерживании Na+ гомоионной глины в об¬
менном красителе в течение 1—15 дней, в зависимости от типа глины.
Глава 26. Емкость катионного обмена
593
После перколяции и фильтрации через фильтр Millipore с размером пор 0,45 мкм из¬
меряют поглощение:
• при 662 нм для метиленового синего;
• при 588 нм для кристаллического фиолетового.
Вытесненные неорганические катионы анализируют методом спектрометрии с ин¬
дуктивно связанной плазмой (Rytwoи др., 1991). Насыщение ионом Na+ лучше всего
проводить в присутствии метиленового синего (Hoffman и Dammler, 1969). Калий задер¬
живает поглощение этого окрашивающего реагента.
Димеризация метиленового синего может вызвать искажение концентрационных
кривых, что приведет к ошибочным результатам. Концентрацию водного раствора сле¬
дует тщательно контролировать. Интенсивность полос поглощения мономеров умень¬
шается, а при уменьшении длины волны возрастает интенсивность полос поглощения
димеров и триммеров (метахроматизм). Мономерный метиленовый синий поглощает
при 673 и 653 нм, димер — при 600 нм, а триммер — при 570 нм (Cenens и Schoonheydt,
1990; Bergmann и O’konski, 1963).
26.4. Измерения ЕКО в буферной среде
26.4.1. Методики измерения ЕКО в буферных средах — общая
информация
Определение зарядов почвенных обменных комплексов при постоянном pH применя¬
ется в присутствии одно- или двухвалентных катионов при различных pH (см. «Влияние
pH» в разделе 26.1.2). В хорошо забуференной среде нивелируются колебания, связан¬
ные с pH почвы, однако если pH буферной системы выше, чем pH почвы, отрицатель¬
ные заряды в глинистых минералах и органическом веществе могут возникать из-за дис¬
социации слабых органических кислот. В этом случае результаты измерений окажутся
завышенными, в особенности для кислых почв с переменными зарядами.
Для одновалентных катионов наиболее распространенными являются буферные си¬
стемы, включающие:
• ацетат аммония (CH3COONH4, М = 77,08) с pH 4,0, 7,6или 9,0;
• ацетат натрия (CH3COONa • ЗН20, М = 136,09) с pH 4,0, 8,0 или 8,2;
• ацетат калия (СН3СООК, М = 98,14) с pH 7,0 или 8,3;
• ацетат лития (CH3COOLi ♦ 2Н20, М = 102,02) с pH 8,2.
Для двухвалентных катионов:
• ацетат кальция (СН3СОО)2Са • Н20, М = 176,18) с pH 4,8, 7,0 или 8,2;
• хлорид кальция (СаС12, М = 110,99) + триэтаноламин (ТЭА, N(CH2CH2OH)3,
М = 149,19) с pH 7,0;
• ацетат бария (СН3СОО)2Ва • Н20, М = 273,47) с pH 7,0;
• хлорид бария (ВаС12 • 2Н20, М = 244,31) + триэтаноламин с pH 8,1-8,2;
• ацетат магния ((СН3СОО)2 Mg- 4Н20, М = 214,47);
• ацетат стронция ((СН3СОО)2 Sr- 1/2Н20, М = 214,73).
С учетом комбинаций различных солей, применяемых для насыщения обменного
комплекса (противокатион), концентраций, длительности контакта, отношений поч¬
ва : раствор, pH, процессов устранения избытка противокатиона и его удаления, а также
методов определения характеристик, только между 1960 и 1980-м годом было предло¬
жено более 200 альтернативных методик (Gautheyrou и Gautheyrou, 1981). Тем не менее,
практически на международном уровне регулярно применяются лишь несколько:
594
Часть 3. Неорганический анализ
• Методика, использующая 1 М раствор ацетата аммония с pH 7,0, считающаяся эта¬
лонной методикой при исследованиях в области картографии и таксономии почв.
• Методика, использующая раствор бариевой соли при pH 8,1 или 8,2, забуферен-
ный триэтаноламином.
• Отдельные методики приемлемы для почв, в которых ЕКО особенно трудно из¬
мерить, например, для почв, сформировавшихся в аридном климате, как правило,
обогащенных известняком, гипсом или растворимыми солями.
Андисоли и почвы с переменным зарядом обычно анализируют с помощью методик,
использующих незабуференные системы.
Методики с применением катиона, не присутствующего в почве, делает возможным
измерение содержания обменных катионов (Са, Mg, К, Na) в фазе насыщения.
Выбор методики зачастую определяется доступностью необходимого лабораторного
оборудования, простотой исполнения процедур и возможность проведения серийных
анализов по приемлемой цене. Например, в общем, можно сказать, что ион аммония
можно дистиллировать и количественно определить волюмометрически; анализ иона
калия пламенно-фотометрическим методом в пламени воздух — пропан — более чув¬
ствителен по сравнению с ионом натрия (ионы кальция и бария не поддаются измере¬
нию); диспергирование глин чаще наблюдается в присутствии солей натрия, что затруд¬
няет разделение без ультрацентрифугирования, а высокое содержание натрия создает
помехи при анализе методом эмиссионной спектрометрии и т. д.
Некоторые исследования сосредоточиваются на потребностях агрономии и почвове¬
дения, таких как оценка рисков фиксации ионов калия и аммония, например, в иллитах
и вермикулитах; преобладание двухвалентных катионов кальция и магния в большин¬
стве некислых почв; сомнительная эффективность ионов аммония при замещении про¬
тонов и ионов алюминия в глинах 1:1; использование иона натрия в качестве проти¬
воиона в засоленных почвах для ограничения вытеснения зарядов; проблема подбора
методики, использующей системы с высоким pH, приближенным к равновесному pH
некоторых почв (pH > 7,0), для ограничения процесса солюбилизации.
Очевидно, что для рутинных анализов важно соотношение цены и качества, кроме
того, следует учесть, что с помощью выбранной методики предстоит анализировать
большое количество проб (при картографических исследованиях и агрономическом
контроле). Знания, приобретенные в ходе исследований, позволили разработать мето¬
дики, применяемые в течение многих лет. Статистический анализ результатов позволяет
разрабатывать модели или создавать консультативные системы по сельскохозяйствен¬
ным технологиям (например, системы DRIS1 2 или PARADES1 в штате Калифорния, охва¬
тывающие около 60 культур во множестве различных климатических условиях с приме¬
нением (или без применения) ирригации).
26.4.2. Методика с применением ацетата аммония при pH 7,0
Основные положения
Почву насыщают противоионом аммония в буферной среде при pH 7,0. Происходит
поглощение аммония и вытеснение эквивалентного количества катионов. Анализ об-
1 DRIS, Diagnosis and Recommendation Integrated System — Интегрированная система диагности¬
ки и рекомендаций.
2 PARADES, Plant Analysis Recommendations and Diagnosis System — Система диагностики и ре¬
комендаций в растениеводстве.
Глава 26, Емкость катионного обмена
595
менных катионов в перколяционном растворе проводят с помощью пламенно-фото¬
метрической, атомно-адсорбционной или ИСП-спектрометрии (Pansu и др., 2001). Из¬
быток противоиона устраняют с помощью растворителя (например, 80%-ного этанола),
останавливая таким образом процесс гидролиза. Противоион вытесняется ионом калия,
содержащимся в стандартном небуферном растворе хлорида калия (или ацетатом на¬
трия при pH 7,0, или хлоридом натрия). Чтобы измерить ЕКО (суммарное количество
противокатиона, которое может удерживать почва), можно провести дистилляцию ам¬
мония с последующим волюмометрическим анализом или автоматизированной спек-
троколориметрией (рис. 26.5). Отметим, что при анализе обменных катионов возможное
удерживание аммония в глинах не оказывает существенного влияния на результат из¬
мерения, однако может занизить значение ЕКО.
Усовершенствования методики
Методика определения ЕКО с помощью буферного раствора ацетата аммония имеет не¬
сколько существенных модификаций (рис. 26.5). Первоначально методику использова¬
ли для определения обменного калия (Prianishnikov, 1913), затем для анализа реакции
почвы (Schollenberger, 1927) и, наконец, для определения ЕКО (Schollenberger и Simons,
1945; Kelley, 1948; Peech и др., 1947), а в последние 50 лет методика стала признанным
международным эталоном измерения ЕКО.
Комбинации первичной обработки для насыщения раствора с различными видами
обработок, направленных на устранение избытка иона аммония, природой противо¬
ионов, необходимых для вытеснения аммония, и методов итогового анализа привели
к появлению различных аналитических процедур, которые, как правило, приводят к су¬
щественно различающимся результатам. На рис. 26.5 перечислены различные модифи¬
кации, внесенные в методику, применяемые в настоящее время.
Реагенты
• 1 М раствор ацетата аммония с pH 7,0 взять навеску 770,08 г CH3COONH4
(М = 77,08) или развести 600 мл ледяной уксусной кислоты (СН3СООН, М = 60,05)
приблизительно в 9 л деионизированной воды и постепенно добавить 750 мл ги¬
дроксида аммония (NH4OH, d = 0,90); охладить и проконтролировать pH; довести
pH до 7,0 добавлением гидроксида аммония или уксусной кислоты, довести объем
до 10 л деионизированной водой.
• Этанол: 80% этанола 96° + 20% деионизированной воды.
• 1 М раствор хлорида калия: растворить 745 г КС1 (М = 74,5) в приблизительно 9 л
деионизированной воды; после достижения температурного равновесия довести
объем до 10 л деионизированной водой.
• Реактив Несслера (для анализа): (а) взять навески 45,5 г йодида ртути (II) (Hgl2,
М = 454,45) и 35,0 г йодида калия (KI, М = 166,02); растворить навески в малом
объеме воды; (Ъ) 112 г гидроксида калия (КОН, М = 56,10) растворить в 500 мл
деионизированной воды (из которой удален С02 путем кипячения в течение 1 ч,
при охлаждении не допускать контакта воды с воздухом); смешать а и Ъ, и довести
объем до 1 л; хранить раствор в светонепроницаемой коричневой бутыли, не до¬
пуская контакта с воздухом; готовить свежий раствор еженедельно; в присутствии
иона аммония реагент придает раствору желто-коричневую окраску (или вызывает
выпадение коричневых хлопьев, если содержание аммония очень велико).
• Реагент Тасниро: смешать одну часть 0,1%-ного метилового красного в этаноле
и три части 0,1%-ного бромкрезола зеленого в этаноле.
596
Часть 3. Неорганический анализ
Почва
^ Prianishnikov (1913)
Обработка насыщением Kelley (1927)
Chapman (1965)
На воронку
Бюхнера
На фильтр На колонку
/
Жидкая фракция
Колба Мариотга
В восходящем потоке
Вакуумная экстракция
Этап 1: насыщение
1 М раствором ацетата
аммония при pH 7
Jaynes и Bigham (1986)
Kalra (1986,1990,1991)
ISRIC( 1987)
Определение обменных
катионов Na, К, Са, Мд
Удаление избытка
противоиона
Этап 2: Удаление избытка
80%-ный этанол (1)
80%-ный этанол (2)
ISRIC( 1987)
РеесЛидр. (1962)
Почвенно-аммонийная
матрица
Отходы фильтрата
Прямая перегонка
с водяным паром
в присутствии
NaCI, NaOH,
Mg(OH)2
Na + HCI
Peech (1945)
Прямая перегонка
с водяным паром
в присутствии
NaOH, Mg(OH)2
1 MKCI
Прямая перегонка
с водяным паром
в присутствии
NaOH, Mg(OH)2
Этап 3: вытеснение
противоиона NH* другим
противоионом (Na* или К+)
Волюмометрия
Peech(1947)
USDA (1972)
£40(1972)
Волюмометрия
USDA (1972)
Волюмометрия
Jackson (1958)
USDA (1972)
£40(1972)
автоматизированная автоматизированная
Schuman (1973) Кеепу и Nelson (1982)
Skjemstad ^976)
Technicon (1973)
Tecator (1985)
Kalra и Maynard (1986,1991)
Этап 4: Определение
противоиона NH4+
Рис. 26.5. Основные методики с применением ацетатно-аммонийного буферного раствора
Глава 26. Емкость катионного обмена
597
• Жженая тяжелая магнезия: (Mg(OH)2, М = 58,34).
• 2%-ный водный раствор борной кислоты (Н3В03, М = 61,84).
• 0,05 н. стандартный раствор серной кислоты.
• 1 % -ный фенолфталеин в этаноле.
Процедура, включающая дистилляцию с водяным паром
Обмен с ионом аммония
• Измерить влажность проб для коррекции результатов на почву, высушенную при
105 °С.
• Взять навеску 2 г (или 5 г при небольшой ЕКО) воздушно-сухой пробы почвы, про¬
сеянной через сито с размером ячеек 0,5 мм.
• Поместить почву в пробирку объемом 100 мл для центрифугирования с завинчи¬
вающейся крышкой и добавить 30 мл 1 М раствора ацетата аммония при pH 7,0.
• Гомогенизировать с помощью вортекс-миксера в течение 2 мин.
• Выдержать в течение ночи.
• Еще раз перемешать с помощью вортекс-миксера (в течение 2 мин) и центрифуги¬
ровать при 5000 g в течение 5-10 мин (в зависимости от физических характеристик
почвы).
• Декантировать надосадочный раствор, который должен быть прозрачным, не до¬
пуская потери почвы.
• Вновь приготовить взвесь в 30 мл ацетата аммония с помощью вортекс-миксера
и отстаивать в течение 15 мин.
• Центрифугировать и декантировать надосадочный раствор, добавить к предыду¬
щей порции.
• Повторить обработку в третий раз, смешать все порции надосадочного раствора.
• Провести количественное определение обменных катионов (Са, Mg, К, Na) в над-
осадочном растворе.
Отмывка избыточного иона аммония
• Добавить 30 мл 80%-ного этанола.
• Гомогенизировать с помощью вихревой мешалки.
• Центрифугировать и удалить надосадочный спиртовой раствор.
• Повторить эту обработку с осторожностью, не допуская потери почвы.
• С помощью реактива Несслера убедиться в отсутствии ионов аммония в третьей
порции надосадочного спиртового раствора.
Вытеснение иона аммония
• Добавить 30 мл 1 М раствора КС1.
• Взболтать в вортекс-миксере и отстаивать в течение 30 мин.
• Центрифугировать и удалить надосадочный раствор так, чтобы избежать потери
почвы.
• Повторить данную обработку дважды; смешать все порции надосадочного раствора.
• С ополаскиванием осторожно перенести экстракционный раствор в перегонную
колбу Къельдаля емкостью 600 мл (раствор также можно сохранить для определе¬
ния аммония автоматизированной колориметрией).
598 Часть 3. Неорганический анализ
Определение иона аммония путем дистилляции с водяным паром
• После добавления в колбу Къельдаля 5 г жженой магнезии и одной капли фенол¬
фталеина (если раствор не окрашивается в розовый цвет, добавить еще магнезии
для получения щелочной среды) немедленно приступить к перегонке с водяным
паром (см. раздел 14.2.1 в гл. 14).
• Собрать дистиллят (около 100 мл) в 20 мл 2%-ного раствора борной кислоты, в ко¬
торую добавлены 3 капли индикатора Тасчиро.
• Титровать 1/40 М раствором H2S04 до появления розово-серой окраски.
Расчеты
На титрование 0,05 миллиэквивалента ЕКО уходит 1 мл 1/40 М раствора H2S04. При
объеме ушедшего на титрование раствора серной кислоты К, мл значение ЕКО Г, опре¬
деленное в смоль (+)/кг для воздушно-сухой почвы, рассчитывают по формуле:
Т= V• 0,05 • 100.
В полученный результат можно внести поправку на влажность почвы. Для проб по¬
чвы с переменным зарядом, анализируемых без предварительного высушивания, в от¬
личие от проб, высушенных при 105 °С, Трассчитывают по формуле:
Т= V• 0,05 • 100 • /,
где/— поправочный коэффициент, учитывающий влажность почвы.
Альтернативная методика с применением автоматизированной колориметрии
Основные положения
Данный альтернативный метод относится только к конечному определению иона ам¬
мония, при этом такие процедуры, как экстракция или вытеснение, остаются неизмен¬
ными (см. выше раздел «Определение иона аммония путем дистилляции с водяным па¬
ром»). Рекомендуется синее окрашивание индофенола в процессе автоматизированного
непрерывного анализа для определения ЕКО почвы (Nelson, 1982) и {Kalra и Maynard,
1986,1991). В щелочной среде в присутствии гипохлорита натрия ион аммония дает си¬
нее окрашивание, катализируемое нитропруссидом натрия. Интенсивность окрашива¬
ния пропорциональна количеству иона аммония и, следовательно, значению ЕКО.
Реагенты
• Базовый раствор аммонийной соли: растворить 0,4717 г сульфата аммония ((NH4)S04,
М = 132,14) в дистиллированной воде и довести объем до 1 л; раствор содержит
100 мкг (NH4-Ы)/мл; хранить раствор в холодильнике в светонепроницаемой ко¬
ричневой бутыли.
• 2 мкг (ИЩ—Щ/мл раствор аммонийной соли\ разбавить 4 мл базового раствора до
200 мл.
• Набор стандартных растворов аммония: в мерные колбы объемом 25 мл внести 0,
2, 4, 6, 8, 10, 12 мкг NH+-N (0-6 мл раствора аммонийной соли концентрацией
2 мкг (NH4 -NJ/мл) и добавить 15 мл раствора NaCl или КС1, используемого для
вытеснения иона аммония (или количество, эквивалентное действительно взятому
объему, см. «Вытеснение иона аммония» далее); довести до 25 мл деионизирован¬
ной водой.
Глава 26. Емкость катионного обмена
599
• Раствор нитропруссида натрия: (Na2Fe(CN)5NO • 2Н20, М = 297,95): растворить
68 мг соли в 100 мл деионизированной воды; гомогенизировать и хранить в холо¬
дильнике в светонепроницаемой коричневой бутыли.
• Фенольный реактив (М = 94,11): растворить 7 г фенола в 100 мл деионизированной
воды; хранить в холодильнике в светонепроницаемой коричневой бутыли.
• Забуференный раствор гипохлорита натрия: растворить 1.480 г гидроксида натрия
(NaOH) приблизительно в 70 мл деионизированной воды; добавить 4,98 г безвод¬
ного гидрофосфата натрия (Na2HP04, М = 141,98); перемешать и после полного
растворения соли добавить 20 мл свежеприготовленного 5%-ного раствора гипо¬
хлорита натрия (NaCIO); значение pH должно быть в диапазоне от 11,4 до 12; затем
довести объем до 100 мл деионизированной водой.
• Раствор двунатриевой соли ЭДТА: растворить 6 г двунатриевой соли диаминте-
трауксусной кислоты (C10H14O8N2Na2 • 2Н20, М = 336,24) приблизительно в 80 мл
деионизированной воды; после полного растворения соли довести pH до 7,0 с по¬
мощью гидроксида натрия и довести объем до 100 мл.
Процедура
• Взять аликвоту отфильтрованного конечного раствора КС1, полученного при ана¬
лизе ЕКО (от 3 до 5 мл в зависимости от концентрации).
• Внести в 25 мл мерную колбу 1 мл раствора соли ЭДТА, 2 мл фенольного реагента,
2 мл раствора нитропруссида, 4 мл буферного раствора гипохлорита. Гомогенизи¬
ровать после внесения каждого реагента.
• Довести объем до 25 мл деионизированной водой.
• Поместить раствор на водяную баню на 30 мин при 40 °С, затем охлаждать в тече¬
ние 15 мин и измерить поглощение при 636 нм. ч
• Провести измерения в двух холостых пробах (реагенты без пробы почвы) и опреде¬
лить результаты по градуировочному графику в стандартном диапазоне концентра¬
ций, построенному по результатам измерений в тех же условиях.
Метод может быть автоматизирован с помощью сегментного анализа постоянного
потока с применением манифольда, представленного на рис. 26.6.
Примечания
Методика с применением ацетата аммония при pH 7 пригодна для анализа бескарбонат-
ных почв с умеренным pH (5,5-7,5) и постоянными зарядами, однако не подходит для
грунтов органического происхождения, компостов и торфяников из-за растворимости
органического вещества в ацетате аммония и этаноле.
Карбонаты кальция и магния, а также растворимый гипс и соли более или менее рас¬
творимы в ацетате аммония и участвуют в обмене. В почвах с переменными зарядами
в этом случае измеренное значение ЕКО оказывается завышенным.
В минералах 1:1, таких как каолинит и галлуазит, ион аммония не может полностью
вытеснить ионы А13+ и Н+. В минералах 2:1, таких как вермикулит и слюды-иллиты,
ион аммония может быть зафиксирован — в этом случае он перестанет быть обменным
ионом. Поэтому иногда предпочтение отдается ионам натрия и бария, поскольку они
обмениваются более полно и легко определяются современными аналитическими ме¬
тодами.
Происхождение применяемого ацетата аммония (см. «Реагенты») определяется фи¬
нансовыми и практическими соображениями. Коммерческие соли дороже составляю-
600
Часть 3. Неорганический анализ
Рис. 26.6. Определение ионов аммония в NaCI экстрактах для измерения ЕКО почвы методом
автоматизированного колориметрического сегментного анализа постоянного потока (Kalra
и Maynard, 1986,1991)
щих компонентов — гидроксида аммония и уксусной кислоты и склонны к затвердева¬
нию в бутыли при длительном хранении раствора.
Последовательные катионный обмен и стадии промывания являются потенциальны¬
ми источниками погрешностей, связанных, например, с потерей почвы вследствие дис¬
персии или неполнотой обмена. Некоторые авторы предлагают использовать воронки
Бюхнера с двойными фильтрами. 80%-ный этанол должен быть нейтральным, однако
нейтрализации гидроксидом аммония в присутствии индикатора не требуется. Этанол
можно очистить перегонкой с гидроксидом кальция. Смесь 80% этанола: 20% воды по¬
зволяет избежать избыточной дегидратации (ISRIC1987).
Способ проверки полноты удаления избыточного аммония в этанольном фильтрате
основан на реакции Несслера:
2 Hglf + 2 NH3-»2 NH3HgI2 + 41~
2 NH3HgI2 -> NH2Hg2l3 (оранжево-коричневый осадок) + Г + NH4+
Прямая дистилляция почвы позволяет избежать погрешностей, так как сокращает
три подэтапа насыщения противоионом с риском потерь. Однако дистилляция может
привести к положительной погрешности, связанной с высвобождением аммония из ор¬
ганического вещества, в особенности если вместо магнезии используется гидроксид на¬
трия.
26.4.3. Методика с применением буферных систем при pH 8,0-8,(5
Основные положения
Методики этой группы разрабатывались, как правило, для анализа почв, образовавших¬
ся в процессе выветривания, которое в аридных районах приводит, в частности, к вы¬
Глава 26. Емкость катионного обмена
601
свобождению солей щелочных и щелочноземельных металлов. Обменный комплекс мо¬
жет насыщаться ионами Na+, Са2+, Mg2+ в карбонатных (CaC03, MgC03), загипсованных
(CaS04), солонцовых и содовых (содержащих растворимые соли, включающие ионы
Na+, Cl~, SO2-, СО2-) почвах. Некоторые гумусовые и торфяные кислые почвы также
анализируют с помощью методик, использующих буферные растворы при pH > 8,0.
Наиболее распространенные методики основаны на:
• применении буферных растворов при pH 8,0-8,6, близким к pH почвы с низкой
химической растворимостью щелочноземельных карбонатов, способных конкури¬
ровать с противоионом; применение ионообменных смол позволяет удалять рас¬
творенные карбонаты и сульфаты (Bergseth и Abdel-Aal, 1975);
• использовании двухвалентных ионов, отсутствующих в почве, с обменной емко¬
стью, близкой к аналогичному параметру ионов кальция и магния, преобладающих
в основных типах почв, или возможном использовании ионов натрия для засолен¬
ных почв, направленном на ограничение изменений в зарядах;
• применение растворителей, таких как этанол или метанол, обладающих меньшей
полярностью, чем вода, и ограничивающих процессы солюбилизации и гидролиза.
Тем не менее, изменение степени сольватации обменных катионов может повлиять
на некоторые характеристики, в особенности в присутствии цеолитов (стерические пре¬
пятствия, молекулярные сита). Универсальной методики определения ЕКО во всех по¬
чвах умеренных и тропических климатов, аридных и гидроморфных систем не суще¬
ствует.
Методики становятся более сложными и менее прецизионными при определении
ЕКО в средах, содержащих растворимые известняк, гипс и соли одновременно.
Множество изысканий проведено в целях достижения удовлетворительных результа¬
тов исследований процессов генезиса сложных почв, упрощения процедур и улучшения
прецизионности и специфичности за счет исключения4этапов, являющихся потенци¬
альными источниками погрешностей.
Одновалентные катионы, такие как натрий или литий (связанные с анионами, та¬
кими как ацетат, формиат или хлорид), или двухвалентные катионы, такие как барий,
кальций или магний (хлорид, нитрат, сульфат) используют в комбинации с триэтанола¬
мином (ТЭА) для забуферивания среды.
Эти методики были впервые описаны в работах Mehlich (1938,1942,1953), Bower и щ>.
(1952), Yaalon и др. (1962), Bascomb (1964), Bergseth и Abdel-Aal (1975), Polemio и др. (1977),
Sayeh и др. (1978), Gillman (1979), Rhoades (1982), Misopolinos и Kavovoulos (1984); Gupta
и др. (1985), Tucker (1985), Frenkel и др. (1986), Begheyn (1987), Drechsel (1987), Sharma
и Dubey (1988).
Натрий-ацетатный буфер (pH 8f2)
Основные положения
Представленная методика включает два этапа (Rhoades, 1982):
• катионообменные центры обменного комплекса насыщаются ионами натрия
в форме ацетата или хлорида, являющимися маркерами, в растворе, содержащем
60% 95°-ного этанола, для ограничения солюбилизации необменных форм;
• противоион натрия экстрагируют катионом магния в нитратный раствор.
Натрий и хлорид определяют в конечном экстракте (избыточный растворимый на¬
трий в экстракционном растворе можно определить из суммарно определяемого натрия,
дающего обменный натрий, содержание которого эквивалентно ЕКО).
602
Часть 3. Неорганический анализ
Этот метод пригоден для анализа почв, содержащих карбонаты, гипс и цеолиты, и его
можно использовать также для анализа солонцовых и содовых почв. Тем не менее, Гупта
с сотр. (Gupta и др., 1985) упоминали о некоторых затруднениях при анализе засолен¬
ных почв с повышенными pH и предлагали корректировать pH раствора для насыщения
противоионами до значений, соответствующих pH почвы в водной среде, а также вклю¬
чить в методику второй этап с раствором нитрата магния при pH 8,6.
Реагенты
• Насыщающий раствор, 0,4 М ацетата натрия и 0,1 М хлорида натрия в 60%-ном эта¬
ноле: навески 54,4320 г ацетата натрия (CH3COONa • ЗН20, М = 136,09) и 5,8450 г
хлорида натрия (NaCl, М = 58,45) растворить приблизительно в 300 мл деиони¬
зированной воды; добавить 600 мл чистого 95° этанола и довести pH до 8,2 с по¬
мощью 6 М раствора гидроксида натрия; довести объем до 1 л деионизирован¬
ной водой; определить содержание ионов натрия и хлорид-ионов и соотношение
Na+: Cl- в растворе.
• Экстрагирующий раствор 0,25 М нитрата магния: взять навеску 6,411 г нитрата маг¬
ния (Mg(N03)2 • 6Н20, М = 256,43) и растворить приблизительно в 900 мл деиони¬
зированной воды, довести объем до 1 л.
Процедура
• Определить влажность проб с тем, чтобы пересчитать на результаты для почв, вы¬
сушенных при 105 °С.
• Поместить 5 г навески воздушно-сухой просеянной почвы (размер ячеек сита 0,5 м),
в пробирку объемом 100 мл для центрифугирования с завинчивающейся крышкой.
Насыщение
• Добавить 33 мл насыщающего раствора (если электропроводность растворенных
солей выше 4 дСм/см, необходимо провести предварительную экстракцию в 33 мл
деионизированной воды).
• Центрифугировать при 2500 g в течение 5 мин и декантировать надосадочный рас¬
твор, не допуская потери почвы.
• Добавить 33 мл насыщающего раствора и встряхивать пробирку в вортекс-миксере
для отделения осадка после центрифугирования, затем поместить пробирку в уль¬
тразвуковую ванну на 30 с для диспергирования пробы.
• Перемешивать в течение 5 мин, центрифугировать и декантировать надосадочный
раствор.
• Повторить обработку дважды, порции жидкой фракции слить.
Замещение натрия противоионом
• Добавить 33 мл экстрагирующего раствора нитрата магния к осадку после центри¬
фугирования.
• Перемешивать в вортекс-миксере и встряхивать в течение 5 мин.
• Центрифугировать и декантировать надосадочный раствор в мерную колбу объе¬
мом 100 мл.
• Вновь добавить 33 мл экстрагирующего раствора и повторить экстракцию дважды.
• Смешать три надосадочных раствора, довести объем до 100 мл экстрагирующим
раствором и гомогенизировать.
Глава 26. Емкость катионного обмена
603
Количественное определение натрия, замещенного противоионом
Определить общее содержание в экстрактах натрия [Nay — пламенно-фотометрическим
методом, и хлорид-иона [С1~ ] — методами кулонометрии-амперометрии (см. раздел
18.3.6 гл. 18), в сМ, используя градуировочные шкалы, построенные в экстрагирующем
растворе нитрата магния.
Расчеты
ЕКО = (Na+ - растворимый Na+), в смоль (+)/кг (почвы)
ЕКО = ^ ([Naq ] Z)/Na+ - [Cl-0] Dfcr),
где w — вес пробы; Df— коэффициент разведения, т. е. отношение итогового объема
к объему аликвоты.
Хлорид бария - триэтаноламин (ТЭА) при pH 8,1
Основные положения
Почву насыщают ионами бария в среде, забуференной триэтаноламином (ТЭА) при
pH 8,1.
Противоион бария замещается двухвалентным катионом магния. Количественный
анализ вытесненного бария проводится методом эмиссионной спектрометрии при
489 нм или атомно-адсорбционной спектрометрии, таким образом, определяют ЕКО
при данном pH; эта величина представляет количество бария, поглощенного почвой
в присутствии известняка в равновесии с С02 воздуха при нормальном давлении.
Данную методику используют при анализе кислых, органогенных, карбонатных или
глинистых (1:1) почв. В почвах, pH которых составляет менее 8,2, значения ЕКО полу¬
чаются завышенными.
Кроме того, методика позволяет измерить общую потенциальную кислотность для
определения граничных условий (см. гл. 24).
Реагенты
• Буферный раствор хлорида бария — триэтаноламина: взять навеску 61,077 г
ВаС12 * 2Н20 (М = 244,31); растворить приблизительно в 900 мл кипяченой де¬
ионизированной воды, не содержащей С02, добавить 29,84 г триэтаноламина
(N(CH2CH2OH)3, М = 169,19); гомогенизировать и довести pH до 8,1 с помощью
НС1; довести объем до 1 л; хранить реагент в бутыли, закрытой пробкой с трубкой,
заполненной натронной известью, чтобы защитить реагент от контакта с С02, со¬
держащимся в атмосферном воздухе.
• Раствор для замещения противоиона: взять навеску 61,077 г ВаС12 * 2Н20, растворить
приблизительно в 900 мл кипяченой деионизированной воды, не содержащей С02,
добавить 0,4 мл вышеописанного буферного раствора; довести объем до 1 л и за¬
щищать от атмосферного С02 так же, как и буферный раствор.
• Итоговый обменный раствор: навеску 123,21 г нитрата магния (Mg(N03)2 • 6Н20
(М = 256,43), хорошо высушенную в эксикаторе, растворить приблизительно
в 900 мл деионизированной воды; довести объем до 1 л.
• Индикатор бромкрезол зеленый (3,3', 5,5' тетрабром-м-крезол сульфонфгалеин):
приготовить 0,1%-ный водный раствор.
604
Часть 3. Неорганический анализ
• Смешанный индикатор: взять навеску 1,250 г метилового красного (л-диметил-
аминоазобензол-2-карбоновая кислота; М = 269,29); взять навеску 0,825 г мети¬
ленового синего (3,7-бис-диметиламинофенотиоцианит хлорид, М = 373,90); рас¬
творить в 1 л нейтрального 90°-ного этанола.
Процедура
• Измерить влажность почвы для коррекции результатов анализа на почву, высушен¬
ную при 105 °С.
• Взять навеску 5 г почвы, добавить 25 мл буферного раствора и гомогенизировать.
• Выдерживать в контакте в течение 1 ч, не допуская контакта с С02 воздуха.
• Перенести смесь в небольшую воронку Бюхнера диаметром 50 мм, оснащенную
тонким фильтром для количественного анализа, и медленно профильтровать.
• Процедить через осадок 75 мл буферного раствора, добавляемого малыми порция¬
ми через одинаковые промежутки времени.
• Поместить фильтрат в мерную колбу объемом 200 мл.
• Добавить 100 мл раствора замещающего иона (ВаС12) небольшими порциями
и смешать с фильтратом в колбе объемом 200 мл.
• Довести объем до 200 мл замещающим раствором (OK-раствор); хранить влажную
почву в воронке Бюхнера до продолжения определения ЕКО.
• Подготовить холостую пробу, смешивая 100 мл ТЭА-буферного раствора и 100 мл
замещающего раствора.
• Промывать почву метанолом (объемом около 100 мл) до удаления хлорида (тест
с AgN03).
• Удалить избыточный метанол промыванием раствором 0,0005 М (0,001 н.) хлорида
бария.
• Уплотнить почву в воронке Бюхнера, распределив по плоскости, для удаления
0,001 н. ВаС12 (погрешность, вызванная присутствием остаточного 0,001 н. ВаС12,
пренебрежимо мала, однако ее можно скорректировать взвешиванием воронки
Бюхнера вместе с фильтром и почвой).
• Добавить 250 мл обменного раствора Mg(N03)2 малыми порциями, выдерживая
воронку Бюхнера под слабым вакуумом; общая длительность контакта должна со¬
ставить около 16 ч.
• Довести объем раствора обменных ионов до 250 мл (ЕКО раствор).
• Провести количественный анализ ионов бария методом атомно-эмиссионной
(489 нм), абсорбционной или ИСП-спектрометрии (Pansu и др., 2001), с помощью
стандартов, приготовленных в обменном растворе Mg(N03)2.
• Измерение обменной кислотности в ОК-растворе: добавить две капли индикатора -
бромкрезола зеленого и несколько капель смешанного индикатора; тировать 0,1 М
раствором НС1 до изменения окраски раствора на серо-фиолетовую.
Расчеты
Обменная кислотность ОК в смоль (Н+)/кг (почвы), выражается формулой
OK =100( V— V0)/w,
где Vx и Уо — объемы 0,1 М раствора НС1, необходимого для обратного титрования хо¬
лостых и рабочих проб, соответственно (OK-фильтрат); w — вес пробы с поправкой на
влажность почвы, высушенной при 105 °С.
Глава 26. Емкость катионного обмена
605
Величина ЕКО в смоль (+)/кг выражается формулой:
ЕКО = 25—Df,
W
где ТВа — титр бария для ЕКО раствора в ммоль (1/2 Ва2+)/л по калибровочной кривой;
Df— коэффициент разведения.
Примечания
Предложенный метод включает нейтрализацию по нескольким типам кислотности (см.
гл. 23):
• Кислотность, определяемая обменными протонами.
• Кислотность, возникающая вследствие гидролиза в почвах с pH < 5,2.
А13+ + ЗОН- -» А1 (ОН)3
А1* (ОН)*-' + у ОН--» А1* (ОН)*
• Кислотность, определяемая присутствием органического вещества (кислотные
функциональные группы), в особенности при pH < 5,5 (Thomas, 1982).
26.4.4. Методики, использующие буферные растворы с различными pH
Эти методики очень полезны для изучения почв с переменными зарядами. Они по¬
зволяют осуществить стандартизацию повторяющихся процедур, используя методики
определения ЕКО в буерной среде с простой заменой экстрагирующего реагента. Заря¬
ды, образующиеся при различных значениях pH, можно анализировать с использовани¬
ем двух или трех измерений в кислой, основной и нейтральной среде, в зависимости от
типа почвы. Это позволяет в допустимом приближении определить ЕКО при pH почвы
и помогает выделить пробы, для которых следует провести более подробные исследова¬
ния заряда (например, ТНЗ, см. гл. 20).
Обменные свойства аллофановых почв характеризует параметр ДЕКО, представляю¬
щий разность между ЕКО, измеренной при pH 3,5, и ЕКО, измеренной при pH 10,5. Тем
не менее, процесс растворения минеральных и органических фракций, наблюдаемый
при экстремальных значениях pH в этих типах почв, иногда приводит к получению не¬
достоверных результатов измерений (Aomine и Jackson, 1959; Yoshinaga и Aomine, 1962;
Alexiades и Paxinos, 1965; Espinoza, 1969).
Методика, использующая 0,05 М раствор (1/2 СаС12) (Fey и Leroux, 1976), применя¬
лась
• при pH 3, 5 и 7 для измерения поверхностных зарядов, зависящих от pH (ЕКО-
ЕАО; Hendershot и Lavkulich, 1983);
• при pH 3, 4, 5, 6, 7, 8 в системе Ca(N03)2-NH4Cl (Duquette и Hendershot, 1987).
Методику с использованием 1 М раствора ацетата аммония применяли в диапазоне
pH 4-9 при анализе аллофановых почв (Gautheyrou и Gautheyrou, 1981), стабильность па¬
раметров ацетатно-аммонийного буфера позволяет работать в данном диапазоне pH, не
прибегая к другим системам (рис. 26.7).
В андосолях аллофановые вещества, неразличимые для рентгеновских лучей, легко
обнаруживаются тестом со фторидом натрия, однако существенные колебания значе¬
ний ЕКО при pH 7 и, в особенности, при pH 9 характерны для почв с переменными
зарядами. Данная методика позволяет измерять (для одного значения pH) изменения
зарядов почв, сохраняемых при исходной влажности и после длительной сушки на воз-
606
Часть 3, Неорганический анализ
Рис. 26.7. Соотношение между pH и емкостью катионного обмена (измеренной с помощью 1 М
раствора ацетата аммония в воздушно-сухих пробах) аллофановых почв из Гваделупы (Gautheyrou
и Gautheyrou, 1981)
духе. Кроме того, можно исследовать эффект предварительной обработки, например,
насыщения ЕАО реагентом, содержащим РО*_. Действительно, добавление РО*_ приво¬
дит к уплотнению поверхностных отрицательных зарядов в гидрандептах, что вызывает
снижение уровня pH до 0 (El Swaify и Sayegh, 1975).
Схема образования отрицательных зарядов (М = Fe, Al, Si, Tl):
М-ОН2 + Н2РО- 7 ——► м-н2ро4-+ н2о
pH £ 5,5
В почвах с сильной алюминиевой кислотностью противоположный эффект наблюда¬
ется для алюминия (Uehara и Keng, 1975). Таблица 26.2 иллюстрирует воздействие фос¬
фат иона на ЕКО в различных типах почв (Gautheyrou и Gautheyrou, 1981).
Таблица 26.2. Зависимость колебаний значений ЕКО от предварительной обработки —
анионного насыщения фосфат-ионами (Gautheyrou и Gautheyrou, 1981; почвы
с естественной влажностью, не подвергавшиеся воздушной сушке, результаты
приведены к почвам, высушенным при 105 °С).
Почва
Происхождение
Влажность,
%
T(NH,)pH 7,0 Общий Р,
смоль (+)/кг сг/кг
1
2
1
2
Криандепт
Эквадор Е
264 а
35,31
10,50
13,50
100,00
234
Падрандепт
Антильские о-ва В
570 с
189,43
22,25
38,00
177,50
1450
Имоголитная
Антильские о-ва
В 570 f
116,68
19,50
44,00
67,25
1700
Ферралитная
Антильские о-ва
L415 а
4,59
14,00
15,00
42,50
244
Ферралитная
Антильские о-ва
L415b
4,23
11,50
11,50
25,00
310
Галлуазит
Антильские о-ва
L21 b
8,58
16,50
20,00
37,50
275
1 - без предварительной обработки.
2 - с насыщением гидрофосфатом калия.
Глава 26. Емкость катионного обмена
607
Использованная литература
Alexiades С A and Paxinos SA (1965) A method for determination of allophane in soil clays. Agric. Anals.
Aristotelian Univ. (Thessaloniki), 275-301.
Amavis (1959) Comparison des mdthodes de mesure de la capacity d’dchange d’ions d’un sol. Mise au
point d'une methode rapide. Science du Sol, AFES, 8, 317-325.
Aomine S and Jackson ML (1959) Allophane determination in ando soils by cation-exchange capacity
delta value. Soil Sci. Soc. Am. Proc., 23,210-213 .
Bascomb CL (1964) A rapid method for the determination of cation exchange capacity of calcareous and
non calcareous soils. J. Sci. Food. Agric., 15, 821-823.
Begheyn L Th (1987) A rapid method to determine cation exchange capacity and exchangeable bases in
calcareous, gypsiferons, saline and sodic soils. Commun. Soil Sci. Plant Anal., 18,911-932.
Bergmann К and O’Konski CT (1963) A spectroscopic study of methylene blue monomer, dimer and
complexes with montmorillonite. J. Phys. Chem., 67,2169-2177.
Bergseth H and Abdel-Aal Shi (1975) Ion exchange removal of calcium carbonate and gypsum from
mineral material prior to determination of cation exchange capacity using 89 St2*. Colloid Polym.
Sci, 253, 322-324.
Bolt GM (1982) Soil Chemistry - В - physico-chemical models. ELSEVIER, Developments in soil
science 5B, 226-229.
Cenens J and Schoonheydt RA (1990) Quantitative absorption spectroscopy of cationic dyes on clays.
Proc. 9th Intern. Clay Conference, Strasbourg. In Pub. Sci. Geol. Mem., Farmer VC and Tardy Y ed.
85,15-23.
Chabra R, Pleysier J and Cremers A (1976) The measurement of the cation exchange capacity and
exchangeable cations in soils : a new method. Proc. Int. Clay Conf., 1,439-449.
Coleman NT and Thomas GW (1967) The basic chemistry of soil aridity. In Soil Acidity and Liming,
Pearson RN et Adams F ed. Am. Soc. Agr.
Cornell RM and Aksoyoglu ES (1991) Simultaneaous determination of the cation exchange capacity and
the exchangeable cations on marl. Clay Miner., 26, 567-570.
Cornell RM and Aksoyoglu ES (1991) Simultaneous determination of the cation exchange capacity and
the exchangeable cations on marl. Clay Miner., 26, 567-570.
Drechsel P (1987) Determining cation exchange capacity and exchangeable cations in saline, calcareous
and gypserous soils. Z. Pflanzenerndhr. Bodenk., 150, 357.
Duquette M and Hendershot WH (1987) Contribution of exchangeable aluminium to cation exchange
capacity at low pH. Canad. J. Soil Sci., 67, 175-185.
Edmeades DC and Clinton OE (1981) A simple rapid method for the measurement of exchangeable
cations and effective cation exchange capacity. Commun. Soil Sci. Plant Anal, 12, 683-695.
El Swaify SA and Sayegh AW (1975) Charge characteristics of an oxisol and an inceptisol from HawaY.
Soil Sci., 120, 49.
Espinoza WG (1969) Determinacion de alofan en suelos de Nuble mediante el valor delta de la capacidad
total de intercambio cationico. Agricultura Tecnica (Chili), 29, 127-132.
Esquevin J (1954) Mesure de la CEC des argiles par le chlorure de cobalti- hexamine. Museum Histoire
Naturelle, Paris.
Fallavier P, Babre D and Breysse M (1985) Determination de la capacity d’Schange cationique des sols
tropicaux acides. Agronomie Tropicale, 40, 298-308.
Fey MV and Leroux J (1976) Electric charges on sesquioxidic soils clays. Soil Sci. Soc. Am. J., 40,
359-364.
Frenkel H, Gerstl Z and Van de Veen Ж (1986) Determination of gypsum and cation exchange capacity
in arid soils by a resin method. Geoderma, 39, 67-77.
Fripiat JJ and Helsen J (1966) Use of cobalt hexamine in the cation exchange capacity determination of
clays. Clays Clay Miner., 14, 163-169.
Gangaiya P and Morrison RJ (1987) A simple non-atomic absorption procedure for determining the
effective cation exchange capacity of tropical soils. Soil Sci. Plant Anal., 18, 1421-1430.
Gautheyrou J and Gautheyrou M (1981) Contribution & 1 ’etude de la capacity d’echange des sols h allophane:
aspect analytique de la CEC et ses consequences sur V interpretation pedo-agronomique., Notes
laboratoires, Vol. 1, 274 pp., vol. 2, 123 pp. + annexe, 154 profils, IRD (ex-Orstom), Antilles, Paris.
608 Часть 3. Неорганический анализ
Gillman GP (1979) A proposed method for the measurement of exchange properties of highly weathered
soils. Austr. J. Soil Res., 17, 129-139.
Gupta RK, Singh CP and Abrol IP (1985) Determining cation-exchange capacity and exchangeable
sodium in alkali soils. Soil Sci., 139, 326-332.
Hendershot WH and Lavkulich LM (1983) Effect of sesquioxide coatings on surface charge of standard
mineral and soil samples. Soil Sci. Soc. Am. J., 47, 1252-1260.
Hoffinan U and Dammler J (1969) Die methylenblauadsorption on mont- morilonite. Chimia, 23, 476-480.
ISRIC (1987) Procedures for Soil Analysis, 9-5/9-7. International soil reference and information center,
2e edition.
Johanson A (1961) Cobalt - an expedient agent in soil testing for T, S and exchangeable Ca, Mg and Mn.
Soil Sci., 364-368.
Johnson CE Jr (1957) МёЛу1ёпе blue adsorption and surface area measurements. 131st National Meeting
Am. Chem. Soc., 7-12.
Juo ASR, Ayanjala SA and Ogunwale JA (1976) An evaluation of cation exchange capacity measurements
of soils in the tropics. Commun. Soil Sci. Plant Anal., 1, 751-761.
Kamprath EJ (1970) Exchangeable A1 as a criterium for liming leached mineral soils. Soil Sci. Soc. Am.
Proc., 34, 252-254.
Keita MK and Van Der Pol F (1987) Comparison of exchangeable bases and CEC by the cobalti-hexamine
method and the standard ammonium acetate method on some malinese soils. ISRIC, 87-98.
Kelley WP (1948) Cation exchange in soils., Reinhold. ACS Monograph no. 109, 144 pages.
Lagaly G (1981) Characterization of clays by organic compounds. Clay Minerals, 16,1-21.
Mantin I and Glaeser R (1960) Fixation des ions cobalti-hexamine par les montmorillonites acides. Bull.
Groupe Fr. Argiles, 12, 83-88.
Mantin I (1969) Mesure de la capacity d^change des ттёгаих argileux par l’6thyl£ne diamine et les ions
complexes et l^thytene-diamine. C.R. Acad. Sc. Paris, 269, 815-818.
Mas A, Peigneur P and Cremers A (1978) Stability of metal (uncharged) ligand complexes in ion
exchanges. Part 2 : The copper-ethylene-diamine complex in montmorillonite and sulphonic acid
resin. J. Chem. Soc. Faraday Trans., 1 (74), 182-189.
Mehlich A (1938) Use for triethanolamine acetate-barium hydroxyde buffer for determination of some
base exchange properties and lime requirements of soil. Soil Sci. Soc. Am. Proc., 3,165-166.
Mehlich A (1942) Rapid estimation of base-exchange properties of soils. Soil Sci., 53,1-14.
Mehlich A (1953) Rapid determination of cation and anion exchange properties and pH of soils. J. Assoc.
Agr. Chem., 36, 445^157.
Misopolinos ND and Kalovoulos JM (1984) Determination of CEC and exchangeable Ca and Mg in non¬
saline calcareous soils. Soil Sci., 35, 93-98.
Morel R (1957) Etude exp6rimentale des ph6nom£nes d’&hange sur diff<6rents ттёгаих argileux. Ann.
Agron., 6, 5-90.
Nevins MJ and Weintritt DJ (1967) Determination of cation exchange capacity by methylene blue
adsorption. Am. Ceram. Soc. Bull., 46, 587-592.
NF ISO 11260 (1994) D£termination de la сараскё d’£change cationique effective et du taux de saturation
en bases ёchangeables & l’aide d’une solution de chlorure de Baiyum. In Qualite des sols, 1999,
AFNOR, Paris, 415-428.
NF ISO 14254 (1997) Determination de Vacidite echangeable dans un extrait au chlorure de baryum.,
AFNOR, Paris.
Oliver R (1984) Etude comparative de deux n^thodes d’extraction et de dosage des bases et de la capacity
d^change sur les sols du Sinegal. Agronomie Tropicale, 39, 14-21.
Orsini, L. & Remy, J.C. 1976. Utilisation du chlorure de cobaltihexammine pour la dёtermination
simulta^e de la сараскё d^change et des bases ёс1ш^еаЫе8 des sols. Science du Sol, 4, 269-275.
Pansu M, Gautheyrou J and Loyer JY (2001) Soil Analysis - Sampling, Instrumentation and Quality
Control., Balkema, Lisse, Abington, Exton, Tokyo, 489 pp.
Peech M, Alexander LT, Dean LA and Ree JF (1947) Methods of Soil Analysis for Soil Fertility
Investigations., US Dept. Agr., 757, 25 p.
Peigneur P (1976) Stability and Adsorption Affinity of Some Transition Metalamine Complexes in
Alumino-Silicates., Thfcse Univ. Louvain.
Глава 26. Емкость катионного обмена
609
Phelps GW and Harris DL (1967) Specific surface and dry strength by methylene blue adsorption. Am.
Ceram. Soc. Bull., 47, 1146-1150.
EKO с органическими катионами
Pleysier J and Cremers A (1973) Adsorption of silver thiourea complexe in montmorillonite. J. Nat., 243,
86-87.
Pleysier J and Cremers A (1975) Stability of silver-thiourea complexes in montmorillonite clay. Chem. J.
Soc. Faraday Trans., I (71), 256-264.
Pleysier JL and Juo ASR (1980) A simple extraction method using silver. Thiourea for measuring
exchangeable cations and effective CEC in soils with variable charges. Soil Sci., 129,205-211.
Polemio M and Rhoades JD (1977) Determining cation exchange capacity : a new procedure for
calcareous and gypserous soil. Soil Sci. Soc. Am. J., 41, 524-528.
Prianishnikov D (1913) Quantitative bestimmung der in boden vorhanden absorptiv gebunden basen.
Landw. Vers. Stat, (79/80), 667-680.
Rhoades JD (1982) Cation exchange capacity. In Methods of Soil Analysis, part 2, Page AL, Miller RH
and Keeney DR ed. Am. Soc. Agronomy, 2,154-157.
Rytwo G, Serban C, Nir S and Margulies L (1991) Use of methylene blue aid crystal violet for
determination of exchangeable cations in mont- morillonite. Clays Clay Miner., 39, 551-555.
Sayeh AH, Khan NA, Khan P and Ryan J (1978) Factors affecting gypsum and cation exchange capacity
determinations in gypsiferous soils. Soil Sci., 125, 294-300.
Schollenbeiger CJ and Simons RH (1945) Determination of exchange capacity and exchangeable bases
in soils. Soil Sci., 59,13-24.
Schollenberger CJ (1927) Exchangeable hydrogen and soil reaction. Science, 65, 552-553.
Searle PL (1986) The measurement of soil cation exchange properties using the single extraction, silver
thiourea method : an evaluation using a range of New Zealand soils. Austr. J. Soil Res., 2,193-200.
Sharma OP and Dubey DD (1988) Evaluation of suitable method for estimating CEC of sodic black soils.
J. Indian Soc. Soil Sci., 36, 546-549.
Siegried CH, Weinert W and Strelow FWE (1986) The influence of thiourea on the cation-exchange
behaviour of various elements in dilute nitric and hydrochloric acids. Talanta, 33,481-487.
Stuanes PO, Ognes G and Орет M (1984) Ammonium nitrate as extractant for soil exchangeable cations,
exchangeable acidity and aluminum commun. Soil Sci. Plant Anal., 15, 773-778.
Stucki JW, Golden DC Roth CB, (1984) Effects of reduction and reoxydation of structural iron on the
surface charge and dissolution of diodahedral smectites. Clays Clay Miner., 32, 350-356.
Thomas GW (1982) Exchangeable cations. In Methods of Soil Analysis, part 2, Page AL, Miller RH and
Keeney DR ed. Am. Soc. Agronomy, 2e edition, 9 (2), 159-165.
Tucker BM (1985) A proposed New Reagent for the measurement of cation exchange properties of
carbonate soils. Aust. J. Soil Res., 23, 633-642.
Uehara G and Keng J (1975) Management implications of soil mineralogy in Latin America. In Soil
Management in Tropical America, Bomemisza E et Alvarado A ed. North-Carolina Univ. Press, 351.
Van Rosmalen HA (1980) Evolution and Modification of the Determination of Exchangeable Bases and
Cation Exchange Capacity of Calcareous and Gysiferous Soils by Using Silver-Thiourea., Royal
Tropical Inst. Dept. Agric. Res. Project A0B039 (Amsterdam), Report BO, 80-83.
Yaalon DH, Van Schuylenborgh J and Slager S (1962) The determination of cation exchange characteristics
of saline and calcareous soils. Nether. J. Agr. Sci., 10, 218-222.
YoshinagaN and Aomine S (1962) Imogolite in some ando soils. Soil Sci. Plant Nutr., 8, 22-29.
Yuan TL (1959) Determination of exchangeable hydrogen in soils by a titration method. Soil Sci., 88,
164-167.
Дополнительная литература
Общая теория EKO
Chu CH and Johnson LJ (1979) Cation-exchange behavior of clays and synthetic aluminosilica gels.
Clays Clay Miner., 27, 87-90.
610 Часть 3. Неорганический анализ
Dyer A, Shaheen Т and Newton GWA (1995) Speciation observed by cation exchange. Sci. Total Environ.,
173-174, 301-311.
Effron D, Jimenez MP, Horra AM de la (2000) Cation exchange capacity at the soil pH level to be applied
to acid soils: methods and determination. Capacidad de intercambio cationica al pH del suelo, para
suelos acidos: metodo de determinacion. Agrochimica., 44,61-68.
Erp PJ van, Houba VJG and Beusichem ML van (2001) Actual cation exchange capacity of agricultural
soils and its relationship with pH and content of organic carbon, Communications-in-Soil-Science-
and-Plant-Analysis, 32, 19-31.
Fauziah Cl, Jamilah I and Omar SRS (1997) An evaluation of cation exchange capacity methods for acid
tropical soils, PertanikaJ. Tropical Agric. Sci., 20, 113-119.
Kalra YP and Maynard DG (1994) A comparison of extractants for the determination of cation exchange
capacity and extractable cations by a mechanical vacuum extractor. Commun. Soil Sci. Plant Anal.,
25, 1505-1515.
Khan NA (1994) Comparison of CEC values with and without pretreatment of gypsiferous soils. Sarhad
Journal of Agriculture, 10, 713-720.
Liu CL, Wang MK and Yang CC (2001) Determination of cation exchange capacity by one-step soil
leaching column method. Commun. Soil Sci. Plant Anal., 32,2359-2372.
Pleysier JL, Jansens J and Cremers A (1986a) A clay suspension stability and point titration method for
measuring cation exchange capacity of soils. Soil Sci. Soc. Am. J., 50, 887-891.
Pleysier JL, Jansens J and Cremers A (1986b) Extraction of cations from some kaolinitic soils of the
tropics. 1SRIC - Proceedings of International Workshop on Laboratory Methods and Data Exchange
Programme, 51-65.
Ralchev 1’ and Toncheva R (1997) Kinetics of the desorption of cations. I. A mathematical model testing.
Pochvoznanie, Agrokhimiya у Ekologiya., 32, 34-36.
Stuck i JN, Golden DC and Roth CB (1984) Effects of reduction and reoxydation of structural iron on the
surface charge and dissolution of dioctahedral smectites. Clays Clay Miner., 32, 350-356.
Sumner ME and Miller WP (1996) Cation exchange capacity and exchange coefficients, (1996). In
Methods of Soil Analysis, part 3, Chemical Methods, Bigham JM and Bartels JM ed. SSSA-ASA,
Madison, WI Etats-Unis, 1201-1229.
Zhao BJ, Lam MT, Back MH, Gamble DS and Wang C (1997) Soil cation exchange capacity measurements
using ultrafiltration techniques: comparison of different metal ions as substitutes. Commun. Soil Sci.
Plant Anal., 28, 161-171.
Zhi ZL, Rios A and Valcarel M (1994) Direct determination of the cation- exchange capacity of soils with
automatic sample pretreatment in a flow system. Anal. Chim. Acta., 298, 387-392.
Методика применения солей бария при естественном pH почвы
Gillman GP, Bruce RC, Davey BG, Kimble JM, Searle PL and Skjemstad JO (1983) A comparison of
methods used for the determination of cation exchange capacity. Commun. Soil Sci. Plant Anal., 14,
1005-1014.
Hendershot WH and Duquette M (1986) A simple Barium chloride method for determining cation
exchange capacity and exchangeable cations. Soil Sci. Soc. Am4, 50, 605-608.
Lambert K, Vanderdeelen J and Baert L (1988) An improved method for cation exchange capacity
determination of peat soils. Pedologie, XXXVIII(l), 5-14.
Matsue N and Wada К (1985) A new equilibrium method for cation exchange capacity measurement. Soil
Sci. Soc. Am. J., 49, 574-578.
Методика с применением буферных систем при pH 7,0
Arbelo CD and Hemandez-Moreno JM (1992) Cation exchange capacity of Andosols as determined
by the ammonium acetate (pH7) method. Capacidad de cambio en Andosoles pro los metodos del
acetato amonico (pH7) у cloruro de basio no tamponado. Agrochimica, 36: 1-2, 53-62.
Chapman HD (1965) Cation-exchange capacity. In Methods of Soil Analysis. Part 2. Chemical and
Microbiological Properties, Black C.A. et al. ed. American Society of Agronomy, 9, 891-901.
FAO (1972) Physical and Chemical Methods of Soil and Water Analysis., FAO Soils Bulletin no. 10.
Глава 26. Емкость катионного обмена
611
Jackson ML (1958) Soil Chemical Analysis., Prentice-Hall, New York.
Jaynes WF and Bigham JM (1986) Multiple cation-exchange capacity measurements on standard clays
using a commercial mechanical extractor. Clays Clay Miner., 1, 93-98.
Kalra YP and Maynard DG (1986) An evaluation of automated and manual methods for NH4-N analysis
in the determination of cation exchange capacity of soils. In Proceedings of International Workshop
on the Laboratory Methods and Data Exchange Programme (Wageningen), Pleijsier LK ed. 67-76.
Kalra YP and Maynard DG (1990) An evaluation of a mechanical vacuum extractor for the determination
of cation exchange capacity and extractable cations in calcareous soils. Trans. 14th Int. Cong. Soil.
Sci.t Kyoto II, 451-452.
Kalra YP and Maynard DG (1991) Methods manual for forest soil and plant analysis. Forets Canada -
Information Report NOR-X-319, 84-94.
Kelley WP (1927) A general discussion of the chemical and physical properties of alkali soils. 1st Int.
Congr. Soil Sci. Proc483-489.
Metson AT (1956) Methods of Chemical Analysis for Soil Survey Samples., USDA- Soil Bureau, Bull. 12.
Peech M (1945) Determination of exchangeable cations and exchange capacity of soils. Rapid
micromethods utilizing centrifuge and spectro photometer. Soil Sci., 59,25-38.
Schuman GE, Stanley MA and Knudsen D (1973) Automated total nitrogen analysis of soil and plant
samples. Soil Sci. Soc. Am. Proc., 37, 480-481.
Skjemstad JO and Reeve R (1976) The determination of nitrogen in soils by rapid high-temperature
kjeldahl digestion and autoanalysis. Commun. Soil Sci. Plant Anal., 7,229-239.
TECHNICON (1973) Method for NH4 N 154-71 W {colorimetry)., Technicon Instrument Corporation
Industrial Methods.
USDA (1972) Soil Survey Laboratory Methods and Procedures for Collecting Soil Samples., USDA -
Soil survey investigations, report no. 1.
Определение ЕКО с помощью хлорида гексааминокобальта(Ш)
Gautheyrou J and Gautheyrou M (1958) Ditermination des cations echangeables et de la CEC des sols a
allophane developes sur cendres volcaniques recentes (soufriere de Guadeloupe) avec le chlorure de
cobalti-hexamine., IRD (ex-Orstom), Guadeloupe, Paris, rapport multigraphte, 1-4.
Maes A, Tits J, Mermans G and Dierckx A (1992) Measurement of the potentially available charge and the
dissociation behaviour of humic acid from cobalti-hexamine adsorption. J. Soil Sci.t 43,669-677.
Ciesielski H and Sterckeman T (1997) Determination of cation exchange capacity and exchangeable
cations in soils by means of cobalt hexamine trichloride. Effects of experimental conditions.
Agronomie, 17: 1, 1-7.
Метод, с комплексом серебро — тиомочевина
Pleysier J (1976) Silver uncharged ligand complexes in aluminosilicates: adsorption and stability., These
Univ. Louvain.
Van Reeuwijk LP (1987) Procedures for soil analysis cation exchange capacity and exchangeable bases
(silver-thiourea method). ISRIC, International Soil Reference and Information Center, 2e edition,
10-U10-6.
ЕКО с органическими катионами (окрашенные реагенты)
Brindley GW and Thompson TD (1970) Methylene blue adsorption by montmorillonite. Determination
of surface areas and exchange capa cities with different initial cation saturations. Isr. J. Chem., 8,
409-415.
Cenens J and Schoonheydt RA (1988) Visible spectroscopy of methylene blue on hectorite, laponite В
and barasym in aqueous suspension. Clays Clay Miner., 36, 214-224.
Hang PT and Brindley GW (1970) Methylene blue absorption by clay minerals. Determination of surface
areas and cation-exchange capacities (clay- organic studies XVII). Clays Clay Miner., 18,203-212.
Marguliers L, Rozen H and Nir S (1988) Model for competitive adsorption of organic cations on clays.
Clays Clay Miner., 36,270-276.
612 Часть 3. Неорганический анализ
Phelps GW and Harris DL (1967) Specific surface and dry strength by methylene blue adsorption. Am.
Ceram. Soc. Bull., 47, 1146-1150.
Santoni S, Bonifacio E and Zanini E (2001) Indophenol blue colorimetric method for measuring cation
exchange capacity in sandy soils. Commun. Soil Sci. Plant Anal, 32,2519-2530.
Методики с применением буферных систем при pH 8,0-8,6
Bower СА, ReiteMeier PF and Fireman M (1952) Exchangeable cation analysis of saline and alkali soils.
Soil Sci., 73,251-261.
Хлорид бария — триэтаноламин при pH 8,1
Bradfields R and Allison WH (1933) Criteria of base saturation in soils. Trans. 2d Comm. lnt. Soil Sci.,
A, 63-79.
Mehlich A (1938) Use of triethanolamine acetate-barium hydroxyde buffer for determination of some
base exchange properties and lime requirement of soil. Soil Sci. Soc. Am. Proc., 3,162-166.
Mehlich A (1953) Rapid determination of cation and anions exchange properties and pHO of soils.
J. Assoc. Agric. Chem., 36, 445-457.
Peech M (1965) Exchange acidity. In Methods of Soil Analysis Black C.A. et al. part 2. Am. Soc.
Agronomy, 9, 905-913.
USDA (1972) Soil Survey Laboratory Methods and Procedures for Collecting Soil Samples., USDA. Soil
survey investigations report no. 1, p. 23.
Глава 27. Емкость анионного обмена
27.1. Теория
Положительные заряды в почве образуются либо при возникновении краевых зарядов
в результате разрушения атомных плоскостей структурных единиц почвы, либо из-за
оксидов железа и алюминия, покрывающих некоторые глины кристаллической струк¬
туры или занимающих межслоевое пространство в кристаллической решетке. Наличие
этих зарядов приводит к поглощению анионов (Zelazny и др., 1996).
До 1975 года емкость анионного обмена (ЕАО) меньше интересовала исследователей,
главным образом потому, что в основных типах почв умеренных зон влияние анионов
слабее, чем влияние катионов, связывающихся с отрицательно заряженными поверх¬
ностями. Это особенно справедливо для глин 2:1, в которых плохо определяется точка
нулевого заряда (ТНЗ, см. гл. 20), поскольку постоянный отрицательный заряд слишком
велик и не может быть уравновешен менее значительными положительными зарядами
(рис. 27.1), возникающими в краевых зонах разрушенных структурных единиц, приводя
к очень высокому отношению ЕКО к ЕАО (Bingham и др., 1965).
Поскольку в глинах 1:1 величина ЕКО относительно низка, влияние краевых зарядов
приводит к снижению отношения ЕКО и ЕАО, и следовательно, влияние ЕАО стано¬
вится более значительным, в особенности если частицы малы и имеют высокую степень
разупорядочения. В некоторых кислых тропических почвах, содержащих оксиды метал¬
лов и органические анионы, эти факторы играют существенную роль (Theng, 1979; Tate
и Theng, 1980; Eick и др., 1999). В сподосолях, ультисолях, оксисолях и андосолях эти па¬
раметры могут варьировать от 1 до 10 ммоль/кг при отношениях ЕКО к ЕАО < 1. В андо¬
солях присутствие аллофанов-имоголитов и органического вещества может резко изме¬
нить обменные свойства. В этих почвах с соотношением кремния к алюминию, близким
к единице, при естественном pH почвы идет активное формирование положительных
зарядов (Wada и Окатит, 1977,1980; Окатит и Wada, 1978; Cruz Huerta и Kientz, 2000).
С другой стороны, если эти почвы содержат алюминиево-гуминовые комплексы, то
положительные заряды слабы, что позволяет различать комплексы А1-ОН, содержащие¬
ся в гуминовой и минеральной фракциях (у органических материалов низкая рНО, сни¬
жающая рНО почвы при фиксации больших органических анионов).
Значительный уровень поглощения фосфора в андосолях обусловлен обменными ме¬
ханизмами, а также структурными замещениями (Rajan, 1975; Parfitt и Henmi, 1980).
Высокую ЕАО индуцируют полимеры оксидов железа и алюминия (Schwertmann
и Taylor, 1989).
В сильно выветренных почвах величина ЕАО может превышать ЕКО; например,
при измерениях по методу Шлмана (Gillman, 1979) в акрогумоксе ЕКО может составить
0,6 смоль(+)/кг, а ЕАО — 3,7 смоль(—)/кг.
Долгое время измерение АЕС считалось менее важным по сравнению с ЕКО, по¬
скольку различные анионы, участвующие в обмене, редко удерживаются простыми
электростатическими связями; как правило, связывание происходит за счет более слож¬
ных взаимодействий. Лишь обмен анионов С1~, NO~ и ClOj возможен без селективной
фиксации; замена одного аниона на другой анион, аналогичный по ионной силе, не
меняет электрофоретической подвижности частиц. Не наблюдается изменений специ-
614
Часть 3, Неорганический анализ
Рис. 27.1. Зависимость заряда поверхностей аллофана и монтмориллонита от pH (Clark
и McBride, 1984; McBride, 1989; ТНЗ, точка нулевого заряда; ЕАО — емкость анионного обмена,
ЕКО — емкость катионного обмена)
фичности, связанных с размером аниона и, вероятно, со стерическими факторами
(Schwertmann и Taylor, 1989).
Другие анионы могут более прочно удерживаться на поверхности оксидов за счет об¬
разования координационных связей или хемосорбции (например, различные силикаты,
молибдаты, арсенаты, селенаты, органические анионы). Ацетат-ион адсорбируется за
счет кулоновского взаимодействия, тогда как цитрат-ион в качестве лиганда алюминия
удерживается сильнее. Поведение анионов описывается моделью адсорбции (Bowden
идр., 1980).
В случае полианионов, чьи экономические последствия в агрономии значительны
(напр.,РО*_, SO2"), ЕАО не определяется обменными формами. В дополнение к про¬
цессу обмена следует принимать во внимание и другие процессы, такие как выпадение
в осадок нерастворимых солей железа, алюминия или щелочноземельных элементов.
Удержанные фосфаты можно эмпирически разделить на две фракции:
* Подвижная фракция, которую можно экстрагировать различными кислыми или
основными реагентами с помощью ряда методик. Эту подвижную фракцию зача¬
стую ошибочно называют «фосфором, доступным для растений», или «легко до¬
ступным фосфором», или «экстрагируемым фосфором» (см. гл. 29).
• Фракция, которая фиксируется по различным механизмам, в частности, таким как
осаждение или включение в комплексы, и может быть количественно измерена
методами дифференциального анализа в насыщающей среде, содержащей избыток
фосфора, методами удержания (см. раздел 29.4).
Почвы с переменными зарядами, обогащенные оксидами железа и алюминия,
с pH < рНО (т. е. более кислые в сравнении с равновесным pH, где положительные
и отрицательные заряды присутствуют в равных количествах (см. гл. 20)), имеют по¬
ложительно заряженную поверхность. Из-за высоких значений рНО гидроксидов железа
Глава 27. Емкость анионного обмена
615
и алюминия (на уровне pH 7-8) почвы, в которых эти гидроксиды преобладают, имеют
высокие АОЕ.
Специфическое связывание анионов, таких как фторид-ион, может привести к зна¬
чительному высвобождению ионов ОН", что позволяет проводить анализ почв, содер¬
жащих активные формы алюминия (аллофановые почвы, почвы, содержащие металло¬
органические алюминиево-гуминовые комплексы) с помощью NaF теста (Kawaguchi,
и др., 1954; Fieldes и Perrott, 1966; US DA, 1975; Shoji и Ono, 1978; Parfitt и Henmi, 1980;
Лшкидр., 2001).
Косвенный эффект насыщения положительных зарядов обменного комплекса иона¬
ми РО*~ может выразиться в повышении ЕКО аллофановых почв, о чем сообщается во
многих публикациях (Mekarus и Uehara, 1972; Schalscha и др.,1972, 1974; Juo и Madukar,
1972; Sawhney, 1974; Rajan, 1976; Byden и Syrs, 1975, 1976; Parfitt и Atkinson, 1976; Galindo
и Bingham, 1977; Garcia-Miragaya, 1984). Вытеснение связанной с металлом молекулы
воды фосфатом приводит к уменьшению положительного заряда и, таким образом,
к повышению общего отрицательного заряда. Дополнительные отрицательные заряды
могут формироваться в окультуренных почвах при внесении фосфатных удобрений.
В полях реакции протекают медленно и непрерывно, и подвижность фосфора низка
(рис. 27.2). Структура связей на поверхностях коллоидных частиц до сих пор не впол¬
не изучена, в особенности для оксидов алюминия, железа или марганца, а также для
аморфных веществ. Одно- или двухъядерные комплексы могут образовываться с желе-
Рис. 27.2. Фиксация и доступность для растений фосфата в почве в зависимости от pH почвы
(согласно Brady, 1974)
^он о
Fe II
/ \о-|_о
\ о
Fe^
1\ж
I он
FcC
\
1\>Н
Рис. 27.3. Одноядерные и двухъядерные комплексы фосфора с железом
616
Часть 3. Неорганический анализ
зом (рис. 27.3; Schwertmann и Taylor, 1989). Обменная емкость может измениться за счет
процессов, происходящих на поверхности раздела вода — коллоидная частица (Charlet
и Schlegel, 1999), в частности, под влиянием переувлажнения (Triana и др., 1995).
Если осаждения нерастворимых солей не происходит, лиотропные ряды анионов,
играющих важную роль в агрономии, выглядят следующим образом (Bolt и de Наап,
1982):
S044- > Р043- > SO3- > NO: > Cl-
38 А 36 А 27 А
На рис. 27.4 представлено поглощение фторидов, силикатов и фосфатов на гетите как
функция pH.
Рис. 27.4. Поглощение анионов—фосфата, силиката и фторида—на гетите (по Hingston и др., 1972)
27.2. Измерения
27.2.1. Основные положения
Методы измерения ЕАО основаны на тех же принципах, что и методы определения
ЕКО, и имеют те же ограничения. Требуется только изменить знак валентного заряда на
противоположный. Если поверхность заряжена положительно, любой избыток анионов,
сохраняющийся за счет электростатических связей в двойном слое, является обменным.
ЕАО представляет максимальное количество вероятно связанных анионов на единицу
веса почвы.
Анионообменный комплекс насыщают противоанионом, не способным к специфи¬
ческому связыванию в условиях определенного pH и ионной силы (как правило, в ка¬
честве противоанионов используют хлорид- или нитрат-анионы, однако использование
нитратов при выраженных окислительно-восстановительных превращениях невозмож¬
но). Затем удаляют избыток анионов, поглощенный противоанион вытесняют другим
противоанионом и проводят количественный анализ, представляя результы в виде ко¬
личества молей поглощенного аниона на единицу массы.
При определении ЕАО необходимо соблюдать те же правила, что и при определении
ЕКО: учитывать отношение pH растворов к количеству поглощенных ионов; необходи-
Глава 27. Емкость анионного обмена
617
мость введения повторных добавок для снижения буферной емкости и сдвига равнове¬
сия; влияние длительности контакта и температуры, влияющих на скорости достижения
равновесия.
Поскольку определение ЕАО связано с применением легко перемещаемых ионов,
нельзя использовать поливалентные анионы, такие как сульфат и фосфат, из-за их
способности к специфической адсорбции (координации), а также из-за осаждения не¬
растворимых солей обменных катионов в реакционной среде. ЕАО обычно составляет
1-5% величины ЕКО (Bolt и de Наап, 1982).
Методика, описанная ниже, предложена Вадой и Окамурой (Wada и Окатига, 1977).
27.2.2. Методика
Реактивы
• 1 М раствор хлорида аммония (NH4C1, М = 53,50).
• 0,1 М раствор хлорида аммония.
• 1 М раствор нитрата калия (KN03, М = 101,10).
Процедура
• Измеряют содержание влаги в пробе, чтобы внести поправку в результаты измере¬
ний на пробы почвы, высушенной при 105 °С.
• Отвешивают 0,5 г почвы, измельченной до размера частиц 0,5 мм, помещают
в предварительно взвешенную пробирку для центрифугирования.
• Добавляют 10 мл 1 М раствора NH4C1 (pH этого раствора должен быть около 6,0).
• Встряхивают в течение 30 мин; центрифугируют при 5000 g, надосадочный раствор
сливают.
• Суспендируют осадок после центрифугирования в 10 мл раствора 0,1 М NH4C1, за¬
тем проводят центрифугирование; обработку повторяют пять раз.
• Взвешивают пробирку вместе с осадком, содержащим почву, насыщенную ио¬
нами С1~, для определения количества удержанного 0,1 М раствора NH4C1:
уС в смоль(-)/кг (почвы).
• Проводят вытеснение иона С1~ путем пятикратного добавления порций 1 М рас¬
твора KN03 объемом 10 мл, между каждым добавлением центрифугируют раствор;
смешивают экстракты и доводят объем до 50 мл. Это раствор для определения сум¬
марного С1".
• Проводят количественный анализ содержания С1~ в экстракте спектрофотометри¬
чески; оС выражают в смоль (-)/кг (почвы).
• ЕАО выражают в смоль(-)/кг (почвы) через оС — уС.
27.3. Одновременное измерение ЕАО, ОКат, ЕКО и чистой ЕКО
27.3.1. Цель
В целях подбора аналитической методики, позволяющей одновременно определять по¬
тенциал и заряд почвы, являющиеся причиной обменной активности в сильно выве¬
тренных почвах, было проведено тестирование ряда методик. Для достижения условий,
приближенных к естественным (pH и ионная сила, соответствующие почвенному рас¬
твору), применялись небуферные и сильно разбавленные среды.
618
Часть 3, Неорганический анализ
Многие методики включают использование катиона бария — Ва2+ и его солей (напри¬
мер, Bascomb, 1964; Gillman, 1979; Gillman и Bakker, 1979; Uehara и Gillman, 1981; Gillman
и Sumpter, 1986).
