/






















































































































































































































































































































































































































































































































































































Автор: Карякин Ю.В.
Теги: продукты основной химической технологии химия лабораторная химия чистые химические реактивы
Год: 1947




Текст
Ю. В. КАРЯКИН
ЧИСТЫЕ ХИМИЧЕСКИЕ
РЕАКТИВЫ
РУКОВОДСТВО
по
ЛАБОРАТОРНОМУ ПРИГОТОВЛЕНИЮ
НЕОРГАНИЧЕСКИХ ПРЕПАРАТОВ
ВТОРОЕ ИЗДАНИЕ
Допущено Министерством высшего образования СССР
в качестве учебного пособия для химических специальностей
высших учебных заведений
ГОСУДАРСТВЕННОЕ НАУЧНО-ТЕХНИЧЕСКОЕ ИЗДАТЕЛЬСТВО
ХИМИЧЕСКОЙ ЛИТЕРАТУРЫ
МОСКВА 1347 ЛЕНИНГРАД
Настоящая книга представляет собой
справочник по приготовлению чистых химических
реактивов, необходимых для лабораторных ра
бот.
В ней указываются как методики
приготовления и очистки важнейших неорганических
препаратов, так и требования, предъявляемые к
препаратам различной степени чистоты. Описание
свойств препаратов дополнено таблицами их
растворимости и удельных весов получаемых
растворов.
Книга охватывает большое количество
важнейших неорганических препаратов, часто
требующихся в лабораторной практике, и поэтому
может найти широкий круг читателей.
К ЧИТАТЕЛЮ
Издательство просит присылать Ваша замечания
и отзывы об этой книге по адресу: Москва,
Новая площадь, 10, подъезд II, Госхимиздат.
СОДЕРЖАНИЕ
Стр.
7
Предисловие .
Принятые сокращения 8
Периодическая система элементов ... 11
Атомные веса элементов 13
Азот *
двуокись и четырехокись . , . . . 19
закись - • }*
окись с 1в
Алюминий бромистый . . . . ; 19
гидрат окиси 20
карбид • • 22
окись 22
сернистый • 23
сернокислый 23
хлористый ,,..,•...••.-• 25
Аммиак 27
Аммоний
азотистокислый ". . . . 33
азотнокислый ... * 33
амальгама 35
бромистый 35
ванадиевокислый (мета) 37
двухромовокислыи . 38
йодистый * 40
молибденовокислый 41
надсернокислый 43
пятисернистый • 45
роданистый 46
сернистый 48
сернистый кислый 49
сернокислый 50
углекислый вд
углекислый кислый 54
уксуснокислый 55
фосфорнокислый однозамещенный • 66
фосфорнокислый трехзамещенный 57
фтористый • . . • 58
хлористый » 59
ХРОМОВОКИСЛЫЙ а\
щавелевокислый б!
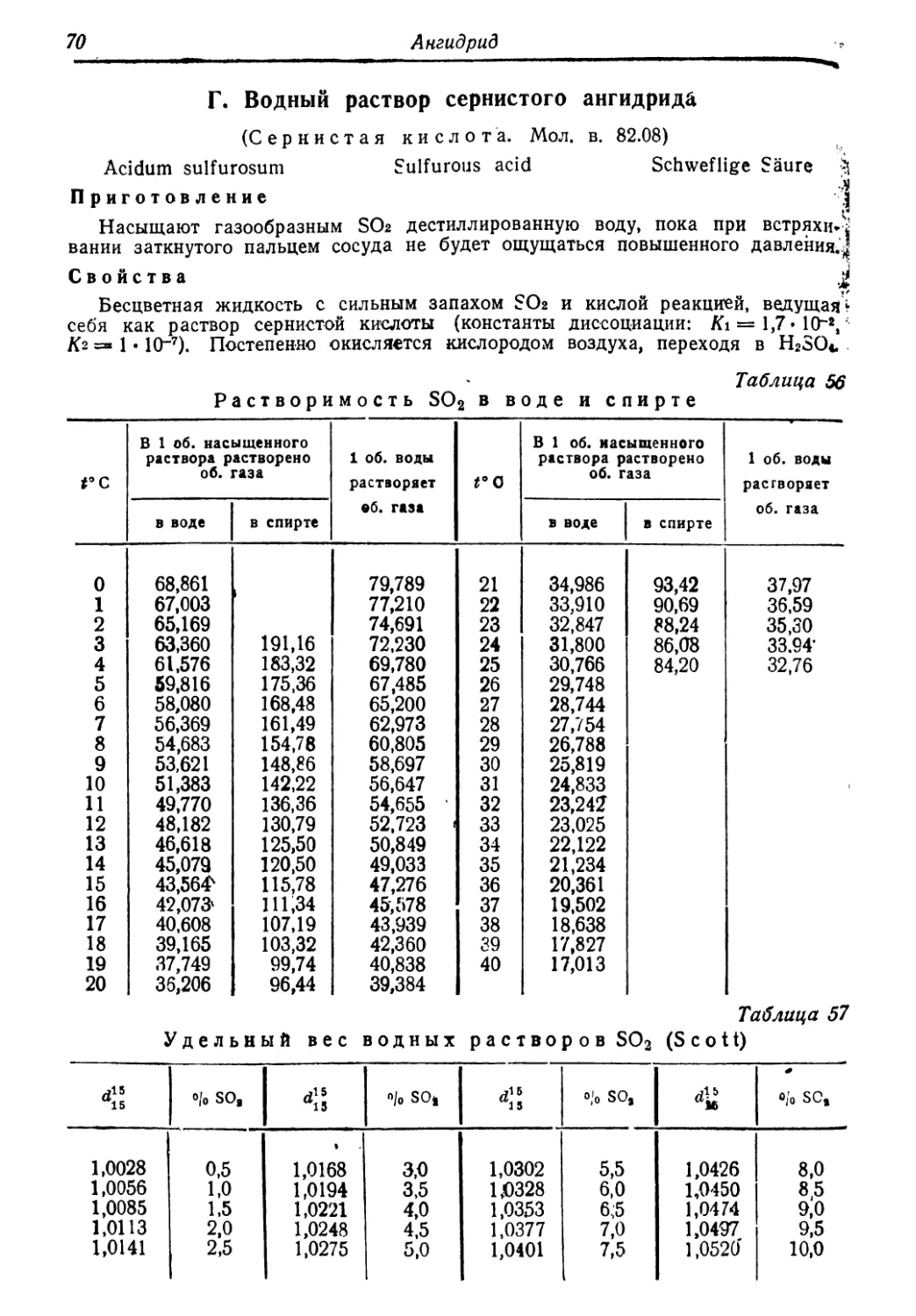
Ангидрид
борный . . . . • * . . . 63
вольфрамовый 63
йодноватый 04
молибденовый 65
мышьяковистый 65
мышьяковый 67
оловянный 67
селенистый ........',..., 67
сернистый .......'. 68
серный ...!...."! 71
сурьмянистый ........... \ 71
сурьмяный ....... 72
теллуристый *....•.....*. " 73
теллуровый 73
угольный t 74
фосфорный !!!!!. 76
хромовый ! . . • 71
Стр.
Асбест платинированный 80
Барий
азотнокислый 80
гидрат окиси . 82
гидрат перекиси , 84
окись : 81
перекись ...... - 86
платинистосинеродистый . . . . ' 87
сернокислый '. 87
углекислый . . .■« 88
хлористый . • . • . 89
хлорноватокислый S2
Бериллия окись 92
Бром ^з
Бумага
иодкрахмальная 97
конго 97
куркумовая 97
лакнусовая • 97
свинцовая 98
тропеолиновая S8
фенолфталеиновая 98
Висмут 99
азотнокислый основной 100
азотнокислый . 101
окись 103
хлористый 104
Вода 105
Водород 11Q
много сернистый . • 114
перекись 115
сернистый 118
хлористый 121
Гидразин 121
гидрат 122
сернокислый 123
солянокислый 125
Гидроксиламии солянокислый 136
Гопкалит » 127
Железо 128
азотнокислое окисное 130
бромистое 132
бромное * 133
гидрат окиси 134
закись-окись . 136
окись 136
сернистое 138
сернокислое записное • 140
сернокислое окисное 142
хлористое 144
хлорное 146
Золото , • 149
хлорное 151
4
Содержание
Стр.
Известь
натронная J52
хлорная 1&*
Иод !53
однохлористый » fc . . 156
КадмиА * .. 156
азотнокислыи » •
опись If?
сернокислый ***»
углекислый • . . J60
уксуснокислый 160
хлористый • 16*
Калий
азотистокислый • . • • • J63
азотнокислый J65
бромистый 167
бромноиагокислый 1Ь9
гидрат окиси .....•«. i .. . 170
двухромовокислый 173
железистое инеродистый 175
железосинеродистый 176
йодистый 178
иоднокатокислый 181
иодноватокислый кислый ....*. 183
марганцовистокислый • . 184
марганцовокислый 185
метабисульфнт 187
надсернокислый 187
нагрий виннокислый 189
натрий углекислый 190
пиросурьмянокнслый кислый .... 191
роданистый 192
сернокислый .... * 195
сернокислый кислый 196
Орьмяновиннокислый 197
тиоуглекислый 198
углекислый 198
углекислый кислый ......... 200
фосфорнокислый кислый однозаме-
щенный 201
фтористый 203
хлористый 205
хлорноватокислый ... ...... 207
хлорнокислый , 209
хромовокислый . . 4 210
цианистый 211
щавелевокислый 212
Кальций
азотнокислый *...*,. 213
гидрат окиси . . 215
окись 217
сернистый 217
сернокислый • . 218
углекислый 219
хлористый 220
Карбосиликагель 222
Квасцы
алюмо-аммонийные . . 223
алюмо-калиевые 224
железо-аммонийные 226
хромо-калиевые 227
Кислород 229
Кислота
азотная 232
борная -238
бром истово дородная 240
бромноватая . • 244
вольфрамовая 244
аоднетоводородная 245
йодная 248
йодноватая • . . . . 249
Стр.
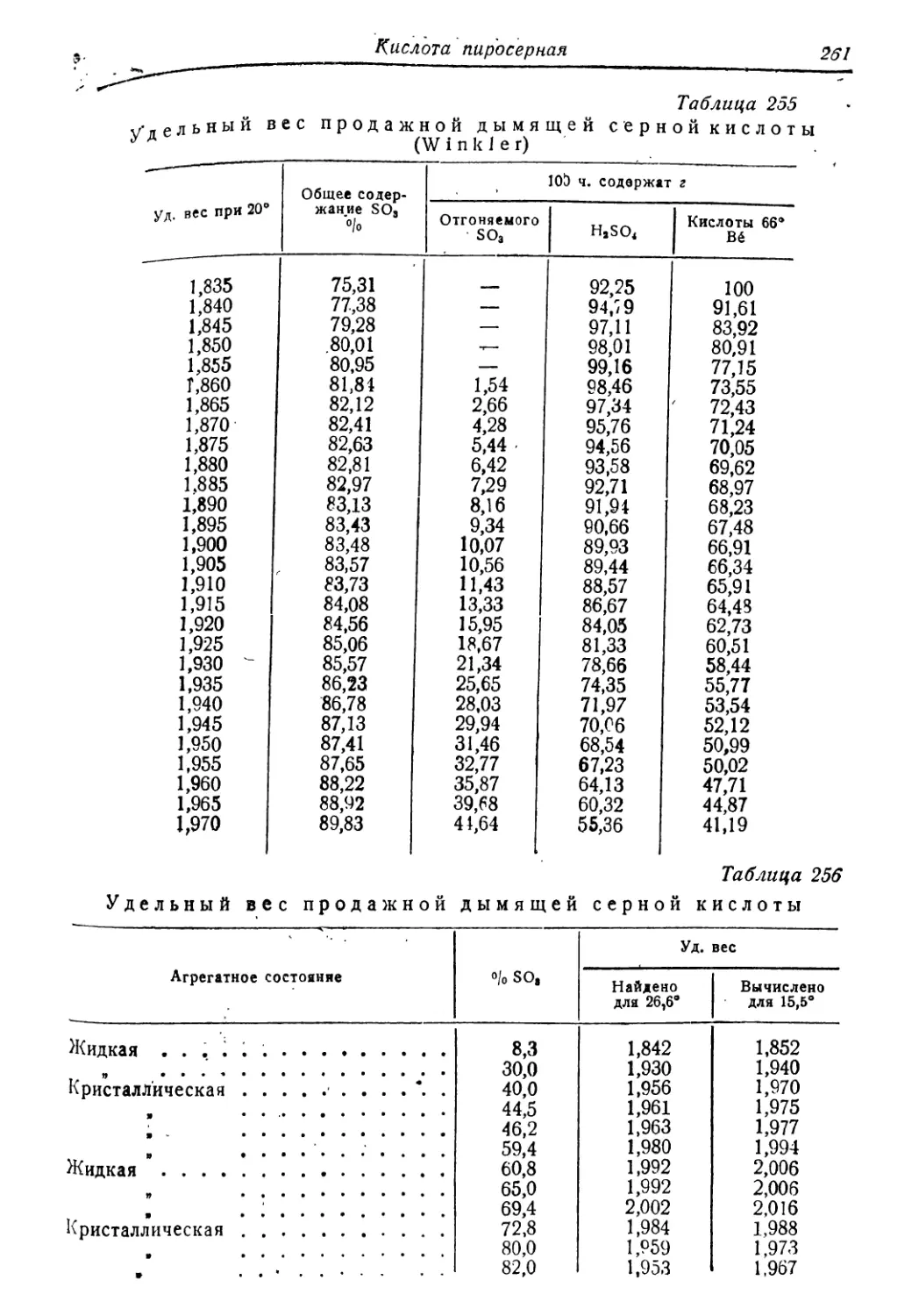
Кислота
Квро 251
кременевая • 252
кремнефтористоводородная ..... 254
метафосфорная 265
молибденовая 256
мышьяковая 257
нитрозил серная 258
оловянная 258
пиросерная • 259
пирофосфорная 262
плагинохлористоводородная 263
селенистая 265/
селеновая 265
серная 267
соляная 276
теллуровая 282
уксусная 283
урановая 285
фосфористая '.•'... 285
фосфорная 286
фосфорноватая .-...• 289
фосфорноватистая 290
фосфорновольфрамовая • 291
фосфорномолибденовая .... . , . 292
фтористоводородная 293
хлорная 295
хлорноватая • . . 300
хлорноватистая 300
хлорсульфоновая 301
цианистоводородная 302
щавелевая . . . . • . 303
Клейстер
иодокрахмальный 304
иодцинккрахмальный 305
крахмальный 305
Кобальт
азотнокислый 805
гидрат закиси • 307
сернокислый 308
углекислый 310
хлористый 311
Крахмал 312
Литий хлористый 312
Магнезиальная смесь . 314
Магний
окись 314
перекись 315
сернокислый 315
углекислый «... 317
хлористый . . . 318
хлорнокислый 320
Марганец
азотнокислый 321
двуокись - ... 323
закись . . 325
перекись 326
сернокислый . 326
углекислый 328
хлористый 328
Медь
азотнокислая • 332
аммоний хлористый . . . . ... . . Зд4
бромистая 336
бромная 386
гидрат, окиси з*б
закись 387
йодистая 330 ;
окись 389 j
сернокислая ••■»•.. 341 I
Содержание
5
Стр.
Медь »:■•»■ 34з
хлористая 34б
хлорная
Мышьяк
трехсернистый _
треххлористыи °™
349
Натрий • • •
язотистокислый е?У
азотнокислый Jg*
амальгама | • 2^
амид . . '
аммоний фосфорнокислый *й>
борнокислый * ?S
бромистый ™
ванадиевокислый ■***
висмутовокислый 5S
вольфромо окислый «О!
гидрат окиси . . «£
гидросерн исто кислый a°j>
двухромовокислый W
йодистый • J;»
ко^алыоазотистокислый o/i
кремнекислый 372
кремнефторитый J/4
метаванад! евокислый 374
мегафосф >риокислый 375
мышьнковочислый трехзамещенный . 376
надиор :окис 1ый 376
нигропругсидный 377
перекись * 379
пирофосфорнокистый 380
пятисернистый 382
сермиС) окислый 382
сер истокислый кислый . , 384
сернистый £85
сер оватистокислый 387
сернокислый 389
сер окислый кислые 391
тиосурьмяюкислый (орто) 392
углекислый 3j2
углекислый кислый ......... 3J5
уксуснокислый 397
фосфорноватистокислый 398
фосфогновольфрамовокя|пый .... 399
фосфорнокислый кислый однозаме-
щенный 400
фосфорнокислый' кислый
двузамещенный 401
фосфорнокислый трехзамешенный . 4J2
фосфорномолибденовокислый .... 403
фтористый 405
хлористый 406
хлорноватистокислый 408
хлормоватокислый 4о9
щавелевокислый 410
Никель
азотнокистый 411
аммоний сернокислый , . 412
окись ; 414
серно-ислый 414
хлористый «... 416
0зон 418
Олово . . • ^ 420
двуокись 421
закись 422
серное . ...\ .';;;;';;;' • 4>э
сернокисюе закисное 423
хлористое * 424
горное !.'!!!!! 423
Платина 428
хлористая 433
хлорная ........ . .' . ] . * | 434
Стр.
Реактив
Дениже 435
Каро 435
Несслера 435
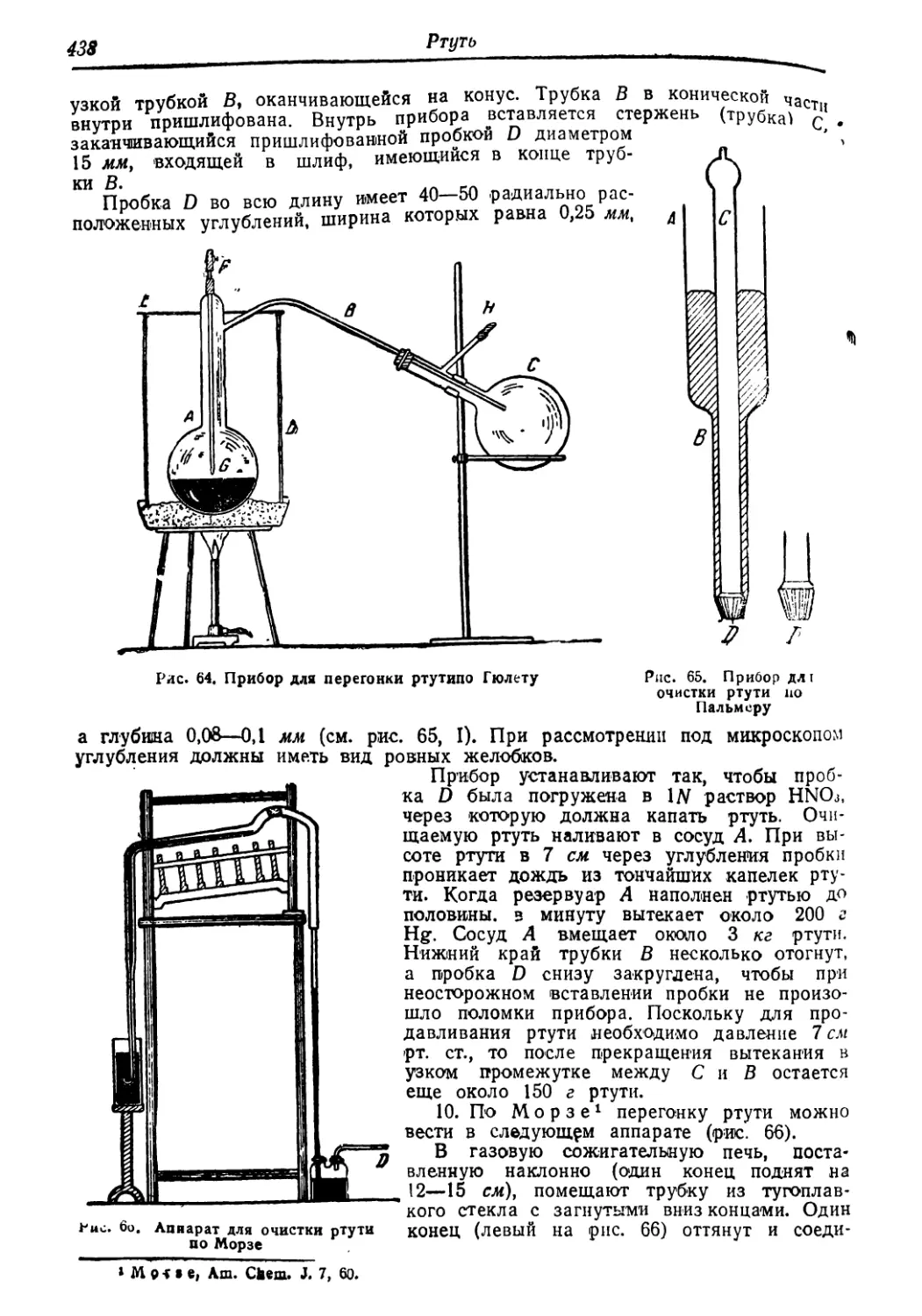
Ргуть . 436
азотнокислая закисная 442
азотнокислая окисная 443
бромистая 443
бр мная 4 4
закись 444
йодистая 445
йодная 445
окись 446
рода ювая . 448
сернистая 448
сернокислая закисная ....... 449
сернокислая окисная ........ 450
хлористая 451
хлорная 452
циановая 454
Свинец 455
азотнокислый 457
амальгама 4^8
гидрат окиси 459
двуокись (перекись) 4г>9
йодистый 4^1
окись 461
сернистый 463
сернокислый 463
углекислый '. 464
укс\снжислый . . . . • 4S6
хлогистый 467
хромовокислый 469
хромовокислый основной 469
Селен 470
Сера 473
бромистая (однобромистая) 478
хлористая (однохлористая) 478
Серебро * 479
азотигтокислое . 483
.азотнокислое 484
бромистое 486
двухоммовокислое 487
йодистое 487
окись 488
сернокислое 488
фтористое 489
хлористое 44)
хромовокислое 491
цианистое • .... 491
Соль
Мора 493
Пелиго 494
Сплав
Вуда 494
Деварда 494
Стронций
азотнокислый 495
гидрат окиси 496
окись 497
хлористый 497
Сульфурил хлористый . . 499
Сурик свинцовый 501
Сурьма 502
пятисернистая 504
пятихлористая • • . . . £05
трехсернистая ......••,... 605
5
Содержание
Стр.
Сурьма
треххлорнстаа. 507
хлористая основная 508
Теллур 609
Тионил хлористый 510
Титан
двуокись 511
серн жислый закисный 512
треххлористый 513
■^ тчетырсххлористыЛ 515
Торий
азотнокислый 516
двуокись 517
сернокислый 519
Углерод v 520
окись 522
сернистый 523
сереокнсь 525
хлорокись 526
четырехллористый. . . • 527
Уран
двуокись 528
закись-окись 528
трехокись , 529
Уранил
азот окислый , . 530
сернокислый 531
>ксуснокнслый 531
Фосфоний йодистый 532
Фосфор 533
бр >мокись 536
патиоромистый 537
Стр.
Фосфор
патихлористый 537
грехбромистыА 538
трехиодистый 641
трехокись 641
трехсернистый 642
треххлористый • 543
хлорокись • 544
Хлор 545
Хром 549
азотнокислый 5£f0
гидрат окиси 551
окись 558
сернокислый 554
хлористый . . . . • Б56
хлорный 556
Хромил хлористый 559
Царская водка 560
Цинк - . . 560
амальгама 562
аммоний хлористый 552
гидрат окиси 563
йодистый 563
окись 565
сернистый 566
сернокислый 568
углекислый. 569
уксуснокислый 570
уранил уксуснокислый 571
хлористый, - .... 571
ПРЕДИСЛОВИЕ
Первое издание настоящей книги, появившейся в 1936 г., разошлось
в течение нескольких месяцев, что является наглядным доказательством
интереса, проявляемого работниками наших лабораторий к пособиям по препарат
тивной химии.
В настоящее время потребность в руководстве по приготовлению возможно
простыми методами широкого ассортимента чистых реактивов значительно
возросла и это обстоятельство побудило нас приступить к переизданию
книги.
Предлагаемая вниманию читателей книга значительно отличается от первого
издания как по объему материала, так и по рассмотренному ассортименту
препаратов. Добавлены методики изготовления новых препаратов,
получивших широкое применение в аналитической практике последних лет.
Пересмотрены, уточнены и частично исключены устаревшие методики, не всегда
приводящие экспериментатора к положительным результатам. Дополнительно
введены синтезы, описанные в новейшей советской и иностранной литературе.
Значительное место отведено впервые публикуемым методикам,
разработанным автором и его сотрудниками в реактивной группе Уральского
индустриального института. Большое внимание уделено также обновлению физико-
химических констант, таблиц растворимости и удельных весов растворов по
работам последних лет. При подборе нового материала советская и
иностранная периодика просмотрена до 1942 г.
В соответствии с пожеланиями читателей для каждого препарата
приведены его названия на латинском (слева), английском (посередине) и
немецком (справа) языках. Молекулярные веса пересчитаны по данным
Интернациональной комиссии на 1943 г.
Автор надеется, что книга окажется полезной не только для заводских
лабораторий, но и для лабораторий вузов, втузов и научно-исследовательских
институтов.
Автор просит читателей сообщить ему (Свердловск 9, Втуз-городок, Уралы
ский индустриальный институт им. С. М. Кирова) свои замечания о резуль*-
тэтах использования описанных в книге методик, а также новые методики^
ускоряющие и облегчающие синтез того или иного препарата.
Ю. Карякин
ПРИНЯТЫЕ СОКРАЩЕНИЯ
f. Русская и перевбдная литература
Аналит химия —Тред вел л (Tredwell). Курс аналитической химии,
-. ГГИ, 1932.
ЖОХ— Журнал общей химии.
ЖПХ —Журнал прикладной химии.
ЖРФХО — Журнал Русского Физико-Химического Общества, часть
химическая.
ЖФХ — Журнал Физической химии.
ЖХП — Журнал Химической Промышленности.
Завод, лаб. — Журнал «Заводская лаборатория»".
Изв. ИРЕА — Известия Института Чистых Химических Реактивов, НТО,
ВСНХ.
Испытание хим. реактивов — А. Зильберберг, Е. Пржевальский и
М. Рождественский, Химические реактивы. Свойства,
применение, хранение, испытание, М. 1935.
Колл. химия — В. Оствальд, Краткое практическое руководство по
коллоидной химии, Гос. Научно-Техническое Издательство, Ленхимсектор,.
Ленинград, 1931.
Лабор. Практика—Журнал «Лабораторная Практика».
Неорг. практ. — Е. Ризенфельд (Е. Riesenfeld), Качественный анализ и;
неорганический практикум, Госиздат, 1930.
Неорг. преп. — Г. Борнеман, Неорганические препараты; Госхимтехиздат,,
Л., 1934.
Неорг. синтез—Л. К. Лепинь, Неорганический синтез, Госхимтехиздат, M.v
1932.
Объемный анализ — И. Кольтгоф (J. M. Kolthoff), Объемный анализ,,
т. II (Практика' объемного анализа), Госхимтехиздат, Л., 1932.
Приг. неорг. преп. — А. Бендер (A. Bender), Приготовление и испытание
неорганических препаратов, изд. Тихомирова, М. 1904.
Реакт. и преп. — Реактивы и препараты лабораторного назначения. Сборник
ИРЕА под ред. проф. В. В. Логинова, вып. I. ГОНТИ М-Л., 1938.'
Сборник работ ИРЕА— см. Изв. ИРЕА.
Соли редких и цветных металлов — Н. Сингаловский, Соли редких и'
цветных металлов, Госхимтехиздат, Л., 1932.
Спутник химика — Справочник «Спутник химика» по «Chemiker Kalender»,
Госхимтехиздат, 1932.
Химич. реактивы—В. Палаузов, Химические реактивы: их свойства,
получение и методы испытания, М., 1935.
Хим. преп. — Г. Эрдман (Н. Erdmann), Руководство к приготовлению и
исследованию химических препаратов, 1899.
Хим. реактивы — Коренблит, Химические реактивы, 1902.
II. Иностранная литература
Abegg — R. Abegg u. F. Auerbach, Handbuch der anorganlschen Chemlo,
Verl. G. Hirzel, Leipzig.
Am. Chem. J. — American Chemical Journal, Baltimore.
Anieit. z. Darst. anorg. Рга\о, — C. Rust, Anleitung zur Darstellung anorga-
nischen Praparaten.
Ann. — Annalen der Chemle und Pharmazie.
Ann. Chim. appl. — Annalt di Chimica appljcata,
Принятые сокращения
9
Ann. Chim. Phys. —Annates de Chimie et de Physique, Paris.
Ann Soc esparT. Fis. Quim. — Annates de la Sociedad espanola de fisica у quimica.
Arch Pharm.— Archiv der Pharmazie, Hannover und Halle, начиная с 1924 г.
именующийся: Archiv der Pharmazie und Berichte der deiUschen
pharmazeutischen Gesellschaft; издатели: H. Beckurts, J. Qadamar, H. Thomas
und P. Siedler; изд. Chemie, Leipzig und Berlin.
Ausfuhr. Lehrb. d. pharm. Gh. — E. Schmidt, Ausfuhrliches Lehrbuch der
pharmazeutischen Chemie, 5, Aufl., 1907.
ger Bericrne der deutschen chemis^hen Gesellschaft, Berlin.
Ber. Wien. Akad. — Sitzungsberichte der K> K. Akademie der Wissenschaften,
Mathematisch-naturwissenschaftliche Klasse, Wien.
Bull. Acad. Belg.-—Bulletin de I'Academie royale des Sciences et Belles-Lettres
de Belgique, Bruxelles.
Bull. Assoc. Beige —Bulletin de Г association Beige de chimisies.
Bull. Soc. Chim. Belg. — Bulletin de la Societe" chimique de Belgique.
Bull. Soc. chim. France—Bulletin de la Societe chimique de France, Paris.
Гг. Arch. — Archiv des Apoihekervereins im nordlichen Deutschland von Rud.
Brandes, 1822—1631, 39 Bande, Schmalkaiden, Lemgo.
C. — Chemisches ZentralbJatt, Leipzig und Berlin,
Chem. Abstr. — Chemical Abstracts.
Chem. Ind. — Du chemische Industrie, Berlin.
Chem. Met. Eng. — Chemical and Metallurgical Engineering, New York.
Chem. News —The Chemical News, London.
Chem. Weekb. — Chemische Weekblad.
Chem. Ztg — Chemikcr Ztitung, COlhtn.
С. г. — Ccmptts rendus des seances de I'Academie des Sciences, Paris.
Deut. Arzneibuch — Deutsches Arzneibuch, 5 Aufl. (1910).
Dingier — Dinglers poiytechnisches Journal, Stuttg'irt.
Gazz. chim. Ital. — Gazzetta chimica italiana, PaLrmo.
Gmelin's Handbuch— Gmelin's Handbuch der anorganischen Chemie.
Handb. d. anorg. Ch.-r-Ab egg, Handbuch der anorganischen Chemie.
Helv. chim. Acta — Helvetica chimica acta.
Herst. ko!l. Lsgg. — The Svedberg, Die Methoden zur Herstellung kolloider
Losungen anorganische Sloffe, издательство: Т. Steinkopf, Dresden und
Leipzig, 1920.
Ind. Eng. Chem. Anal. rEd. — Industrial and Engineering Chemistry, Analilical
Edition.
J. Am. Chem. Soc. — Journal of the American chemical Society, New York.
J. B. — Jahresberichte fiber die Fortschritte der Chemie und verwandte Teile
anderer Wissenschaften, begrtindet von T. Liebig und K. Kopp, Giessen.
J. Chem. Educ. — Journal of chemical Education.
~J. Chem. Soc. — Journal of the chemical Society, London.
J. Ind. Eng. Chem. — The Journal of Industrial and Engineering Chemistry.
J. Pharhi. Ch. — Journal de Pharmacie et de Chimie, Paris.
J. phys! Ch. — The Journal of Physical ChermVry.
J. prakt. Ch—Journal fur prakrische Chemie, Leipzig.
J. Res. Nat. Bur. Stand. — Journal of Research of the National Bureau of Standards.
J. Soc. Chem. Ind. — Journal of the Society of chemical Industry, London.
Journ. f. Gasbel. — Journal fur Gasbeleuchte.
К oil. Z. — Koiioid-Zeitschrift, изд. Steinkopff, Dresden — Leipzig.
Komm. z. Deut. Arzneib. — Kommentar zur Deutschen Arzneibuch, 5 Aufl. (1910).
Landolt-Tabellen — Landolt-Burnsteln, Physikalisch-chemische Tabellen.
V Aufl., J. Springer, Berlin.
Lehrb —В erzelius J.f Lehrbuch der Chemie, Dresden, 1826.
Lehrb. d. anorg. Ch. — Erdmann. Lehrbuch d. ancrganischen Chemie.
Lehrb. d. pharm. Ch. — E. Schmidt, Ausftihrliche Lehrbuch der pharm a z.
Chemie, 5 Aufl. (1907).
Merck —E. Merck, Prufung der chem. Reagentien, Darmstadt, 1922.
Monatsh. — Monatshefte fur Chemie, Wien.
10
Принятые сокращения
Pharm. J. — The pharmaceutical Journal and Transaction, London.
Pharm. C.-H, —Pharmazeutische Zentralhalle fur Deutschland, Dresden.
Pharm. Weekbl. — Pharmazeutisch Weekblad.
Phil. Mag. —Philosophical Magazine and Journal by Tilloch (and Taylor)*
London, 1798—1826, 68 томов; снова стал выходить с 1851 г. j
Phil. Trans.—Philosophical Transactions of the Royal Society of London, ]
pogg. — Annalen der Physik und Chemie, 1824—1873 гг. издавался Т. С. Poggen-
derf'oM, Leipzig 1*877—1899, Wiedemann. С 1900 г. —Drudes Annalen.
Polyt. Journ. — Polytechnisches journal.
Plug. Ch.— L. Vanino, Handbuch d. praparativ. Chemie, 3 Aufl., 1925.
ProcTAm. Acad. — Proceedings of the American Academy of arts and sciences,
Boston.
Proc. Chem.. Soc. — Proceedings of the Chemical Society of London.
Proc. Roy. Ъсс. — Proceedings of the Royal Society of London.
Rec. trav. chim Pays-Bas.— Recueil de travaux chimiques de Pays-Bas, Leiden.
Schweig. — Journal fur Physik und Chemie (Scnweigger) Nurnberg Halle
(1811-33).
Sci. Pap. Inst. phys. chem. Res. — Scientific Papers of the Institution of physical
and cnemical Research, Tohoku, Japan.
St. u. Eisen — Stahl und Eisen.
Svensk. Kem. Tidskr. — Svensk kemisk Tidskrift.
Trans. Faraday Soc. — Transactions of the Faraday Society.
UebungsbeispieJe — H. und W. Blitz, UebungsbeispieJe aus der anorganischen
Experimentalchemie; изд. W. Engelmann, Leipzig.
Z. anal. Ch. — Zeitschrift fur analytiscne Chemie. Wiesbaden.
Z. ang. Ch. — Zeitschrift fur angewandte Chemie. Berlin.
Z. anorg. Ch. — Zeitschrift fur anorganische Chemie, Hamburg und Leipzig;
начиная с 1915 г. выходит под названием:; Zeitschrift ftir anorganlsche
und allgemeine Chemie, изд. S. Voss, Hamburg — Leipzig.
Z. f. Elektrochemie — Zeitschrift fur Elektrochemie und angewandte physikalische;
Chemie, Leipzig.
Z. phys. Ch. — Zeitschrift fur physikalische Chemie, Sttfcbiometrie und Vermahdt-;
schaftslehre. Akad. Verlagsgtsellschaft, Leipzig.
Периодическая система элементов
U
Г
1
СО
IO
Н
1 3=
W
1ч
U h
ПЕРИОДИЧЕСКАЯ
г-
J™
о
1м
р
4
!•*
«1 ■
'1
11 1
4
г
г
м]
tor f
mud
1 ^
1 J- о
j О
j
г
- со
CD
p.
•
Ь g
1 °^
I 0»
fog
1 °
1 S2
[со
пг£
t*
Pi
1 CM
CO
та,
о
7~\
j-J CD?
W 1
^н 1 gst
2 v
1 ^. <o
1 *
1 ^
\ui g
1 c\f
1 CO
|ЬТ"!
о*
со
мл
'СО о.
со
CV
со*
СМ
W 1
Oocol
^ СМ|
я7|
О)
ев oil
►у CMl
< cvf_
а
с^ 1
06
h~m GO
1^
Г *
о ?
1< ^ °°
1 V
Г «я
1 A3 °°
1 ю
Iе4 со
1.£5 °*
1 *^ ю
г*
I И
О)
К. о'
и о
- 5
Jr.?
51
* J
о_§
ч СО
о
а
(А
Г*
IsS s
L
Ift ^
J ф СО
[•'-/ CD
5 5
Н*
со
Ф О
CD»
03 w
со
S j
N3
о 4
«
3 M
OS
>* 1
Vf J
-
9
a,2
' -
1 —, °>
CC 2
t ^
1 ^
ОС °
1 CO
1 "*
I1
k»
(^ ю
15
^ CN
CM
U см
CN
0)
<m 001
* 1
CO
00
й
LT!
f4M>-te-'
1*
lift
1 ф °^
1 •-« см
ел
со*
L 2
0> £
см
г* -
1Л
}£Г\й
см
ел й
в» ^,
"Ч со
5 s
в |
) J
М tftt.L
1 о
I** со
1 сч
1*-» ю"
ID- от
1 *""
1 L. ^
1 м ^
|«В
1 г»
1 см
1 Л
1 **
I 1* CM
1 ^
1»
^ со
г4 °?
со
»* см
*5 «I
О S3
» 1
и» со
«а
j г
8 ^|
О S1
ту
■MMiMi
Ф
PC см
1
со
1 **■• о
СО о
^ CD-
CD
ft см
со
|эО см
Ом Г*
1* О
сч см
со
о
5 И,
Х- о
о
Д смГ
^^ col
0» "I
N 1
И
«о Г
и ■ • ^
м
1 00
1 »-J CSJJ
1» 1
а 5
° 1
™ СМ I
7П
0 1
о ^
^ см 1
Об 1
со mi
о
eg щ
ас 1
X 1
^J
^
1^
ее»
1Л
'«я
Л
S
а
<
h
S
*
^
1 v
I1"*
ю
w
с
3
1с5
м
о
Е
сл
«4
<о
о
ф
пэ
^
А
л
С
ik
ее
и»
г»
О^
СП
со'
ю
О
о*
in
го
о
1Л
г*.
см
^1
S
см
0>
-
со
2|
ill liiiiiM
Г* ^
я?
U а
®
•* ^
^ о
Г° ^
>- ^
еъ
со
L3 3
£—' ^
со 1
1^0 1
Ы £
^ 1
• *|
СГ)|
О чг*
X 21
ео 1
» со
Jr4 см|
О g|
к> I
CO I
£l^
12
Атомные веса элементов
АТОМНЫЕ ВЕСА ЭЛЕМЕНТОВ
(Данные Международной комиссии по определению атомных весов на 1943 г.)
Элемент
ч
\ ___
Азот
Актиний .
Алюминий
Аргон . .
г Барий .и .
Бериллий
Бор . . . '
Бром . • •
Ванадий .
Висмут . .
Водород .
Вольфрам
Гадолиний
Галлий . .
Гафний '.
Гелий . .
Германий
Гольмий * .
Диспрозий .
Европий . .
Железо . .
Золото . . .
«Индий • . ,
Иод . . .
Иридий .
Иттербий
Иттрий
Кадмий . .
t Калий . .
Кальций .
Кислород .
Кобальт .
Кремний .
Криптон .
Ксенон . .
Лантан , .
ч Литий . .
Лютеций .
Магний
Мазурий .
Марганец .
Медь . . .
Молибден
Мышьяк .
Натрий .
1 *
i *
я
(X
1 u
1 7
89
13
18
56
4
5
35
23
83
1
74
64
31
72
2
32
67 •
66 '
63
26
79
49
53
77
70
39
1 48
19
20
8
27
14
36
54
57
з
71
12
43
25
29
42
33
-11
вол
S
я
N
Ас
А1
Аг
В-1
Be
Ва
Вг
V
Bi
н
W
Gd
Ga
Hf
Не
Ge
Но
Dy
Eu
Fe
Au
In
J
Ir
Yb
Y
Cd
К
Ca
0
l Co
i Si
Kr *
Xe
La
Li
Lu
Mg
Ma
Mn
Cu
Mo
As
Na
MHblft
2 о
«8
14,008
("27)
24,97
39,°44
137,36
9,02
10.Я2
79,91в
50,95
209,00
1.СГ8С
183,92
156,9
^9,72,
178,6
4,003
72,60
164,94
162,46
152,0
55,85
197,2
114,76
126,92
193,1
173,04
88,92
112,41
39,096
40,08
16,0000
58,94
2Я.06
83,7
131,3
138.92
6,940
174,99
24,32
—
54,93
63,57
95,95
74,91
22,997
Элемент
Неодим . .
Неон . .
1икель .
Ниобий
Олово ..
Ос> ий .
Палладий
Платина
Полоний
Празеодим .
Протактиний
Радий . . .
Радон
(Эманация, Нитон)
Рений . . .
Родий .
Ртуть .
Рубидий
Рутений .
Самарий
Свинец .
Селен .
Сера . .
Серебро
Скандий
Стронций
Сурьма .
Таллий .
Тантал .
Теллур .
Тербий .
Титан .
Торий .
Тулий .
Углерод
Уран . .
Фосфор
Фтор . .
Хлор . .
Хром . .
Цезий .
Церий .
: Цинк . .
Цирконий
Эрбий .
*
о.
60
■ ю
28
41
50
76
46
. 78
. 84
. 59
. 91
. 88
86
. 75
. 45
. Р0
. 37
44 '
.62.
. 82
. 34
. 16
■ 47
. 21
38
. 51
. 81
. 73
• 52
. 65
. 22
. 90
. 69
• б
. 92
. 15
9
• 17
. 24
. 55
. 58
J 30
. 40
. 68
ч
о
09
2
К
Nd
Ne
Ni
Nb
Sn
Os
Pd
Pt
Po
Pr
Pa
Ra
Rn
(Em NO
Re
Rh
Hg
Rb
Ru
Sm
Pb
Se
S
Ag
Si
Sr
Sb
Ti
Ta
Те
Td
Ti
Th
| Tu
1 С
и 1
р
F
CI
Cr
Cs
Ce
Zn
Zr
Kr
томный
вес
■<
144,27
I 20,183
58,69
92,91
И8.70
190,2
106,7
1195,23
(210,0)
140,92
(231)
..226,05
(222)
166,31
102,91
200,61
85,48
101,7
150,43
207,21
78,96
32,06
107,880
45,10
87,63
121,76-
204,39
180,88
127,61
150,2 '
47,90
232,12
169,4
12,010
238.07
30,98
19,00
35,457
52,01
132,91
140,13
65,38
91,22
167,2
АЗОТ
Nitrogenmm Nitrogen Stickstoft
Asote
N2 At. b. 14,008; мол. в. 28,016
Приготовление
1. Азот, пригодный для большинства работ, содержащий только примесь
инертных газов, получается поглощением из атмосферного воздуха кислорода
и С02.
В сожигательную трубку (тугоплавкую) помещают зерненую окись 'меди
и, накаливая в струе водорода, восстанавливают ее полностью до металли-.
ческой меди. Над полученной тонко раздробленной медью пропускают
воздух, накаливая трубку до яркокрасного каления. Выходящий из трубки
газ, уже не содержащий . кислорода, пропускают через 50%-ный раствор КОН,
затем через концентрированную H2SO4 и, наконец, через колонку,
наполненную CaQh.
Для получения более чистого азота рекомендуется вторичное пропускание
газа через трубку с раскаленной медью.
Метод особенно пригоден для удаления небольших количеств кислорода из
95—99%-ного N*. '
Некоторые авторы рекомендуют, после восстановления Си из СиО, нагреть
ее в вакууме; в противном случае азот может содержать примесь водорода \
2. В круглодонную колбу наливают насыщенный холодный раствор
(NH4)2S04 я закрывают колбу пробкой, через которую проходят капельная
воронка» и газоотводная трубка.
Колбу нагревают на водяной бане, после чего из капельной воронки
приливают по каплям холодный насыщенный раствор NaNC>2. Количество
выделяющегося газа можно регулировать скоростью приливания раствора NaNO*.
Выделяющийся вначале газ еще смешан с находившимся в колбе
воздухом; поэтому собирать газ в газометр следует только после вытеснения из
прибора всего воздуха.
Для совершенной очистки газа от воздуха (попавшего из воды газометра)
и от небольших количеств окислов азота его пропускают вад раскаленной
медью, как описано в способе 12.
Получение этим методом вполне чистого N2 (99,99%} требует особых
предосторожностей. Отделение паров воды и окислов азота проводится в
особой ловушке — стеклянном сосуде, погруженном в жидкий воздух
U1 С. Р уденко8).
3. По Ванино4 пропускают струю воздуха сначала через
концентрированный ЫНЮН, затем над длинной нагретой медной спиралью; реакция про*
текает по уравнению:
4NH» + 30* = 2N* Hi 6НзО.
•?егм89»2069 <1906);А**. и 126.
• fe<bY°iV £k ?Ie Relnaarstellung von Gasen,«0 (1920)-
v a n I n 0, Prap. Ch., 131.
14
Азот
^R\
р-*~
Рис. 1. Колонка для очистки азота
от кислорода
Газ очищается от воды и избыточного NHs пропусканием последовательно
через 50%-ную Ш304, твердый КОН, концентрированную H2SO4 и, для
совершенного осушения, через Р2О5.
4. По К н и ч у * для получения азота, совсем не содержащего кислорода,
пропускают воздух с избытком водорода через тампон из платинированного
асбеста, обернутый в медную сетку и помещенный в железной трубке; при
этом весь кислород превращается в воду без
внешнего нагревания. Оставшийся в избытке
водород полностью сжигается в воду
пропусканием через такую же железную трубку,
содержащую накаленную СиО.
Выходящий газ содержит теперь, кроме
азота, только водяные пары; его пропускают
через холодильник, высушивают и получают
чистый азот
5. По Тичборну2 нагревают 10 г KNOa
и 10 г (NH^SO* с 40 мл глицерина » 60л*л
воды в реторте емкостью 500 мл, приподняв
ее горло наклонно вверх. Выделение N2
начинается ОКОЛО -100° И ПРОИСХОДИТ бЫСТрО 'И
равномерно. Следует избегать слишком
быстрого нагревания реторты во избежание
выделения ]МНз. Газ пропускают через
разбавленный раствор КМп04 для очистки от
следов N02.
6. Равномерный ток азота можно
получить следующим простым путем: прессуют
кубики из хорошей хлорной извести и,
поместив их в средний шар аппарата Киппа, заливают последний
концентрированным NH*OH. Выделяющийся газ содержит избыток ЫНз, от которого
освобождаются пропусканием через 50%-ную H2SO4.
7. По Бэкстеру и Гику чистый N2 получается введением чистой. NO'
в колбу с концентрированным чистейшим, не содержащим аминов NfuOH
(уд. в. максимум 0,92). Смесь NO с избытком NHa пропускают через
тугоплавкую трубку, в которой помещен слой (3—5 см) платинированного .асбеста
или восстановленные медные стружки.
Трубку нагревают в печи для сжигания, как можно сильнее, в результате
чего по реакции
6NO '+ 4NHs = 6H2O + 5N2
получается свободный от 02 азот, если только смесь содержала достаточный
избыток NHs. При этом медь должна оставаться совершенно светлой.
8. Очень хороший метод очистки технического азота от примеси ОГ
заключается в пропускании газа через насыщенный раствор NH4CI в NEUOH
(уд. в. 0,95) в присутствии металлической меди. Отличные результаты
получаются при пропускании газа из баллона последовательно через три колонки
(высота 1 м, диаметр 8 см), содержащие спирали из медной проволоки и
заполненные аммиачным раствором NH4C1 (рис. 1). По мере поглощения
кислорода раствор синеет вследствие окисления меди до Си' ". Обычно раствор в
последней колонке остается почти бесцветным, так как кислород полностью
задерживается в первых двух поглотителях. После прекращения работы синий
раствор быстро регенерируется вследствие восстановления Си' *
металлической медью до Си". При ежедневной очистке нескольких десятков литров Nt
одной зарядки поглотительных колонок хватает на месяц.
J R-К п 1 е t s с h, Berichto v. Internet. Kongress. f. angew, Chem, 1, 674 (1904).
1 lad. Eng. Chera. Anal. Ed. 12, 161 (1920).
Азота двуокись и четырехокись /5
тный газ без запаха и вкуса. Мало растворим в воде (1 об. воды
ВеСо1° оастворяет 0,0152 об. N*) *, несколько лучше в спирте. Т. пл.
при If'о т кип. —-195,67°. 1 л азота весит 1,25056 г (при нормальных усло-
2Ю.5^ » ь
ВИЯ^' т —газ инертный при обычной температуре; из газовых смесей азот
быть поглощен пропусканием над нагретым магнием или над смесью
М°сЮеТс металлическим кальцием.
АЗОТА ДВУОКИСЬ И ЧЕТЫРЕХОКИСЬ
Nitrogenium peroxydatum
N02
Nitrogen peroxide St ickstoffdiox yd
Nitrogen' dioxide §tickdioxyd
Мол. в. 46,008. N2O4 Мол. в. 92,016
Рис, 2. Прибор для очистки fyO*
Приготовление
1. По Гей-Люссаку смешивают совершенно сухой РЬ(ЫОз)а с равным
объемом предварительно прокалённого кварцевого песка и нагревают смесь
в реторте, соединенной с сильно
охлаждаемым приемником. Вначале,
вследствие выделения незначительных
количеств воды, сгущается слабо
зеленоватая, через некоторое время
совершенно бесцветная жидкость. '
2. По Хазенбаху обливают 300 г
iAs-.Оз в кусочках, величиной с
горошину, 200 мл HN03 уд. в. 1,38—1,40
или красной дымящейся HNOs и
выделяющийся газ собирают в приемник,
окруженный охладительной смесью, где
он сжижается. В полученную темно-
зеленую жидкость (смесь N2O3 и N2O4)
пропускают ток совершенно сухого
кислорода, после чего продукт реакции
подвергают перегонке.
Если получение N2O4 ведут в
небольших количествах, то приемником
может служить, например, пробирка В с. двумя трубками (рис. 2, Г).
Прекратив получение N2O4, по трубке А пропускают кислород, после чего заменяют
пробку другой —с одной трубкой, соединенной с сушильной трубкой С,
наполненной стеклянной ватой и P^Os (рис. 2, //). Трубка С соединяется
с другой пробиркой D, погруженной в охладительную смесь. Резиновых
соединений следует избегать. Нагревая пробирку В рукой или погружая ее в
тепловатую воду, заставляют N2O4 перегнаться в приемную пробирку.
3. Для получения сравнительно больших количеств чистой N264 И. Е.
Иошпа и О. П. Спиридонова2 предлагают использовать реакцию
взаимодействия НЫОз и NaN02:
2HN03 + 2№NO,> = 2NaNOs + Н2О + N203
ио^СЛедуюш;нм окислением N2Q3 кислородом. Установка для получения N>Oi
изображена на рис. 3.
№ыг^РУГЛ0Л011ную колбу А емкостью 1,5 л загружают 300—400 г чистого
14U2 и приливают постепенно из склянки В х. ч. НЫОз (50—60%). Газ, про-
> Жпу^Л °v Растворимости азота в воде под давлением см. ЖФХ 6, 1320 (1935).
т11А 12, № 6, 953 (1939).
16
Азот
пускаемый последовательно через дрексель С с 5%-ным раствором AgNO^
колонку D с Р2О5 на стеклянной вате, колонку Е со стеклянной ватой и черЛ
реометр F с вазелиновым' маслом, поступает в окислительный сосуд G \ куш
одновременно подводится кислород из баллона К через реометр L, Образукя
щаяся ЫОг поступает в холодильник Н со смесью льда и NaCl, где проиа
ходят полимеризация NO2 и конденсация N2O4 собираемого в приемнике 1
Подачу кислорода регулируют не только по показаниям реометра, но и в з|
Рис. 3. Получение четырехокиси азота
висимости от цвета жидкой N2O4 (при недостатке Ог продукт имеет
голубовато-зеленый цвет). Производительность установки — при объеме окислитель*
ного сосуда G около 28 л и поверхности охлаждения в холодильнике fl
0,19 м2 — составляет 700 мл жидкой N2O4 за 7 час.
Незначительная влажность жидкой N2O4 обусловливает образование при*
меси HNOa. Для окончательной очистки жидкость обрабатывают Р2Об и пере*
гоняют при 23—24°.
Свойства
При обыкновенной температуре — краснобурый, чрезвычайно ядовитый газ^
представляющий смесь ЫОг и N2O4, причем между ними устанавливается
равновесие:
.N204^2N03,
в сильной степени зависящее от температуры. Степень диссоциации N2O4
видна из табл. 1.
Жидкая N2O4 — желтого цвета при комнатной температуре, бесцветная при
—20°. Уд. в. 1,49 при 0°. Т. кип. + 21,2°. При сильном охлаждении
затвердевает в кристаллическую массу, имеющую т. пл. —10°. Водой разлагается
с образованием NO и Н1ЧОз. От прибавления небольшого количества воды
становится яркозеленои. Пары удушливы, вызывают кашель и при длительном
вдыхании воспаление легких.
» В качестве такого сосуда можно применить стеклянную бутыль.
Азота закись
11
Таблица 1
*яь диссоциации N204 в зависимости оттемпературы
Стелен» ^д Naumann)!
г с
26,7
35,4
39,8
49,6
Степень
диссоциации
Ne04 в Oj'o
19,96
25.65 •
29.23
40,04
/'С
60.2
! 70,0
• 80,6
90,5
Степень
диссоциации
N»04 в «/о
52.84
65,57
76.61
84,83
f°C
100,1
111,3
121,5
| 135,0
Степень
диссоциации
Nt04 в о/о
89,23
92,67
?6,23
98,69
Nitrogenium
oxycttilatum
АЗОТА ЗАКИСЬ (ВЕСЕЛЯЩИЙ ГАЗ)
Nitrous oxide
N20 Мол. в. 44,016
Stickoxydul
Lachgas
Приготовление
1. По Бендеру2 закись азота удобнее всего получать нагреванием
NH4NO3. Соль предварительно осторожно плавят для обезвоживания и
помещают куски в объемистую' реторту, которую слабо нагревают на проволочной
сетке.
- Соль легко плавится и около 170° разлагается:
NH*NOs = N2O + 2ШО.
При начале газообразования следует уменьшить пламя так, чтобы
происходило лишь легкое кипение. Сильное нагревание может повести к взрыву;
кроме того, при перегреве может частично образоваться NO. Поэтому
нагревание прекращают- прежде чем разложится вся соль. Из 4 г NH4NO3
получается больше 1 л №0. Газ собирают над теплой водой (в холодной воде
ЫгО .значительно растворяется) или над раствором NaCl.
Для получения совершенно чистой N2O следует применять NH*NOs, не
содержащий примеси NKUCl, чтобы одновременно с N2O не выделялся хлор.
Для очистки NaO от возможных примесей СЬ и NO газ промывают
последовательно растворами КОН и FeS04.
2. При отсутствии NH4NO3 по Смиту3 употребляют смесь 17 вес. ч.
NaN03, 20 вес. ч. KN03 и 13-14 вес.
ч. (NH^SO*. Смесь помещают в
реторту и нагревают сначала при 230°,
затем при 300°. Реторта должна быть
помещена так, чтобы образующаяся
вода, конденсируясь, не стекала обратно.
При этом условии получается очень
Равномерный ток N2O.
Свойства
Бесцветный газ со слабым
сладковатым запахом и вкусом, довольно
легко сжижаемый. Т. кип. —90,7°, т. пл.
—102,4°. Уд. в. жидкой N20 1,226.- \л
w2u весит при нормальных условиях 1,9778 г. Плотность 1,5299 (по воздуху).
^^ри 520° медленно распадается на N* и О*. Несколько растворим в воде и не-
Таблица 2
Растворимость N20 вводе
t°c
ОСЛО
1 об. воды
растворяет при
760 мм об. N*0
1,2469
1,0480
0,8778 1
ГС
15
20
25
1 об. воды
растворяет ПРИ
76U мм об. N,0
0,7378
0,6294
0,5443
а ^пп. Suppi. 6, 203 (1ЙБ8).
IIриг. неорг. преп., 3j2.
w- S m i t h, J. Soc Ch. IndM 11 (1892).
IS
Азот. Алюминий
много лучше в спирте (1 об. спирта растворяет 4 об. Ni>0). Сухой газ подо( \
кислороду воспламеняет тлеющую лучинку.
Вдыхание N2O вызывает шум в ушах и потерю сознания. Эти явле] а
быстро проходят при доступе свежего воздуха. Вдыхание №0, разбавлен: |
4 объемами воздуха, в течение нескольких минут вызывает опьянение.
Nitrogenium
oxydatum
АЗОТА ОКИСЬ
Nitric oxide
NO Мол. в. 30,008
Stickoxyd
Stickstoffmonoxyd
Salpeterg.as
Рис. 4. Прибор для
получения окиси азота
Приготовление
1. Действуют НЫОз на медь; для того чтобы газ содержал возмо>;
меньше других окислов азота, следует употреблять HNOs уд. в. не выше
и вести реакцию, не допуская заметного разогревания. Обрезки меди (про
лока, стружки и пр.) помещают в склянку Вульфа (рис. 4) и медленно при*-
ливают через воронку разбавленную HNOs. Если
смесь разогреется, охлаждают склянку холодной во>
дой. После удаления бурых паров N02
(образовавшихся от соприкосновения N0 с кислородом воздуха)
собирают газ над водой. Для осушки газ пропускаю*
через раствор NaOH и затем через трубку с тверд
КОН (Миллон).
1а. Кеммерер рекомендует для лучшего регули]
вания тока N0 поместить *в склянку Вульфа полоски
медной жести и налить на 1/з высоты склянки ко%
центрированного раствора NaNOe. Через воронку,
зависимости от желаемой скорости газовыделeHi ■.
приливают более или менее сильную струю H2SO4 уД.
в. 1,84.
2. Реньо рекомендует для получения очень чи»
стой N0 нагревать раствор KNOa с FeCk в
присутствии кислоты. Можно, например, поступать следу»*
щим образом: определенное количество НС1 делят на две части, в одной
растворяют до насыщения железо и затем приливают вторую. Полученный рас»
твор нагревают с KNOs и, после вытеснения КОг и воздуха, получают чисту
N0. Для очистки от увлеченного НС1 газ пропускают через раствор NaOl
3. Джонстон1 предлагает нагревать раствор 40 г KCNS с 30 г азотн^
кислого кобальта, в результате чего получается около 12 л N0:
4KCNS + Co(NOs)2 = 6NO + 4С + 2K*S + CoS + S,
4. По Тиле2 очень равномерная струя совершенно чистого N0 получаетсА
если к солянокислому раствору FeCb или FeS04, находящемуся в колбе, прк
пивать по каплям из капельной воронки крепкий раствор NaNOs:
FeCb + NaN02 + 2HC1 = РеСЬ.+ NaCl + H20 + NO.
Выделяющийся газ промывают раствором NaOH.
5. По В а н-Д евентеру к смеси концентрированных растворов K4Fe(CN)
и NaNOo (причем первый должен быть в избытке) прибавляют из капельно!
воронки 80%-ную СНзСООН. Для начала реакции рекомендуется содержимся
колбы несколько подогреть и тогда уже медленно капать СНзСООН. (боле<
быстрое прибавление уксусной кислоты всегда вызывает образование примесц
1 Johnston, Chem. News, 45, 159.
2 Т h i e I e, Ann., 253, 246 (1889).
Азота окись. Алюминий бромистый
19
их окислов азота). Выделяющийся газ промывают раствором NaOH для
у"?еСаПСе№ениой Чистки NO пропускают медленной струей через
6' ^FeSO. помещенный в объемистой колбе. Здесь N0 поглощается
^амТ0Вжет быть ' вновь выделен при на- Таблица 3
гревании. Первые порции выделяюще- растворимость NO вводе
г^ся газа отбрасывают, так как они г (iT M. W In k 1 ег)
содержат примесь NU*.
Свойства
Бесцветный газ, трудно сгущаемый
в жидкость. Т. пл. — 160,9°, т. кип.
150,2°. Уд. в. жидкой N0 1,27. 1 л
N0 при нормальных условиях весит
1,3402 г. Плотность 1,0367 (по воздуху).
В воде растворим плохо, в спирте
несколько лучше.
*°с
ОСЛО
1 об. воды
растворяет об.
N0
0,07381
0,06461
0,05709
ГС
15
1 20
25
1 об. воды
растворяет об.
NO
0,05147
0 04706
0,04323
АЛЮМИНИЙ БРОМИСТЫЙ (БРОМИД АЛЮМИНИЯ)
Aluminium bromatum Aluminium bromide Aluminiumbromid
АШгз Мол. в. 266,72
Приготовление
По Густавсону1 при помоги капельной воронки (рис. 5) льют каплями
бром в нагретую до 100° колбу; образующиеся пары брома проходят над
листовым' алюминием
(алюминиевой жестью),
помещенным в
наклонно расположенной
стеклянной трубке.
Бромистый алюминий стекает
в реторту, служащую
приемником. Реакция
начинается при
нагревании алюминия; после
начала реакции подо-
гревание можно
прекратить, наблюдая, однако,
чтобы 'А1Вгз не
затвердевал в реакционной
трубке, а стекал в
реторту. Охлаждать
последнюю нет
необходимости.
Получившийся АШгз
Целесообразно
подвергнуть перегонке из
реторты, которая служила
приемником. Первая фракция дестиллата окрашена и содержит свободный
ором; следующие порции дестиллата совершенно чисты. Для хранения
расплавленный АШгз разливают в сухие банки, которые закрывают пробками и
заливают парафином.
Рис 5. Прибор для получения бромистого алюминия
* О ц s t а у s о n, J. prakt. Ch. (2) 63, 110 (1910),
20 Алюминий
Свойства
Бесцветные блестящие листочки уд. в. 2,54 *, плавящиеся при 97° в nti
зрачную, как вода, подвижную жидкость, кипящую при 260°. Дымит на *вс1
духе. С водой реагирует настолько бурно, что происходит подобие взрыд|
Растворим в CSs' и ацетоне.
АЛЮМИНИЯ ГИДРАТ ОКИСИ (ГИДРООКИСЬ АЛЮМИНИЯ)
Argilla pura Aluminium hydroxide lAluminiumhydrox}
'Alumina hydrata Tonerdehydrat."
Aluminium hydroxydatum
А1(ОН)з Мол. в. 77,99
А. Гидрат окиси алюминия обыкновенный
Приготовление
1. Для лабораторного получения чистого гидрата окиси алюминия р<
мендуется способ, предложенный Шмидтом2. Приливают горячий раса
алюминиевых квасцов или Ah(S04)3 (1:20) к такому количеству pa36aej
ного NH4OH (1 об. крепкого аммиака + 3 об. воды), чтобы последний нах<
дился в избытке (например, на 1 об. вышеуказанного раствора квасцов 1,1 щ
10%-ного аммиака).
По осаждении выпавшего гидрата окиси алюминия сливают возможно поЛ
находящуюся над ним жидкость, промывают осадок горячей водой, раствор5
осадок и, вторично осадив, декантируют. Эту операцию повторяют до |
пор, пола слитая прозрачная жидкость при прибавлении раствора хлорист |
бария, подкисленного НС1, не перестанет мутнеть. Осадок переносят на ну
фильтр или колоторку с некрашенным холстом, осторожно отжимают и пос.
предварительного измельчения высушивают при умеренном нагревании (по|
кснец при 100°).
При получении А1(ОН)з из квасцов необходима млогократная декантан
ция; для совершенного освобождения от калия нужно осадок еще раз раство|
рить в НС1, раствор при нагревании вновь осадить небольшим избытк
аммиака и выпавший осадок промыть декантацией до отрицательной реакн
на ион СГ в промывных водах.
2. При получении А1(ОН)з из соли, загрязненной железом, холодный насыГ
щенный раствор квасцов вливают при постоянном помешивании в горяч!
раствор NaOH (10—20%), пока не получится значительный осадок, котор:
растворяют прибавлением некоторого количества NaOH. Оставшийся нерастг
ренным осадок Fe(OH)3 отфильтровывают через волокнистый асбест.
Фильтрат, нагретый до кипения, смешивают, с избытком раствора NH4
и некоторое время нагревают почти до кипения. При этом алюминат расл
дается и выпадает свободный от основной соли А1(ОН)з, который xopoi
промывают декантацией и сушат при 100°. Получается рассыпчатая мае
состава АЬО(ОН)4 (Л ё в е).
3. По методу, разработанному в ИРЕА, растворяют металлический алюм
ний в водном растворе NaOH. Полученный раствор алюмината отфильтров1
вают от нерастворившихся примесей и осторожно нейтрализуют Н1\Юз. Выпа
ший осадок А1(ОН)з отфильтровывают, тщательно промывают и сушат, повь
шая постепенно температуру до 100°.
4. Для очистки продажного А1(ОН)з Р. Ф р и к е рекомендует растворит
его при 50—60° в насыщенном на холоду растворе NaOH, затем разбавить
* Плотность жидкого А1Вга при температуре t может быть вычислена по уравнена!
rfs= 2,905-0,00249 t (Д. И. Ж у р а в л е в, Жф£, 10, 325 (1037).
'Schmidt, Ausfuhr. l^enrb. d. paarm. Co. 1, 928.
Алюминия гидрат окиси 21
— 9
vR в 1,15 и отфильтровать осадок, состоящий в основном из Fe(OH>.
водой до' У£ ' пропускают С02 в течение трех дней. Осадок несколько дней
Через Ф декантацией холодной водой, затем горячей водой до удаления
промыва а на лакмусу. Препарат, высушенный сначала над СаСЬ, а затем
щел0р"о5, почти точно отвечает формуле А1(ОН)з (потеря при прокаливании
чвП0% теоретически 34,59%).
* Аналогичный метод рекомендует применять В. М. Мухэтев1 для
ингтки от органических веществ технического АКОН^з, окрашенного в розо-
й или коричневый цвет. Технический препарат размешивают в течение часа
^Ыопо/ -ным раствором чистого NaOH при 50—60° и отфильтровывают нераство-
пившийся остаток. В интенсивно-бурый фильтрат при 20—40° пропускают СОг.
Через некоторое время- появляется студенистый осадок А1(ОН)з; этот момент
тппювождается характерным ' изменением окраски раствора (опалесценция).
Тпгяа прерывают пропускание С02 и тотчас отфильтровывают черно-бурый
осадок состоящий из А1(ОНЬ, адсорбировавшего все органические примеси.
Совершенно бесцветный фильтрат насыщают СО2, причем выделяется ^
зернистый AlfOH^. После высушивания препарат принимает снежно-белый цвет,
что свидетельствует о полной чистоте.
Свойства
Белая зернистая масса уд. в. 2,423, почти совершенно нерастворимая
в воде (при 20° растворяется 1,5-10-4%). Свежеосажденный А1(ОН)з,
находившийся долгое время при обыкновенной температуре под водой, теряет
способность растворяться в щелочах и кислотах. Подобное изменение
наблюдается .и при высушивании А1(ОН)з выше 130°. Из раствора в КОН иногда
выделяется А1(ОН"]|з в виде мелких кристаллов.
Испытание 2
1. Растворимые в воде примеси. 1 г испытуемого препарата кипятят с 50 мл
воды и отфильтровывают от нерастворившегося остатка. Фильтрат не должен
показывать кислой или щелочной реакции и при выпаривании не должен
оставлять весомого остатка.
2. Кремнекислота. Оставшийся после промывания водой остаток должен
полностью растворяться в НС1.
3. Сульфаты. Раствор, полученный при испытании на Si02, при
прибавлении ВаСЬ не должен давать даже незначительной м*ути.
4. Соли бария. К раствору, полученному при испытании на БЮг,
прибавляют разбавленную H2SO4. Даже при длительном стоянии не должно быть
никакой мути.
5. Соли калия и натрия. Солянокислый раствор препарата нагревают и
л??Дца!0Т ^ебольшим избытком раствора (NH^COs. Отстоявшийся осадок
АнОН)з отфильтровывают и фильтрат выпаривают досуха. Полученный таким
образом осадок солей при прокаливании должен оставлять только очень
незначительный остаток.
6. В растворе NaOH испытуемый препарат должен легко и полностью
растворяться; магниевые соли и сильно высушенный А1(ОН)з остаются нерас-
творенными. . у
7. Цинк и железо. В щелочном растворе прибавление (NH^S не должно
вызывать выпадения осадка. Слабо зеленое окрашивание указывает на
присутствие очень малых количеств железа.
Б. Гидрат окиси алюминия коллоидальный
Приготовление
ный' м?Ди^0Лучения коллоидального раствора А1(ОН> по Гофману влаж-
А1(ОН)з, осажденный из 100 г кристаллического хлористого алюминия
\ ^ав«~Лаб- 8« № 7» 7'6 (1939).
9 Ausfur. Lehrb. d. pharm. ChM 1,
930.
22
Алюминий
(АЮз-бНгО) и подвергавшийся промыванию в течение нескольких дней, для
тельное время нагревают с раствором 4 г Ледяной уксусной кислоты в 100
воды. Образуется молочно-белый, очень постоянный коллоидный раствор
гидрата окиси алюминия.
2. По К. И. Шей д ту1 на дно стеклянного цилиндра наливают 3—5 \
возможно более чистой ртути и туда же помещают тщательно очищенную
полоску алюминия (еще лучше, пучок проволоки диам. 0,1 м), прижимаем >
ко дну стеклянной палочкой. В цилиндр наливается свежеперегнанная вода
и прибор оставляется в покое на 12—24 часа. По прошествии этого времен^
около дна замечается обильное выделение хлопьев А1(ОН)з, в то время как
верхняя часть цилиндра занята почти прозрачным золем. В первый раз зола
обычно, не образуется, а выпадают только хлопья [А1(ОН)я увлекает с собой
все загрязнения и вторичное образование золя происходит уже из
значительно более чистых веществ].
Полученный золь можно отфильтровать, но удобнее отцентрофугировать- и
отобрать верхний, прозрачный слой декантацией или пипеткой. Концентрация
золя 0,8 — 2,383 г/л. Размер частиц 20—340 (j.jx. Автор отмечает высокую
чувствительность золя к электролитам: сохранять его можно только тща!
тельно закрытым в посуде из иенского стекла.
АЛЮМИНИЯ КАРБИД
Aluminium Aluminium carbide Aluminiumcarbid
carbidum
AI4C3 Мол. в. 143,91
Приготовление
1. ПоМатиньону2 тщательно приготовленную при помощи скипидар
смесь 24 ч. сухой сажи и 70—140 ч. порошка алюминия' нагревают в течение
20 мин. в тигельной печи при 1000—1100° и удаляют избыток алюминия
холодной НС1 или, лучше, холодным раствором КОН.
2. О. Руфф рекомендует прокаливать в токе чистого Нг при 2000° смесь
3 ч. алюминиевых стружек и 1 ч. порошка беззольного угля. Нагревание
проводится в течение 30 мин. в закрытом угольном тигле.
Свойства
Красивые большие светложелтые прозрачные кристаллы длиной 5—6 НЩ
уд. в. 2,36; иногда — толстые правильные шестиугольники. Водой медленно
разлагается с выделением метана:
AUCs + 12Н20 = ЗСШ + 4А1(ОН)3.
АЛЮМИНИЯ ОКИСЬ (ГЛИНОЗЕМ)
Aluminiuim Alumina Aluminiumoxyd
oxydatunn Aluminium oxide Tonerde
АЬОз Мол. в. 101,94
Приготовление
1. Для получения АЬОз прокаливают смесь 4 ч. сернокислого алюминия ,
1 ч. безводной соды. Остаток промывают водой до полного удаления NasSO-i.
2. Прокаливают Ab(S04)33 или А1(ОН)з при 120О— 1300° до постоянного
веса. Гюттнг4 рекомендует прокаливать чистый продажный АЬОа при
температуре не ниже 1050°.
1 ЖРФХО (4) 50, 617 (1^.28).
• М a t i я п о п, С. г., 145, Ь76 (1907).
» Неорг. преп. 131.
4 G. F. Н й 11 i g. Z. anorg. Ch., 224, 230 (1935).
Алюминия карбид. Окись. Алюминий сернистый
23
указанию Г. Борне-мана1 возможно чистые алюмоаммонийные
/г измельчают в тонкий порошок и осторожно нагревают в фарфоровом
квасцы 920 квасцы плавятся в кристаллизационной воде, при 190° обез-
ТИГ£тяются сильно вспучиваясь при этом. Повысив температуру до светло-
В° ного каления, продолжают прокаливание довольно продолжительное
Кра^я Охлажденная проба препарата при действии разбавленной НС1 не
1,Рпжна давать реакции на H2SO4. В противном случае вещество растирают в
ступке'и еще раз прокаливают. Выход около 11 г А1(ОН)з из 100 г квасцов.
Ов о йс тва
Белый порошок без вкуса,( уд. в. 3,85, жадно поглощающий воду, но при
этом не растворяющийся. Влажный воздух, пропущенный через трубку сАЬОз,
по выходе содержит всего лишь 0,003 мг/л НгО (Боуэр). Растворимость
в кислотах зависит от температуры, при которой препарат получен: сильно
прокаленный не растворяется в минеральных кислотах и может быть
переведен в раствор только сплавлением в шнкоизмельченном виде с КОН или
KHSO« или же длительным нагреванием с концентрированной H2SO4. Т. пл.
2050°, т.-кип. 2980°.
АЛЮМИНИЙ СЕРНИСТЫЙ (СУЛЬФИД АЛЮМИНИЯ)
Aluminium Aluminium sulfide Aluminiumsulfid
sulfuratum
AI2S3 Мол. в. 150,12
Приготовление
По Фонцес-Диакону2 смешивают вычисленные количества порошка
алюминия и серы и, поместив смесь в объемистый тигель, зажигают ее при
помощи ^ ленты магния. Когда начинается бурная реакция, тигель закрывают
крышкой. При реакции выделяется столько тепла, что избыток алюминия
плавится.
По охлаждении получается желто-серая масса.
Свойства '
ип^еЛ£?'"серая« плотная масса УД- в- 2.37 с запахом сероводорода. Т ил.
1Ш0 . При действии горячей воды происходит бурное выделение
сероводорода
АЛЮМИНИЙ СЕРНОКИСЛЫЙ (СУЛЬФАТ АЛЮМИНИЯ)
Aluminium sulfate
Ab(S04)H8H20 Мол. в 666,41
sliKUri Aluminium sulfate Aluminiumsulfat
Приготовление
AKOmMeDH 189 г HaS°4 (уд' B- ]'84> и 139 »* во*ы прибавляют 100 г
корт Реакция происходит с сильным разогреванием. По охлаждении жид-
в банкиаСВыВаеТ 427беЛУЮ Пористую массу' п°следнюю истирают и всыпают
Свойства
в плпрЧИСТОМ виде белые гексагональные чешуйки или иглы. Легко растворим
w, очень трудно — в спирте. Вкус кислый и горький. При нагревании соль
1 Неорг. преп. 133.
г о n z ё з - D i а с о п. С. г., 130, 1314 (1900).
24
Алюминий
сильно вспучивается и превращается в губчатую массу. При красном календ
теряет SO».
Таблицаf
удельный вес водных растворов AI^SOJb
e/oA!,(SO«),
1
2
4
6
8
d
4
1,009
1,019
1,040
1,061
1,083
Wo Ale(S04)3
10
12
14
16
18
1,105
1,129
1,152
1,176
1,201
о/о Ala(S04)f
20
22
24
26
28
18
1,226
1,252
1,278
1,306.
1,333 J
Растворимость A12(S04)3 вводе
Таблица 5
t°C
0
10
20
°/cA!,(S04)3
23,8
25,1
26,6
t°c
30
40
50
°/oAI,(S04)3
28,8
31,4
34,3
t°c
60
80
100
*/оА1а(80<)»
37Д
42,2
47,1
Испытание
Наибольшие количества примесей, допустимых по ОСТ НКТП 7175/469»
указаны в табл. 4.
Таблица б
Допустимые примеси в A12(S04)3 • 18Н20
Примеся
Допустимое содержание
В о/о
чистом для^
анализа
1. Нерастворимые в воде вещества
2. Хлориды (СГ)
3. Аммонийные соли (NH4') . . • . . •.
4. Металлы, осаждаемые H2S
5. Железо (Fe)
6. Щелочные и щелочноземельные металлы (в виде
сульфатов) ... „ „
0,02
0.002
0,005
0,001
0,0СЗ
0,25
0,05
0,01
0.02
0!002
0,01
0,5
Алюминий сернокислый и хлористый
25
АЛЮМИНИЙ ХЛОРИСТЫЙ (ХЛОРИД АЛЮМИНИЯ)
Aluminium chloratum Aluminium chloride Aluminiumchlorid
А. Безводный
Aids Мол. в. 133,34
приготовление
i по Штокгаузену и Гаттерману получение AlCb ведут в воз-
LI> широкой, тугоплавкой трубке А для сожигания (рис. 6), один конец
М пппй нескочько оттянут, а другой при помощи пробки с двумя отверстиями
пинрн с широкогорлой ба'нкой. Во второе отверстие пробки вставляется
'рТчень узкая газоотводная трубка. В трубку Л, помещенную в^сожигатель-
ню печь, загружают от 10 до 40 г так называемой алюминиевой «крупы> и
^ЯЯ^?=Р^^^^^Ч^^.
Ркс. б# Схема прибора для получения хлористого алюминия
пропускают через аппарат довольно сильную струю сухого хлористого
водорода, высушенного H2SO4.
Когда .весь воздух вытеснится {не раньше\)у нагревают трубку по всей
длине, однако не настолько сильно, чтобы алюминий сплавился в шарики.
Нужную температуру легко подобрать, если пламя увеличивать постепенно;
При должной высоте пламени начинают возгоняться в приемник значительные
количества хлористого алюминия. Выступающую из печи часть реакционной
трубки следует делать -возможно короче,-чтобы избежать значительного
оседания на ней !А1С1з, так как может произойти засорение трубки. Чтобы пробка
приемника не обугливалась, ее защищают толстым асбестовым листом, в
котором проделаны отверстия для трубок. Трубку охлаждают в токе хлористого
водорода' и только по охлаждении вытесняют НС1 воздухом.
2. А. И. К р я г о в а г предлагает подвергать алюминиевые стружки
воздействию хлора или хлористого водорода при 650—700° с последующей
возгонкой А1С1з. .
3. Смешивают АЬОз или А1(ОН)з с тонким порошком угля при помощи
клейкого вещества, способного обугливаться (клейстер, сахарный сироп),
формуют из тестообразной массы шарики и сильно прокаливают в хорошо
закрытом тигле. Полученные пятнистые черные шарики помещают в аппарат,
аналогичный описанному в способе 1, и соединяют реакционную трубку
с аппаратом для получения хлора. Трубку нагревают до умеренно красного
каления и пропускают ток хлора, осушенного пропусканием через FhSO* и
LaCl2; хлористый алюминий возгоняется в приемник в виде массы, состоящей
из кристаллических пластинок.
1 ЖОХ 9, № 19, 1756 (1939),
26
Алюминий. Аммиак
I
4. А. И. Крягова1 рекомендует надежный метод очистки техническое
А1С1з от примеси железа. Метод основан на восстановлении FeCls
металлическим алюминием при нагревании в запаянной трубке при 200°;
FeCh + Al = AlCla 4- Fe.
Затем продукт подвергается возгонке. Недостатком метода является его д
тельность и возможность разрыва запаянных трубок2.
Полученный тем или иным способом AlCh. тотчас же переносят в суху
горячую банку и плотно закупоривают (резиновой пробкой).
Свойства
Бесцветные прозрачные гексагональные таблички уд. в. 2,4)1, плавящш
при 190° (при 2,5 атм). По А. И. Кряговой уд. в. А1СЬ в расплавленном <
стоянии при температуре t может быть вычислен по уравнению: d=\fil
—0,0026 L tann тт
При атмосферном давлении возгоняется, не плавясь, при 183°. На возду.
притягивает воду и образует пары хлористого водорода. Растворим в во|
спирте и эфире со значительным выделением тепла.
Таблица 7
Растворимость AlClg в воде
(Malquori, 1928)
*°с
0
20
40
о/о А1С18
31,03
31,36
31,63
t°c
60
80
o/oAlCl,
31,73
32,32
Таблица
Удельный вес водных р а <
т в о р о в А1С13 (М. Констант!
нов и И. Казарновский, 1933J
Б. Водный
Aids • 6НгО
Мол. в. 241,44
Приготовление
1. Чистый А1С1з*6Н20 легко получается из содержащего железо продукта
По Деннису растворяют неочищенный безводный хлористый алюминий ]
воде, фильтруют через стеклянную вату и смешивают с концентрированно!
НС1. Раствор наливают в склянку Дрекселя, помещенную в смесь льд|
с солью, и насыщают сухим хлористым водородом» Во время пропускани!
последнего образуются мелкие белые кристаллы (если кристаллы не выпа!
дают, добавляют к раствору немного эфира); когда получится нужное количе!
ство кристаллов, их промывают, декантируя холодной концентрированной HClj
до удаления железа (испытание — отсутствие красного окрашивания при доба!
влении KCNS к пробе жидкости).
Отсасыванием по возможности освобождаются от соляной кислоты.
Кристаллы оставляют на глиняной тарелке до исчезновения запаха НС1 и высу-j
шивают при 20°.
2. 28 г гранулированного металлического алюминия растворяют в 460 <
х. ч. НС1 (уд. в. 1,125). Растворение ведется вначале на холоду, затем при
подогревании. Испаряющуюся воду следует восполнять, чтобы раствор не заЗ
густевал: объем жидкости должен быть все время около 400 мл. Указанные
1 ЖОХ 9, № 19. 1755 (1039).
' Об очистке А1С13 электролизом см. В.
R о в с к и ft, ЖФХ, 4 (5), 745 (1933).
Котов, М. Константинов, И. К а з а р-
Алюминий хлористый. Аммиак
27
тва Л1 и НС1 можно увеличить, но во всяком случае не следует рас-
количеС в г1ашке больше 200—250 г алюминия, так как растворение протекает
TBOpfb6ypHo.
°чео створу Дают отстояться, после чего фильтруют через стеклянную вату,
тоат выпаривают до появления кристаллической пленки и охлаждают.
Ф|1Л «ллы отсасывают и промывают декантацией концентрированной НС1
КрйСТ ^19). Препарат отсасывают, снова заливают кислотой и снова отсасы-
(УД- в'к;рметаллы помещают на глиняную тарелку и оставляют в сухом месте
ва1°Т'чезновения запаха НС1, после чего ссыпают в сухие банки, которые зали-
Д°,ют парафином К '
3 Наливают в фарфоровую чашку, помещенную на кипящей водяной
* не 200 мл НС1 (уд. в. 1,19) и при помешивании растворяют в ней неболь-
,ми порциями 50 г А1(ОН)з. Раствор упаривают до получения кашицеобраз-
Шой массы, которую отсасывают и промывают чистой НС1 (уд. в. 1,19).
Выход МО г.
Свойства
Белый кристаллический порошок, сильно гигроскопичный. Очень легко
растворяется в воде. Кристаллизационная вода связана комплексно; поэтому
формулу водного хлористого алюминия - следует писать так: [А1(Н20)в]СЬ.
Испытание
По ОСТ НКТП 7667/659 водный препарат (А1С1з • 6НгО) должен
соответствовать требованиям, указанным в табл. 9.
Содержание AlCU-бНгО должно быть не менее 90%.
Допустимые примеси в АЮ8*6НаО
Таблица 9
Примеси
Допустимое содержание .в °'о
в чистом для
анализа
1. Нерастворимые вещества
2. Сульфаты (SO/)
■J. Аммонийные соли (NH4*)
4. Металлы, осаждаемые H2S
5. Железо (Fe)
6. Щелочные и щелочноземельные металлы (в виде
сульфатов)
7. Кремнекислота (Si02)
0,025
0,01
0,002
0.001
0,002
0,02
0,05
0,05
а,оз
0,005
0,002
0,005
0,5
0,2
Ammonia
Риготовление
АММИАК
Ammoniak
NHa Мол. в. 17,032
А. Газообразный аммиак
1- Удобнее всего получать газообразный ЫНз постепенным нагреванием
кРепкого водного раствора аммиака (NH4OH). Так как растворимость газа
с повышением температуры падает, то получается ровная и сильная струя
1 Разработано Ю. В.. К л и м е н к о.
28
Аммиак
газа. Для осушения его пропускают через склянки Тищенко или и-образны$
грубки, наполненные твердым КОН.
Продажный водный аммиак часто содержит органические вещества (пири*|
дин и его основания, пиррол и др.)- По Шерингу от большей части этих
примесей освобождаются, добавив перед нагреванием 1—2% KMriOi.
Еще лучше поступать по С т а с у следующим образом: NH<OH переводя*
добавлением соляной кислоты в NH4C1 и кипятят насыщенный нейтральный
раствор последнего с добавкой Ую объема концентрированной Н1ЧОз, пока на
прекратится выделение хлора. Раствор охлаждают, выделившиеся при этом
кристаллы отсасывают и еще раз кипятят с небольшим количеством HNO*,
как указано выше. Получившиеся кристаллы пере-»
водят в NHs нагреванием с КОН (п. 2).
2. Смешивают в колбе 5 ч. измельченного
NH4CI с 7 ч. хорошей, свежетюгашенной извести!
К смеси добавляют 10 ч. воды и хорошо
перемешивают. При нагревании колбы на песочной баня
получается равномерная струя газа, который про|
мывается в склянке Тищенко небольшим количе|
ством воды и осушается твердым КОН. Вместо]
NH4CI можно употреблять (NH4O2SO4, вместо!
Са(ОН)2 — КОН.
3. Очень постоянную струю газа можно полу-J
чить по Нейману, приливая из капельной
воронки концентрированный NFhOH к твердому KOHJ
помещенному в склянке Вульфа или бунзеновской?
колбе.
4. Удобный аппарат для лабораторного получё-"
ния значительных количеств NHs. из водного ра
твора, позволяющий регулировать скорость выдел
ния газа, предложен М. 'А. Поповым1.
Железный резервуар (рис. 7), снабженный в;
верхней крышке тремя горловинами и имеющий!
сбоку водомерное стекло, изготовлен из куска]
трубы высотой 11 см и диаметром 9 см; крышка,!
дно и горловины приварены.
Стеклянная трубка А служит для наблюдения-;
за скоростью подачи в аппарат концентрированного^
ЫНЮН; винтовой зажим В на трубке С,
соединенной с бутылью, наполненной NH4OH, позволяет
регулировать скорость подачи. Шарик термометра
помещается около дна прибора. В резервуар нали
вают воды до половины объема и нагревдют его,;
Когда температура достигнет 98°, отвинчивают
зажим В и регулируют число капель NH4OH в
трубке А так, чтобы NHs выделялся из аппарата с требуемой скоростью.
Затем приоткрывают зажим D, чтобы уровень жидкости в резервуаре оставался
постоянным. Выделяющийся ЫНз проходит через шариковый холодильник Е
для конденсации паров воды, затем поступает в промывную склянку с дестил-
лированной водой и в сушильную колонку с твердым КОН.
Если желательно получить ЫНз свободным от пиридиновых оснований,
добавляют к исходному NH4OH 1% КМп04 и применяют полученную мутную4
жидкость, не отфильтровывая осадка.
Отрегулированный прибор, по утверждению автора, может работать
часами, причем в отходящей жидкости остается не более 2% NH3. Для обес- |
печения равномерной работы аппарата полезно покрыть резервуар слоем >
асбеста.
Рис. 7. Аппарат Попова дла
получения аммиака (разрез)
••
' ЖПХ 11, J* 7, 1237 (1938).
Аммиак
29
Свойства
Бесцветный газ с сильным характерным
запахом (в совершенно чистом
vr„nnaPT острым колющим запахом, мало похожим на запах обычного
ООЛадас! ^ттк л-^ло „ n^™^™. гтл « л * тА rv v П RQK9 Т тттт __77г7°
вйДе оон а ^ ^Нз весит ^7708 ^ плотность по воздуху 0,5962. Т. пл. ■
аммиак К^^ош При комнатной температуре может быть приведен в жидкое
Т* Шляние давлением 6—7 ат.
состояние ^идкого NHs при Оо равен 0)638.
В атмосфере кислорода аммиак горит зеленоватым пламенем, образуя азот
воду. Смесь 4 об. ЫНз и 3 об. Оз сильно взрывается при зажигании.
Аммиак хорошо растворим в воде (см. табл. 10—12), при растворении
выделяется, большое количество тепла.
Таблица 10
Растворимость NH8 вводе в зависимости от температуры
(R о s с о е, D i 11 m а г, R а о и 11)
/°C
0
2
4
6
8
10
12
14
16
18 1
1 z воды
растворяет
z NH3
0,875
0,833
0,792
0,751
0,713
0,679
0,645
0,612 |
0,582
0,554
1 об. воды
растворяет
об. NHa
1176
1047
947
857
775
/•с
20
22
24
26
28
30
32
34
36
38
1 z воды
растворяет
z NH,
0,526
0,499 1
0,474 1
0,449
0,426
0,403
0,382
"* 0,362
^ 0,343
0,324
1 об. воды
растворяет
об. NHa
702
639
586
t° с
40
42
44
46
48
50 .
52
54
56
1 Z ВОДЫ
растворяет
?NH3
0,307
0,290
0,275
0,259
0,244
0,229
0,214
0,200
0,186
Таблица 11%
Растворимость NH3 в воде п р и 0° в зависимости от
давления (Ros.coe, Dittmar)
Давление
NH8 в мм
рт. ст.
0
38
76
114
152
190
1 Z ВОДЫ
растворяет
z NHa
0,000
0,175
0,275
0,351
0,411
0,465
Давление
NH8 в мм
рт. ст.
228
304
380
456
. 532
608
1 z воды
раетворяет
z NH8 |
0,515
0,607
0,690
0,768
0,840
0,906
Давление
NHa в мм
рт. ст.
684
| 760
836
912 <
988
1064
1 Z ВОДЫ
растворяет
t NH,
0,968
1,037
1,117
1,208
1,310 ,
1,415
Давление
NH8 в мм
рт. ст.
1140
1216
1292
1368
1444
1520
1 z воды
растворяет
z NH.
1,526
1,645
1,779
1,906
2,046
2,19S
a.
H
a
о
с
s
s
H
о
ш к
«а а
со я
О tt
03
К
*a
о
3
s
a,
о
» 5
Я -:
S E
=1 Z
CM rj*
CO©
CON-
ooddd
CO-<ONino>
'©"©"©"ooo"
—-^——
CO —ЮСМ — —<CM — СТ>ШСМ
Г^НЗ^смсоо^оо — ю©
о со юг-noo ад oo ел en ел
©" о" ©" ©~ © о" © о ©" © ©"
СО О СО (МО) CD 00 — СО ©
(MOOCO'-f— Ю Ю СО СО N
— tN СО-^ Ю СО I- ОС^© ©
©C0CM00t*«OC©CM00tJ«O
О СО — CQ СО ^ •— © СО тг СМ
N00©©0 —СмСМСО^Ю
СО 0О ОО
СО Г*- ОО
©©©
©"©©"
^ооо —— t-©©oo
СОСМСО —ЮООСМ^С-
Ол^— CM Cs (N со со сг>т
© ©*©" ©" © сГо" ©* ©
00<МСССОСОЬ-0)Ю*<3«
юго©юо^п»соь»
О*©*© ©"©о ©"©о
О —©co<N©Ot^Oi
00СМ — ©OiCOlOCOCM
CNTpiOCOCOt^OOOi©
©"©"©*© ©©"©сг^"
COCSoOTfOCOCMOO'*
MO(NOOOiOMOOO
•"•CNCOco-^iocO©
s
ffl
о
° о
e .~.
<D
3
4
5 «
О t;
от©^ —смсмс^^ю©г^оо©© —смсот^юсог^оо©©© —смсо^ю
сОтГ^т^^^т!«^тгт^т^тР^ЮЮЮ10 10 101010ЮЮСОСОСОСОСОСОСО
SOO©©©©©©©©©CD©©©©©©©©©©OOOOCOO
= ©o©ooooo©©©oo©©©ooooo<=>o<~:©o©©
0©©©©©©0©0©©©© ©O p©© 0©©©©0©0©0©
©"©"о*©"© о"©* ©"©*©""©© ©©*©*© © ©"© ©*©~©~©"4©~© ©~©~ ©~©~©~
со"4 см4 r-Г csT od со" od ^ oT t^" cT irT сГ со т-Г ^ * of ос" со" ©"ю© со~—^см'оо"4^*—od
т^ЮЮУЭ©С^Г^00СО©©©тН^СЧСМСОС0^^ЮСССО^-^0000©©©
^т^,^^^,^,^ —— ^CMC4<>JCMCMC^CMC^(NCMCM(NCMC*C^CNCN(>icOC^
V I COCMCMCM fOTf lONO) CM Ю © CO 00 CO © Ю i-" 00 Ю CO — © N Ю Ю © Ю © iO
i со см ос ^© cocnoo^ r-.t- cq© cqcq©cqcq ©со<го©^сосол©л^юсм_—©
^^^^^^^^CMCM(NCMCNC4C4CNCMCMO<<>1CMC4C4COCOCOC^
©OOO^C^OOOO^CNOcXJ^^C^COOcOT^CMOOOCO^CNOOOCOTfCM
TfCCCOCOCOCOCNCMCNCMCN»-»'—• — — — ©©©©©©©©©©CCOOOOOO
© © © © v* © cr. © © ©_© © cT^©_©e© O^c^o^© o^a^oo^oqoo^oo^oo^co o<^oo^
о © © © ©"©"©"©*©©"© ©"©©"©"©"'©©"©""©'©"'©"*©'©*©"©"©"©"©"©"'
0000©©©©— ^^<>|(NC0C0Tt«rt<lOlO©©r--00©©r--iC,MCOTt*lO©t^00
_* т—• ~ ~ CMCMCMCMC>1^CMCNCMCMCMCMCMCMC4CMCMCOCOCOCO^COCOCOCO
©©©©©©©©©©©©©©©©©«о©©©©©©©©©©©©
©О©©©©©©©©O0©©©©©©©©©©©©©©©0©©
©©©©©© ©.p © © © © qqo^o оДо p© ©. © ©.© © © о о ©
©"© © © ©"©'©"©'©*©' о©" cf о © © ©©*©"©©" ©* © ©"© о © © ©"©
•я
О lO ^СО CM ©жГ^Ю ^C>l©.^©^^^©^C^^CM^©>---^CO^Tf^г-^О*©.C^CO^iO Г^
©*TfaTc>dodc^t^CM*r^CNN—*со~^со*©~ю©
r^rHW(NCOCO^^iO»OCOCONN0000000500-H^(N(NrtCO^t
2
О iO-hN.tJ« — ©©©©©©©©©— CM 00 т}*Юг-( h, CO© Ь-ч* — 00©^
©лг^о^солоолоэоолсо оо со оо^ со, оо.солослсо oq со оо со © ^г© © ^г^сс> со-** ©
©л© © —'-Гсм'см'сосо т^ тг'ю ю со"©"t< г^оо оо"©с^©"—^(^смсосо'^'ю'
©оо©^см©оо©^см©оо©"^см©оо©-*см©оо©^см©оосо^см
©©©©O)©CX)00000000i>'t^-l^t,^r^©COCOCO©iOiASUjlOLO-^,Tt*"^*^*
©©о>©©©©©©©©©©©©©©©©©©©©©©©©©©©
^ ©©"СГ©*©"©©"©" о" о*4© ©"©""©"о"©" о" ©*©"©* ©"©©*©©*©"©©"©"
Аммиак
В значительном Количестве NHs растворим также в спирте (см. табл. 14\
Б. Водный раствор аммиака
(Водный аммиак, едкий аммоний, гидрат окиси аммония, гидроокись аммо*
ниЯ, нашатырный спирт).
Liquor ammonii caustici Ammonium hydroxide, Wasseriges Ammoniak
Alkali volatile Ammonia solution, Salmiakgeist,
Ammonia water, Aetzammoniak
Caustic ammonia
П риготовление
1. Полученный одним из указанных способов газообразный ЫНз подводится
к приемнику, содержащему . дестиллированную воду, охлаждаемую льдом.
Вследствие чрезвычайно сильной растворимости аммиака следует
остерегаться переброски жидкости в реакционную колбу. Поэтом-у рекомендуется
вести поглощение в склянках Тищенко и разъединять прибор, как только
прекратится выделение газа.
При употреблении в качестве исходного продукта технического
сернокислого аммония получающийся водный аммиак всегда содержит пиридин.
2. По Тредвеллу1 для приготовления аммиака, не содержащего
углекислоты, в колбу емкостью 1 л, снабженную холодильником, помещают 500 мл
концентрированного ЫНЮН, добавляют около 10 г свежепогашенной извести,
закрывают холодильник трубкой с натронной известью и дают смеси стоять
в течение дня, часто помешивая.
Отдельно готовят 300—400 мл воды, не содержащей СОз, для чего через
вскипевшую дестиллированную воду пропускают до охлаждения воздух;
последний предварительно освобождается -от С02 пропусканием через крепкий
раствор КОН и через колонку с натронной известью.
Колбу с NH4OH ставят на водяную баню так, чтобы холодильник был
направлен косо вверх, и соединяют верхний конец холодильника с
приемником — колбой, содержащей 300—400 мл воды, свободной от СО2. При
нагревании на водяной бане аммиак переходит в приемник и здесь полностью
поглощается водой.
Свойства
Водный раствор аммиака представляет собой бесцветную жидкость легче
воды с характерным запахом аммиака и сильно щелочной реакцией. При
нагревании до кипения выделяет весь аммиак в виде газа. Раствор NHs ведет
себя во всех отношениях, как раствор гидрата окиси аммония (NHaOH), но
попытки получить это соединение в чистом виде, при обычной температуре,
"е увенчались успехом.
Однако исследование кривой ' плавкости системы НгО—ЫНз указало на
существование соединений 21МНз • Н20 или (NH^O — окись аммония с т. пл.
—78,9° и NHs • Н2О (или NH40H — гидрат окиси аммония) с т. пл. —79°,
кристаллизующихся в бесцветных кристаллах.
Испытание
По ОСТ 17403-38 препарат ч. д. а. должен удовлетворять следующим
требованиям: а) содержание NHs от 25 до 27%; б) наибольшие количества до-
"Устимых примесей указаны в табл. 16.
Препарат должен выдерживать приведенное в ОСТ испытание на вещества,
восстанавливающие КМп04.
1 Аналит. химия, Ц ц ч.), Ц5 (1931).
32
Аммиак. Аммоний
Таблица
Растворимость NH3 в различных спиртах
(Р a g 1 i a n i, E m о)
Этиловый спирт
t° с
23,00
21,32
21,61
21.70
22.10
23.19
24,60
23,10
20,40
22,75
22,70
22,98
23.16
давление 1
NH8 в мм I
рт. ст.
455,22
433,78
511.05
5?8,27
467,35
629,17
634,36
610,39
457,0Э
474,89
525,49
1 623,65
613,23
об. спирта
растворяет
об. NH3
63,5
68.5
75,4
81,5
70,6
76,6
84,4
87.3
70,9
68.7
75,2
85,3
91,4
Пропплозый спирт
t°Q |
21.74
19,60
19,80
19,90
20,90
21,36
20,62
20,43
20,62
20,96
21,20 <
давление
NH8 в мм
рт. ст.
'i 464,83 •
1 456,59
484,33
525,54
| 588,08
; 722,88
! 416,97
! 453,82
498,77
576.00
70о,00
1 об. спирта
растворяет
об. NH,
53,4
56,6
5 42
62,7
67,5
78,3
50,9
53,3
59,6
66,4
76,8
— -' ■ ■■ ■ ' .4fc>
Изобутиловый спирт
t°c
20,2
20,18
20,49
20.42
20,62
21,19
21,00
21,21
21,25
—
—
давление
NH3 в мм
рт. ст.
479,00
523,11
535,21
659,89
725,30
538,90
587,99
639,33
733,86
—
—
1 об. спирт*
раств^гяе*
об. ППГ
54,3
59,1
64,3
70,3
75,4
51,9.
55,7
60.fr
67,1
—
—
Растворимость NH3 в этиловом спирте
(D е 1 ё р i n е)
Таблица /|
t°c
0
10
20
30
Концентрация спирта
s lOOo/o
ч
п
23
£
*»
130,5
104,5
75,0
51,5
со
ч
>»
0,782
0,787
0,791
0,798
96^0 •
П
п
1 £
*
tv»
146,0
—
87,5
. ~-~"
а
ч
>»
0,783
^-
0,788
—~
90о/о
ч
со
К
£
с*
173,0
137,5
102,0
77,0
?
>»
0,800
0,794
0,795
0,799
SOD/,,
<?
т-1
СО
а?
z.
t\»
206,5
167,0
119,75
81,75
со
ч
! 70а/о
*t
со
£
£
>» | w
0,808
0,?00
0,821
0,826
_
—
137,5
100,3
со
ч
>»
—
0,829
—"
6Jo/0
«<
са
к"
Z
tM
ш
ч
>»
246,0 0,830
198,250,831
152,5 0,842
129,5
0,846
50о/о
««
со
£
z
С\»
304,5
227,0
са i
ч
>, 1
0,833
0,850
182,7.0.86^
152,0
0,883
Таблица 16,
Допустимые п
Примеси
1. Остаток после прокали-
2. Карбонаты (С08") . . .
4. Сера в виде сульфатов,
сульфитов и сульфидов
(в пересчете на S04") .
римеси в
Допустимое
содержание
в о/0
0,003
0,002
0*0001
0,0003
водном аммиаке (ч. д. а.)
Примеси
5. Металлы, осаждае-
6. Железо (Fe) . , . .
7. Кальций (Са) ....
8. Магний (Mg) ....
Допустимое
содержание
ВО/о
0,0001
0,00002
0,0001
0,0001
Аммоний азотистокисЛый и азотнокислый
33
АММОНИЙ АЗОТИСТОКИСЛЫЙ (НИТРИТ АММОНИЯ)
Attitnoninm
nitrosum
Ammonium nitrite
Ammonium nitrit ■
Salpetngsaures
Ammonium
N№N0* Мол. в. 64,043
приготовление
г]0 Нейма-нн-Кюлингу1 препарат получают взаимодействием амми*
азота и окиси азота.
аК большую, емкостью в несколько литров колбу (рис. 8) помещают в
стеклянную воронку так, чтобы поверхность колбы могла все время охлаждаться
пооточной водой. Колбу закрывают
хорошей пробкой, через которую проходят
^еТЫре широкие стеклянные Трубки. Одна
„з них соединена с газометром,
наполненным азотом, другая — с колбой, в
коброй получается газообразный аммиак
(осторожным нагреванием • крепкого
IsJrbOH). Третья трубка служит для ввода
окиси азота, которую можно получать в
отдельной колбе действием медных
стружек на азотную кислоту (удобнее всего
в целях точного регулирования получать
газы из равномерно действующих
аппаратов, например" из бомб). Все *три газо-
подводящие трубки доходят до нижней
части колбы, четвертая оканчивается
непосредственно под пробкой и служит для
удаления избытка газов в тягу при
помощи резиновой трубки. Для проведения
реакции окись азота, аммиак и азот
подводятся в реакционную колбу не слишком
быстрыми струями, причем тотчас же
образуются красно-желтые пары,
переходящие в плотный белый туман. Последний
при надлежащем охлаждении колбы проточной водой оседает на стенках
в виде твердого кристаллического слоя. •
Свойства
Мелкие призматические бледножелтые кристаллы, уд. в. 1,69. При хранении,
особенно в теплом месте, соль постепенно разлагается на азот и воду;
поэтому NH4NO2 нельзя хранить в запаянных трубках. Концентрированный вод-
ный раствор разлагается значительно быстрее сухой соли. Разбавленный
раствор разлагается при нагревании до 50°, выделяя азот и оставаясь нейтральным.
Сухая соль при нагревании на водяной бане несколько мгновений остается без
изменений, затем при 60—70° взрывает с большой силой. NH4NO2 хорошо
растворим в воде, метиловом и этиловом спиртах; трудно растворим в эфире.
АММОНИЙ АЗОТНОКИСЛЫЙ (НИТРАТ АММОНИЯ)
Ammonium nitricum Ammonium nitrate Arnmoniumnitrat
Nitrum flammans Salpetersaures
Ammonium
NH4NO3 Мол. *. 80,048
приготовление
1. 150 e технического азотнокислого аммония (аммиачной селитры)
растворяют при нагревании в возможно малом количестве воды (40—50 мл).
Рис/8. Прибор для получения а 2 от исто-
кислого аммония
1 tf p u m a n n-K u h I i n gt Aoleitung zum Experimentierea, 3 Aufl, 294 (19(H). J. Cbem. Soc
116(1911).
34
Аммоний
Раствор кипятят несколько минут с животным углем для обесцвечивания
фильтруют через складчатый фильтр в фарфоровую чашку. Фильтрат полц§_
щают в холодное место для кристаллизации.
На другой день выпавшие кристаллы отделяют на воронке Бюхнера и пр^
мывают очень малым количеством ледяной дестиллированной воды. В случ^
надобности продукт перекристаллизовывают, но обычно уже первая кристаИ
лизация дает химически чистый препарат.
Выход не велик вследствие значительной растворимости соли. Маточнв
раствор при выпа.ривании дает большое количество NHaNOs, несколько J
грязненного NH<CI и (NHOsSO*.
2. К 100 мл 25%-пого 1МНЮН, помещенного в тонкостенный стакан, охлв
ждаемый снегом, прибавляют очень маленькими порциями азотную кислоте
уд. в. 1,38. Разогревания раствора следует остерегаться во избежание излшцЬ
ней потери аммиака. .
HNOa прибавляют до слабо щелочной реакции (должен чувствоваться з^-
пах аммиака), посл-е чего раствор отфильтровывают от осадков гидрата окис$
железа и других примесей.
Бесцветный фильтрат подкисляют химически чистой HNOs и выпариваю^
на песчаной бане до появления на поверхности жидкости тонкой кристаллот
ческой пленки. Раствору дают постоять в прохладном месте и выделившиеся
кристаллы отделяют, как указано в способе 1.
Таблица Щ
Растворимость NH4N03 в воде и удельный вес
насыщенных растворов
*°с
0
20
30
40
50
60
70
о/о NH.NO,
54,2
63,9
70,8
, 73,3
78,0
80,2
83 J
Уд. в.
1,3115
1,3415
1,3519
t° с
80
90
100
120
140
160
|
о/о NH4NOa
85,9
88Д
91,0
94,7
97,4
94,1
Уд. в.
1,3910
1,4145
1,4260
1,4320
1,4360
Таблица 1&
Удельный вес водных р а ств оров NH4N03
•/о NH4NO,
1
2
4
6
8
10
и
/:
1,0023
1.0С64
1,0147
1,02о0
1,0313
1,0397
| 1,0482
о/о NH4NO,
14
1 16
18
20
22
24
26
<•
1,0547
1,0653
1,0740
1,0828
1,0916
1,1005
1,1095
о/о NH4NO,
28
30
35
40
45
50
65
*Г
1,1186
1,1277
1,1512
1.1754
1,2003
1.2258
1,2520
i
Аммония амальгама. Аммоний бромистый
35
С*
ойства
бесцветные прозрачные гигроскопические кристаллы (шестигранные призмы)
1,73. Т. пл. 165°. При осторожном нагревании разлагается при 200° на
Уд' ^'ц'закись азота (Гыстрое нагревание может повести к взрыву). Соль хо-
В° ю растворима в воде с сильным охлаждением (при смешивании 1 ч. соли
р°Гч воды температура смеси падает с +15° до —10°). Растворима в спирте
с. . 25 при 20°). Вследствие гигроскопичности препарата его следует сохранять
банках с притертыми пробками.
Испытание
По ОСТ 2601 препарат должен удовлетворять следующим требованиям:
а) содержание NH4NO3 не менее 98%; б) наибольшие количества допустимых
примесей указаны в табл. 19.
Таблица 19
Допустимые примеси в NH4N03
" 1
Примеси 1
1. Нерастворимые в воде вещества .
3. Фосфаты (Р04'") . . .
7. Мышьяк (As)
9. Железо (Fe)
10. Иитрнгы (NOa'). v
Допустимое содержание в п'о
в химически
чистом
0.002
0,005
0,0005
0,0005
0,005
0,001
0.00001
0,0003
0,0001
• 0,0005
В ЧИСТОМ ДЛЯ
анализа
0,005
0,020
0,001
0,002
0,02
0,005
0,00005
0.00J5
0,0002
0,С005
в частом
0,01
0,06
0,002
0,004
0,04
0,01
0,0001
0,001
0 001
0,001
Кроме того, препарат должен выдерживать приведенные в ОСТ
испытания на примеси органических оснований и на нейтральность водного раствора.
АММОНИЯ АМАЛЬГАМА
Ammonium amalgam Ammoniumamalgam
Приготовление
Приливают концентрированный раствор NH4CI к 2%-ной амальгаме
натрия, находящейся в стакане. Тотчас же образуется амальгама аммония в
виде рыхлой массы, выделяющей пузырьки газов (На и КНз).
Свойства
Тестообразная масса с металлическим блеском, быстро разлагающаяся на
ртуть, водород и аммиак.
АММОНИЙ БРОМИСТЫЙ (БРОМИД АММОНИЯ)
Ammonium bromatum Ammonium bromide Ammoniumbromid
Bromammonium
NH4B* Мол, в. 97,956
Ириготовление
Получение ЫНдВг можно вести следующим способом *: в колбу А (рис. 9)
наливают 520—530 г 20%-ного ЫШОН (уд. в. 0,923), в капельную воронку
* Кошт. Z. Dent* Arzaelb.
36
Аммоний
С —350 г брома. Склянка В, содержащая небольшое количество воды, слу^
для поглощения аммиака и увлеченных паров NH4Br.
Копбу Л ставят в холодную воду, так как начало реакции идет бурнс^
сопровождается сильным разогреванием. Осторожно открывая кран, з|-
вляют бром капать в МШОН. Падение каждой капли сопровождается иг
нием, причем верхняя часть колбы на
няется белыми парами бромистого ам^
ния, которые вместе с аммиаком уГ
каются выделяющимся азотом в логлр
тельную склянку. Струя газов, иду;
через В, регулируется скоростью прил:
ния брома.
Избытка брома безусловно необход!
избегать, так как в противном слу
образуется) чрезвычайно опасное веГ
ство — бромистый азот ЫВгз1. После п]
ливания всего брома и остывания жил
сти содержимое склянки В переливают
колбу и дают смеси стоять в течей
двух-трех дней. Если во время реакг
(или после нее) образуются кристалл;
их растворяют, прибавляя немного вод!
Надо следить, чтобы жидкость была пр
зрачна и содержала избыточный аммиа
в случае необходимости добавляют ai
миака до сильно щелочной реакции.
К концу операции образуется бесцвет*
Рис. 9. прибор для получения броми- ное Двойное соединение брома с бромф
стого аммония стым аммонием NH-rBrs, непрочное щ
сухом состоянии, но устойчивое в расл
творе. Пока это соединение находится в растворе, проба жидкости при доба4
влении НзЗО* желтеет. Когда после двух-трехдневного стояния раствора
проба при добавлении H2SO4 перестанет давать желтую окраску, реакция за-J
кончена. Жидкость фильтруют и выпаривают досуха на водяной бане (н&
кипятить на голом огне\). Остаток от выпаривания измельчают в студке.
Свойства
Бесцветные столбчатые кристаллы уд. в. 2,327, обладающие остро-соленый
вкусом. В чистом состоянии устойчив на свету и на воздухе. Легко растворил
в воде, очень трудно в спирте.
Растворимость NH4Er в воде
(A. Benratbu. S. Schiffers2)
Таблица 20
ее
-17
0
10
15
25
30
о/о NH.Br
32Д
37,3
39,8
41.1
43,9
44,8
t°C
40
50
55
75
100
Чр NH4Br
47,3
48,5
50,4
53,6
67,4
■ Z. anorg. СЬ., 240, 67 (1<Ш)..
Аммоний ванадиевокислый
37
Удельный вес водных растворов NH4Br
Таблица 21
NH4Br
1
2
4
6
d"
1,0043
1,0103
1,0215
1,0332
elo NH4Br
8
10
14
!. 18
d13
•
1,0451
1,0572
1.08 22
1,1081
»/, NH4Br
22
26
за
34
d»
4
1.П52
1,16*5
1,1933
1,2247
Испытание
По OCT. НКТП 7666/658 содержание Nb^Br в препарате должно быть не
менее 96.5%; наибольшие количества допустимых примесей указаны в табл. 22.
Допустимые примеси в NH4Br
Таблица 22
Допустимое содержание в °'о
Примеси
в химически
чнсгом
в чистом для
анализа
в чистом
1. Нерастворимые в воде вещества
2. Нелетучие вещества
3. Сульфаты (S04")
4. Хлориды (СГ)
5. Иодиды (J')
6. Железо (Fe)
7. Металлы, осаждаемые'H2S . . .
8. Кислотность (НВг; . . • . • . .
0,002
0,01
0U05
0,05
0,02
0,^002
0,0005
0,01
0,005
0.05
0,01
0,2
0,05
0.О'Ю5
0,0005
0,03
0,01
0,2
0,02
0,8
0,1
0,001
0,001
0,05
АММОНИЙ ВАНАДИЕВОКИСЛЫЙ (МЕТА)
(МЕТАВАНАДАТ АММОНИЯ)
Ammonium vanadicura Ammonium vanadate
NH4VO3 Мол. в. 116,99
Ammoniumvanadat
(meta)
Приготовление
1. Продажный препарат содержит обычно примесь солей щелочных
металлов, алюминия, хрома, меди и кремневой кислоты. Для очистки соль
растворяют при 70° в воде с небольшим количеством NbhOH и после фильтрования
осаждают NHjVOs добавлением NH4Q. Осаждение метаванадата аммония
практически полное, если содержание NH*C1 в растворе составляет около
10—12%.
После повторения вышеуказанных операций соль можно считать довольно
чистой. Для удаления' кремнекислоты препарат переводят прокаливанием в
V2O5, последний выпаривают со смесью HF, H2SO4 и HNOs, после чего
растворяют в аммиаке и вновь высаживают NH4VO3 добавлением NH4CI. Если
Желают получить препарат, совершенно свободный от примеси NH4CI,- то
растворяют NH4VO3 в горячей воде и добавляют равный объем спирта. Препарат
промывают я сушат при комнатной теашеоатуре.
38
Аммоний
2. По С и н г а л о вс к о м у1 препарат высокой чистоты может быть пол
чен следующим методом. Растворяют при 70° 100 г продажного NfbVd
в 1500 мл воды, добавляют 15 мл концентрированного NH<OH и остявлЖ
стоять 40 мин. Затем фильтруют и прибавляют к фильтрату 20 г NH;
Выпавшую соль отфильтровывают, промывают и высушивают при комнатн
температуре.
Свойства
Белое или слабо желтоватого цвета кристаллическое вещество, мало раство
римое в воде и спирте. Растворимость в воде при 18° составляет* 6.47 гд
(что соответствует 0,0553 N раствору), при 70° — 63 г/л. Водные раствор|
NH4VOa быстро желтеют вследствие тидролиза. При 30° соль начинав
терять аммиак и при 125° теряет его полностью. При прокаливании перех >
в пятиокись ванадия.
Испытание
Препарат ч.д.а. подвергается испытанию на содержание NH-iVO* (потер!
при прокаливании не более 22,3%), на нерастворимые в воде примеси, хл
риды (проба с AgfNO.i), сульфаты (проба с BaCL>), тяжелые металлы (проб
с (NH-ibS и H-.-S) и щелочи.'
По техническим условиям НКХП 124—40 примеси допускаются в коли|
чествах, приведенных в табл. 23; содержание ванадиевокислого аммония в npe-ii
парате ч. д. а. должно быть не менее 99%, а в препарате чистое — не менее 93%L
Препарат ч. д, а. должен выдерживать испытания на чувствительность к PCV'j
Таблица 23,
Допустимые примеси в NH4VG3
Прямесн
1. Нерастворимые в воде вещества
3. Сульфаты (SO/7)
5, Щелочные и щелочноземельные
металлы 0 • «
Лопустимг?р сод2рж?ние в "о
в чистом для
i анализа
■
0,02
0.02
002
0,005
0,3
в чистом
0.^5
0,05
0,05
0.05
0,5
АММОНИЙ ДВУХРОМОВОКИСЛЫЙ (БИХРОМАТ АММОНИЯ)
Ammonium Ammonium Ammoniumbichromat
bichromicum bichromate Dichromsaures
Ammonium
(NH4)3Cr207 Мол. в. 252,10
Приготовление
1. К раствору 100 г хромового ангидрида в 100 мл воды прибавляют
маленькими порциями, при энергичном помешивании стеклянной палочкой,
80 мл аммиака (уд. в. 0,91):
2СгО* + 2NH40H = (ШОаСгЮт + НЮ.
Реакция экзотермична, и против разогревания смеси необходимо
принимать меры, охлаждая стакан ледяной водой. Разогревания следует избегать,
1 Соли редких и цветных металлов, стр. 131.
* Б. /1. Золотавин, Диссертация, Свердловск, 1941*
Аммоний двухромовокислый "' 39
двум причинам: а) реакция в горячей среде идет настолько бурно, что
п иПивлние аммиака будет вызывать сильное разбрызгивание жидкости;
Гч аммиак, попадая в- горячую среду, сразу улетучивается, не успев вступить
J реакцию. Результатом этого будет значительный избыток СгОз в растворе.
После того как весь ЫНЮН прилит, полученную тёмнокрасную
мутноватую жидкость фильтруют через двойной бумажный фильтр и выпаривают в
небольшой фарфоровой чашечке до начала появления пленки кристаллов.
Последний момент уловить легко: по оставшейся совершенно чистой поверхности
раствора начинают плавать отдельные кристаллы, собираясь в группы. Тогда
необходимо прекратить выпаривание и поставить чашку для кристаллизации в
прохладное место (на ночь). На утро оранжево-красные кристаллы отсасывают,
промывают малым количеством холодной „воды, отжимают между листами
фильтровальной бумаги и сушат при 40—50°. Выход 100 г.
Для окончательной очистки продукт можно перёкристаллизовать.
Полученный препарат содержит 99,2% (NHOsfCrsO?. Необходимо заметить, что для
получения совершенно чистого продукта (без SO*") нужно иметь хромовый
ангидрид, свободный от серной кислоты. В случае же употребления обычного
продажного СгОз, содержащего до 20% H2SO4, его приходится предварительно
очищать.
2. По методу ИРЕА для получения бихромата аммония проводят
обменную реакцию между концентрированными растворами NasCr^O? и NH^Cl,
отделяя выпадающий NaCl.
Свойства
Оранжево-красные моиоклинические кристаллы уд. в. 2,15, хорошо
растворимые в воде. Растворимы в спирте. При нагревании до 190—200° разлагается
с появлением пламени, оставляя объемистый порошок СггОз.
Таблица 2Ф
Растворимость
г с
15
• 20
1С0 г воды
растворяют г
(NH4), Сг,От
30,75
35,39
(NH4)o Сг207 вводе
ГС
25
30
100 г воды
растворяю г г
(NH^C^O,
40,03
47,14
Испытание
/xtJ1?^0^1 17393"39 препарат ч.д.а. должен содержать не менее 99 6%'
©ШОзСгЮг. препарат ч. - не менее 99,0% (ШфСгЮ;. Наибольшие
количества допустимых примесей указаны в табл. 25.
До п устимые примеси в (NH4)2 Сг207
Таблица 25
Примеси
Допустимое содержание в о/0
в чистом для
анализа
1- Нерастворимые в воде вещества
2. Щелочные металла (в виде сульфатов)
3- Хдорилы (СГ) . . • • . . • .
4- Сульфаты. (SO/7)
5. Метпллы, осаждаемые аммиаком {А1, Fe и др.) .
*• Кальций (Са)
0,003
0,5
0,004
0,015
0,005
0,01
в чистом
0,005
1,0
0,01
0,05
0,01
0,02
40
гАммоний
АММОНИЙ ЙОДИСТЫЙ (ИОДИД АММОНИЯ) }
Ammonium jodatum Ammonium iodide Ammoniumjodid
NJW Мол. в. 144,96
П риготов ление
Смешивают возможно более концентрированные растворы 33 г KJ и 26,5 г
(NH4):>S04, прибавляют двойной объем спирта и оставляют на сутки. На дру-,
гой день отфильтровывают осадок K2SO4, а фильтрат выпаривают досуха на
водяной бане.
Свойства
Чрезвычайно гигроскопичные бесцветные (допускается слабое пожелтение)
кубические кристаллы уд. в. 2,50. Очень легко растворимы в воде (125:100
при —27,5°; 167 : 10Q при +15°) и спирте. Водный раствор на свету
разлагается, окрашиваясь выделяющимся иодом в желтый цвет.
Таблица 26
Удельный вес водных растворов NH4J
в/о NH4J
3,355
6,71
10.92
12,67
dlt
1.0202
1,0424
1,0714
1,0847
в/о NIV
13,42
18,58
30,50
**
1,0899
1,1265
1,2341
•/о NH.J
54,64
58,46
60,44
du
1,5109
1,5688
1,5948
И спытание
По ОСТ 10904-40 препарат должен представлять собою белый или слабо
желтоватый порошок. Содержание NH4J в препарате ч.д.а. — не менее 99%,
в препарате чистом — ке менее 96%; наибольшие количества допустимых при*
цесей указаны в табл. 27,
Таблица 27
Допустимые примеси в NH4J
1.
2.
3.
4.
5.
6.
Примеси
Нерастворимые в воде вещества . •
Остаток после прокаливания (в виде
Щелочность [(NHJ?C03]
сульфатов)
Допустимое содержание в °(о
в чистом дла
анализа
0,005
0,01
0,01
0,0001 '
J 0,003
0,0005
в чистом
0,01
0,02
0,02
Не
нормируется
0,01
0,002
"Аммоний йодистый и молибденовокислый
41
дММОНИЙ МОЛИБДЕНОВОКИСЛЫИ (МОЛИБДАТ АММОНИЯ)
Ammonium molybdate
Ammonium
molybdaenicum
(NH4)eMo7024 • 4H20 Мол. в. 1235,95
А. Твердая соль
Ammoniummolybdat
' Molybdansaures
An
immomum
Приготовление
1. Растворяют молибденовую кислоту в 20%-ном ЫНЮН и к раствору
добавляют спирта. Молибденовокислый аммоний осаждается в виде мелких
кристаллов.
2. Продажный молибденовокислый аммоний не является солью нормальной
молибденовой кислоты, но представляет смесь полимолибдатов. Для
получения хорошего препарата соль перекристаллизовывают из NH4OH, причем
следует помнить, что нормальная соль получается только из раствора,
имеющего сильно щелочную реакцию.
3. Довольно часто продажная соль содержит примесь PCV", которая
может сделать препарат непригодным для аналитических целей. Для очистки
от фосфатов можно применить следующий метод: к раствору (NH0eM07O24
прибавляют небольшое количество азотнокислого магния. Раствору дают
постоять при 40°, затем отфильтровывают образовавшийся осадок (МНфРОд*
• 12МоОз и фильтрат выпаривают досуха. Из остатка водой извлекается
чистый препарат, не содержащий PCV",
Свойства
Бесцветные или слегка зеленоватые призматические кристаллы уд. в. 3,31.
При лежании на воздухе значительно выветриваются, теряя часть аммиака.
При нагревании разлагаются около 150°, теряя NHs и НгО и оставляя
молибденовый ангидрид МоОз. Растворим в воде, сильных кислотах и щелочах.
Испытание
По ОСТ 5016 препарат должен представлять собой бесцветные или слегка
окрашенные « зеленоватый или желтоватый цвет кристаллы, растворимые
в воде; наибольшие количества допустимых примесей указаны в табл. 28.
Допустимые примеси в (NH4)6Mo7024• 4Н20
Таблица 28
Принеся
1. Нерастворимые в аммиачной воде вещества . .
2. Хлориды (СК)
5. Сульфаты (SO/')
Допустимое содержаниг в °/0
в чистой для
анализа
0,01
0,005
0,002
0,0005
0,05
в чистой
0,03
0,01
0,005
0,001
ол
Препарат должен выдерживать приведенное в ОСТ испытание ва
количество нелетучего остатка после гидрохлорирования.
42
'Аммоний
Б. Раствор молибдата аммония — «молибденовая жидкость»
Приготовление
1. Для приготовления молибденовой жидкости, пригодной для обычных
аналитических целей, можно применить следующий способ, рекомендованный
ОСТ: 150 г продажного препарата растворяют в \ л дестиллировапной водь*
И вливают этот раствор в 1 л НКОз уд. в. 1,2 (не наоборот!), причем выпа*
дающий вначале белый осадок молибденовой кислоты затем растворяется
с образованием' прозрачной жидкости. Раствор оставляют на 48 ч., после
чего сливают с могущего выпасть осадка.
2. Для получения реактива из природного сернистого молибдена MoS»
(молибденовый блеск) Фрезениус рекомендует следующий метод. Мелко
истолченный молибденовый блеск смешивают с равным по объему количество^
белого (кварцевого) песка, хорошо промытого соляной кислотой, и
прокаливают смесь в платиновой чашке в муфеле при частом помешивании Нагре*
вание следует вести до тех пор, пока масса не примет лимонно-желтой
окраски.
При охлаждении масса становится белой. Ее растворяют в
концентрированном NH4OH, отфильтровывают нерастворившийся остаток, фильтрат
выпаривают досуха и вновь . прокаливают до. пожелтения. Остаток в течение
50—60 час. нагревают на водяной бане с азотной кислотой для окисления
имеющихся в руде соединений фосфора в фосфорную кислоту. Раствор
выпаривают досуха для удаления HNCh, остаток растворяют в 4 ч.
концентрированного МНЮН, фильтруют и вливают раствор в 15 ч. HNCh уд. в. 1.20.
Полученную жидкость ставят в теплое место (около 40°); через несколько
дней сливают бесцветную жидкость с осадка. Полученный раствор
представляет собой готовую молибденовую жидкость.
3. С. Саито рекомендует готовить молибденовую жидкость растворением
25 г молибденовокислого аммония в 1 л ЪЫ HNO». Полученный раствор долгое
время не выделяет осадка. Свет не действует на реактив, поэтому хранение
его в темных склянках излишне.
В. Регенерация лабораторных молибденовых остатков
1. По предложению В. М. Мирошниченко фильтраты, оставшиеся от
определений фосфора (без промывных вод), упаривают в фарфоровой чашке
до небольшого объема. Жидкость сливают, а остаток, состоящий главным
образом из азотнокислых и молибденовокислых солей, обливают небольшим
количеством гсфячей дестиллированной воды и, слегка растирая пестиком,
переносят в эрленмеиеровскую колбу, в которой его обрабатывают чистым
25%-ным NFLOH, избегая избытка последнего. При этом молибдаты
растворяются; выпавший остаток Fe(OH).3 отфильтровывают через бумажный фильтр
и промывают 2—3 раза горячей водой.
К фильтрату для осаждения молибденовой кислоты прибавляют HNOs
уд. в. 1,4 до прекращения образования белого осадка. Дают отстояться,
фильтруют и, не промывая осадка, растворяют его на фильтре небольшим
количеством 25%-ного NH4OH, избегая избытка последнего. По окончании
растворения промывают фильтр 1—2 раза холодной водой, после чего полученный
раствор молибденовокислого аммония разбавляют водой до уд. в. 1,1 и
медленно влив*ают его в равный объем чистой HNCb уд. в. 1,2. Полученная
молибденовая жидкость вполне пригодна для аналитических целей.
2. По Руднику и Куку1 для регенерации молибденовых остатков
раствор, содержащий соединения молибдена, фильтруют через стеклянную
вату и осаждают фосфорнокислым аммонием. Когда собирается таким путем
значительный осадок, его промывают горячей водой, пока промывные воды
не начнут мутнеть (приблизительно 2 л воды на 100 г- осадка), а затем
сушат. Сухое вещество содержит около 85% МоОз.
ljf RudnicH, С о 9 k e, Ind. End. ChM 9, 109 (19l7)r
Аммоний надсёрнокислыЛ
43
510 г сухого осадка растворяют в смеси 620 мл ЫНЮН уд. в. 0,90 и
380 мл воды, • прибавляют раствор 85 г Mg(NCh)2 в 200 мл воды и дают
стоять 2—3 часа. Затем испытывают на фосфорную кнслрту, для чего не-
сКолько мл раствора прибавляют к двойному количеству НГМОз уд. в. 1,2.
Если при этом выпадает осадок, указывающий на присутствие PCV", то
к0 всей жидкости прибавляют еще немного Mg"(N03>2.
По окончании осаждения фильтруют, промывают осадок и доводят раствор
водой до 2 л. Отдельно разбавляют 2 л НЫОз уд., в. 1,4 до объема
в 4450 мл и прибавляют туда . при охлаждении постепенно в течение часа при
постоянном помешивании ранее полученный раствор молибденовокислого
аммония. После. стояния в течение нескольких дней к отфильтрованному
раствору добавляют еще 320 мл концентрированной HNOs. Полученная
молибденовая жидкость готова к употреблению.
3. П. Дике не1 приводит следующий метод, дающий хорошие результаты,
фильтраты от определений фосфора переливают в стеклянную бутыль,
нагревают жидкость до 70° (пропуская пар через стеклянную трубку) и^ переводят
весь молибден в осадок, добавляя, техническую НзР04 или раствор Na^HPOi
(избегая большого избытка осадителя). Через 12 час. проверяют полноту
осаждения, добавляя к прозрачной жидкости немного №*НРО*. Раствор
сливают, осадок переносят в фарфоровый или фаянсовый бак, взмучивают
сильной струей воды и дают отстояться. Слив жидкость, повторяют
промывание, пока проба осадка не будет почти полностью растворяться в
разбавленном NHiOH (удаление Fe* ' '). Промытый осадок высушивают в фарфоровой
чашке на водяной бане.
325 г осадка растворяют в 1100 мл раствора (полученного смешением
485 мл NHiOH уд. в. 0,91 и 615 мл воды). Не обращая внимания на муть,
осаждают PCV' добавлением 190 мл «магнезиальной жидкости»
(приготовленной растворением 300 г MgCb • 6НЮ и 300 г NHiCl в 1 л воды). Осадку,
дают отстояться, после чего фильтруют жидкость через двойной складчатый
фильтр. Вливая 535 мл полученного фильтрата в 2000 мл НЫОз уд. в. 1,2,
получают вполне готовую молибденовую жидкость; рекомендуется дать ей
постоять несколько дней перед употреблением.
АММОНИЙ НАДСЕРНОКИСЛЫИ (ПЕРСУЛЬФАТ АММОНИЯ)
Ammonium Ammonium persulfate Ammoniumpersulfa{
persulfuricum Ueberschwefelsaures
Ammonium
(NH4)2S2Oi Мол. в. 228,20
Приготовление
1. Большой тонкостенный стакан, служащий электролизером, помещают
в ^сосуд с проточной холодной водой. В стакан наливают
насыщенный раствор (NH«bS04, подкисленный 20 мл концентрированной H2SO4.
Электродами служат два куска платиновой проволоки толщиной 0,75 мм.
Начинают электролиз при напряжении 10 в и силе тока 5—8 а. Плотность
тока на аноде около 2 а/см2.
Через 2—3 часа начинают охлаждать стакан непрерывной струей холодной
воды, следя, чтобы температура электролита не превышала 15—20°
(разогревания следует избегать вследствие возможного разложения получающегося
персульфата). С другой стороны, охлаждение в начале электролиза может
повести к выпадению (NRibSO*. Через несколько часов образующийся на
аноде аммиак делает раствор щелочным, что вредит процессу, так как ток
расходуется на окисление аммиака. Поэтому в стакан добавляют 20 мл
концентрированной H2SO4.
1 Р. Р I с k e of, St. & Eieen, 58, Nk 49, 1403 (1938)^
#
'Аммоний
По мере хода электролиза образующийся (NH^SaOe, вследствие своей
плохой растворимости, выпадает на дно стакана. Через сутки прекращают
пропускание тока, кристаллический осадок отсасывают, промывают небольшим
количеством ледяной зоды и сушат при комнатной температуре. Препарат
содержит 4—20% (NH^SCU. Выход по току 60—70%. Отработанный электро*
Лит можно насытить (NH^SCm и снова подвергнуть электролизу.
2. По Эльбсу1 (NH4^S.>08 с 65%-ным выходом по току получается
следующим путем: катодом служит цилиндрически согнутая пластинка свинца,
окружающая стоящий в сосуде-пористый глиняный стакан емкостью в 80—
100 мл. Анодом служит спираль из платиновой проволоки с поверхностью
около 0,5 см2, помещенная внутрь глиняного стакана. Катодная жидкость
состоит из смеси равных объемов воды и чистой концентрированной H2SO4,
анодная же—из насыщенного раствора (ГМНфБС^ в смеси 8 об. ч. воды
И 1 об. ч. концентрированной H2SO4.
Стакан до краев погружен в чашку со льдом. Охлаждения льдом можно
избежать, если катод устроить из охлаждаемого быстрым током воды
свинцового змеевика, к которому припаяна медная проволока для пропускания
тока. Электролиз ведут при силе тока 2—3 а, для получения которого
необходимо напряжение приблизительно 8 в при внутреннем сопротивлении
источника тока около 1 ома.
Через 3—4 часа электролиз кончают, содержимое глиняного стакана
фильтруют через стеклянную вату, осадок отсасывают и кристаллическую массу
сушат на пористых глиняных или фарфоровых пластинках. Выход 20—40 г
при условии, что температура во внутреннем цилиндре постоянно держится
10—20° и исходные материалы чисты. Фильтрат насыщают твердым (NH^aSOi,
сливают раствор с избытка соли и снова начинают электролиз.
Серная кислота на катоде насыщается ионами NH4*, вследствие чего
раствор постепенно становится щелочным. При этом наблюдается резкое
понижение силы тока из-за уменьшения электропроводности раствора и
одновременного возникновения поляризационного тока; с этого момента
следует вводить свежую H2SO4.
С другой стороны, истощается и анодная жидкость вследствие ухода из
нее ионов NH4* и обогащения серной кислотой. Это перемещение не
уравновешивается выделением (NI-U^S'Os и периодическим насыщением жидкости
твердым (NH4)2SO*; поэтому после каждых двух-трех операций в анодную
жидкость при охлаждении льдом вливают из воронки с капиллярной трубкой
столько NH4OH, насыщенного сернокислым аммонием, чтобы жидкость имела
слабокислую реакцию. Выпадающий при этом (NhhJi^Oe вместе с жидкостью
помещается в анодный стакан.
Из сказанного ясно, что производить электролиз в течение 15—20 час.
было бы бесполезно, так как в этом случае быстрое вначале образование
(ЫШ^БгОв вскоре замедляется и через 3—5 час. использование тока
становится настолько неполным, что анодную жидкость приходится обновлять.
Меньший выход персульфата при первой операции по сравнению с
последующими объясняется тем, что, прежде чем наступит выпадение (NRibSaOe,
раствор должен им насытиться. Значительная часть (Nrh^Oe, образующегося
при первой операции, идет на насыщение анодной жидкости.
Полученный по этому способу препарат содержит от 3 до 5% примесей,
главную часть которых составляет (NH^SO*. В примесях имеется также
некоторое количество алюмоаммонийных квасцов (соединения алюминия
содержатся в глине цилиндра). Для очистки препарат перекристаллизовывают из
тепловатой воды. При этом вследствие значительной растворимости (МЬЬЬЗгОв
происходит заметная его потеря, так как часть, остающаяся в растворе,
из-за -разложения почти совершенно пропадает. При очистке больших
количеств перекристаллизовывают небольшую часть препарата,' чистые
кристаллы растворяют в воде и полученным раствором промывают на воронке
Бюхнера остальную часть кристаллов.
» Е 1 Ьз, J. prakt. Ch. [2J, 48, 186 (1893)%
Аммоний пятисернистый
45
Полученный таким образом препарат, хорошо высушенный в эксикаторе
^д СаСЬ, сохраняется месяцами без разложения. ,
3- Возможно получение персульфата аммония электролизом (без диафрагмы)
нейтрального раствора (NH4)2S04 с небольшой добавкой КгСгОа1.
двойства
Бесцветные, иногда слегка зеленоватые, пластинчатые кристаллы. При
нагревании сначала выделяется кислород и образуется пиросернокислый аммоний
(I\lH4)2S207, а затем уже улетучивается остаток. Водные растворы, особенно
лри нагревании, также легко выделяют кислород, оставляя кислый
сернокислый аммоний NH4HSO4.
Разложение водных растворов замедляется при разбавлении, а также при
добавлении сульфатов щелочных металлов или алюмокалиевых квасцов.
Вполне сухая соль сохраняется неограниченное время без разложения; влажная
при комнатной температуре постепенно разлагается, выделяя сильно озониро*
ванный кислород.
Соль значительно растворима в воде: при 0° в 100 ч. воды растворяется
58 ч.; при комнатной температуре — 65 ч. (ЫН-ОгБгОв (в 100 г раствора
содержится 36,7 г соли).
Испытание
По ОСТ НКТП 7846/750 препарат ч.д.а. должен содержать не менее 85%,
препарат чистый — не менее 75.% (NHOaSaOeT Наибольшие количества
допустимых примесей указаны . в табл. 29.
Таблица 29
Допустимые примеси в (NH4)2S208
Приме ев
Допустимое содержание
в %
чистом для
анализа
1. Нерастворимые в воде вещества
2. Остаток после прокаливания . .
3. Хлориды (С1') ,
4. Металлы, осаждаемые H2S . ♦ .
5. Железо (Fe)
6. Марганец (Мп) -
0,005
С.С5
0.002
0,001
0.001
0,0001
0,02
ОД
0,005
0,002
0,003
0.0003
АММОНИЙ ПЯТИСЕРНИСТЫЙ (ПЕНТАСУЛЬФИД АММОНИЯ)
Ammoniumpentasulfid
Ammonium
pentasulfuratum
Приготовление
Ammonium penta-»
• sulfide
(NHO2S5
Мол. в. 196,38
1. Для приготовления обычно применяемого в лабораторной практике
раствора «многосернистого аммония» (содержащего (NHi^Ss наряду с другими
Полисульфидами) насыщают в закрытой колбе 50 мл концентрированного
ЫНЮН сероводородом и прибавляют еще 50 мл ГМШОН такой же крепости.
*0 других методах лабораторного получения (NH4)tStOe см. В. П. Ильинский
Я. Та ра с о в, ЖПХ 8, № 8, 1345 (1930); П. И. Знмнео, Завод. Лаб. 1, М 12, 69 (1932).
46
Аммоний
В полученной жидкости растворяют при 35—40° возможно большее
количество измельченной черенковой серы. После отфильтрования избытка серь?
раствор готов к употреблению.
2. Для. получения твердой соли по Г. и В.^Бильц полученную по спо*
собу 1 жидкость смешивают в эрленмеиеровской колбочке с равным объемом
95%-ного спирта. При стоянии смеси на льду в течение ночи выпадает
значительное количество яркожелтых игольчатых кристаллов (NHOsSs. Их
отсасывают, промывают спиртом и эфиром и, смочив несколькими каплями NbUOH*
оставляют на несколько дней в вакуум-эксикаторе над негашеной известью;
Выход около 40 г.
3. По Бор нем а ну 100 мл 25%-ного NFhOH насыщают сероводородом,
после чего добавляют еще 100 мл NbhOH. К 200 мл раствора добавляют
75 мл воды и избыток тонко истертой серы, после чего нагревают на водяной
бане не выше 80°. Жидкость отфильтровывают от нерастворившейся серы
и фильтруют в плотно закрывающуюся склянку емкостью 250 мл, заполняя
ее раствором до самого верха. После 6—12-часового стояния в прохладном
месте (NFhbSs выкристаллизовывается. Кристаллы отсасывают, промывают
спиртом и эфиром и сушат в вакуум-эксикаторе над СаО, смоченном
несколькими каплями ЫНЮН. Маточный раствор при охлаждении льдом выделяет
еще некоторое количество кристаллов.
4. По Томасу и Райдингу в раствор 14,4 г NH4HS в 100 мл спирта
вносят 18 г тонко измельченной серы и нагревают 6 час. в колбе с обратным
холодильником при 80°, беспрерывно пропуская через жидкость ток водорода.
Полученный тёмнокрасный раствор при охлаждении выделяет кристаллы
(NHO2S5.
Свойства
Желтые до оранжево-желтого цвета прттзмочки или листочки. При
длительном стоянии, особенно во влажном воздухе, разлагается с образованием
серы (без тиосульфатов). Растворим в воде с выделением серы. При
действии кислот на раствор (NhhH'Ss (и других полисульфидов, вообще, (NH^aSa?)
выделяются сероводород и сера:
(NH4)*Se + 2НС1 = 2NH4C1 + H2S + (* - 1)S.
АММОНИИ РОДАНИСТЫЙ (РОДАНИД АММОНИЯ)
Ammonium rhpdanatum Ammonium rhodanate Ammoniumrhodanld
Ammonium Ammonium thiocyanate Rhodanammonium
sulfocyanatum
NH4CNS Мол. в. 76,12
Приготовление
ЬШульце и Клаус дают следующий метод: смешивают 600 ё
95%-ного спирта. 800 г Nb^OH (уд. в. 0,912) и 350—400 г сероуглерода и оста*
вляют жидкость стоять несколько дней, часто встряхивая ее. Реакция закан*
чивается в несколько дней (в теплом месте — уже через 24 часа). Полученный
Темнокоричневыи раствор выливают в фарфоровую чашку и выпаривают
При умеренном нагревании, причем улетучиваются аммиак, сернистый
аммоний и спирт и выпадает большое количество серы. Когда раствор
обесцвечивается, его фильтруют в горячем состоянии. По охлаждении
кристаллическую кашицу отсасывают И сырой продукт перекристаллизовывают Из горя*
чей воды, после чего он вполне чист. Выход —280 г сухой соли.
2. Значительно лучшие выходы получаются по варианту метода Шульце
И Клауса, разработанному В. А. Рекшинским (ИРЕА). В склянку,
емкостью 0,6 л, помещают ПО г CS2 и 330 мл 25%-ного ЫНЮН, закрывают
пробкой и перемешивают, часто открывая пробку, чтобы дать выход скопив-
Аммоний роданистый
47
ся парам. Затем склянку плотно закрывают и оставляют стоять на
&1*ЬоЛъко дней при . 20—22°, часто перебалтывая. Происходит постепенное
^творение ^52 в аммиачном слое, причем последний окрашивается 'в желтый
Ра<1алее — красно-желтый цвет.
И poc;ie того как сероуглеродный слой совершенно исчезнет (что# происхо-
а через 10—12 дней), жидкость переливают в фарфоровую чашку и
нагребают (огнеопасно!) на водяной бане в вытяжном шкафу
Б хорошей тягой (ом. ниже примечание). Водяная баня
дагрезается сначала до \'50°, затем температуру -медленно
повышают до 80°. При этом- улетучиваются NHs, CS2,
(NHO-'S и жидкость закипает. По мере ослабления кипения
температуру повышают до 100°. Нагревание заканчивают,
когда совершенно -прекратится выделение пузырьков из жид-
(кости. При этом последняя почти обесцвечивается.
Горячий .раствор фильтруют и фильтрат выпаривают на
водяной бане до тех пор, пока взятая на стекло проба
жидкости не будет выделять при охлаждении значительное
количество кристаллов. Горячий' раствор еще раз фильтруют
и охлаждают. Выпавшие кристаллы отсасывают и сушат
в теплом и сухом месте. Маточный раствор при
дальнейшем выпаривании дает кристаллы желтоватого цвета.
Такой препарат необходимо перекристаллизовать.
3. Можно значительно ускорить взаимодействие
сероуглерода с аммиаком, применяя постоянное перемешивание
смеси.
В широкогорлую банку емкостью около 1 _л помещают
110 г CSt» и 330 мл 25%-ного NH.*OH. Банка закрыта
-пробкой, через которую проходят обратный (шариковый)
холодильник и стеклянная мешалка, снабжённая ртутным или
водимым затвором для предотвращения потери CSs (рис. 10).
Банку ставят на водяную баню, нагреваемую не выше 30°, ^
и одновременно пускают в ход мешалку. Об окончании
реакции (через несколько часов) узнают по исчезновению ?"яс*П1%че™%"бр°0р.
нижнего (сероуглеродного) слоя. данистого аммония
Дальнейшую обработку ведут, как указано в способе 2.
Примечание. При работе по всем приведенным методам еле*
дует соблюдать чрезвычайную осторожность ввиду легкой
воспламеняемости сероуглерода. Вблизи -приборов не должны находиться
включаемые рубильники и выключатели или за'жженные горелки. Смесь паров
CS2 с воздухом сильно взрывает. Не следует забывать, что
сероуглерод способен воспламеняться даже от нагретых предметов.
Нагревание водяной бани должно производиться вдали от приборов. Кроме
того, ввиду ядовитости CS2 и выделения значительных количеств
NH«HS, H*S и NHs все операции обязательно следует проводить
в шкафу с хорошей тягой.
Свойства \
Бесцветные блестящие моноклиническйе Таблички уД. в. 1,81, р а сплывай^
Щиеся во влажном воздухе. Легко растворим в воде (122,1 : 100 при 0°;
162,2:100 при 20°) и спирте. При растворении в воде происходит значитель*
ное поглощение тепла: так, при смешении 133 ч. NH*CNS с 100 ч. воды
температура смеси Падает с 13,2° До —18°. Т. пл. 149°. При дальнейшем
нагревании разлагается. При нагревании до 70° частично переходит в ИЗО*
мер — тиомочевину:
/S —NH4 yNH,
48
Аммоний
Концентрированные водные растворы NH4CNS на свету постепенно окрц
шиваются в красный цвет; это объясняется, повидимому, образованием п$
сульфоцианата, НСзЫзЕз. Роданистый аммоний не ядовит.
Таблица 3|
Удельный вес водных растворов NH4CNS
•'о
NH4CNS
1
2
4
6
8
10
л18
1.0С09
1,0032
1.С078
1,0124
1.0 J 70
1,0216
г
%
NH4CNS
14
18
22
26
30
34
d1B
ai
1,0309
1,0456
1.04P5
1.0589 '
1.U645
1,0730
e/o
NH4CNS
38
42
46
50
54
53
d13
4
1,0818
1,0910
1,1007
1,1108
1,1214
1,1322
И спытание
По OCT 5179 содержание NFhCNS для препаратов х. ч. и ч. д. а. дол^
жно быть не ниже 98%; наибольшие количества допустимых примесей ука^
заны в табл. 31.
Таблица 31
Допустимые примеси в NH4CNS
Принеся
1. Нерастворимые в воде вещества . .
3. Хлориды (СГ) . . . . *
4. Сульфаты (SO/7)
Допустимое содержание в °/в
в химически
чистом
0,005
0,010
0,002
0,005
0,0001
в чистом
для анализа
0,010
0,020
0,005
0,020
0,0004
в чистом
0,020
0,050
0,020
0,040
0,0008
АММОНИЙ СЕРНИСТЫЙ (СУЛЬФИД АММОНИЯ)
Ammonium sulfuratum
Ammonium
sulfide
Ammoniumsulfid
Ammoniummonosulfid
Schwefelammonium
(NH4)aS Мол. в. 68,14
Приготовление
В твердом виде (NHOaS не получен.
Водный раствор получают насыщением отмеренного объема ЫНЮН серо*
водородом и добавлением к полученному раствору ЫШН? равного объема
NHiOH:
NH4HSI+ ЫНЮН = (NH4)2S + Н2О.
Для концентрированных растворов ЫНЮН этот Метод по Блоксаму
неприменим, так как концентрированный ЫНЮН при насыщении
сероводородом образует смесь ЫШН8, и (NHkJaS. Для получения раствора (NH4)aS
Аммоний сернистый и сернистый кислый.
49
концентрированного ЫНЮН последний насыщают сероводородом как. раз
по образования (NH^S.
Для определения конца нейтрализации время от времени берут пробу
жидкости и добавляют к ней раствор CuSOe. Выпадающую CuS
отфильтровывают и испытывают реакцию фильтрата: присутствие свободной кислоты
указывает на избыточное насыщение сероводородом, чем обусловливается
образование в препарате NFhHS:
2NH*HS + 2CuS04 =* 2CuS + (NH4)2304 + H2SO4.
Если фильтрат не имеет кислой реакции, образовавшийся осадок CuS прямо
на фильтре обрабатывается NH4OH. Синий цвет фильтрата указывает, что
нейтрализация еще не наступила и в растворе имелся NFLiOH, результатом
чего явилось выпадение Си(ОН)2, растворившегося в избытке NH4OH с
образованием комплексного соединения:
2CuS04 + (NH4)^S + 2NH4OH = CuS + Cu(OH). + 2(NH4)2S04;
Cu(OH)2 + 4NH4OH = [Cu(NH3)4](OH)2 + 4H20.
Отсутствие кислой реакции фильтрата и синего цвета раствора после
обработки осадка CuS аммиаком указывает на точную нейтрализацию:
CuS04 +: (NH4)2S = CuS;+ (NH4)2S04.
Свойства
Водный раствор (NH4)2S представляет собой бесцветную жидкость со
щелочной реакцией и запахом аммиака и сероводорода; даже при сильном
охлаждении из раствора не выделяются кристаллы твердой соли.
Соприкасаясь с воздухом, раствор . быстро желтеет, причем образуются
полисульфиды « тиосульфаты. Это объясняют окислением сероводорода с
образованием воды и серы, причем последняя, растворяясь в (NH4)2S, дает
желтый многосернистый аммоний (ЫН*-)*?^. Образование тиосульфатов, очевидно,
происходит вследствие взаимодействия серы с NH4OH:
6NH4OH + 4S = (Ш4)2£20з + 2(NH4)2S + ЗНЮ.
Испытание
ОСТ 5180 допускает для раствора (NH4)2'3, отвечающего квалификации
ч. д. аШу следующие максимальные -количества, примесей (в %):
Нелетучие вещества 0,005
Углекислые соли 0,005
Хлориды 0,002
Кроме tore, раствор должен быть прозрачен, бесцветен, свободен от осадка
и должен содержать не менее 8,0% сульфидной серы.
Препарат должен выдерживать испытания на предельно допустимые по ОСТ
количества мышьяка, сурьмы -и олова.
АММОНИЙ СЕРНИСТЫЙ КИСЛЫЙ (СУЛЬФГИДРАТ АММОНИЯ)
Ammonium hydrosulfuratum Ammonium Ammonium hydrosulfid
Ammonium sulfhydratum sulfhydrate Amrhoniumsulfhydrat
NH4HS Мол, в. 51,11
Приготовление
1. Раствор NH4HS готовят насыщением NH4OH сероводородом в
отсутствии воздуха;
NH4OH +■ H2S = NH4HS + Н30.
SO 'Аммоний
Если желают получить раствор, свободный от fNH^S, то ЫНЮН пщ
должен быть концентрированным (NH«OH уд. в. 0,880 следует разбавить*!
водой, по крайней мере в четыре раза). J
2. Для получения кристаллического NEUHS по Блоксаму1 поступаю^
следующим образом: объемистую склянку закрывают плотной пробкой с про-■,
ходящими через нее тремя стеклянными трубками: одна из них служит для
ввода NHS, другая для ввода H2S, третья для отвода избытка газов. NFUHS
получается при введении в склянку равных объемов газов или при неболь-,
шом избытке сероводорода. Во время реакции склянку охлаждают льдом..
3. По Томасу и Ридингу2 насыщают аммиаком абсолютный -спирт,
охлаждают раствор до 0° и пропускают сероводород, тщательно высушенный,
СаСЬ. При этом происходит выпадения NPhHS. Когда жидкость? насытится;
сероводородом, ее фильтруют, кристаллы отсасывают, отжимают между
листами фильтровальной бумаги, после чего запаивают в трубку. Препарат
содержит в качестве примеси немного (NH^S.
Свойства
Бесцветные иглы или листочки ромбической системы уд. в. 0,89.
Препарат, полученный по Блоксаму, представляет собой белую фарфоро-
видную массу. Т. пл. 120° (под давлением). Уже при обыкновенной
температуре препарат значительно испаряется и возгоняется. Легко растворим
в воде, причем бесцветный вначале раствор на воздухе скоро желтеет >
вследствие образования полисульфидов. Реакция раствора щелочная. NH^HS
растворим также в спирте. Спиртовый раствор при доступе воздуха
разлагается с образованием сернистого этила, аммиака и серы. На воздухе NH4HS
окисляется, поглощая кислород.
АММОНИЙ СЕРНОКИСЛЫЙ (СУЛЬФАТ АММОНИЯ)
Ammonium Ammonium sulfate Ammoniumsulfat
sulfuricum Schwefelsaures
Ammonium
(NH4)2S04. Мол. в. 132,14
Приготовление
1. Очистка технического сернокислого аммония сводится к следующему:
150 г (NH4)2S04 растворяют в 260 мл дестиллированной воды. В раствор,
нагретый до_ кипения, прибавляют 1—2 г надсернокислого аммония
[(Nlb^SaOe] и кипятят несколько минут для полного окисления примеси
закисного железа в окисное. Полноту окисления необходимо 'проверить, что
производится прибавлением к отфильтрованной пробе раствора K3Fe(CN)e.
Появляющееся синее окрашивание указывает на неполное окисление; в этом
случае обработку (N^^SaOe следует повторить.
После перевода всего железа в трехвалентное к раствору прибавляют
NH4OH до сильно щелочной реакции и фильтруют в фарфоровую чашку.
Бесцветный фильтрат упаривают до консистенции жидкой кристаллической
кашицы и дают раствору охладиться до комнатной температуры. Кристаллы
(NHU^SO* переносят на вороаку Бюхнера, хорошо отсасывают и промывают
несколько раз дестиллироаанйой водой.
Обычную примесь в полученном таким путем препарате составляют соли
кальция (CaS04), количество которых может достигать 0.2%. Освободиться
от них не удается, так как хотя CaS04 связан с (NH4>2S04 не в комплексной,
а в двойной соли CaS04»(NH4)2S04. Н2О, но в насыщенном растворе
сернокислого аммония Са' не осаждается никакими -реактивами. Если в сырье
1 В 1 о х a m, J. Chem. Soc, 67. 288 (1898).
J To m a i u. Ri d i ng, С. Ill, 817 (1923};
Аммоний сернокислый
SI
тсТВуют соли меди, то последняя перед окислением железа удаляется
П^ждением сероводородом и последующей фильтрацией.
оса 2 другой метод очистки технического продукта, предложенный Н. И в а-
в ы м заключается в том, что холодный насыщенный раствор (NH4)a304
Н ° одят ' прибавлением NH«OH до слабо щелочной реакции и примеси оса-
^оВ оТ сернистым аммонием. Избытка последнего следует избегать, так как в
^тивном случае выделяется коллоидная сера и раствор получается мутный.
?Р° К(КТИ дают отстояться и фильтруют. Выпаривание раствора ведут, как
казано в способе I.
Ук з Значительно лучшие результаты дает получение (NH^SCh из техниче-
ких NH4OH и H2SO4. 270 г 25%-ного NH*OH при хорошем охлаждении
с дой или снегом нейтрализуют 20%-ной H2SO4, которой расходуется около
850 мл. Раствор в конце нейтрализации должен иметь ясно щелочную
реакцию. Жидкость нагревают до кипения, отфильтровывают в фарфоровую
чашку и выпаривают, как указано в способе 1.
Обычно уже первая фракция кристаллов вполне удовлетворяет
требованиям, предъявляемым к химически чистому препарату.
Свойства
Белый мелкокристаллический порошок, уд.
в воде и нерастворимый в спирте. Т. пл. 513;
т. разложения 356°. • Вкус резко соленый.
й. 1,77, хорошо растворимый
по Каспару1 т. пл. 326—329°,
Таблица 32
Растворимость (NH4)2S04 в воде
/° с
0
10
20
30
(NH^SO,
41,4
42,2
43,0 "
43,8
Р С
40
50
60
70
(NH^SO,
44,8
45,8
46,8
47,9
Г С
80
90
100
(NH^SO.
48,8
49,8
50,8
Таблица 33
Удельный вес водных растворов (NH^SO^
%
(NH4tS04
1
2
4
6
е
d4
1,0041
1,0101
1,0220
1,0338
1,0456
(NHASO,
10
12
14
16
18
.20
1,0574
1,0691
1,0808
1,0924
1,1039
(NH^S04
20
22
24
26
28
.20
d4
1,1154
1,1269
1,1383
1,1496
1,1609
(NHASO,
30
35
40
df
1,1721
1,2000
1,2277
Испытание
^° 0CT 5120 содержание NH* в препарате должно быть не менее 25%;
иоольшие количества допустимых примесей указаны в табл. 34.
При определении реакции водного раствора препарат должен выдержи-
ьать приведенное в ОСТ испытание.
flspar, Вег. 63, 821, 1920.
&
Аммоний
Таблица $
Допустимые примеси в (NH4)2S04
Примеси
1. Нерастворимые в воде вещества
2. Нелетучие вещества
3. Фосфаты (РО4"0
4. Хлориды (GO
5. Роданиды (CNS0
6. Мышьяк (As)
7. Тяжелые металлы ,
8. Железо (Fe)
9. Нитраты (NCV)
Допустимое содержание в °/о
в химически
чистом
в чистом .
для анализа
О,02
0,005
0,0005
0,0005
0,005
0,0002
0,0005
0,0002
0,001
0,005
0,02
0,001
0,001
0,01
0,0005
0,001
0,0005
0,002
в чистом
0,01
0,06
0,002
0,002
0,02
0,001
0,002
0,001
0,005
АММОНИЙ УГЛЕКИСЛЫЙ (КАРБОНАТ АММОНИЯ)
Ammonium carbonicum
Ammonium Ammoniumcarbonat
carbonate Secundares kohlensaures
Ammonium
(NH4)*COs:• HuO Мол. в. 114,106
Обычный продажный углекислый аммоний представляет собой смесь
кислого углекислого аммония, NfcUHCOs, и карбаминовокислого аммония
& • NH4
С -=0
Приготовление
1. Чистый (NH^CO* получается по Див ер су растворением 1 ч.
продажной соли в 4 ч. концентрированного NH4OH путем двухдневного
настаивания при 20—25° в закрытом сосуде, после чего, открывая пробку,
выпускают скопившийся аммиак и охлаждают сосуд. Выпадающая сначала полу-»
прозрачная кристаллическая кашица вскоре переходит в светлые блестящие
игольчатые кристаллы, которые быстро отсасывают, высушивают и тотчас же
закупоривают. Из маточного раствора можно получить препарат, насыщая
раствор аммиаком и смешивая с половинным объемом спирта. При этом
образуется густая кристаллическая кашица, которую отсасывают.
2. Для получения приблизительно 2N раствора (NH^COs, применяемого
обычно в лабораториях, смешивают 78 г продажного углекислого аммония
с 500 мл 2N ЫНЮН (или раствором другой концентрации, взятым в
количестве, соответствующем 17 г NHs) и разбавляют водой до 1 л.
Свойства
Бесцветные кристаллы с сильным запахом аммиака, теряющие на
воздухе свой блеск и переходящие в NH4HC03.
При нагревании раствор (NH4)2CO.i разлагается- при 70° с выделением
NHs и С02. Сухая соль приблизительно при 58° распадается на Н20, СОз
и NHs.
Таблица 36
Растворимость продажного углекислого
аммония в воде
/• с
15
20
25
100 г воды
растворяют
z продажной
соли
25,0
29.1
33,8
./• с
30
40
50
100 г воды
растворяют
г продажно\
соли
37,4
45,7 .
50,4
г с
' 60
65
100 г воды
растворяют
г продажной
соли
62,5
66,7\
Таблица 36
Удельный вес водных растворов продажного
углекислого аммония
(Lunge)
°:'0
(NHJ2COa
1,66
3,18
4,60
6,04
7,49
8,93
10,85
11,86
W*5
1,005
1,010
1,015
1,020
1.025
1,030
1,035
1,040
13,36
14,83
16,16
17,70
19,18
20,70
22,25
45
1,045
1,050
1,055
1,060
1,065
1,070
1,075
°/о
(NH^CO,
23,78
25,31
26,82
28,33
29,93
31,77
^3,45
-
. .15
dl
1,080
1,085
1,090
1,095
1,100
1,155
1,110
(NH^CO,
35 08
36,88
38,71
40,34
42,20
44,29
44,90
w15
1,115
1,120
1,125
1,130
1,135
1,140
1,141
Испытание *''■■■ -V' ;
По OCT 3895- содержание NHa должно быть не менее 28%; наибольшие
количества допустимых прдмесей указаны в табл. 37.
Препарат должен выдерживать приведенное в. QGT испытание на
содержание органических примесей. t !
Таблица 37
Допустимые примеси в (NH4)aC03• Н20
Примеси
1. Нерастворимые в воде вещества . .
3. Хлориды (СЮ 1
*• <-ера в виде сульфатов, тиосульфа- 1
тов, сульфитов и сульфидов (в
пересчете на SO/)
5- Роданиды (CNS0
6. Тяжелые металлы
7.. Железо (Fe) .«....»,...
,*-. *
Допустимое содержание в о/0
в химически
чистом
0,002
0,002
0,0005
0,003
0,005
0,0003
0,0001
1
в чистом
для анализа
0,005
0,005
0,001
0,005
0,005
0,0005
0,0003
в чистом
0,01
0,01
0,003
0,01
001
0,001
0,0005
54
Аммоний
АММОНИЙ УГЛЕКИСЛЫЙ КИСЛЫЙ (БИКАРБОНАТ АММОНИЯ,
Д АММОНИЙ ДВУУГЛЕКИСЛЫЙ)
Ammonium
bicarbonicum
Приготовление
Ammonium
bicarbonate
NH4HCO3 Мол. в. 79,058
Ammoniumbiicarboni
Doppeltkohlensaurei
Ammonium
Наиболее удобным для лабораторного получения хорошего препарата
является приводимый здесь вариант метода Дэви-Сен-Клер - Д э в и л л я|
основанного на насыщении раствора (ЫН4)2СОа газообразными NHs и GOs|
Процесс ведется следующим образом: 200 г измельченного в порошок
продажного углекислого аммония (так называемого «пищевого») растворяю^
в 200 мл дестиллированной воды при нагревании до 50° и частом взбалтыва^
нии (предварительно во взятой пробе исходного продукта желательно
определить содержание SCU"). После растворения всего углекислого аммония
II ПГ 1
\
Ш
6уГ~
i 1
5? .-3
Uf -ZL
I
И II
||
Рис. 11. Прибор для получения двууглекислого аммония:
Л—колба для получения NHS нагреванием NH4OH (лучше иметь
баллон с NHS); В, С и Б—склянки Тищенко с раствором BaCl, (B)t
небольшим количествам дестиллированной воды (С), борной HtS04
(D); IS—трубка, подводящая аммиак; G—реакционная банка с
раствором (NH^COg.
к раствору для полного осаждения SCu" прибавляют вычисленное количество
Ва(ОН)г в небольшом избытке. Поддерживая температуру около 50°,
раствору дают стоять в течение часа, после чего его быстро фильтруют через
складчатый фильтр (еще лучше через воронку для горячего фильтрования)
в стеклянную банку емкостью 0,7—0,8. л. Затем раствор подвергают
насыщению аммиаком, пользуясь прибором, изображенным на рис. И.
В приведенном на рис. 11 приборе подводящая NHs трубка имеет
расширение на конце во избежание засорения выделяющимися кристаллами
(проще всего применить для этой цели воронку с длинной трубкой, вставляя
ее в пробку с нижней стороны).
Насыщение раствора аммиаком ведут до тех пор, пока не окончится
поглощение его, на что указывает частое прохождение пузырьков газа. Газ
следует пускать не слишком сильной струей для более полного его
поглощения. Расход аммиака приблизительно 13,5 л. По окончании насыщения
вместо колбы, выделяющей NHs, ставится аппарат Киппа для подачи ССЪ.
Промывать его следует в двух склянках Тищенко с дестиллированной водой.
Насыщение углекислотой рекомендуется вести при возможно низкой темпе^
ратуре (около 0°). Конец реакции определяется прекращением выпадения
Аммоний углекислый кислый и уксуснокислый 55
исталлов. Маточный раствор сливают, а кристаллы NH4HCO3 отсасывают
КР воронке Бюхнера и сушат при комнатной температуре в течение 2 час.
Выход около 100 г.
Свойства
Белые ромбические кристаллы уд. в. 1,57. На воздухе разлагается и
улетучивается, но остаток все время сохраняет прежний состав. При
хранении в открытой банке соль может улетучиваться полностью, причем скорость
улетучивания в значителъ-
У л QQttWPUT "" пляж- * Таблица 38
Растворимость NH4HC03 в воде
ной мере зависит от
влажности препарата.
Упругость диссоциации
совершенно сухой соли не
больше 1 мм при 18°, в
присутствии же воды доходит
до 135 мм. При 49°
реакция разложения идет
быстро; при более высокой
температуре характер
реакции изменяется и остаток,
после частичного
улетучивания соли, имеет иной
состав.
Испытание
По ОСТ 3894 содержание NHS должно быть не более 21,7%; наибольшие
количества допустимых примесей указаны в табл. 39.
Таблица 39
Допустимые примеси в NH4HC03
г с
0
10
20
NH4HCO,
10,9
13,7
17,5
г с
30
40
60
NH4HCOe
21,3
24,2
30
Примеси
1. Нерастворимые в воде вещества . .
3. Хлориды (СК)
4. Сера в виде сульфатов, тиосульфа-
тов, сульфитов и сульфидов (в
пересчете на SO/7)
5. Роданиды (CNS')
Допустимое содержание в °/о
в химически
чистом
0,С02
0,002
0,0005
0,003
0,005
0,0005
aoooi
в чистом
для анализа
0,005
0,005
0,001
0,005
0,005
0,0005
0,0003
в чистом
0,01
0,01
0,003
0,01
0,01
0,001
0,0005
Препарат должен выдержать приведенное в ОСТ испытание на примесь
органических оснований.
АММОНИЙ УКСУСНОКИСЛЫЙ (АЦЕТАТ АММОНИЯ;
Ammonium aceticum Ammonium acetate Ammoniumacetat
CH3COONH4 Мол. в. 77,084
Приготовление
Нейтрализуют уксусную кислоту аммиаком и дают раствору медленно
испаряться до кристаллизации. Выделившиеся кристаллы тщательно отсасывают
и помещают в банку с хорошо притертой пробкой.
56
Аммоний
4
Свойства
Мелкие бесцветные игольчатые кристаллы с запахом СНзСООН, легко,
растворимые в воде (1:1) и спирте. Чрезвычайно гигроскопичны. Т. пл,.
112,5—1.14°; при более высокой температуре разлагаются. Легко теряет
аммиак vi переходит в кислую соль.
Таблица 40
Удельный вес водных растворов CH3COONH4
CH8COONH4
d&
1,0008
1,0030
1,0074
1,0117
°/о
CHgCOONH,
8
10
12
14
d"
1,0159
1,0200
1,0240
1,0279
CHaCOONH4
16
18
20
22
rfie
1,0318
1,0356
1,0393
1,0429
CHsCOONH4
24
26
28
30
dl*
1,0465
1,0500
1,0535
1,0569
Испытание1
Наибольшее количество допустимых по техническим условиям НКХП
901—41 примесей указаны в табл. 4L
Таблица 41
Допустимые примеси в ОН3СООЫН4
Примеси
1. Нерастворимый в воде остаток . . ,
2. Нелетучий остаток
3. Хлориды (СК)
4. Сульфаты (SO/)
5. Тяжелые металлы (группы H2S) . .
6. Железо (Fe)
7. Кальций (Са) .
Допустимое содержание в °/о
в чистом
для анализа
0,01
0,01
0,001
0,01
0,0005
О.СООб
0,005
в чистом
0,05
0,05
0,005
0,015
0,001
0,001
0,01
АММОНИЙ ФОСФОРНОКИСЛЫЙ ОДНОЗАМЕЩЕННЫЙ
(ДИГИДРОФОСФАТ АММОНИЯ)
Ammonium
phosphoricum
monobasicum
Ammonium
monophosphate
Ammoniumphosphat
einbasisch
NH4H2PO4
Мол. в. 115,04
Приготовление
1. По Митчерлиху к концентрированному NH4OH прибавляют Н3Р04
до тех пор, пока проба раствора не перестанет давать осадок с ВаСЬ. (При
этом раствор приобретает сильно кислую реакцию на лакмус.) Раствор
осторожно выпаривают до начала кристаллизации; выпавшие при охлаждении
кристаллы отсасывают и отжимают между листами фильтррвальной бумаги.
Испытание хим. реактивов, 58,
Аммоний фосфорнокислый 57
Свойства
Крупные прозрачные квадратные кристаллы уд. в. 1,79 или белый
кристаллический порошок; растворим в воде.
Таблица 42
Растворимость
t° с
0
20
40
n«Apo4
18,2
27,3
Ь5,8
NH4H2P04
г с
60
80
100
в воде
°/о
NH4H.P04
45,0
54,5 .
62,7
Испытание
Наибольшие . количества примесей, допустимых по ОСТ 3407, указаны в
табл. 43.
Таблица 43
Допустимые примеси в NH4H2P04
Примеси
Допустимое содержание в °;0
в химически
чистом
в чистом
для анализа
1. Нерастворимые в воде примеси , .
2. Хлориды (СУ)
3. Нитраты (N03')
4. Сера, общее количество (SO/') . .
5. Щелочные металлы (в виде фосфатов)
6. Железо (Fe)
7. Тяжелые металлы
8. Мышьяк (As)
0,005
0,0005
0,0005
0,004
0,05
0,001
0,0005
0,0005
0,01
0,002
0,001
0,008
0,2
0,003
0,001
0,001
0,02
0,004
0,002
0,016
0,3
0,005
0,002
0,002
АММОНИИ ФОСФОРНОКИСЛЫЙ ТРЕХЗАМЕЩЕННЫЙ
(ФОСФАТ АММОНИЯ)
Ammonium
Phosphoricum tribasicum
(NH4)SP04. ЗНгО
При
Ammonium phosphate
tribasic
Мол. в. 203,15
Ammoniumphosphat
dreibasisch
готовление
1. Чистую фосфорную кислоту разбавляют водой до уд. в. 1,13 (22% ШРО4).
Раствор насыщают аммиаком до сильно щелочной реакции, нагревают до
кипения, фильтруют и оставляют на несколько дней в холодном месте для
кристаллизации. Выпавшие кристаллы отсасывают на воронке Бюхнера,
промывают небольшим количаств1?м холодной дестиллированной воды и сушат между
листами фильтровальной бумаги.
58
Аммоний
2. По Шотлендеру1 смешивают растворы 30 г (ЫШ)гНР042 в 300 j^
воды и 37,5 г NH4C1 в 300 мл воды, нагревают смесь до 60°, добавл^
к ней 300 мл NHUOH уд. в. 0,93 (~ 17%) и дают раствору медленно оЩ
ждаться в закрытой банке. Кристаллический осадок соли отделяют от раствоЩ
и промывают концентрированным ШЮН (уд. в. 0,90). Выдерживают <Щ
несколько часов на фильтровальной бумаге, после чего два раза отжимаЦ
между листами фильтровальной бумаги.
Свойства
Бесцветные пластинчатые кристаллы, хорошо растворимые в воде. В спи]
и эфире соль нерастворима. Водный раствор имеет нейтральную или оЩ
слабо щелочную реакцию. При лежании на воздухе соль частично тер
аммиак. А
АММОНИЙ ФТОРИСТЫЙ (ФТОРИД АММОНИЯ)
Ammonium fluoratum Ammonium fluoride
NH«F Мол. в. 37,04
Ammoniumfluorid
Fluorammonium
Приготовление
1. По Б е р цел и усу очень тонко измельченную смесь 1 ч. NH4CI п21/*}
HF возгоняют в платиновой чашке или в платиновом тигле с вогнутой внутр|
крышкой, на которую для охлаждения наливается вода. Эта вода попо
няется по мере испарения ее во время возгонки. При слабом нагревании
тигля NHUF возгоняется и оседает на крышке, температура которой всле
ствие охлаждения водой не может превышать 100°. Для очистки водный рас
твор полученной соли с небольшим избытком аммиака отфильтровывают черев
промытый чистой фтористоводородной кислотой фильтр, поддерживаемый пла*
тиновым кружком с отверстиями.
2. Получение чистого NHuF из плавиковой кислоты и ЫНЮН требуе^
малодоступного оборудования и вряд ли осуществимо в рядовой лаборатории
(см., напр., Реакт. и преп., 26).
3. Можно также по Миллсу8 возгонять NH4F при 350° из тщательна
изготовленной смеси тонко измельченных CaF* и (NH4)2S04.
Свойства
Мелкие призматические или пластинчатые кристаллы, устойчивые в сухом
и расплывающиеся во влажном воздухе. Легко растворим в воде (1:1)
и метиловом спирте, труднее в этиловом спирте. Возгоняется с
предварительным плавлением. При выпаривании водного раствора при 36—40° получается
зернисто-кристаллическая кислая соль NHaF • HF, расплывающаяся во
влажном воздухе.
Испытание
Наибольшие количества примесей, допустимых по ОСТ 7170/464, указаны
в табл. 44.
1 Неорг. преп. 69.
" Для получения (NH^HP04 к 25% ному раствору НаР04 добавляют понемногу NH4OH до
ясного вапаха аммиака и дают раствору спокойно испаряться в месте, защищенном от пыли.
Выпавшие кристаллы тщательно отсасывают, отжимают между листами фильтровальной бумаги
и помещают в банку с хорошей пробкой (во влажном воздухе соль разлагается, теряя NH, и
переходя в Nn4rJtP04). r ■
« DRP 93849; U 11 m a n n, EnzyklopSdie der technishen Cheraie, В. 1, 398Г, 1915.
Аммоний фтористый и хлористый
59
Таблица 44
Допустимые примеси в NH4F
Примеси
5. Тяжелые металлы, осаждаемые H2S
6. Железо (Fe)
7. Кислая соль (NH4F. HF)
Допустимое содержание в °,'о
в чистом
для анализа
0,02
0,003
0,005
0,1
0,0005
0,002
1,0
в чистом
0,1
0,01
0,01
0,5
0,001
0,005
5,0
АММОНИЙ ХЛОРИСТЫЙ (ХЛОРИД АММОНИЯ)
Ammonium chloratum
Ammonium chloride
NH4C1 Мол. в. 53,497
Ammoniumchlorid
Chlorammonium
П риготовление
1. Получение химически чистого NH4CI перекристаллизацией технического
продукта — нашатыря — удается в том случае,- если имеется нашатырь
выработки Донецкого Содового завода. При употреблений же нашатыря с других
заводов не удается перекристаллизацией освободиться от нелетучих примесей,
в результате чего препарат получается недостаточно чистым.
150 г технического хлористого аммония растворяют при нагревании
в 225 мл дестиллированной воды. В нагретый до кипения раствор прибавляют
25%-ного ЫНЮН (до щелочной реакции) и, после нескольких минут
кипячения, фильтруют от выпавшего Fe(OH)s через складчатый фильтр в
кристаллизатор или фарфоровую чашку1. Для кристаллизации раствор оставляют на
ночь в холодном месте. Выпавший мелкокристаллический NH4CI отсасывают
на воронке Бюхнера, промывают небольшим количеством ледяной
дестиллированной воды и сушат на стекле при комнатной температуре. Препарат
химически чист (ОСТ 2602). Маточный раствор,, выпаренный до половины
объема, дает обычно еще одну фракцию чистого препарата.
2. 150 мл 25%-ного NH4OH в хорошо охлаждаемом водой или снегом
стакане осторожно нейтрализуют 300 мл НС1 (уд. в. 1,12). Если после прили-
вания всей кислоты раствор, вследствие улетучивания части аммиака, будет
иметь кислую реакцию, его доводят до слабо щелочной реакции прибавлением
небольшого количества NH4OH. Раствор нагревают до кипения и фильтруют.
Фильтрат выпаривают до появления пленки и дальше поступают по способу 1.
Препарат, полученный из недостаточно чистых исходных материалов, можно
очистить однократной перекристаллизацией из горячей воды.
Свойства
Белый мелкокристаллический порошок2 уд. в. 1,53. При нагревании до
337,8° диссоциирует на NHs и НС1, которые улетучиваются и, соединяясь вне
сферы нагревания, образуют плотный белый «дым», состоящий из мельчай-
1 Рекомендуемое некоторыми авторами осаждение железа сернистым аммонием вредно
отражается на качестве получаемого препарата, так как (NHjiS легко окисляется в сернокислый
аммоний и освободиться от примеси последнего затруднительно.
п™,„„. ри кристаллизации из растворов, содержащих пектиновую кислоту. NH4C1 образует
прозрачные крисимды длиной до 650 мм и весом'до 265 г (F. Е г Г i с h, Z. anorg. Ch., 208, 31 (1931).
60
Аммоний
ших коисталликов (размером 6:2 • 10~3 см). Легко растворим в воде, трудно,
в спирте. Плавится около 400° при нагревании в запаянной трубке (В Метнер).;
Таблица 45*
t° с
— 16
0
+ 10
+ 20
NH*Cl
19,4
231)
25Л)
27,1
Растворимость NH4C1 в воде
г с
30
40
50
1 ' {
NH,C1
29,3
31,4
33,6
/• с
60
1 70
30
°/о
NH4Cl
35,6
Э7,6
.39,6
/• С
90
100
НО
°.'о
NH4C1 ,.
41,6
43,6
45,6
Удельный вес водных растворов NH4C1
Таблица 46
°;о
NH4C1
1
2
4
6
8
.20
1,0013
1,0045
1,0107
1,0168
1,0227
•'о
NH4C1
10
12
14
16
18
.20
dA
1,0286
1,0344
1,0401
1,0457
1,0512
1
NH4C1
20
22
24
26
dl
1,0567
1,0621
1.06M
1,0726
И спытание
По OCT 2602 содержание NH4CI в препарате должно быть не менее
98,5%; наибольшие количества допустимых примесей указаны в табл. 47.
Допустимые примеси в NH4C1
Таблица 47
Примеси
% ' «
1. Нерастворимые в воде вещества .
2. Нелетучие нещества
3. Фосфаты (PCV") . . . '
4. Сульфаты (SO/7) .........
5. Роданиды (CNS') .
6. Мышьяк (As)
7. Тяжелые металлы ...
8. Железо (Fe)
9.-Кальций и магний (Са и Mg) .' \
Допустимое содержание в °/0
в химически
чистом
в чистом
для анализа
0,002
0,005
0,0005
0,002
0,001
0,00001
0,0005
0,00025
0,002,
0,005
0,020
0.001
0ДО5
0,005
0,00005
0,0005
0,0005
0,003
0,01
0,06
0.002
0,02
0,01
0.0С01
0,001
0,001
0,010
Препарат должен выдерживать приведенное в ОСТ испытание на
содержание органических примесей и иметь нейтральную реакцию, проверяемую,
методом, указанным в том же ОСТ.
Аммоний хромовокислый и щавелевокислый 61
АММОНИЙ ХРОМОВОКИСЛЫЙ (ХРОМАТ АММОНИЯ)
дштопшт chromatum Ammonium chromate
Ammoniumchromat
Chromsaures Ammonium
(NH4)2Cr04
Мол. в. 152,09
Приготовление
1. Для получения чистого (NH4)2Cr04 Егер и Крюсс приводят
следующий метод: смешивают раствор хромового ангидрида (не содержащего '304")
с концентрированным ЫШОН и добавляют последнего столько, чтобы при
слабом нагревании образовавшийся (NH4>;>Cr04 растворился. Раствор,
помещенный в охладительную смесь, выделяет золотисто-желтые кристаллы,
которые отсасывают и сушат до исчезновения запаха аммиака. Препарат
получается совершенно чистым.
2. По методу ИРЕА в насыщенный раствор бихромата аммония добавляют
концентрированный NH4OH до сильного запаха аммиака, причем оранжевая
окраска раствора переходит в желтую. При охлаждении раствора снегом или
холодной водой выделяются кристаллы совершенно чистого (NH4)2Cr04,
которые отфильтровывают, промывают ЫШОН, отжимают между листами
фильтровальной бумаги и сушат при комнатной температуре.
Свойства
Красивые длинные золотисто-желтые иглы уд. в. 1Д При лежании на
воздухе или при сушке в теплом месте теряют часть аммиака, переходя
в (NH4)aCr207. Соль легко растворима в воде (при 0° в растворе содержится
28,7% (NH4)2Cr04). При быстром нагревании она разлагается с
воспламенением, оставляя СггОз.
И спытание
По ОСТ 17401-39 содержание (NH4)aCr04 для препарата ч. д. а. не менее
99,5%, для препарата чистого — не менее 99%; наибольшие количества
допустимых примесей указаны в табл. 48.
Таблица 48
Допустимые примеси в (NH4)2Cr04
Примеси
1. Нерастворимые в воде вещества'. .
2. Щелочные металлы, (в виде
сульфатов)
3. Хлориды (СГ)
4. Сульфаты (SO/)
5. Кальций (Са )
Допустимое содержание в °/0
в чистом
для анализа
0,003
0,?
0,001
0,015
0,004
в чистом
0,005
0,4
0,004
0,05
0,01
АММОНИЙ ЩАВЕЛЕВОКИСЛЫЙ (ОКСАЛАТ АММОНИЯ)
Ammonium oxalicum Ammonium oxalate Ammoniumoxalat
(NH4)2C204 • H2O Мел. в. 142,12
Приготовление
К горячему 30%-йому раствору химически чистой щавелевой кислоты
(Н2С2О4 • 2ШО) приливают при помешивании концентрированный NH4OH до
отчетливого запаха аммиака. Раствору дают охладиться, после чего выпавшие
62
Аммоний, Ангидрид
кристаллы отделяют, промывают небольшим количеством холодной воды tfjj;
сушат при комнатной температуре. у Щ
Свойства
Бесцветные кристаллы ромбической системы уд. в. 1,501. Слабо
растворимы в воде, мало растворимы в спирте.
Таблица 49
Растворимость (NH4bC204 в воде
г с
0
10
15
20
25
о|0
(NH4)tC04
2,48
3,46
3,66
4,24
5,01
/• С
30
40
50
60
70
(NH4)tC,04
6,18
6,95
8,11
9,59
11,83
Таблица 50
Удельный вес водных растворов
(NH4)A04
6/о
(NH,fcCt04
1
2
3
4
d4
1,0035
1,0085
1,0135
1,0186
Wo
5
6
7
И15
1,0238
1,0292
1,0346
Испытание
По ОСТ 2758 содержание (NH4)2C204 • ЬЬО не менее 99,8% (для
квалификаций х. ч. и чи д. а); наибольшие количества допустимых примесей указаны
в табл. 51.
Таблица 51
Допустимые примеси в (Nb^QaC^. Н20
Примеси
Допустимое содержание в о/о
химически
чистом щ
в чистом
для анализа
1. Нерастворимые в воде вещества .
2. Нелетучие вещества
3. Хлориды (СГ)
4. Сульфаты (SO/)
5. Тяжелые металлы
6. Железо (Fe) \ \ .
0,003
0 005
0,001
0,010
0,0002
0,0002
0,005
0,015
0,002
0,020
0/0010
0,0005
0,020
0,030
0,005
0,050
0,0015
0,0010
Ангидрид борный и вольфрамовый
63
АНГИДРИД БОРНЫЙ (ТРЕХОКИСЬ БОРА)
Aridum boricum * Boric acid anhydrous Borsaureanhydrid
anhydricum Bortrioxyd
y ШОз Мож в. 69,64
Приготовление
i Чистую борную кислоту прокаливают длительное время в платиновом
ле (порциями по -5 г) на сильном огне до полного удаления воды. Кислота
ТЙпчала вспучивается, затем сплавляется в стекловидную массу. Для
извлечена последней горя.чий ТИГель погружают нижней частью в холодную воду
?ожно также выливать расплавленную кислоту отдельными каплями в
фарфоровую ступку). Затвердевшую массу еще
орячей переносят в плотно закрывающуюся Таблица 32
банку без притертой пробки. Растворимость Во03
2. По Ко у л и Тэйло.ру при обез- в воде
воживании борной кислоты в вакууме при
200—225° в течение 400 час. получается
кристаллический ВгОз. /• С
Свойства
ГО
100
100 2 волы
растворяют г В,Оа
11
22
164,5
Бесцветная, прозрачная, стекловидная
масса, очень твердая (режет стекло) и хрупкая. г}{
Уд. в. 1,84 при 4°; кристаллический В2Оа
имеет уд. ib. 1,805 -при 25°. Весьма
гигроскопичен, притягивая из воздуха влагу, мутнеет.
Влажный воздух, (пропущенный через трубку
с ВгОз, содержит только 0,15 мг НгО на 1 л (Б уз и М а к - И н т а й р). Т. пл.
557° К Растворим в воде и спирте. На вкус слабо горьковат.
Испытание (по Мерку)
Кремневая кислота и щелочи. Помещают 5 г истертого в порошок
препарата в тарированную чашку, прибавляют 50 мл безводного метилового спирта,
насыщенного на холоду хлористым водородом, перемешивают платиновой
проволокой до полного растворения и выпаривают на сетке на маленьком
пламени. Остаток обрабатывают снова 25 мл СНзОН, насыщенного НС1,
выпаривают досуха и слабо прокаливают. Не должно оставаться весомого
'остатка.
АНГИДРИД ВОЛЬФРАМОВЫЙ
Acidum wolframicum Tungsten trioxyde Wolframtrioxyd
anhydricum Wolframsaureanhydrid
W08 Мол. в. 231,92
Приготовление
1. Прокаливают вольфрамовокислый аммоний в фарфоровом тигле сначала
на бунзеновской горелке и затем, окончательно, в горне с дутьем2.
2- По Калишеру3 кипящий раствор вольфрамовокислого натрия оса*
ждают концентрированной НС1 и небольшим количеством НЫОз при
одновременном пропускании водяного пара. Образовавшийся осадок для
освобождения от щелочей следует в течение нескольких недель декантировать
оолылим количеством воды. Влажная вольфрамовая кислота высушивается
а водяной бане, а затем сильно прокаливается, пока масса не станет темно*
желтой.
(Cole ТаТ^ратуРные Данные о свойствах Вя68 сильно противоречивы. Коул и Тейлор
МсС и 11 ?~к V .Ага* Chera- Soc- 57> 55-68' 1935) nP"B0AaT т- пл. ВаОа 294 ± 1?; Мак*Каллоч
" РгЗо гь Аш- Chem- Soc- 59» 2в5°» 1937) Аает т» пл' 46°-47<>0 и уд. в. 2,42.
I Р^Р- Ch. 725.
Диссертация, Берлин, 1902.
64
Ангидрид
Свойства
Нежный желтый порошок уд. в. 6,84, становящийся темнооранжевым- npj|
нагревании. Т. пл. 1476°. Чистый WOs, находящийся ^ под совершенно чисто!
водой, не зеленеет, даже при довольно долгом воздействии солнечных лучёи
незначительные следы пыли вызывают, однако, быстрое позеленение. РаствоЗ
рим в щелочах, воде и кислотах, за исключением плавиковой.
Acidum jodicum
anhydricum
АНГИДРИД ЙОДНОВАТЫЙ (ПЯТИОКИСЬ ИОДА)
Jodine pentaoxide
J0O5 Мол. в. 333,84
Jodpentoxyd
Jodsaureanhydrid
Приготовление
1. Нагревают ШОз при 180—^200° до прекращения выделения воды.
2. По методу Н. В. Вильямса1 чистую НЛОз помещают в стеклянный
кристаллизатор, слоем не толще 3 см, и нагревают постепенно в сушильной
шкафу до 120—140°. При этой температуре сушку ведут 2—2,5 часа при час*
том перемешивании. Конец обезвоживания определяется взвешиванием (цф
теря веса должна составлять 5,11%). Выход теоретический.
Свойства
Белый кристаллический порошок, иногда со слабым розоватым или ж|
товатым оттенком. Литературные данные об его уд. в. весьма разноречив
приводятся значения от 4,799 до 5,278 2. Вкус кислый, горький. При 250—35
J2O5 разлагается с выделением иода, придающего препарату коричневу!
окраску. Появление желтоватой или розоватой окраски наблюдается при ЩШ
гревднии выше 140°. J2O5 растворим в воде; образует различные гидрата
йодноватой кислоты.
Испытание
По ОСТ 5182 содержание J2Os для препарата ч. д. а. — не ниже 99,5$fc
для препарата чистого — 99,0%; наибольшие количества долустимых примесЙ
указаны в табл. 53; препарат должен выдерживать испытания на содержание
свободного иода (Js).
Таблица 53
Допустимые примеси в J206
Допустимые примеси
1. Нерастворимые в воде вещества . .
1 Допустимое содержание в р/0
в чистом
для анализа
0,006-
0,05
0,03 -
в чистом
0,016
0,10
0,05
1 Реакг. и преп. 154. Е. М о 1 е s Я Р. V 111 а п, Ann. Soc. Espan. Fls. Quim. 34, 787, 1936;
С. II, 541 (1937). i
a Так, например, по Бэкстеру иТиллею (G. С. Baxter и Tilley, Z. anorg.
Ch. 61, 9, 1908; J. Am. Chem. Soc. 31, 20, 1909) Jf «= 4,799; по Дитте rf° —5,037, #1 — 5,020;
по Е. М о л е с у и П. Виллану dX* «= 5,278 (Е. М о 1 е s, С. 1, 4725 (1937); Е. М о 1 е в, Р. V i I-
1а п, Ann» Soc. espan. Fis. Quim. 34, 787—801 (1936); С. И, 511 (1937), по Б э к с т'е р у в К ел ли
d в 4,980 (О. Р. -В а х te г, W. М. К е 11 е у, J. Am. Chem. Soc, 62, 1824 (1940); С. 1, 2 (1941).
Ангидрид йодноватый, молибденовый и мышьяковистый 65
АНГИДРИД МОЛИБДЕНОВЫЙ
Aridum rrtfrlibdaenicum Molybdic acid anhydrous Molybdansaure
дс Molybdenum trioxide Molybdantrioxyd
МоОз Мол. в. 143,95
рриготЯвление
По Мутману1 продажный молибдено'вокислый аммоний подвергают
перекристаллизации до тех пор, пока проба соли не будет полностью
улетучиваться при накаливании на платиновой пластинке. Затем подвергают соль
многочасовому нагреванию в платиновой чашке, причем улетучиваются
кристаллизационная вода и 1 молекула ГМНз. Остаток в течение 4—5 час.
прокаливают в тугоплавкой трубке в струе кислорода при тёмнокрасном калении.
Температуру не следует поднимать слишком высоко во избежание
улетучивания МоОз. Приготовленный таким путем препарат свободен от всяких
примесей.
Свойства
Белый, нежный, напоминающий тальк порошок уд. в. 4,5—4,7. В
сплавленном состоянии имеет желтоватый или сероватый оттенок. При сильном
накаливании возгоняется в виде блестящих, бесцветных, прозрачных табличек. При
нагревании окрашивается в лимонно-желтый цвет и при 795° плавится в
коричневую жидкость. При плавлении на воздухе частично улетучивается в
виде белых паров, которые сгущаются над расплавленной массой, образуя
кристаллический возгон. В воде почти нерастворим (0,002 : 100).
Испытание
См. кислота молибденовая (стр. 256).
АНГИДРИД МЫШЬЯКОВИСТЫЙ (ТРЕХОКИСЬ МЫШЬЯКА)
Acidum arsenicosum Arsenic trioxide Arsentrioxyd
Arsenicum album White arsenic Arseniksaureanhydrid
Arsenicous oxide Weisser Arsenik
As203 Мол. в. 197,82
Giftmehl
Приготовление
1. По Генигшмцдту и Цинтлю2 для совершенной очистки
продажного препарата от SbsOaf его переводят в мышьяковистокислый натрий,
растворением в 5—10% NaOH. Раствор последнего отфильтровывают от
нерастворимого сурьмянокислого натрия и насыщают фильтрат сернистым газом. При
этом' выпадает AssOs в виде сильно блестящих октаэдров.
2. Для получения препарата высокой чистоты Фульк и Гортон3
предлагают следующий метод; растворяют 150 г продажного AsaOa в 350 мл
концентрированной НС1 и перегоняют смесь в токе хлористого водорода в
охлаждаемый приемник, где дестиллат разделяется на два слоя — AsCl3 и НС1.
Нижний (мышьяковый) слой для удаления Sb взбалтывают с 2/3 об. кон-
Центрированной НС1. Эту операцию повторяют до тех пор, пока в промывной
Мел11016 нельзя будет обнаружить уже сурьму. Очищенный таким путем AsCh
пеое ННо выливают из капельной» воронки в сосуд с кипящей водой, сильно
Филк ещи>вая- П° охлаждении выпадает белый порошок AS2O3, который от-
Л^^^^Р^вьгвают и промывают до удаления кислоты.
S^lfthman, Ргйр. Ch. 716.
« р ° n i g s с h m i d t, Z in 11, Ann., 433, 214 (1923); Prap. Ch. 224.
0ц1к, Н о г t о n, J. Am. Chcm. Soc, 51, 2416 (1929).
66
Ангидрид
3. Настаивают продажный непрозрачный As2Cb с ГМНЮН при 70—80° л
сливают жидкость декантацией. По охлаждении из нее выпадают блестящ^
октаэдры чистого AS2O3 (Г и р ц е л ь, В е л е р).
4. Неочищенный, содержащий AssOs продукт лерекристаллизовывают из -Щ
рячей 20%-ной НС1 и затем — из горячей воды, пока маточный раствор &(
перестанет показывать кислую реакцию на диметилгельб. Полученный крч,
сталлический порошок высушивают в эксикаторе над H2SO4.
5. Получение (довольно сложным путем) чистого октаэдрического As»Q
описано М. Рэндаллом « Т. ДудомЧ
Свойства
А. Кристаллический AsiOz. Правильные октаэдры или тетраэдры уд. ^
3,86—4,0 или же моноклинические иглы и листочки. В воде растворим трудна
Образует очень слабую мышьяковистую киол©ту с конотантой диссоциац^
8,4 • 10-1*» (Ишикава и Аош)2.
Таблица 54 Таблица $
Растворимость As203
в воде
Удельный вес водных ■ |.
растворов AsjOa * ■ 3
г с
0
15
25
39,8
°/о As»03
1,19
1,63
2,00
2^5
г с
48,2
62
75
98 5
°/о AitOt
3,32
4,25
5,32
7,55
о/о AstO,
0,25
0,50
1,00
1,50
1,0010
1,0032
1,0073
1,0110
°/о А г. О,
2,00
2,50
3,00
,014$
,0221
Б. Аморфный AS2O2. Стекловидная прозрачная масса, довольно скоро перет
ходящая под влиянием влажного воздуха в кристаллическую фарфоровидную
модификацию. В совершенно сухом воздухе (высушенном Р2О5) остаетс|
стекловидным в течение нескольких лет; в воздухе же, насыщенном водяньшя
парами, помутнение начинается уже через 3—4 дня. Под водой стекловиднь$
AsaOaf становится матово-белым; спирт и эфир изменяют его только с поверф
ности; под СБг остается прозрачным, но становится хрупким. ,j-
В воде растворим несколько лучше кристаллического: в 100 г воды при
обыкновенной температуре растворяется 3,77 г АэгОз, после же 3-часового
кипячения растворяется 11,46 г. Уд. в. стекловидного АэгОз 3,6815, фарфоро-;
видного 3,6461 при 12,5°. При 200° улетучивается без предварительного пла?
вления. Стекловидный АэгОз (не фарфоровидный) несколько растворим в с№
пидаре, бензоле, бензине; лучше — в метиловом и амиловом спиртах, хлороз
форме и эфире. As20s — очень сильный яд. В качестве противоядия
употребляют смесь свежеосажденного Fe(OH)s и слабо прокаленной MgO.
Испытание9
Препарат ч. д. а. должен содержать не менее 99,8%; As^Cb', чистый-^
не менее 99,5%. Кроме того, препарат ч. д. а. должен выдерживать испытания
на нелетучие вещества (остаток от прокаливания под тягой 2 г A&Ojj не бо*
лее 1 мгу т. е. 0Д)5%), не растворимые в NH4OH примеси (0,02%), хлориде
(отсутствие мути от AgNOa в 0,4%-ном растворе As2Ojr, подкисленном HNOs);
сульфиды (проба с РЬ(СНзСОО)2 после растворения препарата в Ш NaOH)*
сурьму, железо и другие металлы (испытание сероводородной водой
уксуснокислого раствора остатка после трехкратного выпаривания АвгОз с НС1, ДРУ*
гая часть раствора испытывается на Fe'" при помощи NH*CNS).
1 М. R а п d а 11 и Th. D о о d, J. phys. Ch„ 43, 613 (1939); С. II, 813 (1939).
¥ * " " Pap. \ ' ' ~
«F.Ishikawa, I. A o k i, Scl
8 Испытание хим. реактивов, 212,
Inst. phys. chem. Res. 37, 955/957; С II, 177 (1940).
Ангидрид мышьяковый, оловянный и селенистый
67
Acidum arsenicicum
anhydricum
АНГИДРИД МЫШЬЯКОВЫЙ (ПЯТИОКИСЬ МЫШЬЯКА)
Arsenic pentoxi.de Arsenpentoxyd
Arsensaureanhydrid
А§205 Мол. в. 229.82
Приготовление
1. Нагревают мышьяковую кислоту до 250—300°; при этом она теряет
п0ду и переходит в ангидрид.
2. 100 г As2Os нагревают в колбе с 150 мл HNO* (уд; в. 1,38). Операцию
ведут под тягой вследствие выделения большого количества окислов азота.
Полученный раствор выпаривают и -остаток прокаливают при слаСокрзсном
калении.
Свойства
Белая аморфная масса уд. в. 4,09, расплывающаяся ю «Лажном возДухе.
При нагревании выше 400° распадается на АбзОз и 0§. Медленно растворяется
в воде, образуя мышьяковую кислоту.
АНГИДРИД ОЛОВЯННЫЙ
См. Кислота оловянная (стр. 258).
АНГИДРИД СЕЛЕНИСТЫЙ (ДВУОКИСЬ СЕЛЕНА)
Acidum selenosum Selenium dioxide
anhydricum
SeOa
Мол. в. 110,96
Selendioxyd
Seleniffsaureanhydrid
Приготовление
1. T о м с о н приводит некоторое видоизменение способа Берцелиуса.
Растворяют селен в концентрированной HNOs и выпаривают раствор досуха..
Для полного удаления HNOs остаток
нагревают до начинающейся возгонки
Se02. Для удаления примеси
селеновой кислоты (освободиться от которой
возгонкой не удается) растворяют
продукт в воде и к раствору добавляют
баритовой воды, пока отфильтрованная
проба не перестанет давать осадок
с Ва(ОН)2. Тогда отфильтровывают
выпавший BaSeO*, раствор выпаривают
Досуха и остаток возгоняют.
Полученный таким путем SeCb совершенно чист.
2. По Яннеку и Мейеру1'
окисляют селен кислородом в присутствии
N02, которая служит переносчиком
кислорода. В колбу А емкостью 30—40 мл
(рис. 12) помещают 20 г истертого
селена « нагревают ее на воздушной
бане. Через трубку пропускают
медленную струю высушенного кислорода при
температуре 200—215°. Кислород,
пройдя через шар С, захватывает пары
N02 и снова осушается в трубке D, на-
п°лненной Р2Об. Когда масса
(приблизительно через полчаса) примет
блестящи серый цвет, повышают температу-
РУ бани до 400°, вследствие чего SeOa
возгоняется в верхний пришлифованный Рис.12. Прибор для получения селенистого
J- J a n п е k, J. М е у е г, Вег. 46, 2876 (1913); Ргар. Ch. ИЗ.
63
Ангидрид
сосуд В. Когда взятый селен почти полностью израсходуется, прибору даюй
охладиться, заменяют нижний сосуд новым, удаляют пары N02 просасываниещ?
струи воздуха и возгоняют снова для удаления* окклюдированных окисло^
азота, что удается количественно. После охлаждения заменяют насадку Щ
пришлифованной пробкой для предохранения SeCb от влаги и пыли. Тако^
препарат сохраняет свой белый цвет неограниченное время. ■%
Свойства
Сублимируясь между 400—500°, образует белые блестящие,
четырехсторонние иглы, иногда в несколько сантиметров длиной. Уд. в. 3,9853 при 15,7°.
Т. пл. 340° (под давлением). Вкус кислый и жгучий. Пары ЗеОз имеют цвет
хлора и характерный запах. Легко растворим в воде, спирте и H2SO4.
Гигроскопичен, особенно в присутствии селеновой кислоты. Соприкасаясь с пылью,
окрашивается в красноватый цвет вследствие восстановления до
элементарного селена.
АНГИДРИД СЕРНИСТЫЙ (ДВУОКИСЬ СЕРЫ, СЕРНИСТЫЙ ГАЗ)
Acidum sulfurosum Sulfurous anhydride Schwefeldioxyd
anhydricum Sulfur dioxide
SO* Мол. в. 64,06
А. Газообразный сернистый ангидрид
Приготовление
1. Нагревают в круглодонной колбе 100 г медных стружек с 100 г
концентрированной H2SO4. Когда начинается бурное газовыделение, нагревание
можно прекратить. Если газовыделение закончится или вследствие сильного
разогревания пойдет слишком бурно, через капельную воронку приливают еще
100 г H2SO4. Выделяющийся газ промывают водой я высушивают CaCls.
2. Приливают по каплям крепкую H2SO4 в колбу, содержащую
концентрированный раствор сульфита натрия, Na2SC>3 (или твердый бисульфит натрия,
NaHSOs) и сушат выделяющийся газ СаСЬ.
3. По Нейману* из 3 ч. сернистокислого кальция (СаЭОз) и 1 ч. галса
(CaS04 • 72Н2О) с небольшим количеством воды формуют быстро
затвердевающие кубики. Последние помещают в средний шар аппарата Киппа, где
обрабатывают концентрированной H2SO4. Рекомендуется загружать в аппарат
такое количество кубиков, какое необходимо только для данной работы.
500 г кубиков дают постоянную струю газа в течение 30 час.
4. По Антону при нагревании смеси серы с концентрированной H2SO4
выделяется ровный ток газа (газовыделение начинается после расплавления
серы). Рекомендуется к смеси добавить несколько кусочков пемзы.
5. Осторожно нагревают густую смесь концентрированной H2SO4 с
порошком древесного угля. Выделяющийся газ содержит значительную примесь _CCfe.
6. По Штольба нагревают в колбе смесь 2—4 ч. безводногоCuSO*'(или
FeS04) с 1 ч. серы. При этом методе следует употреблять широкие
газоотводные трубки во избежание закупоривания их возгоняющейся серой.
Свойства
Бесцветный газ с характерным удушливым запахом, обладающий сильными
белящими свойствами. Вес 1 л 2,9266 г. Плотность 2,2638 (по воздуху). Легко
растворим в воде с образованием сернистой кислоты, Ш£Оз (см. Г). Не горюч
и не поддерживает горения. Растворим в спирте, эфире и СЗг; растворяется
в камфоре в отношении 300:1, образуя жидкость.
Растворимость и уд. в. растворов 502 см. ниже (Г).
» N е и ш а п, J. В. 2123 (1886); Z. ang. Ch. 48, 444 (1906); С. г. 85, 225 (1887); Ргар. Ch. 82.
Ангидрид сернистый
69
Б. Жидкий сернистый1 ангидрид
Приготовление
Полученный по одному из описанных выше способов газообразный SO2
осушают в двух промывных склянках с H2SO4 и пропускают в толстостенные
трубки, охлаждаемые смесью снега с солью (рис. 13); здесь SO2 сжижается.
Избыток SO2 поглощается в склянке с пемзой, прокипяченной с КОН, или
в промывной склянке с водой. Для получения более значительных количеств
жидкого SO2 пользуются аппаратом, изображенным на рис. 14.
Свойства
Бесцветная, очень легкоподвижная жидкость; уд. в. 1,4911 при —20°.
Т. кип. —ilO°. При «испарении вызывает сильное охлаждение (до —57°, а при
разрежении воздуха до —68е).
Хранение жидкой SOa в трубочках (рис. 13) безопасно только при
температуре ниже 0е. При комнатной же температуре давление достигает
нескольких атмосфер и не исключена возможность взрыва. ■—•—^^_ ■ >-
При употреблении трубки / (рис. 13) открывают снача- <^=Е55гтТ
ла только кран С и переливают немного жидкости' <2?=S^ г
Рис. 13. Трубки для хранения жидкого SO, Рис. 14. Аппарат для
сжижения сернистого ангидрида
в шарик D. Затем закрывают С, ставят прибор вертикально и, открыв
кран В, выпускают из шарика D воздух. Вновь закрывают В, открывают С
и наклоняют прибор так, чтобы шарик D весь наполнился жидким SOi>,
после чего С закрывают. Отмеренный таким образом объем выливают,
открывая кран В.
При пользовании трубкой // следует помнить, что при выливании
жидкости через один кран, другой также должен быть открыт. Иначе давлением
паров SO2 из сосуда будет выброшено все содержимое.
Хранить подобные трубки, наполненные жидким SOa, следует, по
указанным выше причинам, во льду.
В. Твердый сернистый ангидрид
Приготовление
1- Жидкий SO2 испаряют под колоколом воздушного насоса, причем часть
его застывает.
2- По Митчелю сосуд с жидким SO2 охлаждают смесью твердой СОг
с эфиром.
Свойства
Белые хлопья, тяжелее жидкого SO2. Т. пл. —- 75,24° ± 0,05е (А. Перлик *)•
1 A- Perl lc k, Zcitschrlft dee Verelns Deutsi'her Ingenieure (Berlin) 74, C. II, 2402 (1938).
70
Ангидрид
Г. Водный раствор сернистого ангидрида
(Сернистая кислота. Мол. в. 82.08)
Acidum sulfurosum Sulfurous acid Schweflige Satire \
Приготовление j
Насыщают газообразным SO2 дестиллированную воду, пока при встряхи^
вании заткнутого пальцем сосуда не будет ощущаться повышенного давления..-!
Свойства J
Бесцветная жидкость с сильным запахом SO2 и кислой реакцией, ведущая*
себя как раствор сернистой кислоты (константы диссоциации: К\ = 1,7» 10"V
К2 = 1. Ю-7). Постепенно окисляется кислородом воздуха, переходя в Н2ЗО4.
Таблица 56
Растворимость S02 в воде и спирте
*°с
0
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
В 1 об. насыщенного
раствора растворено
об.