К сожалению, Ba2+ несовместим с такими анионами, как сульфат, который зачастую
присутствует в аллофановых почвах или же применяется для восполнения нехватки пи¬
тательных веществ в почве, а также для коррекции влияния алюминиевой кислотно¬
сти в сильно выветренных почвах (например, суперфосфатные удобрения или добавки
сульфата кальция). Поэтому были протестированы другие анионы, такие как хлорид
и нитрат, поскольку они присутствуют в обменных комплексах в небольших количе¬
ствах и не склонны к связыванию за счет селективного поглощения. Простота и низкая
стоимость количественного анализа позволяет использовать эти методики для рутинных
анализов с удовлетворительной воспроизводимостью.
27.3.2. Описание методики
Основные положения
Описанный здесь метод (Cochrane и De Souza, 1985) применяется для анализа сильно
выветренных почв с переменными зарядами. Систему, состоящую из пробы почвы
и разбавленного раствора для насыщения противокатионами и противоанионами, име¬
ющего ионную силу, близкую к аналогичному параметру почвенного раствора, при¬
водят в равновесное состояние. Затем фиксированные катионы и анионы вытесняют
другими противоионами и проводят аналитические измерения для расчетов ЕКО и ЕАО
с поправкой на вес. Величину чистой ЕКО рассчитывают по разности.
Реагенты
• Раствор для насыщения ионами. 0,5 М раствор нитрата аммония (NH4N03,
М = 80,05): навеску 40,02 г NH4N03 растворяют приблизительно в 900 мл деиони¬
зированной воды, доводят объем до 1 л.
• Раствор для выравнивания ионной силы. 0,0215 М раствор нитрата аммония: наве¬
ску 1,721 г NH4N03 растворяют в 1 л деионизированной воды.
• Вытесненяющий раствор противоиона. 0,02 М раствор хлорида калия (КС1,
М = 74,55): навеску 1,491 г КС1 растворяют приблизительно в 900 мл деионизирован¬
ной воды; доводят объем до 1000 мл после достижения температурного равновесия.
Процедура
Экстрагирование
• Навеску 3 г просеянной почвы с размером частиц 0,5 мм помещают в предвари¬
тельно взвешенную пробирку для центрифугирования объемом 50 мл с завинчи¬
вающейся крышкой.
• Добавляют 30 мл 0,5 М раствора нитрата аммония (NH4N03).
• Встряхивают в течение 2 ч и центрифугируют при 6000 g.
• Надосадочный раствор сливают: это раствор А.
• Готовят взвесь почвенного остатка в 30 мл 0,0215 М раствора NH4N03, что при¬
близительно соответствует осмотическому потенциалу почвенного раствора (эту
концентрацию можно скорректировать в соответствии с конкретной ситуацией).
• Встряхивают в течение 60 мин,
• Центрифугируют при 6000 g.
Глава 27. Емкость анионного обмена
619
• Надосадочный раствор сливают; повторяют обработку дважды,
• Взвешивают пробирку + остаток почвы после центрифугирования для расчета
удержанного 0,0215 М раствора NH4N03, уА — количество удержанного аммония
(или нитрата), выраженное в смоль/кг (почвы).
• Добавляют 30 мл вытесняющего 0,02 М раствора КС1 (осмотический потенциал
которого близок к осмотическому потенциалу 0,0215 М раствора NH4N03).
• Суспендируют осадок и встряхивают в течение 60 мин.
• Центрифугируют при 6000 g.
• Сохраняют надосадочный раствор, содержащий обменные ионы аммония
и нитрат-ионы.
• Повторяют эту обработку дважды и объединяют три экстракта.
• Доводят объем до 100 мл с помощью 0,02 М раствора КС1: это раствор В.
Определение ЕКО и ЕАО
• Определяют общее содержание аммония в растворе В (см. гл. 28 или воспользо¬
ваться методикой, описанной в работе (Вгетпег и Keeney, 1966), которое представ¬
ляет собой сумму обменного аммония и аммония из 0,0215 М раствора NH4N03
(количественный анализ обычно проводят методом автоматической колориметрии
или путем микродистилляции с последующей волюмометрией): это величина оА.
Рассчитывают ЕКО по количеству обменного аммония (см. гл. 26), выражая резуль¬
таты всмоль(+)/кгпочвы:1
ЕКО = оА - уА.
• Определяют общее содержание нитратов (см. гл. 28), включая обменные нитраты
и нитраты из 0,0215 М раствора NH4N03: это величина oN.
Рассчитывают ЕАО по обменным нитратам, результаты выражают в смоль (—)/кг:
ЕАО = oN - уА;
• Чистую ЕКО определяют по уравнению:
Чистая ЕКО = ЕКО - ЕАО.
Если нельзя провести количественный анализ в тот же день, что и экстракцию, добав¬
ляют в раствор две капли толуола, чтобы остановить биохимические процессы, и хранят
экстракты в холодильнике, не допуская воздействия света.
Определение обменных катионов
Измеряют pH раствора А и проводят количественное определение Са, Mg, К, Na, Fe,
Mn, А1 методом атомно-абсорбционной спектрометрии или спектрометрии с индуктив¬
но связанной плазмой, измеряют содержание сульфат- и хлорид-ионов с помощью аб¬
сорбционной спектрометрии (см. гл. 31 или Pansu и др., 2001):
Са + Mg + К + Na = ОКат (обменные катионы)
А1 + Fe + Mn = О Кис (обменная кислотность)
ЕС + ЕА = эффективная ЕКО (расчет суммированием).
Избыток хлорид- или сульфат-ионов указывает на вероятное присутствие в почве
растворимых солей или гипса.
620
Часть 3. Неорганический анализ
Использованная литература
Bascomb CL (1964) Rapid method for the determination of cation-exchange capacity of calcareous and
non-calcareous soils. J. Sci. Food Agric., 15, 821-823.
Bingham FT, Sims JR and Page AL (1965) Retention of acetate by montmorillonite. Soil Sci. Soc. Am. J.,
29, 670-672.
Bolt GH and De Haan FAM (1982) Anion exclusion in soil. In Soil Chemistry, В - Physico Chemical
Models, Bolt GH ed. Elsevier Amsterdam, Development in Soil Sciences, 5 B, 233-257.
Bowden JW, Nagarajah S, Barrow NJ, Posner A and Quirk JP (1980) Describing the adsorption of
phosphate, citrate and selenite on a variable charge mineral surface. Aust. J. Soil Res., 18,49-60.
Brady NC (1974) The Nature and Properties of Soils., MacMillan New York, 8th edition.
Bremner JM and Keeney DR (1966) Determination and isotope ratio analysis of 3- exchangeable
ammonium, nitrate and nitrite by extraction-distilation methods. Soil Sci. Soc. Am. Proc., 30, 577-
583.
Charlet L and Schlegel ML (1999) La capacite d’echange des sols. Structures et charges a l’interface eau/
particule. Comptes-Rendus-de-rAcademie-d’Agriculture-de-France, 85, 7-24.
Clark CJ and McBride MB (1984) Cation and anion retention by natural and synthetic allophane and
imogolite. Clays Clay Miner., 32, 291-299.
Cochrane TT and Desouza GDM (1985) Measuring suface charge characteristics in oxisols and ultisols.
Soil Sci., 140, 223-22.
Fieldes M and Perrott KW (1966) The nature of allophane in soils. Part 3, Rapid field and laboratory test
for allophane. N. Z. J. Sci., 9,623-629.
Galindo GG and Bingham FT (1977) Homovalent and heterovalent cation exchange equilibria in soils
with variable surface charge. Soil Sci. Soc. Am. J., 41, 883-886.
Garcia-Miragaya J (1984) Effect of phosphate sorption of the cation exchange capacity of two Savannah
ultisols from Venezuela. Commun. Soil Sci. Plant Anal., 15, 935-943.
Cruz Huerta L and Kientz DG (2000) Electric charge of andosols of ‘Cofre de Perote’, Veracruz, Mexico
Carga electrica de los andosoles del Cofre de Perote, Veracruz, Mexico. Terra, 18,115-124.
Eick MJ, Brady WD and Lynch CK (1999) Charge properties and nitrate adsorption of some acid
southeastern soils. J. of Environ. Quality, 28, 138-144.
Gillman GP and Bakker P (1979) The Compulsive Exchange Method for Measuring Surface Charge
Characteristics of Soil., CSIRO-Division of Soils, Report 40 .
Gillman GP and Sumpter EA (1986) Modifications to the compulsive exchange method for measuring
exchange characteristics of soils. Aust. J. Soil Res., 24, 61-66.
Gillman GP (1979) A proposed method for the measurement of exchange properties of highly weathered
soils. Aust. J. Soil Res., 17, 129-139.
Hingston FJ, Posner AM and Quirk JM (1972) Anion adsorption by goethite and gibbsite. 1) The role of
the proton in determining adsorption envelopes. J. Soil Sci., 23, 177-192.
Juo ASR and Madukar HP (1974) Phosphate sorption of some nigerian soils and its effect on cation
exchange capacity. Commun. Soil Sci. Plant Anal., 5,479-497.
Kawaguchi KH, Fukutani H, Murakami H and Hattori T (1954) Ascension of pH valves of ventral NaF
extracts of allitic soils and semi quantitative determination of active alumina by the titration method.
Bull. Res. Inst. Food Sci., (Kyoto University, Japan), 14, 82-9.
McBride MB (1989) Surface chemistry of soil minerals. In Minerals in Soil Environments, Dixon JB and
Weed SB ed. Soil Science Society of America, 2, 35-88.
Mekarus T and Uehara G (1972) Anion adsorption in ferruginous tropical soils. Soil Sci. Soc. Am. Proc.,
36, 296-300.
Okamura Y and Wada К (1978) Charge characteristics of Kuroboku soils : effect of pH and ion
concentration. Soil Sci. Soil Manure (Japan), 24, 32.
Pansu M, Gautheyrou J and Loyer JY (2001) Soil Analysis — Sampling, Instrumentation and Quality
Control., Balkema, Lisse, Abington, Exton, Tokyo, 489 p .
Parfitt RL and Atkinson RJ (1976) Phosphate adsorption of goethite (a.FeOOH). Nature, 264, 740-742.
Parfitt RL and Henmi T (1980) Structure of some allophanes from New Zealand. Clays Clay Miner., 28,
285-294.
Rajan SSS (1975) Mechanism of phosphate adsorption by allophanic clays. N. Z. J. Sci., 18, 93-101.
Глава 27. Емкость анионного обмена
621
Rajan SSS (1976) Changes in net surface charge of hydrous alumina with phosphate adsorption. Nature,
262, 45-46.
Ryden JC and Syers JK (1975) Charge relationships of phosphate sorption. Nature, 255, 51-53 .
Ryden JC and Syers JK (1976) Calcium retention in response to phosphate sorption by soils. Soil Sci. Soc.
Am. J., 40, 845-846.
Sawhney BL (1974) Charge characteristics of soils as affected by phosphate sorption. Soil Sci. Soc. Am.
Proc., 38, 159-160.
Schalscha EB, Pratt PF and Soto D (1974) Effect of phosphate adsorption on the cation-exchange capacity
of volcanic ash soils. Soil Sci. Soc. Am. Proc., 38, 539-540.
Schalscha EB, Pratt PF, Kinjo T and Amar JA (1972) Effect of phosphate salts as saturation solutions in
cation-exchange capacity determinations. Soil Sci. Soc. Am. Proc., 39, 912-914.
Schwertmann U and Taylor RM (1989) Iron hydroxydes. In Minerals in Soil Environments, Dixon JB and
Weed SB ed. Soil Science Society of America, 8, 379-438.
Shoji S and Ono T (1978) Physical and chemical properties and clay mineralogy of andosols from
Kitakami (Japan). Japan Soil Sci., 126,297-312.
Tate KR and Theng BKG (1980) In Soils with Variable Charge. Organic Matter and its Interactions with
Inorganic Soil Constituents, Theng BKG ed. New Zealand Society of Soil Science, 225-249.
Theng BKG (1979) Formation and Properties of Clay-Polymer Complexes., Elsevier Amsterdam, 362
pages. 4
Triana A, Leffoy RDB, Blair GJ, Date RA ed. Grundon NJ ed. Rayment GE ed. and Probert ME (1995)
The effect of flooding on S sorption capacity and AEC of variable charge soils. In Proceedings of the
Third International Symposium, Brisbane, Queensland, Australia, 12-16 September 1993, Kluwer
Dordrecht, 135-139.
Uehara G and Gillman G (1981) The Mineralogy Chemistry and Physics of Tropical Soils with Variable
Charge Clays., Westview Tropical Agriculture Series, 4, 170 pages.
USDA (1975) Soil Survey Staff!, A Basic System for Making and Interpreting Soil Surveys., USDA
Handbook, 436 p.
Wada К and Okamura Y (1977) Measurements of exchange capacities and hydrolysis as means of
characterizing cation and anion retention by soils. Proceedings of the International Seminar on Soil
Environment and Fertility Management in Intensive Agriculture. Soc. Sci. Soil Manure (Japan),
811-815.
Wada К and Okamura Y (1980) Electric charge characteristics of ando A and buried A horizon soils. J.
Soil Sci., 31, 307-314.
Zelazny LW, Liming HE and An Vanwormhoudt M (1996) Charge analysis of soils and anion exchange.
In Methods of Soil Analysis, Part 3, Chemical Methods, Bigham JM and Bartels JM ed. SSSA-ASA,
Madison, WI Etats-Unis, 1231-1253.
Глава 28. Неорганические формы азота
28.1. Введение
28.1.1. Ионы аммония, нитрата и нитрита
Разложение азотсодержащего органического вещества приводит к образованию солей
аммония, которые под воздействием нитрифицирующих бактерий окисляются до ни¬
тритов и нитратов. Нитрат-анион обнаруживают в дренажных водах, весенних стоках
и реках после выщелачивания ирригационными или дождевыми водами. В тропических
зонах, где часты грозы, дождевая вода может содержать некоторое количество нитратов,
образующихся в процессе прямого синтеза из атмосферного азота.
В почве соли аммония частично поглощены обменным комплексом, и их, в отличие
от хорошо растворимых нитратов, нельзя экстрагировать в воду напрямую. Поэтому
экстракция обменного аммония требует замещения другим катионом, присутствующим
в солевом растворе. Кроме того, аммоний может быть частично фиксирован в слоях гли¬
ны, что делает его недоступным для обмена.
Считается, что нитрат-анион при концентрации в воде на уровне 80 мг/л представля¬
ет риск для здоровья. Стандарты ВОЗ1 рекомендуют устанавливать предельное содержа¬
ние нитратов на уровне 44 мг/л для взрослых и 20 мг/л для детей. Высокие концентра¬
ции нитратов зачастую являются признаками загрязнения. В озерах нитраты ускоряют
процессы эвтрофикации. В результате воздействия денитрифицирующих микроорга¬
низмов нитраты могут восстанавливаться до нитритов.
Нитрит-анион нестабилен в природных водах. Обнаружение нитрита в воде является
признаком загрязнения и говорит о том, что, вероятно, содержание нитрита в воде более
0,1 мг/л. При стерилизации воды хлораминами накапливается аммоний, который пре¬
вращается бактериями в нитрит.
28.1.2. Проблемы, связанные с отбором проб
Распределение ионов аммония, нитрата и нитрита в почве значительно варьирует. Раз¬
личия результатов анализа проб, отобранных с участка площадью всего лишь 2 га, соста¬
вили до 40%. Следовательно, при анализах такого типа следует уделять особое внимание
отбору проб.
Чтобы результаты были точными, важно следовать строгой процедуре как при поле¬
вом отборе проб, так и при подготовке их к анализу; при этом все операции должны быть
полностью стандартизированы. Вместо спирального бура предпочтительно использо¬
вать приспособление для отбора проб, предложенное Guiot( 1975). Пробоотборник Guiot
напоминает полуцилиндрический бур позволяет отбирать пробы на точно заданной глу¬
бине и в меньшей степени, чем другие устройства, разрушает структуру пробы почвы.
Пробы образцов готовятся во влажном состоянии и хранятся не более 3 дней при тем¬
пературе менее 10 °С. В качестве альтернативы можно проводить экстракцию в полевых
условиях и доставлять растворы в лабораторию. Кроме того для расчета поправочно¬
го коэффициента, по одной аликвоте из каждой пробы следует высушить в сушильном
1 WHO: World Health Organization — ВОЗ (Всемирная организация здравоохранения).
Глава 28. Неорганические формы азота
623
шкафу при 105 °С . Правила отбора и хранения проб почвы для определения неоргани¬
ческого азота в свежей почве изложены в стандарте ХР Л31-115 (1995). По возможности
измерения следует проводить in situ.
28.1.3. Аналитические проблемы
Экстракты могут быть нестабильными, однако их можно стабилизировать добавлением
I мл насыщенного раствора хлорида ртути1 на каждые 100 мл экстракта, в особенности,
если требуется количественное определение нитрита.
Экстракционные растворы обычно содержат множество посторонних ионов с неиз¬
вестной концентрацией, и могут быть окрашенными или мутными, что затрудняет, а то
и делает невозможными точные колориметрические определения.
Классическая методика определения солей аммония путем дистилляции в щелочной
среде даже при работе под вакуумом включает риск образования новых ионов аммония
из растворимого органического вещества, содержащего азот.
Исходя из этого зачастую предпочтительнее сначала отделить неорганические азот¬
содержащие соединения, предназначенные для количественного анализа. Микродиф-
фузионная методика отделения аммония (см. раздел 28.2.2) Легко осуществима и при¬
годна при анализе следовых количеств. После разделения экстракта можно без помех
определить аммоний.
28.2. Традиционные методики
28.2.1. Экстракция обменных форм
Простая водная экстракция теоретически пригодна для хорошо растворимых нитратов,
однако полной экстракции других форм неорганического N, в особенности аммония,
адсорбированного на обменном комплексе, достичь не удается. Следовательно, для экс¬
тракции следует использовать солевые растворы. Наиболее распространенным экстра¬
гентом является 1 М раствор хлорида калия. Тем не менее, в почвах, содержащих гипс,
растворимость сульфата кальция может привести к ошибкам при количественном опре¬
делении, в частности, колориметрическим методом. 0,5 М раствор сульфата калия, по¬
зволяющий снизить растворимость сульфата кальция, столь же эффективен, как и хло¬
рид калия, применяемый для экстракции иона аммония.
Оборудование и реагенты
• Флаконы емкостью 500 мл с крышками.
• Ротационная мешалка, 30 об./мин.
• Экстрагирующий раствор. 0,5 М раствор сульфата калия (KjSO^.
Методика
• 50 г почвы (см. подготовку пробы в разделе 28.1.2) помещают во флаконы вмести¬
мостью 500 мл, содержащие 100 мл экстрагирующего раствора.
- Закупоренные флаконы встряхивают в течение 1 ч на ротационной мешалке; от¬
стаивают до образования прозрачного надосадочного раствора; если раствор не
прозрачен, его фильтруют или центрифугируют.
Каломель. — Примеч. науч. ред.
624
Часть 3. Неорганический анализ
• Фильтрат используют для определения обменного аммония, нитрита и нитрата.
Если немедленное проведение количественного анализа невозможно, хранят рас¬
творы в защищенном от света месте при 4 °С.
Примечание
Рекомендованный вес образца для почв с низким содержанием азота (тропических почв)
составляет пятьдесят граммов. Для почв, обогащенных азотом, вес пробы может быть
ниже. Некоторые исследователи (Keeney и Nelson, 1982 и Mulvaney, 1996) предложили
брать по 10 г почвы при том же объеме (100 мл) раствора для экстракции.
28.2.2. Разделение микродиффузионным методом
Основные положения
Эта методика была разработана и применена в 1851 г. (Schloesing) и в 1889 г. (Fresenius),
и кодифицирована в 1962 г. Конвеем (Conway). Методика рекомендована для анали¬
за почвы, так как она является простой и прецизионной, в особенности при анализе
окрашенных и непрозрачных экстрактов (Mulvaney, 1996; Mulvaney и др., 1997; Mulvaney
и Khan 1999; Khan и др., 2000).
Показано, что при комнатной температуре из слабощелочного раствора, содержаще¬
го ионы аммония, образцы с большой площадью поверхности и малым размером частиц
могут выделить весь аммоний в форме газообразного аммиака. Метод заключается в се¬
лективном перемещении молекул из исследуемого раствора, в котором давление паров
аммиака велико, в раствор с нулевым давлением паров.
Неорганические формы азота необходимо перевести в летучую легко удаляемую фор¬
му — аммиак. Для ионов аммония достаточно простого подщелачивания реакционной
среды. Сначала необходимо восстановлением с помощью сплава Деварда провести пре¬
вращение нитрат- и нитрит-ионов в ионы аммония.
Выделившийся аммиак можно накапливать в растворе борной кислоты, содержащем
индикатор, и титровать 0,01 М раствором серной кислоты методом, аналогичным ме¬
тоду определения общего азота (см. «Методика (макрометод)» в гл. 10) и органических
форм азота (см. раздел 14.2.1). Поскольку уровень содержания неорганических форм
N зачастую низок, лучше воспользоваться нижеописанной методикой, основанной на
высокочувствительном колориметрическом определении.
Оборудование
Для реализации методики — она была разработана на основе методики Конвея и адапти¬
рована для анализа почвы (Blachere и Ferry, 1957) — необходимо простое оборудование.
Описанное устройство позволяет проводить серийный анализ проб (Susini и Gandjui,
1964).
Используются широкогорлые колбы Эрленмейера емкостью 500 мл; колбу закрывают
резиновой пробкой со стеклянной трубкой диаметром около 10 мм, на конце которой
расположен полый стеклянный шарик диаметром около 20 мм. Когда пробка закрывает
колбу, стеклянный шарик должен быть погружен на глубину, равную половине расстоя¬
ния от дна до горла колбы (рис. 28.1).
Реагенты
• Нормальный раствор серной кислоты. Помещают 2,7 мл концентрированной серной
кислоты в 100 мл дистиллированной воды; это раствор для заполнения стеклянно¬
го шарика.
Глава 28. Неорганические формы азота
625
Рис. 28.1. Приспособление для диффузии в колбе Эрленмейера объемом 500 мл
• Насыщенный раствор карбоната калия (приблизительно 112 г на 100 мл при 20 °С).
• Раствор Конвея. Одна часть 40%-ного раствора каустической соды и три части на¬
сыщенного раствора карбоната калия.
• Сплав Деварда в порошкообразной форме (50% порошка алюминия, 45% порошка
меди и 5% порошка цинка). 1 г этой смеси образует приблизительно 50 мл водорода
в момент выделения.
• Сулъфаминовая кислота (NH2S03H). Помещают 2 г в 100 мл воды; хранят в холо¬
дильнике; готовят растворы непосредственно перед каждой серией анализов для
предотвращения гидролиза.
Методика (аммоний, нитрат + нитрит)
Точно отмеряют 20 мл экстракта, полученного согласно «Процедуре» раздела 28.2.1,
в колбу Эрленмейера емкостью 500 мл (см. рис. 28.1). Рекомендуется готовить серии из
10 колб единовременно. Начинают с выдерживания стеклянного шарика в 1 н. раство¬
ре серной кислоты. Тщательно смачивают, вынимают и удаляют избыточную жидкость.
В каждую колбу вносят по 1,5 мл насыщенного раствора карбоната калия; закрывают
пробкой с трубкой со стеклянным шариком. Убеждаются в том, что колба надежно за¬
купорена. Выдерживают при комнатной температуре в течение 48 ч.
По окончании этого времени осторожно открывают колбу, избегая соприкосновения
стеклянного шарика со стенками колбы и потери капли жидкости, находящейся на ша¬
рике. Удерживая стеклянный шарик над воронкой (рис. 28.2), промывают его малым
количеством дистиллированной воды, собирая стекающую промывную воду в мерную
колбу объемом 20 мл. Доводят объем до 20 мл дистиллированной водой и оставляют до
проведения количественного анализа. Добавляют 40-50 мг порошкообразного сплава
Деварда в колбу Эрленмейера, содержащую остаток экстракционной смеси. Как и на
предыдущем этапе, погружают стеклянный шарик в раствор серной кислоты. Добавля¬
ют в колбу Эрленмейера 4,5 мл смеси Конвея, немедленно закрывают пробкой и про¬
должают процедуру так, как описано выше. Теперь количество вытесненного аммония
соответствует содержанию нитратов и нитритов, восстановленных сплавом Деварда.
626
Часть 3. Неорганический анализ
Рис. 28.2. Выделение вытесненного аммония (к рис. 28.1)
Примечание
Сплав Деварда за счет выделения водорода может создать небольшое избыточное давле¬
ние и выдавить пробку; следует убедиться в том, что пробка достаточно надежна и проч¬
но закреплена.
Разложение и количественный анализ нитритов
Термин «нитраты» зачастую используют применительно к сумме нитратов и нитритов;
оба соединения одинаково реагируют на обработку сплавом Деварда. Поскольку ни¬
триты весьма нестабильны, можно предположить, что их концентрация пренебрежимо
мала в сравнении с концентрацией нитратов, однако это не всегда так, и, возможно, по¬
требуется отдельный анализ нитритов.
Кроме того, можно провести разложение нитрита перед анализом нитрата. Подкис¬
ления экстракта до pH 1 и контакта с воздухом в течение 1 ч достаточно для разложения
нитрита. Другой путь: добавить 0,2 мл раствора сульфаминовой кислоты, перемешать
и перед микродиффузионным анализом выдержать смесь в течение 5 мин. Сочетание
методик с разложением и без разложения нитритов позволяет оценить содержание ни¬
тритов по разности результатов измерений.
28.2.3. Колориметрическое определение аммония
Основные положения
Для проведения микроанализа пригодна колориметрическая методика. В щелочной сре¬
де в присутствии гипохлорита и фенола аммиак образует индофенол, придающий раство¬
ру синюю окраску. Этот метод анализа признан эталонным. Если измерения проводятся
в растворе аммиака, выделенного микродиффузионным методом (см. раздел 28.2.2), то
анализ проходит без помех. Методика позволяет измерить:
627
Глава 28. Неорганические формы азота
• содержание аммония в экстракте после первой микродиффузии;
• суммарное содержание нитритов и нитратов после второй микродиффузии в при¬
сутствии сплава Деварда;
• только нитраты, если нитриты были предварительно разложены (см. «Разложение
и количественный анализ нитритов»).
Содержание аммония можно определить и непосредственно в экстракционном рас¬
творе (см. раздел 28.2.1) в диапазоне концентраций от 0,005 до 20 мг/л (Keeney и Nelson,
1982); при этом существует риск внесения дополнительной погрешности, вызванной
присутствием кальция и магния. Эти помехи можно уменьшить, применяя сильно раз¬
бавленные среды и добавляя ЭДТА.
Колориметрическое определение аммония можно автоматизировать, как описано
в гл. 10 («Методика», раздел 10.2.7) и гл. 26 (см. «Методика» раздел. 26.4.2).
Оборудование и реагенты
Колориметрию проводят при монохроматическом излучении. Следует выбрать длину
волны, на которой наблюдается максимальное поглощение в диапазоне 590-650 нм
(оранжевый) — 636 нм согласно (Keeney и Nelson, 1982). Колориметрическая ячейка
с длиной оптического пути 20 мм позволяет достичь хорошей чувствительности. Линей¬
ная зависимость соответствует концентрациям N от 0,05 до 0,3 мг7л.
Реагенты для колориметрического анализа
• Раствор фенолята натрия: растворить 2,5 г гидроксида натрия приблизительно
в 40-50 мл дистиллированной воды; охладить, добавить 5 г высокочистого фенола,
растворить и довести объем до 100 мл; этот раствор должен быть абсолютно бес¬
цветным.
• 0,2Мраствор гидрофосфата натрия (Na2HPOA): растворить 71,65 г Na2HP04 * 12Н20
в 1 л деионизированной воды.
• 0,05%-ный раствор нитропруссида натрия приготовить непосредственно перед ис¬
пользованием из 1%-ного основного раствора.
• Растворы гипохлорита четырех уровней концентраций активного хлора, начиная
с коммерческого раствора при 20-22 °С. Точный титр коммерческих растворов
следует проверять следующим образом: отмеряют 1 мл коммерческого раствора,
добавляют 50 мл дистиллированной воды, несколько кристаллов йодида калия
и 5 мл чистой уксусной кислоты; встряхивают смесь и титруют (до исчезновения
желтой окраски, вызванной выделившимся йодом) раствором гипосульфита на¬
трия, содержащего 24,8 г Na2S203 • 5Н20 в 1 литре воды (0,1 н. раствор). Титр рас¬
твора рассчитывают по формуле:
Активный хлор = мл раствора гипосульфита х 1,12.
• Раствор ЭДТА: растворить 6 г двузамещенной натриевой соли этилендиаминтетра-
уксусной кислоты в 80 мл деионизированной воды, довести pH до 7, довести объем
до 100 мл и хорошо встряхнуть.
Диапазон стандартов
А — Исходный раствор хлорида аммония: навеску 0,191 г чистого NH4C1 растворить в дис¬
тиллированной воде, довести объем до 500 мл. 1 мл раствора А содержит 0,1 мг N.
В — Приготовить разбавленный раствор хлорида аммония разведением раствора А1:100.
1 мл раствора В содержит 0,001 мг N.
628
Часть 3. Неорганический анализ
Примечание:
Раствор В можно хранить лишь несколько часов. Значительное расхождение между ре¬
зультатами настоящей и предыдущих серий указывает на необходимость использования
свежеприготовленных растворов.
Методика
Калибровка
Помещают раствор В хлорида аммония в объемах, указанных в табл. 28.1, в мерные кол¬
бы объемом 20 мл, затем добавляют в указанном порядке, перемешивая раствор после
каждого добавления реагента:
• 2 мл фенолята натрия;
• 2 мл 0,2 М раствора гидрофосфата натрия;
• 0,5 мл 0,05%-ного раствора нитропруссида натрия;
• 2 мл раствора гипохлорита натрия четырех концентраций активного хлора.
Доводят объем до 20 мл дистиллированной водой. Встряхивают, для полноты измене¬
ния окраски выдерживают раствор в темноте по меньшей мере 30 мин. Затем проводят
колориметрические измерения при оптимальной длине волны из диапазона 590-650 нм
(см. «Оборудование и реактивы» в раздел 28.2.3). Закон Ламберта-Бера1 действует в диа¬
пазоне концентраций, представленном в табл. 28.1. Зависимость оптической плотности
от концентрации является линейной, цвет раствора остается стабильным более 24 ч.
Таблица 28.1. Приготовление калибровочных растворов для колориметрического анализа
разбавлением раствора В (концентрация N 1 мкг/мл)
Объем (мл) раствора В
на 20 мл
мг(>0/л
раствора
мкг (N)/r
почвы
мкг (NH^/r
почвы
мкг (NOjJ/r
почвы
1
0,05
0,1
0,13
0,44
2
0,1
0,2
0,26
0,89
4
0,2
0,4
0,51
1,77
6
0,3
0,6
0,77
2,66
10
0,5
1
1,29
4,43
(Соответствует концентрациям неорганического N, экстрагированного из 50 г почвы в 100 мл)
Количественный анализ
Вносят в мерные колбы емкостью 20 мл, содержащие раствор, собранный после опо¬
ласкивания стеклянного шарика, точно такие же количества реагентов, что и при по¬
строении калибровочной прямой (см. выше — «Калибровка»). Дают проявиться цвету
и измеряют поглощение так же, как при построении калибровочной прямой.
В зависимости от применяемой методики микродиффузии (см. раздел 28.2.2), резуль¬
тат измерений соответствует содержанию аммония, или суммарному содержанию ни¬
тратов и нитритов, или же только нитратов.
Слишком интенсивно окрашенные растворы можно разбавить. Контрольную пробу
следует разбавить в точно такой же степени.
Кроме того, возможно увеличение чувствительности методики увеличением интен¬
сивности окраски экстракцией окрашенного комплекса в изобутанол и измерением по¬
глощения при 655 нм.
1 Закон Бугера—Ламберта-Бера. — Примеч. науч. ред,
629
Глава 28. Неорганические формы азота
Примечание
Если микродиффузионный метод (см. раздел 28.2.2) не применяется, помещают точно
отмеренную аликвоту 5 мл экстракта, описанного выше в разделе 28.2.1, в мерные кол¬
бы объемом 20 мл. Добавляют 1 мл раствора ЭДТА концентрации 5 г/л, встряхивают
и выжидают 1 мин, затем продолжают процедуру, представленную в описании «Количе¬
ственный анализ». В этом случае калибровочные растворы (см. «Калибровка») следует
готовить с использованием раствора для экстракции (см. 28.2.1).
28.2.4. Колориметрическое определение нитритов
Основные положения
Данная методика (Rodier, 1984; Chariot, 1974) основана на диазотировании нитритами
при взаимодействии с сульфаниловой кислотой при pH 2,5 с последующим взаимодей¬
ствием образовавшегося соединения с а-нафтиламином (реактивом Пжсса). Образует¬
ся красный азокраситель, величину колориметрического поглощения определяют при
520 нм. Чувствительность методики можно увеличить концентрированием окрашенного
комплекса экстракцией в хлороформ при pH 9,5-10.
Реагенты
Для приготовления растворов и реагентов следует использовать воду, очищенную от ни¬
тритов путем ионного обмена на смешанной смоле (1 объем катионообменной смолы +
+ 2 объема анионообменной смолы).
• Раствор сульфаниловой кислоты: внести 1,2 г чистой сульфаниловой кислоты в при¬
близительно 140 мл горячей очищенной воды, охладить, добавить 40 мл чистой
хлористоводородной кислоты, довести объем до 200 мл деионизированной водой.
• Раствор а-нафтиламина: взять 1,2 г чистого а-нафгиламина, 2 мл чистой хлори¬
стоводородной кислоты, довести объем до 200 м деионизированной водой.
• Натрий-ацетатный буферный раствор: взять навеску 54,4 г CH3COONa • ЗН20 (или
32,8 г безводной соли), растворить, довести объем до 200 м деионизированной водой.
• Раствор ЭДТА с концентрацией 5 г/л. Используется для связывания в комплексы
железа и тяжелых металлов, способных вызвать помехи при измерениях.
Стандартный раствор нитрит-ионов
Приготовление
Исходное соединение для приготовления раствора — нитрит натрия (NaN02) — явля¬
ется гигроскопичной и хорошо растворимой в воде солью с точкой плавления 271 °С,
очень легко окисляющейся при контакте с воздухом или влагой. Поэтому рекоменду¬
ется проверять качество исходного продукта перед использованием. Растворы можно
законсервировать добавлением небольшого количества хлороформа. Взять точную на¬
веску в 150 мг нитрита натрия, растворить и довести объем до 100 мл деионизированной
водой. 1 мл полученного раствора содержит 0,1 мг нитрита.
Поскольку нитриты чрезвычайно нестабильны, концентрацию стандартного раство¬
ра следует проверять.
Проверка
Основные положения
Для проверки концентрации раствора волюмометрическим анализом применяют мето¬
дику, включающую окисление избытком перманганата калия:
630
Часть 3, Неорганический анализ
2MnO- + 5NO- + 6Н+ -> 2Mn02+ + 5N03" + ЗН20
Количество избыточного окислителя определяют методом йодометрического титро¬
вания.
Методика
• Точно отмеряют 10 мл 0,01 н. раствора перманганата и 2 мл 1 н. серной кислоты
и добавляют к пробе раствора нитрит-ионов объемом ПУ = 20 мл, подготовленной
для титрования.
• Встряхивают и добавляют 5 мл 10%-ного раствора йодида калия.
• Титруют йод 0,01 н. раствором гипосульфита натрия, V— объем раствора гипосуль¬
фита, израсходованного на титрование.
• Повторяют титрование с 20 мл дистиллированной воды вместо раствора нитрита;
V0 — новый объем раствора гипосульфита, израсходованного на титрование.
N0- в мг/л = С( V0- V) • 23 OOO/ПУ
Из этого раствора приготовить исходный раствор, содержащий NO~ в концентрации
1 мг/л.
Калибровочные растворы
Подготовить калибровочные растворы в диапазоне концентраций, представленных
в табл. 28.2, начиная с исходного раствора, содержащего 1 мг/л N0".
Таблица 28.2. Калибровочные растворы для колориметрического определении нитритов
Объем раствора 1 мг (NO~)/n, мл)
Соответствующая концентрация в 50 мл
раствора, мг/л
1
0,02
2
0,04
4
0,08
6
0,12
8
0,16
10
0,20
Методика
• Берут пробу почвенного экстракта (см. «Методика» в разделе 28.2.1) в диапазоне
концентраций калибровочных растворов (например, 50 мл или менее); доводят
объем до 50 мл деионизированной водой.
• Добавляют 1 мл раствора ЭДТА; встряхивают.
• Добавляют 1 мл раствора сульфаниловой кислоты; встряхивают и выдерживают
10 мин.
• Добавляют 1 мл раствора а-нафтиламина, 1 мл буферного раствора, встряхивают,
выдерживают раствор в течение 30 мин, проводят колориметрические измерения
при максимальном поглощении (520-540 нм); общий объем анализируемого рас¬
твора составляет 54 мл; поглощение изменяется линейно от концентрации в ис¬
следуемом диапазоне.
Глава 28. Неорганические формы азота
631
Если СЛ. — концентрация N0“ —N в почве в мкг/г; CL — концентрация N0“ -N в по¬
чве в мкг/мл;/— коэффициент поправки на влажность, то
Cs=2CLf.
Примечание
Если раствор почвенного экстракта (см. раздел 28.2.1) окрашен или непрозрачен, его
можно очистить перед измерением следующим путем:
• к 100 мл экстракта добавляют 5 мл раствора сульфата алюминия концентра¬
ции 120 г/л, затем добавляют постепенно подщелачивающий раствор (100 г
Na2C03 • ЮН20 + 50 г NaOH, на общий объем раствора 300 мл) до появления ще¬
лочной реакции;
• отделяют осадок фильтрованием;
• прозрачный раствор фильтрата используют для количественного анализа.
28.2.5. Колориметрическое определение нитратов
Основные положения
Нитраты можно анализировать с помощью селективного микродиффузионного метода
(раздел 28.2.2) из экстрактов почвы (раздел 28.2.1), восстановлением с колориметриче¬
ским определением аммония (раэдел28.2.3). Кроме того, имеется ряд других доступных
колориметрических методик определения нитратов. В основу одной из наиболее чув¬
ствительных и точных методик положена реакция восстановлении нитрата до нитрита
с последующим колориметрическим измерением (описана в разделе 28.2.4). Нитраты
восстанавливают, пропуская раствор почвенного экстракта (раздел 28.2.1) через колон¬
ку с омедненным кадмием.
Оборудование и реагенты
• Омедненный кадмий для восстановления: помещают 50 г крупнодисперсного по¬
рошка или гранулированного (1x2 мм) кадмия в колбу Эрленмейера вместимостью
400 мл; обрабатывают в течение 1 мин 250 мл 6 М хлористоводородной кислоты;
кислоту сливают, хорошо промывают деионизированной водой; обрабатывают
дважды 250 мл 2%-ного раствора сульфата меди (CuS04* 5Н20); раствор сливают
и промывают несколько раз деионизированной водой.
• 20%-ный раствор хлорида аммония: растворяют 100 г NH4C1 в деионизированной
воде и доводят объем до 500 мл.
• Разбавленный раствор NH4CI: отмеряют 50 мл 20%-ного раствора NH4C1 и доводят
объем до 2 л деионизированной водой.
• Другие реагенты. См. «Реагенты» в разделе 28.2.4.
Методика
Восстановление может быть проведено вручную в колонке из стекла Пирекс с диаме¬
тром 1 см, заполненной на высоту 20 см омедненным кадмием, элюированием около
75 мл разбавленного раствора NH4C1. Однако эта операция является длительной, и по¬
этому желательно ее автоматизировать, применяя оборудование, работающее в режиме
непрерывного потока. Хорошо продуманный коллектор позволяет проводить количе¬
ственное определение нитратов и нитритов с прохождением или без прохождения через
восстановительную колонку (редуктор).
632
Часть 3. Неорганический анализ
28.2.6. Экстрагированный органический азот
Основные положения
Экстрагирующий реагент, описанный в разделе 28.2.1 (0,5 М раствор сульфата калия),
способен частично солюбилизировать органический азот. Щелочной реагент, приме¬
няемый для вытеснения аммиака из солей аммония, в процессе микродиффузии (см.
раздел 28.2.2) может воздействовать на N органического вещества в экстракте. Может
образоваться небольшое количество дополнительного аммиака, не связанного с ионами
аммония, содержащимися в почве. Показано, что при увеличении щелочности вытес¬
няющего реагента растет процент растворенного органического вещества, которое пре¬
вращается в аммиак. С гидроокисью калия степень превращения может достигать 50%
(Blachere и Fery, 1957), тогда как с реактивом Конвея — всего лишь 20%, а с насыщенным
раствором карбоната калия — 6%. Рекомендуется использовать это свойство для коли¬
чественной оценки возможных изменений в N органического вещества (в соответствии
с их степенью сложности).
Методика
Экстрагированный раствор (см. раздел 28.2.1) подвергают двойной диффузионной об¬
работке (см. раздел 2.2):
• Диффузию для первой аналитической пробы проводят с помощью насыщенного
раствора карбоната калия.
• Диффузию для второй аналитической пробы — с помощью реактива Конвея.
Количественное определение аммиака проводится в обоих растворах после вытесне¬
ния. Сравнение результатов и расчет отношения результатов двух измерений позволяет
оценить стабильность органического вещества и контролировать его превращение.
28.3. Другие методы
28.3.1. Определение нитратов и нитритов методом УФ фотометрического
поглощения
Основные положения
Растворы, содержащие нитрат- и нитрит-ионы, сильно поглощают ультрафиолетовое
излучение, в особенности при 210 нм. На указанной длине волны закон Ламберта-Бера
применим для концентраций N приблизительно до 10 мг/л.
Суммарное содержание нитратов и нитритов оценивается измерением УФ поглоще¬
ния при 210 нм до и после обработки катализатором Ренея (Ni-Al). Содержание нитрита
измеряют перед и после взаимодействия с сульфаминовой кислотой.
Методика дает удовлетворительные результаты при анализе почвенных экстрактов
(Norman и Stucki, 1981; Norman и др., 1985). Основным преимуществом методики являет¬
ся ее простота, поскольку нет необходимости в окрашивающем реагенте.
Однако для реализации методики необходим УФ-спектрофотометр, более дорогой,
чем простой спектрометр, работающий в видимом свете. Кроме того, предел обнаруже¬
ния здесь выше по сравнению с пределом обнаружения колориметрической методики,
описанной в разделе 28.2.5.
Реагенты
• 20%-ный раствор серной кислоты, приготовленный из концентрированной H2S04,
</=1,84.
Глава 28. Неорганические формы азота
633
• 2%-ный раствор сульфаминовой кислоты, приготовленный из H3N03S (необходи¬
мо хранить в холодильнике).
• Катализатор Ренея (приблизительно 50% Ni и 50% А1).
• Стандартные растворы нитрата. Для приготовления исходного раствора навес¬
ку 3,606 г чистого KN03 растворить в деионизированной воде, довести объем до
1000 мл. 1 мл раствора содержит 0,5 мг N или 2,214 мг NO". 1 мл базового раствора,
разбавленного 1:50, содержит 0,01 мг N или 0,044 мг N0“.
• Стандартный раствор нитрита с концентрацией N0“ — 0,1 мг/мл (см. «Стандарт¬
ные растворы нитрита»).
• Экстракционный раствор. 0,5 М раствор сульфата калия (см. «Оборудование и реа¬
генты» в разделе 28.2.1).
• Обесцвечивающий раствор. 120 г/л сульфата алюминия.
• Подщелачивающий раствор. 100 г Na2C03 + 50 г NaOH в 300 мл деионизированной
воды.
Методика
Подготовка почвенного раствора
Во флаконы вместимостью 100 мл помещают 10 г почвы (подготовка проб описана
в разделе 28.12), затем 30 мл экстрагирующего раствора, закупоривают флаконы, встря¬
хивают на роторном шейкере в течение 10 мин, сливают и фильтруют прозрачную на-
досадочную часть. Если раствор окрашен, добавляют 1 мл раствора сульфата алюми¬
ния, постепенно вводят подщелачивающий раствор до образования гидроксида и затем
фильтруют.
Приготовление стандартных калибровочных растворов
Готовят две серии стандартных растворов нитрата и нитрита в диапазоне концентраций,
представленных в табл. 28.3. Доводят объем до 30 мл экстрагирующим раствором. Изме¬
ряют поглощение в сравнении с холостой пробой, содержащей только экстрагирующий
реагент, в следующих спектрометрических условиях: длина волны 210 нм, ширина щели
приблизительно 0,8 мм, длина оптического пути в кварцевой измерительной ячейке
1 см. Строят два калибровочных графика: один для нитратов, другой для нитритов.
Количественный анализ нитратов
• К 25 мл почвенного экстракта добавляют 1 мл 2%-ного раствора сульфаминовой
кислоты; встряхивают в течение 1 мин для получения раствора, очищенного от ни¬
тритов. Измеряют поглощение при 210 нм в сравнении с холостой пробой объемом
25 мл, содержащей 1 мл раствора сульфаминовой кислоты (величина «А»).
• К 5 мл раствора, очищенного от нитрит-ионов, добавляют последовательно: около
0,3 г катализатора, 0,5 мл 20%-ного раствора серной кислоты; хорошо перемеши¬
вают, помещают сосуд в сушильный шкаф при температуре 60 °С приблизитель¬
но на 40 мин; охлаждают; полученный раствор не содержит нитратов и нитритов.
Измеряют поглощение раствора при 210 нм в сравнении с холостой пробой, со¬
держащей 5 мл экстрагирующего раствора, 1 мл раствора сульфаминовой кисло¬
ты, 0,3 г катализатора и 0,5 мл 20%-ного раствора серной кислоты, высушивают
в сушильном шкафу в том же режиме, что и пробу; «В» — значение измеренного
поглощения.
Величина поглощения, определяемого нитратом, равняется
(1,04 А) — (1,1 В).
634
Часть 3. Неорганический анализ
Таблица 28.3. Диапазон концентраций стандартных калибровочных растворов для
определения нитрата и нитрита методом УФ-спектрометрии
мл раствора нитрата 0,044 мг (1ЧО~)/мл на 30 мл
мкг (NO”)/r почвы на 10 г
0,5
2,2
1
4,4
2
8,8
N0-
3 4
17,6
8
25,2
10
44
мл раствора нитрита 0,1 мг (NO-умл на 30 мл
мкг (NO~)/r почвы на 10 г
0,1
1
0,2
2
0,4
4
NO-
2 1
10
2
20
4
10
Для получения результата для NO-, в мкг/г почвы необходимо нанести величины
поглощения на калибровочную кривую для нитрата. Коэффициенты 1,04 и 1,1 вносят
поправку на небольшое разведение, вызванное добавлением сульфаминовой кислоты
и серной кислоты.
Количественный анализ нитритов
Вносят 5 мл раствора почвенного экстракта в пробирку, измеряют поглощение при
210 нм в сравнении с холостой пробой, содержащей 5 мл раствора для экстракции; по¬
лученный результат — «D».
Добавляют 5 мл раствора сульфаминовой кислоты к 5 мл почвенного экстракта
и встряхивают в течение 1 мин; измеряют поглощение при 210 нм; сравнивают с холо¬
стой пробой, содержащей 5 мл раствора для экстракции и 5 мл сульфаминовой кислоты;
результат — «Е».
Величина поглощения нитритами равняется D - (2Е).
(Коэффициент 2 вносит поправку на разведение при втором измерении).
Расчет результатов
Результаты получают с помощью калибровочных кривых, снятых в рабочих условиях,
описанных выше. В каждый полученный результат необходимо внести поправку на
влажность пробы почвы.
28.3.2. Определение аммония с помощью селективного электрода
Измерительный электрод
В состав электрода газодиффузионного типа (рис. 28.3) входит газопроницаемая гидро¬
фобная мембрана. Аммоний, содержащийся в пробе, необходимо вытеснить щелочным
раствором. Диффузия аммиака через мембрану электрода происходит до установления
635
Глава 28. Неорганические формы азота
равновесия парциальных давлений по обе стороны мембраны. Гкз растворяется во вну¬
треннем растворе с образованием гидроксид-ионов:
NH3 + H2OoNH4+ + OH-
Константа равновесия описывается выражением:
К= [NH4+][OH-]/[NH3]
Концентрацию ионов аммония во внутреннем растворе (0,22 М NH4C1) можно счи¬
тать постоянной, следовательно: [ОН~] = К' [NH3].
Электрод (так же как pH-электрод) чувствителен к гидроксид-ионам. Таким образом,
потенциал электрода оказывается связан с содержанием аммиака в пробе.
^Коаксиальный
кабель
Электрод сравнения Ag/AgCI
Стеклянный электрод
Отверстие для впуска воздуха
Резервуар, заполненный
0,22 М хлоридом аммония
Уплотнительное кольцо
Мембрана
Завинчивающийся колпачок
Рис. 28.3. Схема газодиффузионного электрода для определения содержания ионов аммония
636
Часть 3. Неорганический анализ
Оборудование и реагенты
Реагенты
• 0,22 Мраствор хлорида аммония (для заполнения электрода): навеску 1,17 г чистого
NH4C1 растворить и довести объем до 100 мл дистиллированной водой.
• Исходный стандартный раствор хлорида аммония (А) (концентрация 0,5 мг (И)/л):
навеску 0,955 г чистого NH4C1 растворить и довести объем до 500 мл дистиллиро¬
ванной водой.
• Раствор хлорида аммония (В) (концентрация N 0,01 мг/л): разбавить раствор А1:50.
• Калибровочные растворы: приготовить калибровочные растворы в мерных колбах
объемом 20 мл из стандартных растворов, объемы которых приведены в табл. 28.4.
Ткблица 28.4. Концентрации калибровочных растворов для определения обменного
аммония и нитратов с помощью аммонийного электрода; процедуры
экстракции (раздел 28.2.1) и микродиффузии (раздел 28.2.2) (50 г материала
почвенной пробы, 100 мл экстрагирующих реагентов, 20 мл пробы для
диффузии, калибровочные растворы доводят до 20 мл)
Объем (мл) растворов NH4CI на 20 мл
Раствор А
0,5 мг (1Ч)/мл
Раствор В
0,01мг (1Ч)/мл
мг (1Ч)/л
мкг (N)/r почвы
мкг (NO^/r почвы
0,5
0,25
0,5
2,21
1
0,5
1
4,42
2
1
2
8,84
4
2
4
17,7
10
5
10
44,2
20
10
20
88,7
0,8
20
40
176,6
2
50
100
442
Подготовка электрода
В зависимости от конструкции все электроды могут выглядеть по-разному, однако их
компоненты всегда аналогичны.
• Отвинтить крышку и достать стеклянный электрод (рис. 28.3).
• Погрузить стеклянный шарик электрода для вымачивания в 0,1 М раствор хло¬
ристоводородной кислоты. Кислый раствор не должен достигать встроенного
электрода сравнения, состоящего из серебряной проволоки, покрытой хлоридом
серебра.
• По окончании 24-часового контакта промыть большим количеством дистиллиро¬
ванной воды; оставить электрод погруженным в дистиллированную воду до при¬
менения.
• Закрепить очень тонкую мембрану из тефлона на дне резервуара электрода
(рис. 28.3), затем туго завинтить крышку.
• Погружать стеклянный электрод, пока мембрана не станет слегка выпуклой — это
указывает на контакт мембраны со стеклянным шариком.
• Заполнить резервуар электрода раствором сравнения — в данном случае 1 мл
0,22 М раствора хлорида аммония.
637
Глава 28. Неорганические формы азота
Теперь электрод готов к использованию. Его можно хранить погруженным в 0,1 М
раствор хлорида аммония. Если не планируется использовать электрод в течение более
двух недель, резервуар следует отделить от электрода, стеклянный электрод следует хра¬
нить погруженным в дистиллированную воду.
Методика
Калибровка электрода, расчет угла наклона - «S» калибровочного графика
• Перед использованием электрода следует установить значение угла наклона кри¬
вой отклика; значение параметра выражается в мВ (как правило, при 10-кратном
изменении концентрации величина изменяется в диапазоне от 45 до 60 мВ).
• В мерных колбах объемом 20 мл готовят растворы для пяти точек калибровочного
графика: 0,5, 1, 5,10, 50 мг (NJ/л, добавляя объемы исходного раствора, представ¬
ленные в табл. 28.4. Доводят объем до 20 мл дистиллированной водой; температура
всех растворов во время измерения должна равняться 25 °С.
• Отмеряют в стакан 20 мл дистиллированной воды (25 °С), погружают электрод,
подготовленный согласно разделу 28.3.2 («Оборудование и реагенты»); добавляют
2 мл 40%-ного раствора гидроксида натрия и встряхивают; через 5 мин измеряют
потенциал в мВ. Если возможна регулировка потенциала, то устанавливают нуле¬
вой уровень: если невозможна — фиксируют значение Е0.
• Проводят измерения в первой точке калибровочного диапазона (0,5 мг/л), перенеся
содержимое колбы объемом 20 мл в стакан. Погружают электрод в раствор, добав¬
ляют 2 мл гидроксида йатрия и перемешивают. Через 5 мин измеряют потенциал Еу
Повторяют операции до точки Es и строят калибровочный график мВ =/ (N) для
определения угла наклона (калибровочного коэффициента) для данного электрода.
Измерения
Измерения выполняют в 20 мл почвенного экстракта, приготовленного согласно описа¬
нию в разделе 28.2.2. Можно проводить прямые измерения, рассчитывая результаты по
калибровочному коэффициенту, или воспользоваться общепринятым методом добавок.
Если С( — содержание азота в мг/л в экстракте; / — поправочный коэффициент на
влажность, то содержание аммонийного азота Cs в мкг/г почвы выражается формулой:
Cs = 2C,f.
Содержание NH+ в мкг/г и NO~ в мкг/г (восстановление катализатором Ренея, см.
раздел 28.3.1) рассчитывают по формулам:
С™4 =2,56 С,/,
<^3 = 8,84^/
28.3.3. Анализ нитратов с помощью ион-селективного электрода
Комбинированный электрод
Отклик компактного комбинированного электрода1 с активной пластиковой мембраной
(Ingold) на нитрат-ионы составляет приблизительно 56 мВ на десятикратное увеличение
или уменьшение концентрации. Отклик электрода имеет линейный характер при из-
1 Совмещает в себе и индикаторный электрод, и электрод сравнения. — Примеч. науч. ред.
638
Часть 3. Неорганический анализ
менении содержания нитратов в диапазоне от 1 г/л до 5 мг/л. За пределами диапазона
линейности электрод можно использовать до концентрации 1 мг/л.
Среди анионов, обнаруживаемых в почвенных экстрактах, наиболее серьезные по¬
мехи связаны с присутствием ионов NO~, С1~, НСО" и в меньшей степени с ионами
SO*-. При отношении нитрат/нитрит, равном 1, погрешность составляет 20%. Отноше¬
ние C1/NO" должно быть меньше единицы, отношение HCO~/NO~ должно быть менее
25, отношение SOJ~/N03" должно быть менее 100.
Электрод применим в широком диапазоне pH (от 2 до 12).
Для того чтобы избежать загрязнения среды хлорид-ионами, следует использовать
электрод сравнения на основе сульфата ртути/сульфата калия. При 22 °С электродвижу¬
щая сила этого электрода составляет +402 мВ, что больше, чем у каломельного электро¬
да в насыщенном хлориде калия. Электрод может применяться в температурном диапа¬
зоне от 0 до 60 °С; температурный коэффициент равняется +0,13 мВ/К.
Оборудование и реагенты
• рН/мВ-метр с разрешением ±0,5 мВ для измерений в диапазоне ± 1 В.
• Нитрат-селективный электрод типа Ingold (или аналогичный).
• Ртутно-сульфатный электрод сравнения (или аналогичный).
• Калибровочные растворы. Растворы с концентрацией N03-N 0,5 мг/мл
и 0,01 мг/мл (см. «Реагенты» в разделе 28.3.1).
• Экстрагирующий раствор. 0,005 М раствор сульфата калия.
• 2%-ный раствор сульфаминовой кислоты — NH2S03H.
Калибровка
Помещают анализируемые образцы, перечисленные в табл. 28.5, в стаканы вмести¬
мостью 50 мл, доводят объем до 20 мл экстрагирующим раствором. Добавляют 0,5 мл
раствора сульфаминовой кислоты, встряхивают в течение 1 мин, выдерживают 10 мин.
Отмечают температуру раствора, которая должна быть близка к 25 °С. Погружают элек¬
троды в раствор и проводят измерения без перемешивания. Измеряют потенциал в мВ
после стабилизации результата (около 2 мин) и строят калибровочную кривую.
Готовят холостую пробу из 20 мл экстрагирующего раствора с сульфаминовой кисло¬
той, обработанного по той же схеме, что и пробы.
Количественный анализ
• Проводят экстракцию почвы в соответствии с процедурой, описанной в разделе
28.2.1, используя разбавленный раствор сульфата калия.
• Отмеряют по 20 мл почвенного экстракта в стаканы вместимостью 50 мл.
• Добавляют в каждый стакан по 0,5 мл раствора сульфаминовой кислоты; встряхи¬
вают в течение 1 мин и выдерживают 10 мин для удаления нитритов.
• Отмечают температуру, которая должна быть близка к температуре, использовав¬
шейся при построении калибровочной кривой.
• Погружают электроды в раствор холостой пробы и убеждаются в том, что положе¬
ние нулевой точки не изменилось.
• Погружают электроды в раствор пробы. После стабилизации результатов измере¬
ний (около 2 мин) измеряют потенциал в мВ без перемешивания. Отмечают точки
на калибровочной кривой, соответствующие концентрациям Ср выраженным в мг
(N03—Ы)/л.
Глава 28. Неорганические формы азота
639
Таблица 28.5. Подготовка калибровочных растворов в необходимом диапазоне
концентраций для определения нитратов методом ионометрии
Точки
калибровки
мг (NO-N)/J!
Объем (мл)
раствора 0,01 мг
(NO,—Ь0/мл
Объем,
дополняющий
до 20 мл
Объем (мл)
раствора 0,5 мг
(NO-N)/mh
Объем,
дополняющий
до 20 мл
2
4
16
3
6
14
5
10
10
10
0,4
19,6
50
2
18
100
4
16
Доводят объем до 20 мл 0,00$ М раствором K^SC^ для экстракции.
Используя методику, описанную выше в разделе 28.2.1 (50 г почвы, 100 мл раствора
для экстракции), концентрацию С в мкг (N03-)/r почвы рассчитывают по формуле:
С =
4,428x1,02x100x20
1000
С,/ = 9,032С,/,
где / — коэффициент поправки на влажность; 4,428 — отношение N03 : N;
1,02 = 20,5/20 — коэффициент поправки по сульфаминовой кислоте.
Если концентрация слишком высока, следует разбавить исследуемый образец. Если
концентрация недостаточно велика — увеличить аналитическую пробу почвы.
28.3.4. Измерения in situ
Основные положения
Формы неорганического азота могут быстро изменяться, и скорость его циклических
превращений может быть высокой. В связи с этим измерения in situ могут оказаться
весьма полезными при оптимизации методов культивации или контроле загрязнения
почвы. Прибор Tensionic (SDEO) состоит из тензиометра с пористым наконечником,
адаптированного для анализа почвенных растворов. Обмен происходит за счет простой
ионной диффузии между почвенным раствором и водой внутреннего объема наконеч¬
ника; прибор позволяет одновременно проводить измерения тензиометрических пара¬
метров.
Описание
Датчик
Датчик представляет собой пористый наконечник (рис. 28.4) для измерений в насы¬
щенной влагой почве.3 Наконечник плотно закрыт ПВХ пробкой; до дна наконечника
проходят три тонких капиллярных трубки, изготовленных из нейлона. Одна из трубок
используется для тензиометрических измерений, а другая — для экстракции раствора,
заполняющего наконечник. Третья трубка встроена в верхнюю часть наконечника и по¬
зволяет проводить продувку контура в начале работы и заливать раствор обратно по¬
сле анализа. Датчик целиком размещен в щупе, изготовленном из непрозрачного ПВХ
стержня, длина которого зависит от цели исследования (чаще всего 50 см или 1 м).
1 SDEC, ВР4233, 37 000 Tours, France.
2 Soil Moisture, P.O. Box 30025, Santa Barbara, CA 93105, USA.
3 Soil Moisture, P.O. Box 30025, Santa Barbara, CA 93105, USA.
640
Часть 3 Неорганический анализ
Измерительная аппаратура
Датчик снабжен батареей, перистальтическим насосом, работающим в двух направлени¬
ях и соединенным с трубками для сбора жидкости и продувки контура, и водонепрони¬
цаемым резервуаром с ионселективным электродом, подключенным к милливольтметру
для измерения. Трехходовой кран на насосе позволяет отбирать аликвоты для проведе¬
ния других анализов.
Методика
При продувке наконечник должен быть заполнен дистиллированной водой для того,
чтобы не допустить образования вакуума. По окончании продувки закрывают трубки
для продувки контура и сбора жидкости пробками. Выкапывают в почве цилиндриче¬
ское углубление, диаметр которого несколько превышает диаметр наконечника; выни¬
мают почву секциями, соответствующими определенной глубине. Смешивают почву со
дна углубления с дистиллированной водой и вводят полученную пастообразную смесь
на дно углубления. Вставляют наконечник в отверстие и проталкивают его к дну, покры¬
тому пастообразной смесью; заполняют свободный объем углубления почвой, соответ¬
ствующей глубине, с которой она была извлечена, слегка утрамбовывая почву. Помеща¬
ют защитное кольцо на поверхность почвы для предотвращения инфильтрации вокруг
датчика.
Измерения можно проводить после установления равновесия в почвенном раство¬
ре, на что требуется 8—10 дней. Включают насос для перекачки раствора из пористого
наконечника в герметичный резервуар, содержащий электроды. Методику, описанную
в разделе 28.3.3, можно использовать для определения содержания нитратов без добав¬
ления сульфаминовой кислоты, вносимой для разрушения нитритов, измеряемых вме¬
сте с нитратами.
Герметичный колпачок
Рис. 28.4. Схема устройства Tensionic (SDEC)
Глава 28. Неорганические формы азота
28.3.5. Необменный аммоний
641
Основные положения
Некоторое количество почвенного аммония может находиться в фиксированном состо¬
янии в глинистых прослоях; эти ионы не вступают в обменные взаимодействия в соле¬
вых растворах и поэтому практически недоступны для растений и микроорганизмов. Для
оценки содержания необменного аммония в почве предложено три методики: (1) мето¬
дика Къельдаля — отгонка при температуре 400 °С после удаления органического азота
и обменного аммония, (2) оценка по разности между количеством аммония, отогнанного
в присутствии гидроксида натрия, и аммония, отогнанного в присутствии карбоната ка¬
лия, и (3) определение количества аммония, высвобождающегося при обработке фтори¬
стоводородной кислотой для разрушения глинистых прослоев. Описанный здесь способ
принадлежит к последней категории (Keeney и Nelson, 1982). Перед обработкой HF орга¬
ническое вещество и обменный аммоний разрушают с помощью смеси карбоната калия
и гипобромита калия, которая, как известно, мало воздействует на необменный аммоний.
Оборудование и реагенты
• Раствор гипобромита калия (КВгО): медленно добавить 6 мл брома к 200 мл охлаж¬
денного льдом 2 М раствора карбоната калия при перемешивании; этот раствор
следует готовить непосредственно перед использованием.
• 0,5 Мраствор КС1: растворить 186 г КС1 в 5 л воды.
• Раствор 5 М (HF) — 1 М (НС1) (используйте полиэтиленовую бутыль с градуиро¬
вочной отметкой на уровне 2 л): наполнить около 1,5 л воды, затем при переме¬
шивании добавить 167 мл концентрированной хлористоводородной кислоты (НС1,
d = 1,19) и 325 мл 52%-ной фтористоводородной кислоты (31 М); довести объем до
2 л деионизированной водой и осторожными вращательными движениями пере¬
мещать содержимое бутыли.
Методика
Помещают 1 г тонкоизмельченной почвы в высокий стакан вместимостью 200 мл и до¬
бавляют 20 мл раствора гипобромита калия. Перемешивают содержимое и накрывают
стакан; оставляют взаимодействовать на 2 ч. Добавляют 60 мл воды и кипятят в течение
5 мин. Выдерживают в течение ночи, затем сливают прозрачный надосадочный рас¬
твор. Переносят остаток в полиэтиленовую пробирку для центрифугирования емкостью
100 мл с помощью промывалки, заполненной 0,5 М раствором КС1. Дополняют пробир¬
ку приблизительно до 80 мл 0,5 М раствора КС1 (уравновешивая пробы по весу), закры¬
вают, несколько секунд встряхивают вручную и центрифугируют при 1100 g в течение 10
мин. Сливают надосадочный раствор и повторяют операцию экстракции 0,5 М раство¬
ром КС1.
Добавляют 20 мл раствора HF-HC1 к осадку после центрифугирования с помощью
полиэтиленовой мерной пробирки. Закупоривают пробирку для центрифугирования
и встряхивают в течение 24 ч на механическом шейкере.
Можно анализировать высвободившийся аммоний напрямую — путем отгонки (см.
гл. 10 и 14) или микродиффузионным методом, описанным выше в разделе 28.2.2. В лю¬
бом случае следует избегать контакта между раствором, содержащим фтористоводород¬
ную кислоту, и стеклянными стенками оборудования: для переноса содержимого проби¬
рок для центрифугирования в щелочную среду использовать полиэтиленовые воронки;
процесс микродиффузии проводить в пластиковых колбах Эрленмейера.
642
Часть 3. Неорганический анализ
Использованная литература
Blachfcre Н and Ferry Р (1957) Dosage de 1’azote mineral dans les sols par micro-diffusion. Ann. Agr., 8,
111-118,495-498.
Chariot G (1974) Chimie analytique quantitative. Masson. TII, 347.
Conway EJ (1962) Micro-diffusion analysis and volumetric error., Crosby lockwood, London, 5ёте edit
Djurhuus J and Jacobsen OH (1995) Comparison of ceramic suction cups and KC1 extraction for the
determination of nitrate in soil. Eur. J. Soil Sci., 46, 387-395.
Guiot J (1975) Estimation des reserves azot£es du sol par determination de l’azote mineral. Revue de
l Agriculture, 5, 1117-1132.
Guito J, Goffart JP and Destain JP (1992) Le dosage des nitrates dans le sol. Bull. Reck Agron., Gembloux,
27, 61-74.
Keeney DR and Nelson DW (1982) Nitrogen - inorganic forms. In Methods of Soil Analysis, Page AL,
Miller RH and Keeney DR ed. ASA-SSSA, Agron. Monograph No 9,2nd ed. Madison, WI Etats-
Unis, 643-698.
Khan SA, Mulvaney RL and Hoeft RG (2000) Direct-diffusion methods for inorganic-nitrogen analysis
of soil. Soil Sci. Soc. Am. J., 64, 1083-1089.
Mulvaney RL and Khan SA(1999) Use of diffusion to determine inorganic nitrogen in a complex organic
matrix. Soil Sci. Soc. Am. J., 63, 240- 246.
Mulvaney RL (1996) Nitrogen - Inorganic forme. In Methods of Soil Analysis, Part 3, Chemical Methods,
Bigham JM and Bartels JM ed. SSSA-ASA, Madison, WI Etats-Unis, 1123-1184.
Mulvaney RL, Khan SA, Stevens WB and Mulvaney CS (1997) Improved diffusion methods for
determination of inorganic nitrogen in soil extracts and water. Biol. Fertil. Soils, 24,413-420.
Norman RJ and Stucki JW (1981) The determination of nitrates and nitrites in soil extracts by UV
spectrophotometry, Soil Sci. Soc. Am. J., 45, 347- 353.
Norman RJ, Edberg JC and Stucki JW (1985) Determination of nitrates in soil extracts by dual¬
wavelengths UV spectrophotometry, Soil Sci. Soc. Am. J., 49, 1182-1185.
Rodier J (1984) Analyse chimique etphysico-chimique de l feau. Dunod (Paris Susini J and N’Gandjui C
(1964) Dosage de l’azote mineral. Cah. ORSTOMSdr.Pedol., 2,57-71.
XP X31-115 (1995) Qual№ des sols. Pr£l£vement et conservation des ёс1шпШ1оп5 de sol en vue de la
dёtermination de l’azote ттёга1 sur sol frais, AFNOR, 8 p.
Дополнительная литература
Boltz DF and Howell JA (1978) Colorimetric Determination of Non Metals., Wiley, New York.
Bremmer JM (1987) Laboratoiy techniques for determination of different forms of nitrogen cycling in
agriculture ecosystems. Proc. Symp. Adv. Nitrogen, Brisbane, Australia: 11-15 mai 1987.
Cheverry C (1983) L’extraction de la “Solution du Sol” par le biais de bougies poreuses. Bulletin du
groupe franqais d’humidimetrie neutronique (GFHN), 14,47-71.
Gautheyrou J and Gautheyrou M (1965) Dosage simultan6 de l’azote ammoniacal et nitrique dans les
sols - Contribution & l’6tude de la dynamique de l’azote. Cah. Orstom Ser. Pddol., 4, 367-391.
Morie GP and Ledeford CJ (1972) Determination of nitrate and nitrite in mixtures with a nitrate ion
electrode. Anal. Chim. Acta., 60, 397-403.
Moutonnet P, Guiraud G and Marol C (1989) Le tensiom£tre et la teneur en nitrates de la solution du sol.
Bulletin du groupe franqais d'humidimetrie neutronique (GFHN), 26, 11-28.
prNF ISO 14256-2 (2002) Qualitd du sol - Dosage des nitrates, des nitrites et de l’ammonium dans des
sols bruts par extraction avec une solution de chlorure de potassium. AFNOR, X 31-423-2.
Taras MJ (1971) Standard methods for the examination of water of wastewater. 13e edit. American Public
health association, Washington, DC 20036.
Глава 29. Фосфор
29.1. Введение
Несмотря на то что количество фосфора (Р) в почвах не слишком велико1, он является
важным элементом и играет существенную роль в агрономических и биогеохимических
циклах. Р присутствует во всех живых организмах. Он способен к образованию много¬
численных ковалентных фосфороорганических соединений и к образованию связей с С,
N, О, Al, Fe, Са. Он участвует в фундаментальных процессах преобразования лучистой
электромагнитной энергии в химическую энергию (фотосинтез) и способствует разви¬
тию корневых систем растений.
Описание и классификация различных форм Р в почве необходимы для количе¬
ственного определения потребности растений в фосфоре и внедрения агротехнических
методов, способствующих удовлетворению этих потребностей. Аспекты, связанные
с питанием растений, тесно переплетаются с проблемами педогенных трансформаций
и риском загрязнения окружающей среды.
Обнаружено более 220 фосфорсодержащих минералов, сохраняющих стабильность
на протяжении геологических периодов. Кроме того, множество соединений фосфора
содержат промышленные продукты, такие как удобрения, пестициды, детергенты, умяг-
чители воды, огнезащитныереагенты, топливные присадку и пластики. Эти соединения
широко распространены в среде обитания человека и играют важную роль в экологии.
С ними связаны новые проблемы, встающие перед аналитиками: например, антропиза-
ция воздействует на пахотные земли и отдельные сельскохозяйственные технологии; под¬
сечно-огневая система земледелия приводит к образованию пирофосфатов; применение
активного ила с очистных сооружений добавляет в почву полифосфаты и органические
формы Р, не существующие в естественной среде. Принято считать, что растения спо¬
собны ассимилировать фосфор только в форме ортофосфатов из почвенного раствора.
Основная часть органических форм Р (50-75%) служит в качестве резерва, а осталь¬
ная находится в непрерывном превращении, в цикле иммобилизации и минерализации
органо-неорганического Р в почве под влиянием pH, окислительно-восстановительных
и биохимических (фосфатазы) процессов, определяемых климатическими условиями.
При pH почвы в диапазоне от 3,5 до 10 ортофосфаты, в основном, присутствуют в фор¬
ме Н2РО~ в кислой среде и НРО*- в основной среде. Активности двух ионов при pH 7,2
приблизительно равны, форма Н3Р04 преобладает при pH 3,5, а форма Р043_ — при pH
11-12.
Для оценки содержания общего Р, особенно в формах, доступных в различной степе¬
ни в краткосрочной и среднесрочной перспективе, разработаны различные методики
(Whiten Beckett, 1964; Dalai и Hallsworth, 1976; Roche и 1978; Pierzynski, 2000), позво¬
ляющие определить:
• количество (Q) различных форм Р: экстрагируемый, окклюдированный, суммар¬
ный Р;
1 Фосфор занимает девятое место по распространенности среди элементов, входящих в состав
земной коры. Особенно часто он встречается в виде ортофосфат-иона, связанного с кальци¬
ем (в форме апатитов, где среднее содержание фосфора составляет приблизительно 0,1%).
644
Часть 3. Неорганический анализ
• интенсивность (7), химический потенциал ионов РО*_ в почвенном растворе, обе¬
спечивающий нормальное развитие растений на протяжении всего цикла вегетации;
• способность к поддержанию концентрации Р в почвенном растворе, выражаемая
через емкость фиксации Р, изотермы адсорбции (AQ/A7);
• кинетика десорбции;
• диффузионная подвижность, протяженность зоны, в которой,* вероятно, образует¬
ся почвенный раствор.
Таким образом, химические и биологические изменения, происходящие в ходе дина¬
мического процесса круговорота фосфора, можно измерять и, вероятно, контролиро¬
вать и направлять.
В отличие от циклов С, Н, N, О, S, при круговороте Р не происходит потерь за счет
испарения (возможным исключением является Н3Р, существование которого в есте¬
ственном состоянии еще не доказано, даже в сильно восстанавливающей среде). Эта
особенность повышает риск эвтрофикации загрязненных водоемов, поскольку воздей¬
ствие Р длится в течение многих лет, даже после ликвидации источников загрязнения.
29.2. Общее содержание фосфора в почве
29.2.1. Введение
Термин «общий фосфор» относится ко всем формам Р, содержащегося в почве, т. е. к ор¬
ганическому, неорганическому, окклюдированному и доступному Р. Это общий дебит Р
в определенный момент времени. Содержание Р в почве зависит от природы материн¬
ской породы, климатических факторов и вызванного ими разрушения (степени выве¬
тривания, биологической активности, эрозии, выщелачивания). Содержание Р в почве
сильно варьирует. Почвы, сформировавшиеся на известковых породах, обогащенных
апатитом, в умеренно влажных климатических условиях могут содержать большое коли¬
чество общего фосфора. С другой стороны, кислые почвы, сформировавшиеся на гра¬
нитных породах с низким содержанием Р, характеризуются в их естественном состоянии
низким содержанием Р.
При химическом анализе необходимо сначала разрушить матрицу (неорганическую
и органическую), чтобы солюбилизировать фиксированные и окклюдированные фор¬
мы, в основном, нерастворимые (такие как фосфаты кальция, железа или алюминия),
относящиеся к среднесрочным и долгосрочным почвенным резервам.
Минерализацию можно осуществить (i) «мокрыми» методами, например, воздей¬
ствием кислоты в окислительной среде, чтобы избежать потерь форм фосфора, которые
могут образоваться в восстановительной среде, (й) сухими методами, например, щелоч¬
ным плавлением в печи с подачей кислорода и с последующей обработкой кислотой
продуктов плавления.
Фосфор имеет пять валентных электронов и образует почти исключительно ковалент¬
ные связи. В основном состоянии на каждой из трех р-орбиталей находится только один
электрон. Это состояние характеризуется сферически симметричным распределением
электронов и высокой энергией ионизации. Фосфор часто образует ковалентные соеди¬
нения. Величина свободной энтальпии образования для различных степеней окисления
в растворе при рНО указывает на относительную нестабильность молекул и склонность
к превращению. Результатом гидролиза фосфора в максимально окисленном состоя¬
нии является ортофосфорная кислота — форма пятивалентного фосфора, обладающая
максимальной термодинамической стабильностью, которая является основной формой
фосфора для большинства химических определений (рис. 29.1).