в воде
68,861
67,003
65,169
63,360
61,576
59,816
58,080
56,369
54,683
53,621
51,383
49,770
48,182
46,618
45,079
43,564^
42,073*
40,608
39,165
37,749
36,206
газа
в спирте
(
191,16
183,32
175,36
168,48
161,49
154,78
148,86
142,22
136,36
130,79
125,50
120,50
115,78
ш;з4
107,19
103,32
99,74
96,44
1 об. воды
растворяет
об. газа
79,789
77,210
74,691
72,230
69,780
67,485
65,200
62,973
60,805
58,697
56,647
54,655 ■
52,723 i
50,849
49,033
47,276
45,578
43,939
42,360
40,838
39,384
t°Q
21
22
23
24
1 25
26
27
1 28
29
1 30
31
32
33
34
35
36
37
1 38
39
40
В 1 об. насыщенного
раствора растворено
об. газа
в воде
34,986
33,910
32,847
31,800
30,766
29,748
28,744
27,754
26,788
25,819
24,833
23,242
23,025
22,122
21,234
20,361
19,502
18,638
17,827
17,013
в спирте
93,42
90,69
88,24
86,08
84,20
1 об. воды
растворяет
об. газа
37,97
36,59
35,30
33.94'
32,76
Таблица 57
Удельный вес водных растворов S02 (Scott)
d15
15
1,0028
1,0056
1,0085
1,0113
1,0141
°,'о SOa
0,5
1,0
1,5
2,0
1 2'5
1 '«
1,0168
1,0194
1,0221
1,0248
| 1,0275
°/о SO|
3,0 1
3,5
4,0
4,5
5,0
dn
13
1,0302
1,0328
1,0353
1,0377
1,0401
о/о SO,
5,5
6,0
6,5
7,0
7,5
1
1,0426
1 1,0450
1,0474
1,0497
1,0520'
*
«/о SC,
8,0
8,5
9,0
9,5
10,0
Ангидрид серный и сурьмянистый
71
Испытание*
раствор SO2, отвечающий квалификации ч. д. д., должен содержать не
менее 6% ЗОг и выдерживать испытание: а) на нелетучие вещества (1 мг
остатка из 20 мл раствора после выпаривания на водяной бане и слабого
прокаливания); б) хлориды (10 мл раствора кипятят до удаления SO2,
разбавляют до 50 мл и приливают 1 мл 0,1 N AgNOs и 1 мл HNO* уд. в. 1,15,
причем через 5 мин. не должно быть опалесценции); в) железо (1 мл раствора
разбавляют до 20 мл, приливают НЫОз, кипятят до удаления SO2 и
разбавляют водой до 100 мл\ от прибавления 2 мл AN NH4CNS не должно
появляться красноватого окрашивания); г) тяжелые металлы (1 мл раствора
разбавляют до 10 мл, кипятят до удаления SO2, разбавляют до 50 мл и
прибивают 1 мл СНз€ООН и 10 мл сероводородной воды — не должно быть
буроватого окрашивания) и д) мышьяк (выпаривают смесь 10 мл раствора и
1 мл 60% H2SO4 до объема 2 мл, приливают 23 мл воды и 25 мл Н2ЗО4
1:4). Проба на As по Хе фт и-Гутцейту должна быть отрицательной
в течение 2 час.
АНГИДРИД СЕРНЫЙ (ТРЕХОКИСЬ СЕРЫ)
Acidum sulfuricum Sulfuric anhydride^ Schwefeltrioxyd
anhydricuro Sulfur trioxide * Schwefelsaureanhydrid
SOs Мол. в. 80,06
Приготовление
Получение SOa в лабораторных условиях является задачей довольно
сложной и кропотливой; поэтому обычно приходится пользоваться готовым
препаратом.
Можно привести следующий метод: в реторте, к горлу которой плотно
присоединен охлаждаемый льдом приемник, нагревают дымящуюся серную
кислоту — олеум (SOs1 переходит только вначале). Для очистки полученного
препарата его подвергают многократной перегонке2.
Свойства
Серный ангидрид диморфен, т. е. известен в двух формах: а-форма —
прозрачные призматические кристаллы, плавящиеся при + 16,8° в
легкоподвижную жидкость, кипящую при 44,6°. Уд. в 1,94 при 16°. Ниже 27° при
стоянии эта модификация переходит в (3-форму. /3-форма (устойчивая) —
масса, напоминающая асбест, при нагревании испаряющаяся без
предварительного плавления. Эта модификация вероятно имеет формулу (50з)г.
С водой БОз соединяется чрезвычайно бурно, часто с шумом,
напоминающим взрыв (всегда следует прибавлять £Оз к воде, а не наоборот). В
совершенно сухом состоянии БОз не имеет кислой реакции. SOs чрезвычайно
гигроскопичен. При работе с ним, вследствие его сильной едкости, следует
соблюдать большую осторожность (предохранительные очки, перчатки),
АНГИДРИД СУРЬМЯНИСТЫЙ (ТРЕХОКИСЬ СУРЬМЫ)
Stibium oxydatum Antimonousoxide AntimQntrioxyd
Flores antimonii Antimony oxide Antimonig-saureanhydrid
Antimonious acid
БЬЮ* Мол. в. 291,52
Приготовление
1. Хорошо промытый альгаротов порошок (хлорокись сурьмы SbOCl)
обрабатывают разбавленным горячим раствором ЫагСОз. При этом получается
°чень чистый SD2O3.
Испытание хим. реактитов, 361.
Приг. неорг. преп. 235.
72
Ангидрид
Можно также осаждать горячий раствор SbCl3 небольшим избытков
1Ма2СОз. Смесь кипятят до удаления СОг, фильтруют и осадок многокоатЛ
кипятят с водой до тех пор, пока реакция с AgrNOs в отфильтоованнЯ
пробе жидкости не покажет отсутствия ионов СГ. Отфильтрованный осадв
высушивают в сушильном шкафу при 100°. Выход 85—90% теоретического»
2. В очень чистом виде препарат получается! осаждением кипящегЯ
раствора 1 ч. рвотного камня, K\(SbO)C4H40o, в 10 ч. воды раствором ЫНЮШ
смесь кипятят некоторое время, осадок декантируют, фильтруют и промываюИ
3. По Г юнцу ал ьгаротов порошок кипятят продолжительное время с 'раз|
бавленным NH4OH, декантируют и повторяют эту операцию, пока не уда-1
лится весь СГ. Тогда промывают дестиллированной водой до исчезновение
щелочной реакции. Полученный таким путем препарат свободен от щелочей.
4. Нагревают в реторте альгаротов порошок; при этом перегоняется SbCli
и остается SD2O3 в виде сплавленной кристаллической массы.
5. П р е й с рекомендует постепенно вносить в раскаленный докрасна
тигель смесь 37 ч. сурьмы, 20 ч. KNOa и 17 ч. KHSCh и плавить до прекра|
щения выделения газов. По охлаждении массу выщелачивают водой и'
в остатке получают £ЬгОз.
Свойства
Сурьмянистый ангидрид диморфен: обычно получается в ромбических криЪ
сталлах, возгонкой в атмосфере инертного газа при тёмнокрасном
.калении получается в правильных кубических кристаллах. Полученный
осаждением БЬгОз представляет собой тонкий белый порошок, уд. в. 5,67. При
нагревании желтеет, при остывании вновь становится белым. При 656°
плавится в желтоватую или серую жидкость, которая при охлаждении
затвердевает в белую, асбестоподобную массу с шелковистым блеском. При более
высокой температуре (без доступа воздуха) возгоняется, не изменяя своего
состава. Очень мало растворим в воде, спирте и разбавленных кислотах.
Легко растворим в концентрированной НС1, растворах щавелевой и винной
кислот, а также в дымящихся HNOs и H2SO4.
Испытание2
Препарат ч. д. а. должен выдерживать испытания на мышьяк, тяжелые!
металлы и хлориды.
1. Мышьяк. Раствор 1 г препарата в 3 мл НС1 (уд. в. 1,19) после доба-!
вления Ъмл SnCh не должен темнеть в течение 1 часа.
2. Тяжелые металлы. Раствор 1 г препарата в 30 мл NaOH уд. в. 1,3
разбавляют водой до 50 мл и пропускают НгБ, причем не должно выцадать
ни белого, ни коричневато-черного осадка.
3. Хлор. 1 г препарата растворяют в 30 мл NaOH уд. в. 1,3, не содержа-!
щего хлоридов. Добавляют 70 мл HNOa, уд. в. 1,15, фильтруют и к филь-!
трату приливают 1 мл 0,1 N AgNOs. Допускается появление слабой мути, hoi
не осадка.
АНГИДРИД СУРЬМЯНЫЙ (ПЯТИОКИСЬ СУРЬМЫ)
Acidum stibicum Antimonic acid Antimonpentoxyd
Antimony pentaoxide Antimonsaureanhydrid
ShsOs Мол. в. 323,52
Приготовление
1. Осаждают раствор сурьмянокислого натрия азотной кислотой,
тщательно промывают осадок и сушат при 275° до постоянного веса.
2. Выливают* пятихлористую сурьму, SbCls в 20—25-кратное количество
холодной воды и дают стоять несколько часов. Образовавшийся осадок от-
1 Неорг. преп., 186.
* Испытание хим. рсактивог^ 366.
Ангидрид сурьмяный, теллуристый и теллуровый % 73
фильтровывают, быстро промывают холодной водой и сушат при 275° до
достоянного веса.
3. По П. С. Ко робко в у* порошок металлической Sb обрабатывают при
нагревании концентрированной HNOa до тех пор, пока выделение окислов
азота не станет незначительным. Полученную жидкую кашицу выпаривают
досуха и остаток осторожно нагревают (не прокаливать!) до исчезновения
запаха окислов азота.
Свойства
Бледножелтый неплавкий порошок уд. в. 6,6. На лакмус показывает
кислую реакцию. Мало растворим в воде и растворе винной кислоты,
нерастворим в спирте. Раствор SD2O5 в воде является до известной степени
коллоидным, по данным С. Гликселли и А. Пжищипковского2
раствор, насыщенный ЕЪгСЬ, содержит 1,86 г/л SD2O5. По указанию авторов,
после фильтрования через мембранный фильтр в растворе осталось 1,14 г/л
SbsOs. Растворим в НС1 и КОН. При прокаливании теряет кислород,
переходя при 300° в четырехокись сурьмы.
АНГИДРИД ТЕЛЛУРИСТЫЙ (ДВУОКИСЬ ТЕЛЛУРА)
Acidum tellurosura Tellurium dioxide Tellurdioxyd
anhydricum
T«Oa Мол. в. 159,61
Приготовление
1. Растворяют теллур в разбавленной HNO3, выпаривают и остаток слабо
прокаливают.
2. Сильно прокаливают теллуровую кислоту, НгТе04.
3. Растворяют теллур в HNOs (уд. в. 1,25) и к концу операции
прибавляют немного спирта. Через несколько часов выделяется ТеОг в
кристаллическом виде; если предварительно раствор подогреть, то выпадение
происходит быстрее.
4. Нагревают теллур истую кислоту НгТеОз до 4<Р.
Свойства
Белый кристаллический порошок, уд. в. 5,65—5,91, очень мало растворимый
в воде (6,7 • 10"*,%). Растворим в сильных кислотах и щелочах, нерастворим
в NH4OH. Вкус, сначала едва заметный, вскоре становится неприятным,
металлическим. При начинающемся калении расплавляется в прозрачную темно-
желтую жидкость; в струе воздуха возгоняется при светлокрасном калении.
АНГИДРИД ТЕЛЛУРОВЫЙ (ТРЕХОКИСЬ ТЕЛЛУРА)
Acidum telluricum Tellurium trioxide Tellurtrioxyd
anhydricum
Te02 Мол. в. 175,61
Приготовление
Нагревают теллуровую кислоту, НгТеОз при 160° (при более высокой
температуре образующийся ТеОз распадается на ТеОз и О2).
Свойства
Оранжево-желтые кристаллы, уд. в. 5,0704 при 14,5°, нерастворимые
в воде, разбавленных щелочах и кислотах. Крепкие щелочи с трудом
растворяют ТеОз с образованием теллуратов (солей теллуровой кислоты).
1 Завод, лаб. 2, № 8, 63 (1933).
■ SL G 11 х е П i, А. Р г z у s г с г у р k.o w s к I, Rosznlkl Chemii (Warszawa), 14, 477; CI.
74 ' Ангидрид
АНГИДРИД УГОЛЬНЫЙ i
(ДВУОКИСЬ УГЛЕРОДА, УГЛЕКИСЛЫЙ ГАЗ, УГЛЕКИСЛОТА) |
Acidum carbonicum Carbonic anhydride Ko^lendiojcyd j
Carbon dioxide Konlensanre 'i
Carbonic acid Kohlensaureanbydrid
COI2 Мол. в. 44,010
Приготовление
1. Наиболее удобным и общеизвестным способом получения СОг являете?
действие НС1 на СаСОз. Удобнее всего проводить реакцию в аппарате Крппа:
в средний шар аппарата помещают куски мрамора величиной с орех, после
чего наливают через верхний шар разбавленную НС1 (1 : 1). Для освобожде-
ния газа от следов НС1 и для высушивания его пропускают последовательно
через воду и концентрированную H2SO4. Если необходимо получить СОз
совершенно свободным от воздуха, то кусочки мрамора предварительно
несколько часов кипятят с водой и еще влажными загружают в аппарат
Последние следы кислорода удаляются пропусканием газа через сожигатель-
ную трубку, в которой накаливается медная спираль.
2. Постоянная струя чистого СОг получается при нагревании чистого
перекристаллизованного NaHCOs в вакууме. Операцию проводят в сожигательной
трубке, обернутой проволочной сеткой и закрытой с одного конца.
Нагревание начинают с закрытого конца, чтобы передний слой iNaHCOs поглощал воду,
3. Очень чистый СОг (98-99%) имеется в продаже в жидком виде (в
стальных бомбах). Он содержит небольшие количества кислорода, азота и окиси
углерода и лишь изредка tbS и SO?. Для полной очистки газа Мозер
рекомендует пропускать его сначала через две промывные склянки с уксуснокислым
хромом (двухвалентным), затем для поглощения кислых паров через U-образ-
ную трубку, наполненную кусочками КНСОз; дальше, для удержания .следов
H2S, — через U-образную трубку с кусочками пемзы, пропитанной раствором
CuSO«, и, наконец, для осушения — через промывную склянку с
концентрированной H2SO4. Последние следы кислорода удаляются пропусканием над
раскаленной медью или частой медной сеткой, помещенной в трубке на
протяжении 40 см\ кислород поглощается вполне, если скорость газа не превышает
10 л в час.
Для удаления СО газ пропускают через асбест, покрытый порошком окиси
меди и накаливаемый также до тёмнокрасного каления в широкой сожига-
гельной трубке. ч
4. Стоун и Бизон1 предлагают очищать СОг от последних следов 02,
пропуская газ через сернокислотный раствор2 CrS04 для увеличения
поверхности соприкосновения газа с жидкостью СОг распыляется пропусканием
через пористую стеклянную пластинку.
Свойства
Бесцветный газ без запаха, слабо кисловатого вкуса. Плотность 1,5292
(воздух =1). 1 л СОг весит при нормальных условиях 1,9769 г. Критическая
температура +31°. Т. кип. жидкого СОг —78,5° (при 760 мм). Уд. в.
твердого СОг 1,53. СОг не горит и не поддерживает горения. Довольно хорошо
растворяется в воде, образуя очень слабую кислоту, НгСОя (константы
диссоциации3.4,б: Ki1*0 = 3 -10-7; Яг2Б0 =5,73- 10-"); растворим в спирте. '
* Ind. Eng. Ch. Anal. Ed. 8, J* 3, 188 (1936).
1 Для получения CrS04 раствор хромокалиевых квасцов (0,1—0,4 М) в 0.1 N HfS04 медленно
1ропускают через редуктор Джонса, заполненный амальгамированным цинком. Редуктор Джонса
представляет собой широкую стеклянную трубку .'/~ 1 м, "d ~ 40 — 50 мм), гнабженную внизу
стеклянным краном. Может быть заменен бюреткой емкостью 100 мл с краном.
» У. К a i k о и Н. Е 1 о, Z. phys. Ch. A- 184, 211 (1939).
* У. К a i к о и А. А I г о 1 a, Z. Phys. Ch. A, 179, 307 (1937). А
1 Термодинамическая константа диссоциации К (т, е. вычисленная не из концентрации
ионов, а из активностей) по Т. Шедловскому и Мак-Инессу, [Т. S h е d 1 о w в к у, D. А. М с I п
ne s, J, Am; chem. Soc. 57, 1705 (1936); С. I, 3801 (1936)] имеет величину Ка «3,72 .. Ю""7'
'Ангидрио угольный
75
Растворимость СОа в воде
Таблица 58
t°c
1 л воды поглощает
при 7о0 мм л СО,
(объем СОв при 0° и
760 мм)
ГС
1 Л воды поглощает
при 760 мм л СО,
(объем СО, при 0е и
760 мм)
t°C
\
1 л воды поглотает
при 760 мм 4 СО,
(объем СО. при 0е и
760 мм)
1,7967
1,7207
1,6481
1,5787
1,5126
1,4497
1,3901
7
8
9
10
11
12
13
1,3339
1,2809
1,2311
1,1847
1,1416
1,1018
1,0653
14
15
16
U
18
19
20
Растворимость СОа в воде
(Bohr, Воск)
1,0321
1,0020
0,9753
0,9519
0,9318
0,9150
0,9014
Таблица 59
t°c
0
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
1 об. воды
растворяет
об. СО,
1,713
-. 1,646
1,584
1,527
1,473
1,424
1,377
1,331
1,282
1,237
1,194
1 1,154
1 1,П7
1,083
1,050
1,019
0,985
0,965
100 г воды
растворяют г СО,
0,3347
0,3214
0,3091
0,2979
1 0,2872
0,2774
0.2681
0,2590
0,2494
0,2404
0,2319
0,2240
0,2166
0,2099
0,2033
•0,1971
0,1904
0,1845
t°c
18
19
1 20
21
22
23
24
25
26
27
28
29
30
35
40
45
50
1 60
1 об. воды
растворяет
об. СО,
0,928
0,902
0,878
0,854
0,829
0,804
0,781
0J59
0:738
0,718
0,699
0,682
0,665
0,592
1 0,530
0,479
0,436
0,359
100 г воды
растворяют г СО,
0,1789
0,1736
0,1689
0,1641
0,1591
0,1541
0,1494
0,1450
0,1407
0,1367
0,1888
0,1293
0,1259
0.1106
0#974
0,0862
0,0762
0,0577
Таблица 60
Растворимость С02 в 99%гно м этиловом спирте
(Bohr;
t°c
— 65
-25
— 20
-15
— 10
— 5
1 об. спирта
растворяет
об. СО,
38,41
8,75
7,51
6,59
5,75
5,01
t°c
0
5
10
15
20
1 об. спирта
растворяет
объСО,
4,44
3,69
3,57
3,25
2,98
*°С
, 25
3d
35
40
45
1 об. спирта
растворяет
об. СО,
2,76
2,57
2,41
2,20
2,01
76
Ангидрид
АНГИДРИД ФОСФОРНЫЙ (ПЯТИОКИСЬ ФОСФОРА)
Acidum phosphoricum
anhydricum
Phosphorus fpentoixide
Phosphoric anhydride
P2O5 Мол. в. 141,96
Phosphorpentoxyd
Phosphorsaureanhydri4
Приготовление
1. Для лабораторного получения P2Os можно применять трехгорлую склянку;
через одно горло которой проходит стеклянная палочка с чашечкой, на
которой сжигают фосфор. Через второе горло подводится тщательно высушенньг!
воздух, уходящий затем через третье горло в стоящую рядом большую
склянку Вульфа. При горении фосфо.
ра образуется Р2О5, который оседает
частью в реакционном сосуде, часть*
в склянке Вульфа.
2. Для получения относительно болы
ших количеств Р2О5 Грабов с к и I
рекомендует следующий аппарат, изо»
Сраженный в разрезе на рис. 16.
Жестяной, луженый, хорошо пропаянны!
тугоплавким припоем барабан А укре*
плен при помощи выступа на деревян*
ном треножнике. В крышку вделана'
загнутая внизу дюймовая трубка В,
которая во время работы закрывается
просверленной пробкой С. Для сожига-
ния служит медная ложка D, которая
при помощи железного стержня
укреплена в деревянной четырехугольной
пробке Е (40 мм шириной), 'плотно
подходящей к тубусу барабана.
К стеклянному сосуду F плотно
подходит широкогорлая жестяная
воронка G, которая в верхней части на
7 мм шире барабана А. Вынимая до*
щечку Я, можно увеличивать щель
-.между -А и С, если будет необходимо
возобновить воздух, который (уже
лишенный кислорода) удаляется через С.
При употреблении аппарата сначала
нагревают ложку D и плотно
вставляют цилиндр А в воронку G, закрывающую сосуд F. Затем вносят в аппарат
маленькими кусочками нарезанный сухой фосфор *, для чего, вынув ложку,
помещают на нее кусочек фосфора и вставляют ложку на прежнее место. Приток
воздуха регулируется опусканием воронки G и открыванием пробки С. Только
вначале, когда барабан еще не нагрелся, выделяется некоторое количество
паров из щели, между барабаном и воронкой.
Продукт, особенно при сухом воздухе, всегда порошкообразен и\ при
постукивании по барабану легко ссыпается в; банку.
3. Г. У и тэк ер2, критически сравнив существующие методы очистки
продажного РгОб,' предложил удобный прибор для этой цели (рис. 16),
состоящий из Т-образной железной трубы Ач несущей на себе стеклянный
«питатель» В. Устройство последнего ясно видно на рисунке. Стеклянная
палочка С с поршнем может передвигаться в пробке D, причем поршень
Рис. 15. Аппарат для получения фосфорного
ангидрида
1 Нарезать фосфор нужно под водой острым ножом, придерживая кусок фосфора
пинцетом. Операцию необходимо проводить в толстостенной посуде, удобиее всего в фарфоровой
ступке. ' ■ " ?->
! Н. W h i t a k e r. J. Chcm. Soc, 127, '-2219 (1925).
Ангидрид фосфорный
. 77
оЛьно плотно прилегает к стенкам трубки Е. Поставив поршень в поло*
ДоВ ие^ изображенное на рис. 16, и приподняв пробку D, загружают в пита»
*ль В технический Р2О5. При поднимании палочки С и легком
постукивали по стенкам В, Р2О5 в требуемом количестве подается в трубку А, нагре-
Для охлаждения трубки А вначале служи!
II
С
докрасна печью F.
Н, погруженной
*+
Ш
£
М
Л
НИИ
ваемую
аяжней частью в чашечку G,
содержащую воду.
Через трубку J подается
чистый кислород,
высушенный пропусканием через
H2SO4, твердый КОН и
технический Р2О5, избыток
кислорода 'выходит из
прибора сквозь щели неплотно
сидящей пробки К.
Возгоняемый в токе кислорода
фосфорный ангидрид
собирается в банке L. Изред- 2
ка открывают втулку М и
скопившийся около М лро-
дукт железным гребком
ссыпают в банку L. Из
200 г технического Р2О5
в течение 2 час. было
получено 70 г чистого
препарата, свободного от
низших окислов.
4. Продажный Р2О5
большей частью содержит
трехокись фосфора (РЮв) и, иногда, свободный фосфор; эти примеси могут
быть удалены возгонкой препарата в токе кислорода; реакцию можно
ускорить, прибавляя катализаторы — платинированный асбест или платиновую
чернь. Для возгонки можно применять приспособление, показанное на рис. 17.
Подвергаемый возгонке Р2О5 вводится в расширение Л, которое нагревается,
в расширение С помещают рыхлый комок асбеста или стеклянной ваты,
сужение В поддерживается все время в нагретом состоянии до окончания
возгонки, причем трубка С
соединена с трубкой, содержащей
технический P2Os для
предохранения от влаги.
По окончании возгонки
трубку разрезают в месте В и
присоединяют к установке, в кото-
следует помещать наклонно или
Ы
Рис. 16. Схема прибора для очистки фосфорного ангидрила
по Уитэкеру
Рис. 17. Трубка для возгонки фосфорного ангидрида
рой требуется осушать газы; трубку с РгО
вертикально так, чтобы проходящий через нее газ шел вверх
5. Стекловидный РаСЬ по А. Н. и А. И. Р. К э м п б ел л * получается при
Длительном нагревании обычного препарата в запаянной трубке при 450°.
Свойства*
Снежно-белая рыхлая хлопьевидная масса уд. в. 2,291 при 26° (для
стекловидного Р2О5 2,737, для кристаллического 2,2842, без запаха;
сильно кислого вкуса, жадно поглощающая воду и расплывающаяся во
влажном воздухе. Отличные осушающие свойства РгСЬ могут быть иллю-
стированы, например, опытами Б е к к е р а, нашедшего, что упругость
водяного пара над Р2О5 равна 0,000 Ям рт. ст. В холодной воде растворяется
\ А. N. С a m p b е 11, A. J. R. С a m р Ъ е 11, Trans. Faraday Soc. 31, 1567 (1935).
1 S. О 1 i x e 11 у, К. В о г a t у n s k i, Z. anorg. Ch.f 235, 225 (1938).
78 Ангидрид • " \Щ
,.- ■■— ■». >4а
с шипением, образуя метафосфорную кислоту НРОз; с HBr, HC1 и РС1б Лщ
бром- или хлорокнсь:
2P20s + 3HC1 = РОСЬ + ЗНРОз,
РЮв + ЗРСЬ = 5РОС1з.
При 563° плавится, а при более высокой температуре испаряется. При
этом около 15% PaOs получается в виде бесцветных моноклинических кр*ь
сталлов с алмазным блеском; остаток плавится в стекловидную массу. Пр>ц
нагревании кристаллов до 440° получают аморфный РаОз, который при воэ«
гонке снова превращается в кристаллический. Молекула РЮв сильно поли-
меризована и правильнее было бы писать формулу фосфорного ангидридам
в «виде (Р20з)/г По Е. Брицке и Е. Гофману1 в интервале температур:
670—1100° мол. в. фосфорного ангидрида отвечает формуле РЛ-Iio. :;
Испытание2 %
Препарат, отвечающий квалификации ч. д. а., должен выдерживать еле»-;;
дующие испытания. _:;'.'
1. Мышьяк. Раствор 1 г препарата при испытании по Хефти-Гутцейту*
в течение 2 час. может дать такое же потемнение сулемовой бумажки, какое
получается с типовым раствором (без Р2О5), содержащим 0,02 мг As. ;-
2. Фосфористый ангидрид. 1 г препарата вносят малыми порциями в 10 дд<
воды, прибавляют 5 мл 10%-ной H2SO4, 0,1 мл 0,1 N КМп04 и нагревают
5 мин. на водяной бане. Розовая окраска раствора не должна совершенно
исчезнуть.
АНГИДРИД ХРОМОВЫЙ
Acidum chromicum Chromium Chromsaure
trioxidei Chromsaureanhydrid
Chromtrioxyd
СгОз. Мол. в. 100,01 /
Приготовление
1. По Рюсту в объемистой фарфоровой чашке растворяют 100 г грубо
измельченного ЫагСггО? в 250 мл воды и, в случае надобности, фильтруют.
К прозрачному раствору приливают при помешивании 400 мл
концентрированной H2SO4. Во время прибавления H2SO4 смесь разогревается, t и под
•конец выпадают мелкие тёмнокрасные кристаллы СгОз. Дают смеси
спокойно охладиться, причем количество кристаллов возрастает.
Охлажденную массу фильтруют через стеклянную воронку с платиновым конусом
(или с комочком стеклянной ваты). Когда все перенесено на фильтр (для
смывания кристаллов с чашки применяют часть фильтрата), сильно
отсасывают осадок, подпрессовывая его при этом фарфоровым пестиком. Для
освобождения полученного препарата от захваченной H2SO4 его промывают
концентрированной HNOs (цель достигается довольно легко, если1 поверхность
кристаллов покрыта HNOs) и, многократно отжимая, дают кислоте стечь.
При трех-» пятикратном промывании описанным способом почти вся H2SO4
удалена. Для проверки собирают немного фильтрата и, разбавив erov водой,
испытывают при помощи Ва(Ж)з)г; не должно быть осадка BaS04.
После последнего тщательного отсасывания переносят кристаллы на
пористую глиняную тарелку и хорошо отжимают фарфоровым шпателем для
более быстрого удаления HNOs. Для окончательного осушения кристаллы
помещают в фарфоровую чашку и нагревают на водяной бане при частом
перемешивании до исчезновения запаха HNO3. Совершенно сухую массу
еще теплой помещают в банки и хорошо закупоривают. Выход 58—60% тео-
1 Е. V. В г i t z k e, E. H 0 f f m a 11 n, Ind. Eng. Chem. Anal. Ed. 4, 1 (1933).
1 Испытание хим. реактивов, 402.
8 Б.е Р л ь-Л у н г е, Химико-технические методы исследования, т. И, ч. 2, вып. 1, стр. 419—
420, ОН^И, Гл. ред. хим. лит., 1938.
Ангидрид хромовый
19
етического. Фильтрование, промывание и высушивание следует вести
возможно быстрее ввиду сильной гигроскопичности СгОз.
2. По методу ИРЕА к раствору 300 г К2СГ2О7 в 500 мл воды прибавляют
420 мл H2SO4 (уд. в. 1,84). При этом выпадает KHSCh; его
отфильтровывают и раствор выпаривают так, чтобы СгОз выпадал только при охлажде-
нйИ. Выпавшие при остывании раствора кристаллы СгОз отсасывают на
в0ронке Бюхнера и промывают концентрированной HNOa. Сушат в фарфо-
оовой чашке при помешивании, сначала около 100°, затем в сушильном
Ькафу при 100-120°.
3. Н. Д. Бирюков1' доказал» что даже после (Многократной сушки
0 вакууме при .70° с периодическим продуванием воздуха (для удаления
HNOs) препарат содержит не более 99,87% СгОз. Получить препарат с более
gbicoKHM содержанием СгОз не удается.
Водные растворы СгОз, совершенно не содержащие Cr", могут быть по
Н. Бирюкову получены анодным окислением раствора продажного СгОз.
4. Ш у-Т с у-Л и и Вен-Ч а нг-Кинг2 получают 98%-ный СгОз,
обрабатывая К2СГ2О7 концентрированной HN03 и отделяя KNOs от СгОз
фракционной кристаллизацией. Выход 90% теоретического.
Свойства
Тёмнокрасные блестящие ромбические кристаллы уд. в. 2,70, расплываю*
щиеся на воздухе и растворимые в воде и разбавленном спирте (осторожно!).
При нагревании СгОз делается черным, при 196° плавится в красно-бурую
жидкость; выше 250° наступает разложение с отщеплением кислорода. При
лежании на бумаге иногда вызывает ее воспламенение; спирт также
вспыхивает при соприкосновении с кристаллами СгОз.
Таблица 61
Растворимость СгС3 в воде
(А. В. Раковский и Д. М. Тарасенков)
ГС
0
20
40
60
0/оСгО,
61,94
62,58
63,12
63,78
t°c
80
I 100
127
0;'0 СГ08
65,47
66,54
71,20
Удельный вес водных растворов СгОв
(Менделеев)
Таблица 61
СгО30|0
0
5
10
15
20
17,5
Л7*5
1,0010
1,037
1,076
1,118
1,162
^
0,999 '
1,036
1,076
1,119
1,166
0/оСгО§
25
30
35
40
,17,5
di7 5
1,208
1,258-
1,312
1,373
л15
1,215
1,268
1,324
1,383
°/0СгОв
45
50
55
60
й17,5
1,440
1,512
1,587
1,665
w15
d4
1,415
1,510
1,579
* ЖОХ 10, № 10, 94? (1940).
а С h u-T s u-L е е a. W е п-С h а п g-K i n g, С. II, 276 (1936).
so
Асбест. Барий
Испытание1 \
По ОСТ НКТП 5765/115 содержание СгОз в препарате ч. д. а. должно'
быть не меньше 98°/0, в препарате чистом — не менее 96%; наибольшие
количества допустимых примесей указаны в табл. 63.
% Таблица 63
Допустимые
Примеси
Ь Нерастворимые в воде вещества
2. Хлориды (СК)
3. Сульфаты (SO"4)
5. Железо, алюминий, 6api.fi и др. •
примеси
• • • » • •
в СгОз
Допустимое содержание в °,'о
в чистом для
анализа
0,004
0,002
0.03
0,15
0.03
в чистом
0,01
0,005
0,06
0,30
0,06
АСБЕСТ ПЛАТИНИРОВАННЫЙ
Asbestos platinized Platinierter Astiest
П риготовление
По Эрдману2 прокаленной длинноволокнистый асбест пропитывается
довольно крепким раствором платинохлористоводородной кислоты (HtePtCle)
и опускается затем в концентрированный NHUOH. После этого асбест
помещают на стеклянную воронку, дают стечь избытку жидкости (что ускоряется
осторожным прессованием) и затем постепенно нагревают до каления; при
этом образуется губчатая платина, проникающая глубоко внутрь волокон
асбеста.
БАРИЙ АЗОТНОКИСЛЫЙ (НИТРАТ БАРИЯ)
Barium nitricum
Barium nitrate
Bariumnitrat
Ba(N03)2 Мол. в. 261.38
Приготовление
1. Растворяют углекислый барий (можно пользоваться природным
витеритом JiaCOa) в разбавленной HN03, прибавляя избыток ВаСОз> нагревают
до кипения и. если исходят из нечистого продуктам прибавляют немного
Ва(ОН)г для осаждения примесей. Раствор фильтруют, бесцветный фильтрат
подкисляют чистой азотной кислотой до слабокислой реакции и выпаривают
до начала кристаллизации.
2- Для получения препарата из хлористого бария по Кульману
смешивают горячие растворы £50 г ВаСЬ • 2Н.20 в 500 мл воды и 85 г NaNOs
в 85 мл воды. Выпадающие кристаллы после охлаждения отсасывают и
очищают перекристаллизацией.
3 Для получения Ba(N03)2 из природного BaSO* (тяжелый шпат, барит)
Г. и В. Бильц* рекомендуют следующий метод. Хорошо смешивают 47 г
С М КиоМеТ0Д8К4Я°П^едеЛе"ИЯ ^N0» B препарате см. Труды Казанского хим. тех. ин-та им.
J Jf hrb. d. anorg. Ch. 275.
* Ucbungsbeispiele.
Асбест платинированный. Барий азотнокислый
81
тонко
измельченного BaS04 с 12 г тонкого порошка древесного угля,
пробного через проволочное сито, и помещают смесь в глиняный тигель, снаб
сеЯнный крышкой (наполняя его на 2/з—3А) и засыпав сверху тонким слоем
^ольного порошка. Тигель помещают в угольную печь (например, кузнеч-
Уг а горн) и нагревают в течение двух час. при красном калении. После
хлаждения образовавшуюся красновато-серую массу сернистого бария
0 здробляют и постепенно вносят в разбавленную азотную кислоту (26 г
Рт^Оз УД- в- 1»4 на 575 мл воды). Раствор кипятят, фильтруют и упаривают
кристаллизации. Выпавшие кристаллы отсасывают, промывают небольшим
количеством холодной воды и высушивают в умереннотеплом месте.
Маточный раствор подвергают многократному упариванию и кристаллизации.
Выход 45—50 г.
Свойства
Прозрачные или матовые правильные октаэдры уд. в. 3,24,
растрескивающиеся при нагрев'ании. Т. пл. 592°. Немного гигроскопичен. Растворим
в воде, нерастворим в спирте.
Таблица 64
Растворимость Ba(N08)2 вводе
(Mulder)
t °с
0
10
20
30
о/0 Ba(NOa)a
4,8
6,5
8.1
10,4
t °с
40
50
60
о/о Ba(N03)8
12.4
14,6
15,9
t °0
80
100
•/о B.i(NO,)j
21,3
25,5
Таблица 65
Удельный вес водных растворов Ва(гТО3)г
(К г е m e г)
°/о Ba(NOe)1
1
2
3
4
„19.5
1,009
1.017
1025
1,034
°/о BafNO,^
5
6
7
d19,5
1,042
1,051
Г.060
Р/о Ba(NO,),
8
9
^19,5
а
1,069
1,077
Испытание
Наибольшие количества примесей, допустимых по ОСТ 2611, указаны
в табл. 66. Содержание Ba(NOs)2 для всех трех квалификаций должно быть
не менее 99%.
82
Барий
Допустимые примеси в Ba(N03)2
Таблица к
Примеси
1. Нерастворимые в воде вещества
2. Щелочные металлы в виде
сульфатов
3. Кальций и стронций (сульфаты)
б. Железо (Fe)
Допустимое содержание в о/0
в химически
чистом
0,005
0,02
0,02
0,0005
0,0005
0.С0025 1
в чистом для
анализа
0.01
0,05
0,10
0.002
0,001
0 0005
в чистом
0.02
од
0,2
0,005
0.002
0,001
БЛРИЯ ГИДРАТ ОКИСИ (ГИДРООКИСЬ БАРИЯ, ЕДКИЙ БАРИТ)
Barftim oxydatum Barium hydroxide Bariumhydroxyd1
hydricum
Barium hydroxide
Caustic barita
Ba(OH)2 Мол. б. 171,38
Ba(OH)2-8H20 Мол. в. 315,50
Приготовление /
1. Для получения кристаллического препарата поступают по Эрдману *
следующим образом. К раствору 1 кг кристаллического хлористого бария
(ВаСЬ*2НЮ) в 2 л горячей дестиллированной воды прибавляют 1150 г
раствора NaOH, приготовленного растворением 310 г NaOH в 840 мл воды.
Раствор быстро фильтруют и упаривают до кристаллизации. Выпавшие
кристаллы отсасывают на воронке Бюхнера или стеклянной воронке с
платиновым конусом и дважды перекристаллизовывают из горячей воды. Полученный
препарат быстро отжимают на глиняных тарелках* и складывают в хорошо
закрывающиеся банки, пробки которых затем заливают парафином.
2. Гидрат Ва(ОН)а • 8ШО трудно теряет последнюю молекулу
кристаллизационной воды. Для получения безводного Ва(ОН)з кристаллический
препарат расплавляют в серебряном тигле и держат некоторое время при
температуре каления.
3. По Артису2 в раскаленный докрасна глиняный тигель постепенно
прибавляют небольшими порциями тщательно приготовленную смесь 8 г
Ва(ЫОз)г с 3 г железных опилок. Масса сначала пенится, затем делается
тестообразной и, наконец, хрупкой. Ее вынимают железным шпателем и
кипятят с водой. Раствор после фильтрования упаривают до кристаллизации
(получается около 5 г кристаллов). Маточный раствор может заменять
баритовую воду.
4. Кипятят ВаОг с водой и полученный раствор фильтруют и упаривают
до кристаллизации.
5- По Гагеру3 прокаливают при красном калении смесь 100 г BaCOs
с 10 г угля. Полученную массу выщелачивают водой, фильтруют и
упаривают до кристаллизации.
1 Хим. преп. 25.
2 А г t у в, J. prakt. Ch., 6, 172.
■ Хим. реактивы,, 89.
Бария гидрат окиси
83
Свойства
Ва(ОН)г • 8Н2О представляет собой белые тетрагональные призмы или
аблички уд. в. 1,66, довольно хорошо растворимые в горячей, хуже в
холодной воде. Прибавление NaCl или NaNOa увеличивает растворимость. Рас-
вор имеет сильно щелочную реакцию1:. Соприкасаясь с воздухом, раствор
поглощает С02 и мутнеет от выпадения нерастворимого ВаСОз. Кристаллы
пои сушении над H2SO4 легко теряют 7 молекул кристаллизационной воды;
последняя молекула НгО отщепляется лишь при красном калении.
Безводный Ва(0Н)з представляет собой блестящую белую кристаллическую массу
уд в. 4,495. Выше 780° переходит в ВаО.
Таблица 67
Растворимость Ва(ОН)2 вводе
(Rosenstiehl)
ГС
0
10
15
20
25
в 100 г воды растворяется
г Ba(OhTt
1,67
2,50
3,22
3,78
4,67
2 ВаО
1,5
2,22
2.89
3,48
4,19
t °с
30
40
50.
60
70
в 100 г воды растворяется
Z B:(OH)t
5,58
8,21
13,10
20,92
35,60
г ВаО
5,0
7,36
11,75
18,76
31,90
Таблица 68
Удельный вес водных растворов Ва^ОН)2
о/о Ва(ОН),
1,25
2,50
5,02
9,83
уд. вес
1,0120
1,0253
1,031
1,076
о/о Ва(ОН:§
15,43
20Д2
24,67
30,30
уд. вес
1,152
1,219
1,278
1,368
И
спытание
По ОСТ 5455 содержание Ва(ОН)2-8НЮ для препарата ч. д. а. должно
бьпъ не менее 99%» для чистого — не менее 97%; наибольшие количества
допустимых примесей указаны в табл. 69,
1 Ва(ОН), является сильным основанием, диссоциирующим ступенчато. Для второй ступени
Диссоциации
ГаОН'^гВа'Ч-ОН'
константа диссоциации по Д^вису равна 0,23 [С. W. D а V i e s, J. Ch m. Soc, 349 (ИЗО;].
84
Барий
Таблица
Допустимые примеси в Ва(ОН)2-8Н20
ПримеСи
1. Примеси, не растворимые в НС1
2. Хлориды (СИ)
3. Сульфиды (S")
4. Чяжелые металлы •
6. Соли щелочных металлов (в виде
7. Карбонат бария (ВаС03) ....
— "-—-— -у
Допустимое содержание в °/о
в химически
чисюм
0,005
> 0.002
0,0002
0,0005
0,0005
0,02
1,0
в чистом для
анализа
0,01
0.005
0,0005
0,001
0,002 !
0,05 !
2,0
в чистом
0,05
0,02
0,001
0,002
0,005
0,1
3.0
БАРИЯ ГИДРАТ ПЕРЕКИСИ
Barium peroxidatum
crystallisatum
Barium peroxide
BaCh-SHoO Мол. в. 313,49
Bariumsuperoxyd
wasserhaltig
Приготовление
1. 30 г чистой перекиси бария ВаОг растирают с водой и прибавляют
к 50 мл разбавленной НС1, охлажденной до 0°. Слабокислый раствор
фильтруют и приливают к 500 мл баритовой воды, насыщенной на холоду и
охлажденной до 0°. Выпавший осадок, состоящий из блестящих кристаллических
листочков, отсасывают, промывают небольшим количеством ледяной воды и
хранят в виде пасты.
2. Если исходным продуктом служит продажная перекись бария, то пре-,
дыдущий метод приходится несколько изменить. Растертую с водой ВаОг
прибавляют к избытку очень разбавленной, холодной НО. Полученный
кислый раствор делают щелочным, прибавляя небольшое количество холодной
баритовой воды. При этом выпадают гидраты Fe и А1; их отфильтровывают
и к фильтрату добавляют избыток баритовой воды для выделения ВаОа • 8НгО.
Дальнейшая обработка ведется по предыдущему методу.
3. По Ризенфельду и Ноттебому1 к раствору 15 г Ba(NOs)2
в 120 мл воды прибавляют при температуре 8—10° раствор 14 г перёкцси
натрия (ЫагОг) в 200 мл воды. Осадок ВаОз -8Н20 отсасывают, промывают
ледяной водой, спиртом и эфиром и сушат в эксикаторе над СаСЬ.
4. По Томсону для получения чистого препарата охлаждают 500 мл
раствора продажной Н2О2 до 0°, прибавляют холодную, насыщенную
баритовую воду до образования остающегося осадка и до приобретения жид*
костью щелочной реакции. Фильтруют и фильтрат при дальнейшем
охлаждении и постоянном помешивании выливают в 2 л баритовой воды. Через
некоторое время отсасывают выделившийся Ва02 • 8НгО и промывают
небольшим количеством ледяной воды.
5. Хороший препарат получается по методу ИРЕА действием НгОа и
ЫНЮН на ВаСЬ или Ba(NOa)2.
1 R i е б е n f e I d, N о 11 е Ъ о h in, Z. anorg. Ch., 89, 405 (1914).
Бария гидрат перекиси. Бария окись 85
двойства
Гексагональные чешуйки с жемчужным блеском, почти нерастворимые
в холодной воде. Горячей водой разлагается. Не растворим; в спирте и
эфире. При нагревании до 130° выделяет воду.
Испытание
Определение содержания ВаОг проводится по методу, описанному на
стр. 86 (Бария перекись). Теоретически препарат должен содержать
54% Ва02г
БАРИЯ ОКИСЬ
Bari,um oxydatum Barium oxide Bariumoxyd
anhydricum Baryterde
BaO Мал. в. 153,36
Приготовление
1. В раскаленный добела тигель из настоящего фарфора, снабженный
плотно прилегающей крышкой, постепенно вносят 150 , г продажного
Ba(NOs)2, который тотчас же плавится и разлагается со вспениванием.
Когда пена осядет, добавляют новую порцию Ва(ЫОз)г и так поступают до
тех пор, пока не будет внесено все количество. Для разложения последних
количеств образующегося азота стоки ело го бария Ва(Ы0г)2 возможно сильнее
прокаливают тигель в течение 2 час. Полученная.сероватая масса не
содержит нитритов. По охлаждении тигель разбивают, удаляют верхнюю пленку,
окрашенную в зеленый цвет марганцовистокислым барием, и наполняют
окисью бария банки с притертыми пробками (еще лучше запаять ВаО в
небольшие колбочки). При работе в глиняном тигле продукт получается
загрязненным кремнекислотой, кальцием, железом и др. Платиновый тигель
сильно разрушается.
2. Значительно труднее диссоциирует ВаСОз; для получения из него ВаО
его растирают с 20—30% вара. - Из полученной массы формуют шарики,
которые подвергают сильному прокаливанию.
3. Очень легко и без вскипания переводится в окись бария иодноватокис-
лый барий. Bai(JO»)2.
Свойства
Серовато-белая, пористая масса уд. в. 5,32—5,72, легко растирающаяся
в порошок. Т. пл. 1923° (по другим данным 1580°). Из Ba(NOsb можно
получить кристаллическую ВаО в виде мелких кубиков. ВаО жадно
притягивает из воздуха влагу и СОг, образуя углекислый барий, ВаСОз.
По Бузу и Ма к-И нтайру1 ВаО является замечательным
осушителем для газов, далеко превосходящим такие обычно применяемые
вещества^ как СаО, СаСЬ или алюмог*ель. По данным Бауэра2 влажный
воздух после пропускания через трубку с ВаО содержит всего 0,000085 мг/л НзО.
Испытание (Мерк)
1. Карбонаты (С02). Препарат должен растворяться в разбавленной НС1
без выделения большого количества пузырьков газа.
2. Окислители [ВаО* и Ba(NOs)2]. Полученный раствор не должен
обесцвечивать КМп04 или вызывать посинение иодокрахмальной бумажки.
' Н. S. Booth, L. H. Mc In tyre, Ind. Eng. Chem. Anal. Ed., 2, 12 (1930).
"J. H, Bower.J. Re*. Nat. Bur, Stand., 12, 241 (1934).
86
Барий
БАРИЯ ПЕРЕКИСЬ
Barium peroxydaturn Barium peroxide Bariumperoxyd
ВаОз Мол. в. 169,36
Приготовление
1. Через нагретую до 500—600° тугоплавкую трубку, в которой находится
30 г измельченной омиси бария, пропускают струю кислорода, осво-'
бождённого от примеси СО2 пропусканием через концентрированный раствор
КОН1.
По Броди сушить кислород не рекомендуется, так как незначительные
следы влаги способствуют образованию ВаОг.
В качестве исходного продукта хорошо подходит техническая
(плавленая) окись бария, 88,5 ч. которой для придания пористости предварительно
тщательно смешивают с 10 ч. Ba(NOs)2 и 1,5 ч. угля и смесь сильно
прокаливают. В калильном жару происходит энергичное выделение окислов азота,
разрыхляющее массу2.
2. Гидрат перекиси бария ВаОг ■ 8Н2О высушивают в вакуум-эксикаторе
над концентрированной FbSO*.
3. По Ван клин у3 прокаливают смесь 2 ч. ВаО и 1 ч. СиО. При этом
образуется Си и ВаОг. ,
4. По С. И. Райхштейн и И. А. Казарновскому4 вполне чистьгй
препарат может быть получен следующим путем: 8 г химически чистого
едкого барита Ва(ОН)з • 8Н20 растворяют в 340 мл воды, освобожденной
продолжительным кипячением от СОг и охлажденной в струе кислорода.
Раствор обрабатывают 10 мл 3%-ной Н2О2 при 0°. Полученный бельш
блестящий чешуйчатый порошок Ва02 • 8ШО подвергают 50-кратному
промыванию водой, освобожденной от СОг и охлажденной в струе кислорода. Выход
15—20% теоретического (по Ва).
При употреблении химически чистых исходных препаратов и работе в
особо сконструированном аппарате (описанном в оригинальной статье) \
позволяющем проводить все операции в отсутствие СОг, получают препарат,
содержащий после обезвоживания 99Jj> ВаОг.
Свойства
Белый порошок уд. в. 4,96, теряющий около 800° половину кислорода и
переходящий в ВаО. При обыкновенной температуре очень устойчива. С
водой образует гидрат (см. стр. 84)» причем при растирании препарата, с водой
не должно происходить разогревания, указывающего на примесь окиси бария.
При действии кислот образует соответствующую соль и перекись
водорода.
Испытание9
Препарат ч. д. а. должен содержать не менее 85% ВаОг.
Для определения содержания ВаОг к 0,3 г препарата, помещенного в .колбу
с притертой пробкой, прибивают 25 мл 10%-ного раствора KJ и 5 мл НС1
(уд. в. 1,19) и оставляют на полчаса при частом взбалтывании.
Выделившийся иод титруют 0,1 N гипосульфитом, прибавляя в конце титрования
крахмал. 1 мл 0,Ш Ыа^гОз соответствует 0,008468 г ВаОг.
1 По Бусин ь олю вместо чистого кислоррда можно пропускать воздух.
• S с h u 1 z e, Герм. пат. 171070 (1903).
" Wa nk I Id, Ber., 7, 1029.
« ЖФХ 8, 86 (1932).
» Испытание хим. реактивов, 75,
Бария перекись. Барий платинистосинеродистый
87
БАРИЙ ПЛАТИНИСТОСИНЕРОДИСТЫЙ
(ТЕТРАЦИАНОПЛАТИН[2]-АТ БАРИЯ)
Harium platinocyanatum Barium platincyanide Bariumplatincyantir
BaPt(CN)4 • 4H20 Мол. в. 508,73
Приготовление
1. По Бергсё1 BaPt{CN)4 количественно получается из платинохлористо-
воДО'родной кислоты, гидрата окиси бария, синильной кислоты (яд!) и
сернистого газа по реакции:
H2PtCle + 4HCN + SBafOHb + SO2 = BaPt(CN)4 + ЗВаСЬ + BaS04 + 8H2O.
В водный раствор H2PtCle прибавляют вычисленные по вышеприведенному
уравнению количества Ва(ОН)г и водного раствора синильной кислоты HCN.
Жидкость нагревают и пропускают S02 до полного обесцвечивания.
Отфильтровывают выпавший BaS04 от горячего раствора и фильтрат несколько
выпаривают; при охлаждении выделяется платинистосинеродистый барий, очень
трудно растворимый в растворе ВаСЬ.
2. По Вейнланду суспендируют 3 ч. тонко истертого ВаСОз в растворе
2 ч. НгРЮо в 10 ч. воды и полученную смесь прибавляют к нагретому до
кипения раствору синильной кислоты:
H2PtCle +' 4ВаСОз + 4HCN =|ВаРЦСН)4 + ЗВаСЦ + 4С02 + ЗНгО + 7*02,
Свойства
Лимонно-желтые кристаллы уд. в. 2,076—2,09. Растворим в воде (1:33 при
16°), легче — в горячей. При хранении над H2SO4 или при нагревании до 100°
теряет половину кристаллизационной воды; при 150° полностью'
обезвоживается. Под действием рентгеновских лучей очень сильно флюоресцирует
желто-зеленым светом.
БАРИЙ СЕРНОКИСЛЫЙ (СУЛЬФАТ БАРИЯ)
Barium sulfuricum Barium sulfate Bariumsulfat
Permanentweiss
BaSOi Мол. в. 233,42
Приготовление
Чистый хлористый барий растворяют при нагревании в воде и при
сильном перемешивании медленно осаждают разбавленной H2SO4 (или каким-либо
растворимым сульфатом). При осаждении свободной кислотой осадок BaS04
отстаивается полно и быстро, при осаждении сульфатами — значительно
медленнее. Осадок декантируют, затем отфильтровывают, промывают на фильтре
горячей водой до полного удаления ионов СГ и высушивают в сушильном
шкафу.
Свойства
Снежно-белый порошок уд. в. 4,5, практически нерастворимый в воде и
кислотах (1 л воды растворяет при 18° 0,0023 г BaS04, при 100° —0,0039 г).
При 1580° BaS04 плавится и затем полностью улетучивается. Углем, уже
пРи 600°, восстанавливается до сернистого бария.
1 В е г $ 8 С е, Z, anorg. Ch,, 19, 320 (1899).
88
Барий
Испытание
Препарат ч. д. а. должен выдерживать следующие испытания:
1. Растворимые в НС1 вещества. 12 г препарата кипятят со смесью ПО мл
воды и 10 мл НС1 (уд. в. 1,12) и фильтруют. 50 мл фильтрата выпаривают ^
платиновой чашке досуха, остаток слегка прокаливают и взвешивают. Вес егсг
не должен превышать 0,5 >мг (для препарата чистого—1,0 мг).
2. Тяоюелые металлы. К 20 мл фильтрата, полученного по п. 1, приливают
1 мл ледяной СНзСООН и 10 мл сероводородной воды, причем не должнё
тотчас же появляться буроватого окрашивания или белой мути. (Препарат
чистый должен выдерживать это испытание с 10 мл фильтрата.)
3. Железо. К 20 мл фильтрата (п. 1) прибавляют 5 мл 10%-ного ЫНЮН
и 10 мл сероводородной воды — не должно тотчас появляться зеленоватого;
окрашивания раствора. (Для препарата чистого берут 10 мл фильтрата.) •
4. Хлориды. Нагревают до кипения 2 г препарата с 40 мл воды и филы
труют через фильтр, трижды промытый горячей водой. При добавлении
к охлажденному фильтрату 1 мл HNOs (уд. в. 1,15) и 1 мл 0,Ш AgNOa
не должно появляться М'ути в течение 5 мин. (При анализе чистого препа^
рата берут навеску 1 г).
5. Нейтральность препарата. 3 г BaS04 взбалтывают с 60 мл воды и филь*
труют. Фильтрат должен иметь нейтральную реакцию на метилоранж.
БАРИЙ УГЛЕКИСЛЫЙ (КАРБОНАТ БАРИЯ)
Barium carbonicum Barium carbonate Bariumcarbonat
BaCOj Мол. в. 197,37
Приготовление
1. Очень чистый BiaCOs получается пропусканием СОг в баритовую воду
[насыщенный раствор Ва(ОН)г] и промыванием полученного осадка.
2. При осаждении ВаСОз из раствора соли баоия содой или поташом
получаемый осадок содержит щелочи. Для освобождения от них рекомендуется
отстоявшийся осадок отделить от жидкости, высушить и слаоо прокалить (до
спекания массы). После этого соли щелочных металлов легко отмываются и
препарат получается вполне чистым.
3. Некоторые авторы рекомендуют вести осаждение ВаСОз.из раствора
бариевой соли при помощи углекислого аммония (ЫН^гСОз. Отмытый осадок
хранят в виде полужидкой массы в закрытых банках.
Свойства
Белый кристаллический нежный порошок уд. в. 4,4, отдающий СОг лишь
при сильном белом калении, быстрее — в пламени вольтовой дуги (при 1100°
упругость СОг всего 20 мм) К В воде, даже при кипячении, растворим
чрезвычайно трудно (1,62 • 10-3% при 13°, Lp — 1,9 • 10-9), несколько растворим в
воде, содержащей СОг, вследствие образования бикарбоната бария Ва(НСОз)г.
Уже на холоду растворим в растворах NrhCl и NH4NO3. Т. пл. около 1740°.
Испытание . ъ
Наибольшие количества примесей, допустимых по ОСТ НКТП 7386/540,
указаны в табл 70.
1 Быстрая диссоциация ВаСОа, повидимому, протекает при 1200? См. С. II, 1966 (1937). ;
Барий сернокислый, углекислый и хлористый
.89
Допустимые примеси в ВаСОв
Таблица 70
Придеси
Допустимое содержание в °/0
в химически
чистом
в чистом для
анализа
1, Нерастворимые в НС1 вещества
% Хлориды (СГ)
3. Щелочность (ОН7)
4. Азот (общий из нитратов,
нитритов, аммиака и пр.)
5. Щелочные металлы (в виде
сульфатов) . .
6. Кальций и стронций (сульфаты) .
7. Тяжелые металлы ........
8. Железо (Fe) ; . . . .
9. Сульфиды (S")
0.01
0,001
0,01
0,001
0,05
0,1
0,0005
0,0005
0,0002
0.0?
0,002
0,02
0,002
0.1
0.25
0.001
0.001
0,0005
0,1
0.01
0,05
0,005
0,25
0.5
0.002
0.002
0,001
БАРИЙ ХЛОРИСТЫЙ (ХЛОРИД БА№Я)
Barium chloratum
Barium chloride
Bariumchlorid
ВаСЬ • 2HsO
Мол. в. 244,31
Приготовление
1. Методика получения вполне чистого хлористого бария из технического
продукта полностью зависит от качества исходного сырья: иногда бывает
достаточно подвергнуть технический ВаСЬ простой перекристаллизации, но в
большинстве случаев приходится вести дополнительную обработку.
Растворяют 100 г технического продукта в 200 мл горячей воды и
насыщают ^горячий раствор сероводородом до полного осаждения тяжелых
металлов (отфильтрованная проба жидкости не должна темнеть от прибавления
сероводородной воды). Раствор фильтруют. В присутствии железа прибавляют
баритовой воды до ясно щелочной реакции и отфильтровывают выпадающие
осадки Fe(OH)3 и А1(ОН)з. Фильтрат подкисляют небольшим количеством
чистой соляной кислоты и выпаривают до начала появления кристаллической
пленки *. В случае надобности фильтруют и оставляют для кристаллизации
при комнатной температуре или на холоду. На другой день выпавшие
кристаллы отсасывают, промывают водой и сушат при комнатной температуре на
пергаменте. Маточный раствор при выпаривании дает еще значительное
количество препарата.
Обычно полученный таким путем препарат' удовлетворяет всем
требованиям, предъявляемым к химически чистому хлористому барию. Однако иногда
в техническом продукте содержатся витраты (ЫОз'), избавиться от которых «е
удается.
2. М аргерит рекомендует для получения хорошего препарата насыщать
газообразным хлористым водородом профильтрованный раствор технического
ВаСЬ. При этом почти весь ВаСЬ выпадает в виде мелких кристаллов.
Раствор сливают, осадок отсасывают и промывают чистой концентрированной
НС1 (в которой ВаСЬ почти нерастворим), пока проба фильтрата, после
осаждения Ва серной кислотой, при выпаривании в платиновой чашке не
перестанет оставлять остаток2.
1 При этом может выпасть значительный оранжево-красный осадок селена. В этом случае
Раствор взбалтывают и отфильтровывают от селена, собравшегося в крупные хлопья.
1 Одадко тажям ву*см нельзя освободиться о-т тяжелых металлов (РЬ).
90
Барий
3. Для получения чистого ВаСЬ из витерита (природный ВаСОз) поступают
по Г. и В. Бильц1 следующим образом. Сначала вычисляют какое-
количество чистой концентрированной соляной кислоты необходимо для
растворения 100 г витерита. Требуемое количество кислоты помещают в банку
емкостью 2 л (не тонкостенную колбу)» разбавляют водой до 1,5 л,
прибавляют измельченный витерит и дают стоять для растворения в теплом месте;
•нерастворенными остаются силикаты. Прибавляют 50 мл хлорной воды и еще
5—10 г витерита и оставляют смесь в теплом месте при частом
взбалтывании; на следующий день добавляют еще 2—5 г витерита. Время от времена
отбирают пробу жидкости, подкисляют соляной кислотой и испытывают на
железо роданистым калием.
Когда все железо выпало, фильтруют и выпаривают фильтрат в
фарфоровой чашке до начала кристаллизации. Если при выпаривании выпадает еще
Fe(OH)3, находившийся ранее в коллоидальном состоянии, фильтруют еще,
раз, после того как жидкость выпарится до половины объема. Кристаллы
отфильтровывают и выпариванием маточного раствора извлекают остальной*
хлористый барий. Соединив все порции кристаллов, их перекрнсталлизовывают
из воды, подкисленной несколькими каплями НС1.
4. Можно получить чистый ВаСЬ растворением витерита в НС1 и
осаждением железа баритовой водой. Избыток Ва(ОН)2 удаляется пропусканием
струи СОг. Раствор фильтруют и упаривают до кристаллизации.
5. Для получения ВаСЬ из тяжелого шпата (BaS04) последний переводят
в BaS (см. барий азотнокислый, 3» стр. 80). Полученный сернистый барий
всыпают в 3—4 ч. 'кипящей воды и прибавляют НС1 до нейтральной или
слабокислой реакции, после чего снова добавляют BaS до щелочной реакции
и выпаривают досуха. Из полученного остатка выщелачивают ВаСЬ,
фильтруют и упаривают до кристаллизации (Б е н д е р).
6. Отделить примесь СаСЬ можно кипячением порошка ВаСЬ с 90—!
95%-ным спиртом, растворяющим хлорид кальция.
Свойства
Бесцветные, блестящие ромбические таблички уд. в. 3,1, теряющие при 100е
кристаллизационную воду и поглощающие ее вновь при лежании во влажном
воздухе. На воздухе устойчив. Т. пл. 962°. Сильно ядовит. Хорошо растворим
в воде, почти не растворим в НС1. В абсолютном спирте теряет свою
кристаллизационную воду, но сам почти не растворяется; не растворим также в
эфире.
Таблица 71
Растворимость ВаС12 в воде
/°с
5
10
15
20
30
40
о/о BaClt
24,4
25.0
25,7
26,4
27,7
29,0
/°с
50
60
70
80
90
100
о/о ВаС1
30,0
31,6
33,0
34,3
35,7
37,0
i yebungebeisplele.
Барий хлористый
• 91
Таблица 72
Удельный вес водных растворов ВаС13
(Sch iff)
.;0ВаС.у2Н,О
1
2
3
4
5
6
7
8
9
10
И
12
13
14
15
о/0 ВаС1,
0,852
1,705
2,557
3,410
4,262
5J15
5,967
6,820
7,672
8 525
9,377
10,23
11.08
11,93
12,79
421,5
1,0073
1,0147
1,0222
1,0298
1,0374
1,0452
1,0530
1,0610
1,0692 I
1,0776
1.0F61
1,0947
1,1(34
1,1122
1,1211
о/о ВаС1ш.2Н,0
16
17
18
19
20
21
22
23
24
25
26
27
* 28
29
30
>
°/о BaClj
13,64
14,49
15.34
16,20
17,05
17,9.0
18,75
1961
20.46
21.31
22,16
23/£
23,87
24,72
25.57
цм«ь
1,1302
1,1394
1,1488
1,1584
1,1683
1,1783
1,1884
1,1986
1,2090
1.2197
1,2304
1,2413
1,2523
1,2636
1,2750
Испытание
Наибольшие количества примесей, допустимых по ОСТ НКТП 7394/548,
указаны в табл. 73, Содержание ВаСЬ«2Н'20 для всех трех квалификаций
должно быть не менее 99>5%-
Таблица 73
Допустимые примеси в ВаС12• 2Н20
Примеси
1. Нерастворимые в воде вещества
2. Азот (общий из нитратов, нйтри-
3. Хлораты (ClOsO
4. Щелочные металлы (в виде суль-
7. Железо (Fe) .
Допустимое содержание в °/о
в химически 1 в чистом дчя
чистом анализа
0,005
0.0Э1
0,005
0,02
0,05
0,(005
0,00005
0*010
0.002
0,01
0,05
од
0,001
0,0001
в чистом
0.02
0,005
0,02
0.1
0,2
0,001
0.0002
92
Барий. Бериллий, Бром
„ m ^
БАРИЙ ХЛОРНОВАТОКИСЛЫЙ
Barium chloricum Barium chlorate Bariumchlorat
Ва(СЮ3)о • ШО Мол. в. 322,29
Приготовление
3 ч. (NH4)2$04 и 5 ч. КСЮз растворяют вместе в фарфоровой чашке в
15 ч. горячей воды. Раствор при постоянном помешивании выпаривают jgjk
консистенции жидкой кашицы, охлаждают и, перенеся массу в колбу, обр^
батывают четырехкратным количеством 80%-ного спирта. Остаток <%
фильтровывают, снова промывают спиртом, после чего от фильтрата отгоняют
спирт в дестилляционном аппарате. Значительный остаток от перегонки, Щ,
держащий NthClOs, нагревают на водяной бане в фарфоровой чашке, доб»
вляют насыщенного раствора Ва(ОН)г и нагревают до исчезновения зашЩ
аммиака и приобретения жидкостью яснощелочной реакции. Раствор выпари?
вают досуха; оставшуюся массу растворяют в пятикратном количестве водш
раствор освобождают от избытка Ва(ОН)а пропусканием СОг и после фил*^
трования выпаривают до кристаллизации.
Свойства
Прозрачные, как вода, столбчатые кристаллы уд. в. 3,18 остро горького
вкуса. При 310° соль распадается на Ва(СЮ4)г и ВаСЬ1. С горючими
веществами Ва(С10з)г энергично вспыхивает. Легко растворим в воде; растворил!
в ацетоне и трудно растворим в спирте.
Таблица 74
Растворимость Ва(СЮ8)2 в воде
(К г е m e г s)
/•с
0
20
100 г воды
растворяют г Ва(СЮ,)ш
22,8
37,0
/•С
40
60 |
100 г воды
растворяют г Ba(C10a)t
52,1
77,5
/°с
80
100
100 г вода
растворяю г Ba(CI08)t
98,0
125,4
БЕРИЛЛИЯ ОКИСЬ
BeriJHum oxydatum Berillium oxide Berilliumoxyd
BeO Мол. в. 25,02
Приготовление
Минерал берилл представляет собою в основном силикаты алюминия и
бериллия. По Д ж(о ю2 сплавляют 1 ч. очень тонко истертого берилла с 3 ч.
КгСОа при температуре яркокрасного каления. По охлаждении сплав
разлагают концентрированной H2SO4 (масса должна быть желатинообразной), Я
вновь нагревают, чтобы удалить H2SO4 и перевести кремнекислоту в
нерастворимое состояние. Растворяют, фильтруют, фильтрат выпаривают до появления
пленки и подвергают кристаллизации в течение 1—2 суток (отделение от
•А12(504)з и K2SO4; промывные воды от кристаллов упаривают для
последующей кристаллизации). Кристаллы растворяют, фильтруют и вливают при
постоянном помешивании в теплый насыщенный раствор углекислого аммония
(выделение Fe и А1)> после чего прибавляют избыток NrhOH; осадок настаивают
* В литержтуре приводится также точка плавления 414*.
* Prap. Ch., 433.
Варий хлорноватокислый. Бериллия окись. Бром 93
продолжение нескольких дней в жидкости, после чего отфильтровывают и
срабатывают еще раз раствором углекислого аммония. (По Джою и
к\лаДО обработка (NH^COs не должна продолжаться свыше 10 суток; в
потявном случае может выпасть соль бериллия.)
Аммиачный раствор при продолжительном кипячении (лучше
пользоваться струей пара, чтобы избежать толчков) выделяет основной углекислый
бериллий, содержащий Fe и А1. При прокаливании получается нечистая,
желтоватая окись бериллия. Дальнейшие операции сводятся к удалению
этих примесей. По Мор ату1 растворяют ВеО в НС1, осаждают ее
аммиаком, отфильтровывают, промывают и осадок настаивают с
концентрированным раствором чистого (ЫН^гООз, взятым в количестве, недостаточном для
полного растворения и смешанным с большим избытком NH4OH. Через
до дней осадок отфильтровывают и фильтрат разлагают водяным паром.
Выпадающий осадок вновь растворяют в НС1, раствор осаждают аммиаком,
полученный осадок настаивают с углекислым аммонием и т. д. Эти операции
повторяют, пока не получится снежно-белый кристаллический порошок
углекислого бериллия (не менее 4 раз), который сильным прокаливанием переводят
в ВеО. Описанный способ, по сообщению ряда авторов, является наилучшим.
Полученная окись бериллия все же не являетсЪ совершенно чистой; так,
при растворении в НС1 она придает раствору зеленовато-желтую окраску,
зависящую от незначительных следов примесей. Для получения
безукоризненного препарата Морат рекомендует обрабатывать раствор Ве(ОН)г
в растворе углекислого аммония избытком (NH^S, оставить на сутки, часто
взбалтывая, после чего нагревать, на водяной бане до 90°. Раствор быстро
отфильтровывают от выделившихся черных хлопьев и разлагают сильным
током пара до прекращения образования С02.
Отделенный и промытый углекислый бериллий — снежно-белого цвета и
растворяется в НС1 без окрашивания. Его переводят в ВеО, как описано выше.
Свойства
Белый объемистый порошок уд. в. 3,06,, плавящийся около 2500° и улету*
чивающийся легче, чем MgO.
БРОМ
Bromum Bromine Brom
Br Am. в. 79,916
П риготовление
1. В малых количествах бром можно получить, перегонкой смеси б ч.
КВг, 4 ч. концентрированной H2SO4 и 2 ч. МпОг. Бром собирают в
охлаждаемом приемнике.
2. Для очистки технического брома, содержащего С1 и J, рекомендуют
промыть продукт водой, высушить над H2SO4, перегнать, собирая фракцию,,
кипящую около 58°, и превратить дестиллат в КВг. К раствору КВг
приливают немного брома, который выделяет иод, последний удаляют
встряхиванием раствора с CS2. Из полученного КВг получают чистый бром по
способу 1. v
3. Рекомендуется перегнать бром над небольшим количеством КВг (для
удаления хлора). (
4. По Менделееву растворяют бром в крепком растворе СаВгг.
При разбавлении раствора водой .выделяется чистый бром, без примесей,
так как содержавшийся в нем хлор, вступив^ во взаимодействие с СаВгз,
вытеснил бром и образовал легко растворимый СаС1*.
» М о г a t h, Dissertation, Munchen, 14J90.
94
Бром
5. Для получения чистого препарата по Браун еру (С т а с у) * продажна
бром неоднократно промывают водой, после чего растворяют в концентр|
рованном растворе СаВгг, из которого снова выделяют бром прибавлений
большого количества воды. Освобожденный таким путем от хлора 6щ
сушат над твердым СаВгг, многократно взбалтывают со смесью СаО и воз
гнанного Р2О5 и, наконец, перегоняют в стеклянном аппарате в струе щ
6. Для получения чистейшего препарата по Генигшмидту
Цинтлю2 сначала для удаления хлора перегоняют бром из концентрир
ванного раствора СаВп>. (Раствор СаВгг получается прибавлением по капля
брома в чистое - аммиачное известковое молоко)!
Чистую СаО для приготовления известкового щ
лока получают трехкратной перекристалля
зацией Са(ЫОз)2 из воды, осаждением хим
чески чистым (NH^sCOs и переведением по
ченного СаСОз в СаО накаливанием в элект
ческой трубчатой печи в струе сухого возду
для чего препарат помещают в лодочки из
глазурованного фарфора.
Для перегонки служит круглодонная колб1
(рис. 18) с -пришлифованной капельной
воронкой и изогнутой почти под прямым углом
отводной трубкой, которая проходит через кожух
холодильника и концом опущена в холодную
воду. По мере «перегонки под раствор CaBrt
прибавляют из капельной воронки новые
порции брома. Прибавляя по каплям перегнанный
бром к раствору неоднократно пере
кристаллизованного щавелевокислого калия {КзСэОы
переводят его в КВг. Выпаривают этот рас-
тво-р, прибавляя время от .времени
подкисленный раствор KMnO-i, выделяющий
незначительное количество свободного брома; последний,
в свою очередь, вытесняет содержащийся иод,
который улетучивается с водяными парами.
Выкристаллизованный КВг для разрушения
органических примесей сплавляют в платиновом
тигле.
Полученный бромистый калий, свободный от
хлора и иода, смешивают с H2SO4 и двухромо-
вокислым калием3 (КгСпЮ? многократно пере-
кристаллизовывают, a H2SO4 предварительно
перегоняют над К2СГ2О7 в реторте, обмазанной
асбестом, отбрасывая первую фракцию,
содержащую галоиды). К2СГ2О7 берется в таком количестве, чтобы четверть КВг
осталась неразложеннои; таким образом бром перегоняется еще из раствора КЗг.
Чистый бром промывают водой, сушат сначала чистым плавленым
СаВгг, аатем, для удаления НВг, смесью чистого СаО и СаВгг и, наконец,
фосфорным ангидридом (Р2О5), возогнанным в струе кислорода. (СаВг2 при-,
готовляется вышеописанным способом из чистого СаО, не содержащего
галоидов, и из уже очищенного брома.)
Рис. 18. Прибор, для получения
брома
1 Brauner (также S t a s), Monatsh., 10, 411 (1899;.
а Н 5 n i gs с h m 1 d t, Z i n 11, Ann., 433, 701 (1923); Prap. Ch. 33; О. Н б n i g s с h m i й t Ы
£; ««ih In /}Q™anorg* Ch* 221' 134 (1934)J О. Н б n i gs с h m i dt и R. S a ch tl e b e n, Z. anocg.
LiLl,} 221» /О (1Уо4). *
» Реакция идет по уравнению:
6 КВг -f KiCrgO, + 7H,S04 = ЗВг, + 4K,S04 + Сг, (SOJa +7HtO.
Бром
95
7 Бром высокого качества, свободный от хлора, получается по методу
ИрЕА, изложенному ниже применительно к лабораторным условиям \ Колба
стекла пайрекс емкостью 0,5 л снабжена пришлифованным дефлегмато-
И3м Гемпеля (со стеклянными бусами), который в свою очередь соединен
^о шлифе с змеевиковым холодильником. Загрузив в колбу 1 кг
технического брома, подогревают ее на водяной бане и собирают фракцию 56—61°
/до 56° перегоняется бром со значительным содержанием С12). Полученный
продукт растворяют в бромистоводородной кислоте уд. в. 1,47—1,49 (на 1 кг
г пом а 600 г кислоты) и раствор -оставляют на ночь.
После выстаивания раствор переливают в дестилляционный аппарат,
описанный выше, и подвергают фракционной перегонке. Из \ %кг брома
получают:
а) около 50 г фракции, кипящей до 56°, — нечистый продукт;
б) около 750 г фракции, кипящей при 56—65°, — чистый бром;
в) около 100 г чистого брома, переходящего вместе с некоторым
количеством бромистоводородной кислоты. Водный слой отделяют и промывают
бром 1—2 раза водой.
~ Для получения брома из лабораторных' остатков (бромиды щелочных
Болас и Гровес рекомендуют смешать остатки с КгСгаСЬ и
разбавленной (2:1) H2SO4 и перегнать, собирая бром в охла-
приемник.
Таблица 75 Таблица 76
Удельный вес брома при
различных температурах
металлов)
избытком
ждаемый
Растворимость брома
(в воде по Винклеру)
г°с
0
20
25
30
<
3,1883
3,1193
3,1025
3,0848
/°С
35
40
45
50
Ч
3,0670
3.0497
3,0320
3,0146
t°c
0,00
10,34
19,96
30,17
40,03
49,85
100 г воды
растворяют
г брома
4,167
3,740
3,578
3,437
3,446
3,522
1U0 г
бромной воды
содержат
г брома
4,00
3,62
3,46
3,33
3,34
3,41
9. Бромная вода. Для получения насыщенной бромной воды, сильно
встряхивают в склянке с притертой пробкой 5 г брома с 100 мл воды,
изредка приоткрывая пробку, чтобы выпустить скопившиеся пары брома.
Свойства
Тяжелая жидкость, образующая тёмнокрасные пары. Цвет в
отраженном свете темнофиолетовый, почти черный, в проходящем свете — темно-
красный. Т. кип. 58,80° (Буза, Лелюан2); при — 7,30 застывает *
в желто-зеленою массу с металлическим блеском (напоминающую иод),
которая при — 252° становится бесцветной4. Уд. в. сильно зависит от
температуры, как это видно из табл. 75.
Запах очень сильный и резкий, при вдыхании пары брома вызывают
насморк, кашель, головокружение и, наконец, головную боль (противоядие —
вдыхание NH.3 и паров спирта). Пары брома сильно действуют на глаза,
вызывая слезы. На коже бром вызывает быстро пропадающие желтые
пятна, при больших количествах — сильно болезненные воспаления. Вкус
1 Реакт. и преп., 39.
* В о и г at, L е 1 u a n, С. г. 178, 635 (1924).
•По Бэкеру (Backer, J. Chem. Soc, 123, 1223 (1923) бром, высушенный в течени*
10 лет, имеет т. пл.—4,5°.
* О цвете твердого брэма в литература имеются противоречивые данные; так, ему припи*
сывают желто-зеленый-, желто-коричневый, стально-серый и красно-коричневый цветл (см. напр.
Abegtf.
96
бром. Бумага
противный, вяжущий и сильно жгучий. Хорошо
эфиое, хлороформе, бензоле и сероуглероде, хуже в
и НВг и растворах КВг.
Таблица 17
растворим в cnijj>>^
воде. Растворим в \щ
Удельный весбромной воды
и содержание в ней брома
(Slessor)
°/о Вг,
1,072
1,067
1,205
1,231
d
1,00901
! i,oop3i
1,00995
1,01223
°/о Вг,
1.874—1,906
1,952-2,009
2,089—2,155
3,102-3,169
(насыщ.)
d
1,01491
1,01585
1,01807
1,02367
Таблица 78
Растворимость брома в раствора
КВг
В 100 мл
раствора
содержится
г КВг
0
0,059
0,119
0,238
0,595
1,19
2,38
5,95"
) 9.22
11.90
20,52
23,66
25.82
36,08
(L. M. Winkler)
В 1С0 мл
О'
4,16
4,17
4,26
4,44
5,02 I
5,97 1
7.91
13.86
20.00
24.31
40,23
42,38 1
51.17
73,67
г г створа КЕг \
г брома
18,5е
3.556
3,61
3,7
3,856
4,38
5.22 ,
6,97
12,3
17,87
21,6
— 1
—
—
"—
растворяется
26»
3,4
3,43
3,5
3,65
4,1
4,93
6,73
11,9
17,6
21,65
—
—
—
63,24
I 1
Насыщенный воднц
растшор (бромная вод
желтого цвета и Hie заме
зает даже при —20°. Обд
дает вяжущим, wo не}
лым вкусом и запах
брома. На воздухе,
стрее при нагревании, бр<|
выделяется. При действ
солнечного света пронсх
дит образование НВг.
Испытание.
Наибольшие количест
примесей, допустимых
ОСТ 17381-40 указаны
табл. 79.
Кроме того, препара
должен выдерживать при
веденное в ОСТ 17381-4
испытание на иод.
Испытание бром
ной воды (Мерк)
1. Содержание H2SOa.
50 мл бромнМ воды при*
ливают 1 мл НС1 (уд.
1,12) и 1— 2 мл 10%-ного
раствора ВаСЬ» после чего
кипятят под тягой до
полного удаления брома.
После часового стояния не
должно появляться осадка
BaS04.
2. Содержание брома. К
100 мл 5%-ного раствора
KJ^ приливают 10 г
бромной воды и
выдерживают в течение 30 мин. в
закрытой склянке, после
чего титруют 0,1 W Na2S203.
1 мл 0,lN Na2S203
соответствует
брома.
Допустимые примеси вВг2
0,007992
Таблица 79
Примеси
1. Нелетучие вещества . , . , .
2. Органические вещества . . .
3. Сульфаты (SO/) . ."
Допустимое содержание в °/о
в химически
чистом
0,005
0,05
0,005
0,05
в чистом для
анализа
0,015
0,05
0,01
0.3
в чистом
0,03 v
0,1
0,02 .
0,5
Бумага иод крахмальная], конго, куркумовая, лакмусовая 9t
БУМАГА ИОДОКРАХМАЛЬНАЯ
Прйготовление
3 г крахмала размешивают с 25 мл воды и полученную кашицу вливают/
225 мл кипящей воды. Прибавляют 1 г KJ, 1 г кристаллического ЫаэСОз»
разбавляют 'водой до 500 мл и полученным раствором пропитывают 1це очень
плотную фильтровальную бумагу, которую высушивают в затемненном месте*
Свойства
Иодокрахмальная бумага синеет в присутствии окислителей (в частности
сВободных галоидов) и поэтому применяется для открытия этих веществ»
Хранить бумагу следует в закрытых темных банках или в пакетах из
темной бумаги.
Испытание
При нанесении на бумагу одной капли 0,01 N раствора NaN02 в соляной
кислоте (1 : 10 по объему) должна появиться синяя окраска. От нанесения на
бумагу одной капли соляной кислоты (1 : 10 по объему) посинения не должно
быть. .. ■- .
БУМАГА КОНГО
Приготовление
Растворяют 3 г конго-рот в 400 мл воды и полученным раствором
пропитывают фильтровальную бумагу, которую затем высушивают.
Свойства
От кислот синеет, от «щелочей снова краснеет (переход от синего цвета
к красному совершается при рН = 6).
Испытание.
При нанесении на бумагу одной капли 0,0Ш раствора НС1 цвет бумаги
должен изменяться от яркокрасного к синему.
БУМАГА КУРКУМОВАЯ
П риготовление
5 г куркумы настаивают в 40 мл спирта в теплом месте при частом
взбалтывании. Раствор сливают, разбавляют смесью 120 мл спирта и 160 мл
воды и полученной жидкостью пропитывают фильтровальную бумагу,
которую затем высушивают.
Свойства
Желтая бумага, окрашиваемая щелочами в бурый цвет. Тот же эффект
получается при действии борной кислоты.
Испытание
При нанесении на бумагу одной капли 0,01 N раствора NaOH цвет
бумаги должен изменяться от желтого к бурому.
БУМАГА ЛАКМУСОВАЯ
Приготовление
1. Для приготовления синей лакмусовой бумаги 1 ч. лакмуса йастаивают
в 6 ч. воды, часто взбалтывают, фильтруют и, разделив фильтрат на две
части, прибавляют к одной половике слабой НзР04 (в крайнем случае
98
Бумага. Висмут
Н2ЗО4) до слабого покраснения. Смешивают обе части фильтрата и под£
ченным раствором пропитывают фильтровальную бумагу, которую высуц
вают в затемненном месте, свободном от кислых и щелочных паров.
2. 2 г тонко измельченного азолитмина растворяют в 200 мл вод
отфильтровывают и добавляют к фильтрату 50 мл чистого спирта Ч р§
делив раствор на 2 части, добавляют к одной из них 6—8 мл 0,Ш Na(L
до посинения, к другой — 6—9 мл 0,Ш H2SO4 до красного цвета жидкое^
После стояния в течение суток растворы фильтруют и применяют для пр
готовления красной и синей бумаги как в сп. 1.
Свойства
Синевато-фиолетовая (синяя) или фиолетово-розовая (красная) бумаг.
меняющая свой цвет от кислот и щелочей. Первые вызывают покраснение
вторые — посинение. Переход красной окраски в синюю совершается пр
рН = 6. переход синей окраски в красную при рН = 7. В лабораториях сл|
дует хранить лакмусовую бумагу в плотно закрытых банках.
Испытание
При нанесении на красную бумагу одной капли 0,0Ш раствора NaO£
цвет бумаги должен меняться от розового (красного) к бледносинему. Пр|
нанесении на синюю бумагу одной капли 0,0Ш раствора НС1 цвет бумаг:
должен меняться от синего к бледнорозовому.
БУМАГА СВИНЦОВАЯ
Приготовление
Пропитывают фильтровальную бумагу 10%-ным раствором уксуснокис^
лого свинца и высушивают в помещении, совершенно свободном от серо4
водорода.
Свойства
Свинцовая бумага чернеет в присутствии сероводорода (образование
сернистого свинца, PbS); применяется для открытия следов ШЭ.
БУМАГА ТРОПЕОЛИНОВАЯ (00)
Приготовление
Пропитывают фильтровальную бумагу непрофильтрованным 3%-,ным водным
раствором тропеолина 00. При сушении темная вначале бумага становится
желтой.
1
Свойства
Желтая бумага, меняющая цвет в кислой среде на розово-красный.
Переход красной окраски обратно в желтую совершается в пределах. рН щ
1,3-8,2.
И спытание
При нанесении на бумагу одной капли 0,0lJV раствора H2SO4 цвет,
бумаги должен меняться от желтого к розовому.
БУМАГА ФЕНОЛФТАЛЕИНОВАЯ
Приготовление
Растворяют 1 г фенолфталеина в 100 мл 95%-ного спирта, прибавляют
при взбалтывании 100 мл воды и полученным раствором пропитывают
фильтровальную бумагу, которую затем высушивают.
* Р. М. Содоловская, Лабор. практика, 15* J4 2—3* 23
(1940).
Вумага свинцовая, тропёояиновая и фенолфталеиновая 99
Свойства
Белая фенолфталеиновая бумага в щелочной среде становится малиново-
пясной. Превращение бесцветного вещества в красное происходит в пре-
дРелах РН = 8,2-10,0.
Испытание
При нанесении на бумагу одной капли O.OliV раствора NaOH бумага
должна окрашиваться в розовый цвет.
ВИСМУТ
Bismuthum Bismuth Wismut
Bi At. в. 209,00
А. МЕТАЛЛИЧЕСКИЙ ВИСМУТ
Приготовление
1. Металлический висмут легко получить восстановлением окиси висмута
В1гОз водородом. Восстановление производится совершенно аналогично
получению Fe из ИегОз (см. Железо, I, стр. 128).
2. По Леве для очистки технического металла поступают следующим
образом: растворяют металл в HNCh, фильтруют; к фильтрату прибавляют
воды до прекращения образования осадка и приливают NaOH и глицерина
для растворения всего Bi (остаются нерастворенными соли Fe, Ni и др.).
Щелочной раствор фильтруют, прибавляют несколько капель! раствора
№гСОз и после 12-часового стояния вновь фильтруют. К фильтрату
добавляют глюкозы (в 4—5 раз больше веса взятого висмута) и оставляют стоять
в теплом месте, причем выпадают Ag и Си. Фильтруют и нагревают
жидкость на, соляной бане1, в результате чего. выпадает висмут в виде серого
порошка. Его отфильтровывают, промывают сначала слабым раствором
NaOH, затем водой до полного обесцвечивания промывных вод и после
этого — очень разбавленной H2SO4. Наконец,, окончательно промывают
горячей водой до исчезновения реакции на SO*" (от ВаСЬ не должно появляться
мути) и высушивают. Полученный препарат можно сплавить.
3. Для получения химически чистого металла по Милиусу и Г р о-
шуффу2 растворяют чистый Bi(N03)3• 5ШО при 18° в половинном
количестве 8%-ной HNOa (причем не должно быть мути от метаоловянной
кислоты) и раствор смешивают с равным по весу количеством
концентрированной HNOa (уд- в. 1,4). При охлаждении до 0° или до —10° при частом
помешивании прозрачный раствор затвердевает в кристаллическую кашицу,
из которой легко изолировать чистую соль Bi(NO»)s • 5НгО отсасыванием и
промыванием кристаллов небольшим количеством ледяной HN03.
Полученный нитрат нагреванием в фарфоровой чашке при 110°. переводят в
основную соль, затем прокаливанием — в ВЬОз, которую плавлением в струе
водорода (или лучше сплавлением с % ч. KCN. Яд\) восстанавливают до металла.
Дальнейшая очистка заключается в медленной кристаллизации висмута
из расплавленного металла под слоем парафина, причем в маточной жидкости
остаются все примеси за исключением Sb, которая дает с Bi смешанные
кристаллы. Полученные кристаллы нагреванием в открытом тигле
освобождают от прилипшего парафина, причем расплавленный металл окисляется
с поверхности; тогда его насасывают в предварительно нагретые
эвакуированные стеклянные трубки. Такой препарат может содержать лишь следы
кислорода, от которых освобождаются, в" случае необходимости, нагреванием
в струе водорода.
J Водяная баня, содержащая вместо воды насыщенный раствор NaCl*
* М у 1 i u s, О г о 8 с h u f f, Z. anorg. Ch., 96, 237, 245-250 (1916)»
100
BucMt/f
Свойства
Белый с красноватым оттенком блестящий металл уд. в. 9,80 при. \Ш
кристаллического строения (ромбоэдры). Очень хрупок, не обладает ни к<у[
костью, ни тягучестью. Т. пл. 271°, т. кип. 1490°. На воздухе почти ц^
изменяется, в воде при доступе воздуха окисляется в окись, которая, соедц^
няясь с углекислотой воздуха, образует блестящие иглы углекислого виа.
мута. Металл может перегоняться в атмосфере водорода. Нераствор^л
в НС1 и разбавленной H2SO4. Растворяется в HN03, концентрирован^
H2SO4 и царской водке. Из растворов солей металлический Bi вытесняете*
действием Zn, Cd, Al, Sn5 Mn, Fe.
Испытание1
Металлический висмут ч. д. а. испытывается на сурьму, мышьяк, олов^
свинец, железо, медь, серебро и другие металлы.
1. Сурьма, мышьяк и олово. При растворении 1 г металла в 6 мл HNOk
и удалении окислов азота нагреванием раствор должен быть совершения
бесцветен и прозрачен. Для дальнейших испытаний раствор разбавляется
5 мл HN03 и 10 мл воды.
2. Свинец, б мл раствора, полученного по п. 1, не должны мутнеть от
прибавления 10 мл разбавленной H2SO4.
3. Железо, медь, серебро и другие металлы. 5 мл раствора (п. I) пр#
добавлении 5 мл 10%-ного NHUOH должны выделить чисто белый осадок,
фильтрат от которого должен быть бесцветен и оставаться прозрачным npif
подкислении 3 мл НС1 (Ag). Подкисленный фильтрат от приливания K4Fe(CN)e
не должен тотчас же изменяться (Zn, Fe и др.); допускается слабое побу-
рение, но не помутнение или выделение желтого осадка (Cd, As).
Б. Коллоидальнорастворимый висмут
Приготовление 2
По Лоттермозеру3 поступают следующим образом. 10 г Bi(N03)s
растворяют в воде, подкисленной небольшим количеством HNOa, разбавляют
до 50 мл, добавляют 40 мл 50%-ного раствора лимоннокислого натрия и
делают раствор сильно щелочным, приливая ЫНЮН. В другой посуде
растворяют 7 г 'SnCl2«2H20 в небольшом- количестве воды, добавляют 50 м-4
раствора лимоннокислого натрия и нейтрализуют NKUOH. Второй раствор
вливают в первый, разбавляют водой до 1500 мл и нагревают длительное время
на водяной бане. Жидкость становится темнокоричневой и непрозрачной;
постепенно на дно осаждается коричневый осадок, который с чистой водой
снова дает коллоидный раствор.
Свойства
Черно-коричневый порошок, растворимый в воде. Из полученного
раствора щелочи снова выделяют коллоидально растворимый Bi, а кислоты и
соли — грубо дисперсный металл.
ВИСМУТ АЗОТНОКИСЛЫЙ ОСНОВНОЙ
Bismutum subnitri- Bismuth oxynitrate Basisches Wismutrritrat
cum
Magisterium
bismuti
Приготовление
1. По германской фармакопее равномерйо растирают 1 ч. среднего азотно*
кислого висмута с 4 ч. воды и вносят полученную смесь при деремешива*
^ *
1 Испытание хим. реактивов, 105.
• О получении золя В1 см. также работу А. Ф. Герасимова ЖРФХО 61. выи. 2. 26(1929%
•Lottermoaer. Uber anorganische Kollolde 59 (1901); Ргйр. Ch. 253.
Висмут металлический. Висмут азотнокислый -J01
21 ч. кипящей воды. Дают отстояться, сливают жидкость с осадка и
нИИ лк отфильтровывают. После стекания фильтрата промывают осадок
Годной водой и сушат при 30°.
2 По Корфильду и Шорту1 смешивают раствор 50 г Bi(N03)s•
МО в 50 мл НЫОз (уд. в. 1,2) с 3 л воды. Через 12 час. отфильтровы-
ют кристаллический порошок, отжимают между листами фильтровальной
I ,*гн и сушат на воздухе при комнатной температуре. Препарат отвечает
$5*Гуле 5Bi«03-5N.O.-.9H.a
Свойства
Белый микрокристаллический порошок или чешуйки с перламутровым
блеском уД. в. 4,928, нерастворимый в воде и спирте. Взболтанный с водой
показывает кислую реакцию. При 100° наступает частичное разложение с пот-
терей НгО и HNOs. При прокаливании становится желтым и при высокой
температуре плавится с образованием В12О3. Препарат имеет переменный
состав в зависимости от условий получения.
Испытание (Мерк)'2
1. Свинец, медь, щелочи и угольная кислота (РЬ* ', Си' ", К', Na* и СОз").
0,5 г препарата дол*кны полностью растворяться яа холоду в 25 мл
разбавленной H2SO4 без выделения ССЬ, образуя прозрачный раствор. 10 мл
этого раствора после прибавления избытка ЫНЮН и фильтрования должны
давать бесцветный фильтрат. 10 мл того же сернокислого раствора,
разбавленные 100 мл воды, после полного осаждения висмута сероводородом
должны' давать фильтрат, оставляющий после выпаривания досуха и
прокаливания не более 0,5 мг осадка.
2. Хлор (СГ)- Раствор 0,5 г препарата в 5 мл HNCb (уд. в. 1Л53) от
прибавления AgN03 не должен изменяться.
3. Сульфаты (SO*"). К раствору 2 г препарата в 5 мл HNOs (уд. в. 1,4)
добавляют 20 мл воды и 0.5 мл 10%-ного раствора Ba(NOs)2, причем не
должно появляться мути или осадка.
4. Аммоний (NH4*). При нагревании 1 г препарата с 10 мл раствора
NaOH (уд. в. 1,3) не должен выделяться аммиак.
5. Нелетучий остаток .(В120з). 1 г препарата при прокаливании должен
оставлять 0,79—0,82 г остатка Bi203.
6. Мышьяк. Нагревают il г препарата с НС1. Полученный раствор от
добавления концентрированного раствора SnCU в НС1 (реактив Беттендорфа)
должен оставаться бесцветным.
ВИСМУТ АЗОТНОКИСЛЫЙ
Bismuthum nitricum Bismuth nitrate ~' " Wismutnitrat
Bi(N03)3 • 5Н20 Мол. в. 485,10
Приготовление
1. Для получения чистого В1(ЫОз)з • 5Н20 нет необходимости исходить из
висмута, свободного от As. По нижеприведенному способу можно из
продажного, содержащего As висмута получить препарат высокого качества.
В колбе, емкостью около 3 л, смешивают 625 мл воды с 625 г технической
HNOs. Колбу помещают в наклонном положении на водяную баню и
нагревают последнюю. Как только температура азотной кислоты достигает 80°,
пламя удаляют и постепенно вносят в колбу маленькими порциями 250 г
висмута в порошке. Если выделение окислов азота станет слишком бурным.
1 С о г f i e 1 d, S h о г t, Pharm. J. (4], 59, 81 (1924).
* См, также Испытание хим. реактивов, 102,
102
Висмут
задерживают прибавление дальнейших порций висмута, выжидая, пока ^л
растворится ранее прибавленное количество. (Ввиду сильного выделения яд<£.
витых окислов азота необходимо операцию растворения проводить под хоро*
шей тягой.) Наконец, главная часть висмута растворится и останется незна*.
чительный сероватый остаток, состоящий из металлического и мышьяковв*
стого висмута, почти не растворимого з азотнокислом растворе Bi(NOs)».
Жидкость охлаждают и- дают ей отстояться. Прозрачный раствор сливаю^
и остаток фильтруют через двойной фильтр. Выпариванием раствора в фар,
форовой чашке до кристаллизации получают чистый ВЦгЮз)з - бШО.
2. По Эрдману1 100 г технического висмута истирают в порошок jf
нагревают в никелевой чашке с 50 г NaNOa до слабого каления. После
полного окисления висмута массу выщелачивают кипячением с раствором 20 а
NaOH в 400 мл воды. При этом 'As и Sb переходят в раствор; окись висмуте
отсасывают на платиновом конусе и растворяют при кипячении в смеси
140 г концентрированной HNOs с 200 мл воды. Раствор фильтруют через
асбестовый фильтр и выпаривают до кристаллизации. (Маточный раствор
можно использовать, выпаривая его и выливая в 500-кратный объем воды.
Выпадающую основную соль отсасывают и высушивают на фильтровальЕГой
бумаге.)
3. Об очистке продажного препарата см. Висмут, 3 (стр. 99).
Свойства
Бесцветные триклинические призмы уд. в. 2,830, плавящиеся при 78° в
своей кристаллизационной воде с образованием основной соли. При 150°
полностью теряет кристаллизационную воду и при 200° разлагается с
выделением N02. В эксикаторе над КОН уже при обычной температуре
распадается, теряя воду и HNOs. Водой разлагается с выпадением основной соли.
В смеси с измельченным сахаром растворяется в воде значительно лучше.
Растертый с маннитом может дать чрезвычайно концентрированный раствор.
Растворим в кислотах и глицерине при нагревании. Растворим в ацетоне
(при 0° в отношении 48,66 : 100, при 19° в отношении 41,70 : 100).
Испытание
Согласно ОСТ НКТП 2855 содержание Bi2Os в препарате ч. д. а. должно
быть не менее 47,5%; в препарате чистом не менее 47,0%. Наибольшие
количества допустимых примесей указаны в табл. 80. , _
Таблица 80
Допустимые примеси Bi(N03)3• 5Н20
Примеси
1. Вещества, нерастворимые в HN03. . .
2. Хлориды (СК)
3. Щелочные и щелочноземельные ме-
4. Железо (Fe)
Допустимое содержание в °/0
в чистом для
анализа
0,003
0#02
0,01
0,0002
ч
в чистом
0.005
0,005
0,03
0,0005 •
* Хим. преп. 46,
Кроме того, препарат должен выдерживать приведенные в ОСТ
испытания на медь (Си) и свинец (РЬ). -
а) На ме^ь- Ю г препарата растворяют в 40 мл HNOs (уд. в. 1,15), до-
. лЯЮт 40 ла 10%-ного NH40H, хорошо перемешивают и дают отстояться.
Затем 50 жл прозрачного раствора переносят в цилиндр на 50 мл. Раствор
ои рассмотрении по оси цилиндра должен быть бесцветным (без голубой
окраски).
б) На свинеЧ- * г препарата при растворении в 50 мл H2SO4 (уд. в. 1,10)
пРи комнатной температуре должен давать прозрачный раствор без осадка.
При стоянии из раствора могут выделяться соли висмута.
ВИСМУТА ОКИСЬ (ОКИСЬ ВИСМУТА [3])
Bismutum oxydatum Bismuth oxyde Wismutoxyd
Bi2Oj Мол. в. 466,00
А. Аморфная окись висмута
Приготовление
1. Bi20s получается при прокаливании до постоянного веса углекислого
или основного азотнокислого висмута. При высокой температуре (например в
горне) В1гОз сплавляется и разрушающе действует на фарфоровый тигель.
2. По Штегеру1 продажный ВЦЫОз)з взмучивают с большим
количеством воды, нагревают полученную молочно-белую жидкость и добавляют
твердого КОН.
Образующийся сначала белый Bi(OH)3 выше 70° переходит в желтый
осадок Bi20s. Последний отфильтровывают, промывают горячей водой до тех
пор, пока фильтрат -не перестанет показывать щелочную реакцию на фенол*
фталеин, и сушат абсолютным спиртом или в шкафу при 250°.
Свойства
Желтовато-белый порошок уд. в. 8,756, становящийся при 704° красно-
желтым и плавящийся при 860° в красно-коричневую жидкость уд. в. 8,929.
Расплавленная Bi2Os при охлаждении закристаллизовывается. Не растворима
в воде; растворима в сильных кислотах.
Б. Кристаллическая окись висмута
Приготовление
По Мьюэр»у и Хетчинсону2 слабо подкисленный НЫОз раствор 1 ч.
ВЦгЮз)з смешивают с 2—2,5 ч. KCN (сильная тяга — яд!) и жидкость, вместе
с выпавшим бурым осадком, кипятят 10---15 мин. и фильтруют. Осадок
обрабатывают несколькими порциями кипящего раствора КОН, пока
промывная жидкость не станет совершенно бесцветной. Препарат представляет собой
темносерый кристаллический порошок, содержащий еще незначительные
количества углеродистых и азотистых веществ. Окончательно очищается
нагреванием до красного каления в токе воздуха или кислорода. После очистки —
ромбические кристаллы.
1 S t e g е г, Диссертация, Берлин, 1913.
2 М u i г, Н ц t с h i п s о n, J. Chem. Soc. 55, ИЗ (1889). ,
Висмут. Вода
ВИСМУТ ХЛОРИСТЫЙ (ХЛОРИД ВИСМУТА [3])
Bismuthum chloratum Bismuth chloride Wismutchlorid
Bids Мол. в. 315,37
П риготовление
1. Растворяют окись висмута (Bi203) в НС1 (или металлический Bi в цар*.
ской водке), раствор выпаривают досуха и сухой остаток перегоняют npg
220—230° в струе СОэ или хлористого водорода, очищенного от пыли и влагчс
2. Для получения препарата высшей чистоты можно воспользоваться
методом, которым получали BiCls Генигшмидт и Биркенбанх1 при опре»
делении атомного веса висмута.
Фарфоровую лодочку с о г металлического Bi вносят в трубку очен|
тугоплавкого стекла, помещенную в печи для элементарного анализа таким
образом, что оба конца ее немного выступают из печи и оттянуты. Аппарат
заполняют абсолютно сухим воздухом, перед хлорированием через трубку
пропускают сухой азот для вытеснения всего воздуха. (Азот получают обычным
путем, т. е. пропускают воздух, насыщенный парами 1МНз, через раскаленную
медь, поглощают жбытк NHa 50%-мой H2SO4 и удаляют всегда
содержащиеся Не и О» пропусканием через раскаленную докрасна трубку, одна по4
ловина которой наполнена металлической медью, а другая — окисью меди).
После вытеснения вовдуха начинают пропускать чистый сухой хлор. Для
нагревания лодочки с Bi служит электрическая печь, состоящая из кварцевой3
трубки, снабженной нихромовой обмоткой и легко передвигаемой по длине'
сожигательной трубки.
Уже при сравнительно низкой температуре начинается образование BiCW
который конденсируется на холодных частях трубки. Во избежание возмож^:
ных потерь эти места охлаждают, обматывая их влажной шерстяной ниткой.
Приблизительно через час образование BiCta заканчивается. Аппарату дают>
остывать в токе азота, после чего трубку наполняют сухим воздухом. При
перенесении образовавшегося BiCU в другие сосуды следует работать
тщательно и возможно быстро, оберегая препарат от доступа влажного воздуха.
3. По Сингаловскому2 к 100 г металллического Bi в 1500 мл НС1
(уд. в. 1,19), постепенно при помешивании, прибавляют 500 мл HN03 (уд. в.
1,4) до полного растворения. Жидкость фильтруют и, упарив фильтрат вдвое,
оставляют на холоду для кристаллизации. Выпавшие кристаллы отсасывают
и промывают смесью 80 мл НС1 (уд. в. 1,19) и 120 мл воды.
4. Растворяют ВЬ*03 в крепкой HC1. Выпарив раствор до густоты сиропа,
оставляют его на 2—3 дня для кристаллизации. Кристаллы отсасывают,
просушивают и сохраняют в плотно закупоренной банке. Препарат высокого
качества получается только из химически чистых исходных веществ3.
Свойства
Возогнанный BiCh представляет собой снежнобелые, с алмазным блеском
кристаллы уд. в. 4,755. При 232,5° плавится в жидкость, кипящую при 447°.
На воздухе расплывается. Водой разлагается с образованием основной соли
(BiOCl). Растворяется в спирте, ацетоне и соляной кислоте. При4 возгонке на
воздухе образуется BiOCl.
Испытание4
Препарат ч. д. а. должен содержать не менее 73% ВЬОз, не больше
0,004% не растворимых в НС1 примесей и не более 0,02% солей щелочных
металллов. Кроме того, препарат должен выдерживать испытания на
сульфаты (проба с БаСЬ после растворения препарата в НС1), медь (отсутствие
iHonig.echmidt, Bircketibanch, Z. i. Elektroch. 19/20, 403 (1920)! Pr2p. Chu, 250.
* Соли редких и цветных металлов, 153*
■ Химич. реактивы, 54.
"* ДОспытагие хим. реактивов, 106.
голубого окрашивания жидкости при обработке препарата" 10%-ным ЫНЮН),
свинец (проба с Л H2SO4 после растворения BiCh в i-ЩОз), мышьяк (проба
с реактивом Беттендорфа; см. стр. 101) и железо (проба с NKhCNS).
Aqua
ВОДА
Water
Н,0 Мол. в. 18,016
Wasser
Очистка
1. К обычной водопроводной или колодезной воде прибавляют несколько
кристаллов КМп04 и кипятят в круглодонной колбе или реторте, проводя
образующиеся пары через стеклянный холодильник и собирая конденсат в
охлаждаемом водой стеклянном приемнике.
2. Для получения «абсолютно чистой» воды возможно более чистую
дестиллиров)анную воду перегоняют еще раз из платиновой ил,и серебряной
реторты с таким же холодильником
(стекло и фарфор постепенно
разъедаются) V отбрасывая первую часть
. дестиллата.
Если употребляемая вода содержит
аммонийные соли, хлориды и
органические вещества, то их перед
перегонкой «обезвреживают» следующим
образом. Если вода содержит следы
аммонийных солей, особенно (NKU^COs, и
не содержит хлоридов, то достаточно
прибавить для связывания Nth' 0,5—
1 г квасцов на каждый литр воды.
При одновременном содержании
хлоридов добавляют на 1 л 0,66 г Na2HP04.
Если вода богата органическими
веществами, их разрушают, добавляя 0>025 г
КМП04 на 1 л.
Вначале добавляют к воде раствор
&МпС>4, через полчаса прибавляют
раствор квасцов [KAl(S04)a* 12K?0], еще
часом позднее раствор Na2HP04. По
истечении еще получаса жидкость
фильтруют в круглодонную колбу,
кипятят около 10 мин. и затем начинают
перегонку.
3. Для приготовления
стерилизованной дестиллированной воды Л. В а-
нино2 рекомендует употреблять прибор, изображенный на рис. 19.
Холодильник этого прибора состоит из трех частей, благодаря чему уменьшается
возможность поломки. Колпачок А служит для предохранения от попадания
в препарат микробов во время перегонки.
4. О. Генигшмидт3 в своих точнейших работах по определению
атомных весов элементов для получения вполне . чистой воды исходит из обычной
дестиллированной воды, перегоняя ее дважды со щелочным раствором КМгЮ4
в стеклянной аппаратуре. Затем дестиллат перегоняется третий раз из колбы
стекла пайрекс с оловянным холодильником, непосредственно соединенным
Рис. 19. Прибор для перегонки воды
1 Некоторые авторы [Honlgschmidt, Вег., 54, 1853 (1921) считают, что вода,
перегнанная в серебряной аппаратуре, содержит некоторое количество ионоз Ag. „
1 Ргйр. Сп., 8.
8 О. HunigschmidtH R. Sachtleben, Z. anorg. Ch„ 221, 70 (1534); О. Н б п I г-
s с h m i dt и R. S с h 1 e e, Z. anorg. Ch. 221, 134 (1934). *
У
106 ; ' Вода.
с горлышком колбы, без каких-либо уплотняющих материалов. Чистая водJ
собирается в колбу иенского стекла, в течение многих лет служащую w
этой4 цели. о г
5. Для получения воды, вполне свободной от металлов, перегоняют ее
с многократно изогнутым кварцевым холодильником *.
6. Очень чистая вода, пригодная для измерений электропроводности, полу,
чается дробным вымораживанием дестиллированной воды. Первые порцнн
льда наиболее чисты 2.
7. Удаление С02 из воды по Нидалю и Арнфельту3 легко достн*
гается продуванием чистого воздуха через воду при пониженном давлении.
Так, при пропускании 2,7 л воздуха через 5 л воды в течение 40 мин. при
20—50 мм содержание ССЬ в воде было снижено до 1 • 10~5 молей на 1 л,
8. А. Гунц4 указывает на возможность получения очень чистой вод^
с удельной электропроводностью около 1 • Ю-6 мо, пропуская пар из колбы
через электрический пароперегреватель (с нихромовой спиралью) и затем через
пористую стеклянную пластинку.
Дестиллат содержит только СОг и следы ЛМНз. Если исходная вода
содержит значительные количества NHs, рекомендуется при перегонке добавлять
к ней немного (NH^SsOe.
9. Получение значительных количеств воды с электропроводностью 0,3 • 10-*—.
0,3-Ю-6 мо может быть осуществлено в аппарате, описанном Ф. Штрау-
б о мб и изображенном на рис. 20. Через трубу А в перегонный куб В
подается дестиллированная вода, полученная конденсацией пара (например из
парового отопления) при температуре немного ниже 100° (очень важно, чтобы
часть пара не конденсировалась). Скорость подачи воды около 15 л /час.
Через трубку С непрерывно приливается для связывания NH3 раствор
хромовой смеси (10 г Na2Cr2'07 и 9 мл конц. H2SO4 на 5 л воды) со скоростью
30 мл/мин.. Избыток воды, не успевающий дестиллироваться, стекает по
сливу D, который служит одновременно регулятором уровня. Оригинальным
устройством, обеспечивающим высокое качество дестиллата, является трубка Е,
подводящая непосредственно под поверхность воды в кубе воздух,
очищенный от СО» и NHa. Электрическим нагревателем поддерживается скорость
дестилл.яции около 9 л/час, причем холодильник отрегулирован так, чтобы
дестиллат имел температуру выше 70°. Полученная чистая водав имеет
рН = 6,8.
10. Получение еще более чистой воды с электропроводностью7, близкой
к теоретической величине (5,52* Ю-8 мо), является весьма сложной операцией.
Лабораторная установка для этой цели, предложенная П. Тиссеном и
К. Германом8 схематически изображена на рис. 21. Исходным сырьем
является обычная вода «для измерения электропроводности» с % ^ 2 • Ю-6 мо,
полученная перегонкой дестиллированной воды в колбе иенского стекла с
оловянным холодильником.
В колбу К\ через трубку А, снабженную пришлифованной пробкой,
наливают 5 л такой воды. Печь Oi регулируют так, чтобы температура воды была
80—90°. Затем через трубку Ri, оканчивающуюся пористой стеклянной
пластинкой Р, пропускают чистый азот, очищенный от СОг и NH3 (для этой цели
его пропускают последовательно через три склянки с концентрированным КОН,
через концентрированную H2SO4 и через три промывных склянки, наполненные
водой «для измерения электропроводности», распыляя газ на мельчайшие
пузырьки с помощью пластинок пористого стекла S). Краны Hi и #з открыты,
1 I. S, М с Н а г g u е а. Е. В. О f f u 11, Ind. Eng. Chem. Anal. Ed. 12, 157 (1940).
■ Ф. Э ф р а и м Неорганическая химия, ч. 1, стр. 349, Госхимтехиздат 1932.
8 F. N у d a h 1, Н. А г п f е 11, Svensk. kem. Tidskr. 62, 17 (1940). С. 1940, II, 797.
4 A. A. Gu n t z. С. I, 2561 '1941); II, 2058 П940).
* F. G. S t г a u b, Ind. Eng. Chem. Anal. Ed. 7, 433 (1935).
• Для отделения капелек масла, содержавшихся в первичной дестиллированной воде,
рекомендуется включить перед трубкой А сепаратор F, устройство которого ясно из рисунка*
7 Уместно напомнить здесь, что наиболее чистая вода (удельная электропроводность х«б° =э
= 5,8.10—8 мо) была получена Кольраушом в 1894 г. 42-кратной перегонкой воды под
уменьшенным давлением в стеклянной аппаратуре, применявшейся для перегонки в течение 10 лет.
e P. A. T i e s s е п, К. Herrmann, Z. f. Elektroch., 43, № 1, G6 (1937).
Вода
107
н tf2 закрыт. Кран 'Я8 соединен с промывной склянкой с чистой водой.
^выходному отверстию этой склянки присоединена трубка с натронной
изолью Для предохранения от С02 Через 20—30 час, закрывая кран Яз,
подавлением газа выдавливают воду через R2 в кварцевую
Когда воды в Кг будет достаточно, открывают кран Я2 и
„U£i-
Воздух
Рис. 20. Схема прибора для получения дестиллированной
воды по Штраубу
вышенным
Кем прекращают
дальнейший ДО^У11 воды из К\\
коан Н\ закрывают.^
Нагревая колбу К2
электропечью Ог, перегоняют воду
в токе азота. Кварцевая
трубка Ra и кварцевая
трубка холодильника Яз
,приплавлены к колбе Кг
(чем определяется в
основном значительная
стоимость установки).
Соединение с колбой К\
производится при помощи
шлифов из кварца или
иенскогс) стекла. Также
при помощи шлифов к
холодильнику #з
присоединяется сосуд для
измерения электропроводности, служащий приемником. Все кварцевые и стеклянные
части перед сборкой очищают теплой хромовой смесью, много дней
промывают и по крайней мере 10 час. пропаривают. Несмотря на это, установка
должна быть много дней в эксплоатации, прежде чем вода начнет получаться
достаточно чистой. Горло
колбы Кг нагревают печью
Оз выше 100®, чтобы
вызвать испарение капель
воды и избежать
переброса капель >в холодильник.
Скорость дестилляции
около 400 мл I час.
Электропроводность полученной
воды %= 6,5 • J (И—8 • 10~8 мо.
И. По указанию А. И.
Б а й б а ев а 1 можно
получить очень чистую воду в
следующем приборе.
Исходная дестиллированная
вода имела *-=* 8 • Ю-6—;
13-Ю"* мо. Холодильник
из трубки иенского стекла
(d = 8 мм, / = 50 см)
соединен с дефлегматором*
а дефлегматор — с. колбой
при помощи
гидравлических затворов.
Применяется дефлегматор Г е н н и н-
г е р а-Л е б е л я с пятью
шарами или колонка Гемпеля с обрезками стеклянных трубок;
последняя дает воду лучшего качества» но работает медленнее. Перегонку ведут
так, чтобы температура конденсата была не ниже 50—70°.
Решающее значение имеет чистота стекла; промывка горячей хромовой
смесью, пропаривание в течение нескольких часов и двухкратное ополаскива-
Рис. 21.
Аппарат Тиссена для получения чистейшей дестил-
. лированной воды
1 ЖПХ 13, N 4, 499 (1940).
108
Вода
ние приемника горячим дестиллатом совершенно необходимы. Благоприятш
влияет добавка 0,3 г KHS04 на каждый литр перегоняемой воды. При (Ш
рости перегонки 500—600 мл/час из 10 л воды получается около 1,5 л пщ
вой фракции (недостаточно чистой, так как' *= 8,5-10-* — 1,6- 10-6 цЛ
остальной дестиллат имеет *.= 0,8: Ю-6 — 0,46- Ю-6 мо.
12. Г. А. Бабичев1 описывает автоматический дестиллятор2 для пощ
производительностью 1 л/час. (рис. 22).
В дно колбы А емкостью 1,5 л впаяна изогнутая трубка Т, присоединена»
к холодильнику Либиха длиной 50—60 см. Нагреватель состоит из двух кусксЙ
угольных электродов для вольтовой дуги (диаметр 10 мм, длина 70 мм). Пр|
помощи толстых медных проводов ВВ (d = 2,0—2,5 мм) угли закреплены-1
резиновой пробке С. На один из проводов надета тонкая резиновая трубка*
Чтобы избежать соприкосновения угольных электродов во время работы, на
один из них надевают кусок пробирки. Трубка D является регулятором уровад|
Рис. 22. Автоматический дестиллятор по Бабичеву
воды в колбе. Достаточно открыть водопроводный кран М, чт^Зы вода
заполнила холодильник, колбу и регулятор D. Избыток воды стекает через Н в
воронку Е, укрепленную на упругой латунной пластинке К. Под тяжестью воды
ворокка опускается и замыкает контакт U (проволока V погружается в
чашечку со ртутью). Через 3—5 мин. вода в колбе закипает. По указанию
автора аппарат, работая в течение года, показал себя очень удобным и
рентабельным. Получаемая вода3 имеет рН = 7,1—7,15.
Свойства
•■
Бесцветная (в толстом слое голубоватая) прозрачная жидкость. Т. пл.
льда 0°; т. кип. воды сильно зависит от атмосферного давления. Для
интервала 660—860 мм рт. ст. температура кипения воды может быть вычислена
с точностью до ±0,0004° по уравнению:
tp = 100 + 0,0368578 (р — 760) — 0,000020159 (р — 760)2 + 0,071621 (р — 760)3,
где р — давление в мм рт. ст. Вода является очень слабым электролитом:
ионное произведение воды при 22°
•W>=(h')(oh') = io-14>
что соответствует степени диссоциации около 0,000001%.
* Лабор. практика 14, № 1, 20 (1937).
♦ Электрифицированные дестилляторы иной конструкции описаны, напр. М. Ф.
Скоковым, Загод. Лаб\ 8, 333 (1939), П. Мельником, Лабор. практика, 13, ЛЬ 3, 30 (1938).
К' А. Гр о х о в с к и м, Сахар, 1в, М 4, 24 (1938). Производительность этих аппаратов \,Ь—2лЫас.
» Получение простой перегонкой водопроводной воды дестиллата с рН«=7,1 является мало
вероятным и должно быть поставлено под сомнение (Ю, Я.).
Вода
109
Ис
пЫтание (Мерк)
Нелетучие примеси. При выпаривании на водяной бане 100 мл испытуе-
- воды не должно оставаться весомого остатка.
MoF* Хлориды {СУ). При добавлении к 100 мл испытуемой воды нескольких
ль HNOs и (AgNOs не должно происходить никакого изменения.
каПез. Сульфаты (SOO- После Таблица 81
Изменение плотности воды
зависимости от температуры
^явления к 100 мл воды НС1
?°п в U2) и Раств0Ра ВаС1г в
ечёние 15 час. не должно обра*
1пвываться осадка.
4. Нитраты (1ЧОз). При
осторожном добавлении к 5 мл
раствора дифениламина 10 мл
воды (на границе соприкосновения
жидкостей не должно
появляться синего окрашивания.
5. Аммиак и соли аммония.
Добавление к 50 мл воды 10—
15 мл реактива Несслера не
должно вызывать изменения.
6. Тяжелые металлы и кальций. От добавления к 100 мл воды
сероводородной воды, ЫНЮН, (NH^S и (NH4)sC204 не должно происходить изменения.
7. Вещества, окисляемые КМп04. К 100 мл воды приливают 1 мл
разбавленной H2SO4, нагревают до кипения, прибавляют 1 каплю 0,1%-ного
раствора КМп04 и кипятят 3 мин., причем розовая окраска не должна
исчезнуть.
Таблица 82
Изменение объема воды в зависимости
от температуры
ГС
—10
- 5
0
4
10
<
0,99815
0,99930
0,999868
1,000000
0,999727
/°с
20
40
70
I 100
<
0,998230
0,99224
0,9778
, 0,95839
/°с
0
4
10
20
30
1 л воды,
измеренный при 4°, имеет
при г объем (мл)
1000,131
1000,000
1000,260
1001,741 ,
1004,310
(•С
33
'40
70
100 1
1 л воды, измерен*
ный при 4°, имеет
при tb объем (мл)
1005,266
1007,82
1022,7
1043,4
Таблица 83
Упругость водяного пара п
t°C
0
5
10
11
12
13
14
15
16 ,
р (мм рт. ст.)
4,579
6,543
9,209
9,844
10,518
11,231
11,987
12,788
13,634
tec
17
18
19
20
21
22
23
24
25
р (мм рт. ст.)
14,530
15,477
16,477
1 17,535
18,650
19,827
21,068
22,377
23,756
рн различных темпе
t°c
30
40
50
60
70
80
90
91
92
93 ,
р (мм рт. ст.)
31,842
55,334
92,51
149,38
233,7
355,1
525,76
546,05
566,99
588,60
t°c
94
95
96
97
98
99
100 !
105 1
по !
ратура х
р (мм рт. ст.)
610,90
633,90
657,62
682,07
707,27
733,24
760,00
906,1 .
107,46
110
Вода. Водород
Поступающую в лабораторию дестиллированную воду обычно пробуют^
хлориды и сульфаты. Необходимо всегда приводить контроль рН воды: -^
добавлении к 10 мл испытуемой воды одной капли фенолрота окраска долз)
быть желтой или желторозовой; при добавлении одной капли метилр|
окраска должна быть чисто желтой.
Таблица Щ
Поглощение водяных паров различными осушителям!
(Becker, Booth, Mclntire)
Осушитель
CuS04
ZnBr2
ZnCl2
CaCi2
CaBr2 1
CaO
NapH
BbOg
Упругость
пара над
осушителем
мм рт. ст.
—
—
0,14-0,25
—
—
0,160
Остаточная
влажность мг
Н»0 на 1 л
воздуха при 25°
1,40
1,10
0,80
0,36
0.20
0,20
0,16
0,15
Осушитель
MgO
КОН
ккОз
HjSOt
Ba(CIQ0,
Mg{Cio4)2 ;
Р2о,
Упругость
пара над
осушителем
мм pt. ст.
0,008
0,002
0,003
0,0014
0/ХЮ
0,000
Остаточна!
влажно :ть щ
Н,0 на 1 л
воздуха приЩ
0,003,
0,003
__
_*
—«.
—
Hydrogenium
Н2
ВОДОРОД
Hydrogen
At. в. 1,008 Мол. в. 2,016
Wasserstoff
Приготовление
1. Обычный способ получения водорода состоит в действии на цинк HU
(1 об. НС1 уд. в. 1,19 на 1 об. воды) или H2SO4 (1 об. H2SO4 уд. в. 1,84 §
8 об. воды). Операцию удобнее всего проводить в аппарате Киппа. Если цин
очень чист, то выделение газ
происходит медленно. Для ускс
рения добавляют несколько ка|
пель раствора C11SO4 (при пр|
Рис. 23. Прибор для получения водорода
(С. К. Дэвилль)
Рис. 23а. Деталь тубусной
склянки
Мененйи НС1—"несколько капель СиС1г). Медь выделяется на цинке и?
образует с ним гальваническую пару.
При отсутствии аппарата Киппа можно воспользоваться прибором,
составленным из двух тубусных склянок, соединенных резиновой трубкой
(1>ис. 23).
Водород ^ . //;
Пои открывании крана А кислота из сосуда С переливается в сосуд В и
гирует с находящимся там гранулированным цинком. По миновании на-
Ре^ности кран А закрывают и давлением выделяющегося водорода кислота
^тесняется обратно в склянку С. При пользовании аппаратом необходимо,
ВЫ бы тубус склянки В находился у самого дна; если же он помещен выше,
ЧТ° вставляют в тубус изогнутую стеклянную трубочку, как изображено на
*?ИС2 Водород высокой степени чистоты может быть получен лишь методом
ктролиза. для этой цели Вез и Лабатю1 предложили следующий
аппарат (рис. 24). В стеклянный сосуд, содержащий в качестве электролита
30°/ -ный раствор NaOH, помещен колокол, соединенный верхней частью
с трехходовым краном. Электродами1 служат два листа никелевой жести.
Один из них (катод) помещен внутри, другой (анод) снаружи колокола.
Борис. 24. Аппарат для электролитичв^
ского получения водорода
Рис.
25. Электролитическое
получение водорода по Сивкову
круг колокола жидкость покрывают тонким слоем масла для предохранения
от испарения.
Сначала отсасывают воздух из колокола так, чтобы электролит заполнил
весь колокол и кран. Затем кран закрывают и включают постоянный ток
(МО б), регулируя реостатом силу тока в пределах 10 а. Вследствие
выделения водорода уровень электролита внутри колокола понижается, и, наконец,
весь катод выходит из жидкости, вследствие чего ток прерывается. (При 10 а
и емкости колокола — до нижней части катода — в 1 л это наступает через
20—15 мин.)2.
1 Ve z e s, Т. La bat ut, Z. anorg. Ch., 32, 464 (1902); Prap. Ch. 2.
f В момент, когда вследствие понижения уровня электролит* катод едва соприкасается с
жидкостью, жесть мгновенно накаляется и слышится шум, как при употреблении
прерывателя Венельта. Если аппарат плохо уплотнен, так что внутрь может попасть воздух, или
полюсы переключены без предупреждения, то в колоколе образуется взрывчатая смесь, которая
от раскаленной жести воспламеняется. Поэтому употребляют следующее приспособление: квтод
соединен с вертикальным никелевым стержнем, опускающимся на несколько сантиметров ниже
край электрода. Нижний конец стержня укреплен в дне фарфоровой чашки, отверстие
которой (несколько сантиметров шириной) находится на 1 см ниже края катода, так что если даже
жесть и не соприкасается с электролитом, то ток продолжает итти через стержень, сечение
которого подбирают так, чтобы сила тока не уменьшалась значительно.
При дальнейшем опускании электролита ток прерывается в момент, когда уровень жид-
ости опустится ниже отверстия фарфоровой чашки.
112
Водород
3. И. Сивков1 описал несколько упрощенный аппарат Веза и Лабатю*
легко изготовляемый в лаборатории. Электродами служат железные сет^
Электролит —30%-ный раствор КОН. Производительность аппарата 15 л/Щ
(рис. 25).
4. Еще более простой аппарат для получения водорода электролизом
3— Ю%-ного раствора NaOH, описанный К. И. А з и х и н ы м2, изображен
на рис. 26. Железный анод А помещается внутри трубки С. Катод -Щ
из железной сетки окружает трубку С. Выделяющийся водород, собираясь в
банке Д вытесняет избыток электролита в
сосуд Е. Скорость электролиза регулируется
ламповым реостатом. По мере надобности водород
расходуется через трубку F.
5. Для получения больших количеств газа
Пар со не предложил аппарат (рис.27),
вполне пригодный для получения и других газов
(H2S, С02).
Г
1
°\
п\
Шр-
Рис. 26. Прибор для
получения водорода
по Азихину
Рис. 27. Аппарат Парсонса
Аппарат состоит йЗ Трех частей: резервуара с кислотой А, приёмника В
для использованной кислоты и газовыделителя С, наполненного
гранулированным цинком. Подходящие размеры аппарата следующие: для сосуда В —
высота 350 мм и диаметр 280 мм\ колонка С, представляющая собственно
газовыделитель, состоит из колокола 220 X 250 мм* служащего сборником газа
и имеющего внизу несколько отверстий для свободного вытекания использо*
ванной кислоты. Вверху колокол переходит в цилиндр высотой в 450 мм и
диаметром просвета в 100 мм\ четырьмя имеющимися внизу цилиндра пере"
тяжками удерживается сетчатая пластинка D, поддерживающая, в свою
очередь, загруженный в цилиндр цинк. Цилиндр с колоколом лучше всего делать
из одного куска; однако если материалом для аппарата служит стекло, То
можно газовыделитель изготовить из двух частей, пришлифованных друг
к другу. Сверху слоя цинка помещают немного стеклянной ваты для равно*
1 Завод, лаб. 3, № 2, 165 (1934).
» Завод, лаб. 2, № 5, 47 (1933).
Водордд
113
г0 распределения кислоты, которая, в противном случае, образует не-
^ ько каналов, по которым стекает вниз, без достаточного использования.
сК°Отводная трубка наверху колонки С должна иметь диаметр около 10 мм.
сосуд Л сделан из непрозрачного материала, то находящаяся снаружи
уда А кислотомерная трубка показывает расход кислоты, концентрация
С°торой берется, как указано в способе 1. Во время работы аппарата давле-
KZe газа определяется высотой (ху) столба отработанной кислоты в сосуде В,
пичем эта высота при указанных на чертеже размерах аппарата не должна
превышать-250 мм.
Как только давление превысит эту величину, газ, вытеснив кислоту,
заполнит весь колокол и, выходя из отверстий, имеющихся внизу, начнет бар-
£ОТИ.ровать через кислоту в атмосферу.
6. Очень чистый водород получается действием крепкого раствора NaOH на
меТаллический алюминий. ^
Очистка
При употреблении недостаточно чистых цинка й кислоты выделяющийся
водород может содержать следующие примеси: фосфористый и мышьяковистый
водород, сернистый ангидрид, закись и окись азота, угольный ангидрид, окись
углерода, азот, сероводород, кислород и углеводороды. Пригодный Для всех
целей газ получается пропусканием неочищенного водорода через раствор 100 £
К2СГ2О7 и 50 г концентрированной H2SO4 в 1 л воды (или через щелочный
раствор КМп04) и сушением газа СаСЬ или Р2О5. Очищенный Таким спосо*
бом водород содержит еще незначительную примесь воздуха; для удаления
кислорода, часто оказывающего вредное действие, газ пропускают через рас*
каленную стеклянную трубку, содержащую восстановленную медь1* Приме*
шанный азот удаляется с большим трудом»
Свойства
Бесцветный газ без запаха и вкуса, горящий бледйоголубым пламенем*
Сгущается в прозрачную жидкость (уд. в. 0>07), кипящую при —#52,79°. При
еще более низкой температуре затвердевает в прозрачные кристаллы,
плавящиеся при —257,14°. В жидкостях растворим очень мало: 1 об. спирта
поглощает при комнатной температуре 0,069 об. Н2 (растворимость в воде
приведена ниже в табл. 85).
1 л водорода весит 0,08995 г при нормальных условиях. Многие металлы
в раскаленном состоянии (Pd, Pt, Fe, Си) поглощают значительные коли*
чества водорода. С кислородом при 180° соединяется медленно, при 600° — со
взрывом. С фтором бурно соединяется при всех условиях, с хлором энергично
реагирует только при освещении солнечным светом. Вообще для водорода
характерны пассивность на холоду и чрезвычайная активность при высокой
температуре.
Примечание 1. При нагревании или поджигании водорода еле*
дует соблюдать большую осторожность ввиду возможного взрыва.
Всегда необходимо собрать газ сначала в пробирку и поджечь:
нечистый газ взрывает; более чистый дает вспышку, сопровождаемую резким
звуком; чистый загорается спокойно.
Примечание 2. Для предохранения от проскакивания
водородного пламени внутрь газовыделительного прибора Фрезениус
применяет стеклянную трубку длиной 10 см и диаметром 10 см, в которой
помещены 10 кружков из мелкой проволочной сетки, плотно прилегающих
к стенкам трубки и отделенных друг от друга комочками ваты. Трубка
ставится перед газовыделительным аппаратом.
1 Деберейнер рекомендует удалять кислород, помещая в газ на некоторое время
губчатую ппатину.
114
Водород
Таблица g$
Растворимость Н2 в воде
(Winkler)
гс
0
5
10
15
20
25
30
35
1 об. воды
поглощает
об. газа,
привел, к норм,
условиям
2,148-10-2
2 014.10-2
1,955-10-2
1,883-10-2
1,819-10-2
1,754-10-2
1,699-10-2
1,666-10-2
100 г воды
поглощают при 760 мм
г газа
1,922*10-*
1,824-10-*
1,740-10-*
1,668-10-*
1,603-10-*
1,535. i0~*
1,474.10-*
, 1,425- 1Q-*
/«С
40
45
50
60
70
80
90
100
1 об. воды
поглощает
Об. таза,
привел, х норм.
условиям
1,644-10-2
1,624.10-2
1,608-10-2
1,600-10-2
1,60). 10-2
1,600-10-2
1,600-10-2
1,600-10-2
100 г воды погдо»
щают при 763 M*i
с газа
1,384.10-*
1,388 • 10-*
1,287- Ю-*
1,178-10-*
1,021-10-*
0,790-10-*
1 0,461 • 10-*^
""""
ВОДОРОД МНОГОСЕРНИСТЫЙ
Hydrogen polysulfides Wasserstoffpolysulfide
HsSn
Приготовление
1. Э. А. Остроумов и Г. С. Масленникова1 описывают разрабо*
тайный ими удобный метод получения многосернистого водорода- В ка*
честве исходного материала берется многосернистый натрий, легко
получаемый из ЫагБ и S по Блоху и Гену следующим образом. В
фарфоровый стакан емкостью 2 л, снабженный стеклянной мешалкой и термоме*
тром, помещают 500 г чистого кристаллического сернистого натрия (Na2S •
• 9Н20) и» нагревают на электроплитке, пока соль не расплавится в своей
кристаллизационной воде. В однородный плав при механическом
размешивании вводят маленькими порциями 250 г тонкого порошка серы в течение
3—4 час. Температуру поддерживают в интервале 70—90°. После прибавле*
ния всей серы продолжают нагревание еще некоторое время, затем
добавляют 400 мл горячей воды» перемешивают и переливают раствор в
нагретую склянку. Охлаждение проводят, закрыв склянку часовым стеклом; затем
плотно закрывают склянку резиновой пробкой и оставляют на двое суток.
Получение H2Sn проводят в' большом фарфоровом стакане емкостью 4 л,
помещенном в смесь льда и соли. В стакан наливают 700 мл НС1 (уд. в. 1,19),
всыпают 150—200 г чистого толченого льда, пускают в ход мешалку и
охлаждают до —5°. Затем из капельной воронки начинают прибавлять по каплям
профильтрованный через вату раствор Na2Sn, полученный, как указано выше.
Реакцию проводят в течение 4—5 час. (скорость приливания, приблизительно,
1 капля в сек.), поддерживая температуру —4° или —5°. (Если температура
повысится, добавляют толченого льда, но не больше 300—350 г.)
По окончании процесса реакционную смесь переливают в два больших
стеклянных стакана, предварительно ополоснутых концентрированной НС!, и
дают смеси отстояться.
Верхний (водный) слой сливают и отделяют тяжелое желтое масло в дели*
тельной воронке, также ополоснутой предварительно НС1. Препарат,
приблизительно отвечающий формуле H2S5. промывают НС1 (уд, в.' 1,19) и хранят
в темной склянке. Выход около 200 г.
i Завод, лаб. 9. Н 5-6 540 (1940).
Водород многосернистый. Водорода перекись
115
2 По Л и б и х у кипятят 1 ч. извести и 1 ч. серы с 16 ч. воды и быстро
ив'ают профильтрованный раствор в 8 ч. смеси НС1 (уд. в. 1,19) и воды
/Л отношении 2 : 1). Отделяют выпавшее масло и дальше поступают, как ука-
**н0 в способе 1.
Сво йства
Многосернистый водород не представляет собой определенного
соединения, так как содержит несколько полисульфидов.; поэтому он только лишь
приблизительно отвечает формуле HsSs. Светложелтая (цвета прованского
масла) жидкость уд. в. 1,71 с запахом, напоминающим хлористую серу и
камфору. При стоянии на воздухе постепенно разлагается» выделяя H2S и, под
конец, осаждая серу. Подожженный — горит светлосиним пламенем. Примесь
разбавленных кислот (особенно НС1) в значительной степени предохраняет
^ногосерни-стый водород от разложения. Щелочи, напротив, вызывают мгновенное
разложение его. H2Sn несколько растворим в воде. Полная растворимость
препарата в бензоле (а также в хлороформе, толуоле, ксилоле) служит
признаком чистоты, так как препарат с большим содержанием серы растворяется не
полностью. Даже чистый препарат при стоянии в течение нескольких дней
при 0° (или сутки при 20°) перестает полностью растворяться. H2Sa
обесцвечивает лакмусовую бумагу и раствор индиго; под влиянием воздуха окраска
восстанавливается. Совершенно сухой препарат можно хранить в запаянной
трубке; если же он влажен, то наступает разложение, которое может быть
причиной разрыва трубки. Фракционированной перегонкой сырого НгБп в
вакууме можно получить H2S2 и H2S3.
ВОДОРОДА ПЕРЕКИСЬ (ПЕРГИДРОЛЬ *)
Hydrogenium Hydrogen dioxide Wasserstoffsuperoxyd
peroxydatum Perhydrol, Hydrogen Perhydrol
peroxide
H2O2 Мол. в. 34,016
Приготовление
1. К охлажденной разбавленной H2SO4 прибавляют Ва02, почти до
насыщения. Образовавшийся раствор перекиси водорода сливают с осадка BaS04
и, в случае нужды, концентрируют, для чего к раствору прибавляют соды
до щелочной реакции и извлекают Н2О2 эфиром. Эфирная вытяжка
выпаривается сначала на водяной бане, а затем выдерживается в эксикаторе над
парафином. Таким путем получается 50%-ная Н2Ог. (При выпаривании
раствора НЮ2 следует соблюдать большую осторожность ввиду опасности
взрыва концентрированной Н202. Необходимо употреблять при работе,
защитные очки и перчатки).
2. По Томсену2 смесь 20 мл крепкой H2SO4 и 200 мл воды сильно
охлаждают смесью снега с солью и время от времени туда прибавляют
такое количество чистого, свежеприготовленного (еще влажного) гидрата
перекиси бария, ВаОг • 8Н2О, чтобы жидкость еще имела слабокислую
реакцию. Дают осесть осадку BaS04> фильтруют и точно нейтрализуют сильно
охлаждаемую жидкость несколькими каплями баритовой воды так, чтобы
раствор не содержал ни Ва'\ ни SO4". Испытание проводится капельным
методом: при помощи капиллярной • трубки переносят каплю быстро
светлеющей жидкости на часовое стекло, помещенное на черном фоне, и испытывают
прибавлением капли разбавленной H2SO4 или ВаСЬ. По окончании
нейтрализации жидкость снова фильтруют и оставляют испаряться в вакууме над
H2S04.
3. Разложение ВаОг можно проводить также по Дюпрецу8; пропу-
екают через воду сильную струю СО* и при хорошем перемешивании и охла-
1 Пеогидролем называют 30%-ный раствор Н,0*.
* Thorasen, Вег., 7, 74.
» Prap. Ch„ 10.
116
Водород
ждении вносят маленькими порциями ВаОг. Когда осядет довольно значитель..
ное количество ВаСОз, сливают раствор и продолжают внесение в него ВаО^
Образующийся в незначительном количестве в растворе бикарбонат бария*
Ва(НСОз)г удаляют прибавлением нескольких капель H2SO4. Наконец
фильтруют и фильтрат концентрируют ib вакууме.
4. По патентному способу Фридериха1 в раствор НзРСЦ или NatbPO*
вносят при охлаждении перекись натр,ия ЫагОг в количестве, требуемом'
уравнениями:
Na202 + H3PO4 = Na2HP04 +' Н2О2,
Na202 + 2NaH2P04 = 2Na2HP04 + Н2О2.
Выкристаллизовывается Na2HP04* I2H21O. Для более полного осаждения
можно охлаждать смесь» После фильтрования получается Н2О2 концентрации
10—20%» содержащая только несколько процентов фосфатов. Совершенно
освободиться от Р04"' можно перегонкой жидкости в вакууме после подкисле-
ния H2SO4 или НзР04.
5. Для приготовления концентрированного раствора Н2Ог Килыпатрик,
Р е й ф и Райе2 рекомендуют следующий метод: чистую перекись натрия
постепенно, при помешивании, вносят в 20%-ную H2SO4, охлаждаемую льдом.
Раствор отфильтровывают от выделившихся кристаллов Na2S04 • ЮН2О и
перегоняют в вакууме порциями по 100 мл сначала при 60—65°, затем при 85°,
Выход 20%-ной Н2О2 около 80% (общий выход 96%). Для удаления следов
хлора (обычно присутствующих в Na202) препарат перегоняют еще раз 'над ,
Ag2S04. Концентрирование раствора НЮ2 производится в вакуум-эксикаторе
над концентрированной H2SO4 при обыкновенной температуре. В течение трех
дней удается сконцентрировать 25%-ный раствор до 88%, теряя при этом
19% имеющегося количества Н202.
6. Очистка продажной Н2О2 от примеси серной кислоты может быть
проведена в установке для перегонки под уменьшенным давлением. В колбу
наливают подлежащий очистке раствор НЮ2 (не больше половины .колбы),
прибавляют около 0,5 г лимоннокислого натрия, закрывают колбу пробкой
с капилляром и перегоняют Н2О2 при 20—25 мм рт. ст. Приемником служит
парафинированная внутри колба, охлаждаемая водой. Отбрасывая первые
порции дестиллата, получают более, концентрированный раствор Н2О2.
7. Разбавленные растворы НЮ2 можно концентрировать, вымораживая
воду3. Устойчивость растворов к нагреванию. и выпариванию зависит от сте- •
пени чистоты препарата. Особенно ускоряют разложение Н2О2 примеси со
щелочными свойствами. 3%-ную НгОг, совершенно свободную от щелочных
примесей, от малейших следов тяжелых металлов и от всякого рода твердых
веществ, можно на водяной бане при 75° довести до содержания о0;7% НаОг
с выходом в 64%. При дальнейшем выпаривании выход уменьшается, так как
значительное количество Н2О2 улетает с водяными парами.
При концентрировании НЮг перегонкой под вакуумом необходимо
соблюдать большую осторожность, так как неоднократно случались сильные взрывы
(защитные очки и перчатки).
8. Методика получения препарата, содержащего выше 99% Н2О2, из про*
дажного (35%-ного) продукта основана на двухступенчатой фракционной
перегонке при 20 мм рт. ст. Необходимо применение целого ряда
предосторожностей 4.
Свойства
Чистая Н2О2 — бесцветная густая жидкость со слабо кислой реакцией,
разлагающаяся при хранении на воду и кислород. Т. пл. —- 0,89°. Т. кип. 80,2°
(при 47 мм). Запах напоминает азотную кислоту. Вкус жгучий, вяжущий»
Стойкость увеличивается с разбавлением. Н2О2 является сильным окислителем.
1 Ргар. Ch. 10.
» РгУ Р Ch Г Ю * * е l f f' R * С 6' J# Am* Ch* S°Cm 48' 3°19 (1926)*
« H. R eg'nault и R. Le Noir de Car Ian, C. II, 3030 (1939).
Водорода перекись
117
3 некоторых случаях может восстанавливать, например окись серебра
восстанавливается перекисью водорода до металлического Ag:
Ag20 + Н2О2 = 2Ag + H2O +■ 02.
Высоко концентрированная Н2О2 воспламеняет органические вещества,
взаимодействие ее с МпОг сопровождается взрывом. На коже Н2О2 дает
белые пятна, которые после короткого жжения и зуда через несколько
часов исчезают. Н2О2 смешивается с водой во всех отношениях.
Таблица 86
Приготовление растворов Н202 различной
концентрации из продажного пергидроля (30% Н2О2)
Крепость пригототовляемого
раствора в о/о вес.
Частей дестиллированной
воды на 1 часть 30%-ного
пергидроля
1
39
2
14
3
9
5
5
10
2
15
1
20
V*
Таблица 87
Удельный вес водных растворов Н202
°/о Нао,
1
2
4
6
8
10
12
14
16
18
18
di
1,0022
1,0058
1,0131
1,0204
1,0277
1,0331
1,0425
j 1,0499
1,0574
1,0649
о/о ITaOs
20
22
24
26
28
30
35
40
45
50
18
di
1,0725
1,0802
1,0880
1,0959
1,1040
1,1122
1,1327
1,1536
1,1749
1 1,1966
°/о H2Ot
55
60
65
70
75
80
85
1 90
'95
I 100
18
di
1,2188
1,2416
1,2652
1,2897
1,3149
1,3406
! 1,3667
1 1,3931
! 1,4197
1,4465
Хранение
Растворы Н2О2 при хранении в стеклянной посуде извлекают из стекла
щелочи, что приводит к быстрому разложению препарата; поэтому
целесообразно применять сосуды, покрытые внутри парафином. Вследствие
возможного саморазложения сосуды нельзя закупоривать слишком плотно (однако
необходимо вполне защищать препарат от попадания пыли).
Испытание ("Мерк)1
1, Серная кислота. 1 МЛ препарата смешивают с 20 млш воды и 1 мл
HNOtf (уд. в. 1,124), нагревают до кипения и прибавляют раствора ВаСк.
После 12-часового стояния не должно образовываться осадка BaS04.
2. Фосфорная кислота. 5 мл. препарата выпаривают на водяной бане и
остаток после. добавления 3 мл воды смешивают с 1 мл магнезиальной
смеси (см. стр. 314) и 3 мл гШЮН (уд. в. 0,96). После 12-часового стояния
не должно быть никакого осадка.
1 Дополнительно ,Исг ытан ie хим. реективов", 106.
118 -
Водород
3. Соляная кислота, 1 мл препарата смешивают с 20 мл воды и 1 щ
HNOa (уд. в. 1,153); прибавляют несколько капель AgNOs, причем не доляед^
выпадать осадка AgCl.
4. Фтористоводородная кислота. Выпаривают 10 г препарата с несколь*
кими каплями раствора NaOH, переносят сконцентрированную жидкость щ
часовое стекло, где высушивают. Остаток смачивают концентрированной H2SQ1
и дают стоять в теплом месте 2—3 часа. По истечении этого времени смьи*
вают массу и стекло высушивают, причем на нем не должно обнаруживаться
разъедания.
5. Щавелевая кислота. Раствор 2 г препарата в 10 мл воды от прибавлю
ния СаСЬ не должен изменяться.
6. Окиси магния, железа, алюминия и др. 5 мл препарата смеишвавд
с ЫНЮН и несколькими мл Na2HP04. Даже после длительного стояния щ
должно образовываться осадка.
Также не должно образовываться осадка от прибавления к препарату
NH4OH и ШШЬСОз.
7. Остаток от выпаривания (H2SO4, НзР04 и др.) 10 мл препарата пр$
выпаривании на водяной бане должны улетучиваться без остатка.
8. Свободные кислоты. 10 мл препарата разбавляют 100 мл воды, вносят
в раствор несколько крупинок МпОг и оставляют стоять (изредка взбалты'^
вая) до прекращения выделения кислорода. Фильтруют и добавляют к филь|
трату 2 капли фенолфталеина. От прибавления 1 капли децинормальноЙ
щелочи должно появиться розовое окрашивание.
ВОДОРОД СЕРНИСТЫЙ (СЕРОВОДОРОД)
Acidum sulfuratum Hydrpsulfuric acid Schwefelwasserstoff
Hydrogen sulfide
Sulfurated hydrogen
H2S Мол. в. 34,08
А. Газообразный сернистый водород
Приготовление
1. Обычный лабораторный способ получения H2S — действие НС1 уд. в. 1,1
(или H2SO4 уд. в. 1,18) на сернистое железо в аппаэате Киппа. Выделяющийся
газ пропускают для удержания примесей (НС1 и др.) через склянку Тищенко
с водой.
2. По Б е р т ь е чистый, свободный от As и Нг газ получается
нагреванием сурьмяного блеска (природная трехсернистая сурьма SD2S3) с чистой,
концентрированной НС1. Для освобождения от увлеченного НС1 газ
пропускают через промывную склянку с водой и затем, для высушивания, —
через трубку с зерненым СаСЬ.
Мен рекомендует действовать горячей ЫС1 на смесь 3 ч. SbsSs и 1 ч.
■кварцевого песка.
3. По указанию Ванино1 очень чистый FLS получается при действии
воды на сернистый алюминий (AbSs). При этом следует брать 'АЬБз в
количестве, необходимом для немедленного расходования.
4. По Габерману смешивают в колбе 1 ч. сернистого кальция (CaS)
и 2 ч. хлористого магния (MgCk • 6Н2О) с таким количествам воды, чтобы
получилась жидкая кашица. Через пробку, закрывающую колбу, кооме
газоотводной трубки проходит шариковая предохранительная воронка (Вельтера)
с небольшим количеством раствора NaOH. Воронка проходит почти до > дна
колбы. При нагревании колбы можно регулировать силу и быстроту тока Нг£
в широких пределах — увеличивая или уменьшая пламя; достаточно отнять
J Ргйр. Ch. 7в,
Водород сернистый 119
елку, чтобы выделение газа почти прекратилось. Примесь — исключительно
шипьяковистый водород AsHs.
5. По Фрезениусу с помощью воды замешивают кашицу из 4 ч.
серистого кальция (CaS) и 1 ч. гипса (CaSCb ^/ШгО). После затвердевания масса
взбивается на куски и помещается в аппарат Киппа. При действии
разбавленной 1 :1 НС1 уд. в. 1,12 происходит равномерное выделение сероводорода.
В б. Очень равномерный ток сероводорода получается при нагревании смеси
1 ч.' серы и 1 ч. парафина (К о р е н б л ит)Л
7. Действуют разбавленной НС1 на технический сернистый натрий
(Na2*S • 9НгО). Ввиду чрезвычайно бурной реакции нельзя пользоваться обычно
применяемыми аппаратами Киппа и др. Помещают сернистый натрий в склянку
Вульфа и медленно, по каплям, прибавляют из капельной воронки
разбавленную НС1, часто покачивая склянку для более равномерного смешения.
Прибавление сразу значительных количеств кислоты всегда вызывает выбрасывание
пробок и иногда даже разрыв аппарата. Для очистки от С02 газ пропускают
через известковую воду.
8. Для получения большого количества H2S можно применять аппарат
Парсонса (Водород, Приготовление, способ 3, см. стр. М2), загружая
вместо цинка сернистое железо, или генератор, предложенный П. П о р х у-
новым2, работающий на техническом NasS.
9. Л. Паласкиано8 рекомендует нагревать до 60° смесь 2 ч. парафина,
1 ч. кизельгура (инфузорная земля), 1 ч. асбеста и 4 ч. серы.
Очистка
1. Для очистки от кислорода газ рекомендуют промывать раствором
хлористого хрома, СгСГг.
2. Простой и хороший способ очистки газа предлагает Якобсен —
пропускание высушенного СаСЬ газа через U-образную трубку, наполненную
сухим иодом. Последний поглощает АбНз, образуя йодистый мышьяк AsJs.
Очищенный газ «пропускают через склянку с водой.
3. Для очистки H2S от мышьяковистого водорода АэНз, являющегося
обычной примесью, Л е н ц предлагает следующий способ. Газ проводят через
систему из четырех промывных склянок, помещенных в нагретую до 60—70°
водяную баню. Каждая из склянок содержит по 20 мл жидкости: первая
содержит 1 ч. НС1 (уд. в. 1,12) с 2 ч. воды; вторая—1 ч. НС1 с 4 ч. воды;
третья — 1 ч. НС1 с 8 ч. воды; четвертая — дестиллированную воду. Склянки
закрывают корковыми, а не резиновыми пробками. Следует также избегать
резины и в соединениях, почему лучше систему промывных склянок
монтировать стационарно, соединив склянки изогнутыми стеклянными трубками.
Трубка, подводящая газ, берется из черной, не вулканизированной резины.
После такой очистки газ совершенно свободен от AsFL. Сушку газа следует
производить над Р2О5, но не над H2SO4, которая его разлагает.
4. Рекомендуемый иногда способ собирания £hS в газометры, наполненные
теплой (для уменьшения растворимости газа) водой, нецелесообразен, так как
меньшая растворимость H2S в горячей воде с избытком компенсируется
большой скоростью растворения. Холодная вода в течение 30 мин. поглощает
всего 0,2—0,3 мл H2S. на 1 см2 поверхности соприкосновения (С т а л о н и-
Добржанский)4.
Можно также собирать H2S в газометре, заполненном водой, покрытой
слоем парафинового масла 5—10 мм .высотой5. Нг£ подводится через
газоотводную трубку газометра; одновременно с той же скоростью вода
выпускается из газометра через нижний тубус. Равные скорости подвода газа и
выливания воды контролируются посредством водяного манометра,
включенного в газоподводящую линию.
263 (1938).
Ctemii (Warszawa) IT, 353 (1937); С. 1, 3598 (1938).
(1938), ' v ;'
120
BodopQd. Гидразин
Свойства
Бесцветный газ с отвратительным запахом тухлых яиц; сильно ядовит
даже в незначительных дозах (в качестве противоядия рекомендуется вды£
хание очень разбавленного хлора; некоторые авторы считают полезным табач*
ный дым). При зажигании горит, образуя SO2 и НгО (при недостатке воздуха
образуется Н2О и S). Сгущается в жидкость при 15—16 ат при обыкновенной
температуре. Т. пл. —183°, т. кип. — 60,2Р. Вес 1 л—1,63922 г (при нормаль*
ных условиях). Растворим в воде и спирте. Жидкий H2S подвижен, как эфир,
уд. в.1 0,938 при —81°. Твердый H2S — белая кристаллическая
просвечивающая масса, тяжелее жидкого сероводорода,
Б. Сероводородная вода (раствор H2S в воде)
Приготовление
Насыщают очищенным сероводородом прокипяченную и затем охлажденную
дестиллированную воду, пока при взбалтывании склянки, заткнутой пальцем*
не будет ощущаться повышенное давление. Чтобы избежать больших потерь
H2S, происходящих вследствие неполного растворения газа, можно
рекомендовать следующий метод. Газ подводят в плотно закрытую склянку,
заполненную до половины водой. При частом взбалтывании газ нацело растворяется»
Таблица 88
Растворимость H2S вводеи спирте
t°c
0
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
Коэф. растворимости» «
в воде
4,3706
4,2874
4,2053
4,1243
4,0442
3,9652
3,8872
3,8102
3,7345
3,6596
3,5858
3,5132
3,4415
3,3708
3,3012
3,2326
3,1651
3,0986
3,0331
2,9687
2,9053
в спирте
17,891
17,242
16,606
15,983
14,373
14,776
14Л93
13,623
13,066
12,523
11,992
11,475
10,971
10,480
10,003
9,539
9,088
8,650
8,225
7,814
7,415
t*c
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
Коэф. растворимости8 а
в воде
2,8430
2,7817
2,7215
2,6624
2,6041
2,5470
2,4909
2,4357
2,3819
2,3290
2,2771
2,2262
2,1764
2,1277
2,0799
2,0322
1,9876
1,9430
1,8994
1,8569
в спирте
7,030
6,659
6,800
5,955
5,625
,
1 J. P. Baxter, L.J. Burr age *. С. С. Tinner, J. Soc. Chera. Ind. 53, Trans. 410
(1934); С II, 2790 (1935).
• Показывающий число объемов HtS, растворяющегося в 1 об, жидкости при 760 цц*
Водород хлористый. Гидразин
121
способствует также некоторое избыточное давление в системе. Этот спо-
Ч6^ получения сероводородной воды можно рекомендовать как очень быстрый.
Пля хранения склянку наливают возможно полнее, плотно закупоривают
.бкой и, для предохранения от доступа воздуха, заливают парафином или
П^огУЧ°м- ' Закупоренная таким образом сероводородная вода при хранении
холодном, защищенном от света' месте очень постоянна.
Свойства
Сероводород в водном растворе является очень слабой двухосновной
кислотой, окрашивающей лакмусовую бумажку в красный цвет. Константы
диссоциации Ki = 3,313 • 10~7 (Э п п р е х т)1, Кг = 2 • Ю-16. Соприкасаясь
с воздухом, сероводородная вода постепенно окисляется и мутнеет, выделяя
серу. По Шильтону добавление 2% сахара или 1 % салициловой кислоть
чялерживает разложение препарата. Уд. в. сероводородной воды (3,32 г/л
^0,99994.
Hydrazinum
ВОДОРОД ХЛОРИСТЫЙ
См. Кислота соляная, стр. 276
ГИДРАЗИН (ДИАМИД)
Hydrazine
N2H4 Мол. в. 32,048
Hydrazin
Приготовление
По Н. Путохииу2 (вводят гидразин-гидра*? гюрцйями по 5 г в избыток
твердой ВаО при хорошем охлаждении *и дестиллируют образовавшийся
гидразин. Препарат сохраняют в запашных стеклянных трубках, из которых
воздух вытеснен водородом. "" '
Свойства
Бесцветная, сильно дымящая на воздухе жидкость уд. в. 1,011,
затвердевающая в кристаллы, которые плавятся при +1,4°. Т. кип. гидразина 113,5° при
761,5 мм и 56° при 71 мм. Не взрывает, очень постоянен. Распадается лишь
выше 350° на азот и аммиак. Горит фиолетовым пламенем. Смешивается во
всех отношениях с водой и спиртами: метиловым, этиловым, пропиловым,
бутиловым и амиловым. Не растворим или мало растворим в других органических
растворителях.
Таблица 89
Удельны.й вес водных растворов N2H4
о/о N.H,
1
2
4
6
8
10
14
1Б
1,0002
1,0013
1,0034
1,0056
1,0077 .
1,0099
1,0143
°/oNtH4
18
22
26
30
35
40
45
is
di
1,0186
1,0228
1,0267
1,0305
1,035
1,038
1,042
0/oNjH4
50
55 •
60
70
80
90
100
15
1,044
1,046
' 1,047
1,046
1,038
1,030
1,011
i A. G. E p p г е с л t, Helv. chim. Acta, 21, 205 (1938).
« Изв. ИРЕА, вып. 2, стр. 60 (1923).
122
Гидразин
Hydrazinum hydricum
ГИДРАЗИН-ГИДРАТ
Hydrazine hydrate
Hydrazinhydrat
N2H4 • H2O *
Мол. в. 50,064
Приготовление
По Н. Путохину1 смешивают в медной реторте 100 г сернокисло
гидразина (N2H4-H2S04) и 100 г сухого, истертого в порошок КОН.
Прибавляют 5 мл воды, закрывают реторту и дестиллируют. Вйачале перегонка идет
за счет теплоты, выделяющейся при реакции, но под конец приходится подо*
гревать.
Дестиллат, содержащий воду, перегоняют еще раз. При 117—118° nepfcv
гоняется почти чистый гидразин-гидрат, несколько загрязненный БЮг.
Свойства
Мало подвижная жидкость уд. в. 1,03, дымящая на воздухе, со специфичен
ским слабым запахом (непохожим на запах ЗЧНз); имеет сильно щелочную
реакцию2. Т. кип. 118,5° при 740 мм и 47° при 26 мм. При охлаждений
ниже — 40° застывает.
Высушенный при 100° отвечает формуле N2H4 • НгО. Смешивается с вод oft
и спиртом; нерастворим в эфире, хлороформе и бензоле; гигроскопичен. Н$
воздухе поглощает СОг. Сильный восстановитель.
Испытание
По ОСТ 10955—40 содержание N2H4 • Н20 для препарата ч.д.а. должно
быть не менее 99,5—101 %, а для чистого—99—101%.
Допустимые наибольшие количества примесей приведены в табл. 90.
Таблица 90
Допустимые примеси в N2H4. Н20
Прймеов
Допустимое содержание в о/0
в чистом для
анализа
1. Остаток после прокаливания .
2. Хлориды (СУ)
3. Сульфаты (SO*") .
4. Железо (Fe)
5. Тяжелые металлы группы H2S
0,005
0/Ю2
0;()01
0,0001
0,0003
0.02
0,005
0,005
0,0005
0,002
NH8v
* Или NH,-NH3.OH, или "| 8\о
1 Изв. ИРЕА, вып. 2, стр. 50 (1923).
■ Константа диссоциации гидразин-гидрата, как кислоты при 15° Кк = 5,75-10 в, как
основания /Со = 7,85-10-' (О. С. W а г е, J. P. S p u I n 1 с к, Е. С. О 11 b е г t, J. Am. Chem. Soc. 68,
1605 (1936); С. II, 536 (1937). По другим источникам константы диссоциации гидразин гидрата?
как основания К\ — 8,5.10—*, К% = 8,4-10—1« (О. S с h w а г z% п Ъ а с h, Helv. chlra. Acta, 19>
178 (1936j; С 1, 4$85 (1936),
Гидразин-гидрат. Гидразин сернокислый
123
ГИДРАЗИН СЕРНОКИСЛЫЙ (ГИДРАЗИН-СУЛЬФАТ)
Hydrazinum Hydrazine sulfate Hydrazinsulfat
sulfuricum
N2H4H2SO4 Мол. в. 130,12
Приготовление
По Рашигу гидразин образуется при взаимодействии аммиака и хлорно-
ватнстокислого натрия. При этом происходит следующая реакция:
NaCIO + МНз = NH2CI + NaOH.
Образовавшийся монохлорамин вступает в реакцию, с избытком аммиака:"
NH2C1.+ NH8 = ЫгШ . НС1.
Одновременно происходят побочные реакции:
^ 3NH2C1 + 2(NHs .= N2 + 3NH4C1,
4NHs + ЗСЬ = NCI* + 3NH4C1.
Хлористый азот в момент образования разлагается избытком аммиака:
NCI* + 4NHs = N2 + 3NH4.C1.
Увеличение вязкости жидкости повышает выход препарата. Р а ш и г
рекомендует прибавлять к реакционной смеси для увеличения вязкости
тростниковый сахар, декстрин, глицерин и, особенно, белки, желатин и столярный
клей.
Сначала приготовляют раствор NaCIO, растворяя 100 г NaOH в 450 мл
воды и при сильном охлаждении насыщая раствор хлором до тех пор, пока
не будет поглощено стехиометрическое количество хлора (88,7 г). Полученный
раствор приливают в течение 20—30 мин. при сильном охлаждении снегом
с солью к 1500 -мл 25%-ного NH4OH (уд. в. 0,91), к которому прибавлен
раствор 2,5—3,5 г столярного клея в 75 мл воды. Температура раствора не
должна подниматься выше 5—6°. Если раствор разогреется, происходит
выделение пузырьков азота >и вспенивание — результат побочных реакций,
понижающих выход препарата. Смесь оставляют на сутки, затем отгоняют аммиак,
поглощая его водой.
Оставшийся раствор, содержащий N2H4 ■ 2НС1, NaCl, NH4CI и остатки
NH4OH, нейтрализуют креп-кой H2SO4 и упаривают до половины объема. При
этом выпадают NaCl, NH4CI, Na2S04 и (NH4)2S04. Отфильтровав осадок,
фильтрат оставляют для дальнейшей обработки, а осадок для извлечения
дополнительного количества N2H4 обрабатывают несколькими миллилитрами
слабого NH4OH. Через 2—3 часа аммиачный раствор отфильтровывают и сильно
подкисляют H2SO4. При охлаждении и стоянии выпадают кристаллы
сернокислого гидразина в количестве около 10 г. К главной массе раствора
прибавляют H2SO4 при помешивании и охлаждении. Выпадает N2H4 • H2SO4,
который отсасывают и перекристаллизовывают из горячей воды. Выход 60—70 г 1.
Можно поступить и иначе2. Сразу по окончании реакции раствор
переливают в колбу и отгоняют NHs. Затем реакционную смесь выпаривают в
фарфоровой чашке до уд. в. 1,14—1,16 (объем сокращается приблизительно втрое).
Раствор переносят в колбу и приливают 80 г ацетона, причем образуется
ацетон-гидразон:
N2H4 + (СНзЪСО =* (СНз)2С = N • NH2 + НЮ.
Смесь перемешивают и отгоняют гидразон до тех пор, пока дестиллат уже не
будет иметь характерного запаха гидразона (лучше контролировать процесс
1 Н. П у т ох и н, Изв. ИРЕА 2, 60 (1923),
• Реакт. и рреп. 69.
124
Гидразин
перегонки по показателю преломления дестиллата, который в конце опера*^
должен быть равен 1,33 при 20°). ^^
К дестиллату приливают 250 г 30%-ной H2SO4 для разложения гидраз^
(СНз)2С = N • NH2 + H2SO4 + Н2С = N2H4 • H2S04 + (СНз)2СО
и отгоняют ацетон (до 90—95°). К жидкости, оставшейся в колбе, приливу
300 мл воды, раствор фильтруют и выпаривают фильтрат на водяной бане
образования кристаллической пленки. Выпавшие при охлаждении кристал!
отсасывают, промывают спиртом и высушивают -при ПО—115°. Выход 60щ
Маточный раствор при выпаривании дает еще около 10 г менее чистого п$
парата *.
Свойства
Толстые стекловидные плитки уд. в. 1,378 или длинные призмы ромб*]
ской системы, плавящиеся при 254° с разложением. Растворим в. воде,
растворим в спирте. Сильный восстановитель.
Таблищ
Растворимость N2H4• H2S04 в воде
(Somraer, Weise)2
fC
20
25
30
40
Wo
NsH4.H§S04
2,794
3,302
3,746
3,987
ГС
50
60
70
80
°/о
6,538
8,322
10,495
12,580
Испытание
По ОСТ 8011/933 препарат ч.д.а. должен содержать при иодометрическом
определении не менее 98,5% N2H4 • H2S04, препарат чистый—не менее 98%.
При ацидиметрическом определении для препаратов обеих квалификаций — не
выше 101%.
Наибольшие количества допустимых примесей указаны в табл. 92.
Допустимые примеси в N2H4-H2S04
Таблица 92
Примеси
Допустимое содержание в °/0
я чистом для
анализа
1. Нерастворимые в воде вещества
2. Остаток после прокаливания . .
3. Хлориды (СК)
4. Металлы, осаждаемые H2S . . .
5. Железо (Fe)
0,002
0,05
0,002
0.001
0,0005
0,005
0,1
0,005
0,0023
0,001
* См. также Синтезы органических препаратов (Organic Svntesei), Госхимтехиздат, т. 1,
г'тр. 70, 1932.
*Z. anorg. Ch. 94, 51 (1916).
Гидразин солянокислый
125
ГИДРАЗИН СОЛЯНОКИСЛЫЙ
Hydrazinum Hydrazine chloride Hydrazinchlorhydrat
hydrochloricum
NaH4-2HCl Мол. в. 104,978
Приготовление
Солянокислый гидразин легко получается при взаимодействий сернокислого
гИдразина с хлористым барием.
К кипящему раствору 50 г N2H4 • H2SO4 в 150 мл воды приливают
постепенно, при помешивании, горячий раствор 93,9 г ВаСЬ • 2Н20 в 150 мл воды.
При этом выпадает BaS04. Кипятят несколько минут и оставляют стоять
в теплом месте. После отстаивания пробуют на полноту осаждения; если
ВаСЬ было прибавлено недостаточно, то вновь нагревают раствор и
прибавляют по каплям раствор ВаСЬ до прекращения образования осадка. Если
же в растворе имеется избыток ионов Ва * ', их удаляют прибавлением
нескольких капель разбавленной H2SO4.
Осевший BaS04 отфильтровывают, промывают водой и промывные воды
присоединяют к фильтрату. Выпаривают последний на водяной бане до начала
кристаллизации.
По охлаждении выкристаллизовывается в октаэдрах N2H4 • 2НС1, который
отсасывают и сушат в эксикаторе. Выпариванием маточного раствора можно
получить еще некоторое количество препарата. Выход 35 г1.
Свойства
Октаэдрические иши игольчатые бесцветные кристаллы уд. в;. 1,423.. Т. пл.
198°, при более высокой температуре разлагается. Хорошо растворим в воде
(при 23° в насыщенном растворе содержится 73% N2H4*2HC1).
Таблица 93
Удельный вес водных растворов N2H4• 2НС1
о/0 N,H4.2HC1
1
2
4
6
8
80
*<
1,0062
1,0098
1,0170
1,0255
1,0340
о/0 NaH4.2HCl
10
15
20
25 ■
«0
d
4
1,0436
1,0675
1,0923
1,1183
Испытание
По ОСТ НКТП 8295/1486 содержание Ntlh • 2НС1, определяемое; иодо-
метрически, для препарата ч. д. а. не менее 99%, для препарата чистого
не менее 98%; определяемое ацидиметрически — для обеих квалификации —
не выше 101%. Наибольшие количества допустимых примесей указаны
в табл. 94.
*Н. Пут ох и н, Изв. И PEA, 2, 50 (1923).
№
Гидроксиламин. Гопкалит
Допустимые примеси в N2H4-2HC1
ТаблиЩ^
Примеси
Допустимое содержание в «^
в чистом для
анализа
в чистом
1. Нерастворимые в воде вещества
2. Остаток после прокаливания . .
3. Сульфаты (SO/)
4. Тяжелые металлы .......
5. Железо (Fe)
0,005
0,05
0002
0,001
0,0005
0,01
0,'0
0,005
0,002
0,001
ГИДРОКСИЛАМИН СОЛЯНОКИСЛЫЙ
Hydroxylaminum
hydrochloricum
Hydroxylamin
hydrochloride
Hydroxylaminchlor*
hydrat
NH2OH • HC1
Мол. в. 69,497
П риготовление1
143 г кристаллического углекислого натрия (№гСОз • ЮНгО) растворяют^
в 100 мл воды и насыщают SO2; полученный раствор NaHSOs приливают
к охлаждаемому льдом и солью раствору 36 г NaN02 в 60 мл воды. Во
время приливания температура не должна подниматься выше 0°, причем
смесь нужно сильно перемешивать, лучше всего механической мешалкой
(если в растворе окажется избыток NaN02, его разрушают
пропусканием SO2).
Раствор разбавляют до 1 л, перелив в круглодонную колбу, нагревают»
на воронке Бабо до кипения и добавляют раствор 250 г кристаллического
ВаСЬ • 2НгО в 300 мл воды до тех-пор, пока отфильтрованная проба не пока
жет отсутствия SO-i".
Реакции получения солянокислого гидроксиламина могут быть выраженШ
следующими уравнениями:
NaHSOs + NaN02 + SO2 = (S03Na)2NOH
(гидроксиламиндисульфонат
натрия)
'(S03Na)2NOH + 2ВаС13 + 2Н20 = ЫНгОН , НС1 + 2BaS04 + 2NaCl + НС1.
Дают осесть Ва304, фильтруют и выпаривают фильтрат сначала на голом!
огне, под конец — на водяной бане, досуха. Образующиеся корки
кристаллов извлекают на край чашки и Тщательно разбивают. Сухой остаток,
состоящий из смеси NaCl и NH2OH "• НС1, кипятят последовательно с 200,
100 и 50 мл спирта. Последний, извлекая солянокислый гидроксиламин, не
растворяет NaCl. Спиртовые вытяжки соединяют вместе и выпаривают до
начала кристаллизации. Выпавшие при охлаждении кристаллы прекристал-1
лизовывают из половинного количества воды (по отношению к весу
препарата).
Свойства
Бесцветные игольчатые кристаллы моноклинической системы, легко
растворимые в воде (при 17° 45 : 100) и метиловом спирте; нерастворим в эфире.
1 Подробное описание метода ИРЕА, пригодного для получения больших количеств
препарата (до 200 z) (см. Ре акт. и преп., 62),
Гидроксиламин солянокислый. Гопкалит
127
^ пл. 151° (с разложением). Является сильнейшим восстановителем, сам
окисляясь до N2O. Ядовит
испытание
По ОСТ НКТП 7176/470 содержание NH2OH • НС1 для препарата ч.д.а.
,ie менее 98,5%, для чистого — не менее 96%; наибольшие количества
допустимых примесей указаны в табл. 95.
Допустимые примеси в NH2OH • НС1
Таблица 95
Примеси
Допустимое содержание в о/о
в чистом для
анализа
0,002
0,01
0.3
0,002
0,0005
0,0005
| 0,0001
в чистом
0 005
0,05
1,0
0,005
0,001
0.001
0,0001
1. Нерастворимые в воде вещества
2. Остаток после прокаливания
3. Хлористый аммоний (NH4C1)
4. Сульфаты (SO/) . . . .-
5. Мета <лы, осаждаемые H2S
6. Железо (Fe)
7. Мышьяк (As)
ГОПКАЛИТ
Hopcalite
Гопкалит представляет собою смесь активных окислов металлов,
являющуюся отличным катализатором для окисления СО в СОа кислородом воздуха
при низких температурах. Различают двухкомпонентный и четырехкомпонент*
ный гопкалит.
А. Приготовление двухкомпонентного гопкалита *
Тщательно перемешивают 60 г активной МпО« (приготовленной, напр., по
Фреми, см. стр. 324) и 40 г СиО (высушенной при ПО—120° в течение
6 час). Смесь прессуют в таблетки на гидравлическом прессе под давле*
нием 250 ат.
Б. Приготовление четырехкомпонентного гопкалита
Препарат представляет собой смесь, состоящую из 50%' МпОг, 30% СиО»
15% СогОз2 и 5% Ag20.
Для приготовления гопкалита смешивают 30. г активной МпСЬ, 30 г СиО
и 16 г С02О3, приливают 240 мл 3%-Hoiro раствора AgNOs, йагрейают на
водяной бане и добавляют при энергичном перемешивании 35 мм 5%-ного
раствора NaOH. Через час промывают осадок водой, сначала
декантированием, а затем на воронке Бюхнера до исчезновения щелочной реакции в про*
мывных водах. Препарат сушат при 110а, после чего истирают в тонкий
1 Е. В. А л е к с е е в с к и й, Общий курс химии защиты, т. II, стр. 202, 1939 и слеД4
Е. В. Алекс еевский, Г. М» Белоцерковский и Т. Г. Плачено в,
Практические работы по химии защиты, Оборонгиз, стр. 48—55, 1940.
* Необходимую для приготовления гопкалита СоаОе готовят, приливая к раствору 50 i
CoS04 в 200 мл воды 10о/0-ного раствора NaOH до полного осаждения Со(ОН в. Добавлением
60 мл насыщенной бромной воды Со(ОН), переводится в Со(ОН)3, после чего смесь нагревается
до удаления запаха ирома. Осадок СовОэ отмывают декантацией, отсасывают, промывают водой
до удаления SCV в промывных водах и высушивают при 11J—120е в течение 6 чао.
128
Железд
порошок и прессуют в таблетки под давлением 250 атм. Таблетки дроби»
с помощью набора сит отбирают фракцию 2—2,5 мм, которую высушиващ
ори 180° и хранят в банках с притертой пробкой.
ЖЕЛЕЗО
Ferrum Iron Eisen
Fe At. в. 55,85
Приготовление
1. Для приготовления порошка железа, восстановленного водородом («Fer*
rum metallicum hydr. reductum» \ чистый высушенный при/ 110—120° |
истертый в тонкий порошок Fe(OH)s помещают тонким слоем в трубку |$|
фарфора или тугоплавкого стекла. Пропуская чистый сухой водород,
вытесняют воздух из прибора и затем, не прекращая тока водорода, доводят
постепенно трубку до тёмнокрасного каления. Восстановление ведут до тех
пор, пока в трубке не перестанет образовываться вода (для испытания выхой
дящие из трубки газы направляют на холодное стекло: отсутствие
запотевания последнего указывает на окончание восстановления). Дают трубке
совершенно остыть в токе водорода и полученный порошок железа
помещают в банку с хорошо притертой стеклянной пробкой. ■■■•_
Важно вести восстановление при правильной температуре. Если трубка^
нагрета ниже тёмнокрасного каления, то получается пирофорное железо^
которое, соприкасаясь с воздухом, немедленно раскаливается и окисляется^
Наоборот, при повышении - температуры выше тёмнокрасного каления вмеЧ
сто, тонкого порошка получается спекшаяся масса.
Употребляемый для восстановления водород предварительно очищают, для
чего пропускают его сначала через щелочной раствор РЬ(СНзСОО)2, затем
через раствор CuS04 и, наконец, для осушки через концентрирован*
ную H2SO4.
П. М. Завеле;вич2 рекомендует при навеске в 20 г нагревать FesOs в
в токе водорода в течение 40 мин., повышая постепенно температуру от 400
до 700° и затем 20 мин. вести восстановление при 700°. Охлаждение ведут
в токе Ш до 400°, затем трубку герметически закрывают. Препарат
содержит 95—96% Fe. Напротив, И. П. Кис ля ко в3 не рекомендует поднимать
температуру выше 630—650°, проводя процесс восстановления 100 г РегОз'
в течение 6 час. Препарат содержит 0,05—0,15% кислорода.
2. Для получения небольших количеств совершенно чистого железа,
вполне пригодного для установки титров, поступают по Мозеру и Шенин-
геру4 следующим образом. Подвергают электролизу раствор хлористого
железа в приборе, изображенном на рис. 28. В стакан емкостью в 200' мл
помещают электролит, приготовленный растворением 3—4 г чистого
хлористого железа, 12—15 г хлористого натрия и 1 г борной кислоты в 100 мл
(воды и подкислением полученного раствора 5 мл AN НО, В качестве
катода употребляют платиновый или танталовый диск К, к которому припаян
стержень, соединенный с источником тока. [Анодом служит укрепленный
шляпкой вниз железный гвоздь А (толщиною около 5 мм), предварительно
обработанный НЖ)з и промытый водой. Между электродами помещается
диафрагма, например, фарфоровый тигель с пористым дном. Назначение
диафрагмы — предохранение катода от шлама, падающего с анода-Л.
Электролиз ведут при силе тока 0,3—0,4 а, что отвечает катодной
плотности тока 0,5—0,7 а на см2. Напряжение 1,4—1,6 в, температура электро-,
лита 60°. При этих условиях в течение часа получается 0,2—0,3 г
чистейшего железа. Когда осаждено требуемое количество железа, не "прекращая
1 Komm. Z. Deut. Arzneib., Pr2p. Ch. 626.
я Изв. ИРЕА, вып. 15, стр. 51, 1937.
» ЖПХ 12, Ко и, 1668 (1939).
« М о s е г, W. S с h б п I п g e r, Z. anal. Ch., 70, 235 (1927),
Железо
№
V
ка> вынимают катод из электролита, обмывают водой; после этого опускают
т° на несколько секунд в 0,17V раствор КОН, снова обмывают водой, спола-
екИвают спиртом и высушивают в струе сухого воздуха. Осажденное железо
соХраняется на диске до момента употребления.
с з. Н. А. Т а н а н а е в х описывает несколько видоизмененный метод С к р а-
5 а л я, основанный на электролизе1 двойной соли щавелевокислого аммония и
железа (катод — платиновая чашка, анод — платиновая спираль). После
выделения осадка железа повторяют процесс электролиза, сделав чашку
с осадком Fe анодом, а чистую платиновую пластинку — катодом.
Электролит— 10%-ный раствор соли Мора. Полученный препарат обладает^ сереб-
ристо-белым цветом и очень постоянен.
4. В. В. Стендер, И. Я. С и рак и Е. И. Кириченко2 указывают
условия получения больших количеств электролитного железа .из FeCb.
Католиком является 35—40%-ный раствор FeCb с добав- -■ ' '
кой NaCl; содержание НС1 в растворе должно
соответствовать 0,0025—0,005ЛГ. Анолит — 25—30 % -
ный раствор NaCl. Анодом служит электрогр^фит,
катодом — пластинка чистого железа. Электролиз
ведут при 80—95°. Катодная плотность тока
250 а/м2. Диафрагмой служит кислотостойкий
асбест.
5. Получение железа высокой чистоты описано
Томпсоном и Кливсом3. Чистая РегОз
восстанавливается водородом при 500°; температуру
повышают до 1000° для спекания железа.
Полученный продукт расплавляют и затем вторично
подвергают сплавлению в вакууме. Препарат был
исследован на 55 различных примесей, из которых
были обнаружены только 6—9 (главным образом
О и S, следы С, Р, N» Н; в некоторых пробах
обнаружены Си, Si, lAl, Be). Общее количество
примесей меньше 0,010%.
6. А. Гаттерер4 исследовал
спектроскопически довольно чистое железо, полученное
разложением карбонила железа Fe(CO)s> и обнаружил в
нем примеси 31 элемента. Автор рекомендует в
качестве исходного продукта применять чистое FeCls,
извлекая его из солянокислого раствора эфи.ром
(для отделения Си, Mn, Ni, Co, A1, V, Ti).* Далее FeCh переводится в Fe(OH)3,
затем в РегОз (см. напр. стр. 134) и восстанавливается водородом.
Полученный таким путем препарат содержит незначительные количества только
четырех элементов.
7. Получение очень чистого железа в больших количествах может быть
проведено лр методу Ф. Эд.кокаб из тонкой жести электролитного железа.
Жесть растворяют в чистой НС1 и раствор выдерживают над избытком
железа (удаление Си и Р). . •
Полученный при выпаривании раствора хлорид помещают в лодочки из
Si02 или АЬОз и" обработкой перегретым паром при 250° переводят в FeaOs.
Пурпурно-красный порошок РегОз восстанавливают водородом при 750°,
затем нагревают в токе N2 до 900° и спекшуюся массу Fe сплавляют в
атмосфере азота в тигле из MgO.
Полученное железо вторично сплавляют в тигле из АЬОз в токе Нг под
Давлением 150 мм рт. ст.; в конце плавления вакуум увеличивают до 30 мм
Рт. ст. Охлаждение проводят так, чтобы верхняя часть расплава затвердевала
Н
Рис. 28. Прибор для
получения железа электролизом
(1939),
1 Н. А. Тан а на ев, Объемный анализ, 6 изд. Гояти, стр. 327, 1939.
* Металлург, 10, № 7, 114 (1935).
3 Т. G. Thompson, Н. Е. S 1 е a v e s, J. Res. Nat. Bur. Stand. 23. 133 (1939); С II, 3681
4 A. Qatterer, С I 1745 (1938).
G F. A <t с о с k, J. Soc. Chem. Ind. 59, 28 (1940); C. I, 1903 (1940).
130
Железо
последней. Препарат содержит около 0,0005% углерода. Сумма всех прб^
примесей составляет 0,001—0,006%. Метод позволяет получить до 50Q :Щш
в день.
Свойства
Мягкий серый металл уд. в. 7,86 или полученный по способу 1 — матсйвд
серый порошок (получающаяся иногда темная окраска зависит от пример
Рез04). Т. пл. очень чистого Fe, содержащего только 0,039% примесей
1533 i 5° (Бристоу1), т. кип. 2450°. Во влажном воздухе медленно окис»
ляется. Растворимо в НО, H2SO4 и разбавленной HNOa. Крепкая дымящая
HNOs делает железо «пассивным»,, покрывая его пленкой окислов, после
чего железо теряет способность растворяться в кислотах.
Испытание (М е р к)
1. Примеси, не растворимые в H2SO4. 10 г препарата должны почти пщ
ностью растворяться в смеси 20 мл H2SO4 и 200 мл воды Нераствор и вшийс*
осадок после отфильтровывания, промывания и высушивания при 100° :-Щ
должен быть больше 0,05 г.
2. Сера (S"). 1 г препарата, помещенного в пробирку, обливают смесью
10 мл НО (уд. в. 1,124) и 10 мл воды. Выделяющийся водород не должен;
в течение 10 сек. вызывать потемнения бумажки, смоченной раствором
РЬ(СНзСОО)2.
3. Углекислый натрий. Взбалтывают 5 г препарата с 50 мл воды и филь«
труют. Фильтрат не должен окрашивать красную лакмусовую бумажку
в синий цвет и при выпаривании не должен оставлять никакого остатка.
4. Азот. 10 г препарата растворяют при нагревании в смеси 200 мл
концентрированной H2SO4 и 200 мл воды. По охлаждении прибавляют 100 мл
раствора NaOH (уд. в. 1,3) и, поместив смесь в перегонную колбу, отгоняют
(применяя холодильник) около 50 мл, собирая дестиллат в приемник,
содержащий 20 мл воды и 3 в 0,Ш НО. Содержимое приемника титруют,
0,1 N КОН, применяя в качестве индикатора метилоранж. Расход КОН не
должен больше чем на 0,2 мл отличаться от количества, необходимого для
нейтрализации 3 мл 0,Ш НО.
5. Мышьяк. Смесь 1 г препарата и 1 г КООз обливают 10 мл НО (уд,
в. 1,124). Когда реакция закончится, нагревают смесь для удаления
свободного хлора и фильтруют. К 5 мл фильтрата прибавляют 15 мл раствора'
. SnCh. В течение часа не должно появляться темного окрашивания.
ЖЕЛЕЗО АЗОТНОКИСЛОЕ ОКИСНОЕ (НИТРАТ ЖЕЛЕЗА [3])
Ferrum nitricum Ferric nitrate Salpetersaures Eisen-
oxydatum oxyd
Ferrinitrat
Fe(NOs)8 • 9H20 Мол. в. 404,02
Приготовление
Составляют прибор, изображенный на рис. 29. В круглодонную колбу^
йенского стекла емкостью 1 л, помещенную на водяной бане, вставляют
рогатый форштосс, к горлышку В которого присоединяют обратный холодильник
Шиффа (шариковый). Верхнее отверстие холодильника закрыто просверлен*
ной резиновой пробкой с проходящей через нее стеклянной газоотводной
трубкой, соединенной с двумя склянками Дрекселя емкостью 100 мл каждая.
.Через горлышко В форштосса пропущена также стеклянная трубка С,
доходящая почти до уровня жидкости и служащая для подвода воздуха внутрь,
колбы. Наливают в колбу 120 мл воды и 330 мл HNOa уд. в. 1,38 и начй?;
нают понемногу прибавлять через горлышко А форштосса мелкие железнью
» СЬ. А. В г 1« t .9 w, С. II, 799 (1939).
Железо азотнокислое окиендё
131
0брезки Ч Предварительно' наливают в склянки Дрекселя по 60 мл HNOa
/п0глощая окислы азота; кислота сильно увеличивается в объеме). Склянки
соединяют с водоструйным или иным насосом, выкачивающим воздух. Таким
образом, бурно выделяющиеся окислы азота просасываются через азотную
кислоту и растворяются в ней.
Реакция длится 2—2^ часа, в течение которых прибавляют 60 г
железных обрезков. Полученную жидкость
фильтруют через бумажные фильтры
в фарфоровую чашку емкостью 1200—
1500 мл. Раствор упаривают до
образования толстой пленки, прекращают
нагревание и тотчас же приливают из
склянок Дрекселя 225 мл азотной:
кислоты, насыщенной окислами азота.
Толстой стеклянной палочкой
размешивают содержимое чашки, раздавливая
приставшую к стенкам кристаллическую
корку. -.;..
Чашку покрывают стеклом и
оставляют для кристаллизадии при
температуре ниже 0°.
Если на другой день кристаллы не
появились, то необходимо бросить
затравку— маленький кристаллик
готового препарата и оставить еще на 3—
4 часа на холоду. После этого сливают
маточный раствор» кристаллы
отсасывают, промывают несколько раз .
небольшими количествами 20%-нойНЫОз
(в общей сложности не больше 25—
<Ю мл) и тотчас же складывают в
сухую банку с плотно закрывающейся
пробкой, которую заливают
парафином.
Если процесс необходимо
повторить, то маточные растворы от первой
операции употребляют для приливания Рис. 29. Прибор для получения азотнокислого
к кристаллизуемому раствору вместо железа-
кислоты, насыщенной окислами азота.
Свойства
Лиловые (в совершенно чистом виде — бесцветные) моноклиническйе
кристаллы уд. в. 1,68. Плавится при 47,2° в своей кристаллизационной воде,
образуя красную жидкость, теряющую при 50° часть HNOs и кипящую при
125° с дальнейшим разложением. Расплавленный препарат при охлаждении
до 21° еще остается жидким. По Малькори при 25° 100 г насыщенного
раствора содержат 46,57% Fe(NChK Препарат необходимо хранить в
прохладном месте.
Испытание
По ОСТ НКТП 2854 содержание Fe в препарате должно быть не
менее 13,6%; наибольшие количества допустимых примесей указаны в
табл. 96.
* Когда требуетзя получить чистый препарат, необходимо пользоваться кислотой, не содер*
жащей хлора. Если имеется только техническая кислота, содержащая 0,4— 0,80/» СГ, то определив
содеожание последнего анализом, добавляют 2J<VHoro раствора AgNOa с расчетом, чтобы после
выпждени* Age* в кислоте оставалось ничтожное количество СГ, который легко отделяете!
процессе получения препарата. Кислоте дают отстояться и сливают с осадка AgCU
132
Железо
Допустимые примесив Fe(N03)3• 9Н20
Таблица д$
Примес»
Допустимое содержание в о|0
в чистом для
анализа
в чистом
1. Нерастворимые вещества
2. Хлориды (СК)
3. Сульфаты (S04") •
4. Щелочные и щелочноземельные металлы (в виде
сульфатов)
5. Металлы, осаждаемые H2S
6. Цинк (Zn)
0,005
0,001
0,01
0,05
Q00J
0,001
0,01
OJ002
0,02
ОД
0,003
0,003
Таблица ty
Удельный вес водных растворов Fe(N03)3
о/о Fe(N03),
5
10
15
20
25
30
d*7,5 |
1,0398
4 1,0770
1,1182
1,1612
1,2110
1,2622
о/о Fe (N08)8
35
40
45
50
55
60
65
d*7,6
1,3164
1,3746
1,4338
1,4972
1,5722
1,6572
1,7532
ЖЕЛЕЗО БРОМИСТОЕ (БРОМИД ЖЕЛЕЗА[2])
Ferrum bromatum Ferrous bromide
А. Безводное
FeBr2 Мол. в. 215,68
Eisenbromur
Ferrobromid
Приготовление
1. Пропускают пары брома над хорошо очищенной от ржавчины железу
ной проволокой, помещенной в нагретую до слабого каления тугоплавкуй
стеклянную трубку. Трубку охлаждают в токе сухого СОг и помещают пре*
парат в сухие банки с притертой пробкой, наполненные С02. У
2. Через раскаленное железо, помещенное в фарфоровой• или стеклянной
трубке, пропускают ток азота, содержащего бромистый водород.
Одновременно получившееся FeBr2 возгоняется.
3. В. Бильц и Е. Б ир к1 для очистки продажного препарата
рекомендуют возгонять его в токе НВг.
Свойства
Желто-зеленые гигроскопические чешуйки уд. в. 4,79. По О. Г е н и г-'
шм^идту и Ш у Чу а н-Л<ианг2 уд. вес FeBr2 4,636; т. пл. 684°. При на--
1 Чг ЪЛ t z, E. В i r k, Z. anbrg. Ch. 134, 131 (1924).
• О. Jiouigschmidtu. Shu-Chuan Liang.Z. anorg, Ch. 241,368 (1939).
Железо бромистое и бромное
133
певании на воздухе при 310° постепенно распадается на FesOa1 и Вгг. Легко
растворим'в воде, трудно—в пиридине (0,49:100 при 25°).
Б. Гидрат
FeBr2 • 6H2Q Мол. В. 323,78
Приготовление
растворяют железные рпилки в бромистоводородной кислоте, фильтруют
от избытка железа и выпаривают раствор, предохраняя от доступа воздуха
до кристаллизации. По охлаждении выпавшую соль отсасывают и перекри-
сталлизовывают из горячей воды.
Свойства
Светлозеленые гигроскопические таблички, растворимые в воде и спирте.
Из водных растворов между 90—100° выкристаллизовывается гидрат FeBr2 •
. 2Н2О в сине-зеленых кристаллах (Ш и м м е л ь), между 50 w 80° — оветло-
зеленый FeBr2 • 4Н2О (Фолькма н).1 ч
Растворимость FeBrt в воде
(Ё t а г d, S с h i m m e 1)
Таблица 98
г с
- 1,8
+12
21
30
41,8
48,5
°/о FeBra
50,3
52,2
53,9
55,4
57,2
68,4
t° С
49
52
65
75
83
°/о FeBr,
58,45
58,6 1
59,5
61,5
63,3
t° с
88
100
116
132
°/о FeBr»
63,6
64,8
66,6
70,2
Таблица 99
% FeBr,
5,39
10,74
Удельный
rt18
1,0469
1,0932
вес водных р-аство
% FeBr,
21,57
32,35
d18
1,1850
1,2756
ров FeBr2
°/о FeBr*
43,14
53,32
dls
a18
1,3660'
1,4560;
Ferrum sesquibroftia
turn
ЖЕЛЕЗО БРОМНОЕ (БРОМИД ЖЕЛЕЗА [3])
Ferric bromide
Eisenbromid
Ferribromid
FeBra
Мол. в. 295,60
Приготовление
1. Обрабатывают железные опилки избытком брома и воды, избегая
разогревания, чтобы не «получить FeeBrs. Пропускают через раствор- СОг, пока
выделяющийся газ не перестанет окрашивать в синий цвет иодокрахмальную
бумажку. Полученному раствору, не содержащему теперь Fe' ' и избьт а
брома, дают испаряться в эксикаторе над серной кислотой.
134
Железо
2. Нагревают 1 часть безводного FeBr2 с 2 ч. брома в запаянной труб^
в течение 6 час. при 170—180°. После удаления избытка брома умеренны^
нагреванием остается FeBra в виде коричневых гигроскопических листочков
с зеленоватым отливом.
^3. Растворяют железо в( бромистоводородной кислоте, добавив избыток
брома, и пропускают через раствор воздух или СОг до тех пор, пока отхо.
дящие газы не перестанут окрашивать в синий цвет влажную иодокрахмаль.
ную бумажку (доказательство полноты удаления брома). Выпариванием рас»
твора в эксикаторе над H2SO4 получают кристаллы FeBrs • 6Н2О.
4. Для приготовления раствора FeBrs по Шмидту1 сначала из 2,7 ц,
порошка железа, 50 ч. воды и 5,14 ч. брома приготовляют раствор FeBr* a
отфильтровывают его от избытка железа. Остаток промывают небольшим
количеством воды, и соединив все порции фильтрата, растворяют в теплой
жидкости еще 2,7 ч. брома. Полученный раствор следует защищать от света,
Свойства
Красно-бурые шестигранные таблички, легко растворимые в воде, спирту
и эфире. Гидрат FeBra • 6Н2О — тонкие темнозеленые кристаллы — плавите^
при 27°.
ЖЕЛЕЗА ГИДРАТ ОКИСИ (ГИДРООКИСЬ ЖЕЛЕЗА [3])
Ferrum oxydatum Ferric hydroxide Eisen hydroxyd
hidricum Eisenoxydhydrat
Ferrihydroxyd
Fe(OH)» Мол. в. 106,87
А. Обыкновенный гидрат окиси железа
Приготовление
1. По Гюттигу и Гарсайду2 270 г чистого кристаллического
хлорного железа обливают водой и, прибавив 100 мл HCL (уд. в. 1,19). разбавляют
водой до 1 л. 50 мл полученного раствора смешивают с раствором 2,6 г
NH4C1 в 50 мл воды и, разбавив до 1 л, подогревают до 70°. К полученному
раствору при хорошем перемешивании прибавляют по каплям 5%-ного NtbOH
в очень незначительном избытке. Осадок декантируют 2—3 раза теплой (50°)а;
водой, хорошо отсасывают на мембранном фильтре, снова 5—б раз
взмучивают в воде и отсасывают. Когда AgNOe перестанет давать муть в промыв*
ных водах, промывание "прекращают. Полученный препарат содержит около
0,12% хлора (СГ).
2. По Симону и Шмидту* ион СГ отмывается очень трудно. Для'
получения хорошего препарата авторы рекомендуют подогретый до 40°
раствор чистого Fe(N03)3 осаждать избытком NFUOH.^
Кирпично-красный гель декантируют около 30 раз, употребляя каждый раз
по литру горячей воды, и отфильтровывают на мембранное фильтре.
Свойства
Свеже осажденный гель отвечает формуле РеЮз'ЛгН20. При высушивании
сильно сокращается в объеме и образует пористые кусочки, блестящие в
изломе. Удельный вес препарата, высушенного на воздухе, равен 2 436 В
воде почти нерастворим (4,8- 10-*% при 18°). Растворимость в. кислотах'
зависит от «возраста» препарата: свежеосажденный легко растворим в минераль-
1 Ausfuhr. Lehrb. d. pharm. I, 839.
! ?' «Г/ H u l tlgV 4: Q,Ysl d e» Z- *nor8* Ch-™, 49 (1929),
'A. Simon, ТЪ. Scltmidt, «toll. 2. M, в6(№&>»
Железа гидрат окиси
135
пых и органических кислотах, постоявший же некоторое время растворим
весьма трудно. При нагревании постепенно теряют воду, переходя в РегОз.
Таблица 100
Обезвоживание Fe(OH)3 в зависимости от
температуры (A ami о)
/•С
100
200
•/о «tO
в препарате
10
1,5
С С
\ 300
700
Wo Н,0
'В препарате
1.0
0
Испытания
По ОСТ 10954—40 содержание Fe в препарате должно быть в пределах
52—60%. Наибольшие количества допустимых примесей указаны в табл. 101.
Таблица 101
Допустимые примеси в Fe (ОН)3
Примеси
Допустимое
содержанке
В °/о
1. Нерастворимые в соляной кислоте вещества . . 0,1
2. Хлориды (СУ) 0,005
3. Сульфаты (SO/) Ю,05
4. Медь (Си) • . I 0,05
5. Вещества, ке осаждаемые аммиаком (в виде
сульфатов) I 0,1
6. Азот (N) — общее количество, ........ . | 0,06
Б. Активный гель Fe(OH)3 (ферригель)
Приготовление
По Белоцерковскому1 к 1%-ному раствору ИеСЬ'бШО приливают
танкой струей при сильном перемешивании 15—20%-ный р-р NthOH, взятый
в очень небольшом избытке.
Перемешивают еще 3—5 мин. и промывают гель декантацией водой до
удаления СГ в промывных водах. Отмытый гель отсасывают и просушивают
сначала при 30° (5—6 час), затем длительное время при 50—60° (50—100 час.)
и наконец при 100—110° (6—8 час).
Свойства
Твердые зерна с блестящим изломом. Цвет от темного красно-бурого до
черного с антрацитовым блеском.
В. Коллоидный раствор (золь) гидрата окиси железа
Приготовление
1. По Ризенфельду2 растворяют 15 г хлорного железа в 50 мл воды
и фильтруют. Отдельно растворяют 12 г истертого в порошок (ЫН^гСОз в
• ^.В.АлексеевскийД.М. Белоцерковский и Т. К. Плачено в,
Практические работы по химии защиты, Оборонгнз, стр. 42, 1940,
8 Неорг. практ. 155.
136
Железо
50 мл горячей воды, охлаждают и 40 мл этого раствора приливают к раств<*
FeCb. От полученной жидкости отделяют */ю часть, а остальное нейтрализу1_
возможно точнее, прибавляя по каплям оставшийся раствор (NH^COa. fcw
падающий при этом Fe(OH)a растворяется при перемешивании снача^
быстро, затем медленнее и наконец остается в виде мути. Для растворен^
этой мути прибавляют намного отлитого вначале слабокислого раствора FeC'i*
Жидкость фильтруют и помещают в диализатор, где выдерживают до почт»
ТТЛ» ТТТТЛ-ГЧ-к 1Г7ТОПЛТТНП «г^чттА1-> /"" 1 ' ГТ/Л TTTTITZStltTt-TTjr 1ГГ\ ТТ ТТЛП ТТ ТТ 1_Тт5г ЛОЛФПЛП ТТ ГЛ TI -Vnn.._' •' "
полного- удаления ионов СГ. Полученный коллоидный раствор при хранений
на холоду в закрытом сосуде сохраняется неопределенно долго.
2. Для быстрой очистки золя Fe(OH)3 по Е. Гофману1 горячий золь
полученный гидролизом FeCh, вносят в ацетон; при этом выпадает Fe(OH&
a FeCb и НС1 остаются в растворе. Коагуляты растворяют в воде, и в
случае необходимости повторяют очистку еще раз.
ЖЕЛЕЗА ЗАКИСЬ-ОКИСЬ (МАГНЕТИТ)
Ferrum oxydato-oxy- Ferroso-ferric acid Eisenoxyduloxyd
dulatum Magnetic oxide of iron Magneteisen
Fe304 Мол. в. 231,55
Приготовление
1. По С. Гильперту и Т. Б е й е р у £ восстанавливают FesOs' при 4(0
водородом, предварительно насыщенным водяными парами пропусканием,
через несколько промывных склянок с теплой (30—50°) водой. Перед
лодочкой с Fe20s полезно поместить медную спираль для связывания кислорода,
Продолжительность реакции при навеске в 5 г FeaCb составляет около 5 час.
2. Смешивают растворы эквивалентных количеств FeSCU и Fea(S04)s и
вливают смесь в избытбк кипящего раствора КОН. Выпавший осадок быстро
промывают горячей водой (декантацией), отсасывают, отжимают и сушат в
атмосфере водорода над СаС1г или Щ$04. Состав препарата приблизительно
отвечает формуле гидрата FeeCh • Н2О.
Свойства
Безводный Fea04 — черный тяжелый порошок уд. в. 5Д6. Т. пл. 1540°. Во
влажном состоянии легко окисляется на воздухе до Fe203. При действии
небольших количеств кислоты образует* РегОз и раствор соли Fe' ';
продолжительное воздействие кислоты приводит к полному растворению препаратов
с образованием солей Fe*' и Fe'". Состав препарата, полученного
описанными выше методами, никогда не соответствует точно формуле Fe304. Гидрат
Fe304 • Н2О — темнобурый, иногда черный порошок, при нагревании
переходящий в РегОз.
ЖЕЛЕЗА ОКИСЬ (ОКИСЬ ЖЕЛЕЗА[3])
Ferrum oxydatum Ferric oxide, Eisenoxyd
Iron peroxide Ferrioxyd
Iron sestjffioxyde
Fe203 Мол. в. 159J0
Приготовление
1. РегОз образуется при прокаливании на воздухе всех гидратов и
кислородных соединений железа, а также Fe(NOa)3 и FeSCU- Так, н-апример3,
прокаливают в течение 2 час. на полном пламени буняеновской горелки Fe(OH)3.
(полученный по методу Г. Гюттигаи Г. Гарсайда (см. стр. 134).
1 Е. Н off man тл. Roll. Z. 73, 36Q П»35).
в S. НИ pert и Т. Beyer., Ber. »4, t608 (191K
» G. F, Н u 11 i g, H. E. T с ft a Is е г и Н'. К i 11 e 1, Z. anorg. Qi. 223, 240 (1936);
\i, Kitt el, Z. anorg, Ch. 221, 50 (1934}.
Железа закись-окись. Железа окись
137
2- По указанию Д. Н. Финкельштейна 100 г Ре(1ЧОз)з • 9НЮ
нагревают в большом фарфоровом тигле на электрической плитке. Вначале соль
спокойно плавится, образуя бурую жидкость, постепенно испаряющуюся. При
121° жидкость начинает кипеть, выделяя постоянно кипящую 68%-ную HNOs.
Постепенно жидкость начинает загустевать и необходимо частое
перемешивание, чтобы избежать толчков и разбрызгивания. Начиная со 130°,
непрерывно перемешивают жидкость фарфоровым шпателем, причем она
загустевает, образуя пасту (без перемешивания жидкость внезапно затвердевает в
сплошную массу). При 132° паста сразу рассыпается в порошок,
продолжая выделять пары HNOs.
Не переставая перемешивать, продолжают нагревание до полного
высушивания; весь процесс занимает 20—25 мин. Сухую массу растирают, переносят
в тигель и прокаливают в муфеле при 600—700° в течение 8—10 час. При
достаточной чистоте исходного нитрата железа полученный продукт отвечает
квалификации х. ч. Выход 95—98% теоретического-, т. е. около 19 г.
3. Для приготовления чистого препарата к нагретому до кипения раствору
закисной соли железа прибавляют вычисленное количество горячего раствора
щавелевой кислоты, причем выпадает закисное щавелевокислое железо. Его
отфильтровывают, тщательно промывают водой, высушивают и прокаливают
при доступе воздуха, непрерывно перемешивая. Выход 90—93%
теоретического. Получаемый препарат содержит 99,79—99,96% Fe20.3.
Можно исходить из продажной соли Мора, нагревая раствор последней
с металлическим железом для восстановления Fe* * " (испытание на полноту
восстановления — отсутствие покраснения пробы раствора от прибавления
KCNS). После фильтрования раствор осаждают Н2С2О4, как указано выше.
4. В фарфоровый котелок емкостью 4 л, снабженный крышкой, помещают
раствор 500 г Fe(N03)s • 9НгО в 2 л воды. Через трубку, проходящую до
дна котелка, пропускают не слишком сильный ток NH3, промытого щелочью и
водой. Время от времени перемешивают жидкость газотюдводящей трубкой.
По окончании осаждения жидкости дают отстояться, раствор декантируют и
промывают осадок горячей водой до удаления NOs' в промывных водах
(реакция с дифениламином). Отмытый Fe(OH).3 просушивают в фарфоровых
чашках, после чего прокаливают в течение 5—6 час. при 550—600°. Выход
96 г (96—97% теоретического)1.
5. При получении РегОз, служащего сырьем для приготовления Fe
высокой чистоты, исходный нитрат железа должен быть исключительно чист.
Путем многократной перекристаллизации Fe(NOs)s • 9Н2О К л и в с и Томпсон2
получили препарат, содержащий всего 0,005% Si и менее 0,001% других
примесей.
6. По Брандту3 целесообразнее всего исходить из химически чистого
железа. Последнее растворяют в НС1, раствор при нагревании обрабатывают
сероводородом, фильтруют и в фильтрате двухвалентное железо окисляют
в трехвалентное кипячением с небольшим количеством HN03. Смесь дважды
выпаривают с концентрированной НС1 и, растворив остаток в избытке
разбавленной НС1, несколько раз взбалтывают раствор с эфиром в большой
делительной воронке. Если исходный материал содержал Со, то содержимому
воронки дают отстояться, спускают через кран нижний (водный) слой и к
оставшейся в воронке эфирной вытяжке прибавляют Vio часть по объему смеси,
полученной встряхиванием НС1 (уд. в. 1,104) с эфиром. Сильно встряхивают,
снова сливают нижний слой и операцию повторяют.
Очищенную эфирную вытяжку фильтруют, эфир отгоняют (или просто
удаляют нагреванием на водяной бане), и» оставшийся раствор FeCb несколько
раз выпаривают с НЫОз. Последнее выпаривание ведут с добавлением NH4NO3.
Выпаривание целесообразно проводить в плоской фарфоровой чашке.
1 Реакт. и преп., 123-
2 Препарат был спектроскопически исследован на 61 элемент, из которых в наличии
оказались лишь Si, Al, Ca, Mg и Си (Н. Е. С le ve s а. Т. G. T horn pson, J. Pves. Nat, Bur.
Stand. 18, 595 (1937); С. 1937, II, 373.
»L. Brandt. Ch. Ztg. 32, 843
138
Железо
После выпаривания остается хрупкая соляная масса, легко отделяющаяся
от чашки. Ее истирают в ступке и порциями по 40—50 г умеренно прокали^
вают в платиновой чашке. Остаток несколько раз смешивают с сухим угле-;
кислым аммонием и вновь прокаливают при слабом красном калении, часто
перемешивая. Эту операцию повторяют до приблизительно постоянного веса
(точно постоянный вес не может быть достигнут, так как незначительное
количество FeaOs уносится парами (МНфСОз).
Свойства
В зависимости от метода получения РегОз представляет собой матовый или
блестящий красный аморфный порошок уд. в. 5,04—5,274 (Так называемая*
8-модификация FesCb имеет уд. в. 4,7). Т. пл. 1565°,т. возгонки 2000° FesG*
очень трудно растворима в кислотах; лучше всего в 16-кратном количеству
кипящей смеси 8 ч. H2SO4 и 3 ч. воды.
Испытание
По ОСТ НКТП 2853 препарат ч. д. а. должен содержать 69,8—70,1 %l Feb
(после высушивания при 120°). Наибольшие количества допустимых примесей
указаны в табл. 102.
Таблица 102
Допустимые примеси в Fe203
Примеси
1. Нерастворимые в НС1 вещества
2. Растворимые в воде вещества
3. Сульфаты (S04") .
4. Азог, общий из NO'3, NO'3 и пр
5. Щелочные и щелочноземельные металлы
(в виде сульфатов)
б Металлы, осаждаемые H2S
7. Влага ,
Допустимое
содержание
0,02
0J1
0,08
0,005
0,1
0,01
0,2
ЖЕЛЕЗО СЕРНИСТОЕ (СУЛЬФИД ЖЕЛЕЗА [2])
Ferrum sulfuratum
Ferrous sulfide
Iron sulfide
FeS Мол. в. 87,91
Eiserisulfid
Ferrosulfid
Приготовление
1. Для синтеза FeS следует брать мелко нарезанное листовое железо и
черенковую серу. По В. Рекшинскому2 в небольшой гессенский тигель
(около 10 см высоты и б—7 см в диаметре) помещают 150 г железной мелочи
и укрепляют его посредством звездообразной глиняной подставки в другом
тигле большего размера с отбитым дном. Размеры внешнего, служащего
муфелем, тигля подбираются такие, чтобы расстояние между стенками - тиглей
составляло 25—30 мм. Верхнее отверстие внешнего тигля прикрывается
асбестовым конусом с отверстием диаметром около 45 мм для выхода пламени.
Общий вид прибора изображен на рис. 30. Нагревание ведется на паяльной
газовой горелке.
Расположение частей прибора и высота пламени должны быть подобраны
так, чтобы пламя, нагревая непосредственно внутренний тигель, охватывало.
1 О. G 1 е m s е г, Е. G
• Изв. ИРЕА 1, 239 (1922}
Железо сернистое
139
со всех сторон и выходило через отверстие асбестового конуса. Нагре-
6яют железо до светлокрасного каления и с этого момента, не прекращая
накаливания, вводят через отверстие конуса серу кусочками величиной с
лесной орех. Прибавлять серу следует с такими промежутками, чтобы она
успевала прореагировать с железом. В противном случае железо охлаждается до
температуры, при которой уже не происходит реакции соединения, и сера
бесполезно испаряется. После прибавления приблизительно 75 г серы
содержимое тигля принимает вид подвижной жидкости — это расплавленное FeS
с примесью металлического железа.
Жидкость выливают на массивную чугунную плиту и дают затвердеть.
Тигель загружают новой порцией железа. Надо отметить, что при
повторных операциях тигель скоро трескается. Для лучшего его использования
следует операции проводить одну за другой, не дожидаясь охлаждения тигля.