Глава 29. Фосфор
645
Рис. 29.1. Пятивалентный фосфор в составе ортофосфорной кислоты (слева):
Н3Р04-> Н2РО"-> НРО*"-> Р04~; пирофосфорная кислота (Н4Р205) (справа)
29.2.2. Минерализация мокрым озолением при общем анализе
Основные положения
Для анализа общего Р можно использовать методики минерализации мокрым озоле¬
нием (см. раздел 31.2 гл. 31). При необходимости измерения только общего фосфора
процедуры можно упростить. Почву подвергают окислительной кислотной обработке,
т. е. длительному кипячению с азотной или хлорной кислотой. Все различные формы Р
переходят в состояние орто-аниона. Органическое вещество разрушается, и высвобож¬
даются ранее связанные соединения Р. Апатиты растворяются наряду с фосфатами же¬
леза и алюминия, фосфитами (НРО*~), триполифосфатами (Na5P3O10), метафосфатами
и пирофосфатами. '
Тем не менее, остаток почвы все еще может содержать первичные минералы с необ¬
работанными включениями Р, которые можно разрушить только воздействием фтори¬
стоводородной кислоты (см. раздел 31.2.3 и 31.2.4).
Оборудование
• Аналитические весы (0,1 мг).
• Электрическая песчаная баня или электрическая плитка.
• Колбы Къельдаля из стекла Пирекс вместимостью 150 мл.
• Штатив для фильтрования и воронки.
• Усиленные аналитические фильтры.
• Лабораторная стеклянная посуда.
Реагенты
• Азотная кислота — HN03, d- 1,4 (температура кипения 120 °С).
• 60%-ная хлорная кислота для анализа (может воспламеняться).
• Деионизированная вода.
Методика с применением азотной кислоты
• Навески 5 г почвы, высушенной при 105 °С, измельченной до размера частиц 0,2
или 0,1 мм, помещают в колбы Къельдаля вместимостью 150 мл.
• Добавляют 30 мл концентрированной азотной кислоты.
• Закрывают микроворонками с оттянутыми концами; помещают на песчаную баню
и кипятят в течение 3 ч (не допуская пересыхания).
646
Часть 3. Неорганический анализ
• Охлаждают и осторожно разбавляют деионизированной водой.
• Фильтруют через усиленный аналитический фильтр, помещенный в воронку с от¬
тянутым концом.
• Промывают фильтр кипящей деионизированной водой и собирают весь фильтрат
в стеклянный (Пирекс) химический стакан вместимостью 250 мл.
• Выпаривают почти досуха.
• Растворяют остаток в 1 мл хлорной кислоты (или серной кислоты).
• Разбавляют деионизированной водой до 50 или 100 мл (в зависимости от типа по¬
чвы и предполагаемого состава); гомогенизируют.
Измерения проводят в аликвотах спектроколориметрическим (или другим) методом (раз¬
дел 29.5 или гл. 31). Результаты выражают относительно почвы, высушенной при 105 °С.
Примечания
Обработка азотной кислотой не позволяет полностью минерализовать почвенную ма¬
трицу и оставляет сравнительно большой остаток.
В органических почвах (например, гистосолях) или гумусовых почвах полное раз¬
рушение органического вещества маловероятно. В таком случае остаток выпаривают
практически досуха и добавляют пергидроль до полного обесцвечивания, затем вновь
обрабатывают 1 мл азотной кислоты.
Раствор для обработки содержит кремний. Если его высушить, возможно спекание
кремния, из-за чего он с трудом поддается ресолюбилизации, однако можно частично
удалить его фильтрованием.
Методика с применением азотной кислоты имеет ряд недостатков, однако менее ри¬
скованна по сравнению с методикой минерализации хлорной кислотой (см. следующий
раздел).
Методика с применением хлорной кислоты
• Навеску 2-5 г почвы сушат при 105 °С и измельчают до размера частиц 0,2 или
0,1 мм.
• Помещают ее в колбу Къельдаля для кипячения из стекла Пирекс объемом 150 мл
и добавляют 30 мл 60%-ной хлорной кислоты.
• Продолжают процедуру в соответствии с описанием, представленным в 2.2.4, осто¬
рожно кипятят на протяжении 30 мин до появления белого дымка и обесцвечива¬
ния.
• Без кипячения выпаривают жидкость, уменьшая объем раствора до получения
почти белого шлама.
• Фильтруют и доводят объем до 250 мл, гомогенизируют.
• Проводят анализ аликвоты (см. раздел 29.5 или гл. 31).
Примечания
Поскольку перхлораты нестабильны, нельзя допускать их высыхания.
Кипятят раствор осторожно, в особенности в начале минерализации, когда органи¬
ческое вещество все еще присутствует в большом количестве (для почв, богатых орга¬
ническим веществом, разрушение органического вещества может оказаться неполным;
в этом случае требуется добавление азотной кислоты, затем концентрирование раствора
практически досуха и обработка остатка 1 мл хлорной кислоты).
647
Глава 29. Фосфор
29.2.3. Сухая минерализация
Основные положения
Щелочное плавление может разрушить структурную матрицу, что приведет к образова¬
нию аморфных твердых растворов, которые легко поддаются обработке в кислой среде.
Обработка, как правило, очень интенсивная и может использоваться для общих ана¬
лизов, включая тугоплавкие элементы. В дополнение к методике, описанной в этом
разделе, для анализа общего Р можно воспользоваться методиками, представленными
в разделе 31.2.6.
Почву смешивают с карбонатом натрия в платиновом тигле и сплавляют в открытой
окислительной среде. Различные формы Р переходят в форму ортофосфата, который со¬
любилизируется при обработке кислотой. Слишком длительного нагрева после плавле¬
ния следует избегать из-за возможности образования пероксомонофосфорной (Н3Р05)
и пероксодифосфорной (Н4Р208) кислот.
Оборудование
• Платиновые тигли вместимостью 50 мл.
• Электрическая печь с возможностью работы с открытой дверцей (окислительная
атмосфера).
• Тигельные щипцы.
• Лабораторная стеклянная посуда.
Реагенты v
• Безводный карбонат натрия (Na2C03, температура плавления — 851 °С);
• Деионизированная вода.
• 5 М раствор азотной кислоты (HN03).
• 5 н. раствор серной кислоты (H2S04).
Методика
• Навеску 1 г почвы, высушенной при 105 °С и измельченной до размера частиц 0,2
или 0,1 мм, помещают в платиновый тигель и смешивают с 5 г карбоната натрия.
• Медленно нагревают пробу, передвигая ее в печке круговыми движениями, пока не
начнется плавление (-850 °С), не допуская выброса содержимого тигля.
• Наполовину закрывают тигель крышкой для сохранения окислительной атмосфе¬
ры и поддерживают температуру в печи на протяжении 20 мин.
• Удерживают тигель щипцами и круговыми движениями распределяют содержимое
продукта плавления по стенкам тигля; выдерживают для охлаждения.
• Помещают тигель вместе с крышкой в стеклянный (Пирекс) химический стакан
вместимостью 250 мл, постепенно добавляют 30 мл 5 н. раствора серной кислоты,
не допуская выброса содержимого стакана (избегать воздействия НС1, разрушаю¬
щей платиновые тигли).
• Кипятят до полного растворения.
• При необходимости добавляют 5 мл 1 н. раствора серной кислоты.
• Фильтруют через усиленный аналитический фильтр.
• Доводят объем до 250 мл деионизированной водой и гомогенизируют.
• Отбирают одну аликвоту для анализа Р (см. раздел 29.5).
• Рассчитывают результаты по отношению к почве, высушенной при 105 °С.
648
Часть 3. Неорганический анализ
Примечание
Содержание общего Р напрямую связано с составом материнской породы и процессами,
повлиявшими на формирование почвы; данные об общем Р не дают представления о
доступности Р для растений. Содержание общего Р описывает общий баланс фосфора
природной почвы и изменения возделываемой почвы, в которую регулярно вносятся
удобрения. Он позволяет проводить мониторинг обогащения почвы Р или его потери
в ходе различных процессов, таких как уборка урожая, эрозия или выщелачивание.
29.3. Разделение различных форм фосфора
29.3.1. Введение
Анализ общего Р не проясняет сложных химических и микробиологических механизмов
изменения форм Р. Доля органической фракции может изменяться от 20 до 80%. Раз¬
ные формы Р, как правило, присутствуют в очень малых количествах, и различия между
ними настолько малы, что их нелегко обнаружить такими инструментальными мето¬
дами, как РД, ИК-спектрометрия (см. гл. 4 и 5) или дифференциальный термический
анализ (см. гл. 7). Методы прямого обнаружения, такие как электронная микроскопия
и ЭД-РСМА (см. гл. 8), полезны для подробных исследований минералогических пре¬
вращений, однако не вполне надежны для анализа потенциального плодородия.
Непрямые методы используют экстрагирующие реагенты в соответствии с четко опре¬
деленной процедурой, позволяющей проводить измерения солюбилизированных форм
Р. Некоторые методы, состоящие из последовательных этапов, позволяют изолировать
различные репрезентативные группы соединений. Реагенты смещают химические равно¬
весия почвы. Можно использовать буферные и небуферные среды. Селективность этих
методик зависит от типа почвы и практикуемых сельскохозяйственных технологий, что
позволяет выявлять общие тенденции в реакциях с участием Р в зависимости от климати¬
ческих и биогеохимических процессов. Сельскохозяйственные эксперименты являются
предпочтительными методами для выявления корреляций между урожайностью растений
и экстрагированными формами Р, а также для оценки усвояемости Р в данном регионе. Из¬
вестно, что большинство растений усваивают Р в формах Н2РО~ и/или НРО*~, присутству¬
ющих в почвенных растворах, в зависимости от pH. Иногда считают, что органический
Р является прямым источником Р для растений, однако эта концепция является спор¬
ной. В комплексном процессе усвоения Р растениями участвуют ферменты (например,
фосфатазы), находящиеся в контакте с корневой системой растений, которые способ¬
ны высвобождать ортоформы Р. В ризосфере происходит весьма быстрый обмен между
группами соединений, до тех пор пока не достигнуты равновесия почвенного раствора.
29.3.2. Последовательные методики
Основные положения
Эти методики позволяют описать некоторые петрологические, минералогические, био-
геохимические или агрономические механизмы путем измерения различий в раство¬
римости неорганического и органического Р в «селективных» реагентах. Было пред¬
принято множество попыток (в частности, метод Chang и Jackson, 1957) измерения
неорганических форм Р и описания превращений Р, вносимого в форме мелиорирую¬
щих добавок или удобрений. Применяемые реагенты позволили разделить фракции Р,
предположительно связанные с алюминием, железом или кальцием, и две фракции Р,
окклюдированного железо-алюминиевыми комплексами. Однако некоторые реагенты
Глава 29. Фосфор
649
недостаточно специфичны. В частности, использование фторида аммония в карбонат¬
ных почвах часто приводит к осаждению фторида кальция из карбоната кальция, а так¬
же к выщелачиванию Р, который впоследствии обнаруживают в других формах.
Существуют методики (Williams и др., 1967; Syers и др., 1972), обладающие улучшен¬
ной специфичностью за счет изменения природы и концентрации некоторых реаген¬
тов. К сожалению, эти методики весьма затратны по времени и не дают информации о
содержании органического Р, преобладающего в почвах и играющего важнейшую роль
в круговороте Р.
Была разработана более сложная методика (Hedley и др., 1982), которая позволяет раз¬
делить почвенный Р на шесть групп, включая органические и неорганические формы.
Обработка хлороформом вызывает лизис микробных клеток, придавая методике допол¬
нительное биохимическое направление. Методику можно использовать не только для
изучения равновесий между различными формами почвенного Р в длительно некульти-
вируемых землях и долгосрочных экспериментах, но и в краткосрочных инкубационных
анализах или исследованиях сельскохозяйственных культур в оранжереях. Таким обра¬
зом, можно дать количественную оценку краткосрочной динамики процессов, которую
затем можно расширить и попытаться дать прогноз долгосрочных трансформаций.
Оборудование
• Сито с размером ячеек 0,1 мм.
• Мелкоячеистые нейлоновые пакеты (для ионообменной смолы).
• Центрифуга и 50 мл пробирки для центрифугирования с завинчивающимися
крышками.
• Лабораторный шейкер.
• Ультразвуковая ванна.
• Микроволновая печь для минерализации с тефлоновым флаконом на 50 мл.
• Лабораторная стеклянная посуда.
Реагенты
• Деионизированная вода.
• Анионообменная смола в форме бикарбоната — Dowex 1 SX50 (или аналогичная).
• 0,5 М раствор гидрокарбоната натрия.
• Хлороформ, СНС13.
• Аналитические фильтры.
• 0,1 М раствор гидроксида натрия.
• 1 М раствор хлористоводородной кислоты.
• Пергидроль.
• Концентрированная серная кислота.
Проба
Органический фосфор можно модифицировать воздушной сушкой и длительным из¬
мельчением. Рекомендуется использовать пробы почвы, хранившиеся при их естествен¬
ной влажности при -40 °С. Пробы следует быстро высушить на воздухе, распределив
почву тонким слоем, затем измельчить до размера частиц 2 мм. Одну аликвоту измель¬
чить до размера частиц 0,1 мм.
В другой пробе следует измерить остаточную влажность, чтобы пересчитать все ре¬
зультаты относительно почвы, высушенной при 105 °С.
650
Часть 3. Неорганический анализ
Методика
• Берут две навески по 0,5 г почвы, измельченной до размера частиц 0,1 мм (см. «Ре¬
агенты» в разделе 29.3.2) и помещают две пробы (А и В) в пробирки для центрифу¬
гирования объемом 100 мл с завинчивающимися крышками.
• Вносят нейлоновый пакет, содержащий 0,4 г анионообменной смолы.
• Добавляют 30 мл воды; встряхивают в течение 16 ч при 24 °С.
• Вынимают нейлоновый пакет и ополаскивают его для удаления остатков почвы.
• Центрифугируют смесь вода — проба и сливают надосадочный раствор.
• Фосфор, фиксированный смолой, является наиболее биологически доступным
неорганическим Р, так же как и Р, присутствующий в почвенном растворе. Смолу
можно (i) разрушить обработкой сильной кислотой (хлорной кислотой), или (ii)
провести обменную экстракцию в 10 мл 10%-ного раствора NaCl при 80 °С (см.
«Методика» в разделе 29.3.2); завершить экстракцию или обработать 50 мл деиони¬
зированной воды. Это проба 1, содержащая «Р, экстрагированный смолой».
Остаток почвы используют для следующей экстракции. Его можно хранить во влаж¬
ном состоянии в течение 24 ч при 24 °С, не закрывая пробирки, чтобы обеспечить инку¬
бацию в среде с достаточным содержанием кислорода.
Проба А
• К остатку А после экстракции смолой добавляют 30 мл 0,5 М раствора NaHC03
и встряхивают в течение 16 ч при 24 °С.
• Центрифугируют и отделяют надосадочный раствор, содержащий лабильный не¬
органический и органический Р с небольшим количеством Р микробиологическо¬
го происхождения. Остаток почвы удаляют.
• Доводят объем жидкости до 50 мл при нейтрализации добавлением 5 мл 4 М рас¬
твора серной или хлорной кислоты. Это проба 2А, «Р, экстрагированный бикарбо¬
натом».
Проба В
• К остатку пробы В после экстракции смолой добавляют 1 мл хлороформа.
• Закрывают пробирку и встряхивают в течение 1 ч.
• В течение ночи выпаривают хлороформ.
• Добавляют 30 мл 0,5 М раствора бикарбоната натрия (NaHC03).
• Встряхивают в течение 16 ч при 24 °С.
• Центрифугируют и отделяют надосадочный раствор.
• Доводят объем до 50 мл при нейтрализации добавлением 5 мл 4 М раствора серной
или хлорной кислоты. Это проба 2В, «Р, экстрагированный хлороформ-бикарбо-
натным раствором».
• Рассчитывают содержание Р, образующегося при разрушении клеток микробов по
разности: [проба 2В-Р] - [проба 2А-Р].
• Проводят последовательную экстракцию остатка почвы В:
• Добавляют 30 мл 0,1 М раствора NaOH; встряхивают в течение 16 ч при 24 °С.
• Центрифугируют и отделяют надосадочный раствор, содержащий неорганиче¬
ский и органический Р, удерживаемый за счет хемосорбции соединениями железа
и алюминия на поверхности частиц; доводят объем до 50 мл при нейтрализации
651
Глава 29, Фосфор
раствора, как описано выше. Это проба 3 — «Р, экстрагированный разбавленным
раствором гидроксида натрия».
• Вновь приводят остаток почвы в контакт с 20 мл 0,1 М раствора NaOH и обраба¬
тывают ультразвуком в течение 2 мин в резервуаре, заполненном тающим льдом.
Доводят объем до 30 мл 0,1 М раствором NaOH и встряхивают в течение 16 ч при
24 °С. Центрифугируют и отделяют надосадочный раствор, содержащий неоргани¬
ческий и органический Р, удерживаемый на внутренних поверхностях почвенных
агрегатов. Доводят объем до 50 мл при нейтрализации. Это проба 4 — «Р, экстраги¬
рованный разбавленным раствором гидроксида натрия с помощью ультразвука».
• Обрабатывают остаток почвы 30 мл 1 М раствора НС1. Встряхивают в течение 16 ч,
центрифугируют и отделяют надосадочный раствор, содержащий апатиты и не¬
которое количество неорганического и органического Р, окклюдированного вы¬
ветренными почвами. Доводят объем до 50 мл деионизированной водой. Это про¬
ба 5 — «Р, экстрагированный соляной кислотой».
• Остаток почвы подвергают минерализации с помощью смеси серной кислоты
и пергидроля (или хлорной кислоты, см. раздел 29.2.2) в течение 3 ч. Такая обработка
позволяет солюбилизировать стабильные формы органического Р и труднораство¬
римые формы неорганического Р. Охлаждают, фильтруют и доводят объем филь¬
трата до 50 мл деионизированной водой. Это проба 6, содержащая «остаточный Р».
Для анализа каждого экстракта отбирают:
• одну аликвоту с концентрацией Р, пригодной для измерения спектроколориметри¬
ческим методом (см. раздел 29.5.2); эту методику следует использовать только для
анализа фосфора в ортоформе; х
• одну аликвоту, которая будет подвергнута минерализации смесью серной кислоты
и пергидроля перед измерением общего Р (органического + неорганического; ми¬
нерализацию не нужно проводить при анализе органического + неорганического
Р методом спектрометрии с индуктивно связанной плазмой). В этом случае орга¬
нический Р можно рассчитать по разности. Анализ следует проводить немедленно
для ограничения гидролиза.
В пробе 1 — «Р, экстрагированного смолой» и в последнем почвенном остатке труд¬
но разделить органические и неорганические формы, относящиеся к среднесрочным
и долгосрочным резервам.
Примечания
Описанная методика является затратной по времени, из-за чего не используется для ру¬
тинных агрономических анализов, однако может оказаться полезной при проведении
исследований, включающих проверку состояния растений или инкубацию почвы.
Поправочный коэффициент, учитывающий микробиологический Р, рассчитанный
для данного типа почвы, часто используется для оценки общей бактериальной и гриб¬
ковой микрофлоры.
Применяется методика спектроколориметрических измерений (Murphy и Riley, 1962),
возможно, с принятыми позднее усовершенствованиями. Эта методика специфична
по отношению к ортофосфатам и позволяет различать органические и неорганические
формы Р. Несмотря на риск гидролиза органических форм, удается достичь удовлетво¬
рительного разделения.
Прямое измерение общего содержания Р в почве (см. раздел 29.2) позволяет прове¬
рить результат, полученный суммированием экстрагированных форм.
652
Часть 3. Неорганический анализ
Молекулярные веса различных форм органического Р, экстрагированных растворами
бикарбоната, гидроксида натрия, а также раствором гидроксида натрия с помощью уль¬
тразвука, составляют < 30 000. В пробе «остаточный Р» органический Р включен в моле¬
кулы, вес которых варьирует в диапазоне от 30 000 до 70 000, что может соответствовать
гуминовым фракциям (см. гл. 11).
29.3.3. Селективная экстракция — индексы доступности
Общее описание методик
Представленные методики основаны на однократной экстракции неорганического Р
в ортоформе тщательно подобранным реагентом, растворяющим специфические до¬
ступные формы Р. Эти методики пригодны только для кислых и щелочных сред; их мож¬
но применять в исследованиях химии почв или же для простых анализов плодородности
почв.
Пробы почвы могут поступать с некультивируемых земель или с интенсивно куль¬
тивируемых земель с регулярной вспашкой, где предусмотрено (или не предусмотрено)
повторное использование растительных остатков, внесение более или менее раство¬
римых удобрений (для устранения, поддержания состояния и коррекции дефицитов),
серьезно нарушающих динамику циклов Р и влияющих на аграрное управление по¬
чвами. Терминология, используемая для описания различных экстрагированных форм,
значительно различается, что отражает сложность процессов распределения и обмена Р
в почве, а также интенсивность превращений форм фосфора, определяющих динамику
системы.
Эти методики позволяют проводить количественную оценку двух категорий форм Р:
* подвижных форм Р, соединенных слабыми химическими связями, называемых
различными авторами активными, легко замещаемыми, доступными, обменными,
экстрагируемыми, легко гидролизуемыми, нестабильными, растворимыми или по¬
лезными формами Р;
• трудноудаляемых и/или прочно адсорбированных форм Р, удерживаемых хелатны¬
ми связями; форм Р в составе включений в минеральные соли железа и кальция;
окклюдированных органических и неорганических форм Р; Р в составе биомассы
и Р, связанного с кристаллической решеткой.
Существует ряд методик, позволяющих оценить объемы фиксации, обменную ем¬
кость, потенциал, способность к удержанию, ретроградации или адсорбционной емко¬
сти фосфора в почвах.
Некоторые термины отражают подвижный характер Р (например, «обменный фос¬
фор»), однако не все процедуры приводят к селективному выделению его форм. Тео¬
ретически существует прямая зависимость между количеством доступного Р и потреб¬
ностью растений; доступный Р может вызвать усиленный рост растений и повышение
урожайности (отклик на внесение удобрений). Тем не менее, если не установлена корре¬
ляция между состоянием растений, типом почвы и экстрагированным Р, использование
термина «доступный Р» является произвольным.
Существует ряд методик, основанных на применении кислых, щелочных и комплек¬
сообразующих реагентов с различными механизмами действия, такими как простое рас¬
творение, восстановление железа, комплексообразование или осаждение алюминия,
железа или кальция. Наиболее распространенными реагентами являются забуференные
или незабуференные сильные или слабые неорганические или органические кислоты,
ряд неорганических или органических оснований, а также соли этих кислот и основа-
Глава 29. Фосфор
653
ний (табл. 29.1). Каждая методика имеет заданное значение pH. pH реагентов обычно
поддерживают в диапазоне существования Н2РО~ и НР02_, т. е. между pH 2,0 и 6,0 при
анализе кислых почв, и pH 8,5 — щелочных. В других случаях величина pH почвы опре¬
деляется методом экстракции (например, водная экстракция, электродиализ, электро¬
ультрафильтрация, экстрация ионообменной смолой).
Для данного реагента на процесс растворения может повлиять ряд переменных, опи¬
сывающих условия эксперимента: pH, концентрация, отношение почва : раствор, дли¬
тельность контакта, температура, размер частиц, интенсивность перемешивания (воз¬
действие ультразвука). При необходимости сравнения результатов надо строго следовать
рекомендуемой процедуре.
Водная экстракция позволяет получить раствор с концентрацией Р, близкой к кон¬
центрации почвенного раствора. Основные реагенты экстрагируют органические и не¬
органические лабильные формы и небольшое количество микробиологического Р при
Таблица 29.1. Некоторые реагенты, используемые для экстракции Р из почвы
ТЪш
Метод, экстрагирующий реагент
Авторы
Смешанный
Вода, анионообменная смола, 32Р или 33Р
изотопное разбавление, электродиализ,
электроультрафильтрация
Экстракция
комплексообразующими
реагентами
EDTA, NH4HC03, + DTPApH 7.6
Soltanpour
Экстракция в щелочной
NaOH, КОН, NH4OH, Na2C03,
Olsen
среде
NaHC03 pH 8,5
KjCOj, (NH4)2C03
Michigin
Экстракция
Уксусная кислота — ацетат аммония или
Morgan
органическими кислотами
натрия при pH 2,5 и pH 4,8
Dyer(\m)
и их солями
Лимонная кислота — цитрат аммония
Demolon (1932)
7V/^31-160(1993)
Молочная кислота - лактат аммония или
кальция
Щавелевая кислота — оксалат аммония
Joret и
Hebert (1955)
7^*31-161 (1993)
Тиогликолевая кислота + NH4F
Уксусная кислота + лактат аммония
Egner-Riehm
Экстракция сильными
НС1 + H2S04
Mehlich (no. 1)
неорганическими кислотами
НС1 + NH4F
(1953)
и их солями
H2S04 0,001 М pH 3,0
Bray и Kurtz (1)
0,01 М pH 2,0
(1945)
0,005 М
Truog
0,2 М
Peech
CaS04 0,005 М
Keer
СаС12 0,005 М
Stieglitz
654
Часть 3. Неорганический анализ
pH около 8,5. Нерастворимые формы, связанные с кальцием, в действительности не
поддаются обработке реагентом при этом значении pH. При высоких pH формы Р, свя¬
занные с гумусом, солюбилизируются, и происходит экстракция соединений, удержи¬
ваемых за счет хемосорбции.
Гуминовые кислоты, фульвокислоты и некоторые гумины экстрагируют горячим 5 М
раствором NaOH, получая интенсивно окрашенные экстракты.
Слабые органические кислоты и их соли могут использоваться для приготовления
хорошо забуференных сред. pH экстрагирующего раствора, в зависимости от методи¬
ки, варьирует от 2 до 8. Реагент щавелевая кислота — оксалат аммония может связывать
и алюминий и железо, а также осаждать активный кальций. Реагент лимонная кисло¬
та — цитрат аммония может разрушать кристаллическую решетку силикатов, осаждать
кальций, взаимодействовать с соединениями железа при pH около 5,0 и с соединениями
алюминия при pH 3-8.
При обработке смесью винной кислоты и тартратов образуются комплексы железа
и алюминия (pH 3,2-7,5). Смесь молочной кислоты и лактатов предпочтительно экстра¬
гирует Р, связанный с кальцием, и лишь малое количество Р, связанного с железом или
алюминием (pH 3,5-3,7). Смеси борной кислоты и боратов, а также уксусной кислоты
и ацетатов при pH 2,5 или 4,8 экстрагируют лишь малое количество Р.
Разбавленные сильные неорганические кислоты способны солюбилизировать Р, свя¬
занный с кальцием, и, в различных количествах — Р, связанный с железом и алюминием.
Как правило, pH варьирует в диапазоне от 1 до 3 (Р в составе апатита, окклюдированный
Р, выветренные почвы). Эффективность экстракции (не связанной с агротехническими
практиками) зависит от положения кислоты в лиотропном ряду H2S04 > НС1 > HN03.
Например, НС1 способствует выделению Р из органических коллоидов (за счет разру¬
шения связей в поливалентных солях). Сильные кислоты вызывают гидролиз органи¬
ческого Р, контроль которого затруднителен. Фтористоводородная кислота способна
солюбилизировать органический и неорганический Р при взаимодействии с кристалли¬
ческой решеткой силикатов. Фторид аммония и фтористоводородная кислота образуют
комплексы с алюминием и железом в кислых почвах, однако их нельзя использовать
в известковых средах из-за возможности неконтролируемого осаждения фторида каль¬
ция (Syersnjip., 1972):
СаС03 + 2 NH4F -► CaF2^ + (NH4)2C03
Образование фторида кальция приводит к занижению результатов содержания Р, не
окклюдированного железом, и Р, связанного с алюминием, и завышению результатов
анализа Р, окклюдированного оксидами и гидроксидами железа, а также Р, связанного
с кальцием.
Изолирующие реагенты, такие как ЭДТА или ДТПА, позволяют солюбилизировать
Р, связанный с оксидами и гидроксидами алюминия и железа, и с различными формами
кальция. Реакция обычно протекает медленно, за исключением лабильных форм.
Оборудование
Согласно всем методикам, представленным в разделе 29.3.3, для анализов требуется
одинаковое оборудование, различающееся лишь незначительными деталями (напри¬
мер, размерами пробирок для центрифугирования).
• Аналитические весы (±0,1 мг).
• Пробирки для центрифугирования с завинчивающимися крышками объемом 100
и 250 мл.
Глава 29. Фосфор
655
• Шейкер.
• Центрифуга.
■ Стеклянные (Пирекс) воронки для фильтрования, с оттянутым концом.
• Шприц для фильтрования объемом 20 мл.
• Лабораторная стеклянная посуда.
• Сито из нержавеющей стали с диаметром ячеек 50 мм и 0,25 мм стандарта AFNOR
NF25.
• Восстановление смолы — см. далее в описании «Методика».
Водорастворимый Р
Основные положения
Методика водной экстракции (или экстракции 0,01 М раствором хлорида кальция или
сульфата кальция) позволяет измерить концентрацию Р, близкую к его концентрации
в почвенном растворе и на уровне возможного порога дефицита.
Непрерывный круговорот Р в почвенном растворе играет важную роль в питании рас¬
тений на протяжении всего цикла вегетации. Он обеспечивается жизнедеятельностью
микроорганизмов и ферментами типа фосфатазы. Если скорость поступления Р в по¬
чвенный раствор ниже скорости переноса к растениям в количестве Р, необходимом для
нормального роста (порог дефицита), будет происходить нарушение развития растений
(например, карликовость). ✓
Оборудование и реагенты
■ Деионизированная вода.
• Диск-фильтры или мембраны с размером пор 0,22 мкм для шприцев.
• Очень тонкие аналитические фильтры.
• 0,01 М раствор хлорида кальция.
• 0,01 М раствор сульфата кальция (растворимость 2,09 г/л).
Методика
• Навеску 5 г воздушно-сухой почвы (размер частиц 2 мм) помещают в пробирку для
центрифугирования с завинчивающейся крышкой; добавляют 50 мл деионизиро¬
ванной воды и встряхивают в течение 5 мин.
• Центрифугируют в течение 15 мин при 10 000 g до получения прозрачного надо-
садочного раствора; фильтруют через очень тонкий аналитический фильтр (синяя
лента) или через фильтр с размером пор 0,22 мкм, помещенный в шприц емкостью
20 мл; отбирают аликвоту, содержащую около 10 мкмоль Р для анализа содержания
водорастворимого Р (см. раздел 29.5).
Примечания. Если почвенный раствор получен in situ с помощью микропористых низ¬
конапорных трубок, его концентрация очень близка к концентрации раствора, кон¬
тактирующего с корнями растений; однако этот метод требует установки специального
оборудования на полевых станциях, что ограничивает его применение. В определенные
периоды года отбор проб становится невозможным из-за климатических условий, в осо¬
бенности в пустынных областях, где равновесие между «Р в почвенном растворе» и «ла¬
бильным неорганическим Р» смещается или не может установиться из-за жизнедеятель¬
ности микроорганизмов.
656 Часть 3. Неорганический анализ
Р, экстрагированный бикарбонатом натрия при pH 8,5 (Olsen)
Основные положения
Р, экстрагированный по описанной методике, соответствует наиболее биологически до¬
ступным формам Р, т. е. .лабильным органическим и неорганическим формам и включа¬
ет фракцию микробиологического Р.
Данную методику можно применять для анализа щелочных, нейтральных или кислых
почв. Экстрагирующий реагент снижает концентрацию кальция в растворе из-за осажде¬
ния нерастворимого карбоната кальция. Растворимость фосфатов кальция повышается
с уменьшением активности кальция (влияние известкования на доступность Р). Реагент
экстрагирует часть лабильного органического Р и неорганического Р, связанного с каль¬
цием. В кислых почвах растворимость фосфатов железа и алюминия (стренгит - варис-
цит) возрастает с повышением pH, оптимальная растворимость наблюдается при при¬
ближении к pH 6-7, где уровни содержания Н2РО~ и НРО*- форм одинаковы. Согласно
модифицированной (Dabin, 1967) методике Олсена, в раствор добавляют фторид аммония.
Эти вещества образуют комплексы с железом и алюминием, что позволяет солюбилизи¬
ровать большее количество Р. В некоторых тропических почвах с низким содержанием
кальция количество экстрагированного Р лучше коррелирует с урожайностью растений.
Реагенты
• 0,5 М раствор бикарбоната натрия (NaHC03, М = 84,01): доводят pH до 8,5 добав¬
лением 1 М раствора NaOH; растворы должны быть свежеприготовленными, их
следует хранить в герметичных бутылях.
• Активированный уголь, не содержащий Р (DARCO 60 или аналогичный), при необ¬
ходимости контроля очищают 2 М раствором НС1, затем раствором NaHC03 и опо¬
ласкивают деионизированной водой.
Методика
• Навеску 5 г воздушно-сухой почвы (размер частиц 2 мм) помещают в пробирку для
центрифугирования объемом 250 мл.
• Добавляют 100 мл раствора бикарбоната натрия; встряхивают в течение 30 мин.
• Центрифугируют в течение 5 мин при 10 000 g и фильтруют (добавление не содер¬
жащего Р активированного угля позволяет получить прозрачный и бесцветный
фильтрат для колориметрического анализа; тем не менее, не следует добавлять
уголь систематически; добавки вносят в пробы, экстракты которых сохраняют мут¬
ность после фильтрации); отобрают одну аликвоту для количественного анализа
после нейтрализации (см. раздел 29.5).
Р( экстрагированный анионообменной смолой
Основные положения
Смолы позволяют экстрагировать наиболее биологически доступные формы неоргани¬
ческого Р. Варьируя время контакта с анионитом, можно получить информацию о ки¬
нетике экстракции Р. Следует использовать анионообменные смолы с размером частиц,
превышающим 0,5 мм.
Реагенты
• Анионообменная смола в бикарбонатной форме (напр., Dowex 1X8-50).
• Анионообменная смола в хлоридной форме (Dowex 2).
Глава 29. Фосфор
657
• Размеры частиц используемой фракции смолы должны быть равны или выше
0,5 мм, проверить мокрым просеиванием. Отбросить более мелкие частицы.
• 10%-ный раствор хлорида натрия.
Методика
• Навеску 5 г воздушно-сухой почвы, измельченной до размера частиц 0,1 мм, по¬
мещают в пробирку объемом 100 мл для центрифугирования.
• Добавляют 5 г просеянной смолы.
• Добавляют 50 мл деионизированной воды; встряхивают в течение 16 ч.
• Отделяют смолу просеиванием через сито с размером ячеек 0,250 мм (AFNOR
NF25); промывают водой и собирают смолу, оставшуюся на сите.
• Струей воды из колбы-промывалки переносят смолу в химический стакан объемом
50 мл; сливают воду декантацией и добавляют 25 мл 10%-ного раствора NaCl; на¬
гревают на водяной бане при 80 °С в течение 45 мин.
• Охлаждают и декантируют раствор в мерную колбу на 50 мл; ополаскивают смолу
10%-ным раствором NaCl и доводят объем до 50 мл; гомогенизируют и отбирают
одну аликвоту для измерения содержания обменного Р.
Примечание
Эта методика позволяет проводить экстракцию Р без разрушения пробы и изменения pH
почвы; кроме того, методика позволяет моделировать работу корневой системы. Остав¬
ляя пробу в контакте с анйонитом на различные периоды времени, можно получить
ценную информацию о количестве, обменной емкости и кинетике Р. В этом случае ме¬
тодика предусматривает длительный контакт с ионитом — от 48 ч до трех недель и более.
Р, экстрагированный оксалатом аммония
Основные положения
Представленная методика (Jorret и Hebert, 1955) является частью стандарта AFNOR NF
Л31-161 (1993). Методика применима для анализа всех типов почвы, за исключением
почв, очень богатых органическими веществами.
Реагенты
• Деионизированная вода.
• Оксалат аммония (NH4)2C204* Н20.
• 0,1 М раствор гидроксида аммония (NH4OH).
• Экстрагирующий раствор (следует готовить свежий раствор ежедневно): растворя¬
ют 14,21 г оксалата аммония в 900 мл деионизированной воды при перемешивании
раствора с помощью магнитной мешалки с тефлоновым якорем; поддерживают
pH на уровне 7,0 с помощью раствора аммиака; переносят раствор в мерную колбу
вместимостью 1 л; ополаскивают стакан водой и доводят объем до 1000 мл; гомо¬
генизируют.
Методика
• Навеску 5 г воздушно-сухой почвы (размер частиц 2 мм) помещают в пробирку
для центрифугирования с завинчивающейся крышкой объемом 250 мл; добавляют
100 мл раствора оксалата аммония; встряхивают в течение 2 ч при 20 °С.
658
Часть 3. Неорганический анализ
• Фильтруют экстракт (он должен стать прозрачным) и определяют содержание Р
в аликвоте (если в экстракте содержится As5+, его влияние можно устранить, вос¬
становив As5+ до As3+ добавлением гипосульфита натрия и формальдегида (для
осветления раствора).
Р, экстрагированный кислотами (HCI-H2S04)
Основные положения
Методика разработана (Mehlich, 1953) для анализа кислых почв с низким содержанием
органического вещества (Северная Каролина, США), прочно фиксирующих Р. Методи¬
ка является базовой при проведении исследований плодородия почв наряду с методика¬
ми для известковых почв (Bray, 1945 и Olsen).
В качестве экстрагирующего раствора для экстракции Р, вызывающего отклик рас¬
тений, выращиваемых на почвах, богатых фосфатом железа, используется смесь соля¬
ной и серной кислот (смесь считается более эффективным реагентом, чем одна соляная
кислота). Методика является простой, быстрой и воспроизводимой, однако ее нельзя
использовать для анализа известковых почв из-за риска неконтролируемой нейтрализа¬
ции экстрагирующего раствора.
Реагенты
• Деионизированная вода.
• Концентрированная серная кислота (H2S04), rf=l,84.
• Концентрированная соляная кислота (НС1), d= 1,19.
• Экстрагирующий раствор: смешивают 6 мл концентрированной серной кислоты
и 36 мл концентрированной соляной кислоты приблизительно в 7,5 л деионизи¬
рованной воды; доводят объем до 9 л (концентрация экстрагирующего раствора:
0,05 М НС1 и 0,025 н. H2S04).
• Активированный уголь (DARCO G6 или аналогичный), не содержащий Р.
• Аналитические фильтры.
Методика
• Навеску 5 г воздушно-сухой почвы (размер частиц 2 мм) помещают в пробирку для
центрифугирования объемом 50 мл; добавляют 200 мг активированного угля, затем
20 мл экстрагирующего раствора.
• Встряхивают в течение 5 мин и фильтруют через аналитический фильтр (синяя
лента) или с помощью шприца, оборудованного фильтр-мембраной с размером
пор 0,45 или 0,20 мкм; отбирают одну аликвоту для измерения содержания Р.
Р, экстрагированный в раствор фторида аммония в соляной кислоте
Основные положения
Методика (Bray, 1945) основана на солюбилизации соединений Р в кислоте и воздей¬
ствии фторид-иона, снижающего активность катиона А13+, а также катионов Fe3+ и Са2+
за счет комплексообразования.
Методика используется для анализа нейтральных и кислых почв, однако она не под¬
ходит для известковых почв (неконтролируемая нейтрализация реагента, растворение
карбоната кальция и осаждение фторида кальция). Воспроизводимость методики зави¬
сит от того, насколько тщательно исполняется процедура.
Глава 29. Фосфор
659
Реагенты
• Деионизированная вода.
• 0,5 М раствор соляной кислоты: добавляют 20,3 мл НС1 в 500 мл деионизирован¬
ной воды.
• Фторид аммония NH4F.
• 1 М раствор NH4R растворяют 37 г фторида аммония в 400 мл деионизированной
воды в пластиковом стакане; разбавляют и доводят объем до 1000 мл; гомогенизи¬
руют и хранят в полиэтиленовом флаконе.
• Экстрагирующий раствор: смешивают 30 мл 1 М раствора NH4F с 50 мл 0,5 М рас¬
твора НС1. Доводят объем до 1000 мл деионизированной водой. Раствор содержит
0,025 М НС1 и 0,03 М NH4F, pH 2,6; реагент сохраняет стабильность при хранении
в полиэтиленовом флаконе.
Методика
• Помещают навеску 2 г воздушно-сухой почвы (размер частиц 2 мм) в пробирку для
центрифугирования объемом 50 мл.
• Добавляют 20 мл экстрагирующего раствора с pH 2,6 и встряхивают в течение 5 мин
на вибрационном шейкере (180 колебаний в минуту).
• Сразу фильтруют через тонкий аналитический фильтр (синяя лента) или с помо¬
щью шприца с фильтрующей мембраной (размер пор 0,45 или 0,20 мкм). Отбирают
одну аликвоту раствора для количественного анализа Р.
Примечания
При воздействии фторида аммония возможно растворение фосфатов алюминия и желе¬
за (III) за счет образования комплексов алюминия и железа; формы, связанные с каль¬
цием, следует экстрагировать кислотой.
Ряд модификаций методики направлен на улучшение корреляции содержания Р
в экстракте с урожайностью растений за счет повышения объема экстрагирующего реа¬
гента или повышения времени экстракции с 1 до 5 мин. Обеспечение воспроизводимо¬
сти затруднительно при времени контакта до 5 мин; процедура трудно исполнима без
автоматизации, например, автоматизированной аспирации и фильтрации через микро¬
мембрану с размером пор 0,20 мкм.
29.3.4. Методы изотопного разбавления
Методики изучения плодородности почв, основанные на изотопном разбавлении 32Р,
появились в 1950-х годах (Dean и др., 1947).
Были разработаны различные методики, позволяющие количественно оценить изо¬
топный обмен Р посредством испытаний растений (величина L, Larsen, 1952) или изме¬
рения потока фосфат-ионов в единицу времени в системе почва — раствор (величина Е,
Gunnarson-Frederickson, 1952).
Методика дает хорошую оценку биодоступности Р в почве и осадках. Биодоступный
фосфор определяется (Fardeau, 1997) как «весь фосфор, способный к перемещению
в почвенный раствор в форме фосфат-ионов за время, совместимое с потребностями
развития растений».
Методика, основанная на анализе кинетики обмена 32Р со стационарными система¬
ми, позволяет измерять активность (концентрацию Р в почвенном растворе), количе-
660
Часть 3. Неорганический анализ
ство и обменную емкость измерением количества ионов, перемещающихся из твердой
фазы в жидкую фазу за 1 мин, один день, три месяца, один год и более, что не дает, одна¬
ко, точной информации о минерализации органического вещества.
Внедрение этих методик требует особых подходов к организации лаборатории из-за
использования меченых соединений. Для достижения высокой точности необходи¬
мо удалить тонкие частицы из суспензии, для чего требуется центрифугирование при
100 000 g или фильтрование через мембраны 0,01 мкм. Измерения проводятся без изме¬
нения состояния системы, что позволяет количественно определять лабильные формы.
Кроме того, данные методики предусматривают использование специального обо¬
рудования, такого как жидкостной сцинтилляционный счетчик или ультрацентрифуга
до 120 000 g. Работа по методикам радиоактивных индикаторов должна проводиться от¬
дельно от других работ, в которых применяют изотопы естественного содержания, во
избежание возможного загрязнения. Процедуры с использованием меченых атомов Р
подробно описаны в работах (Fardeau, 1988-1993 и Gachon, 1988).
29.3.5. Анализ органического фосфора
Введение
Исследование органического Р является чрезвычайно сложным из-за разнообразия ор¬
ганических форм, а также из-за непрерывного изменения форм вследствие деятельно¬
сти микроорганизмов. Биологический круговорот проявляется в биохимических и хи¬
мических реакциях между растениями и почвой. В реакциях, происходящих в процессе
минерализации и иммобилизации, участвуют глины и оксиды, а в процессе формиро¬
вания почвы образуются различные фракции, в которых Р существует в разнообразных
формах, различающихся по степени доступности.
В зависимости от типа почвы, климатических условий и активности микроорганиз¬
мов органический фосфор может составлять от 20 до 80% от общего содержания фос¬
фора. В составе этой фракции Р встречаются специфические соединения; наиболее
распространенные формы — фосфат инозитола, на который может приходиться более
половины органического Р, фосфолипиды (около 5%) и нуклеиновые кислоты (около
2%).
Анализ всех этих форм затруднен из-за процессов гидролиза. Такие методы, как
ядерный магнитный резонанс (31Р ЯМР), позволяют идентифицировать молекулярные
структуры некоторых простых продуктов, если они присутствуют в достаточном коли¬
честве. Однако ЯМР не является достаточно чувствительным методом (приблизительно
10"5); он требует больших затрат и, следовательно, не пригоден для рутинных анализов.
Разработаны и другие методики, которые можно разделить на две основные группы:
общая оценка содержания органического Р путем прокаливания пробы, в ходе которо¬
го происходит разрушение органического вещества (см. «Измерение органического Р
методом термической деструкции») или солюбилизация с последующей экстракцией
в кислых и щелочных растворах (см. «Экстракция органического Р кислыми и щелоч¬
ными реагентами»).
Измерение количества органического Р методом термической деструкции
Основные положения
При прокаливании органический Р превращается в неорганический Р и его содержание
оценивается по разнице между неорганическим Р в прокаленной почве и неорганиче¬
ским Р в необработанной почве.
Глава 29. Фосфор
661
Эта методика не сложна и быстра в исполнении, однако ее прецизионность невысо¬
ка из-за (i) гидролиза органического Р при экстракции в необработанной пробе почвы
и (й) переноса ошибки при расчетах по разности (Pansu и др., 2001).
Оборудование
• Муфельная печь.
• Фарфоровые или кварцевые тигли 45 мм в диаметре.
• Пробирки для центрифугирования объемом 100 мл.
• Центрифуга.
• Штатив для фильтрования.
• Аналитические фильтры усиленной прочности.
• Лабораторная стеклянная посуда.
Реагент
• 0,5 М раствор серной кислоты (H2S04).
Методика
(1) Экстракт прокаленной почвы
• Помещают навеску 1 г почвы, измельченной до размера частиц 0,1 мм, в фарфоро¬
вый или кварцевый тигель.
• Ставят тигель в холодную печь и постепенно поднимают температуру до 550 °С
в насыщенной кислородом среде; поддерживают указанную температуру на про¬
тяжении 2 ч.
• Дают тиглю охладиться, переносят остаток почвы в пробирку для центрифугирова¬
ния вместимостью 100 мл (А); добавляют 50 мл 0,5 М раствора H2S04.
(2) Прямая экстракция
• Навеску 1 г почвы, измельченной до размера частиц 0,1 мм, помещают в пробирку
для центрифугирования объемом 100 мл (В).
• Добавляют 50 мл 0,5 М раствора H2S04.
(3) Экстракция
• Закрывают крышками пробирки А и В и встряхивают в течение 16 ч.
• Центрифугируют при 5000 g в течение 10 мин.
• Фильтруют на усиленном фильтре, отбирают необходимую для анализа Р аликвоту
и хранят в пластиковом флаконе. Количество органического Р рассчитывают по
разности: Р в прокаленной почве минус Р в необработанной пробе (эти результаты
сравнивают с общим содержанием Р для оценки содержания неорганического Р).
Примечания, Прокаливать почву следует в печи, в среде с высоким содержанием кис¬
лорода, чтобы избежать потерь при восстановлении, возможных при температуре выше
400 °С (Н3Р).
Следует точно соблюдать указания методики по конечной температуре и длительно¬
сти прокаливания, чтобы избежать изменений относительной растворимости продук¬
тов, образующихся при данной температуре, поскольку это может привести к случай¬
ным погрешностям при оценке содержания органического Р.
662
Часть 3. Неорганический анализ
Экстракция органического Р кислыми и щелочными реагентами
Основные положения
Эти методики (Mehta и др., 1954) предусматривают химическую экстракцию органиче¬
ских и неорганических форм Р (последовательная обработка сильной кислотой и силь¬
ным основанием).
При предварительной обработке сильными кислотами разрушаются поливалентные
связи и становится возможной экстракция связанных катионов (в основном, Fe, А1
и Са); в результате неорганический Р становится нерастворимым, побочной реакцией
является гидролиз органических соединений. Обработка щелочью, позволяющая солю¬
билизировать органическое вещество, также вызывает гидролиз органического Р.
Минерализация проводится в одной аликвоте экстракта; органический фосфор пере¬
водят в форму ортофосфата и его содержание оценивают по разности между неоргани¬
ческим Р после минерализации и неорганическим Р в исходном экстракте.
Оборудование
• Алюминиевый блок для минерализации или автоклав.
• Полиэтиленовые пробирки для центрифугирования с завинчивающимися крыш¬
ками объемом 100 мл.
• Водяная баня, нагретая до 70-90 °С.
• Мешалка типа Vortex.
• Вентилируемый сушильный шкаф, нагретый до 100 °С.
Реагенты
• Концентрированная хлористоводородная кислота.
• 0,5 М раствор NaOH.
• Деионизированная вода.
Методика
Экстракция
• Навеску 1 г воздушно-сухой почвы (размер частиц 0,1 мм) помещают в пробирку
для центрифугирования объемом 100 мл.
• Добавляют 10 мл концентрированной хлористоводородной кислоты; гомогенизи¬
руют и нагревают на водяной бане в течение 10 мин при 70 °С.
• Добавляют еще 10 мл концентрированной хлористоводородной кислоты, оставля¬
ют взаимодействовать на 1 ч при температуре окружающей среды; добавляют 50 мл
воды, гомогенизируют и центрифугируют при 5000 g в течение 5 мин.
• Декантируют надосадочный раствор в мерную колбу объемом 250 мл; промывают
осадок после центрифугирования небольшим количеством воды и центрифугиру¬
ют в течение 5 мин; декантируют в ту же мерную колбу.
• Обрабатывают остаток почвы 30 мл 0,5 М NaOH; переводят во взвешенное состоя¬
ние энергичным встряхиванием на миксере типа Vortex и оставляют в контакте на
1 ч при температуре окружающего воздуха.
• Центрифугируют и декантируют надосадочный раствор в мерную колбу, содержа¬
щую хлористоводородный экстракт (декантируют медленно, перемешивая рас¬
твор).
Глава 29. Фосфор
663
• К остатку добавляют еще 60 мл 0,5 М раствора NaOH, переводят во взвешенное
состояние путем энергичного встряхивания на миксере типа Vortex и нагревают на
водяной бане, поддерживая температуру 90 °С в течение 8 ч (или в вентилируемом
сушильном шкафу.
• Центрифугируют; после полного охлаждения переносят надосадочный раствор
в мерную колбу на 250 мл, содержащую другие экстракты; доводят объем до метки
деионизированной водой и гомогенизируют; это раствор (А).
Минерализация общего экстрагированного Р
• Встряхивают раствор А, чтобы перевести осадок во взвешенное состояние и бы¬
стро отбирают аликвоту объемом 5 мл в пробирку из стекла Пирекс, подходящую
для данного блока минерализации (озоления) (можно использовать высокий хи¬
мический стакан).
• Добавляют 2 капли концентрированной серной кислоты и 1 мл 72%-ной хлорной
кислоты.
• Гомогенизируют, накрывают часовым стеклом и проводят минерализацию до по¬
явления белого дымка; выдерживают для разложения в течение 30 мин при 200 °С.
• Охлаждают и переносят в мерную колбу вместимостью 50 мл, ополаскивая сосуд
деионизированной водой, доводят объем до 50 мл.
Если содержание Р в почве велико, отбирают аликвоту соответствующего объема для
количественного анализа; цри низком содержании Р используют для анализа все содер¬
жимое мерной колбы.
Анализ неорганического и органического Р
• Декантируют раствор А; аликвоту надосадочного раствора фильтруют и определя¬
ют содержание неорганического фосфора, как описано выше.
• Содержание органического Р рассчитывают по разности между общим Р в минера¬
лизованном экстракте и неорганическим Р в экстракте А (измеренным спектроко¬
лориметрическим методом, см. раздел 29.5.2).
Примечания
• Экстракты почв с высоким содержанием гумуса могут быть окрашены. В этом слу¬
чае их следует обработать активированным углем (Darco 60, не содержащим Р).
• Обработка хлористоводородной кислотой может вызвать гидролиз эфиров фос¬
форной кислоты, например, глицерофосфатов.
• Эта методика позволяет экстрагировать около 80% Р (сравнимо с общим Р, экстра¬
гируемым при обработке хлорной кислотой).
• 16-часовую экстракцию можно заменить сокращенной обработкой с применением
ультразвука.
• Результаты оценки количества органического Р оказываются заниженными вслед¬
ствие гидролиза; точность результатов, рассчитанных по разности, невелика.
• Процентная доля общего органического Р может быть связана с биомассой почвен¬
ных микроорганизмов и активностью микроорганизмов; и может выявить перенос
Р растениями с вертикальным распределением Р, который зависит от содержания
органических остатков на поверхности почвы.
664
Часть 3. Неорганический анализ
29.4. Удержание фосфора
29.4.1. Введение
Возделывание почвы приводит к выведению фосфора из почвы в культурные растения
и в долгосрочной перспективе к дефициту элементов, обусловливающих плодородие по¬
чвы. Снижение количества доступных элементов происходит отчасти из-за ускорения ми¬
нерализации углеродсодержащих веществ, что сопровождает сельскохозяйственную дея¬
тельность, вследствие чего содержание органического вещества существенно уменьшается.
Внесение удобрений позволяет поддерживать удовлетворительный уровень доступ¬
ного Р, необходимый для развития растений и регулирования потоков фосфора. Для
сохранения равновесия при внесении удобрений, обогащенных фосфатами, необходи¬
мо оценить объем выведения Р, а также определить способность почвы к удерживанию
и восстановлению Р.
Анионы могут связываться с обменными центрами и вносить вклад в величину емко¬
сти анионного обмена (ЕАО, см. гл. 27). Как правило, величина ЕАО намного меньше
ЕКО (см. гл. 26), и зависит от pH почвы, уровня электролита и типа глины. Лиотропный
ряд: SiOJ" > РО*" > > SO2- > NO" ^ Cl" показывает, что SiO*~ и РО*" прочно сорбированы
в кислых почвах вследствие связывания РО*~ с октаэдральным алюминием. Фосфаты
остаются нерастворимыми и труднодоступными для растений. Это явление (i) «удержа¬
ния Р», которое можно измерить путем экстрагирования сильно разбавленной кислотой
или (И) «фиксации Р», относящееся к формам Р, которые невозможно экстрагировать
разбавленными кислотами.
Чем ниже pH, тем выше концентрация поливалентных катионов (Al, Fe, Мп) и выше
степень удержания Р. Процесс подкисления может привести к образованию в почве не¬
растворимых форм Р: варисцита (А1Р04* 2Н20) и стренгита (FeP04* 2Н20). Силикаты,
арсениты, селениты и фториды могут замещать фосфатные формы. В щелочной среде
фосфаты могут реагировать с различными формами кальция (в частности, с карбоната¬
ми), образуя нерастворимые фосфаты кальция, представляющие серьезные проблемы
при возделывании аридных карбонатных почв.
Некоторые почвы, такие как андосоли, могут адсорбировать Р в большом количе¬
стве. В подобных почвах наблюдается отсутствие отклика на внесение удобрений и из¬
менения ЕКО (ЕКО может возрасти в два или три раза) при их культивировании. Таким
образом, необходимо измерять степень удержания Р в почве для определения коэффи¬
циентов адсорбции и построения изотерм адсорбции по уравнению Лэнгмюра или урав¬
нению Фрейндлиха. В таком случае почвы можно классифицировать в соответствии с их
адсорбционными характеристиками и установленными связями между количеством ад¬
сорбированного Р (в г/м2) и равновесными концентрациями в почвенном растворе.
29.4.2. Определение параметров удержания Р
Основные положения
Методика (Blakemore и др., 1981) использует уравновешивание образца почвы с раство¬
ром, содержащим растворимый Р, и измерение содержания фосфата, остающегося в рас¬
творе. При pH 4,6 удержание Р приближено к максимальному.
Оборудование
• Центрифуга.
• Шейкер.
Глава 29. Фосфор
665
Реагенты
• Деионизированная вода.
• Раствор для удержания Р (1000 ppm Р): растворяют 8,79 г дигидрофосфата калия
и 32,8 г безводного ацетата натрия приблизительно в 1 л воды; добавляют 23 мл ле¬
дяной уксусной кислоты; доводят объем деионизированной водой до 2 л в мерной
колбе.
Методика
• Навеску 5 г воздушно-сухой почвы (размер частиц 2 мм) помещают в пробирку для
центрифугирования с завинчивающейся крышкой объемом 50 мл; добавляют 25 мл
раствора, содержащего удержанный Р.
• Встряхивают в течение 24 ч при 20 °С.
• Центрифугируют при 5000 g в течение 15 мин.
• Фильтруют, гомогенизируют и отбирают одну аликвоту для количественного ана¬
лиза оставшегося в растворе Р и идентичную аликвоту реагента (холостую пробу)
для расчета удержанного Р по разности. Анализируют оба раствора спектроколо¬
риметрическим методом.
Расчет
Расчет удержанного Р по разности: Р в холостой пробе минус Р в равновесном растворе,
а результат выражают в виде процентной доли.
Примечания
В оксисолях и латеритных почвах Р очень прочно фиксирован при pH < 4,0 (связи P-Fe
и Р-А1), что приводит к сильному дефициту по фосфору. Оксиды железа и алюминия за¬
ряжены положительно ниже точки нулевого заряда. В аллофанных почвах Р фиксирован
на поверхности оксидов и образует комплексы Р-органического вещества и мостиковые
связи с алюминием в неорганических соединениях.
29.5. Анализ Р в экстрактах
29.5.1. Введение
Методы
Для количественного определения Р разработано множество методик. Они различаются
по чувствительности: одни методики пригодны для анализа экстрактов с высоким со¬
держанием Р, другие — для экстрактов, содержащих Р только в следовых количествах.
Редко применяемые в наше время гравиметрические методики основаны на осажде¬
нии аммонийно-магниевых фосфатов или фосфомолибдата аммония. В титриметриче-
ских методиках можно использовать этот осадок для растворения в гидроксиде натрия
и обратного ацидиметрического титрования; кроме того, можно использовать такие
методики, как комплексонометрия или манганатометрия. Наиболее распространены
методы спектроколориметрического поглощения с применением комплекса фосфора
с ванадием и молибденом, поглощающего при 430 нм, или комплекса с молибденовой
синью, поглощающего в диапазоне от 650 до 890 нм, в зависимости от их состава. Ино¬
гда эти измерения автоматизируют или даже роботизируют.
Другие физико-химические методы, такие как атомная абсорбция, амперометрия,
потенциометрия, кондуктометрия, кулонометрия и полярография, используют не-
666
Часть 3. Неорганический анализ
прямые измерения, например, после взаимодействия с молибденом и образования
фосфомолибдатного комплекса. Измерения пламенно-эмиссионным методом или
спектрометрия с индуктивно связанной плазмой позволяют определять органические
и неорганические формы одновременно, однако не могут анализировать их по отдель¬
ности. Чувствительность этих методик к Р невелика (см. гл. 31).
Осветление экстрактов
Какой бы ни была методика анализа, экстракты должны содержать только растворимые
формы; в них не должны присутствовать минеральные или органические взвешенные
частицы.
Фильтрования через очень тонкие (синяя лента) аналитические целлюлозные филь¬
тры (или фильтры повышенной прочности для очень кислых экстрактов) зачастую
оказывается недостаточно, однако можно улучшить эффективность фильтрации, воз¬
вращая в процесс первые отфильтрованные фракции после частичной закупорки пор
фильтра. Приемлемы мембраны с размером пор около 0,45-0,20 мкм, установленные на
шприцевых фильтрах типа Люэр. Пропускная способность этих мембран очень низка,
они легко закупориваются, но, если система оборудована предварительным фильтром,
можно быстро получить 1-2 мл экстракта, что достаточно для рутинного анализа и обе¬
спечивает достаточную прецизионность результатов. В сложных случаях, например,
при анализе белков может потребоваться параллельное ультрацентрифугирование при
50 000-100 000 g.
Иногда применяется электродиализ (или электроультрафильтрация), предел измель¬
чения составляет приблизительно 0,02 мкм. Тем не менее, существует риск изменения
pH около электродов и избыточного разбавления пробы. В ходе детальных исследова¬
ний некоторые авторы подсчитывали остаточное содержание взвешенных частиц с по¬
мощью счетчиков лазерного излучения (Nanosizer) и определяли природу частиц с по¬
мощью электронного микроскопа.
Типы анализов
После очистки экстракта оптимальные области анализа, диапазоны концентраций, мак¬
симальную чувствительность и возможные помехи следует принимать во внимание при
выборе методики измерения.
Как правило, предпочтение отдают прямому анализу экстракта. Однако иногда необ¬
ходимо использовать минимальный объем экстракта, который можно проанализировать
с достаточной точностью, чтобы исключить влияние очень тонких частиц в суспензии
или в растворе.
Для спектроколориметрических измерений предпочтительны узкие колориметриче¬
ские ячейки при условии достаточной чувствительности. Применение 50- или 100-мил¬
лиметровых ячеек усиливает влияние микрочастиц суспензии, что делает необходимой
предварительную осторожную ультрафильтрацию. Реакции могут происходить в хо¬
лодных и горячих средах с различными степенями кислотности или щелочности. Для
ограничения гидролиза органического Р проводится анализ в холодных средах с малой
концентрацией и контролем pH. Тем не менее, фитины нельзя определять в холодных
средах, некоторые фитины анализируют спектроколориметрическими методами при
60 °С. То же относится к неорганическому Р в форме пирофосфата и в других формах.
По возможности следует избегать промежуточных этапов, поскольку некоторые спо¬
собы разделения вызывают невоспроизводимое влияние на результаты (например, из¬
менения pH, загрязнение, очищение экстрактов смолой, экстракцию окрашенных со-
Глава 29. Фосфор
667
единений, добавки при фильтровании, активированный уголь, который редко бывает
свободен от Р).
Важным фактором является селективность анализа:
• спектроколориметрический анализ (ручной, проточно-инжекционный, анализ
сегментного потока) является единственным селективным методом анализа орто¬
фосфатных форм и органических форм, не определяемых из экстрактов;
• атомная спектрометрия (с индуктивно связанной плазмой или пламенная спек¬
трометрия) позволяет измерить содержание всех форм Р; результаты анализа могут
в два-три раза превышать результаты, полученные с помощью колориметрических
методик;
• 31Р ядерный магнитный резонанс (31Р ЯМР) позволяет дифференцировать различ¬
ные формы Р; орто-, пиро- и органический Р можно определять одновременно без
разрушения пробы.
29.5.2. Анализ ортофосфатного Р спектроколориметрическим методом
Спектроколориметрия с использованием молибденовой сини
Основные положения
Глубокое понимание природы химических взаимодействий и возможных помех очень
важно для контроля качества. Точное знание химического состава соединения и кон¬
троль условий эксперимента позволяют проводить измерения интенсивности спектра
на оптимальной длине волны {Gautheyrou и Gautheyrou, 1989).
Реакция с молибденовой синью должна проводиться в строго определенных условиях:
pH, концентрация кислоты, окислительно-восстановительный потенциал, температура
и длительность взаимодействия должны быть подобраны так, чтобы сдвинуть равнове¬
сие в сторону образования наиболее конденсированных форм — таких, где максималь¬
ное число атомов молибдена, окружающих атом Р, равняется 12. При образовании таких
форм оптическая плотность раствора остается постоянной. Помехи происходят только
от высокого содержания кремния, мышьяка и германия.
В ходе химической реакции происходит взаимодействие молибденовой кислоты с ор¬
тоформами фосфора. Конденсация такого типа может происходить только в кислой сре¬
де с высокой ионной силой. Восстанавливающее соединение в присутствии катализато¬
ра — антимонилтартрат-иона образует интенсивно окрашенный в синий цвет комплекс
фосфомолибденового аниона.
НР042- + 12Мо042- + 23Н+ -► [Р04(Мо03)12]3-+12Н20
Синяя восстановленная
форма
Примечания
Окислительно-восстановительный потенциал зависит от pH. Поэтому необходимо
стандартизировать процедуры, чтобы обеспечить максимально возможное постоянство
количества кислоты и восстановителя, а также температуры. Скорость образования
фосфомолибдата пропорциональна концентрации Р.
Может применяться множество минеральных и органических восстановителей1.
В холодной кислой среде высокой каталитической активностью обладает аскорбино-
1 Например, 1-амино-2-нафтол-4-сульфоновая кислота, аскорбиновая кислота, SnCl2, диаминофе-
нол, гидразин, гидрохинон, тиомочевина.
668
Часть 3. Неорганический анализ
вая кислота в сочетании с антимонилтартратом калия (Murphy и Riley, 1962). Методика
позволяет получить интенсивно окрашенный синий комплекс, сохраняющий стабиль¬
ность на протяжении 24 ч. Для достижения максимальной интенсивности поглощения
необходимо соблюдать соотношение Sb:P, равное 1:1, но это соотношение не должно
быть превышено во избежание выпадения осадка1. Максимальное поглощение наблю¬
дается на границе видимой и ближней инфракрасной области спектра: 880—890 нм.
В солянокислой среде чувствительность ниже по сравнению с сернокислой средой.
В среде хлорной кислоты соединения отлично сохраняют стабильность. Концентрации
серной кислоты в диапазоне от 0,15 М до 0,25 М позволяют работать при pH, не превы¬
шающем 1.
Помехи
В структуре полианиона Р позиции атомов кислорода или центрального атома могут
замещаться элементами с близкими ионными радиусами и аналогичными структурны¬
ми свойствами, такие как Si, Ge, As, Ti. Могут образовываться такие соединения, как
(SiMo12O40)*" или (AsMo12O40)3_. Оптимальные условия образования соединений Р, As
или Si не являются одинаковыми: для Р и As устойчивое соединение с молибденом об¬
разуется строго в диапазоне pH от 0,8 до 1,4. Комплексы Si и Ge образуются только при
pH от 1,8 до 2,5. Поэтому реакцию следует проводить в достаточно кислой среде, что¬
бы ограничить помехи, связанные с присутствием кремния. Как правило, присутствие
мышьяка не вызывает помех. Кроме того, причинами помех могут стать:
• элементы, способные к замещению молибдена, например, вольфрам, ионный ра¬
диус которого (0,60 А) близок к ионному радиусу молибдена (0,59 А) или ванадия
(0,58 А);
• элементы, катализирующие процесс реакции (Sn, Sb, Bi), обладающие синерге¬
тическим действием при восстановлении (поскольку содержание этих элементов
в почве весьма мало, помехи такого типа не являются существенными);
• элементы с окрашенными солями: присутствие Сг3+, Ni2+, Ni4+, Cu2+, Mn2+, Mn4+,
Fe3+допустимо приблизительно до уровня 1000 мг/л (несмотря на то что органиче¬
ское вещество поглощает излучение другой длины волны, они могут вызвать по¬
мехи, связанные со снижением энергии излучения переноса, поэтому следует до¬
биваться их полного разложения);
• элементы, склонные к образованию осадков и выделяющие в реакционной среде
нерастворимые соединения;
• элементы, обладающие окислительными свойствами, влияющие на ход реакции
восстановления;
• органические кислоты, способные к образованию комплексов с молибденом (на¬
пример, щавелевая кислота и оксалаты, винная кислота и тартраты, лимонная кис¬
лота и цитраты); если предстоит анализ смеси, экстрагированной с помощью этих
реагентов, следует разрушить матрицу, чтобы избежать помех при реакции образо¬
вания фосфомолибдатного комплекса;
• спирты, в которых антимонилтартрат слаборастворим;
• белки, до некоторой степени способные к осаждению.
1 Растворимость повышается при нагревании, однако при этом возникает риск гидролиза ор¬
ганических P-содержащих соединений.
669
Глава 29. Фосфор
Оборудование
• Спектроколориметр, работающий в области 800-900 нм.
• Измерительные кюветы (10—100 мм).
Реагенты
• 2,5 М раствор серной кислоты (H2S04): осторожно добавить 140 мл концентриро¬
ванной серной кислоты к 900 мл деионизированной воды; охладить и довести объ¬
ем до 1000 мл.
• Раствор молибдата аммония: взять навеску 20 г (NH4)6Mo7024 и растворить в 500 мл
деионизированной воды (готовить свежий раствор еженедельно).
Раствор полугидрата антимонилтартрата калия KSb0C4H406 • 0,5Н2О
/О—с=0 ‘
Н20—►Stb- о с—н
\
-н
:=о
I
М = 333,93: навеску 1,375 г растворить в 500 мл воды (готовить свежий раствор еже¬
недельно).
• Раствор аскорбиновой кислоты
О-
Н QH
I I
'С' vc— с —
/
он
I I I
с—с J
\
он
н
СН2ОН :
растворить 8,75 г аскорбиновой кислоты в 500 мл деионизированной воды (готовить
свежий раствор ежедневно).
• Смешанный реагент: смешать в нижеописанном порядке при встряхивании и го¬
могенизации между внесением каждой добавки: 165 мл 2,5 М раствора H2S04,
50 мл раствора молибдата аммония, 100 мл раствора аскорбиновой кислоты, 16 мл
раствора антимонилтартрата калия, довести объем до 1000 мл деионизированной
водой (готовить свежий раствор ежедневно).
Стандартные растворы фосфатов: для каждого типа экстракта лучше готовить кали¬
бровочные растворы в тех же средах, что и анализируемые пробы, и систематически (i)
готовить холостую пробу с реагентами, подвергающимися той же обработке, что и про¬
бы, (Н) регулярно воспроизводить калибровочную кривую и сравнивать с ранее постро¬
енной кривой для выявления временных сдвигов или возможных изменений, которые
могут снизить точность.
• Исходный раствор — 100 мкг (Р)/мл: растворить 0,4393 г дигидрофосфата калия
(КН2Р04) в деионизированной воде и довести объем до 1000 мл; 1 мл содержит
100 мкг Р; хранить в холодильнике в светонепроницаемом сосуде.
• Срединный раствор (А) — 10 мкг (Р)/мл (хранится в холодильнике одну неделю):
разбавить 100 мл базового раствора до 1000 мл деионизированной водой.
• Срединный раствор (В) — 1 мкг (Р)/мл: разбавить 100 мл промежуточного раствора
(А) и довести объем до 1000 мл деионизированной водой (готовить перед каждой
серией анализов).
670
Часть 3. Неорганический анализ
• Калибровочные растворы, пригодные для работы по любой методике (каждый день
готовить свежий раствор в объеме, необходимом для используемого аналитическо¬
го прибора, 50 или 100 мл.
0,0 (холостая проба), 0,1; 0,2; 0,4; 0,6; 0,8; 1,0; 2,0 мкг(Р)/мл.
Методика
• Точно отмеряют объем экстрагирующего раствора V0 (мл), содержащий от 10 до
20 мкг Р, т. е. аликвоту объемом от 1 до 15 мл максимум (при экстракции водой,
смолой или при изотопной экстракции 32Р отбирают аликвоту максимального
объема; при экстракции подвижных форм в кислой или щелочной среде объем
аликвоты меньше; при измерении общего Р используют промежуточные степени
разбавления).
• Помещают аликвоту в мерную колбу объемом 25 мл; добавляют 5 мл смешанно¬
го реагента и доводят объем до 25 мл деионизированной водой; гомогенизируют;
ожидают 30 мин перед проведением измерений (цвет остается стабильным на про¬
тяжении около 24 ч).
• Измеряют поглощение при 890 нм на спектроколориметре, используя колориме¬
трическую кювету размером 10 мм (или кювету большего размера, в зависимости
от интенсивности цвета, возможно использование кюветы размером 100 мм для
измерений в водных экстрактах). Повторяют процедуру для каждой точки кали¬
бровочного диапазона.
• Строят график с концентрацией Р по оси X и величиной поглощения по оси Y.
Определяют концентрацию Р в растворе пробы: х, мкг/мл. Если V — объем экс¬
трагирующего раствора, мл; Р— вес пробы почвы, г; /— возможный поправочный
коэффициент на влажность, то содержание в почве рассчитывают по формуле (ре¬
зультаты выражают по отношению к почве, высушенной при 105 °С):
или
C—fx V/P вмг(Р)/кгпочвы
С= 2,29 fxV/P в мг Р205/кг почвы.
Альтернативно можно автоматизировать процесс, например, в системе непрерывного
поточного анализа в сегментированном потоке (см. манифольд для определения общего
фосфора на рис, 29.2).
Спектроколориметрический анализ Р с использованием желтого
фосфомолибдатного комплекса
Данная методика менее чувствительна по сравнению с методикой «Спектроколориме-
трия с использованием молибденовой сини» и часто используется для анализа Р в экс¬
трактах с высоким содержанием Р для определения общего, общего органического или
удержанного Р. Ортофосфатные формы анализируют спектрометрическими методами
с использованием реакции образования желтого комплекса, поглощающего при 420—
466 нм:
80 °С
Н3Р04 + 12(NH4)2Mo04 + 2IHNO3 -► ((NH^PMOj^ + 21NH4N03+ 12Н20
В присутствии арсенатов может образовываться комплекс (NH4)3AsMo12O40, мешаю¬
щий колориметрическим измерениям. Помехи могут быть связаны с наличием железа;
если железо окрашивает экстракт, его следует удалять (Salvage и Dixon, 1965 и Ruf \ 1966).
Глава 29. Фосфор
671
Потоки, мл/мин
■vwwv^
rA/V\AA/WAAA/WV4
b\/W
Водяная
Замед-
баня
ление
95 °С
Колориметри¬
ческая ячейка
"1 г-
Отходы
1,00 Раствор пробы
1.50 Воздух
2.00 н. серная кислота
1,20 Разбавленная проба
1,60 Воздух (обратный затвор)
0,80 1,65 н. серная кислота|
0.43 Молибденовый оеагент
I
2,00 Аскорбиновая кислота (реагент)
2,00 (Обратный затвор)
—^Обратный насос
Рис. 29.2. Определение суммарного фосфора в ортоформе методом автоматизированной
колориметрии с регистрацией поглощения при 625 нм (Gautheyrou и Gautheyrou, 1978)
29.5.3. Анализ Р методами атомной спектрометрии
Измерение Р методом атомно-адсорбционной спектрометрии с индуктивно связанной
плазмой (см. раздел 31.2.14) позволяет получить информацию о содержании всех форм
Р в экстракте, однако не может их дифференцировать (например, не может отдельно
определить органический и неорганический Р). Чувствительность метода недостаточно
высока для прямого анализа Р. Как правило, для анализа используют линии при 213,62
и 214,91 нм. Анализу мешает присутствие меди, хрома, железа, ванадия и титана. Кор¬
рекция межэлементного взаимодействия снижает прецизионность методики.
Метод выделения комплекса РМо12 (см. раздел 29.5.1 и 29.5.2) и анализ молибдена по
спектральным линиям УФ при 177,50 и 178,77 нм, и в особенности при 178,29 нм, явля¬
ется чувствительным и прецизионным. Ванадий, титан, никель, медь, марганец и хром
не мешают измерениям при концентрациях до 200 мг/л. Железо, алюминий, кальций,
кремний не мешают измерениям при концентрациях приблизительно до 1000 мг/л.
На результаты измерений методом пламенно-эмиссионной спектрометрии исходное
состояние Р существенно не влияет; интенсивность эмиссии отражает концентрацию
общего Р. Спектры эмиссии являются сложными. Можно использовать полосы при
5249, 5597 или 5097 А (Н-РО) и полосы при 2478, 2464, 2540 А (РО). Предел обнаруже¬
ния - около 1 мг/л, в зависимости от источника возбуждения.
29.5.4. Определение различных форм Р методом 31Р-ЯМР
Метод ЯМР (см. раздел 12.3.4) позволяет получить различающиеся сигналы, соответ¬
ствующие орто- и пирофосфатным формам и более или менее сложным органическим
формам Р.
672
Часть 3. Неорганический анализ
Ядро 31Р характеризуется наличием спинового магнитного момента. При возбуж¬
дении радиоволнами ядра, помещенного в магнитное поле, выделяется энергия, обу¬
словленная энергетическими переходами. Полученный спектр содержит информацию
о ближайшем химическом окружении атома Р без разрушения образца (рис. 29.3). Ме¬
тоды ЯМР высокого разрешения, применяемые для анализа жидкой фазы, в настоящее
время являются наиболее распространенными методами исследования почвенного Р;
твердофазный ЯМР применяется реже. Чувствительность метода ниже по сравнению
с протонным ЯМР, т. е. около 10-5 г.
Пики, соответствующие различным формам неорганических фосфатов с коротки¬
ми линейными цепями (орто-Р, пиро-Р, триполифосфаты), четко дифференцированы.