Выход 210 г. При пользовании кузнечным горном вместо горелки можно
при достаточном объеме тигля увеличить
количества реагентов в 7—10 раз.
2. По В. Рекшинскому1 более
дешевым способом можно получить FeS из
пирита и железа по уравнению:
FeSs + Fe = 2FeS.
В прибор, подобный описанному в
способе 1, загружают около 100 г пирита (в
небольших кусочках), сверх которого
помещают рыхлым слоем- 70 г мелко
нарезанного листового железа. Подбирая более
пологий асбестовый конус» добиваются того,
чтобы пламя отражалось сверху внутрь
тигля. При этих условиях вначале
раскаливается железо и пары серы, выделяющиеся
из ггиптгта, соприкасаются с железом при
достаточно высокой температуре, образуя
FeS, которое стекает на дно тигля в виде
подвижной жидкости. Дальнейшие операции
проводятся так же, как в способе 1.
3. Приготовление вполне чистого FeS, не содержащего примеси окислов и
полисульфидов, является довольно сложной задачей. По методу Ю. В. К 9 р я-
к и н а 2 нагревают в фарфоровом тигле Z5 г порошка Fe и 30 г S до начала
бурной реакции. Полученный неочищенный продукт растирают и помещают
ровным слоем в фарфоровую трубку М, помещенную в электрическую печь
(рис. 31). Из газометра А пропускают смесь rbS и Нг (10—Лб% rhS),
которая перед реометром сушится СаСЬ. Скорость газа .(5—б л/час)
контролируется по реометру С, а давление в системе — по манометру D. После
вытеснения " воздуха из прибора включают печь и повышают температуру до
950—1050°. Через 3—4 часа прекращают нагревание и при помощи крана В
выключают газометр А и включают газометр Е, наполненный азотом. Азот
перед поступлением в прибор очищается от примеси кислорода в промывал-
ках F (с раствором CrS04> и С (с аммиачным раствором NFUCl,. в который
погружены спирали из медной проволоки) и сушится H2SO4.
После полного охлаждения трубки М в /токе азота слабо спекшуюся
серую массу выталкивают из трубки. Содержание Fe и S в препарате отвечает
формуле FeS в пределах точности анализа3.
Свойства
Хрупкая сероватая масса, легко растирающаяся в зеленовато-серый
порошок. Уд. в. 4,84. Т. пл. 1193—1197°. Для того чтобы FeS легко растворялось
* Из». ИРЕА 1, 240 (1922).
• ЖПХ 11, № 12, 1575 (1938).
•Включенный в систему газоанализатор R может быть удален из установки, если не пред.
еодагаетс» анализировать газообразные продукты реакции,
Рис. 30. Прибор для приготовления
сернистого железа
140
Железо
кислотах с выделением H2S, необходимо, чтобы оно содержало некоторь^
зоыток металлического железа. Хотя в этом случае к сероводороду прим^
в
изоыток металлического железа. Хотя в этом случае к сероводороду пример
шивается небольшое количество водорода, но при обычных лабораторных |
аналитических работах эта примесь не мешает. \
Испытание1
Сернистое железо, применяемое для получения сероводорода, должно
содержать не менее 65% FeS.
Рис. 31. Схема установки для получения чистого сернистого железа
ЖЕЛЕЗО СЕРНОКИСЛОЕ ЗАКИСНОЕ
(СУЛЬФАТ ЖЕЛЕЗА [2], ЖЕЛЕЗНЫЙ КУПОРОС)
Ferrum sulfuricum
oxydulatum
Green vitriol
Copperas
Ferrous sulfate
Schwefelsaures.
Eisenoxydul
Ferrosulfat, Eisenvitriol
FeSQ4 • 7H>0
Мол. в. 278,02
Приготовление
1.^Растворяют железо в виде проволоки или опилок в 15—т20%-но#- H2SO4
и нагревают, пока оставшееся железо совершенно не перестанет
растворяться. Раствор фильтруют в колбу, подкисляют H2SO4 и дают охладиться.
Затем насыщают жидкость сероводородом и, плотно закрыв колбу, оставляют
ее на 2—Э дня. Жидкость, сохранившую запах H2S, нагревают на водяной
бане и отфильтровывают от осадка (С, CuS, SnS, AS2S3 и др.). Над фильтра-:
i Испытание хим. реактивов, НО,
Железо сернокислое закисное
Ш
он пропускают чистый С02 до вытеснения воздуха, кипятят до испарения
половины объема, после чего дают раствору кристаллизоваться в атмосфере СОг,
На следующий день сливают маточный раствор, промывают кристаллы
сначала водой, затем спиртом и, поместив препарат между листами
фильтровальной бумаги, возможно быстрее высушивают его при 30°. Если получились
крупные кристаллы, их измельчают в ступке и образовавшийся порошок
отнимают между листами фильтровальной бумаги.
2. Для очистки технического железного купороса его растворяют в воде,
подкисляют H2SO4 и дают стоять несколько дней без доступа воздуха с
кусками железа или FeS. '(освобождение от меди и восстановление
трехвалентного железа в двухвалентное). Затем жидкость для освобождения от As, Sn
и др. насыщают сероводородом и, после стояния в течение нескольких дней,
нагревают и фильтруют. К раствору добавляют немного металлического
железа и выпаривают до кристаллизации ('или осаждают FeS04«7H20 спиртом,
как указано в способе 3).
3. Почти свободный от трехвалентного железа и довольно устойчивый на
воздухе препарат получают прибавлением к раствору FeS04 Эб^ного спирта
до прекращения выпадения осадка. Сильно перемешивают, собирают
кристаллический порошок на фильтр, промывают спиртом и сушат на листе
фильтровальной бумаги при комнатной температуре до исчезновения запаха спирта
(Тананаев рекомендует кристаллическую муку промыть для удаления
спирта прокипяченной водой, насыщенной угольным ангидридом). Содержание
НЮ в препарате обычно несколько меньше теоретического.
4. Для получения безводного FeS04 Кеппелер и Д'А н с * рекомендуют
нагревать осажденное спиртом FeS04 • 7Н2О ib сильной струе водорода, сначала
при 70°, затем при 150—200°, причем получается моногидрат FeS04« H26. При
повышении температуры до 250—300°, не прекращая тока водорода, получают
безводный препарат.
Свойства
Гидрат FeSCU • 7Н2О представляет собой моноклинические косые призмы
уд. в. 1,895—1,898. Совершенно свободная от Fe' ** соль — голубая -и в сухом
воздухе выветривается с образованием белого порошка, снова голубеющего от
действия воды. Растворим в воде и глицерине, нерастворим в спирте; По
Паунду2 раствор FeS04 очень мало окисляется кислородом воздуха.
Зеленый цвет препарата указывает на содержание окисного железа; такой
препарат притягивает из воздуха влагу, переходя в желтую основную соль.
Соль при 73° белеет и при 90° плавится; при 250° начинает разлагаться, теряя
£Оз (Форкранд3). Водный раствор при стоянии на воздухе выделяет
желтый осадок основной соли. Безводный FeS04 имеет уд. в. 3,4.
Растворимость FeSO • 7Н20 в воде
(Fran с k el)
Таблица 103
/•с
0
10
20,1
30,03
40,05
о/о FeS04
15,53
17.02
21,00
24,87
28,67
ГС
50,21
| 52
54,03
60,01
65,0
о/о FeSO,
92,71
33,42
3*,25
35,46
35,73
/°с
70,04
77
80
85,02
90,13
°/о FeSO,
35ДО
31,46
30,35
28,8 '
27,5
1 К е р р е 1 е г, D'A n s, Z. phys. Ch. 62. 89 (1908).
* J rT Pound, J. Sec. Снега, md. 55, Trans. 327 (1936); C. I, 2566 U937).
■ ■ • R. <ie Forcrand, C. r. 158, 21 (1014).
ш
Железа
Удельный вес растворов FeS04
Таблица /0|
о/о FeSO,
1
2
4
6
<? |
1,0085
3,oi8a
1,0375
1,0575
в/о FeS04
8
10
12
14
<У
1,07*5
1,1000
1,1220
1,1445,
°/о FeSO«
16
18
20
1,1675
1,1905
1,2135
Испытание
Наибольшие количества допустимых примесей, по ОСТ 3409> указаны
в табл. 105.
Таблица 10$
Допустимые примеси в FeS04• 7Н20
Примеси
Допустимое содержание в °/о
химически
чистом
в чистом для
анализа
в чистом
1. Нерастворимые в воде вещества . .
2. Хлориды (С1')
3. Медь (Си)
4. UHHK(Zn)
5. Мышьяк (As)
6. Щелочные и щелочноземельные
металлы ♦ . . • . . .
0.0D5
0,0005
0,002
0,005
0,00002
0,05
0.01О
0,002
0,005
0,010
0,00005
0,1
0,020
0,005
0,010
0,020
0,00005
0,2
ЖЕЛЕЗО СЕРНОКИСЛОЕ ОКИСНОЕ (СУЛЬФАТ ЖЕЛЕЗА [3])
Ferrum sulfuricum
oxydatum
Ferric sulfate
Fes(S04)3
Мол. в. 399,88
Schwefelsaures Eisen-
cxyd,
Ferrisulfat
Приготовление
1. По Г. Бор неману1 к нагретому раствору 100г FeS04 в 300мл воды
добавляют 18 г H2SO4 (уд. в. 1,84) и небольшими порциями приливают 8 г
концентрированной НЫОз, каждый раз выжидая окончания выделения окислов
азота. По окончании реакции делают пробу на присутствие Fe* * и, если
потребуется, добавляют еще немного HNOa.
Окисленный раствор выпаривают до консистенции сиропа (под конец на
песочной бане); при охлаждении жидкость затвердевает. Полученную массу
растворяют в воде (процесс растворения идет очень медленно) и снова
выпаривают. Эту операцию повторяют до получения соли, не содержащей HNOs.
Для удаления H2SO4 и воды соль растирают в порошок, помещают в
стеклянную трубку и нагревают в токе СО» до прекращения выделения паров.
Соль должна быть в нагретом состоянии канареечно-желтого цвета, а после
1 Неорг. nptn. 228; См. также A. R е с о и г a, Ann. СЫш. Phye. [8], П, 263 (1907); С. г. 144,
Железо сернокислое окйсноё
143
лаждения — белой; грязно-желтая окраска указывает на присутствие H2SO4.
выход 80-90%.
2 По Мильбауэ ру и Квадрату1 к 10 г истертого в тонкий поро-
шок FeS04 • 7Н2О приливают 100 мл концентрированной H2SO4 и, поместив
в горло колбы маленькую воронку, кипятят смесь около часа. При этом FeSOi
переходит постепенно в тонкие шестиугольные таблички Fe2(S04)s. Время от
времени пробу смеси рассматривают под микроскопом: если осадок полностью
состоит из указанных табличек, реакция закончена. Осадок отсасывают через
асбестовый или стеклянный фильтр, промывают спиртом и эфиром и сушат
пои Ю0° до постоянного веса. Состав полученного препарата близок к
формуле Fe2(S04)$.4tL*0.
Свойства
Белый или желтоватый порошок уд. в. 2,937—3,097, на воздухе постепенно
расплывающийся в коричневую жидкость. Fe2(S04)s способен образовывать
очень концентрированные водные растворы, но растворение идет медленно;
растворим в спирте, нерастворим в концентрированной H2SO4. Водный
раствор вследствие гидролиза [образование золя Fe(OH)3] окрашен в красно-
бурый цвет; добавление H2SO4 подавляет гидролиз, и раствор становится
почти бесцветным. При кипячении разбавленного раствора осаждается
основная соль.
Таблица 106
Удельный вес водных растворов Fe2(S04)3
(На get)
•fo Fet(S04)3
2
4
6
8
10
12
14
16
18
20
e/o Fe
0,56
1,12
1,68
2,24
2,80
3,36
3,92
4,48
5,04
5,60
d"
1,017
1,036
1,057
1,077
1,097
1,118
1,140
1,162
1,184
1,208
о/о Fet(S04)3
22
24
26
28
30
32
34
36
38
40
0/0 Fe
6,16-
6,72
7,28
7,84
8,40
8,96
9,52
10,08
10,67
11,20
di»
1.232
1,258
1.284
1,310
1,337
1,365
1,395
1,427
1 1,458
1,490
Испытание
1. Хлориды (СГ). Раствор препарата (1 : 50) после добавления 1 мл HNO*
(уд. в. 1,15) и 1 мл 0,Ш AgNOs должен в течение 1 мин. оставаться
прозрачным.
2. Нитраты (NOa). Раствор препарата (1:10) смешивают с равным объемом
концентрированной H2SO4 и после охлаждения добавляют кристаллик FeSCh»
• 7НгО; не должно появляться бурого окрашивания.
3. Двухвалентное железо (Fe*'). При прибавлении к раствору препарата
(1 : 10), подкисленному НС1 (уд. в. 1,12), нескольких капель
свежеприготовленного 10%-ного раствора K3Fe(CN)e не должно тотчас же появляться синего
окрашивания.
4. Тяжелые металлы. К раствору препарата (1 :50) прибавляют избыток
NH40H и фильтруют. Прибавление к фильтрату (NH4)2$ не должно вызывать
темной окраски или выпадения осадка.
• МПЬаиегн. Quadrat, Z. anal. Ch. 50, 601 (1911).
т
Железо
ЖЕЛЕЗО ХЛОРИСТОЕ рСЛОРИД ЖЕЛЕЗА [2])
Ferrum chloratum Ferrous chloride Eisencjilorur
Ferrochforid
А. Безводное
FeCb Мол. в. 128,76
Приготовление
1. По Эрдману1 .в тубулированную реторту, горло которой снабжено
газоотводной трубкой, возможно быстро помещают 20 г безводного FeCb и
пропускают через тубус реторты быструю струю тщательно высушенного
водорода (огнеопасно!). Реторту устанавливают на газовой печи, которую
можно зажечь лишь,, когда весь воздух из прибора будет 1вытеснен. Тотчас же
при зажигании печи начинается сильное выделение хлористого водорода; газ
поглощают водой, для чего удобнее всего присоединить газоотводную трубку
к склянке Тищенко, наполненной водой на 1/з.
Конец восстановления определяют по ослаблению выделения НС1 и по
превращению FeCb, находящегося в реторте, в белую кристаллическую массу^
Тогда нагревание прекращают и дают прибору остывать, не прекращая
медленного тока водорода. Еще не совсем остывшую реторту разбивают и
FeCU быстро переносят в заранее приготовленную сухую и теплую банку
с притертой пробкой, которая тотчас заливается парафином. (Еще лучше
запаять препарат в небольшие стеклянные трубки).
2. Очень удобный метод получения безводного FeCb разработан Д. И.
Рябчиковым и В.. М. Шульманом2. 100—200 г железных опилок
загружают в фарфоровую трубку (диам. 30 мм, длина 850 мм), помещенную в
электрическую трубчатую печь с платиновой обмоткой. Трубка соединена
с аппаратом для получения хлористого водорода. Другой конец трубки,
выступающий из печи на 16—20 см, вставлен в шамотный цилиндр, служащий
для конденсации сублимата. Одновременно со включением печи пускают
слабый ток хлористого водорода, скорость которого постепенно увеличивают до
200—250 мл/мин. Температуру поддерживают в интервале 890—900°.
Сублимат конденсируется частью в шамотном цилиндре, частью в выступающем
конце реакционной трубки; необходимо поэтому каждые 20—30 мин.
прочищать трубку длинным шпателем.
Свойства
Белые кубические кристаллы или чешуйки, напоминающие тальк, и
шестиугольные таблички, уд. в. 3,162. Обычный препарат обладает серовато-белой
окраской; при стоянии на воздухе становится чисто белым (гидратируясь) и
только после этого начинает окисляться, окрашиваясь в желтый цвет. Т. пл.
673—674°, т. кип. 1023°. Расплавленный FeCb хорошо проводит электрический
ток. Соль легко растворима в воде, метиловом и этиловом спиртах, ацетоне;
плохо растворима в пиридине.
Б. Гидрат
FeCl2.4H2C Мол. в. 198,83
Приготовление
1. Растворяют чистое металлическое железо в соляной кислоте (уд. в. 1,12),
употребляя избыток железа, чтобы избежать окисления получающегося FeCb.
По окончании реакции (выделение водорода — огнеопасно]) жидкость
нагревают до кипения и фильтруют в чашку, предварительно ополоснутую
концентрированной НС1. Во время охлаждения и кристаллизации к поверхности
раствора подводят медленную струю СОг. Выпавшие кристаллы отжимают
■между листами фильтровальной бумаги и сушат в токе СОг при температуре
не выше 30°. Выход 70% теоретического.
1 Хим. преп. 63.
* ЖПХ 7, 1162 (1934).
Железо хлористое
146
9 Л Г. Берг1 указывает метод получения очень чистого препарата
f?/,.4H2O, свободного от Fe*'*. Продажную соль, растворяют принагрева-
FeU2* Rnne' почти до насыщения и добавляют немного НС1 и х. ч. железа
нии в *и^
_ пооошке.
Колбу закрывают пробкой с бунзеновским клапаном, и когда жидкость
ио-бретет бледноголубой цвет, быстро отсасывают раствор на воронке
к охнера, налив в приемник 2—3 мл эфира. Горячий профильтрованный рас-
вор быстро переливают в круглодонную колбу, содержащую 2—3 мл эфира,
закрывают колбу пробкой с бунзеновским клапаном и охлаждают (не ниже
12 3°, так как в противном случае выпадает неустойчивый гидрат с большим
-содержанием воды). '
Выпавший мелкокристаллический осадок соли отсасывают на бюхнеровскои
воронке, направив на слой соли струю СОг (или создав атмосферу эфира).
Препарат высушивают в вакуум-эксикаторе над H2SO4 до начала
выветривания; вместо создания вакуума можно вытеснить воздух из эксикатора с
помощью СОг. Чем лучше высушен препарат, тем он устойчивее. Описанный
метод позволяет получить кристаллы бледноголубого цвета без зеленоватого
оттенка. При растворении они не дают реакции на Fe'-'c KCNS.
Свойства
Прозрачные голубоватые кристаллы #д. в. 1,937, принимающие на воздухе
травянисто-зеленый цвет вследствие частичного окисления. При легком
нагревании или при хранении над H2SO4 соль переходит в FeCh • 2Н2О. Во
влажном воздухе FeCb • 4НгО расплывается. Очень легко растворима в воде,
легко в спирте. Не растворима в эфире. Соль рекомендуется хранить в
запаянных стеклянных трубках, из которых воздух вытеснен инертным газом.
Растворимость FeCl2 в воде
(A b e g g)
Таблица 107
/• с
1,5
5
8
12,3
16
20,5
25
о/о FeCIi
33,6
34,7
36,7
37,6
ЗР.О
38,6
39,2
Г С
28,5
35,5
42,5
52,0
56
60
68,5
о/о FeClt
39,4
40,4
40,9
42,6
43,4
43,9
45,5
t • С
73
76,5
82
86
90,5
96
117,5
о/о FeClt
46,6
47,4
47,5
47,7
47,9
48,3
50,4
Таблица 108
Удельный вес водных ра створов FeCla
о/о FeCla
1
2
4
6
л18
d4
1,0075
1,0165 J
1,0348
1.05J5
0/0 FeCla
8
10
12
14
*?
1 0726
1,0923
1,1126
1,1336
0/0 FeCla
16
18
20
25
d18
4
1,1551
1,1771
1,1996
1,2596
1 Заводск. лаб. 5,235 (1936).
146
Железо
Испытание
По ОСТ 7397/551 препарат должен иметь содержание FeCh • 4НгО не мен^
99,7% и растворяться без образования мути в равном по весу количеству,
воды, подкисленной 1 каплей НС1 (уд. в. 1,12). Наибольшие количества д<>,
пустимых примесей указаны в табл. 109.
Таблица 109
Допустимые примеси в FeCl2 • 4Н20
Примеси
2. Сульфаты (SO/)
6. Щелочные и
щелочноземельные металлы (в виде сульфатов) .
Допустимое содержание в °/0
в чистом для
анализа
0,01
002
0,015
0,025
0,0002
0,04
в чистом
0,02
0,04
0,03
0,05
0,0002
0,10
ЖЕЛЕЗО ХЛОРНОЕ (ХЛОРИД ЖЕЛЕЗА [3])
Ferrum sesquichloratum Ferric chloride
Eisenchlorid
Ferrichlorid
А. Безводное
FeCb Мол. в. 162,23
Приготовление
1. По Эрдману1 поступают следующим образом-. В реторту с тубусом
емкостью 750 мл помещают .50 г хорошо очищенной от ржавчины (наждачной
бумагой) железной проволоки толщиной в 1 мм, нарезав ее на куски по б—
8 см. Через тубус реторты, закрытый корковой пробкой, проходит возможно
широкая стеклянная трубка, доходящая до слоя железа. Сильно нагрев
реторту на газовой плите, пропускают по трубке энергичную струю хлора,
высушенного промыванием серной кислотой в двух склянках Тищенко. Горло-
реторты закрыто корковой пробкой, через которую проходит трубка,
отводящая избыток газа; последний выпускают в тягу или же поглощают в
промывной склянке раствором NaOH.
После 1—2-часовой реакции прекращают нагревание и пропускание хлора,
вытесняют оставшийся в реторте хлор струей сухого СОг, разбивают реторту
на листе глянцевой бумаги и отбирают FeCh от обломков стекла и непроре-
агировавшего железа. Препарат быстро помещают в заранее приготовленные
сухие и горячие пробирки, которые тотчас же запаивают на паяльной лампе,
следя за тем, чтобы содержащие влагу продукты горения не попали внутрь
трубки. Выход около 130 г.
2. По методу И. Е. Ададурова2 в железную трубку, нагреваемую в
трубчатой электропечи, помещают смесь 20 г железных опилок и 8
г'древесного угля. Через трубку пропускают сильную струю хлора, содержащего
небольшую примесь воздуха. При 450—500° в приемнике собираются фиолетово-
черные кристаллы чистого FeCls. Выход около 45 г.
1 Хим. преп. 64.
» ЖХП, 203 (1929).
Железо хлорное №7
Свойства
Черно-коричневые кристаллические корки или большие гексагональные пла-
инки уд. в- 2,898, гранатово-красные в проходящем и зеленые с металличе-
им блеском в отраженном свете. При 306° плавится в подвижную красную
СК\дкость, КИпящую при 317° с частичным разложением. Однако уже при
Т(Ю° FeCls заметно, улетучивается/ На воздухе жадно притягивает влагу и
оасплывается. В воде растворимо. очень легко со значительным выделением
тепла. Легко растворимо в спирте, глицерине, эфире и ацетоне, трудно в
бензоле. Раствор FeCh в органических растворителях на свету восстанавливается
FeCh, причем растворитель окисляется или хлорируется; например, раствор
FeCh в этиловом спирте образует альдегид. :'
Б. Гидрат
FeCb-бНгО Мол. в, 270,3а
Приготовление
1. FeCh-6H20 можно получить, дав безводной соли расплыться на" воздухе
и поместив ее затем на длительное время в эксикатор над концентрированной
H2SO4.
2. Растворяют железо при нагревании в соляной кислоте (уд. в. 1,12).
Полученный раствор FeCh насыщают хлором до полного окисления
двухвалентного железа в трехвалентное. Когда проба жидкости перестанет давать
синий осадок с K3Fe(CN)e, пропускание хлора прекращают и жидкость,
фильтруют. Фильтрат выпаривают, добавляя время от времени немного крепкой
соляной кислоты (или пропуская в жидкость газообразный хлористый
водород), пока проба при охлаждении не будет полностью затвердевать.
3. По Энгелю для получения весьма чистого препарата обрабатывают
продажный продукт газообразным хлористым водородом, причем соль
расплывается. Прозрачную жидкость сливают с осадка основной соли и похМ^е-
щают под колоколом рядом с твердым КОН, где она вскоре затвердевает
в кристаллическую массу.
Таблица НО
г с
-27
0
10
20
30
371
Растворимость FeCl3 в
о/о FeCl8
38,3
.42,7
45,0
47,9
51,6
60,0
t° С
27,42
32,5 3
30*
50*
566
°/о FeCI,
68,6
72.0
73,2
75,9
78,9
воде
t° с
55?
73,5*
66»
80Ю
100Ю
°/о FeCl,
78,6
81,8
84,0
84,0
84,3
1 FeCl3 • 6Н,0, темп. пл.
• FeCl. • 6Ht J-f FeCla • 3,5H,0.
» FeCl, • 3,5 H,0, темп. пл.
• FeCl3 • 3,5 НаО + FeCl3 • 2,5Н,0.
в FeCla • 2,5 Н,0.
• FeCla • 2,5HtO, темп. пл.
« FeCl3 • 2.511,0 + Fe(V2H,0.
■ FeCl8 • 2-V-V темп. ил.
• FeC!3 • 2HtO -j- FeClf.
10 FsCla.
10:;
т
Железо
Удельный вес водных растворов FeCl3
Таблица /|
d17,5
1,012
1,060
1,078
1,087
1,095
1,113
1,131
1,150
1,170
1,191
1,212
1/34
1,256
1.268
1,280
1,292
1,304
1,316
°/о FeCl,
5
7
9
10
11
13
15
17
19
21
23
25
27
28
29
30 1
31
32
o/oFeCIa-GHjO
8,3
11.6
14,9
16.6
19,3
21,6
24,9
28,3
31,6
34.9
38.3
41,6
44,9
46.6
48.2
49,9
51,6
wO|2
1*17,5
1,328
1.34 J
1,352
1,с64
1.376
1,389
1,403
1,415
1,428
1,441
1,454
1,4 69
1,481
1.494
1,5'J7
1,520
U33
1,574
°/oFeCl3
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
1 в/о FeCl8.6H,0)
54,9
56,6
58,2
59,9
61,5
63,2
64,9
66,6
68,3
69,9
71,6
73,2
74,9
76,5
78.2
79,9
81,5
83,2
Таблица 112
Растворимость FeCl8 в этиловом
спирте (абсолютном)
t°,c
0
15
20
г FeCl3 на 1 г
сач6он
1,36
1.41
1,44
t°c
30
40
50
г FeCl3 на 1 г
свн&он
1,49
1.55
1,76
Таблица 113
Допустимые примесив FeCl3 • 6Н20
Примеси
1. Нерастворимые вещества . . , . !
Ь. Цинк (Zn)
8, Мишьяк (As) .
9. Щелочные и щелочноземельные
металлы (в виде сульфатов) . . .
Допустимое содержание в */0
в чистом для
анализа
0,010
аою
0,01
0.01
0,005
0/J03
0,002
0,002
1 0Д
в чистом
0.05
0,03
0,03
0,03
0,01
0,01
0,005
0,01
0,5
Золото
149
Свойства
Красно-коричневая, влажная на-ощупь, довольно мягкая кристаллическая
'масса, на вкус сильно горькая. Из водных растворов кристаллизуется иногда
t пОЛ'ушаровидных агрегатах лучисто-кристаллического строения. Во влажном
воздухе быстро расплывается. Рчень легко растворим в воде, растворим
(также в спирте, глицерине и эфире. Т. пл. 37°; улетучивается уже при 100°,
Выше 250° распадается на РеЮз, FeQ3 н Clg.
Испытание
Наибольшие количества примесей, допустимых по ОСТ НКТП 6280/265,
указаны в табл. 113.
ЗОЛОТО
Aurum Cold Cold
Au At. в. 197,2
А. Металлическое золото
Приготовление*
1. К р ю ее, работая с металлическим золотом франкфуртской очистки
(почти не содержащим платины), получил чистый препарат следующим путем.
.100 г золота растворяют в царской водке и выпаривают с НО, пока не
начнут выделяться незначительные количества Au или АиСЬ; тогда,
прибавляя по каплям царскую водку, выпавший осадок снова переводят в раствор.
Прозрачную жидкость (содержащую все же хлористое серебро) разбавляют
10 л воды и после суточного стояния отфильтровывают AgCI (около 0,025 г).
; Восстанавливают AuCh до металлического золота/ действием FeCl* или
[Н2С2О4 (при этом примесь Pt остается в растворе). Выпавшее золото
промывают сначала водой, содержащей НС1, затем чистой водой и сушат при 170—
'l80°. Затем смешивают его в платиновой чашке с чистой "концентрированной
H2SO4 и нагревают до такой температуры, чтобы выделялись густые белые
пары. Кислоту отделяют, Au кипятят с водой, сушат и несколько раз
сплавляют с KHS04 (500 г KHS04 на 55 г Au) для удаления небольшого
количества палладия. После этого препарат промывают и высушивают. Наконец
[золото для удаления Ir и Rh сплавляют с селитрой (остающийся бесцветным
'плав указывает на отсутствие этих примесей).
2. По Бендеру2 золотой лом по возможности измельчают и обливают
|В колбочке крепкой НС1. Нагревают на песочной бане и время от времени
'приливают по каплям концентрированную НМОз. После растворения
выпаривают раствор на водяной бане, тщательно предохраняя от пыли, во избежание
'^восстановления, до тех пор, пока проба красно-бурой жидкости не будет при
охлаждении затвердевать.
I Извлекают большим количеством воды, отфильтровывают выделившееся
AgCl и осаждают в горячем состоянии большим избытком раствора FeCte.
Золото, выпавшее в виде порошка, декантируют и несколько раз кипятят
с разбавленной НС1 до тех пор, пока кипящая жидкость не будет иметь даже
и следов желтого окрашивания (зависящего от Fe'*). Осадок
отфильтровывают через беззольный фильтр и сплавляют (в случае необходимости, если
требуется металл в слитке) с прибавлением буры или селитры (Эрдман).
При повторении сплавления вес не должен изменяться.
Если обрабатываются большие количества золотого лома, то можно из
фильтрата при помощи металлического цинка или железа выделить Pt, Pd и,
иногда. Те. Для отделения Pt можно сплавить золотой лом с КМОз; Pt вместе
с некоторым количеством Au переходит в шлак, который по выщелачивании
водою дает остаток, содержащий Au и Pt (П етт е н к о ф е р, Крюсс). Для
1 О получе ии предельно чистого Au см. Т. S. St as, Bull. Soc» Chim. Belg. 46, 377 (1Э37).
* Приг. иеорг. преп. 6U3.
150
Золото
восстановления Au можно пользоваться также разбавленным раствором чистота
FeSCX содержащим НС1. или H2SO4. Au, получающееся при вливании раствор!
золотой соли в раствор FeSCX выделяется в более раздробленном состояние
чем при смешивании растворов в обратном порядке. Л е воль рекомендуй
в качестве восстановителя треххлористую сурьму, которую растворяют в таков*
количестве НС1, чтобы прибавление воды не вызывало мути; этого раствора
берут 2 мл на 1 г Au и восстановление ведут при нагревании.
Щавелевая кислота (Н2С2О4) восстанавливает золото при нагревании, при,
чем., получаются прекрасные блестки или. губчатая масса с золотистым бщ
ском. Так как при реакции выделяется ССЬ и происходит вспенивание, сл$
дует пользоваться большим сосудом.
Т о м с е н и К р ю с с рекомендуют для восстановления применять SC%
Особенно нежный порошок золота получают, вливая раствор AuCts в раствор
HgNOe при 100°. Осадок для полного высушивания нагревают при 170—180°»
3. Для использования золота из отработанных гальванических ванн П лаге
рекомендует следующий способ. Глиняную или пористую фарфоровую
пробирку, наполненную раствором NaCi, помещают в раствор, содержащий Auf
В пробирку погружают цинковый стержень, соединенный медной проволокой
с медным листом, погруженным в золотой раствор. По мере разъедачия
медного листа его заменяют другим. После 3—4-недельного стояния Au полностью
выпадает на дно.
4. Для использования фотографических остатков, содержащих Au
(виражные ванны), Хоок предлагает довести раствор до щелочной реакции, при*
бавляя Na2COs и, смешав со спиртовым раствором анилина, оставить стоять
8 час. на солнечном свету. При этом Au полностью выпадает на дно; его соби/
рают и очищают одним из вышеуказанных методов.
5. Крафт1 получил крупные кристаллы Au (6 мм) 8-дневным нагреванием
5%-ной амальгамы золота при 80° и растворением ее в горячей HNOa
(уд. в 1,35). Кристаллы получаются вначале матовыми от приставшей ртути,
при осторожном нагревании становятся блестящими.
Свойства
В сплавленном состоянии золото — мягкий желтый металл1 способный вытя-
гиваться в тончайшие нити. Au, восстановленное из разных растворов разлйч»;
ными восстановителями, имеет и различные физические свойства. В порошке
Au — бурого цвета, в состоянии тончайшего раздробления — красного. Весьма:
тонкие листочки золота просвечивают синим и зеленым цветом, оставаясь жел*
гыми в отраженном свете.
Уд. в. плавленого золота 19,3336 при 17,5°. Т. пл. 1063°; т. кип. 2677°.
При охлаждении ниже красного каления в случае присутствия посторонних
металлов, появляется внезапно сильный зеленый свет (бликование). Застывая,
металл значительно сжимается.
Б. Коллоидальный раствор золота
П риго тогв л ение
1. Красный гидрозоль золота. По Оствальду2 нагревают до кипения
100 мл дестиллированной воды, к которой добавлено 5—10 мл 0,05%-ного
раствора АиСЬ, осторожно нейтрализованного содой. Прекратив кипячение,
добавляют 95%-ного спирта порциями по 5—10 мл при взбалтывании. Затем
продолжают нагревание 15—20 мин., пока окраска не станет вишнево-красной.
Раствор можно выпаривать, причем он не теряет окраски; к электролитам
раствор очень чувствителен: от прибавления, например, НС1 он в несколько
секунд принимает сине-фиолетовую окраску.
При употреблении более крепких растворов АиСЬ (напр. 0,01%) получаются
грубо дисперсные золи, яркозеленые в проходящем свете и мутные, желто-
коричневые в отраженном. -
*Krafft, Dingier, 168, 283.
• Кодл. химия, 7.
Золото хлорное 151
2. Голубой гидрозоль золота, а) По Гутбиру1 к смеси 100 мл воды и
Ю мл 0,05%-ного раствора АиСЬ „прибавляют на холоду несколько капель
сильно разбавленного раствора гидразин-гидрата* (1 мл продажного 50%-ного
раствора на 1 л воды).
б) К. Зенгелис и Е. Ста тис2 рекомендуют добавить к разбавленному
раствору АиСЬ 10%-ного раствора гуммиарабика и подвергнуть его
восстановлению водородом in statu nascendi. Полученный золь очень устойчив; он не
коагулирует даже при добавлении равного объема Ш НС1 или 0,5iV Ва(1ЧОз)г.
Частицы несут на себе отрицательный заряд.
в) По методу Б ри н им н ге р а3 1 мл раствора Аи'*' (содержащего
0,25 г Аи в 100 мл воды) смешивают со 100 мл возможно лучше очищенной
дистиллированной воды. К раствору приливают 6 мл 0,Ш КгСОз, нагревают
до кипения, добавляют 5 мл 0,5%-ного раствора аскорбиновой или изоаскорби-
новой кислоты и снова кипятят несколько минут. Получается очень прочный,
совершенно прозрачный красный или фиолетовый золь, свободный от невос*
становленного золота 4,
ЗОЛОТО ХЛОРНОЕ
Aurum chloratum Auric chloride Goldchlorid
Gold chloride
АиСЬ Мол. в. 303,6
Приготовление
1. По Дебрею действуют хлором на золотую фольгу при нагревании
около 300°. Хлорное золото сублимируется в виде крупных красивых кри-»
сталлов. Золото в виде порошка при 300° еще не переходит в АиСЬ.
2. По В инк л еру5 при 140° в течение нескольких часов пропускают?
струю хлора (промытого несколько раз ЫагСОз и высушенного) над фарфо-
ровой лодочкой, содержащей золото (полученное осаждением SO2 и
высушенное при 180°), и держат образовавшееся хлорное золото несколько дней
в эксикаторе над КОН.
3. Раствор золота в царской водке при выпаривании нельзя совершений
освободить от избытка кислоты, без того чтобы не образовалось некоторое
количество AuCl; поэтому получить нейтральный, свободный от кислоты раствор
АиСЬ можно только кипячением с водой остатка после выпаривания и от-
фильтровыванием выпавшего AuCl.
4. Кусочки золота растворяют в царской водке; если присутствует Си
(коричневый осадок при действии K4Fe(CN)e на пробу раствора), то разбавляют
водой и смешивают с избытком раствора FeS04. При этом Аи выпадает в виде
буро-черного порошка. Его промывают водой, растворяют в царской водке
и выпаривают досуха на водяной бане.
5. По Сингаловскому6 порошок химически чистого золота обливают
х. ч. НС1 и, нагрев раствор в колбе на водяной бане, пропускают сухой хлор.
По мере растворения Аи раствор сливают, остаток обливают свежей НС1 и
снова пропускают хлор. Операции повторяют, пока все Аи не растворится.
Раствор выпаривают при 50—60°, пропуская хлор и добавляя изредка
несколько миллилитров насыщенной хлорной воды. Упаривание продолжают до
тех пор, пока капля, охлажденная на часовом стекле, не будет быстро
кристаллизоваться. Тогда чашку переносят на полчаса в сушильный шкаф,
нагретый до 95°. Затем охлаждают чашку льдом при постоянном помешивании
фарфоровым шпателем. Кристаллы отсасывают и запаивают в ампулы.
1 Коля, химия, 7.
* К. Z e n gh е 1 i s, E. S t a ti s, Praktika (гр?ч.) 12, 474 (1937); С. II, 1938.
» Н. Brln tzin ger, Koll. Z. 73; № 1, 22 (П37).
4 Если восстановление проводить на холоду, то золь всегда содержит соединения Аи.
О зеленых и кяасных золях Аи см. также Н. Friedman а. Т. Т. Davis (J. phys. Ch.42, 1149
(1938); С. I, 1946 (1939); L. LBIrcumshaw, Trans. Faraday Soc, 34, 1236 (19b8); C, II, 1013 (1939).
к Ргар. Ch. 345.
9 Соли редких и цветных металлов, 48,
152 Известь
Свойства
Темная рубиново-красная или красно-коричневая гигроскопическая масса]
уд. в. 2,44, растворимая в малом количестве воды с красно-коричневой, а
в большом — с красно-желтой окраской. Т. пл. 196° *.
Легко растворимо в спирте и эфире. Совершенно нейтральный водный
раствор постепенно выделяет осадок металлического золота; кислый раствор
сохраняется без изменения.
И спытание
Препарат должен содержать не меньше 48,5% Аи Й не содержать примесе$
Ag, Си и тяжелых металлов.
ИЗВЕСТЬ НАТРОННАЯ
Natrium hydricum calce Lime soda Natronkalk
Приготовление
1. Равные части NaOH и СаО смешивают и прокаливают. По охлаждений
полученную массу раздробляют и хранят в плотно закупоренной банке.
2. По указанию Я. Н. Иващенко хорошая натронная известь получается'
следующим образом.
В большую фарфоровую чашку помещают раствор 135 г NaOH в 600 мл
воды и сразу всыпают из широкого стакана 1 кг свежепрокаленной СаО.
Тотчас же приливают раствор 65 г NaOH в ЭОО мл воды (Осторожно! Остер$*
гаться брызг/)
Свойства
Белая пористая масса в кусочках разной величины. Жадно поглощает влагу
и СОг, переходя в смесь Na^COs и СаСОз.
Испытание
По ОСТ ВКС 5454 препарат для обычного анализа должен состоять из
зерен диаметром 7—\0 мм (для анализа по Преглю 2—3 мм). Поглоти-.
тельная способность не менее 20% СОг по. -весу. Содержание азота
допускается не более 0,002%.
ИЗВЕСТЬ ХЛОРНАЯ (БЕЛИЛЬНАЯ ИЗВЕСТЬ)
Calcaria chlorata Calcium hypochlorite Calciumhypochlorit
Calcium hypochlorosum Chlorkalk
Bleichkalk
Приготовление
К 100 г чистой гашеной извести добавляют 3,5 г воды и хорошо
перемешивают. Полученную смесь помещают в широкую стеклянную трубку,
расположенную горизонтально; смесь должна заполнять не более половины сечения
трубки. Охлаждая трубку холодной водой, пропускают через нее хлор до
тех пор пока вес не перестанет увеличиваться. Теоретически должно
получиться 195,73 г хлорной извести. Препарат сейчас же помещают в плотно
закрывающуюся банку2.
Свойства
Белый или желтоватый порошок с запахом хлора, частично растворимый
в воде. При нагревании или на ярком свету хлорная известь разлагается,
* По „Спут.ику химика- на 1935 г. ч. II уд. в. АиС13 4,67 и т. пл. 288в„
9 Неррг. пред., 42.
Известь натронная и хлорная. Иод
153
рляя Ог и СЬ; необходимо поэтому хранить препарат в темном и холод*
3 месте. Кислоты (даже угольная) разлагают хлорную известь с
выделением СЬ.
Испытание
По ГОСТ В-1692-42 содержание активного хлора в процентах не менее:
сорте А — 35 (и не более 38), в сорте Б — 32, в сорте В — 28. Разрыв между
Содержанием общего и активного хлора — не более 2%
иод
Jodum
Iodine
At. в. 126,92
Jod'
Приготовление
1. Для получения чистого препарата растирают 1 ч. KJ и 2 ч. СаО с 6 ч.
продажного иода и помещают смесь в тонкостенный стакан,' находящийся на
песочной бане. Сверху стакан покрывают часовым стеклом, на которое
налито немного воды. При нагревании иод возгоняется и оседает на часовом
стекле в виде игольчатых кристаллов, в то время как
примеси хлора и брома, вытесняя иод из KJ, связы- ' ■—•
ваются калием. Для лучшей очистки возгонку повторяют
при возможно более низкой температуре и высушивают
препарат над СаСЬ в эксикаторе, шлиф которого не
смазан жиром. Операцию можно проводить также в
фарфоровой чашке, накрытой воронкой; еще удобнее
пользоваться прибором с охлаждением проточной водой
(рис. 32).
2. Для получения чистого иода по Конин ку1
нагревают в реторте с приемником 1 ч. высушенного и
истертого К J с 1,5—2 ч. К2СГ2О7 до прекращения
выделения фиолетовых паров иода. При этом получается иод,
не содержащий примеси KJ и с почти теоретическим
выходом.
3. Серуллас рекомендует промывать J водой,
высушивать между бумагой и: возгонять. Можно его
также растворить в алкоголе, профильтровать и осадить
водой.
4. Стае предлагает очищать J от С1 и Вг следую- Рис 32. Пр„бор для
щим^ образом. Взбалтывают 50%-ныи водный раствор возгонки иода
1 ч; KJ с 4 ч. иода, сливают жидкость и прибавляют
к ней пятикратный объем воды, причем значительная часть иода осаждается.
Выделившийся J промывают водою декантацией и перегоняют с водяным
паром. Дают стечь воде и сушат J под стеклянным колпаком над азотнокислым
барием (последний следует несколько раз менять). Для удаления последних
следов воды и KJ перегоняют иод два раза из смеси с измельченным в
порошок Ва(ОН)г, которого каждый раз следует прибавлять по 7*> от количества
иода, взятого для перегонки. •«,
5. Получение чистого J, свободного от хлора, посредством возгонки его
из смеси с KJ требует много времени, так как для успеха работы требуется
возгонку вести медленно и повторять не один раз. Поэтому Мюссе
предложил обливать в стакане произвольное количество J крепким раствором
KJ и, закрыв стеклянной пластинкой, нагревать до сплавления иода. После
охлаждения его легко вынуть; он будет совершенно свободным от примесей
С1 после того, как стечет маточный раствор. Последний можно употреблять
несколько раз.
6. 'Аппарат для возгонки больших количеств иода (до 500 г) описанный
»L UdeKoninck, Bull. Assoc. Beige, 17, 157; Ргар. Ох. Б9,
154
Иод
Рис, 33. Аппарат Корногя и Олсона для
возгонки иода
Корногом и Олсоном1, изображен на рис. 33. Он состоит из зкеа
катора, в крышку которого плотно вмазана парижской замазкой (plaster |
Paris) электрическая лампочка на 40—60 ватт. Провода припаяны к контай
там лампы; следует остерегаться короткого замыкания через замазку.
Металлическая поверхность цоколя должна быть целиком покрыта замазкой та^
чтобы металл не был обнажен внутри эксикатора. На черепицу или фарфор
ровую плитку С помещают стакан А с возгоняемым иодом и стакан В с P2Q,
(осушитель). При включении тока лампочка нагревает верхнюю часть экси*
катора (для регулировки нагрева полезно
включить реостат); испаряющийся нот
осаждается на более холодной нижней чт
\\ \fc\ сти эксикатора и на дне.
'' та 3 ' Для ускорения процесса возгонки реко*
мендуется изолировать крышку асбесто^
а нижнюю часть эксикатора охлаждать
водой.
7. Для удаления присутствующего иног*.
да йодистого циана по Герцогу смеши»
вают нечистый иод с железом, водой и
поташом, причем весь циан связывается
железом.
8. Для получения иода из йодных
„остатков (если они состоят из одних иод и-
дов) можно- применить также следующий
метод. Раствор концентрируют, переносят
■в круглодонную колбу, подкисляют H2SO4
и прибавляют избыток железоаммонийных
квасцов. В полученную смесь пропускают
струю водяного пара, причем иод перегоняется и собирается в приемнике
вместе с водой:
2HJ + Fe2(S04)3 • (NH4)2S04 - 24H20 =
= 2FeS04 + (NH4)2S04 + H2SO4 + 24H20 + J2.
Иод переносят на стеклянную воронку, дают стечь воде, после чего сушат
над СаСЬ или H2SO4.
Регенерация лабораторных йодных остатков
1. По Бейльштейну2 для получения иода из лабораторных остат,
ков сначала насыщают раствор Ыа2СОз и выпаривают; полученный остаток
прокаливают (в случае почернения продолжают прокаливание, цока масса
не побелеет), и обрабатывают избытком H2SO4. После этого смесь
подвергают воздействию окислов азота, получаемых действием Н1М0з на крах-мал3.
Пропускание окислов азота ведут до прекращения выделения иода.
Выпавший иод промывают, сушат над H2SO4 и очищают одним из указанных
методов.
2. По П. Д и к е н с у4 остатки выпаривают до небольшого объема,
разлагают КОН и сильно концентрируют. Полученный раствор помещают
в колбу для перегонки, подкисляют H2S04 f(l : 1) и добавляют К2СГ2О7,
после чего отгоняют иод перегретым водяным паром. Приемником служат
две соединенные последовательно эрленмейеровские колбы, охлаждаемые
водой. Иод собирают и очищают возгонкой или сушат длительное время
над СаСЬ.
Можно поступать и иначе. Сконцентрированный раствор подкисляют H2SO4
и окисляют иодиды, подводя по трубке, доходящей до дна сосуда, раствор
1 J. С о г п о g a. L. Olson, Ind. En?. Chem. Anal. Ed. 11, 551 (1939).
* В e i 1 s t e i n, Zeitlchift fur Ch?mie und Pharmazie, 13, 528 (1870).
» В объемистую колбу помещают 15г крахмала и i,0 г HN08 и нагревают до появления
красных паров. Тогда прекращают нагревание и, если реакция становится слишком бурной,
охлаждают колбу водой.
• St. & Eisen, 58, Jft 49, 1405 (1938).
Иод
155
NO Осевший иод через некоторое время отфильтровывают и очищают
гонкой или перегонкой с водяным паром.
в035 По методике, рекомендуемой М. К. Чукавиным1, к 25—30 л
атков добавляют 20—25 мл H2SO4, охлаждают и приливают К2СГ2О7
ло1_40 мл насыщенного раствора на каждый литр). Через 4—6 час. сливают
лкость сифоном, отсасывают выделившийся иод на воронке Шотта, про-
Жвают 3 4 раза водой и сушат в эксикаторе над СаСЬ или H_>SC>4. Полу-
Менный технический иод вполне пригоден для переработки на KJ. О
получении KJ из лабораторных остатков см. -стр. 179.
4. Скопившийся в аналитической лаборатории осадок СшЛг (после иодо-
метрического определения меди) может быть по Л. Н. Слотинцеву2
легко переработан на Лг обработкой осадка C112J2 серной кислотой и К2С1Ю7,
как описано выше (метод П. Дике не а3).
Свойства
Хрупкие темносерые пластинчатые кристаллы уд. в. 4,9424 с
металлическим блеском. Т. пл. 113,5°, т. кип. 184,35°. Образует пары фиолетового
цвета. Запах неприятный, несколько похожий на хлор; в очень
разбавленном состоянии напоминает запах свежих орехов. Иод плохо растворим
в воде (0,0181 г в 100 мл воды при 10°), хорошо растворим в спирте, еще,
лучше в эфире, сероуглероде, бензоле, хлороформе и других органических
растворителях.
Растворы иода имеют следующую окраску:
фиолетовую — в сероуглероде, четыреххлористом углероде, хлороформе;
красно-бурую — в толуоле, бромистом и йодистом этиле, параксилоле;
красную — в бензоле, хлористом и бромистом этилене, бензине;
бурую — в эфире, уксусной кислоте, спирте, ацетоне.
Хорошо растворим в HJ и растворах KJ с темнобурой окраской, образуя
вначале соединение Юз; однако раствор KJ может растворять значительно
большее количество иода, чем это следует по реакции
KJ + J2 = KJs.
Мало вероятно, впрочем, что при этом образуются полииодиды с еще
более высоким содержанием иода; скорее всего КЛз как вещество, сходное
с иодом, сильно повышает растворимость последнего5. Важным является
то обстоятельство, что вследствие непрочности полииодидов раствор иода
в К J ведет себя как раствор свободного иода.
Таблица 114
Растворимость иода в растворах йодистого калия
(iVLerz и Weith)
Удельный вес
при 7,9е
1,0234
1,0133
1,0668
1,Рс81
1,1112
.Содержание в
KJ
1,802
3,159
4,628
5,935
7,201
растворе в о/0
J
1,173
2,303
3,613
4,778
6,037
Удельный вес
при 7,9е
1,1332
1,1637
1,1893
1,2110
1,2203
Содержание в растворе в°/о
KJ
8,^63
10,036
11,031
■11.833
12,643
J
7,368
'8,777
9,°49
11,'82
12,060
1 Завод, лаб. 4, № Ц, 1408(1935).
• Завод, лаб. 3, № 5, 461 (1934).
» О выделении иода в виде CufJt см. С. Ю. Фа й н б е р г, Завод, лаб. 4, № 2, 236 (1935).
• Уд. в. жидкого/иода для люоой температуры (/; между 115 —1сб° может быть вычислен
по уравнению
4—3,94916+0,003267(120— t) Т. N а у d е г, C.I, 2961(1935).
• Э ф р а и м, Неорганическая химия, т. 1, Госгимтекиздат, стр. 209, 1932.
155
Иод. Кадмий
Иод сильно ядовит; пары его действуют разъедающе на слизистые оЩ
лочки; вкус сильно вяжущий и острый. Иод плохой проводник тепла и зд|
ктричества.
Испытание
По ОСТ НКТП 6277/257 содержание J в препарате ч, д. а. не ниж*
99,8%, в препарате чистом —не ниже 99,5%; наибольшие количеству
допустимых примесей указаны в табл. 115.
Таблица 115
Допустимые примеси в J2
Примеси
2. Хлор + бром (СИ + Вг')
V"
Допустимое содержание в °/о
в чистом для
анализа
0,02
0,005 |
в чистом
0,05
0.015
Jodum monochloratum
ИОД ОДНОХЛОРИСТЫЙ
Iodine chloride
JC1 Мол. в. 162,38
Jodmonochlo^id
Приготовление
Для приготовления раствора JC1,. пригодного в качестве окислителя, по
Лангу1 27,7 г KJ растворяют в небольшом количестве воды и к раствору
добавляют 400 мл НС1 (1 : 1). Затем прибавляют концентрированный
раствор 17,5 г йодновато кислого калия (КЛОз), 10 мл хлороформа и, по каплям,
0,5%-ный раствор KJ03 до обесцвечивания слоя хлороформа при взбалтьь
вании. Разбавленный до 500 мл раствор очень постоянен.
Свойства
Красное кристаллическое вещество с резким запахом, известное в двух
модификациях. Одна из них (д) имеет уд. в. 3,86 и т. пл. 27,17°, другая (р)
имеет уд. в. 3,66, т. пл. 13,92°. Будучи расплавлены, обе . модификации
вполне идентичны; т. кип. жидкого JC1 101.3°. JC1 растворим в спирте,
эфире, CCU, ледяной СНзСООН и концентрированной HG1. Водой частично
разлагается. Сильный окислитель.
КАДМИЙ АЗОТНОКИСЛЫЙ (НИТРАТ КАДМИЯ)
Cadmium nitricum Cadmium nitrate Cadmiumnitrat
Cd(N03)2 • 4Н20 Мол. в. 308,49
Приготовление
1. Растворяют чистый металлический кадмий, или его окись (CdO), или
углекислую соль (CdCOa) в разбавленной НМОз при нагревании.
Полученный раствор выпаривают на водяной бане и охлаждают для кристаллиза-
зации.
2. В чашку, помещенную в вытяжном шкафу, наливают 600 мл воды,
240 мл х. ч. HN0.1 (уд. в. 1,4) и постепенно прибавляют 130 г чистого метал-
1 L a n g, Z, anorg. Cb., 142, 280 (1925),
Иод однохлорйстый. Кадмий 'азотнокислый
157
ческого кадмия. По окончании бурной реакции чашку нагревают некото-
Л е время при 70—80°, пока не появятся бурые хлопья Fe(OH)3, что ука-
*вает на полную нейтрализацию. Фильтруют и упаривают фильтрат, пока
Згюба при охлаждении не будет кристаллизоваться. Охлаждают, кристаллы
тсасывают, промывают 10 мл дестиллированной воды и сушат при
комнатной температуре. Маточные растворы при упаривании дают еще
значительное количество кристаллов.
Свойства
Бесцветные иглы или призмы уд. в. 2,46, расплывающиеся на воздухе.
jjpH 59—60° плавится в своей кристаллизационной воде. Выше 100° отдает
одновременно воду и азотную кислоту. Безводный плавится около 350°; при
белее высокой температуре разлагается, переходя в CdO. Легко растворим
в воде (см. таблу 116).
Таблица 116
Растворимость Cd(N08)2
в воде
ГС
0
so
о/о Cd (N08).
52,3
58,4
/eC
40
1 59,5
о/о Cd (N03),
61,4
76.6
Таблица 117
Удельный вес водных растворов Cd(N03)2
•/(,Cd(NO,)t
2
4
6
8
IQ
12
1
1,0154
1,0326
1,0502
1,06£3
1.0869
1,1061
о/о Cd (N03)t
14
16
18
20
25
,18
1,1261
1,1468
1,1682
1,1904
1.2488
о/о Cd (NO,)!
30
35
40
45
50
,»
1,3124
1,3822
1,4590
1,5438
1,6356
Испытание
В препарате, в зависимости от степени его чистоты, допускаются
следующие наибольшие количества примесей (табл. 118).
Допустимые примеси в Cd(N03)2• 4Н20
Таблица 118
Примеси
Допустимое содержание в о/0
в химически
чистом
в чистом для
анализа
1. Хлор (СГ)
2. Сульфаты (S04';) . . . .
3» Железо (Fe)
4. Алюминий (А1) . • • •
5. Медь (Си) ♦
6..Другие тяжелые металлы
0,001
0,005
0 001
0,010
0,001
0,000
0.005
0,010
0,004
0,010
0,002
0,000
0,01
0,05
0,008
0,100
0,005
0,002
138
Кадмий
КАДМИЯ ОКИСЬ
Cadmium oxydatum Cadmium oxide Cadmiumoxyd
CdO Мол. в. 128,41
Приготовление
1. Чистую CdO поЛоримеруи Смису1 можно получить следующц
путем. К сильно разбавленному раствору Cd(NCh)2 прибавляют немкоЯ
NHUOH и осаждают избытком чистого (NH^CCh. Выпавший CdC03 пять щя
декантируют большими количествами воды, фильтруют через гладкий филь^ш
тщательно промывают и высушивают прямо на фильтре. Высушенный CdC»
снимают с фильтра (для предохранения от попадания волокон бумаги, щД
ставшую к фильтру часть порошка оставляют), истирают и прокаливаютИ
платиновом тигле до постоянного веса. Тигель во время прокаливания помщ
щают в отверстие, проделанное в листе асбеста; эта предосторожность им§|щ
целью защитить CdO от возможного восстановления продуктами горения.
2. Сушат Cd(N0.3)2 сначала на водяной, а затем на -песочной бане, посте!
пенно повышая температуру, до полного прекращения выделения окисл<щ
азота. Остаток растирают и прокаливают при 500—600°.
Свойства
Коричневый неплавкий порошок уд. в. 8,15. На воздухе постепенно белее$|
притягивая С02 и переходя в CdCOa. Около 900° диссоциирует. Водородощ
восстанавливается уже при 300°.
КАДМИЙ СЕРНОКИСЛЫЙ (СУЛЬФАТ КАДМИЯ)
Cadmium sulfuricum Cadmium sulfate Cadmiumsulfat
CdSQ4.8/зШ02 Мол. в. 256,51
Приготовление
1. Растворяют 16 ч. чистого металлического кадмия в смеси 15 ч. Н2$(Ц
(1:5) с 24 ч. 25%-ной НЫОз, фильтруют и испаряют жидкость в тей|
лом месте; выпадают большие бесцветные кристаллы. Перекристаллизациещ
соли из горячей воды получают препарат, свободный от HNOs. Вместе
металлического кадмия можно применять CdO или CdCOs» растворяя т
в H2SO4, без добавления HN03.
2. По С и н г а л о в с к о м у 3 для приготовления химически чистого трщ
парата, свободного от As, 100 г гранулированного металлического кадмия^
вносят в раствор 50 мл H2SO4 (уд. в. 1,84) в 250 мл воды и к смеси постер
пенно приливают раствор 40 мл HNOs (уд. в. 1,4) в 100 мл воды, Посл^
растворения кадмия раствор выпаривают досуха, остаток растворяют в Ь00.Щ
воды и отфильтровывают от нерастворившегося PbS04. Раетвор упариваю!
до начала кристаллизации, дают охладиться, и "смешивают с равным объемом^
спирта. Выпавшую соль отфильтровывают, промывают спиртом до удаления
HNOs и сушат. Полученный таким путем препарат не содержит ни As, ни Си.
3. Безводный CdS04, не содержащий Н2ЗО4, по Б р и н к м а н у и
Шмедингу* лолучается при медленном нагревании кристаллогидрата
до 500°.
Свойства
Большие бесцветные моноклинические таблички уд. в. 3,1, легко раствори*
мые в воде (см. табл. 119) и нерастворимые в спирте. При 100° теряет только
iLV]cmdse^.8SHTo.th,Z- aHOrg- СЬ- '' 364 (Ш2)-
• Соли редких и цветных металлов, стр. 78.
* О . В г i n k m a n п, W. S с h ш е d d In g, Z. anal. Ch, 114, 161 (1938).
Кадмия окись. Кадмий сернокислый
159
чекулу кристаллизационной воды, остальные = при слабом калении. Уд. в.
' Годной соли 4,7. Т. лл. 1000*. - ^^ ^
Растворимость CdS04 вводе
t° с
0
15
20
о/0 CdS04
43,0
43,2
43,4
Vе С
| 40
1 60
! 74
о/о CdS04
44,0
45,0
4$,7
f С
85
100
112
о/о CdS04
39,6
377
37,0
Таблица 120
Удельный вес водных растворов CdS04
в/. CdS04
2
4
6
8
10
<^8
4
1,0182
1,0383
1,0590
1,0803
1,1023
о/о CdS04
12
. 14
16
18
20
<8
1,1250
1,1485
1,1729
1,1982
1,2243
о/о CdS04
25
30
35
40
d18
4
1,2940
1,3714
1,4551
1,5470
Испытание
По ОСТ 5015 наибольшие количества допустимых примесей указаны
в табл. 121.
Таблица 121
Допустимые примеси в CdS04• 8/3 Н20
Примеси
Допустимое содержание в о/0
химически
чистом
в чистом для
анализа
1. Нерастворимые в воде вещества.
2. Щелочные и щелочноземельные
металлы (в виде сульфатов) . .
3. Цинк (Zn)
4. Алюминий (А1)
5. Хлориды (CV)
6. Свинец (РЬ)
7. Медь (Си)
8. Железо (Fe)
9. Таллий (Т1)
10. Мышьяк (As) ■ . .
П. Азот (N) общий из нитратов,
нитритов, аммиака и т. д
0,003
0,05
0,005
0,02
0,002
0,01
0,002
0,0002
0,005
0,00005
од
- 0,01
0,05
0,005
0,02
0,005
0,0005
0,01
0,0001
0,002
0,005
0,004
0,01
0,2
0,02
ОД
0,01
0,05
0,01
0,002
0,02
0,0002
0,006
160
Кадмий
КАДМИЙ УГЛЕКИСЛЫЙ (КАРБОНАТ КАДМИЯ)
Cadmium carbonicum Cadmium carbonate Cadmiumcarbonat
© CdCOs Мол. в1 172,42
Приготовление
1. По Крауту водный раствор 256,5 а чистого кристаллического серноки^,
пого кадмия (CdS04 • V3H2O) приливают к кипящему раствору 106 г ЫагСО^
2. По Эрдману1 100 г измельченного или гранулированного технич^
ски чистого кадмия обрабатывают в колбе 400 мл холодной H-N'Ck
(уд. в. 1,20). Растворение следует вести в вытяжном шкафу вследствие
выделения значительного количества ядовитых окислов азота. По окончания
растворения сливают раствор с остатка, содержащего все примеси, кроме Р$
и Zn, в фарфоровую чашку емкостью около 6 л, разбавляют 4 л горячед
воды и обрабатывают жидкость небольшим количеством (NHO2CO3 для оса*
нсдения железа. Фильтруют и добавляют к раствору около 120 г (NHOaCQfc
выпадает CdC03, причем Zn остается в растворе. Осадок углекислого Kajfr
мия промывают горячей водой декантацией в высоком цилиндре и
сохраняют в виде пасты или высушивают на водяной бане.
Свойства
Снежно-белый порошок уд. в. 4,238 при 40°. Нерастворим в воде, легко
растворим в кислотах. При прокаливании, теряя СОи, переходит в CdO.
Испытание
По Эрдману препарат испытывается обычными аналитическими
методами на Sb, Bi, Cu, Pft, Fe и Zn.
КАДМИЙ УКСУСНОКИСЛЫЙ (АЦЕТАТ КАДМИЯ)
Cadmium aceticum Cadmium acetate Cadmium acetat
Cd(CH3GOO)2 Мол1. в. 230,50
П риготовление
1. Растворяют CdO в горячей 30%-нОй СНзСООН, прибавляя CdO неболь*!
шими порциями. После внесения избытка CdO раствор фильтруют, фильтрат
выпаривают до консистенции сиропа и ставят для кристаллизации в
холодное место. Ввиду того, что Cd(CH3COO)2 склонен к образованию
пересыщенных растворов, рекомендуется после охлаждения внести в сиропообразную
жидкость затравку — кристаллик Cd(CH3COO)2. Выделившиеся кристаллы
отсасывают, промывают очень небольшим количеством ледяной воды и сушат
между листами фильтровальной бумаги.
Свойства
Большие прозрачные кристаллы, плавящиеся при 255°. Очень легко
растворим в воде, не растворим в эфире. Во влажном воздухе расплывается*
Испытание
Наибольшие количества примесей, допустимых по „ОСТ НКТП 7385/539,
указаны в табл. 122. ....
« Хны. open. 24.
Кадмий углекислый, уксуснокислый и хлористый
161
Таблица 122
Допустимые примеси в Cd (СН3СОО)2
Примеса
Допустимое содержание в о/0
в химически
чистом
1. Нерастворимые в воде вещества
2. Щелочные металлы (в виде
сульфатов)
3. Uhhk(Zii)
4. Алюминий (А1)
5. Хлориды (СГ) . • .
6. Сульфаты (SO/)
7. Свинец (РЬ)
8. Медь (Си)
9. Железо (Fe)
10. Таллий (П)
11. Мышьяк (As)
12. Азот (N) общий из нитратов,
нитритов, аммиака и пр
0,003
0,002
в чистом для
анализа
0,005
0,05
0,005
0,02
0,002
0,005
0,01
0,002
0,0003 1
0,005
0,00005
од
0,01
0,05
0,005
0,01
0,02
0,005
0.U01
0,01
0,0001
0,2
0,02
0,1
0,010
0,02
0,05
0,01
0,002
0,02
0,0002
0,004
0,01
0,006
КАДМИЙ ХЛОРИСТЫЙ (ХЛОРИД КАДМИЯ)
Cadmium chloratum Cadmium chloride Cadmiumchlorid
CdCL
Мол. в. 183,32
Приготовление
1. Для очистки технического CdCb, всегда содержащего цинк, поступают по
Грегеру1 следующим образом. Слабо кислый раствор продажного CdCh
осаждают сероводородом, выпавший CdS отфильтровывают и растворяют в
горячей концентрированной НС1. Фильтруют, если требуется; прозрачный
фильтрат разбавляют и осаждают кадмий большим избытком раствора чистого
(ЫН4)2СОз. После продолжительного отстаивания отфильтровывают выпавший
CdC03, тщательно промывают для удаления маточного раствора,
содержащего цинк, сушат и переводят прокаливанием в CdO. Последнюю растворяют
в НС1; раствор выпаривают досуха и остаток обезвоживают сильным
прокаливанием. Если хотят совершенно освободиться от образующейся основной
соли, то держат вещество в расплавленном состоянии в течение 6 час.
в токе сухого хлористого водорода.
2. По Сингаловскому2 200 г металлического Cd в ленте разрезают
на куски, погружают в раствор NaOH для освобождения от жира, обмывают
водой, погружают на несколько секунд в 10—12%-ную НС1 для удаления Fe
и снова промывают водой. Промытый металл обрабатывают в фарфоровой
чашке 500 мл НС1 (уд. в. 1,19). Когда пройдет бурный период реакции,
чашку подогревают на водяной бане до 40—50°. Раствор охлаждают,
фильтруют и фильтрат выпаривают в фарфоровой чашке на водяной бане,
возможно чаще разбивая корку соли, образующуюся на поверхности жидкости.
Кристаллы вместе с раствором переносят на воронку с фарфоровым или
платиновым конусом и тщательно отсасывают. Сушат, нагревая соль в
фарфоровой чашке на водяной бане, и переносят препарат в банки с притертыми
1 О г б g е г. Monatsh. 23, 520 (1904); Рг5р. Ch. Б64.
■ Соли редких и цветных металлов, 71.
№
Кадмий
пробками. Если препарат не чисто-белого цвета или сод'ержит Fe, его пер^$
кристаллизовывают из воды. щ
3* По Б э кет ер у и Г айн с у для получения безводного CdCb см^
шивают крепкие растворы 53,5 г NH4CI и 227 г CdCb • 2,5Hi>0. При
испарении смеси выпадают белые игольчатые кристаллы двойной соли (CdCb • NH4Cl)
После высушивания кристаллов над H2SO4 их нагревают в токе сухого хлей
ристого водорода до прекращения выделения NH4CI. Оставшийся распла-у
вленный CdCb при охлаждении застывает в кристаллическую массу.
Свойства
Безводный CdCb представляет собой белую массу уд. в. 4,05. Т. пл. 570<\
Т. кип. 964°. Легко растворим в воде (см. табл. 123), несколько раствору
также в метиловом и этиловом спиртах. Водная соль кристаллизуется с 2,5
мол. воды и имеет уд. в. 3,33. При 33,8° CdCV2,5H20 переходит в CdClaV
•2НзО; «иже—5,6° устойчив тетрашдрат CdCfe^bMX.
Таблица 1Щ
Растворимость CdCl2 вводе
t°c
-7
+1
+6
% CdCl2
43,5
47,6
49,7
t°C
10
19
25
°/о CdCl8
51.6
52,6
52,9
*ec
61
82
°,'о CdCl*
57,9
68,8
Испытание
По ОСТ НКТП 7400/554 в водном препарате CdCb • 2,5Н20 наибольшие
количества' допустимых примесей указаны в табл. 124.
Таблица 124
Допустимые пр
Примеси
1. Нерастворимые вводе вещества
2. Азот (N) общий из нитратов,
3. Щелочные металлы (в виде суль-
5. Свинец (РЬ)
6. Медь (Си)
7. Цинк (Zn)
8. Алюминий (А1) .
10. Железо (Fe) ....
11. Таллий (Т1)
>имеси в Cd(
2\% • 2.5Н20
Допустимое содержание.
в химически
чистом
0,003
0,001
0,05
0,005
0,01
0,002
0,005
0,02
0,00005
0,0001
0,02
в чистом для
анализа
0,005
0,002
0,1
0,01
0,02
0,005
0,01
0,05
0,0001
0,0002
| 0,03
В о/о
в чистом
0,01
0,005
0,2
0,02
0,05
0,01
0,02
0,1
0,0002
0,001
0,05
Калий азотистдкислый
163
КАЛИЙ АЗОТИСТОКИСЛЫЙ (НИТРИТ КАЛИЯ)
Kalium nitrosum Potassium nitrite Kaliumnitrit
KNO2 Мол. в. 85,104
Приготовление
1. По Рюсту1 в тигл'е нагревают до плавления 100 г КЖ)з и вносят
0рЦиями при хорошем размешивании железным шпателем 204 г
нарезанного свинца (можно применять также медь или цинк). Последний энергично
окисляется и реакция обычно заканчивается в 25—30 мин. Еще жидкую
маСсу выливают в железную чашку или на большой железный лист для
более легкого последующего измельчения. Хорошо измельченную массу
несколько раз извлекают кипящей водой и вытяжку фильтруют через
складчатый фильтр. В еще горячий фильтрат пропускают в течение пяти минут
(не дольше!) струю СО, после чего находящиеся в растворе со-единения
свинца выпадают в виде карбоната. После повторного фильтрования раствор
выпаривают на водяной бане, точно нейтрализуют разбавленной HN03 и
охлаждают. Выпадающие кристаллы К1МОз отфильтровывают и раствор
выпаривают досуха. Оставшуюся соль расплавляют и выливают на чистый желез*
ный лист; после затвердевания массу разбивают на куски и еще теплой
помещают в банки.
2. Высокопроцентный препарат, содержащий только небольшую примесь
серебра [в виде комплексного жжа Ag(N02)2], может быть получен по
методу Д. С. Славиной2.
К тонкой суспензии AgNCb постепенно прибавляют при перемешивании
теоретическое количество КС1; если проба фильтрата показывает избыток
КС1, то добавляют в смесь еще немного суспензии AgN02 и оттитровывают
из бюретки разбавл'енным раствором КС1, пока раствор не перестанет'
содержать kg* и СГ. Фильтруют, выпаривают фильтрат в вакууме при 200 мм
рт. ст. и перекристаллизовывают продукт из воды. По указанию автооа
анализ первой фракции кристаллов показал состав: 99,66% KNCb, 0,32% Н2О,
0,007% kg и отсутствие SO/'.
3. Другой метод Д. С. Славиной3 заключается в том, что раствор
КГ\Юз обрабатывается в течение 2 час. при нагревании на водяной бане
избытком губчатого свинца [полученного действием Zn на раствор РЬ(СНзСОО)г].
На 1 ч. К1МОз берут 2 ч. РЬ. По окончании процесса в раствор пропускают
струю СОг для осаждения свинца, причем СОг рекомендуется готовить
действием HNOs на мрамор. Фильтруют, фильтрат выпаривают до консистенций
масла и, отделив кристаллы КЫОз, обрабатывают масло 90%-ным спиртом
До прекращения выпадения кристаллов.
Раствор КЫОг выпаривают на водяной бане. Выпавшие кристаллы
отсасывают на воронке Бюхнера и сушат в вакуум-эксикаторе. Препарат
свободен от СГ, SO/' и тяжелых металлов, но содержит всего 80% KNO2'
(остальное KNOs). Выход 60%.
4. Наиболее удобный метод для получения KNO2 в больших масштабах,
разработанный Е. Никитиной4 в ИРЕА, основан на равновесной реакции:
К2СО3 + 2NaN02 ^ 2KN02 + Na2CO*
и различной растворимости образующихсяoKN02 и ЫагСОз.
Сырьем служит х. ч. (или ч. д~ а.) нитрит натрия и карбонат калия,
легко получаемые перекристаллизацией технических продуктов. Необходимо
знать содержание КгСОз и NaNO? в применяемых препаратах, так как важно
соблюдать определенное соотношение между компонентами —4% избытка
ИаЫОг против вычисленного по уравнению количества. К нагретому рас-
1 С. R й s t, An at. Z. Darst. anorg. Ргйр. 1903.
* Изв. ИРЕА, 10, 21 (1930).
9 Изя. ИРЕА, 10. 21 П930).
* ЖПХ 6, 12 (1933); Реакт. в препм 159.
164
Калий
твору 1500 г 80%-ного КгСОз в 1100 мл воды приливают горячий раств<ш
1323 г NaN02 в 1340 мл воды, после чего жидкость выпаривают на водяно^
бане до уд. в. 1,47 при 70° (или 1,5 при 20°). Образующуюся на поверхноет!
пленку соды размешивают, выпадающие кристаллы ЫагСО.тЮНгО времяДИ
времени вычерпывают, отсасывают и фильтраты присоединяют к основном^
раствору.
По охлаждении жидкости отделяют кристаллы Ыа2СОз'ЮНгО и подвер*
гают фильтрат вторичному выпариванию до уд. в. 1,62 (при 20°); теперь
при охлаждении выпадает уже KNCK загрязненный №2СОз и требуюцщ|
перекристаллизации. Осадок отфильтровывают и фильтрат вновь выпаривают
до тех пор, пока дно фарфоровой чаши не будет покрыто значительным
количеством кристаллов KNO2. Охлаждают, отсасывают кристаллы KNO2 и из
раствора последующими выпариваниями1 выделяют еще несколько фракции
кристаллов. После просушки при 60—70° при частом перемешивании препШ
рат, отвечающий квалификации х* ч. и ч. д. а., быстро охладив, пере*
носят в банку с притертой пробкой. Выход около 1,5 кг KN02 (75—80%),
Сложность методики позволяет рекомендовать ее только для получения
больших количеств препарата.
5. По Гагеру1 в раскаленный докрасна гессенский тигель вносят нет
большими порциями тщательно приготовленную смесь 100 г КЖ)з и 15 г
пшеничного крахмала. Прокаливание ведут до тех пор, пока взятая проба не
будет застывать в белую массу. Тогда плав выливают в фарфоровую ступку
или чашку, дают застыть, после чего экстрагируют разбавленным спиртом к
лолученный спиртовый раствор выпаривают досуха. Дальнейшую обработку
ведут по способу 1.
Таблица 125
Растворимость KN02 в воде
(А. В. Раковский и Д. С. Славина)2
/вс
0
20
40
о/о KNO,
73.65
75.41
77,0
*вС
60
80
28
о/о KNO,
77,75
78,98
• 80,35
Таблица 126
Удельный вес водных растворов KN02
•/о KNO,
1
2
4
6
8
10
12
1 Л17'5
1 **
1,005
1,011
1,024
1,036
1,049
1,062
1,075
о/о KNOt
14
16
18
22
26
30
35
40
4
1,088
1,102
1,116
1,144
1,172
1,203
1,242
1,284
о/о KNO,
45
50
55
60
65
70
,17,5
d4
' 1,329
1 1,378
1,430
1,484
1,540
1,598
1 Хим. реэктияы, 1.
* Иав. ИРЕА, 11, 20 (1930).
Калий азотнокислый
163
Свойства
Белая волокнисто-кристаллическая масса, уд. в. 1,92; в продажу обычно
поступает в виде белых или слегка желтоватых палочек; притягивает из
воздуха влагу и расплывается. (По М. Освальду совершенно чистый KNOa
не гигроскопичен.) Т. пл. 297,5°. Очень легко растворим в воде (см. табл. 125)#
£{е растворим в У4—100%-ном этиловом спирте; в разбавленном спирте
растворим- Не растворим в ацетоне, мало растворим <в метиловом спирте.
Водный раствор имеет щелочную реакцию. Насыщенный водный раствор KN02
кипит при 13'2°.
Испытание
По ОСТ 4962 содержание KN02 для препаратов квалификаций дг. ч. и
ч д. а. должно быть не менее 90%, для* препарата чистого — не менее 85%*
наибольшие количества допустимых примесей указаны в табл. 127.
Допустимые примеси в KN02
Таблица 127
Примеси
1. Не растворимые в воде вещества
Допустимое содержание в °/0
в химически
чистом
0,003
0,005
0,005
0,0002
0,0002
в чистом для
анализа
0,005
0,010
0,010
0,0005
0,0004
в чистой
0,010
0,020
0,020
0,001
0,001
КАЛИЙ АЗОТНОКИСЛЫЙ (НИТРАТ КАЛИЯ, КАЛИЕВАЯ СЕЛИТРА)
Kalium nitricum
Potassium nitrate Kaliumnitrat
Nitre Kalisalpeter
KNO3 Мол. в. 101,104
Приготовление
1. Разбавленную HNOs нейтрализуют при помощи КОН или КзСОз;
раствор выпаривают до кристаллизации и полученный продукт перекристаллизо-
вывают.
2. Техническую калиевую селитру можно легко очистить двукратной
перекристаллизацией, если она не содержит больших количеств ЫаЫОз, СГ
и SO/'. От первой примеси избавиться чрезвычайно трудно, а СГ и SO/'
можно удалить, прибавляя по каплям сначала раствор Ва(Ж)з)2 до
прекращения образования BaS04, затем раствор AgNO.3 до прекращения выпадения
AgCl. Раствор фильтруют и для удаления возможно присутствующих в
избытке Ва* * и Ag' * нагревают с небольшим количеством КаСОз. Выпавшие
осадки ВаСОз и AgzC03 отфильтровывают, раствор слегка подкисляют чистой.
HN03 и выпаривают до кристаллизации.
Свойства
Бесцветные, прозрачные, устойчивые на воздухе призматические кристаллы
уд. в. 2,10, легко растворимые в воде и разбавленном спирте (см. табл. 128
и 129). и почти нерастворимые в абсолютном ^спирте. При 336,5° 'плавится в
подвижную жидкость*, которая при дальнейшем нагревании разлагается2
с выделением кислорода и образованием KNO?.
1 N. А. Р u s с h i n и М. R a d о ic i с, Z. anorg. Ch., 233. 41 (1937); С. 11,3125 (1937).
» О механизме разложения KNO, при нагревании см. С. 11, 3274 (1939).
166
Калий
Таблица 1Щ
Растворимость KN03 в воде
t°c
-3
0
+10
20
°/о KN03
11,0
11,6
17,7
24,1
t°c
30
40
50
60
Wo KNO,
31,5
39,1
46,2
52,5
t°c
70
80
90
100
°/o KN03
5&Q
03.8
67,1
71,1
Таблица №
Растворимость KN03 в этиловом спирте при 15°
Вгс. о/0 :пирта ■
10
20
30
о/о KN03
13,2
В35
5,6
Вес. о/о спирта
| 40
50
°/о KN08 J
4,3
2,8
Вес. °/о спирта
60
80
о/о KNOa
1.7
0,4
Таблица 130
Удельный вес водных р а створ о в *Щ03
о/о KN03
1
2
3
4
5
6
<*f
1,0045
1,0108
1,0171
1,0284
1 0299
1,0263
•/о KN08
7
8
9
10
12
14
w20
d4
1,0429
1,0494
i;0560
1,0627
1,0782
1,0899
' о/о KN03
16
18
20
22
24
,20
1,1039
1,1181
1,1326
1,1473
1,1023
Испытание
По OCT 956 содержание KNOe в безводном препарате должно быть не
менее 99,8%; наибольшие количества допустимых примесей указаны в табл. 131»
Калий бромистый
167
Таблица 131
Допустимые примеси в KNOe
Примеси
Допустимое содержание в о/0
1. Не растворимые в воде вещества
2. Хлориды (СГ) . .
3. Хлораты и перхлораты
4*. Сульфаты (SO/) ■., .
5. Тяжелые металлы . • . • . •. .
6°. Железо (Fe)
7. Кальций и магний (Са и Mg)
8. Фосфаты (Р04'")
9. Нитриты (NO/)
10. Иодаты (J03')
в химически
чистом
0,004
0,0005
0,001
0,005
0,0003
0,0005
0.0С5
0.0U05
0,0001
0,001
в чистом для
анализа
0.С05
0,001
0.002
0,010
0,0005
0,0008
0,010
0,001
0.0005
0,002
в чистом
0,010
0,002
0,005
0,030
0,001
0,001
0,020
0,001
0,001
0,004
КАЛИЙ БРОМИСТЫЙ (БРОМИД КАЛИЯ)
Kalium bromatum Potassium bromide Kaliumbromid
КВг Мол. в. 119,012
Приготовление
1. По Фишеру1 к раствору КОН уд. в. 1,33 прибавляют столько брома,
чтобы жидкость приобрела желтоватый цвет. Раствор, содержащий теперь
КВг и КВгОз, выпаривают досуха и остаток прокаливают в тигле с Vs частью
(по весу) угольного порошка. При этом КВгОз переходит в КВг. По
охлаждении массу выщелачивают водой, раствор фильтруют, выпаривают и охлаждают
до кристаллизации.
2. По Эльснеру2 суспендируют в 400 мл воды 20 г тонкого порошка
железа и прибавляют при взбалтывании 40 г брома. Полученный раствор
FeBr2 нагревают до кипения и смешивают с КгСОз; выпавший карбонат
железа отфильтровывают, а раствор упаривают и кристаллизуют.
3. Н. Петрова и 'А. Ильина3 приводят простой метод очистки КВг,
содержащего 0,8—1,2% СГ. К насыщенному раствору КВг приливают
концентрированную бромистоводородную кислоту. Выпадающие кристаллы быстро
отсасывают и высушивают. Содержание СГ в препарате не превышает 0,09—
0,15%. Троекратная очистка таким методом позволяет получить препарат
с содержанием СГ менее 0,005%.
4. Чистый КВгОз плавят в платиновой чашке на спиртовой горелке; соль,
сыделяя кислород, переходит в КВг. Кристаллы измельчают и прокаливают
в платиновой чашке при 500—600° до постоянного веса.
Свойства
Бесцветные, блестящие кубические кристаллы уд. в. 2,756, устойчивые на
воздухе. Т. пл. 728°, т. кип. 1380°. Вкус горько-соленый. Растворим в воде
(см. табл. 132), трудно .растворим в спирте (0,5:100). Водные растворы имеют
нейтральную реакцию.
1 В. Fischer, Lehrbuch der Chemie fur Pharmaceuten, 64 стр., 1885.
■ Хим. реактивы, 48.
8 Изв. ИРЕА. 16, 14 (1939).
168
Калий
Таблица щ
/°с
0
10,5
20
Растворимость
«/о КВг
34.5
38,1
39,4
t°c
40
60
80
i
КВт в воде
о/о КВг
43,2
46,2
48,8
t°c
100
140
180
о/о КВг
51,2
55,4
59
Таблица Щ
в/о КВг
1
2
4
6
8
10
Удельный вес водных растворов КВг
d4
1,0054
1,0127
1,0275
1,0426
1.05S1
1,0740
о/о КВг
12
14
16
18
20
22
d20
1,0903
1,1070
1,1242
1,1419
1,1601
1,1788
о/о КВг
24
26
28
30
35
40
<°
1,1980
1,2178
1,2383
1,2593 ;
1,3147
1,3746 .
И спытание
Наибольшие количества примесей, допустимых по ОСТ 2599, указаны в
табл. 134.
Таблица 134
Допустимые примеси в КВг
Примеси
1. Не растворимые в воде вещества
б. Азот (N) общий из нитратов,
6. Барий (Ва) ..,..«....
7. Кальций (Са)
8. Магний (Mg)
9. Тяжелые металлы .,'..■• . .
10. Железо (Fe)
11 Хлориды (СГ)
12. Иодиды (J') ......."...
Допустимое содержание в °/о
в химически
чистом
0,005
0,01
0,0005
0,002
0,001
1 0,002
0,(.02
0,001
0,0002 i
0,0002
0,05
0,02
в чистом для
анализа
0,01
0,03
0Ь002
0,005
0,002
одн
0,005
0,003
0,00П5
0,С005
0,2
0,05
в чистом
0,02
0.05
0.005
0,01
0,002
0,008
0,01
0,005
0,001
0,001
0,8
0,1
Калий бромноватокислый 169
КАЛИЙ БРОМНОВАТОКИСЛЫЙ (БРОМАТ КАЛИЯ)
Kalium bromicum Potassium bromate Kaliumbromat
КВгОэ
Жол, в. 167,012
Приготовление
1 По Г. и В. Бильц1 в раствор 62 г КОН в 62 мл воды при охлажде-
ии водой добавляют по каплям 80 г брома (осторожно!). Раствор вскоре
н аШИВается в желтый цвет и позднее из него выделяется кристаллический
°орошок КВгОз. Выпавшие кристаллы отсасывают и перекристаллизовывают
из 130 мл кипящей воды. Маточные растворы целесообразно переработать
На КВг. Для^ этого их выпаривают до образования полутвердой массы,
смешивают с о г просеянного порошка древесного угля и высушивают.
Полученный остаток прокаливают в большом фарфоровом тигле на горелке Флет-
чера, окружив тигель для сохранения тепла асбестовой трубкой. Затем
'Выщелачивают массу 120 мл воды, фильтруют, остаток промывают на фильтре еще
20 мл воды и фильтрат выпаривают до кристаллизации.
2. По методу А. Ильина2 в большой фарфоровый стакан наливают
1050 мл раствора КОН (содержащего 450 г КОН). Из капельной воронки
с трубкой, доходящей до дна стакана, очень медленно, при постоянном
помешивании приливают ПО г брома; затем раствор насыщают хлором. Окончание
насыщения раствора хлором определяют следующим методом: 10 мл
жидкости разбавляют 10 мл воды, кипятят до полного удаления Bra и СЬ (проба
иодокрахмальной бумажкой паров жидкости) и добавляют фенолфталеина —
не должно быть покраснения. Раствор охлаждают до 15°, отделяют 300—350 г
кристаллов (КВгОз + КС1) и промывают их 150 мл воды в течение
нескольких часов. Кристаллы отсасывают и повторяют промывку, взяв на этот раз
100 мл воды. После вторичного отсасывания получают 200—240 г сырого
КВгОз; для перекристаллизации его растворяют в 1 л горячей воды,
фильтруют, выпаривают до появления кристаллической пленки и охлаждают.
Выделившиеся кристаллы (около 150 г) вторично перекристаллизовывают из
800 мл воды. Полученные теперь кристаллы высушивают при 80—85°; выход
100—125 г. Маточные растворы от первой и второй кристаллизации при
выпаривании дают еще некоторое количество менее чистого препарата3,
Свойства
Бесцветные кристаллы уд. в. 3,24. При нагревании сначала не изменяется,
при 434° плавится и выделяет кислород, оставляя КВг. Растворим в воде
(см. табл. .135), мало растворим в спирте. Насыщенный водный раствор кипит
при 104°.
Растворимость КВЮ3 в воде (Seidell)
Таблица 135
t°c
1
ia
20
25
30
z КВгОз в 100 г
воды
3,1 '
4,8
6£
8,0
9,5
•/о КВгО,
3,0
4,6
6,5
7,4
8,7
ГС
40
50
60
1 80
100
г КВгОз в 100 г
воды
13,2
17,5
22,7
34,0
50,0
о/о КВгО,
11,7
14,9
18,5
25,4
33,3
1 Uebungsbeispiele.
1 Реакт. и иреп., 174.
9 Детальное описание метода получения КВг03 по этому же принципу дело С, Ктяьмя
Ь,вой (Реда. и преп., 173),
170
КйЛий
Удельный вес водных растворов КВг08
(К г е m e r s)
*—^
Таблица щ
о/о КВгО,
1
2
3
4
d19,5
19,5
1,009
.1,016
1,024
1,031
о/о KBrOg
5
6
7
dl9,5
19,5
1,039
1,046
1,054
о/о КВгОз
8
9
10
а™*
19,5
1,062
1,070
1,079
J/Lt nju тан и е
"По ОСТ 17383—39 препарат квалификаций х. ч. и ч. д. а. должен
держать КВгОз не менее 99,8%, препарат чистый — не менее 99,5%; наибод
шие количества допустимых примесей указаны в табл. 137.
Таблица Я
Допустимые примеси в КВг03
Примеси
1. Не растворимые в воде вещества
2 Бромиды (Вг')
3. Сульфаты (SO/0 .
4. Хлориды и хлораты (СК) . . . .
5. Тяжелые металлы
6. Железо (Fe) . . .
Допустимое содержание в °/о
химически
чистом
в чистом для
анализа
0 002
0.005
0,003
0.03
0.0005
0,0002
0.005
0.02
0 005
0.05
0,0005
0,0005
'—
001
0.О4
0,01
од
0.001
0,001
Препарат должен удовлетворять требованиям ОСТ на щелочность или
кислотность. 5 г препарата растворяют в 100 мл воды; прибавление
к раствору 2 капель 1%-ного раствора фенолфталеина не должно вызывать
розовой окраски; от 1 капли 0,Ш едкой щелочи должна появиться розовая
окраска. (Необходимо вводить поправку на взятое количество воды.)
КАЛИЯ ГИДРАТ ОКИСИ
(ГИДРООКИСЬ КАЛИЯ, ЕДКОЕ КАЛИ, «ЕДКИЙ КАЛИЙ»)
Caustic potash-
Potassium hydroxide
Kaliumhydrat
Kaliumhydroxyd
Aetzkali
KOH
Kalium hydricum
Kalium hydroxydatum
Kalium oxydatum
hydricum
Kalium causticum
Приготовление
1. По Шуберту1 растворяют в железной чашке 300 г кристаллического
Ва(ОН)г в 1 л воды и прибавляют торячий концентрированный раствор 120 г
K2SO4 до тех пор, пока капля отстоявшейся жидкости уже не будет давать
осадка ни с H2SO4, ни с баритовой водой. (Проба производится на часовом
Мол. в. 56,104
» Schubert, J. prakt. Ch. 26, 117.
Далия гидрат окиси J71
сТекле, помещенном на черной бумаге.) Жидкость быстро фильтруют через
складчатый фильтр в стеклянную колбу и фильтрат малыми порциями воз-
'ложно быстрее выпаривают в серебряной чашке, пока не наступит спокойное
давление. Жидкую массу переливают в большую серебряную или никелевую
цашку, наклоняя'последнюю кругообразно—так, чтобы масса застыла ровным
слоем. Полученные корки еще в горячем состоянии помещают в банки с плотно
закрывающимися пробками, которые заливают парафином.
Примечание. При всех работах с расплавленными щелочами
следует защищать глаза плотно прилегающими (автомобильными) очка-
ми или стеклянным экраном.
2. По Бендеру1 обливают негашеную известь водой так, чтобы уровень
воды был на несколько сантиметров выше извести. По окончании гашения
извести образовавшуюся нежную кашицу вносят малыми порциями в -раствор
100 г КгСОз в 1000—1200 мл воды, нагретой до кипения в чугунном котле,
совершенно очищенном от ржавчины. Прибавку следующей порции извести
производят, когда предыдущая порция превратится в зернистый порошок.
Добавление извести прекращают, когда отфильтрованная проба жидкости не
выделяет более ССЬ при вливании в НС1. Смесь кипятят еще некоторое время,
дают охладиться (без доступа воздуха) и отстояться, после чего прозрачную
жидкость слийают сифоном (кипячением остатка с водой можно получить еще
некоторое количество раствора). Для удаления небольшого количества
растворенного Са(ОН)2 к раствору добавляют немного КгСОл. Раствор упаривают
сначала в железном котелке до уд. в. 1,16, а затем в серебряной или
никелевой чашке, как указано в способе 1.
3. Для очистки продажного КОН его настаивают некоторое время
с 96%-ным этиловым спиртом; прозрачный спиртовый раствор сливают с
примесей, остающихся на дне в виде Твердой и сиропообразной массы. Главную
массу сгтирта отгоняют, в остальное количество выпаривают в серебряной
чашке до сплавления КОН. Дальнейшая обработка производится, как описано
в способе 1.
4. Чистый КОН можно получить вымораживанием концентрированных
растворов продажного продукта. При этом КОН выделяется в виде твердого
гидрата, в то время как примеси остаются в маточном растворе.
Свойства
Белая кристаллическая просвечивающая, чрезвычайно едкая масса уд. в.
2,12, т. пл. 360,4° (безводн.), т. кип. 1324°. Жадно поглощает из воздуха
влагу и СОг, расплываясь при этом и переходя в К2СОз. Свойства твердого
КОН как осушителя можно характеризовать следующими данными. Упругость
паров ШО над КОН по Б е к е р у при комнатной температуре составляет
0,002 мм рт. ст. Воздух, просушенный пропусканием через твердый КОН,
содержит при 25° 0,002 мг/л, а при 50° 0,007 мг/л НгО2. В воде растворим
чрезвычайно легко с сильным разогреванием (см. табл. 138). КОН не
рекомендуется хранить © банках с притертыми пробками, так как последние быстро
«заедает».
Таблица 138
Растворимость КОН в воде
*°с
— 9
+ 8,8
15
22,5
«/о КОН
48,1
50,5
51,7
53,4
ГС
32,8
33
49
88,5
о/о КОН
5Д,7
57,0
53.5
62,5
ГС
110
134,6
143,0
о/о КОН
66,4
70,2
75,7
1 Неорг. преп. 424.
» Ц. S, Booth a. L. H.Mac I n t у г е, Ind. Eng. Chem. Anal. Ed. 2, 12 (1930).
172 Калий
Таблица /J
Удельный вес водных растворов КОН
4
1,007
1,014
1,022
1,029
1,037
1,045
1,052
1,060
1,067
1,075
1,083
1,091
1,100
1,108
1,116
1,125
1,134
1,142
1,152
1,162
1,171
1,180
1,190
1,200
1,210
1,220
1,231
1,241
1,252
1,263
lf274
1,285
1,297
1,308
1,320
1,332
1,345
1,357
1,370
1,383
1,397
1,410
1,424
1,438
1,453
1,468
1,483
1.498
1.514
Градусы Боме
(В6)
Г
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26 .
27
28
29
30
31
32
33
34
1 35
36
' 37
1 38
| 39
1 40
1 4*
42
43
44
45
46
47
48
49
Градусы Твед-
дела
(Tw)
1,4
2,8
4,4
5,8
7,4
9,0
10,4
12,0
13,4
15,0
16.6
18,2
20,0
21,6
23,2
25.0
26,8
28,4
30,4
32.4
34 2
36,0
38,0
40,0
42,0
44,0
45 2
48,2
50,4
52,6
54,8
57,0
59,4
61,7
64,0
66,4
69,0
71,4
74,0
76,6
79,4
82,0
84,8
87,6
90,6
93,6
96,6
99,6
Ю2,8
1С0 вес. ч. раствора
содержат вес. ч.
К.0
0,7
;1,4
2,2
2,9
3,8
4,7
5,4
6,2
6.9
7,7
8,5
9,2
10.1
10,8
11,6
12,4
13,2
13,9
14,8
15,6
16,4
17,2
18,0
18,8
19.6
20,3
21,1 |
21,9
22,7
23,5
24 2
25,0
25,8
26,7
27,5
28,3
29,3
30,2 -
31,0
31,8
32,7
33,5:ф.
34,4%
3S,4
36,5
37,5
38,5
39.6
40,6
кон
0,9
1,7
2,6
3,5
4,5
5,6
6,4
7,4
82
9,2
10,1
10,9
12,0
12,9
13,8
14,8
15.7
16,5
17,6
18,6
19,5
20,5
21,4
22,4
23,3
24,2
25.1
26,1
27,0
28,0
28.9
29,8
30,7
318
32.7
33,7
34,9
35.9
36,9
37,8
38,9
39,9
40,9
42,1
43,4
44,6
45,8
47,1
48,3
1 •»•»
1 л раствора
содержит t
к4о 1 кон
7
14
22
30
39
49
57
66
74
83
92
100
111
119 .
129
140
150
159
170 n
181
192
203
214
226
237
248
260
272
284
297
308
321
335
349
363
377
394
410
' 425
440
457
472
490
509,
$Щ
549''
571
593
615
^
17
26.
36
46
58
67
78
88
99
109
119
1о2
143
153
167
178
188
203
216
228
242
255
269
- 282
295
309
. 324
338
353
368
385
398
-416
432
449
469
487
506
522
543
563
582
605
631
675
679
706
731
Калий дву хромовокислый 173
Продолжение табл. 139
d19
1,530
1,546
1.S63
1,581
1.597
1,615
1,634
Градусы Боме
(Be)
50
51
52
£3
54
55
56
Градусы Твед-
дела
(Tw)
106,0
109,2
112,6
110,0
119.4
123.0
126,8
100 вес. ч* рагтвора
содержат вес. ч.
К|0
41,5
42,5
- 43,6
44,7
45,8
47,0
48,3
кон
49,4
1 50,6
51,9
53.2
54,5
55,9
57,5
1 л раствора
содержит t
KgO
635
655
681
706
731
754
789
кон
756
779
811
840
870
902
940
Испытание
По ОСТ 17374—40 содержание едкого кали (КОН) должно быть в
препарате квалификаций х. ч. и ч. д. а. не менее 82%; для чистого — не менее
80%; наибольшие количества допустимых примесей указаны в табл. 1/1А
140.
Таблица 140
Допустимые примеси в КОН
Примеси
2. Сульфаты (SO/0
5. Азот (N) — общее количество из
нитратов, нитритов, аммиака и пр.
6. Железо (Fe)
8. Кальций (Са) • . .
10. Вешества, осуждаемые аммиа-
Допустимое содержание в °/о
в химически
чистом
0,005
0.005
0,01
0,003
0,001
0.0005
0,003
1 0,006
1.0
0,01
в чистом для
анализа
0,01
0.01
0,02
0,005
0,001
0,001
0,003
0,012
2,0
0,02
в чистом
0.025
0,03
0,1
0.07
0,001
0 002
0.003
0,03
4,0
0,1
КАЛИЙ ДВУХРОМОВОКИСЛЫЙ
(БИХРОМАТ КАЛИЯ, КАЛИЕВЫЙ ХРОМПИК)
Kalium bichromicum Potassium bichromate Kaliumbichromat
K2Cr20?
Мол. в. 294,21
П риготовление
1. В лабораторных условиях для приготовления чистого К2СГ2О7
целесообразно применять очистку легко доступного технического продукта (калиевый
хромпик). Для этого его дважды перекристаллизовывают из горячей воды,
быстро охлаждая. Полученный мелкокристаллический порошок отсасывают»
т
Калий
„^
промывают небольшим количеством холодной воды и расплавляют для удал$*
ния влаги. По охлаждении полученный слиток растирают в ступке и хранят*
банке с притертой пробкой. Очищенный таким путем продукт является вполад
пригодным для аналитических целей (установка титра и др.),
2. По указанию Ю. В. Клименко для приготовления препарата, при.
годного для установки титра, растворяют 100 г технической соли (хромпике
в 1 л горячей воды, фильтруют и, упарив фильтрат до V7 части первоначалу
ного объема, охлаждают при энергичном перемешивании для получения воз«
можно мелких кристаллов, так как они более -чисты. Кристаллы отсасывают,
промывают 2—3 раза ледяной водой и трижды перекристаллизовывают из ки«
пящей воды. Для перекристаллизации растворяют 100 г соли в 150 мл кипя»
щей воды и при энергичном размешивании выливают раствор тонкой струей
в фарфоровую чашку. По охлаждении кристаллы отсасывают на платиново}|
конусе или, в крайнем случае, на воронке с пористой стеклянной пластинкой,"
сушат 2—3 часа при 100—105°, измельчают и высушивают окончательно при
200° в течение 10—12 час. Сплавлять препарат не рекомендуется во
избежание образования следов СгЮз. Анализы полученной таким путем соли пока*
зали следующее содержание К2Сг207: 99,97%; 99,98%; 100,00%.
Свойства
Оранжево-красные, безводные триклинические иглы или пластинки уд. в.
2,70, растворимые в воде (см. табл. 141) и не растворимые в спирте. Желто*
красный раствор К2О2О7 слабо окрашивает лакмусовую бумагу в красный
цвет и обладает горьковатотерпким вкусом. При/ кипячении раствор
приобретает интенсивную Тёмнокрасную окраску. При 395° КгС?Ю7 плавится без раз?
ложения в темнокоричНевую Жидкость, застывающую по охлаждении в
кристаллическую массу. При белом калении распадается на СгяОз, Ог и К2СЮ4.
К2СГ2О7 сильно ядовит.
Таблица 141
Растворимость КзСг20? в воде
"III-
0
10
20
30
о/о КзСг^О,
4,43
7,5
11,1
15,4
t°c
40
50
60
70
% К2Сга07
20,6
25,9
31,2
36,2
t°c
80
90
100
о/о К2Сг807
41Л
•45,2
50,5
Таблица 142
Удельный вес водных растворов КгС^О?
°/о КйСг407
1
2
3
4
4
1,0052
1.Q122
1,0193
1,0264
% КйСгй07
5
6
7
8
*20
4
1,0336
1,0408
1,0481
1,0554
! о/0 К»Сг*07
9
10
И
12
d20
4
1,0628
1,0703
1,0779
1,0855
Испытание
По ОСТ 17392—39 содержание КгСггО? должно быть не менее 99,8% для
всех квалификаций; наибольшие количества допустимых примесей указаны
ь табл. 143*
Калий железисто синё род истый
175
Таблица 143
Допустимые
"""" ~~
Примеси
1 Нерастворимые вещества . . .
% Хлориды (С17)
4, Металлы, осаждаемые аммиаком
примеси 1
в К2СГ2О7
Допустимое содержание в Р/0
в химически
чистом
orooi
0,002
0,01
0,002
0,005
в чистом для
анализа
0,003
0,005
0,025
0,01
0,01
в чистом
0,01
0,01
0,05
0,025
0,02
КАЛИЙ ЖЕЛЕЗИСТОСИНЕРОДИСТЫЙ
(ГЕКСАЦИАНОФЕРР[2]-АТ КАЛИЯ, ЖЕЛТАЯ КРОВЯНАЯ СОЛЬ)
Kalium ferrocyanatum
Kalium borussicum
flavum
Potassium ferrocyanide Kaliumferrocyanid
Jellow prussiate of Gelbes Blutlaugensalz,
potash Ferrocyankalium.
K4Fe;(CN)e • ЗН2О Мол. в. 422,39
Приготовление
1. Технический продукт дважды перекрйсталлизовывают из горячей воды.
2. По указанию Н. А. Тананаева1 очень чистый и устойчивый препа»
рат получается при добавлении спирта к- раствору K4Fe(CN)e. Желтоватый
мелкозернистый осадок отфильтровывают, промывают спиртом й высушивают
при комнатной температуре до постоянного веса. Хранить препарат
рекомендуется в плотно закрытой банке.
Свойства
Светложелтые квадратные таблички или октаэдры уД. в. 1,88, горького
вкуса, устойчивые на воздухе. При нагревании До 100° теряют
кристаллизационную воду и белеют. При сильном накаливании выделяется азот й
остаются KCN и FeaC Растворим в воде (см. табл. 144), нерастворим в спирте
и эфире; водный раствор при.продолжительном хранении, особенно на солнеч*
ном свету, постепенно разлагается.
Таблица 144
Растворимость K4Fe(CN)6 в воде
/°с
0
10
20
30
о/0 K4Fe(CN)6
13,0
17,5
22,4
26,9
t°C
40
60
70
о/о K^FeCCNje
29,9
35,9
36,0
t°c
80
90
100
% К4Ре(СМ),
40,7
42,8
~43t6
1 Объемный анализ, 6 изд. ГОНТИ, стр. 17-4, 1939*
176 Калий
Таблица щ
Удельный вес водных растворов KiFefCN)^
°1б
1,0058
1,0116
1,0175
1,02.°4
1,0295
1,0356
1,0417
1,0479
1,0.42
1,0605
% K^e(CN)e.
•ЗНаО
1
2
3
4
5
6
7
8
9
10
о/о I<4Fe(CN)6
0,872
1,744
2,616
3,488
4,360
5,232
6.ЮФ |
6,976
7,848
8,720
«*15
15
1,0669
1,0734
1.0300
1,0866
1,0932 .
1,0999 i
1,1067 i
1,1136
1,1205
1,127S
•зн,о
11
12
13
14
15
16
17
18
19
20
% К^е(Сщ
9,592
10,464
11,336
12,208
13,080
13.952
14,824
15,696
16,568
17,440
Испытание
Наибольшие количества примесей, допустимых по ОСТ 3697, указаны
в табл. 146.
Таблица 14$
Допустимые примеси в K4Fe(CN)e• ЗН20
Прнмеса
1. Нерастворимые в воде вещества
Допустимое содержание в %
в химически :
чистом
0.005
0.005
0,005
0,01
в чистом для
анализа
0,01
0,01
0,01
0,01
• В ЧИСТОМ
0.02
0,02
0,02
0,02
КАЛИЙ ЖЕЛЕЗОСИНЕРОДИСТЫЙ
(ГЕКСАЦИАНОФЕРР[3]-АТ КАЛИЯ, КРАСНАЯ КРОВЯНАЯ СОЛЬ)
Kalium ferricyanatum
Kalium borussicum
rubrum
Potassium ferricyanide
Red prussiate of potash
Kaliumferricyanid
Rotes Blutlaugensal^;
Ferricyankalium.
K3Fe(CN)e
Мол. в. 329,25
Приготовление
1. По Уокеру К раствору 26 г K4Fe(CN)e в 200 мл холодной воды при*
бавляют 8 мл НС1 и медленно приливают холодный раствор 2 г КМп04 в
300 мл воды. Окисление можно считать законченным, когда капля раствора
с FeCls перестанет давать синий осадок и будет вызывать лишь
красно-коричневое окрашивание. Избыточную кислоту нейтрализуют при помощи СаСОз
или ВаСОз, фильтруют и выпаривают раствор на водяной бане. Первая
фракция кристаллов представляет собой чистый препарат, остальные отделяются
от хлоридов дробной кристаллизацией.
2. В раствор K4Fe(CN)e пропускают на холоду хлор, пока проба раствора
не перестанет давать синий осадок с FeCh. Жидкость выпаривают на водяной
Калий железосинеродистый
1?7
пе и дают закристаллизоваться. Препарат содержит большое количество
сеидов и должен быть 1—2 раза перекристаллизован из горячей воды.
Свойства
Тёмнокрасные ромбические призмы или квадратные пластинки уд. в. 1,85
/ измельчённом состоянии»— желтый порошок). Растворим в воде с образова-
^ем желто-зеленого (оливкового) раствора, не растворим в спирте. Из вод-
ного раствора осаждается спиртом.
Таблица 147
t°c
4,4
10
Растворимость
. 7о K,Fe(CN)e
24,8
26,8
t°c
13
15,6
K3Fe(CN)6 в
% K3Fe(CN)e
27,5
29,0
воде
гс
37,8
100
°/о K3Fe(CN)e
37.0
43,7
Таблица 148
Удельный вес водных растворов KsFe (CN)6
d20
4
1
2
4
6
%K3pe(CN)c
1,003
1,009
1,020
1,031
d20
4
8
10
12
14
o/0 K3Fe(CN)6
1,043,
1.054
' 1,066
1,077
rf20
4
16
18
20
°/o KBFe(CN)e
1,089
1,101
1,113
Испытание
По OCT 4103 содержание K3Fe(CN)e должно быть не менее 99%;
наибольшие количества допустимых примесей указаны в табл. 149. #
Таблица 149
Допустимые
Примеси
1. Нерастворимые в воде вещества
2. Железистосинеродистая соль
[Fe(CN)6'"'] .-......-.
примеси в
K3Fe(CN)6
Допустимое содержание в%
в химически
чистом
0,005
0,05
0,005
0,005
в чистом для
анализа
0,01
0,05
0,01
0,02
в чистом
0,02
0,10
0,02
0,04
178
Калий
КАЛИЙ ЙОДИСТЫЙ (ИОДИД КАЛИЯ) , \
Kalium jodatum Potassium iodide Kaliumjodid
KJ Мол. в. 166,02
Приготовление
1. По Г# и В. Бильц1 поступают следующим образом. К смеси 7—8 г
железных опилок с 50 мл воды, помещенной в эрленмейеровскую колбу
емкостью 250 мл, прибавляют небольшими порциями 25 г иода при частом
взбалтывании. Смесь немного нагревают до полного растворения иода, причем
раствор приобретает темножелтый или зеленый цвет.
Слитую с избытка железных опилок жидкость нагревают до кипения и
вливают в кипящий раствор 17 г КгСОз в 50 мл воды. Получившаяся довольно
густая смесь, помещенная в фарфоровую чашку, при дальнейшем нагревании
становится жидкой, выделяя осадок закись-окиси железа2 (FesO^:
3Fe + 4J2 = FesJe,
FesJe + 4К2СОз = Fe304 + 8KJ + 4СОг.
Небольшая отфильтрованная проба должна быть бесцветной и не содержать
железа (в противном случае добавляют к кипящему раствору еще немного
КгСОз). Фильтруют, фильтрат в фарфоровой чаше доводят до сильного кипения,
снова фильтруют, выпаривают до образования кристаллической пленки и
помещают в теплое место для кристаллизации. Наконец, дают раствору совершенно
остыть. Выпавшие кристаллы отсасывают и промывают малым количеством
ледяной дестиллированной воды. Промывные воды соединяют с маточным
раствором и вновь выпаривают до получения кристаллов. Выход 25—30 г.
2. В теплый растлор КОН (уд. в. 1,33) прибавляют иод до тех пор, пока
жидкость не приобретает желтоватую окраску. При этом образуются KJ и
К ЛОз :
6КОН + 3 J2 = 5К J + КЛОз + ЗН2О.
Для разрушения КЛОз к раствору добавляют крахмал (10% от веса
взятого иода), выпаривают досуха и остаток прокаливают в железном тигле.
Оставшуюся массу выщелачивают водой, раствор фильтруют и упаривают до
начала кристаллизации.
3. Нейтрализуют иодистоводородную кислоту поташом и полученный
раствор выпаривают.
4. По Кольтгофу3 осторожно сплавляют чистый KJ03 в платиновой
чашке на спиртовом пламени, причем он разлагается, переходя в KJ с
выделением кислорода. Когда масса затвердеет, приливают по охлаждении
немного воды (для растворения брызг солц, могущих содержать КЛОз),
выпаривают и снова нагревают, не доводя, однако, до плавления.
5. Технический иод, промытый 3—4 раза водой, растворяют4 в 30%-ном
растворе КОН. Для восстановления КЛОз пропускают в раствор H2S до
насыщения:
КЛОз + GHaS = КЛ + 3S '■+ ЗН20.
Жидкость подкисляют H2SO4 до слабо кислой реакции, нагревают на
водяной бане до удаления запаха H2S и обрабатывают суспензией ВаСОз для
связывания SO/'. Осадок отфильтровывают и промывают. Фильтрат вместе
с промывными водами можно либо непосредственно применять в качестве
1 Uebungsbeispiele.
" Fe3J8 является смесью эквивалентных количеств FeJ. и Fe.Je (P. О. Paternosto
* Е' t £2 U i n» Rev- Fac- Scienc. quim. 13, 57, 1938).
8 Объемный анализ, т. II, 289.
« M. К. Ч у к а в и н. Завод, даб. 4, № И, 1408 (1935).
Калий йодистые 129
а j{j( либо выпариванием до начала кристаллизации получить пре-
Р!п1т в сухом виде.
п р<Ггтпплажный KJ для очистки перекристаллизовывают 3—4 раза из спирта
„ с6уШа? при 400-500?
Регенерация KJ из лабораторных йодных остатков
(сЫ также Иод, стр. 154).
1 По указанию Б. Н. Райского1 к раствору добавляют СаСОз до
оекрапдения выделения СОг, вводят в раствор для связывания S04" и S-iOe"
порошок ВаСОз (5% от веса СаСОз), хорошо перемешивают и оставляют
а Ю 15 час. Затем сливают раствор с осадка и промывают последний
декантацией.
Фильтрат вместе с промывными водами сильно упаривают, еще раз
фильтруют и добавляют к нему раствор fo в KJ до бурой окраски, после "чего
нагревают до обесцвечивания (если окраска не исчезает в течение 20—30 мин.,
добавляют несколько капель раствора NaaSOe). Раствор фильтруют через
активированный или животный уголь, фильтрат выпаривают досуха, слегка
прокаливают (для разрушения органических примесей) и снова растворяют
в воде. После фильтрования раствор готов к употреблению.
2. Накапливающийся в аналитических лабораториях осадок C112J2 (при
иодометрическом определении меди) может быть по С. Ю. Файнберг2
легко переработан на KJ. Собранный в количестве 800—1000 г C112J2
помещают в фарфоровый стакан или чашку емкостью 2—3 л и обрабатывают при
нагревании и перемешивании 20%-ным раствором КОН, взяв его на 50—
100 мл меньше, чем требуется по реакции:
Cu2J2 -f 2КОН = С112О + 2KJ + НЮ.
Образуется нраснобурый осадок СигО, причем необходимо, чтобы
жидкость после 10-минутного кипячения оставалась еще слабо щелочной (на
фильтровальную бумагу помещают каплю 1%-ного раствора тимолфталеина
и каплю испытуемой жидкости — на другой стороне бумаги должно появиться
синее пятно).
Осадок отфильтровывают, фильтрат выпаривают до объема 1 л и при 60—
70° добавляют к нему растертый иод до слабо коричневой окраски, избегая
избытка (4—5 г). Не прекращая нагревания, насыщают раствор
сероводородом и дают отстояться. На другой день отфильтровывают осадок (CuS + S),
промывают его водой и нагревают фильтрат до удаления запаха HsS.
Полезно для обесцвечивания прокипятить 10—15 мин. с 1—2 г кровяного
угля и затем отфильтровать. Фильтрат выпаривают на водяной бане,
добавляют очейь немного Лг до буроватой окраски, которую уничтожают
добавлением нескольких капель раствора ЫагБОз. Наконец выпаривают* до
образования густой пленки соли и оставляют на несколько дней, после чего
выпавшие кристаллы отсасывают. Из маточных растворов можно получить
еще значительное количество KJ.
С. К. Файнберг рекомендует полученный из лабораторных остатков
препарат испытывать:
а) На окислители. 1 г препарата растворяют в 20 мл воды, добавляют
0,5 мл крахмала и 0,5 мл AN раствора H2SO4 — в течение 5 мин. не
должно быть пдсинения.
б) На восстановители. Раствор, выдержавший предыдущее испытание, от
добавления 1 капли OfilN раствора J2 должен приобрести устойчивую
синюю окраску.
Свойства
Твердые прозрачные или матовые кубики уд. в. 3,115, устойчивые в сухом
воздухе. При 693° соль плавится, затвердевая при охлаждении в
кристаллическую массу с перламутровым блеском. Т. кип.3 1331°, Очень легко рас*
1 Завод, лаб, 4, № 6, 706 (1935).
» Завод лаб, 4, № 2, 236 (1935).
» Waftenberg u. Schultz, Z. 1. Eiektrochemie, 27,568 (1921).
w
Калий
творим в воде с поглощением тепла. Водный раствор на свету постепенно'
желтеет, выделяя свободный иод.
Водный раствор KJ растворяет иод, образуя непрочное соединение KJa. •'
Растворимость KJb воде
Таблица Щ
ГС
— 22,65
— 11,53
0
+ 9,55
12,9
%KJ
51,8
53,8
55,7
56,4
57,9
t°G
21,05
29,1
37,3
45,75
55,05
%kj
58,9
59,9
61,0 '
62,1
62,8
ГС
65
74,75
86,35
11Р.2
120,0
% KJ
64,5
65,1
66.0
68,3
68,8
Таблица 15f,
Удельный вес водных растворов KJ
%kj
1
2
4
6
8
10
12
14
1 d20
4
1,0055
1,0130
1,0261
1,0487
1,0597
1,0761
1,0930
1.1104
% kj
16
18
20
22
24
26
28
30
d20
4
1,1284
1,1469
1,1660
1,1857
1,2060
1,2270
1,2487
1,2712
°;e kj
35
40
45
50
55
60
ri20
4
1,3308
1.3959
1,4672
1,545.7
1,6327
1,731
Таблица 152
Вес. % спирта
5,2
9,8
23
Растворимость KJ в спирте при 18°
%KJ
56,7
54,4
50
Вес. % спирта
■
29
28
45
%KJ
47,3
43,4
39,9
Вес. % спирта
59
91
%KJ :
32,5
10,2
5,8
Таблица 153
Растворимость KJ
в ацетоне
t°C
—2,5
+22
+56
10Э г ацетона
растворшют
3,08
2,38
1,21
Калий иодноватокислый 181
сТворим в спирте и ацетоне. Горячий спирт растворяет очень большое
„тип KJ; по охлаждении последний осаждается в виде игл. Растворим
количество ине (адб . 100 при 1qo. u . 100 при U6o). .
также ° с
Испытание
Наибольшие количества примесей, допустимых по ОСТ 7384/538, указаны
в табл. 154.
Допустимые примеси bKJ
Таблица 154
Примёсн
1. Нерастворимые в воде вещества .
2. Щелочность (КгСОз)
3. Сульфаты (SO/')
4. Азот(М)-(общий из нитратов,
нитритов, аммиака и пр.)
5. Барий (Ва) . , •
6. Кальций (Са)
7. Магний (Mg)
8. Тяжелые металлы
9. Железо (Fe)
10. Иодаты (JCV) . , , . .
11. Хлориды (О')
Допустимое содержание в °/о
химически
чистом
в чистом для
анализа
0,005
0,01 *
0,003
0,001
0,002
0,002
0,001
0,0002
0,0002
0002
0,02
0,01
0,03
0,01
0,002
0,004
0,005
0,003
0,0005
0,0005
0,005
0,05
0,02
0,05
0,02
0,002
0,008
0,01
0,005
0,001
0,001
0,01
0,1
Об испытании KJ, полученного из лабораторных остатков, см, стр. 179.
КАЛИЙ ИОДНОВАТОКИСЛЫЙ (ИОДАТ КАЛИЯ)
Kalium jodicum ■ Potassium iodate Kaliumjodat
Potassium iodate
KJO* Мол. jj. 214,02
Приготовление
1. По Г. и В. Бильц1 растворяют в колбе емкостью 0,2 л 30 г
хлористого калия (КС1) в 60 мл теплой воды, вносят туда 35 г иода и в
полученную тепловатую смесь вливают 1—2 мл горячей концентрированной HNOs.
Через 1—2 мин. начинается энергичная реакция с выделением хлора с
примесью иода (осторожно]). По окончании бурного периода реакции нагревают
До полного удаления хлора и после этого прибавляют еще 1 г иода.
Вновь нагревают и дают остыть; при этом выкристаллизовывается почти
весь КЛОз. Кристаллы отсасывают на платиновом конусе или, в^ крайнем
случае, на воронке с пористой стеклянной пластинкой, а маточный раствор
выпаривают досуха.
Сырой продукт, обычно содержащий кислую соль (КЛОз • НЛОз),
растворяют в 150 мл горячей воды и точно нейтрализуют раствором КОН. По
охлаждении выкристаллизовывается чистая соль с выходом, близким к
теоретическому.
2. По Н. Пут о хин у2 удобно готовить препарат, исходя из кислого
иодноватокислого калия КЛОз^НЛОз. Сырой КЛОз НЛОз растворяют при
* Uebungsbeispiele.
*Изв. ИРЕА, 4, 67.(1927).
182
Калий
нагревании в фарфоровой чайке, к горячему раствору прибавляют концепт^
рованного раствора КОН до щелочной реакции, фильтруют и охлаждают. №
этом выпадает почти чистый КЛОз. "*
Для окончательной очистки продукт перекристаллизовывают из дестилдй
рованной воды и высушивают при 180°. Препарат содержит 109% KjQp
Маточный раствор упаривают и получают еще некоторое количество п,р5
парата.
Выход около 60%, считая на взятый иод1.
Свойства
Бесцветные матовые или молочно-белые кубические кристаллы уд. в. ЗЖ
При нагревании плавится при 560° и при значительно более высокой тем'п&.
ратуре разлагается, подобно КСЮз, выделяя 02 и оставляя KJ. Водный ра&,
твор вполне нейтрален. В спирте не растворяется.
k.IOs является сильным окислителем; в смеси с органическими веществами
взрывает при ударе.
Таблица /Д|
/°с
0
20
30
40
Растворимость KJ03 в ]
100 г воды
растворяют
*KJ03
4,74 -
8,14
12,88
% KJQ3
в растворе
4,5
7.5
10,5
П.4
аоде
1 100 г болы
t°C I растворяют
| г KJO,
60
80
100
18,52
24,88
32,26
% KJ03
в растворе
15,6
19,9
24,4
i
Таблица I5S
Удельный вес водных растворов KJ03
о/о KJO,
1
2
<8
1,0071
1,0157
о/о KJ03
3
4
4в
1,0245
1,0333
•/о KJOe
5
6
<*
1,0424
1,0515
Испытание
По ОСТ 17382—.39 содержание КЛОз для препаратов квалификаций х. ч.
и ч. д. а не менее S9,8%, для чистого — не мед«е 95,5%.
Наибольшие количества допустимых примесей указаны в табл, 157; раствор
препарата должен выдерживать приведенное в ОСТ испытание на кислотность.
- Маточный раствор можно использовать для растворения новой порции сырого KJ03-HJ(V
При работе с большими ж>личествами можно, используя маточные растворы, довести выход
до 90—95°/о теоретического.
Калий иод но зато кислый кислый
183
Таблица 157
Допустимые примеси в KJOa
Примеси
Не растворимые в воде вещества .
i Иодиды (У) • -
3 Сульфаты (S04 ) •
4' Тяжелые металлы .........
5. Железо (Fe) .... . . .....
5 Хлориды и хлораты (U)
Допустимое содержание в °/0
i .химически
чистом
0,002
0,0Э1
0,005
0,0005
0,0002
0,005
в чистом для
анализа
0,005
0,002
0.01
0,001 ,
0,0005
0,02
0,01
0,005
0,02
0,002
0 001
0,05
КАЛИИ ИОДНОВАТОКИСЛЫИ КИСЛЫЙ (БИ.ИОДАТ КАЛИЯ)
Kalium bijodicum Potassium biiodate Kaliumbijodat
KJO3HJO3 Мол. в. 389,94
Приготовление
1. H. Путохин1 приводит несколько измененный метод Шаффера и
Гартмана2. 100 г КСЮз растворяют при нагревании в 250—300 мл воды
в колбе А (рис. 34), затем вливают
через боковое отверстие" рогатого форштос-
са 3—4 мл НС1 (уд. в. 1,12) и тотчас же
вносят 5—6 г иода3.
Вначале реакция почти не идет,
однако иод растворяется с образованием JC1.
По мере растворения вносят новые
порции иода (по 5 г). Постепенно реакция
начинает идти очень бурно с большим
выделением тепла, причем иод частично
возгоняется4. Прибавление иода следует
вести достаточно быстро, чтобы смесь все
время оставалась жидкой. Всего иода
вносят 115 г, иногда немного больше.
О конце реакции судят по неисчезающей
желтой окраске. Прибавляют несколько
кристалликов КСЮз, причехМ раствор
обесцвечивается; .и быстро выливают
жидкость в большую фарфоровую
чашку. Через несколько часов стояния
отсасывают выпавший КЛОз • НЛОз.
Если необходимо получить чистый препарат, сырой продукт перекристал-
лизовывают два раза из воды и высушивают при температуре не выше 120°.
2. По Кенингу 26 г KJO* растворяют в 125 мл горячей воды и
добавляют раствор 21 г йодноватой кислоты (НЛОз) в 45 мл воды, поДкислейной
шестью каплями концентрированной НС1. По охлаждении ввд&ШетЕя
втдгу
Рис. 34. Прибор для получения кислого
иодноватокислого калия
1 Язв. ИРЕА, 4, 66 (1927).
* P. A. Schaffer u. A. F. Hartmann, Journal of biological Chemistry, 45, 376 (1920).
3 Необходима предварительная проверка иода на отсутствие вспышкр с КСГО8. Описание
аппаратуры и методики для получения KJ03-HJ08 в больших масштабах—ck. Реакт. и преп. 18.
4 Газообгазн^е продукты реакции улавливаются водой в бунзенбвской колбе.
184 '
Калий
КЛ0а»Ш0з, который отсасывают, промывают холодной водой и трижды
перекристаллизовывают из троекратного количества воды. Препарат содет^
жит 99,97—100,00% KJOs-HJOa. ■
Свойства
Бесцветные кристаллы, растворимые в воде (3: 100 при 100°).
Испытание1
По техническим условиям НКХП 59—40 содержание КЛОз • НЛОз дл#
препарата х. ч. и ч. д. а. должно быть 100+0;2%, а для чистого 99,5%?
Наибольшие количества допустимых примесей указаны в табл. 158.
Таблица 158
Допустимые примеси в KJ03• HJQ3
Примеси
Не растворимые в воде ве-
Сульфаты (SO/'')
Допустимое содержание в а 0
в химически
чистом
0,002
0,001
0,005
0,0002
0,005
в чистом для
анализа
0.005
0.002
0,01
0,0005
0,02
в чистом
, 0.01
0,005
0,02
0.001
0,05
КАЛИЙ МАРГАНЦОВИСТОКИСЛЫЙ (МАНГАНАТ КАЛИЯ, ХАМЕЛЕОН2)
Kalium manganicum Potassium manganate Kaliummanganat
К2М1Ю4 Мол. в. 197,12
Приготовление
1. В тигель помещают небольшое количество КОН, смачивают водой и
вносят туда мелкоистертый KMnQi. Тигель постепенно нагревают,
размешивая массу стеклянной палочкой. Температуру доводят до слабого красного
каления и поддерживают ее в продолжение двух часов-.
Сухую порошкообразную массу охлаждают и помещают в хорошо
закупоривающуюся оапку [Ж о л л е с).
2. По Цвенгеру прокаливают 1 ч. МпОг с-3 ч* КЫОз, выщелачивают
массу и выпаривают темнозеленыи раствор в вакууме.
3. Насыщенный раствор 10 г КМп04 кипятят в фарфоровой чашке с 60 г
50%-ного КОН до 'прекращения выделения кислорода. При охлаждении из
раствора выделяется кристаллический порошок* Кристаллы переносят на-
тарелку из пористой глины. Препарат перекристаллизовывают. растворяя
в малом количестве воды или 10—15%-ном растворе КОН и дают раствору
испаряться в вакуум-эксикаторе над HsSO*. Выделившиеся почти черные
кристаллы отжимают на пористой пластинке и сохраняют в банке без
доступа воздуха.
4. В железном тигле сплавляют 30 г КОН и 15 г КСЮз. Прекратив
нагревание, постепенно всыпают в расплавленную массу 30 г МпОг, размешивая
: Исжцтан^ие ^им. Ц^зктивов, 17а
'В последнее время хамелеоном чаще называют ыарганцевокислий калий, КМ и О*.
Калий марганцовистокислый а марганцовокислый 185
0g пооволокой. Затем нагревают тигель несколько минут при красном
ЖеЛ «ии и охлаждают. Сплав выщелачивают водой, фильтруют через асбест
^выпаривают фильтрат в вакууме.
Свойства
Мелкие темнозеленые ромбические кристаллы. Растворим в воде; обра-
цийся зеленый раствор весьма нестоек. При стоянии, а также под дей-
зуКием кислот (даже ССЬ>) или большого количества воды цвет раствора
СТоеходит э красный вследствие образования КМп04.
КАЛИЙ МАРГАНЦОВОКИСЛЫЙ (ПЕРМАНГАНАТ КАЛИЯ)
Kalium permanganicum Potassium permanganate Kaliumpermanganat
Kalium hypermanga-
nicum
KMnO* Мол. в. 158,03
Приготовление1
1. Для очистки продажного препарата, его перекристаллизовывают,
отсасывают на 'платиновом конусе и сушат на стекле при обыкновенной температуре.
2. Выпаривают 500 г раствора КОН (уд. в. 1,475) с 105 г КСЮз; во время
выпаривания прибавляют постепенно при сильном перемешивании 180 г Мп02:
6КОН + ЗМпОг + КСЮз = 3K2Mn04 + KC1 + ЗНгО.
Выпаривание ведут до полного загустения. Охлаждают при размешивании,
истирают в порошок и нагревают в железном тигле до тёмнокрасного
каления. Сплав охлаждают, разбивают на мелкие куски и кипятят с большим
количеством воды, причем протекает процесс внутреннего окисления —
восстановления:
ЗК2Мп04 + 2НЮ = 2КМп04 + Мп02 + 4KOH.
Пропускание в раствор СОг облегчает прохождение реакции, поскольку
образующийся КОН связывается с C02t давая К2СОз:
ЗК2Мп04 + 2С02 = 2КМп04 + Мп02 + 2К2СОз.
Раствор КМп04 нагревают и пропускают СОг до малинового окрашивания
жидкости. Дав смеси отстояться, осторожно сливают малиновую жидкость
с осадка, фильтруют через асбест и выпаривают до кристаллизации. При
выпаривании получаются мелкие кристаллы КМп04.
Свойства
Хорошо образованные красновато-фиолетовые, почти черные кристаллы
с металлическим блеском уд. в. 2,71> растворимые в воде с
интенсивно-фиолетовой окраской. Концентрированные растворы имеют фиолетовый или
синевато-красный цвет, разбавленные растворы — чисто красные. Окраска настолько
интенсивна, что заметна при разведении 1 : 500 000 (в слое в 20 см).
Растворим также в метиловом спирте, ацетоне и СН3СООН. КМп04 очень
сильный окислитель; в щелочной и нейтральной среде окисляет по уравнению:
2KMnOi = К*0 + 2Мп02 + 1^02,
в кислой среде:
2КМп04 = КЮ + 2МпО + 2%02.
|. ...» ■..■>■ ...i...pi,uwr •?-*■■■•'■ '-'■'
» Приготовление КМпО, в значительных количествах целесообразно лишь в эаюдскиж
Условиях.
18ff
Калий
Таблица 159
t°c
0
5
10
15
20
25
30
35
40
45
Растворимость KMnO
100 мл
раствора содержат
г КМпО,*
3,0
л,з
4,0
4,9
5,8
7,0
8,2
9,7
10,6
13,1
°/о
КМп04*
2,75
—
4,01
4,95
6,00
7,1
8,3
9,7
11,2
12,7
ГС
£0
55
60
65
70
75
80
85
90
95
I
4 в воде
100 мл
раствора содержат
г КМп04 *
13,7
15,6
Ш
19,2
22,2
22,8
24,8
26,7
28,6
32,8
°/о
КМпО<*
14,4
16,2
—
20,0
—
—
—
—
—
—""
Таблица 160
Удельный вес водных раство-
о/о КМпО,
1
2
3
ров]
<5
1,0060
1,0130
1,0200
КМп04
о/о КМп04
4
5
6
<5
1.0271
1,0342
1,0414
Испытание
По ОСТ 4780 содержание КМп04 для препаратов квалификаций х. ч. и
ч.д.а. должно быть не менее 99,5%, для чистого препарата — не менее 99%;
наибольшие количества допустимых примесей указаны в табл. 161,
Таблица Щ
Допустимые примеси в КМп04
Приме-
Допустимое содержание в %
химически
чистом
в чистом для
анализа
1. Хлориды и хлораты (С1')
2. Сульфаты (SO/7)
3. Азот (N). . ... .
4. Мышьяк (As)
5. Нерастворимые вещества, исключая
Мп02
0,002
0,005
0,002
0,00002
0,005
0,005
0,020
0,U05
ОДЮ02
0,010
0,010
0,040
0,010
О,00ДО5
0,030
* По Брэдбери..
Калия метабисульфит. Калий над сернокислый, 187
КАЛИЯ МЕТАБИСУЛЬФИТ (ПИРОСУЛЬФИТ КАЛИЯ)
Kalium metabisulfu- Potassium pyrosulphite Kaliummetabisulf.it
rosum
К2З2О5 Мол. в. 222,31
Приготовление
Пропускают SO2 в горячий концентрированный раствор КгСОз или КОН.
При охлаждении выпадает K2S2O5 в виде белого кристаллического осадка.
Свойства
Мелкие белые кристаллы с запахом сернистого ангидрида, разлагающиеся
при 190° с выделением SO2. Легко растворим в ©оде; раствор по&азывает
кислую реакцию, на лакмус. Кислород воздуха постепенно окисляет K2S2OS
до K2SO4; в водном растворе окисление идет быстрее.
Таблица 162
Растворимость K2S2O5 в воде
t°c
0
10
20
40
% K8S406
22,1
26,6
30,9
39,0
*ec
60
80
90
% К^О,
45,4
51,6
54,4
Испытание *
Препарат испытывают: на хлориды (допускается слабая опалесценция от
AgNOs после выпаривания досуха 1 г препарата с Н2О2 и КОН и растворения
остатка в разбавленной HNOs), тяжелые металлы и железо (пссле
выпаривания досуха навески препарата с НС1 и растворения осадка в разбавленной
НС1 от добавления 'сероводородной воды, а также NH40H и (ЫШЪЗ не
должно появляться ни мути, ни окрашивания) и мышьяк (в препарате ч. д. а.
Допускается не более 0,0005% As при определении в аппарате Зангер-Блэка,
в препарате чистом — яе более 0,001%). Содержание K2S2O5 для препарата
ч. д. а.— не менее 95%, для чистого 90%.
КАЛИЙ НАДСЕРНОКИСЛЫЙ (ПЕРСУЛЬФАТ КАЛИЯ)
Kalium Dersulfuricum Potassium persulfate Kaliumpersulfat
K2S2O8 Мол. в. 270,31
Приготовление
1. По Эльбсу2 в большой сосуд, наполненный ледяной водой, помещают
широкую пробирку (или стакан) высотой приблизительно 14 см и диаметром
5 см. Внутри его подвешивают на проволочном* треугольнике открытую с обоих
концов стеклянную трубку диаметром около 2 см и длиной 11 см (можно
воспользоваться пробиркой с обрезанным дном). . .
Катодом служит петля из платиновой проволоки, окружающая
внутреннюю трубку и находящаяся недалеко от поверхности электролита. Анодом
1 Испытание хим. реактивов, 175.
VElbs, Z. f,- Elektrochemie. 2, 165П895;.
188
Калий
служит вплавленная в стеклянную трубку платиновая проволока, проходящее
через узкую внутреннюю трубку и доходящая почти до дна широкого сосщ
(рис. Э5).
Стакан наполняют до половины насыщенным раствором кислого сернок^
лого калия (KHS04). Плотность тока на аноде берут 100 а на 1 дм2. {ИзЩ
ряют длину и толщину выступающего из впая конЩ
анода и вычисляют площадь цилиндрической поверх
ности проволоки. Силу тока при умеренной толщиЩ
проволоки берут ~ 1 а). Температура электролит^
должна быть ниже 16°; в описанном аппарате <Щ
обычно не превышает 6—8°.
Через несколько минут после включения тока,г#р
чинают осаждаться кристаллы трудно растворимого
K2S2O8. Электролиз в первый раз ведут около
40 мин., в следующий раз — 1 час; выпавший препгК
рат отсасывают на плотном фильтре и промывают
спиртом и эфиром.
При работе по указанному методу при электро*;
лизе в продолжение 40—60 мин. получается около
1,5 г 92—95%-ного K2S2O8, что составляет выход по
току 41—45%. Чтобьь очистить соль,' ее перекристал*
лизовывают, для чего растворяют в горячей воде|
раствор быстро фильтруют и охлаждают.
2. По Сагайдачному K2S2O8 легко получается при сливании конценд
трированных растворов вычисленных количеств K2SO4 и. (ЫНфБгОб. Трудно1
растворимый персульфат калия выпадает при этом в кристаллах.
Свойства
Мелкие призмы или, при медленной кристаллизации, большие пластинча-'
тые белые кристаллы. При ыагревании выделяет Ог и SOs. При действии нд
растворы солей наступает разложение на H2SO4 K2SO4, и Ог, причем металлы,
способные окисляться до перекисей (как Mn» Ni, Co, Pb), в присутствии
щелочей образуют черные осадки перекисей.
Испытание
По ОСТ 10160-39 содержание K2S2O8 в препарате ч. д. а, должно быть не
менее 99,5%, в препарате чистом не менее 98%; наибольшие количества до*
пустимых примесей указаны в табл. 163.
Fhc. 35. Прибор для ПОД.У-,
чеиия персульфата калия
Таблица 16$
Доп устимые
Примеси
примеси
в K2S208
Допустимое содержание в %
в чистом для
анализа
в чистом
1. Не растворимые в воде вещества
2. Хлориды (СИ) . . .
3. Металлы, осаждаемые H2S ..:.- .
4. Железо (Fe) t
5. Марганец СМп)
6. Азот (N)
0,005
0,(Ю5
0,002
0,0005
0,0004
0,005
0,01
0,01
0,005
0,001
0,0006
0,0^2
Примечание: Препарату ч. д. а., содержащему не более 0>002°/0 азота,
присваивается квалификация „без азота".
Калий-натрий виннокислый
m
КАЛИЙ-НАТРИЙ ВИННОКИСЛЫЙ (СЕГНЕТОВД СОЛЬ)
Kalium-natrium
tartaricum
Tartarus natronatus
Potassium sodium
tartrate,
Rochelle salt
Weinsaures
Kalium-Natrium
Seignettesalz
Kaliumnatriumtartrat
KNaC^HiOo. 4H2p
Мол. в. 282,229
Приготовление
К смеси 100 г кислого виннокислого к-алия (КНС4Н40в) и 77гЫа2СОз« IOH2O
(или 27,5 г безводного (NasCOe) приливают 400—450 мл воды и
нагревают до прекращения выделения СОа. Полученный раствор фильтруют и
выпаривают фильтрат до начала кристаллизации.
Свойства
Большие бесцветные кристаллы уд. в. 1,767; плавящиеся при 70—80° в своей
кристаллизационной воде. При 100° переходит в моногидрат ЮМаС4ШОв • НгО>
при 130° теряет последнюю молекулу воды. Более сильное нагревание
вызывает разложение с выделением СО и НЮ; оставшаяся обугленная масса
при прокаливании образует KNaCOs.
Сегнетова соль легко растворима в воде и не растворима в спирте.
Таблица 164
Растворимость KNaC4H406.4H20 в воде
f°C
— 4,3
0
10
~в 1000 г водч!
растворяется
г КЫаС4Н406.4НйО
349,93
423,3
632,13
ГС
20
го
в 1000 г воды
растворяется
г KNaC^OHr^O
911.50
1382,78
Таблица 165
Удельный вес водных растворов KNaC4H406
о/0 соли в
растворе
1
2
4
6
8
10
12
<Г
1,0049
1,0116
1,0252
1,0390
1,0530.
1,0673
1,0818
% соли в
растворе
14
16
18
. 20
22
24
26
а20
1,0965
1,1114
1,1265
1,1419
1,1576
1,1735
1,1896
% соли в
1 растворе
28
30
32
34
36
d20
4
1.2059
1,2225
1,2394
1.2566
1,2742
Испытание
Наибольшие количества примесей, допустимых по ОСТ НКТП 5763/113,
указаны в табл. 166.
т
Калий
Таблица [ф
Допустимые примеси в KNaC4H406♦ 4Н20
Прямее?
Допустимое содержание в %
чистом для
анализа
1. Не растворимые в воде вещества. ..'•..
2. Хлорид» (СГ) . .
3. Сульфаты 'SO/) . .
4* Азот общий (N) из нитратов, нитритов и
аммиака
5. Кальций (Са) . . .
6. Металлы, осаждаемые H2S
7. Железо (Fe) . . . .
0,005
0,002
0,010
0,01
0,002
0.0005
0,001
0,010
0,005
0,05
0,02
0,005
0,001
0,002
Кроме того, препарат должен выдерживать приведенные в ОСТ испьг<-
гания на реакцию водного оаствооа и содержание восстанавливающих
веществ.
КАЛИЙ-НАТРИЙ УГЛЕКИСЛЫЙ
Kalium-natrium
carbonicum
Potassium
sodiiirn carbonate
Kaliumnatriumcarbonat
KNaCO*
Мол. в. 122,103
Приготовление
1. Прокаливают сегнетову соль, вначале медленно повышая температуре
Черный остаток (KNaCOs -f- С) выщелачивают горячей водой, раствор
фильтруют и выпаривают досуха. Полученные корки соли растирают в ступке.
2. Пригодный для обычных аналитических целей (сплавление нерастворим
мых силикатов) препарат получается тщательным смешением тонких
порошков КгСОз (13 г) и NaeCOs (10 г).
Свойства
Белый порошок, растворимый в воде. Свойства препарата аналогичны
свойствам составляющие его компонентов (КгСОз и ЫагСОз), кроме
температуры плавления, которая лежит значительно ниже температуры плавления
КгСОз и №гСОз в отдельности.
Испытание
По ОСТ НКТП 2852 препарат, прокаленный при 270—300°, должен
содержать 99,0—101,0% карбонатов (в пересчете на KNaCOs). Наибольшие
количества допустимых примесей указаны в табл. 167,
Калий-натрий углекислый. Калий пиросурьмянокислый кислый 191
Таблица 167
Допустимые примеси в KNaC03
примеси
i He растворимые в воде вещества
о Хлориды (СГ)
3 Сера, общее количество из сульфатов, суль-
' фитов, тиосульфатов и сульфидов (в
пересчете на S04")
4. Кремнекислота и вещества, осаждаемые
аммиаком
5 Кальций и магний (CaO +Mg2P207) . . •" .
б! фосфаты (Р02'") ' . * .
7. Азот iN) (общий из нитратов, нитритов,
аммиака и пр.)
8. Металлы, осаждаемые H2S
9. Железо (Fe)
10. Мышьяк (As) ,
11. Потеря при прокаливании
Допустимое содержание в %
в чистом для
анализа
0,01
0,005
0,005
0,01
0,01
0,003
0,0005
0,0005
0,0005
0,00005
е;о
0,05
0,02
0,005
0,001
0.001
0.001
0,00005
6,0
0,02.
0,01
0,01
КАЛИЙ ПИРОСУРЬМЯНОКИСЛЫЙ КИСЛЫЙ
(ПИРОАНТИМОНАТ КАЛИЯ)
Kalium stibicum
Potassium
pyroantimonate
k2H2Sb207. 4H20 *
Kaliumpyroantimoniat
saures
Мол. в. 507,79
Приготовление
1. Сплавляют равные части рвотного камня [K(SbO)C4H40e] и азотнокис*
лого калия (KNOs). Прокаленную массу выщелачивают теплой водой,
фильтруют и раствор выпаривают. Из сгущенной жодкости через несколько дней
выделяется тестообразная масса, которую разбавляют тройным количеством
воды и размешивают стеклянной палочкой; при этом образуется мелкий
зернистый порошок. Его промывают несколько раз горячей водой и высушивают
на фильтровальной бумаге.
2. По Тредвеллу1 для очистки продажного препарата его кипятят с
концентрированной НЫОз до исчезновения красных паров, кислоту сливают,
осадок метасурьмяной кислоты (Н£ЬОз) промывают водой (декантацией), после
чего кипятят его несколько минут с 2N раствором КОН. Таким путем
получается совершенно чистый . препарат. По охлаждении раствор взбалтывают и
фильтруют. Фильтрат представляет собой готовый раствор K2H2SD2O7; он
тотчас же реагирует с каплей раствора соли натрия (IN).
3.цДля приготовления раствора препарата, пригодного для аналитических
Делей, сурьмяный ангидрид, полученный по методу П. С. Коробкова2 (см.
СТР- 73 п. 3), кипятят 30 мин. с 20%.-ным раствором КОН. Раствор
разбавляют равным объемом воды, по охлаждении взбалтывают и фильтруют.
* * *т,По Асмуссену препарату должна быть приписана формула К [Sb(OH)e]. (Rw. Asmu j-
«en, Kem. Maanedsbl. nord. Handelsbl. kem. Ind. 20, 237 (1939); С I, 2296) (1940).
1 Аналит. химия, 1, 254 П93*).
1 Завод, лаб. 2, № 8, 63 (1933>
192
Калий
*****
Свойства
Белый зернистый порошок, довольно плохо растворимый в воде (в гов^
чей 1 :90, в холодной 1 : 250).
Испытание
По ОСТ 5121 препарат должен выдерживать испытания на реакцию рас%
твора, чувствительность и пригодность в качестве реактива на ион натрия.
КАЛИЙ РОДАНИСТЫЙ (РОДАНИД КАЛИЯ)
Kalium rhodanatum Potassium rhodanide Kaliumrodanid
Kalium sulfocyanatum Potassium thiocyanate KaliumsuIfocyana|
Kaliumthiocyanat
KCNS Мол. в. 97Д7
приготовление
1. По Бабкоку смесь 2 ч. цианистого калия (яд1) и 1 ч. S
прокаливают в железном тигле на бунзеновской горелке до тех пор, пока масоа
совершенно не расплавится и не исчезнет голубое пламя горящей серы Даю*
охладиться настолько, чтобы капля плава, помещенная в воду, не шипела,
встряхивают еще горячую массу с 3 ч. воды и фильтруют. Фильтрат содёр^
жит кроме KCNS еще K2S, KCNO « K2S2O3.
Для разложения примесей к раствору прибавляют разбавленной (1 : 4)
H2SO4 до слабокислой реакции и отфильтровывают выпавшую серу; выиа<
оивают до уз объема, дают выкристаллизоваться K2SO4 и прибавляют равнъгй
объем 90%-ного спирта, вследствие чего почти весь растворенный K2SO4 выЩ
ляется. Раствор, фильтруют и выпаривают до образования кристаллическое
лленки; при охлаждении получается довольно чистый KCNS. Совершение
Чистый препарат получается перекристаллизацией соли из спирта.
2. Удобно получать KCNS сплавлением железистосинеродистого калия
с серой 1:
K4Fe(CN)e + 5S = 4KCNS + FeS + (CN)2.
Сплавление удобнее всего вести в алюминиевой кастрюле или кружке,
снабженной крышкой. - "4
Хорошо истертую смесь 72 г серы и 160 г K4Fe(CN)e. ЗН2О распределяю*
в кастрюле, так, чтобы слой на дне был не толще 1—1% см. Поставив каО
рюлю на медную сетку, начинают нагревание на газовой горелке. Выделяю*
щийся вначале едкий дым (сера и пары воды) выпускают, открыв крышк^
Вскоре масса начинает плавиться и при этом вспучивается; в это время и$
обходимо энергичное размешивание массы, так чтобы объем массы не прев^
шал 2/s кастрюли (если нужно, уменьшают пламя). Плавление продолжают
1—VJ2. часа, после чего масса оседает и перемешивание можно вести времй
от времени. Пламя поддерживают так, чтобы при открывании крышки пари
и сама смесь вспыхивали. К концу реакции вспыхивание уменьшается; спл!в1
ление можно закончить, когда смесь горит слабо и только в отдельных
местах.
Реакционному сосз'ду дают остыть, приливают к полученной комковатой
массе 150 мл горячей дестиллированной воды и дают стоять 30—40 мин.,
размешивая стеклянной палочкой. Жидкость сливают в цилиндр емкостью 1 ;Л
и небольшими порциями воды (по 30—75 мл) смывают туда же оставшуюся
массу. Общий объем жидкости должен составлять около 650—750 мл.
Дав отстояться в течение нескольких часов, декантируют жидкость, осадок
промывают 2—3 раза горячей дестиллцрованнрй водой (по 50—80 мл) и,
соединив все растворы, фильтруют Получается около 1 л раствора, к которому
прибавляют небольшими порциями 20%-ный раствор КОН до слабощелочное
1 Метод ИРЁА.
Калий роданистый
193
пии (все время испытывают лакмусовой бумажкой). Фильтруют, упаривают
реатакой концентрации, чтобы небольшая проба при охлаждении выделяла
Д° * [KCNS и Fe(OH)2], снова добавляют воды и отфильтровывают гидрат
аякисн железа.
Фильтрат выпаривают до образования значительной пленки на поверхности
ют 0СТыть в прохладном месте. Выпавшие кристаллы отсасывают,
помелют в колбу и, облив 80—96%-ны'м спиртом1, нагревают с обратным холо-
^льником на водяной бане до полного растворения. Раствор фильтруют го-
д чим в стеклянный стакан и сильно охлаждают смесью льда и соли.
Выпавшие кристаллы отсасывают, промывают небольшим количеством спирта и
гушат при 80° до образования рассыпчатого порошка. Высушенный препарат
немедленно помещают в горячую банку и плотно закрывают. Выход 60%
теоретического.
3. Метод получения KCN3, детально разработанный в ИРЕА2, основан на
взаимодействии NthCNS с КОН:
NH4CNS + КОН = KCNS + NHs + Н20.
К горячему (70°) раствору 500 г NH4'CNS в 250 мл воды приливают при
помешивании раствор 368 г чистого КОН в 400 мл воды. Работу ведут в
хорошем вытяжном шкафу. Нагревают смесь при 80—85° до полного
удаления 1МНз (проба раствора, при нагревании с КОН не должна выделять NHa,
обнаруживаемого по запаху).
Если требуется получить препарат, отвечающий требованиям ОСТ, то,
отобрав пипеткой пробу раствора, количественно определяют в нем содержание
SO/' и, на основании данных анализа, добавляют к раствору вычисленное
количество Ва(ОН)г. Нагревают раствор в течение 3—4 час, после чего
убеждаются качественными реакциями в отсутствии SO/' и Ва* \ В крайнем
случа^ допускается ничтожная примесь SO/'.
Чистый раствор отфильтровывают через двойные складчатые фильтры и
выпаривают фильтрат до образования пленки соли на поверхности. Выпавшие
при охлаждении кристаллы отсасывают и сушат при 70—80°. Выход 400—440 г
(около 70% теоретического). Упаривание и кристаллизация маточного раствора
дают еще 100—140 г чистой соли.
4. По указанию И. Кольтгофа* очистка продажного препарата может
быть осуществлена путем перекристаллизации из воды, особенно в том случае,
когда исходный продукт содержит хлориды. После трех последовательных
кристаллизации содержание KCI падает с 1% до 0,005%. Отделение примеси
NH4CNS может быть достигнуто перекристаллизацией KCNS из воды или
спирта.
Кольтгоф указывает также, что чистый KCNS осаждается при
добавлении эфира к насыщенному спиртовому раствору продажной соли.
Кристаллы отсасывают на бюхнеровской воронке (без бумажного фильтра) и
сушат в эксикаторе при комнатной температуре. Сухой препарат нагревают в
течение часа при 150°, затем повышают температуру до 200° и выдерживают
расплавленную соль при этих условиях 10—20 мин. для удаления
последних следов растворителя.
Свойства
Бесцветные призматические кристаллы уд. в. 1,886, расплывающиеся во
влажном воздухе. Легко растворим в воде4 с сильным'охлаждением (при
смешении 150 г KCNS со 100 мл воды температура падает с 10,8 до —23,7°)
и спирте. О температуре плавления KCNS литературные Данные разноречивы
1 На каждые Д00 г кристаллов следует брать 420 мл спирта.
• Ре акт. и преп. 186*
• Г. М. К он h off a. J. J. Lf ngane, J. Am. Chem. Soc. 57, 2126 (1935).
• Кристаллогидрат KCNS-Vt HtO, устойчивый между—29,5е н + 6,8% получаетея при
кристаллизации растворов роданида калия при низкой температура.
194
Калий
(161.2°, 172°, 172,3°); по новейшим данным1 т. пл. KCNS 176,8°. При
становится синим, при охлаждении снова обесцвечивается.
Таблица 168
Растворимость KCNS в воде
430$
t*C
— 31,2
0
+ 20
25
100 г воды
растворяют
г KCNS
177,2
217
239
% KCNS
50.25
63,9
68,5
•70,9
Таблица 169
Удельный вес водных растворов KCNS
в/о KCNS
1
2
4
.6
8
10
12
! 1,0035
1,0085
1,0186
1,0288
1.0391
1.0495
1,0601
о/о KCNS
14
16
18
22
26
30
35
л18
1,0708
1,0817
1 1,0927
1,1152
1.1382
1.168
1,1899
•/о KCNS
40
45
'50
55
60
65
70
л18
1,2200
1.2517
1.2849
1,3195
1,3554
1,3925
1,4307
Испытание
По ОСТ 7380/534 содержание KCNS для препаратов квалификаций х; ч. и
ч д. а. должно быть не менее 99%; наибольшие количества допустимых
примесей указаны в табл. 170.
Допустимые примеси в KCNS
Таблица 170
Примеси
1. Не растворимые в воде
вещества
2. Хлориды (С1')
3. Сульфаты (SO/O
4. Тяжелые металлы группы H2S
5. Железо (Fe)
6. Аммонийные соли (NH4) . . .
Допустимое содержание в °/о
в химически
чистом
0.003
0.005
0.005
0,0002
0,03005
0,001
в чистом
для анализа
0,005
0.01
0,01
0,0005
0,0001
0,002
в чистом
0.01
0,02
0.02
0,001
0.ОС02
0,005
1 Р. С. Kracek, J. Warsh. Acad. Scl. 26, 307 (1936); С. И,
Калий сернокислый
195
КАЛИЙ СЕРНОКИСЛЫЙ (СУЛЬФАТ КАЛИЯ)
Kalium sulfuricum Potassium sulfate Kaliumsulfat
K2SO4
Мол. в. 174,25
Приготовление
Лабораторным путем K2.SO4 легко получить, нейтрализуя 10%-ную НгБО*
и помощи КгСОз или КОН. Раствор фильтруют и выпаривают до
кристаллизации.
Свойства
Белые твердые ромбические кристаллы уд. в. 2,67, устойчивые на воздухе.
растворим в воде, не растворим в спирту Т. ад 1067°,
Таблица 171
. Растворимость K2SO4 в воде
ч/вс
0
10
20
25
30
о/о K,SO<
6,87
8,47
10,03
10,76
11,49
*°с
40
50
60
70
80
°/о K„S04
13,1
14,2
15,4
16,6
17,6
i°c
90
100
120
143
170
о/о KeS04
18,6
19,4
20,9
22,4
24,7
Таблица 172
Удельный вес водных растворов K2SO4
°/о KaS04
1
2
3
4
5
«15
1,00820
1,01635
1,02450
1,03277
1,04105
d4
1,0063
1,0145
1,0227
1,0310
1,0393
% K,S04
6
Г 7
; 8
^
9,92
<*15
1,04947
1,05790
1,06644
1,07490
1,08305
и20
1,0477
1,0531
1,0646
1,0^31
1,0817
Испытание
Наибольшие количества примесей, допустимых по ОСТ 5119, указаны в
Табл. 173. Препарат должен выдерживать испытание на примесь натрия (Na)
По интенсивности желтой окраски пламени при - внесении в него препарата;
реакция водного раствора должна быть нейтральна на метилоранж (условия
Испытания см. ОСТ).
196
Калий
Допустимые примеси в K2S04
Таблица Ц
Примеси
I. Не растворимые в воде
вещества
2. Аммонийные соли (NH4*) . . .
3. Нитраты (NCY)
4. Хлориды (СГ) ... • . . . .
5. Кальций, магний и вещества,
осаждаемые аммиаком ....
7. Железо (Fe)
Допустимое содержание в °/о
в химически
чистом
0,005
0,001
0,0010
0,0005
0,01
0,001
0,0002
0,0001
в чистом
для анализа
0,01
0,002 ■
0,002
0,001
0.02 ,
0.001
0,0005
0,0002
в чистсц
0.02
0,004
0.004
0,002
0,05
0,002
0,0010
0,0004
КАЛИЙ СЕРНОКИСЛЫЙ КИСЛЫЙ (БИСУЛЬФАТ КАЛИЯ)
Kalium bisulfuricum Potassium bisulfate Kaliumbisulfat
KHSO4
Мол. в. 136,16
Приготовление
1. В платиновой чашке осторожно нагревают смесь 87 г TGS04 и 49 г чис*<
той H2SO4 (уд. в. 1,84), пока масса не сделается прозрачной, как вода;
жидкость выливают в фарфоровую или платиновую чашку, помещенную в холод?
ную воду. По затвердевании массу разбивают на куски и хранят в плотно
закрытых банках.
2. 20 г чистого КС1 обливают 28 г чистой конц. H2SO4 и нагревают в
платиновой чашке на небольшом пламени до прекращения выделения газа.
По охлаждении плав застывает в кристаллы.
Свойства
Ромбоэдрические кристаллы уд. в. 2,36, растворимые в воде с кислой
реакцией (из растворов выкристаллизовывается K2SO4). О температуре пла>
вления KHS04 литературные данные весьма противоречивы К
Выше 600° теряет H2SO4, причем образуется K2SO4.
Таблица 174
Растворимость KHS04 в воде
/•с
2°0
100 t воды
растворяю!
г KHS04
33,9
48,0
t°c
40
1 100
100 г воды
растворяют
i KriSO,
62.9
113,6
1 Литературные источники указывают на т. пл. KHS04, равную 100*, 120% 200* и даже 315*;
по новаАшим данным т. пл. KHS04 211,7е (Н. Hagieawa, Т. Takai, Sd. Jap. last рЬУ»
сйеш. Ret. 31, /* 677/Ш; С II, 1631 (1937).
Калий сернокислый кислый и сурьмяновиннокислый
197
Таблица 175
Удельный вес водных растворов KHS04
o/e KHS04
1
2
4
6
8
w18
1.0067
1,0142
1,0285
1,0425
1,0568
Wo KHS04
10
12
14
16
18
d4
1,0720
1,0865
0,10-Ю
1,1175 1
1,1335 1
k> khso4
20
22
24
26
27
и18
1.1510
U670
1,1840
1,2018
1,2110
Испытание
Но ОСТ НКТП 2851 содержание действующего начала (в пересчете на
ftHSOO в препарате должно быть 99,0—102,0%; наибольшие количества
допустимых примесей указаны в табл. 176.
Таблица 176
Допустимые примеси в KHS04
Примеси
Допустимое содержание в °/0
в чистом
дл-1 анализа
1. Не растворимые в воде вещества
2 Хлориды (СГ)
3. Кремнекислота и вещества, осаждаемые
аммиаком
4. Кальций и магний (СаО + MgoP207) ....
5. Фосфаш (Р04'") .............
6. Металлы, осаждаемые H2S
7. Железо (Fe)
0,002
0,001
0,01
0,005
0,001
0,0005
0,001
0,005
0,002
0,02
0,01
0.Г02
0,002
0,002
КАЛИЙ СУРЬМЯНОВИННОКИСЛЫЙ
(АНТИМОНИЛТАРТРАТ КАЛИЯ, ВИННОКИСЛЫЙ АНТИМОНИЛКАЛИЙ,
РВОТНЫЙ КАМЕНЬ)
Tartarus stibiatum Potassium Brechweinstein,
Tartarua emeticus antimonyltartrate, Weinsaures
Tartar emetic Antimon-Kalium
K(SbO)C4H4Oe. V2H2O Мол. в. 333,94
Приготовление
100 г хорошо промытого сурьмянистого ангидрида (БЬгОз) кипятят с 125 г
Кислого виннокислого калия (КНС^НЮв) в 1 л воды. Во время кипячения
слабят, чтобы уд. в. раствора не превышал 1,08—1,09; по окончании растворения
Жидкость выпаривают до уд. в. 1,125 и оставляют кристаллизоваться.
Выделившиеся кристаллы отсасывают и слегка сушат между листами
фильтровальной бумаги.
Свойства
Ромбические пирамидальные кристаллы, выветривающиеся на воздухе,
неприятного вкуса. ' -
198 Калий
4 ' "' ' ' *" ~ ^ ~~™ "—*,
КАЛИЙ ТИОУГЛЕКИСЛЫЙ (ТИОКАРБОНАТ КАЛИЯ)
Kalium thiocarbonicum Potassium thiocarbonate Kaliumthiocarbona|
K2CSs Мол. в. 186,38
П риготовление
По Ванино1 в закрытой склянке встряхивают концентрированный раствор
сернистого калия (KaS) с избытком сероуглерода (С8г) до насыщения. Отде.
ляют избыток CS2 в делительной воронке и выпаривают раствор в вакууме
при 30р.
Свойства
Красно-коричневая, гигроскопическая масса, очень легко растворимая &
воде и трудно в спирте,
КАЛИЙ УГЛЕКИСЛЫЙ (КАРБОИАТ КАЛИЯ, ПОТАШ)
Kalium carbonicum Pot-ashes, Potash Kaliumcarbonat
Sal tartari caustic, Pottasche
Potassium carbonate
K2CO3 Мол. в. 138,202
Приготовление
1. В платиновой или серебряной чашке прокаливают при слабокрасном Щ*-,
лении кислый виннокислый калий, КНСШЮв (винный камень2). Остатокобрай
батывают водой, фильтруют и выпаривают досуха. Дальнейшая очистка см. :■:$*!
2. Для очистки от загрязнений КгСОз растворяют в небольшом количестве
воды, фильтруют и переводят в кислый углекислый калий КНСОз (см. стр 206)>
Прокаливая КНСОз в платиновом тигле, получают вполне чистый препара$
3. Чистый препарат легко может быть получен в лаборатории из древес*
ной золы. Золу кипятят с водой в чугунном котелке в течение 2 час,
пополняя испаряющуюся воду. Отфильтровав золу через бумазею, выпаривают рф?|*
твор досуха. Полученный сырой КяСОз очищают, как указано в п. 2. р
4. Препарат чистый легко может быть доведен до более высокой
квалификации перекристаллизацией из воды3. 200 г КгСОз растворяют в 500 ЛИ
воды, фильтруют, выпаривают до объема 350 мл и охлаждают. Выпавшие
кристаллы гидрата КгСОз. РДНгО отделяют, а маточный раствор снова
упаривают и кристаллизуют.
5. Для очистки продажной соли от СГ можно применить метод Н. А. П'ет-
рова и М. В. Л а з а р ево й4, заключающийся в электролизе 15—20%-нб'ГО
раствора К2СО3 (с серебряными электродами). При объеме электролита в 2 Л
и напряжении в 0,8—1,3 в авторы рекомендуют поверхность анода в 1540 СМ*
(т. е. анодная плотность тока составляет 0,02—0,03 ма/см2). Через 50—60 час*
электролиз заканчивают. Раствор содержит суспендированное серебро, оседав
щее через несколько часов. Признак конца очистки — значительное почернен^
анода (образование А^гО).
1 Pr&p. ChM 386.
» Стае рекомендует винный камень предварительно обработать 6%-hqb HCL затеи ottfW
кислоту и высушить. г щ '
8 Реакт. и преп. 160.
« Изв. ИРЕА, 14, 76 (193§). ,
Калий тиоуглекислый и углекислый
199
Для удаления растворенного серебра раствор подвергают электролизу в те-
т^ие одного часа с платиновыми электродами (напряжение тока 6 в).
Содержание CV в результате очистки падает' с 1,6—1,8% до 0,000%. Серебряные
ноды легко регенерируются прокаливанием при 700°,
Свойства
Белый кристаллический порошок уд. в. 2,29, расплывающийся во влажном
воздухе. Т. пл. 891°. Очень легко растворим в воде со щелочной реакцией,
не растворим в спирте и эфире. Из насыщенного горячего водного раствора
При охлаждении выпадает гидрат КаСОз.1,5Н20 в виде блестящих
стекловидных кристаллов, теряющих кристаллизационную воду при 100°,
Таблица 177
t "С
0
10
20
30
40
Растаоримост!
в/оК,С03
51,9
52,2
52,8
53,4.
53,9
t°c
50
60
70
80
90
> К2СО3 в воде
и/оКяСОв
54,8
55>9
57,1
58,3
59,6
| * °С
100
110
120
130
) %К,С03
60,9
62,5
64,4
66V*
Таблица 178
Удельный вес водных растворов К2СО3
•/о К»С08
1
2
3
4
5
6
7
8
9
10
dl°
1,0072
1.0163
1,0254
1,0345-
1,0437
1,0529
1.0622
1,0715
1.0809
1,0904
о/о К,СОэ
11
12
13 ,
14
15
16
17
18
19
20
. «Г
1,1000
1Д096
1,1193.
1,1291
1,1390
1,1490
1,1591
1.1692
1,1795
1,1898
°/о К,С03
22
24
26
28
30
35
40
45
50
55
4°
1,2107
Л,?320
1,2536
1,2756
1,2Q79
1,3548
1,4141
1,4759
1,5404
1,5673
Испытание
По ОСТ 7402/556 содержание углекислого калия в препарате должно быть
для квалификаций х. ч. и чш д. а. не менее 99%, для чистого не менее 97%;«
Наибольшие количества допустимых примесей указаны в табл. 179; препарат
должен выдерживать испытание на натрий — отсутствие желтой окраски пламени
при внесении в него препарата (условия испытания — см. ОСТ).
200
Калий
Допустимые примеси в К2С03
Таблица jyg
Примеси
Допустимое содержание в °/0
в химически
частом
в чистом
для анализа
в чистом
1. Не растворимые в воле вещества , . . .
2. Хлориды и хлораты (СК)
3. Сера, общее количество из сульфатов,
сульфитов, сульфидов (в пересчете на
SO/')
4. Кремнекислота (SiOo) и вещества,
осаждаемые аммиаком (А120з, Fes03) . . . .
5. Фосфаты (РО/")
6. Азот (NX общее количество из нитратов,
нитритов, аммиака и пр
7. Железо (Fe)
8. Тяжелые металлы группы H2S ......
9. Кальций и магний (Са и MgJ ......
10. Мышьяк (As)
П. Потери при прокаливании . ,
0,005
0,001
0,002
0,005
0,005
0,001
0,0005
0,000
0.005
0,01005
1
0.010
0,002
0,004
0,010
0,005
0.001
0,и01
ОлиО
0,015
0,00005
1
0,030
0,010
0,010
0.050
0,020
0,010
0.002
0.000
0.100
0,00005
3
КАЛИЙ УГЛЕКИСЛЫЙ КИСЛЫЙ
(БИКАРБОНАТ КАЛИЯ, ДВУУГЛЕКИСЛЫЙ КАЛИЙ)
Kalium bicarbonicum Potassium bicarbonate Kaliumbicarbonat
КНСОз Мол. в. 100,114
Приготовление
1. В насыщенный раствор КгСОз пропускают СО2, пока взятая. пооба от
прибавления нейтрального раствора соли магния не перестанет образовывать
осадок. Часть КНСОз выделяется уже при пропускании СО2. Маточные
растворы при испарении в эксикаторе над H2SO4 выделяют остальной КНСОз.
Если исходный КгСОя был недостаточно чист, то препарат необходимо
перекристаллизовать; для этого растворяют 100 г КНСОз в 200 мл воды при 70°,
фильтруют и фильтрат медленно охлаждают. Выпавшие .кристаллы отсасывают,
промывают 80%-ным спиртом и высушивают в токе СО2. Растирать кристаллы
не рекомендуется, во избежание быстрого разложения (Б о р н е м а н) К
2. Очень чистый препарат получается при пропускании СОг в раствор
чистого КОН в 80,% -ном спирте. Часть КНСОз выпадает уже во время
пропускания газа.
Свойства
Бесцветные моноклинические кристаллы уд. в. 2,17, устойчивые на воздухе.
Вкус соленый, слабо щелочный. Растворим в воде с очень слабой щелочной
реакцией. В спирте растворим трудно (1:2000). При нагревании при 100°,
быстрее при 200°, КНСОз распадается, на-КгСОз, Н20 и СОз. Водный раствор
КНСОз не осаждает основной соли из солей магния -(отличие от КзСОз).
J Неорган. преп„ Но,
Калий углекислый кислый и фосфорнокислый 201
(Учтена
Таблица 180
КНС03
потеря СС>2 при повышении/0)
растворимость КНС03
в воде
Таблица 181
Удельный вес водных
растворов КНС03
t°c
0
Ю
?о
зо
• о/0 КНСО,
18,4
21,7
24,9
28,1
/°С
40
50
60
о/о КНСО,
31,2
34,2
37,5
о/о КНСО,
1
2
4
<■
1,0058
1,0125
1,0260
о/о КНСОз
6
8
10
w1*
1,0396
1,0534
0,0674
Испытание
По ОСТ НКТП 7370/524 содержание КНСОз для препарата квалификаций
х. ч. и ч. д. а. должно быть не менее 98,5%; наибольшие количества
допустимых примесей указаны в табл. 182; препарат должен выдерживать испытание
на примесь натрия (условия испытания см. ОСТ),
Таблица 182
Допустимые примеси в КНС03
Примеси
Допустимое содержание в °/0
а химически
чистом
в чистом
для анализа
1. Не растворимые в воде вещества . . . .
2. Хлориды и хлораты (СК) . .
3. Сера, общее количество из сульфатов,
сульфитов, тиосульфатов, сульфидов (в
пересчете на S04")
4. Азот (N) (общий из нитратов, нитритов,
аммилка и пр.)
5. Фосфаты (РО/")
6. Кремнекислота и вещества, осаждаемые
аимиаком
7. Кальций и магний (Са и Mg) .......
8. Тяжелые металлы группы H2S
9. Железо (Fe) . . .
0.002
0,002
0,003
0,0005
0,001
0.01
0.005
0.О005
0,0003
0,005
0,005
0,005
0.001
0,103
0,04
0.02
0,001
0,0005
0,02
0,01
0,01
0.002
0,005
0,1
0,05
0,002
0,001
КАЛИЙ ФОСФОРНОКИСЛЫЙ КИСЛЫЙ ОДНОЗАМЕЩЕННЫЙ
Kaliumphosphat
einbasisch
Kalium phosphoricum
monobasicum
Potassium phosphate
monobasic
П
Риготовление
KH2PO4
4
Мол. в. 136,09
По Ванино1 к раствору НзР04 прибавляют такое количество КгСОзг,
Чтобы раствор вызывал только слабое покраснение лакмусовой бумажки.
Раствор выпаривают и, охлаждают, причем соль выкристаллизовывается.
* Ргар. СЬ. 383,
202
Свойства
Калий
Раство
Тетрагональные кристаллы уд. в. 2,33, сильно кислого вкуса, *~ нетверд
г, воде, нерастворим в спирте. При 96° кристаллы плавятся в прозрач^р1
жидкость, застывающую при охлаждении в непрозрачную, стекловц
массу метафосфорнокислого калия.
КН2РО4 -+ НзО + KPOs
Таблица 183
Растворимость КН2РС>4
в воде (А. А. Казанцев)
/•с
0
5
10
15
20
25
30
85
0;0 КН,Р04
12,Р8
14,00
15,50
16.Р7
18,45
20,04 1
21.90
23,65 1
ГС
40
45
50
60
70
80
90
о/о КН.РО,
25.10
26,90
29,00
33,40
37,05
41,30
45,5
Таблица^
Удельный вес
водных растворов КН2!
в/о КН,Р04
1
2
4
6
<Г
1,0070
1,0142
1,0284
1,0425
о/о KHfP04
8
10
12
14
1 1,058?
1,071
1,0863
1,1028
Испытание
По ОСТ 3695 наибольшие количества допустимых примесей указаны в
табл. 185.
Потеря при прокаливании должна составлять для препаратов х. ч. я
ч. д. а. 13,15—13,4%. Концентрация водородных ионов рН для тех же
препаратов должна быть от 4,4 до 4,7.
Таблица Ш
Допустимые примеси в КН2Р04
Примеси
Допустимое содержание в %
химически
чистом
в чистом
для анализа
1. Не растворимые в воде вещества
2. Вещества, осаждаемые аммиаком
3. Влажность
4. Хлориды (СГ)
6. Сульфаты (SO/)
5. Железо (Fe)
7. Тяжелые металлы
8. Нитраты (NO/)
9. Мышьяк (As)
0.002
0 005
0,2
0,002
0,002
0,001
0,001
0,002
0,0005
0,005
0,01
0,5
0,005
0,005
0,002
0,002
0,005
0,001
0,01
0,02
1,0
0,01
0,01
0,005
0,005
0,01
0,002
Калий фтористый
203
S КАЛИЙ ФТОРИСТЫЙ (ФТОРИД КАЛИЯ)
Kalium fluoratum Potassium fluoride Kaliumfluorid
А. Средняя соль
KF
Мол. в. 58,10
Приготовление
« & \00 мл чистой 15%-ной HF, помещенной в платиновой чашке,
прибавляют КгСОз до слабо кислой реакции (около 52 г). Раствор отфильтровывают
выпаривают до объема 80 мл. При охлаждении до 20—22° выпадают
кристаллы гидрата KF. 2НгО. Отфильтрованную соль отжимают между листами
фильтровальной бумаги.
2. Для приготовления больших количеств KF по методу ИРЕА техническую
плавиковую кислоту, помещенную в эбонитовые тазы, нейтрализуют КОН.
раствор фильтруют на медных воронках и выпаривают в медных тазах.
Выпавшие кристаллы отфуговывают на фарфоровой центрофуге и сушат на
воздухе между листами
фильтровальной бумаги. Таблица 186
3. Безводный KF получается Растворимость KF в воде
обезвоживанием гидрата при г
температуре не ниже 130°. (В. С. Ятлов и Е.М.Полякова1)
Свойства
При кристаллизации из
водных растворов ниже 40,2°
выпадают бесцветные кубические
кристаллы KF'2Hj>0 уд. в. 2,481,
плавящиеся дри 46° в
кристаллизационной ©оде. При .лежании
на воздухе кристаллы быстро
расплываются. Очень легко
растворим в воде, не растворим в
спирте. Водный раствор обладает остросоленым вкусом и имеет щелочную
реакцию; разъедает стекло. Т. пл. безводной сани 646°, т. кип. 1505°, уд. в. 2,369.
t°C
9
0
10
15
20
25
30
%KF
30,90
34,87
38.13
48,70
50,41
51,95
fC
35
40
45
60
80
% KF
54,67
58.08
58,62
58,72
60,01
Таблица 187
Удельный вес водных растворов KF
%KF
1
2
4
6
8
<8
1,0072
1,0159
1.П334
1,0512
1,0693
%КР"
10
12
1 14
16
18
dA
1,0877
1,1064
1,1254
1,1448
1,1616
%KF
20
22
24
26
Л9
1,1847
1,2051
1,2260
1,2471
Испытание
По ОСТ НКТП 7169/463 препарат должен растворяться в воде в
отношении 1 :2, причем раствор должен быть прозрачен и свободен от
механических примесей; наибольшие количества допустимых примесей указаны в
табл. 188.
* ЖОХ, 9. Н 8, 774 (1938).
204 Калий
Допустимые примеси в KF-2H20
Таблиц^ щ
Примеси
Допустимое содержание в %
в чистом
дли анализа
0002
0,02
0,002
0.0005
0.2
0,2
0,1
0,05
в чистом
0.005
0.05
0,005
0.001
0,5
0,5
0,2
од
1. Хлориды (СК)
2. Сульфаты (S04")
3. Металлы, осаждаемые H2S
4. Железо (Fe)
5. Кислотность (HF) . . ♦
6. Щелочность (К2СО3)
7. Соли H2SIF6 для препарата с кислой
реакцией (K2SiF)e
8. Кремнекислота (Si02) для препарата со
щелочной реакцией . ,,.
Kalium bifluoratum
Б. Кислая соль
Potassium bifluoride
Kaliumbifluorid
KHF2 Мол. в. 78,11
Приготовление
1. Нейтрализуют чистую HF, добавляя КгСОз, после чего приливают HF в
количестве, равном взятому первоначально. Оставляют стоять на несколько
часов, отфильтровывают осадок K*SiFe и выпаривают фильтрат в платиновой
чашке до начала кристаллизации. После охлаждения соль отделяют и
высушивают в медной чашке при тем-
Таблица 189
Растворимость KHF2 в воде
(В. С. Я т л о в и Е. М. П о л я к о в а)
в токе сухо«
/°с
-7,6
0
10'
20
% KHF,
16,5 1
19,70
23,14 1
28,15
/°с
45
60
80
% KHF,
38,03
44 08
53,28
в.
Свойства
Твердая кристаллическая масса уд.
вакууме) не теряющая кислоты. Во влажном
гогда выделяет пары HF. Разлагается водой
соли и плавиковой кислоты.
пературе ниже 150°
го воздуха \
2. По И. В. Та нана ев у*
смешивают КОН или KF с
теоретическим количеством HF,
выпаривают до кристаллизации и
охлаждают. Кристаллы
-промывают спиртом и высушивают при
комнатной температуре. При
перекристаллизации получается очень
чистый препарат.
3. По указанию И. В. Тана-
н а е в а - KHF2 может быть легко
Получен добавлением HF к
насыщенному раствору KF,
соль выпадает в виде
кристаллов 8.
причем
мелких
5,369, в сухом воздухе (даже в
воздухе притягивает воду и
с образованием нормальной
J G melin, s HandbiKh , Syst. № 19, F, стр. ,-3.
■ ЖПХ U, № 2, 214 ( 98).
• О лолуче ии кислой соли К". 2 HF с т. пл. 80° см. Е. В. R. Prideau* и К. R.
J. chem. Soc. I (1937); С. 1, 3292 (х9о7).
Webb,
Калий хлористый
206
Л
КАЛИЙ ХЛОРИСТЫЙ (ХЛОРИД КАЛИЯ)
Kalium chloratum
Potassium chloride
KC1 Мол. в. 74,553
Kaliumchlorid
Приготовление
1. Очистку технического продукта, содержащего Mg° *, Fe**e, SO*' и
другие примеси, производят по Бендеру следующим образом: 500 г соли
оастирают в большой ступке в 1,5 л холодной воды и фильтруют через
складчатый фильтр в фарфоровую чашку емкостью 3 л. Несколько
нагревают и прибавляют к раствору известковое молоко (приготовленное из 5 г
СаО) и небольшой избыток чистого ВаСЬ (приблизительно 12 г; конец
реакции определяют во взятой пробе).
После отсасывания осадка жидкость фильтруют в стакан и осаждают
избыток Са' • и Ва' ', прибавляя 15 г чистого КгСОз. Фильтруют и фильтрат
при кипячении нейтрализуют разбавленной НС1, после чего возможно быстро
выпаривают на сильном огне до объема 0,5 л. Выпавший КС1 отсасывают на
воронке Бюхнера и высушивают нагреванием в фарфоровой чашке при
помешивании стеклянной палочкой.
2. Для приготовления КС1 из технических КОН или К2СО3 в фарфоровую
чашку помещают 0,5 л НС1 (уд. в. 1,06) и вносят КОН или КгСОз до слабо
щелочной реакции. Отфильтровав выпавший осадок, подкисляют раствор НС1,
добавляют немного ВаС1г и, вновь прибавив чистого К*СОз, нагревают до
кипения. Выпавший осадок (ВаСОз, BaS04) отфильтровывают и, подкислив
фильтрат чистой НС1, выпаривают, как в способе 1.
-3. Можно получить чистый препарат, насыщая концентрированный
раствор КС1 газообразным НС1 (см. Натрий хлористый А, 1, стр. 406).
Свойства
Бесцветные кубические кристаллы (часто — удлиненные столбики), уд. в.>
1,989, соленого вкуса, устойчивые на воздухе. При нагревании растрескивается
и плавится при 768° в жидкость, кипящую при 1415° и затвердевающую при
охлаждении в стекловидную массу.
Растворим в воде с нейтральной реакцией, нерастворим в абсолютном
спирте и эфире.
Таблица 190
Растворимость КС! в воде
/•С
0
10
ю
30
40
% КС1
22,2
23,8
25.5
27.2
28,7
/•С
50
60
70
80
% КС1
30,1
31,3
32,6
33,8
ГС
90
100
130
180
% КС1
34,9
36,0
3^,8
i 43,8
1 П© А. Саппсру, уд. в. 1,987, Z. anorg. Gb. 203> 310 (1932),
ж
Калий
Таблица 191
Удельный вес водных растворов КС1
%КС1
1
2
3
4
5
6
7
8 1
<°
1,0046
1.0П0
1.0175
1,0239
1,0304 ,
1,0369
1,0435 1
1,0500
•/oKCl
•9
10
И
12
13
14
15
16
т
1.0567
1,0633 1
1,0701
1.U768
1.0836
1,0905
1,0974
1,1043
% КС1ч
17
18
19
20
21
22
23
24
«Г
1,1114
1,1185
1,1256
1,1328
1,1401
1.1474
1,1548
1,1623
Таблица 192
Температура кипения водных растворов КО
»/оКС1
9,2
16,7
23.4
t9c
101
102
103
•/о КС1
29,9
36,2
42,4
/°С
104
105
106
*/о КС1
48,4
54,5
57.4
t*c
107
108
108,5
Таблица 193
Растворимость КС1
в разбавленном спирте
При 15е
Bed о/о
с л ир га
10
20
30
40
50
60
80
•/«Да
19,8
14,7
10,7
7.7
5,0
2,8
0,45
При 30е
Вес. в/о .
[ спирта
9.4
25,1
43,1
55.9
65,9
78,1
86,2
' ~% КС1
1 23,2
16,1
10,0
6,4
3.5
1.3
0
По ОСТ 4568 содержание КС1 в препарате должно быть не менее 99,8%
наибольшие количества допустимых примесей указаны в табл. 194,
Калий хлорноватокислый
207
Таблица 194
Допустимые примеси в КС1
Примеси
Допустимое содержание в %
химически
чистом
Не растворимые в воде вещества . . . .
i Хлораты (CKV) .
о сульфаты (SU4 )
а Азот (N) (общий из нитратов, нитритов и
аммиака) • ♦
5 фосфаты (РОЛ
6 Кальций (Са) . . .
l\ Магний (Mg)
g'# Тяжелые металлы
i Железо (Fe)
0.003
0,0012
0,002
0,0005
0,001
0,005
0,002
0,0005
0,0001
В ЧИСТОМ
для анализа
0.005
0,0012
0,005
0,001
0.002
0,007
0,003
0,0005
0,0003
0,020
0,0024
0,010 '
0,001
0.005
0,010
0,005
00010
0,0005
КАЛИЙ ХЛОРНОВАТОКИСЛЫЙ
(ХЛОРАТ КАЛИЯ, БЕРТОЛЕТОВА СОЛЬ)
Kalium chloricum Potassium chlorate Kaliumchlorat
KClOs Мол. в. 122,553
Приготовление
1. Из продажного продукта (бертолетова соль), содержащего СГ, чистый
препарат легко получить двукратной перекристаллизацией из горячей воды.
2. По Эрдману1 100 г КгСОз растворяют в возможно малом количестве
горячей воды и пропускают при кипячении хлор до исчезновения щелочной
реакции. Жидкость разбавляют горячей водой до объема 350 мл> фильтруют
и дают кристаллизоваться (КС1 остается в растворе).
3. По В. и Г. Бильц2 КСЮз может быть получен электролизом теплого
раствора КС1, содержащего К2ОО4. Растворяют 100 г КС1 в 150 мл воды,
добавляют 1 г К2СЮ4 и подвергают электролизу в течение 14 час. при
40—60°, дополняя испаряющуюся воду. Электродами служат платиновые
пластинки: анод, поверхностью около 25 см2, катод — несколько меньшего
размера. Напряжение б—10 в\ анодная плотность тока 0,2 a/см2. По оконча
нии электролиза раствор охлаждают, выпавшие кристаллы отсасывают,
промывают и перекристаллизовывают из горячей воды.
Свойст ва
Бесцветные листочки с перламутровым блеском или 4- или 6-угольные
Моноклинические таблички, устойчивые на воздухе и растворимые в воде с
Нейтральной реакцией. Уд. в. 2,344. Плохо растворим в 95%-ном спирте
(1 : 130) и глицерине (1 : 30); почти не растворим в абсолютном спирте. Пла-
вится при 370° без разложения. При быстром нагреваний плавление с
выделением кислорода начинается при 352°. При 550° весь кислород выделяется.
Если в КСЮз имеются горючие примеси (уголь, сера, фосфор, крахмал,
бумага), то такой препарат нельзя ни нагревать, ни растирать в ступке (воз*
Кожин сильный взрыв!).
1 Хим. пррп., 14.
' UebimgsbeisQielo, L&
208
Калий
Таблица 195
/•с
0
10
20
30
Растворимость KCJOs i
% КСЮз
3,2
4.8
6,8
9,2
/•с
40
50
60
70
•/о. КСЮ,
12,7
16,5
20,6
24,5
i воде
/°с
80
90
100
% КСЮ,
28,4
32,3
36,0
Таблица 196
Удельный вес
водных растворов КС108
•/о КСЮ,
1
2
3
<'
1,0049
1,0113
1,0178
%ксю8
4
5
6
<Ш
1,0245
1,0312
1,0380
Таблица 197
Растворимость КС103 в разбавленном спирте
при 30°
Вес. о/0
спирта
10
20
30
о/о КСЮ,
6,5
4,5
3,2
Вес. о/0
спирта
• 40
50
60
% КСЮ,
2,4
1,6
1,0
Вес. о/0
спирта
70 1
80
90
о/0ксю,
0,54
0,24
0,06
Испытание
По ОСТ НКТП 7174/468 содержание КСЮз в препарате не менее 99%;
наибольшие количества допустимых примесей указаны в табл. 198.
Таблица 198
Допустимые примеси в КСЮ3
Примеси
Допустимое содержание в %
1. Не растворимые в воде вещества
2. Хлориды (СП
3. Сульфаты (S(V)
4. Броматы (вгОзО
5. Металлы, осаждаемые H2S . . .
6. Железо U*e)
7. Кальций |'С'а)
8. Магний (Mg) 4 .
9. Мышьяк (As)
в чистой
для анализа
0,005
0,002
0,005
0,005
0,001
0,0005
0,002
0,002
0,00005
в чистой
0,01
0,005
0,01
0,01
0.002
0,001
0,005
0,005
0,0001
Калий хлорнокислый 209
КАЛИЙ ХЛОРНОКИСЛЫЙ (ПЕРХЛОРАТ КАЛИЯ)
Kalium perchloricum Potassium perchlorate Kalium perchlorat
Galium hyperchloricum
КСЮ4 Мол. в. 138.553
Приготовление
50—80 г КСЮз помещают в шамотный тигель емкостью 100—150 мл и на*
гревают сначала медленно, а затем, увеличив пламя, быстро доводят до
плавления. Если масса слишком вспучивается, пламя уменьшают. Через
30—35 мин. масса загустевает и покрывается коркой; как только выделение
кислорода прекратится, нагревание прерывают и тигель охлаждают. Массу
истирают в порошок. 150 г такого порошка кипятят с 45()мл воды до
прекращения растворения и раствор фильтруют горячим. Фильтрат быстро
кристаллизуют, выпавший кристаллический порошок отсасывают, дважды промывают
на фильтре холодной водой и испытывают на чистоту. Готовый препарат
сушат в сушильном шкафу при 80°,
Свойства
Прозрачные кристаллы ромбической системы, уд. в. 2,52, плавящиеся около
610° с разложением на КС1 и Ог. Растворы имеют нейтральную реакцию и
освежающий вкус. В отличие от NaC104, KQO4 нерастворим в спирте.
Таблица 199
Растворимость КС104 в воде
/•с
0
10
15
% ксю4
0,7
1,1
1,4
/°с
20,5
25
50
о/о КС10<
1,7
2,2
5,1
/°С
70
100
% ксю4
10,9
18,2
Таблица 200
Удельный вес водных растворов КСЮ4
% ксю4
0,2
0,4
0,6
4
1,0004
1,0016
1,0029
°/оКСЮ4
1 0,8
1,0
1,2
«i5
1,0041
1,0054
1 1,0067
°/о КСЮ,
1,4
1,6
1,8
1,0079
1,0092
1,0105
Испытание
1. Раствор препарата при действии концентрированной НС1 не должен
окрашиваться в желтый цвет.
2. Раствор препарата, подкисленный HNOs, не должен давать осадка
с AgNO*.
SI О
Калий
КАЛИЙ ХРОМОВОКИСЛЫЙ (ХРОМАТ КАЛИЯ)
Kalium chromicum Potassium chromate Kaliumchromat
K2C1O4 Мол. в. 194,20
Приготовление
2 ч. измельченного К2СГ2О7 обливают 4 ч. кипящей воды и при помещу
вании прибавляют к раствору чистый КгСОз с избытком 2—3% против ст&
хиометрического количества.. Фильтруют горячий желтый раствор и дают охла*
диться, причем выпадает К2СЮ4. Выпариванием маточного раствора мо^ао
получить еще значительное количество препарата.
Свойства
Желтые ромбические кристаллы уд. в. 2,74, устойчивые на воздуху
Т. пл. 975°. Растворяясь в воде, образуют интенсивно-желтый раствор, щ
действующий на лакмус и куркуму.
Таблица 201
Растворимость КгСг04 в воде
0
10
20
за
% KtCrO,
36,4
37,9
38,6
39,5
ГС
40
50
60
70
%КяСгО<
40,1
40,8
42,1
43,6
t°C
80 .
90
100
о/0 К8Сг04
44,5
45,5
46,5
Таблица 202
Уделььный вес водных растворов К*Сг04
% К8Сг04
1
2
4
6
8
10
12
<?
1,0066
1,0147
1,0311
1.0477
1,0647
1,0821
1,0999
% К,СгО<
14
16
18
20
22
24
'?
1,1181
1,1366
1,15о5
1,1748
1,1945
1,2147
• %К,СгО,
26
28
30
32
36
40
/#18
1,2354
1,2566
1,2784
1,3010
1,3478
1,3963
Окраски раствора настолько ярка, что при разбавлении 1:40 000 еще ясно
замечается желтый цвет. От прибавления кислот цвет раствора переходит в
оранжевый вследствие образования К2СГ2О7:
2К2Сг04 + H2SO4 = К2СГ2О7 + K2SO4 + Н20.
При нагревании КгСг04 приобретает красную окраску, при охлаждении сноя*
становится желтым. Нерастворим в спирте и эфире.
•ч
Калий хромовокислый и цианистый •
211
Ис3 ост 4779 содержание К2СЮ4 в препарате должно быть не менее 99%;
большие количества допустимых примесей указаны в табл. 203.
Таблица 203
Допустимые примеси в КгСг04
Примеси
Допустимое содержание в %
в химически!
чистом
в чистом
для анализа
I Не растворимые в воде вещества
% Свободные щелочи (ОН') . . . .
3. Хлориды (СГ)
4. Сульфаты (SO*77) .
5! Соли алюминия (А1)
6. Соли кальция (Са)
0,002
0,03
0,001
0,010
0,002
0,002
0,004
0»03
0,003
0,025
0,004
0,005
0,010
0,06
0,005
0,05
0,010
0,010
КАЛИЙ ЦИАНИСТЫЙ (ЦИАНИД КАЛИЯ)
Kalium cyanatum Potassium cyanide Kaliumcyanid
KCN Мол. в. 65,114
ITp иготов ление
Сильный яд! Работать очень осторожно\
1. В закрытом железном тигле сплавляют 8 ч. частого K4Fe(CN)e с 3 ч.
КгСОз при температуре красного каления:
K4Fe(CN)e + КгСОа = 5KCN + KCNO+Fe + CO2.
Сплавление считается законченным, когда проба представляет собой белую,
а не желтую массу. Расплавленную соль выливают в плоскую чашку; по
охлаждении разбивают на куски и хранят в плотно закрытых банках (ябЧ).
Железо, получающееся при реакции, собирается в виде порошка на дне
тигля, а циановокислый калий (KCNO) остается в виде примеси к KCN
Однако препарат годится для обычных аналитических целей (К о р е н б л и т1).
2. В подогретый до 40° раствор КОН в абсолютном спирте пропускают
пары синильной кислоты HCN (яд!), получаемые нагреванием K4Fe(CN)e
с H2SO4. Выпадающую соль отсасывают на воронке Бюхнера, споласкивают
абсолютным спиртом и сушат в вакуум-эксикаторе над СаСЬ.
Свойства
Бесцветный, чрезвычайно ядовитый, кристаллический порошок уд. в. 1,52,
легко растворимый в воде. При медленном охлаждении расплавленной соли
кристаллизуется в кубиках; из растворов кристаллизуется в октаэдрах.
Б сухом состоянии не имеет запаха; притягивая влагу из воздуха, начинает
пахнуть синильной кислотой (горьким миндалем) вследствие разложения
углекислотой и влагой воздуха.
^ Хим. реакт., 309.
212
Калий. Кальций
Легко растворим в кипящем спирте. Растворим в абсолютном С1)ирТе
(0,88 : 100) и в метиловом спирте (4,91 : 100). Водный раствор имеет щелочную
реакцию (рН = 9,8 для \N раствора, 9,2 для 0,Ш раствора1) и при хранение
разлагается (быстрее при кипячении), выделяя NHs и переходя в муравьино.
кислый калий: - -
KCN + 2Н20 = NHs + НСООК.
При 623,5° KCN плавится в подвижную, как вода, жидкость; при бол^
сильном нагревании частично испаряется. При плавлении на воздухе или црд
добавлении легко восстанавливающихся металлических окислов попоща^
кислород, переходя в KCNO.
Таблица 04
Удельный вес водных растворов KCN
% KCN
1
2
4
6
л15
1,0041
1,0092
1,0194
1,0297
°/aKCN
8
№
12
d4
1,0401
1.050Я
1,0612
о/о KCN
14
16
18
—*%
d4
1,0718
1,0825
1,0841
Испытание (Мерк)
1. Сернистый калий (K2S). Раствор 1 г препарата в 20 мл воды при
добавлении РЬ(СНзСООЬ должен давать чисто белый осадок. !?
2. Кремнезем (Si02). 5 г препарата выпаривают под сильной тягой с pafc
бавленной НС1 досуха (осторожно/ Выделение чрезвычайно ядовитой синиль*
ной кислоты!). Остаток высушивают в течение получаса при 100° и раствоч
ряют в 250 мл воды, подкисленной небольшим количеством НС1. Раствор
должен быть совершенно прозрачен.
3. Хлор (СГ). 1 г препарата прокаливают с 2 г KNOs и 10 г КгСОз ДЛ$Г
разрушения циана. Остаток растворяют в воде, доводят азотной кислотой щ
кислой реакции и прибавляют AgNOs: не должно быть никакого осадка AgCjt
4. Углекислые соли (СОз"). Раствор 1 г препарата в 20 мл воды от прибШ
вления 5 мл НС1 не должен сильно вскипать. ^
о. Роданистый калий, железистосинеродистый калий и сернокислый калий
[KCNS, K4Fe(CN)e, K2SO4]. Раствор препарата, доведенный прибавлением ИЩ
до кислой реакции, не должен: а) от FeCb давать красного (примесь KCN$f
или синего [примесь K4Fe(CN)e] окрашивания; б) от ВаСЬ образовывай
белой мути (примесь K2SO4).
КАЛИЙ ЩАВЕЛЕВОКИСЛЫЙ (ОКСАЛАТ КАЛИЯ)
Kalium oxalicum Potassium oxalate Kaliumoxalat
Oxalsaures Kalium
K2C2O4H2O Мол. в. 184,228
П риготовление
Горячий раствор щавелевой кислоты нейтрализуют чистым КгСОз до слабо
щелочной реакции, выпаривают до начала кристаллизации и охлаждают. Кр#*
сталлы отсасывают и высушивают на листе фильтровальной бумаги при КО***
натной температуре.
* Н. Л. Тднанмев и Ю. В. Каря^нн, ЖПХ, 13, N 2,304 (1940).
Калий -щавелевокислый. Кальций азотнокислый
21$
с тва
С*ой
Бесцветные ромбические кристаллы уд. в. 2,08. горьковатого освежающего
йКуса. При 100° начинает терять кристаллизационную воду, становясь
непрозрачным; полностью теряет воду только при 160°. Насыщенный водный раствор
^еет слабо щелочную реакцию (рН = 8,2).
Таблица 205
Растворимость К2С204
гс
0
10
20
30
40
50
%К,с,о4
20.3
23,7
2\4
28,6
30,8
33,0
*°с
60
70
80
. 90
; 100
% К«с,о4
35,1
37 2
39,5
41,3
44,0
Таблица 206
Удельный вес
водных растворов К2С204
/оК,С,04
1
2
4
6
л18 1
4
1/4)61
10136
1,< 2*8
1,0441
°/о КяСа04
8
10
12
14
d4
1,0596-
1,и753
1,0912
1,1072
Испытание
По ОСТ 2757 содержание К2СЮ4 • НЮ для препаратов х. ч. и ч. д. а.
не менее 99,8%; наибольшие количества допустимых примесей указаны
в табл. 207,
Таблица 207
Допустимые примеси в КоС204 • Н20
Примеси
Допустимое содержание в %
в химически
чистом
0,003
0,002
0,0 >5
0 0002
0,0002
0,01
. 0,01
в чистом
| для анализа
0,005
0,005
0.015
0/010
о,ооо5
0,025
0,025
в чистом
0,020
0.010
0,030
0,(015
0,0010
0,05
0,05
1. Не растворимые в воде вещества
2. Хлориды (СП
3. С>льфагы(504")
4. Т желые мета лы
5. Железо (Fe) . •
6. Кислот' ость (НС204') .-
7. Щелочность (С03")
КАЛЬЦИЙ АЗОТНОКИСЛЫЙ (НИТРАТ КАЛЬЦИЯ)
Calcium nitricum Calcium nitrate Calciumnitrat
Са(Ж)з)2-4Н20
Мол. в. 236,16
Приготовление i
1. Разбавленную HN03 нейтрализуют СаСОз или Са(ОН)5| добавляют
Са(ОН)2 до слабощелочной реакции и оставляют стоять в теплом месте. Вы-
павшие Fe(OH)3 и А1(ОН)з отфильтровывают, пропускают в раствор струю СОа
Для осаждения избытка Са(ОН)г, отфильтровывают выпавший СаСОз и
выпаривают раствор до образования кристаллической пленки на поверхности. При
охлаждении выпадают кристаллы Са(ЫОз)2*4ШО.
2. Безводную соль получают нагреванием гидрата Са(ИОз)в • 4НаО при 130°.
214
Кальций
Свойства
Водная соль представляет собой прозрачные призматические кристаллы
уд. в. 1,82, расплывающиеся на воздухе. Т. пл. 42,5°. Безводная соль — бела
масса уд. в. 2,4, при прокаливании переходящая в СаО. Т. пл. 561°* я
Таблица 208
Растворимость Ca(N03)2 в воде
t°c
0
18
42.7
о/0 Ca(.N08)t
48,2
54,8
69,5 1
/°с
51,1
151
о/0 Ca(N08)t
75,2
78,4
Таблица 209
Удельный вес водных растворов Ca(NO.i)2
в/о CatNO,),
2
4
6
8
10
л18
1,0137
1,0291
1,0448
1,0608
1,0771
% Ca(N08)s
12
14
16
18
20
И18
1 4
1.0937
1,111*6
1 1279
1,1455
1,1636
%Ca(N03)»
25
30
35
40
45
1 <J8
1,211
1,259
1.311
1,366
1,423
Испытание
Наибольшие количества примесей, допустимых по ОСТ 5462, указаны
в табл. 210. Препарат должен быть испытан на кислотность по приведенному
в ОСТ методу.
Допустимые примесив Ca(N03)2 • 4Н20
Примеси
1. Не растворимые в воде вещества
2. Хлориды (СК)
4. Желеоо (Ft)
5. Тяжелые металлы . .
6. Щелочные металлы и магний
(в виде сульфатов)
Допустимое содержание в °|0
в химически
чистом
0,002
0,002
0.010
0,0001
0,0002
0,05
в чистом для
1 анализа
0,005
0 005
0,020
0,0002
0,0005
0,20
в чистом
0,010
о,ою
о.озо
0,0005
0,001
0,40
Кальция гидрат окиси
215
КАЛЬЦИЯ ГИДРАТ ОКИСИ (ГИДРООКИСЬ КАЛЬЦИЯ, ГАШЕНАЯ
ИЗВЕСТЬ)
Calcium hydroxydatum
Calcium hydroxide
Slaked lime
Calciumhydroxyd,
Kalkhydrat,
Geloschter Kalk
Ca(OH)2
Мол. в. 74Л0
приготовление
1. Д. Н. Финкельштейн предлагает следующий метод: 250 г
СаСЬ'бЬЬО растворяют в 250 мл воды, нагревают до 80° и обрабатывают
несколько приемов профильтрованным раствором 120 г NaOH в 250 мл воды
(операцию ведут в сосуде емкостью 1 л). Горячую кашицу немедленно
переносят на большую воронку Бюхнера и при отсасывании промывают дестилли-
рованной водой до исчезновения реакции на СГ в промывных водах
(отсутствие мути от добавления AgNCb в подкисленный НЬЮз фильтрат). Вначале
промывание ведут водой, содержащей 0,1% NaOH1, затем чистой водой.
Отмытый осадок, высушенный при 80—100°, достаточно чист для большинства
целей.
2. Приготовление известковой воды. 1 ч. хорошо прокаленной СаО
обливают 4 ч. воды и после распадения кусков образовавшийся тонкий порошок
размешивают с водой до получения однородной массы. Через некоторое время
возможно полнее сливают прозрачную жидкость с осадка. Последний вновь
перемешивают, с 50 ч. воды до образования однородной массы, котирую
переливают в закрывающуюся банку, дают ей стоять до полной прозрачности
раствора, после чего последний сливают с осадка.
Свойства
Са(ОЙ)2 — нежный белый порошок, уд. в. 2,078, не теряющий гидратной
воды при 100° и обезвоживающийся только при прокаливании. На воздухе
притягивает СОг, переходя в СаСОз.
В воде растворим довольно плохо. Суспензия Са(ОН)2 в воде носит
название известкового молока.
Таблица 211
ГС
0
10
15
20
30
40
50
60
Растворимость СаО
В растворе содержится:
на 1 ч.
СяО
ч. воды
759
770
791
862
932
1019
1136
на 1 ч.
воды
ч. СаО
0,131
0,129
—
0,126
0.116
0,107
0.098
0,088
Ъ \ МЛ
раствопа
г СаО
0,131
—
0,129
0,123
0,113
0 104
0,096
0,086
или Са(ОН>2 в
ГС
70
80
90
95
99
120
150
190
воде
В растворе содержится:
'Рчя 1 Ч.
СаО
ч. воды
1235
1362
1579
—
1650
—
—
"
на 1 ч.
в^дм
ч. СаО
0,0С0
0,073
0,063
—
0,060
—
—
—*
в 1 мл рас
тво'.а
J СаО
0,075
0,067
—
0,058
—
0,031
0,017
0,008
1 Пои реакции образуется значительное количество NaCl: CaCla-f 2NaOH=Ca ЮН)в + 2NaCl.
Примесь NaCl значительно повышает растворимость Са(ОН),. Прибавление же NaOH, наоборот,
поепятствует растворению.
216
Кальций
Удельный вес известкового молока1
Таблица
а СаО на
1 л
% СаО
%
Са(ОН)|
и20
г СаО
на 1 л
% СаО
%
Са(ОН)а
10
20
30
40
60
60
70
80
90
100
ПО
120
130
140
150
0,99
1,96
2,93
3,88
4,81
5,74
6,65
7,54
8,43
9,30
10,16
11,01
11,86
12,68
13,50
1,31
2,59
3.87
5,13.
6,36
7,58
8,79
9,96
11,14
12,29
13,43
14,55
15,67
16,76
17,84
1,0085
1,017
1,0245
1,0315
1,039
1.046
1,0535
1,0605
1,0675
1,075
1,0825
1,0895
1,0965
1,104
1,111
160
170
180
190
200
210
220
230
240
250
2^0
270
280
290
300
14,30
15,10
15,89
16,67
17,43
18,19
18,94
19,68
20,41
21,12
21,84
22,55
23,24
23,92
24,60
18,90
19,95
21,00
22,03
23,03
24,04
25,03
26,01
26,96
27,91
28,86
29,80
30,71
31,61
32,51
ДЛ А
1,125$
мая
1,140
1Л47|
U 54»
1,161$
1,1685
1,176
1,183§
1,1 Ж
1,197
1,205
1,21
1,21
Известковая вода — прозрачная жидкость, без цвета и запаха, щелочной;
реакции [Са(ОН)2 является сильным основанием5], жадно поглощающая щ
воздуха СО2, причем выделяется белый осадок СаСОз.
При нагревании несколько мутнеет вследствие выпадения Са(ОН)г,
который в горячей воде растворим хуже, чем в холодной; при охлаждении муть'
постепенно исчезает.
Испытание3
Препарат ч. д. а. испытывается на следующие примеси:
1. Карбонаты и нерастворимые в НС1 примеси. К 5 г препарата добавляют
30 мл воды и 20 мл НС1 (уд. в. 1,125), причем допускается лишь очень слабое
выделение газа. Нерастворившегося остатка отфильтрованного, промытого,
высушенного и прокаленного не должно быть более 0,005 г.
2. Хлориды (СГ). Раствор 1 г препарата в 15 мл воды и 5 мл НЫОз
(уд. в. 1,15) при добавлении 1 мл 0,17V раствора lAgNOs может давать через
5 мин. лишь слабую опалесценцию.
3. Сульфаты (SO*"). Профильтрованный раствор 4 г препарата в 16 мл НС1
(уд. в. 1,12) и 30 мл воды осаждают при кипячении 5 мл 10%-ного раствора
ВаСЬ. Выпавший через 18—20 • час. осадок BaSCh после фильтрования,
промывания и прокаливания не должен превышать 0,01 г (что соответствует
0,1% SO/').
4. При добавлении к раствору 2,5 г препарата в разбавленной соляной
кислоте NH-iOH до слабощелочной реакции допускается образование весьма
незначительного количества хлопьев.
« Lenart, С. II, 561 П919).
* Как основание, содержащее 2гидроксильных иона, Са (OH)t диссоциирует двухступенчат*:
Са(ОН)3^±СаОН*+ОН'; СаОН' г± Са'Ч- ОН'.
Первый процесс в водных растворах протекает практически нацело; для второй схемы опредс»
лена константа диссоциации. /Г = 0,03К С. W. Da vies, J. Chem, Soc. 349 (1939).
8 Испытание хим. реактивов, 195,
КАЛЬЦИЯ ОКИСЬ (НЕГАШЕНАЯ ИЗВЕСТЬ)
Cal
Kalk,
Kalk, Aetzkalk
Calcium oxydatum Calcium oxide Lime, Calciumoxyd,
Gatoana usia nslaKed lime, Quicklime Kalk, Gebranntei
CaO Мол. в. 56,08
Приготовление
Ji ^^^'ynSS^nLF^™ c продырявленным
g* (0 полном удалении СО, можно суд^ а> T^Zutllm^ZZ-
87уТри не должно обнаруживаться кристаллическое слоения КУС0ЧК0В-
2 Значительно легче и в более чистом виде ,. рт п г. „»
KS^sa- к "*pa"м, *&!Г5& TSSbZZ
4. Химически чистый Ca(NOa)s осаждают п,
(NH4)2CO,. Осадок СаСО, промывают специально СТВ°Р°М х.имичес„ки чистого
вают и прокаливают в токе сухого воздуха ТпиТп^Тп ^^ ?ЫСуШ"'
путем препарат совершенно чист». У При 1000°- Полученный таким
Свойства
Белый порошок или [при получении из СаГМл ч ■■
кубики уд. в 3,2-3.4. (Последние меньше поада,п0з)г] м.аленькие прозрачные
£ пл. 2572°, т. кип. 2850°. На воздухе притаг'ТСЯ деиствию в°ды и С0£
со значительным увеличением объема в Са(ОН)? ает ВЛЗГУ г гп'' переХ0ДЯ
Благодаря своей способности жадно поглоша далее в ^ri
для осушения газов; влажный воздух, вылет» влагу применяется
СаО, сохраняет только 0,2 мг НЮ на 1 л (Б ус »"ЛЫИ н^отоР°.е в?ем.я над
творимости СаО в воде см. стр. 215. ^ У с и М а к - И н т а и р) *. О рас-
И спытание
5 г препарата, смешанные с 4 г воды обп*,
густое тесто, которое должно растворяться в п,3у?т через. ™™л™° МИНУ*
тельного выделения пузырьков С02 и оставлявзбавленнои HCI без ?начи"
растворившийся осадок (Si02). ЯТь Лишь незначительный не-
Если, ■ разделив раствор на две части, к одной * * *т« ^.и
то может образоваться лишь незначительный „П^и6аВт/пн?Т0Кп1Й?Ж
прибавление к другой части раствора ВаСЬ не п?СаД0К la1(°h'3' Fe(OHM;
Раствор 1 г препарата в разбавленной HNO* п0с!ЛЖН°* вызь,вать4 помутнения,
дать только незначительную опалесцендию. дооавления AgNUs может
КАЛЬЦИЙ СЕРНИСТЫЙ (СУЛЬфИд КАЛЬЦИЯ)
Calcium sulfuratum Calcium sulfide Calciumsulfid
CaS Мол, в. 72,14
Приготовление
Большинство методов получения ■ СаЗ сводите, к восстановлению CaS04
углем.
1 Нет. г. преп. 450,
* НС I 1. I1JJCII. 4UV/.