Молярные отношения определяют путем измерения площади пиков. Легко поддают¬
ся анализу сложные эфиры одно- и двухосновных фосфорных кислот. Неорганические
фосфаты с длинными структурными цепями и конденсированные органические фосфа¬
ты могут иметь сложные спектры. Для правильной интерпретации могут потребоваться
одновременно 31Р- и протонный ЯМР-спектры.
Метод высокоспецифичен и может применяться для анализа Р в различных почвен¬
ных экстрактах (Gautheyrou и др., 1990).
Рис. 29.3. Спектр 31Р ЯМР форм Р в почвенном экстракте (0,5 М NaOH) (Gautheyrou и др., 1990)
29.5.5. Разделение фосфорсодержащих соединений методом
жидкостной хроматографии
Пробу экстракта инжектируют в поток подвижной фазы (например, 0,5 мМ фгалевой
кислоты при pH 2,7), доставляющий ее в колонку, заполненную анионообменной смо¬
лой (Karlson и Frankenberger, 1987). Метод используется, в частности, для определения
содержания фосфорорганических пестицидов (см. раздел 13.3.4), а также для разделения
анионных форм Р. Хроматографический метод позволяет также проводить исследова¬
ния фосфолипидов после экстракции растворителями (см. исследования липидов в раз¬
делах 13.2.5 и 13.3.3).
673
Глава 29. Фосфор
29.6. Прямое определение форм Р in situ или
в экстрагированных частицах
Определение Р в тонком шлифе (см. раздел 8.2.2) или в минералах, выделенных при
фракционировании частиц по размеру (см. гл. 3), представляет чрезвычайно сложную
задачу. Известно около 220 природных минералов, содержащих Р с бесчисленными
атомными замещениями. Несмотря на преобладание форм, связанных с кальцием, алю¬
минием и железом, идентификация частиц почвы является сложным делом.
Минералы, включающие Р, как правило, концентрируются в глине и оксидных фрак¬
циях (например, апатиты, алюмофосфаты, варисцит, вивианит). Магнитная фракция,
экстрагированная из некоторых тропических почв, содержит формы оксида железа
с упорядочением ближнего порядка в большем количестве, чем с кристаллическими
формами. Это объясняет механизмы поглощения Р на поверхности оксида и гидрокси¬
дов железа, например, на поверхности гетита, гематита, лепидокрокита или аморфных
гелей (Schwertmann, 1964).
Внесение фосфорных удобрений может привести к очень высоким локальным кон¬
центрациям Р и перераспределению в почве за счет вытеснения органического Р. Про¬
дукт этой реакции можно определить с помощью физико-химических методов, таких
как микродифракция (Belle и Black, 1970). Игольчатые кристаллы сульфата кальция, об¬
разовавшиеся на микрокристаллах апатита, можно наблюдать с помощью оптического
микроскопа или сканирующего электронного микроскопа, оснащенного ЭД-РС-ми-
кроанализатором, на непокрытых тонких шлифах, обработанных разбавленной серной
кислотой. Диаграммы распределения кальция и фосфора могут быть построены при до¬
статочных локальных концентрациях (Subrarao и Ellis, 1975). В процессе растровой про¬
свечивающей электронной микроскопии и сканирующей электронной микроскопии
с энергодисперсионным рентгеновским микроанализатором при бомбардировке по¬
верхности образца электронами образуются рентгеновские лучи, что позволяет наблю¬
дать распределение Р вокруг частиц и идентифицировать химические вещества в зоне,
площадь которой составляет приблизительно 1 мкм2. Метод микродифракции позволяет
исследовать структуру минеральных скоплений и идентифицировать формы Р в мине¬
ралах. Термодинамические свойства этих соединений могут быть представлены в виде
значений растворимости, позволяющих оценить взаимодействия с Р, содержащимся
в почвенном растворе. Можно рассчитать соотношения P/Al, P/Fe, Р/Са, однако рас¬
четы органических форм требуют более сложных подходов. Подобные методы востребо¬
ваны при проведении подробных исследований минералогических и петрографических
механизмов, в частности процессов фиксации удобрений почвой (El Zahaby и Chien,
1982, Freeman и Rowell, 1981; Henstra и др., 1981).
Использованная литература
Bell ВС and Black С А (1970) Comparison of methods for identifying crystalline phosphate produced by
interaction of orthophosphate fertilizers with soils. Soil Sci. Soc. Am. Proc., 34, 579-582.
Blackemore LC, Searle PL and Daly BK (1981) Methods for chemical analysis of soils., N.Z. Soil Bur.
Sci., Rep. 10A.
Bray RH and Kurtz LT (1945) Determination of total, organic and available forms of phosphorus in soils.
Soil Sci., 59, 39-45.
Chang SC and Jackson ML (1957) Fractionation of soil phosphorus. Soil Sci., 84, 133-144.
Dabin В (1967) Application des dosages automatiques & l’analyse des sols (3° partie) - 3. Analyse du
phosphore assimilable dans les sols tropicaux. Cah. Orstom Ser. Pedol., V, 278-286.'
674
Часть 3. Неорганический анализ
Dalai RC and Hallsworth EG (1976) Evaluation of the parameters of soil phosphorus availability factors
in predicting yield response and phosphorus uptake. Soil Sci. Soc. Amer. J.9 40, 541-546.
Dean LA (1947) Application of radioactive tracer technique to studies of phosphatic fertilizer utilization
by crops. I - Greenhouse experiments. Soil Sci. Soc. Am. Proc.912,107-112.
Demolon A (1932) La dynamique du sol. Dunod, 262.
Dyer В (1894) On the analytical determination of probably available “mineral” plant food in soils.
J. Chem. Soc.9 65, 115-167.
El Zahaby EM and Chien SH (1982) Effect of small amounts of pyrophosphate sorption by calcium
carbonate and calcareous soil. Soil Sci. Soc. Am. P, 46, 38-46.
Fardeau JC and Jappe J (1988) Valeurs caracteristiques des cinetiques de dilution isotopique des ions
phosphate dans les systfcmes sols-solution. In Phosphore et Potassium dans les relations sol-plante,
consequence sur la fertilisation Gachon L. ed. Lavoisier-INRA, 79-99.
Fardeau JC! (1993) Le phosphore assimilable des sols : sa representation par un module fonctionnel h
plusieurs compartiments. Agronomie, 13, 317- 331.
Fardeau JC (1997) Biodisponibilite du phosphore dans les sols, les dechets et les sediments: des approches
isotopiques. In Le phosphore dans les sols, les dechets et les eaux, AFES, Journees thematiques de Mars.
Freeman JS and Rowell DL (1981) The adsorption and precipitation of phosphate on calcite. J. Soil Sci.9
32, 75-84.
Gachon L (1988) Phosphore et Potassium dans les relations sol-plante, consequence sur la fertilisation.,
Lavoisier-INRA, 79-99.
Gautheyrou J and Gautheyrou M (1978) Mdthodologies mecanisees - Introduction a Vautomatisation
des operations analytiques dans les sols, les vegetaux et les eaux dfirrigation., IRD (ex-Orstom),
Guadeloupe, Paris, Notes laboratoire, 113 p.
Gautheyrou J and Gautheyrou M (1989) Dosage du phosphore ortho. la reaction ceruieo molybdique. In
Compte-rendu Journees laboratoires IRD9 Bondy, France, 134-154.
Gautheyrou M, Gautheyrou J and Quantin P (1990) La spectroscopie RMN haute resolution de 3,P -
Etude des formes de phosphore d’un Andosol soumis & Pecobuage. Actes Congrks Int. Sci. du Sol9
Kyoto, Japan.
Gunnarson О and Frederickson L (1951) A Method for determining plantavailable phosphorous in
soil by means of 32P. Proc. isotope technical conf9 Oxford, 1, 427-431.
Hedley MJ, Stewart WB and Chauhan BS (1982) changes in inorganic and organic soil phosphorous
fractions induced by cultivation practices and by laboratory incubations. Soil Sci. Soc. Am. J., 46,
970-976.
Henstra SD, Eijk van der, Boekestein A, Thiel F and Plas van L (1981) Compositional change in triple
superphosphate fertilizer granule. Scanning Electron Microscopy, 1,439-446.
Joret G and Hebert J (1955) Contribution & la determination du besoin des sols en acide phosphorique.
Ann. Agron., 2, 233-299.
Karlson IJ and Frankenberger Jr.WT (1987) Single column ion chromatography. Ill - Determination of
orthophosphate in soils. Soil Sci. Soc. Am. J., 51,12-14.
Larsen S (1952) The use of 32P in studies on the uptake of phosphorous by plants. Plant and Soil, 4,
1-10.
Mehlich A (1953) Determination ofP, Ca, Mg, K, Na, NH4. North Carolina Soil Test Division, Rapport
multigraphie.
Mehta NC, Legg JD, Goring CAI and Black CA(1954) Determination of organic phosphorous in soil. 1.
Extraction method. Soil Sci. Soc. Am. Proc.9 18, 443-449.
Murphy J and Riley JP (1962) A modified simple solution method for the determination of phosphates in
natural waters. Anal. Chim. Acta.9 27, 31-36.
NF X31-160 (1993) Determination du phosphore soluble dans une solution & 20 g L-l d’acide citrique
monohydrate. In Qualite des sols9 3rd ed. 1996, AFNOR, 147-154.
NF X31-161 (1993) Determination du phosphore soluble dans une solution d’oxalate d’ammonium & 0.1
mol L-l. In Qualite des sols, 3rd ed. 1996, AFNOR, 157-165.
Pansu M, Gautheyrou J and Loyer JY (2001) Soil Analysis - Sampling, Instrumentation and Quality
control, Balkema, Lisse, Abington, Exton, Tokyo, 489 p.
675
Глава 29. Фосфор
Pierzynski GM ed. (2000) Methods of Phosphorus Analysis for Soils, Sediments, Residuals, and Waters,
South. Coop. Ser. Bull. 396, Dep. of Agronomy, 2004 Throckmorton Plant Sciences Ctr., Kansas
USA, http://www.soil.ncsu.edu/seral 7/publications/seral 7-2/pm_cover.htm.
Roche P, Grtere L, Babre D, Calba H and Fallavier P (1978) La carence en phosphore des sols
intertropicaux et ses mdthodes d’apprdciation. Science du sol., 4,251-268.
Ruf F (1966) The conditions for spectrophotometric determination of orthophosphate with molybdovanado
phosphate. C.R. Geol. Com. Nat. Malgache Geol., 70,4.
Salvage T and Dixon JP (1965) The colorimetric determination of phosphorus in organic compounds on
the microgram scale. Analyst., 90,24-28.
Schwertmann U (1964) The differentiation of iron oxide in soils by a photochemical extraction
with acid ammonium oxalate. Z. Planzenenahr. Dueng Bodenkd, 105, 194-292.
Subbarao YV and Ellis R (1975) Reaction products of polyphosphates and orthophosphates with soils and
influence of uptake of phosphates by plants. Soil Sci. Soc. Am. Proc., 39,1085-1088.
Syers JK, Smillie GW and Williams JDH (1972) Calcium fluoride formation during extraction of
calcareous soils with fluoride. I - Implications to inorganic P fractionation schemes. Soil Sci. Soc.
Am. Proc., 36,20-25.
White RE and Beckett PT (1964) Studies on phosphate potentials of soils. 1 - The measurement of
phosphate potential. Plant and Soil, 20,1-16.
Williams JDH, Syers Ж and Walker TN (1967) Fractionation of soil inorganic phosphate by a modification
of Chang and Jackson’s procedure. Soil Sci. Soc. Amer. Proc., 31, 736.
Дополнительная литература
Kurtz LT (1942) Elimination of fluoride interference in molybdenum blue reaction. Ind. Eng. Chem.
Anal., 14, 855.
Palache C, Berman M and Frondel C (1951) The systems of mineralogy., Wiley Chapman, New York/
London.
Olsen SR (1952) Measurement of surface phosphate on hydroxylapatite and phosphate rock with
radiophosphorus. J. Phys. Chem., 56,630-632.
Nelson WL, Mehlich A and Winters E (1953) The development, evaluation and use of soil tests for
phosphorus availability. In Soil and fertilizer phosphorus, Pierre W.H. and Norman A.G. ed., A.S.A.
No. 4.
Watanabe FJ and Olsen SR (1965) Test of an ascorbic acid method for determining phosphorus in water
and NaHC03 extracts from soils. Soil Sci. Soc. Am. Proc., 29, 677.
Lehr JR, Brown EH, Frazier AW, Smith JP and Thrasher RD (1967) Crystallographic properties of
fertilizer compounds. Chem. Eng. Bull., (Alabama, Etats-Unis) No. 6.
Fox RL and Kamprath EJ (1970) Phosphate sorption isotherms for evaluating the phosphate requirements
of soils. Soil Sci. Soc. Am. Proc., 34, 902- 907.
Povarennykh AS (1972) Crystal chemical classification of minerals., Plennum New York, Vols. I and II.
Franzen DW and Peck TR (1995) Spatial variability of plant analysis phosphorus levels. Commun. in Soil
Sci. Plant Anal., 26, 2929-2940.
Frossard E, Brossard M, Hedley MJ and Metherel A (1995) Reactions controling the cycling of P in soils.
In Phosphorus in the global environment., Wiley New York 107-137.
Fardeau JC, Guiraud DG and Morel C (1996) The role of isotopic techniques on the evaluation of
effectiveness of P fertilizers. Fert. Res., 45,101-109.
Kuo S (1996) Phosphorus. In Methods of soil analysis, part 3, chemical methods, Bigham JM and
Bartels JM ed., SSSA-ASA, Madison, WI Etats-Unis, 869-919.
Cade-Menun BJ and Preston CM (1996) A comparaison of soil extraction procedures for 31P NMR
spectroscopy. Soil Science, 161, 770-785.
Condron HJ and Frossard LME (1997) Isotopes techniques to study phosphorus cycling in agricultural
and forest soils : a review. Fert. Res., 24,1-12.
Robinson JS and Johnston CT (1998) Combined chemical and 31P-NMR spectroscopic analysis of
phosphorus in wetland organic soils. Soil Sci., 163, 705-713.
676
Часть 3. Неорганический анализ
Isik Y and Ekiz Н (2000) The phosphorus demand of durum wheat grown in Konya and the calibration of
Olsen phosphorus analysis. Konya yoresinde yetistirilen makamalik bugdayin fosforlu gubre istegi
ve Olsen fosfor analiz metodunun kalibrasyonu. Ministry of Agriculture and Rural Affairs, Bahri
Dagdas International Winter Cereals Research Center; Konya; Turkey 230-239.
Steegen A, Govers G, Beuselinck L, Oost К van, Quine ТА, Rombaut A, Stone M (2000) The use of
phosphorus as a tracer in erosion/sedimentation studies. In The role of erosion and sediment transport
in nutrient and contaminant transfer. Proceedings of a symposium held at Waterloo, Ontario, Canada
in July 2000, IAHS Wallingford, UK.
Elrashidi MA (2001) Testing methods for phosporus and organic matter, http://soils.usda.gov/technical/
methods.
Boruvka L, Rechcigl JE (2003) Phosphorus retention by the AP horiszon of a spodosol as inflenced by
calcium amendments. Soil Sci.9 168, 699-706.
Escudey M, Galindo G and Briceno M (2004) Influence of particle size on 31PNMR analysis of extracts
from volcanic ash-derived soils in Chile. J. Chil. Chem. Soc49, 5-9.
Глава 30. Сера
30.1. Введение
30.1.1. Соединения серы
Источники появления серы в почве различны: вулканические выбросы соединений
серы, разрушение скальных пород, трансформации метаморфических и осадочных
пород, влияние поверхностных или подземных вод или морской воды, биологические
источники — различные животные и растения. Сельскохозяйственная, промышленная
и бытовая деятельность, в особенности внесение в почву добавок, удобрений, компоста
и пестицидов, также приводит к обогащению почвы соединениями серы. Использова¬
ние ископаемого топлива загрязняет атмосферу, и впоследствии загрязнители попадают
в почву в составе дождевой воды. Содержание серы в большинстве видов почв, как пра¬
вило, невелико (несколько десятых мг/кг), за исключением кислых сульфатных, заболо¬
ченных гистосолей или гипсоносных почв.
Сера существует как в органической, так и в минеральной форме. Этот элемент ха¬
рактеризует разнообразие простых и комплексных, аморфных или кристаллических
молекулярных структур соединений серы в почвах. Существование и стабильность раз¬
личных серосодержащих производных в значительной мере зависит от физико-химиче¬
ских свойств среды (в частности, окислительно-восстановительного потенциала и pH
почвы). Различные формы серы можно определить при анализе микробиологических
реакций с участием сульфатовосстанавливающих и сульфоокисляющих бактерий. За
исключением некоторых относительно стабильных, малорастворимых сульфатиро-
ванных форм, таких как гипс или ярозит, большинство соединений серы подвержены
трансформациям, являющимся частью круговорота серы в почвах и живых организмах.
С агрономической точки зрения сера является основным элементом, необходимым
для развития растений. Потеря урожая могут произойти как из-за дефицита серы, так
и из-за токсичности ее соединений в почвенной среде. Пороговые уровни зависят от
чувствительности различных видов культурных растений.
Специальные анализы требуются для двух типов почв, особенно богатых серой: (1)
для кислых сульфатных, сформировавшихся из морских или речных наносов (напри¬
мер, мангровые или польдерные), и (2) гипсовых. Для анализа каждого из этих двух
типов почвы разработаны специальные аналитические методики (некоторые представ¬
лены в этой главе). Большую часть методик можно использовать для анализа и других
почв, содержащих соединения серы в меньшем количестве.
30.1.2. Минералогические исследования
В природной среде обнаружено двести видов минеральных сульфатов и 228 сульфидов.
Количество природных и промышленных органических соединений, в состав которых
входит сера, не поддается исчислению. Самые распространенные неорганические фор¬
мы, встречающиеся в почвах, представлены в табл. 30.1. В почвах пустынных и полупу¬
стынных климатических зон участки поверхностной кристаллизации часто сохраняются
в естественном состоянии. Окисленные формы стабильны, и их хранение не связано
с особыми проблемами.
Таблица 30.1. Основные серосодержащие минералы, присутствующие в осадочных отложениях (эвапоритах*), выветрившихся
породах, корках и почвах
Степень окисления
Связь
Минерал
Химическая формула
Межплоскостное
Растворимость, г/л воды
расстояние,
A
о°с
30 °с
100 °с
Гипс
CaS04 • 2Н20
7,56
3,05
4,27
2,41
2,6
2,22
Са2+
Полугидрат
CaS04 0,5Н2О
3,00
6,01
2,80
3,0
Бассанит
CaS04 0,5Н2О
3,00
6,01
2,80
^Ангидрит
CaS04
3,49
2,85
2,32
2,09
1,62
Сульфаты
Эпсомит
MgS04*7H20
4,21
5,35
2,68
710,0
910,0 (40 °С)
Mg2*
Гексагидр ит
MgS04 • бн2о
4,43
4,04
2,94
*Кизерит
MgS04 н2о
3,41
4,84
3,33
684,0
Тенардит
Na2S04
2,78
4,66
3,18
47,6
427,0
Na+
Мирабилит
Na2S04 • 10H2O
5,48
3,28
3,26
110,0
927,0
Бледит
Na2Mg(S04)2 4H20
3,25
4,56
3,29
*Ловеит
Na^SO^ • 5/2H20
3,17
4,29
4,04
*Вантгоффит
Na2Mg(S04)2 • 5/2H20
2,91
3,44
3,43
Натроярозит
NaFe3(0H)6(S04)2
5,04
3,05
3,12
*Лангбейнит
K.Mg.CSO,),
3,14
2,65
4,05
Окисленные
формы
*Пикромерит
K2Mg(S04)2 6H20
3,70
3,04
2,38
Смешанные
*Полигалит
K2MgCa2(S04)4 • 2H20
2,90
3,18
2,85
Ярозит
KFe3(0H)6(S04)2
3,11
3,08
5,09
Кокимбит
Fe2(S04)3,5 или 9H20
8,26
2,76
5,45
Алунит
KA1j(S04)2(0H)6
2,99
2,89
2,29
Натро-алунит
NaAl3(S04)2(0H)6
2,96
4,90
2,97
Базалуминит
Al4(SO4)(OH)10 • 5H20
9,40
4,68
3,68
Джурбанит
Al(SO„)(OH) • 5H,0
—
—
—
678 Часть 3. Неорганический анализ
S элементарная
Сера S°
s„
3,29
6,65
3,74
Н
Сероводород
H2S
—
—
—
Fe
Макинавит
FeS
5,03
2,97
2,31
0,0062
Сульфиды
Fe
Пирит
FeS2
1,63
2,70
2,42
0,0049
(восстановленные
Fe
Марказит
FeS2
2,71
1,76
3,44
0,0049
формы)
Fe
Грейгит
Fe3S4(Fe2+Fe3+S4)
2,98
2,47
1,74
Fe
Cu
Халькопирит
CuFeS2
3,03
1,85
1,59
Глава 30. Сера 679
680
Часть 3. Неорганический анализ
С другой стороны, пробы, содержащие восстановленные формы (например, сульфи¬
ды железа), следует хранить в закрытых светонепроницаемых флаконах и измельчать
непосредственно перед анализом.
Минералогические наблюдения можно проводить с помощью оптического микро¬
скопа или электронного сканирующего микроскопа, сопряженного с ЭД-РС-микроана-
лизатором (см. гл. 8). В последнем случае следует учитывать возможные трансформа¬
ции, связанные с условиями аналитического инструментального измерения (например,
глубоким вакуумом или интенсивностью электронного луча).
Распространенные минеральные формы и классификация почв
*Эвапориты. Геологические отложения - осадки, накапливающиеся в водных бассейнах
при дренировании и испарении или гидротермальном воздействии. Наиболее раство¬
римые соли накапливаются в выветренных формах, образующихся под воздействием
восходящих капиллярных потоков и поверхностного испарения. Хорошо растворимые
сульфаты магния или натрия обнаруживают на поверхности почвы только в пустынных
климатических зонах.
Тенардит (Na2S04) более стабилен, чем мирабилит (Na2S04 4 ЮН20) и другие суль¬
фаты, такие как бледит (Na2Mg(S04)2 • 4Н20), гексагидрит (MgS04 * 6Н20), эпсомит
(MgS04 • 7Н20). Следует помнить о том, что температура влияет на растворимость. Гипс
(CaS04 • 2Н20) является наименее растворимым из почвенных сульфатов и наиболее
распространенным в окружающей среде. Гипсовые отложения зачастую возникают вме¬
сте с отложениями карбоната кальция, если растворимость сульфата кальция превышена.
Первые полевые испытания показали, что анализы надежны и безопасны. Почвы,
свойства которых в основном определяются присутствием соединений серы, внесены
в международные системы классификации почвы.
Во французской системе классификации (CPCS1967) соединения серы присутствуют
в подклассе (гипсовые почвы) и группируются в соответствии с наличием индивиду¬
альных форм сульфата кальция; в классе (солонцовые почвы) и подклассе (засоленные
почвы) в почвах, содержащих сульфидные вещества в восстановленном состоянии в ги-
дроморфной-галоморфной среде, или в окисленном состоянии в аэробной дренирован¬
ной среде.
По классификации ГАО (1968), большие группы (иермосоли и ксеросоли) включа¬
ют гипсовые подгруппы, а глеесоли и флувисоли - тионовые подгруппы для мангровых
почв.
Согласно Таксономии почв (Soil Taxonomy) США (USDA1975), соединения серы пред¬
ставлены в больших группах (гипсиортиды, натраргиды, натриборолли) и в группах почв,
содержащих неорганические и органические восстановленные компоненты (sulfaquents,
sulfihemists), а также в группах кислых сульфатных почв (sulfaquepts, sulfohemists); и, нако¬
нец, в группе горизонтов с преобладанием процессов, протекающих в течение длитель¬
ных временных периодов: гипсовые, петрогипсовые, сернокислые почвы и даже засоленные
или натриевые почвы в присутствии различных растворимых солей.
На практике можно идентифицировать лишь ограниченное число достаточно ста¬
бильных минералов из-за недостаточной чувствительности методов и трудностей, свя¬
занных с разделением различных компонентов без структурных изменений (в особен¬
ности в случае соединений с низким содержанием серы в солевых матрицах с очень
высоким содержанием серы), а также с климатическими ограничениями (например,
температурой или режимом выпадения осадков) или условиями дренированности
почвы.
Глава 30. Сера
681
30.2. Общее содержание серы и соединений серы
30.2.1. Характеристики почв, сформировавшихся на морских и речных
наносах (Fluviomarine Soils)
Почвы такого типа особенно богаты серой, и они формируются в дельтах, эстуариях или
польдерах в различных климатических условиях. В экваториальном или тропическом
климате, в частности, солончаковые почвы и осадочные отложения заселены мангровой
растительностью с очень плотной корневой системой, в изобилии продуцирующей ор¬
ганические вещества.
В условиях затопления в восстановительной среде морские сульфаты восстанавлива¬
ются до сульфидов под действием микроорганизмов, из которых при реакции с желе¬
зосодержащими материалами образуются пириты. При сохранении анаэробной среды
аккумуляция пиритов в почвенных материалах остается стабильной. Падение уровня
подземных вод из-за засухи или искусственного осушения нарушает равновесие, вызы¬
вая окисление пирита и высвобождение серной кислоты и оксогидрооксидов железа.
Почвенная среда закисляется; в условия засушливого тропического климата засоление
может быть ускорено за счет концентрирования солей. При интенсивном закислении
может высвобождаться алюминий в результате воздействия кислот на кристаллические
решетки глин (ацидолиз). Результатом таких процессов является образование смеси
сульфата алюминия, железа и магния, входящей в состав ярозита, натроярозита и раз¬
личных форм квасцов (Vieillefon, 1974; Marius, 1980; Le Brusq и др., 1987; Montoroi, 1994;
Giinin и др., 2001; Montoroi и др., 2002).
Быстрые изменения, влияющие на состояние этих почвенных сред и их компонентов,
требуют особого внимания при отборе проб и их хранении, так же как и при экстракции
и измерении содержания различных форм серы, присутствующей в почве в данный мо¬
мент времени (Pansu и др., 2001). Элемент S представляет собой смесь различных изо¬
топов (32S и 34S). Соединения S существуют в газообразных формах (например, сульфид
водорода), растворимых формах (сульфиды, сульфаты), малорастворимых или нераст¬
воримых формах (полисульфиды, пириты, элементарная сера, гипс, смешанные суль¬
фаты) и в виде органических соединений, в различной степени способных к окислению.
Чтобы правильно понимать процессы почвообразования, при анализе следует четко
различать формы серы в свежеотобранной пробе, репрезентативные для состояния in
situ (сульфидные формы), и формы S, которые могут образоваться в результате окисле¬
ния (сульфатные формы) в высушенной на воздухе почве (Marius и др., 1976).
30.2.2. Отбор и подготовка проб почвы
В почвах, содержащих окисленные продукты (например, гипс, см. раздел 30.3), отбор проб
производится стандартным способом, и пробы почвы готовят к анализу обычным путем.
В восстановительных средах при отборе проб почвы часто возникают специфические
проблемы. Необходимо не допустить окисления соединений серы. Сероводород выс¬
вобождается очень быстро. Следует регулировать превращения сульфидов (даже в ме-
тастабильных формах): защищать пробы от контакта с воздухом и хранить при низкой
температуре, защищая от света.
Отбор проб мангровых почв и почв с заиленных участков проводится с помощью по¬
лого почвенного бура (диаметром 60-100 мм) с острым краем для разрезания волокнис¬
тых материалов и корней (Pansu и др., 2001). Набор удлиняющих стержней (длиной 1 м)
позволяет отбирать пробы с разных глубин. Как правило, для агрономических иссле¬
682
Часть 3. Неорганический анализ
дований достаточная глубина отбора пробы составляет 1 м. Почвенный бур вводят вер¬
тикально в слой подвижных отложений, затем слегка поворачивают, чтобы освободить
основание колоны почвы и вынимают, не допуская разрушения пробы. Затем почвен¬
ный бур кладут на землю и отделяют пробу с помощью ножа. Следует отметить каждый
уровень почвы, отобрать необходимые образцы и провести прямые измерения на коло¬
не почвы (pH и некоторые полевые анализы).
При необходимости отбора проб с больших глубин применяют пробоотборник со ста¬
ционарным плунжером, заключенным в металлическую или пластиковую трубу (длиной
от 1-3 м), позволяющим извлекать пробы без разрушения. Конец трубы, содержащий
пробу, следует немедленно надежно герметизировать пластиком. По возможности, тру¬
бы следует хранить в холодном помещении; в лаборатории их раскрывают по длине для
извлечения пробы. Пробы, отобранные на различных глубинах, обычно разделяют на
четыре фракции;
• Сектор неразрушенной пробы, используемой для изучения морфологии почвы,
должен храниться в холодильнике в герметичном контейнере.
• Еще одну фракцию следует высушить на воздухе и просеять до размера частиц
2 мм; часть этой фракции следует измельчить на сите AFNOR NF2\ (100 мкм) и ис¬
пользовать для общего анализа, поскольку восстановленные формы, как правило,
окисляются.
• Третью фракцию следует хранить во влажном состоянии для последующего ана¬
лиза восстановленных форм; после быстрой гомогенизации на 4-миллиметровом
сите для удаления корней и мелких камней, образцы можно непродолжительное
время хранить в холодильнике в герметичных флаконах, защищенных от света,
а для продолжительного хранения используют морозильную камеру. Следует изме¬
рить содержание влаги.
• Четвертую фракцию — высушить методом лиофильной сушки, чтобы избежать за¬
твердевания образца и восстановленных метастабильных форм.
Пробы надо хранить в холодильнике в защищенных от света флаконах. Содержание
влаги измеряют перед каждым отделением аналитической пробы.
В полевых условиях следует отбирать пробы воды из пробоотборного канала для не¬
медленного анализа таких параметров, как pH, электропроводность, содержание H2S;
одну пробу воды помещают в герметичный флакон и доставляют в лабораторию. Уста¬
новка пьезометров в наиболее репрезентативных пробоотборных каналах позволяет
проводить мониторинг состояния подземных вод (например, уровень pH, концентра¬
цию солей).
30.2.3. Анализ растворимых форм серы
Исследование растворимых форм серы, в частности сульфидов, in situ можно провести
в пробе воды, выделенной из заболоченной почвы (например, полевая проба Hack Н-5-6
п° 223800, диапазоны: 0-0,6 или 0-5 мг H2S).
Присутствие сероводорода ощущается по запаху даже при очень низких концентрациях
(0,025 мг/кг), а его растворимость в воде равна 0,102 моль/л.
Присутствие растворимых сульфидов обнаруживается при обработке кислотой
с последующей пробой ацетатом свинца:
• Обрабатывают несколько граммов почвы 6 М хлористоводородной кислотой в ана¬
литической пробирке:
SJ- + 2НС1 -» H2St + 2С1-
Глава 30. Сера
683
• Накрывают отверстие пробирки индикаторной бумагой с ацетатом свинца на 5 с;
появление темного красно-коричневого окрашивания указывает на высокий уро¬
вень содержания сероводорода в соответствии с реакцией:
H2S + РЬС1г -¥ PbS'l' + 2 НС1
30.2.4. Анализ общей серы
Многочисленность органических и неорганических соединений S может быть обуслов¬
лена (1) множеством степеней окисления S (от -2 до +6), (2) способностью S к образо¬
ванию различных типов связи (ковалентная, координационная, ионная) и (3) различной
геометрией координационных полиэдров. Следовательно, для общего анализа необхо¬
димо перевести различные формы S в стабильные:
• окислением до состояния S6+ (сульфат), являющегося очень стабильным;
• восстановлением до S2- (сульфид); поскольку термодинамически стабильные суль¬
фиды очень легко окисляются, анализ следует проводить, избегая контакта с воз¬
духом; эта методика применяется для анализа, в частности, после окисления до
сульфата.
Можно воспользоваться методами сухого или мокрого окисления. Методы сухого
окисления включают:
• щелочное сплавление с добавкой окислителя или без: Na2C03, Na2C03 + NaN03,
Na2C03 + Na202;
• прокаливание (1) в присутствии NaHC03 + Ag20, (2) в потоке кислорода, (3) в за¬
мкнутом пространстве в присутствии карбоната натрия и (4) методом Шёнигера
(или аналогичным) - в потоке кислорода в замкнутом пространстве.
Мокрое окисление можно проводить в:
• кислой среде:
Н3Р04 + К2Ст207
HC104+HN03
КМп04 + Н3Р05 (пероксофосфорная кислота);
• щелочной среде в присутствии гипобромита натрия (снижается риск выделения
H2S, S02, S03).
Выбор метода анализа серы определяется ее количеством, присутствующими форма¬
ми S и составом почвенной матрицы. Различные методы включают гравиметрию, тур-
бидиметрию, колориметрию, ионную хроматографию (ИХ), непрямую спектрографию.
30.2.5. Солюбилизация общей серы методом окислительного щелочного
плавления
Основные положения
Метод основан на переведении соединений серы в сульфаты путем сплавления при
700 °С со смесью карбоната натрия и нитрата калия (окислительный реагент нитрат ка¬
лия можно заменить нитратом натрия или пероксидом натрия Na202).
Оборудование
• Высокие платиновые или никелевые тигли с крышками (40 мл).
• Муфельная печь на 1000 °С.
• Водяная баня.
684
Часть 3. Неорганический анализ
• Лабораторная посуда из стекла Пирекс.
• Устройства для центрифугирования и фильтрования.
• Лабораторные весы (±0,1 мг).
Реагенты
• Карбонат натрия (Na2C03).
• Нитрат калия (KN03).
• Смесь для сплавления: смешивают 100 г Na2C03 и 10 г KN03.
• Концентрированная хлористоводородная кислота НС1 (37%).
• « 2 Мраствор хлористоводородной кислоты (НС1): в аналитической пробирке (из
стекла Ругех) смешивают 800 мл деионизированной воды и 160 мл концентриро¬
ванной хлористоводородной кислоты; охлаждают смесь и доводят объем до 1000 мл
деионизированной водой, гомогенизируют.
Методика
Сушат пробу почвы на воздухе или (предпочтительно) методом лиофильной сушки и из¬
мельчают до размера частиц 0,1мм.
• На прецизионных весах берут навеску от 200 до 500 мг почвы, помещают в высо¬
кий платиновый тигель объемом 40 мл.
• Навеску 1 г смеси для плавления смешивают с пробой; добавляют 200 мг смеси для
плавления на поверхность.
• Закрытый тигель ставят в открытую муфельную печь и постепенно нагревают до
700 °С; поддерживают температуру 700 °С в течение 40 мин.
• Круговыми движениями распределяют расплавленную массу по стенкам тигля;
выдерживают до охлаждения.
• Осторожно добавляют 10 мл 2 М раствора хлористоводородной кислоты; при необ¬
ходимости разрушают агрегированную массу стеклянной (Пирекс) палочкой.
Когда выделение пузырьков газа прекращается, помещают тигель в кипящую водя¬
ную баню для завершения реакции. Охлаждают и переносят смесь в пробирку для цен¬
трифугирования. Центрифугируют и отделяют надосадочный раствор в мерную колбу
вместимостью 100 мл, ополаскивая осадок после центрифугирования.
Доводят объем до 100 мл деионизированной водой и гомогенизируют смесь.
Кислый раствор, содержащий соединения S в окисленной сульфатной форме, готов
для анализа общего содержания S.
Примечания
Основным компонентом остатка является осажденный белый диоксид кремния.
Если масса окрашена в зеленоватый цвет и становится розовой при контакте с соля¬
нокислым раствором, можно сделать предположение о присутствии марганца (платино¬
вые тигли слегка взаимодействуют с соляной кислотой).
Если масса не прозрачна и интенсивно окрашена, это означает, что обработка была
неполной.
30.2.6. Полная солюбилизация гипобромитом натрия в щелочной среде
Основные положения
Органические и неорганические формы серы окисляются до сульфата гипобромитом
натрия (Tabatabai и Вгетпег, 1970а).
685
Глава 30. Сера
Оборудование
• Стеклянная лабораторная посуда и специальное оборудование {Johnson и Nishita,
1952).
• Магнитная мешалка с тефлоновыми перемешивающими якорями.
• Микропипетка из тефлона емкостью 5 мл.
• Песчаная баня.
• Вытяжной шкаф.
Реагенты
• Гкдроокись натрия (NaOH) в гранулированной форме.
• » 2 М раствор гидроксида натрия (NaOH): растворяют 80 г гидроксида натрия
в 1000 мл деионизированной воды, при хранении необходимо защитить раствор от
контакта с диоксидом углерода, содержащимся в воздухе.
• Бром чистый для анализа (Вг2).
• Раствор гипобромита натрия {NaBrO), приготовленный в вытяжном шкафу не¬
посредственно перед использованием: вводят 100 мл 2 М раствора соды в колбу
Эрленмейера объемом 150 мл; перемешивают с помощью магнитной мешалки
с тефлоновым якорем и добавляют микропипеткой по каплям 3 мл брома; колбу
накрывают, используют раствор как можно быстрее.
• Муравьиная кислота (НСООН).
Методика
Используют воздушно-сухую почву (или лучше высушенную методом лиофильной суш¬
ки), измельченную до размера частиц 0,1 мм.
• Навеску 100-200 мг почвы, содержащей 20-50 мкг S, помещают в колбу для кипя¬
чения вместимостью 50 мл (2? на рис. 30.1) в установке Johnson и Nishita; аналити¬
ческие процедуры проводят в вытяжном шкафу.
• Добавляют 3 мл раствора гипобромита; взбалтывают вращательными движени¬
ями содержимое колбы в течение нескольких секунд и оставляют в контакте на
5 мин.
• Вновь взбалтывают для переведения осадка во взвешенное состояние.
• Нагревают в песчаной бане до температуры 255 ± 5°С, не допуская утечки пены;
выпаривают досуха; поддерживают температуру на протяжении 30 мин; охлаж¬
дают.
• Добавляют 1 мл воды и нагревают короткое время для переведения остатка во взве¬
шенное состояние; охлаждают.
Измерения в растворе проводятся как можно быстрее по методике {Johnson и Nishita,
1952) с использованием метиленового синего (см. раздел 30.2.7).
30.2.7. Анализ S колориметрическим методом с использованием
метиленового синего
Основные положения
В соответствии с методикой {Johnson и Nishita, 1952), сульфаты, образовавшиеся в про¬
цессе окислительной минерализации, восстанавливают до сероводорода смешанным
686 Часть 3. Неорганический анализ
раствором йодистоводородной (HI), фосфиновой (гипофосфорная кислота, HjPO^1 и
муравьиной кислоты (Н-СООН)2.
Выделившийся сероводород поглощается цинк-ацетатным и натрий-ацетатным бу¬
фером с образованием сульфидов. Обработка смесью л-аминодиметиланилин сульфата
и двойного сульфата железа и аммония приводит к образованию метиленового синего
(в реакции участвуют 2 моля и-аминодиметиланилина + 1 моль сульфида).
Интенсивность цвета измеряют при 66—670 нм. Цвет остается стабильным в течение
24 ч, если растворы защищены от контакта с воздухом и хранятся в темном месте.
Оборудование
Компоненты установки (Johnson и Nishita, 1952) включают:
• Баллон с азотом (99,90% N2) с регулятором давления (редуктором).
• Промывная бутыль емкостью 125 мл (А на рис. ЗОЛ).
• Круглодонная стеклянная (Ругех) колба для кипячения вместимостью 50 мл (для
разложения и дистилляции, 2? на рис. 30.1).
• Барботажное устройство, ловушка, предохраняющая от переброса жидкости, и хо¬
лодильник (Сна рис. 30.1).
• Барботажное устройство с запорным краном (D на рис. 30.1) и выпускная трубка
сверху со съемной трубкой для отведения газа (Е на рис. 30.1).
• Мерная колба вместимостью 100 мл с притертой стеклянной пробкой (/на
рис. 30.1).
• Электрический колбонагреватель.
Все стеклянные притертые пробки должны быть стандартизированы; их следует
использовать вместе с тефлоновыми уплотнителями, предназначенными для смачивания
деионизированной водой без использования силиконовых смазок.
• Спектроколориметр.
• Установка для приготовления восстанавливающего реагента (рис. 30.2).
Реагенты
• Деионизированная вода (не содержащая S).
• Перманганат калия (КМп04).
• Хлорид ртути (HgCl2).
• Раствор для очистки азота. Смешивают 100 мл 2%-ного раствора перманганата ка¬
лия в деионизированной воде с 10 г хлорида ртути.
• Йодистоводородная кислота (HI), d= 1,70.
• Гйпофосфорная (или фосфиновая) кислота (50%-ная Н3Р02).
1 Гйпофосфорная кислота — кислота, восстанавливающая оксокислотная группа которой
способна к ионизации и является донором протонов.
2 Муравьиная кислота подкисляет среду и восстанавливает остатки брома по реакции:
НСООН + Вг2 C02t + 2НВг.
Соли муравьиной кислоты играют аналогичную роль, например,
HCOONa + Вг2 C02t + НВг + NaBr.
При проведении общего анализа, в ходе которого гидролиз разрушенных органических сое¬
динений маловероятен, муравьиная кислота предотвращает помехи, вызываемые нитратами.
Глава 30, Сера
687
• 90%-ная муравьиная кислота (НСООН).
• Восстановительная смесь: работая под вытяжкой, смешивают 400 мл йодистоводо¬
родной кислоты, 100 мл гипофосфорной кислоты (фосфиновой кислоты) и 200 мл
муравьиной кислоты в трехгорлой круглодонной колбе объемом 1 л (рис. 30.2); по¬
мещают в термостатируемый колбонагреватель; соединяют колбу для кипячения
с барботажной трубкой, термометром и насадкой, предохраняющей от переброса
перегоняемой жидкости; соединяют предохранительную насадку с колбой Эрлен-
мейера, содержащей приблизительно 150 мл холодной воды; открывают редуктор
баллона с азотом и регулируют скорость барботирования (два пузырька в секунду)
в колбе для очистки газа (между баллоном с азотом и колбой для кипячения); че¬
рез 2-3 мин начинают медленно нагревать до 116 ± 1 °С и поддерживают данную
температуру в течение 10 мин, не допуская повышения температуры свыше 117 °С
(выделение токсичного фосфина, Н3Р); охлаждают при непрерывном барботиро-
вании азота; переносят в бутыль вместимостью 1 л; хранят в темноте.
• 1,2,3 тригидроксибензол (пирогаллол).
688
Часть 3. Неорганический анализ
Азот
Рис. 30.2. Установка для приготовления восстановительного реагента
• Дигидрофосфат натрия (NaH2P04 • Н20).
• Раствор для поглощения газообразных продуктов окисления. Растворяют 10 г дигид¬
рофосфата натрия и 10 г пирогаллола в 10 мл деионизированной воды; в процессе
растворения смесь продувают азотом; свежий раствор следует готовить ежедневно.
• Дигидрат ацетата цинка (CH3COO)2Zn • 2Н20.
• Тригидрат ацетата натрия CH3COONa • ЗН20.
• Раствор для поглощения сероводорода. Растворяют 50 г ацетата цинка и 12,5 г ацетата
натрия в 800 мл деионизированной воды; доводят объем до 1000 мл; смесь должна
быть прозрачной; в противном случае ее следует профильтровать через аналити¬
ческий фильтр (синяя лента).
Глава 30. Сера
689
• Серная кислота H2S04, d = 1,80.
• Раствор п-аминодиметиланилин сульфата: растворяют 2 г я-аминодиметиланилин
сульфата в 1,5 л деионизированной воды в двухлитровой колбе для кипячения из
стекла Ругех; помещают колбу в холодную воду со льдом и медленно добавляют
400 мл концентрированной серной кислоты, перемешивая раствор стеклянной
(Ругех) палочкой; охлаждают и доводят объем до 2000 мл.
• Двойной сульфат аммония и железа, Fe2(S04)3(NH4)2S04 • 24Н20: растворяют 25 г
двойного сульфата в смеси 195 мл деионизированной воды и 5 мл серной кислоты;
выдерживают до охлаждения; перемешивают с помощью магнитной мешалки до
полного растворения.
• Сульфат калия (KjSO^.
• Базовый стандартный раствор: навеску 5,434 г сульфата калия растворяют в 800 мл
деионизированной, свободной от S воды, доводят объем до 1 л. 1 мл раствора со¬
держит 1 мг S в форме SO*-.
• Калибровочные растворы: вносят 1, 2, 3, 4, 5 мл стандартного базового раствора
в мерные колбы объемом 100 мл и доводят объем до 100 мл дистиллированной во¬
дой, не содержащей S. Один миллилитр раствора содержит 10, 20, 30, 40, 50 мкг S
в форме SO*-.
Методика
• Запуск установки Johnson и Nishita. Все стеклянные шлифованные соединения сле¬
дует устанавливать с у!тлотнительной тефлоновой манжетой, смоченной деиони¬
зированной водой для лучшей герметизации. Пипеткой отмеряют 10 мл раствора
пирогаллола — фосфата натрия и вносят в газоуловитель (D на рис. 30.1). Вводят
10 мл раствора ацетата цинка и ацетата натрия в мерную колбу объемом 100 мл
(^на рис. ЗОЛ), добавляют 50 мл деионизированной воды и гомогенизируют смесь.
Погружают трубку (Е на рис. 30.1) в мерную колбу до соприкосновения с дном
и соединяют с выходной трубкой газоуловителя (D на рис. 30.1) с помощью гиб¬
кого шланга производства Tygon. Запускают циркуляцию воды в холодильнике
(Снарис. 30.1).
Анализ
• После подготовки пробы к анализу методом окислительной плавки (методика опи¬
сана в разделе 30.2.61) смешивают пробу с 1 мл муравьиной кислоты в колбе для ки¬
пячения В (рис. 30.1). ГЪмогенизируют и выдерживают в контакте в течение 30 мин,
два или три раза перемешивая вращательными движениями. Быстро вносят пипет¬
кой 4 мл раствора восстановительной смеси (HI + Н3Р02 + НСООН) и соединяют
колбу для кипячения Вс установкой, используя тефлоновую манжету, подходящую
для данного стеклянного шлифа. Регулируют расход азота в промывной бутыли А
до двух пузырьков в секунду, т. е. около 200 мл в минуту. Азот вытесняет воздух
и после этого используется в качестве нейтрального носителя сероводорода. Бар-
ботируют в течение 5 мин. Осторожно кипятят в течение 1 ч. Отсоединяют гиб¬
1 При применении методики окислительной плавки (см. раздел 30.2.5) отбирают аликвоту
солянокислого раствора объемом от 1 до 10 мл, содержащую около 50 мкг S в форме SO*-,
и упаривают ее при осторожном кипячении приблизительно до 2 мл. В этом случае обработка
муравьиной кислотой необязательна.
690
Часть 3. Неорганический анализ
кое соединение (Tygon) от трубки (Е на рис. ЗОЛ) и отставляют мерную колбу (F).
Быстро ополаскивают струей из промывалки трубку (£) (если на трубке имеются
остатки сульфида цинка, трубку следует использовать для перемешивания при по¬
следующей обработке п-аминодиметиланилином и ополоснуть ее позже).
• С помощью прецизионной тефлоновой пипетки вносят в мерную колбу (/) 10 мл
раствора я-аминодиметиланилина. Колбу закрывают и встряхивают для гомогени¬
зации смеси. Добавляют 2 мл раствора двойного сульфата аммония и железа. За¬
крывают колбу и гомогенизируют смесь. Доводят объем до 100 мл деионизирован¬
ной водой и гомогенизируют. Выдерживают до проявления цвета в течение 30 мин
(цвет раствора остается стабильным приблизительно на протяжении 24 ч, если не
контактирует с воздухом и хранится в защищенном от света месте).
• Обрабатывают аликвоты объемом 1 мл, содержащие от 0 до 50 мкг S/мл в форме
(калибровочный диапазон), и холостую пробу с 4 мл восстановительной смеси
в тех же условиях, что и пробы. Строят калибровочные кривые зависимости вели¬
чины поглощения от концентрации S в форме S02-.
Примечания
Максимальное поглощение наблюдается при 667,8 нм. В диапазоне концентраций
1-50 мкг/мл величина поглощения изменяется в соответствии с законом Бэра-Ламбер-
та; калибровочная кривая имеет линейный характер. При более высоких концентрациях
можно проводить измерения после разбавления пробы тем же раствором (я-аминоди-
метиланилина и двойного сульфата железа и аммония). Однако, если я-аминодимети-
ланилин присутствует в количестве, недостаточном для того, чтобы прореагировать со
всеми сульфидами, добавление иона железа (III) позволяет окислить избыток сульфи¬
дов, при этом в результаты измерений вносится погрешность, связанная с разбавлением
раствора. Второй анализ с использованием пробы почвы меньшего размера позволяет
получить более надежные результаты.
Эту методику можно также применять для анализа растений и воды.
Некоторые авторы предпочитают проводить итоговые измерения после окисления
гипобромитом турбидиметрическим методом с использованием коллоида сульфата вис¬
мута (Kornienko и Lowe, 1972; Kowalenko, 1985; Виигтап идр., 1996).
30.2.8. Анализ сульфатов колориметрическим методом с использованием
метилтимолового синего
Основные положения
Эти методики позволяют проводить количественный анализ форм S, окисленных до
сульфата. Метилтимоловый синий (МТС) содержит две N.Nr группы (дикарбоксиме-
тил) аминометил, связанные в положениях 3 и Зг (рис. 30.3), поэтому он может связы¬
вать по одному атому металла у каждой из его вершинных групп.
Реагент представляет собой эквимолекулярную смесь МТС и хлорида бария. В от¬
сутствие сульфата все ионы бария изолированы, раствор имеет темно-синий цвет, по¬
глощающий при 610 нм в сильнощелочной среде:
pH 12
Ва2+ + МТС6" < » ...ВаМТС4"
В присутствии ионов SO2- образуется сульфат бария и высвобождаются МТС6- ионы,
поглощающие при 460 нм (серый цвет).
Глава 30. Сера
691
ВаМТС4... + S02-*-
рН 12
- BaS04i + МТС6-
Постепенно проявляются оттенки синего, интенсивность которых указывает на при¬
сутствие несвязанного метилтимолового (серого цвета), количество которого зависит
от концентрации сульфата. Наличие остаточного окрашивания, поглощающего при
610 нм, существенно не влияет на результаты измерения поглощения при 460 нм.
Помехами являются ионы кальция, которые следует удалять непосредственно в ма-
нифольде при помощи катионообменной смолы или заранее — путем фильтрации ка¬
ждой пробы через микрофильтр, содержащий 1,0 г катионообменной смолы (например,
Dowex 5Q1V-X&).
R2 4*2
соонсн2
n-h2c
CHjN
СООНСНг
РН2-СООН
СН2-СООН
Рис. 30.3. Метилтимоловый синий (МТС)
Оборудование
• Аналитическая цепь для непрерывного проточного анализа в сегментированном
потоке с манифольдом и фильтром для колориметрических измерений при 460 нм
(рис. 30.4).
• Лабораторная стеклянная посуда.
Реагенты
• Раствор хлорида бария: растворяют 1,526 г дигидрата хлорида бария (ВаС12 • 2Н20)
в 800 мл деионизированной воды; гомогенизируют и доводят объем до 1000 мл; при
необходимости фильтруют через аналитический фильтр (синяя лента).
• Падроокись натрия в гранулированной форме.
• 0,18 М раствор гидроксида натрия (NaOH): растворяют 7,2 г гидроксида натрия
в 1000 мл деионизированной воды.
• Метанол СН3ОН (или этанол С2Н5ОН).
• Концентрированная хлористоводородная кислота и 1 М раствор НС1.
• Метилтимоловый синий1 (МТС или 3,3'-Bis[N,М-ди(карбоксиметил)аминометил]ти-
молсульфонфталеин, натриевая соль): растворяют 0,1182 г МТС в 500 мл метанола;
1 Чистота коммерческих красящих веществ, таких как МТС, составляет всего лишь 90 или даже
80%. Следовательно, необходимо проверять их чистоту, чтобы не нарушить эквимолярности
реагента, определяющего линейность зависимости величины поглощения от концентрации
сульфата (Colovos и др., 1976). Если чистота МТС недостаточна, то реагент будет содержать
избыток Ва2+ и двухъядерные комплексы будут образовываться по реакции:
Ва2МТС2~ + SO2- <=> BaS04 + ВаМТС4-.
Поглощение двухъядерного комплекса при 460 нм слабее, чем поглощение ожидаемого мо-
ноядерного соединения и, следовательно, величина поглощения определяется только частью
сульфат-ионов, вступивших в реакцию.
692
Часть 3. Неорганический анализ
добавляют 25 мл раствора хлорида бария, 4 мл 1 М раствора НС1, 71 мл деионизи¬
рованной воды; метанолом доводят объем до 1000 мл (чистоту МТС следует прове¬
рять после приобретения каждой новой партии).
• Сульфат калия (KjSO^.
• Калибровочные растворы. 0-10 мкг (5042_)/мл, в зависимости от используемого ма-
нифольда.
Дегазация
Колориметрическая |
auatii/o . *
ячейка
50 мм
460 нм
S = solvaf lex
или
силиконовая
трубка
Скорость
потока,
мл/мин
0,42
NaOH
1,20
Г Воздух
т .. .
TS 1,37 Метанол
S 1,37 МТВ
г-А
Колонка с ионообменной смолой
0 2 мм L = 30 см 1,00
-► Сброс
Дегазация
0,42 I
Проба
S 0,70
Рециркуляция в ячейку
^ Ополаскивание
2,90
Вода
Рис. 30.4. Измерение содержания сульфатов в экстрактах автоматизированным колориме¬
трическим методом в процессе непрерывного проточного анализа в сегментированном потоке
Методика
Если органическая матрица была разрушена при анализе по методике Шёнигера, то,
чтобы избежать разложения органических окрашивающих реагентов, в пробах не долж¬
но быть избыточного пергидроля. Экстракт, содержащий серу из пробы почвы в фор¬
ме сульфата, анализируют автоматизированным спектроколориметрическим методом
в процессе непрерывнопроточного анализа в сегментированном потоке (рис. 30.4)
путем сравнения величин поглощения с аналогичными параметрами калибровочных
растворов сульфатов в калибровочном диапазоне концентраций (McSwain и др., 1974;
Со/смиидр., 1976).
Чувствительность анализа можно увеличить за счет использования более длинной ко¬
лориметрической измерительной ячейки, например, 50 мм ячейки вместо 15 мм ячейки;
однако это связано с риском повышения уровня фонового шума; или же можно изме¬
нить расход входящего потока пробы, например, установить расход 2,00 мл/мин вместо
1,00 мл/мин.
При одинаковой индексации параметры потока в трубках Л4000 Solvaflex (рекомендо¬
ванных для растворителей) немного отличаются от параметров потока в трубках из ПВХ
типа Tygon 3603. Силиконовые трубки более прочны и менее подвержены деформации
в сравнении с трубками Solvaflex. При использовании измерительной ячейки размером
50 мм, которая легче загрязняется, увеличение скорости рециркуляции помогает огра¬
ничить образование отложений.
Глава 30. Сера
693
30.2.9. Определение общей серы автоматизированным сухим CHN(OS)
элементным анализом
В присутствии соответствующего катализатора аппаратура CHN(OS) позволяет анали¬
зировать пробы, содержащие различные формы S, с использованием сухих процессов
(см. гл. 10). Автоматизация делает возможным проведение быстрых анализов суммарно¬
го содержания S в пробах, измельченных до размера частиц 0,1 мм.
Кроме того, если прибор соединен с масс-спектрометрическим детектором, возмо¬
жен анализ изотопов 34S и 32S.
Для анализа следовых количеств серы можно использовать очень чувствительный
и селективный детектор электронного захвата, поскольку диоксид серы имеет сродство
к свободным электронам.
Каждый производитель пред лагает свою собственную систему ловушек и катализато¬
ров: вольфрамовый ангидрид (W03) делает возможной конверсию S в S02, для окисле¬
ния S02 до S03 часто используется пентаоксид ванадия (V205).
Аппаратура типа Fisons 1108 CHN-OS включает две печи для термической обработки
проб. Печь для анализа S содержит вольфрамовый ангидрид. Различные формы S окис¬
ляются в печи, нагретой до 1100 °С. При возгорании оловянной капсулы, содержащей
пробу, температура в печи повышается до 1600 °С. Поток гелия переносит газообразные
продукты сгорания в медную колонку, затем в хроматографическую колонку, где проис¬
ходит отделение S в форме S02. Затем проводится количественный анализ с помощью
термокондуктометрического детектора (катарометра). Аппаратура NA 1500 (NCS) по¬
зволяет анализировать пробы до 100 мг и проводить полный анализ приблизительно за
10 мин.
В анализаторе Perkin Elmer CHNS применяется водяная ловушка для измерения Н
и катарометр для измерений содержания С в форме С02, N в форме N2 и S в форме S02.
Для измерений общего содержания S, в анализаторе LECO-Sиспользуются ускорите¬
ли сгорания (Сг03, Мо03, порошок железа) в присутствии кислорода, температура ин¬
дукционной печи составляет 1600 °С. Высвобождающийся S02 анализируют йодометри¬
ческим методом с помощью автоматического титрометра:
S02 + I2 + 2Н20 -> H2S04 + 2 HI.
В модели SC132, оборудованной печью, нагреваемой до 1400 °С в атмосфере кисло¬
рода, для количественного анализа газообразной S в форме S02 измеряют ИК-поглоще-
ние при прохождении через измерительную ИК-ячейку.
В приборе Herraeus (Foss-UIQ пробу помещают в печь, нагретую до 1150 °С, содержа¬
щую оксид вольфрама. Газ-носитель, содержащий 90% гелия и 10% кислорода, перено¬
сит S в виде S02 в колонку с перхлоратом магния, где происходит дегидратация. В хро¬
матографической колонке (например, Рогарак) отделяются примеси, такие как метан.
В итоге S измеряют в форме S02c помощью детектора ИК-излучения.
В аппаратуре Antek при общем анализе применяется хемилюминесцентное детекти¬
рование для измерения общего азота и пирофлуоресцентное детектирование для изме¬
рения общей S. Можно анализировать пробы почвы от 50 мг до 1 г, вес пробы зависит от
содержания серы. Пробу окисляют в печи, нагретой до 1000 °С, в присутствии катализа¬
тора для выделения диоксида серы. После высушивания в газовой ловушке, газ подвер¬
гают обработке УФ-излучением: S02 + hv -> S02 + hv'. В фотоумножителе происходит
усиление сигнала флуоресценции частотой v'. Эта реакция специфична по отношению
к S в форме S02, зависимость отклика детектора от концентрации носит линейный ха¬
рактер.
694
Часть 3. Неорганический анализ
Wosthoff предложил специальный прибор — сульфограф. Анализ общей серы произ¬
водится путем сжигания пробы в потоке кислорода при 1350 °С с образованием диок¬
сида серы. Затем S в форме S02 поглощается окислительным водным раствором серной
кислоты, в котором не удерживается диоксид углерода. Концентрацию серы измеряют
по изменению электропроводности в ячейке, содержащей окислительный раствор, до
и после прохождения газа. Это оборудование не автоматизировано, что делает его ме¬
нее привлекательным, несмотря на удовлетворительную прецизионность (Marius и др.,
1976).
Эти примеры показывают, что существует множество методик, использующих сухие
процессы для измерения содержания S. В каждом случае необходимо следовать специ¬
альной процедуре. Как правило, наиболее просты измерения, производимые с помощью
автоматизированной аппаратуры с компьютеризованной процедурой измерения. Пробу
измельчают до размера частиц 0,1 мм, сушат над фосфорным ангидридом в сушильном
шкафу при 105 °С, помещают навеску в серебряную или оловянную капсулу и вводят
непосредственно в систему с помощью дозатора. Соответствующее программное обес¬
печение позволяет получать результат измерений в распечатанном виде.
Для обеспечения удовлетворительных результатов необходимо правильное обслужи¬
вание и управление работой оборудования: обновление катализаторов, замена газовых
ловушек, очистка печей, фильтров, уплотнительных прокладок и т. д. При анализе вос¬
пламеняющихся и летучих продуктов сухими методами существует риск взрыва.
Для исследования серы в почвах используется аппаратура для термического анали¬
за (см. гл. 7), в особенности, в сочетании с аппаратурой для анализа выделившегося
газа (АВГ). Многие измерения, такие как потери веса, температуры фазовых перехо¬
дов и разложения, определение природы выделяющихся газов, таких как S02, S03 или
H2S, можно проводить одновременно. Описание методик измерений см. в публикаци¬
ях Gerzabek и Schaffer, 1986; David и др., 1989; Bremnern Tabatabai, 1971; Bergheijn и van
Schuylenborgh, 1971; Tabatabai и Bremner, 1970b, 1991.
30.2.10. Анализ общего содержания S в форме SO*~ методом ионной
хроматографии
ИХ является быстрым, селективным и очень чувствительным методом, позволяющим
проводить одновременный анализ ряда анионов и катионов в многокомпонентных
растворах (см. раздел 18.3.8). Несмотря на эти преимущества, ИХ не является
универсальным методом, рабочий диапазон метода при общем анализе ограничен. В ес¬
тественном окружении метод представляет полезную альтернативу химическому ана¬
лизу для не слишком засаленных почв, поскольку пределы обнаружения составляют
приблизительно 0,2 мг (S-SO*~)/Oi, а диапазон измеряемых значений — от 0,5 до 100 мг/л
(см. гл. 18).
Оборудование и основные положения метода
ИХ — метод высокоэффективной жидкостной хроматографии (ВЭЖХ), предназна¬
ченный специально для анализа ионов (Pansu и др., 2001). Прибор состоит из ионооб¬
менной колонки, в которой находится неподвижная фаза (для анализа SO*~ можно ис¬
пользовать анионообменную смолу в НСО"-форме), через которую с помощью насоса
с постоянным расходом пропускают поток подвижной фазы, Анализируемую пробу ин¬
жектируют в подвижную фазу. Ионы удерживаются неподвижной фазой в соответствии
с их сродством; затем по истечении воспроизводимого времени удержания компоненты
Глава 30, Сера
695
раствора выходят из колонки, и их количество определяется кондуктометрическим
детектором.
Прямая инжекция растворов полцэлектролитов в ионообменную колонку может со¬
кратить срок службы колонки и снизить ее эффективность из-за загрязнения. Для защи¬
ты разделительной колонки рекомендуется устанавливать форколонку с той же самой
аналитической обменной смолой. В качестве альтернативы для разделения основных
анионов матрицы можно воспользоваться диализом Доннана. Это позволяет ограничить
уменьшение эффективности колонки и связанных с ним последствий (постепенным из¬
менением времен удержания и ухудшением разрешения).
Поскольку детектирование проводится кондуктометрическим методом, ионы
элюирующей фазы необходимо удалить физико-химическими или электрохимическими
методами.
Сульфат-анион обладает сравнительно большим временем удержания, зависящим от
изменения pH. В этом случае следует воспользоваться забуференной подвижной фазой
при pH, позволяющем в достаточной степени сократить время удерживания сульфата.
При анионной хроматографии для измерения количества сульфата подвижной фазой,
как правило, является карбонатный-бикарбонатный буфер с изократным потоком.
Области применения
Аналитическая проба не должна содержать твердых частиц, поэтому растворы следует
профильтровать через дисковые фильтры с размером пор 0,45 или 0,22 мкм.
Для анализа общей серы все формы S, присутствующие в почве, должны быть окис¬
лены до сульфатов. Вследствие этих операций возникают трудности, связанные с тем,
что окисление методом щелочного плавления (см. раздел 30.2.4) или с применением ги-
побромита (см. раздел 30.2.5) приводит к образованию значительного количества солей
сильных кислот (нитратов, хлоридов, перхлоратов). Даже после сильного разбавления
эти растворы нельзя анализировать методом ИХ из-за слишком больших зарядов ио¬
нов на колонках и возникающих помех, связанных с чрезмерно интенсивными пиками.
Вместо этого следует воспользоваться окисляющим методом с применением пергидро¬
ля. Все сульфиды и S-формы, связанные с органическими веществами, количественно
переходят в раствор, не вызывая появления избытка анионов.
Органические вещества необходимо разложить для высвобождения органических
форм S, однако остаточное окрашивание этих органических материалов не создает по¬
мех при кондуктометрическом определении.
Концентрация экстрагирующих реагентов (например, реагента Моргана), необходи¬
мая для анализа некоторых форм S, делает измерение невозможным. С другой стороны,
анализ почвенного раствора простой фильтрацией и разбавлением достаточно прост,
если суммарное содержание растворимых солей не слишком высоко (см. гл. 18).
При неполной солюбилизации погрешности, приводящие к занижению результатов,
могут оказаться значительными, например, в пробах почв, содержащих нерастворимые
соли бария (BaS04).
В природных средах высокое содержание в образцах основного аниона минерального
образца, например, хлорида в морской воде, польдерных, мангровых почвах, в естест¬
венных солончаках, может помешать проведению точных измерений. Окисление важно
в гидрогеотермальных средах, а также в кислых сульфатных почвах в присутствии пири¬
та, богатого сульфидами.
Следует отметить, что эквивалентная электропроводность органических анионов
ниже по сравнению с аналогичным параметром для неорганических анионов, и соот-
696
Часть 3. Неорганический анализ
ветствующие им пики более узкие. Следует отдать предпочтение уксусной кислоте перед
щавелевой кислотой из-за того, что последняя образует нерастворимые соли с кальцием.
Порядок элюирования анионов может отличаться в зависимости от условий процес¬
са. В большинстве случаев возрастание времени удерживания наблюдается в ряду:
F- < Cl- < NO-< N03- < РО,3" < Br < SO,2".
Пик SO2- обычно не отличим от пика SO2- Редко регистрируют пик S20*~.
Дополнительная информация содержится в работах Tabatabai и Dick, 1979; Tabatabai
идр., 1988; Tabatabai и Bremner, \99\;ArtiolanAli, 1990; Tabatabai и Basta, 1991; Kamarkar
и Tabatabai, 1992; Aswan Tabatai, 1993; Tabatabai и Frankenberger, 1996.
30.2.11. Анализ общей S методом плазменной эмиссионной
спектрометрии
Анализ проводится в пробах почвы весом от 0,5 г до 2 г. Почву сушат на воздухе и из¬
мельчают до размера частиц 0,1 мм, затем минерализуют щелочным плавлением (см.
раздел 31.2.6) или прокаливанием с NaHC03/Ag20 или NaBrO. Общую серу в форме
сульфата растворяют в 1 М растворе НС1, фильтруют и доводят до требуемого объема.
Эмиссионная линия при 182,03 нм используется для исключения помех от кальция
при измерении фонового шума при 182,08 нм.
Следует контролировать помехи, связанные с присутствием железа и алюминия.
Железу соответствует слабая эмиссионная линия при 182,04 нм и широкая диффузная
эмиссионная полоса при 182,02-182,12 нм. Алюминию соответствует эмиссионная
полоса от 193 до 181,9 нм.
Концентрация соли не должна быть чрезмерно высока, чтобы не допустить засорения
небулайзера (Ferrott идр., 1991).
30.2.12. Анализ методом рентгеновской флуоресценции
Следует отметить, что данная методика может быть внедрена лишь в тех геохимических
лабораториях, которые способны оправдать стоимость массивного оборудования. Эта
методика как эталонная описана в разделе 31.3.2. Процедура анализа общей S включает
плавление и гранулирование. При высоком содержании органического вещества в про¬
бе (гистосоли, андосоли, торфяники) снижение массы означает, что необходимо внести
поправку на эффекты матрицы (Tabatabai и Вгетпег, 1970b).
30.2.13. Анализ методом атомно-адсорбционной спектрометрии
В данном случае проводятся непрямые измерения. После окислительной минерализа¬
ции полученные сульфаты осаждаются виде сульфата бария. После отделения осадка,
промывания и растворения в кислоте определяют количество бария, соответствующее
исходному содержанию сульфат-ионов, проводя измерение при 553,5 нм, используя
пламя смеси ацетилен - закись азота и внося поправку на фоновый шум.
30.2.14. Аналитическое фракционирование соединений серы
Формы серы и их биогеохимический цикл
В естественной среде минерализация почвы и иммобилизация соединений S регулиру¬
ется активностью микроорганизмов, климатическими условиями и растительностью.
Биогеохимический цикл S связан с циклами С и N, а знание характеристических
Глава 30. Сера
697
соотношений C:S и N:S позволяет определять чистые потери и прирост содержания
компонентов.
Выветривание минеральных соединений, вклад газообразных форм серы, присутст¬
вующих в атмосфере, попадающих в почву через системы ирригации и в составе до¬
ждевой воды, внесение удобрений в сельскохозяйственных зонах позволяет растениям
получать энергию, необходимую для роста. Они преобразуют сульфаты (наиболее ста¬
бильные окисленные минеральные формы) в набор органических соединений, содер¬
жащих С и N. Растительные остатки затем участвуют в ряде биотических процессов
окисления и восстановления. Происходит превращение органической и неорганиче¬
ской S, частичное включение в микробную и грибковую биомассу. Интенсивность по¬
токов от различных фракций S значительно варьирует в зависимости от типа почвы
и растительности. В состоянии равновесия на органические формы S приходится до
90% общей S.
Органическая S
Химическая природа соединений серы в составе органического вещества почвы сложна
и до сих пор не вполне изучена. Надежных методов селективного отделения общей ор¬
ганической части, содержащей S, в настоящее время не существует. В первую очередь,
количественную оценку органической S производят по разности между общим содер¬
жанием S (измеренным методами щелочного плавления или мокрого озоления) и со¬
держанием неорганической S, измеренным путем экстракции водой, разбавленными
кислотами (НС1) или растворами солей (NaHC03).
Изучение химической природы соединений S в почве привело к выделению двух
групп соединений, характеристики и свойства которых оказались различными; тем не
менее, четких границ между группами установлено не было (Tabatabai и Вгетпег, 1970b;
/теиеуидр., 1970).
Органические соединения, содержащие S, связанную с С (S“C)
Эта группа включает соединения, содержащие сульфоаминокислоты, такие как:
H2C-SH
HjC-CH-S-CH,
H2C-S03H
H2C-S-S-CH2
1 1
нс—nh2
1
нс -nh2
н2с- nh2
1 1
PLN-CH НС- NH
2 1 I
соон
1
соон
1 1
СООН соон
Цистеин
Метионин
Таурин
Цистин
Эти соединения устойчивы к микробному воздействию и очень медленно
гидролизуются с образованием сульфатов. Они восстанавливаются при помощи сплава
Ренея (Ni — А1), в то время как S, связанная с гуминовыми и фульвовыми кислотами,
или не восстанавливается, или восстанавливается лишь частично.
Органические соединения, содержащие S, не связанную с С напрямую
К этим соединениям относятся, в основном, сульфатные сложные эфиры (такие
как С—О—S03H, фенольные эфиры, полисахариды), или например холина сульфат.
Они гидролизуются до сульфатов под действием кислот и оснований. Соединения
с высокими и низкими молекулярными весами можно считать переходными формами,
698
Часть 3. Неорганический анализ
образующимися при краткосрочной минерализации. Они восстанавливаются
йодистоводородной кислотой (HI) до сульфида водорода.
Могут оставаться и другие соединения, не восстанавливаемые ни HI, ни сплавом
Ренея. Содержание этих соединений можно оценить по разности балансов, вероятно,
они относятся к соединениям со связью типа С—S, хотя это до сих пор не доказано.
30.2.15. Анализ органической S, связанной с С
Основные положения
Оценку содержания этих соединений можно произвести путем восстановления сплавом
Ренея (никель — 50% алюминия). Сплав в виде тонкоизмельченного порошка быстро
вступает в реакцию в сильнощелочной среде и делает возможной гидрогенизацию
S-содержащих органических соединений. Восстановление катализируется никелем:
2NaOH + 2А1 + 2Н20 2NaA102 + 6 Н
Этот метод используют не только для оценки количества S, связанной с С (CS), но
и для измерения содержания элементарной S и некоторых редких неорганических форм
S в почвах (S2032- тиосульфат-, S202- дитионит-, S402- тетратионит-ионы). Например,
восстановление элементарной серы происходит по реакции:
S° + 2Н -> H2St
H2S, выделяющийся в кислой среде, анализируют колориметрическим методом по
реакции с метиленовым синим (см. раздел 30.2.7).
Количество S, связанной с С, можно оценить по разности между общей серой и S,
восстанавливаемой HI, несмотря на то что присутствие неизвестных продуктов может
исказить результат.
Оборудование
• Оборудование, перечисленное в разделе 30.2.7.
• Адаптер для установки Johnson и Nishita (рис. 30.1) для введения хлористоводород¬
ной кислоты (рис. 30.5).
• Колба для кипячения объемом 150 мл (В на рис. 30.1) с тефлоновой манжетой.
Реагенты
• Гкдроксид натрия (NaOH) в гранулированной форме.
• 5%-ный раствор гидроксида натрия: растворяют 50 г NaOH в 800 мл воды; гомоге¬
низируют, выдерживают до охлаждения и доводят объем до 1000 мл деиоизирован-
ной водой.
• 20%-ный раствор хлористоводородной кислоты (НС1, d= 1.17).
• Активированный катализатор — сплав Ренея в порошкообразной форме; сплав Ре¬
нея постепенно теряет восстановительные свойства после шести месяцев хранения;
вещество классифицируется как «подозреваемое в канцерогенной активности».
Методика
• Сушат почву на воздухе и измельчают до 0,1 мм. Измеряют остаточную влажность.
Навеску почвы 0,2-0,5 г, содержащей от 10 до 50 мкг С, связанного с S, помеща¬
ют в колбу для кипячения (В на рис. 30.1) с целью разложения. Добавляют 100 мг
катализатора Ренея, 5 мл 5%-ного раствора гидроксида натрия, 25 мл деионизиро¬
Глава 30. Сера
699
ванной воды и гомогенизируют. На стеклянное шлифованное соединение отвода
колбы для кипячения надевают тефлоновую манжету и соединяют колбу с уста¬
новкой. Запускают поток воды в холодильнике; подают азот в реакционную смесь
со скоростью 200 мл/мин. Осторожно кипятят 30 мин, не допуская образования
избытка пены. Охлаждают в потоке азота.
Помещают реагенты в промывочную колонку D и приемную колбу /г(рис. ЗОЛ)
установки Johnson и Nishita, в соответствии с описанием «Методика» в разделе
30.2.7. С помощью адаптера, изображенного на рис. 30.5, добавляют в колбу для
кипячения (В) 5 мл 20%-ного раствора хлористоводородной кислоты. Продолжая
барботирование, осторожно кипятят в течение 30 мин до полного вытеснения се¬
роводорода. Проводят реакцию с образованием метиленового синего согласно
описанию «Методика» в разделе 30.2.7. Убеждаются в отсутствии следов осажден¬
ного сульфида на стенках колбы.
Рис. 30.5. Адаптер для добавления хлористоводородной кислоты при анализе S, связанной с С,
с помощью установки Johnson и Nishita (см. рис. 30.1)
Примечание
Результат анализа можно проверить добавлением стандартных соединений известного
состава. Например для образца аминокислоты (L-метионин) — навеску 116,3 мг
L-метионина растворяют приблизительно в 300 мл воды. Доводят объем до 500 мл
деионизированной водой. Раствор содержит 50 мг (S)/n. Отмеряют 600 мкл и переносят
в колбу для кипячения (В на рис. 30.1).
30.2.16. Анализ органической S, не связанной с С
Основные положения
Этот метод основан на восстановлении йодистоводородной кислотой; он позволяет
проводить экстракцию наиболее нестабильных форм органической S, которая может
гидролизоваться до сульфатной S в сильнощелочных и сильнокислых средах. Серосо¬
держащие органические соединения в форме сульфатных эфиров (С—О—S— связи) вос¬
станавливаются так же, как S-полисахариды, S-липиды, S-холин. Кроме того, восста¬
700
Часть 3. Неорганический анализ
навливаются некоторые компоненты гуминовых и фульвовых кислот. Неорганические
формы серы, растворимые в воде или 0,1 М растворе UC1, также восстанавливаются, что
указывает на необходимость внесения поправок в результаты:
Органическая S, _ Общая S, _ Растворимая
восстанавливаемая HI ~~ восстанавливаемая HI неорганическая S
Основные окислительно-восстановительные реакции между соединениями серы
и йода были описаны химиками довольно давно:
H2S + 6HI о 3I2 + 4Н20 + S
S02 + 4HI о 2I2 + 2Н20 + S
H2S + 12 <-* 2HI + S
S + 2HI <-» H2S + 12
Оборудование
• Установка Johnson и Nishita (рис. ЗОЛ).