«GUser, Indikitor*n der Acldlmetrie. u. Alkalimetrb „T„ *. 1nnn
•A. H6nigschmldtu. Horowitz, Mitteilungen i'nfJ&V d1 J.900'
« H. S, Booth a. L. H. Mclntyre, Ind. Eng. Chem. AnJ^Ed^lfw^
Wien, 86, 10, 1910.
(1930).
218
Кальций
А у е р1 например, рекомендует накаливать смесь 25 г алебастп»
(€aS04 • У2Н2О) с 17 г сажи в течение 14 час. при 600—800° без дост.у£|
воздуха.
По Фелею2 через рыхлый слой Са(ОН)2, помещенный в стеклянную
трубку, пропускают H2S, промытый водой. Выход почти теоретический.
Свойства
Желтовато-белая непрозрачная масса уд. в. 2,25, не имеющая запаха.
Растворима в воде в отношении 1 : 500. При употреблении меньших количеств
воды в раствор переходит сульфгидрат [Ca(SH)2], причем остается нераство,
ренным Са(ОН)2:
2CaS + 2НЮ = Ca(SH)2 + Са(ОН)2.
КАЛЬЦИЙ СЕРНОКИСЛЫЙ (СУЛЬФАТ КАЛЬЦИЯ, ГИПС)
Calcium sulfuricum Calcium sulfate Calciumsulfat
Gypsum Gips
CaSOi Мол.в. 136,14; CaS04 • У2Н2О Мол. в. 145,15, Са504-2НгО Мол. в. 172,17
Приготовление
1. Чистый препарат Са304 • 2Н2О получается осаждением его из 20%-ного
раствора СаСЬ при помощи 20%-ной H2SO4. Жидкости дают отстояться
5—6 час, декантируют несколько раз небольшим количеством чистой воды, до
удаления ионов СГ, фильтруют, промывают спиртом и быстро сушат при
температуре н!е выше 40° при помешивании на водяной бане до удаления запаха
спирта. Окончательная сушка проводится при комнатной температуре.
2. По Г. и В. Бильц3 для получения химически чистого полугидрата,
CaS04 •/^НгО, поступают следующим образом: 20 г осажденного CaS04 • 2ШО
(см. способ 1) замешивают в густую кашу с 50 мл Н1МОз (уд. в. 1,4) и
нагревают ла кипящей водяной бане, время от времени покачивая. Для
наблюдения за ходом реакции берут каплю смеси и помещают ее под микроскоп
между предметным и покровным стеклами. Приблизительно через 10 мин.
красивые заостренные иглы дигидрата исчезают и появляются твердые призмы
прямоугольного очертания. Одновременно кашеобразная масса разжижается.
После получасового нагревания охлаждают, дают массе осесть, сливают
жидкость возможно полнее, споласкивают осадок 50%-ным спиртом и тотчас
же отсасывают. Осадок на воронке промывают спиртом и высушивают
в шкафу, обогреваемом водяным паром. Полученный препарат имеет
крупнокристаллическое строение и поэтому с водой твердеет медленнее продажного
гипса.
3. Безводный CaS04 получают нагреванием дигидрата выше 150°.
Свойства
Осажденный CaS04 • 2Н2О — микроскопические, заостренные игольчатые
кристаллы уд. в. 2,32. Переход Са304 * 2НаО в СаБОя'УгНЮ происходит4
при 82°. Полугидрат (гемигидрат, «жженый гипс»)* Сал04*%Н20 — белый
порошок уд. в. 2,6—2,75; при замешивании в жидкую кашицу с небольшим
количеством воды быстро затвердевает, переходя в дигидрат.
1 М. А и е г, J. prakt. Ch. 100, 115 (1920).
* Неорг. преп. Ц5.
• Uebungsbeispiele.
4 Л. А. Турцев, Изв. Акад. Наук СССР, Геолог, серия, М 4, 180 (1939).
, Кальций сернокислый и углекислый
219
Обезвоженный CaS04 — белый сухой порошок уд. в. • 2,97, притягивающий
rv- из воздуха. На этом свойстве основано применение CaS04 в качестве
вДа,тпителя. Влажный воздух, пропущенный через трубку с CaS04, сохраняет
?олько 0,005 иг ШО на 1 л (Бауэр1). Т. пл. 1450°,
Таблица 213
Растворимость CaS04 в воде*
■
t9C
0
10
18
25
% CaSO,
0,176
0,193
0 202
0,208
ГС
30
40
55
60
% CaS04
0,209
0,211
0,208
0,200
ГС
100
151
180
200
о/0 CaSO,
0,155
G.049
0027
0,016
Испытание2
Препарат ч. д. а. должен содержать примесей, не растворимых в НС1, не
более 0,025%, магния и щелочных металлов — не более 0,2%.
Профильтрованный раствор 2 г препарата в НС1 от добавления избытка ЫНЮН и (NH4)2S
не должен мутнеть или окрашиваться (Fe). При добавлении AgN03 к раствору
1 г препарата в разбавленной HNOa допустима лишь слабая опалесценция.
КАЛЬЦИЙ УГЛЕКИСЛЫЙ (КАРБОНАТ КАЛЬЦИЯ)
Calcium carbonicum
Calcium carbonate
Calciumearbonat
СаСОз
Мол. в. 100,09
Приготовление
1. Горячий раствор СаСЬ вливают в 10%-ный раствор (NHU^CCh, к
которому прибавлено 10% по объему NH4OH (уд. в. 0,96). После тщательного
перемешивания жидкость декантируют в высоких
цилиндрах; осадах фильтруют, снова промывают и
осторожно высушивают в никелевых чашках.
2. Чистый препарат, не содержащий. щелочных
металлов (пригодный для определения К* и Na*
110 Л о у р е н с-С м и т у), может ' быть* получен
растворением чистого СаСОз в НС1 и осаждением
Са' *• из разбавленного горячего раствора
добавлением NH4OH и (NH^COe в избытке. Осадок
СаСОз тщательно промывают горячей водой и
высушивают*
Таблица 21
Растворимость
СаС03 вводе
/°с
1С тва
25
50
75
о/о СаСОа
0,001445
0,001515
0,001816
.Белый кристаллический порошок уд. в 2,71,
растворимый в кислотах и почти не растворимый в
воде ,"(zp=8,2-10-8)3.
1 Т. Н. "Bower, J. Res. Nat. Bur. Stand., 12, 2413 (1934); Chem. Abst. 28, 3283(1934).
■ Испытание хим. реачтивов, 197.
• F. Wenz, Deutsch. Zuckerind. 63, 748 (1938); С II. 2039 (1938). По другим данным
р (СаСО.)= 3'83'10 <Р- На11' Z- Pbys. Ch. A. 175, 63 (1935); С. II, 2848 (1936); £>(ClCoifz
=4t8.10~e)' (Undolt-Tabellen),
220
Кальций
Вода, насыщенная СОг, растворяет значительные количества СаСОз
вследствие образования бикарбоната (0,156 г СаСОз в 100 мл воды при 0°). Т.. tm
1339° при 1025 ат. ^
При нагревании до 420° начинает диссоциировать1 на СаО и СОг;
упругость С02 достигает 1 ат при 894,4±0,3°,
И спытание
Наибольшие количества примесей, допустимых по ОСТ НКТП 6279/254,
указаны в табл. 215. Препарат ч. д. а. должен, кроме того, выдерживав
«спытание на щелочность, приведенное в ОСТ.
Таблица 215
Допустимые примеси в СаС03
Присвой
1. Вещества, не растворимые в НС1
2. Хлориды (СГ)
4. Металлы, осаждаемые H2S . . . .
5. Железо (Fe)
6. Щелочные металлы (в виде
сульфатов)
7. Магний и щелочные металлы
■»
Допустимое содержание в °/о
в чистом для
анализа
0,01
0,002
0,01
0,002
0,005
0,05
i
в чистом
0,03
0,01
0,05
0,01
0;010
1,0
КАЛЬЦИЙ ХЛОРИСТЫЙ (ХЛОРИД КАЛЬЦИЯ)
Calcium chloratum Calcium chloride Calciumchlorid
CaCh Мол. в. 110,99
СаСЬ • 6H20
Мол. в. 219,09
Приготовление
Обрабатывают 400 г мрамора в виде мелких кусочков 800 мл технической!
НС1 (можно по Б е н д е р у. использовать жидкость, остающуюся в аппарате
Киппа при получении СОг из мрамора и НС1, добавив к ней СаСОз до
насыщения).
Слив жидкость с нерастворившегося осадка, добавляют к ней хлорной воды
(или хлорной извести) для окисления железа, кипятят и приливают избыток
известкового молока до слабощелочной реакции. Смеси дают стоять некоторое
время в теплом месте, отфильтровывают выпавший Fe,(OH)3, точно
нейтрализуют фильтрат чистой НС1 и, выпарив до уд. в. 1,44, охлаждают. Через
2—3 часа вносят затравку (кристаллик СаСЬ-бНЮ) и дают кристаллизоваться
при 16—18°.
Кристаллы переносят на воронку Бюхнера и оставляют (не отсасывая) на
30 мин. для освобождения от маточного раствора. После этого их промывают
минимальным количеством холодной воды и вставляют на несколько часов
1 Т. С. Southard a. P. H. Royster, J. phys. Chem. 4t>, *35 f 1936); о кинетике диссоциаций
CaC08 ьри различии темдератур* см. A. Oliver!о, С. 1, Ь2\ (.936).
Кальций хлористый
221
хом помещении. Сухие кристаллы переносят в банки и закрывают плотно
3 погнанными пробками, которые заливают парафином.
2 Для получения пористого препарата по Эрдману1 раствор СаСЬ,
полутени по способу 1, выпаривают до получения на поверхности пленки сухой
че и Тогда уменьшают пламя, прибавляют немного концентрированной НС1
С° продолжают нагревание еще 4—5 час. при температуре, не превышающей
200°, Д° тех П0Р» пока СаСЬ не сделается совершенно сухим. Массу еще в го-
пЯче'м состоянии разбивают на куски и складывают в хорошо закрывающиеся
банки. Препарат по составу близок к дигидрату, СаСЬ • 2НгО.
3-. Для получения сплавленного СаСЬ пористый препарат, полученный по
способу 2, нагревают до 800° и расплавленную соль выливают в охлаждаемую
водой никелевую чашку (или на железный лист), где СаСЬ застывает в белую
кристаллическую массу. По охлаждении до 110—120° ее разбивают на куски
й помещают в банки с хорошо пригнанными пробками.
Свойства
СаСЬ'бШО—большие бесцветные ромбические призмы уд. в. 1,65, горько-
соленого вкуса. Т. пл. 29,5°. При нагревании теряет 4НгО и переходит в ди-
гидрат СаСЬ • 2ШО, представляющий собой белую пористую массу.
При красном калении теряет остальную воду, переходя в сильно
гигроскопический безводный СаСЬ —белую кристаллическую массу уд. в. 2,15,
плавящуюся при 774°. Вследствие^частичного разложения сплавленный СаСЬ
всегда содержит СаО и поэтому имеет щелочную реакцию.
Хорошо растворим в воде со значительным выделением тепла; хуже
растворим в спирте и ацетоне. Упругость водяного пара над СаСЬ по Беккеру2
составляет 0,14—0,25 мм рт. ст.; остаточная влажность воздуха3 над СаСЬ
при 25° не превышает 0,36 лга.ШО на 1л.
Таблица 216
Растворимость СаС12вводе
t°c
-55
-25
0
+10
20
29#
% CaClj
29,9
33,3
37,5
39,4
42,7
60,1
/°с
40
45,3
60
70
80
90
% СаС1в
53,5
56,6
57,8
58,6
59,5
60,4
t°c
100
120
140
170 ■
175,5
«/о CiClt
61,4
63,4
65,6
71,8
74,8
Таблица 217
о/о Са€1в
1
2 .
4
6
8
10
Удел ьный
"*°
1,0070
1,0148
1,0316
1,0486
1,0659
1,0835
вес водных растврров СаС12
% СаО,
12
14
1 16
18
20
22
л20
1,1015
1,1198
1,1386
1,1578
1,1775
1,1968
' % CaCl,
24
26
28
30
35
1 40 |
«2,°
1,2175
1,2382
1,2597
1,2816
1,3373
1 1,3957
1 Хим. првп. 20.
"Becker, Ind. Eng. Chem. Anal. Ed. 4. 1 (1933).
'H. S. Booth Я. L. H. Mclntyre, ibid. 2, 12(1930).
Испытание
По ОСТ содержание СаС1г в гранулированном препарате должно быть ае
менее 74% (ОСТ 5458), в плавленом —не менее 93% (ОСТ 5457); содер...
кание СаС12«6НгО в кристаллическом препарате —не менее 95% (ОСТ 5459);
1аиболыпие количества допустимых примесей указаны в табл. 218.
Допустимые примеси в СаС1а
Таблица 2/$
Примеси
Допустимое содержание в %
ОСТ 5458
в хлористом
кальции
гранулированном
ч. д. а
чистом
ОСТ 5457
в хлористом
кальции
плавленном
ОСТ 5459
в хлористом каль»
ции кристаллическом
в
X. Ч.
0,0001
0.1
0,002
0,002
0,002
0,0002
0,004
в
ч. д. а.
0,0002
0,2
0,005
0,005 1
0,005
0,0005
0,004
в
чистом
1. Щелочность [Са(ОН2)] . . .
2. Железо (Fe)
3. Магний и щелочные металлы
(в виде сульфатов)
4. Не растворимые в воде
вещества
5. Сульфаты (SO/0
6. Барий (Ва)
7. Тяжелые металлы
8. Кислотность (НС1)
0,02
0,002
0,3
0,05
0,005
0,5
1
0,5
0,0005
0,5
0,010
0,010
0.010
0,001
0,008
КАРБОСИЛИКАГЕЛЬ
Приготовление
По методу А. П. П а л к и н а и А. Л. Колесникова продажное жидкое
стекло разбавляют водой до содержания 4,1% БЮг. К 50 мл раствора
добавляют требуемое количество активированного угля, просушенного при 105°
и измельченного до 0,06—0,125 мм\ наиболее активный гель получается при
содержании 10% угля.
Смесь размешивают 20 мин., после чего добавляют из бюретки 4%-ной НС1.
Перемешивание продолжают до момента коагуляции, наступающего через
2—5 мин. после прибавления всей необходимой кислоты. Стакан с гелем
помещают для созревания в эксикатор на 2—3 дня; степень созревания геля
определяют, надавливая на него пальцем. Созревший гель оказывает
достаточное сопротивление, образуя трещины вокруг углубления.
Гель разламывают на кусочки, подсушивают на стекле при комнатной
температуре в течение двух суток, затем — в термостате при 40—50° до
содержания влаги не выше 75—80%. Далее промывают препарат в течение 5—6
суток водой до удаления СГ и высушивают 60—70 час. в термостате при
постепенном повышении температуры от 40 до 120°, после чего проводят
активирование геля нагреванием при 240—250° в фарфоровом тигле, закрытом
крышкой К
Свойства
Плотные черные кусочки, обладающие большой адсорбционной способностью
но отношению к парам воды и органических веществ.
1 ЖПХ, 10, №10-11, 1879 (1937).
Карбосиликагель. Квасцы алюмо-аммонийныё
223
квасцы алюмо-аммонийные
алюмо-аммйачные квасцы, двойная соль сернокислого
( алюминия и сернокислого аммония)
,Alumen ammoniacale
Ammonium alum
Aluminium ammonium
sulfate
Ammoniakalaun
(NH4)2S04 • Ab(S04)s • 24HsO *
Мол. в. 906,44
Приготовление
Смешивают горячие профильтрованные растворы 29 г (NH4)2S04 в 50 мл
воды и 147 г сернокислого алюминия в 150 мл воды. При охлаждении
выпадает около 180 г квасцов.
По данным А. Хил.ла и Н. Каплана1 можно не придерживаться
особенно точно стехиометрических соотношений, так как квасцы выпадают
чистыми без избытка одного из компонентов в широком интервале сотноше-
ний между Ab(S04)3 и (NH4)23C>4. *
Свойства
Бесцветные октаэдрические кристаллы уд. в. 1,645, растворимые в воде,
При 92° квасцы плавятся в своей кристаллизационной воде, при 190°
образуют пористую массу безводных квасцов. Сильное прокаливание приводит
к образованию АЬОз, в то время как НаО, SOs и ]МНз улетучиваются.
Таблица 219
Растворимость (NH4)2S04• A12(S04)3• 24Н20 в воде
t°C
0
10
15
20
25
% 'NH4fcS(V
•AlafS04)8-24HeO
4,9
8,4
10,2
12,1
14,2
t*c
30
40
50
60
% WH4)iS04. 1
•AIe(SOi)8.24HlO
16,2
21,6
26\7
33,9
/°c
70
80
90
100
VofNH^SO,.
'(AltS04j..24HtO
41,8
50,8
65,3
80,8
Таблица 220
Удельный вес водных растворов
(NH4)2S04.A12(S04)3
о/0 (NH4)tS04.
•AljlSOJ,
1
2
1 <<
1,0079
1,0167
1 %(NH4)eSCV
•Al,(S04)a
4
6 .
! **
1,0348
1,0533
.* Или NH4A1(S04). '12 H.O.
1 A. E. Hill *. N. Kaplan, J. Am. Cheni. Soc. 60,558 61938); C. I, 4696 (1938).
224
Квасцы
Испытаниеь
Наибольшие) количества примесей, допустимых по ОСТ 2903, указац^
в таОл. 2,2i\*
Таблица 22!
До пустим ые примеси в (NH4)2S04 • A12(S04)3 • 24H20
Примеси
1. Не растворимые в воде вещества
2. Хлориды (СГ) .
3. Железо (Fe) .
5. Мышьяк (As) .
6. Щелочные и щелочноземельные
[ Допустимое содержание в %
в чистом для
анализа
0.0С5
0,001
0.0005
0,0005
0,00005
ол
в чистом
0.01
0,004
0.001
owoi
0,0001
0,2
КВАСЦЫ АЛЮМО-КАЛИЕВЫЕ
(КАЛИЙНЫЕ КВАСЦЫ ДВОЙНАЯ СОЛЬ СЕРНОКИСЛОГО АЛЮМИШ
И СЕРНОКИСЛОГО КАЛИЯ)
Alumen1 kalicum
Potassium alum
Aluminium potassium
sulfate
Kalialaun
Alaun
K2S04Al2fS04)3«24H20
Мол. в. 948,76
Приготовление
1. Смешивают горячие профильтрованные растворы 18,4 г K2SO4 в 70 мЩ
воды и 70,2 г Ah(S04)3 в 60 мл воды. При охлаждении выпадает около 86}'$
квасцов. ;;j
2. Для очистки технических квасцов 200 г загрязненного продукта растврЙ
ряют в 200 мл горячей воды. Раствор фильтруют и охлаждают, причем выпа^
дает около 175 г чистых "квасцов. Кристаллы слегка промывают холодной;
дестиллированной водой и высушивают.
3. Для приготовления безводных («жженых») квасцов нагревают водную
соль в платиновом тигле. Квасцы сначала плавятся в своей кристаллизацион-:;
ной воде, затем обезвоживаются, вспучиваясь при этом. Образовавшуюся
легкую пористую массу растирают в ступке.
Свойства
Большие, прозрачные, бесцветные, октаэдрические кристаллы уд. в. 1,751,
еладкого^вяжущего ,вкуса. Плохо растворимы в холодной воде и очень хорошо
в горячей. На воздухе почти не выветриваются, но легко теряют
кристаллизационную воду при нагревании, превращаясь в белый порошок «жженых*
квасцов, трудно растворимый в воде. Т. пл. 95° (в кристалдизационной воде)'
* Или KAl(S04)i Л2НаО
Квасцы алюмо-калиевые
225
Таблица 222
•
г с
о
10
15
20
25
30
40
'Растворимость K2S04• AI2(S04)3
% K2SCV
•A!e'S04)8
3.1
4,4
4,8
5,7
6,6
9,2
12,0
100 г воды
растворяют г
K8S04.A12(S04V
•24НаО
(Р о g g i a 1 е)
3,9
9.52
N
15,13
—
22,01
30,92
t°C
50
60
70
80
90
100
•
24Н20 в воде
% kbso4.
•Ai»-;b04j3
26,1
—
51,5
—
.—
ЮЭ г воды
растворяют г
K4S(VAla(S04)3.
-Г4Н.О
(Р о g g i a 1 е)
44,11
66,65
90,67
134,47
209,33
457,48 \
Таблица 223
Удельный вес водных растворов
KaS04.Al2(S04)3
.о/о KA1(S04)3
1
2
3
л19
4
1,С079
1,0174 •
1,0270
о/о KA1(S<V,
4
5
6
Ч9
1,0369
1,0465
1,0565
Испытание
По ОСТ 17389-39 квасцы должны содержать при пересчете на безводные
_не менее 19,5% и не более 20,2% АЬОз; потеря при высушивании должна
быть не более 46%; наибольшие количества допустимых примесей при расчете
на водный препарат указаны в. табл. 224.
Таблица 224
Допустимые примесив K2S04 A12(S04)3 • 24H20
Принеси
1. Не растворимые в воде вещества
2. Хлориды (СК)
5. Металлы, осаждаемые H2S . . .
6. Мышьяк (As)
7. Свободная серная кислота (H2S04)
Допустимое содержание в %
в чистом для
анализа
0,005
0,001
0,005
- 0,001
0,001
0,00005
0,2
в чистом
0,01
0,004
0,01
0,002
0.002
0,0001
0,2
226
Квасцы
КВАСЦЫ ЖЕЛЕЗО-АММОНИЙНЫЕ
(ЖЕЛЕЗО-АММИАЧНЫЕ КВАСЦЫ, ДВОЙНАЯ СОЛЬ СЕРНОКИСЛОГО
АММОНИЯ И ОКИСНОГО СЕРНОКИСЛОГО ЖЕЛЕЗА)
Ferrum sulfuricum Ferric ammonium alum Eisenammonalaun
oxydamum Eisenammoniumsulf at
ammoniatum Eisenalaun
(NHO2SO4 • Fe2(S04)3 • 24H20 * Мол. в. 964,40
Приготовление
1. По Эрдману1 в 400 м воды растворяют 400 г FeS04 • 7Н2О, приб^
вляют к раствору 70 г концентрированной H2SO4 и нагревают до кипени^;
Кипящую жидкость обрабатывают небольшими порциями концентрированна
HNOs (всего около 120 г), пока разбавленная водой проба раствора с КзРе(СЩ
не будет давать бурое или зеленое кольцо, а не синий осадок. Жидко$|
выпаривают почти досуха и остаток разбавляют водой до уд. в. 1,317—l,3tjjj
К 300 мл полученного таким путем раствора Fe2'(S04)3 прибавляй^,
раствор 28 г (NH4).2S04 и 100 мл воды и смесь медленно охлаждают. Выпи
дающие кристаллы слегка промывают водой и высушивают без нагревания-.
2. По Ризенфельду2 в колбу емкостью 1 л вносят 75 мл разбавлен*4
ной H2SO4 и растворяют в ней при нагревании 23 г соли Мора и 10 а.
FeS04-'7H20; в случае надобности добавляют воды. Горячий раствор
фильтруют в фарфоровую чашку и, добавив 5 мл концентрированной HNQs
(уд. в. 1,4), кипятят до исчезновения красных паров. Тогда испытывают на
Fe"—проба жидкости не должна давать синего осадка с K3Fe(CN)e.
Если двухвалентное железо еще присутствует, .то кипятят, добавив еще
2—3 мл HNOs. По окончании окисления раствор выпаривают до половины
объема (капля жидкости на часовом стекле от прибавления кристаллика
любых квасцов должна кристаллизоваться; в противном случае продолжают:
выпаривание). Раствор выливают в кристаллизатор и по охлаждении вносят"':
затравку — кристаллик квасцов, полученных при пробе на часовом стекле.
Выпавший препарат отсасывают на воронке Бюхнера и сушат между
листами фильтровальной бумаги.
3. Для получения квасцов из железа 25 г железной проволоки
растворяют в 620 мл 10%-ной H.2SO4. Раствор фильтруют и прибавляют к
фильтрату при нагревании понемногу 30 г HN03 (уд. в. 1,2). По окончаний
окисления закисного железа к горячему раствору добавляют 29—30 г
(NH4)2S04 и 40—45 г воды и охлаждают; в остывший пересыщенный раствор
вносят кристаллик готовых квасцов, чем вызывают кристаллизацию.
Препарат переносят на воронку, дают стечь жидкости, промывают
небольшим количеством холодной дестиллированной воды и сушат при
комнатной температуре.
Свойства
Бесцветные в чистом виде, но обычно светлоаметистовые3 кристаллы
уд. в. 1,719. При стоянии на воздухе становятся с поверхности светлокорич-
невыми. При 33° окрашиваются в коричневый цвет, при 39 41° плавятся.
При 150° происходит потеря 11%НЮ, при 750° образуются безводные
квасцы.
Растворимы в воде (1:3 при 15°); нерастворимы в спирте.
* Или NH4Fe(SOj„ . 12НйО.
1 Хим. преп., 66.
a Иеорг. практ., 184-.
лиПт»7те1Нлер объясняет окраску присутствием сложного иона [Fe (ОаНД;Г\ Другие авторы
считают, что окраска зависит от следов марганца.
Квасцы оюелезо-аммонийные и хромо-калиевые 227
Таблица 225
Удельный,вес водных растворов Ре2(504)з • (NH4)2S04
o/„F<VS04V
'°(NHJ,S04
1
2
4
6
и1Ъ
и !
Г,С07
1,016
1,032
1,050
% Fe.'SOJa-
•(Nh,)9S04
8
10
12
14
и15
d4
1 1,068
1,086
1Д04
1,122
o/0Fe>(S04V
•(NH^SO,
16
18
20
w15
d4
1,141
1,161
1,181
Испытание
Наибольшие количества примесей, допустимых по ОСТ 2904, указаны
в табл. 226.
Таблица 225
Допустимые примеси в Fe2(S04)3 • (NH4)2S04 • 24Н20
Приме и
Допустимое содержание в %
в химически
чистом
0,005
0,0005
0.001
0,002
0.С03
0,05
в чистом для
анализа
0,01
0,001
0,002
0,005
0,005
0,05
в чистом
0,015
0,005
0,005
0,01
0,01
0,2
1. Нерастворимые в воде вещества
2. Хлориды (С1') .
3. Железо закисное (Fe") '
4. Медь (Си)
5. Цинк (Zn)
6. Щелочные и щелочноземельные
соли (в виде сульфатов) . • • •
КВАСЦЫ ХРОМО-КАЛИЕВЫЕ
(ДВОЙНАЯ СОЛЬ СЕРНОКИСЛОГО ХРОМА И СЕРНОКИСЛОГО КАЛИЯ)
Alumen chromicum Chrom alum
Cr2(S04)3 • K2SO4 • 24H20 *
Chromalaun
Kaliumchromalaun
Мол! в. 998,84
Приготовление
1. Растворяют 39,5 г КгСгаО? в 120 мл воды, прибавляют осторожно 9 г
H2SO4 (уд. в. 1,84), охлаждают и насыщают S02. Раствор оставляют
кристаллизоваться при комнатной температуре.
2. По Бёттгеру к холодному раствору 3 ч. К2СГ2О7 в 12 ч. воды и
4 ч. H2S04 (уд. в. 1,84) в фарфоровой чашке при охлаждении прибавляют
80%-ный спирт (следя, чтобы температура не поднималась выше 40°) до
появления запаха ацетальдегида; смесь оставляют стоять сутки.
С осевшего кристаллического порошка сливают маточный раствор и
отфильтровывают. Кристаллы промывают холодной водой до чисто фиолетовой
окраски препарата и промывных вод. Наконец квасцы растворяют в
возможно малом количестве воды при температуре не выше 35° и жидкость
оставляют в холодном месте для кристаллизации.
1 Или KCr(S04,yi2HaO.
228
Квасцы. Кислород
3. По Т р а у б е 1 растворяют 1 ч. К2СГ2О7 при умеренном нагревании в'£&.
H2SO4 и таком количестве воды, чтобы при обыкновенной температуре ж
происходило выделения кристаллов. Полученный раствор вливается пос%
пенно в спирт, находящийся в чашке, погруженной в холодную воду. Бол^
шая часть кристаллов квасцов выпадает при этом в виде кристаллическое
муки.
Маточный раствор выпаривают в присутствии НЖ)з (чтобы избежать пер^,
хода в зеленую модификацию) и смешивают с равным объемом спирт^
Через сутки квасцы выкристаллизовываются; их растворяют в теплой (щ
выше 40°) воде, и раствор оставляют для медленного испарения в тепло,
ватом месте.
Свойства
Темно-фиолетовые октаэдры уд. в. 1,842 при 2,0,8°, просвечивающие pyg
ново-красным цветом. Растворимы в воде (1:7 при 15°). При 75° раств
становится зеленым, однако после охлаждения через несколько дней сно
приобретает фиолетовую окраску.
На воздухе квасцы выветриваются, покрываясь лиловым налетом,
нагревании квасцов до 100° они, теряя часть воды, зеленеют. Полное об!,,
воживание достигается только при 350°. Нагретая выше 350° соль принимает
зеленовато-желтую окраску и теряет способность растворяться в воде.
Таблица 227
Удельный
вес водных растворов
K2S04.Cr2(r04)3
%K3S04.Cra(S04)3
1
2
4
-?
L00F6
1,0182
1,0376
% k9so4.
6
8
а15
4
1,0573
1,0773
Испытание
По ОСТ 4959 содержание K2SO4 • Сг2(304)з • 24В-20 должно быть не менее
98%; наибольшие количества допустимых примесей указаны в табл. 228.
Таблица 228
Допустимые примеси в K2S04• Cr2(S04)3• 24H20
Примеси
1. Не растворимые вещества . . . .
2. Хлориды (СГ)
3. Ж елезо (Fe)
4. Вещества, осаждаемые аммиаком
с (A!203 + Fe203)
5. Кальций (Са)
Допустимое содержание в %
в чистом для
анализа
0,005
0,005
0,01
0,05
0,005
0,01
0,01
0,05
0,15
0,01
1 Traube, Ann., 66, 155.
Кислород
229
Oxygenium
Приготовление
КИСЛОРОД
Oxygen
Q2 Мол. в. 32,00
А. Из КСЮз
Sauerstoff
j# Помещают КСЮз в стеклянную реторту и постепенно нагревают,
причем ' вначале рекомендуется водить горелкой во избежание местного
перегрева.
Необходимо следить, чтобы употребляемый КСЮз не содержал примеси
горючих веществ (сера, уголь, бумага и т. д.) в противном случае возможен
сильный взрыв.
При употреблении вместо чистого КСЮз смеси его с MnO*, Fe^Os, CuO,
платиновой чернью и др. выделение кислорода происходит значительно более
легко и равномерно.
Чаще всего употребляют в
качестве катализаторов МпОг
или РегОз. Так как эти
вещества иногда содержат,
горючие примеси, то необходимо
провести предварительные
испытания, для чего
прокаливают небольшую порцию оме-
си в железной ложке,
обращая внимание, происходит ли
спокойное выделение
кислорода {незначительные искорки,
появляющиеся в смеси, не
представляют опасности).
Обычно применяю/г смесь 1 ч.
МпОг {или ИегОз) и 2 ч.
КСЮз. Выделяющийся
кислород, особенно при
употреблении в качестве катализатора
МпОг, содержит хлор, который поглощается при прохождении газа через
раствор КОН. Реторта для получения кислорода подбирается такой величины,
чтобы смесь не занимала в ней больше гА объема.
2. Бабо рекомендует для равномерного выделения газа применять смесь
2 ч. КСЮз, 2 ч. NaCl и 3 ч. Fe203 или 12 ч. КСЮз, 6 ч. NaCl и 1 ч. МпО*.
М. М е й е р * советует на каждые 10 г КСЮз добавлять 2 г тонко
измельченного К2СГ2О7.
3. М ю н к е для получения значительных количеств кислорода предлагает
употреблять металлический аппарат (рис. 36). Железная труба А 40—50 см
длиной, с внутренним диаметром- 4 см и стенками толщиной 2 мм. с одной
стороны -заделана наглухо, а с другой может запираться винтом С,
прижимающим хорошо пригнанную крышку а.. Наглухо вделанная железная
трубка В служит для отвода газа. Трубу А наполняют до половины растертой
в порошок КСЮз, запирают С и начинают подогревать передвижной тройной
бунзеновской горелкой, которая по мере надобности передвигается слева
направо. По указанию автора работа в этом аппарате вполне безопасна, тг:<
как разложение соли производится в одном месте и опасаться взрыва те
приходится. Регулирование, прерывание и возобновление тока газа легко
достижимо. Очистка и зарядка прибора совершаются легко и быстро. Вместо
Рис. 36. Аппарат для получения кислорода по Мюнкз
1 М. Meyer, J. chem. Educ, 17, 494 (194^; С. I, 2089 (1911).
230
Кислород
винта С можно применять завинчивающуюся крышку, снабженную проклад.
кой из асбеста (но не резины!).
Примечание. Железная труба не долокна быть внутри окрашена^
иначе неизбежен взрыв.
Б. Из хлорной извести и И2О2
Приводимый дорого стоящий способ Фольгарда удобен тем, что ^ дает
постоянный ток кислорода без нагревания. Формуют кубики из хлорной из?
вести и, поместив их в средний шар аппарата Киппа, заливают 3%-ной:
Н2О2, к которой добавлено немного НС1 или НЫОз (для связывания свобод!
ного основания хлорной извести). На 300 г 35%-ной хлорной известщ
(СаОСЬО берут 350 мл 3%-ной Н2О2 с добавлением 57 мл НС1 (уд. в- 1,17|
или 53 мл HNOa (уд. в. 1,36). В противном случае осаждающиеся окиси жЩ
леза и марганца вызывают каталитическое разложение Н2О2.
Выделяющийся газ содержит обычно С02 и СЬ; для очистки его npony-j
екают через раствор КОН. ' ч
В. Из двухромовокислого калия (К2СГ2О7)
По Бальмэну хорошо регулируемый ток газа получается нагреванием
в большой реторте 3 ч. К2СГ2О7 и 4 ч. H2SO4. 1 кг КгСг-О? дает около
114 л Оз.
Г. Из перекиси бария
По Касснеру к смеси 1 ч. Ва02 с небольшим количеством воды при--
ливают понемногу 2,5 ч. насыщенного раствора KsFe(CN)6. При комнатной
температуре выделяется равномерный ток газа. 50 г K3Fe(CN)o дают около
2 л кислорода.
Д. Из Н2О2
1. По Банману грубо измельченную, возможно более чистую Мп02
отсеивают от пыли и помещают в средний шар аппарата Киппа на
подостланный волокнистый асбест. Аппарат заливают смесью, полученной
прибавлением 150 мл концентрированной H2SO4 к 1 л Н2О2 при охлаждении.
Выделяющийся кислород довольно чист. При работе с малыми количествами
вместо аппарата Киппа можно употреблять эрленмейеровскую колбочку.
2. Вартенберг1 рекомендует следующий метод получения чистого
кислорода. Пластинку никелевой жести подвергают платинированию и затем
прокаливают до светлосерого цвета поверхности. Погружая эту пластинку
в 30%-ную Н2О2 на ту или иную глубину, вызывают более или менее
интенсивное каталитическое разложение Н2О2, и, следовательно, процесс получения
Ог можно вести с регулируемой скоростью. Газ пропускают через
вертикальную трубку, заполненную стеклянными бусами (для освобождения от капель
жидкости) и через платинированную медную сетку (для разложения примеси
Н.2О2) и просушивают в промывалке с концентрированной H2SO4.
Выход около 90% (реакция прекращается, когда концентрация Н2Ог
упадет до 1,5%).
Е. Из перманганата. калия (КМп04)
Н. С. Руденко2 для получения вполне чистого О* нагревает КМп04
в вакууме при 120° (для удаления адсорбированных газов), а затем
повышает температуру до начала разложения КМп04. Из 318 г КМп04
получается 17 л чистого кислорода. (Подробное описание аппаратуры и методики
см. в оригинале.)
1 \}г Л?n-oW*a/ctenber8' z- ап<>г£- Ch- 233> 297 (1938)-
2 ЖФХ, «2, №5-6, 668 (1938). .
Кислород
231
Ж. Электролизом воды
В отличие от большинства других методов кислород, полученный электр».*
зом чист и свободен от азота.
1 ho Г а б-е р м а н у в качестве электролита употребляют 20%-ный
аствор СгОз в.10%-ной H2SO4 или насыщенную истертым КМпО* 10%-ную
Рто304. Электролиз ведут в обычном аппарате, позволяющем разделять
выделяющиеся газы. . •
2. Водный раствор Ва(ОН)2 несколько выпаривают в аппарате для перь*
гонки под вакуумом, чтобы удалить растворенные газы. Охлажденный раствор
подвергают электролизу; выделяющийся на аноде кислород очищают от при.^
меси Н2 пропусканием через губчатую платину. Полученный таким методом
кислород почти совершенно чист; он содержит1 только около 0,003% N2.
По указанию Е. Стоддарта2 выделяющийся кислород свободен от Н%
если применяют никелевые электроды, покрытые слоем черной NiO.
Свойства
Бесцветный газ, без запаха и вкуса, уд. в. 1,1053 (по воздуху). Вес 1 я
0-2 при нормальных условиях 1,42892 г. Очень трудно сгущается в жидкость
с т. кип.8—183° (уд. в. жидкого кислорода4 яри 87—59,5° К, d = 1,158 —
1,29).
Очень мало растворим в воде (см. табл. 229); растворимость при
температуре t° выражается формулой: 1 об. воды растворяет (0,04115 + 0,0010899^ +
+ 0,000022'563£2) объема О*.
Кислород быстро поглощается щелочным раствором пирогаллола и
раствором уксуснокислого хрома [2] в соляной кислоте. Расплавленное серебро
растворяет почти 10 об. Ог, выделяя его при затвердевании.
Кислород энергично поддерживает горение; поэтому в атмосфере чистого
Обгорят такие вещества, которые не удается зажечь на воздухе (железо и др.).
Таблица 229
Растворимость кислорода в воде (Winkler)
ГС
0
/ 5
10
12
14 •
16
18
20
25 •
1 об. воды
растворяет об. -Oj
(привел, к
нормальным условиям)
0,04890
0,04286
0,03802
' 0,03637
0,03486
0,03347
0,03220
0,03102
0,02831
100 г воды
растворяют г 02 при
760" м м
0,0 "6948
0,006074
0,005370
0,005129
0,004908
0,004703
0,004515
0,004339
0,003932
t°c 1
30
| 35
40
50
60
70
80
90
100
1 об. воды
растворяет об. 02
(привод, к
нормальным условиям)
0,02608
0,02440
0,02306
0,02090
0,01946
0,01833
0,01761
0,01723
0,01700
100 г воды
растворяют г
02 при 760 мм
0,003588
0,003315
0,003081
0,032657
0,002274
0,001857
0,001381
0,0.0787
~~^
1 М. S t e p h о г d, E. R. W e a v e r, S. F. P i с k e r i п g, J. Rec. Nat. Bur. Stand., 22, 301
(1939); С II; 46 (1939).
2 E. M. St odd art, Proc. Roy. Soc, A, 152, 273 (1935); С I, 4071 (1937).
3 Т. пл. твердого кислорода при 48,2 am £4,90° К, лри 167,7 ат 1Ъ,2Ъ"К« L i s m а п,
С. I, 97L (1936).
* Е. К an da, Bull. chem. Soc. Japan, 12, 473 (1937); C. I, 1141 (1939).
232
Кислота
КИСЛОТА АЗОТНАЯ
Acidum nitricum Nitric acid Salpetersaure
HNOs Мол. в. 63,016
Приготовление
1. Для получения чистой НЖ)з из технической добавляют к последи^
раствор AgNCb до прекращения образования осадка AgCl, хорошо
взбалтывают и дают отстояться в теплом месте. Прозрачную жидкость переливайЦ
сифоном в реторту^ соединенную тампонами из стеклянной ваты с холодияЩ
ником и охлаждаемым приемником (без употребления резиновых или кощ
ковых пробок) и осторожно нагревают на песочной бане (рис. 37). Вначал§
Рис. 37. Прибор для перегонки азотной кислоты
перегоняется жидкость, содержащая значительные количества N02 й нередко
хлора. Время от времени пробу дестиллата ^разбавляют водой и испытывают.
AgN03. на содержание СГ; когда последнего обнаруживаться не будет, ме^
няют приемник и ведут перегонку, пока в реторте не останется
незначительное количество жидкости *.
Перегнанная кислота вследствие небольшого разложения обычно бывает
окрашена окислами азота в желтый цвет. Для удаления последних через
кислоту продувают сильную струю воздуха, свободного от пыли, до полного,
обесцвечивания.
2. По Менделееву прибавляют к технической кислоте РЬ(ЫОз)г для
осаждения СГ. и SO4". Затем добавляют К2СЮ4 для окисления низших окислов
азота и перегоняют кислоту, собирая средние фракции.
3. Можно также прямо перегонять техническую кислоту, собирая фракции,
не содержащие СГ; однако в этом случае выход чистой кислоты не велик.
4. О. Гёнигшмидт и Р. Шлее2 для получения чистейшего препарата
HN03, пригодного для работы по определению атомных весов, рекомендуют
перегонять чистейшую продажную азотную кислоту в колбе иенского стекла
с кварцевым холодильником.
1 Для связывания свободной F2S04 рекомендуется добавить перед перегонкой немного KNOg
* О. Honigschmidl u. R. S с h lc e, Z. anorg. Ch. 221, ^34 (1934).
Кислота азотная 233
z Для * приготовления красной, дымящейся НЫОз перегоняют обычную
%ту с избытком H2SO4 или смешивают 2 ч. КЫОз с 1 "ч. концеитрирован-
^ • HS04.
Н°ИПРИ нагревании полученной смеси сначала образуется обычная HN03; при
льнейшем нагревании выделяются бурые пары и содержимое реторты
датвердевает. Тогда усиливают нагревание.
зЕ Для перегонки следует брать реторту хорошего прочного стекла и доста-'
'точно большого размера.
6. Красную дымящуюся HNOs можно получить также пропусканием дву-
кйси азота N02 в концентрированную HN03 или перегонкой НЫОз с
небольшим количеством крахмала. Последний восстанавливает часть НЫОз и
образующийся N02 растворяется в перегоняющейся кислоте.
7. Для получения безводной HNOs по Эрдману1 в реторте с
припаянной коленчатой трубкой (рис. 38) нагревают смесь 500 мл концентрированной
Р*.с. 38. Аппарат для получения безводной азотной кислоты
HNOs' и 500 мл H2SO4 (уд. в. 1,84). Первые фракции собирают отдельно, пока
взятая проба дестиллата уже не будет давать осадка с AgN03. Тогда
перегоняют основную массу кислоты, оставляя в реторте не меньше 550 мл
жидкости.
Перегнанную кислоту дестиллируют еще раз в таком же аппарате с
равным объемом чистой концентрированной H2SO4, нагревая возможно осторожно
и защищая прибор от яркого света. Кислоту собирают в колбу, закрытую
стеклянной ватой. Дестиллат нагревают на водяной бане и, заткнув
резиновой пробкой с двумя трубками, просасывают воздух, высушенный серной
кислотой и профильтрованный через вату. Полученную 99%-ную кислоту
хранят в темной посуде.
8. Совершенно безводная HNO3 (100%) получается по Гире б ах у и
Кесслеру смешиванием 99,1%-ной HNOe с вычисленным количеством
ангидрида N2O5.
Свойства
Чистая HNO3 — бесцветная жидкость уд. в. 1,526 при 15°. При сильном
охлаждении затвердевает в бесцветные кристаллы. Т. пл. — 41,3°, т. кип. 85°.
При перегонке обычной HN03 вначале переходит вода и при 120,5° начинает
перегоняться гидрат уд. в, 1,4, содержащий около 70% HNO^.
1 ^им. преп., 38.
ГЧЮ1ЭИ>1
1чхо1;эи>1
?9 о9£
1ЧЮ1ГЭИЯ
EONH
'05N
?9 0S*8fr
1ЯХ01ГЭИ>1
1Я101СЗИ>1
?а о98
1ЧХ01ГЭИ>{
сОМН
£о8.м
втгэшгэах
NDjCtfedj
(?9).экод
noifrrudj
(*явя а)
tr
с^©^ю^©ся^^^<>1сос^сх>сосо^сосо©^©ю©со^^со©©с^сх)^
оЪЪЪЪЪЪЪЪЪЪЪЪЪЪЪЪ'сЪЪЪЪЪЪЪЪЪЪЪЪЪ'ооо'ооооо'ооооо
'о о
Сч10><ОСО^С7>СЮЮС\1^об1^^СП01^сОсООО}СОЮС^^©^с£>Ю^сОС^
©^С^Ю^00©<>1^СО1^©^СОЮ©00©С^ГОЮГ~©^сО^СОСО©СЧ^©00О:>^СГ)Ю
OOOOOO'^^^r^1^1^^^C^lC^O^<^^C^IC0г0C0C000C0^,^,'^ч^t,'^, Ю_Ю^Ю^1О_Ю_Ю_©_СО_©^©^со^^^^^^СО^00_
©~ ©~ о* ©~ о~ ©~ о~ ©* о" ©* ©~ ©" о* ©~ ©~ о~ о~ о" ©~ о~ ©~ о~ ©~ ©~ о~ ^
^©©cooor^t^©iOio^^cococ>i^^o©©coco^^^ocococo©©©coco©©
^^^(7^^CO^ЮЮNOOO>OrsCSrt^ЮCOЮNOO<^OrнC^COrrl^5ЮNOOO^O^CNCO^ЮЮNOOФOC^^COтr
) ©,©,© ©©©©©©©1—i^i^^ihhh»-ihi-1 *—< СЧ CN (N <M C^CN C^W С^С^СО С^С^СО СО СО^СО С^СО СО ^^^тг^
©"о"о*©*©о"©"©"©"©"© © ©©"©"'©*"© ©"©"©"©о"©"©"©*©*© ©~©~©*©~©~о*"©" ©о"©Г©*©"©©*о"©©"©"©
8
-^СОСО^С0©©^-^С0--©^ЮС0^©^ЮСО1^©^<ОЮСЧ^©«>Г^1Лт^
©©^С^СТ>^^ЮЮ^ООС©0>©1^<^0*<^^Ю©Г^^СО©©^^СЧСО^»ОЮ1^^
© © ©'© ©©©©©©©©©r-ir-^,^^,^,-.»-!,—i^,-.,—iC^CMOICNCN С^С^С^С^С^С^С^СО^СО^СО СО^СО^СО^СОСО^СС
©"©"©"©'©'©"©"©"©"©"©'©"©"©*©*©"©"©"©"©"©"©"©*©*©*©"©*©"©"©'©"©*©©" ©"©*'©*©'' ©~ ©"©"©©" ©"©"©"
©оою1^©с^т*«^ю-*с^а^10^10©^с^сот^Ю1010юю^сос^
o't^^cn аэ^ю©*^ об с^сГ©~^с^ со ^юю со ^ об©
со — ^с>^оососю©с^юс^ет>ю©^^со©оо1осо©ю^^^©©ю©счю^©с^
*-* со© ю©. rf оо cqo-f-^iooqc^io © с^юоо © со^со ^^^©fc^^^^^^cq^^^^^^^^^^^^^,_t
©~ ^ схГ ^ю ^ об ©" »^ сгГ ^ irj ^ сх5" © ^ сч сгГ i^ ©" ^ od сГ
^,^,^,^,^^^^<>l<^CS|(^<NCN<NrocOC^C^jCOCOCOCO^^
©©©©^т^С100Ю©С0т1«00©Ю^С^СЧ^^^©От^!^1^те©,1О^Ю©СЧсО©<^
г-. СЗО^СО -J© t^T^©_«^^© СОжСЧСЮ СО €^T^C^^©T^oqcv3CX5C^^<^CO^
©~^со ю~^схэ©~с>Гco~i^i>^oo~©~^c6*^co t>-~©~©
г^,^^^.^^с/чС^СЧСЧСЧС>1С^ГОСОСОС^СОСОСО^^тР^^
-©^©"©~о©_©~сГс£Г<о_со~с^^
— © СО 00 Г^О^Ю СО^С^^СЛ СО ©^ЮС^^©^10^СО^г^
О" *-* »-«' CS со" "^Ю СО* 1>Гоб"об"О^©*4»-ГciCO CO "^io'co"1>Г^00 0$С?^^ЫоО^^\Г$<0^^аоо$С>С?^ЫсОСОт&\]Ъ1а
ПэбТп~ёТспТ^^71^ГсТТ^^^
© aq^o со_^© ^^^©^^^оою см^аэсо со о^© co©^©co©_cocq©coc4© сосч©ю^оо ^«—t^co©coc^oo
©*©*^(>f СО^СО т^Ю СО*СО ^00~©"© ©"i^"^ С^ СО^
©т^^СОт^Ю©1^О0©©^С^СО^Ю©^СЮ©©^С^С0^ЮНЭ^00©©^(>^С0^^©^
^^^„^^^„^^(^(^(^(^(^(^(^(^OICMC^COCOCOCOCOCOCOC^
o©"^c^c^co ^^ю со"со*1чГобобоГ©"с^^^с^Гсо"со^ ^ csf c^ сб~ со* т^^ю^^сГ со" со"4
©Ю©Ю©Ю©Ю©Ю©Ю©Ю©10©10©10©Ю©10©Ю©10©Ю©10©Ю©10©10©10 010©10©1П
O©^ — С^С^С^ГОт^^ЮЮС-ОССГ^^оООООэО)©© —'<-iCN(NC000,1iViOiOC0CDNN0000050)OO^-'CN(M
Кислота азотная
235
Продолжение табл. 230
3S
Си О
100 вес, ч. содержат вес. ч.
,2 ч
1230
1,235
1240
1,245
1,250
1,255
1,260
1,265
1,270
1,275
1,280
1,285
1,290
1,295
1,300
1,305
1,310
1,315
1,320
1,325
1,330
1,332,5
1,3.5
1,340
1,345
1,350
1,355
1,360
1,365
1,370
1,375
1,380
1,3833
1,385
1,390
1,395
1,400
1,405
1,410
1,415
1,420
,425
,430
1,435
,440
,445
,450
,455
26,9
27,4
27,9
28,4
28,8
29,3
29,7
30,2
30,6
31,1
31,5
32,0
32,4
32,8
33,3
33,7
34,2
34,6
35,0
35,4
35,8
36,0
36,2
36,6
37,0
37,4
37,8
38,2
38,6
39,0
39,4
39,8
40,0
40,1
40,5
40,8
41,2
41,6
42,0
42,3
42,7
43,1
43,4
43,8
44,1
44,4
44,8
45,1
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
66,5]
67
68
69
70
71
72
73
74'
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
о
о
X
31,53
32,17
32,82
33,47
34,13
34,78
35,44
36,09
36,75
37,41
38,07
38,73
39,39
40,05
40,71
41,37
42,06
42,761
43,47
44,17
44,89'
45,26
45,62
46,35
47,08
47,82
48,57
49,35
50,13
50,91
51,69
52,52
53,08
53,35
54,20
55,07
55,97
56,92i
57,86
58,83]
59,83
60,84]
61,86
62,91
64,01
65,13
66,24
67,38
36,78
37,53]
38,29
39,05
39,82
40,58
41,34
42,Ю
42,87
43,64
44,41
45,18
45,95
46,72
47,49
48,26
49,07
49,89
50,71
51,53
52,37
52,80]
53,22
54,07
54,93]
55,79
56,66
57,57
58,48
59,39
60,30]
61,27
61,92
62,24]
63,23
64,25
65,30
66,40
67,50
68,63
69,80
70,98
72,17
73,39]
74,68
75,98'
77,28
78,60]
69,66
71,08
72,52]
73,96
75,42]
76,86
78,30
79,74
81,20
82,65
84,11
85,57
87,03
88,48
89,94]
91,40
92,94
94,49
96,05
97,60
99,19]
100,00
100,80
102,41
104,04]
105,67
107,31
109,03]
110,75
112,48
114,20
116,04
117,27
117,88]
119,75
121,68
123,67
125,75]
127,84
129,98]
132,19
134,43
136,68
138,991
141,44
143,90
146,36
148,86
59,43
60,61
61,84
63,07
64,31
65,54
66,76]
67,99
69,23
70,48
71,72]
72,96;
74,21
75,45
76,70
77,94
79,25
80,57
81,90]
83,22
84,58
85,27
85,95
87,32
88,71
90,10]
91,51
92,97
94,44
95,91
97,38
98,95
100,00
100,5»!
102,12
103,76
105,46
107,24
109,01
110,84
112,73
114,63]
116,55
118,52
120,61
122,71
124,81
126,94
1 л содержит кг
О
О
37,72
38,49
39,27
40,05
40,84 I
41,62
42,40
43,18
43,97
44,76
45,55
46,34
47,13
47,92
48,71
49,50
50,33
51,17
52,01
52,85
53,71
54,15
54,58
55,46
56,.34
57,22
58,11
59,05
59,98
60,91
61,85
62,84
63,51
63,?4
64,85
65,90
66,97
63,10
69,23
70,39
71,59
72,80
74,02
75,27
76,59
77,93
79,26
80,62
0,387]
0,397
Г0,407
0,417
0,427
0,437
0,447
0,457
0,467]
0,477
0,487
0,498
0,508
0,519
0,529]
0,540
0,551
0,562
0,573]
0,585
0,597
0,603
0,609
0,621
0,633!
0,645
0,658
0,671
0,684
0,698
0,711
0,725]
0,735
0,739]
0,753
0,768
0,783
0,800
0,816
0,832
0,849
0,867
0,885
0,903
0,921
0,941
0,961
0,981
0,452
0,463
0,475
0,486
0,498
0,509|
0,521
0,5331
0,544
0,556
0,668]
0,581
0,593
0,605
0,617
0,630]
0,643
0,656]
0,669
0,683
0,697
0,704
0,710
0,725
0,739
0,753
0,768]
0,783
0,798
0,814
0,829]
0,846
0,857
0,862
0,879
0,896
0,914
о;9зз
0,952
0,971
0,991
1,011
1,032]
1,055]
1,075
1,098]
1,121
1,144
0,856
0,877
0,900
0,921
0,943
0,965
0,987
1,009
1,031
1,054
1,077
1,100
1,123
1,146
1,169
1,193
1,218
1,243
1,268
1,294
1,320
1,333
1,346
1,373
1,400
1,427
1,455
1,483
1,513
1,543
1,573
1,603
1,623
1,633
1,665
1,697
1,731
1,767
1,803
1,839
1,877
1,915
1,955
1,995
2,037
2,080
2,123
2,167
0,730
0,748
0,767
0,785
0,804
0,822
0,841
0,860
0,879
0,898
0,918
0,938
0,957
0,977
0,997
1,017
1,038
1,059
1,080
1,103
1,126
1,137
1,148
1,171
1,193
1,216
1,240
1,265
1,289
1,314
1,339
1,366
1,383
1,392
1,420
1,447
1,476
1,507
1,537
1,568
1,6С0
1,633
1,667
1,701
1,736
1,773
1,810
1,848
я я
Я g СО О OVJ3 О ,
а^ s p s ^ rad^'o
н о £ со n q со н w ЯЗ
to -Ре „aw й*< S
ТОЧНЫЙ
о н 2 a хз £,
Со *< S нС о
2 s п> я
Co S
^ E, ^- со
я 5 о 5 a
« л w я w a
J^o-P н со S Я
Co н Co ^ --
s|E^
Co
! Co
5ea
J-^^g ^ Co
Co J4 >D "^ ж мд
sg ю a S )=» - >=i н
Не - СО Н. " О *< ^
л SE л я ь 5 а
О СП 5 О О -£ ^
* о. g я ov0 g ^
JO-T3-=HOCoCr.^sS
о со
О О ^J Н
П> g О
£ *> к
« со ^
н « s £ S
^ * ДО' я
■ t) л ^. ° а>
= я S 3- Р и | S
Sc9 а гс п> g t, я
я о о
я
•К <ъ
Д д
СО CD
я й
о
* Д
^os|«a = S5se
О) ^ Н щ О CD ««^ Ж р^
Ю СЛ ЯДЯ * &> *0
- ^ ~ О « О СО
St) К * О о £ со
Я Яе
СО
1 G
> я
о
«зк-з^е**^-
tr1 Й 2w* и " ^ г
О СО 00 ->ГСПСл ^СОЮи-ОСОООЧС5Сл^СОЮ'-1ОСлОСлОСлОСЛО
.«1111*11
^ о
-°° I
jX> J<! J-4 ^ ОЭ О) р5 СЛ СЛ
W- Ов V- "*-» 00 rf^ "и-00 "*4^
I I I О I I I | О I I I I О СО СО СО СО СО СО СО СО
I I I Ю I I 1 1 и- | | 1 | ОСООО^ОСЛ4^СОЮ
ООСОООООСОООООООООООООООССОООООООООООЭСХОО^^^^^^СПОЭ
,сл сл слсл сл^-^^^^j^S^^S^^^^^^^S^S^P^S^S^i^T*^0^0
4^СлСГаС^4^^00СОО^СССО^аэС7а>^СОСО|-^ОСОСЛЮО ОЭ C7i СО СТ> СО C75
cocococococococococococococococococococococooooooooooooo-<i
jOjO^}O}O}Op0Q0 ОЭ£0 СО^J^^&i ОЪ С75СЛ СЛ ►^^.^-'^Oj^ O)^ Ю^^О
спел 4^Ъо ю'оХо^сл'со^-' оосл^-Vj со о ел ЬЬЬЬЬч о'^'соТ^'о
ОЧС7)^^<1^СОСОЮО^ОСОО^СООСДОООСОС-ООСЛСЛОЮОО
ОСПСПСПСЛО^СЛСЛСЛСЛСЛСЛСЛСЛСЛСЛСЛСЛСЛСЛСЛ4^^^С1ЭСОС^
<Z> O.CO&CD О СО^СО СО ОО 00p0^Apipi^J\^J\^.QObO^-*^-A rf^^—JX) Oi^CJO —.JO .
"со oovOi*4^1o'oVjТ^^^^^^^оо^Ъ^^Ъосл^^^со^о^^сосоЪо 4^^-*
Ч^СОСОМОЮС>1СО(РСОн»у1СТ|^^>^^СЛООСРСООСОу1--£)ООЮ ">1_
ооооооооооооососососососососососооооооооооооо
ю ю ^о ^- и- ^-» ^-* ^— ^- j3> ^> cd otoio 00 00 00 г-а^ р>$я^$р оо en ел сою
~о"— oooVi'oi'j^lo'o'co о^Ъо'о'сп'Кэоо^ ослЪ>сл^^ю1ос)0"ело
СОЮ»-^СОСЛ^Й^СГ>СГ> й^ЮСДОЮЧ 05050ЮСООСЛОСЛ05ЮСО»- СО
гОЮЮЮЮЮГОгОЮс0с0Г0»ОЮГ0Юг0«0Г0ЮЮ^-<—"—ООООО
cocoocococ«<»oo^-o^cпaiCл4^rf^coco^o^-»—i-^rf^h-- «осл^юо
сост>4^юсо^соо^^>^ осл о сл со ньь оо „* 4>-s о ^. >^ ел кэ оо ел со •—
СлСлСлСлСлСл4^^»1^^4^4^^<^Л'^»^^|^»^4^СаСОСОЮ101\Э'--»—*
•—»—*>— ОООСОСО«00000->1^СПСЛСЛ^С01>ОЮ»-'CDCOO-<lrf^^-COCr)
СлЬОО~-д>й>'^^14^0СП'~*0>О>Р»^'-'>^СП00Он^с0СлЮ>4^ОС0С000
Ъэ Ъо Ъо Ъо оо Ъо Ъо оо оо оо Ъо "^ V^ Vj Vi-Vj Vj Vj Vq "сп сл сл сл j^V'co oo Volo
СПСПСПСЛ^^'СОЮЬО^- ОСОООЧСл^СОМООО^сОЮО'-' СП'О СЛ •—
С04^0^00С001СОЮ^СЛСЛ^СОСОООСЛ0 4^СОЮСОООСПСООСОСОЮ
JO|OjOjOjOJO^WWIOJObO^^|0^tO^JO^|OWjO|OW^
^^^^Т^^^^'^'^'со'соЪэ'со со "со со'со саэ'юТо'Кэ^'^'оо^оТоЪо
^^COCOtOK3^*«—'ОС>СООО,<»СПСл4^СО--'С>СО--4^-СлОСли-сПКЭОО
-vjt^cO^CO^C?OCOCnOlNjrf^4i«>4^COCO»OCOCnCOOOt--'СПСОООКЭСО*ЧСП
а15
4
(В вак.)
Градусы
Боме (Вё)
Градусы
Тв.едделя
(Tw)
N20s
HNO,
Кислоты
40° he
Кислоты
48,5° Вё
N,0S
HN0,
Кислоты
36° Вё
Кислоты
40° Вё
СЛСЛСЛСЛСЛСЛ ^СлСЛ01С^СлСлСЛ^4^^Я^^^кР-^С0<^С0£0^.О--
5<_ К_ ...„ X ч СО СО СО Ю Ю — •— ООС00000^С75СЛ*.ОС5^С^|СЛ(О50
СЛ СЖ **?► *|Sfc4^COCOCOtOtO — ■— <
rf*..»—-CftС»Со СОСЛЮОО*-СО^<
£^> ... I .К*
Кислоты
У
С1»
о
3
л
г
Кислота азотная
237
Таблица 231
~ ппавк» для удельного веса HN03, содержащей N02
Попр (Lunge, March l e w s k i)
о/о NO,
0,25
0,50
0,75
1,00
1,25
1,50
1,75
2,00
2,25
2,50
2,75
3,00
3,25
3,50
3,75
4,00
4,25
Изменение
уд. веса 1
0,0005
0,0008
0,0015
0,0030
0,0018
0,0058
0,0078
0,0105
0,0125
0;0143
0,0163
0,0180
0,0199
0,0217
0,0235
0,02оЗ
0,0269 |
% NOa
4,50
4,75
5,00
5:25
Д50
5,75
6,00
6,25
6,-50
6,75
7,00
7,25
7,50 j
7,75
8,00
8,25
8,50 |
Изменение
уд. веса *
0,0288
0,0305
0,0323
0,0337
0,0360
0,0378
0,0^95
0,0418
0,0430
0,0448
0,0465
0,0482
0,0500 1
0,0517
0,0533
0,0550
0,0566 1
% N02
8,75
9,00
9,25
9,50
1 9,75
10,00
10,25
10,50
10,75
11,00
11,25
11,50
11,75
12,00
12,25
12,50
12,75 ■;
Изменение
уд. веса *
0,0583
0,0630
1 0,0616
0,0633
0,0650
0.0666
00682
0,0698
0,0714
0,0730
0,0745
0,0760
0,0775
0,0785
0,0805
0,0820
0,0835
Таблица 232
Поправки для удельного веса HN03 для температур
между 13 и 17°
Уд. вес
1,000—1,020
1,021-1,040
1,041—1,070
1,071—1,100 j
1,101-1,130
1,131-1,161
Поправка
на± 1°
=t 0,0001
0,0002
0,0003 |
0,0001
0,0005
0,0006 1
Уд. вес
1,162—1,200
1,201—1,245
1,246—1,280
1,281-1,310
1,311-1,350
1,351-1,365 |
Поправка
на± Г
± 0,0007
0,0008
0,0009
0,0010 ,
0,0011
0,0012 |
Уд. вес
1,366—1,400
1,401—1,435
1,436-1,490
1,491—1,500
1,501—1,520
Поправка
на± 1°
± 0,0013
0,0014
0,0015
0,0016
0,0017
Таблица 233
Степень разложения HN03 от нагревания
(С а г i u s)
Температура
разложения °С
—• 86
100 !
130
160
190
220
255
Уд. вес
(по водороду)
^
29,6
■ 29,1
27,6
25,8
23,0 j
20,4
1?,0
1 0/°
разложения
9,53
11,17
18,78
2Р,96
49,34
72,07
100,0
1 г HN03 дает
мл кислорода
8,43
10,41
16,62
26,22
43,69
63,79
88,47
1 Если HNOa уд. в. 1,49 и выше содержит известное количество NOa, to надо для получе-
уд. в., соответствующего действительно имеющейся HN03, отнять от удельного веса,
зисимости от содержания NOs, величины, указанные во второй графе-таблицы.
ния
в зависимости
238
Кислота
Испытание
Азотная кислота вырабатывается «крепкая» и «слабая». По OCT HKJ
2689 кислота должна представлять собой бесцветную или слегка желтоват^
прозрачную жидкость; при уд. в. d1^ кислоты крепкой от 1,390 до 1,410,
лоты слабой — от 1350 до 1,385; содержание HN03 в крепкой кислоте"
63,2% до 67,47%, в слабой —от 57,54% до 62,21%;
допустимых примесей указаны в табл. 234.
Таблица I
наибольшие количее
Допустимые примеси в HN03
Примеси
Допустимое содержание в %
химически
чистом
в чистом для
анализа
в чистоВ
1. Нелетучий остаток
2. Серная кислота (S04") ....
3. Хлориды (СГ)
4. Йодноватая кислота и иод (J')
5. Тяжелые металлы группы H2S
6. Железо (Fe) ........
7. Мышьяк (As) , . .
8. Кальций (Са)
9. Окислы азота (N02')
0,0015
0,0003
0,0001
0,0000
0,0000
0,00005
0,000003
0,1 ~~
0,003
0,002
0,0002
0,0005
0,0005
0,0001
0,000003
од-
0,005
0,005
0,0005
0,0005
0,0005
0,0003
0,ОС001
0,002 '
ОД
Примечания. 1. Испытание на кальций производится только для кк|
слоты, содержащей более 0,004% нелетучего остатки
2. В слабой азотной кислоте йодноватая кислота не опре|
деляется.
3. Окислы азота определяются в пробе, отобранной на
заводе-производителе.
КИСЛОТА БОРНАЯ
Acidum boricum Boracic acid Borsaure
Boric acid
Приготовление
А. Обычная борная (ортоборная) кислота
НзВОз Мол. в. 61,84
Растворяют 100 г чистой буры (ЫагВЮт; ЮНгО) в 250 мл кипящей воды-
и раствор смешивают с разбавленной НС1 (115 мл 25%-ной НС1 на каждые;.
100 г ЫагВЮ?). После суточного стояния отсасывают выделившуюся борную,
кислоту *, промывают небольшим количеством воды и дважды перекристалли-
зовывают из кипящей воды (3,5 ч. воды на 1 ч. НзВОз). Сушить препарат
следует между листами фильтровальной бумаги при комнатной температуре
или в сушильном шкафу при нагревании не"выше 70°.
Б. Метаборная кислота
НВОз Мол. в. 4а,83
При нагревании НзВОз до 107
в НВ02:
она теряет молекулу воды, переходя
НзВОз = НВ02 + Н80.
1 Некоторые авторы рекомендуют употреблять вместо бумажных фильтров стеклянную вату»
Кислота борная 239
г А" Тилю и Г. Зибенеку1 для получения вполне чистой НВОг,
^°: рп'жащей Н^ВОз и НгВЮ?, следует сушить НзВОз при 100—111° в токе
не с0Д ^ ерЖащего определенное количество водяных паров.- Парциальное
га3а"' ние НгО в газе должно быть в пределах между 4. и 113 мм рт. ст.
даВ^нее всего для соблюдения необходимых условий сушить НзВОз при
УД° jIqo B токе воздуха, пропущенного через промывалку с водой, имеющей
температуру 14°.
В. Пироборная (тетраборная) кислота
Н2В40?
Мол. в. 157,30
д. Тиль и Г. Зи'бенек2 указывают, что Н2ВЮ7 может быть получена
пои сушении НзВОз при 100—110° в токе газа, в котором парциальное
давление HsO меньше 4 мм рт. ст. Следовательно, необходимо воздух,
проходящий над препаратом, подвергать предварительному осушению. Авторы
получили вполне чистый препарат Н2ВЮ7, нагревая НзВОз в течение 240 час. при
112° в токе воздуха, пропущенного через 50%-ную H2SO4 при 20°.
Свойства3
Ортоборная кислота представляет собой шестиугольные триклинические
белые чешуйки с перламутровым блеском, уд. в. 1,46, жирные на ощупь.
При 107°, теряя часть воды, переходит в метаборную кислоту, НВОг, при
140—160° — в пиро- или тетраборную кислоту, Н2В4О7. При красном калении
теряет всю воду4, оставляя борный ангидрид, ВЮз.
Растворима >в воде, в спирте (1 : 25), в глицерине (20: 100 при 0°, 73 : 100
при 100°) и эфире.
Борная кислота относится к числу слабых кислот; ее константа
диссоциации б составляет 6,31 • 10~10. В водном растворе борная кислота образует
одновременно ионы, отвечающие орто-, мета- и пиро-формам; поэтому
растворы НзВОз;, Н2ВЮ7 и НВОг совершенно тождественны.
Таблица 235
ГС
0
5
10
15
20
25
30
35
% н3В0з
(„Спутник
химика"
1935)
2,59
—
3,45*
—
4,74*
—
6,26*
~~
Р а с т в о
о/о Н3В03
(пересчитано
по данным
W. S. Blasdale и
С. М. Slansky)R
2,50
3,14
3,52
4,17
4,65
5,44
6,34
7,19
t°c
40
45
50
55
,60
65
70
75
римость Н3В03 в
°;0 н3во3
(„Спутник
химика"
1935)
8,0.2
—
10,35
—
12,90
—
15,86*
""~"
о(о Н3В03
(пересчитано
по данным
W. S. Blasdale и
С. М. Slansky)e.
8,17
9,33
10,22
11,54
12,96
14,42
15,75
17,40
1
воде
ГС
80
85
90
95
100
107,5
115
120
°/о Н3В03
(„Спутник
химика"
1935)
19,11
—
23,30
—
—
36,7
45,0
52,4
°/о Н3В03
(пересчитано
по данным
W. S. Blasdale и
С. JVLJlansky) e
19,06
21,0
23,26
25,21
27,52
— •-.
—
* Интерполированные данные.
1 А. Т h i е 1 и. Н. S i е b е п е с к, 1. anorg. Ch. 220, 238 (1934).
8 loc. cit.f стр. 240.
■ О строении борных кислот см., например, С. II, 18 (1938).
поо-,4 Согласно М. Stackelberg, F. Quatram, J. Dressel, Z. f. Elektrochemie, 43, 14
U937) т. пл. Н3ВО3 170°, т. пл. HBOa 203°.
•J. E. Thygesen. Z. anorg. Ch. 237, 101 (1938); см. также J. chem. Soc, 1&48 (1937).
e J. Am. chem. Soc, 61, 917 (1939).
240
Кислота
Таблица
Удельный вес водных растворов Н8В03
°/о Н3В03
0,5
1,0
1,5
2,0
2,5
л15
(„Спутник
химика" 1935)
1,0038
.—
1,0074
d4
(интерпелиро- i
вано no A. von
Endredy) i
0,99990
1,00204
1.С0380
1,00556
1,00733
о/о НаВ03
!~"
3,0
3,5
4,0
4,5
л15
d4
(„Спутник
химика" 1935)
1,0110
—
1,0147
—
—-*ч
4°
(Интерполира
вано по А.-щ
Endredy) t
1,00912
1,01090
1,01270
1,01490,
Испытание2
По СТ 27—1830 содержание НзВОз в препаратах должно быть не мек
х. ч. 99,5%, ч. д. а. 99,0% и чистом 98,0%; препарат х. ч. должен отвеча
борной кислоте по Зеренсену; наибольшие количества допустимых примесё
приведены в табл. 237. Борная кислота должна выдерживать приведенные
в стандарте испытания на растворимость в 95%-ном этиловом спирте.
енее:
гчать
ieceg
Допустимые примеси в Н3В03
Таблицы 2Ш
Примеси
Допустимое содержание в %
химически
чистом
в чистом для
анализа
1. Не растворимые в воде вещества .
2. Хлориды (СГ) . . . .-
3. Сульфаты (SO/)
4. Фосфаты (Р04'")
5. Кальций (Са)
6. Тяжелые металлы группы H2S . . .
7. Железо (Fe)
8. Мышьяк (As)
9. Вещества не летучие при обработке
HF •
0,005
0,001
0,005
0,001
0,005
0,0005
0,0005
0,0002
0,05
0,01
0,002
0,010
0,001
0,010
0,^01
0,001
0,005
0,100
0,02
0,0J5
0,030
0,003
0,030
0,003
0,002
0,005
0,300
Acidum hydrobro-
micum
Bromwassersfoff-
saure
КИСЛОТА БРОМИСТОВОДОРОДНАЯ
Hydrobromic acid
HBr Мол. в. 80,924
Бромистоводородная кислота представляет собой водный раствор
бромистого водорода.
Приготовление
1. ПоВильгероду в колбу или вульфову склянку, снабженную про*
парафинеыной пробкой, в которую вставлены капельная воронка и газоотвод*
1 Z. anorg. Ch. 222, 285 (1935).
8 Испытанье хим. реактивов, 89,
Кислота бромистоводородная 241
трубка,% помещают 100 г сухого бензола и несколько граммов бромного
1' леза (FeBre) или порошка железа и понемногу из капельной воронки
прибалт ^ мл брома. Происходит энергичная реакция (вначале колбу сле-
^т охлаждать водой), сопровождающаяся равномерным выделением броми-
$of0 водорода. Газ пропускают через U-образпую трубку, одно колено кото-
с\ наполнено FeBr3 (для поглощения бензола), а другое—антраценом (для
Р° Зивания увлеченного брома). Для растворения газ проводят в колбу,
сильно
00*
охлаждаемую смесью льда и соли. Время от времени в колбу подли-
воду. Следует наблюдать, чтобы газоотводная трубка не была погру-
' на в воду, так как вследствие чрезвычайно большой растворимости НВг
* 0оде последнюю может перебросить в реакционный сосуд. Можно также
всТй поглощение в охлаждаемой склянке Тищенко.
2. По Густавсону в приборе, аналогичном предыдущему, бром поне-
н0гу приливают к продажному антрацену (СиНю). Выделяющийся газ про:
:оДят через трубку с антраценом (для поглощения увлеченного брома) и рас-
Т0Оряют в воде, как описано в способе 1. Если желают получить газообраз-
Рис. 39.
Прибор для получения НВг по
Филети и Кроза
Рис. 40. Прибор для получения НВг по
Рекора
>ый бромистый водород, то его для высушивания пропускают через смесь
РЮб с асбестом.
3. Т1о Филети Jk Кроза в колбу (рис. 39) помещают 10 г красного
фосфора и 20 мл воды. Из капельной воронки постепенно приливают 35 мл
брома; при этом происходят следующие реакции:
2Р + 5ВГ2 = 2РВг5;
2РВГ5 + 6Н20 = 2НРОз + ЮНВг.
Для очистки газ можно пропустить через U-образную трубку, наполненную
;месью желтого фосфора с мокрым толченым стеклом или смесью асбеста
1 влажным (не мокрым) красным фосфором.
Выход около 30 л газообразного НВг.
4. Р е к о р а предлагает следующий простой метод. Высокий сосуд
Рис. 40) наполняют бромом, наливают сверху слой воды и пропускают ШЗ.
^аз, выделяющийся согласно уравнению реакции:
H*S + Br* = 2HBr+S,
■роходит через воду, в которой суспендирован красный фосфор. Здесь пол-
■остью поглощается увлеченный бром; выходящий газ совершенно не содер4
Кит H2S и достаточно чист даже при быстром пропускании сероводорода*
Zf±2 Кислота
4ФЦ
5. По Г а г е р у в колбу помещают 100 г гипосульфита (Na2&?03« 5)щ
50 г брома и 17 г воды и слабо подогревают. Выделяющийся газ поглохц^
140 мл дестиллированной воды с соблюдением предосторожностей, указа?
ных в способе 1. '
6. По Кекуле капают водой на трехбромистый фосфор и выделяющие
. газ проводят через промывную склянку с небольшим количеством воды.
7. Ионеско и Радулеско1 рекомендуют следующий практич^ь
метод получения НВг. К сухому керосину, обработанному предварительно
концентрированной H2SO4 и промытому водой до нейтральной реакции, шн
ливают по каплям бром. Катализатором служит AlBrs (с катализатором выход
составляет 80—90%, без него — 45%). Выделяющийся НВг током еоздщ
прогоняется через три промывных склянки с водой.
8. По Гей с игу и Эмдеру2 к раствору 120 г КВг в 200 мл воду
медленно добавляют 90 мл концентрированной H2SO4, следя за тем, чтаВ
температура раствора не поднималась выше 75°. После охлаждения отфиЕГ8
тровывают выпавший KHSO4 и перегоняют фильтрат из колбы емкост
500 мл, собирая постоянно кипящую фракцию (около 126°).
П. Друк3 рекомендует добавлять к жидкости перед перегонкой 0,051
SnCb, чтобы избежать частичного разложения НВг. Препарат содерА
0,01—0,05% H2S04.
9. По методу, разработанному в ИРЕА 'А. Ильиным4, для воеста§
вления брома применяется SO2 в приборе, аналогичном описанному при сщ
собе 4 (Ре кора). Через см'есь 200 мл воды, 500 мл брома и 650 г толчЩ
ного льда пропускают струю SO2 (из -бомбы) с максимальной скоростью, при
которой газ полностью поглощается. Во время пропускания газа реакциоя*
ный сосуд охлаждают водой.
Когда слой брома исчезнет и жидкость просветлеет, насыщение газет
прекращают и добавляют небольшими порциями бром (для связывания
избыточного SO2), пока иодокрахмальная бумажка не начнет синеть в парах жи-дл
кости. Разбавив раствор водой до -уд. в. 1,3—1,28, перегоняют его. собирая
фракции 115—122° (слабая кислота, около 60 мл) и 122—126° (44—45%-ная
кислота, около 700 мл).
Для очистки от примеси H2SO4 в 60 мл последней фракции растворяют^
25 г ВаСОз и полученный раствор смешивают со всем количеством дестил-
лата.
После 10-часового стояния сливают жидкость с осадка BaS04 и перегоняю!
ее, собирая опять фракцию 122—126° (550—590 мл), представляющую собой
чистую кислоту $, уд. в. 1,47—1,49.
Свойства
Бромистый водород — бесцветный газ с запахом, напоминающим НС1;
кислого вкуса, сильно дымящий во влажном воздухе. Т. пл. —87°, т. кип.в —68°-
В воде чрезвычайно легко растворяется, выделяя большое количество тепла.
При нагревании такой раствор отдает часть НВг и при 126° перегоняется
гидрат уд. в. 1,486, содержащий около 46,85% НВг, что соответствует при*
близительно формуле НВг'бгЪО.
При нагревании слабого раствора происходит обратный процесс: сначала
отгоняется вода, затем при 126° переходит гидрат указанного состава. На
коже НВг вызывает зуд и воспаление.
Водная кислота должна сохраняться в хорошо закупоренных склянках
ж темном месте.
1 С. N. I о п е s с о и. V. R a d u 1 е s с о, С. I, 3933 (1937).
8 G. В. Н е i s i g a. E. A m d u г, J. chem. Educ. 14, 187 (1937).
8 О. D г ц с е, J. chem. Educ. 14, 394 (1937).
4 Реакт. и преп. 45; см. также Н. Петроваи А. Ильин, Изв. ИРЕА, 16, 114 (1939).
5 О получении сухого НВг из тетралина и брома см. работу J. Но ill en, J. В о е d 1 е r J*'
W. Fi s ch ег, Ber., 69, 1772 (1936); о чистейшем НВг, полученном синтезом из элементов, см<
О. Н С» n I g s ch m i d t u. R. S а с h 11 e b en, Z. anorg. Ch., 221, 70 (1934),
"ПоБэтсут. кип. НВг —66,8° [J. R. Bate s, C. II, 3474 (1933)].
Кислота бромистоводородная
Ш
Таблица 238
ГС
-25
-20
-15
— 10
— 5
0
Растворимость НВг
1 об. воды
растворяет
об. НВг
—
—
644,5
63Г,0
611,6
100 МЛ ВОДЫ
растворяют
г НВг
255,0
247,3
239,0
233 5
228,0
I 221,2
г°С
+ ю
25
50
75
100
в воде
1 об. воды
растворяет
об. НВг
581,4
532,1
468,6
406,7
344,6
100 мл воды
растворяют
г НВг
210,3
193,0
171,5
150,5
130,0
Таблица 239
Уд ел ьн ы й
о/о НВг
1
2
4
6
8
10
12
14
.20
d4
1,0053
1,0124
1,0269
1,0417
1,0568
1,0723
1,0883
1,1048
вес водных растворов
о,'о НВг
16
18
20
22
24
26
28
30
И20
d 4
1,1219
1,1396
1,1579
1,1767
1,1961
1,2161
1,2367
1,2580
о/о НВг
35
40
45
50
55
60
65
НВг
.20
d4
1,3150
1,3772
1,4446
1,5173
1,5953
1,6787
1,7675
Испытание
По ГОСТ 2062—43 бромистоводородная кислота должна представлять собой
прозрачную бесцветную или слегка желтоватую жидкость. Содержание НВг
для препаратов обеих квалификаций (ч. д. а. и чистый) должно быть nfc
менее 40%. Наибольшие количества допустимых примесей приведены
в табл. 240.
Таблица 240
Допустимые примеси в НВг
Примеси
2. Сульфаты (S04")
3. Хлориды (СК)
54 Фосфаты (РО/'О
6. Железо (Fe) ,
Ъ Тяжелые металлы группы H2S . . .
Допустимое содержание в о/0
в чистом для
анализа
0,005
0,005
од
0,02
0,001
0,0001
0,0005
0,00002
в чистом
0,02
0,03
0,5
0,05
0,005
0,0005
0,001
0,00005
244
Кислота
Acidum bromicum
КИСЛОТА БРОМНОВАТАЯ
Bromic acid
НВгОз Мол. в. 128,924
Bromsaure
Приготовление водного раствора
По Раммельсбергу сначала приготовляют бромноватокислый барис
[Ва(ВгОз)г], для чего сливают кипящие насыщенные растворы 10 ч. бромновато1
кислого калия (КВгОз) и, 8 ч. уксуснокислого бария [Ва(СНзСОО)2]. Обливают
100 г тонко истертого Ва.(ВгОз)а смесью 24 г концентрированной H2SO4 у
240 мл воды и настаивают некоторое время в умеренно теплом месте. После
отделения выпавшего ~ "~
содержание НВгОз не
пает разложение).
В вакууме можно
Таблица 241
Допустимые примеси в НВгОз (я.)
Примеси
1. Свободный бром ....
3. Сульфаты (S04") ....
4, Железо (Fe)
Допустимое
содержание
в %
0,5
0,01
0,02
0,005
BaS04 фильтрат упаривают на водяной бане, пока
достигнет 42,6% (при дальнейшем выпаривании насту.
упарить раствор до содержания 50% НВгОз,
дальнейшее выпаривание вызывает раз*
ложение.
Свойства
Бесцветная, очень кислая
жидкость, почти без запаха. При
перегонке разлагается. J
Испытание
По техническим условия^
НКХП (61—40) содержали!
НВгОз в брО'Мноватой кислотг;
чистой должно быть не мене<#
12%; наибольшие количества доЯ
пустимых примесей приведена,
в табл. 241.
КИСЛОТА ВОЛЬФРАМОВАЯ
Acidum wolframicum Tungstic acid
H2WO4 Мол. в. 249,94
Приготовление
Wolframsaure
m
1. По Г. и В. Бильц1 в течение 3.—5 мин. нагревают 3 г вольфрамовой*'
кислого аммония со смесью 10 мл концентрированной H2SO4 и 5 мл концен^|
трированной HNOs в фарфоровой чашке при слабом кипении. Охлаждают^
разбавляют водой, отсасывают, промывают горячей водой, пока промывна^Ь
вода не покажет нейтральную реакцию, и высушивают в сушильном шкафу,^-
обогреваемом водяным паром. '^
2. Раствор 100 г Na2W04 в 250 мл кипящей воды вливают тонкой струей:;
в горячую смесь НС1 и HN03 (175 мл НС1 и 3,5 мл HN03) при сильном/
перемешивании и непрерывном подогревании. Через 20 мин. всю смесь вли-;.
вают при перемешивании в 1400 мл горячей воды. Осадок H2WO4 промывают/
декантацией, отфильтровывают и сушат при 130°. -;
3. Для получения H2WO4 10 г тонко истертого минерала вольфрамита^
(FeW04 • MnW04) обрабатывают царской водкой до совершенного исчезно-;■■■
вения желтой окраски. Затем выпаривают досуха и несколько раз экстра-^
гируют осадок разбавленной НС1 для удаления Fe и Мп. Наконец, раство-Ду
ряют оставшуюся вольфрамовую кислоту в концентрированном NH*OH,
выпаривают досуха и слегка прокаливают.
1 Uebungebeispiele.
Кислота бромноватая и вольфрамовая 245
Свойства
Желтый порошок, устойчивый до 200°. При прокаливании теряет . воду,
пПЯя вольфрамовый ангидрид, WO*. Вольфрамовая кислота очень плохо
ocTaB_trvr,, в воде; почти нерастворима в H2SQ4, HNOs, разбавленных
порима в воде; почти нерастворима в ri2SU4, ш\и3, разбавленных
Ра^[ j^j? HBr; несколько растворима в концентрированной . НС1; довольно
шо растворяется в плавиковой кислоте (HF) и спиртовом растворе НО.
Таблица 242
Растворимость H2W04 в кислотах
(Bernhardi-Grisson)
Растворитель
i°Q
о/о WC3
25
50
50
80
25
78
44,75
53,7
0,36
0,75
5,8
9,8
Фтористоводородная кислота (40% HF)
. " . (400/0 HF)
Соляная кислота (38°/0 НС1)
„ (380/0 НСГ)
Спиртовый раствор хлорист«г# водореда
(концентрация неизвестна)
То же • . . .
Испытание (Сингаловский)
Для химически чистого препарата: а) содержание V/Оз ъ прокаленной
кислоте должно составлять не менее 99,90%; 6) наибольшие количества долу-
стимых примесей указаны в табл. 243.
Таблица 243
Допустимые примеси в Н2\\Ю4 {х. ч.)
Примеси
1. Окись кальция (СаО) . .
2. Кремнекислота (Si02) . .
3. Сумма окислов (Fe203 +
4. Окись алюминия (А1203)
Допустимое
содержание
В Wo
0,030
0,030
0,010
! о.ою
Примеси
5. Молибден (Мо) . . .
i 9. Тяжелые металлы . .
Допустимое
содержание
В о/0
0,003
0,003
0,003
0,005
0,000
КИСЛОТА ИОДИСТОВОДОРОДНАЯ
Acidum hydrojodioum Hydriodic acid Jodwasserstoffsaure
HJ Мол. в. 127,93
Иодистоводородная кислота представляет собой водный раствор йодистого
водорода.
Приготовление
1. В круглодонную колбу емкостью 100 мл помещают 44 г иода и через
рогатый форштосс понемногу прибавляют 4^ г желтого фосфора в виде кусоч- *
ков, обсушенных фильтровальной бумагой. Прибавление первого кусочка
246
Кислота
*ш&
сопровождается энергичной реакцией с выделением пламени, дальнейшее «*
прибавление идет более спокойно. Необходимо следить, чтобы фосфор щШ
дал в середину колбы.
После внесения всего фосфора получается темная масса трехиодистог
фосфора PJ3.
Тогда заменяют рогатый форштосс пробкой с капельной воронкой и газ$
отводной трубкой и приливают понемногу 6 мл воды, слабо подогрева*
колбу; при этом происходит реакция:
Р J8 + ЗНЮ = НзРОз + 3HJ.
Нагревание колбы ведут до полного осветления жидкости*. Выделя.^
щийся газ пропускают для очистки от увлеченного иода через U-образнЗ
трубку, наполненную смесью 6 г красного фосфора с 1 мл воды и бит
стеклом.
Для получения водной кислоты отводную трубку опускают в колбу, со|
жащую 45 мл воды. Трубку не погружают в воду во избежание перетя
вания жидкости, а оставляют на 8—10 мм над пове
ностью воды.
Для концентрирования полученной кислоты ее не
•гоняют, причем сначала, при 100°, переходит нек®
рое количество воды, затем температура быст
повышается до 125—130° и начинает перегонять
гидрат, содержащий около 57% HJ. Выход 75—80|
теоретического.
2. По Л. М е й е р у выгоднее действовать избыткор
иода и фосфора на воду, чем, наоборот, водой и
иодом на фосфор, так как в последнем' случае ч§|
стично образуются фосфин (РНз) и йодистый фосфо-
ний (РШЛ).
В реторту с тубусом (рис. 41) помещают 100 г иод%
омоченного 10 мл воды. В тубус реторты вставлена
особая капельная воронка, имеющая вместо кращ
длинную палочку, пришлифованную к концу трубки.
В воронку помещают 5 г фосфора с 10 мл воды.
Осторожным поднятием палочки пропускают в ре*
торту каплю воды с фосфором, выжидают окончания реакции и затем при!
бавляют остальное количество фосфора. Таким образом реакция проходит
спокойно, в то время как прибавление сразу большого количества фосфора
может вызвать взрыв.
Иод, увлеченный выделяющимся йодистым водородом, почти весь
осаждается в горле реторты. Чтобы его по возможности совершенно удалить,
соединяют реторту с приемником посредством длинной прямой, не слишкокг
узкой трубки и, кроме того, пропускают газ через небольшое количество
воды в U-образной трубке.
Когда выделение HJ- ослабеет, реторту слегка подогревают; при этом
улетучивается некоторое количество воды, смывающей осевший на стенках
иод. Если после продолжительного нагревания цвет иода не пропадает,
прибавляют еще немного фосфора. После полного прекращения выделения газа
отгоняют водную кислоту.
Из указанных количеств получают 74,4 г газообразного HJ и 23,7 г HJ
в дестиллате. Если же взять 35 г воды,, то получится 47,5 г газообразного
HJ и 57,3 г HJ в дестиллате.
Рис. 41. Прибор для
получения Шпо л. Мейеру
1 Внутри газоотводной трубки часто получаются блестящие кристаллы йодистого фосфония
PH4J, образующегося согласно следующим реакциям:
4Н3Р03=ЗН3РО,+ РН3;
РН3 -f- HJ = PH4J.
■ Хим. реактивы, (
Кислота иодистоводородная
247
S По^Бендеру1 для получения водной кислоты уд. в. 1,7 суспенди-
i немного порошкообразного иода в воде и пропускают FhS; реакцию
РУ10 дят в опрокинутой реторте с широким горлом (рис. 42). Постепенно
П^°бавляют новые порции иода; образующаяся кислота растворяет его и
ПКг получающаяся по реакции
H*S +. Js = 2HJ + S,
не облекает иод плотным слоем.
Когда весь иод прибавлен, взбалтыванием собирают серу в комок, уда-.
ют pbS кипячением и перегоняют кислоту уд. в. 1,7 при 126—128°.
Другой вариант этого метода приводит Ю. Швицер2. В широкогорлую
склянку темного стекла емкостью 400 мл загружают 50 г иода и 250 мл
воды и пропускают FUS. до полного растворения иода. Дальнейшую
обработку проводят, как указано выше.
4. По П етт.енко ф.ер у обливают 1 ч. красного фосфора 24 ч. воды при
60—70°, прибавляют при помешивании 2 ч. иода, сливают жидкость с осадка
в сосуд, содержащий 14 ч. иода, и после
насыщения иодом раствор снова переливают
на фосфор, где держат до обесцвечивания.
Такое переливание повторяют до тех пор,
пока весь иод не перейдет в раствор и
жидкость не обесцветится.
Раствор отфильтровывают от красного
фосфора и отгоняют до сиропообразной
консистенции.
Раствор уд. в. 1,39—1,40 слегка окрашен
иодом в желтый цвет.
5. По Баня-о в у8 растворяют в 10 ч.
иодистоводородной кислоты около 20 ч. иода
и постепенно приливают этот раствор через капельную воронку в колбу,
содержащую 2 ч. красного фосфора, смоченного той же кислотой.
Вначале колбу следует подогреть. Образующийся йодистый водород для
восстановления увлеченного иода пропускают через трубку, наполненную
битым стеклом, красным фосфором и раствором HJ.
Для поглощения HJ служит приемник с водой, охлаждаемый льдом;
подводящая газ трубка не должна быть погружена в воду4.
Свойства
Йодистый водород5 — бесцветный удушливый газ уд. в. 4,375 (воздух = 1).
I л весит при нормальных условиях 5,66 г. Легко сгущается в жидкость.
Т. пл. —52°, т. кип. —35,6°.
Водный раствор (иодистоводородная кислота) — бесцветная жидкость
острого и сильно кислого вкуса, на воздухе постепенно окисляющаяся до
свободного .иода, окрашиваясь при этом в бурый цвет. Насыщенная при 0° вода
содержит 90% HJ. При нагревании до 40° (энергичнее при 55°)
выделяется HJ; остается кипящий при 126°' гидрат уд. в. 1,7, содержащий около
57% HJ.
Испытание
По ОСТ .10093—39 препарат должен иметь плотность (с/420) 1,50—1,55 и
содержание HJ не менее 45,0%; наибольшие количества допустимых
примесей указаны в табл. 245.
Рис. 42. Прибор для получения HJ
по А. Бендеру
1 Неорг. преп. 140.
•2 „По ..извздетво хим.- iармац^втических и техно-химических препаратов", ОИТИ М.— Л,
1934, стр. 38.
3 В a n n о w, Вег. 7, 1498 (1874).
4 О получении сухого HJ из и ;да и тетралина см. J. Н о и 1 е n, J. В о е d 1 е г ц. W. F i s с h e г
Вег., 69, 1772 (I93S).
5 J. I:.. Bates, С. II, 250 (1930).
248
Кислота
Таблица 244'
Удельный вес водных растворов HJ
°/о HJ
1
2
4
6
8
10
12
14
.20
1,0054
1,0127
1,0277
1,0431
1,0589
1,0751
1,0918
1,1091
• о/о Н J
16
18
20
22
24
26
28
30
4
1,1270
1,1456
1,1649
1,1850
1,2059
1,2270
1,2503
1,2737
°/о HJ
35
40
45
50
55
1 60
65
.20
d4
1,3357
1,4029
1,4755
1,560
1,655
1,770
1,901
Таблиц
Допустимые примеси
Примеси
3. Иод (J2)
4. Хлориды и бромиды (в пересчете на СК) . .
5. Сульфаты (SO/0
6. Фосфаты (PCVO
7. Железо (Fe)
в HJ
Допустимое содержание в Щ
в чистом для
анализа
0,015
0,003
0,2
0,005
0,0005
0,0025
0,0001
0,0002
в чистом
0,03
0,01
0,3
1 0,005
0,005
; 0,005 .
0,0003:
0,0002
Acidum perjodicum
КИСЛОТА ЙОДНАЯ
Periodic acid1
HJ04-2H20; [H5JO0]
Perjodsaure
Ueberjodsaure
Мол. в. 227,96
Приготовление
По Уэллсу1 12,7 г иода вносят в 600 г 10.%-ною раствора NaOH;
жидкость нагревают до кипения и пропускают в нее энергичную струю хлора.
Если в жидкости начинаются толчки вследствие образования осадка, удаляют
пламя и пропускают хлор до прекращения выпадения белого осадка
Na2H3JOe.
Осадок промывают холодной водой и высушивают в шкафу,
обогреваемом водяным паром. Получается около 22 г натриевой соли (80% теорет.).
Соль взмучивают в избытке воды; прибавляют 51 г AgNOs, жидкость
нагревают, фильтруют горячей, и черный осадок периодата AgsJOs
промывают водой. Серебряную соль еще во влажном состоянии взмучивают в
небольшом количестве воды и пропускают при помешивании хлор, пока осадок
не станет почти белым. Образовавшееся хлористое серебро отфильтровывают, "
1 W е П s, Am. Cheni. J. 26, 278 (1901),
Кислота йодная it йодноватая
249
оат выпаривают на водяной бане и держат для кристаллизации^ в экси-
фцльтр ^^ H2SO4. Кристаллы йодной кислоты высушивают в эксикаторе над
Свойства
Безводная йодная %кислота состава HJd не получена.
Получаемая приведенным методом кислота отвечает формуле HJ04 • 2НгО
HsJOo. Она кристаллизуется в бесцветных призмах, плавящихся при
?оп° с частичным разложением.
Сильно гигроскопична и легко растворима в воде. Водный раствор жел-
т на воздухе и сильно пахнет озоном. Кислота растворима в спирте; трудно
растворима в эфире.
Таблица 246
Удельный вес водных растворов HJ04• 2Н20 при 17°
°/о H5J06
3,8
7,3
13,7
Уд. вес
1,0288
1,0570
1,1121
а/о H5JOe
24,0
33,7
Уд. вес
1,2165
1,4008
КИСЛОТА ЙОДНОВАТАЯ
Acidum jodicum
HJO3
Jodie acid
Мол. в. 175,93
JodsSure
Trioxyjodsaure
Приготовление
1. По Эрдману1 в реторте из стойкого стекла обливают 30 г иода
160 г чистой 98—100%-ной HNOs (уд. в. 1,54) и при взбалтывании слегка
нагревают. Реторта при этом наполняется красными парами окислов азота,
которые вытесняют сильной струей воздуха (последний вдувают через
стеклянную трубку, конец которой, обмотанный асбестовым шнуром, плотно
закрывает собой тубус реторты). В реторте все же остается некоторое
количество окислов, которые восстанавливают часть образовавшейся HJ0.3' до
иода. Последний улетучивается и вместе с перегоняющейся Н1ЧОз сгущается
•в охлаждаемом приемнике. Поэтому время от времени прекращают
нагревание и дестиллат переливают обратно в реторту. В конце концов в реторте
получается белый осадок; его растворяют в небольшом количестве воды и
выпаривают в фарфоровой чашке до кристаллизации. Выход близок к
теоретическому.
2. Растворяют йодноватый ангидрид J2O5 в небольшом количестве воды
и выпаривают раствор на водяной бане при 50—60° до начала
кристаллизации.
3. По: методу, разработанному Н. Вильямсом2 в ИРЕА, к горячему
раствору 100 г KJOe в 460 мл воды нриливают при перемешивании горячий
раствор 75 г ВаСЬ-2Н20 в 160 мл воды. По охлаждении^ отфильтровывают
выпавший осадок Ва( JO3)» • 2Н2О и промывают его теплой водой до
удаления СГ в промывной жидкости.
Пасту иодата бария помещают в толстостенную стеклянную банку
емкостью 1 л, добавляют разбавленную H2SO4 (47,2 г H2SO4 уд. в. 1,84
1 Хим. преп. 6!о
* Реакт. и прем. 150.
250
Кислота
в 250 мл воды) и перемешивают жидкость механической мешалкой в TeW
нескольких часов; отфильтровав осадок BaS04, промывают его горячей вод11?
Фильтрат вместе с промывными водами (500—600 мл) выпаривают до обт^Эг
250—300 мл, фильтруют и снова упаривают до появления пояска Кристи
лов около стенок чашки. Тогда прекращают нагревание и вызывают &&ш
нейшую кристаллизацию растиранием пестиком кристаллов на дне чащ»|
После полного охлаждения добавляют 60—70 мл ледяной СНзСООН (в щж
рой HJOs почти нерастворима), растирают смесь пестиком, отсасывают щ?
сталлы на воронке Бюхнера и промывают их 2—3 раза ледяной СНзСОш
и 2 раза этиловым эфиром (по 15—20 мл). После сушки в эксикаторе щ
H2SO4 до исчезновения запаха эфира и над КОН до удаления згщщ
СНзСООН препарат растворяют в 150 мл воды.
Выпарив раствор снова до появления кольца кристаллов, растворяют]!
прибавлением небольшого количества горячей воды и добавляют 80 мл леШ»
ной СНзСООН. При этом1 HJ03 выделяется в виде густого теста, которое
после получасового растирания пестиком превращается в белоснежную 'щш
сталлическую массу. Промывку и сушку препарата проводят так же, как п^
первой кристаллизации.
Выход 75—78 г (около 95% теоретического).
Свойства
Белый блестящий порошок уд. в. 4,66s1, кислого вяжущего вкуса,
нагревании до 110° начинает переходить в гидрат ЗЛаОб-НгО (ангидроиод
ватая кислота); полный переход происходит выше 120°.
Очень легко растворима в воде, растворима также в H2SO4; не раст
рима в СНзСООН, спирте, эфире, хлороформе, сероуглероде. В водн
растворах образует полимеры (ШОз)г и (ШОз)з; степень полимеризац
Таблица 247
Растворимость Ш03 вводе и
t° с
0
16
40
Wo HJOa
70.3
71,7
73,7
/°с
60
80
£5
о/о HJ03
75,9
78,3
78,7
t°c
101
ПО
125
°/о Ш03
80,8
82,1
82,7
Таблица 248
Удельный вес водных растворов Ш03
о/о Ш08
1
2
4
6
8
10
d4
1,0071
1,0157
1,0334
1,0517
1,0706
1,0900
°/о HJO,
12
14
16
18
20
22
И™
1,1100
' 1,1306
1,1519
1,1740
1,1969
1,2206
°/о HJ03
24
26
28
30
35
40
dA
1,2450
1,2700
1,2956
1,8218
1,3900
1,4640
* Е. М о 1 е s u. P.
V i 1 1 a n, An. Soc. espan. Fis. Quim. 34, 787 (1936); C. II, 541 (1937),
Кислота .Каро
251
т с разбавлением К НЛОз относится к сильным кислотам — ее истинная
*аД^анта диссоциации * К а =0,163.
кон^1нгИД.роиодноватая кислота ЗЛгОб-НаО (иначе ШзОв) устойчивая до 196°,
^т уд в. 5,054, образуется как при нагревании HJO*, так и при кипячении
йМсь1тенного водного раствора (т. кип. 111°).
Испытание
П0 ОСТ ВКС 5181 содержание НЛОз для препаратов квалификаций х, ч.
ч д. а. не менее 99,90%; для препарата чистого — не менее 99,50%;
наибольшие количества допустимых примесей указаны в табл. 249.
Таблица 249
Допустимые примеси в НЛ03
——
Примеси
1. Нерастворимые в воде вещества . .
3. Серная кислота (SO/)
Допустимое содержание в о/0
в химически
чистом
0,004
0,01
0,01
в чистом для
анализа
0,006
0,03
0,03
в чистом
0,015
0,06
0,05
КИСЛОТА КАРО
Peroxymonosulfuric
H2SOs
acid
Sulfomonopersaure
Carosche Saure
Мол. в. i 14,08
Приготовление
По Бэйеру и Велигеру3 постоянный раствор кислоты Каро
(содержащий H2SO4) получается следующим путем: растирают 10 г персульфата
калия, K2S2O8, с 20 г концентрированной H2SO4 и оставляют смесь на час.
Хотя за это время прореагирует не все количество надсерной кислоты, смесь
не оставляют на больший срок, так как при длительном стоянии с
концентрированной H2SO4 персульфат разлагается с выделением Ог и Оз. Смесь
выливают на лед и разлагают раствором однозамещенного фосфорнокислого
бария [Ва(НгР04)2]. 4 (Общий объем около 1,5 л).
Содержащий некоторое количество Ва' ' раствор отсасывают на воронке
с пористой стеклянной пластинкой от BaS04 и продувают через него воздух
До удаления запаха озона.
Получившаяся жидкость, представляющая собой разбавленный раствор
кислоты Каро, чрезвычайно постоянна и только через несколько месяцев
выделяет следы BaS04.
Свойства
Водный раствор кислоты Каро медленно гидролизуется с образованием
H2SO4 и Н2О2. Растворы в серной и фосфорной кислотах более постоянны.
После введения Ва(ОН)г наступает немедленное разложение. По сравнению
с надсерной кислотой (H2S2O8) кислота Каро более устойчива в кислых
и менее прочна в нейтральных и щелочных растворах.
1 М. R. N а у а г (с сотрудниками), Z. anorg. Ch., 240, 127 (1939).
"Naidichu R i с с i, С. h 2445 (1940). ^ _
'Bayer, Villiger, Ber. 34, 355 (1901); Prap. Ch. 97.
«Для получения Ва (Н.Р04).. прибавляют Н3РО, к вычисленному количеству горячей
концентрированной баритовой воды.
252
Кислота
КИСЛОТА КРЕМНЕВАЯ
Acidum silicicum Silicic acid Kieselsaure
S1O2 Мол. в. 60,06
А. Ангидрид
Приготовление
1. Прокаливают водную кремневую кислоту, полученную осаждением ра^
творимого стекла концентрированной НС1 и тщательным промыванием осад^
2. Пригодный для многих целей порошок SiC>2 может быть получен измели
чением природного кварца или горного хрусталя. Для этой цели минерал
сильно накаливают и погружают в холодную воду.
3. Довольно чистый Si02 получается из чистого кварцевого песка (белого)
кипячением с НС1 и промыванием водой. Еще лучше пользоваться тонко |Ц
мельченным горным хрусталем.
Свойства
Аморфный белый тонкий порошок, растворимый в плавиковой кислоте
щелочах. Т. пл. 1470°, т. кип. 2590°.
Б. Водная (метакремневая) кислота
H2Si03 Мол. в, 78,08
Приготовление
По Жордису1 для получения чивтой кислоты может быть применен
следующий метод.
К продажному жидкому стеклу .с содержанием 35% Si02 добавляют кон<:
центрированную НС1. Выпадающий беловатый зернистый продукт от добавлен
ния половинного количества кислоты, необходимого для связывания присутШ
ствующей щелочи, затвердевает и снова разжижается при добавлении
остатка НС1. Вследствие присутствия железа масса обычно окрашена в ярко-
желтый цвет. Ее отсасывают и неоднократной обработкой концентрирован?!
ной НС1 освобождают от примеси металлов.
При последующем вымыва-нии кислоты водой наблюдаются потери;
одновременно масса становится слизистой и отсасывается с большим трудом.
Лучше всего растереть продукт в ступке, внести его в большое количество
холодной или теплой воды и после длительного настаивания слить через
колоторку. Последняя устроена из стакана с обрезанным дном, обвязанным
полотном. Стакан наполняют полученным студнем -и помещают в воду так/
чтобы она его закрывала. В стакан медленно подводят проточную воду, отводя
ее из внешнего сосуда, и промывают студень до удаления главной массы
солей, т. е. до исчезновения реакции на СГ; затем обезвоживают пробу студня
и испытывают на присутствие Fe, Ca, 'A1 и т. д. Полученный таким путем
очищенный продукт, высушенный на песочной или воздушной бане, служит
исходным материалом для дальнейшей переработки.
Очищенную кремневую кислоту растворяют в вычисленном количестве
теплого раствора NaOH (1 моль NaOH на 1 моль SiO*) и разбавляют до
содержания 3—5% ЗЮг. Затеем смешивают раствор с рассчитанным на
нейтрализацию примененного NaOH количеством НС1 и таким количеством воды, чтобы
смесь содержала 1—3% Si02.
Через некоторое время (при подогревании немедленно) масса
желатинируется. Ее перемешивают или встряхивают и тотчас переносят в колоторку. j
Даже длительным промыванием не удается удалить всех щелочей, так как
последние удерживаются чрезвычайно прочно. Поэтому отмытый от главной
массы примесей студень настаивают многократно с разбавленной НС1, . давая
каждый раз возможность последней на другой день хорошо стечь на свободно
J о г d i s, Z. anorg. Ch. 34, 457 (1903^; Prap. Ch. 292; см также F. Krauss, С Oettnef,
гг. Ch. 222. 367 MQ3SV
Z. anorg. Ch. 222, 367 (1935).
Кислота кремнёвая 2,53
ршени^й колоторке. Эту обработку повторяют до тех пор, по^а проба
по#в внесенная на платиновой проволоке в пламя горелки, не перестанет
стУ^ ' ать пламя в желтей цвет натрия. Тогда промывают студень или кипя-
окрац~о с водой до исчезновения реакции на СГ.
t5jTr6 очищенном указанным образом продукте обычными путями не удается
оУжить посторонних веществ. Студень содержит около 95%' воды; его
0 1 по в случае нужды, обезводить.
Свойства
Желатинообразная масса, плохо растворимая в воде. При выпариваний
вакуумом полученного диализом раствора остается блестящая прозрачная
■Птеклрвидная масса состава ШБЮз.
с в водных растворах, ведет себя как очень слабая кислота (константа1
ассоциации К = 4-\0г7—5«Ю-7), образуя ионы различного типа, главным
образом HsSiCV и H2Si206,/ (Роллер и Э р в и н)2.
Испытание
По ОСТ 10159-39 реактивная кремневая кислота должна представлять
собой белый порошок, теряющий при прокаливании 20—28%. Остаток после
удаления БЮг (выпаривание со смесью H2SO4 -f HF) не должен превышать
для препарата ч. д. а. 0,1%, для препарата ч. 0,3%.
Извлекаемых водой хлоридов (СГ) в препарате ч. д. а. должно быть не
более 0,005%, в чистом — не более 0,01%. При обливании 2 г препарата 30 мл
х. ч. H2SO4 (уд. в. 1,84) кислота в течение 15 мин. должна оставаться
бесцветной, что свидетельствует об отсутствии органических примесей.
В. Коллоидальный SiCh
Приготовление
По Ризенфельду3 растворяют 60 г Ыаг8Юз в 200 мл горячей воды,
фильтруют, если нужно, и дают охладиться. 40 мл НС1 (1 : 1) приливают при
помешивании к раствору Na2SiCh и переносят все в диализатор, пока (через
2—3 дня) проба на С1' (с AgNOe) не даст никакого осадка, а только молоч^
ную муть. Если произошло выделение хлопьев кремневой кислоты, их
отфильтровывают. Затем окончательно точно нейтрализуют раствор ранее
приготовленной разбавленной соляной кислотой и снова подвергают его диализу.
Г. Активный силикагель
Приготовление
Е. В. . А л ек с е е в.с к и й 4 описывает получение активного силикагеля по
Фелльсу и Форсу.
Смешивают равные объемы жидкого стекла (уд. в. 1,15) и НС1 (уд. в. 1,165);
в течение 10—15 мин. наступает коагуляция. Гель оставляют на сутки, затем
разрезают на крупные куски и промывают декантацией водой до удаления СГ.
Следует избегать энергичного перемешивания, так как желательно, чтобы
к концу промывания зерна геля имели размер 4—6 мм.
Промытый гель помещают на большое часовое стекло и высушивают в
течение 18—30 час. при 40—50°, часто перемешивая. Затем сушат полученные
зерна' 10—12 час. при 50—100°. Наконец препарат разделяют с помощью сит
на фракций различной крупности и для активирования подвергают силикагель
просушке при 120—130° (4—5 час.) и прокаливанию при 300—320° (2 часа).
Свойства
Твердая зернистая стеклообразная масса уд. в. 2,0—2,5. Полученная из
чистых продуктов — бесцветна и прозрачна. При продолжительном стоянии
1 Wo. P a u 11 u. L. Р а 1 m г i с h, Koll. Z. 79, 60/193; С. I, 475Э (1937).
« P. Ro 11 е г u. E r v 1 п, С. I, 1525 (1941).
• Неорг. практ. 321.
* Химия защиты, т. 1, стр. 310.
254
Кислота
гель переходит в кристаллическую форму и в значительной мере теря^Гадеор*
ционную способность.
Силикагель является хорошим адсорбентом для летучих растворителей
водяных паров. Воздух, пропущенный для осушки через трубку с силикагелем
содержит по Бауэру1 только 0,03 мг НгО на литр; таким образом силика*
гель является более энергичным осушителем, чем, например, NaOH или Саш
КИСЛОТА КРЕМНЕФТОРИСТОВОДОРОДНАЯ
Acidum hydrosilicio-
fluoricum
Fluosilicic acid,
Silicofluoric acid,
.Hydrofluosilicic acid
Fluorkieselwasserstofft;
saure
H2SiFe
Мол. в. 144,08
Приготовление
1. Для получения водного раствора по Б ер цел и усу смешивают 100 г
сухого кварцевого песка с 100 г CaF2 и смесь осторожно нагревают в колбе
с 350 мл концентрированной H2SO4. Выделяющийся газ пропускают сначала
через склянку с небольшим количеством H2S04, затем газ поступает в фар<]
ровую чашку с водой; на дно чашки помещают небольшую чашечку со ртутью.
Так как SiCb, выделяющаяся по реакции
3SiF4.+ 2ШО = Si02 + 2H2SiFe,b
может засорить газоотводную трубку, то конец последней погружают не в
воду, а в ртуть. Процесс заканчивают по прекращении выделения газа.
Быстро отсасывают выделившийся осадок SiCb, отжимают его и
споласкивают таким количеством воды, чтобы всего получилось около 400 мл
фильтрата. Для очищения последний еще раз фильтруют через обычный
(бумажный) фильтр.
2. По Гемпелю в качестве реакционного сосуда удобно применять
чугунный горшок и выделяющийся газ по широкой (18 мм) трубке пропускать
прямо в воду, не применяя чашки со ртутью. Так как из широкой трубки Si02
легко удаляется, то закупоривания не происходит.
Свойства
Безводная кислота неизвестна. Водные растворы бесцветны и имеют
сильно кислую реакцию. Разбавленные растворы можно хранить в стеклянных
сосудах; последние предпочтительнее все же изнутри парафинировать.
При концентрировании водных растворов после достижения концентрации
13,3% HsSiFo растворы теряют больше SiF4, чем HF, и обогащаются
последним. Продажная thSiFe имеет обычно уд. в. около 1,07.
Таблица 250
Удельный вес водных растворов H2SiF6
о/о H4SiFe
1
2
4
6
8
wl7,5
V
1,007
1,015
1,031
1,048
1,065
°/о HtSiFe
10
12
1.4
16
18
w17»5
1,082
1,100
1,117
1,136
1,154
°/о H2SiF6
20
24
28
32
34
^7,5
1,173
1,212
1,252
1,293
1,314
1 J. H. В о w е г, J. Res. Nat. Bur, Stand, 12, 241 (1934).
Кислота кремнефтористоводородная и метафосфорная $55
исПЬ,таНИе
х\ техническим условиям НКХП 227-40 содержание H2SiFe в препаратах
а. и чистом должно быть не менее 27,5%; наибольшие количества до-
ч' имых примесей приведены в табл. 251.
Допустимые примеси в H2SiF6
Таблица 251
Примеси
Допустимое содержание в °/0
в чистом для
анализа
1 Нелетучий остаток
2. Фтористоводородная кислота
з'. Сульфаты (SO/7) . . .... ,
4, Тяжелые металлы ,
0,02
0,5
0,005
0,001
0,04
1,0
0,025
0,004
КИСЛОТА МЕТАФОСФОРНАЯ
Acidum phosphoricum Metaphosphoric acid Phosphorsaure glasig
glaciale * Metaphosphorsaure
(НРОз)п* Мол. в. (79,99)n
Приготовление
1. По Т а м м а н у1 нагревают концентрированный раствор НзР04 в
золотом тигле (фарфор, серебро и платина сильно разрушаются), причем сначала
при энергичном кипении и разбрызгивании выделяется часть воды. При
дальнейшем нагревании кипение внезапно прекращается; не твердеющий при
охлаждении остаток представляет собой почти чистую НзР04. При дальнейшем
нагревании остатка последний начинает дымить; в этот момент он почти
полностью состоит из пирофосфорной кислоты Н4Р2О7. При продолжающемся
нагревании выделяется еще вода и образуется метафосфорная кислота.
Однако никаких внешних признаков, по которым можно было бы судить о
полноте перехода, не имеется..
В то же время при дальнейшем нагревании вода улетучивается в большей
степени, нежели РгОг,, и продукт становится богаче фосфорным ангидридом,
чем это требуется, формулой НРОз.
Поэтому,' если желают получить кислоту, свободную от Р2О5, необходимо
время от времени производить анализ прокаливаемой массы; если же
препарат предполагают растворить в воде, то избыток PsOs не вредит, так как
он с холодной водой образует НРОз.
2. К- И. Загвоздкин, Ю. М. Рабинович и Н. А. Барилко2,
изучавшие процесс дегидратации Н3РО4, указывают, что полный переход
ортофосфорной кислоты в НРОз может быть достигнут при 300° и упругости
паров воды 0,5 мм. рт. ст.
Свойства
Белая стекловидная масса, расплывающаяся во влажном воздухе. В воде
очень легко растворима, причем раствор, присоединяя воду, постепенно
превращается в НзР04, быстрее — при нагревании с HNOs. При красном калении
улетучивается.
И с п ЫчТ а н и е
По ГОСТ 841-41 содержание щелочных и щелочноземельных металлов в
*' Вероятно (НРОз>6, т. е. Нв (Р03)6. В этой кислоте 4 иона водорода соответствуют сильной
кислоте, а последние два—слабой (R. S alih, Bull. Soc. chim. France [5], 3» 1391 (1936).
1 T a m m a n, J. prakt. Ch. (2) 45, 428 (1892); Prap, Ch. 202.
* ЖПХ 13, № 1, 29 (1940).
255
Кислота
пересчете на NaPO* должно быть не более 0,4%; наибольшие количе^
допустимых примесей указаны в табл. 252; препарат должен выдержи
испытание с раствором белка, а также испытание на отсутствие веществ,
стаиавливающих КМп04.
Таблица q52
Допустимые примеси в гиЮ3
Примеси
1. Кислоты фосфорные орто- и пиро- и
их соли (в пересчете на Н3РО±) . •
2. Хлориды (СГ)
3. Сульфаты (SO/)
4. Нитраты (N03')
5. Железо (Fe)
6. Тяжелые металлы (в пересчете на РЬ)
Допустимое
содержание
5
0,005
0,01
0,001
0,005
0,002
Acidum molybdaenicum
КИСЛОТА МОЛИБДЕНОВАЯ
Molybdic acitf
Molybdensaure
Н2М0О4Н2О или Н4Мо05 Мол. в. 179,98
А. Кристаллическая молибденовая кислота
Приготовление
1. По Муспратту растворяют 3 ч. молибденовокислого аммония в 20 ч.
воды и 20 ч. HNOa (уд. в. 1,16) и дают раствору стоять несколько дней.
Образуется желтая кристаллическая масса, переходящая при стоянии над H2SO4
в безводную кислоту Н2М0О4.
2. Желтый осадок, выпадающий при стоянии молибденовой жидкости,
состоит из Н4М0О5. (О получении этой кислоты в чистом виде см. также
работу Оже1).
Свойства ^
, Полученная осаждением из соли кислотой, молибденовая кислота
представляет собой желтый порошок уд. в. 3,12, растворимый в 500 ч. холодной
или в 960 ч. горячей воды. Имеет сильно металлический вкус, вызывает
покраснение лакмусовой бумажки и, особенно в кислом растворе, побурение
куркумовой бумажки. Концентрированная HNOs разлагает препарат с
осаждением молибденового ангидрида МоОз.
Б. Растворимая молибденовая кислота
Приготовление
По Уллику растворяют продажный молибденовокислый аммоний в воде,
осаждают раствором ВаСЬ, промывают выпавшую смесь бариевых солей
декантацией горячей водой и сушат на водяной бане. Затем размешивают
с водой в жидкую кашицу, разлагают точно необходимым (для осаждения Ва' ')
количеством H2SO4, дают отстояться и фильтруют; фильтрат должен быть
бесцветен и не мутнеть ни от НС1 и ВаСЬ, ни от H2SO4.
При концентрировании раствора последний окрашивается сначала в слабо
зеленоватый, затем в голубовато-зеленый цвет и при испарении над H2SO4
оставляет растворимую молибденовую кислоту в виде растрескавшейся аморф-
1 A u g е г, С. г. 206, 913 (1938); С. I, 1738 (1939). О получении р-формы см« М. М и г g I еГ'
U. D о и с е t, там же, 208, 185, (1939); С. 1, 1454 (1939),
Кислота молибденовая и мышьяковая
25?
,аггы всегда окрашенной в голубоватый цвет вследствие частичного
Jcra-яовления.
Свойства
г ежеприготовленная кислота легко растворима в холодной воде. После
льного хранения становится почти нерастворимой в холодной воде, од-
Длите легко растворяется при нагревании. Фильтровальная бумага, смоченная
иаК°нцентрированно.м раств0ре препарата, быстро окрашивается на солнечном
свету в яркосиний цвет.
Испытание^
Препарат' ч. д. а. не должен содержать более 0,02%' примесей,
нерастворимых в НС1 (для препарата чистого допускается 0,04%); потеря при прокали-
ании — не более 20%; остаток после нагревания препарата в токе хлори^
стого водорода при 500° — не более 0,1% (для чистого не более 0,4%).
Кроме того, раствор препарата в 10%-ном NEUOH испытывается на
тяжёлые металлы (сероводородной водой), фосфаты (отсутствие желтого осадка
при добавлении HNOs и стоянии в течение 18 час. при 40°), хлориды (проба
AgNCb после добавления HN03) и сульфаты (проба ВаСЬ).
КИСЛОТА МЫШЬЯКОВАЯ
Aciduin arsenicicum Arsenic acid Arsensaure
HsAsO* • V«H.O Мол. в. 151,94
Приготовление
200 г As20s' в кусочках величиной с горошину нагревают с 200 мл HNOs
{уд. в. 1,38) в реторте с коленчатой трубкой и тубусом до прекращения
выделения окислов азота. Получившийся раствор сливают с непрореагировавшего
'АэгОз и, прибавив еще 100 мл НЫОз, выпаривают досуха. Охлажденный
остаток растворяют длительным нагреванием с небольшим количеством воды и
выпаривают до густоты сиропа.
Таблица 253
Удельный вес водных растворов H3ASO4
о/о H3AsO,
1
2
4
6
8
10
12
14
16
18
20
И15
1,007
1,012
1,025
1,038
1,051
1,065
1,080
1,095
1,110
1,125
1,141
о/о H3As04
22
24
26
28
30
34
38
42
46
50
w!5
I 1,157
1,174
1,191
1,209
1,227
1,266
1,308
1,352
1,399
1,448
о/о H3AsO,
54
*58
62
66
70
•' 74
78
1 82
86
90
1 wl5
4
1,503
1,562
1,624
1,691
1,765
1,849
1,944
2,043
2,147
2,262
Полученный раствор оставляют в закрытом сосуде на холоду (ниже 15°),
прибавив, если возможно, кристаллик мышьяковой кислоты (затравка). Если
Концентрация раствора достигнута правильная, получаются красивые большие
кристаллы гидрата. НзАэО^ У:>Н«А 'которые высушивают на пористой
тарелке. " ' " """*"
1 Испытание хим. реактивоз, 256.
258
Кислота
Если кристаллизация производилась быстро, отсасывают получи
массу, кристаллы расплавляют при слабом нагревании и дают медленно ?
диться; при этом также образуются крупные кристаллы.
Свойства
Полугидрат HsAs04 • V2H2O кристаллизуется в прозрачных ромби.^
табличках или столбиках, расплывающихся на воздухе и легко раствор^
в воде с поглощением тепла. При длительном нагревании до 100° получа
безводная H3ASO4. Нагревание до 140—180° приводит к образованию ц
кислоты H4AS2O7; при 200—206° получается метамышьяковая кислота Н/
Наконец при 300° остается ангидрид AS2O5.
Раствор, даже разбавленный, обладает кислым вкусом и окрашивает
мусовую бумажку в красный цвет. На коже вызывает пузыри, как от ожс
КИСЛОТА НИТРОЗИЛСЕРНАЯ
Nitrosylsulfuric acid Nitrosylschwefelsa
Nitrosulfonsaure
Bleikammerkrists
HSOa-O-NO Мол. в. 127,08
Приготовление
1. По Веб ер у пропускают сухой SO2 в^дымящую НЫОз, находящ^
в широкогорлой колбе, сильно охлаждаемой во время реакции. Пропуск
SO2 ведут до тех пор, пока содержимое колбы не примет кашицеобра<
консистенцию, однако чтобы еще не все количество НЫОз разложилось (в
тивном случае разрушится и HSOs • О • NO). Полученную массу намазьи
тонким слоем на глиняные тарелки и сушат в эксикаторе над H2SO4 в
должение 2—3 дней.
Хранить препарат следует' в плотно закупоренных банках или запаяь
трубках.
2. Фридберг1 предлагает растворять ЫОг в сероуглероде и получен
раствором действовать на 80з, причем выпадают кристаллы нитрозилсе
кислоты.
3. По Вельтцину в дымящуюся H2SO4, содержащую большое кол
ство избыточного БОз, пропускают окислы азота, получающиеся при наг{
нии AS2O3 с НЫОз (смесь NO и NO2). Предварительно сосуд с серной ки
той взвешивают и операцию заканчивают, когда вес увеличивается согл
уравнению:
H2S2O7 + NO + N02 = 2HS03 • О • NO.
Свойства
Снежно-белая листовидная, пористая или зернистая кристаллическая w<
разлагаемая водой на H2SO4, НГ\тОз и NO. В концентрированной H2SO4
творима без разложения. Плавится при 73°, разлагаясь. Кожу окраши
в желтый цвет.
КИСЛОТА ОЛОВЯННАЯ
Acidum stannicum Stannic acid Zinnsaure
Stannihydroxyd
А. а-Оловянная кислота
П риготовление
По Ванино2 к щелочному раствору станната натрия3 прибавляют
бавленную кислоту (избегая избытка последней). Образовавшийся осадок
1 F r i d b u r g, Chem. News, 47, 52.
8 Prap. Ch.v 589.
8 Полученному прибавлением к SnCl4 раствора NaOH до растворения выпадающего в»
осадка.
f. Кислота нитрозилсернаЯу оловянная и пиросерная 259
ной очистки многократно декантируют дестиллированной водой. При
совеР0 ия воздухе частично получается ]3-кислота,
сушена на
Й с Т В Э
■^В мистый аморфный порошок, несколько растворимый в воде и окраши»
■Ъ" лакмусовую бумажку в красный цвет. При сушении на воздухе пре-
раЮ^йй рчает формуле - SnC>2 • 2Н2О [='Sn(OH)4]; в вакууме образуется
*"PiT.ftO[-H.SnO,].
^ Пшенный стекловидный гидрат не является одинаковым по своему со-
^ЫСтак как содержит переменные количества J3-кислоты.
Б. ^-Оловянная (метаоловянная) кислота
Приготовление
ц0 Энгелю1 растворяют олово в HNCV (уд. е. 1,3—1,4), образовав-
ттийся белый осадок (смесь а- и |3-кислот) промывают, растворяют в
небольшом количестве раствора NaOH и осаждают метастаннат натрия (Na2Sn03)
прибавлением избытка NaOH. Выпавший осадок отфильтровывают, промывают
и обрабатывают HNO3; полученную ^-оловянную кислоту высушивают на
воздухе.
Свойства
Высушенная на воздухе J3-оловянная кислота представляет собой белые
аморфные кусочки, легко растираемые в порошок. Содержание воды
переменно. При красном калении переходит в Sn02.
В. Коллоидальная оловянная кислота
Приготовление
1. По Зигмонд-и2 подвергают диализу SnCU с добавлением щелочи или
станнат натрия [SnO(ONa)2] с добавлением НС1; в обоих случаях сначала в
диализаторе получается студень, который, после того как отдиффундируют
соли, пептизируется небольшим количеством оставшейся щелочи. Щелочь
удаляется при дальнейшем диализе (добавление 1—2 капель йодной тинктуры
облегчает отделение).
При нагревании образуется метаоловянная кислота. Замечательна легкость,
с которой коллоид пептизируется от добавления НС1 или солей.
2. По В. Меклен бургу8 150 г гранулированного олова вносят
порциями пр.и сильном взбалтывании в охлажденную смесь 750 мл: HN03
(УД. в. 1,4) и 750 мл воды, регулируя скорость прибавления олова так, чтобы
температура не поднималась выше 0°. При этом олово растворяется, образуя
прозрачный в проходящем свете золь характерного серого цвета. Если вылить
этот раствор в 10 л воды комнатной температуры, то выпадает оловянная
кислота. Последней дают осесть, промывают декантацией, фильтруют через
полотно и снова промывают горячей водой до нейтральной реакции на
лакмус. Влажный гель, содержащий около 10%' оловянной кислоты, сохраняют в
хорошо закупоренных банках.
КИСЛОТА ПИРОСЕРНАЯ
Acidum pyrosulfuricum Pyrosulfuric acid Pyroschwefelsaure
H2S2O7 Мол. в. 178,14
Приготовление
Смешивают концентрированную H2SO4 с вычисленным количеством серного
ангидрида (БОз). Последний берут в небольшом избытке. Смесь помещают под
1-Е ng el, С. г. 124, 785 (1897); С. I, 709 (1897); Ргар. Ch. 590.
а R. Z s i g m о n d у, Zur Kenntnis der Kolloide (Jena, 1905); T. Svedberg, Herst. koll. Lsg., 303
(имеется русский перевод: С в е д б е р г, Образование коллоидов, 1927 г., МХТИ).
8 W. М е с к 1 е n b u г g, Z. anal. Ch. 52, 293 (1913); В, Б е т г е р, Основы качественного анализа.
гОНТИ, стр. 164, 1931.
„ СМ ГО ^Ю CO Г- 00 OS О г-гСЧС0^ЮС0^00а>О^ <М СО ^lOCOt^OOC^O^CV|CY3^ir)COI^OOO^O»--«CSCO^iOCDb-OOa5 О
О / Юи$1Я ЮЮЮ10 35 Ю СО СО СО СО СО СО СО СО COCO t-t^« t-t^^b-t^ ^ t^ t>-00 00 СО 0000 000000000005O)Oi050)0^030)050)O
c7)ooi^coio^cocM'—«оо- оо^соют^сос^^ооэоог^соютрсо^^оо^схэ^еою^со^^оо^да^сою^ i
Tt'^^TTrt'^Tr^TrxrcOt^cOCOCOCOCOCOCOCOCNlC^C^C^CNCVJCNC^ I
нС^С0трЮО^00С7>О^<>1С0^*ЭС0^00ОО^С^С0т*ЮС0^000>©.^<!>1С0^
^^^^^^^^^^(>IC4CNIC4CN1C4C4<NC4CNCOCOCOCOC^CO
оо^оо^сою^со(^^ос^сог^со1Г5т^сосч--оа>ад^сою^со(^'^ос^сх)^
O0>0^0^0^0iCi0^a>0^0^000000CX)00W00000000t>.t^^t^t^^^C^t^(>.C0C0C0^C0C0C0
C0*0OO^O<OrtOSC0ON^OS^^N^iHN^F4«)^^(»^^00OOl00^W«WW0)^(N05O(N(^CCrt0^^00
(NЮOCO^гчтt'OOC^ЮOiCOOO^SHЮOOCS«oa5WN0^001Hl005(NOOтаN^^OO<NЮa)MCDO•^^^-'ЮOO(NCO
cOI^OC0OЮC0rнO00C0Ю00(NO00^ЮCЧC^O00NЮ^C^OONЮтfCS^0)SO^CS^0)^^O^Mr«O00co^C0^
^^^^^^^^^^^^^^^^^^^^^^^дал^^т1^1Г^1^0>> r-^CO^iO^CO 00лО C^^O^t^O^rM CO^iO^^OO^C^CN^CO^OO^
^^C^Tc^CN~(>f(>f<>fcO*CO*CO*CO~CO*^T^^
оооооеоооо»ооооа*сооосюооооооооооооооаооооооовоооооооФоооозоооэд
Кислота пи ро серная
261
--""""-
удельный в
уд. вес при 20°
1,835
1,840
1,845
1,850
1,855
Г,860
1,865
1,870
1,875
1,880
L885
1,890
1,895
1,900
1,905
1,910 |
1,915 |
1,920
1,925
1,930 ^
1,935
1,940
1,945
1,950
1,955
1,960
1,965
1,970
Удельный ве
Агрегатное с
■ *
Таблица 255
ее продажной дымящей серной кислоты
(Winkler)
Общее
содержание S03
"°/о
75,31
77,38
79,28
.80,01
80,95
81,84
82,12
82,41
82,63
82,81
82,97
83,13
83,43
83,48
83,57
83,73 |
84,08
84,56
85,06
85,57
86,23
86,78
87,13
87,41
87,65
88,22
88,92
89,83
105 ч. содержат г
Отгоняемого
S03
__
—
—
-—
—
1,54
2,66
4,28
5,44
6,42
7,29
8,16
9,34
10,07
10,56
11,43
13,33
15,95 |
18,67
21,34
25,65
28,03
29,94
31,46
32,77
35,87
39,68
44,64
H2SO,
92,25
94,/9
97,11
98,01
99,16
98,46
97,34
95,76
94,56
93,58
92,71
91,94
90,66
89,93
89,44
88,57
86,67
84,05 1
81,33
78,66
74,35
71,97
70,06
68,54
67,23
64,13
60,32
55,36
Кислоты 66*
В6
100
91,61
83,92
80,91
77,15
73,55
' 72,43
71,24
1 70,05
69,62
68,97
68,23
67,48
66,91
66,34
65,91
64,48
62,73
60,51
58,44
55,77
53,54
52,12
50,99
50,02
47,71
44,87
41,19
Таблица 256
с продажной дымящей серной кислоты
остояняе
*
°/о SO,
8,3
30,0
40,0
44,5
46,2
59,4
60,8
65,0
69,4
72,8
80,0
82,0
Уд. вес
Найдено
для 26,6°
1,842
1,930
1,956
1,961
1,963
1,980
1,992
1,992
2,002
1,984
1,959
1,953
Вычислено
для 15,5°
1,852
1,940
1,970
1,975
1,977
1,994
2,006
2,006
2,016
1,988
1,973
1,967
262
Кислота
колокол возле 90-—93% -ной H2SO4, причем избыток БОз улетучивается; ^Ж
при этом закристаллизовывается- ' ^
Свойства
Прозрачная кристаллическая масса, дымящая на воздухе. Т. пл. 35°. п
нагревании разлагается на 50з и H2SO4. Продажная дымящая серная кисло
(олеум) представляет смесь переменных количеств H2SO4 и H2S2O7 (см *Й
254 и 255). ' ^
КИСЛОТА ПИРОФОСФОРНАЯ
Pyrophosphoric acid
Pyrophosphorsaur^
Acidum
pyrophosphoricum
H4P2O7* Мол. в. 177,99
Приготовление
1. По Абеггу1 растворяют пирофосфорнокислый натрий (Ыа4Рг07) в вода
и осаждают уксуснокислым свинцом [РЬ{СНзСО.О)г], причем выпадает труЩ
растворимый PD2P2O7, который легко может быть получен в чистом виде. Щщ
суспендируют в воде и пропускают сероводород; при этом происходит еле,
дующая реакция:
Pb2P207 + 2H2S = 2PbS + Ц4Р2О7.
Черный PbS отфильтровывают и кислоту концентрируют, испаряя воду а
вакууме при низкой температуре.
2. По Бендеру2 нагревают в платиновой чашке сиропообразную орто*
фосфорную кислоту (Н3РО4) выше 200°, пока во взятой пробе, растворенной
в воде и нейтрализованной NH4OH, от прибавления AgN03 не -будет
образовываться чисто белый осадок (А^РгСЬ) 3, а не желтоватый (Ag3P04).
Свойства
Бесцветная стекловидная масса. Водный раствор на холоду довольно
постоянен, при кипячении с HN03 очень быстро переходит в НзР04.
Испытание
По техническим условиям НКХП 748-41 содержание ШРгСЬ в препарате
(чистый) должно быть не менее 50%; наибольшие количества допустимых
примесей приведены в табл. 257.
Препарат должен выдерживать испытание на метафосфорную кислоту,
приведенное в ТУ.
Таблица 257
Допустимые п-рнмеси в Н4Ро07 (чистая)
Примеси
1. Хлориды (СГ)
2. Сульфаты (S04")
3. Железо (Fe)
Допустимое
содержание
в%
0,002
0,0005
0,005
* Есть указания (К о I i t о w s k а, С. II, 2059, 1936), что H4Ps07 существует в двух изомерньц
формах, строение которых
HOv ,ОН НО. ПН
>Р-Р<
но' и и \ш
о о
но
Р-О —Р;
он
i Handb. d. anorg. Ch., Ill, 3, 444.
* Неорг. преп. 341.
* По исследованию К, И. Загвоздкина, Ю. М. Рабинович и Н. А. БорилкО
дожить таким методом совершенно чистую Н4Ра0г невозможна (ЖПХ, 1Э, № 1Т 29, 1940).
Р Кислота пирофосфорная и платинохлористоводородная 263
\У КИСЛОТА ПЛАТИНОХЛОРИСТОВОДОРОДНАЯ
I
Chlorplatini'C acid Platinchlorwasserstoffsaure
H2PtClo-6H20 Мол. в. 518,08
Приготовление
1 По Ванино1 растворяют платину в царской водке и для удаления
имбз несколько раз выпаривают раствор с концентрированной НС1. Если в
ячестве исходного материала применяют нечистую платину (лом или остатки),
* к ее необходимо предварительно очистить. Следует отметить, что в препа-
\*те полученном по этому методу, легко может содержаться нитрозоплатино-
Ср'ид PtCl»(N03)2.
2. По Диттмару и Артуру помещают платину (лучше всего губчатую)
в склянку емкостью 2 л (бесцветного стекла), снабженную притертой пробкой,
й добавив достаточное количество дымящей НС1, наполняют склянку хлором
(предварительно промытым водой и пропущенным через слой асбеста).
Закрыв склянку пробкой, дают ей спокойно стоять на свету. Через 12 час.
хлор полностью поглощается; -склянку снова наполняют хлором, дают стоять
и повторяют эту операцию, по,ка металл полностью или почти полностью не
растворится. Тогда декантируют, если нужно, и повторяют обработку раствора
хлором, чтобы tLPtCU перешла в ШРЮо. Полученный раствор для удаления
избытка хлора и НС1 выпаривают на водяной бане.
3. ПоТредвеллу2 платиновые обрезки сначала кипятят с
концентрированной НС1 и промывают водой. Затем помещают в колбу, обливают
концентрированной НС1 и постепенно приливают HNOs, слабо нагревая на водяной
бане. Платина и часть иридия растворяются, большая же часть иридия
остается в виде черного порошка.
Раствор, не фильтруя, переливают в фарфоровую чашку, выпаривают до
густоты сиропа, разбавляют водой, прибавляют соды и муравьинокислого
натрия (HCOONa) и нагревают -до кипения, причем Pt и 1г в несколько минут
выпадают в виде черного порошка (нагревание следует вести в объемистой
чашке вследствие вспенивания от сильного выделения СОг).
Жидкость сливают, а остаток промывают сначала НС1, и затем водой до
полного удаления кислоты. Порошок сушат, сильно прокаливают в фарфоровом
тигле на паяльной горелке (после чего 1г становится нерастворимым в царской
водке) и взвешивают для определения выхода чистого металла. Металл
растворяют при возможно низкой температуре в НС1, подливая постепенно HN0.3.
Полученную жидкость фильтруют и выпаривают попеременно с водой и с НС1
до прекращения выделения окислов азота.
После этого раствор, содержащий платинйстохлористоводородную кислоту
HaPtCU, насыщают хлором при слабом нагревании, причем окраска
значительно светлеет, и выпаривают до густоты сиропа. По охлаждении жидкость
затвердевает в желто-бурую лучисто-кристаллическую массу, которую
растворяют в небольшом количестве воды и отфильтровывают от нерастворив-
шегося иридия.
4. В Институте чистых реактивов (ИРЕА) разработан следующий метод
получения химически чистой H^PtCle • 6Н2О:
Растворяют металлическую Pt в смеси из 3 об. НС1 и 1 об. НЫОз при
нагревании на водяной бане; полученный раствор упаривают попеременно с
водой и с ,НС1 до сиропообразной консистенции. К охлажденному
сиропообразному раствору добавляют половинный объем спирта и приливают
насыщенный раствор NH4CI до прекращения выпадения желтого осадка хлороплатината
аммония'' (NH4)2PtCle. Осадок отфильтровывают, промывают 30%-ным
раствором NHUCl, отжимают и после просушивания при 100—110° прокаливают
в муфельной печи при 800°:
(NH^PtCh = Pt + 2NHs + 2НС1 + 2С1*.
1 Prap. Ch. 775.
« Аналиг. химия, 279.,
254
Кислота
Полученную таким путем чистую губчатую платину снова растворяют в с,
НС1 и HNOs, раствор фильтруют и упаривают до получения кристалличЗИ
препарата (в конце упаривания необходимо раствор насыщать хлором), %Ш
от времени от раствора отбирают пробу в пробирку и охлаждают холоде
водой: если при этом раствор закристаллизовывается, то упаривание раств^
прекращают и дают ему охладиться до комнатной температуры, непрер'^Р*.
размешивая стеклянной палочкой. Препарат получается в виде кристаллц|°
ской массы.
Свойства
Оранжево-красные кристаллы или красно*бурая плотная масса, расплыва^
щаяся
эфире;
теряет
вании
во влажном воздухе. Препарат легко
при 1
часть ]
растворим в воде, спир?4Х
высушивании над hUSOi даже при
кристаллизационной воды. Всю
около температуры разложения.
У д е л ьн ы й
%
РЮ,
1
2
3
4
5
6
7
8
9
10
Уд. вес
1,009
1,018
1,027
1,036
1,046
1,056
1,066
1,076
1,086
1,097
вес водных растворов
о/о
PICI,
11
12
13
14
15
16
Ч
1*
19
20
Уд. вое
1,108
1,119
1,130
1,141
1,153
1,165
1,176
1,188
1,201
1,214
' о/о
PtCl,
21
22
23
24
25
26
27
28
29
30
Уд. вес
1,227
1,242
1,256
1,270
1,285
1,300
1,315
1,330
1,346
1,362
комнатной температур
воду теряет только при на'гё.
H2PtCl6
•/в
PtCI4
31
32
33
34
35
36
37
38
39
40
]
Таблица $№
(в пересчете на Ptdfi)
Уд. вес
1,378
1,395
1,413
1,431
1,450
1,469
1,488
1,505
1,523
1,546
°,'о
PtCt4
41
42
43
44
45
46
47
48
49
50
У А. Щ
1,568
1,591
1,615
1,641
1,666
1,688
1,712
1,736
1,766
him
Испытание (Мерк)
1. Растворимость в абсолютном спирте. 1 г препарата должен
растворяться в 10 г абсолютного спирта без всякой мути. Также должен быть раство*
рим © воде; раствор должен иметь чисто желтую окраску, но не красную или:
красно-бурую. Последние окраски указывают на присутствие НгРЮЫ или 1г. -"
2. Испытание остатка от прокаливания на примеси, растворимые в HNOs.
2 г препарата прокаливают, остаток выдерживают гА часа на водяной бане
с 5 мл НЫОз (уд. в. 1,2) и 20 мл воды. Фильтруют, фильтрат выпаривают
и прокаливают; остаток не должен быть больше 0,005 г.
3. Азотная кислота. Для испытания на HNOs 2 мл 10%-ного раствора
препарата смешивают с равным объемом концентрированной H2SO4 « на
полученную ■ жидкость осторожно наливают раствор FeS04. Бурое кольцо,
появляющееся между слоями тотчас или после некоторого стояния, указывает на
присутствие HNOs (важно для применения препарата в количественном
анализе!).
4. Сульфаты (SO/'). Раствор 1 г препарата в 20 мл воды не должен
давать мути от прибавления ВаСЬ.
5. Соли бария (Ва**). Раствор 1 г препарата в 20 мл воды после,
добавления нескольких капель H2SO4 в течение 3 час. не должен мутнеть.
6. Действующее начало. Раствор 0,5 г препарата в 50 мл воды нагревают
на водяной бане с 10 ял NaOH (уд. в. 1,3) и 2 г хлоральгидрата
СН (ОН)з • ССЬ до полного восстановления хлорной платины. Осадок филь-
r\ \ Кислота селенистая и селеновая 255
промывают горячей водой до нейтральной реакции промывных вод,
трУ10-' сжигают фильтр, прокаливают до постоянного веса и взвешивают.
суШаосаДка должен быть около 0,188 г1.
КИСЛОТА СЕЛЕНИСТАЯ
Acidum selenosum Selenious acid - Selenigsaure
H2Sep3 Мол. в. 128,98
Приготовление
1. По Веберу 5 ч. 8еОг растворяют в 1 ч. воды; при охлаждении
саждаются длинные кристаллы, которые отжимают между листами
фильтровальной бумаги.
2. По Клаусницеру2 растворяют селен в теплой HNOs (1:1),
раствор фильтруют, выпаривают досуха, и белый сухой остаток нагревают
0 колбе с коротким широким горлом на бане из железных опилок до тех
пор, пока на помещенном сверху холодном часовом стекле уже не будет
конденсироваться влага и пока некоторое количество Se02 не возгонится
на горло колбы. Тогда остаток растворяют в избытке горячей воды и быстро
выпаривают на голом огне (в случае выпадения Se добавляют несколько
капель НЫОз), до тех пор, пока внезапно не прекратится выделение
водяных паров и жидкость не станет сиропообразной.
При медленном охлаждении образуются крупные кристаллы селенистой
кислоты, которые отжимают между листами фильтровальной бумаги.
3. Метод, разработанный в ИРЕА, предусматривает полную возгонку
сухого остатка и растворение в воде полученного £еОг. Однако такой прием
не является необходимым; отличного качества препарат получается по
методу Клаусницера.
4. Соли селенистой кислоты получаются одновременно с селенистыми
металлами при нагревании селена со щелочами или углекислыми солями.
Свойства
Большие продольно исчерченные призматические кристаллы уд. в. 3,00.
В эксикаторе над H2SO4 выветривается, во влажном воздухе расплывается.
Селенистая кислота легко растворима в горячей воде почти во всех
отношениях. Легко растворима в спирте. При нагревании сначала выделяется вода,
затем возгоняется SeCb. FUSeOs относится к числу довольно слабых кислот
(константы диссоциации3 К1 = 2,37 • 10"3, К* = 4,8 • К)-»).
Как сама кислота, так.и ее соли сильно ядовиты»
КИСЛОТА СЕЛЕНОВАЯ
Acidum selenicum Selenic acid Selensaure
H?Se04 Мол. в. 144,98
А. Водный раствор НгБеО*
Приготовление
1. По Вани но4 окисляют раствор 10 a Se02 в 100 мл воды при помощи
6 г бромноватокислого калия (КВгОз) и 5 мл концентрированной HNOs.
После внесения реагентов осаждают из раствора действием Ba(NOs)2 селеново-
кислый барий (BaSe04), который по отделении разлагают вычисленным
количеством H2SO4.
1 Рекомендуется также проверка препарата на содержание Ац и Pd (Испытание хим. реак«
тивов. 334).
a Prap. Ch., 114.
« Н. II a g i s a w а, С. II, 3792 (1939).
1 Ргар. Сп., 114.
266
Кислота
2. По X а у е р у сплавляют SeO-2 с селитрой до прекращения выдел Ъ
красных паров. Растворяют сплав в воде, кипятят с HNCb до удад?^
азотистой кислоты и смешивают с раствором Са(Ж)з)г. Полученный CaS*!?*-
осаждающийся частью немедленно, частью при дальнейшем выпаривай *•
очищают растворением в возможно малом количестве холодной воды и. **>
греванием раствора (CaSe04 в горячей воде растворим хуже, чем в холо
нпй^ Кипяириирд/г гп пчбп.птянным в воле избытком тттавелевокиглпгл ..А*
ной). Кипячением со взболтанным в воде избытком щавелевокислого Ка
мия* (CdC204) соль переводят в . растворимый селеновокислый кадмий
(CdSe04) и раствор отделяют фильтрованием от образовавшегося СаСгО*
избытка CCIC2O4. и
В полученную жидкость пропускают H2S для осаждения кадмия, фИл,
труют и' раствор свободной H2Se04 кипятят для удаления сероводорода.
3. По Т о м с е н у 25 г чистого селена растворяют в Н1ЧОз уд. в. £«
и раствор выпаривают до полного удаления чОкислов азота. Остаток раств&
ряют в 75 мл воды, фильтруют и осаждают раствором 54 г AgNOa в 75 щ
воды в виде почти нерастворимого 'Ag2Se03.
Осадок взбалтывают с водой и бромом, причем сначала прибавляют бро^
а под конец бромную воду, пока раствор не окрасится небольшим избытком
брома в оранжево-желтый цвет.
Отфильтровывают AgBr, кипячением удаляют избыток брома и выпаривает
раствор сначала на водяной бане, а затем при 140—150°.
4. По методу ИРЕА для получения химически чистого препарата
подвергают раствор НгЭеОз электролизу со свинцовыми анодами и свинцовыми (или
из нержавеющей стали) катодами. К полученному в результате анодного
окисления раствору Нг$е04 добавляют РЬ(ЫОз)2, причем выпадает
нерастворимый PbSe04.
Осадок отфильтровывают, тщательно промывают и, суспендировав в воде,
обрабатывают сероводородом при непрерывном механическом перемешивании:
PbSe04 + H2S =_PbS + H2Se04.
Ф
После отфильтрования черного осадка PbS раствор Нг£е04 выпаривают на
водяной бане до содержания 80%. Дальнейшим упариванием в вакууме
можно получить кристаллический препарат.
Б. Безводная HsSeCh
1. Следующий способ предложен Камероном и Макалланом2.
Водный раствор H2Se04 сначала упаривают на водяной бане и затем
отгоняют воду в вакууме, повышая постепенно температуру до 180°. Как
только температура достигает этой точки, отгон прекращают. При
охлаждении остаток в колбе затвердевает в твердую массу, представляющую,
собой почти безводную селеновую кислоту. Насос, создающий вакуум, должен
действовать безукоризненно; между колбой и насосом необходимо включить
трубку с КОН.
2. По Л. Д жил ьбертсону и Д. Кингу3 к 500 г 30%-ной Н2О2
добавляют 150 г возогнанного S-еОг. Через сутки смесь кипятят в течение
12 час. с обратным холодильником, пропуская кислород. Полноту окисления
проверяют пробой с сероводородной водой (не должно выпадать красного
осадка Se); если окисление не закончено, добавляют еще немного Н2О2 и
нагревают.
Полученный раствор подвергают перегонке при уменьшенном давлении,
причем отогняется основная масса воды и избыточная Н2О2. Пропуская сухой
воздух над продуктом, . содержащим еще немного воды (при 150—160° И
1 Получается действием (NH4)eC804 на раствор любой соли кадмия,
а С a m е г о n, Macallan, Ber., 22, 477 (1889); Chetn. News, 59, 219.
a L. J. G i I b e 11 s 0 n a. G. В. К i n g, J. Am. Chera. Soc.r 58, 180 (1936).
\
Кислота серная
267
z мм Рт- ст-)' п0ЛУча1°т вязкую жидкость, из которой при 58° кристалли-
*"" а Нгсе04 в форме длинных игл.
ауеПроДукт содержит 99,8% H2Se04.
Свойства
Белая кристаллическая масса уд. в. 2,9э, плавящаяся при 58°. Очень
ооскопична, чрезвычайно легко растворима- в воде. Обладает, как и H2SO4,
Г1*ень большим сродством к воде и способна разлагать многие органические
°Ч инения с выделением углерода (обугливать).
Испытание^
Препарат должен содержать не более 3% нелетучего остатка; испытания
проводятся на хлориды и бромиды (не должно быть опалесценции при
добавлении AgNOs к прозрачному подкисленному НЫОз раствору препарата),
сульфаты (не должно быть осадка от добавления ВаСЬ к подкисленному
раствору препарата), селенистую кислоту (H2S не должен образовывать
осадка в растворе FbSeO^ и тяжелые металлы (при добавлении (NH^S
раствор препарата не должен изменять окраски или выделять осадка).
Acidum sulfuricum
КИСЛОТА СЕРНАЯ
Sulfuric acid
H2SO4
Мол. в. 98,08
Schwefelsaure
1. Для получения совершенно чистого препарата Цинтль рекомендует
перегонять кислоту с К2СГ2О7.
Для этой цели реторту иенского стекла облепляют мокрым асбестовым
картоном (3 мм) до самого горла. После просушки эту «обмуровку» для
прочности промазывают еще концентрированным раствором Na2SiOa (жидкое стекло)
и вновь высушивают. Реторту устанавливают на кольцо
штатива .и слабо закрепляют зажимом ее горло,
обернув его предварительно полоской асбеста.
В реторту загружают концентрированную H2SO4, в
которой предварительно растворен К2О2О7, до интенсивно
красного окрашивания. Кроме того, для спокойного
кипения в кислоту добавляют, несколько небольших
кусочков неглазурованной глины. Кислоту перегоняют,
пользуясь сильной горелкой и отбрасывают первую четверть
дестиллата.
Для предохранения получающегося препарата от пыли
применяют следующее приспособление (рис. 43): в
колбочке емкостью 100 мл выдувают боковое отверстие, в
которое вставляют горло реторты. Колбу устанавливают
горлом вниз в склянку, предназначенную для собирания
кислоты.
2. Для обесцвечивания коричневой или черной кислоты Розенштан(д и
Вёльдике предлагают прибавлять ШОг. При добавлении всего 0,01—0,03%'
30%-ной Н2О.2 и последующем перемешивании кислота приобретает окраску
от светложелтои до янтарно-желтой. Совершенно че;рная кислота требует до
1% Н202.
Применяя минимально необходимое для обесцвечивания количество
перекиси водорода, можно пренебречь образующейся кислотой Каро H2SO5;
в противном случае рекомендуется разрушить ее (обесцветить) прибавлением
сернистой кислоты.
Рис. 43.
Приспособление для
предохранения от пыли при
перегонке серной
кислоты
1 Испытание хим. реактивов, 358.
263
Кислота
Табли,
Удельный вес водных растворов H2S 04 (Lunge, J s 1 е г, N а е f)
«^;
100 вес. ч. соответствуют
при х. ч. кислоте вес. ч.
1 л содержит кг х
кислоты
1,000
1,005
1,010
1,015
1,020
1,025
1,030
1,035
1,040
1,045
1,050
1,055
1,060
1,065
1,070
1.075
1,080
1,085
1,090
1,095
1,100
1,105
1,110
1,115
1,120
1,125
1,130
1,135
1,140
1,145
1,150
1,155
1,160
1,165
1,170
1,175
1,180
1,185
1,190
1,195
1,200
1,205
1,210
1,215
1,220
0
0,7
1,4
2,1
2,7
3,4
4,1
4,7
5,4
6,0
6,7
7,4
8,0
8,7
9,4
ю,о
10,6
11,2
11,9
12,4
13,0
13,6
14,2
14,9
15,4
16,0
16,5
17,1
17,7
18,3
18,8
19,3
19,8
20,3
20,9
1 21,4
1 22,0
22,5
23,0
23,5
24,0
24,5
25,0
25,5
26,0
0
1
2
з
4
5
6
7
8
9
10
и
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
4(
42
43
44
0,07
0,77
1,28
1,88
2,47
3,07
3,67
4,27
4,87
5,45
6,02
6,59
7,16
7,73
8,32
8,90
9,47
10,04
10,60
11,16
11,71
12,27
12,82
13,36
13,89
14,42
14,95
15,48
16,01
16,54
17,07
17,59
18,11
18,64
19,16
19,69
20,21
20,73
21,26
21,78
22,30
22,82
23,33
23,84
24,36
0,09
0,95
1,57
2,30
3,03
3,76
4,49
5 23
5,96
6,67
7,37
8,07
8,77
9,47
10,19
10,90
11,60
12,30
12,99
13,67
14,35
15,03
15,71
16,36
17,01
17,66
18,31
18,96
19,61
20,26
20,91
21,55
22,19
22,83
23,47
24,12
24,76 I
25,40'
26,04
26,68
27,32
27,95,
28,58 I
29,211
29,84 I
0,12
1,21
2,01
2,95
3,88
4,82
5,78
6,73
7,64
8,55
9,44
10,34
11,24
12,14
13,05
13,96
14,87
15,76
16,65
17,52
18,39
19,26
20,13
20,96
21,80
22,63
23,47
24,29
25,13
25,96
26,79
27,61
28,43
29,25
30,07
30,90
31,73
32,65
33,37
34,19
35,01
35,83
36,66
37,45
38,23
0,14
1,52
2,51
?,68
4,85
6,02
7,18
8,37
9,54
10,67
11,79
12,91
14,03
15,15
16,30
17,44
18,56
19,68
20,78
21,87
22,96
24,05
25,14
26,18
27,22
28,26
29,30
30,34
31,38
32,42
33,46
34,48
35,50
36,53
37,55
38,59
39,62
40,64
41,66
42,69
43,71
44,72
45,73
46,74
47,74
0,001
0,008
0,013
0,019
0,025
0,032
0,038
0,044
0,051
0,057
0,063
0,070
0,076
0,082
0,089
0,096
0,103
0,109
0,116
0,122
0,129
0,136
0,143
0,149
0,156
0,162
0,169
0,176
0,183
0,189
0,196
0,203
0,210
0,217
0,224
0,231
0,238
0,246
0,253
0,260
0,268
0,275
0,282
0,290
0,297
0,001
0,009
0,016
0,023
0,031
1 0,039
1 0,046
1 0,054
0,062
0,071
0,077
0,085
0,093
0,102
0,109
0,117
0,125
,0,133
0,142
0,150
0,158
0,166
0,175
0,183
0,191
0,199
0,207
0,215
0,223
0,231
0,239
0,248
0,257
0,266
0,275
0,2^3
0,292
0,301
0,310
0,319
0,328
0,337
0,346
0,355
0,364
0,001
0,013
0,020
0,030
0,040
0,049
0,059
0,070
0,079
0,089
0,099
0,109
0,119
0,129
0,140
0,150
0,161
0,171
0,181
0,192
0,202
0,212
0,223
0,234
0,245
0,255
0,265
0,276
0,287
0,297
0,308
0,319
0,330
0,341
0,352
0,363
0,374
0,386
0,397
0,409
0,420
0,432
0,444
0,455
0,466
ш.
0,015
о,о&
о.оз/
0,050
0,062
0,074
0,087
0,093
0,112
0,124
0Д36
0,149
0,161
ОД74
0,188
0,201
0,213
0,227
0,240
0,253
0,265
0,279
0,292
0,305
0,318
0,331
0,344
0,358
0,371'
0,385^
0,398
0,412
0,426
0,439
0,453
0,467
0,481
0,496
0,511
0,525
0,539
0,553
0,568
0,533
Кислота серная
2Ю
■— "
<ЗГ
S
Градусы Бом
а»
Градусы Тве
(Tw)
1
10
при
6
сл
вес- ч. соответствуют
х. ч. кислоте шее. ч-
О
X
кислоты 60е
Вё
о
ю
а
н
о
ч
и
Продолжение табл. 259
1 .* содержит кг х. ч.
кислоты
О
со
сл
X
а
н
Очи
а
н
О su
4pQ
«5
26,4
26,9
27,4
27,9
28,4
28,8
29,3
29.7
30,2
30,6
31,1
31,5
32,0
32,4
32,8
33,3
33,7
34,2
34,6
35,0
35,4
35,8
36,2
36,6
37,0
37,4
37,8
38,2
38,6
39,0
39,4
39,8
40,1
40,5
40,8
41,2
41,6
42,0
42,3
42,7
43,1
43,4
.43,8
44,1
4Ф,4
44,8
45,1
45
46
47
48
49
50
51
52
53
54
55
55
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
24,88
25,39
25,88
26,35
26,83
27,29
27,76
28,22
28,69
29,15
29,62
30,10
30,57
31,04
31,52
31,99
32,46
32,94
33,41
33,88
34,35
34,80
33,27
35,71
36,14
36,58
37,02
37,45
37,89
38,32
38,75
39,18
39,62
40,05
40,48
40,91
41,33
41,76
42,17
42,57
42,96
43,36
43,75
44,14
44,53
44,92
45,31
30,48
31,11
31,70
32,28
32,86
33,43
34,00
34,57
35,14
35,71
36,29
3?,87
37,45
38,03
38,61
39,19
39,77
40,35
40,93
41,50
42,08
42,66
43,20
43,74
44,28
.44,82
45,35
45,88
46,41
46,94
47,47
48,00
48,53
49,06
49,59
50,11
50,63
51,15
51,66
52,15
52,63
53,11
53,59
51,07
54,55
55,03
55,50
39,05
о9,86
40,61
41,37
42,11
42,84
43,57
44,30
45,03
45,76
46,50
47,24
47,99
48,73
49,47
50,21
50,96
51,71
52,45
53,18
53,92
54,67
55,36
56,05
56,74
57,43
58,11
58,79
59,48
60,15
60,83
61,51
62,19
62,87
63,55
64,21
64,88
65,55
66,21
66,82
67,44
68,06
68,68
69,29
69,90
70,52
71,12
48,77
49,73
50//2
51,65
52,58
53,49
54,40
55,31
56,22
57,14
58,С6
58,99
59,92
60,85
61,78
62,70
63,63
64,56
65,45
66,40
67,33
68,26
69,12
69,98
70,85
71,71
7255
73,41
74,26
75,10
75,95
76,80
77,65
78,50
79,34
80,18
81,01
81,86
82,66
83,44
84,21
84,98
85,74
86,51
87,28
88,05
88,80
0,305
0,312
0,320
0,327
0,334
0,341
0,348
0,356
0,363
0,370
0,377
0,385
0,393
0,400
0,408
0,416
0,424
0,432
0,439
0,447
0,455 I
0,4Я2
0,471
0,479
0,486
0,494
0,502
0 509
0,517
0,525
0,533
0,541
0,549
0,557
0,564
0,573
0,581
0,589
0,597
0,604
0,612
0,620
0,628
0,636
0,6^3
0,651
0,659
0,373
0,382
0,391
0,410
0,409
0,418
0,426
0,435
0,444
0,454
0.462
0,472
0,481
0,490
0,500
0,510
0,519
0,529
0,538
0,548
0,557
0,567
0,577
0,586
0,596
0,605
0,614
0,624
0,633
0,643
0,653
0,662
0,672
0,682
0,692
0,702
0,711
0,721
0,730
0,740
0,750
0,759
0,769
0,779
0,789
0,798
0,808
0,478
(',490
0,502
0,513
0,524
0,535
0,547
0,558
0,570
0,581
0,593
0,605
0,617
0,629
0,641
0,653
0,665
0,677
0,689
0,702
0,714
0,727
0,739
0,751
0,763
0,775
0,787
0,800
0,812
0,824
0,836
0,849
0,861
0,^73
0,886
0,899
0,912
0,924
0,937
0,949
0,961
0,973
0,986
0,998
1,010
1,023
1,035
270
Кислота
Продолжение табл, о*
£-!
1,460
1,465
1,470
1,475
1,480
1,485
1,490
1,495
1,500
1,505
1,510
1,515
1,520
1,525
1,530
1,535
1,540
1,545
1,550
1,555
1,560
1,565
1,570
1,575
1,580
1,585
1,590
1,595
1,600
1,605
1,610
1,615
1,620
1,625
1,630
1,635
1,640
1,645
1,650
1,655
1,660
1,665
1,670
1,675
1,680
1,685
1,690
I
н
я
10Э вес. ч. соответствуют
при х. ч. кислоте вес. ч.
45,4
45,8
46Д
46,4
46,8
47,1
47,4
47,8
48,1
48,4.
48,7
49,0
49,4
49,7
50,0
50,3
50,6
50,9
51,2
51,5
51,8
52,1
52,4
52>7
1 53,0
1 53,3
53,6
53,9
54,1
54,4
54,7
55,0
55,2
55,5
55,8
56,0
56,3
56,6
56,9
57,1
57,4
57,7
57,9
58,2
58,4
58,7
1 58,9
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
НО
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
45,69
46,07
46,45
46,83
47,21
47,57
47,95
48,34
48,73
49,12
49,51
49,89
50,28
50,66
51,04
51,43
51,78
52,12
52,46
52,79 ,
53,22
53,59
53,95
54,32
54,65
55,03
55,37
55,73
56,09
56,44
56,79
57,15
57,49
57,84
58,18
58,53
58,88
59,22
59,57
59,92
60,26
60,61
60,95
61,29
61,63
61,93
62,29 1
55,97
56,43
56,90
57,37
57,83
58,28
58,74
59,22
59,70
60,18
60,65
61,12
61,59
62,06
62,53
63,00
63,43
63,85
64,26
64,67
65,20
66,65
66,09
66,53
66,95
67,40
67,83
68,26
68,70
69,13
69,56
70,00
70,42
70,85
71,27
71,70
72,12
72,55
72,96
73,40 I
73,81
74.24
74,66
75,0-
75,50
75,94
76,38
1 л содержит кг х. Нш
кислоты
а
2CQ
71,72
72,31
72,91
73,51
74,10
74,68
75,27
75,88
76,50
77,12
77,72
78,32
78,93
79,52
80,13
80,73
81,28
81,81
82,34
82,87
83,50
84,08
84,64
85,21
85,78
86,34
86,88
87,44
88,00
88,55
89,10
89,66
90,20
90,74
91,29
91,83
92,38
92,92
93,45
94.02
94,54
95,08
95,62
96,16
96 69
97,21
97,77
89,55
90,29
91,04
91,79
92,53
93,25
93,98
94,75
95,52
96,29
97,04
97,79
98,54
99,30
100,05
100,80
101,49
102,16
102,82
103, \7
104,30
105,03
105,73
106,42
107,10
107,85
108,52
109,21
109,92
110,61
111,30
112,00
112,68
113,35
114,02
114,71
115,39
116,06
116,72
117,44
118,11
118,77
119,36
120,11
120,50
121,38
122,08
0,667
0,675
0,683
0,691
0,699
,0,707
0,715
0,723
0,731
0,739
0,748
0,756
0,764
0,773
0,781
0,789
0,797
0,805
0,813
0,821
0,830
0,839
0,847
0,856
0,864
0,872
0,880
0,889
0,897
0,906
0,914
0,923
0,931
0,940
0,948
0,957
0,966
0,975
0,983
0,992
1,000
l,0(i9
1,017
1,027
1,035
1,043
1,053
0,817
0,827
0,837
0,846
0,856
0,865
0,876
0,885
0,896
0,906
0,916
0,926
0,936
0,946
0,957
0,967
0,977
0,987
0,996
1,006
1,017
1,027
1,038
1,048
1,058
1,068
1,078
1,089
1,099
1,110
1,120
1,131
1,141
1,151
1,162
1,172
1,182
1,193
1,204
1,215
1,225
1,230
1,246
1,259
1,268
1278
1,289
1,047
1,059
1,072
1,084
1,097
1,109
1,122
1,134
1,147
1,160
1,174
1,187
1,199
1,213
1,226
1,239
1,252
1,564
1.27«
1,289
1,303
1,316
1,^29
1,343
1,356
1,369
1,382
1,395
1,409
1,422
1,435
1,449
1,462
1,473
1,489'
1,502
1,516
1,529
1,543
1,557
1,570
1,584
1,598
1,611
1,625
1,638
1,652
I
1,307
1,323
1,33$
U3&
1.3W
ЫЫ
1.4I&
1,433
1,448
1,465
1,481
1,498
1,514
1,531
1,547
1,563
1,579
1,593
1,609
1,627
1,644
1,660
1,677
1,692
1,709
1,726
1,742
1,759
1,775
1,792
1,810
1,825
1,842
1,859
1,875
1,892
1,909
1,926
1,944
1,960
1,977
1,995
2,012
2,029
2,046
2,064
Кислота серная
щ
Продолжение табл. 259
° f-c
100 вес. ч. соответствуют
при х. ч, кислоте вес. ч.
1 л. содержит кг х. ч*
кислоты
I
59,2
59,5
59,7
60,0
60,2
60,4
60,6
60,9
61,1
61,4
61,6
61,8
62,1
62,3
62,5
62,8
63,0
63,2
63,5
63,7
64,0
64,2
64,4
64,6
64,8
65,0
* 65,1
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
65,2
'65,3
65,4
' 65,5
65,6
65,7
' 65,8
' 65,9
*165*
166
167
'168*
62,64
63,00
63,35
63,70
64,07
64,43
64,78
65,14
65,50
65,86
66,22
66,58
66,94
67,30
67,76
68,17
68,60
68,98
69,47
69,96
70,45
70,96
71,50
72,08
72,96
73,51
73,63
73,80
73,96
74,12
74,29
74,49
74,69
74,86
75,03
75,19
75,46
75,69
75,89
76,12
76,38
76,57
76,90
77,23
77,55
78,04
78,33
76,76
77,17
77,60
78,04
78,48
78,92
79,36
79,80
80,24
80,68
81,12
81,56
82,00
82,44
83,01
83,51
84,02
84,50
85,10
85,70
86,30
86,92
87,60
88,30
89,16
90,05
90,20
90,40
90,60
90,80
91,00
91,25
91,50
91,70
91,90
92,10
92,43
92,70
92,97
93,25
93,56
93,80
94,25
94,60
95,00
95,60
95,95
98,32
98,89
99,44
100,00
1С0,56
101,13
101,69
102,25
102,82
103,38
103,95
104,52
105,08
105,64
106,31
106,91
107,62
108,27
109,05
109,82
110,58
111,32
112,25
.113,15
114,21
115,33
115,59
115,84
116,10
116,35
116,61
116,93
117,25
117,51
117,76
118,02
118,41
118,73
119,07
119,43
119,84
120,19,
120,711
121,22
121,74
122,51
122,96
122,77
123,47
124,16
124,86
125,57
126,27
126,98
127,68
128,38
129,09
129,79
130,49
131,20
131,90
132,80
133,61
134,43
135,20
136,16
137,14
138,08
139,06
140,16
141,28
142,65
144,08
144,32
144,64
144,96
145,23
145,60
146,00
146,40
146,72
147,04
147,36
147,88
148,32
148,73
149,18
149,70
150,08
150,72
151,36
152,00
152,96
153,52
1,062
1,071
1,080
1,089
1,099
1,108
1,118
1,127
1,136
1,145
1,156
1,165
1,175
1,185
1,196
1,207
1,218
1,228
1,240
1,252
1,265
1,277
1,291
1,305
1,322
1,338
1,341
1,345
1,348
1,352
1,356
1,360
1,364
1,368
1,372
1,376
1,382
1,386
1,391
1,396
1,402
1,405
1,412
1,419
1,426
1,436
1,441
1,301
1,312
1,323
1,344
1,346
1,?57
1,369
1,381
1,392
1,404
1,416
1,427
1,439
1,451
1,465
1,478
1,491
1,504
1,519
1,534
1,549
1,564
1,581
1,598
1,618
1,639
1,643
1,647
1,651
1,656
1,661
1,666
1,671
1,676
1,681
1,685
1,692
1,698
1,704
1,710
1,717
1,722
1,73(1
1,739
1,748
1,759
1,765
1,667
■1,681
-1,696
■1,710
1,725
1,739
1,754
1,769
1,784
1,799
1,814
1,829
1,845
1,859
1,877
1,894
1,911
1,928
1,947
1,965
1,983
2,004
2,026
2,048
2,074
2,099
2,104
2,110
2,116
2,122
2,128
2,135
2,142
2,148
2,154
2,159
2,169
2,176
2,184
2,191
2,200
2,207
2,217
2,228
2,239
2,254
2,262
'272
Кислота
Продолжение табд
100 вес. ч* соответствуют
цри х' ч. кислоте вес. ч.
1,8410
1,8415
1,8410
1,8405
1,8400
1,8395
1,8390
1,8385
78,69
79,47
80,16
80,43
80,59
80,63
80,93
81,08
о
«а
96,38
97,35
98,20
98,52
98,72
98,77
99,12
99,31
123,45
124,69
125,84
126,18
126,44
126,50
126,99
127,35
154,20
155,74
157,12
157,62,
157,94
158,00
158,60
158,90
1 л содержи кг х
1,448
1,463
1,476
1,481
1,483
1,484
1,488
1,490
1,774
1,792
1,808
1,814
1,816
1,817
1,823
1,826
Ко
«9
2,273
2,296
2,317
2,325
2,327
2,328
2,336
2,339
Свойства
Бесцветная маслообразная жидкость уд. в. 1,859 при 0° и 1,837 при \Щ
При охлаждении застывает в кристаллы, которые плавятся при 10,49°. Однако/
жидкая кислота легко может быть переохлаждена ниже 0° без замерзания.*
При 30—40° начинает дымить и при дальнейшем нагревании образует парц;';
SO3. При 290° начинается кипение и температура быстро поднимается, пок$;
не прекратится выделение S03. Оставшийся гидрат кипит при 338°. |
Концентрированная H2SO4 жадно поглощает влагу и является поэтому
отличным осушителем; по Беккеру1 упругость пара НзО над H2SO4
составляет всего 0,003 мм рт. ст. (см. также табл. 264).
О свойствах дымящейся H2SO4 см. стр. 259—264, Кислота пир 04
серная.
Таблица 260
Содержание H2S04 в очень концентрированных растворах
(Domke)
°/oH,S04
90
91
92
93
94
<fi°
1,8252
1,8302
1,8346
1,8384
1,8415
f/15
Л,8198
1,8248
1,8293
1,8331
1,8363
«! 1
1,8144
1,8195
1,8240
1,8279
1,8312
°/oHaSO,
95
96
97
98
99
di°
1,8439
1,8457
1,8466
1,8463
1,8445
rfl5
1,8388
1,8406
1,8414
1,8411
1,8393
фо
1,8337
1,8355
1,8364
1,8361
1,8342
1 В е с k e r, Ind. Eng. Chem, Anal. Ed., 4, 1 (1933).
Кислота серная
2J3
Таблица 2€1
«павка для удел ьного веса H2S04 в зависимости от
П°п^ температуры
УД- веС КИп°
1,04
1,07
1,10
Уменьшение
уд. веса при
повышении
температуры
на 1*
Уд. вес
кислоты при 0°
0,0002
0,0003
0,0004
1,15
1,20
1,30
Уменьшение
уд. веса при
повышении
температуры
на 1°
Уд. вес
кислоты при 0°
0,0005
0,0006
0,0007
1,45
1,70
1,85
Уменьшение
уд. веса при
повышении
температуры
на 1°
0,0008
0,0009
0,00096
Таблица 252
Таблица для приготовления H2S04 любой концентраций
смешением H2S04 уд. в. 1,85 с водой
100 ч. воды 1
(15-20°)
смешивают с
Участями HfS04
уд. в. 1,85
ЛГ=1
* 2
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
ПО
120
Получают
кислоту уд.
веса
1,009
1,015
1,035
1,060
1,090
1,113
1,140
1,165
1,187
1,210
1,229
1,248
1,265
1,280
1,297
1,312
1,326
1,340
1,357
1,372
1,386
1,398
1,420
1,438
100 ч. воды
(15—20°) смеши
ваютс X
частями На504уд. в.
1,85
ЛГ=130
140
150
160
170
180
190
200
210
220
230
240
250
1 260
270
280
290
300
310
320
330
340
350
350
Получают
кислоту уд.
веса
1,456
1,473
1,490
1,510
1,530
1,543
" 1,556
1,568 !
1,580
Д.593
1,606
1,620
1,630
1,640
1,648
1,654
1.667
1,678
1,689
1,700
1,705
1,710
1,714
1,719
100 ч. воды 1
(15—20°)
смешивают с X 1
частями HtS04|
уд. в. 1,85
*=370
380
390
400
410
420
430
440
450
460
470
480
490
| 500
510
520
530
540
550
560
570
580
590
600
Получают
кислоту уд.
веса
1,723
1,727
1.730
1,733
1,737
1.740
1,743
1,746
1,750
1,754
1,757
1,760
1,763
1,766
1,768
1,770
1,772
1,774
1,776
1 1,777
1,778
1,779
1,780
1,782
274 Кислота ?ЩП
... _.^. **:^ . , и | ■■■-■ I
Таблица 2бЗ I
Температура замерзания и плавления H2S04
различной концентрации (Lunge)
Уд. вес
при 15°
1,671
1,691
1,712
1,727
1,732
1,749
1,767
1,790
1.807
1,822
1,842
Градусы
Боме (Вё)
58,00
59,00
60,05
60,75
61,00
61,80
62.65
63,75
64,45
65,15
66,00
Температура
замерзания °С
жидк. при—20
. -20
н . -20
-7,5
—8,5
-0,2
+1,6
+4,9
—9,0
жидк. при—^0
. -20
Температура
плавления СС
—
—
—7,5
—8,5
+4,5
+6,5
+8,0
-6,0
—
—■
Таблица Щ
Упругость водяного пара над р а с тв ора ми H2SO,
Уд. вес '
H8SO,
1,344
1,361
1,380
1,398
1,417
1,438
1,459
1,479
1,503
1,524
1,546
1,569
1,592
1,615
1,639
1,662
1,690
1,710
1,732
1,754
% h2so4
44
46
48
50
52
54
56
58
60
62
64
66
68
70
72
74
76
78
80
82
Упругс
10°
4,4
4,0
3,7
3,3
3,0
2,6
2,2
1,9
1,6
1,4
1,2
1,1
0,9
0,8
0,7
0,5
0,4
0,3
0,2
0,1
сть паров Н80
15°
6,1
5,5
5,0
4,5
4,0
3,6
3,1
2,6
i 2д
1 1,8
1,6
1.4
1,2
1,0
0,8
0,6
0,4
0,3
0,2
0,1
в мм рт. от. при темп.
20°
8,5
7,7
7,1
6,5
5,8
5,0
4,3
3,6
3,0
, 2.6
2,2
1,8
1 1,5
1,3
1,0
0,6
0,5
0,4
0,3
0,2
25°
11,5
10,5
9,6
8,8
7,9 °
7,0
6,0
5,1
4,3
3,6
3,0
2,5
2,1
1,8
М
1,2
1,0
0,8
0,6
0,4
Кислота серная
275
Таблица 265
Температура кипения растворов H2S04 (Lunge)
W»b"vt
**\ 15°
1 8400
1*8334
1*8245
1,8140 1
1 7990 1
1,7800
1,7554
1,7400
1,7203
1,7037
1,6786
1,6599
1,6328
%
H8S04
95,3
92,8
90,4
88,7
86,6
84,3 I
81,8
80,6
78,9
77,5
75,3
73,9
71,5
Температура кипе»
ния °С
297 .
280
264
257
241,5
228
218
209
203,5
197
185,5
180
1 173
Барометрическое давление'
(в мм),
приведенное к 0°
718,8
723,9
720,6
726,0
720,1
720,5
726
720,6
725,9
725,2
725,2
725,2
725,2
1
Удельный
вес,
приведенный
к 15°
1
1,6072
1,5825
1,5617
1,5437
1,4960
1.4635
1,4015
1,3554
1,3194
1,2633
1,2042
1,1128
1,0580
%
H,S04
69,5
67,2
65,4
64,3
59,4
56,4
50,3
45,3
41,5
34,7
27,6
15,8
, 8,5
Температура
кипения °С
169
160
158,5
151,5
143
133,5
124
118,5
115
110
, 107
103,5
101,5
Барометрическое давление
(в мм),
приведенное к 0°
730,1
728,8
730,1
730,1
730,1
730,1
730,1
730,1
730,1
732,9
732,9
732,9
735
Испытание
По ОСТ НКТП 3573 кислота должна представлять собой бесцветную
маслянистую прозрачную жидкость; уд. в. d^ от 1,835 до 1,840; содержание серной
кислоты, (H2SO4) должно быть от 93,56 до 95,60%; наибольшие количества
допустимых примесей указаны в табл. 266; кислота должна выдерживать по
ОСТ испытание на определение примеси веществ, восстанавливающих KMnCh;
кислота должна выдерживать испытание по методу Савалля, приведенное
в ОСТ.
Таблица 266
Допустимые примесив H2S04
Примеси
Допустимое содержание в о/0
в химически
чистом
0,001
0,0001
0,0001
0,0002
0,00005
0,000003
0,0002
0,0001
В ЧИСТОМ ДЛЯ
анализа
0,002
0,0002
0,0002
0,0005
0,0001
0,000003
0,0005
0,0003
в чистом
0,01
0,0005
0,0005
0,0005
0,0003
0,00001
0,001
0,001
1. Нелетучий остаток ,
2. Соляная кислота (СК) ....
3. Азотная кислота (N03') • . .
4. Тяжелые металлы группы H2S
5. Железо(Ре) . • ........
6. Мышьяк (As) ,
7. Селен (Se)
8. Аммонийные соли (NH4) • .
276
Кислота
КИСЛОТА СОЛЯНАЯ
(ХЛОРИСТОВОДОРОДНАЯ КИСЛОТА)
Acidum hydrochloricum Hydrochloric acid Chlorwasserstoff*
Acidum muriaticum Muriatic acid Salzsaure
HC1 Мол. в. 36,465
Соляная кислота представляет собой водный раствор газообразного xin6.
ристого водорода.
Приготовление
1. 25 ч. NaCl обрабатывают в реторте 45 ч. H2SO4 (уд. в. 1,84), разбавлЩ*
ной 10 ч. воды. Газ промывают, пропуская через концентрированную На^Ц
и проводят в приемник, содержащий 40 ч. воды 1. ^
2. К 2,5 об. НС1 (уд. в. 1,19) пр-иливают по. каплям концентр.ировайнЩ
H2SO4 (уд. в. 1,84). Этот метод некоторые авторы рекомендуют несколь]^.
видоизменять. Довольно большую склянку Вульфа наполняют на 1/s поваре^
ной солью, приливают чистой концентрированной НС1 так, чтобы уровень ^
был на 5 см выше слоя соли и к полученной смеси приливают из капельн($|
воронки H2SO4 (уд. в. 1,84). Выделяющийся хлористый водород с помощьй.
стеклянной трубки пропускают для осушения через концентрированную се|р
ную кислоту. /;|#
При употреблении технической НС1 рекомендуется добавить к ней немно|$
FeS04 во избежание возможного выделения хлора. 'д
Если желают получить НС1, свободный от кислорода, то смесь NaCl и Htifc
предварительно кипятят 30 мин.2, после чего весь аппарат промывают вода-
родом, затем хлористым водородом и выделяющийся газ пропускают через
нагретую до 500° трубку, содержащую вольфрам в проволоке или порошке;;
наконец, газ пропускают через промывную склянку с концентрированной
H2SO4 (В. Спицын и Каштанов)5.
3. Удобный метод получения хлористого водорода приводит Эрдман4.
Помещают куски нашатыря (хлористый аммоний NH4CI) в средний шар
аппарата Киппа и заливают аппарат концентрированной H2SO4.
4. Из технической соляной кислоты можно получить хлористый водорода
нагревая 10 ч. кислоты с 6 ч. технического хлористого кальция5.
5. По Нейману, действуя концентрированной H2SO4 на карналлит,
получают не сильную, но очень равномерную струю хлористого водорода. При
употреблении 0,5 кг карналлита газ выделяется в течение 50—70 час.
6. Для очистки технической соляной кислоты можно нагревать ее в
аппарате, изображенном на рис. 44.
В охлаждаемую склянку В наливают дестиллированную воду. При
нагревании кислоты сначала выделяется газообразный НС1, растворяющийся в воде,
затем начинает перегоняться гидрат уд. в. 1,12 (25% НС1), сгущающийся
в склянке А.
Перегонку заканчивают, когда в реторте останется 10% взятой кислоты.
Однако, приведенный метод не позволяет освободиться от примеси6 'As.
1ПоФрезениусук серной кислоте добавляют немного окислителей (К2Сгг07, КМп04»
МпОв) и газ промывают, пропуская через металлическую ртуть. Этим путем полностью
освобождаются от могущих образоваться HNOj, Cl^B, и J2.
а В этом случае уп >требляют НС1 ул. в. 1,12.
• V. S p i t z i n u. L. К a s с h t a n 0 f f, Z. anorg. Ch., 157,142 (1926); Rodebnclj, Ynte ma,
». Am. Ch. Cos., 45, 332 (1923).
4 Lehrb. d. anorg. Ch., 302.
в В конце операции может перегоняться FeCl8.
• Газообразный НС1 можно полностью освободить от прнмеси As, промывая его
пропусканием через концентрированный раствор хлористого слова, SnClg,
Кислота соляная
277
7-Я K„n£ui«£, нппи£™*ет простои автоматически действующий при-
для получения НС1. В колбу Вюрца помещают NaCl, в шариковую
йб0ронку- концентрированную Ш304. Нижний конец воронки соединен
псобым затвором, изображенным на рис. 45, А.
с затвор состоит из трубки, нижняя часть которой . представляет собой
лйИдрическос расширение, снабженное несколькими отверстиями по окруж-
Рис.44. Прибор для перегонки соляной кислоты
1
ности и запаянное снизу. В расширении затвора находится полый
стеклянный поплавок С, верхняя часть которого пришлифована к верхней части
расширения трубки. При вливании H2S04 через воронку в колбе создается
повышенное давление хлористого водорода и поплавок, всплывая,
прекращает доступ кислоты.
Отводная трубка колбы Вюрца соединена с
поглотительной стеклянной трубкой, снабженной
автоматическим затвором аналогичного устройства, но
незапаянным снизу (рис. 45, В). При
соприкосновении хлористого водорода с водой в системе
создается вакуум и поплавок С запирает трубку,
предохраняя от переброса жидкости в реакционную колбу.
В этот момент вследствие разрежения в колбе
возобновляется приток H2SO4 через затвор
(рис. 45. Л). Прибор, по указанию автора, работает
очень четко и не требует наблюдения.
8. Установка для лабораторного получения х. ч.
НС1 производительностью 3—5 кг в сутки
предложена Т. В. Полянским2 (рис. 46).
В колбе Л, нагреваемой на плитке В, получают
газообразный хлористый водород любым из
описанных выше способов, — например, из технических
НС1 и H2SO4. Газ пропускают через промывалку
С с чистой НС1, затем — через колонку D,
содержащую 50 г активированного угля ((размеры
колонки: высота 90 см, диаметр 3,5 см), и
фарфоровую трубку G со 100 г активированного угля, нагреваемую электропечью до
250—300°-.
Газ полностью очищается от Fe и As, но содержит еще некоторое
количество H2SO4. Для освобождения от последней газ пропускают через
колбу Е емкостью 5 л, содержащую 3 л дестиллированной воды и 50 г актн-
Рис. 45. Автоматические за«
творы к прибору для
получения НС1 по Сенюта
» Завод, лаб. 9, N 5—6, 648 (1940).
« ЖХП. *2, N 9в 924 (1936).
278
Кислота
вированнрго угля. Для удаления брызг служат обратный холодильник р
колонка, с активированным углем f# (аналогичная колонке D). Наконец
Рис. 46. Прибор для получения соляной кислоты по методу Полянского
поступает в серию поглотителей R с дестиллированной водой. Для ускоренад
процессов трубка L присоединяется к водоструйному насосу. Автор указт^
вает, что установка работала безо*
казно несколько месяцев без сменш
угля и получаемая кислота все1чр
являлась химически чистой соотве^
ственно требованиям ОСТ.
9. Автоматический генератор для
получения газообразного ч хлористого
водорода, работающий без присмотра
и не требующий тяги, описан В. Щ
Живовым * (рис. 47).
Исходными материалами служа!
технические концентрированные НС1 в
H2SO4. Последнюю помещают в
склянку А емкостью 10—15 л с воронкой Q
и краном В для сообщения с внешней
атмосферой. Трехгорлые склянки N и
D, емкостью 8—10 л, работают
попеременно; H2SO4 может быть
направлена в любую из склянок через
тройник Е 'и краны Т и R. Техническую
НС1 наливают в склянки N к D через
воронки Z и К, отработанная кислота
отводится через краны L и М и
трубку Я, присоединенную к 'водоструйно-
му насосу.
Выделяющийся хлористый водород
*?ерез кран О поступает в
газоотводную трубку Р и далее в осушители
с концентрированной H2SO4.
Для увеличения срока службы
аппарата пробки в тубусах склянок Л, N'
и D необхаднмо покрыть парафином. Наиболее ответственное соединение:
трубок, где легче всего разрушается резина — это С; автором предложена особая.
Рис. 47. Прибор для получения хлористого
водорода по Живову
4 Завод лаб, 4, № 13, 1504 (1935).
Кислота соляная
279
гтюукния этой детали, изображенная на рис. 48. Концы тройника Е слегка
януты, верхние концы кранов Г и R немного расширены. Трубка Е вхо-
оТ в эти расширения на 80—120 мм. Снаружи соединение закрыто куском
д11Тчуковой трубки, пропитанной предварительно парафином. Внутри соеди-
^ L образуется воздушный буфер, препятствующий соприкосно-
йе"ию H2SO4 с каучуком.
Пользование аппаратом весьма простое. Сначала в склянку А
открытом кране В и закрытом С наливают через воронку
Пп серной кислоты почти доверху. В склянки N и D через во-
онки Z я К наливают до половины высоты концентрированную
&С1; в это вРемя кРаны Tf R, L и М закрыты. Затем краны на
ронках Q, Z и К закрывают, открывают кран 7\ поворачивают в
надлежащее положение кран О и регулируют приток H2SO4 с
помощью крана С.
В нужный момент переключают кран О на другую склянку D,
а из N удаляют жидкость, открывая кран L и пуская в ход
водоструйный насос Ч
Свойства
Бесцветный газ с острым запахом, уд. в. 1,264 при 17° (по
•воздуху)2. Т. пл.— 114,7°, т. кип. — 85,2°. Критическая
температура— 51,25°, критическое давление 86 ат. Во влажном воздухе
дает туман вследствие образования мельчайших капелек соляной
кислоты. Очень хорошо растворим1 в воде с выделением тепла 5.
Растворим в спирте, в бензоле (2% при 18°), в эфире (35%
при 0°). Лед .растворяет хлористый водород, плавясь при этом.
При нагревании концентрированной НС1 сначала выделяется
газообразный хлористый водород; если же кислота сильно
разбавлена, то сперва выделяются пары воды -и концентрация
кислоты увеличивается.
В обоих случаях, когда содержание НС1 в кислоте достигает
20,3% (при 760 мм рт. ст.) перегоняется раствор постоянного состава,
кипящий при И0°.
Рис. 48.
Деталь
соединения
трубок в
аппарате Жи-
воза
Таблица 267
Растворимость хлористого водорода в воде (Deicke)
и
а,
С
0
4
8
12
МЛ ВОДЫ
астворяет
л НС1 (газ
тч а=?
525,2
497,7
480,3
471,3
>.6
д. вес пол
аемой кисл
ы
>» V Н
1,2257
1,2215
1,2185
1,2148
о содержа-
ие НС1
■о-В
45,148
44,361
43,828
43,277
s
О.
Е
14
18
18,25
23
"с?
« « •
о о,1-;
05 О^
а о ^
~ * *
,-н CU
462,4
451,2
450,7
435,0
>>о
д. вес пол
аемой кисл
ы
лун
1,2074
1,2064
1,2056
1,2014
2*
О 4,
о S
о"' Я
42,829
42,344
42,283
41,536
1 Для получения чистого хлористого водорода следует исходить из чистых НС1 и H8S04.
» Плотность жидкого НС1 при 160° К 1.267; при 163° К 1,206 [(Е. Kand a, Bulletin chemica
of the Society of Japan 12, 473 (1937); С 1, 1141 1939)1-
«Константа диссоциации НС1 при 0° по Уин-Джонсу равна 2,5-107 (W. F. W у п п е*
Johnes.J, Chem. Soq., 1940 (1064); С. II, 2349 (1930),
ЪЗО ' Кислота .
i у 1 ■ г"..:•- -у-.-" -т " • л. i -' у-■-•--^^•^•----•■' ,„
Таблица 26$
Растворимость хлористого водорода в воде
(Roscoe, Dittmar)
/°с
0
4
8
12
16
20
1 г воды
растворяет при
760 мм г НС1
0,825
0,804
0,783
0,762
0,742
0,721
/°с
<*
! 24
28
32
Э6
40
1 г воды
растворяет при
760 мм г НС1
ж
0,665
0,649
0,633
/•с
1 44
48
52
56
60
1 г воды
растворяет при
760 мм г HC1
0,618
0,603
0,589
0,575
0,561
Таблица 259
Удельныивес с оляной кислоты различной концентрации
(Lunge, Marchlewski)
возд.
стр.)
ю^ « о
H4Sc
1,000
1,005
1,010
1,01$
1,020
1,025
1,030
1,035
1,040
1,045
1,050
1,055
1,060
1,065
1,070
1,075
1,080
1,085
1,090
1,095
1,100
1,105
1,110
цусы Боме
Гра.
(Вё)
0,0
0,7
1.4
2,1
2,7
3,4
4,1
4,7
5,4
6,0
6,7
7,4
8,0
8,7
9,4
* 10,0
10,6
11,2
11,9
12,4
13,0
13,6
14,2
2 КИСЛОТЫ
ержат 2
1
© 3°
0,16
1,15
- 2,14
3,12
4,13
5,15
6,15
7,15
8,16
9,16
10,17
11,18
12,19
13,19
14,17
15,16
16,15
17,13
18,11
19,06
20,01
20,97
21.92
кислоты
ержат
С1
1,6
12
22
32
42
53
63
74
85
96
107
118
129
140
152
163
174
186
197
209
220
232
243
возд.
стр.)
1,115
1,120
1,125
1,130
1,135
1,140
1,1425
1,145
1,150
1,152
1,155
1,160
1,163
• 1,165
1,170
1,171
1,175
1,180
1,185
1,190
1,195
1 1,200
цусы Боме
2.^
14,9
15,4
16,0
16,5
17,1
17,7
18,0
18,3
18,8
19,0
19,3
19,8
20,0
20,3
20,9
21,0
21,4
22,0
22,5
23,0
23,5
24,0
2 КИСЛОТЫ
;ржат г
100
СОД€
HC1
22,86
2ЗД2
24,78
25.75
26,70
27,66
28,14
28,61
29,57
29,95
30,55
31,52
32,10
32,49
33,46
33,65
34,42
35,39
36,31
37,23
38,16
39,11
«8.
т-4 UX
255
267
279
291
302
315
321
328
340
345
353
366
373
379
391
391
404
418
430
443
456
469
P'W.'' Кислотд соляная . 281
Таблица 270
Удельный вес растворов хлористого
водорода в этиловом спирте
о/о НС1
0,00
1,27
45
0,7851
0,7907
о/о НС1
5,22
| 13,77
Л25
0,8174
0,8642
Водный раствор НС1 (соляная кислота) в продаже обычно бывает
в, 1,19, что соответствует содержанию 37% НС1 К
Таблица 271
Состав постоянно кипящих смесей Н20 -f Н С1
(Fourk, Holingsworth, W. p. Bonner, R. E. Wallace).
Давление в мм
рт. ст.
&• 800
: : 780
- 770
760
;' 750
740
730
700
680
640
600
Содержание
НСГ в о/0
20,155
20,173
20,197
20,221
20,245
20,269
20,293
20,360
20,413
20,507
20,638
Колич. г дестиллата,
содержащего 1 моль
НС1 (36,457 г)
180,621
180,407
180,193
179,979
179,766
179,551
—.
—
—.
-^
Температура
кипения
°С
110,007
—
—
108,584
—
107,859
—
106,424
105£64
103,967
102,209
Уд. вес
дестиллата
1,0955
—
—
1,095с)
—
1,0962
—
1,0966
1,0968
1,0973
1,0980
Испытание
По ОСТ НКТП 7398/552 кислота представляет собой дымящую,
прозрачную бесцветную жидкость; уд. вес ее d1^ должен быть от 1,180 до 1,190;
^Держание НС1 — от 35,39% до 37,23%.
Наибольшие количества допустимых примесей указаны в табл. 272.
1 Существует очень простой мнемонический прием для вычисления содержания HCI в
соляной кислоте по уд. весу — первые два десятичные знака умножают на два.
' Пример. Имеется кислота уд. в. 1,19; содержание НС1: 19x2 9* 3S вес. °/0. Имеется кис*
*0т» УД. в. 1,1?; содерж*ни# UQU t?x2^24 etc. »/с
282
Кислота
Допустимые примеси в НС1
Примеси
1. Нелетучий остаток • . • . .
2. Серная кислота (SO/) • * •
3. Сернистая кислота (S03*") . .
4. Хлор свободный (С12) ....
5. Тяжелые металлы Ji2S-rpynnbi
6. Железо (Fe)
7. Мышьяк (As) ........
Допустимое содержание в
в химически
чистом
0,001
0,0002
0,0006
0,0002
0,00 0
0,00 05
0,0000 5
В ЧИСТОМ Д1Я
анализа
0,002
0,0005
0,001
0,0002
0,0005
0,0001
.0,00001
0.0С4
0,002
0,001
0,0002
0,0005
0,0003
0,00002
КИСЛОТА ТЕЛЛУРОВАЯ
Acidum telluricum Telluric acid Tellursaure
№Te04-2H20 Мол. в. 229,66
Приготовление
1. По Гуибиру и Вагенкнехту1 в 30%-ный раствор чистейшего
КОН вносят при 60—70° порошок теллур истого ангидрида ТеОг, после чего
к смеси добавляют 15%-ный раствор Н2О2. При кипячении происходит
энергичное газовыделение и ТеОг растворяется; лучше всего добавлять
попеременно ТеОг и Н2О2. Если ТеОг не растворяется даже после
значительной добавки окислителя, это зависи* от недостатка щелочи, которая
добавляется тогда в 30%-ном растворе.
Если ТеОг или КОН не совершенно чисты, то раствор окрашивается
в зеленый до черного цвет и возможно даже образование осадка. Когда
весь ТеОг перейдет в раствор, прибавляют еще 10 мл 15-ной Н2Ог и
кипятят жидкость снова, после чего фильтруют через уплотненный фильтр.
Жидкость в случае необходимости упаривают, охлаждают и осторожно
прибавляют концентрированную HNOs для разложения КгТеО^ Выпадающий
незадолго до нейтрализации белый творожистый осадок КгТе04 при
дальнейшем прибавлении концентрированной HNOe (которая должна находиться
в большом избытке) снова растворяется.
Жидкость охлаждают, причем выпадает НгТе04 • 2НгО в виде красивых
кристаллов. Ее отсасывают, промывают крепкой HNOs, растворяют в
небольшом количестве горячей воды и снова осаждают концентрированной HNOs-
После трехкратного повторения такой операции препарат высушивают па
водяной бане для удаления адсорбированной HNOs и, наконец, еще раз
перекристаллизовывают из небольшого количества воды.
2. По Штауденмайеру окисляют теллур при помощи HNOe и
хромового ангидрида (см. стр. 509, Теллур, Приготовление, способ 4).
3. ПоМейеру и Мол ьденгауеру2 в колбе Эрленмейера емкостью
200 мл, снабженной пришлифованной стеклянной трубкой в 1 м длиной.
нагревают 10 г измельченного теллура с 10 мл концентрированной HNOs "
3 мл концентрированной НС1 до полного растворения. В случае
необходимости добавляют еще немного HCI. В полученную жидкость вливают
маленькими порциями раствор 9 г хлорноватой кислоты3 НСЮз. После каждого
1 О u t Ы е г, WagenHnecht, Z. anorp. Ch , 40, 261 (1904); Ргар. Ch., 126.
■Т. Meyer, H.Moldenhauer, Z. anorg. Ch., 119, 132 (1921).
3 Получается обработкой насыщенного раствора хлорноватокислого бария Ва(СЮ/а серн >и
кислотой; небольшой и бьпок одного из реактивов не влияет на чистоту получаемой i:aTi'Ov
Кислота теллуровая и уксусная
283
-ления НСЮз раствор кипятят и встряхивают, пока выделение хлора
0Р,1лабеет; только тогда прибавляют новую порцию НСЮз.
„е ^[ таком методе удается избежать образования взрывчатых окислов
Когда все количество НСЮз прибавлено и выделение хлора, в основ-
хЛ0ра' закончится, мутный желтоватый раствор НгТе04 фильтруют через
ноМ» в дестилляционную колбу емкостью 0,5—0,75 л и концентрируют
8<^ кууме на водяной бане. При этом выделяется значительное количество
0 и оставшийся раствор обесцвечивается.
хЛ°^огда будет заметно начало кристаллизации, содержимое колбы выли-
т в фарфоровую чашку и упаривают на водяной бане; охлаждением или
Заеданием концентрированной HNOs заставляют выпасть Н2Те04, отсасы-
dof и сушат кристаллы в вакууме до удаления хлора, окислов хлора и
■«слов азота. Маточные растворы при дальнейшем концентрировании дают
^незначительное количество кристаллов Н>Те04, которые обрабатывают ука-
йя&ым способом и сушат. Получившийся продукт представляет собой мелко*
^пмсталлический снежно-белый порошок. Выход почти теоретический.
Свойства
Бесцветная кристаллическая масса, уд. в. 3,05 при 25,5°. При 90° еще
устойчива, при 140° теряет 2 мол. воды, при 160° полностью переходит
в ангидрид ТеОаг. Растворима в воде (см. табл. 273).
? Таблица 273
Растворимость Н2ТеС>4 в воде
(Mylius)
ГС
10
18
30
40
о/о Н,Те04
25,29
28,90
33,36
36,38
ГС
60
80
100
ПО
о/о Н»Те04
43,67
51,55
60,84
-67
КИСЛОТА УКСУСНАЯ
Acidum aceticum Acetic acid1 Essigsaure
СНзСООН Мол. в. 60,052
Приготовление
1. Перегоняют смесь 100 г безводного ацетата натрия и 800 г
концентрированной H»S04. К дестиллату добавляют 1 г К2СГ2О7 и 5 г безводного
CHsCOONa, взбалтывают и отстоявшуюся жидкость снова перегоняют.
2. Для очистки продажной кислоты от примеси НО ее перегоняют,
добавив небольшое количество безводного CHsCOONa.
3. Если техническая кислота содержит БОг, то ее настаивают некоторое
*ре»мя с РЬОг, изредка взбалтывая, после чего перегоняют..
4. Очень концентрированная (так называемая ледяная) уксусная кислота
Получается дробным вымораживанием . продукта, содержащего некоторое
Количество воды. Кислоту охлаждают до 0°, затем выдерживают некоторое
Время п-рп ^'4° и- сливают жидкость с кристаллов безводной кислоты.
После легкого нагревания до плавления кристаллов:. операцию, шымора-
Живания повторяют еще раз.
284
Кислота
Свойства
Бесцветная жидкость уд. в. 1,0553 (при 16°) с резким характерным За -
хом. При небольшом понижении температуры затвердевает в кристаллцЧегЛа'
массу1 с т. пл. 16,635° ±0,002°. Т. кип. 118°. кУ*о
С водой, спиртом и эфиром смешивается в любых отношениях. СНзСОпи
является слабой кислотой; ее константа диссоциации К =1,82-10-5 при j^H
Таблица 274
Удельный вес водных растворов СН3СООН
н,соон
.,*>
сн.сос н
^20
•/о
сн.соон
^о
сн,соон
„20
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
0,9997
1,0012
1,0026
1,0041
1,0055
1,0069
1,0084
1,0098
1,0112
1,0126
1,0140
1,0154
1,0168
1,0181
1,0195
1,0208
1,0222
1,0235
1,0248
1,0261
1,0274
1,0287
1,0299
1,0312
1,0324
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
1 43
44
45
46
47
48
49
50
1,0336
1,0348
1,0360
1,0372
1,0383
1,0394
1,0405
1,0416
1,0426
1,0437
1,0448
1,0458
1,0468
1,0478
1,0488
1,0498
1,0607
1,0516
1,0525
1,0534
1,0543
1,0551
1,0559
1,0567
1,0575
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
1,0583
1,0590
1,0597
1,0604
1,0611
1,0618
1,0624
1,0630
1,0636
1,0642
1,0648
1,0653
1,0658
1,0663
1,0667
1,0671
1,0675
1,0679
1,0683
1,0686
1,С689
1,0691
1,0693
1,0695
1,0697
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
1,0699
1,0700
1,0700
1,0700
1,0699
1,0698
1,0695
1,0694
1,0691
1,0688
1,0684
1,0679
1,0674
1,0668
1,0*60
1,065 2
1,0643
1,0632
1,0620
1,0606
1,0589
1,0570
1,0549
1,0525
1,0197
Примечание. Своеобразная зависимость между концентрацией
уксусной кислоты и ее плотностью приводит к тому, что уд. весу
выше 1,0497 отвечают две различных концентрации кислоты. Если
требуется по уд. весу кислоты найти ее концентрацию, то ввиду
сказанного выше надо знать, крепче или слабее кислота, чем 78%-ная
(максимум плотности). Это может быть легко установлено добавлением
небольшого количества воды; если при этом уд. вес возрастает, значит
кислота была крепче 78%-ной, в противном случае она слабее 78%-ной.
Испытание
По ГОСТ 61—40 содержание СНзСООН в препарате ледяная х. «■
должно быть не менее 99,8%, в препарате х. ч. или ч. д. а.—не менее98%;
температура затвердевания препарата ледяная х. ч. должна лежать между
16,3 и 16,7°; наибольшие количества допустимых примесей указаны
в табл. 275. г
«К Пени II. Ilibtr, Bfr, 70, 3305 (1937).
Кислота урановая и 'фосфористая
285
Таблица 215
Допустимые примеси в СН8СООН
Примеси
I Нелетучий остаток . . .
0 Серная кислота (SO4'0 . .
1 Хлориды (СУ)
4 Металлы, осаждаемые H2S
i Железо (Fe)
Допустимое содержание в о/0
в ледяной хим.
чистой
0,001
0,0003
0,0002
0,0002
0,0002
в химически
чистой
0,001
0,0003
0,0002
0,0002
0,0002
в чистой для
анализа
0,005
0,0005
0,0004
0,0005
0,0005
Кроме того, препарат должен выдерживать приведенные в ОСТ
испытания на примесь веществ, окисляемых КМп04 (альдегиды, муравьиная ки-
вяота), на гомологи и на отсутствие мути при разведении.
Acidum uranicum
КИСЛОТА УРАНОВАЯ
(ГИДРАТ ОКИСИ УРАНИЛА)
Uranic acid
Uransa*ure
Uranylhydroxyd
U02(OH)s или НДК>4 Мол. в. 304,09
Приготовление
По указанию Г. и В. Ъильц1 нагревают 20 г азотнокислого уранила
[UO*(NOs)2 • €НгО] с 50 г безводного спирта на водяной бане так, чтобы
*пирт медленно испарялся, но не закипал. Через некоторое время выпадает
желтый осадок; тогда прибавляют еще спирта и выпаривают досуха. Оса-
Док промывают водой и сушат при умеренной температуре.
Свойства
Кирпично-красная масса, теряющая при нагревании воду и часть
кислорода.
КИСЛОТА ФОСФОРИСТАЯ
Acidum • phosphorosum
Phosphorous acid
Phosphorige Suure
НзРО*
Мол. в. 82,00
Приготовление
1. По Гроссгейнцу2 через нагретый до 60° треххлористый фосфор
(PCls) пропускают струю воздуха, которую затем вместе с увлеченными па-
Рами РС1з пропускают через две последовательно соединенные промывные
склянки, содержащие по 100 мл ледяной воды. При поддержании
температуры воды около 0° приблизительно через 4 часа содержимое первой склянки
застывает в кристаллическую кашицу, которую отсасывают на воронке
Бюхнера, промывают несколько раз малыми порциями ледяной воды и сушат
в вакууме. Маточные растворы и содержимое второй склянки при
повторной работе наливают в первую склянку.
1 Ue'jugsbeispiele, 224.
■Groeeheintz, Bull. aoc. chlm. France (2) 27, 433 (1887)' С. I. 933 (1918); Pr&p. СЬ. 199.
286
Кислота
2. По Гюрцигу и Це Итеру1 чистый препарат получается
3 мол. щавелевой кислоты (Н2С2О4 • 2НгО) с 1 мол. РСЬ нагревать с обо
ным холодильником, пока пенистая масса не сделается прозрачной и *т"
движной; по охлаждении она затвердевает. °*
Свойства
Бесцветные кристаллы уд. в. 1,65, расплывающиеся при лежании на во3
духе. Легко растворима в воде. Т. пл.2 74,4°. При нагревании Н3Р03 ИЛи
ее концентрированного раствора происходит распадение на РНз и Н3РО4:
4НзРОз = РНз + ЗНзР04.
КИСЛОТА ФОСФОРНАЯ
(ОРТО-ФОСФОРНАЯ КИСЛОТА)
Acidum phosphoricum Phosphoric acid Phosphorsaure
Orthophosphoric acid Orthophosphorsaure
H3PO4 Мол. в. 98,00
Приготовление
1. В объемистую реторту с тубусом помещают 240 г HNOa уд. веса 1,2
и, нагрев ее до 50—60°, прибавляют 20 г желтого фосфора.
Примечание. Уд. вес HNCh ни в коем случае не должен
превышать 1,2 во избежание опасных взрывов. В то же время при
применении гШОз уд. в. 1,18 реакция идет слишком медленно.
Нагревают реторту на песочной бане, не доводя до кипения, пока в
приемник «е начнут переходить краснобурые пары3 N02 и HNOs. Тогда
содержимое приемника вновь вливают в реторту и, когда весь фосфор
растворится, доводят до кипения. Затем содержимое реторты выпаривают в
открытой фарфоровой чашке до полного удаления бурых паров N02.
2. По Эрдману4 нагревают в реторте, снабженной приемником, 127 г
желтого фосфора с 1400 мл НМОз уд. в. 1,2. Фосфор плавится и дальнейшая
реакция идет спокойно. Когда через 10—12 час. фосфор полностью
растворится, выпаривают раствор в фарфоровой чашке до полного удаления HNOs
(проба с FeS04 и НаЗОО; температура при выпаривании не должна
превышать 188°. Также следует убедиться в отсутствии фосфористой кислоты
(НзРОз) посредством приливания к пробе раствора HgCU (НзРОз
восстанавливает HgCl2 до металлической ртути).
Затем полученную кислоту обрабатывают при нагревании сероводородом
до полного осаждения мышьяка, разбавляют водой, фильтруют, и снова
медленно выпаривают, пока погруженный в жидкость термометр не покажет
160°. Полученная жидкость представляет собой НзР04.
3. В объемистую колбу помещают красный фосфор, смоченный слегка
водой, и постепенно начинают приливать маленькими порциями HNOs. Каждый
раз происходит энергичная реакция, которой дают окончиться, прежде чем
добавлять следующую порцию кислоты. Когда, наконец, вспышки
прекратятся, колбу помещают на водяную баню, и пробуют, идет ли реакция при
добавлении новой порции HNOs. Если этого не наблюдается, раствор
нагревают до прекращения выделения окислов азота, после чего прибавляют
воды и фильтруют.
1 Hurtzig, Ceuther. Ann., Ill, 159.
* H. L. R e d f i e 1 d а. О. В. К i n g, Journ. phya. Ch. 40, 919 (1936).
■ПоЦиглеру для ускорения растворения Р в HNOa (для чего обычно требуется 1—*
дня) прибавляют небольшие количества иода (0,3—0,6 г J на 100 г Р). Иод прибавляют во время
нагревания Р с HNOe, причем реторту снимают с бани и выжидают, когда пройдет бурная
реакция.
* Хим. преп,, 41,
Кислота фосфорная 287
••Так как-обычно красный фосфор содержит значительные примеси, препа-
^*Получается невысокого качества.
Pjfc По Уотсону1 для получения НзРОд из фосфата натрия поступают
-адуюшим образом.
сД£начала кристаллический Na^HPO* • 12НЮ обезвоживают, выдерживая
^олько дней в эксикаторе2 над H2SO4. Растворяют 29 г безводной соли
* 500 мл воды и насыщают раствор при охлаждении хлористым
водородом Д° п0ЛУчения дымящейся жидкости. По окончании реакции
отфильтровывают NaCl, почти нерастворимый в насыщенном растворе НС1, и филь-
2ат выпаривают на водяной бане до получения густого сиропа. Жидкость
охлаждают, добавляют равный объем абсолютного спирта и
отфильтровывают выпавшие кристаллы NaCl. Из фильтрата медленно отгоняют спирт.
Слученную сиропообразную фосфорную кислоту испытывают на чистоту и,
^случае необходимости, очищают по способу 5. Бурый цвет препарата
изъясняется химическим взаимодействием НзР04 со спиртом в процессе
приготовления.
•: 5. Для очистки технической НзР04 от As и тяжелых металлов ее
помелют в склянку, насыщают при нагревании сероводородом, плотно
закупоривают и дают стоять 2 суток, после чего испытывают на полноту осажде-
■jbta. Если она достигнута, осадок сернистых соединений отфильтровывают
g раствор нагревают для удаления сероводорода.
■I 6. Практичный метод очистки технической фосфорной кислоты от As и РЬ'
описывают 3. А. Иофа, Е. В. Бруцкус и В. Я. Венгеров3.
Разбавляют кислоту до 70—75%, добавляют СиО (по крайней мере пятикратное
Количество относительно присутствующего As) и подвергают раствор
электролизу с платиновым анодом и медным катодом, поддерживая температуру
электролита около 50°.
\ Анодная плотность тока 0,5—1 а/см2, катодная плотность 0,008—0,01 а/см*,
Лолное выделение As- (в виде CusAs) в 1 кг кислоты требует 10—12 а-ч\
Ьатем начинает осаждаться РЬ. Через 15—20 час. электролиз заканчивают
% раствор фильтруют через шоттовскую воронку для отделения взвешенных
гдастиц (As).
'% Расход тока 0,07—0,09 квт-ч на 1 кг кислоты. Полученная таким путем
А«Р04 содержит менее 5*10-5% РЬ.
^Свойства
;Ч Бесцветная сиропообразная жидкость уд. в. 1,88 (по СТ 15—1874 с^должен
быть не менее 1,7) без запаха, с приятным кислым вкусом. Легко
растворима в воде (уд. в. водных растворов см. табл. 276) и спирте, не ядовита.
Из раствора, выпаренного до уд. в. 1,75—1,85, при охлаждении выделяется
кристаллическая кислота в виде твердых хрупких 4- или 6-сторонних
столбиков, плавящихся при 41,75°. Кристаллизация идет легче при внесении
затравки— кристаллика НзРО-i. При нагревании выше 150° фосфорная кислота,
не достигнув 100% содержания Н3РО4, начинает терять связанную воду,
переходя в Н4Р2О7 и далее в НРОз.
Фосфорная кислота стоит между сильными и слабыми кислотами. Ее
константы диссоциации 4 Кх = 1,1 • 10"2, К2 = 1 • 10"в — 7 • 10Л Кз = 3,6 • 1СИ3.
Гидрат Н3РО4 • %НгО также может быть выделен в виде мелких белых
кристалликов, весьма гигроскопичных, с т. пл. 29,35°.
Испытание
По СТ 15—-1874 содержание Л3РО4 в препарате ч. д. а. и чистом должно
"Ыть не менее 85%; наибольшие количества допустимых примесей указаны
в табл. 277.
1 W a t s о п , С. I, 809 (1892).
* Соль можно также обезводить нагреванием до 200°.
а ЖПХ 8, № б, 840 (1935).
4 Меняется * зависимости от концентрации; см. К. S Ы m а, С. I, 4161 (1939)»
288
\ i Кислот!
Препарат должен выдерживать испытания на примеси веществ, восста
вливающих КМп04, и на содержание метафосфорной кислоты. • 1а-
Таблица 2?й
Удельный вес водных растворов Н3Р04
(Schiff)
rfi3
н,ро4
Р.О,
du
Н,Р04
P.Os
du
•/о
Н.РО.
P.O.
1,0054
1,0109
1,0164
1,0220
1,0276
1,0333
1,0390
1,0449
1,0508
1,0567
1,0627
1,0688
1,0749
1,0811
1,0874
1,0937
1,1001
1,1065
1,1130
1,1196
1
2
3
4
5
6
7
8
9
10
И
12
13
14
15
16
17
18
19
20
0,726
1,452
2,178
2,904
. 3,630
4,356
5,082
5,808
6,534
7,2в0
7,986
8,712
9,438
10,164
10,890
11,616
12,342
13,068
13,794
14,520
1,1262
1,1329
1,1397
1,1465
1,1534
1,1604
1,1674
1,1745
1,1817
1,1889
1,1962
1,2036
1,2111
1,2186
1,2262
1,2338
1,2415
1,2493
1,2572
1,2651
21
22
23
24
25
26
27
23
29
30
31
32
33
34
35
36
37
38
39
40
15,246
15,972
16,698
17,424
18,150
18,876
19,602
20,328
21,054
21,780
22,506
23,232
23,958
24,684
25,410
26,136
26,862
27,588
28,341
29,040
1,2731
1,2812
1,2894
1,2976
1,3059
1,3143
1,3227
1,3313
1,3399
1,3486
1,3573
1,3661
1,3750
1,3840
1,3931
1,4022
1,4114
1,4207
1,4301
1,4395
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
29,766
30,492
31,218
31,944
32,670
33,496
34,222
34,948
35,674
36,400
37,126
37.F52
38,578
39,304
40,0 Ю
40,756
41,482
42,208
42,934
43,660
Таблица 277
Допустимые примеси в Н3Р04
Примеси
1. Хлориды (СГ)
2. Сульфаты (SO/')
3. Нитраты (N03)
4. Железо (Fe)
5. Тяжелые металлы группы H2S . . .
7. Щелочные и щелочноземельные ме-
8. Летучие кислоты (СН3СООН) . . .
Допустимое содержание в %
в чистом для
анализа
0,0002
0,002
0,00С5
0,002
0,001
0,0001
ОД
0,0015
0,002
в чистом
0,0005
0,003
0,002
0,005
0,001
0,0002
0,2
0,0015
0,005
Кислота фосфорно*атая
219
КИСЛОТА ФОСФОРНОВАТАЯ
icium hypophos-
phoricum
Hypcphosphoric acid UnterphosphorsEure
H4P206-2HjO или H2POJ.H2O Мол. в. 198,02 или 99,01
;1Готовление
jflo Зальцеру помещают палочки фосфора в стеклянные, суженые
о'у, открытые с обоих концов, трубки А (см. рис. 49). Несколько таких
v6ok помещают в воронку, которую вставляют в стеклянную банку, содер-
лдую воду. Сверху воронку накрывают колпаком так, чтобы внутрь сво-
^00 входил воздух. Прибор ставят на продолжительное время в холодное
,есто (в противном случае Р может расплавиться и вспыхнуть). Фосфор
^вольно быстро окисляется с образованием фосфорной (Н3РО4),
фосфористой (НэРОз) и фосфорноватой (ШРгОв) кислот, которые стекают через во-
1ояМ и растворяются в воде.
Для выделения Н4Р2О6 к полученной смеси сырых кислот добавляют по
каплям насыщенный раствор уксуснокислого натрия (СНзСООЫа), пока не
7WT
1.1 ^ LI
=Ч==
м , «",
Рис. 49. Прибор для получения фосфор-
новатой кислоты
Рис. 50. Прибор для
ускоренного получения фосфорноват-
той кислоты
ЦЙкратится образование осадка кислой соли (ЫагНгРгОе); последнюю очи-
рШг перекристаллизацией (1 ч. соли растворима в 45 ч. холодной и в 5 ч.
горячей воды). Очищенную соль растворяют и к раствору добавляют уксусно-
ЗДслого свинца [РЬ(СНзСОО)2]; тотчас выпадает нерастворимый» РЬзРгОв,
КОтоРый промывают водой.
- Промытый, свежеполученный белый осадок взмучивают в воде и обра-
«тывают сероводородом; для ускорения реакции полезно применять механи-
"ьское перемешивание, так как свинцовая соль быстро садится на дно, вслед-
твие чего выходит из сферы реакции.
Отфильтрованная кислая жидкость не имеет ни запаха, ни цвета и ее
Ни*110 Кипятить без разложения. Однако чтобы довести кислоту выпарива-
нем д0 сиропообразной консистенции, температуру не следует поднимать
*Не 30°; лучше всего испарять воду в вакууме над CaCls.
Ни ^анза1 приводит видоизменение метода 3 а л ь ц е р а, позволяющее
доводить операцию в течение всего 3—4 дней.
Иом 0ЧКи Ф°сФ°Ра 5~6> см длиной просверливают под водой вдоль при
ад°Щи длинной иглы; это удается без труда, если фосфор некоторое время
■.'..• * В a n z a, Z. an org. Gh. 6, 132 (18Э4); Вег. 39, 2837 (1905).
296
Кислота
лежал в теплой (40°) воде. Через палочки протягивают нитки, завязаннк
одном конце узлом, во избежание проскакивания. Свободные концы !|:
пропускают наружу через отверстия в дне конического фарфорового сЙИт°*
с дырчатым дном и выступающим торцом. Концы нитей пропускают ^'^
маленькие пробочки так, чтобы нити можно было передвигать взад и Вп %
Так подвешивают 20 палочек и закупоривают остальные отверстия ве^.
сосуда, оставляя 10—15 дырочек для подвода воздуха. Как показано,Дн*
рис. 50, покрывают этим аппаратом цилиндрический сосуд, содержащие i"a
25%-ного раствора уксуснокислого натрия (СНзСОСЖа). Нити подтягива 4
так, чтобы палочки фосфора на 1 см выступали из жидкости. ^
Когда весь фосфор до уровня жидкости окислится и дно сосуда покр0ет
слоем соли, фарфоровый конус с палочками фосфора извлекают и помеща,Ся
над другим сосудом, содержащим 1 л 25%-ного раствора СНзСОО\та; J,JJ
этом нити подтягивают так, чтобы палочки фосфора опять выступили ]1
жидкости на 1 см. Скорость окисления тем больше, чем больше число
палочек фосфора окисляется одновременно. Самовозгорания избегают, помещая
аппарат в холодное место; густые белые пары, наполняющие сосуд с самого
начала, препятствуют воспламенению.
Полученный таким путем кристаллический ЫагНгРгОв очищают и перера-
батывают, как указано в способе 1.
3. Ф. Милобецкий1 с сотрудниками указывает, что при окислении
красного фосфора раствором h в KJ около 35% Р переходит в HiP2Oo.
Процесс окисления должен проходить при рН = 5,7 — 4,6; эта кислотность
поддерживается применением буферного раствора из СНзСООН +; СНзСОСЖа.
Свойства
Бесцветные ромбические кристаллы, плавящиеся при 35°. Водный раствор
вполне постоянен на воздухе и не изменяется кислотами на холоду. При
кипячении с разбавленной H2SO4 распадается на НзРОз и НзРС>4.
КИСЛОТА ФОСФОРНОВАТИСТАЯ
Acidum hypophos- Hypophosphorous acid Hypophosphorlge Saure
phorosum
H3PO2 или Н2РО(ОН) Мол. в. 66,00
Приготовление
1. По Томсену кипятят с водою 1 ч. фосфора с 3 ч. Ва(ОН)2 Д°
растворения и полного прекращения выделения самовоспламеняющегося
фосфина (РНз). Раствор фосфорноватистокислого бария фильтруют и
освобождают от избытка Ва(ОН)2 пропусканием СОг и последующим
фильтрованием; при испарении получаются кристаллы соли.
285 г (моль) фосфорноватистобариевой соли растворяют в 5 л воды и
разлагают 98 г (1 моль) 100%-ной H2SO4, что соответствует приблизительно
101 г продажной концентрированной H2SO4, разбавленной 300—400 мл воды-
Тщательно перемешав содержимое сосуда, смесь оставляют стоять в тече
ние суток для лучшего осаждения ВаЗС>4, после чего сливают прозрачную
жидкость сифоном и кипятят в фарфоровой чашке, выпаривая до % объема
Затем продолжают выпаривание в платиновой чашке, нагреваемой на сетке
при помешивании термометром, пока температура не достигнет 105° (шариь
термометра должен быть полностью погружен в жидкость, но не касаться
дна). Жидкость фильтруют горячей и подвергают дальнейшему выпариванию
не доводя до кипения. Когда температура повысится до 110°, нагревают гА часа
!F. Milobedzkl, Roczniki Chem. 17, 620 (1937); С. 1, 3756; (1938) Sprawozdania Posied-
zen Towarzyetwa naukowego warszawskiego, Ser. Ill, 30, 107 (1937); С II, 2j07 (1938). См. также
работу Колитовской, посвященную образованию Н4РвОв пря окислении иодом продукта
гидролиза РС1, или РВга и при гидролизе P.J4.( J. Н. К о 1 i t о w s k a. Roczniki Chem. U>i 3lb
(1936); 15, 29 (1935); С II, 2689 (1936); I, 1828; Z. anorg. Ch. 230, 310 (1937).
1Н
Кислота фосфорноватистая й фосфорновольфрамовая 291
J^teneHHO повышая температуру до 130°, также избегая кипения. Теперь
йллота не содержит пузырьков воздуха и не имеет запаха РНз, но дымит
jJjeACTBHe частичного улетучивания. При осторожном нагревании температура
, JfjojceT быть поднята даже до 138° без разложения препарата. После 10-«ми-
>^гяого нагревания при 130° удаляют пламя, дают массе несколько охладиться
[/фильтруют в склянку с Притертой пробкой.
Охлаждают продукт на несколько градусов ниже нуля и, если кристал-
лизация не наступает, потирают дно стеклянной палочкой и оставляют стоять.
При правильной работе получают почти чистую НзРОг с очень незначитель-
' 0ЫМ содержанием НзРОз и НзР04.
. 2. По Мари1 фосфорноватистокислый барий (см. способ 1) осаждают из
распора спиртом и полученный продукт разлагают в кипящем растворе точно
вычисленным количеством кипящей H2SO4; фильтруют, кипятят фильтрат до
Излучения 25%-ной кислоты и удаляют дополнительно выпавший Ва304
иодом фильтрованием. Наконец, раствор концентрируют в вакууме при 80—90°
jlp постоянного веса.
X 3. Для очистки продажного раствора НзРОг, содержащего кальций,
выпаривают 'его сначала на водяной бане, прибавляют затем абсолютного спирта
я избыток сухого эфира до прекращения выпадения объемистого осадка
соли кальция. От фильтрата отгоняют спирт и эфир и остаток выпаривают
jia водяной бане, причем получают довольно чистую кислоту уд. в. 1,4625.
Свойства
■■*' Бесцветная листоватая масса уд. в. 1,49, смешивающаяся с водой во всех
отношениях. Т. пл. 17,4°. Легко расплывается на воздухе в сиропообразную
жидкость, имеющую кислую реакцию. Сильный восстановитель.
Испытание (Бендёр)
Ни от гипсовой воды, ни от растворов HgCh или (NH4)aC204 жидкость
Йе должна изменяться. После нейтрализации NH4OH и подкисления СНзСООН
жидкость при кипячении с уксуснокислым кальцием может давать лишь
слабую муть.
КИСЛОТА ФОСФОРНОВОЛЬФРАМОВАЯ
Acidum phosphowolf- Phospho-tungstic acid1 Phosphorwolframsaure
ramicunn
H3PO4I2WO311H2O*
Приготовление
. 1. E. А. Никитина2 рекомендует применять несколько измененный метод
Дрекселя.
500 мл раствора фосфорновольфрамовокислого натрия (1 :5) смешивают
в большой делительной воронке со 100 мл НС1 (уд. в. 1,19) и 200 мл эфира,
охлажденных льдом. При взбалтывании получаются 3 слоя: верхний
(эфирный), средний (водный раствор NaCl, HC1 и др.) и нижний (тяжелая
маслянистая жидкость — эфират фосфорновольфрамовой кислоты).
После отделения нижнего слоя отгоняют от него эфир на водяной бане
{огнеопасно!). Слегка восстановившуюся кислоту окисляют добавлением
2—4 мл концентрированной НЫОз и выкристаллизовывают при охлаждении.
Полученный таким путем продукт содержит натриевую соль (около 0,5%
ЫагО). Для очистки его растворяют в воде, снова экстрагируют эфиром и
повторяют описанные выше операции.
Препарат точно отвечает формуле и содержит очень небольшую примесь
Na\ Выход около 74% теоретического.
* На [P(WaO10)4lnH,O или Н7 [P(W,07)6]. n H.O;
J Marie, С. г. Ш, 1216 (1904); С. 11, 12 (1904).
• ЖОХ 7, № 20-21, 2609 (1937). |д*
292 ... Кислота f
2. Для приготовления раствора фосфорновольфрамовой кислоты, в кип*
щ;ий водный раствор продажного вольфрамовокислого натрия прибавляло*
фосфорной кислоты до кислой реакции, после охлаждения жидкость сильно
подкисляют уксусной или соляной кислотой и после суточного стоянця
фильтруют.
3. Для приготовления раствора препарата, в 'случае отсутствия НяРо4
можно рекомендовать следующий метод. К раствору 200 г вольфрамовокис-
лого натрия и 120 г фосфорнокислого натрия (NasHPCh • 12НгО) в 1 л воды,
прибавляют 100 мл концентрированной H2SO4.
Свойства
Блестящие кристаллы, легко растворимые в воде. В кислом растворе
устойчива. При кипя'чении со щелочами распадается на фосфат и вольфра-
мат. Азотнокислая закисная ртуть [Hg2(N03)2] дает с раствором
фосфорновольфрамовой кислоты желтый почти нерастворимый в воде осадок.
Содержание воды в препарате колеблется обычно между 9 и 17%.
Испытание1
Препарат ч. д. а. должен при прокаливаний терять в весе от 9 до 17%;
наибольшие количества допустимых примесей: сульфатов не более 0,1%;
солей натрия (в пересчете на NasO) не более 0,1%. Кроме того, препарат
испытывают на присутствие хлоридов, молибдена, азотной кислоты, солей
аммония и пооверяют на чувствительность к алкалоидам.
КИСЛОТА ФОСФОРНОМОЛИБДЕНОВАЯ
Acidum phosphomolyb- Phosphomolybdic acid* Phosphorrnolyfcdensaure
daenicum
НаР04-12МоОз.12Н.О Мол. в. 2041,60
Приготовление
По Шмидту2 кипятят фосфорномолибденовокислый аммоний с царской
водкой до разрушения аммиака. При испарении полученного раствора
выпадает свободная кислота, отвечающая формуле:
НзР04-12МоОз*12НоО.
Из чистой воды выпадает кислота, содержащая около 24% воды.
2. По Сингаловскому3 100 г молибденового ангидрида, свободного
от NH4*, взмучивают с 300 мл воды и 6 мл 90%-ной НзР04, медленно
нагревают и затем кипятят 3—4 часа, причем окраска жидкости становится
золотисто-желтой. На следующий день фильтруют раствор в делительную
воронку, приливают 120 мл охлажденного льдом эфира и добавляют в
несколько приемов 60 мл НС1 (уд. в. 1,19). Смесь осторожно встряхивают,
после отстаивания отделяют нижний слой, содержащий фосфорномолибдено-
вую кислоту, в колбу и нагреванием на водяной бане отгоняют эфир
(огнеопасно!). Раствор переносят в фарфоровую чашку и выпаривают до
кристаллизации.
Полученные кристаллы, содержащие небольшое количество
восстановленной кислоты, растворяют в горячей воде и окисляют 2—3 мл НГМОз уд. в.
Л ,4. Полученный раствор выпаривают в вакууме.
3. По С. Маловану4 5 г фосфорномолибденовокислого аммония
помещают в фарфоровый тигель и нагревают до красного каления дна тигля.
Когда масса станет черно-синей, добавляют 20 мл 3%-нвй НгОа и кипятят
1 Испытание хим. реактивов, 4Э6.
■ Ausfuhrl. Lthrb. d. pharra. Ch. 955; Prep. Ch. 734.
8 Соли редких и цветных металлов,
4 S. L. М а I о w a n, Z. anal. Ch. Ш, 7 (1937),
"«u',': Кислота фосфорномолибденовая и фтористоводородная 293
•j& тех пор, пока раствор не примет золотисто-желтого цвета. Затем доба-
JgjiiOT несколько капель концентрированной HNOs и выпаривают прозрачный
раствор до удаления избытка Н^СЬ.
■ Для получения раствора реактива по Файглю к полученному остатку до-
' Являют воды до объема 100 мл.
4. О приготовлении раствора фосфорномолибденовой кислоты см. Натрий
ф о с ф Q$ н о м о л и б д е н о в о к1й с л ы й, стр. 403.
Двойства
Желтые, блестящие триклинические призмы, легко растворимые в воде.
'Хранить препарат следует в темных банках с притертыми пробками.
А
испытание1
■Ш-. Препарат ч. д. а. должен выдерживать следующие испытания.
.:?•;:;■''.1. Растворимость. 1 г препарата должен полностью растворяться в 10 мл
Щолм.
%>■• 2. Тяжелые металлы. 10%-ный раствор препарата при добавлении
избытка NH4OH и (NH^aS может приобрести лишь слегка зеленоватую
^раску.
£Й' 3. Кальций (Са*'). 10%-ный раствор препарата не должен изменяться при
Добавлении избытка NFUOH и (NH-OsCsO-i.
{;£ 4. Хлориды (СГ). 5%-<ный раствор препарата, подкисленный HNOs, может
^давать лишь слабую опалесценцию при добавлении AgNOs.
К- 5. Сульфаты (SCV). 5%-ный раствор препарата при добавлении НС1 и
|:;'н ВаСЬ не должен изменяться.
I- 6. Аммонийные соли (NH4 *). При добавлении к 1 г кислоты избытка NaOH
;; и нагревании не должны выделяться пары, окрашивающие красную
лакмусовую бумажную в синий цвет.
;: кислота фтористоводородная (плавиковая кислота)
;: Acidum hydrofluoricum Hydrofluoric acid Fluorwasserstoffsaure
HF Мол. в. 20,01
Приготовление
1. Обезвоженный кислый фтористый калий (полученный просушиванием
в токе Н2 продажного KHF2) нагревают в медной или платиновой реторте на
голом огне. Первые 10—20 мл дестиллата отбрасывают, остальное количество
собирают в медный сосуд, охлажденный снегом. Для очистки2 полученный
препарат еще раз перегоняют на водяной бане, нагретой до 3!5—39°.
2. Для получения водной кислоты нагревают 1 ч. плавикового шпата
(свободного от S1O2) в платиновой или свинцовой (свинец не должен быть
пропаян оловом) реторте с 2 ч. концентрированной H2SO4 и улавливают
выделяющуюся HF водой в платиновом или свинцовом приемнике,
охлаждаемом льдом3. Во избежание образования трудно удаляемой из реторты
спекшейся массы можно плавиковый шпат смешать предварительно с равной
частью гипса и затем уже действовать серной кислотой. Горло реторты
только немного погружают в воду приемника; еще лучше в широкогорлыи
приемник поставить платиновую чашку с водой, которая быстро растворяет
переходящую HF.
Вместо плавикового шпата можно употреблять криолит; в? этом случае на
1 ч. криолита берут 2^ ч. H2SQ4.
1 Испытание хим. реактивов, 407
■ I. S 1 m о n s, J. Am. chem. Soc. 46, 218Э (1924).
8 Получение безводном HP в серебряной реторте подробно описано В. Кланом (W.
Klatt), Z. anon?. Ch. 222, 236 (1935). Классический метод получения HF перего:?кой КНГ, в
платиновой реторте см. О. Ru f t. и VV. Plato, Ber. 37, 675 (1901).
294
Кислота
«М>
. . j^
3. Для очистки водной кислоты Бендер1 рекомендует следующий
тод. Так как плавиковый шпат обычно содержит ЗЮг, то вместе с HF Пеп
ходит и фтористый кремний (SiF4), выделяющийся уже из холодного р^
твора в виде пузырьков. Поэтому удобнее удалять его, давая жидкое*"
стоять несколько дней до перегонки. Можно также прибавлять по каплям раси
твора фтористого калия (KF) и сливать чистую жидкость с осадка кремн!Г
фтористого калия (foSiFe).
Для удаления As, Fe, Pb, Ca, H2SO4, ЗОги FUSiFe разбавляют кислоту
до уд. в. 1,15—1,2, пропускают избыток сероводорода и добавляют углекис-
лого калия (КгСОз) больше, чем это требуется для нейтрализации имею!
щихся2 H2SO4 и H2SiFe. Наконец, по Гамильтону3 для удаления избытка
FhS прибавляют Ag^COs или AgF или Ag20 и перегоняют кислоту из свинцовой
реторты с платиновым или золотым шлемом и холодильником, нагревая на
масляной бане при 160—240°.
4. И. В. Тананаев4 рекомендует хранить безводную HF в серебряном
сосуде, охлаждаемом льдом. Даже при длительном хранении препарат
свободен от Ag.
Свойства
Безводная кислота представляет собой бесцветную легкоподвижную, сильно
гигроскопическую жидкость уд. в. 0,9879 при 12,8° (dj^jj), кипящую5 при
19,54°. Т. пл. — 92,3°. На воздухе дымит. Низкая точка кипения может быть
причиной взрыва, если кислоту сохраняют в платиновых бутылках с
завинчивающимися пробками, залитыми парафином.
Пары HF чрезвычайно едки и ядовиты, вызывают боль под ногтями.
Небольшая капля даже разбавленной кислоты вызывает на коже белое, сильно
болезненное гноящееся пятно. При попадании безводной кислоты на руки
необходимо тотчас же погрузить обожженное место на некоторое время в
3%-ный NH4OH или в 10%-ный раствор (NH^COs. При работе с HF лучше
всего применять резиновые перчатки
Водный раствор HF не замерзает еще при —30°; уже небольшая примесь
HF сильно понижает точку замерзания воды.
При перегонке разбавленной кислоты крепость ее постепенно повышается,
пока, наконец, при 120° не начинает перегоняться гидрат уд. в. 1,15, что
отвечает 35,37% HF. Остаток содержит 36—38% HF,
Таблица 278
Удельный вес водных растворов HF
•/о HF
2
4
6
8
10
12
14
16
18
.20
1,005
1,012
1,021
1,028
1,036
1,043
1,050
1,057
1,064
о/о HF
20
22
24
26
28
30
32
34
d4,
1,070
1,077
1,084
1,090
1,096
1,102
1,107
1,114
% HF
36
38
40
42
44
46
48
50
.20
d4
1,118
1,123
1,128
1,134
1^39
1,144
1,150
1,155
1 Неорг. преп. 154.
* Содержание HaS04 и H2SiF6 определяется предварительным анализом.
■ На m 1 11 о n.Chem. News 63, 252 (1889).
4 ЖПХ И, 2,214 (1938).
5 W. К I a t t, Z. anorg. Ch. 222, 236 (1935J.
Кислота хлорная
"295
для
хранения водной
парафина или
кислоты применяют
парафинированного
клянных парафинированных сосудах мало надежно,
сосуды из платины, каучука,
стекла. Однако хранение HF
ытание
" по техническим условиям НКХП 289—41 содержание HF в препарате
i а. должно быть не менее 40% и в чистом — не менее 35%; наибольшие
*■ ячества допустимых примесей указаны в табл. 279; препарат должен вы-
|: -ивать испытание на сернистую кислоту (разбавленная HF не должна
1 каплю 0,1 Л/ раствора Лг).
еР#йВ
$сцвечивать
Ж
ш
Таблица 279
Допустимые примеси в HF
Примеси
1. Нелетучий остаток в виде сульфатов
3 Сульфаты (S04")
4. Кремнефтористоводородная кислота
(HoSiFfi)
Допустимое содержание в °/
в чистом для
анализа
0,005
0,005
0,005
0,25
0,001
0,0005
в чистом
0,01
0,01
0,02
1,0
0,001
0,001
КИСЛОТА ХЛОРНАЯ
Acidum perchloricum
Acidum hyperchloricum
Perchloric acid
HC104
Мол. в. 100,465
Ueberchlorsaure
Perchlorsaure
JB'p иготовление1
1. Для приготовления 70%-ного раствора чистой НСЮ* Уилярд2
предлагает следующий способ. В колбу емкостью 2 л помещают 500 г хлорно-
кислого аммония (NH4CIO4), прибавляют 600 мл воды, 410 г 68—70%-ной
HNOs и нагревают до энергичного кипения. Тогда через капельную воронку
£ длинной трубкой приливают сначала быстро, под конец медленнее в
течение 25 мин. 105 г НС1 (уд. в. 1,19), разбавленной 400—500 мл воды. Во
•Время приливания жидкость должна энергично кипеть. Кипячение производят
* течение часа, изредка пополняя испаряющуюся воду. Затем смесь
переливают в большую фарфоровую чашку и быстро нагревают до появления
тяжелых белых паров НС104.
Получившаяся кислота свободна от ЫШ", но обычно содержит еще
некоторое количество HNOs. Для удаления последней снова прибавляют
небольшое количество НС1 а нагревают до появления дыма. Когда жидкость
начинает сильно дымить, она отвечает двухводному гидрату НС104 • 2НгО. Этот
гйдрат может быть получен также при перегонке раствора HCICU в виде
Постоянно кипящей фракции.
Для получения абсолютно чистого препарата кислоту еще раз перегоняют,
лучше всего в вакууме при 200 мм давления. Для этого служит литровая
Колба с боковой трубкой и пришлифованным холодильником (см. рис. 51).
трубка между колбой и холодильником не должна быть короче 40 см, чтобы
обеспечить достаточное воздушное охлаждение (в противном случае может
1 См. также работу A. S I mon, Z. anorg. Ch. 239, 330 (193В).
1 Н. Н. W i 1 1 а г d I, Am Ch; Soc; 34 1480 Г1912).
296
Ки$лота
лопнуть форштосс холодильника). Горло колбы оттянуто так, чтобы м
было укрепить в нем термометр при помощи кусочка резиновой трубки ре°Ч
Рис. ь\, прибор для перегонки хлорной кислоту
вследствие сильного разъедания парами НС104 должна находиться не ближе
20 см от отводной трубки. Нижний * кон*ец форштосса холодильника соеди-
чя10т с приемником при помощи
резиновой пробки. В колбу, чтобы
избежать толчков, помещают
несколько стеклянных капилляров.
В течение часа в таком приборе
можно легко перегонять до 500 г
кислоты. Дестиллат содержит
небольшое количество СЬ и СЬО,
которые удаляются легким
нагреванием и просасыванаем воздуха.
Полученная кислота не взрывчата и
не ядовита.
2. Для получения безводной
кислоты Форлендер и
Шиллинг1 в колбе емкостью 300 мл
перегоняют 50 г чистого хлорнокис-
лого калия, КС104, с 150—175 мл
концентрированной 96 — 97,5% -ной
H2SO4, получаемой смешением чистой
93—94%-нои H2SO4 с 80%-ным
серным ангидридом (S03); возможная в
последнем примесь SCb не вредит.
Колбу, снабженную
75-сантиметровой коленчатой трубкой (см. рис. 52),
вертикальное колено которой несет
на себе кожух холодильника, нагре-
Рис. 52. Аппарат для получения безводной НСЮ< ВаЮТ На масляной бане. МеЖДУ
А * охлаждаемым смесью льда и соли
приемником и насосом помещена
ТРУа*а наполненная TBfPAUM КОН или NaOH. Части аппарата между
колоой я трубкой с КОН соединяют пробками из асбеста с жидким
Ннсшосу
Vorlander, Schillin
S> Ann. 310, 3G9 (10O)); Am. Chcni. J. 23, 44 (19ЭО).
v-
Кислота хлорная
297
Стеклом \, все остальные части — резиновыми трубками и пробка-ми. Пробки
[||з- асбеста с жидким стеклом обеспечивают плотное соединение даж*е при
частом употреблении для вакуум-перегонки.
Перегонная колба сверху закрыта асбестовой пробкой, снабженной
запаянной с одной стороны или оттянутой в капилляр трубкой, внутри которой
находится термометр.
Когда температура бани достигает 135—145° при 50—-70 мм давления, в
колбе образуются пары НС104 и безводная кислота начинает переходить в
приемник. Повышают температуру бани постепенно до 160—190°, причем баня
должна быть опущена так, чтобы поверхность масла была ниже уровня
кислоты в колбе. Через \Уч—2 часа получают 22—24 г сырой безводной HCICU
■окрашенной примесью двуокиси хлора (СЮг) в желтоватый цвет и
содержащей около 1% H2SO4 и, иногда, следы хлора.
Кислоту для очистки тотчас дважды дестиллируют из помещенной на во-
:дяную баню колбы с капилляром и термометром. При просасывании сухого
воздуха даже при переливании желтоватая кислота обесцвечивается. При
нагревании на водяной бане до 45—65° и 50—70 мм давления в приемник
переходит безводная кислота, в то время как Нг304 и некоторое количество
гидрата НС104«НЮ остаются в колбе. Выход 18—21 г чистой бесцветной
кислоты.
Невозможно избежать постепенного разложения безводной кислоты даже
при хранении на холоду и в темноте; поэтому ее разбавляют водой, для
чего, сильно охладив, выливают на лед (из дестиллированной воды) или
сливают с концентрированным раствором НС104.
3. Для получения водного раствора НС104 по Каспар и2 растворяют
хлорнокислый калий (КСЮ-i) в 7-кратном количестве теплой воды, прибавляют
избыток кремнефтористоводородной кислоты (HtSiFe), нагревают в течение
часа и, прибавив еще FhSiFe, кипятят с водой. При охлаждении выпадает
KsSiFe и некоторое количество неизменившегося КС104. Разбавляют раствор
равным объемом воды и, прибавив небольшой избыток ВаСЬ, удаляют
содержащуюся в растворе примесь ffiSiFe. Прозрачный отстоявшийся раствор
выпаривают до появления белых паров НСЮ4, разбавляют после охлаждения
водой, прибавляют по каплям H2SO4 до прекращения выпадания Ва304 и
после стояния в течение нескольких дней, фильтруют. Получается довольно
чистый раствор НСЮ4.
4. Пригодный для аналитических целей раствор HClCh получают по
Крейдеру следующим образом. В колбе постепенно нагревают 100—300 г
хлорноватокислого натрия (ЫаСЮз) так, чтобы началось медленное выделение
кислорода. Нагревание продолжают, не повышая температуры, до полного
затвердевания массы (1%—2 часа).
Полученный сплав (смесь NaCl и NaC104) растворяют в воде, прибавляют
достаточное количество НС1 для разложения имеющегося еще NaClCb и
выпаривают досуха, часто помешивая. Сухую массу растворяют3 и
обрабатывают в высоком сосуде концентрированной НС1; при этом твердый NaCl
осаждается в течение нескольких минут. Раствор, содержащий НО и НС104,
фильтруют через тигель Гуча. Осадок промывают 1—2 раза
концентрированной НС1 и выпаривают жидкость на водяной бане до появления тяжелых
белых паров НСЮ4.
5. По методу, разработанному в Институте чистых реактивов * 250 г NaC10<
обрабатывают 400 мл НС1 (уд. в. 1,19) и оставляют смесь на несколько ча-
1 Последние приготовляются так: для того чтобы соединить, например, трубку
холодильника с приемником, тонкую полоску асбестового картона шириной около 25 мм при помоТци
сиропообразного жидкого стекла прилепляют к трубке и наматывают вокруг до требуемое
толщины. Конец полоски и всю пробку смачивают еще небольшим количество* жидкого стекла.
Через 10—20 мин. сушения пробка готова к употреблению.
• С а • р а г i, Z. ang>w. Ch. 6, 68 (1893).
» Если в исходных продуктах содержится калий, то рекомендуется массу тщательно
промыть 97°[о-ным спиртом, не расгворяющим солей кали!; спиртовый раствор перегоняют в колбе
до начала выпадения солей и затем, перелив в фарфоровую чашку, упаривают досуха.
* Реакт. и преп. 356. Для получеиия NaCIO* по методике ИРЕА профильтрованный рас
твор 200 г NaClOj в 400 мл воды подвергают электролизу в течение нескольких часов при анод
ной плотности тока 0,7—0.8 а/см*. Температуру электролита поддерживают между 10—16°.
Когда 92—96»/о NaCIO, перейдет в NaClOj раство» выпаривают в фарфоровой чашке досуха.
сов, изредка перемешивая ее. Раствор отсасывают через пористый стеклянный
фильтр от осадка NaCl и промывают осадок 30—40 мл НС1. Фильтрат и гьр0.
мывные воды нагревают в фарфоровой чашке до появления густых белых ца.
ров НС104, тщательно оберегая содержимое чашки от попадания органических
веществ — бумаги, дерева и пр. (опасность взрыва!).
Раствор НС104 охлаждают, отфильтровывают небольшое количество нераз-
ложившегося NaClCh и получают таким образом около 250—270 г сырой
кислоты уд. в. 1,6.
Для очистки кислоту перегоняют при 15—20 мм рт. ст. в колбе из. стекла
«пирекс», причем применение корковых или резиновых пробок недопустимо;
все соединения, включая капилляры и термометр, устраиваются на шлифах.
При 48—64° перегоняется веда со следами хлорной кислоты; при 107—111°
отгоняется 70—72% HCICU. Если не отбирать водяную фракцию отдельно, то
получают около 220—230 г 65%-ной HCICk
6. По К. Ньюмену и Ф. Мэзерсу1 для получения 2N раствора НС104
подвергают электролизу насыщенный раствор ЫаСЮз при возможно низкой
температуре и плотности тока 3,5 а/дм2. Катод железный, анод платиновый.
Катодной диафрагмой служит асбестовая бумага, анодной — пористый
фарфор.
Выход НС104 по току составляет около 60%, по NaClOs 80%.
Свойства
Безводная кислота — сильно дымящая, очень гигроскопичная, подвижная
жидкость уд. в. 1,77. Т. пл. — 112°, т. кип. 39°. На коже производит
болезненные и опасные раны. При соприкосновении с горючими веществами (уголь,
бумага, дерево и г. д.) сильно взрывает. При нагревании, также часто при
хранении (даже в темноте) разлагается со взрывом. Вследствие сильной
взрывчатости кислоты и причиняемых ею болезненных, трудно заживающих
ран, обращаться с кислотой необходимо с большой осторожностью. Работа
с водными растворами1 кислоты совершенно безопасна.
При смешении с малым количеством воды кислота затвердевает в кои-
сталлическую кашицу, образуя гидрат НСЮ4 • НгО (т. пл. 50°). С большим
количеством воды кислота соединяется с шипением, образуя маслообразную
жидкость уд. в. 1,65—'1,85, кипящую около 200°. Раствор — без цвета и
запаха, сильно^ окрашивает лакмусовую бумажку в красный цвет. Вкус очень
разбавленной кислоты сильно и приятно кислый.
При перегонке разбавленных' растворов хлорной кислоты сначала
испаряется вода, затем при 203° перегоняется раствор, содержащий около 72%
НС104. Такая кислота дымит на воздухе, но вполне устойчива. Безводную
кислоту нельзя перегонять под атмосферным давлением; она при 50°
окрашивается в тёмнокрасный цвет, при дальнейшем нагревании темнеет и
становится непрозрачной. При 75° начинается разложение; температура долгое
время держится на 92°, причем выделяются густые белые пары и желтый
газ. Одновременно в приемнике собирается несколько капель тёмнокрасной
жидкости, содержащей 94,77%-ную НС104, которая внезапно взрывает с
громадной силой.
Остающаяся в колбе жидкость содержит 87,75%-ную НС104, затвеодеваю-
щую в белые кристаллы.
Испытание2
По стандарту Американского химического общества реактивная хлорная
кислота должна отвечать следующим требованиям: содержание НСЮ4 не
менее 59%.
Наибольшие количества допустимых примесей приведены в табл. 281.
»1уКС. И 603 М939а " *' F* С" ^ а * h е Г s' 75, 27, 341 (193Э); Gransatlons of the electrochemioa,
» Регкт. и поеп., ;;1$.
4* СО JO ж«—
п>х&я
«< w ш о Q
г* о о £ g
о- н ТЗ 2 Л
-в- о я rt н
0» Ovfcf *<
Н £2 Е ' «С
s -5 s
~ :г ^- о
^Sen- «
а " О О
• и
ОЛ П
о • oj
« « &а
п> ' Я
ооо о
bob-
ОООСл
СЛ >й> •—
^ р сл
2 -к8*
г g g s|
0 n E и
d° 2 • »
ТЗ SS **
о • о» •
м # ft» • z
2*. е . w
2 ^
« С? И
ОО О
§8 8
ОО 4*.
tOui
/
[
Я
TJ
2
О
К
2*=*
о о
Ё 9
S О
Л (V
Я
X
К
А
■
Я*3
О о
У* я
0 » Я
s 5
Я О
л л
1
*=1
О
о
н
^
я
43
S
S
я
со
ГС
Q
О
*•
■
М
^
?>
^
45
^
to
ОО
СЛОСЛОСЛОСЛОСЛОСЛОСЛОС
is^s \
.-.oBS'S'S
no сл о сл о> сл
\
ООС0 0000^ООСЛ4>С0С0КЭ^-ОО<000-<105СЪСЛ4*»С0ГО«—'»--
ЮЮЮЮЮЮЮГОГОЮЮЮЮЮЮи-^ —>и 1—н-и— ^- .— — ,— ^*
-^1005СЛСЛ^^СОСОЮЮ>—•—©ОСОСООООО*^->4СлСлСлСЛ4*.4Ь.
ОСЛО СЛОСЛОСЛОСЛОСЛОСЛОСЛОСЛОСЛОСЛОСЛОСЛО
СО0ЭСОСаЭ0эСОСаЭ<^<^СОСОС0СОЮЮЮЮКЭЮГОКЭЮ1ОЮ1ОЮКЭ
j-«J pi Р> $S* ** ** $Я Ы^Э ЪЭ ~ ~ OfOjOjXjXj-J 05 Сл^СЛ 4*- 4^ COJvJ Ю ^~
о>сл о<4^соТиЪо"ю-^^--,сл о "*►**. Ъо'гоЪ» о^Т^ЪоТо'сл'соЬо'сл coboo*
000)СОСОСЛОСЛ04^00»-^СЛОС^05СЛ^1СОЧ|^ОСЛСОЮ^
J^^COCoCOCoCOCOCOCOCOCoCoCOCOCOCOW^COCOCOlOlOtOhOrO
OOcD«COOOOvi«sjcna5Cncn^4^COCOtOlO^-«-OOtf)cDOOOO^
ОЮСЛОСЛОСЛОСЛО СЛ О СЛОСЛОСЛОСЛОСЛОСЛОСЛОСЛ
^4^4^4^4^4^^^^4^4^^4^^^4^^^^>^4^^COO:CoCoCO
СО О 00J»J<l;<l'j^^p5,Cn Сл_4^ 4* ОО COKOJ-OJO^j— 0>jDjO COJ3© 00^
"сл "to bo bo "lo 45*. o'oi^—'^ТоЪо со oot^ То^ осл с> сл^—Ъэ^—'"сп^Ъэ
ООСОО^СО(ОСЛ»-0)^05Н^ЛСОСОЧСОМО)ООСООООООО
СлСлСЛСлСлСЛСлСлСЛ4^4^^4^4^^Лк4^^-»^-4*-4^Д^4*-4^4^^.4^
ЛОЗСОЮЮ»-и- О СО СО 00 00 -Ч -О СЛ <У> СЛ СЛ 4*> Д*. СО СО Ю Ю >— »—
ОСЛОСЛОСЛОСЛОСЛОСЛОСЛОСЛОСЛОСЛОСЛОСЛОСЛО
СЛСЛСЛСЛСЛСЛСЛСЛСЛСЛСЛСЛСЛСЛСЛСЛСЛСЛСЛСЛСЛСЛСЛСЛСЛСЛСЛ
<D}OjOp0O0CO^4 ^^Р^СЛ СЛ СЛ ^^^^СОСОЮЬОЮ^-,^- ООО
о ою'соЪГ»— ооТи о оЬо'сосл^-bo ел Т— Vj со о сл^-'^'со'со сл"^*
4*. зэ 00 •— t^^H-J^OcDKJCaaioocOO'—'>—*-•—к >-ц«- ^ ^- н- н-©
о> ст> сл с>Ъ5Ъ>Ъэ о/елЪ>"сл о "сл о*сл а> сл'сл'сл'сл сл'сп слЬл сл"сл сл
^4^СЛО^СЛСЛ4^^СОСОЮ10^-^ООСООООС»*^^СЛСЛСЛСЛЛ.
СлОСл©СлОСл©СлОСлоСл©слОСлОСлОСлОСл©СлОСл
*^JO0»C75C75O05C7JC750iCTiC7i05OOO Oi О О Oi О* О* О 05 С7> Оэ Сл
О СО^О СО 00 OO^j-J -^ $Яр*р>&&^^*ьОЭОэС0ЬЭЪЭ~?-~ О О
Т— Vj 1u оoj"гоbo сл^-»~-<Гсо ©Ъ^юоо сл1-*Vj"со"ооТо оо сл1—Va""^.
СЛ^ОЬ04^СлО^С0ООь-С*>ОС»ОЮ^^ОС0С75С0К)СЛ00и-
я
о
о
X
о
о
я
Г)
о
я
Q
о
За
а
с
a
<■)
ш
о
о
о
о
W
о
о
S
о
2
. о
N
S
300
Acidurn chloricum
Кислота
КИСЛОТА ХЛОРНОВАТАЯ
Chloric acid
НСЮа Мол. в. 84,465
Chlor
saure
Приготовление1
1. Берн ар2 предлагает следующий простой способ получения раствоп
НСЮз. Растворяют 800 г хлорноватокислого бария Ва(С10з)г в 1700 мл ^
стилированной воды, фильтруют раствор в колбу емкостью Зли прибавляв*'4
при охлаждении малыми порциями 343 мл концентрированной H2SO4, поел!
чего фильтруют. Таким путем получают 1750 мл почти бесцветного раствора
НСГОз, содержащего только незначительное количество H2SO4.
Свойства
В чистом виде кислота не известна. ВодныЯ раствор можно сгущать в
вакууме до уд. в. '1,282 при 14.20, (что соответствует формуле НСЮз • 7Нг0
с 40,1% НСЮз). При дальнейшем иснареиии в пустоте над H2GO4 раствор
постепенно разлагается с образованием зеленого газа; НС104 при этом не
образуется. При достижении концентрации 51,86% НСЮз (соответствующей
гидрату НСЮз • 4^ НзО) внезапно начинается энергичное выделение газа.
Водные растворы кислоты довольно стойки на свету, но разлагаются при
назревании выше 40°. При —20° растворы НСЮз различных концентраций
загустевают, не выделяя льда и не затвердевая. Крепкая кислота имеет
желтоватый цвет, разбавленная же бесцветна. Запах концентрированной
кислоты, особенно при нагревании, острый, напоминающий HNOs.
Разбавленная кислота на холоду запаха не имеет. Растворы НСЮз имеют кислый,
вяжущий вкус
Таблица 282
Удельный вес водных растворов НСЮ3
°;0 нею.
1
2
4
6
8
w18
1,0044
1,0103
1,0222
1,0344
1,0468
о/в НСЮ,
IS
14
16
w18
1,0594
1,0723
1,0856
1,0S91
о'о НСЮ,
18
:о
22
24
л18
1,1130
1,1273
1,1419
1,1568
КИСЛОТА ХЛОРНОВАТИСТАЯ
Acidurn hypochlorosum Hypochlorous acid Unterchlorige Saure
НСЮ Мол. в. 52,465
Приготовление
1. По Балару в наполненную хлором склянку вносят избыток
суспендированной в воде окиси ртути (HgO), отфильтровывают желто-коричневук-
хлорокись ртути и очищают полученный водный раствор НСЮ перегонкой
в вакууме, причем в первой фракции переходит вода.
1 2 "обучении НСЮ, из КСЮа и H,SiFe см. Ann. Chim Phys- (2), 45, 203 (1830); М u n г о е,
Chem. Met. Eng. 23, 88 (1920).
• Bernard, Z.f chem. Apparalenkunde 1, 81 (1903).
Кислота хлорноватая, хлорноватистая и хлорсульфоновая
301
a-fto Ге-Люссаку к продажной хлорной извести приба!ляют
понежу сильно разбавленную HNCb в количестве, достаточном для насыще-
**° почти половины СаО, и смесь подвергают перегонке.
'^избыток НМОз здесь вреден, так рак получается свободная НС1, которая
ЯС10 реагирует с образованием хлора и воды:
Hci + некого + сь.
л^той реакции можно избежать, применяя вместо НГМОз какую-либо ела»
'* кислоту — лучше всего борную.
3. Пригодный для многих целей раствор НСЮ получается по Волю
, Швейцеру1 следующим образом. В защищенный от яркого дневного
света раствор 50 г двууглекислого натрия (NaHCOs) в 600 мл воды при
охлаждении льдом и постоянном перемешивании пропускают сильную струю
хлора, пока проба жидкости при нагревании с ВаС1г не перестанет давать
осадок ВаСОз.
4. По С. Реформатскому пропускают хлор в колбу емкостью
500мл, охлаждаемую снегом и содержащую 1 объем окиси ртути (HgO) и
5 объемов воды. Колба закрыта пробкой с двумя отверстиями: через одно
проходит почти до дна трубка, подводящая газ, через другое — короткая
трубка, отводящая избыток хлора. Содержимое колбы встряхивают только
перед концом реакции, которая считается законченной, как только исчезнет
HgO'. Полученный водный раствор отгоняют от HgCb. Для удаления
растворенного хлора через дестиллат пропускают струю СОг.
Очень простой способ получения НСЮ описывают Уриссон и
М. Кастнер2. Подвергают обработке хлором суспензию HgO в четырех -
хЛористом углероде. Образовавшийся раствор CUO в CCU дает с водой
тую НСЮ высокой концентрации, очень устойчивую и, по указанию авто-
|, практически свободную от Hg' ', CU и СГ.
Свойства
Концентрированный видный раствор кислоты окрашен в желтый цвет,
разбавленный — бесцветен. Жидкость обладает чрезвычайно едким запахом,
таким же, как у ангидрида (С1гО). Вкус вяжущий, не кислый. В темноте
разлагается медленно и тем скорее, чем концентрированнее раствор и * выше
температура. На прямом солнечном свету происходит быстрое разложение
(до указанию некоторых авторов — в несколько секунд). Разбавленные
растворы относительно постоянны.
НСЮ относится к числу слабых кислот; по последним данным константа
ассоциации НСЮ составляет 3,16• 10"8 при 20° (Е. А. Шилов8).
КИСЛОТА ХЛОРСУЛЬФОНОВАЯ (ХЛОРИСТЫЙ СУЛЬФОНИЛ)
Chlorosulfonic acid Chlorsulfonsaure
Monochlorschwefelsaure
SC2OHCI Мол. в. 116,53
[риготовление
1. По Бендеру4 нагревают в закрытой колбе 3 ч. возможно крепкой
H2SO4 и вносят малыми порциями 2 ч. пятихлористого фосфора (РСЬ). По
прекращении выделения хлористого водорода перегоняют.
2. По Бекуртсу и Отто6 в реторту, снабженную хорошо
охлаждаемым приемником, помещают свежесплавленную пиросерную кислоту
(продажная дымящая серная кислота с 38—42% 50а) и пропускают сухой хло*
> A. W о h I, H S с h w e i t г е г, Вег. 40, S2 (1907). л _ ппп _
. ■ J. О и г is s о п, М. К a s U е г, Cong. chlm. ind. Nancy 18,11, 983 (1938); G. II»€030 (1939)
" E. A. S h i 1 0 v, J. Am. Chera. Soc. 80, 490 (1928). С 1, 1731 (1939). *
S1 4 Приг. неорг. иреп. 255.
i Б H. Beckurts, R. О tt о, Вег. И, 205, 1878; также Sanger ц Ri egel, Z. апогв. Ch.76, 7»
1912;.
302
Кислотй
ристый водород до полного насыщения. Затем отгоняют образовавшую
хлорсульфоновую кислоту и дестиллат, обычно слабоокрашенный, очища£ч
перегонкой, причем почти ©се количество переходит при температур
149—151°. Выход почти теоретический. ур^
Л. Н. Голдырев рекомендует при перегонке препарата вообще р,
пользоваться пробками, применяя подвешенный термометр, и длинную отвод
ную трубку колбы Вюрца непосредственно вставлять в холодильник ЛибИХа
Горлышко колбы прикрывают в этом случае листочком асбеста.
3. По Михаэли с у целесообразно пропускать хлор в охлаждаемую
водой колбу, содержащую 15 ч. концентрированной H2SO4, и затем медленно
по каплям, прибавить туда еще 7 ч. треххлористого фосфора (РСЬ). Пр0;
дукт реакции отгоняют и перегоняют еще раз с небольшим количеством
H2SO4 (Б е н д е р) \
Свойства
Бесцветная, дымящая на воздухе жидкость уд. в. 1,79, кипящая при
155—156°. С водой реагирует чрезвычайно бурно, образуя H2SO4 и HQ.
И с пытание
По техническим условиям НКХП (385—41) содержание хлорсульфоновой
кислоты не нормируется, но указывается на этикетке; сумма свободных
SOs + HC1 + H2SO4 не должна превышать 4%; препарат должен
выдерживать испытания на хлориды серы и сернистый ангидрид.
КИСЛОТА ЦИАНИСТОВОДОРОДНАЯ (СИНИЛЬНАЯ КИСЛОТА)
Acidum Hydrocyanic acid Cyanwasserstoffsaure
hydrocyanicum Prussic acid
HCN Мол. в. 27,026
Приготовление
Чрезвычайно ядовита! Работа с HCN требует величайшей осторожности!
Вдыхание паров №CN или попадание ее в случайную царапину на коже
грозит смертью!
По Вёлеру в реторте под хорошей тягой нагревают смесь 500 г
грубоизмельченного железистосинеродистого калия K4Fe(CN)e, 350 г
концентрированной H2SO4 и 700 мл воды. Горло реторты соединено с
холодильником и затем с двумя склянками Вульфа, погруженными в лед. Пары,
выходящие из последней склянки, пропускают в холодную воду.
Получаемый в склянках дестиллат почти совершенно чист. Если желают получить
абсолютно безводную HCN, то первую склянку Вульфа наполняют до
половины СаСЬ. В конце работы ее погружают в теплую воду, причем
безводная HCN перегоняется во вторую склянку.
Свойства
Очень летучая, легкоподвижная бесцветная жидкость уд. в. 0,691. Т. пл.
—13°, t. кип. +26,5°.
Хранить ее следует в разведенном состоянии; водные растворы прочнее
при добавлении на каждые 100 мл 1 капли слабой минеральной кислоты.
HCN смешивается во всех отношениях с водой, спиртом и эфиром.
1 Приг. неорг. преп. 255.
Кислота цианистоводородная и щавелевая 303
Таэшца 283
Удельный вес водных растворов HCN
% HCN
2
4
6
8
10
15
20
25
w18
d4
0,996
0,993
0,990
0,986
0,982
0,972
0,958
0,943
% HCN
30
35
40
45
50
55
60
65
rf4
0,925
0,908
0,8°2
0,876
0,^60
0,844
0.826
0,809
°/, HCN
70
75
80
85
90
95
100
18
4
0,7< 2
0,775
! 0,758
, 0,741
; 0,724
0,707
0,691
КИСЛОТА ЩАВЕЛЕВАЯ
Acidum oxalicum Oxalic acid Oxalsaure
Hl.C204-2H20 Мол. в. 126,068
Приготовление
1. В колбе, снабженной обратным холодильником, кипятят 500 г сахара
с 2,5 л HNOs уд. в. 1,25 до прекращения выделения окислов азота. Затем
отгоняют HNOs и выделившуюся щавелевую кислоту
перекристаллизовывают из горячее воды до удаления NOe'.
2. По Винклеру для получения чистой Н2С2О4 • 2НгО продажный продукт
дважды перекристаллизовывают из равного по весу количества кипящей НС1
(уд. в. 1,07), причем охлаждение ведут при помешивании для получения мел*
ких кристаллов. Затем перекристаллизовывают препарат из горячей воды
До удаления С1', сушат между листами фильтровальной бумаги и, наконец,
выдерживают до постоянного веса в эксикаторе над кристаллическим
хлористым кальцием (СаС1г • 6НгО).
Генигшмидт и Шлее1 при перекристаллизации щавелевой кислоты
Для особо ответственных целей (определение атомных весов) применяли
платиновую посуду.
3. Для получения безводной Н2С2О4 кристаллическую кислоту растирают
в тонкий порошок и высушивают в течение 2 час. при 98—99°, после чего
подвергают возгонке при 140° в вакууме, создаваемом водоструйным насосом.
Сублимат нагревают еще раз при 100° и охлаждают в вакуум-эксикаторе
над кристаллическим СаСЬ • 6НгО.
Свойства
Бесцветные призматические кристаллы, теряющие кристаллизационную
в°Ду при хранении над концентрированной H2SO4 или при нагревании выше
^0°, около 100° происходит полное обезвоживание. Хорошо растворима
jj воде (см. табл. 284) и спирте (23,7: 100), труднее в эфире (1,47: 100).
Водные растворы при хранении постепенно разлагаются по реакциям
Н2С2О4 = НСООН + С02
2НСООН = СО* + СО + Но + Н20.
1 О. Н б n i g с с h ш i d t и R. S с li 1 е е, Z. anorg. Ch. 22, 134 (1934),
№
Клейстер
Таблица 2%4
Растворимость Н2С204
в воде
Таблица 2<Jj
Удельный вес водных
растворов Н2С404
'/• с
0
10
20
30
40
% НЛО,
3,42
5,73
8,69
12,5
17,7
/° с
50
60
70
80
90
% нас»о4
23,9
30,7
37,9
45,8
54,5
% н,е2о4
1
2
3
4
5
*17'»
1,0035
1,0082
1,0132
1,0181
1,0231
% н,с*о4
6
7
8
9
^
1,0278
1,0326
1,0375
1,0424
Нагревание, а также наличие некоторых примесей (Fe*'\ Мп' ') ускоряет
разложение. Безводная Н2С2О4, полученная возгонкой, представляет собой
весьма гигроскопичные игольчатые кристаллы, плавящиеся при 189° и
разлагающиеся выше этой температуры на СО, СОг и НЮ. Безводная
щавелевая кислота хорошо растворима в эфире (23,6: 100).
Щавелевая кислота является довольно сильной; ее термодинамические
константы диссоциации, определенные при 25° Г. Пэртоном и Р. Д ж и б-
бонсом», tfi = 0,05 (рК =1,30), К* = 5,012• 10~5 (рК = 4,300).
Испытание
Согласно ОСТ В КС 4104 препарат х. чх
99,8% НзС204»2Н20; наибольшие количества
в табл. 286.
Кроме того, препарат должен выдерживать указанное
ние на органические примеси.
должен содержать не менее
допустимых примесей указаны
в ОСТ и с п ы т а-
Таблица 28?
Допустимые примеси в Н2С204*2Н20
Примеси
Допустимое содержание в %
в химически
чистой
в чистой
для анализа
в чистой
1# Нерастворимые в воде вещества .
2. Нелетучий остаток
3. Хлориды (СУ)
4. Сульфаты (SCV)
5. Тяжелые металлы
6. Железо (Fe)
7. Азот (N) общий из нитратов и
солей аммония
0,002
0,01
0,001
0,001
0,0002
0,0002
0,001
0,005
0,01
0,002
0,002
0,0005
0,0005
0,002
0,01
0,05
0,005
0,005
0,001
0,002
0,005
КЛЕЙСТЕР ИОДОКРАХМАЛЬНЫЙ
Jodide starch solution Jodstarkelosung
Приготовление
По Мюллеру для приготовления очень устойчивого иодокрахмального
клейстера поступают следующим образом. В эрленмейеровскую колбу
емкостью 1 л отвешивают 5 г крахмала, прибавляют 30 мл воды,
взбалтывают и приливают пипеткой 25 мл 30%-ного растюра КОН. Колбу 1збалты-
» Н. N. Р а г t о п, R. С. О i ь b о ж s, Trans. Faraday Sac. 35, 512 (1930); С. II, 1857 (1939).
Клейстер иодокрахмальный, иодцинккрахмальный и крахмальный 305
пока внутри не образуется однородная желатинозная масса, после
'добавляют 50 мл воды и 2 г йодистого калия (KJ). Смесь кипятят,
^ем получается вполне прозрачный раствор. По охлаждении его разба-
!1Сот водой до 1 Л и Фильтруют. ;Д"
КЛЕЙСТЕР ИОДЦИНККРАХМАЛЬНЫЙ
Zinc-jodide starch solution Jodzinkstarke losung
[кой готовление
T^> крахмала, 20 г хлористого цинка (ZnCh) и 100 мл воды кипятят до
«одного растворения крахмала,. время от времени пополняя испаряющуюся
воДУ- Прибавляют 2 г йодистого цинка (ZnJ2), разводят раствор водой до
I л и фильтруют. Фильтрат сохраняют в склянке с притертой про(щрй в
темном месте, так как от действия света реактив быстро портится.
КЛЕЙСТЕР КРАХМАЛЬНЫЙ
Starch solution Starkelosung
■v
готовление
1. 1 г крахмала растирают с небольшим количеством дестиллированной
воды и полученную жидкую кашицу тонкой струей вливают в стакан с 100 мл
кипящей воды. Кипятят, дают отстояться и сливают (или фильтруют) с
могущего образоваться хлопьевидного осадка.
1ля придания большей прочности полученному быстро портящемуся
растру некоторые. авторы (Мор, Мюллер) рекомендуют добавлять немного
салициловой кислоты или КОН.
2. Б. Нейман1 рекомендует следующий метод приготовления раствора
крахмала, пригодного в качестве индикатора при иодометрии. 5 г
картофельного крахмала растирают с 10 г красной HgJ2 и небольшим количеством
воды до получения жидкой пасты, которую при постоянном
встряхивании вливают в 1 л кипящей воды. Кипятят еще 2 мин., дают охладиться
и сливают прозрачный раствор с осадка. На 200 мл титруемого раствора
берут 2 мл крахмального клейстера.
КОБАЛЬТ АЗОТНОКИСЛЫЙ (НИТРАТ КОБАЛЬТА [2])
Cobaltum nitricum Cobalt nitrate Cobaltnitrat
Со(Ж>з)2-6Н;>0 Мол. в. 291,05
Приготовление
ВЦ. Свежеполученный Со(ОН)2 или СоСОз (приготовленные из х. ч.
сернокислого кобальта) растворяют при нагревании в HNOs уд. в. 1,15—1,20.
Раствор выпаривают до образования легкой пленки и кристаллизуют в
холодном месте. Выпадающие кристаллы отсасывают и высушивают на стекле
в теплом месте. Выход 95% теоретического.
\ 2. Тригидрат Со(ЫОз)2 • ЗШО получают нагреванием шестиводной соли
при 60°.
? 3. Для получения кобальтовых солей, абсолютно не содержащих никеля,
по указанию Ф. Файгля2 к водному раствору соли кобальта приливают
концентрированный раствор KCN (яд\) до образования прозрачного желто-зеленого
Раствора. Затем добавляют 3%-ной Н2О2 и кипятят несколько минут, до
Получения чисто желтого раство»ра КзСо(СЫ)в. Если выпадает осадок, его
Отфильтровывают, после чего раствор упаривают до получения вязкой жид-
Кости, добавляют твердого диметилглиоксима и при усиленном
перемешивали приливают 40%-ный формалин. Через час жидкость фильтруют и рас-
1 J. chem. Educ. 14. 133 (1937); С. TI, 442 (1937).
* Ф. Ф а й г л ь, Капельный анализ, ГХТИ М.—Л. 1933, стр. 294.
с06
Кобальт
досуха. 0|
ста.
)и баи,'
твор, не содержащий никеля, выпаривают на песочной бане
ток слабо прокаливают до полного почернения.
Дав массе немного охладиться, ее смешивают с теплой водой в
добавляют концентрированной НС1 и нагревают 1—2 часа на водяной
после чего разбавляют водой и фильтруют. Осадок после прокаливай^'
снова подвергают такой же обработке соляной кислотой. Из фильтра 0?}
ждают щелочью Со(ОН)г, который тщательно промывают и прокаливаю5
Полученная окись может быть легко переведена в любую соль кобальт*'
4. Наиболее удобным методом получения кобальтовых солей без hhW
является метод, предложенный Морганом и С ми-сом1. 80 г хло;.й'Ст^
кобальта и 55 г NbhQ растворяют в 140 мл воды и добавляют к раствору
210 мл ЫНЮН уд. в. 0,91. После перемешивания в течение часа раствор нагре
вают, причем выпадают тёмнокрасные кристаллы хлоропентаминокобальт*
хлорида [Со(г4Нз)5С1]СЬ; никель полностью остается в растворе. Из филь.
трата, после добавления 75 мл ЫНЮН и 10 мл Н2О2, перемешивания в
течение часа и кипячения получают еще некоторое количество кристаллов. Общий
выход 77-78 г (92% теор.).
Полученный [Со(ЫНз)йС1]СЬ смешивают с
воды, доставляют NaOH до сильно щелочной
удаления NHs, пополняя испаряющуюся воду.
Таблица 287
равным по весу количеством
реакции и кипятят до полного
Черный осадок Со(ОН)з
отделяют и промывают
декантацией горячей водой до
исчезновения щелочной реакции
промывных вод.
Приготовленный таким
путем Со(ОН)з непосредственно
(или после восстановления до
металла) может быть
применен для получения чистых
солей кобальта.
Свойства
Красные призмы или (при
кристаллизации из
пересыщенных растворов) моноклинические таблички уд. в. 1,83. Во влажном
воздухе расплывается. При хранении над H*S04 теряет одну молекулу воды. При
Таблица 288
Удельный вес водных растворов Co(N03)2
Растворимость
t° с
18
55
о/0 Co(N03)«
49,7
61,7
Co(N03)2
t° с
70
91
в воде
о/0 Co(N03),
64,9
77,2
о/0 Co(N08)a
1
2
3
4
5
6
7
8
9
10
11
12
13
14
4?i5
1,0092
1,0184
1,0276
1,0368
1,0462
1,0551
1,0640
1,0729
1,0818
1,0906
1,1000
1,1094
1,1188
1,1282
о/о Cc(N03)a
15
16
17
18
19
20
21
22
23
24
25
[ 26
27
$*
1,1378
1,1490
1,1602
1,1714
1,1826
1,1936
1,2056
1,2176
1,2296
1,2416
1/2538
1,2668
1,2798
о/о Co(N03)2
28 '
29
30
31
32
33
34
35
i 36
37
38
39
40
d17,5
1,2928
1,3058
1,3190
1,3331
1,3472
1,3613
1,3754
1,3896
1,4049
1,4202
1,4355
1,450£
1,4662
J J. Soc. Chetn. Ind. 43, 131.
Кобальт азотнокислый. Кобальта гидрат закиси 307
нагревании плавится около 55°, затем теряет воду и, наконец, распадается
с образованием закиси кобальта СоО.
Й слытание
Наибольшие количества примесей, допустимых по ОСТ НКТП 6415/323,
^азанц в табл. 289.
Таблица 289
Допустимые примеси в Co(N03)2• 6Н20
Примеси
Допустимое содержание в %
в химически
чистом
в чистом для
анализа
1. Нерастворимые в воде вещества
2. Хлориды (СГ)
3. Сульфаты .(S(W
4. Железо (Fe)
5. Никель (Ni)
6. Цинк (Zn)
7. Металлы, осаждаемые Н£ ....
8. Щелочные и щелочноземельные
металлы (в виде сульфатов) . . .
0,002
0,С02
0,005
0,0005
0.02
0,02
0,005
0.1
0,005
0,005
0,01
0,0005
0,05
0,05
0,02
0,2
0,01
0,01
0,02
0,001
0,5
ОД
0,05
0,5
Примечание. Препарат х. ч. или ч. д. а., содержащий не более
0,005°/0 Ni, носит название .кобальт азотнокислый без никеля*.
КОБАЛЬТА ГИДРАТ ЗАКИСИ (ГИДРООКИСЬ КОБАЛЬТА[2])
Cobalt hydroxide
Cobaltum oxydulatum
hydricum
Cobaltohydroxyd
Cobalthydroxydul
Co(OH)2
Мол. в. 92,96
Приготовление
1. Горячий раствор 100 г х. ч. сернокислого кобальта (CoS04»7H20)
осаждают крепким раствором 42 г NaOH. Осаждение для лучшего отстаивания
рекомендуется проводить в высоком цилиндре. Образующаяся ©начале основная
соль сине-фиолетового цвета быстро переходит при нагревании в розовато-
красный Со(ОН)г. Отстоявшийся осадок промывают несколько раз декантацией,
затем отсасывают на бюхнеровской воронке и промывают водой до
нейтральной реакции промывной воды К
2. Для получения кристаллического препарата по Шультену2
нагребают раствор 10 г хлористого кобальта (СоСЬ-бНгО) в 60 мл воды с 250 г
КОН (в колбе иенского стекла, причем воздух вытесняют током светильного
газа. Образующийся гидрат растворяется в щелочи с синей окраской. После
суточного стояния (в атмосфере светильного газа) выделившиеся кристаллы
пРй« легком помешивании промывают небольшим количеством воды и таким
пУгем отделяют от аморфных примесей.
Свойства
Аморфный гидрат Со(ОН)2«хНгО представляет собой розово-красный
порошок; препарат часто получается коричневым вследствие незначительного
'УспенекийиПутохин, Изв. ИРЕА, 1, 250 (1928).
1 S с h ul t e n, С. г. 109, 266 (1889); РгЗр. Ch. 658.
SOS-
Кобальт
окисления. Кристаллический гидрат — темнофиолетовый порошок, состоящий
из микроскопических полихроматических призм. Устойчив та воздухе, в то вдеМя
как аморфная форма медленно окисляется, переходя в гидрат окиси Lo(OH),.
Уд. в. 3,6t). Нерастворим в воде, растворяется в кислотах и в горячих
растворах щелочей.
КОБАЛЬТ •СЕРНОКИСЛЫЙ (СУЛЬФАТ КОБАЛЬТА[2])
Cobaltum sulfuricum Cobalt sulfate Cobaltosulfat
CoS04-7HiO Мол. в. 28U1
Приготовление .
1. По 'А. Успенскому и Н. Путохину1 для очистки продажного
C0SO4 • 7Н2О, содержащего примесь Fe, поступают следующим образом.
100 г соли растворяют в возможно , малом количестве воды, прибавляют
несколько мл 10%-ной H2SO4 и нагревают до кипения; к кипящему раствору
постепенно прибавляют 3 мл концентрированной НЫОз Для окисления
железа 2.
Дальнейшую обработку можно вести двумя способами* .
А. После окисления железа приступают .«. его отделению, основанному на
том, что при действии №гСОз железо выпадает из раствора в первую очередь.
Кислый (от добавленной кислоты) раствор соли кобальта разбавляют водой
с расчетом получить 25%-ный раствор и нагревают до кипения. К кипящему
раствору прибавляют понемногу 60—70%-ный раствор соды. Выделение
железа происходит полностью прежде, чем наступит нейтрализация и начнется
выделение СоСОз. Прибавление №гСОз рекомендуется вести до тех пор, пока
не начнет появляться характерный розово-фиолетовый осадок СоСОз. Раствор
вместе с осадком нагревают некоторое время при помешивании, отсасывают
осадок и испытывают фильтрат на содержание железа [проба раствора не
должна выделять синего осадка от прибавления K4Fe(CN)e].
В случае присутствия Fe повторяют снова обработку содой. Если же Fe
не обнаружено, раствор* нагревают и осаждают весь кобальт прибавлением
раствора №гСОз (из расчета: на 100 г первоначально взятой сол-и 38 г
Ыа2СОз или 100 г 1Ма2СОз • ЮНгО). Выпавший осадок после
непродолжительного нагревания отсасывают на воронке Бюхнера и промывают горячей
водой, пока проба фильтрата не перестанет давать реакцию, на S(V (с ВаС1г).
Промытый осадок растворяют при нагревании на водяной бане в * 10—
15%-ной х. ч. H2SO4, причем прибавляют последнюю понемногу, избегая ее
избытка. Сернокислотный раствор выпаривают до 2/3 объема на водяной бане
и фильтруют. Фильтрат продолжают упаривать до появления на поверхности
пленки соли, после чего оставляют раствор стоять при комнатной
температуре. Выпавшие кристаллы отсасывают и сушат на листе фильтровальной
бумаги. Выход 50—60 г.
При упаривании маточного раствора можно получить еще 25—30 г
препарата.
Б. Из V10 части раствора осаждают смесь СоСОз и Fe(OH)s, добавляя
раствор соды (около 4 г ЫагСОз); выпавший осадок отсасывают на бюхнеров-
ской воронке и хорошо промывают горячей водой, пока проба фильтрата
не перестанет мутнеть от добавления ВаС1г (отсутствие SCV).
Главную массу кислого раствора (полученного после окисления железа)
нагревают до кипения и прибавляют к нему промытый осадок [СоСОз -f-
4- Fe(OH)s], взмученный в небольшом количестве воды. Прибавлением осадка
1 Изв. И PEA, 1, 244 (1922).
1 Окисление можно вести инач?; растворив соль в воде, прибавляют немного HCI,
нагревают почти до кипения и прибавчяют несколько кристалликов 1<С103 Запах хлора
свидетельствует о законченном окислении. Можно, наконец, окислять, пропуская непосредственно хлор
в раствор соли.
Кобальт сернокислый
309
д0Лжна быть достигнута полная нейтрализация раствора; рекомендуется
ввести даже некоторый избыток СоСОз. Железо, полностью выпадающее при
этом ,в виде Fe(OH)3, отфильтровывают. Фильтрат совершенно свободен от
ре, но содержит некоторое количество ионов N(V, оставшихся от окисления
железа азотной кислотой. Для удаления этой примеси раствор выпаривают
досуха и остаток прокаливают па асбестовой сетке. При этом Со(ЫОз)з
распадается по реакции, схематично выражаемой уравнением:
2Co(NOs)> = 2СоО ;*- 4N02 + О2.
Остаток от прокаливания обрабатывают при нагревании на водяной бане
20%-ной H2SO4, выпаривают досуха и снова прокаливают, после чего
смачивают серной кислотой и растворяют в воде при нагревании. Раствор
фильтруют, выпаривают до небольшого объема и кристаллизуют. Выход 9о%
теоретического.
2. Для получения безводного CoSCU по Клоббу бросают некоторое
количество водной соли (C0SO4 • 7Н2О) в расплавленный, сернокислый
аммоний (NH4)2S04 и затем удаляют воду и (NH4)2$C>4 осторожным
прокаливанием.
3. О получении CoS04»7H*0, не содержащего никеля, см. стр. 306.,
Свойства
Устойчивые на воздухе розово-красные моноклинические кристаллы
уд. в. 1,924, изоморфные с сернокислым . железом (FeSCh • 7НЮ). Т. пл. 96—
98°. При нагревании теряет воду и мутнеет, но даже в калильном жару
не разлагается. Легко растворим в воде и метиловом спирте. Безводный
C0SO4 — красный кристаллический порошок, очень мало растворимый в воде К
Таблица 290
Растворимость CoS04• 7Н20 вводе
/• с
0
5
10
% CoS04
20,35
" 21,88
23,37
t° с
15
20
25
% CoS04
24,83
26,58
28,16
t* С
30
35
40
% CoS04
29,76
31,44
32,81
Таблица 291
Растворимость CoS04 • 7Н20 в метиловом спирте
Вес. о/0
спирта
100
100
г с
18-19
15
100 г спирта
растворяют г
соли
54,5
50,9
Вес. %
.спирта
90,5
50
г с
3-4
3-4
100 г спирта
растворяют г
соли
13,3
1.3
1 О низших кристаллогидратах CoS04 см. R. Rohmer, С." г. 206, 1573 (1Г38>
310
Кобальт
И спытание
По ОСТ НКТП 16416-40 содержание CoS04-7H20 должно быть не меНе
99,5% Для препарата ч. д. а. и 98,5% для чистого; наибольшие количества
допустимых примесей указаны в табл. 292.
Таблица 292
Допустимые примеси в Со804-7Н20
Примеси
Допустимое содержание в п/п
в чистом для
анализа
0,005
0,002
0,005
0,02
0,01
0,005
в чистом
0,01
0,005
0,01
од
од
0,02
1. Нерастворимые в воде вещества ......
2. Хлориды (СГ)
3. Железо (Fe)
4. LUfflK(Zn)
5. Никель (Ni)
6. Металлы, осаждаемые H2S
7. Щелочные и щелочноземельные металлы
(в виде сульфатов)
Примечание. Препарат носит название
никеля менее 0,005%.
0,1
0,5
,без никеля", если содержит
КОБАЛЬТ УГЛЕКИСЛЫЙ (КАРБОНАТ КОБАЛЬТА[2])
Cobaltum carbonicum Cobalt carbonate Cobaltcarbonat
СоСОз • 6H20
Мол. в. 227,05
СоСОз
Мол. в. 118,95
Приготовление
Получить СоСОз осаждением содой нельзя вследствие образования
основной соли. По А. Успенскому и Н. Путохину1 крепкий водный раствор
100 г Co(NOs)2 • 6НЮ в плотно закупоренном сосуде осаждают насыщенным
СОг раствором 35 г двууглекислого натрия (NaHCOs).
Выпавший осадок отсасывают на бюхнеровской воронке, промывают до
нейтральной реакции промывных вод и высушивают. Выход близок к
теоретическому. ,
Свойства
Свежеосажденный СоСОз • 6НгО представляет собой красновато-розовый
порошок, постепенно переходящий в микроскопические призмы. Кроме шести-
водного углекислого кобальта известны еще СоСОз «Р/гНгО и
безводный СоСОз.
Испытание2
Препарат ч. д. а. должен содержать растворимых в воде веществ не более
0,1%, щелочей и щелочных солей не более 0,1%, никеля не более 0,1%.
Кроме того, препарат должен выдерживать испытание на хлориды (после
выпаривания с HNOs и растворения остатка в воде), сульфаты, цинк и тяжелые
металлы (после выпаривания с НС1 досуха на водяной бане и растворения
остатка в воде).
1 Изв. ИРЕА, 1, 253 (1923).
» Испытание хим. реактивов, 215.
Кобальт углекислый и хлористый
311
Cobaltu
КОБАЛЬТ ХЛОРИСТЫЙ (ХЛОРИД КОБАЛЬТА[2])
m chloratum Cobalt chloride Cobaltchlorid1
СоСЬ-бНзО Мол. в. 237,95
приготовление
1 Свеже приготовленный гидрат закиси кобальта Со(ОН)г растворяют
W°/-ной НС1 при нагревании на водяной бане до насыщения; затем добавляют
^■^ хлорной воды и Со(ОН)а для окисления железа. Раствор кипятят,
fbTpoBbisaioT и, выпарив до уд. в. 1,4, оставляют для кристаллизации \
Тетрагидрат СоСЬ • 4НгО получают нагреванием СоСЬ«оНгО до 116°.
3. Дйгидрат СоСЬ • 2НЮ получают нагреванием СоСЬ^НгО до 121°.
|* Моногидрат СоСЬ'НоО можно получить, выпаривая при 100° досуха
паствор СоСЬ • 6НЮ в абсолютном спирте.
б. Безводный СоСЬ получают непосредственно соединением хлора с
кобальтовой пылью или высушиванием водной соли при 140°. При этом в не*
шаяйтельном количестве образуется нерастворимая в воде основная соль.
6. Получение безводного СоСЬ по Д. И. Р я б ч и к о в у и В. М. Шуль-
и а н2 проводится в наклонно поставленной трубчатой печи. В трубку
помещают СоСЬ • 6НЮ и обезвоживают его в токе сухого хлористого водорЮда при
температуре не выше 250°. Выделяющаяся вода, растворяющая часть СоСЬ
(и НС1), стекает в приемник. Препарат чрезвычайно гигроскопичен; перед
извлечением его из трубки последнюю необходимо прогреть по всей длине
Для удаления следов влаги. Целесообразно хранить препарат в
эвакуированном сосуде.
7. О получении СоСЬ, не содержащего никеля, см. стр. 306,.
Свойства
а) СоСЬ • 6Н2О — красные моноклинические кристаллы уд. в. 1,84, стойкие
при комнатной температуре и плавящиеся значительно ниже 100°. При 30—
35° начинает выветриваться и при этом мутнеет. После 4 ч-ас. нагревания
при 45—52° почти нацело переходит в СоСЬ • 4ШО.
При нагревании соль синеет, при охлаждении принимает прежний цвет.
СоСЬ-бШО очень легко растворим в воде, растворим в спирте (насыщенный
спйртовый раствор имеет уд. в. 0,792 » содержит 23,7% СоСЬ; в проходящем
свете раствор синего цвета, в отраженнохМ — черного).
б) СоСЬ — светлосиний порошок уд. е. 3,35, жадно поглощающий воду
с образованием устойчивого при обыкновенной, температуре СоСЬ*6НгО;
Таблица 293
Растворимость СоС12 в воде
*° с
0
Ю
17,5.
20
30
% СоС1а
30,2
31,0
33,82 *
34,9
36,1
/° с
40
43
47,5
50
55
% CoCl,
39,4
42,7*
44,63 *
48,3
47,64 *
t° С
60
75
80
99
100
% CoCf*
48,4
49,53 *
49,0
51,48*
50,7
Примечание. Данные таблицы, помеченные *, определена
Бенратом3.
1 А. Успенский и П. Путохин, Изв. ИРЕА, 1, 250 (1923).
• ЖПХ 7, №7, 1162 (1934).
• Н. Benrath, Z. anorg. Ch. 220, 144 (1934).
312
Литий
реакция сопровождается разогреванием. Т. пл. 722°. Способен возгоцЯТь
особенно в струе хлора. Растворим в спирте с синей окраской; раеТвоп?'
также в ацетоне, бензонитриле, пиридине, хинолине и пиперидине. р ^
Таблица 29t
Удельный вес водных растворов СоС12
в/о СоС?,
1
2
4
6
8
л18
d4
1,008
1,017
1,036
1,055
1,075
% СоС'я
10
12
14
16
.18
4
1,095
1,116
1,137
1,159
% CoCl!
18
20
W13
1,182
1,205
Испытание
По ОСТ 10901-40 содержание СоСЬ • 6Н2О в препарате ч. д. а. не менее
99,0%, в препарате чистом — не менее 97,0%; наибольшие количества
допустимых примесей указаны в табл. 295.
Таблица 295
Допустимые примеси в СсС12• Ш^О
Примеси
Допустимое содержание в %
в чистом ддя
анализа
1.
2.
3.
4.
5.
6.
7.
Нерастворимые в воде вещества . . . .
Сульфаты (S04")
Железо (Fe)
Металлы, осаждаемые H2S
Цинк (Zn)
Никель (Ni) . . . .
Щелочные и щелочноземельные металлы
(в виде сульфатов)
0,01
0,005
0,0005
0,005
0,02
0,05
ОД
0,03
0,02
0,002
0,01
0,06
0,2
0,3
Примечание: Препарат, имеющий марку ,без никеля", должен содер
жать не более 0,002% Ni.
КРАХМАЛ
См. клейстер крахмальный
ЛИТИЙ ХЛОРИСТЫЙ (ХЛОРИД ЛИТИЯ)
Lithium chloratum
Приготовление
LiCl
Lithium chloride
Мол. в. 42,397
Lithiumchlorid
1. Осторожно растворяют 23 г углекислого лития (Li2COs) в 50 г чистой
НС1 (уд. в. -1,19) и, выпарив до половины объема, охлаждают. При этом
выпадает кристаллический LiCl • 2НгО. Из сильно щелочного раствора,
получающегося при большом избытке Li2COa, выкристаллизовывается
кристаллогидрат LiCbH20. V
Литий хлористый
313
2. Для получения безводного LiCl по Ванино1 водный раствор
выпаривают досуха в струе хлористого водорода. Применение IICI необходимо по-
лМУ» чт0» с одной стороны, дигидрат отщепляет воду только при довольно
«Осокой температуре, а, с другой стороны, при красном калении начинается
Гидролитическое расщепление. Можно также раствор LiCl смешать с iNHiCl,
выпарить досуха и из остатка удалить NH4CI прокаливанием.
Свойства
бесцветные октаэдры, уд. в. 2,068, плавящиеся при 606° и после того
доказывающие в водном растворе слабо-щелочную реакцию. Т. кип. 1382°.
.jfa воздухе расплывается (даже сильнее, чем СаСЬ). Растворим в воде (см.
ядбл. 296) и абсолютном спирте.
Таблица 296
Растворимость LiCl в воде
г с
0
12,5
20
25
<\'о LICI
38,9
40,5
44,7
44.9
/° с
40
65
80
100.5
°'о LiCl
47,4
51,0
53,1
56,7
t° с
120
МО
160
°;0 LiCl
57,4
58 2
59,0
Удельный вес водных растворов LiCl
Таблица 297
о/о LiCl
1
2
4
6
8
10
Л
1,0041
1.0099 |
1,0215
1,0330
1,0444
1,0559
о/о LiCl
12
14
16
18
20
24
л18
d 4
1,0675
1,0792
1,0910
1,1029 !
1,1150
1,1399
о/о LiCl
28
32 1
36
1 40
42
,/13
d 4
1,1658
1,1947
1,224
l.S-54
I 1.269
1
Испытание
В препарате, в зависимости от степени его чистоты, допускаются
следующие наибольшие количества примесей (табл. 298).
Таблица 298
Допустимые примеси в LiCl
Примеси
1. Сульфаты (SO/) . .
2. Щелочи
3. Железо (Fe) . . . .
4. Тяжелые металлы . .
Допустимое содержание в 0,'о
) химически
чистом
0,005
0,005
0,002
0,000
чистом для
анализа
0,01
0,01
0,005
0,000
0,05
0,05
0,01
0X0
1 PraD. Ch. 300.
314
Магний
МАГНЕЗИАЛЬНАЯ СМЕСЬ
Magnesia mixture Magnesiamixtur
Приготовление
1. Растворяют 100 г кристаллического хлористого магния (MgCh • 6H,.Q)
и 125 г NH4C1 в 500 мл дестиллироваиной воды И приливают к полученному
раствору 500 мл ЫНЮН (уд. в. 0,928).
2. По другому рецепту 550 г MgCl* • 6Н2О и 1050 г NH4C1 растворяют
в воде, приливают к раствору 3,5 л ЫНЮН (уд. в. 0,91) и разбавляют водой
до 10 л.
МАГНИЯ ОКИСЬ (ЖЖЕНАЯ МАГНЕЗИЯ)
Magnesium oxydatum Magnesium oxide Magnesiumoxyd
Magnesia usta
MgO Мол. в. 40,32
Приготовление
1. Основной углекислый магний набивают в гессенский тигель при по-
мошл пестика и, закрыв тигель крышкой (во избежание засорения угольной
пылью и золой), помещают в горн. Сначала прогревают тигель на слабом
огне, затем в течение %—1 часа при слабокрасном калении.
Выделение СОг заметно, если масса уплотнена, по выскакиванию шариков
на гладкой поверхности и по пыли MgO, захваченной выделяющимся газом.
Тотчас же по прекращении этого явления берут пробу из середины массы,
смешивают с водой и приливают разбавленной H2SO4. Когда не будет
заметно никакого выделения газа, тиглю дают остыть.
2. По Вурцу — обрабатывают продажный углекислый магний (Magnesia
alba) таким количеством НЫОз, чтобы часть соли осталась нераствореннон,
дают смеси стоять некоторое время, часто помешивая, фильтруют,
прибавляют к фильтрату немного MgS04 и спирта и оставляют на сутки. Затем
отфильтровывают от выпавшего CaS04, фильтрат выпаривают и прокалывают.
Остаток тщательно промывают водой и еще раз прокаливают.
Свойства
Снеж<но-белый, нежный рыхлый порошок, уд. в. 3,19—3,71 (.в зависимости
от температуры получения). Т. пл. выше 2500°, т. кип. 2800°. В воде почти
нерастворима (8,4 • 10~4% при 18°); постепенно притягивает из воздуха влагу
И СОг.
Бус и Мак-Интайр1 указывают, что воздух, высушенный над MgO,
содержит 0,008 мг Н2О на л при 25°. Смешанная с 10—«12 частями воды
переходит через некоторое время в кашицеобразную массу Mg(OH)2.
Испытание
По ОСТ НКТП 6766/373 потеря при прокаливании для препаратов ч.д.си
и чистого должна быть не более 10%; наибольшие количества допустимых
примесей указаны в табл. 299.
1 Н. S. Booth a. H. Mc Intyre, Ind. Eng. Chera., Anal. Ed, 2, 12 (1930).
Магнезиальная смесь. Магния окись и перекись 315.
Таблица 299
Допустимые пр
Примеси
и м е с и в MgO
Допустимое содержание з о:0
в чистсм для
анализа
в чистой
, Нерастворимые в-HCl вещества . . .
•у растворимые в воде вещества ....
I Хлориды (С1') .
4 Сера — оэщее количество из S04",
'SCV,S203"hS"(b пересчете на S04") .
5/ Кремнекислота и вещества,
осаждаемые аммиаком •
6. Железо (Fe)
7. Металлы, осаждаемые H2S
8. Барий (Ва)
0,03
0,3
0,02
0,02
0,05
0,005
0,003
0,003
0,10
0,5
0,05
0,20
0,10
0,01
0,01
0,005
Примечание: Препарат, содержащий не более 0,005% SO/', носит
название .окись магния для анализа без серы".
.'Magnesium
|peroxydatum
Magnesium
perhydrol
МАГНИЯ ПЕРЕКИСЬ
Magnesium peroxide Magnesiumperoxyd
MgO* Мол. в. 56,32
Приготовление
К 360 мл раствора MgCl2 уд. в. 1,22 (240 г MgCb • 6Н2О на 200 мл воды),
охлажденного до 5°, при хорошем перемешивании добавляют маленькими
порциями 80 г №262; температура при реакции не должна подниматься
выше 8°. После прибавления всего количества ЫаЮг продолжают
размешивать еще V2 часа и приливают 40 мл спирта. Через час выпавший осадок
отсасывают и сушат при 50—60° (Г м е л и н-К раут).
Сухой остаток рекомендуется истереть в ступке и, смочив 70 мл раствора
MgCl2 уд. в. 1,11, промывать водой до исчезновения реакции на хлор.
Остаток высушивают при 50—60°. Выход 85% теор.
МАГНИЙ СЕРНОКИСЛЫЙ (СУЛЬФАТ МАГНИЯ, ГОРЬКАЯ СОЛЬ)
Magnesium sulfuricum Magnesium sulfate Magnesiumsulfat
Epsom salt Epsomsalz
Bittersalz
MeS04.7H20 Мол. в. 246,50
Приготовление
30%-ному раствору H2SO4 прибавляют при хорошем размеши-
жий магнезит (или MgO) до выпадения Fe(OH)s и
прекращена ^ПтгЬипкТПППЯННЯЯ ПППбп ЖИЛ КОСТИ HP nnnwua TranQTL irr»o/»_
К горячему __ ,„
вании технический магнезит »или ivigw; ди ошиадсшш 1 \z\\jli.)% и
прекращения вспенивания. (Отфильтрованная проба жидкости не должна давать крас-
316
Магний
ного окрашивания с роданистым аммонием NHXNS). Раствор фильтру^
оставляют для кристаллизации в холодном месте. На другой день ивыпавщии
кристаллы отсасывают, промывают небольшим количеством ледяной водЬ1 е
перекристаллизовывают из горячей воды. п
Безводный MgS04 получается в виде белого порошка уд. в. 2,61
осторожном нагревании кристаллической соли до 238°,
Пр.,
Свойства
Прозрачные призматические кристаллы уд. в. 1,68, горько-соленого вкуСа
слегка -выветривающиеся на воздухе. При дегидратации образуются кристалло'.
гидраты с 6; 2,5; 2; 1 и 0,5 молекулами НгО \ MgSCh • 7НЮ легко растворйм
в воде, нерастворим в спирте.
•
Таблица 300
t'C
0
5
10
15
20
25
30
S5
Растворимость MgS04 в
1С0 г воды
раствори ют г MgSO,
26.9
29,3
31,5
33,8
36.2
38,5
40,9
43,3
ГС
40
45
50
55
60
65
70
75
100 г воды
растворяют г MgSO*
45,6
48,0
50,3
52,7
55
| 57.3
59,6
61,9
воде
f С
80
85
so
95
100
105
108,4
10С г воды
растворяют г MgSO,
64.2
66,5
68,9
71,4
73,7
76,2
77,9
Таблица 301
Удельный вес водных р а с т вор о в MgS04
o.'oMgSO,
2
4
6
8
rf20
4
1,0186
1,0392
1,0602
1,0816
о/о MgSO,
10
12
14
16
1
-*20
4
1,1034
1,1256
1,1484
1,1717
»!„ MgSO,
18
; 20
22
1 24
^20
d4
1,1955
1,2198
! 1,2447
1,2701
Испытание
По OCT НКТП 6278/263 содержание MgSOi • 7H2O в препарате должно
быть не менее 99%; наибольшие количества допустимых примесей указаны
в таол. 302.
* N.DemassIeux е. В. Р е d о г о И, С. II, 1018 (1939).
Магний сернокислый, и углекислый
31?
Таблица 302
Допустимые примеси в M;>S04 • 7НЮ
***
Примеси
1 Нерастворимые в
'воде вещества ....
9 X ориды (С1') ....
3, фосфаты (Р04'") . .
^Железо (Fe) ....
5. Металлы, осаждае-
6. Кальций (Са) ....
7. Мышьяк (As) ....
Допустимое содержание в °/0
в химически
чистом
0,002
0,001
0,001
0,0005
0,0002
0,01
0,00005
в чистом для
анашза
0,005
0.С02
0,002
0,0010
0,0005
0,02
0,0001
в чистом
0,01
0.005
0.005
0.002
0,001
0.05
0.0002
МАГНИЙ УГЛЕКИСЛЫЙ (КАРБОНАТ МАГНИЯ)
Magnesium carbonicum
Magnesium carbonate
/
А. Нормальный углекислый магний
Magnesiumcarbonat
MgCOa • ЗН2О
Мол. в. 138,38
Приготовление
1. Смешивают раствор MgSOi с раствором двууглекислого калия (КНСОз)
или натрия (NaHCOj). Вначале прозрачная смесь при стоянии выделяет
кристаллический ссадок углекислого магния (Роз е).
2. По С е и а р м о и у смешивают, эквивалентные количества безводных
MgSOa и Na2CO:* и нагревают смесь на песочной бане при 160—175°, часто
Помешивая. Приблизительно через час охлаждают, промывают*массу до
ударения SO/' и высушивают, повышая постепенно температуру до 300°.
3. По методу Б рил л я пропускают чистый сухой С02 над основным
углекислым магнием (Magnesia alba), нагретым до 150—220°. Реакцию ведут до
тех пор, пока полученный белый • рыхлый порошок не приобретет
постоянного веса.
С в о»]
и с т в а
Блестящие маленькие иглы уд. в. 3,04, спекшиеся в шарики или комочки,
Медленно выветривающиеся на воздухе. Теряет воду и С02 при нагревании
До 100°; СО2 теряется также при кипячении соли с водой. В воде растворим
плохо (1,1-10-*%; Lp= 1,8-Ю-8 при 25°) К Г
Испытание
См. Б. Основной углекислый магний.
1 F. На 11 a, Z. phys. Ch 173, 63 (1935); С II, 2848 (193S).
318
Магний
Б. Основной углекислый магний (белая магнезия)
Magnesia alba Magnesium carbonate Magnesiumcarbonat
basic. basisch
Приготовление
Нагретый до 50—60° раствор MgSCh или MgCb осаждают р?.ств0ро
Na2COs. Отстоявшийся осадок промывают водой, отжимают и высушивает в
воздухе в теплом месте. а
При употреблении MgSOi необходимо брать его в небольшом избытке
в противном случае препарат содержит соли натрия.
Свойства
Белый легкий порошок, обычно спекшийся в легко измельчаемые ко-
мочки. Почти нерастворим в воде (0,02% при 15°). Растворяется в растворах
аммонийных солей. Состав приблизительно соответствует формуле 5MgO • 4С02
• 5Н20.
Испытание
Примесей, нерастворимых в горячей воде, должно быть не больше 0,75%
для препарата ч. д. а. Раствор 1 г препарата в 20 мл СНзСООН не должен
изменяться от прибавления сероводородной воды (соли тяжелых металлов).
Раствор от прибавления ВаСЬ, также от прибавления НЫОз и 'AgNCb, через
5 мден. может давать лишь опалесценцию (сульфаты и хлориды).
Раствор 1 г препарата в 20 мл разбавленной НС1 от добавления 0,5 мл
5%-ного раствора K4Fe(CN)e не должен тотчас синеть {Fe' " •); 1 г препарата
при прокаливании должен оставлять не меньше 0,4 г остатка. Последний
смешивают с 20 мл воды и фильтруют; добавляют к фильтрату (МНфСгСЬ,
причем через 5 минут может появиться лишь опалесценция, «о не осадок (соли
кальция).
МАГНИЙ ХЛОРИСТЫЙ (ХЛОРИД МАГНИЯ)
Magnesium chloratum Magnesium chloride . Magnesiumchlorid
MjrCl2-6H20 Мол. в. 203,33
MgCh Мол. в. 95,23
Приготовление
1. Нейтрализуют HCl избытком MgO (или магнезитом), приливают
хлорной воды и »дают стоять с избытком MgO. Раствор отфильтровывают от
осадка Fe(OH)a, Si02 и др., осторожно выпаривают, добавляя изредка
несколько мл НС1 (уд. в. 1,19), и кристаллизуют. Рекомендуется кристаллы
перекристаллизовать из спирта.
2. А. П. Обухов и В. П. Лавров1 рекомендуют, получив раствор
MgCh, как указано в способе 1, выпаривать его до уд. в. 1,3, после чего
насыщать раствор сухим хлористым водородом. Тотчас начинают выпадать
кристаллы очень чистого MgCh • 6Н2О. Соль быстро отсасывают, промывают
небольшим количеством холодной воды и сушат между листами
фильтровальной бумаги.
3. Для получения безводной соли по Эрдману2 500 г водного
препарата (MgCl2»6H20) и 500 г NFLiCl растворяют в возможно малом количестве
воды; -раствор фильтруют и в фарфоровой (или, лучше, в серебряной) чашке
выпаривают досуха. Оставшуюся твердую массу измельчают и небольшими
порциями тщательно высушивают в платиновых тиглях. Операцию
продолжают до тех пор, пока масса при нагревании не перестанет давать
спекающегося порошка. Сушка считается законченной, когда взятая проба при на-
1 ЖПХ № 7- 8, 994 (1931).
■ Хим. преп. 19.
Магний хлористый
319
гревании в пробирке не выделяет влаги и по улетучивании NbkCl оставляет
йрозрачную жидкость, застывающую при охлаждении в прозрачные кристаллы.
Тогда полученным, еще теплым, порошком наполняют объемистый
платиновый тигель, плотно закрывают его крышкой и ставят в сильно разогретУю
^гельную печь. По выделении NPhCl тигель вынимают, извлекают
оставшийся безводный MgCb и наполняют тигель новой порцией порошка.
Затвердевший MgCh еще теплым переносят в плотно закрывающиеся сосуды.
4. Изучая способы получения чистого MgCU, В. Тред в ел л, А. Коэ^н
й Т. Ц ю р е р * нашли, что наилучшие результаты получаются при
воздействии смеси СО и СЬ на MgO, помещенную в кварцевую трубку. Пр<>Цесс
йдет при 740—750°.
При загрузке 400—500 г MgO скорость СО должна составлять около
340 мл/мин, скорость СЬ — около 200 мл/мин. MgCk получается в виде
снежно-белой массы; содержание воды от 0 до 0,16%. £,'?'
Свойства
MgCl2«6H20—игольчатые или столбчатые кристаллы, уд. в. 1,56, горько-
соленого вкуса, очень легко растворимые в .воде (см. табл. 303). При
нагревании теряет одновременно воду и НС1, оставляя MgO. Т. пл.2 117,5°, т-
разложения 151°. Соль чрезвычайно гигроскопична и должна храниться в плотно
закупоренных банках.
MgCb — пластинки с перламутровым блеском, уд. в. 2,32, растворимые
в воде (с сильным разогреванием) и в спирте. Расплывается во влажном
воздухе. При белом калении в атмосфере водорода возгоняется.
Таблица 303
Растворимость MgCl2 в воде
t° с
— 10
0
+ 10
+ 20
°/о MgC!2
11,1
34,5
34,8
35.3
t° с
40
60
80
100
o/oMgCIj
36,5
37,9
39,7
42,2
t° с
116,7
152,6
181,5
186
0,'oWgC',
46,1
49,0
55,8
56,1
Таблица 304
Удельный вес водных растворов MgCI2
0/oMgCI,
2
4
6
8
10 |
^20
4
1,0146
1,0311
1,0478
1,0646
1,0816
°;о MgC'a
12
14
16
18
20
w20
1,0989
1,1164
1,1342
1,1523
1,1706
o|oMgCl,
22
24
26
28 i
30
1 w20
4
1,1897
1,2088
1,2285
1,2487
1,2688
Испытание
Наибольшие количества примесей, допустимых по ОСТ НКТП 7377/531,
указаны в табл. 305; препарат должен выдерживать испытание на
нерастворимые в спирте примеси.
1 W. D. Treadwell, Л. Cohen u. Th. Ziirrer, Helv. Chim. Acta 22, 449 093*)).
* А. Э. Аужбекович, Ж11Х 9, № 4, 594 (1936).
с 20 Магний
Таблица 305
Допустимые примеси вMgCl• 6Н^О
Примеси
1. 1lepacruopii;. ые в
воде вещества ....
2. Сульфаты (S04") . .
3. Барий (Ва)
4.. Кальций (Са) ....
5. Фосфаты (РО4"0 . .
6. Железо (Fe) ....
7. Тяжелые металлы, .
осаждаемые H2S . .
8. Мышьяк (As) ....
9. Соли аммония (NH4*)
| Допустимое содержание в
в химически
] чистом
0,003
0,005
0,002
0,050
0,0005
0,0002
0,0002
0,00005
0,005
в чистом для
анализа
0,005
0,010
0,002
0,050
0,001
0,0005
0,0005
, 0,0001
0,010
°|0
в чистом
— ^5 -
0,010
0,020
0,005
0,100
1 0,002
0,001
0,0010
0,0002
0,02
МАГНИЙ ХЛОРНОКИСЛЫЙ
(ПЕРХЛОРАТ МАГНИЯ, АНГИДРОН)
Magnesium Magnesium perchlorate Magnesiumperchlorat
pcrchloratum
Мк(СЮ4)г Мол. в. 223,23
Приготовление
иг^П° !?етоду И- П- Алимарина1 в стаКан, содержащий 30%-ную
HClOi, добавляют понемногу х. ч. MgO до насыщения, отфильтровывают
избыток MgO через фильтр типа Щ0тта (с пористой стеклянной пластинкой) и,
нейтрализовав щелочной раствор с помощью НС104 до слабокислой реакции
на копгорот, выпаривают раствор до начала кристаллизации. Охладив чашку
холодной водой, отсасывают выпавшие игольчатые кристаллы на воронке
Бюхиера (без фильтра!), растворяют их в горячей воде и снова выпаривают
до появления пленки соли на поверхности. Из маточных растворов
выпариванием получают еще некоторое количество соли v v
„„^УпИНу1^^ОЛЛ7оМг(С10422*6Н20 нагРевают' в фарфоровой чашке на
„литке При 145-147° кристаллы плавятся а кристаллизационной воде; по
м£п№Леянп • п„р„?„Т»^ 3«вердевает в пористую массу-тригидрат
Mg(C10>)2-3H20; препарат необходимо в это время энергично перемешивать.
Затем температуру повышают до 170-200°, причем тригидрат плавится; при
этой ^температуре (во всяком случае не выш/230°) соль выдерживают 1-
После охлаждения соль измельчают до зере„ диаметром 3^4 мм и поме-'
ш.агат в круглодонную колбу, соед„„€Нную с хорошим масляным насосом,
дающим разрежение около 0,1 л* рт. „. МеЖд ролбой » включают
сушильную колонку с СаСЬ Пустив насос в ход, помещают колбу в
сушильный шкаф и нагревают 2-3 часа при 170°, п«ие чего повышают
температуру до 220-240° и высушивание продолжают ^ з часа
в ».&£ aSaV^^ * ПЛ°ТН° *™»Ы™ баНКаХ ИЛИ' ™ ^
I Завод, лаб. 9, №, 8 915 (1940).];
Магний хлорнркислый. Марганец азотнокислый 321
Щ По германскому патенту (DRP 571385) сливают горячие
концентрирование растворы NaC104 и MgCh. He давая жидкости остыть, отфильтровывают
йЕрший NaCl; при охлаждении фильтрата выкристаллизовывается М^ССЮф •
ЩШ& с примесью 1% NaCl. После перекристаллизации из горячей воды
Удержание NaCl падает до 0,05%. Обезвоживание препарата проводится, как
Указано в способе Г.
у 3. Для регенерации Mg(C104)2, служащего осушителем, его подвергают
дегидратации в вакууме, как описано в способе 1.
Свойства
Белая пористая масса, чрезвычайно жадно притягивающая влагу1; при
смачивании водой наблюдается разогревание и шипение, как с РгОб. Приме-
1яется в качестве отличного осушителя. По Беккеру2 упругость водяного
8ра над Mg(CI04)2 равна 0,000 мм рт. ст. Соль способна поглощать воду в
оличестве до 60% от своего веса, следовательно, и в. этом отношении цер-
аорат магния имеет преимущество перед Р2О5.
Будучи нейтральным веществом, пригоден для осушения различных газов
(Нз, Ог, СЬ, НС), СОз, ЫНз, H2S и др.) и органических жидкостей.
Испытание
Водный раствор препарата должен быть прозрачным и не действовать на
лакмусовую бумагу. При добавлении AgNOe в азотнокислом растворе не
должно выпадать осадка (хлортеды).
МАРГАНЕЦ АЗОТНОКИСЛЫЙ (НИТРАТ МАРГАНЦА[2])
Manganum Manganese nitrate Mangannitrat
nitricum Manganous nitrate
Мп(МОз)2. 6НоО Мол. в. 287,04
Приготовление
■' 1. В 200 мл химически чистой НЫОз уд. в. 1,2 вносят небольшими порциями
100 г углекислого марганца, часть последнего должна остаться нёрастворенной.
Отфильтровывают выпавший Fe(OH)3 и раствор выпаривают при 60—70°,
слегка подкислив Н1ЧОз. Выпаривание ведут до тех пор, пока проба
жидкости при охлаждении не будет закристаллизовыватъся. Раствор оставляют на
ночь в прохладном месте, после чего выпавшие кристаллы отсасывают и
помещают в банку с притертой пробкой.
Маточный раствор при упаривании дает еще значительное количество
кристаллов.
2. При получении химически чистого препарата по указанию В. М. Пещ-
к о в о й3 можно исходить из ферро-марганца.
1 кг измельченного ферро-марганца (76% Мп) добавляют понемногу к 5 л
НЫОз (уд. в. 1,2); по прекращении выделения окислов азота смесь нагревают
и отфильтровывают. К 1 л фильтрата добавляют (гШ^СОз до слабо
щелочной реакций и выпавшую смесь Fe(OH)s и МпСОз отфильтровывают и
тщательно промывают. Затем полученную пасту вносят в оставшиеся 4 л раствора
и нагревают смесь некоторое время почти до кипения. При этом полностью
осаждается Fe* " * в виде Fe(OH)3i
2Fe(N03)3 + ЗМпСОз + ЗНЮ = 3Mn(N03)2 + 2Fe(OH)3 + ЗСО2.
После фильтрования раствор подкисляют чистой HNOs и выпаривают При
температуре не выше 70° до концентрации около 150 г Мп(ЫОз)г на 100 г
1 Наиболее полное высушивание происходит при скорости газа, не превышающей 5 л/час*
2 В е с к е г, Ind. Eng. Chem,, Anal. Ed. 4, 1 (1933).
• Изв. ИРЕА, вып. 9, 91 (1930).
322
Марганец
воды. При последующем охлаждении выделяются хорошо образованные „
сталлы Mn(N03)2 - 6H2O. к^
Последние отсасывают на воронке Бюхнера и выдерживают сутки в э*
каторе над NaOH для удаления свободной Н1ЧОз. Выход 80% -теор. Кс,ь
Свойства
Слабо розовые длинные игольчатые призмы уд. в. 1,82, легко раствопи
мые в воде (табл. 306) и спирте. При 160—200° разлагается с образован*
Мп02. Ием
Таблица
Растворимость Mn(N03)2 в воде
306
t °с
— 16
0
+ И
o|0Mn(N03).
45,5
50,5
54,6
t °с
25,8
23,5
i 27
o('0Mn(N03)2
62,4
64,6
65,6
/ °с
30
35,5
°/oMn;NO
67,6
76,8
Таблица 307
Удельный вес растворов Mn(N03)2
о;0 Mn(N(Ve
1
2
4
6
8
10
12
1,0063
1,0140
1,0298
1,0459
1,0624
1,0794
1,0969
<\'0Mn(NO3)»
14
16
18
20
22
24
26
л18
*4
1,1149
1,1333
1,1522
1,1717
1,1918
1,2125
1,2333
о/о Mn(NCV,
28
30
35
40
45
50
55
л18
1,2557
1,2781
1,3367
1,3993
1,4662
1,5378
1,6146
Испытание
В препарате в зависимости от степени его чистоты допускаются
следующие наибольшие количества примесей (табл. 308).
Таблица 308
Допустимые примеси в Mn(N03)2• 6Н20
Примеси
1. Нерастворимые в воде примеси
2. Сульфаты (SO/)
3. Цинк (Zn)
4. Тяжелые металлы
5. Железо (Fe)
6. Магний и щелочи
7. Кальций (Са)
Допустимое содержание в о/0
в чистом для
анализа
0,005
0,010
0,050
0,0005
0,002
0,10
0,02
в чистом
0,01
0,02
0,05
0,001
0,002
0,25
0,05
*.'*
Марганца двуокись
323
МАРГАНЦА ДВУОКИСЬ
(ОКИСЬ МАРГАНЦА [4] «ПЕРЕКИСЬ» МАРГАНЦА)
Manganum
hyperoxydatum
Manganese dioxide, Manganperoxyd
Manganese peroxide,
Pyrolusite Mangandioxyd
А. Безводная двуокись марганца
MnOa Мол. в. 86,93
Приготовление
... .■■ 1. По Бейльштейну и Явейну растворяют чистую соль марганца в
^HNOs, нагревают и при постоянном кипении постепенно прибавляют
маленькими порциями КСЮз. В короткое время весь марганец осаждается в вида
МпОг; в отдельной пробе жидкости испытывают полноту осаждения, нагревая
с НЫОз и КСЮз.
Выпавшую МпОг, после разбавления жидкости водой, отфильтровывают,
тщательно промывГают и высушивают.
2. Многократно обрабатывают МпО или Mns04 горячей
концентрированной HN03.
3. Нагревают Mn(N03)2 при 200°.
. 4. Осторожно нагревают водную двуокись марганца (гидрат).
Свойства
Черный кристаллический или аморфный порошок уд. в. 5,03. Полученный
по способу 1 — кристаллический порошок из микроскопически малых
пластинок серо-стального цвета, просвечивающих в тонком слое пурпурово-крас-
ным цветом. При сдавливании принимает металлический блеск, довольно
хорошо проводит электричество. При накаливании выше 530° переходит в
Мпз04.
Испытание
По ОСТ НКТП 7110/447 содержание МпО* в препарате ч. д. а. должно
быть не менее 85%, в препарате чистом не менее 72%; наибольшие
количества допустимых примесей указаны в табл. 309.
Таблица 209
Допустимые примеси в MnQ2
Примеси
1. Вещества, не растворимые в НС1 . .
2. Хлориды (СГ)
4. Щелочные и щелочноземельные
металлы (в виде сульфатов) ....
Допустимое содержание в °/о
в чистом для
анализа
0,06
0,002
0,05
2,0
0,005
0,02
0,01
в чистом
0,2
0,01
0,1
5,0
0,01
ОД
0,01
3'2>-
МаргйнёЦ
Б. Водная двуокись марганца
(Гидрат перекиси марганца, марганцоватистая кислота)
МпО(ОН)2 или МпОг-НгО Мол. в. 104,95
Приготовление1
1. По От то к 20%-ному раствору МпСЬ приливают 10%-ный раствоп
ЫагСОз, сначала до полного осаждения МпСОз, затем еще некоторый избыток'
в полученную смео* пропускают хлор до тех пор, пока жидкость при стоянии
в течение суток не будет сохранять запаха хлора. Для окончательного уДа.
ления оставшегося МпСОз прибавляют немного HNOa (до кислой реакции)
Фильтруют, осадок промывают и сушат при 100°.
2. К раствору соли марганца добавляют ЫНЮН и Н2О2 и отделяют выпав-
ший осадок.
3. К 0,5%-ному раствору MnS04 прибавляют разбавленной НЬЮз, нагре-
вают до кипения и приливают раствор КМп04 до тех пор, пока прибавляемая
капля не перестанет о бесцвета в аться (на 669 г MnS04«4H20 требуется 316 г
КМп04).
Свойства
Черный или черно-коричневый, очень рыхлый, пачкающий порошок.
В. Активная двуокись марганца
Приготовление
1. К 250 мл 5%-ного раствора MnSOi при 50—60° приливают по каплям
при перемешивании 200 мл 5%-ного раствора КМп04*: приливание проводится
в течение 2 час. Смесь оставляют еще на 2 часа, после чего помещают ее на
сутки в теплое место. Выпавший осадок отфильтровывают* на воронке Бюх-
нера, промывают водой до удаления SO4" и высушивают в течение 8—10 час.
при 110—120°.
2. В фарфоровом стакане смешивают 150 г тонко измельченного MnSCh
с 142 мл воды. При хорошем перемешивании медленно добавляют к смеси
675 г 93%-Ной H2SO4. Снижают температуру до 50° и прибавляют в течение
2 мин. небольшими порциями 150 г растертого КМп04, следя, чтобы темпера-
Тура не превышала 75°.
Через 10 мин. после прибавления последней порции КМп04 реакция
закончена:
2КМп04 + 3MnS04 + 2Н2О = 5МпОг + K2SO4 -f 2H2'S04.
Содержимое стакана выливают при перемешивании в сосуд с 25 л воды.
Осадок двуокиси марганца промывают сначала декантацией, затем на воронке
Бюхнера до удаления 304". Промытый осадок высушивают в течение 6 час,
постепенно повышая температуру до 180°.
Г. Коллоидальная двуокись марганца
Приготовление
Для получения коллоидальной Мп02 по Бреди ту и Мерку2
пользуются следующей реакцией взаимодействия КМп04 с Н2Ог без прибавления
кислоты или щелочи:
2КМп04 + ЗН2О2 = 2MnO(OH)2 + 2KOH + ЗОа.
1 О получении Y-модификации МпО, см. О. О 1 е m s е г, СИ, 3958 (1939). Е. В. Алексеев-
ский, Г. М. Б е л о ц е р к о в с к и й, Т. Г. Плачено в. Практические работы по химии
защит.*. Оборонгиз, 1940, стр 48—54, также Е*В. Алексеевский. Химия защиты, т. II,
стр. 2Ш н ел. '
* Prap. Ch. 696.
щ;?..
Марганца закись
325
я О* применяют не крепче 3%, "раствор КМп04 берут с содержанием не
' J!ne 16 2 на л. К раствору КМп04 прибавляют Н202 до, исчезновения фио-
с**5мтй окраски и появления коричневой окраски коллоида.
♦леш1Я испытания в пробе жидкости коагулируют МпОг прибавлением не-
«ьких калель раствора NaCl и нагреванием; при этом коричневая окраска
сК!0-Л1Т71а пропадает и становится заметной фиолетовая окраска непрооеаги-
!Й2шего КМп04.
Р°для освобождения от образующегося при реакции КОН подвергают жид-
^JjJ, диализу, лучше всего употребляя особо чистую воду («вода для изме-
*2яия «электропроводности»). Во время диализа испытывают гидрозоль и про-
Цфундировавшую воду на щелочь. Вначале достаточно пробовать
фенолфталеином; когда с ним покраснения уже не будет, применяют метод
электропроводности.
Диализ ведут до тех пор, пока удельная проводимость коллоидного раствора
gtf перестанет уменьшаться. При этом получается коллоид высокой степени
зястоты. Вся стеклянная посуда, служащая для приготовления и хранения
Зялоида, должна быть пропарена до ^совершенной чистоты.
С в о й с т в а
Гидрозоль МпОг— жидкость, кажущаяся прозрачной; в разбавленном
состоянии светложелтая, с увеличением концентрации приближается к темно-
коричневой.
От прибавления" большинства электролитов (кроме КМпОО выпадает
хлопьями гидрат двуокиси марганца МпО(ОН)г.
МАРГАНЦА ЗАКИСЬ (ОКИСЬ МАРГАНЦА[2])
fdanganum oxydulatum Manganous oxide Manganoxydul
Й MnO Мол. в. 70,93
Приготовление
1. По К ее ел ер у насыщенный при 60° раствор КМп04 смешивают
0 избытком горячего насыщенного раствора .щавелевой кислоты (Н2С2О4), к
которому предварительно добавлена СНзСООН. Образовавшийся осадок
щавелевокислого марганца помещают в закрытую банку и взбалтывают с таким
количеством воды, чтобы отстоявшийся осадок занимал около Vs объема всей
смеси».
Жидкость сливают, заменяют ее дестиллированной водой, взбалтывают,
Дают отстояться, снова сливают и повторяют промывание таким образом
около 20 раз.-
Соль «высушивают в платиновой чашке и при помешивании прокаливают
в платиновой лодочке (помещенной в платиновую трубку) в струе чистого
и сухого водорода.
2. МпСОз или, лучше, Мп(ЫОз)г прокаливают сначала без доступа воз-
Духа, а под конец в струе водорода К
Свойства
Зеленовато-серый порошок уд. в. 4,73, легко растворимый в кислотах. При
накаливании на воздухе переходит в MnsOi. В воде нерастворима.
1 О получении МпО восстановлением МпО. водородом при 300° см. Физич. журна i СССР,
Спец. вып. 91 (1936).
325
Марганец
МАРГАНЦА ПЕРЕКИСЬ
См. Марганца двуокись.
МАРГАНЕЦ СЕРНОКИСЛЫЙ (СУЛЬФАТ МАРГАНЦА [2])
Manganum sulfuricum Manganese sulfate Manganosulfat
Manganous sulfate .
MnS04'5H20 Мол. в. 241,07
Приготовление
1. По Сингаловскому1 250 г тонко истертого и просеянного Мп02
смешивают с 250 г Ш304, прокаливают при 300°, перемешивают полученную
массу с 125 г H2SO4 и продолжают обжиг при температуре не выше 750°.
Остывшую массу обливают 500 мл воды и нагревают в течение получаса при
перевешивании. Дают отстояться, декантируют, наливают свежую воду и
продолжают выщелачивание, пока вода будет извлекать лишь незначительные
количества Мп304.
Раствооы соединяют вместе и, подогрев, насыщают сероводородом для
осаждения меди. В случае обнаружения Fe раствор подкисляют, кипятят
с тонко истертым МпОг для окисления Fe* * в Fe* и прибавляют МпСОз
(15—20% от веса имеющегося Мп'304). Тщательно перемешивают; когда все
железо осядет (профильтрованная проба раствора с NFhCNS не должна
давать красного окрашивания), фильтруют, подкисляют 1—1,5%-ной H2SO4 и
выпаривают до уд. в. 1,48—1,53.
Дают раствору стоять в холодном месте сутки, после чего кристаллы
отсасывают, промывают холодной дестиллированной водой, отсасывают насухо
и складывают в банки с притертыми пробками. Маточный раствор
целесообразно упарить еще раз.
2. По методу ИРЕА порошкообразный пиролюзит (МпСЬ), предварительно
промытый 5%-ной НЫОз (для удаления солей щелочных металлов) и
дестиллированной водой, помещают в объемистую банку, заливают водой и при
непрерывном механическом перемешивании насыщают сернистым газом до
перехода черного цвета осадка в светлосерый. Затем, не прекращая
перемешивания, добавляют небольшими порциями МпОг (до почернения осадка) для
окисления и осаждения примесей. Раствор отфильтровывают, выпаривают
досуха и в течение 2—3 час. прокаливают сухую массу при 400—500°.
Растворяют полученную соль в воде, добавляют 5—6 капель
концентрированной H2SO4, нагревают до 50—60° и для осаждения Си и Со насыщают
сероводородом в течение 20—30 мин. Раствор кипятят для удаления HsS,
отфильтровывают темный осадок серы и сульфидов и выпаривают фильтрат
до уд. веса 1,5 (измерение при 17—18°). Выпаренный раствор, оставленный
при комнатной температуре, медленно выделяет кристаллы MnS04 • 5ШО,
которые отсасывают, промывают небольшим количеством холодной воды и
сушат на воздухе.
В случае необходимости быстрого получения кристаллического препарата
к 1 л раствора Мп304, выпаренному до уд. в. 1,5, добавляют 800 мл
96%-ного этилового спирта; через 2—3 часа выделяются крупные кристаллы
MnS04 • 5Н*0.
3. Гептагидрат Мп£04«7НгО получается при кристаллизации соли из
растворов в интервале температур от —4 до +6°. Тетрагидрат MnS04 • 4Н2О
выкристаллизовывается между 20 и 40°.
4. Безводный MnS04 получается нагреванием водной соли при 280° до
постоянного веса.
1 Соли редких и цветных металлов 221.
Марганец сернокислый
327
•*с
тва
tfflS04 • 5Н20 — красноватые, триклинические кристаллы уд. в. 2,1, довольно
^растворимые в воде и нерастворимые в спирте1.
t^tim 54° плавится в кристаллизационной воде. Безводный MnS04 — бела*
ыпчатая хрупкая масса горьковатого металлического вкуса, уд. в. 2,9.
Ра слабом прокаливании устойчив» при сильном калении разлагается.
2вляя"Мпз04.
Растворимость MnS04 в воде
Таблица ЗМ
ГС
0
5
9
15
°/о MnS04
34,7
36,0
37,2
37.9
t °G
20
25
30
о/о MnS04
38.6
39.3
39,4
t*C
50
1 ™
100
°/o MnS04
37,3
34.2
24.9
Удельный вес растворов MnS04
Таблица 311
о/о MnS04
1
2
4
6
8
10
w15
1,0089 •
1,0188
1,0389
1,0595
1,0807
1,1025
о/о MnS04
12
14
16
18
20
w15
4
1,1248
! 1,1478
I 1,1714
1,1956
1,2205
o/o MnS04
22
24
26
28
30
d4
1,2461
1,2725
1,2997
1,3277
1,3565
Испытание
По ГОСТ 435—41 содержание MnSOi в обезвоженном препарате ч. д. а.
'Должно быть не менее 99% и в препарате чистом не менее 98%; потери при
обезвоживании обоих препаратов должны быть не более 39%; наибольшие
количества допустимых примесей указаны в табл. 312.
Таблица 312
Допустимые примеси в MnS04• 5Н20
Примеси
Допустимое содержание в %
в чистом для
анализа
Л. Нерастворимые в воде вещее 1ва
£• Хлориды (СГ)
3- Цинк (Zn) .
4. Металлы, осаждаемые H2S
5. Железо (Fe)
6. Щелочные и щелочноземельные металлы
(в виде сульфатов)
0,005
0,002
0,02
0,0002
0,0005
0,5
0,01
0,005
0,05
0,001
0,0015
1.0
1 О кристаллогидратах MnS04 см. С. II, 4201; 3958;
1002 (1939).
330
Медь
Свойства
MnCb • 4НгО — красноватые моноклинические гигроскопичные кристаллы
уд. в. 1,913, растворимые в воде и спирте. При 58° теряет часть воды,
переходя в МпС12«2НгО. При 200—230° происходит частичное разложение соли
с выделением НС1. Безводный МпС1г — красноватые, легкоплавкие листочки
уд. в. 2,478 (по другим данным 2,977), плавящиеся при 650° и возгоняющиеся
при более высокой температуре.
При нагревании на воздухе разлагается влагой, выделяя НС1 и оставляя
Мпз04. Растворим в абсолютном спирте (2: 1), нерастворим в эфире.
** Таблица 315
Удельный вес водных растворов МпС12
о/0 MnCl,
1
2
4
6
8
10
А9
d4,
1,0069
1,0153
1,0324
1,0498
1,0676
1,0859
Уо MnCli
12
14
16
18
20
W18
1,1046
1,1238
1,1435
1,1638
1,1846
о/о MnCl»
22
24
26
28
30
л18
d4
1,2061
1,2283
1,2541
1,2746
U988
препарате
ч. д. а. должно
Испытание
По ГОСТ 612—41 содержание МпС12-4Н20 в
быть не менее 99%, а в препарате чистом не
чества допустимых примесей указаны в табл. 316; препарат должен
выдерживать испытание на примесь окисляющих <и восстанавливающих веществ.
Допустимые примеси в МпС12- 4Н20
Таблица 316
Примеси
Допустимое содержание в "/•
в чистом дла
анализа
1. Нерастворимые в воде вещества . . • .
2. Сульфаты (SO/)
3. Цинк (Zn)
4. Металлы, осаждаемые H2S
5. Железо (Fe)
6. Щелочные металлы (в виде сульфатов)
7. Барий (Ва) Г . . '
0,005
0,005
0,02
0,0005
0,С002
0,2
0,005
0,01
0,02
0,05
0,001
0,001
0.5
0,01
Cuprum
Си
МЕДЬ
Copper
At. в. 63,57
Kupfer
А. Порошок меди
Приготовление
1. По Лассар-Кону1 к помещенному в фарфоровую чашку холодному
насыщенному раствору CuS04 добавляют цинковую пыль; чтобы избежать
1 L a s s а г-К о h a, Arbeits-Methoden der organischen Chemie 360 (1903); Prap. Ch.
Медь
331
/рззования коиков, Zn прибавляют, просеивая его через тонкое сито.
Прибавление Zn заканчивают, когда раствор, нагреты! до 80°, еще имеет слабо-
^дубой цвет. Получившийся чрезвычайно раздробленный порошок меди
осечёт на дно в виде тяжелого тёмнокрасного слоя. После возможно
тщательного промывания декантацией осадок для удаления следов цинка (всегда
Присутствующего в виде примеси) обливают водой и прибавляют при поме-
вдевании НС1 до прекращения вскипания. Снова промывают осадок декан-
яадиёй, отсасывают и промывают на воронке до нейтральной реакции. Чтобы
0орошрк не спекался и сохранял тонкое раздробление и большую поверх-
йость,'его не высушивают, а употребляют в виде пасты. Из-за неизбежной
окисляемости металла в таком виде пасту хранят в хорошо закупоренной
банке. Полученный препарат вполне пригоден для диазотирования.
2. В и г у р о 1 рекомендует осаждать Си при помощи алюминия. В
фарфоровой чашке смешивают СиС1г с вычисленным количеством крупного порошка
алюминия и небольшим количеством воды и перемешивают; в конце
добавляют еще немного^ воды. Когда жидкость над осадком станет бесцветной
и прозрачной, удаляют избыток алюминия, остаток декантируют водой и
отсасывают.
Полученную кашицу меди переносят в колбу с водяным или ртутным
затвором и, облив частой НС1,. вытесняют воздух водородом и нагревают
до прекращения растворения алюминия. По охлаждении осадок промывают
декантацией до исчезновения кислой реакции, переносят на фильтр и
отсасывают. Затем промывают спиртом и эфиром н сушат сначала на воздухе,
а под конец] слабо нагревая в струе водорода.
3. В очень раздробленном состоянии медь получается при восстановлении
СиО водородом. Для этого в трубку с шариком посредине помещают
порошкообразную СиО и пропускают водород до полного вытеснения воздуха. Затем,
не прекращая тока водорода, трубку осторожно подогревают до 135—150°,
до полного перехода черной СиО в розовый порошок «меди. Тогда
прекращают нагревание и охлаждают трубку в токе водорода.
4. По указанию И. П. Кислякова* восстановление СиО водоррдом
удобно проводить в трубчатой печи при температуре 500—520°. Навеску СиО
берут около 50 г. Медь получается в виде спекшейся легко измельчаемой массы,
довольно устойчивой на воздухе (побуренне начинается через 16—20 час).
5. По Л и б и х у и Вёлеру прокаливают смесь из 5 ч. хлористой меди
(CuCl) и 6 ч. смеси безводного Na?COj и NH4C1. Массу выщелачивают
водой, остаток отжимают между листами фильтровальной бумаги и сушат
при 75° в струе водорода.
Б. Коллоидальная медь
1. По Лоттермозеру3 8,5 г СиСЬ растворяют в небольшом
количестве воды, добавляют 16 г винной кислоты (НвСЮв) и постепенно приливают
30 мл NaOH (уд. в. 1,365); получившийся темносиний раствор разбавляют
водой до 1 л. Отдельно готовят раствор 15 г хлористого олова (SnCb • 2Н2О)
и 50 г винной кислоты (НвСЮо), который делают слабо щелочным, прибавляя
постепенно 85 мл NaOH (уд. в. 1,365) и разбавляют до 300 мл.
Оба раствора смешивают и нагревают смесь на водяной бане, пока
выпавший осадок не сделается совершенно черным. Препарат, как понятно
из способа получения, неизбежно содержит примесь олова.
2. По Герасимову и Матвееву4 30 мл смеси Пааля5 раство-
1 V f gr о и г а и х. Bull. Soc. Chim. 1, 17 (1907).
* ЖПХ № 11 (1939).
•Lottermoser, Ueber anorganische Kolloide 62 (1901); Т. Сведберг, Образование
коллоидов, НХТИ (1927).
« ЖРФХО 62 (4), 839 (1930). хт _
8 Для приготовления смеси Пааля в колбу с раствором 15 г NaOH в 500 мл воды вносят
маленькими порциями ЮЭ г яичного белка, встряхивая каждый раз колбу до получения почти
однородной жидкости. Затем нагревают смесь на водмой бане в течение часа, причем белок
почти полностью растворяется с выделением NH,. Незначительный хлопьевидный осадок
отфильтровывают (С. Р а а 1, Вег. 35, 2195, 2206, 2224 (1102;.
332
Медь
ряют в 27 мл ледяной СНзСООН и нагревают 4—5 мин. на водяной ба
К смеси добавляют 4 мл 15,68%-ного раствора CuSCU, снова наград •
4—5 мин., прибавляют 5 мл 0,3%-ной .ф°сФ°Рноватистой кислоты (НзРо!
и еще раз нагревают в продолжение 5—6 мин. При этом раствор стан01Шт ' '
рубиново-красным в проходящем свете и черным в отраженном. При 0хл°Я
ждении раствор мутнеет и через некоторое время выпадает коричневы* 0ся
док (частичная коагуляция). Раствор осаждают нацело раствором Na,cn'
и осадок 3—4 раза промывают декантацией. " 3
Промытый осадок растворяют в воде, к которой добавлено несколько
капель NaOH, и раствор выпаривают досуха на кипящей водяной бане."
3. Надежный метод получения устойчивого красного золя Си описан
впервые Фрейндлихом и Штейнером1. 3 мл 0,01 молярного раствора
Си в Ш ЫНЮН разбавляют 10 мл воды и нагревают до кипения. К кипя-
щему раствору приливают 3 мл 0,5%-ного гидразин гидрата (ЬШ^НгО) и
тотчас же вслед за ним 1,5 мл раствора желтого фосфора в эфире
(насыщенный раствор, разбавленный эфиром в 10 раз). При добавлении Ы2Ш • Н20
голубой раствор обесцвечивается ©следствие восстановления Си* * до Си*;
при вливании раствора фосфора окраска становится ж|елггой, быстро
переходя в красную.
Частицы золя заряжены отрицательно.
Свойства
Розово-красный, очень мягкий и ковкий металл уд. в. 8,93. Т. пл. 1083°,
т. кип. 2360°. Устойчива по отношению к воздуху и его составным частям;
сильно накаленная металлическая медь на воздухе сгорает.
Нерастворима в разбавленных НС1 и H2SO4, растворима в HNOs и
концентрированных H2SO4 и НС1 (в последней в присутствии платины). При
доступе воздуха растворима в ЫНЮН.
Коллоидальная медь — тончайший черный порошок, несколько
растворимый в воде с образованием красновато-черного раствора.
Коллоидный раствор меди при доступе воздуха быстро окрашивается с поверхности
в светлозеленый цвет.
И с пытаниё
1. Серебро, железо, свинец и другие тяжелые металлы. 2 г меди должны
полностью растворяться в HNOs, образуя прозрачный раствор, в котором
прибавление НС1 или ЫНЮН не должно вызывать помутнения. После
осаждения меди сероводородом, фильтрования, выпаривания и прокаливания и с
должно оставаться заметного остатка.
2. Мышьяк — испытание производят в приборе Марша3.
3. Закись меди — испытание производят по методу Гампе3.
МЕДЬ АЗОТНОКИСЛАЯ (НИТРАТ МЕДИ[2])
Cuprum nitricum Copper nitrate Kupfernitrat
Cu(N03)2 • ЗНгО Мол, в. 241,63
Приготовление4
50 г металлической меди в небольших кусочках обливают в колбе (под
тягой) 450 г 30%-ной HNOs и слегка нагревают смесь на песочной бане.
Когда выделение окислов азота ослабеет, нагревают несколько сильнее до
растворения меди. По окончании растворения разбавляют жидкость равным
1 Н. F г е и в d 11 с h a. D. S t е 1 п е г. J. chera Soc. Ю83 (1937).
» Ф. Тредввлл, Курс аналитической химии т. 1, стр. 233. ГОНТИ. М.-Л, (1931).
» Chem. Ztg. 9в (1889), г л ,
4 О получении безводной Си (NOe)i из СиО и окислов азота. См. В. N е u m a n n, A. Son-
U g, Z. f. Eltktroch. 41, 611 (1935).
Медь азотнокисЛаА
333
мом дестиллированной воды, фильтруют и, выпарив фильтрат до появле-
".кристаллической пленки, дают ему охладиться.
Выпавшие кристаллы отжимают между листами фильтровальной бумаги и,
rfbictpo высушив в умеренно теплом месте, помещают в банку с хорошо
приятой пробкой. Небольшое количество Fe, содержащееся в меди, остается
^последних порциях маточных растворов.
свойства
Темносиние призматические кристаллы уд. в. 2,05, очень легко растворимые
в воде (см. табл. 317) и спирте. Растворима в концентрированном ЫНЮН
J интенсивносиней окраской. Т. пл. 114-,5°; при сильном нагревании
разлается, давая сначала основную соль 4СиО • ЗЫгОв, а затем СиО. Во влажном
здухе расплывается. Бумага, пропитанная спиртовым раствором Cu(NOs)2,
высушивании самовоспламеняется.
Таблица 317
Растворимость Cu(N03)2 в воде
/ °с
0
20
40
% Cu(N08),
45,0
55,6
61,5
/•с
60
80
•/о Cu(N08)t
64,2
67,5
Таблица 318
Удельный вес водных растворов Cu(NOa)2
о/0 Cu(N08),
1
2
4
6
.20
1,006
1,015
1,032
1,050
' о/0 Cu(N03),
8
10
12
14
w20
di
1,068
1,087
1,106
1,126
■/о Cu(NCVi
16
18
20
25
,20
1,147
1,168
1,189
1,247
Испытание
Наибольшие количества примесей, допустимых по ОСТ 539, указаны
в табл. 319.
Таблица 319
Допустимые примесив Cu(N03)2 • ЗН20
Примеси
Допустимое содержание в %
химически
чистом
в чистом для
анализа
в чистом
1. Нерастворимые вещества
2. Хлориды (СГ)
3. Сульфаты (SCV)
4 Железо (Fe)
х5 Соли металлов, не осаждаемых H2S
0,001
0,001
0,005
0,001
0,05
0,005
0,002
0,01
0,005
0,1
0,01
0,005
0,05
0,02
0,2
334
Медь
МЕДЬ-АММОНИЙ ХЛОРИСТЫЙ (ДВОЙНАЯ СОЛЬ ХЛОРНОЙ MFn
И ХЛОРИСТОГО АММОНИЯ; СОЛЬ ГЕЙНЕ) . Ди ч
Kupferammoniumchlo
rid
Мол. в. 277,48
Cuprum-ammonium Copper ammonium
chloratum chloride
CuCh • 2NH4CI • 2H20
Приготовление
1. В фарфоровой чашке смешивают 200 мл воды, 165 мл НС1 (уд. в i \^
и 50 мл HNOs (УД. в.. 1,4) и в полученную смесь понемногу вносят' 60 I
электролитной меди (медной проволоки). Для ускорения растворения* чащКу
можно слегка подогреть. По окончании реакции жидкость подогревают до
полного удаления окислов азота и затем прибавляют 275 мл воды и 85 г
NH4C1, который растворяется при помешивании стеклянной палочкой.
Горячую жидкость фильтруют в фарфоровую чашку и, добавив несколько
мл НС1 (уд. ». 1,19) *, выпаривают на .водяной бане. Образующуюся на
поверхности кристаллическую .пленку время от времени опускают на дно
стеклянной палочкой. Упаренный раствор подвергают кристаллизации при
комнатной температуре. Кристаллы отсасывают, промывают 30—40 жлдестил-
лированной воды и сушат на пергаменте, после чего складывают в банку
# Таблица 320 '«^притертой пробкой. Выход
Растворимость СиС12• 2NH4C1 • 2Н20 Маточный раствор при вы-
в воде паривании может дать еще
около 50 г препарата.
2. 100 г хлорной меди
(СиСЬ *в2Н20) растворяют на
водяной бане в возможно
малом количестве воды,
фильтруют и к фильтрату
добавляют при помешивании
насыщенный профильтрованный
раствор 63 г NHUCl. Раствор
выпаривают до появления кристаллической пленки и охлаждают; выпавшие
кристаллы отсасывают и высушивают на фильтровальной бумаге при
комнатной температуре.
Свойства
Голубые кристаллы уд. в. 1,97, растворимые в воде.
И спытание
Наибольшие количества примесей, допустимых по ОСТ 5463, указаны
в табл. 321.
Таблица 321
Допустимые примесив СиС12 • 2NH4C1 • 2Н20
ГС
0
20
40
% соли
22,02
25,95
30,47
/°с
60
80
0/о СОЛИ
36,13
43,36
Примеси
4. Щелочные и щелочноземельные металлы
Допустимое содержание в %
в чистом для
анализа
0,005
0,05
0,005
0,10
0,005
в чистом
0,010
0,05
0,010
0,25
0,010
* Без добавления HCI препарат получается буроватым.
Медь-аммоний хлористый. Медь бромистая
335
-МЕДЬ БРОМИСТАЯ (МЕДЬ ОДНОБРОМИСТАЯ, БРОМИД МЕДИШ)
Cuprous bromide Kupferbromur
CuBr Мол. в. 143,49
'г Cuprum bromatum
Cuprum monobromatum
Приготовление
По Бодлендеру и Сторбеку* пропускают SO2 в нагретый
разбавленный раствор 259,5 г CuS04«5H20« 119 г КВг, пока не выпадет белый
осадок. Последний промывают многократной декантацией водой,
прокипяченной'и охлажденной в атмосфере СОг (или раствором SO2), быстро
фильтруют и сушат на пористой глиняной или фарфоровой пластинке в вакуум-
эксикаторе над КОН.
Свойства
Бесцветные прозрачные тетраэдры уд. в. 4,72, плавящиеся при 480° в
жидкость, кипящую около 1000°. Очень мало растворима в воде; шо влажном
состоянии быстро окисляется. Аналогично CuCl бромистая медь растворима
в NH4OH и концентрированной НС1.
Испытание
По техническим условиям НКХП 86—40 содержание CuBr в препаратах
ч. д. а, и чистом должно быть не менее 99%; наибольшие количества
допустимых примесей указаны в табл. 322.
Таблица 322
Допустимые примеси в CuBr
Примеси
1. Нерастворимые в НС1 вещества .
2. Железо (Fe)
3. Вещества, не осаждаемые H2S . .
Допустимое содержание в %
в чистом для
анализа
0,01
0,003
0,1
0,001
в чистом
0,05
0,02
0,3
не
испытываете*
МЕДЬ БРОМНАЯ (МЕДЬ ДВУБРОМИСТАЯ, БРОМИД МЕДИ[2])
Cuprum bibromatum Cupric bromide Kupferbromid
CuBr2 _. Мол. в. 223,40
Приготовление
1. Насыщают бромистоводородную кислоту (НВг) окисью меди, раствор
фильтруют и сильно выпаривают на водяной бане до получения почти
насыщенного раствора. Сгущенный раствор помещают в вакуум-эксикатор над
H2SO4, где он в несколько дней почти совершенно затвердевает (для
ускорения высыхания массу ежедневно растирают).
2. 100 г порошка меди взмучивают в дестиллированной воде и при
постоянном помешивании приливают постепенно 275 г брома, пока на дне
не останется лишь незначительный остаток. По охлаждении темный раствор
СиВгг фильтруют через вату и выпаривают, лучше всего в вакууме.
Кристаллы быстро отсасывают на воронке иенского стекла и немедленно
переносят в банки с притертыми пробками.
iBodiander, Storbeck, Z. anorg. Ch. 31, 459 (1902); Prlp. Ch. 511,
336
Медь
Если в качестве исходного продукта применялся технический бр0
раствор СиВгг перед выпариванием следует прокипятить с древесные М' То
для удаления органических примесей. ' У^ъц
И с пытание
По техническим условиям НКХП 85—40 содержание CuBr2 пе Но
руется, а указывается лишь на этикетках упаковки; наибольшие количее
допустимых примесей: 0,02% сульфатов (SCV) и 0,05%' железа. Тва
МЕДИ ГИДРАТ ОКИСИ (ГИДРООКИСЬ МЕДИ[2])
Cuprum Cupric hydroxide Kupferhydroxid
hydroxydatum
Cu(OH)2 Мол. в. 97,59
Приготовление
1. 100 г CuS04»5H20 растворяют в 1500 мл воды, фильтруют, добавляют
к фильтрату 2 мл глицерина и осаждают Си(ОН)г профильтрованным
раствором 34—36 г NaOH в 2 л воды. Прибавление NaOH ведут до тех пор, пока
серо-зеленый, вначале, осадок не приобретает яркоголубой окраски.
Дав осадку осесть, по возможности быстро удаляют декантацией
жидкость и вновь заливают осадок холодной дестйллированной водой, к которой
добавлено 1 мл глицерина. После отстаивания осадок продолжают промывать
декантацией (добавляя каждый раз к промывной воде 1 мл глицерина), пока
слитая жидкость не перестанет давать мути от прибавления ВаСЬ (отсутствие
SOi").
Осадок отсасывают и промывают на фильтре, пока проба, растворенная
в tfiCl, не перестанет давать реакцию на SO4" в течение 3—5 мин. (к
промывным водам попрежнему добавляют по 1 мл глицерина). После
окончательного удаления SO/' дают жидкости стечь, полученную густую пасту
растирают в фарфоровой ступке с 2 мл глицерина и в таком виде сохраняют.
Выход 120 г пасты *.
Назначение глицерина — препятствовать обезвоживанию Си(ОН)2. При
обычном применении этого препарата (для отделения протеинов) примесь
глицерина не вредит. При необходимости же употребления препарата без
примеси глицерина последний легко удаляется двукратным промыванием пасты
дестйллированной водой.
2. По Бётгеру2 прибавляют по каплям 5—8%-ный ЫНЮН в кипящий
10%-ный раствор CuS04, пока образовавшийся зеленый осадок не примет
голубой окраски. Выпавший кристаллический основной сульфат тщательно
промывают водой до удаления SO4" в промывных водах и настаивают
с 10%-ным раствором NaOH в течение 10 мин., причем образуется Си(ОЫ)^.
Осадок промывают тепловатой (30°) водой до удаления щелочи и SOj",
после чего отсасывают и сушат в вакууме над СаО или H2SO4. Выход
90—95%. I
Свойства
Cu(OH)2f полученная по способу 1, — темноголубая, иногда синяя или
сине-зеленая густая паста, устойчивая при хранении. В виде сухого порошка
постепенно обезвоживается, причем скорость разложения зависит от
температуры.
Полное разложение наступает:
При 15° ... . . .-.-.. через 9 мес.
п 30° • . . , . 86 час.
„ 45° ...♦...♦._ 45 час.
\ Разработано Д. Н. Фннкельштейн.
• В 6 11 g е г, J. prakt. Ch. 73, 491 a888); L. V a n I n о u. E. E n g e г t, Ch:m Zt2. 48, 142 (1924).
Меди закись
33/
ше 54° гидрат окиси меди вовсе не образуется. Даже под водой посте-
В*1 происходит обезвоживание, особенно быстро под растворами солей и
п^^йй причем образуется черная ЗСиО»НЮ. Примесь сахара, глицерина и
деД^ ых солей (СаСЬ, M11SO4) замедляет обезвоживание. Сухой препарат
йсКоТОР^ыть нагрет без разложения до 85°; выше этой температуры начи-
*Р*е* 0безвоживание. В избытке щелочи Си(ОН)г растворяется с образова-
0астся псевд0.КОЛЛОИдНого раствора [100 мл ЗЭ%-ного NaOH растворяет
&%г Си<ОН)«].
^Растворимость Си(ОН)2 в сильных щелочах указывает на амфотерный
ак^ер гидроокиси. Мак^Доуэлл и Джонстон1 определили вторую
«гтанту диссоциации (Си(ОН)а как кислоты (т. е.
^Н + CuQ»*) и нашли ее равной 7,9 - 10-»
для процесса НСиОг';
Испытание
По Мерку препарат «для анализа по Fassbender'y» должен иметь
нейтральную реакцию и не содержать сульфатов (SCV').
МЕДИ ЗАКИСЬ (ОКИСЬ МЕДИ [1])
Cuprum oxydulatum Cuprous oxide Kupferoxydul
O12O Мол. в. 143,14
Приготовление
1. По Л ё в е к находящемуся в стакане раствору Си(ЫОз)г концентрации
от 5 до 1*5% прибавляют при охлаждении отмеренный объем NaOH, пока не
осядет Си(ОН)» и отстоявшаяся жидкость не будет иметь ясно-щелочную
реакцию.
Тогда приливают еще объем NaOH, в 1%—2 раза превышающий ранее
прилитый, и, время от времени помешивая, прибавляют к смеси чистый
глицерин до совершенного растворения осадка и просветления жидкости.
Полуденный интенсивно-синий раствор смешивают с 12—15%-ным .раствором чи-
iboro виноградного сахара (взятым из расчета 3—4 ч. сахара на 1 ч.
содержащейся меди). Стакан закрывают стеклянной пластинкой и оставляют
раствор на 8—10 час. в холодном и темном месте. По истечении этого
времени синяя окраска жидкости исчезает и на дне сосуда собирается рыхлый
яркокрасный осадок. Раствор, не перемешивая, сливают через фильтр и
промывают осадок декантацией водой, к которой добавлено немного NaOH
И глицерина. Осадок переносят на фильтр и хорошо промывают, пока
промывная вода не перестанет показывать щелочную реакцию (проба лакмусовой
бумажкой); наконец, осадок высушивают при 100° на воздушной бане.
2. По Вёл еру и Либиху сплавляют смесь 3 вес. ч. сухого №гСОз
и 5 вес. ч. хлористой меди (CuCl) и. сплавленную массу выщелачивают водой;
СигО остается в виде красного порошка.
3. По методу, разработанному Н. Долбоносовым и В. Сеслави-
ным2 действуют инвертированным сахаром на щелочной раствор
комплексного соединения, получаемого действием тростникового сахара на раствор
CuS04.
Для работы приготовляют:
A. Насыщенный при 45—50° раствор CuS04.
Б. 50%-ный раствор NaOH.
B. Насыщенный раствор неинвертированного сахара, содержащий 2 ч.
сахара в 1 ч. горячей (80—90°) воды.
1 L. A. Mc D о w е 11, Н. L. Johnston, J. Am. Chem. Soc.58, 20u9 (1936); C. 1, 3125 (1937).
* Соли редких к цветных металлов, стр. 26.
ззд
Медь
Г. Раствор инвертированного сахара, получаемый растворением ю
тростникового сахара в 50 ч. нагретой до 80° воды, подкисленной i ч-
H2SO4. Раствор оставляют на ночь при умеренном нагревании; утром 0п *"
деляют его объем и содержание инвертированного сахара. Ре- '
В фарфоровую чашку на 1—2 л помещают ПО мл раствора В и 152
раствора Б, хорошо перемешивают и добавляют в три приема 360 %Л
раствора А. После каждого прибавления раствора CuS04 перемешивают ж^4
кость до растворения образующегося осадка Си(ОН)г. Когда температур
смеси поднимется до 70—75°, прибавляют раствор Г в количестве, сфтвет
ствующем 13,2 г инвертированного сахара. Через 20—30 мин. в чашку снова
добавляют в том же порядке те же количества растворов £ и Л, подогре.
вают до 70—75°, приливают прежний объем раствора Г и повторяют операции
в том же порядке до наполнения жидкостью всего сосуда. Через 40 мин
сливают маточный раствор и немедленно размешивают осадок СигО с 10-крат-
ным количеством 0,5%-ной H2SO4. Дальнейшие промывки проводят 0,1%-ным
раствором H2SO4; * осадок, наконец, отсасывают и сушат при комнатной
температуре.
4. Смесь 10 ч. порошка Си и 12,5 ч. СиО нагревают в фарфоровом тигле,
помещенном в вакуированную печь в течение 5 час. при 1000°. После
охлаждения препарат растирают и подвергают вторичному нагреванию *.
5. М. Штраумайс и А. Цирулис2 применяют в качестве
восстановителя гидразин-гидрат, ЫгШ-НЮ. К 50 мл концентрированного раствора
Си(СНяСОО)г добавляют 3—5 мл 20%-ного раствора ЫгНЬ-НгО; при стоянии
жидкости выделяются пузырьки N2 и выпадает желтый или оранжевый
осадок СигО.
4Си(СНзСОО)2 + 5NfeH4 • НЮ «= 2СигО + 4N2H4 • (СНзСООН)2 + N2 + ЗН2О.
Осадок промывают водой, спиртом и эфиром. Выход 5 г. Содержание Си
в препарате 88,1% (теор. 88,82%)г
6. Очень простой метод получения красной СигО высокого качества
разработан Д. Н. Финкельштейном3: 120 г NaOH растворяют в 480 мл
горячей (50—70°) дестиллированной воды; после отстаивания раствор
фильтруют через двойной фильтр и оставляют на сутки. Если по истечении этого
времени раствор помутнеет, необходимо еще раз его профильтровать. 190 г
CuS04«5H20 растворяют в 1100 мл теплой воды я добавляют раствор 48 г
глюкозы4 в 32 г воды. Смесь фильтруют, следя, чтобы фильтрат был
совершенно прозрачным.
К фильтрату, имеющему температуру 32—35° (точное соблюдение
температурного режима оказывает существенное влияние на качество препарата),
быстро приливают раствгьр NaOH, имеющий комнатную температуру. Смесь
перемешивают и оставляют спокойно стоять. Через час осаждение СигО
заканчивается, на что указывает обесцвечивание раствора. Осадок промывают
водой декантацией, отсасывают на воронке Бюхнера, промывают горячей
водой до удаления S04" и высушивают при комнатной температуре. Сушку
можно также проводить в вакууме при 40-—75°.
Препарат обладает карминово-красным цветом и очень чист.
Свойства
Карминово-красный кристаллический порошок уд. в. 6,11 (продажная СигО
имеет уд. в. 4,07—6,11). Нерастворим в воде, растворяется в NH4OH.
Соляная кислота в отсутствии воздуха превращает его в белый кристаллический
порошок CuCl. Плавится выше 1230°. Получаемый иногда аморфный
препарат СигО имеет желтую или оранжевую окраску.
1 W. В I 11 z, о. R о h I f f u. H. U. v.V о g e 1, Z. anorg Ch. 220, 136 (1934).
■ M. S t r a u m a I $ и. А. С 1 r u 1 i a, Z. anorg. Ch-224, 107 (1935).<
■ Авт- свяд. * 47685 от 31 июля 1936 г.
* Можно aaiTb 80 I 6«%-мого гдюкомого сиропа, юддющагос! прг хзводстааяяьш отбросок*
Медь йодистая. Меди окись
339
«питание
Высокое качество продукта определяется его темным карминово-красным
дветом.
В препарате, в зависимости от степени его чистоты, допускаются наиболь*
gpe количества примесей, указанные в табл. 323.
Таблица 323
Допустимые примеси в Cu20
Примеси
I
Окись меди (СиО) . . • ,
Железо (Fe)
Другие тяжелые металлы
Щелочи ,
Сульфаты (SO/) . . . .
Допустимое содержание в %
в чистом для
анализа
0,200
0.025
0,000
0,025
0,015
в чистом
1.0
1,10
0,05
0,25
0,15
МЕДЬ ЙОДИСТАЯ (ИОДИД МЕДИ [1])
Cuprous iodide
CuJ Мол. в. 190,49
Kupferjodiir
Cuprum jodatum
Приготовление
По Дюфлосу раствор 132 г CuS04«5rLO в 300 мл воды насыщают
сернистым газом SO2, добавляя понемногу, при помешивании 87 г KJ в виде
50%-ного раствора. Сначала раствор и осадок буреют, но по мере
добавления $0* раствор обесцвечивается и осадок белеет. Выпавшую CuJ
отфильтровывают и промывают на бюхнеровской воронке до удаления
сульфатов (промывные воды не должны мутнеть после добавления НС1 и ВаС1г).
Промытый осадок высушивают в эксикаторе над CaCU, или нагревая в
фарфоровой чашке не свыше 50°, при помешивании шпателем. Выход 98%
теоретического.
Все операции получения CuJ должны проводиться в темноте.
Свойства
Белый кристаллический порошок уд. в. 5,3—5,6, прочно адсорбирующий воду.
Почти нерастворим в воде; растворяется в концентрированной H2SO4 с
фиолетовой окраской. Растворим также в NH4OH, HC1 и растворах KCN. При
осторожном нагревании становится тёмнокрасным, коричневым и, наконец,
черным; по охлаждении возвращается первоначальный цвет. Т. пл. 602°,
т. кип. 770°.
МЕДИ ОКИСЬ (ОКИСЬ МЕДИ [2])
Cuprum oxydatum
Copper
Cupric
oxide
oxide
Kupferoxyd
CuO Мол. в. 79,57
Приготовление
1. В профильтрованный горячий (80—90°) раствор 34,5 г NaOH в 600
воды вливают горячий раствор 100 г CuS04 • 5НгО в 400 мл воды (не
оборот). Нагревают смесь еще 10—15 мин. при помешивании и
мл
н а-
перели-
340
Медь
вают раствор с осадком в банку емкостью 2—3 л. По возможности бьгст
промывают осадок 2 раза декантацией, выбрасывают его на полотняньг
фильтр и дают жидкости стечь возможно полнее. Затем снова перенося
осадок в банку и промывают до удаления SO4" (гтоба поомывной жит,Лп>Т
в течение 2 час. не должна мутнеть от ВаСЬ).
Декантируют, прибавляют 40 мл ЫНЮН (уд. в. 0,91) и дают стоять iy2^
2 часа при частом взбалтывании. Разбавляют водой, декантируют, повторяв
промывание ЫНЮН и снова промывают водой 5—6 раз до полного удаления
из осадка S04" (проба осадка, растворенная в НС1, по добавлении ВаСЬ не
должна выделять осадка даже по истечении суток).
Окончательно промытую окись меди отсасывают, высушивают на песочной
бане при 200—300° и полученные куски измельчают.
2. Растворяют 100 г CuS04«5H20 в 800 мл воды, фильтруют и добавляют
60—65 мл NH4OH (уд. в. 0,91) до ясно голубой окраски. Выпавший Си(ОН)2
промывают так же, как в способе 1, сначала водой, затем ЫНЮН1. Промытый
Си(ОН)г выбрасывают на полотняный фильтр, дают стечь жидкости, осадок
перекладывают в форфоровую чашку и нагреванием на песочной бане
переводят в СиО.
3. По указанию- Сингаловского2 500 г технического CuS04 • 5Н20
растворяют в 2 л воды, добавляют 2,5 мл Н2О2, нагревают, добавляют 5 г
окиси меди (СиО) и кипятят, причем окисленное железо выпадает в виде
Fe(OH)s. Фильтруют, прибавляют к горячему фильтрату прозрачный,
отстоявшийся раствор 180 г NaOH в 900 мл воды и в течение 20—25 мин. нагревают
при 80—90°. Выпавший осадок принимает черно-бурый цвет и довольно
быстро оседает.
Жидкость сливают, осадок промывают декантацией до удаления сульфатоп
(S04") и щелочи (ОН') в промывных водах, отсасывают и сушат при 300°.
4. Азотнокислую медь [Ca(NOs)2- ЗН2О] помещают в тигель я
прокаливают сначала осторожно, затем более сильно, перемешивая палочкой.
Оставшуюся черную СиО растирают в фарфоровой ступке.
о. Для получения гранулированной СиО полужидкую массу, приготовлен-
иую по способу 1, 2 или 3, намазывают на медный лист с углублениями и
высушивают при 100—150°. Полученные гранули прокаливают на глиняных
тарелках в электрической печи при 920—950° в течение 2 час. (Указанный
предел температуры необходимо точно соблюдать.)
6. По А. А. Казамцеву3 можно избежать длительной промывки СиО,
.необходимой при получении препарата из CuS04 и NaOH. Нагретый до 60—
70° раствор 50 г CuS04 • 5ШО в 200 мл воды осаждают небольшим избытком
NH4OH. Выпавшую основную соль помещают на полотняный фильтр. На
следующий день влажный осадок помещают в муфельную печь, возможно
быстрее нагревают до 800—900° и выдерживают при этой температуре
2—3 часа. Примесь S04" в препарате не превышает 0,02%.
7. По методу ИРЕА4 к горячему раствору 400 г CuS04 • 5НЮ в 2 л воды
добавляют 25 мл 10%-ной H2SO4 и 100 г чистых обрезков листового железа.
Смесь время от времени перемешивают. Когда Си будет полностью выделена,
зеленый раствор сливают, а осадок дважды промывают декантацией горячей
10%-ной H2SO4 для растворения Fe. После того как отделение железа
закончено (пробу Си растворяют в HNOs, пересыщают NH4OH и фильтруют — на
фильтре не должно оставаться желтоватого^ нал-ета), осадок промывают
на воронке Бюхнера горячей дестиллированной водой, следя, чтобы
поверхность меди была все время покрыта водой.
Когда промывные воды дадут отрицательную реакцию на S04", влажный
яорошок меди прокаливают 2—3 часа при 700—800° в фарфоровой чашке,
помещенной в электрическую печь, изредка перемешивая. По охлаждении
1 Промывают водой возможно тщательнее, так как находящийся в растворе
NH4)aSO, сильно повышает растворимость Cu(OHJa в NH4OH.
■ Соли редких и цветных металлов. 180.
• ЖПХ 11, № 7-8, 1108 (1938).
* Раакт. и преп„ 226.
Медь сернокислая
341
рудные гранулн разбивают в фарфоровой ступке, просеивают через сито
^отверстиями не более 1 мм и вновь прокаливают 1 час. Чем суше порошок
Си перед прокаливанием и чем чаще перемешивание, тем мельче получаются
гранули.
г Для получения активной СиО влажный препарат, полученный по методам
j или 3, высушивают в продолжении 6 час. при 110—120° и хранят в плотно
закупоренных банках
С в о й с т в а
Коричнево-черный порошок уд. в. 6,32—6,43 или (полученный по способу 1
я 5) твердые черные кусочки. Т. пл. 1148°. Несколько растворима в NH4OH;
рочти нерастворима в воде (2,3 • 10~3%) *.
Препарат, полученный при низкой температуре, легко растворяется в раз-
Евленных кислотах; сильно прокаленная СиО медленно растворяется даже
горячих концентрированных кислотах. При нагревании в струе водорода,
иси углерода или паров органических веществ СиО легко восстанавливается
металлической меди.
Испытание
Наибольшие количества примесей, допустимых по ОСТ НКТП 7111/448 и
12/449 в препарате квалификации ч. д- а., указаны в табл. 324; окись меди
порошке должиа выдерживать испытание на присутствие свободной щелочи.
Таблица 324
Допустимые примеси в СиО
Примеси
Допустимое содержание в %
ОСТ НКТП 7U1/448
(окись меди в
порошке)
ОСТ НКТП 7112/449
(окись меди
гранулированная)
1* Нерастворимые в НС1 вещества . .
w* Растворимые в воде вещества . . .
Р1 Хлориды (С1')
1 Сера (общее содержание в пересчете
J» на SO/)
5. Вещества, не осаждаемые H2S , . .
б.Азот (N)
7.Углеродистые соединения (С) . . .
0,02
0,10
0,003
0,05
0,25
0,002
0,020
0,05
0,003
0,05
0,25
0,002
0.020
МЕДЬ СЕРНОКИСЛАЯ (СУЛЬФАТ МЕДИ [2], МЕДНЫЙ КУПОРОС2)
Cuprum sulfuricum
Copper sulfate
Blue vitriol
Kupfersulfat
Kupfervitriol
CuSO 6H2O
C11SO4
Мол. в. 249,71
Мол. в. 159,63
П риготовление
1. Наиболее легким путем чистый препарат получают из электролитной
металлической меди.
1 L. A. Mc D о w е 11, Н. L. J о h n s t о n, J. Am. chem. Soc. 58, 2009 (1936).
• Термин „медный купорос" относится только к б-водному препарату.
342
Медь
В фарфоровую чашку наливают 120 мл воды, прибавляют 46 мл х
H2SO4 (уд. в. 1,80 и помещают в смесь 40 г чистой медной проволоки в в'ид1'
спирали (например, звонковой). Нагревают до 70—80° и при этой температур
в течение часа постепенно порциями по 1 мл прибавляют 11 мл концентри!
рованной HNOs. (Если спирали покроются кристаллами, добавляют 10—20 Ml %
воды.)
Когда реакция закончится (что бывает заметно по прекращению выделения
пузырьков газа), вынимают оставшиеся нерастворенными куски проволоки
и, выпарив раствор до появления тонкой кристаллической пленки, дают
охладиться. Выпавшие кристаллы отсасывают и 2—3 раза перекристаллизовы-
вают из дестиллированной воды. Наконец, препарат высушивают на стекле
при комнатной температуре.
2. Часто приходится очищать технический медный купорос. Так как даже
многократная перекристаллизация не дает возможности освободиться от
железа, то прибегают к очистке химическим путем. Для этого раствор CuSO*
кипятят с перекисью свинца РЬОг (или перекисью бария ВаСЬ), пока
отфильтрованная проба жидкости не покажет отсутствия Fe *)• Тогда раствор
выпаривают до образования легкой пленки и охлаждают для кристаллизации.
Этим методом получают препарат, почти свободный от железа, но
содержащий некоторое количество щелочей (всегда имеющихся в РЬОг и ВаОг).
По Н. Шоорлю2 к горячему раствору CuS04 добавляют небольшие
количества Н2О2 и NaOH, кипятят и отфильтровывают осадок. Выпавшие из
фильтрата кристаллы C11SO4 • 5Н20 дважды перекристаллизовывают.
Полученный препарат свободен от Fe и содержит не более 0,01% H2SO4. Соль
нагревают 24 часа при 105°, причем образуется моногидрат CuSO* • Н2О. Затем
выдерживают препарат в эксикаторе над 13%-ной H2SO4 до постоянного веса,
на что требуется 1—2 дня.
Полученный препарат точно отвечает формуле CuSOi • 5НгО. Он очень
устойчив; дает с водой совершенно прозрачные растворы и вполне пригоден
в качестве исходного вещества при установке титра.
3. Для приготовления безводного препарата нагревают
мелкокристаллическую C11SO4 • 5НзО в плоской чашке на песочной бане не выше 220° при
постоянном перемешивании.
Полученный белый порошок растирают в горячей ступке и помещают
в сухую, плотно закрывающуюся банку. Серый цвет препарата указывает
на перегрев.
Свойства
CuS04 • 5НгО — триклинические синие кристаллы уд. в. 2,29, неприятного
металлического вкуса. На воздухе несколько выветриваются. Выше 100°
начинает терять кристаллизационную воду13, переходя последовательно в голубые
гидраты CuS04-4H20, CuS04-3H20 и Cu304 • Н2О; при 220° образует
безводную C11SO4 — белый порошок уд. в. 3,606, жадно притягивающий воду,
с образованием гидратов. Остаточная влажность воздуха над безводной CuS04
при 25° составляет 1,40 мг НЮ на л 4-
При 653° начинается распад на СиО и SOs, который заканчивается при
720°. Растворима в воде и разбавленном спирте, нерастворима в абсолютном
спирте. Водные растворы имеют слабокислую реакцию. Растворима в
концентрированной НС1 со значительным поглощением тепла. Такой раствор при
выпаривании оставляет хлорную медь, CuCls.
1 Испытание на Ре производится следующим образом. Прибавляют к раствору избыток
NH4OH до растворения выпадающего вначале Си (ОН),. Полученный интенсивно-снний рас.вор
фильтруют и тщательно промывают фильтр водой. В случае присутствия железа на фильтре
ясно заметен буры t налет Fe(OH„
■ N Schoorl, Pharm. W.ek-Л. 76, 1841 (1939); С. 1, 433 П940).
• По Н. Д е м а с ь е и Б. Федорову CuS04' бНаО начинает терять воду при 60°; при
100е образуется CuS04. 3 НаО.
При 14и° он переходит в CuS04. Н„0; при 240° образуется безводный CuS04 (M. De та**
sleue. В. Fedoroff, С.ч. 20о, 4э7 (1937);. С. 1,42 (1938,1.
4 Н. S. В о о t h, L. Н. J4cl n t v г е Ind. En$. Chemn Anal. Ed. 2, 12 (1930).
Медь хлористая
343
Растворимость OtS04 в воде
Таблица 325
ГС
0
15
25
31
°/о CuS04
12,9
16.2
18,7
20.3
ГС
40
[ 50
60
70
о/о CuS04
22,8
25,1
28,1
31.4
ГС
80
90
100
°/о CuS04
34.9
38,5
42.4
Таблица 326
Удельный вес водных растворов CuS04
о/о CuS04
1
2
4
6
н*0
1,009
1,01е)
1.040
1,062
°(о CuSO«
8
10
12
.20
rf4
1,084
1,107
1,131
о/о CuS04
14
16
18
4°
1,154
1.180
1.206 .
Испытание
По ОСТ 10538-39 содержание CuS04 • 5НЮ должно быть не менее 99%'
для препарата х. ч. и ч. д. а. и не менее 98% Для препарата чистый,
наибольшие количества допустимых примесей указаны в табл. 327.
Таблица 321
Допустимые примеси в CuS04 • 5Н20
Примеси
Допустимое со ерж. ние в 0,0
. имически
чистом
1. Нерастворимые вещества
2. Хлориды (С1')
3. Железо iFe)
4. Соли металлов, не осаждаемых H2S
С.0'2
СО il
0,001
0,05
в чистом для
ан лиза
0.005
0/4)2
0,01
0,1
0,01
0,005
0,03
0,2
МЕДЬ ХЛОРИСТАЯ (МЕДЬ ОДНОХЛОРИСТАЯ, ХЛОРИД МЕДИ [1])
Cuprous chloride Kupferchlorur
CuCI Мол. в. 99,03
Cuprum chloratum
Cuprum monochloratum
Приготовление
По методу, разработанному в ИРЕА П. И. К у в а л д и н ы м и В. С. Н е-
чаевой1, в фарфоровой чашке при подогревании до 70° растворяют в 2 л
воды 240 г медного купороса и 480 г NaCl. Поддерживая ту же температуру,
* Режкт. и преп^231.
344
МёдВ
и раствор подвешивают марлевый мешок с 240 г осажденной меди *. i|
10 мин. жидкость обесцвечивается; тогда раствор быстро фильтруют в с^ез
клянную бутыль, содержащую 5 л воды и 20 мл НС1 (уд. в. 1,19). При сбпп^
косновении фильтрата с водой немедленно образуется белый осадок СцгГ
После двухчасового стояния жидкость декантируют и осадок CuCl быстп *
отсасывают на воронке Бюхнера с полотняным фильтром. Ро
Хорошо отсосанный осадок размешивают в воронке с 40 мл 2%-ной ftp,
ii снова отсасывают; такую промывку повторяют до полного удаления $0»
Затем осадок промывают несколько раз этиловым спиртом, помещают тонким
слоем в фарфоровую чашку и сушат пря 100° при частом перемешивании
Выход 90—95 г (94-47% теор.)2.
Свойства
Белые тетраэдрические кристаллы уд. в. 3,53—3,68, быстро зеленеющие на
воздухе вследствие образования основной соли.
Т. пл. 425°, т. кип. около 1000°. В воде растворима очень плохо;
растворяется в NH4OH, горячей концентрированной НС1 и растворах щелочных
хлоридов (КС1, NaCl).
Из этих растворов CuCl снова осаждается водой,
Таблица 328
Растворимость CuCl в соляной кислоте
г НС1 в 100 мл
раствора
4.96
14,12
18.29
г CuCl в 100 мл
раствора
1Л
7.5
12,2
г HCI в 10Э мл
раствора
22,98
25.6
г CuCl в 100 мл
раствора
18,7
21,8
Таблица 329
Растворимость CuCl в растворах NaCl
г NaCl в 100 мл
раствора
z CuCl в 100 мл
раотвора
г NaCl в 100 мл
раствора
г CuCl в 100 мл
раствора
0,93
4,7
8.0
12,3
0,12
0,5
1,2
2.9
17,5
24,3
37,0
5,4
12,9
29,8
испытание
По ОСТ 5013 содержание CuCl в препарате квалификации ч. д. а. должно
быть не менее 95% и в чистом — не менее 90%; наибольшие количества
допустимых примесей указаны в табл. 330.
1 Можно поместить в раствор спирали из электролитной меди, например, авонковой
проволоки, так, чтобы вся жидкость была ими пронизана. Однако в этом случае юсстановленне
продолжается несколько часов.
■ О применении инвертированного сахара для получения CuCl см. В. П а н а с ю к, Завод,
лаб. № 4,43 (1933); получение CuCl из CuO, CuHCl описано в работе Сп. В. De Witt, Chemist-
Analyet 24. № 4,15 (1935/, С. 1,1196 (1936).
Медь хлорная
345
Таблица 330
Допустимые примеси в CuCl
Примеси
Допустимое содержание в о;0
в чистом для
анализа
0,01
0,2
0,15
0.002
0,001
в чистом
0,03
! 0,4
0,3
, 0,005
0,005
Нерастворимые в кислоте вещества
Сульфаты (SO/)
$ щелочные и щелочноземельные металлы
1 Нерастворимые в
ГСульфаты (SO/)
«.щелочные и —
| Железо (Fe)
£ Мышьяк (As)
МЕДЬ ХЛОРНАЯ (МЕДЬ ДВУХЛОРИСТАЯ, ХЛОРИД МЕДИ[2])
fftjprum bichloratum Copper chloride Kupferchlorid
Cupric chloride
CuCh • 2H20
Мол. в. 170,52
приготовление
1. В фарфоровую чашку наливают 135 мл воды, 140 мл НС1 (уд. в. 1,19)
ж 43 мл HNOs (уд. в. 1,4) и вносят в полученную смесь 50 г медной прово-
коки, нарезанной мелкими кусками. Реакция сначала идет очень бурно, затем
ослабевает; в конце полезно смесь немного подогреть.
Жидкость фильтруют и, выпарив до Vs объема, дают охладиться.
Выпавшие кристаллы отсасывают и, растворив в воде, снова упаривают раствор до
«рявления тонкой кристаллической пленки, после чего охлаждают. Выпадаю-
РЙие чистые кристаллы СиСЬ • 2Н20 сильно отсасывают и промывают спир-
JitoM. После этого препарат рассыпают тонким слоем на стекле и сушат при
30°, изредка размешивая.
2. По Шмидту1 1 ч. окиси меди или 1,4 ч. основной углекислой меди
•растворяют при нагревании в 4 ч. чистой НС1 (уд. в. 1,12). Фильтруют и
выпаривают фильтрат до появления кристаллической пленки. Охлаждают и
выпавшие кристаллы тщательно отжимают между листами фильтровальной
•бумаги .или, лучше,, между пористыми фарфоровыми пластинками.
Если желают иметь препарат в хорошо образованных кристаллах, то
полученный кристаллический порошок перекристаллизовывают из кипящего
спирта, подкисленного несколькими каплями НС1.
3. Для получения безводной СиСЬ нагревают дигидрат (СиСЬ • 2Н20) в токе
сухого хлористого водорода при 140—150°.
Полученную массу переносят в эксикатор с H2SO4 и NaOH и выдерживают
до удаления адсорбированного хлористого водорода. -
Свойства
Зеленые призматические кристаллы уд. в. 2,50, расплывающиеся на
воздухе. Легко растворима в воде, метиловом (1:7,3 при 20°) и этиловом
спиртах. Растворима в эфире и пропиловом спирте (С3Н7ОН) и несколько
растворима в амиловом спирте (CsHnOH).
Насыщенный водный раствор СиСЬ кипит при 169°. Безводная CuClg —
желто-бурая гигроскопическая масса уд. в. 3,054.
i Ausfuhrl. Lehrb. d. pharm, Ch, 1, 994.
346 Мышьяк
Таблица 331
Растворимость СиС1а
вводе
ТаблцЦа 332
Растворимость СиС12 в 9т
ловом спирте и*
(Etard)
/'С
0
17
31,5
55,0
°/о CuCl,
41,4
43,Сб
44,7
46,5
/•С
68
73
91 .
•/о CuCIt
47,9
49,6
51,0
ГС
CuCl,
о
19
20
32
35,7
35,9
ГС
"'■•> CuCl,
38
50
"ЗР,5
41,7
Таблица 333
Удельный вес водных растворов CuCl?
0|o CuCl,
1
2
4
6
8
1,007
1.017
1,036
1,056
1,075
•/» CuClj
10
12
14
16
18
d4.
1.096
1.116
1.138
1,159
1.182
»l'o CuCl,
20
22
24
26
d\
1,205
1.230
1,253
1,278
И спытание
Наибольшие количества примесей, допустимых по ОСТ НКТП 6414/322,
указаны в табл. 334.
Таблица 334
Допустимые примеси в СиС12• 2Н20
Приыесв
Допустимое содержание в °|0
в чистом для
анализа
в чистом
1. Нераствогимые вещества
2. Сульфаты (SO^)
3. Щелочные и щелочноземельные металлы
(в виде сульфатов)
4. Железо (Fe)
5. Мышьяк (As)
0,01
0,005
0,05
0,003
0,002
0,02
0,02
0,2
0,005
0,005
мышьяк
Arsenicum Arsenic Arsen
As At. в. 74,91
Приготовление
1. По Орлову1 совершенно свободный от сурьмы препарат получают
следующим путем. К раствору чистою мышьяковокислого натрия (NasAsCV
прибавляют магнезиальной смеси (см. стр. 314); выпавший осадок MgNthAsCb
* О г I о w, Ch«m. Zt*. 25, 290 (1901). РгЗр. Ch. 219,
Мышьяк. Мышьяк трехсернистый
34?
-Аяльтровывают, растворяют в НС1, осаждают раствор осторожным
прибавлением NH4OH, собирают среднюю фракцию кристаллов, промывают и вы-
*У0авъют. Полученный продукт смешивают с сахарным углем и нагревают,
рцчем возгоняется чистый мышьяк.
" 2. Растворяют -мышьяковистый ангидрид (АвгОз) в крепкой НС1, приба-
^лот хлористого олова (SnCU • 2Н2О) и нагревают; осаждающийся ' мышьяк
вбирают на фильтр и промывают водой до нейтральной реакции. Высушен-
^Й остаток возгоняют в струе СОг.
3. Для освобождения продажного мышьяка от обычных примесей висмута,
цеди « железа его возгоняют в реторте или колбе; освободиться от сурьмы
*аким путем не удается.
I 4. Для очистки продажного мышьяка от серого налета окислов его
кипятят с 10—15%-ным раствором двухромовокислого калия (К2СГ2О7) и
небольшим количеством H2SO4, затем хорошо промывают водой, спиртом и эфиром,
высушивают и хранят в сухом месте. Можно также удалить легко летучий
АвгОз, нагревая препарат в токе СОг ниже температуры возгонки мышьяка
(360-365°).
Свойства
Серовато-белые, очень хрупкие кристаллы с сильным металлическим блес-
iM уд. в. 5,726 при 14°. Ниже 360° не испаряется; точка сублимации лежит
и 449,5° 1. На воздухе As быстро покрывается слоем окислов, теряя блеск.
од давлением сплавляется при 817°. При 200° фосфоресцирует при доступе
Еисл рода. Не растворяется в воде и кислотах, кроме НМОз и царской водки.
Как сам мышьяк, так и все его соединения чрезвычайно ядовиты.
МЫШЬЯК ТРЕХСЕРНИСТЫЙ (СУЛЬФИД МЫШЬЯКА[3])
Arsenicum trisulfuratura Arsenic sulfide Arsentrisulfid
A82S3 Мол. в. 246,00
А. Обыкновенный трехсернистый мышьяк
Приготовление
Пропускают сероводород в солянокислый раствор мышьяковистой кислоты-.
(As203).
Для очистки осадок растворяют в NH4OH, NaOH или Na2S и
отфильтровывают нерастворившуюся часть. К фильтрату добавляют НС1 до кислой
реакции для разрушения сульфосоли, например:
2Na3AsS3 + 6НС1 = AS2S3 + 6NaCl + ЗН*3.
Осадок As2Ss, промытый водой и спиртом, высушивают при 90—95°, после
чего для удаления серы подвергают его экстрагированию сероуглеродом в
аппарате Сокслета.
Наконец, препарат высушивают на воздухе до исчезновения запаха CSi
(огнеопасно\).
Свойства
Желтая аморфная рассыпчатая масса уд. в. 3,46, при кипячении с водой
выделяющая некоторое количество H2S и образующая следы мышьяковистой
кислоты. Вводе почти нерастворим (5,17 • К)-50/,, при 18°). Плавится при 310°
и, без доступа воздуха, испаряется выше 700°,
1 По другим данным при 630е,
348
Мышьяк, Натрий
Б. Коллоидальный трехсернистый мышьяк
Приготовление
1. Очень устойчивый коллоидный раствор получается по Ваниц0 •
пропускании сероводорода в раствор 1 ч. AS2O3 в 80 ч. прокипяченной
стиллированной воды. Де*
Для получения концентрированного раствора Шульце рекомендуй
к приготовленному вышеописанным способом разбавленному раствору приб
вить новую порцию АэгОз, снова пропустить H2S и, повторив эти оЛеращ?
несколько раз, отфильтровать жидкость от твердого AsaSs. По Шульце таким
путем была достигнута концентрация 37,46% AsaSs.
2. Очень устойчивый золь As2Ss получается при пропускании H*S в раствпп'
AseSs в CHsCOOH1. P
Свойства
Разбавленный коллоидный раствор, будучи чисто желтым в проходящем
свете, в отраженном флюоресцирует красноватым цветом. Концентрированный
коллоидный раствор под микроскопом прозрачен: невооруженному же глазу
представляется в виде непрозрачной, интенсивно желтой жидкости.
При добавлении к раствору осажденного BaS04 выделяется осадок As2Sa,
МЫШЬЯК ТРЕХХЛОРИСТЫЙ (ХЛОРИД МЫШЬЯКА[3])
Arsenicum trichloratum Arsenic trichloride Arsenchlorur
AsCh Мол. в. 181,28
Приготовление
1. По Дюма2 пропускают сухой хлор над помещенным в реторту
мышьяком и слабым нагреванием отгоняют образовавшийся AsCls в приемник. Де-
стиллат подвергают вторичной перегонке над порошком мышьяка для
освобождения от избыточного растворенного хлора.
Вследствие ядовитости паров AsCb работа должна производиться с
осторожностью и непременно в вытяжном шкафу с сильной тягой.
2. По Ризенфельду3 в трубку иенского стекла помещают 30 г
мышьяковистого ангидрида (AS2O3) и соединяют, с одной стороны, с холодильником
и приемником (склянкой Вульфа), а с другой — с аппаратом для получения
хлористого водорода (НС1).
Подогревая трубку (горелкой или электрической печью) до 200°,
пропускают медленный ток хлористого водорода; в приемнике собираются два слоя
жидкости, из которых нижний представляет собой AsCls. Его отделяют в
делительной воронке и дважды перегоняют в вакууме, отбрасывая первую и
последнюю фракции.
Выход около 40 г.
Свойства
Бесцветная, маслообразная, чрезвычайно ядовитая жидкость, уд. в. 2,16,
дымящая на воздухе. При —18° закристаллизовывается в игольчатые
кристаллы с перламутровым блеском. Т. пл. —13°, кипит при +Ь30,4° без
разложения. 1 моль AsCls требует для своего растворения 9 молей воды, полу-
i A. L. Elder, P. N. Burkard, J. phys. Ch. 41, 622(1937).
■ Po ggr, 9, 308; Prep. Ch. 227; Ind. En£. Chem. 31, 109 (1919).
• Неорг. практ., 247.
Мышьяк треххлористый. Натрий
349
titft раствор допускает смешение еще с 9 молями воды; при дальнейшем
Давлении происходит гидролитическое расщепление с выделением AS2O3.
^Чистый AsCU растворяет йодистые соли щелочных металлов; растворяет
^е в значительных количествах серу, фосфор и масла.
&**
НАТРИЙ
Natrium Sodium Natrium
Na Уд. в. 22,997
Mjkc т к а
1. Для очистки натрия, который покрылся под керосином коркой, Розен-
|&льд рекомендует следующий способ. Очищаемый кусок натрия помещают
\ смесь 3 ч. керосина и 1 ч. амилового спирта (С5Н11ОН) и трут его
тряпочки, пока металл не примет серебряный цвет. Тогда погружают его в керосин
5% СбНпОН и, наконец, моют очищенным керосином с 0,5—1% СвНиОН.
Через некоторое время металл покрывается желтым слоем амилата
натрия, который легко стирается фильтровальной бумагой. (Таким же методом
УЬкно очистить калий и литий.)
2. По указанию ГЪрундеану1 можно хранить натрий с серебряно-белой
Й&верхностью неограниченное время.
Для этого расплавляют Na под бензином при температуре около 110°
опасно!). Затвердевшие капли, переносят в свежую порцию горячего бен-
,, после чего помещают их в расплавленный парафин (нагретый до 60—70°)
дают парафину затвердеть. В таком виде натрий хранится любое время.
случае необходимости расплавляют парафин нагреванием до 60°, блестя-
е корольки натрия пинцетом переносят в бензин и затем высушивают
льтровальной бумагой.
3. Для переработки лабораторных остатков натрия, их отжимают между
[стами фильтровальной бумаги и расплавляют под толуолом. Блестящий
алл отделяют от нерасплавившихся частичек.
Приготовление тонко'измельченного натрия*
1. В колбе нагревают натрий под керосином или ксилолом до 120° (огне*
еЬсно\), закупоривают колбу пробкой и, обернув полотенцем, энергично
тряхивают. Удалив пробку, дают спокойно охладиться, причем Na остается
в тонко гранулированном виде. Жидкость сливают и промывают металл пет-
ролейным эфиром, нагретым на водяной бане выше 50° (огнеопасно). Для
■го чтобы иметь возможность быстро высушить натрий перед
употреблением, его сохраняют под лигроином, имеющим низкую точку кипения (Л а с*
$ а р - К о н)8.
2. По Б р ю л ю * лучше и совершенно безопасно приготовлять тонко раз*
Дробленный натрий (натриевую пыль) следующим образом:
В круглодонной колбе нагревают натрий под слоем ксилола до
энергичного кипения последнего, закупоривают колбу пробкой, обвертывают
полотенцем и сильно встряхивают. При этом Na получается в чрезвычайно
раздробленном, очень реакционноспособном состоянии. Величину зерен можно широко
варьировать в зависимости от силы встряхивания, вплоть до частиц, неразли^
чаемых невооруженным глазом. Благодаря тому, что полученная пыль не
Прилипает к стеклу, ее легко смыть в другой сосуд при помощи толуола;
i последний сливают декантацией и заливают натрий, например, эфиром. Автор
[утверждает, что качество получаемого таким путем препарата делает
ненужным употребление натриевого пресса и резальной машины.
»
1 J. J. Р г u n d e a n и, С. 1, 731 (1936),
а РгЗр. Ch. 312.
• L а • s а г-С о h n, Arbeitsmethoden d, or*» Ch., 3 Aufl. 976 (1907)*
4 Br Uhl.Ber. 35, 351в (1»02),
350
Натрий
3. По Леви и 'Андроччи1 расплавляют натрий под парафином,.в.к *
натрий (по 100 г) в шестикратное количество расплавленного парафина
нагревают смесь до 120—125°. Жидкий натрий собирается на дне сосуЙ'
и может быть превращен в мельчайшие шарики (диам. 0,2—0,1 мм), ec^a
содержимое колбы в течение 10 минут беспрерывно сильно и довольно быстр
перемешивать.
Большую часть жидкого парафина удаляют декантацией, остаток промы
ванием теплым (50°) лигроином. Получающийся Na, по указанию автора
раздроблен тоньше и равномернее, чем при плавлении под высококнпящи^
керосином.
Свойства '/
Серебристо-белый, сильно блестящий металл, при обычной температура
мягкий, как воск. Т. пл. 97,6°. Начинает испаряться уже при 100°, хотя кипит
при 877,5°. Во влажном воздухе быстро окисляется, покрываясь коркой NaOH.
С водой реагирует чрезвычайно бурно, плавясь при этом и выделяя водород-
большой кусок натрия, брошенный в воду, сильно взрывает и может
причинить опасные повреждения. Пары натрия разъедают стекло. Хранить натрий
следует под керосином (имеющим нейтральную реакцию) или под
вазелиновым маслом.
Испытание (Мерк)
Соединения азота и посторонние металлы» а) 1 г натрия отжимают от
Керосина фильтровальной бумагой и, нарезав на мелкие кусочки, бросают
последние в 20 мл холодной воды. Полученный раствор NaOH при
нагревании не должен иметь запаха аммиака и от прибавления (NH^S не должен
изменяться, б) Приготовленный, как выше указано, из 1 г Na и 20 мл воды
раствор после добавления 10 мл НС1 (уд. в. 1,124—1,126) не должен изменяться
от сероводородной воды*
НАТРИЙ АЗОТИСТОКИСЛЫЙ (НИТРИТ НАТРИЯ)
Natrium nitrosum. Sodium nitrite Natriumnitrit
NaNOa Мол. в. 69.005
Приготовление
1. По Вейнландсу плавят в фарфоровом тигле 50 г азотнокислого
натрия (NaNOa) с 60 г металлического свинца; по охлаждении сплав
выщелачивают водой и осаждают растворившуюся окись свинца пропусканием СОг.
Дают выкристаллизоваться №гСОз и NaNOa и, выпарив досуха, кипятят
остаток с 96%-ным спиртом. Отфильтровывают от нерастворившегося NaNOs
и отгоняют от фильтрата спирт, причем остается NaNO* в виде белого
порошка.
2. Для очистки продажного (96—97%-ного) продукта растворяют 500 г
соли в 750 мл воды при подогревании, фильтруют и, упарив фильтрат до
начала выделения кристаллов, охлаждают. Кристаллы чистого препарата
отсасывают, а маточные растворы последовательно 1—2 раза выпаривают до
начала кристаллизации.
Выход чистого (100%-ного) препарата около 400—420 г.
С во йства
Бесцветные или слабожелтоватые микроскопические кристаллы уд. в. 2,17
или лучисто-кристаллическая масса. Хорошо растворим в воде, очень плохо
в абсолютном спирте.
Натрий азотистокислыа
351
годный раствор обнаруживает щелочную реакцию и, поглощая из воздуха
^ород, постепенно переходит в NaNOs. Сухая соль на воздухе устойчива.
дл: 271°,
Таблица 335
Растворимость NaN02 в воде
» /°0
- 8
0
+ 19
о/о NaNOt
40,8
41,9
44,9
*°с
52,5
65
81
о/0 NaNO,
51,4
54,6
57,9
/вс
92
103
128
в/о NaNOu
59,7'
62,6
68,7
Таблица 336
Удельный вес водных растворов NaN02
о/о NaNO,
1
2
4
6
8
10
,*20
1,005
1,011
1,024
1,(38
1,052
1,065
о/о NaNOe
12
И
16
18
20
И20
1,078
1,092
1,107
1,122
1,137
»/о NaNOi
24
28
32
36
40
-Г
1,168
1.198
1,230
1,264
1,299
Испытание
По ОСТ НКТП 7378/532 содержание NaNOi в препаратах квалификаций
х. ч. и ч. д. а. должно быть не менее 98%; наибольшие количества
допустимых примесей указаны в табл. 337..
Таблица 337
Допу ст имы е п
Примеси
1. Не растворимые в воде вещества . .
4. Тяжелые металлы группы H2S . . .
5. Железо (Fe) •
6. Калий (Ю
римеси в
NaNO*
Допустимое содержание в °/о
в химически
чистом
0,002
0,005
0,005
0,0002
0,0002
0,000
в чистом для
анализа
0,005
0,02
0,015
0.U005
0,0004
0,005
в чистом
0.01
0,04
0,03
0,001
0,001
0,01
352
Натрий
НАТРИЙ АЗОТНОКИСЛЫЙ (НИТРАТ НАТРИЯ) ,
Natrium nitricum Sodium nitrate Natriumnitrat
Chill salpetre Natronsalpeter
Chilesalpeter
NaNOa Мол. в. 85,005
Приготовление ф
Для очистки продажного продукта Лайти рекомендует к кипящему На.
сыщенному раствору NaNOs прибавить 1/ю часть по весу чистой HNOs
(уд. в. 1,35), тщательно перемешивать до охлаждения и промыть выпавщщ /
кристаллический порошок 10%-ной HNOs. Избыток кислоты удаляют осторож-
ным нагреванием.
2. Можно также к раствору нечистой соли добавить ЫаяСОз до слабо-
щелочной^реакции, прокипятить и отфильтровать выпавшие гидроокиси
посторонних металлов. Фильтрат слабо подкисляют HNOs, выпаривают до уд. в.
1,38—1,40 и охлаждают. Выпавшие кристаллы отсасывают, промывают малым
количеством холодной воды и сушат между листами фильтровальной бумаги.
Свойства
Бесцветные ромбоэдры гексагональной системы уд. в. 2,25,
расплывающиеся в сыром воздухе. Т. пл. 308°'; при дальнейшем нагревании выделяет
сначала кислород (переходя в NaN02), затем смесь кислорода, азота и ЫОг
(К. Лешевский) *. Хорошо- растворим в воде, значительно труднее в спирте.
Таблица 338
Растворимость NaNOa в воде
ГС
0
10
20
30
•/0 NaN08
42,2
44,6
46,8
49,0
ГС
40
50
60
70
% NaNO.
51,2
53,3
55,5
1 57,6
/вс
80
90
100
% NaN03
59,7
61,7
63,5
Таблица 39
Растворимость NaN03 в разбавленном этиловом спирте
Вес. %
спирта
0
10
20
30
40
о/о NaNOa
при 15°
45,7
39,5
32,8
26,2
20,5
при 30°
49,1
43,4
37,4
31,3
25,1
Вес. °,'о
спирта
50
60
70
80
90
•/о NaNO.
при 15е
10,2
—
2,6
—"■
при 30°
18,9
13,0
7,8
1,2
1 О реакции, протекающих при термическом разложении в NaNO. см. например, К. La
«с k e w a k i, Ber.72, 1768 Г1939); С. II 3233 (1939).
Натрий азотнокислый. Натрия амальгама
353
Таблица 340
ty NaN03
1
2
4
6
8
10
12
Удельный
-?
1,0049
1,0117
1,0254
1,0392
1,0532
1,0674
1,0819
вес водных растворов NaN03
% NaNOj
14
16
18
20
22
24
.20
d4
1,0967
1,1118
1,1272
1,1429
1,1589
1,1752
% NaNO,
26
28
30
35
40
45
w20
1,1917
1,2085
1,2256
1,2701
1,3175
1,3683
Испытание
По OCT 5110 содержание NaNCh в безводном препарате для всех
квалификаций должно быть не менее 99,8%; наибольшие количества допустимых
примесей указаны в табл. 341.
Таблица 341
Допустимые примеси в NaN03
Примеси
Допустимое содержание в °/0
в химически
чистом
0»004
0,0005
0,001
0,005
0,0003
0,0001
0,005
0,0005
0,0001
0,0005
0,005
в чистом
для анализа
0,005
0,002
0,003
0,010
0,0005
0,0005
0,010
0,001
0,0005
0,002
Не опре/
в чистом
0,01
0,002
0,006
0,020
0,001
0,001
0,020
, 0,001
0,001
0,004
шляется
1. Нерастворимые в воде вещества
2. Хлориды (СУ)
3. Хлораты и перхлораты(О') . . .
4. Сульфаты (SO/') .
5. Тяжелые металлы
6. Железо (Fe)
7. Кальций и магний (Са и Mg) . .
8. Фосфаты (Р04'")
9. Нитриты (NCV)
10. Иодаты (JCY) .
И. Калий (К)
НАТРИЯ АМАЛЬГАМА
Sodium amalgam Natrium amalgam
Приготовление
1. По Ванино1 для приготовления низкопроцентной (¥i—2%) амальгамы
вносят вычисленное количество очищенного от корки металлического натрия
в нагретую до 50° сухую и чистую ртуть, налитую в фарфоровую чашечку.
Свеже нарезанные тонкие ломтики натрия насаживают на длинную
заостренную стеклянную палочку и при ее помощи прижимают под ртутью ко дну
чашки. Соединение сопровождается выделением пламени и, зачастую, брыз-
1 Ргар. Ch. 313.
354 Натрий
гами металла. (Осторожно! Необходимы предохранительные очки /■< Пе
После внесения всего натрия амальгаму хорошо перемешивают ст^Чаткч)
палочкой. tKJI*HHoi
2. Для приготовления 5%-ной амальгамы Лассар-Кон1 3т»екомА
следующие способ. Помещают ртуть порциями по 10 мл в фарс|( Оров ^Уетч
шечку и вносят туда довольно быстро 7 г чистого натрия, нарезаемого\1 Ча*
ками. Еще теплая амальгама легко вынимается из чашки, так как п Ч*
добавления последних порций натрия и последующего перемешивания °Cj*e
сбивается в шарики. В течение часа можно приготовить 10 так1}^ попц С{?
Для хранения рекомендуется запаять амальгаму в стеклянные ампулы, напо
ненные водородом или азотом. Можно также применить . баночка с' прите
тыми пробками. р*
3. Для получения 10%-ной амальгамы Эрдман2 предлагает Сильно на '
греть в железном котле под тягой 3 кг ртути и внести туда 30'' г натрия
кусками по 5 г, бросая куски возможно быстро один за другим Внесение
каждого куска должно сопровождаться появлением пламени; в противном
случае усиливают нагревание и помешивают массу железной пало^ч<ой. Жид.
кую массу выливают на железный лист, по затвердевании разбиваю^ на куски
которые еще горячими помещают в плотно закрываемую банку.
4. Для получения амальгамы в листочках Гиршфельдер ь<н Гарт3
предложили следующий простой способ: горячую, еще жидкую Амальгаму,
выливают при энергичном перемешивании механической мешалко* : в сосуд
с ксилолом; затем отфильтровывают через сухой фильтр и быстра высуши-
вают.
Свойства
'Амальгама с содержанием Na до 1% —жидкая, от 1 до 2,5% —
Тестообразная, от 2,5% и выше —твердая и кристаллическая. При стоянии !^а воздухе
амальгама покрывается пленкой NaOH вследствие реакции с кислородом и
водяными парами воздуха. Воду разлагает с выделением водороАа, однако
значительно медленнее, чем чистый натрий.
Испытание
10 г амальгамы вносят в 100 мл воды и оставляют смесь ^.-стоять до
прекращения выделения водорода, изредка встряхивая. Затем титрую : раствор
Ш ЙС1 с метилоранжем в качестве индикатора. \ мл IN HC1 соответствует
0,023 г Na.
НАТРИЯ АМИД
Sodium amide Natrium^rnid
NaNH* Мол. в. 39,021
Приготовление
По Тизерлею4 в железную полированную реторту помещает 500 г
металлического натрия и через газопроводящую трубку, оканчивающуюся
почти у поверхности натрия, пропускают быстрый ток NHs. Аммиа . должен
быть тщательно высушен и очищен от. кислорода; последнее моА:ет быть
достигнуто, например, пропусканием газа через трубку, заполненную \
натриевой проволокой.
Когда весь воздух из реторты будет вытеснен, подогревают натрий до
300—400°; реакция идет весьма быстро и почти заканчивается в Sr'<-4 часа.
* Arbeltemethod. d. org. Ch. 3 Aufl., 939 (19ЭЗ).
* Хим. преп., 7.
■Hirschfelder. Hart, Ind. Eng. Chem. 12, 449 1929).
* T 11 h e г 1 e y, J. chem. Soc. 65, 604 (1884).
Натрий амид- Натрий-аммоний фосфорнокислый 355
ллследние количества натрия вступают в реакцию медленно и требуют боль-
£?£о избытка ЫНз. Когда выделяющийся из реторты газ уже не будет содер-
ть H*i реакция закончена. Охлаждают реторту в медленном токе NHs;
полуденный NaNHg разбивают на куски и хранят под сухим эфиром или
бензином.
двойства
Белая лучисто-кристаллическая масса, плавящаяся при 208° и возгоняю-
щаясЪ выше 400°.
,Водой разлагается с образованием ЫНз и NaOH. При нагревании в
вакууме до 300—330° распадается на Na, N2, Н2 и NHs.
л Препарат, пожелтевший при хранении, следует уничтожить, так как при
работе с ним возможен взрыв. ,.,..,.
НАТРИЙ-АММОНИЙ ФОСФОРНОКИСЛЫЙ
Natrium phosphoricum Ammonium sodium Nafriumammonium-
ammoniatum phosphate phosphat
NaNH*HP04 • 4H20 Мол. в. 209,09
Приготовление
1. По Берцелиусу 60 г кислого фосфорнокислого натрия (Na2HP04 •
• 12Н«0) и 10 г NH4CI растворяют в 20 мл кипящей воды и охлаждают.
Выпавшие кристаллы переносят на простую воронку, дают стечь маточному
раствору, отжимают между листами фильтровальной бумаги (или, лучше,
отсасывают на воронке Бюхнера) и для окончательной очистки еще раз пере-
кристаллизовывают из возможно малого количества кипящей воды, добавляя
немного NH4OH.
2. 50 г 50% -ной Н3РО4 нейтрализуют в фарфоровой чашке при - помощи
NH4OH и растворяют в полученной жидкости 80 г измельченного Na*HP04 •
• I2H2O. Добавляют еще немного NH4OH до щелочной реакции, фильтруют и
кристаллизуют.
Препарат очшцают, как указано в способе 1.
3. К 500 г НзР04 (уд. в. 1,12), нагретой в большой фарфоровой чашке,
прибавляют постепенно 55 г NauCOs и выпаривают до половины объема.
Затем прибавляют 180 г 10%-ного NH4OH, горячий раствор фильтруют,
выпаривают до появления пленки кристаллов и, прибавив немного NHiOH,
оставляют для кристаллизации.
Свойства
Бесцветные моноклипические кристаллы уд. в. 1,55, легко растворимые
в воде. Раствор не действует на фенолфталеин, но на лакмус показывает
слабо щелочную реакцию. При 79° плавится, отдавая NHs и воду и образуя
ЫагНР04; при 200° переходит, теряя воду, в ЫагНгРгО? и затем (выше 243°)
в стекловидную массу мета фосфорнокислого натрия (NaPOs)e. На воздухе
выветривается и частично теряет NHs.
Испытание
Наибольшие количества примесей, допустимых по ОСТ 4417, указаны
п табл. 342; водный раствор препарата должен выдерживать испытание,
приведенное в ОСТ.
356
Натрий
Допустимые примесив NaNH4HP04 • 4Н20
Таблица зд
Принеся
1. Нерастворимые в воде вещества
3. Нитраты (NCV)
5. Mai ний (Mg)
6. Железо (Fe)
Допустимое содержание в %
в химически
чисюм
0.002
0.001
0,0005
0,005
0,001
0,0005
0,0005
0,0005
в чистом
для анализа
0,005
0.003
0.001
0,02
0,001
0,001
0,001
0.001
■ ЧИСТОМ
0,01
0,005
0,002
0,04
0,003
0,003
0,002
0.002
НАТРИЙ БОРНОКИСЛЫЙ (НАТРИЙ ТЕТРАБОРНОКИСЛЫЙ,
ТЕТРАБОРАТ НАТРИЯ, БУРА)
Natrium biboricum
Borax
Natriumpyroborat
Natriumbiborat
Borax
Borate of soda
Borax
Sodium tetraborate
Na2B407-ЮНЮ Мол. в. 381,43
А. Кристаллический
Приготовление
1. 140—150 г продажной кристаллической буры растворяют при нагревании
не выше 60° в 300 мл воды и полученный раствор фильтруют через
складчатый фильтр в фарфоровую чашечку, охлаждаемую снегом. Непрерывно
помешивая фильтрат стеклянной палочкой, получают препарат в виде тонкой
кристаллической муки. Отсасывают, промывают кристаллы небольшим
количеством холодной воды и повторяют перекристаллизацию, после чего
высушивают на воздухе в течение 2—3 дней. Полученный препарат отвечает
формуле ЫагВЮт • 10Н2О и вполне пригоден для установки титров.
2. 100 г борной кислоты растворяют в 10% -ном растворе 120 г №2СОз.
Фильтруют, выпаривают до уд. в. 1,16 и охлаждают. Выпавшие кристаллы
очищают по способу 1.
Б. «Жженая бура»
Нагревают кристаллическую буру (ЫагВЮ?* ЮНгО) в тигле или чашке;
соль сначала плавится, затем, теряя воду, переходит во вспученную пористую
массу. Тогда прекращают нагревание; массу по охлаждении растирают и
помещают в плотно закрывающиеся банки.
В. Безводный NazBiOr
Мол. в. 201,27
Истертую жженую буру помещают в огнеупорный тигель и довольно сильно
прокаливают. Соль плавится при 878° и при охлаждении затвердевает в
стекловидную массу. По другим источникам 1 т. пл. 748°.
* Тот о-о-Sit 6, С. 1, 1171 (1935).
Натрий борнокислый
357
Двойства
Na*B407« IOH2O — твердые белые призматические кристаллы уд. в. 1,72.
' Цри нагревании сначала плавится, затем вспучивается, теряя
кристаллизационную воду, и, наконец, переходит в стекловидную массу. До 200° устой-
qiiB моногидрат ЫагВЮ7 • НгО; последняя молекула воды по И. Кольт-
гофУ1 полностью удаляется при 400—450°.
Соль хорошо растворима в воде; значительно растворима в глицеоине;
.дочти не растворима в спирте. Водный раствор показывает на лакмус
щелочную реакцию и при добавлении НС1 до слабо кислой реакции окрашивает
|уркумовую бумажку в бурый цвет.
J: При кристаллизации соли из концентрированного раствора при 56—80°
%*падают октаэдрические кристаллы состава Na2B407-,5H20. Безводный
fJatB407 — бесцветные стекловидные кусочки уд. в. 2,37, поглощающие влагу
AiiipH лежании на воздухе и при этом мутнеющие.
>: Таблица 343
Растворимость Na2B407 в воде
(Blasdale, Slansky)2
leC
0
5
10
15
20
25 '
30
35
•/• N f-\07
1,18
1,44
1,76 1
2,12
2/8
3,13
3,85
4,76
t°c
40
45
50
55
60,8
65
70
75
0/0 Na,B407
6,0
7,5*
9,55
12,25
16,(5
Г,8
19,49
21,30
/°c
80
85
90
95
100
102,8
(т. кип.)
0/oNa,B407
23,38
25,73
2S37
31,28
3 ,63
36,73
Таблица 344
Удельный вес водных растворов Na2p4^7
о/о NatB407
0,5
1,0
1,5
2,0
*У
1,0042
1,0084
1,0131
1,0179
°/о NatB407
2,5
3,0
3,5
w20
1,0226
1,0274
1,0321
Испытание
По ОСТ 4102 для кристаллического препарата содержание №2ВЮ7 • ЮН2О
Должно быть для всех квалификаций не менее 99,5%; наибольшие количества
допустимых примесей указаны в табл. 345; препарат должен выдерживать
приведенное в ОСТ испытание на карбонаты.
\ I- М К о 1 t h > f f, J. Am. chem. Soc. 48, 14.7 (1926); H.Menzel, Z. amr*. Ch. 224.2 (1935).
• J. Am. chem. Soc. 61, 917 (1939J.
358
Натрий
Допустимые примеси в Na2B407.10Н2О
Таблица 345
Принеся
1. Нерастворимые в соляной
кислоте вещества
5. Железо (Fe)
Допустимое содержанке в о;0
в химжчески
чистом
0,003
0.0005
0,005
0,0005
0,0003
0,0001
в чистом
для анализа
0,005
0,002
0,01
0,001
0,0005
0,0005
в чистом
•
0,02
0,005
0,03
0,002
0,001
0,001
НАТРИЙ БРОМИСТЫЙ (БРОМИД НАТРИЯ)
Natrium bromatum Sodium bromide Natriumbromid
NaBr. 2H20
Мол. в. 138.945
Приготовление
1. По Ванино1 смешивают в колбе 50 г порошка железа и 400 г воды
и прибавляют к смеси постепенно маленькими порциями при хорошем
охлаждении и взбалтывании 96 г брома. Затем фильтруют слабо-зеленоватую
жидкость через маленький гладкий фильтр в фарфоровую чашку и добавляют
к раствору еще 32 г брома. Смесь нагревают до кипения и прибавляют затем
постепенно при помешивании раствор 232 г ЫагСОз в 1 л воды. Нагревают
еще некоторое время для лучшего осаждения Fe(OH)3. Жидкость над
осадком должна иметь только слабо-щелочную реакцию. Если осаждение не
закончилось, добавляют еще немного ЫагСОз до слабо-щелочной реакции и
нагревают в течение нескольких минут.
Раствор фильтруют, осадок на фильтре промывают горячей водой и
выпаривают фильтрат до кристаллизации; выпавшие кристаллы сильно отсасывают
и помещают в банку с притертой пробкой. При выпаривании маточных
растворов можно получить еще некоторое количество соли, которую для очистки
необходимо перекристаллизовать.