• Лабораторная стеклянная посуда.
• Прецизионные весы (±0,1 мг).
Smith и Mayer {1924)
Volhard{№l)
Sell {те)
Worris и Cottrell {1896)
Реагенты
См. «Реагенты» в разделе 30.2.
Методика
Сушат на воздухе недавно отобранный образец почвы, измельчают до размера частиц
0,1 мм. Измеряют остаточную влажность.
• Навеску 0,2—0,5 г почвы, содержащей от 20 до 100 мкг общей S, помещают в пе¬
регонную колбу (В на рис. 30.1). Добавляют 2 мл воды и 4 мл восстановительной
смеси (HI + Н3Р02 + НСООН), в соответствии с описанием, представленным
в разделе 30.2.15, проводят восстановление с последующим колориметрическим
измерением содержания метиленового синего (см. раздел 30.2.7).
• Вносят поправку на содержание влаги и неорганической S, экстрагируемой водой
или 0,1 М раствором UC1 (см. «Основные положения» в разделе 30.2.16).
Примечания
• Использование свежих проб, хранившихся в морозильной камере, ограничивает
разложение соединений S, восстанавливаемых HI.
• В пробах, высушенных на воздухе, богатых органическим веществом, легкие час¬
тицы могут всплывать и слабо контактировать с водой; следует хорошо взболтать
содержимое колбы для гомогенизации.
• Эффективность восстанавливающей обработки может быть протестирована на
пробе 4-нитрофенил сульфата (калиевая соль, 02NC6H40S03K • Н20).
30.2.17. Экстракция и анализ содержания растворимых сульфидов
Основные положения
Сульфид водорода в замкнутой среде вытесняется хлористоводородной кисло¬
той и накапливается в растворе ацетата цинка и кадмия. Образовавшиеся сульфиды
Глава 30. Сера
701
цинка и кадмия анализируют методом йодометрического титрования в соответствии
с реакциями:
сн.соо
(CH3COO)2Zn + H2S 2 ► ZnS + 2 СН3СООН
S2~ +12 Кислота* > s + 21- (Chariotи Bezier, 1955)
I2+ 2S20 ► S40*~ + 2Г обратное титрование тиосульфатом
♦Окислительно-восстановительный потенциал I не зависит от pH вплоть до pH 9, однако
йод может быть более сильным окислителем при более высоких значениях pH по сравнению
с другими системами, окислительно-восстановительный потенциал которых снижается при
повышении pH. Поэтому необходимо использовать кислую среду. В щелочной среде может
происходить следующая реакция:
S2- + 412 + 4Н20 °Н > SO2- + 81- + 8Н+
Оборудование
• Аналитические весы (±0,1 мг).
• Установка для перегонки (рис. 30.1).
• Промывные бутыли.
• Бюретка для титрования (±0,1 мл).
• Лабораторная стеклянная посуда.
Реагенты
• Ацетат цинка (Zn(CH3COO)2).
• Ацетат кадмия (Cd(CH3COO)2).
• Уксусная кислота (СН3СООН).
• Смешанный раствор: навеску 17 г ацетата цинка и 8 г ацетата кадмия растворяют
в смеси 200 мл уксусной кислоты и 600 мл дистиллированной воды; доводят объем
до 1000 мл дистиллированной водой
• Хлористоводородная кислота (НС1, d = 1,16).
• * 1Мраствор НС1: отмеряют 100 мл концентрированной хлористоводородной кис¬
лоты и доводят объем до 1000 мл дистиллированной водой.
• Крахмал растворимый порошкообразный.
• Йодид ртути (Hgl2).
• Крахмальный индикатор. Навеску 10 г порошка крахмала растворяют в 50 мл дис¬
тиллированной воды. Добавляют 40 мг йодида ртути (или 40 мг гидроксибензойной
кислоты) в качестве консерванта. Выливают смесь в 950 мл кипящей дистиллиро¬
ванной воды. Кипятят в течение 2 мин. Охлаждают и доводят объем до 1000 мл.
Хранят в коричневом флаконе с притертой пробкой. Раствор сохраняет стабиль¬
ность в течение приблизительно 6 месяцев. Индикатор окрашен в синий цвет при
pH до 8,0 и обесцвечивается при дальнейшем повышении pH.
• 0,1 н. йод, коммерческий титрованный раствор.
• 0,1 н. гипосульфит натрия (Na2S203 • 5Н20), коммерческий титрованный раствор.
• Диоксид углерода, С02 в газовом баллоне с газовым редуктором и регулятором дав¬
ления.
702
Часть 3. Неорганический анализ
Методика
Согласно методике (Gony и Parent, 1966), навеску свежего образца (50 г) помещают
в колбу для кипячения со стеклянным шлифованным соединением (в другой пробе
из того же образца измеряют влажность почвы). Добавляют 50 мл 1 М раствора НС1.
Присоединяют холодильник (рис. 30.1) и устанавливают у газоотводного патрубка две
последовательные промывные бутыли, заполненные 50 мл цинкового и кадмиевого рас¬
твора. Осторожно кипятят в течение 1 ч. В это время происходит перенос сероводорода
в потоке диоксида углерода (3—4 пузырька в секунду) в первую промывную бутыль.
Вторая промывная бутыль позволяет контролировать полноту поглощения сероводорода
в первой промывной бутыли.
Титрование
В первую промывную бутыль добавляют 5 мл 0,1 н. раствора йода (или 10 мл 0,01 н. рас¬
твора йода при слабом выпадении осадка сульфида), затем 2 мл крахмального индикато¬
ра. Титруют избыток йода 0,1 н. (или 0,01 Н) раствором гипосульфита натрия.
Расчеты
1 мл 0,1 н. Na2S203 = 1,603 мг S.
Результаты выражают в г (S)/kt почвы, высушенной при 105 °С.
Примечания
Под воздействием холодного раствора хлористоводородной кислоты из аморфного
сульфида железа выделяется свободный сероводород и S в форме H2S. Макинавит
и грейгит — наиболее стабильные из железосодержащих минералов, вступают в реак¬
цию только при нагревании. Микрокристаллы пирита с солянокислой средой в реакции
не вступают.
Прочие летучие соединения можно идентифицировать в переувлажненных почвах по
соединениям, например, соединениям S и С, синтезированным микроорганизмами:
CS2 дисульфид углерода (СН4 (метан) + 4S CS2 + 2H2S);
СН3—SH (метилмеркаптан);
CH3-S—СН3 (диметилсульфид);
CH3-S-S-CH3 (диметилдисульфид);
СН3—СН2—S—СН2—СН3 (диэтил сульфид);
С—О—S (карбонилсульфид).
30.2.18. Анализ серы в пиритах
Введение
Пириты, сульфиды и полисульфиды, образующиеся в восстановительных средах, встре¬
чаются в почве в виде небольших конкреций черного цвета. Поскольку плотность пири¬
та равняется 5,02, его можно выделить промывкой и флотацией. Эти соединения быстро
разлагаются в окислительной среде. В переувлажненных почвах протеолитические бак¬
терии способны высвобождать сероводород при гидролизе соединений серы, таких как
сульфоаминокислоты (например, цистеин, цистин, метионин). Этот процесс может
происходить и при восстановлении сульфата.
Глава 30. Сера
703
Исследования диагенеза пиритов позволили изучить сложную кинетику реакции,
в ходе которой сера, восстановленная до сульфида водорода, взаимодействует с аморф¬
ными восстановленными соединениями железа с образованием метастабильного суль¬
фида железа, который кристаллизуется в форме тетрагонального макинавита (FeS),
термически стабильного гексагонального пирротина (FeS), грейгита (Fe3S4), ортором¬
бического марказита в древних осадочных образованиях или кубического пирита в со¬
временных морских отложениях. Эта форма пирита является термически стабильной
и ее необходимо анализировать, поскольку она представляет собой конечный продукт
превращений.
Пирит нерастворим в горячей хлористоводородной кислоте, что позволяет отделить
его от других растворимых сульфидов при анализе по методике, описанной в разделе
30.2.17.
Анализы
Метод рентгеновской дифракции (РД) — отличный способ измерения содержания пи¬
ритов, отделенных путем флотации. Однако, как правило, методики РД недостаточно
чувствительны для определения промежуточных фаз без концентрирования. В табл. 30.1
перечислены характеристики рефлексов РД для пирита, марказита, грейгита и макина¬
вита.
При дифференциальном термическом анализе (ДТА) эндотермические реакции
с участием пирита регистрируют при 354, 443, 551 и 613 °С, кроме того, регистрируют
экзотермический эффект прр 450 °С.
При сканирующей электронной микроскопии (+ЭД-РС-микроанализ) пирит легко
обнаруживается в виде зерен размером 1-10 мкм, имеющих характерную форму, напо¬
минающую ягоду малины. Это позволяет отличать пириты биологического происхожде¬
ния от кристаллических пиритных кластеров, обнаруживаемых в вулканических средах.
Основные положения
При анализе почв, не содержащих сульфата или ярозита, следует использовать пробы,
в которых восстановленные соединения серы (различные непиритные сульфиды) и со¬
единения железа (II) и железа (III) уже экстрагированы из среды хлористоводородной
кислоты. Затем эту пробу окисляют горячей азотной кислотой. Железо, переведенное
в раствор, анализируют методом атомно-адсорбционной спектрометрии.
Если почвы содержат как окисленные (сульфат-ярозит), так и восстановленные (пи¬
риты) минералы, следует использовать пробу, из которой уже проведена экстракция
форм, растворимых в Ка3-ЭДТА. Сульфаты, полученные окислением, анализируют тур-
бидиметрическими или колориметрическими методами (см. раздел 30.2.21).
Оборудование
• Лабораторная стеклянная посуда.
• Лиофильная сушка.
• Термостатированная водяная баня.
• Атомно-адсорбционный спектрометр (ААС).
Реагенты
• Азотная кислота (70%-ная HN03).
• Хлористоводородная кислота (НС1, d = 1,17).
704
Часть 3, Неорганический анализ
• 4 Мраствор НСк в мерную колбу вместимостью 1 л вносят около 500 мл деиони¬
зированной воды и добавляют 320 мл хлористоводородной кислоты; охлаждают;
доводят объем до 1000 мл деионизированной водой и гомогенизируют.
• Коммерческий стандартный раствор железа для ААС, содержащий 1 мг (Ре)/мл.
• Хлорид натрия (NaCl).
• Раствор для разбавления при определении содержания железа (1%-ный NaCl — 0,2 М
НС1): растворяют 10 г хлорида натрия приблизительно в 500 мл воды; добавляют
50 мл 4 М НС1, доводят объем до 1000 мл и гомогенизируют.
• Стандартный базовый раствор железа с концентрацией 100 мг/л: с помощью пре¬
цизионной пипетки переносят 10 мл стандартного раствора железа, содержащего
1 мг (Ре)/мл, в мерную колбу объемом 100 мл; доводят объем до метки и гомогени¬
зируют раствор.
• Стандартные калибровочные растворы: в мерные колбы объемом 100 мл вносят 0,5,
10, 15, 20, 25 мл раствора железа, содержащего 100 мг (Ре)/л; добавляют в каждую
колбу по 2,5 мл 4 М раствора НС1, доводят объем до 100 мл деионизированной
водой и гомогенизируют. Концентрации калибровочных растворов составляют:
0 (холостая проба); 5,0; 10,0; 15,0; 20,0 и 25,0 мг (Fe)/n.
Методика
• Промывают остаток пробы после экстракции сульфидов (см. раздел 30.2.17), затем
проводят дегидратацию методом лиофильной сушки; при необходимости измель¬
чают в агатовой ступке до размера частиц 0,1 мм.
• Навеску пробы 250-500 мг помещают в колбу для кипячения из стекла Пирекс
объемом 50 мл.
• Добавляют 10 мл 70%-ной азотной кислоты и помещают в кипящую водяную
баню.
• Выпаривают досуха, затем добавляют к остатку 5 мл 4 М НС1 и нагревают в течение
5 мин в водяной бане.
• Переносят в пробирку для центрифугирования и центрифугируют в течение 10 мин
при 5000 g.
• Декантируют надосадочный раствор в мерную колбу на 100 мл.
• Добавляют приблизительно 40 мл деионизированной воды в пробирку для центри¬
фугирования и вновь центрифугируют в течение 10 мин при 5000 g.
• Добавляют надосадочную жидкость к первой фракции; фильтруют и промывают
почвенный остаток; доводят объем до 100 мл и гомогенизируют.
• Проводят измерение содержания железа в этом растворе; готовят смесь 1%-ного
NaCl и 0,2 М раствора НС1 (в равных объемах) для разбавления и проводят анализ
методом ААС при 248,3 нм в воздушно-ацетиленовом пламени.
30.2.19. Анализ элементарной серы
Происхождение
В некоторых почвенных горизонтах или в ризосфере подъем воды в почве, вызванный
капиллярными силами, с уровня подземных вод, богатых сульфидами, может приве¬
сти к кристаллизации элементарной серы (участки соломенно-желтого цвета) около
поверхности почвы, или в определенных горизонтах почвы, или около корней расте¬
ний.
705
Глава 30. Сера
Анализы
• Серу плотностью 2,07 можно обогатить флотационным методом.
• ДТА выявляет энантиотропный переход из орторомбической в моноклинную фор¬
му при 113 °С в инертной атмосфере. Плавление наблюдается при 124 °С, последу¬
ющие превращения и кипение при 179 °С и 446 °С, соответственно.
• Сканирующая электронная микроскопия в некоторых случаях делает возможным
обнаружение кристаллических систем (орторомбических), однако стабильность
при наблюдениях в электронном пучке низка.
Основные положения
После удаления сероводорода с помощью хлористоводородной кислоты (см. раздел
10.2.17) экстрагируют элементарную серу ацетоном.1
Далее проводится турбидиметрический анализ коллоидной серы в воде (кроме того,
возможна экстракция по Сокслету в присутствии металлической меди с последующим
измерением количества образовавшегося сульфида меди, например, йодометрическим
методом).
Оборудование
• Шейкер.
• Центрифуга.
• Спектрофотометр-турбидиметр.
• Аналитические весы (±0,1 мг).
Реагенты
Не использовать резиновые пробки для любых реагентов, содержащих серу.
• Ацетон (СН3-СО-СН3).
• 99,99%-ная (или 99,5%-ная) сера — S.
• Калибровочные растворы элементарной серы. Навеску 62,5 мг серы в виде тонко
измельченного порошка переносят в колбу для кипячения вместимостью 250 мл;
добавляют 150 мл ацетона и встряхивают смесь до полного растворения серы; пе¬
реносят в мерную колбу объемом 250 мл, доводят объем до метки ацетоном и го¬
могенизируют; 1 мл раствора содержит 0,25 мг элементарной серы; вносят (0), 1,
2, 3, 4, 5 мл этого стандартного раствора в мерные колбы на 100 мл, содержащие
по 80 мл воды; выравнивают содержание ацетона в колбах, добавляя 5 мл ацетона
в колбу 0,4 мл в колбу 1, 3 мл в колбу 2, 2 мл в колбу 3,1 мл в колбу 4,0 мл в колбу
5; доводят объем раствора до метки водой и гомогенизируют.
Концентрации калибровочных растворов: 2,5; 5,0; 7,5; 10,0; 12,5 мг (S)/n.
1 Ацетон (2-пропанон) — наименее опасный из растворителей элементарной серы, несмотря
на то что он воспламеняется и может вызвать раздражение дыхательных путей при вдыхании.
Растворимость S в ацетоне составляет 2,65 г на 100 мл при 25 °С.
Другие растворители более опасны, их применения следует избегать:
- бензол (растворимость S 24 г/л при 30 °С) токсичен при вдыхании и является канцероген¬
ным веществом;
- трихлорметан (хлороформ) СНС13, (растворимость S — 15 г/л при 18 °С) токсичен при
вдыхании, обладает анестетическим эффектом.
- СН212 — дийодметан (метилен йодид) (растворимость S — 91 г/л при 10 °С), вязкое веще¬
ство высокой плотности;
- Пиридин C5H5N, токсичен при вдыхании.
706
Часть 3, Неорганический анализ
Методика
• Анализ твердого остатка после экстракции сульфидов (см. раздел 30.2.17). Остаток
промывают, сушат методом лиофильной сушки и вновь измельчают до размера ча¬
стиц 0,1 мм при необходимости. Навеску 250 мг остатка помещают в пробирку для
центрифугирования с полипропиленовой завинчивающейся крышкой объемом
в 20 мл. Точно отмеряют 10 мл ацетона, встряхивают в течение 30 мин и центрифу¬
гируют в течение 15 мин при 5000 g.
• Вносят 80 мл деионизированной воды в мерную колбу на 100 мл. С помощью пре¬
цизионной пипетки вносят 5 мл ацетонового экстракта. Раствор встряхивают и до¬
водят объем до 100 мл деионизированной водой. Выстаивают в течение 3 ч, время
от времени встряхивая. Измеряют поглощение при 420 нм.
Зависимость поглощения от концентрации раствора в калибровочном диапазоне но¬
сит линейный характер. Органическое вещество может создавать помехи из-за соосаж-
дения и окрашивания раствора.
30.2.20. Анализ водорастворимых сульфатов
Формы
Сульфатные соли имеют морское происхождение, однако они претерпевают изменения
в процессе эволюции почвы, т. е. восстановление, повторное окисление, выщелачивание
и осаждение. Сульфаты обнаруживают в форме сульфата кальция в почвах с большим
содержанием известняка (см. раздел 30.3). В тропических почвах, как правило, бедных
кальцием, сульфат натрия с большей вероятностью обнаруживается в зонах истощения
почвы; кроме того, в них встречаются легкорастворимые смешанные сульфаты алюми¬
ния (квасцы), железа и магния (Le Brusq и др., 1987; Montoroi, 1994).
Экстракция
Растворимые сульфаты можно анализировать из 1:10 водного экстракта для определения
растворимых солей (см. гл. 20). Этот экстракт будет количественно оценивать содержа¬
ние сульфата кальция лишь в случае низкой концентрации сульфатов. То же самое отно¬
сится к сульфатам бария или стронция, соосажденным с карбонатом кальция. Основные
сульфаты железа и алюминия, кокимбит (Fe2(S04)3 • 5Н20) и ярозит KFe3(0H)6(S04)2,
в раствор не переходят и, следовательно, не учитываются при анализе растворимых
сульфатов.
При агрономических исследованиях можно использовать ряд солевых экстрактов для
описания характеристик обменных и растворимых сульфатов (см. раздел 30.4.2).
Оборудование
• Лабораторная стеклянная посуда.
• Аналитические весы (±0,1 мг).
• Муфельная печь (1000 °С).
Реагенты
• 10%-ный раствор хлорида бария (ВаС12).
• 10%-ный раствор хлористоводородной кислоты (НС1).
• 1%-ный раствор нитрата серебра (AgN03).
Глава 30- Сера
707
Методика
Берут аликвоту водного экстракта 1:10 (см. гл. 18), объемом в зависимости от общей за¬
соленности почвы (20—50 мл). Аликвоту вносят в стакан на 250 мл и добавляют 5 мл
10%-ного раствора хлористоводородной кислоты. Доводят до кипения и по каплям
добавляют в кипящую жидкость раствор хлорида бария. Охлаждают и выстаивают
в течение 24 ч. Фильтруют через лабораторный фильтр (синяя лента) диаметром 20 мм,
промывают осадок до исчезновения хлорид-ионов (реакция с нитратом серебра). Поме¬
щают фильтр во взвешенный тигель из тугоплавкого материала и нагревают до 900 °С
в муфельной печи на воздухе. Охлаждают, взвешивают и рассчитывают результаты
в мн. SO^ и мг (S)/kt.
30.2.21. Анализ сульфатов, экстрагированных №3-ЭДТА
Основные положения
С помощью комплексообразующего реагента №3-ЭДТА экстрагируют гипс, небольшое
количество растворимых и обменных сульфатов. Экстрагированную S в форме SO*-
измеряют турбидиметрическим методом после осаждения сульфата бария.
Оборудование
• Лабораторная стеклянная посуда.
• Шейкер.
• Термостатируемая водяная баня.
• Спектрофотометр.
Реагенты
• Тринатриевый этилендиаминтетрауксусной кислоты (Ыа3-ЭДТА) моногидрат,
М = 376,21.
• 0,1 Мраствор Ыаг-ЭДТА: растворяют 18,8 г №3-ЭДТА приблизительно в 250 мл
воды, доводят объем до 500 мл и гомогенизируют.
• 65%-ная азотная кислота (HN03).
• 37%-ная хлористоводородная кислота (НС1).
• Царская водка: под вытяжкой смешивают в химическом стакане 180 мл 37%-ной
НС1 и 60 мл 65%-ной HN03, стакан накрывают часовым стеклом.
• 25%-ный раствор азотной кислоты, в мерный цилиндр емкостью 1 л добавляют
600 мл воды и 360 мл азотной кислоты; охлаждают, доводят объем до 1000 мл деио¬
низированной водой и гомогенизируют.
• 85%-ная фосфорная кислота (Н3Р04).
• Ледяная уксусная кислота (СН3СООН).
• Раствор уксусной и фосфорной кислот: смешивают 180 мл ледяной уксусной кисло¬
ты и 60 мл 85%-ной фосфорной кислоты.
• Раствор гуммиарабика и уксусной кислоты: навеску 0,5 г гуммиарабика растворя¬
ют в 50 мл деионизированной воды; добавляют 50 мл ледяной уксусной кисло-
н2
NaOOC-C Н2 Н2
^n-c-c-n;
NaOOC-C
н2
н2
X-COONa
<
н2с-соон
708 Часть 3. Неорганический анализ
ты; смешивают и фильтруют через усиленный кислотоустойчивый лабораторный
фильтр.
• Сульфат натрия (Na2S04).
• 0,3%-ный раствор сульфата натрия: растворяют 0,3 г сульфата натрия в 100 мл де¬
ионизированной воды.
• Хлорид бария (ВаС12* 2Н20).
• Раствор хлорида бария: растворяют 18,0 г хлорида бария в 44 мл воды, нагретой до
80 °С; добавляют 1,5 мл 0,3%-ного раствора сульфата натрия; доводят до кипения;
быстро охлаждают и добавляют 4 мл уксуснокислого раствора гуммиарабика; рас¬
твор готовят перед каждой серией анализов.
• Стандартные калибровочные растворы S в форме SO*- (см. раздел 30.2.7 и 30.2.8).
Методика
Экстракция
Методика представляет собой адаптацию известного метода (Виигтап и др., 1996). Про¬
бу почвы сушат методом лиофильной сушки и измельчают до размера частиц 0,1 мм
(влажность почвы измеряют в другой пробе, взятой из образца).
• Навеску 250 мг пробы помещают в полипропиленовую пробирку объемом 20 мл
для центрифугирования. Добавляют 10 мл раствора Ыа3-ЭДТА и перемешивают на
роторной мешалке в течение 3 ч. Центрифугируют в течение 15 мин при 5000 g
(остаток после промывания при необходимости можно сохранить для анализа яро-
зита).
• Помещают 2 мл надосадочной жидкости в ампулу объемом 50 мл из стекла Пирекс.
Добавляют 2 мл царской водки и 1 мл 85%-ной фосфорной кислоты. Выпаривают
раствор в кипящей водяной бане почти досуха, затем добавляют 2 мл царской вод¬
ки и вновь выпаривают. Добавляют 10 мл деионизированной воды и ставят в водя¬
ную баню на несколько минут. Охлаждают, гомогенизируют и переносят в мерную
колбу объемом 50 мл. Промывают, доводят объем до 50 мл деионизированной во¬
дой и гомогенизируют.
Анализ
• С помощью прецизионной пипетки переносят 20 мл экстракта (см. «Анализ» в раз¬
деле 30.2.21) в мерную колбу на 50 мл с притертой пробкой. Добавляют 10 мл воды,
5 мл 25%-ного раствора азотной кислоты и 3 мл ледяной уксусной кислоты. Го¬
могенизируют, добавляют 1 мл раствора хлорида бария и сразу же добавляют 0,5 г
порошкообразного ВаС12 • 2Н20, взбалтывая содержимое колбы после добавления
каждого реагента. Встряхивают в течение 15 мин, затем через 5 мин повторяют
встряхивание. Добавляют 2 мл уксуснокислого раствора гуммиарабика. Добавле¬
ние реагента должно быть строго равномерным1. Доводят объем до 50 мл деиони¬
1 Скорость осаждения сульфата бария из растворов, содержащих хлористоводородную кисло¬
ту, следует контролировать путем строгого соблюдения процедуры анализа (чтобы избежать
осаждения сульфата кальция и сульфатов тяжелых двух- или четырехвалентных элементов,
которые могут помешать измерениям). Осаждение замедляет хлористоводородная кислота.
Концентрация кислоты, температура, соотношение раствор: ВаС12, концентрация ВаС12, до¬
бавки солей и режим введения реагентов могут нарушить однородность распределения заря¬
да и повлиять на кинетику образования центров кристаллизации и роста кристаллов.
Глава 30. Сера
709
зированной водой и гомогенизируют. Выдерживают в течение 90 мин, затем пере¬
мешивают и проводят спектрофотометрические измерения при 438 нм.
• Процедура приготовления калибровочных стандартных растворов: вносят 0, 1,2,
3, 4, 5 мл базового раствора с концентрацией 500 мг (SO42")/0i в мерные колбы на
50 мл. Разбавляют приблизительно до 30 мл, добавляют 4 мл смеси уксусной кис¬
лоты и фосфорной кислоты. Гомогенизируют и следуют процедуре, предписанной
для анализа проб. Строят калибровочный график зависимости поглощения от кон¬
центрации раствора для расчета результатов.
Примечание
Органическое вещество может создавать помехи за счет осаждения или окрашивания
реакционной среды.
30.2.22. Анализ ярозита
Введение
В почвах, содержащих ярозит, образовавшийся под влиянием биотических и абиотиче¬
ских явлений, процесс разложения состоит из двух фаз, затрагивающих формы серы,
железа, калия или натрия.
В фазе восстановления активность сульфатредуцирующих бактерий в гидроморфной
(иногда в галоморфной) среде делает возможным восстановление сульфата и органиче¬
ской серы до состояния гидросульфида HS-. В то же время происходит восстановление
иона железа (III) до железа (II).
ЧП2- —ня- —w АмппгЬимй РрЯ Ярозит и натроярозит + H2S04
В фазе окисления, наступающей из-за естественного или искусственного падения
уровня подземных вод, богатые S осадочные отложения оказываются в аэробной среде.
Пириты более или менее быстро окисляются в зависимости от степени усыхания почвы
с образованием трещин и повышением скорости дренирования. pH почвы может сни¬
зиться до pH 3,5, pH 3 и даже ниже (Le Brusq и др., 1987). Если почва содержит карбонат
кальция, снижение уровня pH будет меньше. Карбонат разлагается с образованием
диоксида углерода и растворимого гидрокарбоната, который можно выделить из смеси.
Одновременно образуется гипс.
Ярозит (KFe3(0H)6(S04)2) образуется одновременно с серной кислотой, которая мо¬
жет придать осушенной почве очень высокую кислотность (серные горизонты, sulfaquept,
sulfohemist). Некоторые кристаллические структуры глины могут стать нестабильными.
Может появиться алюминиевая кислотность.
Источником калия, необходимым для формирования ярозита в этой среде, яв¬
ляется разложение глауконита. В морской среде может встречаться натроярозит
(NaFe3(0H)6(S04)2). Присутствие иона аммония затрудняет образование этих соедине¬
ний. Ярозит метастабилен в почве при pH < 4,5 и может подвергаться гидролизу с обра¬
зованием гетита-FeCHOH), что приводит к появлению пятен, окрашенных в цвет ржав¬
чины, в верхней части почвенных профилей.
Пшс CaS04 • 2Н20
Восстановительная среда (Disulfovibrio)
Окислительная среда (Thiobacillus)
(van Вгеетеп и Harmsen, 1973-1975)
710
Часть 3. Неорганический анализ
Ярозит и натроярозит вызывают появление желтых выцветов в почве при высушива¬
нии или около корней растений. Были описаны серии химических реакций окисления
пирита в этих почвах (Chamayou и Legros, 1989). Один моль пирита образует один моль
Н+, когда все железо окисляется до состояния Fe (III):
FeS2 + 15/402 + i/2ho <=> Fe3+ + 2S02" + Н+.
При недостаточном насыщении реакционной среды кислородом железо остается
в форме Fe (И) при высокой кислотности среды:
FeS2 + 7/202 + Н20 <=> Fe2+ + 2SOJ- + 2Н+ .
Кислотность также высока, если железо превращается в гидроксид железа (III).
В сильно увлажненных и хорошо аэрируемых почвах pH почвы может достигать 2.
FeS2 + 15/402 + 7/2н20 <=> Fe(OH)3 + 2S02~ + 4Н+ .
FeS2 + 15/402 + 5/гН20 + 1/3К+ о >/3KFe3(0H)6(S04)2 +V3SO*- + ЗН+
Ярозит
Анализы
Желтые выцветы на поверхности почвы или около корней растений можно собрать
и хранить в герметически закупоренных флаконах после быстрого высушивания на воз¬
духе. Содержание вещества составляет около 80%.
Дифрактограммы можно получить после насыщения катионами магния или ка¬
лия, обработки глицерином и нагревания до 300 и 550 °С (см. гл. 4). Измерение
межплоскостных расстояний позволяет проводить идентификацию:
К-ярозита —3,08—3,11—2,29 А (излучение меди, по порошку).
Na-ярозита — 5,06-3,06-3,12 А (излучение меди, по порошку).
При ДТА в атмосфере азота (см. гл. 7) наблюдают экзотермический эффект око¬
ло 500 °С, соответствующий разложению KFe3(S04)2(0H)6 с образованием KjSO^
Fe2(S04)3. Второй эффект около 800 °С соответствует разложению Fe2(S04)3. Кроме того,
наблюдается эндотермический эффект при 416 °С.
При сканирующей электронной микроскопии (см. гл. 8) обнаруживают кубические
частицы с маленькими октаэдрическими гранями. Измерение pH воды (1:2,5) сразу же
после смешивания воды и почвы и далее через 24 ч позволяет выявить кислотность, выз¬
ванную гидролизом ярозита.
Основные положения анализа
После определения количества сульфатов, растворимых в Ыа3-ЭДТА (см. раздел 30.2.21),
твердый остаток пробы почвы используют для количественного анализа ярозита.
Ярозит растворяют в горячей хлористоводородной кислоте, которая не взаимодействует
с пиритом. Содержание экстрагированного сульфата измеряют турбидиметрическим
методом с применением бария.
Оборудование
• Лабораторная стеклянная посуда.
• Термостатируемая водяная баня.
• Спектрофотометр.
• Центрифуга.
Глава 30. Сера
711
Реагенты
• 37%-ная хлористоводородная кислота (НС1).
• 4 Мраствор НС1: работая под вытяжкой, в мерный цилиндр объемом 1 л вносят
около 500 мл деионизированной воды, затем добавляют 320 мл 37%-ной хлорис¬
товодородной кислоты; охлаждают, доводят объем до 1000 мл деионизированной
водой и гомогенизируют.
• См. также реагенты для анализа с №3-ЭДТА («Реагенты» в разделе 30.2.21).
Методика
• После анализа сульфатов, растворимых в Ыа3-ЭДТА, дважды промывают остаток
центрифугированием в течение 10 мин при 5000gc 10 мл деионизированной воды.
Сливают промывную воду.
• Переносят остаток в химический стакан на 50 мл и добавляют 10 мл 4 М раствора
НС1. Стакан накрывают часовым стеклом и помещают в кипящую водяную баню
на 2 ч. Охлаждают, переносят в пробирку для центрифугирования и центрифуги¬
руют в течение 10 мин при 5000 g. Декантируют надосадочный раствор в мерную
колбу на 50 мл, доводят объем до метки деионизированной водой и гомогенизи¬
руют. Анализируют сульфат в этом растворе турбидиметрическим методом в соот¬
ветствии с описанием «Анализ», представленным выше в разделе 30.2.21. Остаток
почвы можно использовать для анализа пирита (см. раздел 30.2.18).
30.2.Z3. Последовательный анализ форм S
Аналитические задачи и состояние окружающей среды определяют выбор методик,
описание которых представлено выше. Эти анализы можно проводить последователь¬
но, в зависимости от показателей растворимости и порядка выделения форм S. Метод,
представленный на рис. 30.6, рекомендован для анализа кислых сульфатных почв Де¬
партаментом почвоведения и геологии сельскохозяйственного университета Вагенин-
гена (Виигтап и др., 1996).
ИХ является приемлемым методом анализа сульфат-анионов, однако жидкий
носитель может вступать в реакции с некоторыми формами S. Например, H2S и H2S203
не стабильны в кислой среде; полисульфиды являются источниками элементарной серы
или сероводорода.
Оригинальный химический подход был предложен для анализа неорганических форм
S в зонах керогена (Sonne и Dasgupta, 1991). Одновременный анализ сульфида, полисуль¬
фидов, сульфита, тиосульфата и сульфата проводится методом автоматизированного
проточно-инжекционного анализа в непрерывном потоке (ПИА) для водных экстрак¬
тов почвы и соленой воды при турбидиметрическом или колориметрическом детекти¬
ровании. Комплексный манифольд позволяет проводить одновременные измерения
содержания:
• сульфидов и полисульфидов путем умеренного подкисления среды и вытеснения
серы; турбидиметрический анализ коллоидной серы позволяет оценивать количе¬
ство полисульфидов; колориметрический анализ с применением пентацианони-
троферрата (II) в щелочной среде позволяет оценить количество образовавшегося
сероводорода (сульфит не создает помех при измерениях);
• сульфитов путем вытеснения диоксида серы; измерения проводятся колориметри¬
ческим методом, специфичным по отношению к сере в валентном состоянии S™
(обесцвечивание трифенилметана в нейтральном растворе);
Четыре 250 мг 100 мг 250 мг 200 мг
пробы
из образца 0 0 (3)0
Экстракция
раствором ЭДТА
pH 8,0
Раствор Твердая фаза
Гидролиз
в присутствии
4MHCI
Неокислительная
минерализация
Фтористоводородная + серная кислота
Раствор Твердая фаза
Окисление
азотной
кислотой
Экстракция
ацетоном
Раствор
Твердая
фаза
I
Отбросить
Окислительное плавление
с карбонатом натрия
и нитратом калия
при 700 °С
Растворение Твердая
в растворе фаза
2 МНС |
Отбросить
Раствор Твердая
| фаза
Расщепление Расщепление
царской царской
водкой водкой
II "
Турбидимет- Турбидимет- ААС
рический рический анализ
анализ анализ ионов
сульфатов сульфатов железа (III)
и железа (II)
Раствор
Твердая
фаза
▼
ААС
анализ
железа (III)
(пирит)
Турбидиметри-
ческий анализ
сульфатов
Пирит + органика +
+ элементарная S
Турбидиметри-
ческий
анализ
элементарной
S
ir
Анализ
сульфата
VI
s
С S
■ч
Общее
железо
Железо (II)
Железо (III)
(кроме пирита)
—>
—► (пирит)
(- элементарная S)
▼ у
Экстрагируемые + арозит
сульфаты*
У v
+ S (пирит) + Органическая S + Элементарная S
(по разности)
Общая S
Рис. 30.6. Последовательный анализ форм серы в кислых сульфатных почвах (*гипс, малорастворимые или экстрагируемые сульфаты)
Часть 3. Неорганический анализ
Глава 30. Сера
713
• тиосульфатов по обесцвечиванию раствора перманганата калия в слабокислой сре¬
де после осаждения сульфида и полисульфида и образования комплекса сульфита
с гидроксиметилсульфонатом;
• сульфатов с помощью турбидиметрического анализа после осаждения хлоридом
бария; соосаждение полисульфида компенсируют дифференциальным измерени¬
ем поглощения до и после добавления хлорида бария.
30.3. Сера в гипсоносных почвах
30.3.1. Гипсоносные почвы
В мангровых почвах сульфат кальция может образовываться из исходных наносов или
в результате превращений различных соединений серы в процессе образования почвы
в этой специфической среде. Однако количество сульфата кальция, в общем, невелико;
он определяется одновременно с нерастворимыми сульфатами типа ярозита (см. разде¬
лы 30.2.20-30.2.22).
Настоящие гипсоносные почвы (гипсосоли, гипсисоли) характерны для пустынных
и полупустынных зон и часто встречаются в Северной Африке и на Ближнем Востоке.
Основным источником гипса являются эвапоритовые отложения. В этих почвах проис¬
ходит перераспределение сульфата кальция при переносе его водой. Кроме того, вновь
образовавшийся гипс может накапливаться глубоко в некоторых видах почв в часто за¬
топляемых зонах; процесс связан с генезисом гидроморфных или засоленных почв.
Формы сульфата кальцир, обнаруживаемые в этих почвах, относятся к трем основ¬
ным химическим компонентам, различающихся степенью дегидратации соли:
• гипс, CaS04 • 2Н20, представляющий наиболее стабильную фазу;
• бассанит, CaS04 • 0,5Н2О, полугидратная фаза;
• ангидрит, CaS04, не содержащий молекул воды.
Растворимость гипса в чистой воде равняется 2,6 г/л при 25 °С, но на его раствори¬
мость сильно влияет температура и присутствие других солей. Такие соли, как сульфат
натрия или карбонат кальция, снижают растворимость сульфата кальция, в то же время
такие соли, как хлорид натрия, значительно повышают его растворимость. Разные по¬
казатели растворимости, связанные с взаимодействием солей, представляют серьезные
проблемы при экстракции и количественном анализе.
С точки зрения количественных измерений, содержание гипса, определенное
в почве, может быть очень высоким (>50%), несмотря на то что его аккумуляции явно
не видны (например, корка, крустифицированные образования, кристаллы «розы пу¬
стыни», кластерные конкреции, псевдомицелии). Множество микроскопических форм
(Pouget, 1995) рассеяно в почвенной массе.
30.3.2. Предварительные анализы
Кристаллизационная вода
Основные положения
Пробы почвы, содержащей гипс, из-за низкой растворимости сульфата кальция по
сравнению с другими растворимыми солями, экстрагированными водой, необходимо
подвергнуть особой обработке (см. гл. 30). При отсутствии визуального подтверждения
присутствия гипса рекомендуется провести предварительный анализ для выбора наибо¬
лее подходящего отношения почва: вода с целью более полной экстракции пробы.
714
Часть 3. Неорганический анализ
Метод основан на измерении потери веса пробы гипсосодержащих образцов при вы¬
сушивании в сушильном шкафу до постоянного веса в сравнении с потерей веса при
выдерживании над фосфорным ангидридом.
Этот анализ позволяет оценить содержание гипса с разумной точностью. Анализ вы¬
полняется довольно быстро и может быть использован для серийных измерений. Луч¬
шие результаты получают при анализе проб из гипсовых или петрогипсовых горизонтов
(которые могут содержать 60-85% гипса).
Методика
• Навеску 5 г воздушно-сухой почвы помещают в эксикатор с фосфорным ангидри¬
дом или силикагелем и оставляют на 48 ч.
• Взвешивают на лабораторных весах (±0,1 мг); помещают пробу в сушильный шкаф
при 125 °С (или 150 °С) на 24 ч; переносят в эксикатор, охлаждают и взвешивают.
Рассчитывают потерю веса. Второй результат соответствует влажности почвы. Третий
результат — потере кристаллизационной воды, содержащейся в гипсе.
Примечание
В процессе ДТА при низких температурах наблюдают двойной эндотермический эф¬
фект, соответствующий потере 1,5 моль Н20 с образованием полугидрита и далее, потере
0,5 моль Н20 с образованием ангидрита:
CaS04 • 2Н20 CaS04 • 0,5Н2О —
Гипс Бассанит
Лолугидрат
-£yCaS04-
Ангидрит
растворимый
;600
£ р CaS04
«1100°С
>СаО + SO,T
Ангидрит
нерастворимый
Помехи минимальны, если температуру поддерживают ниже (1) точки разложения
органических молекулярных структур и (2) точек дегидратации глин и оксидов (напри¬
мер, гиббсита). При 150 °С результаты оказываются несколько завышенными.
Рентгенодифракционный анализ (РДА)
Рефлексы рентгеновской дифракции при 7,56; 3,059; 4,27,
ИК-анализ
На рис. 30.7 представлены характеристические спектры гипсосодержащих материалов.
Проба с ацетоном
Этот метод основан на различиях в растворимости. К определенному объему
разбавленного водного экстракта (1:2 или 1:5) добавляют равный объем ацетона (СН3-
СО—СН3). На наличие гипса в пробе указывает немедленное образование кремово¬
го осадка (Richards, 1954), количество которого зависит от содержания растворенного
сульфата кальция, оседающего в ацетоне. Этот метод можно также использовать для
количественного анализа содержания гипса (см. раздел 20.3.4).
30.3.3. Экстракция и количественный анализ при многоступенчатой
экстракции
Основные положения
Весь гипс, содержащийся в пробе гипсоносной почвы, не растворяется при простой
экстракции. Для полной экстракции гипса необходимо несколько экстракционных про-
Глава 30, Сера
715
CaS04
Ангидрит
CaS04:0,5Н2О
Бассанит
CaS04: 2I-LO
Гипс 4 2
Рис. 30.7. ИК-спектры гипсосодержащих материалов (таблетированный КВг); слева - валентные
колебания связи в ОН-зоне: молекулы воды находятся в эквивалентных позициях кристаллической
решетки, каждая молекула образует две водородные связи с атомом кислорода сульфат-иона.
цессов с применением все более разбавленных растворов. Высушенные на воздухе про¬
бы используются чаще, чем пробы, высушенные в сушильном шкафу. Рекомендуется
(Loeppert и Suarez, 1996) последовательное использование трех экстрактов: насыщенно¬
го, 1:4 (почва: вода) и 1:40 (гипс — наименее растворимый материал из всех сульфатиро-
ванных минералов, встречающихся в почвах в значительном количестве). Содержание
иона кальция измеряют с помощью атомно-адсорбционной спектрометрии (ААС), а со¬
держание сульфат-аниона - гравиметрическим методом после осаждения барием.
Экстракция и анализ
Подготавливают пробу: 100 г воздушно-сухой почвы измельчают до размера частиц
200 мкм и гомогенизируют. Отделяют три части по 25 г каждая:
• первую часть используют для приготовления насыщенного экстракта (см. гл. 18);
• вторую часть весом 25 г экстрагируют 100 мл деионизированной воды;
• третью часть экстрагируют 1 л деионизированной воды.
Встряхивают две последние суспензии почвы в воде в течение ночи, затем фильтру¬
ют. Измеряют содержание кальция методом ААС при 422,7 нм. Сульфат анализируют
взвешиванием осадка сульфата бария, образовавшегося при добавлении хлорида бария.
Расчеты
Молярная концентрация гипса приравнивается к концентрации ионов кальция
и сульфат-ионов. Причиной избытка кальция может быть высвобождение кальция из
обменных комплексов, поэтому лучше пользоваться концентрацией сульфата. Если Vs
и VI (мл) — объемы насыщенного и разбавленного экстрактов, а Г8 s0 и 7^ so — соот¬
ветствующие концентрации сульфатов в экстрактах в М, количество экстрагированного
гипса в миллимолях рассчитывают по формуле:
япшс “ Тх^ VI— Ts so( Vs,
а концентрация гипса в почве в г/кг равняется:
С = 0,172 х я х 1000/25 = 6,88л.
ГИПС 7 ГИПС ' 7 гипс
716
Часть 3. Неорганический анализ
Примечания
Если почву нагревают до 105 °С в течение ночи перед измельчением, гипс превращается
в бассанит (Rivera и др., 1982), у которого растворимость выше и он переходит в раствор
быстрее, чем гипс, что приводит к более полному растворению пробы.
Малорастворимые сульфаты (сульфаты щелочноземельных металлов), а также
сульфаты свинца, хрома, железа, ртути и висмута не вызывают существенных помех. Тем
не менее, при необходимости эти элементы могут быть определены в экстракте методом
ААС.
30.3.4. Анализ путем осаждения ацетоном
Основные положения
Пробу на присутствие гипса (см. «Проба с ацетоном») можно использовать для коли¬
чественного анализа (Вииппап и др., 1996). Содержащийся в почве гипс растворяется
в воде в разной степени, в зависимости от количества гипса в пробе. Осадок гипса
отделяют, затем вновь растворяют в воде. Содержание кальция в конечном растворе
определяют методом ААС.
Реагенты
• Контрольный образец чистого гипса (CaS04 • 2Н20).
• 37%-ная хлористоводородная кислота (НС1).
• Раствор хлорида бария: 60 г ВаС12 * Н20 растворяют в 25 мл воды.
• Ацетон (СН3СОСН3).
• 2%-ный раствор оксида лантана, Ьа2Ог: смешивают 100 мл 5%-ного лантансодержа¬
щего раствора, 50 мл 1 М раствора хлористоводородной кислоты и доводят объем
до 250 мл деионизированной водой.
• Калибровочные растворы, точно отмеряют 5 мл стандартного коммерческого рас¬
твора кальция с концентрацией 1000 мг (Са)/л в мерную колбу на 100 мл, доводят
объем до 100 мл деионизированной водой, получая концентрацию базового рас¬
твора в 50 мг (Са)/л; последовательно добавляют 0 (холостая проба), 5, 10, 20 мл
базового раствора в мерные колбы на 50 мл; добавляют 10 мл 5%-ного лантансо¬
держащего раствора, доводят объем до 50 мл; получают растворы калибровочного
диапазона концентраций 0, 5, 10, 20 мг (Са)/л.
Методика
• Навеску 10 г образца почвы (размер частиц 200 мкм) помещают в мерную колбу
объемом 250 мл. Добавляют 100 мл деионизированной воды, встряхивают в тече¬
ние ночи.
• Вносят 35 мл взвеси в пробирку для центрифугирования вместимостью 50 мл, из¬
готовленную из прозрачного стекла, и центрифугируют в течение 15 мин при 3500 g
(раствор А).
• Проверка присутствия (качественный анализ) сульфата кальция. Отмеряют 3 мл
надосадочного раствора и добавляют десять капель 1 М раствора НС1 и 2 мл рас¬
твора ВаС12. Если раствор сохраняет прозрачность, гипс в почве отсутствует; если
образуется осадок, продолжают анализ.
• К 20 мл надосадочного раствора А добавляют 20 мл ацетона, гомогенизируют и вы¬
держивают в течение 10 мин. Центрифугируют 10 мин при 2500 g и сливают над-
осадочную жидкость.
Глава 30. Сера
717
• Сушат пробирку для центрифугирования в вентилируемом сушильном шкафу при
50 °С. Охлаждают, затем добавляют 40 мл деионизированной воды для солюбили¬
зации осадка. К 5 мл добавляют 5 мл 2%-ного раствора лантана и гомогенизируют.
• Анализируют кальций методом ААС при 422,7 нм после разбавления 1%-ным рас¬
твором лантана при необходимости.
Расчеты
Си В — концентрации кальция в экстракте и холостой пробе соответственно; F— объем
воды, использованной для экстракции (100 мл); D — коэффициент разбавления (D = 4
в большинстве случаев: 20 мл аликвоты водного экстракта в 40 мл воды, 5 мл пробы,
смешанной с 5 мл раствора лантана);/ — поправочный коэффициент на влажность; Р—
вес пробы почвы; содержание гипса в мг/г почвы рассчитывают по формуле:
G = (C~B)VD'f A/CaS042H20 о 172(С- В)/.
1000/> Ма ”
Примечание
Если содержание гипса в почве превышает 2%, для экстракции следует изменить отно¬
шение почва: раствор в сторону большего разбавления.
30.4. Потребность почвы в сере и гипсе
30.4.1. Введение
Следует четко различать методы, направленные на выявление (1) потребности и де¬
фицита соединений S, являющейся элементом, необходимым для роста растений и (2)
потребности в гипсе, который вносят в засоленные почвы, чтобы сделать возможной их
культивацию.
S играет очень важную роль в обеспечении плодородности почвы. Ее дефицит при¬
водит к серьезным нарушениям развития растений, таким как карликовость, дефекты
листьев, уменьшение фотосинтетической активности и снижение плодоношения.
Во влажных и полувлажных зонах большая часть S встречается в органических фор¬
мах. Роль многих органических соединений в питании растений до сих пор не вполне
ясна. Биохимический синтез необходимых для жизнедеятельности компонентов, на¬
пример, некоторых протеолитических ферментов, аминокислот и белков, гормонов или
гликозидов может происходить только в присутствии S в реакционной среде.
Высокое содержание общей S в почве не несет информации о количестве каждой от¬
дельной формы S и не всегда свидетельствует о том, что почва содержит S в количестве,
достаточном для питания растений. Общее содержание S может быть низким, напр.,
около 0,05% в торфяниках и почвах засушливых зон.
Простые соотношения между содержанием С, N и S можно рассчитать для описа¬
ния связей между этими тремя элементами в биогеохимическом цикле. Параметр «от¬
ношение C:N» (см. введение к гл. 10) широко используется в почвоведении. Кроме
того, полезную информацию представляют отношения C:S и N:S. Низкий уровень N.S
указывает на высокое содержание небелковой S. Считается, что растворимые сульфаты
наиболее репрезентативно отражают способность почвы к обеспечению потребности
растений в питании. Однако возможность почв накапливать эти формы определяется
многими факторами, такими как (1) природа и свойства почвенной матрицы (например,
pH, ЕКО, содержание глины), (2) наличие растительных соединений и S-содержащих
718
Часть 3. Неорганический анализ
растительных остатков, (3) климатические ограничения (в особенности температура
и влажность), регулирующие активность микроорганизмов, и (4) изменения в аэробной
или анаэробной среде.
Сера участвует в годичном цикле, который, в контрастных климатических условиях
включает в себя сильные колебания, например, малая активность наблюдается при тем¬
пературе ниже 10 °С и высокий уровень выщелачивания — в период дождей.
30.4.2. Потребность растений в сере
Подготовка почвы не должна значительно изменять распределение различных форм
S. Тем не менее, простая сушка воздухом может привести к повышению содержания
доступных сульфатов.
Кроме того, содержание S варьирует в зависимости от пространственного распре¬
деления (вертикального в почвенном профиле или горизонтального на поверхности
возделываемой почвы) и временного распределения (сезон пробоотбора, длительность
хранения пробы и продолжительность контакта с воздухом). Поэтому зачастую интер¬
претация результатов является затруднительной.
Микробиологические методики
Данные подходы основаны на моделировании микробиологических и биохимических
процессов (аналогичных процессам, описанным Неубауэром), протекающих в почве
в естественных условиях. Как правило, для оптимизации микробной активности про¬
бы почвы подвергают инкубации при постоянной температуре, зависящей от типа сред
и климатических зон существования почвы.
Содержание образовавшихся растворимых сульфатов измеряют по прошествии опре¬
деленного времени инкубации. Оценку потребности в S для данной культуры следует
связать с (1) с собираемыми урожаями в сравнении с мировыми данными и (2) по воз¬
можности, с результатами анализа состояния растений (состояния листьев), что позво¬
лило бы восполнить возникший дефицит.
Микробиологические методики требуют особого внимания при выборе:
• репрезентативных проб почв и растений;
• даты пробоотбора, позволяющей оценить потребность растений на различных эта¬
пах цикла вегетации (посев, развитие корневой системы и рост, цветение и плодо¬
ношение) и микробиологическую активность (зависящую от влажности и темпера¬
туры), связанную с минерализацией и иммобилизацией S.
Следует принимать во внимание и другие питательные вещества, необходимые расте¬
ниям, в особенности азот. Эти анализы позволяют установить масштабы необходимой
коррекции с помощью простых, быстрых и воспроизводимых методик, включающих
экстракцию химическими реагентами.
Химическая экстракция
Чтобы перевести в раствор различные формы S, необходимо использовать селективные
экстрагирующие реагенты, однако на практике экстрагенты редко бывают селективны¬
ми; в зависимости от применяемого реагента возможно переведение в раствор различ¬
ных количеств адсорбированной S в форме SOили гидролизованной органической
серы. Нерастворимые сульфаты (такие как сульфат бария или смешанный сульфат алю¬
миния и железа) количественно не определяют. Элементарную серу необходимо удалить
из раствора.
Глава 30. Сера
719
В простейшем анализе применяется водная экстракция, однако она зачастую приво¬
дит к дисперсии почвы.
Экстракция разбавленными солевыми растворами (такими как 0,01 М СаС12, MgCl2,
0,15%-ным LiCl) ограничивает дисперсию, однако при этом происходит экстракция не¬
которого количества адсорбированной S. Кроме того, литий действует как ингибитор
микробиологической активности.
Экстракция в буферной среде (0,25 М уксусная кислота + 0,5 М ацетат аммония) ог¬
раничивает гидролиз органических серосодержащих соединений.
Экстракция щелочным реагентом Ольсена (0,5 М NaHC03 с pH 8,5) более пригодна
для почв с pH > 7, однако гидрокарбонат натрия может солюбилизировать небольшое
количество органической S и вызвать засорение пор фильтра, что затрудняет разделение
твердой и жидкой фазы.
При экстракции растворами фосфатов (0,01 М КН2Р04 или Са(Н2Р04)2) фосфат-ани¬
он замещает сульфат-анион в центрах адсорбции, увеличивая таким образом отрица¬
тельный поверхностный заряд. Некоторое количество лабильной S, кратковременно
доступной, можно экстрагировать с помощью солей фосфорной кислоты благодаря
элюирующей способности P-содержащих анионов. Ряд анионов, расположенных в по¬
рядке снижения элюирующей способности, выглядит следующим образом:
гидроксилы > фосфаты > сульфаты = ацетаты > нитраты = хлориды.
Количественные измерения проводятся после экстракции с помощью методов, опи¬
санных в предыдущих разделах: колориметрия (раздел 30.2.7 и 30.2.8), турбидиметрия
(«Анализ» в разделе 30.2.21) или ААС (раздел 30.2.13).
Примечание
Такие почвы, как андосоли, с высоким содержанием соединений с упорядочением
ближнего порядка (например, аллофаны) способны сильно фиксировать S в форме SO2-
за счет абсорбции.
30.4.3. Потребность в гипсе
Введение
Солонцовые почвы (содисоли), структура которых нарушена избытком обменного на¬
трия (Na/T > 15%), можно восстановить внесением добавок гипса (или непосредственно
серной кислоты в случае карбонатных почв). При попадании дождевой или
ирригационной воды натрий, фиксированный обменным комплексом (см. гл. 22), по¬
степенно замещается кальцием, содержащимся в гипсе, при этом структура и гидроди¬
намические характеристики почвы постепенно улучшаются. Образовавшийся сульфат
натрия выводится из почвы путем выщелачивания:
Na2C03 + CaS04 о СаС03 + Na2S04
2 Глина-Na* + CaS04 -> (Глина)2-Са2+ + Na2S04 (дренажные воды)
Оборудование
• Лабораторная стеклянная посуда.
• Механический шейкер.
• Лабораторные весы.
• Атомно-адсорбционный спектрометр.
720
Часть 3. Неорганический анализ
Реагенты
• Сульфат кальция (CaS04 • 2Н20 (гипс)).
• Насыщенный раствор: 5 г CaS04 • 2Н20 в 1 л деионизированной воды перемешива¬
ют в течение 1 ч, фильтруют через аналитический фильтр (синяя лента) и измеряют
точную концентрацию кальция — Са2+ в мн. методом ААС.
Процедура
• Навеску 5 г воздушно-сухой почвы помещают в колбу из стекла Ругех объемом
250 мл.
• С помощью точного объемного дозатора добавляют ровно 100 мл насыщенного
раствора CaS04. Встряхивают в течение 30 мин на роторном шейкере.
• Фильтруют через аналитический фильтр (синяя лента) (без промывания).
• Измеряют содержание кальция методом ААС в аликвоте прозрачной фракции
фильтрата.
Расчет
Потребность в гипсе (Я) в мн. Са2+/кг (почвы):
Bg=(A-B)
1001000
5 1000
= 20 (А-В),
где А — концентрация исходного раствора; В — концентрация фильтрата в мн. Са2+.
Полученные результаты можно выразить в тоннах (гипса)/га как функцию глубины
обрабатываемой почвы. Например, для глубины 10 см (1500 тонн (почвы) Да при объем¬
ной плотности почвы « 1,5) потребность в гипсе в моль (0,5Са2+)/га составляет:
D,= 1500 Я,
g g’
т. е. в тоннах (гипса)/га:
/>=0,129Я.
g 7 g
Примечания
На практике при контакте твердая фаза — раствор в полевых условиях замещается мень¬
шее количество натрия на кальций, чем в лабораторных. Следовательно, количество
натрия, реально замещенного, меньше количества, рассчитанного по лабораторным ре¬
зультатам, и расчетное количество гипса будет завышено приблизительно на 25%.
Например, для обмена 10 ммоль (Ыа+)/кг (почвы) необходимо внести добавку 1300 кг
(гипса)/га. Как правило, количество гипса, необходимое для улучшения засоленных
почв, составляет 5—10 тонн (гипса)Да.
Используемая литература
Artiola FF and АН AMS (1990) Determination of total sulphur in soil and plant samples using sodium
bicarbonate/silveroxide, dry ashing and ion chromatography. Commun. Soil Sci. Plant Anal, 21,
941-949.
Aswa HA and Tabatabai MA (1993) Comparison of some methods for determination of sulfate in soils.
Commun Soil Sci. Plant Anal., 24, 1817-1832.
Begheijn L and Van Schuylenborgh J (1971) Methods for the analysis of soils used in the laboratory of
soil genesis of the Department of Regional Science, Wageningen.
721
Глава 30. Сера
Bremner JM and Tabatabai MA (1971) Use of automated combustion techniques for total carbon, total
nitrogen and total sulfur in soils. In Instrumental Methods for Analysis of Soils and Plant Tissue,
Walsh L.M. ed., SSSA, 1-15.
Buurman P, Van Lagen В and Velthorst EJ (1996) Manual for Soil and Water analysis., Backhuys, Leiden,
The Netherlands, 314 p.
Chamayou H and Legros JP (1989) Les bases physiques et mineralogiques de la Science du Sol., Presses
Universitaires de France, 485-486.
Chariot G and Bezier D (1955) Analyse quantitative miner ale, Masson, Paris.
Colovos G, Panesar MR and Parry EP (1976) Lime arizing the calibration curve in determination of
sulfate by the methylthymol blue method. Anal Chem., 48,1693-1696.
Commission de pddologie et de cartographie des sols (CPCS), (1967). Classification des sols,, Lab. Geol.
Pedol., Ecole Nat. Sup. Agron., Grignon, France, 87 p.
David MB, Mitchell MJ, Aldcom D and Harrison RB (1989) Analysis of sulfur in soil, plant and sediment
materials. Sample handling and use of an automated analyser. Soil Biol Biochem., 21,119-123.
FAO, (1968) Definition of Soil Units for the Soil Map or the World, no. 33.
Freney JR, Melville GE and Williams CH (1970) The determination of carbon bounded sulphur in soils.
Soil Sci., 109,310-318.
G6nin JMR, Refait P, Bourrte G, Abdelmoula M and Trolard F (2001) Structure and stability of Fe (II) -
Fe (III) green rust “fougerite” mineral and its potential for reducing pollutants in soil solutions.
Appl. Geochem., 16, 559-570.
Gerzabek MH and Schaffer К (1986) Determination of total sulphur in soil. A comparison of methods.
Bodenkultur, 37, 1-6.
Gony J and Parent Ch (1966) Etude gdochimique d’une tranche de sediments fins actuels. Bull BRGM,
5, 28-31.
Johnson CM and Nishita H (1952) Microestimation of sulfur in plant materials, soils and irrigation waters.
Anal Chem,, 24,736-742. *
Karmarkar SV and Tabatabai MA (1992) Eluent composition effect on ion chromatographic determination
of oxyanions in solution equilibrated with soils. Chromatographia, 34, 643-648.
Kowalenko CG and Lowe LE (1972) Observations on the bismuth sulphide colorimetric procedure for
sulphate analysis in soil. Commun. Soil Plant Anal, 3, 79-86.
Kowalenko CG (1985) A modified apparatus for quick and versatile sulphate sulphur analysis using
hydroiodic acid reduction. Commun. Soil Sci. Plant Anal., 16,289-300.
Le Brusq JY, Loyer JY, Mouguenot В and Cam M (1987) Nouvelles paragen^ses к sulfates d’aluminium,
de fer et de magnesium, et leur distribution dans les sols sulfates acides du S6n6gal. Sc. du Sol., 25,
173-184.
Loeppert RH and Suarez DL (1996) Carbonate and Gypsum. In Methods of Soils Analysis. Part 3,
Chemical Methods, Sparks DL et al. ed., SSSA book series no. 5, 437-474.
Marius C (1980) Les mangroves du Senegal Ecologie, pedologie et utilisation., IRD (ex. Orstom) 6d.,
Paris.
Marius C, Paycheng C and Lopez J (1976) La determination du soufre et de ses composes au laboratoire
Orstom de Dakar, S£n6gal. Documentation IRD, Dakar, Paris, 16 p.
McSwain MR, Watrous RJ and Douglas, JE (1974) Improved methyl thymol blue procedure for automated
sulfate determinations. Anal Chem., 46, 1329-1331.
Montoroi JP (1994) Dynamique de Veau et geochimie des sels d’un bassin versant amenage de Basse-
Casamance, S6n6gal. Th. Univ. Nancy I, 349 p.
Montoroi JP, Grunberger О and Nasri S (2002) Groundwater geochemistry of a small reservoir catchment
in central Tunisia. Appl Geochem., 17, 1047-1060.
Pansu M, Gautheyrou J and Loyer JY (2001) Soil Analysis - Sampling, Instrumentation and Quality
control., Balkema, Lisse, Abington, Exton, Tokyo, 489 p.
Perrott KW, Kerr BE, Kear MJ and Sutton MM (1991) Determination of total sulphur in soil using
inductively coupled plasma atomic emission spectrometry. Commun. Soil Sci. Plant Anal, 22,1477-
1487.
Pouget M (1995) Gypsosols. In Referentiel Pedologique, INRA, France, 161-165.
Richards LA (1954) Diagnosis and Improvement of Saline and Alkali Soils.,USD A Handbooks, 60, US
Gov. Print Office, 160 p.
722
Часть 3. Неорганический анализ
Rivera ED. Hallmark СТ, West LT and Drees LR (1982) A technique for rapid removal of gypsum from
soil samples. Soil Sci. Soc. Am. J., 46, 1338- 1340.
Sonne К and Dasgupta PK (1991) Simultaneous photometric flow injection determination of sulfide,
polysulfide, sulfite, thiosulfate and sulfate. Anal. Chem., 63, 427-432.
Tabatabai MA and Basta NT (1991) Ion chromatography. In Soil Analysis, Smith KA ed., Dekker, New
York, 229-259.
Tabatabai MA and Bremner JM (1970a) An alkaline oxidation method for determination of total sulfur in
soils. Soil Sci. Soc. Am. Proc., 34, 62-65.
Tabatabai MA and Bremner JM (1970b) Comparison of some methods for determination of total sulfur in
soils. Soil Sci. Soc. Am. Proc., 34, 417-420.
Tabatabai MA and Dick WA (1979) Ion chromatographic analysis of sulfate and nitrate in soils. In Ion
Chromatographic Analysis of Environmental Pollutants, Mulik JD and Sawickie ed., Ann. Arbor Sci.
Publ.,2, 361-370.
Tabatabai MA and Frankenberger WT Jr (1996) Liquid chromatography. In Methods of Soil Analysis,
Part III Chemical Methods, Sparks DL et al., SSSA Book, 5,225-245.
Tabatabai MA (1996) Sulfur. In Methods of Soil Analysis, Part 3, Chemical Methods, Bigham JM and
Bartels JM ed., ASA-SSSA Book, Serie no. 5, Madison, WI Etats-Unis, 921-960.
Tabatabai MA, Basta NT and Pirela HJ (1988) Determination of total sulphur in soils and plant materials
by ion chromatography. Commun. Soil Sci. Plant Anal., 19,1701-1714.
USDA, (1975) Soil Taxonomy. A Basic System of Soil Classification for Making and Interpreting Soil
Surveys., USDA, Agriculture Handbook no. 436, 754.
van Breemen N and Harmsen К (1975) Translocation of iron in acid sulfate soils. I - Soil morphology
and the chemistry and microbiology of iron in a chronosequence of acid sulfate soils. Soil Sci. Soc.
Am. Proc.. 39, 1140-1148.
Vieillefon J (1974) Les sols de mangroves et de tannes de Basse Casamance, Sirngal., IRD (ex. Orstom)
ed. Paris.
Дополнительная литература
Johnson CM and Ulrich A (1959) Analytical Methods for Use in Plant Analysis., California Agric. Exp.
Str. Bull, 766.
Gustafsson L (1960) Determination of ultra micro amounts of sulphate as methylene blue, lfcre partie:
Reaction. Talanta, 4,222-235.
Gustafsson L (1960) Determination of ultra micro amounts of sulphate as methylene blue. 2£me partie :
Reduction. Talanta, 4,236-243.
Kilmer VJ and Nearpass DC (1960) The determination of available sulfur in soils. Soil Sci. Soc. Proc.,
337-339.
Sherman GD, Schultz F and Alway FJ (1962) Dolomite in soils of the red rivervalley, Minnesota. Soil
Sci„ 94, 304-313.
Steinbergs A, Iismaa O, Freney JR and Barrow NJ (1962) Determination of total sulphur in soil and plant
material. Anal. Chim. Acta, 27, 158-164.
Bardsley CE and Lancaster JO (1965) Sulfur. In Methods of Soil Analysis, American Society of
Agronomy, 1102-1116.
Dean GA (1966) A simple colorimetric finish for the Johnson-Nishita micro-distillation of sulphur.
Analyst, 91, 530-532.
Beaton JD. Bums GR and Platou J (1968) Determination of Sulphur in Soils and Plant Material, Sulphur
Institute (Washington), Technical Bulletin no. 14.
Khan SU and Webster GR (1968) Determination of gypsum in solonetz soils by an XR technique. Analyst,
93, 400-402.
Lowe LE (1969) Sulfur fraction of selected alberta profiles of the gleysolic order. Can. J. Soil Sci., 49,
375-381.
Tabatabai M and Bremner JM (1970) An alkaline oxydation method for determination of total sulphur in
soils. Soil Sci. Soc. Am. Proc., 34, 62-65.
Wakayama FJ (1971) Calcium complexing and the enhanced solubility of gypsum in concentrated
sodium-salt solutions. Soil Sci. Am. Proc., 35, 881-883.
Глава 30, Сера
723
Hesse PR (1972) A Textbook of Soil Chemical Analysis., Chemical Publishing Co., 520 p.
Darmody RG, Fanning DJ, Drummond WJ Jr and Foss J. (1977) Determination of total sulfur in tidal
marsh soils by X-Ray spectroscopy. Soil Sci. Soc. Am. J., 41, 761-765.
Nor YM and Tabatabai MA (1977) Oxidation of elemental sulfur in soils. Soil Sci. Soc. Am. J., 41, 736-
741.
Begheijn LTh, Van Breemen N and Velthorst EJ (1978) Analysis of sulphur compounds in acid sulphate
soils and other recent marine soil. Commun. Soil Sci. Plant Anal., 9, 873-882.
Nelson RE, Klameth LC and Nettleton WD (1978) Determining soil gypsum content and expressing
properties of gypsiferous soils. Soil Sci. Soc. Am. J., 42,659-661.
Cronan CS (1979) Determination of sulfate in organically colored water samples.4w<z/. Chem., 51, 1333—
1335.
Siemer DD (1980) Reduction-distillation method for sulfate determination. Anal. Chem., 52, 1271—
1274.
Rivera ED, Hallmark CT, West LT, Drees LR (1982) A technique for rapide removal of gypsum from soil
samples. Soil Sci. Soc. Am. J., 46, 1338- 1340.
Freney JR, Jacq VA and Baldensberger JF (1982) The significance of the biological sulfur cycle in rice
production. In Microbiology of Tropical Soils and Plant Productivity, Dommergues YR and Diem
AG ed. Martinus Wijhoff, 10,271-317.
Adams TMCM and Lane PW (1984) A comparison of four methods of analysing aqueous soil extracts for
sulphate. J. Sci. Food Agric., 35, 740-744.
Lebel A and Teh Fu Yen, (1984) Ion chromatography for determination of metabolic pattern of sulfate-
reducing bacteria. Anal. Chem., 56,807- 808.
Wainwright M (1984) Sulfur oxidation in soils. In Advances in Agronomy, Academic, New York, 37,
350-396.
Lee R, Blakemore LC, Daly BK, Gibson EJ, Speirt W and Orchard VA (1985) Sulphur supply to ryegrass
during a pot trial and corrfelations with soil biological activity: The influence of two different methods
of deter-mining the adsorbed sulphate status of soils. Commun. Soil Sci. Plant Anal., 16, 97-117.
Kowalenko CG (1985) A modified apparatus for quick and versatile sulphate sulphur analysis hydroiodic
acid reduction. Commun. Soil Sci. Plant Anal., 16, 284—300.
Scott NM (1985) Sulphur in soils and plants. In Soil Organic Matter and Biological Activity, Vaughan D
and Malcolm RE ed., Martinus Njhoff/Junk, 379-401.
Gimeno Adelantado JV and Bosch Reig F (1986) Mineralization of some organic sulphur compounds by
fusion with molteno alkali. Talanta, 33, 757- 759.
Keller LP, Me Carthy GF and Richardson JL (1986) Mineralogy and stability of soil evaporites in North-
Dakota. Soil Sci. Soc. Am. J., 50,1069-1071.
Krupa SV and Tabatabai MA (1986) Measurement of sulfur in the atmosphere and in natural waters. In
Sulfur in Agriculture, Tabatabai MA ed. ASA, CSS A, Agron. Monogr. 27,491-548.
Bansal KN and Npal AR (1987) Evaluation of a soil test method and plant analysis for determining the
sulphur status of alluvial soil. Plant and Soil, 98, 331-336.
Vaugh CE, Junes MB and Center DM (1987) Sulfur tests on Northern California sub-clones annual grass
pasture surface soils. Soil Sci., 143,184-191.
Bolan NS, Syers JK, Tillman RW and Scotter DR (1988) Effect of liming and phosphate additions on
sulphate leaching in soils. J. Soils Sci., 39, 493-504.
David MB, Mitchell MJ, Aldcom D and Harrison RB (1989) Analysis of sulfur in soil, plant and sediment
materials: sample handling and use of an automated analyser. Biol. Biochem., 21, 119-123.
Sharp GS, Hoque S, Killham K, Sinclair AH and Chapman ST (1989) Comparison of methods to evaluate
the sulphur status of soils. Commun. Soil Sci. Plant Anal., 20, 1821-1832.
Hue NV, Fox RL and Wolt JD (1990) Sulfur status of volcanic ash-derived soils in Hawa’f. Commun. Soil
Sci. Plant Anal., 21, 299-310.
Hauge S and Maroy К (1991) Detection of sulphate by flamme emission spectrometry. Anal Chim. Acta.,
243,227-237.
Morante C (1991) Determination of plant sulphur and sulphate-sulphur by flow-injection analysis using a
two-live manifold. Anal. Chim. Acta., 249,47*M88.
Singh RP, Pambid ER and Abbas NM (1991) Determination of sulfate in deen subsurface waters by
suppressed ion chromatography. Anal. Chem., 63, 1897-1901.
724
Часть 3. Неорганический анализ
Tabatabai МА and Bremner JM (1991) Automated instruments for determination of total carbon nitrogen
and sulfur in soils by combustion technique. In Soil Analysis, Smith K.A. ed., Dekker, New York,
261-285.
Michel JP and Fairbridge RW (1992) Dictionnary of Earth-Sciences., Wiley, New York, 300 p.
Blanc GJ, Lefroy RDB, Chinoim N, Anderson GC and Barrow NJ (1993) Sulfur soil testing. Plant Soil,
383-386.
Boruah RK and Ghosh P (1993) Quantitative estimation of available sulphur in tea soils Two and a Bud,
40, 26-30.
Jansson H (1994) Sulphur status of soils - a global study Norwegian J. Agric. Sci., SN 15,27-30.
Tan Z, McLaren RG and Cameron КС (1994) Forms of sulfur extracted from soils after different methods
of sample preparation. Aust. J. Soil Res., 32, 823-834.
Trivedi BS, Garni RC and Patel KG (1994) Standardization of method for determining available sulphur
and its critical limit for lowland paddy. Gujarat Agric. Univ. Res. J., 20, 35-41.
Zhao F and McGrath SP (1994) Extractable sulphate and organic sulphur in soils and their availability to
plants. Plant Soil, 164, 243-250.
Santoso D, Lefroy RDB and Blair GJ (1995) A comparison of sulfur extractants of wealthered acid soils.
Aust. J. Soil Res., 33, 125-133.
Shaw Xiao-Quan and Chen Bin (1995) Determination of carbon-bonded sulfur in soils by hydroiodic acid
reduction and hydrogen penoxide oxidation. FreseniusJ. Anal. Chem., 351, 762-767.
Simo R and Grimalt JO (1996) Determination of volatile sulphur species in soil samples of interest for
prospecting for metal sulphide deposits. J. Chromatog., A, 726,161-166.
Zhao FJ, Loke SY, Crosland AR and McGrath SP (1996) Method to determine elemental sulphur in soils
applied to measure sulphur oxidation. Soil Biol. Biochem., 28, 1083-1087.
Prochnow LI, Boaretto AE and Vitti GC (1997) Ion-exchange resin to evaluate sulphur availability in
soils. Utilizacao da resina trocadora de ions para avaliacao do enxofre disponivel do solo. Revista
Brasileira de Ciencia do Solo, 21, 335-339.
Gowrisankar D and Shukla LM (1999) Evaluation of extractants for predicting availability of sulphur to
mustard in Inceptisols. Communi. Soil Sci. Plant Anal., 30,19-20,2643-2654; 33 ref.
Matula J (1999) Use of multinutrient soil tests for sulphur determination. Commun. Soil Sci. and Plant
Anal., 30, 1733-1746.
Zbiral J (1999) Comparison of some extraction methods for determination of sulphur in soils of the Czech
Republic. Porovnani vybranych extra-kcnich postupu pro stanoveni siry v pudach cr. Rostlinna
Vyroba, 45, 439-444.
Prietzel J and Hirsch C (2000) Ammonium fluoride extraction for determining inorganic sulphur in acid
forest soils. Eur. J. Soil Sci., 51, 323-333.
Crosland AR, Zhao FJ and McGrath SP (2001) Inter-laboratory comparison of sulphur and nitrogen
analysis in plants and soils. Commun. Soil Sci. Plant Anal, 32, 685-695.
Глава 31. Анализ экстрагируемых элементов
и полный элементный анализ
31.1. Элементы почвы
31.1.1. Основные элементы
Почвы содержат химические элементы, присутствующие в литосфере, т. е. стабильные
элементы периодической таблицы, в соответствии с геохимическим распределением
и происхождением почвы. В этой главе рассмотрены методы анализа только твердой
фазы. Самым распространенным элементом в почвах и минералах является кислород,
однако его количественный анализ, как правило, не проводят; приблизительное со¬
держание кислорода рассчитывают по количеству других основных элементов, участ¬
вующих в реакциях превращения элементов в оксиды (табл. 31.1). Углерод является
основным химическим элементом органического вещества и карбонатных минералов,
о них речь идет в части 2 и гл. 17, соответственно. Водород, другой важный элемент
минералов, воды и органического вещества, обычно анализируют в органической фор¬
ме (плюс в составе воды), если используется автоматический CHN-анализатор (см. гл.
10), в процессе термического анализа (см. гл. 7) или при изучении протонного обме¬
на (см. гл. 15 и 23). Азот — еще один из основных элементов биосферы и атмосферы;
анализ азота описан в гл. 10 (общий N), 14 (органический N) и 28 (неорганический N).