2. Для очистки продажного NaBr, содержащего значительные количества
NaCI, А. В. Раковский и В. В. Полянский2 предлагают следующий
способ. Продажную соль растворяют в абсолютном метиловом опирте
(растворимость, приблизительно, 17 : 100), отгоняют Vs спирта и отделяют выпавшие
кристаллы, содержащие почти все количество NaCI. Раствор после отгонки
оставшегося спирта дает чистый NaBr, содержащий лишь следы NaCI.
Свойства
Бесцветный кристаллический порошок уд. в. 2,18, щелочно-соленого вкуса,
притягивающий на воздухе влагу- При 15—20° кристаллизуется с 2 мол. воды,
выше 30° — безводный (уд. в. 3,2). Кристаллизационная вода легко отщепляется
как при нагревании выше 30°, так и при обыкновенной температуре при
стоянии над H2SO4 или СаС1г. Т. пл. 740°, т. кип. 1393°. Растворим в воде
(см. табл. 346), в абсолютном этиловом спирте (2,21 : 100 при 19,5°) и в
метиловом спирте (17,4: 100 при 19,5°).
1 Ргйр. Ch. 330
* Изв. ИРЕА, вып. в,б (Ю27).
Натрий бромистый
359
Растворимость NaBr в воде
Таблица 346
гс
0
20
40
50
о/о NaBr
44,3
47,5
51,4
53,7
гс
50,7
80
100
ПО
o/oNaBr
53,9
54,2
54,8
55,1
/•с
140
155
177
199
248
0,'oNaBr
55,6*
56,5*
57,3*
58.1*
60,8*
Таблица 347
o/oNiBr
1
2
4
6
8
10
12
Удельный вес водных растворов NaBr
И20
1
1,0060
1.0139
1.0298
1.0462
1.0631
1.0803
1,0981
ojoNaBr
14
16
18
20
( 22
24
w20
4
1,1164
1.1352
1,1546
1.1745
1,1951
1,2163
o/oNaBr
26
28
30
35
40
,20
dA
1,2382
1,26^8
1,2*41
1,3462
1,4138
Испытание
По ОСТ 4418 для безводного препарата содержание NaBr должно быть
Для всех квалификаций не менее 95%; наибольшие количества допустимых
примесей при расчете на безводный препарат указаны в табл. 348.
Таблица 348
Допустимые примеси в NaBr
t
Примеси
1. Нерастворимые в воде вещества
4. Сульфаты (SO/)
5. Азот общий (N) из нитратов,
12. Иодиды (J')
Допустимое содержание в °,'о
в химически
чистом
0.005
0.01
0,0005
0,002
0,001
0,002
0,0*2
0,001
0,0002
0,0002
1 0,05
0,02
в чистом
для анализа
0.01
0.03
0.002
0,005
0,002
0,004
0,005
0,003
0,0005
0,0005
0,2
0,05
в чистом
0,02
0,05
0.005
0,01
0.02
0.08
0,01
0,005
0,001
0.001
0,8
0,1
* По данным Г. К, Д и с т а и о в а, ЖОХ 7,
№ 3-4,676 (1937).
360
Натрий
НАТРИЙ !ВАНАДИЕВ0КИСЛЫЙ(ОРТО) (ОРТО-ВАНАДАТ НАТРИЯ)
Natrium vanadatum Sodium vanadate Natriumvanadat (ortho^
<ortho) j .
Na3V04-16H20 Мол. в. 472,20
Приготовление
18,2 г ванадиевого ангидрида (V2O5) нагревают с 31,8 г ЫагСОз до
полного прекращения выделения СОг. Сплав имеет сначала зеленый цвет,
который затем переходит в желтый и, наконец, при совершенном охлаждении в
белый. Остывшую массу растворяют в возможно малом количестве холодной
воды и тотчас же приливают большое количество спирта, избегая смешения -
жидкостей; спирт должен образовать сверху слой. Через несколько часов ^
выпадают бесцветные кристаллы; их промывают спиртом и высушивают ц
вакууме.
Свойства
Бесцветные игольчатые кристаллы, очень непостоянные. Водный раствор
показывает щелочную реакцию и постепенно разлагается. При кипячении
распадается на NaA^O? и NaOH. Т. пл. 866°.
НАТРИЙ ВИСМУТОВОКИСЛЫЙ (ВИСМУТАТ НАТРИЯ)
Sodium bismuthate Wismutsaures Natrium
NaBiOs Мол. в. 280,00
Приготовление
1. По И. Вахромйеву 200 з NaOH нагревают до красного каления
в железном или никелевом тигле и очень осторожно небольшими порциями
всыпают в тигель 100 г основного азотнокислого висмута. После полного
растворения так же осторожно добавляют в расплавленную массу маленькими
дозами 20 г Ыаг02.
При работе глаза обязательно защищать очками, а руки — толстыми
перчатками! Брызги расплавленного NaOH очень опасны!
Массу перемешивают железным прутом и выливают на железный лист;
после охлаждения разбивают продукт на мелкие куски и растворяют в 1,5 л
воды, размешивая деревянной палочкой. Осадок виомутата отфильтровывают
через двойной фильтр на воронке Бюхнера и промывают холодной водой до
полного удаления щелочи. Промытый осадок высушивают в фарфоровой
чашке при 100—105°, тонко растирают и пересыпают в банку. Содержание
NaBiOs в препарата1 около 79%.
2. По методу П. Н. Григорьева и Е. М. Осадченко2 90 г
азотнокислого висмута перемешивают с 180—200 мл воды, дают осесть основной
соли, отфильтровывают ее и нагревают влажную массу до 40—50°.
Одновременно в 500 мл 50%-ного раствора NaOH пропускают сильную струю
хлора до буровато-красного цвета (15—20 мин.). Нагревают полученный
раствор до кипения (не прерывая пропускания хлора) и приливают его к теплой
пасте полученной основной соли, причем белый цвет последней быстро
переходит в тёмнокрасный:
BiONOa + 4NaOH + СЬ = NaNOs + NaBiOa + 2NaCl + 2H20.
Осадку висмутата дают отстояться, промывают его до удаления CV (сначала
декантацией, затем на воронке Бюхнера) и высушивают. Выход 97%
теоретического. По утверждению авторов препарат точно отвечает формуле NaBiOs.
1 Завод, лаб. 3, М 3. 269 (1934).
> Завод, лаб. 4, J* 11,14 9 (1935).
Натрий ванадиевокислый, висмутовокислый и вольфрамовокислый 361
с*ойства
Делтый, тёмнокрасный или красно-коричневый аморфный порошок, являю-
шЙся сильным окислителем. Слегка гигроскопичен. Нерастворим в воде;
оудно растворяется в НЫОз на холоду, легко при нагревании.
Висмутат, повидимому, не представляет собой определенного соединения и
му только условно приписывают формулу №ВЮз; Э ф р а и м * считает
возможным, что висмутат является продуктом адсорбции NaOH на Bi20s.
Напротив, Цинтль2 считает, что висмутат имеет определенный состав.
Испытание3
Содержание NaBiOs должно быть не менее 70%' (иодометрическое или
ерманганатометрическое определение); раствор 1 г препарата в 10 мл HNCfo
и нагревании не должен приобретать розового или фиолетового оттенка
».
Кипятят 1 г препарата с 10 мл воды в течение 10 мин., добавляют 40 мл
>ды и фильтруют. Фильтрат при добавлении HNCh и AgNOs может показать
ь легкую опалесценцию (С1').
НАТРИЙ ВОЛЬФРАМОВОКИСЛЫЙ
(НОРМАЛЬНЫЙ ВОЛЬФРАМАТ НАТРИЯ)
Natrium wolframicum 'Sodium tungstate Natriumwolframat
Na2W04-2H20 Мол. в. 329,95
Приготовление
Растворяют вольфрамовый ангидрид (WCb) в концентрированном растворе
NaOH и выпаривают до кристаллизации.
Двойства
Просвечивающие листочки с перламутровым блеском или тонкие таблички
уд. в. 3,25 (безводный — 4,2). На воздухе выветривается; в вакууме или при
нагревании до 100° теряет всю воду. Вкус горько-соленый, вяжущий.
Безводная соль при 698° плавится в прозрачную жидкость. Растворим в воде:
нерастворим в спирте.
Таблица 349
Растворимое ть Na2W04 вводе
ГС
0
5
10
20
o:0NaaW04
36,5
41,0
41,9
42,2
ГС
40
80
100
o/oNaeW04
43,8
47,4
49,2
1 Неорг. химия, т. 2, стр. 194.
■ Z i a 11, Z. anor*. Ch. 245, 32, 1940.
• Испытание_хнм. реактивов, 270,
362
Натрий
Удельный вес водных растворов Na2W04
Таблица 35о
o[0Na1W04
ЛО
o('0NaaW04
^20
o'oNa^WO*
w^O
1
2
4
6
8
1.С074
1,0166
1,0354
1,0546
1,0742
10
14
18
12
1,0944
1,1372
1,1833
1,2328
26
30
34
38
1,2862
1,3444
1,4084
1,4786
Испытание (Мерк) ч
/. Внешний вид и растворимость. Препарат представляет собой бесцветные
призмы или ромбические пластинки, растворимые в 4 ч. воды со щелочной
реакцией.
2. Вода. 1 г препарата после сушки и слабого прокаливания должен
оставлять около 0,88 г остатка.
3. Хлориды {СГ). К раствору 1 г препарата в 20 мл воды прибавляют
5 мл HNOs (уд. в. 1,2), фильтруют и к фильтрату добавляют 5 капель 10%-
ного раствора AgNOs. Допускается появление лишь слабой опалесценции.
4. Сульфаты (SOS'). К раствору 1 г препарата в 20 мл воды прибавляют
5 мл HNOs (уд. в. 1,2), кипятят 10-«-15 мин., фильтруют и к фильтрату
добавляют 1 мл 10%-ного раствора ВаСЬ. Жидкость должна оставаться про
зрачной. *
5. Соли аммония {NHS). При нагревании 1 г препарата с 10 мл воды и
5 мл 30%-ного раствора NaOH не должен выделяться NHs.
6. Определение действующего начала. К раствору 1 г препарата в 10 мл
воды прибавляют 10 мл НС1 (уд. в. 1,125), выпаривают на водяной бане
досуха « сушат 30 мин. при 120°. К остатку добавляет 10 мл НС1 (уд. в. 1,125)
и повторяют выпаривание, сушку и добавление кислоты и воды еще 3—4 раза.
Остаток обрабатывают 20 мл 10%-ного раствора NH4NO3, смешанного
с 2 мл НЖ)з, на водяной бане в течение 15 мин., фильтруют, промывают
полученную вольфрамовую кислоту слабой HNOs, сушат, прокаливают и
взвешивают. Вес осадка должен быть не меньше 0,69 г.
НАТРИЯ ГИДРАТ ОКИСИ (ГИДРООКИСЬ НАТРИЯ, ЕДКИЙ НАТР)
Natrium causticum
Natrium hydricum
Natrium oxydatum
hydricum
Sodium hydroxide
Caustic soda
NaOH
Мод. в. 40,005
Natriumhydroxyd
Aetznatron
Приготовление
1. Растворяют 4 ч. №гСОз в 16 ч. воды и нагревают в чугунном котелке
до кипения. Одновременно гасят 1 ч. хорошо прокаленной СаО 4 ч. воды;
полученную жидкую кашицу вносят небольшими порциями в раствор NasCOs
до тех пор, пока отфильтрованная проба жидкости уже не будет шипеть
от прибавления НС1. В противном случае продолжают кипячение, добавив
немного Са(ОН)г.
Натрия гидрат окиси 368
Цо окончании реакции (каустификации) смеси дают отстояться,
'ро выпар!
1,33, (что
дуаают прозрачную жидкость с осадка СаСОз и быстро выпаривают в чугун
или, лучше, серебряной чашке до
уд. в. \>бб, (что соответствует
Явному раствору).
2. Можно также приливать к раствору Ва(ОН)2 раствор Na^SO*, пока
фильтрованная проба, подкисленная НС1, не перестанет давать осадок
sja^304, а с BaCh не будет давать лишь незначительную муть. Жидкости
от отстояться, сливают прозрачный раствор сифоном и выпаривают
^серебряной чашке.
3. Для приготовления раствора NaOH, свободного от ЫагСОз, по Зерен-
Кну1 растворяют продажный NaOH в равном количестве воды при энер-
ном 1встряхивании в склянке, закрытой резиновой пробкой.
После длительного отстаивания сливают прозрачный раствор с осевшего
IfcCOs.
4. Для получения 0,Ш раствора NaOH, совершенно не содержащего кар-
натов, можно поступать по Н. А. Шилову2 следующим образом. 2—3 г
сто го металлического натрия, очищенного от корочки окислов и взвешен-
под керосином, вносят постепенно в стакан со 100 мл этилового спирта.
пе того как весь натрий растворится с образованием этилата (СгНбО№),
вствор переливают в мерную колбу\ на 1 л и доливают прокипяченой водой
метки.
Следует иметь в виду, что при разбавлении водой раствора этилата
ногда происходит взрыв (защитные очки!).
Свойства
Бесцветная кристаллическая масса уд. в. 2,02, быстро поглощающая на
воздухе СОг и воду, расплываясь при этом и переходя в №зСОз.
Т. пл.3 327,6 + 0,9°, т. кип. 1388°. Легко растворим в воде (уд. <в. водных
растворов NaOH см. табл. на стр. 364), труднее в спирте и эфире. Из
концентрированных водных растворов*при —8° выделяются крупные моноклини?
ческие кристаллы гидрата NaOH • ^ШгО.
Упругость водяного пара над NaOH составляет всего около 0,16 мм
рт. ст., что характеризует это вещество как хороший осушитель; при
пропускании влажного воздуха через трубку с NaOH остаточная влажность
при 25° составляет всего 0,16 мг НгО на 1 л4.
Таблица 35J
Растворимость NaOH в воде
ГС
12,3
18
40,25
57,8
ojoNaOH
50,8
51,7
56,4
62,8
ГС
64,3
62
80
o/oNaOH • !
68,5
74,2
75,8
ГС
ПО
159
192
o/0NaOH
78,2
81,1
83,9
«Sorensen, Biochemisctn Zeftschrift, 21, 186 (1919).
* Объем i ый ai ализ, стр. 66, 1931.
• F. H a 11 a u. H. T о m p a, Z. anorg. Ch. 219, 331 (1934).
« Н. S. В о о t h a. L. II. Мс I п t у г е, Ind. Eng. Chem., Anal. Ed. 2, 12 (1930)
364
Натрий
Таблица Зчо
Удельный вес водных растворов NaOH (Lunge)
Градусы
Боме (Вё)
Градусы
Тв^дделя
(Tw)
o/0NaaO
o/oNaOH
1 л раствора содержит
*Na,0
1
2
3
4
5
6
7
8
9
Ю
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
1,4
2,8
4,4
5,8
7,2
9,0
10,4
12,0
13,4
15,0
16,6
18,2
20,0
21,6
23,2
25,0
26,8
28,4
30,4
3?,4
34,2
36,0
38,0
40,0
42,0
44,0
46,2
48,2
50,4
52,6
54,8
57,0
59,4
61,6
64.0
66,4
69,0
71,4
74,0
76,6
79,4
82.0
84,8
87,6
90,6
93,6
66,6
99,6
102,8
106,0
0,46
0,°3
1,43
1,Р4
2,44
2,94
3,49
4,03
4,54
5,10
5/6
6,25
6,81
7,36
7,98
8,57
9.^2
9,84
10,46
11,12
11.74
12,40
13.11
шо
14,50
15,23
15,97
16,70
17 43
18,21
18,97
19,77
20,60
21,43
2^3)
23,25
24,18
25,19
26,14
27,13
28,18
2918
30,27
31,37
32,57
33,77
35,00
36,22
37,52
38,83
0,59
1,20
1,85
2,50
3 15
3,79
4,50
5,20
586
6,58
7,30
8,0"
8,78
9,50
10,30
11,06
11,90
12,69
13,50
1-1,35
1515
•6,00
16,91
17.Н1
1Р,71
19.65
20,60
21.55
22,50
23.50
24, 8
25,50
26,58
27,65
28,83
?0,00
31,20
32,5Q
3Q,73
35,00
36,36
37,6i
39,06
40,47
42,02
43,58
45,16
46,73
48,41
50,10
4,6
9,4
14,S
20,0
25,3
30,7
36,7
42,7
48,4
54,8
61,3
68,3
74,9
П,5
89,0
96,4
104,6
112,5
120.5
129,2
137,5
146,3
15о,0
165,6
175,5
185,8
196,6
2o7 2
218,2
2d0,0
241,7
2o4,0
267,2
280,0
295,0
309,7
325,2
341,8
^58,1
^75,2
393,7
411,4
431,0
451,1
473,2
495,7
519.1
542,6
568,1
594,1
^Натрий гидросернистокислый
365
|спытание
• flo OCT 17375—40 содержание едкого натра (NaOH) в препаратах
квалификаций х. ч. и ч. д. а. должно быть не менее 95%; в чистом не менее
Жу0; наибольшие количества допустимых примесей указаны в табл. 353.
Таблица 353
% Допустимые примеси в NaOH
*г- \
& Примеси
1.
fe Хлориды (CI7)
4. Фосфаты (РО/")
5, Азот (N), общее количество из
нитритов, нитратов, аммиака и пр.
7. Тяжелые металлы группы H2S . .
8. Кальций (Са)
9. Сода (Na2CO*) .
10. Вещества, осаждаемые аммиа-
Допустимое содержание в °/0
в химически
чистом
0,005
0.005
0,01
0,003
0,001
0,0005
0,003
0,012
1
0,01
в чистом для
анализа
0,01
0,01
0,02
0,005
0,001
0,001
0,003
0,024
2
0,02
в чистом
0,025
0,03
0,1
0,01
0,001
0,002
0,003
0,06
4
0,1
НАТРИЙ ГИДРОСЕРНИСТОКИСЛЫЙ
(ГИДРОСУЛЬФИТ НАТРИЯ)
Natrium hydrosulfuro- 'oodium hydrosulfite Natriumhydrosulfit
sum Sodium hyposulfite
Na2S204«2H20 Мол. в. 210,15
Приготовление
1. Для лабораторного получения ЫагЗгС^ можно применять патентный
способ, принадлежащий Badische 'Anilin- und Soda Fabrik1. 25 ч. кислого
сернистокислого натрия (ЫагЗЮв) в растворе уд. в. 1,38 смешивают с 54 ч.
водного раствора ЗОг (уд. в. 1,03)2. Затем, при перемешивании, медленно
прибавляют 4,2 ч. цинковой пыли3, охлаждением поддерживая температуру
в пределах 30—40°. После 1—^2-часового стояния смешивают жидкость
с известковым молоком, полученным из 4,2 ч. хорошо прокаленной СаО и
20 ч. воды.
Дают стоять 6 час, после чего осадок отфильтровывают и отжимают.
Раствор насыщают поваренной солью, пока высолится Na^SaOi. Кристаллы
отфильтровывают без доступа воздуха, промывают сначала водным,, затем
чистым ацетоном и сушат в вакууме.
Принимая во внимание легкую окисляемость препарата, все операции
следует производить при возможно полном отсутствии воздуха.
Выход около 4 ч.
1 Герм. пат. 119676 (1899).
1 Или раствор NatetOb насыщают соответствующим количеством SOf
•Т. Муроока рекомендует вместо применения цинковой пыли вс,
*м"ьгамой цинка. T.Murooka Scl Jap. Inst. phys. chem. Res. 28, № 603,606 (1935); С
4 У об)
тряхивать раствор с.
... ^ з8(|у
366
Натрий
2. Значительно удобнее получать Na2S204 по методу ИРЕ А, согласно кот
рому сначала готовят гидросульфит цинка, насыщая смесь воды и цинково-
пыли сернистым газом. Операция проводится без доступа воздуха при н
прерывном перемешивании (механическая мешалка с водным затвором) 6 *
охлаждении водой или снегом. Затем, не прекращая перемешивания, чере*!
капельную воронку добавляют вычисленное количество раствора Na2CO
Отсасывают выпавший ZnCOs и насыщают фильтрат газообразным NH3 до
выпадения кристаллов Na*S204 • 2Н2О.
3. Ввиду нестойкости Na2S204 • 2НгО обычно предпочитают готовить без-
водный препарат. Для обезвоживания кристаллы Na2S204-2H20 "быстро
отделяют от маточного раствора и промывают несколькими порциями
этилового спирта, оберегая препарат от соприкосновения с воздухом.
Кристаллическую массу, вместе с последней порцией спирта нагревают ца
водяной бане 5—6 час. при непрерывном перемешивании, следя, чтобы
температура не поднималась выше 70°. Не прекращая перемешивания,
охлаждают смесь, отсасывают кристаллы на воронке Бюхнера, промывают
96%-ным спиртом и просушивают в вакуум-эксикаторе при 66—60° в
течение 2—3 час. Охлаждение препарата проводят в вакууме.
Полученный таким путем гидросульфит устойчив Даже при продолжи*
тельном хранении.
Свойства
Na2S204»2H20 представляет собой блестящие, как стекло, призматические
кристаллы. В сухом состоянии устойчив на воздухе в течение нескольких
дней; будучи влажным — чрезвычайно быстро с окисляется. Безводный
Na2S*04 — мелкий, очень твердый белый порошок.
Очень легко растворим в воде, не растворим в спирте. Водный раствор
жадно поглощает -кислород. t ,
Испытание
По техническим условиям НКХП 158—40 содержание №г£а04 в
препарате ч. д. а. должно быть не менее 85%; наибольшие количества
допустимых примесей указаны в табл. 354.
Таблица 354
Допустимые примеси в Na2S204
Примеси
1. Гипосульфит (Na2S20y)
2. Сульфат (Na2S04)
3. Сульфит (Na2SOa)
4. Хлористый натрий (NaCJ) . . . .
5. Тяжелые металлы группы hUS
(NH,)2S • ". .
Допустимое
содержание
2,5
5
5
1,5
0,03
НАТРИЙ ДВУХРОМОВОКИСЛЫЙ
(БИХРОМАТ НАТРИЯ, НАТРИЕВЫЙ ХРОМПИК)
Natrium bichromicum Sodium bichromate Natriumbichromat
МиСггОт. 2Н2О Мол. в. 298,05
Приготовление
1. По способу Гуда растворяют 100 г хромовокислого калия (КгСгОО
и 166 г кристаллического сернокислого натрия (Na*S04' IOH2O) при 100°.
в возможно малом количестве воды и полученный раствор охлаждают до #
Натрий двухромовокислый
367
wip. быстро отсасывают осадок K2SO4, к фильтрату добавляют разбавленной
f?S04 (в количестве, соответствующем 25,2 г 100%-ной Н2ЗО1), выпаривают
начинающейся кристаллизации и сильно охлаждают. Выпавший осадок
SL'S04 • ЮН2О отфильтровывают, фильтрат, снова выпаривают до появления
!1исталлической пленки и охлаждают. Выделившиеся кристаллы препарата
2тгтельно отсасывают и перекристаллизовывают из очень малого количества
апДЫ.
2. Процесс очистки продажного (технического) натриевого хромпика
довольно сложен, и количества реактивов, применяемых для удаления тех или
^иых примесей, зависят от количества этих примесей1. Обычными
примерши в ЫагСгаО? являются NaCl и Na2SC>4, количества которых колеблются
i различных партиях сырья в довольно широких пределах. Указываемые
Щиже количества реактивов относятся к сырью, содержащему 90% ЫагСггО,
Щ% Na2S04, 2% NaCl.
•$ТТаким образом, при загрузке в 200 г такого сырья в переработке нахо-
|ртся: 180 г ЫагСггО?, 1,2 г Na2S04 и 4 г NaCl.
|ГВ случае применения сырья иного состава, необходимо произвести
предварительный пересчет. Рекомендуется производить анализ уже
приготовлении) и профильтрованного раствора ЫагСггО?.
Щ- а) 200 г ЫагСггО? растворяют в 200 мл воды в фарфоровой чашке и
нйфильтровывают от нерастворимых примесей через двойной бумажный
фильтр. Для удаления Са' * фильтрат упаривают до половины объема и
ФЬлаждаЪт. При этом выпадает 12—15 г кристаллов, загрязненных солями
(йльция. Их отсасывают на воронке Бюхнера и фильтрат подвергают даль-
|)Вёйшей очистке.
'#f б) Следующая операция сводится к удалению из раствора ионов СГ. Так
жак некоторое количество NaCl выпало вместе с кристаллами ЫагСггСЬ (при
отделении Са"), то в растворе осталось около 3,75 г NaCl. Для удаления
СГ употребляют вычисленное количество (9,9 г) хромовокислого серебра
AgfiCr04. (Получение Ag2Cr04 см. стр. 491). Раствор ЫагСггСЬ, объем
которого составляет теперь около 120 мл, разбавляют равным объемом воды и
переносят в склянку, куда небольшими порциями при разбалтывании
прибавляют £goCr04 (всего около .10 г): при этом красно-бурое !Ag2Cr04 переходит
в белый осадок AgCl: •
Ag2Cr04 + 2NaCl = Na2Cr04 + 2AgCl
Ф
Прибавление ведут до тех пор, пока не будет ясно заметно прекращение
перехода Ag2CrC>4 в AgCl. Тогда производят контрольную пробу на СГ (2 мл
раствора разбавляют 8 мл воды, подкисляют 4 мл HNOs уд. в. 1,2,
подогревают до 50° и добавляют 1—2 капли 5%-ного раствора AgNOs: не должно
быть мути в течение 10 мин.). В случае помутнения прибавляют еще не большое
количество Ag*CrC>4.
в) Теперь приступают к удалению ионов SCV, применяя для этой цели
раствор двухромовокислого бария2 ВаСг207.
В фарфоровую чашку с раствором NaoCttCb вливают раствор ВаСггСЬ,
хорошо перемешивают и нагревают \Уъ—2 часа, после чего производят
контрольную пробу на SCV (2 мл раствора отфильтровывают, разбавляют 6—8 мл
воды, подкисляют 2—3 мл НС1 уд. в. 1,12 и прибавляют 1—2 капли 5%-ного
раствора BaCU; после 2-часового стояния не должно быть помутнения).
Если SCV еще присутствует, снова прибавляют небольшое количество
BaCrjCh. После удаления SO*" производят пробу на Ва ** (2 мл отфильтро-
1 Приведенный метод очистки разработан Е. А. II и к и т и н о и, Изв. ПРЕД, вып. 9, 161
1930).
■ Для приготовления раствора ВаСг207 растворяют отдельное г хромового ангидрида (СгО$)
в 4 мл горячей дестиллированной воды и 3 г едкого барита [Ва(ОН)а • 8HtO] в 12 мл горячей
воды и фильтруют. Горючий ра-твор Ва (ОН)в (если он остыл, подогревают) приливают к
горячему же раствору СгО,. При сливании частично выпадает осадок ВаСгО*, который не мешает
реакции.
368
Натрий
ванного раствора разбавляют 4 мл воды и прибавляют 1—2 капли Ю0/^
H2SO4 — в течение получаса не должно появляться мути). Прибаш,
ВаСггСЬ необходимо очень осторожно, чтобы достигнуть удаления S04" ь
вводя избытка Ва' *. ' н^
г) Раствор ЫагСггСЬ фильтруют и выпаривают на водяной бане до Появл
ния кристаллической пленки, после чего охлаждают до комнатной Те**
пературы, помешивая раствор. Выпавшие кристаллы (130—135 г) отсасывав
сушат при 35—40° при частом перемешивании и помещают в банку с при
тертой пробкой1.
Свойства
Желтовато-красные иглы или таблички, теряющие кристаллизационную воду
при температуре выше 30°; при 84° плавится в кристаллизационной воде-
при 110° полностью переходит в безводную соль. Последняя представляет
собой оранжево-красный порошок уд. в. 2,5, плавящийся при 320° и при
400° разлагающийся с выделением кислорода. Сильно гигроскопичен; очень
легко растворим в воде; образует оранжевый раствор, обладающий кислой
реакцией и резко охлаждающим горьким металлическим вкусом.
Таблица 355
Растворимость Ка2Сгг07 в воде
PC
0
10
20
30
о/о NajCrt07
62,0
63,0
64,3
66,3
t°C
40
50
60
70
о/в NaaCr,07
68,8
71,3
73,9
76,4
гвС
80
83
1 98
о/о N3,0,0,
79,4
80,6
81,24
Таблица 356
Удельный вес водных растворов Ка2Сг207
% Ка,Сг407
1
2
4
6
8
10
<5
1,006
1,013
1,027
1,041
1,056
1,070
о/о NaaCr,0,
12
14
16
18
22
26
* 1
1,084
1,098
1,112
1,126
1,153
1,179
% NaeCrs07
30
35
40
45
50
4
1,207
1,244
1,279
1,312
1,342
Испытание
По ОСТ 10540—39 содержание Na2Cr207 • 2Н20 должно быть для всех
квалификаций не менее 99%; количества допустимых примесей указаны
в табл. 357.
1 Приведенный метод, ввиду его сложности целесообразно применять только при очистке
иначигельных количеств Na,Cr,07.
Натрий йодистый
369
Таблица 357
Допустимые примеси в Na2Cr207-2H20
Примеси
Допустимое содержание в %
В ЧИСТОМ ДЛ1
анализа
I Нерастворимые вещества
£ Хлориды (СГ)
$И:ульфаты (S04")
^Вещества, осаждаемые аммиаком (Al, Fe, Сг
'й др.)
ЕуСоли кальция (Са)
0,003
0,005
0,025"
0,01
0,01
0,01
0,02
0,05
0,025
0,02
НАТРИЙ ЙОДИСТЫЙ (ИОДИД НАТРИЯ)
Natrium jodatum 'Sodium iodide Natriumjodid
NaJ • 2H20
Мол. в. 185,95
lip иготовление
В объемистой фарфоровой чашке обливают 1 ч. железа (в виде порошка
Зйли опилок) 8 ч. воды и прибавляют при постоянном помешивании малень-
жимп порциями 3 ч. твердого иода. Последний, вступая в реакцию с
железом, образует растворимое йодистое железо, FeJ2.
Получившийся зеленоватый раствор отфильтровывают от непрореагиро-
©агашего железа и к фильтрату добавляют еще 1 ч. иода, причем FeJ2
переходит в FeeJe. Приготовленный таким образом раствор вливают тонкой струей
в кипящий раствор 4,5 ч. кристаллической соды (ЫагСОз • ЮНгО) в 10—12 ч.
воды и несколько минут кипятят. Фильтруют слабощелочной раствор и
фильтрат выпаривают досуха К
10 ч. иода дают теоретически 11,81 ч. безводного NaJ.
Свойства
При обыкновенной температуре кристаллизуется с 2 мол. воды в
моноклинических призмах уд. в. 2,45, выветривающихся в сухом воздухе. Выше
40° кристаллизуется без воды в кубических кристаллах уд. в. 3,7. Т. пл. без-
Таблица 358
t°c
0
10
20
30
Растворимость NaJ в воде
%N aJ
61,4
62,8
64,2
65,5
t°C
40
50
60
65
% NaJ
67,2
69,5
72,0
74,4
t°C
80
1C0
120
140
% NaJ
74,7
75,2
76,3
77,0
1 Чтобы не выпаривать щелочмого раствора, фильтрат рекомендуется предварительно
нейтрализовать иоднотоводородной кислотой.
370
Натрий
водной соли 661,4°, т. кип. 1300°. Безводная соль притягивает из во3дУх
влагу и во влажном состоянии постепенно разлагается с выделением ИОд
Безводную соль следует защищать от действия света. • •
Хорошо растворим в воде; насыщенный раствор кипит при 141°. Раство
рим также в спирте (100 ч. 96%-ного спирта растворяют 33,3 ч. NaJ).
Таблица 35$
Удельный вес водных растворов NaJ
о/о NaJ
1
2
4
6
8
10
12
14
.20
1,0060
1,0138
1,0298
1,0463
1,С6 3
1,08 Л
1,0988
1,1174
о/о NaJ
16
18
20
22
24
26
28
1 *»
4
1.П66
1,1564
1,1769
1,1981
1,°201
1,2-128
1,2663
о/о NaJ
30
35
40
45
50
55
60
J" a? "
1,2907
1,3556
1,4271
1,5082
1,5942
1,6927
1,8038
Испытание
По техническим условиям НКХП 38—40 содержание NaJ в сухом прс
парате для квалификаций ч, д. а. и чистый должно быть не менее 99%;
наибольшие количества допустимых примесей указаны в табл. 360.
Таблица 360
Допустимые примеси в NaJ
Примеси
Допустимое содержание в °/0
в чистом для
анализа
0,01
0,03
0,01
0,002
0,004
0,005
0,003
0,0005
0,0005
0,005
0,05
2,5
в чистом
0*02
0,05
0,02
0,002
0,008
0,01
0,005
0,001
0,001
0,01
0,1
5
1. Нерастворимые в иоде вещества
2. Щелочность (Na2C03)
3. Сульфаты (SO/)
4. Азот (N) пбщий
5. Барий (Ва)
6. Кальций (Са)
7. Магний (Mg)
8. Тяжелые металлы (РЬ)
9. Железо (Fe) . .
10. Иодаты (JCV)
11. Хлориды (СК)
12. Влага
-?."■• Натрий кобальтоазотистокислый S71
р f НАТРИЙ КОБАЛЬТОАЗОТИСТОКИСЛЫЙ
^ /ГЕКСАНИТРОКОБАЛЬТ.-[3]-АТ НАТРИЯ, КОБДЛЬТИНИТРИТ
НАТРИЯ)
Katrlum cobaltinitrosum Cobalt sodium nitrit Natriumcobaltinitrit
*. Na3Co(N02)e • ^HsO Мол, в. 412,99
Приготовление
1. Растворяют 50 г хлористого кобальта (СоС1г«6НЮ) при нагревании
• ^ 60 мл воды и одновременно 150 г азотистокислого натрия (NaNOt)
ъ "600 мл воды; охлажденные растворы сливают вместе. К смеси при
достоянном перемешивании (под тягой) прибавляют 50 мл 50%-ной СНзСООН.
Жидкости дают отстояться, после чего сливают с осадка и фильтруют; филь-
дат представляет собой готовый реактив.
/2. По Бильману1 поступают следующим образом. Раствор 150 г NaNO*
i 160 мл теплой воды охлаждают до 40—50° (причем выпадает некоторое
Количество NaNOa); к раствору прибавляют 50 г кристаллического хлористого
кобальта (C0CI2 * 6НгО) и затем небольшими порциями приливают 50 мл
60%-ной СНзСООН. После этого в течение % часа пропускают сильную
струю воздуха для удаления окислов азота. При этом образуется
значительный осадок, для осаждения которого дают раствору стоять в течение 2 час.
Жидкость отфильтровывают, а осадок собирают на фильтр и тщательно
отсасывают. Если фильтрат не совершенно прозрачен, то его еще раз
фильтруют через тот же фильтр, однако этой фильтрации следует, по
возможности, избегать, так как она происходит очень медленно. Осадок, состоящий
наряду с ЫазСо(ЫОг)в также и из K3Co(N02)e, вследствие обычной примеси
соли калия в продажном NaNOa, выщелачивают 50 мл воды при 70—80°.
Получившийся раствор отфильтровывают от КзСо(ЫОг)в и присоединяют к
главной массе раствора; общий объем жидкости составляет теперь около 300 мл.
Добавляя к раствору 250 мл 96%-ного спирта, осаждают твердый
NaaCo(N02)e, которому дают осаждаться в течение нескольких часов (чтобы
осадок не выпадал в виде чрезвычайно тонкого порошка, лучше всего
приливать спирт тонкой струйкой при постоянном перемешивании). Выпавшую
соль отсасывают, промывают 100 мл спирта (порциями по 25 мл) и 50 мл
эфира (также в два приема, порциями по 25 мл) и сушат на воздухе. Выход
60--63 г. Полученный таким путем препарат еще не совершенно чист, и,
обычно, не дает с водой прозрачного раствора.
Для окончательной очистки поступают по Г. и В. Б и л ь цг следующим
образом: делят сырой продукт на три части и каждую смешивают с
полуторным количеством холодной воды. Незначительный нерастворившийся осадок
отсасывают и в полученный прозрачный фильтрат тонкой струйкой вливают
смесь 70 ч. спирта с 1 ч. ледяной уксусной кислоты.
Выпавший осадок отсасывают, промывают спиртом и эфиром и сушат при
температуре не свыше 80°. Выход 40 г.
3. Для приготовления раствора, могущего служить реактивом на соли
Калия, рубидия и цезия, Бухнер3 рекомендует приготовить растворы: 112 г
сернокислого кобальта (CoS04e7HiO) в 500 мл воды, 180 г NaN02 в 350 мл
воды и 20 мл ледяной уксусной кислоты в 50 мл воды и постепенно их
смешать. После многочасового стояния при 30—40° охлаждают смесь до 0°,
отсасывают выпавший сернокислый натрий и фильтрат разбавляют до 1 л.
4. Для некоторых анализов Л. Ванино4 рекомендует приготовлять
Уксуснокислый раствор NasCo(N02)e: к раствору 10 г кристаллического
Уксуснокислого кобальта прибавляют раствор 20 г чистого NaNOa в 40-—50 мл
1 В и 1 m a n n, Z. f. anal. Ch. 39, 286 (1900).
■ Uebungsbeispiele.
• В u с n n • г, DiuertatiQO, Munchen, 1909; Prap. Ch. 367.
4 Prlp. Ch. 127,
24*
372
Натрий
воды, предварительно подкисленной небольшим количеством СНзСоои
После двухчасового стояния жидкость фильтруют и раствор употребляют и
реактив. Ка*
Свойства
Тонкий желтый порошок, устойчивый на воздухе. Удалить кристаллиза.
ционную воду нагреванием не удается, так как препарат при этом разла*
гается. Очень легко растворим в воде, не растворим в спирте и эфНре
Водные растворы неустойчивы.
Испытание
По ОСТ 10158—39 содержание Со в препарате ч. д. а. должно лежать
в пределах 13,5т—44,5%; содержание примесей не должно превышать следую-
щих норм: 0,05% нерастворимых в воде веществ, 0^021% сульфатов (S04")
Кроме того, раствор препарата дрлжен отчетливо открывать ион К' при
концентрации его 0,05 мг в 25 мл.
НАТРИИ КРЕМНЕКИСЛЫЙ (СИЛИКАТ НАТРИЯ, РАСТВОРИМОЕ СТЕКЛО)
Natrium silicicum Sodium silicate Natriumsilikat
Water glass Natronwasserglass
NasSiOa Мол. в. 122,05
Приготовление
1. Для получения силиката натрия сплавляют смесь 45 ч. тонкого
кварцевого песка, 23 ч. безводного NaaCOs и 3 ч. угольного порошка. Плав
выливают в воду (для удобства измельчения), причем он распадается на
мелкие частицы. Для переведения в раствор полученного твердого продукта, его
измельчают и кипятят с небольшим количеством воды под повышенным
давлением К
2. Для получения препарата состава Na23iOs*9H20 (мол. в. 284,20)
смешивают 1 об. обычного продажного жидкого стекла, 1 об. воды, 2 об.
раствора NaOH (уд. в. 1,26) и 2 об. спирта. Смесь разделяется на два слон,
из которых нижний представляет пересыщенный раствор Na*SiOs • 9НгО;
последний через некоторое время выкристаллизовывается. Для очистки
продукт перекристаллизовывают из смеси 10 об. воды, 1 об. раствора NaOH
(уд. в. 1,26) и 2 об. спирта. Кристаллизация происходит очень медленно и
ускоряется прибавлением небольшого количества твердой соли2.
3. По Краусу и Этнеру3к 1 кг продажного жидкого стекла (38—40°
Вё) добавляют раствор 420 г NaOH в 600 мл воды. Смесь мутнеет и через
некоторое время осаждаются Fe(OH)s и А1(ОН)з. Прозрачный раствор
декантируют и оставляют на несколько дней для кристаллизации. Маточный
раствор сливают возможно полнее и растворяют кристаллические корки в 600 мл
горячей воды. После охлаждения вносят «затравку» — кристаллик №г£Юз •
•9НЮ и оставляют на 2—3 дня.
Выпавшие кристаллы отфильтровывают, промывают небольшим
количеством воды и разбавленным спиртом. Препарат почти свободен от Fe и А1
н по составу близок к формуле Na^SiO* • 9НЮ.
4. Продажное жидкое стекло разлагают соляной кислотой,
отфильтровывают кремневую кислоту, тщательно промывают ее водой и растворяют в
концентрированном растворе NaOH. Полученный раствор сильно упаривают и
оставляют в холодном месте для кристаллизации.
» Komm. z. Deut. Arzneib. II, 69; Ргар. Ch. 353.
а РгЗр. СЬ. 353.
1 Р. К г а и » s, С. О е 11 d e r„ Z. anorg. Ch. 222, 357 (1935); Р. К г a u s •, Z, anorg. Ch. 204,
313 (193)0.
Натрий кремнекислые
373
ойства
Аморфная стекловидная масса уд. в. 2,4. Т. пл. 1088°. Растворим в воде,
створим в спирте. Водный раствор ЫагЗЮаг (жидкое стекло) — прозрач-
L густая, бесцветная или слабожелтая жидкость, имеющая щелочную реак-
«ЙК»1- Разлагается кислотами (даже углекислотой воздуха) с выделением
^уденистого осадка кремнекислоты. Раствор Na2SiOs следует хранить в сосу-
£а£ с резиновыми, пробками, так как стеклянные и корковые пробки сильно
вклеиваются к горлышку.
Таблица 361
Удельный вес водных растворов Na2Si03
fl/0KtSiO3
1
2
4
6
8
1 <8
1,0094
1,0203
1,0425
1,0652
1,0884
°/0N,SiO3
10
12
14
16
18
-Г
1,1122
1,1365
1,1613
1,1866
1,2123
o/oNtSlOa
20
22
24
26
<s
1,2385
1,2653
1,2926
1,3204
Еспытание
По ОСТ 10154-39 раствор 5 г препарата, ч. д. а. или 2 г препарата чистого
100 мл воды комнатной температуры должен быть прозрачным и не
содержать механических примесей; допускается легкая опалесценция; содержание
№аО в препарате ч. д. а. должно составлять 19,3—22,8%, в препарате
чистом—18,7—23,4%; отношение П/° С-Д—в препарате ч. д. а. должно быть
и/0ыи2
|*авным 1,03 + 0,03, в препарате чистом—* 1,03 ±0,06; раствор 5 г препарата
^точностью до 0,1 г) в 25 мл воды при добавлении 5 мл НС1 (уд. в. 1,12)
*fe должен вспениваться от выделяющегося СОг; наибольшие количества допу-
Зйшых примесей указаны в табл. 362.
Таблица 362
Допустимые примеси вNaaS103• 9Н20
Примеси
Допустимое содержание в °/0
в чистом для
анализа
"1. Хлориды (С10 • . .
2. Сульфаты (SO/)
3. Металлы, осаждаемые H2S ....
4. Полуторные окислы (А1203 + Fe203)
0,01
0,01
0,001
0,05
0,02
0,02
0,002
0,1
1 Растворы N0gSIO3 характеризуются иногда как растворы свободного NaOH, содержащие
Пептизированную кремневую кислоту. Однако имеются возражения против такого
представления. См., например, О. D е г n ie г, С. II, 48 (1939).
374
Натрий
НАТРИЙ КРЕМНЕФТОРИСТЫЙ (КРЕМНЕФТОРИД НАТРИЯ) {
Natrium silicofluoricum Sodium fluosilicate NatriumsilicofiuoV-н-
Fluorkieselwasserstoff
saures Natrium
NasSiFe
Мол. в. 188,05
Приготовление
Чистую аморфную кремневую кислоту растворяют в избытке 40%-ной HF
пересыщают раствор хлористым натрием и отсасывают после непродол житель
ного стояния через парафинированную воронку. Затем промывают соль холспЛ
ной водой до удаления СГ.
Препарат высушивают в платиновой чашке при 120° и сохраняют в
парафинированной внутри банке.
Свойства
Белый кристаллический порошок, уд. в. 2,68, мало растворимый в воде
и не растворимый в спирте.
Таблица 363
Растворимость Na2SiF6 вводе
(Н .С. Николаев, Н. А. Иванов, С. Г. Калтынин)1
/°с
10
20
о/о Nsf,SiFe
0,53
0,66
ГС
50
100
о/о NafSiFe
1,27
2,4
НАТРИЙ МЕТАВАНАДИЕВОКИСЛЫЙ (МЕТАВАНАДАТ НАТРИЯ)
Natrium vanadatum
(meta)
Приготовление
Sodium metavanadate
Natriummetavanadat
NaVO»
Мол. в. 121,95
Плавят 182 г пятиокиси ванадия, V2O5 (или 234 г ванадиевокислого
аммония, NH4VOs) с 212 г безводного №гСОз; растворяют плав в воде, насыщают
раствор С02 и дают закристаллизоваться. Сначала выпадает №гСОз • ЮНгО,
затем NaVOs; для очистки последний перекристаллизовывают из горячей
воды'
Свойства
Микроскопические белые или желтоватые призмы, легко плавящиеся и при
охлаждении образующие белую кристаллическую массу, только медленно
растворимую в воде.
1 ЖПХ 9. Ж 7, 1183 (1936).
'Roicol, Phil. Trent. 160, II, 317; Ana. Suppl. 8,E6.
Натрий кремнефтористый, ванадиевокислый и фосфорнокислый 375
КАТРИЙ МЕТАФОСФОРНОКИСЛЫЙ (МЕТАФОСФАТ НАТРИЯ)
jfatrium phosphoricum Sodium metaphosphate Natriummetaphosphat
л (meta)
NaPOa Мол. в. 101,98
(NaP03)e Мол. в. 611,88
Приготовление
1. Постепенно нагревают натрий-аммоний фосфорнокислый (NaNH4HP04«
4Н*0) в вакууме, причем по М е й е р у * сначала теряется вода и аммиак.
^Йежду 160—170° соль начинает плавиться, затем вспенивается и снова
ставится твердой. Тогда следует истереть соль в порошок и снова нагревать
$ри 320°, пока проба, прокаленная на тройной горелке, не перестанет
показывать потерю веса. Полученный таким образом препарат растворим в воде
лишь на 50%. Растворившуюся часть можно выделить в кристаллах,
осаждающихся при добавлении спирта.
2. По А. X. Бронникову и В. Ф. Постникову2 нагревают одно-
замещенный кислый фосфорнокислый натрий в течение 10—15 мин. при 620°,
после чего быстро охлаждают. Получается гексаметафосфат (ЫаРОз)в.
3. По Кнорре3 для получения NaPOs растворяют 25 г NaNOs в 25 мл
воды, добавляют 42 г Н.1РО4 (уд. в. 1,3) и выпаривают на водяной бане.
Остаток нагревают 4 часа при 330°.
NaNOs + НзР04 = NaPOs + HNOs + Н20
Свойства
Белый порошок уд. в. 2,48, трудно растворимый в воде; водный раствор
имеет слабокислую реакцию. Т. пл. 610°.
Метафосфат весьма склонен к полимеризации; в зависимости от
температуры получения образуются различные полимеры. По В. Тредвеллу и
Ф. Лейтвайлеру4 процесс образования метафосфатов может быть
выражен следующей схемой:
250е lWie fifl7e
[NaNH4HP04]-^NaH2P04i^(NaP03)2^(NaP03)3-^(NaP034^(NaP03)6
Структурная формула гексаметафосфата (Тредвелл и Лейтвайлер
там же) следующая: 1
0 ONa О
II I II
NaO—Р-О—Р— О—Р—ONa
1 /\ I
00 О О
1 * \/ I
NaO—P—О—Р—О—P-ONa
К I II
О ONa О
1 Е. М е у е г, Вег. 45, 3760 (1912).
1 ЖПХ 11, 1295 (1938).
• Неорг. преп. 74.
4 W. D. Tread well u. P. Leutwyl! er Helv. chim. Acta 21, 1450 (1938).
376
Натрий
Таблшщ 364
Удельный вес водных растворов NaP03 - „
o/oNaPO.
1
2
4
-20
rf4
1,0064
1,0145
1,0304
o/oNaPO,
6
8
1,0461
1,0614
НАТРИЙ МЫШЬЯКОВОКИСЛЫЙ ТРЕХЗАМЕЩЕННЫЙ
(АРСЕНАТ НАТРИЯ)
Natrium arsenicum Sodium arsenate Natriumarsenat
Na3AsQ4 • 12H20 Мол. в. 424,09
Приготовление
1. К раствору HaAs04 или Na2HAs04 добавляют NaOH, пока рН раствора
не достигнет величины 11,5—12,5. При испарении выпадают кристаллы арсе-
ната тем крупнее, чем выше рН раствора (Ю. В. К а р я к и н и О. С.
Воробьева).
2. Растворяют 20 г AS2O3 в растворе 25 г NaOH, добавляют 17,5 г NaNOt
и выпаривают раствор. Остаток прокаливают при 500—600° до полной сухости
As203 + 2NaNOa + 6NaOH = 2Na3As04 + 2NaNOs + ЗН2О.
После охлаждения смесь солей растворяют в воде и концентрируют до
кристаллизации (Г. Борнеман1).
Вначале выкристаллизовывается чистый NasAs04* I2H2O. Азотистокислый
натрий практически полностью остается в маточном растворе.
Свойства
Бесцветные кристаллы ромбической или гексагональной системы уд. в. 1,76,
легко растворимые в воде (28: 100). На воздухе не изменяется. При 85,5°
плавится в кристаллизационной воде. Водный раствор Na3As04, имеющий
сильно щелочную реакцию, постепенно поглощает СОг из воздуха, переходя
в Na2HAs04.
НАТРИЙ НАДБОРНОКИСЛЫЙ (П'ЕРБОРАТ НАТРИЯ)
Natrium perboricum Sodium perborate Natriumperbotat
NaB03*4H20 Мол. в. 153,88
Приготовление
. 1. ПоТанатару растворяют 20 г буры и 4 г NaOH в небольшом
количестве воды и к раствору прибавляют 120 л л 3%-ной Н2Ог. Через некоторое
время при стоянии на холоду осаждаются мелкие, трудно растворимые
кристаллы пербората. Их отфильтровывают и* промывают сначала холодной
водой, затем спиртом и эфиром. Из 20 г буры получается 15 г чистого
препарата.
2. В охлажденный льдом до —8° раствор 62 г борной кислоты в 500 мл
воды вносят небольшими порциями 78 г перекиси натрия (№гОг), избегая на-
1 Неорг. прем. 79
Натрий мышьяковокислый, надборношслый и Штропруссидный 377
^евания раствора выше +10°. После суточного стояния на холоду отсасы-
Зйот осадок, промывают спиртом и высушивают в эксикаторе. Выход
небольшой (Д° 35% теоретического). ь
3. По Арндту1 NaBOs получают анодным окислением раствора смеси
Лфнокислого (Na2&407) и углекислого натрия (№гСОз).
В качестве анода служит толстая зигзагообразная платиновая проволока,
груженная зигзагообразным катодом из оловянной трубки так, что
расстояние между электродами составляет всего несколько миллиметров. Электро-
яИТ—раствор 30 г борнокислого натрия и 120 г №2СОз в 1 л воды, к кото-
,му добавлены 1 г №гСг04 и 1 капля ализаринового масла. Оловянная
убка (катод) охлаждается водой во все время электролиза.
При температуре 10—15° и анодной плотности тока 10-—20 а/см2 перборат
лучается (вначале) с выходом по току 60%; в дальнейшем выход падает,
рез некоторое время выделяются кристаллы чистого препарата.
Свойства
Бесцветные кристаллы, трудно растворимые в воде. В сухом состоянии
препарат может сохраняться неограниченное время. Водный раствор при
продолжительном стоянии выделяет кислород; при кипячении раствора наблю-
Еется энергичное выделение кислорода. При нагревании препарата до 70°
рбразуется моногидрат, NaBOa • НЮ — белая, твердая, фарфоровидная,
гигроскопическая масса.
Таблица 365
Растворимость NaB03 в воде
(Lion)
t°c
15
21
°'о NaB03
2,49
2,62
t°c
26
32
о/о NaB03
2,77
3,*4
НАТРИЙ НИТРОПРУССИДНЫЙ
(НИТРОЗОПЕНТАЦИАНОФЕРРИАТ НАТРИЯ, НИТРОПРУССИД НАТРИЯ)
Natrium nitroprussicum
Sodium nitroprussiate
Nitroprussidnatrium
Nitroferridcyannatrium
Na2Fe(CN)3NO. 2H20
Мол. в. 297,97
Приготовление
1. Для получения нитропруссида натрия необходимо иметь сильную тягу
вследствие чрезвычайной ядовитости выделяющихся при реакции газообразных
продуктов (циан и окислы азота). В широкогорлой колбе (рис. 53) растворяют
при нагревании 120 г железистосинеродистого калия [K4Fe(CN)e« ЗН2О]
в 240 мл воды и при помешивании вносят туда же 78 г азотистокислого
натрия (NaNCb).
Колбу помещают на водяную баню, закрывают пробкой, через которую
проходят термометр, капельная воронка и широкая стеклянная трубка,
служащая для отвода в тягу выделяющихся газов. Нагревают до 85° и очень
1А г n d t, Z. f. Electrochem. 22, 63 (1916).
378
Натрий
медленно, маленькими порциями начинают прилий *
из капельной воронку раствор 68 г H2SO4 (уд. в ^
в 240 мл воды. Приливание следует регулирова >
— в 240
температура не поднималась
Рис. 53. Прибор для
получения нитропруссида
i атрия
.84)
ать»
85-90°. ВЫШе
Когда будет прилита вся необходимая по расчетл/
кислота, приливают еще 30 мл такого же раствоп
H2SO4 и «агревают жидкость до 100° в течение
1—2 час. К По истечении этого времени .испытывают
раствор на присутствие K4Fe(CN)e, для чего 0,5 л*л
раствора разбавляют 2,5 мл воды и смешивают *
с несколькими каплями FeS-04 — не должно получаться *
синего окрашивания; в противном случае добавляют
еще 2—3 г NaN02 и повторяют нагревание2.
Реакция образования нитропруссида может быть вьь
ражена следующими уравнениями:
4NaNO* + 2H2SO4 = 2Na2S04 + 4HN02
2K4Fe(CN)e + 4HN02 + 2H2S04=2K2Fe(CN)5NO + 2NO + <CN)2 + 2K2SO4 + 4H20.
Дальнейшая операция имеет целью получение нерастворимого
нитропруссида меди, для чего жидкость фильтруют в фарфоровую чашку и добавляют
при перемешивании нагретый до 80° раствор 73 г CuS04«5H20 в 150 мл
воды, причем происходит реакция:
K2Fe(CN)5NO + CuS04 = CuFe(CN)5NO + K2S04.
I
Выпавшему светлозеленому осадку дают отстояться, декантируют жидкость
и промывают осадок декантацией тепловатой (25—30°) водой до исчезновения
реакции на SO4" в промывных водах (добавление ВаСЬ не должно вызывать
помутнения).
Отмытый осадок переносят в фарфоровую чашку и там разлагают раство
ром 24—25 г Na2COs согласно уравнению:
CuFe(CN)5NO + Na2C03 = Na2Fe(CN)6NO + CuCQ3.
Фильтруют и осадок CuCOa промывают на фильтре водой. Фильтрат,
содержащий нитропруссид натрия, вместе с промывными вода-ми подкисляют 6—8 ли
ледяной СНзСООН и выпаривают до половины объема. В полученном растворе
окончательно осаждают примесь S04", добавляя 1^5—2 г уксуснокислого бария
[Ва(СНзСОО)2]. Раствор фильтруют, выпаривают на водяной бане до
появления толстой кристаллической пленки и кристаллизуют в прохладном месте.
Выпавший продукт перекристаллизовывают из горячей воды и сушат при тем-
1ературе не выше 40°3.
2. По Рюсту4 поступают следующим образом: 40 г K4Fe(CN)e • ЗНгО
растворяют при нагревании в 60 мл воды в стакане емкостью 0,5 л. К раствору
при помешивании прибавляют 64 мл HNOa (уд. в. 1,24) и держат при
умеренном нагревании на водяной бане до окончания реакции (для определения
конца реакции несколько капель коричневого раствора смешиваю?1
с FeS04 или FeCh, причем должен получиться темнозеленый, но ие синий
осадок).
водяную баню заменяют воронкой Бабо или баней с насиненным раствором NaCl,
■ Другая реачци : проба жидюсти от добааления CuS04 должна давать светлозеленый, но
не коритевый оса пч.
■ Разработано ИРЕЛ.
4 Anleit. 2. Darst. anorg. Ргар. 1903; РгЗр. Ch. 355.
Натрия перекись
379
So время реакции, кроме СОг и N*, выделяются циан и синильная кис-
цха, поэтому необходимо работать под хорошей тягой.
Смеси дают спокойно стоять в течение I—2 дней и затем нейтрализуют
вяой, тщательно избегая избытка последней. Нейтрализованный раствор
нагревают до кипения, фильтруют и прозрачный фильтрат быстро выпаривают
на голом огне, после чего охлаждают и смешивают с равным объемом спирта;
при этом большая часть образовавшегося КЫОз выпадает в осадок.
Фильтруют и фильтрат снова быстро выпаривают с целью возможно быстро
удалить спирт. Из полученного тёмнокрасного раствора выпадают при
охлаждении кристаллы, которые отсасывают, промывают небольшим количеством хо-
эдной воды и сушат между листами фильтровальной бумаги.
Маточный раствор при выпаривании дает еще некоторое количество
препарата.
Свойства
Гранатово-красные ромбические кристаллы уд. в. 1,71, не теряющие даже
при 100° кристаллизационной воды. При 400° разлагается с выделением N0
и {CN)2. Имеет строение [Fe(Q^*]Na2. Растворим в воде (1:2,5), не
растворим в абсолютном спирте. Водный раствор довольно быстро разлагается даже
на рассеянном свету. В щелочном растворе — сильный восстановитель. Нитро-
пруссид натрия сильно ядовит.
Испытание
По ОСТ 4101 содержание Na2Fe(CN)sNO • 2НгО должно быть для всех
квалификаций не менее 97,5%; наибольшие количества допустимых примесей
указаны в табл. 366.
Таблица 366
Допустимые примеси в Na2Fe(CN)a • NO • 2Н20
Прммеси
Допустимое содержание в °/0
в чистом для
анализа
0.005
0.01
0,01
в чистом
0,01
0.03
0,03
НАТРИЯ ПЕРЕКИСЬ
Sodium peroxide
Na2Os
Мол. в. 77,994
Natriumperoxyd
Natrium peroxydatum
Natrium
hyperoxydatum
Приготовление
1. Ванино1 предлагает поместить металлический натрий в алюминиевый
с°суд, снабженный трубками для подвода и отвода газов, и, нагрев до 300°,
пропускать сухой, свободный от СОг воздух до полного сгорания натрия.
1 РгВр. Сп. 320,
380
Натрий
2. Для приготовления гидрата №г02*8Н20, мол. в. 222,12 растворяют
1 ч. Na202 в 4 ч. ледяной воды, тщательно наблюдая, чтобы температура
жидкости не поднималась выше 40°. При охлаждении полученного раствора*
до 0° выкристаллизовывается ЫагОг-вНгО, который отсасывают и сушат
в атмосфере, свободной от СОг.
Свойства
Совершенно чистая №гОг белого цвета. Однако продажный препарат
обычно • имеет желтоватую окраску.. При нагревании желтеет. Соприкасаясь
с водой, сильно разогревается и растворяется с частичным разложением. При
кипячении раствора выделяется кислород. При употреблении ледяной воды
или же измельченного льда разложение незначительно. Не растворим
в спирте. Гидрат, NaaCte. 8HzO — белые таблички, плавящиеся при 30°
в кристаллизационной воде. При длительном хранении расплывается,
разлагаясь на NaOH, воду и кислород. •
Испытание (Мерк)
1. Сульфаты (SCh"). 5 г препарата вносят маленькими порциями в смесь
25 мл НС1 (уд. в.М,124) и 100 мл воды. Полученный прозрачный раствор
после прибавления ВаСЬ и стояния в течение 12 час. не должен выделять
даже ничтожного осадка.
2. Галоиды (С/\ Br', J'). 3 г препарата вносят малыми порциями в смесь
20 мл НЫОз (уд. в. 1,153) и 100 мл воды. Полученный прозрачный раствор
от прибавления AgNOs может давать лишь слабую опалесценцию.
3. Фосфаты (Р04'"). 2,5 г препарата вносят маленькими порциями в смесь
20 мл HNOj (уд. в. 1,153) и 100 мл воды. От прибавления к полученному
раствору молибденовой жидкости (см. стр. 42) и стояния в течение 2 час.
при 30—40° не должно наблюдаться выпадения хотя бы незначительного
желтого осадка.
4. Азот. 1 г препарата с большой осторожностью смешивают с 0,2 г
глюкозы в объемистом никелевом тигле. Слабо подогревая дно закрытого крышкой
тигля, вызывают вспышку смеси. Охлажденный остаток растворяют в 5 мл
воды и подкисляют 10 мл разбавленной H2SO4. Несколько мл полученной
жидкости осторожно наливают поверх 5 мл раствора дифениламина
(полученного растворением 0,2 г дифениламина [(CeHsfeNH] в смеси 2 мл воды и
20 мл H2SO4 уд. в. 1,84). На границе соприкосновения слоев жидкости не
должно появляться синей окраски.
5. Тяжелые металлы. 5 г препарата при осторожном внесении в 100 мл
воды должны давать совершенно прозрачный и почти бесцветный раствор,
с которым производят следующие испытания:
а) 40 мл раствора от прибавления 10 мл НС1 (уд. в. 1,124) и
сероводородной воды не должны изменяться;
б) 40 мл раствора от прибавления нескольких капель (ЫН4)гЗ не должны
принимать бурой или зеленой окраски или выделять даже ничтожный осадок.
НАТРИЙ ПИРОФОСФОРНОКИСЛЫЙ (ПИРОФОСФДТ НАТРИЯ)
Natrium pyro- Sodium pyrophosphate Natriumpyrophosphat
phosphoricum
Na4P2(>7 Мол. в. 265,95
Приготовление
Обычный кислый фосфорнокислый натрий (Na*HP04 • 12НаО) прокаливают,
пока взятая проба, растворенная в воде, не будет давать с AgNOs чисто
белый (не желтоватый) осадок. Для получения кристаллического препарата
(Na4P*07' ЮН2О, мол. в. 446,11) полученную при прокаливании массу
растворяют в горячей воде, фильтруют и выпаривают до кристаллизации.
Натрий пирофосфорнокислый
381
jgoftcTBa
Безводный №4Рг07 — белая масса уд. в. 2,45, плавящаяся при 988°.
:|&драт Na4P207 • ЮН2О — бесцветные, блестящие кристаллы уд. в. 1,85,
растворимые в воде и нерастворимые в спирте. Водный раствор имеет щелоч-'
Щую реакцию; при кипячении в присутствии кислот переходит в Na2HP04.
Таблица 367
Растворимость Na4P207 вводе
ГС
0
10
20
о/о Na^PjO,
3,1 1
3,8
5,8
/°с
30
40
50
°/о Na4P,0,
9,0*
11.9
14,8
t°c
60
70
80
°/о Na4P,07
18,0
20,3
23,1
Таблица 368
Удельный вес водных растворов Na4P207
о/о Na4P,07
1
2
4°
1,009
1,019
°/о Na4Pt07
3
4
.20
d4
1,028
1,037
Испытание',
По ГОСТ 342—41 содержание Ыа4Рг07 • ЮНгО в препарате ч. д. а. должно
быть не менее 99% й в препарате чистом — не менее 97%; наибольшие
количества допустимых примесей указаны в табл. 369.
Таблица 369
Допустимые примеси в Na4P207• 10Н2О
Приме»
Допустимое содержание в 0;0
в чистом для
анализа
0,005
0,002
0,05
0,001
0,001
0,0001
0,3
0,15
в чистом
0,01
0,005
0,05
0,003
0,002
0,0005
0,3
0,3
1. Нерастворимые в воде вещества .
2. Хлориды (СЮ . . .
3. Сульфаты (SO^)
4. Железо (Fe)
5. Тяжелые металлы, осаждаемые H2S
6. Мышьяк (As) \
7. Ортофосфаты (РО/'О
8. Натрий углекислый (Na2C03) . . .
382
Натрий
НАТРИЙ ПЯТИСЕРНИСТЫЙ (ПЕНТАСУЛЬФИД НАТРИЯ)
Natrium persulfuratum Sodium pentasulfide Natriumpentasulfid
Na2S5-5H20 Мол. в. 296,37
Приготовление
1. По В а ни но1 растворяют 8 г NaOH в возможно малом количестве *
спирта, делят раствор пополам и одну часть насыщают сероводородом. Сме- <3
шав затем обе части, растворяют в смеси при нагревании 12,8 г истертой
черенковой серы.
При стоянии на холоду в течение нескольких дней сначала выпадает
Na2S4»8H20, а затем при 5° — Na2Ss • 5Н20.
2. Для приготовления раствора так называемого «многосернистого» натрия,
представляющего собой омесь различных полисульфидов, настаивают теплый
раствор сернистого натрия (№гЗ) с серой и фильтруют.
Свойства
Оранжево-красные кристаллы, легко растворимые в воде.
НАТРИЙ СЕРНИСТОКИСЛЫЙ (СУЛЬФИТ НАТРИЯ)
Natrium sulfurosum Sodium sulfite Natriumsultit
Na2S03.7H20 Мол. в. 252,17
Приготовление
1. Профильтрованный раствор 160 г ЫагСОз'ЮНгО (или эквивалентного
количества NaOH) в 320 мл воды делят пополам и одну часть насыщают SO*
при нагревании до 40° на водяной бане. К жидкости (содержащей теперь
NaHSOa) приливают вторую половину раствора, причем происходит
взаимодействие согласно следующему уравнению:
_L
2NaHSOs + Na2C08 = 2Na*'SOs + Н2О + СО2.
Раствор оставляют кристаллизоваться, по возможности защищая от
действия воздуха2. Выход 80—90% теоретического.
Свойства
Большие бесцветные, призматические кристаллы уд. в. 1,56, легко
растворимые в воде со щелочной реакцией. На воздухе выветривается и окисляется,
переходя в Na2S04. При 150° теряет кристаллизационную воду; при более
сильном нагревании плавится в смесь ЫагЗ и NaaSOi.
При хранении в банках верхний слой Na2SOs отлично защищает от
окисления нижний слой: кроме того, найдено, что безводный NazSOa окисляется
гораздо медленнее кристаллического.
* Ргар. сь. 334.
■ О получении N-.SO, в присутствии отрицательных катализаторов, замедлжющих окисла»
яла препарата, см. Я. И. 3 и л ь б • р ы а и ■ П. Т. И ■ а но в, ЖПХ, 13, N 4, Ш (1840).
Натрий пятисернистый и сернистокислый 383
Таблица 370
Растворимость Na2SOa в воде
ГС
0
10
20
30
о/о NatS08
12,5
16,0
20,7
26,1
/°с
33,4
40
50
60
о/о Na,S03
28,0
27,0
25,7
24,5
/°С
70
80
90
100
о/о NatS03
23.5
22,6
21.7
21,2
Таблица 371
Удельный вес водных растворов Na2S03
о/0 Na,SO,
*—-—
1
2
4
6
"I9
1,0078
1,0172
1.03НЗ
1,0556
о/0 NaaSO,
8
10
1 12
<9
1,0751
1,0948
1,1146
о/о NatSO.
14
16
18
«Г
1,1346
1,1549
1,1755
По ГОСТ 195—41 содержание Ыа*^Оз в безводном препарате ч. д. а.
должно быть не менее 94,0% и в препарате чистом не менее 91,0%;
наибольшие количества допустимых примесей указаны в табл. 372.
По ГОСТ 429—41 содержание Na2SOa • 71-ЬО в кристалличгском препарате
■ д. а. должно быть не менее 96,0% и в препарате чистом не менее 91,0%;
наибольшие количества допустимых примесей указаны в табл. 372.
Таблица 372
Допустимые примесив Na2S08
Примеси
1. Нерастворимые в воде вещества
«. Хлс риды (CY)
»• Тяжелые металлы, осаждаемые
Допустимое содержание в о»0
для безводного Na|S04
в чистом
для анализа
0,01
0,01
0,001
0,001
0,1001
в чистом
0,02
0,02
0,002
0.002
0.0005
отсутствие
0,3 | 0,3
для NagSOe.yHjO
в чистом
для анализа
0.001
0.005
0.001
0,001
0,0001
отсу
0,3 |
в чистом
0,01
0,01
0,001
0.002
0,000
тствие
0,3
384
Натрий
НАТРИЙ СЕРНИСТОКИСЛЫЙ КИСЛЫЙ
(БИСУЛЬФИТ НАТРИЯ, ПИРОСУЛЬФИТ НАТРИЯ
о
Natrium Sodium bisulfite Natriumbisulfit
bisulfurosum
Na2S20s Мол. в. 190,11
Приготовление1
1. В колбу помещают 80 г чистого двууглекислого натрия (NaHCO.j) * и
400—600 мл воды и пропускают в полученную смесь струю SO2 (промытого
водой) до полного прекращения выделения СОг. Раствор, сильно пахнущие
SO2, сохраняют в закупоренной склянке. Для получения препарата в твердом
виде к (раствору прибавляют равный объем 96%-ного спирта и отсасывают
выпавший кристаллический порошок. Выход 80—90%! теоретического.
2. Сухим путем можно приготовить препарат следующим образом. В хлор-
кальциевую сушильную колонку (см. рис. 55), нижняя часть которой
наполнена стеклянной ватой, загружают смесь NaHCOs с толченым стеклом
(последнее должно быть тщательно отсеяно от пыли). Через трубку С нижнего
отверстия колонки пропускают ток SO2, пока не прекратится выделение СО*
и сернистый газ не будет проходить колонку, не поглощаясь. Тогда
содержимое колонки отделяют от осколков стекла и помещают в плотно
закупоренную склянку.
Свойства
Белый порошок с сильным запахом SO*. При лежании на воздухе теряет
SOa и поглощает кислород, переходя в Na2S04. Водный раствор имеет кислую
реакцию и разлагается выше 65° на NaaSOs и SOa. Раствор ЫагБгОб
растворяет серу с образованием Na2Ss03.
Таблица 373
Растворимость Na2S205 в воде
t°c
0
10,4
15
°/о NaaS|0B
37,4
38,8
39,2
t°c
22,8
31,4
40,2
°;0 Na2Sa06
39,8
40,8
41,6
t°c
59
81,4
97,2
о/о Na8Sx03
44,3
47,4
49,1
Испытание3.
По 3 а н г е р у-Б л э к у препарат ч. д. а. должен содержать не менее
95% действующего начала и не более 0,0005% As.
Раствор 1 г препарата в 10 мл воды после добавления 5 мл HNOs (уд. в.
1,3) выпаривают досуха на .водяной бане. Остаток, растворенный в50и*Л'воды,
после приливания НЫОз и AgNOs должен оставаться в течение 1 мин.
совершенно прозрачным (хлориды).
Раствор 2 г соли в 10 мл воды и 20 мл НС1 (уд. в. 1,12) выпаривают
досуха на водяной бане. Остаток растворяют в 20 мл воды, добавляют 1 мл
НС1 и 10 мл сероводородной воды. Жидкость должна остаться прозрачной и
бесцветной; осадок не должен выпадать также после приливания 5 мл ЫНЮН
и (NH4)*S.
1 При получении NaHS03 следует иметь в виду, что эта соль существует только в
растворе; при кристаллизации выделяется пиросульфит Na,SaOB (т. е. 2 NaHSO, — HtO«= Na|SM06)
■ NailCOa можно заменить 140 г NaaCOa • 10 HtO или 40 г NaOtt.
8 Испытание хам. реактивов, 267.
Натрий сернистокислый кислый. Натрий сернистый 385
HAtPHH СЕРНИСТЫЙ (СУЛЬФИД НАТРИЯ, МОНОСУЛЬФИД НАТРИЯ)
Natrium sulfuratum Sodium sulfide Natriumsulfid
Na2S-9H20 Мол. в. 240,20
Na2S.5H20 Мол. в. 168,13
Приготовление
1. По Сабатье продажный сернистый натрий растворяют в небольшом
(рличестве теплой воды; при этом основная масса тиосульфата
(присутствующего в виде примеси) остается нерастворенной. При охлаждении профиль-
Кованного раствора осаждаются крупные призматические и октаэдрические
|ристаллы. Как указывает автор, маточный раствор при испарении в вакууме
|{ает абсолютно чистые кристаллы1.
2. По Блоксаму2 небольшое количество чистого препарата можно
поручить, пропуская сероводород в раствор 100 г NaOH в 100 г воды (если
юри этом происходит побурение раствора, то ему дают отстояться и
фильтруют через стеклянную вату).
Во время пропускания газа осаждается значительное количество
игольчатых кристаллов соединения Na2S с NaOH; при дальнейшем пропускании Нг5,
еще прежде чем наступит насыщение раствора, выпавший осадок снова
растворяется. Тогда оставляют жидкость спокойно стоять в течение 3 суток в
холодном месте, плотно закрыв сосуд во избежание окисления. За это время
1©саждаются большие бесцветные октаэдрические кристаллы Na2S»9H20. Их
переносят на воронку Бюхнера, дают стечь маточному раствору, промывают
1—2 раза небольшим количеством спирта и сушат фильтровальной бумагой.
3. По Тиде и Рейнике3 для получения чистейшего препарата
поступают следующим образом: растворяют при комнатной температуре чистейший
NaOH («purissimum» de Haen) в 5—6-кратном количестве абсолютного
рдирта, часто энергично взбалтывая. Через 3 часа раствор насыщен:
жидкость становится тягучей и при фильтровании раствора падающие капельки
некоторое время скользят по поверхности жидкости в виде маленьких
шариков4. Около половины полученного раствора насыщают сероводородом. Если
раствор не был насыщен, осадок не выпадает ни при каких обстоятельствах,
в противном случае тотчас начинается осаждение. Рекомендуется пользоваться
энергичной струей газа и широкой газопроводящей трубкой. Прежде чем
весь выпавший осадок снова растворится при дальнейшем пропускании
сероводорода, прерывают ток газа и жидкость встряхивают; при этом вступает
в реакцию физически растворенный H2S и остаток Na2$ растворяется с
образованием NaHS. Полученный раствор фильтруют в оставшийся спиртовый
раствор NaOH, причем встряхиванием и растиранием стеклянной палочкой
Добиваются получения возможно мелких кристаллов Na2S • 5Н20 (в противном
случае получаются крупные плитчатые кристаллообразования). Осадок
отсасывают, многократно промывают небольшими количествами абсолютного
спирта и хранят в вакуум-эксикаторе над хлористым кальцием.
4. Для получения безводного Na2S высушивают кристаллогидрат в су-
Шильном пистолете Фишера над P2Os, применяя в качестве нагревающей
Жидкости ацетон.
Устройство сушильного пистолета Фишера видно из рис. 54. Осушающее
вещество (например, Р2О5) помещается в «пистолет» Л, соединенный шлифом
с широким цилиндром В. Высушиваемый препарат помещается в открытой
1 Следует заметить, что при проверке применения этого метода очистки технического NaaS
°н оказался далеко не простым; кроме того, препарат, получаемый :-тил путем, всегда содержал
значительные количества тиосульфата и полисульфидов. Возможно, что окисления удастся
Избежать при испарении раствора в вакууме или в атмосфере инертного газа.
> В 1 о х a m, J. Слет. Soc. 77, 761 (1900).
*Tiede, Reinlcke, Ber. 66, 667 (1923).
* При стоянии раствора приблизительно через •сутки образуются вследствие окисления
сильно окрашенные продукты уплотнения спирта.
386
Натрий
пробирке в цилиндр В и аппарат вакуируется через кран С, соедИцен \
с насосом. • ^ i
Таким образом, принцип устройства тот же, что и у вакуум-эксикатоп , i
Отличительным является приспособление, позволяющее высушивать при ™?
гревании. Для этого в колбе D кипятят ^j,
кость с подходящей температурой кипени*
(напри-мер, толуол с т. кип. ИР). Пары ТоЯ
луола обогревают цилиндр В, а следо1з:1Тел,
«о, и высушиваемый препарат и, кондецси*
руясь в холодильнике Е, стекают в ВИд*
жидкости обратно в колбу D. щ
Пистолет Фишера позволяет вести высув
шивание при строго определенной темпера-
туре и чрезвычайно ускоряет процесс сушки
оригинально используя одновременно дей.'
ствие осушителя, вакуума и нагревания.
По К у р т у а 1 при таком методе полу-
чения препарат удерживает около 4% воды;
поэтому рекомендуется дополнительно
прокалить полученную соль до 700° в токе
водорода.
Препарат содержит 99,5—99,8% Na:S.
По С а б а т ь е можно сразу нагревать
ЫагБ'бНгО в реторте (в токе Fh) выше точки
плавления. Процесс заканчивают, когда
выходящий в реторты водород уже не будет
содержать воды. Красная масса, оставшаяся
в реторте, после охлаждения становится белой.
Реторту разбивают, препарат еще теплым
помещают в банку и плотно закрывают.
Свойства
Кристаллогидрат NasS • 9НгО — большие
прозрачные бесцветные или
слабо-окрашенные квадратные призмы, растворимые в воде
с сильно щелочной реакцией.
Раствор Na2S на воздухе быстро
окисляется, поэтому некоторые авторы
рекомендуют хранить его под слоем прованского
масла. На воздухе препарат становится влажным, но не расплывается;
одновременно происходит медленное окисление.
Рис. 64* Сушильный пистолет
Фишера
Уд. в. безводного NasS 1,86. Т. пл.2 1180°±10°в
Таблица 374
Растворимость Na2S вводе
/•с
10
18
28
37
о/0 Na,S
13,4
15,3
17,7
21,0
t°C
45
48
60
о/о Na.S
24,2
26,3
28,1
feC
70
80
90
о/о Na,S
30,2
32,9
36,4
l G. С о и г t о i s, С. г. 207, 1220 (1938).
I О. С о и г t о i s, С. г. 2С8, 277 (1939).
Натрий серноватистокислый
387
Таблица 375
Удельный вес водных растворов Na2S
о/0 NatS
1
2
4
6
rf413
1,010
1,021
1,044
1,067
о/о NatS
8
10
12
<*41в
1,091 1
1,115
1,139
о/о NatS
14
16
18
<у»
1,163
1,1*8
1,214
'
испытание
^v ПО ГОСТ 2053—43 содержание Na2S • 9НгО в препарате ч. д. а. должно
£,$ыть не менее 97% и в препарате чистом — не менее 92%; наибольшие коли-
;$ества допустимых примесей указаны в табл. 376.
Допустимые примесив Na2S • 9Н20
Таблица 376
1. Нерастворимые в воде вещества
2. Вещества, окисляемые иодом . .
(в пересчете на Г^Г^гО^ -5Н20)
3. Соли аммония (NH*4) ......
4. Металлы группы H2S
0,002
1.0
0,002
Отсутствие
0,005
3,0
0,006
Natrium hyposulfu-
rosum
"Natrium thyosulfuricum
НАТРИЙ СЕРНОВАТИСТОКИСЛЫЙ
(ТИОСУЛЬФАТ НАТРИЯ, ГИПОСУЛЬФИТ)
Natriumhyposulfit
Sodium hyposulfate
Sodium thiosulfate
Na£S203 • 6Н2О
Мол. в. 248.19
Приготовление
1- Для получения препарата кипятят в колбе с обратным холодильником
раствор /00 г сернистокислого натрия (Na2SOs) в 200 мл воды с 14 г тонко
Истертой черенковой серы, предварительно смоченной спиртом (иначе сера не
смачивается раствором и плавает на поверхности). Нерастворившуюся серу
отфильтровывают и раствор выпаривают до кристаллизации.
2. О. Дейнес и Г. Грассман1 приводят следующий метод лаборатор-
fioro получения ЫагЕаОз. В раствор 30 г NaOH в 200 мл воды пропускают
сильный ток HaS и SO» так, чтобы скорость FhS была немного большей
^контроль по числу пузырьков в промывных склянках).
О. v. D i n e s, H. Orassmin, Z. anorg. Gh. 220, 343 (1934).
38 8 Натрий
Когда начнет выделяться свободная сера (признак скорого окончания п л
цесса), колбу взбалтывают, причем сера растворяется. Тогда прекращают 'г0*
Нг£ и продолжают пропускание SCte, пока желтый раствор не станет бесцве
ным и не покажет нейтральной реакции на лакмус: ет* *
6NaOH + 4S02 + 2H2S = 3Na2S203 + 5Н20.
Раствор фильтруют, выпаривают и оставляют для кристаллизации в холод,
ном месте. Выход около 180 г ЫагБгОз • 5НЮ.
3. Для получения вполне чистого препарата, пригодного для ч всех анали
тических целей, Мейнеке рекомендует следующий метод. Растирают очи". •
щенную перекристаллизацией соль с 96%-ным спиртом, выливают на фильтр V
и, дав стечь спирту, промывают соль абсолютным спиртом и эфиром.
Препарат оставляют на сутки, покрыв фильтровальной бумагой, после чего
ссыпают в сухую банку. По свидетельству автора очищенный таким путем
препарат содержал 99,99%' гипосульфита и даже после 5-летнего хранения
дал содержание 99,9—99,94%.
Свойства
Большие прозрачные моноклинические призмы уд. в. 1,73, не изменяющиеся
на воздухе. Легко растворим в воде со слабощелочной реакцией; не
растворим в спирте. В водном растворе постепенно распадается на Na^SOs и S.
Существуют указания, что твердый №23гОз ведет себя аналогично *. При 56°
плавится в кристаллизационной воде, при 100° обезвоживается. При
прокаливании распадается на Na2S и Na2S04. Разлагается сильными кислотами с
выделением S и ЗОг.
Таблица 371
Растворимость Na2S203 вводе
ГС
•
0
10
20
30
о/о Na.StO,
34,4
37,9
41,2
45,9
t°c
40
45
50
1 60
°/о N?tS,Oa
50,6
54,5
62,9
67,4
t°c
72
80,5
90,5
100
°/о N28S,03
70,4
71,3
71,8
72,7
Таблица 378
Удельный вес водных растворов Na2S203
0/tNa,S,O6
Г
2
4
6
8
10
d?
1,0065
1,0148
1,0315
1,0483
1,0654
1,0827
*/• Na,S,0,
12
14
16
18
20
22
dA*>
1,1003
1,1182
1,1365
1,1551
1,1740
1,1932
0.0 N3,8,03
24
26
28
30
35
40
л
1,2128
1,2328
1,2532
1,2739
1,3273
1,3827
i Y. К. L а М е г а. Н. М.ТртИпзоп, lad. Eng. Chem., Anal. Ed. 9, 588 (1937).
Натрий сернокислый
389
0р ы т а н и е
Щи> ОСТ 10900-40 содержание №2£20з • 5Н20 должно быть для препарата
, д <*• не менее "»°% и Для препарата чистого — не менее 98%; наибольшие
' *лячества допустимых примесей указаны в табл. 379; препарат должен
выуживать приведенное в ОСТ испытание на реакцию водного раствора.
Таблица 379
Допустимые примесив NaaS203
Примеси
Допустимое содержание в о/0
ЧИСТОМ ДЛ1
анализа
^Нерастворимые в воде вещества . .
& Кальций (Са)
Ж Сульфиды (S")
Р^Сульфаты и сульфиты (SO/' + S03")
0,01
0,005
0,0005
0,15
0,015
0,01
0,001
0,3
НАТРИЙ СЕРНОКИСЛЫЙ
(СУЛЬФАТ НАТРИЯ, ГЛАУБЕРОВА СОЛЬ»
'Natrium sulfuricum Sodium sulfate Natriumsulfat
Sodium sulfate
Glauber's salt
Na2SO410H2O Мол. в. 322,21
Na2S04 Мол. в. 142,05
Claubersalz
Приготовление
Нейтрализуют раствор ЫагСОз разбавленной H2SO4, фильтруют w
выпаривают до кристаллизации.
4 Для получения безводного препарата декагидрат (Na2SC>4 • ЮН2О)
нагревают в фарфоровой чашке при температуре около 100°, пока не останется
рыхлый белый порошок.
Свойства
Большие бесцветные моноклинические призмы уд. в. 1,46, горько-соленого
ькуса. На воздухе быстро выветривается, переходя в белый порошок
безводной соли. При 32,4° Na2$04 • 10НЮ плавится в кристаллизационной воде.
Безводный Na2S04 имеет2 т. шь 885°, т. кип. 1430°.
Таблица 380
ГС
I
1
0
10
15
20
Растворимость Na2S04 вводе
о/о Na9S04
4,5
8,2
11,7
16,1
/°с
25
30
32,28
40
°,'о Na,S04
21,9
28,8
33,2
32,5
(°С
50
70
90
100
о/0 Na,S04
31,9
30,5
30,0
29,9
1 Термин глауберова соль" относится только к 10-водному препарату.
1 G. С о и г t о i s, Сотр. rend. 208, 199 (1939),
390
Натрий
Удельный вес водных растворов Na2S04
(S с h i f I)
Таблица 3Sj
d »•
1,0040
1,0079
1.0Р8
1,0158
1,0198
1,0238
1,0278
1,0318
1,0358
1,0298
1.С439
1,0479
1,0520
1,^560
1,0601
И спытани
°/oN.",S04
0,441
0,881
1,323
1,7'4
2,205
2,646
3,087
3,528 ,
3,969
4,410
4,851
5,292
5,773
6,374
6,615
е
°/oN2,SOY10PsO
1
2
3
4
5
.-' 6
7
8
9
Ю
И
12 |
13
14
15
1
d19'
1,0642
1,0083
1,0725
1,0766
1,0807
1,0849
1.0Я90
1,0931 ,
1 1,0973
1,1015 I
1,1057
1,1-00
1Л42
1,11*4
1,1226
о/о Na,S04
7,056
7,497
7.Q38
8,379
8,820
9,261
9,702
10,143
10,584
11,025
11, 65
11,007
12,.: 48
12,. 89
13,230
k>NrsS04.ioHa0
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
По ОСТ НКТП 7371/525 и 7372/526 наибольшие количества допустимых при»
месей указаны в табл. 382; оба препарата должны выдерживать приведенные
в ОСТ испытания на реакцию водного раствора,
Таблица 382
Допустимые примеси в Na2S04
Примеси
Допустимое содержание в °/0
для безвотного NaeS04
(OCT 7372/t>2o)
s я о
s а* н
£ О о
к 2J *
X ЕР у
S А
с s
Н <Л W
и со
S « S
для N«,SO4.10H,O
(ОС1 737 J/526)
ее 2
1 я о
£ Z о
1. Нерастворимые в воде вещества . .
2. Хлориды (СИ) . •
3. Аммонийные соли (NH4*)
4. Нитраты (N03')
5. Железо (Fe)
6. Кальций, магний и вещества,
осаждаемые аммиаком
7. Тяжелые металлы, осаждаемые H2S .
8. Мышьяк (As)
9. Влажность . „
0,005
0,001
0,002
0,002
0,0002
0,02
0,001
0,00021
0,2
0,01
0,003
0,002
0,002
0,0008
0,04
0,002
Д,0'05
0,2
0,02
0,005
0,006
0.С06
0,0015
0,1
0,004
0,0005
0,5
0,002
0,0005
и,001
0,001
0,0001
0,01
0,0005
0,0001
0,005
0,001
0,001
0,'01
0,0U04|
0,02
0,001
0,0003
0,01
0,002
0,003
0,003
0,0008
0,05
0,002
0,0003
Натрий сернокислый кислый
391
НАТРИЙ СЕРНОКИСЛЫЙ КИСЛЫЙ (БИСУЛЬФАТ НАТРИЯ)
Natrium bisulfuriciim Sodium bisulfate Natriumbisulfat
NaHS04 Н20 Мол. в. 138,08
NaKS04 Мол. в. 120,07
Приготовление
1. растворяют 90 ч. кристаллического сернокислого натрия Na2$O4«10H2O
смеси 1 ч. H2SO4 и 13 ч. воды. Раствор выпаривают на водяной бане (не
доводя до кипения) и охлаждают. Кристаллы отсасывают и помещают в
плотно закрывающиеся банки.
2. Для получения безводного NaHSCh нагревают до полной прозрачности
смесь 71 ч. Na2S04 и 49 ч. чистой H2SO4 (уд. в. 1,84). Расплавленную массу
выливают в платиновую или фарфоровую чашку, охлаждаемую водой. По
затвердении массу разбивают на куски и складывают в банки.
Свойства
NaHS04 • Н2О — крупные, бесцветные гигроскопические кристаллы,
растворимые в воде с сильно кислой реакцией. Т. пл. 315°. При нагревании сначала
дает безводную соль, затем, теряя еще молекулу воды, переходит в пиросер-
нокислый натрий.
Безводный NaHSC>4 — прозрачные призматические кристаллы уд. в. 2,74,
мутнеющие на воздухе, но не гигроскопичные.
Таблица 383
Удельный вес водных ра ст в оро в NaHS04
°/0NaHSO4
J
1
2
4
6
d4»°
1,0059 '
1,0137
1,0293
1,0451
0/0 NaHS04
8
10
12
14
<*4*>
1.Г611
1,0773
1,0937
1,1103
о/о NaHSO«
16
18
20
22
dp
1,1271
.1,1441
1,1614
1,1789
Испытание
1. Хлориды (СУ). Раствор препарата (1 : 20), подкисленный HNOs (уд. в. 1,15),
Может давать лишь слабую опалесценцию от прибавления 0AN раствора
AgN03.
I 2. Калий (К'). При наблюдении через кобальтовое стекло (или индиговую
призму) пламени горелки, окрашенного препаратом в желтый цвет, допускается
лишь быстро исчезающее фиолетовое окрашивание.
Р 3. Тяжелые металлы и железо. Раствор препарата (1 : 20) не должен
изменяться от прибавления сероводородной воды. При добавлении к раствору
NH4OH (до щелочной реакции) и (NH4)2S не должно появляться темного
окрашивания или осадка.
4. Мышьяк (As). К 3 мл насыщенного раствора SnCb в НС1 (уд. в. 1,19)
прибавляют 1 г растертого препарата; в течение часа не должно появляться
Темного окрашивания.
5. Кальций (Са'*). К раствору препарата (1:20) прибавляют NH4OH до
Мелочной реакции и (ЫН4)2Сг04. В течение 10 мин. раствор должен оставаться
Прозрачным.
6. Содержание NaHS04 • Н2О в препарате должно быть для всех
квалификаций не менее 95,% яри титровании щелочью по методу насыщения.
392
Натрий
НАТРИЙ ТИОСУРЬМЯНОКИСЛЫЙ(ОРТО)
(СУЛЬФОАНТИМОНАТ НАТРИЯ, СОЛЬ ШЛИППЕ) ]
Natrium Sodium Natriumsulfoantim * i
thiostibicum thioantimonate Schlippesdies sa?at *
Na3SbS4-9H20 Мол. в. 481,14
Приготовление
1. По Шлиппе нагревают до наступления спокойного плавления в
крытом гессенском тигле тщательно приготовленную смесь 8 ч. безводно3^?
Na2S€)4 и 3 ч. угля. Затем вносят при перемешивании смесь 6 ч. тонко и
тертой сернистой сурьмы (Sb^Ss) с 1,2 ч. серы и продолжают нагревание°"
закрытом тигле' до вторичного спокойного плавления и исчезновения серой
окраски, зависящей от присутствия непрореагировавшей £ЬгЗз; следует избе-
гать слишком высокой температуры.
Полученный сплав выливают на железный лист, после затвердевания
измельчают и кипятят с 10-кратным количеством воды. Когда жидкость несколько
просветлеет, фильтруют (остаток на фильтре промывают небольшим
количеством горячей воды) и фильтрат выпаривают до кристаллизации. Выпавшие
кристаллы собирают на воронку, обмывают разбавленным раствором NaOH
и сушат между листами фильтровальной бумаги. Маточный раствор, после
добавления небольшого количества NaOH и упаривайия, дает еще порцию
кристаллов.
2. По Рюсту1 к раствору 7,7 г NaOH в большой фарфоровой чашке
прибавляют 18 г тонко истертой сернистой сурьмы (8Ьг5з) и 3 г порошка
серы и смесь нагревают до кипения при постоянном перемешивании,
пополняя испаряющуюся воду. Кипячение продолжают до тех пор, пока серая
вначале окраска не перейдет в желтоватую. Тогда раствор декантируют через
фильтр и оставшуюся в чашке массу выщелачивают 50 мл воды при
кипячении. После фильтрования все растворы объединяют и выпаривают на водяной
бане до начала кристаллизации; если при этом раствор мутнеет и
приобретает коричневую окраску, добавляют немного NaOH. Выпадающие при
охлаждении кристаллы собирают «а воронке; из стекающего маточного раствора
можно извлечь выпариванием еще порцию кристаллов.
Полученную соль сушат между листами фильтровальной бумаги. Если
необходимо сохранить кристаллы без побурения, их хранят в спиртовом
растворе NaOH.
Свойства
Бесцветные или слегка желтоватые тетраэдры уд. в. 1,81, на воздухе
покрывающиеся с поверхности красно-бурым налетом ЗЬгЯз. Растворим в в°де
(1:3), не растворим в спирте. Водные растворы имеют щелочную реакцию^
Для сохранения кристаллов их рекомендуется хранить под спиртовым Рас"
твором NaOH.
НАТРИЙ УГЛЕКИСЛЫЙ (КАРБОНАТ НАТРИЯ, СОДА)
Natrium carbonicum Sodium carbonate, Natriumcarbonat
Soda Soda
Na2COs Мол. в. 106,004.
Na2CO3-10H2O Мол. в. 286,164
Приготовление
1. Для получения кристаллического препарата растворяют 1 ч. безводи
соды (Na2COa) в 5 ч. горячей воды, фильтруют и охлаждают до комнат*
Anleit. z. Daret. wig. РгЯр. 1903.
Натрий тиосурьмянокислый и углекислый
393
оатуры. Выпавшие кристаллы быстро отсасывают и помещают в плотно
TiUnbiTVK) банку.
"ЧГПо Ста с у технический Na2CO.i прокаливают и кипятят с таким коли-
вом в°Ды» чтобы часть взятого продукта осталась нерастворенной. Филы-
*бС*уг и фильтрат испытывают на присутствие Fe (пропускание H2S не должно
Р ять появления зеленого окрашивания или осадка); в случае обнаружения
^операции повторяют.
v Если в сырье содержится силикат натрия NasSiOa, то раствор выпаривают
Incvxa с кусочками (NH^CCb, сначала на водяной, затем на песочной бане.
¥vzou остаток растворяют в воде, отфильтровывают SiO* и проводят
повторную очистку, как указано выше.
Г 3. Совершенно чистый безводный ЫагСО* получают прокаливанием двуугле-
йсяелого натрия (NaHCOa) в платиновой чашке до постоянного веса. При упо-
Еоеблении хорошей бунзеновской горелки достаточно 15-минутного нагревания.
Г?4 однако Л у н г е рекомендует, во избежание образования окиси натрия
fNa*0). весту: нагревание при температуре не выше 270—300°, при постоянном
'перемешивании платиновой проволочкой. Полученный №гСОз охлаждают в
Эксикаторе.
Другие авторы рекомендуют еще более осторожное нагревание, применяя
Нйтробензольную баню1 (т. кип. 206°) или продолжительное выдерживание при
160° до постоянного веса; однако такая обработка требует много времени.
• 4. Можно получить2 безводный ЫагСОз, нагревая декагидрат Na2CO* •
• H)HsO в фарфоровой чашке на песочной бане, не поднимая температуры
выше 308°.
5. Для приготовления моногидрата (№гСОз • НгО) нагревают десятиводную
соль (№гСОз- ЮНгО) до 60—70°. При этом соль плавится в
кристаллизационной воде и частью остается в растворе, частью выпадает в виде мелких
кристаллов моногидрата. Еще теплую жидкость фильтруют через воронку для
горячего фильтрования и промывают кристаллический порошок -горячим
спиртам.
Свойства
ЫагСОз • ЮНгО — бесцветные, прозрачные, моноклинические кристаллы
уд. в. 1,5, быстро выветривающиеся на воздухе с образованием белого
порошка ЫагСОз • бНгО. При 35,37° плавится в кристаллизационной воде,
выделяя гидраты с меньшим содержанием воды (№гСО * НгО и, возможно, №гСО.ч •
•2НгО и ЫагСОз • ЗНгО). Растворим в воде (с охлаждением); максимальная
растворимость наблюдается при 34°. Водные растворы имеют сильно
щелочную реакцию. (Для 6Д/ раствора рН = 12,8, для IN рН = 12,27, для 0,1Л/
рН = 10,9 3.
Таблица 384
Растворимость Na2C03 вводе
/•с
0
5
10
15
20
о;0 NatC03
6,63
8,6
П,2
14,1
17,8
ГС
1 25
30
40
50
i
•о Na8C03
22,8
29,0
33,2
32,2
t°c
60
70
88,4
104,8
<Ч Na,C03
31,7
31,4
31,1
31,1
1 S t а 1 о n y-D о b r z а п с k i, С. 1, 107, 1268 (1 Jo).
9 Q 1 a s e r, Indicatoren der Alcali- u. Acidimetrb, 1900, стр. 83,
Wnvfn,RobertsiG'Man«old, C- 1, 2449 (194Э>; Н. А. Т а н а н а е в и Ю. В. К а р я к и н,
*КПХ 13, 306 (1940).
394
Натрий
Ыа2СОз • НгО — мелкие, ромбические кристаллы уд. в. 2,259 \ начинающт I
терять воду уже при 47° на воздухе. ^ 1
ЫагСОз — белый порошок уд. в. 2,5, легко растворимый в воде с разогп"*« '
нием (теплота гидратации). Т.* пл. 852° (по другим авторам 856°). ре* % i
Таблица 385
Удельный вес водных растворов Na2C03
ванием
A VT л С* Г\
*\о NaaL.Ua
1 '
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
о/0КааСОз-ЮН80
2,7
5,4
8,1
10,8
13,5
16,2
18,9
21,6
24,3
27,0
29,7
32,4
35,1
37,8
40,5
43,2
45,9
48,6
51,3
54,0
56,7
59,4
62,1
64,8
67,5
70,2
72,9
75,6
78,3
81,0
</4зо
—
—
—
—
—
—
—
—
—
—
—
—
1,1417
1,1526
1,1636
1,17-7
1,1859
1,1972
1,2086
.1,2201
1,2317
1,2434
1,2552
1,2671
1,2790
1,2910
1,3031
1,3152
1,3274
dp
1,0096
1,0201
1,0306
1/411
1,0516
1,06 2
1,0728
1, 834
1,0941
1,1048
1,1156
1,1265
1,1375
1,1485
—
—
—
—
—
—
—
—
■"~
1 л раствора при 3J° С
содержит
NajGO.
10,09
20,38
Зи,8Я
41,59
52,51
63,64
74,98
86,53
9^,20
110,3
122,5
134,9
147,6
160,5
—
—
NajCOa-lOHjO
27,22
55,03
83,37
11 ,3
141,8
171,2
202,5
233,6
265,8
297,8
330,8
364,3
39\6
433,3
—
—
—
—
—
—
—
—
—.
—.
—
—
—.
—
—
~
Испытание
По ГОСТ 83—41 содержание ЫагСОз в безводном препарате (считая на
прокаленный) для всех квалификаций должно быть не менее 99,8%;
наибольшие количества допустимых примесей приведены в табл. 386.
По ГОСТ 84—41 содержание 1Ма>СОз в кристаллическом препарате (считая
на прокаленный) для всех квалификаций должно быть не менее 99,8%;
наибольшие количества допустимых примесей приведены в табл. 386.
»Н. Braeeeur, Zeitschrift Krlstallographie A 90, 282 (1935).
Натрий углекислый кислый
395
Допустимые примесив Na2CO;?
Таблица 386
Примеси
Допустимое содержание
для безводного N?-C03
ГОСТ«3-41
Н 5
5 Я О
м о о
9 х
— а эт
Э* * S
33 с< Ч
ГОС1 81 — 41
2 = 5
к «2
t Потери при прокаливании . .
*'% Нерастворимые в воде вещества
&Хло>иды (CV)
4, Сера — общ е количество в сул ьфа-
- тах, сульфитах, тиосульфатах, суль-
' фидах и пр. — в пе-n сч те на SO/'
5. Азот — общее количество в
нитратах, нитритах, аммиже и пр. . . .
& KpcMHtкислота tSi03") ....
% Фосфаты (РО/")
f Вещества, осаждаемые аммиаком
Кальций и магний (Са и Mg) .
40, Тяжелые металлы, осаждаемые H2S
t Железо (Fe)
Мышьяк (As)
13. Калий (К)
1
0,005
0,001
(J.004
0,001
0,0 J 3
0,002
[0,005
0,01
0,0005
0,00)1
0,ОМ°5
0,005
1
0,01
0,002
0,С0б
|0,001
(,0 3
0,002
0,01
0,02
0,0005
0,'ЮОЗ
1,5
0,02
0,005
0,01
0,002
0,01
0,005
,0,(3
|о/>2
0,001
0,001
1,00005 0,00005
0,005 ,0,02
0,002
0,0J05
0,002
0,0^05
0,001
0,0005
0,002
0,005
0, ,003
0, 0005
0,00 03
0,003
0,005
0,001
0,003
0,0005
0,001
0,0005
0,004
0,01
0,('003
0,000*5
0,00003
0,003
0,01
0,003
0,005
0,001
0,005
0,002
0,01
0,01
|0,0005
0,0и03
[0,00003
0,01
НАТРИЙ УГЛЕКИСЛЫЙ КИСЛЫЙ
(БИКАРБОНАТ НАТРИЯ, ДВУУГЛЕКИСЛЫЙ НАТРИЙ)
Natrium bicarbonicum Sodium bicarbonate Natriumbicarbonat
NaHCOs
Мол. в. 84,015
Приготовление
1. В большую сушильную колонку (см. рис. 55)
помещают десятиводную соду (ЫагСОз * ШНгО),
удерживая ее в верхней части колонки при помощи
рыхлого тампона из стеклянной ваты или сетки Л.
Через трубку С подводят медленный ток СОг,
промытого пропусканием через раствор NaHCOs.
Одновременно с поглощением СОг содой
выделяющаяся кристаллизационная вода стекает вниз и может
быть выпущена через трубку В.
Реакция происходит согласно уравнению:
№2СОз • 10Н2О + СО* = 2№НСОз + 9ШО.
Сверху колонку прикрывают часовым стеклом для
Предохранения от пыли. Когда совершенно
прекратится выделение воды, полезно еще в течение часа
Пропускать СОз для перевода последних остатков соды
В NaHCOa. Полученный рыхлый порошок тщательно P^. 55 Колонка для получе-
% отсасывают, промывают небольшим количеством ледя- ння бикарбоната натрия
3%
Натрий
ной воды, насыщенной СОг, и высушивают на стекле при комнатной темп
ратуре. ^
2. По П е с ц и очень чистый NaHCOa получается при насыщении yi Ле "*
лым газом раствора №:>СОз в 80%-ном спирте.
лекис:
Свойства
Снежно-белый кристаллический порошок, растворимый в воде и ц0Чти
не растворимый в спирте. При 350—400° теряет воду и СОг, количественно
переходя в ЫаэСОз. Даже при лежании на воздухе при комнатной темпера,
туре соль теряет заметные количества СОг.
NaHCOs кристаллизуется также в виде маленьких четырехугольных
табличек. Й
4
ГС
0
10
15
Растворимое ть
о/о NaHCOe
6,45
7,58
8,09
У дел ь н ы й
о/о NaHCOa
1
2
3
4
/°с
20
30
40
вес водны
rf418
1,0059
1,0132
1,0206
1,0280
NaHC03 вводе
о/о NaHC03
8,76
9,96
11,27
х раствор
о;0 NaHC03
1 5
6
7
8
/°с
45
50
60
о в NaHCOg
<J*lB
1 1,0354
1,0429
1,0505
| 1,0581
Таблица 387^
о/о NaHCO,
12,17
12,67
14,09
Таблица 388
Испытание
а) По ОСТ НКТП 7379/533 содержание NaHCOs для препаратов х. ч. и
ч.д.а. должно быть не менее 98,5%; наибольшие количества допустимых при
месей указаны в табл. 389.
Таблица 389
Допустимые примеси в NaHC08
Примеси
1. Нерастворимые в воде вещества
2. Хлориды и хлораты (С1) ....
3. Сера (общее количество из
сульфатов, сульфитов, тиосульфатов,
сульфидов в пересчете на SO/')-
4. Азот (общий из нитратов, нит-
6. Кремнекислота и вещества, оса-
8. Тяжелые металлы
9. Железо (Fe)
10. Калий (К)
Допустимое содержание в °/0
в химически
чистом
0,005
0,002
0,005
0,0005
0,001
0,005
0,005
0,0005
0,0005
0,01
в чистом для
анализа
0,01
0,005
0,005
0,001
0,002
0,01
0,01
0,001 \
0,001 р
0,02 *
в чистом
0,02 -
0,01
0,01
0,002
0,005
0,05
0,02
0,002
0,002
0,04
Натрий уксуснокислый
397
НАТРИЙ УКСУСНОКИСЛЫЙ (АЦЕТАТ НАТРИЯ)
^atrium aceticum Sodium acetate
CHsCOONa * ЗН2О
CHaCOONa
Natriumacetat
Essigsaures Natrium
Мол. в. 136,089
Мол. в. 82,041
Приготовление
1. 400 мл чистой 30%-ной СНзСООН нейтрализуют, добавляя понемногу
Ег ЫагСОз • ЮНгО; под конец нейтрализация проводится при нагревании на
яной бане. Горячий раствор отфильтровывают, выпаривают фильтрат до
,ема 250 мл и дают охладиться. Выпавшие кристаллы отсасывают и
высуживают при комнатной температуре.
2. Для очистки продажного препарата от органических примесей (для
объемного определения марганца) по Б. Рейницеруи П. Конрату1 к
раствору 30—40 г соли в 500 мл горячей воды приливают 0,1N КМ11О4 до неис-
Чезающей в течение нескольких часов слабо-розовой окраски. Горячий
раствор фильтруют через стеклянную вату, выпаривают на водяной бане до
объема 50—60 мл и дают закристаллизоваться.
3. Безводный CHsCOONa получают нагреванием кристаллической соли при
120° в фарфоровой чашке до получения сухой белой массы.
Свойства
Бесцветные прозрачные кристаллы уд. в. 1,528. При 58° плавятся в
кристаллизационной воде, выше ЮО0* обезвоживаются. Легко растворим в воде
£см. табл. 390), труднее в спирте (2,1 : 100). Водные растворы имеют слабо
щелочную реакцию (для IN раствора рН = 8,2).
Безводный СНзСООЫа плавится при 324°; при более высокой температуре
разлагается на углекислый натрий и ацетон:
2CH*COONa = Na2COs + (СНз^СО.
Таблица 390
Растворимость CH3COONa в воде
/° с
0
10
20
30
о;0 CH8COONa
26,6
29,0
31,7 !
35,2
г с
40
50
58
o;0CH3COONa
39,5
45,3
58,0
t° с
60
1 80
100
о/о CHjCOONa
58,2
60,5
63,0
Таблица 391
Удельный вес водных растворов CH3COONa
CHjCOONa
1
2
4
6
8
4°
1,0033
1,0084
1,0186
1,0289
1,0392
°,'о
CH3COONa
10
12
14
1 1'6
18
-У
1,0495
1,0598
1,0702
1,0807
1,0913
| CH.COONa
20 1
22
24
26
28
а20
4
1,1021
1,1130
1,1240
1,1351
1,1462
1 В, R e i n i t х е г, Р. С о п г a t h. Z. analyt. Ch. 68, 129 (1926).
398
Натрий
%
Испытание
По ГОСТ 199—41 содержание CHsCOONa. ЗНЮ в препарате ч. д. а. „•
чистом должно быть не менее 99,0%; наибольшие количества допустимых При.
месей приведены в табл. 392; препарат ч. д. а. должен выдерживать
испытание на вещества, восстанавливающие КМп04; препарат ч. д. а. с надписью на
этикетке: «Пригоден для определения марганца» должен выдерживать прнве.
денную в ГОСТ пробу Рейнитцера.
Таблица 392
Допустимые примеси в CH3COOONa • ЗН20
Примеси
Допустимое содержание в«
в чистом
для анализа
0,005
0,0Г»2
0,005
0,001
0,0005
0,0005
0,002
0,003
в чистом
0,01
0,005
0,01
0,002
0,001
0,002
0,005
0,01
1. Нерастворимые в воде веществе
2. Хлориды (С1')
3. Сульфаты (S04")
4. Тяжелые металлы, осаждаемые H2S
5. Железо (Fe)
6. Фосфаты (РО/")
7. Вещества, осаждаемые аммиаком
8. Кальций и магний (Са и Mg)
9. Кисло ность (СН3СООН) или щелочность
(Na^Ou)
0,02
0,05
НАТРИЙ ФОСФОРНОВАТИСТОКИСЛЫЙ (ГИПОФОСФИТ НАТРИЯ)
Natrium
hypophosphorosum
Sodium hypophosphite
monobasic
Natriumhypophosphit
Unterphosphorigsaures
Natrium
NaH2PQ2 • H2O
Мол. в. 106,01
Приготовление
К раствору гипофосфита бария1, освобожденному от Ва(ОН)2 пропуска
нием СОг, добавляют понемногу раствор Na»COs до прекращения выпадения
осадка ВаСОз. Избытка Na2C03 следует избегать. Жидкость нагревают до
кипения, отфильтровывают и выпаривают на водяной бане досуха.
При выпаривании следует соблюдать осторожность (не наклоняться над
чашкой, беречь глаза), так как иногда происходят взрывы.
Свойства
Бесцветные, расплывающиеся на воздухе кристаллы, легко растворимые в
воде и спирте. Сильный восстановитель. При нагревании распадается по
уравнению:
4NaH2P02 = 2РНз + 2Na2HP04.
Получение безводной соли нагреванием кристаллогидрата, поэтому,
невозможно.
i См. стр. 290. Можно пользоваться также раствором гипофосфита кальция, приготовленным
аналогичным путем.
Натрий фосфорноватистокислый а фосфорновольфрамовокислый 399
испытание
По ГОСТ 200—41 содержание NaH2PC>2 • Н2О в препарате ч. д. а. должно
0ЫТЬ не менее 98,0% и в препарате чистом —по. менее 95,0%; наибольшие
количества допустимых примесей приведены в табл; 393; препараты обеих
квалификаций должны выдерживать приведенные в ГОСТ испытания на
щышьяк и на пригодность их для определения мышьяка, а также на реакцию
5%-ного водного раствора.
Таблица 393
Допустимые примеси в NaH2P02-H20
Примеси
Допустимое содержание в о/0
в чистом
для анализа
1. Нерастворимые в воде вещества
2. Фосфиты (Na2HP03 • 5Н20) . . . .
3. Кал» ций (Са)
4. Сульфаты (SO/)
0,005
0,5
0,02
0,02
0,01
1,5
ОД
0J
НАТРИЙ ФОСФОРНОВОЛЬФРАМОВОКИСЛЫЙ
(ВОЛЬФРАМАТ-ФОСФАТ НАТРИЯ)
Natrium phosphowolf-
ramicum
Sodium phosphotung-
state
Phosphorwolframsaures
Natrium
Na2H5[P(W207)e] Мол. в. 2961,05
Приготовление
•По методу, разработанному в ИРЕА, 50 г вольфрамовокислого натрия
(Na2W04 • 2Н2О) сушат 4 часа при 150°, растворяют в 100 мл дестиллирован-
ной воды (не содержащей NH4", Mg*', Са** и восстановителей) на холоду
и оставляют на сутки, после чего отфильтровывают от образовавшегося
осадка. Фильтрат разбавляют вдвое водой, нагревают до кипейия и
нейтрализуют разбавленной (2:1) НО, применяя в качестве индикатора
лакмусовую бумажку. Образовавшийся осадок вольфрамовой кислоты (H2WO4)
должен при этом раствориться. Нейтрализованный раствор кипятят еще 15 мин.,
пополняя испаряющуюся воду, и оставляют на сутки, в течение которых
выпадает белый нерастворимый осадок.
К прозрачному раствору (паравольфрамат натрия) прибавляют 78 г
фосфорнокислого натрия (Na2HP04 • 12НгО), нагревают до кипения и небольшими
порциями добавляют 45 мл НС1 (уд. в. 1,19). Раствор фильтруют и
выпаривают до половины объема. Через 12—14 час. выпавшие кристаллы
отсасывают, растворяют при нагревании в 2 ч. воды, фильтруют и выпаривают до
кристаллизации. По охлаждении кристаллы отсасывают, сушат при 40—50° и
хранят в плотно закрытой банке темного стекла.
Свойства
Мелкие белые кристаллы, выветривающиеся на воздухе. Хорошо
растворим в воде (47,39% при 20°).
400
Натрий
Испытание
Препарат квалификации ч. д. а. должен удовлетворять Следующим требо-"
ваниям: кристаллы должны быть белого цвета; раствор препарата после
3—5-минутного кипячения должен оставаться бесцветным и прозрачнцм;
должен содержать не более 0,3% SOs и 0,003% С1; содержание составных
частей: 94—95% W03, 2,3—2,45% Р2О3 и 2,15—3,3% Na20.
НАТРИЙ ФОСФОРНОКИСЛЫЙ КИСЛЫЙ ОДНОЗАМЕЩЕННЫЙ
(ДИГИДРОФОСФАТ НАТРИЯ)
Natrium phosphoricum
monobasicum
Sodium phosphate
monobasic
Natriumphosphat
primares (einbasisch)
NaH2P04 * 2Н2О
Мол. в. 156,03
Приготовление
По Митчерлиху к раствору двузамещенного фосфата натрия, ЫагНРСЬ,
приливают Н3РО4 до тех пор, пока капля смеси не перестанет давать осадок
с раствором ВаСЬ. Раствор выпаривают до начала кристаллизации и
охлаждают *.
Свойства
Бесцветные ромбические кристаллы уд. в. 2,04. При 57,4° плавятся в
кристаллизационной воде, при 100° обезвоживаются. Дальнейшее нагревание
приводит при 200° к образованию пирофосфата, ЫагНгРгСЬ.
Испытание
По ГОСТ 245—41 содержание NaFbPCh • 2НгО в препарате ч. д. а. должно
быть не менее 99,0% и в препарате чистом — не менее 98,0%; наибольшие
количества допустимых примесей указаны в табл. 394. рН раствора должен
быть 4,2—4,6.
Таблица 394
Допустимые примеси в NaH2P04 • 2Н20
Примеси
Допустимое содержание в °'0
в чистом
для анализа
1. Нераствориые в воде вещества
2. Хлориды (СГ)
3. Сульфаты (S04")
4. Железо (Fe)
5. Вещества, осаждаемые аммиаком
6. Тяжелые металлы, осаждаемые H2S
7. Азот — общее содержание в нитратах,
нитритах, аммиаке и пр
8. Мышьяк (As)
0,01
0,005
0,01
0,002
0,01
0,001
0,002
0,0002
0,02
0,01
0,02
0,005
0,02
0,005
0,005
0,0005
чаются
1 Наилучшие кристаллы по данным Ю. В. К а р
ся при рН раствора ^ 4,2.
якина и О. С. Воробьевой полу-
Щу^". Натрий фосфорнокислый кислый 401
!» НАТРИЙ ФОСФОРНОКИСЛЫЙ КИСЛЫЙ ДВУЗАМЕЩЕННЫЙ
^1* (ФОСФАТ НАТРИЯ)
■jjitrium phosphoricum Sodium phosphate Natriumphosphat
dibasic zweibasisch
;: Na2HP04 • I2H2O Мол. в. 358,17
Приготовление
Помещают в объемистую фарфоровую чашку 20%-ную фосфорную кислоту
ШдРОО и, нагрев ее на паровой бане, прибавляют раствор NaaCOs, пока
?о£ячая жидкость не приобретет слабо щелочную реакцию. Тогда фильтруют
,щ- фильтрат выпаривают до кристаллизации. Выделившиеся по охлаждении
Кристаллы помещают на воронку и, после того как жидкость стечет,
высушивают между листами фильтровальной бумаги при комнатной температуре.
" Из 100 ч. 20%-ной НзР04 и 60 ч. ЫагСОз-ЮНгО теоретически должно
получиться 73 ч. препарата.
для получения дигидрата №гНР04-2НгО по Зеренсену 12-водную соль
растирают в порошок и оставляют на 2 суток при 36—38°.
Свойства
£■ Бесцветные, прозрачные, моноклииические призмы уд. в. 1,63, быстро вы-
?;йетривающиеся на воздухе. Растворим в воде со щелочной реакцией (для
ШЛ—1.0N раствора рН около 9,0 *) и не растворим в спирте. Т. пл. 38°. При
||0ОО теряет кристаллизационную воду, при 250° распадается с образованием
?|1йрофосфорнокислого натрия, Ыа4Рг07. При кристаллизации из водных
растворов при температуре выше 30° выпадает гептагидрат, №гНР04 • 7НгО.
Таблица 395
Растворимость Na2HP04 в воде
к;
*°С
9
10
16
20
25
o/oN2»HP04
1,6
3>7
5,1
7,2
10,5
ГС
Эб
40
48,4
59
70
o/oNatHP04
30,0
35,6
44,1
47,6
48,7
ГС
78,5
85
95
o;oNa,HPO#
48,9
49,3
51,0
*
Таблица 396
Удельный вес водных
растворов Na2HP04
°!oNatHP04
1
2
3
*Ч
1,009
1,020
1,031
o/0NatHPO,
4
5
6
4
1,043
1,055
1,067
1 Н. А. т\ н а н а е в и Ю. В. К а р я к и н, ЖПХ 18, 303 (1940).
402
Натрий
И спытание
По ОСТ НКТП 7369/523 наибольшие количества допустимых примесей v
заны в табл. 397; при определении реакции водного раствора препарат/*а'
должен выдерживать испытание, приведенное в, ОСТ.
Допустимые примеси в Na2HP04« 12H?0
Телица 39?
Допустимые примеси
Допустимое содержание в о;0
1. Нерастворимые в в:де вещества
2. Хлориды (СГ)
3. Нитраты (NO{')
4. Сульфаты (S04")
5. Магний (Mg)
в. Железо (Fe) • . . • .
7. Тяжелые металлы
8, Мышьяк (As) . . . . •
в химически
чистом
0,002
0,001
0,0005
0,005
0.001
0.С005
0,< 005 1
0,0005
в чистом
для анализа
0,005
0.003
0,001
0,02
0,001
0,001
0,001
0,001 |
в чисто
0,01
0,005
0. 02
0,04
0/03
0,003
0,002
0,002
НАТРИЙ ФОСФОРНОКИСЛЫЙ ТРЕХЗАМЕЩЕННЫЙ'
(ОРТОФОСФАТ НАТРИЯ, ТРЕТИЧНЫЙ)
Natrium phosphoricum
tri]fc>asicum
Sodium phosphate
normal
Na3P04 • 12H20
Na3P04
Natriumphosphat drei-
basisch
Natriumorthophosphat
tertiar
Мол. в. 380,16
Мол. в. 163,97
Приготовление
1. По Холлу* к водному раствору 100 г кислого фосфорнокислого
натрия (Na2HP04> добавляют раствор 100 г NaOH в 100 мл воды. Затем
стакан с раствором погружают в другой большего размера стакан, содержащий
глицерин; внутренний стакан помещают так, чтобы уровень глицерина был
несколько выше уровня раствора. Оба стакана закрывают и нагревают на
водяной бане при температуре около 77° до тех пор, пока (примерно через
10 час.) не получится достаточное количество кристаллов. Маточный раствор
быстро сливают, а мелкие кристаллы промывают спиртом и хранят под слоем
спирта или бензина.
2. По Грэму к концентрированному раствору ЫагНР04 прибавляют
концентрированный раствор NaOH (на 10% меньше количества, требуемого
теорией). Жидкость выпаривают и охлаждают. Выпавшую соль перекристаллизо-
вывают из двойного количества воды, отсасывают, отжимают и сушат при
обыкновенной температуре.
Чистый NasP04 * 12НгО выделяется при испарении раствора NazHPOh
к которому добавлен NaOH до рН = 13,0 (Ю. В. К а р я к и н и О. С.
Воробьева).
* Н а 11, Chem. Soc. 51, 94 (1887); Prap. Ch. 344.
Натрий фосфорнокислый и фосфорномолибдёновокислый 403
Цойства
Бесцветные кристаллы уд. в. 1,63, в сухом воздухе выветривающиеся. При
734° плавится в кристаллизационной воде; полное обезвоживание не дости-
яется даже при 200°. В водном растворе соль почти полностью расщепляется
[я N42HPO4 и NaOH и имеет сильную щелочную реакцию. Уд. вес
безводной соли 2,54.
Таблица 398
Растворимость Na3P04 в воде
ГС
0
"10
15
20
0('oNa3P04
1.5
3,9
9,5
9.9
t°c
30
40
50
60
°/oN'8P04
16,7
23,7
30,1
35,5
t°c
70
80
P0
100
o/0Na3P04
40,8
44,8
48,7
51,9
Таблица 399
°loNa,P04
1
2
3
4
Удельный
л15
1,0087
1,0194
1,и2°9
1,0405
вес водных растворов NajP04
o/oNa3P04
5
6
7
И15
1,0515
1,0624
1,0737
o/0Na8P04
8
9
10
И15
dA
1,0850
1,0962
1,1083
НАТРИЙ ФОСФОРНОМОЛИБДЁНОВОКИСЛЫЙ
(24-МОЛИБДАТ-2-ФОСФАТ НАТРИЯ1, РЕАКТИВ ЗОННЕНШТЕЙНА)
Sodium phosphomolibdate
Natrium phospho-
molybdaenicum
Phosphormolybdansaures
Natrium
3Na2Q' P2O5 • 24МоОз + (58 + x)H2Q
Приготовление
1. По Керману при нагревании растворяют 100 г продажного молиб-
Деновокислого натрия и 15 г фосфорнокислого натрия (Na2HP04 • 12НгО) в
Возможно малом количестве воды и приливают к раствору при энергичном
Перемешивании концентрированную НС1 до прекращения образования осадка.
Полле охлаждения прибавлением НС1 проверяют полноту осаждения.
Получаемый по этому методу препарат содержит значительное
количество СГ.
2. По Брандхорсту и Крауту2 раствор 1000 г молибденовокислого
Натрия и 100 г фосфорнокислого натрия (Na2HP04 • 12Н20) в 4 л воды
смешивают с 350 г НС1, разбавленной в 4 раза, выпаривают досуха и
экстрагируют остаток 2,5 л смеси спирта и эфира. После отгонки спирта и эфира на
Водяной баке (огнеопасно\) остаток обрабатывают небольшим количеством
"NOa и кристаллизуют.
1 Номенклатура Розенгейиаи Коппеля.
•Brandhorst, Kraut. Ana. 249, 373.
404 Натрий *
3. А. С. Шахов1 предлагает следующий способ, дающий возможность,
исходя из более доступных материалов, получать препарат высокого качеств*
Прибавляют постепенно 100 г молибденовой кислоты к смеси 500 мл NH4o?f
(уд. в. 0,906) с 400 мл воды, находящейся в фарфоровой чашке. В начале
растворения смесь приходится несколько подогревать; в дальнейшем, вслед!
стпие выделяющегося тепла, растворение идет без применения нагревания
извне. ^
но окончании операции мутный темносерый раствор фильтруют горячим
причем получается светлый, но недостаточно прозрачный фильтрат.
Полученный фильтрат смешивают с одновременно приготовляемым горя.
чим раствором 100 г №гНРС>4 • 12НЮ в возможно малом количестве воды и
при 60° и постоянном перемешивании приливают 400 мл НС1 (уд. в. 1,19).
Выпавшему осадку дают отстояться, жидкость испытывают на полноту осаждения
прибавлением небольшого количества НС1 и, если требуется, снова добавляют
НС1, подогрев раствор до 60°.
Когда полнота осаждения будет достигнута, промывают осадок декантацией.
Вначале отстаивание идет быстро, но постепенно, по мере отмывания
примесей, осадок приобретает способность давать коллоидальный раствор;
промывание поэтому ведут до тех пор, пока осадок не перестанет быстро
отстаиваться. Тогда по возможности полно сливают промывную воду, а осадок в
виде кашицы прибавляют постепенно к раствору 200 г NaOH в возможно
малом количестве воды.
После растворения осадка образуется прозрачная голубовато-зеленая
жидкость; добавляют еще немного NaOH, горючем раствор мутнеет вследствие
выпадения гидратов окисей металлов, содержащихся в продажной
молибденовой кислоте. Раствор упаривают. После того как осадок гидратов
превратится в крупные хлопья и раствор станет прозрачным, фильтруют и фильтрат
выпаривают досуха* на песочной бане; в конце выпаривания рекомендуется
непрерывное перемешивание массы во избежание разбрызгивания.
Полученную массу для удаления NHs нагревают на песочной бане,
возможно глубже погружая в песок сосуд с осадком (в противном случае в
верхней части не будет достаточного нагревания). Температура прокаливаемой
массы не должна превышать 140°. Нагревание ведут до тех пор, пока проба,
растворенная в воде, не перестанет давать реакцию на NH4* (с NaOH при
нагревании не должен выделяться NHs).
По удалении NHs соль растворяют в воде, прибавляют еще немного NaOH
и упаривают, причем выпадает еще некоторое количество гидратов окисей
металлов (возможно из NaOH); их отфильтровывают, слабо зеленоватый
прозрачный раствор выпаривают до появления толстой пленки кристаллов и дают
ему закристаллизоваться. Выпавшие кристаллы отсасывают и сушат на водяной
бане.
Выход почти количественный.
4. Для приготовления раствора препарата по Коренблиту* раство*
ряют 20 г молибденовокислого аммония [(NH4)eMo?024 • 4Н*0] в 20 мл
горячей воды и прибавляют 100 г HNOs (уд. в. 1,2). К жидкости, нагретой на
водяной бане, добавляют раствор Na*HP04 до прекращения выпадения осадка.
Отстоявшуюся жидкость декантируют, осадок промывают горячей водой,
отсасывают и растворяют при нагревании на водяной бане в растворе NasCOs.
Раствор выпаривают досуха; остаток прокаливают до полного удаления
NHs, смачивают несколькими каплями HNOs н снова прокаливают до
сплавления. Наконец, белую соль растворяют в 20 мл воды и добавляют немного
HNOs (раствор при этом желтеет).
Свойства
Белый порошок, состоящий из мелких чешуек. Очень легко растворим н
воде. Хранить препарат следует в плотно закрытых банках оранжевого
стекла.
1 Сборник работ И PEA, 4, 7в (1927).
• Хим. реактивы, Ш1.
Натрий фтористый
405
циытание^
Препарат ч. д. а. испытывается на хлориды (с 'AgNOa), сульфаты (с ВаСЬ)
й-.чувствительность к алкалоидам.
4 г препарата растворяют в 80 мл воды, отфильтровывают осадок молмб-
ggBQBoPi кислоты и делят фильтрат на две части:
™а) Хлориды (СГ). Раствор, подкисленный 10 "мл 30%-ной НЫОз, после до-
деления 'AgNOs может дать лишь слабую опалесценцию.
б) Сульфаты (504"). Раствор, подкисленный 10 мл НС1 (уд. в. 1,12), после
^рил'ивания 1 мл 10%-ного раствора ВаСЬ в течение 3 час. не должен
образовывать mvth или осадка.
Для испытания на чувствительность к алкалоидам к 2 мл 5%-»ного
раствора " препарата (отфильтрованного от Н2М0О4) добавляют 2 мл 30%-ной
jffjOs и затем по стенке пробирки приливают 1 мл раствора солянокислого
|1ншна (1:400 000). Через 10 мин. в месте соприкосновения слоев должно
Дфявиться белое кольцо.
НАТРИЙ ФТОРИСТЫЙ (ФТОРИД НАТРИЯ)
Natrium fluoratum Sodium fluoride Natriumfluorid
NaF Мол. в. 42,00
Приготовление
V 1, Чистую плавиковую кислоту нейтрализуют ЫазСОз в платиновой чашке,
раствор выпаривают досуха и остаток прокаливают.
'■*' 2. Для очистки продажного препарата от примеси Na2SiFe по Н. А. Т а-
нанаеву2 растворяют 45 г соли в 1 л горячей воды, добавляют 13—15 г
КС1 и оставляют на ночь. Выпавший K»3iFe отфильтровывают, а раствор
выпаривают на водяной бане до объема 100 мл. Полученные кристаллы
отсасывают на воронке Бюхнера, промывают холодной водой до удаления СГ и
Высушивают при 120—150°.
Препарат следует хранить в парафинированной изнутри банке.
Свойства
Белый кристаллический
Щелочной реакцией. Т. пл
Таблица 400
Растворимость NaF в воде
порошок уд. в. 2,77,
988, т. кип. 1693°.
растворимый в воде со
Таблица 401
Удельный вес водных
растворов NaF
*°с
15
18
o/oNaF
3,85
4,22
/°с
21
25
o/oNaF
4 00
4,03
o/oNaF
1
2
3
<«
1,00«2
1.0198 J
1,0304 1
o/oNaF
4
5
4
104^9
1,0515
Испытание
По ОСТ НКТП 6760/367 содержание NaF в препарате ч. д. а. должно
быть не менее 98%; наибольшие количества допустимых примесей указаны
в табл. 402.
1 Испытание хим. релкивол, 2 8.
1 Объемный анализ, 6 изд., ГОНТИ, J939, стр. 2431
Натрий
Допустимые примеси в NaF
Табу,
Шца
Примеси
Допустимое содержание 8 0
в ччстом
для анал за
чистом
1. Нерастворимые в воде вещества
2. Хлориды (СК)
3. Сульфаты (SO/')
4. Тяжелые металлы (группы H2S)
5. Железо (Fe)
6. Кислотность (HF)
7. Щелочность (Na2C03)
8. Соли H2SiF6 для препарата с кислой реакцией
Na2SiF6
9. Кг емнек fслота для препарата со щелочной
реакцией (Si02)
0,05
0,005
0,05
0,005
0,005
0,2
1,0
0,4
0,05:
0,10
0,01
0,1
0,01
0,01
0,5
1,5
0,8
0,1
НАТРИЙ ХЛОРИСТЫЙ
(ХЛОРИД НАТРИЯ, ПОВАРЕННАЯ СОЛЬ)
Natrium chloratum
Приготовление
Sodium chloride,
Common salt
Natriumchlorid
Kochsalz
NaCl
Мол. в. 58,454
А. Обыкновенный хлористый натрий
1. Для очистки технического продукта (поваренной соли), содержащего
Ыаг304, MgSC>4 и CaS04, приготовляют концентрированный раствор соли,
фильтруют его, если нужно, и насыщают газообразным хлористым
водородом. Конец газоподводящей трубки, погруженный в раствор, рекомендуется
снабдить воронкой, чтобы выделяющаяся кристаллическая масса не закупорила
выходного отверстия. Так как NaCl очень плохо растворим в воде,
насыщенной НС1, то постепенно выпадает значительное количество мелких
кристаллов. Препарат отсасывают на платиновом конусе, промывают несколько раз
чистой концентрированной НС1 и, высушив, прокаливают соль в платиновой
чашке при 500—600° до постоянного веса. Хранить препарат рекомендуется
в плотно закрытых банках.
Приведенным методом можно очистить NaCl от S04"; однако некоторое
количество КС1 и MgCh может остаться в препарате.
2. Очень чистый препарат можно получить следующим способом. К
водному раствору NaCl при нагревании прибавляют по каплям ВаСЬ до
полного осаждения S04". Осадок отфильтровывают и добавляют Na2COs Д°
щелочной реакции; дают стоять, после чего отфильтровывают выпавшие
углекислые соли магния, бария и кальция, гидроокиси железа и алюминия и
др. Наконец, раствор подкисляют чистой НС1 и выпаривают до консистенции
жидкой кашицы. КС1 остается в маточном растворе.
Б. Сплавленный хлористый натрий
Высушенный NaCl нагревают в платиновой чашке до плавления (800°).
избегая излишнего перегрева массы. Чашку с расплавленной солью ставят
Натрий хлористые
401
полную каменную плитку, прикрыв сверху куском жести. По затверде-
#-мэЛЬ отскакивает от чашки.
• В. Коллоидный хлористый натрий
отовление
п Л. Каргачу (упрощено В. Оствальдом)1 довольно прочный
п<МаС1* получается следующим путем,
^тщательно высушивают в сушильном шкафу 15—20 г салициловокислого
тоия (CethOHCOONa) (от степени высушивания зависит
Гоочность геля!).
В сосуд с притертой пробкой помещают 20 г хлористого тионила (ЗОСЬ)
всыпают туда же высушенный салициловокислый натрий. Сосуд со слабо
вставленной пробкой помещают под тягу (выделение SO2), не взбалтывая.
Через сутки содержимое сосуда превращается в красивый зелено-оранжевый
студень, консистенции жидкого мыла. При хорошей укупорке студень дер-
дагся несколько недель, после чего мутнеет.
Свойства
Белые кубические кристаллики или мелко-крйсталлический порошок уд. в.
2,1675 (dl). Т. пл. 800°, т. кип. 1440°. Растворим в воде, не .растворим
Таблица 403
Растворимость NaCl в воде
ГС
-15
0
+ 5
9
14
о/о NaCl
24,66
26,21
26,27 !
26,33
26,41
t°c
25
40
50
60
70
o|0NaCl
26,54
26,81
27,00
27,14
27,47
t°c
so
90
100
109,7
o|0NaCl
27,65
27,99
28,38
28,75
Таблица 404
Удельный вес водных растворов
(G е г 1 а с h)2
NaCl
0/oNaCl
1
2
3
4
5
6
7
8
9
а
1,00725
1,01450
1,0^174
1.02S99
1,03)24
1,0ч 366
1,051(8
l,0fS851
1,06593
o/oNaCl
10
11
1 12
13
14
15
• 16
17
18
d
1,07335
1,08097
1,08859
1,09622
1,10384
1,11146
1,11938
1,12730
1,13523
°/0NaCl
19
20
21
22
23
24
25
26
26,395
d
1,14315
1,15107
1,15931
1,16755
1,17580
1,18404
1,19228
1,20098
1,20433
1 К')лл. химия, 12.
ЛЛЛ ш Новейшие данные см. Н. В.
Jf*113* Академии Наук ССС ", 9,82 (1936) Удел]
очного знака для концентраций 0,11—2,9о/0; Г. В а
Г е в е л и н г, «Известия сектора физико-химического
ьные веса с точностью до шестого деся-
tuecai. Z. phye. Ch. A, 182 (1938).
408
Натрий
в^спирте. При кристаллизации из водных растворов при температуре _10о
выпадают большие шестиугольные таблички гидрата NaCl • 2НЮ.
Испытание
По ОСТ НКТП 7396/550 содержание NaCl для всех квалификаций должно
быть не менее 99,8%; наибольшие количества допустимых примесей указаны
в табл. 405,
Таблица 405
Допувтимые примеси в NaCl
Примеси
Допустимое содержание в °/о
химическч
чистом
в чистом
для анализа
1. Нерастворимые в воде
вещества
2. Сульфаты (SO4'0
3. Азот (N) общий (из нитратов,
яитритов и аммиака)
4. Кальций (Са)
5. М»гний (Mg)
6. Мышьяк (As)
7. Металлы, осаждаемые H2S« . .
8. Железо (Fe) • . . .
9. Калий (К) .... •
10. Иод (У)
0,003
0,002
0,0005
0,005
0,002
0,10002
0,0105
0,0001
0,010
0,008
0,005
0.005
0,001
0,007
0,003
0,00005
0,0005
0,0003
0,020
0,008
0,020
0,010
0,001
0,010
0005
0,0001
0/01
0,0005
0,040
0,012
НАТРИЙ ХЛОРНОВАТИСТОКИСЛЫЙ (ГИПОХЛОРИТ НАТРИЯ)
Natrium hypochloroeum Sodium hypochlorite
NaClO
Мол. в. 74,454
Natrium hypochlorit
Unterchlorigsaures
Natrium
Приготовление
1. Для приготовления раствора средней концентрации Ванн и о1
приводит следующий метод. Смешивают в большом высоком сосуде 2 кг крепкого
раствора NaOH (1:1) с 2 кг толченого льда и при возможно сильном охла-
ЖДешад сосуда холодильной смесью пропускают струю газообразного хлора,
пока вес раствора не увеличится на 800 г. Температура должна быть все
время ниже—10°.
Приготовленный таким путем раствор можно довольно долго сохранять
в темном и холодном месте без потери им реакционной способности.
2. По Рашигу2 в склянку емкостью 10 л помещают 1800 мл 35%-ного
раствора NaOH и 6 кг льда и, уравновесив сосуд на чашке весов,
пропускают струю хлора до тех пор, пока вес сосуда не увеличится «а 710 г.
Затем разбавляют раствор, остававшийся все время холодным, до 10 л.
В полученном растворе приблизительно 10% примененного NaOH остаются
без изменения; в случае употребления технического NaOH раствор
отфильтровывают через стеклянную вату (не через бумагу!) от выделившихся хлопьев
Fe(OH)s, 'А1(ОН)з и Si02 * хНгО. Приготовленный таким путем раствор NaClO
сохраняется месяцами без изменения.
1 Ргёр. Ch. 32).
» Вег. 40, 4386 (1907).
Щ Натрий хлорноватистокислый и хлорноватокислый 409
вдовства
• NaClO — сильный окислитель. Раствор NaCIO — бесцветная жидкость со
' лочной реакцией, постепенно разлагающаяся на NaCl, NaClCh и О*; ско-
5^ь разложения зависит от концентрации раствора и примеси свободной
Р^очи. На свету, а также при нагревании, разложение протекает особенно
раствор NaCIO носит название лабарраковой воды.
НАТРИЙ ХЛОРНОВАТОКИСЛЫЙ (ХЛОРАТ НАТРИЯ)
Natrium chloricum Sodium chlorate Natriumchlorat
Chlorsaures Natrium
r NaClOs Мол. в. 106,454
Приготовление
1. По В а ни но1 100 г КСЮз и 60 г безводного №г304 растворяют при
йагревании в 600 jo воды. Тотчас же при охлаждении смешивают раствор
100 г КСЮз и 60
600 мл воды. Тотчас же
С;300 г спирта. После двухдневного стояния жидкость сливают с осадка и
Йбследннй промывают 30%-ным спиртом. Промывную жидкость соединяют
е главной порцией спиртового раствора, переливают в колбу и отгоняют
спирт. Остаток выпаривают и охлаждают до кристаллизации. Для очистки
срль подвергают вторичной перекристаллизации из воды.
■:.. 2. Другой метод, указываемый Ванино1, заключается в следующем.
К крепкому раствору 19,5 г винной кислоты (НгСчНЮв) добавляют раствор
18,33 г кристаллической соды (№гСОз • 10НЮ) и равное количество горячей
Воды. К полученному горячему раствору кислой соли, №НС4НЮв,
прибавляют горячий насыщенный раствор 16 г КСЮз в 50—60 мл воды и
оставляют омесь на сутки. При этом
осаждается тр^д нор а ет копимый
KHC^Oe, a NaClOs остается в
растворе. Последний выпаривают
Досуха, остаток растворяют в
возможно малом количестве
горячей воды, фильтруют, если
вужно, и охлаждают для
кристаллизации.
Свойства
Бесцветные кристаллы
правильной системы уд. в. 2,5. Т. пл.
«48°; при более высокой
температуре разлагается с выделением
* 90%-ном спирте.
Растворимость
Таблица 406
NaC103 в воде
/°с
0
15
20
40
o^NaCIO.
45,1
47,6
49,7
56,5
ГС
60
80
100
o/gNaCIOa
59,5
63,6
• 67,1
Ог. Хорошо растворим в воде, очень плохо
Таблица 407
Удельный вес водных растворов NaC103
.foNaClO,
1
2
4
6
8
10
1,0053
1,0121
1,0258
1,0397
1,0538
1,0681
o/0NaC103
12
14
16
18
22
rf4
1,0827
1,0977
1,1131
1,1288
1,1614
°loNaC10a
"26
30
34
38
40
V8
d4
1,1953
1.2307
1,2680
1,3085
1,3285
* Prlp. Ch.
330.
410
Натрий. Никель
НАТРИЙ ЩАВЕЛЕВОКИСЛЫЙ (ОКСАЛАТ НАТРИЯ)
Natrium oxalicum Sodium oxalate
Na2C204 Мол. в. 134,014
Natriumoxalat
Oxalsaures Natrium
Приготовление
К горячему концентрированному раствору щавелевой кислоты добавляют
Na2C03 до нейтральной реакции. Выпавший белый кристаллический осадок
№гСг04 отфильтровывают, растворяют в воде и, добавив несколько капель
NaOH, оставляют на ночь.
Отфильтровывают выпавшие примеси (главным образом СаСгО^, фильтрат
выпаривают до */ю первоначального объема и охлаждают. Осадок ЫагСгО*
отсасывают, промывают холодной водой и перекристаллизовывают до полной
чистоты. Высушивание препарата должно происходить при температуре не
выше 240°.
Свойства
Белый кристаллический порошок, мало растворимый в воде и не
растворимый в спирте. При прокаливании разлагается на №гСОз и СО.
Таблица 408 Таблица 409
Растворимость Удельный вес
Na2C204 в воде водных растворов
Na2Co04
/°с.
15
20
25
o;oN2,cec4
3,12
3,30
3,47
o/oNa.C^O*
1
2
3
4
4
1,0064
1,0147
1,0229
1,0312
Испытание
По ОСТ 2759 препарат х. ч. должен содержать не менее 99,8% №гСг04;
наибольшие количества допустимых примесей указаны в табл. 410; препарат
должен выдерживать указанное в ОСТ испытание на присутствие
органических веществ.
Таблица 410
Допустимые примеси в Na2C204
Примеси
1. Нерастворимые в воде вещества
2. Хлориды (СГ)
3 Сульфаты (SO/)
4. Железо (Fe)
5. Калий (К)
6. Гигроскопическая влажность . .
7. Кислотность (НоС204) ...-.•
8. Щелочность (СО/)
Допустимое содержание в °/о
в химически
чистом
0,005
0,002
0,004
0,0002
0,006
0,010
0,010
0,020
в чистом для
анализа
0,010
0,005
0,008
0,0005
0,010
0,020
0,025
0,050
в чистом
0,020
0,010
0,015
0,001
0,020
0,030
0,050
0,100
Натрий щавелевокислый. Никель азотнокислый
411
НИКЕЛЬ АЗОТНОКИСЛЫЙ (НИТРАТ НИКЕЛЯ)
jflccolum nitricum Nickel nitrate
Ni(N03)2 6Н2О Мол. в. 290,80
Nickelnitrat
Nickelonitrat
Приготовление
1. 46 г чистого углекислого никеля взбалтывают с 140 мл воды и в
полученную смесь прибавляют постепенно 84 мл HNOs (уд. в. 1,4). Общий объем
Жидкости («240 мл) выпариванием доводят до 140 мл; раствор охлаждают,
1 выпавшие кристаллы отсасывают и помещают в плотно закрывающиеся
банки. Выход 35—38 г.
Маточные растворы при выпаривании дают еще значительное количество
Кристаллов.
2. Разбавленную HNOs нейтрализуют закисью никеля (NiO), раствор
фильтруют, выпаривают и кристаллизуют.
3. Металлический никель обливают небольшими порциями HNOs (1 :3);
Когда останется 10—15% металла, нагревают до 50—60° в течение часа.
Раствор фильтруют, -выпаривают, пока проба жидкости, взятая стеклянной
палочной, не будет закристаллизовываться, снова фильтруют, подкисляют раствор
HNOs (10 нл на *1 л раствора) и оставляют на сутки. Выпавшие кристаллы
Отсасывают, промывают небольшим количеством холодной воды и помещают
Ьлажными в банку с притертой пробкой.
Свойства
Изумрудно-зеленые кристаллы уд. в. 2,05, слегка выветривающиеся -в сухом
Воздухе и быстро расплывающиеся во влажном. При 57° плавится в
кристаллизационной воде; при дальнейшем нагревании теряет 3 мол. воды и, позднее,
HNOs. При прокаливании распадается, оставляя №Оз. Растворим в воде и
спирте.
Таблица 411
Растворимость NI(NOs)2 вводе
*°с
0
20
40
°,'oNi(NOl)f
44,3
49,1
54,8
ГС
55
70
95
0(oNi(N03)3
61,1
63,9
77,2
Таблица 412
Удельный вес водныу растворов Ni(N03)2
—
°/oNi(NOs)e
1
2
4
6
8
<1
1,007
1,016
1,033
1,051
1,069
°/oNI(NO»4
10
12
14
16
18
-и
1,088
1,108
1,128
1,148
1,170
(o/0Ni(NO,)1
20
25
30
35 >■ j
w18
1,191
1,249
1,311
1,379
412
Никель
И спытание
Наибольшие количества примесей, допустимых по ОСТ НКТП 67б5/37о'
указаны в табл. 413. 2»
Допустимые примеси в Ni(N03)2 • 6Н?0
Таблица 413
Примеси
1. Нерастворимые в воде вещества
4. Ж' лезо (Fe)
6. Кобальт (Со)
7. Металлы, осажаемые H2S . . . .
8. Щелочные и щелочи земельные
металлы (в виде сульфатов) . .
Допустимее содержание в °/0
в химически
чистом
0.002
0,002
0,005
0,С002
0.0u5
0.01
0.002
ОДО
в чистом для
анализа
0.005
0.005
0:1
0.0005
0,01
0.02
0,005
1 0,20
в чистом
0,01
0,01
0.05
0.001
0,02
02
■л 0,01
0,50
Примечание. Препарат, содержащий меньше 0,005% Со, носит
название «без кобальта».
Niccolum sulfuricum
ammoniatum
НИКЕЛЬ-АММОНИЙ СЕРНОКИСЛЫЙ
Nickelammoniumsulfat
Nickel ammonium
sulfate
NiSO,. (NH4)2S04 • 6H.O
Мол. в. 394,99
Приготовление
1. По Зереноену1 сильно взбалтывают смесь 100—125 г (ЫН^гСОз,
60 г Na2HP04 и 10 г NH4C1 в 300 мл теплой (40—50°) воды. При этом
большая часть солей растворяется, после чего жидкость охлаждают до комнатной
температуры и насыщают ее СОг.
К приготовленному таким образом раствору прибавляют раствор 50 г
азотнокислого никеля и смесь постепенно нагревают до кипения. При этом
образовавшийся вначале осадок почти полностью растворяется; в противном
случае добавляют по каплям ГМНЮН до почти полной прозрачности раствора.
Кипятят 15—30 мин., причем выпадает кобальт в виде CoNH4P04, в то время
как Ni *" остается в растворе. Трудность заключается в том, что при
недостаточно долгом кипячении не полностью осаждается кобальт, если же
кипятить слишком долго, то частично выпадает и никель.
После кипячения раствору дают стоять час и фильтруют; к фильтрату
прибавляют около 100 мл 50%-ной Н2304 и выпаривают до объема 100—150 мл.
Уже в горячем растворе частично выпадает NiS04 • (NH4)-2S04 • 6НгО, а при
охлаждении выпадение почти полное (при неполном отделении Со' '
маточный раствор имеет розовый оттенок).
Маточный раствор сливают и кристаллы несколько раз промывают на
фильтре водой. Еще влажную кристаллическую массу перекристаллизовывают
из возможно малого количества кипящей воды. Выделение кристаллов начи-
« S б г е п s е о, Z. anorg. Ch. 5, 361 (1894).
Никель-аммоний сернокислый
413
tc5i после прибавления 100 мл насыщенного раствора (NH4)2S04, подкислен-
Щ^ Ю мл разбавленной H2SO4. По охлаждении фильтруют, кристаллы
многопромывают холодной водой и, наконец, спиртом, не содержащим кис
л^Выход 48—50 г (около 80% теорет.).
2 Если примесь Со' * не является вредной, то препарат можно получить
аяачительно более простым методом — сливая горячие насыщенные растворы
So г сернокислого никеля (NiS04«7H20) и 150 г (NH4)2S04, подкисленные
ffg04. Сразу начинает выпадать кристаллическая мука двойной соли. По
охлаждении препарат отсасывают и промывают холодной водой и спиртом.
Выход близок к теоретическому.
Сюйства
Сине-зеленые кристаллы, уд. в. 1,91, изоморфные с соответствующими сое»
синениями Со и Mg. Почти нерастворим в холодном насыщенном растворе
{Nft^SO*. При нагревании теряет воду без предварительного плавления.
Таблица 414
Растворимость NiS04 • (NH4)2S04 • 6H20 вводе
ГС
3,5
10
16
20
О/о СОЛИ
1,8
3,1
5,5
5,6
ГС
30
40
50
0/о СОЛИ
7,2
10,3
12,6
/°с
59
68
85
°jo СОЛИ
14,3
15,8
22,2
Испытание
По ОСТ 10163—39 препарат ч. д. а, должен содержать не менее 98,0%
Ni(NH4)2(S04>2 • 6Н2О, препарат чистый — не менее 96%; наибольшие
количества допустимых примесей указаны в табл. 415.
Таблица 415
Допустимые примеси в NiS04 • (NHJjSOi • 6H20
Примеси
Допустимое содержание в °/0
в чистом для
анализа
1. Нерастворимые в воде вещества
2. Хлориды (СК)
3. Железо (Fe) ■
4. Цинк (Zn) ,
5. Кобальт (Со) . . . . •
6. Металлы, осаждаемые H2S . #
7. Щелочные и щелочноземельные металлы
(в виде сульфатов)
0,005
0.002
0,0005
0,005
0,05
0,002
0,05
0,01
0,005
0,002
0,02
0,1
0.00$
0.1
П ри м е ч а и и е. Препарат,
название „без кобальта".
содержащий меньше 0,002% Со не сит
414
Никель
НИКЕЛЯ ОКИСЬ (ОКСИД НИКЕЛЯ [3])
Niccolum oxydatum
Nickel sesquioxide
N12O3 Мол. в. 165,38
Nickeloxyd
По методу, разработанному Д. Н. Ф и н ке л ьштейно м, к теплом
профильтрованному раствору 400 г NiS04 • 7НгО в 1,5 л воды приливают пъ!
помешивании раствор 130 г чистого NaOH в 800 мл воды и понемногу при
перемешивании добавляют раствор 450 г хлорной извести в 1800—1900 Мл
воды. (Раствор хлорной извести должен быть отфильтрован, оставлен на
сутки и снова отфильтрован через двойной фильтр перед прибавлением •
к NiS04.)
Смесь оставляют на несколько часов до прекращения выделения пузырь-
«ов О2, затем нагревают до исчезновения запаха хлора, дают отстояться и
сливают прозрачную жидкость с осадка. Осадок промывают сначала
декантацией, а затем на воронке Бюхнера до удаления СГ и SO/' в промывных
водах.
Высушив осадок при 100°, его растирают и снова промывают до уда.
ления СГ и SO/'. Окончательно препарат высушивают при 130°.1
Свойства
Блестящие черные комочки, легко растирающиеся в порошок. Растворим
в H2SO4 и Н1МОз с выделением Ог й в НС1 с выделением СЬ.
Испытание
По ОСТ 10152—39 препарат должен содержать не менее 60,0% Ni;
наибольшие количества допустимых примесей указаны в табл. 416.
Таблица 416
Допустимые примеси в №2Оз
Принеси
1. Нерастворимые в НС1
2. Об цая сера (в
пересчете на S04") ...
3. Железо (Fe) . . . .
4. Кобальт (Со) ....
Допустимое
содержание в °/0
0,2
0,05
| 0.003
0,2
Примеси
5. Щелочные и
щелочноземельные металл».
(в виде сульфатов) .
6. Металлы, осуждаемые
H2S
7. Хлориды (СК) ....
8. Нитраты (NO/) . . .
Допустимое
содержание в°|0
0,3
0,01
0,01
0,05
Nickelsulfat
Nickelosulfat
НИКЕЛЬ СЕРНОКИСЛЫЙ
Niccolum sulfuricum Nickel sulfate
NiS04*7H20 Мол. в. 280,86
Приготовление
1. 100 г технического сернокислого никеля растворяют при нагревании до
80° в 250 мл воды, прибавляют 150 мл холодной воды и, охладив до 40°,
1 Если препарат содержит повышенные количества щелочных и щелочноземельных
металлов, то его размешивают с 250 мл б^о-ной HNO, в течение 1—2 час, затем отсасывают и
промывают водой.
Никеля окись. Никель сернокислый
415
;<аябавляют 1,5 г перекиси бария (ВаОг), замешанной с водой в жидкую
;;Si0HUy- Кипятят полчаса, пополняя испаряющуюся воду (реакция смеси
•*лэкна быть слабо кислая). Испробовав на полноту осаждения железа
^фильтрованная проба жидкости с KCNS не должна давать красного
окрашивания), отфильтровывают осадок BaS04 и Fe(OH).3 \ пропускают в филь-
-трат H2S, фильтруют, слабо подкисляют H2SO4 и, подогрев до 60°, снова
пропускают H2S.
Убедившись в полноте осаждения Си*' и Zn"', фильтруют, выпаривают
фильтрат до уд. в. 1,43 и кристаллизуют.
2. Для получения NiS04 • 7H2O из продажного металлического никеля
можно поступать по методу Террейля.
Растворяют металл в 7—8-кратном количестве царской водки, выпаривают
досуха и выщелачивают водой, причем" мышьяковое железо остается нерас-
творенным. Осадок отфильтровывают, раствор настаивают с металлическим
железом; при этом находившаяся в растворе медь количественно выпадает
в виде металла. Растворенное железо окисляют хлором или НЫОз и к раствору
добавляют H2SO4. Выпаривают досуха для удаления НС1, остаток извлекают
.водой и раствор смешивают со свежеосажденным ВаСОз, причем Fe и As
"выпадают количественно. Фильтруют; фильтрат, содержащий теперь чистый
NiSO* выпаривают и кристаллизуют.
Свойства
' Безводный NiSOa, получаемый при легком прокаливании NiS04-7H20,
представляет собой светложелтую массу, зеленеющую на воздухе ^следствие
поглощения воды. Растворим в воде, нерастворим в спирте и эфире. f
Из водного раствора выкристаллизовываются в зависимости от условий
различные гидраты:
а) NiS04 • 7НгО — кристаллизуется между 15 и 20°. Темноизумрудные
ромбическо-гемиэдрические кристаллы уд. в. 1,98.
б) NiS04 • 6Н2О — кристаллизуется между 30 и 40° в кристаллах
квадратной системы.
в) NiS04 • 4НгО — кристаллизуется между 50 и 70° в виде
моноклинических кристаллов, при обыкновенной температуре вскоре становящихся
непрозрачными.
Таблица 417
Растворимость N'S04 вводе
/°с
0
15
30
31,5
o/0NiS04
21,4
25,5
29,8
30,2
t°c
32,3
44,7
50
53,3
OjoNiSO*
30,4
32,4
33,4
34,5
t°C
60
70
80
99
°/0NiSO4
35,4
37,3
38,7
43,4
Испытание
По OCT 10910—40 содержание Ni в препаратах ч. д. а. и чистом должно
быть от 20,3 до 22,4% (первая цифра относится к семиводному, а вторая —
к шестиводному кристаллогидрату); наибольшие количества допустимых
примесей указаны в табл. 418.
1 Значительно лучшие результаты дает метод отделения железа, проверенный Д. Н. Ф и н-
к е л ь и; т е й н о м.
ВбО^о-ный расгвор НО г NiS04 • 7Н^О, подкисленный H,S04, вносят 2 г РиОа, нагревают до
полного ок.-ел ния Fe" в Fe", пос ie чего нейтрал зуют раовор, ввода небольшими порциями
избыток N1C08. Кипятят до полного осаждения Fe(OH;3 и фильтруют.
416
Никель
Допустимые примесив NiS04• 7НаО
ТаблиЦа
41в
Примеси
Допустимое содержание в о
в чистом для
анализа
1. Нерастворимые в воде вещества
2. Хлориды (СК)
3. Железо (Fe)
4. Кобальт (Со)
5. Металлы, осаждаемые H2S
6. Цинк (Zn)
7. Щелочные металлы, магний и кальций (в виде
сульфатов)
0,005
0,002
0,0005
0,02
0,002
0,01
ОД
в чистом
0,02
0,01
0,005
0,2
0,01
0,05
0,5
Примечание. Препарат, содержащий менее 0,002%Со носит
название «без кобальта».
Nickel chlorii r
Nickefchlorid
НИКЕЛЬ ХЛОРИСТЫЙ (ХЛОРИД НИКЕЛЯ)
Niccolum chloratum Nickel chloride
NiCh Мол. в. 129,60
А* Безводный хлористый никель
Приготовление
1. По Д. И. Рябчикову и В. М. Шульман1 к концентрированному
раствору NiS04 приливают при энергичном перемешивании концентрированный
NHtOn, насыщенный NH4CI.
Раствор синеет и выделяются сине-фиолетовые кристаллы гексамминникель*
хлорида [Ni(NHs)e]Cla. Кристаллы отсасывают на воронке Бюхнера,
промывают NH4OH, насыщенным NH4CI, затем чистым NrhOH. Промытый осадок
просушивают при 70°, после чего нагревают, разложив возможно более
тонким слоемч при температуре не выше 200° (иначе частично образуется NiO).
2. Тонкий порошок чистого металлического никеля помещают в
фарфоровые лодочки, которые нагревают в трубчатой печи до 500—600° в токе сухого
хлора. Реакция сильно экз.отермична; после прокаливания остается
кристаллическая масса NiCh.
3. Можно также обезводить NiCh • 6Н2О нагреванием, а затем возогнать
полученный продукт в вакууме2.
Свойства
Желтый порошок или желто-коричневые блестящие листочки уд. в. 2,56,
при* нагревании в вакууме возгоняющиеся без изменения. Т. пл. 1001°.
Гигроскопичен, во влажном воздухе расплывается и зеленеет вследствие образова
ния кристаллогидратов.
Растворим в воде, спирте, гликоле.
1 ЖПХ 7.J* 7, 1162 С1934).
1 W. Р 1 • с h е г u, R. О е w e h г, Z, aoorg. Ch. 222, 308 (1935).
Никель хлористый
417
Б. Водный хлористый никель
NiCl2.6H20 Мол. в. 237,70
Приготовление
растворяют NiO, Ni(OH)2 или NiCOa в чистой НС1 до насыщения, раствор
упаривают, фильтруют и охлаждают. Кристаллизуя при комнатной
температуре, получают кристаллы гексагидрата NiCb • 6Н2О.
Свойства
Кристаллы травянисто-зеленого цвета, выветривающиеся в сухом и
расплывающиеся во влажном воздухе. Растворим в воде и спирте.
Таблица 419
Растворимость NiCl2 в воде
г
0
20
о/о NiCl»
35,3
37,5
39,0
t° с
30
40
50
°/о NiClt
40,5
41,9
43,2
t° С
60
78
96
о/о NiClj
44,8
46,5
46,8
Удельный вес водных растворов NiCl2
Таблица 420
ЦоШС!,
1
2
4
6
w18
4
1,008
1,018
1,038
1,058
°/о NiCl,
8
10
12
. 14
«1<8
1,079
1,100
1,122
1,144
о/о NiCI,
16
18
20
rf18
1,167
1,192
1,216
Испытание
Наибольшие количества примесей, допустимых по ОСТ НКТП 6761/368 для
кристаллогидрата ЫЮЬ'бНгО, указаны в табл. 421.
Таблица 421
Допустимые примеси в NiCl2• 6Н20
Примеси
1. Нерастворимые вещества ....
3. Железо (Fe) .
В. Металлы, осаждаемые H2S . . .
?. Щелочные и щелочноземельные
металлы (в виде сульфатов). . .
Допустимое содержание в о/0
в химически
чистом
0,005
0,005
0,0005
0.01
0,01
0,002
0,1
в чистой
для анализа
0,01
0,02
0,002
0,02
0,02
0,005
0,2
в чистом
0,02
0,05
0,01
0,05
0,2
0,01
0,5
Примечание. Препарат, содержащий менее 0,005 % Со, нос :ст
название „без кобальта*.
418
Озон
Ог
озон
Мол. в. 48,00
Приготовление
Из простых аппаратов для получения озона действием тихого электрИЧе.
ского разряда на кислород можно указать следующие1:
1 Аппарат Мюллера состоит из трех стеклянных цилиндров, впаянных
один в другой, как указано на рис. 56. Внутренний цилиндр А и наружный
С наполнены слабым соляным раствором, служащим обкладкою. Средний
цилиндр В служит реакционным пространством, где кислород под влиянием
тихого разряда превращается в озон. Газ поступает через трубку D и отво- *
дится через трубку '£. Подвод тока от индукционной катушки (или перемен-
иого тока) производится через проволоки F и G,
погруженные в соляной раствор. Отвод газа из
трубки Е может быть произведен через пришлнф0.
ванную к ней трубку, так как практика
показывает, что резиновые трубки сильно разрушаются
озоном и становятся хрупкими.
2. Аппарат Либермала изображен на рис. 57,
В одно колено U-образной трубки опущена и
закреплена толстостенная пробирка; через пробку
последней проходит платиновая проволока, оканчи-
Рис. 56. Аппарат Мюллера
(озонатор)
Рис. 57. Аппарат Либермана
(озонатор)
вающаяся кусочком платиновой жести. Другое колено U-образной трубки
плотно закрывают пробкой. Весь прибор вставляется в прорез в асбестовом
картоне (на рис. 57 не показан), служащем крышкой для толстостенного
стакана (батарейного). Стакан и пробирка наполняются серной кислотой уд. в.
■1,4—1,5.
В кислоту, налитую в стакан, погружают платиновую проволоку, также
заканчивающуюся кусочком платиновой жести. Обе проволоки соединяются
с индукционной катушкой. Для подвода и отвода воздуха оба конца
U-образной трубки снабжаются длинными просверленными пробками. Для осушки и
очистки поступающего воздуха и озонированного воздуха (последний при
наличии влаги содержит окислы азота) можно при помощи тех же
просверленных пробок присоединять любую необходимую аппаратуру.
Все пробки рекомендуется предварительно проварить в парафине во
избежание сильного разрушения их озоном.
3. Шихэн и Кэрмоди2 предложили простой лабораторный озонатор,
изображенный на рис. 58. Для устройства озонатора использован либихов-
1 Об установке лля регулярного получения озона см., например, Н. В г а с h, Chem. Zte.
86, 1325 (1912); Bull. Soc. chim. France (5), 3, 701 (1936).
■ W. Sbtchan i.W. Carmody, Ind. Erg. Chem. Anal. Б'. 9, 8 (1937).
Озон
419
- холодильннк длиной 600 мм из стекла «пайрекс» с припаянной боковой
^л^тунная проволока А, укрепленная в резиновых пробках а и С, прохо-
' через форштосс Холодильника, заполненный 5%-ной H2SO4. Проволока
^„«щрна с одной клеммой транс-
fSSopa О на 10-15 тыс. в (от
%LoBoft лампы). Другая клемма
|[!зНСформатора присоединена к
алюминиевой фольге £, служащей
наружной обкладкой кожуха
холодильника. Рекомендуется обкладку
заземлять с помощью провода F.
Кислород вводится в озонатор через
отросток С; озониоованный гая
отводится через второй отросток и
трубку Я.
В случае необходимости можно
монтировать серию озонаторов
такого типа, соединяя их
последовательно с помощью изолированных
Проводов.
Свойства
Слабо-голубой1 газ уд. в. 1,658
(по воздуху) с характерным запахом,
вызывающим головную боль и при
больших концентрациях
напоминающим хлор. Запах ощутим еще при
разбавлении 1 : 500 000. Т. пл. — 250°,
т. кип.—112,3°. В воде растворим
лучше кислорода (1 ч. воды
растворяет при 0° и 760 мм давления
0,49 ч. озона). Воздух, растворенный
в воде, в 20 раз богаче озоном, чем
атмосферный; растворимость озона
в воде зависит от парциального
давления озона В воздухе. Рис. 58.'Озонатор Шихэн и Кэрмоди
Таблица 422
Растворимость озона в воде при 20° (интерполировано
по Л. И. Каштанову и О. Н. Олещук2)
Оа в газ* (в об. °/о)
0,05
0,10
0,25
0,50
0,75
1,00
100 мл раствора содержат
Молей 0|
6-1Q-6
9-10-6
3,4-10-5
6,5- 10-б
9,4 • Ю-ь
1,26-10-*
г О,
0,000288
0,000432
0,00163
0,00322
0,00452
0,00605
1 По некоторым источникам —бесцветный.
• Ж О X 7, 839 11936).
420
Олово
Легко растворим в ледяной СНзСООН и ССЦ с темносиней окраской к
центрированный озон при быстром нагревании взрывает с большой сил-' -*
Жидкий озон —темнофиолетовая жидкость уд. в. 1,78 при —182°. Тверд?!?
озон черного цвета с фиолетовым отблеском. ^ УДЫа»
Характерная реакция на озон — почернение предварительно нагретого куск
серебряной жести.
ОЛОВО
Stannum Tin Zinn
Sn At. в. 118,70 %
Приготовление
1. По Б ухнер у1 чистое олово можно получить следующим путем: 400 г
хлористого олова (SnCla*2H20) растворяют в 6 л воды и полученный раствор
постепенно приливают к смеси 6 л воды, 4 л раствора NaOH (уд. в. 1,33) и
60 г цианистого калия {яд\).
В полученную жидкость погружают листы цинка; выделившееся через
некоторое время олово промывают водой и высушивают.
2. Для получения металла высокой чистоты применяется нижеследующий
электрохимический метод.
Сначала приготовляют сернокислое олово, для чего раствор 240 г х. ч.
кристаллической сернокислой меди (СиЗО^бЬЬО) настаивают с 120 г мелко
истолченного металлического олова. После полного обесцвечивания раствор
фильтруют и употребляют для электролиза. Очень хорошие результаты
получаются при следующем составе ванны: 50 г SnS04, 50 г Na2$04, 10 г мета-
крезола (СвНЮНСНз), 10 г столярного клея, 30—50 г ШЗОв, 1 л воды.
Плотность тока 1,5—2,5 а/см*. Напряжение тока при расстояний между
электродами 5 см должно составлять 0,(3—0,8 в. Наилучшая температура 18—20°. Во
время электролиза необходимо энергичное перемешивание электролита, лучше
всего при помощи механической мешалки. Электроды изготовляются из
оловянных пластин.
Получаемый металл содержит лишь тысячные доли процента 'As и Sb;
содержание Sn = 99,995%.
3. Гранулированное олово для очистки обрабатывают НС1, не доводя до
полного растворения металла; при этом Си, Pb, Sb и часть As остаются
с нерастворившимся металлом, часть As улетучивается в виде АбНз и в
раствор переходит Sn и следы Zn.
Раствор после фильтрования нагревают до кипения и осаждают ЫагССЬ;
выпавший осадок обрабатывают HNOa, причем Sn переходит в нерастворимую
метаолоеянеую кислоту, a Zn остается в растворе. Белый порошок SnOj
промывают и восстанавливают измельченным сахаром или KCN (яд\).
По Г. и В. Бильц2 плавят в фарфоровом тигле 20 г истертой 8пОг с 20 г
порошка цианистого калия до тех пор, пока смесь не уменьшится на %
первоначального объема, для чего требуется приблизительно получасовое
нагревание в сильном жару. После охлаждения олово (12—15 г) очищается
водой.
Можно также смешать 30 г Sn02 с 10 г сахарного угля и 6 г (ЫНОгСОз»
поместить смесь в тигель и прокалить до начинающегося белого каления.
4. Крупные правильные кристаллы олова можно получить, наливая на
крепкий подкисленный раствор SnCk слой воды так, чтобы жидкости не
смешивались. При погружении в жидкость оловянной пластинки последняя »
нижней части будет растворяться, в то время как на границе слоев буду"1'
нарастать кристаллы.
1 В и с h n e г, Chem. «tg. 1894 (1905).
* Ujbungsbelspiele.
Олово. Олова двуокись
421
Порошок олова можно получить, если расплавить его в железной
jce, й> дав остыть Д° 200°, быстро измельчить пестиком.
Губчатое олово по Ванино1 можно получить следующим методом.
Л„лтворяют 200 г хлористого олова (SnCl2«2H20) в 3 л воды, выливают рас-
1 | в 2 л NaOH (уд. в. 1,33), разбавляют 3 л воды и, прибавив 30 г KCN
*?/) погружают в жидкость полоски листового цинка. Выпадающее губча-
е олово тщательно промывают водой, причем препарат получается в виде
Сероватого, очень устойчивого порошка.
Свойства
Блестящий серебристо-белый кристаллический металл уд. в. 7,287 (при 15°)
очень мягкий и тягучий. При сгибании трещит; выше 195° становится
хрупким вследствие перехода в другую модификацию (ромбическое олово).
Т. пл. 231,54°, т. кип. 2275°. Ниже +18° устойчива другая модификация —
серое олово уд. в. 5,7: поэтому, находясь долгое время на холоду, олово
превращается в рыхлый серый порошок («оловянная чума»).
При обычной температуре олово почти не изменяется от действия воздуха
и очень разбавленных кислот. Очень концентрированная HNOs не действует
на олово, кислота уд. в. 1,4 окисляет его в метаоловянную кислоту.
Разбавленная НЫОз растворяет олово с образованием Sn(NOa)2. Крепкая thSOi
переводит Sn в SnS04, сама частично раскисляясь до SO2. Зп легко растозо*
римо в концентрированней НС1 при кипячении с образованием SnCb;
присутствие платины способствует растворению.
Олово растворяется также в щелочах (при кипячении), образуя соли
оловянной кислоты '(станнаты), МегБпОз.
Испытание (Мерк)
Свинец, медь, железо и цинк. 5 г металла обрабатывают 40 мл HNOs при
нагревании на водяной бане до полного превращения металла в белый
порошок метаоловянной кислоты. Выпаривают досуха, остаток смешивают
с 10 мл НЫОз (уд. в. 1,153) и 50 мл воды и фильтруют. К фильтрату
добавляют 1 мл разбавленной H2SO4 и выпаривают возможно сильнее на
водяной бане. После фильтрования на фильтре не должно оставаться весомого
остатка (PbS04). К фильтрату прибавляют ЫНЮН.до щелочной реакции,
причем жидкость совершенно не должна принимать, голубой окраски (Си).
Прибавляют (NH^aS и дают смеси стоять 4—5 час. при 50°; могущий
выпасть осадок (Fe233, ZnS) после прокаливания не должен весить больше 0,002 г.
Сурьма. 5 г олова нагревают на водяной бане с 75 мл НС1 (уд. в. 1,12),
Добавляя понемногу КСЮз до полного растворения металла. В нагретый до
кипения раствор вносят куски чистой фортепианной проволоки (полностью
растворимой в НС1); допускается образование лишь ничтожных количеств
черного осадка.
Мышьяк. Содержание мышьяка, определенное по 3 а н гер-Б л эку3
после перевода Sn в метаоловянную кислоту 'не должно превышать 0,0001%.
ОЛОВА ДВУОКИСЬ
(ОКИСЬ ОЛОВА [4], ОЛОВЯННЫЙ АНГИДРИД)
Stannum oxydatum Stannic oxide Ziiinsaureanhydrid
Stannioxyd
SnOz Мол. в. 150,70
Приготовление
Прокаливают метаоловянную кислоту (ЗпОг . Н20) при 500—600°*
1 РгЗр. Ch. 587
1 РгЗр. Ch. 5S7.
a Sanger a. В I e а с k, Proc. Am. Acad. Afls a. Sc. ff, 1907; Ф. Тредвелл, Курс
аналитической химии, ГОНТИ, М.-Л. 1931, т. II, кн. 1, стр. 157, 159. уу чи
422
Олово
Свойства
Белый порошок уд. в. 6,95, не растворимый в воде, кислотах и щелочах' '
при сплавлении со щелочами образует соли оловянной кислоты (станнаты), :%
Испытание1 ,
Для чистого препарата допустимы следующие наибольшие количества при.
месей: 0,100% щелочи (NaOH); 0,015% сульфатов (ЗОз); 0,005% хлоридов (Cl);
0,020% тяжелых металлов.
ОЛОВА ЗАКИСЬ (ОКИСЬ ОЛОВА [2]) |
Stannum oxydulatum Stannous oxide Zinnoxydul
Stannooxyd
SnO Мол. в. 134,70
Приготовление
1. По Микстеру2 прибавляют к раствору ЗпСЬ избыток ЫНЮН и
нагревают смесь в течение нескольких дней на паровой бане. Образовавшуюся
темную кристаллическую массу промывают, сушат при 100° и нагревают при
400° в струе СОг до тех пор, пока совершенно не прекратится выделение Шз
и воды.
Полученный таким путем почти черный кристаллический порошок
содержит некоторое количество ЗпОг и следы аммиака и воды.
2. По Д и т т е к раствору SnCh прибавляют ЫНЮН и выпавший белый
осадок очищают многократной декантацией. Наконец, нагревают водную
суспензию до кипения и прибавляют кристаллик ЗпС1г • 2НзО. Осадок тотчас
окрашивается в розово-красный цвет и в течение нескольких минут
кипячения переходит в зеленые или фиолетовые кристаллы SnO. Прибавление ЗпСЬ
имеет целью образование хлорокиси Sn(OH)Cl, которая легко распадается
по уравнению:
Sn(OH)Cl = SnO + НС1.
Образовавшаяся свободная соляная кислота с Sn(OH)2 снова дает хлор-
окись и т. д.
3. По Либиху к раствору SnCh приливают концентрированный раствор
щавелевой кислоты. Выпавший белый кристаллический осадок оксалата
ЗпСг04 • ЗНгО отфильтровывают, промывают и высушивают при 160°.
При сильном прокаливании полученной соли в закрытом тигле (или лучше
в тугоплавкой трубке в токе СОг) образуется SnO, окрашенная в
светлосерый цвет благодаря примеси угля.
Свойства
В зависимости от метода изготовления SnO получается в различных
видах: по Микстеру образуется почти черный порошок, по Дитте темно-
зеленый или фиолетовый. При пользовании другими методами получаются
также синевато-черные или красные кристаллы. Уд. в. 6,3.
SnO не растворима в воде и щелочах; растворима в кислотах с
образованием солей Зп *". При нагревании на воздухе сгорает с выделением пламени,
образуя SnO*.
1 Испытание хим. реактивов, 320.
• М i х t е г, Am. J. Soc. (Sill). [4], 27, 229; С. 1, 1792 (1909),
Олова закись. Олово серное и сернокислое 423
ОЛОВО СЕРНОЕ
(СУЛЬФИД ОЛОВА[4], «СУСАЛЬНОЕ
' ctannum bisulfuratum Tin bisulfide
Mosaic gold
SnS2 Мол. в. 182,82
Приготовление
1. По Л а гуту для приготовления красноватого «муссивного золота»
можно поступать следующим образом. Тщательно смешивают 50 г оловянной
амальгамы1 (1:1), 25 г хлористого олова (ЗпСЬ • 2НгО), 35 г серного цвета
й 35 г NH4C1. Смесь помещают в реторту, обернутую тройным слоем
асбестовой бумаги, и сильно нагревают на большой горелке с 'дутьем, поворачивая
реторту для равномерного прогревания всей массы.
Нагревание ведут в течение 3—зу2 час; сначала улетучивается NH4CI,
затем HgS, в то время как SnS2, в основном, остается. (Вследствие плохой
теплопроводности S11S2 операцию можно производить лишь с количествами, не
выше указанных.)
2. Светложелтое SnS2 получается при аналогичной обработке тщательно
приготовленной смеси 50 г ЗпСЬ • 2ШО и 25 ч. серного цвета.
ЗоЗолотистое SnS* получается следующим образом2. 12 ч. чистого олова,
свободного от РЬ, переводят в амальгаму при помощи 6 ч. ртути. Амальгаму
смешивают с 7 ч. серного цвета и 6 ч. NH4CI, после чего подвергают массу
многочасовому нагреванию (в стеклянной реторте или в глиняном тигле),
сначала не доводя до каления, а позднее, когда прекратится выделение паров,
повышая температуру до тёмнокрасного каления. Вначале улетучивается
NH4C1, затем возгоняется HgS и некоторое количество ЗпСЬ, a SnS2 остается
в реторте. Верхний слой полученного таким образом препарата состоит из
золотистых, нежных, прозрачных листочков.
4. Для приготовления аморфного SnS2 пропускают H2S через слабо
подкисленный раствор хлорного олова (SnCU). Осадок промывают водой, затем
'спиртом и эфиром и быстро высушивают в вакууме. При длительном сушении
на воздухе препарат выделяет tbS и при этом темнеет.
Свойства
Кристаллическое SnS2 — блестящие прозрачные желтые чешуйки или
6-угольные листочки уд. в. 4,51, мягкие и жирные на-ощупь, как графит. При
нагревании становится тёмнокрасным, иногда почти черным; однако при
охлаждении желтый цвет возвращается. При очень сильном нагревании SnS2
распадается на SnS и серу.
Аморфное SnS2 — желтая масса, нерастворимая в разбавленных кислотах;
растворимо в NH40H (с образованием красной жидкости) и в растворе
№гСОз. Особенно легко растворяется в NaOH и многосернистом аммонии
[(NH4)23n]. В воде почти нерастворимо (при 18Q растворяется 1,36 • 10_в%).
ОЛОВО СЕРНОКИСЛОЕ ЗАКИСНОЕ (СУЛЬФАТ ОЛОВА [2])
Stannum sulfuricum Stannous sulfate Stannosulfat
oxydulatum Zinnoxydulsulfat
S11SO4 Мол. в. 214,76
Приготовление
1. По Букэ растворяют при нагревании свежеосажденный гидрат закиси
олова, Зп(ОН)г, в разбавленной H2SO4 до насыщения. При охлаждении
профильтрованного раствора выпадают кристаллы SnS04.
1 Амальгаму олова получают, добавляя при перемешивании вычисленное количество
ртути к расплавленному олову при температуре около 250е. Ввиду ядовитости паров ртути
необходимо получение амгльгамы проводить под тягой.
I Prap. Ch. 600,
ЗОЛОТО»)
Stannisulfid
Zinnsulfid
Musivgold
424 Олово
2. Хороший метод получения SnS04 указан выше (см. Олово, метод о
стр. 420). 4 #
Свойства
Мелкие бесцветные иглы или1 листочки с перламутровым блеском. pa(S
творимо в воде; водный раствор при стоянии выделяет осадок основной сопи"
При 19° в воде растворяется 15,83% SnS04, а при 100° 15,33%.
ОЛОВО ХЛОРИСТОЕ '
(ХЛОРИД ОЛОВА[2], ОЛОВЯННАЯ СОЛЬ)
Stannum chloratum Stannous chloride Zinnchlorur
Stannochlorid
А. Безводное хлористое олово
SnCb Мол. в. 189,61
Приготовление
1. По Эрдману1 нагревают гидрат (ЗпС1г• 2Н20), который сначала
плавится в кристаллизационной воде, затем, теряя ее, твердеет и, наконец,
снова плавится. Тогда слегка охлаждают, быстро измельчают и, поместив
з. тугоплавкую реторту, обмазанную глиной, возможно быстрее перегоняют
почти до конца на сильном пламени (перегонка идет при 620°).
2. Нагревают гидрат (SnCk • 2ШО) в токе сухого хлористого водорода при
120° до полного обезвоживания.
Свойства
Хлористое олово — бесцветная просвечивающая кристаллическая масса
уд. в. 3,95, плавящаяся при 241° и кипящая при 603,25°. Растворимо в воде,
спирте, эфире, ацетоне, пиридине и^-уксусно-этиловом эфире. В твердом виде
довольно устойчиво на воздухе.
Б. Гидрат хлористого олова
SnCb-2H20 Мол. в. 225,65
Приготовление
Растворяют металлическое олово в чистой HCI (уд. в. 1,14) при
нагревании, выпаривают раствор с добавкой еще некоторого количества Sn и дают
закристаллизоваться, по возможности добавив «затравку» — кристаллик SnCb •
•2НгО. Растворение Sn в платиновой чашке идет значительно быстрее, чем
в фарфоровой (Pt образует с Sn гальваническую пару); поэтому за
неимением достаточно большой платиновой чашки растворение ведут в фарфоре,
но в присутствии куска платиновой жести.
Свойства
Бесцветные иглы2 или таблички уд. в. 2,7, металлического вкуса,
растворимые в воде и спирте. Растворимость в воде чрезвычайно велика:
например, 1 л насыщенного при 15° раствора содержит 1333 г SnCb и 494 г воды,
имея уд. в. 1,827.
При 40° плавится в кристаллизационной воде, теряя воду при более
сильном нагревании.
1 Хим. прег., 34.
» Игольчатые кристаллы SnCJ»-2HaO достигают иногда в длину 150 мм при толщине
20 -30 мм,
Олово хлористое
425
Таблица 423
o/oSriCl*
1
2
4
6
8
10
12
14
Удельный вес в
и15
1,0068
1,0146
1,0306
1,0470
1,0638
1,0810
1,0986
1,1167
°,'о SnC'a
16
18
20
22
24
26
28
30
одных раст во р ов SnCl2
л15
dA
1,1353
1,1545
1,1743
1,1948
1,2159
1,2377
1,2603
1,2837
0,'oSnC!,
35
40
45
50
55
60
65
'Г
1,3461
1,4145
1,4897
1,5729
1,6656
1,7695
1,8865
В. Раствор SnCb (реактив Беттендорфа)
Приготовление
1. Для приготовления раствора SnCb, применяемого в качестве реактива
fca As, можно смешать 50 г SnCb-2H20 с 100 мл НС1 (уд. в. 1,124) в тесто
И насыщать последнее сухим хлористым водородом.
' 2. Для этой же цели д е-Д ж о н г рекомендует следующий метод. 25 г
фзпСЬ'ЗШО помещают в склянку, куда налито 150 мл эфира. После того
|ак большая часть соли растворится, прибавляют такое количество НС1
!$д. в. 1,124), чтобы эфирный раствор стал совершенно прозрачным, и
заем сливают прозрачную жидкость с осадка.
,.' Для пробы на As, испытуемую жидкость разбавляют 5 мл НС1 и,
прибавив 5 мл реактива, приготовленного вышеуказанным путем, хорошо
перемеривают и оставляют на несколько минут при 40°. Предел чувствительности
реакции для As111 — 0,00f5 мг, для Asv —0,03 мг.
Испытание
г По ГОСТ 36—40
опОг • 2НгО, чистый -
примесей указаны в
*а As.
препарат ч. д. а. должен содержать не менее 97%
-не менее 95%; наибольшие количества допустимых
табл. 424; препарат должен выдерживать испытания
Допустимые примеси в SnCl2• 2Н20
Таблица 424
Примеси
1. Нерастворимые в солян й кисло-
#3. Вещества, осаждаемые H2S в ще-
4. Щелочные и щелочноземельные
металлы в виде сульфатов ....
Я Железо (Fe)
Допустимое содержание в °/0
в чистом для
анализа
0,005
0,003
0,02
0,02
0,003
в чистом
0,01
0,01
0,1
од
0,01
426
Олово
ОЛОВО ХЛОРНОЕ (ХЛОРИД ОЛОВА[4])
Stannum tetrachloratum Stannic chloride Zinnchlorid ' *
Tin tetrachloride Stannichlorid
А. Кристаллическое хлорное олово
SnCl4*5H20 Мол. в. 350,61
Приготовление
Насыщают крепкий водный раствор SnCl2 хлором, упаривают при умерен.
ном нагревании (пока проба при охлаждении не будет давать кристаллы)!
и выставляют раствор на холод. Если раствор сконцентрирован до
консистенции сиропа, то кристаллизация сильно замедляется; в этом случае при-
бавляют затравку —. кристаллик SnCh • 5НгО.
Свойства
Белые полупрозрачные или мутные кристаллы, плавящиеся около 60° н
расплывающиеся на воздухе. В* вакууме теряет 3 молекулы
кристаллизационной воды. Очень легко растворимо в воде (см. Б).
Б. Безводное хлорное олово
SnCh Мол. в. 260,53
Приготовление
1. По Эрдману1 150 г чистого гранулированного олова нагревают
в реторте с тубусом, обмазанной глиной, на газовой печи до плавления и
через трубку, погруженную в расплавленный металл, пропускают сильный ток
Рис. 59. Прибор для получения хлорного олова
сухого хлора (некоторые авторы рекомендуют не погружать трубку, а лишь
подвести ее к самой поверхности металла).
Реторту соединяют с возможно более длинным холодильником, который
соединен с двумя склянками Вульфа (рис. 59). Последние погружены в
ледяную воду для лучшего охлаждения крайне летучего SnCU. Полученный
продукт перегоняют еще раз из колбы Вюрца, снабженной термометром
причем для удаления избытка хлора, растворенного в препарате, в колбу
добавляют немного листового олова (станиоля).
* Хим. преп. 34.
Олово хлорное
427
л По Лоренцу* большую пробирку (3—4 см диаметром, 20—25 см
яы) заполняют на % объема гранулированным оловом. Пробирку закры-
*Д*лт пробкой с двумя отверстиями: через одно проходит газоподводящая
ь «бка, через другое — форштосс обратного холодильника (рис. 60).
^ Налив в пробирку несколько мл SnCU, пропускают в нее сухой хлор
такой скоростью, чтобы газ успевал прореагировать с оловом. Происходит
^ояая реакция, иногда сопровождаемая пламенем. Когда слой SnCU на дне
™ ятт пробирки значительно увеличится, трубку, подводящую хлор,
несколько поднимают, оставляя ее погруженной в жидкость.
По окончании реакции SnCU сливают, выдерживают 1 час
в закрытой склянке, добавив несколько гранулей Зп для
связывания свободного хлора, после чего перегоняют2,
собирая фракцию, кипящую при 112—114°.
Если исходное олово содержало Fe, то рекомендуется
перегонку SnCU не доводить до конца во
избежание перехода примеси FeCls.
Если требуется приготовить большое
количество SnCU, то можно рекомендовать
применение прибора, изображенного на
рис. 61, позволяющего получать до 1 кг
SnCU в день. Две пробирки Л и Б с
тубусами F (длиной 200—250 мм,
диаметром 40 мм) соединены
трубкой С. К пробирке В
присоединяют обратный
холодильник D. Пробирки наполняют
гранулированным оловом и
пропускают через трубку Е
ток сухого хлора, сначала
медленно во избежание
чрезмерного разогрева, затем
(когда трубка Е окажется
погруженной в SnCU) быстрее.
Когда пробирка А
окажется почти заполненной SnCU
(в пробирке В к этому
времени собирается SnCU
приблизительно до половины вы-
соты)> отключают газоподво-
дящую трубку от источника
хлора, подставляют под
трубку Е сухую склянку и через верхнее отверстие холодильника продувают
аппарат воздухом с помощью резиновой груши.
При этом SnCU почти полностью переливается в подставленную склянку.
Загрузив через тубусы F новую порцию олова, продолжают хлорирование.
Очистку и перегонку препарата проводят, как описано выше.
3. По методу ИРЕА пропускают газообразный хлор в сосуд,
содержащий безводное SnCU в порошке. Когда почти вся соль превратится
в жидкое SnCU, последнее фильтруют и перегоняют, собирая фракцию
112—114°.
4. Из заводских отходов руд, содержащих олово, или лабораторных
остатков (метаоловянная кислота) можно получить SnCU по методу ИРЕА
следующим образом. Смешивают перерабатываемый материал с углем,
помещает смесь в трубчатую печь и хлорируют при 500—600°, собирая SnCU
в охлажденный приемник.
^
V4^. Т
тя
т
Рис. 60. Прибор
для получения
хлорного олова по
Лоренцу
Рис. 61. Прибор для получения
хлорного олова
в больших количествах
1 L о г е n z, Z. anorg. Ch. 10, 44 (1895).
1 Пробки перегонного аппарат! должны быть проварены в ларафине,
428
Олово. Платина
0°. Т.
Свойства
Бесцветная легкоподвижная жидкость уд. в. 2,278 при и~. i. Пл __«
т. кип. +114°. На воздухе сильно дымит. При действии влажного возл »■
или вычисленного количества воды образует кристаллы гидрата SnCu.3fK?
При смешении 22 ч. SnCU с 7 ч. воды образуется кристаллический Г11д *)-
FnCl4«5H20. В большем количестве воды растворяется. Со спиртом Реа*т
с образованием эфира и выделением хлорокиси. С эфиром дает J?"
,,_,„_ SnCh • 2(С2Н5)20.
чарует
раты,
например,
Таблица п
Удельный вес водных растворов SnCl4npH 15°
1,012
1,024
1,036
1,048
1,059
1,072
1,084
1,097
1,110
1,1236
1,137
1,151
1,165
1,180
1,195
1,210
stk
SnCI4.5HtO.
1,49
2,98
4,46
5,95
7,43
8,91
10,40
11,88
13,87
14,86
16,35
17,83
19,32
20,80
22,28
23,78
2
4
6
8
10
12
14
16
18
20
22
24
26
28
30
32
1,2268
1,242
1,259
1,2755
1,293
1,310
1,329
1,347
1,366
1,386
1,406
1,426
1,447
1,468
1,491
1,514
SnC14
SnCl,-5H40
SnCI4
SnCV5HtQ
25,24
26,76
28,22
29,74
31,20
32,72
34,18
35,68
37,16
38,65
40,14
41,62
43,11
44,60
46,09
47,58
34
35
38
40
42
44
46
48
50
52
54
56
58
60
62
64
1,588
1,563
1,587
1,614
1,641
1,669
1,698
1,727
1,759
1,791
1,824
1,859
18ЭЗ
1,932
1,969
1,988
49,06
50,55
52,03
53,52
55,00
56,49
57,98
59,47
60,95
62,44
63,92
65,10
66,89
68,37
69,86
71,35
66
68
70
72
74
76
78
80
82
84
86
88
90
92
94
96
Испытание1 (для SnCU• 5НгО ч. д. а.).
Препарат может содержать следующие наибольшие количества примесей:
1. нерастворимые в НС1 вещества . . . 0,005%
2. железо и цинк (Fe203 + ZnO) 0,01%
3. щелочи 0,01%
Кроме того, водный раствор препарата испытывают:
а) на сульфаты (SO/') — не должно быть мути от BaCIz в течение 4 час;
б) на SnCh — добавление HgCh не должно вызывать помутнения;
в) на тяжелые металлы — при добавлении избытка ЫШОН и (NHO-'S
раствор должен оставаться бесцветным и прозрачным;
г) «а нитраты (ЛЮз')— кристаллик FeSC>4, помещенный в испытуемый прс
парат, не должен вызывать краснобурого окрашивания.
Platinum
Pt
ПЛАТИНА
Platinum
At. в. 195,23
Platin
А. Чистая платина
Приготовление
1. По методу Девилля и Стаса отделение платины от других
металлов может быть произведено следующим путем2.
1 Испытание хим. реактивов, 322
» М у 1 i u s u. F о е г s t е г, Вег. 25, 665 (1892); Ргар. Ch. 764.
Платина
4*29
Около 5 2 металла сплавляют с 10-кратным количеством свинца в уголь-
-j тигле, снабженном крышкой и помещенном в большой глиняный тигель,
гюлненный древесным углем. Сплавление производится в течение 4—5 час.
' **» 1000°. Охлажденный сплав обрабатывают теплой, сильно разбавленной
ijOa Д° полного прекращения газовыделения. Получившийся раствор
SSep**11 около 98,4% взятого свинца, а также небольшие количества пла~-
ЙяЫ, родия и железа, весь палладий и всю медь.
■Оставшийся нерастворимым черный металлический порошок
отфильтровывают, промывают и при нагревании обрабатывают разбавленной царской
водкой (2 об. HNOa, 8 об. НС1 и 90 об. воды). После 5—7-часовой
обработки царская водка растворяет оставшийся свинец, платину и родий,
оставляя серые листочки сплава, содержащего Ir, Ru и, иногда, Fe; этот
остаток собирают на фильтр. Раствор металлов в царской водке освобождают
яосле выпаривания от РЬ при помощи Ня£04, снова выпаривают, добавляют
хлорной воды и выливают полученную жидкость (приблизительно 100 мл)
на холоду в насыщенный раствор NH4CI. Смесь нагревают до 100° и
охлаждают.
л Выпадающий при этом хлороплатинат аммония (NH^PtCle (содержащий
родий) промывают декантацией и при помощи насыщенного раствора NFhCl
Омывают на фильтр.
Фильтрат вместе с промывными водами выпаривают, пока при охлаждении
раствора снова не выпадет (NH^PtClo, который, после промывания,
присоединяют к главнбй порции осадка.
Фильтр с хлороплатинатом высушивают, осадок переносят в фарфоровую
чашку и там, при возможно низкой температуре, осторожно восстанавливают
в струе светильного газа. Получившийся губчатый металл в той же чашке
шеишвают с КН304 и нагревают постепенно до тёмнокрасного каления.
Обработку бисульфатом повторяют до тех пор, пока соль,
соприкасающаяся с металлом в течение 10 мин., не перестанет окрашиваться. Массу
после охлаждения обрабатывают водой (причем растворяются все примеси»
перешедшие в растворимое состояние при сплавлений с KHS04) и оставшуюся,
^.платину тщательно промывают водой. Для удаления еще присутствующих
небольших количеств PbS04 металл последовательно обрабатывают
растворами (ЫН4)гСОз и HNOs, промывают разбавленной фтористоводородной
кислотой, затем тщательно промывают водой и прокаливают. Выход
количественный.
2. По Коллари и Баталья1 платину кипятят сначала с НС1, затем
С HNOa (для удаления поверхностных загрязнений) и растворяют в царской
водке. Выпарив раствор несколько раз досуха с НС1, остаток растворяют
в 8-кратном количестве воды, приливают х/3 объема спирта и добавлением
NH4C1 осаждают (NbhJaPtCle. Промытый осадок прокаливают в токе Нг;
металлическую платину снова растворяют в царской водке и повторяют
Описанные выше операции. Полученный металл уже очень чист.
Для окончательной очистки растворяют Pt еще раз в царской водке,
выпаривают раствор с НС1 досуха, растворяют остаток в 0,4%-ной H2SO4 и,
нагрев до 60°, подвергают раствор электролизу при плотности тока
0,05 а/дм2 (катодом служит платиновая пластинка).
Растворив еще раз металл в царской водке и переходя через (NH4)2PtCle
к Pt, получают платину исключительной чистоты. Сплавление готового
металла рекомендуется вести в лодочках из СаО или Zr02.
Свойства
Серовато-белый блестящий, очень тягучий металл, по мягкости напомй*
нающий м£дь. Т. пл.2 1773,3 ±1°, т. кип. 3800°. Уд. в. 21,4—21,48. Очень
Устойчив по отношению к большинству химических реагентов; растворим
Только в царской водке.
.* N;. С о 11 а г I е. А. В a 11 a g I i a, Ann. Cfalm. appl. 28, 310 (1938); C. 1, 363 (1939)*
■F.H. Scno'rUl d.C. 1,1187(1935). v '
4зй
Платина
Б. Платиновая чернь
Приготовление ^
1. По Лёву раствор 50 г хлорной платины (PtCU) в 50—60 Мл в
смешивают с 70 мл 40—45%-ного формалина (НСОН) и в полученную Смес
вносят при хорошем охлаждении раствор 50 г NaOH в 80 Мл воды, rjp*
этом тотчас .выпадает бблыпая часть металла. После 12-часового стояни
осадок отсасывают на воронке Бюхнера (полученный желтоватый фильтре
кипятят, причем выпадает еще некоторое количество Pt) и промывают. Кс-гда
бблыпая часть солей отмоется, прерывают отсасывание, так как жидкость
начинает проходить черной вследствие частичного растворения чрезвычайно
тонко раздробленных частиц Pt и образования коллоидального раствора. г
Вскоре происходящий в осадке окислительный процесс заканчивается, после
чего фильтрат снова начинает прдходить бесцветным. Рыхлую пористую
массу теперь снова промывают для удаления следов NaCl, затем отжимают
и высушивают в эксикаторе над H2SO4.
2. По Гутбиру и Маишу1 нейтрализованный посредством №*СОч при
кипячении раствор платинохлористоводородной кислоты (HsPtCle)^ вливают
в кипящий раствор муравьинокислого натрия (HCOONa). Выпавший черный
осадок промывают декантацией горячей водой, отсасывают и возможно
тщательнее отжимают между листами фильтровальной бумаги. Наконец,
порошок высушивают в вакуум-эксикаторе над H2SO4 или Р2О5; однако
совершенно безводный препарат получить не удается.
Свойства
Черный порошок, адсорбирующий при 0° громадные количества водорода;
в этом отношении он стоит выше губчатой платины.
В. Губчатая платина
Приготовление
1. По Гутбиру и Маишу нагревают платиновую чернь в струе
водорода до 400° и охлаждают в токе СОг, свободного от кислорода.
2. Губчатая платина получается также при слабом прокаливании хлоро-
платината аммония (NH^PtCle.
Свойства
Серая губчатая масса с сильно развитой поверхностью; применяется
в качестве катализатора.
Г. Коллоидальная платина
Приготовление
1. По Генриху к 50 мл OfilN раствора платинохлористоводородной
кислоты, H2PtClo, доведенного прибавлением Na2COs до слабощелочной
реакции, приливают при нагревании 7,5 мл Q,\N раствора пирокатехина, причем
появляется темная желто-коричневая окраска. После короткого Стояния
раствор подвергают диализу.
2. Лоттермозер предлагает следующий способ. К 75 мл 0,06%-ного
раствора H2PtCle прибавляют 4 мл 0.2N раствора NaOH и нагревают до
кипения. После удаления пламени прибавляют 5 мл раствора формалина,
НСОН (полученного разбавлением 1 мл продажного 40%-ного формалина до
100 мл), вновь нагревают и во время энергичного кипения прибавляют еще
% A. G ц t b i е г, О. М а I в с h, Вег. 52, 1370 (1919).
Платина
431
такого же раствора формалина. При дальнейшем кипячении желтова-
£-**вначале раствор постепенно темнеет до полной непрозрачности. Чтобы
^#?ясать выпадения нерастворимой Pt, которое происходит при спокойном
'^«шии раствора, жидкость разбавляют равным объемом воды и подвергают
^ЛиЗУ через пергаментную гильзу, предварительно в течение продолжитель-
fiz! времени выдержанную в дестиллированной воде.
^^Упариванием можно значительно сконцентрировать гидрозоль, не опасаясь
радения платины.
3. Гутбир1 предлагает восстановление HsPtCle гидразин-гидратом с
прибавлением предохранительного агента или без него.
а) С предохранительным агентом. Смешивают разбавленный раствор PtCU
/лучше всего 0,1%) с равным объемом 1%-ного раствора гуммиарабика2,
Предварительно нагретого при 95° в течение 3 час, и несколькими каплями
0,05%-ного раствора гидразин-гидрата ЫгШ-НгО, причем раствор уже при
обыкновенной температуре становится темнокоричневым; если окраска не
усиливается от дальнейшего прибавления N2H4 • НгО, смесь подвергают
диализу.
. Полученный раствор фильтруется через бумагу без разложения, и его
110ЖНО даже несколько сконцентрировать выпариванием. Этот чрезвычайно
устойчивый раствор дает при испарении в вакууме над H2SO4 твердый
гидрозоль, обладающий способностью нацело растворяться в теплой воде.
' б) Без предохранительного агента. Смешивают в чистом сосуде без
нагревания при хорошем перемешивании приготовленный на свежеперегнанной
воде 0,075%-ный раствор 1 г PtCU с несколькими каплями сильно
разбавленного раствора гидразин-гидрата. Полученную жидкость, окрашенную в
темный цвет, выливают в диализатор, которым может служить пергаментная
гильза, продолжительное время выдержанная в дестиллированной воде.
4. По Б р и нтци н ге р у3 1 мл 0,5%-ного раствора HaPtCIe раабавляют
водой до 200 мл и добавляют 4 мл 0,\N NaOH. Затем (на холоду или при
нагревании) приливают 2 мл 0,5%-ного раствора аскорбиновой (или изоаскор-
4$иновой) кислоты, причем тотчас же образуется прозрачный коричневый золь.
;■ 5. К. 3 енг-ил ,ис и Е. Стати с4 получают очень прочный золь Pt,
подвергая разбавленный раствор PtCU воздействию водорода in statu nascendl,
Получаемого при взаимодействии металлического А1 и NaOH. Предварительно
К раствору добавляют 10%-ного раствора гуммиарабика. Коллоидные частицы
заряжены отрицательно. Золь катализирует разложение Н2О2 и не
коагулирует даже при добавлении равного объема 1JV НС1 или 0,5N Ba(NOa)2.
6. По Бред игу5 можно получить гидрозоль путем катодного
распыления платины. Стеклянную чашку емкостью 100—150 мл наполняют свобод*
ной от СОг водой, к которой прибавлено незначительное количество КОН.
Чашку охлаждают снаружи измельченным льдом и затем под поверхностью
Жидкости получают возможно спокойную вольтову дугу между двумя
платиновыми проволочками (d = 1 мм). Рекомендуемая сила тока 8—10 а при
напряжении 30—40 в. Ток пропускают до тех пор, пока жидкость не примет
глубоко черного цвета. Образовавшиеся в некотором количестве твердые
Частицы платины отфильтровывают.
, Р.изенфельд предлагает брать для получения гидрозоля путем
катодного распыления особо чистую воду («для измерения электропроводности»,
см. стр. 107) и применять ток силою не выше 10 а и напряжением 70—220 в.
Свойства
Коллоидальный раствор Pt черного цвета; от прибавления электролитов
Или при кипячении происходит обесцвечивание раствора и выпадение пла*
* А. О u t b i е г, Z. anorg. Ch. 32, 352 (1903); J. prakt. Ch. 71, 359 (1905); Prep. Ch. 768.
* В качестве предохранительных агентов иногда применяются также лизальбиыэвая и
ЧротальбЪновая кислоты.
» Н. Brintzinger, Koll. Z. 73, № 1, 22 (1937).
* К. Z e n g h e 1 i s u. E. 3 t a t h i s, Praktica 12, 474 (1937) (греч.).
л « Bredig, Z. f. Elektroch. 4, 514 (1898); Z. ang. Ch. 941 (1828); Z. phyi. Ch. 31, 271 (18S9);
Pr3p. Ch. 769. x
432
Платина
тины в виде черного порошка. Если каплю коллоидального раств
прибавить к Н2О2, то последняя моментально разлагается с вылЛ1)а ^
кислорода. * деление^
Для удачного восстановления платиновые солей в гидрозоль Пл
необходимо точное выполнение рецептурных условий и применение TUfibl
дельно вымытых сосудов и свежеперегнанной воды. Щь
Д. Получение платины из лабораторных остатков
1. При длительном хранении платиновых остатков они нередко загряз
каются; кроме того, при хранении остатков, содержащих спирт, зачастую
происходит восстановление платиновых солей до платиновой черни, пл
Бертольду1 сначала отфильтровывают осадок, обрабатывают его царской
водкой до растворения платины и снова фильтруют. Последний фильтрат для
удаления избытка царской водки выпаривают и остаток выщелачивают горя-
чей водой и смешивают с первым фильтратом, т. е. с главной порцией рас-
твора.
В качестве примесей в платиновых остатках получающихся в
аналитических лабораториях, содержатся, главным образом, соли К', Na", b\g" и
NH4* (возможны также соли Ва'*). Остальные катионы при употреблении
tbPtCle как реактива заранее удаляются методами групповых осаждений.
Кроме того, могут присутствовать спирт и эфир, не мешающие выделению
платины.
Жидкость (от которой предварительно отогнаны спирт и эфир) смешивают
с чистым цинком и небольшим количеством концентрированной HCI; для
восстановления foPtClo смесь подогревают до растворения соли. По
окончании восстановления (которое легко определяется по обесцвечиванию
жидкости) осторожно сливают отстоявшийся раствор и кипятят остаток с крепкой
НС1. Наконец, порошок платины промывают декантацией горячей водой до
полного удаления СГ в промывных водах.
2. По де-Джонгу2 раствор платиновых, остатков выпаривают до
содержания приблизительно 100 г Pt в 700 мл, подщелачивают NaOH и добавляют
муравьинокислый натрий (из расчета 50 г на 100 г Pt), поместив чашку на
кипящую водяную баню. Образующуюся пену (СОг) уничтожают добавлением
NaOH.
Подкисляют жидкость 25%-ной НС1 и нагревают час на водяной бане;
остаток отсасывают, промывают дважды соляной кислотой, затем декантируют
водой, снова отсасывают, тщательно промывают водой и прокаливают.
Прокаленный осадок нагревают в колбе сначала с 25%-ной HCI, затем
с водой; добавляют 50%-ной HN03 до прекращения выделения газов. HNOs
удаляют нагреванием с соляной кислотой; для удаления НС1 осадок
выпаривают с водой, отсасывают платину и промывают водой.
Получается свободный от калия оранжево-желтый препарат H2PtCh.
3. Для выделения Pt из лабораторных остатков, содержащих foPtCle и
спирт, Тредвелл3 предлагает, выпарив их досуха, растворить в воде
и вылить в раствор NaOH (уд. в. 1,2), прибавив к нему 8% глицерина. Смесь
нагревают до кипения, причем Pt выпадает в виде черного порошка; его
промывают сначала еодой, затем НС1, снова водой и прокаливают.
4. Для выделения платины из остатков после определения К* в виде
KsPtCle можно применить следующий метод4.
Из спиртовых промывных вод осаждают H2PtCle в виде (NH^PtCle,
добавляя избыток NH4CI. Собранные осадки foPtCle и (NH^PtCle помещают
в большую банку, содержащую 2 л дестиллированной воды и 25 мл 85%-ной
1 В е г t h о 1 d, Z. angew. Ch. 121 (1G01); Prap. Ch. 765.
» D. J. de J о n g, Chem. Weekb. 10, 883 (1913); Prap. Ch., 766.
8 Аналит. химия, 1, 279 (1930).
4 M. С. S w i s h e r a. F. F. H ц m m e 1, Ind. Erg Chem Anal. Ed. 11, 162 (1939).
Платина хлористая
433
' «вьйной кислоты. Банку неплотно закрывают и оставляют на 3—5 дней
*#**омнатной температуре.
>11^ЯЫДеЛИВШИЙСЯ осадок металлической Pt промывают горячей водой до уда-
#■* Q
Е. Очистка платиновых сосудов
ВаНЙНо1 °™ечает быструю' порчу платиновых сосудов при чистке тон-
им морским песком и рекомендует проводить очистку сплавлением с KHS04,
SH такой температуре, чтобы выделялись белые пары серного ангидрида
12е ограничиваясь плавлением соли в кристаллизационной воде).
}■ рекомендуется также применять для сплавления буру ЫагЕШт или борно-
^Аюристый калий KBF4.
^Зонштадт приводит следующий метод, дающий хорошие результаты, но
Йебующий применения печи с высокой температурой нагрева. В подлежащем
листке сосуде нагревают хлористый магний-аммоний MgCla • 2NH4C1 до
tilCO—1200° применяя криптольную, каскадную или высокочастотную
тигельную печь; по охлаждении кипятят в сосуде воду. Очищенная таким путем
платина становится белой и блестящей, как новая.
; Штольба рекомендует сплавлять в сосудах смесь равных частей бор-
фй кислоты НзВОз и борнофтрристого калия, KBF4, причем масса легко рас-
Йоряется после охлаждения тигля, в то время как один KBF4 крепко
придает к стенкам и растворяется только после длительного кипячения,
с,Для удаления из тигля затвердевшего сплава можно поступать следую-
jjfcfcM образом. Втыкают в еще незатвердевшую массу спираль из платиновой
Дюволоки и после застывания подвешивают тигель к лапке штатива при
Йшощи проволоки, захватывающей платиновую спираль. Проволоку
подтягивают так, чтобы дно тигля приходилось несколько выше кольца штатива.
Щя непродолжительном нагревании тигля последний падает, в то время как
<|плав остается на спирали.
ПЛАТИНА ХЛОРИСТАЯ (ХЛОРИД ПЛАТИНЫ [2])
'Platinum chloratum Platinum dichloride Platinchlorur
Platinous chloride Platinochlorid
PtCh Мол. в. 266,14
Приготовление
1. По Берцелиусу водный раствор платинохлористоводородной кислоты
выпаривают досуха, оставшуюся массу измельчают и нагревают в
фарфоровой чашке, постоянно перемешивая, при 220—230° до полного удаления хлора.
2. По Ш юц.ен б ер ге р у2 препарат мождо получить прямым
соединением элементов; для этого пропускают до прекращения поглощения сухой
Хлор через губчатую платину, находящуюся в стеклянной трубке, нагретой
йа масляной бане3 до 240—250°.
Свойства
• Коричневый или серо-зеленый порошок уд. в. 6,05, нерастворимый в воде
;** растворимый в горячей НС1 с образованием платинистохлористоводород-
#ой кислоты, H2PtCU.