Силикатные минералы, формирующиеся из пород вулканического происхождения,
состоят, по большей части, из кислорода и основных элементов третьего и четвертого
периода периодической таблицы. Эти металлы могут образовывать основные оксиды;
наиболее выраженными основными свойствами обладают оксиды щелочных металлов
(натрия и калия); за ними следуют оксиды щелочноземельных металлов (магния и каль¬
ция) и, наконец, оксиды переходных металлов (железа, титана и марганца) и алюми¬
ния. Кроме того, силикатные минералы включают неметаллы (в частности, кремний
и фос^юр), образующие кислотные оксиды. Разделение магм на два типа основано на
содержании в них оксидов кремния. Один тип описывается как кислая гранитная магма
с высоким содержанием оксида кремния (>60%) и сравнительно высоким содержани¬
ем натрия и калия. Другой тип — основная базальтовая магма с содержанием кремния
ниже 50% и сравнительно высоким содержанием железа, магния и кальция. Более точ¬
ная классификация представлена в табл. 31.1.
Состав почвы (Greenlandи Hayes, 1983) изменяется в зависимости от происхождения
почвы, влияния различных погодных условий, а также деятельности человека. Основ¬
ные элементы вулканических пород обнаруживают в почвах в различных пропорциях.
Как показано в табл. 31.1, первым способом проверки точности анализа является сло¬
жение рассчитанных процентных долей содержания наиболее стабильных оксидов ос¬
новных элементов. Следует учесть влагу и органическое вещество, а в отдельных случаях
и прочие элементы, присутствующие в больших количествах. Сумма должна составить
около 100%.
31.1.2. Микроэлементы и примеси
Концентрации микроэлементов в почвах (Baize, 1997), так же как и содержание основ¬
ных элементов, зачастую связаны с концентрациями в подстилающей материнской
726 Часть 3. Неорганический анализ
Таблица 31.1. Усредненные элементные составы некоторых вулканических пород (по
данным Тигкеап и Wedepohl, 1961)
Элемент Ста^**^ь”ый к Процентная концентрация
ультраосновные базальтовые гранитные кислые
породы породы породы породы
SiOz< 45%
45% < SIO,<
<52%
52% < SI02 <
<68%
SI02>66%
El. Ox.
El. Ox.
El. Ox.
El. Ox.
Si
Si02
2,139
20,5
43,9
23,0
49,2
31,4
67,2
34,7
74,3
A1
A1203
1,889
2,0
3,8
7,8
14,7
8,2
15,5
7,2
13,6
Fe
Fe203“
1,430
9,4
13,4
8,6
12,3
3,0
4,3
1,4
2,0
Ca
CaO
1,399
2,5
3,5
7,6
10,6
2,5
3,5
0,5
0,7
Mg
MgO
1,658
20,4
33,8
4,6
7,6
0,9
1,5
0,2
0,3
Na
Na20
1,348
0,4
0,5
2,0
2,7
2,8
3,8
2,6
3,5
К
K20
1,205
0,004
0,005
0,8
1,0
2,5
3,0
4,2
5,1
Ti
ТЮ2
1,668
0,030
0,050
1.4
2,3
0,34
0,6
0,12
0,2
Mn
MnO
1,291
0,160
0,207
0,15
0,2
0,054
0,1
0,04
0,1
P
P2O5
2,291
0,022
0,050
0,11
0,3
0,092
0,2
0,06
0,1
Общий %
99,2
101,0
99,6
99,9
0
%
43,8
44,9
47,8
48,9
Е1. - элемент, Ох. — стабильный оксид, к — множитель = отношению М оксида / М элемента.
а В восстановительной среде принимает форму FeO {к — 1,286).
породе, хотя встречаются и значительные аномалии. Подстилающая порода не всегда
имеет решающее влияние, почвенные материалы могут формироваться из аллохтонных
гетерогенных материнских пород или привнесенных примесей.
В табл. 31.2 представлены диапазоны концентраций (по Aubertu Pinta, 1971) некото¬
рых микроэлементов, встречающихся в почвах. Содержание отдельных элементов, та¬
ких как хром, ванадий или цинк, обычно хорошо коррелирует с составом материнской
породы. Содержание других элементов, таких как бор, кобальт или молибден, зависит от
типа и происхождения почвы. Такие элементы, как йод или свинец, как правило, обна¬
руживают в почвах в концентрациях, значительно превышающих концентрации в под¬
стилающих породах (в особенности в осадочных породах морского происхождения, как
в случае йода). Органические почвы могут содержать в большом количестве некоторые
элементы, такие как селен.
При органическом загрязнении молекулы примеси, как правило, не присутствуют
в почве изначально, поэтому их идентификация и количественный анализ вызывают за¬
труднения (см. гл. 13), однако их происхождение не вызывает сомнений. Этого нельзя
Глава 31. Анализ экстрагируемых элементов и полный элементный анализ
727
Таблица 31.2. Концентрации некоторых микроэлементов в почве (по данным Aubert и Pinta,
1971); общее содержание элементов (мг/кг) и легко экстрагируемых элементов
(% от общего содержания элементов)
Элемент
Минимальная
концентрация,
мг/кг
Максимальная
концентрация,
мг/кг
Средняя
концентрация,
мг/кг
Средняя %-ная доля
легко экстрагируемой
фракции (% от суммарной
концентрации)
В
1-2
250-270
20-50
0,1-Ю и более
(1)
(подзолистые
(торфяники
(натриевые почвы)
почвы, Беларусь)
эвтрофные,
Израиль)
Сг
Следы
3000-4000
100-300
0,01-0,4
(2)
0,1-1
(3)
Со
0,05
300 (вертисоли,
10-15
0,5-50
(2)
(подзолистые
Центральная
0,3-21
(3)
почвы, Россия)
Африка)
Си
следы
200-250
15-40
0,05-5
(2)
(вертисоли,
7-17
(4)
Индия)
18-60
(5)
I
0,1
25 (перегнойно-
1-5
(гидроморфные,
глеевые почвы,
Амурдарья)
Латвия)
Мо
Следы
24 (бурые лесные
1-2
2-20(3,2,6)
(3,
почвы, Россия)
2,
6)
Ni
Следы
>5000
2
(2)
(уплотненные
7-20(5)
(5)
горизонты, Новая
Каледония)
Pb
Следы
1200 (подзолистые
15-25
1-30
(2)
почвы, Канада)
Se
0,1
1000 (торфяники,
1-7
Ирландия)
V
Следы
400
100-200
0,4-0,6
(2)
Zn
Следы
900
50-100
0,2-20
(3)
Li
5
200
Rb
10
500
Ba
100
3000
500
Sr
50
1000
350
Ga
2
100
30
Правая колонка: экстрагирующий реагент: (1) горячая вода, (2) 2,5% СН3СООН pH 2,5, (3) 1 М
CH3COONH4 при pH 7, (4) ЭДТА, (5) 1 М НС1, (6) буферный раствор щавелевой кислоты — оксалата
аммония при pH 3,3 (реагент Григга)
728
Часть 3. Неорганический анализ
сказать о неорганических элементах: различить элементы геохимического и антропо¬
генного происхождения не всегда просто (Bourreliem Berthelin, 1998).
Опытный геохимик, владеющий информацией о типе почвы и материнской породы,
отметит, что некоторые компоненты присутствуют в слишком высоких концентрациях.
Иногда этому находятся очевидные объяснения: например, медь, содержание которой
обычно коррелирует с типом почвы и материнской породы, но в избытке обнаруживает¬
ся в почвах виноградников.
Однако в большинстве случаев идентификация происхождения почв более сложна
и требует статистических сравнений состава почв в предполагаемом источнике загряз¬
нения и с прилегающих участков (с одинаковой материнской породой). Кроме того,
состав почвы связан со способностью элементов к обмену с почвенными обменными
комплексами (см. гл. 19).
31.1.3. Биогенные и токсичные элементы
Некоторые основные элементы (см. раздел 31.1.1) и микроэлементы (см. раздел 31.1.2)
особенно важны для жизнедеятельности организмов, населяющих Землю; анализ этих
элементов требуется проводить более часто, а процессы, протекающие в почвах с их уча¬
стием, необходимо тщательно исследовать.
К основным элементам, входящим в состав тканей растений, относятся углерод, кис¬
лород, водород, азот, фосфор, сера, калий, кальций, магний и иногда натрий (в соле¬
выносливых растениях) или кремний (в злаковых растениях), а также другие элементы,
аккумулируемые отдельными растениями в высоких концентрациях.
К другим элементам, играющим важную роль в физиологии растений, относятся
медь, железо, марганец, цинк, бор и молибден. Несмотря на то, что эти элементы при¬
сутствуют в клеточной ткани в очень малом количестве (микроэлементы или следовые
элементы) — от нескольких мг/кг до нескольких г/кг, их дефицит может замедлить раз¬
витие растений. В то же время, слишком высокие концентрации делают их токсичными
(Coppenet и Juste, 1982; Abo, 1984). Поэтому необходима подробная информация о кон¬
центрации и доступности биогенных и токсичных элементов.
В почвах эти элементы (медь, цинк, бор и молибден) обычно встречаются в следо¬
вых количествах, однако могут присутствовать и в качестве основных элементов (железо
или марганец). Их доступность определяется не только их концентрацией, но и физи¬
ко-химическим равновесием с молекулярными структурами почвы (см. «Минералогия»
в части 1 и «Органическое вещество» в части 2); таким образом, содержание основных
элементов связано с pH почвы (см. гл. 15), окислительно-восстановительным потенци¬
алом (см. гл. 16), зарядом обменного комплекса (см. гл. 20 и 21), емкостью катионного
обмена (см. гл. 26) и емкостью анионного обмена (см. гл. 27). Другие элементы, такие
как алюминий, связанные с формированием обменной кислотности (см. гл. 23), в опре¬
деленных условиях также могут оказаться токсичными для растений.
Другие элементы также важны для жизнедеятельности организмов, несмотря на
то что они присутствуют в почве на уровне минимальных следовых количеств (Aubert
и Pinta, 1971; Baize, 1997). Например, для образования гемоглобина животным необхо¬
дим кобальт. Из-за дефицита кобальта в почвах и, вследствие этого, в кормовых расте¬
ниях у коров и овец может развиться анемия. Важным элементом для человека является
йод, он играет важную роль в выработке гормона щитовидной железы. Из-за дефицита
йода может появиться зоб, в прошлом являвшийся обычным заболеванием в районах
с дефицитом йода. Молибден играет важную роль в жизнедеятельности растений и жи¬
Глава 31. Анализ экстрагируемых элементов и полный элементный анализ
729
вотных, например, в круговороте азота, где он ускоряет процесс восстановления двуоки¬
си азота до азота. Аналогичную функцию выполняет ванадий. Селен способен накапли¬
ваться в растениях и становиться токсичным для живых организмов.
31.1.4. Полный элементный анализ
Для полного элементного анализа используют множество химических и физико-хими¬
ческих методов (Smith, 1991; Tan, 1996; Не etXiang, 1999; Pansu и др., 2001). Эти методы
включают (i) анализы, требующие отделения элементов от органических и минеральных
матриц путем переведения в раствор, (и) анализы, которые можно проводить непосред¬
ственно в твердых средах.
Анализ с переведением элементов в раствор
Первые методики, разработанные для анализа природных силикатных материалов,
включали переведение элементов в раствор. В самом деле, классические химические
методы анализа основаны на свойствах элементов, находящихся в растворе.
Эти методики впоследствии были дополнены атомно-спектрографическими измере¬
ниями и широко применяются до сих пор, поскольку методы атомно-абсорбционной
спектрометрии (ААС), атомно-эмиссионной спектрометрии с индуктивно связанной
плазмой (ИСП-АЭС) и масс-спектрометрии с индуктивно связанной плазмой (ИСП-
МС) легче применять в растворах, чем в твердых средах.
Анализ в твердых средах
Для анализа в твердых средах материал обрабатывают потоком излучения соответству¬
ющей частоты. Цель облучения — индуцировать трансформации в атомных структурах
на уровне электронных оболочек или атомного ядра. Измерение энергий излучения во
время процессов релаксации позволяет произвести качественный и количественный
анализ элементного состава.
Если применяемые энергии не слишком высоки, соответствующие методы являют¬
ся недеструктивными: атомы возвращаются в основное состояние; исходное состояние
вещества не изменяется. Это не всегда верно для ядерных переходов, например, при
нейтронно-активационном анализе, или для ситуаций, когда вещество подвергается
воздействию слишком высокой термической энергии, например, при спектроскопии
с дуговым или искровым источником излучения, или эмиссии ИСП в твердой среде.
В зависимости от источника излучения и способа измерения испускаемого излучения
для анализа твердой среды доступно множество методик.
Самыми распространенными являются рентгенофлуоресцентные методы с исполь¬
зованием возбуждения глубоких электронных оболочек в потоке рентгеновских лучей
(см. раздел 31.3.2), электронного микрозондирования с использованием энергии потока
электронов (см. гл. 8) и нейтронно-активационного анализа, в процессе которого веще¬
ство подвергается воздействию потока нейтронов (см. раздел 31.3.3).
31.1.5. Экстрагируемые элементы
Полный элементный анализ не всегда позволяет получить достаточное количество ин¬
формации о доступности питательных веществ почвы для растений. Кроме того, для ма¬
лых лабораторий с ограниченным набором оборудования полный анализ больших ана¬
литических серий может оказаться затруднительным. Используемые реагенты зачастую
очень агрессивны, поэтому требования безопасности строги и лабораторное оборудова-
730
Часть 3. Неорганический анализ
ние стоит дорого. В связи с этим в течение многих лет агрохимики пытаются заменить
полный анализ простыми, но достаточно точными химическими или биохимическими
тестами, позволяющими идентифицировать пороги дефицита и токсичности для ра¬
стений (Реек и Soltanpour, 1990). Поскольку задачей подобного тестирования зачастую
является выявление потребности в удобрениях, методики анализа разрабатывают для
конкретных условий, и их нельзя использовать во всех системах земледелия.
Эффективность экстракции меняется в зависимости от типа почвы. Необходимо уста¬
новить корреляцию между экстрагируемостью данного элемента и влиянием этого эле¬
мента на конкретное растение в полевых условиях либо в контролируемых тепличных
условиях. Различные степени доступности можно оценить с учетом экстрагирующей
способности применяемого реагента. Элементы, наиболее часто исследуемые по пара¬
метру доступности для растений, являются основными элементами, входящими в состав
питательных веществ, потребляемых растениями: неорганический азот (см. гл. 28), по¬
тенциально доступный азот (см. гл. 14), различные формы фосфора (см. гл. 29), формы
серы (см. гл. 30), обменные катионы (см. гл. 22). В этой главе описаны дополнительные
процедуры, позволяющие исследовать другие доступные или потенциально доступные
формы встречающихся в почве элементов.
В1.2. Методы, включающие солюбилизацию
31.2.1. Методы полной солюбилизации
Методы солюбилизации при полном анализе почв аналогичны методам более общего
геохимического анализа. Иногда необходима адаптация методик к условиям определен¬
ных почвенных сред, например, сред с высоким содержанием органического вещества.
Полный анализ требует разрушения как структур органического вещества, так и ми¬
нералогических решеток алюмосиликатов. Обработка должна быть агрессивной и весь¬
ма длительной; она является потенциально опасной. Поэтому к выбору методик следует
подходить со вниманием (Kawasaki и Arai, 1996). Основными источниками погрешности
при полном анализе с солюбилизацией являются: (i) загрязнение реагентов и оборудо¬
вания, (И) неполное вскрытие почвенной матрицы и (Ш) помехи при измерениях, созда¬
ваемые реагентами, применяемыми для вскрытия пробы.
Загрязнение может повлиять на результаты анализа микроэлементов. Холостые пробы
(включающие, за исключением пробы, все реагенты и полный процесс обработки) зача¬
стую необходимы для вычитания результата измерений в холостой пробе из результата
измерений в аналитической пробе образца. При расчетах учитывается дисперсия изме¬
рений для холостой и аналитической пробы. Когда концентрация аналита в аналитиче¬
ской пробе приближается к концентрации в холостой пробе, измерения с применением
солюбилизации становятся невозможными; в этом случае необходимо переходить на
реагенты более высокой очистки или же проводить анализ непосредственно в твердой
среде. Постоянный прогресс в приборостроении и новые аналитические потребности,
возникающие в процессе изучения окружающей среды и геохимических исследований,
определяют новые требования в отношении чистоты и выбора реагентов.
Еще одним возможным источником погрешности является неполнота деструкции
почвенной матрицы. Отдельные минералы чрезвычайно сложно перевести в раствор
полностью даже в случаях, когда твердые остатки не видны после обработки. Среди
элементов, с трудом поддающихся солюбилизации, отметим хром, титан и цирконий.
Чтобы полностью перевести в раствор эти элементы, иногда возникает необходимость
изменить тип и пропорцию реагентов, способ вскрытия пробы (например, проводить
731
Глава 31. Анализ экстрагируемых элементов и полный элементный анализ
вскрытие пробы в открытом или закрытом сосуде, нагревать на горячей плитке, СВЧ-
излучением и т. д.).
Некоторые типы помех могут вызывать реагенты, в зависимости от применяемой
аналитической методики. Типичная физическая помеха, потенциально приводящая
к серьезным ошибкам при измерениях методами атомно-абсорбционной спектроме¬
трии (ААС) и пламенной или плазменно-эмиссионной спектрометрии, возникает из-
за наличия твердых частиц аналитической матрицы, засоряющих систему ввода пробы.
Один из главных источников погрешностей при ИСП-АЭС (Butman, 1987) связан с от¬
ложениями твердых веществ в распылителе, воздействующих на поток, попадающий
в горелку. В ААС реагенты могут оказаться источниками химических помех различных
типов. Однако тщательный подбор реагентов для обработки пробы помогает избежать
помех (Jeanroy, 1974). В эмиссионной спектрометрии следует учитывать возможные
спектральные помехи. В ИСП-МС следует принимать во внимание возможность об¬
разования различных полиатомных структур из атомов плазмы и молекул реагентов
и вероятные помехи при измерении анализируемого элемента, связанные с близостью
величин их масс.
Методы солюбилизации можно разделить на три группы:
• разложение кислотами в открытом сосуде;
• кислотное вскрытие в закрытом сосуде;
• щелочное сплавление.
Для разложения пробы можно использовать различные кислоты: выбор кислоты за¬
висит от типа обрабатываемого материала. Кроме того, выбор реагента зависит от мето¬
дики последующих аналитических измерений. Важно изучить свойства основных анали¬
тических реагентов с точки зрения их пригодности в данной аналитической методике.
31.2.2. Основные реагенты для полного растворения
Хлористоводородная кислота
При высоких температурах концентрированной хлористоводородной кислотой (36%,
12 М, rf= 1,18) вскрывают пробы с минералами, содержащими силикаты, оксиды, суль¬
фаты и фториды. Хлористоводородная кислота обладает слабой восстанавливающей
способностью и, как правило, для разложения органического вещества непригодна, за
исключением особых случаев, таких как гидролиз белков для выделения аминокислот
(см. гл. 14). Часто ее используют в качестве итоговой среды при приготовлении рас¬
творов для измерений методом ААС, поскольку она снижает уровень отдельных помех.
Однако она непригодна для анализа методом ИСП-МС, поскольку некоторые поли-
атомные структуры, такие как АгСГ, С10+ или С10Н+, способны создать значительные
помехи при анализе As, V, Сг, Fe, Ga, Ge, Se, Ti, Zn (Jarvis, 1994b). Поскольку точка кипе¬
ния азеотропной смеси НС1-Н20 ниже точки кипения азеотропной смеси HN03-H20,
хлористоводородную кислоту можно эффективно удалять серией последовательных вы¬
париваний в присутствии азотной кислоты.
Азотная кислота
Азотная кислота является одним из реагентов, наиболее часто используемых для под¬
готовки проб. Она способна высвобождать микроэлементы в форме нитратов, сильно
растворимых в комплексных матрицах. Сильным реагентом, рекомендованным для со¬
любилизации микроэлементов в почвах (стандарт AFNOR NFISO 11466 1995), является
так называемая «царская водка» (один объем концентрированной HN03 (16 М), сме-
732
Часть 3. Неорганический анализ
шанный с тремя объемами концентрированной НС1 (12 М)). Концентрированная азот¬
ная кислота (68%, 16 М, d— 1,42) является сильным окислителем, способным разрушить
органическое вещество в почвах с низким содержанием органики. Тем не менее, окис¬
лительный потенциал азотной кислоты снижен, поскольку точка кипения азеотропной
смеси (122 °С) ограничивает температуру процесса вскрытия пробы в открытом сосуде.
Для полного разложения органической матрицы зачастую необходимы реагенты, обла¬
дающие большей окислительной способностью, такие как пероксид водорода или хлор¬
ная кислота. Во избежание взрыва при вскрытии проб с очень высоким содержанием
органических веществ рекомендуется проводить предварительную обработку азотной
кислотой перед применением более сильных окислителей.
Как и хлористоводородная кислота, азотная кислота является подходящей средой
для анализа растворов методами ААС и эмиссионной спектрометрии, несмотря на бо¬
лее слабое корректирующее воздействие на некоторые виды помех. Азотнокислая сре¬
да особенно подходит для работы по методикам с ИСП-МС-измерениями, поскольку
полиатомные структуры, которые может образовать HN03 в аргоновой плазме при вы¬
соких температурах, не отличаются от структур, возникающих при постоянном присут¬
ствии в плазме элементов Н, N и О.
Фтористоводородная кислота
Это единственная кислота, которая может растворять материалы с силикатной основой
за счет образования гексафторсиликат-ионов, способных трансформироваться в лету¬
чий тетрафторид кремния в соответствии с полными реакциями следующего типа:
Si02 + 6HF->H2SiF6 + 2H20
H2SiF6-> SiF4+ 2HF
Из-за летучести SiF4 фтористоводородную кислоту нельзя использовать для вскры¬
тия проб в открытых сосудах при количественном анализе оксида кремния. Кроме того,
фториды других элементов, таких как бор, мышьяк, сурьма и германий, могут также об¬
ладать летучестью, в зависимости от степени окисления. Вскрытие проб обычно прово¬
дят смесями концентрированной фтористоводородной кислоты (48%, 29 М, d = 1,16)
и кислот-окислителей (таких как азотная или хлорная кислоты), завершающих процесс
растворения; в результате элементы, растворенные в аналитической среде, имеют высо¬
кие степени окисления.
Если в количественном анализе оксида кремния нет необходимости, предпочтитель¬
но удалить этот основной элемент для уменьшения количества твердых веществ в ана¬
литическом растворе. Это минимизирует риск серьезных погрешностей, возникающих
из-за засорения систем распыления в аппаратуре для эмиссионной и абсорбционной
спектрометрии (Вигтап, 1987). Сравнительно высокое давление паров фтористоводо¬
родной кислоты приводит к образованию азеотропной смеси с водой с относительно
низкой температурой кипения: 112 °С. Следовательно, эта кислота легко удаляется из
реакционной среды выпариванием.
Одним из неудобств, связанных с применением фтористоводородной кислоты, яв¬
ляется ее способность разъедать стекло и силикаты даже при невысоких концентра¬
циях. Этим определяется необходимость использования исключительно пластиковых
контейнеров, предпочтительно ПТФЭ. Кроме того, при плазменно-эмиссионной спек¬
трометрии эта кислота может повредить некоторые типы распылителей и кварцевую
горелку, применяемую в аппаратуре для ИСП-спектрометрии, поэтому кислоту следу-
Глава 31. Анализ экстрагируемых элементов и полный элементный анализ
733
ет полностью удалить из итоговой реакционной среды. Фтористоводородную кислоту
можно связать добавлением насыщенного раствора борной кислоты, однако это может
привести к увеличению количества твердых частиц в аналитической среде. Если коли¬
чественный анализ силикатов не проводится, кремний и фтористоводородную кислоту
удаляют последовательными выпариваниями с риском потенциальной потери других
элементов.
Техника безопасности
Фтористоводородная кислота обладает высокой коррозийной активностью и токсич¬
ностью; работа с этим веществом требует чрезвычайной осторожности. Даже кратко¬
временный контакт с фтористоводородной кислотой может вызвать необратимые по¬
вреждения кожи и глаз. Лаборатория должна быть оборудована вытяжными шкафами
в рабочем состоянии. Необходимо использование защитной одежды. В случае контакта
с кожей следует обильно промыть место контакта водой и, по возможности, нанести на
пораженную кожу гель глюконата кальция. При попадании кислоты в глаза следует без¬
отлагательно вызвать службу оказания экстренной медицинской помощи.
Хлорная кислота
Хлорная кислота - одна из самых сильных неорганических кислот и очень сильный
окислитель. В концентрированном виде (НС104 72%, d= 1,67) горячая кислота может
взрываться при контакте с органическими веществами (в частности, с жирами), однако
этого не происходит при использовании холодной и разбавленной кислоты. Поэтому
следует работать исключительно с хлорной кислотой, разбавленной, по меньшей мере,
в четыре раза в другой кислоте, как правило, в азотной кислоте. Кроме того, проводят
предварительную обработку пробы азотной кислотой.
Хлорная кислота образует азеотропную смесь с водой при 72% НС104 с температурой
кипения 203 °С. В сочетании с фтористоводородной кислотой хлорная кислота повы¬
шает интенсивность обработки тугоплавких минералов за счет увеличения температу¬
ры кипения. В этом случае увеличивается способность смеси к растворению минералов
с одновременной минерализацией органического вещества. Кроме того, повышение
температуры кипения помогает удалять HF и SiF4 на этапе выпаривания.
Другим преимуществом этой кислоты является способность образовывать раствори¬
мые соли с большинством элементов, что свойственно не всем кислотам, в частности,
серной кислоте. Одним из неблагоприятных факторов использования хлорной кислоты
при анализе методом ИСП-МС являются помехи, создаваемые при измерении малых
количеств мышьяка и ванадия: ^Аг^СГ против 75As, 35С1160+ против51V (Jarvis, 1994b).
Меры предосторожности
Не рекомендуется использовать хлорную кислоту для прямого вскрытия проб с большим
содержанием органического вещества, в особенности, с высоким содержанием жиров
(например, некоторых видов активного ила). В этом случае необходимо провести пред¬
варительную обработку пробы по другой методике. Масла и смазки не следует обраба¬
тывать смесью хлорной и азотной кислот, так как при этом возникает опасность взрыва
при выпаривании азотной кислоты. Многие твердые перхлораты взрывоопасны.
При работе с хлорной кислотой следует строго соблюдать правила техники безопасно¬
сти. Лаборатория должна быть оснащена системой вытяжной вентиляции и вытяжными
шкафами, изготовленными из инертного пластика. Внутренние поверхности необходи¬
мо регулярно очищать, чтобы избежать накопления потенциально взрывоопасных перх-
734
Часть 3. Неорганический анализ
лоратов. Следует выделить специальный вытяжной шкаф исключительно для обработки
кислотами; в нем ни в коем случае нельзя работать с органическими веществами.
Реагенты для щелочного сплавления
В этих методиках используются щелочные реагенты, способные разрушать силикатные
решетки при высоких температурах и образовывать твердые растворы (стекла) при ох¬
лаждении. В этом случае можно легко растворить эти твердые растворы в разбавленных
кислотах. Эти методики пригодны для вскрытия геологических образцов, но не под¬
ходят для обработки биологических материалов. Их преимущество состоит в том, что
обычно происходит полное растворение без улетучивания, что позволяет проводить ко¬
личественный анализ, например, кремния. Их главным недостатком является необхо¬
димость введения большого количества растворенных твердых веществ в аналитические
растворы, что зачастую требует высоких разведений и повышает порог обнаружения при
анализе микроэлементов.
Рекомендованы к использованию различные реагенты для сплавления: метаборат
стронция (Sr(B02)2), метаборат лития (UB02), тетраборат лития (U2B407), карбонаты
(Na2C03, I^COj), гидроксиды (например, NaOH, КОН), пероксиды (например, Na202),
фториды (KHF2). Метаборат стронция рекомендован для анализа методом пламенной
атомно-абсорбционной спектрометрии (ПААС) (Jeanroy, 1974), поскольку стронций об¬
легчает коррекцию взаимовлияний.
Эти методики плохо адаптированы к ИСП-МС-анализу из-за большого числа пере¬
крывающихся спектральных сигналов атомов реагента, вводимого при сплавлении. На¬
илучшим субстратом для ИСП-МС-анализа считается метаборат лития (Jarvis, 1994а).
31.2.3. Кислотное вскрытие пробы в открытом сосуде
Основные положения
Данная методика пригодна для анализа многих элементов, включая Zn, Си, Ni, Со, Ti,
Mn, Mo, Sc и редкоземельные элементы. Методику нельзя использовать для анализа
кремния или летучих элементов, таких как Pb, Cd, As или Hg. Она особенно пригод¬
на для спектрометрических измерений микроэлементов, поскольку в ней используется
меньшее количество растворенных твердых веществ в составе реагентов, и общее коли¬
чество растворенных твердых веществ снижается за счет улетучивания кремния. Кроме
того, в этом случае удается избежать ошибок, возникающих при засорении системы рас¬
пыления спектрометра (см. раздел 31.2.1).
Для кислотного вскрытия предложено несколько типов растворов. Стандарт AFNOR
NF X 31-151 (1993) рекомендует два вида вскрытия проб почв, осадочных отложений
и шлама сточных вод: (i) вскрытие пробы царской водкой в системе с обратным холо¬
дильником, (И) озоление при 450 °С с последующим разложением смесью фтористо¬
водородной и хлорной кислот на плитке. Некоторые рекомендуют разложение смесью
фтористоводородной, серной и хлорной кислот(Hossner, 1996). Французский AFNOR NF
^ 31-1471 (1996) и международный Pr ISO CD 14869-part 1 (1998) стандарты рекомен¬
дуют проводить разложение смесью фтористоводородной и хлорной кислот, возможно
с предварительной обработкой при анализе почв с высоким содержанием органики.
Нижеописанная методика основана на международном стандарте. Фтористоводород¬
ная кислота (см. «Фтористоводородная кислота») способна разрушать решетки силика¬
тов в комбинации с хлорной кислотой (см. «Хлорная кислота»), повышающей темпе¬
ратуру кипения. Хлорная кислота является очень сильным окислителем, позволяющим
Глава 31. Анализ экстрагируемых элементов и полный элементный анализ
735
полностью разрушать органическое вещество. При анализе почв с очень высоким содер¬
жанием органического вещества реакция может проходить через взрывоопасные стадии;
в этом случае рекомендуется более мягкая обработка. Французский стандарт AFNOR NF
ХЗ1-147 (1996) рекомендует три методики, выбор которых зависит от содержания орга¬
нического углерода (С):
• С < 20 г/кг, без предварительной обработки;
• 20 < С < 40 г/кг, предварительная обработка — разложение азотной кислотой;
• С > 40 г/кг, без озоления перед обработкой.
Эти диапазоны указывают на то, что (i) предварительной обработки азотной кисло¬
той, как правило, достаточно для большинства почв, сформировавшихся в холодном
или умеренном климате, (И) вскрытие пробы без предварительной обработки возможно
при анализе многих видов тропических почв. В любом случае, следует избегать приме¬
нения серной кислоты в составе смесей для кислотного вскрытия, поскольку она обра¬
зует нерастворимые соли, является вязким реагентом, с трудом удаляется путем выпари¬
вания и несовместима с измерениями ИСП-МС (Jarvis, 1994а).
Реагенты
• Ультрачистая деионизированная вода (электропроводность менее 0,5 мкСм/см).
• Фтористоводородная кислота (HF, d= 1,16).
• Хлорная кислота (НС104, d = 1,67).
• Хлористоводородная кислота (НС1, d= 1,19).
• Азотная кислота (HN03, d= 1,41).
• Раствор ViHCl: смешивают 500 мл НС1 (d = 1,19) с 450 мл воды, встряхивают, ох¬
лаждают, доводят объем до 1 л с одновременным гомогенизированием.
• Раствор HHNC^: готовят в соответствии с представленным выше описанием, но
с 500 мл HN03 (d = 1,41).
Все кислоты должны быть высокой степени чистоты, рекомендованной для спект¬
рографического анализа. Для проверки чистоты реагентов проводят анализ холостой
пробы.
Оборудование
• Тигли из политетрафторэтилена (ПТФЭ1 (внутренний диаметр 5 см, высота 2 см,
приблизительный объем 40 мл)); тигли необходимо заполнить разбавленной азот¬
ной кислотой и выдерживать, по меньшей мере, в течение ночи, затем ополоснуть
деионизированной водой. Обработка более эффективна, если тигли кипятят в те¬
чение нескольких часов с 8 М HN03 с последующим обильным ополаскиванием
деионизированной водой и сушкой при 105 °С для полного удаления сорбирован¬
ной кислоты (Jarvis, 1994а). Изношенные тигли следует заменять, поскольку они
активнее сорбируют и десорбируют элементы.
• Альтернативно используют платиновые тигли2 (внутренний диаметер 4 см, высота
2,5 см, приблизительный объем 30 мл).
• Кислотоустойчивая нагревательная плитка (следует помнить, что оборудования,
способного выдерживать очень длительный контакт с применяемыми смесями, не
существует).
1 Techniverre, 93 380 Pierrefltte sur Seine, France.
2 Lyon Alemand Louyot SA, 75 139 Paris Cedex 03, France.
736
Часть 3. Неорганический анализ
• Эффективно работающий, легко очищаемый, пластиковый лабораторный вытяж¬
ной шкаф (см. «Меры предосторожности» в разделе «Хлорная кислота»), оборудо¬
ванный системой очистки отходящих паров кислот. При анализе микроэлементов
обработку кислотами необходимо проводить в атмосфере, не содержащей пыли.
Одним из решений проблемы является использование ламинарного вытяжного
шкафа. Также можно установить вытяжной шкаф в чистой комнате с системой воз¬
душной фильтрации. Обязательным является использование спецодежды и защит¬
ных приспособлений (нейлоновые рабочие халаты, пластиковые очки и перчатки)
и соблюдение мер предосторожности, в особенности при работе с фтористоводо¬
родной и хлорной кислотами (см. «Фтористоводородная кислота» и «Хлорная кис¬
лота»).
• Мерные колбы на 50 мл.
• Муфельная печь с температурным программированием до 500 °С.
Методика
Почвы с умеренно высоким содержанием органики
Пробы почв готовят стандартным способом: измельчают почву до размера частиц 0,2 мм
(Pansun др., 2001).
• (Е1) Точную навеску 0,5 г (±0,1 мг) пробы помещают в ПТФЭ капсулу или плати¬
новый тигель.
• (Е2) Добавляют несколько капель деионизированной воды для увлажнения пробы.
• (ЕЗ) Добавляют в следующем порядке: около 10 мл концентрированной HF и 5 мл
концентрированной НСЮ4.
• Накрывают пробы ПТФЭ пластиной (оставляют свободное пространство меж¬
ду пробой и крышкой, чтобы избежать конденсации) и оставляют в контакте на
ночь.
• Снимают крышку, нагревают на плитке при 40—50 °С до появления белого дыма,
указывающего на начало испарения хлорной кислоты.
• Добавляют 5 мл концентрированной фтористоводородной кислоты. Поднимают
температуру приблизительно до 150 °С.
• Сушат до прекращения выделения белого дыма.
• (R1) Добавляют 10 мл раствора НС1 к остатку.
• Медленно выпаривают, не допуская дополнительного озоления.
• (R2) Добавляют 5 мл раствора НС1.
• Добавляют кипящую деионизированную воду, при необходимости нагревают в те¬
чение нескольких минут для ускорения процесса растворения.
• Охлаждают и переносят количественно в мерную колбу вместимостью 50 мл.
• Доводят объем до 50 мл деионизированной водой.
Примечания. Эта среда (5%-ная хлористоводородная кислота) подходит для измере¬
ний методом молекулярной и атомно-абсорбционной спектрометрии, а также пламен¬
ной или ИСП-АЭС-спектрометрии.
Платиновые капсулы очень дороги, однако более просты в обращении по сравнению
с ПТФЭ капсулами. Их применение позволяет сократить время нагрева и лучше конт¬
ролировать температуру процесса.
737
Глава 31. Анализ экстрагируемых элементов и полный элементный анализ
Альтернативная методика для ИСП-МС-измерений
При работе по этой высокочувствительной методике следует соблюдать следующие
меры предосторожности: работать в чистом помещении, не содержащем пыли, защи¬
щать тело, волосы и обувь, применять только ультрачистые реагенты.
Вместо хлористоводородной кислоты для разбавления итоговой реакционной среды
используют азотную кислоту (см. раздел 31.2.2). Хлорная кислота должна быть полно¬
стью удалена из аналитической матрицы.
До стадии R1 последовательность действий аналогична описанной в методике «Поч¬
вы с умеренно высоким содержанием органики».
• (R1) Добавляют к остатку 10 мл Уг HN03.
• Медленно выпаривают досуха, не допуская дополнительного озоления.
• Добавляют 10 мл Уг HN03 и повторяют предыдущую стадию.
• (R2) Добавляют 5 мл Уг HN03 и продолжают согласно методике «Анализ почв
с умеренно высоким содержанием органики».
В качестве итоговой аналитической среды для других спектрометрических измерений
можно также использовать 5%-ную азотную кислоту, несмотря на то что ее способность
корректировать отдельные взаимные влияния снижена.
Альтернативная методика анализа почв с умеренным содержанием органики
Для почв, содержащих С менее 40 г/кг, на стадиях Е1 и Е2 работают в соответствии с ме¬
тодикой «Почвы с умеренно высоким содержанием органики», затем добавляют 10 мл
концентрированной азотной кислоты; нагревают на плитке в течение 30 мин. Охлажда¬
ют, добавляют несколько капель деионизированной воды и продолжают в соответствии
с методикой «Анализ почв с умеренно высоким содержанием органики», начиная со
стадии ЕЗ.
Альтернативная методика анализа почв с очень высоким содержанием органики
• (Е1) Навеску 0,5 г (±0,1 мг) пробы помещают в платиновую капсулу (при отсут¬
ствии платинового используют фарфоровый тигель).
• Ставят тигель в муфельную печь и программируют медленное повышение темпера¬
туры до 450 °С приблизительно за 1 ч (медленный подъем температуры позволяет
избежать потерь за счет выбросов вещества). Поддерживают заданную температуру
в течение Зч.
• Охлаждают. Если пользуются платиновой капсулой, продолжают в соответствии
методикой «Почвы с умеренно высоким содержанием органики», начиная со ста¬
дии Е2, разделив объем кислоты на две части (если капсулы недостаточно велики).
При отсутствии платиновых капсул количественно переносят содержимое фарфо¬
рового тигля в ПТФЭ капсулу, затем продолжают в соответствии методикой «Поч¬
вы с умеренно высоким содержанием органики», начиная со стадии Е2.
Альтернативная методика анализа тугоплавких минералов
Тугоплавкие минералы, такие как хромит, гранат, магнетит или оксид циркония, лишь
частично вскрываются с помощью описанной выше процедуры. На первой стадии обра¬
ботки R1, описанной в методике «Почвы с умеренно высоким содержанием органики»,
следует проверить пробу на возможное присутствие невскрытых твердых фрагментов.
Вскрытие можно завершить повторным высушиванием и повторением стадии ЕЗ. Не¬
полное вскрытие пробы не всегда приводит к неизбежным ошибкам при количествен-
738
Часть 3. Неорганический анализ
ном анализе, в особенности если не проводится анализ таких элементов, как Cr, Hf, Мо,
Sc, Zr или тяжелых редкоземельных элементов от Gd до Lu (Totland и др., 1992). В этом
случае необходимо декантировать или (предпочтительнее) центрифугировать итоговую
аналитическую среду перед спектрометрическими измерениями, чтобы избежать помех,
связанных с твердым остатком. О наличии твердого остатка следует обязательно упомя¬
нуть в итоговом аналитическом отчете.
Разложение серосодержащих минералов при обработке смесью HF-HC104 может
оказаться неполным. Рекомендуется (Jarvis, 1994а) удалять их путем предварительной
обработки 10 мл «царской водки» (1 объем HN03 + 3 объема НС1). Пробу выдержива¬
ют при комнатной температуре до окончания выделения газа, затем нагревают до 60 °С
и выстаивают в течение одного часа перед сухим испарением при 150 °С. Окончательно
завершают разложение с помощью смеси HF-HC104b соответствии с описанием, пред¬
ставленным в методике «Почвы с умеренно высоким содержанием органики».
Предлогаются и другие модификации аналитической процедуры (Vemetn Govindaraju,
1992): добавление азотной кислоты к смеси HF-HC104 для растворения некоторых се¬
росодержащих минералов, таких как галенит или пирит; ограничение температуры до
100 °С во избежание превращения фосфора в фосфатную форму и применение «царской
водки» для анализа ртути.
31.2.4. Кислотное вскрытие в закрытом сосуде
Основные положения
Вскрытие пробы в закрытом сосуде проводится, главным образом, при количественном
анализе летучих элементов, таких как Pb, Cd, As, Sc, В, Hg, Sb, Se или Sn на микро- или
ультрамикроуровне. При вскрытии такого типа становится возможным растворение ве¬
щества в тугоплавкой фазе и сокращается длительность вскрытия пробы. Процесс поз¬
воляет увеличить температуру и давление благодаря повышению точки кипения кислот.
Поскольку не происходит испарения, расходуются меньшие объемы реагентов. Умень¬
шение количества реагентов снижает риск загрязнения и повышает защиту от проник¬
новения в систему частиц пыли. Кроме того, снижается количество кислот, а также ток¬
сичных и обладающих коррозионной активностью испарений.
В зависимости от требуемой температуры процесса используют различные контей¬
неры для вскрытия проб (или «бомбы»). Применение бомбы Парра для кислотного
вскрытия позволяет достигать температуры свыше 250 °С при внутреннем давлении
12,4 МПа (предохранительный клапан срабатывает при 24 МПа). В комплект входит
ПТФЭ чашка объемом 23 мл с ПТФЭ защитным кожухом, устройство целиком заклю¬
чено в стальной контейнер с завинчивающейся крышкой, обеспечивающей герметич¬
ное соединение между чашкой и крышкой. Бомбы такого типа дороги и не просты
в использовании. Если вскрытия проводятся при более низкой температуре и давле¬
нии, можно использовать более простые приспособления. Согласно описанной здесь
методике, контейнеры целиком изготовлены из тефлона с завинчивающейся ПТФЭ
крышкой.
Различные авторы предлагают разные составы смеси для вскрытия проб, например:
HN03-HC104-HF (Totland и др., 1992), HN03-HF (Jarvis, 1994а), «царская водка»-
HF (Hossner, 1996), HN03-HC1-HF (см. ниже). Процесс вскрытия пробы можно за¬
вершать удалением кислоты (в особенности фтористоводородной кислоты) серией
последовательных процессов сухого испарения с добавлением разбавленной азотной
или хлористоводородной кислоты. Так же как и методика, описанная в разделе 31.2.3,
Глава 31. Анализ экстрагируемых элементов и полный элементный анализ
739
данная методика позволяет уменьшить количество растворенных твердых веществ
и фтористоводородной кислоты, способных помешать аналитическим измерениям.
Тем не менее, она не позволяет проводить количественный анализ кремния и есть
риск потери некоторых фторидов за счет улетучивания и образования нерастворимых
форм.
Предотвратить улетучивание можно добавлением избытка борной кислоты после
кислотного вскрытия. Борная кислота вступает в реакцию с фтористоводородной кис¬
лотой с образованием тетрафторобората водорода (HBF4); реакция является двухстадий¬
ной, экзотермической:
Н3В03 + 3HF о HBF3OH + 2Н20
HBF3OH + HF HBF4 + Н20
В присутствии избытка борной кислоты равновесие процесса сильно сдвигается
вправо. Фтористоводородная кислота связывается и не создает проблем при анализе
в течение, по меньшей мере, двух часов (Bemas, 1968). Добавление борной кислоты за¬
вершает разложение за счет растворения нерастворимых гексафторидов металлов, и сре¬
да, содержащая «HBF4- Н3В03- силикатный ионный компонент», становится пригод¬
ной для количественного анализа методом атомно-абсорбционной спектрометрии (Urn
и Jackson, 1982).
Данная методика менее пригодна для анализа методом плазменной эмиссионной
спектрометрии (Jarvis, 1994а). Даже нейтрализованная фтористоводородная кислота
может разъедать стекло, в особенности в хрупкой конструкции атомайзера Мейхарда.
Кроме того, необходимое количество борной кислоты вызывает образование твердых
частиц в количестве, превышающем количество частиц при анализе по методике щелоч¬
ного сплавления, что может мешать определению многих микроэлементов.
Реагенты
Чистота всех реагентов должна соответствовать чистоте, необходимой для конкретного
типа спектрографического анализа. Анализ холостой пробы проводится для проверки
чистоты реагентов:
• концентрированной фтористоводородной кислоты;
• концентрированной азотной кислоты;
• концентрированной хлористоводородной кислоты;
• борной кислоты;
• ультрачистой деионизированной воды (электропроводность менее 0,5 мкСм/см).
Оборудование
• Контейнеры для вскрытия проб с завинчивающимися ПТФЭ крышками, внутрен¬
ний диаметр 45 мм, высота 60 мм (Techniverre, 93,380 Pierrefltte sur Seine, France).
• Водяная баня с регулируемым термостатом.
• Нагревательная плитка.
• Ультразвуковая ванна.
• Мерные колбы на 25 мл.
• Пластиковый вытяжной шкаф с системой промывки газов.
740
Часть 3. Неорганический анализ
Методика
Традиционная методика
(А) Навеску пробы около 250 мг (±0,1 мг) (размер частиц 200 мкм) помещают в ПТФЭ
реактор:
• добавляют несколько капель деионизированной воды для увлажнения всей пробы;
• добавляют 1 мл концентрированной HF, 1 мл концентрированной HN03 и 1 мл
концентрированной НС1;
• закрывают реактор и оставляют в контакте на ночь;
• обрабатывают ультразвуком в течение 10 мин;
• выдерживают в водяной бане при 60 °С в течение 24 ч;
• дают охладиться, открывают реактор и проверяют полноту вскрытия пробы или
наличие твердых остатков.
(В) При полном вскрытии пробы выпаривают раствор досуха на плитке при темпе¬
ратуре около 40 °С, защищая пробу от попадания пыли с помощью ПТФЭ крышки,
размещенной над реактором на высоте нескольких сантиметров:
• добавляют 1 мл концентрированной азотной кислоты;
• выпаривают досуха;
• добавляют 1 мл концентрированной азотной кислоты и горячую ультрачистую де¬
ионизированную воду;
• переносят количественно в мерную колбу на 25 мл;
• дают охладиться и доводят объем до 25 мл ультрачистой деионизированной водой.
Итоговая среда содержит 4%-ную азотную кислоту и пригодна для анализа микроэ¬
лементов методом атомно-абсорбционной спектрометрии с гидридной приставкой или
электротермической атомизацией. Кроме того, методика пригодна при анализе методом
ИСП-АЭС.
Альтернативная методика анализа тугоплавких материалов
При неполном вскрытии пробы по «Традиционной методике», по завершении стадии А:
• добавляют 1 мл концентрированной HF и 1 мл концентрированной HN03;
• останавливают реакцию в ПТФЭ реакторе и выдерживают его в водяной бане при
температуре 60 °С в течение 24 ч;
• продолжают в соответствии с «Традиционной методикой», стадия В.
Альтернативный метод предотвращения улетучивания фторидов
• После вскрытия пробы методом, описанным в «Традиционной методике», стадия
А, или «Альтернативная методика анализа тугоплавких материалов», добавляют
в реактор борную кислоту (2 г Н3В03 растворяют в малом объеме воды).
• Останавливают процесс, нагревают при 130 °С в течение 15 мин; дают охладиться.
• Доводят объем до 200 мл 1 М раствором HN03 и, если измерение не проводится
немедленно, хранят раствор в полиэтиленовых бутылях.
• При анализе методом ИСП-МС перед измерением растворы следует разбавить де¬
сятикратно (Jarvis, 1994а).
31.2.5. Микроволновая минерализация
При впечатляющем развитии аналитического оборудования стадия подготовки пробы
все еще является фактором, ограничивающим производительность аналитической ла¬
Глава 31. Анализ экстрагируемых элементов и полный элементный анализ
741
боратории. Современный спектрометр позволяет получать результаты измерения содер¬
жания нескольких элементов в течение трех или четырех минут. Кислотное растворение
аналогичной пробы может занять три или четыре дня. Исследованы два способа ускоре¬
ния операций, производимых вручную, выполняемых перед аналитическими измерени¬
ями (иногда их комбинируют): (1) автоматизация и роботизация процессов, (2) совер¬
шенствование оборудования, применяемого для подготовки проб к анализу
Микроволновую аппаратуру для минерализации, используемую в целях сокраще¬
ния длительности процесса растворения пробы, впервые стали производить в 1975 году.
Подобное оборудование часто используют в сочетании с закрытыми контейнерами для
вскрытия проб, оборудованными предохранительным клапаном. Комбинированное
воздействие высоких давлений и микроволнового излучения позволяет значительно со¬
кратить время, необходимое для переведения пробы в раствор. Органическое вещест¬
во быстро разрушается и исчезает необходимость в обработке хлорной кислотой (Vernet
и Govindaraju, 1992), что сокращает время от начала обработки до анализа.
Системы микроволновой обработки можно использовать в открытых системах. Они
наиболее пригодны там, где процедура полностью автоматизирована, начиная от вне¬
сения реагентов до окончательного растворения. Система позволяет программировать
сложные циклы, в том числе множество вскрытий проб, проводящихся без вмешатель¬
ства оператора. Таким образом, минимизируются риски для здоровья, связанные с при¬
менением опасных кислот (см. раздел 31.2.2).
Микроволновая аппаратура была впервые использована при анализе биологических
проб. Затем ее стали применять при анализе геологических материалов, первоначально
при разработке месторождений (в 1970 г.), а затем и в других геохимических исследова¬
ниях (Lamothe и др., 1986; Totland и др., 1992; Le Согпес и др., 1994). Сравнительные ис¬
следования показывают, что классические методики вскрытия проб в открытых сосудах
и обработка микроволнами приводят к аналогичным результатам для большинства эле¬
ментов, присутствующих в образцах геологических материалов. При применении обеих
методик возникают проблемы, связанные с точностью анализа Сг, Hf и Zr в определен¬
ных типах проб. Следовательно, лучше проводить количественный анализ этих элемен¬
тов после щелочного вскрытия сплавлением с метаборатом лития (Totland и др., 1992).
Различные стадии кислотного разложения легче контролировать в открытой системе
(см. раздел 31.2.3 и 31.2.4) (Le Сотес и др., 1994). Кроме того, процесс финального упа¬
ривания в микроволновых системах контролировать труднее, чем в системах, исполь¬
зующих нагревание на плитке. Полагают (Zischa и Knapp, 1997), что будут разработаны
автоматизированные закрытые системы для микроволновой минерализации твердых
веществ, в то время как при анализе жидких и полужидких проб предпочтительнее сис¬
темы минерализации непрерывных потоков.
При разнообразии микроволновых систем трудно рекомендовать универсальную ме¬
тодику микроволнового кислотного расщепления. Мы рекомендуем адаптировать для
конкретных условий классические системы, описанные в разделах 31.2.3 и 31.2.4, в со¬
ответствии с индивидуальными требованиями.
31.2.6. Щелочное сплавление
Основные положения
Эта методика обладает тремя основными преимуществами:
• пригодна для анализа всех основных элементов, поскольку в ней отсутствует риск
потери кремния;
742
Часть 3. Неорганический анализ
• выполняется быстрее по сравнению с классическими методиками кислотного
вскрытия, включающими нагревание на плитке или в водяной бане в открытом
или закрытом сосуде;
• как правило, приводит к полному растворению пробы и, таким образом, является
наиболее эффективной методикой растворения при анализе тугоплавких элемен¬
тов, таких как V, Сг, Zr или Y.
Основным недостатком методики является образование в большом количестве твер¬
дого остатка. При спектрометрическом анализе для предотвращения засорения распы¬
лителей, приводящего к снижению чувствительности при анализе микро- или ультра¬
микроэлементов, требуется большее разбавление.
Кроме того, уровень нагрева, необходимый для щелочного сплавления, может вы¬
звать потерю некоторых летучих микроэлементов.
Несмотря на то что для щелочного сплавления предложен ряд различных реагентов
(см. «Реагенты для щелочного сплавления» в разделе 31.2.2), в настоящее время наибо¬
лее распространенным является метаборат натрия. Этот реагент пригоден для измере¬
ний различными методами, в частности ИСП-МС {Jarvis, 1994а). Другая, более ранняя,
методика сплавления с применением метабората стронция {Jeanroy, 1974) использова¬
лась при анализе методом атомно-абсорбционной спектрометрии {Riandey и др., 1982),
однако в настоящее время метаборат лития считается лучшим реагентом для сплавления
{Le Сотес, IRD Bondy, France, персональное сообщение). Первая процедура исключает
возможность измерения содержания лития, а вторая делает невозможным определение
стронция, что может быть полезным при анализе почв. Кроме того, обе методики не
позволяют анализировать бор — микроэлемент, который может быть в дефиците или
являться токсичным для растений. Общее содержание бора можно измерить спектроко¬
лориметрическим методом или плазменно-эмиссионной спектрометрией (ИСП-АЭС)
после солюбилизации сплавлением с гидроксидом натрия {Abo, 1984).
Реагенты
• Ультрачистая деионизированная вода (электропроводность менее 0,5 мкСм/см).
• Реагенты для сплавления должны быть высокочистыми с сертифицированными
концентрациями анализируемых элементов.
• Метаборат лития (UB02), или, при его отсутствии, эквимолярная смесь:
- оксида лития (Li20) или карбоната лития (Ы2С03);
- оксида бора В203.
• Метаборат стронция (Sr(B02)2), или, при его отсутствии, эквимолярная смесь:
- карбоната стронция (SrC03);
- оксида бора (В203).
• Концентрированная азотная или хлористоводородная кислота.
• 5%-ная НС1 или 5%-ная HN03: в мерную колбу объемом 2 л добавляют 100 мл кон¬
центрированной НС1 или HN03 приблизительно к 1,8 л ультрачистой воды, встря¬
хивают, дают охладиться и доводят объем до 2 л.
• 2%-ная НС1 или 2%-ная HN03: в мерную колбу объемом 2 л, содержащую 1,8 л
воды, добавляют 20 мл концентрированной НС1 или HN03, встряхивают, дают ох¬
ладиться и доводят объем до 2 л.
Примечания. Если Sr(B02)2 недоступен, можно воспользоваться смесью SrC03 и В203
в эквивалентном количестве (1 моль каждого вещества на 1 моль Sr(B02)2). Когда тем¬
Глава 31. Анализ экстрагируемых элементов и полный элементный анализ
743
пература тигля сравняется с температурой в печи, происходит образование метабората
стронция в соответствии с уравнением реакции:
SrC03 + В203 -> Sr(B02)2 + С02
То же справедливо и для UB02, который является эквивалентом эквимолярной смеси
Li20-B203 или Li2C03-B20r
Тетраборат лития (У2В407 или смесь Li20-2B203, точка плавления 915 °С) исполь¬
зуют в качестве реагента для сплавления вместо метабората (точка плавления 845 °С),
обладающего более выраженными щелочными свойствами. Метаборат больше подхо¬
дит для обработки более кислых кремнийсодержащих материалов, в то время как те¬
траборат предпочтителен для более щелочных материалов (глиноземных, тугоплавких,
карбонатных и т. д.). Смесь метабората и тетрабората, образующаяся в точке эвтектики
при 832 °С, признана почти универсальным реагентом для сплавления при анализе проб
геологических материалов (Vemetи Govindaraju, 1992). Реагент можно получить, смешав
73% В203 и 27% 1л20.
Оборудование
• Круглодонные цилиндрические тигли, изготовленные из графита1 (высота = вне¬
шнему диаметру = 24 мм, глубина и внутренний диаметр составляет 18 мм).
• Муфельная печь, нагреваемая до 1100 °С, или (предпочтительно) индукционная
печь с подачей азота (рис. 31.1).
Рис. 31.1. Индукционная печь для щелочного сплавления
1 Sodemi, 95 370 St Ouen l’Aumone, France.
744 Часть 3. Неорганический анализ
• Стаканы химические объемом 200 мл, с ПТФЭ магнитными якорями для переме¬
шивания.
• Мерные колбы вместимостью 200 мл.
• Мерные колбы вместимостью 50 мл.
Примечания. Индукционная печь предпочтительнее муфельной печи по двум причи¬
нам: во-первых, режим нагрева в индукционной печи повышает интенсивность обра¬
ботки и ускоряет растворение пробы (менее 5 мин вместо 15—30 мин); во-вторых, конс¬
трукция индукционной печи делает возможной проведение процесса в атмосфере азота,
что существенно продлевает срок эксплуатации графитовых тиглей. В муфельной печи
риск разрушения тиглей из-за воспламенения можно снизить, поместив их в графито¬
вые контейнеры.
Графитовые тигли коммерчески доступны в очень чистых формах, совместимых с из¬
мерениями микроэлементов, и имеют слабое поверхностное натяжение расплавленной
массы на графите, что облегчает количественный перенос содержимого тигля в кислот¬
ный реагент для растворения. Однако в некоторых тиглях могут возникать проблемы
с удерживанием; изношенные тигли следует заменять.
В процессе сплавления отдельные восстановительные процессы могут привести к по¬
тере таких элементов, как железо или кобальт. Однако такие явления не характерны для
процессов сплавления силикатных материалов, в особенности там, где применяются
индукционные печи, сокращающие время растворения.
Интересной альтернативой являются тигли, изготовленные из сплавов платина — зо¬
лото или золото — платина — родий, несмачиваемых сплавов, которые, как правило,
используют при сплавлении в муфельных печах или с помощью газовой горелки Ме-
кера. В этом случае для предотвращения образования сплавов между материалом тигля
и элементами пробы необходимо контролировать условия окислительного процесса.
Методика с метаборатом стронция
• В графитовый тигель помещают точную навеску около 100 мг (±0,1 мг) пробы поч¬
вы (размер частиц не более 200 мкм).
• Добавляют 1 г метабората стронция (или эквивалентное количество SrC03 + В203)
и тщательно перемешивают.
• При анализе органической почвы (С > 40 г/кг) помещают тигель с навеской в печь
с регулируемой температурой и медленно (приблизительно в течение 1 ч) повыша¬
ют температуру до 450 °С. Выдерживают при этой температуре в течение 1 ч.
• Повышают температуру до 1100-1200 °С на 5 мин в индукционной печи и на
15 мин — в муфельной печи.
• Удерживая тигель с помощью тигельных щипцов, проверяют однородность рас¬
плава и переносят его горячим в химический стакан вместимостью 200 мл, содер¬
жащий около 150 мл 2%-ной азотной кислоты
• Дают тиглю охладиться около стакана. Затем проверяют на наличие твердых фраг¬
ментов и переносят их в стакан при необходимости.
• Помещают ПТФЭ якорь в стакан и перемешивают с помощью магнитной мешалки
до полного растворения (около 30 мин).
• Количественно переносят содержимое стакана в мерную колбу и доводят объем
до 200 мл 2%-ным раствором азотной кислоты. В состав матрицы аналитической
пробы входят 0,5% Sr(B02)2, 2% HN03.
Глава 31. Анализ экстрагируемых элементов и полный элементный анализ
745
Примечания. Этот раствор пригоден для определения содержания основных элемен¬
тов и некоторых микроэлементов методами атомно-абсорбционной спектрометрии, на¬
пример:
• Fe, Mn, Mg, Na, К, Си, Ni в воздушно-ацетиленовом пламени;
• Si, Al, Т1, Са в пламени закиси азота-ацетилена;
• и других микроэлементов с помощью электротермической атомизации.
Si, Al, Ti и Р также можно измерять спектроколориметрическими методами с удовлет¬
ворительными результатами.
Методика с метаборатом лития
Основные элементы
• В графитовый тигель помещают навеску приблизительно 60 мг (±0,1 мг) почвы
(размер частиц не превышает 200 мкм).
• Добавляют 0,5 г UB02 и тщательно перемешивают шпателем.
• При анализе органической почвы (С > 40 г/кг) помещают тигель с пробой в печь
с регулируемой температурой и медленно (приблизительно в течение 1 ч) повыша¬
ют температуру до 450 °С. Поддерживают эту температуру в течение 1 ч.
• Нагревают до 1100 °С в высокочастотной индукционной печи (или в муфельной
печи).
• Быстро переносят сплавленную массу в химический стакан вместимостью 250 мл,
содержащий 100 мл 2%-ного раствора НС1, с магнитным якорем из ПТФЭ. При
необходимости переносят все твердые фрагменты, оставшиеся после охлаждения.
• Перемешивают до полного растворения (15—20 мин).
• Количественно переносят содержимое в мерную колбу вместимостью 200 мл и до¬
водят объем до метки ультрачистой водой. В состав матрицы аналитической пробы
входят 1% НС1, 0,25% LiBOr
Примечания: Количественный анализ этого раствора позволяет одновременно опре¬
делять содержание основных элементов: Ti, Fe, К, Na, Mg, Са, Al, Si и, возможно, Р, если
его содержание велико (Р205 > 1%), методами плазменно-эмиссионной спектрометрии
(ИСП-АЭС).
Альтернативно можно применять пламенно-эмиссионные или ААС-методы, описан¬
ные в «Методике с метаборатом стронция», с добавлением в пробу лантана для коррек¬
ции интерференций. Фосфор и Si, Al и Ti также могут быть точно измерены спектроко¬
лориметрическими методами.
В итоговой аналитической среде вместо 2%-ной НС1 можно использовать 2%-ный
раствор азотной кислоты.
Альтернативные методики анализа микроэлементов (например, V, Cr, Zr, Y)
Последовательность действий аналогична представленному в описании «Основные эле¬
менты», однако для анализа отбирают большую порцию образца — 0,1 г или более. Пос¬
ле щелочного сплавления полученную массу растворяют в 5%-ном растворе НС1 и дово¬
дят объем до 50 мл.
Примечания. Измеряют содержание микроэлементов в полученном растворе мето¬
дами ИСП-АЭС и электротермической ААС (ЭААС). Методика наиболее полезна для
анализа микроэлементов, таких как V, Сг, Zr, Y, стойких по отношению к кислотному
вскрытию.
746
Часть 3. Неорганический анализ
Поскольку концентрация обработанной пробы в сравнении с пробой после кислот¬
ного вскрытия меньше, итоговая среда разбавлена сильнее, а количество растворенных
твердых веществ выше, при анализе других микроэлементов данная методика менее
удобна, чем кислотное вскрытие (см. раздел 31.2.3 и 31.2.4).
Методика пригодна для измерения содержания ультрамикроколичеств элементов
методом ИСП-МС, однако в этом случае аналитическую среду, представляющую собой
5%-ную НС1, следует заменить 5%-ной HN03.
31.2.7. Селективная экстракция
Экстрагирующие реагенты
Большая часть химических экстрагирующих реагентов, применяемых в агрономическом
анализе, позволяет получить информацию только о равновесных концентрациях эле¬
ментов в почве в определенный момент времени. Другие методики (например, инкуба¬
ция почвы) позволяют количественно оценить влияние активности микроорганизмов
на доступность определенного элемента, например, азота (см. гл. 14), фосфора (см. гл.
29) или серы (см. гл. 30). Кроме того, корневая система растений оказывает комплекс¬
ное влияние на экстракцию питательных веществ (Callot и др., 1982); при простых испы¬
таниях эти процессы трудно смоделировать с достаточной точностью.
Водная экстракция (см. гл. 18) позволяет получить информацию о реальной доступ¬
ности элементов, присутствующих в почвенном растворе. Кипящая вода является более
агрессивным реагентом, который можно использовать для анализа доступности бора
(см. например, S8 здесь, в описании «Экстрагирующие растворы», или стандарт AFNOR
NFX 31-122, 1993).
Воздействие других реагентов, применяемых в холодном или горячем состоянии,
протекает по различным механизмам, таким как гидролиз, ионный обмен, изменение
pH или окислительно-восстановительного потенциала, комплексообразование и т. д.
Существует множество различных реагентов для ионообменной экстракции в буфер¬
ных или небуферных средах. Среды, содержащие хлориды калия или кальция, фторид
натрия, позволяют определять содержание обменных протонов или гидроксил-ионов
(см. гл. 15). Ацетаты аммония или натрия, а также хлорид бария являются типичными
реагентами, применяемыми для анализа обменных катионов или емкости ионного об¬
мена (см. гл. 22 и гл. 26). Для исследований обменного железа, связанного с хлорозом
растений, используют буферный раствор ацетата аммония или ацетата натрия с pH 4,8
или 3, а также раствор оксалата аммония (стандарт FDX31-146,1996). Применение хло¬
рида калия и ацетата аммония позволяет проводить количественный анализ обменного
алюминия (см. гл. 23) и обменного магния (Martens и Lindsay, 1990). Реагенты, участ¬
вующие в ионообменных взаимодействиях, используются в исследованиях различных
форм азота (см. гл. 28), фосфора (см. гл. 29) или серы (см. гл. 30). Кроме того, ионы
могут участвовать в реакциях, протекающих в твердой фазе, например, в ионообменных
смолах.
Комплексообразующие реагенты также широко применяются для селективной солю¬
билизации (при которой время достижения равновесного состояния, как правило, весь¬
ма велико) за счет образования хелатов, зачастую в сочетании с другими реагентами,
работающими по механизму ионного обмена, окислительно-восстановительного или
кислотного воздействия. Самыми распространенными комплексообразующими реа¬
гентами являются этилендиаминтетрауксусная кислота (ЭДТА), диэтилентриаминпен-
тауксусная кислота (ДТПА) и триэтаноламин (ТЭА). Стандарт NFX31-120 (1992) реко-
Глава 31. Анализ экстрагируемых элементов и полный элементный анализ
747
мендует проводить экстракцию меди, марганца (Gambrell и Patrick, 1982) и цинка смесью
ацетата аммония и ЭДТА. Стандарт NFXS1-121 (1993) используется для количественной
оценки содержания тех же элементов (плюс железо) с применением ДТПА. Кроме того,
в этом стандарте рекомендуется использовать реагент ДТПА-СаС12-ТЭА для экстрак¬
ции токсичных металлов {Risser и Baker, 1990).
Окисляющие или восстанавливающие реагенты позволяют экстрагировать некоторые
элементы в различных валентных состояниях. Например, некоторые формы железа
экстрагируют 0,5%-ной щавелевой кислотой, 0,2%-ным гидрохиноном и ацетатом ам¬
мония. Одну из форм легко восстанавливаемого марганца экстрагируют в присутствии
дитионита натрия.
Кислотные реагенты зачастую используют для вытеснения потенциально доступных
трудно экстрагируемых форм. Уксусная кислота при pH 2,5 позволяет солюбилизиро¬
вать всю фракцию «суммарных обменных катионов». Кипящая концентрированная ук¬
сусная кислота растворяет сидерит. Для экстракции некоторых форм Си, Ni, Zn, Cd, Сг,
Hg или Pb используют хлористоводородную кислоту в различных разведениях (0,1, 0,5,
1 М); то же относится и к фосфорной кислоте (например, для экстракции некоторых
форм Мп). Вскрытие силикатных решеток фтористоводородной кислотой позволяет
проводить оценку содержания необменного аммония (см. гл. 28).
Вскрытие пробы с помощью трех концентрированных кислот (1 объем HN03, 2 —
НС1, 4 — H2S04) в разные времена было рекомендовано для отделения практически не¬
растворимых первичных минералов от растворимых минералов, сформировавшихся в не
столь давнем прошлом {Hardy и Follet-Smith, 1931; Claisse, 1968; Njopmuo и Orliac, 1979).
Экстрагирующие растворы
Здесь описываются только стандартизированные растворы и наиболее широко исполь¬
зуемые экстрагирующие растворы; описать все возможные варианты не представляется
возможным из-за их многочисленности.
• (S1) Стандарт NFZ31-120 (1992): 1 М раствор ацетата аммония и 0,01 М ЭДТА (экс¬
тракция Си, Mn, Zn).
Растворяют 3,723 г ЭДТА (C10H14Na2O8 • 2Н20) и 77 г ацетата аммония (CH3COONH4)
в мерной колбе объемом 1 л, содержащей около 400 мл деионизированной воды.
Доводят объем до 800 мл. Измеряют значение pH и при необходимости регулируют
pH до 7,0 с помощью 1 М раствора аммиака или уксусной кислоты. Доводят объем
до 1 л деионизированной водой при перемешивании раствора.
• (S2) {Lindsay и Norvell, 1978; Risser к Baker, 1990), стандарт NFX31-121 (1993) и NF
ISO 14870 (1998): смешивают 0,1 М ТЭА, 0,01 М СаС12,0,005 М ДТПА (экстракция
Си, Mn, Zn, Fe, биогенных микроэлементов, токсичных металлов).
В мерной колбе объемом 1 л растворяют в деионизированной воде: 14,92 г ТЭА,
1,967 г ДТПА и 1,47 г дигидрата хлорида кальция. Доводят объем до 800 мл деиони¬
зированной водой и регулируют pH на уровне 7,3 с помощью раствора V2HCI при
перемешивании. Дают охладиться и доводят объем до 1 л деионизированной водой
при перемешивании.
• (S3) {Risser и Baker, 1990): разбавленный раствор хлористоводородной кислоты, 0,1
М НС1 (Zn, щелочность, токсичные металлы).
Вносят 8,3 мл концентрированной хлористоводородной кислоты в мерную колбу
объемом 1 л, содержащую 800 мл деионизированной воды. При перемешивании
доводят объем до 1 л.
748
Часть 3. Неорганический анализ
• (S4) (Сох, 1968; Risser и Baker, 1990): две кислоты (токсичные металлы, почвы с низ¬
ким pH, низкой величиной ЕКО и низким содержанием органического вещества).
Растворяют 8,3 мл концентрированной хлористоводородной кислоты и 1,4 мл кон¬
центрированной серной кислоты в 2 л деионизированной воды.
• (S5) Легко восстанавливаемый марганец (Gambrellи Patrick, 1982). Смесь 1 М рас¬
твора ацетата аммония (СН3СО(ЖН4) с pH 7 и 0,2%-ного гидрохинона (или ги-
дроксиламина).
Растворяют 77,1 г ацетата аммония в химическом стакане на 1 л, содержащем
750 мл деионизированной воды. Добавляют 2 г хлористоводородного гидроксила-
мина (NH2OH • НС1) или гидрохинона. Регулируют pH до 7 с помощью раствора */2
аммиака или 1/2 уксусной кислоты. Доводят объем до 1 л.
• (S6) Общее содержание абсорбированных металлов (USЕРАА, 1986): концентри¬
рованная азотная кислота, концентрированная хлористоводородная кислота, 30%-
ный пероксид водорода.
• (S7) «Свободные оксиды» железа (Deb, 1950; Petard, 1993). Раствор ацетата и тарт¬
рата натрия.
В химическом стакане вместимостью 400 мл растворяют 136 г CH3COONa • ЗН20
в минимально необходимом объеме деионизированной воды; в химическом стака¬
не вместимостью 250 мл растворяют 23 г Na2C4H406 • 2Н20 в минимально необхо¬
димом объеме деионизированной воды; смешивают два раствора и доводят объем
до 1л.
• (S8) Бор — экстракция кипящей водой (NF2Г31-122, 1993), 0,01 М раствор СаС12.
Используют деионизированную воду, не содержащую бора (проверить путем анализа
холостой пробы), растворяют 1,47 г СаС12 • 2Н20 в мерной колбе объемом 1 л, доводят
объем до 1 л водой, не содержащей бора, при перемешивании.
Оборудование
Здесь перечислено только наиболее широко распространенное оборудование.
• Весы с верхней загрузкой (±1 сг) с пластиковой ложечкой для порошкообразных
веществ.
• Пластиковые колбы (пробирки) для экстракции объемом 50, 100 и 200 мл с за¬
винчивающимися крышками, которые, по возможности, можно использовать для
центрифугирования.
• Колбы для консервации экстрагирующего раствора с объемными дозаторами.
• Шейкер, качающий платформу в горизонтальной плоскости, или ротационный
шейкер, переворачивающий платформу в вертикальной плоскости, в который
можно загружать необходимое количество колб для экстракции вместимостью 50,
100 или 200 мл. Режим взбалтывания устанавливают так, чтобы условия процесса
были воспроизводимы при скорости, позволяющей перемещать всю массу твердой
пробы; при этом скорость взбалтывания не должна быть чрезмерно высока. Пере¬
мешивание производят в изотермических условиях, температуру следует устано¬
вить на уровне 20 °С (±1 °С).
• Центрифугируют (при 5000 g) в пробирках, соответствующих объему экстрактов.
4 US ЕРА = US Environment Protection Agency (Управление по охране окружающей среды
США).
Глава 31. Анализ экстрагируемых элементов и полный элементный анализ
749
• Воронки и фильтры из а-целлюлозы (Whatman No. 42 или аналогичные) или
устройство для фильтрования с фильтрующей мембраной (размер пор 0,45 мкм).
• Круглодонные колбы для кипячения объемом 250 мл с шлифованными соедине¬
ниями и холодильниками подходящей конструкции для экстракции бора кипящей
водой.
• Стаканы химические вместимостью 100 мл с соответствующими крышками.
Методика
Общая методика
Пробу почвы подготавливают в соответствии с выбранной процедурой (как правило,
почву просеивают до размера частиц 2 мм и сушат на воздухе).
• Берут навеску пробы требуемой массы (в соответствии со специальным стандар¬
том, см. ниже) (±0,01 г) с помощью пластиковой ложечки для порошкообразных
материалов.
• Помещают пробу в колбу для экстракции соответствующего объема (см. далее).
• Добавляют необходимый объем экстрагирующего реагента.
• Колбу закрывают и ставят на шейкер с установленным режимом встряхивания или
встряхивают вручную в термостатируемом помещении при температуре 20 °С в те¬
чение определенного времени (см. далее).
• Центрифугируют или фильтруют (или проводят обе операции) и сохраняют экс¬
тракт до анализа.
Примечания. Все условия Процесса и в особенности длительность экстракции должны
быть стандартизированы. Для обработки экстрагентами и уравновешивания системы
требуется довольно много времени; в особенности долго протекают реакции комплек-
сообразования.
Всегда проводят анализ холостой пробы (реагенты в отсутствии пробы обрабатывают
по всей процедуре).
Готовят стандартные калибровочные растворы для спектрометрических измерений;
для разведения используют растворы, применяемые для экстракции.
Для экстрактов с нейтральной средой срок хранения экстракта до анализа ограничен
и не превышает двух суток.
Условия экстракции реагентом S1
Колба 100 мл; масса пробы 5 г; объем экстрагирующего реагента 50 мл; время экстра¬
кции 2 ч.
Условия экстракции реагентом S2
Колба 100 мл; масса пробы 10 г; объем экстрагирующего реагента 20 мл; время экстра¬
кции 2 ч.
Условия экстракции реагентом S3
Колба (или закрытая пробирка для центрифугирования) 50 мл; масса пробы 2 г; объем
экстрагирующего реагента 20 мл; время экстракции 5 мин; 3 последовательных экстра¬
кции; итоговый объем доводят до 100 мл.
Условия экстракции реагентом S4
Колба 50 мл; масса пробы 5 г; объем экстрагирующего реагента 25 мл; время экстракции
15 мин.
750
Часть 3. Неорганический анализ
Условия экстракции реагентом S5
Колба 200 мл; масса пробы 10 г; объем экстрагирующего реагента 100 мл; время экстра¬
кции 30 мин (при перемешивании); время контакта 6 ч (при периодическом переме¬
шивании). Содержание легко восстанавливаемого марганца рассчитывают вычитанием
количества, обнаруженного в экстракте, из количества обменного марганца, экстраги¬
рованного 1 М ацетатом аммония при pH 7 (см. гл. 22).
Альтернативная методика определения общего содержания абсорбированных
металлов - S6 (US ЕРА 1986)
2 г пробы помещают в стеклянный (Пирекс) стакан объемом 100 мл, добавляют 10 мл
концентрированной азотной кислоты, накрывают стакан и нагревают в течение 25 мин
на плитке при 95 °С без кипячения. Дают охладиться, добавляют 5 мл азотной кислоты
и вновь нагревают в течение 30 мин. Операцию повторяют. Открывают поверхность на чет¬
верть и выпаривают до объема 5 мл без кипячения. Охлаждают, осторожно добавляют 2 мл
деионизированной воды и 3 мл 30%-ного пероксида водорода. Нагревают с осторожно¬
стью, чтобы избежать бурною выделения газа. Продолжают добавлять пероксид водорода
порциями по 1 мл до окончания выделения пузырьков газа. Добавляют 5 мл концентриро¬
ванной хлористоводородной кислоты и 10 мл деионизированной воды. Накрывают стакан
и вновь нагревают в течение 15 мин без кипячения. Фильтруют через бумажный фильтр
(Whatman No. 41 (или аналогичный)) и доводят объем до 50 мл деионизированной водой.
Итоговая хлористоводородная среда не пригодна для анализа такими методами, как
ЭААС или ИСП-МС. В этих случаях следует нагревать смесь HN03-H202 до объема
приблизительно 5 мл. Фильтруют и доводят объем до 50 мл, как в предыдущем опи¬
сании. Эта методика не рекомендована для анализа Hg. Методика предназначена для
измерения концентраций обменных микроэлементов или микроэлементов, адсорбиро¬
ванных компонентами почвы (например Cd, Ni, Pb, Сг, As, Se); используется, главным
образом, для обнаружения загрязнителей промышленного происхождения. Методика
непригодна для оценки общего содержания элементов, связанных с силикатами, и не
рекомендована для общего анализа.
Альтернативная методика определения свободных оксидов железа
Эта методика представляет альтернативу методике (Deb, 1950), применяемой во многих
лабораториях (Petard, 1993).
1 г пробы помещают в пробирку для центрифугирования объемом 100 мл, добавляют
50 мл ацетатно-тартратного раствора (S7), интенсивно перемешивают стеклянной па¬
лочкой, добавляют 2 г гидросульфита натрия и хорошо перемешивают. Ставят на водя¬
ную баню, нагретую до 40 °С, выдерживают 40 мин, перемешивая содержимое каждые
5 мин. Центрифугируют в течение 3 мин при 3000 g и декантируют надосадочную жид¬
кость в мерную колбу на 1 л. Добавляют 50 мл 0,05 М раствора НС1 к остатку после цент¬
рифугирования и вновь помещают пробирку на водяную баню на 15 мин, периодически
перемешивая содержимое. Центрифугируют и декантируют надосадочный раствор в ту
же самую мерную колбу. Повторяют процедуру экстракции (тартрат-ацетат Н- гидросуль¬
фит и промывание разбавленной хлористоводородной кислотой) дважды, каждый раз
сливая экстракт в ту же мерную колбу вместимостью 1 л. Добавляют 10 мл концент¬
рированной хлористоводородной кислоты в колбу и доводят объем до 1 л деионизиро¬
ванной водой при перемешивании раствора. После выдерживания раствора в течение
ночи фильтруют раствор перед анализом на железо методами атомной спектрометрии
или спектроколориметрии.
751
Глава 31. Анализ экстрагируемых элементов и полный элементный анализ
Альтернативная методика определения экстрагируемого бора
Навеску 25 г образца помещают в колбу для кипячения объемом 250 мл с притертой проб¬
кой. Добавляют 50 мл 0,01 М раствора CaClj (S8). Гомогенизируют и кипятят с обратным
холодильником в течение 5 мин. Охлаждают, фильтруют при низкой скорости филь¬
трации через беззольную фильтровальную бумагу. Измеряют содержание бора мето¬
дом ИСП-АЭС или спектроколориметрическим методом по реакции с азометином Н.
31.2.8. Методы измерений
До появления усовершенствованных методик атомно-спектрометрических измерений
даже анализ основных элементов в почвах или породах представлял собой длительный
процесс. Анализ одного лишь элемента зачастую был востребован чаще, чем анализ всех
элементов. Методики вскрытия проб при многоэлементном анализе не были стандар¬
тизированы, в отличие от методик, описанных в разделе 31.2.3-31.2.6, и зачастую были
пригодны для анализа одного конкретного элемента. Например, концентрированная
азотная кислота использовалась для обработки проб при анализе общего фосфора (см.
раздел 29.2), анализ известняка проводили путем волюмометрического измерения дву¬
окиси углерода, выделившейся при обработке пробы кислотой (см. гл. 17).
Количественное определение общего кремния часто проводят после щелочного
сплавления (см. раздел 31.2.6), чтобы избежать его улетучивания в присутствии фто¬
ристоводородной кислоты. Измерение количества улетучившегося компонента можно
использовать для приблизительной гравиметрической оценки содержания кремния, од¬
нако для более точных результатов используют спектроколориметрию. В кислой среде
диоксид кремния вступает в реакцию с молибдатом аммония с образованием кремнемо¬
либденовой кислоты, окрашивающей реакционную среду в желтый цвет; чтобы избе¬
жать наложения сигналов фосфора, добавляют щавелевую кислоту и проводят измере¬
ния при 420 нм. Количественное определение кремния можно проводить методом ААС
в пламени оксида азота(1) — ацетилена, однако в этом случае довольно высок предел
обнаружения. Более точен анализ кремния методом ИСП-АЭС.
Фосфор практически не определяется с помощью ААС, а его анализ методом ИСП-
АЭС не обладает достаточной точностью и чувствительностью. Лучшим методом анали¬
за фосфора в растворе является спектроколориметрия, основанная на образовании фос¬
форномолибденового комплекса с молибдатом аммония в кислой среде. Содержание
комплекса желтого цвета можно определить прямыми спектрофотометрическими изме¬
рениями на волне около 420 нм, или же измерить содержание восстановленной формы
комплекса синего цвета, в этом случае измерения проводят при 830 нм (см. гл. 30).
Алюминий и титан можно измерять методом ААС в пламени оксида азота(1) — ацети¬
лена, однако этот метод не отличается высокой чувствительностью, поскольку эти эле¬
менты являются тугоплавкими. Лучше воспользоваться методом ИСП-АЭС; тем не ме¬
нее, спектроколориметрия является точной и экономичной альтернативной методикой
определения алюминия и титана. Алюминий образует комплекс красного цвета с эри-
охром цианином, а титан образует желто-оранжевый комплекс с пероксидом водорода
в сернокислой среде.
Пламенно-эмиссионная спектрометрия рекомендована для анализа щелочных метал¬
лов, в частности, натрия и калия; возможно применение классических атомно-абсорб¬
ционных или эмиссионных спектрометров и даже небольших недорогих спектрометров,
которые следует держать в резерве для анализа этих элементов (Pansu и др., 2001). Со¬
держание остальных основных элементов можно точно измерить классической ААС
752
Часть 3. Неорганический анализ
в воздушно-ацетиленовом пламени при анализе Fe, Mn, Mg, Ni (Na или К) и в пламени
оксида азота(1) — ацетилена при анализе Са. В большинстве почв ИСП-АЭС позволя¬
ет проводить одновременное количественное определение всех основных элементов, за
исключением фосфора.
Анализ следовых элементов более проблематичен, но на протяжении многих лет спе¬
циалисты занимались исследованиями в этой области (Pinta, 1962). Прогресс в области
ААС позволил усовершенствовать методики измерений: предложены гидридный метод,
метод холодных паров и метод электротермической атомизации. Многоэлементный
анализ методами ИСП-АЭС также был усовершенствован, однако для многих микроэ¬
лементов ИСП-АЭС остается менее чувствительным методом измерений по сравнению
с модифицированным ААС-анализом, например, с электротермической ионизацией.
Новая эра в анализе микроэлементов наступила с появлением ИСП-МС-аппаратуры.
Этот метод позволил исследовать почти все элементы периодической таблицы, причем
предел обнаружения часто оказывался ниже предела, достигаемого другими методами.
Кроме того, метод позволил изучить ряд изотопов, представляющих большой интерес
для геохронологических и экологических исследований. Тем не менее, высокая стои¬
мость аппаратуры и сложная методика приводят к тому, что обычной практикой явля¬
ется резервирование этого оборудования для отдельных исследовательских программ.
В методиках, описанных в этой главе, представлены следующие способы аналитичес¬
ких измерений:
• спектроколориметрическое определение Р, Si, Al, Ti (см. раздел 31.2.9);
• пламенно-эмиссионный спектрометрический анализ щелочных элементов (см.
раздел 31.2.10);
• метод ПААС (см. раздел 31.2.11);
• метод ААС в системах, использующих летучие гидриды или холодное испарение
(см. раздел 31.2.12);
• метод ЭААС (см. раздел 31.2.13);
• метод ИСП-АЭС (см. раздел 31.2.14);
• метод ИСП-МС (см. раздел 31.2.15).
31.2.9. Спектроколориметрический анализ
Фосфор
В форме ортофосфорной кислоты фосфор реагирует с молибденовой кислотой с образо¬
ванием желтого фосфоромолибдатного комплекса. Поглощение измеряют при 420 нм.
При восстановлении образуется комплекс синего цвета, который определяют при
830 нм. Метод весьма чувствителен и позволяет селективно измерять ортофосфатную
форму фосфора. Методика описана в гл. 29.
Кремний
Основные положения
При pH 1,2 оксид кремния вступает в реакцию с молибдатом аммония с образованием
кремнемолибденовой кислоты, имеющей желтую окраску. Добавление щавелевой кис¬
лоты предотвращает помехи от фосфора. Для приготовления аналитического раствора
при щелочном сплавлении рекомендуется брать пробу весом 60 мг (см. раздел 13), ана¬
литическую матрицу образует 1%-ная НС1, итоговый объем составляет 200 мл.
Глава 31. Анализ экстрагируемых элементов и полный элементный анализ
753
Реагенты
• 1,5%-ный раствор хлористоводородной кислоты.
• 10%-ный раствор молибдата аммония: растворяют 50 г в 500 мл деионизированной
воды, встряхивают и фильтруют. Раствор сохраняет стабильность лишь на протя¬
жении нескольких дней.
• Раствор щавелевой кислоты с концентрацией 50 г/л.
• Стандартный раствор: поскольку раствор оксида кремния не стабилен, стандарт¬
ный раствор следует готовить заново для каждой аналитической серии; для щелоч¬
ного сплавления (см. раздел 31.2.5) в графитовый тигель помещают 60 мг чистого
сухого оксида кремния; сплавление производят по той же процедуре, что и при
анализе пробы почвы.
• Растворы калибровочного диапазона, приготовленные разведением стандартного
раствора 1%-ным раствором НС1:
мл на 100 мл 0 10 20 50 100
% Si02 в твердой пробе почвы 0 10 20 50 100
В качестве альтернативы можно воспользоваться эталонными образцами с известным
содержанием оксида кремния.
Методика
В колбы объемом 50 мл добавляют:
• 10 мл калибровочного раствора или раствора пробы;
• 30 мл 1,5%-ного раствора НС1;
• 5 мл 10%-ного раствора молибдата аммония;
• 5 мл 5%-ного раствора щавелевой кислоты.
Гомогенизируют, переносят в колориметрическую ячейку и измеряют поглощение
при 420 нм. По калибровочному графику измеряют содержание оксида кремния в пробе
почвы без поправки на влажность. Методику можно автоматизировать, установив сис¬
тему непрерывно-проточного анализа (рис. 31.2).
Реагент
ЛЛЛ
гг
Мл/мин
Воздух
Образец
Поток,
мл/мин
2,00
2,00
0,80
3\ЛМ
420 нм
Яг
Сброс
колориметрическая ячейка
Молибдат 0,32
"аммония
_ Щавелевая 0,80
"кислота
] Перекачивание 2,00
-Промывка 1,20
Рис. 31.2. Автоматизация спектроколориметрических измерений кремния непрерывно-про¬
точным анализом в сегментированном потоке (Paycheng, 1980): SC — малый контур смесителя;
LC — большой контур; длина волны: 420 нм, ополаскивание дистиллированной водой, скорость
30 проб/ч
754
Часть 3. Неорганический анализ
Алюминий
Основные положения
С эриохром цианином алюминий образует соединение красно-фиолетового цвета; ре¬
акция очень чувствительна к условиям и протекает нестабильно. Следует строго соблю¬
дать аналитическую процедуру. pH необходимо отрегулировать на уровне 6,3. Помехи от
Fe (III) устраняют путем восстановления до Fe (II) аскорбиновой кислотой.
Реагенты
• Раствор эриохром цианина с концентрацией 1 г/л\ растворяют 1 г в 800 мл деиони¬
зированной воды, добавляют 1 мл концентрированной азотной кислоты и доводят
объем до 1 л.
• 0,5%-ный раствор аскорбиновой кислоты.
• Буферный раствор с pH 6,3: растворяют 250 г ацетата натрия (безводная форма или
эквивалентная кристаллогидратная форма) в 500 мл деионизированной воды, до¬
бавляют 10 мл уксусной кислоты. При необходимости, регулируют pH с помощью
pH-метра и доводят объем до 1 л деионизированной водой.
• Базовый раствор А1гОг с концентрацией 100 мг/л: обрабатывают 52,9 мг чистого
порошкообразного алюминия 10 мл концентрированной хлористоводородной
кислоты, доводят объем до 1 л деионизированной водой (кроме того, можно вос¬
пользоваться коммерческими стандартными растворами с более точными концен¬
трациями).
• Кшибровочныерастворы: от 0 до 50 мг АЦО/л; разбавляют базовый раствор 1 %-ной
хлористоводородной кислотой.
Методика
Рекомендуется использовать растворы, полученные растворением массы, образовав¬
шейся при щелочном сплавлении, в среде 1%-ной НС1 (см. раздел 31.2.6).
• В колбу объемом 50 мл вносят 5 мл пробы или калибровочного раствора, добавля¬
ют 15 мл раствора аскорбиновой кислоты.
• Выдерживают несколько минут для завершения реакции, затем добавляют 25 мл
буферного раствора и 5 мл эриохром цианина.
• Проводят колориметрические измерения при 535 нм.
Примечания
Если буферной емкости раствора недостаточно для поддержания pH, то обрабатывают
раствор аммиаком для нейтрализации среды до значений pH, приближенных к тем, при
которых будут проводиться аналитические измерения.
В качестве связывающего и восстанавливающего реагента вместо аскорбиновой кис¬
лоты можно использовать тиогликолят.
Анализ холостой пробы можно провести, связав железо и алюминий ЭДТА, что поз¬
воляет определить фоновое поглощение, связанное с присутствием других элементов,
которое вычитают из окончательного результата (Chariot, 1984).
Альтернативно можно приготовить эриохром цианин (Chariot, 1984): к 200 мл воды
добавляют 1 г эриохром цианина, 25 г хлорида натрия, 25 г нитрата аммония, 2 мл азот¬
ной кислоты, растворяют, доводят объем раствора до 1 л деионизированной водой. Этот
реагент оказался более эффективным при анализе экстрактов засоленных почв по срав¬
нению с описанным выше в разделе «Реагенты» (Paycheng, 1980).
Глава 31. Анализ экстрагируемых элементов и полный элементный анализ 755
Титан
Основные положения
Измеряют интенсивность желто-оранжевой окраски, в который окрашивается серно¬
кислая среда при взаимодействии титана с пероксидом водорода. Добавление фосфор¬
ной кислоты позволяет исправить смешение цветов, связанное с высоким содержанием
железа (III). Раствор не должен содержать фторидов.
В реакции с образованием окрашенных комплексов могут вступать и другие ионы,
однако их концентрации, как правило, малы (U, Mo, Nb). Исключение представля¬
ет V, который может вызывать серьезные помехи. Ванадий можно измерять одновре¬
менно с Б™; колориметрические измерения проводятся при двух длинах волн: 410 (Ti)
и 460 (V) нм. Добавление фторида маскирует комплекс титана, что позволяет измерить
содержание ванадия.
Реагенты
• 3%-ный раствор пероксида водорода: 30%-ный пероксид водорода десятикратно
разбавляют деионизированной водой.
• Серная кислота, десятикратно разбавленная деионизированной водой.
• Фосфорная кислота, трижды разбавленная деионизированной водой.
• 1 % -ный раствор хлористоводородной кислоты.
• Стандартный базовый раствор ТЮ2 с концентрацией 1 г/л: навеску 600 мг K^IiFg
помещают в платиновый тигель, добавляют несколько капель деионизированной
воды и около 3 мл концентрированной серной кислоты, выпаривают досуха; по¬
вторяют эту операцию, растворяют остаток в 5%-ном растворе H2S04 и доводят
объем до 200 мл деионизированной водой.
• Диапазон концентраций стандартных растворов: от 0 до 100 мг (ТЮ2)/л, для раз¬
бавления используют 1%-ный раствор НС1.
Методика
Для анализа основных элементов рекомендуется (см. раздел 31.2.6) использовать экстра¬
кты в 1%-ной НС1 после щелочного сплавления.
В колбу объемом 50 мл вносят 20 мл раствора пробы или калибровочного раствора,
10 мл 1/8 серной кислоты, 15 мл V3 фосфорной кислоты, 5 мл 3%-ного раствора перок¬
сида водорода. Проводят колориметрические измерения при 410-420 нм.
31.2.10. Пламенно-эмиссионная спектрометрия
Условия
Несмотря на то что атомно-абсорбционная спектрометрия часто применяется вместо
пламенно-эмиссионной спектрометрии (ПЭС), ПЭС является полезным методом из¬
мерения содержания щелочных элементов, в частности, натрия и калия. Эти элементы
легче остальных поддаются атомизации и ионизации. Чтобы добиться максимальной
атомизации и минимальной ионизации, пламя не должно быть слишком горячим. Мож¬
но использовать пламя, образующееся при горении смесей воздух - бытовой газ, воз¬
дух - бутан, воздух - пропан или воздух - ацетилен. Щелочные элементы можно также
анализировать методом плазменно-эмиссионной спектрометрии (см. раздел 31.2.14),
однако чувствительность будет ниже из-за высокой температуры плазмы.
756
Часть 3. Неорганический анализ
Эмиссия, соответствующая переходу между основным электронным уровнем и пер¬
вым возбужденным уровнем, является наиболее информативной при анализе щелочных
элементов. При низших длинах волн часто регистрируются дублетные спектральные
линии с приблизительно удвоенной интенсивностью (табл. 31.3). Дублетные линии не
всегда разделяются коммерческими спектрометрами, однако это не мешают удовлетво¬
рительному количественному анализу, когда используется весь спектр излучения.
Другие условия процесса зависят от типа аппаратуры. Элементы, отличные от щелоч¬
ных металлов, можно определять и методом ПЭС, однако, как правило, ААС предпоч¬
тительней.
Таблица 31.3. Основные длины волн щелочных элементов в пламенно-эмиссионном
спектре
Элемент
Li
Na
К
Rb
Cs
X (нм)
670,79
589,00
589,59
766,49
766,90
780,02
794,47
852,11
894,35
Как правило, спектрометрические измерения проводят при наименьших длинах волн (верхняя строка),
поскольку интенсивность соответствующих им линий почти вдвое превосходит интенсивность прочих
линий.
Калибровочный диапазон и расчеты
Состав калибровочных растворов определяется природой элемента и субстратом. Диа¬
пазон концентраций от 0 до 100 мг (К^/л обычно выбирают при анализе калия. Диа¬
пазон концентраций от 0 до 50 мг (Ыа20)/л — при анализе натрия. Можно использовать
коммерческие стандартные растворы, а также чистые коммерческие реактивы, такие
как Ь^СОз или Na2C03, для приготовления растворов по методике, аналогичной приме¬
няемой для приготовления стандартов для ААС (см. «Калибровочные растворы»). Те же
стандарты можно использовать при анализе методами АЭС и ААС.
31.2.11. Пламенная атомно-абсорбционная спектрометрия
Анализ основных элементов методом ПААС
Параметры процессов
В табл. 31.4 перечислены основные параметры процесса анализа основных элементов
в почвенных растворах; элементы классифицируют в соответствии с величинами пре¬
делов обнаружения. На практике метод не пригоден для анализа фосфора, а чувстви¬
тельность его при анализе Si, И и А1 ниже по сравнению со спектроколориметрией (см.
раздел 31.2.9). Более подробно параметры оборудования и рабочие условия процессов
описаны в отраслевых изданиях (например Wright и Stuczynski, 1996; Рати и др., 2001)
или в документации к коммерческому оборудованию.
Калибровочные растворы
Продукты
• Для приготовления калибровочных растворов можно воспользоваться коммерчес¬
кими калибровочными растворами (см. «Многоэлементная калибровка при анали¬
зе основных элементов») или коммерческими ультрачистыми реактивами. Послед¬
ние представлены в табл. 31.5.
• Оксид лантана высокочистый (La203).
Глава 31. Анализ экстрагируемых элементов и полный элементный анализ
757
Таблица 31.4. Основные рабочие условия анализа основных элементов методом ПААС в по¬
чвах (классификация в соответствии с пределами обнаружения элементов (ПО)
в: (1) чистой воде (документация Varian), (2) почвенном экстракте, получен¬
ном щелочным сплавлением со смесью, содержащей 0,5% Sr(B02)2 и 2% HN03
(Jeanroy, 1972).
Элемент
А,, нм
ПО, мкг/л (1)
ПО, мкг/л (2)
Пламя
Na
589,0
0,2
1
Воздух—С2Н2
Mg
285,2
0,3
0,8
Воздух—С2Н2
Са
422,7
1
4
N20 —С2Н2
Мп
279,5
2
5
Воздух — С2Н2
К
766,5
3
2
Воздух — С2Н2
Fe
248,3
6
12
Воздух — С2Н2
А1
309,3
30
140
n2o — с2н2
Ti
365,4
50
250
n2o—С2Н2
364,3
100
Si
251,6
300
400
N20 —С2Н2
Р
213,6
40000
X
и
1
о
сч
X
• Ультрачистая концентрированная хлористоводородная кислота.
• Ультрачистая концентрированная азотная кислота.
• Метаборат лития (см. раздел 31.2.6).
• Метаборат стронция (см. раздел 31.2.6).
• Матричный раствор (А) по процедуре для щелочного сплавления, представленной
в «Методике с метаборатом стронция» (раздел 31.2.6): растворяют 25 г метабората
стронция в 200 мл деионизированной воды и 100 мл концентрированной азотной
кислоты. Дают охладиться и доводят объем до 1 л. Образовавшийся раствор содер¬
жит 2,5% метабората и 10% азотной кислоты (в пять раз превышает концентрацию
кислотной матрицы согласно описанию «Методика с метаборатом стронция»).
• Матричный раствор (А') по процедуре для щелочного сплавления, представлен¬
ной в разделе «Методика с метаборатом лития» (раздел 31.2.6): растворяют 12,5 г
метабората лития в 200 мл деионизированной воды и 50 мл концентрированной
хлористоводородной кислоты. Дают охладиться и доводят объем до 1 л. Образовав¬
шийся раствор содержит 1,25% метабората и 5% хлористоводородной кислоты (в
пять раз превышает концентрацию кислотной матрицы согласно описанию «Ме¬
тодика с метаборатом лития»).
• Раствор лантана (L): растворяют 23,45 г La203 в 10 мл 2%-ной хлористоводородной
кислоты (10 мл воды + 2 мл концентрированной хлористоводородной кислоты),
доводят объем до 100 мл. Этот раствор содержит 20% лантана.
Методика приготовления базового раствора
Процедура для приготовления калибровочных растворов описана в работах Jeanroy, 1972
nRiandeynup., 1982. Втабл. 31.5 перечислены ингредиенты для приготовления базовых
758
Часть 3. Неорганический анализ
Таблица 31.5. Приготовление базовых растворов (Е или ЕО для анализа основных элементов
методом атомно-абсорбционной спектрометрии.
Элемент
Оксид
Максимальный
% оксида
в пробе
Концентрация
раствора,
мкг оксида/мл
Обработка
по 21.2.6
Е Е’
Используемый
продукт
Вес в мг/л
Обработка
по 21.2.6
Е Е'
А1
АЦО,
50
1250
750
А1 (порошок)
661,7
397,0
Fe
FeA
30
750
450
Fe (порошок)
524,8
314,9
Мп
МпО
5
125
75
Мп02
153,2
91,9
Mg
MgO
30
750
450
MgO
750,0
450,0
Са
СаО
30
750
450
СаС03
1339,3
803,6
Na
Na20
20
500
300
Na2C03
854,8
512,9
К
К20
30
750
450
KjC03
1101,1
660,7
Ti
ТЮ2
10
250
150
Ti (порошок)
150,0
90,0
Si
Si02
100
2500
1500
SiO,
2500,0
1500,0
Концентрации базовых растворов (колонка 4) соответствуют пятикратной максимальной концентрации
раствора для обработки проб (см. раздел 21.2.6), т. е.
♦ для обработки Е, описанной в «Методике с метаборатом стронция»: 100 мг пробы, 200 мл итогового
раствора, содержащего 0,5% Sr(B02)2 и 2% HN03;
* для обработки Е', описанной в «Методике с метаборатом лития»: 60 мг пробы, 200 мл итогового рас¬
твора, содержащего 0,25% 1лВ02 и 1% НС1.
стандартных растворов в диапазонах калибровки для двух типов вскрытия проб почвы
щелочным сплавлением, представленных в описаниях «Методика с метаборатом строн¬
ция» (раствор Е) и «Методика с метаборатом лития» (раствор Е'), с концентрацией,
в пять раз превышающей максимальное содержание элементов, обычно наблюдаемое
в почвах. Можно приготовить растворы для калибровки по одному элементу или по не¬
скольким элементам в составе комплексных калибровочных растворов. Следует избегать
присутствия силикатов в комплексных растворах, поскольку это приводит к осаждению
алюмосиликатов. Раствор диоксида кремния готовят отдельно при щелочном сплавле¬
нии чистого диоксида кремния по той же процедуре, что и при сплавлении пробы (см.
«Кремний»). Калибровочные растворы следует готовить в матрице, аналогичной матри¬
це раствора, использовавшегося при вскрытии пробы. Можно использовать коммерче¬
ские стандартные растворы для ААС. Альтернативно можно готовить растворы из высо¬
кочистых реактивов (металлов, оксидов, карбонатов, сульфатов и т. д.), как в примере,
представленном ниже.
В стакан объемом 400 мл помещают точные навески каждого стандартного продукта
из перечисленных в табл. 31.5, соответствующих используемому методу обработки про¬
бы. Добавляют несколько мл деионизированной воды, затем 20 мл азотной кислоты (для
способа обработки согласно описанию «Методика с метаборатом стронция») или 10 мл
хлористоводородной кислоты (для способа обработки согласно описанию «Методика
с метаборатом лития»). Накрывают стакан и выдерживают до полного растворения при
759
Глава 31. Анализ экстрагируемых элементов и полный элементный анализ
легком нагревании в случае необходимости. Дают охладиться, переносят в мерную кол¬
бу и доводят объем до 1 л деионизированной водой.
Методика приготовления калибровочных растворов
В мерные колбы объемом 250 мл вносят объемы базового раствора Е или Е', перечис¬
ленные в табл. 31.6. Добавляют 50 мл матричного раствора А («Методика с метаборатом
стронция») или А' («Методика с метаборатом лития»).
Таблица 31.6. Объемы базового раствора Е или Е' (табл. 31.5) на 250 мл и соответствующее
содержание оксида в % в пробах почвы для двух способов обработки (вскрытия
проб), описанных в разделе 31.2.6
Номер
Объем базового
раствора Е или Е’, мл
AljO,,
МпО,
%
MgO,
%
СаО,
%
Т’
Т
TIOJ(
%
SiO,,
%
0
0
0
0
0
0
0
0
0
0
0
1
1
1
0,6
0,1
0,6
0,6
0,4
0,6
0,2
2
2
5
5
3
0,5
3
3
2
3
1
10
3
10
10
6
1
6
6
4
6
2
20
4
25
25
15
2,5
15
15
10
15
5
50
5
50
50
30
5
30
30
20
30
10
100
К растворам, соответствующим способу обработки, описанному в разделе «Методика
с метаборатом лития», добавляют 1,25 мл раствора лантана L (при обработке, описанной
в разделе «Методика с метаборатом стронция», для коррекции помех достаточно самого
стронция). Доводят объем до 250 мл деионизированной водой.
Измеряют концентрации компонентов в почве обычно в % содержания оксида по
калибровочным кривым, построенным в соответствии с табл. 31.6, для двух способов
обработки, согласно описанию в разделе «Методика с метаборатом стронция», исполь¬
зуя пробы весом соответственно 100 мг (Е) и 60 мг (Е') на 200 мл раствора. Для других
проб почвы весом />(мг) умножают итоговый результат на 100//ЧЕ) и 60/Р(Е'), соответ¬
ственно. Кроме того, может потребоваться поправка на влажность.
В качестве альтернативы представленной методике внешней калибровки (табл. 31.6)
можно использовать базовые растворы, перечисленные в табл. 31.5, для измерений по
методу стандартных добавок.
Пределы обнаружения (ПО) измерены для чистой воды, поэтому они несколько ниже
пределов обнаружения элементов в почвенных экстрактах.
Анализ микроэлементов методом ПААС
Многие микроэлементы можно определять с помощью ПААС, если их концентрации
не слишком малы. В табл. 31.7 перечислены основные параметры процесса анализа этих
элементов.
Диапазоны концентрации калибровочных растворов определяются анализируемым
материалом. Для таких элементов, как Си, Со, Ni, диапазон обычно составляет от 0 до
2% оксида в почве. Как правило, при анализе микроэлементов (см. «Анализ основных
элементов методом ПААС» выше) нет необходимости в добавлении лантана, однако до¬
полнительные исследования должны проводиться как с добавлением, так и без добавле¬
ния лантана. Калибровочные растворы готовят по той же процедуре, что калибровочные
760 Часть 3. Неорганический анализ
Таблица 31.7. Микроэлементы, измеряемые методом ПААС в пламени воздух -
и оксид азота(1) — ацетилен.
- ацетилен
Воздух — С2- Н2 пламя
NzO - CjHj пламя
Элемент
X, нм
ПО, мкг/л
Элемент
X, нм
ПО, мкг/л
Си
324,7
3
Mo
313,3
20
Со
240,7
5
Ba
553,6
20
Ni
232,0
10
Be
234,9
1
Cd
228,8
2
Eu
459,4
1,5
Zn
213,9
1
Sr
460,7
2
Ag
328,1
2
Tm
371,8
20
Cr
357,9
6
Yb
398,8
4
Cs
852,1
4
Pb
217,0
10
Pd
244,8
10
Rb
780,0
10
Rh
343,5
5
Au
242,8
10
растворы для ИСП-АЭС (см. «Комплексные калибровочные растворы при анализе ми¬
кроэлементов» в разделе 31.2.14).
31.2.12. Анализ микроэлементов ААС с генерацией гидридов и методом
холодного пара
Гидри дная ААС
Основные положения
Некоторые элементы (см. табл. 31.8) могут образовывать гидриды при взаимодействии
с борогидридом натрия (NaBH4) в кислой среде (НС1 или H2S04). Для элемента Е можно
записать уравнение реакции:
Таблица 31.8. Элементы, определяемые гидридным методом
Элемент
ХК, мкг/л
X,HM
As
0,2
193,7
Bi
0,2
223,1
Hg
0,4
253,7
Sb
0,15
217,6
Se
0,3
196,0
Sn
0,4
235,5
Те
0,15
214,3
ХК — характеристическая концентрация, которой соответствует сигнал поглощения 0,0044 (1% погло¬
щения от фонового излучения), ХК в 5-20 раз превышает ПО; X — рекомендуемая длина волны в нм.
Глава 31. Анализ экстрагируемых элементов и полный элементный анализ
761
NaBH4 + ЗН20 + НС1 -> Н3В03 + NaCl + 8Н
Ел+ + иН -> EH Т
Л
Гкзообразные гидриды попадают в поглощающую ячейку, состоящую из кварцевой
трубки (рис. 31.3). Умеренный нагрев этой трубки до 800-900 °С воздушно-ацетилено¬
вым пламенем позволяет разрушать гидриды с образованием атомных форм элементов.
Таким образом, уровень поглощения этих элементов выше по сравнению с поглощени¬
ем при классической атомизации в пламени (см. раздел 31.2.11), и предел обнаружения
существенно снижается.
Большую часть элементов, перечисленных в табл. 31.8, можно перевести в летучие
формы при высоких температурах. Для определения их содержания в почвах следует
воспользоваться определенным способом обработки проб почвы. Мы рекомендует про¬
водить кислотное вскрытие в закрытой системе, описанное в разделе 31.2.4.
Реагенты
• 0,6%-ный раствор борогидрида натрия: в 50 мл ультрачистой воды растворяют 0,5 г
гидроксида натрия, затем 0,6 г борогидрида натрия и доводят объем до 100 мл уль¬
трачистой водой. Свежий раствор готовят ежедневно.
• V2 хлористоводородная кислота: растворяют 50 мл концентрированной хлорис¬
товодородной кислоты спектрографической чистоты в 40 мл ультрачистой воды,
встряхивают, дают охладиться и доводят объем до 100 мл.
• 10%-ный йодид калия *в ультрачистой воде.
Стандарты
Измерения в диапазоне концентраций 0-100 мкг/л можно проводить для всех элемен¬
тов, перечисленных в табл. 31.8. Иногда калибровочные кривые имеют наиболее вы-
Нагреваемая измерительная ячейка
Рис. 31.3. Атомно-абсорбционная спектрометрия с генерацией гидридов
762
Часть 3, Неорганический анализ
раженный линейный характер в диапазоне от 0 до 20 мкг/л. Проще начинать с ком¬
мерческих стандартных растворов. Например, при анализе мышьяка можно начинать
с разбавления коммерческого раствора с концентрацией 1000 мг (As)/л до концентрации
1 мг (As)/n (два последовательных разбавления 10 мл в 100 мл); в результате получают
базовый раствор, который используют для приготовления калибровочных растворов.
В четыре мерных колбы объемом 100 мл вносят 0,0,5,1 и 2 мл базового раствора мышь¬
яка с концентрацией 1 мг (As)/n. Объемы доводят до 100 мл; получают калибровочные
растворы с концентрациями 0, 5, 10 и 20 мкг (As)/r. Итоговая среда должна быть такой
же, как и раствор реальной пробы (при вскрытии пробы, описанном в разделе 31.2.4,
добавляют 4 мл концентрированной азотной кислоты и получают 4%-ную азотнокислую
среду).
Для отдельных элементов, таких как мышьяк, перед анализом рекомендуется прово¬
дить восстановление, чтобы в аналитической среде присутствовала только одна форма
элемента, а именно As111. Чувствительность анализа зависит от выхода образующихся
гидридов, который связан со степенью окисления этого элемента в растворе. Смешива¬
ют пробу раствора As с 5 мл 1/2 НС1 + 5 мл раствора 10%-ным KI. Нагревают в течение
20 мин в водяной бане при 60 °С, дают охладиться и доводят объем до 50 мл ультрачи-
стой водой.
Расчет
Содержание анализируемого элемента в нг (элемента)/г почвы Трасчитывают по урав¬
нению
T=(X-V-D)fP,
где X — содержание данного элемента, определенное по калибровочной кривой, мкг/л;
V — объем раствора, использованного для обработки пробы, мл; D — вероятный коэф¬
фициент разбавления перед анализом; Р— вес аналитической пробы почвы, г.
Метод холодного пара
Основные положения
Этот весьма чувствительный метод используется исключительно для определения рту¬
ти. Конструкция прибора аналогична конструкции, применяющейся для гидридного
метода, представленной на рис. 31.3. Единственное отличие состоит в геометрии изме¬
рительной трубки, позволяющей улавливать испарения на выходе. Перистальтический
насос позволяет добавлять в систему растворы дихлорида олова и гидроксиламина гид¬
рохлорида, восстанавливающие ртуть до простого состояния. Пар& элементарной рту¬
ти — Hg° переносятся в измерительную трубку потоком инертного газа, где измеряется
поглощение при 253,7 нм.
Реагенты
• 10%-ный раствор дихлорида олова: растворяют 10 г дихлорида олова в 10 мл уль-
трачистой хлористоводородной кислоты, нагревают до полного растворения, дают
охладиться, доводят объем до 100 мл ультрачистой водой; свежий раствор готовят
ежедневно.
• 10%-ный раствор гидроксиламина гидрохлорида: растворяют 10 г в 100 мл ультра¬
чистой воды.
• 1/2 раствор хлористоводородной кислоты в ультрачистой воде.
Глава 31. Анализ экстрагируемых элементов и полный элементный анализ
763
• Калибровочные растворы, содержащие 0, 5, 10, 20 мкг (Hg)/n, приготовленные из
базового раствора с концентрацией 1 мг (Hg)/n: в мерные колбы объемом 100 мл
вносят 50 мл ультрачистой воды, добавляют 4 мл концентрированной азотной кис¬
лоты спектрографической чистоты и 0, 0,5, 1 и 2 мл базового раствора, соответ¬
ственно. Доводят объем до 100 мл ультрачистой водой.
• Раствор для заполнения ловушки паров ртути: растворяют 3 г йодида калия и 0,25 г
йода в 100 мл деионизированной воды.
Формулы расчетов аналогичны формулам, представленным в разделе «Расчеты».
31.2.13. Анализ микроэлементов электротермической ААС
В настоящее время ЭААС является одним из самых чувствительных методов анализа ми¬
кроэлементов. Для многих элементов предел обнаружения методом ЭААС ниже предела
обнаружения методом ПААС в 100-1000 раз. Оборудование для ЭААС особенно эф¬
фективно работает в комбинации с системами коррекции неселективного поглощения,
в особенности с системами коррекции на основе эффекта Зеемана (Рати и др., 2001).
Далее, метод наиболее пригоден для микроанализа в жидких средах, поскольку требует¬
ся лишь несколько мкл раствора для каждого анализируемого элемента (обычно 20 мкл).
Успешная работа ЭААС-оборудования основана на оптимизации параметров прибо¬
ров для анализа каждого элемента в конкретной аналитической среде и очень точной
регулировке оптимальных температур атомизации и разложения. Химические помехи
можно минимизировать с помощью растворов, корректирующих матрицу, добавленных
в капиллярную трубку прй инжекции (например, 1 мкл на 10 мкл пробы). В качестве
модификатора матрицы часто рекомендуют использовать нитрат палладия (Pd(N03)2).
Можно пользоваться и другими модификаторами, такими как Mg(N03)2, Ni(N03)2 или
NH4H2P04 (см. например, Hoenig и Kersabiec, 1990). В табл. 31.9 перечислены основ¬
ные микроэлементы, которые можно анализировать методом ЭААС. Кроме того, чув¬
ствительность этого метода при анализе основных элементов значительно превышает
чувствительность ПААС (см. «Анализ основных элементов методом ПААС» в разделе
31.2.11); тем не менее, анализы такого типа не оправдывают применения ЭААС, которая
является более сложной в исполнении и отличается меньшей повторяемостью по срав¬
нению с ПААС. Поэтому эти элементы не включены в табл. 31.9.
Калибровочный диапазон изменяют в зависимости от анализируемого элемента;
растворы и диапазон концентраций зачастую аналогичны описанным в разделе 31.2.12.
Формулы расчеты идентичны представленным в «Расчетах» в разделе 31.2.12.
31.2.14. Анализ методом атомно-эмиссионной спектрометрии
с индуктивно связанной плазмой (ИСП-АЭС)
Преимущества и ограничения
ИСП-АЭС получает все более широкое распространение в геохимических исследова¬
ниях, благодаря тому что анализ является многоэлементным. При анализе большин¬
ства элементов чувствительность метода аналогична чувствительности ПААС (см. раз¬
дел 31.2.11), однако ИСП-АЭС менее чувствительна по сравнению с ЭААС (см. раздел
31.2.13), гидридной ААС и ААС холодного пара (см. раздел 31.2.12) и ИСП-МС (см. раз¬
дел 31.2.15).
Большинство помех возникает из-за спектральных интерференций, вызванных на¬
ложением нескольких эмиссионных линий, наблюдаемых в сложных средах при высо-
764
Часть 3. Неорганический анализ
Таблица 31.9. Микроэлементы в почвах, анализируемые методом ЭААС
Элемент
X, нм
ХК, мкг/л
Элемент
X, нм
ХК, мкг/л Элемент
X, нм
ХК, мкг/л
Ag
328,1
0,035
As
193,7
0,5
Au
242,8
0,22
Ва
553,6
0,85
Be
234,9
0,025
Bi
223,1
0,45
Cd
228,8
0,01
Co
240,7
0,21
Cr
357,9
0,075
Cs
852,1
0,55
Cu
324,7
0,3
Ga
294,4
0,23
Ge
265,1
0,45
In
303,9
0,35
Li
670,8
0,2
Mo
313,3
0,35
Ni
232,0
0,24
Pb
217,0
0,28
Pd
244,8
0,43
Pt
265,9
3,5
Rb
780,0
0,05
Rh
343,5
0,4
Ru
349,9
0,75
Sb
217,6
0,5
Se
196,0
0,7
Sn
235,5
0,5
Sr
460,7
0,1
Tb
432,7
0,18
Те
214,3
0,45
T1
276,8
0,75
V
318,5
1.1
Yb
398,8
0,15
Zn
213,9
0,0075
ХК — характеристическая концентрация в чистой воде, дающая сигнал поглощения на уровне 0,0044
(1% от фонового излучения). ХК больше, чем ПО; X - рекомендуемая длина волны в нм. Основные эле¬
менты почв не включены в данную таблицу.
ких температурах. Механические помехи могут возникать из-за изменений во входящем
потоке анализируемого раствора и закупорки распылителей там, где растворы содержат
слишком много растворенных материалов.
Другая проблема связана с выбором стратегии многоэлементной калибровки. Диа¬
пазоны концентраций калибровочных растворов должны приблизительно соответство¬
вать ожидаемым значениям концентраций в анализируемых средах. Влияние матрицы,
как правило, несущественно, однако может проявиться изменениями чувствительности
и изгибом калибровочной кривой, который может отличаться от изгиба кривой, постро¬
енной с помощью стандартных растворов. Две основные причины изменений сигнала
эмиссии: (i) изменение температуры плазмы, (И) изменения во входящем аналитиче¬
ском растворе в процессе анализа. Два возможных способа коррекции помех такого типа
основаны на применении внутренних стандартов. Рекомендуется (Ramsey и Coles, 1992)
использовать корректирующую программу, основанную на введении двух внутренних
стандартов: рубидия — для коррекции изменений во входящем потоке раствора про¬
бы, и ртути — для коррекции температуры эмиссии. При анализе основных элементов
(Si, Al, Fe, Mg, Са и Na) предлагается использовать в качестве внутренних стандартов
галлий, кадмий и литий для снижения относительного стандартного отклонения при¬
близительно до 0,5% (Walsh, 1992) Элементы, добавляемые в качестве внутренних стан¬
дартов, не анализируют; их содержание в исходном анализируемом растворе считают
несущественным.
Случайная погрешность при применении классической ИСП-МС для анализа гео¬
логических материалов составляет около 2% (Walsh, 1992), т. е. порядок величины коле¬
баний такой же, как и для других аналитических методик. В большинстве случаев мож¬
но воспользоваться классической методикой внешней калибровки. Для аналитических
сред, содержащих небольшое количество растворенных твердых материалов, точность
измерений увеличивается при применении многоэлементных калибровочных растворов
с концентрацией элементов, максимально приближенной к их концентрации в анали¬
тической среде. Кроме того, анализ эталонных образцов методом ИСП-МС (так же, как
Глава 31. Анализ экстрагируемых элементов и полный элементный анализ
765
и другими методами) при межлабораторных сравнениях (Abbey, 1992) позволяет прове¬
рить точность измерений в аналитических сериях.
Многоэлементная калибровка при анализе основных элементов
Среда для многоэлементных калибровочных растворов должна быть максимально при¬
ближена по составу к раствору пробы. Описанная здесь процедура приготовления рас¬
творов связана с общим анализом по методике с использованием метабората лития,
представленной в разделе «Методика с метаборатом лития». Вес аналитической пробы рав¬
няется 60 мг на 200 мл итогового объема. Итоговая среда содержит 1% НС1, 0,25% УВ02.
В табл. 31.10 представлены калибровочные диапазоны для элементов Al, Fe, TI, Са,
Mg, Na, К и Мп, количественный анализ элементов Si и Р проводится отдельно. Для
каждого элемента готовят холостую пробу (0) и 3 калибровочных раствора (I, II, III
в табл. 31.10), соответствующих вероятному содержанию оксида в анализируемой почве
или породе.
Вносят соответствующие объемы каждого из базовых растворов с концентрацией 1
г/л в мерную колбу вместимостью 1 л (см. табл. 31.10), добавляют 200 мл раствора, содер¬
жащего 12,5 г (1лВ02)/л, в 5%-ный раствор НС1. Доводят объем до 1 л деионизированной
водой для получения итоговой среды, аналогичной растворам проб после обработки (см.
«Методика с метаборатом лития»), т. е., содержащей 0,25% UB02, 1% НС1. Эти много¬
элементные растворы калибровочного диапазона позволяют получать результаты непо¬
средственно в процентных долях оксидов в пробах почвы из исследуемых образцов ве¬
сом 60 мг (табл. 31.10), при итоговом объеме экстракционного раствора, составляющем
200 мл. Для анализируемой пробы, вес которой равен Л/мг, результат следует умножить
на 60/М
Таблица 31.10. Приготовление многоэлементных растворов калибровочного диапазона
при анализе основных элементов; содержание элементов, выраженное в %
оксидов в пробе почвы (методика вскрытия пробы описана в разделе 31.2.6
«Методика с метаборатом лития»: 60 мг пробы, 200 мл итоговый объем),
коммерческие базовые растворы с концентрацией 1 г (элемента)/л
Раствор оксцца
AljOj,
%
FeA>
%
НО,
*2
СаО,
%
MgO,
%
Na20,
%
к2о,
%
МпО,
%
Мл раствора
0
0
0
0
0
0
0
0
0
с концентрацией
I
12,5
15
2,5
15
12,5
10
20
2,5
1 г (эл.)/л
п
25
30
5
30
25 ,
20
40
5
ш
50
60
10
60
50
40
80
10
Состав раствора, мг
0
0
0
0
0
0
0
0
0
(оксида)/л
I
23,62
21,45
4,17
20,99
20,73
13,48
24,09
3,23
II
47,24
42,89
8,34
41,98
41,45
26,96
48,18
6,46
III
94,47
85,78
16,68
83,95
82,90
53,92
96,37
12,91
%-ное содержание
0
0
0
0
0
0
0
0
0
оксида в пробе почвы
I
7,87
7,15
1,67
7,00
6,91
4,49
8,03
1,08
II
15,75
14,30
2,78
13,99
13,81
8,99
16,06
2,15
III
31,49
28,59
5,56
27,98
27,63
17,97
32,12
4,30
766
Часть 3. Неорганический анализ
Анализ фосфора методом ИСП-АЭС является не очень точным и не слишком чув¬
ствительным. Тем не менее, метод позволяет определить содержание Р, если оно не
слишком низкое. Растворы калибровочного диапазона готовят, начиная с базового рас¬
твора с концентрацией Р в 1 г /л; концентрация Р составляет 0, 10, 25, 50, 75 мкг/мл,
что соответствует содержанию Р205 в количестве 0, 22,90, 57,26, 114,52 и 171,77 мкг/мл
в растворе и 0; 7,63; 19,09; 38,17; 57,25% в исходной пробе.
Растворы, содержащие кремний, не очень стабильны. При анализе кремния следует
использовать стандартные эталонные образцы (например Geostandard, CRPG-CNRS 54
501 Vandoeuvre les Nancy, France) и проводить обработку, аналогичную обработке проб.
Альтернативно можно использовать точную навеску чистого коммерческого диоксида
кремния. Эти растворы долго не хранятся, анализ кремния должен быть проведен в те¬
чение двух дней после обработки.
Комплексные калибровочные растворы для анализа микроэлементов
Метод ИСП-АЭС позволяет определять 70 элементов периодической таблицы.
Стандартные растворы для многоэлементной калибровки для анализа микроэлементов,
так же как и при анализе основных элементов (см. «Многоэлементная калибровка для
анализа основных элементов»), следует готовить в определенной аналитической среде.
Калибровочные растворы, представленные в табл. 31.11, соответствуют восьми микро¬
элементам, которые чаще всего анализируют в почвах и породах. Растворы готовят в той
же среде, что и растворы для полного вскрытия пробы в открытом сосуде (см. «Методи¬
ка» в разделе 31.2.3), и, следовательно, они не включают элементы, которые могут уле¬
тучиваться при обработке кислотой, например, Pb, Cd, As или Se. Результат измерения
Сг может оказаться заниженным при неполном вскрытии некоторых тугоплавких ми¬
нералов. Так же, как и в методике «Многоэлементная калибровка для анализа основных
элементов», калибровочные растворы готовят из коммерческих базовых растворов.
Растворы готовят в мерных колбах объемом 1 л путем разбавления базовых растворов,
перечисленных в табл. 31.11. Вносят в колбу 500 мл ультрачистой деионизированной
воды, затем 50 мл концентрированной хлористоводородной кислоты, доводят объем до
1 л ультрачистой водой при температуре 20 °С. Раствор 0 (холостой) можно приготовить
по той же процедуре, но без добавления базового стандартного раствора.
Таблица 31.11. Примеры многоэлементных калибровочных растворов для анализа
микроэлементов
Элемент
Мл базового раствора
с концентрацией 1 г/л
на 1 л раствора
Раствор III, мг/л
Раствор II, мг/л
Раствор I, мг/л
Ва
15
15
7,50
3,75
Be
1
1
0,50
0,25
Со
5
5
2,50
1,25
Сг
5
5
2,50
1,25
Си
2,5
2,5
1,25
0,62
Ni
5
5
2,50
1,25
Sr
15
15
7,50
3,75
Zn
5
5
2,50
1,25
767
Глава 31. Анализ экстрагируемых элементов и полный элементный анализ
Расчет
Для пробы почвы весом Р г (0,5 г) в итоговом объеме К(мл) раствора (50 мл) концентра¬
цию Cs (мкг/г) каждого элемента образца определяют по соответствующей точке калиб¬
ровочной кривой С{ (мкг/мл) и рассчитывают по формуле С8 = С{ V/P.
Калибровочные растворы для анализа редкоземельных элементов
Готовят калибровочные растворы так же, как и для других микроэлементов (см. выше
«Комплексные калибровочные растворы для анализа микроэлементов»), разбавлением
базовых растворов в среде 5%-ного раствора хлористоводородной кислоты, аналогич¬
ной используемой для полного вскрытия проб в открытых системах (см. «Методика»
в разделе 31.2.3). Можно использовать коммерческие базовые растворы. Объемы наибо¬
лее концентрированного раствора III перечислены в табл. 31.12 (Le Cornec, IRD, Bondy,
France, персональное сообщение). Все стандартные растворы приготовлены в среде 5%-
ного раствора хлористоводородной кислоты.
Рабочие условия для ИСП-АЭС-оборудования
Поскольку для каждого типа оборудования существуют специальные требования, реко¬
мендуется ознакомиться с описаниями в специальной литературе и соблюдать инструк¬
ции производителя. В табл. 31.13 перечислены некоторые значения длин волн, которые
можно использовать при плазменно-эмиссионном спектрометрическом анализе эмис¬
сионных линий элементов почвы. При необходимости проведения других измерений
следует обратиться к более полным публикациям (например, Boumans, 1981; Winge и др.,
1982).
Таблица 31.12. Приготовление многоэлементных калибровочных растворов для анализа
редкоземельных элементов (+ иттрий)
Элемент
Базовый раствор, г/л
Мл базового
раствора для 1 л
раствора III
Раствор
III, мг/л
Раствор II,
мг/л
Раствор I,
мг/л
Се
1.0
2,0
2,00
1,000
0,500
Dy
0,1
5,0
0,50
0,250
0,125
Ей
0,1
2,5
0,25
0,125
0,062
Ег
0,1
5,0
0,50
0,250
0,125
La
1,0
1,0
1,00
0,500
0,250
Nd
1,0
1,0
1,00
0,500
0,250
Sm
0,1
5,0
0,50
0,250
0,125
Yb
0,1
5,0
0,50
0,250
0,125
Y
1,0
2,0
2,00
1,000
0,500
31.2.15. Анализ методом масс-спектрометрии с индуктивно связанной
плазмой
Преимущества и недостатки
ИСП-МС представляет собой масс-спектрометрический анализ ионов, образующихся
в плазме, в которую инжектируют раствор пробы (рис. 31.4). Теоретически все элементы
768 Часть 3. Неорганический анализ
Ткблица 31.13. Некоторые длины волн (X, нм) эмиссионных линий, используемых в анализе
почв методом ИСП-АЭС, и соответствующие пределы обнаружения (ПО)
Элемент
X) нм
ПО, мг/л
Элемент
X, нм
ПО, мг/л
Элемент
X, нм
ПО, мг/л
А1
396,152
4
Ti
337,280
i
Si
288,158
18
167,081
1,5
334,941
0,6
251,611
5
Fe
259,940
1,5
Mn
257,610
0,3
Са
317,933
6
Na
588,995
1
P
213,618
19
393,366
0,03
177,499
18
Mg
285,213
0,9
К
769,896
20
279,553
0,1
766,490
10
Sr
421,552
0,06
Cu
324,754
2
Co
228,616
5
407,771
0,02
Ba
455,403
0,07
Cr
267,716
4
Ni
231,604
5,5
Zn
213,856
0,9
La
408,672
0,02
Ce
413,380
9
Y
371,030
0,2
379,478
0,02
418,660
7,5
Nd
430,358
2,5
Dy
353,603
Sm
359,262
8
401,225
2
353,170
0,3
442,434
7
Yb
328,937
0,3
Eu
381,967
0,3
Примечания: ПО (мкг/л) измеряют в чистой водной матрице, для почвенных экстрактов ПО может быть
выше.
- регистрация линии А1 (при 167,081 нм) и Р (при 177,499 нм) требует проведения продувки оптического
канала и монохроматора (вакуумом, Аг, N2), чтобы предотвратить поглощение 02.
- линия Са (при 393,366 нм) может быть слишком чувствительна к высоким концентрациям;
- линия К (при 766,490 нм) может перекрываться линией Mg;
- линия La (при 379,478 нм) перерывается линиями V и Fe;
- для линии Sm (при 442,434 нм) спектральный фон возмущен линией аргона, расположенной рядом.
периодической таблицы и их изотопы можно определять масс-спектрометрией; кроме
того, этот тип регистрации отличается чрезвычайной чувствительностью.
Однако на практике эксплуатация оборудования такого типа весьма сложна и тре¬
бует присутствия опытного оператора. Стоимость оборудования высока, настройка его
занимает много времени. Все анализы и обработки должны проводиться в обеспылен¬
ном помещении. Персонал должен носить защитную одежду, лабораторная стеклянная
посуда — тщательно вымыта, а реагенты — высокой степени чистоты. Эти требования
аналогичны требованиям к исполнению других методик анализа микроэлементов, та¬
ких как ЭААС (см. раздел 31.2.13), однако в данном случае соблюдение этих правил еще
более важно.
Другая важная проблема состоит в выборе изотопов и комбинаций изотопов, которые
следует учитывать при анализе данного элемента. Масс-спектр представляет собой за¬
пись сигнала детектора (в единицу времени), соответствующего параметру отношения
m/z для каждой ионизированной частицы с массой т и зарядом z. Благодаря высокой
температуре аргоновой плазмы (>5000 К), как правило, все химические компоненты
подвергаются атомизации и ионизации.
Тем не менее, температура очень быстро снижается на выходе плазмы и входе в спек¬
трометр, и может происходить рекомбинация ионов, в особенности, в пробах, содержа¬
щих в большом количестве такие компоненты, как аргон, кислород, азот, ионы, про-
Глава 31. Анализ экстрагируемых элементов и полный элементный анализ
769
Рис. 31.4. Масс-спектрометр с индуктивно связанной плазмой
исходящие из кислой среды, и основные матричные ионы. Чрезвычайно важно учесть
все атомные и молекулярные компоненты, которые могут попадать в спектрометр даже
в нестабильном состоянии. Это может вызвать изобарические ошибки (одинаковое зна¬
чение m/z для двух компонент), которые необходимо установить.
Многие авторы предлагают решения проблем помех. Большая часть элементов имеют
по несколько изотопов, о распространенности которых в природе имеется информация;
это можно использовать для устранения помех при анализе. Труднее найти решение
проблемы для моноизотопных элементов (со 100%-ным естественным содержанием,
табл. 31.14).
В табл. 31.14 перечислены наиболее широко применяемые изотопы и некоторые по¬
мехи, однако в зависимости от типа исследования возможен выбор других вариантов.
Например, при палеоклиматологических исследованиях (Le Сотес и Cortege, 1997) од¬
новременно измерялось содержание U, Са и Sr в кораллах. Существует значительная
разница между концентрациями урана, присутствующего в ультрамикроколичестве,
и основных элементов Са и Sr. При анализе урана используют наиболее распространен¬
ный изотоп 238U. Изотоп 40Са использовать нельзя из-за помех от 40Аг. Из-за высокой
концентрации кальция изотопы 44Са (природное содержание — пс: 2,086% от общего
кальция) и 42Са (пс: 0,647%) не используются, поскольку происходит насыщение детек¬
тора. Вместо них используются изотопы 43Са (пс: 0,135%) и^Са (пс: 0,187%), однако
следует проверять взаимовлияния между 43Са-48Са и 86Sr.
Другая проблема ИСП-МС связана с нестабильностью измерений при использова¬
нии аналитических растворов с большим количеством растворенных твердых веществ
или твердых частиц. Схема на рис. 31.4 иллюстрирует трудности, связанные с настрой¬
кой и эксплуатацией оборудования. Внутри горелки образуется аргоновая плазма при
атмосферном давлении с температурой более 5000 К, в то время как масс-спектрометр
функционирует при обычной температуре и под вакуумом. Вакуумные насосы должны
обеспечивать сильный поток. Интерфейс между горелкой и спектрометром состоит из
двух конусов с небольшими центральными отверстиями, через которые газообразные
вещества покидают плазму со сверхзвуковой скоростью. Колебания потока в интер¬
фейсе могут привести к серьезной нестабильности в системе. Конусы следует регулярно
очищать. Для контроля стабильности в ходе измерения рекомендуются к применению
770 _ Часть 3. Неорганический анализ
1кблица 31.14. Изотопы, часто применяемые в ИСП-МС анализе (по Jarvis, 1994b; Navez, 1997)
Элемент'
m/z
Распростра¬
ненность
Помехи
Элемент
m/z
Распростра¬
ненность
Помехи
Li
1
92,5
Be
9
100
В
11
80
Na
23
100
Mg
24
79
A1
27
100
Si
29
4,7
NH
P
31
100
NO, NOH
s
33
0,75
OjH
Cl
35
75,8
Малая
чувстви¬
тельность
к
39
93,3
Ca
44
2,08
n2o
Sc
45
100
CaH
Ti
47/49
7,3/5,5
SOH, CaH
V
51
99,7
CIO
Cr
52/53
83,8/9,5
C10H
Mn
55
100
Fe
56/57
91,7/2,2
ArO, ArOH
Co
59
100
Ni
60
26,1
Cu
63/65
69,2/30,8
Zn
66/68
27,9/18,8
S2
Ga
71
39,9
Ge
72/73
27,4/7,8
FeO, FeOH
As
75
100
ArCl
Se
77
7,6
ArCl
Br
79/81
50,7/49,3
Rb
85
72,2
Sr
88
82,6
Y
89
100
Zr
90
51,4
Nb
93
100
Mo
95
15,91
Ru
99/101
12,7/17,0
Rh
103
100
Pd
105
22,3
Ag
107/109
51,8/48,2
Cd
111
12,8
In
115
95,7
Sn
Sn
118
24,2
Sb
121
57,3
Те
125
7,1
I
127
100
Cs
133
100
Ba
137
11,2
La
139
99,9
Ce
140
88,5
Pr
141
100
Nd
143/145/146
44,4
Sm
147/149/152
28,8/26,7
BaOH, CeO
Eu
151/153
47,8/52,2
BaO
Gd
157
15,7
PrO, CeOH
Tb
159
100
Dy
163
24,9
NdOH, SmO
Ho
165
100
SmO
Er
166/167
33,4/56,3
SmO, NdO
Tm
169
100
Yb
172/173
21,8/38,1
GdO
Lu
175
97,4
GdOH
Hf
178
27,2
DyO
Та
181
99,9
W
182
26,3
Re
185
37,4
Os
189
16,1
Ir
193
62,7
Pt
194/195
32,9/33,8
Au
197
100
Hg
202
29,8
T1
205
70,5
Pb
206/207/208
98,6
Сумма
изотопов
Bi
209
100
Th
232
100
U
238
99,3
771
Глава 31, Анализ экстрагируемых элементов и полный элементный анализ
такие методы, как внутренняя калибровка и изотопное разбавление. Необходимо уста¬
новить временной интервал и число ополаскиваний между инжекциями, позволяющие
избежать эффекта памяти при переходе от одной пробы к другой. При анализе матрич¬
ных растворов с небольшим количеством растворенных твердых веществ стабильность
измерений сравнима со стабильностью плазменно-эмиссионной спектрометрии, а чув¬
ствительность приблизительно в 1000 раз выше.
Несмотря на вышеупомянутые трудности, плазменная масс-спектрометрия является
очень перспективным методом благодаря ее высокой чувствительности и, в особенно¬
сти, благодаря доступности большого числа изотопов-природных элементов. Иссле¬
дования диапазона возможностей применения метода в геохимии (Falkner и др., 1995),
космохимии (Shinotsuka и Ebihara, 1997), педологии (Soltanpour и др., 1996), различных
экологических дисциплинах (Trolard и др., 2002), седиментологии и т. д. только начина¬
ются. Несомненно, метод способствует прогрессу во всех областях почвоведения.
Исполнение
Приготовление растворов проб для ИСП-МС должно быть особенно тщательным, что¬
бы избежать изобарических помех от матричного раствора. В качестве итоговой анали¬
тической среды рекомендуется использовать разбавленную азотную кислоту (см. раздел
31.2.2), начиная с кислотных разложений (см. раздел 31.2.3 и 31.2.4) после тщательного
удаления других кислот. В качестве реагента для щелочного сплавления рекомендуется
также использовать метаборат лития (см. раздел 31.2.6). Приготовление многоэлемент¬
ных растворов для калибровки аналогично приготовлению растворов для плазменно¬
эмиссионной спектрометрии (см. раздел 31.2.14). Кроме того, можно воспользоваться
микроволновым расщеплением (Das и др., 2001).
Методы внешней калибровки (Falkner и др., 1995) позволяют обеспечить приемлемую
прецизионность измерений (коэффициент вариации 3-8%) вдали от предела обнаруже¬
ния (ПО) при условии применения аналогичных сред для разбавления калибровочных
растворов и растворов проб. Измерения должны производиться через достаточные про¬
межутки времени, необходимо обеспечить адекватное промывание системы, чтобы из¬
бежать проявлений эффекта памяти. В IRD лаборатории Bondy (France) степень преци¬
зионности метода внешней калибровки, как правило, ниже 3% для концентраций выше
ПО и около 5-10% для концентраций, близких к ПО (Le Сотес, IRD Bondy, France,
персональное сообщение). Метод стандартных добавок также помогает улучшить ре¬
зультаты в случае сложных и переменных матриц. Использование метода внутренних
стандартов позволяет улучшить прецизионность как метода внешней калибровки, так
и метода стандартных добавок на 2—5%. Следует применять внутренние стандарты,
близкие к аналиту по массе и потенциалу ионизации. Это делает возможной коррек¬
цию измеряемого сигнала путем соотнесения его с сигналом внутреннего стандарта,
а также с сигналами стандартных растворов или проб. В методе изотопного разбавления
используют один из изотопов анализируемого элемента в качестве внутреннего стандар¬
та, что минимизирует погрешность метода внутренних стандартов, возникающую из-за
близости значений массы и потенциала ионизации двух изотопов. В известные объемы
титрованных растворов необходимо внести определенные объемы с известными кон¬
центрациями добавок, обогащенных желательным изотопом. Затем можно рассчитать
концентрацию анализируемого элемента с высокой степенью точности (0,1-1%).
Метод ИСП-МС можно использовать для определения изотопных отношений, хотя
и с меньшей точностью в сравнении с методом термоионизационной масс-спектроме-
трии (ТИМС).
772
Часть 3. Неорганический анализ
31.3. Анализ в твердых средах
31.3.1. Методы
Химический анализ твердых веществ всегда был более сложным по сравнению с анали-
зом растворов. Атомная структура твердых веществ очень сложна, электроны внешних
оболочек атомов участвуют в процессах электропроводимости и в валентных взаимодей¬
ствиях, поэтому прямой анализ твердых тел весьма затруднителен. Раньше существовал
один путь: требовалось разрушить решетку, нарушить целостность атомных структур,
после чего проводить обычный химический анализ.
Однако ситуация совершенно изменилась после ряда открытий в области строения
атома, квантовой химии и взаимодействий между веществом и излучением. Эти фун¬
даментальные открытия привели к росту количества доступных методик анализа, что
позволило наблюдать различные энергетические состояния материи (Pansu и др., 2001).
Изучение структуры твердых веществ методами минералогии связано с низшими
энергетическими уровнями (см. часть 1). Оптическая и электронная микроскопия ис¬
пользует фотоны или электроны, пропускаемые или отражаемые поверхностными сло¬
ями, для формирования изображений. Рентгеновские лучи или электронное излучение
с более глубоких кристаллических слоев позволяют охарактеризовать кристаллические
структуры. Колебания, вызываемые инфракрасным излучением, делают возможным из¬
учение свойств некоторых молекулярных связей.
Высшие энергетические уровни возбужденного состояния используются в процессе
физико-химического анализа твердых веществ. В проводящих материалах применение
потоков электронов высокой интенсивности позволяет атомизировать вещество с пере¬
ходом в газообразное состояние и возбуждением внешних электронных оболочек ато¬
мов. Излучение, испускаемое в процессе релаксации, позволяет охарактеризовать атомы
и оценить их количество. Методы дуговой или искровой спектрометрии напоминают
методы, использующие для атомизации другие энергетические источники, такие как
пламенная или плазменная эмиссионная спектрометрия, описанные в разделе 31.2.
Возбуждение может также затронуть внутренние электронные оболочки атомов с бо¬
лее сложными процессами релаксации. Существует ряд доступных спектроскопических
методов, в частности рентгенофлуоресцентных, выбор которых зависит от источника
возбуждения и излучения для целевого материала. Микрозондирование позволяет по¬
лучить точную карту химического состава твердых материалов на микроскопическом
уровне. И наконец, возбуждение может оказать воздействие на само атомное ядро, как
правило, это наблюдается при бомбардировке нейтронами, что делает возможным ана¬
лиз испускаемого излучения при релаксации атомного ядра (нейтронно-активацион¬
ный анализ).
Большую часть спектрометрических методик, описанных в разделе 31.2, теоретически
можно использовать и для анализа твердых материалов. Некоторые из них, например,
ЭААС с использованием эффекта Зеемана, применяются на практике. Тем не менее, ме¬
тодологические проблемы ограничивают их применение при анализе твердых матери¬
алов. Основной трудностью при пламенной или плазменной спектрометрии, ААС или
плазменной масс-спектрометрии является достижение воспроизводимости при вводе
твердой пробы в источник атомизации. Для решения этой проблемы проведено множе¬
ство исследований и выдвинут ряд предложений. Проблема была отчасти решена путем
ввода пробы в жидкость в виде суспензии (взвеси). И все же анализ твердых проб ни¬
когда не был особенно успешным. Вследствие этого большинство аналитиков предпо¬
читают методы с переведением пробы в раствор, описанные в разделе 31.2, поскольку
773
Глава 31. Анализ экстрагируемых элементов и полный элементный анализ
они хорошо изучены и намного лучше воспроизводимы. Это предпочтение зачастую
подкреплялось трудностью с получением стандартов с твердой матрицей, аналогичной
матрице пробы. В наши дни этот аргумент утратил актуальность благодаря доступности
эталонных образцов.
Для анализа методами дуговой и искровой спектрометрии требуются проводящие об¬
разцы, и поэтому его нельзя непосредственно использовать для основных почвенных
материалов. Пробу, как правило, готовят смешиванием с порошком графита, чтобы ма¬
териал стал проводящим. Несомненно, это одна из причин того, что эти методы получи¬
ли более широкое распространение в металлургии, чем в геологии и почвоведении, где
они часто останавливались на стадии качественного или полуколичественного анализа,
несмотря на многообещающие перспективы (Voiovitch, 1988).
Основными методами количественного анализа, применяемыми в настоящее время
для исследования твердых геологических материалов, являются: (i) методы с примене¬
нием микрозондирования (на микроскопическом уровне, см. гл. 8), (ii) на макроскопи¬
ческом уровне — рентгенофлуоресцентный (см. раздел 31.3.2) и нейтронно-активацион¬
ный анализ (см. раздел 31.3.3).
31.3.2. Рентгенофлуоресцентный анализ
Основные положения
При облучении пробы рентгеновским излучением высокой энергии возникает вторич¬
ное рентгеновское излучение с различными длинами волн, характеристичными для
каждого элемента пробы. Спектры эмиссии такого типа наблюдаются при использова¬
нии других источников возбуждения, например, потока электронов в электронном мик¬
розонде (Castaing, 1961). Рентгеновский эмиссионный спектр возникает из-за возбуж¬
дения электронов внутренних оболочек атома, которые временно переходят на более
высокие квантовые энергетические уровни: с уровня к на уровень 1, с к на m, с 1 на m
и т. д. Возвращаясь в основное состояние, возбужденный электрон испускает энергию Е
(эВ) определенной длины волны X (нм), в соответствии с уравнением Планка:
Е= 1240/Х.
Диапазон рентгеновского излучения соответствует длинам волн от 0,01 до 20 нм и лежит
на шкале электромагнитного излучения между УФ- и у-излучением, однако применяе¬
мый в рентгенофлуоресцентном анализе диапазон ограничен длинами волн от 0,02 до
2 нм. Самые легкие элементы, Н и Не, не имеют рентгенофлуоресцентных спектров.
Число возможных переходов резко возрастает при увеличении атомного числа, однако
для анализа, как правило, используют простые переходы, Они обычно происходят в глу¬
боких энергетических уровнях, в особенности на ^-уровень. Согласно терминологии
Сигбана (Siegbahn) линии, соответствующие таким переходам, обозначают символом Sy:
Syfotp SyfcP2, Sy/a, и т. д. Эти обозначения обозначают положение электронной вакансии
(например, оболочка к, /), тип перехода (а: на вакантный уровень с соседнего уровня, Р:
на вакантный уровень с последующих уровней, и т. д.) и относительную интенсивность
перехода (1 более интенсивен, чем 2, и т. д.). Интенсивность линий к$ приблизительно
в шесть раз ниже по сравнению с соответствующими линиями ка.
Оборудование
Схема прибора для рентгенофлуоресцентного анализа представлена на рис. 31.5. Ис¬
точником возбуждения является рентгеновская трубка, принцип работы которой хо-
774 Часть 3. Неорганический анализ
Рентгеновская ВД-РСА
трубка
Рис. 31.5. Схема рентгенофлуоресцентного оборудования, иллюстрирующая два способа де¬
тектирования: волнодисперсионный рентгеноспектральный анализ с использованием плоского
или искривленного кристалла (ВД-РСА — справа) и энергодисперсионный рентгеноспектральный
анализ (ЭД-РСА — слева)
рошо известен. Электроны, испускаемые разогретой до высокой температуры нитью,
перемещаются к металлическому аноду (антикатоду), ускоряясь в электрическом поле
(потенциал может достигать 50 кВ). При контакте с анодом электроны замедляются
и происходит эмиссия излучения непрерывного рентгеновского спектра с линиями ме¬
таллов с большими атомными номерами, из которых изготовлен анод (например, родий,
вольфрам). Минимальная длина волны в спектре может быть рассчитана по уравнению
Планка (см. «Основные положения» в разделе 31.3.2).
Оборудование для рентгенофлуоресцентного анализа различается, главным образом,
типом идентификации вторичного рентгеновского излучения, испускаемого пробой:
одноканальная система с фильтром, волновая дисперсия, рассеяние энергии. В ге¬
ологических дисциплинах чаще всего используется спектрометрическое оборудова¬
ние с дисперсией по длине волны. Работа этих приборов основана на законе Брэгга (см.
гл. 4), связывающем параметры длины волны X, угол падения луча 0 и межплоскостное
расстояние d: X = 2dsin0. Для кристалла с известным значением d длина волны прямо
связана с величиной угла падения луча на вращающийся кристалл. Для изготовления
кристалла можно использовать несколько различных материалов {Jones, 1991). Выбор
приемлемого оборудования зависит от анализируемых длин волн, в идеале длина волны
составляет около 2d (двойное расстояние между параллельными плоскостями кристал¬
лической решетки). Системы с изогнутым кристаллом применяются для оптимизации
фокусировки детектора. Коллиматор заменен щелью, кристалл и детекторы расположе¬
ны в определенной позиции фокуса на дуге окружности Роуланда. Энергодисперсионные
системы совершенствуются с 1960-х гг. в связи с развитием электроники. Применение
полупроводников позволяет определять энергию каждого фотона и подсчитывать их
в измерительном канале (многоканальные детекторы).
775
Глава 31. Анализ экстрагируемых элементов и полный элементный анализ
Преимущества и ограничения
Еще одним интересным аспектом метода является то, что при анализе не происходит
разрушения пробы, поскольку после релаксации атомы возвращаются в основное со¬
стояние.
Рентгенофлуоресцентный анализ представляет собой быстрый аналитический метод,
обладающий двумя преимуществами: (i) анализ является многоэлементным и (ii) анализ
проводится непосредственно в твердом веществе. Он позволяет детектировать более 80
элементов с атомными номерами выше восьми (начиная со фтора). Метод использовали
даже для анализа более легких элементов: углерода, азота и кислорода в почвах (Jones,
1991). Некоторые элементы, такие как Вг, S и Р, можно с достаточной чувствительно¬
стью анализировать с помощью рентгенофлуоресцентной спектрометрии, в то время
как анализ другими методами может оказаться затруднительным.
Рентгенофлуоресцентный метод позволяет анализировать микроэлементы. Однако
чувствительность его ниже, чем у более мощных методов, таких как атомно-абсорб¬
ционный (см. раздел 31.2.12 и 31.2.13), плазменно-эмиссионный (см. раздел 31.2.14
и 31.2.15) или нейтронно-активационный (см. раздел 31.3.3) методы. Существует спо¬
соб (Jones, 1991) приблизительной оценки минимальных детектируемых концентраций,
сильно варьирующих в зависимости от атомного номера анализируемого элемента. От
натрия (Z= 11) до кальция (Z= 20) значения детектируемого минимума уменьшаются
экспоненциально с увеличением атомного номера: от 1000 мкг/г для Na до приблизи¬
тельно 2 мкг/г для Са. Метод наиболее чувствителен к переходным металлам от Сг до Zn
с пределом обнаружения около 1 мкг/г. Порог чувствительности повышается с повыше¬
нием атомного номера; предел обнаружения для Ва (Z= 56) составляет приблизительно
10 мкг/г.
Основные трудности рентгенофлуоресцентной спектрометрии связаны с влиянием
матрицы; эта проблема зачастую ограничивает применение метода качественным или
полуколичественным анализом. Некоторые помехи имеют физическую природу и свя¬
заны с однородностью и размером пробы, качеством поверхности и плотностью пробы.
Другие помехи имеют химическую природу: (i) наложение спектров элементов, (ii) меж¬
элементные эффекты, связанные поглощением или усилением из-за переизлучения. Се¬
рьезным источником потенциальной ошибки является фоновый шум от матрицы про¬
бы. При расчете содержания элемента из величины пика элемента вычитают фоновый
шум по обеим сторонам пика.
Твердые пробы следует тщательно готовить к анализу: пробу необходимо очень тонко
измельчить, проба должна иметь очень гладкую поверхность и быть не слишком плот¬
ной, чтобы снизить фоновый шум, однако достаточно плотной, чтобы избежать сниже¬
ния флуоресценции. Состав калибровочных стандартов должен быть приближен к со¬
ставу пробы.
При анализе основных элементов лучшим способом подготовки пробы является
приготовление твердого раствора или щелочно-боросиликатного стекла по методике,
аналогичной используемой при вскрытии пробы щелочным сплавлением (см. раздел
31.2.6). При этом пробы и стандарты трансформируют в гомогенные тонкие плоские
диски. В работах (Norrish и Hutton, 1969) описывается данный вопрос для геологических
материалов. Те же авторы предложили метод расчетов, включающий коррекцию влия¬
ния матрицы, что сделало рентгенофлуоресценцию методом, подходящим для количес¬
твенного анализа.