5 PrBp. Ch. 771.
? Ргйр. СГГ. 774.
1 При 200° реакция идет еще чрезвычайно медленно
434
Платина. Реактивы
Platinum tetrachloratum
Приготовление
ПЛАТИНА ХЛОРНАЯ
Platinic chloride
PtCU Мол. в. 337,06
Platinchlorid
1. По Пижону1 препарат легко получается нагреванием платинохлоп
стоводородной кислоты HsPtCle в струе хлора до 360°. ри"
При нагревании кристаллов HaPtCle-6H20 до указанной температип
кислота сначала плавится в своей кристаллизационной воде; расплавленна
масса вспучивается и разбрызгивается. Поэтому лучше кристаллы выдеп*
жать сначала несколько дней в вакуум-эксикаторе, причем теряется 2 моле
кулы воды. При нагревании полученного гидрата до 360° в токе хлора он
отдает воду и НС1 без плавления, оставляя PtCh а виде порошкообразной
коричневой массы.
2. По Эрдману2 для получения PtCU из лабораторных остатков
содержащих платину, их прокаливают в фарфоровой чашке до удаления
органических оримесей, затем размешивают с небольшим количеством слабой НС1
и добавляют металлического цинка.
При этом платина восстанавливается до металла; ее отделяют
декантацией и кипятят несколько раз с водою и НС1. После этого растворяют
металл в царской водке, выпаривают до малого объема и осаждают
крепким раствором NFUCl. Выпадающий хлороплатинат аммония (NH^PtCle
отфильтровывают и нрокаливают в фарфоровом тигле. Получившуюся
губчатую платину вновь растворяют в царской водке и выпаривают досуха на
водяной бане, добавляя время от времени несколько капель НС1. Остаток
НгГЧСЬ'бШО обрабатывают по сп. 1.
Свойства
Красно-бурые гигроскопические кристаллы. Растворима в воде, спирте и
ацетоне; из водного раствора кристаллизуется с 5 молекулами воды.
Таблица 426
Удельный вес водных растворов PtCI4 (P г е с h t)
°/о PtCl4
1
2
3
4
5
6
7
8
9
10
И
12
13
14
15
16
17
уд. вес
1,009
1/ 18
1,027
1,036
1,046
1,056
1,066
1,076
1.086
1,097
1,108
1,119
1,Ш
1 141
1,153
1,165 1
1,176
°/о PtCl4
18
19
20
21
22
53
24
25
26
27
28
29
30
31
32
33
34
уд. вес
1,188
1,201
1,214
1,228
1,242
1,256
1,270
1,285
1,300
1,315
1,330
1,346
1,362
1,378
1,3 5
U13
1,431
°;'о PtCl4
35
36
37
38
1 39
40
41
42
43
44
45
46
47
48
49
50
уд. вгс
1,450
1,469
1,488
1,505
1,525
1,546
1,568
1,591
1,615
1,641
1,664
' 1,688
1,712
1,736
1.76)
1,785
1 Р 1 g е о п, Сг. 110, 77 (1890); Z. anorg. Ch. 81, 378 (1913).
■ Хим. преп. 6, 7.
Щ± Платина хлорная. Реактивы Дениже, Каро и Несслера 435
испытание (Бен дер)
* 1 растворимость в спирте. 1 г препарата должен полностью растворяться
* jQ г абсолютного спирта.
* д. Остаток от прокаливания. 2 г препарата сильно прокаливают: остаток
Лпабатывают на водяной бане 5 мл НЫОз (уд. в. 1,2) и 20 мл воды, филь-
• уют фильтрат выпаривают и прокаливают; остаток не должен быть больше
РЕАКТИВ ДЕНИЖЕ
Приготовление1
!".;■' 2 5 г <молибденовокиелого аммония раствоояют в 25 мл воды, добавляют
' % мл H2SO4 (уд. в. 1,84) и по охлаждении смеси вносят 0,25 г опилок
Электролитной меди. Жидкость взбалтывают в течение 30 мин., разбавляют
водой до объема 250 мл и оставляют на сутки. Хранить реактив следует
£j темной склянке.
%
]г'р в о й с т в а
$1 Жидкость темнобурого цвета; применяется для определения As и Р по
Молибденовой сини.
W РЕАКТИВ КАРО
приготовление
Ь По Бэйеру и Виллигеру2 при сильном охлаждении смешивают 1 ч*
^6%-ной НЮг с 5 ч. концентрированной H2SO4.
С в о й с т в а
;• Жидкость, применяемая в качестве реактива на кетоны, с которыми она
Дает соединения перекисного характера. Действующим началом реактива
является кислота Каро, H2SO5.
РЕАКТИВ НЕССЛЕРА
K2HgJ4 • лКОН
Приготовление
1. По Несслеру 32 г двуиодистой ртути (HgT2) и 20 г KJ растворяют
* 50 мл воды и разбавляют раствор до 200 мл, причем выпадает
незначительное количество Hgj2, находящейся в избытке. 20 мл полученного раствора
смешивают с 30 мл концентрированного КОН.
2. Взбалтывают смесь 35 г К J и 13 г хлорной ртути {яд!) с 800 мл
Ьоды и нагревают до полной прозрачности жидкости. Затем прибавляют по
каплям насыщенный раствор HgCh до начала образования неисчезаю.щего
осадка, после чего добавляют раствор 160 г КОН (или 120 г NaOH) в 1 д
Ьоды и е^це несколько капель HgCh. Жидкости дают отстояться, фильтруют,
Сели есть осадок, и сохраняют в склянке с притертой пробкой.
3. По Винклеру 6 г HgCh растворяют в фарфоровой чашке в 50 мл
Зрячей (80°) воды, свободной от NH.i3, добавляют раствор 7,4 г KJ в 50 мл
воды, охлаждают и дают отстояться. Слив жидкость, осадок Hgj2 трижды
промывают декантацией холодной водой по 20 мл для удаления
образовавшегося КС1. К промытому осадку добавляют 5 г KJ и немного воды для
1 G. D 6 n i g e s, С. г. 184, 687 (1927).
1 В а е у е г, Villiger. Вег. 33, 14(10 i.
8 Получаемый перегонкой ооы *ной воды с неб;льшим количеством NagCOj. Первую ш
Последнюю чьтверъи погона отбрасывают.
436
Ртуть
растворения; к полученному раствору добавляют концентрированный растп
20 г NaOH и по охлаждении разбавляют водой до 100 мл. Дают отстоять^
сливают прозрачную жидкость сифоном в темную склянку с притертой проб* *
кой и сохраняют в темном месте. °* '
Свойства
Бледножелтая жидкость, дающая с NH4* красно-бурый осадок. Чувству
тельность реакции 0,00005—0,000025 мг NHs.
Hydrargyrum
Hg
РТУТЬ
Mercury
At. в. 200,61
Quecksilber
А. Обыкновенная ртуть
Приготовление
Из методов лабораторного получения ртути можно упомянуть лишь опере-
гонке в тугоплавкой реторте смеси киновари с порошком железа. Гораздо
чаще в лабораторной практике встречается необходимость очистки ртути,
загрязненной различными примесями. Ниже рассматриваются несколько таких
методов.
Очистка
1. По Пфаундлеру ртуть хорошо очищается фильтрованием через
замшу. Предложенный им аппарат состоит из узкой железной трубки,
оканчивающейся цилиндрическим расширением с винтовой нарезкой. В
расширение вкладывается несколько кружков замши, которые
зажимают навинчиваемой муфтой с отверстием посредине (рис. 62).
Наливаемая в трубку ртуть своей тяжестью продавливается
через замшу и стекает мелкими каплями.
2. Для очистки ртути от примесей (Zn, Pb, Sn, Си и др-)
рекомендуется химический метод—взбалтывание с различными
реагентами. В качестве последних разные авторы предлагают
разбавленную НЖ)з, концентрированный раствор FeCh,
0,5%-ный раствор КгСггО?, подкисленный H2SO4, и др. Можно
также настаивать нечистую ртуть с крепкой H2SO4 -в течение
нескольких часов.
3. Можно рекомендовать следующий быстрый и довольно
простой метод очистки, пригодный для переработки
значительных количеств ртути. Две воронки, размер которых зависит
от количества очищаемой ртути, помещают в штативе одну
над другой (рис. 63). В воронки кладут фильтры из
фильтровальной бумага, причем верхний фильтр протыкается
обычной, а нижний — самой тонкой иглой. Назначение верх-н£г<-
фильтра — задержать наиболее крупные механические
примеси, могущие засорить отверстие нижнего фильтра. Под нижнюю воронку
помещается высокая (около метра) широкая стеклянная трубка, наполненная
0,5%-ной HNOs.
В верхнюю воронку наливают очищаемую ртуть. Последняя, проходя
через два фильтра, выходит из трубки нижней воронки в виде мельчайшие
капель. Капли ртути проходят через высокий столб НЫОз, где очищаются
от примесей. Внизу трубки собирается чистая ртуть. По окончании операции
ртуть переливают в фарфоровую чашку и промывают декантацией водой Д°
полного удаления НЫОз. Остающиеся на поверхности ртути капли воды
собирают при помощи полосок хорошей фильтровальной бумаги.
Рис. 62.
Аппарат Пфаундле-
ра для очистки
ртути
Ртуть
437
i КРа Фт рекомендует для окисления посторонних металлов продолжи-
*кЯое время просасывать через ртуть ток воздуха. Это можно производить
* яяклонной железной трубке. При небольшом загрязнении ртути для этого
''Паточно одного рабочего дня; при очень грязном металле окисление
забивается в течение суток.
Дня удаления олова Бакен родер рекомендует облить ртуть соляной
дслотой, содержащей SCb, и подвергнуть смесь двействию солнечного света при
ястом взбалтывании. При последующем нагревании смеси
о 80° в течение нескольких часов олово переходит в раствор.
5. По Ризенф ел ьд у 1 загрязненную ртуть (помещен-
^асалась к поверхности ртути. Через несколько часов
перевешивания (рекомендуется время от времени сменять
раствор HgsS04) ртуть мож,но считать очищенной от
неблагородных металлов, которые, вытесняя Hg из Hg2S04, сами
переходят в раствор.
6. По А. Расселю и Д. Эвансу2 взбалтывают ртуть
С 35%-ной H2SO4 (уд. в. 1,26) и -насыщенным раствором
КМп04, добавляя последний до прекращения
обесцвечивайся. Промыв ртуть водой, встряхивают ее с 0,1 Д/ КМпО*,
иодкисленным 2N Н2ЗО4, до перехода ртути в дисперсное
сйстояние. Затем снова тщательно промывают, взбалтывают*
й %N H2SO4 для коагуляции и продавливают очищенную
ртуть через замшу.
~ 7. По !А. Д о б р о в с к о м у3 взбалтывают ртуть с
насыщенным раствором KMnCV, цвет последнего вскоре
переходи! .в зеленый и коричневый; ртуть эмульгируется. Очистка
закончена, если -розовый цвет КМп04 не исчезает в течение
30 сек. Добавляют к смеси HNCb, причем ртуть собирается
:.* сплошную массу. Отделив ртуть в делительной воронке,
усушат ее фильтровальной бумагой и затем фильтруют через
яисчую бумагу, проколотую тонкой «иглой.
v Для очистки от благородных металлов (Ag, Au) ртуть
/Необходимо перегнать.
'■$: 8. Для перегонки Гюлет предлагает следующий аппарат
'.Эщис. 64). Очищаемая ртуть помещается в длинногорлую
.^Вол-бу Вюрца А с длинной отводной трубкой В и перего-
"Мяется нагреванием на песочной или асбестовой бане в кол-
Sf Вюрца С. Для лучшего использования тепла рекомен-
Муется окружить колбу А стаканом D с отбитым дном, при-
|Ррытым сверху асбестовым кругом Е.
i; Через каучуковую трубку, снабженную винтовым зажимом F, проходит
|&еклянная трубка G, оканчивающаяся капилляром около поверхности ртути.
Що этой трубке медленно пропускается струя высушенного (СаСЬ) и
профильтрованного (вата) азота или ССЬ. Перегонка ведется под возможно
низшем давлением, для чего приемник С соединен трубкой Я с хорошим
водоструйным насосом. Высоту пламени регулируют так, чтобы ртуть
конденсировалась в отводной трубке ниже изгиба (у места, отмеченного на рис. 64
Фуквой В); при этом перегоняется около 200 г Hg в час. При перегонке
рЮгда наблюдаются яркозеленые вспышки в трубке В, особенно когда вслед-
$твие усиления нагревания граница конденсации ртути передвигается ниже.
■■ При перегонке ртути следует помнить, что пары ее сильно ядивиты.
;j 9. П а л ь м е р 4 рекомендует следующий очень хороший прибор (риЕ. 65),
'Представляющий собой цилиндрический сосуд А с припаянной к нему более
Рис. 63. Очитгка
ртути
Фильтрованием
1 Неорг. практ. 216.
• A S R ц s « е 1 a. D. С. Е v а п в, J. cliem. Soc. 127, ^298 (i925).
•A. DoDrow*ky, Спеш. Zls: 64, 32 (.94 J).
4 W. P a I m e r, Bw. 32, J391 (189J); Prip. Ch. 570.
438
Ртуть
узкой трубкой В, оканчивающейся на конус. Трубка В в конической част,,
внутри пришлифована. Внутрь прибора вставляется стержень (трубка^ с .
заканчивающийся пришлифованной пробкой D диаметром
15 мм, входящей в шлиф, имеющийся в конце труб-
Ш Пробка D во всю длину имеет 40-50 радиально
расположенных углублений, ширина которых равна 0,25 мм, А \
Рас. 64. Прибор для перегонки ртутило Полету
PilC. 65. Прибор ДЛ1
очистки ртути но
Пальмсру
а глубина 0,08—0,1 мм (см. рис. 65, I). При рассмотрении под микроскопом
углубления должны иметь вид ровных желобков.
Прибор устанавливают так, чтобы
пробка D была погружена в 1Д/ раствор HNCb,
через которую должна капать ртуть.
Очищаемую ртуть наливают в сосуд А. При
высоте ртути в 7 см через углубления пробки
проникает дождь из тончайших капелек
ртути. Когда резервуар А наполнен ртутью до
половины, в минуту вытекает около 200 г
Hg. Сосуд А вмещает около 3 кг ртути.
Нижний край трубки В несколько отогнут,
а пробка D снизу закруглена, чтобы при
неосторожном вставлении пробки не
произошло поломки прибора. Поскольку для про-
давливания ртути необходимо давление 7 см
рт. ст., то после прекращения вытекания в
узком промежутке между С и В остается
еще около 150 г ртути.
10. По Морзе1 перегонку ртути можно
вести в следующем аппарате (рис. 66).
В газовую сожигательную печь,
поставленную наклонно (один конец поднят на
12—15 см), помещают трубку из тугоплав-
' кого стекла с загнутыми вниз концами. Один
tuc. бо. Аппарат для очистки ртути конец (левый на рис. 66) оттянут и соеди-
по Морзе
'Мни, Am. Ckem. J. 7, 60.
Ртуть
439
с ДРУг°й трубкой (d = "10 мм)\ последняя должчп быть тят<ой длины.
. ^loW образовалась вертикальная трубка длиной в 700 мм. конец которой
•'*ИгРУжеН в у3!К11И стеклянный цилиндр со ртутью, подлежащей очистке.
Я° Лп^тт, ТПУбкИ. НаХОЛЯШЯЯГСТ п npuu гЛГНУТа ТаК. ЧТОбы ППГ.ЛРпиаа mi
Часть трубки, находящаяся в печи, согнута так, чтобы последняя горелка
печй приходилась под самой высшей точкой1. Отсюда, не утоньшаясь, трубка
агйбается вниз на 200 мм. В этот конец при помощи каучуковой трубки
плотно вставляют более узкую стеклянную трубку длиной 800 мм (в месте
соединения устраивается надежный герметический затвор, изо- _
Сраженный на фис. 67). Нижний конец трубки доходит до дна
склянки Вульфа, соединенной с водоструйным насосом.
Перегонка ведется следующим образом:
пустив в ход насос, добиваются поднятия ртути в
I горизонтальную часть трубки. Как только ртуть
примет вид длинного языка, протянувшегося от
одного колена трубки до другого, запирают
зажимом каучуковую трубку D, ведущую от
аппарата к насосу, и нагревают печь очень слабым
пламенем.
- Ртуть слабо закипает и перегоняется через
Асбестовую пробку. Когда половина правой
вертикальной трубки наполнится ртутью, в склянку
^вульфа впускают воздух, осторожно открывая
>зажим на трубке D, и удаляют эту трубку
^совсем. Теперь разрежение внутри аппарата
Поддерживается столбом ртути. С этого
времени аппарат действует автоматически, если в
стеклянный цилиндр подливать очищаемую ртуть2.
11. Кар стен предложил для перегонки
ртути следующий аппарат (рис. 68).
В горло склянки А с отрезанным дном
плотно вставляется на пробке трубка В наружного Ри.# 67. деТаль
диаметра 10 мм, длиною 1450 мм с загнутым затвора аппа-
нижним концом. Снаружи трубка в верхней Рата Морзе
части окружена другой стеклянной трубкой С
Диаметром 15 мм, длиною 800 мм и наверху
раздутой в шар. Трубка В на расстоянии 400 мм от верхнего конца
суживается в капилляр. Сосуд А наполняют нечистой ртутью, В соединяют с
насосом и осторожно выкачивают воздух, одновременно подливая в А такое
количество ртути, чтобы она из шара переливалась по каплям во внутреннюю
трубку В. Последняя своим сужением действует аналогично ртутному насосу
Ш п р е н г е л я, в результате чего в аппарате достигается -высокий вакуум.
Удалив насос, нагревают шар кольцевой горелкой; начинается перегонка
(около 250 г Hg в час), которая продолжается непрерывно, если подливать
ртуть в сосуд Л 3.
Рис. 68.
Аппарат Карстена
для очистки
ртути
Свойства
Белый с синеватым оттенком, жидкий металл уд. в. 13,551. (По другим
данным d4° = 13,59539 ±0,00001*. Т. пл. — 38,89°, т. кип. 356,66 + 0,02°. При
застывании становится ковким, как свинец, и кристаллическим (октаэдры,
1 В высшую точ-ry трубки помешается рыхлая асбестовая пробка.
» О применении электрического обогрева при перегонке ртути по Морзе см. В. А. Захарьев-
с к ий, Завод. Лаб. 7 № 3, 89S (1938).
8 Метод очисгки ртути кипячением с NaOH см. В. R e s с е, Ann. Chlra. appl. 25, 354 (1936).
С. 1, 3987 (1936); P. Dickens, Stahl u. Eisen 58, № 49, 1403 (1j38); аппарат для очистки ртути
промывкой, аэрацией и перегонкой см. W. А. С a risen a. L. F. Borchardt, Fnd. Eng.
Chem. Anal. Ed. 10, № 2, 94(1938). О других аппаратах для перегонки ртути см., например,
Ш и в о в, Завод. Лаб. 4, 46Э (1935); W. С. J о h n s о п, J. Soc. Cbem. Ind. 5?, 10, 360 (193% С. I,
4073 /1937)
«Т. в'а t u в с a i, u. F. L С a s a d ot J. СЦега. physique 33, 41 (1936); С. U 4123 (193*1.
440 Ртуть
срастающиеся в иглы). При обыкновенной температуре не окисляется на во*
духе; окисление происходит только при длительном нагревании около Зобо
Совершенно чистая ртуть при выливании образует круглые, блестящи* '
легкоподвижные капли; нечистая ртуть покрывается пленкой и оставляет дЛНн* '
ные белые полосы,
Испытание
1. При нагревании 20 г препарата в фарфоровом тигле не должно 0СТа.
ваться весомого остатка (под тягой! Пары ртути сильно ядовиты]).
2. Кипятят в пробирке в течение минуты 5 г ртути с 5 мл воды и 4,5 г
гипосульфита (Na2S20a • 5Н20). При этом ртуть не должна терять своего
блеска и, самое большее, может принимать слабо желтый оттенок.
3. 5 г препарата при растворении в 20 мл HNOs (уд. в. 1,4) должны дать
прозрачный раствор.
4. При встряхивании ртути с чистым, свободным от пыли воздухом поверх-
ность ее должна оставаться блестящей.
5. По Мэтью с у определение количества примесей в ртути может быть
проведено следующим путем.
100—200 г ртути вносят в склянку емкостью 250 мл, добавляют 100 мл
воды и 25 мл 10%-ной H2SO4 и титруют при сильном встряхивании 0,1—0,02/V
КМп04, пока ртуть внезапно не превратится в эмульсию. Чем меньше
затрачено KMnCU для этого, тем чище ртуть. (Метод основан на том, что высокое
поверхностное натяжение чистой ртути чрезвычайно сильно снижается в
присутствие примесей.)1
Б. Коллоидальная ртуть
Приготовление
1. Лоттермозер2 рекомендует сливать при перемешивании
разбавленные растворы азотнокислой закиси ртути (HgNOa) и азотнокислого олова
[Зп(ЫОз)2] 3. Оба раствора должны содержать лишь столько свободной
кислоты, чтобы не выпадали основные соли. В результате получается темно-
коричневая жидкость; ее смешивают с концентрированным раствором
лимоннокислого аммония [(NH^CeHsCb], вследствие чего коллоидальная ртуть
высаливается. Коричневая окраска жидкости переходит в черную и отстаивается
тонкий черный порошок. Теперь нейтрализуют жидкость аммиаком при
помешивании, избегая сильного разогревания. После того как осадок отстоялся,
сливают раствор, удаляют еще некоторое количество жидкости отсасыванием
на пористом глиняном фильтре и все еще довольно жидкую пасту
высушивают в вакуум-эксикаторе над H2SO4.
Получаются кусочки с серебряным блеском, растворимые в воде с темно-
коричневой окраской.
2. По Герасимову и Козыреву4 30 мл белковой смеси Пааля (см.
сноску 5 на стр. 331) растворяют в 24 мл ледяной СНзСООН и нагревают
несколько минут на водяной бане. Если белковая смесь свежая, то
продолжают нагревание до исчезновения запаха H2S. Затем при постоянном и
тщательном взбалтывании приливают по каплям раствор 0,375 г HgCte.
Полученный коллоид коагулируют при помощи ЫагСОз, осадок не менее четырех раз
промывают водой, растворяют в очень слабом растворе NaOH и выпаривают
досуха на водяной бане. Остается блестящий черный порошок, раствор
которого в закрытом сосуде сохраняется неопределенное время. При 17° уд. в.
1%-ного рпаствора 1,01-00. Частицы коллоида заряжены отрицательно.
1 Н. Р, Matthews, Soc chem. Ind. Proceed. W, 1104 (1936); Z. anal. Ch. ИЗ, 855 (1938).
•Lottermoser, J. pr.ikt. Ch. (2), 57, 486 (1898).
• Соотношение между реагентами вычисляется по уравнению реакции 2HgN03 • H*Odj-Sn
(NO,>t=2Hg-f Sn(N04)4-T-^H20, но необходимо употреблять значительный избыток 5h(NO,)i,
так как иначе коллоидачьная ртуть легко видоизменяется,
' ЖРФХО 4, 92, 833 Ц930).
Ртуть
44 J
ш Высоко дисперсная порошкообразная ртуть получается при восстановлен
Л-' водной суспензии Hg20 гидроксиламином, гидразин-гидратом или фор-
Цьдегидом. Препарат, содержащий 99,6—99,8% Hg, очень ядовит1.
В. Переработка ртутных остатков
|, Для простой переработки лабораторных ртутных остатков на соли ртути
УО&с'но рекомендовать следующий метод.
Остатки выпаривают и нагревают с НЫОз до полного растворения. Раствор
«ыпаривают дссуха, выщелачивают водой и фильтруют, после чего, добавив
царской водки, снова выпаривают досуха. Остаток растворяют в горячей
^одё, фильтруют и, подкислив НС1, насыщают сероводородом. Полученный
о^аток сернистых соединений обрабатывают при нагревании многосернистым
-щюиием [(NH«>2Sn] и снова фильтруют. Оставшееся сернистые соединения
Йгайлов IV группы промывают и нагревают со слабой НС1 до прекращения
^деления H2S.
Смесь декантируют, осадок^ HgS промывают» до исчезновения кислой
«Аакции и растворяют в царской водке. Раствор выпаривают досуха, выщела-
Втргу
Рис. 69. Прибор дл» uvpepaouTKH лабораторных ртутных
остатков.
Витают водой, фильтруют и осаждают окись ртути (HgO) при помощи КОН*
Выпавший желтый осадок хорошо промывают водой и перерабатывают на
юол,и ртути обычным путем2.
/ 2. По Эрдману ртутные остатки обливают царской водкой и выпаривают
Досуха на водяной бане. Остаток помещают в большую фарфоровую чашку
я нагревают на песочной бане, прикрыв чашку большой воронкой. При этом
разгоняется HgCk (яд/), которая оседает на стенках воронки. Кристаллы
собирают с воронки и, омешав с 0,1 ч. HgO, еще раз возгоняют. При этом способе
Йолучают довольно чистую HgCb.
3. По Диккеису3 10 г лабораторных остатков, содержащих ртуть
(например, после определения железа по Циммерману-Рейнгардт у),
Помещают в стеклянную банку, добавляют 8 г FeS и оставляют смесь стоять,
изредка перемешивая. Окончание реакции осаждения определяется по
появлению запаха HsS. После отстаивания жидкость сливают, удаляют куски FeS
И отсасывают осадок сернистой ртути. HgS просушивают при 100°, смешивают
с половинным количеством СаО, помещают смесь в лодочку, нагреваемую
в трубчатой печи (рис. 69), и прокаливают, собирая ртуть з охлаждаемом
водой приемнике. Выход 85—90% теоретического.
1 А. О а 1 at г к у, Bull. Soc. chim. France, (5) 2, 1301 (1935); С. 1, 3476 (1936).
■ По методу Таверне массу, полученную при осаждении сероводородом, можно
переработать на ртуть иначе, а именно —смешать с металлическим жедезом и смесь подвергнуть
Черегочке, лучше всего в вач-ууме. С. II, 121 (191.4).
• ?. D I с )г t о •, St. о. Eiaao W, М 49, 1405 (1938).
442
Ртуть
РТУТЬ АЗОТНОКИСЛАЯ ЗАКИСНАЯ
(НИТРАТ (РТУТИ [1])
Hydrargyrum
nitricum oxydulatum
Mercurous nitrate
HgNOs НЮ
Мол. в. 280,63
Mercuronitrat
Quecksilbernitrat
(oxydul)
Приготовление
По Е. Шмидту1 смесь 1 ч. металлической ртуш с 1,5 ч. чистой 25%-ной
HNOs ставят на несколько дней в холодное место, защитив от пыли.
Находящаяся в избытке Hg постепенно растворяется, выделяя N0, которая
с кислородом воздуха образует бурые пары N02. Через некоторое время на ^
поверхности ртути выделяются белые кристаллы HgNOs. Когда масса не-
растворившейся ртути перестанет уменьшаться, смесь слегка нагревают, чтобы
кристаллы снова растворились. Отфильтровывают кислый раствор от нераство-
рившейся ртути через асбест и оставляют на сутки в холодном месте.
Выпавшие кристаллы высушивают при обыкновенной температуре между
листами фильтровальной бумаги (осторожно, яд!). Если препарат окрашен, его
растворяют в равном объеме теплой воды с прибавлением нескольких капель
HNOs и оставляют раствор для кристаллизации.
Маточные растворы можно употреблять для повторного получения HgNOs,
добавив к ним концентрированной HNOs с расчетом, чтобы содержание
последней в растворе составляло 25%.
Свойства
Бесцветные, моноклинические, сильно ядовитые кристаллы, уд. в. 4,79 со
слабым запахом азотной кислоты. В сухом воздухе выветриваются. Т. пл. 70°;
при более высокой температуре распадается на HgO и N02. При действии
воды разлагается, выделяя желтый осадок основной соли. Раствор HgNOs
легко окисляется, переходя в соль окиси; поэтому его рекомендуется хранить
в склянках, на дно которых налито немного ртути. Легко (растворяется в воде,
подкисленной HNOs.
Испытание
По ОСТ НКТП 6758/365 содержание HgNOs • Н2О, устанавливаемое по
общему содержанию Hg' и Hg*', для препарата х. ч. должно быть не
менее 99,5%, для я. д. а. — 99,0% и чистого 98,0%; наибольшие количества
допустимых примесей указаны в табл. 427.
Таблица 427
Допустимые примеси в HgN03H20
V
Примеси
& Соли окиси ртути (Hg") ....
. Вещества, не растворимые
в HNOa
Допустимое содержание в °'0
в химически
чистом
0,005
0.0002
0,2
0,03
в чистом для
анализа
0,02
0,0005
05
0,05
в чистом
0,05
0,0 Л
1,0
0,1
Или Hg. (N03)t • 2НвО, мол. в. 5, 61,26.
» Ausftfhr. Lehrb. d. pharm. Cii. 1, 1076.
Ртуть азотнокислая и бромистая
443
РТУТЬ АЗОТНОКИСЛАЯ ОКИСНАЯ
(НИТРАТ РТУТИ [2])
Hydrargyrum
pitricum oxydatum
Mercuric nitrate
Hg(NO,)2. V2W2O Мол. в. 333,63
Mereurinitrat
Quecksilbernitrat
(oxyd)
дригото вление
1. 12,5 ч. окиси ртути (HgO) слабо нагревают в колбочке с 30 ч. чистой
25%-ной HNOa, раствор фильтруют, выпаривают и дают закристаллизоваться
йа холоду.
2. Можно также исходить из металлической ртути, нагревая ее с 4 ч.
25%-ной HN0.3 до тех пор, пока проба раствора не перестанет давать осадок
с NaCl (отсутствие Hg"),
Двойства
Бесцветные кристаллы уд. в. 4,3 (безводная соль), очень легко раствори*
мые в воде (в воде гидролизуются). Т. пл. безводной соли 79°.
Испытание
Наибольшие количества примесей, допустимых по ОСТ НКТП 6759/366,.
указаны в табл. 428.
Таблица 428
Допустимые примеси в Hg(N03)2• xI^O
Примеси
3. Хлориды (СО
Допустимое содержание в °/0
в химически
ЧИСТОМ '
0,005
0,0002
0,001
0,005
1,0
В ЧИСТОМ ДЛЯ
анализа » чистом
0,02
0,0005
0,003
0,02
3,0
0,05
0,001
0,005
0,05
5,0
. Кроме того, 2 г препарата должны полностью растворяться в 4 мл НС1
(уд. в. 1,12), давая прозрачный раствор (что свидетельствует об отсутствии
закисной соли).
Hydrargyrum
bromatum
Приготовление
РТУТЬ БРОМИСТАЯ
(БРОМИД РТУТИ [1])
Mercurous bromide
HgBr
Мол. в. 280,53
Mercurobromid
Quecksilberbromur
По Саа и Чудхури1 препарат получается прибавлением раствора КВг
к раствору HgN03, содержащему небольшое количество HNOs (2 мл HNOs
на 200 мл воды). Через сутки твердый белый осадок отмывают декантацией,
l Saba, Choudhur , Z. anorg. Ch. 77, 42 (1012); Prap. Ch. 579.
444
Ртуть
фильтруют и высушивают над СаСЬ. Так как препарат чувствителен
ствию света, все эти операции следует проводить в темноте. к Дей-
Свойства
Белый порошок уд. в. 7,3, значительно менее чувствительный к свету
HgJ, но все же частично разлагающийся при непосредственном дейетЧем
солнечных лучей. При 345° возгоняется. В воде почти нерастворим (поиоЙ
растворяется 3,9- 10-*%). * ^
РТУТЬ БРОМНАЯ (БРОМИД РТУТИ [2])
Hydrargyrum Mercuric bromide Mercuribromid
bibromatum Quecksilberbromid
HgBra Мол. в. 360,44
Приготовление
По Рейндерсу1 обливают ртуть водой и постепенно приливают по
каплям бром, пока последний не перестанет обесцвечиваться. Тогда нагревают
фильтруют и дают закристаллизоваться. Для очистки препарат перекристал-'
лизовывают. Полученные кристаллы сушат при возможно низкой температуре
и для окончательной очистки подвергают возгонке. Последняя проводится
в нагреваемой на песочной бане фарфоровой чашке, покрытой перевернутым
кристаллизатором. HgBr2 возгоняется в виде красивых белых игл, после чего
в чашке остается лишь незначительный остаток*.
Свойства
В возогнанном виде — красивые белке игольчатые кристаллы, уд. в. 5,73;
из водных растворов кристаллизуется листочками. Растворим в воде (1:200
на холоду, 1:25 при кипячении) и значительно лучше в спирте (23,1% при
25°) и эфире. Т. пл. около 236°, т. кип. 325°.
Препарат, загрязненный следами HgBr или органическими веществами,
темнеет на свету; хранить его следует в темных, плотно закупоренных банках.
Испытание3
В препарате ч. д: а. допускается содержание нелетучих примесей до 0,05%
и до 0,0002% As (испытание по 3 а и ге р.Б л* эк у4). При растворении 2 г
препарата в 100 мл 2%-ного раствора КВг не должно быть мути или осадка.
РТУТИ ЗАКИСЬ (ОКИСЬ РТУТИ[1])
Hydrargyrum Merctirous oxide Mercurooxydul
oxydulatum Quecksilberoxydul
HgsO Мол. в. 417,22
Приготовление
По Шмидту5 закись ртути можно получить растиранием тонко
измельченной каломели (HgCl) с избытком КОН или NaOH или осаждением из
водного раствора закисной соли ртути избытком щелочи.
* W. R e i n d е г в, Z. pbys. Ch. 32, 495 (1900); РгЗр. Ch. 579.
■ По литературным дааным г>ч*нь чистую HgBra, содержащую ничтожную примесь
HcrtBiOaV мо"но получить, обрабатываа HgO раствором брома в СС14. М. L&uira-
c'hands е. Р. Р 1 е г г о п. С. г. 204, 1Ь4о (1937); С. 4024 (193«). К сожалению, авторы не указываю^
условий проведения реакции.
» Испытание хим. р активов, 340.
«Ф. Тредвелл, Kvpc аналитической химии, т. II. кн. I, стр. 68. ГОНТИ, 1931.
1 AuafUlir. Lenrb. d. pharw. Си. 1, 1060.
I
Ртуть бромная. Ртути закись. Ртуть йодистая 445
Щ['± ч азотнокислой закисной ртути (HgNOa • Н20) тонко измельчают в фар-
Й яой ступке и смешивают с 1,8 ч. чистой 25%-ной HNOs. Смесь выли-
'|2£г (избегая нагревания) в такое количество дестиллировавдюй воды, чтобы
•rf** объем составлял 100 ч. Полученный раствор вливают в раствор 4 ч.
2ЙЯ в 50 ч. спирта.
Куйбразующуюся коричневатую массу, после тщательного промывания водой
Темноте, высушивают при комнатной температуре.
Коричнево-черный порошок уд. в. 9,8, нерастворимый в воде. Препарат
# .дрочен и разлагается при хранении на HgO и металлическую ртуть (раз-
мясени* Л}ет быстрее на свету или при нагревании). .Частичное разложение
/уступает уже в процессе получения HgzO. „
у;'
РТУТЬ ЙОДИСТАЯ (ИОДИД РТУТИ [1])
.' - Hydrargyrum Mercurous iodide Alercuroiqdid
jodatum Green jodide Quecksilbefjodur
of mercury
; HgJ Мол. в. 327,53
А. Обыкновенная йодистая ртуть
Приготовление
По Варе разбавленный раствор KJ приливают при непрерывном поме*
Щивании к эквивалентному количеству раствора HgNOs, содержащему HNOs
(1 моль HNOs на 2 моля HgNOs). Полученный желто-зеленый осадок про-
Иывают, оберегая от действия дневного света, пока он не примет красивого
Желтого цвета. Тогда переносят на фильтр, хорошо промывают и
высушивают в сушильном шкафу при 110°, все время защищая от света. Полученный
таким образом препарат совершенно чист.
Свой
ст ва
• Желтый порошок без вкуса,, совершенно не растворимый в спирте и
эфире и почти не растворимый в воде. При действии света распадается на
"gfe и Hg. При нагревании окрашивается около 70° в красный цвет, три
•Дальнейшем нагревани>и—в темнофиолетовый. При 290° плавится в черную
Жидкость, из которой при осторожном охлаждении получают прозрачные
Желтые кристаллы HgJ. Умеренно нагретая HgJ при охлаждении принимает
Первоначальную желтую окраску. Возгонка начинается уже при 120°.
Б. Коллоидальная йодистая ртуть
Приготовление
По Гей дену поступают так же, как для приготовления коллоидальной
Днохлористой ртути (HgCl), употребляя KJ вместо КС1 (см. стр. 452).
Свойства
Зеленовато-желтый порошок, растворимый в воде с образованием желто-
зеленой жидкости с нейтральной реакцией.
446 Ртуть
РТУТЬ ЙОДНАЯ (ИОДИД РТУТШ2])
Hydrargyrum Mercuric iodide Mercurijodid
bijodatum Red iodide Quecksilberjodid
of mercury
Приготовление
HgJi Мол. в. 454,45
А. Красная модификация
1. Растворяют 4 ч* хлорной ртути (яд\) в 80 ч. воды и 5 ч. KJ в 15 "
воды и выливают оба раствора одновременно тонкими струйками в'юо ч' *
холодной воды. Образующийся^ при этом чрезвычайно мелкий осадок отфиль- \
тровывают и промывают водой до удаления Cl'(AgN03 в присутствии Н1М03
в промывных водах должно давать лишь слабую опалесценцию). Наконец
препарат высушивают при 70°.
2. П. Па тер но сто1, для получения хорошо фильтрующегося осадка
HgJ2 .рекомендует слить кипящие 0,02^ растворы HgCh и К J и оставить смесь
на 24 часа.
Б, Желтая модификация
Растворяют красную HgJ2 в спирте и полученный бесцветный раствор
выливают в холодную воду. Образующаяся сначала светложелтая эмульсия
постепенно выделяет пластинчатые желтые кристаллы.
Свойства
Красная модификация представляет собой тонкокристаллический порошок,
уд. в. 6,28, без вкуса, почти не Растворимый в воде (1:25000). Растворима
в этиловом спирте (1:б0 при 20°) и в метиловом спирте (1:25). Кроме того,
растворима в эфире, хлороформе, глицерине, ацетоне, сероуглероде и др.
1. ПЛ. 2ио , Т. КИП. о4У .
Желтая модификация имеет удч в. 6,27; при обыкновенной температуре
неустойчива. Через несколько часов переходит в красную, стабильную форму
(быстрее, в тонко истертом состоянии).
'Испытание
1. Хлорная ртуть (HgCh). Водная вытяжка препарата не должна
вызывать покраснения лакмусовой бума>*<ки.
2. Хлор (СГ). Водная вытяжка препарата не должна давать никакого
•осадка от AgNOs.
3. Растворимость в спирте. 0,2 г Препарата должны полностью растворяться
:в 15 мл этилового спирта.
4. Нелетучий остаток. При прокаливании навески препарата в платиновом
тигле (под тягой) остаток должен составлять не больше 0,5%.
РТУТИ ОКИСЬ (ОКИСЬ РТУТИ [2])
Merciuric oxide
Red oxicie of mercury
HgO Мол. в. 216,61
Hydrargyrum Mercuric oxide Mercurioxyd
oxydatum Red oxicde of mercury Quecksilberoxyd
Приготовление
А. Красная окись ртути
1. Осторожно прокаливают (не вь^ше 340°) азотнокислую закисную ртуть
HgNOs [или смесь окисной соли H;g(NOs)2 и ртути] до полного удаления
1 Р. О. Р a t е г п о 9 t о, Rec. Fac. Cienc. quim< La Plata 10, 11, 1935; C. II 3151 П936).
Ртуть йодная. Ртути окись 447
в азота. Остаток промывают сначала слабым раствором NaOH, затем
о*0* и высушивают.
'j^fno Дюфану1 к раствору 100 г HgCla (яд\) в 500 мл воды приливают
' воо 180 г КгООз в 500 мл воды и кипятят до тех пор, пока образовав-
Р*?гя ко,Рич,невый осаА°,к не станет красным. Дают осесть, декантируют, при-
^ йЮТ раствор 15—20 г КОН и .несколько минут кипятят. Наконец промы-
^а водой до полного удаления СГ в промывных водах.
***? По Бросатти2 горячий концентрированный раствор Ва(ОН)г вливают
I ^пящий раствор HgCk до тех пор, пока коричневый вначале осадок не
\ * ??ет принимать яркокрасную окраску. Затем разбавляют смесь кипящей
*а Й И| дав отстояться, промывают осадок водой.
1 Б. Желтая, окись ртути
f 1 л 10%ного раствора HgCb3 вливают при размешивании в 650 мл 1,5Л/
««атвора NaOH (не наоборот). Оба раствора рекомендуется перед сливанием
отладить До 30° для получения более тонкого осадка, кроме того, в тепле,
осадок принимает красноватый оттенок.
Жидкости с осадком дают постоять около часа в умеренно теплом месте;,
изредка помешивая, после чего промывают водой до удаления СГ. Промытый,
осадок высушивают в темноте при температуре не выше 40° или лучше в
вакуум-эксикаторе над H2SO4. Хранить препарат следует в темных банках.
(Свойства
':;> Окись ртути — ярко красный или желтый порошок уд. в. 11,27, темнеющий
'$т действия света. Ядовита. При осторожном нагревании чернеет, при охла-
"Звдении принимает первоначальную окраску. Почти не растворима в воде
д^юрасная модификация 1 :20500, желтая 1 : 19500) и спирте. Растворима
Щ HNOs и НС1. Препарат рекомендуется хранить в плотно закупоренных
фанках. Красная модификация под микроскопом обнаруживает
кристаллическое строение4.
испытание красной окиси ртути (Мерк) '
Е, 1. Содержание желтой окиси ртути. 1 г препарата в течение часа часто
Йзбалтывают с 10 мл 10%-ноге раствора щавелевой кислоты (Н2С2О4). При
Фгом не должно наблюдаться никакого изменения цвета (посветления).
;. 2. СГ, SO4", NOz и нелетучие примеси. Испытание проводится, как
указано для желтой HgO.
Испытание желтой окиси ртути (Мерк)
1. Хлориды (СГ). Раствор 1 г препарата в 5 мл НЫОз (уд. в. 1,150—Г, 152)?
И 15 мл воды от прибавления AgNCb может давать лишь слабую опалесцен-
цию.
2. Сульфаты (SO/'). Раствор 1 г препарата в 5 мл HNOs (уд. в. 1,150—г
1,152) и 15 мл воды от прибавления ВаСЬ не должен изменяться.
3. Нитраты (NO/). К смеси 1 г препарата, 2 мл воды и 2 мл H2SO4
(уд. в. 1,84) после охлаждения, избегая смешения, приливают крепкий раствор
FeS04. На границе слоев не должно появляться бурого кольца.
4. Нелетучие примеси. 2 г препарата при прокаливании не должны
оставлять весомого остатка.
5. Содержание красной окиси ртути. При взбалтывании с раствором
щавелевой кислоты (Н2С2О4) препарат должен постепенно превратиться в белый-
кристаллический порошок (красная модификация при этом не изменяется).
1 Dufan, J. pharm. Ch. I ], 16, 439, 442; С. II, i5l!) (19,2).
2 Prap. Ch. 673.
■По Фольгарду, если ж лают получить сонер и нно л 'чу шй пг/ па; ат, необходимо*
применяемую HgClj очистить возгонкой с Ш|0 HgO.
« В водном )астмре HgO гидратируется, дав я Н ;рИ)а, обладающий амфотерными
свойствами. О величинах констант диссоциации, см. А. В. Н а г г е 11 a. H i г 8 с h 1 е i С. 1. 4302^
[1938); А. В. Н а г г е t a. W. W. Н о w е 11, J. Am. chem. Soc. 61 173U (1939).
448 РтутВ
РТУТЬ РОДАНОВАЯ (РОДАНИД РТУТИ [2])
Mercuric thiocyanate
Mercuric rhodanide
Hg(CNS)i Мол. в. 316,77
Hydrargyrum rhodana- Mercuric thiocyanate Mercurirhodanid
turn Mercuric rhodanide Quecksilberrhodani
Приготовление
Смешивают растворы эквивалентных количеств роданистого калия, KCN<s
и азотнокислой окисной ртути Hg(NOs)2; выпадающий белый осадок отфи^.'
тровывают и промывают.
Свойства
Белый порошок, очень плохо растворимый в воде (0,069%), легче в спирте
и эфире. Заметно «растворима в НС1 и растворах KCNS. При нагревании
или поджигании начинает тлеть, образуя одновременно с парами ртути
{сильно ядовиты!) чрезвычайно объемистую пористую массу («фараоновы
змеи»).
РТУТЬ СЕРНИСТАЯ (СУЛЬФИД РТУТИ[2], КИНОВАРЬ)
Hydrargyrum sul- Mercuric sulfide, Mercurisulfid
furatum Cinnabar, Vermilion Quecksilbersulfid
Zinnober
HgS Мол. в. 232,67
А. Обыкновенная сернистая ртуть
Приготовление
1. По В а ни но1 хорошо смешивают 1 ч. серы с 6 ч. ртути, нагревают
смесь в железном сосуде до плавления и полученную массу возгоняют на
глубокой песочной бане в глиняном, неплотно закрытом сосуде.
Красно-коричневый возгон при растирании принимает яркокрасную окраску.
2. По Эрдману2 60 г ртути растирают в ступке с 23 г серного цвета
до полного исчезновения капель ртути. Полученный продукт обливают
раствором 15 г КОН в 80 мл воды и оставляют на несколько дней при 45°,
энергично помешивая и пополняя испаряющуюся воду. Когда масса
покраснеет, отделяют ее отмучиванием от избытка ртути и непрореагировавшей серы
и «кипятят с раствором гипосульфита. Раствор отсасывают и промывают осадок
горячей водой. Выход 40 г.
3. Хороший выход препарата получается по методу, предложенному Г е и-
теле3. Для работы по этому методу необходим пятисарнистый калий K2S5,
получаемый прокаливанием 15 ч. КгСОз с 10 ч. серы и растворением сплава
в воде.
В этом растворе при нагревании растирают пестиком ртуть до тех пор,
пока она не превратится в красно-бурую порошковатую массу, после чего
жидкость сливают. Массу переносят на железную сковородку и там
размешивают с небольшим количеством 15%-ного раствора КОН, нагревая до
40—50°, пока масса не примет огненно-красного цвета. Тогда ее охлаждают и
промывают.
Следует избегать более высокой температуры, так как иначе препарат
может приобрести красно-бурый цвет.
4. Крустинеоне4 для получения красного препарата предлагает нагр^
вать черную HgS выше 313° или выдерживать HgS неделю в эксикаторе над
РгОб, а затем нагревать при 200—290°.
1 РгЗр. Ch. 582.
■ Хим. преп. 29.
• I. О. С е n t е 1 е, Lehrb. d. Farbenfabrication, 11, 224(1909), Verb A. Buntrock. Braunschweig
«J.Krustinson», Z. anorg. Ch. 245, i$52 (1941).
Щ Ртуть родановая, сернистая и сернокислая 449
л По Б а р ф у чрезвычайно тонко растирают 50 г ртути с 20 г серы и
гяивают смесь с раствором 12 г NaOH в 60 мл воды при температуре не
• *е 45° (пополняя испаряющуюся воду), пока препарат не примет ярко-
fl>agHfI0 JI и б и х у длительное время обрабатывают многосернистым
лходит очень медленно. Красивый красный цвет получается после обработки
0£2парата КОН, последующей промывки и сушки*.
Т*По Л и б и х у длительное время обрабатывают многосернистым аммо-
JJj (NH4)2Sn «белый преципитату (NHsHgCl) * при 45—50°. Перемена цвета
прохо-
йрепарата
Свойства
Красный кристаллический порошок уд. в. 8,09, нерастворимый в воде и
спирте. При нагревании становится сначала коричневым, затем черным; при
охлаждении возвращается красный цвет. Т. возгонки 580°, т. пл. 1450° (под
давлением). На свету HgS темнеет вследствие выделения тонкораздроблен*
йой ртути.
Испытание
Чистота препарата легко определяется по полному улетучиванию препа*
рата при накаливании в платиновом тигле.
* Б. Коллоидальная сернистая ртуть
1. По Лоттермозеру3 несколько капель насыщенного раствора циа^-
новой ртути, Hg(CN)2, (яд[) прибавляют к 200 мл воды и В полученный раствор
пропускают ШБ; получается темнокоричневый золь, трудно поддающийся
Диализу.
2. Прозрачный коричневый золь получается также при прибавлении
сероводородной воды к очень разбавленному раствору Hg(CN)2.
3. Наиболее чистый золь HgS получается конденсацией паров ртути/ серы
,и воды на поверхности, охлаждаемой жидким воздухом, и плавлением
полуденного льда4.
РТУТЬ СЕРНОКИСЛАЯ ЗАКИСНАЯ (СУЛЬФАТ РТУТИ [1])
Mercurosulfat,
Schwefelsaures
Quecksilberoxydul
Hydrargyrum sulfuri- Mercurous sulfate Mercurosulfat,
cum oxydulatum Schwefelsaures
Hgi&Oi Мол. в. 497,28
Приготовление
1. По указанию Ван и но5 нагревают 1 ч. ртути с %—1 ч.
концентрированной H2SO4, пока приблизительно половина ртути не перейдет в твердую
соль. Неиспользованную ртуть сливают, а соль промывают небольшим
количеством холодной воды.
2. К раствору HgNOs прибавляют разбавленной H2SO4. Выпадающий
Мелкокристаллический осадок промывают небольшим количеством воды и
высушивают.
* Хлористый меркур-аммониЙ получается в виде белого осадка при действии NH4OH на
раствор HgCl2.
'Гаузаман приводит интересный метод получения яркокрасной HgS нагреванием
NHjjHgCl с NaaS203. Ввиду чрезмерной краткости описания работы Гаузамана метод нуждается
■ проверке и уточнении (Hausama nn, Bet. 7, 1747, (1884).
■Во. Оствальд, Краткое практическое руководство по коллоидной химии, стр. 99, Лен-
имсекюр ГОНТИ, Л. 1931.
« Р. Черннцкая и В. К а р г и н, ЖФХ 9 (4), 481,(1937).
■ Ргйр. СП. 583.
450
Ртуть
Свойства
Белая кристаллическая мука или мелкие бесцветные призмы уд. в 7- ч
мало растворимые в воде (1 : 500) и легко растворимые в горячей концентр?* '
рованной H2SO4. На свету легко принимает серую окраску. Плавится п^
красном калении.
Испытание
Препарат ч. д. а. должен оставлять не более 0,02% нелетучего остатка i
(при прокаливании под хорошей тягой 5 г препарата). Этот остаток после
растворения в НС1 уд. в. 1,12 и разбавления до 100 мл не должен давать *
красноватого окрашивания с NHfCNS (железо). Раствор препарата в раз.
бавлеиной HN03 не должен мутнеть при добавлении AgNOa (хлориды). *
РТУТЬ СЕРНОКИСЛАЯ ОКИСНАЯ (СУЛЬФАТ РТУТИ [2])
Hydrargyrum Mercuric sulfate Mercurisulfat
tulfuricum oxydatum Schwefelsaures
Hydrargyrum Quecksilberoxyd
sulfuricum neutrale
HgS04 Мол. В. 296,67
Приготовление
1. По Коксу1 чистый нейтральный препарат получается многократным
выпариванием ртути с концентрированной H2SO4, пока весь металл не
перейдет в HgS04. Соль промывают 7N раствором H2SO4 и отсасывают на тигле
Гуча до совершенной сухости.
2. 30 г чистой ртути обливают о фарфоровой чашке 50 г H2SO4 (уд. в. 1,84).
Прибавляют 1 мл HNOs (уд. в. 1,40) и нагревают на песочной бане до
прекращения выделения SO2, часто перемешивая. Затем выпаривают досуха и
помещают препарат в плотно закрывающуюся банку.
Выход 40—42 г.
ш
Свойства
Белая кристаллическая масса уд. в. 6,47 или (закристаллизованная при
избытке серной кислоты) серебристые листочки, сгруппированные в звездочки.
При нагревании сначала желтеет без разложения, затем становится
коричневой; при охлаждений окраска исчезает. При более сильном нагревании
разлагается. С небольшим количеством воды безводный препарат образует белые
кристаллы гидрата HgS04 • Н2О. Большим количеством воды HgS04
разлагается (особенно при нагревании), образуя желтую основную соль и
свободную H2S04.
Испытание (для препарата ч. д. а.)2
1. Препарат испытывают на нелетучие примеси, железо и хлориды так же,
как описано для Hg2304.
2. Раствор препарата в НС1 не должен быть мутным (соли закиси ртути).
3. Раствор 2 г препарата в 10—15 мл насыщенного раствора NaCl после
Добавления фенолфталеина титруют 0,2/V раствором NaOH до розовой
окраски. Должно пойти не более 0,5 мл NaOH, что соответствует 0,25%
H2SO4.
1 С о х, Z. anorg. Ch. 40, 165 (1904); Priip. Ch. 583
» Испытание хим. реактивов, 451.
w
Ртуть сернокислая и хлористой
Ш
РТУТЬ ХЛОРИСТАЯ
(ХЛОРИД РТУТИ [1], КАЛОМЕЛЬ)
Hydrargyrum Mercurous chloride, Mercurochlorid,
chloratum Calomel Quecksilberchlorur,
Hydrargyrum Kalomel
jnuriaticum mite
Hg^Ch Мол. в. 472,13 или HgCl Мол. в.%236,07
А. Обыкновенная хлористая ртуть
fj p иготовление
> 1. В нагретый до кипения раствор сулемы (яд!) пропускают SO2 до
превращения выпадения осадка. Реакция протекает по уравнению:
^ - 2HgCU + SO2 + 2Н20 = 2HgCl + 2HC1 + H2SO4.
-I . Выпавшую HgCl промывают водой до исчезновения в промывных водах
реакции на 30/' (от ВаСЬ не должно быть помутнения) и высушивают.
*7: 2. По Ш мидту-1 в профильтрованный раствор 15 г NaCl в 75—80 мл
;зоды вливают при помешивании раствор 50 г азотнокислой закисной ртути
iifHgNOs) в 445 мл воды, к которой добавлено 8 мл HNOs. Во избежание
образования основной соли необходимо соблюдать указанный
порядок1—приливать HgNOs к NaCl, но не наоборот.
Выпавшей HgCl дают отстояться в темном месте, декантируют, осадок
.смешивают с холодной водой, снова дают отстояться и опять декантируют.
Это повторяют до тех пор, пока проба промывных вод с AgNOa не будет
давать только небольшое помутнение. Промытый осадок отфильтровывают и
высушивают.
Свойства v
Белый порошок уд. в. 7,15, без вкуса. Нерастворима в воде, спирте, эфире
Я разбавленных кислотах. Растворяется в горячих концентрированных HNOs
I1H2SO4 с образованием солей Hg*'. Кроме того, растворяется при кипячении"в
JHC1, растворах NH4CI и щелочей, выделяя ртуть и образуя HgCta.
Растворима в бензоле и пиридине. При постепенном нагревании возгоняется при
^63,2° без предварительного разложения2. Т. пл. 543°. При продолжительном
Щ^йствии света темнеет вследствие выделения металлической ртути.
/Испытание3 (для препарата ч. д. а.)
1. Нелетучий остаток и железо. 5 г препарата, смоченные 1 мл H2SO4
(уд. в. 1,84), при прокаливании под хорошей тягой не должны оставлять
больше 1 мг остатка (0,02%). Нелетучий остаток, растворенный в НС1
уд. в. 1,12, не должен давать красного окрашивания с NH4CNS (железо).
2. Хлорная ртуть (HgCh). Взбалтывают 1 г препарата с 20 мл эфира,
фильтруют и выпаривают 10 мл фильтрата досуха. Остаток растворяют в 50 мл воды
и добавляют 1 мл 0,1 N 'AgNOa; в течение 5 мин. не должно быть опалесценции.
3. Сульфаты (504"). 4 г препарата обливают 20 мл НС1 (уд. в. 1,095); через
10 мин. фильтруют и выпаривают 10 мл фильтрата досуха на паровой бане,
добавив 0,01 г NaoCOa. Остаток обрабатывают 2 мл НС1 (уд. в. 1,12),
приливают 40 мл воды и 2 мл 10%-ного раствора ВаСЬ. Через 20 мин. не должно
быть мути мли осадка.
4. Аммонийные соли (NH*'). При нагревании 1 г препарата с 10 мл
10%-ного раствора КОН не должен выделяться NHs, обнаруживаемый по
посинению влажной красной лакмусовой бумажки, внесенной в пары жидкости.
1 Autfiihr. Lehrb. d pharm. Ch. 1, 1033.
■ Плотность пара HgCl при 375—425° отвечает формуле HgCl, но не Hg„Clt.
* Испытание хим. реактивов, 342.
452
Ртуть
Б# коллоидальная хлористая ртуть
По Г ей дену1 лл^ получения твердой коллоидальной HgCi можно м \
ствовать раствором К& на растворимые соли одновалентной ртути в ПрИс£
ствии, например яиччню го белка; при испарении полученного раствора octaefi
твердая коллоидальна* "^обой сероватый порошок, растворимый в йл
(1^0°) "'^зГани^ молочной жидкости. В с'пирте. э$ире. Р« смеси, U
золе и хлорРофопме SS растворима. Из водного раствора от соляной кисло*
золе и хлороформе шег г астворяющийся при добавлении щелочей. ты
выпадает осадок, снсовя p*^dvf . *** у
РТУТЬ ЖЛ«°РНАЯ (ХЛОРИД РТУТИ[2], СУЛЕМА)
н .„..^^ к|.1лл Mercuric chloride Mercurichlorid
/1^ Bichloride of mercury Quecksilberchlorid
ratum corrosivum Corrosive sublimate Sublimat
HgCb Мол. в. 271,52
Приготовление
Сильный яд, работсаГЬ>°ст°Р°жно! „ Л /и сг, ч
1 Сухим путем HteC^2 получается возгонкой сернокислой (HgSOO или
сернистой (HffS) ртутж с NaC1 B Реторте, нагреваемой на песочной бане.
2 20 г HgO расти'оак°т с водои в жидкую кашицу, которую постепенно,
порциями, вносят в 2^7 ? кипящей НС1 (уд. в. 1,134), разбавленной двойным
объемом воды. По раствсРРении н£° жидкость фильтруют, выпаривают до
появления кристаллическое ™**™ ***™ закристаллизоваться.
3. 100 г ртути облшв^^т 45 МЛ НС1 (Уд- в- 1»19)» слаб° нагревают и
добавляют 7 МЛ HNOs (у7Д. в* ^ ''
По окончании реак'ЦиИ нагревают еще час, после чего раствор сливают,
к оставшейся ртути снго»* приливают 45 мл НС1 и 7 ^ HNOs и повторяют
эти операции три ра<за <всего расходуется 135 мл НС1 и 21 лл HNOs).
Все порции раствора «со-е^иняют» выпаривают досуха, добавляют 10 мл НС1
и упаривают досуха на в°ДЯН0И бане' сухой остаток перемешивают с 10 мл
НС1 и 200 мл горячей вюАы- Добавляют несколько граммов HgO и кипятят;
отфильтровывают HgO и! Fe(OH)3, выпаривают раствор до объема 100 лм и
охлаждают. Выпавшие кристаллы отсасывают, промывают 10 мл ледяной воды
и высушивают при 20—ЗЮР- Выход около 128 г (94% теорет.) *.
4. Для получения HigCl* из ртутных остатков по Эрдману3 растворяют
последние в смеси техжич^ских HCl и НЫОз, выпаривают досуха и остаток
возгоняют. 10—20% поломанного возгона растворяют в горячей воде, доба;
вляют NaOH и выпавопую Н2° после промывания растирают с остальной
частью возгона.
Черный (от присутстти'я хлорокиси) порошок помещают в колбу, закрывают
часовым стеклом и напревают на песочной бане. Возгоняющаяся HgCl2
оседает в верхней части коолбЬ1 в виде шелковистых игл. По окончании возгонки
к еше горячему дну колгбь* прикасаются мокрым полотенцем и через
образовавшееся отверстие собшра*°т кристаллы при помощи гусиного пера.
5. Для получения зн'ач^тельных количеств HgCb можно рекомендовать
метод описанный Шви ц е ром4, затруднительный лишь в том отношении, что
для него требуется кварщев**я колба с четырьмя тубусами. Через один из них
проходит трубка капелыно» воронки содержащей чистую ртуть, через вто;
рой —трубка, подводящая СУХ0Й сь> чеРез третий — высокотемпературный
термометр (на 500°). Четтв«ртый тУбУс соединен широкой стеклянной трубкой
с большой эрленмейеровсскюА колбой, служащей камерой для осаждения воз-
1 Герм. пат. К1. 12 р. 1652882.
я Соли редких и цветных ;иет£лЛ0В*
« DIeMFabr!cation oharm u cJi*m-,technIlcner Pfoducte (имеется русскййперевод: Ю. Шви-
пер, яПроизводствохим.-фарм;; и т*хн°-Х1|МНЧвских препаратов", стр.55 М. Р. Х.Л. М.-Л., 1934).
Ртуть хлорная
453
люшейся HgCh. Прибор помещают под хорошую тягу и, нагрев кварцевую
Л, на песочной бане до 450—500°, пускают ток хлора; выждав немного,
'***1йвают из капельной воронки ртуть (по каплям). Встречаясь с хлором при
^кой температуре, ртуть образует HgCU, которая возгоняется и осаждается
•стенках эрленмейеровской колбы. При избытке хлора и чистой ртути
подается препарат очень высокого качества.
По указанию автора при емкости кварцевой колбы 500 мл и эрленмейе-
^1гой колбы 4—7 л аппарат может в день дать несколько килограммов
SS частой HgCl2.
Свойства
Бесцветные иглы (ромбические призмы) уд. в. 5,42, едкого металлического
«куса. Хорошо растворима в горячей воде, спирте (1:3) и эфире (1:12—14),
рачительно хуже в холодной воде. Сильно ядовита! Уже 0,2—0,4 г являются
вертельной дозой.
Т. пл. 275°, т. кип. 301°. Летуча с парами воды. Водные растворы
постепенно разлагаются под действием воздуха и света, образуя HgCl, HC1 и О*.
Таблица 429
Растворимость HgCI2 в воде
/• с
0
10
20
25
30
40
°/о HgCl,
4,1
53
6,2
6.8
7,8
8,8
Г С
50
60
70
Р0
90
100
°/о HgCl,
10,2
12.2
Н,7
19,5
27.1
25,1
Таблица 4°0
°/о HgCl,
1
2
Удельный вес водных раст в оро в HgC'2
4°
•
1,0072
l,01i8
&,'о HgCl,
3
4
«4°
1,02*6
1,0 J23
Wo HgCl,
5
>6
и20
1,0411
1,0500
Таблица 431
Удельный вес спиртовых растворов HgCl2
°/о HgCl,
0
1.22
2,38
rfao
0,81435
0,fl22<
0,8314
°/о HgCl,
4,42
8,56
12.43
а»
0,8463
0.Я789
0,9119
о/о HgCl,
15,01
1^,32
22,46
rfao
0,9425
0,°753
1.0083
454
Ртут$. Свинец
И спытание
По ОСТ НКТП 6281/266 содержание HgCU в препарате ч. д. а. Дол
быть не менее 99,5%, в препарате чистом —не менее 99,0%; наибольщИе Но
личества допустимых примесей указаны в табл. 432, Ко*
Таблица 433
Допустимые примесив HgCl2
Примеси
1. Вещества, нерастворимые в го-
3. вещества, не осаждаемые H2S .
4. Железо (Fe)
Допустимое содержание в о/0
в чистом для
анализа
0,02
0,02
0.02
0,0005
| 0,0005
в чистом
0.05.
0,04
0,04
0,0 Л
0,001
РТУТЬ ЦИАНОВАЯ (ЦИАНИД РТУТИ [2])
Mercuricyanid
Quecksilbercyanid
Hydrargyrum cyanatum Mercuric cyanide
Hg(CN)« Мол. в. 252,65
Приготовление
Все работы проводить под хорошей тягой!
1. По указанию Эрдмана1 к разбавленному раствору синильной
кислоты {осторожно! Сильнейший яд!) прибавляют желтой окиси ртути до ясно
щелочной реакции. Фильтруют и, добавив к фильтрату еще HCN до слабо
кислой реакции, выпаривают под хорошей тягой до кристаллизации.
Отделив кристаллы, добавляют к маточяому раствору еще HCN и снова
выпаривают.
2. По Ванино2 тщательно растирают 3 ч. осажденной окиси ртути
с 4 ч. берлинской лазури, постепенно прибавляя 20 ч. воды, и нагревают
смесь сначала на водяной бане, пополняя испаряющуюся воду, затем на
открытом огне, до исчезновения голубого цвета. Если посветление не наступит
после 10-минутного кипячения, то необходимо добавить еще немного HgO.
Отфильтровывают выпавшую Рез04, промывают осадок горячей водой,
присоединяют фильтрат к главной порции раствора и, добавив синильной кислоты
HCN (яд\)> выпаривают под хорошей тягой до кристаллизации.
3. Ванино3 приводит еще следующий метод. Вычисленное количество
киновари HgS г#чосят в 10%-ный раствор цианистого натрия NaCN (яд!).
Жидкость нагревают, причем осевшая вначале Hg(CN)& переходит в раствор и
вновь выкристаллизовывается при охлаждении.
4. По указанию Сийгаловского4 в профильтрованный и нагретый до
40° раствор 10 г цианистого натрия NaCN в 75 мл воды вносят (порциями
по 1—3 г) 30 г сернокислой окисной ртути HgS04. Кашицеобразную массу по
остывании обрабатывают теплым спиртом, причем Hg(CN)2 растворяется,
a Nas304 остается. Раствор выпаривают; остаток растворяют в горячей воде,
1 Хим. преп. 29.
« РгЗр. Ch. 584; Apot. Ztg. (1908).
• loc. cit.
* Соли редких и цветных металлов, стр. 97.
Ртуть циановая. Свинец
455
|^пзуют и, выпарив фильтрат до половины объема, охлаждают для кри-
действа
Белые квадратные призмы и пирамиды уд. в. 3,99 без запаха, с
неприятным металлическим вкусом. Очень ядовита! При нагревании в пробирке
кристаллы сначала раскалываются, затем плавятся и, наконец, распадаются на
ртуть и циан; часть последнего полимеризуется и остается в пробирке в виде
{роркчяево-черного порошка парациана.
растворима в воде (9,3% при 20°), в этиловом спирте (1 : 12) и метило-
до спирте (1 : 3); плохо растворима в эфире.
1Р&..П ы т а н и е
I. Нелетучие примеси. При осторожном нагревании препарат должен
совершенно улетучиваться. Разбрасывания кристаллов можяо избежать, -истирая
gK в порошок и помещая его на платиновую жесть небольшими порциями
(0,1 г). Операцию необходимо проводить под сильной тягой.
■ 2. Хлорная ртуть (HgCh). Раствор 1 г препарата в 14 мл воды не должен
вызывать покраснения лакмуса.
; 3. Хлориды (СУ). Раствор препарата, подкисленный HNOs, от прибавления
AgNOa не должен давать осадка.
СВИНЕЦ
Plumbum Lead Blei
Pb At. в. 207,21
А. Чистый свинец
Приготовление
1. Совершенно чистый свинец по Стасу может быть получен из
уксуснокислого свинца следующим путем.
Раствор уксуснокислого свинца РЬ(СНзСОО)г • ЗНгО настаивают в
свинцовом котле при 40—50° с очень тонким листовым свинцом для удаления
находящихся в жидкости примесей kg и Си. После этого раствор фильтруют и
вливают в разбавленную H2SO4. Выпавший PbS04 тщательно промывают и!
настаивают со смесью (NH^COs и NUiOII, причем PbSOa переходит в РЬСОз •
•лРЬ(ОН)г (испытание на полноту перехода: хорошо промытая проба осадка
должна полностью растворяться в разбавленной НЖ)з). Осадок промывают
до удаления SO4" [проба промывной воды после подкисления НЫОз не должна
мутнеть от прибавления Ва(МОзЬ] и часть его осторожным нагреванием в
платиновом тигле переводят в окись.
Главную часть осадка нагревают до кипения с HNOs (взятой в количестве,
недостаточном для полного растворения) и для осаждения железа к
полученному раствору добавляют отдельно приготовленную РЬО. Нагревают до
кипения, фильтруют и к фильтрату добавляют (NH^sCOs. Выпавший совершенно
чистый углекислый свинец промывают, высушивают и восстанавливают до
металла сплавлением с цианистым калием (яд!) в неглазуровакном
фарфоровом тигле. После вторичного сплавления с KCN (причем металл должен иметь
выпуклую поверхность) свинец разливают в полированные стальные формы.
2. Для получения металлического РЬ из химически чистого хлористого
свинца PbCU смешивают 3 ч. последнего с 2 ч. безводного ЫагСОз и
полученную смесь вносят в расплавленный KCN (яд\).
3. По Ванино1 для быстрого получения свинца можно исходить из
глета (РЬО). Для этого 40 г РЬО сплавляют с 50 г KCN (яд\) в закрытом
1 Ргар. СП. 603, г
456
Свинец
фарфоровом тигле на сильной газовой горелке в течение 15 мин. По охлЗ
ждении королек металла промывают водой.
4. Для получения свинца в крупных кристаллах-октаэдрах, его распла
вляют, дают частично застыть и сливают оставшийся еще жидким металл
с крупнокристаллической массы.
0. В кристаллическом виде РЬ также получается действием цинка на рас.
творы свинцовых солей.
Б. Губчатый свинец
По Бендеру1 на цинковую пластинку помещают 25-миллиметровый
слой густого теста из PbSCU и воды, покрывают другой пластинкой цинка и
все погружают в раствор NaCl. Через 9-—10 дней РЬ304 превращается в губ»
чатый металлический свинец.
Коллоидальный свинец
1. По Сведбергу2 коллоидный раствор РЬ в метиловом спирте
может быть получен искровым распылением свинцовой фольги, взвешенной в
метиловом спирте; искру получают между электродами из трудно
распыляемого материала (Fe, A1). Получение золя совершенно аналогично получению
коллоидальной сурьмы (см. стр. 503).
Приготовленный таким путем коллоидный раствор РЬ в проходящем свете
интенсивно синий, в отраженном сине-черный.
2. По Сведбергу3 очень быстро можно приготовить золь свинца
распылением, путем облучения ультрафиолетовыми лучами.
Свинец, очищенный с поверхности от пленки окислов, помещают в плоскую
фарфоровую чашку, заливают этиловым спиртом и подвергают облучению
кварцевой ртутной лампой на расстоянии нескольких саитиметров от свинца.
Через 5 мин. получается прекрасный коллоидальный раствор.
3. Для получения коллоидального РЬ, растворимого в воде, по А. Ф. Г е-
расимову и Б. М. Козыреву4 нагревают 30 мл смеси Пааля5 с 30 л*л
ледяной СНзСООН на водяной бане до удаления запаха НгЗ, после чего
добавляют по каплям при встряхивании 3 мл 25%-ного раствора РЬ(ЫОз)г.
Смесь нагревают до почернения и коагулируют добавлением ЫагСОз.
Осадок отфильтровывают, промывают 5—6 раз водой, затем снова растворяют в
воде, добавив несколько капель 20%-«ого раствора NaOH, и дают раствору
испариться при комнатной температуре. Осадок высушивают при 105°.
Свойства
Сильно блестящий, синевато-серый металл уд. в. 11,352—11,356 при 23°.
Т. пл. 327°, т. кип. 1540°. Образующиеся при кипении пары чрезвычайно
ядовиты.
Во влажном воздухе свинец теряет свой блеск, покрываясь пленкой окиси,
которая защищает остальной металл от окисления. Свинец, находящийся в
воде, при доступе воздуха и СОг постепенно выделяет крупные блестящие
кристаллы основной углекислой соли.
При плавлении свинец теряет блеск и покрывается серой пленкой, быстро
переходящей в желтую РЬО.
Губчатый свинец — темносерая пористая масса, легко окисляющаяся на
воздухе.
Испытание
Методика испытаний на примесь Ag, Bi, Fe, Zn, As, '3n, Sb может быть
заимствована из ОСТ ЦМ 36—4а ГОСТ 635—41 и ГОСТ 860—41.
1 Неорг. преп. 532.
■ Вег. 38, 3616 Ц905).
■ Вег. 42, 4276 (1909).
4 Труды Казанского хим.-технол. института им. Бутлерова 1, 119 (1934),
в О приготовлении смеси Пааля си. сноску Ь на стр. 831.
Свинец азотнокислый
457
СВИНЕЦ АЗОТНОКИСЛЫЙ (НИТРАТ СВИНЦА [2])
I plumbum nitricum Lead nitrate Bleinitrat
Pb(N03)j Мол. в. 331,23
Приготовление
1. Растворяют чистый металлический свинец в горячей концентрированной
tfNOa- При охлаждении почти вся соль осаждается в виде кристаллической
Куки. Ее отсасывают на платиновом конусе (или на воронке с тампоном *нз
стеклянной ваты), промывают небольшим количеством ледяной воды и сушат
при 125—130°.
2. Глет РЬО или углекислый свинец РЬСОз растворяют при нагревании в
11йстой разбавленной HNOs, раствор фильтруют и осторожно выпаривают
досуха. |
Свойства ;
Правильные бесцветные; или белые кристаллы уд. в. 4,5, довольно легко
•растворимые в воде и не растворимые в спирте. Плохо растворим в
метиловом спирте (1,35:100). При нагревании выше 200° распадается на РЬО, NO»
И Ог.
Таблица 433
Растворимость Pb(NOy)2
/ °с
0
10
20
30
40
о/о Pb (NOa)t
26,7
30,8
34,3
37,8
41,0
/вс
50
60
70
80
100
о; о Pb(NOe).
44,0
46,8
49,4
51,8
56,0
Таблица 434
Удельный вес водных растворов Pb (N03)2
о/о Pb (NCV.
1
2
4
6
8
10
-?
1,0074 '
1,0163
1,0344
1,0529
1,0720
1,0918
о/0 Pb (N03>,
12
14
16
18
20
<
1,1123
1,1336
1,1557
1,1789
1,2030
о/о Pb (NO,).
22
24
26
28
*30
d4
1,2277
1,2529
1,2783
1,3037
1,3289
Испытание
Наибольшие количества примесей, допустимых по ОСТ НКТП 6763/370,
указаны в табл. 435.
458
Свинец
Допустимые примеси в Pb(N03)2
Таблица 435
Примеси
1. Нерастворимые в воде вещества
3. Железо (Fe)
4. Медь (Си)
5. Щелочные и щелочноземельные
металлы (в виде сульфатов) . . .
Допустимое содержание в о-0
в чистом для
анализа
0,005
0,002
0f001
0,001
0,05
в чистом
0,02
0,005
0,005
0,003
0,10
Приготовлен" и е
СВИНЦА АМАЛЬГАМА
Lead amalgam
Bleiamalgam
1. Расплавляют свинец и осторожно смешивают его с вычисленным
количеством предварительно подогретой ртути. Образование амальгамы
происходит с поглощением тепла.
Можно также нагревать очищенный от пленки свинец со ртутью до
полного растворения.
2. М. Холт и А. Грей1 рекомендуют готовить амальгаму свинца
следующим путем:
40 г гранулированного свинца обливают концентрированной НС1,
перемешивают в течение 10 мин., затем промывают водой и сушат несколько минут
при 100°. Прилив к свинцу 60 г чистой ртути, нагревают смесь 1 час при
перемешивании, дают охладиться и сливают жидкость. Амальгаму промывают
водой и высушивают фильтровальной бумагой.
3. В случае необходимости амальгамировать свинец лишь с поверхности
лист свинца, хорошо очищенный от пленки окиси, натирают ртутью до
приобретения всей поверхностью характерного блеска.
Свойства
Амальгама свинца может быть твердой
температуре в зависимости от состава2.
или жидкой при обыкновенной
Температуры
плавления амальгамы свинца
сти от сост ава (П у ш ин)
Таблица 436
зависимо-
Атомн.
°/о Hg
2,6
6,3
11
16,6
Т. пл.
318,5
305,25
288,0
267,5
Атомн.
°,о Hg
27,0
35.8
40,6
50,0
Т. пл.
232,0
204,0
189,5
162,5
Атомн.
°/о Hg
54,7
63,5
68,4
75,0
Т. пл.
149,5
129,5
120,25
110,5
Атомн.
°/о Hg
83,0
86,4
89,7
98,2
Т. пл*
101
96,75
90,75
ниже 23°
1 М. Z. Н о 11 a. A. G. О г а у, Ind. Eng. Chem. Anal. Ed. 12, 144 (1940).
а О растворимости Pb в ртути в зависимости от температуры см. Н. Б.
J. phy§. Ch. 39, 655 (1936).
Thompson*
Свинца амальгама, гидрат окиси и двуокись 459
•и Твердая амальгама — хрупкая, белая, зернистая, кристаллическая масса,
Щ воздухе покрывающаяся синевато-желтой побежалостью. По Ф э ю и
Жорзу содержит соединение Pb'Hg наряду со сплавом РЬ и Hg.
| СВИНЦА ГИДРАТ ОКИСИ (ГИДРООКИСЬ СВИНЦА [2])
■;;.. plumbum oxydatum Lead hydroxide Bleihydroxyd
hydricum
;i РЪОлМгО
■риготовление
f. К раствору азотнокислого [РЬ(ЫОз)г] или уксуснокислого [РЬ(СНзСОО)г]
]$винца прибавляют избыток NHtOH; выпадающий белый осадок тщательно
Промывают и высушивают. Состяч -осадка различен в зависимости от условий
Получения (2РЬО • НЮ, ЗРЬО • Н20).
С в о й с т в а
£■■ Белый, микрокристаллический порошок со слабо щелочной реакцией; на
"воздухе поглощает СОг, переходя в углекислую соль. При 100° постоянен,
При 130° начинается обезвоживание, заканчивающееся при 145°.
СВИНЦА ДВУОКИСЬ (СВИНЦА ПЕРЕКИСЬ, ОКИСЬ СВИНЦА[4])
Plumbum peroxydatum Lead peroxide Bleiperoxyd1
Lead dioxide Bleidioxyd
РЬОа Мол. в. 239,21
Приготовление
1. Нагревают сурик РЬз04 со слабой НЫОз; черный осадок промывают
разбавленной НЫОз, затем водой и высушивают. Из 4 ч. РглСЬ получается
1,5 ч. РЬ02.
2. К раствору 100 г .уксуснокислого свинца [РЬ(СНзСОО)2 • ЗНгО]
прибавляют крепкий раствор 30 г ЫаяСОз и пропускают сильную струю хлора
до тех пор, пока масса не приобретет темнокоричневого цвета. Подводящая
хлор трубка должна доходить почти до дна сосуда. По окончании реакции
колбу нагревают \Уч часа на водяной бане, затем выливают все содержимое
колбы в стакан. Дав отстояться, декантируют и нагревают осадок с
разбавленной (24—25%-ной) НЫОз для удаления примеси РЬСОз; наконец, осадок
промывают несколько раз декантацией, отсасывают на воронке Бюхнера и
промывают водой до удаления ОХ (подкисленная HNOs проба промывных
вод от добавления AgNOe не должна мутнеть). Выход 90—96%
теоретического.
3. Некоторые авторы {Овенбек, Рейнозо) предлагают окислять
РЬО или РЬО • *НгО в щелочном растворе при помощи КМп04 или KsFe(CN)e.
4. По методу, разработанному Д. Н. Финкельштейном1, сначала
готовят раствор окислителя, замешивая 750 г хлорной извести с 2250 мл воды.
Тщательно раздавливают ,комочки извести, дают отстояться и фильтруют через
плотный фильтр, промывая осадок 500 мл воды. Через несколько часов
раствор полезно еще раз профильтровать.
К прозрачному раствору хлорной извести, помещенному в большую
фарфоровую чашку, приливают при перемешивании теплый (~50°) прозрачный
раствор 325 г чистого РЬ(СНзСОО)г • ЗНгО в 500 мл воды. Выпадающий
желтый осадок быстро приобретает коричневый цвет:
РЬ(СНзСОО)* + CaOClf -f- H2O = РЬОг + CaCl* + 2СШСООН.
1 Улучшенный метод Бёттгера (BSttger, Po^g. 49, 4ЭЗ) и Ризенфельда
(ft i e s в n f e 1 d, Неорг. практ. 225).
460
Свинец
Смесь нагревают при 100—110° и через 20—30 мин. испытывают на пол;'
ноту осаждения РЬ*'; если имеется непрореагировавший РЬ' ', то добавляй»
еще 250—500 мл раствора хлорной извести и продолжают кипячение em*
2 часа. Осадок РЬО» промывают декантацией горячей водой (при хороши
перемешивании) до удаления СГ, затем заливают осадок 500—750 мл HNo[
(УД. в. 1,08), оставляют его стоять на 2 часа, после чего снова промываю*
2—3 раза водой. Осадок отсасывают на воронке Бюхнера, помещают полу,
ченную пасту на пергаментную бумагу слоем в 4—5 мм и сушат при IoqV
Выход около 200 г препарата ч. д. а.
По «методике ИРЕА раствор 250 г ацетата свинца в 375 мл воды прили.
вают к раствору хлорной извести (250 г в 1250 мл воды) и добавляют к этой
смеси еще 60 г NaOH, растворенного в 100 мл воды. Дальше поступают как
указано выше.
5. По указанию Л. А. Ушакова для получения РЬОз в лабораторном
«масштабе размешивают в воде 100 г РЬО и 60 г СаО и в полученную сус^
пензию пропускают хлор до получения темнокоричневого осадка. Так как
РЬО быстро осаждается на дно и выходит из сферы реакции, рекомендуется
жидкость энергично перемешивать.
Осадок промывают горячей водой до исчезновения реакции на СГ в про-
мывных водах и высушивают.
6. Для препарата, «гранулированного по Пр«глю»л полученную по одному
из вышеописанных способов кашицеобразную массу намазывают на
пергаментную бумагу слоем в несколько миллиметров и высушивают. При этом слой
растрескивается на отдельные кусочки — «гранули», которые отсеиваются от
примеси порошкообразного препарата.
Свойства
Темнокоричневый порошок уд. в. 8,93.' Почти не растворим в воде и
разбавленных кислотах (кроме щавелевой). При нагревании выше 280°
распадается на сурик (РЬзОО и Ог. Сильный окислитель. Поглощает SO2,
переходя в белый PbS04. При взаимодействии с горячей концентрированной
H2SO4 выделяет кислород, с НС1 выделяет хлор.
Н2З вызывает самораскаливание влажной РЬОг. Кристаллическая РЬО*
имеет уд. в. 9,36.
Испытание
По ОСТ НКТП 7847/751 содержание РЬОг в препарате по активному
кислороду должно быть не менее 90%; наибольшие количества допустимых
примесей указаны в табл. 437.
Таблица 437
Допустимые примеси в РЬ02
Примеся
1. Нерастворимые в кислоте веще-
2. Хлорилы (СГ)
3. Сульфаты (SO/7)
5. Металлы, осаждаемые H2S (кроме РЬ)
6. Вещества, не осаждаемые H2S . . .
7. Азот общий (N)
8. Карбонаты (СОв")
Допустимое содержание в о/0
В ЧИСТОМ Д1Я
анализа
0,1
0,02
0,05
0,0003
0,01
1 0,5
0,01
0,03
в чистом
0,3
0,1
од к
0,0С05
0,03
1.0
0,02
0,06
Свинец йодистый. Свинца окись
461
СВИНЕЦ ЙОДИСТЫЙ (ИОДИД СВИНЦА [2])
Plumbum jodatum Lead iodide Bleijodid
PbJ2 Мол. в. 461,05
Приготовление
К 10%-ному раствору KJ, содержащему незначительное количество
СНяСООН, прибавляют вычисленное количество уксуснокислого свинца
[РЬ(СНзСОО)2-ЗНгО]. Перекристаллизацией полученного желтого осадка из
ледяной СНзСООН получают особенно хорошо кристаллизующийся препарат.
Свойства
Желтый порошок; в кристаллическом виде — блестящие золотистые ли*
сточки уд. о. 6,10. При нагревании окрашивается сначала в желто-красный
цвет, затем в крипично-красный и, наконец, в коричнево-черный; при
охлаждении, однако, возвращается первоначальный желтый цвет. Т. пл. 393°,
т. кил. около 900°. В .воде растворим плохо (0,17% при 20°, 0,39% при
100°); значительно растворяется в концентрированных растворах KJ и NaJ,
образуя соединения типа Me[PbJs] К При разбавлении таких растворов
водой PbJ2 выпадает снова.
СВИНЦА ОКИСЬ (ОКИСЬ СВИНЦА [2], ГЛЕТ, МАССИКОТ)
Plumbum oxydatum Lea.d monooxide Bleioxyd
Bleiglatte
PbO Мол. в. 223,21
А. Желтая модификация
Приготовление
1. Растворяют гидрат окиси свинца РЬО«*НгО в расплавленном КОН
или же, по Г е й т е р у, растворяют 1 ч. РЬО • *НгО в нагретом почти до
кипения растворе 7 ч. КОН в 14 ч, воды. При медленном охлаждении
выделяются желтые кристаллы РЬО.
2. По методу ИРЕА2 для получения чистого РЬО 555 г технического
глета помещают в фарфоровую чашку, заливают 100 мл воды и 1,1 л HNOs
(уд. в. 1,05) и нагревают до растворения. К прозрачному горячему раствору
добавляют 5 мл концентрированной НС1 для осаждения Ag* и оставляют
омесь на ночь. Отстоявшийся раствор отфильтровывают, нагревают до 80°
и осаждают РЬ(ОН)* добавлением 2,5 л прозрачного 10%-ного раствора
NaOH. При кипячении белый осадок РЬ(ОН)2 переходит в желто-зеленый
PbO. Смеси дают отстояться, отсасывают РЬО на воронке Бюхнера,
промывают водой до удаления NOs' (проба с дифениламином), сушат и
прокаливают 30—50 мин. при 400—500°. Выход около 500 г (90% теорет.).
3. Д. Н. Финкельштейн предлагает следующий метод, ^позволяющий
получать препарат высокой чистоты. 400 г РЬ(СНзСОО)г« ЗШО растворяют
в 1,2 л воды, добавляют 1 мл НС1 (уд. в. 1,19) и через час фильтруют в
большую фарфоровую чашку.
1 Me —одновалентный металл.
■ Реак. и пред. 297.
462
Свинец
Нагрев раствор до 60°, приливают к нему тонкой струей, слегка помещ»
вая прозрачный раствор 120 г КОН (или 90 г NaOH) в 1,1 л воды, кипятят '
-*до перехода РЬ(ОН)2 в хорошо отстаивающийся РЬО и промывают последний "
горячей водой до удаления С1'. После сушки препарат помещают в фарфоро.
вую чашку, прокаливают в течение 1 часа при 600—650° и дают остыть на
воздухе. Выход около 200 г.
4. Прокаливают углекислый свинец при 650° в токе азота.
Б. Красная модификация
Приготовление
1. Растворяют 1 ч. РЬО^хНЮ в 5 ч. расплавленного КОН и сплав очень к
медленно охлаждают.
2. Нагревают РЬО • jtfbO продолжительное время при 150°.
3. Растворяют избыток РЬО-хШО в кипящем 33%-яюм растворе NaOH;
жидкость кипятят до тех пор, пока образовавшаяся вначале желтая РЬО
не перейдет в красную.
4. Прокаливают углекислый свинец в токе азота при 450°.
б. Прокаливают РЬ(гЮз)г при 400—450°.
Свойства
Желтые кристаллы ромбической системы уд. в. 9,28—9,38 или красные
Тетрагональные кристаллы уд. в. 9,5 (цвет, в зависимости от метода
получения, колеблется от зеленовато-желтого до гранатово-красного). Выше 587°
красная «модификация переходит в желтую1. Т. пл. 880, т. кип. 1470°. Окись
свинца почти не растворима в воде (0,012 г/л; водная вытяжка имеет
слабощелочную реакцию), растворима в горячих растворах КОН и NaOH с
образованием плюмбитов, растворима также в HNOs и СНзСООН; соляной
кислотой переводится в РЬС1г.
На воздухе РЬО медленно поглощает СО*. Расплавленная РЬО сильно
действует на фарфор.
Испытание (Мерк)
1. Примеси, нерастворимые в СНзСООН. 2 г препарата обливают 5 мл
воды и 10 мл разбавленной СНзСООН, причем не должно наблюдаться
газовыделения. Нерастворившийся остаток после промывания и высушивания
при 100° не должен быть больше 0,005 г.
2. Углекислые соли (СО&"). 5 г препарата при нагревании до плавления не
должны терять .в весе больше 0,005 г.
3. Хлор (СУ). Раствор 1 г препарата в 5 мл НЫОз (уд. в. 1,150—1,152) и
20 мл .воды от прибавления 'AgNOs может давать лишь слабую опал ее-
ценцию.
4. Азотнокислые и азотистокислые соли (NO* и NO*'). Раствор 1 г
препарата в 5 мл разбавленной СНзСООН (уд. в. 1,040—1,042) и 5 мл воды
смешивают с 0,2 мл раствора индиго (1 : 1000) и 10 мл концентрированной
H2SO4 (уд. в. 1,84). Синяя, окраска раствора не должна исчезать.
5. Щелочные и щелочноземельные металлы, В раствор 1 г препарата
в 10 мл разбавленной СНзСООН (уд. в. 1,040—1,042) и 50 мл воды
пропускают H2S до полного осаждения свинца и фильтруют. Фильтрат после
выпаривания и прокаливания не должен оставлять больше 0,003 г остатка.
6. Медь, железо и алюминий (Си' ' ', Fe' * \ АГ **). К раствору 2 г
препарата в 10 мл НЫОз и 5 мл воды прибавляют 3 мл концентрированной H2SO4
и отфильтровывают выпавший PbS04. К фильтрату добавляют избыток NHUOH,
причем не должно появляться ни синей окраски, ни осадка.
I Koblschfitter u. Roseti, Ber. 56, 285 (1923),
Свинец сернистый и сернокислый , 463
СВИНЕЦ СЕРНИСТЫЙ (СУЛЬФИД СВИНЦА [2])
..plumbum sulfuratum Lead sulfide Bleisulfid
| PbS Мол. в. 239,27
А. Аморфный сернистый свинец
приготовление
Аморфный PbS получают пропусканием H2S в раствор РЬ(СНзСОО)г или
pb(NOsV> выпадающий осадок промывают и высушивают.
•; Для получения совершенно чистого препарата по Родуэллу
осажденный PbS после промывания водой суспендируют в воде, обрабатывают серо*
водородом и снова промывают. Промытый препарат высушивают при 100°,
^умеренно прокаливают' в струе H*S и, наконец, в токе водорода.
Свойства
; Черный, с коричневатым оттенком порошок уд. в. 7,1. Т. пл. 1110°; уже
-йри 860° начинает частично улетучиваться. Растворим при нагревании в
разбавленной HNOa; почти не растворим в воде (8,67. \0г9 г/л при 25°, Lp*
s= 7,00- 10^°Н
Б. Кристаллический сернистый свинец
Приготовление
По Ванино2 для получения кристаллического PbS прокаливают тща*
тельно растертую смесь 1 ч. сухого осажденного PbS, 6 ч. сухого СаСОз и
6 ч. серы при светлокрасном калении. После охлаждения выщелачивают
массу водой.
Свойства
Свинцово-серая кристаллическая масса, состоящая из кубиков и октаэдров,
УД. в. 7,5.
В. Коллоидальный сернистый свинец
'Приготовление
По Винсингеру в очень разбавленный раствор РЬ(СНзСОО)г (к
которому прибавлено, во избежание образования основных солей», необходимое
количество СНзСООН) пропускают несколько пузырьков сероводорода. При
этом жидкость превращается в темнокоричневый, прозрачный, очень
постоянный коллоидный раствор. Насыщение жидкости сероводородом, а также
кипячение вызывают быструю коагуляцию.
СВИНЕЦ СЕРНОКИСЛЫЙ (СУЛЬФАТ СВИНЦА [2];
Plumbum sulfuricum Lead sulfate Bleisulfat
PbS04 Мол. в. 303,27
Приготовление
PbS04 получается в виде тонкого порошка осаждением из раствора какой-
либо соли свинца, при помощи разбавленной H2SO4.
В виде кристаллического порошка получается сплавлением PbSOi с PbCl»
и выщелачиванием охлажденного сплава горячей водой.
* S. P. R a v i t z, J. phys. Ch. 40, 61 (1936).
■ Prap. Ch. 612.
464
Свинец
Свойства
Белый порошок уд. в. 6,(6, плавящийся около 1100° и застывающий
охлаждении в кристаллическую массу. Очень трудно растворим в вПри
(0,0046% при 18°). Растворим в щелочах (образуя плюмбиты) и в аммиак
ных растворах уксуснокислого и виннокислого аммония (давая двойные соли
Испытание
По техническим условия* НКХП 127—40 содержание РЬ304 в препа-
ратах ч. д. а. и чистом должно быть не менее 99%; наибольшие количества*
допустимых примесей указаны в табл. 438.
Таблица 438
Допустимые примеси в PbS04
Примеси
1. Растворимые в воде вещества . .
2. Нерастворимые в COCOON H4
4. Хлориды (СК) ...
6. Потеря при прокаливании ....
t
Допустимое содержание в °[0
в чистом для
анализа
ОД
0,05
0.002
0,002
не нормиг.
0,3 |
в чистом
0,25
0,1
0,006
0,005
>уется
0,8
Примечание. В свя;ш с применением PbS04 в тигельной пробе для
определения золота и серебра, на этикетке препарата ч. д. а. должно быть
указано содержание серебру
СВИНЕЦ УГЛЕКИСЛЫЙ (КАРБОНАТ СВИНЦА[2])
Lead carbonate Bleicarbonat
РЪСО, Мол. в. 267,22
Plumbum
carbonicum
А. Нормальный углекислый свинец
Приготовление
Вливают раствор какой-либо нейтральной свинцовой соли [РЬ(ЫОз).\
РЬ(СНзСОО)г] в избыток холодного раствора №гСОз или К2СОз;
выпадающий осадок тщательно промывают.
Свойства
Белый порошок уд. в. 6,4—6,6; при кипячений с водой медленно теряет
СОг, переходя в основную соль. Очень плохо растворим в воде. При 300°
полностью распадается на РЬО и СОг.
И спытание
иЛ??6?™™^ количества примесей, допустимых по техническим условиям
НКХП 128—40, указаны в табл. 439,
Свинец углекислый* 463
Таблица 439
Допустимые примеси в РЬСОд
ж. —-*■■ .
Примеси
1. Нерастворимые в HN03 веще-
2. Нерастворимые в СНьСООН ве-
3. Щелочные и щелочноземельные
Л металлы (в виде сульфатов) . , .
4. Остаток после прокаливания » .
Допустимое содержание в о/0
й чистом для
анализа
0,05
0,08
0,4
83
в чистом
0,1
0,15
0,8
83
Б. Основной углекислый свинец
Приблизительно отвечает формуле 2РЬСОз • РЬ(ОН)г.
Приготовление
По Лёве1 в раствор 50 г уксуснокислого свинца РЬ(СНзСОО)г• ЗНЮ
43,5 г Pb(NOab в 250—300 мл воды добавляют постепенно 28 г твердого
^аНСОз (или раствор 8 г ЫагСОз в 8 мл воды и 9—10 г NaHCOs). Выпав-
йй осадок углекислого свинца промывают водой. Одновременно готовят рас-
ор основного уксуснокислого свинца, для чего к раствору 25 г РЬ(СНзСОО)г
125—150 мл воды добавляют 15 г тонко истертого глета РЬО. Полученный
раствор добавляют к приготовленному раньше РЬСОз. Жидкость хорошо
размешивают, дают осесть основной соли, отфильтровывают последнюю, тща-
льно промывают и отжимают.
Свойства
Белый, плотный, аморфный, тяжелый порошок, нерастворимый в воде и
спирте. Растворяется в разбавленных HNOs и СНзСООН, а также в
растворах КОН и NaOH.
При нагревании выше 180° переходит в РЬО.
Испытание (для препарата ч. д. а.)1
1. Потеря при прокаливании должна быть не больше 15% .
2. Раствор 5 г препарата в смеси 5 мл Н1ЧОз (уд. в. 1,4) и 46 мл воды
Должен быть прозрачным и не содержать осадка. После пропускания в этот
раствор H2S до полного осаждения РЬ*' его фильтруют и 20 мл фильтрата
выпаривают досуха; остаток прокаливают; вес остатка не должен превышать
2 мг (щелочные и щелочноземельные металлы, цинк).
3. Растворяют 2 г препарата в горячей смеси 10 мл 36%-ной СНзСООН
и 40 мл воды, раствор фильтруют, остаток промывают горячей водой и
прокаливают; вес его не должен быть больше 0,5 мг (силикат свинца).
* Ргйр. Си. 613;'герм. пат» 42307.
* Испытание хим. реактивов, 353.
466 Свинец
СВИНЕЦ УКСУСНОКИСЛЫЙ (АЦЕТАТ СВИНЦА. СВИНЦОВЫЙ rAv
САХАР-САТУРН) ^AXApf
Plumbum Lead acetate, Diacetate Bleiacetat
aceticum lead, Salt of Saturn, Essigsaures Blei
Sal Saturni Sugar of lead Bleizuckcr
РЬ(СНзСОО>2 • ЗН2О Мол. в. 379,35
Приготовление
1. Растворяют РЬО в горячей 50%-ной СНзСООН до насыщения. Раствор
фильтруют, добавляют немного СН2СООН, выпаривают до уд. в. 1,4 и
оставляют для кристаллизации. Выпавшую соль отделяют от маточного раствора
и сушат между листами фильтровальной бумаги {ядовит).
2. Для очистки технического ацетата свинца (свинцового сахара)
растворяют при нагревании 100 г соли в 100 мл 1%-ной СНзСООН, фильтруют
горячий раствор и дают фильтрату охладиться до комнатной температуры.
После отделения кристаллов маточный раствор выпаривают до половины
объема и снова кристаллизуют.
Если сырье содержит примеси железа, то раствор кипятят с РЬОг, затем
добавляют РЬО, снова * кипятят, отфильтровывают осадок, содержащий
Fe(OH)s, и, подкислив раствор СНзСООН, выпаривают до кристаллизации.
Свойства
Большие прозрачные моноклинические кристаллы сладковатого вкуса
уд. в. 2,49, быстро выветривающиеся на воздухе и покрывающиеся слоем
РЬСОз. В вакууме над H2SO4 теряет кристаллизационную воду уже при 40°.
При 75° плавится в кристаллизационной воде, при дальнейшем нагревании
переходит в белый порошок с т. пл. 280°. Легко растворим в воде и
глицерине (1:5); нерастворим в эфире, трудно растворим в спирте (1:28).
Ацетат свинца ядовит.
Таблица 440
Растворимость РЬ (СН3СОО)2 в воде
г°С
0
10
20
25
Pb(CH8COO)i
16,45
22,65
30,68
35,26 |
t°C
30
40
50
РЬ (СН.СОО),
41,02
53,79
68,85
Таблица 441
Удельный вес водных растворов РЬ(СН3СОО)2
РЬ (СН3СОО),
1
2
4
6
10
48
1,0061
1,0137
1,0290
1,0446
1,0605
1,0768
°/о
РЬ(СНвСОО)э
12
14
16
18
20
22
<
1,0936
1,1109
1,1283
1,1473
1,1663
1,1860
°/о*
РЬ (СН,СОО),
24
26
28
30
35
40
-?
1,2063
1,2273
1,2489
1,2711
1,3304
1,3994
'*№№■:
Свинец уксуснокислый и хлористый
46?
Испытание
^ По ГОСТ 1027—41 содержание Pb(CHsCOO)2 • ЗН20 должно быть в пре-
'щрате ч. д- а- не менее 99% и в препарате чистом не менее 98%;
наибольшие количества допустимых примесей указаны в табл. 442.
Таблица 442
Допустимые примеси в РЬ(СН3СОО)2 • ЗН20
Приме ев
Допустимое содержание в о/0
в чи:том для
анализа
U Нерастворимые вещества • • •
2. Хлориды (С1') •
3. Меди (Си)
4. Железо (Fe)
5. Щелочные и щелочноземельные металлы
(в виде сульфатов)
0,005
Ю.0005
©,0005
0,001
0,02
0,01
0,002
0,001
0,002
0,05
СВИНЕЦ ХЛОРИСТЫЙ (ХЛОРИД СВИНЦА[2])
Plumbum
chloratum
Lead chloride
РЬСЬ Мол. в. 278,12
Bleichlorid
Приготовление 1
При действии на раствор сол,и свинца раствором НС1 или ее солей
выпадает РЬСЬ в виде белого (Микрокристаллического осадка. При
перекристаллизации соли из горячей воды получают РЬСЬ в хорошо образованных
игольчатых кристаллах.
Препарат промывают небольшими порциями ледяной воды.
Свойства
Мелкие ромбические иглы или листочки с шелковистым блеском уд. в. 5,91.
Z пл. 500°, кип.~ 900°.
Расплавленный РЬСЬ при охлаждении затвердевает в белую роговидную
массу.
В воде, особенно холодной, растворим плохо.
Присутствие в раствор© НС1 увеличивает растворимость; насыщенный
раствор РЬСЬ в концентрированной НС1 при разбавлении выделяет некоторую
часть соли в твердом виде.
Нерастворим в спирте и эфире.
1 О получении коллоидального хлористого свинца см. Van der Velde, Ch. Ztg. 17,1908 (1893).
О получении чистейшего PbCU (для определения атомных весов) см. В. М. Пермяков.
ЖОХ 9, 5, 381 (1939).
468
Свинец
Таблица 443
Растворимость РЬС12 вводе
/вс
0
15
25
45
о/о PbCIg
0,64
0,91
1,05
1,55
t°c
65
80
100
°/о PbCli
2,08
2,54.
3,20
Таблица 444
Растворимость РЬС12 в соляной кислоте
Bee 0/0.
НС1
0
5,3
9,1
15,25
18
23,95
0Q
0,74
0,273
0,118
0,234
0,45
1,064
•/о I
20°
1,06
0,291
0,138
0,458
0,583
1,233
i
^bCIt при
40°
1,45
0,43
0,31
0,67
0,94
1,60
55°
1,73
0.61
0,52
0,893
0,143
1,935
86°
2,36
1,10
1,07
1,65
1,92
2,76
1
Испытание
По ОСТ НКТП 3265 содержание РЬСЬ» в препарате ч. д. а. должно быть
не менее 99,5%, в препарате чистом —не менее 98,0%; наибольшие
количества допустимых примесей указаны в табл. 445.
Таблица 445
Допустимые примеси в РЬС!2
1 ■ ф
Примеси
4. Щелочные и щелочноземельные металлы
Допустимое содержание в о/о
в чистом для
анализа
0,05
0,02
0,01
0,01
0,0005
в чистом
0,1
0,05
0,02
0,02
0,001
Свинец хромовокислый
469
СВИНЕЦ ХРОМОВОКИСЛЫЙ (ХРОМАТ СВИНЦА [2])
plumbum chromicum Lead chromate Bleichromat
Chrom yellow
РЬСгО* Мол. в. 323,22
приготовление
1. Смешивают водные растворы 33 г РЬ(СНзСОО)г и 20 г К2СЮ4 (или
20 г К2СГ2О7); выпавший желтый осадок тщательно промывают водой и
высушивают, при 110°.
2. По Манросу для получения кристаллического PbCrO/j сплавляют
foCrOi н РЬС12.
Сильно нагрев печь, охлаждают ее до слабокрасного каления и помещают
туда гессенский тигель, содержащий вышеуказанные соли (РЬС1« в избытке
против теорет.); печь закрывают возможно плотнее и дают охладиться.
Остывшая масса внизу окрашена окисью хрома в зеленый цвет, выше —
красноватая, пронизанная красными прозрачными кристаллами РЬСЮ*
Чтобы отделить их, массу кипятят с водой для удаления КС1 и РЬОг и
отмучивают от CraOs1.
Свойства
Осажденный PbCrOi — яркий лимонно-желтый порошок; кристаллический
РЬСЮ4 — красивые, прозрачные, красные или темнокоричневые кристаллы
уд. в. 6,12. При нагревании хромат становится красньгм, однако при
охлаждении возвращается желтая окраска. При 844° плавится, застывая при
охлаждении в темнокоричневую массу. При более сильном нагревании отдает
кислород. Нерастворим в воде, растворяется в HNOs и щелочах.
Испытание
По ОСТ НКТП 7108/445 препарат квалификации ч. д. а. должен
содержать не менее 98% РЬСЮ4; наибольшие количества допустимых примесей
указаны в табл. 446.
Таблица 446
Допустимые примеси в РЬСЮ4
Примеси
1. Растворимые в воде вещества .
Допустимое
содержание в -о/о
0,3
0,006
0,002
СВИНЕЦ ХРОМОВОКИСЛЫЙ ОСНОВНОЙ
Plumbum chromicum Lead chromate basic Chromrot
basicum
РЬСг04.РЬО Мол. в. 546,43
Приготовление
1. По Либиху и Велеру2 осажденный хромовокислый свинец (РЬСЮО
вносят небольшими порциями в слабо накаленный KNOs, пока большая часть
1 О получении золен РбСг04, положительно и отрицательно заряженных, см. С. 11,3952 (1939Х
1 Fogg. 23, 580.
470
Селен
последнего не разложится. Происходит вспенивание, причем сплав Им
черный цвет (цвет PbCrCU • РЬО в горячем состоянии). Снимают ТИр т
с огня, через несколько минут сливают еще жидкие KNOs и К2СЮ4 и быСтЛЬ
промывают остаток в тигле водой (при длительном стоянии порошка ПР°
водой он становится светлокрасным). Полученный при соблюдении всех пеп?
численных условий препарат имеет чистый киноварно-красный цвет (nPD *
сильном накаливании KNOs получают коричневый продукт). т
2. По В а н и н о для получения PbCrCU • РЬО мокрым путем кипятят Пр0
должительное время средний хромовокислый свинец (РЬСгО^ с раствором
К2СЮ4; последний при этом переходит в КгСгЮт.
Свойства
Полученный по методу 1 препарат представляет собой порошок яркого
красного цвета, состоящий из тонких блестящих кристаллов. Не растворим
в воде; растворим в разбавленной НЫОз с образованием красной жидкости*
С СНзСООЦ образует РЬСЮ4 и раствор РЬ(СНзСОО)*.
Испытание
Проводится аналогично РЬСг04 (ом. стр. 469).
СЕЛЕН
Selenum Selenium Selen
Se At. в. 78,96
Приготовление
Для получения селена из селеновой грязи сернокислотных заводов можно
итти следующими путями.
1. По Вёл ер у грязь замешивают в жидкую кашицу с разбавленной
H2SO4 (1:1); смесь кипятят, прибавляют постепенно HNOs или КСЮз до
исчезновения красноватого оттенка, разбавляют водой, фильтруют и,
прибавив % объемна дымящей НС1 или соответственное количество NaCl (для
восстановления селеновой кислоты в селенистую), выпаривают до гА объема.
По охлаждении жидкость сливают с выделившихся NaCl и K2SO4, насыщают
SO2 и отфильтровывают выпавший селен (для выделения небольшого
количества Se, оставшегося в растворе, последний еще раз кипятят с НС1 и
насыщают SO2). Полученный Se для очистки от Fe, Pb и Си перегоняют. Для
удаления Hg продукт растворяют © царской водке, выпаривают для
удаления HNOs и пересыщают содой. Получившийся раствор селеновохислого
натрия (NatSeO^ выпаривают, остаток прокаливают, затем кипятят с НС1
и осаждают Se пропусканием SO2.
2. Ра-ботая с грязью из Лукавица, Бе р цел и ус предложил следующий
способ. Грязь растворяют в горячем КОН, раствор фильтруют и оставляют
стоять на воздухе при 22°, причем на дно выпадает селен (около 11%). Для
выделения остатков Se (около 0,25%) из жидкости последнюю кипятят с
кусочками серы; при этом методе некоторая часть серы остается примешанной
к селену.
3. По Розе пропускают медленный ток сухого хлора над высушенной
грязью, нагреваемой в фарфоровой трубке. При этом масса не должна
плавиться. Образующиеся летучие хлористые соединения серы и селена
улавливают в приемнике водой; полученную жидкость отфильтровывают от серы
(содержащей некоторое количество Se) и осаждают селен раствором
сернистого калия,
Селен
471
4, Для получения Se из селеновых отбросов или из других селенсодержа-
й)/материалов по Эрдману1 сплавляют .эти вещества в гессенском тигле
* смесью .равных частей Na2C03 и KNO». Массу охлаждают и вышел ачи-
°^ют кипящей водой. Вытяжку выпаривают до небольшого объема, сильно
Влдкисляют концентрированной НС1 и кипятят в колбе с обратным холодиль-
иком до прекращения выделения хлора. Оставшуюся жидкость переливают
большую колбу, разбавляют большим количеством горячей воды и снова
кипятят, 'прибавляя при этом по каплям раствор NaHSOs до прекращения
образования красного объемистого осадка Зе, быстро превращающегося
й черноватую массу. Последнюю собирают на фильтр и высушивают.
Очистка
1. Для очистки технического селена, содержащего обычно серу и
незначительные количества теллура, по Гюго2 обрабатывают селен разбавленной
ЙДООз, выпаривают досуха, возгоняют получившийся SeCfe, снова растворяют
его в воде, осаждают следы H2SO4 при помощи баритовой воды и выделяют
селен в виде коричневого порошка пропусканием $Ог в профильтрованный
в подкисленный НС1 раствор.
2. Очистку технического селена можно также провести следующим
способом, предложенным Дивер сом и Шимозе. Обрабатывая Se крепкой
Н2ЗО4, переводят его в раствор; последний разбавляют водой и насыщают SO2,
причем селен выпадает. Его отфильтровывают, тщательно промывают и-сушат
на воздухе. Полученный таким путем Se свободен от Те и> содержит лишь
незначительные количества S,
Аллотропические видоизменения селена3
Приготовление
1. Аморфный красный порошкообразный Se. Получается восстановлением
селенистой кислоты при помощи SO2, Zn, Fe, SnCta РЮв и др. на холоду
или осаждением водой раствора Se в концентрированной H2SO4.
2. Аморфный стекловидный Se. При медленном охлаждении
расплавленного селена последний остается жидким значительно ниже точки плавления
и, наконец, при 50° в условиях равномерного охлаждения застывает в
аморфную ' массу.
3. Кристаллический моноклинический Se. Кипятят несколько граммов
красного аморфного селена с 1 л CS2 в течение 2 час. в колбе, снабженной
обратным холодильником (осторожно! огнеопасно!). Полученную светлооран-
жевую с зеленоватым оттенком жидкость подвергают медленному испарению
в стакане, закрытом несколькими слоями фильтровальной бумаги и
обвязанном бечевкой. Получаются крупные хорошие кристаллы наряду с
мелкокристаллической массой.
4. Серый «металлический» Se. Расплавленный селен охлаждают до 180°
и держат некоторое время при этой температуре. Вскоре масса становится
кристаллической, причем этот переход сопровождается повышением
температуры.
Можно также нагревать аморфный селен в течение нескольких часов при
150—200°.
Свойства
Красный аморфный Se — чрезвычайно тонкий порошок, при нагревании
становящийся клейким и. около 50° темнеющий и размягчающийся. Уд. в. 4,259
при 20°. После охлаждения — хрупкая и ломкая масса. Истертый аморфный
Se несколько (растворим в CS2 (0,016% при 0°, 0,1% при 46,6°).
-— i
1 Хим. преп. 55.
1 Ann. Chim. Phys. (7), 21, 34 (1900); Prap. Ch. 108,
1 РгЗр. Ch. Ю9,
472
Селен. Сера
Стекловидный аморфный Se — хрупкая, масса уд. в. 4,28 при 20°, л^*
царапающаяся и измельчающаяся. Обладает поверхностью с металлически0
блеском « раковистым изломом. При измельчении превращается в темн^1
красный порошок. Не имеет определенной т. пл.; выше 100° разжижается
при охлаждении тянется ъ упругие .нити; при 250° вполне жидок, при. 5оо
вполне тверд. Почти не растворим «в CS2.
Кристаллический моноклинический Se существует в двух модификациях• "
красные листочки уд. в. ^4,47 и тёмнокрасные прозрачные короткие призмы!
Почти не растворим в СЗг.
«Металлический» Se, получаемый очень медленным охлаждением стекло- i
видного селена, имеет свинцово-серую зернистую поверхность и тонкозерни- 1
стый матовый излом. Уд. в. 4,8 К Т. пл. 220,2°, т. кип. 688Р. При истирании
получается серый порошок. Не растворим в CS?. ^ m •
Испытание
По техническим условиям НКХП 75—40 селен металлический
(реактив) должен представлять собой порошок или пластинки серо-черного цвета;
содержание селена в препарате должно быть не менее 99,5%; наибольшие
количества допустимых примесей указаны в табл, 447.
Таблица 447
Допустимые примеси в Se
Примеси
1. Сера (S)
2. Нелетучий остаток
3. Влага
4. Теллур
5. Азот .......
Допустимое сод ер»
жание в °/0
0,2
0,2
0,2
|не нормируются,
но содержание
указывается
на этикетке
Коллоидальный селен
Приготовление
1. По Шульце при смешении растворов H2SO3 и НгЕеОз в
количествах, вычисленных по уравнению:
Se02 + 2S02 + 2H2O = Sd + 2Н?304,
цвет жидкости быстро переходит в желтый, затем в оранжевый и, наконец, —
в кроваво-красный. Если растворы не очень разбавлены, образуется темно-
красный осадок; жидкость становится прозрачной, оставаясь тёмнокрасной.
Осадок представляет собой растворимый селен; вскоре после получения он
способен полностью растворяться в воде, образуя прозрачную красную
жидкость.
При хранении (даже под водой) постепенно становится нерастворимым.
Подвергая жидкость диализу, получают оаствор, свободный от всех примесей.
» Об уд. в. жидкого Se при 228-348° см. С. I, 4616 (1937).
Серй
473
2 По Гутбиру для получения очень прочного коллоидного раствора
жно итти следующим путем. К раствору 1 г очищенного SeOs в 500 мл
иловатой дестиллированной воды прибавляют по каплям раствор гидразин-
Тддрата (1 :2000). Обычно тотчас же появляется желтое окрашивание; при
Sjjge сильном нагревании окраска переходит в тёмнокрасную. Для
совершенной очистки раствор подвергают диализу.
3. По Мей ер у1 растворяют истертый селен в нескольких миллилитрах
гйдразин-гидрата до насыщения, быстро наступающего при энергичном
встряхивании; образуется густой сиропообразный раствор, окрашенный в темный
Прибавляя несколько капель этого раствора при быстром размешива-
НЙИ к нескольким литрам воды, получают интенсивно красный, чрезвычайно
устойчивый раствор.
- 4. К 100 мл теплого ((50°) 0,05%-ного раствора ЗеОг добавляют 2 мл
0,6%-ного раствора аскорбиновой (или изоаскорбиновой) кислоты. Образуется
•красный слабомутный золь (Б »р и н т ц и н г е р)2.
5. Наиболее простой метод предложен А. Эльдером и П. Бур к ар-
дом8.
К раствору красного Se в сероуглероде добавляют различные количества
ледяной СНзСООН. Полученные золи почти бесцветны в проходящем свете;
в отраженном свете окраска их варьирует от красной и розовой до
желтоватой в зависимости от концентрации. Золи устойчивы в течение 3 дней.
Таким методом были получены золи с концентрациями Se 0,00179—
0,447 г/Л и СЗг 0,2—50 об. %.
СЕРА
Sulfur Sulfur Schwefel
S At. в. 32,06
А. Очищенная сера
Приготовление
Возогнанную серу измельчают и просеивают. 10 ч. истертой серы
смешивают с 7 ч. воды и- 1 ч. ЫНЮН и оставляют смесь на час при частом
взбалтывании. Затем серу тщательно промывают водой, сушат при температуре
не выше 30° и истирают в порошок.
Свойствам испытание
Сухой, желтый порошок без запаха и вкуса, почти полностью растворяю*
щийся при кипячении с раствором NaOH (минеральные примеси); влажную
лакмусовую бумажку не должен окрашивать в красный цвет (свободная
кислота).
Для испытания на содержание As смешивают 1 г очищенной серы с 20 мл
нагретого до * 3(5—40° ЫШОН и дают смеси стоять час, изредка встряхивая.
Фильтруют; фильтрат ни от подкислеыия НС1, ни от добавления НС1 и
сероводородной воды не должен окрашиваться в желтый цвет или выделять
осадок 4.
Б. Чистейшая сера
Приготовление
1. Для получения совершенно чистой серы Треф о л л, Брирли и
Аллеи5 предлагают следующий «метод: возможно чистую серу расплавляют,
* Т. М е у е г, Вег. 46, 3089 (1913).
* Н. Brintzinger. Koll. Z. 78, № 1, 22 (1937).
* А„ L. Е 1 d e г а. Р. N. В и г к а г d, J. phys. Ch. 41, 622 (1937).
4 Komm. 7. Deut. Arzneib, II, 127.
* T h г e f a 11, Brearley, Allen, Proc. Roy. Soc. 56, 32 (1894).
474
Сера
процеживают через стеклянную вату и , платиновую сетку и для освобожл
ния от пыли дважды] ^перегоняют, лучше всего ib вакууме. Де*
Препарат, «полученный таким способом, не имеет запаха, улетучиваете
без остатка и при быстром охлаждении ^сильно нагретого сплава затвердевав
в кристаллическую массу. Получающийся при этом кристаллический Пр0!
дукт растворим в СЗг без остатка.
2. Для получения чистой серы можно применить также следующий метод i
Около 200 г серы измельчают в кусочки величиной в горошину и сущат'
несколько часов в сушильном шкафу. Полученную серу перегоняют Из
реторты без тубуса, применяя в качестве приемника стакан с дестиллировац.
ной водой, накрытый асбестовой пластинкой, в которой проделано отверстие
для горла реторты (последнее не должно касаться воды).
Перегнанную серу разбивают на кусочки, высушивают, расплавЛягот. и,
дав остыть, истирают в порошок. Взвесив серу, помещают ее в
полулитровую склянку, содержащую 1,25%-ный раствор NaOH, из расчета 8 ч.
раствора на 10 ч. серы. Склянку закупоривают и, поместив в аппарат для
механического встряхивания, взбалтывают в течение 12 час. Серу, уже не
содержащую м!ышьяк?а, отфильтровывают, промывают до нейтральной реакции
и сушат при температуре не выше 30°; полученный препарат испытывают на
растворимость гори нагревании в NaOH и CS2.
Если при этом получаются мутные растворы, то необходимо провести
•дополнительную очистку, для чего серу с равным по весу количеством
СЗг нагревают на водяной бане в колбе с обратным холодильником до
прекращения растворения (осторожно! CS2 и его пары крайне огнеопасны) -.
Раствор фильтруют горячим через складчатый фильтр и фарфоровую чашку;
по охлаждении большая часть серы выкристаллизовывается.
Маточный раствор сливают, а серу раскладывают на
фильтровальной бумаге и сушат на воздухе в защищенном от огня месте, до
исчезновения запаха С$2. От маточного раствора отгоняют на водяной бане CS2,
^л^ оставшуюся серу расплавляют и выливают на
стеклянную или фарфоровую пластинку.
В. Серный цвет
Приготовление
Рис. 70. Прибор для получе
ния серного цвета
Для получения серного цвета можно поступать
следующим образом. Горло реторты (рис. 70) при
помощи асбестового картона укрепляется в боковом
тубусе большой .колбы так, чтобы оно было
направлено кверху, иначе расплавленная сера может
стечь «в колбу и последняя лопнет. Наполнив
реторту до половины серой, начинают ее осторожно
и медленно нагревать. Пламя должно охватывать
весь баллон реторты во избежание значительной
•конденсации серы в верхней части. Пары серы,
проходя в колбу конденсируются и оседают на стенках в виде мельчайшего
порошка серного цвета.
Г. Серное молоко (осажденная сера)
Приготовление
1. По Векенродеру3 в колбе при кипячении растворяют 2 ч. серного
цвета и 1 ч. прокаленной извести (погашенной 3 ч. воды) в 13 ч. воды.
К профильтрованному красно-бурому раствору маленькими порциями доба-
1 Deut. Aizneibuch, V, 502.
* Вблизи прибора не должно быть огня. Водяную баню следует нагреть в другой комнате.
9 W а с k e n г о d е г, Br. Arch. 26, 180; Ргар. С1ь 72,
Сера
475
йют ПРЙ помешивании разбавленную НС1 так, чтобы CaS разложился не
оЛй°сТЬЮ' Т* е* до слаб° блочной реакции. Осадок S собирают на фильтр и
«ошывают, так как в противном случае из находящихся в растворе тиосуль-
!ьатов может выделиться желтая, грубо-дисперсная сера, засоряющая серное
молоко.
2. Можно привести еще следующий метод1, дающий хорошие "результаты,
.об ч. свежепрокаленной извести, помещенной в железный котел, гасят 75 ч.
*%ь и к полученной кашице добавляют 25 ч. очищенной серы и затем 250 ч.
вОДЫ. Смесь кипятят в течение часа при непрерывном перемешивании (изредка
пополняя испаряющуюся воду), после чего сливают жидкость через
полотняный мешок. Остаток кипятят еще полчаса с 160 ч. воды; выливают
снова в мешок и промывают осадок горячей водой. Фильтрату дают отстояться
,в течение нескольких дней, затем фильтруют и разбавляют водой до объема
$00 ч. К полученному раствору, помещенному в объемистый сосуд,
постепенно прибавляют при перемешивании 33 ч. чистой НС1, разбавленной 67 ч.
►ды (т. е. до тех пор, пока жидкость над отстоявшимся осадком серы не
*дет уже иметь желтого цвета и щелочной реакции). После осаждения серы
озрачную жидкость сливают, а осадок промывают сначала дестиллирован-
й водой декантацией, затем на колоторке, пока промывные воды не пере-
ганут показывать щелочную реакцию и давать муть с AgNOs. Тогда оса-
рк отжимают и сушат при температуре не выше 30°.
Если осажденная сера содержит железо, что узнается по серо-зеленой
' аске, то ее .очищают, для чего, слив маточный раствор, промывают осадок
кантацией и выщелачивают в течение нескольких часов смесью 3 ч. чистой
1 и 12 ч. воды. Дальше поступают, как указано выше.
йства
Осажденная S представляет собой желтый или сероватый аморфный, чрез-
чайно тонко раздробленный порошок уд. в. 1,92, почти полностью
растворимый в СЗг, без вкуса и запаха. Хорошо высушенный препарат при
хранении в закрытой банке почти не изменяется; лишь после длительного
хранения, особенно в присутствии влаги, приобретает кислую реакцию и слабый
|апах.
Д. Модификации серы
ёиготовление
а) Ромбическая сера
1. При кристаллизации серы из CS2 или других растворителей при
невысокой температуре выделяются острые ромбические кристаллы. Например,
растворяют серу в равном количестве СЗг, нагревая в колбе с обратным
холодильником на водяной бане (осторожно! CS2 крайне огнеопасен!), и
полученный, раствор очень медленно охлаждают.
I 2. Моноклиническая сера в течение нескольких дней переходит в
ромбическую форму. Переход совершается быстрее на свету.
3. По А р е н с у насыщают пиридин сероводородом. При стоянии раствора
на воздухе в течение нескольких дней сера выделяется в прекрасных
октаэдрах с острыми ребрами и блестящими гранями.
б) Моноклиническая сера
1. Расплавляют серу в глиняном тигле и дают несколько остыть. После того
как на поверхности образуется достаточно толстая корка, ее пробивают
гвоздем и выливают серу, оставшуюся жидкой. При этом стенки тигля ,
оказываются покрытыми тонкими косыми столбиками моноклинической серы. .
2. По Д е в и л л ю растворяют серу в кипящем бензоле и очень медленно
охлаждают. Между 80 и 75° выпадает сера в моноклинических кристаллах
(при более низкой температуре выделяется смесь ромбической и моноклини,-
ческой серы).
i Komm. ?. Deut. Arzneibuqh II, 427; Ргар. СЬ. 72,
476
Сера
3. По Г. и В. Бильц1 расплавляют серу в пробирке до начинают^
потемнения (140—150°) и, опустив в расплавленную массу термометр, пог
жают пробирку в стакан с концентрированной H2S04, нагретой д0 '80—с££ '
Дав сере остыть до 110°, прекращают переохлаждение, погружая в с *
стеклянную палочку; при этом сера затвердевает в желтые, цвета воска ПпРУ
зрачные моноклинические иглы; температура застывшей массы повышается
на несколько градусов.
4. Нагревают несколько прозрачных кристалликов ромбической сеоьт
в пробирке, погруженной в кипящий концентрированный раствор NaCl (\^ .
112°), в течение 2—3 час, изредка пополняя испаряющуюся воду; постепенно i
кристаллики мутнеют и, наконец, полностью переходят в светложелтую^ массу '
кристаллики мутнеют
моноклинической серы,
массу
в) Пластическая сера
Нагретую почти до кипения серу выливают в холодную воду. Получаю-
щаяся резинообразная масса в течение нескольких дней теряет свою
пластичность и становится хрупкой.
Свойства
Ромбическая сера — крупные, ломкие, ромбические кристаллы уд. в. 2,07.
Т. пл. 112,8°. Растворима в сероуглероде {CS2), хлористой сере (S2CI2),
углеводородах, эфирных маслах и жидком SO2. Не растворима в воде; в спирте
и эфире почти не растворима.
Моноклиническая сера — бледные, голубовато-желтые кристаллы уд. в. 1,96.
В течение нескольких дней переходит с выделением тепла в ромбическую
серу. Точка перехода при обычном давлении 95,8°. Т. пл. 118,95°,
т. кип,г 444,55°. Растворима в CS* и толуоле.
Таблица 448
Растворимость серы в органических
растворителях
Растворитель
Бензол ....
Толуол ....
*
г с
26
71
23
Частей серы
на 100 частей
растворителя
0,965
4,377
1,479
,
Растворимость се
t° с |
-11
- 8
0
+ 15
100 г CSf
растворяют
* серы
16,54
17,75
28,99
41,65
Растворитель
Эфир этиловый
Хлороформ . .
Фенол ....
Анилин . . .
t° с
23,5
22
174
130
Частей серы
на 100 частей
растворителя
0,972
1,208
16,35
85,96
Таблица 449
>ры в сероуглерод
Г С
22
38
48
55
106 г CSg
е
растворяют
г серы
1 46,05
94,57
146,21
18
1,34
J H. u. W. В i 11 z, Uebungsbeispiele (1920), 28.
'-° J* |а!п* .S при Различном атмосферном давлении см. J. А. В е а 11 i e, M. Benedict
*, В. Е. В 1 а i 1 d e 11, рГОс. Аш. Acad. Art» S. 71, 32, (1937); C.l, 4073 (1937),
Cepd
477
Таблица 450
удельный вес растворов серы в сероуглероде
*&
ш
W
4,9
2,1
' 2,4.
2,6
2,9
3,1
3,4
3,6
3,9
4,1
4.4
4,6
4,8
5.0
5,3
,5,6
.5.8
rfis
°I0S
ф*
1,271
1,272
1,273
1,274
1,275
1,276
1,277
1,278
1579
1,280
1.281
1,282 !
1,283
1,284
1,285
1,2*6
1,287
1,288
1,289
1,290
1,291
1,292
1,293
1,294
1,295
6,0
6,3*
6,5
6,7
7,0
7,2
7,5
7,8
8,0
8,2
8,5
8,7
8,9
9,2
9,4 |
9,7 1
9,9
10,2
10,4
10,6
10,9
1 "'*
11,3
11,6
11,8
1,296
1,297
1,298
1,299
1,300
1,301
1,302
1,303
1,304
1,305
1,306
1,307
1,с08
1,309
1,310
1,311
1,312
1,313
1,314
1,315
1,316
1,317
1,318
1,319
1,320
o.'oS
12,1
12,3
12,6
12,8
13,1
13,3
13,5
13,8
14,0
14,2
14,5
14,7
15.0
15,2
15,4
15,6
15,9
16,1
16,4
16,6
16.9
17,1
17,4
17,6
17,9
фЬ
1,321
1,322
1,323
1,324
1,325
1,326
1,327
1,328
1,329
1,330
1,331
1,332
1,3*3
1,334
1,335
1,336
1,337
1,338
1,339
1,340
1,341
1,342
1,343
1,344
1,345
°,'о5
18,1
18,4
18,6
18,9
19,0
19,3
19,6
19,9
?0,1
20.4
20,6
21,0
21,2
21,5
21,8
22,1
22,3
22,7
23,0
23,2
23,6
24,0
24,3
24,8
23,1
d\%
°;<>s
1,346
1,347
1,348
1,349
1,350
1,Ь51
1,352
1,353
1,354
1,355
1,356
1,357
1,358
1,359
1,360
1,361
1,362
1,363
1,364
1,365
1,365
1,367
1,368
1,369
1,370
25,6
26,0
26,5
26,9
27,4
28,1
28,5
29,0
29,7
30,2
30,8
31,4
31,9
32,6
33,2
33,8
34,5
35,2
36,1
36,7
37,2
фЬ
1,371
1,372
1,373
1,374
1,375
1,376
1,377
1,378
1,37!
1,31
1,381
1,382
1,383
1,384
1,385
1,386
1.3Я7
1,388
1,389
1,390
1,391
Приготовление
Е. Коллоидальная сера
1. По Л о бри де Брюйну1 светложелтый коллоидный раствор серы
получают следующим путем: смешивают 10 мл 20%-ного раствора желатина
с 5 мл 0.4JV раствора ЫагБгОз и одновременно 10 мл 20%-ного раствора
желатина с 5 мл НС1. Оба раствора, имеющие температуру 25—30°,
сливают и быстро охлаждают, выливая в охлажденную льдом чашку.
Приготовленный по этому методу прозрачный раствор устойчив в течение 15 мин.
2. По М. Раффо2 в- охлажденные льдом 70 г H2SO4 (уд. в.- L84) при-
ливают по каплям при размешивании раствор 50 г гипосульфита (ЫагЗгОз*
• 5НЮ) в 30 мл воды. К полученной густой желтоватой массе прибавляют
30—50 мл воды и удаляют образующийся SO2 нагреванием на водяной бане
в течение 10—'15 мин. Получается' прозрачная серовато-желтая жидкость,
которую после охлаждения отфильтровывают от хлопьев серы через
стеклянную вату или полотно. Фильтрат представляет собой концентрированное
коллоидное серное молоко, содержащее значительные количества Na2SOi.
Золь становится прозрачным при нагревании и мутнеет при охлаждении.
Разбавленные растворы довольно постоянны. ~~
*LobrydeBruyn, Rec. trav. chim. Paye-Bas 19, 236 (1900); С. It,
■ Т. Сведберг .Образование коллоидов", НХТИ, 1927*
> (1900)
478
Сера. Серебро
Свойства
Коллоидный раствор с частицами, не различимыми даже в ультрамн
скоп, представляет собой светложелтую жидкость. По мере увелич^0*»
размеров частиц раствор мутнеет и наконец становится совсем непр0зТИя
ным. Одновременно наблюдается и изменение окраски. Раствор иринимар"
цвет от красноватого до красновато-коричневого, когда величина части
доходит до границы ультрамикроскопической видимости. При еще большем
увеличении частиц раствор принимает оттенок от пурпурового до синего
однако с укрупнением частиц чистота окраски уменьшается, так что почти
нельзя указать характерного цвета. При добавлении желатины в качестве
предохранительного агента раствор принимает слабую, чисто голубую
окраску. и
СЕРА БРОМИСТАЯ (ОДНОБРОМИСТАЯ)
Sulfur bromatum Sulfur monobromide Schwefelbromtir
S'Br* Мол. в. 223,95
Приготовление
Для получения очень чистого препарата по Руффу и Винтерфельду1
в стеклянной трубке запаивают 24 г чистой (перекристаллизованной из CSa)
серы и открытую пробирку, содержащую 19 мл чистого брома. После
запаивания трубку -помещают в защитный кожух {изготовленный из куска
водопроводной трубы диаметром 25—30 мм) и напревают в течение двух часов на
водяной бане. Получается гранатово-красная жидкость, которую для очистки
перегоняют при высоком вакууме (при 0,22 мм, т. кип. 57—58°). Дестилля-
ция должна производиться в абсолютно сухой аппаратуре и полученную S2Br*
необходимо тотчас же поместить в плотно закрывающиеся склянки или
запаянные трубки, так как препарат чрезвычайно чувствителен к влаге воздуха.
Свойства
Красная маслообразная тяжелая жидкость уд. в. 2,64, не омачивающая
стекло (подобно ртути). Т. пл. — 46°, т. кип. 54° при давлении 0,18 мм; 58°
при 0,22 мм\ 200—210° при нормальном давлении. По запаху напоминает
БгСЬ. Растворяет серу. Водой тотчас разлагается, причем выпадает S.
СЕРА ХЛОРИСТАЯ (ОДНОХЛОРИСТАЯ)
Sulfur chloratum Sulfur monochloride Schwefelchlorur
S2CI2 Мол. в. 135,03
Приготовление
1. По В амин о2 расплавляют в реторте с тубусом серу и, поворачивая
реторту, дают сере расплыться по стенкам, после чего охлаждают. Затем
устанавливают реторту так, чтобы горло ее было направлено вертикально
вниз в подставленную склянку и впускают через тубус струю хлора,
высушенного пропусканием через H2SO4.
Образующаяся S2CI2 стекает из реторты в склянку; реакцию прекращают
прежде, чем будет израсходована вся взятая сера. Полученный сырой про-
.дукт содержит серу и несколько раз очищается перегонкой, пока почти весь
препарат не будет перегоняться при 138—139°.
2. По Эрдману3 в реторте с тубусом нагревают до 125—130° 300 г
серы. К расплавленной сере подводят струю сухого хлора. Трубка, подво-
1 R u f f, Winterfeld, Ber. 36, 2438 (1903).
■ PraPk Ch. 97.
8 Хим. преп. 49*
Сера бромистая и хлористая. Серебро
479
яЯ газ, должна оканчиваться около поверхности серы. Образующийся
A^vKT быстР° изгоняется и собирается в охлаждаемом приемнике; реак-
№ не доводят до израсходования всей серы.
•аИКгТля очистки препарат перегоняют с дефлегматором, собирая фракцию,
яерсХОДЯЫУ10 около 138°. w
,СИЙсТва
Темножелтая маслянистая жидкость уд. в. 1,68 при 0° с отвратительным
удушливым запахом, сильно дымящаяся на воздухе. Т. пл. — 80% т. кип.
Пары S2CI2 Раздражают глаза, вызывая слезы; вкус —»кислый,
горький. Растворяет серу (до 67%) и иод. Смешивается во всех отношениях
с бромом и CS2. Водой тотчас разлагается с образованием НС1, S и H*SOa.
При насыщении хлором при 25° образует четыреххлористую серу SCU —
светлокоричневую жидкость, затвердевающую при — 30°.
При насыщении хлором около 0° образуется «двухлористая сера»,
которая по Р у ф ф у представляет смесь S2CI2 и SCU.
СЕРЕБРО
Argentum Silver Silber
At. в*. 107,880
Приготовление
А. Чистое серебро
а) Из сплавов, содержащих медь
По С т а с у сплав растворяют в разбавленной кипящей HNOs, раствор
Выпаривают досуха и оставшуюся соляную массу расплавляют. По охлаждении
срлав растворяют в избытке сильно (разбавленного ЫНЮН и раствор
оставляют на двое суток. Затем жидкость фильтруют через двойной фильтр
В' разбавляют водой с расчетом, чтобы содержание (Ag не превышало
1%. Прежде чем приступить к восстановлению полученного раствора при
НОмощи сернистокислого аммония (NH^SOs, необходимо точно установить
рОтребное количество этого реактива. Для этой цели берут определенный
Объем раствора (МНфБОз, нагревают до кипения и устанавливают количество
ведносеребряного раствора, обесцвечивающегося от этого количества соли.
Ватем весь серебряный раствор обрабатывают вычисленным количеством
(аммиачного раствора (NH^SOs и смесь оставляют на 48 час. в плотно
закрытом стекляннохм сосуде. При этом */з kg выделяется в виде сероватого,
часто сильно блестящего кристаллического «дождя». Осадок декантируют и
рсаждают остальное серебро нагреванием раствора до 50—70°. После
промывания водой, содержащей NH4OH, осажденное серебро нагревают несколько
|Лней с NH4OH и, наконец, тщательно промывают водой.
б) Из сплавов, содержащих АиУ Си, Pb, Sn, As, In и др.
1. По Керлю2 сплав растворяют в разбавленной НЫОз, раствор
разбавляют дестиллированной водой и оставляют его спокойно стоять, причем
осаждаются Au, S11O2, БЬгОз и др. После отстаивания фильтруют через
двойной или тройной фильтр в объемистый стакан и осаждают соляной кислотой
(не NaCl!), взятой в незначительном избытке. Жидкость с осадком
нагревают, причем посторонние металлы растворяются в образовавшейся царской1
1 По другим данным т. кип. 137,1°.
■ В. К е г 1 а. М ц 9 р г a 11, 7, 1634 (1900).
480
Серебро
водке (при употреблении NaCl в осадке вместе с AgCl могут соярпм,
РЬСЬ, BiOCl и SbOCl). идеРЖатьСя
Осевшее AgCl отфильтровывают и повторно кипятят с разбавленной Нг
затем промывают горячей дестиллированной водой декантацией до исчезн '
ния кислой реакции <и до прекращения реакции «а Си" в промывных воВе*
[K4Fe(CN)e не должен давать красно-бурого осадка]. Полученное АеП
восстанавливают, как указано ниже {см. в). г^'
2. Чистейший металл, применяемый О. Генигшмидтом и Р. ЩЛее1
при определении атомных весов» получается из рафинированного серебря
Последнее переводят в AgNOs, пятикратно осаждают концентрированной'
HNOa из насыщенного водного раствора и восстанавливают муравьинохислым (
аммонием (HCOONH4) до металла. Порошок kg тщательно промываю^, высу- <
шивают и сплавляют в лодочке из СаО. Полученные крупные капли служат '
анодом при последующем электролизе, проводимом при 1,34 в.
Кристаллический катодный осадок сплавляется в токе Нг в лодочках из СаО. Корольки
промывают разбавленной НЫОз, затем водой и сушат в вакууме при 400°1
в) Из хлористого серебра
1. Ванино2 приводит следующий способ. Смешивают чистое AgCl с водой
в фарфоровой чашке, прибавляют NaOH, нагревают до кипения и время от
времени добавляют кусочки виноградного сахара. При этом следует избегать
избытка последнего; жидкость должна все время оставаться щелочной.
Металлическое серебро осаждается на дно в виде серой зернистой массы.
Конец восстановления определяют по полному растворению в разбавленной
HNOs тщательно промытой пробы серебра. Тогда массу промывают в чашке
декантацией до исчезновения щелочной реакции, фильтруют, снова
промывают, высушивают при 100° и, если нужно, сплавляют.
2. По Ван,и но3 смешивают AgCl с разбавленным NaOH и добавляют
формалин. Через короткое время (быстрее при нагревании на водяной бане)
выделяется металлическое серебро в виде черного, рыхлого порошка,
принимающего при растирании металлический блеск.
3. Некоторое видоизменение этого способа рекомендовано А. В. Р а к о в*
схим и С. И. Добр ж и не км м 4.
В цилиндрический сосуд с приводной мешалкой помещают 90 г 40%-ного
формалина и 100 г AgCl в виде жидкого теста; пускают мешалку в ход и
постепенно, порциями добавляют 'раствор 60 г NaOH в 150 мл воды.
Выпавшее Ag промывается сначала слабой H2SO4, затем водой, наконец, раствором
NH4OH и снова водой.
По указанию авторов описанный способ дает значительно лучшие резуль*
таты, чем другие.
4. По Мульдеру и1 Варрентрапу смешивают 6 ч. влажного AgCl
с 3 ч. сухого №гСОз; смесь высушивают и растирают с 1 ч. KNOs. В то же
время накаливают фарфоровый тигель, помещенный в гессенский тигель и
окруженный тонким белым песком; в раскаленный тигель вносят серебряной
ложкой приготовленную смесь, избегая переливания через край вследствие
вспенивания; все же из-за энергичной реакции часть вещества разбрыз*
гйвается.
Когда плавление массы будет происходить спокойно, ее перемеши*
вают глиняной палочкой, причем Ag опускается на дно; после некоторого
охлаждения выливают содержимое тигля в глиняные формы. Чтобы
избежать одновременного высыпания в воду песка, его покрывают слоем
плавленной буры, образующей твердую корку. Сплавленный металл кипятят с
разбавленной H2SO4. Слитки, полученные выливанием в формы, должны быть
очищены щеткой.
10. H3ni*echmidt u. fc. Schlee, 2. anofg. Ch. 221, 134 (1934); см. также О. Н 3*
ftigschmidtu. Horowitz. loc. cit 56, 5 (1914); В. М. Пермяков, ЖОХ. 8, М 18,
1881 (19&8).
» РгВр. Ch. 521.
» loc cit.
* И». ИРЕА, 1. 200 (1923).
Щ:-
Серебро
481
^ Вели не требуется получение абсолютно чистого Ag, то плавление можно
ааэодить в гессенском тигле, прикрытом стеклом, внутренняя сторона
которого покрыта бурой или натерта мелом или каолин ом.
Р 5. Сплавленное AgCl обливают очень разбавленной НС] и восстанавливают
„истым металлическим железом или цинком. Слив жидкость, удаляют
избыток Fe или ^п и кипятят осадок серебра с разбавленной НС1.
6. Мор предлагает следующий метод восстановления железом или
цинком. AgC1 обливают в стакане разбавленной (1:60) H2SO4. Кусок цинка,
К которому прикрепляется серебряная или платиновая проволока,
завязывают в животный пузырь таким образом, чтобы часть проволоки осталась
сяаружи, и когружают в AgCl. Восстановление постепенно распространяется
ох проволоки на всю массу AgCl.
7. Смесь 180 г AgCl, 80 г №*СОз и 70 г угля сплавляют в шамотном
тигле при 950—10000 в течение 4—6 час1
г) Из азотнокислого серебра
i ; По Тананаеву2 чистое серебро с высокой реакционной способностью
^можно получить электролизом нагретого до 40° 1%-ного раствора AgNOs,
;-содержащего на 1 л 50 мл HN0.3 (уд. в. 1,4). Электролиз ведут при силе
тока 0,5—0,7 а и напряжении 4,5 в.
f. Выделившееся Ag промывают водой и высушивают в 'эксикаторе над
"РЮ5 до постоянного веса.
д) Из серебряных остатков (лабораторных, фотографических и др.)*
у- 1. Остатки кипятят с гранулированным цинком в присутствии НС1.
Восстановившееся серебро отделяют от избытка Zn, промывают декантацией и,
растворив в НГМОз, осаждают соляной кислотой в виде AgCl. Последнее
восстанавливают одним из вышеуказанных способов (ом. в).
2. Выпаривают остатки досуха и сухую массу кипятят с
концентрированной НС1 и КСЮз до прекращения выделения хлора. Осадку дают осесть,
тщательно промывают декантацией и полученное AgCl восстанавливают, как
указано выше (см. в).
3. Для выделения Ag из фотографических остатков {фиксажные ванны)
добавляют NH4OH до появления запаха NH3 и действуют небольшим избытком
(NH4)2S.
После отстаивания жидкость сливают, осадок отсасывают,
промывают горячей водой, высушивают н прокаливают при 950°, добавив 1—2 г
безводной буры. Для удаления буры гранули металла кипятят с водой.4.
Свойства
Мягкий, очень тягучий белый металл уд. в. 10,49, кристаллизующийся
в кубах и октаэдрах. Т. пл. 960,5°, т. кип. 2152°. Совершенно постоянен
в отношении воды; на воздухе медленно покрывается тонким слоем Ag2S.
Расплавленное при доступе воздуха серебро поглощает .кислород, выделяя
его при ""охлаждении.
HF, HC1 и H2SO4 при обыкновенной температуре не действуют на серебро;
горячая концентрированная Н2ЗО4 растворяет ею с образованием AgsSO^
выделяя SOa. Расплавленные щелочи и селитра на серебро не действуют.
Лучшим растворителем является HNOa, переводящая Ag в AgNO.3.
Испытание
Раствор 2 г металла в возможно малом количестве HN03 (уд. в. 1,2)
Должен быть бесцветным и не содержать нерастворившегося остатка (Sn, Sb).
1 Мухина, Анализ черных металлов, стр. 120, ОЗоронгиз, М.-Л. (1939).
2 Chem. Ztg. 32, 385 (1908).
3 См. также В. Я. Тартаковский Завод, лаб. 9, 112 (1940).
* 4 См. также P. D I с k e n s, St. u. Eiseu, 58, № 49, 1403 (1938).
482
Серебро
От прибавления воды раствор не должен мутнеть (Bi). После осаж
всего серебра в виде AgCl и выпаривания фильтрата не должно остацДеНИя
никакого весомого остатка. ать>Ся
Незначительный остаток после растворения в кислоте испытываете*
помощи H2S, NH4OH и (NHOsS. при
Б. Молекулярное серебро
Приготовление
1. По Впслиценусу осажденное на холоду AgCl. прзмывают
декантацией дестиллированной водой и восстанавливают под водой цинком.
Выделившееся серебро отделяют от избытка цинка и постепенно нагревают до l£o°.
2. Очень хороший метод предложен Гомбергом и Коне1.
Хорошо промытое AgCl помещают в батарейный стакан, заливают водой
и погружают туда мелкопористую глиняную пробирку, содержащую несколько
палочек металлического цинка. Затем погружают в AgCl кусок платиновой
жести и замыкают ток, соединяя платиновой проволочкой жесть с цинком.
Ускоряя реакцию прибавлением нескольких капель НС1 к воде в пробирке,
можно переработать в несколько дней до 250 г AgCl. При помощи сифона
поддерживают уровень жидкости внутри пробирки ниже уровня в стакане,
чем до минимума уменьшают диффузию ZnCh наружу.
Серый порошок серебра промывают водой, затем для удаления следов
невосстановившегося AgCl аммиаком и, наконец, спиртом и эфиром.
Промытый металл сушат сначала в вакууме над H2SO4, затем в шкафу при 150°.
В. Коллоидальное серебро (колларгол)
Приготовление
1. По >Кэри Ли к 200 мл 10%-<ного раствора AgNOe прибавляют смесь
200 мл раствора FeS04, 280 мл 40%-ного раствора лимоннокислого натрия
(СвНбОтЫаз), и 50 мл 10%-ного раствора NaOH. Образовавшийся осадок
имеет переходящий лиловый цвет; он сохраняется некоторое время под
маточным раствором и переходит на фильтре в синий, причем осадок
остается растворимым. Промывают его раствором какой-либо соли, лучше
всего NH4NO3. После этого он тотчас растворяется в чистой воде, образуя
раствор кроваво-красного цвета; от прибавления 5—10% соли снова
становится нерастворимым. Очищенный повторным растворением в чистой воде,
осаждением с помощью NH4NO3 и промыванием 95%-ным спиртом осадок
'еще растворим, однако несколько труднее, чем прежде. Он содержит,
в среднем, 97,2% Ag, остальное — FesOs и лимонная кислота.
2. По другому способу К э р и Л и к раствору 40 г NaOH и 40 г
декстрина в 2 л воды, прибавляют понемногу концентрированный раствор 28 г
AgNOs. Хотя содержание Ag меньше 1%, жидкость имеет глубокий черный
цвет; при разведении становится красной, при еще более сильном
разбавлении —■ желтоватой.
Из различных сортов декстрина лучше всего действует обычный, палевый.
3. По Ван и но2 нагревают смесь 100 мл воды, 8 капель 0,Ш раствора
AgNOs, 2 мл 0,Ш раствора КОН с 1 мл 2,5%-ного раствора цитартша3.
Получается раствор красного цвета.
4. По Оствальду4 к 100 'мл 0,0027V раствора AgN03 прибавляют
|несколько капель свежеприготовленного 1%-ного тапнина и несколько капель
1%-«ого раствора NasCOs. При нагревании получается золь, интенсивно окра-
1 М. О о m b е г g, L. H.Cone, Ber. 39, 3287 (1906); Prap. Cli. 522.
» Koll. Z. 20, 122 (1917); Prap. Ch. 523.
• Цитарин- C7He07N3;>, ангидрометилепцитрат натрия.
4 Колл. химия, 8.
чГЩ Серебро. Серебро азотистокислое 483
ННЫЙ Б красно-коричневый цвет, не особенно чувствительный к электро-
^бЛ1о Гутбиру 100 им 0.0001N раствора AgNOs по прибавлении по
' йпЛЯМ разбавленного (1:2000) раствора гидразин-гидрата и нагревании дает
**?£ яркожелтый в проходящем свете и зеленоватый в отраженном.
*$. Г. Фрейндлих и Д. Штейнер1 к 100 мл аммиачного раствора
• А«0 (соотношение Ag : ЫНз = 1 : 4), содержащего 0,01 г Ag, добавляют смесь
Ш мл В°ДЫ и 2 мл 0,1%-ного раствора гидразин-гидрата; полученный
жёлтый золь совершенно прозрачен.
: 7. Для получения золя Ag можно применять также метод К- Зенгелиса
J й К* Статиса (см. Платина, стр. 431).
I 8. По Г. Б р и нтцин г ер у2, 0,1 мл 0,1/V AgNOe разбавляют водой до
Г 200 мл, добавляют 1 мл 0,5%-ной аскорбиновой (или изоаскорбиновой)
кислоты и полученный желтовато-белый, слегка мутный золь подвергают диализу.
9. М. Маньяни3 рекомендует получать очень устойчивый бордово-крас-
рй золь Ag распылением металла" при помощи вольтовой дуги, питаемой
переменным током. Автор описывает особое устройство электродов для их
равномерного использования. Наиболее устойчивые концентрированные
высокодисперсные золи ^ образуются в растворах галловокислого натрия (рН = 6^8),
.получаемого нейтрализацией NaOH галловой кислотой.
-V;
Г. Получение серебряного зеркала
>' 1. По Кайзеру готовят два раствора: а) 10 г AgNCb растворяю^ в 50мл
-воды, прибавляют NH4OH до слабой опалесценции и разбавляют водой до
1 л; б) в течение получаса кипятят раствор 20 г сегнетовой соли (МаКСШЮв)
В 26 г леденцового сахара в 200 мл воды и разбавляют до 1 л. Для серебрения
смешивают равные объемы обеих жидкостей и быстро выливают на стекло.
Через 15—20 мин. зеркало готово.
2. По У ад с во р з у берется раствор 90 г сахара в 1 л воды, к которому
добавлено 4 мл HNOs (уд. в. 1,22) и 175 мл спирта. Вторая жидкость —
аммиачный раствор AgNOs (3 ч. NH4OH уд. в. 0,88 на 4 ч. AgNOs), к которому
.перед употреблением прибавляют еще КОН из расчета 5 г КОН% на 1 г AgNOs.
i Употребление — как в способе 1.
3. По Беттгеру для быстрого серебрения стекла следует употреблять
смесь следующих растворов: 4 г AgNOe, растворенного в возможно малом
количестве NHaOH и смешанного с 1 г (NH4>2S04 в 350 мл воды; 1,2 г
чистого крахмала или глюкозы, растворенных в 350 мл воды и смешанных
с 3 г чистого КОН.
4. Для серебрения стекла пригодна также смесь 20 мл 3%-ного раствора
AgNOs с 0,5 мл метилового спирта и 0,5 мл формалина.
Стекло, подвергающееся серебрению, должно быть
тщательно вымыто и обезжирен'о промыванием спиртом.
СЕРЕБРО АЗОТИСТОКИСЛОЕ (НИТРИТ СЕРЕБРА)
Argentum nitrosum Silver nitrite Silbernitrit
AgN02 Мол. в. 153,888
Приготовление
Сливают тепловатые концентрированные растворы 20 г AgN03 и 10 г х. ч.
NaN02 (или 12,5 г KNO2); выпавший осадок AgN02 промывают декантацией
или перекристаллизовывают из горячей (70°) воды.
1 Н. F г е и л d 1 i с h u. D. S t e i n е г, J. chem. Soc. 1083 (1937).
1 Ч. В г i n t z i n g e r, Koll. Z. 78, № 1, 22 (1937).
• M Magnani, Chim. с I- d. (Mila o) 19, 5 (1УС7); С II, 539 (1937).
3i*
434
Серебро
Свойства
Тонкие, как волос, светложелтые, почти белые иглы уд. в. 4,45, чернеюши
под влиянием света и воздуха вследствие образования AgNOe и Ag. в во '
при обычной температуре растворимо довольно плохо: 1 л воды раствопя*6
3,18 г AgN02 (Науман, Рюкер). Легко растворимо в ШЮН и ацет?
нитриле. Разлагается при нагревании.
СЕРЕБРО АЗОТНОКИСЛОЕ (НИТРАТ СЕРЕБРА, ЛЯПИС)
Argentum nitricum Silver nitrate Silbernitrat
Hollenstein
AgNOs Мол. в. 169,888
Приготовление
а) Из химически чистого серебра
Кусочки чистого Ag растворяют при нагревании в возможно малом
количестве НЫОз (уд. в. 1,25) и выпаривают профильтрованный раствор досуха на
песочной или водяной бане. Затем перекристаллизовывают препарат из
небольшого количества горячей воды.
б) Из сплавов, содержащих медь
1. Сплав растворяют в HNOs умеренной концентрации до насыщения
В части раствора при помощи КОН осаждают Ag20 с примесью Си(ОН)2;
ее промывают и смешивают с остальной частью раствора, причем количественно
выпадает Си(ОН)г. Профильтрованная жидкость после выпаривания дает
чистое AgN03. Для использования Ag, оставшегося в осадке Си(ОН)г,
последний обрабатывают НС1; оставшееся нерастворенным AgCl
восстанавливается в металлическое серебро (см. Серебро А, в, стр. 480).
2. Для извлечения серебра из сплавов, более бедных серебром, в которых
все же Ag больше, чем Си, можно по А. В. Раковскому и С. И. Добр-
жинскому1 поступать следующим образом. После растворения сплава
в HNOe, последующего фильтрования, выпаривания и кристаллизации
кристаллы AgNOs содержащие Си(ЫОз)г, промывают водой или в случае
значительных количеств меди 5—10%-ным раствором HNOs.
Промытые кристаллы перекристаллизовывают из горячей воды, причем
получают х. ч. препарат. Промывные воды, содержащие значительное
количество AgNOs, могут быть применены при повторной работе для промывания
кристаллов. Ф
3. Сплавы, в которых меди больше, чем серебра, нуждаются в
дополнительной очистке.
Промытые по предыдущему методу кристаллы AgNOs, содержащие Си(Ж)з)2,
плавят в платиновой чашке в течение часа при 290—300°. При этом
Cu(NOs)2 разлагается, оставляя СиО, в то время как AgNOs почти не
изменяется. После охлаждения сплавленную массу растворяют в воде,
отфильтровывают СиО, фильтрат подкисляют HNOs и упаривают до кристаллизации.
4. Для очень бедных серебром (до 30%) сплавов целесообразнее всего,
растворив сплав в HN03, осадить соляной кислотой все серебро в виде
AgCl и последнее после промывания восстановить до металла (см. Серебро
А, в, стр. 480). Чистое серебро растворяют в HN03 и выпаривают.
5. По Бендеру2 при кипячении смеси AgN03 и Cu{N03>2 с 90%-ным
спиртом последний извлекает почти только одну медную соль.
Свойства
Бесцветные ромбические пластинки уд. в. 4,3551, плавящиеся при 208,5°
и разлагающиеся в калильном жаре. Не изменяется под действием света,
1 Изв. ИГЕА, 1, :05. 2С8 (1923).
* Не орг. npen. 5CC.
""ЩГ Серебро азотнокислое 485
1£м не присутствуют органические вещества; в противном случае чернеет
5£ этой причине оставляет черные пятна на руках, материи и т. д. Вкус
^уший. металлический, едкий; ядовито. Легко растворимо в воде (раствор
•■'Ееет нейтральную реакцию) и глицерине. Растворимо в этиловом метиловом
•Гйзопропиловом спиртах, труднее в ацетоне и бешоле. Очень легко раство-
pifMO в бензонитриле. Почти не растворимо в концентрированной HNOa
Таблица 451
Ра створимость AgN03 в воде
/°с
0
10
20
30
40
50
60
70
o/oAgNOa
53,5
61,5
68,3
73,8
77,0
80,0
82,5
84;6
/вс
80
90
100
ПО
155
133
159
о/0 AgN03
86,7
88,4
90,1
91,7
94,2
95,1
100,0
Таблица 452
Удельный вес водных ра ст в о р о в AgNOa
•/о AgN03
5
10
15
2а
d»4
1,0422
1,0893
1,1404
1,1958
°/о AgNO,
25
30
35
40
1
d\
1,2555
1,3213
1,3945
1.4773
0/oAgN03
1
45
50
55
ео
Ф\
1,5705
1,6745
1,7895
1,9158
Таблица 453
Растворимость AgN03 в водном спирте при 15°
■
°/о спирта в
растворителе
"*v ■ ■
100
95
80
70
1 100 г
растворителе рас*
твбряют
г AsNOj
3.1
ЗД
10,3
22,1
о/0 спирта в
растворителе
60
50
40
100 г
растворителя
растворяют
2 AgN08
30,5
35 Л
56Д
о/0 спирта в
растворителе
30
20
1Q
100 г
растворителя
растворяют
г AgNOa
73,7
107
158
Испытание
По ГОСТ 1277-41 содержание AgNOs в препарате ч.д.а. должно быть
не менее 99,80%, а в чистом — 99,75%; наибольшие количества допустимых
примесей указаны в табл. 454.
486
Серебро
Допустимые примеси в AgN03
Таблиц
454
Примеси
Допустимое содержание и г
в чистом дхя
анализа
в чистом
1. Нерастворимые в воде вещества
2. Вещества, не осаждаемые НС1 .
3. Сульфаты (SO/')
4. Медь, висмут и свинец . . . .
5. Свободная азотная кислота . . .
0,005
0,04
0,004
0,01 *
v 0,06
0,006
[качественное испытание
I поТОСТ
Argentum
bromatum
СЕРЕБРО БРОМИСТОЕ (БРОМИД СЕРЕБРА)
Siver bromide
AgBr Мол. в. 187,796
Silberbromid
Bromsilber
А. Обыкновенное бромистое серебро
Приготовление
К раствору AgNOe прибавляют бромистоводородной кислоты (НВг) или:
раствора щелочного бромида (KBr, NaBr) в количестве, достаточном для пол-*
ного осаждения AgBr. Выпадающий осадок хорошо промывают и сушат
в отсутствии света. Растворяя препарат в растворе азотнокислой ртути,
получают при охлаждении AgBr в виде кристаллов *.
Свойства
В зависимости от метода получения — порошковатый желтый или зерни*
стый желтовато-белый осадок, почти не растворимый в -воде (3,57 • 10~5%) и
разбавленных кислотах. Растворим в ЫНЮН (100 г NH4OH уд. в. 0,959
растворяют 0,4 г AgBr) и пиперидине. При 422° плавится в красноватую
жидкость уд. в. 6,47.
Испытание
Наибольшие количества допустимых примесей указаны в табл. 455.
Таблица 455
Допустимые примеси в AgBr
Примеси
Допустимое содержание в°/<>
1. Вещества, нерастворимые в 25°/0-ном NH4Orf
2. М.дь (Си)
3. Железо (Fe)
4. Щелочи
5. Нитраты (NO/)
0,001
0,0005
0,0000
0,001
0,0002
1 Pra \ Ch. 533.
Серебро, бромистое, двухромовокислое и йодистое 487
Б. Коллоидальное бромистое серебро
По Оствальду1 к смеси 2--3 мл 0,017V раствора КВг я 10° мл -воды
■„рябавляют 1—2 мл 0,1 N раствора AgN03. Образуется слегка сероватый
гиДРоЗОЛЬ' довольно непрочный.
СЕРЕБРО ДВУХРОМОВОКИСЛОЕ (БИХРОМАТ СЕРЕ5Р А)
Argentum Silver bichromate Silberbifbiromat
bichromicum
Ag2Cr207 Мол. в. 431,78
Приготовление
По А у те ирит у* к 20 мл 5%-ного раствора AgNOe прибавляют 10 мл
10—12%-ного раствора HNOa; нагревают смесь ■ до кинения и приливают
раствор К2СГ2О7 или К2СЮ4. Тотчас начинается осаждение блестящего
красного осадка. Как только жидкость остынет и прекратится образование
кристаллов, осадок отсасывают, промывают небольшим количеством холодной
воды и сушат, в эксикаторе над Н2ЗО4.
Свойства
Блестящие красные ромбические листочки* уд. в. 4,77. При кипячении с
водой образует Ag2Cr04; из раствора при охлаждении снова осаждаются
кристаллы Ag2Cr207. Хорошо растворимо в HN03 и NHUOH.
СЕВЕБРО ЙОДИСТОЕ (ИОДИД СЕРЕБРА)
Argentum Silver iodide Silberjodid
jodatum Joflsilber
AgJ Мол. в. 234,80
А. Обыкновенное йодистое серебро
Приготовление
К сильно разбавленному раствору AgNOe прибавляют при энергичном
перемешивании . некоторый избыток раствора KJ. Светложелтый осадок хорошо
промывают и высушивают.
Свойства
Светложелтый аморфный порошок уд. в. 5,68, почти нерастворимый в воде
(2,1-7%, по другим данным a 3,46-10-e%), NH4OH (0,04% в IOVVhom ШЮН)
и разбавленных кислотах. Растворим в концентрированных растворах KJ,
Na2S203, и в метиламине СНзЫН2. При 552° плавится в^ желтую жидкость,
при более высокой температуре приобретает красно-бурый цвет.
Б. Коллоидальное йодистое серебро
По Лоттермозеру4 к смеси 2—3 мл 0f\N раствора KJ и 100 мл
воды прибавляют 1—2 мл 0,1/V раствора AgN03. Получается желтовато-
зеленый гидрозоль, который без диализа может сохраняться месяцами.
Полная очистка раствора путем диализа не удается.
Более концентрированный, но менее прочный золь получается при влива-,
пии 7—8 мл 0,Ш раствора AgNOa в 20 мл 0,057V раствора KJ.
* Колл. химия, 10.
a W. A u t e n r i e s th., Вег. 35, 2057 (1902); PrSp. Ch. 534.
» Ch. В е d e 1, С. г. 207, 632 (1938). я 9mA -,_.
« A. L о 11 е г щ о s е г, Z. phys. Ch. 62, 370 (19С8); J. prakt. Ch. 73, 374 J1906).
488
Серебро
СЕРЕБРА ОКИСЬ
Argentum oxydatum Silver oxide Silberoxyd
AgoO Мол. в. 231,760
•Приготовление
1. Осаждают раствор чистого AgNOa раствором КОН или Ва(ОН)2 не
содержащим СГ; выпавший осадок промывают и высушивают в теплом месте.
2. К концентрированному раствору 500 г AgNOs прибавляют разбавлен-
ный раствор 125 г NaOH (свободного от СГ) и осадок промывают
декантацией1. Применяемая ©ода предварительно освобождается от растворенного
СОг, для чего через нее длительное время пропускают свободный от^с'О*
воздух.
После промывания большую часть воды, удержанной осадком, отделяют от-
сасыванием, после чего влажную еще массу центро фугируют! Затем препарат
переносят в реторту с тубусом, присоединенную к склянке Вульфа, которая
другим горлом соединяется с водоструйным насосом.
Поместив реторту в нагретую до 85—8$° водяную баню и пустив в ход
насос, просасывают через аппарат сухой, лишенный СОг воздух; сушка
заканчивается через 3 суток.
Свойства
Коричневый порошок уд. в. 7,52, после сушки между 60 и 80° — почти
черный; по Хёст-Мадсену темкокор-ичиевый с темнофнрлетовым оттенком.
Окись серебра несколько растворяется в воде (0,006—*0,()09%), придавая ей
металлический вкус и щелочную реакцию. При хранении как в сухом, так
и во влажном состоянии постоянна; однако на свету и в калильном жаре
разлагается на Ag и Ог. При нагревании в струе воздуха разлагается уже
при 300°.
Формалин в присутствии щелочи восстанавливает Ag20 до металлического
серебра.
СЕРЕБРО СЕРНОКИСЛОЕ (СУЛЬФАТ СЕРЕБРА)
Argentum Silver sulfate Silbersulfat
sulfuricum
AgiSOi Мол. в. 311,82
Приготовление
1. К очень концентрированному раствору AgN03 прибавляют 40%-ную
H2SO4 и выделяющийся кристаллический порошок промывают холодной
водой.
2. Растворяют металлическое серебро в горячей концентрированной H2SO4
и выпаривают избыток кислоты.
3. К ' раствору 100 г AgNOe в 75 мл воды прибавляют раствор 110 г
кристаллического сернокислого натрия (Na2S04 • ЮН2О) ib 75 мл воды и
промывают осадок до полного удаления NaNOa. Выход 80 г.
Свойства
Мелкие ромбические кристаллы уд. в. 5,40, довольно -плохо растворимые
в воде («при 18°—0,75%, при 100°-^ 1,5%). легко растворимые в NH40"H и
нерастворимые в спирте. Сравнительно легко растворимо в концентрированной
H2SO4, но снова выпадает при разбавлении раствора водой.
1 Z. anorg. Ch. 79, 195 (1913); РгЗр. Ch. 524.
Серебра окись. Серебро сернокислое и фтористое 489
рыгание!
;Д^ раствор 0,5 г препарата в 100 мл воды должен быть нейтрален на
f* % Медь (Си''). Раствор 2 г препарата в 5 мл воды и ю мл NH-ЮН
/уд. в. W6) не Должен иметь синего окрашивания.
3. Нитраты. (NOi). Встряхивают 0,4 г препарата с 10 мл воды, фильтруют
й добавляют к фильтрату 2 мл 0,5%-ного раствора дифениламиша в I-hSO*.
Йе должно быть синего окрашивания.
СЕРЕБРО ФТОРИСТОЕ (ФТОРИД СЕРЕБРА)
Argentum Silver fluoride Silberfluorid
fluoratum Fluorsilber
А. Дигидрат
AgF2H20 Мол. в. 162,91
Яржготовление
У; По Саксу и Вани и о2 лучше всего получать дигидрат из тетрагидрата
jjjjjrF • 4НгО. Последний уже при комнатной температуре плавится в своей кри-
^ашшзационной воде. Расплавленную соль помещают в вакуум-эксикатор
И выкачивают из него воздух. При этом быстро, в течение часа, осаждаются
Довольно крупные шестиугольные таблички. Ускоряя испарение
непрерывным откачиванием воздуха из эксикатора, получают препарат в виде
тонких, прозрачных как стекло, кристаллов.
Свойства
Бесцветные, довольно непостоянные кристаллы уд. в. 5,85, расплывающиеся
„во влажном воздухе. При длительном стоянии над H2SO4, особенно в вакууме,
теряет воду и HF, чернеет, вследствие выделения Ag и превращается с
поверхности в основную соль. Растворимо в воде (1 : 1,5); безводная соль
плавится при 435г
о
Б. Тетрагидрат
AgF-4H20 Мол. в. 198,94
Приготовление
По Саксу и Вани но3 приготовленное из AgNOs л NasCCb углекислое
серебро тщательно отсасывают, промывают и сушат (по возможности в
отсутствии света). Полученное Ag^COs вносят небольшими порциями в чистую
концентрированную плавиковую кислоту (HF) почти до нейтрализации. Небольшая
яасть Ag2COs при этом разлагается. Раствор выпаривают досуха на песочной
бане, причем образуется коричневая твердая, довольно компактная масса
безводного AgF. При добавлении к ней небольшого количества воды часть
AgF растворяется с большим выделением тепла, а часть переходит в твердый
гидрат; последний, поглотив кристаллизационную воду, становится настолько
твердым, что с трудом поддается измельчению и только очень медленно
растворяется в воде.
Фильтруют раствор, не применяя стеклянной посуды; дают раствору капать
в охлажденную льдом платиновую чашку и получают AgF • 4Н2О в виде
прекрасных, длинных игольчатых кристаллов.
Свойства
Соль постоянна только при 0°; при более высокой температуре
расплывается в своей кристаллизационной воде.
■ш
1 Испытание хим. реактивов, 360.
1 R S а с b », Dissertation, Mimchei (1913); Prap. Ch. 530.
'R.Sachs, Dissertation, Miinchen (1913).
490
Серебро
СЕРЕБРО ХЛОРИСТОЕ (ХЛОРИД СЕРЕБРА)
Argentum Silver chloride Silberchlori'd
chloratum Chlorsilber
AgrCI Мол. в. 143,337
А. Обыкновенное хлористое серебро
Приготовление
К раствору AgNOs прибавляют раствор НС1 или NaCl до прекращения
выделения осадка. Выпавшее AgCl фильтруют, промывают и сушат в -Темноте
при 80—90°.
Свойства
Светочувствительный оелый аморфный порошок уд. в. 5,56, плохо раство-
римый в воде (см. табл. 456) и разбавленных кислотах, значительно
растворим при кипячении ъ конц. НС1; несколько растворим в крепких растворах
NaCl и NHiCl, хорошо растворим в NH4UH. Пр.и 455° плавится и при
охлаждении застывает в роговидную массу.
Таблица 456
Раств ор имость AgCl вводе
/°с
1,6
10
18
о/» AgCl
0,56-10-4
0,8-9 • 10-4
1,6 -Ю-4
ГС
25
50
100
'„ AgCl
1,9-10-4
5,2- 10-4
21,7-10-4
Испытание (для препарата ч.д.а.)1 ^
1. Реакция водной вытяжки. 20 г препарата взбалтывают со 100 мл воды
и фильтруют; фильтрат должен иметь нейтральную реакцию на лакмус.
2. Растворимые в воде вещества. 50 мл фильтрата (п. 1) выпаривают на
водяной бане и высушивают остаток при 110°; вес его не должен превышать
0,05 мг.
3. Растворимые хлориды (CV). 5 мл фильтрата (п. 1) разбавляют водой до
50 мл, добавляют 2 мл HNOs (уд. в. 1,15) и 1 мл 0*1 N AgNOs. He должно
тотчас появляться опалесценции.
4. Аммиак. При кипячении б г препарата с 5 мл 10%-ной NaOH
выделяющиеся пары не должны окрашивать красную лакмусовую бумажку в синий
цвет.
Б. Коллоидальное хлористое серебро
Приготовление
1. По Оствальду2 к смеси 2—3 мл 0,1Л/ раствораHCI в 100 мл воды
добавляют 1—2 мл 0y\N AgNOa: образуется довольно непрочный, молочно-
белый гидрозоль.
* Испытание хим. реактивов, 361.
* Коал, химия, 10.
'-^ Серебро хлористое, хромовокислое и цианистое 491
2. fi° способу химической фабрики Г е й д е н а * к темному раствору кол-
лйДН°г0 сеРе^Ра прибавляют хлорной воды до обесцвечивания. Получается
Лелая или светложелтая жидкость, в достаточно разбавленном состоянии
• прозрачная и опалесцирующая.
" От прибавления ^небольших количеств минеральных кислот или солей AgCl
выпадает в Зычной, нерастворимой форме. При осаждении уксуснокислым
' йатрием (CHsCOONa) или желатиной и лимоннокислым натрием получается
д$С1, несколько растворимое в воде.
СЕРЕБРО ХРОМОВОКИСЛОЕ (ХРОМАТ СЕРЕБРА)
Argentum Silver chromate Silberchromat
ch ro mi cum
Ag-Cr04 Мол. в. 331,77
Приготовление
1. По Егеруи Крюссу многократно кипятят двуххромовокислое серебро
'(AgaC^O?) с водой до тех пор, пока при длительном кипячении со свежей
водой последняя не перестанет окрашиваться в желтый цвет. Осадок
высушивают при 100—110°.
2. По Маргошесу2 можно получить красную модификацию Ag2Cr04,
если аммиачный раствор Ag2Cr04 или А^гСггО? испарять или кипятить до
удаления NH3. После этого остается Ag2Cr04 в виде темнозеленых корок
с металлическим блеском, при истирании превращающихся в красный порошок.
Свойства
Полученный по способу 1 — яркозеленый кристаллический порошок уд. в. 5,63,
немного растворимый в H2SO4, НЫОз и NH4OH. Полученный по способу
2— красный, аморфный (или неясно кристаллический) порошок. В ледяной
СНзСООН не растворимо; в разбавленной растворяется в бблыыей или
меньшей степени в зависимости от концентрации кислоты.
В воде растворяется 2,5» К)-3-—3- 10-3% Ag2Cr04.
СЕРЕБРО ЦИАНИСТОЕ (ЦИАНИД СЕРЕБРА)
Argentum Silver cyanide Silbercyanid
cyanatum Cyansilber
AgCN Мол. в. 133,898
Приготовление
По Глассфорду и Напиру осаждают AgCN из раствора AgNCfo
чистым KCN (яоЧ), избегая избытка последнего. Если применяемый KCN
содержит KCNO и К2СО», то очистить получаемый препарат от AgCNO и Ag2COs
можно настаиванием с разбавленной HNCh; однако в KCN не должно
содержаться СГ и Fe(CN)e"", так как эти ионы перейдут в препарат.
Полученное AgCN сушат при температуре не выше 126° (при более
высокой температуре оно буреет).
Свойства
Белый порошок уд. в. 3,96, растворимый в NH4OH, Na2S203 и K4Fe(CN)e.
На свету буреет. При нагревании в отсутствии воздуха около 325° плавится
в красно-бурую жидкость и выделяет часть циана; при затвердевании
получается серая масса, оставляющая при накаливании на воздухе чистое се-
1 Герм. пат. 103,106 (1897); С. II, 497 (1899.
» В. М. М а г g о i с д e s, Z. aiior*. Ch. 41, 68 (1904).
492
Срли
ребро. Разбавленная (1:1) ILoCh разлагает при кипячении AgCN с образо
нием 'Ag2S04 (Осторожно, пары яоовиты!). , Qa-
Испытание
Наибольшие количества допустимых примесей в реактивном препарате
указаны в табл. 457.
Таб.
шца 4$
Допустимые примеси в AgCN
Примеси
Допустимое содержание в ш.
1. Вещества, не растворимые в 25%-ном NH4OH
2. Медь (Си) • .
3. Железо (Fe)
4 Щелочи . • . .
5. Нитраты (N0/)
0,0011
0,0005
0,0000
0,001
0,0002
СОЛЬ МОРА
(ДВОЙНАЯ СОЛЬ СЕРНОКИСЛОГО ЗАКИСНОГО ЖЕЛЕЗА
И СЕРНОКИСЛОГО АММОНИЯ)
Ferro-Ammonium sul-
furicum
Ferrous ammonium
sulfate
Mohr's salt
Mohrsches Salz
Ferroammoniumsulfat
FeS04 • (NH4)2S04 • 6Н2О или Fe(NHi)2(S04)2 • 6Н2О Мол. в. 392,15
Пр и г отовление
1. По Мору 139 г чистого (светлоголубого) железного купороса (FeSOi*
• 7НгО) и 66 г чистого (NH4)2S04 растворяют отдельно в возможно малых
количествах воды; нагревают оба раствора до 60^-70°, сливают вместе в
фарфоровую чашку и, подкислив небольшим количеством H2S04, охлаждают при
непрерывном перемешивании. На другой день выпавшую кристаллическую
муку отсасывают, промывают разбавленным спиртом, отжимают между
листами фильтровальной бумаги и сушат в прохладном месте, пока кристаллы
не перестанут приставать к стеклянной палочке.
2. По И. К о л ьтго ф у * железо-аммонийные квасцы иерекристаллизовы-
вают до полной чистоты, насыщают H2S в течение нескольких дней (до
полного восстановления Fe' ' *), фильтруют и выпаривают до начала
кристаллизации.
После перекристаллизации препарат высушиъают /над расплывающимся
NaBr до постоянного веса. Препарат содержит 99,97% FeS04 • (NH4):>S04 •
• 6Н2О.
Свойства <-
Прозрачные, синевато-зеленые моноклинические кристаллы уд. в. 1,87, не.
изменяющиеся при хранении. Около 100° теряет кристаллизационную воду.
1 Объемный анализ, т. II., Л. Госхимтехиздат. стр. 296.
Соль Мора 493
Таблица 458
Растворимость FeS04 -"(NH^SO* • 6Н20 в воде
(Н. Т. Meyer)
ее
<^~~
0
10
20
100 г волы
растворяют
2 С'ОЛИ
15.1
18,1
21,2
/°С
30
40
50
100 г воды
растворяют
г Соли
24,5
37,8
31,3
1°С
60
70
80
100 г воды
растворяют
г соли
34,8
38,5
42,2
Таблица 459
Удельный вес водных растворов FeS04 • (NH4)2S04
безводной
соли
1
2
4
6
d16,5
4
•1,008
1,016
1,032
1,048
°,'о безводной
соли
8
10
п
d16,5
4
1,065
1,083
1,100
°/о безводно!
соли
14
16
18
d16,5
4
1,118
1,136
1,155
Испытание
По ОСТ 3007 препарат должен представлять собой голубовато-зеленые
Кристаллы или кристаллический порошок того же цвета; содержание FeS04»
(NH4)2S04 • 6Н2О для препаратов квалификаций х. ч. и ч. д. а. не менее 99,7%;
Наибольшие количества допустимых примесей указаны в табл. 460.
Таблица 460
Допустимые примеси в FeS04 • (NH^SC^ • 6H20
Примеси
Допустимое содержание в °/0
химически
чистом
в чистом для
анализа
1. Нерастворимые в воде вещества
2. Хлориды (CV)
3. Железо (окисное)
4. Медь (Си)
5. Цинк (Zn)
6. Щелочные и щелочноземельные
металлы • . .
0,С05
0,001
0,005
О,003
0,005
0,05
0,010
0,002
0,010
0,005
0,010
0,10
0,020
0,005
0,020
0,010
0,020
0,20
494
Сола. Сплавы. Стронций
СОЛЬ ПЕЛИГО (ХЛОРОХРОМОВОКИСЛЫЙ КАЛИЙ)
Peligot's salt Kaliumchlorochromat
Peligotsalz
КСгОзС1 или С1Сг020К Мол. в. 174,56
Приготовление
По Эрдману1 в колбу, содержащую смесь 100 мл воды и 130 г чистой
НС1 (уд. в. 1,19), вносят 100 г тонко «стертого К2СГ2О7 и слабо нагревают
до полного растворения, после чего фильтруют. При спокойном стоянии
фильтрата через сутки выделяются кристаллы, которые высушивают на глиняной
тарелке. Выход 65 г.
Свойства
Крупные красные призмы или таблички, выделяющие при нагревании до
100° чистый хлор.
СПЛАВ ВУДА (МЕТАЛЛ ВУДА)
Wood's metal Woods Metal)
Приготовление
В объемистый фарфоровый тигель, вставленный в отверстие в куске
асбестового картона, помещают немного парафина (для предохранения от
окисления) и 20 г металлического висмута и нагревают до плавления последнего;
последовательно добавляют 5 г свинца, 2,5 г олова № 8,5 г кадмия. Когда
металлы сплавятся, в чем убеждаются, перемешивая железной проволокой,
выливают смесь в воду.
Последние следы парафина удаляют, промывая сплав бензолом или
эфиром. Очищенный сплав гранулируют, выливая по каплям в холодную воду.
Свойства
Блестящий металл уд. в. 9,765, плавящийся при 71°. Применение сплава
Вуда основано на его легкоплавкости.
СПЛАВ ДЕВАРДА
Devarda's alloy Devarda'sche Legierung:
Приготовление
По Деварду смешивают 50 г медных стружек, 45 г алюминиевой
«крупы» и 5 г гранулированного цинка; смесь помещают в глиняный тигель,
замазывают глиной (во избежание окисления) и накаливают в тигельной печи
с платиновой обмоткой около часа. Еще горячий расплавленный металл
медленно выливают в чашку с холодной водой, где он застывает.
Свойства
Хороший восстановитель. Отличительным свойством этого сплава является
его хрупкость, благодаря которой он легкоу как стекло, измельчается в
тонкий порошок. Его применение основано на том, что при восстановлении этим
сплавом остается медная пыль, обеспечивающая ровное, без толчков кипение
при перегонке.
Испытание
Препарат испытывают на содержание азота перегонкой с NaOH.
Выделившийся из 2 г сплава аммиак должен соответствовать не более, чем 0,2 мл
0,2/V кислоты.
1 Хим. npe.i. 55
Соль Пелиго. Сплавы В уда и Деварда. Стронций азотнокислый 495
СТРОНЦИЙ АЗОТНОКИСЛЫЙ (НИТРАТ СТРОНЦИЯ)
ctrotttium nitricum Strontium nitrate Strontiumnitrat
Sr(N03)2 Мол. в. 211,65
дрнготовл ение
Обрабатывают Sr(OH)2 или SrCOa азотной кислотой; раствор фильтруют и
выДаРивают на водяной бане досуха или выпаривают раствор до появления
йа поверхности пленки соли и дают закристаллизоваться.
Свойства
[Прозрачные октаэдртгческие кристаллы уд. в. 2,93. При нагревании соль
растрескивается и при 645° плавится, образуя нитрит и окись. В воде
<Ко растворим; не растворим в спирте. При кристаллизации из холодного
раствора (ниже 31,3°) выпадают моноклинические кристаллы тетрагидрата,
Sr(N03)2 • 4Н2О, быстро выветривающиеся на воздухе и теряющие воду
при 100°.
Таблица 461
Растворимость Sr(N03)2 в воде
(Mulder)
ГС
0
5
10
20
30
40
50
100 г воды
растворяют
2Sr(NCVa
39,5
47,3
64,9
i 70,8
1 87,6
91,3
92,6
°[oSr(N03),
28,3
—
35,5 1
41,5
4<\7
47,7
ГС
60
70
i 80
90
| 100
107,9
•
100 2 воды
растворяю? i
*Sr(N<Vt
94,0
95,6
97,2
99,0
101,1
102,9
•JoSrfNO.1!
44,5
—
49,3
—
50,3
—
Таблица 462
Удельный вес водных растворов Sr(NOb)2
(К г е m e r s)
0/oSr(NOj)a
2
4
5
. 6
'М9.5
rfi9,5
1,017
1,031
1,041
1,049
<Vo St (NO,),
1 •
10
15
20
,19,5
d19,5
1,068
1,085
U31
1,181
Vo Si (N03),
25 •
30
35
40
.19,5
rf19,5
1,235
1,292
1,354
1,422
Испытание
1. Хлориды (СГ). Раствор препарата (1:50), подкисленный IWO*, должен
в течение 5 мин. после прибавления 0,1/V раствора AgNOe оставаться
прозрачный.
2. Сульфаты (SO*"). Раствор препарата (1 : 10), подкисленный НС1 (уд. в.
1>12), от прибавления BaCls не должен в течение часа давать мути или осадка.
496
Стронций
3. Тяжелые металлы. Раствор препарата (1:20), подкисленный Hci
(уд. в. 1,12), должен оставаться бесцветным после прибавления сероводород
ной воды.
4. Соли бария (Ва' ') К 10 мл раствора препарата (1 : 5) прибавляют 9
CHsCOONa, 1 мл СНзСООН (уд. в. 1,04) и 5 капель 10%ного раствора*
КгСггСЬ; испытуемый раствор в течение 10 мин. должен оставаться прозрач.
ным.
СТРОНЦИЯ ГИДРАТ ОКИСИ (ГИДРООКИСЬ СТРОНЦИЯ)
Strontium oxydatum Strontium hydroxide Strontiumhydroxyb
Л hydricum
J\. Кристаллический
Sr(OH)2-8H20 Мол. в. 265,77
Приготовление
1. Смешивают окись стронция, SrO, с эквивалентным количеством воды
(реакция, аналогичная гашению извести).
2. Осаждают насыщенный раствор ЗгС1г или $r{NOs)2 25%-ным раствором
NaOH, не содержащим сульфатов и карбонатов. При этом выпадает большое
количество кристаллов Sr(OH)2 • 8Н2О. Их быстро отсасывают, промывают
холодной водой, не содержащей СОг, и перекристаллизовывают из горячей
воды. Выход 60—70% теоретического.
3. По Эрдману1 150 г порошкообразного .минерала целестина (SrSO-i)
смешивают с 50 г древесного угля; смесь плотно набивают в тигель и
покрывают слоем угольного порошка. Тигель закрывают железной крышкой
с загнутыми краями и накаливают в течение часа при белом калении в
тигельной печи.
Проба полученного продукта при 'растворении в разбавленной НС1 должна
оставлять лишь незначительный остаток угля.
Массу суспендируют в 1 л воды в фарфоровой чашке, нагревают до
кипения и прибавляют окиси меди (100—150 г) до тех пор, пока отфильтрованная
проба жидкрсти не перестанет давать бурый осадок с раствором РЬ(СНзСОО)2
(окись меди предварительно смачивают азотной кислотой и прокаливают).
После этого горячую жидкость отфильтровывают, остаток кипятят с 100 мл
воды, фильтруют и объединенный фильтрат оставляют для кристаллизации.
Через сутки сливают маточный раствор, кристаллы быстро сушат на
глиняной пластинке и помещают в плотно закрытый сосуд. Пробку заливают
парафином.
Маточный раствор для вторичной кристаллизации выпаривают до объема
300 мл.
Таблица 463
Растворимость Sr(OH)2 в воде
t° с
0
10
15
20
30
40
°'о SrO
0,35
0,48
0,57
0,68
1,00
1,48
n/oSr(OH)t 8HeO
0,90
1,23
1,46
1,74
2,57
3,80
1 ГС
50
60
70
80 1
90
1С0
101,2
•/о SrO
2,13
3,03
4,36
6,56
12,00
18,60
19,40
U/oSr(OHV8H,0
5,46
7,77
11,16
16,83
30,78
47,71
49,75
1 Хим. npei
in. 23.
W---- Стронция гидрат окись и окись. Стронций хлористый 49?
ройства
' ■ Прозрачные тетрагональные кристаллы уд. в. 1,4. При нагревании пла-
,^тся и выше 100° медленно теряют кристаллизационную воду. На воздухе
.^эДНО поглощают СОг, переходя в БгСОз.
Б. Безводный
Sr(OH)2 Мол. в. 121,65
Приготовление
Осторожно нагревают гидрат, Sr(OH)2 • 8Н2О, до 400°; при этом получается
безводный Sr(OH)2 в виде светлой, как вода, жидкости, застывающей при
Охлаждении В сероватую массу уд. в. 3,63. Жадно поглощает СОг.
СТРОНЦИЯ ОКИСЬ
.у. Strontium oxydatum Strontium oxide Strontiumoxyd
% SrO Мол. в. 103,63
Приготовление
[;■■' 1. Получение SrO из Sr(NOs)2 совершенно аналогично получению ВаО из
$Ja(N03)2 (см. стр. 85, Бария окись, Приготовление, способ 1).
2. Сильно прокаливают (выше 710°) 5г(ОН)г или SrCOs (в токе Нг при
1350—1400° до полного удаления СОг1).
Свойства
v Сероватая пористая масса уд. в. 4,5, аморфного или кристаллического
.строения. Т. пл. 2430°. При смачивании водой переходит в Sr(OH)a се
значительным выделением тепла.
СТРОНЦИЙ ХЛОРИСТЫЙ (ХЛОРИД СТРОНЦИЯ)
Strontium chloratum Strontium chloride Strontiumchlorid
SrCh-6H20 Мол. в. 266,64
Приготовление
1. Барте И Фальер2 предлагают следующий метод. Растворяют
стронцианит (SrCOe) или полученный восстановлением углем или серой
целестина (SrS04) сернистый стронций в таком количестве НС1, чтобы часть
соли осталась нерастворенной. Полученный прозрачный раствор содержит,
кроме Зг, еще Fe* ' ', А1* ' *, Mg* *, Ca*:' и Ва* ".
Для удаления Fe и А1 к слитому с остатка соли раствору прибавляют
небольшой избыток NH4OH и отфильтровывают выпавшие Fe(OH)s и А1(ОН)з,
к фильтрату добавляют избыток H2SO4, причем выпадает осадок BaS04,
SrS04 и CaS04. Осадок промывают декантацией сначала 1—2%-ной H2SO4,
затем чистой водой до удаления следов Mg'' и Са" '.
Остаток переносят в банку, обливают 10%-ным раствором (МШ^СОз и
оставляют стоять два дня на холоду, часто взбалтывая. Полученную омесь
Углекислых и сернокислых солей обрабатывают разбавленной НС1,
растворяющей SrC03 и немного ВаСОз. Прозрачный раствор сливают, дают стоять
сутки, фильтруют через фильтр, смоченный НС1, и прибавляют ещ» НС1
1 A. G ц n t z е. F. В еп о i t, Bull. Soc. chim. France (4), 35, 712 (1924).
* В а г t h 6, Failures, Bull. Soc. Chim. (3), 7, 104 (1892).
498
Стронций. Сульфурил
(уд. в. 1,17) из расчета 200 г на 1 л раствора. Добавляют 2—3 г осажд
ного SrS04 (может содержать ВаСОз) и дают смеси стоять несколько час
при частом взбалтывании; при этом Sr* ' будет переходить в раствор, оса>°В
дая BaS04. Жидкость фильтруют и выпаривают досуха. Остаток растворяв"
в тройном количестве воды, дают стоять сутки, после чего фильтруют еще паТ
и выпаривают до кристаллизации. Выпадающая соль не содержит ни Ва''3
ни Са' '. *
2. По Б о р н е м а н у1 растворяют БгСОз в разбавленной НС1 при
нагревании, отфильтровывают нерастворившийся остаток и насыщают фильтрат
хлором. Затем добавляют Sr(OH)2 до щелочной реакции, кипятят и отфцЛь.
тровывают выпавшие Fe(OH)s, Al(OH)s, МпОг и др. Фильтрат подкисляют
чистой НС1, кипятят до удаления запаха хлора, выпаривают и кристаллизуют
Препарат полезно еще перекристаллизовать, но полное удаление Ва'* иСа"'
этим методом не достигается.
3. Для получения безводного SrCh нагревают кристаллическую соль д0
удаления большей части кристаллизационной воды, смешивают остаток с
чистым NH4C1 и медленно нагревают в токе сухого хлористого водорода до
полного удаления аммонийных солей. Полученный препарат еще раз
подвергают нагреванию с NFUCl в тех же условиях. Препарат содержит 99,75—
99,80% SrCb2.
Свойства
Длинные прозрачные шестигранные гексагональные иглы уд. в. 1,96,
расплывающиеся во влажном воздухе. При 61,5° переходит в дигидрат, SrCla •
• 2НЮ; при 100° теряет остальную кристаллизационную воду и при 868°
сплавляется в стекловидную массу уд. в. 3,08. Т. кип. ^ 1250°
(экстраполировано). Легко растворим в воде, растворим в 99%-ном спирте (0,86% на
холоду, 0,38% при кипении).
Таблица 454
Растворимость SrCl2 вводе
t°c
0
10
20
30
40
50
60
100 г воды
растворяют
г SrCIi
44,2
48,3
53,9
60,0
66,7
74,4
83,1
о/о SrC!t
30,7
32,6
35,0
37,5
40,0
42,7
45,4
I
ГС
70
80
90
100
ПО
118,8
100 г воды
растворяют
г SrCIi
89,6
92,4
96,2
101,9
109,1
116,4
•/о SrC'2
47,3
48,0
49,0
50,5
52,2
—
Таблица 465
а"
■ 4
1,028
1,054
1,083
1,112
1,143
У
о/о SrC!a
2,97
5,95
8,92
11,90
14,88
дельный вес
o/oSrCV
•6HsO
5
10
15
20
25
d13
4
1,175
1,209
1,244
1,280
1,318
водных
о/о SrCl,
17,85
20,83
23,80
26,78
29,76 j
pa ctbg
°/o SrCl.»
•6HtO
30
35
40
45
50
|ров SrCl2
d15
4
1,358
1,374
°/o SrCla
32,73
33,92
0/0 SrCf2.
•бР^О
55
57
i Heopr. npen., 120.
Ю. H. Huttig, Ch.Slonim,Z. anorg. Ch. 181, 74 (1929).
С у ль фу рил хлористый
499
Испытание
По ОСТ НКТП 3264 содержание SrCk • 6Н2О — не менее 99,7% для всех
квалификаций; наибольшие количества допустимых примесей указаны в
табл. 466; препарат должен выдерживать приведенные в ОСТ испытания на
кислотность.
Таблица 466
Допустимые примеси в SrCl2-6H20
Примеси
^.Нерастворимые в воде вещества . .
2. Сульфаты (SO/')
5. Кальций (Са)
6. Железо (Fe)
7. Щелочные металлы и магний
Допустимое содержание в о/0
в чистом для
анализа
0,003
0,0013
0,0002
0,02
0,03
0,00003
0,012
В ЧИСТОМ
0>005
0,0025
0,001
од
0,05
0,00009
0,024
СУЛЬФУРИЛ ХЛОРИСТЫЙ
Sulfuryl chloride
SO2CI2
Мол. в. 134,97
Sulfurylchloride
Schwefelsaurechlorid
Приготовление
1. Шульце использует для получения препарата реакцию соединения SOa
и С1г в присутствии камфоры, применяемой в качестве катализатора. Для этой
цели в колбу средней величины вносят 10 г грубо измельченной камфоры й
закрывают колбу пробкой, через которую проходит газоприводная трубка,
доходящая почти до дна колбы, и газоотводная трубка, оканчивающаяся сразу
под пробкой.
Поместив колбу в ледяную воду, пропускают газообразный SCb,
высушенный серной кислотой, причем камфора, абсорбируя газ, плавится. Когда
камфора почти насытится (1 ч. камфоры абсорбирует при 725 мм около
0,88 вес. ч. SCb), прекращают приток SO2 и начинают пропускать хлор,
который быстро поглощается жидкостью, обесцвечиваясь. После насыщения
хлором пропускают вновь 30>, затем снова СЬ. Когда таким путем будет
получено приблизительно 30 г SO2CI2 то, снабдив газоподводящую трубку
тройником, пропускают в колбу смесь SCh и СЬ одновременно, регулируя скорость
газов так, чтобы жидкость в колбе не окрашивалась в зеленый цвет. В этих
условиях реакция протекает быстро и спокойно.
Получив необходимое количество препарата, перегоняют его на водяной
бане при 69—70°. После первой перего'нки продукт еще содержит камфору,
повторной ректификацией получают вполне чистый препарат.
Вес употребляемой камфоры должен составлять около 2% от веса
приуготовляемого ЗО2О2.
2. По Мельзе-нсу пропускают одновременно СЬ и ЗОг в ледяную
уксусную кислоту. Получающийся продукт очищают дробной перегонкой.
3. Е. В. Алексеевский, Г. И. Белоцерковский и Т. Г.
Плаче но в описывают удобную аппаратуру для получения SO2CI2 по методу
500
Сурик
Попе1 (соединена SO2 и СЬ в присутствии катализатора—акти'вированног
угля2). °
Хлор и сернистый газ, высушенные пропусканием через концентрированную
H2SO4, поступают в равных объемах в смеситель — склянку Дрекселя А через
тройник В (рис. 71). Для дозировки газов, служат реометры, включаемые
перед тройником В\ скорость поступления БОг и СЬ в смеситель поддержу
вается в пределах 150—200 мл/мин.
Газовая смесь проходит по трубке С в реактор D — широкую стеклянную
трубку (диаметр 20 мм, длина 300 мм), заполненную зерненым активирован-
ным углем и окруженную -водяной муфтой Е. Температура воды контроЛ1ь
руется по термометру F и должна все время поддерживаться около 30°.
Реакция начинается тотчас же и SO2CI2 собирается в приемник G, охлаждав
Рис. 71. Аппарат для получения хлористого сульфурила
мый льдом. Непрореагировавшие газы удаляются через рогатый форштосс //,
обратный холодильник /, промывную склянку с водой К и две (/-образные
трубки L, наполненные натронной известью.
Соединения форштосса Н с реактором D, приемником G и холодильником /
делаются на шлифах.
По окончании работы SO2CI2 переливают в пробирки, которые немедленно
запаивают. Препарат совершенно чист.
Свойства
Подвижная бесцветная или слегка желтоватая жидкость уд. в. 1,67 при 20°,
слабо дымящая на воздухе и обладающая чрезвычайно резким запахом.
Т. пл. — 54,1°, т. кип. 69,1°. Медленно 'разлагается холодной водой, быстрее —
горячей; разлагается также кислотами и спиртом.
Испытание
По техническим условиям НКХП 45—40 при разгонке препарата
фракция 67—70,2° должна составить по объему не менее 95%; d2£ 1,655—1,675.
1 Е. В. Алексеевский, Г. М. Б е л о к е р к о в с к и й и Т. Г. Плач в, нов
„Практические работы по химии зашиты", Оооронгиз 1910, стр. 68.
8 Для активации костаной уголь кипятят с HG1, фильтруют, промывают водой, сушат и
прокаливают.
Сурик свинцовый 501
СУРИК СВИНЦОВЫЙ (ОРТОПЛЮМБАТ СВИНЦА)
plumbum oxydatum Minium Menniffe
™b™m Red lead Bleierthoplumbat
Minium r
РЬз04 или РЬРЬО* Мол. в. 655,63
Приготовление
1. Для получения сурика в небольших количествах можно Жфстумать
следующим образом *. Сначала приготовляют РЬОг, для чего к смеси 500 мл
$%-ного раствора уксуснокислого свинца [РЬ(СНзСОО)г] и 100 мл 3%-ной
jfj&s постепенно прибавляют при размешивании и охлаждении 100 мл
:Щ%-110го раствора КОН. Выпадающий осадок декантируют и промывают хо-
.^юдной водой. Так как осадок получается слизистым, рекомендуется
растереть ехо в ступке, продекантировать и снова растереть. Обрабатывая
полученную пасту горячим насыщенным раствором КОН, переводят ее в сурик. Для
гэтрй цели обрабатывают 10 г пасты 150 г КОН и 50 мл воды. При этом
поручается превосходный красный препарат с содержанием лерекисного кислорода
•2,32% (вместо теоретических 2,33%, соответствующих формуле РЬзОО.
2. По М и л ь б а у е р у2 лучше всего получать РЬз04 в лабораторном
масштабе следующим методом: в плоскую платиновую чашку, нагретую до
: начинающегося красного каления, помещают чистый углекислый свинец,
размешивая его шпателем. После разложения РЬСОз начинается окислительный
процесс, течение которого контролируется анализом на содержание РЫ04.
Для этого пробу вещества (0,3 г) помещают в колбочку, добавляют 1 мл
концентрированной СНзСООН и 10 мл воды и приливают по каплям 4 мл
20%-ного раствора KJ. При энергичном встряхивании проба растворяется,
причем выделяется J2. Жидкость титруют Q.IN раствором NaiSzOa (1 мл 0,Ш
ЫагЗгОз соответствует 0,03428 г РЬзОД
Когда количество РЬз04 перестанет возрастать (что достигается через
несколько часов), кипятят массу несколько раз с 10%-ным раствором
РЬ(СНзСОО)г, декантируют еще теплую жидкость, осадок фильтруют и
промывают горячей водой. При употреблении чистых исходных продуктов
получают чистый препарат.
3. Для очистки су*рика Лёве рекомендует длительное время нагревать в
закрытой колбе технический сурик с 10—15-кратным количеством 10—12%-ного-
раствора Pb(NOs)2. Смесь затем кипятят и оставшуюся массу промывают до
исчезновения реакции на РЬ*' в фильтрате. Потери составляют 16—31%.
Свойства
Красный кристаллический порошок уд. в. 9,07, при нагревании сначала
принимающий еще более яркую окраску, а затем выше 550° разлагающийся
с отдачей кислорода. Нерастворим в воде, растворим в избытке ледяной
СНзСООН. Азотной кислотой разлагается, соляную кислоту окисляет с
выделением хлора.
Испытание (Шмидт)
Примеси (песок, PbSO*, FezOs и др.). 4 г препарата вносят малыми
порциями в нагретую смесь 10 г НЫОз с 10 мл воды и прибавляют 0,2—0,3 г
щавелевой кислоты или сахара, причем сурик должен полностью раствориться.
Испытание на Са* ", Fe''' и Си' * проводят с полученным раствором
аналогично испытанию РЬО (см. стр. 462. Свинца окись, Испытание,- пункты
5 и 6).
1 Pr&p. Ch. вОв.
•Milbiuer, Chem. Ztg.. 38, 474 (1914).
502
Сурьма
СУРЬМА
Stibium Antimony Antimon
Sb At. b. 121,76
А. Сурьма без мышьяка
Приготовление
1. По Ар туе у смешивают 1 ч. возможно тонко измельченного сурьмяного
блеска (Sb2Ss) с 2 ч. NaCl, обливают массу смесью 3 ч. концентрированной
H2SO4 с 2 ч. воды, настаивают в течение 6—8 час. и затем нагревают в
течение часа до кипения. При этом почти вся ЗЬгБз растворяется. Смешивают
раствор с равным объемом спирта (или с таким количеством горячей воды,
чтобы не оставалось осадка) и дают стоять несколько часов. Фильтруют и
прибавляют к фильтрату воды до прекращения образования осадка (т. е.
пока отфильтрованная проба жидкости не перестанет мутнеть от добавления
воды). Осадок отфильтровывают, тщательно промывают дестиллированной
водой, отжимают и сушат при легком нагревании.
100 ч. полученной основной хлористой сурьмы смешивают с 80 ч. Na2CO$
и 20 ч. угольного порошка, и смесь прокаливают в течение 15—20 мин.;
получается сурьма, свободная от мышьяка.
2. По Келеру и Мейеру смешивают 1 ч. продажной сурьмы с 1,25 ч.
NaNOs и 0,5 ч. ЫагСОз, массу нагревают до слабого каления, выщелачивают
после охлаждения водой и оставшийся сурьмянокислый натрий после
промывания и высушивания сплавляют с равным количеством чистого винного камня
(битартрат калия, КНСШЮв); полученный остаток представляет собой сурьму,
свободную от As, К* и Na*, но содержащую некоторое количество РЬ, Си и Fe.
^ Б. Чистая сурьма
Приготовление
1. По Грошуфу1 чистую сурьму можно приготовить восстановлением
сурьмяной кислоты, получаемой в чистом виде, из хлорсурьмяной кислоты.
100 г возможно чистой пятихлористой сурьмы (SbCb) постепенно,
маленькими порциями вносят в 70 мл концентрированной НС1; бурную реакцию
умеряют, охлаждая колбу снаружи холодной водой2. Для получения
однородного, свободного от хлорокиси продукта, по окончании внесения SbCls. смесь
нагревают на водяной бане до 45-—50°, причем осадок растворяется, затем
раство'р охлаждают до комнатной температуры. Кристаллизацию удобнее всего
вызывать затравкой (для получения последней небольшую пробу раствора
сильно охлаждают льдом). Во время кристаллизации жидкость перемешивают,
во избежание образования кристаллических корок. Кристаллическую кашицу
отфильтровывают через воронку Б ю х н е р а (без бумажного фильтра) или
стеклянную воронку с дырчатой фарфоровой пластинкой и несколько раз
промывают концентрированной НС1, охлажденной льдом.
Из фильтрата получают вторую фракцию кристаллов, насыщая раствор
газообразным хлористым водородом при охлаждении холодильной смесью.
Получаемый фильтрат выпаривают на водяной бане до *4 объема и из него
таким же способом получают третью фракцию кристаллов.
Отдельные фракции перекристаллизовывают из концентрированной НС1,
пока ни в кристаллах, ни в маточном растворе не будет обнаруживаться
никаких примесей.
Полученную чистую хлорсурьмяную кислот*' растворяют в возможно
малом количестве холодной воды и осаждают ббльшую часть Sb в виде сурь-
iQroschuff, Z. ас*. Ch. 103. 169 (1918).
' Если исходят из SdCI3 to 10j г ее растворяют в92.*л концентрированной ПО, пропускают
хлор до насыщения к с полученным раствором SbCI5 поступают, как указано аыше.
Сурьма
5СЗ
мЯнои кислоты, добавляя воды из расчета 100 мл на 100 г хлорсурьмяной
кислоты; осаждение заканчивают, прибавляя ГМНЮН до прекращения
образования осадка. Смесь нагревают на водяной бане до начала отстаивания
Кладка; п'ромывают последний несколько раз декантацией горячей водой
отсасывают через бумажный фильтр, высушивают, нагревая в плоской чашке
№ водяной бане, и восстанавливают.
I Для восстановления цианистым калием (яд\) по Б рун к у смешивают равные
части сурьмяной кислоты и порошка KCN и нагревают в фарфоровом тигле
до плавления, затем повышают температуру до сплавления сурьмы. По
охлаждении сплав промывают водой и для удаления вкрапленных кусочков
шлака и KCN переплавляют.
| Лучше восстанавливать водородом (Г е н и г ш м и д т, Ц и нтль, Л и н-
хард), для чего получены ю сур'мяную кислоту переводя"* нагреванием в
фарфоровом тигле в SD2O4 и послетг восстанавливают рг ^ом при 500°.
2. Декстер получил вполне чистую Sb при сильном накаливании
гидрата сурьмяной кислоты ,, <ом тигле с набойкой из сажи.
Образовавшийся королек переплавлялся с небольшим количеством плавленой
сурьмяной кислоты.
Свойства1
Белый, как олово, сильно блестящий металл ясно ристаллической
(листовой или зернистой) структуры уд. в. р "9 Кристаллизуется в гексагональных
.ромбоэдрах.
Sb тверда и хрупка, легко измельчается в порошок. Т. пл. 630°, при 1440°
кипит, давая пары, легко сгорающие в окись. При обычных условиях Sb не
изменяется.
Нерастворима в НС1, HF, разбавленной H2SO4 и щелочах; растворяется в
царской водке и смеси азотной и винной кислот.
Испытание
Продажная сурьма нередко содержит примесь As, Pb, Fe и Си. По
Шмидту хорошее качество сурьмы определяется по листовато-кристаллическому
строению и по легкости, с которой она плавится в блестящие металлические
зерна.
При действии пламени паяльной трубки, королек должен совершенно
улетучиваться, не оставляя желтого налета (РЬ) и не распространяя
чесночного запаха (As). Содержание «различных тяжелых металлов определяется
обычными аналитическими методами после растворения тонко истертого
металла в НС1 с прибавлением небольшого количества HNOs. Присутствие SD2S3
можно определить по запаху H2S, появляющемуся при нагревании истертого
металла с концентрированной НС1.
В. Коллоидальная сурьма
Приготовление
1. По Сведбергу2 золь сурьмы можно получать методом катодного
распыления. Вторичную обмотку индукционной катушки с искрой в 12 см
соединяют параллельно со стеклянным конденсатором с площадью обложенной
поверхности 225 см2, ьторичные полюса соединяют с электродами из сурьмы,
погруженными в наполненную эфиром фарфоровую чашку. При пропускании
тока начинается энергичное проскакивание искр и образование черного золя.
Одновременно с золем часто образуется значительное количество грубой
суспензии.
1 О модификациях сурьмы: „черной Sb" и .взрывчатой Sb' см. Вег. 3», 3837 (1905); 37,898
); Нецтап п, Anleitunsr zum Experimentieren> 3 Aufl. 498; также Prap. Ch. 236.
а Т. s v e d b е г g, Вег, 38, 3625 (1905),
504
Сурьми
2. Герасимов и Морозов1 предлагают следующий метод получен^
растворимой в воде коллоидной сурьмы: 54 мл кислой белковой смеси (На
1 мл СНзСООН—1,25 мл смеси Пааля; см. сноску на стр. 331), нагревают
в течение 10 мин. на водяной бане. К нагретой смеси добавляют 3 мл рас.
твора SbCh следующего состава: 14,69 г SbCb, 14 мл ледяной СНзСООН
14,4 з винной кислоты (НгС^Ов) и 25 мл воды. Затем прибавляют 4,5 Мл
10%-ного раствора фосфорноватистой кислоты (НзРОз) и продолжают
нагревать 15—20 мин. до потемнения раствора. По охлаждении коллоид частично
коагулирует.. Полная и быстрая коагуляция вызывается раствором NasCO*.
Коагулят промывают декантацией и растворяют в воде, в присутствии 2—3
капель 20%-ного раствора NaOH.
3. По Герасимову и Морозову к 54 мл кислой белковой смеси
(на 1 мл СНзСООН —1,25 мл смеси Пааля) прибавляют 7 мл ледяной
СНзСООН, нагревают около 10 мин. и добавляют 20 мл 5%-ного раствора
рвотного камня [K(SbO)C4H4Oe], 4 мл 10%-ного раствора фосфорноватистой
кислоты (НзРОг) и по каплям п'риливают 0,5 мл НС1 (уд. в. 1,19).
Нагревание при помешивании продолжают еще около 10 мин., раствор становится
красным, вишневым и, наконец, почти черным.
Свойства
Блестящий черный порошок, устойчивый в хорошо высушенном виде.
Быстро растворяется в воде, образуя коллоидный раствор, тёмнокрасный п
проходящем свете и почти черный в отраженном. Раствор устойчив в закры
том сосуде; при доступе воздуха окисляется, обесцвечиваясь при этом.
Коллоидные частицы заряжены отрицательно.
СУРЬМА ПЯТИСЕРНИСТАЯ (СУЛЬФИД СУРЬМЫ [2])
Stibium sulfuratum Antimony pentasulfide Antimonpentasulfid
auranticum . Fiinffach-Schwefelan-
Sulfur auratum timon
antimonii Goldschwefel
Sb2Ss Мол. в. 403,82
Приготовление
1. Растворяют 24 г соли Шлиппе (тио-ортосурьмянокпелый натрий
NasSbS4 • 9НгО) в 100 мл воды, и фильтруют, если нужно; разбавляют
раствор 600 мл дестиллированной воды, и вливают его в охлажденную смесь
9 г H2SO4 (уд. в. 1,84) и 200 мл воды. Получившийся осадок Sb2Ss
обрабатывают по Шмидту2 следующим образом.
По возможности без доступа воздуха и как можно полнее удаляют избы*
ток жидкости декантацией. Осадок снова замешивают с водой и промывают
на колоторке, пока промывные воды не будут показывать лишь слабую
реакцию на H2SO4. Фильтруют и осадок снова Промывают дестиллированной
водой. Получившуюся ЗЬгБз осторожно отжимают, осадок измельчают,
высушивают при слабом нагревании (не выше 50°) в отсутствии света и истирают.
2. Пропускают Нг£ в спиртовый раствор пятихлористой сурьмы (SbCb).
Свойства
Темнооранжевый порошок сладковатого вкуса. При нагревании в
отсутствии воздуха распадается на S и черную трехсернистую сурьму (SteSs).
Не растворим в воде, спирте и растворе (ЫН4>2СОз. Чистый препарат не
растворим также в винной кислоте. Растворим в щелочах, (NH4)eS и
(NH4>aSn. Несколько улетучивается при 100—110° без разложения. Ряд
» ЖРХО 62 (4), 827 П930).
• Ausftthr. Uhrb. d. pharm. Ch. I, 440.
Сурьма пятисернистая, пятихлористая и трехсернистая 505
Шторов указывает, что получаемая обычно пятисернистая сурьма в
действительности представляет собой Sb234 + S.
Хранить Sb*Ss следует в сухих банках, защищенных от света; на свету
Препарат светлеет.
испытание
Мышьяк. Встряхивают при 50—60е в течение 2 мин. 0,5 г препарата
| о мл насыщенного при 15° раствора (ЫН^СОз. Фильтрат после
подписания НС1 в течение 6 час. не должен выделять желтого хлопьевидного
осад; а.
СУРЬМА ПЯТИХЛОРИСТАЯ (ХЛОРИД СУРЬМЫ [5])
Stibium Antimony pentachloride Antimonpentachlorid
и pentachloratum
SbCb Мол. в. 299,05
П риготовление
В колбе, снабженной газоподводящей и отводной трубками, расплавляют
SbCls и пропускают через нее сухой хлор, пока последний не перестанет
поглощаться. Тогда удаляют избыток хлора, просасывая через жидкость
воздух, тщательно высушенный пропусканием через РгОз. Наконец перегоняют
SbCb в вакууме.
Свойства
Бесцветная или слабожелтоватая легкоподвнжная жидкость уд. в. 2,39
при 20°, сильно дымящая на воздухе. При 44 мм давления перегоняется (при
68°) без разложения. При охлаждении затвердевает в кристаллическую массу,
имеющую т. пл. + 4°. При атмосферном давлении не может быть перегнана
вследствие распадения на SbCb и СЬ.
При смешении с эквивалентными количествами воды образует
кристаллические гидраты SbCb • НгО и SbCb • 4НгО; большим количеством воды
разлагается. Из воздуха притягивает влагу и затвердевает в гидрат.
СУРЬМА ТРЕХСЕРНИСТАЯ (СУЛЬФИД СУРЬМЫ [3])
Stibium sulfuratum Antimony sulfide Antimontrisulfid
nigrum Antimony trisulfide Antimonsulfur
Antimonite
ShiSz Мол. в. 339,70
А. Аморфная трехсернистая сурьма
Приготовление
1. Пропускают H2S в подогреваемый, солянокислый раствор SbCb, к
которому добавлена винная кислота. Образовавшийся оранжевокрасныи осадок
после промывания теплой водой свободен от сульфохлорида. Подогревание-
раствора во время пропускания НгБ облегчает получение зернистого, легко
фильтрующегося осадка.
2. Особенно интенсивно-красная форма Sb?S3, так называемая «сурьмяная
киноварь» (Antimonzinnober) получается по Штр'олю при осаждении
раствора 5 ч. твердой SbCb в 25 ч. воды раствором б ч. кристаллического
ИагЗгОз в 25 ч. воды и нагреванием точно до кипения. Выпавший оса-
Док отфильтровывают, промывают разбавленной НС1, аатем чистой водой и
высушивают rtpH' 100°.
506
Сурьма
Для удаления незначительной примеси S можно рекомендовать экстРаг^
рование CSt (огнеопасно!).
Свойства
Объемистый оранжево-красный порошок, при нагревании уменьшающийся
в объеме и приобретающий красно-бурый цвет. Уд. в. 4,12—4,65. «СурЬМя.
ная киноварь» — тонкий огненно-красный порошок. Т. пл. 548°; в сильном
калильном жаре п^ри отсутствии воздуха БЬгБз может ^перегоняться без раз.
ложения. В воде не растворима.
Б. Кристаллическая трехсернистая сурьма
Приготовление
1. Осажденную и высушенную S'^jSs нагревают при 210—220° в струе индц.
ферентного газа (СО2, N2). Кристаллизация, сопровождаемая ивтемнением,
начинается в одном месте и распространяется затем по всем направлениям
(Кук).
2. Сплавляют смесь 1 ч. металлической ЗЬ и 0,4 ч. серы. Оба вещества
должны быть тонко истерты.
Для получения искусственного "«сурьмяного блеска» по Л. Лигабо1
через ЗЬгОз пропускают при 450° сначала хлористый водород (% часа),
затем смесь 3 об. H2S и I об. НС1 в течение нескольких часов.
Свойства
Серый, нежно-кристаллический порошок, напоминающий металл.
В. Коллоидальная трехсернистая сурьма
Приготовление
ч
1. По Шульце 4,3 г свежеосажденной ЗЬгОз растворяют в растворе
12,9 г винной кислоты2 и разбавляют прокипяченной дестиллированной водой
до 1 л. Крепость раствора берется такая, чтобы получаемый золь SbsS3
имел концентрацию 1 : 200.
При насыщении раствора сероводородом получается интенсивно-красный,
почти кровавый раствор, прозрачный в проходящем свете и кажущийся
бурым и мутным в отраженном свете. Изменяя концентрацию раствора БЬгОз,
получают, после насыщения сероводо'родом, золи следующих цветов;
1:200 —тёмнокрасный
1:400 — малиново-красный
1:800 —интенсивно красно-желтый
1:1000 —желто-красный, кажущийся в склянке желтым
1:10000 —желтый (напоминающий раствор FeCl3), слабо флкюо-
ресцирующий; в склянке—светложелтый
1:100 000 — слабо-желтоватый
1:1 000 000 — только в литровой колбе заметен желтоватый цвет
2. По А. Эльдеру и П. Буркарду3 получаются очень прочные
золи БЬгЗз при пропускании, H2S через раствор БЬгОз в СНзСООН.
i L. L i g a b о, С. I, 2328 (1938).
* Можно употреблять раствор 2—3 г рвотного камня [K(SbO)C4H4OJ в 1 л иоды
• A. L. Е 1 d е г а, Р. N. В u r k a r d, J. phys. Chem, 41, 622 (1937).
Сурьма треххлористая
501
СУРЬМА ТРЕХХЛОРИСТАЯ (ХЛОРИД СУРЬМЫ [3])
:■ stibium chloratum Antimony trichloride Antimonitrichlorid
Putyrum antimonii Antimonchlorur
Antimonbutter
SbCh Мол. в. 228,13
fta* яготрвленис
1. По Г е н г с е н у * очень чистую SbCb можно получить следующим путем.
J3 длинногорлую колбу Вюрца А (рис. 72) погружают оттянутый (до 8 мм
l|jyfrp. диам.) конец трубки В (легкоплавкого стекла) диаметром около 15 мм,
j/Ш толщине стенок 2 мм. Трубку неплотно набивают довольно крупными
Зеками сурьмы и закупоривают неплотно сидящей пробкой С. Аппарат
Щтанавливается так, чтобы трубка В была несколько наклонна к колбе;
йубку^ помещают на асбестовую подставку или на глиняный желоб,
употребимый в сожигательных печах. После этого пропускают через отросток D
$рую сухого хлора. Сначала некоторое количество хлора, содержащего воз-
Рис. 72. Прибор для получения треххло- Рис. 73. Прибор для возгонки треххлори-
ристой сурьмы стой сурьмы
Щаются (если ток хлора не слишком силен) и образующаяся SbCh стекает в колбу.
Опасаться закупоривания трубки В в оттянутой части не приходится,
так как здесь ЗЬОз, поглощая хлор, переходит в жидкую SbCls. Если
поддерживать непрерывную струю хлора, то аппарат работает автоматически,
необходимо лишь время от времени вводить в трубку свежие куски сурьмы.
По окончании реакции нагревают содержимое колбы с кусками сурьмы и,
наконец, для восстановления последних остатков SbCb еще с порошком
сурьмы. Затем содержимое колбы перегоняют, причем ЗЬСЬ переходит
приблизительно около 220°.
Особенно хороший препарат получается, если очищенную дестилляцией
ЗЬСЬ подвергнуть еще возгонке. Для этой цели SbCb помещают в колбу
Вюрца А, емкостью 1%—2 л, соединенную с приемником В и хлоркальцие-
вой трубкой С (рис. 73); колбу А помещают на водяную баню так, чтобы
часть ее, где находится расплавленная SbCb, была полностью в водяных
парах. Одновременно противоположную стенку охлаждают, для че,го на нее
помещают кусок фильтровальной бумаги, постоянно смачиваемой, холодной
водой.
1 Henien, Rec. Trav. China.. Pays-Bas, 9, 301 (1890).
508 Сурьма. Теллур
В таких условиях ЗЬСЬ испаряется и осаждается на охлаждаемой стенке
в виде длинных ланцетовидных кристаллов. Когда образуется довольно
большое количество кристаллов, колбе дают медленно охладиться, не трогая
прибора. Расплавленная ЗЬСЬ затвердевает и тогда возогнанные кристаллы
стряхивают в приемник В легкими толчками. Затем повторяют весь процесс
В течение дня таким путем легко получить 200 г возопнашюй SbCb.
2. По Эрдману1 смешивают в колбе 100 г порошка ЗЬгЗз (сурьмяный
блеск) с 500 мл НС1 и смесь нагревают под хорошей тягой, постепенно при.
бавляя 4 г бертолетовой соли (КСЮз). Когда ЗЫБз перейдет в раствор
последний отфильтровывают от серы через стеклянную вату и перегоняют
из рето'рты. При этом собирают три фракции: сперва идет соляная кислота
затем желтоватый (от FeCh) концентрированный раствор SbCh и, наконец'
при 215—222° чистая SbCb, застывающая в красивых белых кристаллах'.
Все фракции собирают отдельно; кол-бочки, содержащие чистую ЗЬСЬ,
немедленно запаивают.
Свойства
По способу 1 — красивые сухие кристаллы, по способу 2 — мягкая,
просвечивающая, кристаллическая масса. Уд. в. 3,064 при 26°. Т. пл. 73,2;,
т. кип. 221°. При перекристаллизации из сухого CS* получаются ромбические
кристаллы. На воздухе слабо дымит и жадно поглощает влагу; водой
разлагается, образуя хлорокись (альгаротов порошок). На кожу SbCb
действует чрезвычайно едко. В абсолютном спирте при обычной температуре
растворяется без разложения, при нагревании же вступает со спиртом >в
реакцию с образованием основной соли. Легко растворима в НС1 и растворах
вивдюй кислоты.
СУРЬМА ХЛОРИСТАЯ ОСНОВНАЯ (ХЛОРОКИСЬ СУРЬМЫ,
АЛЬГАРОТОВ ПОРОШОК)2
Stibium oxychloratum Antimony Algarotpulver
oxychloride Antimonoxychlorid
Antimonychlorid
A. SbOCl Мол. в. 173,22
Приготовление
1. По Сабанееву смешивают 10 ч. твердой SbCb с 7 ч. воды. Через
несколько дней осадок полностью кристаллизуется. Его отфильтровывают,
отжимают и промывают эфиром, причем получаются ромбоэдрические
кристаллы до 1,5 мм ширины. Выход меньше теоретического, так как
значительная часть сурьмы остается в растворе.
2. По Шефферу нагревают 114 г SbCb с 69 г этилового спирта до
140—150°; как побочные продукты образуются НО и хлористый этил.
C2H5CI.
Свойства
Бесцветные кристаллы, разлагаемые водой. Растворима в СЗг и в
растворе винной кислоты (НгСЛЮв). При 170° разлагается.
Б. Sb203-2SbOCl Мол. в. 637,95
Приготовление
По Сабанееву обрабатывают SbCls таким количеством воды, чтобы
на 1 ч. ЗЬСЬ приходилось от 5 до 50 ч. воды. При употреблении больших
1 Хим. преп. 44.
■ Альгаротов порошок представляет собой основную хлористую сурьму с переменным со
держанием SbC!| и SbgCV
Сурьма хлористая основная. Теллур $09
да
^дичеств воды или при длительном промывании препарат получается смень-
Здо,м содержанием хлора.
двойства
Белый порошок либо призматические или пластинчатые кристаллы. При
довторной обработке горячей водой или разбавленным раствором ЫагСОз
меряет весь хлор, переходя в БЬгОз. Не растворим в воде, спирте и эфире,
растворим в CS2 и хлороформе (СНСЬ).
ТЕЛЛУР
'к Telluruin Tellurium Tellur
р Те At. в. 127,61
^ А, Чистый теллур
Приготовление
1. Для получения совершенно чистого теллура Гутбир1 рекомендует
Поступать следующим образом. Возможно чистая теллуровая кислота,
помещенная в платиновый тигель (который в свою очередь стоит в несколько
большем платиновом тигле для предохранения от восстановительного
действия газов пламени), нагревается " при медленном повышении температуры
сначала в сушильном шкафу до полного обезвоживания (если нагревать
быстро, расплавившаяся в кристаллизационной воде кислота разбрызгивается
при дальнейшем нагревании), затем на сильной бунзеновской горелке; при
этом кислота разлагается, оставляя ТеОг. Последнюю кладут в маленькие
фарфоровые лодочки, помещаемые в сожигательную трубку, и восстанавливают
накаливанием в сильной ctfpye чистого водорода. Чтобы иметь возможно
более чистый водород, рекомендуется получать его из чистого цинка и
перегнанной H2SO4 и пропускать через несколько промывных склянок,
содержащих по порядку КОН, KMn04, AgNOa и концентрированную H2SO4. При
восстановлении следует пропускать довольно энергичную струю водорода и
лодочки в трубке поместить наклонно (для стекания образующегося
теллура).
Получившийся препарат содержит еще следы ТеОг, которая заключается
в шариках металла. Для дальнейшей очистки его ещё несколько раз
перегоняют в струе водорода. Получающийся теллур отвечает самым строгим
требованиям.
2. Для очистки сырого теллура по Штауденмайерсу2 его окисляют
азотной кислотой, удаляют избыток последней выпариванием с НС1,
обрабатывают остаток разбавленной НС1, фильтруют и осаждают из раствора тел-
лур при помощи SO2. Осажденный теллур отфильтровывают и промывают,
после чего снова растворяют в избытке разбавленной HNOa; затем для
окисления НгТеОз в НгТе04 прибавляют СгОз в количестве, несколько большем
вычисленного, и сильно выпаривают на водяной бане. При этом осаждается
крупнокристаллическая Н2Те04.
Осадок промывают азотной кислотой, растворяют в возможно малом
количестве воды, прибавляют немного спирта для восстановления избыточного
СгОз и осаждают затем теллуровую кислоту прибавлением значительного
количества НЫОз. Повторяя эту операцию дважды, получают чистую НгТе04
в виде бесцветного кристаллического порошка. Его сушат на водяной бане,
переводят нагреванием в ТеОг и восстанавливают ЗОг или нагреванием
в струе водорода, как указано выше.
3. По X а й м л и для получения чистого теллура плавят неочищенный
продукт и погружают в него платиновую п'роволоку до средины жидкой
массы. После застывания слиток завертывают в кусок шерстяной материи или
1 Gutbier, Z. anorg. Ch. 32, 41, (1939); Prap. Ch. 122.
•Staudenmaiers, Z. anorg. Ch. 10, 189 (1ь95); также A. StShler и В Teich Z
anorg. Ch. 98, 8 (1916); О. Н б n 1 s с h ra i d t u. H. Bandrexler, Z anorg. Ch. 223, 92, (19!5ь
610
Теллур. Тионил. Титан
полотна, как в мешок. Затем устраивают электролитную ванну; анодом ел у-
жит платиновая пластинка, катодом — завернутый теллур, электролитом — раз.
бавленный раствор КОН. При пропускании тока на дно сосуда выпадает К2Те .
окрашивая жидкость в коричневато-фиолетовый цвет. '
Выделяющийся на аноде кислород восстанавливает его до элементарного
теллура.
Свойства
Полученный восстановлением осажденный теллур представляет собой
тяжелый коричневый порошок уд. в. 6,1. После плавления и охлаждения
получается в кристаллической форме в виде оловянно-белой, довольно хрупкой
массы с сильным металлическим блеском. Форма кристаллов — гексагональные
ромбоэдры. Уд. в. 6,24. Не растворим во всех индиферентных растворителях.
Растворим только в жидкостях, образующих химическое соединение с Те,
например, в концентрированной H2SO4.
Т. пл. 452,5°, т. кип. 1390°.
Б. Коллоидальный теллур
1. По Гутбиру1 растворяют 2—3 г возможно чистой кристаллической
теллуровой кислоты в 1 л чистейшей дестиллированнои воды и нагревают
раствор на водяной бане до 40—50°; более высокая температура, хотя и не
приносит непосредственного вреда, но вызывает желтое окрашивание, если
вода и сосуды не совершенно чисты.
Приготовленный таким образом раствор смешивают с сильно разбавленным
раствором гидразин-гидрата (1:2000). При этом уже при добавлении первых
капель восстановителя быстро появляется окрашивание и происходит
образование гидрозоля. Прибавляют раствор N2H4 • НгО по каплям до тех пор,
пока окраска жидкости не перестанет изменяться,ш и выливают гидрозоль
в заранее приготовленный диализатор, где и оставляют его до совершенной
очистки.
Неочищенный гидрозоль нельзя фильтровать, так как он при этом частью
осаждается в виде геля.
2. По Г. Б р ищцин гер у2 к 100 мл 0,5%-ной НгТеС)4 добавляют 0,Ш
КгСОз, кипятят и добавляют 5 мл 0,5%-ного раствора аскорбиновой (или изо-
аскорбиновой) кислоты; образуется прозрачный красновато-коричневый золь.
ТИОНИЛ ХЛОРИСТЫЙ
Thionylchlorid
Thionyl chloride Schwefeligsaure-
chlorid
SOCb Мол. в. 118,97
Приготовление3
1. Помещают 50 г PCls в реторту с тубусом, снабженную обратным
холодильником, и пропускают 30*. Вначале PCU превращается в жидкость с
выделением тепла; для окончания реакции необходимо реторту нагревать и
взбалтывать ее содержимое. Реакцию прерывают, когда последняя часть РСЬ
превратится в жидкость.
1 G u t b i е г, 7. anorg. Ch. 32, 51 (1£02); 42, 177 (1904); также Р а а 1 u. Koch, Вег. 38, 534
(1905); Prap. Ch. 124.
■ Н. В г i n t z i 11 я е г, Koll. Z. 78, № 1, 22 (1937):
8 См. также метод П. Каррэ и Д. Либерман, основанный на переведении F.rL ч п-бу-
/\N=SO
тилсульфит [(C4HeO)eS01, затем в тиониланилнн, I | и наконец в SOC?t, выход ьЗ-94%
теор. Р. С а г г 6 e.D. Libermann, Bull. Soc. chim. France (3), 6, J* 3, 579 (1939).
Тионил хлористый. Титана двуокись
511
Несмотря на большое различие в температурах кипения хлористого тио-
w#ia и хлорокиси фосфора (79° для SOCls, 107,5° для РОС1з), разделение
|£Их веществ фракционированной перегонкой является довольно кропотливой
Операцией. Удобнее всего поступать следующим образом.
Разгоняют продукт реакции в колбе, снабженной хорошим дефлегматором,
до фракции: до 80°, 80—85°, 85—90°, 90—95°, 95—100°, 100—105°. Первой и
Последней фракции получается меньше всего. Каждую из фракций, кроме
Йе'рвой и последней, подвергают снова разгонке, собирая такие же фракции.
После 20—25 перегонок средних фракций почти не остается—весь
продукт находится во фракциях до 80° и 100—105°.
•' Первую фракцию подвергают окончательной разгонке (с высокой
ректификационной колонкой, наполненной обрезками стеклянных трубок), собирая
фракцию 77—80°. Препарат очень чист.
По Ш и ф ф у, для того чтобы легко выделить ЗОС1*, следует
переработать не меньше 1 кг РСЬ.
2. По Михаэлису в однохлористую серу (S2CI2), помещенную лучше
всего, в высокие цилиндры, соединенные последовательно, пропускают, при
охлаждении смесью льда и соли, хлор до тех пор, пока прибавка веса не
будет соответствовать вычисленной для образования двуххлористой серы
(SCh). Полученную холодную SCh переливают в большую колбу,
снабженную обратным холодильником и газоотводной трубкой, и перегоняют туда
1 моль SOa, получаемого нагреванием в реторте соответствующего количества
продажного олеума, содержащего до 40% избыточного ангид(рида. Колбу при
этом охлаждают ледяной водой.
При достаточном выделении SO2 и обычно вначале при выделении
твердого вещества (S2O3CI4?) реакция идет сама собой. Твердое вещество, кал
правило, исчезает, когда перегонится весь SCb (если все же остается твердый
остаток, нагревают смесь на водяной бане, пока не образуется подвижная
жидкость):
3SC12 + 4S03 = SOCb + S2O3CI4 + 4S02.
S2O3CI4 + БОз = SOCb + (S02C1)20.
Жидкость подвергают дробной перегонке, Причем сначала улетучиваются
значительные количества абсорбированного SO2 (необходимо иметь хорошую
тягу!). Дестиллат содержит SOCb, немного SCh и некоторое количество
(SO2CO2O, главная часть которого остается в перегонной колбе. Для
удаления SCh дестиллат несколько раз перегоняют с добавкой серы, которая
переводит низкокипящую SCh в S2CI2 с более высокой точкой кипения.
При тщательной работе метод дает около 80% теоретического выхода,
причем препарат очень чист и содержит, самое большее, следы хлористой серы.
Свойства
Бесцветная, сильно преломляющая свет жидкость уд. в. 1,675 при 0°,
с запахом, напоминающим SO2. Т. пл. —105°, т. кип. 78,8°. Водой (еще
быстрее щелочами) разлагается на SO2 и НС1. Растворим в сильных кислотах,
Щелочах и спирте.
ТИТАНА ДВУОКИСЬ
(ОКИСЬ ТИТАНА[4], ТИТАНОВЫЙ АНГИДРИД)
Titanium oxydatum Titanium dioxide Titandioxyd
Titansaureanhydrid
ТЮ2 Мол. в. 79,90
Приготовление
1. По Бертье минерал рутил (представляющий собой нечистый ТЮг)
возможно тонко истирают и отмучивают. Приготовленный таким путем
тончайший порошок сплавляют с тройным количеством КгСОз, причем выделяется
512.
Титан
СОг и образуется титанат калия. Охлажденную массу обрабатывают водой-
при этом титанат калия, РеЮз и SnO* остаются нерастворенными, в то время
как foSiOs, K2M11O4 и K2S11O2 переходят в раствор. Нерастворимый остаток
переносят на фильтр, промывают водой, пока не начнет стекать молочно-
белая жидкость, и растворяют в холодной концентрированной НС1.
Раствор сильно разбавляют водой и пропускают HeS до осаждения Sn
в виде SnS; осадок SnS отфильтровывают и к фильтрату добавляют NHiOH
до Прекращения образования осадка метатитановой кислоты, FeS и MnS.
Если жидкость не имеет теперь запаха HaS, то прибавляют немного (NH4)>S
до полного осаждения железа.
После того как жидкость над осадком после сильного взбалтывания и
стояния станет совершенно прозрачной, декантируют и обливают осадок
крепким водным раствором ЗОг. При этом FeS и MnS полностью растворяются.
Оставшуюся титановую кислоту отфильтровывают и промывают.
При прокаливании кислота, теряя воду, переходит в ТЮг.
2. По Вёле'ру сплавляют в платиновом тигле, помещенном внутри
глиняного, очень тонко истертый рутил с двойным по весу количеством КгСОз.
Сплавленную массу истирают и растворяют точно в необходимом количестве
плавиковой кислоты (HF). Образуется трудно растворимый foTiFe (титано-
фтористый калий), который начинает выпадать в виде чешуек. Потом
нагревают, прибавляя, в случае необходимости, немного воды до растворения соли
и кипящую жидкость фильтруют (стеклянную посуду -можно употреблять
только в том случае, если не было взято избытка HF).
При охлаждении большая часть foTiFe выпадает в виде блестящих
чешуек, так что жидкость обращается в кашицу. Собирают соль на бюхнеров-
скую воронку, осторожно прессуют ее при помощи стеклянной пробки,
промывают несколько раз холодной водой и отжимают между фильтровальной
бумагой. Для окончательной очистки перекристаллизовывают соль из кипящей
воды. После сушения получают листочки с перламутровым блеском. Их
растворяют в го'рячей воде и добавляют ЫНЮН до прекращения выпадения
снежно-белого осадка титановой кислоты. Осадок отфильтровывают и
прокаливают, причем остается чистая ТЮз.
3. В очень чистом виде ТЮг получается осаждением титановой кислоты
при действии ЫНЮН на растворы TiCU или ТЦБОф; осадок промывают,
высушивают и прокаливают1.
Свойства
Снежно-белый аморфный порошок без вкуса, уд. в. 4,13—4,25. Плавится
при 1560° (по новым данным — около 1800°). Не растворим в воде; после
прокаливания не растворим также в НС1 и разбавленной H2SO4 даже при
кипячении. v
ТИТАН СЕРНОКИСЛЫЙ ЗАКИСНЫЙ (СУЛЬФАТ ТИТАНА[3])
Titanium sulfuricum Titanium sulfate Titansulfat
oxydulatum (oxydul)
Tb(S04)a Мол. в. 383,98
Приготовление
По методу ИРЕ А 100 г двуокиси титана (ТЮг) вносят в 250 мл H2SO4
(уд. в. 1,84), нагревают смесь на песочной бане до 240° и выдерживают при
этой температуре 6 час.
Массу охлаждают до 50—60°, осторожно добавляют 2 л воды,
подогревают до 60—70° и в течение 2 час. размешивают механической мешалкой.
Добавляют еще 1 л воды, фильтруют и насыщают фильтрат сероводородом
1 О получении геля TiO, см. ЖПХ 11, 4 (1933),
Титан сернокислый и треххлориСтый 513
*
г
/
I течение 2,5—3 час. После отфильтровывания осадка сульфидов раствор
Одаривают при 80—90° до уд. в. 1,29 и подвергают электролизу.
™ В большую банку (рис. 74) помещают глиняный или
фарфоровый пористый стакан, служащий диафрагмой,
0 банку наливают раствор Ti(S04)2 и погружают катод —
свинцовую пластинку А.
В пористый сосуд наливают 20%-ную H2SO4 и
помещают свинцовый анод В.
Ток регулируется с таким расчетом, чтобы его
плотность на катоде составляла 0,02—0,03 а/см2. Во время
электролиза электролит желательно энергично
перемешивать, лучше всего механической мешалкой.
Через несколько часов испытывают полноту
восстановления; для этой цели к налитому в пробирку раствору
яимола и концентрированной H2SO4 приливают
осторожно, чтобы жидкости не смешивались, немного
испытуемого раствора. Если восстановление не закончено, на
границе раздела появляется яркокрасное кольцо. Когда
весь Ti'V" перейдет в ТГ'*, кольцо принимает
желтый цвет. Можно также контролировать течение
процесса, отбирая изредка пробу электролита и титруя ее
раствором FeCls.
Добившись полного восстановления, раствор Ti2(SO-i)3 переливают в банки
с притертыми Пробками, наливая жидкость до самых пробок во избежание
окисления.
В
Рис. 74. Прибоо лля пэ-
дучения сернокислого
титана
И с п ы*т а н и е
По техническим условиям НКХП 390—41 15%-ный раствор Tia(304)s должен
быть темнофиолетового цвета; содержание сернокислого титана в растворе
должно быть не менее 15%; наибольшие количества допустимых примесей
указаны в табл. 467.
Таблица 467
Допустимые примеси в растворе Ti2(S04);{
Примеси
1..Вещества, не осаждаемые аммиаком
Допустимое
в чистом для
анализа
0,02
0,101
содержание в °/0
в чистом
' 0,08
0,005
ТИТАН ТРЕХХЛОРИСТЫЙ (ХЛОРИД ТИТАНА'[3])
Titanium trichlora- Titanium trichloride Titantrichlorid
turn
T1CI3 Мол. в. 154,27
Приготовление
1. Штелер и Бахран1 рекомендуют нагревать смесь TiCU с
водородом; для этой цели авторы предлагают следующий аппарат (рис. 75).
Силундовая трубка А длиной в 25 см, с внешним диаметром 12 мм и
внутренним 4 мм закреплена обоими концами в угольных стержнях В, которые
„ * Stabler, Bachran, Вег. 42, 3215 (1909); 44, 2907 (1911); Monateh. 34, 182? (1913); Prap.
Ch. 620.
514
Титан
соединены с источником тока высокого напряжения толстыми медными пп0
волоками С. Весь нагревательный прибор окружен двойной медной муф^л
между стенками которой циркулирует холодная вода. Концы угольных степ'
жней приблизительно на Vs выступают из медного холодильника и покрыты *
стеклянными колпачками D, благодаря которым пространство между
нагревательным прибором и холодильником герметически закрыто. Части прибора
соединены резиновыми кольцами, покрытыми коллодием.
После того как аппарат собран, вытесняют из него воздух и влагу сухим
водородом, причем трубку А слабо нагревают. Затем готовят совершенно
сухую колбу, наполненную TiCU. Водород (Предварительно тщательно
освобожденный от кислорода пропусканием через платинированный асбест и
высушенный при помощи Р2О5) проходит через склянку с TiCU. нагретым До
50—60°, и поступает в аппарат (через трубку £), насыщенный парами TiCl.,. '
Рис. 75. Прибор для получения хлористого титана
Нагревают силундовую трубку до светлокрасного каления и пропускают
водород с такой скоростью, чтобы следы TiCU выходили из аппарата еще
неразложенными. Медный кожух в течение 2 час. покрывается толстым слоем
TiCb. По окончании работы отъедшгяют колбу с TiCU и дают аппарату
охладиться в токе водорода.
2. Для получения раствора TiCb, пригодного для аналитических целей
(титанометрия), раствор TiCU пропускают через редуктор Джонса,
наполненный амальгамированным цинком или алюминием1.
3. М. Билли и П. Брассёр2 нашли, что наилучшие результаты
получаются при восстановлении TiCU металлической Sb (полученной осаждением
цинком из раствора SbCls).
Описание специально сконструированлого авторами прибора для восстано
вления * см. в оригинале.
Свойства
Блестящие, интенсивнофиолетовые кристаллические чешуйки. Расплывается
во влажном воздухе. В воде растворяется со вспышкой,. При стоянии на
воздухе раствор обесцвечивается и из него осаждается метатитановая кислота.
Испытание
По ГОСТ 311-41 препа'рат (15%-ный раствор в разбавленной соляной
кислоте) по внешнему виду должен представлять собой темнофиолетовую
жидкость, обесцвечивающуюся при окислении на воздухе; содержание TiCb
должно быть не менее 15%; наибольшие количества допустимых примесей
указаны в табл. 468.
1 Об электролитическом восстановлении раствора TiCb см., например, D i с t h e 1 m u.
F о е г s t е г, Z. phye. Cli. 62, 129 (lb08).
* M. В i 11 у* . Р. В г а в s e u г, С. г. 200, № 21, 1765 (1935)
i~
Титан четыреххлористый
Допустимые примеси в в растворе TiCJ8
Таблица
1Г 1
Примеси
-1; Нерастворимые вещества
Ж Сульфаты (SO*')
3. Железо (Fe)
4. Ill/личные и щелочноземельные
металлы (в виде сульфатов) ....
Допустимое содержимое в о/о
в чистом для
анализа
0,002
0,ои1
0,0(Л
0,01
в чистом
0,008
о.осз
0,003
0,02
ТИТАН ЧЕТЫРЕХХЛОРИСТЫЙ (ХЛОРИД ТИТАНЛ[4])
Titanium chloratum Titanium tetrachloride Titantetrachlorid
TiCl4 Мол. в. 189,73
Приготовление
1. По Пьеру из 4 ч. порошкообразной титановой кислоты и 1 ч. сажи
при- помощи масла формуют шарики величиной с орех, обсыпают их
угольным порошком и прокаливают в тигле. Затем нагревают их в ctfpye хлора
в тугоплавкой стеклянной или фарфоровой трубке *. TiCh улетучивается и
собирается в охлаждаемом, хорошо высушенном приемнике. Полученный
продукт очищают ректификацией.
Если вместо титановой кислоты применяют природный рутил, то
одновременно получается твердый FeCb, от которого TiCU отделяют сливанием и
перегонкой (Пфордтер). Для очистки от хлора и SiCU перегонку ведут
над ртутью. Для получения препарата, свободного от ванадия, TiCU
встряхивают до полной прозрачности с амальгамой натрия при температуре около
48—50°.
2. Для переработки более значительных количеств минералов Ш тел ер2
рекомендует следующие способы, в зависимости от того, какое сырье
подвергается переработке — рутил или ильменит.
а) Из рутила. 1 кг минерала омешивают с 350—400 г угля и сплавляют
маленькими порциями в вольтовой дуге при силе тока в 200—250 а и
напряжении в 35 в.
Процесс длится 40—60 мин. После охлаждения хорошо сплавленный
карбид тонко измельчают и обрабатывают сухим хлором при начинающемся
красном калении. Отгоняющийся TiCU сначала нагревают на водяной бане
До удаления главной части растворенного хлора и затем дестиллируют. После
обработки дестиллата амальгамой натрия для освобождения от ванадия (см.
способ 1) продукт еще раз перегоняют в вакууме, причем получается около
2 кг совершенно чистого TiCh. Нелетучий остаток после хлорирования
прокаливают на воздухе и снова сплавляют в электрической печи для перевода
в карбид.
б) Из ильменита. 1 кг ильменита тонко измельчают и сплавляют с
угольным порошком в вольтовой дуге при силе тока в 150—200 а и напряжении
30 е. Получают ковкий твердый королек железа с небольшой примесью Ti
1 При ббльших масштабах в глиняной или фарфоровой реторте или стеклянной реторте.
обмазанной глиной. '
* • S11 h I е г, Вег. 38, 2<19 (1906).
516
Торий
и С; ббльшая же часть титана* образует шлаковистый карбид, который
перерабатывается тем же путем, как и карбид из рутила (см. а).
3. Для очистки продажного продукта (от on и Fe) Вагнер рекомендует
перегнать его 1—2 раза над амальгамой натрия и 2—3 раза подвергнуть
дробной перегонке.
Свойства
Прозрачная, как вода, жидкость, уд. в. 1,76 с характерным запахом
Т. пл. —23°, т. кип. +136.5°. На воздухе при обыкновенной температуре
образует туман; во влажном воздухе разлагается. В воде растворим только
с разложением, в концентрированной НС1 дает раствор титанохлористоводо-
родной кислоты (FhTiCle).
ТОРИЙ АЗОТНОКИСЛЫЙ (НИТРАТ ТОРИЯ)
Thorium nitricum Thorium nitrate Thoriumnitrat
Th(NOa)4 • 4H20 Мол. в. 552,22
Приготовление
1. По Коппелю и Гольткампу1 100 г продажного сернокислого
тория осаждают аммиаком. Кипятят жидкость несколько минут на голом
пламени, отсасывают осадок на нутч-фильтре и промывают водой, пока
фильтрат не перестанет давать реакцию на SCV'.
Остаток растворяют в концентрированной Н1МОз и выпаривают досуха на
водяной бане.
Полученный продукт, по утверждеиию авторов, отвечает формуле тетрагид-
рата Th(NO*)4 • 4НЮ.
2. По Браунеру2 чистый щавелевокислый торий, Тп(Сг04)2• 6Н2О
кипятят с избытком концентрированной HNCh, пока не получится светлый
раствор, уже не выделяющий красных паров при кипячении.
Выпаривают бблыпую часть Н1ЧОз при 100°, затем продолжают
упаривание около 80°, пока из кислого раствора не начнут выделяться кристаллы.
Тогда раствор охлаждают до комнатной температуры, выпавшие кристаллы
отсасывают и сушат между листами фильтровальной бумаги.
Свойства
Бесцветные кристаллы, легко расплывающиеся на воздухе. Хорошо
растворим в воде (с кислой реакцией) и спирте. При 500° соль полностью
переходит в ТпОг.
Испытание8
1. Препарат ч.д.а. должен давать после прокаливания не менее 48%
остатка (ТпОг) и может содержать не больше чем 0,004% Р2О5 и 0,05%
суммы окисей Al, Ca. Mg и щелочных металлов.
2. Растворимость. 26 г Препарата должны полностью растворяться в 25 мл
воды при взбалтывании в течение 40 мин.; допускается лишь слабая
желтоватая окраска раствора.
3. Сульфаты. (SOS'). К профильтрованному раствору 1 г препарата
в 20 мл воды добавляют 2 мл HN03 (уд. в. 1,15) и 1 мл 10%-ного раствора
BaCU — через 5 мин. не должно быть мути или осадка.
* Корре 1. Holtkamp, Z. алог?. Ch. 67, 290 <19Ю); Ргар. Ch. 760.
2 В. В г a u n е г, J. Chem. Soc. 78, 984 (1898).
1 Испытание хим. реактивов, 377.
Щ/ Торий азотнокислый. Тория двуокись 517
ь ,-. 4. Хлориды. (СУ). При добавлении к раствору 1 г препарата в 20 мл
^оды 2 мл HNOs (уд. в. 1,15) и 1 мл Q,IN А^ГМОз может появиться лишь
'^абая опалесценция.
V 5. Железо и тяжелые металлы. При добавлении к 30%-ному раствору
"Препарата равного объема 2%-ного раствора NFUCNS может появиться лишь
слаборозовое окрашивание.
При омешении 20 мл 30%-ного раствора препарата с 50 мл
сероводородной воды допускается лишь слабое потемнение жидкости.
I 6. Церий (Се'ш). К раствору 2 г препарата в 10 мл воды добавляют
КаСОз до растворения образующегося вначале осадка.
После приливания к пол учении .л у раствору «нескольких капель 3%-ной
НгОа жидкость не должна принимать заметного желтого оттенка.
ТОРИЯ ДВУОКИСЬ (ОКИСЬ ТОРИЯ)
Thorium oxydatum Thorium oxide Thoriumdioxyd
Thoria Thoriumoxyd
Thorerde
ТЪОг Мол. в. 264Л2
Приготовление
1. Р. Мей ер1 рекомендует в платиновой или железной чашке облить 50г
монацитового песка 75 г концентрированной H2SO4 и, постепенно повышая
температуру, нагревать до 200° в течение 3—5 час. При этом масса
превращается в белую или сероватую кашицу (смесь растворимых сульфатов).
К концу операции пробуют на полноту разложения, для чего небольшое
количество массы переносят стеклянной палочкой на часовое стекло, растирают
с несколькими каплями воды и рассматривают при помощи лупы, имеются
ли еще желтые зерна монацита. Если они обнаруживаются, то продолжают
нагревание.
Когда разложение закончено, массу охлаждают и маленькими порциями
переносят при тщательном размешивании в 750 мл ледяной воды, причем
происходит весьма медленное растворение. Дают жидкости стоять до
растворения всех сульфатов; нерастворившийся остаток (циркон, кварц, кремневая
и титановая кислоты и др.) отфильтровывают и к фильтрату приливают
концентрированный раствор щавелевой кислоты (Н2С2О4) до полного выпадения
оксалатов. Последние отсасывают, промывают теплой водой и, поместив в
чашку, обливают концентрированной HNOs (с примесью дымящейся HNOs)
и окисляют 0,Ш раствором КМп04а.
После этого нагревают на голом опне до полного растворения и раствор
выпаривают на водя-ной бане досуха.
Остаток обливают концентрированной НС1 и снова выпаривают, причем
нитраты переходят в хлористые соли. Последние растворяют в воде и
к кипящему, почти нейтральному раствору прибавляют ЫагБгОз. При этом
выпадает основной тиосульфат тория, содержащий небольшое количество
церитовых окислов. Его отфильтровывают (фильтрат может быть применен
для получения солей церия) и растворяют в концентрированной НС1. Раствор
отфильтровывают от серы и выпаривают досуха; остаток растворяют в \0 мл
воды и из раствора при 60° осаждают Th, прибавив 1 мл 30%-ной Н2О2.
При этом выпадает гидрат перекиси тория в виде желатинозного осадка; его
отфильтровывают через беззольный фильтр, промывают водой с добавлением
NH4NO3 и Прокаливают осадок вместе с фильтром. Таким путем получается
около 0,5 г сравнительно чистой ТпОг.
1 Ризенфельд, Неорг. практ. 287.
■ Можно перерабатывать оксалаты иначе—по способу Фронштейна и Мэ (М. Frons tein.
Т. М t i, Z. tn$ew. Ch. 642 (1в97); Prup. Ch. 757.
518
Торий
2. Вырубов и Вернейль1 предлагают растворить монацит в H2S04)
разбавить и дать стоять 12—18 час. Нерастворившийся остаток отфильтровьь
вают и прибавляют к раствору щавелевую кислоту в количестве,
необходимом для осаждения половины всех металлов. Выпавшие оксалаты,
содержащие немного фосфатов, отфильтровывают, промывают до исчезновения
реакции на РО/" в промывных водах и обрабатывают при нагревании 10%-ным
раствором ЫагСОз, вследствие чего оксалаты полностью переходят в угле-
кислые соли. К раствору прибавляют немного NaOH для полного
осаждения Th.
Осадок карбонатов промывают до удаления С2О4", растворяют в
возможно малом (без избытка) количестве НС1 и в полученный почти
нейтральный раствор вносят малыми порциями ВаОг до прекращения образования
осадка. Получившийся к'расноватый осадок содержит весь Th и, кроме того,
на 20—30% состоит из окислов других металлов. После промывания осадок
на холоду растворяют в концентрированной НС1, удаляют Ва* "" прибавлением
H2SO4 и после фильтрования разбавляют раствор так, чтобы получился
15%-ный раствор кислоты, из которого щавелевой кислотой снова осаждают
оксалаты. Осадок отфильтровывают и по возможности полнее вымывают из
него железо, после чего обрабатывают его возможно концентрированным
раствором (ЫНОгСОз, содержащим ЫНЮН. В растворе получают весь торий,
содержащий около 7% других металлов.
К жидкости прибавляют NaOH и образовавшийся осадок промывают до
удаления С2О4". Растворяют его в точно необходимом количестве HNOe,
разбавляют до 2% содержания и осаждают пе(рекисыо водорода (на 1 кг Th
7—8 л 10%--ной ЬЬОг). После фильтрования объемистый осадок промывают
до тех пор, пока в фильтрате ЫНЮН не перестанет давать осадок
(фильтрат, содержащий немного Th, осаждают NH4OH и осадок ©новь
употребляют).
В полученном таким путем соединении Th еще содержится около 1%
церия. Для удаления последнего растворяют осадок снова в HNOs и вторично
обрабатывают раствор Н2О2. Выпавший осадок теперь свободен от Се,
однако содержит еще H2SO4, НзР04 и Са*'. Его растворяют в НС1,
осаждают щавелевой кислотой и разлагают оксалат возможно чистым NaOH.
Наконец осадок, содержащий значительное количество щелочи, растворяют
в НС1, осаждают ГМШОН и полученный осадок прямо или через нитрат
переводят в чистую ТпОг.
3. Для получения ТпОг из ториевых солей прокаливают до постоянного
веса Th(NOs)4 или Th(S04)2, или соли органических кислот.
4. Дл.я получения кристаллической ТЬОг по Норденскьольду и Хи-
д е н и у с у аморфную ТпОг прокаливают в платиновой чашке в фарфоровой
печи с четырехкратным по весу количеством безводной буры; массу по
охлаждении выщелачивают водой.
Свойства
Аморфная ТпОг — белый порошок, кристаллическая — коричневые
кристаллы. Уд. в. 9,87. Прокаленная и кристаллическая ТпОг медленно
растворяется только в концентрированной H2SO4. При накаливании ТпОг не
плавится даже при употреблении в качестве плавней углекислых солей или
щелочей (т. пл. 3050°). Ни углем, ни калием не восстанавливается до
металла.
Испытание2 (для препарата ч. д. а.)
Препарат испытывают на летучие вещества, церий и празеодим, карбонаты,
мышьяк, титан, железо а тяжелые металлы.
ч iWyrouboff, Verneull, С. г. 127. 412,863 (1898); см также. Bull. Soc. Шт. [3l, 19,
219 (189в); М. К ass, Dissertation, Berlin (1904); Ргар. Ch. 758.
9 Испытание ХИМ. оеактмппп. Д7Я.
yiv^^l, *■»• '\ и» »р uioj^Jiqu Ш, JJC1 II
9 Испытание хим. реактивов, 378.
Торий сернокислый
519
1. Летучие вещества. Потеря в весе при прокаливании 1 г препарата в
й^чение 5 мин. в платиновом тигле не должна превышать 0,01 г.
I 2. Церий и празеодим (Се и Рг). При выпаривании смеси 0,5 г ТЬОг с \ мл
Ю^Оз (уд. в. 1,3) и последующем прокаливании остаток может иметь только
рлабо желтую или розоватую окраску.
;" ■ 3. Карбонаты (СОз"). При действии «а препарат НС1 (уд. в. 1Д2)
допускается лишь очень слабое выделение газа.
4. Мышья*. (As). 2 г препарата, растертые с 10 мл реактива Беттендорфа
{(см. стр. 425), не должны в течение часа приобретать темной окраски.
I 5. Сплавляют 0,5 г препарата с 5 г NaHS04 и сплав растворяют «в 50 мл
медяной воды.
| а) Железо и тяжелые металлы. При добавлении к 10 мл полученного
распора KCN3 может появиться только слаборозовая окраска, а от
сероводородной воды не должно быть потемнения.
б) Титан (Ti). К Ю мл раствора добавляют несколько капель 3%-ной
НгОз, причем не должно появляться желтой или красной окраски.
ТОРИЙ СЕРНОКИСЛЫЙ (СУЛЬФАТ ТОРИЯ)
Thorium sulfuricum Thorium sulfate Thoriumsulfat
Th(S04)2 • 9НгО Мол. в. 586,38
Приготовление
1. Из перегоревших ауэровских колпачков можно получать Th(S04)2 по
Мануэлли и Гаспаринетти1 следующим способом.
Золу от колпачков долгое время обрабатывают концентрированной НС1,
после чего остаток промывают и сплавляют с 3 частями NaH304. Сплав
растворяют в воде подкисленной НС1, и Th осаждают из раствора щавелевой
кислотой.
рсадок оксалата тория растворяется в растворе (NH4)2Cs04 и из раствора
прибавлением НС1 онова высаживают Th(G?C>4)2. Последний или прямо
растворяют в H2SO4 или же сначала переводят в окись и ее уже переводят
в сульфат действием H2SO4. . При медленном испарении раствора,
содержащего значительное количество H2SO4, получают Th(S04>2 • 9НгО в крупных
кристаллах.
2. По М е й е р у и Г у м п е р ц у 2 тонко истертую TI1O2 смачивают водой
и' выпаривают несколько раз в платиновой чашке с крепкой H2SO4, не доводя,
однако, до каления. Полученную соль, несколько загрязненную бисульфатом,
тонко истирают и, при размешивании механической мешалкой, вносят в
ледяную воду. Если при этом остается нерастворимый остаток, то его снова
обрабатывают H2SO4.
Раствор упаривают при 30—35° до кристаллизации, причем -выпадает
ThKSOO* • 9Н20.
Свойства
Светлые, как вода, хорошо образованные моноклинические призмы, уд. в.
4,23. Несколько растворим в воде (1,38% при 13°, 1,85% при 25°). При
нагревании до 400° обезвоживается, при красном калении переходит в основную
соль.
1 Manuelli, Gasparinetti, Gazz. chim, ital. 32, 11,523 (190P); Prap. Gil. 763.
« R. J. Meyer, A. Gumperz, Ber. 38, 820 (19.5); 15, 2619 (1882); Prap. Ch, 764,
520
Углерод
УГЛЕРОД
Carboneum Carbon Kohlenstoff
С At. в. 12,010
А. Древесный уголь
Carbo ligni Wood charcoal ., Holzkohle
Приготовление
1. Для получения древесного угля в порошке1 наполняют горн кусками
продажного древесного угля величиной в кулак и зажигают. Как только уголь
раскалится, закрывают печь железной крышкой. Когда уже ни дым, ни пар
не выделяются, уголь тушат, для чего прекращают доступ воздуха, или
высыпают уголь на холодную металлическую (или каменную) плиту, или же
помещают его слоями в плотно закрываемый горшок.
Затем сдувают при помощи мехов золу с поверхности кусков и
измельчают еще теплый уголь в ступке до грубого по'рошка, который тотчас
складывают в плотно закрываемые банки. Просеиванием получают еще более
тонкий порошок. Охлаждение накаленного угля, измельченще, просеивание и
складывание в банки должны быстро следовать одно за другим, так как
уголь постепенно адсорбирует из воздуха влагу и газы.
2. Для получения беззольного угля по Миллеру2 уголь измельчают,
просеивают через сито 300 меш. и замешивают в кашицу с чистой HF.
Полученную массу нагревают сначала слабо до исчезновения паров HF, затем
сильнее.
Сухой уголь кипятят с концентрированной НС1, разбавляют водой и
отсасывают на воронке Бюхнера.
Обработка соляной кислотой проводится дважды, после чего отмывают
водой от СГ.
Для кровяного угля трехкратная очистка по приведенному методу снижает
содержание золы с 8,39 до 0,06%; для активированного сахарного угля уже
однократная очистка понижает зольность с 0,1 до 0,00% V
Свойства
Хорошее качество угля определяется по черному блестящему излому,
чистому звуку при ударе о твердый предмет и по сгоранию без дыма и Пламени.
Истинная плотность4 аморфного углерода 1,475. Хороший уголь содержит
87,5—91% углерода. 1,3—2% золы, 5—7% воды, 1J8—2,5% водорода и
},5-Ч),5% кислорода.
Т. пл. и возгонки: угля 5 — около 3500°.
Испытание
Порошок древесного угля не должен иметь примесей, извлекаемых
этиловым спиртом. Он должен сгорать без пламени, оставляя не больше 5%
остатка.
1 Komm. z. Deut. Arzneib. 1, 329.
2МП1ег, J. phys. Ch. 32. 2967; E. В. А л е к сее в с к и й, Общий курс химии мщлты,
ОНТИ, 1935, ч. 1, стр. 236.
8 О специальной установке для получения чистого аморфного углерода из ацетилена см.
J. Е. D а у, Ind. Eng. Chem. 28, 234 (1936).
« I. Mat б una ga, С I, 731 (1936).
' Т. кип. графита лежит около 3180° [С. I, 3392 (1930)).
Углерод 521
В. Сахарный уголь
Charcoal from Zuckerkofile
sugar
Приготовление
Нагревают истертый сахар сначала осторожно, при ограниченном доступе
воздуха, до прекращения выделения паров, затем повышают температуру до
начинающегося красного каления. Если препарат предназначается для
приготовления безводных хлоридов или бромидов из окисей, то целесообразно
прокалить его еще раз в струе хлора (или, соответственно, брома). Продукт
следует тотчас поместить в закупоренные банки, так как он легко притягивает
влагу.
В. Кровяной уголь
Carbo sanguinis Charcoal from Blutkohle
Carbo animalis blood
Приготовление
По указанию Ваяино1 для получения кровяного угля выпаривают смесь
8 ч. крови и 1 ч. КгСОз и прокаливают остаток без доступа воздуха при
.550—950°, обугленную массу истирают, обрабатывают водой для удаления
растворимых примесей, затем — разбавленной НС1 и, наконец, горячей водой
до нейтральной реакции.
Выход 10%' от веса крови,
Свойства
Сухой легкий черный порошок. Чем меньше удельный вес препарата, тем
выше его адсорбционная способность.
Г. Активированный уголь
При готовленне
По Дубинину2 обыкновенный древесный уголь измельчают, просеивают
через сито 12—15 меш и осторожно прокаливают (для удаления смолистых
веществ) в медном или железном тигле в течение часа. Затем 30 г угля
обливают 500 мл 4N HNOs и оставляют на сутки; на другой день кипятят
смесь в течение 1 часа, отсасывают на воронке Бюхнера, промывают водой
и кипятят 1 час с 2 л дестиллированной воды. После высушивания при 110°
уголь прокаливают в закрытом фарфоровом тигле в течение получаса.
Затем повторяют обработку HNOs, промывают водой, сушат при 110е* и
прокаливают в фарфоровой трубке, обогреваемой электропечью, при 600—800°
в течение часа, откачивая выделяющиеся газы масляным насосом. После
охлаждения в вакууме впускают в аппарат сухой воздух, переносят уголь в
банку с притертой пробкой и хранят в эксикаторе над СаСЬ или Р2О53.
Свойства
Мелкие черные кусочки, отличающиеся значительной пористостью.
Поверхность 1 г активированного угля колеблется между 10 и 1000 м2, что
обусловливает высокую адсорбционную способность препарата.
1 Ргар. Ch 269.
я Е. В. Ал е кс еев ский, Общий курс химии защиты, ОНТИ, 1935, т. 1, стр. 237.
• ■ О других методах получения активированного угля см. Е. В. А л е к сее век ий, loc. eft,
втр. 238 и ел.
522
Углерод
Испытание1 ^
Ана'лиз активированного угля производится в зависимости от его
назначения (см. подробнее описание в книге Е. В. Ал ек сееве кого). Качественно
препарат испьгшвается на содержание* N, S, Zn" ', Си' ', Со*', S04", СГ на
примесь смол, на кислотность и др. Ниже Приводится только испытание на
обесцвечивающую способность угля.
В цилиндре с притертой пробкой взбалтывают 0,1 г высушенного при 120°
и просеянного через тонкое сито испытуемого угля с 25 мл 0,15%-ного
раствора метиленовой сини. После обесцвечивания добавляют новую порцию
метиленовой сини (5 мл), снова встряхивают и продолжают испытание, пока
раствор не перестанет обесцвечиваться. Хороший^ медицинский уголь должен
обесцветить не менее 35 мл раствора метиленовой сини.
УГЛЕРОДА ОКИСЬ
(ОКИСЬ УГЛЕРОДА[2], УГАРНЫЙ ГАЗ)
Carbon monoxide Kohlenoxyd
Kohlenmonoxyd
CO Мол. в. 28,010
Приготовление
I. Из муравьиной кислоты и HzSOa. По Рулпу2 в колбу, снабженную
капельной воронкой и газоотводной трубкой, помещают концентрированную
H2SO4, нагревают до 100° и из воронки приливают по каплям 98%-ную
муравьиную кислоту (НСООН). Скорость газовыделения регулируют быстротой
приливания кислоты.
По маре расходования кислоты газовыделение замедляется; тогда
помещают колбу на проволочную сетку и нагревают очень маленьким пламенем,
Причем реакция идет совершенно гладко до конца.
2С Из железистосинеродистого калия [K\Fe(CN)b\ и H2SO4. По Гримму и
Р а м д о р у нагревают в объемистой колбе, снабженной газоотводной трубкой,
1 ч. тонко измельченного K4Fe(CN)e с 9 ч. концентрированной H2SO4;
газоотводную трубку соединяют с промывной склянкой, содержащей NaOH (или
известковое молоко) для поглощения одновременно образующейся СОг.
Когда смесь нагреется до температуры начала реакции, что легко заметить
по начинающемуся вспениванию в колбе, выделение СО довольно долгое
время происходит само собой и нагревание следует прекратить. Когда
выделение газа замедлится, снова осторожно подогревают.
Из 15 г K4Fe(CN)e получается около 4 л СО.
3. Из щавелевой кислот и HtSO*. Нагревают смесь 100 г кристаллической
щавелевой кислоты (НгСг?^ • 2ШО) и 500 г концентрированной H2SO4 в кругло-
донной колбе, на сетке. Как только начнется выделение газа, пламя удаляют,
так как реакция идет бурно.
Выделяющаяся СО содержит СОг (в эквивалентном количестве), воздух и
влагу. СО» полностью поглощается при пропускании газа через 2 склянки
с КОН (1:2) и через трубку длиной 40 см, до половины наполненную зер-
неной натронной известью и твердым КОН. Кислород воздуха поглощается
при пропускании газа через раствор гидросульфита натрия. Осушка газа
производится' при помощи СаСЬ, концентрированной H2SO4 и Р2О5.
4. Из щавелевокислого кальция и извести. Смешивают равные количества
СаСг04 и СаО и нагревают в тугоплавкой сожигательной трубке. При
незначительном почернении оксалата начинается выделение СО, загрязненной С02,
парами ©оды и незначительным количеством воздуха; эти примеси удаляются
IE. В. Алексеевскнй, Г. М. Белоцерковский и Т. Г. Плачено в, Практические
работы по химии защиты, Обор^нгиз, 1940, стр. 119.
■ Ru рр, Спещ. Ztg. 32, 983 (1908); Z. f. Elekt.och. 15, 506 (1909).
Углерода окись» Углерод сернистый 523
Щ^уированием трубки, непродолжительным выделением газа и вторичным
йакуированием.
Щ Затем газ пропускают через твердый КОН и сушат обычным путем СаСЬ
^р2о5.
Двойства
Бесцветный, сильно ядовитый газ, без запаха и вкуса, очень мало
растворимый в воде (в 1 объеме воды при 0° растворяется 0,03537 объема СО, а при
2$° -—0,02142 объема). Сгущенный в жидкость кипит при —190°; т. пл. —207°
{при давлении в 100 мм),
■Г Горения не поддерживает; горит на воздухе синим пламенем, образуя СОг.
Еоглощается растворами CuCl в НС1 или ЫШОН. Плотность 0,9672 (по
воздуху); 1 л газа при нормальных условиях весит 1,2504 г.
Ничтожная примесь СО к воздуху вызывает сильную головную боль.
УГЛЕРОД СЕРНИСТЫЙ (СЕРОУГЛЕРОД)
Carboneum sulfuratum Carbon disulfide Schwefelkohlenstoff *
CSs Мол. в. 76,13
Очистка
Все работы с CS2 крайне огнеопасны!
1. По Пальмиери очистку технического CS2 ведут следующим путем.
На каждые 100 г CS2 прибавляют 2—3 г безводной CuS04 и взбалтывают.
Когда осядет почерневший порошок и исчезнет неприятный запах, CS2
декантируют. Для получения совершенно чистого препарата его перегоняют над
безводной CuS04.
2. По А л л а р й CS2 обливают водой и понемногу прибавляют, при
сильном встряхивании, раствор КМп04, пока водный слой не приобретет красной
окраски. Тогда CS2 отделяют в длительной воронке и промывают дестиллиро-
ванной водой. По утверждению автора таким путем получается очень чистый
препарат.
3. По У н р у для получения совершенно сухого и чистого препарата
взбалтывают 500—700 мл CS2 с 6—8 мл сухой ртути и с зерненым
(пористым) СаСЬ; фильт'руют и перегоняют в темноте с вертикально поставленным
/Шариковым холодильником.
Эти операции повторяют до тех пор, пока ртуть не перестанет чернеть.
При каждой перегонке отбрасывают низкокипящий погон (влажный CS2,
составляющий 15—20% от всего дестиллата) и высококипящий погон,
переходящий после того как CS2 в перегонной колбе пожелтеет. Эти фракции
смешивают- с техническим СЗг и подвергают очистке, как указано выше.
4. Г. Борнеманн1 рекомендует сочетать методы Аллери и Унру —
сначала взбалтывать 300 мл CS2 с 5—6 мл Hg в течение получаса, затем
обрабатывать порциями по 50 мл 10%-ного раствора КМп04 в течение 15 мин.,
пока водный слой не будет иметь ясно заметной вишневой окраски.
.Отфильтровывают CS2 через сухой фильтр в колбу, добавляют 30 г СаСЬ,
' кипятят в течение получаса с обратным холодильником (на водяной бане),
после чего перегоняют CS2, собирая фракцию, кипящую при 46—48°.
Свойства
Бесцветная, подвижная, сильно преломляющая свет жидкость уд. в. 1,2661
~(^2о ) с эфирным запахом. Т. пл. —112°, т. кип. 46,25°.
Растворяет иод, бром, серу, жиры, воск, гуттаперчу, смолы, каучук,
камфору, желтый фосфор. Смешивается во всех отношениях с абсолютным
спиртом, эфиром, бензолом, хлороформом, четыреххлористым углеродом, эфирными
1 Hejpr. преп. 58»
524
Углерод
и жирными маслами. Растворим в едких и сернистых щелочах, почти не рас
творим в воде. Пары С8г поглощаются иодом, парафином, раствором бром
в КВг и, особенно хорошо, льняным маслом, если последним смазана поверх-.
ность стеклямиой трубки.
В высшей степени огнеопасен; воспламеняется даже от горячего предмета
(температура воспламенения 232°).
Пары СЗг с воздухом или кислородом взрывают со страшной силой.
При действии хлора разлагается по уравнению
CS* + ЗСЬ = S2CI2 + CCU. ^.
На солнечном свету несколько разлагается и приобретает неприятный з
пах гнилой редьки, свойственный продажному сорту СЗг.
л
Растворимость CS2 вводе
(Chancel, Palmentier)
Таблица 469
t°c
0
5
10
15
г CS,
в 00 мл
раствора
0,204
0,199
0,194 1
0.187
/•с
20
25
30
35
г CS,
в loo мл
раствора
0,179
0,169
0.15S
0,137
t °с
40
45
49
г CS,
В 100 мл
раствора
0,111
0.070
©,014
Таблица 470
Растворимость CS2 в разбавленном спирте
(Tuchschidt, Follenius)
Вес. о/о
спирта
$8,5
98,15
96,95
10 мл сиирта
растворяют
г CS,
13,2
12,2
10,0
Вес. о/о
спирта
93,54
91,37
84,12
10 мл спирта I Вес. р.
растворяют С11„рта
7,0
5.0
3,0
76,02
48,40
47,90
J0 мл спирта
растворяют
г CS,
2.0
02
0,0
Испытание
1. Нелетучий остаток. При испарении на водяной бане 50 г препарата
остаток не должен быть больше 0,5 мг.
2. Растворенная сера (S). При взбалтывании препарата в сухом сосуде
с сухой ртутью последняя не должна покрываться темной пленкой.
5. Сероводород (H2S). При встряхивании препарата с углекислым свинцом
последний не должен принимать коричневой окраски.
4. Сернистая и серная кислоты. (H*SO% и H2SO4). Взболтанный с водой
препарат не должен показывать кислой реакции.
5. Влажность. Безводная C11SO4 при стоянии с препаратом в течение
нескольких часов не должна синеть.
Углерода сероокись
525
УГЛЕРОДА СЕРООКИСЬ
Carbonyl sulfide Kohlenoxysulfid
Carbon oxysulfide
COS Мол. в. 60,07
Приготовление
По Класону прибавляют к 1000мл воды 2080г концентрированной H2SO4
g вносят смесь после охлаждения в колбу емкостью 5 л вместе с 200 мл
насыщенного раствора роданистого аммония (содержащего 138 г NFhCNS).
Очень равномерное выделение газа получают при нагревании смеси на водяной
бане. Рекомендуется шачале нагреть до 42°' и, как только начнется
газовыделение, охладить до 20°; при ослаблении газовыделения снова повышают
температуру до 35° (при более высокой температуре выделяющийся COS
сильно загрязнен №»S и CS2. Работать под тягой!).
Полученный неочищенный газ содержит кроме COS значительные
количества H-S, HCN, CS2 и других продуктов разложения NH4CNS. Газ очищают,
Рис. 76. Прибор для получения сероокиси углерода
пропуская последовательно через две промывных склянки с 30%-ным
раствором КОН склянку с концентрированной H2SO4, трубку с триэтилфосфином
[Р(СгН5)з] и собирают в газометре над водой. Однако приготовленный и
хранимый таким образом газ быстро разлагается.
Для получения совершенно чистого, прочного газа применяют
предложенный Гемпелем метод очистки, а именно — берут вместо чистого Р(СгНз)з
смесь 1 ч. Р(СгНй)з, 9 ч. пиридина и 10 ч. нитробензола. Пары этих
жидкостей, примешанные к газу, удаляются охлаждением последнего до
температуры от —10° до —20°. Для отделения трудно конденсируемых газов (N2,
Ог) сероокись углерода сжижают.
Общий вид прибора для получения COS по Класону, с применением
метода очистки Г ем пел я, изображен на рис;. 76, где А — литровая колба,
нагреваемая на водяной бане, В и С — промывные склянки с КОН, D —
промывная склянка с H2SO4, Е—трубка, наполненная смесью триэтилфосфина,
пиридина и нитробензола, F — холодильник, в котором сгущаются пары этих
жидкостей.
Холодильник состоит из трубки а диаметром в 10 мм, наполненной битым
стеклом и помещенной в жестяной сосуд b со смесью толченого льда и соли.
Жестяной сосуд окутан очищенной овечьей шерстью и помещен в деревянный
ящик с. Газ, очищенный в холодильнике от паров промывных жидкостей,
пропускают для сгущения через U-образную трубку G, окруженную смесью
твердой СОа с эфиром и находящуюся в сосуде Н из цинковой жести.
S26
Углерод
Этот сосуд также заключен в деревянный ящик и для сохранения теплп
обернут овечьей шерстью. Конденсирующийся COS стекает в пробирку /у, По.
мещенную в стакан, наполненный смесью твердой СОз с эфиром; стакан оку-
тан шерстью и вставлен во второй стакан большего размера.
По исследованию Мицека1 COS довольно сильно абсорбируется КОН
По утверждению автора целесообразно концентрировать газ в сильно
охлажденном (смесью эфира и твердого С02) приемнике и несколько раз
подвергнуть дробной перегонке, причем СОэ удаляется с начальным, a HCN, Cs.
и НСООН —с конечными фракциями. Наконец газ высушивают
концентрированной H2SO4 или СаС1г.
Свойства
Чистый COS —газ с очень слабым запахом. При 300° распадается на СО
И S; на воздухе горит синим пламенем, образуя СОг и SO2. Т. кип.
сжиженного газа 47,5°. Значительно растворим в воде (1 мл воды при 13,5 и Да.
влении 756 мм растворяет 0,8 мл газа). Смесь с воздухом взрывает при
зажигании, если содержание COS в смеси составляет от 11,9 до 26,5 объемных
процентов.
Сильно действует на нервную систему; вдыхание незначительных количеств
уже вызывает оду'рманивание.
,
УГЛЕРОДА ХЛОРОКИСЬ (ХЛОРИСТЫЙ КАРБОНИЛ, ФОСГЕН)
Carbon oxychloride
Phosgene
сось
Мол. в. 98,924
Kohlenoxychlorid
Carbonylchlorid
Chlorkohlenoxyd
Phosgen
f% f
Рис. 77. Прпбор для получения
фосгена
Приготовление
1. По Эрдману 100 мл четыреххларисто-
го. углерода (CCU) нагревают в круглодонной
колбе емкостью 300 мл, снабженной
шариковым холодильником (рис. 77) на водяной
бане до энергичного кипения; затем из
капельной воронки с остро оттянутым концом
начинают приливать 120 мл дымящей H2SO4(c80%
свободного SOe), причем приливание
регулируют так, чтобы каждая капля сначала
пришла в тесное соприкосновение с парами ССЬ
внутри шарикового холодильника и затем уже
падала в реакционную колбу.
Получающуюся равномерную струю СОСЬ
для удержания паров SOs и (SO2CO2O (пиро-
сульфурилхлорид) проводят в промывную
склянку Дрекселя, наполненную
концентрированной H2SO4, затем газ поступает в приемник
(U-образную трубку с кранами), окруженный
охладительной смесью. Промывная Склянка
несколько разогревается; ее нужно охлаждать
холодной -водой. Соединения стекла должны
производиться в стык; необходимая плотность
достигается при помощи каучуковых трубок.
Фсгсген легко сжижается в приемнике и
благодаря этому отделяется от других газов.
1 Mi tz ek, J. f. Oasbeleucht. 46, 26 (1902).
Углерода хлорокись. Углерод четырех хлористый 527
Жогда вся H2SO4 прилита и газовыделегьие ослабеет, жидкость кипятят,
рггобы выделить растворенный фосген.
;^; Сырой продукт (90% теоретического выхода) перегоняют, наг'ревая колбу
Шгкой; газообразный фосген еще раз пропускают через промывную склянку
Гф H2SO4 и снова конденсируют в холодильной смеси. Небольшие количества
^CGU и пиросульфурилхлорида, содержащиеся в сыром фосгене, удаляются при
L^grperoHKe.
Ф) •.
ЗС в о й с т в а
■ Бесцветный ядовитый газ с запахом гнилого сша. Т. пл. —126°, т. кип.
|!+8,2 при 760 мм. Уд. в. 1,436 при 0°. Б воде растворим немного; лучше
-'растворяется в углеводородах, как бензол, толуол и др. Водой разлагается
tea С02 и НС1.
'■:■':■ В продажу СОСЬ поступает в виде 30%-ного раствора в толуоле.
. .При вдыхании очень незначительного количества СОС1г утрачивается
способность различать тонкие вкусовые ощущения. Уже в ничтожных дозах
СОС12 проявляет себя энергично действующим ядом, вызывающим удушье.
Лучшим противоядием является вдыхание паров спирта.
УГЛЕРОД ЧЕТЫРЕХХЛОРИСТЫЙ (ТЕТРАХЛОРМЕТАН)
Carboneum СатЬоп tetrachloride Kohlenstofftetrachlorid
tetrachloratum
CCli Мол. в. 153,838
Приготовление
1. По Реньо пропускают сухой хлор в спокойно кипящий хлороформ
(СНСЬ), помещенный в колбу с обратным холодильником. Реакцию
рекомендуется начать на ярком солнечном свету; прибавление треххлористой сурьмы
(ЗЬСЬ) или хлористого иода (JC1) делает реакцию независимой от света.
Продукт освобождают от хлора встряхиванием с металлической ртутью и
очищают перегонкой.
2. Пропускают сухой хлор в CS2, в котором растворено немного иода
(катализатор):
CS2 + ЗС12 = CCU + S2CI2.
Получившийся CCU отгоняют при 77° от SsCb, кипящей при 136°.
Свойства
Бесцветная жидкость уд. в. 1,632 при 0° с приятным эфирным запахом.
Т. пл. — 23,77°, т. кип. 76,6°. Почти, не растворим в воде (0,08%); со спиртом,
эфиром, жирными и эфировыми маслами смешивается во всех отношениях.
Четыреххло'ристый углерод не горюч. —-
Испытание
По ОСТ 10153—39 препарат должен представлять собой бесцветную
прозрачную жидкость уд. в. 1,595 (d|°) с т. кип. при 760 мм 76,7—77,2° для
препарата ч. д. а. или 76,7—77,7° для препарата чистого; в этом интервале
Должно перегоняться не менее 95% препарата по объему; наибольшее
количество допустимых примесей указано в табл. 471.
Кроме того, препарат должен выдерживать испытания на кислотность,
свободный хлор, органические примеси и восстановители.
528
Уран
Допустимые примеси в CCU
Таблица 47у
Примеси
■
Допустимое содержание в »/0
в чистом для
анализа
0,00С5
0,и002
0,С002
0,0002
в чистом
0,001
0,0005
0,001
0,0000
'
УРАНА ДВУОКИСЬ (ОКИСЬ УРАНА[4], ЗАКИСЬ УРАНА)
Uranum oxydulatum
Uranium dioxide
UO2 Мол. в. 270,0?
Uranoxydul
Uranooxyd
Приготовление
1. Окснер де Конин к1 нашел, что при нагревании бромистого ура-
нила (иОгВгг) происходит легкое отщепление брома и остается UO2 в виде
кирпично-красной массы; последняя при нагревании в струе водорода, не
изменяясь химически, становится черной.
2. По Колани2 получают кристаллическую UO2 действием воды на
двойную соль Na2UCle. Для этого водяной пар в смеси с СОг пропускают
через нагреваемую стеклянную трубку, в которой в фарфоровых лодочках
находится указанная соль. Реакция начинается ниже точки плавления соли и
делается заметной по обильному выделению хлористого водорода. Немного
ниже температуры красного каления реакция протекает очень энергично.
Хлорид вспучивается и постепенно стекает навстречу водяному пару, причем
масса превращается в кристаллическую 1Юг и NaCl. По окончании
реакции массу промывают водой; 1Юг остается в виде чериого кристаллического
порошка. Тонкие кристаллы красноваты и при рассматривании под
микроскопом прозрачны.
Свойства
Черный кристаллический порошок уд. в. 10,95. В пламени гремучего газа
сгорает, разбрасывая искры; при умеренном нагревании на воздухе
превращается в UsOs. Т. пл. 2176°. Не растворима в воде; из кислот хорошо
растворяется в НЫОз.
Uranium oxydato*
oxydulatum
Приготовление
УРАНА ЗАКИСЬ-ОКИСЬ
Uranoso-uranio oxide
UsOs
Мол. в. 842,21
Uranoxyduloxyd
Uranouranioxyd1
Uranoctoxiyd
По Циммерману, теплый солянокислый раствор гидрата окиси ура-
нила [урановой кислоты — UCMOH)^] насыщают сероводородом и оставляют
чт
* Oecksner de К о n i n к', С. 135, 900 (1902); Ргар. Ch. 734*
• С о 1 а п 1, An. Chim. Phys. (7), 12, 76 (1907)*
Урана двуокись, закись-окись и трехокись
529
го'ять продолжительное время. Образующийся осадок сернистых соединений
.^шьяка и» других металлов отфильтровывают и добавляют к раствору ЫНЮН
{ |ЫН4)аСОз в избытке. После непродолжительного нагревания к жидкости
фибавляют еще (NH^S. Уран в виде двойной аммонийной соли остается в
растворе, «в то время как остальные металлы III аналитической группы
осаждаются.
Осадок отфильтровывают, добавляют к раствору избыток НС1, кипятят
до удаления С02 и, прибавляя МШОН и (NH^zS, осаждают сернистый уран
^ виде осадка шоколадного цвета.
Нагревают до разрушения большей части (NH^S,* отфильтровывают смесь
Ш)г и серы, промывают водой и прокаливают на сильной газовой горелке.
Получившуюся UaOs растворяют в HNOa, :раствор фильтруют и выпаривают
ф- кристаллизации. Кристаллическую массу обрабатывают эфиром, в котором
а|отнокислый ура] : ^ЫОф] легко растворяется. После фильтрования
п'аствор выпари) на водяной бане досуха и остаток сильно прокаливают.
Образовавшуюся ; jOs для очистки снова растворяют в НЫОз и еще 'раз
проводят все указанные выше операци
Свой с г ва
Темнозеленый порошок уд. в. 8,2, растворимый в кислотах и не раствори*
0ый в воде.
При длительном прокаливании в атмосфере азота или СО* переходит в
JO*.
УРАНА ТРЕХОКИСЬ
УРАНОВЫЙ АНГИДРИД, ОКИСЬ УРАНИЛА)
Uranium trioxide Urantrioxyd
Uranyloxyd
Uransauranfrydrid
UO3 Мол. в. 286,07
Up иготовление
1. По Розенгейму1 сушат урановую кислоту (H2UO4 — гидрат окиси
уранила) в сушильном шкафу при 100°. Полученный порошок нагревают при
перемешивании в пробирке на парафиновой или масляной бане при 250—300°
под тягой до тех пор, пока он не примет кирпично-красного цвета.
2. По Л и но2 осаждают раствор азотнокислого уранила аммиаком,
выпадающий желтый осадок ураната аммония [(NH^lbO?] сушат, тонко
измельчают и нагревают на песочной бане в противнях при перемешивании (или,
при малых количествах, в эрленмейеровской колбе) в течение 30 час. при
250—280°.
Так как песочная баня нагревает неравномерно, то при недостаточно
частом перемешивании можно легко получить неоднородный продукт
Свойства
Оранжево-желтый порошок уд. в. 5,92—6,0, при накаливании распадаю*
Щийся на UsOe и Ог.
Не растворим в воде; растворяется в сильных кислотах и растворах угле*
кислых щелочей.
1 A. R о s e n h e i m,*Z. anori?. Ch. 20, 285 (1899); Uebungsbeispiele.
1 Li e n a u, Dissertat. Berlin (1898;.
(ОКИСЬ УРАНА[6],
Uranium oxydatum
630
Уран
УРАНИЛ АЗОТНОКИСЛЫЙ
Uranium Uranyl nitrate -Uranylnitrat
nitricum Urannitrat
U02(NOs)2 • 6H2O Мол. в. 502,18
Приготовление
1. Растворяют U02, UsOs или UOs в чистой НШз и выпаривают до
кристаллизации.
2. Для очистки растворяют продажный препарат в воде и фильтруют,
В фильтрат пропускают БОг, который восстанавливает HaAsCh до HsAsOa.
Жидкость нагревают до 60° и насыщают серовододдД |№^SPn этом выпадг1от
сернистые соединения Pb, As, Sn и др. ОеадокЛ^ * ;j^ ^вают, филы ат
подкисляют HNOa и выпаривают до кристаллизяг# .Цв^ий при с а-
ждении кристаллический продукт пере-кристаллизовшШб'т Т^^фира.
Свойства
Прозрачные желтовато-зеленые призмы или таблички уд. в. 2,807,
флюоресцирующие зеленым цветом. Расплывается во влажном и выветривается
в сухом воздухе. При 59,5° плавится в кристаллизационной воде; при
более высокой температуре теряет HNOs и воду, образуя UOs и дальше
изОв. Растворим в воде; очень легко растворим в спирте, эфире и ацетоне,
Не растворим в бензоле, толуоле и ксилоле.
Препарат и его растворы следует хранить в темных плотно закрытых
банках.
Таблица 472
Растворимость U02 (N03)2 вводе
г с
0
5,5
12,3
UO,(NCvi
49,5
50,6
52,9
г с
21,1
25,6
1 36,7
Wo
UOt(N08),
56,0
57,2
61,3
t° С
45,2
51,8
fr',5
•'о
UCVNO.),
65,1
67,8
78,5
Испытание (Мерк)
1. Сульфаты (SO*"). Раствор 1 г препарата в 20 мл воды не должен
в течение 15 -мин. изменяться от прибавления ВаСЬ.
2. Соли щелочных металлов. 1 г препарата прокаливают, остаток
обрабатывают 20 мл воды и фильтруют. Фильтрат после выпаривания ие должен
оставлять весомого остатка.
3. Соли щелочноземельных металлов. Раствор 1 г препарата в 20 мл
йоды должен оставаться прозрачным при добавлений избытка ЫНЮН и
(>Ш4)2СОз.
4. Соли закиси урана. Раствор 1 в препарата в 20 мл воды и 1 мл
разбавленной H2SO4 должен принимать розовую окраску от прибавления 0,1—
0,2 мл 0,Ш раствора КМп04.
5. Прочие металлы, а) Раствор, полученный при испытании 3, от
добавления 2—3 капель (NH4)?S не должен принимать темнокоричневой окраски или
давать осадка.
б) Нагретый до кипения раствор 5 г препарата в 100 мл воды,
подкисленный 5 мл НС1.(уд. в. 1,124), не должен изменяться при пропускании
сероводорода.
Уранил азотнокислый, сернокислый и уксуснокислый 531
Ф *" "■"■' ■ ■ .11,1 I ■ "■- .ПИШИ
УРАНИЛ СЕРНОКИСЛЫЙ
Uranium Uranyl sulfate Uranyisulfat
sulfUricum
U02(S04) Мол. в. 366,13
Криготовле н ие
По Эбельмеиу раствор азотнокислого уранила [U02(NOa)s] выпари-
ают досуха с H2SO4, удаляют избыток кислоты, продолжая нагревание рас-
оряют остаток в воде и, выпарив до консистенции сиропа, дают закристал-
зоваться. Кристаллизация проходит медленно.
Свойства
Зеленые кр-иУрпШк.*^.', J3,28, немного выветривающиеся на воздухе. Рас*
таорим в воде (14,82°/ i 15 ), спирте; трудно растворим в H2SO4.
УРАНИЛ УКСУСНОКИСЛЫЙ (АЦЕТАТ УРАНИЛА)
Uranium aceticum Uranyl acetate Uranylacetat
и02(СНзСОО)2 • 2H20 Мол. в. 424,19
Приготовление
1. По Вертгейму нагревают нитрат уранила до начинающегося
выделения кислорода; растворяют оранжевую массу, содержащую еще NOs'*
,-В теплой концентрированной СНзСООН и выпаривают раствор до
кристаллизации. Примесь нитрата остается в маточном растворе.
2. Прокаливают уранат аммония (NH4)2U207 при 250—280° и полученный
1Юз растворяют в 4 ч. горячей 20%-ной СНзСООН. Раствор
отфильтровывают и выпаривают до кристаллизации. Кристаллы ацетата уранила
отсасывают, промывают 1%-ной СНзСООН и сушат при комнатной температуре.
Свойства
Желтые ромбические кристаллы уд. в. 2,89. При 100° теряет
кристаллизационную воду. Растворим в воде, спирте и эфире. Водные растворы на
свету восстанавливаются, выделяя фиолетовый осадок. Препарат следует
растворять в воде, подкисленной СНзСООН.
Испытание1
Препарат ч. д. а. должен содержать:
1. Нерастворимых веществ не более 0,01%.
2. Солей натрия (ЫагО) >не более 0,06%.
3. Солей четырехвалентного ypatfa (U** "•) не более 0,1%.
4. Ацетата уранила иОз(СНзСООЬ * 2Н20 не менее 98%. Кроме того,
препарат испытывается на содержание хлоридов, сульфатов, железа и солей
Щелочноземельных и тяжелых металлов.
Хлориды (СУ). 0,5 г препарата растворяют в 20 мл воды, добавляют 1 м..
НЫОз (уд. в. 1,15) и, если нужно, фильтруют через промытый горячей водой
фильтр. К фильтрату добавляют 1 мл 0,1N AgNOs. Через 5 мин. не должно
быть опалесценции.
Сульфаты (SOS'). 2 г препарата растворяют в 25 мл воды, добавляют 0,5 мл
ледяной СНзСООН, фильтруют, если раствор получается мутным, и
приливают к фильтрату 1 мл 10%-ного раствора ВаСЬ. Через 5 мин. не должно
быть осадка или мути.
1 Испытание хим. реактивов, 387.
532
Уран. Фосфор
Натрий (Na'). 5 г препарата растворяют в 200 мл воды и ЮжлСНзСООН
(1 :2), нагревают до кипения и добавляют избыток ЫНЮН (уд. в. 0,91);
фильтруют и фильтрат выпаривают досуха. Сухой остаток осторожно
прокаливают, растворяют после охлаждения в 10—20 мл воды и титруют 1NHQ
с метилоранжем в качестве индикатора, до розовой окраски. Должно быть
израсходовано не более 0,1 мл Ш НС1.
Щелочноземельные металлы. 1 г препарата растворяют в 20 мл воды и
3 мл СНзСООН (1:2) и добавляют NH4OH (уд. в. 0,96) до появления осадка,
не исчезающего при взбалтывании. Затем приливают раствор (NH^CCh д0
полного растворения — в течение 1 мин. не должно появляться мути или
осадка.
Тяжелые металлы. 1 г препарата растворяют у им. воды и 2 мл НС1
(уд. в. 1,12), нагревают до кипения и добавляют Щ мл, свежеприготовленной
сероводородной воды. Не должно тотчас появляться О * атого
окрашивания.
Железо (Fe' ' *). 0,5 г препарата растворяют В см > I мл' H2SO4 (уд. в. 1,10)
и 4 мл воды и выпаривают раствор на песочной б * удаления паров
H2SO4. К остатку добавляют 0,5 мл HNOs (уд. в. 1,1 10 мл воды,
нагревают до кипения, разбавляют водой до 100 мл и добавляют 1 мл H2SO4
(уд. в. 1,10) и 5 мл 4N NH4CNS. Не должно тотчас появляться красноватою
окрашивания раствора.
Соединения четырехвалентного урана (V ' '). Растворяют 6 г препарата
в 400 мл воды и 4 мл H2SO4 (уд. в. 1,10). Раствор делят «а две равные части,
одна из которых остается для сравнения. К другой части добавляют 0,1 N
раствор КМп04, причем для получения заметного изменения окраски должно
пойти не более 0,2 мл KMnO-i.
Phosphonium
jodatum
ФОСФОНИЙ ЙОДИСТЫЙ
Phosphonium jodide
PH4J Мол. в. 161,93
Phosphoniumjodid
Приготовление
По Бэйеру в реторту с тубусом вносят 50—100 мл сухого СЗа,
растворяют в нем 100 г фосфора и при охлаждения вносят маленькими
порциями 170 г иода. Затем присоединяют к реторте холодильник, отгоняют CS*
Рис. 78. Прибор для получения йодистого фосфония
и удаляют последние количества его, пропуская ток СОг при слабом
нагревании реторты. После охлаждения к реторте вместо холодильника присоеди-^
няют длинную и широкую тонкостенную стеклянную трубку, конец которой
соединен с аллонжем (рис. 78). Последний опущен в стакан с водой так, чтобы
Фосфоний йодистый. Фосфор
533
|юнец его не касался воды. Затем через тубус реторты при помощи оттянутой
#а конце капельной воронки прибавляют маленькими порциями 50 мл воды.
Jflpn каждом приливании происходит энергичная реакция. РЬЫ сгущается
внутри реторты и в широкой трубке; выделяющиеся одновременно
незначительные количества HJ поглощаются водой в приемнике. После приливания
всей воды нагревают 'реторту сначала осторожно, затем до слабого каленич
ц полностью перегоняют PH4J в холодильную трубку. Последнюю по охла
ясдении разбивают и извлекают РЬЫ в виде толстых корок, напоминающих*
NH4CI. d
Выход около 120 г.
С в о й с тв а
Крупные прозрачные кристаллы с алмазным блеском. Т. возгонки 61,8°;
частично возгоняется уже при комнатной температуре. Осторожным
нагреванием в закрытом сосуде можно возгонять РЬЫ без предварительного
плавления.
ФОСФОР
Phosphorus Phosphorus Phosphor
Р At. в. 30,98
А. Белый фосфор
Очистка
1. Продажный фосфор, упакованный в жестяные банки, часто покрывается
черным порошковатым налетом. Перед употреблением такого фосфора его
выдерживают в течение суток в 5% -ной HNOa и затем промывают дестилли-
рованной водой.
2. Для того чтобы сделать бесцветным фосфор, ставший желтым
вследствие долгого хранения, по Бендеру можно:
а) расплавить Р под водой, содержащей 3,5 ч. К2СГ2О7 и 3,5 ч. H2SO4 на
каждые 100 ч. фосфора;
б) настаивать Р с очень разбавленной НЫОз в колбе с газоотводной
трубкой, погруженной в воду;
в) обработать Р хлорной водой, затем КОН и NH40H.
3. Для удаления As, часто встречающегося в продажном фосфоре,
поступают по одному из следующих методов:
а) настаивают 1 ч. фосфора с 2 ч. HNOs (уд. в. 1,1);
б) дважды перегоняют фосфор с водяным паром. Для этой цели по
Нёллингу и Фейе р. штейну1 помещают 100 г продажного желтого
фосфора в колбу емкостью 3—4 л, содержащую 500 мл воды, соединенную
с парообразователем, с аппаратом для получения СОг и с холодильником;
форштосс последнего оканчивается в приемнике под поверхностью теплой
(30°) воды. Воздух из аппарата вытесняют струей С02 и во все время
перегонки пропускают через аппарат медленный ток СО*. Затем пропускают пар
так, чтобы содержимое колбы непрерывно кипело. Фосфор перегоняется с
водяным паром в виде бесцветных, сильно светопреломляющих капель,
падающих на дно приемника и там через некоторое .время затвердевающих.
Перегоняющуюся воду время от времени удаляют из приемника сифоном.
В течение 8 час. получается приблизительно 50 г Р.
Если после первой перегонки продукт содержит еще следы As, то,
повторяя операцию, получают абсолютно чистый препарат.
Свойства
Бесцветное прозрачное вещество с жирным блеском уд. в. 1,83. Т. пл.
44,5°, т. кип. 280,5°. Летуч с водяными парами.
1 Е. N б I! in g, W. F e u e r s t e i п, Вег. 33. 2684 (1900).
534
Фосфор
I
На воздухе воспламеняется при 60°. В воде почти нерастворим; очень
плохо растворяется в спирте и глицерине; значительно лучше — в эфире,
бензоле, скипидаре и твердых маслах. Легко растворим в S2CI2, РСЬ, РВг3 и .
CS* (1 ч. растворяет 18 ч. Р).
Бумага, смоченная раствором фосфора в CS2, при высыхании
самовоспламеняется.
Из растворов в летучих маслах при медленном охлаждении Р
кристаллизуется в октаэдрах и ромбических додекаэдрах правильной системы.
Запах Р похож на чесночный. Сильно ядовит! Ожоги от горящего
фосфора чрезвычайно опасны и болезненны.
Б. Гранулированный желтый (белый) фосфор
Приготовление
1. Тонко раздробленный гранулированный фосфор по Михаэлису1
получается следующим путем: обливают кусочки фосфора теплой водой или
водным раствором мочевины [СО(ЫНг)2] в толстостенном стеклянном сосуде
и, плотно закупорив его, встряхивают (лучше в механической болталке) до
тех пор, пока расплавленный фосфор не затвердеет. При этом получают Р
в виде мелких шариков.
2. Помещают куски фосфора в спирт 36° Вё, расплавляют и встряхивают
до затвердевания.
3. В толстостенную ступку наливают воду, /нагретую до 38—4(Р, и
помещают небольшие куски фосфора.
Выше 34,3° фосфор становится настолько хрупким, что может быть
осторожно истолчен в порошок.
В. Красный фосфор
Приготовление
Красный фосфор может быть получен нагреванием белого фосфора при
250°. Для проведения процесса в лабораторном масштабе помещают сухой
фосфор в колбу А (рис. 79),
по груженную в масляную
баню и закрытую пробкой
с двумя отверстиями. Через
одно из них проходит дважды
изогнутая трубка В, одно
колено которой 76 см длины
погружено в стакан с ртутью.
Через другое отверстие
проходит трубка С,
соединенная с хлор кальциевой
трубкой D, второй конец трубки
которой присоединен к
аппарату Килпа Е, где получается
СОг из мрамора и НС1.
Сначала в колбу пускают ток СО»,
пока воздух из прибора не
Рис. 79. Прибор для получения красного фосфора
будет полностью вытеснен. Тогда запаивают трубку С и нагревают -масляную
баню.
1 М i с h a e 11 s, Ann. 310, 56 (1900).
Фосфор
535
При нагреваиии в течение SO—40 час. при 250—260° расплавленный фосфор
дочти полностью превращается в красную форму.
Для отделения его от неизменившегося белого фосфора Бендер
'рекомендует прибавить «немного CS2 .и затем раствора СаСЬ уд. в. 1,349—1»384.
Раствор белого Р в CS2 всплывает наверх, юраоный же фосфор остается
на дне.
(С сероуглеродным раствором белого фосфора следует обращаться с
осторожностью, так как при испарении растворителя остающийся мелко
раздробленный Р самовозгорается),
Г. Светлокрасный фосфор
Приготовление
По Шенку1 нагревают раствор 100 г белого фосфора в 1 кг трехбро-
мистого фосфора (РВгз) в колбе с обратным холодильником до кипения 10
часов. При этом фосфор почти полностью переходит в светлокрасную форму.
Отсасывают и порошок несколько раз кипятят с CS2 (осторожно! огнеопасно!)
для удаления растворителя и небольших количеств белого фосфора. После
фильтрования на фильтре остается светлокрасный фосфор. Из 100 г белого
Р получается в среднем 130 г светлокраоного препарата. Привес состоит из РВгз
(при растворении небольшой пробы в НЫОз и Прибавления AgNOs
происходит значительное выделение AgBr). Удержанный РВгз не удаляется при
кипячении с индиферентными растворителями (СНСЬ, CCU СЗг); препарат также
не дымит на воздухе, несмотря на содержание РВгз.
Удалить большую часть РВгз удается кипячением препарата с водой;
последние количества РВгз извлекаются с большим трудом.
Д. Металлический (кристаллический) фосфор
Приготовление
По Гитторфу2 широкую трубку тугоплавкого стекла, запаянную с одного
и суженную с другого конца, наполняют при постоянном токе СОг на гА
высоты белым фосфором и до % кусками металлического свинца. (Лучше
применять свинец, уже употреблявшийся для этой цели и, благодаря этому
ставший настолько тугоплавким, что в сильном жару он не плавится, а только
размягчается).
Удалив посредством вакуум-насоса воздух и гигроскопическую воду, трубку
запаивают и помещают в более широкую железную трубку, заполнив п'ро^
странство между ними магнезией (MgO). Нагревают, постепенно повышая
температуру, в течение 8—10 час. до умеренного краонокалильного жара, при^
чем в продолжение первого часа трубку время от времени поворачивают во
"-избежание возгонки Р в одном м'есте.
Фосфор раство'ряется в свинце и кристаллизуется на его поверхности.
Кристаллы Р, образовавшиеся внутри свинцовой массы, извлекаются при
растворении свинца в HNOs уд. в. 1,1. Для удаления примеси РЬ и РЬО
кристаллы кипятят с концентрированной НС1; однако полностью освободиться
от примеси РЬ не удается.
Свойства
Тонкие, черные, кристаллические листочки с сильным металлическим бле*
ском, просвечивающие красным цветом.
Кристаллы, полученные после растворения свинца, представляют собой
ромбоэдры уд. в. 2,34.-
1 Sch e n к, Вст. 36, 979 (1903),
• Pogff, 126, 193.
636
Фосфор
Е. Коллоидальный фосфор
Приготовление
1. По Сведбергу1 в стеклянную воронку с плоско пришлифованной
к верхнему краю стеклянной пластинкой помещают алюминиевый конический
сосуд, загруженный красным фосфором и изобутиловым спиртом (рис. 80).
Стеклянная пластинка имеет по середине отверстие, через которое проходит
алюминиевая проволока. Воронка изолируется
и укрепляется, как указано на рис. 80.
Алюминиевый сосуд и проволоку соединяют
с источником тока через индукционную
катушку, причем искровой разряд происходит
внутри жидкости. Целесообразно заземлить
алюминиевую проволоку, чтобы удобнее было
устанавливать ее незащищенной рукой.
Получающийся коллоидный раствор в
проходящем свете почти бесцветен (слабо
желтый), в отраженном имеет мясокрасный цвет.
2. По А. Эльдеру и П. Б у р х а р д у 2
к раствору 0,03 г желтого Р в 2 мл бензола
очень медленно добавляют 998 мл ледяной
СНзСООН, причем получается прочный золь.
Можно менять соотношения между
количествами фосфора, бензола и СНзСООН вплоть
до 3:200:800.
3. Золь красного фосфора по Лотте р-
м о з е р у3 удобно наблюдать следующим
образом: нагревают непродолжительное время
в запаянной трубке при 140° белый фосфор
с трехбромистым фосфором (РВгз). Жидкость
окрашивается в интенсивно желтый цвет, оставаясь, однако, совершенно
прозрачной. Желтая окраска зависит от коллоидально растворенного фосфора,
который при дальнейшем нагревании осаждается в виде светлокрасного масла.
4. По Веймарну и Б. Малышеву4 быстро выливают 5—25 мл
насыщенного при температуре кипения спиртового раствора фосфора в 1 л
холодной воды и энергично размешивают жидкость. В зависимости от
количества спиртового раствора получают золи различной степени дисперсности.
Рис. 80. Прибор для получения
коллоидального фосфора
ФОСФОРА БРОМОКИСЬ (ОКСИБРОМИД ФОСФОРА)
Phosphorus oxybromatus Phosphorus oxybromide Phosphoroxybromid
РОВгз
Мол. в. 286,73
Приготовление
По Будримону смешивают 5 ч. пятибромистого фосфора (PBrs)
в кусках и 1 ч. высушенной щавелевой кислоты (Н2С2О4); смесь назревают
в реторте сначала осторожно, затем сильнее. Образующуюся РОВгз,
смешанную с некоторым количеством РВгз, перегоняют при 193°.
Свойства
Крупнолистоватая кристаллическая масса уд. в. 2,82, Т. пл. около 55°,
т. кип. 193°. Растворима в эфире, хлороформе, сероуглероде и скипидаре.
Водой постепенно разлагается с образованием НзР04 и НВг; с этиловым
спиртом дает бромистый этил.
1 Herst. koll. Lsgg. 490.
■ A L. Elder a P. N. Burkard, J. phys. Ch. 41, 622 (1937).
• Abegg, III, 389.
« P. von Weimiro, B. Malyschew, C. II, 271 <1910).
Фосфора бромоокись. Фосфор пятибромистый и пятихлористы'и 537
ФОСФОР ПЯТИБРОМИСТЫЙ (БРОМИД ФОСФОРА[5])
Phosphorpentabromid
Phosphorus
pentabromatus
Phosphorus pentabro-
mid'e
PBr5
Мол. в. 430,56
Приготовление
По Эрдману1 растворяют желтый фосфор в сероуглероде (CS2),
охлаждают ледяной водой и прибавляют по каплям бром. Через некоторое
время осаждается трудно растворимый в CS2 пятибромистый фосфор.
Свойства
Кристаллическая насса цвета от желтого до оранжево-красного,
плавящаяся при умеренном нагревании и при более высокой температуре
распадающаяся на РВг* а Вг2. Т. кип. 106°. Водой разлагается с образованием
НзР04 и НВг.
Испытание
По техническим условиям НКХП 57—40 содержание брома >в препарате
чистом должно быть не менее 91%, хлоридов (СГ) не более 0,7%.
ФОСФОР ПЯТИХЛОРИСТЫЙ (ХЛОРИД ФОСФОРА[5])
Phosphorus
pentachloratus
Priosphorus
pentachloride
Phosphorpentachlorid
РСЬ
Мол. в, 208,27
Приготовление
1. Из PCh и хлора. Подводят совершенно сухой хлор к треххлористому
фосфору (РСЬ), помещенному в широкогорлой толстостенной, хорошо
охлаждаемой банке. Газоподводящую трубку следует брать
широкую и конец ее не должен быть погружен в жидкость во
избежание засорения. Когда масса сделается кашицеобразной,
ее перемешивают толстой стеклянной палочкой, вновь
пропускают хлор и так поступают до тех пор, пока не получится
совершенно сухой кристаллический продукт.
Реакцию можно считать законченной, если хлор,
находящийся в банке над растертым препаратом, остается непоглр-
щенным в течение суток. Избыток хлора удаляют
пропусканием СОг.
Ризенфельд2 рекомендует проводить получение РСЬ из
РСЬ ъ следующем аппарате (рис. 81). Банку, имеющую
притертую пробку, закрывают каучуковой пробкой с боковьгм
прорезом и двумя отверстиями; через одно проходит
капельная воронка, из которой приливают по каплям РСЬ, через
другое пропущена трубка, подводящая хлор. -^
•Внутри этой трубки может передвигаться в отрезке
резиновой трубки стеклянная палочка, дающая возможность
прочищать нижний конец трубки, закупоривающийся
образующимся РСЬ. Рис. 81. Прибор
Каждая падающая капля РСЬ должна встречать избыток пятихлористого
хлора. По окончании реакции пропускают хлор еще в течение фосфора
1 Н. Erdmann. Lehrb. d. anorg. Ch. 360 (1910).
1 Hecnr. пракг. 3Q4.
£38
Фосфор
3 мин., затем вынимают резиновую пробку и закупоривают байку притертой
пробкой, смазав шлиф вазелином.
2. Из фосфора и хлора. Поступают так же, как в способе 1, но вместо
PCls в толстостенную банку помещают раствор 150 г желтого фосфора
в 600 мл CS2 (огнеопасноХ). При пропускании хлора склянку несколько
охлаждают водой. Однако следует помнить, что при сильном охлаждении в
полученном препарате может оказаться примесь свободного фосфора, способная
вызвать взрыв. Сначала образуется РС1з, а затем из раствора последнего
в сероуглероде выпадает РСЬ в виде желтовато-белой массы. Когда хлор
перестанет поглощаться, реакция закончена. Реакционную массу переносят
в колбу и нагреванием «а водяной бане осторожно отгоняют CS* а
оставшийся PCU хранят в тщательно высушенных банках.
Свойства
Твердая белая или слабожелтоватая кристаллическая масса уд. в. 2,114
(при 25°), .возгоняющаяся при 100° без предварительного плавления. Жадно
притягивает влагу из воздуха, 'разлагаясь на РОСЬ и НС1. Под давлением
плавится при 163°, возгоняется при 169° (при 760 мм) с частичным
разложением; при 300° полностью разлагается на РСЬ и СЬ.
Допустимые примесив РС15 Таблица 473
(ч.д.а)
Испытание
Примеси
Допустимое
содержание
в о/о
1. Фосфор треххлористый (РС13) . .
2. Нелетучий остаток
3. Железо (Fe) . . .
0,03
ОД
0,002
По техническим
условиям НКХП 386—41
содержание РСЬ в препарате
ч. д. а. должно быть не
менее 99,5%; наибольшие
количества допустимых
примесей указаны в табл. 473;
препарат должен
выдерживать испытания на
отсутствие сульфатов.
Хранение
Хранить PCls следует в тщательно высушенных, плотно закупоренных
стеклянных сосудах, залитых парафином или снабженных шлифом, смазанным
вазелином. Однако если препарат был недостаточно защищен от влаги, то
вскоре наступает полное разложение; образующийся хлористый водород
создает внутри сосуда повышенное давление, которое приподнимает пробку,
вследствие чего влага воздуха попадает в сосуд и действует на препарат.
ФОСФОР ТРЕХБРОМИСТЫЙ (БРОМИД ФОСФОРА[3])
Phosphortribromid
Phosphorus
tribromatus
РВгз
Phosphorus
tribromide
Мол. в. 27073
Приготовление
1. По В. Ре к шине к ому1 100 г сухого красного фосфора помещают
в трехгорлую колб^ таких размеров, чтобы Р занимал не больше % объема.
Горло А прибора, составленного, как указано на рис. 82, соединяют с
прибором для получения СОг. Заполнив колбу газом отъединяют трубку,
подводящую СОг, и горло А закрывают пробкой, свернутой из асбеста2.
Из капельной воронки начинают приливать бром, сначала медленно, по
каплям, так как первые капли вызывают сильную вспышку; постепенно п'рили-
» Изв. ИРЕА, 3, 46 (1924).
8 Необходимо следить, чтобы пробка возможно меньше входила внутрь горла А, так как
« условиях реакции асбест очень плотно пристает к стеклу.
Фосфор трехбромистый
539
Звание ускоряют. Колба вскоре заполняется бурыми парами брома, вследствие
.-того что фосфор обволакивается слоем твердого PBrs, препятствующим
реакции. Тогда необходимо подогреть реакционную колбу, удобнее всего на во,-
ронке Бабо' (воздушная баня),
Протекает реакция;
ЗРВгз + 2Р = 5РВгз.
По мере того как слой РВгз покрывает фосфор, реакция начинает итти
равномерно и гладко с образованием РВгз. Если в -верхней части колбы
осядет твердый PBrs, прекращают приток брома и, нагревая колбу до
кипения РВгз, смывают PBrs вниз, где он переходит снова в РВгз.
Когда значительная часть взятого фосфора вступит в реакцию,
прекращают назревание и приливание брома, вносят {в случае, если нужно получить
большее количество РВгз) через горую А новую
порцию фосфора так, чтобы весь фосфор был
покрыт жидким PBrs, и повторяют операцию,
как указано выше.
Полученный РВгз для очистки и
окончательного освобождения от PBrs
перегоняют из колбы Вюрца..
прибавив немного фосфора;
вся аппаратура
должна быть совершение
суха. Выход 80%
теоретического.
2. По X р и с т о м а н о с у *
РВгз с очень хорошим
выходом получается при действии
брома на фосфор,
помещенный под бензолом.
В колбу емкостью 1 л с
пробкой, в которой
укреплена воронка (конец воронки
косо срезай) и трубка,
изогнутая, как показано на рис. 83,
помещают чистый бензол,
свободный от тиофена. Взяв 90—
120 мл бензола, можно перевести в РВгз больше 200 г
фосфора.
20 г фосфора (или большее количество в зависимости
от потребности в РВгз) нарезают кусочками, хорошо
высушивают и помещают в бензол, после чего колбу закрывают пробкой. В
скляночке отвешивают бром из расчета 155 г на 20 г железного фосфора и нали-.
вают- его по мере надобности в шарик воронки.
При употреблении чистого, безводного бензола и чистого фосфора сразу
получают чистый продукт. Однако можно применять и обычные продажные
сорта фосфора и бензола, очищая РВгз дробной перегонкой; нежелательной
является примесь As в исходных материалах, так как хотя AsBre кипит г/ри
220°-, но он в значительной степени летуч с парами РВгз.
Реакцию можно проводить без тяги, так как выделяется лишь
незначительное количество бромистого водорода. Только к концу реакции
необходимо работать под тягой, так как при действии брома на полученный РВгз
наблюдается сильное повышение температуры и, как следствие этого, бурное
выделение НВг, растворенного в бензоле и РВгз.
При непрерывном встряхивании приливают в колбу бром маленькими
каплями; хотя заметно повышение температуры и резкое потрескивание от
взаимодействия фосфора с парами брома, все же температура не повышается^
Рис. 82. Прибор для получения
трехЗромистого фосфор^
Рис.83. Прибор дл*
получении трех-
бром истого фосфо
pi по Христом*
носу
* Chris to man os, Z. an org. Ch. 4', 276 (1904), Prap. Ch. 200.
540
Фосфор
до кипения бензола. Разогревание ослабляют, погружая колбу в чашку
с холодной водой.
Если употреблять 20 г фосфора, то реакция идет меньше часа. С первыми
каплями брома прозрачная ©начале жидкость мутнеет, кусочки фосфора
становятся темножелтыми, плавятся с поверхности и распределяются в жидкости
в виде хлопьев. Смесь нагревают, причем хлопья исчезают, бензол
окрашивается в яркожелтый цвет, фосфор плавится и объем его быстро уменьшается
до незначительного красного или темного остатка. В колбе и на конце
газоотводной трубки показываются белые характерные пары^ НВг.
Жидкость становится красноватой и, наконец, красной, как бромная вода;
эта окраска больше не исчезает. Теперь реакция закончена. Если прибавить
еще несколько капель брома и дать смеси охладиться, то на стенках колбы
осаждаются маленькие хорошо образованные кристаллы РВгл, появление
которых свидетельствует об отсутствии избытка фосфора или раствора его
в РВгз. Если колбу теперь погрузить в горячую воду, то кристаллы^ плавятся
и исчезают, выделяются пары НВг, окрашенные бромом в красный цвет, и
жидкость несмотря на перемешивание остается оранжевой. Обесцвечивание
достигается длительным нагреванием до кипения. Еще горячую жидкость
(80—100°) отфильтровывают от оранжевого кашицеобразного тягучего осадка 1
в колбу для перегонки (если нагревать вместе с этим осадком, то еще
прежде, -чем перегонится весь РВгз, может произойти сильный и опасный
взрыв).
Перегонка отфильтрованной жидкости легко идет при нагревании на
сетке. Сначала выделяется НВг и окраска жидкости переходит в желтую;
после полного удаления довольно прочно удерживаемого брома, жидкость
становится почти бесцветной. При 75° переходят первые капли бензола;
температура тотчас повышается до 81° и остается постоянной, пока не отгонится
ббльлпая часть бензола. Фракцию, кипящую до 120°, отделяют. От 120°
температура быстро поднимается до 167° и при 167—173° переходит почти все
оставшееся содержимое колбы. Светлокрасныи аморфный фосфор оседает на
стенках колбы в виде налета и в колбе остается только совсем маленький
остаток цвета от о'ранжево-красного до темнокоричневого.
Для получения из первого дестиллата почти чистого РВгз к нему
прибавляют несколько капель брома до красного окрашивания, причем с
вскипанием образуются желтые хлопья PBrs. Нагревают на масляной бане до
кипения; при этом растворенный фосфор с образовавшимся PBrs дает РВгз.
Продукт перегоняют; между 160—169,3° переходит незначительное количество
пахнущего бензолом РВгз; при употреблении дефлегматора почти чистый
РВгз перегоняется при 170—171°, и только очень незначительный остаток,
состоящий из РВгз и фосфенила2, образует третью фракцию, кипящую при
172-473°.
Если дефлегматор достаточно велик (с 4—5' шариками), то этот остаток
составляет 2—4 г.
Свойства
Бесцветная, подвижная, остро пахнущая, чрезвычайно едкая жидкость
уд. в. 2,85, сильно дымящая на воздухе. Т. пл. —40°, т. кип. 172,9°. На коже
оставляет оранжевые пятна. Растворяется в эфире, ацетоне, хлороформе,
бензоле, сероуглероде и четыреххлористом углероде. Водой разлагается с
сильным выделением тепла и образованием фосфористой кислоты и НВг. Спиртом
^разлагается на НзРОз и CsHsBr.
Испытание
По техническим условиям НКХП 58—40 содержание брома в препарате
чистом должно быть не менее 85%, хлоридов — не более 0,8%.
1 Этот осадох (несколько дециграмм) состоит в большей чисти из фосфора, РВг6, РВг,
г производных бензола.
' С,,Н5РВ , бромаигидрид фосфенилооой кислоты.
Фосфор трехиодистый. Фосфора трехокись 541
ФОСФОР ТРЕХИОДИСТЫЙ (ИОДИД ФОСФОРА[3])
Phosphorus trijodatus Phosphorus triiodide Phosphortrijodid
PJs Мол. в. 411,74
Приготовление
1. По Бессону пропускают сухой йодистый водород в раствор треххло*
ристого фосфора (РСЬ) в четыреххлористом углероде и выпаривают жидкость
под вакуумом досуха, тщательно оберегая от водяных паров.
Реакция обмена происходит уже при температуре холодильной смеси.
2. По Ф. Д ж ё р м а « у и Р. Трэкслеру1 к раствору 5,4 г желтого
фосфора в 90 мл CSa добавляют постепенно 66 г иода, вытеснив воздух- из
реакционного сосуда струей СОг. Раствор фильтруют через фильтр Шотта
также ь атмосфере СОг и выпаривают «на водяной бане (огнеопасно!) почти
досуха. Отделив незначительное количество оставшегося маточного раствора,
РЛз высушивают при 80° <в вакууме, создаваемом водоструйным насосом.
Свойства
Интенсивно-красные кристаллы, плавящиеся при 61°. Во влажном воздухе
быстро разлагается.
ФОСФОРА ТРЕХОКИСЬ (ОКИСЬ ФОСФОРА[3])
Phosphorus Phosphorus trioxide Phosphortrioxyd
trioxydatus
P4Oe Мол. в. 219,92
Приготовление
По Торпе и Тёттону2 пользуются прибором, изображенным на рис. 84.
Сухой фосфор, которого для каждой операции идет приблизительно 2
палочки, помещают кусочками в 25 мм в сожигательную трубку А диаметром
Рис. 84. Прибор для получения трехокиси фосфора
около 35—40 мм. Трубка изогнута, как указано на 'рисунке, чтобы
расплавленный фосфор не выливался. Один конец трубки открыт для доступа
воздуха, который не подвергается предварительной просушке. К другому концу
присоединена медная трубка, окруженная более широким медным кожухом,
образующим холодильник. Последний заливают водой через трубку С. Тем-
> F. Е. Е. Oermanu a. R. N. Traxler, J.Am. Chem. Soc. 49, 307 (1927); W. Blitz u.
A. Sapper, Z. anorg. Ch. 203, 28 i (193*).
■ Torpe, Tut ton, J. С hem. Soc. 57, 547(1890); также Kraft u. Neumann, Ber. 34, 566(1001)
Prap. Ch. 191.
mi
Фосфор
ператуфу воды можно измерять термометром В. Около В помещают рыхлую
пробку из стеклянной ваты длиною в 50 мм («а рисунке не видно).
Медная (внутренняя) трубка соединяется с U-образной конденсационной
трубкой. В месте изгиба U-образной трубки (длина колена которой должна
быть не менее 300 мм) впаяна отводная трубка, соединенная плотной пробкой
с маленькой банкой, где собирается расплавленный препарат. При помощи
водоструйного насоса через Прибор можно просасывать воздух. Между
насосом и конденсационной трубкой поставлена промывная склянка Дрекселя
с концентрированной H2SO4. Серная кислота препятствует попаданию
водяных паров в прибор; по той же склянке контролируется скорость струи
воздуха. U-образная трубка погружена в холодильную смесь; для более
совершенной конденсации можно применить две такие трубки, соединенные
последовательно.
Когда прибор собран, пускают в ход насос, несколько мгновений
маленьким пламенем нагревают фосфор у переднего конца трубки! для его
воспламенения и просасывают энергичную струю воздуха, так чтобы температура
воды в холодильнике все время была около 60°. Лишь к концу операции
допускается повышение до 60°.
Че*рез полчаса после воспламенения фосфора РЮв начинает переходить
в конденсационную трубку. Если пробка из стеклянной ваты достаточно
плотна и температура воды не превышает 60°, то в приемник переходят
только следы Р2О5 и очень малые количества свободного фосфора, в то
время как РЮв собирается там в виде снежно-белой, воскообразной массы.
'Операцию во 'избежание окисления продукта-заканчивают, когда израсходовано
л/ъ взятого фосфора. U-образную трубку отнимают и нагревают.
Расплавленная РЮв стекает в подставленную склянку.
Хранят РЮв в закрытых стеклянных трубках, воздух из которых
вытеснен СОг.
Свойства
Белая кристаллическая масса уд. в. 2,14, плавящаяся при 23,8° в
бесцветную, прозрачную, очень подвижную жидкость. При отсутствии кислорода
кипит при 173° без разложения. Растворяется, не изменяясь, в эфире,
бензоле и tS2. В холодной воде медленно растворяется с образованием НзРОз.
С горячей водой бурно, наподобие взрыва, превращается в красную недокись
фосфора (РЮ), красный фосфор, Н3РО4 и самовозгорающийся РНз. На
рассеянном дневном свету медленно Желтеет; на прямом солнечном свету
быстро окисляется, при 70° даже загораясь. Плотность пара 7,7.
РЮв почти так же ядовит, как и белый фосфор.
ФОСФОР ТРЕХСЕРНИСТЫЙ (СУЛЬФИД ФОСФОРА[3])
Phosphorus sulfuratus Phosphorus sulfide Phosphortrisulfid
P2S3 Мол. в. 158,14
Приготовление
По Эрдману1 310 г красного фосфора омешивают с 430 г серного
Цвета и смесь вносят ложками в гессенский тигель, предварительно
нагретый на бунзеновской горелке.. По внесении каждой порции тигель закрывают
Крышкой; тотчас же начинается реакция. После внесения всей смеси тигель
охлаждают так, чтобы масса еще оставалась жидкой, и выливают
содержимое на железный лист.
Затвердевший продукт еще теплым разрезают на куски и складывают
В плотно закрывающуюся банку.
i Хим. преп. 42.
Фосфор трехсернистый и треххлористый
513
Свойства
I Твердая черная хрупкая масса уД< в. 2,03, ясно кристаллического
строения. Т. пл. 172,5°, т. кип. 407°.. При сильном нагревании на воздухе
загорается. При лежании на воздухе становится липким и разлагается, выделяя
H2S. He растворим в воде, НС1 и H2SO4; растворяется в НМОз,
сероуглероде и растворах щелочей*
ФОСФОР ТРЕХХЛОРИСТЫЙ (ХЛОРИД ФОСФОРА[3])
Phosphorus trichloratus Phosphorus trichloride Phosphortrichlorid
РСЬ Мол. в. 137,35
Приготовление
В. Рекшинский1 предлагает следующий способ Получения РСЬ.
100 г сухого красного фосфора помещают в реторту с тубусом (рис. 85, а).
Реторта А должна быть таких размеров, чтоПы фосфор занимал не более
половины объема. Через тубус пропускают стекл..плую трубку В, диаметром
8—10 мм, конец которой
приблизительно на 3 см не достигает
поверхности фосфора. Загнутое
вертикально вниз горло р-;топты
соединено с приемником — колбой
Су имеющей отросток,
направленный вверх под углом 45° (при
отсутствии такой колбы можно
воспользоваться круглодонной А rffc
колбой и рогатым форштоссом
(рис. 85, б).
Отросток -колбы соединен с
обратным холодильником D,
закрытым хлоркальциевой трубкой
Е для предохранения от
попадания водяных паров. Колба С
помещена в холодную воду.
Сначала вытесняют из
прибора «воздух, подводя через
трубку В ток сухого СОз, после
чего «нагревают реторту до
появления в верхней ее части
желтоватого возгона. Тогда по трубке
В начинают пропускать ток
совершенно сухого хлора.
" Начало реакции заметно по .появлению бледного пламени у конца
трубки В; нагревание и ток хлора регулируют таким образом, чтобы, с одной
стороны, не было возгона желтого фосфора (в этом случае увеличивают ток-
хлора и ослабляют нагревание), а с другой стороны, не происходило бы
образования твердого РСЬ (необходимо усилить нагревание и уменьшить ток
хлора)2.
Реакцию ведут до израсходования приблизительно % всего фосфора.
Иногда Р спекается и пристает к стеклу; этого можно избежать, если фос*
фор предварительно промыть и высушить на водяной бане. Получаемый
РСЬ перегоняется в колбу С. В нем может быть примесь желтого фосфора,
переходящая в РСЬ при пропускании хлора. Если имеется примесь РСЬ,
Рис.. 85. Прибор для получения треххлористого
фосфора
1 Изв. ИРЕА, 8, 46, (19:4).
* По Михаэлису мочено регулировать лроцесс, пгредвигая^трубку В: для удаления
желтого фосфора трубку поднимают выше, для удаления PUu —трубку опускают. Темпеэатура
поддерживается за счет теплоты реакции.
544
Фосфор. Хлор
то прибавляют немного красного фосфора и подогревают до 60°.
Находящийся в жидкости и на стенках колбы твердый РСЬ при взбалтывании
с красным фосфором переходит в жидкий РСЬ.
Для окончательной очистки РСЬ перегоняют из колбы Вюрца, причем
избыток фосфора остается в колбе.
Хлорокись фосфора, РОСЬ, могущая образоваться в присутствии влаги
{вследствие недостаточной сушки газов—. СОг и СЬ—-или негерметичности
соединений), отделяется при перегонке, как выше кипящая фракция.
Выход 60% теоретического.
Свойства
Бесцветная, напоминающая воду, легкоподвижная жидкость уд. в. 1,57,
дымящая на воздухе и вызывающая раздражение слизистых оболочек
(слезотечение).
Т. пл.— 92°, т. кип. 76,6° три давлении 760 мм*.
С эфиром, хлороформом, бензолом и сероуглеродом смешивается во всех
отношениях; водой и спиртом разлагается.
Испытание
По ГОСТ 91-41 реактивный препарат должен иметь удельный вес {df)
1,58, т. кип. 75—77° (при 760 мм рт. ст.) и содержание РСЬ не менее 95,5%.
ФОСФОРА ХЛОРОКИСЬ
Phosphorus oxychloride Phosphoroxychlorid
РОСЬ Мол. в. 153,35
Приготовление
1. В круглодонную колбу емкостью 250 мл помещают 100 г РСЬ,
закрывают колбу пробкой с укрепленным в ней обратным холодильником и
понемногу, очень маленькими порциями (не больше 0,5—1 г) вносят 33 г тонко
растертого КСЮз. Реакция протекает очень бурно, сопровождается сильным
потрескиванием внутри жидкости и даже появлением пламених. При
соблюдении указанных выше условий (Метод не представляет опасности. По
окончании реакции (последние порции КСЮз остаются непрореагировавшими)
РОСЬ сливают в колбу Вюрца и перегоняют, собирая фракцию 105—108°.
2. Если в распоряжении экспериментатора имеется небольшое количество
готового РОСЬ, то описанный выше метод можно по указанию Ульмана2
несколько видоизменить с тем, чтобы' обеспечить более спокойное течение
реакции.
В небольшую круглодонную колбу (емкостью 100—150 мл) помещают
16 г тонко растертого сухого КСЮз и 20 г РОСЬ. Закрывают колбу пробкой
с обратным (шариковым) холодильником и в верхний конец холодильника
вставляют свободно (без пробки) капельную воронку с 50 г РСЬ. Вначале
РСЬ добавляют редкими каплями, каждая из которых вызывает энергичную
реакцию и вскипание жидкости; когда будет прилито 15—20 мл РСЬ,
скорость приливания можно несколько увеличить.
По окончании реакции РОСЬ отгоняют на масляной бане, нагретой до
150°.
Выход 72,5 г (95,5% теоретического).
1 Добавлять новую порцию КС108 можно только п,сле окончания бурной реакции, вызванной
добавлением предыдущей порции КСЮ..
» Ullmann, Вег. 34, 2172 (1901).
Фосфора хлороокись. Хлор
545
I
Э. В качестве побочного продукта POCU получается при синтезе
хлористого тионила (см. стр. 510, метод 1). Полученную этим методом фракцию,
^кипящую выше 100°, состоящую из неочищенной POCh, достаточно перегнать,
'собирая фракцию 104—108°.
Свойства
Бесцветная, сильно преломляющая свет жидкость уд. в. 1,69 с резким
запахом, напоминающим РСЬ. Т. пл. 1,3°, т. кип. 107,23°. Во влажном
воздухе дымит.
ХЛОР
1 Chlorum Chlorine Chlof
>', Cl At. в. 35,467 Мол. в. 70,914
Приготовление
А. Из МпОа и НС1
1. В .колбу емкостью 1 л (рис. 86) помещают двуокись марганца, МпОг,
Кусочками величиной с орех и приливают через воронку 500 мл
концентрированной НС1. Нагревают сначала осторожно, затем сильнее до превращения
выделения хлора. Для очистки от брызг
кислоты газ пропускают через промыв*
ную склянку с -водой.
Чтобы не разбить колбу при загрузке
МпОг, рекомендуется .налить
предварительно © колбу воды, а перед
добавлением НС1 слить воду.
2." При частой потребности в хлоре
удобно пользоваться аппаратом Орлов-
с ко г о, видоизмененным А р е« д то м.
На общей железной подставке
устанавливаются 3 больших колбы и
укрепляются так, чтобы при переноске
аппарата они не могли упасть. Собственно
аппарат, выделяющий хлор, представляет
собой среднюю колбу А (рис. 87); ее
емкость 3 л или немного больше; две
другие — соответствующей величины. Все три
колбы закрыты просверленными
резиновыми пробками; для А и В — с тремя,
С — с двумя отверстиями.
От В отходят 3 стеклянные тр.убки,
из которых с оканчивается под пробкой,
а и b доходят до дна, причем концы последних трубок срезаны вкось; cub
снабжены короткими резиновыми трубками с зажимами и могут соединяться
с более длинными трубками.
Колба А имеет трубку а, доходящую до дна и срезанную вкось, она
согнута под прямым углом и -может быть соединена с трубкой а колбы В.
Трубка d служит предохранительной трубкой. Она обрезана под пробкой,
а снаружи согнута U-образно и доходит до дна высокого цилиндра D,
содержащего концентрированную H2SO4; трубка е также оканчивается под
пробкой и соединена с колбой С, где эта трубка доходит до дна. Наконец,
трубка / оканчивается под пробкой колбы С и служит газоотводной трубкой.
Чтобы п'ривести аппарат в действие, сначала в колбу А накладывают
немного больше чем до половины ее объема кусочков двуокиси марганца
величиной не меньше горошины и не больше ореха, вставляют
соответствующую пробку с тремя трубками, причем держат колбу горизонтально (чтобы
трубки могли достигнуть дна колбы). Затем наливают в В около 1 л кон цен-
Рис. 86. Прибор для получения хлора
35 *3ак. 1364. Чистые химические реактивы.
546 Хлор
трированной НО, и закупоривают соответствующей пробкой. На трубку ь
надевают зажим и соединяют обе трубочки а толстостенным, плотно надеваю-
щимся каучуком.
В колбу С наливают 200—300 мл концентрированной H2SO4, закрывают
ее соответствующей пробкой и одну трубку соединяют с е, а другую —
с приборами, в которых О должен с чем-либо реагировать. Нужно иметь
еще два-три коротких кусочка каучуковой трубки в 3—4 см длиной для
замыкания наружных стеклянных трубок колбы В, причем их закрывают
с одного конца стеклянными пробочками или зажимают зажимом (все за-
жимы должны быть такого устройства, чтобы их можно было сдвигать
с каучука на стеклянную трубку для открывания соответствующего каучука;
можно употреблять винтовые зажимы Гофмана).
Для выделения хлора запирают b и с, освобождают от зажима аа и слабо
подогревают В горелкой (колбы А и В стоят на сетках). Находящаяся в В
крепкая НС1 выделяет газообразный НС1, который своим давлением вытес-
Рис. 87. Прибор ддя получения хлора по Арендту
няет жидкую кислоту в колбу А. Как только это достигнуто, замыкают
зажимом трубку а и передвигают горелку под Л, вследствие чего тотчас
начинается выделение хлора> длящееся до тех пор, пока НС1 не истощится (через
несколько часов). Выделяющийся С1 промывается в С и оттуда направляется
для использования через /. Чтобы прекратить реакцию, достаточно запереть
зажимом трубку / и отпереть аа\ b остается п,ри этом запертой, а с
отпирается и соединяется с длинной трубкой, погруженной в раствор КОН.
Горелка остается под А, пока давлением газа вся кислота не перегонится в В.
Хлор, который при этом переходит туда же, поглощается КОН. Затем
уби'рают горелку и запирают из предосторожности аа\ через несколько минут
действие аппарата вполне прекращается. Чтобы снова началось выделение
газа через любое время, открывают а, запирают Ь и с, открывают /, ставят
горелку под В и поступают далее, как было уже описано; через несколько
минут начинается выделение С1.
Когда соляная кислота истощается, ее можно заменить свежей
следующим простым способом. После того как выделение хлора вполне
прекратилось, запирают аа и с, отпирают трубку Ь, соединив ее с длинной
стеклянной трубкой, затем подставляют под нее большой (более 1 л) стакан и
нагревают В. подставив горелку. Жидкость давлением собственных паров
вытесняется через трубку Ь, которая действует при этом как сифон.
Хлор
547
При удалении горелки следует, конечно, открыть с, чтобы мог войти воз-
Е&ух. Когда таким образом баллон В опорожнен, запирают с (аа остается
акрытой, Ь открытой) и нагревают оставшуюся в в жидкость, чтобы парами
, we отчасти 'вытеснить воздух. В то же время снимают с Ь каучук с зажимом,
^быстро пододвигают большой стакан Е с крепкой НС1, помещенный заранее
fjia подставку соответствующей высоты; затем убирают горелку.
|; Вследствие конденсации паров в В образуется разрежение и наружный
^воздух вдавливает кислоту через Ь; когда кислота начнет течь через эту
[трубку, открывают аа или с и кислота вся сама перетекает в В через
сифон Ь. Аппарат снова готов к действию; опоражнивание и наполнение
кислотой при некотором навыке экспериментатора занимают всего несколько минут.
Предохранительное приспособление D устраняет опасность, которая могла"
бы возникнуть, если бы какая-нибудь из трубок а, е, f или прибор, в
который поступает О, оказались закупоренными. Газ выделяется тогда
беспрепятственно через H2SO4 в цилиндре D.
Этот аппарат, предложенный впервые Орловским (без
предохранительного приспособления D), может при хороших каучуках стоять месяцами
без ^потребления и без всяких предварительных подготовок давать
равномерный ток С1, как только подставят горелку под Л.
Б. Из MnOa, NaCl и H2S04
К смеси б ч. МпОг и 3 ч. NaCl прибавляют 20 ч. теплой H2SO4,
полученной смешением равных количеств концентрированной H2SO4 (уд. в. 1,84)
и воды.
После смешения начинается выделение
почти безводного хлора; к концу реакции
полезно смесь подогреть. Выделяющийся газ
по М о з е р у * содержит некоторое количе*
ство хлористого водорода.
в. из кмп04 и на*
Очень удобно и быстро можно получить
струю хлора, приливая по каплям
концентрированную НС1 «а твердый перманганат
(KMnCh), помещенный в колбу Бунзена.
Выделяющийся газ промывают, пропуская через
концентрированную НС1.
Равномерная струя хлора получается этим
методом >в аппарате Левиса и Ведекин-
да3, снабженном регулирующим
приспособлением для изменения давления (рис. 88).
Баночка С с концентрированной H2SO4
закрыта пробкой, в которой может
передвигаться пипетка Р.
При выдвигании пипетки из кислоты газ
выходит в тягу; при погружении пипетки
хлор идет в реакционный сосуд и
выделяется в тягу только тогда, когда
соответствующий положению пипетки столбик
кислоты не сможет уравновешивать давления газа.
К 10 г чистого истертого КМп04 приливают по каплям 60—65 мл НС1
(уд. в. 1,17); получают равномерную струю хлора, совершенно свободного от
кислорода 4.
1 L. Moser, Di> Reindarstellun? von Oasen, Verb P. Enke, Stuttgart, 1920,
• iQ.Oraebe, Ber. 34, 645 (1901); 35 43 (i902j.
•Levis, Wedekind. Z. an?. Ch. 21 580 (1909).
4Велери Штрейхер утверждают, что полученный этим методой хлор содержит
кислород и рекомендуют полому для получения особо чистого хлора действовать НС1 на гидрат
перекиси марганца <86о|0-иый по М е р к у).
Реанционньш
^= сосуд
Рис. 88. Аппарат для получения
хлора по Левису
548
Хлор. Хром
Г. Из КаСгаОт и НС1
Нагревают на водяной бане 180—200 г тонко измельченного двухромсжо-
кислого калия с 1 л технической НС1.
Выделяющийся газ может содержать пары кислоты, незначительные
количества окиси хлора, следы СОг, воздуха и водяных паров. Для очистки газ
пропускают последовательно через две промывные склянки с водой, через
концентрированную H2SO4 и для разрушения окислов хлора через
накаленную докрасна трубку длиною в 30 см, наполненную асбестом. Очищенный
таким образом хлор пропускают через U-образную трубку, охлаждаемую
смесью льда и соли, и, наконец, высушивают при помощи СаСЬ или P2Os.
t Д. Из хлорной извести и НО
Для получения больших количеств хлора (и для частого получения
небольших количеств) целесообразно применять метод Буассено.
В средний шар аппарата Киппа помещают кубики из хлорной извести
и наливают в аппарат разбавленную НС1 (уд. в. 1,12). Для очистки газ
промывают, пропуская через воду и концентрированную H2SO4. При длительных
перерывах в работе кислоту из аппарата рекомендуется выливать, так как,
поглощая хлор, она постепенно заполняет средний шар, вследствие чего
вновь начинается выделение хлора. При коротких перерывах в аппарат для
удаления хлора вдувают воздух.
Кубики из хлорной извести можно приготовлять прессованием свежей
хлорной извести или замешиванием 1 ч. хлорной извести с % ч. гипса и
небольшим количеством воды. По Классону нет надобности изготовлять
кубики, так как хлорную известь можно применять в том виде, в каком она
имеется в продаже.
Е. Очистка хлора от примеси О*
Для получения хлора, не содержащего даже следов Ог, О. Ген иг-
шмидт и К. Винтерсбергер1 рекомендуют пропускать газ через
кварцевую трубку, наполненную кусочками древесного угля и нагретую до
красного каления.
Ж. Приготовление хлорной воды
По Л и б и х у наполняют реторту холодной дестиллированной водой и,
перевернув, кладут ее на толстый слой материи или соломы. Через
стеклянную трубку вводят хло'р, который частично растворяется, частично
собирается над водой, вытесняя последнюю в горло реторты. Взбалтывая время
от времени жидкость, вызывают растворение хлора, собравшегося над водой.
Свойства
Зеленовато-желтый ядовитый лаз с резким удушливым запахом
уд. в. 2,486 (по воздуху). При 0° и 6 ат давления сгущается в золотисто-
желтую жидкость. Т. пл. —100,5°, т. кип. —33,9°. Растворим в воде, и
вследствие этого его следует собирать над ртутью (за неимением ее — над
горячей водой или над насыщенным раствором NaCl). Вода, растворяя хлор,
окрашивается в зеленоватый цвет и приобретает запах хлора (хлорная вода).
Растворим в четыреххлористом углероде, бромистом этилене и
концентрированной НС1 (1 л НС1 растворяет 7,3 г хлора).
Хлор не горит. Зажженная свеча, опущенная в хлор, продолжает гореть
коптящим пламенем. Пламя светильного газа или спирта окрашивается в ат-
мосфере хлора в зеленый цвет.
Вдыхание небольших количеств хлора вызывает удушливый кашель; при
больших количествах наступает кровохарканье.
1 О. H6nigschmidt ц. К. Wintersberger, Z. а юг*. Ch. 219, 166 (1934).
Хром
549
Таблица 414
Растворимость хлора в воде
/°с
10
И
12
13
14
15
16
17
18
19
20
21
22
23
24
25
1 об. воды
растворяет
об. хлора
2,5852
2,5413
2 4У77
' 2,4543
2,4111
2,3681
2,3253
2,2827
2,2405
2,1984
2,1565
2,1148
2,l>734
2,0322
1.9912
1,9504
100 мл
хлораой воды
содержат
г хлора
0,82038
0,» 646
0.79264
0,77884
0,76515
0,75159
0.73792
0,72443
0,71102
0,69765
0,68435
0,67112
0,65797
0,64490
0,63189
0,61894
/°с
26
27
1 28
29
30
31
32
33
34
35
36
37
38
39
40
1 об. воды
растворяет
об. хлора
.1,9099
1,8695
1,82°5
1,7895
1,7499
1 1,7104
1,6712
1.63V2
1,5934
1,5550
1,5166
1,4785
1,4406
1,4029
1,3656
100 мл
хлорной воды
содержат
г хлора
0,60610
0,593*7
0,58057
0,56789
0,55531
0,54279
0,53035
0.517У6
0,50567
0 4 )34/
0.4 S128
0 46919
Ю.45717
0.44320
0,43336
ХРОМ
Cromium Chromium Chrom
Cr At. в. 52,01
Приготовление
1. По Гольдшмидту1 омесь 600 г окиси хрома (СггОз), 120 г
плавленого и растертого хромового ангидрида (СгОз) и 270 г порошка
алюминия (или 60,9 г СггОз, 24 г металлического кальция и 10,8 г А1) помещают
в тигелв^с набойкой из магнезии и зажигают.
Некоторую разновидность указанного алюмотермического метода приводит
Ризеяфельд2.
В глиняный тигель высотой 10 см всыпают сначала 10 г
порошкообразного фтористого кальция (CaF2) и затем тщательно приготовленную смесь
35 г СггОз, 25 г двухромовокислого калия (К2СГ2О7) и 20 г алюминиевой
пыли. Эту смесь покрывают тонким слоем зажигательной массы,
приготовляемой смешиванием 5 г А1 с 15 г пер-еюгои бария (ВаСЬ). Тигель вставляют
в -чашку, наполненную песком, и удаляют находящиеся поблизости горючие
вещества, так как раскаленные частицы разбрасываются в стороны на метр
и более.
Для зажигания пользуются полоской селитряной бумага3, которую
зажигают спичкой. Если воспламенение не удалось, увеличивают количество
зажигательной смеси или подогревают предварительно тигель до 100—200°.
По окончании бурной реакции (необходимо защитить глаза очками) и
охлаждении^ тигель разбивают и королек хрома очищают от приставшего
шлака.
*Goldschmldt, Aim 301, 19 (1898); Bull. Soc. Chim. (4), 1, 10; W. Prandln B. Bleyer
Z. anorg. Ch. 64, 217 (1909).
■* Неорг. практ., 169.
• Фильтровальная бумага, пропитанная 10%-ным раствором KN09 и высушенная.
550
Хром
2. По Гляцелю растворяют 100 г КгСгиСЬ в возможно малом
количестве воды, прибавляют 400 мл НО (уд. в. 1,124) и затем постепенно
прибавляют 100 мл 80%-ного спирта. Добавив к жидкости 160 г КС1, фильтруют,
выпаривают досуха и обезвоживают остаток умеренным нагреванием.
Отделив полученную соль от зеленых частиц, ее смешивают с 50 г порошка
магния и накаливают полчаса в гессенском тигле при красном калении.
После охлаждения тигель разбивают; сплавленную массу очищают с
поверхности от зеленых частиц и бросают в воду; при этом масса распадается
в тонкий порошок. Последний очищают промыванием водой с последующей
декантацией, кипячением с HNOs и вторичной декантацией, после чего
сушат на водяной бане. Выход 27 г.
3. По методу Грубе в большой стакан помещают раствор 120 г С Юз,
1,5 г Cr2(S04)3 и 4,4 г Сг(ОН)з в 4 л воды; смесь подогревают [причем
Сг(ОН)з полностью растворяется] и подвергают электролизу.
Катодом служит медная проволока, помещенная в центре стакана,
анодом—свинцовый змеевик (холодильник) с проточной водой. Сила тока 0,1 а,
напряжение 3,2 в.
Электролиз ведут в течение нескольких дней без перемешивания
электролита, после чего отделяют слой Сг и нагревают его в высоком вакууме при
600° для удаления Ш. Полученный серо-стальной металл очень чист1.
Свойства
Серый, кристаллический порошок уд. в. 7,1, частично состоящий из
микроскопических ромбоэдров. Т. пл.2 1765°, т. кип. около 2200°.
Полученный алюмотермическим путем—твердая, серовато-белая
блестящая масса, листовато-кристаллического строения.
На воздухе металл окисляется очень медленно даже при красном
калении. Растворим в НС1 с выделением водорода; в H2SO4 растворяется при
нагревании; НЫОз, даже горячая и концентрированная, на хром не
действует, так как последний, подобно железу, при этом «пассивируется».
ХРОМ АЗОТНОКИСЛЫЙ (НИТРАТ ХРОМА[3])
Cromium nitricum Chromic nitrate Chromnitrat oxyd
Cr(NOs)3 -9HoO Мол. в. 400,18
Приготовление
По С. К. Чиркову8 к 100 г CrOs приливают 260 мл HNOa (уд. в. 1,3—
1,4) и смесь осторожно восстанавливают сахаром или крахмалом, добавляя
восстановитель до появления устойчивой желтой окраски окислов азота.
К охлажденному раствору добавляют HNOs (120—200 мл на 1 л раствора)
и оставляют на сутки для кристаллизации. Кристаллы Cr(NOs)a • 9НгО
отсасывают и сушат в токе сухого воздуха при температуре не выше 36°.
Свойства
Фиолетовые кристаллы, очень легко растворимые в воде и спирте. Т. пл.
36,5°.
i H. Haraldsenu. E. Kowalskl, Z. anon*. Ch. 224, 330 (1935).
»Фроиман и Зауэрвальд предполагают, что т пл. совершенно чистого хрома лежит
около 1 2) ; незначительная прим сь Н- или N. сильно снижают температуру плавления.
Е. Frit in an i* u. F. Sauerwald, Z. an org. Ch. 209, 73 (1931). * JVJ
»и,Изнеков, пНовостд техники-. № J ,34 (193. )f
Хром азотнокислый. Хрома гидрат окиси
551
Таблица 475
Удельный вес водных растворов Cr(N03)3
о/0 Cr(N03)a
1
2
4
6
8
10
'"
1,0073
1,0155
1,0322
1,04°2
1,0666
1,0844
о/о Cr(NO,)8
12
14
16
18
20,
-У
1,1027
1,1214
1,1407
1,1606
1,1810
о/о C^NO,),
22
24
26
28
30
-?
1,2020
1,2236
1,2459
1,2690
1,2929
Испытание
Наибольшие количества примесей, допустимых по ОСТ НКТП 7171/465;
указаны в табл. 476.
Таблица 476
Допустимые примеси в Сг (N03)3• 9Н20
Примеси
1. Нерастворимые вещества ....
2. Хлориды (СИ)
4. Железо (Ре)
5. Щелочные и щелочноземельные
металлы (в виде сульфатов) . . .
Допустимое содержание в °/0
в чпстом для
анализа
0,0^5
0,005
0,005
0,005
0,20
в чистом
0,03
0,01
0.01
0,01
0,50
ХРОМА ГИДРАТ ОКИСИ (ГИДРООКИСЬ ХРОМА[3])
Chromium oxydatum Chromic hydroxide Chromhydroxyd
hydricum
Сг(ОН)з
Мол. в. 103,03
Приготовление1
1. 100—110 г двухромовокислого натрия (ЫагСггО? • 2Н2О) растворяют в
270—300 мл воды в фарфоровой чашке и фильтруют2. Прозрачный раствор
нагревают до 600° в фарфоровой чашке (под тягой), добавляют к нагретому
раствору ) мл НС1 (уд. в. 1,19) и после перемешивания вносят небольшими
порциями заранее приготовленную смесь 50 мл 32—35%-ного формалина
с 60 мл НС1 (уд. в. 1,19).
1 Детальк разработано ИРЕА.
8 Хромпик должен содержать не меньше 90% NatCr,07 и не больше 1% Na,S04. если
сырь° не удовлетворяет этим требованиям, приготовляют насыше ный п и 15—20° раствор
NaeCr207 так, чтоиЫ часть соли ос.алась нерастыоренн >й. В *тих условиях почтя весь
Na^SUi останется в осадке. 160 г такого раствора разбавляют 240 мл воды.
552 Хром
Реакция восстановления протекает бурно и быстрое прибавление
формалина ведет к выбрасыванию жидкости. Коричневый цвет раствора к концу
реакции переходит в темный сине-зеленый (СгСЬ). Необходима ripo6a на
полноту восстановления, для чего несколько мл раствора нагревают,
прибавляют ЫНЮН до слабого запаха1 и, доведя до кипения, оставляют стоять
для осаждения Сг(ОН)з.
Если над осадком отстаивается вполне бесцветный раствор, это указывает
на полноту восстановления.
Следующая операция заключается в переводе СгСЬ в Сг(ОН)з.
Для этого раствор разбавляют 200 мл дистиллированной воды и, нагрев
почти до кипения, прибавляют небольшими порциями чистый ЫНЮН до
слабого запаха (приблизительно 190 мл). ГГри осаждении надо следить, чтобы
количество ЫНЮН было достаточное (раствор над осевшим Сг(ОН).-* должен
быть бледнофиолетовым). Если осадок получается плотный и липкий, следует
прибавить еще некоторое количество ЫНЮН.
По окончании осаждения Сг(ОН)з переносят на бязевый или бумажный
фильтр и несколько раз промывают горячей дестиллированной водой для
удаления главной массы растворимых солей. Дав жидкости стечь, переносят
осадок в склянку емкостью 2—3 л и промывают дестиллированной (или
профильтрованной водоп'роводной) водой. При первых промываниях
рекомендуется добавлять к воде по 10—15 мл ЫНЮН. Промывку ©едут, хорошо
взбалтывая осадок с водой и после отстаивания сливая воду сифоном. После
10—15-кратного промывания осадок переносят в фарфоровую чашку и
несколько раз промывают, нагревая почти до кипения с дестиллированной
водой. Окончание промывки контролируется растворением п'робы осадка в НЫОз
и добавлением AgNCb; отсутствие мути (AgCl) указывает на достаточную
чистоту продукта2.
Тогда Сг(ОН)з переносят на фильтр и дают стечь промывной воде. Мокрый
осадок перекладывают в стеклянную или фарфоровую чашку и высушивают
при 40—60°, часто перемешивая.
Высушенный продукт растирают в ступке и просеивают через закрытое сито
[сухой Сг(ОН)з сильно пылит].
Полученный гидрат отвечает не формуле Сг(ОН)з, а формуле Сг(ОН)з •
• А'НзО.
Выход около 100 г.
2. По Кольшюттеру3 к Ъ л 0,Ш раствора Сг(ЫОз)з при сильном
перемешивании (лучше механической мешалкой) приливают по каплям 2,5 л 0,1 N
ЫНЮН. При дальнейшем перемешивании в течение 2 час. осадок Cr(OH):i
исчезает (пептизируясь солью Трехвалентного хрома). Тогда медленно
приливают еще 2,5 л 0,Ш ЫНЮН, причем выпадающий Сг(ОН)з легко отмывается
от ЫОз'. Промытый препарат высушивают около 100°.
Свойства
Серо-зеленый или серо-голубой аморфный порошок уд. в. 2,9, не
растворимый в воде. После сушения над H2SO4 отвечает формуле Сг(ОН)з • 2НгО;
высушенный при 100°—Сг(ОН)з.
Выше 100° разлагается на, СгаОз и воду, далее масса раскаливается и
окрашивается в темнозеленый цвет, теряя способность растворяться в кислотах.
Растворим в щелочах с образованием хромитов.
Испытание
По ОСТ 10165-39 чистый препарат (реактив) может содержать следующие
наибольшие количества примесей (табл. 477).
1 Если NH4OH был взят в избытке, то оы частично растворяет Сг(ОНЬ» придавая
раствору фиолетовый цвет.
2 Д. Н. Финкельштейн, указывая на невозможность полной отмывки Сг(ОН)3 от С1',
рекомендует вести восстановление формалином в азотнокислом растворе, предварительно
удалив пэимесь CU в NajCi^O, добавлением рассчитанного количества AgNO,.
3 Н. W. К о h 1 s с h u t t е г, Z. anorg. Ch. 220, 372 (1934).
Хрома окись
553
Таблица 477
Допустимые примеси в Сг(ОН)3- 2Н20
Примеси
1. Нерастворимые в НС1 вещества
2. Хлориды (СГ)
4. Щелочные и щелочноземельные
металлы (в виде сульфатов) . . .
5. Железо (Fe)
Допустимое со-
держание в °[0
0,06
0,01
0,2
0,3
0,003
Потеря при прокаливании должна быть не меньше 45,3% и не больше
57%. Содержание СгЮз должно составлять не меньше 43% и не больше 54%.
ХРОМА ОКИСЬ (ОКИСЬ ХРОМА[3])
Chromium oxydatum Chromic oxide ' Chromoxyd
Chromioxyd
Cr2Os Мол. в. 152,02
Л. Аморфная окись хрома
Приготовление
1. Прокаливают в гессенском тигле смесь 50 г К2СГ2О7 (не содержащего
железа) и 10 г серы. После охлаждения тигля массу отделяют от его стенок,
промывают водой для удаления K2SO4, отфильтровывают и сушат (Д и т р и х).
2. По Шефферу растворяют 250 г тонко истертого ЫагСггСЬ в
глицерине при нагревании на водяной бане. Полученная сиропообразная масса при
зажигании сгорает с энергичным газообразованием, оставляя СггОз.
3. Назревают в плоской чашке хромовокислый или двухромовокислый
аммоний [(NH^CKX или (ЫН4)гСг207]; наступает энергичная реакция,
сопровождающаяся самораскаливанием, и1 СгаОз бьет фонтаном в виде зеленых
рыхлых хлопьев. Поместив чашку перед началом реакции над листом бумаги,
можно собрать СггОз без потерь.
Однако по Хэрборду и Кингу1 полученный таким путем СгЮз
содержит некоторый избыток кислорода; повидимому в препарате имеется
примесь СгОз.
4. Нагревают Cr(OH)g при 120° до постоянного веса.
Свойства
Зеленый порошок, постоянный на воздухе.
Б. Кристаллическая окись хрома
Приготовление
По Шифу и Дитте смешивают равные весовые части истертых К2СГ2О7
и NaCl и омесь сильно прокаливают в гессенском тигле, засыпав сверху
слоем NaCl. После охлаждения массу выщелачивают водой, причем остается
кристаллическая СггОз.
»Harbord а. К i n g, С. II, 1921 (1938;.
554
Хром
Свойства
Черное с металлическим блеском или1 темнозеленое вещество уд. в. 5,21,
не растворимое в кислотах и щелочах. Т. пл. 2140°. Обладает магнитными
свойствами.
Испытание
По техническим условиям НЩМ 1273-43 содержание СггОз в препаратах
я. д. а. и чистом должно быть не менее 99%; наибольшие количества
допустимых примесей указаны в табл. 478.
Таблица 478
Допустимые примесив Сг203
Примеси
2. Сульфаты (S04")
Допустимое содержание в °/а
в чистом для
анализа
0,005
0,02
в чистом
0,01
0,05
ХРОМ СЕРНОКИСЛЫЙ (СУЛЬФАТ ХРОМА[3])
Chromium sulfuricum Chromic sulfate Chromsulfat
Cr2(S04)3 • I8H2O
Мол. в. 716,49
Приготовление
1. По Вейнланду и Кребсу1 растворяют 6 г х.ромовокалиевых
квасцов [KCr(S04)2« 12HiO] в 45 мл воды, прибавляют при охлаждении 45 г
концентрированной H2S04 (95%-ной) и дают раствору испариться в
эвакуированном эксикаторе при 10°.
2. Значительно лучшие результаты дает метод Хиджли, проверенный и
видоизмененный Д: Н. Финкельштейном.
В фарфоровую чашку емкостью 1 л наливают 500 мл дестиллированной
воды и туда же постепенно при помешивании стеклянной палочкой вносят
125 мл H2SO4 (уд. в. 1,84). Жидкости дают охладиться до комнатной
температуры и растворяют в ней 125 г чистых хромовокалиевых квасцов [KCr(S04)2-
• I2H2O]. Раствор фильтруют от нерастворившихся квасцов через
бумажный фильтр в большой стеклянный или фарфоровый стакан емкостью
1,2—1,5 л.
К раствору квасцов медленно, по каплям приливают из капельной воронки
320 мл H2S04 (уд. в. 1,84), 'регулируя приливание кислоты и охлаждая
стакан так, чтобы температура жидкости не поднималась выше 15—20°. Чем
ниже температура, тем выше выход продукта. Рекомендуется во время
реакции перемешивать жидкость механической мешалкой. По прибавлении всей
кислоты выпавший осадок фиолетового сернокислого хрома перемешивают
дают ему отстояться и отсасывают на воронке Бюхнера, дно которой покрыто
слоем длинноволокнистого асбеста. Продукт сильно отсасывают и промывают
абсолютным спиртом, пока промывная жидкость не будет стекать бесцветной
Тогда еще раз сильно отсасывают; затем препарат высушивают на листах
Weinland, К г eb s, Z. tnorg. Ch. 49, 165 (1906).
Хром сернокислый
555
пергаментной бумаги при комнатной температуре до удаления запаха спирта и
складывают в байку с притертой пробкой. Выход 50—55 г (65% теор.).
3. По Лёве л ю к раствору 25 г Сг(ОН)з в 63 г НГчЮз (уд. в. 1,425)
прибавляют 63 мл воды и кипятят 15 мин. По охлаждении к раствору добавляют
113 г H2SO4 (уд. в. 1,23); к холодной смеси приливают 600 г 99%-ного спирта
и сильно взбалтывают.
Выпавший фиолетовый кристаллический порошок через сутки
отфильтровывают и промывают 99%-ным спиртом. Высушивают препарат сначала в
эксикаторе над СаСЬ, затем в сушильном шкафу при 90°.
Свойства
Мелкие фиолетовые чешуйки уд. в. 1,86, растворимые в воде и не
растворимые в спирте. При нагревании до 70° фиолетовый раствор сульфата быстро
принимает зеленый цвет..
Высушенный при 100° Препарат содержит 5—6 молекул воды; полное
обезвоживание наступает только при красном калении.
Таблица 479
Удельный вес растворов Cr2(S04)3
о/о Cr.lSO*)! I
1
2
4
6
8
10
12
14
16
1
фиолетовая
модификация
1,0091
1,0191
1,0395
| 1,0604
1 1,0Я17
1 1,1034
1,1257
1,1486
1,1722
зеленая
модификация
1,0172
1,0358
1,0551
1,0751
1,09^8
1 1,1172
1,1392
1,1618
о/0 Cr,(SO<)3
18
20
22
24
26
28
30
35
40
фиолетовая 1
модификация 1
1
1,1966
, 1,2218
1 2479
1,2750
1,3032
1,3325
1 —
1 —
1 "
*\5
зеленая
модификация
1,1851
1,2091
1,2339
1,2594
1 1,2856
1,3125
1,3401
1,4123
1,4893
Испытание
По ОСТ 10909-40 препарат должен содержать 20,7—21,0% Сг и 53,7—
57,6% SCV; наибольшие количества допустимых примесей указаны в
табл. 480.
Таблица 180
Допусти 1иые примеси в Cr2(S04)3• 6Н20
Примеси
1. Нерастворимые вещества ....
2 Хлориды (СК)
3. Щеючние и щелочноземельные
металлы (в виде сульфатов) . . .
4. Железо (Fe)
Допустимое содержание в о/0
в чистом для
ана шза
0,01
0,005
од
0,003,
в чистом
0,02
0,005
0,3
0,006
556
Хром
ХРОМ ХЛОРИСТЫЙ (ХЛОРИД ХРОМА[2])
Chromium bichloratum Chromous chloride Chromchlorur
Chromochlorid
А. Безводный хлористый хром
СгСЬ Мол. в. 122,92
Приготовление
По Д ё р и н г у 1 восстанавливают совершенно высушенный в струе хлора
CrCls абсолютно сухим» свободным от Ог, водородом при 420°.
Свойства
Бесцветные блестящие иглы уд. в. 2,75. Т. пл. 824°. Соль растворяется в
воде, свободной от кислорода, с синим цветом. На -воздухе раствор жадно
поглощает кислород и окрашивается в зеленый цвет, зависящий от
растворенной хлорокиси хрома.
Б. Гидрат хлористого хрома
СгС12-4Н20 Мол. в. 194,99
Приготовление
1. По Ре кур а растворяют уксуснокислый хром (закисный) в
эквивалентном количестве дымящей НС1, охлаждают до 0° и прибавляют 1—1,5 об.
дымящей НС1 (или насыщают раствор чистым хлористым водо'родом, свободным
от кислорода). Выпадающие кристаллы СгСЬ^НгО отфильтровывают и высу«
шивают.
2. Для приготовления раствора СгС12 восстанавливают сильно солянокислый
раствор CrCls гранулированным цинком. Так как раствор быстро поглощае*
кислород, необходимо хранить его под тонким слоем бензина. Однако и за*
щищенный таким путем раствор со временем разлагается.
Свойства
Прозрачные синие кристаллы, легко окисляющиеся при доступе кислорода,
ХРОМ ХЛОРНЫЙ (ХЛОРИД ХРОМА[3])
Cromium chloratum Chromic chloride Chromchlorid
Chromichlorid
А. Безводный хлорный хром
CrCh Мол. в. 158,38
Приготовление
1. По Бурьону2 пропускают SO* над СггОз, полученной прокаливанием
осажденного Сг(ОН)з при 400°, повышают температуру постепенно до красного
каления и охлаждают в токе сухого хлористого водорода.
2. 125 г влажной3 СггОз смешивают с 50 г сажи и густым крахмальным;
клейстером в тесто, из которого формуют шарики диаметром <в 10—15 мм
1 D 6 г i n g, J. prakt Ch. 66, 77 (1902).
■ р. В о ur i о n, Ann. Chim. Pliys. (8) 21, 56 <J010).
8 См. стр. 553. Хрома окись, Приготовление, способ 1.
Хром хлористый и хлорный 557
Последние высушивают при умеренном шагревании, помещают в гессенский
тигель и обильно засыпают угольным порошком.
Тигель плотно закрывают железной крышкой и накаливают 15 мин. на
сильной газовой горелке или в каскадной тигельной печи. По остывании
кусочки отделяют от угольной пыли, складывают в гессенский тигель и
накрывают другим тиглем такой же величины, в дне которого просверлены два
отверстия. Через одно из них проходит фарфоровая трубка Л, доходящая почти
до дна нижнего тигля; через второе проходит короткая
изогнутая стеклянная трубка В (рис. 89).
Обе трубки обмотаны асбестовым шнуром и
вставляются в отверстия вполне плотно. .Плотность между краями
тиглей достигается • прокладкой между ними полоски
асбестовой бумаги, тщательной обмоткой асбестовым шнуром,
пропитыванием обмотки ж^идким стеклом и просушиванием.
Тигли помещают в каскадную или криптольную
тигельную печь, закрытую двумя кусками толстой жести с
полукруглыми вырезами так, чтобы верхний тигель выдавался
из печи наружу. Сначала нагревают прибор в токе ССЬ,
пока «а стеклянной трубке не останется следов влаги.
Тогда повышают температуру насколько возможно и
пропускают по трубке А хлор, избыток которого, выходящий
из Ву поглощают раствором NaOH.
По охлаждении в верхнем тигле оказывается возошан-
к ли СгСЬ в виде фиолетовых блестящих кристаллов, не
растворимых в воде. Его быстро промывают водой и высу-
• шивают при 100° (Вёлер).
3. Д. И. Рябчиков и В. М. Шульман1
рекомендуют действовать смесью СО и СЬ на СгЮз при
температуре ниже 950°. Можно также подвергать действию СЬ смесь 2 ч. СггОз и
1 ч. животного "угля.
Рис.89.11 риоор для
получения
хлорного хрома
Свойства
Фиолетовые, сильно блестящие листочки уд. в. 2,9. Т. пл. около 1150°.
Почти не растворим в воде; при длительном кипячении об'разует зеленый
раствор. При прокаливании на воздухе переходит в СггОз; в струе хлора
возгоняется.
Б. Гидрат хлорного хрома
СгСЬ-6Н20 Мол. в. 266,48
Различают три модификации водного хлорного хрома общей формулы
СгС1з-6Н20:
а) темнозеленую — [СгС12(ШО)4]С1 • 2НЮ;
б) светлозеленую — [СгСЦНгСЭДСЬ- ШО;
в) фиолетовую— [Сг(Н20)о]С1з.
Все три модификации в водном растворе превращаются в смесь темнозе-
леной и фиолетовой модификаций. Этот переход длится от 18 до 40 дней в
зависимости от внешних условий. При 25° через 14—15 дней растворы
содержат около 42% фиолетовой и около 58% темнозеленой модификаций.
а) Темнозеленая модификация
Получение
- Предварительно проанализированный гель окиси хрома (содержащий свыше
90% воды) растворяют в крепкой НС1 (уд. в. 1,19), взяв последнюю из
расчета 350 мл на каждые 100 г СггОз, содержащиеся в геле. Через несколько
1 ЖПХ 7 N ', 1162 (1934),
$58
Хром
часов после растворения всего количества геля раствор отсасывают через
полотно на воронке Бюхнера. Фильтрат выпаривают на водяной бане до
Vs объема (жидкость должна иметь консистенцию сиропа). Упаренный раствор
переливают в стеклянный сосуд, охлаждаемый водой или снегом, и насыщают
сухим хлористым водородом, пропущенным через промывную склянку с
концентрированной H2S04 и через U-образную трубку с СаСЬ. Трубка,
подводящая НС1, должна оканчиваться расширением в виде воронки во избежание
засорения кристаллами СгСЬ-бНгО. Насыщение ведут до тех пор, пока
раствор не станет очень густым от выпавших кристаллов.
Смесь пе'реносят на лед и оставляют на сутки для окончания
кристаллизации.
Выделившиеся кристаллы тщательно отсасывают и высушивают между
30 и 40° (при более высокой температуре кристаллы расплываются). Сухие
кристаллы хранят в банке с притертой пробкой.
Свойства
Изумрудно-зеленые кристаллы, растворимые в воде (раствор сине-зеленого
цвета). Не растворим в концентрированной НС1. Т. пл„ 83°.
б) Светлозеленая модификация
Получение
-■- По В е р н е р у и Г у б з е р у * 100 г хромового ангидрида (СгОз)
смешивают с 400 мл концентрированной НС1, нагревают в течение часа и
выпаривают при беспрестанном пропускании хлористого водорода.
Затвердевающий через несколько часов сырой хлорид промывают
небольшим количеством ледяной воды, растворяют в равном по весу количестве
воды и полученный раствор насыщают хлористым водородом. Через 3—4 часа
отсасывают выпавшие кристаллы и высушивают их над H2SO4. После
1—2-дневного сушения растирают кристаллы с сухим ацетоном для удаления
удержанной НС1, отсасывают и промывают ацетоном.
Свойства
Светлозеленые гигроскопические кристаллы, легко растворимые в воде и
трудно в спирте.
Не растворим в ацетоне, эфире и концентрированной НС1.
в) Фиолетовая модификация
Получение
Раствор 50 г зеленой модификации в 50 мл воды кипятят полчаса в
колбе с обратным холодильником, затем охлаждают в охладительной омеси и
насыщают хлористым водородом. После сливания сине-зеленого раствора
оставшиеся кристаллы промывают концентрированной НС1 (уд. в. 1,20—1,22),
отсасывают на асбесте и отжимают на глиняной пластинке; наконец растирают
кристаллы с ацетоном, фильтруют^ и промывают им же, пока фильтрат не
перестанет окрашиваться в зеленый цвет.
После испарения удержанного ацетона продукт растворяют в 20 мл воды,
фильтруют й снова насыщают хлористым водородом.
Кристаллы, выделившиеся после стояния раствора на льду, отсасывают на
полотняном фильтре, промывают ацетоном и сушат на глиняной пластинке в
эксикаторе над H2SO4.
Выход 12 г.
1 A. W е г п е г, AI. O-u b s е г. Вег. 34, 1691 (1901).
Хромал хлористый
559
Свойства
Серо-фюлетовый, весьма гигроскопичный кристаллический порошок. Не
растворим в ацетоне. Т. пл. 95°. Растворим в воде с тусклой
фиолетово-зеленой окраской.
Таблица 481
Удельный вес водных ра ство р ов СгС13- 6Н20
°/о CiCl8
1
2
4
6
<*18
фиолетовая
модификация
1,0076
1,0166
1,0349
1,0535
темно-
зеленая
модификация
1,0071
1,0157
1,0332 /
1,0510
о/о СгС!.
8
10
12
1 н
d4
фиолетовая
модификация
1,0724
1,0917
1 1,1114
1,1316
темно-
зеленая модифи*
кация
1,0691
1,0876
1,1065
Испытание
По ОСТ НКТП 7172/466 наибольшие количества допустимых примесей
указаны в табл. 482.
Таблица 482
Допустимые прямее и в СгС13• 6Н20
Примеси
1. Нерастворимые вещества ....
3. Железо (Fe)
4. Щелочные и щелочноземельные
металлы (в виде сульфатов) . . .
Допустимое содержание в °/0
в чистом для
анализа
0,005
0.05
0,002
0,2
в чистом
0,03
0.2
0,005
0,5
ХРОМИЛ ХЛОРИСТЫЙ
(ХРОМИЛХЛОРИД, ХЛОРОХРОМОВЫЙ АНГИДРИД)
Chromyl chloride
Chromium oxychloride
Chromylchlorid
Chromoxychlorid
CrOfCl»
Мол. в. 154,92
Приготовление
i 1.Молес и Гомец1 предлагают помещать смесь хорошо высушенных и
тонко истертых 50 г NaCI и 80 г КгСггО? в тубулированную реторту емкостью
1,5 л, снабженную тубулированным приемником, соединенным с промывной
* Е. М о 1 е в, L. О о m e z, Z. physlk. Ch. 80, 613; РгЗр. Ch. 713.
660
Царская водка. Цинк
склянкой с H2SO4. К смеси малыми порциями прибавляют 150 г дымящей
H2SO4. Все части прибора должны быть предварительно тщательно высушены.
Уже на холоду начинается реакция с обильным выделением хлора и па- '
ров СгОгС12. Приемник следует охлаждать проточной водой или снегом. При
легком нагревании реторты на воронке Бабо реакция ускоряется и
продолжается без вспучивания массы. Как только образование пены превратится,
нагревание прерывают и аппарат охлаждают.
Выход (по хлору) 50%.
2. Дрэйк-Ло и Моллуо-Перкин1 приводят следующий метод:
50 г хромового ангидрида (СгОз) растворяют в 170 мл концентрированной
НС1 и при охлаждении добавляют к раство'ру 100 мл концентрированной
H2SO4 порциями по 20 мл. .Цают смеси стоять 20 мин. в^ делительной
воронке, после чего отделяют нижний слой, представляющий собой Сг02СЬ,
просасывают через него воздух в течение часа и перегоняют.
Для получения больших количеств хлористого хромила лучше всего
приготовлять его порциями, исходя каждый раз из 50 г СгОз.
3. По ЭрДману2 в гессенском тигле сплавляют при умеренной
температуре 200 г Хромовокислого калия (К2СЮ4) с 122 г NaCl. Сплавленную
массу выливают на железный лист и после остывания разбивают на
небольшие куски.
Последние помещают в объемистую реторту, снабженную обратным
холодильником, и обрабатывают смесью 66 мл дымящей H2SO4 (уд. в. 1,906) и
134 мл концентрированной H2SO4 (уд. е. 1,84)3, приливая ее по каплям.
Реакция вначале идет очень бурно; когда она ослабеет, присоединяют
холодильник -и нагревают реторту, пока в приемник не перестанут капать
бурые капли. Для очистки препарат перегоняют вторично.
Хранить СгОгСЬ следует в запаянных сосудах.
Свойства
Тёмнокрасная, дымящая на воздухе жидкость уд. ib. 1,92. Т. пл. — 96,5°, #
т. кип. 116,7°. Водой быстро разлагается с образованием СгОз и HCI; с
горючими веществами (сера, фосфор, спирт) взрывает со вспышкой.
ЦАРСКАЯ ВОДКА
Acidum chloronitrosum Aqua regia Konigwasser
Acidum chloronitricum Nitrohydrochloric acid Salpeter-Salzsaure
Aqua regia
Приготовление
Смешивают 3 ч. концентрированной НС1 (уд. в. 1,19) с 1 ч. HNCb (уд. в. ,
1,38—1,40). Смесь следует готовить перед самым употреблением.
Свойства
Бесцветная, быстро желтеющая, очень едкая жидкость с запахом хлора.
ЦИНК
Zincum ' Zinc Zink
2n At. в. 65,38
Приготовление
1. Для получения цинка, свободного от As, Л'От рекомендует сплавлять в
гессенском тигле 1 кг продажного цинка и погружать в расплавленный металл
(на самое дно тигля) куски безводного MgCk, удерживая их плотной желез-
* Н. Drake-Low, F. М о 11 w о-Р е г k i n, J. Chem. Soc. 91. 191 (1907).
■ Хим. преп. 63.
« Или 200 мл 100%-иой HeSO,.
Царская водка. Цинк 661
■ой спиралью до полного растворения. Мышьяк при этом улетучивается в
виде lAsCla.
£ Ни всю операцию расходуется 15 г MgCb. Тигель несколько охлаждают и
юыливают металл тонкой струей в сосуд с водой; полученные гранули высу*
рпивают в теплом месте *.
§:■■.. 2. Для удаления As M е й л ь е п'редлагает сплавлять в гессенском тигле
Йинк, смешанный послойно с 25% по весу КЫОз.
у 3. Для получения химически чистого цинка рекомендуют применять
комбинацию методов 1 и 2.
<?. 500 г- цинка расплавляют в шамотовом титле и в жидкую массу вносят
ф г сухого NH4CI. Затем повышают температуру и добавляют небольшими
Порциями 10 г сухого KNOs, после чего снова прибавляют 5 г NFhCl. Ждут
пока шлак не затвердеет, снимают его и расплавленный металл гранулируют.
Высушенные гранули смешивают с 25% (по весу) чистого KNOs и
помещают смесь в шамотовый тигель, дно которого покрыто слоем KNOs; массу
засыпают сверху KNOs и нагревают. Когда горение превратится, окисление
примесей можно считать законченным; дают шлаку затвердеть, снимают его
и металл выливают в холодную воду. Для очистки от слоя окиси металл
вносят в тигель с расплавленной бурой (ЫащВЮ?).
, Выход около 250 г.
4. Для получения цинка в порошке выливают расплавленный металл в
нагретую железную ступку и тотчас по затвердевании превращают в пыль
сильными ударами пестика.
5. Хороший метод получения цинка без мышьяка предложил Н. Петров;
метод основан на адсо'рбции As из раствора ZnS04 свежеосажденным Fe(OH)s.
К 50 мл 48%-ного раствора ZnS04 добавляют 1 мл 0,2/V раствора Fe^SO*)*,
Перемешивают и прибавляют затем 0,25 г чистого ZnCO*. После тщательного
перебалтывания отфильтровывают осадок Fe(OH).3 и ZnCOe, содержащий весь
мышьяк, и подвергают раствор электролизу. Полученный металл
удовлетворяет требованиям ОСТ для препарата ч. д. а.г
6. Для получения активированного цинка, легко 'растворимого в кислотах»
покрытие его слоем Pt или Си (например, добавлением к кислоте
нескольких капель раствора PtCU или CuSOi) нецелесообразно.
Лучше всего сплавить Zn с 1—1,5% безводного MgCb; полученный цинк,
содержащий Мя, очень активен и рекомендуется, например, для определения
As (Шэ).
Свойства
Блестящий голубовато-белый металл уд. в. 6,88—7,19 (в зависимости от
Обработки), постоянный в сухом воздухе. Во влажном воздухе покрывается
пленкой Zn(OH)2 или — при доступе СОг — основной углекислой соли. Образую*
щаяся тонкая серая пленка защищает металл от дальнейшего окисления.
Т. пл. 419,44°, т. кип. 907°.
Легко раство'рим в кислотах и в горячем растворе КОН, особенно в при*
Сутствии порошка железа.
Цинковая пыль содержит обычно 80—90% Zn, 5—15% ZnO, переменные
количества Cd, Pb и Fe, иногда небольшие количества As, Sb, Cu, SiOt;
всегда присутствует около 0,4% нитрида цинка, ZmNs (Гмелин)*
Испытание
По ГОСТ 989-41 цинк металлический без мышьяка должен представлять
собой гранули весом не более 2 г каждая; препарат должен выдерживать
приведенные в ГОСТ испытания на отсутствие мышьяка и на пригодность
для определения мышьяка; наибольшие количества допустимых примесей
указаны в табл. 483.
* Достичь пол того удаления As этим методом не удается*
а Изв. ИРЕА 16, 1Ш (193У)*
$62
Цинк
Таблица 483
Допустимые примесив Zn
Примеси
1. Фосфор
2. Сера •
3. Вещества, окисляемые КМп04,
(в пересчете на железо)
4. Вещества, не растворимые в
серной кислоте
Допустимое
содержание в °/о
0,0005
0.01
0,006
0,04
Испытание цинковой пыли (Мерк)
Азот (N). 20 г препарата растворяют в смеси 20 мл H2SO4 (уд. в. 1,84) и 200 мл
воды. Иногда растворение идет с большим трудом; в этом случае
целесообразно добавить каплю раствора хлорной платины, не содержащей HNOa. К
раствору добавляют 100 мл 30%-ного раствора NaOH, не содержащего азота,
и перегоняют около 75 мл жидкости, собирая дестиллат в приемнике,
содержащем 10 мл воды и 2—3 мл 0.27V раствора НС1. Содержимое приемника
титруют 0,27V раствором КОН с метилоранжем в качестве индикатора. Для
нейтрализации аммиака должно итти не больше 0,3 мл 0,2Д/ раствора НС1.
ЦИНКА АМАЛЬГАМА
Zinc amalgam
Zinkamalgam
Приготовление
1. Зг гранулированного цинка нагревают до 90-^100° со 100 г Hg«3—5 мл
разбавленной H2SO4. После охлаждения отделяют твердые куски сплава Zn-Hg,
а жидкую амальгаму промывают водой.
2. Для получения амальгамированного цинка 240 г мелко
гранулированного цинка обрабатывают ЗЛ/ НС1 в течение полуминуты; затем добавляют
100 мл воды и б мл насыщенного раствора HgCb. Вскоре выделение Нг
прекращается. Продолжают перемешивать цинк с раствором еще 3 <мни., после*
чего амальгамированный металл промывают дестиллированной водой1.
Свойства
Жидкая или тве'рдая масса (в зависимости от содержания Цинка) с ме*
таллическим блеском. Сильный восстановитель2.
Zincum-Ammonium
chloratum
ЦИНК-АММОНИЙ ХЛОРИСТЫЙ
Zinkammoniumchlorld
Zinc ammonium
chloride
ZnCk • 3NH4CI Мол. Ь 296,79
Приготовление
По Бэкстеру и Лэмби8 для приготовления двойной соли ZnCb • 3NH4C1
растворяют в воде эквивалентные количества ZnCh (136,29 ч.) и NH4C1
» Н. W. S t о пе, С. В е е э о n, Ind. Eng. Chera., Anal. Ed. 8, *& 3, 138 (1936).
■ Исследора ие восстановительных свойств цинковой амальгамы см. Н. W. Stone,
D.N.Hume, Ind. Eng. Chera., Anal. Ed. 11, 5<J8 (L939).
•Baxter, Lamby, Am. Cbem. J. 31, 229 (1904); Prep. CU. 652*
Цинка амальгама* Цинк* аммоний хлористый. Цинка гидрат окиси 663
$160,5 ч.), выпаривают раствор до насыщения и охлаждают. Выпавшие
кристаллы отсасывают и высушивают на воздухе*
Свойства
Красные ромбические кристаллы, устойчивые «а воздухе.
ЦИНКА ГИДРАТ ОКИСИ (ГИДРООКИСЬ ЦИНКА)
Zincum hydroxy- Zinc hydroxide , Zinkhydroxyd
datum
2n(OH)* Мол. в. 99,40
А* Аморфный гидрат окиси цинка
Приготовление
£ Осаждают Zn(N03)2 несколько меньшим, чем требуется по расчету, количе-
{CfrBOM КОН и тщательно промывают образовавшийся осадок. При
употреблении избытка КОН или при замене Zn(NCh)2 хлористой или сернокислой солью
препарат получается недостаточно чистым.
Свойства
Белый рыхлый порошок с переменным содержанием воды. При
нагревании до красного каления переходит в ZnO.
Б. Кристаллический гидрат окиси цинка
Приготовление
1. По Вилю вносят тонко истертый средний углекислый цинк (ZnCOa),
Полученный действием СОг на ZnO в присутствии воды, в двойное против
"теоретического количество 0,Ш или 0.05N раствора КОН. После взбалтывания
[через .несколько минут начинают выпадать мелкие призматические кристаллы,
ьрост которых удобно наблюдать под микроскопом. Кристаллизация
заканчивается через 20—30 мин. Если употреблять основной углекислый цинк, то
Образование 'кристаллов происходит значительно медленнее.
2. По Гуд рману1 к 50 мл Ш раствора КОН из бюретки медленно
прибавляют по каплям IN раствор ZnS04j выпадающий вначале осадок снова
растворяется; затем жидкость мутнеет и при внесении затравки—>кристал«
Лика Zn(OH)2 — оседает тяжелый зернистый порошок.
Свойства
Мелкие длинные призматические кристаллики уд. в. 3,08, после
высушивания при 40—50° отвечающие формуле Zn(OHb П*ри 100° уже начинает
терять-воду. В воде почти не растворяется (l^'lCr^/o при 29°)
ЦИНК ЙОДИСТЫЙ (ИОДИД ЦИНКА)
Zincum jodatum Zinc jodide Zinkjodid
ZnJa Мол. в. 319,22
fl р-иготов л ение
По Ванино2 настаивают 1 ч. порошка цинка и 3 ч. иода с 10 ч. воды
До растворения иода. Раствор упаривают до консистенции сиропа «и затем
Дают ему испаряться в вакуум-эксикаторе над H2SO4.
1 G о u d г I a n, Rec. Trav. chlm. Pays-Baa 39» 505 (1920).
• Prap. Ch. 653.
564
Цинк
Свойства
Прозрачные гигроскопические октаэдрические кристаллы уд. в. 4,70, очень
легко растворимые в воде, спирте и эфире (в спирте легче, чем в эфире).
Т. пл. 446°.
Таблица 484
Растворимость ZnJ2 в воде
(D i e t г)
Р С
0
18
40
э/о ZnJt
81,11
81.20
81,66
Р С
60
80
100
о/о ZnJt
82,37
83,п5
83,62
Таблица 485
Удельный вес водных растворов ZnJ2
(К г е m e г s)
о/о ZnJa
17,7
31,69
42,65
55.81
19,5
19,5
1,1715
1,3486 1
1,5121
1.7815
о/о ZnJ,
' 63,5
69.F8
76.0
Л9.5
*19.5
1,9746
2,1^53
2,3976
Испытание
По ОСТ 3008 в препарате, в зависимости от степени чистоты, допускаются
наибольшие количества примесей, указанные в табл. 486.
Таблица 486
Допустимые примеси в ZnJ2
Примеси
Допустимое содержание в */«
в чистом для
анализа
0,005
0.10
Од 05
0,^05
0,01
0.0005 1
в чистом
0,01
0.2
0,01
0,02
0,05
0,001
1. Нерастворимые в воле вещества .
2. Щелочные соли (в виде сульфатов)
3. Сульфаты (SO/7)
4. Вещества, восстанавливающие иод
5. Свобсдный иод и иодаты
6. Железо (Fe)
^ 'Цинк йодистый. Цинка окись 565
ЦИНКА ОКИСЬ (ОКСИД ЦИНКА)
Zincum oxydatum Zinc oxide Zinkoxyd
ZnO Мол. в. 81,38
Приготовление
1. В фарфоровой чашке нагревают до кипения профильтрованный раствор
11 ч. кристаллической соды (№2СОз« ЮНгО) в 100 ч. воды и постепенно
прибавляют к нему при непрерывном кипячении и перемешивании
профильтрованный раствор 10 ч. ZnS04-7H20 в 40 ч. воды. Если отстоявшаяся
жидкость не показывает щелочной реакции, добавляют немного NaaCOs и
снова нагревают. Когда осадок хорошо отстоится, промывают его несколько
раз декантацией, пока промывная вода не перестанет мутнеть от добавления
Ba(N03)2.
• Основной углекислый цинк отжимают, высушивают и переводят в ZnO
прокаливанием в тонкой фарфоровой чашке при перемешивании шпателем.
Реакция закончена, когда охлажденная проба растворяется в разбавленной
H2SO4 без выделения СОг.
Выход 20% по весу от взятого ZnSCVTtbO.
2. Мил и ус и Ф р о м для получения чистой ZnO предлагают следующий
метод. Из капельной воронки приливают профильтрованный раствор Zn(NOs)t
в ЫНЮН, содержащий 10% Zn*', к 8—10-кратному количеству кипящей
воды, которая струей пара поддерживается в непрерывном движении.^олба
снабжена холодильником, в котором конденсируются пары.
Получившийся вследствие гидролиза плотный кристаллический осадок
желтоватого цвета представляет собой ZnO, содержащую немного NHs и NOa'. При
прокаливании осадка получают чистую ZnO. Иногда препарат имеет красноватый
цвет; окраска зависит, однако, не от примеси посторонних металлов, а от
образующегося соединения неизвестного состава (вероятно, содержащего азот),
которое только при сильном прокаливание переходит в ZnO (см. п. 4).
3. По Деферу 125 г металлического Zn растворяют в 500 г НС1
(уд. в. 1,4), прибавляют 8 г концентрированной HNOs и кипятят для
окисления железа. Затем выпаривают досуха, растворяют остаток в воде и, добавив
10 г свежеосажденного СаСОз, оставляют на сутки.
Отфильтровывают осадок, содержащий все железо, и прибавляют к
фильтрату NH4OH до прекращения выпадения осадка Zn(OH)2.l Фильтруют,
промывают осадок Zn(OH)2, высушивают его без доступа СОг и нагревают до
красного каления, причем остается чистая ZnO.
4. При растворении обычной ZnO в расплавленном NH4NO3 получают
красную модификацию окиси цинка2.
Свойства
Белый с желтоватым оттенком порошок уд. в. 5,42. Термостоетс; при
нагревании становится лимонно-желтым, принимая при охлаждении прежний цвет.
Возгоняется при 1800°. Водородом не восстанавливается.
И спытание
М ю р р е й указывает следующие наибольшие количества допустимых
примесей в окиси цинка (табл. 487).
1 Избыток NH4OH недопустим вследствие образования растворимых комплексных
аммиакатов цинка.
■ Иолр>бно см. A. Kutzelni?, Z. an->rg. Ch. 203, 23(1932); Н. В е г g, Z. anorg. Ch.
2С9, 328(1932); A. Kut zelnie, Z. anorg. Ch. 224. 46 (ИЗ)).
О других методах получения чисюй ZnO см. R. Sch older u. О. Hendrlch. Z.
•norg. Ch. 241, 77 (19!9); O. H. H fl t t i g, H. E. T s ch a k er t u. H. KI t t e 1. Z. anorg. Cn. 223.
248 (1935); A. Scbleede, JVLRichter, W. S с h m i d t, Z. anorg. Ch. 223, 60 (1936).
Цинк
Таблица 48?
Допустимые примесив ZnO
Примеси
1. Вещества, не растворимые в H2S04
2. Мышьяк (As)
3. Сульфаты (S04")
4. Хлориды (СП
5. Нитраты (N0'3)
6. Карбонаты (СО/7)
7. Кальций (Са) ,
8. Магний (Mg)
9. Тяжелые металлы
10. Щелочи (NaOH)
11. Фосфаты (Р20Б)
Допустимое содержание в о/0
в чистом для
анализа
0,0001
0.CU075
0,0050
0,0j25
0.001
0.000
0020
0,0050
0,0'Ю
0,0001
0,0020
в чистом
0.0001
0.0015
0.050
0,010
0,003
—
0.020
0,010
0,000
0,4000
0,002
ЦИНК СЕРНИСТЫЙ (СУЛЬФИД ЦИНКА)
Zincum sulfuratum Zinc sulfide Zinksulfid
ZnS Мол. в. 97,44
А. Обыкновенный сернистый цинк
Приготовление
1. По Тредвеллу1 к слабокислому раствору соли цинка в эрленмейе-
ровской колбе прибавляют ЫагСОз до появления неисчезающей мути.
Последнюю растворяют в нескольких каплях ЫНЮН, прибавляют на каждые
100 мл жидкости 5 г роданистого или уксуснокислого аммония и небольшой
избыток ввежеприготовленного бесцветного (ГМНфБ; колбу наполняют почти
до верха прокипяченной водой, закрывают пробкой и оставляют на
12—24 часа.
Затем жидкость декантируют; к осадку приливают раствор, содержащий
в 100 мл 5 г роданистого (или уксуснокислого) аммония и 2 мл (NHO2S;
взбалтывают, дают осесть и фильтруют. Осадок промывают на фильт'ре
сначала вышеуказанным раствором, затем водой, содержащей (NH^S.
Промытый осадок высушивают.
2. Химически чистый ZnS по Томашеку2 можно получить следующим
методом: 10%-ный раствор продажного ZnS04 смешивают с небольшим
количеством HN0.3 и, подогрев, пропускают H2S до сильного равномерного
помутнения жидкости от выпадающего ZnS. После непродолжительного
отстаивания раствор фильтруют, лучше всего через мембранный фильтр; совершенно
чистый фильтрат подкисляют небольшим количеством HNOa и некоторое
время кипятят.
Затем нейтрализуют аммиаком до начала выпадения осадка, обычно
имеющего грязно-белый цвет. Его отфильтровывают и фильтрат подкисляют
небольшим количеством H2SO4 (до слабокислой реакции) и, подогрев,
подвергают электролизу током около 2 в и 0,1 а
1 Аналит. химия, И П), 111 (1931),
5 Ann, Pbys. 65, 189 (1921),
Цинк сернистый
867
Профильтрованный раствор смешивают с избытком ЫНЮН (до
растворения выпадающего сначала осадка), дают длительное время стоять и
фильтруют.
В нагретый фильтрат пропускают №S до насыщения; после некоторого
стояния отфильтровывают горячий раствор и осадок ZnS промывают горячей
дестиллированнои водой до тех пор, пока через фильт'р не начнет проходить
мутная жидкость. После высушивания получается белый пылеобразный
порошок вполне чистого ZnS.
3. Растворяют окись цинка в разбавленной НС1, кипятят раствор с
небольшим количеством HNOs и осторожно насыщают чистым аммиаком.
К теплому раствору добавляют многосернистый аммоний (из расчета 5 мл
на 100 г ZnO), причем выпадают тяжелые металлы и немного ZnS. После
отстаивания жидкость декантируют и фильтруют через мембранный фильтр
для полного удаления нерастворимых примесей. В полученный прозрачный
раствор пропускают H*S до полного осаждения ZnS. Дают отстояться,
прозрачную жидкость сливают сифоном, осадок отсасывают на нутч-фильтре и
высушивают при 40—50° в сушильном шкафу.
Во все время работы препарат следует оберегать от пыли.
Свойства
Белый порошок уд. в. 2,9—3.1 (аморфный) или 4,06—4,09
(кристаллический), почти не растворимый в воде (6,88* 10^%, по другим данным 1,43*
♦ Ю-60/,). Т. пл. около 1800° (под давлением 100—150 ат)% т. возгонки 1182°
(по другим данным 1064°). Темнеет на солнечном свету; при длительном
стоянии во влажном воздухе переходит в ZnS04.
Б. Фосфоресцирующий сернистый цинк
Приготовление
Добавляют к 200 г сухого ZnS 65 г смеси, полученной из следующих
компонентов: 20 г КО, 20 г NaCl, 8 г NH4CI, 48 г S, 0,04 г хлористой меди
в виде аммиачного раствора.
Все препараты должны быть безукоризненной чистоты и хорошо высушены.
После тщательного перемешивания помещают смесь в кварцевый тигель,
крышку которого плотно замазывают глиной, и прокаливают 21/6—3 часа
сначала при 550—650°, затем при 800° и, наконец, при 1000°.
Спекшийся королек вынимают из тигля горячим, очищают острым шпате*
лем от шлака, осторожно растирают и просеивают через шелковое сито.
Свойства
После освещения дневным или искусственным светом препарат светится
в темноте бледнозеленым светом. Продолжительность и сила свечения
зависят от чистоты исходных продуктов. ^
В. Коллоидальный сернистый цинк
Приготовление
По Мюллеру к Ъ мл раствора 7,5 г чистого ZnS04»7H20 в 1 л воды
добавляют 20 мл глицерина. К раствору прибавляют смесь из 10 мл глицерина
и 10 мл жидкости, полученной взбалтыванием 130 мл чистого глицерина
(уд. в. 1,265) с 25 мл желтого сернистого аммония до исчезновения
слизистого осадка.
При взбалтывании всей смеси происходит обесцвечивание, причем
жидкость остается совершенно прозрачной и в отсутствии воздуха прозрачность
сохраняется долгое время.
Описанный метод дает хорошие результаты только в том случае, если
не было взято избытка сернистого аммония. Если поместить жидкость в
неплотно закрытый сосуд, то уже через % часа замечается вверху слабая
опалесценция; через сутки вся1 жидкость делается молочно-белой и через
трое суток начинается выпадение хлопьев ZnS.
668
Цинк
ЦИНК СЕРНОКИСЛЫЙ (СУЛЬФАТ ЦИНКА, ЦИНКОВЫЙ КУПОРОС1)
Zincum sulfuricum Zinc sulfate Zinksulfat
ZnS04.7H20 ' Мол. в. 287,55
Приготовление
1. К 5—6 частям воды о фарфоровой чашке приливают 5 ч. концентр иро-
ванной H2SO4, прибавляют 31/а—4 ч. обрезков цинка ri дают смеси
стоять на воздухе (вне помещения) или под хорошей тягой («может
выделяться чрезвычайно ядовитый AsHa). Когда пройдет бурный период реакции,
жидкость слегка подогревают до прекращения выделения водорода, после чего
фильтруют.
Обычно жидкость кроме ZnS04 содержит Fe304 и поэтому с KsFe(CN)e
дает голубоватое или зеленое окрашивание. Остальные примеси (Pb, Cu, Cd,
As) остаются нерастворенными в виде черных хлопьев; As также частью
удаляется в виде Asbta.
Для окисления Fe" "н осаждения Fe,,# раствор отфильтровывают от
избытка цинка и прибавляют растертую с водой перекись свинца (РЬОг), пока
отфильтрованная проба жидкости не покажет отсутствия железа. При этом
FeS04 и РЬОг образуют нерастворимый PbS04 и основной1 сульфат железа.
Дают твердой фазе осесть, фильтруют, подкисляют фильтрат серной
кислотой и выпаривают до кристаллизации.
2. Растворяют ZnO в разбавленной H2SO4 до насыщения, затем добавляют
небольшой избыток ZnO, кипятят до осаждения железа в виде Fe(OH)a и
фильтруют. Фильтрат слабо подкисляют H2SO4, выпаривают и кристаллизуют.
Свойства
Бесцветные ромбические кристаллы уд. в. 1,96, постепенно
выветривающиеся в сухом воздухе. При 39° плавится в кристаллизационной воде,
переходя в ZnS04 • 6Н2О; при 250—270° обезвоживается; при яркокрасном кале*
нии разлагается на ZnO и SOa. Очень легко растворим в воде; не растворим
в спирте.
Таблица 488
Растворимость ZnS04 в воде
Iе С
0
10
15
25
о/о ZnS04
29,4
32,0
33,4
36,6
ГС
35
39
50
|
о/о ZnS04
39,9
41,2
43,1
t9 С
70
80
100
о/о ZnS04
47,1
46,2
44,0
Таблица 489
Удельный вес водных растворов ZnSO<
о/о ZnS04
2
4
6
8
d20
а4
1.0190
1.О103
1,0620
1,0842
о/0 ZdS04
10
12
*4
16
-7
1,1071
1,1308
1,1553
1,1806
| о/0 ZnS04
20
25
30
■ *
1,232
1,304
1,378
Название относится только к еми одному гидрату.
Цинк сернокислый и углекислый
569
Испытание
А: Наибольшие количества примесей, допустимых по ОСТ 5460, указаны
в табл. 490.
Допустимые примеси в ZnS04• 7Н20
Таблица 490
1
1 Прнмеси
1
г 1. Нерастворимые в воде вещества
1 2. Щелочные соли и Mg (в виде
Г'З. Хлориды (СК) '. . . .
! 4. Вещества, осаждаемые аммиаком
6. Медь (Си)
Допустимое содержание в °/0
в химически
чистом
0.003
0.05
o.coi
0.005
0,001
0.002
О.0ОЭ05
в чистом для
анализа
0,01
0.1
0.002
0,02
0.002
0,005
О.ОСОЗ
в чистом
0,02
0,2
0,005
0.04
0,005
0.01
0.0005
Кроме того, препарат испытывается па кислотность,
ЦИНК УГЛЕКИСЛЫЙ (КАРБОНАТ ЦИНКА)
Zincum carbonicum
Zinc carbonate
А. Нормальный углекислый цинк
Zinkcarbonat
ZnCOs
Мол. в. 125.39
Приготовление
1. По Кр ауту к охлажденному до 3—4° раствору 100 г ZnS04 в 3,5 л
воды приливают также охлажденный и насыщенный СОз раствор 140 г
КНСОз (или 117 г №НСОз).
Полученный плотный зернистый осадок возможно быстро отсасывают,
промывают ледяной водой и отжимают.
2; К 700 мл охлажденного до 3° 0,1/V раствора ZnSOi (приливают при
помешивании 300 мл также охлажденного насыщенного раствора КНСОз,
через который предварительно в течение длительного времени пропускался СОг.
Дают смеси стоять 3—4 дня на холоду (ниже 10°) и 2—3 дня при
комнатной температуре, пока рыхлый вначале осадок не превратится в
кристаллический порошок. Осадок промывают водой декантацией до удаления SO/' и
высушивают1 при комнатной температуре или при 130°.
Свойства
Аморфный порошок, при промывании водой частично, а при кипячении
с водой полностью переходящий в основную соль.
Б. Основной углекислый цинк
Приготовление
Вливают тонкой струей при постоянном помешивании раствор 2 ч. ZnSOi-
• 7Н20 в кипящий раствор 9 ч. кристаллической соды (Na2COs • 10НЮ). Смесь
iQ.P.Hflttlg, A.ZCrner u.O. H h e vK о vek у, Mo natsh. Ch. 72,31 (1938); С. И, 3256 (1W9)*
570 Цинк
кипятят 5—10 мин.» затем дают отстояться и фильтруют. Осадок промывают
посредством декантации горячей водой до удаления SO4" (в промывных водах
не должна появляться муть от прибавления раствора ВаСЬ), отсасывают
отжимают и сушат при 100°.
Свойства
Белый порошок уд. в. 4,40, очень трудно растворимый в воде (0,05—0,03%),
При 140° диссоциирует, причем образуются ZnO, C02 и Н20.
ЦИНК УКСУСНОКИСЛЫЙ (АЦЕТАТ ЦИНКА)
Zincum aceticum Zinc acetate Zinkacetat
Essigsaures Zink
Zn(CH3COO)**2HaO Мол. в. 219,50
Приготовление
1. Растворяют Zn, ZnO или ZnCOa в разбавленной CKUCOOH при 60°.
2. Смешивают растворы 100 г ZnS04-7H*0 и ПО г РЬ(СНзСОО)2 • ЗШЭ.
Отфильтровав PbS04, осаждают избыток РЬ' ' сероводородом.
3. Для получения безводного гп(СНзСОО)г нагревают смесь -10,2 г
азотнокислого цинка Zn(N03>2 • 6Н2О и 40 мл уксусного ангидрида [(СНзСОО)20];
после окончания реакции кипятят 15 мин. Охлаждают, отсасывают белую
кристаллическую массу, промывают ее уксусным ангидридом и эфиром и
сушат в вакууме над КОН или H2SO4. Выход 95% теоретического.
Свойства
Шестисторонние блестящие мелкие чешуйки или таблички уд. в. 1,73. При
100° теряет воду, переходя в гп(СНзСОО)2 уд. в. 1,840. Около 244°
плавится, при более высокой температуре разлагается с выделением ацетона. Под
уменьшенным давлением (150 мм) ацетат цинка возгоняется без разложения
п,ри 200°.
Испытание
Наибольшие количества примесей, допустимых по ОСТ 3006, указаны
в табл. 491.
Таблица 491
Допустимые примеси в Zn(CH3COO)2.2Н20
Примеси
1. Нерастворимые в воде вещества
2. Щелочные металлы (в виде суль-
фаюв)
3. Хлориды (СК)
4. Сульфаты (SO/)
5. Свинец (РЬ)
6. Медь (Си)
7. Железо.(Fe)
8. Мышьяк (As)
Допустимое содержание
в химически
чистом
0,003
0,05
0,001
0,^05
1 0,002
0,002
0,0002
| 0,00005
в чистом для
анализа
0,005
0,1
0,002
0.01
0,005
0,005
0,0005
0,0001
В о/о
ч *
в чистом
0,01
0,2
0.005
0,02
0 01
0/;(
0,001
0,00.02
Цинк уксуснокислый. Цинкгуранил уксуснокислый S7f
ЦИНК-УРАНИЛ УКСУСНОКИСЛЫЙ
(АЦЕТАТ ЦИНКА И У РАНИ Л А, ЦИНК-УРАН И Л-АЦЕТАТ)
Zinc-uranyl acetate Zinkuranylacctat
Приготовление1
1. Растворяют при нагревании 10 г уксуснокислого уранила в б г 30%-ной
СНзСООН и разбавляют раствор 50 мл воды. Отдельно растворяют 30 г
уксуснокислого цинка в 30 г 30%-ной СНзСООН и 50 мл воды. Смешав обе
жидкости горячими, оставляют омесь на сутки, после чего прозрачный желтый
раствор отфильтровывают.
2. Для регенерирования ацетата цинк-уранила из остатков после
определения Na' В. Я. Тартаковский2 предлагает следующий метод. Остатки
помещают в большой фарфоровый стакан, нагревают и, добавив HNOs (уд. в.
1,4) до растворения осадка, выпаривают до появления кристаллической корки.
На другой день выделившиеся кристаллы отсасывают, снова растворяют
при нагревании в воде (взятой в таком количестве, чтобы она только
покрывала кристаллы) и оставляют на ночь для кристаллизации.
Кристаллы отбрасывают, а фильтраты от 1 и 2 кристаллизации соединяют,
добавляют к ним 10—15% по объему концентрированной HNOs, разбавляют
водой и осаждают при нагревании избытком NH»OH. Фильтруют через
воронку Бюхнера, промывают осадок разбавленным ЫНЮН и, растворив в HNOs,
снова переосаждают NH4OH. Переосаждение проводят 3 раза; бесцветные
фильтраты выливают, а осадок, состоящий из ураната аммония, загрязненного
гидроокисями Fe'" * и АГ ", высушивают при 60—70°.
Для очистки полученную массу растирают, обливают в фарфоровом стакане
800 мл воды и при нагревании добавляют 120—150 г (NH^COa, продолжая
нагревать и перемешивать до полного растворения ураната.
Осадок Fe(OH)s и А1(ОН)з отфильтровывают; к фильтрату добя*ит*гпТ
понемногу HNOe до кислой реакции и осаждают при нагревании ЫНЮН.
Полученный уранат аммония (NHO2U2O7 высушивают при 60—70°.
Для приготовления раствора ацетата цинк-уранила к 20 г (NH^lbCh
приливают 150 мл воды и 22 мл 60%-ной СНзСООН и нагревают при
перемешивании до полного растворения. Одновременно растворяют 68 г ацетата цинка
в смеси 75 мл воды я 5 мл СНзСООН. Оба раствора смешивают горячими,
составляют на сутки и отфильтровывают от осадка.
Свойства
В растворах, содержащих Na*, реактив дает желтый кристаллический
осадок тройного ацетата, NaZn(UC>2)3 • (СШСОО)в • 9Н20. Рекомендуется
сохранять раствор в темном месте.
ЦИНК ХЛОРИСТЫЙ (ХЛОРИД ЦИНКА)
Zincum chloratum Zinc chloride Zinkchlorid
А. Безводный хлористый цинк
ZnCb Мол. в. 136,29
Приготовление
1. Совершенно чистый нейтральный безводный ZnCb получается по Б эк-
стеру и Лэмби3 нагреванием цинк-аммония хлористого (ZnCb• 3NH4C1)
в струе хлористого водорода. Предварительно освобождают ZnCl2*3NH4Cl от
1 I. M. Kolthoff, Z. anal. Cb. 70, 397 (1927); I. M. Kolthoff a. Barber, J. Am.chem. Soc
;a=. ли9Я\« И М Кплктгпт П^^иныи miiai ГУТЫ Ю(0 - тт —_ ел
* 1. iU. l\OHBUii, «-• enai. \-.u. iv, u^/ yiati), i. m. I\ О I l П О I I 8. DlTI
SO, 1623 (1928); И. М. Кольтгоф, Объемный анализ, ГХТИ. 19J2, т. II, сто
* Завод. Лаб. 9, № 11-12, 1356 (19 Ш). ' V
«Baxter, Lamb у, Am. cbera. J. 31, 229 (1904\ Z. an?, cb. 20, 1662 (19(
641.
(1907),
572
Цинк
удержанной воды, для чего его тонко истирают и сушат в эксикаторе в
течение нескольких дней, сначала над H2SO4, затем над Р2Ов.
Высушенную соль нагревают 1—2 часа в фарфоровой лодочке, помещенной
в трубку тугоплавкого стекла, в токе сухого хлористого водорода. Вначале
нагревание ведут осторожно, позднее —на сильном пламени двух бунзеновских
горелок. Оставшаяся в лодочке соль представляет собой чистейший
безводный ZnCb.
2. Окись цинка прокаливают при 800—900° и обрабатывают при
нагревании 5 ч. НС1 (1:1). В раствор для полного удаления меди подвешивают
пластинку металлического цинка.
Через 2 дня фильтруют, насыщают в течение получаса хлором и кипятят
с чистой ZnO (на 100 г ZnCl* 2 г ZnO). Раствору дают отстояться и
фильтруют; фильтрат подкисляют чистой 2%-ной НС1 и выпаривают до уд. в.
1,25—1,3. По охлаждении отфильтровывают осадок РЬС12, фильтрат
выпаривают и нагревают при 300—G600 до полной прозрачности жидкости, после
чего безводный ZnCl2 разливают в формы.
3. Препарат, содержащий только немного воды, получается следующим
методом *:
Вносят в колбу 7—8 ч. цинковых обрезков, 30 ч. воды и маленькими
порциями добавляют 30 ч. НС1.
Так как одновременно с водородом выделяется крайне ядовитый
мышьяковистый водород AsH3, то колбу на время растворения помещают под тягу.
Когда реакция замедлится, ее ускоряют, нагревая колбу на водяной или,
лучше, на паровой бане (осторожно с огнем! Водород!).
Количества цинка и НС1 должны быть такие, чтобы часть металла
осталась нерастворенной и содержащиеся в цинке РЬ, Си и Cd остались в шламе.
Дают колбе стоять сутки в теплом месте, изредка встряхивая; затем
фильтруют. Фильтрат, содержащий ZnCb и FeCU, для окисления железа насыщают
хлором и дают ему стоять в плотно закупоренной банке 12—24 часа. По
истечении этого времени после открывания п-робки должен ощущаться сильный
запах хлора, и проба жидкости -не должна давать синего осадка с KsFe(CN)e.
При нагревании на водяной бане прибавляют к жидкости такое количество
растертой с водой окиси цинка, чтобы все железо выпало в виде Fe(OH)a.
Затем фильтруют и фильтрат после сильного подкисления НС1 выпаривают
досуха в фарфоровой чашке на песочной бане при помешивании фарфоровым
шпателем. Вначале можно выпаривать быстро, однако, как только масса'
станет кашеобразной, следует уменьшить пламя, чтобы избежать распадения
ZnCb • 2Н2О и образования значительных количеств основной соли.
В этой стадии выпаривания прибавляют немного чистой НС1, продолжают
выпаривание досуха при умеренном нагревании и складывают полученную
соль в заранее нагретые, плотно закрывающиеся банки. Если препарат
окрашен в серый цвет (что зависит от попадания органических примесей — пыли,
волосков фильтра и др.), то растворяют его в царской водке и снова
выпаривают досуха.
4. В фарфоровую лодочку помещают гранулированный металлический цинк
и нагревают лодочку, помещенную в тугоплавкую стеклянную трубку, в
трубчатой печи под сильной струей сухого хлора. Свободный конец трубки
соединяют с баночкой, предназначенной для хранения препарата. При температуре
красного каления образуется безводный ZnCb, возгоняющийся в
присоединенный приемник. По окончании реакции пропускают через аппарат ток
сухого воздуха, после чего банку с ZnCU плотно закрывают.
v.
Свойства
Белый порошок уд. в. 2,92 или (плавленый) прозрачная фарфоровидная
масса. Чрезвычайно гигроскопичен (влажный воздух, пропущенный через
трубку с ZnCU, содержит только 0,98 мг НЮ на 1 л2); легко растворим в воде,
» Komml. z.Deutsch.Arznelb. II, Б55.
t J. H. Bo v е г, Bur. Stand. J. Research. 12, 241 (1934).
Цинк хлористый
673
спирте, эфире и глицерине. Т. пл. 283°, т. кип. 730°; при калильном жаре
образует густой белый дым возгоняющегося ZnCb.
Таблица 492 .
Растворимость ZnCla вводе (D let z) '
f С
0
10
20
26
°/о ZnClg
67,5
73,1
78,6
80,9
Г с
40
60
80
1 100
о/в ZnCla
81,9
83,0
84,4
86,0
Таблица 493
•/о ZnCIt
2
4
6
8
10
12
14
Удельный вес водных растворов ZnCl?
л20
! 4
1,0167
1,0350
1,0532
1,0715
1,0819
1,1085
1,1275 1
°/с ZnCl,
16
18
20
25
30
35
40
| -Г
1,1468
1,1665
1,1866
1,2380
1,2928
1,3522
1,4173
о/о ZnClt
45
50
55
60
65
70
*
1,4890
1,5681
1,655
1,749
1,851
1,962
Испытание
По ОСТ НКТП 7173/467 содержание цинка в препарате (в пересчете на
ZnCU) не менее 98%; наибольшие количества допустимых примесей указаны
в табл. 494.
Препарат должен выдерживать следующее испытание на хлорокись цинка:
10 г препарата растворяют в 80 мл воды (в цилиндре с притертой
пробкой), прибавляют 10 мл \N HC1, 5 капель 0,1%-ного раствора конгорот и от-
титровывают избыток НС1 щелочью, добавляя ее по каплям при сильном
встряхивании.
Должно пойти для препарата ч. д. а. не более 3,0 мл, IfiN HC1, а для
препарата чистого не более 6,0 мл.
Таблица 494
Допустимые примеси в ZnCl?
ПрвмеСж
1. Нерастворимые вещества ... *
4. Металлы, осаждаемые H2S . ♦ . .
5. Щелочные и щелочноземельные
металлы (в виде сульфатов) . . .
Допустимое содержание в »/0
в чистом для
анализа
0,005
0 01
0,001
0,001
0,05
в чистом
0,01
0,03
0,002
0,002
0*2
т
Цинк
Б. Гидрат хлористого цинка
ZnCl2.iy2H20 Мол. в. 163,32
Приготовление
Кристаллический ZnCh^l^HsO получается, если сиропообразный раствор
ZnCU смешать с концентрированной НС1 и оставить для кристаллизации.
Свойства
Крупные, хорошо образованные бесцветные призмы, расплывающиеся на
воздухе. Т. пл. 26°. Устойчив только в интервале 12,5—26°. Очень легко рас*
творим в воде, спирте и глицерине. *
Отв. редактор Л # Вахаровский
Техн. редактор М. С, Лурье
Сдано в производство 15/1 1946 г. А 05305 Подписано к печати 26V/I1 1946 г.
Формат бумаги 60X92Vie Печ. л, 36 Учетноизд. л. 61.5*
Типогр. знаков в 1 печ. листе 67080. Цена 40 руб. Тираж 15000 экз. Зак. № 1364-
4-я типография им. Евг. Соколовой треста сПолиграфкнига» ОГИЗа
црн Совете Министров ООСР. Ленинград, Измайловский щр., 20.
К читателю
Прежде чем пользоваться книгой, исправьте следующие опечатки
Стр.
52
73
87
120
144
180
204
215 1
215
$23
276
298
306
328
334
348
394
466
504
510
513
539
Строка или
таблица
табл. 34 п. 1
11 снизу
25 снизу
табл. 88, столбец 3
5 сверху
табл. 152, столбец 3
33 снизу
табл. 211, столбцы 3 и 7
табл. 211, столбцы 4 и 8i
7 сверху
сноска 1
23 сверху *
6 сверху
7 снизу
5 сверху
12 сверху
табл. 385
9 сверху
3 снизу
5 сверху
Рис. 74
16 снизу
Напечатано
0,02 _
Те02
BaPt (CH)4
14,373
Мол. в. 128,76
28
Мол. в. 78,11
на 1 ч. воды
в 1 мл
' Мол. в. 906,44
HN02, Cl2B2 и J2
т. кип. 39°
Из фильтра
Мол. в. 197,88
Мол. в. 277,48
AsaSs в СН3СООН
содержит
СН2СООН
(NH4)2Sn
восстанавливает
1 буквы Л и В поменять
; железного фосфора
Должно бить
0,002
ТеОв
ВаР1(СГ*)4
15,373
Мол. в. 126,76
38
Мол. в. 78,10
на 100 ч. воды
в 100 мл
Мол. в. 906,64
HN08, Cl* Brj и J3
т. кип. 39° при 50 мм
Из фильтрата
Мол. в. 197,91
Мол. в. 277,51
I AsjA в СН8СООН
содержит г
СН8СООН
(NH^
окисляет
местами
1 желтого фосфора