В щелочных твердых растворах происходит разбавление пробы, поэтому они не
пригодны для анализа микроэлементов и даже основных элементов, таких как натрий
776
Часть 3- Неорганический анализ
(и даже магний в некоторых пробах), к которым метод недостаточно чувствителен. При
анализе этих элементов рекомендуется использовать прессованные диски, при изго¬
товлении которых к тонкоизмельченному образцу прилагают очень большое давление
и в результате получают тонкий плоский диск.
Анализ основных элементов
Описанная здесь методика была первоначально предложена в работах Norrish и Hutton,
1969, а затем доработана (Jones, 1982 и Karathanasis и Hajek, 1996).
Оборудование
• Тигли объемом 15 мл, изготовленные из сплава 95% Pt и 5% Аи.
• Тигельные щипцы с платиновыми концами для работы с горячими тиглями или
кольцо, изготовленное из платиновой проволоки, для перемешивания расплавлен¬
ных смесей.
• Пресс-форма для изготовления дисков (рис. 31.6).
• Горелка Мекера или муфельная печь (1000 °С).
• Нагревательная плитка, регулируемая до 200 °С.
Продукты
• Смесь для сплавления: используют коммерческие смеси или готовят синтетичес¬
кую смесь следующим образом: для получения 100 г смеси смешивают 46,53 г тет-
Деревянная рукоятка
Алюминиевый плунжер
Медное кольцо
Графитовый диск
Рис. 31.6. Пресс-форма для изготовления проб твердых растворов (боратных стекол) в виде тон¬
ких дисков для анализа основных элементов методом рентгенофлуоресцентной спектрометрии
согласно Norrish и Hutton, 1969; размеры в мм
777
Глава 31. Анализ экстрагируемых элементов и полный элементный анализ
рабората лития (Li2B407), 36,24 г карбоната лития (Li2C03), 16,17 г оксида лантана
(La203) и 1,07 г нитрата лития (LiN03).
• Карбонат натрия и разбавленный раствор хлористоводородной кислоты для очис¬
тки тиглей.
• Чистый диоксид кремния и другие оксиды основных элементов, присутствующих
в почвах, для изготовления дисков — холостых проб и калибровочных стандартов.
Методика
• Пробы измельчают до размера частиц менее 200 мкм и сушат при 110 °С.
• Нагревают пресс-форму (рис. 31.6) на нагревательной плитке при 220 °С.
• Вносят точную навеску 340 мг пробы и 1,8 г смеси для сплавления в предваритель¬
но взвешенный Pt/Au тигель. Тщательно перемешивают шпателем, затем медленно
сплавляют с помощью горелки Мекера или муфельной печи при 1000 °С. Переме¬
шивают сплавляемую массу платиновой проволокой или вращают тигель, удержи¬
вая его щипцами, для перемешивания и устранения пузырьков.
• Помещают графитовый диск в медное кольцо (рис. 31.6). Сливают расплавленную
смесь в центр диска и немедленно выравнивают поверхность алюминиевым плун¬
жером. Убирают плунжер и кольцо и помещают пробу в виде диска между двумя
теплоизоляторами на нагревательную плитку. По окончании процедуры медленно
охлаждают плитку, затем помещают дисковидные пробы в небольшие пронумеро¬
ванные конверты, которые хранят в эксикаторе до анализа.
• Таким же способом, кДк и пробы, изготавливают (i) диск из чистого оксида крем¬
ния, который используют в качестве холостой пробы для измерения фонового
шума; (и) стандартные калибровочные диски из оксидов, смешанных с чистым
оксидом кремния. Эталонные образцы также можно использовать в качестве стан¬
дартов.
• Для количественного анализа отдельно измеряют уменьшение веса пробы путем
нагревания ее до 1000 °С в тех же условиях.
Методика измерения интенсивности ренгенофлуоресцентного излучения зависит
от применяемого оборудования. При анализе дисковидных проб в качестве источника
возбуждения используют рентгеновскую трубку с анодом из хрома с напряжением 40—
50 кВ, что приемлемо для всех основных элементов, за исключением Мп, для которого
рекомендуется трубка с вольфрамовым анодом.
Расчеты
Для определенного элемента Nn — измеренная интенсивность излучения, соответ¬
ствующего пику пробы или пику стандарта (с коррекцией на время задержки прибора);
Аф — пик фонового шума или холостой пробы.
Рассчитывают интенсивность излучения, соответствующего пику пробы (А) или
пику стандарта (А), по разности между соответствующим пиком и пиком холостой
пробы (Nn — АфЬ) или разности между соответствующим пиком и фоновым шумом
(Nn — (Аф+ + Аф_)/2, где ЛГф+ и N^_ обозначают значения фонового шума, измеренного
с обеих сторон пика.
Сс — процентная концентрация в стандартном образце (в процентах оксида), перво¬
начальная оценка концентрации С в пробе выражается уравнением
C3 = CclVNc.
778
Часть 3. Неорганический анализ
Сумма процентных долей основных элементов и потери веса при сплавлении должна
быть близка к 100%. Для более точных результатов в расчеты вводят поправку на влия¬
ние матрицы (Norrish и Hutton, 1969):
С> = С3 \М + Ji(plml +pnmjj,
где — скорректированная концентрация оксида в виде процентного содержания эле¬
мента э; р{ — массовая доля каждого из оксидов {pt = С,/100); рп - массовая доля потерь
через улетучивание в процессе подготовки пробы; Л/ — коэффициент коррекции вли¬
яния матрицы (матрица - боратное стекло) для элемента э; mi и тп — коэффициенты
коррекции для элемента э каждого оксида i, присутствующего в пробе, и потерь через
улетучивание, соответственно.
Коэффициенты коррекции М, т( и тп определены Норришем и Хаттоном (Norrish
и Hutton, 1969) и Джонсом (Jones, 1982). Например, выражение для расчета скорректиро¬
ванного процентного содержания Fe203 для приблизительной концентрации Сэ = 8,4%
при потере веса 9,5% принимает вид:
C'e]0i = 8,4[1,046+(0,84 х (-0,027)) - 0,031рМп0 + 0,146рТЮг +
+0,134/>СаО +0,126^0 — 0,06/?SOj —0,06/>р2о5 —0,065/?SiO2 —
-0,074pMi0y -0,09/>MgO -0,110/?Na2O + (0,095x(-0,163))].
Аналогичным образом формула применяется ко всем оксидам, присутствующим
в пробе. Исходя из скорректированного процентного содержания С*э, рассчитывают
новые значения массовых долей и вновь применяют формулу поправок с целью опреде¬
ления нового значения процентного содержания С\. Процесс поправок продолжают до
достижения пренебрежимо малой разности между двумя последовательными оценками.
В лучшем случае, сумма скорректированных значений и потери веса должна составить
100 + 0,1%. Ручные расчеты коэффициентов коррекции весьма времязатратны, однако
их можно выполнить очень быстро с помощью компьютера, что позволит к тому же из¬
бежать ошибок при введении коэффициентов.
Анализ неосновных элементов и микроэлементов
Существует множество альтернативных методик анализа неосновных элементов и мик¬
роэлементов с помощью рентгенофлуоресцентной спектрометрии. Описанная ниже ме¬
тодика была предложена и модифицирована в работах (Norrish и Chappell, 1977), (Jones,
1982) и (Karathanasis и Hajek, 1996). Задача состоит в получении при подготовке пробы
к анализу таблетированной пробы с однородной поверхностью, максимально плоской
и гладкой. Это возможно не для всех почв, поэтому иногда требуются альтернативные
решения, например, внесение добавок для оптимизации измельчения и гранулирова¬
ния, или же применение пленок в качестве основы.
Оборудование и реагенты
• Незагрязняющий измельчитель для измельчения пробы до размера частиц, не пре¬
вышающих 50 мкм. Возможно несколько альтернативных вариантов (см. Тап, 1996;
Pansun др., 2001).
• 25-тонный гидравлический пресс.
• Пресс-форма, аналогичная изображенной на рис. 31.7.
779
Глава 31. Анализ экстрагируемых элементов и полный элементный анализ
* Борная кислота или целлюлоза, или другое покрытие для сохранения формы таб¬
леток.
Методика
Соединяют цилиндр со стальной полированной пластиной-основанием (рис. 31.7).
Вставляют в алюминиевый цилиндр мундштук для заполнения формы. Вносят около
2 г пробы или стандарта в виде тонкоизмельченного порошка с размером частиц менее
50 мкм. Придают плоскую форму, уплотняют, поворачивая прессующий поршень. Вы¬
нимают поршень и внутренний цилиндр так, чтобы между цилиндром сжатия и пробой
оставалась пустое пространство. Осторожно вносят порошок для покрытия над пробой
и вокруг нее.
30
А
Плунжер
из полированной
стали 75
Цилиндр
из полированной
стали
Пластина-основание : | I
пресса из полированной
стали ;
Пластина-основание
с пресс-формой
3
1
f
; 0 33
< ►
60
Внутренний
плунжер
100
70
25.5
26
29.7
+ ►»
Алюминиевый
внутренний
цилиндр
;*■
65
Рис. 31.7. Примерная схема пресса для изготовления таблеток для анализа неосновных
и микроэлементов методом рентгенофлуоресцентной спектрометрии
780
Часть 3. Неорганический анализ
Вводят стальной полированный поршень в цилиндр и с помощью гидравлического
пресса прилагают давление, достаточное для получения таблетки с удовлетворительны¬
ми характеристиками (как правило, давление 5-10 т прилагается в течение 1-2 мин).
Отсоединяют основание с пресс-формой (рис. 31.7) и вынимают таблетку из пресс-
формы. Хранят таблетированные пробы и стандарты в небольших пронумерованных
конвертах в эксикаторе до проведения анализа.
Расчеты
Для проб и стандартов считают число измеренных импульсов (см. «Расчеты» в разделе
«Анализ основных элементов»). Для каждого элемента строят калибровочные графики
зависимости числа импульсов от концентрации элемента в калибровочном стандарте
и по графику определяют концентрацию элемента в пробе. Кроме того, может понадо¬
биться коррекция влияния матрицы. Это влияние следует оценивать отдельно для каж¬
дого типа пробы и элемента, и процедура оценки по сравнению с «Расчетами» раздела
«Анализ основных элементов» в меньшей степени стандартизирована.
31.3.3. Нейтронно-активационный анализ
Преимущества и ограничения
В отличие от основных спектроскопических методов, использующих переходы, проис¬
ходящие во внешних (см. раздел 31.2) или внутренних (см. раздел 31.3.2) электронных
оболочках атомов, нейтронно-активационный анализ (НАА) основан на переходах,
происходящих в атомном ядре. В этом случае результаты измерений не зависят от изме¬
нения состояния химических связей между элементами.
НАА является очень чувствительным и точным методом, позволяющим анализи¬
ровать более 70 элементов периодической таблицы. Его часто использовали в качест¬
ве эталонного метода при сравнения с другими методами для проверки их точности.
В отличие от многих спектроскопических методик, требующих растворения проб (см.
раздел 31.2), НАА особенно подходит для анализа твердых материалов, таких как почвы
и геологические породы. Таким образом, удается устранить потенциальный источник
погрешности, возникающей при растворении пробы. Жидкие пробы также можно ана¬
лизировать методом НАА, однако проведение таких анализов не рекомендовано из-за
опасностей, связанных с расширением сред такого типа при облучении.
Метод был впервые предложен в 1936 году, всего лишь через четыре года после от¬
крытия нейтронов. Первоначально в процессе анализа использовали детекторы неболь¬
шой селективности, вследствие чего требовалось химическое разделение компонентов;
ряд различных методик предложен пионерами в этой области (Albert, 1964). Основное
преимущество применяемых методов разделения состояло в том, что они применялись
после введения радиоактивной метки путем нейтронной активации. Загрязнение от не
помеченных реагентов не учитывалось, поскольку измерялись только меченые элемен¬
ты. И наоборот, добавление не помеченных форм анализируемого элемента позволяло
отделить меченую форму с гораздо большей эффективностью по сравнению с традици¬
онным химическим разделением. Эти процессы легли в основу метода изотопных носи¬
телей.
Современное развитие электроники и систем детектирования позволяет проводить
количественный анализ многих элементов без химического разделения даже на уров¬
не следовых количеств. Инструментальный нейтронно-активационный анализ (ИНАА)
дает возможность проводить рутинные измерения около 30 элементов в почвах, поэтому
Глава 31, Анализ экстрагируемых элементов и полный элементный анализ
781
здесь мы ограничиваемся описанием только этого метода. Однако в некоторых случаях
в процессе ультрамикроколичественного анализа элементов требуется химическое раз¬
деление: предел детектирования удается снизить более чем в 1000 раз при химическом
разделении, проведенном после облучения. Таким образом, методы разделения с помо¬
щью изотопных носителей до сих пор применяются в геохимических и экологических
исследованиях.
Один из недостатков метода связан со склонностью элементов к образованию радио¬
активных изотопов с измеряемым уровнем излучения. Основная техническая трудность
заключается в получении доступа к ядерному реактору для проведения нейтронной ак¬
тивации. Кроме того, серьезным ограничением является необходимость соблюдения
правил техники безопасности при работе с радиоактивными изотопами.
Основные положения
Общее описание
Нейтронно-активационный анализ на первом этапе заключается в облучении образца
потоком активирующего излучения, способного вызвать трансформацию атомного ядра
с образованием искусственных радиоизотопов. Анализ излучения, испускаемого этими
искусственными радиоизотопами, позволяет проводить идентификацию и измерение
количества элементов, содержащихся в образце. Искусственные радиоизотопы всех
элементов можно получить путем бомбардировки частицами различных типов: нейтро¬
нами, протонами, а-частицами (jHe2*), дейтонами (!2Н+) или фотонами — у. Однако
наиболее распространен метод нейтронной активации. Захват нейтрона элементом Е
с атомной массой т и атомным номером Zописывается следующей схемой:
Для простоты специалисты часто записывают реакцию как: mE(n, y)m + *Е, например,
для калия: 41К (п, у)42К.
Существует пять различных видов радиоактивной релаксации активированных ато¬
мов: испускание электрона ф-) из ядра, испускание позитрона ф+), испускание частицы
из двух протонов и двух нейтронов (\ Не, а), захват электрона и деление. Самым распро¬
страненным механизмом распада изотопов, образовавшихся при нейтронной актива¬
ции, является эмиссия р~ частиц, компенсирующая увеличение, вследствие нейтронной
активации, соотношения нейтроны/протоны в ядре. Радиоактивный распад сопровож¬
дается эмиссией у-излучения различных энергий, происходящей при релаксации ак¬
тивированных форм в соответствии со схемой релаксации, специфичной для каждого
элемента. Для активированного элемента Е схема реакции записывается следующим
образом:
Наиболее распространенным подходом к нейтронно-активационной спектрометрии
является у-спектрометрия, поскольку детектирование у-излучения наиболее просто.
Каждый активированный элемент обладает соответствующим спектром радиоактивно¬
го распада, которые хорошо описаны в справочниках и публикациях (Lederer и Shirley,
1978; ErdtmannnSoyka, 1975а, 1975b; /,&идр., 1975а, 1975b). При выборе линии у-спектра
для анализа необходимо учитывать период полураспада изотопа, интенсивность линии
и соответствующую энергию, а также вероятные спектральные помехи от других радио¬
4
Z
Е + \п -> ;+1Е«Чу.
782
Часть 3. Неорганический анализ
изотопов. В обзорных работах, таких как (Koons и Helmke, 1978; Helmke, 1982 или Helmke,
1996), представлена полезная информация по основным типам почв.
Количественный анализ
После облучения скорость распада атома m+1zE* пропорциональна N (количеству атомов
этого типа в пробе) и X (константе скорости распада), т. е. dN/dt = — XN. После интегри¬
рования закон распада принимает вид
N=N0e~Xt,
где N0 — количество атомов в пробе в начальный момент времени /0.
Константу скорости часто заменяют периодом полураспада радиоактивного элемен¬
та: Т1/2 — момент, когда N= NJ2 и, таким образом, Т[/2 = X/ In 2.
Если бы все атомы анализируемого элемента полностью трансформировались в из¬
меряемые изотопы, знание закона распада позволило бы определить их концентрацию
сразу же по изначальному количеству атомов N0. На практике количество радиоактив¬
ных атомов, образующихся при облучении элемента, пропорционально сечению захвата
и потоку нейтронов. НАА проводится путем сравнения измеренного излучения пробы
с излучением калибровочного стандарта. Поскольку считывание данных проводится
не одновременно для пробы и стандарта, следует тщательно фиксировать точную дату
и время считывания данных. Затем все измерения приводят к единому моменту начала
отсчета /0 с помощью представленного выше уравнения перед расчетом концентрации.
Аппаратура и измерения
Нейтронная активация
Несмотря на существование небольших нейтронных генераторов, лучшим источником
излучения для НАА геохимических материалов является исследовательский ядерный ре¬
актор. Он позволяет проводить анализ с высокой чувствительностью, поскольку реактор
обеспечивает гомогенный поток тепловых нейтронов с плотностью от 1013 до 1015 п /(см2 • с).
Пробы и стандарты помещают в капсулы, изготовленные из материалов с малым и од¬
нородным сечением захвата нейтронов. Контейнеры переносят автоматически (обычно
пневматической почтой) в поток нейтронов в центре реактора. Длительность облуче¬
ния зависит от выполняемого анализа. Длительность облучения, равная нескольким пе¬
риодам полураспада измеряемого элемента, требуется для достижения максимального
уровня наведенной радиоактивности. Поиск компромиссного времени и оптимизация
длительности облучения требуются при многоэлементном анализе. На практике время
облучения геологических материалов варьирует от нескольких секунд для короткоживу-
щих элементов до 4-12 ч для долгоживущих элементов.
Обычно проводится облучение тепловыми нейтронами, поскольку в большинстве
реакторов (например, Osiris, Сакль, Франция) энергетический спектр нейтронного из¬
лучения находится в зоне 0,01 эВ. В зависимости от требований к анализу возможна мо¬
дификация относительной активности активированных элементов. На самом деле, се¬
чение захвата нейтронов элементами зависит от энергии нейтронов. Поэтому тепловые
нейтроны можно удалить из потока излучения, попадающего на пробу, если обернуть
ее кадмиевым листом. В этом случае в активации будут участвовать только надтепловые
нейтроны с энергией, превышающей 0,5 эВ (при этом длительность облучения, как пра¬
вило, возрастает, например, до 25 ч). В этих условиях активность некоторых элементов,
таких как 46Sc, создающих высокий фоновый шум, значительно уменьшается; в резуль¬
783
Глава 31. Анализ экстрагируемых элементов и полный элементный анализ
тате анализ других элементов, таких как Nd, Zr и Ni, становится намного более точным
(Сйяу/д идр., 1973).
у-спектрометрия
Функция детектора состоит в конвертации регистрируемого излучения в электрический
сигнал, амплитуда которого пропорциональна энергии излучения. В у-спектрометрии
старые сцинтилляционные детекторы, такие как кристаллический Nal, активирован¬
ный таллием (П), заменены современными полупроводниковыми детекторами. Среди
них Ое(1л)-детектор (германий, допированный литием), который применялся в течение
многих лет. Этот детектор обладал хорошими рабочими характеристиками, однако у него
был существенный недостаток — хрупкость, вызванная подвижностью лития при ком¬
натной температуре. Кристалл приходилось постоянно охлаждать жидким азотом; в от¬
сутствии жидкого азота детектирующие свойства исчезали за несколько минут. Поэтому
Ge(Li)-детекторы в настоящее время заменили детекторами на основе сверхчистого гер¬
мания, называемыми также детекторами с собственной проводимостью. Этот детектор
обладает также очень хорошей разрешающей способностью по энергиям. Однако, хотя
детектор не разрушается при комнатной температуре, при измерениях его необходимо
охлаждать жидким азотом.
Разрешающая способность у-спектрометра зависит от уровня энергии соответству¬
ющей спектральной линии, который определяется как «полная ширина линии на по¬
ловине минимальной высоты (FWHM)» пика. Разрешение 1,6 или 1,9 кэВ для 1330 кэВ
(60Со) позволяет различать очень близкие пики нескольких основных элементов в поч¬
вах (например, Sc и Zn, Br, As и Sb). Эффективность детектора возрастает с увеличением
его размера, особенно при детектировании у-излучения высокой энергии. Стоимость
оборудования также повышается при увеличении его размеров, поэтому приходится ис¬
кать компромиссные варианты. Детектор должен быть смонтирован в комплекте с крио¬
статом, содержащим около 30 л жидкого азота. Устанавливать детектор следует вдали
от любых источников излучения; он должен быть защищен от космических и земных
излучений листами свинца.
Многоканальные анализаторы необходимы для дискриминации импульсов по ампли¬
тудам и подсчета их числа в каждом канале, связанном с источником излучения. В ана¬
лизаторе должно быть минимум 2048 каналов, однако в ИНАА предпочтительны ана¬
лизаторы с 4096 каналами и более. Доступны как аналоговые, так и цифровые системы.
Время, необходимое для обнаружения сигнала (время счета) варьирует от нескольких
минут до 1-10 ч, в зависимости от анализируемого элемента, типа облучения и задерж¬
ки между облучением и измерением. Геометрию и размещение образцов и стандартов
необходимо воспроизводить в точности.
После обнаружения сигнал обрабатывают с помощью компьютера (ручная обработка
возможна, однако она требует много времени): определяют положение пиков и распре¬
деляют их по энергетическим каналам анализатора, отделяют плохо идентифицирован¬
ные пики путем деконволюции сигнала и измеряют площади пиков с вычитанием фо¬
нового шума. Перед расчетами концентраций проводится коррекция массы и времени
для проб и стандартов.
Методика
Основные положения
Существует множество альтернативных процедур, выбор которых зависит от целей ана¬
лиза. Оптимальная длительность облучения зависит от анализируемого элемента (см.
784
Часть 3. Неорганический анализ
«Нейтронная активация»). То же можно сказать о промежутке времени между облучени¬
ем и измерением. В работе (Salmon и Came, 1991) была продемонстрирована важность
этого промежутка времени, зависящего от анализируемого элемента. Через несколько
минут после облучения средне- и долгоживущие элементы маскируются значительным
фоновым шумом, в то время как очень короткоживущие элементы, такие как Al, Mg или
V, доступны для измерений. По мере снижения радиоактивности образца фоновый шум
уменьшается и другие элементы становятся доступными для обнаружения и измерения:
Са через 30 мин; Мп и Na через 3 ч; Вг, La, Sc, К через 3-4 дня; Tb, Th, Сг, Eu, Fe, Со
через десять дней и т. д. Описанная ниже процедура применяется в лаборатории Пьера
Сю (Pierre Sue) в Сакле, Франция, для анализа редкоземельных элементов, а также сред¬
не- и долгоживущих микроэлементов в породах и почвах.
Оборудование и реагенты
• Стандартные образцы почв и пород, полученные от национальных и международ¬
ных организаций, занимающихся контролем качества и стандартизацией (Pansu
идр., 2001).
• Алюминиевые или пластиковые ампулы для облучения длиной 10 см и диаметром
2 см, герметизированные опрессовкой краев.
• Бумага с покрытием из ультрачистого алюминия.
• Ядерный реактор или источник нейтронов.
• Автоматизированная система транспортировки между лабораторией и источником
нейтронного излучения со специальным радиохимическим шкафом, оснащенным
дистанционными манипуляторами для приема облученных проб.
• Перчаточный бокс с герметичным плексигласовым передаточным шлюзом.
• Цилиндро-конический пластиковый контейнер вместимостью 1 см3 с плотными
пробками.
• Свинцовые кирпичи для защиты.
• у-детектор на основе ультрачистого германия (или Ое(Ы)-детектор) с разрешением
менее 2 кэВ при 1333 кэВ и полезным выходом 15-25%.
• Дополнительное охлаждающее оборудование, содержащее около 30 л жидкого азота.
• Свинцовый лист для защиты от радиации.
• Многоканальный анализатор на 4096 энергетических каналов.
• Микрокомпьютер с соответствующим программным обеспечением для сбора и об¬
работки информации.
Подготовка и облучение пробы
Рекомендуется использовать два стандарта на одну ампулу для облучения: первый (Е)
для расчета концентрации по калибровке, второй (С) для контроля точности измерений.
Пробы и стандарты следует высушить одинаковым способом (на воздухе или в сушиль¬
ном шкафу) и тонко измельчить до размера частиц менее 100 мкм. Нельзя использовать
измельчающие устройства, содержащие такие материалы, как карбид вольфрама, из-за
риска загрязнения следовыми элементами. Рекомендуется использовать агатовые из¬
мельчители, что связано с их высокой чистотой.
Ножницами вырезают квадраты из алюминизированной бумаги с размерами сторон
около 3 см. Складывают квадраты вдвое и фломастером надписывают номер пробы или
стандартного образца. Разворачивают и берут точную навеску приблизительно 100 мг
(±0,1 мг) (в зависимости от состава) пробы или стандарта на квадрат. С помощью тон-
Глава 31. Анализ экстрагируемых элементов и полный элементный анализ
785
ких лабораторных щипцов аккуратно загибают кромку с трех сторон так, чтобы закрыть
пробу внутри.
Помещают пробы и стандарты в ампулу (10-20) — по одному стандарту Е с каждого
конца и один в середине, и стандарт С в середине. Для облучения надтепловыми нейтро¬
нами заворачивают все пробы в кадмиевый лист. Герметизируют ампулу опрессовкой.
Помещают ампулу в поток нейтронов (2 • 1014 п/(см2 • с) - в реакторе Osiris в Сакле,
Франция) для выбранной длительности облучения: приблизительно 6 ч в полном потоке
для определения редкоземельных и некоторых переходных элементов, 12—25 ч в потоке
надтепловых нейтронов.
Измерения
Извлекают облученную ампулу и выдерживают за свинцовыми кирпичами в радиохи¬
мическом вытяжном шкафу в течение 10 дней. Распечатывают ампулу и переносят ее
в перчаточный бокс с герметичным шлюзом между двумя свинцовыми кирпичами.
С помощью тонкого лабораторного пинцета осторожно открывают каждый конверт
из алюминизированной бумаги, содержащий пробы или стандарты, и количественно
переносят порошкообразные пробы в цилиндро-конические пластиковые контейнеры
емкостью 1 см3, на который наносят тот же номер, что и на бумагу с алюминиевым по¬
крытием. Эти операции следует проводить рядом с увлажненной фильтровальной бума¬
гой для удерживания летучей радиоактивной пыли.
Осторожно закрывают пластиковые контейнеры пробками. Собирают ампулу и ис¬
пользованную алюминизированную бумагу, влажную фильтровальную бумагу и чистя¬
щую бумагу, складывают их1 в небольшой герметичный пакет и помещают в контейнер
для радиоактивных отходов. С помощью счетчика Гейгера проверяют радиоактивность
перчаточного бокса и при необходимости вновь очищают его влажной фильтровальной
бумагой.
Все анализируемые пластиковые контейнеры последовательно помещают в каме¬
ру измерения в строго одинаковом положении относительно детектора (от расстояния
между пробой и детектором зависит амплитуда сигнала, поэтому его следует отрегулиро¬
вать в самом начале работы), регистрируют точное время и дату проведения измерений.
Подсчет импульсов продолжают на протяжении приблизительно 50 мин; время подсчета
импульсов для каждой пробы и стандарта должно быть одинаковым (фиксируют время).
Вновь выдерживают пробы за свинцовыми блоками до снижения активности при¬
близительно 1 месяц. Ведут подсчет так же, как и раньше, но на этот раз в течение 10 ч.
Эти измерения проводятся через 40 дней после облучения и позволяют получить более
полные и точные результаты по долгоживущим элементам по сравнению с первыми ре¬
зультатами, полученными через 10 дней после облучения.
Расчеты
Проводят поиск и идентификацию элемента, соответствующего каждому пику энергии
у-излучения. Измеряют площади пиков методом численного интегрирования «площадь
пика минус фоновый шум». Необходимо принимать во внимание возможность наложе¬
ния пиков, поскольку это может исказить результаты измерений. Существуют методы,
позволяющие избежать погрешности из-за перекрытия пиков: например, локализация
пика по нулевой точке первой производной (вершина пика) или по нулевой точке вто¬
рой производной (точки перегиба), описание Гауссовой кривой (или аналогичными)
и деконволюция, преобразования Фурье. Кроме того, у-спектры всех элементов хорошо
изучены, и возможные перекрытия спектров легко идентифицировать. Каадый элемент
786
Часть 3. Неорганический анализ
можно рассчитать по несколько раз с помощью разных пиков его у-спектра. Поскольку
относительные интенсивности этих пиков хорошо известны, для анализа полной раз¬
вертки сигнала можно воспользоваться библиотеками спектров.
Для данного элемента вводятся обозначения: S{ — скорость подсчета импульсов от
пробы i массы т., подсчет начался в момент времени t(, £, те и te — соответствующие
значения для стандарта е с концентрацией С; X — константа скорости уменьшения ак¬
тивности анализируемого элемента, активности корректируют по формуле уменьшения
активности (см. «Количественный анализ в разделе 31.3.3) с учетом массы пробы. В ре¬
зультате получают Scl — скорректированную величину активности пробы i и Sce— скор¬
ректированную величину активности стандарта е. Выбрав условную временную точку t0
как точку отсчета (например, дату и время первого измерения для данной пробы), запи¬
шем:
Sce — Sj^~^/me.
Sc. рассчитывают для каждого повторного измерения стандарта Е (см. «Подготовка
и облучение пробы» в разделе 31.3.3). Далее необходимо использовать среднюю величи¬
ну или величину, усредненную после исключения сомнительных результатов. Результат
измерения контрольного стандарта С может помочь в выборе исключений. Наконец,
рассчитывают концентрацию в пробе С{ по формуле:
Cr(Sc/Sce)Ce.
Аналогичные расчеты можно выполнить для нескольких пиков одного элемента. Сле¬
дует использовать среднее значение или результат, который представляется наиболее на¬
дежным (наиболее интенсивный пик, результат для стандарта Сит. д.). Относительная
погрешность анализа составляет менее 1%.
Используемая литература
Abbey S (1992) Evaluation and application of reference materials for the analysis of rocks and
minerals. Chem. GeoL, 95, 123-130.
Abo F (1984) Influence du bore et du manganese sur la production et le developpement du ble sur sols
de regions tempiree et aride., Thfcse doctorat £s-sciences, University Paris VII, 89-90.
Albert P (1964) Vanalyse par radioactivation. In Applications des sciences nucleaires, Lefort M. ed. Ade
Visscher, Gauthier-Villars, Paris.
Aubert H and Pinta M (1971) Les У laments traces dans les sols. ORSTOM, Paris Baize D (1997) Teneur
totale en elements traces dans les sols., INRA, Versailles, France.
Bemas В (1968) A new method for decomposition and comprehensive analysis of silicates by atomic
absorption spectrometry. Anal. Chem., 40, 1682-1686.
Boumans PWJM (1981) Conversion of «Tables of Spectral-Line Intensities» for NBS copper arc into
tables for inductively coupled argon plasmas. Spectrochim. A eta., 36B, 169-203.
Bourrelier PH and Berthelin J ed. (1998) Contamination des sols par les elements en traces: les
risques et leur gestion., Acadymie des sciences, France, rapport No 42, Lavoisier (Technique et
documentation), Paris.
Burman JO (1987) Applications: geological. In Inductively Coupled Plasma Emission Spectroscopy,
Part 2. Boumans PWJM ed. Wiley, 27-47.
Callot G, Chamayou H, Maertens C and Salsac L (1982) Mieux comprendre les interactions sol-racine.,
INRA, Paris.
787
Глава 31, Анализ экстрагируемых элементов и полный элементный анализ
Castaing R (1961) The fundamentals of quantitative electron probe micro-analysis. Adv. X-Ray Anal.,
4,351-369.
Chariot G (1984) Chimie analytique quantitative, tome II., Masson, Paris.
Chayla B, Jaffrezic H and Joron JL (1973) Analyse par activation dans les neutrons dpithermiques.
Application к la determination d’eiements en trace dans les roches. C.R. Acad. Sci. Pans, 277, D,
273-275.
Claisse G (1968) Etude experimental de Г analyse aux trois acides - comportement du quartz pur
к l’attaque triacide. Cah. Orstom ser. Pedol., VI, 129-149.
Coppenet M and Juste C (1982) Trace elements essential to the growth of plants and toxicity phenomena.
In Constituents and properties of soils, Bonneau M and Souchier В ed. Masson, Paris, 458-466.
Cox (1968) Development of a yield response prediction and manganese soil test interpretation for
soybeans. Agron. J., 60, 521-524.
Das AK, Chakraborty R, Guardia M de la, Cervera ML and Goswami D (2001) multielement
determination in fly ash after microwave-assisted digestion of samples. Talanta., 54,975-981.
Deb BC (1950) The estimation of free oxides in soils and clays and their removal. J. Soil Sci., 1, 212—
220.
Erdtmann G and Soyka W (1975a) The gamma-ray lines of radionuclides, ordered by atomic and mass
number, part 1. J. Radioanal. Chem., 26, 375-495.
Erdtmann G and Soyka W (1975b) The gamma-ray lines of radionuclides, ordered by atomic and mass
number, part 2. J. Radioanal. Chem., 27, 137-286.
Falkner KK, Klinkhammer GP, Ungerer CA and Christie DM (1995) Inductively coupled plasma mass
spectrometry in geochemistry. Annu. Rev. Earth Planet. Sci., 23,409-449.
FD X31-146, (1996) Determination de l’indice de pouvoir chlorosant (IPC) selon Juste and Pouget. In
Qualite des sols, AFNOR, 117-125.
Gambrell RP and Patrick WH (1982) Manganese. In Methods of soil analysis, part 2 - chemical and
microbiological properties 2nd ed., Page AL, Miller RH and Keeney DR ed. ASA-SSSA.
Greenland DJ and Hayes MHB (1983) Soils and soil chemistry. In The chemistry of soil constituents,
Greenland DJ and Hayes MHB ed. Wiley, 1-27.
Hardy F and Follet-Smith RR (1931) Studies in tropical soils. - II. Some characteristic igneous rocks
soil profiles in British Guiana, South america. J. Agric. Sci., 739 p.
Helmke PA (1982) Neutron Activation Analysis. In Methods of soil analysis, part 2 - chemical and
microbiological properties, ASA-SSSA.
He Li Yuan and Xiang YaLing (1999) Soil sampling error in agricultural environment. Chinese J. Appl.
Ecol., 10,353-356.
Helmke PA (1996) Neutron Activation Analysis. In Methods of soil analysis, part 3 - chemical
Methods, Bigham JM and Bartels JM ed. SSSA- ASA, Madison, WI Etats-Unis,SSSA, 141—
159.
Hoenig M and Kersabiec AM de (1990) L'atomisation electrothermique en spectrometrie d’absorption
atomique., Masson, Paris.
Hossner LR (1996) Dissolution for total elemental analysis. In Methods of Soil Analysis, Part 3,
Chemical methods, Bigham JM and Bartels JM ed. SSSA-ASA, Madison, WI Etats-Unis, 49-64.
Jarvis I (1994a) Sample preparation for 1CP-MS. In Handbook of inductively coupled plasma mass
spectrometry, Jarvis KE, Gray AL and Houk RS ed. Blackie academic. & professional, 172-224.
Jarvis I (1994b) Elemental analysis of solutions and applications. In Handbook of Inductively Coupled
Plasma Mass Spectrometry, Jarvis KE, Gray AL and Houk RS ed. Blackie Academic and Professional,
225-264.
Jeanroy E (1972) Analyse totale des silicates naturels par spectrometrie d’absorption atomique.
Application au sol et к ses constituants. Chim.Anal., 54,159-166.
Jeanroy E (1974) Analyse totale par spectrometrie d’absorption atomique, des roches, sols, minerais,
ciments apres fusion au metaborate de strontium. Analusis, 2,703-712.
Jones AA (1982) X-ray Fluorescence Spectrometry. In Methods of Soil Analysis - part 2, Page AL,
Miller RH and Keeney DR ed. ASA-SSSA, 85-121.
Jones AA (1991) X-Ray Fluorescence Analysis. In Soil analysis - modern instrumental techniques, 2nd
ed. Smith K.A. ed, Dekker.
788
Часть 3. Неорганический анализ
Karathanasis AD and Hajek BF (1996) Elemental Analysis by X-ray Fluorescence Spectroscopy. In
Methods of soil analysis, part 3, Bigham JM and Bartels JM ed. SSSA-ASA, Madison, WI Etats-
Unis, 161-223.
Kawasaki A and Arai S (1996) Evaluation of digestion methods for multielemental analysis of organic
wastes by inductively coupled plasma mass spectrometry. Soil Sci. Plant Nutr., 42,251-260.
Koons RD and Helmke PA (1978) Neutron Activation Analysis of Standard Soils. Soil Sci. Soc. Am.
J., 42, 237-240.
Lamothe PJ, Fries TL and Consul JJ (1986) Evaluation of a microwave oven system for the dissolution
of geological samples. Anal. Chem., 58, 1881-1886.
Le Comec F and Corrdge T (1997) Determination of uranium to calcium and strontium to calcium
ratios in corals by Inductively Coupled Plasma Mass Spectrometry. J. Anal. Atom. Spectrom., 12,
969-973.
Le Comec F, Riandey C and Richard ML (1994) Mineralisation par micro-ondes de matfriaux
g£ologiques (roches et sols) et comparison avec les m£thodes classiques de mise en solution. In
L'echantillonnage, du preldvement a l *analyse, Rambaud D ed. Orstom, Paris.
Lederer CM and Shirley VS (1978) Table of isotopes, 7th ed. Wiley, New York.
Lim CH and Jackson ML (1982) Dissolution for total elemental analysis. In Methods of soil
analysis, part 2, Page AL, Miller RH and Keeney DR ed. ASA-SSSA.
Lindsay WL and Norvell WA (1978) Development of a DTPA test for zinc, iron, manganese and
copper. Soil Sci. Soc. Am. J., 42, 421-428.
Lis SA, Норке PK and Fasching JL (1975a) Gamma-ray tables for neutron, fast-neutron and photon
activation analysis - 1 - List of all the nuclides with their associated gamma-rays in order of
increasing atomic number and mass. J. Radioanal. Chem., 24,125-247.
Lis SA, Норке PK and Fasching JL (1975b) Gamma-ray tables for neutron, fast-neutron and photon
activation analysis - 2 - list of all the nuclides with their associated gamma-rays in order of
increasing atomic number and mass. J. Radioanal. Chem., 25, 303-428.
Martens DC and Lindsay WL (1990) Testing soils for copper, iron, manganese an zinc. In Soil testing
and plant analysis 3nd ed., Westerman RL ed. SSSA book series 3,231-260.
NF ISO 11466 (1995) Etements en traces solubles dans l’eau r£gale. In Qualite des sols, AFNOR,
283-292.
NF ISO 14870 (X 31-427), 1998) Extraction des oligo-ilements par une solution tamponnie de DTPA,
AFNOR, к l’gtude.
NF X 31-147 (1996) Sols, sediments - Mise en solution totale par attaque acide. In Qualite des sols,
AFNOR, 127-138.
NF X 31-151 (1993) Sols, sediments, boues de stations depuration - Mise en solution d’£l£ments
m&alliques en traces (Cd, Co, Cr, Cu, Mn, Ni, Pb, Zn) par attaques acides. In Qualite des sols,
AFNOR, 139-145.
NF X 31-120 (1992) Determination du cuivre, du manganese et du zinc - Extraction par Г acetate
d’ammonium en presence d’EDTA. In Qualite des sols, AFNOR, 75-81.
NF X 31-121 (1993) Determination du cuivre, du manganese, du zinc et du fer - Extraction en presence
de DTPA. In Qualite des sols, AFNOR, 83-89.
NF X 31-122 (1993) Extraction du bore soluble к l’eau bouillante. In Qualite des sols., AFNOR, 91-95.
Njopwouo D and Orliac M (1979) Note sur le comportement de certains mineraux к l’attaque triacide.
Cah. Orstom ser. Pedol., XVII, 283-328.
Norrish К and Chappell BW (1977) X-ray fluorescence spectrometry. In Physical methods in
determinative mineralogy, 2nd ed. Zussman J ed. Academic Press Inc., 201-272.
Norrish К and Hutton JT (1969) An accurate X-ray spectrographic method for the analysis of a wide range
of geological samples. Geochim. Cosmochim. Acta., 33,431—453.
Pansu M, Gautheyrou J and Loyer JY (2001) Soil analysis - sampling, instrumentation and quality
control, 489 p, Balkema, Lisse, Abington, Exton, Tokyo.
Paycheng C (1980) Methodes d'analyse utilisees au laboratoire commun de Dakar., Document
Orstom - Dakar - Paris, 103 p.
Peck TR and Soltanpour PN (1990) The principles of soil testing. In Soil testing and plant analysis 3nd ed.,
Westerman RL ed. SSSA book series 3, 3-9.
Глава 31. Анализ экстрагируемых элементов и полный элементный анализ
789
Petard J (1993) Les methodes d 'analyse. Tome 1: analyses de sols., Notes techniques No. 5, Orstom,
Noumea, Paris.
Pinta M (1962) Recherche et dosage des eliments traces., Dunod, Paris, 726 p.
Pr ISO CD 14869 (1998) Soil quality - determination of total trace elements content - part 1:
digestion with hydrofluoric and perchloric acids for the determination of total contents - part 2:
solubilisation by allkaline fusion., AFNOR, Paris.
Ramsey MH and Coles BJ (1992) Strategies of multielement calibration for maximising the
accuracy of geochemical analysis by inductively coupled plasma-atomic emission spectrometry.
Chemi. Geol., 95, 99-112.
Riandey C, Alphonse P, Gavinelli R and Pinta M (1982) Dёtermination des ё1ётет8 majeurs des
sols et des roches par spectrom6trie demission de plasma et spectron^trie d’absorption atomique.
Analusis, 10, 323-332.
Risser JA and Baker DE (1990) Testing soils for toxic metals. In Soil testing and plant analysis 3nd ed.,
Westerman RL ed. SSSA book series 3,275-298.
Shinotsuka К and Ebihara M (1997) Precise determination of rare earth elements, thorium
and uranium in chondritic meteorites by inductively oupled plasma mass spectrometry - a
comparative study with radiochemical neutron activation analysis. Anal. Chim. Acta., 338, 237-
246.
Smith KA ed. (1991) Soil analysis - modern instrumental techniques, 2nd ed. Dekker.
Soltanpour PN, Johnson GW, Workman SM, Jones JB and Miller RO (1996) Inductively coupled
plasma emission spectrometry and inductively coupled plasma mass spectrometry. In Methods of
soil analysis, part 3t chemical methods, Bigham JM and Bartels JM ed. SSSA-ASA, Madison,
WI Etats-Unis, 91-139.
Tan KH (1996) Soil sampling, preparation and analysis, Dekker.
Totland M, Jarvis I and Jarvis К (1992) An assessment of dissolution techniques for the analysis of geo¬
logical samples by plasma spectrometry. In Plasma spectrometry in the earth sciences, Jarvis I
and Jarvis К ed. Chemical geology, special issue, 35-62.
Trolard F, Воитё G, Jaffrezic A (2002) Distribution spatiale et тоЫШё des ETM dans les sols
en tfgion d^levage intensif en Bretagne. In: Les Aments traces n^talliques dans les sols -
approches fonctionnelles et spatiales, (coord. M. Тегсё & D. Baize), pp. 183-199. INRA,
Collection un point sur., Paris.
Turekian KK and Wedepohl KH (1961) Distribution of the elements in some major units of the earth’s
crust. Geol. Soc. Am. Bull, 72, 175-191.
US Environmental Protection Agency, 1986) Acid digestion of sediment, sludge and soils. In Test
methods for evaluating solid waste, SW-846. USEPA, Cincinnati.
Vemet M and Govindaraju К (1992) Mise en solution des matiriaux avant analyse., Techniques de
l’inglnieur, P 222,1-16.
Voinov itch LA (1988) Analyses des sols, roches et ciments - mithodes choisies., Masson, Paris.
Walsh JN (1992) Use of multiple internal standards for high-precision, routine analysis of geological
samples by inductively coupled plasma-atomic emission spectrometry. Chem. Geol., 95,113-121.
Winge RK, Fassel VA, Peterson VJ, Floyd MA (1982) ICP emission spectrometry: on the selection of
analytical lines, line coincidence tables, and wavelength tables. Appl. Spectrosc., 36,210-221.
Wright RJ and Stuczynski T (1996) Atomic absorption and flame emission spectrometry. In Methods of
soil analysis, part 3, chemical methods, Bigham JM and Bartels JM ed. SSSA-ASA, Madison,
WI Etats-Unis, 65-90.
Zischa M and Knapp G (1997) Microwave-assisted sample decomposition progress and challenges.
Analysis Europa, Novembre, 18-23.
Предметный указатель
|3С 314, 342, 358, 361-367,439, 443
|4С 256,272,275,314,439,443
l5N 361-362, 283,286, 314-315, 317,419—
420,439,443,451
Л
Агрегаты 37, 53, 59, 73,100,103,106, 113,
130,185, 225-226, 256-258, 260,
262-263, 265-266,268, 270-273, 275,
281,323, 434, 461,542, 532,651
Адсорбированная вода 25, 143
Азовскит 163
Акаганеит 163,190
Актиномицет 255
Алдрин 407
Аллофаны 31, 81, 97, 116, 127-128, 148, 161,
169, 174,185,205,216, 218, 246,587,719
Алунит 678
Алюминий 31, 41, 83, 117—118, 134, 144-145,
147, 158, 163-164, 166, 169, 172-174,
177-178, 181-182, 184-189, 193-198,
213, 298, 416, 448, 459-460, 474, 500,
541, 553- 558, 560-561, 573, 577, 579—
580, 587, 591, 605-606, 608, 613, 615,
619, 632-633, 643, 654, 662, 664-665,
671, 673,678,681,697,726,728, 745, 752,
754, 756-758, 764-765, 768, 770, 784
обменный 554, 557
необменный 554
Алюмосиликаты 119, 121, 148, 158,174, 185,
186,519, 540
Амидные формы белка 420
Аминокислоты 382, 417, 420,421, 423, 425,
426, 444, 445-449
Аминосахара 420,425
Анализ
гранулометрического состава 35, 37, 39,
46,67
динамический механический 200
дифференциальный термический (ДТА)
30,92, 94,116-118,127,139,153,178,
189,195, 200-201, 205, 210-222, 312,
357, 648, 703,705,710,714
количественный 148,151,218, 365, 325,
603,628-629, 633-634, 638, 745, 782,
786
микроскопический 73, 225
морфологический 37, 73
общей серы 683,694
путем осаждения ацетоном 716
рентгеноспектральный 774
рентгенофлуоресцентный 773,775
спектроколориметрический 667
твердых тел 772
термический 30, 94, 122, 153, 165, 178,
200, 205, 210, 357, 359,312, 494, 648
термогравиметрический 30, 94,153,165,
200,204,209, 312,494
термомеханический 200
элементный 8,11,17, 249, 286, 457, 725,
729
Анатаз 119
Андезин 83
Андосоли 41, 50
Андосоль 54
Анкерит 492
Анне 295
Антигорит 95
Арагонит 308
Арканит 502
Атомно-абсорбционная спектрометрия
(ААС) 94,127, 172,177,181,184,188,
192, 550, 556-557,571,585-586, 589,
590,619, 703-704,712, 715- 717,719—
720, 729, 731-732,734, 736,739-740,
742, 751-752, 745,755-756, 758, 760,
763,772
с индуктивно связанной плазмой (ИСП-
АЭС) 556, 729,731, 736,740,742, 745,
751-752, 760,763,766-768
Атразин 412
Б
Базалуминит 678
Байерит 83
Бактерии 622, 702
Бассанит 678,714,715
Бейделлит 83
Бёмит 187
Бернессит 163
Биогенные и токсичные элементы 728
Биогенный кремнезем 163
Биологические методы 429, 431,436,454
Биотит 116,192
Предметный указатель
791
Бледит 680
Ближний инфракрасный диапазон (БИК)
133, 138-139, 151-153,313,352
Ближняя инфракрасная спектрометрия
(БИКС) 151,313,314
Боксит 83
Брукит 163
Брюхоногие моллюски 255
Бурые гуминовые кислоты (БГК) 320,
345-346
Буферная емкость 460,465,481,617,754
Буферный метод Шумейкера-Маклина-
Пратга 562
В
Валентные колебания 147
Вантгоффит 678
Вермикулит 31, 95,110,128, 521
диоктаэдрический 95
Вернадит 163
Вискозиметрия 373
Влажность 25-26, 33, 50, 61,172-173,178,
205, 208, 248, 285, 288, 310, 434, 441—
442, 461, 474-476, 484, 488, 498-499,
504, 534, 561, 585, 590, 597-598,602,
604,631, 634,637, 639,649,670,698,
700, 702, 708, 717-718, 753,759
Влияние известкования 560,570,578,656
Внешнесферные комплексы 524,532
Внутрисферные поверхностные комплексы
524,531,571
Водная экстракция 623,653, 719
Водорастворимое вещество 324
Водорастворимый Р 655
Водоудерживающая способность 27,29,
227,321
Волнодисперсионный рентгеноспектраль¬
ный микроанализ (ВД-РСМА) 158,
233,240,249,250
Воды набухания 81
Волюмометрия 510, 596
Воски 383
Восстановительная фрагментация 356
Вторичные электроны 242
Выветривание 697
Высокоэффективная жидкостная
хроматография (ВЭЖХ) 354,400-401,
407-408,445,694
Вязкость 39,44,55, 57,81-82,229,373
Вязкоупругость 201
Г
Газожидкостная хроматография 446
Галит 501
Галлуазит 95, 109,113,116,118, 202,218,599
Гантит 492
Г&усманит 83
ГЬксагидрит 501,680
Гексаметафосфат 41,53-55,269
Гексозамин 382,421-422
Гексозамины 381,420,426
Гексозные сахара 385
Гекторит 95
Гель 92, 143, 158-159, 163, 166, 168-170,
221, 225, 344, 347, 353-354, 368,
371-372, 390, 486, 554, 583, 673, 733
декстрановый 344
Гель-проникающая хроматография 286
Гетит 83, 110, 118,127,167,219
Гйббсит 31,82-83,110, 118,127,219
Гйгрометрическая вода 25-26, 218
ГКцрофилит 502
Гизингерит 163
Пшатомелановые кислоты 319
Гйпс 31, 84, 110, 127, 219, 579,678,680,709,
714-715
Гистосоли 174,204,310,590,696
Глауконит 116
1)школипиды 401
Глинистые минералы 118,120,205, 217,
288-289, 307,463,539, 561
Глицериды 401
Гломалин 449,453
Гомоионное насыщение 208,216
Гониометр 98-99
Гравий 35-36,259, 260
Гранулометрический состав 35, 37, 48, 69,
72-73, 258, 262
Грейгит 702
Грибы 255, 382,449
Гроутит 83
ГУмин 366,319,337,338
ГУминовые
вещества 341,348,353, 355, 370, 291, 319,
337
кислоты 341,342-345, 355, 357, 367-368,
298, 320, 324-325, 327,334, 382
остатки 331, 356
фракции 652
ГУмус 168, 290, 319, 341, 343, 346, 350, 261,
286,287,319,321,323,383,418, 541,663
792
Предметный указатель
Д
Дальний инфракрасный диапазон 134,135,
142,145,146
Дальняя тонкая структура рентгеновских
спектров поглощения (ДТС РСП) 94,
158,162
Датирование 256
Двойной электронно-ядерный резонанс
(ДЭЯР) 161
ДДЦ 407
ДЦТ 384,407,411
ДДЭ 407,411
Дегидратация 30-31,117,205,166,693
Дегидроксилирование 30-31,147
Дезоксигексозы 381-382,385,386
Дельтаметрин 407
Дефицит железа 500
Джурбанит 560, 678
Дзета-потенциал 53, 73, 165
Диатомеи 73,81, 96,121, 185
Диккит 83,127,219
Дилатометрия 201
Динамическая экстракция 551
Диоксины 384
Диоктаэдрические
минералы 120, 145-146
силикаты 110
Диспергирование 41, 52, 54, 76-77, 108, 187,
261, 263, 265-267, 269, 278-279, 323,
594
Диспергирующие реагенты 79, 261, 263, 269,
282
Диурон 407
Дифрактограммы 99-104, 108, 113,116,
118-119,125, 127,160, 231,243-244
Дифференциальная
сканирующая калориметрия (ДСК) 30,
94, 200-201, 205, 210, 212-214, 217,
219-220
термогравиметрия (ДТГ) 200, 204,209,
210, 220, 222,357
Диффузное отражение 138, 143,313
Диэлдрин 407
Доли
адсорбированного натрия 508, 519, 546,
570
обменного натрия 519
Доломит 492, 497
Доступность фосфора 648
биологическая 560, 660
£
Емкость
анионного обмена 285,450,472, 519,532,
613-14,664,728
катионного обмена (ЕКО) 53, 94, 128,
152, 194, 196, 285, 321,472, 520-521,
532, 539,541-543, 545, 549, 551, 554-
556, 558, 560-561, 567, 573, 576-580,
582-588,590-593, 595, 597-606,614,
664, 717, 728, 748
Ж
Железистые конкреции 35
Жидкая фаза 71,159,162,484,522, 547, 568,
579, 582
Жидкостная хроматография 343, 354, 392,
400,403,406,408, 413,447, 672,694
Жидкофазная хроматография 402
Жирные кислоты 357, 383,402—406,410,412
Закон
действия масс 522, 576
Стокса 39, 44-47,55,79, 81,105,256, 278
Затвердевшие почвы 492
Звуковое и ультразвуковое диспергирование
261, 263
Извлечение корней растений 257
Измерение
pH 348, 422, 459, 461, 467-468,470, 474,
509
содержания сульфатов в экстрактах
автоматизированным
колориметрическим методом 692
общей и карбоксильной кислотности 348
проводимости, окислительно¬
восстановительного потенциала 474
удельной электропроводности 505
Изотерма катионного обмена 567
Изотопное разбавление 653, 771
Изотопный анализ 94
Изоэлектрическая точка (ИЭТ) 530,
532-533
ИК-микроскоп 137, 153
ИК-спектрометрия 134,137-138, 145,151-
154, 353,334, 648
Иллит 109,110,116,119,218,521,590
Ильменит 83, 110
Имоголит 116, 148,161, 174,176, 185, 520
Индекс
хлорозного потенциала 500
доступности 652
Предметный указатель
793
Индикаторы 291,465
Индукционная печь 743
Индуктивносвязанная плазма (ИСП) 94,
127, 172,176, 595, 604, 729, 731-737,
740, 742, 745-746, 750-752, 760, 763,
764, 766-771
Инструментальный нейтронно¬
активационный анализ 780
Интеркаляционные комплексы 92
Интерферометр Майкельсона 136
Инфракрасная микроскопия 153
Инфракрасная спектрометрия см. ИК-
спектрометрия
Ионная хроматография 399,508-509,515,
694
Ионный баланс 475,516
Ионометрии 305,474, 480, 508,639
Ионообменная экстракция 746
Ионообменные смолы 53, 79
Ион-селективный электрод 637
Искусственные радиоизотопы 781
Исходная точка нулевого заряда 530
К
Каломельный электрод 468,477,485,486
Кальциметр Бернарда 495
Кальцит 102,122,144, 308, 313,492, 508
Камни 35, 259, 288,682
Каолинит 73, 86, 90-91,96,113,115-116,
119-122,124,128, 145,146,148,
188-189,201, 205, 217-218,243, 245,
307, 539, 590, 599
Капиллярная вода 27
Карбаматы 409,411,413
Карбоксильные группы 349
Карбонаты 31, 35,61,147,148, 177, 278,
290, 292, 308-309, 323-324,492, 494,
501, 503, 508-509,513,546,601-602,
734
Карбофуран 407
Кварц 46,79, 91,96,102,115,119,121-122,
124,128,144,151,163,186,214,
216-217, 302
Кизерит 678
Кислотное вскрытие 734, 738
Кислотность 194, 342, 348,349, 356, 320,
334, 460,461, 462,463,467, 472, 519,
553-558, 560-562, 564, 578, 580,
583-584, 586, 588, 603-605, 618-619,
666, 709-710, 728
Кислотные
гербициды 384
реагенты 747
Кислотный гидролиз 319, 357, 383-384,424,
426, 443-444, 460
и кипячение 357
реагент 744
Кислые
гербициды 408,410,412
сульфатные почвы 293,459-460,680,
695,711-712
Классификация глин 520
Клеточный автолиз 341
Клиахит 163
Клинохлор 202
Клинтонит 95
Когезия 79
Кокимбит 706
Коллигативное свойство 371
Коллоиды 62,79,85,88-89,175,183,244,273,
343,255-256,523, 542, 576,615-616, 690
Колориметрические методы385, 391-392,
428, 400, 428, 445, 510, 623, 628,
630-631, 666,670,685,690,692,698,
700,711,754-755
Колориметрическое определение
Eh 489
хлоридов 511
Компартмент растительных остатков 256
Комплексообразователи 112,175,180,445,
483, 500
Комплекс
обменный 463, 520, 573, 579,601
органоминеральный 49
поглощающий 519
фосфомолибденового аниона 667
фосфора с железом 615
Конституционная вода 26-27,51, 168
Корни растений 257^ 280,259, 281, 555,561,
704, 710
Корунд 125-126,163,202,228
Коэсит 163
Коэффициент
Пшона 569
селективности 567
Кремний 111,146,186-187, 646,671, 725,
728, 733, 766
Криптокристаллические соединения 148
Крупнодисперсные
глинистые частицы 68
794
Предметный указатель
пески 36, 59
Крупный ил 272, 573
Кукеит 95
Кулонометрия 665
Къельдаль 274, 289, 298-299, 301-302,
304,306-307, 314-315, 422-424,438,
597-598,641,645-646
Л
Лангбейнит 678
Лансфордит 492
Лейкоксен 163
Лепидокрокит 163,190
Лизардит 95
Лизис клеточных стенок 341
Линдан 407
Линурон 407
Лиотропный ряд 570, 654,664
Лиофилизатор 227
Липиды 319, 383, 396-398,401-402,406,
412,415,672
Литиофорит 163, 189
Ловеит 678
М
Маггемит 168
Магнезит 31, 219, 313
Магнетит 31, 83, 110, 118,195, 219
Макинавит 679, 702
Максимальная водоудерживающая
способность 27
Манганит 163
Манганозит 83
Маргарит 95
Марказит 194
Масс-спектрометр 221, 312, 314,769
Масс-спектрометрия с индуктивно¬
связанной плазмой (ИСП-МС) 94,
729,731-735, 737, 740, 742, 746, 750,
752,763-764, 767, 769-771
Межпакетная вода 107,115, 146,540
Межплоскостные расстояния 107,109-110,
123
Межслоевое пространство 122,613
Межслоевые промежутки 581,588
Мелкий ил 272
Мелкодисперсная глина 68
Мелкодисперсные глинистые частицы 90
Мелкозем 28, 536
Мелкозернистые пески 35-36
Мессбауэровская спектроскопия 162,158,
165,174,374
Метагаллуазит 116,205
Металлизация 234-235, 373
Метилпаратион 407
Метод
денсиметрический 62
измельчения 255-256
Къельдаля 289, 298
основанный на использовании дитионит-
цитрат-бикарбоната (ДЦБ) 174
основанный на использовании
этилендиаминтетрауксусной кислоты
(ЭДТА) 179
очистки 322—333,335
полной солюбилизации 730
рассева 256
с использованием известковой воды 562
Тамма 170-171
холодного пара 762
электрометрический 464, 466
Механизмы дубления 341
Меченые элементы 780
Микробиологические методики 718
Микробное дыхание 442
Микробный синтез 341
Микродифракция 94, 225, 240,673
Микрозонд 246-247
Микрозондирование 729, 772
Микроэлементы 460, 546,725, 728,731,
759-760, 763-764, 775
Минерализация 94,285, 287, 291—292, 295,
298-299, 300, 302-307, 418,419,426,
429, 430,432, 434-436, 439-443, 560,
643, 645-646,649,651,660,662-664,
685,696, 698, 741,455
мокрым озолением 645
Минералогическая экстракция 159
Мирабилит 501,680
Многоножки 255
Многоэлементная калибровка 764,766
Модель Гуя-Чепмена 523
Молекулярная структура 341
Моллюски 255
Монтмориллонит 31, 95, 108, 116, 127, 202,
218, 288, 547, 590
Мусковит 91, 116, 202, 219
Н
Накрит 116,219
Предметный указатель
795
Нанесение покрытий и пропитывание 226
Напыление углерода 233-234
Насекомые 255
Насыщение катионами 107
Натрон 492, 501
Натроярозит 681
Негуминовые вещества 381
Нейтральные липиды 402
Нейтронно-активационный анализ 772—
773, 780
Нематода 255
Необменный аммоний 641,747
Неомыляемые вещества 403-404
Неорганический кислород 311
Неорганический углерод 290
Неотокит 163
Неперегоняемая фракция 426
Непрерывная экстракция 550
Неупругое рассеяние 242
Нитраты 306, 417,434,450, 501, 511, 619,
622, 626- 627,719
Нитриты 299, 306, 307,417,434, 626-627,
629
Номографический метод 84
Нонтронит 83, 219
Нордстрандит 83,118
Нсутит 83
Нулевая точка титрования (НТТ) 530, 533,
536,537
О
Обменная кислотность 194,320,460,461,
519,553-558, 561,580,583-584, 586,
604,619,728
Обменные
катионы 107,475,547,549, 556, 584-585,
588,619,730
основания 546
Оборудование для металлизации 235
Обработка
глицерином 111
фтористоводородной кислотой 334
этиленгликолем 112
Общая сера 311,683,693-695
Общее
органическое вещество 288,290
содержание абсорбированных металлов
748
Общие
липиды 398
карбонаты 493,494
Общий
азот 311, 432
органический кислород 311
Окислительная фрагментация 356
Окислительно-восстановительный
потенциал 483, 489,667, 701
Окислительное щелочное плавление 683
Оксигидроксиды 100,158,162-163, 540
Октаэдрический слой 117, 539, 540
Олигоклаз 83
Омыление 403,411
Опал 82, 84
Определение
(а-аминокислоты)-Ы 425
(NH3 + аминосахара)-Ы 425
NH; -N 425
РНКиРНСА 472
водорода 311
кислорода 311
молекулярной массы 370
нитратов 516,631
общего азота в гидролизате 424
общей серы 693
растворимых анионов в почвах методом
ионной хроматографии 515
(серин + треонин)-Ы 426
серы 311
содержания мочевины 427,453
содержания общих карбонатов 493
сульфата 512
углеводородов методом жидкостной
хроматографии 400
фосфатов 512
флсфора 752,673
функциональных групп 354
Оптическая и электронная микроскопия 73,
225, 772
Оптическая микроскопия 236
Органические поллютанты (загрязнители)
383
Органические формы азота 417,451
Органический фосфор 648-649,650, 651,
652, 660, 667
Органический углерод 289, 290-291, 295,
296, 308-309,312
Органоминеральные
связи 92, 151,186, 261, 269
коллоиды 255
Ортоклаз 83
796
Ортофосфаты 643,647,651,662
Ортофосфорная кислота 480
Осветление экстрактов 666
Основные единицы
глин 522
глинистых минералов 144
Остаточные пестициды 406
Отбор проб воды из почв 504
Отмучивание 38,255, 258-259, 273,281
Отношение С: N 441
Отношение Е4: Е6 350-351, 369
Отношение Касагранда 62
Отражательные сканирующие микроскопы
247
Очистка гуминовых веществ 333
П
Парагонит 95
Паукообразные 255
Пеннинит 95
Пентозные сахара 381-382, 385, 391-392
Перегонка с паром 421
Переменные заряды 519, 530-534
Перенос тяжелых металлов 159
Перераспределение зарядов 523
Периодический кислотный гидролиз 443
Пестициды 285, 321, 353, 383-384, 406-408,
410,412-413,416,643,672, 677
Пигменты 402
Пикромерит 678
Пипетка Робинсона 55
Пиретриноиды 409, 412-413
Пириты 194,681,703
Пиролюзит 83
Пирофосфатный метод 181
Пирохлорит 83
Плазменная масс-спектрометрия 771
Пламенная атомно-абсорбционная
спектрометрия (ПААС) 734,752,
756-757, 759-760, 763
Пламенно-эмиссионная спектрометрия 17,
751,742,755
Пластичность 26, 27, 35, 174, 285
Платиновый электрод 467
Плодородие 79, 159, 398, 426,439, 460,461,
519, 648, 658, 664
Поверхностный заряд 44, 73, 524, 531-534,
719
Полевая влагоемкость почвы 26
Полевой шпат 83
Предметный указатель
Полевые измерения 474-476,488
Полигалит 678
Полисахариды 258,260, 347, 336-338, 357,
381-385, 398, 697,699
Полифосфаты 643
Полихлорированные бифенилы (ПХБ) 384
Полициклические ароматические
углеводороды 384
Поллютанты 381,383-384,406-408,412
Полное внутреннее отражение 138, 143
Полный элементный анализ 249, 729
Положительные заряды 613
Полугидрат 678, 714
Полуторные оксиды 35
Поляризационный микроскоп 236
Полярные и неполярные липиды 383
Последовательный анализ форм серы 712
Постоянные заряды 533, 539-540
Потенциал ионов 644
Потенциальная кислотность 460, 554
Потенциально минерализуемый азот 429,
430, 436
Потенциометрический метод 467
Потенциометрическое титрование 349
Потребности
в извести 461,560-561
в гипсе 719,720
веере и гипсе 717
Почвенные сахара 382
Предел Аттерберга 27
Преобразование Фурье 136,362
Припороговая спектроскопия высокого
разрешения 161,162
Продукты пиролиза 358, 360
Проницаемость 94, 321,533,540,582
Пропазин 407
Просвечивающая растровая электронная
микроскопия 158, 161,240,246
Прохлорит 95
Псевдопески 35
Р
Равновесие Доннана 522, 576
Разбавленные экстракты 503, 504
Разделение
перегонкой 424
по плотности 257, 275
центрифугированием 548
Разложение кислотами в открытом сосуде 731
Рамсделит 83
Предметный указатель
797
Распределение
зарядов 94, 520
частиц 260, 262
Рассев 56,127, 256, 273,277, 279-281
Рассеяние 42, 94, 98-99,138,154, 216, 239,
242,371,774
Растворенные вещества 196, 407, 504, 507,
516
Растворимость гидроксидов 186
Растворимые
соли 29, 32, 37, 61, 76, 127,177, 208-209,
290,487, 501, 502, 546, 549, 550, 577,
582,584, 601, 619,680, 695, 706, 733
сульфаты 680,706,717-718
сульфиды 682, 700,703
Растрескивание 35, 68, 79, 90, 105,115, 216
Реакция Майяра 341
Регистрация выделяемого газа 153,200, 220
Редкоземельные элементы 734
Релаксация 158, 239, 362-363, 365, 367, 729,
772,775, 781
Рентгеновская дифракция 86,88,91-92,
94, 97,100, 115, 117-120,123-124,
127-128, 132, 143, 147, 158, 160-161,
165,174,176, 188, 231, 244, 703,714
Рентгеновская фотоэлектронная
спектроскопия 374
Респирометрия 286
рН-метр 304, 470, 475-476, 485
Рутил 119,163,186
С
Сапонит 95,108
Сауконит 95
Свободное органическое вещество 274
Свободные жирные кислоты 383,402
СГК 345,346
СДК 483-484,488,489
Седиментационный цилиндр 46, 55, 57
Седиментометрии 59
Селадонит 116
Селективная экстракция 187, 504,652, 746
Селективное растворение 92,94,122,128,
165,188,189
Селективность 155,159,171,179,180,182,
187,192, 407, 522, 567-571, 573, 579,
581,590-591,648, 667, 780
Селективный электрод 638
Сепиолит 91,117, 219
Сера 111,175, 290, 308, 311-312, 554,677,
680-684, 693-698, 700, 702-705, 709,
711-713,718, 728, 730, 746
органическая 697-700,712,719
Серые гуминовые кислоты 345, 320
Сидерит 209-210, 289-290, 308, 310, 492,
494,497-498, 747
Силгидрит 84
Силикагель 163, 205, 354
Силикаты 110,142, 144, 179,185, 509, 614,
664,731,732
Силы Ван-дер-Ваальса 52, 54
Сильвинит 501
Система пор 79
Сканирующая электронная микроскопия
(СЭМ) 87, 158-159, 192,229,233-236,
238-240, 243,247-249,494,705
с режимом естественной среды 248
Скорость диффузии кислорода 483-484,489
Слюды 81, 90, 95,109,110,116,122,128,
182, 202, 243, 520-521, 599
Смолы 53, 54, 79, 194, 227-231, 263,
269-271, 275, 278-280, 291, 293, 308,
334-335, 383, 388, 401,445, 486,629,
649-650,655-657,691
Сольватация 92,105,112,113,131, 571,601
Спектроколориметрический метод 651,663,
665-667,692,742,745,751,753
Спектрометрия
характеристических потерь энергии
электронами (СХПЭЭ) 240
УФ-видимого диапазона 349
Спиновые переходы 162
Средневзвешенная молекулярная масса 371
Среднечисловая молекулярная масса 371
Стеклянный электрод 467, 474,636—637
Стерины 401—403
Стероиды 383
Стильпносидерит 163
Субмикрометрический анализ 72
Судоит 95
Сульфаты 31, 272, 275,292-294,300-303,
305, 325, 327, 333, 388,404,409-412,
459,486, 501, 511-512,519, 524, 560,
584-585, 598, 601, 618,623,631-633,
638, 655, 673, 680-681, 683-686,
689-692,695-697, 700, 702-703,
706-719,731
Сульфиды 31, 33, 135, 175, 178,210, 211, 312,
357,460, 497, 677, 680-682, 686, 690,
695,698, 699,700, 702-706,711, 713
798
Предметный указатель
Сульфонилмочевина 407
Сурьмяный электрод 468
Сухая минерализация 647
Т
Тальк 95, 540
Тенардит 501
Тербутилазин 407
Термическая обработка 105,116, 121,693
Термическое разложение 296
Термовесы 206, 208
Термопары 135,208,215
Тиокарбаматы 384
Типы излучений 242
Титан 671,730, 751
Титратор 509
Тодорокит 163
Тонкие фракции 91, 268, 383
Точка нулевого заряда (ТНЗ) 519, 530-533,
537, 578,580, 605,613,614
Точка нулевого солевого эффекта (ТНСЭ)
530, 532- 535, 537
Точка нулевого чистого заряда (ТНЧЗ) 530,
532-533, 540
Точка нулевого чистого протонного заряда
(ТНЧПЗ) 530, 532-533
Триазины 410, 412
Тридимит 84
Триметилсилильные производные 194
Триметилсилирование 193
Триоктаэдрические
минералы 120, 145
силикаты ПО
Триоктаэдрический вермикулит 95
Тяжелая неорганическая жидкость 274—276
У
Углеводороды 381, 384, 399, 400,413
Удельная электропроводность 503, 505—507
Ультразвуковая обработка 265, 267, 270, 275,
277,279
Ультразвуковой зонд 231, 264, 279
Ультрамикровесы 137
Ультрафильтрация 189,371-372,653,666
Ультрацентрифугирование 87, 106, 124, 149,
159,183,278,334-354,364,371,373,666
Упругое рассеяние 154,242
Уравнение
Керра 568
селективности 567
Уроновые кислоты 381,388,391, 392, 399
УЭП 503,505-506,516
Ф
Файткнехтит 83
Фероксигит 161,163-164,169,190
Фиксация фосфора 159,561
Филлосиликаты 107, 111, 119, 146,186, 334,
520,540
Фильтрование 55,194, 280,325, 388, 434,
507, 548, 645-655,661, 684, 749
Флогопит 95, 150
Флокуляция 37,52, 53,61,73, 76, 81,90,
176, 177, 320, 325, 327, 336,519
Флуоресцентная спектроскопия 10, 367
Формы
воды в почвах 201
Формы
фосфора 643, 645,647-648,651- 652,
667, 673,730
серы 554, 677, 681,684, 700, 709,730
Фосфатиды 402
Фосфаты 312, 509, 511,519,614-615,644,
664,672,719
Фосфорорганические соединения 384, 407,
409,411,672
Фрагменты волокон 255
Фракционирование 238
гуминового остатка 9,336
гуминовых кислот 343
макроагрегатов 268
метиловых эфиров жирных кислот 406
по плотности 255, 272
растворителями 402
Фульвокислоты 186, 341, 350, 367-368, 370,
298, 324-325, 327, 334, 654
Фурье-ИК-спектроскопия 158, 208
X
Халцедон 163
Халькопирит 679
Хелатообразование 321
Хемосорбция 165, 614, 650, 654
Химическая
полимеризация 341
экстракция 718
Химические методы 7, 10—11,41,118,160,
355,436, 455,665, 729
Химическое диспергирование 269
Хлоридсеребряный электрод 468,486
Предметный указатель
799
Хлориты 95,105,108-109,122,189, 218,243,
520
Хлорорганические соединения 384,409,
410,412
Хризолит 95
Хроматограмма альдитолацетатов 392
Хроматографические методы 20, 383,420
Ц
Цианазин 407
Цикл азота 297, 417,435
Цитокамера Хетгиха 139
Ч
Чилийская селитра 502
Ш
Шлиф 227-231,236-238
щ
Щелочная плавка 194
Щелочной гидролиз 357
Щелочность 12,460,509,510,747
Э
Эвансит 163
Экстрагируемые
карбонаты и бикарбонаты 12, 509
катионы 507, 547
растворы 343, 335, 746, 747
элементы 17, 729
Экстрагируемый
бор 512
фосфор 662-663
хлорид 510
Экстракции
в аппарате Сокслета 388
насыщенной пасты 502
горячей водой 12, 504
фосфора 181, 653
Электротермическая атомно-абсорбционная
спектрометрия (ЭААС) 750,752,763,
764,768,772
Электронная микроскопия 8,73, 122,158,
161,225, 240,246-248,648,705,772
Электронный
парамагнитный резонанс (ЭПР) 158,
161-162,174,178,351,369 377
спиновый резонанс (ЭСР) 92,369,370
Электроны Оже 239, 247,250
Электропроводность 475, 501, 503-507, 550,
602,682, 695, 735,739,742
Электроультрафильтрация (ЭУФ) 419,
450-451,455,653
Электрофорез 343-346, 353
Элементы, определяемые гидридным
методом 760
Энергодисперсионный
рентгеноспектральный микроанализ
(ЭД-РСМА) 20, 73,158,159,229,231,
233-234, 238, 240, 249-250,494,648
Эпсомит 680
Эффект ослабления массы 125
Эффективная ЕКО 555, 578,619
Я
Ядерный магнитный резонанс (ЯМР) 9,
16, 20, 92, 94, 155, 161-162,178,189,
342, 356, 358, 361-367, 377, 660, 667,
671-672
‘Н 362-363, 367
13С 342, 362-367
3,Р 660,667,671
твердотельный 362
Ярозит677,681, 703
М. Пансю, Ж. Готеру
Анализ почвы
Справочник
Минералогические, органические
и неорганические методы анализа
Перевод с английского языка 2-го издания
под редакцией Д. А. Панкратова
ISBN 978-5-91884-060-3
9 785918 840603
Ответственный редактор В. Земских
ЦОП «Профессия»
Санкт-Петербург, 190020, а/я 140
Тел./факс: (812) 313-54-14
URL: www.epcprof.ru
E-mail: info@epcprof.ru
Интернет-магазин и on-line заказ книг издательства: www.epcprof.ru
Подписано к печати 10.02.2014
Формат 70x100 1/16. Уел. печ. л. 64,81
Бумага офсетная. Заказ N2 915
Отпечатано способом ролевой струйной печати
в ОАО «Первая Образцовая типография»
Филиал «Чеховский Печатный Двор»
142300, Московская область, г. Чехов, ул. Полиграфистов, д 1
Сайт: www.chpd.ru, E-mail: sales@chpd.ru
8(495)988-63-76, т/ф. 8(496)726-54-10